WO2018193482A1 - Improved method for the synthesis of 1,2-ethanediamine, n, n'-bis(2-aminoethyl)-dihydrochloride(trientine dihydrochloride) - Google Patents
Improved method for the synthesis of 1,2-ethanediamine, n, n'-bis(2-aminoethyl)-dihydrochloride(trientine dihydrochloride) Download PDFInfo
- Publication number
- WO2018193482A1 WO2018193482A1 PCT/IN2018/050243 IN2018050243W WO2018193482A1 WO 2018193482 A1 WO2018193482 A1 WO 2018193482A1 IN 2018050243 W IN2018050243 W IN 2018050243W WO 2018193482 A1 WO2018193482 A1 WO 2018193482A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- trientine
- ethane
- dihydrochloride
- treating
- diylbis
- Prior art date
Links
- ZFOZEAYEEUISFP-UHFFFAOYSA-N CC(C)(C)OC(N(CCN(CC#N)C(OC(C)(C)C)=O)CC#N)=O Chemical compound CC(C)(C)OC(N(CCN(CC#N)C(OC(C)(C)C)=O)CC#N)=O ZFOZEAYEEUISFP-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/44—Preparation of compounds containing amino groups bound to a carbon skeleton by reduction of carboxylic acids or esters thereof in presence of ammonia or amines, or by reduction of nitriles, carboxylic acid amides, imines or imino-ethers
- C07C209/48—Preparation of compounds containing amino groups bound to a carbon skeleton by reduction of carboxylic acids or esters thereof in presence of ammonia or amines, or by reduction of nitriles, carboxylic acid amides, imines or imino-ethers by reduction of nitriles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/62—Preparation of compounds containing amino groups bound to a carbon skeleton by cleaving carbon-to-nitrogen, sulfur-to-nitrogen, or phosphorus-to-nitrogen bonds, e.g. hydrolysis of amides, N-dealkylation of amines or quaternary ammonium compounds
Definitions
- the present invention describes an improved process for the preparation of substantially pure N, N'-bis (2-aminoethyl)-l,2-ethanediamine dihydrochloride, also known as Trientine dihydrochloride.
- Trientine dihydrochloride (I) is N, N'-bis (2-aminoethyl)-l,2-ethanediamine dihydrochloride. It is marketed in US as SYPRINE (250 mg) capsules for oral administration. Trientine dihydrochloride is a chelating compound for removal of excess copper from the body to treat Wilson's disease, particularly in those who are intolerant to penicillamine.
- Trientine dihydrochloride was reported in many patents and non-patent literature. The contents of which are hereby incorporated as reference in their entirety.
- Trientine dihydrochloride by treating 70% triethylenetetramine with di-isopropyl ether and ethanol, followed by drop wise addition of ethanolic hydrochloride at 0-10°C, and filtered the salt.
- the salt was washed with di-isopropyl ether: ethanol (1: 1) and dried in desiccator at 50°C-60°C to obtain 70-75% of trientine dihydrochloride with purity 99.6% having melting point range between 118°C-
- Trientine quaternary salt to the desired secondary salt in the prior art processes suffer limitation such as application of high temperature, use of large volumes of solvent and repeated distillation and washing, making the process very tedious. Also, use of excess amount of hydrochloric acid may result in formation of inorganic impurities.
- the present inventors have reported a simple, improved and efficient method for the synthesis of pharmaceutical grade of Trientine dihydrochloride with desired purity and the process being feasible on commercial scale.
- Trientine dihydrochloride (I) having a purity of greater than 99.5% by HPLC.
- Another object of the invention is to provide novel process for the preparation of Trientine dihydrochloride by blending pure Trientine tetrahydrochloride with pure Trientine free base.
- an improved process for the preparation of substantially pure Trientine dihydrochloride (I) having a purity of greater than 99.5% comprising the steps of: a) reacting 2-chloroacetonitrile (VI)
- the base employed in step a) and step e) of the above described process for the preparation of Trientine dihydrochloride is selected from the group comprising of sodium hydroxide, potassium hydroxide, potassium carbonate, sodium carbonate, potassium bicarbonate, sodium bicarbonate, triethylamine, pyridine or the like.
- the protecting group employed in step b) of the above described process for the preparation of Trientine dihydrochloride is selected from the group comprising of fluorenylmethyloxy carbonyl (Fmoc), ie/t-butyloxycarbonyl (Boc), phthalimide, toluene sulfonyl (Ts), methane sulfonyl (Ms), triphenylmethyl (Trityl), carboxybenzyl (CBZ) or the like
- step c) of the above described process for the preparation of Trientine dihydrochloride is carried out in presence of Raney nickel and methanolic ammonia.
- the protic solvent used in the above described process for the preparation of substantially pure Trientine dihydrochloride (I) having a purity of greater than 99.5% is selected from the group comprising of acetone, water, methanol, ethanol, isopropyl alcohol (IPA), n-propanol, n-butanol or mixture thereof.
- a crystalline form of Trientine dihydrochloride characterized by X-ray powder diffraction (XRPD) pattern having peaks expressed as 2 ⁇ values at about 9.91, 13.36, 16.62, 19.92, 20.26, 20.72, 23.38, 23.79, 24.01, 24.55, 24.78, 25.07, 25.8310, 26.76, 27.03, 27.34, 27.70, 28.67, 28.89, 29.91, 30.08, 31.26, 31.75, 33.68, 33.89, 34.25, 37.39, 38.00, 40.50, 40.70, 40.98, 41.84, 42.69, 42.90, 44.49, 45.04, 46.53, 48.26 and 49.49+ 0.2 degrees.
- XRPD X-ray powder diffraction
- a crystalline form of Trientine dihydrochloride characterized by X-ray powder diffraction (XRPD) pattern having peaks expressed as 2 ⁇ values at about 9.91, 13.36, 16.62, 19.92, 20.26, 20.72, 23.38, 23.79, 24.01, 24.55, 24.78, 25.07, 25.8310, 26.76, 27.03, 27.34, 27.70, 28.67, 28.89, 29.91, 30.08, 31.26, 31.75, 33.68, 33.89, 34.25, 37.39, 38.00, 40.50, 40.70, 40.98, 41.84, 42.69, 42.90, 44.49, 45.04, 46.53, 48.26 and 49.49+ 0.2 degrees, wherein the said crystalline form is prepared by a process comprising steps of: a) reacting 2-chloroacetonitrile (VI)
- the inventors of the present invention have surprisingly carried out an improved process for the preparation of Trientine dihydrochloride (I) having a purity of greater than 99.5% by
- an improved process for the preparation of substantially pure Trientine dihydrochloride (I) having a purity of greater than 99.5% comprising the steps of: a) reacting 2-chloroacetonitrile (VI)
- Step a) involves the addition of 2-chloroacetonitrile (VI) to ethane- 1,2-diamine (VII) in the presence of a base in a suitable aprotic solvent at 25-30°C to obtain 2,2'-(ethane-l,2- diylbis(azanediyl) diacetonitrile (V).
- Step b) proceeds by protecting the amino groups of intermediate (V) using suitable protecting groups.
- the reaction filtrate obtained in step a) containing intermediate (V) was cooled and reacted with a suitable protecting group.
- the final residue was washed with different volumes of a suitable aprotic solvent and dried under vacuum to obtain Di-tert-butyl ethane- l,2-diylbis(cyanomethylcarbamate) (IV).
- the protecting group employed in step b) of the above described process for the preparation of Trientine dihydrochloride is selected from the group comprising of fluorenylmethyloxy carbonyl (Fmoc), ie/t-butyloxycarbonyl (Boc), phthalimide, toluene sulfonyl (Ts), methane sulfonyl (Ms), triphenylmethyl (Trityl), carboxybenzyl (CBZ) or the like
- Step c) proceeds with the reduction of the cyano group of intermediate (IV) with a suitable base and catalyst, preferably Raney-nickel was used as a catalyst.
- a suitable base and catalyst preferably Raney-nickel was used as a catalyst.
- the residue so obtained was treated with fumaric acid dissolved in a protic solvent and heated at 50-70°C.
- the reaction mixture was cooled and the solid so formed was washed with a mixture of protic solvents and an aprotic solvent to obtain Di-tert-butyl ethane- l,2-diylbis(2- aminoethylcarbamate) difumarate (III).
- hydrogenation process of step c) of the above described process for the preparation of Trientine dihydrochloride is carried out in presence of Raney nickel and methanolic ammonia.
- Step d) involves deprotection of amino group from intermediate (III),and conversion offumarate salt to Trientine tetrahydrochloride.
- Intermediate (III) was reacted with concentrated hydrochloric acid and heated at 50-70°C. On completion of reaction, reaction mass was cooled to yield Nl, Nl '-(ethane- 1,2-diyl) bis(ethane-l,2-diamine) tetrahydrochloride (II).
- Intermediate (II) was purified by treating with mixture of solvents selected from the group comprising methanol, ethanol, isopropyl alcohol, n-butanol, tert- butanol, acetonitrile, water or the like or combination of any of these solvents.
- Step e) proceeds with the preparing Trientine free base (Ila) by dissolving intermediate (II)in a suitable protic solvent and in presence of a suitable base.
- the reaction mass was cooled, stirred for 15 to 16 hrs at 25-30°C and filtered.
- a suitable protic solvent was added to the filtrate and distilled the solvent.
- the residue was washed with a suitable aprotic solvent, filtered through Hyflo and distilled under vacuum to obtain Trientine free base (Ila).
- Step f) proceeds with reacting intermediate (II) with Trientine free base (Ila).
- the reaction mass was then distilled off and the crude was cooled to 25-30°C.
- the crude was further treated with suitable protic solvents and isolated to obtain pure 1,2-ethanediamine, N, N'- bis(2-aminoethyl)-dihydrochloride (I) which is greater than 99.5% pure by HPLC.
- the base employed in step a), step c) and step e) of the above described process for the preparation of Trientine dihydrochloride is selected from the group comprising of potassium carbonate, sodium carbonate, potassium hydroxide, sodium hydroxide, sodium bicarbonate, potassium bicarbonate, triethylamine, pyridine, aqueous ammonia, methanolic ammonia, group of metal alkoxides comprising of sodium methoxide, sodium ethoxide; potassium methoxide, potassium ethoxide, sodium butoxide, potassium butoxide, potassium-t-butoxide, sodium tertiary butoxide or the like .
- potassium carbonate, methanolic ammonia, sodium hydroxide, sodium carbonate and sodium methoxide were used in the present invention.
- the conversion of the Trientine tetrahydrochloride (II) to Trientine dihydrochloride (I) using Trientine free base (Ila) is innovative and advantageous over other processes as less undesired impurities are formed resulting in formation of 1,2-ethanediamine N, N'-bis (2-amino ethyl)-dihydrochloride (I) with purity greater than 99.5% and with 88% yield, and thus avoiding any tedious purification process.
- the advantage of the present invention is the preparation of Trietnine dihydrochloride by adding of Trientine free base toTrientine tetrahydrochloride.
- Trientine free base can be prepared by using Trientine tetrahydrochloride, which is subsequently used to convert Trientine tetrahydrochloride salt to dihydrochloride salt.
- Trientine tetrahydrochloride was converted to its free base and re-converted to dihydrochloride by adding two equivalents of hydrochloric acid.
- the other advantage of the present invention is that, we could control the hydrochloric acid content in the final dihydrochloride salt of Trientine.
- the suitable aprotic solvent used in step a), step b), step c), step d) and step e) is selected from the group comprising acetone, acetonitrile, 1,4- dioxane, diethyl ether, dichloromethane, ethyl acetate, ⁇ , ⁇ -dimethylformamide, methyl tertiary butyl ether, hexane, cyclohexane, toulene, tetrahydrofuran or the like; preferably acetone, acetonitrile, dichloromethane, ethyl acetate and cyclohexane.
- suitable protic solvent used in step b), step c), step d) step e) and step f) were selected from a group comprising of acetone, water, methanol, ethanol, isopropyl alcohol (IPA), n-propanol, n-butanol or the like or mixture thereof; preferably water, methanol, ethanol and isopropyl alcohol.
- IPA isopropyl alcohol
- Trientine dihydrochloride characterized by X-ray powder diffraction (XRPD) pattern, as shown in Figure 1, having peaks expressed as 2 ⁇ values at about 9.91, 13.36, 16.62, 19.92, 20.26, 20.72, 23.38, 23.79, 24.01, 24.55, 24.78, 25.07, 25.8310, 26.76, 27.03, 27.34, 27.70, 28.67, 28.89, 29.91, 30.08, 31.26, 31.75, 33.68, 33.89, 34.25, 37.39, 38.00, 40.50, 40.70, 40.98, 41.84, 42.69, 42.90, 44.49, 45.04, 46.53, 48.26 and 49.49 ⁇ 0.2 degrees.
- XRPD X-ray powder diffraction
- a crystalline form of Trientine dihydrochloride characterized by X-ray powder diffraction (XRPD) pattern having peaks expressed as 2 ⁇ values at about 9.91, 13.36, 16.62, 19.92, 20.26, 20.72, 23.38, 23.79, 24.01, 24.55, 24.78, 25.07, 25.8310, 26.76, 27.03, 27.34, 27.70, 28.67, 28.89, 29.91, 30.08, 31.26, 31.75, 33.68, 33.89, 34.25, 37.39, 38.00, 40.50, 40.70, 40.98, 41.84, 42.69, 42.90, 44.49, 45.04, 46.53, 48.26 and 49.49+ 0.2 degrees, wherein the said crystalline form is prepared by a process comprising steps of: a) reacting 2-chloroacetonitrile (VI)
- reaction mass was allowed to settle for 20-30 min at 25-30°C and then filtered through Hyflo.
- the filtrate was distilled under vacuum and 200 mL of isopropyl alcohol was added to the residue.
- the solvent was distilled under vacuum below 45° C and the residue obtained was cooled to 25-30°C.
- 700 mL of isopropyl alcohol and lOOg of fumaric acid dissolved in 300mL of methanol were added to the residue at 25-30°C.
- the reaction mass was heated for 45-60 min at 60-65°C and then cooled to 25-30°C. It was further cooled to 0-5°C for 5-6 hrs.
- the precipitated solid was filtered and washed with chilled isopropyl alcohol: methanol solvent mixture (7:3) and 200 mL of chilled acetone and dried under vacuum.
- the solid so obtained was further dissolved in a mixture of (7:3) isopropyl alcohol: methanol at 25-30°C, heated for 60-90 min at 65-70°C.
- the reaction mass was then cooled to 25-30°C and stirred for 90-120 min. Further the reaction mass was cooled to 0-5°C for 3-4hrs.
- the solid so obtained was washed with a mixture of chilled isopropyl alcohol: acetone (1: 1), then with chilled acetone at 10-15°C and dried. 8 volumes of methanol were added to the wet cake at 25-30°C and heated at 70-75°C. The reaction mixture was cooled to 0-5°C and stirred for 60-90 mins. The solid so obtained was filtered under vacuum and washed with chilled acetone and dried under vacuum. Again, the solid was heated with 3 volumes of water at 45-50°C for 20-30 min. To the mixture 10 volumes of ethanol was added and cooled to 0- 5°C.
- reaction mass was maintained for 10-12 hrs at 0-5°C.
- the solid so formed was filtered, washed with 2 volumes of chilled acetone to obtain Nl, Nl'-(ethane-l,2-diyl) bis (ethane- 1 ,2-diamine)tetrahydrochloride (II) .
- Nl Nl '-(ethane- 1,2-diyl) bis (ethane- 1,2-diamine) tetrahydrochloride (II) was dissolved in 1000 mL methanol at 25-30°C.
- the reaction mixture was cooled to 10- 15°C, and 68.4(1.71 mol) of sodium hydroxide was added to the reaction mixture.
- the reaction mass was stirred for 15 - 16hrs at 25-30°C, then filtered through Hyflo bed and the filtrate was distilled off under vacuum. 200 mL of methanol was added to the residue and the solvent was distilled off under vacuum.
- Trientine free base (Ila) was dissolved in 2000mL methanol and 82.7g (0.283 mol) Trientine tetrahydrochloride (II) was added to the reaction mass. The reaction mass was stirred for 3 hrs at 25-30°C and the pH of the reaction mass was maintained at 7.0-8.5 with addition of Trientine free base. On completion of the reaction the solvent was completely distilled off under vacuum below 45°C, degassed the crude and cooled to 25-30°C. 1200 mL ethanol was charged to the crude and stirred for 16-20 hrs at 25-30°C. The solid so obtained was cooled to 0-5°C, filtered and suck dried under vacuum at 0-5°C.
- the obtained solid was taken in 1200 mL of ethanol at 0-5°C, refluxed at 75-80°C followed by addition of water to get a clear solution. lOg of activated charcoal was added to the clear solution, stirred and filtered through Hyflo at 75-80°C. The filtrate was cooled to 25-30°C, stirred and was maintained for 16-17 hrs at 25-30°C. The solid formed was filtered, washed with chilled ethanol and dried under vacuum below 50°C to obtain 1,2-Ethanediamine, N, N'-bis(2-aminoethyl)- dihydrochloride (I).
- Trientine tetrahydrochloride (II) 500 g, 1.71 mol
- methanol (6 L) 20-25°C and stirred the reaction mass for 2 hrs at 25-30°C.
- the suspension was filtered, and the filtered cake was washed with methanol (500 mL).
- the filtrate was concentrated under reduced pressure.
- Dichloromethane (3L) was added to the mass and stirred for 10 minutes.
- the suspension was filtered, washed the filter cake with dichloromethane (500 mL).
- the filtrate was distilled off under vacuum and the mass was dissolved in methanol (7.5 L).
- Trientine tetrahydrochloride (II) 500 g, 1.71 moles
- Trientine tetrahydrochloride (II) (500 g, 1.71 moles) was added and stirred for 30 minutes at 25-30°Cand evaporated the solvent at reduced pressure, then isopropyl alcohol (2.5 L) was added and filtered the precipitated solid to give pure Trientine dihydrochloride (I)
- Trientine tetrahydrochloride (II) 500g, 1.71 mol was added and stirred for 60 minutes at 25-30°C.
- the solvent was distilled off up to 80% and cooled to 0-5 °C to precipitate pure Trientine dihydrochloride (I).
Abstract
The present invention relates to an improved method for the synthesis of substantially pure 1,2-ethanediamine, N, N'-bis(2-aminoethyl)-dihydrochloride (I). Formula (I). Ν,Ν'-bis (2-aminoethyl)-l,2-ethanediamine dihydrochloride (Trientine dihydrochloride)
Description
IMPROVED METHOD FOR THE SYNTHESIS OF 1,2-ETHANEDIAMINE, N, N'- BIS^- AMTNOETHYL)-DIHYDROCHLORIDE(TRIENTINE DIHYDROCHLORIDE)
RELATED PATENT APPLICATION(S)
This application claims the priority to and benefit of Indian Provisional Patent Application No. 201741014337 filed on April 22, 2017; the disclosures of which are incorporated herein by reference.
FIELD OF THE INVENTION
The present invention describes an improved process for the preparation of substantially pure N, N'-bis (2-aminoethyl)-l,2-ethanediamine dihydrochloride, also known as Trientine dihydrochloride.
BACKGROUND OF THE INVENTION
Trientine dihydrochloride (I) is N, N'-bis (2-aminoethyl)-l,2-ethanediamine dihydrochloride. It is marketed in US as SYPRINE (250 mg) capsules for oral administration. Trientine dihydrochloride is a chelating compound for removal of excess copper from the body to treat Wilson's disease, particularly in those who are intolerant to penicillamine.
The synthesis of Trientine dihydrochloride was reported in many patents and non-patent literature. The contents of which are hereby incorporated as reference in their entirety.
US7582796 reported process for the synthesis of Trientine dihydrochloride. The general route of synthesis reported basically involves synthesis of a dinitrile intermediate 3,3'-(ethane-l,2- diylbis(azanediyl))dipropanenitrile followed by tert-butyloxycarbonyl (Boc) protection and reduction to form tert-butyl ethane- l,2-diylbis(2-aminoethylcarbamate). Deprotection of this intermediate resulted in a mixture of triethylenetetramine primary, secondary, tertiary or quaternary salt. Further, the tertiary or quaternary salts were converted to the desired secondary salt by use of sodium methoxide, hydrochloric acid, methyl tertiary butyl ether and precipitating with ethanol.
Proc. R. Soc. Med. 1977; 70(3)10-12 describes formation of Trientine dihydrochloride by treating 70% triethylenetetramine with di-isopropyl ether and ethanol, followed by drop wise
addition of ethanolic hydrochloride at 0-10°C, and filtered the salt. The salt was washed with di-isopropyl ether: ethanol (1: 1) and dried in desiccator at 50°C-60°C to obtain 70-75% of trientine dihydrochloride with purity 99.6% having melting point range between 118°C-
122°C.
The conversion of Trientine quaternary salt to the desired secondary salt in the prior art processes suffer limitation such as application of high temperature, use of large volumes of solvent and repeated distillation and washing, making the process very tedious. Also, use of excess amount of hydrochloric acid may result in formation of inorganic impurities. To overcome these problems, the present inventors have reported a simple, improved and efficient method for the synthesis of pharmaceutical grade of Trientine dihydrochloride with desired purity and the process being feasible on commercial scale.
SUMMARY OF THE INVENTION Accordingly, it is an object of the present invention to provide an improved process for the synthesis of Trientine dihydrochloride (I) having a purity of greater than 99.5% by HPLC.
Another object of the invention is to provide novel process for the preparation of Trientine dihydrochloride by blending pure Trientine tetrahydrochloride with pure Trientine free base.
In one aspect of the invention, there is provided an improved process for the preparation of substantially pure Trientine dihydrochloride (I) having a purity of greater than 99.5%, the said process comprising the steps of:
 a) reacting 2-chloroacetonitrile (VI)
a) reacting 2-chloroacetonitrile (VI)

(VI)
with ethane- 1,2-diamine (VII)
Η,Ν
tvii)
in presence of base to for '-(ethane-l,2-diylbis(azanediyl)) diacetonitrile (V);

(V)
b) protecting free amino groups of intermediate (V) by treating with a suitable protecting group preferably i-butyl carbamate (BOC) to obtain the Di-ie/ -butyl ethane- 1 ,2-diylbis(cyanomethylcarbamate) (IV) ;
 c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-tert-butyl ethane- l,2-diylbis(2- aminoethyl .2HOOCCH=CHCOOH
c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-tert-butyl ethane- l,2-diylbis(2- aminoethyl .2HOOCCH=CHCOOH

(III)
d) deprotecting intermediate (III) by treating with a concentrated hydrochloric acid to obtain Nl, Nl '-(ethane- 1,2-diyl) diethane-l,2-diaminetetrahydrochloride (II);
H
,NH2 .4HC1
H,N *N'
H e) preparing Trientine free base (Ila) by treating intermediate (II) with a suitable base; and
H
.NH,
H,N N
H
(Ha)
f) treating Nl, Nl '-(ethane- 1,2-diyl) diethane-l,2-diaminetetra hydrochloride (II) with Trientine free base (Ila) at pH 7.0 to 8.5 to obtain 1,2-ethanediamine, N, N'- bis(2-aminoethyl)-dihydrochloride (I).
In some embodiment of the invention, the base employed in step a) and step e) of the above described process for the preparation of Trientine dihydrochloride is selected from the group comprising of sodium hydroxide, potassium hydroxide, potassium carbonate, sodium carbonate, potassium bicarbonate, sodium bicarbonate, triethylamine, pyridine or the like.
In some embodiment of the invention, the protecting group employed in step b) of the above described process for the preparation of Trientine dihydrochloride is selected from the group comprising of fluorenylmethyloxy carbonyl (Fmoc), ie/t-butyloxycarbonyl (Boc), phthalimide, toluene sulfonyl (Ts), methane sulfonyl (Ms), triphenylmethyl (Trityl), carboxybenzyl (CBZ) or the like
In some embodiment of the invention, hydrogenation process of step c) of the above described process for the preparation of Trientine dihydrochloride is carried out in presence of Raney nickel and methanolic ammonia.
In another aspect of the invention, there is provided a process for preparation of substantially pure Trientine dihydrochloride (I) having a purity of greater than 99.5%, said process comprising the steps of:
a) adding pure Trientine free base to Trientine tetrahydrochloride;
b) adjusting pH of the Trientine tetrahydrochloride to 7.0 to 8.5; and
c) isolating Trientine dihydrochloride from protic solvent.
In some embodiment of the invention, the protic solvent used in the above described process for the preparation of substantially pure Trientine dihydrochloride (I) having a purity of greater than 99.5%, is selected from the group comprising of acetone, water, methanol, ethanol, isopropyl alcohol (IPA), n-propanol, n-butanol or mixture thereof.
In another aspect of the invention, there is provided a crystalline form of Trientine dihydrochloride (I) characterized by X-ray powder diffraction (XRPD) pattern having peaks expressed as 2Θ values at about 9.91, 13.36, 16.62, 19.92, 20.26, 20.72, 23.38, 23.79, 24.01, 24.55, 24.78, 25.07, 25.8310, 26.76, 27.03, 27.34, 27.70, 28.67, 28.89, 29.91, 30.08, 31.26, 31.75, 33.68, 33.89, 34.25, 37.39, 38.00, 40.50, 40.70, 40.98, 41.84, 42.69, 42.90, 44.49, 45.04, 46.53, 48.26 and 49.49+ 0.2 degrees.
In some embodiment of the invention there is provided a crystalline form of Trientine dihydrochloride (I) characterized by X-ray powder diffraction (XRPD) pattern having peaks expressed as 2Θ values at about 9.91, 13.36, 16.62, 19.92, 20.26, 20.72, 23.38, 23.79, 24.01, 24.55, 24.78, 25.07, 25.8310, 26.76, 27.03, 27.34, 27.70, 28.67, 28.89, 29.91, 30.08, 31.26, 31.75, 33.68, 33.89, 34.25, 37.39, 38.00, 40.50, 40.70, 40.98, 41.84, 42.69, 42.90, 44.49, 45.04, 46.53, 48.26 and 49.49+ 0.2 degrees, wherein the said crystalline form is prepared by a process comprising steps of: a) reacting 2-chloroacetonitrile (VI)

(VI)
with ethane- 1,2-diamine (V

tVII)
in presence of base to for '-(ethane-l,2-diylbis(azanediyl)) diacetonitrile (V);

00
b) protecting free amino groups of intermediate (V) by treating with a suitable protecting group preferably i-butyl carbamate (BOC) to obtain the Di-ie/ -butyl ethane- 1 ,2-diylbis(cyanomethylcarbamate) (IV) ;

(IV)
c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-ie/t-butyl ethane- l,2-diylbis(2- aminoethylcarbamate) difumarate (III);
.2HOOCCH=CHCOOH

(III)
d) deprotecting intermediate (III) by treating with a concentrated hydrochloric acid to obtain N 1 , N 1 '-(ethane- 1 ,2-diyl) diethane- 1 ,2-diaminetetrahydrochloride (II) ;
H
,NH2 .4HC1
H,N
H
CIO
e) preparing Trientine free base (Ila) by treating intermediate (II) with a suitable
H
,NH2
H,N
H
base; (IIa>
f) treating Nl, N 1 '-(ethane- 1 ,2-diyl) diethane- 1,2-diaminetetra hydrochloride (II) with Trientine free base (Ila) at pH 7.0 to 8.5 to obtain 1,2-ethanediamine, N, N'- bis(2- aminoethyl)-dihydrochloride (I); and
g) isolating crystalline Trientine dihydrochloride from protic solvent.
In another aspect of the invention, there is provided a process for the preparation of crystalline form of Trientine dihydrochloride (I), characterized by X-ray diffraction spectrum having peaks expressed as 2Θ values at about 9.91, 13.36, 16.62, 19.92, 20.26, 20.72, 23.38, 23.79, 24.01, 24.55, 24.78, 25.07, 25.8310, 26.76, 27.03, 27.34, 27.70, 28.67, 28.89, 29.91, 30.08, 31.26, 31.75, 33.68, 33.89, 34.25, 37.39, 38.00, 40.50, 40.70, 40.98, 41.84, 42.69, 42.90, 44.49, 45.04, 46.53, 48.26 and 49.49+ 0.2 degrees, said process comprising the steps of:

C
a) reacting 2-chloroacetonitrile (VI)
 with ethane- 1,2-diamine (VII)
Η,Ν
with ethane- 1,2-diamine (VII)
Η,Ν
tvii)
in presence of base to for '-(ethane-l,2-diylbis(azanediyl)) diacetonitrile (V);

(V)
b) protecting free amino groups of intermediate (V) by treating with a suitable protecting group preferably i-butyl carbamate (BOC) to obtain the Di-ie/ -butyl ethane- 1 ,2-diylbis(cyanomethylcarbamate) (IV) ;
 c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-ie/t-butyl ethane- l,2-diylbis(2- aminoethyl .2HOOCCH=CHCOOH
c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-ie/t-butyl ethane- l,2-diylbis(2- aminoethyl .2HOOCCH=CHCOOH

(III)
d) deprotecting intermediate (III) by treating with a concentrated hydrochloric acid to obtain N 1 , N 1 '-(ethane- 1 ,2-diyl) diethane- 1 ,2-diaminetetrahydrochloride (II) ;
H
.N, ,NH2 .4HC1
H,N *N'
H e) preparing Trientine free base (Ila) by treating intermediate (II) with a suitable
H
,N, -NH,
H,N *N*
H
base; (IIaJ
f) treating Nl, N 1 '-(ethane- 1 ,2-diyl) diethane- 1,2-diaminetetra hydrochloride (II) with Trientine free base (Ila) at pH 7.0 to 8.5 to obtain 1,2-ethanediamine, N, N'- bis(2- aminoethyl)-dihydrochloride (I); and
g) isolating crystalline Trientine dihydrochloride from protic solvent.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 : Infrared (IR) Spectrum of Trientine dihydrochloride (I)
Figure 2: X-ray diffractogram (XRD) of Trientine dihydrochloride (I) DETAILED DESCRIPTION OF THE INVENTION
The inventors of the present invention have surprisingly carried out an improved process for the preparation of Trientine dihydrochloride (I) having a purity of greater than 99.5% by
HPLC.
In one aspect of the invention, there is provided an improved process for the preparation of substantially pure Trientine dihydrochloride (I) having a purity of greater than 99.5%, the said process comprising the steps of:
 a) reacting 2-chloroacetonitrile (VI)
a) reacting 2-chloroacetonitrile (VI)

Cvi)
with ethane- 1,2-diamine (V

tVII)
in presence of base to form 2,2'-(ethane-l,2-diylbis(azanediyl)) diacetonitrile (V);

b) protecting free amino groups of intermediate (V) by treating with a suitable protecting group preferably i-butyl carbamate (BOC) to obtain the Di-ie/ -butyl ethane- 1 ,2-diylbis(cyanomethylcarbamate) (IV) ;
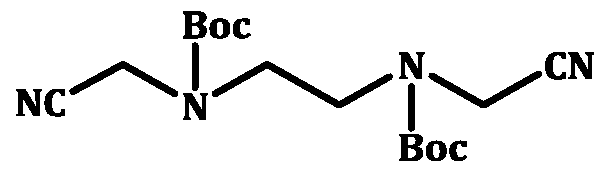 c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-tert-butyl ethane- l,2-diylbis(2- aminoethyl .2HOOCCH=CHCOOH
c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-tert-butyl ethane- l,2-diylbis(2- aminoethyl .2HOOCCH=CHCOOH

CHI)
d) deprotecting intermediate (III) by treating with a concentrated hydrochloric acid to obtain Nl, Nl '-(ethane- 1,2-diyl) diethane-l,2-diaminetetrahydrochloride (II);
H
,NH2 .4HC1
H,N N
H
(II)
e) preparing Trientine free base (Ila) by treating intermediate (II) with a suitable base; and
H
.NH,
H2N-
H
(Ha)
f) treating Nl, Nl '-(ethane- 1,2-diyl) diethane-l,2-diaminetetra hydrochloride (II) with Trientine free base (Ila) at pH 7.0 to 8.5 to obtain 1,2-ethanediamine, N, N'- bis(2-aminoethyl)-dihydrochloride (I).
The above described process for the preparation of Trientine dihydrochloride (I) having a purity of greater than 99.5% by HPLC is shown in Scheme 1.
Step a) involves the addition of 2-chloroacetonitrile (VI) to ethane- 1,2-diamine (VII) in the presence of a base in a suitable aprotic solvent at 25-30°C to obtain 2,2'-(ethane-l,2- diylbis(azanediyl) diacetonitrile (V).
Step b) proceeds by protecting the amino groups of intermediate (V) using suitable protecting groups. The reaction filtrate obtained in step a) containing intermediate (V) was cooled and reacted with a suitable protecting group. On completion of reaction, the final residue was
washed with different volumes of a suitable aprotic solvent and dried under vacuum to obtain Di-tert-butyl ethane- l,2-diylbis(cyanomethylcarbamate) (IV). The two free amine groups of intermediate (V) were protected by treating with i-butyl carbamate (Boc) anhydride to yield Di-tert-butyl ethane- 1,2-diylbis (cyanomethylcarbamate) (IV), which is further purified by preparing slurry in cyclohexane or ethyl diisopropyl ether.
In some embodiment of the invention, the protecting group employed in step b) of the above described process for the preparation of Trientine dihydrochloride is selected from the group comprising of fluorenylmethyloxy carbonyl (Fmoc), ie/t-butyloxycarbonyl (Boc), phthalimide, toluene sulfonyl (Ts), methane sulfonyl (Ms), triphenylmethyl (Trityl), carboxybenzyl (CBZ) or the like
Step c) proceeds with the reduction of the cyano group of intermediate (IV) with a suitable base and catalyst, preferably Raney-nickel was used as a catalyst. On completion of reaction, the residue so obtained was treated with fumaric acid dissolved in a protic solvent and heated at 50-70°C. The reaction mixture was cooled and the solid so formed was washed with a mixture of protic solvents and an aprotic solvent to obtain Di-tert-butyl ethane- l,2-diylbis(2- aminoethylcarbamate) difumarate (III). In some embodiment of the invention, hydrogenation process of step c) of the above described process for the preparation of Trientine dihydrochloride is carried out in presence of Raney nickel and methanolic ammonia.
Step d) involves deprotection of amino group from intermediate (III),and conversion offumarate salt to Trientine tetrahydrochloride.Intermediate (III) was reacted with concentrated hydrochloric acid and heated at 50-70°C. On completion of reaction, reaction mass was cooled to yield Nl, Nl '-(ethane- 1,2-diyl) bis(ethane-l,2-diamine) tetrahydrochloride (II). Intermediate (II) was purified by treating with mixture of solvents selected from the group comprising methanol, ethanol, isopropyl alcohol, n-butanol, tert- butanol, acetonitrile, water or the like or combination of any of these solvents. The conversion of difumarate salt (III) to Nl, Nl '-(ethane- 1,2-diyl) bis (ethane- 1,2-diamine) tetrahydrochloride (II) as described above is not reported in any prior art, which improves the purity of intermediate (II)
Step e) proceeds with the preparing Trientine free base (Ila) by dissolving intermediate (II)in a suitable protic solvent and in presence of a suitable base. The reaction mass was cooled, stirred for 15 to 16 hrs at 25-30°C and filtered. A suitable protic solvent was added to the filtrate and distilled the solvent. The residue was washed with a suitable aprotic solvent, filtered through Hyflo and distilled under vacuum to obtain Trientine free base (Ila).

(V)
Step b

D/'-tert-butyl ethane
-l,2-diylbis(cyanomethyIcarbamate) CH=CHCOOH

Di-tert-butyl ethane-l,2-diylbis
(2-amlnoethylcarbamate) dlfumarate
(III)
Step d
H
NH2 .2HC1 step f H
H, N ,NH2 .4HC1
H N H
Trientine dihydrochloride Ai1,Ai1 -(ethane-l,2-diyl)dlethane-l,2-diamine (I) tetrahydrochloride

.NH,
H2N N
H
Trientine free base
(Ila)
Scheme 1
Step f) proceeds with reacting intermediate (II) with Trientine free base (Ila). The reaction mass was then distilled off and the crude was cooled to 25-30°C. The crude was further treated with suitable protic solvents and isolated to obtain pure 1,2-ethanediamine, N, N'- bis(2-aminoethyl)-dihydrochloride (I) which is greater than 99.5% pure by HPLC.
In some embodiment of the invention, the base employed in step a), step c) and step e) of the above described process for the preparation of Trientine dihydrochloride is selected from the group comprising of potassium carbonate, sodium carbonate, potassium hydroxide, sodium hydroxide, sodium bicarbonate, potassium bicarbonate, triethylamine, pyridine, aqueous ammonia, methanolic ammonia, group of metal alkoxides comprising of sodium methoxide, sodium ethoxide; potassium methoxide, potassium ethoxide, sodium butoxide, potassium butoxide, potassium-t-butoxide, sodium tertiary butoxide or the like . Preferably potassium carbonate, methanolic ammonia, sodium hydroxide, sodium carbonate and sodium methoxide were used in the present invention.
In another aspect of the invention, there is provided a process for preparation of substantially pure Trientine dihydrochloride (I) having a purity of greater than 99.5%, said process comprising the steps of:
a) adding pure Trientine free base to Trientine tetrahydrochloride;
b) adjusting pH of the Trientine tetrahydrochloride to 7.0 to 8.5; and
c) isolating Trientine dihydrochloride from protic solvent.
In another embodiment, the conversion of the Trientine tetrahydrochloride (II) to Trientine dihydrochloride (I) using Trientine free base (Ila) is innovative and advantageous over other processes as less undesired impurities are formed resulting in formation of 1,2-ethanediamine N, N'-bis (2-amino ethyl)-dihydrochloride (I) with purity greater than 99.5% and with 88% yield, and thus avoiding any tedious purification process.
The advantage of the present invention is the preparation of Trietnine dihydrochloride by adding of Trientine free base toTrientine tetrahydrochloride. Trientine free base can be prepared by using Trientine tetrahydrochloride, which is subsequently used to convert Trientine tetrahydrochloride salt to dihydrochloride salt. Whereas in the literature all the Trientine tetrahydrochloride was converted to its free base and re-converted to dihydrochloride by adding two equivalents of hydrochloric acid. The other advantage of the
present invention is that, we could control the hydrochloric acid content in the final dihydrochloride salt of Trientine.
In some embodiment of the invention, the suitable aprotic solvent used in step a), step b), step c), step d) and step e) is selected from the group comprising acetone, acetonitrile, 1,4- dioxane, diethyl ether, dichloromethane, ethyl acetate, Ν,Ν-dimethylformamide, methyl tertiary butyl ether, hexane, cyclohexane, toulene, tetrahydrofuran or the like; preferably acetone, acetonitrile, dichloromethane, ethyl acetate and cyclohexane. In some embodiment of the invention, suitable protic solvent used in step b), step c), step d) step e) and step f) were selected from a group comprising of acetone, water, methanol, ethanol, isopropyl alcohol (IPA), n-propanol, n-butanol or the like or mixture thereof; preferably water, methanol, ethanol and isopropyl alcohol. In one aspect, there is provided a pure Trientine dihydrochloride analyzed by IR spectrum as illustrated in Figure 1.
In another aspect of the invention, there is provided a crystalline form of Trientine dihydrochloride (I) characterized by X-ray powder diffraction (XRPD) pattern, as shown in Figure 1, having peaks expressed as 2Θ values at about 9.91, 13.36, 16.62, 19.92, 20.26, 20.72, 23.38, 23.79, 24.01, 24.55, 24.78, 25.07, 25.8310, 26.76, 27.03, 27.34, 27.70, 28.67, 28.89, 29.91, 30.08, 31.26, 31.75, 33.68, 33.89, 34.25, 37.39, 38.00, 40.50, 40.70, 40.98, 41.84, 42.69, 42.90, 44.49, 45.04, 46.53, 48.26 and 49.49± 0.2 degrees. In some embodiment of the invention there is provided a crystalline form of Trientine dihydrochloride (I) characterized by X-ray powder diffraction (XRPD) pattern having peaks expressed as 2Θ values at about 9.91, 13.36, 16.62, 19.92, 20.26, 20.72, 23.38, 23.79, 24.01, 24.55, 24.78, 25.07, 25.8310, 26.76, 27.03, 27.34, 27.70, 28.67, 28.89, 29.91, 30.08, 31.26, 31.75, 33.68, 33.89, 34.25, 37.39, 38.00, 40.50, 40.70, 40.98, 41.84, 42.69, 42.90, 44.49, 45.04, 46.53, 48.26 and 49.49+ 0.2 degrees, wherein the said crystalline form is prepared by a process comprising steps of: a) reacting 2-chloroacetonitrile (VI)

(VI)
with ethane- 1,2-diamine (V

tvii)
in presence of base to for '-(ethane-l,2-diylbis(azanediyl)) diacetonitrile (V);

00
b) protecting free amino groups of intermediate (V) by treating with a suitable protecting group preferably i-butyl carbamate (BOC) to obtain the Di-ie/ -butyl ethane- 1 ,2-diylbis(cyanomethylcarbamate) (IV) ;
 c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-ie/t-butyl ethane- l,2-diylbis(2- aminoethylcarbamate) difumarate (III);
c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-ie/t-butyl ethane- l,2-diylbis(2- aminoethylcarbamate) difumarate (III);
Boc
»NH2 .2HOOCCH=CHCOOH
H,N N
I
Boc
(III)
d) deprotecting intermediate (III) by treating with a concentrated hydrochloric acid to obtain N 1 , N 1 '-(ethane- 1 ,2-diyl) diethane- 1 ,2-diaminetetrahydrochloride (II) ;
H
,NH2 .4HC1
H,N
H
(II)
e) preparing Trientine free base (Ila) by treating intermediate (II) with a suitable
H
\ NH2
H2N
H
base; (IIa)
f) treating Nl, N 1 '-(ethane- 1,2-diyl) diethane-l,2-diaminetetra hydrochloride (II) with Trientine free base (Ha) at pH 7.0 to 8.5 to obtain 1,2-ethanediamine, N, N'- bis(2- aminoethyl)-dihydrochloride (I); and
g) isolating crystalline Trientine dihydrochloride from protic solvent.
In another aspect of the invention, there is provided a process for the preparation of crystalline form of Trientine dihydrochloride (I), characterized by X-ray diffraction spectrum having peaks expressed as 2Θ values at about 9.91, 13.36, 16.62, 19.92, 20.26, 20.72, 23.38, 23.79, 24.01, 24.55, 24.78, 25.07, 25.8310, 26.76, 27.03, 27.34, 27.70, 28.67, 28.89, 29.91, 30.08, 31.26, 31.75, 33.68, 33.89, 34.25, 37.39, 38.00, 40.50, 40.70, 40.98, 41.84, 42.69, 42.90, 44.49, 45.04, 46.53, 48.26 and 49.49+ 0.2 degrees, said process comprising the steps of:
a) reacting 2-chloroacetonitrile (VI)

(VI)
with ethane- 1,2-diamine (V

tVII)
in presence of base to for '-(ethane-l,2-diylbis(azanediyl)) diacetonitrile (V);


(IV)
c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-ie/t-butyl ethane- l,2-diylbis(2- aminoethylcarbamate) difumarate (III);
Boc
H N^— ·Ν·— — NH2 .2HOOCCH=CHCOOH
2 I
Boc
(III)
d) deprotecting intermediate (III) by treating with a concentrated hydrochloric acid to obtain N 1 , N 1 '-(ethane- 1 ,2-diyl) diethane- 1 ,2-diaminetetrahydrochloride (II) ;
H
. ^N. ^\ ^^^NH2 .4HC1
H,N N
H
CIO
e) preparing Trientine free base (Ila) by treating intermediate (II) with a suitable
H
H2N N
H
base; (IIa>
f) treating Nl, N 1 '-(ethane- 1 ,2-diyl) diethane- 1,2-diaminetetra hydrochloride (II) with Trientine free base (Ila) at pH 7.0 to 8.5 to obtain 1,2-ethanediamine, N, N'- bis(2- aminoethyl)-dihydrochloride (I); and
g) isolation of crystalline Trientine dihydrochloride from protic solvent. EXAMPLES
EXAMPLE 1
Preparation of 2,2'-(Ethane-l,2-diylbis(azanediyl))diacetonitrile (V):
lOOg (1.663 mol) of ethane- 1,2-diamine (VII) was dissolved in 1200 mL of acetonitrile at 25- 30°C, to this 460g (3.326 mol)of potassium carbonate was added. The reaction mass was cooled to 15-20°C and 276.8 g (3.658 mol) of 2-chloroacetonitrile (VI) was added to the reaction mass over a period of 40-45 min at 15-20°C. The temperature of the reaction mass was raised to 25-30°C and stirred for 22-24 hrs. On completion of the reaction, the reaction mass was filtered at 25-30°C and the solid washed with 200 mL of acetonitrile. The filtrate so obtained was cooled to 15-20°C to obtain 2,2'-(Ethane-l,2-diylbis(azanediyl)) diacetonitrile(V). The obtained filtrate was used in the next step without isolation.
EXAMPLE 2
Preparation of Di-tert-butyl ethane-l,2-diylbis(cyanomethylcarbamate) (IV):
1088 g (4.99 mol) of tertiary-butyl carbamate (Boc) anhydride was added to the filtrate containing 2,2'-(ethane-l,2-diylbis(azanediyl)) diacetonitrile (V) obtained in the example- 1 over a period of 40-60 min at 15-20°C. The reaction mass was maintained for 12-14 hrs. On completion of the reaction, the solvent was distilled off under vacuum below 55°C and the residue was cooled to 25-30°C. 2000 mL of ethyl acetate was added to the residue and stirred for 15-30 min at 25-30°C. 10% aqueous sodium chloride solution was added to the reaction mass at 25-30°C, stirred for 15-30 min and then allowed stand forl5-30 min. Again, same procedure was followed with 10% of sodium chloride solution. The organic layer was distilled off completely and 1000 mL of cyclohexane was added to the residue. The reaction mass was stirred and the solid so formed was filtered. Finally, the wet cake so obtained was washed with 200 mL of cyclohexane at 25-30°C. Again, the wet cake was charged with 1500 mL of cyclohexane in a clean and dry RB flask, stirred at 25-30°C for 60-90 min. The solid obtained was filtered and washed with 200 mL of cyclohexane followed by suck dried under vacuum below 40°C to obtain pure tert-butyl ethane- 1,2-diylbis (cyanomethyl carbamate) (IV).
Yield % : 71
Purity by Gas chroma tography(GC) % : 99.0
EXAMPLE 3
Preparation of Di-tert-butyl ethane-l,2-diylbis(2-aminoethylcarbamate)difumarate (III):
lOOg of tert-butyl ethane l,2diylbis (cyanomethylcarbamate) (IV) was charged in a clean and dry autoclave and 1000 mL of methanolic ammonia was added to it at 25-30°C. 50g of Raney nickel (rinsed with purified water and methanol) was added to the reaction mass at 25-30°C, under nitrogen atmosphere. A pressure of 2 - 3 Kg of nitrogen gas was applied to the autoclave and the reaction mass stirred for 5 -10 min at 25-30° C. The nitrogen gas was released, and again repeated the same procedure. Then, the reaction mass was maintained under hydrogen gas at a pressure of 2-3 Kg for 24-28 hrs. After completion of the reaction, the reaction mass was allowed to settle for 20-30 min at 25-30°C and then filtered through Hyflo. The filtrate was distilled under vacuum and 200 mL of isopropyl alcohol was added to
the residue. The solvent was distilled under vacuum below 45° C and the residue obtained was cooled to 25-30°C. 700 mL of isopropyl alcohol and lOOg of fumaric acid dissolved in 300mL of methanol were added to the residue at 25-30°C. The reaction mass was heated for 45-60 min at 60-65°C and then cooled to 25-30°C. It was further cooled to 0-5°C for 5-6 hrs. The precipitated solid was filtered and washed with chilled isopropyl alcohol: methanol solvent mixture (7:3) and 200 mL of chilled acetone and dried under vacuum. The solid so obtained was further dissolved in a mixture of (7:3) isopropyl alcohol: methanol at 25-30°C, heated for 60-90 min at 65-70°C. The reaction mass was then cooled to 25-30°C and stirred for 90-120 min. Further the reaction mass was cooled to 0-5°C for 3-4hrs. The solid so formed was filtered, washed with chilled acetone and dried under vacuum below 40°C to obtain di-tert-butyl ethane- l,2-diylbis(2-aminoethylcarbamate) difumarate (III) salt.
Yield% : 35
Purity by HPLC % : 99.5 EXAMPLE 4
Preparation of Nl, Nl'-(Ethane-l,2-diyl) bis (ethane- 1,2-diamine) tetrahydrochloride (Π):
To lOOg (0.172 mol) of di-tert-butyl ethane- l,2-diylbis(2-aminoethylcarbamate) difumarate salt (III), 300mL of concentrated hydrochloric acid was added at 25-30°C. The reaction mass was heated at 65-70°C for l-2hrs, then cooled to 25-30°C and maintained for 8-10 hrs. 300 mL of isopropyl alcohol and 300 mL of acetone were added to the reaction mass, cooled to 10-15°C and stirred for 45-60 min. The solid formed was filtered and dried under vacuum. The solid so obtained was washed with a mixture of chilled isopropyl alcohol: acetone (1: 1), then with chilled acetone at 10-15°C and dried. 8 volumes of methanol were added to the wet cake at 25-30°C and heated at 70-75°C. The reaction mixture was cooled to 0-5°C and stirred for 60-90 mins. The solid so obtained was filtered under vacuum and washed with chilled acetone and dried under vacuum. Again, the solid was heated with 3 volumes of water at 45-50°C for 20-30 min. To the mixture 10 volumes of ethanol was added and cooled to 0- 5°C. The reaction mass was maintained for 10-12 hrs at 0-5°C.The solid so formed was filtered, washed with 2 volumes of chilled acetone to obtain Nl, Nl'-(ethane-l,2-diyl) bis (ethane- 1 ,2-diamine)tetrahydrochloride (II) .
Yield% :79
Purity by HPLC % : 99.90
Example 5
Preparation of Trientine free base (Ha):
lOOg (0.342 moles) of Nl, Nl '-(ethane- 1,2-diyl) bis (ethane- 1,2-diamine) tetrahydrochloride (II) was dissolved in 1000 mL methanol at 25-30°C. The reaction mixture was cooled to 10- 15°C, and 68.4(1.71 mol) of sodium hydroxide was added to the reaction mixture. The reaction mass was stirred for 15 - 16hrs at 25-30°C, then filtered through Hyflo bed and the filtrate was distilled off under vacuum. 200 mL of methanol was added to the residue and the solvent was distilled off under vacuum. 1000 mL of dichloro methane was added to the residue again, stirred for l-2hrs at 25-30°C and filtered through Hyflo, the filtrate was dried over sodium sulphate and filtered. lOg of neutral charcoal was added to the filtrate and stirred at 25-30°C. The reaction mixture was filtered through Hyflo and the filtrate distilled under vacuum to obtain Trientine free base (Ila).
Yield % : 70
Purity by HPLC % : 99.89
EXAMPLE 6
Preparation of 1,2-Ethanediamine, N, N'-bis(2-aminoethyl)-dihydrochloride (I):
Trientine free base (Ila) was dissolved in 2000mL methanol and 82.7g (0.283 mol) Trientine tetrahydrochloride (II) was added to the reaction mass. The reaction mass was stirred for 3 hrs at 25-30°C and the pH of the reaction mass was maintained at 7.0-8.5 with addition of Trientine free base. On completion of the reaction the solvent was completely distilled off under vacuum below 45°C, degassed the crude and cooled to 25-30°C. 1200 mL ethanol was charged to the crude and stirred for 16-20 hrs at 25-30°C. The solid so obtained was cooled to 0-5°C, filtered and suck dried under vacuum at 0-5°C. The obtained solid was taken in 1200 mL of ethanol at 0-5°C, refluxed at 75-80°C followed by addition of water to get a clear solution. lOg of activated charcoal was added to the clear solution, stirred and filtered through Hyflo at 75-80°C. The filtrate was cooled to 25-30°C, stirred and was maintained for 16-17 hrs at 25-30°C. The solid formed was filtered, washed with chilled ethanol and dried under vacuum below 50°C to obtain 1,2-Ethanediamine, N, N'-bis(2-aminoethyl)- dihydrochloride (I).
Yield % : 88
Purity by HPLC % : 99.97
Melting range between 118-122°C
EXAMPLE 7
Alternative process for the preparation of 1,2-Ethanediamine, N, N'-bis(2-aminoethyl)- dihydrochloride (I):
Sodium carbonate (369.9g, 6.85 mol) was added to the slurry of Trientine tetrahydrochloride (II) (500 g, 1.71 mol) in methanol (6 L) at 20-25°C and stirred the reaction mass for 2 hrs at 25-30°C. The suspension was filtered, and the filtered cake was washed with methanol (500 mL). The filtrate was concentrated under reduced pressure. Dichloromethane (3L) was added to the mass and stirred for 10 minutes. The suspension was filtered, washed the filter cake with dichloromethane (500 mL). The filtrate was distilled off under vacuum and the mass was dissolved in methanol (7.5 L). To this Trientine tetrahydrochloride (II) (500 g, 1.71 moles) was added and stirred for 30 minutes at 25-30°C and evaporated the solvent to give pure Trientine dihydrochloride(I)
Yield % : 97
Purity by HPLC % :99.90
Melting range between 118°-122°C
EXAMPLE 8
Alternative process for the preparation of 1,2-Ethanediamine, N, N'-bis(2-aminoethyl)- dihydrochloride (I):
Sodium methoxide (907g, 8.56mol) was added to the slurry of Trientine tetrahydrochloride(II) (500 g, 1.71 mol) in methanol (3 L) at 20-25°C and stirred the reaction mass for 4hrs at 25-30°C. The suspension was filtered, and the filtered cake was washed with methanol (500 mL). The filtrate was concentrated under reduced pressure then dichloromethane (3 L) was added to the mass and stirred for 10 minutes. The suspension was filtered, washed the filter cake with dichloromethane (500 mL). The filtrate was distilled off under vacuum and the reaction mass was dissolved in methanol (10 L). To the reaction mass Trientine tetrahydrochloride (II)(500 g, 1.71 moles) was added and stirred for 30 minutes at 25-30°Cand evaporated the solvent at reduced pressure, then isopropyl alcohol (2.5 L) was added and filtered the precipitated solid to give pure Trientine dihydrochloride (I)
Yield % : 91
Purity by HPLC % :99.95
EXAMPLE 9
Alternative process for the preparation of 1,2-Ethanediamine, N, N'-bis(2-aminoethyl)- dihydrochloride (I):
Sodium hydroxide (287.5g, 7.19mol) was added to the slurry of Trientine tetrahydrochloride (II)(500g, 1.71 mol) in ethanol (5 L) at 20-25°C. The reaction mass was stirred for 4 hrs. at 25-30°C. The suspension was filtered, and the filtered cake was washed with ethanol (600 mL). The filtrate was concentrated at reduced pressure. Methyl tert-butyl ether (6 L) was added to the reaction mass and stirred for 10 minutes. The suspension was filtered, washed the filter cake with methyl tert-butyl ether (500 mL). The filtrate was distilled under vacuum and dissolved in ethanol (5 L), to this Trientine tetrahydrochloride (II) (500g, 1.71 mol) was added and stirred for 60 minutes at 25-30°C. The solvent was distilled off up to 80% and cooled to 0-5 °C to precipitate pure Trientine dihydrochloride (I).
Yield % : 93.00
Purity by HPLC% : 99.96
Claims
1. A process for the preparation of substantially pure Trientine dihydrochloride (I) having a purity of greater than 99.5%, the said process comprising the steps of:
H
H,N 'NH2 .2HC1
H
(I)
a) reacting 2-chloroacetonitrile (VI)

with ethane- 1 ,2-diamine (VII)

CVII)
in presence of base to form '-(ethane-l,2-diylbis(azanediyl)) diacetonitrile (V);

00
b) protecting free amino groups of intermediate (V) by treating with a suitable protecting group preferably i-butyl carbamate (BOC) to obtain the Di-tert-buty\ ethane- 1,2- diylbis(cyanomethylcarba
 c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-feri-butyl ethane- 1 ,2-diylbis(2-aminoethylcarbamate) difumarate (III);
c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-feri-butyl ethane- 1 ,2-diylbis(2-aminoethylcarbamate) difumarate (III);
.2HOOCCH=CHCOOH

d) deprotecting intermediate (III) by treating with a concentrated hydrochloric acid to obtain Nl, Nl'-(ethane-l,2-diyl) diethane- 1 ,2-diaminetetrahydrochloride (II);
H
,NH2 .4HC1
H,N
H
CH)
e) preparing Trientine free base (Ila) by treating intermediate (II) with a suitable base; and
H
.NH,
H2N
H
(Ila)
treating Nl, Nl'-(ethane-l,2-diyl) diethane- 1 ,2-diaminetetra hydrochloride (II) with Trientine free base (Ila) at pH 7.0 to 8.5 to obtain 1,2-ethanediamine, N, N'-bis(2- aminoethyl)-dihydrochloride (I).
2. The process as claimed in claim 1, wherein the base employed in step a) and step e) is selected from the group comprising of sodium hydroxide, potassium hydroxide, potassium carbonate, sodium carbonate, potassium bicarbonate, sodium bicarbonate, triethylamine, pyridine or the like.
3. The process as claimed in claim 1, wherein the protecting group employed in step b) is selected from the group comprising of fluorenylmethyloxycarbonyl (Fmoc), tert- butyloxycarbonyl (Boc), phthalimide, toluene sulfonyl (Ts), methane sulfonyl (Ms), triphenylmethyl (Trityl), carboxybenzyl (CBZ) or the like.
4. The process as claimed in claim 1, wherein the hydrogenation is carried out in presence of Raney nickel and methanolic ammonia.
A process for preparation of substantially pure Trientine dihydrochloride (I) having a purity of greater than 99.5%, said process comprising the steps of:
a) adding pure Trientine free base to Trientine tetrahydrochloride;
b) adjusting pH of the Trientine tetrahydrochloride to 7.0 to 8.5; and
c) isolating Trientine dihydrochloride from protic solvent.
The process as claimed in claim 5, wherein the protic solvent used is selected from the group comprising of acetone, water, methanol, ethanol, isopropyl alcohol (IP A), n- propanol, n-butanol or mixture thereof.
7. A crystalline form of Trientine dihydrochloride (I) characterized by X-ray powder diffraction (XRPD) pattern having peaks expressed as 2Θ values at about 9.91, 13.36, 16.62, 19.92, 20.26, 20.72, 23.38, 23.79, 24.01, 24.55, 24.78, 25.07, 25.8310, 26.76, 27.03, 27.34, 27.70, 28.67, 28.89, 29.91, 30.08, 31.26, 31.75, 33.68, 33.89, 34.25, 37.39, 38.00, 40.50, 40.70, 40.98, 41.84, 42.69, 42.90, 44.49, 45.04, 46.53, 48.26 and 49.49 +0.2 degrees.

8. The crystalline form of Trientine dihydrochloride (I) as claimed in claim 7, wherein
said crystalline form is prepared by a process comprising steps of: a) reacting 2-chloroacetonitrile (VI)

(VI)
with ethane- 1 ,2-diamine (VII)

(VII)
in presence of base to form 2,2'-(ethane-l,2-diylbis(azanediyl)) diacetonitrile (V);

00
b) protecting free amino groups of intermediate (V) by treating with a suitable protecting group preferably i-butyl carbamate (BOC) to obtain the Di-tert-buty\ ethane- 1,2- diylbis(cyanomethylcarba
 c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-feri-butyl ethane- 1 ,2-diylbis(2-aminoethylcarbamate) difumarate ( .2HOOCCH=CHCOOH
c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-feri-butyl ethane- 1 ,2-diylbis(2-aminoethylcarbamate) difumarate ( .2HOOCCH=CHCOOH

(III)
d) deprotecting intermediate (III) by treating with a concentrated hydrochloric acid to obtain Nl, Nl'-(ethane-l,2-diyl) diethane- 1 ,2-diaminetetrahydrochloride (II);
H
,NH2 .4HC1
H,N
H
an
preparing Trientine free base (Ila) by treating intermediate (II) with a suitable base;
H
NH
H,N N
H
[Ha)
f) treating Nl, Nl'-(ethane-l,2-diyl) diethane- 1,2-diaminetetra hydrochloride (II) with Trientine free base (Ila) at pH 7.0 to 8.5to obtain 1 ,2-ethanediamine, N, N'-bis(2- aminoethyl)-dihydrochloride (I); and
g) isolating crystalline Trientine dihydrochloride from protic solvent.
9. A process for the preparation of crystalline form of Trientine dihydrochloride (I), characterized by X-ray diffraction spectrum having peaks expressed as 2Θ values at about 9.91, 13.36, 16.62, 19.92, 20.26, 20.72, 23.38, 23.79, 24.01, 24.55, 24.78, 25.07, 25.8310, 26.76, 27.03, 27.34, 27.70, 28.67, 28.89, 29.91, 30.08, 31.26, 31.75, 33.68, 33.89, 34.25, 37.39, 38.00, 40.50, 40.70, 40.98, 41.84, 42.69, 42.90, 44.49, 45.04, 46.53, 48.26 and 49.49± 0.2 degrees, said process comprising the steps of:
H
H,N -NH2 .2HC1
H
(I)
a) reacting 2-chloroacetonitrile (VI)

vi)
with ethane- 1,2-diamine (VII)
,NH,
H2
(VII)
in presence of base to form '-(ethane-l,2-diylbis(azanediyl)) diacetonitrile (V);

m
b) protecting free amino groups of intermediate (V) by treating with a suitable protecting group preferably i-butyl carbamate (BOC) to obtain the Di-tert-buty\ ethane- 1,2- diylbis(cyanomethylcarba
 c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-feri-butyl ethane- 1 ,2-diylbis(2-aminoethylcarbamate) difumarate (III);
c) reducing intermediate (IV) by hydrogenation and treating the hydrogenated product with fumaric acid to obtain Di-feri-butyl ethane- 1 ,2-diylbis(2-aminoethylcarbamate) difumarate (III);
.2HOOCCH=CHCOOH

d) deprotecting intermediate (III) by treating with a concentrated hydrochloric acid to obtain Nl, Nl'-(ethane-l,2-diyl) diethane- 1 ,2-diaminetetrahydrochloride (II);
H
,NH2 .4HC1
Η,Ν'
H
CH)
preparing Trientine free base (Ila) by treating intermediate (II) with a suitable base;
H
NH
H2N N
H
CHa)
f) treating Nl, Nl'-(ethane-l,2-diyl) diethane- 1,2-diaminetetra hydrochloride (II) with Trientine free base (Ila) at pH 7.0 to 8.5 to obtain 1,2-ethanediamine, N, N'-bis(2- aminoethyl)-dihydrochloride (I); and
g) isolating crystalline Trientine dihydrochloride from protic solvent.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN201741014337 | 2017-04-22 | ||
| IN201741014337 | 2017-04-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018193482A1 true WO2018193482A1 (en) | 2018-10-25 |
Family
ID=63857067
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2018/050243 WO2018193482A1 (en) | 2017-04-22 | 2018-04-21 | Improved method for the synthesis of 1,2-ethanediamine, n, n'-bis(2-aminoethyl)-dihydrochloride(trientine dihydrochloride) |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2018193482A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024018394A1 (en) * | 2022-07-19 | 2024-01-25 | Biophore India Pharmaceuticals Pvt. Ltd | A process for the preparation of crystalline form a of n, n'-bis(2-aminoethyl)-1,2-ethanediamine tetrahydrochloride |
| WO2024033786A1 (en) * | 2022-08-07 | 2024-02-15 | Philera New Zealand Ltd. | Method for triethylenetetramine purification |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8394992B2 (en) * | 2004-07-19 | 2013-03-12 | Philera New Zealand Limited | Synthesis of triethylenetetramines |
| WO2017046695A1 (en) * | 2015-09-18 | 2017-03-23 | Emcure Pharmaceuticals Limited | An improved process for preparation of trientine dihydrochloride |
-
2018
- 2018-04-21 WO PCT/IN2018/050243 patent/WO2018193482A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8394992B2 (en) * | 2004-07-19 | 2013-03-12 | Philera New Zealand Limited | Synthesis of triethylenetetramines |
| WO2017046695A1 (en) * | 2015-09-18 | 2017-03-23 | Emcure Pharmaceuticals Limited | An improved process for preparation of trientine dihydrochloride |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024018394A1 (en) * | 2022-07-19 | 2024-01-25 | Biophore India Pharmaceuticals Pvt. Ltd | A process for the preparation of crystalline form a of n, n'-bis(2-aminoethyl)-1,2-ethanediamine tetrahydrochloride |
| WO2024033786A1 (en) * | 2022-08-07 | 2024-02-15 | Philera New Zealand Ltd. | Method for triethylenetetramine purification |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10421717B2 (en) | Process for preparing brivaracetam | |
| AU2007218140B2 (en) | Process for the preparation of adamantanamines | |
| WO2016016766A2 (en) | A process for the preparation of isavuconazonium or its salt thereof | |
| EP3350154B1 (en) | An improved process for preparation of trientine dihydrochloride | |
| WO2018193482A1 (en) | Improved method for the synthesis of 1,2-ethanediamine, n, n'-bis(2-aminoethyl)-dihydrochloride(trientine dihydrochloride) | |
| EP2368874A1 (en) | Racemisation of (R)-N-Boc-3-hydroxyadamant-1-yl glycine | |
| US10421716B2 (en) | Process for preparing alpha-carboxamide pyrrolidine derivatives | |
| CN106459150B (en) | Method for producing synthetic pentapeptide | |
| CA2810393A1 (en) | Method of preparing 3-amino-4-(2-oxo-piperidin-1-yl)-butyric acid derivative for synthesizing medicament | |
| WO2013177938A1 (en) | Method of preparing cinacalcet hydrochloride | |
| US9598350B2 (en) | Process for preparing cinacalcet and pharmaceutically acceptable salts thereof | |
| AU2022331536A1 (en) | Process for preparing histone demethylase inhibitors | |
| WO2016038560A1 (en) | Process for the preparation of enzalutamide | |
| WO2024018394A1 (en) | A process for the preparation of crystalline form a of n, n'-bis(2-aminoethyl)-1,2-ethanediamine tetrahydrochloride | |
| US9902693B2 (en) | Preparation method for pyrrolidine-2-carboxylic acid derivatives | |
| EP3902779B1 (en) | Process for the preparation exo-tert-butyl n-(3-azabicyclo[3.2.1]octan-8-yl)carbamate | |
| JP2007510695A (en) | Method for preparing gabapentin | |
| US20230174478A1 (en) | Method for preparing brivaracetam and intermediate thereof | |
| KR101628946B1 (en) | Improved Process of Silodosin | |
| EP2736875B1 (en) | Chemical process for opening ring compounds | |
| US20060264671A1 (en) | Process for the preparation of midodrine, pharmaceutically acceptable salts thereof and intermediates | |
| CN111247127B (en) | Process for the production of intermediate compounds for the synthesis of medicaments | |
| WO2020194138A1 (en) | An improved process for the preparation of eliglustat and its intermediate | |
| US20230348417A1 (en) | Process for preparing (s)-1-(1-acryloylpyrrolidin-3-yl)-3-((3,5-dimethoxyphenyl) ethynyl)-5-(methylamino)-1h-pyrazole-4-carboxamide | |
| WO2019073427A1 (en) | A process for the preparation of trientine dihydrochloride |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18787854 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 18787854 Country of ref document: EP Kind code of ref document: A1 |