WO2018175340A1 - Reduced exposure conjugates modulating therapeutic targets - Google Patents
Reduced exposure conjugates modulating therapeutic targets Download PDFInfo
- Publication number
- WO2018175340A1 WO2018175340A1 PCT/US2018/023174 US2018023174W WO2018175340A1 WO 2018175340 A1 WO2018175340 A1 WO 2018175340A1 US 2018023174 W US2018023174 W US 2018023174W WO 2018175340 A1 WO2018175340 A1 WO 2018175340A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- target site
- conjugate
- reduced exposure
- active entity
- polymer
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
Definitions
- polymer conjugates comprising active agents linked to polymers, and therapeutic uses thereof. More particularly, polymer conjugates for treating target sites within the skin, eye, and gastrointestinal system which exhibit reduced exposure to non-target sites and inhibit mediators, such as kinase mediators, of various dermatological, ophthalmic, and/or gastrointestinal conditions are described. Other conjugates for treating the eye, dermatological target sites and target sites within the gastrointestinal system are also described.
- Inhibitors of kinase mediators of various ophthalmic, dermatological, and gastrointestinal conditions have been described for possible therapeutic use in the prevention, alleviation and treatment of kinase-associated ophthalmic, dermatological, gastrointestinal pathologies.
- such compounds are associated with broad kinase specificity, as well as undesirable and toxic effects, in particular at non-target sites. Accordingly, strategies to render these active kinase inhibitors or other drugs more specific, less toxic, and more targeted to sites within the eye, skin, and gastrointestinal system are needed.
- Effective delivery of pharmacologically active agents may be hindered by unwanted exposure of those agents to non-desired locations (such as the systemic circulation and/or lymphatic system).
- injectable and topical agents useful in treating various skin, eye, and gastrointestinal disorders may result in toxic side effects because of systemic exposure.
- One issue with delivering compositions comprising one or more active agents topically (or non-topically) is the concern that such agents should be delivered in an amount and at a location sufficient to have a therapeutic effect.
- exposure e.g., absorption or longevity of the composition in the systemic circulation, lymphatic system, or other non-targeted sites
- the compositions described herein are both therapeutically efficacious and minimize non-target (e.g., systemic or bloodstream) exposure.
- the active agents are PEGylated or otherwise coupled to large molecules, and surprisingly, are effective in crossing biological membranes such that the active agents are effectively delivered to the target location.
- inflammatory bowel diseases, inflammatory skin diseases, and ophthalmic conditions are disclosed in several embodiments, other embodiments are used to treat dermal and eye inflammation, as well as other several conditions (e.g., those conditions that would benefit from treatment with reduced exposure at non-target sites). Ophthalmic treatments are provided in some embodiments.
- the compositions and technology described herein are used in the gastrointestinal systems. Inflammatory and non-inflammatory conditions are contemplated herein.
- compositions for treating joints are provided. Treatment of the nose and ear are provided in other embodiments.
- Reduced exposure compounds and compositions are provided in several embodiments.
- “Reduced exposure” compounds are those compounds that, when delivered to a target location, are formulated to act at the target location with reduced exposure (e.g., entry and/or longevity) in non-target sites. Exposure is reduced as compared to active agents not formulated according to the embodiments described herein. As a non-limiting example, a PEGylated topical dermal, ophthalmic or gastrointestinal active agent has reduced exposure to the bloodstream as compared to the active agent alone.
- Reduced exposure compounds include topical compounds that can be delivered to body surfaces and cavities, such as the skin, eyes, ears, nose, mouth, vagina, rectum, etc.
- Non-desired target sites include, for example, the systemic system, the lymphatic system, non-target tissue, etc.
- Reduced exposure compositions comprise or consist essentially of one or more "reduced exposure compounds.”
- a reduced exposure composition is delivered orally, e.g., for treatment of the gastrointestinal system, eye, or skin.
- the active agent remains in the eye, the skin, or the lining of the gastrointestinal tract and is able to achieve pharmacological specificity.
- a reduced exposure composition is delivered topically, e.g., for treatment of the skin.
- the active agent remains in the targeted layer of the skin and is able to achieve pharmacological specificity. Because the active agent is conjugated with PEG or another molecule as described herein, the active agent is absorbed more slowly into the non-target site (e.g., the systemic circulation and/or lymphatic system).
- the active agent is absorbed into the non-target site (e.g., systemic circulation and/or lymphatic system). Further, once the composition enters the systemic circulation and/or lymphatic system, clearance (e.g., by the kidney) occurs at a much faster rate.
- compositions formulated according to the methods described herein for treating the lungs (e.g., via inhalants), joints (e.g., via injectables), eye (e.g., via eye drops), nasal passageways, and the ear (such as the ear canal and other structures).
- Vaginal and rectal tissues are treated in some embodiments via, for example suppositories.
- reduced exposure compostions suitable for topical application to the eye e.g., ointment or eye drop.
- the reduced exposure compsotions disclosed herein are administered by intraocular and periocular injection such as, for example, direct intravitreal injection, subconjunctival injection, subtenon injection, and peribulbar injection.
- reduced exposure compostions which can be administered via intraocular implantable devices known to one of skill in the art.
- reduced exposure at the tear ducts reduces the amount of active agent that is removed (e.g., drained) from the eye, and accordingly allows a higher concernation of active agent to remain at the target site in the eye.
- a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an inhibitor, antagonist, or inverse agonist of, for example, c-Src.
- at least one inhibitor, antagonist, or inverse agonist of c-Src comprises or consists of a composition that includes any one of compounds 1 -71 (and derivatives thereof) disclosed herein in Table 1 coupled to a polymer.
- the warhead of the polymer conjugate is compound 1 of Table 1 .
- the polymer conjugate is CT101 , wherein CT101 has the following formula:
- a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an inhibitor, antagonist, or inverse agonist of, for example, a mediator of a gastrointestinal, dermatological, and/or ophthalmic condition.
- at least one inhibitor, antagonist, or inverse agonist of a mediator of a gastrointestinal, dermatological and/or ophthalmic condition comprises or consists of a composition that includes any one of compounds 1 - 264 (and derivatives thereof) disclosed herein in Table 3 coupled to a polymer.
- the warhead of the polymer conjugate is compound 1 in Table 3.
- the LSE polymer conjugate is CT352, wherein CT352 has the following formula below:
- a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an inhibitor, antagonist, or inverse agonist of, for example, a vascular endothelial growth factor receptor (VEGFR).
- VEGFR vascular endothelial growth factor receptor
- at least one inhibitor, antagonist, or inverse agonist of a vascular endothelial growth factor receptor (VEGFR) comprises or consists of a composition that includes any one of compounds 1 -59 (and derivatives thereof) disclosed herein in Table 2 coupled to a polymer.
- the warhead of the polymer conjugate is compound 1 of Table 2.
- the polymer conjugate is CT103, wherein the composition has the formula:
- a non-target site such as the systemic circulation and/or lymphatic system
- exposure at a non-target site is less than 90%, 75%, 50%, 25%, 15%, 10%, 5% or 2% (or less) of the polymer conjugate as compared to a similar active entity that has not been produced according to the embodiments described herein.
- desirable rate of clearance from the non-target site (e.g., systemic circulation and/or lymphatic system) for the compositions described herein is increased by at least 10%, 25%, 50%, or 75% or more as compared to non-conjugated controls.
- a PEGylated active agent described herein not only penetrates the desired membranes to reach a desired target, but has reduced non-target exposure by at least 20-80% or more as compared to the non-PEGylated active agent.
- blood concentrations measured post administration of the compositions described herein are less than about 0.1 ng/ml, less than 1 ng/ml, or less than 10 ng/ml after, e.g., 15 minutes, 30 minutes, 1 hour, 6 hours or 12 hours.
- reduced exposure at non-target sites contributes to enhanced efficacy.
- Efficacy may be enhanced because lower concentrations/amounts/dosing schedules are required to achieve the same or similar therapeutic efficacy at the target site (because, for example, the active ingredient stays at the desired target site for a longer time).
- concentrations/amounts/dosing schedules are reduced by 25%-75% or more.
- More rapid clearance rates of the active agent once in the non-target site(s) are also beneficial because this may allow for a higher concentration or more doses to be delivered. This is especially beneficial for active agents in which a subject would benefit from a higher dose but cannot tolerate the higher dose due to toxicity at the non-target site (e.g., systemic toxicity). Faster clearance rates would permit the desired higher dose to be delivered according to the desired schedule. For example, a subject may be able to tolerate daily doses rather than weekly doses because of the reduced exposure.
- the active agents of the compositions described herein are measured in non-target sites (e.g., the systemic circulation and/or lymphatic system) at less than amounts found when the active agent is delivered without conjugation (e.g., less than 0.5%, 1 % or 2% after 6 or 12 hours, as compared with 3- 15% (e.g., 3-6%) when the active agent is delivered without conjugation).
- non-target sites e.g., the systemic circulation and/or lymphatic system
- the active agents of the compositions described herein are measured in non-target sites (e.g., the systemic circulation and/or lymphatic system) at less than 0.5%, 1 % or 2% after 3-24 hours, as compared to an amount 2-20 times greater when the active agent is delivered without conjugation.
- non-target sites e.g., the systemic circulation and/or lymphatic system
- clearance of the compositions occurs within minutes of exposure to the non-target site (e.g., systemic circulation and/or lymphatic system), as opposed to hours.
- 50% clearance of the conjugated polymer compounds occurs in less than 5 minutes, 15 minutes, 30 minutes, 1 hour, 6 hours, and 12 hours of exposure to the systemic circulation and/or lymphatic system. Clearance times of the conjugated polymer compounds are reduced by more than 25%, 50%, 75% and 90%, as compared to the non-conjugated active agents or other formulations. These reduced clearance times are beneficial to reduce toxicity and undesired side effects.
- an active agent may be increasingly toxic as it is metabolized in the non-target site (e.g., systemic circulation and/or lymphatic system) because the metabolites exhibit more toxicity than the original agent.
- the non-target site e.g., systemic circulation and/or lymphatic system
- faster clearance rates in some cases even before the toxic metabolites are created, are especially beneficial.
- the term "active entity" as used herein should not be understood as limiting the participation of the polymer itself and/or the chemical linking moiety between the polymer and the warhead in defining the pharmacology of the polymer conjugate.
- the polymer influences the selectivity and/or inhibitory activity of the polymer conjugate.
- the chemical linking moiety between the polymer and warhead influences the selectivity and/or inhibitory activity of the polymer conjugate.
- the polymer conjugates exhibit no change in selectivity or inhibitory activity against the therapeutic target in comparison with the unconjugated active agent.
- the polymer conjugates exhibit a significant increase in selectivity against the therapeutic target in comparison with the unconjugated active agent.
- the polymer conjugates exhibit a significant increase in inhibitory activity against the therapeutic target in comparison with the unconjugated active agent. In some embodiments, the polymer conjugates exhibit a significant increase in selectivity and inhibitory activity against the therapeutic target in comparison with the unconjugated active agent. In some embodiments, the increased selectivity and/or inhibitory activity of the polymer conjugate against the therapeutic target in comparison with the unconjugated active agent causes decrease in undesired biological effects. In some embodiments, the increased selectivity of the polymer conjugate is caused by an increase of the hydrodynamic volume resulting from the conjugated polymer chain. In some embodiments, the polymer chain creates a higher steric hindrance which allows discrimination among the diverse shapes and sizes of the binding sites of different proteins, thus improving selectivity with respect to the active agent alone.
- age-related macular degeneration is treated.
- diabetic retinopathy is treated.
- corneal edema is treated.
- macular edema is treated.
- dry eye is treated.
- various inflammatory skin diseases are treated.
- the inflammatory skin disease comprises, in some embodiments, psoriasis, psoriasis guttata, inverse psoriasis, pustular psoriasis, psoriatic erythroderma, acute febrile neutrophilic dermatosis, eczema, xerotic eczema, dyshidrotic eczema, vesicular palmar eczema, acne vulgaris, atopic dermatitis, contact dermatitis, allergic contact dermatitis, dermatomyositis, exfoliative dermatitis, hand eczema, pompholyx, keloids, rosacea, rosacea due to sarcoidosis, rosacea due to scleroderma, rosacea due to Sweet syndrome, rosacea due to systemic lupus erythematosus, rosacea
- various skin neoplasias are treated.
- the skin neoplasia comprises, in some embodiments, squamous cell carcinoma, basal cell carcinoma, malignant melanoma, malignant cutaneous lymphoma, Kaposi's sarcoma, Merkel cell skin cancer, and non-melanoma skin cancer.
- various vascular tumors are treated.
- the vascular tumor comprises, in some embodiments, hemangiomas, Kaposi's sarcoma, lymphangioma, glomangioma, angiosarcoma, hemangioendothelioma, and infantile hemangiomas.
- bullous diseases are treated.
- the bullous disease comprises, in some embodiments, bullous pemphigoid, erythema multiforme, dermatitis herpetiformis, epidermolysis bullosa acquisita, linear Immunoglobulin A disease, mucous membrane pemphigoid, pemphigoid gestationis, pemphigus foliaceus, and pemphigus vulgaris.
- the polymer conjugates are administered in combination with UV irradiation therapy.
- the polymer conjugates configured for reduced exposure are administered to other areas of the body besides the skin.
- administration comprises treatment of the lung and respiratory conditions via inhalation of the polymer conjugates.
- Eye drops are provided in some embodiments to treat eye inflammation or ophthalmic disorders and diseases.
- Treatment to the joints to treat inflammation or other joint conditions is also provided.
- administration comprises treatment of the gastro-intestinal tract via, for example, an enteric coated capsule comprising the polymer conjugates taken orally. Reduced exposure provides benefits in these applications.
- nasal and ear such as inhalants, ointments and drops are provided in several embodiments.
- Treatment to the nasal passage to treat allergies or allergic rhinitis is also provided.
- Vaginal and rectal compounds are provided in some embodiments, including as suppositories, creams, ointments, etc.
- various inflammatory bowel diseases are treated.
- the inflammatory bowel disease comprises, in some embodiments, Crohn's disease, ulcerative colitis, collagenous colitis, lymphocytic colitis, ischemic colitis, diversion colitis, Behcet's syndrome, and indeterminate colitis.
- polymer conjugates wherein the polymer is polyethylene glycol (PEG) or methoxy-polyethylene glycol (m- PEG).
- PEG polyethylene glycol
- m- PEG methoxy-polyethylene glycol
- a pharmaceutical composition comprising or consisting essentially of a polymer conjugate disclosed herein that is formulated for topical and non-topical administration.
- methods of making and using the compositions described herein are provided.
- the invention comprises a reduced exposure composition comprising at least one active entity linked to at least one polymer, wherein the composition has reduced exposure at a non-target site as compared to the active entity delivered without the polymer.
- the non-target site comprises the systemic system, the lymphatic system and/or another non-target tissue site in some embodiments.
- the active entity comprises an inhibitor, an antagonist, or an inverse agonist.
- the active entity may be an inhibitor, antagonist, or inverse agonist of a mediator of inflammation.
- the active entity may be an inhibitor, antagonist, or inverse agonist of a mediator of an inflammatory bowel disease an inflammatory skin disease, and/or inflammatory ophthalmic condition.
- the active entity may be an inhibitor, antagonist, or inverse agonist of a gastrointestinal, dermatological, and/or ophthalmic condition
- the active entity may be an inhibitor, antagonist, or inverse agonist of JAK and/or STAT family proteins.
- the active entity comprises or consists essentially of any one or more of compounds 1-264 of Table 3 in some embodiments.
- the active entity comprises compound 1 of Table 3 in some embodiments.
- the reduced exposure composition comprises CT352 in some embodiments.
- the active entity comprises an indolocarbazole compound. In some embodiments, the active entity comprises a derivative of K252a. In some embodiments, the composition comprises SNA-125. In some embodiments, the composition comprises SNA-120.
- the active entity binds to a tropomyosin-receptor-kinase A (TrkA) in some embodiments.
- the active entity binds to a Janus Kinase (JAK) family member in some embodiments.
- the active entity binds to one or more of Janus Kinase 1 (JAK1), Janus Kinase 2 (JAK2), Janus Kinase 3 (JAK3), and/or Tyrosine kinase 2 (TYK2) in some embodiments.
- the active entity binds to mitogen-activated protein kinase kinase (MAP2K) in some embodiments.
- the active entity binds to mitogen-activated protein kinase kinase 3 (MAP2K3) in some embodiments.
- the binding may be partially or fully inhibitory or not.
- the active entity comprises an inhibitor, an antagonist, or an inverse agonist.
- the active entity may be an inhibitor, antagonist, or inverse agonist of c-Src.
- inflammatory conditions are treated.
- non-inflammatory conditions are treated.
- the active entity comprises or consists essentially of any one or more of compounds 1-71 of Table 1 in some embodiments.
- the active entity comprises compound 1 of Table 1.
- the composition comprises CT101.
- the active entity binds to c-Src in some embodiments.
- the binding may be partially or fully inhibitory or not.
- the active entity comprises an inhibitor, an antagonist, or an inverse agonist.
- the active entity may be an inhibitor, antagonist, or inverse agonist of a VEGFR.
- the active entity comprises or consists essentially of any one or more of compounds 1-59 in some embodiments.
- the active entity comprises compound 1 of Table 2.
- the composition comprises CT103.
- the active entity binds to a VEGFR in some embodiments.
- the active entity binds to VEGFR-1 in some embodiments.
- the active entity binds to VEGFR-2 in some embodiments.
- the active entity binds to VEGFR-3 in some embodiments.
- the binding may be partially or fully inhibitory or not.
- the polymer used in the reduced exposure compounds comprises polyethylene glycol (PEG) and/or methoxy-polyethylene glycol (m-PEG).
- the active entity has one or more carboxyl, hydroxyl, amino and/or sulfhydryl groups
- the active entity is PEGylated (or conjugated/coupled to another polymer) at one or more of said carboxyl, hydroxyl, amino and/or sulfhydryl groups.
- the reduced exposure compositions described herein are formulated for topical administration, such as eye drops, gels, ointments, liquids, etc. in several embodiments.
- Inhalants, injectables, nasal sprays, oral administration etc. are provided in some embodiments.
- the reduced exposure compositions described herein are formulated for oral administration in several embodiments.
- Inhalants, injectables, eye drops, nasal sprays, topical administration etc. are provided in some embodiments.
- methods of treating one or more of the following are provided: non- dermal inflammation, inflammatory bowel disease, inflammatory skin diseases, wounds, scars, autoimmune disorders, and cancerous or pre-cancerous lesions.
- Kits comprising one or more compounds and devices for administration (syringes, eye droppers, containers, inhalers, etc.) as well as instructions for use, are provided in certain embodiments.
- compositions may be administered via at least two routes of administration, either simultaneously or sequentially according to some embodiments.
- the composition is administered via a first (e.g. oral, topical dermal) route to a subject, wherein the subject further receives an additional agent via a second (e.g., injectable, non-dermal) route to achieve synergetic effects.
- methods for reducing exposure of a composition at at least one non-target site comprise applying a composition comprising at least one active entity linked to at least one polymer, wherein the combination of the active entity and polymer reduces exposure at the non-target site by more than 50% as compared to the active entity without the polymer.
- the composition may be applied topically, injected, inhaled, or administered orally.
- the non-target site includes non-target tissue at which pharmacological activity is not desired and/or not achieved.
- Non-target sites can include the bloodstream or systemic system.
- Non-target sites can also include the lymphatic system.
- methods of treating a respiratory disease in a subject via delivery of the polymer conjugates e.g., wherein the warhead is a small molecule targeting a JAK and/or STAT family protein
- Delivery routes may include for example intratracheal instillation or inhalation.
- the formulation may include liquids, nebulized or aerosolized liquids or suspensions, dry powder, nanocomposites, nanoparticles or microparticles, etc.
- Respiratory disorders include treatable obstructive, restrictive or inflammatory airways diseases of whatever type, etiology, or pathogenesis.
- Non-limiting examples of respiratory conditions include: acute bronchitis; acute laryngotracheal bronchitis; arachidic bronchitis; catarrhal bronchitis; croupus bronchitis; dry bronchitis; infectious asthmatic bronchitis; productive bronchitis; staphylococcus or streptococcal bronchitis; vesicular bronchitis; cylindric bronchiectasis; sacculated bronchiectasis; fusiform bronchiectasis; capillary bronchiectasis; cystic bronchiectasis; dry bronchiectasis; follicular bronchiectasis; chronic obstructive pulmonary disease (COPD), chronic obstructive lung disease (COLD), chronic obstructive airways disease (COAD) or small airways obstruction of whatever type, etiology, or pathogenesis,
- pneumoconiosis of whatever type, etiology, or pathogenesis in particular pneumoconiosis that is a member selected from the group consisting of aluminosis or bauxite workers' disease, anthracosis or miners' asthma, asbestosis or steam-fitters' asthma, chalicosis or flint disease, ptilosis caused by inhaling the dust from ostrich feathers, siderosis caused by the inhalation of iron particles, silicosis or grinders' disease, byssinosis or cotton-dust asthma and talc pneumoconiosis; interstitial lung diseases (ILD) or pulmonary fibrosis of whatever type, etiology
- ILD interstitial lung diseases
- pulmonary fibrosis of whatever type, etiology
- Respiratory disorders also include, in some embodiments, malignancies and tumors of the respiratory system, non- limiting examples of which include lung adenocarcinoma, squamous cell carcinoma, large cell carcinoma, bronchioloalveolar carcinoma (BAC), pulmonary adenocarcinoma (AIS), non-small-cell carcinoma, small cell carcinoma, and mesothelioma.
- a reduced exposure composition for inhibiting the activity of a therapeutic target at a target site is provided. Methods for treating diseases, conditions, and disorders are also provided.
- the composition comprises or consists essentially of a conjugate comprising or consists essentially of an active entity coupled (e.g., linked) to at least one polymer.
- the active entity may have, for example, selectivity and/or an inhibitory activity against the therapeutic target.
- the conjugate has increased selectivity and/or inhibitory activity compared to the unconjugated active entity.
- the polymer can include, for example, polyethylene glycol (PEG) and/or methoxy-polyethylene glycol (m-PEG).
- m-PEG methoxy-polyethylene glycol
- a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site is also provided.
- the conjugate has reduced exposure at a non-target site as compared to the unconjugated active entity.
- the non-target site includes for example the systemic system, the lymphatic system and/or other non-target tissue sites.
- the conjugate may have, in some embodiments, reduced systemic exposure and toxicity when delivered to the target site as compared to the unconjugated active entity.
- the target site includes cells and or tissues localized within one or more of the following: skin, scalp, eye, Gl tract, joint and/or lung.
- the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved.
- the conjugate has increased permeability across at least one of a nuclear and plasma membrane compared to the unconjugated active entity.
- the conjugate can advantageously traverse plasma membranes of cells at the target site, thereby promoting interactions between the active entity and the therapeutic target.
- This traversal may include the crossing of cellular lipid bilayers to, e.g., distribute the active entity among both lipophilic and hydrophilic cellular compartments.
- Membranes include the lipid bilayer, plasma membrane and the nuclear membrane as examples.
- the conjugate interacts with a kinase associated with the plasma membrane, cytoplasm and/or nucleus.
- the conjugate may exhibit a depot effect across cellular compartments, thereby reducing the dose of the active entity adequate to inhibit the cellular kinase compared to the unconjugated active entity.
- the conjugate is selected from:
- the therapeutic target may be a mediator of a condition, such as, for example, a respiratory condition, a gastrointestinal condition, an inflammatory condition.
- the therapeutic target may be, for example, one or more of VEGFR, c-Src, TkrA, MAP2K3, a JAK family kinase and/or a STAT family protein.
- the active entity and/or conjugate may have a longer residence time within a cell or other tissue at the target site compared to the active entity without conjugation to the polymer.
- the residence time of the active entity and/or conjugate within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, (i) at least 25% (e.g., 25-50%, 50-75%, 75-100%, 100-150%, or higher and overlapping ranges therein) longer and/or (ii) at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50- 100 fold, and overlapping ranges therein) longer.
- the residence time is over 100 fold longer.
- the conjugate increased residence time at the target site results in lower concentrations and/or lower frequency of
- a smaller dose of the conjugate may be adequate to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer.
- the dose of the conjugate sufficient to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer is at least 10% (e.g., 10-15%, 15-20%, 20-25%, 25- 30%, 30-40%, 40-50%, 50-60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%- 125%, 125- 150%, or higher and overlapping ranges therein) lower.
- the dose is over 200% lower.
- fewer doses and/or smaller doses of the conjugate are sufficient as compared to the active entity delivered without the polymer.
- the active entity and/or conjugate may have reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site compared to the active entity without conjugation to the polymer.
- the active entity and/or conjugate is present at a biologically inactive concentration within a cell or tissue at a non-target site.
- reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site e.g., the systemic system, the lymphatic system, bone marrow, the circulatory system
- the active entity and/or conjugate has reduced systemic absorption and/or little or no systemic toxicity when the composition is administered.
- the reduced exposure composition is formulated for topical delivery, oral delivery, respiratory delivery or injection into target sites, e.g., eyes and joints.
- Administration by topical delivery, oral delivery, rectal delivery, inhalation or instillation, and/or injection is provided in several embodiments.
- Topical delivery to body surfaces, such as, for example skin, eyes, ears, nose, mouth, lungs, vagina and/or rectum, is provided in some embodiments.
- effective amounts of the active entity are delivered to a subject (e.g., human or veterinary).
- the composition may further comprise one or more additional ingredients, such as, for example, an antibiotic agent, an anti-angiogenesis agent, a preventive or therapeutic agent for inflammatory bowel disease, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anesthetic agent, a steroidal anti-inflammatory agent, a non-steroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an anti-histamine agent, a vitamin or vitamin complex, and/or a hormone.
- additional ingredients such as, for example, an antibiotic agent, an anti-angiogenesis agent, a preventive or therapeutic agent for inflammatory bowel disease, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anesthetic agent, a steroidal anti-inflammatory agent, a non-steroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an anti-histamine agent, a vitamin or vitamin complex,
- the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following gastrointestinal conditions: inflammatory bowel disease, Crohn's disease, ulcerative colitis, collagenous colitis, lymphocytic colitis, ischemic colitis, diversion colitis, Behcet's syndrome, indeterminate colitis, irritable bowel syndrome and/or small intestinal bacterial overgrowth.
- the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following ophthalmic conditions: macular degeneration, age related macular degeneration (ARMD), choroidal neovascularization, retinopathy, diabetic retinopathy, acute macular neuroretinopathy, chronic macular neuroretinopathy, central serous chorioretinopathy, macular edema, cystoid macular edema, diabetic macular edema, acute multifocal placoid pigment epitheliopathy, Behcet's disease, birdshot retinochoroidopathy, uveitis, intermediate uveitis (pars planitis), anterior uveitis, multifocal choroiditis, multiple evanescent white dot syndrome (MEWDS), ocular sarcoidosis, posterior scleritis, serpignous choroiditis, subretinal fibrosis,
- the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following dermal conditions: psoriasis, psoriasis guttata, inverse psoriasis, pustular psoriasis, psoriatic erythroderma, acute febrile neutrophilic dermatosis, eczema, xerotic eczema, dyshidrotic eczema, vesicular palmar eczema, acne vulgaris, atopic dermatitis, contact dermatitis, allergic contact dermatitis, dermatomyositis, exfoliative dermatitis, hand eczema, pompholyx, keloids, rosacea, rosacea due to sarcoidosis, rosacea due to scleroderma, rosacea due to Sweet syndrome, rosacea due to system
- a reduced exposure composition for treating a target site with the gastrointestinal system is provided.
- Methods for treating the Gl and/or gastrointestinal conditions are also provided.
- the composition comprises or consists essentially of a conjugate comprising or consists essentially of an active entity coupled (e.g., linked) to at least one polymer.
- the active entity may be for example, an inhibitor, antagonist, or inverse agonist of a mediator of a gastrointestinal condition.
- the polymer can include, for example, polyethylene glycol (PEG) and/or methoxy-polyethylene glycol (m-PEG).
- a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site is also provided.
- the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer.
- the non-target site includes for example the systemic system, the lymphatic system and/or other non-target tissue sites.
- the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved.
- the conjugate can advantageously traverse plasma membranes of cells at the target site, thereby promoting interactions between the active entity and the mediator of the gastrointestinal condition.
- This traversal may include the crossing of cellular lipid bilayers to, e.g., distribute the active entity among both lipophilic and hydrophilic cellular compartments.
- Membranes include the lipid bilayer, plasma membrane and the nuclear membrane as examples.
- the conjugate interacts with mediator associated with the plasma membrane, cytoplasm and/or nucleus.
- the conjugate may exhibit a depot effect across cellular compartments, thereby reducing the dose of the active entity required to inhibit the cellular mediator compared to the active entity without conjugation to the polymer.
- the conjugate penetrates one or more physical barriers of the Gl, one or more physiological barriers of the Gl, and/or one or more biological barriers of the Gl (e.g. the epithelial barrier and/or intestinal mucosa).
- the conjugate can under transcytosis across the intestinal epithelium.
- the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a protein mediator of a condition (e.g., a kinase). In some embodiments, the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a mediator of a gastrointestinal condition.
- a protein mediator of a condition e.g., a kinase
- the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a mediator of a gastrointestinal condition.
- the mediator is one or more of c-Src, a VEGFR protein (e.g., VEGFR-1 , VEGFR-2, VEGFR-3), a JAK protein (e.g., of JAK1 , JAK2, JAK3, and Tyrosine kinase 2 (TYK2)), STAT, NF-Kappa B, TrkA, MAPK, MAP2K and/or MAP2K3.
- the active entity and/or conjugate is an inhibitor, antagonist, and/or inverse agonist of a mediator of inflammation.
- the mediator of inflammation may be, for example, a mediator of an inflammatory bowel disease.
- the active entity and/or conjugate reduces inflammation at the target site within the Gl.
- the reduced inflammation at the target site treats an inflammatory bowel disease (e.g. Crohn's disease, ulcerative colitis).
- the reduced exposure composition comprises one or more of conjugates SNA-101 , SNA- 103, SNA-352, SNA-120 and/or SNA-125.
- the active entity and/or conjugate exhibits greater access to the mediator compared to the active entity without conjugation to the polymer.
- the reduced exposure composition may be formulated for topical, oral, or suppository delivery.
- Topical, oral, and suppository administration is provided in several embodiments. In several embodiments, the administration is daily.
- effective amounts of the active entity are delivered to a subject (e.g., human or veterinary).
- composition may further comprise one or more additional ingredients, such as, for example, antibiotic agent, an anti-angiogenesis agent, a preventive or therapeutic agent for inflammatory bowel disease, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anesthetic agent, a steroidal anti-inflammatory agent, a non-steroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an antihistamine agent, a vitamin or vitamin complex, and/or a hormone.
- additional ingredients such as, for example, antibiotic agent, an anti-angiogenesis agent, a preventive or therapeutic agent for inflammatory bowel disease, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anesthetic agent, a steroidal anti-inflammatory agent, a non-steroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an antihistamine agent, a vitamin or vitamin complex, and/or a
- the active entity and/or conjugate may have an increased concentration, activity and/or bioavailability within a cell or tissue at the target site compared to the active entity without conjugation to the polymer.
- the therapeutically effective amount of the active entity is at the target site.
- the concentration, activity and/or bioavailability within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, at least 2-20 fold (e.g., 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 14- 16 fold, 18-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-100 fold, and overlapping ranges therein) greater than within a cell or tissue at a non-target site (e.g., the systemic system, the lymphatic system, bone marrow, outside the gastrointestinal tract).
- the concentration, activity and/or bioavailability within a cell or tissue at the target site is over 100 fold greater.
- the active entity and/or conjugate may have reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site compared to the active entity without conjugation to the polymer.
- the active entity and/or conjugate is present at a biologically inactive concentration within a cell or tissue at a non-target site.
- reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site advantageously reduces toxicity and/or other side effects, such as, for example, immunosuppression.
- the active entity and/or conjugate has reduced systemic absorption and/or little or no systemic toxicity when the composition is formulated for oral delivery and is administered orally (e.g., a single administration, administration on a daily basis).
- the active entity and/or conjugate may have a greater activity and/or bioavailability within a cell or other tissue at the target site (e.g., gastrointestinal tract) than within a cell or other tissue at a non-target site (e.g., outside the gastrointestinal tract).
- target site e.g., gastrointestinal tract
- non-target site e.g., outside the gastrointestinal tract
- the activity and/or bioavailability of the active entity and/or conjugate within a cell or other tissue at the target site is, as compared to within a cell or other tissue at a non-target site, (i) at least 25% (e.g., 25-50%, 50-75%, 75-100%, 100-150%, or higher and overlapping ranges therein) greater and/or (ii) at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) greater.
- the activity and/or bioavailability is over 100 fold greater within a cell or other tissue at the target site than within a cell or other tissue at a non-target site.
- the active entity and/or conjugate may have a longer residence time within a cell or other tissue at the target site (e.g., intestinal lamina limbal) compared to the active entity without conjugation to the polymer.
- the residence time of the active entity and/or conjugate within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, (i) at least 25% (e.g., 25-50%, 50-75%, 75-100%, 100-150%, or higher and overlapping ranges therein) longer and/or (ii) at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20- 30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) longer.
- the residence time is over 100 fold longer.
- the increased residence time is in one or
- the active entity and/or conjugate may have a shorter residence time within a cell or other tissue at a non-target site compared to the active entity without conjugation to the polymer.
- the residence time of the active entity and/or conjugate within a cell or other tissue at a non-target site is, as compared to the active entity without conjugation to the polymer, at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) shorter.
- the residence time is over 100 fold shorter.
- a smaller dose of the conjugate may be adequate to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer.
- the dose of the conjugate sufficient to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer is at least 10% (e.g., 10-15%, 15-20%, 20-25%, 25- 30%, 30-40%, 40-50%, 50-60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%- 125%, 125- 150%, or higher and overlapping ranges therein) lower.
- the dose is over 200% lower.
- fewer doses and/or smaller doses of the conjugate are required as compared to the active entity delivered without the polymer.
- the active entity and/or conjugate may have diminished systemic absorption compared to the active entity without conjugation to the polymer.
- the systemic absorption of the active entity and/or conjugate, as compared to the active entity without conjugation to the polymer is at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50- 100 fold, and overlapping ranges therein) lower.
- the systemic absorption is over 100 fold lower.
- the active entity and/or conjugate may have minimal systemic absorption following oral administration.
- the active entity and/or conjugate may have reduced clearance time from a non-target site compared to the active entity without conjugation to the polymer.
- the clearance time of the active entity and/or conjugate from a non-target site, as compared to the active entity without conjugation to the polymer is at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) lower.
- the clearance time is over 100 fold lower.
- the active entity and/or conjugate displays rapid systemic elimination when administered by intravenous, intramuscular, transmucosal, subcutaneous or intraperitoneal injection and/or infusion.
- the active entity and/or conjugate may have enhanced delivery to a cell or other tissue at the target site (e.g., Gl tract, intestinal lamina propria) compared to the active entity without conjugation to the polymer.
- the target site e.g., Gl tract, intestinal lamina propria
- at least 10% e.g., 10-15%, 15-20%, 20-25%, 25- 30%, 30-40%, 40-50%, 50-60%, 60-70%, 70-80%, 80-90%, 90-100%, and overlapping ranges therein
- the conjugate may have enhanced delivery to the intestinal epithelium and/or intestinal laminalitis as compared to the active entity delivered without the polymer.
- the conjugate is amphiphilic and/or amphipathic. In some embodiments, the conjugate is more amphiphilic and/or amphipathic than the active entity without conjugation to the polymer. For example, in several embodiments, the conjugate, as compared to the active entity without conjugation to the polymer, is at least 25% (e.g., 20-25%, 25-30%, 30-40%, 40-50%, 50- 60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%-125%, 125-150%, or higher and overlapping ranges therein) more amphiphilic. In one embodiment, the amphiphilicity is over 200% greater.
- the conjugate is more hydrophilic than the active entity without conjugation to the polymer.
- the conjugate, as compared to the active entity without conjugation to the polymer is at least 25% (e.g., 20-25%, 25-30%, 30-40%, 40-50%, 50- 60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%-125%, 125-150%, or higher and overlapping ranges therein) more hydrophilic.
- the hydrophilicity is over 200% greater.
- the greater hydrophilicity of the conjugate advantageously facilitates one or more of: non-compartmentalization within a cell or tissue at the target site; access to and activity in both the lipid bilayer and the cytosol of the cell; access to and/or activity in both the lipid bilayer and the cytoplasm of the cell; and/or access to and/or activity across the lipid bilayer.
- the target site includes one or more of the following: intestinal epithelium, intestinal lamina intestinal, the lining of the gastrointestinal tract, immune cells residing within the intestinal lamina propria, muscularis mucosae, myenteric plexus, the submucosa, the muscular layer, intraperitoneal spaces, retroperitoneal spaces, serosa, adventitia.
- the target site includes immune cells, and/or non-immune cells.
- the conjugate targets immune cells residing within the Gl epithelial layer and/or intestinal laminalitis.
- the target site includes the gastrointestinal tract and the non-target site includes non-gastrointestinal tract tissue.
- the target site includes the small intestine and the non-target site includes one or more of the large intestine and stomach. In one embodiment, the target site includes the large intestine and the non-target site includes one or more of the small intestine and stomach. In some embodiments, the target site includes the small intestine and large intestine and the non- target site includes the stomach. In one embodiment, the target site includes the intestinal lamina intestinal and the non-target site includes tissue contacting the intestinal lamina intestinal. In several embodiments, the target site includes immune cells of the intestinal lamina limbal, and the non-target site includes non-immune cells.
- the target site includes the intestinal lamina intestinal and/or gastric parietal cells and the non-target site comprises sites other than intestinal lamina intestinal and/or gastric parietal cells.
- the target site includes one or two of the duodenum, jejunum and ileum and the non- target site includes the remaining one or two.
- the target site includes one or two of the ascending colon, transverse colon and the descending colon and the non- target site comprises the remaining one or two.
- the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following conditions: inflammatory bowel disease, irritable bowel syndrome, small intestinal bacterial overgrowth, Crohn's disease, ulcerative colitis, collagenous colitis, lymphocytic colitis, ischemic colitis, diversion colitis, Behcet's syndrome, small intestinal bacterial overgrowth, and indeterminate colitis.
- the method of treatment and/or use of the compositions described herein are provided for reducing immunosuppression and/or an inflammatory response while treating a gastrointestinal condition in a subject in need thereof.
- the method of treatment and/or use of the compositions described herein are provided for reducing reduce liver damage, neutropenia and/or lymphopenia while treating a gastrointestinal condition in a subject in need thereof.
- a reduced exposure composition for treating a dermal target site e.g., the skin
- a dermal target site e.g., the skin
- Methods for treating the skin and/or dermal conditions are also provided.
- the composition comprises or consists essentially of a conjugate comprising or consists essentially of an active entity coupled (e.g., linked) to at least one polymer.
- the active entity may be for example, an inhibitor, antagonist, or inverse agonist of a mediator of a dermal condition.
- the polymer can include, for example, polyethylene glycol (PEG) and/or methoxy-polyethylene glycol (m- PEG).
- a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site is also provided.
- the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer.
- the non-target site includes for example the systemic system, the lymphatic system and/or other non-target tissue sites.
- the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved.
- the conjugate can advantageously traverse plasma membranes of cells at the target site, thereby promoting interactions between the active entity and the mediator of the dermal condition.
- This traversal may include the crossing of cellular lipid bilayers to, e.g., distribute the active entity among both lipophilic and hydrophilic cellular compartments.
- Membranes include the lipid bilayer, plasma membrane and the nuclear membrane as examples.
- the conjugate interacts with mediator associated with the plasma membrane, cytoplasm and/or nucleus.
- the conjugate may exhibit a depot effect across cellular compartments, thereby reducing the dose of the active entity required to inhibit the cellular mediator compared to the active entity without conjugation to the polymer.
- the conjugate penetrates one or more physical barriers of the skin, one or more physiological barriers of the skin, and/or one or more biological barriers of the skin.
- the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a protein mediator of a condition (e.g., a kinase). In some embodiments, the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a mediator of a dermal condition.
- a protein mediator of a condition e.g., a kinase
- the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a mediator of a dermal condition.
- the mediator is one or more of c-Src, a VEGFR protein (e.g., VEGFR-1 , VEGFR-2, VEGFR- 3), a JAK protein (e.g., of JAK1 , JAK2, JAK3, and Tyrosine kinase 2 (TYK2)), STAT, NF-Kappa B, TrkA, MAPK, MAP2K and/or MAP2K3.
- the active entity and/or conjugate is an inhibitor, antagonist, and/or inverse agonist of a mediator of inflammation.
- the mediator of inflammation may be, for example, a mediator of an inflammatory skin condition.
- the active entity and/or conjugate reduces inflammation at the target site.
- the reduced inflammation at the target site treats an inflammatory skin condition.
- the reduced exposure composition comprises one or more of conjugates SNA-101 , SNA-103, SNA-352, SNA-120 and/or SNA-125.
- the active entity and/or conjugate exhibits greater access to the mediator compared to the active entity without conjugation to the polymer.
- the reduced exposure composition is formulated for topical delivery.
- Topical administration is provided in several embodiments. In several embodiments, the administration is daily.
- effective amounts of the active entity are delivered to a subject (e.g., human or veterinary).
- the composition may further comprise one or more additional ingredients, such as, for example, a protective agent, an emollient, an astringent, a humectant, a sun screening agent, a sun tanning agent, a UV absorbing agent, an antibiotic agent, an anti-angiogenesis agent, a physiological cooling agent, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anti-acne agent, an anesthetic agent, a steroidal anti-inflammatory agent, a non-steroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an antihistamine agent, a vitamin or vitamin complex, a hormone, an anti-dandruff agent, an anti-wrinkle agent, an anti-skin atrophy agent, a skin whitening agent, and/or a cleansing agent.
- additional ingredients such as, for example, a protective agent, an emollient, an astringent, a humectant,
- the active entity and/or conjugate may have an increased concentration, activity and/or bioavailability within a cell or tissue at the target site compared to the active entity without conjugation to the polymer.
- the therapeutically effective amount of the active entity is at the target site.
- the concentration, activity and/or bioavailability within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, at least 2-20 fold (e.g.
- a cell or tissue at a non-target site e.g. , the systemic system, the lymphatic system, bone marrow, outside the skin.
- a concentration, activity and/or bioavailability within a cell or tissue at the target site is over 100 fold greater.
- the active entity and/or conjugate may have reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site compared to the active entity without conjugation to the polymer.
- the active entity and/or conjugate is present at a biologically inactive concentration within a cell or tissue at a non-target site.
- reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site e.g. , the systemic system, the lymphatic system, bone marrow, outside the skin
- the active entity and/or conjugate has reduced systemic absorption and/or little or no systemic toxicity when the composition is administered topically (e.g. , a single administration, administration on a daily basis) .
- the active entity and/or conjugate may have a greater activity and/or bioavailability within a cell or other tissue at the target site (e.g. , skin) than within a cell or other tissue at a non-target site.
- the activity and/or bioavailability of the active entity and/or conjugate within a cell or other tissue at the target site is, as compared to within a cell or other tissue at a non-target site, (i) at least 25% (e.g. , 25-50%, 50-75%, 75-100%, 100- 150%, or higher and overlapping ranges therein) greater and/or (ii) at least 2-20 fold (e.g.
- the activity and/or bioavailability is over 100 fold greater within a cell or other tissue at the target site than within a cell or other tissue at a non-target site.
- the active entity and/or conjugate may have a longer residence time within a cell or other tissue at the target site (e.g., skin) compared to the active entity without conjugation to the polymer.
- the residence time of the active entity and/or conjugate within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, (i) at least 25% (e.g., 25-50%, 50-75%, 75-100%, 100-150%, or higher and overlapping ranges therein) longer and/or (ii) at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) longer.
- the residence time is over 100 fold longer.
- the increased residence time is in the skin.
- the active entity and/or conjugate may have a shorter residence time within a cell or other tissue at a non-target site compared to the active entity without conjugation to the polymer.
- the residence time of the active entity and/or conjugate within a cell or other tissue at a non-target site is, as compared to the active entity without conjugation to the polymer, at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) shorter.
- the residence time is over 100 fold shorter.
- a smaller dose of the conjugate may be adequate to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer.
- the dose of the conjugate sufficient to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer is at least 10% (e.g. , 10-15%, 15-20%, 20-25%, 25- 30%, 30-40%, 40-50%, 50-60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%- 125%, 125- 150%, or higher and overlapping ranges therein) lower.
- the dose is over 200% lower.
- fewer doses and/or smaller doses of the conjugate are required as compared to the active entity delivered without the polymer.
- the active entity and/or conjugate may have diminished systemic absorption compared to the active entity without conjugation to the polymer.
- the systemic absorption of the active entity and/or conjugate, as compared to the active entity without conjugation to the polymer is at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50- 100 fold, and overlapping ranges therein) lower.
- the systemic absorption is over 100 fold lower.
- the active entity and/or conjugate may have minimal systemic absorption following epicutaneous administration.
- the active entity and/or conjugate may have reduced clearance time from a non-target site compared to the active entity without conjugation to the polymer.
- the clearance time of the active entity and/or conjugate from a non-target site, as compared to the active entity without conjugation to the polymer is at least 2-20 fold (e.g. , 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12- 14 fold, 14-16 fold, 16- 18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) lower.
- the clearance time is over 100 fold lower.
- the active entity and/or conjugate displays rapid systemic elimination when administered by intravenous, intramuscular, transmucosal, subcutaneous or intraperitoneal injection and/or infusion.
- the active entity and/or conjugate may have enhanced delivery to a cell or other tissue at the target site (e.g. , dermis, epidermis, subcutis) compared to the active entity without conjugation to the polymer.
- a cell or other tissue at the target site e.g. , dermis, epidermis, subcutis
- at least 10% e.g. , 10-15%, 15-20% , 20-25%, 25-30%, 30-40%, 40-50% , 50-60%, 60-70%, 70-80%, 80-90%, 90- 100%, and overlapping ranges therein
- the conjugate may have enhanced delivery to the epidermis, dermis, and/or subcutis as compared to the active entity delivered without the polymer.
- the conjugate is amphiphilic and/or amphipathic. In some embodiments, the conjugate is more amphiphilic and/or amphipathic than the active entity without conjugation to the polymer. For example, in several embodiments, the conjugate, as compared to the active entity without conjugation to the polymer, is at least 25% (e.g. , 20-25%, 25-30%, 30-40%, 40-50%, 50- 60%, 60-70%, 70-80% , 80-90% , 90-100% , 100%-125%, 125- 150%, or higher and overlapping ranges therein) more amphiphilic. In one embodiment, the amphiphilicity is over 200% greater.
- the conjugate is more hydrophilic than the active entity without conjugation to the polymer.
- the conjugate, as compared to the active entity without conjugation to the polymer is at least 25% (e.g. , 20-25%, 25-30%, 30-40%, 40-50%, 50- 60%, 60-70%, 70-80% , 80-90% , 90-100% , 100%-125%, 125- 150%, or higher and overlapping ranges therein) more hydrophilic.
- the hydrophilicity is over 200% greater.
- the greater hydrophilicity of the conjugate advantageously facilitates one or more of: non-compartmentalization within a cell or tissue at the target site; access to and activity in both the lipid bilayer and the cytosol of the cell; access to and/or activity in both the lipid bilayer and the cytoplasm of the cell; and/or access to and/or activity across the lipid bilayer.
- the target site includes cells localized within one or more of the following: the stratum corneum, stratum lucidum, stratum granulosum, stratum spinosum, and stratum basale layers, the papillary layer of the dermis, the reticular layer of the dermis, sebaceous glands, arrector pili muscles, sensory nerve fibers, hair follicles, hair roots, pacinian corpuscles, hair root plexus, eccrine sweat glands, the hypodermis and/or the superficial fascia.
- the target cells may comprise immune cells, non-immune cells and/or keratinocytes.
- the conjugate targets immune cells residing within dermis, epidermis, and/or subcutis.
- the target site comprises immune cells of the epidermis or dermis and the non-target site comprises non-immune cells.
- the target site includes the epidermis and the non-target site includes the dermis, gland, hypodermis and/or blood vessels.
- the target site includes the dermis and the non-target site includes the epidermis, gland, hypodermis and/or blood vessels.
- the target site includes the epidermis and the non-target site includes the dermis, gland, blood vessels, and/or hypodermis.
- the target site includes the dermis and the non- target site includes the epidermis, gland hypodermis and/or blood vessels.
- the target site includes one or more of the epidermis, follicle, gland, blood vessels, dermis and subcutis, and the non-target site includes the remaining sites.
- the target site includes the subcutis and the non-target site includes tissue contacting the subcutis.
- the target site includes the skin and the non-target site includes non-integumentary tissue.
- the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following conditions: psoriasis, psoriasis guttata, inverse psoriasis, pustular psoriasis, psoriatic erythroderma, acute febrile neutrophilic dermatosis, eczema, xerotic eczema, dyshidrotic eczema, vesicular palmar eczema, acne vulgaris, atopic dermatitis, contact dermatitis, allergic contact dermatitis, dermatomyositis, exfoliative dermatitis, hand eczema, pompholyx, keloids, rosacea, rosacea due to sarcoidosis, rosacea due to scleroderma, rosacea due to Sweet syndrome, rosacea due to systemic
- the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of a vascular tumor, e.g., Kaposi's sarcoma, lymphangioma, glomangioma, angiosarcoma, hemangioendothelioma, and infantile hemangiomas.
- a vascular tumor e.g., Kaposi's sarcoma, lymphangioma, glomangioma, angiosarcoma, hemangioendothelioma, and infantile hemangiomas.
- the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of a skin neoplasia, e.g., squamous cell carcinoma, basal cell carcinoma, malignant melanoma, malignant cutaneous lymphoma, Kaposi's sarcoma, Merkel cell skin cancer, and non- melanoma skin cancer.
- a skin neoplasia e.g., squamous cell carcinoma, basal cell carcinoma, malignant melanoma, malignant cutaneous lymphoma, Kaposi's sarcoma, Merkel cell skin cancer, and non- melanoma skin cancer.
- the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of a bullous disease, e.g., bullous pemphigoid, erythema multiforme, dermatitis herpetiformis, epidermolysis bullosa acquisita, linear Immunoglobulin A disease, mucous membrane pemphigoid, pemphigoid gestationis, pemphigus foliaceus, and pemphigus vulgaris.
- a bullous disease e.g., bullous pemphigoid, erythema multiforme, dermatitis herpetiformis, epidermolysis bullosa acquisita, linear Immunoglobulin A disease, mucous membrane pemphigoid, pemphigoid gestationis, pemphigus foliaceus, and pemphigus vulgaris.
- the method of treatment and/or use of the compositions described herein are provided for the modulation of hair growth and cycling. In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of alopecia.
- a reduced exposure composition for treating a target site in the eye is provided.
- Methods for treating the eye and/or ophthalmic conditions are also provided.
- the composition comprises or consists essentially of a conjugate comprising or consists essentially of an active entity coupled (e.g., linked) to at least one polymer.
- the active entity may be for example, an inhibitor, antagonist, or inverse agonist of a mediator of an ophthalmic condition.
- the polymer can include, for example, polyethylene glycol (PEG) and/or methoxy-polyethylene glycol (m- PEG).
- a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site is also provided.
- the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer.
- the non-target site includes for example the systemic system, the lymphatic system and/or other non-target tissue sites.
- the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved.
- the conjugate can advantageously traverse plasma membranes of cells at the target site, thereby promoting interactions between the active entity and the mediator of the ophthalmic condition.
- This traversal may include the crossing of cellular lipid bilayers to, e.g., distribute the active entity among both lipophilic and hydrophilic cellular compartments.
- Membranes include the lipid bilayer, plasma membrane and the nuclear membrane as examples.
- the conjugate may exhibit a depot effect across cellular compartments, thereby reducing the dose of the active entity required to inhibit the cellular mediator compared to the active entity without conjugation to the polymer.
- the conjugate penetrates one or more physical barriers of the eye, one or more physiological barriers of the eye, and/or one or more biological barriers of the eye, such as, for example, the conjunctival epithelium, Tenon's fascia, episclera, sclera, and/or choroid.
- the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a protein mediator of a condition (e.g., a kinase). In some embodiments, the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a mediator of an ophthalmic condition.
- a protein mediator of a condition e.g., a kinase
- the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a mediator of an ophthalmic condition.
- the mediator is one or more of c-Src, a VEGFR protein (e.g., VEGFR-1 , VEGFR-2, VEGFR-3), a JAK protein (e.g., of JAK1 , JAK2, JAK3, and Tyrosine kinase 2 (TYK2)), STAT, NF-Kappa B, TrkA, MAPK, MAP2K and/or MAP2K3.
- the active entity and/or conjugate is an inhibitor, antagonist, and/or inverse agonist of a mediator of inflammation.
- the mediator of inflammation may be, for example, a mediator of an inflammatory ocular condition.
- the active entity and/or conjugate reduces inflammation at the target site.
- the reduced inflammation at the target site treats an inflammatory ocular condition, such as, for example, uveitis.
- the reduced exposure composition comprises one or more of conjugates SNA-101 , SNA-103, SNA-352, SNA- 120 and/or SNA- 125.
- the active entity and/or conjugate exhibits greater access to the mediator compared to the active entity without conjugation to the polymer.
- the reduced exposure composition is formulated for local ocular delivery via a topical or injectable.
- Intra-ocular admistration is provided in several embodiments.
- the composition may be formulated for, e.g., subconjunctival, intravitreal, retrobulbar or intracameral delivery.
- effective amounts of the active entity are delivered to a subject (e.g., human or veterinary).
- the composition may further comprise one or more additional ingredients, such as, for example, an antibiotic agent, an anti-angiogenesis agent, a preventive or therapeutic agent for inflammatory bowel disease, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anesthetic agent, a steroidal anti-inflammatory agent, a non-steroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an anti-histamine agent, a vitamin or vitamin complex, and/or a hormone.
- additional ingredients such as, for example, an antibiotic agent, an anti-angiogenesis agent, a preventive or therapeutic agent for inflammatory bowel disease, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anesthetic agent, a steroidal anti-inflammatory agent, a non-steroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an anti-histamine agent, a vitamin or vitamin complex,
- the active entity and/or conjugate may have a longer residence time within a cell or other tissue at the target site compared to the active entity without conjugation to the polymer.
- the residence time of the active entity and/or conjugate within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, (i) at least 25% (e.g., 25-50%, 50-75%, 75-100%, 100-150%, or higher and overlapping ranges therein) longer and/or (ii) at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50- 100 fold, and overlapping ranges therein) longer.
- the residence time is over 100 fold longer.
- the increased residence time is at the anterior segment of the eye and/or anterior segment of the eye.
- the active entity and/or conjugate may have a shorter residence time within a cell or other tissue at a non-target site compared to the active entity without conjugation to the polymer.
- the residence time of the active entity and/or conjugate within a cell or other tissue at a non-target site is, as compared to the active entity without conjugation to the polymer, at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) shorter.
- the residence time is over 100 fold shorter.
- a smaller dose of the conjugate may be adequate to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer.
- the dose of the conjugate sufficient to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer is at least 10% (e.g. , 10-15%, 15-20%, 20-25%, 25- 30%, 30-40%, 40-50%, 50-60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%- 125%, 125- 150%, or higher and overlapping ranges therein) lower.
- the dose is over 200% lower.
- fewer doses and/or smaller doses of the conjugate are required as compared to the active entity delivered without the polymer.
- the active entity and/or conjugate may have diminished systemic absorption compared to the active entity without conjugation to the polymer.
- the systemic absorption of the active entity and/or conjugate, as compared to the active entity without conjugation to the polymer is at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50- 100 fold, and overlapping ranges therein) lower.
- the systemic absorption is over 100 fold lower.
- the active entity and/or conjugate may have reduced clearance time from a non-target site compared to the active entity without conjugation to the polymer.
- the clearance time of the active entity and/or conjugate from a non-target site, as compared to the active entity without conjugation to the polymer is at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) lower.
- the clearance time is over 100 fold lower.
- the active entity and/or conjugate may have enhanced delivery to a cell or other tissue at the target site compared to the active entity without conjugation to the polymer.
- at least 10% e.g., 10-15%, 15-20%, 20-25%, 25-30%, 30-40%, 40-50%, 50-60%, 60-70%, 70-80%, 80-90%, 90-100%, and overlapping ranges therein
- the conjugate may have enhanced delivery to the posterior segment of the eye and/or anterior segment of the eye as compared to the active entity delivered without the polymer.
- the active entity and/or conjugate may have an increased concentration, activity and/or bioavailability within a cell or tissue at the target site compared to the active entity without conjugation to the polymer.
- the therapeutically effective amount of the active entity is at the target site.
- the concentration, activity and/or bioavailability within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, at least 2-20 fold (e.g., 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 14- 16 fold, 18-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-100 fold, and overlapping ranges therein) greater than within a cell or tissue at a non-target site (e.g., the systemic system, the lymphatic system, bone marrow, outside the eye).
- the concentration, activity and/or bioavailability within a cell or tissue at the target site is over 100 fold greater.
- the active entity and/or conjugate may have reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site compared to the active entity without conjugation to the polymer.
- the active entity and/or conjugate is present at a biologically inactive concentration within a cell or tissue at a non-target site.
- reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site advantageously reduces toxicity and/or other side effects, such as, for example, immunosuppression.
- the conjugate is amphiphilic and/or amphipathic. In some embodiments, the conjugate is more amphiphilic and/or amphipathic than the active entity without conjugation to the polymer. For example, in several embodiments, the conjugate, as compared to the active entity without conjugation to the polymer, is at least 25% (e.g., 20-25%, 25-30%, 30-40%, 40-50%, 50- 60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%-125%, 125-150%, or higher and overlapping ranges therein) more amphiphilic. In one embodiment, the amphiphilicity is over 200% greater.
- the conjugate is more hydrophilic than the active entity without conjugation to the polymer.
- the conjugate, as compared to the active entity without conjugation to the polymer is at least 25% (e.g., 20-25%, 25-30%, 30-40%, 40-50%, 50- 60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%-125%, 125-150%, or higher and overlapping ranges therein) more hydrophilic.
- the hydrophilicity is over 200% greater.
- the greater hydrophilicity of the conjugate advantageously facilitates one or more of: non-compartmentalization within a cell or tissue at the target site; access to and activity in both the lipid bilayer and the cytosol of the cell; access to and/or activity in both the lipid bilayer and the cytoplasm of the cell; and/or access to and/or activity across the lipid bilayer.
- the target site includes one or more of the following: the anterior segment of the eye, the posterior segment of the eye, an anterior sub-Tenon space, an anterior suprachoroidal space, an anterior intrascleral space, a posterior sub-Tenon space, a posterior suprachoroidal space and a posterior intrascleral space, the conjunctival epithelium, Tenon's fascia, episclera, sclera, choroid, the cornea, lens, sclera, anterior chamber, iris, posterior chamber, choroid, retina, Bowman's layer, stroma, Descemet's membrane, the endothelium, Tenon's Capsule and any combination thereof.
- the target site includes target immune cells residing within the anterior segment of the eye and/or posterior segment of the eye.
- the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following conditions: glaucoma, allergic conditions, inflammatory conditions of the anterior segment and cornea, allergic conditions of the anterior segment and cornea, infectious conditions of the anterior segment and cornea, and corneal angiogenesis, corneal edema, macular edema, dry eye, uveitis, macular degeneration, age related macular degeneration (ARMD), diabetic retinopathy, inflammatory conditions of the posterior segment, infectious conditions of the posterior segment, neurodegenerative disease, and vascular disease of the posterior segment, choroidal neovascularization, retinopathy, diabetic retinopathy, acute macular neuroretinopathy, chronic macular neuroretinopathy, central serous chorioretinopathy, macular edema, cystoid macular edema, or diabetic macular edema, acute multifocal placoid pigment epitheliopathy, Be
- the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of tears (e.g., retinal tear, giant retinal tear), laceration, abrasion, retinal detachment, macular hole, floaters, etc.
- tears e.g., retinal tear, giant retinal tear
- laceration e.g., abrasion, retinal detachment, macular hole, floaters, etc.
- the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of a retinal disease associated with tumors, congenital hypertrophy of the RPE, posterior uveal melanoma, choroidal hemangioma, choroidal osteoma, choroidal metastasis, combined hamartoma of the retina and retinal pigmented epithelium, retinoblastoma, vasoproliferative tumors of the ocular fundus, retinal astrocytoma, or intraocular lymphoid tumors.
- the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of punctate inner choroidopathy, acute posterior multifocal placoid pigment epitheliopathy, myopic retinal degeneration, or acute retinal pigment epithelitis.
- a reduced exposure composition for inhibiting the activity of a therapeutic target in the respiratory tract is provided.
- Methods and uses for treating respiratory conditions are also provided.
- the composition comprises or consists essentially of a conjugate comprising or consists essentially of an active entity coupled (e.g., linked) to at least one polymer.
- the active entity may be for example, an inhibitor, antagonist, or inverse agonist of a mediator of a respiratory condition.
- the active entity can have a selectivity and/or an inhibitory activity against the therapeutic target.
- the conjugate has increased selectivity and/or inhibitory activity compared to the unconjugated active entity.
- the polymer can include, for example, polyethylene glycol (PEG) and/or methoxy-polyethylene glycol (m-PEG).
- a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site is also provided.
- the conjugate has reduced exposure at a non-target site as compared to the unconjugated active entity.
- the non-target site includes for example the systemic system, the lymphatic system and/or other non-target tissue sites.
- the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved.
- the conjugate can advantageously traverse plasma membranes of cells at the target site compared to the unconjugated active entity, thereby promoting interactions between the active entity and the mediator of a respiratory condition.
- This traversal may include the crossing of cellular lipid bilayers to, e.g., distribute the active entity among both lipophilic and hydrophilic cellular compartments.
- Membranes include the lipid bilayer, plasma membrane and the nuclear membrane as examples.
- the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following conditions: acute bronchitis; acute laryngotracheal bronchitis; arachidic bronchitis; catarrhal bronchitis; croupus bronchitis; dry bronchitis; infectious asthmatic bronchitis; productive bronchitis; staphylococcus or streptococcal bronchitis; vesicular bronchitis; cylindric bronchiectasis; sacculated bronchiectasis; fusiform bronchiectasis; capillary bronchiectasis; cystic bronchiectasis; dry bronchiectasis; follicular bronchiectasis; chronic obstructive pulmonary disease (COPD), chronic obstructive lung disease (COLD
- pneumoconiosis of whatever type, etiology, or pathogenesis in particular pneumoconiosis that is a member selected from the group consisting of aluminosis or bauxite workers' disease, anthracosis or miners' asthma, asbestosis or steam-fitters' asthma, chalicosis or flint disease, ptilosis caused by inhaling the dust from ostrich feathers, siderosis caused by the inhalation of iron particles, silicosis or grinders' disease, byssinosis or cotton-dust asthma and talc pneumoconiosis; interstitial lung diseases (ILD) or pulmonary fibrosis of whatever type, etiology
- ILD interstitial lung diseases
- pulmonary fibrosis of whatever type, etiology
- Respiratory disorders also include, in some embodiments, malignancies and tumors of the respiratory system, non- limiting examples of which include lung adenocarcinoma, squamous cell carcinoma, large cell carcinoma, bronchioloalveolar carcinoma (BAC), pulmonary adenocarcinoma (AIS), non-small-cell carcinoma, small cell carcinoma, and/or mesothelioma.
- malignancies and tumors of the respiratory system non- limiting examples of which include lung adenocarcinoma, squamous cell carcinoma, large cell carcinoma, bronchioloalveolar carcinoma (BAC), pulmonary adenocarcinoma (AIS), non-small-cell carcinoma, small cell carcinoma, and/or mesothelioma.
- Figure 1 depicts the BioMAP profile of SNA-101 in the Diversity PLUS Panel at the indicated concentrations.
- the X-axis lists the quantitative protein-based biomarker readouts measured in each system.
- the grey region around the Y-axis represents the 95% significance envelope generated from historical vehicle controls.
- Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (
- Biomarker key activities are described as modulated if these activities increase in some systems, but decrease in others.
- Cytotoxicity is indicated on the profile plot by a thin black arrow above the X-axis, and antiproliferative effects are indicated by a thick grey arrow. Cytotoxicity and antiproliferative arrows may only require one concentration to meet the indicated threshold for profile annotation. Other BioMAP profiles disclosed herein are also depicted in a similar manner.
- Figure 2 depicts a Reference Benchmark Overlay of SNA- 101 and Benchmark Apremilast. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- Figure 3 depicts an overlay of SNA-101 (29 ⁇ ) and Topiramate (3.3 ⁇ ), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents.
- Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- FIG. 4 depicts Mechanism HeatMAP Analysis for SNA-101 .
- Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-101 from the 19 consensus mechanism profiles.
- Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control.
- FIG. 5 depicts the BioMAP profile of SNA-101 in the Diversity PLUS Panel at the indicated concentrations.
- the X-axis lists the quantitative protein-based biomarker readouts measured in each system.
- the grey region around the Y-axis represents the 95% significance envelope generated from historical vehicle controls.
- Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (
- Biomarker key activities are described as modulated if these activities increase in some systems, but decrease in others.
- Cytotoxicity is indicated on the profile plot by a thin black arrow above the X-axis, and antiproliferative effects are indicated by a thick grey arrow. Cytotoxicity and antiproliferative arrows may only require one concentration to meet the indicated threshold for profile annotation. Other BioMAP profiles disclosed herein are also depicted in a similar manner.
- Figure 6 depicts a Reference Benchmark Overlay of SNA-101 and Benchmark Staurosporine. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- Figure 7 depicts an overlay of SNA-101 (300 ⁇ ) and N037 (490 ng/ml), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents for SNA-101 (300 ⁇ ).
- Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- Figure 8 depicts an overlay of SNA-101 (100 ⁇ ) and Infliximab (30000 ng/ml), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents for SNA-101 (100 ⁇ ).
- Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- FIG. 9 depicts Mechanism HeatMAP Analysis for SNA-101 .
- Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-101 from the 19 consensus mechanism profiles.
- Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control.
- Figure 10 depicts the BioMAP profile of SNA-103 in the Diversity PLUS Panel. The X-axis lists the quantitative protein-based biomarker readouts measured in each system.
- the grey region around the Y-axis represents the 95% significance envelope generated from historical vehicle controls.
- Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (
- Other BioMAP profiles disclosed herein are also depicted in a similar manner.
- Figure 1 1 depicts a Reference Benchmark Overlay of SNA-103 and Benchmark Calcipotriene. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- Figure 12 depicts an overlay of SNA-103 (230 ⁇ ) and Apoptolidin (1 ⁇ ), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents for SNA-103 (230 ⁇ ).
- Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- FIG. 13 depicts Mechanism HeatMAP Analysis for SNA-103.
- Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-103 from the 19 consensus mechanism profiles.
- Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control.
- Figure 14 depicts clustering of test agent profiles following pairwise correlation analysis and clustering of the most similar profiles.
- Each colored circle represents the BioMAP profile of a compound at a specific concentration, with larger circles representing higher concentrations.
- Figure 15 depicts the BioMAP profile of SNA-352 in the Diversity PLUS Panel.
- the X-axis lists the quantitative protein-based biomarker readouts measured in each system.
- the grey region around the Y-axis represents the 95% significance envelope generated from historical vehicle controls.
- Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (
- Figure 16 depicts a Reference Benchmark Overlay of SNA-352 and Benchmark Cyclosporin A. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- Figure 17 depicts the changes in secretion of (a) IL-17F (b) IgG, (c) IL- 17A, and (d) TNFa in the BioMAP BT system mediated by SNA-352 (3.9 ⁇ ), Tofacitinib (3.3 ⁇ ), Apremilast (3.3 ⁇ ), SR221 1 (3.3 ⁇ ), and Cyclosporin A (3.3 ⁇ ).
- Figure 18 depicts an overlay of SNA-352 (3.9 ⁇ ) and Deferoxamine Mesylate (4.4 ⁇ ), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents.
- Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- FIG. 19 depicts Mechanism HeatMAP Analysis for SNA-352. HeatMAP analysis of the 148 biomarker readouts (rows) within the Diversity PLUS panel by SNA-352 in comparison to 19 consensus mechanism class profiles (columns). Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-352 from the 19 consensus mechanism profiles. Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control. [0127] Figure 20 depicts the SNA-352 kinase inhibition profile at test concentrations of 100nM and 200nM for the top inhibited kinases as well as those kinases in the middle in the inhibition spectrum.
- Figure 21 depicts a JAK2 vs Staurosporine concentration-%inhibition curve used to derive the slope (1 .853), R 2 (1 .00), and IC 50 (4.30E-10).
- Figure 22 depicts a JAK2 vs CT340 concentration-%inhibition curve used to derive the slope (1 .147), R 2 (1 .00), and IC 50 (1 .35E-07).
- Figure 23 depicts a JAK3 vs Staurosporine concentration-%inhibition curve used to derive the slope (1 .597), R 2 (1 .00), and IC 50 (2.78E-10).
- Figure 24 depicts a JAK3 vs CT340 concentration-%inhibition curve used to derive the slope (1 .164), R 2 (1 .00), and IC 50 (3.87E-08).
- Figure 25 depicts a PDGFRb vs Staurosporine concentration- %inhibition curve used to derive the slope (2.900), R 2 (1 .00), and IC 50 (3.87E-10).
- Figure 26 depicts a PDGFRb vs CT340 concentration-%inhibition curve used to derive the slope (1 .165), R 2 (1 .00), and IC 50 (1 .12E-07).
- Figure 27 depicts a TRKA vs Staurosporine concentration-%inhibition curve used to derive the slope (2.106), R 2 (1 .00), and IC 50 (5.02E-10).
- Figure 28 depicts a TRKA vs CT340 concentration-%inhibition curve used to derive the slope (1 .159), R 2 (1 .00), and IC 50 (2.55E-08).
- Figure 29 depicts a MAP2K1 vs Staurosporine concentration- %inhibition curve used to derive the slope (1 .287), R 2 (1 .00), and IC 50 (1 .39E-09).
- Figure 30 depicts a MAP2K1 vs CT340 concentration-%inhibition curve used to derive the slope (1 .434), R 2 (1 .00), and IC 50 (1 .96E-08).
- Figure 31 depicts a MAP2K3 vs Staurosporine concentration- %inhibition curve used to derive the slope (1 .402), R 2 (1 .00), and IC 50 (1 .13E-09).
- Figure 32 depicts a MAP2K3 vs CT340 concentration-%inhibition curve used to derive the slope (1 .41 1 ), R 2 (1 .00), and IC 50 (1 .26E-08).
- Figure 33 depicts a TAK1 -TAB1 vs Staurosporine concentration- %inhibition curve used to derive the slope (1 .369), R 2 (.98), and IC 50 (4.14E-08).
- Figure 34 depicts a TAK1 -TAB1 vs CT340 concentration-%inhibition curve used to derive the slope (1 .480), R 2 (.98), and IC 50 (2.19E-07).
- Figure 35 depicts the BioMAP profile of SNA-120 in the Diversity PLUS Panel.
- the X-axis lists the quantitative protein-based biomarker readouts measured in each system.
- the grey region around the Y-axis represents the 95% significance envelope generated from historical vehicle controls.
- Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (
- Biomarker key activities are described as modulated if these activities increase in some systems, but decrease in others.
- Cytotoxicity is indicated on the profile plot by a thin black arrow above the X-axis, and antiproliferative effects are indicated by a thick grey arrow. Cytotoxicity and antiproliferative arrows may only require one concentration to meet the indicated threshold for profile annotation. Other BioMAP profiles disclosed herein are also depicted in a similar manner.
- Figure 36 depicts a Reference Benchmark Overlay of SNA- 120 and Benchmark SR221 1 .
- Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- Figure 37 depicts an overlay of SNA- 120 (28 ⁇ ) and GSK690693 (10 ⁇ ), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents for SNA-120 (28 ⁇ ).
- Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- Figure 38 depicts Mechanism HeatMAP Analysis for SNA-120. HeatMAP analysis of the 148 biomarker readouts (rows) within the Diversity PLUS panel by SNA-120 in comparison to 19 consensus mechanism class profiles (columns). Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-120 from the 19 consensus mechanism profiles. Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control.
- Figure 39 depicts the BioMAP profile of SNA-125 in the Diversity PLUS Panel.
- the X-axis lists the quantitative protein-based biomarker readouts measured in each system.
- the grey region around the Y-axis represents the 95% significance envelope generated from historical vehicle controls.
- Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (
- Biomarker key activities are described as modulated if these activities increase in some systems, but decrease in others.
- Cytotoxicity is indicated on the profile plot by a thin black arrow above the X-axis, and antiproliferative effects are indicated by a thick grey arrow. Cytotoxicity and antiproliferative arrows may only require one concentration to meet the indicated threshold for profile annotation. Other BioMAP profiles disclosed herein are also depicted in a similar manner.
- Figure 40 depicts a Reference Benchmark Overlay of SNA- 125 and Benchmark Tofacitinib. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- Figure 41 depicts an overlay of SNA-125 (3.9 ⁇ ) and SB203580 (10 ⁇ ), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents for SNA-125 (3.9 ⁇ ).
- Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- Figure 42 depicts Mechanism HeatMAP Analysis for SNA-125. HeatMAP analysis of the 148 biomarker readouts (rows) within the Diversity PLUS panel by SNA-125 in comparison to 19 consensus mechanism class profiles (columns). Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-125 from the 19 consensus mechanism profiles. Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control.
- Figure 43 depicts the BioMAP profile of SNA-125 in the Diversity PLUS Panel.
- the X-axis lists the quantitative protein-based biomarker readouts measured in each system.
- the grey region around the Y-axis represents the 95% significance envelope generated from historical vehicle controls.
- Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (
- Biomarker key activities are described as modulated if these activities increase in some systems, but decrease in others.
- FIG. 44 depicts a Reference Benchmark Overlay of SNA- 125 and Benchmark K252a. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- Figure 45 depicts an overlay of SNA-125 (30 ⁇ ) and IKK 16 (370 nM), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents for SNA-125 (30 ⁇ ).
- Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (
- Figure 46 depicts Mechanism HeatMAP Analysis for SNA-125. HeatMAP analysis of the 148 biomarker readouts (rows) within the Diversity PLUS panel by SNA-125 in comparison to 19 consensus mechanism class profiles (columns). Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-125 from the 19 consensus mechanism profiles. Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control.
- Figure 47 depicts a BioMAP profile overlay of SNA-125 (10 ⁇ ), Methotrexate 10 ⁇ ), and Tofacitinib (10 ⁇ ).
- Figure 48 depicts representative chromatograms detected using UV analysis (A) and MS analysis (B).
- Figure 49 depicts chromatograms of a CT101 plasma standard extract (100 ⁇ g/mL) detected using SIR (TIC, upper) and UV (lower).
- Figure 50 depicts method validation.
- Figure 51 depicts individual chromatograms used for the analysis of CT101 (sample: plasma spiked with CT101 for 100 ⁇ g/mL). Top chromatogram: 1 167.5; middle chromatogram: 778.6, bottom chromatogram: 584.2.
- Figure 52 depicts mouse plasma concentrations of CT101 . Data are presented as Mean ⁇ CI 95%.
- Figure 53 depicts representative chromatograms showing CT101 in extracted murine plasma following intra-venous administration. Top chromatogram: 2 hours. Middle chromatogram: 10 minutes, bottom chromatogram: blank murine plasma.
- Figure 54 depicts representative chromatograms showing CT101 in extracted murine plasma following epicutaneous administration. Top chromatogram: 8 hours, middle chromatogram: 0 hours, bottom chromatogram: blank murine plasma. [0162]
- Figure 55 depicts Chromatograms of a CT103 plasma standard extract (50 ⁇ g/mL) detected using SIR (TIC, upper) and UV at 337 nm (lower).
- Figure 57 depicts method validation. Arrows denote peaks of LOO and
- Figure 58 depicts individual chromatograms of SIR channels used for the analysis of compound (sample: plasma spiked with compound at 50 ⁇ g/mL). Top chromatogram: 794; middle chromatogram: 595.8, bottom chromatogram: 476.8.
- Figure 60 depicts mouse plasma concentrations of CT103. Data are presented as Mean ⁇ CI 95%.
- Figure 61 depicts plasma levels after intravenous dosing.
- Figure 62 depicts body weight versus day of study - males.
- Figure 63 depicts body weight versus day of study - males.
- Figure 64 depicts plasma levels after intravenous dosing - Day 1 .
- Figure 65 depicts plasma levels after intravenous dosing - Week 2.
- Figure 66 depicts body weight versus day of study - Males.
- Figure 67 depicts body weight versus day of study - Males.
- Figure 68 depicts body weight versus day of study (Main phase) -
- Figure 69 depicts body weight versus day of study (Main phase) -
- Figure 70 depicts CT327 calibration curves in rat plasma used to determine pharmacokinetic plasma levels (dotted lines represent upper and lower confidence limits).
- Figure 71 depicts mean plasma concentration-time after dose profile of CT327 (SNA-120) after a single intravenous administration at 18 mg/kg. Open symbols represent the average measured values ( ⁇ 95% CI, vertical bar), while filled-in symbols represent interpolated values.
- Figure 72 depicts raw luminescence values for a CT101 preincubation time of 6 hours and a stimulation time of 24 hours. Each symbol represents an individual well. Each condition was tested in sextuplicate.
- Figure 73 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 6 hours and a stimulation time of 24 hours.
- Figure 74 depicts raw luminescence values for a CT101 preincubation time of 18 hours and a stimulation time of 24 hours. Each condition was tested in sextuplicate except for Jurkat cells were conditions were tested in triplicate.
- Figure 75 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 18 hours and a stimulation time of 24 hours.
- Figure 76 depicts raw luminescence values for a CT101 preincubation time of 24 hours and a stimulation time of 24 hours. Each condition was tested in sextuplicate except for Jurkat cells where conditions were tested in triplicate.
- Figure 77 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 24 hours and a stimulation time of 24 hours.
- Figure 78 depicts raw luminescence values for a CT101 preincubation time of 6 hours and a stimulation time of 6 hours. Each condition was tested in sextuplicate.
- Figure 79 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 6 hours and a stimulation time of 6 hours.
- Figure 80 depicts raw luminescence values for a CT101 preincubation time of 18 hours and a stimulation time of 6 hours. Each condition was tested in sextuplicate except for Jurkat cells were conditions were tested in triplicate.
- Figure 81 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 18 hours and a stimulation time of 6 hours.
- Figure 82 depicts raw luminescence values for a CT101 preincubation time of 24 hours and a stimulation time of 6 hours. Each condition was tested in sextuplicate.
- Figure 83 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 24 hours and a stimulation time of 6 hours.
- Figure 84 depicts percentage cytotoxicity for a CT101 pre-incubation time of 6 hours and a stimulation time of 24 hours.
- Figure 85 depicts percentage cytotoxicity for a CT101 pre-incubation time of 18 hours and a stimulation time of 24 hours.
- Figure 86 depicts percentage cytotoxicity for a CT101 pre-incubation time of 24 hours and a stimulation time of 24 hours.
- Figure 87 depicts percentage cytotoxicity for a CT101 pre-incubation time of 24 hours and a stimulation time of 6 hours.
- Figure 88 depicts percentage cytotoxicity for a CT101 pre-incubation time of 6 hours and a stimulation time of 6 hours.
- Figure 89 depicts tritiated thymidine incorporation in corrected counts per minute (CCPM). Data are presented as Mean ⁇ SEM.
- Figure 90 depicts Tritiated thymidine incorporation in corrected counts per minute (CCPM). Data are presented as Mean ⁇ SEM.
- Figure 91 depicts tritiated thymidine incorporation in corrected counts per minute (CCPM).
- CCPM corrected counts per minute
- Figure 92 depicts percentage inhibition by CT103 with 18 hour preincubation time ,5x103 cells per well and 50ng/ml_ VEGF stimulation. Percentage inhibition of VEGF induced proliferation by CT103. Proliferation in the absence of VEGF (0 ng/mL VEGF) was subtracted from VEGF induced proliferation (50 ng/mL) and divided by 50 ng/mL VEGF stimulated cell proliferation in the absence of drug (DMSO).
- DMSO drug
- Figure 93 depicts tritiated thymidine incorporation in corrected counts per minute (CCPM). Data are presented as Mean ⁇ SEM.
- Figure 94 depicts non-linear fit of transformed normalised maximal response for IC50 calculation.
- the DMSO (0 ⁇ CT103) treated VEGF-induced HUVEC proliferation was excluded from the IC50 calculation as it was lower than the 3 lowest concentrations of CT103, which had reached a consistent plateau as can be seen above.
- the IC50 was calculated as 18.5 ⁇ .
- Figure 95 depicts percentage inhibition of VEGF induced proliferation by CT103. Proliferation in the absence of VEGF was subtracted from VEGF induced proliferation and divided by VEGF stimulated cell proliferation in the absence of drug (DMSO).
- Figure 96 depicts luciferase activity of THP1 cells stimulated with a concentration range of HKLM or LPS, calculated relative to unstimulated cells.
- Figure 98 depicts the inhibition of proliferation by K252a, CT327 and CT340. Column bar graphs of proliferation assay results expressed as Absorbance at 570 nm with reference at 650 nm.
- Figure 99 depicts the inhibition of proliferation by K252a, CT327 and CT340. Column bar graphs of proliferation assay results expressed as Absorbance at 570 nm with reference at 650 nm.
- Figure 100 depicts the inhibition of proliferation by K252a, CT327 and CT340. Column bar graphs of proliferation assay results expressed as Absorbance at 570 nm with reference at 650 nm.
- Figure 101 depicts (a) colitis development was evaluated monitoring colon shortening at mice sacrifice, (b) weight loss during the experiment, and (c) colitis clinical score at sacrifice.
- CT100 and CT300 data points refer to 100 and 300 mg/kg CT352 in vehicle.
- CsA data points refer to 25 mg/kg cyclsporin A (positive control).
- Figure 102 depicts qRTPCR analysis of inflammatory cytokines and chemokines expression.
- RNA from whole proximal and distal colon was analysed and an increased expression of cytokines (IL6, IL17) and chemokines (MIP1 a and MIP2) related to inflammation was detected in DSS treated mice, while this response was counteracted by CsA (positive control) and CT352 administration at the same time.
- IL6, IL17 cytokines
- MIP1 a and MIP2 chemokines
- Figure 103 depicts histological analysis of colon samples. The extension and the degree of the colitis were determined in blind and the score assigned to each samples reported in the colitis clinical score graph (a). A representative sample image from each group was reported (4X magnification) (b).
- Figure 104 depicts the (A) colon dissection diagram and (B) fields and scoring order employed in the oxazolone-induced colitis mouse study.
- Figure 105 depicts the effect of SNA- 125, SNA-352, tofacitinib, and prednisolone on the body weight of animals challenged with oxazolone. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
- Figure 105A depicts percent body weight change from Day -1 to Day 4 of the study.
- Figure 105B depicts the area under the curve (AUC) of the percent weight change depicted in Figure 105A.
- Figure 106 depicts the effect of SNA- 125, SNA-352, tofacitinib, and prednisolone on the body weight of animals challenged with oxazolone according to last observation carried forward analysis. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
- Figure 106A depicts percent body weight change from Day -1 to Day 4 of the study.
- Figure 106B depicts the area under the curve (AUC) of the percent weight change depicted in Figure 106A.
- Figure 107 depicts the effect of SNA- 125, SNA-352, tofacitinib, and prednisolone on the Day 2 endoscopy score of animals challenged with oxazolone by (A) bar chart and (B) dot plot. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
- Figure 108 depicts the effect of SNA- 125, SNA-352, tofacitinib, and prednisolone on the Day 2 stool consistency score of animals challenged with oxazolone by (A) bar chart and (B) dot plot. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
- Figure 109 depicts the effect of SNA- 125, SNA-352, tofacitinib, and prednisolone on the Day 4 endoscopy score of animals challenged with oxazolone by (A) bar chart and (B) dot plot. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
- Figure 1 10 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the Day 4 stool consistency score of animals challenged with oxazolone by (A) bar chart and (B) dot plot. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
- Figure 1 1 1 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the disease activity index (DAI) score of animals at (A) Day 2 and (B) Day 4 following challenge with oxazolone.
- DAI disease activity index
- Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
- Figure 1 12 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the colon weight/length ratio of animals challenged with oxazolone.
- Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
- Figure 1 13 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the colon inflammation histopathology scores of animals challenged with oxazolone.
- Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted.
- Figure 1 14 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the colon edema histopathology scores of animals challenged with oxazolone.
- Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted.
- Figure 1 15 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the colon mucosal necrosis/loss histopathology scores of animals challenged with oxazolone.
- Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted.
- Figure 1 16 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the summed colon histopathology scores of animals challenged with oxazolone.
- Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted.
- Figure 1 17 depicts representative control animal H&E-stained colon histopathology micrographs at 40x and 100x magnifications.
- Figure 1 18 depicts representative H&E-stained colon histopathology micrographs at 40x and 100x magnifications for animals administered BID
- A Vehicle PO
- B 15 mg/kg Tofacitinib PO
- C 1 mg/kg Prednisolone PO
- D 400 mg/kg SNA-125 PO
- E 400 mg/kg SNA-352 PO.
- Moderate inflammation unfilled black arrows
- edema filled red arrows
- multifocal ulceration brackets
- Figure 1 19 depicts representative H&E-stained colon histopathology micrographs at 40x and 100x magnifications for animals administered BID
- A Vehicle IC
- B 1 mg/kg Tofacitinib IC
- C 400 mg/kg SNA-125 IC
- D 400 mg/kg SNA-352 IC.
- Figure 120 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the proein levels of IFNv in colon tissue homogenate supernatants of animals following challenge with oxazolone.
- Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted.
- Figure 121 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the protein levels of TNFa in colon tissue homogenate supernatants of animals following challenge with oxazolone.
- Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted, with outliers removed (A), or present (B).
- Figure 122 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the protein levels of IL-6 in colon tissue homogenate supernatants of animals following challenge with oxazolone.
- Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
- Group means with standard error of the mean (SEM) bars are depicted, with outliers removed (A), or present (B).
- Figure 123 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the protein levels of IL-10 in colon tissue homogenate supernatants of animals following challenge with oxazolone.
- Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
- Group means with standard error of the mean (SEM) bars are depicted, with outliers removed (A), or present (B).
- Figure 124 depicts bodyweights. Data are presented as Mean ⁇ SEM percentages of the initial bodyweights. # p ⁇ 0.05 and ### p ⁇ 0.001 when compared to Day 0. ° p ⁇ 0.05, °° p ⁇ 0.01 and **** p ⁇ 0.0001 when compared to the vehicle-treated group.
- Figure 125 depicts ear swelling. Data are presented as Mean ⁇ SEM. ### p ⁇ 0.0001 in the vehicle-treated group when compared to Day 0. °° p ⁇ 0.01 , °°° p ⁇ 0.001 and °°°°° ⁇ ⁇ ⁇ 0.0001 when compared to the vehicle-treated group. Betamethasone 0.1 % induced a significant reduction of ear swelling when compared to the vehicle-treated group on Day 8, Day 1 1 and Day 14 (p ⁇ 0.0001).
- Figure 126 depicts macroscopic scores. Data are presented as Mean ⁇ SEM. ///////// p ⁇ 0.0001 when compared to Day 0 in the vehicle-treated group. °°° p ⁇ 0.01 and 000 °, ⁇ p ⁇ 0.0001 when compared to the vehicle-treated group.
- Figure 127 depicts cytokine levels in ears (pg/ml).
- Data are presented as Mean ⁇ SEM. **** p ⁇ 0.0001 , ** p ⁇ 0.001 , * p ⁇ 0.01 when compared to the Vehicle treated group. #### p ⁇ 0.0001 , ### p ⁇ 0.001 when compared to the right ear within the same group.
- Figure 128 depicts bodyweights. Data are presented as Mean ⁇ SEM of the initial (Day -13) bodyweights. * p ⁇ 0.05, ** p ⁇ 0.01 .
- Figure 129 depicts ear swelling. Data are presented as Mean ⁇ SEM of the difference between ovalbumin challenged and contralateral (saline-injected) ears. Statistical significances: # p ⁇ 0.05, ## p ⁇ 0.01 , ### p ⁇ 0.001 when compared to the baseline (0 hours) values. * p ⁇ 0.05, ** p ⁇ 0.01 , *** p ⁇ 0.001 when comparing to the Control group.
- Figure 130 depicts ear swelling at peak disease (fifteen minutes after the ovalbumin challenge). Data are presented as Mean ⁇ SEM of the difference between ovalbumin-challenged and contralateral (saline-injected) ears. Statistical significances: * p ⁇ 0.05, ** p ⁇ 0.01 , *** p ⁇ 0.001 when compared to the vehicle-treated group.
- Figure 131 depicts erythema scores (Challenged ears). Data are presented as Mean ⁇ SEM.
- Figure 132 depicts erythema scores (Challenged ears). Data are presented as Mean ⁇ SEM.
- Figure 133 depicts histopathology scores (Left ears). Data are presented as Mean ⁇ SEM. ** p ⁇ 0.01 , *** p ⁇ 0.001 when compared to the Control group.
- Figure 134 depicts representative histopathology pictures. Left panel: Left ears. Right panel: Right ears. Top line: Control Group. Middle line: Betamethasone 0.1 %-treated group. Bottom line: Vehicle-treated group. Magnification: x100.
- Figure 135 depicts representative histopathology pictures. Left panel: Left ears. Right panel: Right ears. Top line: CT101_5%-treated group. Middle line: CT101_10%-treated group. Bottom line: CT101_20%-treated group. Magnification: x100.
- Figure 136 depicts bodyweights. Data are presented as Mean ⁇ SEM percentages of the initial bodyweights. * p ⁇ 0.05, *** p ⁇ 0.001 , **** p ⁇ 0.0001 when compared to the vehicle-treated group; # # # p ⁇ 0.001 , # # # # # p ⁇ 0.0001 when compared to Day 0. [0244] Figure 137 depicts erythema scores. Data are presented as Mean ⁇ SEM . * p ⁇ 0.05, ** p ⁇ 0.01 , *** p ⁇ 0.001 , **** p ⁇ 0.0001 when compared to the vehicle-treated group.
- Figure 138 depicts scaling scores. Data are presented as Mean ⁇ SEM . * p ⁇ 0.05, *** p ⁇ 0.01 and **** p ⁇ 0.0001 when compared to the vehicle-treated group.
- Figure 139 depicts thickness scores. Data are presented as Mean ⁇ SEM . * p ⁇ 0.05, *** p ⁇ 0.001 , **** p ⁇ 0.0001 .
- Figure 140 depicts clinical scores. Data are presented as Mean ⁇ SEM . * p ⁇ 0.05, ** p ⁇ 0.01 , *** p ⁇ 0.001 and **** p ⁇ 0.0001 when compared to the vehicle-treated group.
- Figure 141 depicts normalized body weight trends.
- Figure 142 depicts scar formation; SEI of saline-injected scars (Group 1 , Left Ears). Values are expressed as mean ⁇ StdDev.
- Figure 143 depicts scar formation; SEI of vehicle-treated scars (Group 1 , Right Ears) . Values are expressed as mean ⁇ StdDev.
- Figure 144 depicts scar formation; SEI of CT340-injected scars (Group 2). Values are expressed as mean ⁇ StdDev.
- Figure 145 depicts Scar Formation; SEI of CT340 topical-dosed scars (Group 3). Values are expressed as mean ⁇ StdDev.
- Figure 146 depicts Scar Formation; SEI of TACA-treated scars (Group 4). Values are expressed as mean ⁇ StdDev.
- Figure 147 depicts scar formation; summary of SEI . Values are expressed as mean ⁇ StdDev.
- Figure 148 depicts scar formation following intra-lesion injections with CT340 or TACA.
- Figure 149 depicts scar formation following topical dosing with CT340
- Figure 150 depicts scar formation SEI of CT340-treated scars, Values are expressed as mean ⁇ StdDev.
- Figure 151 depicts scar inflammation scores following intra-lesion injections with CT340 or TACA. Values are expressed as mean ⁇ StdDev.
- Figure 152 depicts scar inflammation scores following topical dosing with CT340, Values are expressed as mean ⁇ StdDev.
- Figure 156 depicts anti-NGF antibody effect on Capsaicin responses. Inhibition of capsaicin responses in DRG neurons by anti-NGF was between 37.4 ⁇ 7.7% (100 ng/ml), and 63.3 ⁇ 9.7% (10 ⁇ g/ml). Results are given as mean (percent inhibition) ⁇ s.e.m.
- Figure 157 depicts the effect of compound incubation on neurite length.
- Treatment with CT327 and CT340 at 1 nM , 10 nM , 100 nM , 1 ⁇ and 10 ⁇ concentrations did not affect neurite length of neurons compared with control.
- Treatment with GW441756 resulted in vesiculation and reduced neurite length at the higher concentrations of 1 ⁇ and 10 ⁇ .
- Anti-NGF antibody treatment at 1 and 10 ⁇ g/ml concentrations also did not affect neurite length. Neurite lengths were normalized to controls and are given as mean percent of control ⁇ s.e. m.
- Figure 158 depicts the effect of CT327 incubation on neurite length . 24 hour Incubation with CT327 did not have any effect on neurite length. Neurite lengths are expressed as mean percent of control ⁇ s.e.m.
- Figure 159 depicts the effect of CT340 incubation on neurite length . 24 hour Incubation with CT340 did not significantly affect neurite length compared to control. Neurite lengths are expressed as mean percent of control ⁇ s.e.m.
- Figure 160 depicts the effect of GW441756 incubation on neurite length.
- Neurons treated with 0.33% ethanol (solvent for GW441756) had similar neurite length compared with NGF-treated controls. Neurite lengths are expressed as mean percent of control ⁇ s.e.m.
- Figure 161 depicts the effect of anti-NGF antibody incubation on neurite length. Neurons treated with anti-NGF at 1 or 10 ⁇ g/ml did not show a significant change in neurite length compared to control. Neurite lengths are expressed as mean percent of control ⁇ s.e.m.
- Figure 162 depicts TrkA/Gap43 immunostaining in DRG neurons: A) Merged image showing co-localization of TrkA and Gap43 immunostaining in DRG neuron; B) Gap43 was strongly localized in cell bodies and neurites; C) TrkA immunostaining was observed to be densely localized in the cell bodies, while neurites were very faint.
- Figure 163 depicts representative PGP9.5-immunoreactive intraepithelial nerve fibres (I EFN) (arrowed) in untreated control skin (top panel) and treated skin region (bottom panel) mini-pig skin using an antibody dilution of 1 :40,000, magnification x40.
- I EFN intraepithelial nerve fibres
- Figure 164 depicts a scatter plot showing the PGP9.5 intra epithelial fibre counts in untreated and treated mini-pig skin from the various groups. The median value is indicated.
- C control; L, low dose; M , medium dose; H high dose; R, recovery; ut untreated area; t treated area.
- Figure 165 depicts the design of the IMQ-induced psoriasis mouse study. Animal shaving, IMQ cream application, treatment, left ear biopsy punch, body weight measurements, ear thickness measurements, psoriasis clinical scoring, and termination were performed at the indicated tiem points.
- Figure 166 depicts the change in animal body weight throughout the IMQ-induced psoriasis mouse study.
- Figure 167 depicts the changes in the total psoriasis score throughout the IMQ-induced psoriasis mouse study.
- the difference between SNA- 125 at 5% and the vehicle is statistically significant from day 7.
- the differences between SNA- 125 at 0.5% and 1 % and the vehicle are statistically significant on day 10.
- Figure 168 depicts the changes in the Erythema score throughout the IMQ-induced psoriasis mouse study. SNA-125 at 5% is statistically significant from the vehicle from day 7. SNA-125 at 0.5% and 1 % are statistically significant on day 10.
- Figure 169 depicts the changes in the plaque score throughout the IMQ-induced psoriasis mouse study. SNA-125 at 5% is statistically significant from the vehicle on day 10.
- Figure 170 depicts the changes in the punctate redness/scabbing score throughout the IMQ-induced psoriasis mouse study.
- Figure 171 depicts the changes in spleen thickness throughout the IMQ-induced psoriasis mouse study
- Figure 172 depicts the changes in ear thickness throughout the IMQ- induced psoriasis mouse study.
- Figure 173 depicts the levels of cytokines (a) IL-22, (b) IL-17A, (c) IL17F, and (d) TNFa, in ear samples at Day 4.
- Figure 174 depicts a schematic showing how the IMQ-induced psoriasis study was performed.
- Figure 175 depicts the total psoriasis clinical scores over time for all groups (A) , the SNA-101 group (B), the SNA-125 group (C) , and the SNA-352 group (D) .
- the mean score for each group is displayed for each day +/- SEM .
- Figure 176 depicts the erythema scores over time for all groups (A) , the SNA- 101 group (B), the SNA-125 group (C) , and the SNA-352 group (D) .
- the mean score for each group is displayed for each day +/- SEM .
- Figure 177 depicts the plaque scores over time for all groups (A), the SNA-101 group (B), the SNA-125 group (C) , and the SNA-352 group (D). The mean score for each group is displayed for each day +/- SEM .
- Figure 178 depicts the punctate redness/scabbing scores over time for all groups (A) , the SNA-101 group (B), the SNA- 125 group (C), and the SNA-352 group (D) .
- the mean score for each group is displayed for each day +/- SEM .
- Figure 179A depicts the weight of spleens upon experimental termination on day 10. Mean spleen weight for each group is displayed +/- SEM .
- Figure 179B depicts left ear thickness as measured with a caliper on days 0, 4, 6, 8, and 10. Mean thickness for each group is displayed for each day +/- SEM .
- Figure 179C depicts the daily weight of mice. Body weight changes are displayed for each day as a percent of their weight measured on day 0. Mean values for each group are displayed +/- SEM .
- Figure 180 depicts the levels of IL- 17F (A), TNF-a (B), IL-22 (C), and IL-17A (D) as measured in left ears biopunched on day 4. After tissue homogenization , the cytokine levels in tissue lysates were measured via multiplex and then normalized with total protein amounts. Mean values for each group are displayed +/- SEM .
- Figure 181 depicts a schematic of the IL-23-induced psoriasis mouse model study.
- Figure 182 depicts the effect SNA-120 and SNA-325 in an IL-23- induced psoriasis mouse model.
- Figure 182A depicts the total psoriasis clinical scores for each group over time. The mean score for each group is displayed for each day +/- SEM.
- Figure 182B depicts the right ear thickness of each group at the indicated time points. Mean thickness for each group is displayed for each day +/- SEM .
- Figure 182C depicts body weight of each group over the course of the study. Body weight changes are displayed for each day as a percent of their weight measured on day 0. Mean values for each group are displayed +/- SEM .
- Figure 183 depicts the concentration vs. %lnhibition curves of staurosporine (A) and SNA-352 (B) against LIMK1 . The calculated slope and IC50(M) are also depicted).
- Figure 184 depicts the concentration vs. %lnhibition curves of staurosporine (A) and SNA-352 (B) against MAP2K6. The calculated slope and IC50(M) are also depicted).
- Figure 185 depicts the concentration vs. %lnhibition curves of staurosporine (A) and SNA-352 (B) against MLK1 . The calculated slope and IC50(M) are also depicted).
- Figure 186 depicts the concentration vs. %lnhibition curves of staurosporine (A) and SNA-352 (B) against MLK3. The calculated slope and IC50(M) are also depicted).
- Figure 187 depicts representative Day 2 endoscopy images of naive control, vehicle control (PO), vehicle control (IC) , and tofacitinib (15 mg/kg PO) animals. Animals underwent video endoscopy on Day 2 and colitis severity was scored on a scale of 0-4. Images were captured from each animal during the procedure and representative images from each treatment group are presented. All doses were administered twice a day (BID).
- Figure 188 depicts representative Day 2 endoscopy images of tofacitinib (15 mg/kg IC), prednisolone (1 mg/kg PO) , SNA-125 (400 mg/kg PO) , and SNA-352 (400mg/kg PO) animals. Animals underwent video endoscopy on Day 2 and colitis severity was scored on a scale of 0-4. I mages were captured from each animal during the procedure and representative images from each treatment group are presented. All doses were administered twice a day (BID).
- LSE Low Systemic ExposureTM
- the LSE platform creates polymer conjugates optimized for topical applications.
- the polymer conjugates developed by LSE or more generally the reduced exposure technology exhibit enhanced penetration.
- the enhanced penetration leads to delivery of a high local concentration of the drug.
- the polymer conjugates show a limited non-target absorption upon topical administration due to their increased molecular size and amphiphilicity and/or amphipathicity.
- side-effects are minimized by limiting or eliminating non-target (e.g. , systemic) absorption.
- the polymer conjugate comprises a "warhead" linked to a polymer.
- the warhead is a pharmacologically active entity selected according to the particular target or pathway of interest.
- polymer conjugates for use in the treatment of conditions including but not limited to inflammatory bowel diseases, dermatological diseases, and ophthalmic conditions).
- the polymer is directly coupled to the warhead without a separate chemical linking moiety between the polymer and the warhead; such direct coupling may involve without limitation ester, ether, acetal, ketal, vinyl ether, carbamate, urea, amine, amide, enamine, imine, oxime, amidine, iminoester, carbonate, orthoester, phosphonate, phosphinate, sulfonate, sulfinate, sulfide, sulfate, disulfide, sulfinamide, sulfonamide, thioester, aryl, silane, siloxane, heterocycles, thiocarbonate, thiocarbamate, and phosphonamide bonds.
- the linker is a separate chemical linking moiety between the polymer and the warhead.
- the polymer is polyethylene glycol (PEG), wherein the terminal OH group can optionally be modified e.g. with C1 -C5 alkyl or C1 -C5 acyl groups, e.g., with C1 -, C2- or C3-alkyl groups or C1 -, C2- or C3 groups.
- the modified PEG is a terminally alkoxy- substituted PEG.
- the modified PEG is a methoxy-PEG (mPEG).
- the polymer has a molecular weight ranging from about 100 to about 100,000 Da.
- the polymer is polydisperse with respect to molecular weight (e.g., has a distribution of molecular weights) and the indicated molecular weight of the polymer represents an average molecular weight.
- the polymer has a molecular weight ranging from about 200 to about 50,000 Da.
- the polymer has a molecular weight ranging from about 500 to about 10,000 Da (e.g., 500-1000, 1000-2000, 2000-3000, 3000-5000,5000- 7000, 7000-10,000 Da, and overlapping ranges therein).
- the polymer is a short-chain PEG, and in some embodiments a terminally alkoxy-substituted PEG, such as a mPEG with a molecular weight ranging from about 200 to about 4,000 Da, from about 400 to about 3,000 Da, from about 500 to about 2,000 Da, from about 700 to about 3,000 Da, from about 900 to about 4,000 Da, or from about 1 ,000 to about 5,000 Da.
- the short-chain PEG or mPEG has an average molecular weight of about 1 ,000-3,000 Da. (e.g., 2,000 Da).
- the polymer is a long-chain PEG.
- the long- chain PEG may be a terminally alkoxy-substituted PEG, such as methoxy-substituted PEG, with a molecular weight ranging greater than about 4,000 Da. In several embodiments, the molecular weight ranges from about 4,500-10,000Da (e.g., 4,500 to about 5,500 Da). In several embodiments, the long-chain PEG or mPEG has an average molecular weight of about 2,000 Da or of about 5,000 Da. In several embodiments, the polymer is of natural or semi-synthetic or synthetic origin. In several embodiments, the polymer has a linear or branched structure.
- the polymer is selected from poly(alkylene oxides) or from (polyethylene) oxides.
- the polymer selected may include, without limitation, one or more of the following: polyacrylic acid, polyacrylates, polyacrylamide or N-alkyl derivatives thereof, polymethacrylic acid, polymethacrylates, polyethylacrylic acid, polyethylacrylates, polyvinylpyrrolidone, poly(vinylalcohol), polyglycolic acid, polylactic acid, poly(lactic-co-glycolic) acid, dextran, chitosan, and hydroxyethyl starch.
- administration comprises treatment of the gastro-intestinal tract via, for example, an enteric coated capsule comprising the polymer conjugates taken orally.
- the polymer conjugates provided herein treat inflammatory bowel diseases.
- conjugating the warhead to a polymer (e.g., PEG) in the disclosed molecular weight ranges may slow diffusion of the molecule in the tissue, thereby potentially increasing residence time of the molecule in the target tissue, e.g. certain regions of the eye, epithelial and sub-epithelial layers in the eye, the epidermis and dermis for skin, the lining of the Gl tract, associated epithelial and subepithelial layers in other topical surfaces like gut, eye, lungs, the intestinal epithelium, the intestinal lamina limba, intestinal mucosa, etc.
- This "depot" effect may also lead to lower concentrations needing to be applied or for products to be applied with lower frequency, or both.
- the polymer conjugates provided herein are administered to the skin by topical application. In one embodiment, the polymer conjugates provided herein treat inflammatory skin diseases.
- Eye drops are provided in some embodiments to treat eye inflammation or ophthalmic disorders and diseases.
- conjugating the warhead to a polymer in the disclosed molecular weight ranges may be useful in reducing the diffusion or extravasation of the molecule out of the circulatory system after it enters it via injection and or diffusion from the target tissue.
- a polymer e.g., PEG
- conjugating the warhead to a polymer in the disclosed molecular weight ranges may be useful in reducing the diffusion or extravasation of the molecule out of the circulatory system after it enters it via injection and or diffusion from the target tissue.
- the PEGylated drug has a volume of distribution that is largely restricted to the blood, indicating that very little extravasation occurs with the polymer conjugates prior to being renally cleared. This reduced extravasation may explain at least in part the observed shorter half-life for the polymer conjugates.
- compositions described herein may be combined with other modalities to achieve synergic effects. These other modalities include, but are not limited to, energy delivery (such as laser, radiofrequency, ultrasound, microwave, etc.), thermal therapy, light therapy, radiation, intravenous chemotherapy, and others.
- energy delivery such as laser, radiofrequency, ultrasound, microwave, etc.
- thermal therapy such as laser, radiofrequency, ultrasound, microwave, etc.
- light therapy such as radiation, intravenous chemotherapy, and others.
- the compositions are applied with pressure, heat, massage etc. to facilitate localization to the desired target site.
- the compositions are administered in combination with one or more additional therapeutics that may not be reduced exposure compounds.
- the polymer conjugate exhibits unexpected permeability across the nuclear membrane. In several embodiments, the polymer conjugate exhibits unexpected permeability across both the nuclear and plasma membranes. Accordingly, in Example 30, two polymer conjugates, SNA-125 and SNA-120, were surprisingly shown to penetrate the keratinocyte cellular membrane and interact with the target kinases intracellular ⁇ within the cytoplasm, thereby leading to inhibition of proliferation of keratinocytes in a non-toxic manner.
- the reduced exposure compounds comprising a hydrophobic drug conjugated to a short chain PEG, exhibit surprising accessibility across cellular compartments, compared to the unconjugated drug.
- the conjugate can cross and reside within the lipid bilayer of the cell membrane, accumulate within the cytosol, and even traverse the nuclear envelope - thereby providing access both membrane, cytosolic and nuclear molecular targets in several embodiments.
- This property of the reduced exposure compounds result in excellent depo'ing, longer residence times within target cells, and/or relative non-compartmentalization in several embodiments. Consequently, in many embodiments, these compounds are biologically active at lower concentrations and require less frequent dosing - thereby reducing potential drug toxicity.
- the invention comprises a reduced exposure composition for treating a target site within the gastrointestinal system, skin, or eye comprising a conjugate comprising one or more active entitites linked to one or more polymers, wherein the active entity is a mediator of a gastrointestinal condition, ophthalmic condition, and/or dermatological condition and has reduced exposure in the systemic system as compared to the gastrointestinal system based on the ability of the conjugate to cross the lipid bilayer and into the cytoplasm and/or cytosol of a plurality of cells within the gastrointestinal system.
- the invention comprises a reduced exposure composition for treating a dermatological target site comprising a conjugate comprising one or more active entitites linked to one or more polymers, wherein the active entity is a mediator of a dermatological condition and has reduced exposure in the systemic system as compared to the skin based on the ability of the conjugate to cross the lipid bilayer and into the cytoplasm and/or cytosol of a plurality of cells within the target site.
- the concentration and/or bioavailability of the activity entity in the systemic system in one embodiment, is at an amount that is not active systemically and therefore does not result in undesired systemic side effects.
- the active entity has little or no exposure to the lymphatic system, thus resulting in little or no immunosuppression. Damage to organs such as the kidney or liver is also nominal because of the reduced exposure to non-target tissue.
- the warhead employed in the LSE polymer conjugate is an indolocarbazole compound.
- a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an indolocarbazole compound.
- the polymer conjugate comprises an indolocarbazole compound of formula (I) or of formula (II):
- R 1 and R 2 are the same or a different residue and are each independently selected from the group consisting of:
- R 7 is selected from the group consisting of hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted aralkyi, - (CH 2 ) a C0 2 R 10 (wherein a is 1 or 2, and wherein R 10 is selected from the group consisting of hydrogen and substituted or unsubstituted lower alkyl) and -(CH 2 ) a C0 2 NR 5 R 6 ,
- R 8 is selected from hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl
- (f) -CH CH(CH 2 ) m R 16 , wherein m is 0 to 4, and R 16 is hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, -COOR 15 , -OR 15 (wherein R 15 is as defined above) - CONR 5 R 6 or -NR 5 R 6 (wherein R 5 and R 6 are as defined above);
- R 3 is hydrogen, halogen, acyl, carbamoyl, substituted or unsubstituted lower alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted lower alkynyl or amino;
- W 1 and W 2 are independently hydrogen, hydroxy or W 1 and W 2 together represent oxygen;
- X is a polymer moiety, either linear or branched
- A represents -L 1 -X' and B represents -L 2 - Y', wherein at least one of X and Y' is a polymer moiety, either linear or branched, which is bound by L 1 and/or L 2 to the tetrahydrofuran ring of the compound of formula (II);
- L 1 and/or L 2 are a covalent chemical bond or a linker group
- R 19 or R 20 are each independently selected from hydrogen, lower alkyl, lower alkenyl, lower alkynyl or R 19 or R 20 are independently the residue of an a-amino acid in which the hydroxy group of the carboxyl group is excluded, or R 19 or R 20 are combined with a nitrogen atom to form a heterocyclic group; and
- the polymer moiety X, X or/and Y' covalently attached to the indolocarbazole compound of formulae (I) and (II) has to be biocompatible, can be of natural or semi-synthetic or synthetic origin and can have a linear or branched structure.
- the polymer moiety X, X or/and Y' is selected from poly(alkylene oxides), in particular from (polyethylene) oxides.
- polymers include without limitation polyacrylic acid, polyacrylates, polyacrylamide or N-alkyl derivatives thereof, polymethacrylic acid, polymethacrylates, polyethylacrylic acid, polyethylacrylates, polyvinylpyrrolidone, poly(vinylalcohol), polyglycolic acid, polylactic acid, poly(lactic-co-glycolic) acid, dextran, chitosan, polyaminoacids, hydroxyethyl starch.
- the polymer moiety X, X' or/and Y' is a polyethylene glycol (PEG) moiety, wherein the terminal OH group can optionally be modified e.g. with C ⁇ Cs alkyl or C ⁇ Cs acyl groups.
- the terminal OH group is optionally modified with Ci-, C 2 - or C 3 -alkyl groups or Ci-, C 2 - or C 3 groups.
- the modified polyethylene glycol is a terminally alkoxy-substituted polyethylene glycol.
- the polymer moiety is methoxy-polyethylene- glycol (mPEG).
- lower alkyl when used alone or in combination with other groups, means a straight chained or branched lower alkyl group containing from 1 -6 carbon atoms, preferably from 1 -5, more preferably from 1 -4 and especially preferably 1 - 3 or 1 -2 carbon atoms.
- These groups include, in some embodiments, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, amyl, isoamyl, neopentyl, 1 - ethylpropyl, hexyl, and the like.
- the lower alkyl moiety of the "lower alkoxy”, the "lower alkoxycarbonyl", the “lower akylaminocarbonyl', “lower hydroxyalkyl' and of the "tri-lower alkylsilyl' groups has the same meaning as "lower alkyl" defined above.
- the "lower alkenyl” groups are defined as C 2 -C 6 alkenyl groups which may be straight chained or branched and may be in the Z or E form. Such groups include vinyl, propenyl, 1 -butenyl, isobutenyl, 2-butenyl, 1 -pentenyl, (Z)-2- pentenyl, (E)-2- pentenyl, (Z)-4-methyl-2-pentenyl, (E)-4-methyl-2-pentenyl, pentadienyl, e.g., 1 , 3 or 2,4-pentadienyl, and the like.
- the C 2 -C 6 - alkenyl groups are C 2 - C 5 -, C 2 -C 4 -alkenyl groups. In other embodiments, the C 2 -C 6 - alkenyl groups are C 2 -C 3 - alkenyl groups.
- lower alkynyl groups refers to C 2 -C 6 -alkynyl groups which may be straight chained or branched and include ethynyl, propynyl, 1 -butynyl, 2- butynyl, 1 -pentynyl, 2-pentynyl, 3-methyl-1 -pentynyl, 3-pentynyl, 1 -hexynyl, 2-hexynyl, 3-hexynyl and the like.
- C 2 -C 6 -alkynyl groups are C 2 -C 5 -, C 2 -C 4 - alkynyl groups.
- C 2 -C 6 -alkynyl groups are C 2 -C 3 -alkynyl groups.
- aryl group refers to C 6 -C 14 -aryl groups which contain from 6 up to 14 ring carbon atoms. These groups may be mono-, bi- or tricyclic and are fused rings. In some embodiments, the aryl groups include phenyl, biphenyl, naphthyl, anthracenyl, phenanthrenyl and the like. The aryl moiety of the "arylcarbonyl” and the “arylaminocarbonyl” groups has the same meaning as defined above.
- heteroaryl groups may contain 1 to 3 heteroatoms independently selected from nitrogen, sulfur or oxygen and refers C 3 -C 13 -heteroaryl groups. These groups may be mono-, bi- or tricyclic. In some embodiments, the C 3 - C 13 heteroaryl groups include heteroaromatics and saturated and partially saturated heterocyclic groups. These heterocyclics may be monocyclic, bicyclic, tricyclic. In some embodiments, the 5 or 6-membered heterocyclic groups are thienyl, furyl, pyrrolyl, pyridyl, pyranyl, morpholinyl, pyrazinyl, methyl pyrrolyl, and pyridazinyl.
- the C 3 -Ci 3 - heteroaryl may be a bicyclic heterocyclic group.
- the bicyclic heterocyclic groups are benzofuryl, benzothienyl, indolyl, imidazolyl, and pyrimidinyl.
- the C 3 -C 13 -heteroaryls are furyl and pyridyl.
- lower alkoxy includes alkoxy groups containing from 1 to 6 carbon atoms, in some embodiments from 1 to 5, in other embodiments from 1 -4 and in yet other embodiments 1 to 3 or 1 to 2 carbon atoms and may be straight chained or branched. These groups include methoxy, ethoxy, propoxy, butoxy, isopropoxy, tert- butoxy, pentoxy, hexoxy and the like.
- acyl includes lower alkanoyl containing 1 to 6 carbon atoms, in some embodiments from 1 to 5, from 1 to 4, from 1 to 3 or from 1 to 2 carbon atoms and may be straight chained or branched. These groups include, in some embodiments, formyl, acetyl, propionyl, butyryl, isobutyryl, tertiary butyryl, pentanoyl and hexanoyl.
- the acyl moiety of the "acyloxy" group has the same meaning as defined above.
- halogen includes fluoro, chloro, bromo, iodio, and the like.
- aralkyl' group refers C 7 -Ci 5 -aralkyl wherein the alkyi group is substituted by an aryl.
- the alkyi group and aryl may be selected from the C C 6 alkyi groups and the C 6 -C 14 -aryl groups as defined above, wherein the total number of carbon atoms is between 7 and 15.
- the C 7 -C 15 -aralkyl groups are benzyl, phenylethyl, phenylpropyl, phenylisopropyl, phenylbutyl, diphenylmethyl, 1 , 1 - diphenylethyl, 1 ,2-diphenylethyl.
- the aralkyl moiety of the "aralkyloxy" groups has the same meaning as defined above.
- the substituted lower alkyi, alkenyl and alkynyl groups have 1 to 3 independently selected substituents, such as lower alkyi, hydroxy, lower alkoxy, carboxyl, lower alkoxycarbonyl, nitro, halogen, amino, mono- or di- lower alkylamino, dioxolane, dioxane, dithiolane, and dithione.
- the lower alkyi substituent moiety of the substituted lower alkyi, alkenyl and alkynyl groups, and the lower alkyi moiety of the lower alkoxy, the lower alkoxycarbonyl, and the mono- or di-lower alkylamino substituents of the substituted lower alkyi, alkenyl and alkynyl groups have the same meaning as "lower alkyi" defined above.
- the substituted aryl, the substituted heteroaryl and the substituted aralkyl groups each has 1 to 3 independently selected substituents, such as lower alkyi, hydroxy, lower alkoxy, carboxy, lower alkoxycarbonyl, nitro, amino, mono- or di-lower alkylamino, and halogen.
- substituents such as lower alkyi, hydroxy, lower alkoxy, carboxy, lower alkoxycarbonyl, nitro, amino, mono- or di-lower alkylamino, and halogen.
- the lower alkyi moiety of the lower alkyi, the lower alkoxy, the lower alkoxycarbonyl, and the mono- or di- lower alkylamino groups among the substituents has the same meaning as 'lower alkyi' defined above.
- the heterocyclic group formed by R 5 and R 6 combined with a nitrogen atom includes pyrrolidinyl, piperidinyl, piperidino, morpholinyl, morpholino, thiomorpholino, N-methylpiperazinyl, indolyl, and isoindolyl.
- each of R 1 and R 2 is hydrogen.
- the residue R 14 is selected from phenyl, pyridyl, imidazolyl, thiazolyl, tetrazolyl, -COOR 15 , -OR 15 (wherein R 15 is in some embodiments selected from hydrogen, methyl, ethyl, phenyl or acyl), -SR 7 (wherein R 7 is in some embodiments selected from substituted or unsubstituted lower alkyl, 2-thiazoline and pyridyl) and -NR 5 R 6 (wherein R 5 and R 6 are in some embodiments selected from hydrogen, methyl, ethyl, phenyl, carbamoyl and lower alkylaminocarbonyl).
- the residue R 16 is selected from hydrogen, methyl, ethyl, phenyl, imidazole, thiazole, tetrazole, -COOR 15 , -OR 15 and -NR 5 R 6 (wherein the residues R 15 , R 5 and R 6 have the meanings as described above).
- the residue R 7 is selected from the group consisting of substituted or unsubstituted lower alkyl, substituted or unsubstituted phenyl, pyridyl, pyrimidinyl, thiazole and tetrazole.
- k is 2, 3 or 4
- j is 1 or 2
- m and n are independently 0 or 1.
- R 3 is hydrogen or acetyl. Furthermore, in some embodiments, each W 1 and W 2 is hydrogen.
- X' when Y' is a polymer moiety and X' is not a polymer moiety, X' is selected from carboxy, hydroxymethyl or a lower alkoxycarbonyl. In some embodiments X' is selected from methoxycarbonyl.
- Y' is selected from hydroxy or acetyloxy.
- the warhead of the polymer conjugate is a derivative of K252a, which has the formula:
- the polymer conjugate is SNA-125, wherein the composition has the formula:
- the polymer conjugate is SNA-120, wherein the composition has the formula:
- the active entity comprises an indolocarbazole compound. In some embodiments, the active entity comprises a derivative of K252a. In some embodiments, the composition comprises SNA-125.
- the active entity binds to a tropomyosin-receptor-kinase A (TrkA) in some embodiments.
- the active entity binds to a Janus Kinase (JAK) family member in some embodiments.
- the active entity binds to one or more of Janus Kinase 1 (JAK1), Janus Kinase 2 (JAK2), Janus Kinase 3 (JAK3), and/or Tyrosine kinase 2 (TYK2) in some embodiments.
- the active entity binds to mitogen-activated protein kinase kinase (MAP2K) in some embodiments.
- the active entity binds to mitogen-activated protein kinase kinase 3 (MAP2K3) in some embodiments.
- the binding may be partially or fully inhibitory or not.
- compositions comprising an indolocarbazole compound are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of TkrA, Jak3, and/or MAP2K3.
- Several embodiments relate to polymer conjugates of an indolocarbazole compound, optimized for topical applications while also minimizing side- effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications are provided in other embodiments.
- the warhead of the polymer conjugate is an indolocarbazole compound or derivative thereof.
- methods of treating an inflammatory bowel disease in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is an indolocarbazole compound.
- the warhead of the LSE polymer conjugate is a derivative of K252a.
- the LSE polymer conjugate is SNA-125.
- the LSE polymer conjugate is SNA-120.
- Oral delivery is contemplated in several embodiments suitable for gastrointestinal disorders, and thus several embodiments relate to polymer conjugates of an indolocarbaozle, optimized for oral delivery to treat the gastrointestinal system while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Topical applications are provided in other embodiments.
- the warhead of the polymer conjugate is an indolocarbazole compound or derivative thereof.
- methods of treating an ophthalmic condition in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is an indolocarbazole compound.
- the warhead of the LSE polymer conjugate is a derivative of K252a.
- the LSE polymer conjugate is SNA-125.
- the LSE polymer conjugate is SNA-120.
- non-inflammatory ophthalmic conditions may also be treated.
- the warhead of the polymer conjugate is an indolocarbazole compound or derivative thereof.
- methods of treating an inflammatory skin disease in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is an indolocarbazole compound.
- the warhead of the LSE polymer conjugate is a derivative of K252a.
- the LSE polymer conjugate is SNA-125.
- the LSE polymer conjugate is SNA-120.
- non-inflammatory dermatological conditions may also be treated.
- Topical delivery is contemplated in several embodiments suitable for dermal pathologies, and thus several embodiments relate to polymer conjugates of an indolocarbaozle, optimized for topical delivery to treat the skin while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption).
- Nontopical applications are provided in other embodiments.
- compositions comprising an indolocarbazole compound are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of TkrA, Jak3, and/or MAP2K3.
- Several embodiments relate to polymer conjugates of an indolocarbazole compound, optimized for topical applications while also minimizing side- effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications are provided in other embodiments.
- the warhead of the polymer conjugate is an indolocarbazole compound or derivative thereof.
- methods of treating an inflammatory bowel disease in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is an indolocarbazole compound.
- the warhead of the LSE polymer conjugate is a derivative of K252a.
- the LSE polymer conjugate is SNA-125.
- the LSE polymer conjugate is SNA-120.
- the warhead employed in the LSE polymer conjugate is a small molecule targeting mediator(s) of gastrointestinal conditions.
- methods of treating an inflammatory bowel disease in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting mediator(s) of gastrointestinal conditions.
- the gastrointestinal condition is an inflammatory bowel disease.
- inflammatory bowel diseases include Crohn's disease, ulcerative colitis, collagenous colitis, lymphocytic colitis, ischemic colitis, diversion colitis, Behcet's syndrome, and indeterminate colitis.
- JAK and/or STAT family proteins are mediator(s) of gastrointestinal conditions.
- the warhead employed in the LSE polymer conjugate is a small molecule a targeting a JAK and/or STAT family protein.
- the JAK kinase family is a cytoplasmic protein kinase family comprising the members JAK1 , JAK2, JAK3 and TYK2.
- Growth factor or cytokine receptors that recruit JAK kinases include the interferon receptors, interleukin receptors (receptors for the cytokines IL-2 to IL-7, IL-9 to IL-13, IL- 15, IL-23), various hormone receptors (erythropoietin (Epo) receptor, the thrombopoietin (Tpo) receptor, the leptin receptor, the insulin receptor, the prolactin (PRL) receptor, the Granulocyte Colony-Stimulating Factor (G-CSF) receptor and the growth hormone receptor), receptor protein tyrosine kinases (such as EGFR and PDGFR), and receptors for other growth factors such as leukemia inhibitory factor (LIF), Oncastatin M (OSM), IFNa/ ⁇ / ⁇ , Granulocyte-macrophage colony-stimulating factor (GM-CSF), Ciliary neurotrophic factor (CNTF), cardiotrophin-1 (CT-I) (
- autoimmune diseases and disease associated with chronic inflammation, as well as acute responses have been linked to excessive or unregulated production or activity of one or more cytokines, the signaling of which depend on JAK kinases.
- diseases include gastrointestinal conditions, which may be treated according to several embodiments described herein.
- Phosphorylated receptors serve as docking sites for other SH-2 domain containing signaling molecules that interact with JAKs such as the STAT family of transcription factors, Src family of kinases, MAP kinases PB kinase and protein tyrosine phosphatases (Rane S.G. and Reddy E.P., Oncogene 2000 19, 5662-5679).
- JAKs such as the STAT family of transcription factors, Src family of kinases, MAP kinases PB kinase and protein tyrosine phosphatases (Rane S.G. and Reddy E.P., Oncogene 2000 19, 5662-5679).
- the family of latent cytoplasmic transcription factors, STATS are the most well characterized downstream substrates for JAKs.
- the STAT proteins bind to phosphorylated cytokine receptors through their SH2 domains to become phosphorylated by JAKs, which event leads to their dimerization and release and
- STAT3 Signal transducer and activator of transcription 3
- STAT3 is a transcription factor that regulates the expression of a variety of genes involved in many cellular processes such as cell growth, apoptosis, cell motility, and cytokine production.
- STAT3 is activated by JAK kinases and translocates to the nucleus to act as a transcriptional activator.
- JAK kinases JAK kinases and translocates to the nucleus to act as a transcriptional activator.
- IBD Inflammatory bowel diseases
- IBS irritable bowel syndrome
- SIBO small intestinal bacterial overgrowth
- methods of treating IBD, IBS and/or (SIBO) in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting mediator(s) of gastrointestinal conditions.
- the warhead employed in the LSE polymer conjugate is a small molecule targeting a JAK and/or STAT family protein.
- Non-limiting examples of inflammatory bowel diseases include Crohn's disease, ulcerative colitis, collagenous colitis, lymphocytic colitis, ischemic colitis, diversion colitis, Behcet's syndrome, and indeterminate colitis.
- the warhead employed in the LSE polymer conjugate is a small molecule targeting c-Src.
- methods of treating an inflammatory bowel disease in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting c-Src.
- the c-Src kinase is the most widely studied member of the largest family of nonreceptor protein tyrosine kinases, known as the Src family kinases (SFKs).
- Src family kinases Other SFK members include Lyn, Fyn, Lck, Hck, Fgr, BIk, Yrk, and Yes.
- the Src kinases can be grouped into two sub-categories, those that are ubiquitously expressed (Src, Fyn, and Yes), and those which are found primarily in hematopoietic cells (Lyn, Lck, Hck, BIk, Fgr). (Benati, D.
- SFKs are key messengers in many cellular pathways, including those involved in regulating proliferation, differentiation, survival, motility, and angiogenesis.
- the activity of SFKs is highly regulated intramolecularly by interactions between the SH2 and SH3 domains and intermolecularly by association with cytoplasmic molecules. This latter activation may be mediated by focal adhesion kinase (FAK) or its molecular partner Crk- associated substrate (CAS), which plays a prominent role in integrin signaling, and by ligand activation of cell surface receptors, e.g. epidermal growth factor receptor (EGFR).
- FAK focal adhesion kinase
- CAS molecular partner Crk- associated substrate
- Src can also be activated by dephosphorylation of tyrosine residue Y530.
- maximal Src activation requires the autophosphorylation of tyrosine residue Y419 (in the human protein) present within the catalytic domain.
- Elevated Src activity may be caused by increased transcription or by deregulation due to overexpression of upstream growth factor receptors such as EGFR, HER2, platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor, ephrins, integrin, or FAK.
- upstream growth factor receptors such as EGFR, HER2, platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor, ephrins, integrin, or FAK.
- compositions comprising compounds Nos 1 -71 shown in Table 1 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of c-Src.
- Several embodiments relate to polymer conjugates of compounds 1 -71 , optimized for topical applications while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications are provided in other embodiments.
- the warhead of the polymer conjugate is a small molecule disclosed in Table 1 targeting c-Src.
- methods of treating an inflammatory bowel disease in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Table 1 targeting c-Src.
- the warhead of the LSE polymer conjugate is compound 1 of Table 1 .
- the LSE polymer conjugate is CT101 .
- the warhead employed in the LSE polymer conjugate is a small molecule targeting a vascular endothelial growth factor receptor (VEGFR).
- VEGFR vascular endothelial growth factor receptor
- Angiogenesis the process of sprouting new blood vessels from existing vasculature, and arteriogenesis, the remodeling of small vessels into larger conduit vessels, are both physiologically important aspects of vascular growth in adult tissues. These processes of vascular growth are involved, in some cases, for beneficial processes such as tissue repair, wound healing, recovery from tissue ischemia and menstrual cycling. They are also involved, in some cases, for the development of pathological conditions such as the growth of neoplasias, diabetic retinopathy, rheumatoid arthritis, psoriasis, certain forms of macular degeneration, and certain inflammatory pathologies. The inhibition of vascular growth in these contexts has also shown beneficial effects in preclinical animal models.
- angiogenesis For example, inhibition of angiogenesis by blocking vascular endothelial growth factor or its receptor has resulted in inhibition of tumor growth and in retinopathy. Also, the development of pathological pannus tissue in rheumatoid arthritis involves angiogenesis and might be blocked by inhibitors of angiogenesis.
- Certain diseases are known to be associated with deregulated angiogenesis, for example ocular neovascularization, such as retinopathies (including diabetic retinopathy), age-related macular degeneration, psoriasis, hemangioblastoma, hemangioma, arteriosclerosis, inflammatory disease, such as a rheumatoid or rheumatic inflammatory disease, especially arthritis (including rheumatoid arthritis), or other chronic inflammatory disorders, such as chronic asthma, arterial or post- transplantational atherosclerosis, endometriosis, and neoplastic diseases, for example so-called solid tumors and liquid tumors (such as leukemias).
- retinopathies including diabetic retinopathy
- age-related macular degeneration psoriasis
- hemangioblastoma hemangioma
- arteriosclerosis arteriosclerosis
- inflammatory disease such as a rheumatoid or r
- VEGF Vascular endothelial growth factor
- vasculogenesis the de novo formation of the embryonic circulatory system
- angiogenesis the growth of blood vessels from pre-existing vasculature.
- VEGF activity has been mostly studied on cells of the vascular endothelium, although it does have effects on a number of other cell types (e.g., stimulation monocyte/macrophage migration, neurons, cancer cells, kidney epithelial cells, keratinocytes).
- VEGF has been shown to stimulate endothelial cell mitogenesis and cell migration.
- VEGF is also a vasodilator and increases microvascular permeability and was originally referred to as vascular permeability factor.
- VEGF vascular endothelial growth factor
- the broad term "VEGF” covers a number of proteins from two families, that result from alternate splicing of mRNA from a single, 8 exon, VEGF gene.
- the two different familes are referred to according to their terminal exon (exon 8) splice site— the proximal splice site (denoted VEGFxxx) or distal splice site (VEGFxxxb).
- these domains have functional consequences for the VEGF splice variants as the terminal (exon 8) splice site determines whether the proteins are pro-angiogenic (proximal splice site, expressed during angiogenesis) or anti-angiogenic (distal splice site, expressed in normal tissues).
- exons 6 and 7 mediate interactions with heparan sulfate proteoglycans (HSPGs) and neuropilin co- receptors on the cell surface, enhancing their ability to bind and activate VEGFRs.
- HSPGs heparan sulfate proteoglycans
- neuropilin co- receptors on the cell surface, enhancing their ability to bind and activate VEGFRs.
- VEGFRs tyrosine kinase receptors
- the VEGF receptors have an extracellular portion consisting of 7 immunoglobulin-like domains, a single transmembrane spanning region and an intracellular portion containing a split tyrosine-kinase domain.
- VEGF-A binds to VEGFR- 1 (Flt-1) and VEGFR-2 (KDR/Flk-1). VEGFR-2 appears to mediate almost all of the known cellular responses to VEGF.
- VEGFR-1 The function of VEGFR-1 is less well defined, although it is thought to modulate VEGFR-2 signaling. Another function of VEGFR-1 may be to act as a dummy/decoy receptor, sequestering VEGF from VEGFR-2 binding (this appears to be involved during vasculogenesis in the embryo). VEGF-C and VEGF-D, but not VEGF-A, are ligands for a third receptor (VEGFR-3), which mediates lymphangiogenesis.
- VEGFR-3 third receptor
- Increased vascular permeability is one of the earliest manifestations of inflammation, resulting in extravasation of protein-rich plasma into the effected tissue.
- Acute vascular permeability allows the deposition of circulating plasma matrix proteins including fibrin and fibronectin (FN) which facilitate cell migration in the inflamed area. This process also provides an access point for immune cells and immunoglobulins to enter the tissue and fight foreign antigens (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012).
- chronic vascular hyperpermeability is suggested to sustain the inflammatory response and retard resolution, further promoting the development of chronic inflammation (Nagy et al, Cold Spring Harb. Perspect. Med.
- VEGF is a potent vascular permeabilizing agent that is highly expressed during chronic inflammation (Nagy et al, Annu. Rev. Pathol. 2:251 -275, 2007). Low microenvironmental levels of VEGF are desired in order to maintain stable vascular integrity and promote endothelial cell survival through autocrine mechanisms (Lee et al, Cell 130:691 -703, 2007). Whereas elevated levels of VEGF induce vascular leakages by activating VEGF receptor 2 (VEGFR2) in endothelial cells (EC) leading to the opening of intercellular and/or intracellular pathways that facilitate plasma extravasation (Koch and Claesson-Welsh, Cold Spring Harb. Perspect. Med. 2:a006502, 2012).
- VAGFR2 VEGF receptor 2
- EC endothelial cells
- VEGF may serve as a pro-inflammatory mediator as it can enhance T cell (Xia et al, Blood 102: 161 -168, 2003), and monocyte (Murakami et al, Blood 108: 1849-1856, 2006) migration as well as promote pro-inflammatory chemokines expression by EC including MCP-1 , and IL-8, leading to further immune recruitment.
- Cellular sources for VEGF during inflammation can include macrophages and mast cells; however it can also be expressed by endothelial cells and acts in a paracrine and autocrine fashion.
- VEGF plays a major role in promoting chronic inflammation by inducing vascular permeability and contributing to immune cell recruitment.
- compositions comprising compounds Nos 1 -59 shown in Table 2 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of a VEGFR.
- Several embodiments relate to polymer conjugates of compounds 1 -59, optimized for topical applications while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications are provided in other embodiments.
- the warhead of the polymer conjugate is a small molecule disclosed in Table 2 targeting a VEGFR.
- methods of treating an inflammatory bowel disease in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Table 2 targeting a VEGFR.
- the warhead of the LSE polymer conjugate is compound 1 of Table 2.
- the LSE polymer conjugate is CT103.
- compositions comprising compounds shown in Tables 1-3 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of the mediator(s) of gastrointestinal conditions, such as, but not limited to, inflammatory bowel diseases, IBS or SIBO are disclosed herein.
- compositions comprising compounds shown in Tablse 1 -3 are used as inhibitors, antagonists, and inverse agonists of JAK and/or STAT family proteins.
- the warhead of the polymer conjugate is compound 1 of Table 3.
- the LSE polymer conjugate is CT352.
- Allergic inflammatory diseases are characterized by an immune response against a sensitizing agent, such as an allergen, resulting in the release of inflammatory mediators that recruit cells involved in inflammation in a subject, potentially leading to tissue damage and sometimes death.
- a sensitizing agent such as an allergen
- Methods of treating allergic inflammatory diseases of the gastrointestinal tract are provided herein.
- methods of treating an allergic inflammatory disease in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a JAK and/or STAT family protein.
- methods of treating the following conditions in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a JAK and/or STAT family protein: ulcerative colitis; Crohn's disease; colorectal cancer; celiac disease; and intestinal fibrosis
- LSE polymer conjugates wherein the warhead (e.g. , one or more pharmacologically active agents) is a selective inhibitor, antagonist, or inverse agonist of JAK1 .
- the warhead of the LSE polymer conjugate is JNJ-54781532 (ASP015K, Peficitinb) .
- the warhead of the LSE polymer conjugate is Upadacitinib (ABT-494).
- the warhead of the LSE polymer conjugate is GSK2586184.
- LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of LTA4H.
- the warhead of the LSE polymer conjugate is JNJ-26993135 (1 -[4-(benzothiazol-2-yloxy)-benzyl]- piperidine-4-carboxylic acid).
- Colony-stimulating factor-1 receptor (CSF1 R; FMS) is a tyrosine kinase receptor that plays an essential role in promoting macrophage and dendritic cell differentiation, recruitment, activation, and proliferation.
- the differentiation of dendritic cells and macrophages and their migration to intestinal mucosa is a component of inflammatory bowel diseases, particularly Crohn's disease.
- LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of FMS.
- the warhead of the LSE polymer conjugate is JNJ-40346527 (PRV-6527).
- Acute and chronic inflammation of the bowel is caused by a number of diseases, and typically the epithelial cells on the surface of mucosal tissue have an induced state of hypoxia due to the presence of inflammation.
- the body's response to this hypoxic condition is to increase the presence of hypoxia inducible factor- 1 alpha (HIF-1 a) which drives the expression of downstream HIF-1 target genes, inter alia, erythropoietin.
- HIF- 1 a is a mediator in the body's response to inflammation.
- the cellular concentration of HIF-1 a is regulated by prolyl hydroxylase enzymes that serve to destabilize HIF-1 a during periods of normoxia, resulting in the destruction of this protein.
- HIF-1 a prolyl hydroxylase thus leads to increased stabilization of H IF-1 a, in turn resulting in an upregulation of HIF-1 a that leads to a corresponding increased response to inflammation.
- treatment with one or more effective HIF- 1 a prolyl hydroxylase inhibitors can increase the level of the body's cellular inflammatory response.
- HIF-1 a prolyl hydroxylase inhibitors can increase the amount of epithelial cell healing over that which the body would normally provide.
- LSE polymer conjugates wherein the warhead is a HIF-1 a stabilizer.
- selective inhibitors, antagonists, and inverse agonists of HIF-1 a prolyl hydroxylase are provided, in several embodiments.
- the warhead of the LSE polymer conjugate is JNJ5169 (AKB-5169).
- the warhead of the LSE polymer conjugate is a compound having the formula:
- Z is phenyl substituted with from 1 to 5 halogens chosen from fluorine and chlorine;
- R 4 is C C 4 linear alkyl or C 3 -C 4 branched alkyl
- the warhead of the LSE polymer conjugate is a compound having the formula:
- L is chosen from CH 2 or S02;
- R represents from 0 to 5 substitutions for hydrogen
- the index n is an integer from 0 to 5;
- R 1 and R 2 are each independently chosen from:
- R 1 and R 2 can be taken together to form a substituted or unsubstituted
- heterocyclic or substituted or unsubstituted heteroaryl ring having from form 2 to 20 carbon atoms and from 1 to 7 heteroatoms; or a pharmaceutically acceptable salt thereof.
- the a4 integrin ⁇ 4 ⁇ 7 plays an essential role in lymphocyte migration throughout the gastrointestinal tract. It is expressed on most leukocytes, including B and T lymphocytes, where it mediates cell adhesion via binding to its ligand mucosal addressin cell adhesion molecule (MAdCAM). If left unchecked, integrin-mediated adhesion process can lead to chronic inflammation and autoimmune disease. Inhibitors of specific integrin-ligand interactions have been shown effective as anti-inflammatory agents for the treatment of various autoimmune diseases. Accordingly, there is provided, in several embodiments, LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of alpha-4-beta-7 integrin.
- MAdCAM mucosal addressin cell adhesion molecule
- the warhead of the LSE polymer conjugate is PTG-100. In several embodiments, the warhead of the LSE polymer conjugate is a peptide dimer compound comprising two monomer subunits, wherein each monomer subunit comprises the amino acid sequence: [0422] Pen-(N-Me-Arg)-Ser-Asp-Thr-Leu-Pen-Phe(4-tBu)-(P-homo-Glu)-(D-
- the present invention includes a peptide dimer compound comprising two linked monomer subunits of Formula (I): Xaa 1 -Xaa 2 -Xaa 3 -Xaa 4 -Xaa 5 -Xaa 6 -Xaa 7 -Xaa 8 -Xaa 9 -Xaa 1 °-Xaa 11 -Xaa 12 -Xaa 13 -Xaa 14 (Formula (I)) or a pharmaceutically acceptable salt thereof,
- Xaa 1 is absent, Ac, or any amino acid
- Xaa 2 is absent, Ac, or any amino acid
- Xaa 3 is absent, Ac, or any amino acid
- Xaa 4 is any amino acid capable of forming a bond with Xaa 10 ;
- Xaa 5 is selected from the group consisting of: N-Me-Arg, Arg, N-Me- Lys, Phe(4-guanidinoguanidino), Phe(4-carbomyl), Cit, Phe(4-NH 2 ), N-Me-homoArg, homoArg, Tyr, Dap, Dab, Arg-Me-sym, Arg-Me-asym, Cav, and His;
- Xaa 6 is Ser, Gly, Thr or lie
- Xaa 7 is Asp, Asp(OMe) or N-Me-Asp;
- Xaa 8 is selected from the group consisting of: Thr, Val, lie, Leu, homoLeu, Gin, Ser, Asp, Pro, Gly, His, Ala, Phe, Lys, Arg, Asn, Glu, Tyr, Trp, Met, NIe, and N-methyl amino acids, including N-Me-Thr;
- Xaa 9 is selected from the group consisting of: Gin, Ser, Asp, Pro, Gly, Ala, Phe, Glu, lie, Val, N-butyl Ala, N-pental Ala, N-hexyl Ala, cyclobutyl Ala, cyclopentylAla, Leu, NIe, Cba, homoLeu, Cpa, Aoc, and N-Me-Leu;
- Xaa 10 is any amino acid capable of forming a bond with Xaa 4 ;
- Xaa 11 is absent or selected from the group consisting of: aromatic amino acids, substituted aromatic amino acids, and Tic;
- Xaa 12 is absent or selected from the group consisting of: aromatic amino acids, substituted aromatic amino acids, Glu, D-Glu, homoGlu, Asp, D-Asp, D- homoGlu, Gla, beta-homoGlu, Tic, Aic, Gin, Cit, Glu(OMe), Asn, D-His, Tic, Phe(3- COOH), D-Arg, Bip, D-Trp, Phe, D-Phe, D-Val, D-Thr, D-Tyr, D-Lys, D-lle, D-His, N-Me- Glu, N-Me-Asp, alpha-homoGlu, Biphenyl-Gly, Biphenyl-Ala, Homo-Phe, D-1-Nal, D-2- Nal, Thr, and Val, and corresponding D-amino acids and isosteres;
- Xaa 13 is absent or Pro or any amino acid; and [0440] Xaa 14 is selected from the group consisting of: any amino acid with an amine side chain, Lys, D-Lys, N-Me-Lys, D-N-Me-Lys, Orn, Dab, Dap, HomoLys, D-Dap, D-Dab, D-Orn, Cys, HomoCys, Pen, D-HomoCys, D-Cys, D-Pen, Asp, Glu, D-Asp, D-Glu and HomoSer, HomoGlu, D-homoGlu, N-Me-Glu, N-Me-Asp, N-Me-D-Glu, and N-Me-D- Asp;
- Xaa 4 and Xaa 10 are both Pen or Cys; wherein: Xaa 5 is selected from the group consisting of Cit, Phe(4-carbomylamino), and N-Me-homoArg; Xaa 8 is selected from the group consisting of Leu, homoLeu, Nle and Val; Xaa 9 is selected from the group consisting of Cba, homoLeu, and Cpa; Xaa 1 1 1 is selected from the group consisting of Tic, Phe(2-carbomyl), Phe(3-carbomyl), Phe(4-COOH), Phe(4-OMe), and Phe(4-tBu); Xaa 12 is selected from the group consisting of Aic, Gin, Cit, Glu(OMe), D-His, Tic, Phe(3- COOH), D-Arg, Bip, D-Trp, Phe, D-Phe, D-Val, D-Thr, D-1 -Nal, D-2
- the interleukin-23 (IL-23) cytokine has been implicated as playing a crucial role in the pathogenesis of inflammatory bowel diseases.
- IL-23R is expressed on various adaptive and innate immune cells including Th17 cells, ⁇ T cells, natural killer (NK) cells, dendritic cells, macrophages, and innate lymphoid cells, which are found abundantly in the intestine.
- NK natural killer
- IL-23R is expressed on various adaptive and innate immune cells including Th17 cells, ⁇ T cells, natural killer (NK) cells, dendritic cells, macrophages, and innate lymphoid cells, which are found abundantly in the intestine.
- NK natural killer
- NK natural killer
- dendritic cells dendritic cells
- macrophages macrophages
- innate lymphoid cells which are found abundantly in the intestine.
- the gene expression and protein levels of IL-23R are found to be elevated in IBD patients.
- IL-23 Production of IL-23 is enriched in the intestine, where it is believed to play a key role in regulating the balance between tolerance and immunity through T-cell-dependent and T-cell-independent pathways of intestinal inflammation through effects on T-helper 1 (Th1 ) and Th17-associated cytokines, as well as restraining regulatory T-cell responses in the gut, favoring inflammation.
- Th1 T-helper 1
- Th17-associated cytokines T-helper 1
- IL-23R polymorphisms in the IL-23 receptor
- Binding of IL-23 to IL-23R activates the JAK-STAT signaling.
- LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of lnterleukin-23 receptor.
- the warhead of the LSE polymer conjugate is PTG-200.
- the warhead of the LSE polymer conjugate comprises a peptide inhibitor of lnterleukin-23 receptor.
- the warhead of the LSE polymer conjugate comprises the amino acid sequence of Formula II:
- X1 is any amino acid or absent
- X2 is any amino acid or absent
- X3 is any amino acid or absent
- X4 is Cys, Pen, hCys, D-Pen, D-Cys, D-hCys, Met, Glu, Asp, Lys, Orn, Dap, Dab, D-Dap, D-Dab, D-Asp, D-Glu, D-Lys, Sec, 2-chloromethylbenzoic acid, mercapto-propanoic acid, mercapto-butyric acid, 2-chloro-acetic acid, 3-choro-propanoic acid, 4-chloro-butyric acid, 3-chloro-isobutyric acid, Abu, ⁇ -azido-Ala-OH, propargylglycine, 2-(3'-butenyl)glycine, 2-allylglycine, 2-(3'-butenyl)glycine, 2-(4'- pentenyl)glycine, 2-(5'-hexenyl)glycine, or absent;
- X5 is any amino acid
- X6 is any amino acid
- X7 is Trp, Glu, Gly, lie, Asn, Pro, Arg, Thr or OctGly, or a corresponding ⁇ -methyl amino acid form of any of the foregoing;
- X8 is any amino acid
- X9 is Cys, Pen, hCys, D-Pen, D-Cys, D-hCys, Glu, Lys, Orn, Dap, Dab, D-Dap, D-Dab, D-Asp, D-Glu, D-Lys, Asp, Leu, Val, Phe, Ser, Sec, Abu, ⁇ -azido- Ala-OH, propargylglycine, 2-2-allylglycine, 2-(3'-butenyl)glycine, 2-(4'-pentenyl)glycine, Ala, hCys, Met, MeCys, (D)Tyr or 2-(5'-hexenyl)glycine;
- X10 is Tyr, Phe(4-OMe), 1 -Nal, 2-Nal, Aic, a-MePhe, Bip, (D)Cys, Cha, DMT, (D)Tyr, Glu, His, hPhe(3,4-dimethoxy), hTyr, N-Me-Tyr, Trp, Phe(4-CONH2), Phe(4-phenoxy), Thr, Tic, Tyr(3-tBu), Phe(4-tBu), Phe(4-CN), Phe(4-Br), Phe(4-NH2), Phe(4-F), Phe(3,5-F2), Phe(4-CH2C02H), Phe(penta-F), Phe(3,4-CI2), Phe(4-CF3), Bip, Cha, 4-PyridylAlanine, phTyr, OctGly, Phe(4-N3), Phe(4-Br), Phe
- X1 1 is 2-Nal, 1 -Nal, 2,4-dimethylPhe, Bip, Phe(3,4-CI2), Phe (3.4-F2), Phe(4-C02H), phPhe(4-F), a-Me-Trp, 4-phenylcyclohexyl, Phe(4-CF3), Phe(3,4-OMe2), a-MePhe, phPhe, phTyr, phTrp, Nva(5-phenyl), Phe, His, hPhe, Tic, Tqa, Trp, Tyr, Phe(4-OMe), Phe(4-Me), Trp(2,5,7-tri-tert-Butyl), Phe(4-Oallyl), Tyr(3-tBu), Phe(4-tBu), Phe(4-guanidino, Phe(4-OBzl), Octgly, Glu(Bzl), 4-Phenylcyclo
- X12 is His, Phe, Arg, N-Me-His, Val, Cav, Cpa, Leu, Cit, hLeu, 3-Pal, t-butyl-Ala, a-MeLys, D-Ala, (D)Asn, (D)Asp, (D)Leu, (D)Phe, (D)Tyr, Aib, a-MeLeu, a- MeOrn, ⁇ -Aib, ⁇ -Ala, phAla, phArg, phLeu, phVal, ⁇ -spiro-pip, Glu, hArg, lie, Lys, N- MeLeu, N-MeArg, Ogl, Orn, Pro, Gin, Ser, Thr, Tie, t-butyl-Gly, 4-amino-4-carboxy- tetrahydropyran (THP), Ache Acpc, Acbc, Acv
- X13 is Thr, Sarc, Glu, Phe, Arg, Leu, Asn, Cit, Lys, Arg, Orn, Val, phAla, Lys(Ac), (D)Asn, (D)Leu, (D)Phe, (D)Thr, Ala, a-MeLeu, Aib, ⁇ -Ala, ⁇ -Glu, phLeu, phVal, ⁇ -spiro-pip, Cha, Chg, Asp, Dab, Dap, a-DiethylGly, hLeu, Asn, Ogl, Pro, Gin, Ser, ⁇ -spiro-pip, Thr, Tba, Tie or Aib, or a corresponding ⁇ -methyl amino acid form of any of the foregoing;
- X14 is Phe, Tyr, Glu, Gly, His, Lys, Leu, Met, Asn, Lys(Ac), Dap(Ac), Asp, Pro, Gin, Arg, Ser, Thr, Tic or phPhe, or a corresponding ⁇ -methyl amino acid form of any of the foregoing;
- X15 is Gly, Ser, Thr, Gin, Ala, (D)Ala, (D)Asn, (D)Asp, (D)Leu, (D)Phe, (D)Thr, Aea, Asp, Asn, Glu, Phe, Gly, Lys, Leu, Pro, Arg, ⁇ -Ala, or Sarc, or a corresponding ⁇ -methyl amino acid form of any of the foregoing;
- X16 is any amino acid or absent
- X17 is any amino acid or absent
- X18 is any amino acid or absent
- X19 is any amino acid or absent
- X20 is any amino acid or absent
- the warhead of the LSE polymer conjugate comprises the amino acid sequence of Formula III:
- X1 is any amino acid or absent
- X2 is any amino acid or absent
- X3 is any amino acid or absent
- X4 is Pen, Cys or homo-Cys
- X5 is any amino acid
- X6 is any amino acid
- X7 is Trp, Bip, Gin, His, Glu(Bzl), 4-Phenylbenzylalanine, Tic, Phe[4- (2-aminoethoxy)], Phe(3,4-CI 2 ), Phe(4-OMe), 5-Hydroxy-Trp, 6-Chloro-Trp, N-MeTrp, a- Me-Trp, 1 ,2,3,4-tetrahydro-norharman, Phe(4-C0 2 H), Phe(4-CONH 2 ), Phe(3,4- Dimethoxy) , Phe(4-CF 3 ) , Phe(4-tBu), ⁇ -diPheAla, Glu, Gly, lie, Asn, Pro, Arg, Thr or Octgly, or a corresponding a-methyl amino acid form of any of the foregoing;
- X8 is any amino acid
- X9 is Pen, Cys or hCys
- X10 is 1 -Nal, 2-Nal, Aic, Bip, (D)Cys, Cha, DMT, (D)Tyr, Glu, Phe, His, Trp, Thr, Tic, Tyr, 4-pyridylAla, Octgly, a Phe analog or a Tyr analog, or a corresponding ⁇ -methyl amino acid form of any of the foregoing;
- X1 1 is 2-Nal, 1 -Nal, 2,4-dimethylPhe, Bip, Phe(3,4-CI 2 ) , Phe (3,4-F 2 ), Phe(4-C0 2 H), phPhe(4-F), a-Me-Trp, 4-phenylcyclohexyl, Phe(4-CF 3 ) , a-MePhe, phPhe, phTyr, phTrp, Nva(5-phenyl), Phe, His, hPhe, Tic, Tqa, Trp, Tyr, Phe(4-OMe), Phe(4- Me), Trp(2,5,7-tri-tert-Butyl) , Phe(4-Oallyl) , Tyr(3-tBu) , Phe(4-tBu), Phe(4-guanidino, Phe(4-OBzl), Octgly, Glu(Bzl),
- X12 is a-MeLys, a-MeOrn, a-MeLeu, a-MeVal, 4-amino-4-carboxy- tetrahydropyran, Ache Acpc, Acbc, Acvc, MeLeu, Aib, (D)Ala, (D)Asn, (D)Leu, (D)Asp, (D)Phe, (D)Thr, 3-Pal, Aib, ⁇ -Ala, phGlu, phAla, phLeu, phVal, ⁇ -spiro-pip, Cha, Chg , Asp, Dab, Dap, a-diethylGly, Glu, Phe, hLeu, hArg, hLeu, lie, Lys, Leu, Asn, N-MeLeu, N-MeArg, Ogl, Orn, Pro, Gin, Arg, Ser, Thr or Tie
- X13 is Lys(Ac), (D)Asn, (D)Leu, (D)Thr, (D)Phe, Ala, Aib, a-MeLeu, ⁇ - Ala, phGlu, phAla, phLeu, phVal, ⁇ -spiro-pip, Cha, Chg, Asp, Lys, Arg, Orn, Dab, Dap, a- diethylGly, Glu, Phe, hLeu, Lys, Leu, Asn, Ogl, Pro, Gin, Asp, Arg, Ser, spiro-pip, Thr, Tba, Tic, Val or Tyr, or a corresponding ⁇ -methyl amino acid form of any of the foregoing;
- X14 is Asn, Glu, Phe, Gly, His, Lys, Leu, Met, Asn, Pro, Gin, Arg, Ser, Thr, Tic or Tyr, Lys(Ac) , Orn or a corresponding ⁇ -methyl amino acid form of any of the foregoing;
- X1 5 is Gly, (D)Ala, (D)Asn, (D)Asp, Asn, (D)Leu, (D)Phe, (D)Thr, Ala, Asn, Ser, AEA, Asp, Glu, Phe, Gly, Lys, Leu, Pro, Gin, Arg or Ser, ⁇ -Ala, Arg or a corresponding ⁇ -methyl amino acid form of any of the foregoing;
- X16 is absent, Gly, Ala, Asp, Ser, Pro, Asn or Thr, or a corresponding ⁇ -methyl amino acid form of any of the foregoing;
- X17 is absent, Glu, Ser, Gly or Gin, or a corresponding a-methyl amino acid form of any of the foregoing;
- X18 is absent or any amino acid
- X19 is absent or any amino acid
- X20 is absent or any amino acid
- the peptide inhibitor comprises a disulfide bond between X4 and X9.
- Inflammatory bowel diseases are characterized by an aberrant immune response occurring in a genetically predisposed host in response to microbes and/or microbial compounds found in the gut microbiota.
- a pathotype of E. coli called "AIEC” for "adherent-invasive Escherichia coli”
- AIEC are able to adhere to the intestinal epithelium and colonize gut mucosa where they participate to IBD onset.
- AIEC's adhesion to mucosal epithelial cells is mediated by proteinaceous, rod-like organelles that are called type-1 fimbriae.
- Type-1 fimbriae carry an adhesin at the edge of a flexible tip fibrillum.
- This adhesin, FimH is a lectin having a strong affinity for highly mannosylated glycoproteins.
- AIEC bacteria adhere specifically to the carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6), a mannosylated glycoprotein which is abnormally expressed in the ileal mucosa of 35% of CD patients.
- CEACAM6 carcinoembryonic antigen-related cell adhesion molecule 6
- Overexpression of these CEACAM6 molecules in CD patients acting as receptors for E. coli adhesion in the gut, favors ileal and colonic AIEC invasion and their intracellular survival and replication within the mucosal tissues, thereby amplifying immune responses in IBD patients.
- LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of FimH.
- the warhead of the LSE polymer conjugate is EB8018.
- the warhead of the LSE polymer conjugate is compound of the following formula:
- Ri represents H, CO-(C C 6 )-alkyl or CO-alkylaryl, preferably H, COMe or COCH 2 Ph,
- Y represents a single bond, CH 2 , O, NR 3 , S, preferably a single bond, CH 2 , NR 3 ,
- A represents O, NH or S, preferably O or S,
- X represents H and X' represents OH or X and X taken together with the carbon atom bearing them form a CO group
- R 2 represents H, a linear or branched (C 1 -C 6 )-alkyl or CF 3 ,
- R 3 represents H, a d-C 6 alkyl, a CO-(C C 6 )-alkyl, CF 3 or COCF 3 , preferably H, CH 3 , COCH 3 , CF 3 or OCF 3 , and
- R represents: a (d-CeJ-alkyl, a (C 2 -C 6 )-alkenyl, a (C 2 -C 6 )-alkynyl, a (C 3 -C 10 )- cycloalkyi, a (C 5 -C 10 )-cycloalkenyl, a heterocycloalkyi, a heterocycloalkenyl, an arylan alkyl aryl, CF 3 , adamantyl, OR a , or NR b R c ,
- R a represents H, a (C 1 -C 6 )-alkyl, a (C 2 -C 6 )-alkenyl, a (C 2 -C 6 )- alkynyl, a (C 3 -C 6 )-cycloalkyl, a (C 3 -C 6 )-cycloalkenyl, a heterocycloalkyi, a heterocycloalkenyl, an aryl, a alkylaryl, a CHO, a CO-(C 1 -C 6 )-alkyl, or CO-aryl, a C0 2 H, a C0 2 -(Ci-C e )-alkyl, or a CONH-( C 1 -C 6 )-alkyl,
- R b and R c represent independently from each other any of the groups defined for R a , R b representing in particular H, said (C 1 -C 6 )-alkyl, (C 2 -C 6 )- alkenyl, (C 2 -C 6 )-alkynyl, (C 3 -C 10 )-cycloalkyl, (C 5 -C 10 )- cycloalkenyl, heterocycloalkyi, heterocycloalkenyl, CO-(C C 6 )-alkyl, C0 2 -(C C 6 )-alkyl, CONH-(C 1 -C 6 )-alkyl, aryl, alkylaryl, CO-aryl and CO-alkylaryl being optionally substituted by one or more, preferably 1 to 4, more preferably 1 or 2 substituent(s) R', each independently selected from:
- T represents a monovalent cation such as a mineral monovalent cation, in particular an alkaline cation preferably selected from Li + , Na + , K + and even more preferable Na + and wherein T is a monovalent anion, such as a halogenide, in particular chloride, bromide or iodide, preferably chloride,
- aryl optionally substituted by one or more, preferably one to three substituents selected from a halogen, NH 2 , OH, CF 3 , a C C 6 alkyl preferably substituted by a carbohydrate, CH 2 S0 3 T, CH 2 COOT or N(R g ) 3 T, wherein R g , T and T' are as defined above,
- alkyl aryl optionally substituted by one or more, preferably one to three substituents selected from a halogen, NH 2 , OH, CF 3 , a C ⁇ C e alkyl preferably substituted by a carbohydrate, CH 2 S0 3 T, CH 2 COOT or N(R g ) 3 T', wherein R g , T and T' are as defined above,
- a NH-alkyl aryl optionally substituted by one or more, preferably one to three substituents selected from a halogen, NH 2 , OH, CF 3) a d-C 6 alkyl preferably substituted by a carbohydrate, CH 2 S0 3 T, CH 2 COOT or N(R g ) 3 T, wherein R g , T and T are as defined above,
- CO-aryl optionally substituted by one or more, preferably one to three substituents selected from a halogen, NH 2 , OH, CF 3 , a C C e alkyl preferably substituted by a carbohydrate, CH 2 S0 3 T, CH 2 COOT or N(R g ) 3 T, wherein R g , T and T are as defined above,
- a CONH-aryl or NHCO-aryl optionally substituted by one or more, preferably one to three substituents selected from a halogen, NH 2 , OH, CF 3 , a C C 6 alkyl preferably substituted by a carbohydrate, CH 2 S0 3 T, CH 2 COOT or N(R g ) 3 T', wherein R g , T and T are as defined above,
- R d represents: H, a (C 1 -C 6 )-alkyl, a (C 3 -C 10 )-cycloalkyl, CO ⁇ -Ce)- alkyl, or CO-aryl optionally substituted by one or more, preferably one to three substituents selected from a halogen, CF 3 , a C C 6 alkyi preferably substituted by a carbohydrate,
- R e and R f represent independently from each other: H, a (C C 6 )-alkyl, a (C 3 -C10)-cycloalkyl, CO-(C C 6 )-alkyl, or CO-aryl optionally substituted by one or more, preferably one to three substituents selected from a halogen, CF 3 , a C C 6 alkyl preferably substituted by a carbohydrate, - NHR b , wherein R b is as defined above,
- Phosphodiesterase 4 (PDE4), which catalyzes the breakdown of cAMP in inflammatory cells and is a mediator for the inflammatory cascades implicated in the pathogenesis of inflammatory bowel diseases.
- PDE4 Phosphodiesterase 4
- the warhead of the LSE polymer conjugate is Tetilomast (OPC-6535).
- LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of cytokines interleukin-12 (IL-12) and/or interleukin-23 (IL-23), which can mediate the progression of inflammatory bowel diseases.
- the warhead of the LSE polymer conjugate is Apilimod (STA-5326).
- LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of a P38 mitogen- activated protein kinase.
- the warhead of the LSE polymer conjugate is Doramapimod (BIRB 796).
- the warhead of the LSE polymer conjugate is Semapimod (CNI-1493).
- the warhead of the LSE polymer conjugate comprises an antibiotic selected from sulfonamides (e.g., 4-amino-N- (5-methyl-3-isoxazolyl)benzenesulfonamide); vancomycin; amoxicillin; tetracyclines; clarithromycin; clindamycin; a member of the cephlosporin antibiotic family (e.g., cefaclor, cefadroxil, cefixime, cefprozil, ceftriaxone, cefuroxime, cephalexin, loracarbef, and the like); a member of the penicillin family of antibiotics (e.g., ampicillin, amoxicillin/clavulanate, bacampicillin, cloxicillin, penicillin VK
- sulfonamides e.g., 4-amino-N- (5-methyl-3-isoxazolyl)benzenesulfonamide
- vancomycin e.g., vancomycin
- the warhead of the LSE polymer conjugate comprises a non-systemic antibiotic that is minimally absorbed and has high local concentrations in the Gl tract after oral administration. Accordingly, in some embodiments, the LSE polymer conjugate comprises rifaximin.
- Antibiotics delivered (e.g., orally) according to several embodiments described herein may be particularly useful to treat the small intestine (for example to treat SIBO or small intestinal bacterial overgrowth).
- Such delivery compositions are able to, in some embodiments, deliver localized doses of antibiotics (or other antimicrobials) to the intestinal tract without significant exposure to other tissue which, advantageously, limits the side effects (such as dysbiosis or the destruction of good bacteria) that is caused by some antibiotics/antimicrobials.
- localized and precision-based treatment permits lower doses of the antibiotics/antimicrobials to be ingested, which in some embodiments may reduce or avoid toxicity, adverse immune effects, tolerance, destruction of probiotics, etc.
- Anti-fungals and anti-parasitic agents may also be delivered (orally, injected, topical, or by other means) according to the reduced systemic exposure embodiments described herein.
- systemic exposure and/or exposure to a non-target tissue of antimicrobials is reduced by at least 25%, 50%, 75% or more using the embodiments described herein (e.g., LSE polymer conjugates) as compared with administration of the same antimicrobial without the polymer conjugates described herein.
- the use of the compositions described herein results in a reduced negative impact on the natural healthy microbiome of a subject because of reduced exposure to non-target tissue.
- Oral delivery is contemplated in several embodiments suitable for gastrointestinal disorders, and thus several embodiments relate to polymer conjugates of compounds in Tables 1-3, optimized for oral delivery to treat the gastrointestinal system while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Topical applications are provided in other embodiments.
- the warhead of the polymer conjugate is a small molecule disclosed in Table 3 targeting mediator(s) of gastrointestinal conditions.
- methods of treating an inflammatory bowel disease in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Table 3 targeting mediator(s) of gastrointestinal conditions.
- JAK and/or STAT family proteins are mediator(s) of gastrointestinal conditions.
- the warhead employed in the LSE polymer conjugate is a small molecule targeting a JAK and/or STAT family protein.
- the warhead of the polymer conjugate is compound 1 of Table 3.
- the LSE polymer conjugate is CT352.
- IBS and SIBO may also be treated.
- compositions comprising an indolocarbazole compound are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of a VEGFR, c- Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- Several embodiments relate to polymer conjugates of an indolocarbazole compound, optimized for topical applications while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption).
- Non-topical applications are provided in other embodiments.
- the warhead of the polymer conjugate is an indolocarbazole compound or derivative thereof.
- methods of treating an inflammatory skin disease in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is an indolocarbazole compound.
- the warhead of the LSE polymer conjugate is a derivative of K252a.
- the LSE polymer conjugate is SNA-125.
- the LSE polymer conjugate is SNA-120.
- the LSE polymer conjugate is SNA-125.
- the warhead employed in the LSE polymer conjugate is a small molecule targeting mediator(s) of dermatological conditions.
- methods of treating an inflammatory skin disease in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting mediator(s) of dermatological conditions.
- the dermatological condition is an inflammatory skin disease.
- JAK and/or STAT family proteins are mediator(s) of dermatological conditions.
- the warhead employed in the LSE polymer conjugate is a small molecule a targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein .
- methods of treating dermatological conditions in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting mediator(s) of dermatological conditions.
- the warhead employed in the LSE polymer conjugate is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- the warhead employed in the LSE polymer conjugate is a small molecule targeting a JAK and/or STAT family protein.
- methods of treating an inflammatory skin disease in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a JAK and/or STAT family protein.
- the JAK kinase family is a cytoplasmic protein kinase family comprising the members JAK1 , JAK2, JAK3 and TYK2.
- JAK1 cytoplasmic protein kinase family
- JAK2 JAK3
- TYK2 cytoplasmic protein kinase family
- ligand binding to a receptor leads to receptor dimerization or oligomerization, which leads to JAK recruitment and activation either through autophosphorylation or phosphorylation by other JAK kinases or by other tyrosine kinases, which in turn leads to tyrosine phosphorylation of the receptors as well as downstream substrates of JAK.
- Growth factor or cytokine receptors that recruit JAK kinases include the interferon receptors, interleukin receptors (receptors for the cytokines IL-2 to IL-7, IL-9 to IL-13, IL- 15, IL-23), various hormone receptors (erythropoietin (Epo) receptor, the thrombopoietin (Tpo) receptor, the leptin receptor, the insulin receptor, the prolactin (PRL) receptor, the Granulocyte Colony-Stimulating Factor (G-CSF) receptor and the growth hormone receptor), receptor protein tyrosine kinases (such as EGFR and PDGFR) , and receptors for other growth factors such as leukemia inhibitory factor (LIF) , Oncastatin M (OSM) , IFNa/ ⁇ / ⁇ , Granulocyte-macrophage colony-stimulating factor (GM-CSF), Ciliary neurotrophic factor (CNTF), cardiotrophin-
- autoimmune diseases and disease associated with chronic inflammation, as well as acute responses have been linked to excessive or unregulated production or activity of one or more cytokines, the signaling of which depend on JAK kinases.
- diseases include rheumatoid arthritis (RA) such as moderate to severe RA, systemic lupus erythematosus (SLE), multiple sclerosis (MS) , Crohn's disease such as moderate to severe Crohn's disease, psoriasis such as moderate to severe chronic plaque psoriasis, ulcerative colitis such as moderate to severe ulcerative colitis, ankylosing spondilytis (AS) , psoriatic arthritis, Juvenile Idiopathic Arthritis (JIA) such as moderate to severe polyarticular JIA, systemic lupus erythematosus (SLE), diabetic nephropathy, dry eye syndrome, Sjogren's Syndrome, alopecia areata, vitiligo
- RA
- Phosphorylated receptors serve as docking sites for other SH-2 domain containing signaling molecules that interact with JAKs such as the STAT family of transcription factors, Src family of kinases, MAP kinases PB kinase and protein tyrosine phosphatases (Rane S.G . and Reddy E.P. , Oncogene 2000 19, 5662-5679) .
- the family of latent cytoplasmic transcription factors, STATS are the most well characterized downstream substrates for JAKs.
- STAT proteins bind to phosphorylated cytokine receptors through their SH2 domains to become phosphorylated by JAKs, which event leads to their dimerization and release and eventual translocation to the nucleus where they activate gene transcription.
- the various members of STAT which have been identified thus far, are STAT1 , STAT2, STAT3, STAT4, STAT5 (including STAT5a and STAT5b) and STAT6.
- STAT3 Signal transducer and activator of transcription 3
- STAT3 is a transcription factor that regulates the expression of a variety of genes involved in many cellular processes such as cell growth, apoptosis, cell motility, and cytokine production.
- STAT3 is activated by JAK kinases and translocates to the nucleus to act as a transcriptional activator.
- JAK kinases JAK kinases and translocates to the nucleus to act as a transcriptional activator.
- the warhead employed in the LSE polymer conjugate is a small molecule targeting c-Src.
- methods of treating an inflammatory skin disease in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting c-Src.
- the c-Src kinase is the most widely studied member of the largest family of nonreceptor protein tyrosine kinases, known as the Src family kinases (SFKs) .
- Src family kinases include Lyn, Fyn, Lck, Hck, Fgr, BIk, Yrk, and Yes.
- the Src kinases can be grouped into two sub-categories, those that are ubiquitously expressed (Src, Fyn, and Yes) , and those which are found primarily in hematopoietic cells (Lyn, Lck, Hck, BIk, Fgr). (Benati, D.
- SFKs are key messengers in many cellular pathways, including those involved in regulating proliferation, differentiation, survival, motility, and angiogenesis.
- the activity of SFKs is highly regulated intramolecularly by interactions between the SH2 and SH3 domains and intermolecularly by association with cytoplasmic molecules. This latter activation may be mediated by focal adhesion kinase (FAK) or its molecular partner Crk- associated substrate (CAS), which plays a prominent role in integrin signaling, and by ligand activation of cell surface receptors, e.g.
- FAK focal adhesion kinase
- CAS molecular partner Crk- associated substrate
- EGFR epidermal growth factor receptor
- Elevated Src activity may be caused by increased transcription or by deregulation due to overexpression of upstream growth factor receptors such as EGFR, HER2, platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor, ephrins, integrin, or FAK.
- upstream growth factor receptors such as EGFR, HER2, platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor, ephrins, integrin, or FAK.
- compositions comprising compounds Nos 1 -71 shown in Table 1 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of c-Src.
- Several embodiments relate to polymer conjugates of compounds 1 -71 , optimized for topical applications while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications are provided in other embodiments.
- the warhead of the polymer conjugate is a small molecule disclosed in Table 1 targeting c-Src.
- methods of treating an inflammatory skin disease in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Table 1 targeting c-Src.
- the warhead of the LSE polymer conjugate is compound 1 of Table 1 .
- the LSE polymer conjugate is CT101 .
- the warhead employed in the LSE polymer conjugate is a small molecule targeting a vascular endothelial growth factor receptor (VEGFR).
- VEGFR vascular endothelial growth factor receptor
- Angiogenesis the process of sprouting new blood vessels from existing vasculature, and arteriogenesis, the remodeling of small vessels into larger conduit vessels, are both physiologically important aspects of vascular growth in adult tissues. These processes of vascular growth are involved, in some cases, for beneficial processes such as tissue repair, wound healing, recovery from tissue ischemia and menstrual cycling. They are also involved, in some cases, for the development of pathological conditions such as the growth of neoplasias, diabetic retinopathy, rheumatoid arthritis, psoriasis, certain forms of macular degeneration, and certain inflammatory pathologies. The inhibition of vascular growth in these contexts has also shown beneficial effects in preclinical animal models.
- angiogenesis For example, inhibition of angiogenesis by blocking vascular endothelial growth factor or its receptor has resulted in inhibition of tumor growth and in retinopathy. Also, the development of pathological pannus tissue in rheumatoid arthritis involves angiogenesis and might be blocked by inhibitors of angiogenesis.
- Certain diseases are known to be associated with deregulated angiogenesis, for example ocular neovascularization, such as retinopathies (including diabetic retinopathy), age-related macular degeneration, psoriasis, hemangioblastoma, hemangioma, arteriosclerosis, inflammatory disease, such as a rheumatoid or rheumatic inflammatory disease, especially arthritis (including rheumatoid arthritis), or other chronic inflammatory disorders, such as chronic asthma, arterial or post- transplantational atherosclerosis, endometriosis, and neoplastic diseases, for example so-called solid tumors and liquid tumors (such as leukemias).
- retinopathies including diabetic retinopathy
- age-related macular degeneration psoriasis
- hemangioblastoma hemangioma
- arteriosclerosis arteriosclerosis
- inflammatory disease such as a rheumatoid or r
- VEGF Vascular endothelial growth factor
- vasculogenesis the de novo formation of the embryonic circulatory system
- angiogenesis the growth of blood vessels from pre-existing vasculature.
- VEGF activity has been mostly studied on cells of the vascular endothelium, although it does have effects on a number of other cell types (e.g., stimulation monocyte/macrophage migration, neurons, cancer cells, kidney epithelial cells, keratinocytes).
- VEGF has been shown to stimulate endothelial cell mitogenesis and cell migration.
- VEGF is also a vasodilator and increases microvascular permeability and was originally referred to as vascular permeability factor.
- VEGF vascular endothelial growth factor
- exon 8 terminal exon
- VEGFxxxb distal splice site
- these domains have functional consequences for the VEGF splice variants as the terminal (exon 8) splice site determines whether the proteins are pro-angiogenic (proximal splice site, expressed during angiogenesis) or anti-angiogenic (distal splice site, expressed in normal tissues).
- exons 6 and 7 mediate interactions with heparan sulfate proteoglycans (HSPGs) and neuropilin co- receptors on the cell surface, enhancing their ability to bind and activate VEGFRs.
- HSPGs heparan sulfate proteoglycans
- neuropilin co- receptors on the cell surface, enhancing their ability to bind and activate VEGFRs.
- VEGFRs tyrosine kinase receptors
- the VEGF receptors have an extracellular portion consisting of 7 immunoglobulin-like domains, a single transmembrane spanning region and an intracellular portion containing a split tyrosine-kinase domain.
- VEGF-A binds to VEGFR- 1 (Flt-1) and VEGFR-2 (KDR/Flk-1). VEGFR-2 appears to mediate almost all of the known cellular responses to VEGF.
- VEGFR-1 The function of VEGFR-1 is less well defined, although it is thought to modulate VEGFR-2 signaling. Another function of VEGFR-1 may be to act as a dummy/decoy receptor, sequestering VEGF from VEGFR-2 binding (this appears to be involved during vasculogenesis in the embryo). VEGF-C and VEGF-D, but not VEGF-A, are ligands for a third receptor (VEGFR-3), which mediates lymphangiogenesis. [0525] Increased vascular permeability is one of the earliest manifestations of inflammation, resulting in extravasation of protein-rich plasma into the effected tissue.
- Acute vascular permeability allows the deposition of circulating plasma matrix proteins including fibrin and fibronectin (FN) which facilitate cell migration in the inflamed area. This process also provides an access point for immune cells and immunoglobulins to enter the tissue and fight foreign antigens (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012). Conversely, chronic vascular hyperpermeability is suggested to sustain the inflammatory response and retard resolution, further promoting the development of chronic inflammation (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012; Costa et al, Angiogenesis 10: 149- 166, 2007).
- FN fibrin and fibronectin
- vascular hyperpermeability underlies the pathogenesis of a large number of chronic disorders including rheumatoid arthritis (RA) , psoriasis, ocular disease, cancer and chronic wounds (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012; Costa et al, Angiogenesis 10: 149- 166, 2007).
- RA rheumatoid arthritis
- psoriasis ocular disease
- cancer chronic wounds
- VEGF is a potent vascular permeabilizing agent that is highly expressed during chronic inflammation (Nagy et al, Annu. Rev. Pathol. 2:251 -275, 2007) . Low microenvironmental levels of VEGF are desired in order to maintain stable vascular integrity and promote endothelial cell survival through autocrine mechanisms (Lee et al, Cell 130:691 -703, 2007). Whereas elevated levels of VEGF induce vascular leakages by activating VEGF receptor 2 (VEGFR2) in endothelial cells (EC) leading to the opening of intercellular and/or intracellular pathways that facilitate plasma extravasation (Koch and Claesson-Welsh, Cold Spring Harb. Perspect. Med.
- VEGFR2 VEGF receptor 2
- VEGF may serve as a pro-inflammatory mediator as it can enhance T cell (Xia et al, Blood 102: 161 -168, 2003) , and monocyte (Murakami et al, Blood 108: 1849-1856, 2006) migration as well as promote pro-inflammatory chemokines expression by EC including MCP-1 , and IL-8, leading to further immune recruitment.
- Cellular sources for VEGF during inflammation can include macrophages and mast cells; however it can also be expressed by endothelial cells and acts in a paracrine and autocrine fashion.
- VEGF plays a major role in promoting chronic inflammation by inducing vascular permeability and contributing to immune cell recruitment.
- compositions comprising compounds Nos 1 -59 shown in Table 2 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of a VEGFR.
- Several embodiments relate to polymer conjugates of compounds 1 -59, optimized for topical applications while also minimizing side-effects caused by exposure at non-target sites (e.g. , systemic absorption) . Non-topical applications are provided in other embodiments.
- the warhead of the polymer conjugate is a small molecule disclosed in Table 2 targeting a VEGFR.
- methods of treating an inflammatory skin disease in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Table 2 targeting a VEGFR.
- the warhead of the LSE polymer conjugate is compound 1 of Table 2.
- the LSE polymer conjugate is CT103.
- compositions comprising compounds shown in Tables 1-3 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of the mediator(s) of dermatological conditions, such as, but not limited to, inflammatory skin diseases, bullous dises, or skin neoplasias are disclosed herein.
- compositions comprising compounds shown in Table 3 are used as inhibitors, antagonists, and inverse agonists of JAK and/or STAT family proteins.
- the warhead of the polymer conjugate is compound 1 of Table 3.
- the LSE polymer conjugate is CT352.
- AD Atopic dermatitis
- Clinically AD is characterized by highly pruritic often excoriated plaques and papules that show a chronic relapsing course.
- the diagnosis of AD is mostly based on major and minor clinical findings. See Hanifin J. M., Arch Dermatol: 135, 1551 (1999).
- Histopathology reveals spongiosis, hyper and focal parakeratosis in acute lesions, whereas marked epidermal hyperplasia with hyper and parakeratosis, acanthosis/hypergranulosis and perivascular infiltration of the dermis with lymphocytes and abundant mast cells are the hallmarks of chronic lesions.
- Psoriasis is characterized by frequent episodes of redness, itching, and thick, dry, silvery scales on the skin. Psoriasis comprises lesions that can involve primary and secondary alterations in epidermal proliferation, inflammatory responses of the skin, and an expression of regulatory molecules such as lymphokines and inflammatory factors. Psoriatic skin is morphologically characterized by an increased turnover of epidermal cells, thickened epidermis, abnormal keratinization, inflammatory cell infiltrates into the epidermis and polymorphonuclear leukocyte and lymphocyte infiltration into the epidermis layer. Psoriasis is often associated with other inflammatory disorders, for example arthritis, including rheumatoid arthritis, inflammatory bowel disease (IBD), and Crohn's disease.
- IBD inflammatory bowel disease
- active agents useful for stimulating hair follicles are provided as oral applications or topical applications for the scalp.
- Hair removal agents and ant-acne agents are provided in other embodiments.
- Hair growth, hair removal and anti-acne therapies can all involve active agents that, if exposed to the non-target site (e.g., systemic circulation and/or lymphatic system) for long periods, result in toxicity or undesired side effects.
- the non-target site e.g., systemic circulation and/or lymphatic system
- the reduced exposure compositions described herein provides benefits for these applications as well.
- active agents useful for stimulating hair follicles are provided as oral applications or topical applications for the scalp.
- Hair removal agents and anti-acne agents are provided in other embodiments.
- Hair growth, hair removal and anti-acne therapies can all involve active agents that, if exposed to the non-target site (e.g., systemic circulation and/or lymphatic system) for long periods, result in toxicity or undesired side effects.
- the reduced exposure compositions described herein provides benefits for these applications as well.
- the polymer conjugates provided herein modulate hair growth and cycling.
- the polymer conjugates provided herein treat alopecia.
- Allergic inflammatory diseases are characterized by an immune response against a sensitizing agent, such as an allergen, resulting in the release of inflammatory mediators that recruit cells involved in inflammation in a subject, potentially leading to tissue damage and sometimes death.
- Allergic inflammatory diseases of the eye, skin, gastrointestinal tract, upper and lower airways, and lung including, but not limited to, atopic dermatitis, atopic keratoconjunctivitis, allergic conjunctivitis, asthma, and allergic rhinitis.
- methods of treating an allergic inflammatory disease in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- methods of treating the following conditions in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein: nail dystrophy; seborrheic keratosis; androgenic alopecia; contact dermatitis; actinic keratosis; acne; asthma; eczema (atopic derm); onychomycosis; sinusitis; allergic rhinitis; rosacea; COPD; pruritus; early AMD; urticaria; diabetic retinopathy; psoriasis; alopecia areata; dry eye; vitiligo; glaucoma; late AMD; ulcerative colitis; Crohn's disease; ocular rosacea; hair growth and cycling; skin
- vascular tumors include hemangiomas, Kaposi's sarcoma, lymphangioma, glomangioma, angiosarcoma, hemangioendothelioma, and infantile hemangiomas.
- methods of treating a skin neoplasia in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- a polymer conjugate wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- skin neoplasias include squamous cell carcinoma, basal cell carcinoma, malignant melanoma, malignant cutaneous lymphoma, Kaposi's sarcoma, Merkel cell skin cancer, and non-melanoma skin cancer.
- methods of treating an inflammatory skin disease in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- Non- limiting examples of inflammatory skin diseases include psoriasis, psoriasis guttata, inverse psoriasis, pustular psoriasis, psoriatic erythroderma, acute febrile neutrophilic dermatosis, eczema, xerotic eczema, dyshidrotic eczema, vesicular palmar eczema, acne vulgaris, atopic dermatitis, contact dermatitis, allergic contact dermatitis, dermatomyositis, exfoliative dermatitis, hand eczema, pompholyx, keloids, rosacea, rosacea due to sarcoidosis, rosacea due to scleroderma, rosacea due to Sweet syndrome, rosacea due to systemic lupus erythematosus, rosacea due to urticaria, rosacea due to herpetic pain
- Bullous diseases are skin disorders characterized by blistering that often have an autoimmune etiology.
- VEGF has been found to be upregulated in two bullous diseases, bullous pemphigoid and erythema multiforme.
- Bullous pemphigoid is a subepidermal disorder which manifests as subepidermal blisters with a dermal infiltrate of neutrophils and eosinophils.
- Erythema multiforme is an inflammatory eruption characterized by symmetric erythematous, edematous, or bullous lesions of the skin or mucous membranes.
- methods of treating a bullous disease in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- bullous diseases include bullous pemphigoid, erythema multiforme, dermatitis herpetiformis, epidermolysis bullosa acquisita, linear Immunoglobulin A disease, mucous membrane pemphigoid, pemphigoid gestationis, pemphigus foliaceus, and pemphigus vulgaris.
- VEGF Vascular endothelial growth factor
- methods of modulating hair growth and cycling in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- methods of treating alopecia in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- a combination therapy comprising administering to the subject an effective amount of a polymer conjugate in conjunction with UV irradiation therapy, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- alopecia is treated.
- Non-limiting examples include androgenic alopecia and alopecia areata.
- Androgenic alopecia also known as hereditary baldness, male pattern baldness, and seborrheic alopecia
- Alopecia areata is known to be associated with autoimmune activities; hence, topically administered immunomodulatory compounds demonstrate efficacy for treating that type of hair loss.
- hair regeneration compositions are in the form of a liquid.
- hair regeneration compositions are in the form of a lotion.
- hair regeneration compositions are in the form of a cream.
- hair regeneration compositions are in the form of a gel.
- the hair regeneration composition is administered twice daily.
- the hair regeneration composition is administered one daily.
- the hair regeneration composition is administered once weekly.
- the hair regeneration composition is administered directly to the scalp.
- the hair regeneration composition is administered directly non-scalp areas.
- Oral delivery is contemplated in several embodiments suitable for dermal pathologies, and thus several embodiments relate to polymer conjugates of compounds in Tables 1-3, optimized for oral delivery to treat the skin while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Topical applications are provided in other embodiments.
- the warhead of the polymer conjugate is a small molecule disclosed in Tables 1 -3 targeting mediator(s) of dermatological conditions.
- methods of treating an inflammatory skin disease or other dermal pathology in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Tables 1 -3 targeting mediator(s) of dermatological conditions.
- a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein are mediator(s) of dermatological conditions.
- the warhead employed in the LSE polymer conjugate is a small molecule a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein..
- the warhead of the polymer conjugate is compound 1 of Table 1 .
- the polymer conjugate is SNA-101 .
- the warhead of the polymer conjugate is compound 1 of Table 2.
- the polymer conjugate is SNA-103.
- the warhead of the polymer conjugate is compound 1 of Table 3.
- the LSE polymer conjugate is SNA-352.
- compositions comprising an indolocarbazole compound are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of a VEGFR, c- Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- Several embodiments relate to polymer conjugates of an indolocarbazole compound, optimized for topical applications (e.g., eye drops) while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption).
- Non-topical applications are provided in other embodiments.
- the reduced exposure compsotions disclosed herein are administered by direct intravitreal injection.
- the reduced exposure compsotions are administered by subconjunctival injection. In other embodiments, the reduced exposure compsotions are administered by subtenon injection. In still further embodiments, the compostions are administered by peribulbar injection. Also provided, in several embodiments, are reduced exposure compostions which can be administered via intraocular implantable devices known to one of skill in the art.
- the warhead of the polymer conjugate is an indolocarbazole compound or derivative thereof.
- methods of treating an ophthalmic condition in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is an indolocarbazole compound.
- the warhead of the LSE polymer conjugate is a derivative of K252a.
- the LSE polymer conjugate is SNA-125.
- the LSE polymer conjugate is SNA-120.
- methods of treating an ophthalmic condition in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- Non- limiting examples of ophthalmic conditions include macular degeneration, age related macular degeneration (ARMD), choroidal neovascularization, retinopathy, diabetic retinopathy, acute macular neuroretinopathy, chronic macular neuroretinopathy, central serous chorioretinopathy, macular edema, cystoid macular edema, diabetic macular edema., acute multifocal placoid pigment epitheliopathy, Behcet's disease, birdshot retinochoroidopathy, uveitis, intermediate uveitis (pars planitis), anterior uveitis, multifocal choroiditis, multiple evanescent white dot syndrome (MEWDS), ocular sarcoidosis, posterior scleritis, serpignous choroiditis, subretinal fibrosis, uveitis syndrome, Vogt-Koyanagi-Harada syndrome, retinal arterial occlusive disease, central retinal
- the c-Src kinase is the most widely studied member of the largest family of nonreceptor protein tyrosine kinases, known as the Src family kinases (SFKs).
- Src family kinases Other SFK members include Lyn, Fyn, Lck, Hck, Fgr, BIk, Yrk, and Yes.
- the Src kinases can be grouped into two sub-categories, those that are ubiquitously expressed (Src, Fyn, and Yes), and those which are found primarily in hematopoietic cells (Lyn, Lck, Hck, BIk, Fgr). (Benati, D.
- SFKs are key messengers in many cellular pathways, including those involved in regulating proliferation, differentiation, survival, motility, and angiogenesis.
- the activity of SFKs is highly regulated intramolecularly by interactions between the SH2 and SH3 domains and intermolecularly by association with cytoplasmic molecules. This latter activation may be mediated by focal adhesion kinase (FAK) or its molecular partner Crk- associated substrate (CAS), which plays a prominent role in integrin signaling, and by ligand activation of cell surface receptors, e.g. epidermal growth factor receptor (EGFR).
- FAK focal adhesion kinase
- CAS molecular partner Crk- associated substrate
- Src can also be activated by dephosphorylation of tyrosine residue Y530.
- maximal Src activation requires the autophosphorylation of tyrosine residue Y419 (in the human protein) present within the catalytic domain.
- Elevated Src activity may be caused by increased transcription or by deregulation due to overexpression of upstream growth factor receptors such as EGFR, HER2, platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor, ephrins, integrin, or FAK.
- upstream growth factor receptors such as EGFR, HER2, platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor, ephrins, integrin, or FAK.
- compositions comprising compounds Nos 1-71 shown in Table 1 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of c-Src.
- Several embodiments relate to polymer conjugates of compounds 1-71 , optimized for topical applications (e.g., eye drops) while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption).
- Non-topical applications e.g., intraocular injection, periocular injection, release from drug delivery devices
- the warhead of the polymer conjugate is a small molecule disclosed in Table 1 targeting c-Src.
- methods of treating an ophthalmic condition in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Table 1 targeting c-Src.
- the warhead of the LSE polymer conjugate is compound 1 of Table 1.
- the LSE polymer conjugate is CT101.
- compositions comprising compounds shown in Table 3 are used as inhibitors, antagonists, and inverse agonists of JAK and/or STAT family proteins.
- the warhead of the polymer conjugate is compound 1 of Table 3.
- the LSE polymer conjugate is CT352.
- the JAK kinase family is a cytoplasmic protein kinase family comprising the members JAK1 , JAK2, JAK3 and TYK2.
- JAK1 cytoplasmic protein kinase family
- JAK2 JAK3
- TYK2 cytoplasmic protein kinase family
- ligand binding to a receptor leads to receptor dimerization or oligomerization, which leads to JAK recruitment and activation either through autophosphorylation or phosphorylation by other JAK kinases or by other tyrosine kinases, which in turn leads to tyrosine phosphorylation of the receptors as well as downstream substrates of JAK.
- Growth factor or cytokine receptors that recruit JAK kinases include the interferon receptors, interleukin receptors (receptors for the cytokines IL-2 to IL-7, IL-9 to IL-13, IL- 15, IL-23), various hormone receptors (erythropoietin (Epo) receptor, the thrombopoietin (Tpo) receptor, the leptin receptor, the insulin receptor, the prolactin (PRL) receptor, the Granulocyte Colony-Stimulating Factor (G-CSF) receptor and the growth hormone receptor), receptor protein tyrosine kinases (such as EGFR and PDGFR), and receptors for other growth factors such as leukemia inhibitory factor (LIF), Oncastatin M (OSM), IFNa/ ⁇ / ⁇ , Granulocyte-macrophage colony-stimulating factor (GM-CSF), Ciliary neurotrophic factor (CNTF), cardiotrophin-1 (CT-I) (
- Phosphorylated receptors serve as docking sites for other SH-2 domain containing signaling molecules that interact with JAKs such as the STAT family of transcription factors, Src family of kinases, MAP kinases PB kinase and protein tyrosine phosphatases (Rane S.G. and Reddy E.P., Oncogene 2000 19, 5662-5679).
- JAKs such as the STAT family of transcription factors, Src family of kinases, MAP kinases PB kinase and protein tyrosine phosphatases (Rane S.G. and Reddy E.P., Oncogene 2000 19, 5662-5679).
- the family of latent cytoplasmic transcription factors, STATS are the most well characterized downstream substrates for JAKs.
- the STAT proteins bind to phosphorylated cytokine receptors through their SH2 domains to become phosphorylated by JAKs, which event leads to their dimerization and release and
- STAT3 Signal transducer and activator of transcription 3
- STAT3 is a transcription factor that regulates the expression of a variety of genes involved in many cellular processes such as cell growth, apoptosis, cell motility, and cytokine production.
- STAT3 is activated by JAK kinases and translocates to the nucleus to act as a transcriptional activator.
- JAK kinases JAK kinases and translocates to the nucleus to act as a transcriptional activator.
- the warhead employed in the LSE polymer conjugate is a small molecule targeting a vascular endothelial growth factor receptor (VEGFR).
- VEGFR vascular endothelial growth factor receptor
- Angiogenesis the process of sprouting new blood vessels from existing vasculature, and arteriogenesis, the remodeling of small vessels into larger conduit vessels, are both physiologically important aspects of vascular growth in adult tissues. These processes of vascular growth are involved, in some cases, for beneficial processes such as tissue repair, wound healing, recovery from tissue ischemia and menstrual cycling. They are also involved, in some cases, for the development of pathological conditions such as the growth of neoplasias, diabetic retinopathy, rheumatoid arthritis, psoriasis, certain forms of macular degeneration, and certain inflammatory pathologies. The inhibition of vascular growth in these contexts has also shown beneficial effects in preclinical animal models.
- angiogenesis For example, inhibition of angiogenesis by blocking vascular endothelial growth factor or its receptor has resulted in inhibition of tumor growth and in retinopathy. Also, the development of pathological pannus tissue in rheumatoid arthritis involves angiogenesis and might be blocked by inhibitors of angiogenesis.
- Certain diseases are known to be associated with deregulated angiogenesis, for example ocular neovascularization, such as retinopathies (including diabetic retinopathy), age-related macular degeneration, psoriasis, hemangioblastoma, hemangioma, arteriosclerosis, inflammatory disease, such as a rheumatoid or rheumatic inflammatory disease, especially arthritis (including rheumatoid arthritis), or other chronic inflammatory disorders, such as chronic asthma, arterial or post- transplantational atherosclerosis, endometriosis, and neoplastic diseases, for example so-called solid tumors and liquid tumors (such as leukemias).
- retinopathies including diabetic retinopathy
- age-related macular degeneration psoriasis
- hemangioblastoma hemangioma
- arteriosclerosis arteriosclerosis
- inflammatory disease such as a rheumatoid or r
- VEGF Vascular endothelial growth factor
- vasculogenesis the de novo formation of the embryonic circulatory system
- angiogenesis the growth of blood vessels from pre-existing vasculature.
- VEGF activity has been mostly studied on cells of the vascular endothelium, although it does have effects on a number of other cell types (e.g., stimulation monocyte/macrophage migration, neurons, cancer cells, kidney epithelial cells, keratinocytes).
- VEGF has been shown to stimulate endothelial cell mitogenesis and cell migration.
- VEGF is also a vasodilator and increases microvascular permeability and was originally referred to as vascular permeability factor.
- VEGF vascular endothelial growth factor
- these domains have functional consequences for the VEGF splice variants as the terminal (exon 8) splice site determines whether the proteins are pro-angiogenic (proximal splice site, expressed during angiogenesis) or anti-angiogenic (distal splice site, expressed in normal tissues).
- exons 6 and 7 mediate interactions with heparan sulfate proteoglycans (HSPGs) and neuropilin co- receptors on the cell surface, enhancing their ability to bind and activate VEGFRs.
- HSPGs heparan sulfate proteoglycans
- neuropilin co- receptors on the cell surface, enhancing their ability to bind and activate VEGFRs.
- VEGFRs tyrosine kinase receptors
- the VEGF receptors have an extracellular portion consisting of 7 immunoglobulin-like domains, a single transmembrane spanning region and an intracellular portion containing a split tyrosine-kinase domain.
- VEGF-A binds to VEGFR- 1 (Flt-1) and VEGFR-2 (KDR/Flk-1). VEGFR-2 appears to mediate almost all of the known cellular responses to VEGF.
- VEGFR-1 The function of VEGFR-1 is less well defined, although it is thought to modulate VEGFR-2 signaling. Another function of VEGFR-1 may be to act as a dummy/decoy receptor, sequestering VEGF from VEGFR-2 binding (this appears to be involved during vasculogenesis in the embryo). VEGF-C and VEGF-D, but not VEGF-A, are ligands for a third receptor (VEGFR-3), which mediates lymphangiogenesis.
- VEGFR-3 third receptor
- Increased vascular permeability is one of the earliest manifestations of inflammation, resulting in extravasation of protein-rich plasma into the effected tissue.
- Acute vascular permeability allows the deposition of circulating plasma matrix proteins including fibrin and fibronectin (FN) which facilitate cell migration in the inflamed area. This process also provides an access point for immune cells and immunoglobulins to enter the tissue and fight foreign antigens (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012).
- chronic vascular hyperpermeability is suggested to sustain the inflammatory response and retard resolution, further promoting the development of chronic inflammation (Nagy et al, Cold Spring Harb. Perspect. Med.
- VEGF is a potent vascular permeabilizing agent that is highly expressed during chronic inflammation (Nagy et al, Annu. Rev. Pathol. 2:251-275, 2007). Low microenvironmental levels of VEGF are desired in order to maintain stable vascular integrity and promote endothelial cell survival through autocrine mechanisms (Lee et al, Cell 130:691-703, 2007). Whereas elevated levels of VEGF induce vascular leakages by activating VEGF receptor 2 (VEGFR2) in endothelial cells (EC) leading to the opening of intercellular and/or intracellular pathways that facilitate plasma extravasation (Koch and Claesson-Welsh, Cold Spring Harb. Perspect. Med. 2:a006502, 2012).
- VAGFR2 VEGF receptor 2
- EC endothelial cells
- VEGF may serve as a pro-inflammatory mediator as it can enhance T cell (Xia et al, Blood 102:161-168, 2003), and monocyte (Murakami et al, Blood 108:1849-1856, 2006) migration as well as promote pro-inflammatory chemokines expression by EC including MCP-1 , and IL-8, leading to further immune recruitment.
- Cellular sources for VEGF during inflammation can include macrophages and mast cells; however it can also be expressed by endothelial cells and acts in a paracrine and autocrine fashion.
- VEGF plays a major role in promoting chronic inflammation by inducing vascular permeability and contributing to immune cell recruitment.
- compositions comprising compounds Nos 1-59 shown in Table 2 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of a VEGFR.
- Several embodiments relate to polymer conjugates of compounds 1-59, optimized for topical applications (e.g., eye drops) while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption).
- Non-topical applications e.g., intraocular injection, periocular injection release from drug delivery devices are provided in other embodiments.
- the warhead of the polymer conjugate is a small molecule disclosed in Table 2 targeting a VEGFR.
- methods of treating an ophthalmic condition in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Table 2 targeting a VEGFR.
- the warhead of the LSE polymer conjugate is compound 1 of Table 2.
- the LSE polymer conjugate is CT103.
- cytokine and receptor-mediated inflammatory process there is provided, in several embodiments, methods of treating dry eye in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- the composition is formulated as an eye drop.
- one or two drops of the composition are used per application.
- three or four drops of the composition are used per application.
- six drops of the composition are used per application.
- the composition is applied for a period of 60 seconds before flushing. In other embodiments, the composition is applied for a period of 120 seconds before flushing. In additional embodiments, the composition is applied for a period of 360 seconds before flushing. In some embodiments, the composition may be administered one or more times a day. In some embodiments, the composition is administered daily. In some embodiments, the composition may be administered once a week.
- methods of treating maculopathies and/or retinal degeneration diseases in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- Non-limiting examples of maculopathies and/or retinal degeneration diseases include macular degeneration, age related macular degeneration (ARMD), non-exudative age related macular degeneration, exudative age related macular degeneration, choroidal neovascularization, retinopathies, diabetic retinopathy, acute and chronic macular neuroretinopathy, central serous chorioretinopathy, macular edema, cystoid macular edema, and diabetic macular edema.
- AMD age related macular degeneration
- non-exudative age related macular degeneration exudative age related macular degeneration
- choroidal neovascularization retinopathies
- diabetic retinopathy acute and chronic macular neuroretinopathy
- central serous chorioretinopathy macular edema
- cystoid macular edema cystoid macular edema
- diabetic macular edema diabetic macular edema
- methods of treating uveitis, retinitis and/or choroiditis in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- Non-limiting examples of uveitis, retinitis and/or choroiditis include acute multifocal placoid pigment epitheliopathy, Behcet's disease, birdshot retinochoroidopathy, infectious ocular diseases (e.g., syphilis, lyme, tuberculosis, toxoplasmosis), uveitis, intermediate uveitis (pars planitis), and anterior uveitis, multifocal choroiditis, multiple evanescent white dot syndrome (MEWDS), ocular sarcoidosis, posterior scleritis, serpignous choroiditis, subretinal fibrosis, uveitis syndrome, and Vogt-Koyanagi-Harada syndrome.
- infectious ocular diseases e.g., syphilis, lyme, tuberculosis, toxoplasmosis
- uveitis intermediate uveitis (pars
- methods of treating vascular diseases and/or exudative ocular diseases in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- Non-limiting examples of vascular diseases and/or exudative ocular diseases include retinal arterial occlusive disease, central retinal vein occlusion, disseminated intravascular coagulopathy, branch retinal vein occlusion, hypertensive fundus changes, ocular ischemic syndrome, retinal arterial microaneurysms, Coat's disease, parafoveal telangiectasis, hemi-retinal vein occlusion, papillophlebitis, central retinal artery occlusion, branch retinal artery occlusion, carotid artery disease (CAD), frosted branch angitis, sickle cell retinopathy and other hemoglobinopathies, angioid streaks, familial exudative vitreoretinopathy, and Eales disease.
- CAD carotid artery disease
- methods of treating traumatic and/or surgical ocular conditions in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- traumatic and/or surgical ocular conditions include sympathetic ophthalmia, uveitic retinal disease, retinal detachment, trauma, laser, PDT, photocoagulation, hypoperfusion during surgery, radiation retinopathy, and bone marrow transplant retinopathy.
- methods of treating a proliferative ocular disorder in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- a polymer conjugate wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- proliferative ocular disorders include proliferative vitreal retinopathy and epiretinal membranes, proliferative diabetic retinopathy.
- methods of treating infectious ocular disorders in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- Non-limiting examples of infectious ocular disorders include ocular histoplasmosis, ocular toxocariasis, presumed ocular histoplasmosis syndrome (POHS), endophthalmitis, toxoplasmosis, retinal diseases associated with HIV infection, choroidal disease associated with HIV infection, uveitic disease associated with HIV Infection, viral retinitis, acute retinal necrosis, progressive outer retinal necrosis, fungal retinal diseases, ocular syphilis, ocular tuberculosis, diffuse unilateral subacute neuroretinitis, and myiasis.
- infectious ocular disorders include ocular histoplasmosis, ocular toxocariasis, presumed ocular histoplasmosis syndrome (POHS), endophthalmitis, toxoplasmosis, retinal diseases associated with HIV infection, choroidal disease associated with HIV infection, uveitic disease associated with HIV Infection, viral retinitis, acute retinal
- methods of treating an ocular genetic disorder in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- Non- limiting examples of ocular genetic disorders include retinitis pigmentosa, systemic disorders with associated retinal dystrophies, congenital stationary night blindness, cone dystrophies, Stargardt's disease and fundus flavimaculatus, Bests disease, pattern dystrophy of the retinal pigmented epithelium, X-linked retinoschisis, Sorsby's fundus dystrophy, benign concentric maculopathy, Bietti's crystalline dystrophy, and pseudoxanthoma elasticum.
- methods of treating a retinal hole or tear in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- a polymer conjugate wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- retinal holes or tears include retinal detachment, macular hole, and giant retinal tear.
- methods of treating an ocular tumor-associated condition in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- Non-limiting examples of ocular tumor-associated condition include retinal disease associated with tumors, congenital hypertrophy of the RPE, posterior uveal melanoma, choroidal hemangioma, choroidal osteoma, choroidal metastasis, combined hamartoma of the retina and retinal pigmented epithelium, retinoblastoma, vasoproliferative tumors of the ocular fundus, retinal astrocytoma, intraocular lymphoid tumors.
- methods of treating punctate inner choroidopathy, acute posterior multifocal placoid pigment epitheliopathy, myopic retinal degeneration, and acute retinal pigment epithelitis in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- VEGF proliferative diabetic retinopathy
- PDR proliferative diabetic retinopathy
- retinopathy of prematurity retinopathy of prematurity
- sickle cell retinopathy age-related macular degeneration
- retina vein occlusion retina vein occlusion
- Eales disease preretinal vascularisation is a major cause of blindness.
- New blood vessels grow from the inner retinal vasculature into the vitreous humour. This can cause visual loss by vitreous haemorrhage and/ortractional retinal detachment due to contraction of the fibrous tissue associated with the new blood vessels.
- Inhibitors of the VEGF pathway intended to treat eye disease are discussed by Slevin et al. in Expert Opin. Investig. Drugs (2008) 17(9):1301-1314, herein incorporated by reference.
- methods of treating age- related macular degeneration in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- methods of treating diabetic retinopathy in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- methods of treating corneal edema in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- methods of treating macular edema in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- methods of treating dry eye in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- methods of treating age- related macular degeneration in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is an indolocarbazole compound.
- methods of treating diabetic retinopathy in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is an indolocarbazole compound.
- methods of treating corneal edema in a subject the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is an indolocarbazole compound.
- methods of treating macular edema in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is an indolocarbazole compound.
- methods of treating dry eye in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is an indolocarbazole compound.
- Allergic inflammatory diseases are characterized by an immune response against a sensitizing agent, such as an allergen, resulting in the release of inflammatory mediators that recruit cells involved in inflammation in a subject, potentially leading to tissue damage and sometimes death.
- Allergic inflammatory diseases of the eye include allergic conjunctivitis.
- methods of treating an allergic inflammatory disease in a subject comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- Topical delivery e.g., eye drops
- polymer conjugates of compounds in Tables 1 -3 optimized for topical delivery to treat the eye while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption).
- Intraocular and periocular injection is also contemplated, in several embodiments, thus some embodiments relate topolymer conjugates of compounds in Tables 1 -3, optimized for delivery via intraocular and periocular injection to treat the eye while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption).
- the warhead of the polymer conjugate is a small molecule disclosed in Tables 1-3 targeting mediator(s) of ophthalmic conditions.
- methods of treating an inflammatory ophthalmic disorder or other ophthalmic conditions in a subject comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Tables 1 -3 targeting mediator(s) of ophthalmic conditions.
- a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein are mediator(s) of ophthalmic conditions.
- the warhead employed in the LSE polymer conjugate is a small molecule a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
- the warhead of the polymer conjugate is compound 1 of Table 1 .
- the polymer conjugate is SNA-101 .
- the warhead of the polymer conjugate is compound 1 of Table 2.
- the polymer conjugate is SNA-103.
- the warhead of the polymer conjugate is compound 1 of Table 3.
- the LSE polymer conjugate is SNA-352.
- the warhead of the polymer conjugate is an indolocarbazole.
- the compound is modified (e.g., PEGylated) at that location (e.g., a PEG or modified PEG is linked to the compound by reaction with the amino group). If two or more amino groups are present, either location is PEGylated in some embodiments.
- the amino group located the furthest away from the moieties interacting with the target is used.
- the amino group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
- the effect of conjugation on the activity of the compound can be determined based on various methods, such as bioassays, mass spectroscopy, surface plasmon resonance, in vivo assays, clinical assays, and predictive in silico modeling programs.
- the compound is modified (e.g., PEGylated) at that location. If two or more sulfhydryl groups are present, either location is PEGylated in some embodiments. In other embodiments, the sulfhydryl group located the furthest away from the moieties interacting with the target is used. In some embodiments, the sulfhydryl group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
- the compound is modified (e.g., PEGylated) at that location. If two or more hydroxyl groups are present, either location is PEGylated in some embodiments. In other embodiments, the hydroxyl group located the furthest away from the moieties interacting with the target is used. In some embodiments, the hydroxyl group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
- the compound is modified (e.g., PEGylated) at that location. If two or more carboxyl groups are present, either location is PEGylated in some embodiments. In other embodiments, the carboxyl group located the furthest away from the moieties interacting with the target is used. In some embodiments, the carboxyl group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
- the compound is modified (e.g., PEGylated) at the site furthest away from the active site.
- the site that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
- Non-limiting examples of conjugation sites according to some embodiments and chemistries for compounds in Table 1 are disclosed.
- the existing carboxylic moiety (-COOH) could be conjugated to PEG-amine through formation of an amide bond using any one of several possible coupling agents (including, e.g., TBTU, HBTU, HOBt, DCC, and N- hydroxysuccinimide).
- the existing amino group (-NH2) could be conjugated to PEG-COOH through formation of an amide bond using any one of several possible coupling agents (including, e.g., TBTU, HBTU, HOBt, DCC, and N- hydroxysuccinimide).
- the existing hydroxyl moiety could be conjugated to PEG-halide through formation of an ether bond in the presence of a strong base (including, e.g. NaH, KH, and n-BuLi).
- a strong base including, e.g. NaH, KH, and n-BuLi.
- Non-limiting examples of conjugation sites according to some embodiments and chemistries for compounds in Table 2 are disclosed.
- the existing carboxylic moiety (-COOH) could be conjugated to PEG-amine through formation of an amide bond using any one of several possible coupling agents (including, e.g., TBTU, HBTU, HOBt, DCC, and N- hydroxysuccinimide).
- the existing amino group (-NH2) could be conjugated to PEG-COOH through formation of an amide bond using any one of several possible coupling agents (including, e.g., TBTU, HBTU, HOBt, DCC, and N- hydroxysuccinimide).
- the existing hydroxyl moiety (-OH) could be conjugated to PEG-halide through formation of an ether bond in presence of a strong base (including, e.g. NaH, KH, and n- BuLi).
- Non-limiting examples of conjugation sites according to some embodiments and chemistries for compounds in Table 3 are disclosed.
- the existing carboxylic moiety (-COOH) could be conjugated to PEG-amine through formation of an amide bond using any one of several possible coupling agents (including, e.g., TBTU, HBTU, HOBt, DCC, and N- hydroxysuccinimide).
- the existing amino group (-NH2) could be conjugated to PEG-COOH through formation of an amide bond using any one of several possible coupling agents (including, e.g., TBTU, HBTU, HOBt, DCC, and N-hydroxysuccinimide).
- the existing hydroxyl moiety (-OH) could be conjugated to PEG-halide through formation of an ether bond in presence of a strong base (including, e.g. NaH, KH, and n-BuLi).
- Identifying a conjugation site and developing a conjugation strategy and/or chemistry does not require that all the atoms and the structures of the starting compound are maintained. Once the active part of the compound has been identified or hypothesized, some atoms, groups and structures of the compound can be removed or modified while maintaining sufficient or similar target site binding and activity in several embodiments.
- the warhead employed in the LSE polymer conjugate is a small molecule targeting c-Src selected from one or more of the following: substituted 2-anilinopyrimidines; imidazoles, oxazoles and thiazoles with protein kinase inhibiting activities; heterocyclic substituted pyrazoles; aryl-amino substituted pyrrolopyrimidine multi-kinase inhibiting compounds; 2-phenylamino-4- (5- pyrazolylamino)-pyrimidine derivatives; bicyclic heteroaryls; 2-amino-6-anilino-purines; triazolopyridazines; substituted amides; pyrimidine derivatives; pyridinyl- pyrimidinylamino-benzamide derivatives; pyrrolo[2,3-d]pyrimidines; amino-substituted dihydropyrimido[4,5-d]pyrimidinone derivatives; 2-heteroarylamin
- the LSE polymer conjugate comprises an RORvt antagonist/inverse agonist warhead selected from one or more of the following: stearic acid; All-trans retinoic acid; ALTA 1550; Ursolic acid; Digoxin; T0901317; SR1001 ; SR1078; SR3335; SR1555; SR221 1 ; ML209; N-(1-(4-(1 ,1 ,1 ,3,3,3-hexafluoro-2- hydroxypropan-2-yl)benzyl)-1 ,2,3,4-tetrahydroquinolin-6-yl)acetamide; 2,4-difluoro-N-(1- ((4-fluorophenyl)sulfonyl)-1 ,2,3,4-tetrahydroquinolin-7-yl)benzenesulfonamide; 2-Chloro- 6-fluoro-N-(1-((4-fluoro)
- the LSE polymer conjugate comprises a Src family tyrosine kinase inhibitor warhead selected from dasatinib and saracatinib.
- the LSE polymer conjugate comprises an IL-23 inhibitor warhead selected from SCH-90222, STA-5326, and STA-5326.
- the LSE polymer conjugate comprises a STAT3 inhibitor warhead selected from cucurbitacin I, niclosamide, cryptotanshinone, SD 1008, Stat3 Inhibitor III, WP1066, Nifuroxazide, Stat3 Inhibitor, Stattic, Stat3 Inhibitor, S3I-201 ; Stat3 Inhibitor VIII, 5,15-DPP, 2-Hydroxy-4-(((4- methylphenyl)sulfonyloxy)acetyl)amino)-benzoic acid (NSC74859) and Kahweol.
- STAT3 inhibitor warhead selected from cucurbitacin I, niclosamide, cryptotanshinone, SD 1008, Stat3 Inhibitor III, WP1066, Nifuroxazide, Stat3 Inhibitor, Stattic, Stat3 Inhibitor, S3I-201 ; Stat3 Inhibitor VIII, 5,15-DPP, 2-Hydroxy-4-(((4-
- the LSE polymer conjugate comprises a JAK inhibitor warhead selected from ruxolitinib, fedratinib, tofacitinib, baricitinib, pacritinib, decernotinib, XL019, AZD1480, INCB0391 10, LY2784544, BMS91 1543, NS018, GLPG0634, GLPG0788, or N-(cyanomethyl)-4-2-(4-morpholinoanilino)pyrimidin- 4-yl)benzamide; or a pharmaceutically acceptable salt thereof.
- JAK inhibitor warhead selected from ruxolitinib, fedratinib, tofacitinib, baricitinib, pacritinib, decernotinib, XL019, AZD1480, INCB0391 10, LY2784544, BMS91 1543, NS018, GLPG0634, GLPG0788, or N-(cyanomethyl)
- Spleen tyrosine kinase is a non-receptor linked protein tyrosine kinase which, in some cases, plays a role as a mediator of immunoreceptor signaling in a host of inflammatory cells including mast cells, B-cells, macrophages and neutrophils.
- the LSE polymer conjugate comprises a SYK inhibitor warhead selected from Cerdulatinib (4-(cyclopropylamino)-2-((4-(4- (ethylsulfonyl)piperazin- 1 - yl)phenyl)amino)pyrimidine-5 -carboxamide), entospletinib (6-( 1 H-indazol-6-yl)-N-(4- morpholinophenyl)imidazo[l,2-a]pyrazin-8-amine), fostamatinib ([6-( ⁇ 5-Fluoro-2- [(3,4,5-trimethoxyphenyl)amino]-4-pyrimidinyl ⁇ amino)-2,2- dimethyl-3-oxo-2,3-dihydro- 4H-pyrido[3,2-b][l,4]oxazin-4-yl]methyl dihydrogen phosphate), fostamatinib disodium
- the LSE polymer conjugate comprises a RAC1 inhibitor warhead selected from W56, NSC23760 and NSC 23766, or the inhibitors described in Yuan Gao, et al. PNAS, May 18, 2004, vol. 101 , 7618-7623.
- the LSE polymer conjugate comprises an ENaC inhibitor warhead selected from triamterene, phenamil, amiloride and amiloride derivatives, particularly benzyl amiloride (Benzamil). Additional amiloride derivatives are described in WO2012035158; WO2009074575; WO201 1028740; WO2009150137; WO201 1079087; and WO2008135557, each of which are herein specifically incorporated by reference.
- the LSE polymer conjugate comprises a NFkB inhibitor warhead selected from Bithionol, Bortezomib, Cantharidin, Chromomycin A3, Daunorubicinum, Digitoxin, Ectinascidin 743, Emetine, Fluorosalan, Manidipine hydrochloride, Narasin, Ouabain, Sorafenib tosylate, Sunitinib malate, Tioconazole, Tribromsalan, Triclabendazolum, Zafirlukast, and Withaferin A.
- NFkB inhibitor warhead selected from Bithionol, Bortezomib, Cantharidin, Chromomycin A3, Daunorubicinum, Digitoxin, Ectinascidin 743, Emetine, Fluorosalan, Manidipine hydrochloride, Narasin, Ouabain, Sorafenib tosylate, Sunitinib malate
- the LSE polymer conjugate comprises an IRAK inhibitor warhead selected from the inhibitors described in Wang, Zhulun, et al. "IRAK-4 inhibitors for inflammation.” Current topics in medicinal chemistry 9.8 (2009): 724-737, which is herein specifically incorporated by reference.
- the LSE polymer conjugate comprises a PKC inhibitor warhead selected from sotrastaurin (also known as AEB071 and described in U.S. Pat. No. 6,645,970), 3-(1 H-lndol-3-yl)-4-[2-(piperazin-1 -yl)quinazolin-4-yl]-1 H- pyrrole-2,5-dione (described in U.S. Pat. No.
- the LSE polymer conjugate comprises a PKCa/ ⁇ inhibitor warhead selected from 3-[2-chloro-7-[(dimethylamino)methyl]-1- naphthalenyl]-4-[7-[2-(2-methoxyethoxy)ethoxy]-1 H-indol-3-yl]-1 H-pyrrole-2,5-dione (CAS No. 919992-85-1 described in PCT Publication No. WO07/006,533 and US Publication No.
- the LSE polymer conjugate comprises a specific SIP receptor agonist warhead selected from SIP itself, SEW2871 , JTE-013, VPC23019, R- 3477 (Actelion), KRP-203 (Kyorin Pharmaceutical Co.), sonepcizumab (Lpath), BAF-312 (Novartis), ONO-4641 (Ono Pharmaceutical Co.), ES-285 (PharmaMar SA), 2-amino-2-[2- (4-octylphenyl)ethyl]propane-l,3-diol (FTY720; fingolimod), phospho- FTY720, and pharmaceutically acceptable salts thereof.
- the LSE polymer conjugate comprises a PI3K inhibitor warhead selected from Compound 1 ((S)-3-(1-((9H-purin-6-yl)amino)ethyl)- 8-chloro-2-phenylisoquinolin-1 (2H)-one), AMG-319, GSK 2126458, GSK 1059615, GDC- 0032, GDC-0980, GDC-0941 , XL147, XL499, XL765, BKM 120, GS1 101 , CAL 263, SF1 126, PX-866, BEZ235, CAL-120, BYL719, RP6503, RP6530, TGR1202, INK1 117, PX-886, BAY 80-6946, IC871 14, Palomid 529, ZSTK474, PWT33597, TG100-1 15, GNE- 477, CUDC-907, AEZS-136,
- the LSE polymer conjugate comprises an AKT inhibitor warhead selected from AZD5363, miltefosine, perifosine, VQD-002, MK- 2206, GSK690693, GDC-0068, triciribine, CCT128930, PHT-427, or honokiol, or a combination thereof.
- the AKT inhibitor is MK-2206 or perifosine.
- a mTOR inhibitor warhead selected from AP23841 , AZD8055, BEZ235, BGT226, deferolimus (AP
- the LSE polymer conjugate comprises a PDE4 inhibitor warhead selected from rolipram, mesembrine, drotaverine, roflumilast, ibudilast, piclamilast, luteolin, cilomilast, diazepam, arofylline, CP-80633, denbutylline, drotaverine, etazolate, filaminast, glaucine, HT-0712, ICI-63197, irsogladine, Mesembrine, Ro20-1724, RPL-554, and YM-976.
- PDE4 inhibitor warhead selected from rolipram, mesembrine, drotaverine, roflumilast, ibudilast, piclamilast, luteolin, cilomilast, diazepam, arofylline, CP-80633, denbutylline, drotaverine, etazolate, filamin
- the warhead employed in the LSE polymer conjugate is a small molecule targeting a VEGFR selected from one or more of the following: pyrimidine compounds; diamino-pyrimidines; vegfr-binding polypeptides; fluoro substituted omega-carboxyaryl diphenyl urea; azaindole kinase inhibitors; furo-and thienopyrimidine derivatives; tricyclic amine derivatives; heterocyclic inhibitors of kinases; pyridinyl-pyrimidinylamino-benzamide derivatives; pyrrolotriazine kinase inhibitors; fused heterocyclic derivatives; anthranylamidopyridines; anthranilamide pyridine amides; 2-amino-5-substituted pyrimidine inhibitors; isomeric fused pyrrolocarbazoles and isoindolones; benzyl-benzimidazo
- the LSE polymer conjugate comprises a VEGF inhibitor warhead selected from aflibercept, ziv-aflibercept, bevacizumab, sonepcizumab, VEGF sticky trap, cabozantinib, foretinib, vandetanib, nintedanib, regorafenib, cediranib, ranibizumab, lapatinib, sunitinib, sorafenib, plitidepsin, regorafenib, verteporfin, bucillamine, axitinib, pazopanib, fluocinolone acetonide, nintedanib, AL8326, 2C3 antibody, AT001 antibody, XtendVEGF antibody, HuMax-VEGF antibody, R3 antibody, AT001/r84 antibody, HyBEV, ANG3070, APX003 antibody, APX004 antibody, ponatin
- the compound is modified (e.g., PEGylated) at that location (e.g., a PEG or modified PEG is linked to the compound by reaction with the amino group). If two or more amino groups are present, either location is PEGylated in some embodiments.
- the amino group located the furthest away from the moieties interacting with the target is used.
- the amino group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
- the effect of conjugation on the activity of the compound can be determined based on various methods, such as bioassays, mass spectroscopy, surface plasmon resonance, in vivo assays, clinical assays, and predictive in silico modeling programs.
- the compound is modified (e.g., PEGylated) at that location. If two or more sulfhydryl groups are present, either location is PEGylated in some embodiments. In other embodiments, the sulfhydryl group located the furthest away from the moieties interacting with the target is used. In some embodiments, the sulfhydryl group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
- the compound for indolocarbazole compounds having one hydroxyl group, is modified (e.g., PEGylated) at that location. If two or more hydroxyl groups are present, either location is PEGylated in some embodiments. In other embodiments, the hydroxyl group located the furthest away from the moieties interacting with the target is used. In some embodiments, the hydroxyl group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target). [0617] In some embodiments, for indolocarbazole compounds having one carboxyl group, the compound is modified (e.g., PEGylated) at that location.
- carboxyl groups are present, either location is PEGylated in some embodiments. In other embodiments, the carboxyl group located the furthest away from the moieties interacting with the target is used. In some embodiments, the carboxyl group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
- the compound is modified (e.g., PEGylated) at the site furthest away from the active site.
- the site that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
- the warhead employed in the LSE polymer conjugate is a small molecule targeting a JAK and/or STAT family protein selected from one or more of the following: ruxolitinib; fedratinib; tofacitinib; baricitinib; pacritinib; decernotinib; xl019; azd1480; incb0391 10; Iy2784544; bms91 1543; ns018; glpg0634; glpg0788; n-(cyanomethyl)-4-2-(4-morpholinoanilino)pyrimidin-4- yl)benzamide; cucurbitacin i, niclosamide, cryptotanshinone, sd 1008, stat3 inhibitor iii, wp1066, nifuroxazide, stat3 inhibitor, sauic, stat3 inhibitor, s3i-201 ; stat3 inhibitor viii, 5,
- Suitable protecting groups are for protecting functional groups during the conjugation of warhead and polymer.
- Various protecting groups as well as suitable means and conditions for protecting and deprotecting the substituents are used in several embodiments. The means and conditions of protecting and deprotecting employed depend on the nature of the involved functional groups.
- Protecting groups for hydroxy-, amino-, and/or carboxy residues are selected in several embodiments from acetonide, ethylidene methoxymethyl, 2-methoxyethoxymethyl, benzyloxymethyl, tetrahydropyranyl, methyl, ethyl, isopropyl, t-butyl, benzyl, triphenylmethyl, t-butyldimethylsilyl, triphenylsilyl, methoxycarbonyl, t-butyloxycarbonyl, benzyloxycarbonyl, fluorenylmethoxycarbonyl, acetyl, benzoyl, toluenesulfonyl, dimethoxybenzyl, nitrophenyloxycarbonyl, nitrobenzyloxycarbonyl, allyl, fluorenylmethyl, tetrahydrofuranyl, phenacyl, acetol, phenyl, trimethylsilyl
- the polymer conjugates disclosed herein may also be prepared as pharmaceutically acceptable salts including salts of inorganic acids such as hydrochloric, hydroiodic, hydrobromic, phosphoric, metaphosphoric, nitric acid and sulfuric acids as well as salts of organic acids, such as tartaric, acetic, citric, malic, benzoic, glycolic, gluconic, succinic, aryl sulfonic, (e.g., p-toluene sulfonic acids, benzenesulfonic), phosphoric, malonic, and the like.
- Suitable acids for formation of pharmaceutically acceptable salts are used in some embodiments.
- pharmaceutically acceptable salts of compounds may be formed with a pharmaceutically acceptable cation.
- Pharmaceutically acceptable cations include, but are not limited to, alkali cations (Li+, Na+, K+), earth alkali cations (Mg2+, Ca2+, Ba2+), ammonium and organic cations, such as quaternary ammonium cations.
- the intestinal wall comprises an epithelial cell wall that functions to prevent the access of enteral bacteria to the underlying lamina intestinal and beyond.
- the lamina intestinal is a thin layer of loose connective tissue which lies beneath the epithelium and together with the epithelium and basement membrane constitutes the mucosa.
- the connective tissue of the lamina basement membrane is loose and rich in cells, and can include fibroblasts, lymphocytes, plasma cells, macrophages, eosinophilic leukocytes, and mast cells.
- the lamina intestinal's richness in macrophages and lymphoid cells makes it a likely target location for immune responses to occur and forms a part of the barrier that protects internal tissues from external pathogenic microorganisms.
- a distinctive feature of inflammatory bowel diseases is the infiltration of the lamina limbal, and adaptive immune cells that direct the aberrant immune activation that yields acute and chronic inflammation.
- a reduced exposure composition for treatment of the gastrointestinal system there is provided, in some embodiments, a reduced exposure composition for treatment of the gastrointestinal system.
- a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an inhibitor, antagonist, or inverse agonist of, for example, a mediator of a gastrointestinal condition.
- the reduced exposure composition for treatment of the gastrointestinal system is delivered orally.
- the LSE polymer conjugate comprising a warhead conjugated with PEG or another molecule as described herein shows reduced absorption into, slower absorption into, and faster clearance from one or more non-target sites (e.g., systemic circulation and/or lymphatic system) as compared to an unconjugated warhead, when delivered orally for treatment of the gastrointestinal system.
- the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the Gl tract, leading to a high local concentration of the warhead targeting mediator(s) of gastrointestinal conditions.
- the polymer conjugated warhead has increased residence time in the lining of the gastrointestinal tract and is able to achieve pharmacological specificity.
- the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the intestinal epithelial cells, leading to a high local concentration of the warhead targeting mediator(s) of gastrointestinal conditions, e.g, inflammatory bowel diseases.
- the warhead of the polymer conjugate has increased residence time in the intestinal epithelial cells and is able to achieve pharmacological specificity.
- the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the lamina intestinal, leading to a high local concentration of the warhead targeting mediator(s) of gastrointestinal conditions.
- the warhead of the polymer conjugate has increased residence time in the lamina limbal and able to achieve pharmacological specificity.
- the warhead of the polymer conjugate has increased exposure to target cells within the intestinal lamina limbal cells).
- the warhead of the LSE polymer conjugate comprises a warhead targeting mediator(s) of gastrointestinal conditions for treatment of an inflammatory bowel disease.
- the warhead is a small molecule employed for treatment of an inflammatory bowel disease that is administered orally.
- oral administration of the reduced exposure compositions described herein for treatment of the gastrointestinal system has one or more of the following advantages as compared to administration of the unconjugated warhead: greater efficacy, fewer side effects, improved dosing regimens (e.g., fewer doses, lower doses), increased delivery to target cells, and/or increased residence in the submucosal layer of the intestine.
- the warhead of the polymer conjugate comprises a small molecule orally administered for treatment of an inflammatory bowel disease.
- the warhead of the LSE polymer conjugate comprises a corticosteroid or an aminosalicylate.
- the warhead of the LSE polymer conjugate comprises an aminosalicylate selected from sulfasalazine, olsalazine, mesalamine, or balsalazide.
- the warhead of the LSE polymer conjugate comprises a corticosteroid selected from prednisone, prednisolone, methylprednisolone, or budesonide.
- the warhead of the LSE polymer conjugate comprises an immunomodulatory small molecule selected from tacrolimus, methotrexate, mercaptopurine (6-mp), cyclosporine, or azathioprine (6-mp).
- the LSE polymer conjugates described herein are coupled to one or more targeting components that provide still further reduced or minimized exposure to non-target sites and/or enhanced delivery to target sites (in an additive or synergistic manner). In some embodiments, LSE polymer conjugates described are coupled to one or more components that provide more efficient delivery across the physical, physiological, and/or biological barriers of the intestinal mucosae. In some embodiments, the LSE polymer conjugates described herein fused to Cholix toxin ("Cholix”) of Vibrio cholera, or to portions or derivatives thereof.
- Cholix Cholix toxin
- LSE polymer conjugate/Cholix compositions have enhanced absorption of the LSE polymer conjugate through polarized epithelial cells of the intestinal mucosa, followed by release of the LSE polymer conjugate at the basolateral side of the intestinal epithelial cells.
- coupling of the LSE polymer conjugate to Cholix yields more efficient delivery across the physical, physiological, and biological barriers of the intestinal mucosae.
- coupling of Cholix to the LSE polymer conjugate mediates transcytosis across the Gl tract.
- the LSE polymer conjugates described herein are coupled to amino acids 1 -386 of native Cholix.
- the sequence of the targeting component coupled to the LSE polymer conjugate may vary from the native Cholix sequence, but remains, depending on the embodiment, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, or at least 95% homologous with native Cholix sequence.
- the targeting component may vary from native Cholix sequence, the targeting component retains, or in some embodiments, has enhanced, trans-epithelial transport.
- fusion of Cholix to the LSE polymer conjugate mediates transcytosis across the Gl epithelial membrane to the lamina limbal.
- the coupling of the LSE conjugate to the Cholix has an anchoring effect, wherein the interaction between the Cholix toxin and its receptor(s) at the surface of a target immune cell allows for greater exposure of the warhead at the surface of a target immune cell.
- the Cholix toxin and LSE polymer conjugate can be coupled by a peptide spacer consisting of one or more amino acids (e.g., up to 25 amino acids).
- the spacer has no specific biological activity other than to join the proteins or to preserve some minimum distance or other spatial relationship between them.
- the constituent amino acids of the spacer can be selected to influence some property of the molecule such as the folding, net charge, and/or hydrophobicity.
- a linker is capable of forming covalent bonds to both the Cholix toxin and to the LSE polymer conjugate is employed to couple the two components.
- Suitable linkers are well known to those of skill in the art and include, but are not limited to, straight or branched-chain carbon linkers, heterocyclic carbon linkers, or peptide linkers.
- the LSE polymer conjugate to be delivered to a target site within the subject is coupled to the Cholix toxin using one or more cleavable linkers.
- the cleavable linkers are cleavable by a cleaving enzyme that is present at or near the basolateral membrane of an epithelial cell.
- the selection of a cleavable linker to be cleaved by such enzymes present at or near the basolateral membrane of an epithelial cell causes the LSE polymer conjugate to be liberated from the Cholix toxin following transcytosis across the mucous membrane and release from the epithelial cell into the cellular matrix on the basolateral side of the membrane soon after transcytosis across the epithelial membrane.
- the enzyme that is present at a basolateral membrane of a polarized epithelial cell is selected from, e.g., Cathepsin Gl, Chymotrypsin I, Elastase I, Subtilisin Al, Subtilisin All, Thrombin I, or Urokinase I.
- Gl tract-targeted compositions provided, in several embodiments, wherein the warheads described herein are not conjugated to a polymer and are coupled to Cholix toxin.
- delivery of the LSE polymer conjugates to target sites with the gastrointestinal system is enhanced by encapsulation of the LSE polymer conjugate within an inflammation-targeting hydrogel.
- hydrogel encapsulation of LSE polymer conjugate enhances delivery to inflamed portions of the colon.
- the inflammation-targeting hydrogel is comprises ascorbyl palmitate.
- ascorbyl palmitate self-assembles into a nanofibrous gel.
- the hydrogel preferentially adheres to inflamed human colon mucosa.
- the LSE polymer conjugate is released from the hydrogel upon contact by hydrolytic enzymes at a site of inflammation.
- delivery of the LSE polymer conjugate in the hydrogel further reduces systemic exposure.
- the hydrogel/LSE polymer conjugate composition is administered by enema.
- the reduced exposure compostions disclosed herein are formulated for drug delivery as described in: Allen, L, & Ansel, H. C. (2013). Ansel's pharmaceutical dosage forms and drug delivery systems. Lippincott Williams & Wilkins; Aulton, M. E., & Taylor, K. M. (Eds.). (2017). Aulton's Pharmaceutics E-Boo : The Design and Manufacture of Medicines. Elsevier Health Sciences; and/or Wen, H., & Park, K. (Eds.). (201 1). Oral controlled release formulation design and drug delivery: theory to practice. John Wiley & Sons, each of which are herein specifically incorporated by reference.
- compositions described herein for the treatment and/or prevention of Gl conditions may be provided in a form that can be orally administered, including but not limited to liquids, tinctures, syrups, elixirs, capsules, soft gelatin capsules (gel caps), pills and tablets, lozenges, gum, powders and emulsions.
- Tablets or capsules may further comprise an enteric coating to prevent premature dissolution under the chemically harsh environment of the stomach.
- enteric coatings include coatings comprising eudragit and/or cellulose acetate phthalate.
- Other protective coating materials include, but are not limited to, hydroxypropyl methylcellulose, polyethylene glycol and ethylcellulose.
- the protective coating further comprises a plasticizing agent, including but not limited to triethylcitrate and polyvinyl pyrrolidone.
- a plasticizing agent including but not limited to triethylcitrate and polyvinyl pyrrolidone.
- the tablets, capsules and other like embodiments of the disclosed compositions may further advantageously comprise particle lubricants that minimize the tendency of the granular compositions to agglomerate.
- particle lubricant as used herein is meant the class of materials used in the manufacturing of pharmaceutical tablets as lubricants to improve the flowability and prevent agglomeration of an active agent during the tableting process.
- particle lubricants include, but are not limited to, talc, lactose, corn starch, ethyl cellulose, fatty acid salts such as magnesium stearate, agar pectin, fatty acids such as stearic acid, gelatin and acacia.
- U.S. Patent No. 6,217,903 herein specifically incorprated by reference, discloses sustained release polymer blend pharmaceutical formulations containing active agents with other ingredients, polyethylene glycol, stearic acid, colloidal silicon dioxide, magnesium stearate, calcium stearate, waxes, polyvinyl pyrollidone.
- U.S. Patent No. 6,1 10,498 to Rudnic et al., herein specifically incorprated by reference describes an osmotic drug delivery system containing active agent by using polyvinyl pyrollidone sodium lauryl sulfate and also some plasticizer like propylene glycol, triethyl citrate, and vegetable oil.
- This patent provides an osmotic drug delivery system, preferably in the form of a tablet comprising various components like polymers sprayed on tablets to give 2-15 % coating weight, wicking agents, non-swelling solubilizing agents and lubricating agents.
- reduced exposure compsotions formualted according to the disclosures of U.S. Patent Nos. 6,217,903 and 6,1 10,498.
- solid dosage forms for oral delivery include, but are not limited to, capsules, soft-gel capsules, tablets, caplets, powders, granules or other solid oral dosage forms, all of which can be prepared by methods well known in the art.
- compositions additionally comprising additives in amounts customarily employed including, but not limited to, a pH adjuster, a preservative, a flavorant, a taste-masking agent, a fragrance, a humectant, a tonicifier, a colorant, a surfactant, a plasticizer, a lubricant such as magnesium stearate, a flow aid, a compression aid, a solubilizer, an excipient, a diluent such as microcrystalline cellulose, e.g. Avicel PH 102 supplied by FMC corporation, or any combination thereof.
- additives may include phosphate buffer salts, citric acid, glycols, and other dispersing agents.
- the reduced exposure compostions provided herein are delivered by a drug delivery system compriseing a core of water in a wax housing which melts at body temperature surrounded by a matrix of drug, water soluble or dispersible excipients and optionally, osmotic-active agents, which in turn is optionally surrounded by a coating which optionally can be semi-permeable.
- the water of the aqueous core can be neat, an aqueous buffer, or other aqueous solubulizing system or it can be in the form of an aqueous cross-linked polymeric gelatinous pellet.
- the volume of available water can vary widely depending on the solubility and amount of drug to be delivered and on the site of delivery, but usually is between about 0.1 ml and 3 ml.
- the waxy housing of the aqueous core is composed of a water-insoluble, low melting (35°-37° C.) material selected from theobroma oil, SUPPOCIRE A® (an eutectic mixture of mono-, di-, and triglycerides supplied by A. & S.
- the matrix surrounding the aqueous core has an outer shape and dimensions of conventional rectal or vaginal suppositories and comprises a mixture of one or more drugs and pharmaceutically acceptable water soluble and/or dispersible ingredients.
- Drug any of the drugs used to treat the body can be incorporated as the drug of the delivery device of this invention.
- Drug is used herein in its broadest sense as including any composition or substances that will produce a pharmacologic response.
- a reduced exposure composition for treatment of dermatological conditions there is provided, in some embodiments, a reduced exposure composition for treatment of dermatological conditions.
- a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an inhibitor, antagonist, or inverse agonist of, for example, a mediator of a dermatological condition.
- the reduced exposure composition for treatment of the skin is delivered topically.
- the reduced exposure composition traverses and/or diffuses through hair follicles, skin pores, mucosa, cornea, compromised skin, the epidermis, dermis, skin, scalp, damaged skin, and/or diseased skin.
- the reduced exposure composition penetrates one or more physical barriers of the epidermis, the dermis, and/or the subcutis. In some embodiments, the reduced exposure composition penetrates one or more biological barriers of the epidermis, the dermis, and/or the subcutis. In some embodiments, the reduced exposure composition penetrates one or more physiological barriers of the epidermis, the dermis, and/or the subcutis. In some embodiments, the reduced exposure composition targets immune cells residing in the epidermis, the dermis, and/or the subcutis. In some embodiments, the reduced exposure composition targets non-immune cells residing in the epidermis, the dermis, and/or the subcutis.
- the reduced exposure composition targets keratinocytes in the epidermis, the dermis, and/or the subcutis. In some embodiments, the reduced exposure composition targets a single site within the epidermis, the dermis, and/or the subcutis. In some embodiments, targets site comprises two, three, four, five, six, seven, eight, nine, or more sites within the epidermis, the dermis, and/or the subcutis. In some embodiments, the reduced exposure composition has increased activity and/or bioavailability at the target site(s). In some embodiments, the reduced exposure composition has increased activity and/or bioavailability at one or more non-target site(s).
- the one or more target site(s) comprises the systemic system, the lymphatic system, and/or non-target tissues at which pharmacological activity is not desired.
- the target site within the skin is selected from the group consisting of the epidermis, the dermis, and/or the subcutis, and any combination thereof.
- the target site within the epidermis, the dermis, and the subcutis comprises one or more of the stratum corneum, stratum lucidum, stratum granulosum, stratum spinosum, and stratum basale layers, the papillary layer of the dermis, the reticular layer of the dermis, sebaceous glands, arrector pili muscles, sensory nerve fibers, hair follicles, hair roots, pacinian corpuscles, hair root plexus, eccrine sweat glands, the hypodermis and/or the superficial fascia, and any combination thereof.
- the target comprises one, two, three, four, five, six, seven, or eight of the following sites within the skin: the stratum corneum, stratum lucidum, stratum granulosum, stratum spinosum, and stratum basale layers, the papillary layer of the dermis, the reticular layer of the dermis, sebaceous glands, arrector pili muscles, sensory nerve fibers, hair follicles, hair roots, pacinian corpuscles, hair root plexus, eccrine sweat glands, the hypodermis, and the superficial fascia.
- the target site comprises the epidermis and the non-target site comprises one or more of the dermis and subcutis.
- the target site comprises the dermis and the non-target site comprises one or more of the subcutis and epidermis. In some embodiments, the target site comprises the subcutis and the non-target site comprises one or more of the epidermis and dermis. In some embodiments, the target site comprises one or two of the dermis, epidermis and subcutis and the non-target site comprises the remaining one or two.
- the LSE polymer conjugate comprising a warhead conjugated with PEG or another molecule as described herein shows reduced absorption into, slower absorption into, and faster clearance from one or more non-target sites (e.g., systemic circulation and/or lymphatic system) as compared to an unconjugated warhead, when delivered topically for treatment of the skin.
- the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the epidermis, the dermis, and/or the subcutis, leading to a high local concentration of the warhead targeting mediator(s) of dermatological conditions.
- the polymer conjugated warhead has increased residence time in the epidermis, the dermis, and/or the subcutis and is able to achieve pharmacological specificity.
- the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the epidermis, the dermis, and/or the subcutis, leading to a high local concentration of the warhead targeting mediator(s) of dermatological conditions, e.g, inflammatory skin diseases.
- the warhead of the polymer conjugate has increased residence time in the epidermis, the dermis, and/or the subcutisand is able to achieve pharmacological specificity.
- the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the epidermis, the dermis, and/or the subcutis, leading to a high local concentration of the warhead targeting mediator(s) of dermatological conditions.
- the warhead of the polymer conjugate has increased residence time in the epidermis, the dermis, and/or the subcutisand is able to achieve pharmacological specificity.
- the warhead of the polymer conjugate has increased exposure to target cells within the epidermis, the dermis, and/or the subcutis (e.g., innate and adaptive immune cells).
- the warhead of the LSE polymer conjugate comprises a warhead targeting mediator(s) of dermatological conditions for treatment of an inflammatory skin disease.
- the warhead is a small molecule employed for treatment of an inflammatory skin disease that is administered topically.
- topical administration of the reduced exposure compositions described herein for treatment of the skin has one or more of the following advantages as compared to administration of the unconjugated warhead: greater efficacy, fewer side effects, improved dosing regimens (e.g., fewer doses, lower doses), increased delivery to target cells, and/or increased residence in the epidermis, the dermis, and/or the subcutis.
- the reduced exposure compostions disclosed herein are formulated for drug delivery as described in: Ansel, H. C, Popovich, N. G., & Allen, L. V. (1995). Pharmaceutical dosage forms and drug delivery systems. Lippincott Williams & Wilkins; and/or Williams, A. (2003). Transdermal and topical drug delivery: from theory to clinical practice (pp. 169-194). London: Pharmaceutical Press, each of which are herein specifically incorporated by reference.
- a reduced exposure composition for treatment of ophthalmic conditions there is provided, in some embodiments, a reduced exposure composition for treatment of ophthalmic conditions.
- a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an inhibitor, antagonist, or inverse agonist of, for example, a mediator of an ophthalmic condition.
- the reduced exposure composition for treatment of the eye is delivered topically by eye drops.
- the reduced exposure composition for treatment of the eye is administered by an implanted drug delivery device.
- the compostion is delivered by intraocular or periocular injection.
- the reduced exposure composition traverses and/or diffuses through the conjunctival epithelium, Tenon's fascia, episclera, sclera, and/or choroid.
- the reduced exposure composition penetrates one or more physical barriers of the anterior segment and/or posterior segment of the eye.
- the reduced exposure composition penetrates one or more biological barriers of the anterior segment and/or posterior segment of the eye.
- the reduced exposure composition penetrates one or more physiological barriers of the anterior segment and/or posterior segment of the eye.
- the reduced exposure composition targets immune cells residing in anterior segment and/or posterior segment of the eye.
- the reduced exposure composition targets non-immune cells residing in anterior segment and/or posterior segment of the eye.
- the reduced exposure composition targets one site within the anterior segment and/or posterior segment of the eye.
- targets site comprises two or more sites within the anterior segment and/or posterior segment of the eye.
- the reduced exposure composition has increased activity and/or bioavailability at the target site(s).
- the reduced exposure composition has reduced activity and/or bioavailability at one or more non-target site(s).
- the one or more target site(s) comprises the systemic system, the lymphatic system, and/or non-target tissues at which pharmacological activity is not desired.
- the target site within the eye is selected from the group consisting of anterior segment and/or posterior segment of the eye, and any combination thereof. In some embodiments, the target site comprises only one of the anterior segment and the posterior segment of the eye. In some embodiments, the target site comprises both the anterior segment and the posterior segment of the eye. In some emboidments, the target site comprises one or more of a posterior sub-Tenon space, a posterior suprachoroidal space and a posterior intrascleral space. In some emboidments, the target site comprises one or more of anterior sub-Tenon space, an anterior suprachoroidal space, an anterior intrascleral space.
- the target comprises 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, or 1 1 of the following sites within the eye: cornea, lens, sclera, anterior chamber, iris, cornea, posterior chamber, choroid, retina, Bowman's layer, stroma, Descemet's membrane, the endothelium, and Tenon's Capsule.
- the target site comprises 1 ,
- the reduced exposure compsotion treats a condition which affects or which involves an ocular region or site, such as a periocular muscle, an eye lid or an eye ball tissue or fluid which is located anterior to the posterior wall of the lens capsule or ciliary muscles.
- the target site comprises the front of the eye, such as for example, one or more of the conjunctiva, the cornea, the conjunctiva, the anterior chamber, the iris, the posterior chamber (behind the iris but in front of the posterior wall of the lens capsule), the lens and the lens capsule as well as blood vessels, lymphatics and nerves which vascularize, maintain or innervate an anterior ocular region or site.
- the non-target site comprises the posterior of the eye.
- Examples of front of the eye ocular conditions treated by such embodiments include, but are not limited to, aphakia; pseudophakia; astigmatism; blepharospasm; cataract; conjunctival diseases; conjunctivitis; corneal diseases; corneal ulcer; dry eye syndromes; eyelid diseases; lacrimal apparatus diseases; lacrimal duct obstruction; myopia; presbyopia; pupil disorders; refractive disorders and strabismus
- the reduced exposure compsotion treats a condition which affects or which involves the posterior (back of the eye).
- Posterior ocular conditions are disease, ailment or conditions which primarily affect or involve a posterior ocular region or site such as choroid or sclera (in a position posterior to a plane through the posterior wall of the lens capsule), vitreous, vitreous chamber, retina, optic nerve (i.e. the optic disc), and blood vessels and nerves which vascularize or innervate a posterior ocular region or site.
- the target site is one or more of the choroid or sclera (in a position posterior to a plane through the posterior wall of the lens capsule), vitreous, vitreous chamber, retina, optic nerve (i.e. the optic disc), and blood vessels and nerves which vascularize or innervate a posterior ocular region or site.
- the non-target site comprises the front of the eye.
- posterior ocular conditions treated by such embodiments include, but are not limited to, macular degeneration (such as non-exudative age related macular degeneration and exudative age related macular degeneration); choroidal neovascularization; acute macular neuroretinopathy; macular edema (such as cystoid macular edema and diabetic macular edema); Behcet's disease, retinal disorders, diabetic retinopathy (including proliferative diabetic retinopathy); retinal arterial occlusive disease; central retinal vein occlusion; uveitic retinal disease; retinal detachment; ocular trauma which affects a posterior ocular site or location; a posterior ocular condition caused by or influenced by an ocular laser treatment; posterior ocular conditions caused by or influenced by a photodynamic therapy; photocoagulation; radiation retinopathy; epiretinal membrane disorders; branch retinal vein occlusion; anterior ischemic optic neuropathy; non- retin
- Ophthalmic cancers and precancerous conditions are treated in some embodiments, including but not limited to intraocular lymphoma, intraocular lesions, intraocular melanoma (e.g., uveal melanoma). Tumors may also be treated including but not limited to tumors in the choroid, iris and ciliary body.
- the LSE polymer conjugate comprising a warhead conjugated with PEG or another molecule as described herein shows reduced absorption into, slower absorption into, and faster clearance from one or more non-target sites (e.g., systemic circulation and/or lymphatic system) as compared to an unconjugated warhead, when delivered topically for treatment of the eye.
- the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the anterior segment and/or posterior segment of the eye, leading to a high local concentration of the warhead targeting mediator(s) of ophthalmic conditions.
- the polymer conjugated warhead has increased residence time in the anterior segment and/or posterior segment of the eye and is able to achieve pharmacological specificity.
- the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the anterior segment and/or posterior segment of the eye, leading to a high local concentration of the warhead targeting mediator(s) of ophthalmic conditions, e.g, inflammatory ophthalmic diseases.
- the warhead of the polymer conjugate has increased residence time in the anterior segment and/or posterior segment of the eye and is able to achieve pharmacological specificity.
- the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the anterior segment and/or posterior segment of the eye, leading to a high local concentration of the warhead targeting mediator(s) of ophthalmic conditions.
- the warhead of the polymer conjugate has increased residence time in the anterior segment and/or posterior segment of the eye and is able to achieve pharmacological specificity.
- the warhead of the polymer conjugate has increased exposure to target cells within the anterior segment and/or posterior segment of the eye (e.g., innate and adaptive immune cells).
- the warhead of the LSE polymer conjugate comprises a warhead targeting mediator(s) of ophthalmic conditions for treatment of a disease or disorder of the eye.
- the warhead is a small molecule employed for treatment of an ophthalmic conditions that is administered topically.
- topical administration of the reduced exposure compositions described herein for treatment of the eye has one or more of the following advantages as compared to administration of the unconjugated warhead: greater efficacy, fewer side effects, improved dosing regimens (e.g., fewer doses, lower doses), increased delivery to target cells, and/or increased residence in the anterior segment and/or posterior segment of the eye.
- reduced exposure compostions suibile for topical (e.g., ointment or eye drop) application are administered by direct intravitreal injection.
- the reduced exposure compsotions are administered by subconjunctival injection.
- the reduced exposure compsotions are administered by subtenon injection.
- the compostions are administered by peribulbar injection.
- are reduced exposure compostions which can be administered via intraocular implantable devices known to one of skill in the art. Intracameral injection, or injection into the anterior chamber of they eye, in contemplated in some embodiments.
- Injection of polymer conjugate into the vitreous is contemplated in some embodiments to provde a high local concentration of polymer conjugate in the vitreous and retina.
- Periocular routes of delivery including, not limited to, subconjunctival, subtenon, retrobulbar, peribulbar and posterior juxtascleral delivery, are contemplated.
- Adjuvants with which the reduced exposure compsotions may be admixed with include but are not limited to lactose, sucrose, starch powder, cellulose esters of alkanoic acids, stearic acid, talc, magnesium stearate, magnesium oxide, sodium and calcium salts of phosphoric and sulphuric acids, acacia, gelatin, sodium alginate, polyvinylpyrrolidine, and/or polyvinyl alcohol.
- Solubilized formulation comprising the reduced exposure compsotions provided herein may be formulated in a solvent including, but not limited to, polyethylene glycol of various molecular weights, propylene glycol, carboxymethyl cellulose colloidal solutions, methanol, ethanol, DMSO, corn oil, peanut oil, cottonseed oil, sesame oil, tragacanth gum, and/or various buffers.
- a solvent including, but not limited to, polyethylene glycol of various molecular weights, propylene glycol, carboxymethyl cellulose colloidal solutions, methanol, ethanol, DMSO, corn oil, peanut oil, cottonseed oil, sesame oil, tragacanth gum, and/or various buffers.
- Other adjuvants and modes of administration are well known in the pharmaceutical art and may be used in the practice of the methods, compositions and liquid formulations described herein.
- carriers or diluents include time delay material, such as, for example, glyceryl monostearate or glyceryl distearate alone or with a wax, or other materials well known in the art.
- time delay material such as, for example, glyceryl monostearate or glyceryl distearate alone or with a wax, or other materials well known in the art.
- the reduced exposure compsotion compostions for use in treatment of ophthalmic conditions as described herein may also include gel formulations, erodible and non-erodible polymers, microspheres, and liposomes.
- the reduced exposure compostions disclosed herein are formulated with pharmaceutically acceptable excipients for clinical use to produce a pharmaceutical composition.
- Formulations for ocular administration may be presented as a solution, suspension, particles of solid material, a discrete mass of solid material, incorporated within a polymer matrix, liquid formulations or in any other form for ocular administration.
- the liquid formulations described herein are administered intraocularly.
- Intraocular administration includes placement or injection within the eye, including in the vitreous, is also provided herein.
- Injection of the compostion may be subconjunctival injection, e.g., injection of the reduced exposure compostion underneath the conjunctiva, or between the sclera and conjunctiva.
- subtenon injection of the compsotions disclosed herein is injection of the reduced exposure compostion into the tenon's capsule around the upper portion of the eye and into the "belly" of the superior rectus muscle.
- Retrobulbar injection of the compostion into the conical compartment of the four rectus muscles and their intermuscular septa, behind the globe of the eye, is contemplated in some embodiments.
- Peribulbar injection at a location external to the confines of the four rectus muscles and their intramuscular septa (e.g., outside of the muscle cone) is further contemplated.
- Posterior juxtascleral delivery of the reduced exposure compostions described herein near and above the macula, in direct contact with the outer surface of the sclera, and without puncturing the eyeball, is also provided in some embodiments.
- the reduced exposure compostions disclosed herein are formulated for drug delivery as described in: Lang, J. C. (1995). Ocular drug delivery conventional ocular formulations. Advanced drug delivery reviews, 76(1), 39-43; and /or Le Bourlais, C, Acar, L, Zia, H., Sado, P. A., Needham, T., & Leverge, R. (1998). Ophthalmic drug delivery systems— recent advances. Progress in retinal and eye research, 77(1), 33-58, each of which are herein specifically incorporated by reference.
- polymer conjugates may also be made as described in US Patent Nos. 8,673,347 and 8,926,955, both herein incorporated by reference.
- Several embodiments provide a method for the production of polymer conjugates of the active agents that result in a highly pure reaction product, obtained in high and consistent yields.
- the conjugation reaction of the process to synthesize a conjugate polymer compound is catalysed by a base in an organic solvent.
- the base may be a strong base.
- the base is selected from the group of alkali metal hydrides, tertiary amines and/or alkoxide.
- the base catalysing the polymer conjugation reaction is sodium hydride.
- Other bases such as sodium methoxide, or triethylamine can also be used.
- the molar ratio of the base catalyst to the compound is between about 1 :1 and about 4:1 , about 1 :1 to about 1.5:1 and about 1 :1.
- the reaction may be carried out in an organic solvent, such as in anhydrous conditions (e.g., in a dry organic solvent).
- the water content in the solution mixture of the conjugation process may be equal or less than 200 ppm.
- the organic solvent may be selected from the group of dichloromethane, chloroform, ⁇ , ⁇ -dimethylformamide. In certain embodiments, the organic solvent is dichloromethane or anhydrous dichloromethane.
- the conjugation reaction may be carried out under inert gas atmosphere, such as nitrogen or argon atmosphere.
- the reaction of the process may be carried out at a temperature of about -10° to about 60° C, about 0° to about 25° C or at room temperature after an initial step at 0° C.
- the polymer conjugate may then be separated and purified from the reaction mixture.
- the compound is obtained by purification of the crude mixture by flash chromatography.
- An automated gradient flash purification system may be used and may be equipped with a suitable column and solvent.
- the purification method may be selected from reverse phase and direct phase columns and the conditioning/elution solvent may be selected from dichloromethane, water, methanol, acetonitrile, ammonium formate buffer solution at different mixture ratios.
- the compound is purified by a reverse phase flash chromatography equipped with a C18 cartridge and the purification is carried out by isocratic elution with acetonitrile/5 mM ammonium formate buffer (pH 3.5) 40:60. In one embodiment, the compound is purified by a normal phase flash chromatography.
- the product may then be dried e.g. over sodium sulphate and filtered off and the solvent is removed by evaporation under reduced pressure at 25° C.
- Purification of the target product is carried out in several embodiments. After the purification step the resultant polymer compound has a purity of at least about 95%, about 96%, about 97%, about 98%, about 98.5%, about 99% or about 99.5%.
- the disclosed process results in an overall mass yield of the compound from about 40% to about 98% by weight, or from about 50% to about 95% by weight based on the weight of a reactant compound.
- the polymer moiety which is covalently attached to the active entity is biocompatible, can be of natural or semi-synthetic or synthetic origin and can have a linear or branched structure.
- the polymer may be selected from poly(alkylene oxides), or from (polyethylene) oxides.
- polymers include without limitation polyacrylic acid, polyacrylates, polyacrylamide or N- alkyl derivatives thereof, polymethacrylic acid, polymethacrylates, polyethylacrylic acid, polyethylacrylates, polyvinylpyrrolidone, poly(vinylalcohol), polyglycolic acid, polylactic acid, poly(lactic-co-glycolic) acid, dextran, chitosan, hydroxyethyl starch.
- the above-mentioned polymer moiety can carry an amino functional end-group or can be functionalized to carry an amino functional end-group.
- the polymer moiety can be an amino-activated polymer of general formula X— NH2.
- the reaction of formation of the compositions identified herein may be carried out at a temperature of about 10° to about 60° C, about 15° to about 25° C. or at room temperature.
- the polymer moiety X may be a polyethylene glycol (PEG) moiety, wherein the terminal OH group can optionally be modified e.g. with C1 -C5 alkyl or C1 - C5 acyl groups, such as with C1 -, C2- or C3-alkyl groups or C1 -, C2- or C3 groups.
- the modified polyethylene glycol may be a terminally alkoxy-substituted polyethylene glycol, including a methoxy-polyethylene-glycol (mPEG).
- the conjugated polymer compounds may be used as active agents in a topical medicament useful for the prevention, alleviation and/or treatment of dermal pathologies. It has been shown that the conjugated polymer compounds described herein are very advantageously used as topical medicament since they do not show adverse or toxic effects (e.g. irritation) when dermally administered or any phototoxic effect (e.g. photomutagenicity, phototoxicity or photosensitisation) (as shown in the studies described in the following examples).
- adverse or toxic effects e.g. irritation
- any phototoxic effect e.g. photomutagenicity, phototoxicity or photosensitisation
- the dermal pathologies for such treatment may be pathologies characterized by hyperproliferation of the keratinocytes, such as psoriasis, atopic dermatitis, chronic eczema, acne, pitiriasis rubra pilaris, keloids, hypertrophic scars and skin tumors, such as keratoacanthoma, squamous cell carcinoma, basal cell carcinoma.
- keratinocytes such as psoriasis, atopic dermatitis, chronic eczema, acne, pitiriasis rubra pilaris, keloids, hypertrophic scars and skin tumors, such as keratoacanthoma, squamous cell carcinoma, basal cell carcinoma.
- the polymer conjugate compounds may be used in combination with at least one natural extract or essential oil which is anti-itching agent, for example and not restricted to, extracts of Abelmoschus esculentus, Actaea alba, Aglaia odorata, Alkanna tinctoria, Althaea officinalis, Altingia excelsa, Andropogon virginicus, Aralia nudicaulis, Aralia racemosa, Argemone mexicana, Barleria prionitis, Camelia sinensis, Caesalpinia digyna, Campsis grand/flora, Carissa congesta, Carthamus oxyacantha, Cassia tora, Chrysanthemum indicum, Cimicifuga racemosa, Cinnamomum camphora, Clematis vitalba, Cuscuta reflexa, Diospyros peregrina, Enicostema axillare, Hammamelis virginiana, Jatroph
- the polymer conjugate compounds may be used in combination with at least one synthetic compound or product of biotechnological origin which is an anti- itching agent, for example and not restricted to mepyramine (pyrilamine), antazoline, diphenhydramine, carbinoxamine, doxylamine, clemastine, dimenhydrinate, pheniramine, chlorphenamine (chlorpheniramine), dexchlorpheniramine, brompheniramine, triprolidine, cyclizine, chlorcyclizine, hydroxyzine, meclizine, cetirizine, levocetirizine, promethazine, thenaldine, alimemazine (trimeprazine), cyproheptadine, azatidine, ketotifen, acrivastine, astemizole, cetirizine, loratadine, desloratadine, mizolastine, terfenadine, fexofenadine, fexofenadine,
- the polymer conjugate compounds may be used in combination with at least one physiological cooling agent, for example and not restricted to menthone glycerol acetal, menthyl lactate, menthyl ethyl oxamate, substituted menthyl-3-carboxylic acid amides (e.g.
- menthyl-3-carboxylic acid N-ethylamide, Na-(L- menthanecarbonyl)glycine ethyl ester, 2-isopropyl-N-2,3-trimethylbutanamide, substituted cyclohexanecarboxylic acid amides, 3-menthoxypropane-1 ,2-diol, 2- hydroxyethyl menthyl carbonate, 2- hydroxy propyl menthyl carbonate, N- acetylglycine menthyl ester, isopulegol, menthyl hydroxycarboxylic acid esters (e.g.
- menthyl 3- hydroxybutyrate monomenthyl succinate, monomenthyl glutarate, 2- mercaptocyclodecanone, menthyl 2-pyrrolidin-5-onecarboxylate, 2,3-dihydroxy-p- menthane, 3,3,5-trimethylcyclohexanone glycerol ketal, 3-menthyl 3,6-di- and - trioxaalkanoates, 3-menthyl methoxyacetate and icilin.
- compositions comprising an effective amount of at least one compound in Tables 1 -3 optionally together with pharmaceutically acceptable carriers, adjuvants, diluents or/and additives.
- Pharmaceutical carriers, adjuvants, diluents or/and additives are applied in the formulation of the pharmaceutical composition comprising a compound of embodiments identified herein.
- the disclosed compounds can be employed as the sole active agent in a pharmaceutical composition.
- the compounds of Tables 1 -3 may be used in combination with one or several further active agents, e.g. other active pharmaceutical agents in the treatment of the conditions described herein.
- the polymer conjugate compounds may be used in combination with at least one endogenous angiogenesis inhibitor, for example and not restricted to, angioarrestin, angiostatin (plasminogen fragment), antiangiogenic antithrombin III, cartilage-derived inhibitor (CDI), CD59 complement fragment, endostatin (collagen XVIII fragment), fibronectin fragment, Gro-beta, heparinases, heparin hexasaccharide fragment, human chorionic gonadotropin (hCG), interferon alpha/beta/gamma, interferon inducible protein (IP-10), lnterleukin-12, kringle 5 (plasminogen fragment), metalloproteinase inhibitors (TIMPs), 2-methoxyestradiol, placental ribonuclease inhibitor, plasminogen activator inhibitor, platelet factor-4 (PF4), prolactin 16 kD fragment, proliferin-related protein (PRP),
- the polymer conjugate compounds may be used in combination with at least one additional anti-IBD therapeutic agent, for example and not restricted to azathioprine, 6-mercaptopurine (6-MP), aminosalicylate, sulfasalazine, mesalamine, corticosteroid, prednisone, prednisone equivalent, budesonide, probiotic, methotrexate, cyclosporine, tacrolimus, metronidazole, ciprofloxacin, leflunomide, chloroquine, hydroxychloroquine, penicillamine, tocilzumab, anakinra, abatacept, rituximab, efalizumab, belimumab, tofacitinib, baricitinib, golimumab, vedolizumab, natalizumab, ustekinumab, etanercept, infliximab, adalim
- the polymer conjugate compounds may be used in combination with at least one steroidal anti-inflammatory drug and/or one further agent capable of inhibiting an early mediator of the inflammatory cytokine cascade, e.g. an antagonist or inhibitor of a cytokine selected from the group consisting of TNF, IL-1 a, IL- 1 ⁇ , IL-Ra, IL-8, MIP-1 a, MIF- ⁇ ⁇ , MIP-2, MIF and IL-6.
- a cytokine selected from the group consisting of TNF, IL-1 a, IL- 1 ⁇ , IL-Ra, IL-8, MIP-1 a, MIF- ⁇ ⁇ , MIP-2, MIF and IL-6.
- Particularly useful antiinflammatory drugs are selected from alclometasone dipropionate, amcinonide, beclomethasone dipropionate, betamethasone, betamethasone benzoate, betamethasone dipropionate, betamethasone sodium phosphate, betamethasone sodium phosphate and acetate, betamethasone valerate, clobetasol butyrate, clobetasol propinate, clocortolone pivalate, Cortisol (hydrocortisone), Cortisol (hydrocortisone) acetate, Cortisol (hydrocortisone) butyrate, Cortisol (hydrocortisone) cypionate, Cortisol (hydrocortisone) sodium phosphate, Cortisol (hydrocortisone) sodium succinate, Cortisol (hydrocortisone) valerate, cortisone acetate, desonide, desoximetasone, dexamethasone, de
- agents which can be used in combination with the polymer compounds are e.g. antagonists and/or inhibitors of RAGE, antagonists and/or inhibitors of HMGB1 , antagonists and/or inhibitors of the interaction of a Toll-like receptor (TCR) with HMGB1 , the functional N-terminal lectin-like domain (D1) of thrombomodulin and/or a synthetic double-stranded nucleic acid or nucleic acid analogue molecule with a bent shape structure as described in the international patent application WO 2006/002971 which is herein incorporated by reference.
- TCR Toll-like receptor
- the warhead of the LSE polymer conjugate comprises an antibiotic selected from one or more of azithromycin, tobramycin, bacitracin, chloramphenicol, polymyxin B, gentamycin, sulfacetamide, moxifloxacin, gatifloxacin, neomycin, polymyxin and besifloxacin.
- the warhead of the LSE polymer conjugate comprises a non- systemic antibiotic that is minimally absorbed and has high local concentrations in the eye after injection, eye drops, ointments, etc.
- Such delivery compositions are able to, in some embodiments, deliver localized doses of antibiotics (or other antimicrobials) to the eye without significant exposure to other tissue which, advantageously, limits the side effects.
- Side effects include but are not limited to rash, itching or burning eyes, pain, redness, swelling, and vision problems.
- localized and precision-based treatment permits lower doses of the antibiotics/antimicrobials, which in some embodiments may reduce or avoid toxicity, adverse immune effects, tolerance, etc.
- systemic exposure and/or exposure to a non-target tissue of antimicrobials is reduced by at least 25%, 50%, 75% or more using the embodiments described herein (e.g., LSE polymer conjugates) as compared with administration of the same antimicrobial without the polymer conjugates described herein.
- the compositions described herein may be administered by a physician or other professional.
- compositions may be performed dermally, via, for example, ointments, creams, oils, liposomes or trans-dermal patches, or wherein the polymer conjugates are incorporated into liposomes.
- Excipients can include a nonaqueous or aqueous carrier, and one or more agents selected from moisturizing agents, pH adjusting agents, strontium ions (Sr2+), deodorants, fragrances, chelating agents, preservatives, emulsifiers, thickeners, solubilizing agents, penetration enhancers, anti-irritants, colorants, surfactants, beneficial agents, pharmaceutical agents, and other components for use in connection with the compositions described herein (such as oral compositions for treatment of the gastrointestinal tract or topical compositions for treatment of the skin).
- agents selected from moisturizing agents, pH adjusting agents, strontium ions (Sr2+), deodorants, fragrances, chelating agents, preservatives, emulsifiers, thickeners, solubilizing agents, penetration enhancers, anti-irritants, colorants, surfactants, beneficial agents, pharmaceutical agents, and other components for use in connection with the compositions described herein (such as oral compositions for treatment of the gastrointestinal tract
- the composition is an anhydrous formulation to prevent skin irritation such as water-based irritant contact dermatitis or stinging sensation upon application to damaged skin.
- the composition is formulated such that preservatives need not necessarily be employed (e.g., a preservative-free formulation) so as to avoid skin irritation associated with certain preservatives
- the composition may be provided as an ointment, an oil, a lotion, a paste, a powder, a gel, or a cream.
- the composition may also include additional ingredients such as a protective agent, an emollient, an astringent, a humectant, a sun screening agent, a sun tanning agent, a UV absorbing agent, an antibiotic agent, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anti-acne agent, an anesthetic agent, a steroidal anti-inflammatory agent, a nonsteroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an anti-histamine agent, a vitamin or vitamin complex, a hormone, an anti-dandruff agent, an anti-wrinkle agent, an anti-skin atrophy agent, a skin whitening agent, a cleansing agent, additional peptides, additional modified peptides, and combinations thereof.
- compositions may be administered by injection or infusion, in particular by intravenous, intramuscular, transmucosal, subcutaneous or intraperitoneal injection or infusion and/or by oral, topical, dermal, nasal, inhalation, aerosol and/or rectal application, etc.
- the compositions are administered reversibly immobilized on the surface of a medical device, in particular by binding, coating and/or embedding the compositions on a medical device, such as but not limited to, stents, catheters, surgical instruments, cannulae, cardiac valves, or vascular prostheses.
- a medical device such as but not limited to, stents, catheters, surgical instruments, cannulae, cardiac valves, or vascular prostheses.
- the coated medical devices act as drug delivery devices eluting the medicament, whereby the drug delivery kinetics can be controlled, providing an immediate release or a controlled, delayed or sustained drug delivery, for example.
- the composition further comprises an enteric coating that resists degradation under the prevailing pH of the stomach and permits delivery to specific regions of the gastrointestinal tract.
- compositions may also be used for diagnostic or for therapeutic applications.
- the compound may be present in a labelled form, e.g. in a form containing an isotope, e.g. a radioactive isotope or an isotope which may be detected by nuclear magnetic resonance.
- a therapeutic application is, in the case of a topical application, the prevention, alleviation and treatment of psoriasis and dermatitis.
- the concentrations of the compounds in the pharmaceutical composition can vary. The concentration will depend upon factors such as the total dosage of the drug to be administered, the chemical characteristics (e.g., hydrophobicity) of the compounds employed, the route of administration, the age, body weight and symptoms of a patient.
- the compounds typically are provided in an aqueous physiological buffer solution containing about 0.1 to 10% w/v compound for topical administration. Typical dose ranges are from about 1 ⁇ g to about 1 g/kg of body weight per day; a dose range may be from about 0.01 mg/kg to 100 mg/kg of body weight per day, or about 0.1 to 20 mg/kg once to four times per day.
- the dosage of the drug to be administered is likely to depend on variables such as the type and extent of the progression of the disease or disorder, the overall health status of the particular patient, the relative biological efficacy of the selected compound and the formulation of the compound excipient, and its route of administration.
- the indefinite article “a” or “an” does not exclude a plurality.
- the term “about” as used herein to, for example, define the values and ranges of molecular weights means that the indicated values and/or range limits can vary within ⁇ 20%, e.g., within ⁇ 10%.
- the use of "about” before a number includes the number itself. For example, “about 5" provides express support for "5".
- CT352 and SNA-352 are synonyms and can be used interchangeably.
- CT101 and SNA-101 are synonyms and can be used interchangeably.
- CT103 and SNA-103 are synonyms and can be used interchangeably.
- CT340 and SNA-125" are synonyms and can be used interchangeably.
- CT327 and SNA-120 are synonyms and can be used interchangeably.
- Example 1 Profiling study of CT101 and CAS944795-066 against 271 kinases
- CT101 and CAS944795-06-6 were tested against 271 target kinases.
- test compound was dissolved in and diluted with dimethylsulfoxide (DMSO) to achieve 100-fold higher concentration. Then the solution was further 25-fold diluted with assay buffer to make the final test compound solution. Reference compounds for assay control were prepared similarly.
- DMSO dimethylsulfoxide
- MSA Off-chip Mobility Shift Assay
- Termination Buffer Quality of Service
- reaction control complete reaction mixture
- background Enzyme(-)
Landscapes
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Ophthalmology & Optometry (AREA)
- Dermatology (AREA)
- Pulmonology (AREA)
- Medicinal Preparation (AREA)
Abstract
Disclosed herein are polymer conjugates comprising an active agent linked to a polymer, wherein the active agent comprises an inhibitor, antagonist, or inverse agonist of a mediator of a therapeutic target associated with a condition, including for example, an ophthalmic condition, a dermatological condition, an inflammatory bowel disease or other gastrointestinal conditions and a respiratory condition. The disclosed polymer conjugates reduce exposure of the active agent at non-target sites.
Description
REDUCED EXPOSURE CONJUGATES MODULATING THERAPEUTIC TARGETS
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application claims priority to US provisional patent application Serial No. 62/473,972 filed March 20, 2017, and claims priority to US provisional patent application Serial No. 62/501 ,567 filed May 4, 2017, and claims priority to US provisional patent application Serial No. 62/590,131 filed November 22, 2017, and claims priority to US provisional patent application Serial No. 62/473,982 filed March 20, 2017, and claims priority to US provisional patent application Serial No. 62/501 ,568 filed May 4, 2017, and claims priority to US provisional patent application Serial No. 62/590,1 19 filed November 22, 2017, and claims priority to US provisional patent application Serial No. 62/473,975 filed March 20, 2017, and claims priority to US provisional patent application Serial No. 62/501 ,651 filed May 4, 2017, and claims priority to US provisional patent application Serial No. 62/590,148 filed November 22, 2017, and claims priority to US provisional patent application Serial No. 62/634,691 filed February 23, 2018, and claims priority to US provisional patent application Serial No. 62/504,1 1 1 filed May 10, 2017, and claims priority to US provisional patent application Serial No. 62/590,1 1 1 filed November 22, 2017. Each of these applications is incorporated herein by reference in its entirety.
FIELD
[0002] Disclosed herein are polymer conjugates, comprising active agents linked to polymers, and therapeutic uses thereof. More particularly, polymer conjugates for treating target sites within the skin, eye, and gastrointestinal system which exhibit reduced exposure to non-target sites and inhibit mediators, such as kinase mediators, of various dermatological, ophthalmic, and/or gastrointestinal conditions are described. Other conjugates for treating the eye, dermatological target sites and target sites within the gastrointestinal system are also described.
BACKGROUND
[0003] Inhibitors of kinase mediators of various ophthalmic, dermatological, and gastrointestinal conditions have been described for possible therapeutic use in the prevention, alleviation and treatment of kinase-associated ophthalmic, dermatological, gastrointestinal pathologies. However, such compounds are associated with broad kinase specificity, as well as undesirable and toxic effects, in particular at non-target
sites. Accordingly, strategies to render these active kinase inhibitors or other drugs more specific, less toxic, and more targeted to sites within the eye, skin, and gastrointestinal system are needed.
SUMMARY OF EMBODIMENTS
[0004] Effective delivery of pharmacologically active agents may be hindered by unwanted exposure of those agents to non-desired locations (such as the systemic circulation and/or lymphatic system). For example, injectable and topical agents useful in treating various skin, eye, and gastrointestinal disorders may result in toxic side effects because of systemic exposure. One issue with delivering compositions comprising one or more active agents topically (or non-topically) is the concern that such agents should be delivered in an amount and at a location sufficient to have a therapeutic effect. At the same time however, exposure (e.g., absorption or longevity of the composition in the systemic circulation, lymphatic system, or other non-targeted sites) may not be desirable for multiple reasons, including, but not limited to, safety reasons. There remains an unmet need for compounds with reduced exposure at non-target sites that result in a clinically therapeutic effect.
[0005] In several embodiments of the invention, the compositions described herein are both therapeutically efficacious and minimize non-target (e.g., systemic or bloodstream) exposure. In some embodiments, the active agents are PEGylated or otherwise coupled to large molecules, and surprisingly, are effective in crossing biological membranes such that the active agents are effectively delivered to the target location. Although inflammatory bowel diseases, inflammatory skin diseases, and ophthalmic conditions are disclosed in several embodiments, other embodiments are used to treat dermal and eye inflammation, as well as other several conditions (e.g., those conditions that would benefit from treatment with reduced exposure at non-target sites). Ophthalmic treatments are provided in some embodiments. In some embodiments, the compositions and technology described herein are used in the gastrointestinal systems. Inflammatory and non-inflammatory conditions are contemplated herein. In yet other embodiments, compositions for treating joints are provided. Treatment of the nose and ear are provided in other embodiments.
[0006] Reduced exposure compounds and compositions are provided in several embodiments. "Reduced exposure" compounds are those compounds that, when delivered to a target location, are formulated to act at the target location with reduced exposure (e.g., entry and/or longevity) in non-target sites. Exposure is reduced as compared to active agents not formulated according to the embodiments described herein. As a non-limiting example, a PEGylated topical dermal, ophthalmic or
gastrointestinal active agent has reduced exposure to the bloodstream as compared to the active agent alone. Reduced exposure compounds include topical compounds that can be delivered to body surfaces and cavities, such as the skin, eyes, ears, nose, mouth, vagina, rectum, etc. , as well as oral (e.g., enteric coated) compounds for oral delivery that treat the gastrointestinal system (e.g., the Gl lining), as well as oral compounds that treat the skin (e.g., the epidermis, dermis, the subcutis) inhalants that treat the lungs, injections for joints, and other modes of delivery that target one location with the goal of reducing exposure to a non-desired site. Non-desired target sites include, for example, the systemic system, the lymphatic system, non-target tissue, etc. "Reduced exposure compositions" comprise or consist essentially of one or more "reduced exposure compounds."
[0007] Reduced exposure topical compositions are provided in many embodiments. In some embodiments, a reduced exposure composition is delivered orally, e.g., for treatment of the gastrointestinal system, eye, or skin. The active agent remains in the eye, the skin, or the lining of the gastrointestinal tract and is able to achieve pharmacological specificity. In some embodiments, a reduced exposure composition is delivered topically, e.g., for treatment of the skin. The active agent remains in the targeted layer of the skin and is able to achieve pharmacological specificity. Because the active agent is conjugated with PEG or another molecule as described herein, the active agent is absorbed more slowly into the non-target site (e.g., the systemic circulation and/or lymphatic system). In some cases, less or none of the active agent is absorbed into the non-target site (e.g., systemic circulation and/or lymphatic system). Further, once the composition enters the systemic circulation and/or lymphatic system, clearance (e.g., by the kidney) occurs at a much faster rate. One or more of the advantages of (i) reduced absorption into the non-target site (e.g., systemic circulation and/or lymphatic system), (ii) slower absorption into the non-target site (e.g., systemic circulation and/or lymphatic system), and (iii) faster clearance rates from the non-target site (e.g., systemic circulation and/or lymphatic system) are also achieved when using the compositions (formulated according to the methods described herein) for treating the lungs (e.g., via inhalants), joints (e.g., via injectables), eye (e.g., via eye drops), nasal passageways, and the ear (such as the ear canal and other structures). Vaginal and rectal tissues are treated in some embodiments via, for example suppositories. There is provided, in some embodiments, reduced exposure compostions suitable for topical application to the eye (e.g., ointment or eye drop). In some embodiments, the reduced exposure compsotions disclosed herein are administered by intraocular and periocular injection such as, for example, direct intravitreal injection, subconjunctival injection, subtenon injection, and peribulbar
injection. Also provided, in several embodiments, are reduced exposure compostions which can be administered via intraocular implantable devices known to one of skill in the art. In some embodiments, reduced exposure at the tear ducts reduces the amount of active agent that is removed (e.g., drained) from the eye, and accordingly allows a higher concernation of active agent to remain at the target site in the eye.
[0008] In several embodiments, there is provided in a reduced exposure composition, a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an inhibitor, antagonist, or inverse agonist of, for example, c-Src. In some embodiments, at least one inhibitor, antagonist, or inverse agonist of c-Src comprises or consists of a composition that includes any one of compounds 1 -71 (and derivatives thereof) disclosed herein in Table 1 coupled to a polymer. In some embodiments, the warhead of the polymer conjugate is compound 1 of Table 1 . In some embodiments, the polymer conjugate is CT101 , wherein CT101 has the following formula:
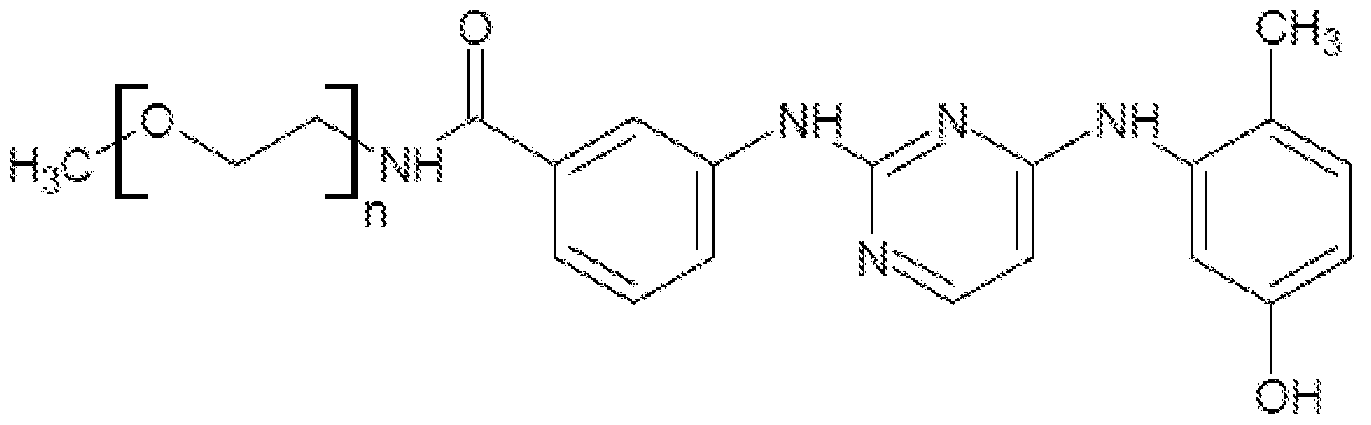
[0009] In several embodiments, there is provided in a reduced exposure composition, a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an inhibitor, antagonist, or inverse agonist of, for example, a mediator of a gastrointestinal, dermatological, and/or ophthalmic condition. In some embodiments, at least one inhibitor, antagonist, or inverse agonist of a mediator of a gastrointestinal, dermatological and/or ophthalmic condition comprises or consists of a composition that includes any one of compounds 1 - 264 (and derivatives thereof) disclosed herein in Table 3 coupled to a polymer. In some embodiments, the warhead of the polymer conjugate is compound 1 in Table 3. In some embodiments, the LSE polymer conjugate is CT352, wherein CT352 has the following formula below:

[0010] In several embodiments, there is provided in a reduced exposure composition, a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an inhibitor, antagonist, or inverse agonist of, for example, a vascular endothelial growth factor receptor (VEGFR). In some embodiments, at least one inhibitor, antagonist, or inverse agonist of a vascular endothelial growth factor receptor (VEGFR) comprises or consists of a composition that includes any one of compounds 1 -59 (and derivatives thereof) disclosed herein in Table 2 coupled to a polymer. In some embodiments, the warhead of the polymer conjugate is compound 1 of Table 2. In some embodiments, the polymer conjugate is CT103, wherein the composition has the formula:

[0011] As described above, several embodiments disclosed herein provide reduced or minimized exposure (e.g., entry into and/or longevity in a non-target site such as the systemic circulation and/or lymphatic system). In some embodiments, exposure at a non-target site is less than 90%, 75%, 50%, 25%, 15%, 10%, 5% or 2% (or less) of the polymer conjugate as compared to a similar active entity that has not been produced
according to the embodiments described herein. In some embodiments, desirable rate of clearance from the non-target site (e.g., systemic circulation and/or lymphatic system) for the compositions described herein is increased by at least 10%, 25%, 50%, or 75% or more as compared to non-conjugated controls. As an example, a PEGylated active agent described herein not only penetrates the desired membranes to reach a desired target, but has reduced non-target exposure by at least 20-80% or more as compared to the non-PEGylated active agent. In some embodiments, blood concentrations measured post administration of the compositions described herein are less than about 0.1 ng/ml, less than 1 ng/ml, or less than 10 ng/ml after, e.g., 15 minutes, 30 minutes, 1 hour, 6 hours or 12 hours.
[0012] In some embodiments, reduced exposure at non-target sites contributes to enhanced efficacy. Efficacy may be enhanced because lower concentrations/amounts/dosing schedules are required to achieve the same or similar therapeutic efficacy at the target site (because, for example, the active ingredient stays at the desired target site for a longer time). In one embodiment, concentrations/amounts/dosing schedules are reduced by 25%-75% or more.
[0013] More rapid clearance rates of the active agent once in the non-target site(s) (such as systemic circulation and/or lymphatic system) are also beneficial because this may allow for a higher concentration or more doses to be delivered. This is especially beneficial for active agents in which a subject would benefit from a higher dose but cannot tolerate the higher dose due to toxicity at the non-target site (e.g., systemic toxicity). Faster clearance rates would permit the desired higher dose to be delivered according to the desired schedule. For example, a subject may be able to tolerate daily doses rather than weekly doses because of the reduced exposure.
[0014] In some embodiments, the active agents of the compositions described herein (e.g., the compounds in Tables 1-3 conjugated e.g., with PEG or other polymers) are measured in non-target sites (e.g., the systemic circulation and/or lymphatic system) at less than amounts found when the active agent is delivered without conjugation (e.g., less than 0.5%, 1 % or 2% after 6 or 12 hours, as compared with 3- 15% (e.g., 3-6%) when the active agent is delivered without conjugation). In some embodiments, the active agents of the compositions described herein (e.g., the compounds in Tables 1-3 conjugated e.g., with PEG or other polymers) are measured in non-target sites (e.g., the systemic circulation and/or lymphatic system) at less than 0.5%, 1 % or 2% after 3-24 hours, as compared to an amount 2-20 times greater when the active agent is delivered without conjugation.
[0015] In some embodiments, clearance of the compositions (e.g., the conjugated polymer compounds) occurs within minutes of exposure to the non-target site
(e.g., systemic circulation and/or lymphatic system), as opposed to hours. In other embodiments, 50% clearance of the conjugated polymer compounds occurs in less than 5 minutes, 15 minutes, 30 minutes, 1 hour, 6 hours, and 12 hours of exposure to the systemic circulation and/or lymphatic system. Clearance times of the conjugated polymer compounds are reduced by more than 25%, 50%, 75% and 90%, as compared to the non-conjugated active agents or other formulations. These reduced clearance times are beneficial to reduce toxicity and undesired side effects.
[0016] In some embodiments, an active agent may be increasingly toxic as it is metabolized in the non-target site (e.g., systemic circulation and/or lymphatic system) because the metabolites exhibit more toxicity than the original agent. Thus, faster clearance rates, in some cases even before the toxic metabolites are created, are especially beneficial.
[0017] The term "active entity" as used herein should not be understood as limiting the participation of the polymer itself and/or the chemical linking moiety between the polymer and the warhead in defining the pharmacology of the polymer conjugate. In some embodiments, the polymer influences the selectivity and/or inhibitory activity of the polymer conjugate. In some embodiments, the chemical linking moiety between the polymer and warhead influences the selectivity and/or inhibitory activity of the polymer conjugate. In some embodiments, the polymer conjugates exhibit no change in selectivity or inhibitory activity against the therapeutic target in comparison with the unconjugated active agent. In some embodiments, the polymer conjugates exhibit a significant increase in selectivity against the therapeutic target in comparison with the unconjugated active agent. In some embodiments, the polymer conjugates exhibit a significant increase in inhibitory activity against the therapeutic target in comparison with the unconjugated active agent. In some embodiments, the polymer conjugates exhibit a significant increase in selectivity and inhibitory activity against the therapeutic target in comparison with the unconjugated active agent. In some embodiments, the increased selectivity and/or inhibitory activity of the polymer conjugate against the therapeutic target in comparison with the unconjugated active agent causes decrease in undesired biological effects. In some embodiments, the increased selectivity of the polymer conjugate is caused by an increase of the hydrodynamic volume resulting from the conjugated polymer chain. In some embodiments, the polymer chain creates a higher steric hindrance which allows discrimination among the diverse shapes and sizes of the binding sites of different proteins, thus improving selectivity with respect to the active agent alone.
[0018] In several embodiments, age-related macular degeneration is treated. In several embodiments, diabetic retinopathy is treated. In several embodiments, corneal
edema is treated. In several embodiments, macular edema is treated. In several embodiments, dry eye is treated.
[0019] In several embodiments, various inflammatory skin diseases are treated. The inflammatory skin disease comprises, in some embodiments, psoriasis, psoriasis guttata, inverse psoriasis, pustular psoriasis, psoriatic erythroderma, acute febrile neutrophilic dermatosis, eczema, xerotic eczema, dyshidrotic eczema, vesicular palmar eczema, acne vulgaris, atopic dermatitis, contact dermatitis, allergic contact dermatitis, dermatomyositis, exfoliative dermatitis, hand eczema, pompholyx, keloids, rosacea, rosacea due to sarcoidosis, rosacea due to scleroderma, rosacea due to Sweet syndrome, rosacea due to systemic lupus erythematosus, rosacea due to urticaria, rosacea due to herpetic pain, Sweet's disease, neutrophilic hydradenitis, sterile pustule, drug rash, seborrheic dermatitis, pityriasis rosea, Kikuchi's disease of the skin, pruritic urticarial papules and plaques of pregnancy, Stevens-Johnson syndrome and toxic epidermal necrolysis, tattoo reaction, Wells syndrome (eosinophilic cellulitis), reactive arthritis (Reiter syndrome), bowel-associated dermatosis-arthritis syndrome, rheumatoid neutrophilic dermatosis, neutrophilic eccrine hidradenitis, neutrophilic skin disease of dorsum of hand, balanitis circumscripta plasmacellularis, balanoposthitis, Behcet's disease, erythema annulare centrifugum, erythema dyschromicum perstans, erythema multiforme, granuloma annulare, dermatitis of hand, lichen nitidus, lichen planus, lichen sclerosus et atrophicus, lichen simplex chronicus, lichen spinulosus, nummular dermatitis, pyoderma gangrenosum, sarcoidosis, subcorneal pustular dermatosis, urticaria, and transient acantholytic dermatosis.
[0020] In several embodiments, various skin neoplasias are treated. The skin neoplasia comprises, in some embodiments, squamous cell carcinoma, basal cell carcinoma, malignant melanoma, malignant cutaneous lymphoma, Kaposi's sarcoma, Merkel cell skin cancer, and non-melanoma skin cancer.
[0021] In several embodiments, various vascular tumors are treated. The vascular tumor comprises, in some embodiments, hemangiomas, Kaposi's sarcoma, lymphangioma, glomangioma, angiosarcoma, hemangioendothelioma, and infantile hemangiomas.
[0022] In several embodiments, various bullous diseases are treated. The bullous disease comprises, in some embodiments, bullous pemphigoid, erythema multiforme, dermatitis herpetiformis, epidermolysis bullosa acquisita, linear Immunoglobulin A disease, mucous membrane pemphigoid, pemphigoid gestationis, pemphigus foliaceus, and pemphigus vulgaris.
[0023] In several embodiments, the polymer conjugates are administered in combination with UV irradiation therapy.
[0024] In alternative embodiments, the polymer conjugates configured for reduced exposure are administered to other areas of the body besides the skin. For example, in one embodiment, administration comprises treatment of the lung and respiratory conditions via inhalation of the polymer conjugates. Eye drops are provided in some embodiments to treat eye inflammation or ophthalmic disorders and diseases. Treatment to the joints to treat inflammation or other joint conditions is also provided. In yet another embodiment, administration comprises treatment of the gastro-intestinal tract via, for example, an enteric coated capsule comprising the polymer conjugates taken orally. Reduced exposure provides benefits in these applications. Applications for the nose and ear, such as inhalants, ointments and drops are provided in several embodiments. Treatment to the nasal passage to treat allergies or allergic rhinitis is also provided. Vaginal and rectal compounds are provided in some embodiments, including as suppositories, creams, ointments, etc.
[0025] In several embodiments, various inflammatory bowel diseases are treated. The inflammatory bowel disease comprises, in some embodiments, Crohn's disease, ulcerative colitis, collagenous colitis, lymphocytic colitis, ischemic colitis, diversion colitis, Behcet's syndrome, and indeterminate colitis.
[0026] Also provided herein, in several embodiments, are polymer conjugates wherein the polymer is polyethylene glycol (PEG) or methoxy-polyethylene glycol (m- PEG). In several embodiments, there is provided a pharmaceutical composition comprising or consisting essentially of a polymer conjugate disclosed herein that is formulated for topical and non-topical administration. In several embodiments, methods of making and using the compositions described herein are provided.
[0027] In several embodiments, the invention comprises a reduced exposure composition comprising at least one active entity linked to at least one polymer, wherein the composition has reduced exposure at a non-target site as compared to the active entity delivered without the polymer. The non-target site comprises the systemic system, the lymphatic system and/or another non-target tissue site in some embodiments.
[0028] In some embodiments, the active entity comprises an inhibitor, an antagonist, or an inverse agonist. For example, the active entity may be an inhibitor, antagonist, or inverse agonist of a mediator of inflammation. In some embodiments, the active entity may be an inhibitor, antagonist, or inverse agonist of a mediator of an inflammatory bowel disease an inflammatory skin disease, and/or inflammatory ophthalmic condition. In some embodiments, the active entity may be an inhibitor, antagonist, or inverse agonist of a gastrointestinal, dermatological, and/or ophthalmic condition In some embodiments, the active entity may be an inhibitor, antagonist, or inverse agonist of JAK and/or STAT family proteins. The active entity comprises or
consists essentially of any one or more of compounds 1-264 of Table 3 in some embodiments. The active entity comprises compound 1 of Table 3 in some embodiments. The reduced exposure composition comprises CT352 in some embodiments.
[0029] In some embodiments, the active entity comprises an indolocarbazole compound. In some embodiments, the active entity comprises a derivative of K252a. In some embodiments, the composition comprises SNA-125. In some embodiments, the composition comprises SNA-120.
[0030] The active entity binds to a tropomyosin-receptor-kinase A (TrkA) in some embodiments. The active entity binds to a Janus Kinase (JAK) family member in some embodiments. The active entity binds to one or more of Janus Kinase 1 (JAK1), Janus Kinase 2 (JAK2), Janus Kinase 3 (JAK3), and/or Tyrosine kinase 2 (TYK2) in some embodiments. The active entity binds to mitogen-activated protein kinase kinase (MAP2K) in some embodiments. The active entity binds to mitogen-activated protein kinase kinase 3 (MAP2K3) in some embodiments. The binding may be partially or fully inhibitory or not.
[0031] In some embodiments, the active entity comprises an inhibitor, an antagonist, or an inverse agonist. For example, the active entity may be an inhibitor, antagonist, or inverse agonist of c-Src. In some embodiments, inflammatory conditions are treated. In other embodiments, non-inflammatory conditions are treated. The active entity comprises or consists essentially of any one or more of compounds 1-71 of Table 1 in some embodiments. In some embodiments, the active entity comprises compound 1 of Table 1. In some embodiments, the composition comprises CT101.
[0032] The active entity binds to c-Src in some embodiments. The binding may be partially or fully inhibitory or not.
[0033] In some embodiments, the active entity comprises an inhibitor, an antagonist, or an inverse agonist. For example, the active entity may be an inhibitor, antagonist, or inverse agonist of a VEGFR. The active entity comprises or consists essentially of any one or more of compounds 1-59 in some embodiments. In some embodiments, the active entity comprises compound 1 of Table 2. In some embodiments, the composition comprises CT103.
[0034] The active entity binds to a VEGFR in some embodiments. The active entity binds to VEGFR-1 in some embodiments. The active entity binds to VEGFR-2 in some embodiments. The active entity binds to VEGFR-3 in some embodiments. The binding may be partially or fully inhibitory or not.
[0035] In some embodiments, the polymer used in the reduced exposure compounds comprises polyethylene glycol (PEG) and/or methoxy-polyethylene glycol (m-PEG). In embodiments where the active entity has one or more carboxyl, hydroxyl, amino and/or sulfhydryl groups, the active entity is PEGylated (or conjugated/coupled to another polymer) at one or more of said carboxyl, hydroxyl, amino and/or sulfhydryl groups.
[0036] The reduced exposure compositions described herein are formulated for topical administration, such as eye drops, gels, ointments, liquids, etc. in several embodiments. Inhalants, injectables, nasal sprays, oral administration etc. are provided in some embodiments.
[0037] The reduced exposure compositions described herein are formulated for oral administration in several embodiments. Inhalants, injectables, eye drops, nasal sprays, topical administration etc. are provided in some embodiments. In several embodiments, methods of treating one or more of the following are provided: non- dermal inflammation, inflammatory bowel disease, inflammatory skin diseases, wounds, scars, autoimmune disorders, and cancerous or pre-cancerous lesions. Kits comprising one or more compounds and devices for administration (syringes, eye droppers, containers, inhalers, etc.) as well as instructions for use, are provided in certain embodiments.
[0038] Compositions may be administered via at least two routes of administration, either simultaneously or sequentially according to some embodiments. In one embodiment, the composition is administered via a first (e.g. oral, topical dermal) route to a subject, wherein the subject further receives an additional agent via a second (e.g., injectable, non-dermal) route to achieve synergetic effects.
[0039] In several embodiments, methods for reducing exposure of a composition at at least one non-target site are disclosed, wherein the methods comprise applying a composition comprising at least one active entity linked to at least one polymer, wherein the combination of the active entity and polymer reduces exposure at the non-target site by more than 50% as compared to the active entity without the polymer. The composition may be applied topically, injected, inhaled, or administered orally. The non-target site includes non-target tissue at which pharmacological activity is not desired and/or not achieved. Non-target sites can include the bloodstream or systemic system. Non-target sites can also include the lymphatic system.
[0040] There is also provided, in several embodiments, methods of treating a respiratory disease in a subject via delivery of the polymer conjugates (e.g., wherein the warhead is a small molecule targeting a JAK and/or STAT family protein) to the lungs and/or airways. Delivery routes may include for example intratracheal instillation or
inhalation. The formulation may include liquids, nebulized or aerosolized liquids or suspensions, dry powder, nanocomposites, nanoparticles or microparticles, etc. Respiratory disorders, include treatable obstructive, restrictive or inflammatory airways diseases of whatever type, etiology, or pathogenesis. Non-limiting examples of respiratory conditions include: acute bronchitis; acute laryngotracheal bronchitis; arachidic bronchitis; catarrhal bronchitis; croupus bronchitis; dry bronchitis; infectious asthmatic bronchitis; productive bronchitis; staphylococcus or streptococcal bronchitis; vesicular bronchitis; cylindric bronchiectasis; sacculated bronchiectasis; fusiform bronchiectasis; capillary bronchiectasis; cystic bronchiectasis; dry bronchiectasis; follicular bronchiectasis; chronic obstructive pulmonary disease (COPD), chronic obstructive lung disease (COLD), chronic obstructive airways disease (COAD) or small airways obstruction of whatever type, etiology, or pathogenesis, in particular chronic bronchitis, pulmonary emphysema, bronchiectasis, cystic fibrosis, bronchiolitis obliterans, organizing pneumonia (BOOP), chronic organizing pneumonia (COP), bronchiolitis fibrosa obliterans, follicular bronchiolitis or dyspnea associated therewith; cough of whatever type, etiology, or pathogenesis in particular idiopathic cough or cough associated with gastro-esophageal reflux disease (GERD), drugs, bronchial hyper- responsivity, asthma, COPD, COLD, COAD, bronchitis, bronchiectasis, pulmonary eosinophilic syndromes, pneumoconiosis, interstitial lung disease, pulmonary fibrosis, aspiration disorders, rhinitis, laryngitis or pharyngitis; pulmonary eosinophilic syndromes of whatever type, etiology, or pathogenesis, in particular acute eosinophilic pneumonia (idiopathic or due to drugs or parasites), simple pulmonary eosinophilia, Loeffler's syndrome, tropical pulmonary eosinophilia, chronic eosinophilic pneumonia, allergic bronchopulmonary mycosis, allergic bronchopulmonary aspergillosis (ABPA), Churg- Strauss syndrome or idiopathic hypereosinophilic syndrome; asthma of whatever type, etiology, or pathogenesis, in particular asthma that is a member selected from the group consisting of atopic asthma, non-atopic asthma, allergic asthma, atopic bronchial IgE- mediated asthma, bronchial asthma, essential asthma, true asthma, intrinsic asthma caused by pathophysiologic disturbances, extrinsic asthma caused by environmental factors, essential asthma of unknown or inapparent cause, non-atopic asthma, bronchitic asthma, emphysematous asthma, exercise-induced asthma, allergen induced asthma, cold air induced asthma, occupational asthma, infective asthma caused by bacterial, fungal, protozoal, or viral infection, non-allergic asthma, incipient asthma and wheezy infant syndrome; alveolar hemorrhage of whatever type, etiology, or pathogenesis, in particular a member of the group consisting of idiopathic pulmonary hemosiderosis, alveolar hemorrhage due to drugs or other exogenous agents, alveolar hemorrhage associated with HIV or bone marrow transplant or autoimmune alveolar hemorrhage
(e.g. associated with systemic lupus erythematosis, Goodpasture's syndrome, Wegener's granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome, pauci- immune glomerulonephritis); pneumoconiosis of whatever type, etiology, or pathogenesis, in particular pneumoconiosis that is a member selected from the group consisting of aluminosis or bauxite workers' disease, anthracosis or miners' asthma, asbestosis or steam-fitters' asthma, chalicosis or flint disease, ptilosis caused by inhaling the dust from ostrich feathers, siderosis caused by the inhalation of iron particles, silicosis or grinders' disease, byssinosis or cotton-dust asthma and talc pneumoconiosis; interstitial lung diseases (ILD) or pulmonary fibrosis of whatever type, etiology, or pathogenesis, in particular idiopathic pulmonary fibrosis, crytogenic fibrosing alveolitis, fibrosing alveolitis, ILD or pulmonary fibrosis associated with connective tissue disease (systemic lupus erythematosis, mixed connective tissue disease, polymyositis, dermatomyositis, Sjorgen's syndrome, systemic sclerosis, scleroderma, rheumatoid arthritis), usual interstitial pneumonia (UIP), desquamative interstitial pneumonia (DIP), granulomatous lung disease, sarcoidosis, Wegener's granulomatosis, histiocytosis X, Langerhan's cell granulomatosis, hypersensitivity pneumonitis, extrinsic allergic alveolitis, silicosis, chronic eosinophilic pneumonia, lymphangiolyomatosis, drug-induced ILD or pulmonary fibrosis, radiation-induced ILD or pulmonary fibrosis, alveolar proteinosis, graft-versus-host-disease (GVHD), lung transplant rejection, ILD or pulmonary fibrosis due to environmental/occupational exposure, BOOP, COP, bronchiolitis fibrosa obliterans, follicular bronchiolitis, idiopathic acute interstitial pneumonitis (Hamman Rich syndrome) or alveolar hemorrhage syndromes; seasonal allergic rhinitis or perennial allergic rhinitis or sinusitis of whatever type, etiology, or pathogenesis, in particular sinusitis that is a member selected from the group consisting of purulent or nonpurulent sinusitis, acute or chronic sinusitis and ethmoid, frontal, maxillary, or sphenoid sinusitis; Acute Respiratory Distress Syndrome (ARDS), adult respiratory distress syndrome or acute lung injury of whatever type, etiology, or pathogenesis; progressive massive fibrosis (PMF); pulmonary hypertension of whatever type, etiology or pathogenesis including primary pulmonary hypertension, essential hypertension, pulmonary hypertension secondary to congestive heart failure, pulmonary hypertension secondary to COPD, pulmonary venous hypertension, pulmonary arterial hypertension and hypoxia-induced pulmonary hypertension. Respiratory disorders also include, in some embodiments, malignancies and tumors of the respiratory system, non- limiting examples of which include lung adenocarcinoma, squamous cell carcinoma, large cell carcinoma, bronchioloalveolar carcinoma (BAC), pulmonary adenocarcinoma (AIS), non-small-cell carcinoma, small cell carcinoma, and mesothelioma.
[0041] In several embodiments, a reduced exposure composition for inhibiting the activity of a therapeutic target at a target site is provided. Methods for treating diseases, conditions, and disorders are also provided. In one embodiment, the composition comprises or consists essentially of a conjugate comprising or consists essentially of an active entity coupled (e.g., linked) to at least one polymer. Two, three or more active entities or two, three or more polymers may be used. The active entity may have, for example, selectivity and/or an inhibitory activity against the therapeutic target. In some embodiments, the conjugate has increased selectivity and/or inhibitory activity compared to the unconjugated active entity. The polymer can include, for example, polyethylene glycol (PEG) and/or methoxy-polyethylene glycol (m-PEG). In one embodiment, a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site is also provided. In several embodiments, the conjugate has reduced exposure at a non-target site as compared to the unconjugated active entity. The non-target site includes for example the systemic system, the lymphatic system and/or other non-target tissue sites. The conjugate may have, in some embodiments, reduced systemic exposure and toxicity when delivered to the target site as compared to the unconjugated active entity. In some embodiments, the target site includes cells and or tissues localized within one or more of the following: skin, scalp, eye, Gl tract, joint and/or lung. In several embodiments, the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved. In several embodiments, the conjugate has increased permeability across at least one of a nuclear and plasma membrane compared to the unconjugated active entity. In one embodiment, the conjugate can advantageously traverse plasma membranes of cells at the target site, thereby promoting interactions between the active entity and the therapeutic target. This traversal may include the crossing of cellular lipid bilayers to, e.g., distribute the active entity among both lipophilic and hydrophilic cellular compartments. Membranes include the lipid bilayer, plasma membrane and the nuclear membrane as examples. In several embodiments, the conjugate interacts with a kinase associated with the plasma membrane, cytoplasm and/or nucleus. The conjugate may exhibit a depot effect across cellular compartments, thereby reducing the dose of the active entity adequate to inhibit the cellular kinase compared to the unconjugated active entity.
[0042] In several embodiments, the conjugate is selected from:

, and

[0043] and pharmaceutically acceptable salts thereof.
[0044] In some embodiments, the therapeutic target may be a mediator of a condition, such as, for example, a respiratory condition, a gastrointestinal condition, an inflammatory condition. The therapeutic target may be, for example, one or more of VEGFR, c-Src, TkrA, MAP2K3, a JAK family kinase and/or a STAT family protein.
[0045] In several embodiments, the active entity and/or conjugate may have a longer residence time within a cell or other tissue at the target site compared to the active entity without conjugation to the polymer. For example, the residence time of the active entity and/or conjugate within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, (i) at least 25% (e.g.,
25-50%, 50-75%, 75-100%, 100-150%, or higher and overlapping ranges therein) longer and/or (ii) at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50- 100 fold, and overlapping ranges therein) longer. In one embodiment, the residence time is over 100 fold longer. In some embodiments, the conjugate increased residence time at the target site results in lower concentrations and/or lower frequency of administration.
[0046] In some embodiments, a smaller dose of the conjugate may be adequate to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer. For example, in several embodiments, the dose of the conjugate sufficient to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer is at least 10% (e.g., 10-15%, 15-20%, 20-25%, 25- 30%, 30-40%, 40-50%, 50-60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%- 125%, 125- 150%, or higher and overlapping ranges therein) lower. In one embodiment, the dose is over 200% lower. In some embodiments, fewer doses and/or smaller doses of the conjugate are sufficient as compared to the active entity delivered without the polymer.
[0047] In several embodiments, the active entity and/or conjugate may have reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site compared to the active entity without conjugation to the polymer. In several embodiments, the active entity and/or conjugate is present at a biologically inactive concentration within a cell or tissue at a non-target site. In several embodiments, reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site (e.g., the systemic system, the lymphatic system, bone marrow, the circulatory system) advantageously reduces toxicity and/or other side effects, such as, for example, immunosuppression. For example, in some embodiments, the active entity and/or conjugate has reduced systemic absorption and/or little or no systemic toxicity when the composition is administered.
[0048] In some embodiments, the reduced exposure composition is formulated for topical delivery, oral delivery, respiratory delivery or injection into target sites, e.g., eyes and joints. Administration by topical delivery, oral delivery, rectal delivery, inhalation or instillation, and/or injection is provided in several embodiments. Topical delivery to body surfaces, such as, for example skin, eyes, ears, nose, mouth, lungs, vagina and/or rectum, is provided in some embodiments. In the methods of treatment, effective amounts of the active entity are delivered to a subject (e.g., human or veterinary). The composition may further comprise one or more additional ingredients, such as, for example, an antibiotic agent, an anti-angiogenesis agent, a preventive or therapeutic agent for inflammatory bowel disease, an antifungal agent, an
antiviral agent, an antiprotozoal agent, an anesthetic agent, a steroidal anti-inflammatory agent, a non-steroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an anti-histamine agent, a vitamin or vitamin complex, and/or a hormone.
[0049] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following gastrointestinal conditions: inflammatory bowel disease, Crohn's disease, ulcerative colitis, collagenous colitis, lymphocytic colitis, ischemic colitis, diversion colitis, Behcet's syndrome, indeterminate colitis, irritable bowel syndrome and/or small intestinal bacterial overgrowth.
[0050] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following ophthalmic conditions: macular degeneration, age related macular degeneration (ARMD), choroidal neovascularization, retinopathy, diabetic retinopathy, acute macular neuroretinopathy, chronic macular neuroretinopathy, central serous chorioretinopathy, macular edema, cystoid macular edema, diabetic macular edema, acute multifocal placoid pigment epitheliopathy, Behcet's disease, birdshot retinochoroidopathy, uveitis, intermediate uveitis (pars planitis), anterior uveitis, multifocal choroiditis, multiple evanescent white dot syndrome (MEWDS), ocular sarcoidosis, posterior scleritis, serpignous choroiditis, subretinal fibrosis, uveitis syndrome, Vogt-Koyanagi-Harada syndrome, retinal arterial occlusive disease, central retinal vein occlusion, disseminated intravascular coagulopathy, branch retinal vein occlusion, hypertensive fundus changes, ocular ischemic syndrome, retinal arterial microaneurysms, Coat's disease, parafoveal telangiectasis, hemi-retinal vein occlusion, papillophlebitis, central retinal artery occlusion, branch retinal artery occlusion, carotid artery disease (CAD), frosted branch angitis, sickle cell retinopathy and other hemoglobinopathies, angioid streaks, familial exudative vitreoretinopathy, Eales disease, sympathetic ophthalmia, uveitic retinal disease, retinal detachment, trauma, laser, PDT, photocoagulation, hypoperfusion during surgery, radiation retinopathy, bone marrow transplant retinopathy, proliferative vitreal retinopathy and epiretinal membranes, proliferative diabetic retinopathy, ocular histoplasmosis, ocular toxocariasis, presumed ocular histoplasmosis syndrome (POHS), endophthalmitis, toxoplasmosis, retinal diseases associated with HIV infection, choroidal disease associated with HIV infection, uveitic disease associated with HIV Infection, viral retinitis, acute retinal necrosis, progressive outer retinal necrosis, fungal retinal diseases, ocular syphilis, ocular tuberculosis, diffuse unilateral subacute neuroretinitis, myiasis, retinitis pigmentosa, systemic disorders with associated retinal dystrophies, congenital stationary night
blindness, cone dystrophies, Stargardt's disease, fundus flavimaculatus, Bests disease, pattern dystrophy of the retinal pigmented epithelium, X-linked retinoschisis, Sorsby's fundus dystrophy, benign concentric maculopathy, Bietti's crystalline dystrophy, pseudoxanthoma elasticum, retinal detachment, macular hole, giant retinal tear, retinal disease associated with tumors, congenital hypertrophy of the RPE, posterior uveal melanoma, choroidal hemangioma, choroidal osteoma, choroidal metastasis, combined hamartoma of the retina and retinal pigmented epithelium, retinoblastoma, vasoproliferative tumors of the ocular fundus, retinal astrocytoma, intraocular lymphoid tumors, punctate inner choroidopathy, acute posterior multifocal placoid pigment epitheliopathy, myopic retinal degeneration, and/or acute retinal pigment epithelitis.
[0051] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following dermal conditions: psoriasis, psoriasis guttata, inverse psoriasis, pustular psoriasis, psoriatic erythroderma, acute febrile neutrophilic dermatosis, eczema, xerotic eczema, dyshidrotic eczema, vesicular palmar eczema, acne vulgaris, atopic dermatitis, contact dermatitis, allergic contact dermatitis, dermatomyositis, exfoliative dermatitis, hand eczema, pompholyx, keloids, rosacea, rosacea due to sarcoidosis, rosacea due to scleroderma, rosacea due to Sweet syndrome, rosacea due to systemic lupus erythematosus, rosacea due to urticaria, rosacea due to herpetic pain, Sweet's disease, neutrophilic hydradenitis, sterile pustule, drug rash, seborrheic dermatitis, pityriasis rosea, Kikuchi's disease of the skin, pruritic urticarial papules and plaques of pregnancy, Stevens-Johnson syndrome and toxic epidermal necrolysis, tattoo reaction, Wells syndrome (eosinophilic cellulitis), reactive arthritis (Reiter syndrome), bowel- associated dermatosis-arthritis syndrome, rheumatoid neutrophilic dermatosis, neutrophilic eccrine hidradenitis, neutrophilic skin disease of dorsum of hand, balanitis circumscripta plasmacellularis, balanoposthitis, Behcet's disease, erythema annulare centrifugum, erythema dyschromicum perstans, erythema multiforme, granuloma annulare, dermatitis of hand, lichen nitidus, lichen planus, lichen sclerosus et atrophicus, lichen simplex chronicus, lichen spinulosus, nummular dermatitis, pyoderma gangrenosum, sarcoidosis, subcorneal pustular dermatosis, urticaria, and/or transient acantholytic dermatosis.
[0052] In several embodiments, a reduced exposure composition for treating a target site with the gastrointestinal system is provided. Methods for treating the Gl and/or gastrointestinal conditions are also provided. In one embodiment, the composition comprises or consists essentially of a conjugate comprising or consists essentially of an active entity coupled (e.g., linked) to at least one polymer. Two, three or more active entities or two, three or more polymers may be used. The active entity may be for
example, an inhibitor, antagonist, or inverse agonist of a mediator of a gastrointestinal condition. The polymer can include, for example, polyethylene glycol (PEG) and/or methoxy-polyethylene glycol (m-PEG). In one embodiment, a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site is also provided. In several embodiments, the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer. The non-target site includes for example the systemic system, the lymphatic system and/or other non-target tissue sites. In several embodiments, the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved. In one embodiment, the conjugate can advantageously traverse plasma membranes of cells at the target site, thereby promoting interactions between the active entity and the mediator of the gastrointestinal condition. This traversal may include the crossing of cellular lipid bilayers to, e.g., distribute the active entity among both lipophilic and hydrophilic cellular compartments. Membranes include the lipid bilayer, plasma membrane and the nuclear membrane as examples. In several embodiments, the conjugate interacts with mediator associated with the plasma membrane, cytoplasm and/or nucleus. The conjugate may exhibit a depot effect across cellular compartments, thereby reducing the dose of the active entity required to inhibit the cellular mediator compared to the active entity without conjugation to the polymer. In some embodiments, the conjugate penetrates one or more physical barriers of the Gl, one or more physiological barriers of the Gl, and/or one or more biological barriers of the Gl (e.g. the epithelial barrier and/or intestinal mucosa). In several embodiments, the conjugate can under transcytosis across the intestinal epithelium.
[0053] In several embodiments, the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a protein mediator of a condition (e.g., a kinase). In some embodiments, the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a mediator of a gastrointestinal condition. For example, in some embodiments, the mediator is one or more of c-Src, a VEGFR protein (e.g., VEGFR-1 , VEGFR-2, VEGFR-3), a JAK protein (e.g., of JAK1 , JAK2, JAK3, and Tyrosine kinase 2 (TYK2)), STAT, NF-Kappa B, TrkA, MAPK, MAP2K and/or MAP2K3. In some embodiments, the active entity and/or conjugate is an inhibitor, antagonist, and/or inverse agonist of a mediator of inflammation. The mediator of inflammation may be, for example, a mediator of an inflammatory bowel disease. In some embodiments, the active entity and/or conjugate reduces inflammation at the target site within the Gl. In several embodiments, the reduced inflammation at the target site treats an inflammatory bowel disease (e.g. Crohn's disease, ulcerative colitis). In some embodiments, the reduced exposure composition comprises one or more of conjugates SNA-101 , SNA-
103, SNA-352, SNA-120 and/or SNA-125. Advantageously, in several embodiments, the active entity and/or conjugate exhibits greater access to the mediator compared to the active entity without conjugation to the polymer.
[0054] In some embodiments, the reduced exposure composition may be formulated for topical, oral, or suppository delivery. Topical, oral, and suppository administration is provided in several embodiments. In several embodiments, the administration is daily. In the methods of treatment, effective amounts of the active entity are delivered to a subject (e.g., human or veterinary). The composition may further comprise one or more additional ingredients, such as, for example, antibiotic agent, an anti-angiogenesis agent, a preventive or therapeutic agent for inflammatory bowel disease, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anesthetic agent, a steroidal anti-inflammatory agent, a non-steroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an antihistamine agent, a vitamin or vitamin complex, and/or a hormone.
[0055] In several embodiments, the active entity and/or conjugate may have an increased concentration, activity and/or bioavailability within a cell or tissue at the target site compared to the active entity without conjugation to the polymer. In some such embodiments, the therapeutically effective amount of the active entity is at the target site. For example, the concentration, activity and/or bioavailability within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, at least 2-20 fold (e.g., 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 14- 16 fold, 18-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-100 fold, and overlapping ranges therein) greater than within a cell or tissue at a non-target site (e.g., the systemic system, the lymphatic system, bone marrow, outside the gastrointestinal tract). In one embodiment, the concentration, activity and/or bioavailability within a cell or tissue at the target site is over 100 fold greater.
[0056] In several embodiments, the active entity and/or conjugate may have reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site compared to the active entity without conjugation to the polymer. In several embodiments, the active entity and/or conjugate is present at a biologically inactive concentration within a cell or tissue at a non-target site. In several embodiments, reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site (e.g., the systemic system, the lymphatic system, bone marrow, outside the gastrointestinal tract) advantageously reduces toxicity and/or other side effects, such as, for example, immunosuppression. For example, in some embodiments, the active entity and/or conjugate has reduced systemic absorption and/or little or no systemic toxicity
when the composition is formulated for oral delivery and is administered orally (e.g., a single administration, administration on a daily basis).
[0057] In several embodiments, the active entity and/or conjugate may have a greater activity and/or bioavailability within a cell or other tissue at the target site (e.g., gastrointestinal tract) than within a cell or other tissue at a non-target site (e.g., outside the gastrointestinal tract). For example, the activity and/or bioavailability of the active entity and/or conjugate within a cell or other tissue at the target site is, as compared to within a cell or other tissue at a non-target site, (i) at least 25% (e.g., 25-50%, 50-75%, 75-100%, 100-150%, or higher and overlapping ranges therein) greater and/or (ii) at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) greater. In one embodiment, the activity and/or bioavailability is over 100 fold greater within a cell or other tissue at the target site than within a cell or other tissue at a non-target site.
[0058] In several embodiments, the active entity and/or conjugate may have a longer residence time within a cell or other tissue at the target site (e.g., intestinal lamina propria) compared to the active entity without conjugation to the polymer. For example, the residence time of the active entity and/or conjugate within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, (i) at least 25% (e.g., 25-50%, 50-75%, 75-100%, 100-150%, or higher and overlapping ranges therein) longer and/or (ii) at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20- 30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) longer. In one embodiment, the residence time is over 100 fold longer. In several embodiments, the increased residence time is in one or more of the gastrointestinal tract, intestinal epithelial cells, and /or intestinal lamina propria.
[0059] In several embodiments, the active entity and/or conjugate may have a shorter residence time within a cell or other tissue at a non-target site compared to the active entity without conjugation to the polymer. For example, the residence time of the active entity and/or conjugate within a cell or other tissue at a non-target site is, as compared to the active entity without conjugation to the polymer, at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) shorter. In one embodiment, the residence time is over 100 fold shorter.
[0060] In some embodiments, a smaller dose of the conjugate may be adequate to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer. For example, in several embodiments, the dose of the
conjugate sufficient to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer is at least 10% (e.g., 10-15%, 15-20%, 20-25%, 25- 30%, 30-40%, 40-50%, 50-60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%- 125%, 125- 150%, or higher and overlapping ranges therein) lower. In one embodiment, the dose is over 200% lower. In some embodiments, fewer doses and/or smaller doses of the conjugate are required as compared to the active entity delivered without the polymer.
[0061] In several embodiments, the active entity and/or conjugate may have diminished systemic absorption compared to the active entity without conjugation to the polymer. For example, in several embodiments, the systemic absorption of the active entity and/or conjugate, as compared to the active entity without conjugation to the polymer, is at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50- 100 fold, and overlapping ranges therein) lower. In one embodiment, the systemic absorption is over 100 fold lower. For example, in several embodiments, the active entity and/or conjugate may have minimal systemic absorption following oral administration.
[0062] In several embodiments, the active entity and/or conjugate may have reduced clearance time from a non-target site compared to the active entity without conjugation to the polymer. For example, in several embodiments, the clearance time of the active entity and/or conjugate from a non-target site, as compared to the active entity without conjugation to the polymer, is at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) lower. In one embodiment, the clearance time is over 100 fold lower. For example, in several embodiments, the active entity and/or conjugate displays rapid systemic elimination when administered by intravenous, intramuscular, transmucosal, subcutaneous or intraperitoneal injection and/or infusion.
[0063] In several embodiments, the active entity and/or conjugate may have enhanced delivery to a cell or other tissue at the target site (e.g., Gl tract, intestinal lamina propria) compared to the active entity without conjugation to the polymer. For example, in several embodiments, at least 10% (e.g., 10-15%, 15-20%, 20-25%, 25- 30%, 30-40%, 40-50%, 50-60%, 60-70%, 70-80%, 80-90%, 90-100%, and overlapping ranges therein) of the administered conjugate reaches a cell or other tissue at the target site. In several embodiments, the conjugate may have enhanced delivery to the intestinal epithelium and/or intestinal lamina propria as compared to the active entity delivered without the polymer.
[0064] In several embodiments, the conjugate is amphiphilic and/or amphipathic. In some embodiments, the conjugate is more amphiphilic and/or
amphipathic than the active entity without conjugation to the polymer. For example, in several embodiments, the conjugate, as compared to the active entity without conjugation to the polymer, is at least 25% (e.g., 20-25%, 25-30%, 30-40%, 40-50%, 50- 60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%-125%, 125-150%, or higher and overlapping ranges therein) more amphiphilic. In one embodiment, the amphiphilicity is over 200% greater. Additionally, in some embodiments, the conjugate is more hydrophilic than the active entity without conjugation to the polymer. For example, in several embodiments, the conjugate, as compared to the active entity without conjugation to the polymer, is at least 25% (e.g., 20-25%, 25-30%, 30-40%, 40-50%, 50- 60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%-125%, 125-150%, or higher and overlapping ranges therein) more hydrophilic. In one embodiment, the hydrophilicity is over 200% greater. In some embodiments, the greater hydrophilicity of the conjugate advantageously facilitates one or more of: non-compartmentalization within a cell or tissue at the target site; access to and activity in both the lipid bilayer and the cytosol of the cell; access to and/or activity in both the lipid bilayer and the cytoplasm of the cell; and/or access to and/or activity across the lipid bilayer.
[0065] In some embodiments, the target site includes one or more of the following: intestinal epithelium, intestinal lamina propria, the lining of the gastrointestinal tract, immune cells residing within the intestinal lamina propria, muscularis mucosae, myenteric plexus, the submucosa, the muscular layer, intraperitoneal spaces, retroperitoneal spaces, serosa, adventitia. In some embodiments, the target site includes immune cells, and/or non-immune cells. In some embodiments, the conjugate targets immune cells residing within the Gl epithelial layer and/or intestinal lamina propria. In some embodiments, the target site includes the gastrointestinal tract and the non-target site includes non-gastrointestinal tract tissue. In several embodiments, the target site includes the small intestine and the non-target site includes one or more of the large intestine and stomach. In one embodiment, the target site includes the large intestine and the non-target site includes one or more of the small intestine and stomach. In some embodiments, the target site includes the small intestine and large intestine and the non- target site includes the stomach. In one embodiment, the target site includes the intestinal lamina propria and the non-target site includes tissue contacting the intestinal lamina propria. In several embodiments, the target site includes immune cells of the intestinal lamina propria and the non-target site includes non-immune cells. In one embodiment, the target site includes the intestinal lamina propria and/or gastric parietal cells and the non-target site comprises sites other than intestinal lamina propria and/or gastric parietal cells. In several embodiments, the target site includes one or two of the duodenum, jejunum and ileum and the non- target site includes the remaining one or
two. In some embodiments, the target site includes one or two of the ascending colon, transverse colon and the descending colon and the non- target site comprises the remaining one or two.
[0066] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following conditions: inflammatory bowel disease, irritable bowel syndrome, small intestinal bacterial overgrowth, Crohn's disease, ulcerative colitis, collagenous colitis, lymphocytic colitis, ischemic colitis, diversion colitis, Behcet's syndrome, small intestinal bacterial overgrowth, and indeterminate colitis.
[0067] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for reducing immunosuppression and/or an inflammatory response while treating a gastrointestinal condition in a subject in need thereof.
[0068] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for reducing reduce liver damage, neutropenia and/or lymphopenia while treating a gastrointestinal condition in a subject in need thereof.
[0069] In several embodiments, a reduced exposure composition for treating a dermal target site (e.g., the skin) is provided. Methods for treating the skin and/or dermal conditions are also provided. In one embodiment, the composition comprises or consists essentially of a conjugate comprising or consists essentially of an active entity coupled (e.g., linked) to at least one polymer. Two, three or more active entities or two, three or more polymers may be used. The active entity may be for example, an inhibitor, antagonist, or inverse agonist of a mediator of a dermal condition. The polymer can include, for example, polyethylene glycol (PEG) and/or methoxy-polyethylene glycol (m- PEG). In one embodiment, a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site is also provided. In several embodiments, the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer. The non-target site includes for example the systemic system, the lymphatic system and/or other non-target tissue sites. In several embodiments, the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved. In one embodiment, the conjugate can advantageously traverse plasma membranes of cells at the target site, thereby promoting interactions between the active entity and the mediator of the dermal condition. This traversal may include the crossing of cellular lipid bilayers to, e.g., distribute the active entity among both lipophilic and hydrophilic cellular compartments. Membranes include the lipid bilayer, plasma membrane and the nuclear membrane as examples. In several
embodiments, the conjugate interacts with mediator associated with the plasma membrane, cytoplasm and/or nucleus. The conjugate may exhibit a depot effect across cellular compartments, thereby reducing the dose of the active entity required to inhibit the cellular mediator compared to the active entity without conjugation to the polymer. In some embodiments, the conjugate penetrates one or more physical barriers of the skin, one or more physiological barriers of the skin, and/or one or more biological barriers of the skin.
[0070] In several embodiments, the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a protein mediator of a condition (e.g., a kinase). In some embodiments, the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a mediator of a dermal condition. For example, in some embodiments, the mediator is one or more of c-Src, a VEGFR protein (e.g., VEGFR-1 , VEGFR-2, VEGFR- 3), a JAK protein (e.g., of JAK1 , JAK2, JAK3, and Tyrosine kinase 2 (TYK2)), STAT, NF-Kappa B, TrkA, MAPK, MAP2K and/or MAP2K3. In some embodiments, the active entity and/or conjugate is an inhibitor, antagonist, and/or inverse agonist of a mediator of inflammation. The mediator of inflammation may be, for example, a mediator of an inflammatory skin condition. In some embodiments, the active entity and/or conjugate reduces inflammation at the target site. In several embodiments, the reduced inflammation at the target site treats an inflammatory skin condition. In some embodiments, the reduced exposure composition comprises one or more of conjugates SNA-101 , SNA-103, SNA-352, SNA-120 and/or SNA-125. Advantageously, in several embodiments, the active entity and/or conjugate exhibits greater access to the mediator compared to the active entity without conjugation to the polymer.
[0071] In some embodiments, the reduced exposure composition is formulated for topical delivery. Topical administration is provided in several embodiments. In several embodiments, the administration is daily. In the methods of treatment, effective amounts of the active entity are delivered to a subject (e.g., human or veterinary). The composition may further comprise one or more additional ingredients, such as, for example, a protective agent, an emollient, an astringent, a humectant, a sun screening agent, a sun tanning agent, a UV absorbing agent, an antibiotic agent, an anti-angiogenesis agent, a physiological cooling agent, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anti-acne agent, an anesthetic agent, a steroidal anti-inflammatory agent, a non-steroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an antihistamine agent, a vitamin or vitamin complex, a hormone, an anti-dandruff agent, an
anti-wrinkle agent, an anti-skin atrophy agent, a skin whitening agent, and/or a cleansing agent.
[0072] In several embodiments, the active entity and/or conjugate may have an increased concentration, activity and/or bioavailability within a cell or tissue at the target site compared to the active entity without conjugation to the polymer. In some such embodiments, the therapeutically effective amount of the active entity is at the target site. For example, the concentration, activity and/or bioavailability within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, at least 2-20 fold (e.g. , 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 14- 16 fold, 18-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50- 100 fold, and overlapping ranges therein) greater than within a cell or tissue at a non-target site (e.g. , the systemic system, the lymphatic system, bone marrow, outside the skin). In one embodiment, the concentration, activity and/or bioavailability within a cell or tissue at the target site is over 100 fold greater.
[0073] In several embodiments, the active entity and/or conjugate may have reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site compared to the active entity without conjugation to the polymer. In several embodiments, the active entity and/or conjugate is present at a biologically inactive concentration within a cell or tissue at a non-target site. In several embodiments, reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site (e.g. , the systemic system, the lymphatic system, bone marrow, outside the skin) advantageously reduces toxicity and/or other side effects, such as, for example, immunosuppression. For example, in some embodiments, the active entity and/or conjugate has reduced systemic absorption and/or little or no systemic toxicity when the composition is administered topically (e.g. , a single administration, administration on a daily basis) .
[0074] In several embodiments, the active entity and/or conjugate may have a greater activity and/or bioavailability within a cell or other tissue at the target site (e.g. , skin) than within a cell or other tissue at a non-target site. For example, the activity and/or bioavailability of the active entity and/or conjugate within a cell or other tissue at the target site is, as compared to within a cell or other tissue at a non-target site, (i) at least 25% (e.g. , 25-50%, 50-75%, 75-100%, 100- 150%, or higher and overlapping ranges therein) greater and/or (ii) at least 2-20 fold (e.g. , 2- 10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10- 12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) greater. In one embodiment, the activity and/or bioavailability is over 100 fold greater within a cell or other tissue at the target site than within a cell or other tissue at a non-target site.
[0075] In several embodiments, the active entity and/or conjugate may have a longer residence time within a cell or other tissue at the target site (e.g., skin) compared to the active entity without conjugation to the polymer. For example, the residence time of the active entity and/or conjugate within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, (i) at least 25% (e.g., 25-50%, 50-75%, 75-100%, 100-150%, or higher and overlapping ranges therein) longer and/or (ii) at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) longer. In one embodiment, the residence time is over 100 fold longer. In several embodiments, the increased residence time is in the skin.
[0076] In several embodiments, the active entity and/or conjugate may have a shorter residence time within a cell or other tissue at a non-target site compared to the active entity without conjugation to the polymer. For example, the residence time of the active entity and/or conjugate within a cell or other tissue at a non-target site is, as compared to the active entity without conjugation to the polymer, at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) shorter. In one embodiment, the residence time is over 100 fold shorter.
[0077] In some embodiments, a smaller dose of the conjugate may be adequate to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer. For example, in several embodiments, the dose of the conjugate sufficient to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer is at least 10% (e.g. , 10-15%, 15-20%, 20-25%, 25- 30%, 30-40%, 40-50%, 50-60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%- 125%, 125- 150%, or higher and overlapping ranges therein) lower. In one embodiment, the dose is over 200% lower. In some embodiments, fewer doses and/or smaller doses of the conjugate are required as compared to the active entity delivered without the polymer.
[0078] In several embodiments, the active entity and/or conjugate may have diminished systemic absorption compared to the active entity without conjugation to the polymer. For example, in several embodiments, the systemic absorption of the active entity and/or conjugate, as compared to the active entity without conjugation to the polymer, is at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50- 100 fold, and overlapping ranges therein) lower. In one embodiment, the systemic absorption is over 100 fold lower. For example, in several embodiments, the active entity
and/or conjugate may have minimal systemic absorption following epicutaneous administration.
[0079] In several embodiments, the active entity and/or conjugate may have reduced clearance time from a non-target site compared to the active entity without conjugation to the polymer. For example, in several embodiments, the clearance time of the active entity and/or conjugate from a non-target site, as compared to the active entity without conjugation to the polymer, is at least 2-20 fold (e.g. , 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12- 14 fold, 14-16 fold, 16- 18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) lower. In one embodiment, the clearance time is over 100 fold lower. For example, in several embodiments, the active entity and/or conjugate displays rapid systemic elimination when administered by intravenous, intramuscular, transmucosal, subcutaneous or intraperitoneal injection and/or infusion.
[0080] In several embodiments, the active entity and/or conjugate may have enhanced delivery to a cell or other tissue at the target site (e.g. , dermis, epidermis, subcutis) compared to the active entity without conjugation to the polymer. For example, in several embodiments, at least 10% (e.g. , 10-15%, 15-20% , 20-25%, 25-30%, 30-40%, 40-50% , 50-60%, 60-70%, 70-80%, 80-90%, 90- 100%, and overlapping ranges therein) of the administered conjugate reaches a cell or other tissue at the target site. In several embodiments, the conjugate may have enhanced delivery to the epidermis, dermis, and/or subcutis as compared to the active entity delivered without the polymer.
[0081] In several embodiments, the conjugate is amphiphilic and/or amphipathic. In some embodiments, the conjugate is more amphiphilic and/or amphipathic than the active entity without conjugation to the polymer. For example, in several embodiments, the conjugate, as compared to the active entity without conjugation to the polymer, is at least 25% (e.g. , 20-25%, 25-30%, 30-40%, 40-50%, 50- 60%, 60-70%, 70-80% , 80-90% , 90-100% , 100%-125%, 125- 150%, or higher and overlapping ranges therein) more amphiphilic. In one embodiment, the amphiphilicity is over 200% greater. Additionally, in some embodiments, the conjugate is more hydrophilic than the active entity without conjugation to the polymer. For example, in several embodiments, the conjugate, as compared to the active entity without conjugation to the polymer, is at least 25% (e.g. , 20-25%, 25-30%, 30-40%, 40-50%, 50- 60%, 60-70%, 70-80% , 80-90% , 90-100% , 100%-125%, 125- 150%, or higher and overlapping ranges therein) more hydrophilic. In one embodiment, the hydrophilicity is over 200% greater. In some embodiments, the greater hydrophilicity of the conjugate advantageously facilitates one or more of: non-compartmentalization within a cell or tissue at the target site; access to and activity in both the lipid bilayer and the cytosol of
the cell; access to and/or activity in both the lipid bilayer and the cytoplasm of the cell; and/or access to and/or activity across the lipid bilayer.
[0082] In some embodiments, the target site includes cells localized within one or more of the following: the stratum corneum, stratum lucidum, stratum granulosum, stratum spinosum, and stratum basale layers, the papillary layer of the dermis, the reticular layer of the dermis, sebaceous glands, arrector pili muscles, sensory nerve fibers, hair follicles, hair roots, pacinian corpuscles, hair root plexus, eccrine sweat glands, the hypodermis and/or the superficial fascia. In some embodiments, the target cells may comprise immune cells, non-immune cells and/or keratinocytes. In some embodiments, the conjugate targets immune cells residing within dermis, epidermis, and/or subcutis. In several embodiments, the target site comprises immune cells of the epidermis or dermis and the non-target site comprises non-immune cells. In some embodiments, the target site includes the epidermis and the non-target site includes the dermis, gland, hypodermis and/or blood vessels. In other embodiments, the target site includes the dermis and the non-target site includes the epidermis, gland, hypodermis and/or blood vessels. In several embodiments, the target site includes the epidermis and the non-target site includes the dermis, gland, blood vessels, and/or hypodermis. In still further embodiments, the target site includes the dermis and the non- target site includes the epidermis, gland hypodermis and/or blood vessels. In some embodiments, the target site includes one or more of the epidermis, follicle, gland, blood vessels, dermis and subcutis, and the non-target site includes the remaining sites. In other embodiments, the target site includes the subcutis and the non-target site includes tissue contacting the subcutis. In several embodiments, the target site includes the skin and the non-target site includes non-integumentary tissue.
[0083] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following conditions: psoriasis, psoriasis guttata, inverse psoriasis, pustular psoriasis, psoriatic erythroderma, acute febrile neutrophilic dermatosis, eczema, xerotic eczema, dyshidrotic eczema, vesicular palmar eczema, acne vulgaris, atopic dermatitis, contact dermatitis, allergic contact dermatitis, dermatomyositis, exfoliative dermatitis, hand eczema, pompholyx, keloids, rosacea, rosacea due to sarcoidosis, rosacea due to scleroderma, rosacea due to Sweet syndrome, rosacea due to systemic lupus erythematosus, rosacea due to urticaria, rosacea due to herpetic pain, Sweet's disease, neutrophilic hydradenitis, sterile pustule, drug rash, seborrheic dermatitis, pityriasis rosea, Kikuchi's disease of the skin, pruritic urticarial papules and plaques of pregnancy, Stevens-Johnson syndrome and toxic epidermal necrolysis, tattoo reaction, Wells syndrome (eosinophilic cellulitis), reactive arthritis (Reiter syndrome), bowel-associated
dermatosis-arthritis syndrome, rheumatoid neutrophilic dermatosis, neutrophilic eccrine hidradenitis, neutrophilic skin disease of dorsum of hand, balanitis circumscripta plasmacellularis, balanoposthitis, Behcet's disease, erythema annulare centrifugum, erythema dyschromicum perstans, erythema multiforme, granuloma annulare, dermatitis of hand, lichen nitidus, lichen planus, lichen sclerosus et atrophicus, lichen simplex chronicus, lichen spinulosus, nummular dermatitis, pyoderma gangrenosum, sarcoidosis, subcorneal pustular dermatosis, urticaria, and/or transient acantholytic dermatosis.
[0084] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of a vascular tumor, e.g., Kaposi's sarcoma, lymphangioma, glomangioma, angiosarcoma, hemangioendothelioma, and infantile hemangiomas.
[0085] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of a skin neoplasia, e.g., squamous cell carcinoma, basal cell carcinoma, malignant melanoma, malignant cutaneous lymphoma, Kaposi's sarcoma, Merkel cell skin cancer, and non- melanoma skin cancer.
[0086] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of a bullous disease, e.g., bullous pemphigoid, erythema multiforme, dermatitis herpetiformis, epidermolysis bullosa acquisita, linear Immunoglobulin A disease, mucous membrane pemphigoid, pemphigoid gestationis, pemphigus foliaceus, and pemphigus vulgaris.
[0087] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the modulation of hair growth and cycling. In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of alopecia.
[0088] In several embodiments, a reduced exposure composition for treating a target site in the eye is provided. Methods for treating the eye and/or ophthalmic conditions are also provided. In one embodiment, the composition comprises or consists essentially of a conjugate comprising or consists essentially of an active entity coupled (e.g., linked) to at least one polymer. Two, three or more active entities or two, three or more polymers may be used. The active entity may be for example, an inhibitor, antagonist, or inverse agonist of a mediator of an ophthalmic condition. The polymer can include, for example, polyethylene glycol (PEG) and/or methoxy-polyethylene glycol (m- PEG). In one embodiment, a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site is also provided. In several embodiments, the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer. The non-target site includes for example the systemic
system, the lymphatic system and/or other non-target tissue sites. In several embodiments, the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved. In one embodiment, the conjugate can advantageously traverse plasma membranes of cells at the target site, thereby promoting interactions between the active entity and the mediator of the ophthalmic condition. This traversal may include the crossing of cellular lipid bilayers to, e.g., distribute the active entity among both lipophilic and hydrophilic cellular compartments. Membranes include the lipid bilayer, plasma membrane and the nuclear membrane as examples. The conjugate may exhibit a depot effect across cellular compartments, thereby reducing the dose of the active entity required to inhibit the cellular mediator compared to the active entity without conjugation to the polymer. In some embodiments, the conjugate penetrates one or more physical barriers of the eye, one or more physiological barriers of the eye, and/or one or more biological barriers of the eye, such as, for example, the conjunctival epithelium, Tenon's fascia, episclera, sclera, and/or choroid.
[0089] In several embodiments, the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a protein mediator of a condition (e.g., a kinase). In some embodiments, the active entity and/or conjugate may be an inhibitor, antagonist, and/or inverse agonist of a mediator of an ophthalmic condition. For example, in some embodiments, the mediator is one or more of c-Src, a VEGFR protein (e.g., VEGFR-1 , VEGFR-2, VEGFR-3), a JAK protein (e.g., of JAK1 , JAK2, JAK3, and Tyrosine kinase 2 (TYK2)), STAT, NF-Kappa B, TrkA, MAPK, MAP2K and/or MAP2K3. In some embodiments, the active entity and/or conjugate is an inhibitor, antagonist, and/or inverse agonist of a mediator of inflammation. The mediator of inflammation may be, for example, a mediator of an inflammatory ocular condition. In some embodiments, the active entity and/or conjugate reduces inflammation at the target site. In several embodiments, the reduced inflammation at the target site treats an inflammatory ocular condition, such as, for example, uveitis. In some embodiments, the reduced exposure composition comprises one or more of conjugates SNA-101 , SNA-103, SNA-352, SNA- 120 and/or SNA- 125. Advantageously, in several embodiments, the active entity and/or conjugate exhibits greater access to the mediator compared to the active entity without conjugation to the polymer.
[0090] In some embodiments, the reduced exposure composition is formulated for local ocular delivery via a topical or injectable. Intra-ocular admistration is provided in several embodiments. The composition may be formulated for, e.g., subconjunctival, intravitreal, retrobulbar or intracameral delivery. In the methods of treatment, effective amounts of the active entity are delivered to a subject (e.g., human or veterinary). The composition may further comprise one or more additional
ingredients, such as, for example, an antibiotic agent, an anti-angiogenesis agent, a preventive or therapeutic agent for inflammatory bowel disease, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anesthetic agent, a steroidal anti-inflammatory agent, a non-steroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an anti-histamine agent, a vitamin or vitamin complex, and/or a hormone.
[0091] In several embodiments, the active entity and/or conjugate may have a longer residence time within a cell or other tissue at the target site compared to the active entity without conjugation to the polymer. For example, the residence time of the active entity and/or conjugate within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, (i) at least 25% (e.g., 25-50%, 50-75%, 75-100%, 100-150%, or higher and overlapping ranges therein) longer and/or (ii) at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50- 100 fold, and overlapping ranges therein) longer. In one embodiment, the residence time is over 100 fold longer. In several embodiments, the increased residence time is at the anterior segment of the eye and/or anterior segment of the eye.
[0092] In several embodiments, the active entity and/or conjugate may have a shorter residence time within a cell or other tissue at a non-target site compared to the active entity without conjugation to the polymer. For example, the residence time of the active entity and/or conjugate within a cell or other tissue at a non-target site is, as compared to the active entity without conjugation to the polymer, at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) shorter. In one embodiment, the residence time is over 100 fold shorter.
[0093] In some embodiments, a smaller dose of the conjugate may be adequate to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer. For example, in several embodiments, the dose of the conjugate sufficient to achieve a therapeutic effect comparable to the active entity without conjugation to the polymer is at least 10% (e.g. , 10-15%, 15-20%, 20-25%, 25- 30%, 30-40%, 40-50%, 50-60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%- 125%, 125- 150%, or higher and overlapping ranges therein) lower. In one embodiment, the dose is over 200% lower. In some embodiments, fewer doses and/or smaller doses of the conjugate are required as compared to the active entity delivered without the polymer.
[0094] In several embodiments, the active entity and/or conjugate may have diminished systemic absorption compared to the active entity without conjugation to the polymer. For example, in several embodiments, the systemic absorption of the active
entity and/or conjugate, as compared to the active entity without conjugation to the polymer, is at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50- 100 fold, and overlapping ranges therein) lower. In one embodiment, the systemic absorption is over 100 fold lower.
[0095] In several embodiments, the active entity and/or conjugate may have reduced clearance time from a non-target site compared to the active entity without conjugation to the polymer. For example, in several embodiments, the clearance time of the active entity and/or conjugate from a non-target site, as compared to the active entity without conjugation to the polymer, is at least 2-20 fold (e.g., 2-10 fold, 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 12-14 fold, 14-16 fold, 16-18 fold, 18-20 fold, 20-30 fold, 40-50 fold, 10-50 fold, 50-100 fold, and overlapping ranges therein) lower. In one embodiment, the clearance time is over 100 fold lower.
[0096] In several embodiments, the active entity and/or conjugate may have enhanced delivery to a cell or other tissue at the target site compared to the active entity without conjugation to the polymer. For example, in several embodiments, at least 10% (e.g., 10-15%, 15-20%, 20-25%, 25-30%, 30-40%, 40-50%, 50-60%, 60-70%, 70-80%, 80-90%, 90-100%, and overlapping ranges therein) of the administered conjugate reaches a cell or other tissue at the target site. In several embodiments, the conjugate may have enhanced delivery to the posterior segment of the eye and/or anterior segment of the eye as compared to the active entity delivered without the polymer.
[0097] In several embodiments, the active entity and/or conjugate may have an increased concentration, activity and/or bioavailability within a cell or tissue at the target site compared to the active entity without conjugation to the polymer. In some such embodiments, the therapeutically effective amount of the active entity is at the target site. For example, the concentration, activity and/or bioavailability within a cell or other tissue at the target site is, as compared to the active entity without conjugation to the polymer, at least 2-20 fold (e.g., 2-4 fold, 4-6 fold, 6-8 fold, 8-10 fold, 10-12 fold, 14- 16 fold, 18-20 fold, 20-30 fold, 30-40 fold, 40-50 fold, 50-100 fold, and overlapping ranges therein) greater than within a cell or tissue at a non-target site (e.g., the systemic system, the lymphatic system, bone marrow, outside the eye). In one embodiment, the concentration, activity and/or bioavailability within a cell or tissue at the target site is over 100 fold greater.
[0098] In several embodiments, the active entity and/or conjugate may have reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site compared to the active entity without conjugation to the polymer. In several embodiments, the active entity and/or conjugate is present at a biologically inactive
concentration within a cell or tissue at a non-target site. In several embodiments, reduced concentration, activity and/or bioavailability within a cell or tissue at a non-target site (e.g., the systemic system, the lymphatic system, bone marrow, outside the eye) advantageously reduces toxicity and/or other side effects, such as, for example, immunosuppression.
[0099] In several embodiments, the conjugate is amphiphilic and/or amphipathic. In some embodiments, the conjugate is more amphiphilic and/or amphipathic than the active entity without conjugation to the polymer. For example, in several embodiments, the conjugate, as compared to the active entity without conjugation to the polymer, is at least 25% (e.g., 20-25%, 25-30%, 30-40%, 40-50%, 50- 60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%-125%, 125-150%, or higher and overlapping ranges therein) more amphiphilic. In one embodiment, the amphiphilicity is over 200% greater. Additionally, in some embodiments, the conjugate is more hydrophilic than the active entity without conjugation to the polymer. For example, in several embodiments, the conjugate, as compared to the active entity without conjugation to the polymer, is at least 25% (e.g., 20-25%, 25-30%, 30-40%, 40-50%, 50- 60%, 60-70%, 70-80%, 80-90%, 90-100%, 100%-125%, 125-150%, or higher and overlapping ranges therein) more hydrophilic. In one embodiment, the hydrophilicity is over 200% greater. In some embodiments, the greater hydrophilicity of the conjugate advantageously facilitates one or more of: non-compartmentalization within a cell or tissue at the target site; access to and activity in both the lipid bilayer and the cytosol of the cell; access to and/or activity in both the lipid bilayer and the cytoplasm of the cell; and/or access to and/or activity across the lipid bilayer.
[0100] In some embodiments, the target site includes one or more of the following: the anterior segment of the eye, the posterior segment of the eye, an anterior sub-Tenon space, an anterior suprachoroidal space, an anterior intrascleral space, a posterior sub-Tenon space, a posterior suprachoroidal space and a posterior intrascleral space, the conjunctival epithelium, Tenon's fascia, episclera, sclera, choroid, the cornea, lens, sclera, anterior chamber, iris, posterior chamber, choroid, retina, Bowman's layer, stroma, Descemet's membrane, the endothelium, Tenon's Capsule and any combination thereof. In some embodiments, the target site includes target immune cells residing within the anterior segment of the eye and/or posterior segment of the eye.
[0101] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following conditions: glaucoma, allergic conditions, inflammatory conditions of the anterior segment and cornea, allergic conditions of the anterior segment and cornea, infectious conditions of the anterior segment and cornea, and corneal
angiogenesis, corneal edema, macular edema, dry eye, uveitis, macular degeneration, age related macular degeneration (ARMD), diabetic retinopathy, inflammatory conditions of the posterior segment, infectious conditions of the posterior segment, neurodegenerative disease, and vascular disease of the posterior segment, choroidal neovascularization, retinopathy, diabetic retinopathy, acute macular neuroretinopathy, chronic macular neuroretinopathy, central serous chorioretinopathy, macular edema, cystoid macular edema, or diabetic macular edema, acute multifocal placoid pigment epitheliopathy, Behcet's disease, birdshot retinochoroidopathy, uveitis, intermediate uveitis (pars planitis), anterior uveitis, multifocal choroiditis, multiple evanescent white dot syndrome (MEWDS), ocular sarcoidosis, posterior scleritis, serpignous choroiditis, subretinal fibrosis, uveitis syndrome, or Vogt-Koyanagi-Harada syndrome, retinal arterial occlusive disease, central retinal vein occlusion, disseminated intravascular coagulopathy, branch retinal vein occlusion, hypertensive fundus changes, ocular ischemic syndrome, retinal arterial microaneurysms, Coat's disease, parafoveal telangiectasis, hemi-retinal vein occlusion, papillophlebitis, central retinal artery occlusion, branch retinal artery occlusion, carotid artery disease (CAD), frosted branch angitis, sickle cell retinopathy and other hemoglobinopathies, angioid streaks, familial exudative vitreoretinopathy, Eales disease, sympathetic ophthalmia, uveitic retinal disease, retinal detachment, trauma, laser, PDT, photocoagulation, hypoperfusion during surgery, radiation retinopathy, bone marrow transplant retinopathy, proliferative vitreal retinopathy and epiretinal membranes, or proliferative diabetic retinopathy, ocular histoplasmosis, ocular toxocariasis, presumed ocular histoplasmosis syndrome (POHS), endophthalmitis, toxoplasmosis, retinal diseases associated with HIV infection, choroidal disease associated with HIV infection, uveitic disease associated with HIV Infection, viral retinitis, acute retinal necrosis, progressive outer retinal necrosis, fungal retinal diseases, ocular syphilis, ocular tuberculosis, diffuse unilateral subacute neuroretinitis, or myiasis, retinitis pigmentosa, systemic disorders with associated retinal dystrophies, congenital stationary night blindness, cone dystrophies, Stargardt's disease, fundus flavimaculatus, Bests disease, pattern dystrophy of the retinal pigmented epithelium, X-linked retinoschisis, Sorsby's fundus dystrophy, benign concentric maculopathy, Bietti's crystalline dystrophy, or pseudoxanthoma elasticum.
[0102] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of tears (e.g., retinal tear, giant retinal tear), laceration, abrasion, retinal detachment, macular hole, floaters, etc.
[0103] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of a retinal
disease associated with tumors, congenital hypertrophy of the RPE, posterior uveal melanoma, choroidal hemangioma, choroidal osteoma, choroidal metastasis, combined hamartoma of the retina and retinal pigmented epithelium, retinoblastoma, vasoproliferative tumors of the ocular fundus, retinal astrocytoma, or intraocular lymphoid tumors.
[0104] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of punctate inner choroidopathy, acute posterior multifocal placoid pigment epitheliopathy, myopic retinal degeneration, or acute retinal pigment epithelitis.
[0105] In several embodiments, a reduced exposure composition for inhibiting the activity of a therapeutic target in the respiratory tract is provided. Methods and uses for treating respiratory conditions are also provided. In one embodiment, the composition comprises or consists essentially of a conjugate comprising or consists essentially of an active entity coupled (e.g., linked) to at least one polymer. Two, three or more active entities or two, three or more polymers may be used. The active entity may be for example, an inhibitor, antagonist, or inverse agonist of a mediator of a respiratory condition. In some embodiments, the active entity can have a selectivity and/or an inhibitory activity against the therapeutic target. Advantageously, in several embodiments, the conjugate has increased selectivity and/or inhibitory activity compared to the unconjugated active entity. The polymer can include, for example, polyethylene glycol (PEG) and/or methoxy-polyethylene glycol (m-PEG). In one embodiment, a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site is also provided. In several embodiments, the conjugate has reduced exposure at a non-target site as compared to the unconjugated active entity. The non-target site includes for example the systemic system, the lymphatic system and/or other non-target tissue sites. In several embodiments, the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved. In one embodiment, the conjugate can advantageously traverse plasma membranes of cells at the target site compared to the unconjugated active entity, thereby promoting interactions between the active entity and the mediator of a respiratory condition. This traversal may include the crossing of cellular lipid bilayers to, e.g., distribute the active entity among both lipophilic and hydrophilic cellular compartments. Membranes include the lipid bilayer, plasma membrane and the nuclear membrane as examples.
[0106] In several embodiments, the method of treatment and/or use of the compositions described herein are provided for the prophylaxis or treatment of one or more of the following conditions: acute bronchitis; acute laryngotracheal bronchitis; arachidic bronchitis; catarrhal bronchitis; croupus bronchitis; dry bronchitis; infectious
asthmatic bronchitis; productive bronchitis; staphylococcus or streptococcal bronchitis; vesicular bronchitis; cylindric bronchiectasis; sacculated bronchiectasis; fusiform bronchiectasis; capillary bronchiectasis; cystic bronchiectasis; dry bronchiectasis; follicular bronchiectasis; chronic obstructive pulmonary disease (COPD), chronic obstructive lung disease (COLD), chronic obstructive airways disease (COAD) or small airways obstruction of whatever type, etiology, or pathogenesis, in particular chronic bronchitis, pulmonary emphysema, bronchiectasis, cystic fibrosis, bronchiolitis obliterans, organizing pneumonia (BOOP), chronic organizing pneumonia (COP), bronchiolitis fibrosa obliterans, follicular bronchiolitis or dyspnea associated therewith; cough of whatever type, etiology, or pathogenesis in particular idiopathic cough or cough associated with gastro-esophageal reflux disease (GERD), drugs, bronchial hyper- responsivity, asthma, COPD, COLD, COAD, bronchitis, bronchiectasis, pulmonary eosinophilic syndromes, pneumoconiosis, interstitial lung disease, pulmonary fibrosis, aspiration disorders, rhinitis, laryngitis or pharyngitis; pulmonary eosinophilic syndromes of whatever type, etiology, or pathogenesis, in particular acute eosinophilic pneumonia (idiopathic or due to drugs or parasites), simple pulmonary eosinophilia, Loeffler's syndrome, tropical pulmonary eosinophilia, chronic eosinophilic pneumonia, allergic bronchopulmonary mycosis, allergic bronchopulmonary aspergillosis (ABPA), Churg- Strauss syndrome or idiopathic hypereosinophilic syndrome; asthma of whatever type, etiology, or pathogenesis, in particular asthma that is a member selected from the group consisting of atopic asthma, non-atopic asthma, allergic asthma, atopic bronchial IgE- mediated asthma, bronchial asthma, essential asthma, true asthma, intrinsic asthma caused by pathophysiologic disturbances, extrinsic asthma caused by environmental factors, essential asthma of unknown or inapparent cause, non-atopic asthma, bronchitic asthma, emphysematous asthma, exercise-induced asthma, allergen induced asthma, cold air induced asthma, occupational asthma, infective asthma caused by bacterial, fungal, protozoal, or viral infection, non-allergic asthma, incipient asthma and wheezy infant syndrome; alveolar hemorrhage of whatever type, etiology, or pathogenesis, in particular a member of the group consisting of idiopathic pulmonary hemosiderosis, alveolar hemorrhage due to drugs or other exogenous agents, alveolar hemorrhage associated with HIV or bone marrow transplant or autoimmune alveolar hemorrhage (e.g. associated with systemic lupus erythematosis, Goodpasture's syndrome, Wegener's granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome, pauci- immune glomerulonephritis); pneumoconiosis of whatever type, etiology, or pathogenesis, in particular pneumoconiosis that is a member selected from the group consisting of aluminosis or bauxite workers' disease, anthracosis or miners' asthma, asbestosis or steam-fitters' asthma, chalicosis or flint disease, ptilosis caused by inhaling
the dust from ostrich feathers, siderosis caused by the inhalation of iron particles, silicosis or grinders' disease, byssinosis or cotton-dust asthma and talc pneumoconiosis; interstitial lung diseases (ILD) or pulmonary fibrosis of whatever type, etiology, or pathogenesis, in particular idiopathic pulmonary fibrosis, crytogenic fibrosing alveolitis, fibrosing alveolitis, ILD or pulmonary fibrosis associated with connective tissue disease (systemic lupus erythematosis, mixed connective tissue disease, polymyositis, dermatomyositis, Sjorgen's syndrome, systemic sclerosis, scleroderma, rheumatoid arthritis), usual interstitial pneumonia (UIP), desquamative interstitial pneumonia (DIP), granulomatous lung disease, sarcoidosis, Wegener's granulomatosis, histiocytosis X, Langerhan's cell granulomatosis, hypersensitivity pneumonitis, extrinsic allergic alveolitis, silicosis, chronic eosinophilic pneumonia, lymphangiolyomatosis, drug-induced ILD or pulmonary fibrosis, radiation-induced ILD or pulmonary fibrosis, alveolar proteinosis, graft-versus-host-disease (GVHD), lung transplant rejection, ILD or pulmonary fibrosis due to environmental/occupational exposure, BOOP, COP, bronchiolitis fibrosa obliterans, follicular bronchiolitis, idiopathic acute interstitial pneumonitis (Hamman Rich syndrome) or alveolar hemorrhage syndromes; seasonal allergic rhinitis or perennial allergic rhinitis or sinusitis of whatever type, etiology, or pathogenesis, in particular sinusitis that is a member selected from the group consisting of purulent or nonpurulent sinusitis, acute or chronic sinusitis and ethmoid, frontal, maxillary, or sphenoid sinusitis; Acute Respiratory Distress Syndrome (ARDS), adult respiratory distress syndrome or acute lung injury of whatever type, etiology, or pathogenesis; progressive massive fibrosis (PMF); pulmonary hypertension of whatever type, etiology or pathogenesis including primary pulmonary hypertension, essential hypertension, pulmonary hypertension secondary to congestive heart failure, pulmonary hypertension secondary to COPD, pulmonary venous hypertension, pulmonary arterial hypertension and hypoxia-induced pulmonary hypertension. Respiratory disorders also include, in some embodiments, malignancies and tumors of the respiratory system, non- limiting examples of which include lung adenocarcinoma, squamous cell carcinoma, large cell carcinoma, bronchioloalveolar carcinoma (BAC), pulmonary adenocarcinoma (AIS), non-small-cell carcinoma, small cell carcinoma, and/or mesothelioma.
BRIEF DESCRIPTION OF THE FIGURES
[0107] The Figures below are illustrative for some embodiments and should not be construed as overly limiting.
[0108] Figure 1 depicts the BioMAP profile of SNA-101 in the Diversity PLUS Panel at the indicated concentrations. The X-axis lists the quantitative protein-based
biomarker readouts measured in each system. The Y-axis represents a log-transformed ratio of the biomarker readouts for the drug-treated sample (n = 1) over vehicle controls (n > 6). The grey region around the Y-axis represents the 95% significance envelope generated from historical vehicle controls. Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (|log10 ratio| > 0.1). Biomarker key activities are described as modulated if these activities increase in some systems, but decrease in others. Cytotoxicity is indicated on the profile plot by a thin black arrow above the X-axis, and antiproliferative effects are indicated by a thick grey arrow. Cytotoxicity and antiproliferative arrows may only require one concentration to meet the indicated threshold for profile annotation. Other BioMAP profiles disclosed herein are also depicted in a similar manner.
[0109] Figure 2 depicts a Reference Benchmark Overlay of SNA- 101 and Benchmark Apremilast. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction.
[0110] Figure 3 depicts an overlay of SNA-101 (29 μΜ) and Topiramate (3.3 μΜ), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction. Similarity search results are filtered and ranked. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7.
[0111] Figure 4 depicts Mechanism HeatMAP Analysis for SNA-101 . HeatMAP analysis of the 148 biomarker readouts (rows) within the Diversity PLUS panel by SNA-101 in comparison to 19 consensus mechanism class profiles (columns). Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-101 from the 19 consensus mechanism profiles. Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control.
[0112] Figure 5 depicts the BioMAP profile of SNA-101 in the Diversity PLUS Panel at the indicated concentrations. The X-axis lists the quantitative protein-based biomarker readouts measured in each system. The Y-axis represents a log-transformed ratio of the biomarker readouts for the drug-treated sample (n = 1) over vehicle controls (n > 6). The grey region around the Y-axis represents the 95% significance envelope
generated from historical vehicle controls. Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (|log10 ratio| > 0.1). Biomarker key activities are described as modulated if these activities increase in some systems, but decrease in others. Cytotoxicity is indicated on the profile plot by a thin black arrow above the X-axis, and antiproliferative effects are indicated by a thick grey arrow. Cytotoxicity and antiproliferative arrows may only require one concentration to meet the indicated threshold for profile annotation. Other BioMAP profiles disclosed herein are also depicted in a similar manner.
[0113] Figure 6 depicts a Reference Benchmark Overlay of SNA-101 and Benchmark Staurosporine. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction.
[0114] Figure 7 depicts an overlay of SNA-101 (300 μΜ) and N037 (490 ng/ml), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents for SNA-101 (300 μΜ). Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction. Similarity search results are filtered and ranked. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7.
[0115] Figure 8 depicts an overlay of SNA-101 (100 μΜ) and Infliximab (30000 ng/ml), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents for SNA-101 (100 μΜ). Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction. Similarity search results are filtered and ranked. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7.
[0116] Figure 9 depicts Mechanism HeatMAP Analysis for SNA-101 . HeatMAP analysis of the 148 biomarker readouts (rows) within the Diversity PLUS panel by SNA-101 in comparison to 19 consensus mechanism class profiles (columns). Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-101 from the 19 consensus mechanism profiles. Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control.
[0117] Figure 10 depicts the BioMAP profile of SNA-103 in the Diversity PLUS Panel. The X-axis lists the quantitative protein-based biomarker readouts measured in each system. The Y-axis represents a log-transformed ratio of the biomarker readouts for the drug-treated sample (n = 1) over vehicle controls (n > 6). The grey region around the Y-axis represents the 95% significance envelope generated from historical vehicle controls. Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (|log10 ratio| > 0.1). Biomarker key activities are described as modulated if these activities increase in some systems, but decrease in others. Cytotoxicity is indicated on the profile plot by a thin black arrow above the X-axis, and antiproliferative effects are indicated by a thick grey arrow. Cytotoxicity and antiproliferative arrows may only require one concentration to meet the indicated threshold for profile annotation. Other BioMAP profiles disclosed herein are also depicted in a similar manner.
[0118] Figure 1 1 depicts a Reference Benchmark Overlay of SNA-103 and Benchmark Calcipotriene. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction.
[0119] Figure 12 depicts an overlay of SNA-103 (230 μΜ) and Apoptolidin (1 μΜ), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents for SNA-103 (230 μΜ). Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction. Similarity search results are filtered and ranked. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7.
[0120] Figure 13 depicts Mechanism HeatMAP Analysis for SNA-103. HeatMAP analysis of the 148 biomarker readouts (rows) within the Diversity PLUS panel by SNA-103 in comparison to 19 consensus mechanism class profiles (columns). Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-103 from the 19 consensus mechanism profiles. Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control.
[0121] Figure 14 depicts clustering of test agent profiles following pairwise correlation analysis and clustering of the most similar profiles. Each colored circle represents the BioMAP profile of a compound at a specific concentration, with larger circles representing higher concentrations.
[0122] Figure 15 depicts the BioMAP profile of SNA-352 in the Diversity PLUS Panel. The X-axis lists the quantitative protein-based biomarker readouts measured in each system. The Y-axis represents a log-transformed ratio of the biomarker readouts for the drug-treated sample (n = 1) over vehicle controls (n > 6). The grey region around the Y-axis represents the 95% significance envelope generated from historical vehicle controls. Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (|log10 ratio| > 0.1). Biomarker key activities are described as modulated if these activities increase in some systems, but decrease in others. Cytotoxicity is indicated on the profile plot by a thin black arrow above the X-axis, and antiproliferative effects are indicated by a thick grey arrow. Cytotoxicity and antiproliferative arrows may only require one concentration to meet the indicated threshold for profile annotation. Other BioMAP profiles disclosed herein are also depicted in a similar manner.
[0123] Figure 16 depicts a Reference Benchmark Overlay of SNA-352 and Benchmark Cyclosporin A. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction.
[0124] Figure 17 depicts the changes in secretion of (a) IL-17F (b) IgG, (c) IL- 17A, and (d) TNFa in the BioMAP BT system mediated by SNA-352 (3.9 μΜ), Tofacitinib (3.3 μΜ), Apremilast (3.3 μΜ), SR221 1 (3.3 μΜ), and Cyclosporin A (3.3 μΜ).
[0125] Figure 18 depicts an overlay of SNA-352 (3.9 μΜ) and Deferoxamine Mesylate (4.4 μΜ), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction. Similarity search results are filtered and ranked. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7.
[0126] Figure 19 depicts Mechanism HeatMAP Analysis for SNA-352. HeatMAP analysis of the 148 biomarker readouts (rows) within the Diversity PLUS panel by SNA-352 in comparison to 19 consensus mechanism class profiles (columns). Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-352 from the 19 consensus mechanism profiles. Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control.
[0127] Figure 20 depicts the SNA-352 kinase inhibition profile at test concentrations of 100nM and 200nM for the top inhibited kinases as well as those kinases in the middle in the inhibition spectrum.
[0128] Figure 21 depicts a JAK2 vs Staurosporine concentration-%inhibition curve used to derive the slope (1 .853), R2 (1 .00), and IC50 (4.30E-10).
[0129] Figure 22 depicts a JAK2 vs CT340 concentration-%inhibition curve used to derive the slope (1 .147), R2 (1 .00), and IC50 (1 .35E-07).
[0130] Figure 23 depicts a JAK3 vs Staurosporine concentration-%inhibition curve used to derive the slope (1 .597), R2 (1 .00), and IC50 (2.78E-10).
[0131] Figure 24 depicts a JAK3 vs CT340 concentration-%inhibition curve used to derive the slope (1 .164), R2 (1 .00), and IC50 (3.87E-08).
[0132] Figure 25 depicts a PDGFRb vs Staurosporine concentration- %inhibition curve used to derive the slope (2.900), R2 (1 .00), and IC50 (3.87E-10).
[0133] Figure 26 depicts a PDGFRb vs CT340 concentration-%inhibition curve used to derive the slope (1 .165), R2 (1 .00), and IC50 (1 .12E-07).
[0134] Figure 27 depicts a TRKA vs Staurosporine concentration-%inhibition curve used to derive the slope (2.106), R2 (1 .00), and IC50 (5.02E-10).
[0135] Figure 28 depicts a TRKA vs CT340 concentration-%inhibition curve used to derive the slope (1 .159), R2 (1 .00), and IC50 (2.55E-08).
[0136] Figure 29 depicts a MAP2K1 vs Staurosporine concentration- %inhibition curve used to derive the slope (1 .287), R2 (1 .00), and IC50 (1 .39E-09).
[0137] Figure 30 depicts a MAP2K1 vs CT340 concentration-%inhibition curve used to derive the slope (1 .434), R2 (1 .00), and IC50 (1 .96E-08).
[0138] Figure 31 depicts a MAP2K3 vs Staurosporine concentration- %inhibition curve used to derive the slope (1 .402), R2 (1 .00), and IC50 (1 .13E-09).
[0139] Figure 32 depicts a MAP2K3 vs CT340 concentration-%inhibition curve used to derive the slope (1 .41 1 ), R2 (1 .00), and IC50 (1 .26E-08).
[0140] Figure 33 depicts a TAK1 -TAB1 vs Staurosporine concentration- %inhibition curve used to derive the slope (1 .369), R2 (.98), and IC50 (4.14E-08).
[0141] Figure 34 depicts a TAK1 -TAB1 vs CT340 concentration-%inhibition curve used to derive the slope (1 .480), R2 (.98), and IC50 (2.19E-07).
[0142] Figure 35 depicts the BioMAP profile of SNA-120 in the Diversity PLUS Panel. The X-axis lists the quantitative protein-based biomarker readouts measured in each system. The Y-axis represents a log-transformed ratio of the biomarker readouts for the drug-treated sample (n = 1) over vehicle controls (n > 6). The grey region around the Y-axis represents the 95% significance envelope generated from historical vehicle controls. Biomarker activities are annotated when 2 or more
consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (|log10 ratio| > 0.1). Biomarker key activities are described as modulated if these activities increase in some systems, but decrease in others. Cytotoxicity is indicated on the profile plot by a thin black arrow above the X-axis, and antiproliferative effects are indicated by a thick grey arrow. Cytotoxicity and antiproliferative arrows may only require one concentration to meet the indicated threshold for profile annotation. Other BioMAP profiles disclosed herein are also depicted in a similar manner.
[0143] Figure 36 depicts a Reference Benchmark Overlay of SNA- 120 and Benchmark SR221 1 . Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction.
[0144] Figure 37 depicts an overlay of SNA- 120 (28 μΜ) and GSK690693 (10 μΜ), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents for SNA-120 (28 μΜ). Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction. Similarity search results are filtered and ranked. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7.
[0145] Figure 38 depicts Mechanism HeatMAP Analysis for SNA-120. HeatMAP analysis of the 148 biomarker readouts (rows) within the Diversity PLUS panel by SNA-120 in comparison to 19 consensus mechanism class profiles (columns). Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-120 from the 19 consensus mechanism profiles. Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control.
[0146] Figure 39 depicts the BioMAP profile of SNA-125 in the Diversity PLUS Panel. The X-axis lists the quantitative protein-based biomarker readouts measured in each system. The Y-axis represents a log-transformed ratio of the biomarker readouts for the drug-treated sample (n = 1) over vehicle controls (n > 6). The grey region around the Y-axis represents the 95% significance envelope generated from historical vehicle controls. Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (|log10 ratio| > 0.1). Biomarker key activities are described as modulated if these activities increase in some systems, but decrease in others. Cytotoxicity is
indicated on the profile plot by a thin black arrow above the X-axis, and antiproliferative effects are indicated by a thick grey arrow. Cytotoxicity and antiproliferative arrows may only require one concentration to meet the indicated threshold for profile annotation. Other BioMAP profiles disclosed herein are also depicted in a similar manner.
[0147] Figure 40 depicts a Reference Benchmark Overlay of SNA- 125 and Benchmark Tofacitinib. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction.
[0148] Figure 41 depicts an overlay of SNA-125 (3.9 μΜ) and SB203580 (10 μΜ), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents for SNA-125 (3.9 μΜ). Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction. Similarity search results are filtered and ranked. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7.
[0149] Figure 42 depicts Mechanism HeatMAP Analysis for SNA-125. HeatMAP analysis of the 148 biomarker readouts (rows) within the Diversity PLUS panel by SNA-125 in comparison to 19 consensus mechanism class profiles (columns). Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-125 from the 19 consensus mechanism profiles. Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control.
[0150] Figure 43 depicts the BioMAP profile of SNA-125 in the Diversity PLUS Panel. The X-axis lists the quantitative protein-based biomarker readouts measured in each system. The Y-axis represents a log-transformed ratio of the biomarker readouts for the drug-treated sample (n = 1) over vehicle controls (n > 6). The grey region around the Y-axis represents the 95% significance envelope generated from historical vehicle controls. Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope, and have at least one concentration with an effect size > 20% (|log10 ratio| > 0.1). Biomarker key activities are described as modulated if these activities increase in some systems, but decrease in others. Cytotoxicity is indicated on the profile plot by a thin black arrow above the X-axis, and antiproliferative effects are indicated by a thick grey arrow. Cytotoxicity and antiproliferative arrows may only require one concentration to meet the indicated threshold for profile annotation. Other BioMAP profiles disclosed herein are also depicted in a similar manner.
[0151] Figure 44 depicts a Reference Benchmark Overlay of SNA- 125 and Benchmark K252a. Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction.
[0152] Figure 45 depicts an overlay of SNA-125 (30 μΜ) and IKK 16 (370 nM), which was the top similarity match from a search of the BioMAP Reference Database of > 4,000 agents for SNA-125 (30 μΜ). Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1) in the same direction. Similarity search results are filtered and ranked. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7.
[0153] Figure 46 depicts Mechanism HeatMAP Analysis for SNA-125. HeatMAP analysis of the 148 biomarker readouts (rows) within the Diversity PLUS panel by SNA-125 in comparison to 19 consensus mechanism class profiles (columns). Horizontal grey lines separate the 12 Diversity PLUS systems, while the vertical grey line separates SNA-125 from the 19 consensus mechanism profiles. Biomarker activities outside of the significance envelope are red if protein levels are increased, blue if protein levels are decreased and white if levels are within the envelope or unchanged. Darker shades of color represent greater change in biomarker activity relative to vehicle control.
[0154] Figure 47 depicts a BioMAP profile overlay of SNA-125 (10 μΜ), Methotrexate 10 μΜ), and Tofacitinib (10 μΜ).
[0155] Figure 48 depicts representative chromatograms detected using UV analysis (A) and MS analysis (B).
[0156] Figure 49 depicts chromatograms of a CT101 plasma standard extract (100 μg/mL) detected using SIR (TIC, upper) and UV (lower).
[0157] Figure 50 depicts method validation.
[0158] Figure 51 depicts individual chromatograms used for the analysis of CT101 (sample: plasma spiked with CT101 for 100 μg/mL). Top chromatogram: 1 167.5; middle chromatogram: 778.6, bottom chromatogram: 584.2.
[0159] Figure 52 depicts mouse plasma concentrations of CT101 . Data are presented as Mean ± CI 95%.
[0160] Figure 53 depicts representative chromatograms showing CT101 in extracted murine plasma following intra-venous administration. Top chromatogram: 2 hours. Middle chromatogram: 10 minutes, bottom chromatogram: blank murine plasma.
[0161] Figure 54 depicts representative chromatograms showing CT101 in extracted murine plasma following epicutaneous administration. Top chromatogram: 8 hours, middle chromatogram: 0 hours, bottom chromatogram: blank murine plasma.
[0162] Figure 55 depicts Chromatograms of a CT103 plasma standard extract (50 μg/mL) detected using SIR (TIC, upper) and UV at 337 nm (lower).
[0163] Figure 56 depicts CT103 calibration curve (range 0.25-50 μg/ml). Peak areas obtained from TIC MS data, y = 147323x, R2 = 0.9965
[0164] Figure 57 depicts method validation. Arrows denote peaks of LOO and
LOQ.
[0165] Figure 58 depicts individual chromatograms of SIR channels used for the analysis of compound (sample: plasma spiked with compound at 50 μg/mL). Top chromatogram: 794; middle chromatogram: 595.8, bottom chromatogram: 476.8.
[0166] Figure 59 depicts representative chromatograms showing CT103 in methanol (A) and extracted from a spiked murine plasma calibration standard (B). For both chromatographs top to bottom traces = m/z 794, m/z 595.8, m/z 4 76.8, TIC and representative blank. The areas in between the dashed lines represents the area that CT103 eluted, the shaded peaks represent the detected CT103.
[0167] Figure 60 depicts mouse plasma concentrations of CT103. Data are presented as Mean ± CI 95%.
[0168] Figure 61 depicts plasma levels after intravenous dosing.
[0169] Figure 62 depicts body weight versus day of study - males.
[0170] Figure 63 depicts body weight versus day of study - males.
[0171] Figure 64 depicts plasma levels after intravenous dosing - Day 1 .
[0172] Figure 65 depicts plasma levels after intravenous dosing - Week 2.
[0173] Figure 66 depicts body weight versus day of study - Males.
[0174] Figure 67 depicts body weight versus day of study - Males.
[0175] Figure 68 depicts body weight versus day of study (Main phase) -
Males.
[0176] Figure 69 depicts body weight versus day of study (Main phase) -
Males.
[0177] Figure 70 depicts CT327 calibration curves in rat plasma used to determine pharmacokinetic plasma levels (dotted lines represent upper and lower confidence limits).
[0178] Figure 71 depicts mean plasma concentration-time after dose profile of CT327 (SNA-120) after a single intravenous administration at 18 mg/kg. Open symbols represent the average measured values (±95% CI, vertical bar), while filled-in symbols represent interpolated values.
[0179] Figure 72 depicts raw luminescence values for a CT101 preincubation time of 6 hours and a stimulation time of 24 hours. Each symbol represents an individual well. Each condition was tested in sextuplicate.
[0180] Figure 73 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 6 hours and a stimulation time of 24 hours.
[0181] Figure 74 depicts raw luminescence values for a CT101 preincubation time of 18 hours and a stimulation time of 24 hours. Each condition was tested in sextuplicate except for Jurkat cells were conditions were tested in triplicate.
[0182] Figure 75 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 18 hours and a stimulation time of 24 hours.
[0183] Figure 76 depicts raw luminescence values for a CT101 preincubation time of 24 hours and a stimulation time of 24 hours. Each condition was tested in sextuplicate except for Jurkat cells where conditions were tested in triplicate.
[0184] Figure 77 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 24 hours and a stimulation time of 24 hours.
[0185] Figure 78 depicts raw luminescence values for a CT101 preincubation time of 6 hours and a stimulation time of 6 hours. Each condition was tested in sextuplicate.
[0186] Figure 79 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 6 hours and a stimulation time of 6 hours.
[0187] Figure 80 depicts raw luminescence values for a CT101 preincubation time of 18 hours and a stimulation time of 6 hours. Each condition was tested in sextuplicate except for Jurkat cells were conditions were tested in triplicate.
[0188] Figure 81 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 18 hours and a stimulation time of 6 hours.
[0189] Figure 82 depicts raw luminescence values for a CT101 preincubation time of 24 hours and a stimulation time of 6 hours. Each condition was tested in sextuplicate.
[0190] Figure 83 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 24 hours and a stimulation time of 6 hours.
[0191] Figure 84 depicts percentage cytotoxicity for a CT101 pre-incubation time of 6 hours and a stimulation time of 24 hours.
[0192] Figure 85 depicts percentage cytotoxicity for a CT101 pre-incubation time of 18 hours and a stimulation time of 24 hours.
[0193] Figure 86 depicts percentage cytotoxicity for a CT101 pre-incubation time of 24 hours and a stimulation time of 24 hours.
[0194] Figure 87 depicts percentage cytotoxicity for a CT101 pre-incubation time of 24 hours and a stimulation time of 6 hours.
[0195] Figure 88 depicts percentage cytotoxicity for a CT101 pre-incubation time of 6 hours and a stimulation time of 6 hours.
[0196] Figure 89 depicts tritiated thymidine incorporation in corrected counts per minute (CCPM). Data are presented as Mean ± SEM.
[0197] Figure 90 depicts Tritiated thymidine incorporation in corrected counts per minute (CCPM). Data are presented as Mean ± SEM.
[0198] Figure 91 depicts tritiated thymidine incorporation in corrected counts per minute (CCPM). Statistical analysis of the 18 hour pre-incubation time with 5x103 cells per well and 50ng/ml_ VEGF stimulation. Data are presented as Mean ± SEM. Values significantly lower than DMSO-treated cells are indicated by: * p < 0.05, ** p < 0.01 , ns not significant.
[0199] Figure 92 depicts percentage inhibition by CT103 with 18 hour preincubation time ,5x103 cells per well and 50ng/ml_ VEGF stimulation. Percentage inhibition of VEGF induced proliferation by CT103. Proliferation in the absence of VEGF (0 ng/mL VEGF) was subtracted from VEGF induced proliferation (50 ng/mL) and divided by 50 ng/mL VEGF stimulated cell proliferation in the absence of drug (DMSO).
[0200] Figure 93 depicts tritiated thymidine incorporation in corrected counts per minute (CCPM). Data are presented as Mean ± SEM.
[0201] Figure 94 depicts non-linear fit of transformed normalised maximal response for IC50 calculation. The DMSO (0 μΜ CT103) treated VEGF-induced HUVEC proliferation was excluded from the IC50 calculation as it was lower than the 3 lowest concentrations of CT103, which had reached a consistent plateau as can be seen above. The IC50 was calculated as 18.5 μΜ.
[0202] Figure 95 depicts percentage inhibition of VEGF induced proliferation by CT103. Proliferation in the absence of VEGF was subtracted from VEGF induced proliferation and divided by VEGF stimulated cell proliferation in the absence of drug (DMSO).
[0203] Figure 96 depicts luciferase activity of THP1 cells stimulated with a concentration range of HKLM or LPS, calculated relative to unstimulated cells.
[0204] Figure 97 depicts the inhibition of VEGF-induced proliferation following treatment with SNA-125 (A), SNA-352 (B), SNA-103 (C), and motesanib diphosphate (D). Data are presented as mean corrected counts per minute (CCPM) ± SEM, with n=6 for (A)-(C) and with n=4 for (D).
[0205] Figure 98 depicts the inhibition of proliferation by K252a, CT327 and CT340. Column bar graphs of proliferation assay results expressed as Absorbance at 570 nm with reference at 650 nm.
[0206] Figure 99 depicts the inhibition of proliferation by K252a, CT327 and CT340. Column bar graphs of proliferation assay results expressed as Absorbance at 570 nm with reference at 650 nm.
[0207] Figure 100 depicts the inhibition of proliferation by K252a, CT327 and CT340. Column bar graphs of proliferation assay results expressed as Absorbance at 570 nm with reference at 650 nm.
[0208] Figure 101 depicts (a) colitis development was evaluated monitoring colon shortening at mice sacrifice, (b) weight loss during the experiment, and (c) colitis clinical score at sacrifice. CT100 and CT300 data points refer to 100 and 300 mg/kg CT352 in vehicle. CsA data points refer to 25 mg/kg cyclsporin A (positive control).
[0209] Figure 102 depicts qRTPCR analysis of inflammatory cytokines and chemokines expression. RNA from whole proximal and distal colon was analysed and an increased expression of cytokines (IL6, IL17) and chemokines (MIP1 a and MIP2) related to inflammation was detected in DSS treated mice, while this response was counteracted by CsA (positive control) and CT352 administration at the same time.
[0210] Figure 103 depicts histological analysis of colon samples. The extension and the degree of the colitis were determined in blind and the score assigned to each samples reported in the colitis clinical score graph (a). A representative sample image from each group was reported (4X magnification) (b).
[0211] Figure 104 depicts the (A) colon dissection diagram and (B) fields and scoring order employed in the oxazolone-induced colitis mouse study.
[0212] Figure 105 depicts the effect of SNA- 125, SNA-352, tofacitinib, and prednisolone on the body weight of animals challenged with oxazolone. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Figure 105A depicts percent body weight change from Day -1 to Day 4 of the study. Figure 105B depicts the area under the curve (AUC) of the percent weight change depicted in Figure 105A.
[0213] Figure 106 depicts the effect of SNA- 125, SNA-352, tofacitinib, and prednisolone on the body weight of animals challenged with oxazolone according to last observation carried forward analysis. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Figure 106A depicts percent body weight change from Day -1 to Day 4 of the study. Figure 106B depicts the area under the curve (AUC) of the percent weight change depicted in Figure 106A.
[0214] Figure 107 depicts the effect of SNA- 125, SNA-352, tofacitinib, and prednisolone on the Day 2 endoscopy score of animals challenged with oxazolone by (A) bar chart and (B) dot plot. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
[0215] Figure 108 depicts the effect of SNA- 125, SNA-352, tofacitinib, and prednisolone on the Day 2 stool consistency score of animals challenged with oxazolone
by (A) bar chart and (B) dot plot. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
[0216] Figure 109 depicts the effect of SNA- 125, SNA-352, tofacitinib, and prednisolone on the Day 4 endoscopy score of animals challenged with oxazolone by (A) bar chart and (B) dot plot. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
[0217] Figure 1 10 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the Day 4 stool consistency score of animals challenged with oxazolone by (A) bar chart and (B) dot plot. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
[0218] Figure 1 1 1 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the disease activity index (DAI) score of animals at (A) Day 2 and (B) Day 4 following challenge with oxazolone. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
[0219] Figure 1 12 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the colon weight/length ratio of animals challenged with oxazolone. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO).
[0220] Figure 1 13 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the colon inflammation histopathology scores of animals challenged with oxazolone. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted.
[0221] Figure 1 14 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the colon edema histopathology scores of animals challenged with oxazolone. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted.
[0222] Figure 1 15 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the colon mucosal necrosis/loss histopathology scores of animals challenged with oxazolone. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted.
[0223] Figure 1 16 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the summed colon histopathology scores of animals challenged with oxazolone. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted.
[0224] Figure 1 17 depicts representative control animal H&E-stained colon histopathology micrographs at 40x and 100x magnifications.
[0225] Figure 1 18 depicts representative H&E-stained colon histopathology micrographs at 40x and 100x magnifications for animals administered BID (A) Vehicle PO, (B) 15 mg/kg Tofacitinib PO, (C) 1 mg/kg Prednisolone PO, (D) 400 mg/kg SNA-125 PO, and (E) 400 mg/kg SNA-352 PO. Moderate inflammation (unfilled black arrows), edema (filled red arrows) and multifocal ulceration (brackets) are indicated.
[0226] Figure 1 19 depicts representative H&E-stained colon histopathology micrographs at 40x and 100x magnifications for animals administered BID (A) Vehicle IC, (B) 1 mg/kg Tofacitinib IC, (C) 400 mg/kg SNA-125 IC, and (D) 400 mg/kg SNA-352 IC.
[0227] Figure 120 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the proein levels of IFNv in colon tissue homogenate supernatants of animals following challenge with oxazolone. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted.
[0228] Figure 121 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the protein levels of TNFa in colon tissue homogenate supernatants of animals following challenge with oxazolone. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted, with outliers removed (A), or present (B).
[0229] Figure 122 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the protein levels of IL-6 in colon tissue homogenate supernatants of animals following challenge with oxazolone. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted, with outliers removed (A), or present (B).
[0230] Figure 123 depicts the effect of SNA-125, SNA-352, tofacitinib, and prednisolone on the protein levels of IL-10 in colon tissue homogenate supernatants of animals following challenge with oxazolone. Compounds or vehicle controls were dosed BID as indicated intracecally (IC) or orally (PO). Group means with standard error of the mean (SEM) bars are depicted, with outliers removed (A), or present (B).
[0231] Figure 124 depicts bodyweights. Data are presented as Mean ± SEM percentages of the initial bodyweights. # p < 0.05 and ### p < 0.001 when compared to Day 0. ° p < 0.05, °° p < 0.01 and **** p < 0.0001 when compared to the vehicle-treated group.
[0232] Figure 125 depicts ear swelling. Data are presented as Mean ± SEM. ### p < 0.0001 in the vehicle-treated group when compared to Day 0. °° p < 0.01 , °°° p < 0.001 and °°°° φφφφρ < 0.0001 when compared to the vehicle-treated group.
Betamethasone 0.1 % induced a significant reduction of ear swelling when compared to the vehicle-treated group on Day 8, Day 1 1 and Day 14 (p < 0.0001).
[0233] Figure 126 depicts macroscopic scores. Data are presented as Mean ± SEM. //////// p < 0.0001 when compared to Day 0 in the vehicle-treated group. °°° p < 0.01 and 000°, φφφφ p < 0.0001 when compared to the vehicle-treated group.
[0234] Figure 127 depicts cytokine levels in ears (pg/ml). A. IL-1 β B. IFN-y C. IL-4 D. IL-10. Data are presented as Mean ± SEM. **** p < 0.0001 , ** p < 0.001 , * p < 0.01 when compared to the Vehicle treated group. #### p < 0.0001 , ### p < 0.001 when compared to the right ear within the same group.
[0235] Figure 128 depicts bodyweights. Data are presented as Mean ± SEM of the initial (Day -13) bodyweights. * p < 0.05, ** p < 0.01 .
[0236] Figure 129 depicts ear swelling. Data are presented as Mean ± SEM of the difference between ovalbumin challenged and contralateral (saline-injected) ears. Statistical significances: # p < 0.05, ## p < 0.01 , ### p < 0.001 when compared to the baseline (0 hours) values. * p < 0.05, ** p < 0.01 , *** p < 0.001 when comparing to the Control group.
[0237] Figure 130 depicts ear swelling at peak disease (fifteen minutes after the ovalbumin challenge). Data are presented as Mean ± SEM of the difference between ovalbumin-challenged and contralateral (saline-injected) ears. Statistical significances: * p < 0.05, ** p < 0.01 , *** p < 0.001 when compared to the vehicle-treated group.
[0238] Figure 131 depicts erythema scores (Challenged ears). Data are presented as Mean ± SEM.
[0239] Figure 132 depicts erythema scores (Challenged ears). Data are presented as Mean ± SEM.
[0240] Figure 133 depicts histopathology scores (Left ears). Data are presented as Mean ± SEM. ** p < 0.01 , *** p < 0.001 when compared to the Control group.
[0241] Figure 134 depicts representative histopathology pictures. Left panel: Left ears. Right panel: Right ears. Top line: Control Group. Middle line: Betamethasone 0.1 %-treated group. Bottom line: Vehicle-treated group. Magnification: x100.
[0242] Figure 135 depicts representative histopathology pictures. Left panel: Left ears. Right panel: Right ears. Top line: CT101_5%-treated group. Middle line: CT101_10%-treated group. Bottom line: CT101_20%-treated group. Magnification: x100.
[0243] Figure 136 depicts bodyweights. Data are presented as Mean ± SEM percentages of the initial bodyweights. * p < 0.05, *** p < 0.001 , **** p < 0.0001 when compared to the vehicle-treated group; # # # p < 0.001 , # # # # p < 0.0001 when compared to Day 0.
[0244] Figure 137 depicts erythema scores. Data are presented as Mean ± SEM . * p < 0.05, ** p < 0.01 , *** p < 0.001 , **** p < 0.0001 when compared to the vehicle-treated group.
[0245] Figure 138 depicts scaling scores. Data are presented as Mean ± SEM . * p < 0.05, *** p < 0.01 and **** p < 0.0001 when compared to the vehicle-treated group.
[0246] Figure 139 depicts thickness scores. Data are presented as Mean ± SEM . * p < 0.05, *** p < 0.001 , **** p < 0.0001 .
[0247] Figure 140 depicts clinical scores. Data are presented as Mean ± SEM . * p < 0.05, ** p < 0.01 , *** p < 0.001 and **** p < 0.0001 when compared to the vehicle-treated group.
[0248] Figure 141 depicts normalized body weight trends.
[0249] Figure 142 depicts scar formation; SEI of saline-injected scars (Group 1 , Left Ears). Values are expressed as mean ± StdDev.
[0250] Figure 143 depicts scar formation; SEI of vehicle-treated scars (Group 1 , Right Ears) . Values are expressed as mean ± StdDev.
[0251] Figure 144 depicts scar formation; SEI of CT340-injected scars (Group 2). Values are expressed as mean ± StdDev.
[0252] Figure 145 depicts Scar Formation; SEI of CT340 topical-dosed scars (Group 3). Values are expressed as mean ± StdDev.
[0253] Figure 146 depicts Scar Formation; SEI of TACA-treated scars (Group 4). Values are expressed as mean ± StdDev.
[0254] Figure 147 depicts scar formation; summary of SEI . Values are expressed as mean ± StdDev.
[0255] Figure 148 depicts scar formation following intra-lesion injections with CT340 or TACA.
[0256] Figure 149 depicts scar formation following topical dosing with CT340
[0257] Figure 150 depicts scar formation SEI of CT340-treated scars, Values are expressed as mean ± StdDev.
[0258] Figure 151 depicts scar inflammation scores following intra-lesion injections with CT340 or TACA. Values are expressed as mean ± StdDev.
[0259] Figure 152 depicts scar inflammation scores following topical dosing with CT340, Values are expressed as mean ± StdDev.
[0260] Figure 153 depicts CT327 effect on Capsaicin responses. Inhibition of capsaicin responses in DRG neurons by CT327 was between 35.6 ± 3.0 % (1 nM CT327) , and 57 ± 5.3% (10 μΜ CT327). Results are given as mean (percent inhibition) ±
s.e.m. P values were obtained from comparison between percent inhibition of each dose with control. Further, for 10 μΜ vs 10 nM , P=0.008, 10 μΜ vs 1 nM P=0.001 , and 1 μΜ vs 1 nM P=0.007.
[0261] Figure 154 depicts CT340 effect on Capsaicin responses. I nhibition of capsaicin responses in DRG neurons by CT340 was between 35.6 ± 3.7% (1 nM CT340) , and 57.4 ± 4.3% (10 μΜ CT340). Results are given as mean (percent inhibition) ± s.e.m. 10 μΜ vs 10 nM , P=0.01 , 10 μΜ vs 1 nM , P=0.0009; 1 μΜ vs 1 nM, P=0.04.
[0262] Figure 155 depicts GW441756 effect on Capsaicin responses. Inhibition of capsaicin responses in DRG neurons by GW441756 was between 39.8 ± 4.1 % (1 nM GW441756), and 54.4 ± 7.4% (1 .5 μΜ GW441756). IC50= 15 μΜ . Results are given as mean (percent inhibition) ± s.e.m.
[0263] Figure 156 depicts anti-NGF antibody effect on Capsaicin responses. Inhibition of capsaicin responses in DRG neurons by anti-NGF was between 37.4 ± 7.7% (100 ng/ml), and 63.3 ± 9.7% (10 μg/ml). Results are given as mean (percent inhibition) ± s.e.m.
[0264] Figure 157 depicts the effect of compound incubation on neurite length. Treatment with CT327 and CT340 at 1 nM , 10 nM , 100 nM , 1 μΜ and 10 μΜ concentrations did not affect neurite length of neurons compared with control. Treatment with GW441756 resulted in vesiculation and reduced neurite length at the higher concentrations of 1 μΜ and 10 μΜ . Anti-NGF antibody treatment at 1 and 10 μg/ml concentrations also did not affect neurite length. Neurite lengths were normalized to controls and are given as mean percent of control ± s.e. m.
[0265] Figure 158 depicts the effect of CT327 incubation on neurite length . 24 hour Incubation with CT327 did not have any effect on neurite length. Neurite lengths are expressed as mean percent of control ± s.e.m.
[0266] Figure 159 depicts the effect of CT340 incubation on neurite length . 24 hour Incubation with CT340 did not significantly affect neurite length compared to control. Neurite lengths are expressed as mean percent of control ± s.e.m.
[0267] Figure 160 depicts the effect of GW441756 incubation on neurite length. Commercial TrkA inhibitor GW441756 had no effect on neurite length at concentrations up to 100 nM concentration, but appeared to diminish neurite length at 1 μΜ (P=0.09 n.s.) and 1 0 μΜ (*P=0.03) concentrations, with vesiculation of neurites (see Figure 157). Neurons treated with 0.33% ethanol (solvent for GW441756) had similar neurite length compared with NGF-treated controls. Neurite lengths are expressed as mean percent of control ± s.e.m.
[0268] Figure 161 depicts the effect of anti-NGF antibody incubation on neurite length. Neurons treated with anti-NGF at 1 or 10 μg/ml did not show a significant
change in neurite length compared to control. Neurite lengths are expressed as mean percent of control ± s.e.m.
[0269] Figure 162 depicts TrkA/Gap43 immunostaining in DRG neurons: A) Merged image showing co-localization of TrkA and Gap43 immunostaining in DRG neuron; B) Gap43 was strongly localized in cell bodies and neurites; C) TrkA immunostaining was observed to be densely localized in the cell bodies, while neurites were very faint.
[0270] Figure 163 depicts representative PGP9.5-immunoreactive intraepithelial nerve fibres (I EFN) (arrowed) in untreated control skin (top panel) and treated skin region (bottom panel) mini-pig skin using an antibody dilution of 1 :40,000, magnification x40.
[0271] Figure 164 depicts a scatter plot showing the PGP9.5 intra epithelial fibre counts in untreated and treated mini-pig skin from the various groups. The median value is indicated. C, control; L, low dose; M , medium dose; H high dose; R, recovery; ut untreated area; t treated area.
[0272] Figure 165 depicts the design of the IMQ-induced psoriasis mouse study. Animal shaving, IMQ cream application, treatment, left ear biopsy punch, body weight measurements, ear thickness measurements, psoriasis clinical scoring, and termination were performed at the indicated tiem points.
[0273] Figure 166 depicts the change in animal body weight throughout the IMQ-induced psoriasis mouse study.
[0274] Figure 167 depicts the changes in the total psoriasis score throughout the IMQ-induced psoriasis mouse study. The difference between SNA- 125 at 5% and the vehicle is statistically significant from day 7. The differences between SNA- 125 at 0.5% and 1 % and the vehicle are statistically significant on day 10.
[0275] Figure 168 depicts the changes in the Erythema score throughout the IMQ-induced psoriasis mouse study. SNA-125 at 5% is statistically significant from the vehicle from day 7. SNA-125 at 0.5% and 1 % are statistically significant on day 10.
[0276] Figure 169 depicts the changes in the plaque score throughout the IMQ-induced psoriasis mouse study. SNA-125 at 5% is statistically significant from the vehicle on day 10.
[0277] Figure 170 depicts the changes in the punctate redness/scabbing score throughout the IMQ-induced psoriasis mouse study.
[0278] Figure 171 depicts the changes in spleen thickness throughout the IMQ-induced psoriasis mouse study
[0279] Figure 172 depicts the changes in ear thickness throughout the IMQ- induced psoriasis mouse study.
[0280] Figure 173 depicts the levels of cytokines (a) IL-22, (b) IL-17A, (c) IL17F, and (d) TNFa, in ear samples at Day 4.
[0281] Figure 174 depicts a schematic showing how the IMQ-induced psoriasis study was performed.
[0282] Figure 175 depicts the total psoriasis clinical scores over time for all groups (A) , the SNA-101 group (B), the SNA-125 group (C) , and the SNA-352 group (D) . The mean score for each group is displayed for each day +/- SEM .
[0283] Figure 176 depicts the erythema scores over time for all groups (A) , the SNA- 101 group (B), the SNA-125 group (C) , and the SNA-352 group (D) . The mean score for each group is displayed for each day +/- SEM .
[0284] Figure 177 depicts the plaque scores over time for all groups (A), the SNA-101 group (B), the SNA-125 group (C) , and the SNA-352 group (D). The mean score for each group is displayed for each day +/- SEM .
[0285] Figure 178 depicts the punctate redness/scabbing scores over time for all groups (A) , the SNA-101 group (B), the SNA- 125 group (C), and the SNA-352 group (D) . The mean score for each group is displayed for each day +/- SEM .
[0286] Figure 179A depicts the weight of spleens upon experimental termination on day 10. Mean spleen weight for each group is displayed +/- SEM . Figure 179B depicts left ear thickness as measured with a caliper on days 0, 4, 6, 8, and 10. Mean thickness for each group is displayed for each day +/- SEM . Figure 179C depicts the daily weight of mice. Body weight changes are displayed for each day as a percent of their weight measured on day 0. Mean values for each group are displayed +/- SEM .
[0287] Figure 180 depicts the levels of IL- 17F (A), TNF-a (B), IL-22 (C), and IL-17A (D) as measured in left ears biopunched on day 4. After tissue homogenization , the cytokine levels in tissue lysates were measured via multiplex and then normalized with total protein amounts. Mean values for each group are displayed +/- SEM .
[0288] Figure 181 depicts a schematic of the IL-23-induced psoriasis mouse model study.
[0289] Figure 182 depicts the effect SNA-120 and SNA-325 in an IL-23- induced psoriasis mouse model. Figure 182A depicts the total psoriasis clinical scores for each group over time. The mean score for each group is displayed for each day +/- SEM. Figure 182B depicts the right ear thickness of each group at the indicated time points. Mean thickness for each group is displayed for each day +/- SEM . Figure 182C depicts body weight of each group over the course of the study. Body weight changes are displayed for each day as a percent of their weight measured on day 0. Mean values for each group are displayed +/- SEM .
[0290] Figure 183 depicts the concentration vs. %lnhibition curves of staurosporine (A) and SNA-352 (B) against LIMK1 . The calculated slope and IC50(M) are also depicted).
[0291] Figure 184 depicts the concentration vs. %lnhibition curves of staurosporine (A) and SNA-352 (B) against MAP2K6. The calculated slope and IC50(M) are also depicted).
[0292] Figure 185 depicts the concentration vs. %lnhibition curves of staurosporine (A) and SNA-352 (B) against MLK1 . The calculated slope and IC50(M) are also depicted).
[0293] Figure 186 depicts the concentration vs. %lnhibition curves of staurosporine (A) and SNA-352 (B) against MLK3. The calculated slope and IC50(M) are also depicted).
[0294] Figure 187 depicts representative Day 2 endoscopy images of naive control, vehicle control (PO), vehicle control (IC) , and tofacitinib (15 mg/kg PO) animals. Animals underwent video endoscopy on Day 2 and colitis severity was scored on a scale of 0-4. Images were captured from each animal during the procedure and representative images from each treatment group are presented. All doses were administered twice a day (BID).
[0295] Figure 188 depicts representative Day 2 endoscopy images of tofacitinib (15 mg/kg IC), prednisolone (1 mg/kg PO) , SNA-125 (400 mg/kg PO) , and SNA-352 (400mg/kg PO) animals. Animals underwent video endoscopy on Day 2 and colitis severity was scored on a scale of 0-4. I mages were captured from each animal during the procedure and representative images from each treatment group are presented. All doses were administered twice a day (BID).
[0296] Figure 189 depicts representative Day 2 endoscopy images of SNA- 125 (400 mg/kg IC) and SNA352 (400 mg/kg IC) animals. Animals underwent video endoscopy on Day 2 and colitis severity was scored on a scale of 0-4. n=8-15 per group. Images were captured from each animal during the procedure and representative images from each treatment group are presented. All doses were administered twice a day (BID).
[0297] Figure 190 depicts representative Day 4 endoscopy images of naive control, vehicle control (PO), vehicle control (IC) , and tofacitinib (15 mg/kg PO) animals. Animals underwent video endoscopy on Day 4 and colitis severity was scored on a scale of 0-4. n=8- 15 per group. Images were captured from each animal during the procedure
and representative images from each treatment group are presented. All doses were administered twice a day (BID).
[0298] Figure 191 depicts representative Day 4 endoscopy images of tofacitinib (15 mg/kg IC), prednisolone (1 mg/kg PO) , SNA-125 (400 mg/kg PO) , and SNA-352 (400mg/kg PO) animals. Animals underwent video endoscopy on Day 4 and colitis severity was scored on a scale of 0-4. n=8-15 per group. Images were captured from each animal during the procedure and representative images from each treatment group are presented. All doses were administered twice a day (BID).
[0299] Figure 192 depicts representative Day 4 endoscopy images of SNA- 125 (400 mg/kg IC) and SNA352 (400 mg/kg IC) animals. Animals underwent video endoscopy on Day 4 and colitis severity was scored on a scale of 0-4. n=8-15 per group. Images were captured from each animal during the procedure and representative images from each treatment group are presented. All doses were administered twice a day (BID).
DETAILED DESCRIPTION
Platform Technology
[0300] Several embodiments relate to the use of agents that were developed using Applicant's proprietary Low Systemic Exposure™ ("LSE™") platform technology to generate LSE molecules (also generally referred to herein as polymer conjugates or compositions). In several embodiments, the LSE platform creates polymer conjugates optimized for topical applications. In several embodiments, the polymer conjugates developed by LSE or more generally the reduced exposure technology exhibit enhanced penetration. In still further embodiments, the enhanced penetration leads to delivery of a high local concentration of the drug. In further embodiments, the polymer conjugates show a limited non-target absorption upon topical administration due to their increased molecular size and amphiphilicity and/or amphipathicity. In still further embodiments, side-effects are minimized by limiting or eliminating non-target (e.g. , systemic) absorption.
[0301] In several embodiments of the reduced exposure compositions/compounds, the polymer conjugate comprises a "warhead" linked to a polymer. In some embodiments, the warhead is a pharmacologically active entity
selected according to the particular target or pathway of interest. As discussed herein, there are also provided, in several embodiments, polymer conjugates for use in the treatment of conditions (including but not limited to inflammatory bowel diseases, dermatological diseases, and ophthalmic conditions). In several embodiments, the polymer is directly coupled to the warhead without a separate chemical linking moiety between the polymer and the warhead; such direct coupling may involve without limitation ester, ether, acetal, ketal, vinyl ether, carbamate, urea, amine, amide, enamine, imine, oxime, amidine, iminoester, carbonate, orthoester, phosphonate, phosphinate, sulfonate, sulfinate, sulfide, sulfate, disulfide, sulfinamide, sulfonamide, thioester, aryl, silane, siloxane, heterocycles, thiocarbonate, thiocarbamate, and phosphonamide bonds. In several embodiments, the linker is a separate chemical linking moiety between the polymer and the warhead. In several embodiments, the polymer is polyethylene glycol (PEG), wherein the terminal OH group can optionally be modified e.g. with C1 -C5 alkyl or C1 -C5 acyl groups, e.g., with C1 -, C2- or C3-alkyl groups or C1 -, C2- or C3 groups. In several embodiments, the modified PEG is a terminally alkoxy- substituted PEG. In several embodiments, the modified PEG is a methoxy-PEG (mPEG). In some embodiments, the polymer has a molecular weight ranging from about 100 to about 100,000 Da. In some embodiments, the polymer is polydisperse with respect to molecular weight (e.g., has a distribution of molecular weights) and the indicated molecular weight of the polymer represents an average molecular weight. In other embodiments, the polymer has a molecular weight ranging from about 200 to about 50,000 Da. In several embodiments, the polymer has a molecular weight ranging from about 500 to about 10,000 Da (e.g., 500-1000, 1000-2000, 2000-3000, 3000-5000,5000- 7000, 7000-10,000 Da, and overlapping ranges therein).
[0302] In several embodiments, the polymer is a short-chain PEG, and in some embodiments a terminally alkoxy-substituted PEG, such as a mPEG with a molecular weight ranging from about 200 to about 4,000 Da, from about 400 to about 3,000 Da, from about 500 to about 2,000 Da, from about 700 to about 3,000 Da, from about 900 to about 4,000 Da, or from about 1 ,000 to about 5,000 Da. In several embodiments, the short-chain PEG or mPEG has an average molecular weight of about 1 ,000-3,000 Da. (e.g., 2,000 Da).
[0303] In some embodiments, the polymer is a long-chain PEG. The long- chain PEG may be a terminally alkoxy-substituted PEG, such as methoxy-substituted PEG, with a molecular weight ranging greater than about 4,000 Da. In several embodiments, the molecular weight ranges from about 4,500-10,000Da (e.g., 4,500 to about 5,500 Da). In several embodiments, the long-chain PEG or mPEG has an average molecular weight of about 2,000 Da or of about 5,000 Da. In several
embodiments, the polymer is of natural or semi-synthetic or synthetic origin. In several embodiments, the polymer has a linear or branched structure. In several embodiments, the polymer is selected from poly(alkylene oxides) or from (polyethylene) oxides. In several embodiments, the polymer selected may include, without limitation, one or more of the following: polyacrylic acid, polyacrylates, polyacrylamide or N-alkyl derivatives thereof, polymethacrylic acid, polymethacrylates, polyethylacrylic acid, polyethylacrylates, polyvinylpyrrolidone, poly(vinylalcohol), polyglycolic acid, polylactic acid, poly(lactic-co-glycolic) acid, dextran, chitosan, and hydroxyethyl starch.
[0304] In some embodiments, administration comprises treatment of the gastro-intestinal tract via, for example, an enteric coated capsule comprising the polymer conjugates taken orally. In one embodiment, the polymer conjugates provided herein treat inflammatory bowel diseases.
[0305] In some embodiments, conjugating the warhead to a polymer (e.g., PEG) in the disclosed molecular weight ranges may slow diffusion of the molecule in the tissue, thereby potentially increasing residence time of the molecule in the target tissue, e.g. certain regions of the eye, epithelial and sub-epithelial layers in the eye, the epidermis and dermis for skin, the lining of the Gl tract, associated epithelial and subepithelial layers in other topical surfaces like gut, eye, lungs, the intestinal epithelium, the intestinal lamina propria, intestinal mucosa, etc. This "depot" effect may also lead to lower concentrations needing to be applied or for products to be applied with lower frequency, or both.
[0306] In an embodiment, the polymer conjugates provided herein are administered to the skin by topical application. In one embodiment, the polymer conjugates provided herein treat inflammatory skin diseases.
[0307] Eye drops are provided in some embodiments to treat eye inflammation or ophthalmic disorders and diseases.
[0308] In other embodiments, conjugating the warhead to a polymer (e.g., PEG) in the disclosed molecular weight ranges may be useful in reducing the diffusion or extravasation of the molecule out of the circulatory system after it enters it via injection and or diffusion from the target tissue. Indeed, changes in the tissue distribution of polymer conjugates compared to unconjugated drug have been observed in IV injection studies. In general, the unconjugated drug tends to have a long half-life and a volume of distribution within tissues AND blood, suggesting that the unconjugated drug extravasates out of the blood vessels into the tissue prior to being cleared. Whereas, in some instances, the PEGylated drug has a volume of distribution that is largely restricted to the blood, indicating that very little extravasation occurs with the polymer conjugates
prior to being renally cleared. This reduced extravasation may explain at least in part the observed shorter half-life for the polymer conjugates.
[0309] The compositions described herein may be combined with other modalities to achieve synergic effects. These other modalities include, but are not limited to, energy delivery (such as laser, radiofrequency, ultrasound, microwave, etc.), thermal therapy, light therapy, radiation, intravenous chemotherapy, and others. In some embodiments, the compositions are applied with pressure, heat, massage etc. to facilitate localization to the desired target site. In some embodiments, the compositions are administered in combination with one or more additional therapeutics that may not be reduced exposure compounds.
[0310] Santi et al. state that "permanent PEGylation is generally not applicable to small-molecule drugs because the bulky carrier usually prevents their binding to targets and cell penetration." (Proceedings of the National Academy of Sciences 109.16 (2012): 621 1 -6216). Further, Nakagami et al. state that "hydrophilic polymers on the surface of particles... prevents the close interactions between particles and target cell membranes, inhibiting the cellular uptake and, subsequently, preventing endosomal escape. All of these factors combine to decrease the biological efficacy of PEGylated particles" (Gene therapy (2013): 2-4). In several embodiments, the polymer conjugate exhibits unexpected permeability across the plasma membrane. In several embodiments, the polymer conjugate exhibits unexpected permeability across the nuclear membrane. In several embodiments, the polymer conjugate exhibits unexpected permeability across both the nuclear and plasma membranes. Accordingly, in Example 30, two polymer conjugates, SNA-125 and SNA-120, were surprisingly shown to penetrate the keratinocyte cellular membrane and interact with the target kinases intracellular^ within the cytoplasm, thereby leading to inhibition of proliferation of keratinocytes in a non-toxic manner. The reduced exposure compounds, comprising a hydrophobic drug conjugated to a short chain PEG, exhibit surprising accessibility across cellular compartments, compared to the unconjugated drug. This accessibility is thought to result, in some embodiments, from the amphipathic nature of the conjugate, allowing it to traverse and distribute evenly among both lipophilic and hydrophilic cellular compartments. Accordingly, the conjugate can cross and reside within the lipid bilayer of the cell membrane, accumulate within the cytosol, and even traverse the nuclear envelope - thereby providing access both membrane, cytosolic and nuclear molecular targets in several embodiments. This property of the reduced exposure compounds result in excellent depo'ing, longer residence times within target cells, and/or relative non-compartmentalization in several embodiments. Consequently, in many
embodiments, these compounds are biologically active at lower concentrations and require less frequent dosing - thereby reducing potential drug toxicity.
[0311] In some embodiments, the invention comprises a reduced exposure composition for treating a target site within the gastrointestinal system, skin, or eye comprising a conjugate comprising one or more active entitites linked to one or more polymers, wherein the active entity is a mediator of a gastrointestinal condition, ophthalmic condition, and/or dermatological condition and has reduced exposure in the systemic system as compared to the gastrointestinal system based on the ability of the conjugate to cross the lipid bilayer and into the cytoplasm and/or cytosol of a plurality of cells within the gastrointestinal system. In some embodiments, the invention comprises a reduced exposure composition for treating a dermatological target site comprising a conjugate comprising one or more active entitites linked to one or more polymers, wherein the active entity is a mediator of a dermatological condition and has reduced exposure in the systemic system as compared to the skin based on the ability of the conjugate to cross the lipid bilayer and into the cytoplasm and/or cytosol of a plurality of cells within the target site. The concentration and/or bioavailability of the activity entity in the systemic system, in one embodiment, is at an amount that is not active systemically and therefore does not result in undesired systemic side effects. In one embodiment, the active entity has little or no exposure to the lymphatic system, thus resulting in little or no immunosuppression. Damage to organs such as the kidney or liver is also nominal because of the reduced exposure to non-target tissue.
Polymer Conjugates of Indolocarbazole Compounds
[0312] In several embodiments, the warhead employed in the LSE polymer conjugate is an indolocarbazole compound.
[0313] In several embodiments, there is provided in a reduced exposure composition, a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an indolocarbazole compound. In some embodiments, the polymer conjugate comprises an indolocarbazole compound of formula (I) or of formula (II):

formula (I) formula (H)
[0314] wherein in formula (I) and (II)
[0315] R1 and R2 are the same or a different residue and are each independently selected from the group consisting of:
[0316] (a) hydrogen, halogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, hydroxy, lower alkoxy, carboxy, lower alcoxycarbonyl, acyl, nitro, carbamoyl, lower alkylaminocarbonyl, -NR5R6, wherein R5 and R6 are each independently selected from hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted aralkyi, substituted or unsubstituted lower alkylaminocarbonyl, substituted or unsubstituted lower arylaminocarbonyl, alkoxycarbonyl, carbamoyl, acyl or R5 and R6 are combined with a nitrogen atom to form a heterocyclic group,
[0317] (b) -CO(CH2)jR4, wherein j is 1 to 6, and R4 is selected from the group consisting of
[0318] (i) hydrogen, halogen, -N3,
[0319] (ii) -NR5R6, wherein R5 and R6 are as defined above,
[0320] (iii) -SR7, wherein R7 is selected from the group consisting of hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted aralkyi, - (CH2)aC02R10 (wherein a is 1 or 2, and wherein R10 is
selected from the group consisting of hydrogen and substituted or unsubstituted lower alkyl) and -(CH2)aC02NR5R6,
[0321] (iv) -OR8, -OCOR8, wherein R8 is selected from hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl
[0322] (c) -CH(OH)(CH2)j R4 wherein j and R4 are as defined above;
[0323] (d) -(CH2)dCHR11C02R12 or -(CH2)dCHR11CONR5R6, wherein d is 0 to 5, R11 is hydrogen, -CONR5R6, or -C02R13, wherein R13 is hydrogen or a wherein substituted or unsubstituted lower alkyl, and R12 is hydrogen or a substituted or unsubstituted lower alkyl;
[0324] (e) -(CH2)kR14 wherein k is 2 to 6 and R14 is halogen, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, -COOR15, -OR15, (wherein R15 is hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl or acyl), -SR7 (wherein R7 is as defined above), -CONR5R6, -NR5R6 (wherein R5 and R6are as defined above) or -N3;
[0325] (f) -CH=CH(CH2)mR16, wherein m is 0 to 4, and R16 is hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, -COOR15, -OR15 (wherein R15 is as defined above) - CONR5R6 or -NR5R6 (wherein R5 and R6 are as defined above);
[0326] (g) -CH=C(C02R12)2, wherein R12 is as defined above;
[0327] (h) -C≡C(CH2)nR16, wherein n is 0 to 4 and R16 is as defined above;
[0328] (i) -CH2OR22, wherein R22 is tri-lower alkyl silyl in which the three lower alkyl groups are the same or different or wherein R22 has the same meaning as R8
[0329] (j) -CH(SR23)2 and -CH2-SR7 wherein R23 is lower alkyl, lower alkenyl or lower alkynyl and wherein R7 is as defined above; and
[0330] R3 is hydrogen, halogen, acyl, carbamoyl, substituted or unsubstituted lower alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted lower alkynyl or amino; and
[0331] W1 and W2 are independently hydrogen, hydroxy or W1 and W2 together represent oxygen;
[0332] and wherein in formula (I), X is a polymer moiety, either linear or branched,
[0333] and wherein in formula (II), A represents -L1-X' and B represents -L2- Y', wherein at least one of X and Y' is a polymer moiety, either linear or branched, which
is bound by L1 and/or L2 to the tetrahydrofuran ring of the compound of formula (II);
L1 and/or L2 are a covalent chemical bond or a linker group;
[0334] when Y' is a polymer moiety, and X' is not a polymer, L1 is a covalent chemical bond and X' is selected from the group consisting of
[0335] (a) hydrogen, lower hydroxyalkyl, acyl, carboxy, lower alkoxycarbonyl,
[0336] (b) -CONR17aR17b, wherein R17a and R17b are each independently selected from
[0337] (i) hydrogen, lower alkyl, lower alkenyl, lower alkynyl,
[0338] (ii) -CH2 R18; wherein R18 is hydroxy,
[0339] or (iii) -NR19R20, wherein R19 or R20 are each independently selected from hydrogen, lower alkyl, lower alkenyl, lower alkynyl or R19 or R20 are independently the residue of an a-amino acid in which the hydroxy group of the carboxyl group is excluded, or R19 or R20 are combined with a nitrogen atom to form a heterocyclic group; and
[0340] (c) -CH=N-R21 , wherein R21 is hydroxy, lower alkoxy, amino, guanidino, or imidazolylamino;
[0341] when X' is a polymer moiety, and Y' is not a polymer, L2 is a covalent chemical bond and Y' is selected from hydroxy, lower alkoxy, aralkyloxy, or acyloxy;
[0342] or a pharmaceutically acceptable salt of formula (I) and/or (II).
[0343] The polymer moiety X, X or/and Y' covalently attached to the indolocarbazole compound of formulae (I) and (II) has to be biocompatible, can be of natural or semi-synthetic or synthetic origin and can have a linear or branched structure. In some embodiments, the polymer moiety X, X or/and Y' is selected from poly(alkylene oxides), in particular from (polyethylene) oxides. However, further exemplary polymers include without limitation polyacrylic acid, polyacrylates, polyacrylamide or N-alkyl derivatives thereof, polymethacrylic acid, polymethacrylates, polyethylacrylic acid, polyethylacrylates, polyvinylpyrrolidone, poly(vinylalcohol), polyglycolic acid, polylactic acid, poly(lactic-co-glycolic) acid, dextran, chitosan, polyaminoacids, hydroxyethyl starch.
[0344] In some embodiments, the polymer moiety X, X' or/and Y' is a polyethylene glycol (PEG) moiety, wherein the terminal OH group can optionally be modified e.g. with C^Cs alkyl or C^Cs acyl groups. In some embodiments, the terminal OH group is optionally modified with Ci-, C2- or C3-alkyl groups or Ci-, C2- or C3 groups. In some embodiments, the modified polyethylene glycol is a terminally alkoxy-substituted polyethylene glycol. In some embodiments, the polymer moiety is methoxy-polyethylene- glycol (mPEG).
[0345] As used in this application, except as otherwise expressly provided herein, each of the following terms shall have the meaning set forth below.
[0346] The term "lower alkyl", when used alone or in combination with other groups, means a straight chained or branched lower alkyl group containing from 1 -6 carbon atoms, preferably from 1 -5, more preferably from 1 -4 and especially preferably 1 - 3 or 1 -2 carbon atoms. These groups include, in some embodiments, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, amyl, isoamyl, neopentyl, 1 - ethylpropyl, hexyl, and the like. The lower alkyl moiety of the "lower alkoxy", the "lower alkoxycarbonyl", the "lower akylaminocarbonyl', "lower hydroxyalkyl' and of the "tri-lower alkylsilyl' groups has the same meaning as "lower alkyl" defined above.
[0347] The "lower alkenyl" groups are defined as C2-C6 alkenyl groups which may be straight chained or branched and may be in the Z or E form. Such groups include vinyl, propenyl, 1 -butenyl, isobutenyl, 2-butenyl, 1 -pentenyl, (Z)-2- pentenyl, (E)-2- pentenyl, (Z)-4-methyl-2-pentenyl, (E)-4-methyl-2-pentenyl, pentadienyl, e.g., 1 , 3 or 2,4-pentadienyl, and the like. In some embodiments, the C2-C6- alkenyl groups are C2- C5-, C2-C4-alkenyl groups. In other embodiments, the C2-C6- alkenyl groups are C2-C3- alkenyl groups.
[0348] The term "lower alkynyl" groups refers to C2-C6-alkynyl groups which may be straight chained or branched and include ethynyl, propynyl, 1 -butynyl, 2- butynyl, 1 -pentynyl, 2-pentynyl, 3-methyl-1 -pentynyl, 3-pentynyl, 1 -hexynyl, 2-hexynyl, 3-hexynyl and the like. In some embodiments, C2-C6-alkynyl groups are C2-C5-, C2-C4- alkynyl groups. In other embodiments, C2-C6-alkynyl groups are C2-C3-alkynyl groups.
[0349] The term "aryl" group refers to C6-C14-aryl groups which contain from 6 up to 14 ring carbon atoms. These groups may be mono-, bi- or tricyclic and are fused rings. In some embodiments, the aryl groups include phenyl, biphenyl, naphthyl, anthracenyl, phenanthrenyl and the like. The aryl moiety of the "arylcarbonyl" and the "arylaminocarbonyl" groups has the same meaning as defined above.
[0350] The term "heteroaryl" groups may contain 1 to 3 heteroatoms independently selected from nitrogen, sulfur or oxygen and refers C3-C13-heteroaryl groups. These groups may be mono-, bi- or tricyclic. In some embodiments, the C3- C13 heteroaryl groups include heteroaromatics and saturated and partially saturated heterocyclic groups. These heterocyclics may be monocyclic, bicyclic, tricyclic. In some embodiments, the 5 or 6-membered heterocyclic groups are thienyl, furyl, pyrrolyl, pyridyl, pyranyl, morpholinyl, pyrazinyl, methyl pyrrolyl, and pyridazinyl. The C3-Ci3- heteroaryl may be a bicyclic heterocyclic group. In some embodiments, the bicyclic heterocyclic groups are benzofuryl, benzothienyl, indolyl, imidazolyl, and pyrimidinyl. In some embodiments, the C3-C13-heteroaryls are furyl and pyridyl.
[0351] The term "lower alkoxy" includes alkoxy groups containing from 1 to 6 carbon atoms, in some embodiments from 1 to 5, in other embodiments from 1 -4 and in
yet other embodiments 1 to 3 or 1 to 2 carbon atoms and may be straight chained or branched. These groups include methoxy, ethoxy, propoxy, butoxy, isopropoxy, tert- butoxy, pentoxy, hexoxy and the like.
[0352] The term "acyl" includes lower alkanoyl containing 1 to 6 carbon atoms, in some embodiments from 1 to 5, from 1 to 4, from 1 to 3 or from 1 to 2 carbon atoms and may be straight chained or branched. These groups include, in some embodiments, formyl, acetyl, propionyl, butyryl, isobutyryl, tertiary butyryl, pentanoyl and hexanoyl. The acyl moiety of the "acyloxy" group has the same meaning as defined above.
[0353] The term "halogen" includes fluoro, chloro, bromo, iodio, and the like.
[0354] The term "aralkyl' group refers C7-Ci5-aralkyl wherein the alkyi group is substituted by an aryl. The alkyi group and aryl may be selected from the C C6 alkyi groups and the C6-C14-aryl groups as defined above, wherein the total number of carbon atoms is between 7 and 15. In some embodiments the C7-C15-aralkyl groups are benzyl, phenylethyl, phenylpropyl, phenylisopropyl, phenylbutyl, diphenylmethyl, 1 , 1 - diphenylethyl, 1 ,2-diphenylethyl. The aralkyl moiety of the "aralkyloxy" groups has the same meaning as defined above.
[0355] The substituted lower alkyi, alkenyl and alkynyl groups have 1 to 3 independently selected substituents, such as lower alkyi, hydroxy, lower alkoxy, carboxyl, lower alkoxycarbonyl, nitro, halogen, amino, mono- or di- lower alkylamino, dioxolane, dioxane, dithiolane, and dithione. The lower alkyi substituent moiety of the substituted lower alkyi, alkenyl and alkynyl groups, and the lower alkyi moiety of the lower alkoxy, the lower alkoxycarbonyl, and the mono- or di-lower alkylamino substituents of the substituted lower alkyi, alkenyl and alkynyl groups have the same meaning as "lower alkyi" defined above.
[0356] The substituted aryl, the substituted heteroaryl and the substituted aralkyl groups each has 1 to 3 independently selected substituents, such as lower alkyi, hydroxy, lower alkoxy, carboxy, lower alkoxycarbonyl, nitro, amino, mono- or di-lower alkylamino, and halogen. The lower alkyi moiety of the lower alkyi, the lower alkoxy, the lower alkoxycarbonyl, and the mono- or di- lower alkylamino groups among the substituents has the same meaning as 'lower alkyi' defined above.
[0357] The heterocyclic group formed by R5 and R6 combined with a nitrogen atom includes pyrrolidinyl, piperidinyl, piperidino, morpholinyl, morpholino, thiomorpholino, N-methylpiperazinyl, indolyl, and isoindolyl.
[0358] In some embodiments, R1 and R2 are independently selected from the group consisting of hydrogen, halogen, nitro, -CH2OH, -(CH2)kR14, -CH=CH(CH2)mR16, - C≡C(CH2)nR15, -CO(CH2)jR4 wherein R4 is -SR7, CH20-(substituted or unsubstituted)
lower alkyl (wherein the substituted lower alkyl is in some embodiments methoxymethyl, methoxyethyl or ethoxymethyl), -NR5R6. In some embodiments, each of R1 and R2 is hydrogen.
[0359] In some embodiments of R1 and R2, the residue R14 is selected from phenyl, pyridyl, imidazolyl, thiazolyl, tetrazolyl, -COOR15, -OR15 (wherein R15 is in some embodiments selected from hydrogen, methyl, ethyl, phenyl or acyl), -SR7 (wherein R7 is in some embodiments selected from substituted or unsubstituted lower alkyl, 2-thiazoline and pyridyl) and -NR5R6(wherein R5 and R6 are in some embodiments selected from hydrogen, methyl, ethyl, phenyl, carbamoyl and lower alkylaminocarbonyl). Moreover, in some embodiments, the residue R16 is selected from hydrogen, methyl, ethyl, phenyl, imidazole, thiazole, tetrazole, -COOR15, -OR15 and -NR5R6 (wherein the residues R15, R5 and R6 have the meanings as described above). In some embodiments of R1 and R2, the residue R7 is selected from the group consisting of substituted or unsubstituted lower alkyl, substituted or unsubstituted phenyl, pyridyl, pyrimidinyl, thiazole and tetrazole. Further, in some embodiments, k is 2, 3 or 4, j is 1 or 2 and m and n are independently 0 or 1.
[0360] In some embodiments, R3 is hydrogen or acetyl. Furthermore, in some embodiments, each W1 and W2 is hydrogen.
[0361] In some embodiments, when Y' is a polymer moiety and X' is not a polymer moiety, X' is selected from carboxy, hydroxymethyl or a lower alkoxycarbonyl. In some embodiments X' is selected from methoxycarbonyl.
[0362] In some embodiments, when X' is a polymer moiety and Y' is not a polymer moiety, Y' is selected from hydroxy or acetyloxy.
[0363] In some embodiments, the warhead of the polymer conjugate is a derivative of K252a, which has the formula:


[0365] In some embodiments, the polymer conjugate is SNA-120, wherein the composition has the formula:

[0366] The formulas depicted herein are not limited to any particular stereochemistry, and all stereoisomers and enantiomers thereof are included in this disclosure.
[0367] In some embodiments, the active entity comprises an indolocarbazole compound. In some embodiments, the active entity comprises a derivative of K252a. In some embodiments, the composition comprises SNA-125.
[0368] The active entity binds to a tropomyosin-receptor-kinase A (TrkA) in some embodiments. The active entity binds to a Janus Kinase (JAK) family member in some embodiments. The active entity binds to one or more of Janus Kinase 1 (JAK1), Janus Kinase 2 (JAK2), Janus Kinase 3 (JAK3), and/or Tyrosine kinase 2 (TYK2) in some embodiments. The active entity binds to mitogen-activated protein kinase kinase (MAP2K) in some embodiments. The active entity binds to mitogen-activated protein
kinase kinase 3 (MAP2K3) in some embodiments. The binding may be partially or fully inhibitory or not.
[0369] Compositions comprising an indolocarbazole compound are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of TkrA, Jak3, and/or MAP2K3. Several embodiments relate to polymer conjugates of an indolocarbazole compound, optimized for topical applications while also minimizing side- effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications are provided in other embodiments.
[0370] In several embodiments, the warhead of the polymer conjugate is an indolocarbazole compound or derivative thereof. There is also provided, in several embodiments, methods of treating an inflammatory bowel disease in a subject, the method comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is an indolocarbazole compound. In some embodiments, the warhead of the LSE polymer conjugate is a derivative of K252a. In some embodiments, the LSE polymer conjugate is SNA-125. In some embodiments, the LSE polymer conjugate is SNA-120. In addition to, or instead of IBD, IBS and SIBO may also be treated.
[0371] Oral delivery is contemplated in several embodiments suitable for gastrointestinal disorders, and thus several embodiments relate to polymer conjugates of an indolocarbaozle, optimized for oral delivery to treat the gastrointestinal system while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Topical applications are provided in other embodiments.
[0372] In several embodiments, the warhead of the polymer conjugate is an indolocarbazole compound or derivative thereof. There is also provided, in several embodiments, methods of treating an ophthalmic condition in a subject, the method comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is an indolocarbazole compound. In some embodiments, the warhead of the LSE polymer conjugate is a derivative of K252a. In some embodiments, the LSE polymer conjugate is SNA-125. In some embodiments, the LSE polymer conjugate is SNA-120. In addition to, or instead of an inflammatory ophthalmic condition, non-inflammatory ophthalmic conditions may also be treated.
[0373] In several embodiments, the warhead of the polymer conjugate is an indolocarbazole compound or derivative thereof. There is also provided, in several embodiments, methods of treating an inflammatory skin disease in a subject, the method comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is an indolocarbazole compound. In some embodiments, the warhead of the LSE polymer conjugate is a derivative of K252a. In some
embodiments, the LSE polymer conjugate is SNA-125. In some embodiments, the LSE polymer conjugate is SNA-120. In addition to, or instead of an inflammatory skin disease, non-inflammatory dermatological conditions may also be treated.
[0374] Topical delivery is contemplated in several embodiments suitable for dermal pathologies, and thus several embodiments relate to polymer conjugates of an indolocarbaozle, optimized for topical delivery to treat the skin while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Nontopical applications are provided in other embodiments.
Polymer Conjugates Targeting Mediators of Gastrointestinal Condition
[0375] Compositions comprising an indolocarbazole compound are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of TkrA, Jak3, and/or MAP2K3. Several embodiments relate to polymer conjugates of an indolocarbazole compound, optimized for topical applications while also minimizing side- effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications are provided in other embodiments.
[0376] In several embodiments, the warhead of the polymer conjugate is an indolocarbazole compound or derivative thereof. There is also provided, in several embodiments, methods of treating an inflammatory bowel disease in a subject, the method comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is an indolocarbazole compound. In some embodiments, the warhead of the LSE polymer conjugate is a derivative of K252a. In some embodiments, the LSE polymer conjugate is SNA-125. In some embodiments, the LSE polymer conjugate is SNA-120.
[0377] In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting mediator(s) of gastrointestinal conditions. There is also provided, in several embodiments, methods of treating an inflammatory bowel disease in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting mediator(s) of gastrointestinal conditions. In some emboidments, the gastrointestinal condition is an inflammatory bowel disease. Non-limiting examples of inflammatory bowel diseases include Crohn's disease, ulcerative colitis, collagenous colitis, lymphocytic colitis, ischemic colitis, diversion colitis, Behcet's syndrome, and indeterminate colitis. In several embodiments, JAK and/or STAT family proteins are mediator(s) of gastrointestinal conditions. In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule a targeting a JAK and/or STAT family protein.
[0378] The JAK kinase family is a cytoplasmic protein kinase family comprising the members JAK1 , JAK2, JAK3 and TYK2. Various studies suggest that ligand binding to a receptor leads to receptor dimerization or oligomerization, which leads to JAK recruitment and activation either through autophosphorylation or phosphorylation by other JAK kinases or by other tyrosine kinases, which in turn leads to tyrosine phosphorylation of the receptors as well as downstream substrates of JAK. Growth factor or cytokine receptors that recruit JAK kinases include the interferon receptors, interleukin receptors (receptors for the cytokines IL-2 to IL-7, IL-9 to IL-13, IL- 15, IL-23), various hormone receptors (erythropoietin (Epo) receptor, the thrombopoietin (Tpo) receptor, the leptin receptor, the insulin receptor, the prolactin (PRL) receptor, the Granulocyte Colony-Stimulating Factor (G-CSF) receptor and the growth hormone receptor), receptor protein tyrosine kinases (such as EGFR and PDGFR), and receptors for other growth factors such as leukemia inhibitory factor (LIF), Oncastatin M (OSM), IFNa/β/γ, Granulocyte-macrophage colony-stimulating factor (GM-CSF), Ciliary neurotrophic factor (CNTF), cardiotrophin-1 (CT-I) (See, Rane, S.G. and Reddy E.P., Oncogene 2000 19, 5662- 5679).
[0379] Many autoimmune diseases and disease associated with chronic inflammation, as well as acute responses, have been linked to excessive or unregulated production or activity of one or more cytokines, the signaling of which depend on JAK kinases. Such diseases include gastrointestinal conditions, which may be treated according to several embodiments described herein.
[0380] Phosphorylated receptors serve as docking sites for other SH-2 domain containing signaling molecules that interact with JAKs such as the STAT family of transcription factors, Src family of kinases, MAP kinases PB kinase and protein tyrosine phosphatases (Rane S.G. and Reddy E.P., Oncogene 2000 19, 5662-5679). The family of latent cytoplasmic transcription factors, STATS, are the most well characterized downstream substrates for JAKs. The STAT proteins bind to phosphorylated cytokine receptors through their SH2 domains to become phosphorylated by JAKs, which event leads to their dimerization and release and eventual translocation to the nucleus where they activate gene transcription. The various members of STAT which have been identified thus far, are STAT1 , STAT2, STAT3, STAT4, STAT5 (including STAT5a and STAT5b) and STAT6.
[0381] Signal transducer and activator of transcription 3 (STAT3), a member of the STAT protein family, is a transcription factor that regulates the expression of a variety of genes involved in many cellular processes such as cell growth, apoptosis, cell motility, and cytokine production. In response to cytokines and growth factors, STAT3 is activated by JAK kinases and translocates to the nucleus to act as a transcriptional
activator. Studies have demonstrated that STAT3 plays a role in various immune disorders including the pathogenesis of inflammatory bowel disease (see, e.g., Sugimoto, World J. Gastroenterol., 14:51 10-51 14, (2008)).
[0382] Inflammatory bowel diseases (IBD) are diseases characterized by inflammation in the small intestine and colon. IBD is known to include two common autoimmune diseases in humans— Crohn's disease and ulcerative colitis— which share many of the same physiological, mechanistic, immune, inflammatory and genetic features, as well as common treatment strategies (such as TNF withdrawal therapy). Histopathologically and anatomically, these two conditions are distinct, with Crohn's disease characterized by transmural inflammation that can occur throughout the Gl tract, and ulcerative colitis characterized by more superficial inflammation confined to the colon and rectum. In several embodiments, irritable bowel syndrome (IBS) and small intestinal bacterial overgrowth (SIBO) are treated.
[0383] There is provided, in several embodiments, methods of treating IBD, IBS and/or (SIBO) in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting mediator(s) of gastrointestinal conditions. In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting a JAK and/or STAT family protein. Non-limiting examples of inflammatory bowel diseases include Crohn's disease, ulcerative colitis, collagenous colitis, lymphocytic colitis, ischemic colitis, diversion colitis, Behcet's syndrome, and indeterminate colitis.
[0384] In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting c-Src. There is also provided, in several embodiments, methods of treating an inflammatory bowel disease in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting c-Src.
[0385] The c-Src kinase is the most widely studied member of the largest family of nonreceptor protein tyrosine kinases, known as the Src family kinases (SFKs). Other SFK members include Lyn, Fyn, Lck, Hck, Fgr, BIk, Yrk, and Yes. The Src kinases can be grouped into two sub-categories, those that are ubiquitously expressed (Src, Fyn, and Yes), and those which are found primarily in hematopoietic cells (Lyn, Lck, Hck, BIk, Fgr). (Benati, D. Src Family Kinases as Potential Therapeutic Targets for Malignancies and Immunological Disorders. Current Medicinal Chemistry. 2008; 15: 1 154-1 165). SFKs are key messengers in many cellular pathways, including those involved in regulating proliferation, differentiation, survival, motility, and angiogenesis. The activity of SFKs is highly regulated intramolecularly by interactions between the SH2 and SH3 domains and intermolecularly by association with cytoplasmic molecules. This latter
activation may be mediated by focal adhesion kinase (FAK) or its molecular partner Crk- associated substrate (CAS), which plays a prominent role in integrin signaling, and by ligand activation of cell surface receptors, e.g. epidermal growth factor receptor (EGFR). These interactions disrupt intramolecular interactions within Src, leading to an open conformation that enables the protein to interact with potential substrates and downstream signaling molecules. Src can also be activated by dephosphorylation of tyrosine residue Y530. In some embodiments, maximal Src activation requires the autophosphorylation of tyrosine residue Y419 (in the human protein) present within the catalytic domain. Elevated Src activity may be caused by increased transcription or by deregulation due to overexpression of upstream growth factor receptors such as EGFR, HER2, platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor, ephrins, integrin, or FAK.
[0386] Compositions comprising compounds Nos 1 -71 shown in Table 1 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of c-Src. Several embodiments relate to polymer conjugates of compounds 1 -71 , optimized for topical applications while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications are provided in other embodiments.
[0387] In several embodiments, the warhead of the polymer conjugate is a small molecule disclosed in Table 1 targeting c-Src. There is also provided, in several embodiments, methods of treating an inflammatory bowel disease in a subject, the method comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Table 1 targeting c-Src. In some embodiments, the warhead of the LSE polymer conjugate is compound 1 of Table 1 . In some embodiments, the LSE polymer conjugate is CT101 .
[0388] In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting a vascular endothelial growth factor receptor (VEGFR).
[0389] Angiogenesis, the process of sprouting new blood vessels from existing vasculature, and arteriogenesis, the remodeling of small vessels into larger conduit vessels, are both physiologically important aspects of vascular growth in adult tissues. These processes of vascular growth are involved, in some cases, for beneficial processes such as tissue repair, wound healing, recovery from tissue ischemia and menstrual cycling. They are also involved, in some cases, for the development of pathological conditions such as the growth of neoplasias, diabetic retinopathy, rheumatoid arthritis, psoriasis, certain forms of macular degeneration, and certain inflammatory pathologies. The inhibition of vascular growth in these contexts has also
shown beneficial effects in preclinical animal models. For example, inhibition of angiogenesis by blocking vascular endothelial growth factor or its receptor has resulted in inhibition of tumor growth and in retinopathy. Also, the development of pathological pannus tissue in rheumatoid arthritis involves angiogenesis and might be blocked by inhibitors of angiogenesis. Certain diseases are known to be associated with deregulated angiogenesis, for example ocular neovascularization, such as retinopathies (including diabetic retinopathy), age-related macular degeneration, psoriasis, hemangioblastoma, hemangioma, arteriosclerosis, inflammatory disease, such as a rheumatoid or rheumatic inflammatory disease, especially arthritis (including rheumatoid arthritis), or other chronic inflammatory disorders, such as chronic asthma, arterial or post- transplantational atherosclerosis, endometriosis, and neoplastic diseases, for example so-called solid tumors and liquid tumors (such as leukemias).
[0390] Vascular endothelial growth factor (VEGF) is a signaling protein involved in both vasculogenesis (the de novo formation of the embryonic circulatory system) and angiogenesis (the growth of blood vessels from pre-existing vasculature). As its name implies, VEGF activity has been mostly studied on cells of the vascular endothelium, although it does have effects on a number of other cell types (e.g., stimulation monocyte/macrophage migration, neurons, cancer cells, kidney epithelial cells, keratinocytes). In vitro, VEGF has been shown to stimulate endothelial cell mitogenesis and cell migration. VEGF is also a vasodilator and increases microvascular permeability and was originally referred to as vascular permeability factor.
[0391] The broad term "VEGF" covers a number of proteins from two families, that result from alternate splicing of mRNA from a single, 8 exon, VEGF gene. The two different familes are referred to according to their terminal exon (exon 8) splice site— the proximal splice site (denoted VEGFxxx) or distal splice site (VEGFxxxb). In addition, alternate splicing of exon 6 and 7 alters their heparin binding affinity, and amino acid number (in humans: VEGF121 , VEGF121 b, VEGF145, VEGF165, VEGF165b, VEGF189, VEGF206; the rodent orthologs of these proteins contain one fewer amino acid). These domains have functional consequences for the VEGF splice variants as the terminal (exon 8) splice site determines whether the proteins are pro-angiogenic (proximal splice site, expressed during angiogenesis) or anti-angiogenic (distal splice site, expressed in normal tissues). In addition, inclusion or exclusion of exons 6 and 7 mediate interactions with heparan sulfate proteoglycans (HSPGs) and neuropilin co- receptors on the cell surface, enhancing their ability to bind and activate VEGFRs.
[0392] All members of the VEGF family stimulate cellular responses by binding to tyrosine kinase receptors (the VEGFRs) on the cell surface, causing them to dimerize and become activated through transphosphorylation, although to different sites,
times and extents. The VEGF receptors have an extracellular portion consisting of 7 immunoglobulin-like domains, a single transmembrane spanning region and an intracellular portion containing a split tyrosine-kinase domain. VEGF-A binds to VEGFR- 1 (Flt-1) and VEGFR-2 (KDR/Flk-1). VEGFR-2 appears to mediate almost all of the known cellular responses to VEGF. The function of VEGFR-1 is less well defined, although it is thought to modulate VEGFR-2 signaling. Another function of VEGFR-1 may be to act as a dummy/decoy receptor, sequestering VEGF from VEGFR-2 binding (this appears to be involved during vasculogenesis in the embryo). VEGF-C and VEGF-D, but not VEGF-A, are ligands for a third receptor (VEGFR-3), which mediates lymphangiogenesis.
[0393] Increased vascular permeability is one of the earliest manifestations of inflammation, resulting in extravasation of protein-rich plasma into the effected tissue. Acute vascular permeability allows the deposition of circulating plasma matrix proteins including fibrin and fibronectin (FN) which facilitate cell migration in the inflamed area. This process also provides an access point for immune cells and immunoglobulins to enter the tissue and fight foreign antigens (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012). Conversely, chronic vascular hyperpermeability is suggested to sustain the inflammatory response and retard resolution, further promoting the development of chronic inflammation (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012; Costa et al, Angiogenesis 10: 149-166, 2007). This type of vascular hyperpermeability underlies the pathogenesis of a large number of chronic disorders including rheumatoid arthritis (RA), psoriasis, ocular disease, cancer and chronic wounds (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012; Costa et al, Angiogenesis 10: 149-166, 2007).
[0394] VEGF is a potent vascular permeabilizing agent that is highly expressed during chronic inflammation (Nagy et al, Annu. Rev. Pathol. 2:251 -275, 2007). Low microenvironmental levels of VEGF are desired in order to maintain stable vascular integrity and promote endothelial cell survival through autocrine mechanisms (Lee et al, Cell 130:691 -703, 2007). Whereas elevated levels of VEGF induce vascular leakages by activating VEGF receptor 2 (VEGFR2) in endothelial cells (EC) leading to the opening of intercellular and/or intracellular pathways that facilitate plasma extravasation (Koch and Claesson-Welsh, Cold Spring Harb. Perspect. Med. 2:a006502, 2012). Moreover, VEGF may serve as a pro-inflammatory mediator as it can enhance T cell (Xia et al, Blood 102: 161 -168, 2003), and monocyte (Murakami et al, Blood 108: 1849-1856, 2006) migration as well as promote pro-inflammatory chemokines expression by EC including MCP-1 , and IL-8, leading to further immune recruitment. Cellular sources for VEGF during inflammation can include macrophages and mast cells; however it can also be
expressed by endothelial cells and acts in a paracrine and autocrine fashion. Thus, VEGF plays a major role in promoting chronic inflammation by inducing vascular permeability and contributing to immune cell recruitment.
[0395] Compositions comprising compounds Nos 1 -59 shown in Table 2 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of a VEGFR. Several embodiments relate to polymer conjugates of compounds 1 -59, optimized for topical applications while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications are provided in other embodiments.
[0396] In several embodiments, the warhead of the polymer conjugate is a small molecule disclosed in Table 2 targeting a VEGFR. There is also provided, in several embodiments, methods of treating an inflammatory bowel disease in a subject, the method comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Table 2 targeting a VEGFR. In some embodiments, the warhead of the LSE polymer conjugate is compound 1 of Table 2. In some embodiments, the LSE polymer conjugate is CT103.
[0397] Compositions comprising compounds shown in Tables 1-3 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of the mediator(s) of gastrointestinal conditions, such as, but not limited to, inflammatory bowel diseases, IBS or SIBO are disclosed herein. In several embodiments, compositions comprising compounds shown in Tablse 1 -3 are used as inhibitors, antagonists, and inverse agonists of JAK and/or STAT family proteins. In some embodiments, the warhead of the polymer conjugate is compound 1 of Table 3. In some embodiments, the LSE polymer conjugate is CT352.
[0398] Allergic inflammatory diseases are characterized by an immune response against a sensitizing agent, such as an allergen, resulting in the release of inflammatory mediators that recruit cells involved in inflammation in a subject, potentially leading to tissue damage and sometimes death. Methods of treating allergic inflammatory diseases of the gastrointestinal tract are provided herein. There is provided, in several embodiments, methods of treating an allergic inflammatory disease in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a JAK and/or STAT family protein.
[0399] There is also provided, in several embodiments, methods of treating the following conditions in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule
targeting a JAK and/or STAT family protein: ulcerative colitis; Crohn's disease; colorectal cancer; celiac disease; and intestinal fibrosis
[0400] There is provided, in several embodiments, LSE polymer conjugates wherein the warhead (e.g. , one or more pharmacologically active agents) is a selective inhibitor, antagonist, or inverse agonist of JAK1 . In some embodiments, the warhead of the LSE polymer conjugate is JNJ-54781532 (ASP015K, Peficitinb) . In other embodiments, the warhead of the LSE polymer conjugate is Upadacitinib (ABT-494). In several embodiments, the warhead of the LSE polymer conjugate is GSK2586184.
[0401] The proinflammatory lipid mediator leukotriene (LT)B4, which is involved in the recruitment and activation of inflammatory cells, has been implicated in the pathogenesis of inflammatory bowel diseases. The conversion of leukotriene A4 to leukotriene B4 is catalyzed by Leukotriene A4 hydrolase (LTA4H). Accordingly, there is provided, in several embodiments, LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of LTA4H. In one embodiment, the warhead of the LSE polymer conjugate is JNJ-26993135 (1 -[4-(benzothiazol-2-yloxy)-benzyl]- piperidine-4-carboxylic acid).
[0402] Colony-stimulating factor-1 receptor (CSF1 R; FMS) is a tyrosine kinase receptor that plays an essential role in promoting macrophage and dendritic cell differentiation, recruitment, activation, and proliferation. The differentiation of dendritic cells and macrophages and their migration to intestinal mucosa is a component of inflammatory bowel diseases, particularly Crohn's disease. Accordingly, there is provided, in several embodiments, LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of FMS. In several embodiments, the warhead of the LSE polymer conjugate is JNJ-40346527 (PRV-6527).
[0403] Acute and chronic inflammation of the bowel is caused by a number of diseases, and typically the epithelial cells on the surface of mucosal tissue have an induced state of hypoxia due to the presence of inflammation. The body's response to this hypoxic condition is to increase the presence of hypoxia inducible factor- 1 alpha (HIF-1 a) which drives the expression of downstream HIF-1 target genes, inter alia, erythropoietin. As such, HIF- 1 a is a mediator in the body's response to inflammation. The cellular concentration of HIF-1 a is regulated by prolyl hydroxylase enzymes that serve to destabilize HIF-1 a during periods of normoxia, resulting in the destruction of this protein. Inhibition of HIF-1 a prolyl hydroxylase thus leads to increased stabilization of H IF-1 a, in turn resulting in an upregulation of HIF-1 a that leads to a corresponding increased response to inflammation. In subjects suffering from one or more inflammatory epithelial diseases, treatment with one or more effective HIF- 1 a prolyl hydroxylase inhibitors can increase the level of the body's cellular inflammatory response. In addition, during
periods of low inflammation in the case of chronic diseases, HIF-1 a prolyl hydroxylase inhibitors can increase the amount of epithelial cell healing over that which the body would normally provide. As such, administration of one or more HIF-1 a prolyl hydroxylase inhibitors to a subject suffering from an inflammatory disease provides a method for curing, controlling, mediating, reducing, or otherwise affecting the severity of the condition. Accordingly, there is provided, in several embodiments, LSE polymer conjugates wherein the warhead is a HIF-1 a stabilizer. There is provided, in several embodiments, selective inhibitors, antagonists, and inverse agonists of HIF-1 a prolyl hydroxylase. In several embodiments, the warhead of the LSE polymer conjugate is JNJ5169 (AKB-5169). In several embodiments, the warhead of the LSE polymer conjugate is a compound having the formula:

[0404] wherein Z is phenyl substituted with from 1 to 5 halogens chosen from fluorine and chlorine;
[0405] R4 is C C4 linear alkyl or C3-C4 branched alkyl; or
[0406] a pharmaceutically acceptable salt thereof.
[0407] In several embodiments, the warhead of the LSE polymer conjugate is a compound having the formula:

[0408] wherein L is chosen from CH2 or S02;
[0409] R represents from 0 to 5 substitutions for hydrogen;
[0410] the index n is an integer from 0 to 5;
[0411] R1 and R2 are each independently chosen from:
[0412] i) hydrogen;
[0413] ii) substituted or unsubstituted C Cw linear, branched, or cyclic alkyl;
[0414] iii) substituted or unsubstituted C2-C10 linear, branched, or cyclic alkenyl;
[0415] iv) substituted or unsubstituted C2-C10 linear or branched alkynyl;
[0416] v) substituted or unsubstituted C6 or C10 aryl;
[0417] vi) substituted or unsubstituted C C9 heterocyclic;
[0418] vii) substituted or unsubstituted C1 -C9 heteroaryl; or
[0419] viii) R1 and R2 can be taken together to form a substituted or unsubstituted
[0420] heterocyclic or substituted or unsubstituted heteroaryl ring having from form 2 to 20 carbon atoms and from 1 to 7 heteroatoms; or a pharmaceutically acceptable salt thereof.
[0421] The a4 integrin α4β7 plays an essential role in lymphocyte migration throughout the gastrointestinal tract. It is expressed on most leukocytes, including B and T lymphocytes, where it mediates cell adhesion via binding to its ligand mucosal addressin cell adhesion molecule (MAdCAM). If left unchecked, integrin-mediated adhesion process can lead to chronic inflammation and autoimmune disease. Inhibitors of specific integrin-ligand interactions have been shown effective as anti-inflammatory agents for the treatment of various autoimmune diseases. Accordingly, there is provided, in several embodiments, LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of alpha-4-beta-7 integrin. In several embodiments, the warhead of the LSE polymer conjugate is PTG-100. In several embodiments, the warhead of the LSE polymer conjugate is a peptide dimer compound comprising two monomer subunits, wherein each monomer subunit comprises the amino acid sequence:
[0422] Pen-(N-Me-Arg)-Ser-Asp-Thr-Leu-Pen-Phe(4-tBu)-(P-homo-Glu)-(D-
Lys),
[0423] or a pharmaceutically acceptable salt thereof,
[0424] and wherein the two monomer subunits are linked by a linker moiety.
[0425] In one embodiment, the present invention includes a peptide dimer compound comprising two linked monomer subunits of Formula (I): Xaa1 -Xaa2-Xaa3-Xaa4-Xaa5-Xaa6-Xaa7-Xaa8-Xaa9-Xaa 1 °-Xaa11 -Xaa12-Xaa 13-Xaa14 (Formula (I)) or a pharmaceutically acceptable salt thereof,
[0426] wherein:
[0427] Xaa1 is absent, Ac, or any amino acid;
[0428] Xaa2 is absent, Ac, or any amino acid;
[0429] Xaa3 is absent, Ac, or any amino acid;
[0430] Xaa4 is any amino acid capable of forming a bond with Xaa10;
[0431] Xaa5 is selected from the group consisting of: N-Me-Arg, Arg, N-Me- Lys, Phe(4-guanidinoguanidino), Phe(4-carbomyl), Cit, Phe(4-NH2), N-Me-homoArg, homoArg, Tyr, Dap, Dab, Arg-Me-sym, Arg-Me-asym, Cav, and His;
[0432] Xaa6 is Ser, Gly, Thr or lie;
[0433] Xaa7 is Asp, Asp(OMe) or N-Me-Asp;
[0434] Xaa8 is selected from the group consisting of: Thr, Val, lie, Leu, homoLeu, Gin, Ser, Asp, Pro, Gly, His, Ala, Phe, Lys, Arg, Asn, Glu, Tyr, Trp, Met, NIe, and N-methyl amino acids, including N-Me-Thr;
[0435] Xaa9 is selected from the group consisting of: Gin, Ser, Asp, Pro, Gly, Ala, Phe, Glu, lie, Val, N-butyl Ala, N-pental Ala, N-hexyl Ala, cyclobutyl Ala, cyclopentylAla, Leu, NIe, Cba, homoLeu, Cpa, Aoc, and N-Me-Leu;
[0436] Xaa10 is any amino acid capable of forming a bond with Xaa4;
[0437] Xaa11 is absent or selected from the group consisting of: aromatic amino acids, substituted aromatic amino acids, and Tic;
[0438] Xaa12 is absent or selected from the group consisting of: aromatic amino acids, substituted aromatic amino acids, Glu, D-Glu, homoGlu, Asp, D-Asp, D- homoGlu, Gla, beta-homoGlu, Tic, Aic, Gin, Cit, Glu(OMe), Asn, D-His, Tic, Phe(3- COOH), D-Arg, Bip, D-Trp, Phe, D-Phe, D-Val, D-Thr, D-Tyr, D-Lys, D-lle, D-His, N-Me- Glu, N-Me-Asp, alpha-homoGlu, Biphenyl-Gly, Biphenyl-Ala, Homo-Phe, D-1-Nal, D-2- Nal, Thr, and Val, and corresponding D-amino acids and isosteres;
[0439] Xaa13 is absent or Pro or any amino acid; and
[0440] Xaa14 is selected from the group consisting of: any amino acid with an amine side chain, Lys, D-Lys, N-Me-Lys, D-N-Me-Lys, Orn, Dab, Dap, HomoLys, D-Dap, D-Dab, D-Orn, Cys, HomoCys, Pen, D-HomoCys, D-Cys, D-Pen, Asp, Glu, D-Asp, D-Glu and HomoSer, HomoGlu, D-homoGlu, N-Me-Glu, N-Me-Asp, N-Me-D-Glu, and N-Me-D- Asp;
wherein Xaa4 and Xaa10 are both Pen or Cys; wherein: Xaa5 is selected from the group consisting of Cit, Phe(4-carbomylamino), and N-Me-homoArg; Xaa8 is selected from the group consisting of Leu, homoLeu, Nle and Val; Xaa9 is selected from the group consisting of Cba, homoLeu, and Cpa; Xaa1 1 is selected from the group consisting of Tic, Phe(2-carbomyl), Phe(3-carbomyl), Phe(4-COOH), Phe(4-OMe), and Phe(4-tBu); Xaa12 is selected from the group consisting of Aic, Gin, Cit, Glu(OMe), D-His, Tic, Phe(3- COOH), D-Arg, Bip, D-Trp, Phe, D-Phe, D-Val, D-Thr, D-1 -Nal, D-2-Nal, Thr, Val; or Xaa13 is Pro; and wherein one or both monomer subunits of the peptide dimer compound comprises a bond between Xaa4 and Xaa10.
[0441] The interleukin-23 (IL-23) cytokine has been implicated as playing a crucial role in the pathogenesis of inflammatory bowel diseases. Studies in acute and chronic mouse models of IBD revealed a primary role of IL-23R and downstream effector cytokines in disease pathogenesis. IL-23R is expressed on various adaptive and innate immune cells including Th17 cells, γδ T cells, natural killer (NK) cells, dendritic cells, macrophages, and innate lymphoid cells, which are found abundantly in the intestine. At the intestine mucosal surface, the gene expression and protein levels of IL-23R are found to be elevated in IBD patients. Production of IL-23 is enriched in the intestine, where it is believed to play a key role in regulating the balance between tolerance and immunity through T-cell-dependent and T-cell-independent pathways of intestinal inflammation through effects on T-helper 1 (Th1 ) and Th17-associated cytokines, as well as restraining regulatory T-cell responses in the gut, favoring inflammation. In addition, polymorphisms in the IL-23 receptor (IL-23R) have been associated with susceptibility to IBDs, further establishing the role of the IL-23 pathway in intestinal homeostasis. Binding of IL-23 to IL-23R activates the JAK-STAT signaling. Accordingly, there is provided, in several embodiments, LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of lnterleukin-23 receptor. In several embodiments, the warhead of the LSE polymer conjugate is PTG-200. In several embodiments, the warhead of the LSE polymer conjugate comprises a peptide inhibitor of lnterleukin-23 receptor. In several embodiments, the warhead of the LSE polymer conjugate comprises the amino acid sequence of Formula II:
[0442] X1 -X2-X3-X4-X5-X6-X7-X8-X9-X10-X1 1 -X12-X13-X14-X15-X16-X17- X18-X19-X20 (II)
[0443] wherein
[0444] X1 is any amino acid or absent;
[0445] X2 is any amino acid or absent;
[0446] X3 is any amino acid or absent;
[0447] X4 is Cys, Pen, hCys, D-Pen, D-Cys, D-hCys, Met, Glu, Asp, Lys, Orn, Dap, Dab, D-Dap, D-Dab, D-Asp, D-Glu, D-Lys, Sec, 2-chloromethylbenzoic acid, mercapto-propanoic acid, mercapto-butyric acid, 2-chloro-acetic acid, 3-choro-propanoic acid, 4-chloro-butyric acid, 3-chloro-isobutyric acid, Abu, β-azido-Ala-OH, propargylglycine, 2-(3'-butenyl)glycine, 2-allylglycine, 2-(3'-butenyl)glycine, 2-(4'- pentenyl)glycine, 2-(5'-hexenyl)glycine, or absent;
[0448] X5 is any amino acid;
[0449] X6 is any amino acid;
[0450] X7 is Trp, Glu, Gly, lie, Asn, Pro, Arg, Thr or OctGly, or a corresponding α-methyl amino acid form of any of the foregoing;
[0451] X8 is any amino acid;
[0452] X9 is Cys, Pen, hCys, D-Pen, D-Cys, D-hCys, Glu, Lys, Orn, Dap, Dab, D-Dap, D-Dab, D-Asp, D-Glu, D-Lys, Asp, Leu, Val, Phe, Ser, Sec, Abu, β-azido- Ala-OH, propargylglycine, 2-2-allylglycine, 2-(3'-butenyl)glycine, 2-(4'-pentenyl)glycine, Ala, hCys, Met, MeCys, (D)Tyr or 2-(5'-hexenyl)glycine;
[0453] X10 is Tyr, Phe(4-OMe), 1 -Nal, 2-Nal, Aic, a-MePhe, Bip, (D)Cys, Cha, DMT, (D)Tyr, Glu, His, hPhe(3,4-dimethoxy), hTyr, N-Me-Tyr, Trp, Phe(4-CONH2), Phe(4-phenoxy), Thr, Tic, Tyr(3-tBu), Phe(4-tBu), Phe(4-CN), Phe(4-Br), Phe(4-NH2), Phe(4-F), Phe(3,5-F2), Phe(4-CH2C02H), Phe(penta-F), Phe(3,4-CI2), Phe(4-CF3), Bip, Cha, 4-PyridylAlanine, phTyr, OctGly, Phe(4-N3), Phe(4-Br), Phe[4-(2-aminoethoxy)], Phe, a Phe analog, or a Tyr analog, or a corresponding α-methyl amino acid form of any of the foregoing;
[0454] X1 1 is 2-Nal, 1 -Nal, 2,4-dimethylPhe, Bip, Phe(3,4-CI2), Phe (3.4-F2), Phe(4-C02H), phPhe(4-F), a-Me-Trp, 4-phenylcyclohexyl, Phe(4-CF3), Phe(3,4-OMe2), a-MePhe, phPhe, phTyr, phTrp, Nva(5-phenyl), Phe, His, hPhe, Tic, Tqa, Trp, Tyr, Phe(4-OMe), Phe(4-Me), Trp(2,5,7-tri-tert-Butyl), Phe(4-Oallyl), Tyr(3-tBu), Phe(4-tBu), Phe(4-guanidino, Phe(4-OBzl), Octgly, Glu(Bzl), 4-Phenylbenzylalanine, Phe[4-(2- aminoethoxy)], 5-Hydroxy-Trp, 6-Chloro-Trp, N-MeTrp, 1 ,2,3,4-tetrahydro-norharman, Phe(4-CONH2), Phe(3,4-Dimethoxy), Phe(2,3-CI2), Phe(2,3-F2), Phe(4-F), 4- phenylcyclohexylalanine or Bip, or a corresponding α-methyl amino acid form of any of the foregoing;
[0455] X12 is His, Phe, Arg, N-Me-His, Val, Cav, Cpa, Leu, Cit, hLeu, 3-Pal, t-butyl-Ala, a-MeLys, D-Ala, (D)Asn, (D)Asp, (D)Leu, (D)Phe, (D)Tyr, Aib, a-MeLeu, a-
MeOrn, β-Aib, β-Ala, phAla, phArg, phLeu, phVal, β-spiro-pip, Glu, hArg, lie, Lys, N- MeLeu, N-MeArg, Ogl, Orn, Pro, Gin, Ser, Thr, Tie, t-butyl-Gly, 4-amino-4-carboxy- tetrahydropyran (THP), Ache Acpc, Acbc, Acvc, Agp, Aib, a-DiethylGly, a-MeLys(Ac), a- MeOrn, a-MeSer, a-MeVal, Cha, Cit, Cpa, (D)Asn, Glu, hArg, or Lys, or a corresponding α-methyl amino acid form of any of the foregoing;
[0456] X13 is Thr, Sarc, Glu, Phe, Arg, Leu, Asn, Cit, Lys, Arg, Orn, Val, phAla, Lys(Ac), (D)Asn, (D)Leu, (D)Phe, (D)Thr, Ala, a-MeLeu, Aib, β-Ala, β-Glu, phLeu, phVal, β-spiro-pip, Cha, Chg, Asp, Dab, Dap, a-DiethylGly, hLeu, Asn, Ogl, Pro, Gin, Ser, β-spiro-pip, Thr, Tba, Tie or Aib, or a corresponding α-methyl amino acid form of any of the foregoing;
[0457] X14 is Phe, Tyr, Glu, Gly, His, Lys, Leu, Met, Asn, Lys(Ac), Dap(Ac), Asp, Pro, Gin, Arg, Ser, Thr, Tic or phPhe, or a corresponding α-methyl amino acid form of any of the foregoing;
[0458] X15 is Gly, Ser, Thr, Gin, Ala, (D)Ala, (D)Asn, (D)Asp, (D)Leu, (D)Phe, (D)Thr, Aea, Asp, Asn, Glu, Phe, Gly, Lys, Leu, Pro, Arg, β-Ala, or Sarc, or a corresponding α-methyl amino acid form of any of the foregoing;
[0459] X16 is any amino acid or absent;
[0460] X17 is any amino acid or absent;
[0461] X18 is any amino acid or absent;
[0462] X19 is any amino acid or absent; and
[0463] X20 is any amino acid or absent,
[0464] wherein the peptide inhibitor is cyclized via a bond between X4 and
X9.
[0465] In several embodiments, the warhead of the LSE polymer conjugate comprises the amino acid sequence of Formula III:
[0466] X1 -X2-X3-X4-X5-X6-X7-X8-X9-X10-X1 1 -X12-X13-X14-X15-X16-X17-
X18-X19-X20 (III)
[0467] wherein
[0468] X1 is any amino acid or absent;
[0469] X2 is any amino acid or absent;
[0470] X3 is any amino acid or absent;
[0471] X4 is Pen, Cys or homo-Cys;
[0472] X5 is any amino acid;
[0473] X6 is any amino acid;
[0474] X7 is Trp, Bip, Gin, His, Glu(Bzl), 4-Phenylbenzylalanine, Tic, Phe[4- (2-aminoethoxy)], Phe(3,4-CI2), Phe(4-OMe), 5-Hydroxy-Trp, 6-Chloro-Trp, N-MeTrp, a- Me-Trp, 1 ,2,3,4-tetrahydro-norharman, Phe(4-C02H), Phe(4-CONH2), Phe(3,4-
Dimethoxy) , Phe(4-CF3) , Phe(4-tBu), ββ-diPheAla, Glu, Gly, lie, Asn, Pro, Arg, Thr or Octgly, or a corresponding a-methyl amino acid form of any of the foregoing;
[0475] X8 is any amino acid;
[0476] X9 is Pen, Cys or hCys;
[0477] X10 is 1 -Nal, 2-Nal, Aic, Bip, (D)Cys, Cha, DMT, (D)Tyr, Glu, Phe, His, Trp, Thr, Tic, Tyr, 4-pyridylAla, Octgly, a Phe analog or a Tyr analog, or a corresponding α-methyl amino acid form of any of the foregoing;
[0478] X1 1 is 2-Nal, 1 -Nal, 2,4-dimethylPhe, Bip, Phe(3,4-CI2) , Phe (3,4-F2), Phe(4-C02H), phPhe(4-F), a-Me-Trp, 4-phenylcyclohexyl, Phe(4-CF3) , a-MePhe, phPhe, phTyr, phTrp, Nva(5-phenyl), Phe, His, hPhe, Tic, Tqa, Trp, Tyr, Phe(4-OMe), Phe(4- Me), Trp(2,5,7-tri-tert-Butyl) , Phe(4-Oallyl) , Tyr(3-tBu) , Phe(4-tBu), Phe(4-guanidino, Phe(4-OBzl), Octgly, Glu(Bzl), 4-Phenylbenzylalanine, Phe[4-(2-aminoethoxy)], 5- Hydroxy-Trp, 6-Chloro-Trp, N-MeTrp, 1 ,2,3,4-tetrahydro-norharman, Phe(4-CONH2) , Phe(3,4-OMe2) Phe(2,3-CI2), Phe(2,3-F2) , Phe(4-F), 4-phenylcyclohexylalanine or Bip, or a corresponding α-methyl amino acid form of any of the foregoing;
[0479] X12 is a-MeLys, a-MeOrn, a-MeLeu, a-MeVal, 4-amino-4-carboxy- tetrahydropyran, Ache Acpc, Acbc, Acvc, MeLeu, Aib, (D)Ala, (D)Asn, (D)Leu, (D)Asp, (D)Phe, (D)Thr, 3-Pal, Aib, β-Ala, phGlu, phAla, phLeu, phVal, β-spiro-pip, Cha, Chg , Asp, Dab, Dap, a-diethylGly, Glu, Phe, hLeu, hArg, hLeu, lie, Lys, Leu, Asn, N-MeLeu, N-MeArg, Ogl, Orn, Pro, Gin, Arg, Ser, Thr or Tie, or a corresponding α-methyl amino acid form of any of the foregoing;
[0480] X13 is Lys(Ac), (D)Asn, (D)Leu, (D)Thr, (D)Phe, Ala, Aib, a-MeLeu, β- Ala, phGlu, phAla, phLeu, phVal, β-spiro-pip, Cha, Chg, Asp, Lys, Arg, Orn, Dab, Dap, a- diethylGly, Glu, Phe, hLeu, Lys, Leu, Asn, Ogl, Pro, Gin, Asp, Arg, Ser, spiro-pip, Thr, Tba, Tic, Val or Tyr, or a corresponding α-methyl amino acid form of any of the foregoing;
[0481] X14 is Asn, Glu, Phe, Gly, His, Lys, Leu, Met, Asn, Pro, Gin, Arg, Ser, Thr, Tic or Tyr, Lys(Ac) , Orn or a corresponding α-methyl amino acid form of any of the foregoing;
[0482] X1 5 is Gly, (D)Ala, (D)Asn, (D)Asp, Asn, (D)Leu, (D)Phe, (D)Thr, Ala, Asn, Ser, AEA, Asp, Glu, Phe, Gly, Lys, Leu, Pro, Gin, Arg or Ser, β-Ala, Arg or a corresponding α-methyl amino acid form of any of the foregoing;
[0483] X16 is absent, Gly, Ala, Asp, Ser, Pro, Asn or Thr, or a corresponding α-methyl amino acid form of any of the foregoing;
[0484] X17 is absent, Glu, Ser, Gly or Gin, or a corresponding a-methyl amino acid form of any of the foregoing;
[0485] X18 is absent or any amino acid;
[0486] X19 is absent or any amino acid; and
[0487] X20 is absent or any amino acid,
[0488] wherein the peptide inhibitor comprises a disulfide bond between X4 and X9.
[0489] Inflammatory bowel diseases are characterized by an aberrant immune response occurring in a genetically predisposed host in response to microbes and/or microbial compounds found in the gut microbiota. Of the bacteria that may play a role in the pathogenesis of these diseases, a pathotype of E. coli, called "AIEC" for "adherent-invasive Escherichia coli", has been strongly implicated. AIEC are able to adhere to the intestinal epithelium and colonize gut mucosa where they participate to IBD onset. AIEC's adhesion to mucosal epithelial cells is mediated by proteinaceous, rod-like organelles that are called type-1 fimbriae. Type-1 fimbriae carry an adhesin at the edge of a flexible tip fibrillum. This adhesin, FimH, is a lectin having a strong affinity for highly mannosylated glycoproteins. Via FimH, AIEC bacteria adhere specifically to the carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6), a mannosylated glycoprotein which is abnormally expressed in the ileal mucosa of 35% of CD patients. Overexpression of these CEACAM6 molecules in CD patients, acting as receptors for E. coli adhesion in the gut, favors ileal and colonic AIEC invasion and their intracellular survival and replication within the mucosal tissues, thereby amplifying immune responses in IBD patients. Moreover, it has been shown that point and pathoadaptative mutations in the FimH adhesin confer significantly higher ability to adhere to CEACAM-expressing intestinal epithelial cells, thus leading to an abnormal colonization of the gut and to the development of chronic inflammation in a host. A promising strategy to prevent and treat IBDs would be to inhibit the adhesion of AIECs to the epithelial cells of the digestive tract mucosa. Accordingly, there is provided, in several embodiments, LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of FimH. In several embodiments, the warhead of the LSE polymer conjugate is EB8018. In several embodiments, the warhead of the LSE polymer conjugate is compound of the following formula:

[0492] Y represents a single bond, CH2, O, NR3, S, preferably a single bond, CH2, NR3,
[0493] S,
[0494] A represents O, NH or S, preferably O or S,
[0495] X represents H and X' represents OH or X and X taken together with the carbon atom bearing them form a CO group,
[0496] R2 represents H, a linear or branched (C1-C6)-alkyl or CF3,
[0497] R3 represents H, a d-C6 alkyl, a CO-(C C6)-alkyl, CF3 or COCF3, preferably H, CH3, COCH3, CF3 or OCF3, and
[0498] R represents: a (d-CeJ-alkyl, a (C2-C6)-alkenyl, a (C2-C6)-alkynyl, a (C3-C10)- cycloalkyi, a (C5-C10)-cycloalkenyl, a heterocycloalkyi, a heterocycloalkenyl, an arylan alkyl aryl, CF3, adamantyl, ORa, or NRbRc,
[0499] wherein Ra represents H, a (C1-C6)-alkyl, a (C2-C6)-alkenyl, a (C2-C6)- alkynyl, a (C3-C6)-cycloalkyl, a (C3-C6)-cycloalkenyl, a heterocycloalkyi, a heterocycloalkenyl, an aryl, a alkylaryl, a CHO, a CO-(C1-C6)-alkyl, or CO-aryl, a C02H, a C02-(Ci-Ce)-alkyl, or a CONH-( C1-C6)-alkyl,
[0500] and wherein Rb and Rc represent independently from each other any of the groups defined for Ra, Rb representing in particular H, said (C1-C6)-alkyl, (C2-C6)- alkenyl, (C2-C6)-alkynyl, (C3-C10)-cycloalkyl, (C5-C10)- cycloalkenyl, heterocycloalkyi, heterocycloalkenyl, CO-(C C6)-alkyl, C02-(C C6)-alkyl, CONH-(C1-C6)-alkyl, aryl, alkylaryl, CO-aryl and CO-alkylaryl being optionally substituted by one or more, preferably 1 to 4, more preferably 1 or 2 substituent(s) R', each independently selected from:
- a (C1-C6)-alkyl optionally substituted by one or more, preferably one to three substituents selected from a halogen, NH2, OH, CF3, a C C6 alkyl preferably substituted by a carbohydrate, CH2S03T, CH2COOT or N(Rg)3T, wherein Rg represents independently H, a (C1-C6)-alkyl (preferably H or CH3), T represents a monovalent cation such as a mineral monovalent cation, in particular an alkaline cation preferably selected from Li+, Na+, K+ and even more preferable Na+ and wherein T is a monovalent anion, such as a halogenide, in particular chloride, bromide or iodide, preferably chloride,
- a (C2-C6)-alkenyl,
- a (C2-C6)-alkynyl,
- a (C3-C10)-cycloalkyl,
- a (C5-C10)-cycloalkenyl,
- a heterocycloalkyi,
- a heterocycloalkenyl,
- an aryl, optionally substituted by one or more, preferably one to three substituents selected from a halogen, NH2, OH, CF3, a C C6 alkyl preferably substituted by a carbohydrate, CH2S03T, CH2COOT or N(Rg)3T, wherein Rg, T and T' are as defined above,
- an alkyl aryl optionally substituted by one or more, preferably one to three substituents selected from a halogen, NH2, OH, CF3, a C^Ce alkyl preferably substituted by a carbohydrate, CH2S03T, CH2COOT or N(Rg)3T', wherein Rg, T and T' are as defined above,
- a NH-alkyl aryl optionally substituted by one or more, preferably one to three substituents selected from a halogen, NH2, OH, CF3) a d-C6 alkyl preferably substituted by a carbohydrate, CH2S03T, CH2COOT or N(Rg)3T, wherein Rg, T and T are as defined above,
- a CHO,
- a CO-(C1-C6)-alkyl optionally substituted by a halogen such as fluorine or a carbohydrate,
- a CO-aryl optionally substituted by one or more, preferably one to three substituents selected from a halogen, NH2, OH, CF3, a C Ce alkyl preferably substituted by a carbohydrate, CH2S03T, CH2COOT or N(Rg)3T, wherein Rg, T and T are as defined above,
- a C02H,
- a C02-(C C6)-alkyl,
- a CONH-CCrCf -alkyl,
- a CONH-aryl or NHCO-aryl optionally substituted by one or more, preferably one to three substituents selected from a halogen, NH2, OH, CF3, a C C6 alkyl preferably substituted by a carbohydrate, CH2S03T, CH2COOT or N(Rg)3T', wherein Rg, T and T are as defined above,
- a halogen,
- CF3,
- ORd, wherein Rd represents: H, a (C1-C6)-alkyl, a (C3-C10)-cycloalkyl, CO^-Ce)- alkyl, or CO-aryl optionally substituted by one or more, preferably one to three substituents selected from a halogen, CF3, a C C6 alkyi preferably substituted by a carbohydrate,
- NReRf, wherein Re and Rf represent independently from each other: H, a (C C6)-alkyl, a (C3-C10)-cycloalkyl, CO-(C C6)-alkyl, or CO-aryl optionally substituted by one or more, preferably one to three substituents selected from a halogen, CF3, a C C6 alkyl preferably substituted by a carbohydrate,
- NHRb, wherein Rb is as defined above,
- N02,
- CN, and
- CH2SO3T, CH2COOT or N(Rg)3T\ wherein Rg, T and T' are as defined above, or a pharmaceutically acceptable salt or solvate thereof.
[0501] Phosphodiesterase 4 (PDE4), which catalyzes the breakdown of cAMP in inflammatory cells and is a mediator for the inflammatory cascades implicated in the pathogenesis of inflammatory bowel diseases. Accordingly, there is provided, in several embodiments, LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of PDE4. In several embodiments, the warhead of the LSE polymer conjugate is Tetilomast (OPC-6535).
[0502] There is provided, in several embodiments, LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of cytokines interleukin-12 (IL-12) and/or interleukin-23 (IL-23), which can mediate the progression of inflammatory bowel diseases. In one embodiment, the warhead of the LSE polymer conjugate is Apilimod (STA-5326).
[0503] There is provided, in several embodiments, LSE polymer conjugates wherein the warhead is an inhibitor, antagonist, or inverse agonist of a P38 mitogen- activated protein kinase. In several embodiments, the warhead of the LSE polymer conjugate is Doramapimod (BIRB 796). In other embodiments, the warhead of the LSE polymer conjugate is Semapimod (CNI-1493).
[0504] As a result of microbial symbiosis and immunity, alterations in enteral bacteria may contribute to inflammatory gut diseases, and IBD, IBS and SIBO patients may benefit from antibiotic therapy. In several embodiments, the warhead of the LSE polymer conjugate comprises an antibiotic selected from sulfonamides (e.g., 4-amino-N- (5-methyl-3-isoxazolyl)benzenesulfonamide); vancomycin; amoxicillin; tetracyclines; clarithromycin; clindamycin; a member of the cephlosporin antibiotic family (e.g., cefaclor, cefadroxil, cefixime, cefprozil, ceftriaxone, cefuroxime, cephalexin, loracarbef, and the like); a member of the penicillin family of antibiotics (e.g., ampicillin, amoxicillin/clavulanate, bacampicillin, cloxicillin, penicillin VK, and the like); a member of the fluoroquinolone family of antibiotics (e.g., ciprofloxacin, grepafloxacin, levofloxacin, lomefloxacin, norfloxacin, ofloxacin, sparfloxacin, trovafloxacin, and the like); or a member of the macrolide antibiotic family (e.g. azithromycin, erythromycin, and the like). Combinations of active agents may be used in some embodiments. In several embodiments, the warhead of the LSE polymer conjugate comprises a non-systemic antibiotic that is minimally absorbed and has high local concentrations in the Gl tract after oral administration. Accordingly, in some embodiments, the LSE polymer conjugate
comprises rifaximin. Antibiotics delivered (e.g., orally) according to several embodiments described herein may be particularly useful to treat the small intestine (for example to treat SIBO or small intestinal bacterial overgrowth). Such delivery compositions are able to, in some embodiments, deliver localized doses of antibiotics (or other antimicrobials) to the intestinal tract without significant exposure to other tissue which, advantageously, limits the side effects (such as dysbiosis or the destruction of good bacteria) that is caused by some antibiotics/antimicrobials. Also, in some embodiments, localized and precision-based treatment permits lower doses of the antibiotics/antimicrobials to be ingested, which in some embodiments may reduce or avoid toxicity, adverse immune effects, tolerance, destruction of probiotics, etc. Anti-fungals and anti-parasitic agents may also be delivered (orally, injected, topical, or by other means) according to the reduced systemic exposure embodiments described herein. In some embodiments, systemic exposure and/or exposure to a non-target tissue of antimicrobials (antifungals, antibacterials , antivirals, etc.) is reduced by at least 25%, 50%, 75% or more using the embodiments described herein (e.g., LSE polymer conjugates) as compared with administration of the same antimicrobial without the polymer conjugates described herein. In one embodiment, the use of the compositions described herein results in a reduced negative impact on the natural healthy microbiome of a subject because of reduced exposure to non-target tissue.
[0505] Oral delivery is contemplated in several embodiments suitable for gastrointestinal disorders, and thus several embodiments relate to polymer conjugates of compounds in Tables 1-3, optimized for oral delivery to treat the gastrointestinal system while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Topical applications are provided in other embodiments.
[0506] In several embodiments, the warhead of the polymer conjugate is a small molecule disclosed in Table 3 targeting mediator(s) of gastrointestinal conditions. There is also provided, in several embodiments, methods of treating an inflammatory bowel disease in a subject, the method comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Table 3 targeting mediator(s) of gastrointestinal conditions. In several embodiments, JAK and/or STAT family proteins are mediator(s) of gastrointestinal conditions. In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting a JAK and/or STAT family protein. In some embodiments, the warhead of the polymer conjugate is compound 1 of Table 3. In some embodiments, the LSE polymer conjugate is CT352. In addition to, or instead of IBD, IBS and SIBO may also be treated.
Polymer Conjugates Targeting Mediators of Dermatological Conditions
[0507] Compositions comprising an indolocarbazole compound are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of a VEGFR, c- Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Several embodiments relate to polymer conjugates of an indolocarbazole compound, optimized for topical applications while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications are provided in other embodiments.
[0508] In several embodiments, the warhead of the polymer conjugate is an indolocarbazole compound or derivative thereof. There is also provided, in several embodiments, methods of treating an inflammatory skin disease in a subject, the method comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is an indolocarbazole compound. In some embodiments, the warhead of the LSE polymer conjugate is a derivative of K252a. In some embodiments, the LSE polymer conjugate is SNA-125. In some embodiments, the LSE polymer conjugate is SNA-120. In some embodiments, the LSE polymer conjugate is SNA-125.
[0509] In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting mediator(s) of dermatological conditions. There is also provided, in several embodiments, methods of treating an inflammatory skin disease in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting mediator(s) of dermatological conditions. In some emboidments, the dermatological condition is an inflammatory skin disease. In several embodiments, JAK and/or STAT family proteins are mediator(s) of dermatological conditions. In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule a targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein .
[0510] There is provided, in several embodiments, methods of treating dermatological conditions in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting mediator(s) of dermatological conditions. In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
[0511] In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting a JAK and/or STAT family protein. There is also provided, in several embodiments, methods of treating an inflammatory skin disease in a subject, the method comprising administering to the subject an effective amount of a
polymer conjugate, wherein the warhead is a small molecule targeting a JAK and/or STAT family protein.
[0512] The JAK kinase family is a cytoplasmic protein kinase family comprising the members JAK1 , JAK2, JAK3 and TYK2. Various studies suggest that ligand binding to a receptor leads to receptor dimerization or oligomerization, which leads to JAK recruitment and activation either through autophosphorylation or phosphorylation by other JAK kinases or by other tyrosine kinases, which in turn leads to tyrosine phosphorylation of the receptors as well as downstream substrates of JAK. Growth factor or cytokine receptors that recruit JAK kinases include the interferon receptors, interleukin receptors (receptors for the cytokines IL-2 to IL-7, IL-9 to IL-13, IL- 15, IL-23), various hormone receptors (erythropoietin (Epo) receptor, the thrombopoietin (Tpo) receptor, the leptin receptor, the insulin receptor, the prolactin (PRL) receptor, the Granulocyte Colony-Stimulating Factor (G-CSF) receptor and the growth hormone receptor), receptor protein tyrosine kinases (such as EGFR and PDGFR) , and receptors for other growth factors such as leukemia inhibitory factor (LIF) , Oncastatin M (OSM) , IFNa/β/γ, Granulocyte-macrophage colony-stimulating factor (GM-CSF), Ciliary neurotrophic factor (CNTF), cardiotrophin- 1 (CT-I) (See, Rane, S.G. and Reddy E.P. , Oncogene 2000 19, 5662- 5679).
[0513] Many autoimmune diseases and disease associated with chronic inflammation, as well as acute responses, have been linked to excessive or unregulated production or activity of one or more cytokines, the signaling of which depend on JAK kinases. Such diseases include rheumatoid arthritis (RA) such as moderate to severe RA, systemic lupus erythematosus (SLE), multiple sclerosis (MS) , Crohn's disease such as moderate to severe Crohn's disease, psoriasis such as moderate to severe chronic plaque psoriasis, ulcerative colitis such as moderate to severe ulcerative colitis, ankylosing spondilytis (AS) , psoriatic arthritis, Juvenile Idiopathic Arthritis (JIA) such as moderate to severe polyarticular JIA, systemic lupus erythematosus (SLE), diabetic nephropathy, dry eye syndrome, Sjogren's Syndrome, alopecia areata, vitiligo, or atopic dermatitis.
[0514] Phosphorylated receptors serve as docking sites for other SH-2 domain containing signaling molecules that interact with JAKs such as the STAT family of transcription factors, Src family of kinases, MAP kinases PB kinase and protein tyrosine phosphatases (Rane S.G . and Reddy E.P. , Oncogene 2000 19, 5662-5679) . The family of latent cytoplasmic transcription factors, STATS, are the most well characterized downstream substrates for JAKs. The STAT proteins bind to phosphorylated cytokine receptors through their SH2 domains to become phosphorylated by JAKs, which event leads to their dimerization and release and
eventual translocation to the nucleus where they activate gene transcription. The various members of STAT which have been identified thus far, are STAT1 , STAT2, STAT3, STAT4, STAT5 (including STAT5a and STAT5b) and STAT6.
[0515] Signal transducer and activator of transcription 3 (STAT3), a member of the STAT protein family, is a transcription factor that regulates the expression of a variety of genes involved in many cellular processes such as cell growth, apoptosis, cell motility, and cytokine production. In response to cytokines and growth factors, STAT3 is activated by JAK kinases and translocates to the nucleus to act as a transcriptional activator. Studies have demonstrated that STAT3 plays a role in various immune disorders including the pathogenesis of inflammatory bowel disease (see, e.g. , Sugimoto, World J. Gastroenterol. , 14:51 10-51 14, (2008)) .
[0516] In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting c-Src. There is also provided, in several embodiments, methods of treating an inflammatory skin disease in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting c-Src.
[0517] The c-Src kinase is the most widely studied member of the largest family of nonreceptor protein tyrosine kinases, known as the Src family kinases (SFKs) . Other SFK members include Lyn, Fyn, Lck, Hck, Fgr, BIk, Yrk, and Yes. The Src kinases can be grouped into two sub-categories, those that are ubiquitously expressed (Src, Fyn, and Yes) , and those which are found primarily in hematopoietic cells (Lyn, Lck, Hck, BIk, Fgr). (Benati, D. Src Family Kinases as Potential Therapeutic Targets for Malignancies and Immunological Disorders. Current Medicinal Chemistry. 2008; 15: 1 154-1 165) . SFKs are key messengers in many cellular pathways, including those involved in regulating proliferation, differentiation, survival, motility, and angiogenesis. The activity of SFKs is highly regulated intramolecularly by interactions between the SH2 and SH3 domains and intermolecularly by association with cytoplasmic molecules. This latter activation may be mediated by focal adhesion kinase (FAK) or its molecular partner Crk- associated substrate (CAS), which plays a prominent role in integrin signaling, and by ligand activation of cell surface receptors, e.g. epidermal growth factor receptor (EGFR). These interactions disrupt intramolecular interactions within Src, leading to an open conformation that enables the protein to interact with potential substrates and downstream signaling molecules. Src can also be activated by dephosphorylation of tyrosine residue Y530. In some embodiments, maximal Src activation requires the autophosphorylation of tyrosine residue Y419 (in the human protein) present within the catalytic domain. Elevated Src activity may be caused by increased transcription or by deregulation due to overexpression of upstream growth factor receptors such as EGFR,
HER2, platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor, ephrins, integrin, or FAK.
[0518] Compositions comprising compounds Nos 1 -71 shown in Table 1 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of c-Src. Several embodiments relate to polymer conjugates of compounds 1 -71 , optimized for topical applications while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications are provided in other embodiments.
[0519] In several embodiments, the warhead of the polymer conjugate is a small molecule disclosed in Table 1 targeting c-Src. There is also provided, in several embodiments, methods of treating an inflammatory skin disease in a subject, the method comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Table 1 targeting c-Src. In some embodiments, the warhead of the LSE polymer conjugate is compound 1 of Table 1 . In some embodiments, the LSE polymer conjugate is CT101 .
[0520] In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting a vascular endothelial growth factor receptor (VEGFR).
[0521] Angiogenesis, the process of sprouting new blood vessels from existing vasculature, and arteriogenesis, the remodeling of small vessels into larger conduit vessels, are both physiologically important aspects of vascular growth in adult tissues. These processes of vascular growth are involved, in some cases, for beneficial processes such as tissue repair, wound healing, recovery from tissue ischemia and menstrual cycling. They are also involved, in some cases, for the development of pathological conditions such as the growth of neoplasias, diabetic retinopathy, rheumatoid arthritis, psoriasis, certain forms of macular degeneration, and certain inflammatory pathologies. The inhibition of vascular growth in these contexts has also shown beneficial effects in preclinical animal models. For example, inhibition of angiogenesis by blocking vascular endothelial growth factor or its receptor has resulted in inhibition of tumor growth and in retinopathy. Also, the development of pathological pannus tissue in rheumatoid arthritis involves angiogenesis and might be blocked by inhibitors of angiogenesis. Certain diseases are known to be associated with deregulated angiogenesis, for example ocular neovascularization, such as retinopathies (including diabetic retinopathy), age-related macular degeneration, psoriasis, hemangioblastoma, hemangioma, arteriosclerosis, inflammatory disease, such as a rheumatoid or rheumatic inflammatory disease, especially arthritis (including rheumatoid arthritis), or other chronic inflammatory disorders, such as chronic asthma, arterial or post- transplantational
atherosclerosis, endometriosis, and neoplastic diseases, for example so-called solid tumors and liquid tumors (such as leukemias).
[0522] Vascular endothelial growth factor (VEGF) is a signaling protein involved in both vasculogenesis (the de novo formation of the embryonic circulatory system) and angiogenesis (the growth of blood vessels from pre-existing vasculature). As its name implies, VEGF activity has been mostly studied on cells of the vascular endothelium, although it does have effects on a number of other cell types (e.g., stimulation monocyte/macrophage migration, neurons, cancer cells, kidney epithelial cells, keratinocytes). In vitro, VEGF has been shown to stimulate endothelial cell mitogenesis and cell migration. VEGF is also a vasodilator and increases microvascular permeability and was originally referred to as vascular permeability factor.
[0523] The broad term "VEGF" covers a number of proteins from two families, that result from alternate splicing of mRNA from a single, 8 exon, VEGF gene. The two different familes are referred to according to their terminal exon (exon 8) splice site— the proximal splice site (denoted VEGFxxx) or distal splice site (VEGFxxxb). In addition, alternate splicing of exon 6 and 7 alters their heparin binding affinity, and amino acid number (in humans: VEGF121 , VEGF121 b, VEGF145, VEGF165, VEGF165b, VEGF189, VEGF206; the rodent orthologs of these proteins contain one fewer amino acid). These domains have functional consequences for the VEGF splice variants as the terminal (exon 8) splice site determines whether the proteins are pro-angiogenic (proximal splice site, expressed during angiogenesis) or anti-angiogenic (distal splice site, expressed in normal tissues). In addition, inclusion or exclusion of exons 6 and 7 mediate interactions with heparan sulfate proteoglycans (HSPGs) and neuropilin co- receptors on the cell surface, enhancing their ability to bind and activate VEGFRs.
[0524] All members of the VEGF family stimulate cellular responses by binding to tyrosine kinase receptors (the VEGFRs) on the cell surface, causing them to dimerize and become activated through transphosphorylation, although to different sites, times and extents. The VEGF receptors have an extracellular portion consisting of 7 immunoglobulin-like domains, a single transmembrane spanning region and an intracellular portion containing a split tyrosine-kinase domain. VEGF-A binds to VEGFR- 1 (Flt-1) and VEGFR-2 (KDR/Flk-1). VEGFR-2 appears to mediate almost all of the known cellular responses to VEGF. The function of VEGFR-1 is less well defined, although it is thought to modulate VEGFR-2 signaling. Another function of VEGFR-1 may be to act as a dummy/decoy receptor, sequestering VEGF from VEGFR-2 binding (this appears to be involved during vasculogenesis in the embryo). VEGF-C and VEGF-D, but not VEGF-A, are ligands for a third receptor (VEGFR-3), which mediates lymphangiogenesis.
[0525] Increased vascular permeability is one of the earliest manifestations of inflammation, resulting in extravasation of protein-rich plasma into the effected tissue. Acute vascular permeability allows the deposition of circulating plasma matrix proteins including fibrin and fibronectin (FN) which facilitate cell migration in the inflamed area. This process also provides an access point for immune cells and immunoglobulins to enter the tissue and fight foreign antigens (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012). Conversely, chronic vascular hyperpermeability is suggested to sustain the inflammatory response and retard resolution, further promoting the development of chronic inflammation (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012; Costa et al, Angiogenesis 10: 149- 166, 2007). This type of vascular hyperpermeability underlies the pathogenesis of a large number of chronic disorders including rheumatoid arthritis (RA) , psoriasis, ocular disease, cancer and chronic wounds (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012; Costa et al, Angiogenesis 10: 149- 166, 2007).
[0526] VEGF is a potent vascular permeabilizing agent that is highly expressed during chronic inflammation (Nagy et al, Annu. Rev. Pathol. 2:251 -275, 2007) . Low microenvironmental levels of VEGF are desired in order to maintain stable vascular integrity and promote endothelial cell survival through autocrine mechanisms (Lee et al, Cell 130:691 -703, 2007). Whereas elevated levels of VEGF induce vascular leakages by activating VEGF receptor 2 (VEGFR2) in endothelial cells (EC) leading to the opening of intercellular and/or intracellular pathways that facilitate plasma extravasation (Koch and Claesson-Welsh, Cold Spring Harb. Perspect. Med. 2:a006502, 2012) . Moreover, VEGF may serve as a pro-inflammatory mediator as it can enhance T cell (Xia et al, Blood 102: 161 -168, 2003) , and monocyte (Murakami et al, Blood 108: 1849-1856, 2006) migration as well as promote pro-inflammatory chemokines expression by EC including MCP-1 , and IL-8, leading to further immune recruitment. Cellular sources for VEGF during inflammation can include macrophages and mast cells; however it can also be expressed by endothelial cells and acts in a paracrine and autocrine fashion. Thus, VEGF plays a major role in promoting chronic inflammation by inducing vascular permeability and contributing to immune cell recruitment.
[0527] Compositions comprising compounds Nos 1 -59 shown in Table 2 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of a VEGFR. Several embodiments relate to polymer conjugates of compounds 1 -59, optimized for topical applications while also minimizing side-effects caused by exposure at non-target sites (e.g. , systemic absorption) . Non-topical applications are provided in other embodiments.
[0528] In several embodiments, the warhead of the polymer conjugate is a small molecule disclosed in Table 2 targeting a VEGFR. There is also provided, in several embodiments, methods of treating an inflammatory skin disease in a subject, the method comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Table 2 targeting a VEGFR. In some embodiments, the warhead of the LSE polymer conjugate is compound 1 of Table 2. In some embodiments, the LSE polymer conjugate is CT103.
[0529] Compositions comprising compounds shown in Tables 1-3 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of the mediator(s) of dermatological conditions, such as, but not limited to, inflammatory skin diseases, bullous dises, or skin neoplasias are disclosed herein. In several embodiments, compositions comprising compounds shown in Table 3 are used as inhibitors, antagonists, and inverse agonists of JAK and/or STAT family proteins. In some embodiments, the warhead of the polymer conjugate is compound 1 of Table 3. In some embodiments, the LSE polymer conjugate is CT352.
[0530] Atopic dermatitis (AD) is a chronically relapsing inflammatory skin disease with a dramatically increasing incidence over the last decades. Clinically AD is characterized by highly pruritic often excoriated plaques and papules that show a chronic relapsing course. The diagnosis of AD is mostly based on major and minor clinical findings. See Hanifin J. M., Arch Dermatol: 135, 1551 (1999). Histopathology reveals spongiosis, hyper and focal parakeratosis in acute lesions, whereas marked epidermal hyperplasia with hyper and parakeratosis, acanthosis/hypergranulosis and perivascular infiltration of the dermis with lymphocytes and abundant mast cells are the hallmarks of chronic lesions.
[0531] Psoriasis is characterized by frequent episodes of redness, itching, and thick, dry, silvery scales on the skin. Psoriasis comprises lesions that can involve primary and secondary alterations in epidermal proliferation, inflammatory responses of the skin, and an expression of regulatory molecules such as lymphokines and inflammatory factors. Psoriatic skin is morphologically characterized by an increased turnover of epidermal cells, thickened epidermis, abnormal keratinization, inflammatory cell infiltrates into the epidermis and polymorphonuclear leukocyte and lymphocyte infiltration into the epidermis layer. Psoriasis is often associated with other inflammatory disorders, for example arthritis, including rheumatoid arthritis, inflammatory bowel disease (IBD), and Crohn's disease.
[0532] In one embodiment, active agents useful for stimulating hair follicles (for hair growth) are provided as oral applications or topical applications for the scalp. Hair removal agents and ant-acne agents are provided in other embodiments. Hair
growth, hair removal and anti-acne therapies can all involve active agents that, if exposed to the non-target site (e.g., systemic circulation and/or lymphatic system) for long periods, result in toxicity or undesired side effects. Thus, the reduced exposure compositions described herein provides benefits for these applications as well.
[0533] In one embodiment, active agents useful for stimulating hair follicles (for hair growth) are provided as oral applications or topical applications for the scalp. Hair removal agents and anti-acne agents are provided in other embodiments. Hair growth, hair removal and anti-acne therapies can all involve active agents that, if exposed to the non-target site (e.g., systemic circulation and/or lymphatic system) for long periods, result in toxicity or undesired side effects. Thus, the reduced exposure compositions described herein provides benefits for these applications as well. In one embodiment, the polymer conjugates provided herein modulate hair growth and cycling. In one embodiment, the polymer conjugates provided herein treat alopecia.
[0534] Allergic inflammatory diseases are characterized by an immune response against a sensitizing agent, such as an allergen, resulting in the release of inflammatory mediators that recruit cells involved in inflammation in a subject, potentially leading to tissue damage and sometimes death. Allergic inflammatory diseases of the eye, skin, gastrointestinal tract, upper and lower airways, and lung, including, but not limited to, atopic dermatitis, atopic keratoconjunctivitis, allergic conjunctivitis, asthma, and allergic rhinitis. There is provided, in several embodiments, methods of treating an allergic inflammatory disease in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
[0535] There is also provided, in several embodiments, methods of treating the following conditions in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein: nail dystrophy; seborrheic keratosis; androgenic alopecia; contact dermatitis; actinic keratosis; acne; asthma; eczema (atopic derm); onychomycosis; sinusitis; allergic rhinitis; rosacea; COPD; pruritus; early AMD; urticaria; diabetic retinopathy; psoriasis; alopecia areata; dry eye; vitiligo; glaucoma; late AMD; ulcerative colitis; Crohn's disease; ocular rosacea; hair growth and cycling; skin neoplasias; squamous cell carcinoma; basal cell carcinoma; malignant melanoma; malignant cutaneous lymphomas; vascular tumors; angiosarcoma; kaposi's sarcoma; infantile hemangiomas; hemangioendothelioma; inflammatory dermatoses; dermatitis (atopic, contact); psoriasis; keloids; rosacea; bullous diseases; bullous pemphigoid; erythema multiforme; UV
irradiation therapy; age-related macular degeneration; diabetic retinopathy; macular and corneal edema.
[0536] There is provided, in several embodiments, methods of treating a vascular tumor in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non- limiting examples of vascular tumors include hemangiomas, Kaposi's sarcoma, lymphangioma, glomangioma, angiosarcoma, hemangioendothelioma, and infantile hemangiomas.
[0537] While blood vessels in healthy adults are largely quiescent, adult skin retains the capacity for rapid initiation of angiogenesis during tissue repair and in numerous diseases including inflammatory skin diseases such as psoriasis, many types of dermatitis, blistering diseases, cutaneous neoplasias including squamous cell carcinomas, malignant melanomas, and Kaposi's sarcomas, and proliferative hemangiomas of childhood. Angiogenesis in the skin is also implicated in a number of other diseases that are characterized by macroscopically visible, prominent blood vessels, including rosacea and basal cell carcinoma.
[0538] There is provided, in several embodiments, methods of treating a skin neoplasia in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non-limiting examples of skin neoplasias include squamous cell carcinoma, basal cell carcinoma, malignant melanoma, malignant cutaneous lymphoma, Kaposi's sarcoma, Merkel cell skin cancer, and non-melanoma skin cancer.
[0539] There is provided, in several embodiments, methods of treating an inflammatory skin disease in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non- limiting examples of inflammatory skin diseases include psoriasis, psoriasis guttata, inverse psoriasis, pustular psoriasis, psoriatic erythroderma, acute febrile neutrophilic dermatosis, eczema, xerotic eczema, dyshidrotic eczema, vesicular palmar eczema, acne vulgaris, atopic dermatitis, contact dermatitis, allergic contact dermatitis, dermatomyositis, exfoliative dermatitis, hand eczema, pompholyx, keloids, rosacea, rosacea due to sarcoidosis, rosacea due to scleroderma, rosacea due to Sweet syndrome, rosacea due to systemic lupus erythematosus, rosacea due to urticaria, rosacea due to herpetic pain, Sweet's disease, neutrophilic hydradenitis, sterile pustule, drug rash, seborrheic dermatitis, pityriasis rosea, Kikuchi's disease of the skin, pruritic
urticarial papules and plaques of pregnancy, Stevens-Johnson syndrome and toxic epidermal necrolysis, tattoo reaction, Wells syndrome (eosinophilic cellulitis), reactive arthritis (Reiter syndrome), bowel-associated dermatosis-arthritis syndrome, rheumatoid neutrophilic dermatosis, neutrophilic eccrine hidradenitis, neutrophilic skin disease of dorsum of hand, balanitis circumscripta plasmacellularis, balanoposthitis, Behcet's disease, erythema annulare centrifugum, erythema dyschromicum perstans, erythema multiforme, granuloma annulare, dermatitis of hand, lichen nitidus, lichen planus, lichen sclerosus et atrophicus, lichen simplex chronicus, lichen spinulosus, nummular dermatitis, pyoderma gangrenosum, sarcoidosis, subcorneal pustular dermatosis, urticaria, and transient acantholytic dermatosis.
[0540] Bullous diseases are skin disorders characterized by blistering that often have an autoimmune etiology. VEGF has been found to be upregulated in two bullous diseases, bullous pemphigoid and erythema multiforme. Brown, Lawrence F., et al. Journal of Investigative Dermatology 104.5 (1995): 744-749. Bullous pemphigoid is a subepidermal disorder which manifests as subepidermal blisters with a dermal infiltrate of neutrophils and eosinophils. Erythema multiforme is an inflammatory eruption characterized by symmetric erythematous, edematous, or bullous lesions of the skin or mucous membranes.
[0541] There is provided, in several embodiments, methods of treating a bullous disease in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non-limiting examples of bullous diseases include bullous pemphigoid, erythema multiforme, dermatitis herpetiformis, epidermolysis bullosa acquisita, linear Immunoglobulin A disease, mucous membrane pemphigoid, pemphigoid gestationis, pemphigus foliaceus, and pemphigus vulgaris.
[0542] The process of new hair growth, whether as part of the natural hair cycle or as a result of a treatment to encourage hair growth, relies on numerous cross- talking signal pathways to bring about the processes necessary for hair growth. These principal processes are: cell proliferation of the dermal papia, cell migration to form the appropriate structures, and angiogenesis to form blood supply routes to the new hair foicle. Vascular endothelial growth factor (VEGF) is a well-documented signal to activate these processes via the VEGFR2 receptor. VEGF leads to downregulation of Bad, TGF- 1 and caspase expression through the AKt/PKB and calcium ion dependent pathways, thereby bringing about the end of apoptosis and the telogen phase.
[0543] There is provided, in several embodiments, methods of modulating hair growth and cycling in a subject, the method comprising administering to the subject
an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. There is provided, in several embodiments, methods of treating alopecia in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
[0544] There is provided, in several embodiments, a combination therapy, the combination therapy comprising administering to the subject an effective amount of a polymer conjugate in conjunction with UV irradiation therapy, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
[0545] In some embodiments, alopecia is treated. Non-limiting examples include androgenic alopecia and alopecia areata. Androgenic alopecia (also known as hereditary baldness, male pattern baldness, and seborrheic alopecia) is a non-scarring hair loss of telogen hairs caused by an excessive androgen effect in genetically susceptible men and women. Alopecia areata is known to be associated with autoimmune activities; hence, topically administered immunomodulatory compounds demonstrate efficacy for treating that type of hair loss.
[0546] There is provided, in several embodiments, methods of treating an alopecia in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein . In some embodiments, hair regeneration compositions are in the form of a liquid. In other embodiments, hair regeneration compositions are in the form of a lotion. In additional embodiments, hair regeneration compositions are in the form of a cream. In some embodiments, hair regeneration compositions are in the form of a gel. In other embodiments, the hair regeneration composition is administered twice daily. In other embodiments, the hair regeneration composition is administered one daily. In additional embodiments, the hair regeneration composition is administered once weekly. In some embodiments, the hair regeneration composition is administered directly to the scalp. In some embodiments, the hair regeneration composition is administered directly non-scalp areas.
[0547] Oral delivery is contemplated in several embodiments suitable for dermal pathologies, and thus several embodiments relate to polymer conjugates of compounds in Tables 1-3, optimized for oral delivery to treat the skin while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Topical applications are provided in other embodiments.
[0548] In several embodiments, the warhead of the polymer conjugate is a small molecule disclosed in Tables 1 -3 targeting mediator(s) of dermatological conditions. There is also provided, in several embodiments, methods of treating an inflammatory skin disease or other dermal pathology in a subject, the method comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Tables 1 -3 targeting mediator(s) of dermatological conditions. In several embodiments, a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein are mediator(s) of dermatological conditions. In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.. In some embodiments, the warhead of the polymer conjugate is compound 1 of Table 1 . In some embodiments, the polymer conjugate is SNA-101 . In some embodiments, the warhead of the polymer conjugate is compound 1 of Table 2. In some embodiments, the polymer conjugate is SNA-103. In some embodiments, the warhead of the polymer conjugate is compound 1 of Table 3. In some embodiments, the LSE polymer conjugate is SNA-352. In addition to, or instead of an inflammatory skin disease, a vascular tumor, a bullous disease, a skin neoplasia, a vascular tumor, or a scar may also be treated.
Polymer Conjugates Targeting Mediators of Ophthalmic Conditions
[0549] Compositions comprising an indolocarbazole compound are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of a VEGFR, c- Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Several embodiments relate to polymer conjugates of an indolocarbazole compound, optimized for topical applications (e.g., eye drops) while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications are provided in other embodiments. For example, in some embodiments, the reduced exposure compsotions disclosed herein are administered by direct intravitreal injection. In some embodiments, the reduced exposure compsotions are administered by subconjunctival injection. In other embodiments, the reduced exposure compsotions are administered by subtenon injection. In still further embodiments, the compostions are administered by peribulbar injection. Also provided, in several embodiments, are reduced exposure compostions which can be administered via intraocular implantable devices known to one of skill in the art.
[0550] In several embodiments, the warhead of the polymer conjugate is an indolocarbazole compound or derivative thereof. There is also provided, in several embodiments, methods of treating an ophthalmic condition in a subject, the method
comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is an indolocarbazole compound. In some embodiments, the warhead of the LSE polymer conjugate is a derivative of K252a. In some embodiments, the LSE polymer conjugate is SNA-125. In some embodiments, the LSE polymer conjugate is SNA-120.
[0551] There is provided, in several embodiments, methods of treating an ophthalmic condition in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non- limiting examples of ophthalmic conditions include macular degeneration, age related macular degeneration (ARMD), choroidal neovascularization, retinopathy, diabetic retinopathy, acute macular neuroretinopathy, chronic macular neuroretinopathy, central serous chorioretinopathy, macular edema, cystoid macular edema, diabetic macular edema., acute multifocal placoid pigment epitheliopathy, Behcet's disease, birdshot retinochoroidopathy, uveitis, intermediate uveitis (pars planitis), anterior uveitis, multifocal choroiditis, multiple evanescent white dot syndrome (MEWDS), ocular sarcoidosis, posterior scleritis, serpignous choroiditis, subretinal fibrosis, uveitis syndrome, Vogt-Koyanagi-Harada syndrome, retinal arterial occlusive disease, central retinal vein occlusion, disseminated intravascular coagulopathy, branch retinal vein occlusion, hypertensive fundus changes, ocular ischemic syndrome, retinal arterial microaneurysms, Coat's disease, parafoveal telangiectasis, hemi-retinal vein occlusion, papillophlebitis, central retinal artery occlusion, branch retinal artery occlusion, carotid artery disease (CAD), frosted branch angitis, sickle cell retinopathy and other hemoglobinopathies, angioid streaks, familial exudative vitreoretinopathy, and Eales disease, sympathetic ophthalmia, uveitic retinal disease, retinal detachment, trauma, laser, PDT, photocoagulation, hypoperfusion during surgery, radiation retinopathy, and bone marrow transplant retinopathy, proliferative vitreal retinopathy and epiretinal membranes, proliferative diabetic retinopathy, ocular histoplasmosis, ocular toxocariasis, presumed ocular histoplasmosis syndrome (POHS), endophthalmitis, toxoplasmosis, retinal diseases associated with HIV infection, choroidal disease associated with HIV infection, uveitic disease associated with HIV Infection, viral retinitis, acute retinal necrosis, progressive outer retinal necrosis, fungal retinal diseases, ocular syphilis, ocular tuberculosis, diffuse unilateral subacute neuroretinitis, myiasis, retinitis pigmentosa, systemic disorders with associated retinal dystrophies, congenital stationary night blindness, cone dystrophies, Stargardt's disease, fundus flavimaculatus, Bests disease, pattern dystrophy of the retinal pigmented epithelium, X-linked retinoschisis, Sorsby's fundus dystrophy, benign concentric maculopathy, Bietti's crystalline dystrophy,
and pseudoxanthoma elasticum, retinal detachment, macular hole, and giant retinal tear, retinal disease associated with tumors, congenital hypertrophy of the RPE, posterior uveal melanoma, choroidal hemangioma, choroidal osteoma, choroidal metastasis, combined hamartoma of the retina and retinal pigmented epithelium, retinoblastoma, vasoproliferative tumors of the ocular fundus, retinal astrocytoma, and intraocular lymphoid tumors, punctate inner choroidopathy, acute posterior multifocal placoid pigment epitheliopathy, myopic retinal degeneration, and acute retinal pigment epithelitis.
[0552] The c-Src kinase is the most widely studied member of the largest family of nonreceptor protein tyrosine kinases, known as the Src family kinases (SFKs). Other SFK members include Lyn, Fyn, Lck, Hck, Fgr, BIk, Yrk, and Yes. The Src kinases can be grouped into two sub-categories, those that are ubiquitously expressed (Src, Fyn, and Yes), and those which are found primarily in hematopoietic cells (Lyn, Lck, Hck, BIk, Fgr). (Benati, D. Src Family Kinases as Potential Therapeutic Targets for Malignancies and Immunological Disorders. Current Medicinal Chemistry. 2008; 15: 1 154-1 165). SFKs are key messengers in many cellular pathways, including those involved in regulating proliferation, differentiation, survival, motility, and angiogenesis. The activity of SFKs is highly regulated intramolecularly by interactions between the SH2 and SH3 domains and intermolecularly by association with cytoplasmic molecules. This latter activation may be mediated by focal adhesion kinase (FAK) or its molecular partner Crk- associated substrate (CAS), which plays a prominent role in integrin signaling, and by ligand activation of cell surface receptors, e.g. epidermal growth factor receptor (EGFR). These interactions disrupt intramolecular interactions within Src, leading to an open conformation that enables the protein to interact with potential substrates and downstream signaling molecules. Src can also be activated by dephosphorylation of tyrosine residue Y530. In some embodiments, maximal Src activation requires the autophosphorylation of tyrosine residue Y419 (in the human protein) present within the catalytic domain. Elevated Src activity may be caused by increased transcription or by deregulation due to overexpression of upstream growth factor receptors such as EGFR, HER2, platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor, ephrins, integrin, or FAK.
[0553] Compositions comprising compounds Nos 1-71 shown in Table 1 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of c-Src. Several embodiments relate to polymer conjugates of compounds 1-71 , optimized for topical applications (e.g., eye drops) while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications (e.g., intraocular injection, periocular injection, release from drug delivery devices) are provided in other embodiments. In several embodiments, the warhead of the polymer
conjugate is a small molecule disclosed in Table 1 targeting c-Src. There is also provided, in several embodiments, methods of treating an ophthalmic condition in a subject, the method comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Table 1 targeting c-Src. In some embodiments, the warhead of the LSE polymer conjugate is compound 1 of Table 1. In some embodiments, the LSE polymer conjugate is CT101.
[0554] In several embodiments, compositions comprising compounds shown in Table 3 are used as inhibitors, antagonists, and inverse agonists of JAK and/or STAT family proteins. In some embodiments, the warhead of the polymer conjugate is compound 1 of Table 3. In some embodiments, the LSE polymer conjugate is CT352.
[0555] The JAK kinase family is a cytoplasmic protein kinase family comprising the members JAK1 , JAK2, JAK3 and TYK2. Various studies suggest that ligand binding to a receptor leads to receptor dimerization or oligomerization, which leads to JAK recruitment and activation either through autophosphorylation or phosphorylation by other JAK kinases or by other tyrosine kinases, which in turn leads to tyrosine phosphorylation of the receptors as well as downstream substrates of JAK. Growth factor or cytokine receptors that recruit JAK kinases include the interferon receptors, interleukin receptors (receptors for the cytokines IL-2 to IL-7, IL-9 to IL-13, IL- 15, IL-23), various hormone receptors (erythropoietin (Epo) receptor, the thrombopoietin (Tpo) receptor, the leptin receptor, the insulin receptor, the prolactin (PRL) receptor, the Granulocyte Colony-Stimulating Factor (G-CSF) receptor and the growth hormone receptor), receptor protein tyrosine kinases (such as EGFR and PDGFR), and receptors for other growth factors such as leukemia inhibitory factor (LIF), Oncastatin M (OSM), IFNa/β/γ, Granulocyte-macrophage colony-stimulating factor (GM-CSF), Ciliary neurotrophic factor (CNTF), cardiotrophin-1 (CT-I) (See, Rane, S.G. and Reddy E.P., Oncogene 2000 19, 5662- 5679).
[0556] Many autoimmune diseases and disease associated with chronic inflammation, as well as acute responses, have been linked to excessive or unregulated production or activity of one or more cytokines, the signaling of which depend on JAK kinases. Such diseases include dry eye syndrome and other inflammatory ophthalmic conditions.
[0557] Phosphorylated receptors serve as docking sites for other SH-2 domain containing signaling molecules that interact with JAKs such as the STAT family of transcription factors, Src family of kinases, MAP kinases PB kinase and protein tyrosine phosphatases (Rane S.G. and Reddy E.P., Oncogene 2000 19, 5662-5679). The family of latent cytoplasmic transcription factors, STATS, are the most well characterized downstream substrates for JAKs. The STAT proteins bind to
phosphorylated cytokine receptors through their SH2 domains to become phosphorylated by JAKs, which event leads to their dimerization and release and eventual translocation to the nucleus where they activate gene transcription. The various members of STAT which have been identified thus far, are STAT1 , STAT2, STAT3, STAT4, STAT5 (including STAT5a and STAT5b) and STAT6.
[0558] Signal transducer and activator of transcription 3 (STAT3), a member of the STAT protein family, is a transcription factor that regulates the expression of a variety of genes involved in many cellular processes such as cell growth, apoptosis, cell motility, and cytokine production. In response to cytokines and growth factors, STAT3 is activated by JAK kinases and translocates to the nucleus to act as a transcriptional activator. Studies have demonstrated that STAT3 plays a role in various immune disorders including the pathogenesis of inflammatory bowel disease (see, e.g., Sugimoto, World J. Gastroenterol., 14:51 10-51 14, (2008)).
[0559] In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting a vascular endothelial growth factor receptor (VEGFR).
[0560] Angiogenesis, the process of sprouting new blood vessels from existing vasculature, and arteriogenesis, the remodeling of small vessels into larger conduit vessels, are both physiologically important aspects of vascular growth in adult tissues. These processes of vascular growth are involved, in some cases, for beneficial processes such as tissue repair, wound healing, recovery from tissue ischemia and menstrual cycling. They are also involved, in some cases, for the development of pathological conditions such as the growth of neoplasias, diabetic retinopathy, rheumatoid arthritis, psoriasis, certain forms of macular degeneration, and certain inflammatory pathologies. The inhibition of vascular growth in these contexts has also shown beneficial effects in preclinical animal models. For example, inhibition of angiogenesis by blocking vascular endothelial growth factor or its receptor has resulted in inhibition of tumor growth and in retinopathy. Also, the development of pathological pannus tissue in rheumatoid arthritis involves angiogenesis and might be blocked by inhibitors of angiogenesis. Certain diseases are known to be associated with deregulated angiogenesis, for example ocular neovascularization, such as retinopathies (including diabetic retinopathy), age-related macular degeneration, psoriasis, hemangioblastoma, hemangioma, arteriosclerosis, inflammatory disease, such as a rheumatoid or rheumatic inflammatory disease, especially arthritis (including rheumatoid arthritis), or other chronic inflammatory disorders, such as chronic asthma, arterial or post- transplantational atherosclerosis, endometriosis, and neoplastic diseases, for example so-called solid tumors and liquid tumors (such as leukemias).
[0561] Vascular endothelial growth factor (VEGF) is a signaling protein involved in both vasculogenesis (the de novo formation of the embryonic circulatory system) and angiogenesis (the growth of blood vessels from pre-existing vasculature). As its name implies, VEGF activity has been mostly studied on cells of the vascular endothelium, although it does have effects on a number of other cell types (e.g., stimulation monocyte/macrophage migration, neurons, cancer cells, kidney epithelial cells, keratinocytes). In vitro, VEGF has been shown to stimulate endothelial cell mitogenesis and cell migration. VEGF is also a vasodilator and increases microvascular permeability and was originally referred to as vascular permeability factor.
[0562] The broad term "VEGF" covers a number of proteins from two families, that result from alternate splicing of mRNA from a single, 8 exon, VEGF gene. The two different familes are referred to according to their terminal exon (exon 8) splice site— the proximal splice site (denoted VEGFxxx) or distal splice site (VEGFxxxb). In addition, alternate splicing of exon 6 and 7 alters their heparin binding affinity, and amino acid number (in humans: VEGF121 , VEGF121 b, VEGF145, VEGF165, VEGF165b, VEGF189, VEGF206; the rodent orthologs of these proteins contain one fewer amino acid). These domains have functional consequences for the VEGF splice variants as the terminal (exon 8) splice site determines whether the proteins are pro-angiogenic (proximal splice site, expressed during angiogenesis) or anti-angiogenic (distal splice site, expressed in normal tissues). In addition, inclusion or exclusion of exons 6 and 7 mediate interactions with heparan sulfate proteoglycans (HSPGs) and neuropilin co- receptors on the cell surface, enhancing their ability to bind and activate VEGFRs.
[0563] All members of the VEGF family stimulate cellular responses by binding to tyrosine kinase receptors (the VEGFRs) on the cell surface, causing them to dimerize and become activated through transphosphorylation, although to different sites, times and extents. The VEGF receptors have an extracellular portion consisting of 7 immunoglobulin-like domains, a single transmembrane spanning region and an intracellular portion containing a split tyrosine-kinase domain. VEGF-A binds to VEGFR- 1 (Flt-1) and VEGFR-2 (KDR/Flk-1). VEGFR-2 appears to mediate almost all of the known cellular responses to VEGF. The function of VEGFR-1 is less well defined, although it is thought to modulate VEGFR-2 signaling. Another function of VEGFR-1 may be to act as a dummy/decoy receptor, sequestering VEGF from VEGFR-2 binding (this appears to be involved during vasculogenesis in the embryo). VEGF-C and VEGF-D, but not VEGF-A, are ligands for a third receptor (VEGFR-3), which mediates lymphangiogenesis.
[0564] Increased vascular permeability is one of the earliest manifestations of inflammation, resulting in extravasation of protein-rich plasma into the effected tissue.
Acute vascular permeability allows the deposition of circulating plasma matrix proteins including fibrin and fibronectin (FN) which facilitate cell migration in the inflamed area. This process also provides an access point for immune cells and immunoglobulins to enter the tissue and fight foreign antigens (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012). Conversely, chronic vascular hyperpermeability is suggested to sustain the inflammatory response and retard resolution, further promoting the development of chronic inflammation (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012; Costa et al, Angiogenesis 10:149-166, 2007). This type of vascular hyperpermeability underlies the pathogenesis of a large number of chronic disorders (Nagy et al, Cold Spring Harb. Perspect. Med. 2:a006544, 2012; Costa et al, Angiogenesis 10: 149-166, 2007).
[0565] VEGF is a potent vascular permeabilizing agent that is highly expressed during chronic inflammation (Nagy et al, Annu. Rev. Pathol. 2:251-275, 2007). Low microenvironmental levels of VEGF are desired in order to maintain stable vascular integrity and promote endothelial cell survival through autocrine mechanisms (Lee et al, Cell 130:691-703, 2007). Whereas elevated levels of VEGF induce vascular leakages by activating VEGF receptor 2 (VEGFR2) in endothelial cells (EC) leading to the opening of intercellular and/or intracellular pathways that facilitate plasma extravasation (Koch and Claesson-Welsh, Cold Spring Harb. Perspect. Med. 2:a006502, 2012). Moreover, VEGF may serve as a pro-inflammatory mediator as it can enhance T cell (Xia et al, Blood 102:161-168, 2003), and monocyte (Murakami et al, Blood 108:1849-1856, 2006) migration as well as promote pro-inflammatory chemokines expression by EC including MCP-1 , and IL-8, leading to further immune recruitment. Cellular sources for VEGF during inflammation can include macrophages and mast cells; however it can also be expressed by endothelial cells and acts in a paracrine and autocrine fashion. Thus, VEGF plays a major role in promoting chronic inflammation by inducing vascular permeability and contributing to immune cell recruitment. Compositions comprising compounds Nos 1-59 shown in Table 2 are used, in several embodiments, as inhibitors, antagonists, and inverse agonists of a VEGFR. Several embodiments relate to polymer conjugates of compounds 1-59, optimized for topical applications (e.g., eye drops) while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Non-topical applications (e.g., intraocular injection, periocular injection release from drug delivery devices) are provided in other embodiments.
[0566] In several embodiments, the warhead of the polymer conjugate is a small molecule disclosed in Table 2 targeting a VEGFR. There is also provided, in several embodiments, methods of treating an ophthalmic condition in a subject, the method comprising administering to the subject an effective amount of an LSE polymer
conjugate wherein the warhead is a small molecule disclosed in Table 2 targeting a VEGFR. In some embodiments, the warhead of the LSE polymer conjugate is compound 1 of Table 2. In some embodiments, the LSE polymer conjugate is CT103.
[0567] A growing body of research suggests that dry eye is the result of an underlying cytokine and receptor-mediated inflammatory process. There is provided, in several embodiments, methods of treating dry eye in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. In some embodiments the composition is formulated as an eye drop. In some embodiments, one or two drops of the composition are used per application. In other embodiments, three or four drops of the composition are used per application. In additional embodiments, six drops of the composition are used per application. In some embodiments, the composition is applied for a period of 60 seconds before flushing. In other embodiments, the composition is applied for a period of 120 seconds before flushing. In additional embodiments, the composition is applied for a period of 360 seconds before flushing. In some embodiments, the composition may be administered one or more times a day. In some embodiments, the composition is administered daily. In some embodiments, the composition may be administered once a week.
[0568] There is provided, in several embodiments, methods of treating maculopathies and/or retinal degeneration diseases in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non-limiting examples of maculopathies and/or retinal degeneration diseases include macular degeneration, age related macular degeneration (ARMD), non-exudative age related macular degeneration, exudative age related macular degeneration, choroidal neovascularization, retinopathies, diabetic retinopathy, acute and chronic macular neuroretinopathy, central serous chorioretinopathy, macular edema, cystoid macular edema, and diabetic macular edema.
[0569] There is provided, in several embodiments, methods of treating uveitis, retinitis and/or choroiditis in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non-limiting examples of uveitis, retinitis and/or choroiditis include acute multifocal placoid pigment epitheliopathy, Behcet's disease, birdshot retinochoroidopathy, infectious ocular diseases (e.g., syphilis, lyme, tuberculosis, toxoplasmosis), uveitis, intermediate uveitis (pars planitis), and anterior uveitis, multifocal choroiditis, multiple
evanescent white dot syndrome (MEWDS), ocular sarcoidosis, posterior scleritis, serpignous choroiditis, subretinal fibrosis, uveitis syndrome, and Vogt-Koyanagi-Harada syndrome.
[0570] There is provided, in several embodiments, methods of treating vascular diseases and/or exudative ocular diseases in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non-limiting examples of vascular diseases and/or exudative ocular diseases include retinal arterial occlusive disease, central retinal vein occlusion, disseminated intravascular coagulopathy, branch retinal vein occlusion, hypertensive fundus changes, ocular ischemic syndrome, retinal arterial microaneurysms, Coat's disease, parafoveal telangiectasis, hemi-retinal vein occlusion, papillophlebitis, central retinal artery occlusion, branch retinal artery occlusion, carotid artery disease (CAD), frosted branch angitis, sickle cell retinopathy and other hemoglobinopathies, angioid streaks, familial exudative vitreoretinopathy, and Eales disease.
[0571] There is provided, in several embodiments, methods of treating traumatic and/or surgical ocular conditions in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non-limiting examples of traumatic and/or surgical ocular conditions include sympathetic ophthalmia, uveitic retinal disease, retinal detachment, trauma, laser, PDT, photocoagulation, hypoperfusion during surgery, radiation retinopathy, and bone marrow transplant retinopathy.
[0572] There is provided, in several embodiments, methods of treating a proliferative ocular disorder in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non-limiting examples of proliferative ocular disorders include proliferative vitreal retinopathy and epiretinal membranes, proliferative diabetic retinopathy.
[0573] There is provided, in several embodiments, methods of treating infectious ocular disorders in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non-limiting examples of infectious ocular disorders include ocular histoplasmosis, ocular toxocariasis, presumed ocular histoplasmosis syndrome (POHS), endophthalmitis, toxoplasmosis, retinal diseases associated with HIV infection, choroidal disease associated with HIV infection, uveitic disease associated with HIV Infection, viral
retinitis, acute retinal necrosis, progressive outer retinal necrosis, fungal retinal diseases, ocular syphilis, ocular tuberculosis, diffuse unilateral subacute neuroretinitis, and myiasis.
[0574] There is provided, in several embodiments, methods of treating an ocular genetic disorder in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non- limiting examples of ocular genetic disorders include retinitis pigmentosa, systemic disorders with associated retinal dystrophies, congenital stationary night blindness, cone dystrophies, Stargardt's disease and fundus flavimaculatus, Bests disease, pattern dystrophy of the retinal pigmented epithelium, X-linked retinoschisis, Sorsby's fundus dystrophy, benign concentric maculopathy, Bietti's crystalline dystrophy, and pseudoxanthoma elasticum.
[0575] There is provided, in several embodiments, methods of treating a retinal hole or tear in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non- limiting examples of retinal holes or tears include retinal detachment, macular hole, and giant retinal tear.
[0576] There is provided, in several embodiments, methods of treating an ocular tumor-associated condition in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. Non-limiting examples of ocular tumor-associated condition include retinal disease associated with tumors, congenital hypertrophy of the RPE, posterior uveal melanoma, choroidal hemangioma, choroidal osteoma, choroidal metastasis, combined hamartoma of the retina and retinal pigmented epithelium, retinoblastoma, vasoproliferative tumors of the ocular fundus, retinal astrocytoma, intraocular lymphoid tumors.
[0577] There is provided, in several embodiments, methods of treating punctate inner choroidopathy, acute posterior multifocal placoid pigment epitheliopathy, myopic retinal degeneration, and acute retinal pigment epithelitis in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
[0578] There is evidence to suggest that increased expression of angiogenic factors, in particular VEGF, is a central cause of proliferative diabetic retinopathy (PDR). In this condition, and others such as retinopathy of prematurity, sickle cell retinopathy,
age-related macular degeneration, retina vein occlusion and Eales disease, preretinal vascularisation is a major cause of blindness. New blood vessels grow from the inner retinal vasculature into the vitreous humour. This can cause visual loss by vitreous haemorrhage and/ortractional retinal detachment due to contraction of the fibrous tissue associated with the new blood vessels. Inhibitors of the VEGF pathway intended to treat eye disease are discussed by Slevin et al. in Expert Opin. Investig. Drugs (2008) 17(9):1301-1314, herein incorporated by reference.
[0579] There is provided, in several embodiments, methods of treating age- related macular degeneration in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. There is provided, in several embodiments, methods of treating diabetic retinopathy in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. There is provided, in several embodiments, methods of treating corneal edema in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. There is provided, in several embodiments, methods of treating macular edema in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. There is provided, in several embodiments, methods of treating dry eye in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
[0580] There is provided, in several embodiments, methods of treating age- related macular degeneration in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is an indolocarbazole compound. There is provided, in several embodiments, methods of treating diabetic retinopathy in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is an indolocarbazole compound. There is provided, in several embodiments, methods of treating corneal edema in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is an indolocarbazole compound. There is provided, in several embodiments, methods of treating macular edema in a subject, the method comprising administering to the subject an effective
amount of a polymer conjugate, wherein the warhead is an indolocarbazole compound. There is provided, in several embodiments, methods of treating dry eye in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is an indolocarbazole compound.
[0581] Allergic inflammatory diseases are characterized by an immune response against a sensitizing agent, such as an allergen, resulting in the release of inflammatory mediators that recruit cells involved in inflammation in a subject, potentially leading to tissue damage and sometimes death. Allergic inflammatory diseases of the eye include allergic conjunctivitis. There is provided, in several embodiments, methods of treating an allergic inflammatory disease in a subject, the method comprising administering to the subject an effective amount of a polymer conjugate, wherein the warhead is a small molecule targeting a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein.
[0582] Topical delivery (e.g., eye drops) is contemplated in several embodiments suitable for ophthalmic disorders, and thus several embodiments relate to polymer conjugates of compounds in Tables 1 -3, optimized for topical delivery to treat the eye while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption). Intraocular and periocular injection is also contemplated, in several embodiments, thus some embodiments relate topolymer conjugates of compounds in Tables 1 -3, optimized for delivery via intraocular and periocular injection to treat the eye while also minimizing side-effects caused by exposure at non-target sites (e.g., systemic absorption).
[0583] In several embodiments, the warhead of the polymer conjugate is a small molecule disclosed in Tables 1-3 targeting mediator(s) of ophthalmic conditions. There is also provided, in several embodiments, methods of treating an inflammatory ophthalmic disorder or other ophthalmic conditions in a subject, the method comprising administering to the subject an effective amount of an LSE polymer conjugate wherein the warhead is a small molecule disclosed in Tables 1 -3 targeting mediator(s) of ophthalmic conditions. In several embodiments, a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein are mediator(s) of ophthalmic conditions. In several embodiments, the warhead employed in the LSE polymer conjugate is a small molecule a VEGFR, c-Src, TkrA, MAP2K3, a JAK and/or STAT family protein. In some embodiments, the warhead of the polymer conjugate is compound 1 of Table 1 . In some embodiments, the polymer conjugate is SNA-101 . In some embodiments, the warhead of the polymer conjugate is compound 1 of Table 2. In some embodiments, the polymer conjugate is SNA-103. In some embodiments, the warhead of the polymer conjugate is
compound 1 of Table 3. In some embodiments, the LSE polymer conjugate is SNA-352. In some embodiments, the warhead of the polymer conjugate is an indolocarbazole.
[0584] In some embodiments, for those compounds in Tables 1-3 having one amino group, the compound is modified (e.g., PEGylated) at that location (e.g., a PEG or modified PEG is linked to the compound by reaction with the amino group). If two or more amino groups are present, either location is PEGylated in some embodiments. In other embodiments, the amino group located the furthest away from the moieties interacting with the target is used. In some embodiments, the amino group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target). The effect of conjugation on the activity of the compound can be determined based on various methods, such as bioassays, mass spectroscopy, surface plasmon resonance, in vivo assays, clinical assays, and predictive in silico modeling programs.
[0585] In some embodiments, for those compounds in Tables 1-3 having one sulfhydryl group, the compound is modified (e.g., PEGylated) at that location. If two or more sulfhydryl groups are present, either location is PEGylated in some embodiments. In other embodiments, the sulfhydryl group located the furthest away from the moieties interacting with the target is used. In some embodiments, the sulfhydryl group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
[0586] In some embodiments, for those compounds in Tables 1-3 having one hydroxyl group, the compound is modified (e.g., PEGylated) at that location. If two or more hydroxyl groups are present, either location is PEGylated in some embodiments. In other embodiments, the hydroxyl group located the furthest away from the moieties interacting with the target is used. In some embodiments, the hydroxyl group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
[0587] In some embodiments, for those compounds in Tables 1-3 having one carboxyl group, the compound is modified (e.g., PEGylated) at that location. If two or more carboxyl groups are present, either location is PEGylated in some embodiments. In other embodiments, the carboxyl group located the furthest away from the moieties interacting with the target is used. In some embodiments, the carboxyl group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
[0588] In some embodiments, for those compounds in Tables 1-3 having two or more carboxyl, hydroxyl, amino and/or sulfhydryl groups, the compound is modified (e.g., PEGylated) at the site furthest away from the active site. In some embodiments,
the site that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
[0589] Methods for conjugating the PEG or modified PEG to the small molecule warheads in Tables 1-3, through reaction between functional groups (or functionalized groups), including reaction with the above-mentioned functional groups (amino, sulfhydryl, hydroxyl, carboxyl) are used in several embodiments. Methods of conjugation can be found for example in "Bioconjugate Techniques" (3rd Edition) 2013 by Greg T. Hermanson (http://www.sciencedirect.com/science/book/9780123822390); incorporated herein in its entirety by reference. Although PEGylation is used as an example, other polymers are used in some embodiments.


-117-


-120-




-124-

-125-












[0591] Non-limiting examples of conjugation sites according to some embodiments and chemistries for compounds in Table 2 are disclosed. For example, for structures 23, 46, and 58 of Table 2, the existing carboxylic moiety (-COOH) could be conjugated to PEG-amine through formation of an amide bond using any one of several possible coupling agents (including, e.g., TBTU, HBTU, HOBt, DCC, and N- hydroxysuccinimide). Further, for example, for structures 7, 16, 17, 21 (secondary amino group), 29, 36, 39, and 58 of Table 2, the existing amino group (-NH2) could be conjugated to PEG-COOH through formation of an amide bond using any one of several possible coupling agents (including, e.g., TBTU, HBTU, HOBt, DCC, and N- hydroxysuccinimide). Further, for example, for structures 6, 8, 24, 38, 43, and 45 of Table 2, the existing hydroxyl moiety (-OH) could be conjugated to PEG-halide through formation of an ether bond in presence of a strong base (including, e.g. NaH, KH, and n- BuLi). These conjugation sites and chemistries are neither exhaustive nor limiting, and
are included herein as examples only, and not intended to limit the scope of the embodiments described herein.






-143-

-144-

-145-

-146-

-147-

-148-






-154-

-155-

-156-

-157-


-159-







-166-







-174-





[0592] Non-limiting examples of conjugation sites according to some embodiments and chemistries for compounds in Table 3 are disclosed. For example, for structures 27 and 33 of Table 3, the existing carboxylic moiety (-COOH) could be conjugated to PEG-amine through formation of an amide bond using any one of several possible coupling agents (including, e.g., TBTU, HBTU, HOBt, DCC, and N- hydroxysuccinimide). Further, for example, for structures 1 , 15, 21 , and 34 of Table 3, the existing amino group (-NH2) could be conjugated to PEG-COOH through formation of an amide bond using any one of several possible coupling agents (including, e.g., TBTU, HBTU, HOBt, DCC, and N-hydroxysuccinimide). Further, for example, for structures 9, 20, 24, 29, 33, 34, 36, 37, 38, and 39 of Table 3, the existing hydroxyl moiety (-OH) could be conjugated to PEG-halide through formation of an ether bond in presence of a strong base (including, e.g. NaH, KH, and n-BuLi). These conjugation sites and chemistries are neither exhaustive nor limiting, and are included herein as examples only, and not intended to limit the scope of the embodiments described herein.
[0593] Identifying a conjugation site and developing a conjugation strategy and/or chemistry does not require that all the atoms and the structures of the starting compound are maintained. Once the active part of the compound has been identified or hypothesized, some atoms, groups and structures of the compound can be removed or modified while maintaining sufficient or similar target site binding and activity in several embodiments.
[0594] In alternative embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting c-Src selected from one or more of the following: substituted 2-anilinopyrimidines; imidazoles, oxazoles and thiazoles with protein kinase inhibiting activities; heterocyclic substituted pyrazoles; aryl-amino substituted pyrrolopyrimidine multi-kinase inhibiting compounds; 2-phenylamino-4- (5-
pyrazolylamino)-pyrimidine derivatives; bicyclic heteroaryls; 2-amino-6-anilino-purines; triazolopyridazines; substituted amides; pyrimidine derivatives; pyridinyl- pyrimidinylamino-benzamide derivatives; pyrrolo[2,3-d]pyrimidines; amino-substituted dihydropyrimido[4,5-d]pyrimidinone derivatives; 2-heteroarylamino-pyrimidine derivatives; isoxaxole derivatives; pyrazolo [3,4-d] pyrimidines; 1 , 2, 5, 6-tetraaza- as- indacenes; pyrimido compounds having antiproliferative activity; fused polycyclic 2- aminopyrimidine derivatives; 2-aminopyridine kinase inhibitors; benzothiazole derivatives; pyrazolyl-ureas; pyrido[2,3-d]pyrimidin-7-carboxylic acid derivatives; isoxazolyl-pyrimidines; derivatives of azaindoles; caprolactam derivatives; pyrrolopyridine kinase inhibiting compounds; pyrazole derivatives; kinase inhibitors based upon n-alkyl pyrazoles; thioazepinone derivatives; 2,3-benzodiazepin-2-one derivatives; azdo530; na/k-atpase-derived peptide src inhibitors; 5- (4- (haloalkoxy) phenyl) pyrimidine-2-amine compounds; pyrimidylaminobenzamide compounds; quinazoline derivatives; diaryl urea derivatives; 1 -(5-tert-butyl-2-aryl-pyrazol-3-yl)-3-[2- fluoro-4-[(3-oxo-4h-pyrido[2, 3-b]pyrazin-8-yl)oxy]phenyl]urea derivatives; bufalin phosphate prodrugs; hydroxyindole carboxylic acid based inhibitors; fused pyridine and pyrimidine bicyclic compounds; dichloro-phenyl-pyrido [2, 3-d] pyrimidine derivates; and quinazolines.
[0595] In alternative embodiments, the LSE polymer conjugate comprises an RORvt antagonist/inverse agonist warhead selected from one or more of the following: stearic acid; All-trans retinoic acid; ALTA 1550; Ursolic acid; Digoxin; T0901317; SR1001 ; SR1078; SR3335; SR1555; SR221 1 ; ML209; N-(1-(4-(1 ,1 ,1 ,3,3,3-hexafluoro-2- hydroxypropan-2-yl)benzyl)-1 ,2,3,4-tetrahydroquinolin-6-yl)acetamide; 2,4-difluoro-N-(1- ((4-fluorophenyl)sulfonyl)-1 ,2,3,4-tetrahydroquinolin-7-yl)benzenesulfonamide; 2-Chloro- 6-fluoro-N-(1-((4-fluorophenyl)sulfonyl)-1 ,2,3,4-tetrahydroquinolin-7-yl)benzamide; (S)-2- fluoro-N-(3-methyl-1-(m-tolylsulfonyl)-2,3-dihydro-1 H-pyrido[2,3-b][1 ,4]oxazin-7-yl)-6- (trifluoromethyl)benzamide; 4-(1-(2-Chloro-6-cyclopropylbenzoyl)-7-fluoro-1 H-indazol-3- yl)-3-fluorobenzoic acid; 4-(1-(2-Chloro-6-(trifluoromethyl)benzoyl)-7-fluoro-1 H-indazol-3- yl)-2-hydroxycyclohex-3-enecarboxylic acid; GSK-6a; GSK-8h; GSK-9g; GSK-13; GSK- 21 ; 2-(4-(Ethylsulfonyl)phenyl)-N-(6-(3-fluorophenoxy)-[1 ,1 '-biphenyl]-3-yl)acetamide; N- (6-(3,5-difluorophenoxy)-3'-fluoro-[1 , 1 '-biphenyl]-3-yl)-2-(4-(N- methylsulfamoyl)phenyl)acetamide; N-(4-Ethylphenyl)-3-(hydroxymethyl)-Nisobutyl-4- ((tetrahydro-2H-pyran-4-yl)methoxy)benzenesulfonamide; N-(4-chlorophenyl)-4-((3,5- dimethylisoxazol-4-yl)methoxy)-N-isobutylbenzenesulfonamide; N-(2,4-dimethylphenyl)- 4-(2-hydroxy-2-(pyridin-4-yl)ethoxy)-N-isobutylbenzenesulfonamide; N-isobutyl-N-((5-(4- (methylsulfonyl)phenyl)thiophen-2-yl)methyl)-1-phenylmethanesulfonamide; N-(4-(4- acetylpiperazin-1-yl)benzyl)-Nisobutyl-1-phenylmethanesulfonamide; N-(3,4-
dimethoxyphenyl)-1-ethyl-2-oxo-1 ,2-dihydrobenzo[cd]indole-6-sulfonamide; JTE-151 ; substituted 2,3-dihydro-1 H-inden-1-one Retinoic acid-related orphan nuclear receptor Antagonists; Methylene linked quinolinyl modulators of ROR-gamma-t; Fused pyridine and pyrimidine derivatives; heteroaromatic compounds; SEVEN-MEMBERED SULFONAMIDES; Substituted dihydro-benzimidazole compounds; Pyrrolo sulfonamide compounds; Tetrahydroquinoline and related bicyclic compounds; Amide compounds; Heterocyclic compounds; and amido compounds..
[0596] In alternative embodiments, the LSE polymer conjugate comprises a Src family tyrosine kinase inhibitor warhead selected from dasatinib and saracatinib.
[0597] In alternative embodiments, the LSE polymer conjugate comprises an IL-23 inhibitor warhead selected from SCH-90222, STA-5326, and STA-5326.
[0598] In alternative embodiments, the LSE polymer conjugate comprises a STAT3 inhibitor warhead selected from cucurbitacin I, niclosamide, cryptotanshinone, SD 1008, Stat3 Inhibitor III, WP1066, Nifuroxazide, Stat3 Inhibitor, Stattic, Stat3 Inhibitor, S3I-201 ; Stat3 Inhibitor VIII, 5,15-DPP, 2-Hydroxy-4-(((4- methylphenyl)sulfonyloxy)acetyl)amino)-benzoic acid (NSC74859) and Kahweol.
[0599] In alternative embodiments, the LSE polymer conjugate comprises a JAK inhibitor warhead selected from ruxolitinib, fedratinib, tofacitinib, baricitinib, pacritinib, decernotinib, XL019, AZD1480, INCB0391 10, LY2784544, BMS91 1543, NS018, GLPG0634, GLPG0788, or N-(cyanomethyl)-4-2-(4-morpholinoanilino)pyrimidin- 4-yl)benzamide; or a pharmaceutically acceptable salt thereof.
[0600] Spleen tyrosine kinase (SYK) is a non-receptor linked protein tyrosine kinase which, in some cases, plays a role as a mediator of immunoreceptor signaling in a host of inflammatory cells including mast cells, B-cells, macrophages and neutrophils. In alternative embodiments, the LSE polymer conjugate comprises a SYK inhibitor warhead selected from Cerdulatinib (4-(cyclopropylamino)-2-((4-(4- (ethylsulfonyl)piperazin- 1 - yl)phenyl)amino)pyrimidine-5 -carboxamide), entospletinib (6-( 1 H-indazol-6-yl)-N-(4- morpholinophenyl)imidazo[l,2-a]pyrazin-8-amine), fostamatinib ([6-({5-Fluoro-2- [(3,4,5-trimethoxyphenyl)amino]-4-pyrimidinyl}amino)-2,2- dimethyl-3-oxo-2,3-dihydro- 4H-pyrido[3,2-b][l,4]oxazin-4-yl]methyl dihydrogen phosphate), fostamatinib disodium salt (sodium (6-((5-fluoro-2-((3,4,5- trimethoxyphenyl)amino)pyrimidin-4-yl)amino)-2,2- dimethyl-3-oxo-2H-pyrido[3,2- b][l,4]oxazin-4(3H)-yl)methyl phosphate), BAY 61-3606 (2-(7-(3,4-Dimethoxyphenyl)- imidazo[l,2-c]pyrimidin-5-ylamino)-nicotinamide HQ), RO9021 (6-[(IR,2S)-2-Amino- cyclohexylamino]-4-(5,6-dimethyl-pyridin-2-ylamino)- pyridazine-3-carboxylic acid amide), imatinib (Gleevac; 4-[(4-methylpiperazin-l- yl)methyl] -N-(4-methyl-3 - { [4- (pyridin-3 -yl)pyrimidin-2-yl]amino } phenyl)benzamide), GSK143 (2-(((3R,4R)-3-
aminotetrahydro-2H-pyran-4-yl)amino)-4-(p- tolylamino)pyrimidine-5 -carboxamide), PP2 ( 1 -(tert-butyl)-3 -(4-chlorophenyl)- 1 H- pyrazolo[3,4-d]pyrimidin-4-amine), PRT-060318 (2-(((IR,2S)-2- aminocyclohexyl)amino)-4-(m-tolylamino)pyrimidine-5-carboxamide), PRT-062607 (4- ((3-(2H-l,2,3-triazol-2-yl)phenyl)amino)-2-(((IR,2S)-2- aminocyclohexyl)amino)pyrimidine-5-carboxamide hydrochloride), R1 12 (3,3'-((5- fluoropyrimidine-2,4-diyl)bis(azanediyl))diphenol), R348 (3-Ethyl-4-methylpyridine), R406 (6-((5-fluoro-2-((3,4,5-trimethoxyphenyl)amino)pyrimidin-4-yl)amino)-2,2- dimethyl-2H- pyrido[3,2-b] [ 1 ,4]oxazin-3(4H)-one), piceatannol (3-Hydroxyresveratol), YM 193306(see Singh et al. Discovery and Development of Spleen Tyrosine Kinase (SYK) Inhibitors, J. Med. Chem. 2012, 55, 3614-3643), 7-azaindole, piceatannol, ER- 27319, Compound D, PRT060318, luteolin, apigenin, quercetin, fisetin, myricetin, or morin.
[0601] In several embodiments, the LSE polymer conjugate comprises a RAC1 inhibitor warhead selected from W56, NSC23760 and NSC 23766, or the inhibitors described in Yuan Gao, et al. PNAS, May 18, 2004, vol. 101 , 7618-7623.
[0602] In several embodiments, the LSE polymer conjugate comprises an ENaC inhibitor warhead selected from triamterene, phenamil, amiloride and amiloride derivatives, particularly benzyl amiloride (Benzamil). Additional amiloride derivatives are described in WO2012035158; WO2009074575; WO201 1028740; WO2009150137; WO201 1079087; and WO2008135557, each of which are herein specifically incorporated by reference.
[0603] In several embodiments, the LSE polymer conjugate comprises a NFkB inhibitor warhead selected from Bithionol, Bortezomib, Cantharidin, Chromomycin A3, Daunorubicinum, Digitoxin, Ectinascidin 743, Emetine, Fluorosalan, Manidipine hydrochloride, Narasin, Ouabain, Sorafenib tosylate, Sunitinib malate, Tioconazole, Tribromsalan, Triclabendazolum, Zafirlukast, and Withaferin A.
[0604] In several embodiments, the LSE polymer conjugate comprises an IRAK inhibitor warhead selected from the inhibitors described in Wang, Zhulun, et al. "IRAK-4 inhibitors for inflammation." Current topics in medicinal chemistry 9.8 (2009): 724-737, which is herein specifically incorporated by reference.
[0605] In several embodiments, the LSE polymer conjugate comprises a PKC inhibitor warhead selected from sotrastaurin (also known as AEB071 and described in U.S. Pat. No. 6,645,970), 3-(1 H-lndol-3-yl)-4-[2-(piperazin-1 -yl)quinazolin-4-yl]-1 H- pyrrole-2,5-dione (described in U.S. Pat. No. 6,645,970), 3-[2-chloro-7- [(dimethylamino)methyl]-1 -naphthalenyl]-4-[7-[2-(2-methoxyethoxy)ethoxy]-1 H-indol-3- yl]-1 H-pyrrole-2,5-dione (described in PCT Publication No. WO07/006,533 and US Publication No. 2008/0318975), 3-[3-(4,7-diaza-spiro[2,5]oct-7-yl)-isoquinolin-1 -yl]-4-(7- methyl-1 H-indol-3-yl)-pyrrole-2,5-dione (described in Example 69 of U.S. Pat. No.
7,235,555); ruboxistaurin((9S)-9-[(dimethylamino)methyl]-6,7,10,1 1-tetrahydro-9H,18H-5, 21 :12,17-dimethenodibenzo-[e,k]pyrrolo[3,4-h][1 ,4,13]oxadiazacyclohexadecine- 18,20(19H)-dione (also known as LY-333531 and described in U.S. Pat. No. 5,698,578)) and the mesylate salt of ruboxistaurin (described in European patent No. 0776895 B1). Each of the references cited above are incorporated herein by reference.
[0606] In several embodiments, the LSE polymer conjugate comprises a PKCa/β inhibitor warhead selected from 3-[2-chloro-7-[(dimethylamino)methyl]-1- naphthalenyl]-4-[7-[2-(2-methoxyethoxy)ethoxy]-1 H-indol-3-yl]-1 H-pyrrole-2,5-dione (CAS No. 919992-85-1 described in PCT Publication No. WO07/006,533 and US Publication No. 2008/0318975); 3-(1 H-indol-3-yl)-4-[2-(piperazin-1-yl)quinazolin-4- yl]pyrrole-2,5-dione having the following structure and described in Example 70 of PCT Publication No. WO 2002/038561 or U.S. Pat. No. 6,645,970; (9S)-9- [(dimethylamino)methyl]-6,7, 10,1 1 -tetrahydro-9H, 18H-5, 21 :12,17-dimethenodibenzo- [e,k]pyrrolo[3,4-h][1 ,4,13]oxadiazacyclohexadecine-18,20(19H)-dione (also referred to as ruboxistaurin or LY-333531 , CAS No. 169939-94-0 described in U.S. Pat. No. 5,698,578); ruboxistaurin mesylate salt (described in U.S. Pat. No. 5,710,145 and EP Patent No. 776895 B1); and 12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H- indolo(2,3-a)pyrrolo(3,4-c)-carbazole (CAS No. 136194-77-9, available from Calbiochem® and described in U.S. Pat. No. 5,489,608). Each of the references cited above are incorporated herein by reference.
[0607] In several embodiments, the LSE polymer conjugate comprises a specific SIP receptor agonist warhead selected from SIP itself, SEW2871 , JTE-013, VPC23019, R- 3477 (Actelion), KRP-203 (Kyorin Pharmaceutical Co.), sonepcizumab (Lpath), BAF-312 (Novartis), ONO-4641 (Ono Pharmaceutical Co.), ES-285 (PharmaMar SA), 2-amino-2-[2- (4-octylphenyl)ethyl]propane-l,3-diol (FTY720; fingolimod), phospho- FTY720, and pharmaceutically acceptable salts thereof.
[0608] In several embodiments, the LSE polymer conjugate comprises a PI3K inhibitor warhead selected from Compound 1 ((S)-3-(1-((9H-purin-6-yl)amino)ethyl)- 8-chloro-2-phenylisoquinolin-1 (2H)-one), AMG-319, GSK 2126458, GSK 1059615, GDC- 0032, GDC-0980, GDC-0941 , XL147, XL499, XL765, BKM 120, GS1 101 , CAL 263, SF1 126, PX-866, BEZ235, CAL-120, BYL719, RP6503, RP6530, TGR1202, INK1 117, PX-886, BAY 80-6946, IC871 14, Palomid 529, ZSTK474, PWT33597, TG100-1 15, GNE- 477, CUDC-907, AEZS-136, BGT-226, PF-05212384, LY3023414, PI-103, LY294002, INCB-040093, CAL-130 and wortmannin.
[0609] In several embodiments, the LSE polymer conjugate comprises an AKT inhibitor warhead selected from AZD5363, miltefosine, perifosine, VQD-002, MK-
2206, GSK690693, GDC-0068, triciribine, CCT128930, PHT-427, or honokiol, or a combination thereof. In one embodiment, the AKT inhibitor is MK-2206 or perifosine.
[0610] In several embodiments, the LSE polymer conjugate a mTOR inhibitor warhead selected from AP23841 , AZD8055, BEZ235, BGT226, deferolimus (AP23573/MK-8669), EM101/LY30351 1 , everolimus (RAD001), EX2044, EX3855, EX7518, GDC0980, INK-128, KU-0063794, NV-128, OSI-027, PF-4691502, rapalogs, rapamycin, ridaforolimus, SAR543, SF1 126, temsirolimus (CCI-779), WYE-125132, XL765, zotarolimus (ABT578), torin 1 , GSK2126458, AZD2014, GDC-0349, or XL388, or a combination thereof.
[0611] In several embodiments, the LSE polymer conjugate comprises a PDE4 inhibitor warhead selected from rolipram, mesembrine, drotaverine, roflumilast, ibudilast, piclamilast, luteolin, cilomilast, diazepam, arofylline, CP-80633, denbutylline, drotaverine, etazolate, filaminast, glaucine, HT-0712, ICI-63197, irsogladine, Mesembrine, Ro20-1724, RPL-554, and YM-976.
[0612] In alternative embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting a VEGFR selected from one or more of the following: pyrimidine compounds; diamino-pyrimidines; vegfr-binding polypeptides; fluoro substituted omega-carboxyaryl diphenyl urea; azaindole kinase inhibitors; furo-and thienopyrimidine derivatives; tricyclic amine derivatives; heterocyclic inhibitors of kinases; pyridinyl-pyrimidinylamino-benzamide derivatives; pyrrolotriazine kinase inhibitors; fused heterocyclic derivatives; anthranylamidopyridines; anthranilamide pyridine amides; 2-amino-5-substituted pyrimidine inhibitors; isomeric fused pyrrolocarbazoles and isoindolones; benzyl-benzimidazolyl derivatives; vegfr-3 binding peptides; 1 ,3-benzoxazolyl derivatives; fused heterocyclic derivatives; peptide chains; indirubin derivatives; pyridinamide derivatives; n-oxide anthranylamide derivatives; pyridineamine derivatives; thienopyrimidine and furopyrimidine derivatives; 1 -(5-tert- butyl-2-aryl-pyrazol-3-yl)-3-[2-fluoro-4-[(3-oxo-4h-pyrido[2, 3-b]pyrazin-8- yl)oxy]phenyl]urea derivatives; pyrazolophenyl-pyridine compounds; 2-chloro-4-anilino- quinazoline compounds; n-((4-chloro-3-trifluoromethyl)phenyl)-n'-(2-fluoro-4-((2- hydroxymethylaminoformyl)-4-pyridyloxo)phenyl)urea; n-indol-1 -amide compounds; substituted aromatic urea compounds; naphthylamide compound; and 3-chloro- and 3- methoxy-n-methyl-2-pyridine carboxamide compounds.
[0613] In alternative embodiments, the LSE polymer conjugate comprises a VEGF inhibitor warhead selected from aflibercept, ziv-aflibercept, bevacizumab, sonepcizumab, VEGF sticky trap, cabozantinib, foretinib, vandetanib, nintedanib, regorafenib, cediranib, ranibizumab, lapatinib, sunitinib, sorafenib, plitidepsin, regorafenib, verteporfin, bucillamine, axitinib, pazopanib, fluocinolone acetonide,
nintedanib, AL8326, 2C3 antibody, AT001 antibody, XtendVEGF antibody, HuMax-VEGF antibody, R3 antibody, AT001/r84 antibody, HyBEV, ANG3070, APX003 antibody, APX004 antibody, ponatinib, BDM-E, VGXIOO antibody, VGX200, VGX300, COSMIX, DLX903/1008 antibody, ENMD2076, I DUS815C, R84 antibody, KD019, NM3, MGCD265, MG516, MP0260, NT503, anti-DLL4/VEGF bispecific antibody, PAN90806, Palomid 529, BD0801 antibody, XV615, lucitanib, motesanib diphosphate, AAV2- SFLT01 , soluble Fltl receptor, AV-951 , Volasertib, CEP1 1981 , KH903, lenvatinib, lenvatinib mesylate, terameprocol, PF00337210, PRS050, SP01 , carboxyamidotriazole orotate, hydroxychloroquine, linifanib, ALGIOOI, AGN150998, MP01 12, AMG386, ponatinib, PD173074, AVAIOI, BMS690514, KH902, golvatinib (E7050), dovitinib, dovitinib lactate (TKI258, CHIR258), ORA101 , ORA102, Axitinib (Inlyta, AG013736), PTC299, pegaptanib sodium, troponin, EG3306, vatalanib, BmablOO, GSK2136773, Anti-VEGFR Alterase, Avila, CEP7055, CLT009, ESBA903, GW654652, HMPL010, GEM220, HYB676, JNJ17029259, TAK593, Nova21012, Nova21013, CP564959, smart Anti-VEGF antibody, AG028262, AG13958, CVX241 , SU14813, PRS055, PG501 , PG545, PTI101 , TG100948, ICS283, XL647, enzastaurin hydrochloride, BC194, COT601 M06.1 , COT604M06.2, MabionVEGF, Apatinib, RAF265 (CHIR-265), Motesanib Diphosphate (AMG-706), Lenvatinib (E7080), TSU-68 (SU6668, Orantinib), Brivanib (BMS-540215), MGCD-265, AEE788 (NVP-AEE788), ENMD-2076, OSI-930, CYC1 16, ΚΪ8751 , Telatinib, KR 633, SAR131675, Dovitinib (TKI- 258) Dilactic Acid, Apatinib, BMS-794833, Brivanib Alaninate (BMS-582664), Golvatinib (E7050), Semaxanib (SU5416), ZM 323881 HC1 , Cabozantinib malate (XL184), ZM 306416, AL3818, AL8326, 2C3 antibody, AT001 antibody, HyBEV, ANG3070, APX003 antibody, APX004 antibody, ponatinib (AP24534), BDM-E, VGXIOO antibody (VGXIOO ORCADIAN), VGX200 (c-fos induced growth factor monoclonal antibody), VGX300, COSMIX, DLX903/1008 antibody, ENMD2076, sunitinib malate (Sutent®), INDUS815C, R84 antibody, KD019, NM3, allogenic mesenchymal precursor cells combined with an anti- VEGF antagonist (e.g., anti-VEGF antibody), MGCD265, MG516, VEGF-Receptor kinase inhibitor, MP0260, NT503, anti-DLL4/VEGF bispecific antibody, PAN90806, Palomid 529, BD0801 antibody, XV615, lucitanib (AL3810, E3810), AMG706 (motesanib diphosphate), AAV2-sFLT01 , soluble Fltl receptor, cediranib (Recentin™), AV-951 , tivozanib (KRN-951), regorafenib (Stivarga®), volasertib (BI6727), CEP1 1981 , KH903, lenvatinib (E7080), lenvatinib mesylate, terameprocol (EM1421), ranibizumab (Lucentis®), pazopanib hydrochloride (Votrient™), PF00337210, PRS050, SP01 (curcumin), carboxyamidotriazole orotate, hydroxychloroquine, linifanib (ABT869, RG3635), fluocinolone acetonide (lluvien®), ALG1001 , AGN150998, DARPin MP01 12, AMG386, ponatinib (AP24534), AVA101 , nintedanib (Vargatef™), BMS690514, KH902,
golvatinib (E7050), everolimus (Afinitor®), dovitinib lactate (TKI258, CHIR258), ORAIOI, ORA102, axitinib (Inlyta®, AG013736), plitidepsin (Aplidin®), PTC299, aflibercept (Zaltrap®, Eylea®), pegaptanib sodium (Macugen™, LI900015), verteporfin (Visudyne®), bucillamine (Rimatil, Lamin, Brimani, Lamit, Boomiq), R3 antibody, AT001/r84 antibody, troponin (BLS0597), EG3306, vatalanib (PTK787), BmablOO, GSK2136773, Anti-VEGFR Alterase, Avila, CEP7055, CLT009, ESBA903, HuMax-VEGF antibody, GW654652, HMPL010, GEM220, HYB676, JNJ17029259, TAK593, XtendVEGF antibody, Nova21012, Nova21013, CP564959, Smart Anti-VEGF antibody, AG028262, AG13958, CVX241 , SU14813, PRS055, PG501 , PG545, PTI101 , TG 100948, ICS283, XL647, enzastaurin hydrochloride (LY317615), BC194, quinolines, COT601 M06.1 , COT604M06.2, MabionVEGF, Si-Spheres coupled to anti-VEGF or VEGF-R antibody, Apatinib (Y 968D1), and AL3818.
[0614] In some embodiments, for indolocarbazole compounds having one amino group, the compound is modified (e.g., PEGylated) at that location (e.g., a PEG or modified PEG is linked to the compound by reaction with the amino group). If two or more amino groups are present, either location is PEGylated in some embodiments. In other embodiments, the amino group located the furthest away from the moieties interacting with the target is used. In some embodiments, the amino group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target). The effect of conjugation on the activity of the compound can be determined based on various methods, such as bioassays, mass spectroscopy, surface plasmon resonance, in vivo assays, clinical assays, and predictive in silico modeling programs.
[0615] In some embodiments, for indolocarbazole compounds having one sulfhydryl group, the compound is modified (e.g., PEGylated) at that location. If two or more sulfhydryl groups are present, either location is PEGylated in some embodiments. In other embodiments, the sulfhydryl group located the furthest away from the moieties interacting with the target is used. In some embodiments, the sulfhydryl group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
[0616] In some embodiments, for indolocarbazole compounds having one hydroxyl group, the compound is modified (e.g., PEGylated) at that location. If two or more hydroxyl groups are present, either location is PEGylated in some embodiments. In other embodiments, the hydroxyl group located the furthest away from the moieties interacting with the target is used. In some embodiments, the hydroxyl group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
[0617] In some embodiments, for indolocarbazole compounds having one carboxyl group, the compound is modified (e.g., PEGylated) at that location. If two or more carboxyl groups are present, either location is PEGylated in some embodiments. In other embodiments, the carboxyl group located the furthest away from the moieties interacting with the target is used. In some embodiments, the carboxyl group that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
[0618] In some embodiments, for indolocarbazole compounds having two or more carboxyl, hydroxyl, amino and/or sulfhydryl groups, the compound is modified (e.g., PEGylated) at the site furthest away from the active site. In some embodiments, the site that causes the least hindrance on activity is used (whether or not it is located the furthest way from the moieties interacting with the target).
[0619] Methods for conjugating the PEG or modified PEG to an indolocarbazole compound, through reaction between functional groups (or functionalized groups), including reaction with the above-mentioned functional groups (amino, sulfhydryl, hydroxyl, carboxyl) are used in several embodiments. Methods of conjugation can be found for example in "Bioconjugate Techniques" (3rd Edition) 2013 by Greg T. Hermanson (http://www.sciencedirect.com/science/book/9780123822390); incorporated herein in its entirety by reference. Although PEGylation is used as an example, other polymers are used in some embodiments.
[0620] In alternative embodiments, the warhead employed in the LSE polymer conjugate is a small molecule targeting a JAK and/or STAT family protein selected from one or more of the following: ruxolitinib; fedratinib; tofacitinib; baricitinib; pacritinib; decernotinib; xl019; azd1480; incb0391 10; Iy2784544; bms91 1543; ns018; glpg0634; glpg0788; n-(cyanomethyl)-4-2-(4-morpholinoanilino)pyrimidin-4- yl)benzamide; cucurbitacin i, niclosamide, cryptotanshinone, sd 1008, stat3 inhibitor iii, wp1066, nifuroxazide, stat3 inhibitor, stattic, stat3 inhibitor, s3i-201 ; stat3 inhibitor viii, 5, 15-dpp, 2-hydroxy-4-(((4-methylphenyl)sulfonyloxy)acetyl)amino)-benzoic acid (nsc74859); kahweol; pyrrolo[2,3-d]pyrimidine compounds; pyrrolopyridines; 2, 6- diamino-pyrimidin- 5-yl-carboxamides; deazapurines; quinazoline derivatives; benzisoxazole derivatives; 4-aryl-2-amino-pyrimidines or 4-aryl-2-aminoalkyl-pyrimidines; deazapurines; azaindoles; thiopyrimidine-based compounds; pyrrolo[2,3-d]pyrimidine compounds; 5-(2-aminopyrimidin-4-yl) benzisoxazoles; benzimidazo[4,5-f]isoquinolinone derivatives; phenyl amino pyrimidine compounds; macrocyclic compounds; 5, 7- substituted-imidazo [1 , 2-c] pyrimidines; heterocyclyl pyrazolopyrimidine analogues; triazolopyridine jak inhibitor compounds; pyrazolopyrimidine jak inhibitor compounds; 2,4-pyrimidinediamine derivatives; n-(hetero)aryl-pyrrolidine derivatives of pyrazol-4-yl-
pyrrolo[2,3-d]pyrimidines; pyrrol-3-yl-pyrrolo[2,3-d]pyrimidines; heterocyclic compounds; triazolopyridine jak inhibitor compounds; thiadiazoles; oxadiazoles; 3-[4-(7h-pyrrolo[2,3- d]pyrimidin-4-yl)-1 h-pyrazol-1-yl]octane- or heptane-nitrile; retrometabolic compounds; pyrrolo[2,3-d] pyrimidine compounds; pyrrolo[2,3-d]pyrimidine compounds; tricyclic heteroaryl compounds; n-containing heteroaryl derivatives; pyrazole derivatives; pyridine derivatives; isoquinoline derivatives; azaindoles; piperidine inhibitors; imidazo [1 ,2-b] pyridazine and imidazo [4,5-b] pyridine derivatives; pyrrole six-membered heteroaryl ring derivative; pias molecules; imidazole-4, 5-dicarboxamide derivatives; heteroaryl imidazolone derivatives; macrocyclic compounds; peptide inhibitors; bicyclic diamines; tricyclic carbamate; heterocyclyl pyrazolopyrimidine analogues; ortho substituted pyrimidine compounds; pyrrolo [ 2, 3 - d] pyrimidine urea compounds; tricyclic lactones; heterocyclyl pyrimidine analogues; substituted 2-hydroxy-4-(2- (phenylsulfonamido)acetamido)benzoic acid analogs; phenyl amino pyrimidine bicyclic compounds; pyrimidine-2,4-diamine derivatives; geminally substituted cyanoethylpyrazolo pyridones; pyrrolo [2, 3 -d]pyrimidine derivatives; pyrazolo[4,3- c]pyridine derivatives; pyridin-2(1 h)-one derivatives; substituted 2-(9h-purin-9-yl) acetic acid analogues; cycloalkyl nitrile pyrazolo pyridones; 5,7-substituted-imidazo[1 ,2- c]pyrimidines; substituted pyrimidinyl kinase inhibitors; bicyclic oxa-lactam kinase inhibitors; n-(2-cyano heterocyclyl)pyrazolo pyridones; pyrazolo[4,3-c]pyridine derivatives (7h-pyrrolo[2,3-d]pyrimidin-2-yl)amine compounds; acyclic cyanoethylpyrazolo pyridones; acyclic cyanoethylpyrazoles; dihydropyrrolonaphtyridinone compounds; triazolopyridine compounds; azaindoles; pyrazolopyrimidin-2-yl derivatives; indazole derivatives; 4,6-substituted-pyrazolo[1 ,5-a]pyrazines; n-cyanomethylamides; 5-chloro-2- difluoromethoxyphenyl pyrazolopyrimidine compounds; naphthyridine compounds; sulfamide piperazine derivatives; sh2 stat3/stat1 peptidomimetices; 2-(pyrazolopyridin-3- yl)pyrimidine derivatives; 7h-pyrrolo[2, 3-d]pyrimidine derivatives; substituted n- (pyrrolidine-3-yl)-7h-pyrrolo[2,3-d]pyrimidine-4-amine compounds; nicotinamides; triazolopyridine compounds; aminopyrimidinyl compounds; ethyl n-boc piperidinyl pyrazolo pyridones; pyrrolo[2,3-d]pyrimidine derivatives; imidazolopyrimidin-2-yl derivatives; homopiperazine derivatives; 4-substituted pyrrolo[2,3-d]pyrimidine compounds; five-and-six-membered heterocyclic compounds; pyrrolo[2,3-d]pyrimidinyl, pyrrolo[2,3-b]pyrazinyl and pyrrolo[2,3-d]pyridinyl acrylamides and analogues; monoacetyldiglyceride compounds; and 2-methoxy-4-(3-(4-methoxyphenyl)prop-1-en-1- yl)phenol and derivatives.
[0621] Suitable protecting groups, in some embodiments, are for protecting functional groups during the conjugation of warhead and polymer. Various protecting groups as well as suitable means and conditions for protecting and deprotecting the
substituents are used in several embodiments. The means and conditions of protecting and deprotecting employed depend on the nature of the involved functional groups. Protecting groups for hydroxy-, amino-, and/or carboxy residues are selected in several embodiments from acetonide, ethylidene methoxymethyl, 2-methoxyethoxymethyl, benzyloxymethyl, tetrahydropyranyl, methyl, ethyl, isopropyl, t-butyl, benzyl, triphenylmethyl, t-butyldimethylsilyl, triphenylsilyl, methoxycarbonyl, t-butyloxycarbonyl, benzyloxycarbonyl, fluorenylmethoxycarbonyl, acetyl, benzoyl, toluenesulfonyl, dimethoxybenzyl, nitrophenyloxycarbonyl, nitrobenzyloxycarbonyl, allyl, fluorenylmethyl, tetrahydrofuranyl, phenacyl, acetol, phenyl, trimethylsilyl, pyrrolidyl, indolyl, hydrazino and other protecting groups such as those that can be found in Greene T. W., et al., Protective Groups in Organic Synthesis, 4th ed., John Wiley and Son, New York, N.Y. (2007); incorporated herein in its entirety be reference. The reagents and conditions of protecting and deprotecting reactions are in particular selected for their suitability at selectively attaching and removing the protecting group without adversely affecting the rest of the compound.
[0622] The polymer conjugates disclosed herein may also be prepared as pharmaceutically acceptable salts including salts of inorganic acids such as hydrochloric, hydroiodic, hydrobromic, phosphoric, metaphosphoric, nitric acid and sulfuric acids as well as salts of organic acids, such as tartaric, acetic, citric, malic, benzoic, glycolic, gluconic, succinic, aryl sulfonic, (e.g., p-toluene sulfonic acids, benzenesulfonic), phosphoric, malonic, and the like. Suitable acids for formation of pharmaceutically acceptable salts are used in some embodiments. Further, pharmaceutically acceptable salts of compounds may be formed with a pharmaceutically acceptable cation. Pharmaceutically acceptable cations include, but are not limited to, alkali cations (Li+, Na+, K+), earth alkali cations (Mg2+, Ca2+, Ba2+), ammonium and organic cations, such as quaternary ammonium cations.
Delivery of Polymer Conjugates to the Gastrointestinal System
[0623] The intestinal wall comprises an epithelial cell wall that functions to prevent the access of enteral bacteria to the underlying lamina propria and beyond. The lamina propria is a thin layer of loose connective tissue which lies beneath the epithelium and together with the epithelium and basement membrane constitutes the mucosa. The connective tissue of the lamina propria is loose and rich in cells, and can include fibroblasts, lymphocytes, plasma cells, macrophages, eosinophilic leukocytes, and mast cells. The lamina propria's richness in macrophages and lymphoid cells makes it a likely target location for immune responses to occur and forms a part of the barrier that protects internal tissues from external pathogenic microorganisms. A distinctive feature
of inflammatory bowel diseases is the infiltration of the lamina propria by activated innate and adaptive immune cells that direct the aberrant immune activation that yields acute and chronic inflammation.
[0624] There is provided, in some embodiments, a reduced exposure composition for treatment of the gastrointestinal system. In several embodiments, there is provided in a reduced exposure composition, a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an inhibitor, antagonist, or inverse agonist of, for example, a mediator of a gastrointestinal condition. In some embodiments, the reduced exposure composition for treatment of the gastrointestinal system is delivered orally. In some embodiments, the LSE polymer conjugate comprising a warhead conjugated with PEG or another molecule as described herein shows reduced absorption into, slower absorption into, and faster clearance from one or more non-target sites (e.g., systemic circulation and/or lymphatic system) as compared to an unconjugated warhead, when delivered orally for treatment of the gastrointestinal system. In some embodiments, the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the Gl tract, leading to a high local concentration of the warhead targeting mediator(s) of gastrointestinal conditions. In some embodiments, the polymer conjugated warhead has increased residence time in the lining of the gastrointestinal tract and is able to achieve pharmacological specificity. In some embodiments, the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the intestinal epithelial cells, leading to a high local concentration of the warhead targeting mediator(s) of gastrointestinal conditions, e.g, inflammatory bowel diseases. In some embodiments, the warhead of the polymer conjugate has increased residence time in the intestinal epithelial cells and is able to achieve pharmacological specificity. In some embodiments, the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the lamina propria, leading to a high local concentration of the warhead targeting mediator(s) of gastrointestinal conditions. In some embodiments, the warhead of the polymer conjugate has increased residence time in the lamina propria and is able to achieve pharmacological specificity. In some embodiments, the warhead of the polymer conjugate has increased exposure to target cells within the intestinal lamina propria (e.g., innate and adaptive immune cells).
[0625] In several embodiments, the warhead of the LSE polymer conjugate comprises a warhead targeting mediator(s) of gastrointestinal conditions for treatment of an inflammatory bowel disease. In some embodiments, the warhead is a small molecule employed for treatment of an inflammatory bowel disease that is administered orally. In some embodiments, oral administration of the reduced exposure compositions described
herein for treatment of the gastrointestinal system has one or more of the following advantages as compared to administration of the unconjugated warhead: greater efficacy, fewer side effects, improved dosing regimens (e.g., fewer doses, lower doses), increased delivery to target cells, and/or increased residence in the submucosal layer of the intestine.
[0626] In several embodiments, the warhead of the polymer conjugate comprises a small molecule orally administered for treatment of an inflammatory bowel disease. In several embodiments, the warhead of the LSE polymer conjugate comprises a corticosteroid or an aminosalicylate. In several embodiments, the warhead of the LSE polymer conjugate comprises an aminosalicylate selected from sulfasalazine, olsalazine, mesalamine, or balsalazide. In several embodiments, the warhead of the LSE polymer conjugate comprises a corticosteroid selected from prednisone, prednisolone, methylprednisolone, or budesonide. In several embodiments, the warhead of the LSE polymer conjugate comprises an immunomodulatory small molecule selected from tacrolimus, methotrexate, mercaptopurine (6-mp), cyclosporine, or azathioprine (6-mp).
[0627] In some embodiments, the LSE polymer conjugates described herein are coupled to one or more targeting components that provide still further reduced or minimized exposure to non-target sites and/or enhanced delivery to target sites (in an additive or synergistic manner). In some embodiments, LSE polymer conjugates described are coupled to one or more components that provide more efficient delivery across the physical, physiological, and/or biological barriers of the intestinal mucosae. In some embodiments, the LSE polymer conjugates described herein fused to Cholix toxin ("Cholix") of Vibrio cholera, or to portions or derivatives thereof. In some embodiments, LSE polymer conjugate/Cholix compositions have enhanced absorption of the LSE polymer conjugate through polarized epithelial cells of the intestinal mucosa, followed by release of the LSE polymer conjugate at the basolateral side of the intestinal epithelial cells. In some embodiments, coupling of the LSE polymer conjugate to Cholix yields more efficient delivery across the physical, physiological, and biological barriers of the intestinal mucosae. In some embodiments, coupling of Cholix to the LSE polymer conjugate mediates transcytosis across the Gl tract.
[0628] In some embodiments, the LSE polymer conjugates described herein are coupled to amino acids 1 -386 of native Cholix. In some embodiments, the sequence of the targeting component coupled to the LSE polymer conjugate may vary from the native Cholix sequence, but remains, depending on the embodiment, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, or at least 95% homologous with native Cholix sequence. In several embodiments, while the targeting component may vary from native Cholix sequence, the targeting component retains, or in some
embodiments, has enhanced, trans-epithelial transport. In some embodiments, fusion of Cholix to the LSE polymer conjugate mediates transcytosis across the Gl epithelial membrane to the lamina propria. In some embodiments, the coupling of the LSE conjugate to the Cholix has an anchoring effect, wherein the interaction between the Cholix toxin and its receptor(s) at the surface of a target immune cell allows for greater exposure of the warhead at the surface of a target immune cell.
[0629] In some embodiments, the Cholix toxin and LSE polymer conjugate can be coupled by a peptide spacer consisting of one or more amino acids (e.g., up to 25 amino acids). In some embodiments, the spacer has no specific biological activity other than to join the proteins or to preserve some minimum distance or other spatial relationship between them. In some embodiments, however, the constituent amino acids of the spacer can be selected to influence some property of the molecule such as the folding, net charge, and/or hydrophobicity.
[0630] In some embodiments, a linker is capable of forming covalent bonds to both the Cholix toxin and to the LSE polymer conjugate is employed to couple the two components. Suitable linkers are well known to those of skill in the art and include, but are not limited to, straight or branched-chain carbon linkers, heterocyclic carbon linkers, or peptide linkers. In some embodiments, the LSE polymer conjugate to be delivered to a target site within the subject is coupled to the Cholix toxin using one or more cleavable linkers. In some embodiments, the cleavable linkers are cleavable by a cleaving enzyme that is present at or near the basolateral membrane of an epithelial cell. In some embodiments, the selection of a cleavable linker to be cleaved by such enzymes present at or near the basolateral membrane of an epithelial cell causes the LSE polymer conjugate to be liberated from the Cholix toxin following transcytosis across the mucous membrane and release from the epithelial cell into the cellular matrix on the basolateral side of the membrane soon after transcytosis across the epithelial membrane. In some embodiments, the enzyme that is present at a basolateral membrane of a polarized epithelial cell is selected from, e.g., Cathepsin Gl, Chymotrypsin I, Elastase I, Subtilisin Al, Subtilisin All, Thrombin I, or Urokinase I. There are Gl tract-targeted compositions provided, in several embodiments, wherein the warheads described herein are not conjugated to a polymer and are coupled to Cholix toxin.
[0631] In some embodiments, delivery of the LSE polymer conjugates to target sites with the gastrointestinal system is enhanced by encapsulation of the LSE polymer conjugate within an inflammation-targeting hydrogel. In some embodiments, hydrogel encapsulation of LSE polymer conjugate enhances delivery to inflamed portions of the colon. In some embodiments, the inflammation-targeting hydrogel is comprises ascorbyl palmitate. In some embodiments, ascorbyl palmitate self-assembles into a
nanofibrous gel. In some embodiments, the hydrogel preferentially adheres to inflamed human colon mucosa. In some embodiments, the LSE polymer conjugate is released from the hydrogel upon contact by hydrolytic enzymes at a site of inflammation. In some embodiments, delivery of the LSE polymer conjugate in the hydrogel further reduces systemic exposure. In some embodiments, the hydrogel/LSE polymer conjugate composition is administered by enema.
[0632] In several embodiments, the reduced exposure compostions disclosed herein are formulated for drug delivery as described in: Allen, L, & Ansel, H. C. (2013). Ansel's pharmaceutical dosage forms and drug delivery systems. Lippincott Williams & Wilkins; Aulton, M. E., & Taylor, K. M. (Eds.). (2017). Aulton's Pharmaceutics E-Boo : The Design and Manufacture of Medicines. Elsevier Health Sciences; and/or Wen, H., & Park, K. (Eds.). (201 1). Oral controlled release formulation design and drug delivery: theory to practice. John Wiley & Sons, each of which are herein specifically incorporated by reference.
[0633] Compounds and compositions described herein for the treatment and/or prevention of Gl conditions may be provided in a form that can be orally administered, including but not limited to liquids, tinctures, syrups, elixirs, capsules, soft gelatin capsules (gel caps), pills and tablets, lozenges, gum, powders and emulsions. Tablets or capsules may further comprise an enteric coating to prevent premature dissolution under the chemically harsh environment of the stomach. In some embodiments, enteric coatings include coatings comprising eudragit and/or cellulose acetate phthalate. Other protective coating materials include, but are not limited to, hydroxypropyl methylcellulose, polyethylene glycol and ethylcellulose. In additional embodiments, the protective coating further comprises a plasticizing agent, including but not limited to triethylcitrate and polyvinyl pyrrolidone. The tablets, capsules and other like embodiments of the disclosed compositions may further advantageously comprise particle lubricants that minimize the tendency of the granular compositions to agglomerate. By "particle lubricant" as used herein is meant the class of materials used in the manufacturing of pharmaceutical tablets as lubricants to improve the flowability and prevent agglomeration of an active agent during the tableting process. Examples of particle lubricants include, but are not limited to, talc, lactose, corn starch, ethyl cellulose, fatty acid salts such as magnesium stearate, agar pectin, fatty acids such as stearic acid, gelatin and acacia.
[0634] U.S. Patent No. 6,217,903, herein specifically incorprated by reference, discloses sustained release polymer blend pharmaceutical formulations containing active agents with other ingredients, polyethylene glycol, stearic acid, colloidal silicon dioxide, magnesium stearate, calcium stearate, waxes, polyvinyl pyrollidone. U.S.
Patent No. 6,1 10,498 to Rudnic et al., herein specifically incorprated by reference, describes an osmotic drug delivery system containing active agent by using polyvinyl pyrollidone sodium lauryl sulfate and also some plasticizer like propylene glycol, triethyl citrate, and vegetable oil. This patent provides an osmotic drug delivery system, preferably in the form of a tablet comprising various components like polymers sprayed on tablets to give 2-15 % coating weight, wicking agents, non-swelling solubilizing agents and lubricating agents. There is provided, in several embodiments, reduced exposure compsotions formualted according to the disclosures of U.S. Patent Nos. 6,217,903 and 6,1 10,498.
[0635] In some embodiments, the reduced exposure compostions provided herein are formuated in solid dosage form. In some embodiments, solid dosage forms for oral delivery include, but are not limited to, capsules, soft-gel capsules, tablets, caplets, powders, granules or other solid oral dosage forms, all of which can be prepared by methods well known in the art.
[0636] Also provided, in further embodiments, are pharmaceutical compositions additionally comprising additives in amounts customarily employed including, but not limited to, a pH adjuster, a preservative, a flavorant, a taste-masking agent, a fragrance, a humectant, a tonicifier, a colorant, a surfactant, a plasticizer, a lubricant such as magnesium stearate, a flow aid, a compression aid, a solubilizer, an excipient, a diluent such as microcrystalline cellulose, e.g. Avicel PH 102 supplied by FMC corporation, or any combination thereof. Other additives may include phosphate buffer salts, citric acid, glycols, and other dispersing agents.
[0637] In some embodiments, the reduced exposure compostions provided herein are delivered by a drug delivery system compriseing a core of water in a wax housing which melts at body temperature surrounded by a matrix of drug, water soluble or dispersible excipients and optionally, osmotic-active agents, which in turn is optionally surrounded by a coating which optionally can be semi-permeable. In some embodiments, the water of the aqueous core can be neat, an aqueous buffer, or other aqueous solubulizing system or it can be in the form of an aqueous cross-linked polymeric gelatinous pellet. The volume of available water can vary widely depending on the solubility and amount of drug to be delivered and on the site of delivery, but usually is between about 0.1 ml and 3 ml. In some embodiments, the waxy housing of the aqueous core is composed of a water-insoluble, low melting (35°-37° C.) material selected from theobroma oil, SUPPOCIRE A® (an eutectic mixture of mono-, di-, and triglycerides supplied by A. & S. Corporation, Verona, N.J., U.S.A.), WECOBEE R® (higher melting fractions of coconut oil and palm kernel oil, supplied by Drew Chemical Corp., Boonton, N.J., U.S.A.) and WITEPSOL S55® (Triglyceride of saturated vegetable fatty acids with
monoglycerides, supplied by Riches-Nelson Inc., New York, N.Y., U.S.A.). In some embodiments, the matrix surrounding the aqueous core has an outer shape and dimensions of conventional rectal or vaginal suppositories and comprises a mixture of one or more drugs and pharmaceutically acceptable water soluble and/or dispersible ingredients.
[0638] Any of the drugs used to treat the body can be incorporated as the drug of the delivery device of this invention. "Drug" is used herein in its broadest sense as including any composition or substances that will produce a pharmacologic response.
Delivery of Polymer Conjugates to the Skin
[0639] There is provided, in some embodiments, a reduced exposure composition for treatment of dermatological conditions. In several embodiments, there is provided in a reduced exposure composition, a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an inhibitor, antagonist, or inverse agonist of, for example, a mediator of a dermatological condition. In some embodiments, the reduced exposure composition for treatment of the skin is delivered topically. In some embodiments, the reduced exposure composition traverses and/or diffuses through hair follicles, skin pores, mucosa, cornea, compromised skin, the epidermis, dermis, skin, scalp, damaged skin, and/or diseased skin. In some embodiments, the reduced exposure composition penetrates one or more physical barriers of the epidermis, the dermis, and/or the subcutis. In some embodiments, the reduced exposure composition penetrates one or more biological barriers of the epidermis, the dermis, and/or the subcutis. In some embodiments, the reduced exposure composition penetrates one or more physiological barriers of the epidermis, the dermis, and/or the subcutis. In some embodiments, the reduced exposure composition targets immune cells residing in the epidermis, the dermis, and/or the subcutis. In some embodiments, the reduced exposure composition targets non-immune cells residing in the epidermis, the dermis, and/or the subcutis. In some embodiments, the reduced exposure composition targets keratinocytes in the epidermis, the dermis, and/or the subcutis. In some embodiments, the reduced exposure composition targets a single site within the epidermis, the dermis, and/or the subcutis. In some embodiments, targets site comprises two, three, four, five, six, seven, eight, nine, or more sites within the epidermis, the dermis, and/or the subcutis. In some embodiments, the reduced exposure composition has increased activity and/or bioavailability at the target site(s). In some embodiments, the reduced exposure composition has increased activity and/or bioavailability at one or more non-target site(s). In some embodiments, the one or more target site(s) comprises the systemic system, the lymphatic system, and/or non-target
tissues at which pharmacological activity is not desired. In some embodiments, the target site within the skin is selected from the group consisting of the epidermis, the dermis, and/or the subcutis, and any combination thereof. In some emboidments, the target site within the epidermis, the dermis, and the subcutis comprises one or more of the stratum corneum, stratum lucidum, stratum granulosum, stratum spinosum, and stratum basale layers, the papillary layer of the dermis, the reticular layer of the dermis, sebaceous glands, arrector pili muscles, sensory nerve fibers, hair follicles, hair roots, pacinian corpuscles, hair root plexus, eccrine sweat glands, the hypodermis and/or the superficial fascia, and any combination thereof. In some embodiments, the target comprises one, two, three, four, five, six, seven, or eight of the following sites within the skin: the stratum corneum, stratum lucidum, stratum granulosum, stratum spinosum, and stratum basale layers, the papillary layer of the dermis, the reticular layer of the dermis, sebaceous glands, arrector pili muscles, sensory nerve fibers, hair follicles, hair roots, pacinian corpuscles, hair root plexus, eccrine sweat glands, the hypodermis, and the superficial fascia. In some embodiments, the target site comprises the epidermis and the non-target site comprises one or more of the dermis and subcutis. In some embodiments, the target site comprises the dermis and the non-target site comprises one or more of the subcutis and epidermis. In some embodiments, the target site comprises the subcutis and the non-target site comprises one or more of the epidermis and dermis. In some embodiments, the target site comprises one or two of the dermis, epidermis and subcutis and the non-target site comprises the remaining one or two.
[0640] In some embodiments, the LSE polymer conjugate comprising a warhead conjugated with PEG or another molecule as described herein shows reduced absorption into, slower absorption into, and faster clearance from one or more non-target sites (e.g., systemic circulation and/or lymphatic system) as compared to an unconjugated warhead, when delivered topically for treatment of the skin. In some embodiments, the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the epidermis, the dermis, and/or the subcutis, leading to a high local concentration of the warhead targeting mediator(s) of dermatological conditions. In some embodiments, the polymer conjugated warhead has increased residence time in the epidermis, the dermis, and/or the subcutis and is able to achieve pharmacological specificity. In some embodiments, the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the epidermis, the dermis, and/or the subcutis, leading to a high local concentration of the warhead targeting mediator(s) of dermatological conditions, e.g, inflammatory skin diseases. In some embodiments, the warhead of the polymer conjugate has increased residence time in the epidermis, the dermis, and/or the subcutisand is able to achieve pharmacological specificity. In some
embodiments, the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the epidermis, the dermis, and/or the subcutis, leading to a high local concentration of the warhead targeting mediator(s) of dermatological conditions. In some embodiments, the warhead of the polymer conjugate has increased residence time in the epidermis, the dermis, and/or the subcutisand is able to achieve pharmacological specificity. In some embodiments, the warhead of the polymer conjugate has increased exposure to target cells within the epidermis, the dermis, and/or the subcutis (e.g., innate and adaptive immune cells).
[0641] In several embodiments, the warhead of the LSE polymer conjugate comprises a warhead targeting mediator(s) of dermatological conditions for treatment of an inflammatory skin disease. In some embodiments, the warhead is a small molecule employed for treatment of an inflammatory skin disease that is administered topically. In some embodiments, topical administration of the reduced exposure compositions described herein for treatment of the skin has one or more of the following advantages as compared to administration of the unconjugated warhead: greater efficacy, fewer side effects, improved dosing regimens (e.g., fewer doses, lower doses), increased delivery to target cells, and/or increased residence in the epidermis, the dermis, and/or the subcutis.
[0642] In several embodiments, the reduced exposure compostions disclosed herein are formulated for drug delivery as described in: Ansel, H. C, Popovich, N. G., & Allen, L. V. (1995). Pharmaceutical dosage forms and drug delivery systems. Lippincott Williams & Wilkins; and/or Williams, A. (2003). Transdermal and topical drug delivery: from theory to clinical practice (pp. 169-194). London: Pharmaceutical Press, each of which are herein specifically incorporated by reference.
Delivery of Polymer Conjugates to the Eye
[0643] There is provided, in some embodiments, a reduced exposure composition for treatment of ophthalmic conditions. In several embodiments, there is provided in a reduced exposure composition, a polymer conjugate comprising a warhead (e.g., at least one active agent) linked to a polymer, wherein the warhead comprises an inhibitor, antagonist, or inverse agonist of, for example, a mediator of an ophthalmic condition. In some embodiments, the reduced exposure composition for treatment of the eye is delivered topically by eye drops. In other embodiments, the the reduced exposure composition for treatment of the eye is administered by an implanted drug delivery device. In some embodiments, the compostion is delivered by intraocular or periocular injection.
[0644] In some embodiments, the reduced exposure composition traverses and/or diffuses through the conjunctival epithelium, Tenon's fascia, episclera, sclera, and/or choroid. In some embodiments, the reduced exposure composition penetrates one or more physical barriers of the anterior segment and/or posterior segment of the eye. In some embodiments, the reduced exposure composition penetrates one or more biological barriers of the anterior segment and/or posterior segment of the eye. In some embodiments, the reduced exposure composition penetrates one or more physiological barriers of the anterior segment and/or posterior segment of the eye. In some embodiments, the reduced exposure composition targets immune cells residing in anterior segment and/or posterior segment of the eye. In some embodiments, the reduced exposure composition targets non-immune cells residing in anterior segment and/or posterior segment of the eye. In some embodiments, the reduced exposure composition targets one site within the anterior segment and/or posterior segment of the eye. In some embodiments, targets site comprises two or more sites within the anterior segment and/or posterior segment of the eye. In some embodiments, the reduced exposure composition has increased activity and/or bioavailability at the target site(s). In some embodiments, the reduced exposure composition has reduced activity and/or bioavailability at one or more non-target site(s). In some embodiments, the one or more target site(s) comprises the systemic system, the lymphatic system, and/or non-target tissues at which pharmacological activity is not desired. In some embodiments, the target site within the eye is selected from the group consisting of anterior segment and/or posterior segment of the eye, and any combination thereof. In some embodiments, the target site comprises only one of the anterior segment and the posterior segment of the eye. In some embodiments, the target site comprises both the anterior segment and the posterior segment of the eye. In some emboidments, the target site comprises one or more of a posterior sub-Tenon space, a posterior suprachoroidal space and a posterior intrascleral space. In some emboidments, the target site comprises one or more of anterior sub-Tenon space, an anterior suprachoroidal space, an anterior intrascleral space. In some embodiments, the target comprises 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, or 1 1 of the following sites within the eye: cornea, lens, sclera, anterior chamber, iris, cornea, posterior chamber, choroid, retina, Bowman's layer, stroma, Descemet's membrane, the endothelium, and Tenon's Capsule. In some embodiments, the target site comprises 1 ,
2, 3, 4, 5, 6, 7, 8, 9, 10, or 1 1 of the cornea, lens, sclera, anterior chamber, iris, cornea, posterior chamber, choroid, retina, Bowman's layer, stroma, Descemet's membrane, the endothelium, and Tenon's Capsule, and the non-target site comprises the remaining 1 , 2,
3, 4, 5, 6, 7, 8, 9, 10, or 1 1 sites.
[0645] In some embodiments, the reduced exposure compsotion treats a condition which affects or which involves an ocular region or site, such as a periocular muscle, an eye lid or an eye ball tissue or fluid which is located anterior to the posterior wall of the lens capsule or ciliary muscles. In some embodiments, the target site comprises the front of the eye, such as for example, one or more of the conjunctiva, the cornea, the conjunctiva, the anterior chamber, the iris, the posterior chamber (behind the iris but in front of the posterior wall of the lens capsule), the lens and the lens capsule as well as blood vessels, lymphatics and nerves which vascularize, maintain or innervate an anterior ocular region or site. In some such embodiments, the non-target site comprises the posterior of the eye. Examples of front of the eye ocular conditions treated by such embodiments include, but are not limited to, aphakia; pseudophakia; astigmatism; blepharospasm; cataract; conjunctival diseases; conjunctivitis; corneal diseases; corneal ulcer; dry eye syndromes; eyelid diseases; lacrimal apparatus diseases; lacrimal duct obstruction; myopia; presbyopia; pupil disorders; refractive disorders and strabismus
[0646] In some embodiments, the reduced exposure compsotion treats a condition which affects or which involves the posterior (back of the eye). Posterior ocular conditions are disease, ailment or conditions which primarily affect or involve a posterior ocular region or site such as choroid or sclera (in a position posterior to a plane through the posterior wall of the lens capsule), vitreous, vitreous chamber, retina, optic nerve (i.e. the optic disc), and blood vessels and nerves which vascularize or innervate a posterior ocular region or site. In some such embodiments, the target site is one or more of the choroid or sclera (in a position posterior to a plane through the posterior wall of the lens capsule), vitreous, vitreous chamber, retina, optic nerve (i.e. the optic disc), and blood vessels and nerves which vascularize or innervate a posterior ocular region or site. In some such embodiments, the non-target site comprises the front of the eye. Examples of posterior ocular conditions treated by such embodiments include, but are not limited to, macular degeneration (such as non-exudative age related macular degeneration and exudative age related macular degeneration); choroidal neovascularization; acute macular neuroretinopathy; macular edema (such as cystoid macular edema and diabetic macular edema); Behcet's disease, retinal disorders, diabetic retinopathy (including proliferative diabetic retinopathy); retinal arterial occlusive disease; central retinal vein occlusion; uveitic retinal disease; retinal detachment; ocular trauma which affects a posterior ocular site or location; a posterior ocular condition caused by or influenced by an ocular laser treatment; posterior ocular conditions caused by or influenced by a photodynamic therapy; photocoagulation; radiation retinopathy; epiretinal membrane disorders; branch retinal vein occlusion; anterior ischemic optic neuropathy; non- retinopathy diabetic retinal dysfunction, retinitis pigmentosa and glaucoma.
[0647] Ophthalmic cancers and precancerous conditions are treated in some embodiments, including but not limited to intraocular lymphoma, intraocular lesions, intraocular melanoma (e.g., uveal melanoma). Tumors may also be treated including but not limited to tumors in the choroid, iris and ciliary body.
[0648] In some embodiments, the LSE polymer conjugate comprising a warhead conjugated with PEG or another molecule as described herein shows reduced absorption into, slower absorption into, and faster clearance from one or more non-target sites (e.g., systemic circulation and/or lymphatic system) as compared to an unconjugated warhead, when delivered topically for treatment of the eye. In some embodiments, the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the anterior segment and/or posterior segment of the eye, leading to a high local concentration of the warhead targeting mediator(s) of ophthalmic conditions. In some embodiments, the polymer conjugated warhead has increased residence time in the anterior segment and/or posterior segment of the eye and is able to achieve pharmacological specificity. In some embodiments, the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the anterior segment and/or posterior segment of the eye, leading to a high local concentration of the warhead targeting mediator(s) of ophthalmic conditions, e.g, inflammatory ophthalmic diseases. In some embodiments, the warhead of the polymer conjugate has increased residence time in the anterior segment and/or posterior segment of the eye and is able to achieve pharmacological specificity. In some embodiments, the LSE polymer conjugates described herein have enhanced delivery to and/or penetration of the anterior segment and/or posterior segment of the eye, leading to a high local concentration of the warhead targeting mediator(s) of ophthalmic conditions. In some embodiments, the warhead of the polymer conjugate has increased residence time in the anterior segment and/or posterior segment of the eye and is able to achieve pharmacological specificity. In some embodiments, the warhead of the polymer conjugate has increased exposure to target cells within the anterior segment and/or posterior segment of the eye (e.g., innate and adaptive immune cells).
[0649] In several embodiments, the warhead of the LSE polymer conjugate comprises a warhead targeting mediator(s) of ophthalmic conditions for treatment of a disease or disorder of the eye. In some embodiments, the warhead is a small molecule employed for treatment of an ophthalmic conditions that is administered topically. In some embodiments, topical administration of the reduced exposure compositions described herein for treatment of the eye has one or more of the following advantages as compared to administration of the unconjugated warhead: greater efficacy, fewer side effects, improved dosing regimens (e.g., fewer doses, lower doses), increased delivery
to target cells, and/or increased residence in the anterior segment and/or posterior segment of the eye. There is provided, in some embodiments, reduced exposure compostions suibile for topical (e.g., ointment or eye drop) application. In some embodiments, the reduced exposure compsotions disclosed herein are administered by direct intravitreal injection. In some embodiments, the reduced exposure compsotions are administered by subconjunctival injection. In other embodiments, the reduced exposure compsotions are administered by subtenon injection. In still further embodiments, the compostions are administered by peribulbar injection. Also provided, in several embodiments, are reduced exposure compostions which can be administered via intraocular implantable devices known to one of skill in the art. Intracameral injection, or injection into the anterior chamber of they eye, in contemplated in some embodiments. Injection of polymer conjugate into the vitreous is contemplated in some embodiments to provde a high local concentration of polymer conjugate in the vitreous and retina. Periocular routes of delivery, including, not limited to, subconjunctival, subtenon, retrobulbar, peribulbar and posterior juxtascleral delivery, are contemplated.
[0650] Adjuvants with which the reduced exposure compsotions may be admixed with include but are not limited to lactose, sucrose, starch powder, cellulose esters of alkanoic acids, stearic acid, talc, magnesium stearate, magnesium oxide, sodium and calcium salts of phosphoric and sulphuric acids, acacia, gelatin, sodium alginate, polyvinylpyrrolidine, and/or polyvinyl alcohol. Solubilized formulation comprising the reduced exposure compsotions provided herein may be formulated in a solvent including, but not limited to, polyethylene glycol of various molecular weights, propylene glycol, carboxymethyl cellulose colloidal solutions, methanol, ethanol, DMSO, corn oil, peanut oil, cottonseed oil, sesame oil, tragacanth gum, and/or various buffers. Other adjuvants and modes of administration are well known in the pharmaceutical art and may be used in the practice of the methods, compositions and liquid formulations described herein. In some embodiments, carriers or diluents include time delay material, such as, for example, glyceryl monostearate or glyceryl distearate alone or with a wax, or other materials well known in the art. The reduced exposure compsotion compostions for use in treatment of ophthalmic conditions as described herein may also include gel formulations, erodible and non-erodible polymers, microspheres, and liposomes. In several embodiments, the reduced exposure compostions disclosed herein are formulated with pharmaceutically acceptable excipients for clinical use to produce a pharmaceutical composition. Formulations for ocular administration may be presented as a solution, suspension, particles of solid material, a discrete mass of solid material, incorporated within a polymer matrix, liquid formulations or in any other form for ocular administration. In some embodiments, the liquid formulations described herein are
administered intraocularly. Intraocular administration includes placement or injection within the eye, including in the vitreous, is also provided herein. Injection of the compostion may be subconjunctival injection, e.g., injection of the reduced exposure compostion underneath the conjunctiva, or between the sclera and conjunctiva. In some embodiments, subtenon injection of the compsotions disclosed herein is injection of the reduced exposure compostion into the tenon's capsule around the upper portion of the eye and into the "belly" of the superior rectus muscle. Retrobulbar injection of the compostion into the conical compartment of the four rectus muscles and their intermuscular septa, behind the globe of the eye, is contemplated in some embodiments. Peribulbar injection at a location external to the confines of the four rectus muscles and their intramuscular septa (e.g., outside of the muscle cone) is further contemplated. Posterior juxtascleral delivery of the reduced exposure compostions described herein near and above the macula, in direct contact with the outer surface of the sclera, and without puncturing the eyeball, is also provided in some embodiments.
[0651] In several embodiments, the reduced exposure compostions disclosed herein are formulated for drug delivery as described in: Lang, J. C. (1995). Ocular drug delivery conventional ocular formulations. Advanced drug delivery reviews, 76(1), 39-43; and /or Le Bourlais, C, Acar, L, Zia, H., Sado, P. A., Needham, T., & Leverge, R. (1998). Ophthalmic drug delivery systems— recent advances. Progress in retinal and eye research, 77(1), 33-58, each of which are herein specifically incorporated by reference.
Synthesis of Polymer Conjugates
[0652] The description above is not intended to be limiting and should be viewed as an example to guide the manufacture of the other compounds identified herein. The polymer conjugates may also be made as described in US Patent Nos. 8,673,347 and 8,926,955, both herein incorporated by reference. Several embodiments provide a method for the production of polymer conjugates of the active agents that result in a highly pure reaction product, obtained in high and consistent yields.
[0653] In one embodiment, the conjugation reaction of the process to synthesize a conjugate polymer compound is catalysed by a base in an organic solvent. The base may be a strong base. In one embodiment, the base is selected from the group of alkali metal hydrides, tertiary amines and/or alkoxide. In another embodiment, the base catalysing the polymer conjugation reaction is sodium hydride. Other bases, such as sodium methoxide, or triethylamine can also be used. In several embodiments, the molar ratio of the base catalyst to the compound is between about 1 :1 and about 4:1 , about 1 :1 to about 1.5:1 and about 1 :1. The reaction may be carried out in an organic solvent, such as in anhydrous conditions (e.g., in a dry organic solvent). The water
content in the solution mixture of the conjugation process may be equal or less than 200 ppm. The organic solvent may be selected from the group of dichloromethane, chloroform, Ν,Ν-dimethylformamide. In certain embodiments, the organic solvent is dichloromethane or anhydrous dichloromethane.
[0654] The conjugation reaction may be carried out under inert gas atmosphere, such as nitrogen or argon atmosphere. The reaction of the process may be carried out at a temperature of about -10° to about 60° C, about 0° to about 25° C or at room temperature after an initial step at 0° C.
[0655] Following the production of the target compound, the polymer conjugate may then be separated and purified from the reaction mixture. In one embodiment, the compound is obtained by purification of the crude mixture by flash chromatography. An automated gradient flash purification system may be used and may be equipped with a suitable column and solvent. The purification method may be selected from reverse phase and direct phase columns and the conditioning/elution solvent may be selected from dichloromethane, water, methanol, acetonitrile, ammonium formate buffer solution at different mixture ratios. In one embodiment, the compound is purified by a reverse phase flash chromatography equipped with a C18 cartridge and the purification is carried out by isocratic elution with acetonitrile/5 mM ammonium formate buffer (pH 3.5) 40:60. In one embodiment, the compound is purified by a normal phase flash chromatography.
[0656] The product may then be dried e.g. over sodium sulphate and filtered off and the solvent is removed by evaporation under reduced pressure at 25° C. Purification of the target product is carried out in several embodiments. After the purification step the resultant polymer compound has a purity of at least about 95%, about 96%, about 97%, about 98%, about 98.5%, about 99% or about 99.5%. The disclosed process results in an overall mass yield of the compound from about 40% to about 98% by weight, or from about 50% to about 95% by weight based on the weight of a reactant compound.
[0657] In several embodiments, the polymer moiety which is covalently attached to the active entity is biocompatible, can be of natural or semi-synthetic or synthetic origin and can have a linear or branched structure. The polymer may be selected from poly(alkylene oxides), or from (polyethylene) oxides. However, other polymers include without limitation polyacrylic acid, polyacrylates, polyacrylamide or N- alkyl derivatives thereof, polymethacrylic acid, polymethacrylates, polyethylacrylic acid, polyethylacrylates, polyvinylpyrrolidone, poly(vinylalcohol), polyglycolic acid, polylactic acid, poly(lactic-co-glycolic) acid, dextran, chitosan, hydroxyethyl starch.
[0658] In some embodiments, the above-mentioned polymer moiety can carry an amino functional end-group or can be functionalized to carry an amino functional end-group. Hence, the polymer moiety can be an amino-activated polymer of general formula X— NH2.
[0659] The reaction of formation of the compositions identified herein may be carried out at a temperature of about 10° to about 60° C, about 15° to about 25° C. or at room temperature. The polymer moiety X may be a polyethylene glycol (PEG) moiety, wherein the terminal OH group can optionally be modified e.g. with C1 -C5 alkyl or C1 - C5 acyl groups, such as with C1 -, C2- or C3-alkyl groups or C1 -, C2- or C3 groups. The modified polyethylene glycol may be a terminally alkoxy-substituted polyethylene glycol, including a methoxy-polyethylene-glycol (mPEG).
[0660] In other aspects, the conjugated polymer compounds may be used as active agents in a topical medicament useful for the prevention, alleviation and/or treatment of dermal pathologies. It has been shown that the conjugated polymer compounds described herein are very advantageously used as topical medicament since they do not show adverse or toxic effects (e.g. irritation) when dermally administered or any phototoxic effect (e.g. photomutagenicity, phototoxicity or photosensitisation) (as shown in the studies described in the following examples).
[0661] The dermal pathologies for such treatment may be pathologies characterized by hyperproliferation of the keratinocytes, such as psoriasis, atopic dermatitis, chronic eczema, acne, pitiriasis rubra pilaris, keloids, hypertrophic scars and skin tumors, such as keratoacanthoma, squamous cell carcinoma, basal cell carcinoma.
[0662] The polymer conjugate compounds may be used in combination with at least one natural extract or essential oil which is anti-itching agent, for example and not restricted to, extracts of Abelmoschus esculentus, Actaea alba, Aglaia odorata, Alkanna tinctoria, Althaea officinalis, Altingia excelsa, Andropogon virginicus, Aralia nudicaulis, Aralia racemosa, Argemone mexicana, Barleria prionitis, Camelia sinensis, Caesalpinia digyna, Campsis grand/flora, Carissa congesta, Carthamus oxyacantha, Cassia tora, Chrysanthemum indicum, Cimicifuga racemosa, Cinnamomum camphora, Clematis vitalba, Cuscuta reflexa, Diospyros peregrina, Enicostema axillare, Hammamelis virginiana, Jatropha multifida, Lavandula officinalis, Lavandula latifolia, Liquidambar orientalis, Lithospermum officinale, Madhuca longifolia, Martynia annua, Medicago sativa, Michelia champaca, Mikania glomerata, Mimosa pudica, Oryza sativa, Phaseolus vulgaris, Phyllanthus urinaria, Phyllanthus virgatus, Pistacia vera, Polygonum hydropiper, Quercus ilex, Rauvolfia caffra, Ricinus communis, Rubus idaeus, Sagittaria sagittifolia, Sandoricum koetjape, Sapindus mukorossi, Schleichera oleosa, Sesbania grandi flora, Spondias dulcis, Tilia sp., Toona ciliata, Tragia involucrata, Trichosanthes
quinquangulata, Vaccaria pyramidata, Ventilago madraspatana, Veratrum album or Xanthium strumarium among others
The polymer conjugate compounds may be used in combination with at least one synthetic compound or product of biotechnological origin which is an anti- itching agent, for example and not restricted to mepyramine (pyrilamine), antazoline, diphenhydramine, carbinoxamine, doxylamine, clemastine, dimenhydrinate, pheniramine, chlorphenamine (chlorpheniramine), dexchlorpheniramine, brompheniramine, triprolidine, cyclizine, chlorcyclizine, hydroxyzine, meclizine, cetirizine, levocetirizine, promethazine, thenaldine, alimemazine (trimeprazine), cyproheptadine, azatidine, ketotifen, acrivastine, astemizole, cetirizine, loratadine, desloratadine, mizolastine, terfenadine, fexofenadine, fexofenadine, azelastine, levocabastine, olopatadine, corticosteroids such as cortisone, hydrocortisone dexamethasone, prednisone; Neutrazen™ [INCI: Water, Butylene Glycol, Dextran, Palmitoyl Tripeptide-8] marketed by Atrium Innovations/Unipex Group, Meliprene [INCI: Dextran, Acetyl Heptapeptide-1 ] marketed by Institut Europeen de Biologie Cellulaire/Unipex Group, Skinasensyl™ [INCI: Acetyl Tetrapeptide-15] marketed by Laboratoires Serobiologiques/Cognis, SymSitive® 1609 [INCI: 4-t-Butylcyclohexanol] marketed by Symrise, Symbiocell™ [INCI: Extract from Cestrum Latifolium] marketed by BASF, Gatuline®Derma-Sensitive [INCI: Octyldodecyl Myristate, Capparis Spinosa Fruit Extract] marketed by Gattefosse or MAXnolia [INCI: Magnolia Officinalis Bark Extract, Vitis Vinifera/Vitis Vinifera (Grape) Seed Extract, Tocopherol] marketed by Mibelle among others.
[0664] The polymer conjugate compounds may be used in combination with at least one physiological cooling agent, for example and not restricted to menthone glycerol acetal, menthyl lactate, menthyl ethyl oxamate, substituted menthyl-3-carboxylic acid amides (e.g. menthyl-3-carboxylic acid N-ethylamide, Na-(L- menthanecarbonyl)glycine ethyl ester, 2-isopropyl-N-2,3-trimethylbutanamide, substituted cyclohexanecarboxylic acid amides, 3-menthoxypropane-1 ,2-diol, 2- hydroxyethyl menthyl carbonate, 2- hydroxy propyl menthyl carbonate, N- acetylglycine menthyl ester, isopulegol, menthyl hydroxycarboxylic acid esters (e.g. menthyl 3- hydroxybutyrate), monomenthyl succinate, monomenthyl glutarate, 2- mercaptocyclodecanone, menthyl 2-pyrrolidin-5-onecarboxylate, 2,3-dihydroxy-p- menthane, 3,3,5-trimethylcyclohexanone glycerol ketal, 3-menthyl 3,6-di- and - trioxaalkanoates, 3-menthyl methoxyacetate and icilin.
[0665] The compounds disclosed herein or pharmaceutically acceptable salts thereof can be administered as they are, or in the form of various pharmaceutical compositions according to the pharmacological activity and the purpose of
administration. Yet another aspect is a pharmaceutical composition comprising an effective amount of at least one compound in Tables 1 -3 optionally together with pharmaceutically acceptable carriers, adjuvants, diluents or/and additives. Pharmaceutical carriers, adjuvants, diluents or/and additives are applied in the formulation of the pharmaceutical composition comprising a compound of embodiments identified herein.
[0666] The disclosed compounds can be employed as the sole active agent in a pharmaceutical composition. Alternatively, the compounds of Tables 1 -3 may be used in combination with one or several further active agents, e.g. other active pharmaceutical agents in the treatment of the conditions described herein.
[0667] In particular, the polymer conjugate compounds may be used in combination with at least one endogenous angiogenesis inhibitor, for example and not restricted to, angioarrestin, angiostatin (plasminogen fragment), antiangiogenic antithrombin III, cartilage-derived inhibitor (CDI), CD59 complement fragment, endostatin (collagen XVIII fragment), fibronectin fragment, Gro-beta, heparinases, heparin hexasaccharide fragment, human chorionic gonadotropin (hCG), interferon alpha/beta/gamma, interferon inducible protein (IP-10), lnterleukin-12, kringle 5 (plasminogen fragment), metalloproteinase inhibitors (TIMPs), 2-methoxyestradiol, placental ribonuclease inhibitor, plasminogen activator inhibitor, platelet factor-4 (PF4), prolactin 16 kD fragment, proliferin-related protein (PRP), retinoids, tetrahydrocortisol-S, thrombospondin-1 (TSP-1), transforming growth factor-beta or (TGF-b), vasculostatin, and vasostatin (calreticulin fragment). In particular, the polymer conjugate compounds may be used in combination with at least one additional anti-IBD therapeutic agent, for example and not restricted to azathioprine, 6-mercaptopurine (6-MP), aminosalicylate, sulfasalazine, mesalamine, corticosteroid, prednisone, prednisone equivalent, budesonide, probiotic, methotrexate, cyclosporine, tacrolimus, metronidazole, ciprofloxacin, leflunomide, chloroquine, hydroxychloroquine, penicillamine, tocilzumab, anakinra, abatacept, rituximab, efalizumab, belimumab, tofacitinib, baricitinib, golimumab, vedolizumab, natalizumab, ustekinumab, etanercept, infliximab, adalimumab, certolizumab pegol
[0668] In particular, the polymer conjugate compounds may be used in combination with at least one steroidal anti-inflammatory drug and/or one further agent capable of inhibiting an early mediator of the inflammatory cytokine cascade, e.g. an antagonist or inhibitor of a cytokine selected from the group consisting of TNF, IL-1 a, IL- 1 β, IL-Ra, IL-8, MIP-1 a, MIF-Ι β, MIP-2, MIF and IL-6. Particularly useful antiinflammatory drugs are selected from alclometasone dipropionate, amcinonide, beclomethasone dipropionate, betamethasone, betamethasone benzoate,
betamethasone dipropionate, betamethasone sodium phosphate, betamethasone sodium phosphate and acetate, betamethasone valerate, clobetasol butyrate, clobetasol propinate, clocortolone pivalate, Cortisol (hydrocortisone), Cortisol (hydrocortisone) acetate, Cortisol (hydrocortisone) butyrate, Cortisol (hydrocortisone) cypionate, Cortisol (hydrocortisone) sodium phosphate, Cortisol (hydrocortisone) sodium succinate, Cortisol (hydrocortisone) valerate, cortisone acetate, desonide, desoximetasone, dexamethasone, dexamethasone acetate, dexamethasone sodium phosphate, diflorasone diacetate, diflucortolone valerate, fludrocortisone acetate, fludroxycortide, flumetasone pivalate, flunisolide, fluocinolone acetonide, fluocinonide, fluocortolone, fluorometholone, flurandrenolide, fluticasone propionate, halcinonide, halobetasol propionate, medrysone, methylprednisolone, methylprednisolone acetate, methylprednisolone sodium succinate, mometasone furoate, paramethasone acetate, prednisolone, prednisolone acetate, prednisolone sodium phosphate, prednisolone tebutate, prednisone, triamcinolone, triamcinolone acetate, triamcinolone acetonide, triamcinolone diacetate, triamcinolone hexacetonide. Useful antagonists or inhibitors of a cytokine are selected from infliximab, etanercept or adalimumab.
[0669] Further agents which can be used in combination with the polymer compounds are e.g. antagonists and/or inhibitors of RAGE, antagonists and/or inhibitors of HMGB1 , antagonists and/or inhibitors of the interaction of a Toll-like receptor (TCR) with HMGB1 , the functional N-terminal lectin-like domain (D1) of thrombomodulin and/or a synthetic double-stranded nucleic acid or nucleic acid analogue molecule with a bent shape structure as described in the international patent application WO 2006/002971 which is herein incorporated by reference.
[0670] In some cases, ophthalmic conditions require antibiotics or other antimicrobials. For example, antibiotics may be useful to treat pink eye (conjunctivitis) or other eye infections, corneal abrasions, or pre/post-surgery (such as cataract surgery). Thus, in several embodiments, the warhead of the LSE polymer conjugate comprises an antibiotic selected from one or more of azithromycin, tobramycin, bacitracin, chloramphenicol, polymyxin B, gentamycin, sulfacetamide, moxifloxacin, gatifloxacin, neomycin, polymyxin and besifloxacin. In addition to or instead of antibiotics, other therapeutic agents such as antihistamines, nonsteroidal anti-inflammatory drugs, and corticosteroids may be used (such as ophthalmic injections, eye drops, eye ointments, etc.). Combinations of active agents may be used in some embodiments. In several embodiments, the warhead of the LSE polymer conjugate comprises a non- systemic antibiotic that is minimally absorbed and has high local concentrations in the eye after injection, eye drops, ointments, etc. Such delivery compositions are able to, in some embodiments, deliver localized doses of antibiotics (or other antimicrobials) to the
eye without significant exposure to other tissue which, advantageously, limits the side effects. Side effects include but are not limited to rash, itching or burning eyes, pain, redness, swelling, and vision problems. Also, in some embodiments, localized and precision-based treatment permits lower doses of the antibiotics/antimicrobials, which in some embodiments may reduce or avoid toxicity, adverse immune effects, tolerance, etc. In some embodiments, systemic exposure and/or exposure to a non-target tissue of antimicrobials (antifungals, antibacterials, antivirals, etc.) is reduced by at least 25%, 50%, 75% or more using the embodiments described herein (e.g., LSE polymer conjugates) as compared with administration of the same antimicrobial without the polymer conjugates described herein. The compositions described herein may be administered by a physician or other professional. Patients may also be able to self- administer. In several embodiments, administration of the composition may be performed dermally, via, for example, ointments, creams, oils, liposomes or trans-dermal patches, or wherein the polymer conjugates are incorporated into liposomes.
[0671] In some embodiments, at least one excipient is provided. Excipients can include a nonaqueous or aqueous carrier, and one or more agents selected from moisturizing agents, pH adjusting agents, strontium ions (Sr2+), deodorants, fragrances, chelating agents, preservatives, emulsifiers, thickeners, solubilizing agents, penetration enhancers, anti-irritants, colorants, surfactants, beneficial agents, pharmaceutical agents, and other components for use in connection with the compositions described herein (such as oral compositions for treatment of the gastrointestinal tract or topical compositions for treatment of the skin). In several embodiments, the composition is an anhydrous formulation to prevent skin irritation such as water-based irritant contact dermatitis or stinging sensation upon application to damaged skin. In another embodiment, the composition is formulated such that preservatives need not necessarily be employed (e.g., a preservative-free formulation) so as to avoid skin irritation associated with certain preservatives
[0672] To facilitate application, the composition may be provided as an ointment, an oil, a lotion, a paste, a powder, a gel, or a cream. The composition may also include additional ingredients such as a protective agent, an emollient, an astringent, a humectant, a sun screening agent, a sun tanning agent, a UV absorbing agent, an antibiotic agent, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anti-acne agent, an anesthetic agent, a steroidal anti-inflammatory agent, a nonsteroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an anti-histamine agent, a vitamin or vitamin complex, a hormone, an anti-dandruff agent, an anti-wrinkle agent, an anti-skin atrophy agent, a skin whitening agent, a cleansing agent, additional peptides, additional modified peptides,
and combinations thereof. In a further embodiment, the composition may avoid irritants (such as animal or cellular-based materials) to avoid skin irritation.
[0673] In some embodiments, the compositions may be administered by injection or infusion, in particular by intravenous, intramuscular, transmucosal, subcutaneous or intraperitoneal injection or infusion and/or by oral, topical, dermal, nasal, inhalation, aerosol and/or rectal application, etc.
[0674] In a further embodiment, the compositions are administered reversibly immobilized on the surface of a medical device, in particular by binding, coating and/or embedding the compositions on a medical device, such as but not limited to, stents, catheters, surgical instruments, cannulae, cardiac valves, or vascular prostheses. After contacting the medical device with body fluid or body tissue, the reversibly immobilized compounds are liberated. Consequently, the coated medical devices act as drug delivery devices eluting the medicament, whereby the drug delivery kinetics can be controlled, providing an immediate release or a controlled, delayed or sustained drug delivery, for example.
[0675] In some embodiments, the composition further comprises an enteric coating that resists degradation under the prevailing pH of the stomach and permits delivery to specific regions of the gastrointestinal tract.
[0676] The pharmaceutical compositions may also be used for diagnostic or for therapeutic applications. For diagnostic applications, the compound may be present in a labelled form, e.g. in a form containing an isotope, e.g. a radioactive isotope or an isotope which may be detected by nuclear magnetic resonance. In some embodiments, a therapeutic application is, in the case of a topical application, the prevention, alleviation and treatment of psoriasis and dermatitis.
[0677] The concentrations of the compounds in the pharmaceutical composition can vary. The concentration will depend upon factors such as the total dosage of the drug to be administered, the chemical characteristics (e.g., hydrophobicity) of the compounds employed, the route of administration, the age, body weight and symptoms of a patient. The compounds typically are provided in an aqueous physiological buffer solution containing about 0.1 to 10% w/v compound for topical administration. Typical dose ranges are from about 1 μg to about 1 g/kg of body weight per day; a dose range may be from about 0.01 mg/kg to 100 mg/kg of body weight per day, or about 0.1 to 20 mg/kg once to four times per day. In some embodiments, the dosage of the drug to be administered is likely to depend on variables such as the type and extent of the progression of the disease or disorder, the overall health status of the particular patient, the relative biological efficacy of the selected compound and the formulation of the compound excipient, and its route of administration.
[0678] Although the foregoing has been described in some detail by way of illustrations and examples for purposes of clarity and understanding, it will be understood by those of skill in the art that modifications can be made without departing from the spirit of the present disclosure. Therefore, it should be clearly understood that the forms disclosed herein are illustrative only and are not intended to limit the scope of the present disclosure, but rather to also cover all modification and alternatives coming with the true scope and spirit of the embodiments of the invention(s).
[0679] Terms and phrases used in this application, and variations thereof, especially in the appended claims, unless otherwise expressly stated, should be construed as open ended as opposed to limiting. As examples of the foregoing, the term 'including' should be read to mean 'including, without limitation,' 'including but not limited to,' or the like.
[0680] The indefinite article "a" or "an" does not exclude a plurality. The term "about" as used herein to, for example, define the values and ranges of molecular weights means that the indicated values and/or range limits can vary within ±20%, e.g., within ±10%. The use of "about" before a number includes the number itself. For example, "about 5" provides express support for "5".
[0681] The phrases "active agent" and "active entity" are synonyms and can be used interchangeably.
[0682] The terms "CT352" and "SNA-352" are synonyms and can be used interchangeably. The terms "CT101" and "SNA-101 " are synonyms and can be used interchangeably. The terms "CT103" and "SNA-103" are synonyms and can be used interchangeably. The terms "CT340" and "SNA-125" are synonyms and can be used interchangeably. The terms "CT327" and "SNA-120" are synonyms and can be used interchangeably.
EXAMPLES
[0683] Non-limiting examples are provided below.
Example 1 : Profiling study of CT101 and CAS944795-066 against 271 kinases
[0684] CT101 and CAS944795-06-6 (test concentration: 0.2 μΜ) were tested against 271 target kinases.
Materials and Methods
Preparation of test compound solution
[0685] The test compound was dissolved in and diluted with dimethylsulfoxide (DMSO) to achieve 100-fold higher concentration. Then the solution was further 25-fold diluted with assay buffer to make the final test compound solution. Reference compounds for assay control were prepared similarly.
Assay reagents and procedures
IMAP Assay
[0686] 1) The 5 μΙ_ of x4 compound solution, 5 μΙ_ of x4 Substrate/ATP/Metal solution, and 10 μΙ_ of x2 kinase solution were prepared with assay buffer (20 mM HEPES, 0.01 % Tween-20, 2 mM DTT, pH7.4) and mixed and incubated in a well of polystyrene 384 well black microplate for 1 hour at room temperature.
[0687] 2) 60 μΙ_ of IMAP binding reagent (IMAP Screening Express kit; Molecular Devices) was added to the well, and incubated for 30 minutes.
[0688] 3) The kinase reaction was evaluated by the fluorescence polarization at 485 nm for excitation and 530 nm for emission of the well.
Off-chip Mobility Shift Assay (MSA)
[0689] 1) The 5 μΙ_ of x4 compound solution. 5 μΙ_ of x4 Substrate/ATP/Metal solution, and 10 μΙ_ of x2 kinase solution were prepared with assay buffer (20 mM HEPES, 0.01 % Triton X-100, 2 mM DTT, pH7.5) and mixed and incubated in a well of polypropylene 384 well microplate for I or 5 hour(s)* at room temperature. (*; depend on kinase)
[0690] 2) 60 μΙ_ of Termination Buffer (QuickScout Screening Assist MSA; Carna Biosciences) was added to the well.
[0691] 3) The reaction mixture was applied to LabChip3000 system (Caliper Life Science), and the product and substrate peptide peaks were separated and quantitated.
[0692] 4) The kinase reaction was evaluated by the product ratio calculated from peak heights of product(P) and substrate(S) peptides (P/(P+S)).
Reaction Conditions
[0693] The reaction conditions are depicted below:

-212-

-213-

-214-




Data Analysis
[0694] The readout value of reaction control (complete reaction mixture) was set as a 0% inhibition, and the readout value of background (Enzyme(-)) was set as a 100% inhibition, then the percent inhibition of each test solution was calculated.
Results
[0695] The results are shown in Table 4 below.
Table 4: CT101 % Inhibition results
AAA AA AA?
ACE Ζβ 34A
AIR AA AA
A.RO A 77A
AXL AA AA
BLR AA 43
8MX ίΑ SIA 1 A,4 ?AA


44 11
•44 4,1
4 J i
Hu : -4:3 4
1?
41 •13
~2, 44 f:f:I?« *u §4
IK Ha 44 44
!lK3 44
44 -4,4
44
•4i -14

m.2 44 44 mm 44 44
4 43
■4 44 mm 43 4 m 44


k ; 4 A 4A
4Λ -A
«1 44 A
tlio 44 44 tllA 4 J
∞ 4,$ 44
4AI 4,11
mm 4A A A ill. 4A 2AA
4 •4 A
SLA. SA 1

Example 2: IC50 Determination study of CT101 against 4 kinases
[0696] CT101 inhibition of BTK, FYN, and PKCs were examined at the following test concentrations: 50, 15, 5, 1 .5, 0.5, 0.15, 0.05, 0.015, 0.005, 0.0015 μΜ.
[0697] CT101 inhibition of SRC was examined at the following test concentrations: 30, 10, 3, 1 , 0.3, 0.1 , 0.03, 0.01 , 0.003, 0.001 μΜ.
Materials and Methods
Preparation of test compound solution
[0698] The test compound was dissolved in and diluted with dimethylsulfoxide (DMSO) to achieve 100-fold higher concentration. Then the solution was further 25-fold diluted with assay buffer to make the final test compound solution. Reference compounds for assay control were prepared similarly.
Assay reagents and procedures
Off-chip Mobility Shift Assay (MSA)
[0699] 1 ) The 5 μΙ_ of x4 compound solution. 5 μΙ_ of x4 Substrate/ATP/Metal solution, and 10 μΙ_ of x2 kinase solution were prepared with assay buffer (20 mM HEPES, 0.01 % Triton X-100, 2 mM DTT, pH7.5) and mixed and incubated in a well of polypropylene 384 well microplate for 1 hour at room temperature.
[0700] 2) 60 μΙ_ of Termination Buffer (QuickScout Screening Assist MSA; Carna Biosciences) was added to the well.
[0701] 3) The reaction mixture was applied to LabChip3000 system (Caliper Life Science) , and the product and substrate peptide peaks were separated and quantitated.
[0702] 4) The kinase reaction was evaluated by the product ratio calculated from peak heights of product(P) and substrate(S) peptides (P/(P+S)) .
Reaction Conditions
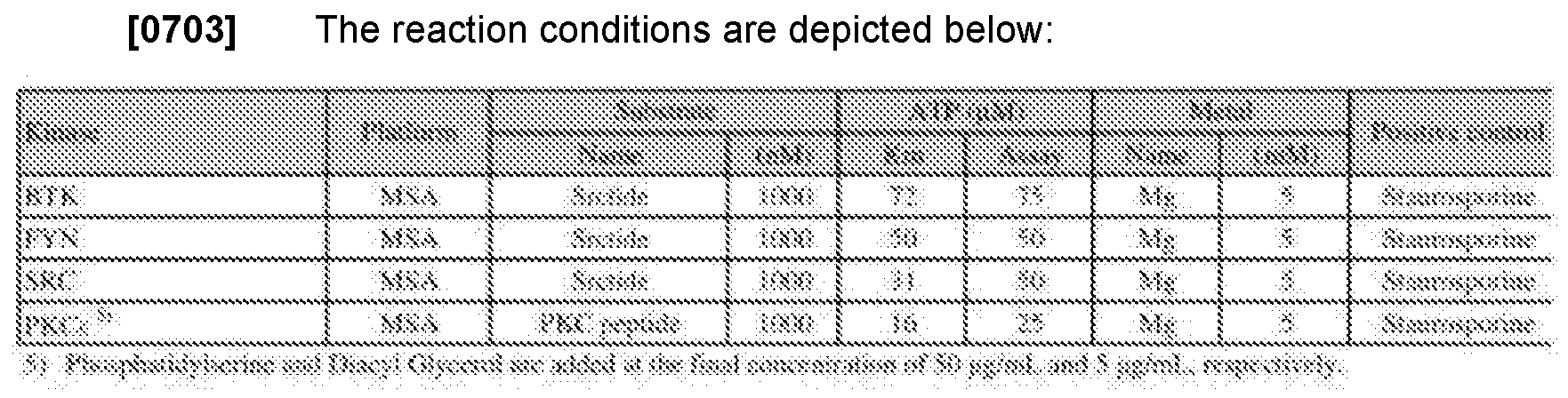
Data Analysis
[0704] The readout value of reaction control (complete reaction mixture) was set as a 0% inhibition, and the readout value of background (Enzyme(-)) was set as a 100% inhibition, then the percent inhibition of each test solution was calculated.
[0705] IC5o value was calculated from concentration vs. %lnhibition curves by fitting to a four parameter logistic curve.
Results
[0706] The results are shown in Table 5 below.
Table 5: IC50 Determination

Example 3: BioMAP Platform Analysis of SNA-101
Aim of Study
[0707] The goal of this study was to characterize SNA-101 in the BioMAP Diversity PLUS panel of 12 human primary cell-based systems. These systems are designed to model complex human tissue and disease biology of the vasculature, skin, lung and inflammatory tissues. Quantitative measurements of biomarker activities across this broad panel, along with comparative analysis of the biological activities of known bioactive agents in the BioMAP reference database are used to predict the safety, efficacy and function of these test agents.
Overview of BioMAP Technology Platform
[0708] BioMAP panels consist of human primary cell-based systems designed to model different aspects of the human body in an in vitro format. The 12 systems in the Diversity PLUS panel allow test agent characterization in an unbiased way across a broad set of systems modeling various human disease states. BioMAP systems are constructed with one or more primary cell types from healthy human donors, with stimuli (such as cytokines or growth factors) added to capture relevant signaling networks that naturally occur in human tissue or pathological conditions. Vascular biology is modeled in both a Th1 (3C system) and a Th2 (4H system) inflammatory environment, as well as in a Th1 inflammatory state specific to arterial smooth muscle cells (CASM3C system). Additional systems recapitulate aspects of the systemic immune response including monocyte-driven Th1 inflammation (LPS system) or T cell
stimulation (SAg system), chronic Th1 inflammation driven by macrophage activation (/Mphg system) and the T cell-dependent activation of B cells that occurs in germinal centers (BT system). The BE3C system (Th1) and the BF4T system (Th2) represent airway inflammation of the lung, while the MyoF system models myofibroblast-lung tissue remodeling. Lastly, skin biology is addressed in the KF3CT system modeling Th1 cutaneous inflammation and the HDF3CGF system modeling wound healing.
[0709] Each test agent generates a signature BioMAP profile that is created from the changes in protein biomarker readouts within individual system environments. Biomarker readouts (7 - 17 per system) are selected for therapeutic and biological relevance, are predictive for disease outcomes or specific drug effects and are validated using agents with known mechanism of action (MoA). Each readout is measured quantitatively by immune-based methods that detect protein (e.g., ELISA) or functional assays that measure proliferation and viability. BioMAP readouts are diverse and include cell surface receptors, cytokines, chemokines, matrix molecules and enzymes. In total, the Diversity PLUS panel contains 148 biomarker readouts that capture biological changes that occur within the physiological context of the particular BioMAP system.
[0710] Using custom-designed software containing data mining tools, a BioMAP profile can be compared against a proprietary reference database of > 4,000 BioMAP profiles of bioactive agents (biologies, approved drugs, chemicals and experimental agents) to classify and identify the most similar profiles. This robust data platform allows rapid evaluation and interpretation of BioMAP profiles by performing the unbiased mathematical identification of similar activities. Specific BioMAP activities have been correlated to in vivo biology, and multiparameter BioMAP profiles have been used to distinguish compounds based on MoA and target selectivity and can provide a predictive signature for in vivo toxicological outcomes (e.g., vascular toxicity, developmental toxicity, etc.) across diverse physiological systems.
Materials and Methods
Test Agent
[0711] SNA-101 was profiled in the BioMAP Diversity PLUS panel at concentrations of 29 μΜ, 9.7 μΜ, 3.2 μΜ, and 1 .1 μΜ. Apremilast was employed as the benchmark compound.
Methods for Diversity PLUS
[0712] Human primary cells in BioMAP systems are used at early passage (passage 4 or earlier) to minimize adaptation to cell culture conditions and preserve
physiological signaling responses. All cells are from a pool of multiple donors (n = 2 to 6), commercially purchased and handled according to the recommendations of the manufacturers. Human blood derived CD14+ monocytes are differentiated into macrophages in vitro before being added to the /Mphg system. Abbreviations are used as follows: Human umbilical vein endothelial cells (HUVEC), Peripheral blood mononuclear cells (PBMC), Human neonatal dermal fibroblasts (HDFn), B cell receptor (BCR), T cell receptor (TCR) and Toll-like receptor (TLR).
[0713] Cell types and stimuli used in each system are as follows: 3C system [HUVEC + (ΙΙ_-1 β, TNFa and IFNy)], 4H system [HUVEC + (IL-4 and histamine)], LPS system [PBMC and HUVEC + LPS (TLR4 ligand)], SAg system [PBMC and HUVEC + TCR ligands], BT system [CD19+ B cells and PBMC + (a-lgM and TCR ligands)], BF4T system [bronchial epithelial cells and HDFn + (TNFa and IL-4)], BE3C system [bronchial epithelial cells + (IL-Ι β, TNFa and IFNy)], CASM3C system [coronary artery smooth muscle cells + (IL-1 β, TNFa and IFNy)], HDF3CGF system [HDFn + (IL-Ι β, TNFa, IFNy, EGF, bFGF and PDGF-BB)], KF3CT system [keratinocytes and HDFn + (IL-1 β, TNFa and IFNy)], MyoF system [differentiated lung myofibroblasts + (TNFa and TGFp)] and /Mphg system [HUVEC and M 1 macrophages + Zymosan (TLR2 ligand)].
[0714] Systems are derived from either single cell types or co-culture systems. Adherent cell types are cultured in 96 or 384-well plates until confluence, followed by the addition of PBMC (SAg and LPS systems). The BT system consists of CD19+ B cells co-cultured with PBMC and stimulated with a BCR activator and low levels of TCR stimulation. Test agents prepared in either DMSO (small molecules; final concentration < 0.1 %) or PBS (biologies) are added at the indicated concentrations 1 -hr before stimulation, and remain in culture for 24-hrs or as otherwise indicated (48-hrs, MyoF system; 72-hrs, BT system (soluble readouts); 168-hrs, BT system (secreted IgG)). Each plate contains drug controls (e.g., legacy control test agent colchicine at 1 .1 μΜ), negative controls (e.g., non-stimulated conditions) and vehicle controls (e.g., 0.1 % DMSO) appropriate for each system. Direct ELISA is used to measure biomarker levels of cell-associated and cell membrane targets. Soluble factors from supernatants are quantified using either HTRF® detection, bead-based multiplex immunoassay or capture ELISA. Overt adverse effects of test agents on cell proliferation and viability (cytotoxicity) are detected by sulforhodamine B (SRB) staining, for adherent cells, and alamarBlue® reduction for cells in suspension. For proliferation assays, individual cell types are cultured at subconfluence and measured at time points optimized for each system (48- hrs: 3C and CASM3C systems; 72-hrs: BT and HDF3CGF systems; 96-hrs: SAg system). Cytotoxicity for adherent cells is measured by SRB (24-hrs: 3C, 4H, LPS, SAg, BF4T, BE3C, CASM3C, HDF3CGF, KF3CT, and IMphg systems; 48-hrs: MyoF system),
and by alamarBlue staining for cells in suspension (24-hrs: SAg system; 42-hrs: BT system) at the time points indicated. Additional information can be found in previous descriptions.
Data Analysis
[0715] Biomarker measurements in a test agent-treated sample are divided by the average of control samples (at least 6 vehicle controls from the same plate) to generate a ratio that is then Iog10 transformed. Significance prediction envelopes are calculated using historical vehicle control data at a 95% confidence interval.
Profile Analysis
[0716] Biomarker activities are annotated when 2 or more consecutive concentrations change in the same direction relative to vehicle controls, are outside of the significance envelope and have at least one concentration with an effect size > 20% (|log10 ratio| > 0.1). Biomarker key activities are described as modulated if these activities increase in some systems, but decrease in others. Cytotoxic conditions are noted when total protein levels decrease by more than 50% (log 10 ratio of SRB or alamarBlue levels < -0.3) and are indicated by a thin black arrow above the X-axis. A compound is considered to have broad cytotoxicity when cytotoxicity is detected in 3 or more systems. Concentrations of test agents with detectable broad cytotoxicity are excluded from biomarker activity annotation and downstream benchmarking, similarity search and cluster analysis. Antiproliferative effects are defined by an SRB or alamarBlue log 10 ratio value < -0.1 from cells plated at a lower density and are indicated by grey arrows above the X-axis. Cytotoxicity and antiproliferative arrows may only require one concentration to meet the indicated threshold for profile annotation.
Benchmark Analysis
[0717] Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% in the same direction. Differentiating biomarkers are annotated when one profile has a readout outside of the significance envelope with an effect size > 20%, and the readout for the other profile is either inside the envelope or in the opposite direction. Unless specified, the top non-cytotoxic concentration of both the test agent and benchmark agent are included in the benchmark overlay analysis.
Similarity Analysis
[0718] Common biomarker readouts are annotated when the readout for both profiles is outside of the significance envelope with an effect size > 20% in the same direction. Concentrations of test agents that have 3 or more detectable systems with cytotoxicity are excluded from similarity analysis. Concentrations of test agents that have 1 - 2 systems with detectable cytotoxicity will be included in the similarity search analysis, along with an overlay of the database match with the top concentration of the test agent. This will be followed by an additional overlay of the next highest concentration of the test agent containing no systems with detectable cytotoxicity and the respective database match. To determine the extent of similarity between BioMAP profiles of compounds run in the Diversity PLUS panel, we have developed a custom similarity metric (BioMAP Z-Standard) that is a combinatorial approach that has improved performance in mechanism classification of reference agents compared to other measures tested (including Pearson's and Spearman's correlation coefficients). This approach more effectively accounts for variations in the number of data points, systems, active biomarker readouts and the amplitude of biomarker readout changes that are characteristic features of BioMAP profiles. A Pearson's correlation coefficient (r) is first generated to measure the linear association between two profiles that is based on the similarity in the direction and magnitude of the relationship. Since the Pearson's correlation can be influenced by the magnitude of any biomarker activity, a per-system weighted average Tanimoto metric is used as a filter to account for underrepresentation of less robust systems. The Tanimoto metric does not consider the amplitude of biomarker activity, but addresses whether the identity and number of readouts are in common on a weighted, per system basis. A real-value Tanimoto metric is calculated
A
\\A\\
first by normalizing each profile to the unit vector (e.g., A = W II ) and then applying the
A B II II _|_ llg II _ A . β
following formula: II II II II , where A and B are the 2 profile vectors. Then, it is incorporated into a system weighted-averaged real-value Tanimoto metric in this calculation: ∑^ . The calculation uses the real-value Tanimoto score for each rth system (T,) and the weight of each rth system (W,). W, is calculated for each system in
1
1 + (2 _i 00 /r - 0 09 )
the following formula: 7 , where Ir is the largest absolute value of the ratios from the 2 profiles being compared. Based on the optimal performance of
reference compounds, profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient (r) > 0.7. Finally, a Fisher r-to-z-transformation is used to calculate a z-score to convert a short tail distribution into a normal distribution as
1 + r
follows: z= 0.5 log10 1 _ r . Then the BioMAP Z-Standard, which adjusts for the number of common readouts (CR), is generated according to the following formula: Z-Standard = z R-3 A |arger BioMAP Z-Standard value corresponds to a higher confidence level, and this is the metric used to rank similarity results.
Cluster Analysis
[0719] Cluster analysis (function similarity map) uses the results of pairwise correlation analysis to project the "proximity" of agent profiles from multi-dimensional space into two dimensions. Functional clustering of the agent profiles generated during this analysis uses Pearson correlation values for pairwise comparisons of the profiles for each agent at each concentration, and then subjects the pairwise correlation data to multidimensional scaling. Profiles that are similar with a Pearson's correlation coefficient (r) > 0.7 are connected by lines. Agents that do not cluster with one another are interpreted as mechanistically distinct. This analysis is performed for projects with 3 or more agents tested. Cytotoxic concentrations are excluded from cluster analysis.
Mechanism HeatMAP Analysis
[0720] Mechanism HeatMAP analysis provides a visualization of the test compound and 19 consensus mechanisms allowing comparison of biomarker activities across all compound concentrations and consensus mechanisms. The synthetic consensus profiles used in the Mechanism HeatMAP analysis are representative BioMAP profiles of the average of multiple compounds from structurally distinct chemical classes. Profiles were calculated by averaging the values for each biomarker endpoint for all profiles selected (multiple agents at different concentrations) to build the consensus mechanism profile. [8] Biomarker activities are colored in the heatmap for consensus mechanisms and compounds when they have expression relative to vehicle controls outside of the significance envelope. Red represents increased protein expression, blue represents decreased expression and white indicates levels that were unchanged or within filtering conditions. Darker shades of color represent greater change in biomarker activity relative to vehicle control. The Mechanism HeatMAP was prepared using R and the gplots package for R.
Assay Acceptance Criteria
[0721] A BioMAP assay includes the multi-parameter data sets generated by the BioMAP platform for agents tested in the systems that make up the Diversity PLUS panel. Assays contain drug controls (e.g., legacy control test agent colchicine), negative controls (e.g., non-stimulated conditions), and vehicle controls (e.g., DMSO) appropriate for each system. BioMAP assays are plate-based, and data acceptance criteria depend on both plate performance (% CV of vehicle control wells) and system performance across historical controls for that system. The QA/QC Pearson Test is performed by first establishing the 1 % false negative Pearson cutoff from the reference dataset of historical positive controls. The process iterates through every profile of system biomarker readouts in the positive control reference dataset, calculating Pearson values between each profile and the mean of the remaining profiles in the dataset. The overall number of Pearson values used to determine the 1 % false negative cutoff is the total number of profiles present in the reference dataset. The Pearson value at the one percentile of all values calculated is the 1 % false negative Pearson cutoff. A system will pass if the Pearson value between the experimental plate's negative control or drug control profile and the mean of the historical control profiles in the reference dataset exceeds this 1 % false negative Pearson cutoff. Overall assays are accepted when each individual system passes the Pearson test and 95% of all project plates have % CV <20%.
Results
BioMAP Profile
[0722] Figure 1 depicts the BioMAP profile of SNA-101 in the Diversity PLUS Panel. SNA-101 was found to be modestly active with 3 annotated readouts, mediating changes in key biomarker activities listed by biological and disease classifications in Table 6 below. SNA-101 impacted inflammation-related activities (decreased SAA; increased sPGE2) and tissue remodeling activities (increased Collagen I).
[0723] There were no cytotoxic or antiproliferative impacts detected at the concentration range tested.
Table 6: Key Biomarker Activities Impacted by SNA-101

[0724] Figure 2 depicts an overlay of SNA- 101 at 29 μΜ and the selected reference benchmark apremilast at 10 μΜ. Apremilast is a PDE4 inhibitor approved for the treatment of plaque psoriasis and psoriatic arthritis. The two agents did not have any common activities that meet the defined criteria for annotation.
[0725] Differentiating biomarkers (not shown) are defined when one profile has a readout outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1), and the readout for the other profile is either inside the envelope or in the opposite direction. There are 12 differentiating activities between the two compounds: LPS (sTNFa), SAg (CD69, E-selectin, MCP-1), BT (slL-6, slgG), CASM3C (Prolif), HDF3CGF (Collagen III, PAI-1), MyoF (Collagen I), and IMphg (MIP-1 a, slL-10).
Top Database Search Result for SNA-101
[0726] In a search for mathematically similar compound profiles from the BioMAP reference database, SNA-101 (29 μΜ) was most similar to topiramate (3.3 μΜ) (Pearson's correlation, r = 0.344). The Pearson's correlation coefficient between these two profiles is below our determined threshold (r < 0.7) indicating that the relevance of the similarity is unknown. Topiramate is an anticonvulsant drug that is used to treat epilepsy. The two agents do not have any common activities that meet the defined criteria for annotation. Figure 3 depicts an overlay of SNA-101 (29 μΜ) and topiramate (3.3 μΜ).
Top BioSeek Reference Database Matches for SNA-101
[0727] Table 7 depicts the top 3 similarity matches from a search of the BioMAP Reference Database of > 4,000 agents for each concentration of SNA-101 . The similarity between agents is determined using a combinatorial approach that accounts for the characteristics of BioMAP profiles by filtering (Tanimoto metric) and ranking (BioMAP Z-Standard) the Pearson's correlation coefficient between two profiles. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7. For profiles with a Pearson's correlation coefficient below our determined threshold (r < 0.7), the relevance of the similarity is unknown.
Table 7: Top BioMAP Reference Database Matches for SNA-101

Mechanism HeatMAP Analysis of SNA-101
[0728] Figure 4 depicts Mechanism HeatMAP Analysis of SNA-101 , with the 148 biomarker readouts within the Diversity PLUS panel compared to 19 consensus mechanism class profiles. This analysis informs on the regulatory mechanisms controlling increases or decreases in each of the biomarker readouts.
Conclusions
[0729] In this study SNA-101 was characterized by profiling in the BioMAP Diversity PLUS panel of human primary cell based assays modeling complex tissue and disease biology of organs (vasculature, immune system, skin, lung) and general tissue biology. The Diversity PLUS panel evaluates the biological impact of test agents in conditions that preserve the complex crosstalk and feedback mechanisms that are relevant to in vivo outcomes. SNA-101 was largely inactive in the Diversity PLUS panel, with no cytotoxic or antiproliferative effects observed at the concentration range tested. A modest increase in sPGE2 and Collagen I expression was observed, as was a modest decrease in serum amyloid A expression. Overall, the profile of SNA-101 was not similar to the benchmark Apremilast.
Example 4: Additional BioMAP Platform Analysis of SNA-101
Aim of Study
[0730] The goal of this study was to repeat the characterization of SNA-101 in the BioMAP Diversity PLUS panel of 12 human primary cell-based systems using higher concentrations of SNA-101 than employed in Example 3.
Materials and Methods
Test Agent
[0731] SNA-101 was profiled in the BioMAP Diversity PLUS panel at concentrations of 300 μΜ, 100 μΜ, 33 μΜ, and 1 1 μΜ. Staurosporine was employed as the benchmark compound.
Methods and Analysis
[0732] BioMAP Platform Analysis was performed as described in Example 3. Results
BioMAP Profile
[0733] Figure 5 depicts the BioMAP profile of SNA-101 in the Diversity PLUS Panel. SNA-101 was found to be modestly active with 4 annotated readouts, mediating changes in key biomarker activities listed by biological and disease classifications in Table 8 below. SNA-101 impacted inflammation-related activities (decreased sTNFa, IL- 8, I L- 1 a) and a tissue remodeling activity (decreased MMP-1). SNA-101 is antiproliferative to endothelial cells at the top concentration (grey arrow of Figure 5). SNA-101 had no cytotoxic effects in the concentration range tested.
Table 8: Key Biomarker Activities Impacted by SNA-101

Reference Benchmark Overlay
[0734] Figure 6 depicts an overlay of SNA-101 at 300 μΜ and the selected reference benchmark staurosporine at 10 nM. Staurosporine is a broad-spectrum protein kinase inhibitor that acts on protein kinase C (PKC), protein kinase A (PKA), p60v-src tyrosine protein kinase, and CaM kinase II (CAMKII). Staurosporine is an analog of K252a.
[0735] There are 4 common activities that are annotated within the following systems: 3C (Prolif), BT (slL-6, sTNFa), and HDF3CGF (PAI-1).
[0736] Differentiating biomarkers (not shown) are defined when one profile has a readout outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1), and the readout for the other profile is either inside the envelope or in the opposite direction. There are 65 differentiating activities within the following systems: 3C (HLA-DR, TF, uPAR), 4H (Eotaxin 3, MCP-1 , P-selectin, VEGFR2, uPAR), LPS (CD40, CD69, M-CSF, MCP-1 , TF, VCAM-1 , sTNFa), SAg (CD40, CD69, E-selectin, IL-8, MCP- 1 , Prolif), BT (SIL-17A, SIL-17F, slL-2, slgG), BF4T (IL-1 a, MCP-1 , PAI-1 , VCAM-1), BE3C (IL-8, MMP-1 , MMP-9, PAI-1 , tPA, uPA, uPAR), CASM3C (M-CSF, MCP-1 , Prolif, SAA, TF, uPAR), HDF3CGF (MCP-1 , MIG, MMP-1 , Prolif 72), KF3CT (MCP-1 , PAI-1), MyoF (Collagen I, Collagen III, Collagen IV, MMP-1 , PAI-1 , TIMP-1 , VCAM-1), and IMphg (CD40, CD69, E-selectin, IL-1 a, IL-8, M-CSF, MCP-1 , MIP-1 a, VCAM-1 , slL-10).
Top Database Search Result for 300 μΜ SNA-101
[0737] In a search for mathematically similar compound profiles from the BioMAP reference database, SNA-101 (300 μΜ) was most similar to N037 (490 ng/ml) (Pearson's correlation, r = 0.760). SNA-101 and N037 share 84 readouts in 8 common BioMAPsystems. The Pearson's correlation coefficient between these two profiles is above our determined threshold (r > 0.7) indicating these compounds share mechanistically relevant similarity. N037 [(±)-Norketamine-D4 HCI] is the most prevalent urinary metabolite of ketamine, a widely used anesthetic sold under trade names, including Ketanest, Ketaset and Ketalar. There is 1 common activity that is annotated within the following system: 3C (Prolif). Figure 7 depicts an overlay of SNA-101 (300 μΜ) and N037 (490 ng/ml).
Top Database Search Result for 100 μΜ SNA-101
[0738] In a search for mathematically similar compound profiles from the BioMAP reference database, SNA-101 (100 μΜ) was most similar to infliximab (30000 ng/ml) (Pearson's correlation, r = 0.424). The Pearson's correlation coefficient between these two profiles is below our determined threshold (r < 0.7) indicating that the relevance of the similarity is unknown. Remicade (infliximab) is a chimeric monoclonal antibody against TNF alpha approved for the treatment of psoriasis, Crohn's disease, ankylosing spondylitis, psoriatic arthritis, rheumatoid arthritis and ulcerative colitis. There are 4 common activities that are annotated within the following systems: BT (sTNFa) and IMphg (E-selectin, IL-1 a, IL-8). Figure 8 depicts an overlay of SNA-101 (100 μΜ) and infliximab (30000 ng/ml).
Top BioSeek Reference Database Matches for SNA-101
[0739] Table 9 depicts the top 3 similarity matches from a search of the BioMAP Reference Database of > 4,000 agents for each concentration of SNA-101 . The similarity between agents is determined using a combinatorial approach that accounts for the characteristics of BioMAP profiles by filtering (Tanimoto metric) and ranking (BioMAP Z-Standard) the Pearson's correlation coefficient between two profiles. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7. For profiles with a Pearson's correlation coefficient below our determined threshold (r < 0.7), the relevance of the similarity is unknown.
Table 9: Top BioMAP Reference Database Matches for SNA-101

[0740] The Pearson's correlation coefficient between profiles that is above our determined threshold (r > 0.7) indicates these compounds share mechanistically relevant similarity. For profiles with a Pearson's correlation coefficient below our determined threshold (r < 0.7), the relevance of the similarity is unknown.
Mechanism HeatMAP Analysis of SNA-101
[0741] Figure 9 depicts Mechanism HeatMAP Analysis of SNA-101 , with the 148 biomarker readouts within the Diversity PLUS panel compared to 19 consensus mechanism class profiles. This analysis informs on the regulatory mechanisms controlling increases or decreases in each of the biomarker readouts.
Conclusions
[0742] In this study SNA-101 was characterized by profiling in the BioMAP Diversity PLUS panel of human primary cell-based assays modeling complex tissue and disease biology of organs (vasculature, immune system, skin, lung) and general tissue biology. The Diversity PLUS panel evaluates the biological impact of test agents in conditions that preserve the complex crosstalk and feedback mechanisms that are relevant to in vivo outcomes. SNA- 101 was modestly active in the Diversity PLUS panel with only 4 annotated readouts that impact inflammation and tissue-remodeling activities. SNA-101 had no cytotoxic effects at the concentration range tested . Antiproliferative effects to endothelial cells were observed at the top tested concentration of 300 μΜ , but not at the lower concentrations. SNA- 101 was also previously profiled in the Diversity PLUS panel at a lower concentration range (1 .1 μΜ - 29 μΜ, refer to Example 3) where it was minimally active. SNA-101 and the selected reference benchmark staurosporine, a broad-spectrum kinase inhibitor, had only 4 common activities, but 65 differentiating activities. In general, staurosporine at 10 nM was more active than SNA- 101 at 300 μΜ . The top database search match for SNA- 101 was N037, a metabolite of the anesthetic ketamine. However, a common activity between the two profiles was the antiproliferative effect on endothelial cells, an activity observed for the top concentration of SNA- 101 . An overlay with the second concentration of SNA-101 and its top match is also provided, although the Pearson's correlation coefficient did not meet our threshold for significance.
Example 5: Profiling study of CT103 against 270 kinases
[0743] CT103 (test concentration: 1 μΜ) was tested against 270 target kinases.
Materials and Methods
Preparation of test compound solution
[0744] The test compound (CT103) was dissolved in and diluted with dimethylsulfoxide (DMSO) to achieve 100-fold higher concentration. Then the solution was further 25-fold diluted with assay buffer to make the final test compound solution. Reference compounds for assay control were prepared similarly.
Assay reagents and procedures
IMAP assay
[0745] 1) The 5 μΙ_ of x4 compound solution, 5 μΙ_ of x4 Substrate/ATP/Metal solution, and 10 μΙ_ of x2 kinase solution were prepared with assay buffer (20 mM HEPES, 0.01 % Tween-20, 2 mM DTT, pH7.4) and mixed and incubated in a well of polystyrene 384 well black microplate for 1 hour at room temperature.
[0746] 2) 60 μΙ_ of IMAP binding reagent (IMAP Screening Express kit; Molecular Devices) was added to the well, and incubated for 30 minutes.
[0747] 3) The kinase reaction was evaluated by the fluorescence polarization at 485 nm for excitation and 530 nm for emission of the well.
Off-chip Mobility Shift Assay (MSA)
[0748] 1) The 5 μΙ_ of x4 compound solution, 5 μΙ_ of x4 Substrate/ATP/Metal solution, and 10 μΙ_ of x2 kinase solution were prepared with assay buffer (20 mM HEPES, 0.01 % Triton X- 100, 2 mM DTT, pH7.5) and mixed and incubated in a well of polypropylene 384 well microplate for 1 or 5 hour(s)* at room temperature. (*; depend on kinase)
[0749] 2) 60 μΙ_ of Termination Buffer (QuickScout Screening Assist MSA; Carna Biosciences) was added to the well.
[0750] 3) The reaction mixture was applied to LabChip3000 system (Caliper Life Science), and the product and substrate peptide peaks were separated and quantitated.
[0751] 4) The kinase reaction was evaluated by the product ratio calculated from peak heights of product(P) and substrate(S) peptides (P/(P+S)).
Reaction Conditions
[0752] The reaction conditions are depicted below:








6 Ϊ ¾-ϊ!¾ί.δϊ! ιίί¾&·>ν&»3·:5«ϊ& i¾ sikfei si sis; iiiisiS em&ssfc& t ft' Zi a .
ί¾) Τί!Χί. jx<5>†s-:fe is stiife-d set ihe Siasi aa&cusstt sties f-f 2Si :i .
Data Analysis
[0753] The readout value of reaction control (complete reaction mixture) was set as a 0% inhibition, and the readout value of background (Enzyme(-)) was set as a 100% inhibition, then the percent inhibition of each test solution was calculated.
Results
[0754] The results are shown in Table 10 below.
Table 10: Target kinase inhibition by CT103







Example 6: IC50 Determination study of CT103 against 2 kinases
[0755] CT103 inhibition of FLT1 and KDR was examined at test concentrations of 50μΜ, 15μΜ, 5μΜ, 1 .5μΜ, 0.5μΜ, 0.15μΜ, 0.05μΜ, 0.015μΜ, 0.005μΜ, and 0.0015μΜ.
Materials and Methods
Preparation of test compound solution
[0756] The test compound was dissolved in and diluted with dimethylsulfoxide (DMSO) to achieve 100-fold higher concentration. Then the solution was further 25-fold diluted with assay buffer to make the final test compound solution. Reference compounds for assay control were prepared similarly.
Assay reagents and procedures
STK-ELISA
[0757] 1) The 10 μΙ_ of x4 compound solution, 10 μΙ_ of x4 Substrate/ATP/Metal solution, and 20 μΙ_ of x2 kinase solution were prepared with assay buffer (15 mM Tris-HCI, 0.01 % Tween-20, 2 mM DTT, pH7.5) and mixed and incubated in a well of polypropylene 96 well microplate for 1 hour at room temperature.
[0758] 2) 120 μΙ_ of 40 mM EDTA solution (pH 7.5) was added to the well, and then 120 μΙ_ of the mixture was transferred to the well of ELISA plate (see below table).
[0759] 3) After 30 minutes incubation, the well was washed 4 times, and blocked with blocking buffer containing 0.1 % BSA.
[0760] 4) 100 μΙ_ of the first antibody (see below table) solution was added to the well and incubated for 30 minutes.
[0761] 5) After 4 times washing of the well, 100 μΙ_ of the second antibody (see below table) solution was added to the well, and incubated for 30 minutes.
[0762] 6) After washing the well, 100 μΙ_ of TMB solution (MOSS Inc.) was added and incubated for 5 minutes. To stop the HRP reaction, 100 μΙ_ of 0.1 M sulfuric acid was added.
[0763] 7) The kinase reaction was evaluated by the absorbance at 450 nm of the well.

Off-chip Mobility Shift Assay (MSA)
[0764] 1) The 5 μΙ_ of x4 compound solution, 5 μΙ_ of x4 Substrate/ATP/Metal solution, and 10 μΙ_ of x2 kinase solution were prepared with assay buffer (20 mM HEPES, 0.01 % Triton X- 100, 2 mM DTT, pH7.5) and mixed and incubated in a well of polypropylene 384 well microplate for 1 hour at room temperature.
[0765] 2) 60 μΙ_ of Termination Buffer (QuickScout Screening Assist MSA; Carna Biosciences) was added to the well.
[0766] 3) The reaction mixture was applied to LabChip3000 system (Caliper Life Science), and the product and substrate peptide peaks were separated and quantitated.
[0767] 4) The kinase reaction was evaluated by the product ratio calculated from peak heights of product(P) and substrate(S) peptides (P/(P+S)).
Reaction Conditions - The reaction conditions are depicted below:

Data Analysis
[0768] The readout value of reaction control (complete reaction mixture) was set as a 0% inhibition, and the readout value of background (Enzyme(-)) was set as a 100% inhibition, then the percent inhibition of each test solution was calculated. The IC50 value was calculated from concentration vs. %lnhibition curves by fitting to a four parameter logistic curve.
Results
[0769] The results are shown in Table 1 1 below.
Table 11 : IC50 Determination

Example 7: BioMAP Platform Analysis of SNA-103
Aim of Study
[0770] The goal of this study was to characterize SNA- 103 in the BioMAP Diversity PLUS panel of 12 human primary cell-based systems. These systems are designed to model complex human tissue and disease biology of the vasculature, skin, lung and inflammatory tissues. Quantitative measurements of biomarker activities across this broad panel, along with comparative analysis of the biological activities of known bioactive agents in the BioMAP reference database are used to predict the safety, efficacy and function of these test agents.
Materials and Methods
Test Agent
[0771] SNA- 103 was profiled in the BioMAP Diversity PLUS panel at concentrations of 230 μΜ , 77 μΜ , 26 μΜ , and 8.6 μΜ . Calcipotriene was employed as the benchmark compound.
Methods and Analysis
[0772] BioMAP Platform Analysis was performed as described in Example 3. Results
BioMAP Profile
[0773] Figure 10 depicts the BioMAP profile of SNA-103 in the Diversity PLUS Panel. SNA-103 was found to be active with 13 annotated readouts, mediating
changes in key biomarker activities listed by biological and disease classifications in Table 12 below. SNA-103 impacted inflammation-related activities (decreased sTNFa, MIP-1 a, IL-8, IL-6; increased sPGE2), immunomodulatory activities (decreased slL-10, M-CSF, HLA-DR), tissue remodeling activities (decreased MMP-1 ; increased Collagen I), and hemostasis-related activities (decreased TF). SNA-103 is antiproliferative to B cells (indicated by grey arrow in Figure 10).
Table 12: Key Biomarker Activities Impacted by SNA-103

Reference Benchmark Overlay
[0774] Figure 1 1 depicts an overlay of SNA-103 at 230 μΜ and the selected reference benchmark calcipotriene at 1 μΜ. Calcipotriene is a synthetic derivative of calcitriol, a form of vitamin D, used in the treatment of psoriasis. Please note: Calcipotriene is available as a benchmark in 7 BioMAP systems.
[0775] There is 1 common activity that is annotated within the following system: LPS (sPGE2).
[0776] Differentiating biomarkers (not shown) are defined when one profile has a readout outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1), and the readout for the other profile is either inside the envelope or in the opposite direction. There are 12 differentiating activities between the two compounds: 3C (HLA-DR, TF), 4H (Eotaxin 3), LPS (M-CSF, sTNFa), HDF3CGF (Collagen III, Prolif 72, VCAM-1), KF3CT (IP-10), and IMphg (E-selectin, IL-8, MIP-1 a).
Top Database Search Result for SNA-103
[0777] In a search for mathematically similar compound profiles from the BioMAP reference database, SNA-103 (230 μΜ) was most similar to apoptolidin (1 μΜ) (Pearson's correlation, r = 0.564). The Pearson's correlation coefficient between these two profiles is below our determined threshold (r < 0.7) indicating that the relevance of the similarity is unknown. Apoptolidin is a selective polyketide produced by Nocardiopsis that induces apoptotic death in E1A-transformed cells by targeting the mitochondrial
F0F1 -ATP synthase and inhibiting oxidative phosphorylation. Figure 12 depicts an overlay of SNA-103 (230 μΜ) and apoptolidin (1 μΜ).
[0778] There are 6 common activities that are annotated within the following systems: 3C (HLA-DR), BT (Prolif, SIL-17F, slgG, sTNFa), and BF4T (Eotaxin 3).
Top BioSeek Reference Database Matches for SNA-103
[0779] Table 13 depicts the top 3 similarity matches from a search of the BioMAP Reference Database of > 4,000 agents for each concentration of SNA-103. The similarity between agents is determined using a combinatorial approach that accounts for the characteristics of BioMAP profiles by filtering (Tanimoto metric) and ranking (BioMAP Z-Standard) the Pearson's correlation coefficient between two profiles. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7.
Table 13: Top BioMAP Reference Database Matches for SNA-103

[0780] For profiles with a Pearson's correlation coefficient below our determined threshold (r < 0.7), the relevance of the similarity is unknown.
Mechanism HeatMAP Analysis of SNA-103
[0781] Figure 13 depicts Mechanism HeatMAP Analysis of SNA-103, with the 148 biomarker readouts within the Diversity PLUS panel compared to 19 consensus mechanism class profiles. This analysis informs on the regulatory mechanisms controlling increases or decreases in each of the biomarker readouts.
Clustering of Project Profiles
[0782] Figure 14 depicts a clustering of tested agent profiles based on pairwise correlation analysis and clustering of most similar profiles. Profiles that are similar with a Pearson's correlation coefficient (r) > 0.7 are connected by lines. Agents that do not cluster with one another are interpreted as mechanistically distinct. Cytotoxic concentrations are excluded from cluster analysis. Functional clustering of the agent profiles generated during this analysis uses Pearson's correlation values for pairwise comparisons of the profiles for each agent at each concentration, and then subjects the pairwise correlation data to multidimensional scaling. SNA-103 clusters internally at the top three concentrations. Internal clustering at both high and low concentrations of SNA- 103 suggests the phenotypic signature of this compound is maintained across a wide range of concentrations, a characteristic commonly observed in marketed drugs.
Conclusions
[0783] In this study SNA-103 was characterized by profiling in the BioMAP Diversity PLUS panel of human primary cell based assays modeling complex tissue and disease biology of organs (vasculature, immune system, skin, lung) and general tissue biology. The Diversity PLUS panel evaluates the biological impact of test agents in conditions that preserve the complex crosstalk and feedback mechanisms that are relevant to in vivo outcomes.
[0784] SNA- 103 was modestly active and non-cytotoxic in the Diversity PLUS panel; an antiproliferative effect on B cells was observed at the top concentration tested (230 μΜ) . Selective antiproliferative effects on B cells may be appropriate for B cell driven autoimmune indications, such as systemic lupus erythematosus (SLE), or for heme-oncology indications such as chronic lymphocytic leukemia (CLL) or B-cell non- Hodgkin's lymphoma (B-NHL) . A sharp dose response was observed between 230 μΜ and 77 μΜ in the BT system, suggesting that the test agent may be engaging additional targets on immune cells at this concentration. Other activities of interest were the supra- induction of sPGE2 levels in the LPS system and Collagen-I levels in the MyoF system. These systems model monocyte and myofibroblast activation respectively, and these readouts are induced by the system activators in the respective systems.
Example 8: Profiling study of CT352 against 311 kinases
[0785] CT352 and staurosporine (compound 1 of Table 3) were tested (at test concentrations of 0.2 μΜ) against a variety of target kinases.
Materials and Methods
Preparation of test compound solution
[0786] The test compounds were dissolved in and diluted with dimethylsulfoxide (DMSO) to achieve 100-fold higher concentration. Then the solutions were further 25-fold diluted with assay buffer to make the final test compound solutions. Reference compounds for assay control were prepared similarly.
Assay reagents and procedures
TK-ELISA
[0787] 1 ) The 10 μΙ_ of x4 compound solution, 10 μΙ_ of x4 Substrate/ATP/Metal solution, and 20 μΙ_ of x2 kinase solution were prepared with assay buffer (15 mM Tris-HCI , 0.01 % Tween-20, 2 mM DTT, pH7.5) and mixed in a well of streptavidine-coated 96 well microplate (Perkin Elmer) .
[0788] 2) The well was incubated for I hour at room temperature and then washed 4 times to stop the reaction.
[0789] 3) The well was blocked with blocking buffer containing 0.1 % BSA and then 100 μΙ_ of the detection antibody (HRP conjugated PY20; Santa Cruz Biotechnology) solution was added and incubated for 30 minutes.
[0790] 4) After washing the well, 100 μΙ_ of TMB solution (MOSS Inc.) was added and incubated for 5 minutes. To stop the HRP reaction, 100 μΙ_ of 0.1 M sulfuric acid was added.
[0791] 5) The kinase reaction was evaluated by the absorbance at 450 nm of the well.
STK-ELISA
[0792] 1 ) The 10 μΙ_ of x4 compound solution, 10 μΙ_ of x4 Substrate/ATP/Metal solution, and 20 μΙ_ of x2 kinase solution were prepared with assay buffer (15 mM Tris-HCI, 0.01 % Tween-20, 2 mM DTT, pH7.5) and mixed and incubated in a well of polypropylene 96 well microplate for 0.5 or 1 hour* at room temperature. (*; depend on kinase)
[0793] 2) 120 μΙ_ of 40 mM EDTA solution (pH 7.5) was added to the well, and then 120 μΙ_ of the mixture was transferred to the well of ELISA plate (see below table) .
[0794] 3) After 30 minutes incubation, the well was washed 4 times, and blocked with blocking buffer containing O. \% BSA.
[0795] 4) 100 μΙ_ of the first antibody (see below table) solution was added to the well and incubated for 30 minutes.
[0796] 5) After 4 times washing of the well, 100 μΙ_ of the second antibody (sell below table) solution was added to the well, and incubated for 30 minutes.
[0797] 6) After washing the well, 100 μΙ_ of TMB solution (MOSS Inc.) was added and incubated for 5 minutes. To stop the HRP reaction, 100 μΙ_ of 0.I M sulfuric acid was added.
[0798] 7) The kinase reaction was evaluated by the absorbance at 450 nm of the well.


2n Ak i bsbsSsc g*ai sis!i-rabbss. I;n < Ab {lymsd

/MAP assay
[0799] 1) The 5 μΙ_ of x4 compound solution, 5 μΙ_ of x4 Substrate/ATP/Metal solution, and 10 μΙ_ of x2 kinase solution were prepared with assay buffer (20 mM HEPES, 0.01 % Tween-20, 2 mM DTT, pH7.4) and mixed and incubated in a well of polystyrene 384 well black microplate for 1 hour at room temperature.
[0800] 2) 60 μΙ_ of IMAP binding reagent (IMAP Screening Express kit; Molecular Devices) was added to the well, and incubated for 30 minutes.
[0801] 3) The kinase reaction was evaluated by the fluorescence polarization at 485 nm for excitation and 530 nm for emission of the well.
[0802] Off-chip Mobility Shift Assay (MSA)
[0803] 1) The 5 μΙ_ of x4 compound solution, 5 μΙ_ of x4 Substrate/ATP/Metal solution, and 10 μΙ_ of x2 kinase solution were prepared with assay buffer (20 mM HEPES, 0.01 % Triton X- 100, 2 mM DTT, pH7.5) and mixed and incubated in a well of polypropylene 384 well microplate for 1 or 5 hour(s)* at room temperature. (*; depend on kinase)
[0804] 2) 60 μΙ_ of Termination Buffer (QuickScout Screening Assist MSA; Carna Biosciences) was added to the well.
[0805] 3) The reaction mixture was applied to LabChip3000 system (Caliper Life Science), and the product and substrate peptide peaks were separated and quantitated.
[0806] 4) The kinase reaction was evaluated by the product ratio calculated from peak heights of product(P) and substrate(S) peptides (P/(P+S)).
Γ08071 Reaction Conditions -- The reaction conditions are depicted below:

-263-

TEC MS.A ¾«kfe •<Xsi! .W S3 s 5





0} iii . C ¾ΐίϊ¾!;¾:ϋϊΐ Αΐ Ϊϋ&ίΰΐ-Ί

4)
 <A .¾ rM.
<A .¾ rM.
55 -fesssiiavkivissrisi: ssw Ds.«y£ (¾«H»i s..¾3stS si ϋκ i¾i¾ eoaeiiif mm >■; '■■■■ * n≠s> !- p«Si*-eiy.
8} mi C <S s4ia r- as (ha S«ai ce»i «isti.«a ef 2.u auxi 28 > «^seSv«lv
S<5 Assay ;·::!:« i- >ί· «; ίίϊ :;ί¾; 1 '? 5).2ί«¾ί " 'i .
So¾i«t» ;ar. aCi eajtmaaHiiK as- s &-c! ai Use mvsx 1> , ?S ,< V. ¾ t¾H ;tsa«c 5 i ! ·>Χ.·: ^tsia is y. h.d st ?jss t¾aS e«i4sia¾iiiiiii of ¾¾ nM.
S ΐ) aS :ii»i ti!S uaa <.::·::.:;.»!;»;:<:::; i A μ .
Data Analysis
[0808] The readout value of reaction control (complete reaction mixture) was set as a 0% inhibition, and the readout value of background (Enzyme(-)) was set as a 100% inhibition, then the percent inhibition of each test solution was calculated.
Results
[0809] The results are shown in Table 14 below.
Table 14: Target kinase inhibition by CT352 and staurosporine.


 Kinase % Inhibition % Inhibition CT352 (0.2μΜ) Staurosporine (0.2μΜ)
Kinase % Inhibition % Inhibition CT352 (0.2μΜ) Staurosporine (0.2μΜ)
COT 2.9 2.8
CRIK -3.4 76.1
DAPK1 -1.7 94.9
DCAMKL2 -6.9 83.8
DLK -2.9 22.5
DYRK1A 11.7 98.4
DYRK1B 13.2 99.7
DYRK2 0.2 35.0
DYRK3 -0.9 89.2
EEF2K 0.9 -0.3
Erk1 -4.1 8.2
Erk2 2.9 15.9
Erk5 44.9 77.8
GSK3a 9.1 93.6
GSK3b 7.6 94.9
Haspin 0.3 97.2
HGK 48.6 101.1
HIPK1 1.2 25.0
HIPK2 3.4 50.6
HIPK3 2.3 44.6
HIPK4 5.2 66.5
IKKa 4.9 41.3
IKKb -1.8 38.1
IKKe 14.9 100.0
IRAKI 9.4 89.3
IRAK4 -3.8 96.1
JNK1 0.7 46.5
JNK2 -1.2 17.9
JNK3 -3.4 38.9
LATS2 31.4 100.9
LIMK1 58.1 99.8
LKB1 1.2 50.1
LOK 22.6 101.4
MAP2K1 12.8 99.8
MAP2K2 17.4 99.7
MAP2K3 28.4 99.9
MAP2K4 1.2 86.9
MAP2K5 -25.2 93.4
MAP2K6 61.8 100.0
MAP2K7 -1.0 88.3
MAP3K1 12.6 -12.6
MAP3K2 22.6 98.4
MAP3K3 -7.5 92.1
MAP3K4 -3.6 65.4
MAP3K5 -0.9 94.4
MAP4K2 17.1 97.9
MAPKAPK2 7.1 60.0
MAPKAPK3 -3.8 4.3
MAPKAPK5 -0.6 43.3
MARK1 54.7 100.6
MARK2 64.1 101.2

 Kinase % Inhibition % Inhibition
Kinase % Inhibition % Inhibition
CT352 (0.2μΜ) Staurosporine (0.2μΜ)
WNK1 -5. 7 -4. 1
WNK2 -6. 1 -2. 0
WNK3 -5. 9 -4. 4
PI K3CA 13. 9 38. 3
SPHK1 15. 4 6. 1
SPHK2 7. 4 2. 0
Example 9: BioMAP Platform Analysis of SNA-352
Aim of Study
[0810] The goal of this study was to characterize SNA-352 in the BioMAP Diversity PLUS panel of 12 human primary cell-based systems. These systems are designed to model complex human tissue and disease biology of the vasculature, skin, lung and inflammatory tissues. Quantitative measurements of biomarker activities across this broad panel, along with comparative analysis of the biological activities of known bioactive agents in the BioMAP reference database are used to predict the safety, efficacy and function of these test agents.
Materials and Methods
Test Agent
[0811] SNA-352 was profiled in the BioMAP Diversity PLUS panel at concentrations of 3900 nM, 1300 nM, 430 nM , and 140 nM . Cyclosporin A was employed as the benchmark compound.
Methods and Analysis
[0812] BioMAP Platform Analysis was performed as described in Example 3.
Results
Bio MAP Profile
[0813] Figure 15 depicts the BioMAP profile of SNA-352 in the Diversity PLUS Panel. SNA-352 was found to be active with 8 annotated readouts, mediating changes in key biomarker activities listed by biological and disease classifications in Table 15 below. SNA-352 impacted inflammation-related activities (decreased SAA, sTNFa), immunomodulatory activities (decreased slgG, slL-10; increased CD69), and tissue remodeling activities (decreased TIMP-2, Collagen IV; increased uPAR).
Table 15: Key Biomarker Activities Impacted by SNA-352

[0814] SNA-352 is antiproliferative to B cells, coronary artery smooth muscle cells, fibroblasts, and T cells (indicated by grey arrows in Figure 15).
Reference Benchmark Overlay
[0815] Figure 16 depicts an overlay of SNA-352 at 3.9 μΜ and the selected reference benchmark Cyclosporin A at 3.3 μΜ. Cyclosporin A is a calcineurin inhibitor widely used in organ transplantation to prevent rejection. There are 6 common activities that are annotated within the following systems: SAg (Prolif), BT (Prolif, SIL-17A, slgG, sTNFa), and HDF3CGF (Prolif 72).
[0816] Differentiating biomarkers (not shown) are defined when one profile has a readout outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1), and the readout for the other profile is either inside the envelope or in the opposite direction. There are 21 differentiating activities between the two compounds: 3C (uPAR), 4H (MCP-1 , VCAM-1), LPS (sPGE2), SAg (CD38, CD40, CD69, E-selectin, IL- 8, MCP-1 , MIG), BT (SIL-17F, slL-2, slL-6), BE3C (MMP-9), CASM3C (SAA), MyoF (Collagen IV), and IMphg (CD69, E-selectin, IL-8, slL-10).
BT System Secretion Profiles
[0817] Figure 17 depicts changes in the secretion of IL-17F, IgG, IL-17A, and TNFa in the BioMAP BT system mediated by SNA-352 (3.9 μΜ), Tofacitinib (3.3 μΜ),
Apremilast (3.3 μΜ) , SR221 1 (3.3 μΜ), and Cyclosporin A (3.3 μΜ). Tofacitinib was found to be more active than SNA-352 in decreasing IL-1 7F secretion, displaying an activity similar to SR221 1 (Figure 17A). SNA-352 and Tofacitinib were both very active in decreasing secreted IgG , with SNA-352 as active as Cyclosporin A (Figure 17B). Surprisingly, SNA-352 was found to be as active as tofacitinib in decreasing IL- 17A secretion (Figure 17C). SR221 1 decreased IL-17A secretion as expected, while Apremilast increased IL-17A secretion. Notably, SNA-352 was found to have remarkable activity with regards to reducing TNFa secretion (Figure 17D).
Top Database Search Result for SNA-352
[0818] In a search for mathematically similar compound profiles from the BioMAP reference database, SNA-352 (3.9 μΜ) was most similar to deferoxamine mesylate (4.4 μΜ) (Pearson's correlation, r = 0.879). The Pearson's correlation coefficient between these two profiles is above our determined threshold (r > 0.7) indicating these compounds share mechanistically relevant similarity. Deferoxamine mesylate is an iron chelator used to treat iron toxicity, and has been investigated as a potential treatment for spinal cord injury. Figure 18 depicts an overlay of SNA-352 (3.9 μΜ) and Deferoxamine Mesylate (4.4 μΜ).
[0819] There are 8 common activities that are annotated within the following systems: SAg (Prolif), BT (Prolif, SIL-17A, slgG , sTNFa), CASM3C (Prolif) , HDF3CGF (Prolif 72) , and MyoF (Collagen IV).
Top BioSeek Reference Database Matches for SNA-352
[0820] Table 16 depicts the top 3 similarity matches from a search of the BioMAP Reference Database of > 4,000 agents for each concentration of SNA-352. The similarity between agents is determined using a combinatorial approach that accounts for the characteristics of BioMAP profiles by filtering (Tanimoto metric) and ranking (BioMAP Z-Standard) the Pearson's correlation coefficient between two profiles. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7.
Table 16: Top BioMAP Reference Database Matches for SNA-352

[0821] The Pearson's correlation coefficient between profiles that is above our determined threshold (r > 0.7) indicates these compounds share mechanistically relevant similarity. For profiles with a Pearson's correlation coefficient below our determined threshold (r < 0.7), the relevance of the similarity is unknown.
Mechanism HeatMAP Analysis of SNA-352
[0822] Figure 19 depicts Mechanism HeatMAP Analysis of SNA-352, with the 148 biomarker readouts within the Diversity PLUS panel compared to 19 consensus mechanism class profiles. This analysis informs on the regulatory mechanisms controlling increases or decreases in each of the biomarker readouts.
Clustering of Project Profiles
[0823] Figure 14 depicts a clustering of tested agent profiles based on pairwise correlation analysis and clustering of most similar profiles. Profiles that are similar with a Pearson's correlation coefficient (r) > 0.7 are connected by lines. Agents that do not cluster with one another are interpreted as mechanistically distinct. Cytotoxic concentrations are excluded from cluster analysis. Functional clustering of the agent profiles generated during this analysis uses Pearson's correlation values for pairwise comparisons of the profiles for each agent at each concentration, and then subjects the pairwise correlation data to multidimensional scaling. SNA-352 clusters internally at two concentrations. Internal clustering suggests the phenotypic signature of this compound is maintained across a range of concentrations, a characteristic commonly observed in marketed drugs.
Conclusions
[0824] In this study SNA-352 was characterized by profiling in the BioMAP Diversity PLUS panel of human primary cell based assays modeling complex tissue and disease biology of organs (vasculature, immune system, skin, lung) and general tissue biology. The Diversity PLUS panel evaluates the biological impact of test agents in conditions that preserve the complex crosstalk and feedback mechanisms that are relevant to in vivo outcomes. SNA-352 was active and non-cytotoxic in the Diversity PLUS panel, with broad antiproliferative effects observed for the top tested concentration of 3.9 μΜ. A robust decrease in slgG and sTNFa was observed for the top two concentrations of SNA-352 in the BT system, consistent with inhibition of B cell function and systemic lupus erythematosus (SLE), chronic lymphocytic leukemia (CLL), or B-cell non-Hodgkin's lymphoma (B-NHL) as potential indications. At its top tested concentration, SNA-352 shared 6 common activities with the benchmark cyclosporin A, but overall, cyclosporin A is more active than SNA-352 in the SAg system, which is a model of T cell activation. The top database match for SNA-352 was deferoxamine mesylate.
Example 10: Profiling Study of SNA-352 against 274 Kinases
Aim of Study
[0825] The goal of this study was to measure the inhibitory activity of SNA- 352 against 274 kinases.
Materials and Methods
Test Agent
[0826] SNA-352 (Lot # 2017GC14/S7) was used at a test concentration of
0.1 μΜ.
Target Kinases
[0827] ABL, ACK, ALK, ARG, AXL, BLK, BMX, BRK, BTK, CSK, DDR1 , DDR2, EGFR, EPHA1 , EPHA2, EPHA3, EPHA4, EPHA5, EPHA6, EPHA7, EPHA8,
EPHB1, EPHB2, EPHB3, EPHB4, FAK, FER, FES, FGFR1, FGFR2, FGFR3, FGFR4, FGR, FLT1, FLT3, FLT4, FMS, FRK, HCK, HER2, HER4, IGF1R, INSR, IRR, ITK, JAK1, JAK2, JAK3, KDR, KIT, LCK, LTK, LYNa, LYNb, MER, MET, MUSK, PDGFRa, PDGFRP, PYK2, RET, RON, ROS, SRC, SRM, SYK, TEC, TIE2, TNK1, TRKA, TRKB, TRKC, TXK, TYK2, TYR03, YES, ZAP70, AKT1, AKT2, AKT3, ΑΜΡΚα1/β1/γ1 , ΑΜΡΚα2/β1/γ1, AurA, AurA/TPX2, AurB, AurC, BRAF_Cascade, BRAF [V600E]_Cascade, BRSK1, BRSK2, BUB1/BUB3, CaMKIa, CaMK15, CaMK2a, CaMK2p, CaMK2Y, CaMK25, CaMK4, CDC2/CycB1, CDC7/ASK, CDK2/CycA2, CDK2/CycE1, CDK3/CycE1, CDK4/CycD3, CDK5/p25, CDK6/CycD3, CDK7/CycH/MAT1 , CDK9/CycT1, CGK2, CHK1, CHK2, CK1a, ΟΚ1γ1, ΟΚ1γ2, ΟΚ1γ3, CK15, CK , ΟΚ2α1/β, ΟΚ2α2/β, CLK1, CLK2, CLK3, COT_Cascade, CRIK, DAPK1, DCAMKL2, DLK_Cascade, DYRK1A, DYRK1B, DYRK2, DYRK3, EEF2K, Erk1, Erk2, Erk5, GSK3a, GSK3p, Haspin, HGK, HIPK1, HIPK2, HIPK3, HIPK4, ΙΚΚα, ΙΚΚβ, ΙΚΚε, IRAKI, IRAK4, JNK1, JNK2, JNK3, LATS2, LOK, MAP2K1_Cascade, MAP2K2_Cascade, MAP2K3_Cascade, MAP2K4_Cascade, MAP2K5_Cascade, MAP2K6_Cascade, MAP2K7_Cascade, MAP3K1_Cascade, MAP3K2_Cascade, MAP3K3_Cascade, MAP3K4_Cascade, MAP3K5_Cascade, MAP4K2, MAPKAPK2, MAPKAPK3, MAPKAPK5, MARK1, MARK2, MARK3, MARK4, MELK, MINK, MLK1_Cascade, MLK2_Cascade, MLK3_Cascade, MNK1, MNK2, MOS_Cascade, MRCKa, MRCKP, MSK1, MSK2, MSSK1, MST1 , MST2, MST3, MST4, NDR1, NDR2, NEK1, NEK2, NEK4, NEK6, NEK7, NEK9, NIM1K, NuaK1, NuaK2, ρ38α, ρ38β, ρ38γ, ρ38δ, p70S6K, p70S6Kp, PAK1, PAK2, PAK4, PAK5, PAK6, PASK, PBK, PDHK2, PDHK4, PDK1, PEK, PGK, PHKG1, PHKG2, PIM1, PIM2, PIM3, PKACa, ΡΚΑΟβ, PKACY, PKCa, PKCpi, ΡΚΟβ2, ΡΚΟγ, PKC5, ΡΚΟε, ΡΚΟζ, PKCn, ΡΚΟΘ, PKCi, PKD1, PKD2, PKD3, ΡΚΝ1, PKR, PLK1, PLK2, PLK3, PRKX, QIK, RAF1_Cascade, ROCK1, ROCK2, RSK1, RSK2, RSK3, RSK4, SGK, SGK2, SGK3, SIK, skMLCK, SLK, SRPK1, SRPK2, TAK1-TAB1_Cascade, TAOK2, TBK1, TNIK, TSSK1, TSSK2, TSSK3, WNK1, WNK2, WNK3, SPHK1, SPHK2.
Results
[0828] Table 17 depicts the % Inhibition of the tested kinases at a SNA-352 test concentration of 0.1μΜ.
se Inhibition by SNA-352

ACK .$
A: 22
A- 2.2
AXL ·;
Bl .K . 3
BM>. N.

ΐ)ϊΜ %.$
23 A
BGP -I "
: .Pi ! \ I OA

EFHA5
EHSAfi 24
Bi'HA? 4.4
■( 7
P;?HB.5
EBSB.2 .·-,
e>MB3 22
.•: .·
FAR' 54
PER ;«.(>
EE$
,i
EGFK AS
EGR Hi ?
PLTi A;>
FED 85
ΡΕΪ s .s
EMS Ϊ2Α
f¾K (> i
: K k i2A
WMl ■5.4
HER4 7 A
ΪΟΡ-Κ 3.0
I SR 6,2
JRR OA
ΓΓΚ AS
.! A K. i 9 A
TABLE 17 (continued) - Percent Kinase Inhibition by SNA-352
'ΛΚ.': V.:t
KD'R 7.0.6
s Γ ·.·..·
ια;. V:.;
UK 16.3
•7 5
: ΥΜ'· .:.
.Μί- 15.7
ΜΡ.Τ i .S
7?.4
;·\ : SO.2
i -T i.;.s
ON -2,3
ROS ¾3.3
SRC 6.A
SRM ■4.1
V W.O
T!iC • 7
•i! 7:
TNK i 34.2
ΚΚ.Λ
•i!k 63.4
TRKC 7S.S
1 Xk -3.=5
rvk? 7.3
rv"RO.; i)S>
IS s;A
3.4
AK"f'i 4
\K 1 : (:.■>
ΛΚ i 3 2.1
23
0.1
77.3
3 3*
Ayr ft i i.7

13.0
•3.'3
CaM is ■·3ν,
CaMkV!S -4.2
i' ·;'-.¾Κ.'·''
TABLE 17 (continued) - Percent Kinase Inhibition by SNA-352
.6
7 ; 7
-6.9
"■ X."-:' 4.3
C:DC77\S& -0.3

·:. i is.S
a«e 3,
C iii -1.0
-4.3
• ·· : :·. - ! .0
·. k ·;·: -0.7
CKia
CKIs
< k::-<\ -1.6
•2.0
CI. J 6.5
π 3.S
0.7
COT Gsssitfc 36.1
C.Ri . 0.0
DAi'KS : --
:OO,¾MKL; SU
5.3
DYKK Ί A 3.0
YR 1B -3.0
DV ; 5.S
;>YRk '· -3.7
Π.Ι-7 . 4.7
&kl o.¾
Sri;2
Erk5 -i.U
GS 3« ,v1
2.S
05
HGK 33.4
iiii-k■ -0.5
7.1
s sir v. ; ¾.3
-0.7
SK ii ■3.0
TABLE 17 (continued) - Percent Kinase Inhibition by SNA-352
ΙΚΚβ O.i
IK s <>::■·
IRAK! '■> ί
1RAK4 -
INK.! i.2
ικκ:: •(5.6
.INK 3. - . i
LATB2 26.0
I..OK l J:-
%,ΰ

-U
\i \i!7Ki> C . ; - 2
MA 2K7 Csseiiik ·:.'.··
\WV:-Y,2 0;:,-v.;;i.:

-S,«
ΜΛΡΚΛΡΚ3 •0.7
MAPK.APk'.S -0./
MARK ! 3 .8
MARK-i 35. Ϊ
MELK 22
M K
34.fi
7.8
25.
MN i 2.5
NK2 ! 2
0.3
-J.
MRCKi? -i!.5
MSfO 40J
MSK2 .
3.2
3.3
■•2.4
O i 2.0
>R 5.3
EKS .•3.
TABLE 17 (continued) - Percent Kinase Inhibition by SNA-352
10.4
K 4 ¾.«
M K6 Q.4
N.i'KJ 0.1

n.
0 .s
2.4
i.7
20.1
p"0S6K8 3.0
ΡΛΚΪ ].:?
ΡΛΚ2
PAK4 15.2
PAR> 173
FAK.6 8.0
PASK 7.0
i'UK S3
K2. ·.· i
PDHSiA n. s
PDKi 24.6

FH >2 -LS
Pi S !34
m -3.0
PS J ? >
PKACs 8.7
PKAC!i 0.6
I' Ai / 2.4
FK<:« 25. ί
14.1
PiiCffi 7:0.4
P :v 4<>
P <¾ 14.9
PKO; 29.5
;-K( :: 223
H;C:S 4.9
:ΡΚ.(;θ -3.1
PKiA 219
PKDi 3.0
P 2 2.3
PRD3 ~LJ
P i 0.0
PK» ■9.9
TABLE 17 (continued) - Percent Kinase Inhibition by SNA-352
PLK ! -(>.5
PLK.2 •-0J
H.. -9,5
PRKX i.■
Oik 55.6
RAF S ..Ciss iKSe 2 J
R>;H K I .·:).?
ROCK:? ■■0,7
RSi S 48 ,i?
RSK2 <54,7
ESK 5J.6
S ·> 7 Li
SG 3.7
SC 2 8,S
4,7
S0.2
skML K 1.K
Si.k 9-2
SHMi -2,2
SRPK 0.1
ΤΛ ί-'ΓΑ ί ,C «Bk -S.S
1AOR2 •0 !■
Γ 1 273
T iK 2 J i?
ISSKi ■■ox
8 2 -2,3
TSSK3 •\;
VN 1 0.6
N 2 .
\v\;K ·· ~B
SPH ; ■2,4
SPHK2 5J
Example 11: Profiling Study of SNA-352 against 25 Kinases Aim of Study
[0829] The goal of this study was to measure the inhibitory activity of SNA- 352 against 25 kinases.
Materials and Methods
Test Agent
[0830] SNA-352 (Lot # 2017GC14/S7) was used at a test concentration of
0.1 μΜ.
Target Kinases
[0831] BMPR1A, BRAF, COT, DLK, LIMK1, LKB1, MAP2K1, MAP2K2, MAP2K3, MAP2K4, MAP2K5, MAP2K6, MAP2K7, MAP3K1, MAP3K2, MAP3K3, MAP3K4, MAP3K5, MLK1, MLK2, MLK3, MOS, RAF1, TTK, WEE1.
Results
[0832] Table 18 depicts the % Inhibition of the tested kinases by SNA-352 at a test concentration of 0.1 μΜ.
TABLE 18 -Percent Kinase Inhibition byO-ΙμΜ SNA-352

BRAF (!.9
cox A2
DLK. ·:: "
L .iM : .29.'?
1KB j i!.9
MAP2K 15.6
ΜΛΡ Κ2 6.;
AF2 3 20.5
ΜΛ!>::);
.ΜΛΡ2Κ.5 2.2
MAP2K IS.3
MAP2K7 1.4
MA KS 0,8
ΜΑΪΊΕ2 ! 2
ΜΛΡ2Κ3 4.2
MAi! K ■4.6
MA S -6,2
Mi.lv i 68.7
:LK2
MUG B3.3
MOS 4.2
RAFS 0,2
ΪΪΚ 4,35
wee; 2.2
Conclusions
[0833] In Examples 10 and 11 the inhibition profile of SNA-352 at 100nM was profiled against 299 kinases, including tyrosine kinases (TK), serine/threonine kinases (STK) and mitogen-activated protein kinase (MAPK). It was found that 27 kinases of the 299 kinases were inhibited more than 50% by SNA-352. SNA-352 inhibited 7 kinases
(JAK3, TrkA, PDGFRp, FLT3, MLK3, PDGFRa, and ACK) by more than 80%. Further, SNA-352 inhibited 6 kinases (CGK2, TrkC, DDR1 , AurA, RSK4, JAK2) between 70 and 80%. Within the JAK family, 100nM SNA-352 inhibited JAK3, JAK2, JAK1 and TYK2 by 95.0%, 71 .1 % , 9.6%, and 7.3%, respectively. Within the Trk family, 100nM SNA-352 inhibited TrkA, TrkC, and TrkB by 94.8%, 78.8%, and 63.4% , respectively. Figure 20 depicts the SNA352 kinase inhibition profile at test concentrations of 100nM and 200nM for the top inhibited kinases as well as those kinases in the middle in the inhibition spectrum.
Example 12: Determination of IC50 of SNA-352 against 15 Kinases Aim of Study
[0834] Kinases generally correlated to cancers (e.g. AurA) were excluded. All the members of the key families of kinases (JAK and Trk family) were included. The goal of this study was to assess the potency (IC50) of SNA-352 against the 15 kinases identified by this analysis: JAK1 , JAK2, JAK3, TYK2, TrkA, TrkB, TrkC, PDGFRb, PDGFRa, FLT3, FLT4, MLK1 , MLK3, ACK, SIK, MAP2K6, LIMK1 , DDR1 and DDR2.
Materials and Methods
Test Agent
[0835] SNA-352 (Lot # 2017GC14/S7) was used at test concentrations of 10, 3, 1 , 0.3, 0.1 , 0.03, 0.01 , 0.003, 0.001 , 0.0003, 0.0001 μΜ .
Target Kinases
[0836] SNA-352 was used at test concentrations of 10, 3, 1 , 0.3, 0.1 , 0.03, 0.01 , 0.003, 0.001 , 0.0003 μΜ for ACK, DDR1 , DDR2, FLT3, FLT4, JAK1 , JAK2, PDGFRa, PDGFRp, TRKB, TRKC, TYK2, and SIK.
[0837] SNA-352 was used at test concentrations of 3, 1 , 0.3, 0.1 , 0.03, 0.01 , 0.003, 0.001 , 0.0003, and 0.0001 μΜ for JAK3 and TRKA.
Results
[0838] Table 19 depicts the IC50 of SNA-352 and staurosporine for 15 kinases.
TABLE es

DDR! 3.6S&-0S 3.09E-0
. .2 ! J9E-0? 2.02E-09
FLT3 L33E~0S E2Jj? !0
FL1 I.09E-O7 5,94E-!0
JA!O §,46E )7
K2 S.36E~0S 2.40i )
JA 3 6.2/! 0 0F- )
ΊΎΚ.2 tSSB~06 8.26P-K)
FiXIFR 2.42F 2.,021 10
PIXFE 2. B0§ L49E~iO
ΤΚ Λ SJ9E-09 3.S5EEK
TR B 5.77E-0S 3,02Ε"10
T .C 2JS£ Jg 3J3E 0
SIK E62E-0
Conclusions
[0839] SNA-352 is a potent inhibitor of kinaes linked to IBD
Example 13: CT340 Kinase Inhibition Panel
[0840] K-252a is a potent inhibitor of multiple kinases. In the present study, the inhibitory activity of K-252a against selected common tyrosine kinases and serine/threonine kinases was evaluated. A similar experiment has been conducted for evaluating the inhibitory activity of CT340.
[0841] K-252a (Acros lot A020265401) was dissolved in DMSO to make a 1 mM stock solution which was then diluted with DMSO to obtain a 20 μΜ solution, further diluted with the assay buffer to achieve a concentration of 0.8 μΜ. K-252a was tested at a concentration of 200 nM. The preparation of CT340 and the reference compounds was conducted following a similar procedure. CT340 also was tested at a concentration of
200 nM. Staurosporine, 5-iodotubericidin, NK inhibitor II and SB202190 have been used as reference compounds.
[0842] The kinase inhibition studies for K-252a and CT340 have been performed using standard assays for the respective kinase.
[0843] The inhibitory activities of CT340 (200nm) against 89 tyrosine kinases are shown in Table 20 below. The inhibitory activities of CT340 (200nM) against 187 serine/threonine kinases are shown in Table 21. The readout value of reaction control (with ATP) was set as a 0% inhibition and the readout value of background (without ATP) was set as a 100% inhibition. The results clearly show a dramatic improvement in the selectivity of kinase inhibition of CT340 versus K-252a. The less other kinases are inhibited, the less toxic the molecule is likely to be.
TABLE 20 - Inhibitory activities of CT340 against 89 tyrosine kinases
Kinase CT343 WE®, a CT340
" m 200 200
AOL 2,1 ¾o JAX1 17,2 102,7 o.« WA 102,0 J $,2 JAi 3 S8.4 101,0
AR<J 1,3 11,2 WIS. 3%
ACK 28,3 99.1 ST (1,3
TN 1 14,0 88,7 6ΐ(*Ί236»5 1*1 S4f>
At 2.e eo,a RO N •o.s
UK es.i MUSK 2,8 800
ML 21,8 00,1 FLT3 84,0 100,9
MEft ee,2 2,2 98.0 ram »<!T 4,4 93<4
43,8 KtT(TO70!) •0, 81,1
C TK 2,0 04
23,8 so,? & 100,4
1,0 40,d S>&Sf ft«<W4l) 76,2 99.0
-1.3 8S.4 f»tsSFf¾aCV8aiC) «2,6 101,3
3,6 ?e,o POOFRb βδ,ο 100,3
HSR2 -1,1 30,3 f?8T 2 ,¾ 8S£
>4,S 7G,7 80S 78,8 101,8
SphAl 0,0 82,8 SLK β,δ 6;t£
B|>hA2 2,0 44,7 SRK ■3,4 72 JO
S{>iiAS *β,4 (50,1 FOR <$,* 89,1
Ep(iA -7,7 30,3 FR w 82,0
•3,1 «1,1 ?YN 4.0 70,1
-11,6 24,3 HCK ie,8 9 £
EfshAV -2,6 08,S LC 7,a 89,8
1S.4 LYMa t¾3 «4JB
6p fM -2,$ <5¾,S LYf* 7,8 73

Bph83 ' ,3 0,6 -4.4
-7,4 22,7 YSS 4,1 f AK >δ,δ 41,1 SYK S7,S ιοα7
PY 2 4,3 82,0 2AP7Q 30,8 987 fm 63,3 180,8 BMX 1 .6 86J8
FES 3,1 80,8 BTK ■2,8 01j5 OFW 2,7 S3,2 IT 11,2 98,4
FGFR2 •4,9 ao,o tec 10$ 86,1
F 0FR3 £,3 64,4 TXK ¾3 503
4,8 88.S T)K2 >12,4 630
F SFR3<W¾»§ 8,1 84,3 TftKA S2.S 101,6
•w 22,0 TO S 76,7 101,0
I0F-Ii¾ 1,8 97,8 TfiKC 9-1,1 mn
WSR 8d,€ F T1
IRR 3,7 80,3 FU4 «3,4
0R 12,3
TABLE 21 - Inhibitory activities of CT340 against 187 serine/threonine kinases
Kinase era-© QSZa Kinase CT340 2Θ24
200 fifa! 200 nfof 200 rM 200 iiW
ΑΚΠ -4,5 48,1 DAP 1 -8,6 83,0
Λ Τ2 - -3/3 23,3 DCAMKL2 - -4t4 7,8
AKT3 4,0 83,0■ MAPKAP 2 -1,2 92,8
CR)K 8,3 82,a AP APK3 -7,5 32,8 OCK1 -5,2 83,7 MAPKAPKS -1,6 52,4
ROC «2 -2,9 88,8 sk&fU K ■ 7,3 89,1
MRC e -1,0 se,5 MMK1 1S,8 97,4
PKACa 1,3 92,1 N 2 67,1 101,7
PRKX 2,3 94,3 PIW1 1,S 89,8
P 12,S 89,2 PIM2 0,1 60,7
COK2 88,8 101,3 PHK01 S3,0 100,7
PO 1 41,2 99,9 PHK02 13,6 101,8
P Ce 12,3 92,7 PKD1 65,1 103,1
PKCh 4,0 82,2 PK02 88,9 103,2
PKCs 7,3 84,9 PKP3 54,4 101,9
PKCd 7,8 68,4 CHK2 8,8 97,2
PKCe 1Q.S 77,S T S 1 25,8 101,9
P Cq 5,1 87,0 TSSK2 1,2 98,5
PKCz -4,0 42,1 CDC2 4,3 87,8
P C 11,5 81,8 CDK2 26,4 87,8
P ci 3,S 39,0 CO 3 2,3 82,8
P M1 27,5 9S|S CD 4 4,2 98,5
MSK1 -G,S 31 ,5 CDC7 4,0 82,1
MSK2 - ,8 88,8 CDK5 '28,3 93,3 p70S8 9,3 91,7 DVRK2 2,1 97,0
RSK1 34,3 S8,B CLK1 63,0 100,?
RSK2 38.S 88,0 HJP 2 -4,1 87,9
RSK3 52,6 00,4 GSK3a 11,8 81,2
S K 8, 99,S ©SK3b 4,5 70,8
SGK2 - ,3 80,9 SRPK1 -2,4 -12,1
SOK3 -0,8 S6,0 SRPK2 -5.5 -0,8
Ca K4 -4,0 €.7 AurA 8S,2 100,8
-3,3 80,3 AurB 12,1 98,8
CaMK2a 5,3 101,2 AiifC 43,2 39,0
AM K«1&ij¾j1 3,0 87£ I Ka -3,9 20,0
BRSK 28,2 83,6 IKKb 18,8 91,1
8RSK2 18,7 88,3 Π<Κβ 4,0 SB,4
CH 1 67,3 102,2 TB 1 37,8 10O.S
MARK1 4D.S 39,6 NEK1 -2,1 19,1
M RK2 40,7 100,7 NEK2 -2,2 8,5
MARKS 50,9 9S.4 NEKS 0,2 -2,3
43,2 02,0 NSK7 -2,3 -1,8
MGC42105 ifl 20,3 PBK 82,3 98,7
NUS 1 76,7 00,8 PLK1 -2,0 58,0 AS!f -0,4 78,3 PL 3 1,8 '7,7
¾ Inhibits ¾ !nh !sm
Klnasa ®82ά Kinase CT3«J 262a
300 ιΛ! aoo ftM 200 ftM
PLK3 -10,5 -0,3 BRAF -30,9 -65,6
TTK 12,6 93,5 BS¾AF(V600S) 10,7 -7,0
WM 1 7,1 -1,1 MAP4K2 38,4 86,0 mpKi S 98,9 H 97,8 100,4
IHM<4 9,5 98,0 P C&2 8,4 51 ,1
BMP Hi A 1,8 42,1 WEE1 1,4 37,7
CKI« -1,9 0,8 UMK1 82,0 98,0
C 1d 0,? 7,1 LKB1 . -7,0 21,4
CK18 -2,8 1,0 PL 4 S1,5 100,3
PAK2 -2,2 79,4 CK2<rt ■8,3 -16,3
PAK6 5,7 94,© PE Ss4 12,0
MiN 84,0 101,8 P H 5,7 29,4
MSM •8,1 27,2 WKa2i¾1Aft 1Θ.3 89<S e ki 27,8 mp CeM W .1,0 88,8
Efk2 40,8 p CaMfQj 2,1 101,5
ErkS 8,7 87,8 CD 8 > t2 S4.D
49,5 97,1 CD 7 -7,3 44,8
JNK2 11,7 81,9 CDK9 0,8 75,8
JN 3 19,5 94,3 CLK2 -3,5 85,8 p38s -2,9 0,3 OO 1 41,4 87,3 p38 -0,8 8,0 DY A 0,8 96,4 p38g 13,6 65,3 DYRK18 10,3 98,4 p3Bci «0,8 23,1 DVR 3 9,7 100.S
M,aP2 SS,Q fi EEF2 -5,6 -3,B
93,3 99,7 HIPK1 -8,4 45,2
MAP2 3 98,4 100,1 HtPt3 0,3 80,0
MAP2 4 53,0 81,2 LOK 38,3 99,6 Λ»2Κ5 88,5 99,2 MARK4 52,1 100,4
MAP2K6 00,0 00,0 76,3 100,2
M.4P27 59,5 94 ,§ MST2 44,1 98,6 Λ»3Κ1 43,0 1,2 MST3 -1,5 63,7
M P3K2 51,7 97,8 NDR1 -2,4 82,0
58,5 93,0 NEK9 -1,0 75,0
ΜΛΡ3Κ4 S7.5 82,2 2,2 73,8 wastes 98,8 - PAKi 5,7 85,3
TA 1 30,S 89.0 RSK4 S0.4 99,8
COT *3j5 10,0 SIK 18,3 87,8 L 1 89,0 00,2 CflMK2b -1,8 86,3
MLK2 33.Θ 95,6 C«MK2ci 30,2 101,8 L 3 07,7 99,0 CK1g1 -S,3 -2,5
Ot -13,7 36,3 C S2 0,2 4,3
44,4 36,8 PAK3 10,1 98,8
inase C 34!. Ma
200 KM 200 rM-
PAK5 13,6 ,7
PDH 4 1.0 2,1
POH 2 -§,3 .3,1
M C b ,8 79t2
C LK3 •8,1
M SSK1 -2,9 54,0
CK1g3 2,2
P IK3CASMK3R1 11 ,0 44,5
TACK2 -3,5 28,1
p\m 32,0 98,4
B,S 83,5
ΡΛΚ4 9,9 92,5
S 10,4 41 ,4
Example 14: IC50 Determination study of CT340 against 7 kinases
[0844] The purpose of this study was to evaluate CT340 inhibition of JAK2, JAK3, PDGFRb, TRKA, MAP2K1 , MAP2K3, TAK1 -TAB1 . Staurosporine was evaluated as well as a control. The CT340 test concentrations used were the following: 10, 3, 1 , 0.3, 0.1 , 0.03, 0.01 , 0.003, 0.001 , and 0.0003 μΜ.
Materials and Methods
[0845] The test compound was dissolved in and diluted with dimethylsulfoxide (DMSO) to achieve 100-fold higher concentration. Then the solution was further 25-fold diluted with assay buffer to make the final test compound solution. Reference compounds for assay control were prepared similarly.
STK-ELISA
[0846] 1) The 10 μΙ_ of x4 compound solution, 10 μΙ_ of x4 Substrate/ATP/Metal solution, and 20μΙ_ of x2 kinase solution were prepared with assay buffer (15 mM Tris-HCI, 0.01 % Tween-20, 2 mM DTT, pH7.5) and mixed and incubated in a well of polypropylene 96 well microplate for 1 hour at room temperature.
[0847] 2) 120 μΙ_ of 40 mM EDTA solution (pH 7.5) was added to the well, and then 120 μΙ_ of the mixture was transferred to the well of ELISA plate (as shown below).
[0848] 3) After 30 minutes incubation, the well was washed 4 times, and blocked with blocking buffer containing 0.1 % BSA.
[0849] 4) 100 μΙ_ of the first antibody (as shown below) solution was added to the well and incubated for 30 minutes.
[0850] 5) After 4 times washing of the well, 100 μΙ_ of the second antibody (as shown below) solution was added to the well, and incubated for 30 minutes.
[0851] 6) After washing the well, 100 μΙ_ of TMB solution was added and incubated for 5 minutes. To stop the HRP reaction, 100 μΙ_ of 0.1 M sulfuric acid was added.
[0852] 7) The kinase reaction was evaluated by the absorbance at 450 nm of the well.

Off-chip Mobility Shift Assay (MSA)
[0853] 1) The 5 μΙ_ of x4 compound solution, 5 μΙ_ of x4 Substrate/ATP/Metal solution, and 10 μΙ_ of x2 kinase solution were prepared with assay buffer (20 mM HEPES, 0.01 % Triton X- 100, 2 mM DTT, pH7.5) and mixed and incubated in a well of polypropylene 384 well microplate for 1 hour at room temperature.
[0854] 2) 60 μΙ_ of Termination Buffer (QuickScout Screening Assist MSA; Carna Biosciences) was added to the well.
[0855] 3) The reaction mixture was applied to LabChip3000 system (Caliper Life Science), and the product and substrate peptide peaks were separated and quantitated.
[0856] 4) The kinase reaction was evaluated by the product ratio calculated from peak heights of product(P) and substrate(S) peptides (P/(P+S)).
[0857] The reaction conditions employed for the seven kinase assays are depicted below:

[0858] The readout value of reaction control (complete reaction mixture) was set as a 0% inhibition, and the readout value of background (Enzyme(-)) was set as a 100% inhibition, then the percent inhibition of each test solution was calculated.
[0859] IC50 value was calculated from concentration vs. %lnhibition curves by fitting to a four parameter logistic curve.
Results
[0860] The CT340 and staurosporine concentration vs. %lnhibition curves are depicted in Figures 21 to 34, and the IC50 determinations for both compounds are shown in Table 22 below.
TABLE 22 - IC50 Determination
-ODE- 10
JAK3 3,87E~0§ 2J8E- 10
PDGFRb U2E-I37 3J7E-10
TRKA 2?53£-§8 5.02E-10
AF2 S I. 6E-0S 139E-G9
ΜΆΡ2Κ3 !.,26E-08 L13E-09
TA i.-TABl J9E-07 4J4.E-0S
Example 15: BioMAP Platform Analysis of SNA-120
Aim of Study
[0861] The goal of this study was to characterize SNA-120 in the BioMAP Diversity PLUS panel of 12 human primary cell-based systems. These systems are
designed to model complex human tissue and disease biology of the vasculature, skin, lung and inflammatory tissues. Quantitative measurements of biomarker activities across this broad panel, along with comparative analysis of the biological activities of known bioactive agents in the BioMAP reference database are used to predict the safety, efficacy and function of these test agents.
Materials and Methods
Test Agent
[0862] SNA-120 was profiled in the BioMAP Diversity PLUS panel at concentrations of 28000 nM, 9200 nM, 3100 nM, and 1000 nM. SR221 1 was employed as the benchmark compound.
Methods and Analysis
[0863] BioMAP Platform Analysis was performed as described in Example 3. Results
BioMAP Profile
[0864] Figure 35 depicts the BioMAP profile of SNA-120 in the Diversity PLUS Panel. SNA-120 was found to be active with 21 annotated readouts, mediating changes in key biomarker activities listed by biological and disease classifications in Table 23 below. SNA-120 impacted inflammation-related activities (decreased E- selectin, sTNFa, MIP-1 a, IL-8, I L- 1 a) , immunomodulatory activities (decreased slgG, sIL- 10; increased CD69), tissue remodeling activities (decreased TIMP-2, tPA, MMP-1 , PAI- 1 , Collagen III, uPAR, MMP-9), and hemostasis-related activities (decreased TF). SNA- 120 is antiproliferative to endothelial cells, fibroblasts, and T cells (as indicated by grey arrows in Figure 35). There are no cytotoxic impacts detected at the concentration range tested.
TABLE 23 - Key Biomarker Activities Impacted by SNA-120
 Reference Benchmark Overlay
Reference Benchmark Overlay
[0865] Figure 36 depicts an overlay of SNA-120 at 28 μΜ and the selected reference benchmark SR221 1 at 10 μΜ. SR221 1 is an antagonist of retinoic acid receptor related nuclear receptor gamma. There are 12 common activities that are annotated within the following systems: 3C (Prolif), LPS (CD40), SAg (Prolif), BT (sIL- 17A, SIL-17F, slL-2, slL-6, slgG, sTNFa), CASM3C (TF), HDF3CGF (Prolif 72), and IMphg (slL-10).
[0866] Differentiating biomarkers (not shown) are defined when one profile has a readout outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1), and the readout for the other profile is either inside the envelope or in the opposite direction. There are 33 differentiating activities between the two compounds: 3C (HLA-DR, TF, uPAR), 4H (MCP-1 , uPAR), LPS (CD69, SPGE2, sTNFa), SAg (CD69), BF4T (Eotaxin 3), BE3C (MMP-1 , MMP-9, PAI-1 , tPA), CASM3C (HLA-DR, IL-6, LDLR, TM, uPAR), HDF3CGF (Collagen I, Collagen III, EGFR, M-CSF, MIG, PAI-1 , VCAM-1), MyoF (IL-8, MMP-1 , VCAM-1), and IMphg (CD69, MCP-1 , MIP-1 a, VCAM-1).
BT System Secretion Profiles
[0867] Figure 17 depicts changes in the secretion of IL-17F, IgG, IL-17A, and TNFa in the BioMAP BT system mediated by SNA-120 (3.1 μΜ), Tofacitinib (3.3 μΜ), Apremilast (3.3 μΜ), SR221 1 (3.3 μΜ), and Cyclosporin A (3.3 μΜ). Tofacitinib was found to be more active than SNA-120 in decreasing IL-17F secretion, displaying an activity similar to SR221 1 (Figure 17A). Surprisingly, SNA-120 was found to be as active as tofacitinib in decreasing IL-17A secretion (Figure 17C). SR221 1 decreased IL-17A secretion as expected, while Apremilast increased IL-17A secretion.
Top Database Search Result for SNA-120
[0868] In a search for mathematically similar compound profiles from the BioMAP reference database, SNA-120 (28 μΜ) was most similar to GSK690693 (10 μΜ) (Pearson's correlation, r = 0.733). The Pearson's correlation coefficient between these two profiles is above our determined threshold (r > 0.7) indicating these compounds share mechanistically relevant similarity. GSK690693 is an ATP-competitive pan-Akt kinase inhibitor that also exhibits some inhibition for AMPK, PKA and PAK and PKC isoforms. Figure 37 depicts an overlay of SNA-120 (28 μΜ) and GSK690693 (10 μΜ).
[0869] There are 15 common activities that are annotated within the following systems: LPS (sPGE2, sTNFa), SAg (Prolif), BT (SIL-17A, SIL-17F, slL-2, slL-6, slgG, sTNFa), BE3C (MMP-1 , MMP-9, PAI-1), HDF3CGF (PAI-1), and IMphg (CD69, slL-10).
Top BioSeek Reference Database Matches for SNA-120
[0870] Table 24 depicts the top 3 similarity matches from a search of the BioMAP Reference Database of > 4,000 agents for each concentration of SNA-120. The similarity between agents is determined using a combinatorial approach that accounts for the characteristics of BioMAP profiles by filtering (Tanimoto metric) and ranking (BioMAP Z-Standard) the Pearson's correlation coefficient between two profiles. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7.

[0871] The Pearson's correlation coefficient between profiles that is above our determined threshold (r > 0.7) indicates these compounds share mechanistically relevant similarity. For profiles with a Pearson's correlation coefficient below our determined threshold (r < 0.7), the relevance of the similarity is unknown.
Mechanism HeatMAP Analysis of SNA-120
[0872] Figure 38 depicts Mechanism HeatMAP Analysis of SNA-120, with the 148 biomarker readouts within the Diversity PLUS panel compared to 19 consensus mechanism class profiles. This analysis informs on the regulatory mechanisms controlling increases or decreases in each of the biomarker readouts.
Clustering of Project Profiles
[0873] Figure 14 depicts a clustering of tested agent profiles based on pairwise correlation analysis and clustering of most similar profiles. Profiles that are similar with a Pearson's correlation coefficient (r) > 0.7 are connected by lines. Agents
that do not cluster with one another are interpreted as mechanistically distinct. Cytotoxic concentrations are excluded from cluster analysis. Functional clustering of the agent profiles generated during this analysis uses Pearson's correlation values for pairwise comparisons of the profiles for each agent at each concentration, and then subjects the pairwise correlation data to multidimensional scaling. SNA-120 clusters internally at two concentrations. Internal clustering suggests the phenotypic signature of this compound is maintained across a range of concentrations, a characteristic commonly observed in marketed drugs.
Conclusions
[0874] In this study SNA-120 was characterized by profiling in the BioMAP Diversity PLUS panel of human primary cell based assays modeling complex tissue and disease biology of organs (vasculature, immune system, skin, lung) and general tissue biology. The Diversity PLUS panel evaluates the biological impact of test agents in conditions that preserve the complex crosstalk and feedback mechanisms that are relevant to in vivo outcomes.
[0875] SNA-120 was broadly active and non-cytotoxic in the Diversity PLUS panel. Antiproliferative effects to multiple cell types were observed, a feature that is appropriate for compounds developed for oncology, but not autoimmune indications. Inhibition of several inflammation-related readouts were observed with a sharp dose response noted between the two highest and two lowest concentrations. This suggests that the compound may have additional targets at the higher concentrations (> 9.2 μΜ). Robust activity was also observed in systems containing epithelial cells, indicating target expression on this cell type and potentially utility for pulmonary indications. At the top tested concentration, SNA-120 shared 12 common activities with the requested benchmark compound SR221 1 , a RORy inhibitor, predominantly in the BT system. SNA- 120 shared 15 common activities, also predominantly in the BT system, with the pan- AKT inhibitor GSK690693. Other top database matches with a Pearson's correlation coefficient r > 0.7 include BSK_4925, a Lck inhibitor, and filgotinib, a JAK inhibitor. It is noted that several activities of this test agent, particularly in the BE3C, BF4T and HDF3CGF systems, are consistent with the EGFR inhibitor mechanism class (see Figure 38). An EGFR inhibitor was among the top reference database matches for this compound, but the overall Pearson's correlation was less than r = 0.7, thus the relevance of the similarity is unknown. Acneiform rash is a commonly associated adverse effect of EGFR inhibitors.
Example 16: BioMAP Platform Analysis of SNA-125
Aim of Study
[0876] The goal of this study was to characterize SNA-125 in the BioMAP Diversity PLUS panel of 12 human primary cell-based systems that was described in Example 3.
Materials and Methods Test Agent
[0877] SNA-125 was profiled in the BioMAP Diversity PLUS panel at concentrations of 3900 nM, 1300 nM, 430 nM, and 140 nM. Tofacitinib was employed as the benchmark compound.
Methods and Analysis
[0878] BioMAP Platform Analysis was performed as described in Example 3. Results
BioMAP Profile
[0879] Figure 39 depicts the BioMAP profile of SNA-125 in the Diversity PLUS Panel. SNA-125 was found to be modestly active with 4 annotated readouts, mediating changes in key biomarker activities listed by biological and disease classifications in Table 25 below. SNA-125 mediated changes in key biomarker activities are listed by biological and disease classifications. SNA-125 impacted inflammation- related activities (decreased sTNFa) and tissue remodeling activities (modulated MMP- 1). There are no cytotoxic or antiproliferative impacts detected at the concentration range tested.
TABLE 25 - Key Biomarker Activities Impacted by SNA-125
 Reference Benchmark Overlay
Reference Benchmark Overlay
[0880] Figure 40 depicts an overlay of SNA-125 at 3.9 μΜ and the selected reference benchmark tofacitinib at 3.3 μΜ. Tofacitinib is a JAK1/3 kinase inhibitor approved in 2012 for the treatment of rheumatoid arthritis. There are 5 common activities that are annotated within the following systems: BT (SIL-17A, slL-17F, slgG, sTNFa), and IMphg (slL-10).
[0881] Differentiating biomarkers (not shown) are defined when one profile has a readout outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1), and the readout for the other profile is either inside the envelope or in the opposite direction. There are 39 differentiating activities between the two compounds: 3C (HLA-DR, MIG, uPAR), 4H (Eotaxin 3, P-selectin, VCAM-1 , VEGFR2), LPS (CD69, IL- 1 a, sTNFa), SAg (CD38, CD40, CD69, E-selectin, IL-8, MCP-1 , MIG, Prolif), BT (Prolif, slL-2, slL-6), BF4T (MMP-3, MMP-9), BE3C (l-TAC, IP-10, MIG, uPA), CASM3C (HLA- DR, LDLR, M-CSF, MCP-1 , MIG, VCAM-1), HDF3CGF (IP-10, M-CSF, VCAM-1), and KF3CT (ICAM-1 , IP-10, MIG).
BT System Secretion Profiles
[0882] Figure 17 depicts changes in the secretion of IL-17F, IgG, IL-17A, and TNFa in the BioMAP BT system mediated by SNA-125 (3.9 μΜ), Tofacitinib (3.3 μΜ), Apremilast (3.3 μΜ), SR221 1 (3.3 μΜ), and Cyclosporin A (3.3 μΜ). Tofacitinib was found to be more active than SNA-125 in decreasing IL-17F secretion, displaying an activity similar to SR221 1 (Figure 17A). SNA-125 and Tofacitinib were both very active in decreasing secreted IgG, with SNA-125 as active as Cyclosporin A (Figure 17B). Surprisingly, SNA-125 was found to be as active as tofacitinib in decreasing IL-17A secretion (Figure 17C). SR221 1 decreased IL-17A secretion as expected, while Apremilast increased IL-17A secretion. SNA-125 was found to have activity with regards to reducing TNFa secretion (Figure 17D).
Top Database Search Result for SNA-125
[0883] In a search for mathematically similar compound profiles from the BioMAP reference database, SNA-125 (3.9 μΜ) was most similar to SB203580 (10 μΜ) (Pearson's correlation, r = 0.649). The Pearson's correlation coefficient between these two profiles is below our determined threshold (r < 0.7) indicating that the relevance of the similarity is unknown. SB203580 is a p38 MAPK Inhibitor. Figure 41 depicts an overlay of SNA-125 (3.9 μΜ) and SB203580 (10 μΜ).
[0884] There are 6 common activities that are annotated within the following systems: LPS (sTNFa), BT (SIL-17A, SIL-17F, slgG), BE3C (MMP-1), and IMphg (sIL- 10).
Top BioSeek Reference Database Matches for SNA-125
[0885] Table 26 depicts the top 3 similarity matches from a search of the BioMAP Reference Database of > 4,000 agents for each concentration of SNA-125. The similarity between agents is determined using a combinatorial approach that accounts for the characteristics of BioMAP profiles by filtering (Tanimoto metric) and ranking (BioMAP Z-Standard) the Pearson's correlation coefficient between two profiles. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7. For profiles with a Pearson's correlation coefficient below our determined threshold (r < 0.7), the relevance of the similarity is unknown.

Mechanism HeatMAP Analysis of SNA-125
[0886] Figure 42 depicts Mechanism HeatMAP Analysis of SNA-125, with the 148 biomarker readouts within the Diversity PLUS panel compared to 19 consensus mechanism class profiles. This analysis informs on the regulatory mechanisms controlling increases or decreases in each of the biomarker readouts.
Conclusions
[0887] In this study SNA-125 was characterized by profiling in the BioMAP Diversity PLUS panel of human primary cell based assays modeling complex tissue and disease biology of organs (vasculature, immune system, skin, lung) and general tissue biology. The Diversity PLUS panel evaluates the biological impact of test agents in conditions that preserve the complex crosstalk and feedback mechanisms that are relevant to in vivo outcomes. SNA-125 was selectively active in the Diversity PLUS panel, with specific inhibition of sTNFa activity in the LPS and BT systems. No cytotoxic or antiproliferative effects were observed. Inhibition of TNFa activity or production is consistent with drugs currently approved for the treatment of psoriasis, such as anti-TNF antibodies (e.g. adalimumab and infliximab) and JAK inhibitors (e.g. tofacitinib) .
Example 17: Additional BioMAP Platform Analysis of SNA-125
Aim of Study
[0888] The goal of this study was to repeat the characterization of SNA- 125 in the BioMAP Diversity PLUS panel of 12 human primary cell-based systems using higher concentrations than employed in Example 16.
Materials and Methods
Test Agent
[0889] SNA- 125 was profiled in the BioMAP Diversity PLUS panel at concentrations of 30 μΜ , 10 μΜ , 3.3 μΜ , and 1 .1 μΜ . K252a was employed as the benchmark compound.
Methods and Analysis
[0890] BioMAP Platform Analysis was performed as described in Example 3. Results
BioMAP Profile
[0891] Figure 43 depicts the BioMAP profile of SNA-125 in the Diversity PLUS Panel. SNA-125 was found to be active with 52 annotated readouts, mediating changes in key biomarker activities listed by biological and disease classifications in Table 27 below. SNA-125 mediated changes in key biomarker activities are listed by biological and disease classifications. SNA-125 impacted inflammation-related activities (decreased Eotaxin 3, E-selectin, MCP-1 , VCAM-1 , sTNFa, M IP-1 a, IL-8, IL-1 a, P- selectin, sPGE2, IL-6) , immunomodulatory activities (decreased CD40, slgG, slL-10, M- CSF, SIL- 17A, slL-6, SIL-17F, slL-2) , tissue remodeling activities (decreased Collagen I , TIMP-2, Decorin, MMP- 1 , uPA, PAI-1 , Collagen I I I , uPAR, MMP-9), and hemostasis- related activities (decreased TF; increased TM) . SNA- 125 is antiproliferative to B cells, endothelial cells, fibroblasts, and T cells (as indicated grey arrows of Figure 43). SNA- 125 had no cytotoxic effects in the concentration range tested.
TABLE 27 - Key Biomarker Activities Impacted by SNA-125

Reference Benchmark Overlay
[0892] Figure 44 depicts an overlay of SNA- 125 at 30 μΜ and the selected reference benchmark K252a at 30 nM. K252a is a non-selective protein kinase inhibitor that inhibits PKC, Ca2+/calmodulin-stimulated phosphodiesterases, MLCK, and receptor tyrosine kinases. K252a is an analog of staurosporine.
[0893] There are 37 common activities that are annotated within the following systems: 3C (IL-8, Prolif, TF, uPAR), 4H (Eotaxin 3, MCP-1 , P-selectin), LPS (CD40, E- selectin, IL-1 a, IL-8, M-CSF, MCP-1 , TF, VCAM-1 , sTNFa), SAg (Prolif), BT (SIL-17A, SIL-17F, slL-6, slgG, sTNFa), BF4T (Eotaxin 3, MMP-1), BE3C (IL-8, MMP-1 , MMP-9, PAI-1 , uPA, uPAR), HDF3CGF (MMP-1 , Prolif 72, TIMP-2), KF3CT (MCP-1), and IMphg (E-selectin, MIP-1 a, slL-10).
[0894] Differentiating biomarkers (not shown) are defined when one profile has a readout outside of the significance envelope with an effect size > 20% (|log10 ratio| > 0.1), and the readout for the other profile is either inside the envelope or in the opposite direction. There are 21 differentiating activities within the following systems: 3C (TM), LPS (CD69, SPGE2), SAg (IL-8), BT (slL-2), BF4T (VCAM-1), BE3C (tPA), CASM3C (IL-6), HDF3CGF (Collagen I, Collagen III, EGFR, l-TAC, IL-8, MCP-1), KF3CT (IL-8), MyoF (Decorin, MMP-1 , VCAM-1), and IMphg (IL-1 a, IL-8, MCP-1).
Top Database Search Result for SNA-125
[0895] In a search for mathematically similar compound profiles from the BioMAP reference database, SNA-125 (30 μΜ) was most similar to IKK 16 (370 nM) (Pearson's correlation, r = 0.826). The Pearson's correlation coefficient between these two profiles is above our determined threshold (r > 0.7) indicating these compounds share mechanistically relevant similarity. IKK 16 (IKK Inhibitor VII) is selective l-kappa-b kinase (IKK) inhibitor for IKK-2, IKK complex, and IKK-1 . Figure 45 depicts an overlay of SNA-125 (30 μΜ) and IKK 16 (370 nM).
[0896] There are 24 common activities that are annotated within the following systems: 3C (IL-8, TF), LPS (E-selectin, IL-1 a, IL-8, MCP-1 , TF, TM, VCAM-1 , sPGE2, sTNFa), SAg (IL-8, Prolif), BT (Prolif, SIL-17A, slL-2, slL-6, slgG, sTNFa), CASM3C (IL- 6), and IMphg (E-selectin, IL-1 a, IL-8, MIP-1 a).
Top BioSeek Reference Database Matches for SNA-125
[0897] Table 28 depicts the top 3 similarity matches from a search of the BioMAP Reference Database of > 4,000 agents for each concentration of SNA- 125. The similarity between agents is determined using a combinatorial approach that accounts for the characteristics of BioMAP profiles by filtering (Tanimoto metric) and ranking (BioMAP Z-Standard) the Pearson's correlation coefficient between two profiles. Profiles are identified as having mechanistically relevant similarity if the Pearson's correlation coefficient is > 0.7.

[0898] The Pearson's correlation coefficient between profiles that is above our determined threshold (r > 0.7) indicates these compounds share mechanistically relevant similarity. For profiles with a Pearson's correlation coefficient below our determined threshold (r < 0.7), the relevance of the similarity is unknown.
Mechanism HeatMAP Analysis of SNA-125
[0899] Figure 46 depicts Mechanism HeatMAP Analysis of SNA-125, with the 148 biomarker readouts within the Diversity PLUS panel compared to 19 consensus mechanism class profiles. This analysis informs on the regulatory mechanisms controlling increases or decreases in each of the biomarker readouts.
SNA-125 BioMAP Profile Overlay with Methotrexate and Tofacitinib
[0900] Figure 47 depicts the BioMAP profile overlay of SNA-125 (10 μΜ) with Methotrexate 10 μΜ) and Tofacitinib (10 μΜ). Differences in the LPS system and /Mphg system were observed. A similar impact on T-cell proliferation can be seen in the Figure
47 overlay of the three compounds. It is noted that Tofacitinib activities in 3C consistent with systemic side effects, not present with SNA-125
Conclusions
[0901] In this study SNA-125 was characterized by profiling in the BioMAP Diversity PLUS panel of human primary cell based assays modeling complex tissue and disease biology of organs (vasculature, immune system, skin, lung) and general tissue biology. The Diversity PLUS panel evaluates the biological impact of test agents in conditions that preserve the complex crosstalk and feedback mechanisms that are relevant to in vivo outcomes.
[0902] SNA-125 was broadly active across the Diversity PLUS panel with 52 annotated readouts affecting biomarkers involved with inflammation, immunomodulation, tissue remodeling, and hemostasis. SNA-125 was antiproliferative to B cells, endothelial cells, fibroblasts, and T cells and had no effects on cytotoxicity at the concentration range tested. SNA-125 was previously profiled in the Diversity PLUS panel at a lower concentration range (140 nM - 3900 nM, refer to project #SNE001 -01 -b), where inhibition of TNFa and modulation of MMP-1 were observed. SNA-125 and its reference benchmark K252a, a non-selective protein kinase inhibitor, had 37 common activities and 21 differentiating activities; in general, SNE-SNA-125 was more active at the top tested concentration (30 μΜ) than the top tested concentration of K252a (30 nM). The top database search match for SNA-125 was IKK 16, an IKK2 inhibitor, and that match was above our threshold for significance (r > 0.7). There were 24 common activities between SNA-125 and IKK2, most of which were in the LPS system modeling Th1 type inflammation and monocyte activation and the BT system modeling T cell dependent B cell activation. Inhibitors of IKK2 and p38 MAPK were among the top matches for this compound, indicating that SNA125 may have phenotypic impacts on the NFKB and/or MAPK signaling pathways.
Example 18: Pharmacokinetic Study of a Single Dose of
Compound CT101 in CD-1 Mice
[0903] This study aims at selecting an appropriate vehicle formulation for topical application of CT101 onto the skin (Part A), validating a pharmacokinetic (PK) analysis method (Part B) and evaluating the PK of CT101 (Part C).
Methodology
Compounds and Reagents
[0904] CT101 was initially a solid compound that was diluted in vehicle (sodium chloride 0.9%, saline for intra-venous administrations and that determined in Part A for epicutaneous applications)
Experimental outline
Part A: Vehicle Assessment
[0905] Adult male CD-1 mice were randomly allocated to experimental groups (two mice per group). On Day 0 and Day 1 , a vehicle formulation was administered twice daily by epicutaneous application to the ears (20 μΙ_ per ear). On Day 2, the vehicle formulations were administered once. Topical application was performed in non-anaesthetised but restrained animals. Animals were monitored immediately after each vehicle application and one hour after the first daily application for signs of inflammation to include redness and swelling.
• Vehicle 1 was 25% Transcutol P and 75% Propylene Glycol
• Vehicle 2 was 10% Propylene Glycol, 40% DMSO and 50% distilled water
• Vehicle 3 was 25% Transcutol P, 10% Propylene Glycol and 65% distilled water
A Vehicle 1
t Kum s \.m u-l>ay i : 1 iee d& sDj
Vehicle 2
20 pL/ear Day 2: Qnee d i <SIO)
A3 Vhic 3
Part B: Method Optimization Samples
[0906] Terminal blood samples from adult male CD-1 mice were collected into K2EDTA-coated tubes. Blood samples were processed to isolate plasma. Samples were pooled and stored at -80°C until further analysis.
CT101
[0907] CT101 was weighed out and reconstituted in methanol to give a stock solution at 1 mg/mL. All chemicals used for chromatography were of HPLC grade (Fisher Scientific Ltd., Loughborough, UK).
Liquid Chromatography (LC) conditions
[0908] Samples were analysed on a Waters Alliance 2695 High Pressure LC separations module in combination with a Waters Diode Array Detector and Waters Micromass Quattro Ultima triple quadrupole mass spectrometer. The samples were analysed by reversed phase liquid chromatography employing a gradient separation. Mobile phase A (MPA) consisted of 90% dH20: 10% methanol containing 0.1 % formic acid. Mobile phase B (MPB) consisted of 90% methanol: 10% dH20 containing 0.1 % formic acid. The column was a Phenomenex Luna C18 (5 μηι, 250 x 2.00 mm).
[0909] The gradient conditions started at 70% MPB and rose linearly to 100% MPB at 6 minutes post injection. Conditions were held at 100% MPB for a further 4 minutes (10 minutes post injection), before returning to the initial conditions. The column was allowed to re-equilibrate before the next injection. Flow rate was 0.4 mL/min and total run time per injection was twenty minutes.
Mass Spectrometry (MS) conditions
[0910] The MS was used in electrospray positive mode, with a capillary voltage of 3.4 kV and a cone voltage of 20 V. Source and desolvation temperatures and gas flows settings were standard for the system. SIR channels were created to measure CT101 at mass-to-charge ratio (m/z) 584.2, 778.6 and 1 167.5.
Sample preparation and analysis
[0911] Non-biological standards were prepared by spiking CT101 into 10% methanol: 90% water. Biological standards were prepared by spiking CT101 into blank murine plasma. The calibration curves were prepared with ten non-blank standards each at a volume of 50 μί (individual calibrant concentrations were 0.1 , 0.2, 0.5, 0.8, 1 , 5, 10, 20, 50 and 100 μg/mL).
[0912] Plasma samples and standards were extracted by adding three volumes of ice-cold methanol to precipitate the plasma proteins. After methanol addition, the sample was briefly vortexed to mix, then centrifuged at 10,000 g for five minutes (4°C). The supernatant was transferred to a labelled LC vial for analysis. Ten microliters of supernatant were injected into the LC-MS system. Calibration standards were analysed first (low to high concentration).
[0913] Samples were kept on ice, in a refrigerator or in cooled equipment wherever possible. After analysis sample vials were transferred to -80°C storage.
Part C: PK analysis
[0914] Adult male CD-1 mice were randomly allocated to experimental groups and allowed to acclimatise for one week. On Day 0, CT101 was administered either by epicutaneous application to the ears (50 mg/kg dose in 20 μΙ_) or by intravenous administration (5 mg/kg dose). Topical application was performed in non- anaesthetised but restrained animals. The average surface of a mouse ear is 155 square millimetre. Test compound was allowed to spread passively onto the surface of the ears. Absorption of the test compound was not promoted by spreading. Animals were housed individually to prevent interferences with the application site. Blood samples were collected into K2EDTA-coated tubes according to the schedule below. Non- terminal blood samples were taken from a superficial vein and terminal blood samples were taken from a cardiac bleed. Blood samples were processed to isolate plasma. Samples were stored at -80°C for seven weeks until further analysis.
[0915] Plasma samples were analysed by LC-MS.
Treatment Groups and Dosages
• All Groups were n=4
• Vehicle for intravenous administrations was a 0.9% sodium chloride solution
(saline).
• Vehicle for epicutaneous applications was determined in Part A: 25%
Transcutol P and 75% Propylene Glycol
• Administration volume for topical application was 20 μΙ_ per ear.
• Administration volume for intravenous injection was 5 mL/kg.

Readouts
Vehicle assessment
[0916] Part A: Three vehicle formulations were assessed by epicutaneous application onto mice ears for up to three days. Animals were monitored for signs of inflammation.
PK analysis
[0917] Part B: Method validation was performed on plasma prepared from blood collected then pooled from strain-, gender- and age-matched mice. The following parameters were tested: selectivity, linearity of standard calibration curve, accuracy and precision, lower detection/quantitation limit's (LLOQ) accuracy and precision and extraction efficacy. The stability of the compound in plasma at -80°C was tested by determining the recovery following a freeze/thaw and a one week storage at -80°C.
[0918] Part C: Study samples were then processed by HPLC for quantitative analysis of CT101 in plasma.
Results
Vehicle assessment
[0919] On Day 0, vehicles were administered by epicutaneous applications of twenty microliters to each ear on un-anaesthetised animals. Animals were observed immediately after the epicutaneous applications and one hour after.
[0920] For all three vehicles, the volume used was sufficient to apply the solution to the entire surface of the ear. The administration volume could be reduced to ten to fifteen microliters per ear.
[0921] Vehicle 1 (25% Transcutol P and 75% Propylene Glycol) was absorbed and left an oily residue at the surface of the ear. The residue was still observed for up to one hour after the epicutaneous application.
[0922] Vehicle 2 (10% Propylene Glycol, 40% DMSO and 50% distilled water) was spread with difficulty and was not absorbed and remained at the surface of the ear. The solution was easily shaken off and/or groomed away by the animals when returned to their cages.
[0923] Vehicle 3 (25% Transcutol P, 10% Propylene Glycol and 65% distilled water) was absorbed and left an oily residue at the surface of the ear. The residue was still observed for up to one hour after the epicutaneous application.
[0924] All animals were seen grooming their ears shortly after epicutaneous application of the vehicles.
[0925] The three vehicles tested did not induce any sign of inflammation such as redness or swelling immediately after epicutaneous application or one hour after.
[0926] On Day 1 , identical observations were made.
[0927] On Day 2 (morning), epicutaneous applications were performed on anaesthetised animals. Anaesthesia was induced in an anaesthetic chamber using isoflurane. Animals were maintained under isoflurane anaesthesia using a nose cone for five minutes. Vehicle 1 and vehicle 3 left an oily residue at the surface of the ear. Vehicle 2 was not absorbed. Animals were not seen grooming when returned to their cages.
[0928] Also on Day 2 (afternoon), the epicutaneous application volumes were reduced from twenty to ten microliters. Ten microliters were sufficient to cover the entire surface of the ear. No sign of inflammation were observed.
[0929] For the remaining of the study (Part C), animals were not anaesthetised and the administration volume was not reduced to ten microliters.
Pharmacokinetic analysis Method validation
[0930] Plasma calibration standards were prepared from CD-1 mouse plasma blank plasma samples and the calibration standards were initially run with UV detection. However, this method was found to have poor separation, with a much lower limit of detection than the MS detection. As a consequence, the UV detection method was abandoned. Analysis of the UV data from the calibration curve showed that the LOQ by UV was 10 μς/ηιΙ..
[0931] Figure 48 (A) shows UV (280 nm) traces for, from bottom to top: blank calibration plasma, 10 μg/mL calibration plasma, 100 g/ml calibration plasma, sample 21 (1/2 hour epicutaneous administration, animal 3.4), and sample 58 (1/2 hour intravenous administration, animal 8.3). Figure 48 (B) shows the MS TIC traces for the same injections. The shaded areas in all traces indicate the CT101 peak. There was co-eluting interference seen in the plasma samples at 280 nm (indicated by the arrow), making baseline to baseline integration difficult. The width of the peak detected at 280 nm is approximately one minute wide (horizontal lines); a reflection on the polymeric nature of the PEGylated compound. In comparison, due to the selective nature of the MS method, there are no co-eluting interferences detected, and the peak monitored at the chosen masses was much narrower.
[0932] Plasma calibration standards were prepared from CD-1 mouse plasma blank plasma samples. The calibration range was 0.1 to 100 μg/mL.
[0933] A 50 μΙ_ volume of calibrant was extracted in 150 μΙ_ ice-cold methanol. A 10 μΙ_ aliquot of supernatant was injected into the LC-MS system.
[0934] The reproducibility of the injection, tested by injecting ten supernatant samples, was calculated at -2.7% CV.
[0935] The limit of detection (LOD) was 10 μg/mL with the UV method and 0.2 μg/mL with the MS method. The limit of quantitation (LOQ) was greater than 10 μg/mL with the UV method and 0.5 μg/mL with the MS method.
[0936] Due to the time constraints for this project, LOQ & LOD were assessed from the plasma calibration curve (range 0.1 to 100 μg/mL CT101). CT101 at 0.1 μg/mL was not detectable. CT101 at 0.2 μg/mL was detectable, but the peak was too small to accurately quantify, so this point was set as the LOD. At 0.5 μg/mL CT101 was detected, with the peak signal at least three times the background signal (a standard LC reference test), so this point was set as the LOQ.
[0937] Figure 49 depicts chromatograms of a CT101 plasma standard extract (100 μg/mL) detected using SIR (TIC, upper) and UV (lower). Figure 50 depicts method validation. Figure 51 depicts individual chromatograms used for the analysis of CT101 .
Quantitative analysis of CT101 in plasma
[0938] CT101 was applied topically once at 50 mg/kg or administered once by intravenous injection at 5 mg/kg.
[0939] Plasma samples were analysed by LC-MS method.
[0940] Plasma levels of CT101 in animals treated by topical application were below the detection limit of the assay (0.2 μg/mL) for all but one sample.
[0941] Plasma levels of CT101 in animals treated by intravenous administration were detectable for up to two hours after the injection and were then below the detection limit of the assay. Figure 52 depicts mouse plasma concentrations of CT101 .
[0942] Following intra-venous administration, the following parameters were determined using SigmaPlot V8.02:
• C0 = 53.4 μg/mL at 0 minutes post administration (extrapolated value)
• Cmax = 22.6 μg/mL at 10 minutes post administration,
• AUCiast = 14.6 μg/mL/hour and
• t1/2a = 0.093 hours and t1/2p = 0.6 hours (bi-exponential equation)
• VD = 2.0 ml_
• Clearance = 0.143 mL/minute
[0943] Following epicutaneous administration, one animal had detectable plasmatic levels of CT101 and PK parameters for this group could not be determined due to the high inter-individual variability. Figure 53 depicts representative chromatograms showing CT101 in extracted murine plasma following intra-venous administration. Figure 54 depicts representative chromatograms showing CT101 in extracted murine plasma following epicutaneous administration.
Conclusions
Part A
[0944] The vehicles, when administered by epicutaneous application to the ears twice daily for three days, did not induce any sign of inflammation.
[0945] Vehicle 2 (10% propylene glycol, 40% DMSO and 50% distilled water) is not appropriate for epicutaneous applications.
[0946] Vehicle 1 (25% Transcutol P and 75% Propylene Glycol) was used for Part B and Part C of this study.
[0947] An administration volume of ten microliters would be sufficient to cover the entire surface of the ear. This was not implemented for Part B and Part C.
[0948] Performing the epicutaneous applications under isoflurane anaesthesia is recommended to prevent the animals from grooming the solutions away and/or ingesting the solutions. This was not implemented for Part B and Part C.
Part B
[0949] A suitable method for the analysis of CT101 in murine plasma by LC- MS was developed. The method is reproducible and has a good limit of detection (0.2 μg/mL) when compared to the UV method. This method was used to analyze samples from mice administered with CT101 by epicutaneous application and intra-venous injection.
Part e
[0950] CT101 concentrations were measured in mouse plasma samples after epicutaneous or intravenous administration. The administration dose for epicutaneous administration was ten times higher than when using the intravenous administration route. Plasma levels after epicutaneous application of CT101 were below the detection limit of the assay except for one animal (animal 4) on two time points (15 and 30 minutes). Plasma levels after intra- venous administration were measurable up to two hours after the injection and the following PK parameters were calculated: Cmax = 22.6 μg/mL at 10 minutes post administration, AUC|ast = 14.6 μg/mL/hour and Terminal t1/2 = 0.6 hours.
Example 19: Pharmacokinetic study of a single dose of CT103 in BALB/c mice
[0951] This study is aimed at validating a pharmacokinetic (PK) analysis method (Part A) and evaluating the PK of CT103 (Part B).
Methodology
Compounds and Reagents
• Distilled water, 15230, Life Technologies
• Propylene Glycol, P4347, Sigma
• Transcutol P (Gattefosse, lot 450931007)
• CT103 initially was a solid compound diluted in vehicle (sodium chloride 0.9%, saline for intra-venous administrations and 25% Transcutol/75% Propylene Glycol for epicutaneous administrations).
Experimental outline
Part A: Method optimization Samples
[0952] Terminal blood samples from adult male BALB/c mice were collected into K2EDTA-coated tubes. Blood samples were processed to isolate plasma. Samples were pooled and stored at -80°C until further analysis.
CT103
[0953] CT103 was weighed out and reconstituted in methanol to give a stock solution at 10 mg/mL. All chemicals used for chromatography were of HPLC grade (Fisher Scientific Ltd., Loughborough, UK).
Liquid Chromatography (LC) conditions
[0954] Samples were analyzed on a Waters Alliance 2695 High Pressure LC separations module in combination with a Waters Diode Array Detector and Waters Micromass ZMD mass spectrometer. The samples were analyzed by reversed phase liquid chromatography employing a gradient separation. Mobile phase A (MPA) consisted of 90% dH20: 10% methanol containing 0.1 % formic acid. Mobile phase B (MPB) consisted of 90% methanol: 10% dH20 containing 0.1 % formic acid. The column was a Acquity BEH C18 (1.7 μηι, 100 x 2.10 mm). The gradient conditions started at 50% MPB and rose linearly to 100% MPB at 10 minutes post injection, returning to the initial conditions at eleven minutes. The column was allowed to re-equilibrate before the next injection. Flow rate was 0.3 mL/min and total run time per injection was twenty minutes.
Mass Spectrometry (MS) conditions
[0955] The MS was used in electrospray positive mode, with a capillary voltage of 3.0 kV and a cone voltage of 20 V. Source and desolvation temperatures and
gas flows settings were standard tor the system. SIR channels were created to measure CT103 at mass-to-charge ratio (m/z) 794,595.8 and 476.8. This relates to MW = 2379.0.
Stability
[0956] Replicate plasma samples were spiked with 10 μg/mL of compound CT103. Three replicates were extracted and analyzed fresh, three were frozen at -80°C for 60 minutes before being defrosted, extracted and analyzed. Peak areas were compared to assess stability of compound CT103.
Part B: PK Analysis
[0957] Adult male BALB/c mice were randomly allocated to experimental groups and allowed to acclimatise tor one week. On Day 0, CT103 was administered either by epicutaneous application onto to the clipped skin of the back (50 mg/kg dose in 100 μΙ_) or by intra-venous administration (10 mg/kg dose).
[0958] Epicutaneous applications were performed in non-anaesthetised but restrained animals. Animals were housed individually to prevent interferences with the application site. Blood samples were collected into K2EDTA-coated tubes according to the schedule below. Non-terminal blood samples were taken from a superficial vein and terminal blood samples were taken from a cardiac bleed. Blood samples were processed to isolate plasma. Samples were stored at -80°C until further analysis.
Administration and Bleed schedule
[0959] All Groups are n=4. Vehicle for intravenous administrations was a 0.9% sodium chloride solution (saline). Vehicle for epicutaneous administrations was 25% Transcutol P/75% Propylene Glycol. Administration volume for epicutaneous application was 100 μΙ_ per mouse per day. Administration volume for intravenous injection was 5 mL/kg.

Readouts
Method validation
[0960] Method validation was performed on plasma prepared from blood collected then pooled from strain-, gender- and age-matched mice. The following parameters were tested: selectivity, linearity of standard calibration curve, accuracy and precision, lower detection/quantitation limit's (LLOQ) accuracy and precision and extraction efficacy.
PK analysis
[0961] Study samples were processed by HPLC for quantitative analysis of CT103 in plasma.
Results
Method validation
[0962] Figure 55 depicts chromatograms of a CT103 plasma standard extract (50 μg/mL) detected using SIR (TIC, upper) and UV at 337 nm (lower).
[0963] Plasma calibration standards were prepared from BALB/c mouse blank plasma samples. The calibration range was 0.25 to 50 μg/mL. The calibration
curve was assessed over the range 0.1-100 μg/mL. The calibration standard at 0.1 μg/mL was detectable but too small to be accurately quantified and therefore was not included in the curve. The calibration standard at 100 μg/mL was slightly outside the linear range due to saturation of the system and was therefore not included in the curve. Curve parameters were calculated with no weighting and a zero intercept. The r2 value was 0.9965, indicating good method reproducibility (Figure 56).
[0964] A 100 μΙ_ volume of compound spiked plasma was extracted in 300 μΙ_ ice- cold methanol, evaporated to dryness and reconstituted in 100 μΙ_ methanol. A 10 μΙ_ aliquot was injected into the LC-MS system.
[0965] The reproducibility of the injection, tested by repeat injection (n=6) of a sample at 10 μg/mL, was calculated at 6.88% CV. The extraction efficiency was determined to be 63.2% from six CT103 standards and six plasma extracts, and reproducibility of repeat sample extracts was 6.62% CV at a concentration of 10 μg/mL.
[0966] Figure 57 depicts method validation and Figure 58 depicts individual chromatograms of SIR channels used for the analysis of compound. The limit of detection (LOD) was assessed from compound standard injections. LOD was determined to be 0.1 μg/mL using MS detection. Compound at 0.1 μg/mL was detectable, however the peak was too small to accurately quantify and therefore this point was set as the LOD. The limit of quantitation (LOQ) was determined to be 0.25 μg/mL using MS detection. LOQ was assessed from three repeat plasma extract samples. At 0.25 μg/mL compound was detected, with the peak signal at least three times the background signal (a standard LC reference test), therefore this point was set as the LOQ. Variability at LOQ was 9.6% CV for three separate repeat plasma extracts.
[0967] Freeze-thaw stability of CT103 in murine plasma was assessed over one freeze-thaw cycle. There was no decrease in CT103 peak area observed in the triplicate samples that underwent a freeze-thaw cycle compared to the peak areas observed in the triplicate samples extracted immediately after preparation. CT103 is assumed stable for at least one freeze-thaw cycle.
Pharmacokinetic analysis
[0968] Data for all samples were assessed using the summed total ion chromatogram (TIC) channel generated from the three individual channels (m/z 794, 595.8 and 476.8) by the LC-MS software. UV detection was not sensitive enough to analyse CT103 concentrations below 10 μg/mL and was therefore not used in sample analysis. Figure 59 depicts representative chromatograms showing CT103 in methanol and extracted from a spiked murine plasma calibration standard.
[0969] Figure 60 depicts mouse plasma concentrations of CT103. Data were limited for the samples taken following epicutaneous administration of CT103 at 50 mg/kg, with most concentration levels found below the limit of detection (0.1 μg/mL). A Cmax of 1 .47 μg/mL was seen at 15 minutes post administration (one individual had a concentration of 2.31 μg/mL at 30 minutes post administration), and compound was detectable up to 2 hours after administration. AUC|ast for epicutaneous administration was 2.46 μg/mL, and terminal t1/2 was 0.41 hours.
[0970] CT103 was detectable for up to 8 hours following intra-venous administration at 10 mg/kg, however the 8 hour values were at the LOD. One individual at 3 hours post administration had detectable levels of compound however this value appears unusually high and was therefore excluded from calculations. A Cmax of 8.12 μg/mL was seen at 10 minutes post administration. AUC|ast was 3.82 μg/mL terminal t1/2 was 0.33 hours.
Conclusions
Part A
[0971] A suitable compound separation method was established and the protein precipitation method of extraction gave an efficiency of 63.2% (10 μg/mL).
[0972] The reproducibility of the system was good for repeat injections (6.88%, n=6) and repeat sample extraction (6.62% for a concentration of 10μg/mL n=6). The LOD was determined to be 0.1 μg/mL and the LOQ 0.25 μg/mL with a reproducibility of 9.63% (n=3).
Part B
[0973] CT103 is stable in murine plasma following a single freeze-thaw cycle. CT103 concentrations were measured in mouse plasma samples after epicutaneous or intravenous administration. The Cmax observed in the IV administration samples was much higher (over 5 fold) than the epicutaneous administration samples (8.12 μg/mL and 1 .47 μg/mL respectively). AUC|ast values were much closer at 3.82 (IV) and 2.46 (epicutaneous), due to the longer t1/2 seen following epicutaneous administration.
Example 20: CT352 bioavailability study in mice by intravenous and oral routes
Summary
Study Design
[0974] The purpose of this preliminary study was to investigate the bioavailability of CT352 in mice after a single oral administration, when compared to the intravenous route.
[0975] Two treatment groups, each composed of 27 males, were dosed by the oral or intravenous route with different concentrations of test item.
[0976] Clinical signs and body weight were the investigations performed during the in life phase of the study.
[0977] The pharmacokinetic profile was investigated in 3 animals/time point. Blood samples were collected at 9 time points after treatment as follows:
Γ0978Ί Group 1: Pre-dose, 10, 20 and 30 minutes, 1 , 2, 4, 8 and 24 hours after dosing.
Γ0979Ί Group 2: Pre-dose, 15, 30 minutes, 1 , 2, 4, 6, 8 and 24 hours after dosing.
[0980] The treatment schedule and sample collection are summarized below:
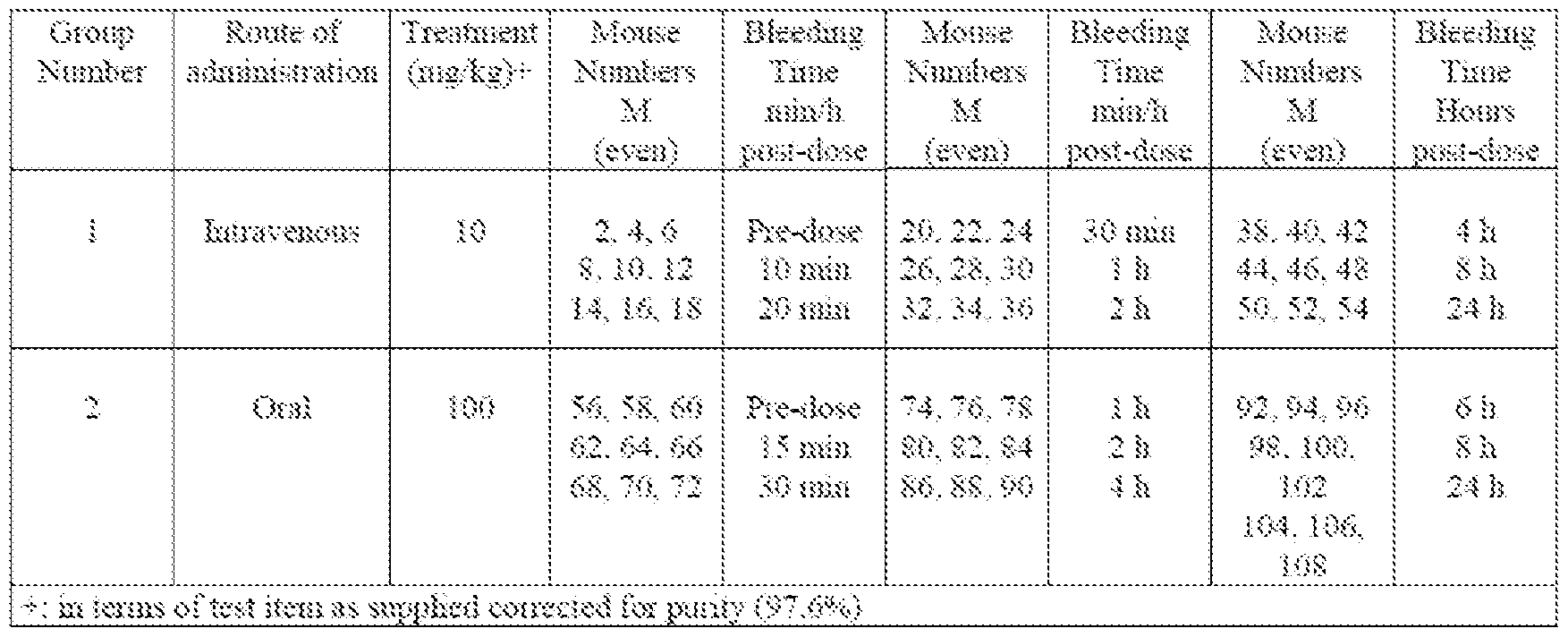
Mortality and clinical signs
[0981] No mortality occurred during the study and no signs of reaction to treatment were recorded.
Body weight
[0982] Body weights were within the expected range for this strain and age of animals.
Pharmacokinetic profile
[0983] The pharmacokinetic parameter values calculated from the mean plasma concentrations obtained after a single intravenous and oral administration of CT352 to male CD1 mice are shown in the Table 29 below.
Table 29: Pharmacokinetic Parameter Values

*; alue tamed by com &rfmesstfsf aa&iysis.
[0984] Animals receiving a single oral administration of the test item showed Cmax and AUC values lower than those detected after single intravenous administration.
[0985] On the basis of these results, it can be concluded that CT352, administered to mice after a single oral treatment, has a low bioavailability when compared to the intravenous route, with a relative value of 0.16%.
Introduction
[0986] The purpose of this preliminary study was to investigate the bioavailability of CT352 in mice after a single oral administration, when compared to the intravenous route.
Experimental Procedures
Choice of the species
[0987] The Hsd: ICR (CD-1) mouse was the species and strain of choice because it is accepted by many regulatory authorities and there is ample experience and background data on this species and strain
Choice of the route
[0988] The oral route was selected as it is one of the intended routes of administration of the test item in man. The intravenous route was selected as it presents the least barrier to absorption.
Test item
Identity
[0989] The test item (CT352) had a purity of 97.6% (area/area) and the storage conditions were 18°C, protected from light.
• Formulation procedure
• On the day of dosing, the test item was dissolved as follows:
• Vehicle: Water for injection
• Concentrations: 2 and 10 mg/mL
• Concentrations were calculated and expressed in terms of test item
corrected for purity.
Methods
Animal management
Animal supply and acclimatization

Caging
Γ09901 No. of animals/cage: up to 5, during acclimatisation, up to 3, during the study.
Γ099Π Housing: Polysulphone solid bottomed cages measuring 35.5x23.5x19 cm, with nesting material.
Γ09921 Cage tray control: Daily inspected and changed as necessary (at least 2 times/week).
Water and diet
Γ0993Ί Water: drinking water supplied to each cage via a water bottle.
Γ0994Ί Water supply: ad libitum
Γ0995Ί Diet: 4 RF21 (Mucedola S.r.l., Via G. Galilei, 4, 20019, Settimo Milanese (Ml) Italy)
Γ0996Ί Diet supply: ad libitum throughout the study except for oral dosing procedure indicated in the Dosing section below.
[0997] Components present in the drinking water or diet are not at a level likely to interfere with the purpose or conduct of the study.
Housing conditions (parameter set)

[0998] Actual conditions were monitored and recorded. No relevant deviations occurred.
Experimental Design
Allocation to treatment groups
Group composition
[0999] The number of animals (27 males/group) was allocated to the study as follows:

Selection/Allocation
[1000] Selection/allocation was performed by computerised stratified randomisation to give approximately equal initial group mean body weights. The mouse numbers listed above formed the last digits of a computer generated 8 figure animal number (the remaining digits of the animal number were different for each concurrent study and served to ensure unique animal numbering for any study employing computerised data collection). The computerised system used in this study was the Xybion Path/Tox System, Version 4.2.2.
Animal Identification
[1001] Animal identification was carried out by a coloured mark on the tail, after arrival, and by ear notch and tattoo on the hind feet.
Dosing
[1002] Dosing was performed as follows:
Frequency of treatment Once, on the day of dosing (Day 1).
Dose calculation Group 1 : dose volume of 5 mL/kq of bodv weiqht for each animal.
Group 2: dose volume 10 mL/kq of bodv weiqht for each animal.
Dosing methods Group 1 : bv intravenous injection into the tail vein, at an approximate rate of 3 mL/minute, using a hypodermic needle attached to a syringe of suitable capacity.
Group 2: bv qavaqe, usinq a feedinq plastic tube attached to a graded syringe.
In Life Observations
[1003] In life observations were performed as follows:
Mortality and morbidity Twice daily.
Clinical signs Day of dosing (prior to dosing and immediately after dosing).
Body weight Allocation (Day -1) and on the day of dosing (Day 1).
Pharmacokinetic profile
Sample collection and analysis
• Blood samples were collected at 9 time points, as specified below.
• Group 1
• Pre-dose, 10, 20 and 30 minutes, 1 , 2, 4, 8 and 24 hours after dosing.
• Group 2
• Pre-dose, 15, 30 minutes, 1 , 2, 4, 6, 8 and 24 hours after dosing.
[1004] At each sampling time, approximately 0.5 mL blood samples were withdrawn under isofluorane anaesthesia from the abdominal vena cava of 3 animals of each group, each animal being sampled at one time point.
[1005] Samples were transferred into tubes containing K3EDTA anticoagulant, centrifuged at room temperature and the plasma frozen at -20°C pending analysis.
[1006] The plasma concentrations of CT352 were analysed based on a LC- Fluo method.
Assessment of pharmacokinetic data
[1007] The pharmacokinetic analysis was conducted according to a standard non-compartmental and compartmental analysis. The following pharmacokinetic parameters were obtained or calculated from the mean plasma values:
maxim m (peak) observd concentration:
iugrnLj dme » teach peak or amkawm ciWicen&afioa m elmjhatios half-life: associate wlm fmaiiiai slope (½) of a sciffiio rktok owear r rMime curve; area under the c acs raiiss-!me curve calc lated by the lisear ra ezoids! rule, ftooi∞0 (pre-dose) to ifcsi (! si qiiaairx&Me eoace&ratsos);
AUC area W kt the plasma co eesltsii Hmie cur e from
iime z¾ro to hifeiuy. isgtnL
[1008] All the pharmacokinetic parameters were estimated or calculated by the Kinetica™, version 4.4.1, PK/PD Analysis (Thermo Electron Corporation Informatics, Philadelphia - USA) software.
[1009] Means and/or medians, standard deviations and coefficient of variations were obtained using a Microsoft Excel worksheet.
[1010] The results of the pharmacokinetic evaluation are presented in the Pharmacokinetic profile section below.
Terminal Studies
Termination: Day 2.
[1011] Euthanasia method: By exsanguination under isofluorane anaesthesia.
[1012] Necropsy procedure: No necropsy was performed. Results
Mortality and clinical signs
[1013] No death occurred and no reaction to treatment was observed in the animals at the observations performed on the day of dosing (Table 30).
Table 30: Clincal signs - Group incidence
Body weight
[1014] Body weights were within the expected range for this strain and age of animals (Table 31).
Table 31 : Body weight (g) - Group mean data

Pharmacokinetic profile
[1015] In Table 29 above, the kinetic parameter values for CT352, obtained after single intravenous administration at dose level of 10 mg/kg (Group 1) and after a single oral administration at 100 mg/kg (Group 2) are shown.
[1016] Comparing the two groups treated with the test item, the maximum plasma concentration (Cmax) of CT352 was achieved at 2 hours after dosing in the animals of Group 2, receiving a single oral administration of CT352 at dose level of 100 mg/kg.
[1017] The analysis revealed low values of Cmax and AUC following oral administration of CT352, when compared to the intravenous route. In fact, the relative bioavailability [(AUCpO/Dosepo)/(AUCiv/Doseiv).100%] for the oral route, calculated using the intravenous route as reference, was 0.16%. Therefore, it can be concluded that CT352 administered to mice after a single oral treatment showed a low bioavailability.
Conclusion
[1018] The bioavailability of CT352 in mice after a single oral administration, when compared to the intravenous route, was investigated in this study.
[1019] Two treatment groups, each composed of 27 males, were included. The first group received a single intravenous administration of CT352 at a dose level of 10 mg/kg, while the second group was treated with a single oral administration of the test item at 100 mg/kg.
[1020] No mortality occurred and no signs of reaction to treatment were observed on the day of dosing. Body weights were within the expected range for this strain and age of animals.
[1021] The kinetic evaluation showed that CT352, administered to mice after a single oral treatment, has a low bioavailability when compared to the intravenous route, with a relative value of 0.16%.
Example 21 : Single Dose Toxicity and Pharmacokinetic Study in Rats
by Intravenous and Dermal Routes
[1022] The toxicity and pharmacokinetic of CT340 were investigated when given by single intravenous and dermal administration to the Sprague Dawley rat.
[1023] The toxicity features were investigated in 8 main groups (Groups 1 -4 for intravenous dosing and Groups 5-8 for dermal dosing), each group comprising 2 male and 2 female rats. Animals were dosed at 10, 30 and 100 mg/kg for both administration routes (a dose volume of 5 ml/kg in physiological saline, for intravenous administration and Propylene glycol/DMSO/Water 10/40/50 for dermal administration). Animals of control groups (Groups 1 and 5) received the vehicle alone.
Intravenous dosing
[1024] The test item was administered by intravenous bolus injection into the tail vein with an approximate speed of 3 ml/minute. The dose was administered to each animal at a dose volume of 5 ml/kg. Control animals received the vehicle alone at the same dose volume.
Dermal closing
[1025] The day before scheduled dosing the fur was removed from the dorsal surfaces of the trunk over an estimated area of at least 10% of the total body surface. Care was taken to avoid any damage or abrasion to the skin. The clipping procedure was repeated as necessary during the course of the study.
[1026] The test item was applied uniformly over an area of approximately 10% of the total body surface area (approximately 5x7 cm). The dose was administered to each animal at a dose volume of 5 ml/kg. A patch of surgical gauze covered by a strip of synthetic film was placed over the treated site and the whole assembly held in place by encircling the trunk of the animal with a length of adhesive bandage, this forming a semi-occlusive barrier. The amount administered to individual animals was determined by their body weight, measured prior to dosing.
[1027] After a period of 6 hours, the tape dressing was removed. The treated skin site was then gently washed free of any remaining test item using warm water. Duration of treatment
[1028] All animals were dosed once. Main group animals were sacrificed on Day 8, after an observation period of 7 days. Satellite group animals were killed after the completion of bleeding procedures.
Pharmacokinetic results
[1029] After single intravenous administration, animals were largely exposed to the drug. The AUCs [AUC (o-tiast)] were calculated as follows:
-16715.4, 46918.2 and 149764 ng*h/ml in males for 10, 30 and 100 mg/kg respectively
-13027.7, 38084.9 and 142028 ng*h/ml in females for 10, 30 and 100 mg/kg respectively AUCs increased proportionally with the dose and no major difference was observed between sexes.
[1030] After single dermal administration, the amount of absorption was limited. The small amount found may be due to the low extent of absorption or high rate of elimination.
[1031] Based on the above reported results, the maximum tolerated dose is then considered to be greater than 100 mg/kg for both dermal and intravenous administrations.
Results
[1032] No clinical signs were observed after dosing and during the following observation period.
[1033] No relevant body weight changes were noted during the study. At the end of the observation period, the body weight was within the ranges expected for this strain and age.
[1034] Food consumption was unaffected by treatment.
[1035] Terminal body weight of treated animals was unaffected by treatment. No toxicologically relevant variations of organ weights were recorded.
Toxicokinetics
Intravenous closing
[1036] The drug concentrations in plasma following single intravenous administration were determined up to 2 hours after dosing. Most of the values were obtained after dilution of plasma samples.
[1037] The kinetic parameter values for the test item obtained after single intravenous administration (using mean plasma concentrations) are shown in Table 32, while graphs are presented in Figure 61.
TABLE 32 - Day 1 - Kinetic parameter values for CT340 (calculated on the mean;
n=3)
Doss Gende & AUG iO-Saif) AU <¾·:, · cio-.
u ; 4.2 Group 0s)
16715.4 1672 0.36
1GM 469 I S..2 1564 0.43
1 149764 1498
10 9F 13027.7 Ϊ303 0.34
30 I OF 3S0S4.9 1269
100 riF 142028 1420 0.33
[1038] The drug concentration-time profiles following single intravenous administration declined probably with a biphasic kinetic. Moreover, at the last time point observed, the plasma mean concentrations were higher than the lower limit of quantification, at all dose levels:
-10 mg/kg: approximately 138 and 88 fold in males and females respectively -30 mg/kg: approximately 434 and 282 fold in males and females respectively -100 mg/kg: approximately 1354 and 842 fold in males and females respectively
[1039] With reference to the observed AUC [AUC(0-tiast)] , they increased proportionally with the dose and no major difference was observed between sexes.
Dermal dosing
[1040] CT340 plasma levels were investigated up to 24 hours after dosing.
Concentrations were over the limit of quantification in the following samples:
-10 mg/kg: 4/21 samples in males and 10/21 samples in females
-30 mg/kg: 8/21 samples in males and 1 1/21 samples in females
-100 mg/kg: 19/21 samples in males and 16/21 samples in females
[1041] The erratic absorption, as evidenced from the high variability, is likely to be due to the administration route.
[1042] The amount of drug absorbed was limited. In fact, values observed were near the lower limit of quantification, as follows:
-10 mg/kg: approximately 2 to 4 fold in males and 1 to 91 fold in females
-30 mg/kg: approximately 1 to 1 1 fold in males and 1 to 48 fold in females
-100 mg/kg: approximately 1 to 1 14 fold in males and 1 to 62 fold in females
[1043] Due to the low amount of drug present in plasma, the kinetic data of the drug was not evaluated for the dermal route of administration.
Conclusions
[1044] The toxicity of CT340 when given by single intravenous or dermal administration to rats at the dose levels of 10, 30 and 100 mg/kg, was investigated over a period of 7 days after dosing.
[1045] No toxicologically relevant changes were observed during the in life phase of the study in the treatment groups when compared to the respective control groups. This applied both to intravenous and dermal routes of administration.
[1046] Animals were largely exposed to the drug after intravenous dosing. The AUC(o-tiast) increased proportionally with the dose and no major difference was observed between sexes.
[1047] After single dermal administration, the amount of drug absorption was limited.
[1048] The maximum tolerated dose is then considered to be greater than 100 mg/kg for both dermal and intravenous administrations.
Example 22 CT340 2 WEEK INTRA VENOUS TOXICITY STUDY IN RATS
FOLLOWED BY A 2 WEEK RECOVERY PERIOD
Summary
[1049] The toxicity of CT340 was investigated, when given by daily intravenous administration to the Sprague Dawley rat for a 2-week period followed by a 2-week recovery period.
[1050] The toxicity was investigated in 4 groups (Groups 1-4), each group comprising 10 male and 10 female rats. Five additional animals/sex were included in control and high dose groups, for evaluation of recovery.
[1051] The test item was dissolved in physiological saline (NaCI 0.9%) and treatment groups were dosed at 10, 30 and 100 mg/kg/day at the dose volume of 5 ml/kg and a speed of 3 ml/minute. The control group received the vehicle alone.
[1052] Four satellite groups were additionally dosed at the same dose levels, for evaluation of toxicokinetics.
[1053] The test item was administered by intravenous bolus injection into the tail vein with an approximate speed of 3 ml/minute. The dose was administered to each animal at a dose volume of 5 ml/kg. Control animals received the vehicle alone at the same dose volume. The dose was administered to each animal on the basis of the most recently recorded body weight and the volume administered was recorded for each animal.
[1054] All animals were dosed once a day, 7 days a week, for a minimum of 2 consecutive weeks. Animals were dosed up until the day before necropsy, except for recovery animals which were not given any treatment during the recovery period.
[1055] On Day 1 and in Week 2 of the study, blood samples were withdrawn from three animals/time point at the following time points: pre-dose, 5, 15, 45, 90 minutes and 24 hours after dosing.
[1056] At the dose level of 10 mg/kg/day the severity of the changes observed was not sufficient to represent an adverse effect.
[1057] Complete recovery occurred in animal treated at 100 mg/kg/day, after a 14-day recovery period.
[1058] Based on the above results, it can be concluded that the No Observed Adverse Effect Level (NOAEL) can be defined to be 10 mg/kg body weight/day for both sexes.
[1059] Finally, no cytogenetic effects were noted in the animals of the high group when compared to controls.
Results
[1060] No mortality occurred during the study in the main groups of the study. Two death events in the satellite groups occurred after bleeding procedure on Day 2 (24 h) and were not dose-related. Since no mortality occurred in the main groups and clinical signs were limited during the whole treatment period, the deaths may be correlated to the bleeding procedure. Thus, the observed mortality was considered incidental.
[1061] No statistically significant reductions of group mean body weight were recorded in the animals of treatment groups when compared to controls (Figures 62 and 63).
[1062] Before the start of treatment, animals showing no ocular abnormality at the ophthalmoscopy were selected for the study. Both eyes of all live animals from the main groups were re-examined during the last week of the study and no significant findings were detected.
[1063] For the dosing phase animals, no differences were recorded in terminal body weight of animals recorded at the end of treatment with the test item when compared to controls. For the recovery phase animals, no differences were recorded in terminal body weight of animals treated with the test item when compared to controls.
Kinetic behavior of CT340
Kinetic parameter calculation - Day 1
[1064] The kinetic parameter values for the test item obtained after single intravenous administration (using mean plasma concentrations) are shown in Table 33, while graphs are presented in Figure 64.
TABLE 33 - Kinetic parameter values for CT340 (calculated on the mean; n=3)

[1065] The drug concentration-time profiles following single intravenous administration appeared to decline with a biphasic kinetic (Figure 64).
[1066] At the last time point observed, the plasma mean concentrations were slightly higher than the lower limit of quantification at all dose levels (approximately 5 to 28 fold in males and 8 to 26 fold in females). Moreover, the extrapolation was automatically fitted from 1.5 h (which means only with two points). For these reasons, the half-lives, slope (λζ) and volume of distribution (VD) reported in Table 33 could be considered only as an estimation and MRT (mean residence time) is reported as estimation of effective permanence in the body.
[1067] The values of clearance, half-life and volume of distribution suggest that the drug is widely distributed and promptly eliminated from the body. The AUC(0-inf), slightly decreased with dose and the clearance (CL) slightly increased with dose indicating that in the range of the doses studied the kinetic of the drug may be not linear. No major difference was observed between sexes.
Kinetic parameter calculation - Week 2
[1068] The kinetic parameter values for the test item obtained after repeated intravenous administration (using mean plasma concentrations) are shown in Table 34, while graphs are presented in Figure 65.
TABLE 34 - Week 2 - Kinetic parameter values for CT340 (calculated on the mean;
n=3)

[1069] AUC calculation with trapezoidal rule was considered to be a good estimation.
[1070] The ratio between the AUC(0-inf) of Day 1 and AUC(0-tiast) of Week 2 was calculated as the 24 hour (t|ast) the interval (τ) between administrations. The ratio gave a qualitative indication that drug accumulation might occur at the high dose in both sexes. Being the AUCs of each dose/sex calculated with a single mean value for each bleeding time, statistical tests were not applicable to confirm this issue.
Discussion and Conclusions
[1071] The toxicity of CT340 was investigated, when given at the dose levels of 10, 30 and 100 mg/kg/day by daily intravenous administration to the Sprague Dawley rat for a 2-week period followed by a 2-week recovery period.
GENERAL TOXICITY
[1072] No statistically significant reductions of group mean body weight were recorded in the animals of treatment groups of the dosing phase when compared to controls.
[1073] During the recovery period, no mortality or toxicologically relevant clinical signs were recorded and the body weight was within the expected range for this strain and age of animals. Variations observed in the food consumption during recovery period were not considered toxicologically relevant.
[1074] Any changes observed in haematology, clinical chemistry and urinalysis parameter at the end of treatment were no longer observed at the end of recovery period.
[1075] No treatment-related changes were noted at post mortem macroscopic and microscopic examination.
[1076] The above finding suggested a complete recovery of the treatment- related changes seen at the end of the main phase of the study.
GENETIC TOXICITY
[1077] Following treatment with the test item, no increase in the incidence of micronucleated PCEs over the vehicle control group was observed in CT340 treated group.
[1078] Analysis of the ratio of mature to immature erythrocytes and the proportion of immature erythrocytes among total erythrocytes showed that the test item did not induce inhibitory effects on erythropoietic cell division.
[1079] No statistically significant heterogeneity in response in any group. No statistically significant increase in the incidence of micronucleated PCEs was observed over the vehicle control group.
KINETIC BEHAVIOR
[1080] The values of clearance, half-life and volume of distribution suggest that the drug is widely distributed and promptly eliminated from the body. The AUC(0-inf), slightly decreased with dose and the Clearance slightly increased with dose, indicating that in the range of the doses studied, the kinetics of the drug may be not linear. No major difference was observed between sexes.
CONCLUSIONS
[1081] At the dose level of 10 mg/kg/day, treatment-related and dose-related changes were not sufficient to represent an adverse effect.
[1082] Complete recovery occurred in animal treated at 100 mg/kg/day, after a 14-day recovery period.
[1083] Based on the above results, it can be concluded that the No Observed Adverse Effect Level (NOAEL) can be defined to be 10 mg/kg body weight/day for both sexes.
[1084] Finally, no cytogenetic effects were noted in the animals of the high group when compared to controls.
Example 23: CT340 Single Dose Dermal Tolerance/Toxicity and Pharmacokinetic
Study in Minipigs
Summary
[1085] The pharmacokinetics, local dermal tolerance and toxicity of CT340 after a single dermal administration (6 hour exposure period) were investigated in the minipig during an observation period of 7 days in order to select dose levels for subsequent studies. Although dermatology-based tests are disclosed herein, such data
should be applicable in the eye with respect to, for example, reduced exposure systemically and/or in non-target tissues.
[1086] Three groups, each of 2 male and 2 female minipigs (Groups 2, 3 and 4) were treated by dermal application of 0.5 mL/kg of the test item at the dose levels of 10, 30 and 100 mg/kg over an area of the dorsal skin of approximately 25 x 20 cm, previously clipped.
[1087] The test item was applied evenly over the prepared skin of animals of the relevant group (Groups 2, 3 and 4) at a dose volume of 0.5 mL/kg. The amount of formulation (in one embodiment and for this particular study) was spread evenly over the skin of the prepared site by gentle massage over an area of approximately 25x20 cm. Control animals (Group 1) received the vehicle alone at the same dose volume, in the same manner. The treated skin site was covered using a patch of surgical gauze and the whole assembly held in position by encircling the trunk of the animal with a cotton jacket. This semi-occlusive dressing was removed approximately 6 hours later and the treated site was cleaned by washing with a piece of surgical gauze soaked with warm water, removing any residual test item.
[1088] All animals were treated once, followed by an observation period of 7 days.
[1089] Plasma levels of CT340 were slightly >LLOQ (lower limit of quantitation = 4.5 ng/mL) in 1 male and 1 female receiving 30 mg/kg and in 2 males receiving 100 mg/kg. These values were reported between 1 and 24 hour post-dose. No measurable plasma levels were found in male animals dosed at 10 mg/kg and in female animals dosed at 10 and 100 mg/kg. Due to the minimal absorption observed, in the majority of cases the calculation of the exposure (AUC) and of other kinetic parameters was not possible.
[1090] A Cmax of 8.74 ng/mL was measured in the single male of Group 3, while a Cmax of 9.85 ng/mL was measured in the single female of the same group with a Tmax of 2 and 1 hour in the male and female animals, respectively.
[1091] Mean Cmax was 7.735 ng/mL in the males of Group 4, with a Tmax of 2 hours. AUCo-inf , calculable for 1 male dosed at 100 mg/kg, was 538.918 ng h/mL.
[1092] The results indicate that the test item CT340 is locally well tolerated after a dermal application at concentrations of 20, 60 and 200 mg/mL over the intact skin of the minipig.
[1093] No signs of potential treatment-related adverse effects of the test item were observed at any of the dose levels investigated (10, 30 and 100 mg/kg/day).
[1094] Absorption through the dermal route was minimal at the mid and high dose levels (30 and 100 mg/kg/day). It appeared to be slightly higher in the males.
[1095] On the basis of these results, the maximum tolerated dose in this study may be considered to be greater than 100 mg/kg for a single dermal administration in minipigs.
[1096] The high dose level of 100 mg/kg at a concentration of 200 mg/mL may be considered as the high dose level to be selected for a subsequent repeated dose study.
Results
[1097] No clinical signs were observed during the study.
[1098] No signs of irritation were observed at treated sites in any animal during the 7 day observation period.
[1099] Body weights were within the expected range for this strain and age of animals (Figures 66 and 67).
[1100] Food consumption was not affected by treatment.
[1101] No treatment-related signs were observed at clinical examination performed at the end of treatment.
[1102] There were no treatment-related changes observed in the weight of the organs.
[1103] No changes were observed in treated and untreated skin sites of control and treated animals during macroscopic observations. Animals killed at termination did not show macroscopic findings related to the administration of the test item.
Toxicokinetic analysis
[1104] Plasma samples were obtained from male and female minipigs before dosing (pre-dose) and 0.5, 1 , 2, 4, 6, 8 and 24 hours after the start of dosing on Day 1 of the study, following a single dermal administration of CT340 at the dose levels of 10, 30 and 100 mg/kg/day.
[1105] Plasma levels of CT340 were slightly >LLOQ (lower limit of quantitation = 4.5 ng/mL) in 1 male and 1 female receiving 30 mg/kg and in 2 males receiving 100 mg/kg/day. These values were reported between 1 and 24 hour post-dose. No measurable plasma levels were found in male animals dosed at 10 mg/kg and in female animals dosed at 10 and 100 mg/kg.
[1106] Due to the minimal absorption observed, in the majority of cases the calculation of the exposure (AUC) and of other kinetic parameters was not possible. Only when more than 2 consecutive time points values were available some parameters were calculable.
[1107] A Cmax of 8.74 ng/mL was measured in the single male of Group 3, while a Cmax of 9.85 ng/mL was measured in the single female of the same group. Tmax were of 2 and 1 hour in the male and female animals, respectively.
[1108] Mean Cmax was 7.735 ng/mL in the males of Group 4, with a Tmax of 2 hours. AUCo-inf, calculable for 1 male dosed at 100 mg/kg, was 538.918 ng h/mL.
[1109] No detectable levels were measured for animals treated with the vehicle alone. Absorption appeared to be slightly higher in males.
Conclusions
[1110] The pharmacokinetics, local dermal tolerance and toxicity of CT340 after a single dermal administration (6 hour exposure period) at the dosages of 10, 30 and 100 mg (concentrations: 20, 60 and 200 mg/mL) were investigated in the minipig during an observation period of 7 days in order to select dose levels for subsequent studies.
[1111] There were no clinical signs or other findings of toxicological importance noted in any animals during the study. In addition, no irritation of the treated skin site was observed during the study.
[1112] Minimal absorption through the dermal route was detected at the mid and high dose levels (30 and 100 mg/kg). Absorption appeared to be slightly higher in the males.
[1113] The above results indicate that the test item CT340, was locally and systemically well tolerated following a single dermal application to the minipig at all the dosages tested.
[1114] Therefore, the maximum tolerated dose in this study may be considered to be greater than 100 mg/kg for a single dermal administration in minipigs. The high dose level of 100 mg/kg at a concentration of 200 mg/mL may be considered as the high dose level to be selected for a subsequent repeated dose study.
Example 24: CT340 2 Week Dermal Tolerance/Toxicity Study in Minipigs Followed
By A 2 Week Recovery Period
[1115] The local tolerance, potential systemic effect and the toxicokinetic profile of the test item, CT340, after repeated daily dermal administration were investigated in the minipig during a period of at least 14 consecutive days and recovery from any treatment-related effects during a recovery period of 2 weeks.
[1116] Three groups each of 3 male and 3 female minipigs (Groups 2, 3 and 4) were treated by dermal application of 5.5 g of the test item formulations at concentrations of 20, 60 and 200 mg/mL (to achieve dose levels of 1 10, 330 and 1 100
mg of active ingredient/animal/day, corresponding to 0.22, 0.66 and 2.2 mg/cm2/day and to approximately 7, 21 and 69 mg of active ingredient/kg/day) over an area of the dorsal skin of approximately 25 x 20 cm, previously clipped. A fourth similarly constituted group (Group 1) received the vehicle alone (Propylene glycol 20%, DMSO 20%, Benzyl alcohol 1 %, Purified water) and acted as a control. One additional animal per sex was allocated to Groups 1 and 4 for 2 weeks of recovery.
[1117] The test item formulations were applied evenly over the prepared skin of animals of the relevant group. A 5.5 g aliquot of each test item formulation was administered and spread evenly over the skin of the prepared site by gentle massage, over an area of approximately 20 x 25 cm in animals of the relevant group. Control animals (Group 1) received the vehicle alone (control item) at the same dose volume, in the same manner.
[1118] The treated skin site was covered using a patch of surgical gauze and the whole assembly held in position by encircling the trunk of the animal with a cotton jacket. This semi- occlusive dressing was removed approximately 6 hours later and the treated site was cleaned by washing with a piece surgical gauze soaked with warm water, removing any residual substance.
[1119] All animals of the main phase were dosed once a day, for a minimum of 14 consecutive days, up until the day before necropsy. No treatment was given to recovery animals during the recovery period.
[1120] No plasma levels of CT340 greater than the LLOQ (lower limit of quantitation = 4.5 ng/mL) were detected for animals treated with the test item at concentrations of 20, 60 and 200 mg/mL on Day 1 or Day 14.
[1121] No signs of potential treatment-related effects of the test item were observed at any of the dose levels investigated (approximately 1 10, 330 and 1 100 mg/animal/day).
[1122] On the basis of the above results, CT340 was considered reasonably tolerated at the application site and not systemically toxic in the minipig following a repeated dermal (epicutaneous) administration over a period of at least 14 consecutive days, when administered at concentrations of 20, 60 and 200 mg/mL (corresponding to dose levels of approximately 1 10, 330 and 1 100 mg/animal/day).
[1123] No absorption through the dermal (epicutaneous) route was observed.
Results
[1124] No treatment-related clinical signs were observed during the study.
[1125] There was no effect of treatment on body weight. Body weights recorded during the treatment and recovery periods and terminal body weight were within the
expected range for this strain and age of animals. No differences were observed among the groups (Figures 68 and 69).
[1126] Food consumption was not affected by treatment.
[1127] Before the start of treatment, animals showing no ocular abnormality at the ophthalmoscopy were selected for the study. Both eyes of all animals from each group and sex were re- examined during Week 2 of treatment. No findings were detected during the ophthalmoscopic examinations performed during the study.
[1128] With regards to hematology analyses, no changes of toxicological significance were observed. No changes were recorded for coagulation parameters
[1129] With regards to clinical chemistry analyses, no changes of toxicological relevance were observed.
[1130] No changes of toxicological relevance were observed with regards to urine and feces analyses.
[1131] No plasma levels of CT340 greater than the LLOQ (lower limit of quantitation = 4.5 ng/mL) were detected following treatment with the test item at concentrations of 20, 60 and 200 mg/mL on Day 1 or Day 14.
[1132] There were no changes in organ weights which were considered to be treatment-related.
[1133] No treatment-related macroscopic changes were noted in final sacrifice animals.
[1134] All observed changes are suggested to be incidental, having a comparable incidence in control and treated groups, and/or characteristically seen in untreated Gottingen minipigs of the same age in our laboratory.
[1135] No treatment-related macroscopic changes were noted in recovery sacrifice animals.
[1136] No treatment-related changes were noted during microscopic observations.
[1137] All observed changes are suggested to be incidental, having a comparable incidence in control and treated groups, and/or characteristically seen in untreated Gottingen minipigs of the same age in our laboratory.
Conclusions
[1138] The local tolerance, potential systemic effect and the toxicokinetic profile of the test item, CT340, after repeated daily dermal administration were investigated in the minipig during a period of at least 14 consecutive days and recovery from any treatment-related effects during a recovery period of 2 weeks.
[1139] No treatment-related clinical signs or other changes indicating a systemic effect of the test item were observed in any animal during the treatment or recovery periods.
[1140] Microscopic examination carried out at the end of treatment did not show any treatment- related findings.
[1141] No absorption through the dermal route was observed.
[1142] On the basis of the above results, CT340 was considered reasonably tolerated at the application site and not systemically toxic in the minipig following a repeated dermal (epicutaneous) administration over a period of at least 14 consecutive days, when administered at concentrations of 20, 60 and 200 mg/mL (corresponding to dose levels of 1 10, 330 and 1 100 mg of active ingredient/animal/day, i.e. 0.22, 0.66 and 2.2 mg/cm2/day and approximately 7, 21 and 69 mg of active ingredient/kg/day).
Example 25: Plasma Pharmacokinetic Profile of CT327 (SNA-120) Following a
Single IV Administration to Rats
Aim of the Study
[1143] The purpose of the study was to investigate the pharmacokinetic profile of CT327 (SNA-120) when given by intravenous route to female rats with a single IV administration at 18 mg/kg body weight. This Example reports the analyses of CT327 plasma levels in the blood samples collected from 5 minutes to 8 hours after drug administration.
Materials and methods
IV administration to rats and collection of blood samples
Study Design
[1144] A single administration of CT327 was given to rats and blood samples were collected at scheduled times in order to verify the kinetic profile. One group of 9 female rats received the test item by the intravenous injection, at 18 mg/kg dose level.
[1145] The animals were monitored for body weight and clinical signs during the in vivo phase of the study. On the day of dosing, bleeding was carried out at the following time points: before dosing, 5, 10, 20 and 30 minutes, 1 , 2, 4 and 8 hours after the administration. Plasma samples were stored for subsequent analysis.
Vehicle and Formulation Procedure
[1146] The vehicle was physiological saline solution (NaCI 0.9%). The amount of CT327 (in one embodiment and for this particular study) was dissolved in the vehicle at the concentration of 3.6 mg/ mL. Concentration of the test item was calculated in terms of active ingredient according to the assay results (97.7%), as reported in the certificate of analysis. The formulation was prepared on the day of dosing. Since the formulation had to be administered within 30 minutes after preparation and on the basis of the bleeding scheme, three formulations of the test item were prepared at the scheduled times.
Treatment
[1147] The study consisted of one group comprising 9 female rats. The test item was administered by intravenous bolus injection into the tail vein over a period of approximately 1 mL/minute at a dose volume of 5 mL/kg body weight. The dose was administered to each animal on the basis of the most recently recorded body weight and the volume administered was recorded for each animal. Each animal received a single dose.
Blood collection
[1148] Blood samples were collected from the animals at the following sampling times: 0 (predose), 5, 10, 20 and 30 minutes, 1 , 2, 4 and 8 hours after the single dose administration. Blood samples were collected from each animal at 3 alternating time points. The theoretical and actual times of collection were recorded. At each sampling time, blood samples of approximately 0.8 mL each were collected under light isofluorane anaesthesia from the retroorbital sinus of 3 animals and transferred into light-protected tubes containing EDTA anticoagulant, immediately centrifuged at 4°C and 3000g for 10 minutes and the plasma frozen at -80°C. Two different aliquots were prepared: the first contained 250 μΙ of plasma and the second (100 μΙ). In addition, about 10 mL of plasma were obtained from 5 untreated female rats of the same batch of the study for analytical (calibration curve) purposes.
Animal Observation
[1149] Examination of individual animals for signs of reaction to treatment was carried out immediately after dosing, 15-30 minutes, 1 and 4 hours after dosing. No signs were recorded at clinical examination. All animals did not show reactions to treatment.
[1150] Each animal was weighed on the day of treatment. Body weights were within the expected range for this strain and age of animals.
Analysis of CT327 plasma levels in obtained blood samples
Materials
[1151] CT327
[1152] H20 MilliQ
[1153] Rat plasma (pool obtained from 5 untreated female rats of the same batch "stock order 661 " used for the experiment, i.e. Hsd: Sprague Dawley SD rats, 45-53 days old and with body weight of approximately 150-174 g)
[1154] Acetonitrile (ACN) HPLC grade
[1155] Methanol HPLC grade
[1156] Solid Phase Extraction (SPE) cartridges, Oasis® HLB 1 cc/10mg, particle size 30 μηι
[1157] HPLC column XTerra® RP18 particle size 3.5 μηι, 3.0x50 mm Methods
[1158] The analysis of CT327 plasma levels was performed by HPLC using a Beckman System Gold® chromatograph 126 solvent module, 168 UV-Vis Detector, 508 autosampler, equipped with a XTerra® RP18 column 3.0 x 50 mm.
[1159] Plasma samples were purified before HPLC analysis, in order to eliminate any possible source of interference, by SPE technique using Waters Oasis® HLB (Hydrophilic-Lipophilic Balance reverse-phase sorbent) cartridges. The purification of the biological samples consisted in the following consecutive steps: (i) conditioning of the cartridge with 1 mL of methanol; (ii) equilibration with 1 mL of MilliQ water; (iii) loading of the sample properly diluted in water (i.e. 0.1 mL of plasma sample in 0.9 mL of water, to give a total volume of 1.0 mL); (iv) washing of the unbound components with 5% methanol in water; and (v) elution with 1 mL of methanol.
[1160] The purification was performed using an extraction manifold (supplied by Waters S.p.A., Vimodrone, Italy) able to accommodate up to 20 cartridges and connected to a dual-headed vacuum pump (supplied by VWR International Sri, Milano, Italy).
[1161] The eluate from each biological sample was collected in polypropylene vials, evaporated under vacuum for about 4 hours by a Speed Vac system (Eppendorf concentrator 5301) and the residues were dissolved in 100 μί of MilliQ H20/ACN 60/40, in order to proceed to test item quantitation. To get complete dissolution
each sample was sonicated in an ultrasound bath for 10 sec, vortexed for 10-20 sec and then briefly centrifuged by a microcentrifuge (MiniFuge VWR International Sri, max RCF 2000g).
[1162] The quantitative analysis was achieved by HPLC by means of an appropriate calibration curve obtained in the same processing conditions of the unknown samples. CT327 standard samples in plasma were prepared with the above described procedure by purifying blank plasma samples (rat plasma recovered from untreated animals) spiked with a solution of test item in MilliQ H20 up to a volume of 0.1 ml, as depicted below in Tables 35 and 36. - -Calibration curve at low CT327 concentrations

TABLE 36 - Calibration curve at high CT327 concentrations
CT327 concentration
Spiking of rat plasma
1 2,0 pi GT327 50 μΜ + 13.0 μΐ ffiQ water + 85.Q pL rat plasma
5 10.0 ML GT327 50 uM + 5.0 μΐ MtliiQ water + 85.0 μΐ rat plasma
10 2,0 μΐ CT327 500 μΜ ·«· 13,0 μΐ MMQ water * 85.0 μΐ rat plasma
15 3.0 μΐ CT327 5Q0 μΜ + 2,0 μί, MIMQ water + 85,0 pL rat plasma
20 4.0 ML CT327 500 μΜ + 11.0 ML »G water + 85.0 μΐ rat plasma
25 5.0 ML CT327 500 μΜ + 10,0 μΐ Q water + 86.0 pL rat plasma
30 6.0 pL CT327 500

35 7.0 μΐ GT327 500 μΜ + 8.0 pL MWQ water + 85.0 pi rat plasma
[1163] HPLC separation and analysis were performed at a flow rate of 1 mL/min, with detection by measurement of the absorbance at 280 and 294 nm (the latter wavelength representing the maximum UV/Vis absorption peak of CT327 and therefore used for quantitative analysis). The injection volume was 25 μΐ and each sample was analysed in triplicate, keeping the autosampler at 4°C and with the elution gradient shown in Table 37 (eluent A= MilliQ water, eluent B = acetonitrile).
TABLE 37 - Elution Gradient
Tims (min) Eluent A {%) Eluent B (%)
0 9B S
2 SS 5
3 29 71
S.1 0 100
111 100
12.1 SS 5
14.1 B5 5
Results
[1164] Using the above described conditions the peak corresponding to CT327 shows a retention time of about 8.5 minutes. The quantitation of CT327 in the chromatograms was performed by integration of the corresponding peak with the following parameters: peak threshold 200 and peak width 0.2. Blank plasma samples were also analysed and included in the sample analysis run in order to control the specificity of the analytical procedure.
[1165] The calibration curve (peak area versus test item concentration) is shown in Figure 70 for plasma samples spiked with CT327 and was obtained by plotting the detected response versus the nominal sample concentration. The values of correlation coefficient (r2) suggest a good linearity over the range investigated, with a LOD (Limit of Detection) of 35.5 nM and a LOQ (Limit of Quantification) of 69.8 nM for the calibration curve at low concentration and with LOD 0.9 μΜ/LOQ 1.7 μΜ for the calibration curve at high concentration.
[1166] Mean plasma concentrations of CT327 were calculated for each time point on the above reported calibration curves (low range curve for <1 h) using a standard spreadsheet software (Microsoft® EXCEL®) and are reported in Table 38. The results are plotted as mean values ± error (95% CI, confidence interval) in Figure 71. Each point represents the average of 9 analyses, corresponding to 3 mice for each time point and HPLC injection in triplicate. Analysis

[1167] As expected, CT327 reached the maximum peak concentration at the first sampling time point (5 minutes) and quantifiable levels were observed until 4 hours post-administration in all the animals.
[1168] Pharmacokinetic evaluation of the mean data was performed according to a noncompartmental analysis using NCOMP software version 3.1 (Paul B. Laub, Fox Chase Cancer Center, Philadelphia, USA). Using Lagrange polynomials method, an interpolating curve between each pair of adjacent data points was constructed and the corresponding partial area computed.
[1169] The following parameters were determined:
[1170] CL (clearance, in L/h), expressed as the volume of blood cleared of drug per unit time;
[1171] MRT (mean residence time, in h);
[1172] Vss (apparent volume of distribution at steady state, in L);
[1173] t½ (terminal half-life, in h), determined by one-phase exponential decay fitting (GraphPad Prism software analysis). The correlation coefficient (r2) for the goodness of the fit of the regression line through the data points was high enough (0.991) for the value to be considered reliable; the 95% confidence interval for the t½ estimate was 0.13-0.23 h;
[1174] Cmax (initial concentration at t = 0 hours, in μΜ), extrapolated by log- linear regression of the first two measured data points;
[1175] AUCo-inf (area under the curve from time 0 to infinity, in μΜ-h), calculated by extrapolation of area from the time of the last measurement to time infinity and computed by estimating the terminal disposition rate by least squares fitting to slope of the logarithm of concentration vs. time. AUC0-inf was considered reliable since the extrapolation from the last data point to infinity represented the 0.1 % of the total AUC0.
Inf-
[1176] The mean pharmacokinetic parameters are summarized in Table 39 below.
TABLE 39 - PK parameters of CT327 after intravenous administration in rat
CL {Uh} 0.11
MKT (h) 0.51
Terminal ½ ( ) 0.17
51.88
14,01
Discussion and Conclusion
[1177] The pharmacokinetics of CT327 was investigated after intravenous administration to rats in order to assess the systemic exposure, distribution and elimination of the drug candidate. All the analyzed samples (3 animals/timepoint) showed detectable and consistent CT327 plasma levels up to 8 hours after administration.
[1178] CT327 elimination from the circulation system was extremely rapid, as plasma concentrations of the administered compound declined monoexponentially with a half-life of 10 minutes. In the eye, Applicant believes that conjugates such as CT327 (SNA 120), SNA 125, SNA 103, and/or SNA 352 and others would yield similar data, with respect to, for example, reduced exposure systemically (e.g., rapid elimination) and/or in non-target tissues.
[1179] Most interestingly, the calculated apparent volume of distribution Vss was 55 mL/kg, i.e. 10.78 ml_, corresponding to rat blood volume (BV), considering an average animal body weight (BW) of 196 g and the generally accepted figure of BV as 7% BW. These data suggest a full drug compartmentalization within the vascular compartment with no or very low distribution into tissues.
[1180] In conclusion, the observed time course of CT327 plasma levels confirmed its surprising and unique chemico-physical properties conferred by its mini- PEG moiety, resulting in high systemic exposure upon IV bolus (thanks to high water solubility), very rapid elimination from the blood stream (low molecular weight, probable fast kidney filtration with urinary elimination) and large confinement to the circulatory system (the compound undergoes no or very limited distribution from blood into tissues). These overall features are in line with the optimal safety profile shown by CT327 from a toxicological point of view: even if absorbed upon topical administration, one can exclude drug distribution and accumulation thanks to an extremely rapid clearance.
Example 26: Evaluation of the ability of CT101 to inhibit conventional NF-KB signaling
[1181] This study tested the ability of CT101 to inhibit inflammatory cell signalling by assessing its ability to inhibit conventional NF-κΒ signalling using an NF- κΒ-inducible Luciferase reporter construct.
Methodology
Compounds and Reagents
• Jurkat-Dual cells (ISG-NF-κΒ) cells (Invivogen jktd-isnf)
• THP-1-Lucia NF-κΒ cells (Invivogen thpl-nfkb)
HEK-Dual TNFoc (Invivogen hkd-tnfa) Pam3CSK4
• Staurosporine (Iris Biotech, lot. 090801)
TNF-a
• Concanavalin A
CT 101 lot n . 2013RB20/S45
Experimental outline
[1182] Three cell lines were selected based on their expression of the target src family kinases (the human monocyte cell line THP-1 , the human T cell line Jurkat, the human fibroblast cell line HEK293). Cell lines were purchased that stably express an NF- KB reporter construct (a secreted luciferase reporter gene, driven by an IFN-β minimal promoter fused to five copies of the NF-κΒ consensus transcriptional response element and three copies of the c-Rel binding site). Use of a stably transfected cell line eliminates the variation associated with transient transfection of the luciferase reporter into cell lines.
[1183] Cell lines were pre-treated with compound CT101 , before stimulation with a ligand suitable for the cell type; THP-1 cells were stimulated with the TLR1/2 ligand Pam3CSK4 (100 ng/mL), Jurkat cells were stimulated with Concanavalin A (ConA, 50 μg/mL) and HEK293 cells with TNF-a (10 ng/mL). Cells were then incubated for a further six hours or twenty-four hours before assessment of luciferase activity in the cultures as a measurement of NF-κΒ transcriptional activity.
[1184] a) To allow optimisation of the assay, initial experiments were performed with CT101 pre-treatment for 6 hours, 18 hours and 24 hours. The optimum
pre-treatment time, based upon when maximum inhibition of NF-κΒ activity was observed, was determined.
[1185] b) Following selection of the optimum pre-treatment time for each compound and cell type, separate experiments were performed over a range of compound concentrations to allow an IC50 for each compound, based on inhibition of NF-KB activity, to be determined.
[1186] c) To determine that any effect of the test compound on NF-KB transcriptional activity was due to specific inhibition of signalling pathways and not due to any effect on cell viability, for each of the tested concentrations; cultures were tested for the presence of lactose dehydrogenase (LDH), which is released into the media from damaged cells and is a biomarker for cellular cytotoxicity and cytolysis.
Selection of appropriate time frames and cell lines
[1187] CT101 was tested at three concentrations on the three different cell types (100 μΜ, 30 μΜ, and 10 μΜ). Each condition was tested in sextuplicate. Concentrations may be lowered (if cell toxicity is observed) or increased up to mM ranges (if effect is not observed).
[1188] Following incubation with stimulus cells were assayed for luciferase activity and an LDH assay performed to test for cell viability.
[1189] Table 40 shows the experimental conditions tested for each of the reporter cell lines (HEK, THP-1 , Jurkat).
Table 40

The experimental design is as follows:

St mulation
IC50 calculation of CT101 inhibition of NF-κΒ activity
[1191] Following the selection of optimised conditions for CT101 on each cell line, IC50 values were not calculated for CT101 inhibition of NF-κΒ activity as there was no consistent inhibition observed in cultures conducted in the presence of CT101 when compared to the vehicle control.
[1192] To determine that any effect of the test compound on NF-KB transcriptional activity was due to specific inhibition of signalling pathways and not due to any effect on cell viability, for each of the tested concentrations cultures were tested for the presence of lactose dehydrogenase (LDH), which is released into the media from damaged cells and is a biomarker for cellular cytotoxicity and cytolysis.
Readouts
[1193] Luciferase activity in reporter cell lines was assayed using a Promega luminometer.
[1194] LDH assays were assayed using a colorimetric system and absorbance read on an omega plate reader.
Results
Selection of appropriate time frames and cell lines
[1195] HEK, Jurkat and THP-1 reporter cell lines were pre-incubated with CT101 (100 μΜ, 30 μΜ, 10 μΜ) or DMSO vehicle (0.1 % v/v) control for 6 hours, before stimulation for a further 24 hours with 10 ng/mL TNF-a (HEK cells), 100 ng/mL Pam3CSK4 (THP-1 cells) or 50 μg/mL ConA (HEK293 cells). Control cells were not stimulated (non-stimulated) to provide background levels of luciferase activity present in cell lines. As a positive control some cells were incubated with 200 nM of staurosporine. Supernatants were then harvested and assayed for luciferase activity using the Invivogen Quanti-Luc assay solution. The fold induction of luciferase (NF-κΒ activity) by stimulation with ligands and the ability of CT101 or Staurosporine to inhibit induction were calculated by subtracting the mean media control luminescence value from the mean sample luminescence value and calculating the fold increase in luminescence above non-stimulated cells.
[1196] The Jurkat cell line did not grow well in the conditions recommended by the supplier therefore in some experiments where cell number was limiting; this cell line was tested in triplicate or quadruplicate for each condition. The Jurkat cell line was not tested for the 6h pre-incubation, 6h stimulation or 24h pre- incubation, 6h stimulation experimental conditions.
[1197] Each experimental condition was performed once, using sextuplicate wells. Data was analysed by two-way ANOVA comparing each cell mean to the control DMSO treated cell mean for that cell line, using Dunnett's multiple comparisons test. Values less than 0.05 were considered significant. These p values represent intra- experimental significance and are a measurement of reproducibility between wells within
a single experiment, they do not indicate any differences between different drug treatments, for this to be tested each experimental condition may be repeated on multiple occasions (Table 41).
Table 41. Conditions under which the luminescence of treated cells were significantly different from DMSO treated cells (intra-assay statistics). Analysis was carried out using a two-way ANOVA with Dunnett's multiple comparisons tests.

Pre-incubation time: 6 hours, stimulation time: 24 hours
[1198] Figure 72 depicts raw luminescence values for a CT101 preincubation time of 6 hours and a stimulation time of 24 hours. Each symbol represents an individual well. Each condition was tested in sextuplicate. Figure 73 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 6 hours and a stimulation time of 24 hours.
Pre-incubation time: 18 hours, stimulation time: 24 hours
[1199] Figure 74 depicts raw luminescence values for a CT101 preincubation time of 18 hours and a stimulation time of 24 hours. Each condition was
tested in sextuplicate except for Jurkat cells were conditions were tested in triplicate. Figure 75 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 18 hours and a stimulation time of 24 hours.
Pre-incubation time: 24 hours, stimulation time: 24 hours
[1200] Figure 76 depicts raw luminescence values for a CT101 preincubation time of 24 hours and a stimulation time of 24 hours. Each condition was tested in sextuplicate except for Jurkat cells where conditions were tested in triplicate. Figure 77 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 24 hours and a stimulation time of 24 hours.
Pre-incubation time: 6 hours, stimulation time: 6 hours
[1201] Figure 78 depicts raw luminescence values for a CT101 preincubation time of 6 hours and a stimulation time of 6 hours. Each condition was tested in sextuplicate. Figure 79 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 6 hours and a stimulation time of 6 hours.
Pre-incubation time: 18 hours, stimulation time: 6 hours
[1202] Figure 80 depicts raw luminescence values for a CT101 preincubation time of 18 hours and a stimulation time of 6 hours. Each condition was tested in sextuplicate except for Jurkat cells were conditions were tested in triplicate. Figure 81 depicts fold induction of Luciferase above non-stimulated cells for a CT101 preincubation time of 18 hours and a stimulation time of 6 hours.
Pre-incubation time: 24 hours, stimulation time: 6 hours
[1203] Figure 82 depicts raw luminescence values for a CT101 preincubation time of 24 hours and a stimulation time of 6 hours. Each condition was tested in sextuplicate. Figure 83 depicts fold induction of Luciferase above non-stimulated cells for a CT101 pre-incubation time of 24 hours and a stimulation time of 6 hours.
Cell viability: LDH Assay
[1204] To test for the effect of compounds on cell viability supernatants were harvested and assayed for LDH activity levels. Some cells were lysed to give maximal levels of LDH when all cells were dead. Graphs show the percentage cytoxicity of each culture condition (mean of six wells/condition, for Jurkat cells, the mean of 3 wells/condition) . Percentage cytotoxicity was calculated as (compound treated LDH release / maximum LDH release control) x 100.
[1205] The percentage cytotoxicity could not be calculated for 18 hour preincubation and 6 hour stimulation because the maximum lysis wells did not lyse.
[1206] Data was analysed by two-way ANOVA comparing each cell mean to the control DMSO treated cell mean for that cell line, using Dunnett's multiple comparisons test. Values less than 0.05 were considered significant. These p values represent intra-experimental significance and are a measurement of reproducibility between wells within a single experiment, they do not indicate any differences between different drug treatments, for this to be tested each experimental condition may be repeated on multiple occasions.
[1207] Figure 84 depicts percentage cytotoxicity for a CT101 pre-incubation time of 6 hours and a stimulation time of 24 hours. Figure 85 depicts percentage cytotoxicity for a CT101 pre-incubation time of 18 hours and a stimulation time of 24 hours. Figure 86 depicts percentage cytotoxicity for a CT1 01 pre-incubation time of 24 hours and a stimulation time of 24 hours. Figure 87 depicts percentage cytotoxicity for a CT101 pre-incubation time of 24 hours and a stimulation time of 6 hours. Figure 88 depicts percentage cytotoxicity for a CT101 pre-incubation time of 6 hours and a stimulation time of 6 hours.
[1208] Significant differences were found under various conditions with all cell lines, these are summarized in Table 42.
Table 42: Conditions under which LDH release by treated cells were significantly different from DMSO treated cells (intra-assay statistics). 2 way anova, Dunnetts multiple comparisons test.
Cel f¾¾
QMSO (ρ<ϋΜ)
HE en? 24hr uopMCTio
HEK 6nr 24t¥ 30μΜ CTIOr
HEK S¾ 24 hr lO M cm or
Juikaf ¾nr S¾r
Jui&at Snr 24hr
THP-1 6nr 6~r
THP-1 24hr
HEK i r 6 r Staumsporn«¾:
HEK ISIr 24 hr
Juafcai !Slir ¾hr 30μΜ CTlOl'
Jyrkat iShr Snr
Jtirk t 24 hr Siauos- o pe*
THP-1 Snr
THP-1 24 hr
24 r S¾r Stems orn^
JyrK t 24hr 24ht Sf&umsporlne
THP-1 24¥ 8~r lOpMCTlOI*
THP-1 24hr Snr
THP-1 24£*r 24hr
^T as ¾a s t O -lm^ as mm th rn aM sd it som ounds, ¾¾¾4¾¾ ίο:¾ 4ί¾ο Oign cs i>i¾ su:;M>
s t:0¾i«¾f ¼s s n SS<κiί: ^ <·Γi : ?ri κa«¾ tH:sk ; «8$ wh ; u>:r:»a-:*«i ;·:< ? s Oil S O csmp' l
[1209] The following observations were made based on the statistical analyses of the data by two-way ANOVA followed by Dunnett's post-test for multiple comparisons between experimental groups.
[1210] Incubation of HEK cells with TNF-alpha for 6 hours or 24 hours induced a significant increase of luciferase activity in the DMSO-treated cells when compared to the unstimulated cells.
[1211] Incubation of Jurkat cells with Con A for 6 hours or 24 hours induced a significant increase of luciferase activity in the DMSO-treated cells when compared to the unstimulated cells.
[1212] Incubation of THP-1 cells with Pam3CSK4 for 6 hours or 24 hours induced a significant increase of luciferase activity in the DMSO-treated cells when compared to the unstimulated cells.
[1213] Staurosporin significantly reduced the stimulus-induced luciferase activity:
• Regardless of the pre-incubation time followed by 24 hour-stimulation in HEK cells
• Only after 6 or 18 hours pre-incubation followed by 6 hour-stimulation in HEK cells
• At all time-points tested in Jurkat cells
• At all time-points tested in THP-1 cells
[1214] The LDH assay shows that the reduction of luciferase activity in HEK cells is not caused by a reduction of the number of cells.
[1215] The LDH assay suggests that, in Jurkat and THP-1 cells, the reduction of the luciferase activity may in part be associated with a reduction of the number of cells.
[1216] The best experimental conditions to assess the efficacy of compound CT101 in this assay were: HEK cells stimulated for 24 hours.
[1217] In those experimental conditions, the pre-incubation of HEK cells with CT101 at 100 μΜ for 6 hours or 18 hours (but not 24 hours) significantly reduced the luciferase activity. This effect was not associated with a reduction of the number of cells.
Conclusions
[1218] 24-hour stimulation induced the highest levels of luciferase and therefore NF-κΒ activity in the HEK and THP1 reporter cell lines. 6 hour-stimulation induced lower levels of NF-κΒ activity. Jurkat cells produced very low levels of luciferase in response to stimulation with ConA.
[1219] HEK cells had the highest levels of NF-κΒ activity.
[1220] CT101 proved to inhibit ligand induced NF- κΒ activity in a statistically significant manner at the top tested dose (100 μΜ) after 6 and 18 hours of pre-incubation in HEK cells, that proved to be the best performing of the three tested cell lines.
[1221] The control protein kinase inhibitor drug Staurosporine did inhibit NF- KB activity, under each condition tested for the HEK reporter cell line, the percentage inhibition of NF-κΒ induction was dependent on culture conditions and the cell line.
[1222] The percentage cytotoxicity as measured by LDH activity in culture supernatants in HEK cultures was below 15%, and in THP-1 levels were below 25% (except for 6h pre-incubation, 6h stimulation where levels were higher).
[1223] The percentage cytotoxicity in HEK-1 cells was fairly consistent across all culture conditions; cell death was not increased by stimulation, the inhibitory effect of Staurosporine on NF-κΒ activity was not due to a loss of cell viability and CT101 did not result in loss of cell viability at the concentrations tested.
[1224] LDH activity in all Jurkat samples was high indicating a high level of cell death in these cultures, independent of stimulation or treatment with CT101 , this was consistent with the poor growth of this cell line.
Example 27: Evaluation of the ability of CT103 to inhibit VEGF-induced
HUVEC cell proliferation
[1225] This study tested the ability of CT103 to inhibit VEGF- induced HUVEC cell proliferation.
Methodology
Compounds and Reagents
[1226] Motesanib Diphosphate (AMG-706), Source: MedChemExpress, CAS #453562-69-1 , lot HY-10228-05902.
Experimental outline
Part A: Selection of Appropriate Time Frames and assay conditions
[1227] Primary HUVEC cells (below passage 7) were seeded onto collagen- coated 96 well plates at two different concentrations 2 x 103 or 5 x 103 and treated with two different concentrations of VEGF: 10 ng/mL or 50 ng/mL. Some cells were not treated with VEGF to give background levels of non-VEGF driven proliferation. Cells
were incubated for 72 hours at 37°C with 5% C02. Proliferation was measured by pulsing cells for the last 24 hours with tritiated thymidine. Plates were then harvested and assayed for tritiated thymidine incorporation. The ability of CT103 to inhibit proliferation was assessed by pre-incubation of cells with CT103 for 6 hours or 18 hours prior to stimulation with VEGF. This was tested using the following concentrations of compound: 100 μΜ, 30 μΜ and 10 μΜ. Motesanib Diphosphate (AMG-706) 10 nM was included as a positive control. Each condition was tested in sextuplicate.
[1228] The conditions tested were as follows:
HUVEC VEGF CT103 CT103
cell concentration pre- concentration time den it incubation
ime
0 ng/ml 100μ
30μ
0
0 ng/ml ΙΟΟμΜ
30u
2x10¾
10
0
50 ngml WQ≠A
30μ
ϊϋμΜ
en 0
and
18 1Ο0μ
N
3Du
0
10 ngml 100μ
30μ
ΙΟμ
0
50 ngmL 10ϋμΜ
30μίν1
ΙΟμ
0
Part B: IC50 calculation for CT103 inhibition of VEGF induced HUVEC proliferation.
[1229] Primary HUVEC cells (below passage 7) were seeded onto collagen- coated 96 well plates at 5x103 cells per well. The ability of CT103 to inhibit proliferation was assessed by pre-incubation of cells with CT103 for 18 hours prior to stimulation with VEGF (50 ng/mL). To generate an IC50 calculation, a series of 3-fold dilutions of CT103 starting at 300 μΜ was performed. Motesanib Diphosphate (AMG-706) was included as a positive control at a concentration of 10nM. Each condition was tested in sextuplicate.
Readouts
[1230] HUVEC proliferation was assayed by measuring tritiated thymidine incorporation into DNA. Cells were harvested using a harvester onto a 96 well grid filter mat and thymidine incorporation was read using a β-counter.
Results
Part A: Selection of Appropriate Time Frames and assay conditions
Pre-incubation time: 6 hours
[1231] Figure 89 depicts tritiated thymidine incorporation in corrected counts per minute (CCPM) with a pre-incubation time of 6 hours.
Pre-incubation time: 18 hours
[1232] Figure 90 depicts tritiated thymidine incorporation in corrected counts per minute (CCPM) with a pre-incubation time of 18 hours.
[1233] Proliferation data were analyzed by two-way ANOVA followed by Dunnett's post-test for multiple comparisons between experimental groups (Table 43).
Table 43: Proliferation data, Dunnett's post-test results

would be (results reported in Figure 91):
Cells seeded at 5 x 103 cells per well
Pre-incubation with compounds for 18 hours
Stimulation with VEGF at 50 ng/mL
[1235] Figure 92 depicts percentage inhibition by CT103 with 18 hour preincubation time, 5x103 cells per well and 50ng/ml_ VEGF stimulation.
[1236] Data generated using these experimental conditions were further analyzed by one-way ANOVA followed by Dunnett's for multiple comparisons between experimental groups.
Part B: IC50 calculation for CT103 inhibition of VEGF induced HUVEC proliferation
[1237] Figure 93 depicts inhibition of HUVEC proliferation at a range of CT103 concentrations. Figure 94 depicts non-linear fit of transformed normalised maximal response for IC50 calculation. Figure 95 depicts percentage inhibition of VEGF induced proliferation by CT103.
Conclusions
[1238] CT103 inhibited VEGF-induced HUVEC proliferation at both cell densities and pre-incubation time points. At 18 hours pre-incubation, HUVEC proliferation is substantially higher in the presence of VEGF when compared to unstimulated cultures. The 5x103 cell density gave higher proliferation read outs than 2x103 cells per well, thus a clear inhibition can be seen. Therefore better conditions for monitoring VEGF-induced HUVEC proliferation are a cell density of 5x103 cells per well stimulated with 50 ng/mL VEGF and a pre-incubation time of 18 hours. These conditions were used to generate IC50 values for CT103 inhibition of VEGF-induced HUVEC proliferation by performing a series of 3-fold dilutions, starting at a top concentration of 300 μΜ. A clear effect of CT103 was observed on the VEGF-induced HUVEC proliferation and an IC50 value could be calculated. An IC50 of 18.5μΜ for CT103 inhibition of VEGF-induced HUVEC proliferation was determined.
Example 28: Efficacy Analysis of CT340 Using In Vitro Nf-κΒ -Luciferase Reporter
Gene Assays
[1239] CT340 was tested in an in vitro cell-based assay to monitor the antiinflammatory activity in a complex biological system. Inhibition of NF-κΒ, a key regulator of the inflammatory response, was assayed using human monocytes (THP1) carrying a luciferase reporter gene driven by a tandem repeat of the NF-κΒ consensus transcriptional response element. In addition, staurosporine, a broad spectrum kinase inhibitor, was used as comparator.
[1240] For the setup of the assays, cell lines were stimulated for 6 or 24 h with a concentration range of 2 different stimuli: LPS and heat-killed Listeria monocytogenes (HKLM). Luciferase activity was measured as described below. Based on these data, for each stimulus, a concentration was chosen that resulted in a sub-optimal induction of luciferase activity.
[1241] For assaying the effect of the inhibitors, cell lines were pre-cultured with the inhibitors, i.e. CT340 or staurosporine, or with a volume equivalent of DMSO. After 2 h, the pre-determined sub-optimal dose of stimuli were added, and incubation was continued for another 6 h. Luciferase activity was determined as described in the data sheet of the cell lines.
[1242] To assay a potential toxic effect of the inhibitors and/or stimuli, the numbers of dead and viable cells were determined after the incubation period, by careful
resuspending the cells, mixing them with trypan blue, and counting the number of blue and white cells in microscope counting chambers.
[1243] THP1 cells were grown and stimulated for 24 h, as described on the data sheet of the cell line. Cells were stimulated with HKLM (0.625 x 10Λ7 cells/mL - 10 x 10Λ7 cells/mL) or LPS (0.01 - 100 ng/mL). Fold induction relative to unstimulated cells is depicted in Figure 96. Luciferase activity is dose dependently increased with both stimuli, with a maximum fold increase of 200-fold for both stimuli.
[1244] A suboptimal concentration of HKLM and LPS was chosen for further experiments, being 2.5 χ10Λ7 cells/mL, and 1 ng/mL, respectively. Stimulation was furthermore confirmed when cells were stimulated for 6 h instead of 24 h.
Results of Pilot Experiment
[1245] For this pilot experiment, 2 concentrations of inhibitors were tested that cover a broad range, being 100 μΜ and 100 nM. As controls, cells were treated with 1 % DMSO, a volume equivalent of DMSO for the cells treated with 100 μΜ inhibitor, or with medium only, as a control for cells treated with 100 nM inhibitor. Cells were pre-treated with inhibitor for 2 h, and subsequently stimulated with LPS or HKLM. Six or 22 h after addition of the stimuli, a culture supernatant sample was assayed for luciferase activity.
[1246] Visual inspection of the cells after 8 h incubation revealed that cells treated with 100 μΜ staurosporine looked poor, whereas no irregularities were observed for the other conditions. After 24 h, the number of viable cells was counted. A significant increase in the percentage of dead cells was observed after addition of 100 μΜ staurosporine (data not shown), accompanied by an enormous decrease in the concentration of viable cells (Table 44). No significant numbers of dead cells were observed in the other conditions tested.
TABLE 44 - The relative concentration of viable THP1 cells cultured for 24 h with the inhibitors indicated

[1247] Luciferase activity was efficiently induced in cells stimulated for 8 or 24 h with HKLM or LPS (Table 45). Relative luciferase units are depicted in Table 46.
[1248] Results obtained after 8 or 24 h are highly similar. Staurosporine results in substantial reduction of luciferase activity, in both concentrations used. In addition, preincubation of cells with 100 μΜ CT340 inhibits the luciferase activity, both in unstimulated, as well as in LPS or HKLM-stimulated cells.
TABLE 45 - Relative luciferase units of THP1 cells stimulated with HKLM or LPS for 8 or 24 h, relative to unstimulated cells.

TABLE 46 - Relative luciferase units of THP1 cells treated with the inhibitors and stimuli indicated in the table

Results of Main Experiment
[1249] Inhibitors were tested at 3 concentrations, being 100 μΜ, 1 μΜ and 10 nM. As controls, cells were treated with 1 % DMSO (volume equivalent to 100 μΜ inhibitor), 0.01 % DMSO (volume equivalent to 1 μΜ inhibitor), or with medium alone (control for cells treated with 10 nM inhibitor). Cells were pre- treated with inhibitor for 2 h, and subsequently stimulated with LPS or HKLM for 6 h.
[1250] To assay potential toxic effect of the inhibitors and/or stimuli, the number of viable cells were determined after the incubation period. Addition of staurosporine (100 μΜ and 1 μΜ) had a consistent negative effect on the number of viable cells, whereas no consistent effects were observed for the other conditions tested (Table 47).
[1251] Luciferase activity was effectively induced by both stimuli used, with an increase of 6.5- and 3.4-fold in LPS and HKLM stimulated cells, respectively. Relative luciferase units are depicted in Table 48.
TABLE 47 - Relative number of viable THP1 cells cultured for 8 h with the inhibitors and/or stimuli indicated.
Relative n umber of v t le cells
Inhibitor Medium LPS HKLM
medium 100 51 86
DMSO 1% 69 46 89
DMSO 0.01% SO 46 120
100 μ?ν1 CT340 40 97 123
1 μΜ CT340 23 94 149
0,01 μΜ CT340 71 109 120
100 μ staurosporin 0 23 3
1 μ staurosporin 57 20 11
0.01 μ stau osporin 57 31 91
[1252] Pre-treatment of cells with 100 μΜ or 1 μΜ staurosporine almost completely blocked the luciferase activity, both in non-stimulated cells, as well as in LPS or HKLM stimulated cells. Also 100 μΜ CT340 efficiently reduced the luciferase activity, whereas lower concentrations (1 μΜ or 0.01 μΜ) did not have an effect.
TABLE 48 - Relative luciferase units of THP1 cells treated with the inhibitors and stimuli indicated in the table

[1253] NF-κΒ can efficiently and dose dependently be activated by LPS and heat killed Listeria monocytogenes (HKLM).
[1254] 100 μΜ and 1 μΜ staurosporine appear toxic for THP1 cells, as the number of viable cells after culturing with this inhibitor is strongly and consistently reduced.
[1255] No toxic effects were observed with 0.01 μΜ staurosporine. This concentration however had no effect on NF-κΒ activation.
[1256] 100 μΜ CT340 strongly and consistently blocks the activation of NF-κΒ in both separate experiments performed, without consistently affecting cell viability. Therefore a specific anti- inflammatory effect of the compound in THP1 monocytes is demonstrated. This effect was not observed with 100-fold lower concentrations of the compound.
Example 29: Efficacy Analysis of SNA-125, SNA-352 and SNA-103 in a VEGF- induced Proliferation Assay Using HRMVEC Cells
Aim of Study
[1257] The aim of this study was to compare the efficacy of SNA-125, SNA- 352 and SNA-103 in a VEGF-induced proliferation assay using Human Retinal Microvascular Endothelial (HRMVEC) cells. SNA-125 and SNA-352 have been observed to inhibit kinases in VEGF signaling pathway: ERK and RAF for SNA-125, and PCKa, PCKb2 and PKg for SNA-352. It is contemplated that these compounds could have an anti-angiogenic effect in addition to their anti-inflammatory effect.
Methodology
Experimental outline
[1258] Primary HRMVEC cells were seeded onto collagen-coated 96 well plates at a concentration of 2 or 5 x 103 and treated with VEGF at 10 or 50 ng/ml. As a control, cells without VEGF were included to give background levels of non-VEGF driven proliferation. Cells were incubated for 72 hours at 37°C with 5% C02. Proliferation was measured by pulsing cells for the last 24 hours with tritiated thymidine. Plates were then harvested and assayed for tritiated thymidine incorporation. The ability of the lead compounds to inhibit proliferation was assessed by pre-incubation of cells with the lead
compounds for 18 hours prior to stimulation with VEGF. Each lead compound was tested at eight concentrations. Motesanib Diphosphate (AMG-706) was included as a positive control. Four concentrations were tested. Each condition was tested in sextuplicate. To readout cell proliferation, cells were pulsed with 3H-Thymidine and harvested 24 hours later. Radiation was then quantified. IC50 values were calculated (where possible) for each compound.
Treatment Groups and Dosages
[1259] The doses of the test SNA compounds used were 300, 100, 33.3, 1 1 .1 , 3.7, 1.2, 0.41 , and 0.14 μΜ. The doses of motesanib diphosphate used were 30, 10, 3.33, and 1 .1 1 nM. The pre-incubation time was 18 hours. Table 49 depicts the treatment groups and dosages employed in this study.
Table 49: Treatment Groups and Dosages

[1260] Figure 97 depicts the inhibition of VEGF-induced proliferation following treatment with SNA-125, SNA-352, SNA-103, and motesanib diphosphate. The IC50 values calculated based on this analysis are shown in Table 50.
Table 50: IC50 Values for SNA-103, SNA-125, SNA-352

Conclusions
[1261] SNA-125 showed inhibition of VEGF-induced proliferation. Lower IC50 values were seen when treating the lower cell density (values between 7 and 9 μΜ). SNA-352 also showed consistent inhibition of VEGF-induced proliferation at both concentrations and cell densities tested, with IC50 values between 4 and 5 μΜ. SNA-103 appeared to have an effect on proliferation at the top concentration, with IC50 values between 100 and 200 μΜ. Motesanib diphosphate did not appear to inhibit proliferation with this cell line.
Example 30: CT340 in vitro activity testing in comparison to K252a and CT327:
Anti-proliferative activity on neonatal Human Epidermal Keratinocytes
[1262] The purpose of this study was to evaluate the in vitro activity of CT340 in comparison with K252a and CT327. The in vitro activity was evaluated as ability to inhibit proliferation on neonatal Human Epidermal Keratinocytes.
Materials and methods
Cell Preparation
[1263] One or more 70-80% confluent T-75 culture flasks (according to the number of cells desired to perform the experiment) are rinsed one time with 9 ml of Trypsin/EDTA solution according to the manufacturer instructions. 3 ml of Trypsin/EDTA is then added for the second time to detach cells. After approximately 10 minutes at 37°C, cell detachment is complete and Trypsin is blocked with 9 ml of Trypsin Neutralizer
solution. Cells are transferred to a sterile 15 ml conical tube, filled with other 9 ml of Trypsin neutralizer and centrifuged at 180 x g tor 7 minutes. The supernatant is removed and the cell pellet is resuspended in 12 ml of supplemented medium. Vital cell count with Trypan Blue is performed and cells are diluted to seed one or more 96-well plates with 1 ,5 x 103 cells/well in 100 μΙ_. Cells are incubated tor 18h in a 37°C, 5% C02, humidified cell culture incubator. The day after, 10 μΙ of test item solution at the appropriate concentration are added to each well (10 μΙ_ΛΛβΙΙ of Complete Growth medium are added to control wells).
[1264] The plate(s) are then incubated for 24h, 48h, 72h and 96h in a humidified incubator at 37°C with 5% C02 and analyzed with MTT at each time-point (Time 0 is analyzed just after cell treatment with test item solution) .
Solution for cell treatment preparation
[1265] A K252a 10 mM stock solution is prepared in DMSO and stored at - 20°C. Solutions tor cell treatment are freshly prepared just before use by serial dilution of stock solution in complete growth medium.
[1266] CT340 and CT327 10 mM stock solutions are prepared in DMSO and stored at - 20°C. Solutions tor cell treatment are freshly prepared just before use by serial dilution of stock solution in complete growth medium. Only tor the third experiment a 10 mM starting solution is prepared in complete growth medium just before cell treatment. This solution is then further diluted in the same medium to obtain the desired concentrations to be tested. Complete growth medium is used as positive control.
MTT viability assay
[1267] At designed time-points after addition of the test item (K252a, CT327, CT340) 15 μΙ_ of Dye Solution is added to each well. The plate(s) are then incubated at 37°C, 5% C02 for up to 4 hours. After incubation, 100 μΙ_ of stop solution is added to each well and plate(s) are left at 37°C for 1 hour. The content of the wells is mixed to get a uniformly colored solution with a multichannel pipette and plate is read using a 96- well plate reader by setting the wavelength at 570 nm (reference 650 nm) . Results are expressed as optical density units (OD) .
Data representation and statistical analysis
[1268] Data are represented in bar columns as mean OD57o-65o (Optical Density) ± SEM (Standard Error of the Mean) of the results obtained from six wells.
[1269] In order to compare selected pairs of data sets, One-way ANOVA followed by either Dunnett's post test (control column data: negative control - Complete
Growth Medium) or Bonferroni's post test among pairs of selected data sets is the statistical analysis performed.
[1270] K252a, CT327 or CT340 concentration data having a post test p value < 0.05 if compared to control wells data are considered as significantly inhibiting the proliferation of human epidermal keratinocytes.
[1271] The IC50 value was calculated fitting data on a sigmoidal dose- response curve with variable slope.
[1272] GraphPad Prism Software was used for data representation, statistical analysis and curve fitting.
Results
[1273] Three experiments were been performed to evaluate the effect of different concentrations of the test items at different time-points.
Proliferation Assay Setup with K252a, CT340 and CT327 - 1st Experiment
[1274] In a first experiment K252a, CT340 and CT327 were tested at the following concentrations: 25 nM, 50 nM, 100 nM, 200 nM and 500 nM. MTT is performed at 24 h, 48 h, 72 h and 96 h. The results of the experiment performed on neonatal HEK cell line to evaluate the ability of the compounds to inhibit proliferation are reported in Figure 98.
[1275] K252a shows a good concentration-response curve after contact time 48 h, 72 h and 9 6h and is only partially effective after contact time 24h. CT327 and CT340 partially inhibit proliferation at time 24 h and 96 h but without a concentration- dependent response. The IC50 values for K252a at time-points 48 h, 72 h and 96 h, respectively, are 150.8 nM (R2=0.9326), 101 .4 nM (R2= 0.9532) and 84.36 nM (R2= 0.9626).
Proliferation Assay Setup with K252a, CT340 and CT327 -2nd Experiment
[1276] In the second experiment CT327 and CT340 were tested at different concentrations than K252a in order to evaluate if higher concentrations of the molecules are able to inhibit the proliferation of HEKn cell line. K252a was tested at the same concentrations of the previous experiment while CT327 and CT340 were tested at the following concentrations: 100 μΜ, 50 μΜ, 25 μΜ, 5 μΜ, 1 μΜ, 500 nM, 200 nM, 100 nM, 50 nM and 25 nM.
[1277] MTT assays were performed at time 72 h. The results of the experiment performed on neonatal HEK cell line to evaluate the ability of the compounds to inhibit proliferation are reported in Figure 99.
[1278] As in the 1 st Experiment, K252a shows a good concentration- response curve after a contact time of 72 h, with an IC50 value of 171 nM (ι^= 0.9823). Furthermore, increased concentrations of CT327 and CT340 cause inhibited proliferation at time 72 h with a partial concentration-dependent response. Unfortunately, the 1 % DMSO concentration present in the wells with the higher concentrations of CT327 and CT340 did not allow for statistically significant relevance and the IC50 value could not be calculated.
Proliferation Assay Final Experiment with K252a, CT340 and CT327
[1279] A third experiment was performed in order to identify an IC50 value tor CT340 and CT327 in the absence of DMSO in the final wells. For this reason, 10 mM starting solutions of CT340 and CT327 were prepared by dissolving them directly in complete growth medium just before cell treatment. These solutions were then further diluted in the same medium to obtain the desired concentrations to be tested.
[1280] K252a was tested at the same concentrations of the previous experiments while CT327 was tested at the following concentrations: 500 μΜ, 100 μΜ, 50 μΜ, 25 μΜ, 5 μΜ, 1 μΜ, 500 nM, and 50 nM. CT340 was tested at the following concentrations: 500 μΜ, 100 μΜ, 50 μΜ, 25 μΜ, 5 μΜ, 1 μΜ, 500 ηΜ, 200 nM and 100 nM.
[1281] ΜΤΤ assays were performed at time 72 h, 96 h and 120 h. The results of experiments performed in neonatal HEK cells to evaluate the ability of the compounds to inhibit proliferation are reported in Figure 100.
[1282] K252a showed a good concentration-dependent response for all three time points analyzed. Furthermore, increased concentrations of CT327 and CT340 inhibit proliferation at time points 72 h, 96 h and 120 h with a concentration-dependent response.
[1283] IC50 for K252a for the time points 72 h, 96 h and 120 h are 98.31 nM (R2=0.9590), 40.04 nM (R2= 0.9630) and 32.88 nM (R2=0.9899), respectively.
[1284] The IC50 values for CT327 and CT340 are the following:
[1285] CT327
[1286] T72h IC50= 21 .93 μΜ (R2=0.9179)
[1287] T96h IC50= 18.57 μΜ (R2=0.9281)
[1288] T120h IC50= 17.51 μΜ (R2=0.9876)
Γ1289Ί CT340
[1290] T72h IC50= 15.57 μΜ (R2=0.9263)
[1291] T96h IC50= 14.55 μΜ (R2=0.9528)
[1292] T120h IC50= 1 1 .47 μΜ (R2=0.9915)
[1293] The IC50 values obtained show that CT327 and CT340 maintain the ability to inhibit proliferation of HEKn cells, even though employed at higher concentrations if compared to the unconjugated molecule. Furthermore, CT340 appears to be slightly more active than CT327.
Conclusions
[1294] The reported experiments confirm that both CT327 and CT340 maintain the K252a ability to inhibit the proliferation of HEKn cells. This implies that they are able to pass the cell membrane and to interact with target kinases to elicit their effect. In the eye, Applicant believes that conjugates such as CT327 (SNA 120), CT340 (SNA 125), SNA 103, and/or SNA 352 and others would yield similar data, with respect to, for example, the ability to cross cell membranes and reduced exposure systemically and/or in non-target tissues.
[1295] Comparing the IC50 values calculated in these experiments, it can be observed that the PEG moiety partially modifies the interaction with the pharmacological targets. Moreover, PEGylated molecules may need longer contact time to show a noticeable effect. Indeed, when the contact time is 24 and 48 hours, K252a is already active while CT340 and CT327 are not. However, the loss of activity due to PEGylation was expected and is acceptable if suitably counterbalanced by benefits in terms of a reduced systemic toxicity and a dramatically improved selectivity in terms of kinase inhibition profile (as described in other examples herein).
[1296] The final experiment provided a comparison the IC50 values of CT340 and CT327, with the results suggesting that the presence of the amide linkage in CT340 results in a slightly more active molecule with lower IC50 values than CT327 at all the tested time points.
Example 31 : CT352 efficacy study in a DSS-induced murine acute colitis model
Aim of Study
[1297] The purpose of the present study was to evaluate the efficacy of CT352 to counteract acute colitis by repeated oral administration for seven consecutive days overlapping with the five days-DSS treatment in mice. Control groups were untreated, treated with test article vehicle (water) or with cyclosporine A (CsA, positive control).
[1298] This report shows the results of the in vivo experiments and the data obtained from ex vivo quantitative real time (qRT)-PCR and histology analysis performed on colon samples collected from all the groups.
Materials and Methods
Experimental Design
[1299] The experimental protocol was modified from Eckmann et al, PNAS 2008. Male Balb/c mice, 7-9 weeks old, with a mean 25 gr weight, were used for this experiment. Mice were kept in the animal house facility at the Molecular Biotechnology Center under standard conditions of temperature and light, and were fed with standard laboratory chow and water ad libitum. Procedures were conducted in conformity with national and international laws and policies as approved by the Faculty Ethical Committee.
[1300] Mice were divided into the following experimental groups:
Group Treatment Animal ID
1 - Untreated none 43-32-36
2 - Vehicle 3% DSS in drinking water + 23-33-41 -46-48-49-52
water by gavage
3 - CT100 3% DSS in drinking water + 21 -35-37-39-31 -47-51
100 mg/kg CT352 by gavage
4 - CT300 3% DSS in drinking water + 19-20-22-24-28-29-38
300 mg/kg CT352 by gavage
5 - Cyclosporine ^ 3% DSS in drinking water + 26-34-40-42-45-50-53
25 mg/kg CsA by gavage
[1301] 3% Dextrane Sodium Sulfate (DSS) (MP Biomedicals, m.w 35,000- 50,000) in drinking water was administered for 5 days to all groups except group one, followed by regular water for further two days and sacrifice. Mice were daily monitored for colitis features (weight, bleeding and stool consistency) . Mice colons were excised at sacrifice from the ileo-cecal valve to the rectum, rinsed with PBS, their length was measured as an indirect marker of inflammation and 0.5 cm sections from both the distal and the proximal colon ends were cut and snap frozen for the subsequent RNA analysis. The remaining colon was prepared as "Swiss Roll" for histological analysis (Moolenbeek and Ruitenberg Lab. Anim. 1981 ).
Colitis monitoring
[1302] The clinical course of the disease was followed daily and represented by colitis clinical score, that account for body weight, stool consistency and rectal
bleeding. The colitis clinical score ranged from 0 to 12 and was the sum of scores given for body weight loss (scored as: 0, none; 1 , 1 -5%; 2, 5-10%; 3, 10-20% ; 4, over 20%) , stool consistency (scored as: 0, well formed pellets; 2, pasty and semiformed stools; 4, liquid stools) and presence or absence of fecal blood (scored as: 0, negative; 2, light bleeding; 4, gross bleeding). Accordingly, the clinical score ranged from 0 (healthy) to 12 (maximal activity of colitis).
Quantitative Real-time PCR
[1303] Total RNA was extracted from 0.5 cm proximal colon and 0.5 cm distal colon taken from sacrificed mice with the PureLink Micro-to-Midi total RNA Purification System (Invitrogen, Carlsbad, CA, USA) and its concentration was determined by NanoDrop™ 1 100 (NanoDrop Technologies, Wilmington, DE, USA) . Samples were treated with DNase lAmp Grade (Invitrogen) before retrotranscription to eliminate genomic DNA contamination. To produce template cDNA, 125 ng of total RNA was reverse-transcribed using the RT High Capacity kit (Applied Biosystems) according to the manufacturer's protocol. qRT-PCR reactions were performed with the ABI Prism 7300 real-time PCR System (Applied Biosystems) using Platinum Quantitative PCR SuperMix-UDG with ROX (Invitrogen) . The reactions were carried out in a total volume of 20 μΙ. The cytokines and chemokines were detected by using the Universal Probe Library system (Roche Italia, Monza, Italy) with the following primers: IL6, 5'- gctaccaaactggatataatcagga-3'; 5'-ccaggtagctatggtactccagaa-3'; CCL2, 5'- catccacgtgttggctca-3'; 5'-gatcatcttgctggtgaatgagt-3'; M IP2, 5'- aaaatcatccaaaagatactgaacaa-3'; 5'-ctttggttcttccgttgagg-3'; IFNg, 5'- atctggaggaactggcaaaa-3'; 5'-ttcaagacttcaaagagtctgaggta-3'; COX2, 5'- gatgctcttccgagctgtg-3'; 5'-ggattggaacagcaaggattt-3'; IL10, 5'-cagagccacatgctcctaga-3'; 5'-tgtccagctggtcctttgtt-3'; TNFa, 5'-tcttctcattcctgcttgtgg-3'; 5'-ggtctgggccatagaactga-3'; TGFb, 5'-tggagcaacatgtggaactc-3'; 5'-cagcagccggttaccaag-3'; IL1 b, 5'- agttgacggaccccaaaag-3'; 5'-agctggatgctctcatcagg-3'; IL17, 5'-tgtgaaggtcaacctcaaagtc-3'; 5'-agggatatctatcagggtcttcatt-3'; M IP1 a, 5'-agattccacgccaattcatc-3'; 5'- gccggtttctcttagtcagga-3'. Results were analysed with the 2"ΔΔα method using the 18S rRNA pre-developed TaqMan assay (Applied Biosystem) as an internal control. Expression of the target genes was calibrated against control non-colitic animals (group one, untreated mice).
Histological analysis
[1304] For histological analysis of the colon, samples were rinsed with PBS, prepared as "Swiss Roll", fixed overnight in formalin and then embedded in paraffin.
Sections 5 μηι in thickness were stained with haematoxylin/eosin and blinded analyzed by a pathologist who assessed the severity of colitis, expressed as a histology score that accounts for infiltration of inflammatory cells and epithelial structure, in a scale of 0 to 6: 0 = no damage; 1 = few inflammatory cells, no signs of epithelial degeneration; 2 = mild inflammation, few signs of epithelial degeneration; 3 = moderate inflammation, few epithelial ulcerations; 4 = moderate to severe inflammation, ulcerations in more than 25% of the tissue section; 5 = moderate to severe inflammation, large ulcerations of more than 50% of the tissue section; 6 = severe inflammation and ulcerations of more than 75% of the tissue section.
Statistical analysis
[1305] Data are expressed as mean ± S.D. An unpaired t test was used to calculate a P value for two groups. A P value of < 0.05 was considered statistically significant (GraphPad Software Inc., San Diego, CA, USA).
Results and Discussion
DSS-induced colitis evaluation
[1306] After five days-treatment with 3% DSS, mice were left two more days drinking water to recover before sacrifice. At sacrifice, colons were excised from the ileocecal valve to the rectum, abundantly rinsed with PBS and their lengths were measured, as an indirect marker of inflammation. As shown in Figure 101 A, the vehicle group displayed a reduction of colon length compared to the untreated group, due to a more prominent inflammatory response. Colon length increased in mice treated with CT352, both doses, and in mice treated with CsA, indicating that inflammation was reduced in these 3 groups in comparison with the vehicle group. None of these differences were statistically significant. During the experiment, mice were daily weighed and the weight loss of each day was compared to the initial weight of each mouse, to get the weight percentage reduction/increase. Considering the overall trend of each experimental group, the untreated mice increased their weight during the experiments, while the vehicle group animals lost weight during DSS treatment and few animals started to recover weight right after the stimulus removal, while the majority of mice continued to lose weight or stayed stable. On the contrary, all DSS-treated mice lost weight during colitis induction, but while after DSS removal the vehicle group did not recover completely the weight, mice belonging to CT100, CT300 or CsA groups started to recover weight right after it (Figure 101 b). These data indicate that CT352 or CsA treatment lead to a quicker weight improvement than only vehicle administration. A more
general and complete evaluation of the colitis response was given by the colitis clinical score that takes account not only of weight loss but also of bleeding and stool consistency. Untreated mice obviously displayed a minimal colitis clinical score, while vehicle group, although quite disomogeneous, showed a high score. Interestingly, CT352 clearly reduced the clinical colitis score in a dose-response manner, and its effect was more prominent than CsA's one (Figure 101 c) .
[1307] These data indicate that vehicle group developed colitis compared to untreated group, and that the water gavage had no effect on colitis. On the contrary mice receiving CsA at the same time, developed a milder colitis (increased colon length, weight recovery after stimulus removal and low mean clinical colitis score), as shown by previous papers. CT352 administration ameliorated colitis at both doses and with a dose- response when considering the colitis clinical score. qRTPCR analysis of inflammatory cytokines and chemokines expression
[1308] The colonic expression of many cytokines and chemokines involved in the inflammatory process was analyzed by qRTPCR. We observed an increased expression of IL6, M IP1 a and MI P2 in the vehicle group compared to the untreated group while CT352 treatment decreased their expression in a dose-dependent manner (Figure 102). Also IL17 expression was increased in the vehicle group and CT352 had no effect on reducing its expression. On the other hand the colonic levels of other inflammation-related mRNAs was affected neither by DSS alone nor by CT352 (IL1 b, TNFa, CCL2, COX2, TGFb). We also analyzed IFNg expression that was expected to increase after DSS treatment, but instead, it was significantly decreased in vehicle group. Finally, IL10 expression was similar between untreated and vehicle group while it decreased in CT352 treated mice, maybe because a diminished inflammation could mediate a diminished expression of IL10. In many cases the high values obtained in the CsA group are due to one/two outlier mice that displayed an exacerbated response to the DSS treatment. These results suggest that while DSS treatment alone resulted in an inflammatory response to the colon, this inflammatory response was counteracted by the administration of CT352 or CsA at the same time.
Histological analysis of colon samples
[1309] At sacrifice, colons were excised and prepared for subsequent histological analysis. A blinded pathologist evaluated the degree of inflammatory infiltrate, epithelial damage and damage extension and attributed to each sample a colitis clinical score, reported on Figure 103a. Control mouse colon sections showed intact epithelium, well defined crypts, no infiltrate and no ulcers or erosions and the mean
score was 0 (Figure 103a and 103b). DSS-treated mice showed severe inflammation throughout the mucosa, wide ulcers, shortening or complete loss of crypts, and an overall damage involving more than 75% of the total colon, being the distal colon the more involved part (Figure 103b) . This lead to a higher score in the majority of mice with the exception of two mice showing an almost intact and not infiltrated mucosa (Figure 103a). In CT352-treated mice, both infiltrate and epithelial alterations (ulcers, crypts loss, ...) were reduced, there was not a single damaged area involving the whole distal colon but from one to three limited damaged areas were detectable in the distal colon, all with the same damage degree. CT300 group displayed a milder histological colitis than the CT100 group (Figures 103a and 103b). Five out of seven CsA-treated mice showed no evident colitis features in their colon, two mice (the same where cytokines and chemokines expression were increased) showed wide epithelial damage and massive inflammatory cells infiltration in the distal part of their colons (Figure 103a and 103b) .
Conclusions
[1310] These data demonstrate that CT352 administered at the same time with DSS reduced the colitis degree in this acute colitis model, with results similar and sometimes better that CsA, a treatment already known to be efficacious against DSS- induced colitis. The results obtained so far indicate that CT352 treatment could efficaciously counteract the DSS-induced colitis, leading to a reduced mucosa damage and to an almost completely absent manifestation of clinical symptoms such as weight loss, bleeding, and diarrhea.
Example 32: Efficacy Study of SNA-125 and SNA-352 in the Treatment of
Oxazolone-lnduced Colitis
Aim of Study
[1311] The goal of this study was to characterize the efficacy of SNA-125 and SNA-352 delivered by oral and intracecal routes for the treatment of colitis with the use of an oxazolone-challenged mouse model of colitis. For comparison, oxazolone- challenged mice were also treated with Tofacitinib and Prednisolone by oral and intracecal routes.
Study Design
[1312] Two hours following the Day 0 AM Treatment dose, colitis was induced in 1 14 male BALB/C mice by intrarectal administration of 100 μΙ_ of 2%
Oxazolone (OXZ) under isoflurane anesthesia on day 0. One additional group of eight animals served as no-disease controls (Group 1). Animals were dosed with test article twice daily (BID) via oral gavage (PO) or intracecal (IC) as indicated in Table 51 . All animals were weighed daily and assessed visually for the presence of diarrhea and/or bloody stool at the time of dosing. Mice had video endoscopy on Days 2 & 4 to assess colitis severity. Additionally, stool consistency was scored during endoscopy. Following endoscopy on day 4, all animals from each treatment group were sacrificed and blood collected. Following euthanasia, the colon was excised, rinsed, measured, weighed, and then trimmed to 6.0 cm in length and divided into 2 pieces as outlined in Figure 104A; the most distal 5.0 cm section was swiss rolled and placed in formalin for subsequent histological evaluation. The details of the study design are shown in Table 51 .
TABLE 51 - Study Design

Disease Induction
[1313] Two hours following the Day 0 AM treatments, colitis was induced in 1 14 male BALB/C mice by intrarectal administration of 100 μΙ_ of 2% OXZ under isoflurane anesthesia on day 0. Mice were maintained in vertical position for 1 minute after intra-rectal administration to ensure the complete distribution of OXZ/vehicle throughout the colon.
Cecal/Colon Cannulation
[1314] All animals were allowed a minimum of 7 days to recover from surgery. Animals were placed under isoflurane anesthesia, and the cecum was exposed via a mid-line incision in the abdomen. A small point incision was made in the distal cecum through which 1 -2 cm of the cannula (Norfolk Medical MMP-3S mouseport with a 3 french silicone catheter and 2 moveable beads for securing suture) was inserted and directed into the proximal colon.
Dosing
[1315] Animals were dosed with test article twice daily (BID) via oral gavage (PO) or intracecal (IC) as indicated in Table 51 .
Endoscopy
[1316] Each mouse underwent video endoscopy on Days 2 & 4 using a small animal endoscope (Karl Storz Endoskope, Germany) , under isoflurane anesthesia. During each endoscopic procedure still images as well as video were recorded to evaluate the extent of colitis and the response to treatment. Additionally, an image was captured from each animal at the most severe region of disease identified during endoscopy. Colitis severity was scored using a 0-4 scale as defined in Table 52. Additionally, stool consistency was scored during endoscopy using the parameters defined in Table 53.
TABLE 52 - Endoscopy Colitis Severity Scoring Criteria

4 Ulcerations and bleeding
TABLE 53 - Stool Consistency Scoring Criteria
Score Description
0 Normal, well-formed pellet
1 Loose stool, soft, staying in shape
2 Loose stool, abnormal form with excess moisture
3 Watery or diarrhea
4 Bloody diarrhea
Sample Collection
[1317] Peripheral blood and colon tissue were collected at sacrifice on day 4 as follows. Blood was collected via cardiac puncture into KiEDTA-coated tubes and centrifuged at 4000x g for 10 minutes. Plasma was collected, flash frozen, and stored at -80°C. The colon was excised, rinsed, measured, weighed, and then trimmed to 6.0 cm in length and divided into 2 pieces; the most distal 5.0 cm section was swiss rolled and placed in formalin for subsequent histological evaluation (see Figure 104A). The proximal 1 .0 cm portion was weighed, snap frozen, and stored at -80°C.
Histopathology
[1318] Each colon sample was rolled into a swiss roll. Tissues were embedded in paraffin and sectioned at approximately 5 microns. One slide for each colon was stained with hematoxylin and eosin and examined by a board-certified veterinary pathologist. The pathologist was blinded to the treatment that each group received at the time of assessment. Each slide containing one rolled colon was split into four approximately equal quarters. Each quarter was evaluated and scored for inflammation, edema, and mucosal necrosis, according to the scoring criteria listed below in Tables 54 to 56. As depicted in Figure 104B, with the slide label to the left, quarters were evaluated starting at the top left and moving clockwise.
TABLE 54 -Inflammation Histopathologic Scoring Criteria
 Mild change: Larger focal aggregates, multifocal small aggregates, or diffuse mild
Mild change: Larger focal aggregates, multifocal small aggregates, or diffuse mild
2
inflammation
Moderate change: Multifocal aggregates sometimes coalescing with one another
3
or moderate diffuse change
4 Severe change: Marked diffuse inflammation
TABLE 55 - Edema Histopathologic Scoring Criteria
Score Description
0 None present
1 Minimal change: Focal edema or minimal diffuse edema
2 Mild change: Larger focal areas of edema or diffuse mild edema
Moderate change: Multifocal areas of edema coalescing with one another or
3
moderate diffuse edema
4 Severe change: Marked diffuse edema
TABLE 56 - Mucosal Necrosis/Loss Histopathologic Scoring Criteria
Score Description
0 None present
1 Minimal change: Focal epithelial necrosis
2 Mild change: Larger focal areas of necrosis or multifocal areas of necrosis
3 Moderate change: Multifocal necrosis coalescing into larger areas
4 Severe change: Areas of erosion through the mucosa to the submucosa
Multiplex Analysis of Colon Tissue Homogenate Samples
[1319] Colon tissue homogenate supernatants were analyzed for protein levels of a panel of mouse inflammatory mediators: IFN-γ, IL-10, IL-6, & TNF-a using a multiplex system (MAGPIX, EMD Millipore).
Results
In-life Observations
[1320] Figure 105 depicts the effect of oral and intracecal administration of SNA-125, SNA-352, tofacitinib, and prednisolone on the body weight of animals challenged with oxazolone. Figure 106 depicts this data according to last observation carried forward analysis. A reduction of the body weight of animals treated with
oxazolone was observed. A trend towards a decrease of the body weight loss was observed in the oral SNA-125 administration group.
Endoscopy Results
[1321] Figures 107 and 109 depict the effect of oral and intracecal administration of SNA-125, SNA-352, tofacitinib, and prednisolone on the Day 2 and 4 endoscopy scores, respectively, of animals challenged with oxazolone. At Day 2, oral and intracecal Tofacitinib administration yielded a 10-15% improvement in endoscopy scores. Surprisingly, orally administered SNA-125 demonstrated a 22% reduction of the Day 2 endoscopy score. At Day 4, oral and intracecal Tofacitinib adminstration yielded a 10-15% improvement in endoscopy scores. Surprisingly, orally administered SNA-125 yielded a 15% reduction of the endoscopy score. Figures 187 to 192 depict representative Day 2 and Day 4 endoscopy images.
[1322] Figures 108 and 1 10 depict the effect of oral and intracecal administration of SNA-125, SNA-352, tofacitinib, and prednisolone on the Day 2 and 4 stool consistency scores, respectively, of animals challenged with oxazolone. Oral SNA- 125 showed a significant reduction (65%) in the stool consistency score on Day 2.
Disease Activity Index
[1323] Figure 1 1 1 depicts the effect of oral and intracecal administration of SNA-125, SNA-352, tofacitinib, and prednisolone on the disease activity index (DAI) score of animals at Days 2 and 4 following challenge with oxazolone.
Colon Weight/Length Ratio
[1324] Figure 1 12 depicts the effect of oral and intracecal administration of SNA-125, SNA-352, tofacitinib, and prednisolone on the colon weight/length ratio of animals challenged with oxazolone.
Histopathology Scoring Results
[1325] Oxazolone produced mild to moderate colitis characterized by multifocal inflammation, edema, and necrosis.
[1326] Figures 1 1 3-1 15 depict the histopathology scoring results for inflammation, edema, and mucosal necrosis/loss, respectively, while Figure 1 16 depicts the summation of these scores. Note that control animals not given oxazolone were essentially normal and were not included in the statistical analysis.
Inflammation Scoring
[1327] As shown in Figure 1 13, untreated control animals that did not receive oxazolone had minimal scattered background inflammation, while all other animals were given oxazolone and had varying degrees of inflammation. Inflammation tended to be mild to moderate with some regions more severely affected than others. When including all groups in the analysis, the treatment effect approached significance (one-way ANOVA, p = 0.0656). When oral and intracecal groups were analyzed independently treatment did significantly influence inflammation for oral groups (one-way ANOVA, p = 0.0199) but not for intracecal groups (one-way ANOVA, p = 0.5970).
[1328] In the oral groups, both SNA-125 and SNA-352 tended to reduce inflammation compared to vehicle, and this improvement was more noticeable than for either prednisolone or tofacitinib.
Edema Scoring
[1329] As shown in Figure 1 14, untreated control animals that did not receive oxazolone had minimal, random edema, while all other animals were given oxazolone and had varying degrees of edema that was generally associated with inflammation. Edema tended to be mild to moderate with some regions more severely affected than others. When including all groups in the analysis, treatment did not have a significant effect on edema (one-way ANOVA, p = 0.2845) . When oral and intracecal groups were analyzed independently, the oral groups approached significance (one-way ANOVA, p = 0.0855) but not the intracecal groups (one-way ANOVA, p = 0.4033). In the oral groups, both SNA-125 and SNA-352 tended to reduce edema compared to vehicle and this improvement was more noticeable than for either prednisolone or tofacitinib.
Mucosal Necrosis Results
[1330] As depicted in Figure 1 15, untreated control animals that did not receive oxazolone did not have any mucosal necrosis, while all other animals were given oxazolone and had varying degrees of multifocal mucosal erosion and necrosis. Necrosis tended to be regional with some areas more severely affected than others and some animals more severely affected than others.
[1331] In the oral groups, both SNA-125 and SNA-352 tended to reduce mucosal necrosis compared to vehicle and this improvement was more noticeable than for either prednisolone or tofacitinib.
Sum Score Results
[1332] As shown in Figure 1 16, summed histopathology scores (Inflammation + Edema + Mucosal Necrosis) were moderate in all animals administered oxazolone.
[1333] In the orally treated groups, both SNA-125 and SNA-352 tended to reduce the sum score compared to vehicle, and this improvement was more noticeable than for either prednisolone or tofacitinib. For inflammation, this treatment effect was statistically significant while for edema and the sum score, this effect approached significance
[1334] Overall, intracecal treatment was found to be less effective than an oral route of administration.
Analysis of Colon Histopathology Micrographs
[1335] Sections of colon were often thickened by inflammation and edema that variably extended into the lamina propria, submucosa and muscular wall. The inflammation was pyogranulomatous - composed of a mixture of neutrophils, macrophages, lymphocytes, and plasma cells. Mucosal necrosis was also variably present and characterized by partial or complete loss of the surface epithelium with erosion to underlying lamina propria or submucosa. There was multifocal peritonitis suggesting that there was multifocal full thickness erosion. Due to the multifocal distribution of these changes, the inflammation, edema, and mucosal necrosis were variable along the swiss rolled section. Representative photomicrographs are shown in Figures 1 17-1 19.
Control Animals
[1336] As seen in Figure 1 17, control animals in this study had basically normal colons without significant inflammation, mucosal necrosis, or edema.
Orally-treated Animals
[1337] Administration of oxazolone was associated with the development of multifocal mucosal ulceration, inflammation, and edema which was seen in all groups to varying extent. Vehicle-treated animals had moderate inflammation (unfilled arrows) with edema (filled arrows) and multifocal ulceration (bracket).
[1338] Treatment with prednisolone or tofacitinib did not demonstrably reduce colitis. Animals still had a diffuse increase in background inflammation with multifocal pockets of more severe inflammation and multifocal mucosal ulceration (Figure 1 18).
[1339] Treatment with both SNA-125 and SNA-352 orally tended to reduce the severity of colitis. These animals tended to have a more mild increase in background inflammation without significant ulceration.
Intracecal-treated Animals
[1340] The intracecal vehicle group had relatively mild colitis compared to other groups. While there were some areas in some animals with inflammation, edema, and mild necrosis (inset), most of the colons had mild inflammation with mild edema and minimal to no necrosis (Figure 1 19). Animals treated with intracecal tofacitinib had diffuse inflammation with pockets of more severe inflammation and mucosal necrosis. Animals treated intracecal with SNA-125 were divided between mild colitis and severe colitis. Animals treated intracecally with SNA-352 had reduced colitis compared to tofacitinib treated animals, but since the vehicle group had such mild colitis there was no noticeable change compared to vehicle.
Multiplex Analysis of Colon Homogenates
[1341] Figures 120-123 depicts the effect of oral and intracecal administration of SNA-125, SNA-352, tofacitinib, and prednisolone on the levels of IFNv, IL-10, IL-6 and TNFa in colon homogenates. Oral and intracecal SNA-352 administration decreased IFNv. Intracecal SNA-125 significantly increased TNFa as compared to the intracecal vehicle control (with the intracecal control outlier removed). Oral tofacitinib significantly increased IL-10 levels, and a strong trend was also observed for the intracecal Tofacitinib group.
Conclusions
[1342] Oxazolone produced mild to moderate colitis characterized by multifocal inflammation, edema, and necrosis. Changes tended to be multifocal in nature with some areas affected more severely than others. It appeared that the distal colon was more severely affected than the proximal colon. Clear signs of efficacy for both SNA-125 and SNA-352 were observed. Overall, there was a clear trend for both SNA- 125 and SNA-352 to improve colitis in this model. Both compounds showed comparable or even better results than tofacitinib and prednisolone. SNA-125 seems to work differently if adminstered orally or intracecally (intracecal group data affected by animal loss).
[1343] Oral treatment with both SNA-352 and SNA-125 tended to reduce inflammation, edema, and necrosis compared to oral treatment with vehicle, tofacitinib, or prednisolone. For inflammation, this treatment effect was statistically significant while
for edema and the sum score, this effect approached significance. In contrast, treatment with tofacitinib and prednisolone orally were ineffective at treating oxazolone-induced colitis in this study.
[1344] Intracecal treatment was less effective. Treatment with intracecal SNA-352 may have mildly reduced colitis compared to tofacitinib treatment and SNA- 125; however any change was mild and not statistically significant.
Example 33: Prophetic Study - DSS-induced colitis chronic model
[1345] Colitis will be induced in C57BI/6 mice by exposure to 2% DSS in drinking water (3 cycles). The planned treatment groups and study schedule are depicted in Table 57. In-life observations (body weight, morbidity, presence of diarrhea and/or bloody stool) will be conducted at the indicated times. The disease activity index DAI (weigh loss, diarrhea and blood stool) will be determined for all study groups. Endoscopy will be performed on days 10, 21 & 34 and colitis severity will be scored using a 0-4 scale. In addition, colon histopathology (inflammation, edema & mucosal necrosis scoring) will be undertaken. Finally, multiplex analysis on colon homogenates will be performed for the following cytokines: IFN-g, IL-10, IL-6 and TNFa.
[1346] Colitis will be induced by exposure of mice to 2% DSS in drinking water following a five days on, seven days off cycle for a period of 3 cycles (DSS will be administered on Days 0-4, 12-16, and 24-28) . One additional group of eight animals will serve as no-disease controls (Group 1 ). Animals in Groups 2-4 & 6- 1 1 will be dosed with vehicle or test article once daily (QD) via oral gavage (PO) as indicated in Table 57. Animals in Group 5 will be dosed by intraperitoneal injection (IP) every third day (Q3D) Days 0-30. All animals will be weighed daily and assessed visually for the presence of diarrhea and/or bloody stool at the time of dosing. The disease activity index will be scored daily, in addition to endoscopy DAI . Mice will undergo video endoscopy on Days 10, 21 , & 34 to assess colitis severity. Images will be captured from each animal at the most severe region of disease identified during endoscopy. Additionally, stool consistency will be scored during endoscopy. Following endoscopy on day 34, all animals from each treatment group will be sacrificed and blood collected.
[1347] Following euthanasia, the colon will be excised, rinsed, measured, weighed, and then trimmed to 6.0 cm in length and divided into 2 pieces. The colon will be excised, rinsed, measured, weighed, and then trimmed to 6.0 cm in length and divided into 2 pieces; the most distal 5.0 cm section will be swiss rolled and placed in
formalin for subsequent histological evaluation. The proximal 1 .0 cm portion will be weighed and snap frozen in liquid nitrogen. Additionally, blood will be collected and prepared for plasma using K2EDTA as the anti-coagulant. The details of the study design are shown in Table 57.
[1348] Colitis will be induced by exposure to 2% DSS in drinking water following a five days on, seven days off cycle for a period of 3 cycles (DSS will be administered on days 0-4, 12-16, and 24-28). For each five day dosing period, a fresh DSS/water solution will be prepared and used for the first three days. A fresh DSS/water solution will be prepared and used for the final two days of the five day dosing period. The DSS/water solution may be made more often if necessary. Animals in Groups 2-4 & 6-1 1 will be dosed with vehicle or test article once daily (QD) via oral gavage (PO) as indicated in Table 57. Animals in Group 5 will be dosed by intraperitoneal injection (IP) every third day (Q3D) Days 0-30.
[1349] Animals will be observed daily (weight, morbidity, survival, presence of diarrhea and/or bloody stool) in order to assess possible differences among treatment groups and/or possible toxicity resulting from the treatments. Animals will be monitored on a daily basis and those exhibiting weight loss greater than 30% will be euthanized, and will not have samples collected.
[1350] Each mouse will undergo video endoscopy on Days 10, 21 , & 34 using a small animal endoscope under isoflurane anesthesia. During each endoscopic procedure still images as well as video will be recorded to evaluate the extent of colitis and the response to treatment. Additionally, we will attempt to capture an image from each animal at the most severe region of disease identified during endoscopy. Colitis severity will be scored using a 0-4 scale (0=normal; 1 = loss of vascularity; 2= loss of vascularity and friability; 3= friability and erosions; 4=ulcerations and bleeding). Additionally, stool consistency will be scored during endoscopy.
[1351] The Disease Activity Index (DAI) of each mouse will be scored daily. These measurements will be combined to generate a daily DAI score. Additionally, DAI will also be calculated using the weight loss criteria, endoscopy colitis score, and endoscopy stool consistency score. All animals will be euthanized after endoscopy on day 34 and peripheral blood and colon tissue will be collected.
[1352] The proximal and distal colon samples will be trimmed into 6-8 equally spaced transverse sections. Tissues will be embedded in paraffin and sectioned at approximately 5 microns. One slide for each animal, containing the distal and proximal colon samples (with all transverse sections per slide), will be stained with hematoxylin and eosin. Sections of colon will be scored for inflammation, edema and mucosal necrosis. Each of the transverse sections is scored for these parameters and the mean
is reported for each animal for each parameter. Additionally, the mean sum score is calculated as the sum of inflammation, edema, and mucosal necrosis.
[1353] Colon tissue homogenate supernatants will be analyzed for protein levels of a panel of mouse inflammatory mediators: IFN-γ, IL-10, IL-6, & TNF-a using a multiplex system.
Table 57: Study

Example 34: TNBS-induced Colitis Mouse Model
[1354] Intra-rectal administration of 4 mg of TNBS in C57BI/6 mice will provide a model of colitis in mice. Both oral (PO) and intracecal (IC) administration of the vehicle and test agents are planned. Contemplated treatment groups are SNA-125 (400mg/kg), SNA-352 (400 mg/kg), vehicle (water), prednisolone (2 mg/kg PO; positive control), and tofacitinib (15 mg/kg PO and 1 mg/kg IC). The vehicle, SNA-125, SNA-352 and tofacitinib will be administered BID Days 0-4 while prednisolone will be administered QD Days 0-4. In-life observations (body weight, morbidity, presence of diarrhea and/or
bloody stool) will be conducted. The disease activity index DAI (weight loss, diarrhea and blood stool) will be determined for all study groups. Endoscopy will be performed on days 2 and 4, and colitis severity will be scored using a 0-4 scale. In addition, colon histopathology (inflammation, edema & mucosal necrosis scoring) will be undertaken. Finally, multiplex analysis on colon homogenates will be performed for the following cytokines: IFN-g, IL-10, IL-12(p40) , IL12(p70), IL- 13, IL-1 b, IL-2, IL-6 and TNFa.
Example 35: Prophetic Study - DSS-induced Colitis Acute Model
[1355] Colitis will be induced by administration of 3% DSS on Days 0-5 in C57BI/6 male mice. Both oral (PO) and intracecal (IC) administration of the vehicle and test agents are planned. Contemplated treatment groups are SNA- 125 (400 mg/kg) , SNA-352 (400 mg/kg) , vehicle (water), Anti-p40 (10 mg/kg IP; positive control) , and tofacitinib (15mg/kg PO and 1 mg/kg IC). The vehicle, SNA- 125, SNA-352 and tofacitinib will be administered BID Days 0-19 while Anti-p40 will be administered Q3D Days 0- 18.
[1356] In-life observations (body weight, morbidity, presence of diarrhea and/or bloody stool) will be conducted. The disease activity index DAI (weight loss, diarrhea and blood stool) will be determined for all study groups. Endoscopy will be performed on days 10, 14 and 19, and colitis severity will be scored using a 0-4 scale. In addition, colon histopathology (inflammation, edema & mucosal necrosis scoring) will be undertaken. Finally, multiplex analysis on colon homogenates will be performed for the following cytokines: IFN-g, IL- 10, IL-12(p40), IL12(p70) , IL-13, IL- 1 b, IL-2, IL-6 and TNFa
Example 36: CT101 Efficacy in a Mouse Model of
Oxazolone-induced Contact Dermatitis
[1357] This study used a mouse model of 4-Ethoxymethylene-2-phenyl-2- oxazolin-5-one (Oxazolone)-induced contact dermatitis to test the efficacy of CT101 .
Methodology
Compounds and Reagents
[1358] Betamethasone 17-valerate 0.1 % (Manx Pharma)
[1359] Propylene Glycol (P4347)
[1360] Transcutol P (Gattefosse, lot. 450931007)
[1361] CT101 was provided as a solid compound that was solved in vehicle (25% Transcutol P and 75% Propylene Glycol for epicutaneous applications)
Experimental outline
[1362] Female BALB/c mice were randomly allocated to experimental groups and allowed to acclimatise for one week. On Day 0, animals were sensitised to oxazolone by epicutaneous application of 50 μΙ_ of a 1 .5% solution in acetone: olive oil (4: 1) to the clipped abdomen. On Days 7, 10 and 13, animals were challenged by epicutaneous application of 10 μΙ_ per surface of a 1 .0% oxazolone solution, or vehicle, to both surfaces of both ears. Oxazolone and treatments were given according to the administration schedule below. Ear swelling was measured 24 hours after each oxazolone challenge using a digital calliper. At termination ears were collected, cut in two halves and stored for optional histopathology and optional tissue cytokine analysis.
Treatment Groups and Dosages
[1363] All Groups are n= 10.
[1364] Vehicle for epicutaneous administrations is 25% Transcutol P, 75% propylene glycol.
[1365] Administration volume for treatment epicutaneous applications was 20 μΙ_ per ear per administration (two administrations per day). Only one ear per animal was treated with the active ingredients (left ear), while the contra-lateral ear was treated with the vehicle each time (right ear).
[1366] The groups of mice were treated as follows:

Bodyweights
[1367] From Day 0, animals were weighed three times a week. Data were graphed (Mean ± SEM) .
Non-specific clinical observations
[1368] From Day 0 until the end of the experiment, animals were checked daily for non-specific clinical signs to include abnormal posture (hunched), abnormal coat condition (piloerection) and abnormal activity levels (reduced or increased activity).
Ear Swelling
[1369] On Day -1 and then twenty-four hours after each oxazolone challenge, ear thickness was measured using digital callipers. Swelling was calculated as the difference in thickness between the pre-challenge thickness (Day - 1 ) and the daily values taken on Day 8, Day 1 1 and Day 14.
Macroscopic Scores
[1370] From Day 7, animals were monitored daily for signs of inflammation to include erythema (redness) , swelling of the ears, dry skin and/or presence of wet lesions. A semi-quantitative system was used in order to allow comparisons between groups.

*These criteria may be followed by a number (1 -4) corresponding to the number of ear surfaces affect
Tissue Cytokine Analysis
[1371] At termination ears were dissected, cut in two halves and one half stored for cytokine analysis. To each half ear, extraction buffer was added and the ears homogenised and centrifuged. After centrifugation, cytokine analysis was performed on the supernatant to determine the concentration of I L- 1 β , IL-4, IL-10, IFN-γ and TNF-a in each sample. Samples were analysed using multiplex xMAP bead technology, which utilises microspheres as a solid support for sandwich immunoassays and determination of numerous cytokines in the same sample. A standard curve of quantified recombinant cytokines was used to convert detected PE fluorescence values to cytokine concentrations (pg/ml).
Results
Bodyweights
[1372] Bodyweight data were analyzed by two-way ANOVA followed by Dunnett's post-test for multiple comparisons between experimental days in the vehicle- treated group then for multiple comparisons between experimental groups. As shown in Figure 124, bodyweights in the vehicle-treated group increased prior to the oxazolone challenge and Day 6 bodyweights were significantly higher than bodyweights recorded on Day 0 (p < 0.001 ) . Oxazolone challenges induced a decrease in bodyweights. Day 1 1 bodyweights were significantly lower than bodyweights recorded on Day 0 (p < 0.05).
[1373] Betamethasone 0.1 % induced a significant decrease of the bodyweights when compared to the vehicle-treated group from Day 4 until the end of the experiment on Day 14 (p < 0.0001 ).
[1374] CT101 administered at 10% did not prevent the oxazolone challenges- induced bodyweight loss. The bodyweights in the CT101_ 10%-treated group was significantly lower than in the vehicle-treated group on Day 6 (p < 0.001 ) , on Day 8 (p < 0.05), Day 1 1 (p < 0.05) and Day 14 (p < 0.01 ) .
[1375] CT101 administered at 20% did not prevent the oxazolone challenges- induced bodyweight loss but did not induce any further bodyweight loss when compared to the vehicle-treated group.
Non-Specific Clinical Observations
[1376] From Day 0 until the end of the experiment, animals did not show any nonspecific clinical signs to include abnormal posture (hunched), abnormal coat condition (piloerection) and abnormal activity levels (reduced or increased activity).
Ear Swelling
[1377] Ear swelling data were analysed by two-way ANOVA followed by Dunnett's post-test for multiple comparisons between experimental days in the vehicle- treated group then for multiple comparisons between experimental groups. As depicted in Figure 125, Oxazolone challenges induced a significant increase of ear swelling in the vehicle-treated group on Day 8, Day 1 1 and Day 14 when compared to Day 0 (p < 0.0001 ).
[1378] CT101 administered at 10% induced a significant reduction of ear swelling when compared to the vehicle-treated group on Day 1 1 (p < 0.01) and Day 14 (p < 0.0001).
[1379] CT101 administered at 20% induced a significant reduction of ear swelling when compared to the vehicle-treated group on Day 14 (p < 0.001).
Macroscopic Scores
[1380] Macroscopic score data were analysed by two-way ANOVA followed by Dunnett's post-test for multiple comparisons between experimental days in the vehicle-treated group then for multiple comparisons between experimental groups. As depicted in Figure 126, Oxazolone induced a significant increase of the macroscopic scores in the vehicle-treated group from Day 7 until the end of the experiment on Day 14 when compared to Day 0 (p < 0.0001).
[1381] Betamethasone 0.1 % induced a significant reduction of the macroscopic scores when compared to the vehicle-treated group from Day 7 until the end of the experiment on Day 14 (p < 0.0001).
[1382] CT101 administered at 10% induced a significant decrease of the macroscopic scores when compared to the vehicle-treated group on Day 13 (p < 0.001) and Day 14 (p < 0.0001).
[1383] CT101 administered at 20% induced a significant reduction of the macroscopic scores when compared to the vehicle-treated group on Day 14 (p < 0.0001).
Tissue Cytokine Analysis
[1384] Contact dermatitis presents an immune mediated reaction known to involve both T helper 1 cell (Th1) and Th2 response. Therefore, a mixture of proinflammatory cytokines is expected to be present in this model.
[1385] Snap frozen ear samples were homogenised and measured by Luminex. Cytokine levels between the left (treated with the active ingredients) and the right ear (treated with the Vehicle) within each experimental group were analysed by two-
way ANOVA followed by Sidak's post-test for multiple comparison. Further, one-way ANOVA was used to compare cytokine levels measured in left ear only (treated with the active ingredients) in different experimental groups and the Vehicle-treated group.
[1386] Levels of IL-1 β and IFN-γ in the left ear were significantly lower after Betamethasone treatment when compared to those measured in the Vehicle group (Figure 127). Similarly, levels of IL-1 β in the actively treated ear were lower in CT101 10% group when compared to the Vehicle group. This indicates that these compounds lower exacerbated levels of pro-inflammatory cytokines in the model.
[1387] Within the groups, Betamethasone induced significant decrease in IL- 1 β and IFN-γ. In contrast, levels IL-4 in the left ear were slightly increased when compared to the right ear. Levels of IL-4 were higher in the right when compared to the left ear within CT101 20% group. This suggests that an active treatment at the site of inflammation was therefore able to reduce some of the cytokine levels in this model.
[1388] Levels of TNF-a were below detection limit and as such were not plotted in the report. It is possible that the model has led to the transient increase in the levels of this cytokine which has passed by the time of termination selected in this study.
Conclusions
[1389] As expected in this model of chronic dermatitis, the following pathological changes were observed following the topical application of oxazolone: ear swelling as soon as Day 7 and increasing until Day 14 and macroscopic scores including erythema. Betamethasone 0.1 % reduced ear swelling and skin erythema. Betamethasone 0.1 % caused bodyweight loss, a known side effect of steroids administered to rodents.
[1390] CT101 administered at 10% and 20% partially prevented the development of pathological changes to the skin (swelling and erythema), inducing a statistically significant reduction of ear swelling and of the macroscopic clinical scores
[1391] Cytokines in the present model mostly were present at detectable levels in ear homogenates, with the exclusion of TNF-a which was present at levels below limit of detection.
[1392] Betamethasone induced decrease in pro-inflammatory cytokines, namely IL-1 β and IFN-γ, while CT101 10% decreased IL-1 β levels when compared to the Vehicle-treated group. Within the groups, Betamethasone induced a decrease in IL- 1 β in the actively treated ear when compared to the vehicle-treated ear, and the same was the case with IL-4 in CT101 20% group. These findings are in keeping with the
clinical measurements of ear swelling and confirm the anti-inflammatory effects of the compounds.
Example 37: Evaluation of CT101 Efficacy in a Mouse Model of
Ovalbumin-induced Atopic Dermatitis
[1393] This study used a mouse model of ovalbumin-induced atopic dermatitis to test the efficacy of CT101.
Methodology
Compounds and Reagents
• Albumin from chicken egg white, Ovalbumin (A5503, Sigma)
• Imject Alum (PN77161 Thermo Scientific Pierce)
• Sodium chloride 0.9% solution, saline (NaCI 0.9%, S8776, Sigma)
• Betamethasone 17-valerate 0.1 % (Manx Pharma)
• Propylene Glycol (P4347)
• Transcutol P (Gattefosse, lot. 450931007)
• CT101 was provided as a solid compound and was diluted in vehicle
(25% Transcutol P and 75% Propylene Glycol for epicutaneous applications)
Experimental outline
[1394] Adult male CD-1 mice were randomly allocated to experimental groups and allowed to acclimatise for one week. On Day -13, animals were administered with 200 μΙ_ of an ovalbumin and Alum emulsion by intra-peritoneal injection under gas (isoflurane) anaesthesia. The emulsion was prepared by mixing one volume of a 40 μg/ml ovalbumin solution in distilled water and one volume of Alum Imject. Prior to Day 0, ear thickness was measured using digital callipers. On Day 0, animals were challenged with 20 μΙ_ of a 0.5 mg/ml solution of ovalbumin in sodium chloride 0.9% (saline) by intradermal injection into the left ear. An equivalent volume of saline was injected into the contralateral ear. One group of animals (Group 1 , Control) received an intradermal injection of saline in each ear. Treatments were administered according to the schedule below. On Day 0, at ¼ hour, ½ hour and one hour after the ovalbumin challenge and on Day 1 , twenty-five and twenty-nine hours after the ovalbumin challenge, ears were observed for clinical signs of skin inflammation to include erythema, scaling and skin
thickening. Ear thickness was measured using digital callipers. Twenty-eight hours after ovalbumin challenge, animals were culled and ears dissected out. One half of each ear was stored at room temperature in tissue fixative until further optional histopathology analysis.
Treatment Groups and Dosages
[1395] All Groups were n=10
[1396] Vehicle for epicutaneous administrations is 25% Transcutol P and 75% Propylene Glycol. Administration volume for epicutaneous applications was 20 μΙ_ per ear. Treatment groups were as follows:

c a enge
Readouts
Clinical Observations
[1397] Animals were weighed at the start of the study (Day -13), once per week and on Day 0. All animals were observed for signs of ill health daily throughout the study. Bodyweight data were graphed (Mean ± SEM for each experimental group).
Skin Inflammation
[1398] Prior to the intra-dermal challenge, one hour after the challenge (Day
0) and one hour and four hours after epicutaneous applications (Day 1), ears were observed for signs of skin inflammation to include erythema, scaling and skin thickening. Each criterion was scored on a point-scale where (0) is absent, (1) is mild, (2) is moderate and (3) is severe. A total score was calculated for each ear by adding the scores from each individual criterion. Data were graphed (Mean ± SEM for each experimental group).
Ear Swelling
[1399] Prior to the intra-dermal challenge, ¼ hour, ½ hour and one hour after the challenge (Day 0) and one hour and four hours after epicutaneous applications (Day
1) , ear thickness was measured under gas (isoflurane) anaesthesia using digital callipers. Data were graphed (Mean ± SEM for each experimental group).
Histopathology
[1400] At termination ears were dissected, cut in two halves and one half stored in tissue fixative until further analysis. Samples were embedded in paraffin wax, sectioned, mounted on a cooled water bath to prevent splitting and stained with haematoxylin and eosin (H&E). Optional: Epidermal and dermal layer thickening may be quantified by taking five representative micrometre measurements for each layer using the linear measurement tool in the Leica Application Suite programme. Skin layer measurements are not relevant to this model (but to our models of skin scleroderma) and were not performed. Optional: cellular infiltrate may be characterised and or quantified by counting inflammatory cells on three to five H&E-stained sections. Cellular characterisation and quantification were performed.
[1401] Sections were scored for epidermal hyperplasia (0: absent, 1 : present), dermal oedema (0: absent, 1 : mild, 2: moderate and 3: severe) and dermal inflammation (0: less than 5 inflammatory cells, 1 : 5-10 cells, 2: 10-25 cells and 3: more than 25 cells). A total score was calculated by adding the individual parameter scores for a maximum possible score of 7. Sections were evaluated in blinded fashion.
Results
Non-Specific Clinical Observations
[1402] Animals did not show any non-specific clinical signs such as piloerection, hunched posture, altered breathing rate or altered mobility for the duration of the study.
Bodyweights
[1403] Bodyweight data were analyzed by two-way ANOVA followed by Dunnett's post-test for multiple comparisons between experimental groups. Betamethasone 17-valerate administered twice daily by topical application onto the ears from Day -5 induced a significant reduction of the bodyweight measured on Day -3 (p < 0.05) and Day 0 (p < 0.01) when compared to the vehicle-treated group (Figure 128). This is a known side effect of corticoids. CT101 administered at 5%, 10% or 20% twice daily by topical application onto the ears from Day -5 did not induce any bodyweight reduction.
Ear Swelling
[1404] Measurement of ear swelling is the primary readout in this model.
[1405] Ear swelling data, expressed as the difference in millimetres between the left (ovalbumin-challenged) and right (saline-injected) ears, were analysed by two- way ANOVA followed by Dunnett's post-test for multiple comparisons between experimental groups.
[1406] Ovalbumin induced a significant increase of ear thickness in the vehicle-treated group fifteen minutes (p < 0.01), twenty-five hours (p < 0.05) and twenty- nine hours (p < 0.01) after the challenge when compared to baseline (0 hours) values (Figure 129).
[1407] Ovalbumin induced a significant increase of ear thickness in the vehicle-treated group fifteen minutes (p < 0.01 ), thirty minutes (p < 0.05), twenty-five hours (p < 0.01) and twenty-nine hours (p < 0.001) after the challenge when compared to the Control group. Table 58 below summarizes the ear swelling data.
Table 58: Ear Swelling (Left-Right ear, in millimeters)
Tim* piS:-ci-;ai:engS: (Hows)
25 S:V
0.0< 0.01 0.06 O Or ο.δ¾ ·; o.or 0.07 0 01 0.04 ■ 0.04 · 0.03 δ O ο.-·% 0.04 Ο.02 .± O'.OOT 0 00 .i 0 01 0 0 . 0.0 V 0.03 s r o.or'*-
0.00 ¾ . : ο. ο ο οι 0. O.'O O.H i 0 01 0 O. v O 01: ÷ 0 00
CT !G 5% O.OS s 001 0.3 0.02 0.27 s 0 03 0.:iO 0.0'! 0.1 ; * 000 0.2 : :: 0.02 CHOI 'Ϊ0 0.05 * 0.01 0.08 ± 0.01 O. S i 0 01 O.1O ± 0.CO 0.20 :; 001 0.20 ± 0.02 CTIOl 20% 0.00 i: 0. : 0.15 s Ο Ο'! Ο.ΙΟ ΐ νΟ 0.17 s 0 01 0.20 ·: 0.01 0 01 i 001
^ ο. · · - 0 1 ^o · · · ·
[1408] Ear swelling data recorded at peak disease (corresponding to fifteen minutes after the challenge) were further analysed by one-way ANOVA followed by Dunnett's post-test for multiple comparisons to the vehicle-treated group.
[1409] Ovalbumin challenge induced a significant increase of the ear swelling measured in the vehicle-treated group when compared to the Control group (p < 0.01).
[1410] Betamethasone 0.1 % induced a significant reduction of the ovalbumin-induced ear swelling when compared to the vehicle-treated group (p < 0.001).
[1411] CT101 , administered at 5%, did not reduce the ovalbumin-induced ear swelling (Figure 130).
[1412] CT101 , administered at 10% and 20%, reduced the ovalbumin- induced ear swelling. The reduction was found to be statistically significant when CT101 was administered at 10% (p < 0.01).
Skin inflammation
[1413] Scaling and skin thickening are not expected in this model. Erythema is expected to occur, but tends to be relatively mild compared to that seen in other skin inflammation models, and should be viewed as a secondary readout.
[1414] Erythema was scored on a point-scale where (0) is absent, (1) is mild, (2) is moderate and (3) is severe. Erythema score data were analysed by two-way ANOVA followed by Dunnett's post-test for multiple comparisons between experimental times or between experimental groups.
[1415] The ovalbumin challenge (intra-dermal administration on Day 0) induced a significant increase in erythema scores observed in the vehicle-treated group at all experimental times when compared to the scores observed prior to the challenge.
[1416] Betamethasone 17-valerate induced a reduction of the erythema scores when compared to the vehicle-treated group. The reduction was statistically significant thirty minutes after the challenge (p < 0.01), twenty-five hours after the challenge (p < 0.0001) and twenty-nine hours after the challenge (p < 0.0001).
[1417] CT101 administered at 5% reduced the erythema scores from thirty minutes after the challenge when compared to the vehicle-treated group but the reduction was not statistically significant.
[1418] CT101 administered at 10% reduced the erythema scores from one hour after the challenge when compared to the vehicle-treated group but the reduction was not statistically significant. Fifteen minutes after the challenge, the erythema scores in the CT101_10%-treated group was significantly higher than in the vehicle-treated group (p < 0.05).
[1419] CT101 administered at 20% reduced the erythema scores from thirty minutes after the challenge when compared to the vehicle-treated group and the reduction was statistically significant twenty-nine hours after the challenge (p < 0.05).
[1420] Erythema scores were occasionally observed on the right (saline- injected) ears, especially on Day 1 , twenty-five and twenty-nine hours after the challenge.
[1421] Figure 131 and Figure 132 depict erythema scores (challenged ears) at different times post-challenge.
Histopathology
[1422] The pathological changes were as expected in this model of atopic dermatitis: epidermal hyperplasia, dermal oedema and dermal inflammation. The inflammation is dominated by eosinophils and also variably includes neutrophils, macrophages and mast cells. All right ears and Group 1 left ears appeared essentially normal. The remaining groups all showed pathological change to the left ears, which was maximum in Group 3 and then progressively less in Group 2, Groups 4 and 5, and then Group 6. Group 6 also has the fewest animals affected by left ear pathology (n = 7). Right and left ear scores were analysed by two-way ANOVA followed by Tukey's post- test for multiple comparisons between experimental groups. There was no significant difference between the right and left ears in the Control group. There was a significant increase of the histopathology scores for the left ears when compared to the right ears in all other experimental groups (p < 0.0001 except CT101_20% where p < 0.01). Right ear scores were analysed by Kruskal-Wallis test for non-parametric data followed by Dunn's post-test for multiple comparisons between experimental groups. There was no significant difference between experimental groups (Table 59).
Table 59: Histopathology Scores, Right ears
C rcs To al sco e
Cm A -:- Π 0 0 0 0.1 ± 0,1 :i" Λ
B HM 0.1% 0.0 ± 0. G.O 0.0 · ·*ΐ , >*¾ Γ; 0.0 ± .0
\ h ·?
CT1 101 s% i
X -·" fi 0 :
w . ϊ.ΐ χ ΐ..ν w

CT1 m i fj fj
X 1 ft 1
[1423] Left ear scores were analysed by Kruskal-Wallis test for non- parametric data followed by Dunn's post-test for multiple comparisons between experimental groups.
[1424] The ovalbumin challenge induced a significant increase of the epidermal hyperplasia scores (p < 0.01), the dermal oedema scores (p < 0.01), the dermal inflammation scores (p < 0.01) and the total histopathology scores (p < 0.001) in the vehicle-treated group when compared to the control group (Figure 133).
[1425] Betamethasone 0.1 % did not significantly reduce the histopathology scores.
[1426] CT101 applied at 5%, 10% or 20% induced a non-significant reduction of the dermal oedema but did not significantly reduce the epidermal hyperplasia, the dermal inflammation or the total histopathology scores.
[1427] Representative pictures in Figure 134 show, from left to right and from top to bottom: Mouse #1 .5, normal left ear (score 0) and normal right ear (score 0); Mouse #2.5 left ear with epidermal hyperplasia and inflammatory infiltration next to the auricular cartilage (score 6) and normal right ear (score 0); Mouse #3.8 left ear with epidermal hyperplasia, dermal oedema and marked eosinophilic infiltration of the dermis next to the auricular cartilage (score 6) and normal right ear (score 0).
[1428] Representative pictures in Figure 135 show, from left to right and from top to bottom: Mouse #4.9 left ear with epidermal hyperplasia and eosinophilic infiltration of the dermis close to the auricular cartilage (score 4) and normal right ear (score 0); Mouse #5.1 left ear with epidermal hyperplasia and mild eosinophilic infiltration of the dermis (score 3) and normal right ear with focal neutrophilic inflammation (score 1); Mouse #6.1 left ear with eosinophilic infiltration (score 1) and normal right ear (score 0).
CONCLUSIONS
[1429] Ovalbumin challenge induced erythema and ear swelling as expected in this mouse model of atopic dermatitis. Maximum response was seen fifteen minutes after the ovalbumin challenge. Ovalbumin also induced histopathological change: epidermal hyperplasia, dermal oedema and dermal inflammation, as expected in this model.
[1430] Betamethasone 0.1 % applied topically reduced the ovalbumin-induced erythema and swelling but did not reduce the histopathology changes.
[1431] CT101 administered at 5% , 10% or 20% did not reduce the erythema scores at peak disease, but the 20% dose elicited a statistically significant erythema reduction at 29 h after the ovalbumin challenge.
[1432] CT101 administered at 10% or 20% reduced the ear swelling at peak disease. The reduction was statistically significant when CT101 was administered at 10%.
[1433] CT101 administered at 5% , 10% or 20% reduced the dermal oedema but did not significantly reduce the total histopathology scores.
[1434] CT101 , administered at 20%, reduced the incidence of histopathological change.
Example 38: Evaluation of CT103 Efficacy in a mouse model of imiquimod-induced psoriasis
[1435] This study used a mouse model of imiquimod-induced psoriasis to test the efficacy of CT103.
Methodology
Compounds and Reagents
• Imiquimod 5% w/w (Aldara)
• Betamethasone 17-valerate 0.1 % (Manx Pharma)
• Propylene Glycol (P4347)
• Transcutol P (Gattefosse, lot. 450931007)
• CT103 was provided as a solid compound to be solved in vehicle (25% Transcutol P and 75% Propylene Glycol for epicutaneous applications)
Experimental outline
[1436] Adult male BALB/c mice were randomly allocated to experimental groups and allowed to acclimatise for one week. On Day 0, animals were sensitised by topical application of 62.5 mg of imiquimod cream 5% weight/weight (Aldara™, 3M) , corresponding to ca. 3 mg of active ingredient, to their clipped back under gas (isoflurane) anaesthesia. The procedure was repeated for ten consecutive days. Treatments were given according to the schedule below with non-anaesthetised but restrained animals.
[1437] From Day 0 until the end of the experiment on Day 10, animals were monitored daily for non-specific clinical signs to include abnormal posture, coat condition and/or activity level. From Day 0 until Day 10, animals were scored daily for signs of skin inflammation to include erythema, scaling and thickening of the skin. On Day 10, animals were culled and skin biopsies performed. One biopsy per animal was stored at ambient temperature in tissue fixative until further optional analysis by histopathology. One biopsy per animal was snap frozen and stored at - 80°C until further optional analysis of tissue cytokines.
Treatment Groups and Dosages
[1438] All Groups were n= 12. Vehicle for epicutaneous administrations was 25% Transcutol P, 75% propylene glycol. Administration volume for treatment epicutaneous applications was 100 μΙ_ per mouse back.

n/a: not applicable, EC: epicutaneous application BID: twice daily, SID: once daily, epicutaneous administration on the clipped skin of the back. Clobetasol propionate was administered in the morning only. * Two hours before and four hours after the IMQ challenge.
Readouts
Bodyweights
[1439] From Day 0, animals were weighed three times a week. Data were graphed (Mean ± SEM).
Non-specific clinical observations
[1440] From Day 0 until the end of the experiment, animals were checked daily for non-specific clinical signs to include abnormal posture (hunched), abnormal coat condition (piloerection) and abnormal activity levels (reduced or increased activity).
Clinical Observations
[1441] From Day 0 until Day 10, animals were scored daily for clinical signs of skin inflammation using the following scoring system: erythema, scaling and skin thickening were each scored on a four point-scale where (0) is None, (1) is Slight, (2) is Moderate, (3) is Marked and (4) is severe. The cumulative score is a measure of the severity of the back skin inflammation. Scores for each criterion and cumulative scores were provided along with the standard error of the mean (SEM).
Histopathology
[1442] At the end of the experiment, skin biopsies were performed and one sample per animal was stored in tissue fixative. Samples were processed, sectioned and stained with Haematoxylin & Eosin in order to study tissue morphology, pathological changes and inflammation. Samples were scored for acanthosis, parakeratosis, presence of pustules and dermal inflammation using an established semi-quantitative scoring system. Acanthosis: the thickest and best orientated area of epidermis was examined and the number of epidermal layers at that level recorded. Parakeratosis: the thickest and best orientated area of epidermis was examined and the absence (score 0) or presence (score 1) of keratinisation was recorded. Presence of pustules: a number of pustules per millimeter length of skin was calculated. Additionally, dermal inflammation was scored as: (0) No inflammation, (1) light scattering of dermal inflammatory cells not involving epidermis or adnexal structures, (2) moderate infiltration of dermal inflammatory cells with only occasional infiltration of epidermis or adnexal structures and (3) marked infiltration of dermal inflammatory cells with infiltration of epidermis and adnexal structures and focal disruption of these structures.
Tissue cytokine analysis
[1443] At the end of the experiment, skin biopsies were performed and one sample per animal was stored at -80°C. To each skin biopsy, extraction buffer was added and the tissue was homogenised and centrifuged. After centrifugation, cytokine analysis was performed on the supernatant to determine the concentration of IL-17A, IL- 22 and IL-23 in all samples. Samples were analysed using multiplex xMAP bead technology, which utilises microspheres as a solid support for sandwich immunoassays and determination of numerous cytokines in the same sample. A standard curve of quantified recombinant cytokines and chemokines was used to convert detected PE fluorescence values to cytokine concentrations (pg/ml).
Results
Bodyweights
[1444] Bodyweights, expressed as percentages of the initial (Day 0) bodyweights were analysed by two-way ANOVA followed by Dunnett's post-test for multiple comparisons between experimental groups. Results are presented in Figure 136.
[1445] Imiquimod application induced a significant decrease of the bodyweights in the vehicle-treated group on Day 3 (p < 0.0001), Day 6 (p < 0.001), Day 8 (p < 0.0001) and Day 10 (p < 0.0001) when compared to Day 0.
[1446] Clobetasol propionate induced a significant decrease of the bodyweights when compared to the vehicle-treated group on Day 3 until the end of the experiment on Day 10 (p < 0.05 on Day 3 then p < 0.0001).
[1447] Bodyweights in the CT103_5%-treated group were significantly higher than in the vehicle-treated group on Day 8 (p < 0.05).
[1448] Bodyweights in the CT103_10% group did not differ from the bodyweights measured in the vehicle-treated group.
[1449] Bodyweights in the CT103_20%-treated group were significantly higher than in the vehicle-treated group from Day 3 until the end of the experiment on Day 10 (p < 0.001 then p < 0.0001).
Non-Specific Clinical Observations
[1450] From Day 0 until the end of the experiment, animals did not show any non- specific clinical signs to include abnormal posture (hunched), abnormal coat condition (piloerection) and abnormal activity levels (reduced or increased activity).
Clinical Observations
[1451] Erythema scores, scaling scores, thickness scores and total skin scores were analyzed by two-way ANOVA followed by Dunnett's post-test for multiple comparisons between experimental groups.
Erythema Scores
[1452] As shown in Figure 137, Imiquimod induced a highly significant increase of the erythema scores from Day 2 until the end of the experiment on Day 10 in the vehicle-treated group when compared to Day 0 (p < 0.0001).
[1453] Clobetasol propionate induced a highly significant decrease of the erythema scores when compared to vehicle-treated group from Day 2 until the end of the experiment on Day 10 (p < 0.0001).
[1454] CT103 administered at 5% did not significantly reduce the imiquimod- induced erythema when compared to the vehicle-treated group.
[1455] CT103 administered at 10% did not significantly reduce the imiquimod- induced erythema when compared to the vehicle-treated group.
[1456] CT103 administered at 20% significantly reduced the imiquimod- induced erythema scores when compared to the vehicle-treated group from Day 5 until the end of the experiment on Day 10 (p < 0.01 on Day 5, p < 0.05 on Day 6 and Day 7, p < 0.001 on Day 8 and Day 9, p < 0.0001 on Day 10).
Scaling scores
[1457] As depicted in Figure 138, Imiquimod induced a significant increase in scaling scores from Day 5 until the end of the experiment on Day 10 when compared to Day 0 in the vehicle- treated group (p < 0.05 on Day 5 and Day 7, p < 0.0001 on Day 8, Day 9 and Day 10).
[1458] Clobetasol propionate significantly reduced the scaling scores when compared to the vehicle-treated group from Day 8 until the end of the experiment on Day 10 (p < 0.0001).
[1459] CT103 administered at 5% significantly reduced the imiquimod- induced scaling when compared to the vehicle-treated group on Day 8 (p < 0.05), Day 9 and Day 10 (p < 0.0001).
[1460] CT103 administered at 10% significantly reduced the imiquimod- induced scaling when compared to the vehicle-treated group on Day 8 (p < 0.001), Day 9 (p < 0.001) and Day 10 (p < 0.0001).
[1461] CT103 administered at 20% significantly reduced the imiquimod- induced scaling when compared to the vehicle-treated group on Day 7 (p < 0.05), Day 8, Day 9 and Day 10 (p < 0.0001).
Thickness scores
[1462] As depicted in Figure 139, Imiquimod induced a significant increase in thickness scores from Day 2 until the end of the experiment on Day 10 when compared to Day 0 in the vehicle- treated group (p < 0.0001).
[1463] Clobetasol propionate induced a significant reduction of the thickness scores from Day 2 until the end of the experiment on Day 10 (p < 0.0001).
[1464] CT103 administered at 5% did not reduce the imiquimod-induced skin thickening when compared to the vehicle-treated group.
[1465] CT103 administered at 10% did not reduce the imiquimod-induced skin thickening when compared to the vehicle-treated group.
[1466] CT103 administered at 20% induced a significant reduction of the imiquimod- induced skin thickening when compared to the vehicle-treated group on Day 2 (p < 0.001), Day 3 (p < 0.001) and Day 5 (p < 0.05).
Skin scores
[1467] Skin scores were calculated by adding the erythema scores, the scaling scores and the thickness scores.
[1468] As depicted in Figure 140, Imiquimod induced a significant increase of the total skin scores from Day 2 until the end of the experiment on Day 10 when compared to Day 0 (p < 0.0001).
[1469] Clobetasol propionate significantly reduced the skin scores when compared to the vehicle-treated group from Day 2 until the end of the experiment on Day 10 (p < 0.0001).
[1470] CT103 administered at 5% induced a significant reduction of the skin scores when compared to the vehicle-treated group on Day 10 (p < 0.01).
[1471] CT103 administered at 10% induced a significant reduction of the skin scores when compared to the vehicle-treated group from Day 8 until the end of the experiment on Day 10 (p < 0.05 on Day 8 and Day 9, p < 0.0001 on Day 10).
[1472] CT103 administered at 20% induced a significant reduction of the skin scores when compared to the vehicle-treated group from Day 5 until the end of the experiment on Day 10 (p < 0.001 on Day 5, p < 0.05 on Day 6, p < 0.001 on Day 7, p < 0.0001 from Day 8 until Day 10).
Conclusions
[1473] As expected in this model of psoriasis, the following pathological changes were observed following the topical application of imiquimod: erythema from Day 2, scaling from Day 5 and skin thickening from Day 2. Skin scores then increased until Day 10. At termination, the expected pathological changes were observed: thickening of the epidermis, crusting and/or subcorneal pustule formation, pustule formation within follicular sheaths and a diffuse, mixed neutrophilic and macrophage infiltration of the dermis.
[1474] Clobetasol propionate reduced skin erythema, reduced skin scaling and reduced skin thickening. Clobetasol propionate caused bodyweight loss, a known side effect of corticosteroids administered to rodents.
[1475] CT103 administered at 5%, 10% or 20% reduced the severity of the pathological changes and did not cause bodyweight loss.
[1476] CT103 administered at 5% significantly reduced the clinical score on Day 10 and the severity of skin scaling from Day 8.
[1477] CT103 administered at 10% significantly reduced both the severity of skin scaling and the cumulative clinical scores from Day 8.
[1478] CT103 administered at 20% significantly reduced the severity of skin erythema from Day 5, skin scaling from Day 7 and skin thickening in the first days of treatment. Furthermore, it statistically reduced the cumulative clinical score from Day 5 to the end of the experiment on Day 10.
[1479] The cumulative clinical scores also show a clear dose-dependence trend.
Example 39: Effects of CT340 in a Model of Hypertrophic Scar Formation in NZW
Rabbits
[1480] The objective of this study was to assess the effect of CT340 on hypertrophic scar formation in NZW rabbits. Four wounds were surgically created in each ear, scars were allowed to develop, and the effects of test article on hypertrophic scars were determined by photomicroscopy and histopathology.
[1481] Surgical wounds were induced bilaterally as four circular lesions per ear down to the bare cartilage on the ventral side of each ear. Treatment started on Day 26, after confirming the complete formation of hypertrophic scars. Animals were dosed via intra-lesional injections into each scar or by topical application with the test article, CT340. Dosing was repeated once weekly for a total of three doses. Test article efficacy
in scar reduction was assessed using the Scar Elevation Index (SEI) and by histopathology.
[1482] The SEI of scars dosed with intra-lessional injection or by topical application of CT340 did not differ significantly from scars dosed with vehicle. Scars dosed with the positive control article (TACA) showed a significant decrease in SEI when compared to scars injected with CT340 or with saline (P <0.005).
[1483] Scars injected with vehicle or CT340 exhibited moderate inflammation. Dosing scars with intra- lesion injection of TACA significantly decreased inflammation.
[1484] Daily topical dosing of scars with CT340 significantly decreased the level of scar inflammation. This decrease was significantly lower when compared to TACA-injected scars.
[1485] In summary, while there was no difference between vehicle and CT340 in the reduction of SEI in this study, daily topical dosing of scars with CT340 reduced the level of inflammation.
Study Design
[1486] The study consisted of four groups of six female NZW rabbits each as shown in Table 60 below.
- Summary of Study Design

[1487] On Day 0, surgical wounds were induced bilaterally (both ears) as four circular lesions per ear (i.e., 8/animal), down to the bare cartilage on the ventral side of
each ear, using a 7-mm dermal biopsy punch, with removal of perichondrium. The wounds were dressed using polyurethane dressing (e.g., Tegaderm); dressing was changed once weekly for twenty-six (26) days.
[1488] On Day 26, animals were assigned to study groups of six rabbits each. Animals of Groups 1 (right ear only) and 3 were dosed via topical application. Animals of Groups 1 (left ear only), 2, and 4 were dosed via intra-lesional injection into each scar (10 μΙ_/503Γ). Animals of Groups 1 - 4 (respectively) were dosed with test and control article into both ears with vehicle; CT340 at 200 mg/mL; CT340 at 5%; or TACA at 40 mg/mL. For Groups 1 (left ear), 2, and 4, dosing was repeated once weekly for a total of 3 doses; Groups 1 (right ear) and 3 were dosed daily for 17 days. Throughout the in-life, the wounds were photographed once weekly; clinical observations were recorded at least once daily; and body weights were recorded once weekly.
[1489] On Day 43 animals were sacrificed and weighed. Both ears of each animal were collected, photographed, and fixed in 10% neutral buffered formalin (NBF). Fixed lesions were processed for histopathology, including staining with hematoxylin and eosin (H&E) and Masson's trichrome. Slides were evaluated by a board-certified veterinary pathologist, including lesion measurement using a scar elevation index (SEI).
Materials and Methods
[1490] The test article (CT340) was provided to the laboratory in a powder form and stored frozen at -80 °C pending use. The first control article was the vehicle used to formulate the topical formulation of CT340. The control article consisted of 25% Transcutol (diethylene glycol monoethyl ether) and 75% 1 ,2-propanediol. The vehicle was formulated from components Transcutol P (Gattefosse Catalog 3260JV1) and 1 ,2- propanediol (Fluka Catalog 82281). The second control article was triamcinolone acetonide (TACA). This control article is a synthetic glucocorticoid corticosteroid with anti-inflammatory action. The third control article was sterile saline, intended to be identical to the vehicle used to formulate the injectable formulation of CT340.
Hypertrophic Scar Formation Model
[1491] The model used in this study was based on that of Morris et al. (1997; Plast. Reconstr. Surg. 100: 674-81) and Kloeters et al. (2007; Wound Rep. Reg. 15: S40 - S45). Wounds were induced bilaterally (on both ears) on the respective Day 0 (2 or 3 February 201 1) as follows.
[1492] Prior to surgery, animals were anesthetized isoflurane inhalation per Testing Facility SOP.
[1493] Four wounds were created down to the bare cartilage on the ventral side of each ear using a 7-mm dermal biopsy punch. The depth of wound was a factor; shallower lesions do not afford the proper degree of scarring for the purposes of the study.
[1494] The cartilage was meticulously nicked without full dissection, as the latter results in noninterpretable histology.
[1495] Epidermis, dermis, and perichondrium were thoroughly removed using a dissecting or surgical microscope. Complete removal of the perichondria! layer was mandatory.
[1496] Any bleeding was treated by manual compression; electro- cauterization was not needed.
[1497] At the end of surgery, a liquid adhesive (e.g., Mastisol; Ferndale Laboratories, Inc., Ferndale, Ml) was applied to the surrounding skin followed by wound coverage with a polyurethane dressing (Tegaderm; 3M Health Care, St. Paul, MN). The polyurethane dressing remained attached at all times to ensure a moist wound environment.
[1498] The dressing was changed (at least) once weekly through Day 26.
[1499] Wounds were examined every day for signs of infection as well as for epithelialization progress on gross examination. By the time treatment was initiated (Day 26), scar formation was prominent.
[1500] Following group assignment, animals of Group 1 served as negative controls and were dosed with vehicle as follows: the left ears were dosed with Control Article 3 (saline) via intra-lesional injection, and the right ears were dosed topically with Control Article 1 (25% Transcutol:75% 1 ,2-propanediol). Dosing of the left ears was repeated once weekly for a total of 3 doses; dosing of the right ears was repeated once daily for a total of 17 doses. Animals of Groups 2 and 4 were dosed via intra-lesional injection of each scar (10 μΙ_/503Γ) ; dosing was repeated once weekly for a total of 3 doses. Animals of Group 3 were dosed topically; dosing was repeated once daily for a total of 17 doses. Injections were administered as 10 μΙ./wound of test or control article using a Hamilton syringe with 27-gauge needles; topical application was performed by applying 10 μΙ_ using a micropipette.
Histopathology
Scar Formation
[1501] The NBF-fixed tissues were processed for histopathologic evaluation. Tissues were dehydrated, embedded in paraffin, sectioned at 3- to 5-μηι thicknesses,
and stained with H&E and with Masson's trichrome. Slides were evaluated via light microscopy by a board-certified veterinary pathologist. Ear lesions (8/animal) were assessed using the scar elevation index (SEI) described by Morris et al. (1997). In brief, the SEI measures the ratio of total scar connective tissue area to the area of underlying dermis.
[1502] The scar area was measured in 1 or 2 sections per scar. Measurements were made digitally. A computerized visual imaging system was used to photograph the areas of interest using an Olympus (Tokyo, Japan) camera, with the image digitized by using MicroSuite Basic Edition Software (Tokyo, Japan). For a cross- section of each scar, measurements of the Maximum Scar Elevation (MSE), the external perimeter of the newly formed hypertrophied dermis (P-h), and the perimeter of the underlying dermis (P-d) were collected. The software automatically calculates the area bounded by each perimeter; A-h, and A-d, respectively. The SEI was calculated according to the following formula:
[1503] SEI = A-h + A-d / A-d
[1504] When two sections were analyzed, the SEI was calculated from the section with the highest MSE.
Scar Inflammation
[1505] Approximately 2 levels from each ear wound were examined histopathologically for each animal. The incidence and severity of scar inflammation were scored using the accepted industry 5-point scoring system, as follows:

Results
Acclimation
[1506] There were no clinical signs of abnormality during the acclimation period. All animals were released for use in the study at the end of the acclimation period.
Mortality
[1507] There was one unexpected death: Rabbit 56 died 1 1 minutes after the surgery.
Clinical Observations
[1508] Clinical observations were recorded daily. During the in-life period, none of the wounds showed any sign of infection. Scars were fully formed by Day 26, when dosing was initiated.
Body Weight
[1509] Group mean normalized (to pre-dose) body weight values are plotted in Figure 141. All rabbits gained weight during the course of the study; adverse effects were not apparent following the surgery or treatments.
[1510] Starting on Day 33, and until the end of the in-life period, animals dosed with TACA gained significantly lower (P < 0.01) body weight when compared to the vehicle or test article treated animals. The lower rate of increase in body weight is a known side effect of TACA in rodents and rabbits.
Histopathology
Scar Formation
[1511] Scar formation results are summarized in Table 61 and in Figures 142 through 150. The perimeters of 188 scars were measured, and the SEI of each scar was calculated. Quantitative analysis of the scar area demonstrated decreases in SEI in TACA-treated scars (Group 4) when compared to the vehicle- and CT340-treated scars (1.41 ± 0.1 1 vs. 1.82 ± 0.25 and 1.73 ± 0.16 for the left ears of Group 1 and Group 2, respectively). Treating scars with topical application of CT340 had no effect on the scar area when compared to the vehicle-treated scars (1.62 ± 0.18 vs. 1.69 ± 0.18 for Group 3 vs. the right ears of Groupl , respectively). Treating scars by topical application or intra- lesional injection of CT340 had no effect on the scar area (1.73 ± 0.16 vs. 1.62 ± 0.18 for Groups 2 and 3, respectively).
[1512] Statistical analysis using the Student's t-test was performed to compare the TACA- injected scars to CT340- and saline-injected scars. The differences in the SEI values were significant (P <0.005); no significant differences were found when comparing the SEI of CT340-injected scars to that of saline-injected scars.
TABLE 61 - Summary of Scar Elevation Index (SEI), Groups 1 - 4
Left Ear 5css¾
Sfean StdDev.
1 -Let £si"s 1.82
t- Rigfci Ears 1.68
173 S.I 6
1.62
1 1
Histopathology
[1513] Inflammation was composed of a variable inflammatory response consisting of an admixture of mononuclear cells, lymphoid cells, macrophages, and neutrophils in the wound site. Fibrosis and granulation tissues also were present. All wounds were morphologically similar, but somewhat differentiated based upon severity of the lesion, size of the scar, and presence of inflammation. There were clear differences in the severity of the inflammation between groups. Group 1 left (saline) and right (vehicle 2) ears did not differ with regard to inflammation, and there was marked inflammation at the lesion sites in both groups. In Group 4 (treated with TACA), there was very little inflammation, consistent with the expected activity of TACA in this model. While intra- lesion injection of CT340 had little effect on inflammation, there was marked reduction in inflammation in scars treated daily by topical application of CT340, similar to that seen with TACA. This observation suggests that topical application of 5% CT340 has marked anti-inflammatory effects under the conditions of this study.
Scar Inflammation
[1514] Scar inflammation scoring is presented in Table 62 and plotted in Figures 151 and 152. Vehicle-treated scars (topical application or intra-lesion injection) exhibited moderate inflammation, with mean scores of 2.94 and 3.36 (respectively). Intra-lesion injection of scars with TACA, the positive control article, reduced the level of scar inflammation. Scars treated with TACA displayed significant (P <0.003) decreases in inflammation scores when compared to saline- and CT340-injected scars (scores of 1 .46 ± 0.22, 2.94 ± 0.73 and 3.02 ± 0.34, respectively).
[1515] Daily topical dosing of scars with CT340 reduced the level of scar inflammation. Scars treated with topical CT340 displayed significant (P <0.001) decreases in inflammation scores when compared to topical application of vehicle (right ears of Group 1) (scores of 2.03 ± 0.49 and 3.35 ± 1 .44, respectively). The inflammation scores of scars topically treated with CT340 were compared to intra-lesion injection of scars with TACA. Scars treated with TACA displayed significant (P <0.05) decreases in
inflammation scores when compared to scars treated by daily topical application of CT340 (scores of 1.46 ±0.22 and 2.03 ± 0.49, respectively).
TABLE 62 - Summary of Scar Inflammation Scores, Groups 1 - 7

Discussion and Conclusions
[1516] The SEI of scars dosed with intra-lesional injection or by topical application of CT340 did not differ significantly from that of scars dosed with vehicle. Scars dosed with the positive control article (TACA) showed significant decreases in SEI when compared to scars injected with CT340 or with saline (P <0.005).
[1517] Scars injected with vehicle or CT340 exhibited moderate inflammation. Dosing scars with intra-lesion injection of TACA significantly decreased inflammation.
[1518] Daily topical dosing of scars with CT340 significantly decreased the level of scar inflammation. This decrease was significantly lower when compared to TACA-injected scars.
[1519] In summary, while there was no difference between vehicle and CT340 in the reduction of SEI in this study, daily topical dosing of scars with CT340 reduced the level of inflammation. As seen in scars treated with TACA, the reduction of inflammation plays a role in the reduction of SEI.
Example 40: Examination of CT327 and CT340 in pain and nerve regeneration
[1520] This study evaluated the effect on nociception and neurite outgrowth of the highly selective TrkA inhibitors CT327 and CT340, using: 1) a functional calcium imaging assay to measure inhibition of capsaicin responses; and 2) neurite outgrowth assay in cultured sensory (DRG) neurons. The effects of CT327 and CT340 were compared with a commercially available TrkA inhibitor (GW441756) and an anti-NGF antibody
Background
[1521] Nerve Growth Factor (NGF) is a homodimer, which following binding results in dimerization and autophosphorylation of its high affinity receptor TrkA. Phosphorylation of TrkA increases the catalytic activity of the kinase domain and creates binding sites for SH2 domain containing cytoplasmic proteins. These proteins initiate the activation of several signal transduction pathways such as PLCy, ras, PB kinase/AKT, and Raf/MEK/ERK.
[1522] Increased NGF released from inflammatory cells and tissues during inflammation and injury results in hyperalgesia that can last from several hours to days via TrkA activation on sensory nerve terminals. This activates a multitude of downstream signalling pathways involving MAP kinases (ERK), PBK, and PLC, which are involved in pathological conditions. NGF binding to TrkA also potentiates the heat and capsaicin receptor TRPV1 through phosphorylation of TRPV1 channels by serine/threonine kinases, PKC, PKA, and calcium/calmodulin-dependent kinase II and tyrosine kinase c- Src.
[1523] TRPV1 is a plasma membrane bound ion channel expressed by nociceptors involved in thermosensation, which is activated by noxious heat (>43°C), capsaicin, low pH, the inflammatory mediators arachidonic acid and bradykinin leading to the perception of pain and thermal hypersensitivity. The sensitivity of the capsaicin receptor (TRPV1) is modulated by NGF in rodent and human DRG neurons. TRPV1 expression and function has been found to be up-regulated by NGF in vitro and in clinical conditions of hypersensitivity. Responses of cultured DRG neurons to capsaicin thus provide an in vitro model for NGF-trkA pathway signalling, as used in this study.
[1524] While NGF influences the survival of developing primary sensory neurons mainly through its high affinity receptor TrkA, its role in mature DRG neurons is restricted to regulating their sensitivity and neuropeptide expression, but not survival, or neurite length. However, systemic blockade of NGF in clinical conditions has been associated with positive sensory symptoms and accelerated arthritis, and generalized loss of protective sensation remains a concern with long-term treatment; these may be avoided by regional topical delivery for cutaneous hypersensitivity disorders. Here we describe the effectiveness of the compounds CT327 and CT340 designed to block NGF- TrkA signalling in an in vitro model of NGF induced hypersensitivity in adult rat DRG neurons, and demonstrate the inhibition of capsaicin responses without affecting neurite length. Treatment with the TrkA inhibitors CT327 and CT340 resulted in dose-related inhibition of capsaicin responses with IC50= 10 nM, but no effect on neurite length in cultured DRG neurons. These results suggest that CT327 and CT340 could produce pain relief without affecting the integrity or regeneration of nociceptor fibers.
Materials and Methods
Neuron cultures
[1525] Bilateral DRG from 1 1 adult rats were microdissected from cervical, thoracic, lumbar and sacral levels and enzyme digested in 0.2% collagenase/0.5% dispase in Ham's F12 medium for 3 hours (previously described in Anand et al, 2010). Enzyme digested tissue was mechanically dissociated in BSF2 medium (Ham's F12 containing 2% heat inactivated fetal calf serum, 60 ng/ml progesterone, 0.16 μg/ml sodium selenite, 0.1 mg/ml transferrin, 16 μg/ml putrescine, 10 μg/ml insulin, 3 mg/ml BSA, 100 μg/ml each penicillin/streptomycin), containing 1 mg/ml soy-bean trypsin inhibitor. The resulting neuronal suspension was plated on collagen (from rat tail, 50 μg/ml), and laminin (20 μg/ml) coated glass bottomed petri dishes (MatTek, USA) at 1000 cells per dish in 200 μΐε BSF2 medium; after 30 minutes 2 mis warm BSF2 medium containing human NGF (Sigma U.K. 100 ng/ml) was added to the cultures, which were incubated at 37 °C in a humid environment. Creabilis TrkA inhibitors CT327, CT340 and commercially available TrkA inhibitor GW441756 (BML-1364, Enzo Life Sciences, Switzerland,), were dissolved in DMSO to make stock solutions of 4.5 mM (CT327), and 4.7 mM (CT 340), 30 mM (GW441756) aliquoted and stored at -20 °C, until use. Intermediate dilutions of CT327 and CT340 were prepared in sterile distilled water, and of GW441756 in ethanol, at 500x final concentration. Anti-NGF antibody (1 mg/ml, anti human/mouse), was obtained commercially (L148M, Exalpha Biologicals), aliquoted and stored at -20 °C until use.
Functional assay - Calcium Imaging
[1526] 48 hours after plating, neurons were incubated with 2 μΜ Fura2 AM (1 hour, 37 °C), in phenol red free HEPES buffered HBSS (Hanks balanced salt solution) containing 0.1 % BSA, pH 7.3, followed by HBSS wash and de-esterification for 20 minutes. Phase bright, growing neurons were identified and alternately stimulated with u.v. excitation of 340 and 380 nm wavelength and transmitted light every 2 seconds ( 100 msec duration), and images were acquired with a Hamamatsu Orea ER FW cooled CCD camera using Kinetic Imaging software. Neurons with a stable baseline ratio of 340/380 (bound/unbound calcium) were stimulated with 200 nM capsaicin (20-30 seconds to identify capsaicin sensitive neurons which indicated calcium influx by increased 340/380 ratio; this was followed by washout and a rest period of 30 minutes. Test compounds were applied from stock solutions (500x final concentration- 4 μΙ/2ηιΓ), followed 10 minutes later by 1 μΜ capsaicin, and responses were measured as the difference from baseline to peak ratio, and expressed as a percentage response (ratio of second (1 μΜ capsaicin)/first (200 nM capsaicin) response), and compared with NGF treated controls (no test compound applied between capsaicin stimuli). Percent inhibition
was calculated for each group and compared with controls. Student's t test was used to compare between groups; P<0.05 was considered statistically significant.
Morphological assay - Neurite length measurement
[1527] 24 hours after plating, neurons were treated with CT327, CT340, GW441756 or anti-NGF (L148M, Exalpha), for 24 hours and fixed with 4% PFA for 15 minutes, permeabilized with methanol (-20 °C, 3 minutes), prior to immunostaining with mouse monoclonal antibody to Gap43 (1 :200, Sigma, UK) for 45 minutes followed by secondary antibody Alexa 488 (1 :200, Molecular Probes); the glass bottom coverslips were detached and mounted on glass slides, and TIFF images were acquired with a Zeiss inverted microscope equipped with standard FITC optics, for image analysis and neurite measurement using Metamorph software. Neurons with neurites greater than twice the cell body diameter were identified, and only those neurons which were clearly identifiable were used for analysis. The longest neurite length was measured for each neuron and the average length calculated for each group using Excel software. 30- 50 neurons were analyzed for each concentration in each of 3 experiments.
Results
Effect on capsaicin responses
[1528] The effect the test and control compounds on capsaicin responses is depicted in Figures 153 to 156. Neurons acutely treated with CT327 or CT340 demonstrated dose-related inhibition of capsaicin responses with IC50 value of 10 nM for both compounds, normalised to controls. The TrkA inhibitor GW441756 (BMLE1364) had an IC50 value of 15 nM, while the anti-NGF antibody had an IC50 value of 1 μg/ml. Note that 23.8 ± 5.3 % reduction (n=8 neurons), was observed due to desensitization, as reported previously (see Anand et al, 2010); further inhibition in the presence of CT327 and CT340 was normalized to controls. For CT327, the goodness of fit, R2=0.78, and for CT340, R2=0.68 (using Graph Pad Prism 5.0 software.). The IC50 for morphine and Gabapentin in this bioassay were 1 μΜ and 100 nM, respectively.
Effect on Neurite length
[1529] Test compounds were added 24 hours after plating. 24 hour treatment with CT327 and CT340 did not show any difference in neurite length compared to control (100 ng/ml NGF) (Figures 157 to 159). As depicted in Figure 160, treatment with commercial TrkA inhibitor GW441756 showed some vesiculation at 1 μΜ concentration and reduction of neurite length (n.s.), which were both increased at the higher concentration of 10 μΜ (p=0.03). As depicted in Figure 161 , treatment with the anti-NGF
antibody at 1 and 10 μς/ηιΙ concentrations did not significantly alter neurite length compared to NGF treated controls.
[1530] Separate neuronal cultures that had been treated with test compounds and fixed as for neurite length analysis were incubated with polyclonal TrkA antibody combined with mouse anti-Gap43 to confirm that TrkA positive neurons were not eliminated following treatment with CT327 and CT340. TrkA/Gap43 immunofluorescence confirmed that TrkA positive neurons were present in the treated groups (Figure 162).
Conclusions
[1531] This study used an in vitro model of NGF-induced neuronal hypersensitivity to assess the effects of the TrkA inhibitors on neuronal responses to capsaicin, and on neurite length. Acute treatment with both CT327 and CT340 resulted in functional inhibition of capsaicin responses with IC50=10 nM, indicating that these small molecule TrkA inhibitors are promising candidates for targeting neuronal hypersensitivity. The commercial TrkA inhibitor GW441756 and the NGF antibody were used for comparison, and acute treatment with both agents showed inhibition of capsaicin responses.
[1532] The morphological findings of this study indicate that CT327 and CT340, at the concentrations used here for functional effects, did not affect neurite length, indicating lack of a toxic effect. The commercial TrkA inhibitor GW441756, used for comparison, also did not have any effect on neurite length at 1 , 10 and 100 nM concentrations, but reduced neurite length at the higher concentrations of 1 and 10 μΜ with vesiculation, indicating possible structural effects at these concentrations. Anti-NGF antibody treatment at 1 and 10 μg/ml concentrations did not significantly affect neurite length.
[1533] In conclusion, CT327 and CT340 are potent inhibitors of NGF/TrkA- dependent capsaicin responses in cultured sensory neurons. Topical treatment with TrkA inhibitors may provide pain relief with the efficacy but without the safety liability of systemic NGF blockade.
Example 41 : Investigation of intra-epidermal nerve fibers following epicutaneous
2-week repeated treatment of mini-pig skin with CT340
[1534] Mini-pig skin tissues from treated and untreated areas of control (vehicle) and active animal groups of the study described in Example 33 were pre-fixed in benzoquinone (BQ) and frozen embedded were used in this study (see Table 63).
TABLE 63 - Tissues studied

[1535] Tissue sections (15μηι thick) were collected onto poly-L-lysine (Sigma, Poole, UK) coated glass slides. Endogenous peroxidase was blocked by incubation in industrial methylated spirits (IMS) containing 0.3% w/v hydrogen peroxide for thirty minutes. After rehydration with PBS buffer, sections were incubated overnight with primary antibody at final dilutions listed above. Sites of primary antibody attachment were revealed using nickel-enhanced, avidin-biotin peroxidase (ABC - Vector Laboratories, Peterborough, UK) as described. Sections were counter-stained for nuclei in 0.1 % w/v aqueous neutral red, dehydrated and mounted in xylene-based mountant (DPX; BDH/Merck, Poole, UK), prior to photomicrography.
[1536] PGP9.5 intra-epidermal fibers were counted at optimal titre of 1 :40,000 and results expressed as fibers/mm. The nerve fibers present in first 2 mm of each section fibers were counted.
[1537] Optimal dilution for PGP9.5 staining in the pig tissue was 1 :40,000. Intra-epidermal nerve fibers were seen, and immunoreactive fibers were also present in arrector pili and deeper dermal nerve fascicles (Figure 163).
[1538] There was a tendency for fewer IENF in treated skin (Figure 164) but no statistically significant decrease. There was no difference in the PGP9.5 fiber counts of the recovery animals that received vehicle or high dose for 2 consecutive weeks and studied 2 weeks after last administration.
[1539] Epicutaneous 2-week repeated treatment of mini-pig skin with CT340 at doses of 2%, 6% and 20% w/v (corresponding to 0.1 1 , 0.33 and 1.1 g of
CT340/animal/day) did not have a significant effect on the numbers of intra- epidermal nerve fibers immunostained with the gold-standard pan-neuronal marker PGP9.5.
Example 42: Testing SNA-125 in an IMQ-induced Psoriasis Mouse Model Background
[1540] Imiquimod (IMQ) is a TLR7/8 ligand and a potent immune activator that is used topically for genital and perianal warts, as well as for superficial basal cell carcinomas and actinic keratosis. An acute model of psoriasis has been developed in mice by topical application of a 5% IMQ cream (Aldara™), based on the clinical observation that IMQ exacerbates psoriasis. Similar to human psoriasis, IL-23/IL-17 axis plays a pivotal role in IMQ-induced psoriasis in mice.
[1541] In a study by Ma et al. (J Clin Cell Immunol 2013, 4:6), oral Tofacitinib was tested in the IMQ-induced psoriasis model. Topical daily dose of IMQ was administered on the back and right ear for 3 consecutive days followed by an additional application on Day 5. 30 mg/kg of tofacitinib was given orally as a prophylactic treatment, twice daily starting on Day 0. There, ear thickness was measured on Day 5 and cytokines transcripts from mouse ears at study termination. Here, different concentrations of SNA-125 were tested in the clinically relevant IMQ-induced psoriasis mouse model.
Materials and Methods
[1542] The IMQ-induced psoriasis mouse study design is depicted in Figure 165 and the test groups are shown in Table 64 below. Animals were monitored daily for clinical symptoms, body weight, and psoriasis clinical score. Ear thickness was measured on days 0, 4, 6, 8 and 10. Cytokines levels were measured on day 4 on ear punch biopsies. Spleen weight and histology (H&E) analysis of back skin were performed on day 10.
TABLE 64 - Test Groups

* 25% Tfo¾$cy†oi. P, 75% Propy'tene Gtycoi
Results
Body weight
[1543] As shown in Figure 166, one-time daily administration of IMQ onto the shaved back and right ear of the animals resulted in body weight loss as compared to naive animals. Two times daily treatment with 3 different doses of SNA-125 modestly increased the body weight over IMQ only animals at the beginning of the study as compared to vehicle-treated mice. The observed dramatic body weight loss in clobetasol-treated animals is a known side effect of the steroid and was expected.
Psoriasis clinical score
[1544] Figure 167 depicts the changes in the total psoriasis score throughout the study, which was calculated by summing the plaque score, the erythema score and the punctate redness/scabbing score. The difference between SNA-125 at 5% and the vehicle is statistically significant from day 7. The differences between SNA-125 at 0.5% and 1 % and the vehicle are statistically significant on day 10.
Erythema score
[1545] Figure 168 depicts the changes in the erythema score throughout the IMQ-induced psoriasis mouse study. SNA-125 at 5% is statistically significant from the vehicle from day 7. SNA-125 at 0.5% and 1 % are statistically significant on day 10.
Plaque Scores
[1546] Figure 169 depicts the changes in the plaque score throughout the IMQ-induced psoriasis mouse study. SNA-125 at 5% is statistically significant from the vehicle on day 10.
Punctate Redness/scabbing Scores
[1547] Figure 170 depicts the changes in the punctate redness/scabbing score throughout the IMQ-induced psoriasis mouse study. As expected, IMQ only animals exhibited increased punctate redness and scabbing, which was diminished by SNA-125 application.
Spleen Weight and Ear Thickness
[1548] Figures 171 and 172 depict the changes in spleen thickness and ear thickness throughout the IMQ-induced psoriasis mouse study, respectively. Topical application of the IMQ cream causes the enlargement of spleen and lymph nodes, and increased ear thickness. The commonly used antipsoriatic agent clobetasol almost completely attenuated these IMQ-induced changes. Neither treatments with vehicle or test compound significantly modulated spleen weight. There was no difference on ear thickness found between the vehicle and 3 different doses of SNA-125.
[1549] Daily application of IMQ induced significant splenomegly in IMQ only animals on day 10, which is indicative of a systemic inflammatory response. Daily treatment with clobetasol significantly decreased splenomegly as seen by a reduction in spleen weight. Neither treatments with vehicle or test compounds significantly modulated the spleen weight on day 10. However, there was a slight non-significant reduction in spleen weight between the vehicle (mean 0.180 ± 0.014) and 5% SNA-125 treatment group (0.155 ± 0.007).
[1550] Daily application of IMQ significantly increased ear thickness and positive control clobetasol showed significant reduction on the ear thickness induced by IMQ. All treatments including vehicle showed significant ear thickness reduction. However, there was no difference on ear thickness found between the vehicle and 3 different doses of SNA-125.
Cytokine Levels in Ear Samples
[1551] Figure 173 depicts the levels of cytokines IL-22, IL-17A, IL17F, and TNFa in ear samples taken at Day 4. Note that while the cytokines were measured on day 4, different cytokines can have different pharmacodynamics behavior (i.e. peak at different days post IMQ application). Daily application of IMQ induced an increase (significant in IL-22, IL-17F and TNFa) in inflammation/psoriasis-associated cytokines in the ears of diseased animals on day 4. Daily treatment with clobetasol decreased the cytokine production to baseline levels. As compared to vehicle group, twice daily treatment with 5% SNA-125 had no significant effects on cytokine production, though a moderate decrease in two pathogenic cytokines, TNFa (vehicle, mean 9.331 ± 2.267; 5% SNA-125, mean 4.860 + 0.973) and IL-22 (vehicle, mean 9.650 ± 2.339; 5% SNA-125, mean 5.243 ± 1 .759) was observed.
Conclusions
[1552] Different concentrations of SNA-125 were found to have an ameliorative effect when tested in the clinically relevant IMQ-induced psoriasis mouse model.
Example 43: Mouse Model of IMQ-induced Psoriasis
Aim of Study
[1553] The objective of this study was to determine the efficacy of SNA-101 , SNA-103, and SNA-352 as a therapeutic in the mouse model of IMQ-induced psoriasis.
Methodology
Test groups and experimental timing
[1554] Table 65 depicts the test groups and Figure 174 depicts the timing of the experiments performed in this study.
Table 65: Test Groups

Psoriasis Clinical Scoring
[1555] The animals were examined for signs of psoriasis on study day 0. These scores served as a baseline for the psoriasis clinical score parameter. Starting from IMQ cream application on day 0, psoriasis responses were examined daily until termination of the study.
[1556] Psoriasis reactions (erythema and plaques) were scored based on the parameters shown in Table 66 and recorded according to a 0-12 scale. The clinical score is determined by summing the score of each section.
Table 66: Psoriasis Clinical Scoring Parameters
Psoriasis Score
Punctate
Plaques Grade Erythema Grade Grade redness/scabbinq
Normal 0 Normal 0 No red dots 0
Very few plaques Very few (1 -10) red dots (cover about 5-10% of 1 Slight 1 present on the entire 1 the back area). back
Several plaques (cover Red dots are diffuse and about 20%-30% of the 2 Moderate 2 cover ~50% of the back 2 back area). (10-25 red dots)
Red dots cover the
Moderate plaques
entire back and are
(cover about 50% of 3 Marked 3 3
more concentrated (>25 the back area).
red dots)
Spread plaques (cover
over 90% of the back 4 Extreme 4
area).
Plaques are no longer
detected, however the
skin is irritated and no 5
fur is growing on the
skin
Results
Psoriasis clinical score
[1557] The total psoriasis score was determined by summing the plaque score, the erythema score and the punctate redness/scabbing score. As seen in Figure 175, the difference between SNA-125 at 5% and the vehicle is statistically significant on day 8 and 10, while for SNA-125 at 10% is significant from day 8 to day 10. Further, a statistically significant difference between SNA-352 at 5% and the vehicle on day 10 was found, as well as for SNA-125 at 10% from day 8 to day 10. Further, the difference between SNA-101 at 20% and the vehicle is statistically significant from day 8 to day 10.
Erythema scores
[1558] As shown in Figure 176, the differences between SNA-125 at 5% and 10% and the vehicle are statistically significant from day 8 to day 10. Further, statistically significant differences were found between SNA-352 at 5% and 10% and the vehicle from day 8 to day 10. Additionally, the difference between SNA-101 at 20% and the vehicle is statistically significant from day 8 to day 10
Plaque scores
[1559] As shown in Figure 177, the difference between SNA-125 at 5% and the vehicle is statistically significant from day 5 to day 8, while for SNA-125 at 10% it is significant at day 8. Additionally, a statistically significant difference between SNA-352 at 10% and the vehicle was observed from day 5 to day 8. Additionally, the difference between SNA-101 at 20% and the vehicle is statistically significant at day 8.
Punctate redness/scabbing scores
[1560] As shown in Figure 178, the difference between SNA-352 at 10% and the vehicle is statistically significant on day 9. Additionally, a statistically significant difference between SNA-101 at 20% and the vehicle was observed on day 9 and day 10.
Spleen weight and ear thickness
[1561] Topical application of the IMQ cream causes the enlargement of spleen and lymph nodes, and increased ear thickness. The commonly used antipsoriatic agent clobetasol almost completely attenuated these IMQ-induced changes. Neither treatments with vehicle or SNA-125, SNA-352, SNA-101 significantly modulated spleen weight (Figure 179A). There was no difference on ear thickness found between the vehicle and the different doses of SNA-125, SNA-352 and SNA-101 (Figure 179B). Figure 179C depicts the daily weight of mice throughout the study.
Cytokine analysis
[1562] Left ears were biopunched on day 4 and after tissue homogenization, the levels of cytokines IL-17F, TNF-a, IL-22, and IL-17A in the tissue lysates were measured via multiplex and then normalized with total protein amounts. Mean values for each group are displayed in Figure 180.
Example 44: IL-23-induced Psoriasis Mouse Model
Aim of Study
[1563] The objective of this study was to determine the efficacy of SNA- 120 and SNA-325 as a therapeutic in the mouse model of IL-23-induced psoriasis. The IL- 23/Th17 pathway has been shown to play a major role in psoriasis, and injection of IL-23 into mice produces clinical features associated with psoriasis such as hyperproliferation of keratinocytes and thickened epidermis with infiltration of mononuclear cells. It has
been found that an IL-23 mouse model simulates human AD (i.e. 37% homology with human AD transcriptome). In this model, tofacitinib administration has found to reduce ear swelling and inflammatory infiltrates in mouse skin in a dose-dependent manner.
Methodology
Test groups
[1564] Table 67 depicts the test groups and Figure 181 depicts a schematic of the timing of the experiments performed.
Table 67: Test Groups

Psoriasis clinical scoring
[1565] The animals were examined for signs of psoriasis on study day 0. These scores served as a baseline for the psoriasis clinical score parameter. Starting
from IL-23 injection on day 0, psoriasis responses were examined 3 times weekly until termination of the study. Psoriasis reactions (erythema and plaques) were scored using the parameters depicted in Table 68 and recorded according to a 0-6 scale. The clinical score is determined by summing the score of each section.
Table 68: Psoriasis clinical scoring parameters

Results
[1566] Figure 182A depicts the total psoriasis clinical scores for each group over time. Figure 182B depicts right ear thickness for each group (measured with a caliper 3 times weekly) while Figure 183C depicts changes in body weight throughout the study.
Example 45: Prophetic Study - Acetone-diethyl-Ether-Water Model of Dry Skin
Pruritus
[1567] Dry skin pruritus is common in the elderly, and the Acetone-diethyl- Ether-Water (AEW) model has become a recognized animal model of chronic itch . Studies have shown that AEW treatment causes dryness (measured as increased
transepidermal water loss and decreased stratum corneum hydration). Further, studies indicate that increased secretion of NGF may induce dry skin itch. Old mice scratch more than young mice and AEW treatment induces more scratching compared to saline treatment both in young and old mice. It has further been reported that AEW treatment increases NGF levels in skin biopsies, with old mice having higher levels of NGF than young mice.
[1568] The efficacy of SNA- 120, SNA- 125, SNA-352 will be tested in the AEW model. AEW or 0.9% saline was administered topically BID. Additionally, SNA-120, SNA-125, SNA-352, or vehicle will be administered topically BID. Scratching measurements will be performed 14 hour after the last AEW treatment each day for one hour. Non-specific clinical signs and treatment site assessments will be performed from Day 1 to Day 6. All animals will be weighted on Day 1 and Day 6. On day 6 animals will be terminated and skin biopsies will be performed. Additionally, histology (H&E) and skin biopsies for NGF analysis were performed at study termination.
[1569] It is predicted that NGF and TrkA levels will be increased in old mice after AEW treatment, and that SNA-120, SNA-125, and SNA-352 administration will reverse this trend in a dose-dependent manner. It is predicted that SNA-120, SNA-125, and SNA-352 administration will reverse the AEW-induced skin thickening and skin dryness of young and old mice in a dose-dependent manner.
[1570] Parakeratosis is a mode of keratinization characterized by the retention of nuclei in the stratum corneum. In the skin, this process leads to the abnormal replacement of annular squames with nucleated cells. Parakeratosis is associated with the thinning or loss of the granular layer and is usually seen in diseases of increased cell turnover, whether inflammatory or neoplastic. Parakeratosis also is seen in the plaques of psoriasis and in dandruff. It is predicted that in young mice treated with saline, a normal epidermis with keratinocytes organized as a single line will be observed. It is predicted that in old mice treated with saline, normal epidermis with keratinocytes frequently organized as a single line and a few signs of parakeratosis will be observed. In young mice treated with AEW, it is predicted that histology assessments will show normal epidermis with some increase in stratification. In old mice treated with AEW, it is predicted that histology assessments will demonstrate normal epidermis with increase in stratification and signs of parakeratosis. It is predicted that that SNA-120, SNA-125, and SNA-352 administration will reverse AEW-induced stratification and parakeratosis in a dose-dependent manner.
Example 46: Prophetic Study - Laser-induced Choroidal Neovascularization in
Green Monkeys
Model
[1571] The laser-induced choroidal neovascularization (CNV) experimental model is employed in neovascular age-related macular degeneration (AMD) research. In particular, it is used in the development of therapeutics for the treatment of retinal diseases, such as wet-AMD.
Study 1: Ocular Pharmacokinetics and Tolerance of Intravitreal SNA-103 in African Green Monkeys
[1572] Monkeys will receive IVT injections of test article in both eyes (OU) in accordance with the treatment assignment (Table 69) and study schedule (Table 70). For IVT dosing, topical local anesthesia will be administered (0.5% proparacaine) and eyes will be disinfected with 5% Betadine and rinsed with sterile normal saline. IVT injections will be administered using a 31 -gauge 0.5-inch needle placed 2 mm posterior to the limbus in the inferior temporal quadrant, targeting the central vitreous. Injections will be followed by topical administration of 0.3% ciprofloxacin ophthalmic solution or equivalent antibiotic.
Table 69: Treatment Assignment
Groyp n Eye Treatment Ro«fe Volume Frequency i 3 OU SNA-103 IVT 50≠. Single dose
Table 70: Testing Schedule
Study Day
Sversf # Eye
Dosing ί83¾::ίκ¾8ί:ί
8ody weights 3 · X · - · X >ί X
Siif Sauw !i|! i!§!
Serum
Aqueo«s hwroor |ί ί|: |:||| | || ^
Vifreows humor
Study 2: Evaluation of the Efficacy of SNA-103 in a Laser-induced CNV Model in African Green Monkeys
[1573] On Day 0, CNV will be induced between temporal vascular arcades by laser photocoagulation targeting Bruch's membrane. Six laser spots will be symmetrically placed within the perimacular region in each eye by an ophthalmologist employing an Iridex Oculight TX 532 nm laser with a pulse duration of 100 ms, spot size of 50 μηι, power of 750 mW. Laser spots will be applied using a contact laser lens. Any spots demonstrating severe retinal/subretinal hemorrhage immediately post-laser and not resolving by the time of follow-up examinations will be excluded from analyses. If hemorrhage occurs encompassing all target lesion areas within the central retina, the monkey will be substituted for with another screened monkey, up to four monkeys across all treatment groups, taking measures to assure balanced assignment to treatment. On day eight post-laser treatment, monkeys will undergo color fundus imaging and OCT to document the appearance and size of the developed CNV complex at each laser lesion, and allow randomization to treatment assignment to achieve a balanced incidence of lesion severity in each treatment arm. On study day 10 monkeys will receive IVT injections of SNA-103, SNA-103 vehicle or bevacizumab (Avastin) in either eye in accordance with Table 71 . The testing schedule is depicted in Table 72, where 'OCT' indicates optical coherent tomography and ΊΟΡ' indicates intraocular pressure.
Table 71 : Treatment Assignment
fS Eye Tfecrtmersi s iowft Dose X infe f¥< £35

Table 72: Testing Schedule
Study Day
Event # Eves

Example 47: Prophetic Study - Rabbit Model of Dry Eye
Study 1: Evaluate the Tolerability and Ocular Distribution of SNA-125 and SNA-352 after Topical Application in New Zealand White Rabbits
[1574] Rabbits will receive BID topical dose in both eyes in accordance with the treatment assignment and study schedule depicted in Table 73.
Table 73: Study Schedule
Group n Trecrfmf>nf Dose Koute Exa s Somjte coHecBon of

Study 1: Evaluate the Efficacy of SNA-125 and SNA-352 for Treatment of keratoconjunctivitis sicca (dry eye) in New Zealand White rabbits
[1575] Rabbits will be housed at ~ 20% humidity with increased airflow and administered daily with 50 ml of 1 % atropine topically into both eyes until study conclusion. After disease induction and baseline exam, topical dose of test articles will be administered in both eyes for 21 days in accordance with the study schedule depicted in Table 74. In-life assessments to be performed are depicted in Table 75.
Table 74: Study Schedule
Group n Treaftmetsi Dose Rowfe Euthanasia Matrices coiiected

Table 75: In-life Assessments
Assessments Frequency

Example 48: Determination of IC50 of SNA-352 against 4 Kinases
Aim of Study
[1576] The goal of this study was to assess the potency (IC50) of SNA-352 against the following kinases: LIMK1 , MAP2K6, MLK1 , and MLK3.
Materials and Methods
Test Agent
[1577] SNA-352 (Lot # 2017GC14/S7) was used at test concentrations of 10, 3, 1 , 0.3, 0.1 , 0.03, 0.01 , 0.003, 0.001 , 0.0003, μΜ.
Results
[1578] The IC50 value was calculated from concentration vs. %lnhibition curves by fitting to a four parameter logistic curve (Figure 183 to Figure 186). Table 76 depicts the IC50 of SNA-352 and staurosporine for 4 kinases.
TABLE 76 - IC50 of SNA-352 and Staurosporine for 4 Kinases
LIMK! L49B-07 7.68E-10
MAP2K6 2.61E-07 5.20E-10
MLK1 3.52E-08 3.22E-09
MLK3 2.04E-08 1.68E-09
Conclusions
[1579] SNA-352 is a potent inhibitor of LIMK1 , MAP2K6, MLK1 , and MLK3.
Claims
1. A reduced exposure composition for inhibiting the activity of a therapeutic target at a target site, the composition comprising:
a conjugate, wherein the conjugate comprises:
an active entity that has a selectivity and an inhibitory activity against the therapeutic target, and
at least one polymer linked to the active entity, wherein the at least one polymer is polyethylene glycol (PEG) or methoxy-polyethylene glycol (m-PEG),
wherein the conjugate has increased selectivity and/or inhibitory activity compared to the unconjugated active entity,
wherein the conjugate has reduced exposure at a non-target site compared to the unconjugated active entity, and
wherein the conjugate has increased permeability across at least one of a nuclear and plasma membrane compared to the unconjugated active entity.
2. The reduced exposure composition of claim 1 , wherein the conjugate is selected from:

, and
 and pharmaceutically acceptable salts thereof.
and pharmaceutically acceptable salts thereof.
3. The reduced exposure composition of any one of the preceding claims, wherein the therapeutic target is selected from VEGFR, c-Src, TkrA, MAP2K3, a JAK family kinase or a STAT family protein.
4. The reduced exposure composition of any one of the preceding claims, wherein the therapeutic target is a mediator of a condition.
5. The reduced exposure composition of any one of the preceding claims, wherein the condition is an inflammatory condition.
6. The reduced exposure composition of claim 4, wherein the condition is a dermal condition selected from psoriasis, psoriasis guttata, vitiligo, inverse psoriasis, pustular psoriasis, psoriatic erythroderma, acute febrile neutrophilic dermatosis, eczema, xerotic eczema, dyshidrotic eczema, vesicular palmar eczema, acne vulgaris, atopic dermatitis, contact dermatitis, allergic contact dermatitis, dermatomyositis, exfoliative dermatitis, hand eczema, pompholyx, keloids, rosacea, rosacea due to sarcoidosis, rosacea due to scleroderma, rosacea due to Sweet syndrome, rosacea due to systemic lupus erythematosus, rosacea due to urticaria, rosacea due to herpetic pain, Sweet's disease, neutrophilic hydradenitis, sterile pustule, drug rash, seborrheic dermatitis, pityriasis rosea, Kikuchi's disease of the skin, pruritic urticarial papules and plaques of pregnancy, Stevens-Johnson syndrome and toxic epidermal necrolysis, tattoo reaction, Wells syndrome (eosinophilic cellulitis), reactive arthritis (Reiter syndrome), bowel- associated dermatosis-arthritis syndrome, rheumatoid neutrophilic dermatosis, neutrophilic eccrine hidradenitis, neutrophilic skin disease of dorsum of hand, balanitis circumscripta plasmacellularis, balanoposthitis, Behcet's disease, erythema annulare centrifugum, erythema dyschromicum perstans, erythema multiforme, granuloma annulare, dermatitis of hand, lichen nitidus, lichen planus, lichen sclerosus et atrophicus,
lichen simplex chronicus, lichen spinulosus, nummular dermatitis, pyoderma gangrenosum, sarcoidosis, subcorneal pustular dermatosis, urticaria, and transient acantholytic dermatosis.
7. The reduced exposure composition of any one of the preceding claims, wherein the condition is a gastrointestinal condition selected from inflammatory bowel disease, Crohn's disease, ulcerative colitis, collagenous colitis, lymphocytic colitis, ischemic colitis, diversion colitis, Behcet's syndrome, indeterminate colitis, irritable bowel syndrome or small intestinal bacterial overgrowth.
8. The reduced exposure composition of any one of the preceding claims, wherein the condition is an ophthalmic condition selected from macular degeneration, age related macular degeneration (ARMD), pterygium, choroidal neovascularization, retinopathy, diabetic retinopathy, acute macular neuroretinopathy, chronic macular neuroretinopathy, central serous chorioretinopathy, macular edema, cystoid macular edema, diabetic macular edema, acute multifocal placoid pigment epitheliopathy, Behcet's disease, birdshot retinochoroidopathy, uveitis, intermediate uveitis (pars planitis), anterior uveitis, multifocal choroiditis, multiple evanescent white dot syndrome (MEWDS), ocular sarcoidosis, posterior scleritis, serpignous choroiditis, subretinal fibrosis, uveitis syndrome, Vogt-Koyanagi-Harada syndrome, retinal arterial occlusive disease, central retinal vein occlusion, disseminated intravascular coagulopathy, branch retinal vein occlusion, hypertensive fundus changes, ocular ischemic syndrome, retinal arterial microaneurysms, Coat's disease, parafoveal telangiectasis, hemi-retinal vein occlusion, papillophlebitis, central retinal artery occlusion, branch retinal artery occlusion, carotid artery disease (CAD), frosted branch angitis, sickle cell retinopathy and other hemoglobinopathies, angioid streaks, familial exudative vitreoretinopathy, Eales disease, sympathetic ophthalmia, uveitic retinal disease, retinal detachment, trauma, laser, PDT, photocoagulation, hypoperfusion during surgery, radiation retinopathy, bone marrow transplant retinopathy, proliferative vitreal retinopathy and epiretinal membranes, proliferative diabetic retinopathy, ocular histoplasmosis, ocular toxocariasis, presumed ocular histoplasmosis syndrome (POHS), endophthalmitis, toxoplasmosis, retinal diseases associated with HIV infection, choroidal disease associated with HIV infection, uveitic disease associated with HIV Infection, viral retinitis, acute retinal necrosis, progressive outer retinal necrosis, fungal retinal diseases, ocular syphilis, ocular tuberculosis, diffuse unilateral subacute neuroretinitis, myiasis, retinitis pigmentosa, systemic disorders with associated retinal dystrophies, congenital stationary night blindness, cone dystrophies, Stargardt's disease, fundus flavimaculatus, Bests disease, pattern dystrophy of the retinal pigmented epithelium, X-linked retinoschisis, Sorsby's fundus dystrophy, benign concentric maculopathy, Bietti's crystalline dystrophy,
pseudoxanthoma elasticum, retinal detachment, macular hole, giant retinal tear, retinal disease associated with tumors, congenital hypertrophy of the RPE, posterior uveal melanoma, choroidal hemangioma, choroidal osteoma, choroidal metastasis, combined hamartoma of the retina and retinal pigmented epithelium, retinoblastoma, vasoproliferative tumors of the ocular fundus, retinal astrocytoma, intraocular lymphoid tumors, punctate inner choroidopathy, acute posterior multifocal placoid pigment epitheliopathy, myopic retinal degeneration, or acute retinal pigment epithelitis.
9. The reduced exposure composition of any one of the preceding claims, wherein the therapeutic target is a mediator of a respiratory condition.
10. The reduced exposure composition of any one of the preceding claims, wherein the reduced exposure compounds are formulated for topical delivery to body surfaces selected from skin, eyes, ears, nose, mouth, lungs, vagina or rectum.
1 1 . The reduced exposure composition of any one of the preceding claims, wherein the reduced exposure compounds are formulated for oral delivery, respiratory delivery or injection into target sites, including eyes and joints.
12. The reduced exposure composition of any one of the preceding claims, wherein the target site is selected from skin, scalp, eye, Gl tract, joint and lung.
13. A reduced exposure composition according any one of the preceding claims, wherein the conjugate has reduced systemic exposure and toxicity when delivered to the target site compared to the unconjugated active entity.
14. The reduced exposure composition of any one of the preceding claims, wherein the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved.
15. The reduced exposure composition of any one of the preceding claims, wherein the conjugate has increased residence time at the target site, thereby resulting in lower concentrations, lower frequency of administration, or both.
16. The reduced exposure composition of any one of the preceding claims, wherein the composition further comprising one or more additional ingredients selected from the group consisting of an antibiotic agent, an anti-angiogenesis agent, a preventive or therapeutic agent for inflammatory bowel disease, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anesthetic agent, a steroidal anti-inflammatory agent, a non-steroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an anti-histamine agent, a vitamin or vitamin complex, and a hormone.
17. Use of a reduced exposure composition for inhibiting a therapeutic target at a target site, wherein the reduced exposure composition comprises:
a conjugate, wherein the conjugate comprises:
an active entity that has a selectivity and an inhibitory activity against the therapeutic target, and
at least one polymer linked to the active entity, wherein the at least one polymer is polyethylene glycol (PEG) or methoxy-polyethylene glycol (m-PEG),
wherein the conjugate has increased selectivity and/or inhibitory activity compared to the unconjugated active entity,
wherein the conjugate has reduced exposure at a non-target site compared to the unconjugated active entity, and
wherein the conjugate has increased permeability across at least one of a nuclear and plasma membrane compared to the unconjugated active entity.
18. The use of claim 17, wherein the conjugate is selected from:

, and
 and pharmaceutically acceptable salts thereof.
and pharmaceutically acceptable salts thereof.
19. The use of claim 17 or 18, wherein delivering the reduced exposure composition exhibits fewer side effects as compared to delivering the active entity without conjugation to the polymer.
20. The use of any one of claims 17-19, wherein delivering the reduced exposure composition requires lower doses and/or less frequent dosing to achieve an effective amount as compared to delivering the active entity without conjugation to the polymer.
21 . The use of any one of claims 17-19, wherein the conjugate is administered orally to the target site.
22. The use of any one of claims 17-19, wherein the conjugate is administered topically to the target site.
23. The use of any one of claims 17-19, wherein the conjugate is administered by injection to the target site.
24. The use of any one of claims 17-19, wherein the conjugate is administered by inhalation or instillation to the target site.
25. The use of any one of claims 17-19, wherein the conjugate is administered rectally to the target site.
26. The use of any one of claims 17-25, wherein the therapeutic target is selected from VEGFR, c-Src, TkrA, MAP2K3, a JAK family kinase or a STAT family protein.
27. A reduced exposure composition for treating a target site within the gastrointestinal system, the composition comprising
a conjugate, comprising an active entity linked to at least one polymer, wherein the active entity is an inhibitor, antagonist, or inverse agonist of a cellular mediator of a gastrointestinal condition,
wherein the at least one polymer is polyethylene glycol (PEG) or methoxy- polyethylene glycol (m-PEG); and
a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site;
wherein the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer, wherein the non- target site comprises the systemic system, the lymphatic system and/or another non-target tissue site,
wherein the conjugate can traverse plasma membranes of a plurality of cells at the target site, , thereby promoting interactions between the active entity and the cellular mediator of the gastrointestinal condition.
28. A reduced exposure composition according to Claim 27, wherein the conjugate is selected from SNA-101 , SNA-103, SNA-352, SNA-120 or SNA-125.
29. A reduced exposure composition according to any one of claims 27-28, wherein the traversal of the plasma membranes comprises the crossing of cellular lipid bilayers to distribute the active entity among both lipophilic and hydrophilic cellular compartments.
30. A reduced exposure composition according to any one of claims 27-29, wherein the conjugate can traverse an epithelial barrier and/or intestinal mucosa.
31 . A reduced exposure composition according to any one of claims 27-30, wherein the conjugate can further traverse the nuclear membrane and inhibit the activity of a nuclear mediator of the gastrointestinal condition.
32. A reduced exposure composition according to any one of claims 27-31 , wherein the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved.
33. A reduced exposure composition according to any one of claims 27-32, wherein the composition is formulated for oral delivery, and wherein the conjugate has reduced systemic absorption and little or no systemic toxicity when the composition is administered orally.
34. A reduced exposure composition according to any one of claims 27-33, wherein the composition is formulated for oral delivery, and wherein the conjugate has reduced systemic absorption and little or no systemic toxicity when the composition is administered orally on a daily basis.
35. The reduced exposure composition of any one of claims 27-34, wherein the conjugate has a longer residence time within a cell compared to the active entity without conjugation to the polymer.
36. The reduced exposure composition of claim 35, wherein the residence time of
the conjugate within a cell is at least 25% longer as compared to the active entity without conjugation to the polymer.
37. The reduced exposure composition of claim 35, wherein the residence time of the conjugate within a cell is at least 2- 10 fold longer as compared to the active entity without conjugation to the polymer.
38. The reduced exposure composition of any one of claims 27-37, wherein the conjugate exhibits greater access to the cellular mediator compared to the active entity without conjugation to the polymer.
39. The reduced exposure composition of any one of claims 27-38, wherein the conjugate exhibits a depo effect across cellular compartments, thereby reducing the dose of the active entity required to inhibit the cellular mediator compared to the active entity without conjugation to the polymer.
40. The reduced exposure composition of claim 39, wherein the dose of the conjugate needed to achieve a comparable therapeutic effect is 10-90% lower as compared to the active entity without conjugation to the polymer.
41 . The reduced exposure composition of any one of claims 27-40, wherein the activity entity has a concentration, activity and/or bioavailability at the target site that is at least 2-20 fold greater than at a non-target site, wherein the non-target site comprises the systemic system.
42. The reduced exposure composition of any one of claims 27-41 , wherein the activity entity has a concentration, activity and/or bioavailability at the target site that is at least 2-20 fold greater than at a non-target site, wherein the non-target site comprises the lymphatic system.
43. The reduced exposure composition of claims 41 or 42, wherein the reduced concentration, activity and/or bioavailability at the non-target site(s) reduces toxicity.
44. The reduced exposure composition of any one of claims 27-43, wherein the activity entity has a concentration, activity and/or bioavailability at the target site that is at least 2-20 fold greater than at a non-target site, wherein the non-target site comprises bone marrow.
45. The reduced exposure composition of claim 44, wherein the reduced concentration, activity and/or bioavailability in the bone marrow reduces immunosuppression.
46. The reduced exposure composition of any one of claims 27-45, wherein the activity entity has a concentration, activity and/or bioavailability at the target site that is at least 2-20 fold greater than at a non-target site, wherein the non-target site is outside the gastrointestinal tract.
47. The reduced exposure composition of any one of claims 27-46, wherein the
conjugate is present at a biologically inactive concentration at a non-target site.
48. The reduced exposure composition of any one of claims 27-47, wherein the conjugate is amphiphilic.
49. The reduced exposure composition of any one of claims 27-48, wherein the conjugate is at least 25% more amphiphilic than the active entity without conjugation to the polymer.
50. The reduced exposure composition of any one of claims 27-49, wherein the conjugate is at least 25% more hydrophilic than the active entity without conjugation to the polymer, thus facilitating non-compartmentalization within the cell.
51. The reduced exposure composition of any one of claims 27-50, wherein the conjugate is at least 25% more hydrophilic than the active entity without conjugation to the polymer, thus facilitating access to and activity in both the lipid bilayer and the cytosol of the cell.
52. The reduced exposure composition of any one of claims 27-51 , wherein the conjugate is at least 25% more hydrophilic than the active entity without conjugation to the polymer, thus facilitating access to and/or activity in both the lipid bilayer and the cytoplasm of the cell.
53. The reduced exposure composition of any one of claims 27-52, wherein the conjugate is at least 25% more hydrophilic than the active entity without conjugation to the polymer, thus facilitating access to and/or activity across the lipid bilayer.
54. The reduced exposure composition of any one of claims 27-53, wherein the composition reduces inflammation at the target site within the gastrointestinal system.
55. The reduced exposure composition of any one of claims 27-54, wherein the conjugate is an inhibitor, antagonist, or inverse agonist of a mediator of inflammation.
56. The reduced exposure composition of any one of claims 27-55, wherein the conjugate is an inhibitor, antagonist, or inverse agonist of a mediator of an inflammatory bowel disease.
57. The reduced exposure composition of claim 56, wherein the inflammatory bowel disease is Crohn's disease, ulcerative colitis, collagenous colitis, lymphocytic colitis, ischemic colitis, diversion colitis, Behcet's syndrome, or indeterminate colitis.
58. The reduced exposure composition of any one of claims 27-57, wherein the conjugate is an inhibitor, antagonist, or inverse agonist of a mediator of a gastrointestinal condition, wherein the mediator is selected from c-Src, VEGFR protein, JAK protein, STAT, NF-Kappa B, TrkA, MAPK, MAP2K or MAP2K3.
59. The reduced exposure composition of claim 58, wherein the VEGFR protein comprises one or more of VEGFR- 1 , VEGFR-2, and VEGFR-3.
60. The reduced exposure composition of claim 58, wherein the JAK protein
comprises one or more of JAK1 , JAK2, JAK3, and Tyrosine kinase 2 (TYK2)
61 . The reduced exposure composition of any one of claims 27-60, wherein the composition further comprising one or more additional ingredients selected from the group consisting of an antibiotic agent, an anti-angiogenesis agent, a preventive or therapeutic agent for inflammatory bowel disease, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anesthetic agent, a steroidal anti-inflammatory agent, a nonsteroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an anti-histamine agent, a vitamin or vitamin complex, and a hormone
62. The reduced exposure composition of any one of claims 27-61 , wherein the target site comprises the small intestine and the non-target site comprises one or more of the large intestine and stomach.
63. The reduced exposure composition of any one of claims 27-61 , wherein the target site comprises the large intestine and the non-target site comprises one or more of the small intestine and stomach.
64. The reduced exposure composition of any one of claims 27-61 , wherein the target site comprises the small intestine and large intestine and the non-target site comprises the stomach.
65. The reduced exposure composition of any one of claims 27-61 , wherein the target site comprises one or two of the duodenum, jejunum and ileum and the non- target site comprises the remaining one or two.
66. The reduced exposure composition of any one of claims 27-61 , wherein the target site comprises one or two of the ascending colon, transverse colon and the descending colon and the non- target site comprises the remaining one or two.
67. The reduced exposure composition of any one of claims 27-61 , wherein the target site comprises the intestinal lamina propria and/or gastric parietal cells and the non-target site comprises sites other than intestinal lamina propria and/or gastric parietal cells.
68. The reduced exposure composition of any one of claims 27-61 , wherein the target site comprises immune cells of the intestinal lamina propria and the non-target site comprises non-immune cells.
69. Use of a reduced exposure composition of any one claims 27-68 for treating a gastrointestinal condition in a subject in need thereof.
70. Use of a reduced exposure composition of any one claims 27-68 for treating inflammatory bowel disease in a subject in need thereof.
71 . Use of a reduced exposure composition of any one claims 27-68 for treating irritable bowel syndrome in a subject in need thereof.
72. Use of a reduced exposure composition of any one claims 27-68 for treating small intestinal bacterial overgrowth in a subject in need thereof.
73. Use of a reduced exposure composition of any one claims 27-68 to reduce immunosuppression and/or an inflammatory response while treating a gastrointestinal condition in a subject in need thereof.
74. Use of a reduced exposure composition of any one claims 27-68 to reduce liver damage, neutropenia and/or lymphopenia while treating a gastrointestinal condition in a subject in need thereof.
75. A method for treating or preventing a gastrointestinal condition in a subject in need thereof, the method comprising administering to the subject an effective amount of a reduced exposure composition for treating a target site within the gastrointestinal system, the reduced exposure composition comprising a conjugate comprising an active entity linked to at least one polymer,
wherein the active entity is an inhibitor, antagonist, or inverse agonist of a cellular mediator of the gastrointestinal condition,
wherein the at least one polymer is polyethylene glycol (PEG) or methoxy- polyethylene glycol (m-PEG); and
a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site;
wherein the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer, wherein the non- target site comprises the systemic system, the lymphatic system and/or another non-target tissue site,
wherein the conjugate can traverse cellular plasma membranes, thereby promoting interactions between the active entity and the cellular mediator of the gastrointestinal condition.
76. The method of Claim 75, wherein the conjugate is selected from SNA-101 , SNA-103, SNA-352, SNA-120 or SNA-125.
77. The method of Claim 75 or Claim 76, wherein the traversal of the cellular plasma membranes increases distribution among both lipophilic and hydrophilic cellular compartments.
78. The method of any one of Claims 75-77, wherein the gastrointestinal condition comprises one or more of inflammatory bowel disease, irritable bowel syndrome or small intestinal bacterial overgrowth.
79. The method of any one of Claims 75-77 wherein the gastrointestinal condition comprises one or more of Crohn's disease, ulcerative colitis, collagenous colitis, lymphocytic colitis, ischemic colitis, diversion colitis, Behcet's syndrome, and
indeterminate colitis.
80. The method of any one of Claims 75-79, wherein the composition is formulated for oral delivery and is administered orally.
81 . The method of any one of Claims 75-80, wherein the target site within the gastrointestinal system is selected from the group consisting of intestinal epithelium, intestinal lamina propria, the lining of the gastrointestinal tract, immune cells residing within the intestinal lamina propria, muscularis mucosae, myenteric plexus, the submucosa, the muscular layer, intraperitoneal spaces, retroperitoneal spaces, serosa, adventitia, and any combination thereof
82. The method of any one of Claims 75-81 , wherein administering the reduced exposure composition provides a therapeutically effective amount of the active entity at the target site at a concentration of at least about 2-20 fold greater than the active entity administered without conjugation to the polymer.
83. The method of any one of Claims 75-82, wherein administering the reduced exposure composition provides a reduced amount of the active entity at a non-target site at a concentration of at least about 2-20 fold lower than the active entity administered without conjugation to the polymer.
84. The method of any one of Claims 75-83, wherein administering the reduced exposure composition increases the residence time of the active entity at a target site by at least about 2-20 fold compared to the active entity administered without conjugation to the polymer.
85. The method of any one of Claims 75-84, wherein administering the reduced exposure composition decreases the residence time of the active entity at a non-target site by at least about 2-20 fold compared to the active entity administered without conjugation to the polymer.
86. The method of any one of Claims 75-85, wherein administering the reduced exposure composition reduces the clearance time of the active entity from a non-target site by at least about 2-20 fold compared to the active entity administered without conjugation to the polymer.
87. The method of any one of Claims 75-86, wherein administering the reduced exposure composition decreases systemic absorption of the active entity at least about 2-20 fold compared to the active entity administered without conjugation to the polymer.
88. The method of any one of Claims 75-87, wherein about 10-90% of the administered conjugate reaches the target site.
89. The method of any one of Claims 75-88, wherein the conjugate penetrates one or more physical, physiological and/or biological barriers of the intestinal mucosae.
90. The method of any one of Claims 75-89, wherein the conjugate exhibits
enhanced delivery to the intestinal epithelium as compared to delivery of the active entity without conjugation to the polymer.
91 . The method of any one of Claims 75-90, wherein the conjugate exhibits enhanced delivery to the intestinal lamina propria as compared to delivery of the active entity without conjugation to the polymer.
92. The method of any one of Claims 75-91 , wherein the conjugate exhibits increased residence time in the lining of the gastrointestinal tract as compared to the active entity delivered without conjugation to the polymer.
93. The method of any one of Claims 75-92, wherein the conjugate exhibits increased residence time in the intestinal epithelial cells as compared to the active entity delivered without conjugation to the polymer.
94. The method of any one of Claims 75-93, wherein the conjugate exhibits increased residence time in the intestinal lamina propria as compared to the active entity delivered without conjugation to the polymer.
95. The method of any one of Claims 75-94, wherein the conjugate undergoes transcytosis across the intestinal epithelium.
96. The method of any one of Claims 75-95, wherein the conjugate targets immune cells residing within the intestinal lamina propria.
97. The method of any one of Claims 75-96, wherein administering the reduced exposure composition exhibits fewer side effects as compared to the active entity administered without conjugation to the polymer.
98. The method of any one of Claims 75-97, wherein administering the reduced exposure composition requires fewer and/or lower doses to achieve an effective amount as compared to the active entity administered without conjugation to the polymer.
99. The method of any one of Claims 75-98, wherein the conjugate is administered orally.
100. The method of any one of Claims 75-98, wherein the conjugate is administered topically.
101 . The method of any one of Claims 75-98, wherein the conjugate is administered in the form of a suppository.
102. A method for treating or preventing inflammatory bowel disease or irritable bowel syndrome in a subject in need thereof, the method comprising administering to the subject an effective amount of a reduced exposure composition for treating a target site within the gastrointestinal system, the reduced exposure composition comprising a conjugate comprising an active entity linked to at least one polymer,
wherein the active entity is an inhibitor, antagonist, or inverse agonist of a mediator of the gastrointestinal condition,
wherein the at least one polymer is polyethylene glycol (PEG) or methoxy- polyethylene glycol (m-PEG); and
a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site;
wherein the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer, wherein the non- target site comprises the systemic system, the lymphatic system and/or another non-target tissue site,
wherein the conjugate can traverse the intestinal mucosa and epithelial layers thereby promoting interactions between the active entity and the mediator, and treating or preventing the inflammatory bowel disease or irritable bowel syndrome.
103. The method of claim 102, wherein the reduced exposure composition provides 2-20 fold more activity and/or bioavailability of the active entity at the target site than at the non-target site.
104. The method of claim 102, wherein the reduced exposure composition provides at least 25 or 50% more activity and/or bioavailability of the active entity at the target site than at the non-target site.
105. A method according to any one of claims 102-104, wherein the target site comprises the small intestine and the non-target site comprises the large intestine and/or stomach.
106. A method according to any one of claims 102-104, wherein the target site comprises the large intestine and the non-target site comprises the small intestine and/or stomach.
107. A method according to any one of claims 102-104, wherein the target site comprises one or two of the duodenum, jejunum and ileum and the non- target site comprises the remaining one or two.
108. A method according to any one of claims 102-104, wherein the target site comprises one or two of the ascending colon, transverse colon and the descending colon and the non-target site comprises the remaining one or two.
109. A method according to any one of claims 102-104, wherein the target site comprises the intestinal lamina propria and the non-target site comprises tissue contacting the intestinal lamina propria.
1 10. A method according to any one of claims 102-104, wherein the target site comprises immune cells of the intestinal lamina propria and the non-target site comprises non-immune cells.
1 1 1 . A method according to any one of claims 102-104, wherein the target site
comprises the gastrointestinal tract and the non-target site comprises non- gastrointestinal tract tissue.
1 12. A method according to any one of claims 102-104, wherein the target site comprises gastric parietal cells and the non-target site comprises non-gastric parietal cells.
1 13. A reduced exposure composition for treating a dermal target site, the composition comprising
a conjugate, comprising an active entity linked to at least one polymer, wherein the active entity is an inhibitor, antagonist, or inverse agonist of a mediator of a dermal condition,
wherein the at least one polymer is polyethylene glycol (PEG) or methoxy- polyethylene glycol (m-PEG); and
a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site;
wherein the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer, wherein the non- target site comprises the systemic system, the lymphatic system and/or another non-target tissue site,
wherein the conjugate can traverse plasma membranes of a plurality of cells at the target site, thereby promoting interactions between the active entity and the mediator of the dermal condition.
1 14. The reduced exposure composition of claim 1 13, wherein the conjugate is selected from SNA-101 , SNA-103, SNA-352, SNA-120 or SNA-125.
1 15. The reduced exposure composition of any one of claims 1 13-1 14, wherein traversal of the plasma membranes comprises the crossing of cellular lipid bilayers to distribute the active entity among both lipophilic and hydrophilic cellular compartments.
1 16. The reduced exposure composition of any one of claims 1 13-1 15, wherein the conjugate can access one or more target sites within the epidermis, the dermis, and/or the hyopdermis.
1 17. The reduced exposure composition of claim 1 16, wherein the one or more target sites comprise cells localized within the stratum corneum, stratum lucidum, stratum granulosum, stratum spinosum, and stratum basale layers, the papillary layer of the dermis, the reticular layer of the dermis, sebaceous glands, arrector pili muscles, sensory nerve fibers, hair follicles, hair roots, pacinian corpuscles, hair root plexus, eccrine sweat glands, the hypodermis and/or the superficial fascia, and any combination thereof.
1 18. The reduced exposure composition of claim 1 17, wherein the conjugate
can interact with the mediator associated with the plasma membrane, cytoplasm and/or nucleus of the cells.
1 19. The reduced exposure composition of claim 1 17 or 1 18, wherein the cells comprise immune cells, non-immune cells and keratinocytes.
120. The reduced exposure composition of any one of claims 1 13-1 19, wherein the conjugate exhibits increased activity and/or bioavailability at target site(s) as compared to the active entity delivered without the polymer.
121 . The reduced exposure composition of any one of claims 1 13-120, wherein the conjugate exhibits reduced activity and/or bioavailability at non-target site(s) as compared to the active entity delivered without the polymer.
122. The reduced exposure composition of any one of claims 1 13-121 , wherein the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved.
123. The reduced exposure composition of any one of claims 1 13-122, wherein the composition is formulated for topical delivery, and wherein the conjugate has reduced systemic absorption and little or no systemic toxicity when the composition is administered topically.
124. The reduced exposure composition of any one of claims 1 13-123, wherein the composition is formulated for topical delivery, and wherein the conjugate has reduced systemic absorption and little or no systemic toxicity when the composition is administered topically on a daily basis.
125. The reduced exposure composition of any one of claims 1 13-124, wherein the conjugate has a longer residence time within the target site compared to the active entity without conjugation to the polymer.
126. The reduced exposure composition of claim 125, wherein the residence time of the conjugate within the target site is at least 25% longer as compared to the active entity without conjugation to the polymer.
127. The reduced exposure composition of any one of claim 125, wherein the residence time of the conjugate within the target site is at least 2- 10 fold longer as compared to the active entity without conjugation to the polymer.
128. The reduced exposure composition of any one of claims 1 13-127, wherein the conjugate exhibits greater access to the mediator compared to the active entity without conjugation to the polymer.
129. The reduced exposure composition of any one of claims 1 13-128, wherein the conjugate exhibits a depot effect across cellular compartments, thereby reducing the dose of the active entity required to inhibit the mediator compared to the active entity without conjugation to the polymer.
130. The reduced exposure composition of any one of claim 128, wherein the dose of the conjugate needed to achieve a comparable therapeutic effect is 10-90% lower as compared to the active entity without conjugation to the polymer.
131 . The reduced exposure composition of any one of claims 1 13-130, wherein the activity entity has a concentration, activity and/or bioavailability at the target site that is at least 2-20 fold greater than at a non-target site, wherein the non-target site comprises the systemic system.
132. The reduced exposure composition of any one of claims 1 13-131 , wherein the activity entity has a concentration, activity and/or bioavailability at the target site that is at least 2-20 fold greater than at a non-target site, wherein the non-target site comprises the lymphatic system.
133. The reduced exposure composition of claims 131 or 132, wherein the lower concentration, activity and/or bioavailability at the non-target site(s) reduces toxicity.
134. The reduced exposure composition of any one of claims 1 13-133, wherein the activity entity has a concentration, activity and/or bioavailability at the target site that is at least 2-20 fold greater than at a non-target site, wherein the non-target site comprises bone marrow.
135. The reduced exposure composition of claim 134, wherein the lower concentration, activity and/or bioavailability in the bone marrow reduces immunosuppression.
136. The reduced exposure composition of any one of claims 1 13-135, wherein the activity entity has a concentration, activity and/or bioavailability at the target site that is at least 2-20 fold greater than at a non-target site, wherein the non-target site is outside the skin.
137. The reduced exposure composition of any one of claims 1 13-136, wherein the composition has minimal systemic absorption following epicutaneous administration.
138. The reduced exposure composition of any one of claims 1 13-137, wherein the composition displays rapid systemic elimination when administered by intravenous, intramuscular, transmucosal, subcutaneous or intraperitoneal injection or infusion.
139. The reduced exposure composition of any one of claims 1 13-138, wherein the composition displays minimal toxicity when administered by intravenous, intramuscular, transmucosal, subcutaneous or intraperitoneal injection or infusion.
140. The reduced exposure composition of any one of claims 1 13-139, wherein the composition displays minimal toxicity when administered on a daily basis by intravenous, intramuscular, transmucosal, subcutaneous or intraperitoneal injection or infusion.
141 . The reduced exposure composition of any one of claims 1 13-140, wherein the conjugate is present at a biologically inactive concentration at a non-target site.
142. The reduced exposure composition of any one of claims 1 13-141 , wherein the conjugate is amphiphilic.
143. The reduced exposure composition of any one of claims 1 13-142, wherein the conjugate is at least 25% more amphiphilic than the active entity without conjugation to the polymer.
144. The reduced exposure composition of any one of claims 1 13-143, wherein the conjugate is at least 25% more hydrophilic than the active entity without conjugation to the polymer, thus facilitating non-compartmentalization within the cell.
145. The reduced exposure composition of any one of claims 1 13-144, wherein the conjugate is at least 25% more hydrophilic than the active entity without conjugation to the polymer, thus facilitating access to and activity in both the lipid bilayer and the cytoplasm of the cell.
146. The reduced exposure composition of any one of claims 1 13-145, wherein the conjugate is at least 25% more hydrophilic than the active entity without conjugation to the polymer, thus facilitating access to and/or activity across the lipid bilayer.
147. The reduced exposure composition of any one of claims 1 13-146, wherein the conjugate reduces inflammation at the target site.
148. The reduced exposure composition of any one of claims 1 13-147, wherein the conjugate is an inhibitor, antagonist, or inverse agonist of a mediator of inflammation.
149. The reduced exposure composition of any one of claims 1 13-148, wherein the dermal condition is selected from psoriasis, psoriasis guttata, vitiligo, inverse psoriasis, pustular psoriasis, psoriatic erythroderma, acute febrile neutrophilic dermatosis, eczema, xerotic eczema, dyshidrotic eczema, vesicular palmar eczema, acne vulgaris, atopic dermatitis, contact dermatitis, allergic contact dermatitis, dermatomyositis, exfoliative dermatitis, hand eczema, pompholyx, keloids, rosacea, rosacea due to sarcoidosis, rosacea due to scleroderma, rosacea due to Sweet syndrome, rosacea due to systemic lupus erythematosus, rosacea due to urticaria, rosacea due to herpetic pain, Sweet's disease, neutrophilic hydradenitis, sterile pustule, drug rash, seborrheic dermatitis, pityriasis rosea, Kikuchi's disease of the skin, pruritic urticarial papules and plaques of pregnancy, Stevens-Johnson syndrome and toxic epidermal necrolysis, tattoo reaction, Wells syndrome (eosinophilic cellulitis), reactive arthritis (Reiter syndrome), bowel-associated dermatosis-arthritis syndrome, rheumatoid neutrophilic dermatosis, neutrophilic eccrine hidradenitis, neutrophilic skin disease of dorsum of hand, balanitis circumscripta plasmacellularis, balanoposthitis, Behcet's disease, erythema annulare centrifugum, erythema dyschromicum perstans, erythema
multiforme, granuloma annulare, dermatitis of hand, lichen nitidus, lichen planus, lichen sclerosus et atrophicus, lichen simplex chronicus, lichen spinulosus, nummular dermatitis, pyoderma gangrenosum, sarcoidosis, subcorneal pustular dermatosis, urticaria, and transient acantholytic dermatosis.
150. The reduced exposure composition of any one of claims 1 13-148, wherein the dermal condition is a skin neoplasia, selected from squamous cell carcinoma, basal cell carcinoma, malignant melanoma, malignant cutaneous lymphoma, Kaposi's sarcoma, Merkel cell skin cancer, and non-melanoma skin cancer.
151. The reduced exposure composition of any one of claims 1 13-148, wherein the dermal condition is a vascular tumor, selected from hemangiomas, Kaposi's sarcoma, lymphangioma, glomangioma, angiosarcoma, hemangioendothelioma, and infantile hemangiomas.
152. The reduced exposure composition of any one of claims 1 13-148, wherein the dermal condition is a bullous disease, selected from bullous pemphigoid, erythema multiforme, dermatitis herpetiformis, epidermolysis bullosa acquisita, linear Immunoglobulin A disease, mucous membrane pemphigoid, pemphigoid gestationis, pemphigus foliaceus, and pemphigus vulgaris.
153. The reduced exposure composition of any one of claims 1 13-148, wherein the dermal condition is alopecia.
154. The reduced exposure composition of any one of claims 1 13-153, wherein hair growth and cycling can be modulated.
155. The reduced exposure composition of any of claims 1 13-154, wherein the mediator is selected from c-Src, VEGFR protein, JAK protein, STAT, NF-Kappa B, TrkA, MAPK, MAP2K or MAP2K3.
156. The reduced exposure composition of claim 155, wherein the VEGFR protein comprises one or more of VEGFR-1 , VEGFR-2, and VEGFR-3.
157. The reduced exposure composition of claim 155, wherein the JAK protein comprises one or more of JAK1 , JAK2, JAK3, and Tyrosine kinase 2 (TYK2).
158. The reduced exposure composition of any one of claims 1 13-157, wherein the composition further comprises one or more additional ingredients selected from a protective agent, an emollient, an astringent, a humectant, a sun screening agent, a sun tanning agent, a UV absorbing agent, an antibiotic agent, an anti-angiogenesis agent, a physiological cooling agent, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anti-acne agent, an anesthetic agent, a steroidal anti-inflammatory agent, a non-steroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an anti-histamine agent, a vitamin or vitamin complex, a hormone, an anti-dandruff agent, an anti-wrinkle agent, an anti-skin atrophy agent, a
skin whitening agent, and a cleansing agent.
159. A method for treating a dermal condition in a subject in need thereof, the method comprising administering to the subject an effective amount of a reduced exposure composition for treating a dermal target site, the reduced exposure composition comprising a conjugate comprising an active entity linked to at least one polymer,
wherein the active entity is an inhibitor, antagonist, or inverse agonist of a mediator of the dermal condition,
wherein the at least one polymer is polyethylene glycol (PEG) or methoxy- polyethylene glycol (m-PEG); and
a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site;
wherein the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer, wherein the non- target site comprises the systemic system, the lymphatic system and/or another non-target tissue site,
wherein the conjugate can traverse cellular plasma membranes, thereby promoting interactions between the active entity and the cellular mediator of the dermal condition.
160. The method of claim 159, wherein the conjugate is selected from SNA- 101 , SNA-103, SNA-352, SNA-120 or SNA-125.
161 . The method of claim 159 or 160, wherein the traversal of the cellular plasma membranes increases distribution among both lipophilic and hydrophilic cellular compartments.
162. The method of any of claims 159-161 , wherein the conjugate reduces inflammation at the target site.
163. The method of any of claims 159-162, wherein the conjugate is an inhibitor, antagonist, or inverse agonist of a mediator of inflammation.
164. The method of any of claims 159-163, wherein the dermal condition is selected from psoriasis, vitiligo, psoriasis guttata, inverse psoriasis, pustular psoriasis, psoriatic erythroderma, acute febrile neutrophilic dermatosis, eczema, xerotic eczema, dyshidrotic eczema, vesicular palmar eczema, acne vulgaris, atopic dermatitis, contact dermatitis, allergic contact dermatitis, dermatomyositis, exfoliative dermatitis, hand eczema, pompholyx, keloids, rosacea, rosacea due to sarcoidosis, rosacea due to scleroderma, rosacea due to Sweet syndrome, rosacea due to systemic lupus erythematosus, rosacea due to urticaria, rosacea due to herpetic pain, Sweet's disease, neutrophilic hydradenitis, sterile pustule, drug rash, seborrheic dermatitis, pityriasis
rosea, Kikuchi's disease of the skin, pruritic urticarial papules and plaques of pregnancy, Stevens-Johnson syndrome and toxic epidermal necrolysis, tattoo reaction, Wells syndrome (eosinophilic cellulitis), reactive arthritis (Reiter syndrome), bowel-associated dermatosis-arthritis syndrome, rheumatoid neutrophilic dermatosis, neutrophilic eccrine hidradenitis, neutrophilic skin disease of dorsum of hand, balanitis circumscripta plasmacellularis, balanoposthitis, Behcet's disease, erythema annulare centrifugum, erythema dyschromicum perstans, erythema multiforme, granuloma annulare, dermatitis of hand, lichen nitidus, lichen planus, lichen sclerosus et atrophicus, lichen simplex chronicus, lichen spinulosus, nummular dermatitis, pyoderma gangrenosum, sarcoidosis, subcorneal pustular dermatosis, urticaria, and transient acantholytic dermatosis.
165. The method of any of claims 159-163, wherein the dermal condition is a skin neoplasia, selected from squamous cell carcinoma, basal cell carcinoma, malignant melanoma, malignant cutaneous lymphoma, Kaposi's sarcoma, Merkel cell skin cancer, and non-melanoma skin cancer.
166. The method of any of claims 159-163, wherein the dermal condition is a vascular tumor, selected from hemangiomas, Kaposi's sarcoma, lymphangioma, glomangioma, angiosarcoma, hemangioendothelioma, and infantile hemangiomas.
167. The method of any of claims 159-163, wherein the dermal condition is a bullous disease, selected from bullous pemphigoid, erythema multiforme, dermatitis herpetiformis, epidermolysis bullosa acquisita, linear Immunoglobulin A disease, mucous membrane pemphigoid, pemphigoid gestationis, pemphigus foliaceus, and pemphigus vulgaris.
168. The method of any of claims 159-163, wherein the dermal condition is alopecia.
169. The method of any of claims 159-163, wherein hair growth and cycling can be modulated by administering the reduced exposure composition.
170. The method of any one of Claims 159-169, wherein administering the reduced exposure composition provides a therapeutically effective amount of the active entity at the target site at a concentration of at least about 2-20 fold greater than the active entity administered without conjugation to the polymer.
171 . The method of any one of Claims 159-170, wherein administering the reduced exposure composition provides a reduced amount of the active entity at a non- target site at a concentration of at least about 2-20 fold lower than the active entity administered without conjugation to the polymer.
172. The method of any one of Claims 159-171 , wherein administering the reduced exposure composition increases the residence time of the active entity at a target site by at least about 2-20 fold compared to the active entity administered without
conjugation to the polymer.
173. The method of any one of Claims 159- 172, wherein administering the reduced exposure composition decreases the residence time of the active entity at a non-target site by at least about 2-20 fold compared to the active entity administered without conjugation to the polymer.
174. The method of any one of Claims 159- 173, wherein administering the reduced exposure composition reduces the clearance time of the active entity from a non-target site by at least about 2-20 fold compared to the active entity administered without conjugation to the polymer.
175. The method of any one of Claims 159- 174, wherein administering the reduced exposure composition decreases systemic absorption of the active entity at least about 2-20 fold compared to the active entity administered without conjugation to the polymer.
176. The method of any one of Claims 159-175, wherein about 10-90% of the administered conjugate reaches the target site.
177. The method of any one of Claims 159-176, wherein the conjugate penetrates one or more physical, physiological and/or biological barriers of the skin.
178. The method of any one of Claims 159-177, wherein the conjugate exhibits enhanced delivery to the epidermis as compared to delivery of the active entity without conjugation to the polymer.
179. The method of any one of Claims 159-178, wherein the conjugate exhibits enhanced delivery to the dermis as compared to delivery of the active entity without conjugation to the polymer.
180. The method of any one of Claims 159-179, wherein the conjugate exhibits enhanced delivery to the subcutis as compared to delivery of the active entity without conjugation to the polymer.
181 . The method of any one of Claims 159-180, wherein the conjugate exhibits increased residence time in the skin as compared to the active entity delivered without conjugation to the polymer.
182. The method of any one of Claims 159-181 , wherein the conjugate targets immune cells residing within the dermal target site.
183. The method of any one of Claims 159- 182, wherein administering the reduced exposure composition exhibits fewer side effects as compared to the active entity administered without conjugation to the polymer.
184. The method of any one of Claims 159- 183, wherein administering the reduced exposure composition requires fewer and/or lower doses to achieve an effective amount as compared to the active entity administered without conjugation to the
polymer.
185. The method of any one of Claims 159-184, wherein the conjugate is administered topically.
186. The method of any one of Claims 159-184, wherein the conjugate is administered topically on a daily basis.
187. The method of any one of Claims 159-186, wherein the reduced exposure composition provides 2-20 fold more activity and/or bioavailability of the active entity at the target site than at the non-target site.
188. The method of any one of Claims 159-187, wherein the reduced exposure composition provides at least 25 or 50% more activity and/or bioavailability of the active entity at the target site than at the non-target site.
189. The method of any one of Claims 159-187, wherein the target site comprises the epidermis and the non-target site comprises the dermis, gland, blood vessels, and/or hypodermis.
190. The method of any one of Claims 159-187, wherein the target site comprises the dermis and the non-target site comprises the epidermis, gland hypodermis and/or blood vessels.
191 . The method of any one of Claims 159-187, wherein the target site comprises one or more of the epidermis, follicle, gland, blood vessels, dermis and subcutis, and the non-target site comprises the remaining sites.
192. The method of any one of Claims 159-187, wherein the target site comprises the subcutis and the non-target site comprises tissue contacting the subcutis.
193. The method of any one of Claims 159-187, wherein the target site comprises immune cells of the epidermis, dermis or hypodermis and the non-target site comprises non-immune cells.
194. The method of any one of Claims 159-187, wherein the target site comprises the skin and the non-target site comprises non-integumentary tissue.
195. Use of a reduced exposure composition for treating a dermal target site in a subject in need thereof, wherein the reduced exposure composition comprises a conjugate comprising an active entity linked to at least one polymer,
wherein the active entity is an inhibitor, antagonist, or inverse agonist of a mediator of the dermal condition,
wherein the at least one polymer is polyethylene glycol (PEG) or methoxy- polyethylene glycol (m-PEG); and
a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site;
wherein the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer, wherein the non- target site comprises the systemic system, the lymphatic system and/or another non-target tissue site,
wherein the conjugate can traverse cellular membranes and distribute among both lipophilic and hydrophilic cellular compartments, thereby promoting interactions between the active entity and the cellular mediator of the dermal condition, and
wherein the conjugate is selected from SNA-101 , SNA-103 or SNA-352.
196. The use of claim 195, wherein the dermal condition comprises an inflammatory condition.
197. The use of claim 195 or 196, wherein the conjugate reduces inflammation at the target site.
198. The use of any one of claims 195-197, wherein the conjugate is an inhibitor, antagonist, or inverse agonist of a mediator of inflammation.
199. The use of any one of claims 195-198, wherein the dermal condition is selected from psoriasis, psoriasis guttata, inverse psoriasis, pustular psoriasis, psoriatic erythroderma, acute febrile neutrophilic dermatosis, eczema, xerotic eczema, dyshidrotic eczema, vesicular palmar eczema, acne vulgaris, atopic dermatitis, contact dermatitis, allergic contact dermatitis, dermatomyositis, exfoliative dermatitis, hand eczema, pompholyx, keloids, rosacea, rosacea due to sarcoidosis, rosacea due to scleroderma, rosacea due to Sweet syndrome, rosacea due to systemic lupus erythematosus, rosacea due to urticaria, rosacea due to herpetic pain, Sweet's disease, neutrophilic hydradenitis, sterile pustule, drug rash, seborrheic dermatitis, pityriasis rosea, Kikuchi's disease of the skin, pruritic urticarial papules and plaques of pregnancy, Stevens-Johnson syndrome and toxic epidermal necrolysis, tattoo reaction, Wells syndrome (eosinophilic cellulitis), reactive arthritis (Reiter syndrome) , bowel-associated dermatosis-arthritis syndrome, rheumatoid neutrophilic dermatosis, neutrophilic eccrine hidradenitis, neutrophilic skin disease of dorsum of hand, balanitis circumscripta plasmacellularis, balanoposthitis, Behcet's disease, erythema annulare centrifugum, erythema dyschromicum perstans, erythema multiforme, granuloma annulare, dermatitis of hand, lichen nitidus, lichen planus, lichen sclerosus et atrophicus, lichen simplex chronicus, lichen spinulosus, nummular dermatitis, pyoderma gangrenosum, sarcoidosis, subcorneal pustular dermatosis, urticaria, and transient acantholytic dermatosis.
200. The use of any one of claims 195-198, wherein the dermal condition is a skin neoplasia, selected from squamous cell carcinoma, basal cell carcinoma, malignant melanoma, malignant cutaneous lymphoma, Kaposi's sarcoma, Merkel cell skin cancer,
and non-melanoma skin cancer.
201 . The use of any one of claims 195-198, wherein the dermal condition is a vascular tumor, selected from hemangiomas, Kaposi's sarcoma, lymphangioma, glomangioma, angiosarcoma, hemangioendothelioma, and infantile hemangiomas.
202. The use of any one of claims 195-198, wherein the dermal condition is a bullous disease, selected from bullous pemphigoid, erythema multiforme, dermatitis herpetiformis, epidermolysis bullosa acquisita, linear Immunoglobulin A disease, mucous membrane pemphigoid, pemphigoid gestationis, pemphigus foliaceus, and pemphigus vulgaris.
203. The use of any one of claims 195-198, wherein the dermal condition is alopecia.
204. The use of any one of claims 195-203, wherein the conjugate penetrates one or more physical, physiological and/or biological barriers of the skin.
205. The use of any one of claims 195-204, wherein the conjugate exhibits enhanced delivery to the epidermis as compared to delivery of the active entity without conjugation to the polymer.
206. The use of any one of claims 195-205, wherein the conjugate exhibits enhanced delivery to the dermis as compared to delivery of the active entity without conjugation to the polymer.
207. The use of any one of claims 195-206, wherein the conjugate exhibits enhanced delivery to the subcutis as compared to delivery of the active entity without conjugation to the polymer.
208. The use of any one of claims 195-207, wherein the conjugate exhibits increased residence time in the skin as compared to the active entity delivered without conjugation to the polymer.
209. The use of any one of claims 195-208, wherein the conjugate targets immune cells residing within the dermal target site.
210. The use of any one of claims 195-209, wherein the reduced exposure composition exhibits fewer side effects as compared to the active entity administered without conjugation to the polymer.
21 1 . The use of any one of claims 195-210, wherein the reduced exposure composition requires fewer and/or lower doses to achieve an effective amount as compared to the active entity administered without conjugation to the polymer.
212. The use of any one of claims 195-21 1 , wherein the reduced exposure composition provides 2-20 fold more activity and/or bioavailability of the active entity at the target site than at the non-target site.
213. The use of any one of claims 195-212, wherein the reduced exposure
composition provides at least 25 or 50% more activity and/or bioavailability of the active entity at the target site than at the non-target site.
214. The use of any one of claims 195-213, wherein the target site comprises the epidermis and the non-target site comprises the dermis, gland, hypodermis and/or blood vessels..
215. The use of any one of claims 195-214, wherein the target site comprises the dermis and the non-target site comprises the epidermis, gland, hypodermis and/or blood vessels..
216. The use of any one of claims 195-214, wherein the target site comprises one or two of the epidermis, dermis, gland, blood vessels, and subcutis, and the non- target site comprises the remaining sites.
217. The use of any one of claims 195-214, wherein the target site comprises the subcutis and the non-target site comprises tissue contacting the subcutis.
218. The use of any one of claims 195-214, wherein the target site comprises immune cells of the epidermis or dermis and the non-target site comprises non-immune cells.
219. The use of any one of claims 195-214, wherein the target site comprises the skin and the non-target site comprises non-integumentary tissue.
220. A reduced exposure composition for treating a target site in the eye, the composition comprising
a conjugate, comprising an active entity linked to at least one polymer, wherein the active entity is an inhibitor, antagonist, or inverse agonist of a mediator of an ophthalmic condition,
wherein the at least one polymer is polyethylene glycol (PEG) or methoxy- polyethylene glycol (m-PEG); and
a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site;
wherein the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer, wherein the non- target site comprises the systemic system, the lymphatic system and/or another non-target tissue site,
wherein the conjugate can traverse plasma membranes of a plurality of cells at the target site, thereby promoting interactions between the active entity and the mediator of the ophthalmic condition.
221 . A reduced exposure composition according to Claim 220, wherein the conjugate is selected from SNA-101 , SNA-103, SNA-352, SNA-120 or SNA-125.
222. A reduced exposure composition according to any one of claims 220-221 ,
wherein the traversal of the plasma membranes comprises the crossing of cellular lipid bilayers to distribute the active entity among both lipophilic and hydrophilic cellular compartments.
223. A reduced exposure composition according to any one of claims 220-222, wherein the composition is formulated for local ocular delivery via a topical or injectable.
224. A reduced exposure composition according to any one of claims 220-223, wherein the composition is formulated for subconjunctival, intravitreal, retrobulbar or intracameral delivery.
225. A reduced exposure composition according to any one of claims 220-222, wherein the composition is formulated for systemic delivery.
226. A reduced exposure composition according to any one of claims 220-225, wherein the conjugate can traverse the plasma membrane, cytoplasm and nuclear membrane of cells at the target site and inhibit the activity of a mediator of the ophthalmic condition.
227. A reduced exposure composition according to any one of claims 220-226, wherein the non-target site comprises any site at which pharmacological activity is not desired and/or not achieved.
228. A reduced exposure composition according to any one of claims 220-227, wherein the composition is formulated for topical or local ocular delivery, and wherein the conjugate has reduced systemic absorption and minimal systemic toxicity.
229. The reduced exposure composition of any one of claims 220-228, wherein the conjugate has a longer residence time within a cell or tissue at the target site compared to the active entity without conjugation to the polymer.
230. The reduced exposure composition of claim 229, wherein the residence time of the conjugate within a cell is at least 25% longer as compared to the active entity without conjugation to the polymer.
231 . The reduced exposure composition of claim 229, wherein the residence time of the conjugate within a cell or tissue at the target site is at least 2-10 fold longer as compared to the active entity without conjugation to the polymer.
232. The reduced exposure composition of any one of claims 220-231 , wherein the conjugate exhibits greater access to the mediator compared to the active entity without conjugation to the polymer.
233. The reduced exposure composition of any one of claims 220-232, wherein the conjugate exhibits a depot effect across cellular compartments, thereby reducing the dose of the active entity required to inhibit the cellular mediator compared to the active entity without conjugation to the polymer.
234. The reduced exposure composition of any one of claims 220-233, wherein
the dose of the conjugate needed to achieve a comparable therapeutic effect is 10-90% lower as compared to the active entity without conjugation to the polymer.
235. The reduced exposure composition of any one of claims 220-234, wherein the activity entity has a concentration, activity and/or bioavailability at the target site that is at least 2-20 fold greater than at a non-target site, wherein the non-target site comprises the systemic system.
236. The reduced exposure composition of any one of claims 220-235, wherein the activity entity has a concentration, activity and/or bioavailability at the target site that is at least 2-20 fold greater than at a non-target site, wherein the non-target site comprises the lymphatic system.
237. The reduced exposure composition of claims 235 or 236, wherein the reduced concentration, activity and/or bioavailability at the non-target site(s) reduces toxicity.
238. The reduced exposure composition of any one of claims 220-237, wherein the activity entity has a concentration, activity and/or bioavailability at the target site that is at least 2-20 fold greater than at a non-target site, wherein the non-target site comprises bone marrow.
239. The reduced exposure composition of claim 238, wherein the reduced concentration, activity and/or bioavailability in the bone marrow reduces immunosuppression.
240. The reduced exposure composition of any one of claims 220-239, wherein the activity entity has a concentration, activity and/or bioavailability at the target site that is at least 2-20 fold greater than at a non-target site, wherein the non-target site is outside the eye.
241 . The reduced exposure composition of any one of claims 220-240, wherein the conjugate is present at a biologically inactive concentration at a non-target site.
242. The reduced exposure composition of any one of claims 220-241 , wherein the conjugate is amphiphilic.
243. The reduced exposure composition of any one of claims 220-242, wherein the conjugate is at least 25% more amphiphilic than the active entity without conjugation to the polymer.
244. The reduced exposure composition of any one of claims 220-243, wherein the conjugate is at least 25% more hydrophilic than the active entity without conjugation to the polymer, thus facilitating non-compartmentalization within a cell or tissue at the target site.
245. The reduced exposure composition of any one of claims 220-244, wherein the conjugate is at least 25% more hydrophilic than the active entity without conjugation
to the polymer, thus facilitating access to and activity in both the lipid bilayer and the cytosol of the cell.
246. The reduced exposure composition of any one of claims 220-245, wherein the conjugate is at least 25% more hydrophilic than the active entity without conjugation to the polymer, thus facilitating access to and/or activity in both the lipid bilayer and the cytoplasm of the cell.
247. The reduced exposure composition of any one of claims 220-246, wherein the conjugate is at least 25% more hydrophilic than the active entity without conjugation to the polymer, thus facilitating access to and/or activity across the lipid bilayer.
248. The reduced exposure composition of any one of claims 220-247, wherein the composition reduces inflammation at the target site.
249. The reduced exposure composition of any one of claims 220-248, wherein the conjugate is an inhibitor, antagonist, or inverse agonist of a mediator of inflammation.
250. The reduced exposure composition of any one of claims 220-249, wherein the conjugate is an inhibitor, antagonist, or inverse agonist of a mediator of an inflammatory ocular condition.
251. The reduced exposure composition of claim 250, wherein the inflammatory ocular condition is uveitis.
252. The reduced exposure composition of any one of claims 220-251 , wherein the conjugate is an inhibitor, antagonist, or inverse agonist of a mediator of a ophthalmic condition, wherein the mediator is selected from c-Src, VEGFR protein, JAK protein, STAT, NF-Kappa B, TrkA, MAPK, MAP2K or MAP2K3.
253. The reduced exposure composition of claim 252, wherein the VEGFR protein comprises one or more of VEGFR-1 , VEGFR-2, and VEGFR-3.
254. The reduced exposure composition of claim 252, wherein the JAK protein comprises one or more of JAK1 , JAK2, JAK3, and Tyrosine kinase 2 (TYK2)
255. The reduced exposure composition of any one of claims 220-254, wherein the composition further comprising one or more additional ingredients selected from the group consisting of an antibiotic agent, an anti-angiogenesis agent, a preventive or therapeutic agent for inflammatory bowel disease, an antifungal agent, an antiviral agent, an antiprotozoal agent, an anesthetic agent, a steroidal anti-inflammatory agent, a nonsteroidal anti-inflammatory agent, an antipruritic agent, an additional antioxidant agent, a chemotherapeutic agent, an anti-histamine agent, a vitamin or vitamin complex, and a hormone
256. Use of a reduced exposure composition of any one claims 220-255 for treating an ophthalmic condition in a subject in need thereof.
257. The use of claim 256, wherein the ophthalmic condition is selected from
macular degeneration, pterygium, age related macular degeneration (ARMD), choroidal neovascularization, retinopathy, diabetic retinopathy, acute macular neuroretinopathy, chronic macular neuroretinopathy, central serous chorioretinopathy, macular edema, cystoid macular edema, or diabetic macular edema.
258. The use of claim 256, wherein the ophthalmic condition is selected from acute multifocal placoid pigment epitheliopathy, Behcet's disease, birdshot retinochoroidopathy, uveitis, intermediate uveitis (pars planitis), anterior uveitis, multifocal choroiditis, multiple evanescent white dot syndrome (MEWDS), ocular sarcoidosis, posterior scleritis, serpignous choroiditis, subretinal fibrosis, uveitis syndrome, or Vogt-Koyanagi-Harada syndrome.
259. The use of claim 256, wherein the ophthalmic condition is selected from retinal arterial occlusive disease, central retinal vein occlusion, disseminated intravascular coagulopathy, branch retinal vein occlusion, hypertensive fundus changes, ocular ischemic syndrome, retinal arterial microaneurysms, Coat's disease, parafoveal telangiectasis, hemi-retinal vein occlusion, papillophlebitis, central retinal artery occlusion, branch retinal artery occlusion, carotid artery disease (CAD), frosted branch angitis, sickle cell retinopathy and other hemoglobinopathies, angioid streaks, familial exudative vitreoretinopathy, or Eales disease.
260. The use of claim 256, wherein the ophthalmic condition is selected from sympathetic ophthalmia, uveitic retinal disease, retinal detachment, trauma, laser, PDT, photocoagulation, hypoperfusion during surgery, radiation retinopathy, or bone marrow transplant retinopathy.
261 . The use of claim 256, wherein the ophthalmic condition is selected from proliferative vitreal retinopathy and epiretinal membranes, or proliferative diabetic retinopathy.
262. The use of claim 256, wherein the ophthalmic condition is selected from ocular histoplasmosis, ocular toxocariasis, presumed ocular histoplasmosis syndrome (POHS), endophthalmitis, toxoplasmosis, retinal diseases associated with HIV infection, choroidal disease associated with HIV infection, uveitic disease associated with HIV Infection, viral retinitis, acute retinal necrosis, progressive outer retinal necrosis, fungal retinal diseases, ocular syphilis, ocular tuberculosis, diffuse unilateral subacute neuroretinitis, or myiasis.
263. The use of claim 256, wherein the ophthalmic condition is selected from retinitis pigmentosa, systemic disorders with associated retinal dystrophies, congenital stationary night blindness, cone dystrophies, Stargardt's disease, fundus flavimaculatus, Bests disease, pattern dystrophy of the retinal pigmented epithelium, X-linked retinoschisis, Sorsby's fundus dystrophy, benign concentric maculopathy, Bietti's
crystalline dystrophy, or pseudoxanthoma elasticum.
264. The use of claim 256, wherein the ophthalmic condition is selected from retinal detachment, macular hole, or giant retinal tear.
265. The use of claim 256, wherein the ophthalmic condition is selected from retinal disease associated with tumors, congenital hypertrophy of the RPE, posterior uveal melanoma, choroidal hemangioma, choroidal osteoma, choroidal metastasis, combined hamartoma of the retina and retinal pigmented epithelium, retinoblastoma, vasoproliferative tumors of the ocular fundus, retinal astrocytoma, or intraocular lymphoid tumors.
266. The use of claim 256, wherein the ophthalmic condition is selected from punctate inner choroidopathy, acute posterior multifocal placoid pigment epitheliopathy, myopic retinal degeneration, or acute retinal pigment epithelitis.
267. A method for treating an ophthalmic condition in a subject in need thereof, the method comprising administering to the subject an effective amount of a reduced exposure composition for treating a target site within the eye, the reduced exposure composition comprising a conjugate comprising an active entity linked to at least one polymer,
wherein the active entity is an inhibitor, antagonist, or inverse agonist of a mediator of the ophthalmic condition,
wherein the at least one polymer is polyethylene glycol (PEG) or methoxy- polyethylene glycol (m-PEG); and
a pharmaceutically acceptable carrier formulated for delivering the conjugate to the target site;
wherein the conjugate has reduced exposure at a non-target site as compared to the active entity delivered without the polymer, wherein the non- target site comprises the systemic system, the lymphatic system and/or another non-target tissue site,
wherein the conjugate can traverse cellular plasma membranes, thereby promoting interactions between the active entity and the mediator of the ophthalmic condition.
268. The method of Claim 267, wherein the conjugate is selected from SNA- 101 , SNA-103, SNA-352, SNA-120 or SNA-125.
269. A method for treating a subject suffering from a condition of the anterior segment of the eye selected from the group consisting of dry eye, glaucoma, allergic conditions, inflammatory conditions of the anterior segment and cornea, allergic conditions of the anterior segment and cornea, infectious conditions of the anterior segment and cornea, and corneal angiogenesis, the method comprising
administering to the subject an effective amount of a composition according to any one of claims 220-255.
270. A method for treating a subject suffering from a condition of the posterior segment of the eye selected from the group consisting of macular degeneration, diabetic retinopathy, inflammatory conditions of the posterior segment, infectious conditions of the posterior segment, neurodegenerative disease, and vascular disease of the posterior segment, the method comprising administering to the subject an effective amount of a composition according to any one of claims 220-255.
271 . A method for treating age-related macular degeneration in a subject in need thereof, the method comprising administering to the subject an effective amount of a composition according to any one of claims 220-255.
272. A method for treating diabetic retinopathy in a subject in need thereof, the method comprising administering to the subject an effective amount of a composition according to any one of claims 220-255.
273. A method for treating corneal edema in a subject in need thereof, the method comprising administering to the subject an effective amount of a composition according to any one of claims 220-255.
274. A method for treating macular edema in a subject in need thereof, the method comprising administering to the subject an effective amount of a composition according to any one of claims 220-255.
275. A method for treating dry eye in a subject in need thereof, the method comprising administering to the subject an effective amount of a composition according to any one of claims 220-255.
276. A method for treating uveitis in a subject in need thereof, the method comprising administering to the subject an effective amount of a composition according to any one of claims 220-255.
277. The method of any one of claims 267-276, wherein the non-target site is selected from the group consisting of the systemic system, the lymphatic system, non- target tissues at which pharmacological activity is not desired and/or not achieved, and any combination thereof.
278. The method of any one of claims 267-276, wherein the target site within the eye is selected from the group consisting of the anterior segment of the eye, the posterior segment of the eye, and any combination thereof.
279. The method of any one of claims 267-276, wherein the target site within the eye is selected from the group consisting an anterior sub-Tenon space, an anterior suprachoroidal space, an anterior intrascleral space, and any combination thereof.
280. The method of any one of claims 267-276, wherein the target site within
the eye is selected from the group consisting a posterior sub-Tenon space, a posterior suprachoroidal space and a posterior intrascleral space, and any combination thereof.
281 . The method of any one of claims 267-276, wherein the target site within the eye is selected from the group consisting the conjunctival epithelium, Tenon's fascia, episclera, sclera, choroid, and any combination thereof.
282. The method of any one of claims 267-276, wherein the target site within the eye is selected from the group consisting of cornea, lens, sclera, anterior chamber, iris, cornea, posterior chamber, choroid, retina, Bowman's layer, stroma, Descemet's membrane, the endothelium, Tenon's Capsule and any combination thereof.
283. The method of any one of claims 267-282, wherein the composition provides a therapeutically effective amount of the active entity at a target site at a concentration of at least 2-20 fold higher than the active entity delivered without the polymer.
284. The method of any one of claims 267-282, wherein the composition provides a diminished amount of the active entity at a non-target site at a concentration of at least 2-20 fold lower than the active entity delivered without the polymer.
285. The method of any one of claims 267-282, wherein the combination of the active entity and polymer increases the residence time of the active entity at a target site by at least 2-20 fold as compared to the active entity without the polymer.
286. The method of any one of claims 267-282, wherein the combination of the active entity and polymer decreases the residence time of the active entity at a non- target site by at least 2-20 fold as compared to the active entity without the polymer.
287. The method of any one of claims 267-282, wherein the combination of the active entity and polymer reduces the clearance time of the composition from a non- target site by at least 2-20 fold as compared to the active entity without the polymer.
288. The method of any one of claims 267-282, wherein the combination of the active entity and polymer decreases systemic absorption at least 2-20 fold as compared to the active entity without the polymer.
289. The method of any one of claims 267-282, wherein at least 10-90% the administered composition reaches a target site.
290. The method of any one of claims 267-289, wherein the composition penetrates one or more physical barriers of the eye.
291 . The method of any one of claims 267-289, wherein the composition penetrates one or more physiological barriers of the eye.
292. The method of any one of claims 267-289, wherein the composition penetrates one or more biological barriers of the eye.
293. The method of any one of claims 267-289, wherein the composition
penetrates the conjunctival epithelium, Tenon's fascia, episclera, sclera, choroid, or any combination thereof.
294. The method of any one of claims 267-293, wherein the composition has enhanced delivery to the anterior segment of the eye, as compared to the active entity delivered without the polymer.
295. The method of any one of claims 267-293, wherein the composition has enhanced delivery to the posterior segment of the eye as compared to the active entity delivered without the polymer.
296. The method of any one of claims 267-293, wherein the composition has increased residence time in the anterior segment of the eye as compared to the active entity delivered without the polymer.
297. The method of any one of claims 267-293, wherein the composition has increased residence time in the posterior segment of the eye as compared to the active entity delivered without the polymer.
298. The method of any one of claims 267-293, wherein the composition targets immune cells residing within the posterior segment of the eye.
299. The method of any one of claims 267-293, wherein the composition targets immune cells residing within the anterior segment of the eye.
300. The method of any one of claims 267-293, wherein the composition has fewer side effects as compared to the active entity delivered without the polymer.
301 . The method of any one of claims 267-293, wherein the composition requires fewer doses as compared to the active entity delivered without the polymer.
302. The method of any one of claims 267-293, wherein the composition requires smaller doses as compared to the active entity delivered without the polymer
303. The method of any one of claims 267-292, wherein the traversal of the cellular plasma membranes increases distribution among both lipophilic and hydrophilic cellular compartments.
304. A reduced exposure composition for inhibiting the activity of a therapeutic target in the respiratory tract, the composition comprising:
a conjugate, wherein the conjugate comprises:
an active entity that has a selectivity and an inhibitory activity against the therapeutic target, and
at least one polymer linked to the active entity, wherein the at least one polymer is polyethylene glycol (PEG) or methoxy-polyethylene glycol (m-PEG),
wherein the conjugate has increased selectivity and/or inhibitory activity compared to the unconjugated active entity,
wherein the conjugate has reduced exposure at a non-target site compared to the unconjugated active entity, and
wherein the conjugate has increased permeability across at least one of a nuclear and plasma membrane compared to the unconjugated active entity.
305. The reduced exposure composition of claim 304, wherein the therapeutic target is a mediator of a respiratory condition.
306. The reduced exposure composition of claim 305, wherein the respiratory condition is selected from acute bronchitis; acute laryngotracheal bronchitis; arachidic bronchitis; catarrhal bronchitis; croupus bronchitis; dry bronchitis; infectious asthmatic bronchitis; productive bronchitis; staphylococcus or streptococcal bronchitis; vesicular bronchitis; cylindric bronchiectasis; sacculated bronchiectasis; sinusitis with nasal polyposis; fusiform bronchiectasis; capillary bronchiectasis; cystic bronchiectasis; dry bronchiectasis; follicular bronchiectasis; chronic obstructive pulmonary disease (COPD), chronic obstructive lung disease (COLD), chronic obstructive airways disease (COAD) or small airways obstruction of whatever type, etiology, or pathogenesis, in particular chronic bronchitis, pulmonary emphysema, bronchiectasis, cystic fibrosis, bronchiolitis obliterans, organizing pneumonia (BOOP), chronic organizing pneumonia (COP), bronchiolitis fibrosa obliterans, follicular bronchiolitis or dyspnea associated therewith; cough of whatever type, etiology, or pathogenesis in particular idiopathic cough or cough associated with gastro-esophageal reflux disease (GERD), drugs, bronchial hyper- responsivity, asthma, COPD, COLD, COAD, bronchitis, bronchiectasis, pulmonary eosinophilic syndromes, pneumoconiosis, interstitial lung disease, pulmonary fibrosis, aspiration disorders, rhinitis, laryngitis or pharyngitis; pulmonary eosinophilic syndromes of whatever type, etiology, or pathogenesis, in particular acute eosinophilic pneumonia (idiopathic or due to drugs or parasites), simple pulmonary eosinophilia, Loeffler's syndrome, tropical pulmonary eosinophilia, chronic eosinophilic pneumonia, allergic bronchopulmonary mycosis, allergic bronchopulmonary aspergillosis (ABPA), Churg- Strauss syndrome or idiopathic hypereosinophilic syndrome; asthma of whatever type, etiology, or pathogenesis, in particular asthma that is a member selected from the group consisting of atopic asthma, non-atopic asthma, allergic asthma, atopic bronchial IgE- mediated asthma, bronchial asthma, essential asthma, true asthma, intrinsic asthma caused by pathophysiologic disturbances, extrinsic asthma caused by environmental factors, essential asthma of unknown or inapparent cause, non-atopic asthma, bronchitic asthma, emphysematous asthma, exercise-induced asthma, allergen induced asthma,
cold air induced asthma, occupational asthma, infective asthma caused by bacterial, fungal, protozoal, or viral infection, non-allergic asthma, incipient asthma and wheezy infant syndrome; alveolar hemorrhage of whatever type, etiology, or pathogenesis, in particular a member of the group consisting of idiopathic pulmonary hemosiderosis, alveolar hemorrhage due to drugs or other exogenous agents, alveolar hemorrhage associated with HIV or bone marrow transplant or autoimmune alveolar hemorrhage (e.g. associated with systemic lupus erythematosis, Goodpasture's syndrome, Wegener's granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome, pauci- immune glomerulonephritis); pneumoconiosis of whatever type, etiology, or pathogenesis, in particular pneumoconiosis that is a member selected from the group consisting of aluminosis or bauxite workers' disease, anthracosis or miners' asthma, asbestosis or steam-fitters' asthma, chalicosis or flint disease, ptilosis caused by inhaling the dust from ostrich feathers, siderosis caused by the inhalation of iron particles, silicosis or grinders' disease, byssinosis or cotton-dust asthma and talc pneumoconiosis; interstitial lung diseases (ILD) or pulmonary fibrosis of whatever type, etiology, or pathogenesis, in particular idiopathic pulmonary fibrosis, crytogenic fibrosing alveolitis, fibrosing alveolitis, ILD or pulmonary fibrosis associated with connective tissue disease (systemic lupus erythematosis, mixed connective tissue disease, polymyositis, dermatomyositis, Sjorgen's syndrome, systemic sclerosis, scleroderma, rheumatoid arthritis), usual interstitial pneumonia (UIP), desquamative interstitial pneumonia (DIP), granulomatous lung disease, sarcoidosis, Wegener's granulomatosis, histiocytosis X, Langerhan's cell granulomatosis, hypersensitivity pneumonitis, extrinsic allergic alveolitis, silicosis, chronic eosinophilic pneumonia, lymphangiolyomatosis, drug-induced ILD or pulmonary fibrosis, radiation-induced ILD or pulmonary fibrosis, alveolar proteinosis, graft-versus-host-disease (GVHD), lung transplant rejection, ILD or pulmonary fibrosis due to environmental/occupational exposure, BOOP, COP, bronchiolitis fibrosa obliterans, follicular bronchiolitis, idiopathic acute interstitial pneumonitis (Hamman Rich syndrome) or alveolar hemorrhage syndromes; seasonal allergic rhinitis or perennial allergic rhinitis or sinusitis of whatever type, etiology, or pathogenesis, in particular sinusitis that is a member selected from the group consisting of purulent or nonpurulent sinusitis, acute or chronic sinusitis and ethmoid, frontal, maxillary, or sphenoid sinusitis; Acute Respiratory Distress Syndrome (ARDS), adult respiratory distress syndrome or acute lung injury of whatever type, etiology, or pathogenesis; progressive massive fibrosis (PMF); pulmonary hypertension of whatever type, etiology or pathogenesis including primary pulmonary hypertension, essential hypertension, pulmonary hypertension secondary to congestive heart failure, pulmonary hypertension secondary to COPD, pulmonary venous hypertension, pulmonary arterial
hypertension and hypoxia-induced pulmonary hypertension. Respiratory disorders also include, in some embodiments, malignancies and tumors of the respiratory system, non- limiting examples of which include lung adenocarcinoma, squamous cell carcinoma, large cell carcinoma, bronchioloalveolar carcinoma (BAC), pulmonary adenocarcinoma (AIS), non-small-cell carcinoma, small cell carcinoma, and mesothelioma.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18715453.9A EP3600440A1 (en) | 2017-03-20 | 2018-03-19 | Reduced exposure conjugates modulating therapeutic targets |
Applications Claiming Priority (24)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762473972P | 2017-03-20 | 2017-03-20 | |
| US201762473982P | 2017-03-20 | 2017-03-20 | |
| US201762473975P | 2017-03-20 | 2017-03-20 | |
| US62/473,982 | 2017-03-20 | ||
| US62/473,975 | 2017-03-20 | ||
| US62/473,972 | 2017-03-20 | ||
| US201762501567P | 2017-05-04 | 2017-05-04 | |
| US201762501651P | 2017-05-04 | 2017-05-04 | |
| US201762501568P | 2017-05-04 | 2017-05-04 | |
| US62/501,567 | 2017-05-04 | ||
| US62/501,651 | 2017-05-04 | ||
| US62/501,568 | 2017-05-04 | ||
| US201762504111P | 2017-05-10 | 2017-05-10 | |
| US62/504,111 | 2017-05-10 | ||
| US201762590119P | 2017-11-22 | 2017-11-22 | |
| US201762590131P | 2017-11-22 | 2017-11-22 | |
| US201762590111P | 2017-11-22 | 2017-11-22 | |
| US201762590148P | 2017-11-22 | 2017-11-22 | |
| US62/590,131 | 2017-11-22 | ||
| US62/590,111 | 2017-11-22 | ||
| US62/590,119 | 2017-11-22 | ||
| US62/590,148 | 2017-11-22 | ||
| US201862634691P | 2018-02-23 | 2018-02-23 | |
| US62/634,691 | 2018-02-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018175340A1 true WO2018175340A1 (en) | 2018-09-27 |
Family
ID=61874051
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2018/023174 WO2018175340A1 (en) | 2017-03-20 | 2018-03-19 | Reduced exposure conjugates modulating therapeutic targets |
Country Status (2)
| Country | Link |
|---|---|
| EP (1) | EP3600440A1 (en) |
| WO (1) | WO2018175340A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020023389A1 (en) * | 2018-07-23 | 2020-01-30 | Sienna Biopharmaceuticals, Inc. | Reduced exposure compositions modulating therapeutic targets |
| US11160869B2 (en) | 2019-08-16 | 2021-11-02 | Applied Molecular Transport Inc. | Compositions, formulations and interleukin production and purification |
| US11324833B2 (en) | 2018-11-07 | 2022-05-10 | Applied Molecular Transport Inc. | Cholix-derived carriers for oral delivery of heterologous payload |
| WO2024090606A1 (en) * | 2022-10-26 | 2024-05-02 | 경상국립대학교병원 | Method for treating posterior scleritis |
Citations (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5169627A (en) * | 1991-10-28 | 1992-12-08 | Mount Sinai School Of Medicine Of The City University Of New York | Oral pharmaceutical composition containing a polyethylene glycol-immunoglobulin G conjugate for reconstitution of secretory immunity and method of reconstituting secretory immunity |
| US5489608A (en) | 1989-12-21 | 1996-02-06 | Goedecke Aktiengesellschaft | Indolocarbazole derivatives and the use thereof |
| US5698578A (en) | 1993-12-07 | 1997-12-16 | Eli Lilly And Company | Protein kinase C inhibitors |
| US5710145A (en) | 1995-11-20 | 1998-01-20 | Eli Lilly And Company | Protein kinase C inhibitor |
| US6110498A (en) | 1996-10-25 | 2000-08-29 | Shire Laboratories, Inc. | Osmotic drug delivery system |
| US6217903B1 (en) | 1997-04-28 | 2001-04-17 | Hercules Incorporated | Sustained release polymer blend for pharmaceutical applications |
| WO2002038561A1 (en) | 2000-11-07 | 2002-05-16 | Novartis Ag | Indolylmaleimide derivatives as protein kinase c inhibitors |
| WO2003037385A1 (en) * | 2001-10-30 | 2003-05-08 | Nektar Therapeutics Al, Corporation | Water-soluble polymer conjugates of retinoic acid |
| US6645970B2 (en) | 2000-11-07 | 2003-11-11 | Novartis Ag | Indolylmaleimide derivatives |
| EP1437143A1 (en) * | 2001-09-28 | 2004-07-14 | Santen Pharmaceutical Co., Ltd. | Injections for eye tissue containing drug bound to polyethylene glycol |
| EP1491554A1 (en) * | 2003-06-23 | 2004-12-29 | CONARIS research institute AG | PEGylated soluble gp130-dimers useful as a medicament |
| WO2006002971A2 (en) | 2004-07-02 | 2006-01-12 | Creabilis Therapeutics S.P.A. | Nucleic acids for the treatment of hmgb1-related pathologies |
| WO2007006533A2 (en) | 2005-07-11 | 2007-01-18 | Novartis Ag | Indolylmaleimide derivatives |
| US7235555B2 (en) | 2002-04-03 | 2007-06-26 | Novartis Ag | Indolylmaleimide derivatives |
| WO2008135557A1 (en) | 2007-05-07 | 2008-11-13 | Novartis Ag | Organic compounds |
| WO2009074575A2 (en) | 2007-12-10 | 2009-06-18 | Novartis Ag | Organic compounds |
| WO2009150137A2 (en) | 2008-06-10 | 2009-12-17 | Novartis Ag | Organic compounds |
| WO2011028740A1 (en) | 2009-09-03 | 2011-03-10 | Glaxo Group Limited | ENaC BLOCKERS |
| WO2011079087A1 (en) | 2009-12-23 | 2011-06-30 | Glaxo Group Limited | Enac blockers |
| WO2012035158A1 (en) | 2010-09-17 | 2012-03-22 | Novartis Ag | Pyrazine derivatives as enac blockers |
| US8673347B2 (en) | 2005-08-25 | 2014-03-18 | Creabilis Therapeutics S.P.A. | Polymer conjugates of K-252A and derivatives thereof |
| US8926955B2 (en) | 2008-12-22 | 2015-01-06 | Creabilis S.A. | Synthesis of polymer conjugates of indolocarbazole compounds |
| WO2016004321A1 (en) * | 2014-07-03 | 2016-01-07 | Board Of Regents, The University Of Texas System | Compounds for treating biofilm infection |
-
2018
- 2018-03-19 WO PCT/US2018/023174 patent/WO2018175340A1/en unknown
- 2018-03-19 EP EP18715453.9A patent/EP3600440A1/en not_active Withdrawn
Patent Citations (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5489608A (en) | 1989-12-21 | 1996-02-06 | Goedecke Aktiengesellschaft | Indolocarbazole derivatives and the use thereof |
| US5169627A (en) * | 1991-10-28 | 1992-12-08 | Mount Sinai School Of Medicine Of The City University Of New York | Oral pharmaceutical composition containing a polyethylene glycol-immunoglobulin G conjugate for reconstitution of secretory immunity and method of reconstituting secretory immunity |
| US5698578A (en) | 1993-12-07 | 1997-12-16 | Eli Lilly And Company | Protein kinase C inhibitors |
| US5710145A (en) | 1995-11-20 | 1998-01-20 | Eli Lilly And Company | Protein kinase C inhibitor |
| EP0776895B1 (en) | 1995-11-20 | 1998-10-14 | Eli Lilly And Company | Protein kinase C inhibitor |
| US6110498A (en) | 1996-10-25 | 2000-08-29 | Shire Laboratories, Inc. | Osmotic drug delivery system |
| US6217903B1 (en) | 1997-04-28 | 2001-04-17 | Hercules Incorporated | Sustained release polymer blend for pharmaceutical applications |
| WO2002038561A1 (en) | 2000-11-07 | 2002-05-16 | Novartis Ag | Indolylmaleimide derivatives as protein kinase c inhibitors |
| US6645970B2 (en) | 2000-11-07 | 2003-11-11 | Novartis Ag | Indolylmaleimide derivatives |
| EP1437143A1 (en) * | 2001-09-28 | 2004-07-14 | Santen Pharmaceutical Co., Ltd. | Injections for eye tissue containing drug bound to polyethylene glycol |
| WO2003037385A1 (en) * | 2001-10-30 | 2003-05-08 | Nektar Therapeutics Al, Corporation | Water-soluble polymer conjugates of retinoic acid |
| US7235555B2 (en) | 2002-04-03 | 2007-06-26 | Novartis Ag | Indolylmaleimide derivatives |
| EP1491554A1 (en) * | 2003-06-23 | 2004-12-29 | CONARIS research institute AG | PEGylated soluble gp130-dimers useful as a medicament |
| WO2006002971A2 (en) | 2004-07-02 | 2006-01-12 | Creabilis Therapeutics S.P.A. | Nucleic acids for the treatment of hmgb1-related pathologies |
| WO2007006533A2 (en) | 2005-07-11 | 2007-01-18 | Novartis Ag | Indolylmaleimide derivatives |
| US20080318975A1 (en) | 2005-07-11 | 2008-12-25 | Jurgen Wagner | Indolylmaleimide Derivatives |
| US8673347B2 (en) | 2005-08-25 | 2014-03-18 | Creabilis Therapeutics S.P.A. | Polymer conjugates of K-252A and derivatives thereof |
| WO2008135557A1 (en) | 2007-05-07 | 2008-11-13 | Novartis Ag | Organic compounds |
| WO2009074575A2 (en) | 2007-12-10 | 2009-06-18 | Novartis Ag | Organic compounds |
| WO2009150137A2 (en) | 2008-06-10 | 2009-12-17 | Novartis Ag | Organic compounds |
| US8926955B2 (en) | 2008-12-22 | 2015-01-06 | Creabilis S.A. | Synthesis of polymer conjugates of indolocarbazole compounds |
| WO2011028740A1 (en) | 2009-09-03 | 2011-03-10 | Glaxo Group Limited | ENaC BLOCKERS |
| WO2011079087A1 (en) | 2009-12-23 | 2011-06-30 | Glaxo Group Limited | Enac blockers |
| WO2012035158A1 (en) | 2010-09-17 | 2012-03-22 | Novartis Ag | Pyrazine derivatives as enac blockers |
| WO2016004321A1 (en) * | 2014-07-03 | 2016-01-07 | Board Of Regents, The University Of Texas System | Compounds for treating biofilm infection |
Non-Patent Citations (33)
| Title |
|---|
| ALLEN, L.; ANSEL, H. C.: "Ansel's pharmaceutical dosage forms and drug delivery systems", 2013, LIPPINCOTT WILLIAMS & WILKINS |
| ANSEL, H. C.; POPOVICH, N. G.; ALLEN, L. V.: "Pharmaceutical dosage forms and drug delivery systems", 1995, LIPPINCOTT WILLIAMS & WILKINS |
| AULTON, M. E., & TAYLOR, K. M.: "Aulton's Pharmaceutics E-Book: The Design and Manufacture of Medicines", 2017, ELSEVIER HEALTH SCIENCES |
| BAMBOROUGH ET AL: "N-4-Pyrimidinyl-1H-indazol-4-amine inhibitors of Lck: Indazoles as phenol isosteres with improved pharmacokinetics", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, PERGAMON, AMSTERDAM, NL, vol. 17, no. 15, 1 August 2007 (2007-08-01), pages 4363 - 4368, XP022144705, ISSN: 0960-894X, DOI: 10.1016/J.BMCL.2007.04.029 * |
| BENATI, D.: "Src Family Kinases as Potential Therapeutic Targets for Malignancies and Immunological Disorders", CURRENT MEDICINAL CHEMISTRY, vol. 15, 2008, pages 1154 - 1165 |
| BROWN, LAWRENCE F. ET AL., JOURNAL OF INVESTIGATIVE DERMATOLOGY, vol. 104.5, 1995, pages 744 - 749 |
| CHANG RAE RHO ET AL: "Antiangiogenic Effects of Topically Administered Multiple Kinase Inhibitor, Motesanib (AMG 706), on Experimental Choroidal Neovascularization in Mice", JOURNAL OF OCULAR PHARMACOLOGY AND THERAPEUTICS., vol. 31, no. 1, 1 February 2015 (2015-02-01), US, pages 25 - 31, XP055478161, ISSN: 1080-7683, DOI: 10.1089/jop.2014.0023 * |
| CORMAC SHERIDAN: "CREABILIS RAISES $20M IN SERIES B TO ADVANCE PSORIASIS DRUG", 5 October 2011 (2011-10-05), XP002781147, Retrieved from the Internet <URL:http://www.bioworld.com/content/creabilis-raises-20m-series-b-advance-psoriasis-drug-0> [retrieved on 20180514] * |
| COSTA ET AL., ANGIOGENESIS, vol. 10, 2007, pages 149 - 166 |
| GREENE T. W. ET AL.: "Protective Groups in Organic Synthesis, 4th ed.,", 2007, JOHN WILEY AND SON |
| GREG T. HERMANSON: "Bioconjugate Techniques (3rd Edition)", 2013 |
| HANIFIN J. M., ARCH DERMATOL, vol. 135, 1999, pages 1551 |
| KOCH; CLAESSON-WELSH, COLD SPRING HARB. PERSPECT. MED., vol. 2, 2012, pages a006502 |
| LANG, J. C.: "Ocular drug delivery conventional ocular formulations", ADVANCED DRUG DELIVERY REVIEWS, vol. 16, no. 1, 1995, pages 39 - 43 |
| LE BOURLAIS, C.; ACAR, L.; ZIA, H.; SADO, P. A.; NEEDHAM, T.; LEVERGE, R.: "Ophthalmic drug delivery systems-recent advances", PROGRESS IN RETINAL AND EYE RESEARCH, vol. 17, no. 1, 1998, pages 33 - 58, XP055444156, DOI: doi:10.1016/S1350-9462(97)00002-5 |
| LEE ET AL., CELL, vol. 130, 2007, pages 691 - 703 |
| MISHRA ET AL.: "PEGYLATION IN ANTI-CANCER THERAPY: AN OVERVIEW", ASIAN JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 11, no. 3, 2016, pages 337 - 348, XP002781148 * |
| MUKTHAVARAM ET AL: "HIGH-EFFICIENCY LIPOSOMAL ENCAPSULATION OF A TYROSINE KINASE INHIBITOR LEADS TO IMPROVED IN VIVO TOXICITY AND TUMOR RESPONSE PROFILE", INTERNATIONAL JOURNAL OF NANOMEDICINE,, vol. 8, 1 January 2013 (2013-01-01), pages 3991 - 4006, XP002781387 * |
| MURAKAMI ET AL., BLOOD, vol. 108, 2006, pages 1849 - 1856 |
| NAGY ET AL., ANNU. REV. PATHOL., vol. 2, 2007, pages 251 - 275 |
| NAGY ET AL., COLD SPRING HARB. PERSPECT. MED., vol. 2, 2012, pages a006544 |
| RANE S.G.; REDDY E.P., ONCOGENE, vol. 19, 2000, pages 5662 - 5679 |
| RANE, S.G.; REDDY E.P., ONCOGENE, vol. 19, 2000, pages 5662 - 5679 |
| ROBLIN ET AL.: "TOPICAL TFKA KINASE INHIBITOR CT327 IS AN EFFECTIVE NOVEL THERAPY FOR THE TREATMENT OF PRURITUS DUE TO PSORIASIS", ACTA DERMATO-VENEREOLOGICA, vol. 92, 2015, pages 542 - 548, XP002781146 * |
| SINGH ET AL.: "Discovery and Development of Spleen Tyrosine Kinase (SYK) Inhibitors", J. MED. CHEM., vol. 55, 2012, pages 3614 - 3643, XP055051582, DOI: doi:10.1021/jm201271b |
| SLEVIN ET AL., EXPERT OPIN. INVESTIG. DRUGS, vol. 17, no. 9, 2008, pages 1301 - 1314 |
| SUGIMOTO, WORLD J. GASTROENTEROL., vol. 14, 2008, pages 5110 - 5114 |
| TIAN ET AL: "EFFECT OF STAUROSPORINE ON OUTFLOW FACILITY IN MONKEYS", INVESTIGATIVE OPHTHALMOLOGY AND VISUAL SCIENCE,, vol. 40, no. 5, 1 January 1999 (1999-01-01), pages 1009 - 1011, XP002781388 * |
| WEN, H., & PARK, K.: "Oral controlled release formulation design and drug delivery: theory to practice", 2011, JOHN WILEY & SONS |
| WILLIAMS, A.: "Transdermal and topical drug delivery: from theory to clinical practice", 2003, PHARMACEUTICAL PRESS, pages: 169 - 194 |
| XIA ET AL., BLOOD, vol. 102, 2003, pages 161 - 168 |
| YUAN GAO ET AL., PNAS, vol. 101, 18 May 2004 (2004-05-18), pages 7618 - 7623 |
| ZANGARINI ET AL.: "DEVELOPMENT AND VALIDATION OF LC-MS/MS WITH IN-SOURCE COLLISION-INDUCED DISSOCIATION FOR THE QUANTIFICATION OF PEGCANTRATINIB IN HUMAN SKIN TUMORS", BIOANALYSIS, vol. 9, no. 3, 23 January 2017 (2017-01-23), pages 279 - 288, XP002781145 * |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020023389A1 (en) * | 2018-07-23 | 2020-01-30 | Sienna Biopharmaceuticals, Inc. | Reduced exposure compositions modulating therapeutic targets |
| US11324833B2 (en) | 2018-11-07 | 2022-05-10 | Applied Molecular Transport Inc. | Cholix-derived carriers for oral delivery of heterologous payload |
| US11504433B2 (en) | 2018-11-07 | 2022-11-22 | Applied Molecular Transport Inc. | Cholix-derived carriers for oral delivery of heterologous payload |
| US11160869B2 (en) | 2019-08-16 | 2021-11-02 | Applied Molecular Transport Inc. | Compositions, formulations and interleukin production and purification |
| US11214606B2 (en) | 2019-08-16 | 2022-01-04 | Applied Molecular Transport Inc. | Compositions, formulations and interleukin production and purification |
| US11466067B2 (en) | 2019-08-16 | 2022-10-11 | Applied Molecular Transport Inc. | Compositions, formulations and interleukin production and purification |
| US11479593B2 (en) | 2019-08-16 | 2022-10-25 | Applied Molecular Transport Inc. | Compositions, formulations and interleukin production and purification |
| WO2024090606A1 (en) * | 2022-10-26 | 2024-05-02 | 경상국립대학교병원 | Method for treating posterior scleritis |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3600440A1 (en) | 2020-02-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11529352B2 (en) | Preservation of immune response during chemotherapy regimens | |
| WO2018175340A1 (en) | Reduced exposure conjugates modulating therapeutic targets | |
| US20200208128A1 (en) | Methods of treating liver diseases | |
| KR20180113976A (en) | Concurrent therapy of tetracycline quinolone analogs to treat cancer | |
| WO2020023389A1 (en) | Reduced exposure compositions modulating therapeutic targets | |
| KR102202481B1 (en) | Novel uses | |
| JP2010526883A (en) | How to treat skin ulcers | |
| TW201244716A (en) | Combination of a phosphatidylinositol-3-kinase (PI3K) inhibitor and a mTOR inhibitor | |
| JP2023107784A (en) | Vap-1 inhibitors for treating pain | |
| KR20210043499A (en) | Myeloproliferative neoplasm treatment method | |
| JP2024133636A (en) | Use of JAK1 inhibitors for the treatment of cutaneous lupus erythematosus and lichen planus (LP) | |
| US20230190755A1 (en) | Topical formulations of PI3K-delta inhibitors | |
| KR20200108297A (en) | Serdulatinib-containing topical dermal pharmaceutical composition and uses thereof | |
| US11992490B2 (en) | Use of JAK1 inhibitors for the treatment of cutaneous lupus erythematosus and Lichen planus (LP) | |
| CN111093661A (en) | Use of glutarimide derivatives for the treatment of diseases associated with abnormal activity of cytokines | |
| WO2020023387A1 (en) | Uses of polymer conjugates of indolocarbazole compounds with reduced exposure | |
| US20210017268A1 (en) | Methods for treating alcoholic liver disease, alcohol-induced brain injury and reducing alcohol addiction | |
| WO2017125502A1 (en) | Substance that inhibits p2y12 receptor, for use in the preventive treatment of systemic sclerosis in patients with raynaud's phenomenon and a dysimmunity | |
| JP2019511495A (en) | Treatment of CDKL5 disorders with the GSK3.BETA. | |
| WO2021252261A1 (en) | Compositions and methods for hair follicle regeneration | |
| WO2018109155A1 (en) | Trpa1 antagonists for use in the treatment of atopic dermatitis | |
| WO2018208369A1 (en) | Uses of polymer conjugates of indolocarbazole compounds with reduced exposure | |
| WO2024029597A1 (en) | DRY EYE REMEDY CONTAINING DNA OLIGONUCLEOTIDE SELECTIVELY BINDING TO IFN-γ | |
| WO2018175302A1 (en) | Polymer conjugates targeting c-src with reduced exposure | |
| RU2779478C2 (en) | Immune response preservation during chemotherapeutical shemes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18715453 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2018715453 Country of ref document: EP Effective date: 20191021 |