WO2018171602A1 - 用于ido和tdo双重抑制剂的脲类化合物 - Google Patents
用于ido和tdo双重抑制剂的脲类化合物 Download PDFInfo
- Publication number
- WO2018171602A1 WO2018171602A1 PCT/CN2018/079718 CN2018079718W WO2018171602A1 WO 2018171602 A1 WO2018171602 A1 WO 2018171602A1 CN 2018079718 W CN2018079718 W CN 2018079718W WO 2018171602 A1 WO2018171602 A1 WO 2018171602A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ring
- group
- alkyl
- compound
- pharmaceutically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- YNAVUWVOSKDBBP-UHFFFAOYSA-N C1NCCOC1 Chemical compound C1NCCOC1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 5
- GLUUGHFHXGJENI-UHFFFAOYSA-N C1NCCNC1 Chemical compound C1NCCNC1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 3
- BRNULMACUQOKMR-UHFFFAOYSA-N C1NCCSC1 Chemical compound C1NCCSC1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 3
- LOZWAPSEEHRYPG-UHFFFAOYSA-N C1SCCSC1 Chemical compound C1SCCSC1 LOZWAPSEEHRYPG-UHFFFAOYSA-N 0.000 description 3
- 0 CS(CCCC1/*2=C\N=C\CC=*=*22)(CC1P=C2NC(NI*)=S)=C* Chemical compound CS(CCCC1/*2=C\N=C\CC=*=*22)(CC1P=C2NC(NI*)=S)=C* 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N C1OCCOC1 Chemical compound C1OCCOC1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- JBYHSSAVUBIJMK-UHFFFAOYSA-N C1OCCSC1 Chemical compound C1OCCSC1 JBYHSSAVUBIJMK-UHFFFAOYSA-N 0.000 description 2
- OJUFHWVBTVWZQR-UHFFFAOYSA-N Brc1cc2ccccc2[n]2c1nnc2 Chemical compound Brc1cc2ccccc2[n]2c1nnc2 OJUFHWVBTVWZQR-UHFFFAOYSA-N 0.000 description 1
- ZGIKWINFUGEQEO-UHFFFAOYSA-N Brc1cnc(cccc2)c2c1 Chemical compound Brc1cnc(cccc2)c2c1 ZGIKWINFUGEQEO-UHFFFAOYSA-N 0.000 description 1
- CJQKWDBCHJFKSV-CIIODKQPSA-N C/C(/N=C(\C)/NN)=C\C=C/Br Chemical compound C/C(/N=C(\C)/NN)=C\C=C/Br CJQKWDBCHJFKSV-CIIODKQPSA-N 0.000 description 1
- ZZSPPTYWLPKIFG-UHFFFAOYSA-N C1CC(C(COC2)SC2C2OCCOC2)SCC1 Chemical compound C1CC(C(COC2)SC2C2OCCOC2)SCC1 ZZSPPTYWLPKIFG-UHFFFAOYSA-N 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N C1CCOCC1 Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- YPWFISCTZQNZAU-UHFFFAOYSA-N C1CCSCC1 Chemical compound C1CCSCC1 YPWFISCTZQNZAU-UHFFFAOYSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N CCN1CCOCC1 Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- SJRJJKPEHAURKC-UHFFFAOYSA-N CN1CCOCC1 Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 1
- MJOMRSZNMRVXGH-UHFFFAOYSA-N Cc(c1ccccc11)c(C)[n]2c1cnc2 Chemical compound Cc(c1ccccc11)c(C)[n]2c1cnc2 MJOMRSZNMRVXGH-UHFFFAOYSA-N 0.000 description 1
- KVMPWHZQGSYKRF-UHFFFAOYSA-N Cc1c(Cc(nc2C)ccc2O)cccc1 Chemical compound Cc1c(Cc(nc2C)ccc2O)cccc1 KVMPWHZQGSYKRF-UHFFFAOYSA-N 0.000 description 1
- KVRLIEFLTYSTCI-UHFFFAOYSA-N Cc1cc(cccc2)c2[n]2c1cnc2 Chemical compound Cc1cc(cccc2)c2[n]2c1cnc2 KVRLIEFLTYSTCI-UHFFFAOYSA-N 0.000 description 1
- GILLCUNOXPSKFS-UHFFFAOYSA-N Cc1cc(cccc2)c2[n]2c1nnc2 Chemical compound Cc1cc(cccc2)c2[n]2c1nnc2 GILLCUNOXPSKFS-UHFFFAOYSA-N 0.000 description 1
- ISAAQXDGJXSKGR-UHFFFAOYSA-N Cc1cc2ccccc2c2cnc[n]12 Chemical compound Cc1cc2ccccc2c2cnc[n]12 ISAAQXDGJXSKGR-UHFFFAOYSA-N 0.000 description 1
- SYHULSKRGVFEHP-UHFFFAOYSA-N Cc1cc2ccccc2c2cnn[n]12 Chemical compound Cc1cc2ccccc2c2cnn[n]12 SYHULSKRGVFEHP-UHFFFAOYSA-N 0.000 description 1
- LRRDYPSCLJGQRQ-UHFFFAOYSA-N Cc1cc2ccccc2c2nnn[n]12 Chemical compound Cc1cc2ccccc2c2nnn[n]12 LRRDYPSCLJGQRQ-UHFFFAOYSA-N 0.000 description 1
- BEFOQKLZMNIYTC-UHFFFAOYSA-N Cc1cc2ncccc2c2cnc[n]12 Chemical compound Cc1cc2ncccc2c2cnc[n]12 BEFOQKLZMNIYTC-UHFFFAOYSA-N 0.000 description 1
- XECMLOGMXSDOCR-UHFFFAOYSA-N Cc1ccc[n]2c1cnc2 Chemical compound Cc1ccc[n]2c1cnc2 XECMLOGMXSDOCR-UHFFFAOYSA-N 0.000 description 1
- AMTVPXODMPYFPU-UHFFFAOYSA-N Cc1cccc2cnc[n]12 Chemical compound Cc1cccc2cnc[n]12 AMTVPXODMPYFPU-UHFFFAOYSA-N 0.000 description 1
- JMQFEGJORFJQDU-UHFFFAOYSA-N Cc1nc2ccccc2c2cnc[n]12 Chemical compound Cc1nc2ccccc2c2cnc[n]12 JMQFEGJORFJQDU-UHFFFAOYSA-N 0.000 description 1
- XZGLNCKSNVGDNX-UHFFFAOYSA-N Cc1nnn[nH]1 Chemical compound Cc1nnn[nH]1 XZGLNCKSNVGDNX-UHFFFAOYSA-N 0.000 description 1
- RWWUFTQVKMNRQN-UHFFFAOYSA-N Clc1nc2ccccc2cc1Br Chemical compound Clc1nc2ccccc2cc1Br RWWUFTQVKMNRQN-UHFFFAOYSA-N 0.000 description 1
- HPLPQYDUAVRXNY-UHFFFAOYSA-N c1nnc2[n]1c1ccccc1cc2N=C(c1ccccc1)c1ccccc1 Chemical compound c1nnc2[n]1c1ccccc1cc2N=C(c1ccccc1)c1ccccc1 HPLPQYDUAVRXNY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
- C07D231/56—Benzopyrazoles; Hydrogenated benzopyrazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the invention belongs to the technical field of medicine, relates to urea compounds of dual inhibitors of IDO and TDO, a preparation method thereof, a pharmaceutical composition containing the same, and the activity thereof in regulating IDO and TDO and treating diseases and disorders mediated by them the use of.
- IDO Indoleamine 2,3-dioxygenase
- TDO tryptophan 2,3-oxygenase
- IDO inhibitors may be an effective treatment for these diseases.
- IDO inhibitors are mainly classified into the following categories: 1) competitive inhibitors such as tryptophan derivatives 1-MT; 2) non-competitive inhibitors such as phenylimidazole; 3) anti-competitive Inhibitors such as alkaloids exiguamine A; 4) inhibitors through other mechanisms of action.
- TDO is expressed in a variety of tumor cells, resulting in increased survival and migration of tumor cells and inhibition of the immune system's ability to respond to tumor cells.
- the effect of TDO on tumor cells and immunosuppression has made it a new anti-tumor drug target. Therefore, screening new IDO/TDO inhibitors and studying their in vivo activities, pharmacokinetics, therapeutic effects and adverse reactions are still a field worth exploring.
- the present invention provides a compound of formula I or a pharmaceutically acceptable salt thereof,
- Ring A is absent or ring A is a benzene ring or contains 1, 2 or 3 5-6 membered heteroaryl rings selected from N, O or S atoms;
- Ring B is a five-membered aromatic ring in which one, two, three or four of W, V, X, Y and Z are selected from N or NH, and others are selected from C or CH, specifically,
- Y and V are N, Z and W are CH, and X is C;
- X is N, Z, V and W are CH, and Y is C;
- Y is N, Z, V and W are CH, and X is C;
- Viii) Z is NH, V and W are CH, and X and Y are C;
- V is NH, Z and W are CH, and X and Y are C;
- x) W is NH, V and Z are CH, and X and Y are C;
- V and W are N or NH, Z is CH, and X and Y are C;
- V and Z are N or NH, W is CH, and X and Y are C;
- Xiii) Z and W are N or NH, V is CH, and X and Y are C;
- Y and W are N, V and Z are CH, and X is C;
- X and W are N, V and Z are CH, and Y is C;
- X and Z are N or NH, V and W are CH, and Y is C;
- Y and Z are N, V and W are CH, and X is C;
- Xix) Z, V and W are N or NH, X and Y are C;
- Xxi) Y, Z and W are N, V is CH, X is C; or
- P is C(R 6 ) or N
- Q is O or S
- D is selected from NH, O, S or CH 2 ;
- E is selected from NH, O or CH 2 ;
- L is selected from the group consisting of a single bond, S, SO, SO 2 , CO, C(O)O, S(O)O or S(O) 2 O;
- Each R 1 is independently selected from halogen, amino, hydroxy, cyano, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 cycloalkyl or C 3-6 heterocycloalkyl;
- R 2 is selected from C 3-12 cycloalkyl, C 3-12 heterocycloalkyl, 6-12 membered aryl or 5-12 membered heteroaryl containing 1, 2 or 3 selected from N, O or S atoms. a group, which may be optionally substituted with one or more groups independently selected from R 3 ;
- R 3 is selected from the group consisting of halogen, cyano, nitro, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 cycloalkyl, C 3-6 heterocycloalkyl, containing 1, 2 or 3 5-6 membered heteroaryl selected from N, O or S atoms, C 3-6 cycloalkyl CH 2 -, C 3-6 heterocycloalkyl CH 2 -, containing 1, 2 or 3 5-6 membered heteroaryl CH 2 -, -OR 4 , -SR 4 , -N(R 4 ) 2 , -C(O)OR 4 , -C(O)N from N, O or S atoms R 4 ) 2 , -C(O)R 4 , -S(O)R 4 , -S(O)OR 4 , -S(O)N(R 4 ) 2 , -S(O) 2 R 4 , -S(O) 2 OR 4 ,
- Each R 4 is independently selected from H, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 cycloalkyl, C 3-6 heterocycloalkyl, phenyl or contains 1, 2 Or 3 5-6 membered heteroaryl selected from N, O or S atoms;
- R 5 is selected from the group consisting of halogen, amino, cyano, hydroxy, -COOH, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 cycloalkyl or C 3-6 heterocycloalkyl;
- R 6 is selected from H, halogen, amino, cyano, hydroxy, -COOH or halogenated C 1-3 alkyl;
- n 0, 1, or 2.
- ring A is absent or ring A is a benzene ring, a furan ring, a thiophene ring, a pyrrole ring, a pyrazole ring, an imidazole ring, a pyridine ring, a pyrimidine ring, a pyridazine ring, a pyrazine.
- ring A is absent or ring A is a benzene ring or a pyridine ring.
- ring A is absent or ring A is
- each R 1 is independently selected from halo, halo C 1-3 alkyl or C 1-6 alkyl.
- each R 1 is independently selected from the group consisting of fluorine, chlorine, bromine, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, Fluoromethyl, difluoromethyl, trifluoromethyl, monofluoroethyl, difluoroethyl, trifluoroethyl, tetrafluoroethyl, pentafluoroethyl, monochloromethyl, dichloromethyl or tri Chloromethyl.
- each R 1 is independently selected from fluoro, chloro or trifluoromethyl.
- m is 0 or 1.
- ring B is an aromatic ring wherein i) Y and V are N, Z and W are CH, X is C; ii) Y, Z, V and W are N, X Is C; iii) X and V are N, Z and W are CH, Y is C; iv) Y, Z and V are N, W is CH, X is C; v) X, Z and V are N, W Is CH, Y is C.
- D is selected from NH, O or CH 2 ;
- E is selected from NH, O or CH 2 .
- -DC(Q)-E- is selected from the group consisting of -NHC(O)NH-, -NHC(S)NH-, -OC(O)NH-, -NHC(O) CH 2 - or -CH 2 C(O)NH-.
- One embodiment of the compounds of the invention of formula I, L is selected from a single bond or SO 2.
- R 2 is selected from C 3-6 cycloalkyl, C 3-6 heterocycloalkyl, phenyl or contains 1, 2 or 3 selected from N, O or S atoms 5-6 membered heteroaryl group, which may optionally be substituted with one or more groups independently selected R 3 groups.
- R 2 is selected from the group consisting of cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and the loss of one hydrogen atom at any position.
- R 2 is selected from cyclohexyl, which loses a hydrogen atom at any position.
- Phenyl, pyridyl or pyrimidyl group which may optionally be substituted with one or more substituents independently selected from R 3 group.
- R 2 is selected from Which may be optionally substituted with one or more groups independently selected R 3 groups.
- R 2 is selected from
- R 3 is selected from the group consisting of halogen, cyano, nitro, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 heterocycloalkyl, containing 1 , 2 or 3 5-6 membered heteroaryl selected from N, O or S atoms, C 3-6 heterocycloalkyl CH 2 -, -OR 4 , -SR 4 , -S(O) 2 R 4 , -S(O) 2 N(R 4 ) 2 , -C(O)OR 4 , -C(O)N(R 4 ) 2 , -N(R 4 )S(O) 2 N(R 4 ) 2 , -P(O)(R 4 ) 2 or -P(O)(OR 4 ) 2 , wherein C 1-6 alkyl, C 3-6 heterocycloalkyl, containing 1, 2 or 3 are selected from The 5-6 membered heteroaryl or C
- R 3 is selected from the group consisting of fluorine, chlorine, bromine, cyano, nitro, monofluoromethyl, difluoromethyl, trifluoromethyl, monofluoroethyl, difluoroethyl , trifluoroethyl, tetrafluoroethyl, pentafluoroethyl, monochloromethyl, dichloromethyl, trichloromethyl, -OR 4 , -SR 4 , -S(O) 2 R 4 ,- S(O) 2 N(R 4 ) 2 , -C(O)OR 4 , -C(O)N(R 4 ) 2 , -N(R 4 )S(O) 2 N(R 4 ) 2 , -P(O)(R 4 ) 2 , -P(O)(OR 4 ) 2 , methyl, ethyl, propyl, iso
- R 3 is selected from fluoro, chloro, bromo, cyano, nitro, trifluoromethyl, -OR 4, -SR 4, -S (O) 2 R 4, - S(O) 2 N(R 4 ) 2 , -C(O)OR 4 , -C(O)N(R 4 ) 2 , -N(R 4 )S(O) 2 N(R 4 ) 2 , -P(O)(R 4 ) 2 , -P(O)(OR 4 ) 2 , methyl, loss of one hydrogen atom at any position Tetrazolyl or Where the methyl group, at any position, loses a hydrogen atom Tetrazolyl or Optionally substituted with one or more groups independently selected from R 5 .
- R 3 is selected from fluoro, chloro, bromo, cyano, nitro, trifluoromethyl, -OCF 3, -SCH 3, -S (O) 2 CH 3, - S(O) 2 NH 2 , -C(O)OCH 3 , -COOH, -C(O)NHCH 3 , -C(O)NH 2 , -NHS(O) 2 NH 2 , -P(O) ( CH 3 ) 2 , -P(O)(OCH 2 CH 3 ) 2 , -C(OH)(CF 3 ) 2 ,
- each R4 is independently selected from H, halo C1-3 alkyl or C1-6 alkyl.
- each R 4 is independently selected from the group consisting of H, monofluoromethyl, difluoromethyl, trifluoromethyl, monofluoroethyl, difluoroethyl, trifluoroethyl , tetrafluoroethyl, pentafluoroethyl, monochloromethyl, dichloromethyl, trichloromethyl, methyl, ethyl, propyl, isopropyl, butyl, isobutyl or tert-butyl.
- each R 4 is independently selected from H, trifluoromethyl, methyl or ethyl.
- R 5 is selected from the group consisting of hydroxy, monofluoromethyl, difluoromethyl, trifluoromethyl, monofluoroethyl, difluoroethyl, trifluoroethyl, tetrafluoroethyl Base, pentafluoroethyl, monochloromethyl, dichloromethyl or trichloromethyl.
- R 5 is selected from hydroxy or trifluoromethyl.
- R 6 is selected from H or halogen.
- R 6 is selected from H or chloro.
- the invention provides a compound of Formula II, or a pharmaceutically acceptable salt thereof,
- Ring A is a benzene ring or a 5-6 membered heteroaryl ring containing 1, 2 or 3 atoms selected from N, O or S atoms;
- Ring B is an aromatic ring in which one, two, three or four of W, V, X, Y and Z are selected from N or NH, and others are selected from C or CH, in particular,
- Y and V are N, Z and W are CH, and X is C;
- X is N, Z, V and W are CH, and Y is C;
- Y is N, Z, V and W are CH, and X is C;
- Viii) Z is NH, V and W are CH, and X and Y are C;
- V is NH, Z and W are CH, and X and Y are C;
- x) W is NH, V and Z are CH, and X and Y are C;
- V and W are N or NH, Z is CH, and X and Y are C;
- V and Z are N or NH, W is CH, and X and Y are C;
- Xiii) Z and W are N or NH, V is CH, and X and Y are C;
- Y and W are N, V and Z are CH, and X is C;
- X and W are N, V and Z are CH, and Y is C;
- X and Z are N or NH, V and W are CH, and Y is C;
- Y and Z are N, V and W are CH, and X is C;
- Xix) Z, V and W are N or NH, X and Y are C;
- Xxi) Y, Z and W are N, V is CH, X is C; or
- P is C(R 6 ) or N
- Q is O or S
- L is selected from the group consisting of a single bond, S, SO, SO 2 , CO, C(O)O, S(O)O or S(O) 2 O;
- Each R 1 is independently selected from halogen, amino, hydroxy, cyano, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 cycloalkyl or C 3-6 heterocycloalkyl;
- R 2 is selected from C 3-12 cycloalkyl, C 3-12 heterocycloalkyl, 6-12 membered aryl or 5-12 membered heteroaryl containing 1, 2 or 3 selected from N, O or S atoms. a group, which may be optionally substituted with one or more groups independently selected from R 3 ;
- R 3 is selected from the group consisting of halogen, cyano, nitro, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 cycloalkyl, C 3-6 heterocycloalkyl, containing 1, 2 or 3 5-6 membered heteroaryl selected from N, O or S atoms, C 3-6 cycloalkyl CH 2 -, C 3-6 heterocycloalkyl CH 2 -, containing 1, 2 or 3 5-6 membered heteroaryl CH 2 -, -OR 4 , -SR 4 , -N(R 4 ) 2 , -C(O)OR 4 , -C(O)N from N, O or S atoms R 4 ) 2 , -C(O)R 4 , -S(O)R 4 , -S(O)OR 4 , -S(O)N(R 4 ) 2 , -S(O) 2 R 4 , -S(O) 2 OR 4 ,
- Each R 4 is independently selected from H, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 cycloalkyl, C 3-6 heterocycloalkyl, phenyl or contains 1, 2 Or 3 5-6 membered heteroaryl selected from N, O or S atoms;
- R 5 is selected from the group consisting of halogen, amino, cyano, hydroxy, -COOH, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 cycloalkyl or C 3-6 heterocycloalkyl;
- R 6 is selected from H, halogen, amino, cyano, hydroxy, -COOH or halogenated C 1-3 alkyl;
- n 0, 1, or 2.
- Ring A is a benzene ring, a furan ring, a thiophene ring, a pyrrole ring, a pyrazole ring, an imidazole ring, a pyridine ring, a pyrimidine ring, a pyridazine ring, a pyrazine ring, a thiazole ring, Isothiazole ring, oxazole ring, isoxazole ring, tetrazole ring or triazine ring.
- Ring A is a phenyl ring or a pyridine ring.
- ring A is absent or ring A is
- each R 1 is independently selected from halo, halo C 1-3 alkyl or C 1-6 alkyl.
- each R 1 is independently selected from the group consisting of fluorine, chlorine, bromine, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, Fluoromethyl, difluoromethyl, trifluoromethyl, monofluoroethyl, difluoroethyl, trifluoroethyl, tetrafluoroethyl, pentafluoroethyl, monochloromethyl, dichloromethyl or tri Chloromethyl.
- each R 1 is independently selected from fluoro, chloro or trifluoromethyl.
- m is 0 or 1.
- ring B is an aromatic ring wherein i) Y and V are N, Z and W are CH, X is C; ii) Y, Z, V and W are N, X Is C; iii) X and V are N, Z and W are CH, Y is C; iv) Y, Z and V are N, W is CH, X is C; v) X, Z and V are N, W Is CH, Y is C.
- L is selected from a single bond or SO 2.
- R 2 is selected from C 3-6 cycloalkyl, C 3-6 heterocycloalkyl, phenyl or contains 1, 2 or 3 selected from N, O or S atoms. 5-6 membered heteroaryl group, which may optionally be substituted with one or more groups independently selected R 3 groups.
- R 2 is selected from the group consisting of cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and a hydrogen atom is lost at any position.
- R 2 is selected from cyclohexyl, which loses a hydrogen atom at any position.
- Phenyl, pyridyl or pyrimidyl group which may optionally be substituted with one or more substituents independently selected from R 3 group.
- R 2 is selected from Which may be optionally substituted with one or more groups independently selected R 3 groups.
- R 2 is selected from
- R 3 is selected from the group consisting of halogen, cyano, nitro, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 heterocycloalkyl, containing 1 , 2 or 3 5-6 membered heteroaryl selected from N, O or S atoms, C 3-6 heterocycloalkyl CH 2 -, -OR 4 , -SR 4 , -S(O) 2 R 4 , -S(O) 2 N(R 4 ) 2 , -C(O)OR 4 , -C(O)N(R 4 ) 2 , -N(R 4 )S(O) 2 N(R 4 ) 2 , -P(O)(R 4 ) 2 or -P(O)(OR 4 ) 2 , wherein C 1-6 alkyl, C 3-6 heterocycloalkyl, containing 1, 2 or 3 are selected from The 5-6 membered heteroaryl or C
- R 3 is selected from fluoro, chloro, bromo, cyano, nitro, fluoromethyl, difluoromethyl, trifluoromethyl group, a fluoroethyl, difluoro ethyl , trifluoroethyl, tetrafluoroethyl, pentafluoroethyl, monochloromethyl, dichloromethyl, trichloromethyl, -OR 4 , -SR 4 , -S(O) 2 R 4 ,- S(O) 2 N(R 4 ) 2 , -C(O)OR 4 , -C(O)N(R 4 ) 2 , -N(R 4 )S(O) 2 N(R 4 ) 2 , -P(O)(R 4 ) 2 , -P(O)(OR 4 ) 2 , methyl, ethyl, propyl, isopropyl
- R 3 is selected from fluoro, chloro, bromo, cyano, nitro, trifluoromethyl, -OR 4, -SR 4, -S (O) 2 R 4, - S(O) 2 N(R 4 ) 2 , -C(O)OR 4 , -C(O)N(R 4 ) 2 , -N(R 4 )S(O) 2 N(R 4 ) 2 , -P(O)(R 4 ) 2 , -P(O)(OR 4 ) 2 , methyl, loss of one hydrogen atom at any position Tetrazolyl or Where the methyl group, at any position, loses a hydrogen atom Tetrazolyl or Optionally substituted with one or more groups independently selected from R 5 .
- R 3 is selected from fluoro, chloro, bromo, cyano, nitro, trifluoromethyl, -OCF 3, -SCH 3, -S (O) 2 CH 3, - S(O) 2 NH 2 , -C(O)OCH 3 , -COOH, -C(O)NHCH 3 , -C(O)NH 2 , -NHS(O) 2 NH 2 , -P(O) ( CH 3 ) 2 , -P(O)(OCH 2 CH 3 ) 2 , -C(OH)(CF 3 ) 2 ,
- each R 4 is independently selected from H, halo C 1-3 alkyl or C 1-6 alkyl.
- each R 4 is independently selected from the group consisting of H, monofluoromethyl, difluoromethyl, trifluoromethyl, monofluoroethyl, difluoroethyl, trifluoroethyl , tetrafluoroethyl, pentafluoroethyl, monochloromethyl, dichloromethyl, trichloromethyl, methyl, ethyl, propyl, isopropyl, butyl, isobutyl or tert-butyl.
- each R 4 is independently selected from H, trifluoromethyl, methyl or ethyl.
- R 5 is selected from the group consisting of hydroxy, monofluoromethyl, difluoromethyl, trifluoromethyl, monofluoroethyl, difluoroethyl, trifluoroethyl, tetrafluoroethyl Base, pentafluoroethyl, monochloromethyl, dichloromethyl or trichloromethyl.
- R 5 is selected from hydroxy or trifluoromethyl.
- R 6 is selected from H or halogen.
- R 6 is selected from H or chloro.
- the present invention provides a compound of Formula II-1, or a pharmaceutically acceptable salt thereof,
- the invention provides a compound of Formula II-2, or a pharmaceutically acceptable salt thereof,
- the present invention provides a compound of Formula II-3, or a pharmaceutically acceptable salt thereof,
- the invention provides a compound of Formula II-4, or a pharmaceutically acceptable salt thereof,
- the invention provides a compound of Formula II-5, or a pharmaceutically acceptable salt thereof,
- the invention provides a compound of Formula III, or a pharmaceutically acceptable salt thereof,
- Ring B is an aromatic ring in which one, two, three or four of W, V, X, Y and Z are selected from N or NH, and others are selected from C or CH, in particular,
- Y and V are N, Z and W are CH, and X is C;
- X is N, Z, V and W are CH, and Y is C;
- Y is N, Z, V and W are CH, and X is C;
- Viii) Z is NH, V and W are CH, and X and Y are C;
- V is NH, Z and W are CH, and X and Y are C;
- x) W is NH, V and Z are CH, and X and Y are C;
- V and W are N or NH, Z is CH, and X and Y are C;
- V and Z are N or NH, W is CH, and X and Y are C;
- Xiii) Z and W are N or NH, V is CH, and X and Y are C;
- Y and W are N, V and Z are CH, and X is C;
- X and W are N, V and Z are CH, and Y is C;
- X and Z are N or NH, V and W are CH, and Y is C;
- Y and Z are N, V and W are CH, and X is C;
- Xix) Z, V and W are N or NH, X and Y are C;
- Xxi) Y, Z and W are N, V is CH, X is C; or
- Q is O or S
- D is selected from NH, O, S or CH 2 ;
- E is selected from NH, O, or CH 2 ;
- L is selected from the group consisting of a single bond, S, SO, SO 2 , CO, C(O)O, S(O)O or S(O) 2 O;
- Each R 1 is independently selected from halogen, amino, hydroxy, cyano, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 cycloalkyl or C 3-6 heterocycloalkyl;
- R 2 is selected from C 3-12 cycloalkyl, C 3-12 heterocycloalkyl, 6-12 membered aryl or 5-12 membered heteroaryl containing 1, 2 or 3 selected from N, O or S atoms. a group, which may be optionally substituted with one or more groups independently selected from R 3 ;
- R 3 is selected from the group consisting of halogen, cyano, nitro, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 cycloalkyl, C 3-6 heterocycloalkyl, containing 1, 2 or 3 5-6 membered heteroaryl selected from N, O or S atoms, C 3-6 cycloalkyl CH 2 -, C 3-6 heterocycloalkyl CH 2 -, containing 1, 2 or 3 5-6 membered heteroaryl CH 2 -, -OR 4 , -SR 4 , -N(R 4 ) 2 , -C(O)OR 4 , -C(O)N from N, O or S atoms R 4 ) 2 , -C(O)R 4 , -S(O)R 4 , -S(O)OR 4 , -S(O)N(R 4 ) 2 , -S(O) 2 R 4 , -S(O) 2 OR 4 ,
- Each R 4 is independently selected from H, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 cycloalkyl, C 3-6 heterocycloalkyl, phenyl or contains 1, 2 Or 3 5-6 membered heteroaryl selected from N, O or S atoms;
- R 5 is selected from the group consisting of halogen, amino, cyano, hydroxy, -COOH, halo C 1-3 alkyl, C 1-6 alkyl, C 3-6 cycloalkyl or C 3-6 heterocycloalkyl;
- n 0, 1, or 2.
- each R 1 is independently selected from halo, halo C 1-3 alkyl or C 1-6 alkyl.
- each R 1 is independently selected from fluoro, chloro, bromo, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, Fluoromethyl, difluoromethyl, trifluoromethyl, monofluoroethyl, difluoroethyl, trifluoroethyl, tetrafluoroethyl, pentafluoroethyl, monochloromethyl, dichloromethyl or tri Chloromethyl.
- each R 1 is independently selected from chloro or trifluoromethyl.
- m is 0 or 1.
- ring B is an aromatic ring wherein i) Y and V are N, Z and W are CH, X is C; ii) X and V are N, Z and W are CH , Y is C.
- D is selected from NH, O or CH 2; E is selected from NH, O or CH 2.
- -DC(Q)-E- is selected from the group consisting of -NHC(O)NH-, -NHC(S)NH-, -OC(O)NH-, -NHC(O) CH 2 - or -CH 2 C(O)NH-.
- L is selected from the group consisting of a single bond.
- R 2 is selected from phenyl or contains 1, 2 or 3 5-6 membered heteroaryl selected from N, O or S atoms, which may optionally be one or A plurality of groups independently selected from R 3 are substituted.
- R 2 is selected from the group consisting of phenyl, furanyl, thienyl, pyrrolyl, pyrazolyl, imidazolyl, pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, thiazolyl , isothiazolyl, oxazolyl, isoxazolyl, tetrazolyl, triazinyl, which may be optionally substituted with one or more groups independently selected R 3 groups.
- R 2 is selected from phenyl, pyridyl or pyrimidyl group, which may optionally be substituted with one or more substituents independently selected from R 3 group.
- R 2 is selected from Which may be optionally substituted with one or more groups independently selected R 3 groups.
- R 2 is selected from
- R 3 is selected from halogen or halogenated C 1-3 alkyl.
- R 3 is selected from fluoro, chloro, bromo or trifluoromethyl.
- the invention provides a compound of formula III-1, or a pharmaceutically acceptable salt thereof,
- the invention provides a compound of formula III-2, or a pharmaceutically acceptable salt thereof,
- the present invention preferably comprises the following compound or a pharmaceutically acceptable salt thereof,
- the invention in another aspect, relates to a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula I, a compound of formula II, and a compound of formula III, or a pharmaceutically acceptable salt thereof, of the invention.
- the pharmaceutical compositions of the present invention further comprise one or more pharmaceutically acceptable carriers or excipients.
- the pharmaceutical compositions of the invention may further comprise one or more additional therapeutic agents.
- the invention relates to a method of treating a mammalian immunosuppressive disorder mediated by indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-oxygenase (TDO), including
- IDO indoleamine 2,3-dioxygenase
- TDO tryptophan 2,3-oxygenase
- a mammal preferably a human in need of such treatment, is administered a therapeutically effective amount of a compound of formula I, a compound of formula II, and a compound of formula III, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof.
- the invention relates to a compound of formula I, a compound of formula II, and a compound of formula III, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, for the prophylaxis or treatment of guanamine 2,3-dual addition
- Uses of oxygenase (IDO) and tryptophan 2,3-oxygenase (TDO) mediated drugs for immunosuppressive diseases for the prophylaxis or treatment of guanamine 2,3-dual addition
- IDO oxygenase
- TDO tryptophan 2,3-oxygenase
- the present invention relates to a compound of formula I, a compound of formula II, and a compound of formula III, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, for use in the prophylaxis or treatment of guanamine 2,3-dual plus Use of oxygenase (IDO) and tryptophan 2,3-oxygenase (TDO) mediated immunosuppressive diseases.
- IDO oxygenase
- TDO tryptophan 2,3-oxygenase
- the immunosuppressive disease is associated with an infectious disease or cancer.
- the immunosuppressive disease may be selected from, but not limited to, pneumonia, malignancy, measles, hepatitis, kidney disease or arthritis.
- the infectious disease is selected from the group consisting of influenza, hepatitis C virus (HCV), human papillomavirus (HPV), cytomegalovirus (CMV), poliovirus , herpes zoster virus, human immunodeficiency virus (HIV), Epstein-Barr virus (EBV) or Coxsackie virus.
- the cancer is selected from the group consisting of colon cancer, pancreatic cancer, breast cancer, prostate cancer, lung cancer, brain cancer, ovarian cancer, cervical cancer, testicular cancer, renal cancer, head or neck cancer, lymphoma, leukemia or melanoma.
- references to “one embodiment” or “an embodiment” or “in another embodiment” or “in some embodiments” throughout the specification are meant to include in the at least one embodiment The specific reference elements, structures or features described.
- the appearances of the phrase “in one embodiment” or “in an embodiment” or “in another embodiment” or “in some embodiments” are not necessarily all referring to the same embodiment.
- the particular elements, structures, or characteristics may be combined in any suitable manner in one or more embodiments.
- a reaction including a “catalyst” includes a catalyst, or two or more catalysts.
- the term “or” is generally used in its meaning including “and/or” unless it is specifically defined otherwise.
- an ethyl group “optionally” means ethyl substituted with halo may be unsubstituted (CH 2 CH 3), monosubstituted (e.g., CH 2 CH 2 F), polysubstituted (e.g. CHFCH 2 F, CH 2 CHF 2, etc.) or completely substituted (CF 2 CF 3 ). It will be understood by those skilled in the art that for any group containing one or more substituents, no substitution or substitution pattern that is sterically impossible to exist and/or which cannot be synthesized is introduced.
- C mn means having mn carbon atoms in this moiety.
- C 3-10 cycloalkyl means that the cycloalkyl group has 3 to 10 carbon atoms.
- the "C 0-6 alkylene group” means that the alkylene group has 0 to 6 carbon atoms, and when the alkylene group has 0 carbon atoms, the group is a bond. It is easy to understand that when a hetero atom is contained, mn represents the sum of the number of carbon atoms and hetero atoms.
- C 1-10 means that the group may have 1 carbon atom, 2 carbon atoms, 3 carbon atoms, 4 carbon atoms, 5 carbon atoms, 6 carbon atoms, 7 carbon atoms, 8 One carbon atom, nine carbon atoms or ten carbon atoms.
- substituted means that any one or more hydrogen atoms on a particular atom are replaced by a substituent as long as the valence of the particular atom is normal and the substituted compound is stable.
- any variable eg, R
- its definition in each case is independent.
- the group may optionally be substituted with at most two R, and each case has an independent option.
- combinations of substituents and/or variants thereof are permissible only if such combinations result in stable compounds.
- linking group When the number of one linking group is 0, such as -(CRR) 0 -, it indicates that the linking group is a single bond.
- one of the variables When one of the variables is selected from a single bond, it means that the two groups to which it is attached are directly linked. For example, when L represents a single bond in A-L-Z, the structure is actually A-Z.
- substituent When a substituent is vacant, it means that the substituent is absent. For example, when X is vacant in AX, the structure is actually A. When a bond of a substituent can be cross-linked to two atoms on a ring, the substituent can be bonded to any atom on the ring. When the recited substituents do not indicate which atom is attached to a compound included in the chemical structural formula including but not specifically mentioned, such a substituent may be bonded through any atomic phase thereof. Combinations of substituents and/or variants thereof are permissible only if such combinations result in stable compounds. For example, a structural unit It is indicated that it can be substituted at any position on the cyclohexyl or cyclohexadiene.
- pharmaceutically acceptable is for those compounds, materials, compositions and/or dosage forms that are suitable for use in contact with human and animal tissues within the scope of sound medical judgment without excessive Toxicity, irritation, allergic reactions or other problems or complications are commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salt refers to a salt of a compound of the present invention, as a pharmaceutically acceptable salt of a compound of the formula I, formula II and formula III, for example, a metal salt, an ammonium salt, a salt formed with an organic base, a salt formed with an inorganic acid, a salt formed with an organic acid, or a salt formed with a basic or acidic amino acid.
- metal salts include, but are not limited to, salts of alkali metals such as sodium salts, potassium salts, and the like; salts of alkaline earth metals such as calcium salts, magnesium salts, barium salts, and the like; aluminum salts and the like.
- the pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound containing an acid group or a base by conventional chemical methods.
- such salts are prepared by reacting a compound of the free acid or base form with a stoichiometric amount of a suitable base or acid in water or an organic solvent or a mixture of the two.
- a nonaqueous medium such as ether, ethyl acetate, ethanol, isopropanol or acetonitrile is preferred.
- the compounds of formula I, formula II and formula III of the present invention may exist in unsolvated or solvated forms, including hydrated forms. In general, the solvated forms are equivalent to the unsolvated forms and are included within the scope of the invention.
- the compounds of formula I of the present invention may exist in polymorph or amorphous form.
- the compounds of the formula I, formula II and formula III of the invention may have asymmetric carbon atoms (optical centers) or double bonds. Racemates, diastereomers, geometric isomers and individual isomers are included within the scope of the invention.
- the compounds of formula I, formula II and formula III of the invention may exist in specific geometric or stereoisomeric forms. All such compounds are contemplated by the present invention, including cis and trans isomers, (-)- and (+)-enantiomers, (R)- and (S)-enantiomers, diastereoisomers , (D)-isomer, (L)-isomer, and racemic mixtures thereof and other mixtures, such as enantiomerically or diastereomeric enriched mixtures, all of which belong to the present Within the scope of the invention. Additional asymmetric carbon atoms may be present in the substituents such as alkyl groups. All such isomers, as well as mixtures thereof, are also included within the scope of the invention.
- optically active (R)- and (S)-isomers as well as the D and L isomers can be prepared by chiral synthesis or chiral reagents or other conventional techniques. If an enantiomer of a compound of the invention is desired, it can be prepared by asymmetric synthesis or by derivatization with a chiral auxiliary wherein the resulting mixture of diastereomers is separated and the auxiliary group cleaved to provide purity The desired enantiomer.
- a molecule contains a basic functional group (such as an amino group) or an acidic functional group (such as a carboxyl group)
- a diastereomeric salt is formed with a suitable optically active acid or base, and then known to those skilled in the art.
- the diastereomeric resolution is carried out by fractional crystallization or chromatography, and then the pure enantiomer is recovered. Furthermore, the separation of enantiomers and diastereomers is generally accomplished by the use of chromatography using a chiral stationary phase, optionally in combination with chemical derivatization (eg, formation of an amino group from an amine). Formate).
- the compounds of Formula I, Formula II, and Formula III of the present invention may contain unnatural proportions of atomic isotopes at one or more of the atoms that make up the compound.
- radiolabeled compounds can be used, such as tritium (3 H), iodine -125 (125 I) or C-14 (14 C). All isotopic compositions of the compounds of Formula I, Formula II, and Formula III of the present invention, whether radioactive or not, are included within the scope of the invention.
- pharmaceutically acceptable carrier refers to any formulation or carrier medium that is capable of delivering an effective amount of an active substance of the present invention, does not interfere with the biological activity of the active substance, and has no toxic side effects to the host or patient, including water, oil, Vegetables and minerals, cream bases, lotion bases, ointment bases, etc. These bases include suspending agents, tackifiers, transdermal enhancers and the like. Their formulations are well known to those skilled in the cosmetic or topical pharmaceutical arts. For additional information on vectors, reference is made to Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005), the contents of which are hereby incorporated by reference.
- excipient generally refers to the carrier, diluent and/or vehicle required to formulate an effective pharmaceutical composition.
- an "effective amount” or “therapeutically effective amount” with respect to a pharmaceutical or pharmacologically active agent refers to a sufficient amount of a drug or agent that is non-toxic but that achieves the desired effect.
- an "effective amount” of an active substance in a composition refers to the amount required to achieve the desired effect when used in combination with another active substance in the composition. The determination of the effective amount will vary from person to person, depending on the age and general condition of the recipient, and also on the particular active substance, and a suitable effective amount in a case can be determined by one skilled in the art based on routine experimentation.
- active ingredient refers to a chemical entity that is effective in treating a target disorder, disease or condition.
- pharmaceutical composition refers to a mixture of one or more compounds of the invention or a salt thereof and a pharmaceutically acceptable adjuvant.
- the purpose of the pharmaceutical composition is to facilitate administration of the compounds of the invention to an organism.
- halo or halogen, by itself or as part of another substituent, denotes a fluorine, chlorine, bromine or iodine atom.
- haloalkyl is intended to include monohaloalkyl and polyhaloalkyl; for example, the term “halo C 1-3 alkyl” is intended to include, but is not limited to, trifluoromethyl, 2,2,2-trifluoro. Ethyl and 3-bromopropyl and the like. Examples of haloalkyl groups include, but are not limited to, trifluoromethyl, trichloromethyl, pentafluoroethyl, and pentachloroethyl.
- hydroxy refers to -OH.
- cyano refers to -CN.
- nitro refers to -NO 2 .
- amino refers to -NH 2 , -NH(alkyl) and -N(alkyl) 2 , and specific examples of the amino group include, but are not limited to, -NH 2 , -NHCH 3 , -NHCH(CH 3 ) 2 , - N(CH 3 ) 2 , -NHC 2 H 5 , -N(CH 3 )C 2 H 5 and the like.
- alkyl refers to a straight or branched saturated aliphatic hydrocarbon group consisting of a carbon atom and a hydrogen atom, such as methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, ⁇ , ⁇ , etc.
- the specific alkyl group includes all isomeric forms thereof, for example, the propyl group includes -CH 2 CH 2 CH 3 , -CH(CH 3 ) 2 , for example, butyl includes -CH 2 CH 2 CH 2 CH 3 ,- CH(CH 3 )(CH 2 CH 3 ), -C(CH 3 ) 3 , -CH 2 CH(CH 3 ) 2 .
- C 1-8 alkyl refers to an alkyl group having from 1 to 8 carbon atoms.
- C1-6 alkyl refers to an alkyl group having from 1 to 6 carbon atoms.
- C 1-4 alkyl refers to an alkyl group having from 1 to 4 carbon atoms.
- C 1-3 alkyl refers to an alkyl group having from 1 to 3 carbon atoms.
- the "alkyl”, “C 1-8 alkyl”, “C 1-6 alkyl”, “C 1-4 alkyl” or “C 1-3 alkyl” may be unsubstituted or one Or a plurality of substituents selected from a hydroxyl group, a halogen or an amino group.
- cycloalkyl refers to a monocyclic saturated aliphatic hydrocarbon group consisting solely of carbon atoms and hydrogen atoms, such as a C3-20 cycloalkyl group, preferably a C3-6 cycloalkyl group, such as a cyclopropyl group, Cyclobutyl, cyclopentyl, cyclohexyl and the like.
- the cycloalkyl group may be unsubstituted or substituted, and the substituent includes, but is not limited to, an alkyl group, an alkyloxy group, a cyano group, a carboxyl group, an aryl group, a heteroaryl group, an amino group, a halogen, a sulfonyl group. , sulfinyl group, phosphoryl group, hydroxyl group and the like.
- hetero denotes a hetero atom or a hetero atomic group (ie, a radical containing a hetero atom), including atoms other than carbon (C) and hydrogen (H), and radicals containing such heteroatoms, including, for example, oxygen (O).
- ring means substituted or unsubstituted cycloalkyl, heterocycloalkyl, cycloalkenyl, heterocycloalkenyl, cycloalkynyl, heterocycloalkynyl, aryl or heteroaryl. So-called rings include single rings, interlocking rings, spiral rings, parallel rings or bridge rings. The number of atoms on the ring is usually defined as the number of elements of the ring. For example, "5-7 membered ring” means 5-7 atoms arranged around. Unless otherwise specified, the ring optionally contains from 1 to 3 heteroatoms.
- 5-7 membered ring includes, for example, phenyl, pyridine, and piperidinyl; in another aspect, the term “5-7 membered heterocycloalkyl ring” includes pyridyl and piperidinyl, but does not include phenyl.
- ring also includes ring systems containing at least one ring, each of which "ring” independently conforms to the above definition.
- heterocycloalkyl refers to a cyclic group that is fully saturated and can exist as a monocyclic, bicyclic or spiro ring. Unless otherwise indicated, the heterocyclic ring is typically a 3 to 7 membered ring containing from 1 to 3 heteroatoms (preferably 1 or 2 heteroatoms) independently selected from sulfur, oxygen and/or nitrogen.
- 3-membered heterocycloalkyl groups include, but are not limited to, oxiranyl, cyclohexylethane, cycloalkylethane, non-limiting examples of 4-membered heterocycloalkyl including, but not limited to, azetidinyl, acetophenan
- Examples of a cyclic group, a thibutyl group, a 5-membered heterocycloalkyl group include, but are not limited to, tetrahydrofuranyl, tetrahydrothiophenyl, pyrrolidinyl, isoxazolidinyl, oxazolidinyl, isothiazolidinyl, thiazolidine
- Examples of the group, imidazolidinyl group, tetrahydropyrazolyl group, pyrrolinyl group, dihydrofuranyl group, dihydrothienyl group, 6-membered heterocycloalkyl group include, but are not limited to
- aryl refers to an all-carbon monocyclic or fused polycyclic aromatic ring group having a conjugated ⁇ -electron system.
- an aryl group can have 6 to 20 carbon atoms, 6 to 14 carbon atoms, or 6 to 12 carbon atoms.
- Non-limiting examples of aryl groups include, but are not limited to, phenyl, naphthyl, anthracenyl, 1,2,3,4-tetrahydronaphthalene, and the like.
- heteroaryl refers to a monocyclic or fused polycyclic ring system containing at least one ring atom selected from N, O, S, the remaining ring atoms being C, and having at least one aromatic ring.
- Preferred heteroaryl groups have a single 4 to 8 membered ring, especially a 5 to 8 membered ring, or a plurality of fused rings containing from 6 to 14, especially from 6 to 10 ring atoms.
- heteroaryl groups include, but are not limited to, pyrrolyl, furyl, thienyl, imidazolyl, oxazolyl, pyrazolyl, pyridyl, pyrimidinyl, pyrazinyl, quinolinyl, isoquinolinyl , tetrazolyl, triazolyl, triazinyl, benzofuranyl, benzothienyl, fluorenyl, isodecyl and the like.
- compositions of the present invention can be prepared by combining the compounds of the present invention with suitable pharmaceutically acceptable excipients, for example, as solid, semi-solid, liquid or gaseous preparations such as tablets, pills, capsules, powders. , granules, ointments, emulsions, suspensions, suppositories, injections, inhalants, gels, microspheres and aerosols.
- suitable pharmaceutically acceptable excipients for example, as solid, semi-solid, liquid or gaseous preparations such as tablets, pills, capsules, powders. , granules, ointments, emulsions, suspensions, suppositories, injections, inhalants, gels, microspheres and aerosols.
- Typical routes of administration of a compound of the invention, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof include, but are not limited to, oral, rectal, topical, inhalation, parenteral, sublingual, intravaginal, intranasal, intraocular, intraperitoneal, Intramuscular, subcutaneous, intravenous administration.
- the pharmaceutical composition of the present invention can be produced by a method well known in the art, such as a conventional mixing method, a dissolution method, a granulation method, a sugar-coating method, a grinding method, an emulsification method, a freeze-drying method, and the like.
- the pharmaceutical composition is in oral form.
- the pharmaceutical compositions can be formulated by admixing the active compound with pharmaceutically acceptable excipients known in the art. These excipients enable the compounds of the present invention to be formulated into tablets, pills, troches, dragees, capsules, liquids, gels, slurries, suspensions and the like for oral administration to a patient.
- Solid oral compositions can be prepared by conventional methods of mixing, filling or tabletting. For example, it can be obtained by mixing the active compound with a solid adjuvant, optionally milling the resulting mixture, adding other suitable excipients if necessary, and then processing the mixture into granules to give tablets. Or the core of the sugar coating. Suitable excipients include, but are not limited to, binders, diluents, disintegrants, lubricants, glidants, sweeteners or flavoring agents, and the like.
- compositions may also be suitable for parenteral administration, such as sterile solutions, suspensions or lyophilized products in a suitable unit dosage form.
- the daily dose is from 0.01 to 200 mg/kg body weight, preferably from 0.01 to 20 mg/kg body weight, more preferably from 0.01 to 10 mg/ Kg body weight, either alone or in divided doses.
- the compounds of the present invention can be prepared by a variety of synthetic methods well known to those skilled in the art, including the specific embodiments set forth below, combinations thereof with other chemical synthetic methods, and those well known to those skilled in the art. Equivalent alternatives, preferred embodiments include, but are not limited to, embodiments of the invention.
- the compound of the formula 1 is reacted with triphosgene in dichloromethane to give the compound of the formula 2; the compound of the formula 2 is reacted with R 2 -L-NH 2 in pyridine to give the compound of the formula 3.
- the compound of the formula 1 is reacted with thiophosgene in dichloromethane to give the compound of the formula 4; the compound of the formula 4 is reacted with R 2 -L-NH 2 in pyridine to give the compound of the formula 5.
- the temperature is Celsius.
- the reagents were purchased from commercial suppliers such as Sinopharm Chemical Reagent Beijing Co., Ltd., Alfa Aesar, or Beijing Belling Technology Co., Ltd., and these reagents can be used directly without further purification unless otherwise stated.
- the following reactions are carried out in an anhydrous solvent, under a positive pressure of nitrogen or argon or using a drying tube; a reaction vessel is provided with a rubber septum to add substrate and reagents via a syringe; glassware is dried and/or Heat and dry.
- the column chromatography was performed using 200-300 mesh silica gel from Qingdao Ocean Chemical Plant; the thin layer chromatography was used to separate the thin layer chromatography silica gel prefabricated panels (HSGF254) produced by Yantai Chemical Industry Research Institute; the measurement of MS was performed by Thermo LCQ.
- ESI Fleet type liquid chromatography-mass spectrometer.
- Nuclear magnetic data ( 1 H-NMR) was run at 400 MHz using a Varian apparatus unless otherwise stated.
- the solvent used for the nuclear magnetic data is CDCl 3 , CD 3 OD, D 2 O, DMSO-d 6 , etc., based on tetramethylsilane (0.00 ppm) or based on residual solvent (CDCl 3 : 7.26 ppm; CD 3 OD : 3.31 ppm; D 2 O: 4.79 ppm; DMSO-d 6 : 2.50 ppm).
- NBS stands for N-bromosuccinimide
- m-CPBA stands for m-chloroperoxybenzoic acid
- NCS stands for N-chlorosuccinimide
- DPPA stands for azidophosphate Diphenyl ester
- Boc- represents t-butoxycarbonyl
- room temperature represents 20-30 °C.
- the compound of the formula 2 was added portionwise to a solution of R 2 -L-NH 2 (0.5 eq.) in pyridine and stirred at room temperature for 12 hours. After the completion of the reaction, the reaction mixture was poured into water, and ethyl acetate was evaporated.
- the compound of the formula 4 was added portionwise to a solution of R 2 -L-NH 2 (1.0 eq.) in pyridine and stirred at room temperature for 12 hours. The reaction mixture was poured into water and extracted with ethyl acetate.
- Triphenylphosphine 49.8 g was added to a solution of 1-azidomethyl-3-chloroisoquinoline (35.6 g) in tetrahydrofuran (300 mL) and water (30 mL). The reaction mixture was poured into water (100 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was evaporated, evaporated, mjjjjjjjj (27.7g).
- Step 7 Synthesis of N-(imidazo[5,1-a]isoquinolin-5-yl)-1,1-benzophenone imide
- N-(imidazo[5,1-a]isoquinolin-5-yl)-1,1-benzophenone imine (15.0 g) was dissolved in tetrahydrofuran (200 mL), and hydrochloric acid was added to the obtained solution. (30 mL, 2N), stirred at room temperature for 2 hours. The reaction mixture was poured into water (100 mL), EtOAc (EtOAc m. 1-a] Isoquinoline-5-amine (6.3 g).
- Step 3 Synthesis of tetrazolo[5,1-a]isoquinoline-5-amine
- 5-Fluoro-2,3-dihydro-1H-inden-1-one (8.0 g) was dissolved in diethyl ether (150 mL), cooled to 0 ° C, dry hydrogen chloride was passed to the solution for 3 hours, then slowly drip Isoamyl nitrite (12.5 g) was added, and after completion of the dropwise addition, the mixture was stirred at room temperature for 3 hours, solid was formed, filtered, and the filter cake was washed with diethyl ether. The filter cake was dried to give 5-fluoro-2-mercapto-2,3 -Dihydro-1H-inden-1-one (7.0 g).
- Steps 3, 4, 5, 6, 7, 8, and 9 refer to steps 1, 2, 3, 4, 5, 6, and 7 in the synthesis of Intermediate 1, respectively.
- step 1 in the synthesis of intermediate 3 5-fluoro-2,3-dihydro-1H-inden-1-one was substituted for 5-fluoro-2,3-dihydro-1H-inden-1-one.
- Steps 2, 3, 4, 5, 6, 7, 8, and 9 refer to steps 2, 3, 4, 5, 6, 7, 8, and 9 in the synthesis of Intermediate 3, respectively.
- 3-Chloro-1-methylisoquinoline (8.3 g) was slowly added to concentrated sulfuric acid (100 mL) at 0 ° C, then potassium nitrate (5.7 g) was added portionwise, and the mixture was stirred at 0 ° C for 1 hour. Pour the reaction solution into crushed ice, adjust the pH to 9 with ammonia water, filter, wash the filter cake with water, and dry the filter cake in vacuo to obtain 3-chloro-1-methyl-5-nitroisoquinoline (6.2 g).
- 3-Chloro-1-methyl-5-nitroisoquinoline (6.0 g) was dissolved in a mixed solvent of methanol and water (1:1, 200 mL), ammonium chloride (14.4 g) was added and heated to 60 After the solution became clear, the reduced iron powder (15.1 g) was added, and the mixture was stirred at 60 ° C for 30 minutes, cooled to room temperature, filtered, and the filtrate was concentrated under reduced pressure to remove methanol and extracted with ethyl acetate (150 mL ⁇ 2). The mixture was washed with brine, dried over anhydrous sodium sulfate -aminoisoquinoline (3.9 g).
- Steps 4, 5, 6, 7, 8, and 9 refer to steps 2, 3, 4, 5, 6, and 7 in the synthesis of Intermediate 1, respectively.
- 3-Bromo-2-indolylquinoline (0.4 g) was added to formic acid (25 mL), and then stirred at room temperature overnight, and the mixture was poured into water and extracted with ethyl acetate (100 mL ⁇ 2). The organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered and evaporated.
- Zoxao[4,3-a]quinoline (0.32 g).
- Steps 1, 2, 3, 4, 5, and 6 refer to steps 2, 3, 4, 5, 6, and 7 in the synthesis of Intermediate 1, respectively.
- Steps 4, 5, 6, 7, 8, 9, and 10 refer to steps 1, 2, 3, 4, 5, 6, and 7 in the synthesis of Intermediate 1, respectively.
- N-chlorosuccinimide (2.7 g) was slowly added to a solution of 1-methylisoquinolin-3-amine (3.2 g) in dichloromethane (50 ml), and the mixture was stirred at room temperature for 1 hour, then cold water was added. The reaction was quenched (5 ml), EtOAcjjjjjjj -Methylisoquinolin-3-amine (3.5 g).
- Step 4 Synthesis of 2-(1-(azidomethyl)-4-chloroisoquinolin-3-yl)isoindole-1,3-dione
- Step 1 Synthesis of 2-methylquinoline-3-carboxylic acid ethyl ester
- Step 3 Synthesis of tert-butyl (2-methylquinolin-3-yl)carbamate
- Step 4 Synthesis of tert-butyl imidazo[1,5-a]quinolin-4-ylcarbamate
- Step 2 Synthesis of ethyl 6-(tert-butoxycarbonylamino)-4-chloropicolinate
- Step 3 Synthesis of (4-chloro-6-(hydroxymethyl)pyridin-2-yl)carbamic acid tert-butyl ester
- Step 4 Synthesis of (4-chloro-6-(chloromethyl)pyridin-2-yl)carbamic acid tert-butyl ester
- Step 5 Synthesis of (4-chloro-6-((1,3-dioxoisoindolin-2-yl)methyl)pyridin-2-yl)carbamic acid tert-butyl ester
- Step 6 Synthesis of (6-(aminomethyl)-4-chloropyridin-2-yl)carbamic acid tert-butyl ester
- Steps 7 and 8 refer to steps 5 and 6 in the synthesis of Intermediate 1, respectively.
- Step 5 (5-Chloro-2-((1,3-dioxoisoindolin-2-yl)methyl)pyridin-3-yl)-tert-butoxyamide
- Steps 7 and 8 refer to steps 5 and 6 in the synthesis of Intermediate 1, respectively.
- Step 3 Synthesis of (2-cyano-5-(trifluoromethyl)pyridin-3-yl)carbamic acid tert-butyl ester
- Step 4 Synthesis of tert-butyl (2-(aminomethyl)-5-(trifluoromethyl)pyridin-3-yl)carbamate
- Steps 5 and 6 refer to steps 5 and 6 in the synthesis of Intermediate 1.
- Steps 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 are referred to the synthesis steps 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 of Intermediate 3, respectively, to give the title compound.
- Test Example 1 Biological activity assay
- PCR amplification of IDO RNA derived from HeLa cells
- TDO RNA derived from U87MG cells
- amplification of the amplified PCR product and then pET28a plasmid and IDO (TDO) gel recovery product
- the restriction endonuclease of NotI and EcoRI was digested (37 ° C, digested for 2 h), and the T4 rapid ligase ligation product was added to E. coli DH5 ⁇ .
- Competent cells were placed on ice for 30 min, heat shocked at 42 °C for 90 s, shaken in LB medium and plated, picked up monoclonal overnight shakes, extracted plasmid and identified by enzyme digestion, and then sent After sequencing, positive clones, ie His-IDO/pET28a and His-TDO/pET28a plasmids were successfully constructed.
- IPTG was induced at 30 ° C for 3.5 h, and after induction, the cells were collected by centrifugation at 6000 rpm for 45 min.
- the collected cells were resuspended with lysate (40 mM Tris-HCl, pH 8.0; 110 mM NaCl; 2.2 mM KCl; 10% glycerol; 0.5% Tween-20; 20 mM imidazole; 1 mM DTT).
- Ultrasonic lysis power 30%, cleavage on ice for 15 min
- the lysed broth was centrifuged at 12000 rpm, 4 ° C for 60 min, and the resulting supernatant was filtered through a 0.22 ⁇ M filter to prepare a sample for use.
- the nickel column was equilibrated with lysate for 5 column volumes, and the prepared supernatant sample was applied to a nickel column.
- the eluent 40 mM Tris-HCl, pH 8.0; 110 mM NaCl; 2.2
- the protein was eluted with a gradient of mM KCl; 10% glycerol; 20-250 mM imidazole.
- the eluted protein solution was dialyzed overnight at 4 ° C, concentrated after dialysis, and stored at -80 ° C until use.
- the enzymatic activity detection platform of IDO and TDO was established by light absorption method.
- Compounds were diluted 10 times with 100% DMSO starting from 10 mM (5 concentrations in total), and 4 ⁇ L of each concentration was added to 96 ⁇ L of reaction buffer.
- the mixture was mixed in a liquid (50 mM K 2 HPO 4 -KH 2 PO 4 phosphate buffer, pH 6.5) and used as a 4* compound.
- 2*IDO enzyme was prepared using reaction buffer at a final concentration of 75 nM (total TDO enzyme concentration of 300 nM), 4* substrate (L-(+)-Absorbate with a final concentration of 20 ⁇ M, respectively, purchased from Alfa Aesar, Cat. No.
- the 384-well plate was placed in an incubator for 13 minutes at 23 ° C (TDO reaction for 120 minutes), then 4 ⁇ L of 6 M TCA was added to each well for 30 min at 55 ° C, centrifuged at 2500 rpm for 10 min, and 40 ⁇ L of supernatant was taken per well.
- 2% 4-dimethylaminobenzaldehyde available from TCI, Cat. No. D495, dissolved in glacial acetic acid
- the absorbance 480 nM light absorption
- mice Male Sprague-Dawley rats were obtained from Beijing Weitong Lihua Experimental Animal Technology Co., Ltd., and the rats were divided into groups of 3, and the test sample suspension (5 mg/kg) was administered orally by a single oral administration. Animals were fasted overnight before the experiment, and the fasting time was from 10 hours before administration to 4 hours after administration. Blood was collected at 0.25, 0.5, 1, 2, 4, 6, 8, and 24 hours after administration. After anesthesia with a small animal anesthesia machine, 0.3 mL whole blood was taken through the fundus venous plexus and placed in a heparin anticoagulant tube.
- the sample was centrifuged at 4 ° C, 4000 rpm for 5 min, and the plasma was transferred to a centrifuge tube and placed in -80. °C is saved until analysis. Samples in plasma were extracted using protein precipitation and the extracts were analyzed by LC/MS/MS.
- Example 51 40 52 Dose (mg/kg) 5 5 5 T 1/2 (hr) 3.28 4.31 3.56 T max (hr) 0.25 0.58 0.33 C max (ng/mL) 721 243 507 AUC 0-inf (hr*ng/mL) 1520 1018 2180
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
如式(I)所示的具有IDO和TDO双重抑制活性的脲类化合物或其药学上可接受的盐及其制备制备方法、药物组合物以及治疗IDO和TDO介导的免疫抑制疾病中的用途。
Description
本发明属于医药技术领域,涉及IDO和TDO双重抑制剂的脲类化合物、其制备方法、含有该化合物的药物组合物、以及其在调控IDO和TDO的活性以及治疗由它们介导的疾病和紊乱的用途。
吲哚胺2,3-双加氧酶(IDO)和色氨酸2,3-加氧酶(TDO)是分解色氨酸的两种重要酶,其在多种肿瘤细胞及周围微环境细胞中表达,通过降解局部组织中的色氨酸,产生色氨酸耗竭环境及升高局部代谢产物犬尿氨酸浓度而影响T细胞的免疫功能,在诱发宿主免疫防御、抑制T细胞免疫和抗肿瘤免疫、诱导母胎免疫耐受和移植物免疫耐受中均发挥重要的代谢性免疫调节作用。
IDO表达或活性的异常增高已被证实与抑郁症、阿尔茨海默病、白内障、癌症等疾病的发病机制密切相关,因此,IDO抑制剂可能成为治疗这些疾病的一种有效方法。尽管国内外有关IDO抑制剂的研究已开展数十年,但目前仍然没有IDO抑制剂作为药物上市。目前已有的IDO抑制剂主要分为以下几个类别:1)竞争性抑制剂,如色氨酸衍生物1-MT;2)非竞争性抑制剂,如苯基咪唑;3)反竞争性抑制剂,如生物碱exiguamine A;4)通过其他作用机制的抑制剂。这几类抑制剂普遍存在抑制效率低下、无法透过细胞膜以及产生吲哚环结构代谢物等问题。20世纪90年代,通过对IDO的底物色氨酸为模板进行结构改造而获得的衍生物1-甲基色氨酸(1-methyl-tryptophan,1-MT)是目前体内外实验中通用的IDO抑制剂,其抑制常数(Ki)为34μM。截至目前,美国New link Genetics公司的NLG919化合物与美国Incyte公司的INCB024360化合物已经进入了临床试验。
最近的研究也发现,TDO在多种肿瘤细胞中表达,导致肿瘤细胞的存活和迁移能力的提高以及免疫系统对肿瘤细胞的应答能力受到抑制。TDO对肿瘤细胞和免疫抑制产生的效应使其成为一个新的抗肿瘤药物靶标。因此,筛选新型IDO/TDO抑制剂,并研究其体内活性、药代动力学、治疗作用和不良反应等仍是一个值得探索的领域。
发明内容
本发明提供通式I的化合物或其药学上可接受的盐,

其中,
环A不存在或者环A为苯环或含有1、2或3个选自N、O或S原子的5-6元杂芳环;
环B为五元芳香环,其中,W、V、X、Y和Z中的1个、2个、3个或4个选自N或NH,其它选自C或CH,具体的,
i)Y和V是N,Z和W是CH,X是C;
ii)Y、Z、V和W是N,X是C;
iii)X和V是N,Z和W是CH,Y是C;
iv)Y、Z和V是N,W是CH,X是C;
v)X、Z和V是N,W是CH,Y是C;
vi)X是N,Z、V和W是CH,Y是C;
vii)Y是N,Z、V和W是CH,X是C;
viii)Z是NH,V和W是CH,X和Y是C;
ix)V是NH,Z和W是CH,X和Y是C;
x)W是NH,V和Z是CH,X和Y是C;
xi)V和W是N或NH,Z是CH,X和Y是C;
xii)V和Z是N或NH,W是CH,X和Y是C;
xiii)Z和W是N或NH,V是CH,X和Y是C;
xiv)Y和W是N,V和Z是CH,X是C;
xv)X和W是N,V和Z是CH,Y是C;
xvi)X和Z是N或NH,V和W是CH,Y是C;
xvii)Y和Z是N,V和W是CH,X是C;
xviii)Y、V和W是N,Z是CH,X是C;
xix)Z、V和W是N或NH,X和Y是C;
xx)X、W和V是N,Z是CH,Y是C;
xxi)Y、Z和W是N,V是CH,X是C;或
xxii)X、Z、V和W是N,Y是C;
P为C(R
6)或N;
Q为O或S;
D选自NH、O、S或CH
2;
E选自NH、O或CH
2;
L选自单键、S、SO、SO
2、CO、C(O)O、S(O)O或S(O)
2O;
每个R
1独立地选自卤素、氨基、羟基、氰基、卤代C
1-3烷基、C
1-6烷基、C
3-6环烷基或C
3-6杂环烷基;
R
2选自C
3-12环烷基、C
3-12杂环烷基、6-12元芳基或者含有1、2或3个选自N、O或S原子的5-12元杂芳基,其可任选地被一个或多个独立地选自R
3的基团取代;
R
3选自卤素、氰基、硝基、卤代C
1-3烷基、C
1-6烷基、C
3-6环烷基、C
3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C
3-6环烷基CH
2-、C
3-6杂环烷基CH
2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH
2-、-OR
4、-SR
4、-N(R
4)
2、-C(O)OR
4、-C(O)N(R
4)
2、-C(O)R
4、-S(O)R
4、-S(O)OR
4、-S(O)N(R
4)
2、-S(O)
2R
4、-S(O)
2OR
4、-S(O)
2N(R
4)
2、-OC(O)R
4、-OC(O)OR
4、-OC(O)N(R
4)
2、-N(R
4)C(O)R
4、-N(R
4)C(O)OR
4、-N(R
4)C(O)N(R
4)
2、-N(R
4)S(O)N(R
4)
2、-N(R
4)S(O)
2N(R
4)
2、-P(O)(R
4)
2或-P(O)(OR
4)
2,其中C
1-6烷基、C
3-6环烷基、C
3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C
3-6环烷基CH
2-、C
3-6杂环烷基CH
2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH
2-可任选地被一个或多个独立地选自R
5的基团取代;
每个R
4独立地选自H、卤代C
1-3烷基、C
1-6烷基、C
3-6环烷基、C
3-6杂环烷基、苯基或者含有1、2或3个选自N、O或S原子的5-6元杂芳基;
R
5选自卤素、氨基、氰基、羟基、-COOH、卤代C
1-3烷基、C
1-6烷基、C
3-6环烷基或C
3-6杂环烷基;
R
6选自H、卤素、氨基、氰基、羟基、-COOH或卤代C
1-3烷基;
m为0、1或2。
需要说明的是,当环A不存在时,通式I为

本发明式I化合物的一个实施方案中,环A不存在或环A为苯环、呋喃环、噻吩环、吡咯环、吡唑环、咪唑环、吡啶环、嘧啶环、哒嗪环、吡嗪环、噻唑环、异噻唑环、噁唑环、异噁唑环、四唑环或三嗪环。
本发明式I化合物的一个实施方案中,环A不存在或环A为苯环或吡啶环。
本发明式I化合物的一个实施方案中,环A不存在或环A为

本发明式I化合物的一个实施方案中,每个R
1独立地选自卤素、卤代C
1-3烷基或C
1-6烷基。
本发明式I化合物的一个实施方案中,每个R
1独立地选自氟、氯、溴、甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基或三氯甲基。
本发明式I化合物的一个实施方案中,每个R
1独立地选自氟、氯或三氟甲基。
本发明式I化合物的一个实施方案中,m为0或1。
本发明式I化合物的一个实施方案中,环B为芳香环,其中,i)Y和V是N,Z和W是CH,X是C;ii)Y、Z、V和W是N,X是C;iii)X和V是N,Z和W是CH,Y是C;iv)Y、Z和V是N,W是CH,X是C;v)X、Z和V是N,W是CH,Y是C。
本发明式I化合物的一个实施方案中,结构单元
 选自
选自


本发明式I化合物的一个实施方案中,结构单元
 选自
选自


本发明式I化合物的一个实施方案中,D选自NH、O或CH
2;E选自NH、O或CH
2。
本发明式I化合物的一个实施方案中,-D-C(Q)-E-选自-NHC(O)NH-、-NHC(S)NH-、-OC(O)NH-、-NHC(O)CH
2-或-CH
2C(O)NH-。
本发明式I化合物的一个实施方案中,L选自单键或SO
2。
本发明式I化合物的一个实施方案中,R
2选自C
3-6环烷基、C
3-6杂环烷基、苯基或者含有1、2或3个选自N、O或S原子的5-6元杂芳基,其可任选地被一个或多个独立地选自R
3的基团取代。
本发明式I化合物的一个实施方案中,R
2选自环丙基、环丁基、环戊基、环己基、任意位置失去一个氢原子的

 苯基、呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基,其可任选地被一个或多个独立地选自R
3的基团取代。
苯基、呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基,其可任选地被一个或多个独立地选自R
3的基团取代。
本发明式I化合物的一个实施方案中,R
2选自环己基、任意位置失去一个氢原子的
 苯基、吡啶基或嘧啶基,其可任选地被一个或多个独立地选自R
3的基团取代。
苯基、吡啶基或嘧啶基,其可任选地被一个或多个独立地选自R
3的基团取代。
本发明式I化合物的一个实施方案中,R
2选自

 其可任选地被一个或多个独立地选自R
3的基团取代。
其可任选地被一个或多个独立地选自R
3的基团取代。
本发明式I化合物的一个实施方案中,R
2选自


本发明式I化合物的一个实施方案中,R
3选自卤素、氰基、硝基、卤代C
1-3烷基、C
1-6烷基、C
3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C
3-6杂环烷基CH
2-、-OR
4、-SR
4、-S(O)
2R
4、-S(O)
2N(R
4)
2、-C(O)OR
4、-C(O)N(R
4)
2、-N(R
4)S(O)
2N(R
4)
2、-P(O)(R
4)
2或-P(O)(OR
4)
2,其中C
1-6烷基、C
3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基或C
3-6杂环烷基CH
2-可任选地被一个或多个独立地选自R
5的基团取代。
本发明式I化合物的一个实施方案中,R
3选自氟、氯、溴、氰基、硝基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基、三氯甲基、-OR
4、-SR
4、-S(O)
2R
4、-S(O)
2N(R
4)
2、-C(O)OR
4、-C(O)N(R
4)
2、-N(R
4)S(O)
2N(R
4)
2、-P(O)(R
4)
2、-P(O)(OR
4)
2、甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基,任意位置失去一个H原子的


 呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、
呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、

 其中甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基,任意位置失去一个H原子的
其中甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基,任意位置失去一个H原子的


 呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、
呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、


 可任选地被一个或多个独立地选自R
5的基团取代。
可任选地被一个或多个独立地选自R
5的基团取代。
本发明式I化合物的一个实施方案中,R
3选自氟、氯、溴、氰基、硝基、三氟甲基、-OR
4、-SR
4、-S(O)
2R
4、-S(O)
2N(R
4)
2、-C(O)OR
4、-C(O)N(R
4)
2、-N(R
4)S(O)
2N(R
4)
2、-P(O)(R
4)
2、-P(O)(OR
4)
2、甲基、任意位置失 去一个氢原子的
 四唑基或
四唑基或
 其中甲基、任意位置失去一个氢原子的
其中甲基、任意位置失去一个氢原子的
 四唑基或
四唑基或
 可任选地被一个或多个独立地选自R
5的基团取代。
可任选地被一个或多个独立地选自R
5的基团取代。
本发明式I化合物的一个实施方案中,R
3选自氟、氯、溴、氰基、硝基、三氟甲基、-OCF
3、-SCH
3、-S(O)
2CH
3、-S(O)
2NH
2、-C(O)OCH
3、-COOH、-C(O)NHCH
3、-C(O)NH
2、-NHS(O)
2NH
2、-P(O)(CH
3)
2、-P(O)(OCH
2CH
3)
2、-C(OH)(CF
3)
2、

本发明式I化合物的一个实施方案中,每个R4独立地选自H、卤代C
1-3烷基或C
1-6烷基。
本发明式I化合物的一个实施方案中,每个R
4独立地选自H、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基、三氯甲基、甲基、乙基、丙基、异丙基、丁基、异丁基或叔丁基。
本发明式I化合物的一个实施方案中,每个R
4独立地选自H、三氟甲基、甲基或乙基。
本发明式I化合物的一个实施方案中,R
5选自羟基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基或三氯甲基。
本发明式I化合物的一个实施方案中,R
5选自羟基或三氟甲基。
本发明式I化合物的一个实施方案中,R
6选自H或卤素。
本发明式I化合物的一个实施方案中,R
6选自H或氯。
另一方面,本发明提供通式II的化合物或其药学上可接受的盐,

其中,
环A为苯环或含有1、2或3个选自N、O或S原子的5-6元杂芳环;
环B为芳香环,其中,W、V、X、Y和Z中的1个、2个、3个或4个选自N或NH,其它选自C或CH,具体的,
i)Y和V是N,Z和W是CH,X是C;
ii)Y、Z、V和W是N,X是C;
iii)X和V是N,Z和W是CH,Y是C;
iv)Y、Z和V是N,W是CH,X是C;
v)X、Z和V是N,W是CH,Y是C;
vi)X是N,Z、V和W是CH,Y是C;
vii)Y是N,Z、V和W是CH,X是C;
viii)Z是NH,V和W是CH,X和Y是C;
ix)V是NH,Z和W是CH,X和Y是C;
x)W是NH,V和Z是CH,X和Y是C;
xi)V和W是N或NH,Z是CH,X和Y是C;
xii)V和Z是N或NH,W是CH,X和Y是C;
xiii)Z和W是N或NH,V是CH,X和Y是C;
xiv)Y和W是N,V和Z是CH,X是C;
xv)X和W是N,V和Z是CH,Y是C;
xvi)X和Z是N或NH,V和W是CH,Y是C;
xvii)Y和Z是N,V和W是CH,X是C;
xviii)Y、V和W是N,Z是CH,X是C;
xix)Z、V和W是N或NH,X和Y是C;
xx)X、W和V是N,Z是CH,Y是C;
xxi)Y、Z和W是N,V是CH,X是C;或
xxii)X、Z、V和W是N,Y是C;
P为C(R
6)或N;
Q为O或S;
L选自单键、S、SO、SO
2、CO、C(O)O、S(O)O或S(O)
2O;
每个R
1独立地选自卤素、氨基、羟基、氰基、卤代C
1-3烷基、C
1-6烷基、C
3-6环烷基或C
3-6杂环烷基;
R
2选自C
3-12环烷基、C
3-12杂环烷基、6-12元芳基或者含有1、2或3个选自N、O或S原子的5-12元杂芳基,其可任选地被一个或多个独立地选自R
3的基团取代;
R
3选自卤素、氰基、硝基、卤代C
1-3烷基、C
1-6烷基、C
3-6环烷基、C
3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C
3-6环烷基CH
2-、C
3-6杂环烷基CH
2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH
2-、-OR
4、-SR
4、-N(R
4)
2、-C(O)OR
4、-C(O)N(R
4)
2、-C(O)R
4、-S(O)R
4、-S(O)OR
4、-S(O)N(R
4)
2、-S(O)
2R
4、-S(O)
2OR
4、-S(O)
2N(R
4)
2、-OC(O)R
4、-OC(O)OR
4、-OC(O)N(R
4)
2、-N(R
4)C(O)R
4、-N(R
4)C(O)OR
4、-N(R
4)C(O)N(R
4)
2、-N(R
4)S(O)N(R
4)
2、-N(R
4)S(O)
2N(R
4)
2、-P(O)(R
4)
2或-P(O)(OR
4)
2,其中C
1-6烷基、C
3-6环烷基、C
3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C
3-6环烷基CH
2-、C
3-6杂环烷基CH
2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH
2-可任选地被一个或多个独立地选自R
5的基团取代;
每个R
4独立地选自H、卤代C
1-3烷基、C
1-6烷基、C
3-6环烷基、C
3-6杂环烷基、苯基或者含有1、2或3个选自N、O或S原子的5-6元杂芳基;
R
5选自卤素、氨基、氰基、羟基、-COOH、卤代C
1-3烷基、C
1-6烷基、C
3-6环烷基或C
3-6杂环烷基;
R
6选自H、卤素、氨基、氰基、羟基、-COOH或卤代C
1-3烷基;
m为0、1或2。
本发明式II化合物的一个实施方案中,环A为苯环、呋喃环、噻吩环、吡咯环、吡唑环、咪唑环、吡啶环、嘧啶环、哒嗪环、吡嗪环、噻唑环、异噻唑环、噁唑环、异噁唑环、四唑环或三嗪环。
本发明式II化合物的一个实施方案中,环A为苯环或吡啶环。
本发明式II化合物的一个实施方案中,环A不存在或者环A为

本发明式II化合物的一个实施方案中,每个R
1独立地选自卤素、卤代C
1-3烷基或C
1-6烷基。
本发明式II化合物的一个实施方案中,每个R
1独立地选自氟、氯、溴、甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基或三氯甲基。
本发明式II化合物的一个实施方案中,每个R
1独立地选自氟、氯或三氟甲基。
本发明式II化合物的一个实施方案中,m为0或1。
本发明式II化合物的一个实施方案中,环B为芳香环,其中,i)Y和V是N,Z和W是CH,X是C;ii)Y、Z、V和W是N,X是C;iii)X和V是N,Z和W是CH,Y是C;iv)Y、Z和V是N,W是CH,X是C;v)X、Z和V是N,W是CH,Y是C。
本发明式II化合物的一个实施方案中,结构单元
 选自
选自


本发明式II化合物的一个实施方案中,结构单元
 选自
选自


本发明式II化合物的一个实施方案中,L选自单键或SO
2。
本发明式II化合物的一个实施方案中,R
2选自C
3-6环烷基、C
3-6杂环烷基、苯基或者含有1、2或3个选自N、O或S原子的5-6元杂芳基,其可任选地被一个或多个独立地选自R
3的基团取代。
本发明式II化合物的一个实施方案中,R
2选自环丙基、环丁基、环戊基、环己基,任意位置失去一个氢原子的

 苯基、呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基,其可任选地被一个或多个独立地选自R
3的基团取代。
苯基、呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基,其可任选地被一个或多个独立地选自R
3的基团取代。
本发明式II化合物的一个实施方案中,R
2选自环己基、任意位置失去一个氢原子的
 苯基、吡啶基或嘧啶基,其可任选地被一个或多个独立地选自R
3的基团取代。
苯基、吡啶基或嘧啶基,其可任选地被一个或多个独立地选自R
3的基团取代。
本发明式II化合物的一个实施方案中,R
2选自

 其可任选地被一个或多个独立地选自R
3的基团取代。
其可任选地被一个或多个独立地选自R
3的基团取代。
本发明式II化合物的一个实施方案中,R
2选自


本发明式II化合物的一个实施方案中,R
3选自卤素、氰基、硝基、卤代C
1-3烷基、C
1-6烷基、C
3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C
3-6杂环烷基CH
2-、-OR
4、-SR
4、-S(O)
2R
4、-S(O)
2N(R
4)
2、-C(O)OR
4、-C(O)N(R
4)
2、-N(R
4)S(O)
2N(R
4)
2、-P(O)(R
4)
2或-P(O)(OR
4)
2,其中C
1-6烷基、C
3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基或C
3-6杂环烷基CH
2-可任选地被一个或多个独立地选自R
5的基团取代。
本发明式II化合物的一个实施方案中,R
3选自氟、氯、溴、氰基、硝基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基、三氯甲基、-OR
4、-SR
4、-S(O)
2R
4、-S(O)
2N(R
4)
2、-C(O)OR
4、-C(O)N(R
4)
2、-N(R
4)S(O)
2N(R
4)
2、-P(O)(R
4)
2、-P(O)(OR
4)
2、甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基,任意位置失去一个H原子的


 呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、
呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、


 其中甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基,任意位置失去一个H原子的
其中甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基,任意位置失去一个H原子的


 呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、
呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、


 可任选地被一个或多个独立地选自R
5的基团取代。
可任选地被一个或多个独立地选自R
5的基团取代。
本发明式II化合物的一个实施方案中,R
3选自氟、氯、溴、氰基、硝基、三氟甲基、-OR
4、-SR
4、-S(O)
2R
4、-S(O)
2N(R
4)
2、-C(O)OR
4、-C(O)N(R
4)
2、-N(R
4)S(O)
2N(R
4)
2、-P(O)(R
4)
2、-P(O)(OR
4)
2、甲基、任意位置失去一个氢原子的
 四唑基或
四唑基或
 其中甲基、任意位置失去一个氢原子的
其中甲基、任意位置失去一个氢原子的
 四唑基或
四唑基或
 可任选地被一个或多个独立地选自R
5的基团取代。
可任选地被一个或多个独立地选自R
5的基团取代。
本发明式II化合物的一个实施方案中,R
3选自氟、氯、溴、氰基、硝基、三氟甲基、-OCF
3、-SCH
3、-S(O)
2CH
3、-S(O)
2NH
2、-C(O)OCH
3、-COOH、-C(O)NHCH
3、-C(O)NH
2、-NHS(O)
2NH
2、-P(O)(CH
3)
2、 -P(O)(OCH
2CH
3)
2、-C(OH)(CF
3)
2、

本发明式II化合物的一个实施方案中,每个R
4独立地选自H、卤代C
1-3烷基或C
1-6烷基。
本发明式II化合物的一个实施方案中,每个R
4独立地选自H、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基、三氯甲基、甲基、乙基、丙基、异丙基、丁基、异丁基或叔丁基。
本发明式II化合物的一个实施方案中,每个R
4独立地选自H、三氟甲基、甲基或乙基。
本发明式II化合物的一个实施方案中,R
5选自羟基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基或三氯甲基。
本发明式II化合物的一个实施方案中,R
5选自羟基或三氟甲基。
本发明式II化合物的一个实施方案中,R
6选自H或卤素。
本发明式II化合物的一个实施方案中,R
6选自H或氯。
另一方面,本发明提供式II-1所示的化合物或其药学上可接受的盐,

其中,基团或原子定义如通式II化合物所述。
另一方面,本发明提供式II-2所示的化合物或其药学上可接受的盐,

其中,基团或原子定义如通式II化合物所述。
另一方面,本发明提供式II-3所示的化合物或其药学上可接受的盐,

其中,基团或原子定义如通式II化合物所述。
另一方面,本发明提供式II-4所示的化合物或其药学上可接受的盐,

其中,基团或原子定义如通式II化合物所述。
另一方面,本发明提供式II-5所示的化合物或其药学上可接受的盐,

其中,基团或原子定义如通式II化合物所述。
另一方面,本发明提供通式III的化合物或其药学上可接受的盐,

其中,
环B为芳香环,其中,W、V、X、Y和Z中的1个、2个、3个或4个选自N或NH,其它选自C或CH,具体的,
i)Y和V是N,Z和W是CH,X是C;
ii)Y、Z、V和W是N,X是C;
iii)X和V是N,Z和W是CH,Y是C;
iv)Y、Z和V是N,W是CH,X是C;
v)X、Z和V是N,W是CH,Y是C;
vi)X是N,Z、V和W是CH,Y是C;
vii)Y是N,Z、V和W是CH,X是C;
viii)Z是NH,V和W是CH,X和Y是C;
ix)V是NH,Z和W是CH,X和Y是C;
x)W是NH,V和Z是CH,X和Y是C;
xi)V和W是N或NH,Z是CH,X和Y是C;
xii)V和Z是N或NH,W是CH,X和Y是C;
xiii)Z和W是N或NH,V是CH,X和Y是C;
xiv)Y和W是N,V和Z是CH,X是C;
xv)X和W是N,V和Z是CH,Y是C;
xvi)X和Z是N或NH,V和W是CH,Y是C;
xvii)Y和Z是N,V和W是CH,X是C;
xviii)Y、V和W是N,Z是CH,X是C;
xix)Z、V和W是N或NH,X和Y是C;
xx)X、W和V是N,Z是CH,Y是C;
xxi)Y、Z和W是N,V是CH,X是C;或
xxii)X、Z、V和W是N,Y是C;
Q为O或S;
D选自NH、O、S或CH
2;
E选自NH、O、或CH
2;
L选自单键、S、SO、SO
2、CO、C(O)O、S(O)O或S(O)
2O;
每个R
1独立地选自卤素、氨基、羟基、氰基、卤代C
1-3烷基、C
1-6烷基、C
3-6环烷基或C
3-6杂环烷基;
R
2选自C
3-12环烷基、C
3-12杂环烷基、6-12元芳基或者含有1、2或3个选自N、O或S原子的5-12元杂芳基,其可任选地被一个或多个独立地选自R
3的基团取代;
R
3选自卤素、氰基、硝基、卤代C
1-3烷基、C
1-6烷基、C
3-6环烷基、C
3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C
3-6环烷基CH
2-、C
3-6杂环烷基CH
2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH
2-、-OR
4、-SR
4、-N(R
4)
2、-C(O)OR
4、-C(O)N(R
4)
2、-C(O)R
4、-S(O)R
4、-S(O)OR
4、-S(O)N(R
4)
2、-S(O)
2R
4、-S(O)
2OR
4、-S(O)
2N(R
4)
2、-OC(O)R
4、-OC(O)OR
4、-OC(O)N(R
4)
2、-N(R
4)C(O)R
4、-N(R
4)C(O)OR
4、-N(R
4)C(O)N(R
4)
2、-N(R
4)S(O)N(R
4)
2、-N(R
4)S(O)
2N(R
4)
2、-P(O)(R
4)
2或-P(O)(OR
4)
2,其中C
1-6烷基、C
3-6环烷基、C
3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C
3-6环烷基CH
2-、C
3-6杂环烷基CH
2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH
2-可任选地被一个或多个独立地选自R
5的基团取代;
每个R
4独立地选自H、卤代C
1-3烷基、C
1-6烷基、C
3-6环烷基、C
3-6杂环烷基、苯基或者含有1、2或3个选自N、O或S原子的5-6元杂芳基;
R
5选自卤素、氨基、氰基、羟基、-COOH、卤代C
1-3烷基、C
1-6烷基、C
3-6环烷基或C
3-6杂环烷基;
m为0、1或2。
本发明式III化合物的一个实施方案中,每个R
1独立地选自卤素、卤代C
1-3烷基或C
1-6烷基。
本发明式III化合物的一个实施方案中,每个R
1独立地选自氟、氯、溴、甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基或三氯甲基。
本发明式III化合物的一个实施方案中,每个R
1独立地选自氯或三氟甲基。
本发明式III化合物的一个实施方案中,m为0或1。
本发明式III化合物的一个实施方案中,环B为芳香环,其中,i)Y和V是N,Z和W是CH,X是C;ii)X和V是N,Z和W是CH,Y是C。
本发明式III化合物的一个实施方案中,结构单元
 选自
选自

本发明式III化合物的一个实施方案中,结构单元
 选自
选自


本发明式III化合物的一个实施方案中,D选自NH、O或CH
2;E选自NH、O或CH
2。
本发明式III化合物的一个实施方案中,-D-C(Q)-E-选自-NHC(O)NH-、-NHC(S)NH-、-OC(O)NH-、-NHC(O)CH
2-或-CH
2C(O)NH-。
本发明式III化合物的一个实施方案中,L选自单键。
本发明式III化合物的一个实施方案中,R
2选自苯基或者含有1、2或3个选自N、O或S原子的5-6元杂芳基,其可任选地被一个或多个独立地选自R
3的基团取代。
本发明式III化合物的一个实施方案中,R
2选自苯基、呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、 吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基,其可任选地被一个或多个独立地选自R
3的基团取代。
本发明式III化合物的一个实施方案中,R
2选自苯基、吡啶基或嘧啶基,其可任选地被一个或多个独立地选自R
3的基团取代。
本发明式III化合物的一个实施方案中,R
2选自
 其可任选地被一个或多个独立地选自R
3的基团取代。
其可任选地被一个或多个独立地选自R
3的基团取代。
本发明式III化合物的一个实施方案中,R
2选自


本发明式III化合物的一个实施方案中,R
3选自卤素或卤代C
1-3烷基。
本发明式III化合物的一个实施方案中,R
3选自氟、氯、溴或三氟甲基。
另一方面,本发明提供式III-1所示的化合物或其药学上可接受的盐,

其中,基团或原子定义如通式III化合物所述。
另一方面,本发明提供式III-2所示的化合物或其药学上可接受的盐,

其中,基团或原子定义如通式III化合物所述。
本发明优选以下化合物或其药学上可接受的盐,




另一方面,本发明涉及药物组合物,其包含本发明的通式I化合物、通式II化合物及通式III化合物或其药学上可接受的盐。在一些实施方案中,本发明的药物组合物还包括一种或多种药学上可接受的载体或赋形剂。本发明的药物组合物可以进一步含有一种或多种额外的治疗剂。
另一方面,本发明涉及治疗哺乳动物由吲哚胺2,3-双加氧酶(IDO)和色氨酸2,3-加氧酶(TDO)介导的免疫抑制疾病的方法,包括对需要该治疗的哺乳动物、优选人类给予治疗有效量的通式I化合物、通式II化合物及通式III化合物或其药学上可接受的盐、或其药物组合物。
另一方面,本发明涉及通式I化合物、通式II化合物及通式III化合物或其药学上可接受的盐、或其药物组合物在制备预防或者治疗由吲哚胺2,3-双加氧酶(IDO)和色氨酸2,3-加氧酶(TDO)介导的免疫抑制疾病的药物中的用途。又一方面,本发明涉及通式I化合物、通式II化合物及通式III化合物或其药 学上可接受的盐、或其药物组合物用于预防或者治疗由吲哚胺2,3-双加氧酶(IDO)和色氨酸2,3-加氧酶(TDO)介导的免疫抑制疾病的用途。
在本发明的一些实施方案中,所述免疫抑制疾病与传染性疾病或癌症相关。作为优选的实施方案,所述免疫抑制疾病可选自但不限于肺炎、恶性肿瘤、麻疹、肝炎、肾病或关节炎。
在本发明的一些实施方案中,所述传染性疾病选自下列病毒感染:流感、丙型肝炎病毒(HCV)、人乳头状瘤病毒(HPV)、巨细胞病毒(CMV)、脊髓灰质炎病毒、带状疱疹病毒、人类免疫缺陷病毒(HIV)、爱泼斯坦-巴尔二氏病毒(EBV)或柯萨奇病毒。所述癌症选自结肠癌、胰腺癌、乳腺癌、前列腺癌、肺癌、脑癌、卵巢癌、子宫颈癌、睾丸癌、肾癌、头或颈癌、淋巴瘤、白血病或黑素瘤。
定义和说明
在以下的说明中,包括某些具体的细节以对各个公开的实施方案提供全面的理解。然而,相关领域的技术人员会认识到,不采用一个或多个这些具体的细节,而采用其它方法、部件、材料等的情况下可实现实施方案。
除非本发明中另外要求,在整个说明书和其后的权利要求书中,词语“包括(comprise)”及其英文变体例如“包括(comprises)”和“包括(comprising)”应解释为开放式的、含括式的意义,即“包括但不限于”。
在整个本说明书中提到的“一个实施方案”或“实施方案”或“在另一实施方案中”或“在某些实施方案中”意指在至少一个实施方案中包括与该实施方案所述的相关的具体参考要素、结构或特征。因此,在整个说明书中不同位置出现的短语“在一个实施方案中”或“在实施方案中”或“在另一实施方案中”或“在某些实施方案中”不必全部指同一实施方案。此外,具体要素、结构或特征可以任何适当的方式在一个或多个实施方案中结合。
应当理解,在本发明说明书和附加的权利要求书中用到的单数形式的冠词“一”(对应于英文“a”、“an”和“the”)包括复数的对象,除非文中另外明确地规定。因此,例如提到的包括“催化剂”的反应包括一种催化剂,或两种或多种催化剂。还应当理解,术语“或”通常以其包括“和/或”的含义而使用,除非文中另外明确地规定。
除非另有说明,本文所用的下列术语和短语旨在具有下列含义。一个特定的术语或短语在没有特别定义的情况下不应该被认为是不确定的或不清楚的,而应该按照普通的含义去理解。当本文中出现商品名时,意在指代其对应的商品或其活性成分。
术语“任选”或“任选地”是指随后描述的事件或情况可能发生或可能不发生,该描述包括发生所述事件或情况和不发生所述事件或情况。例如,乙基“任选”被卤素取代是指乙基可以是未被取代的(CH
2CH
3)、单取代的(如CH
2CH
2F)、多取代的(如CHFCH
2F、CH
2CHF
2等)或完全被取代的(CF
2CF
3)。本领域技术人员可理解,对于包含一个或多个取代基的任何基团,不会引入任何在空间上不可能存在和/或不能合成的取代或取代模式。
本文所用的C
m-n指该部分中具有m-n个碳原子。例如,“C
3-10环烷基”指该环烷基具有3-10个碳原子。“C
0-6亚烷基”指该亚烷基具有0-6个碳原子,当亚烷基具有0个碳原子时,该基团为键。容易理解,当含有杂原子时,m-n代表碳原子和杂原子数量的总和。
本文中的数字范围是指给定范围中的各个整数。例如“C
1-10”是指该基团可具有1个碳原子、2个碳原子、3个碳原子、4个碳原子、5个碳原子、6个碳原子、7个碳原子、8个碳原子、9个碳原子或10个碳原子。
术语“被取代”是指特定原子上的任意一个或多个氢原子被取代基取代,只要特定原子的价态是正常的并且取代后的化合物是稳定的。当取代基为酮基(即=O)(也称为氧代基)时,意味着两个氢原子被取代,酮取代不会发生在芳香基上。
当任何变量(例如R)在化合物的组成或结构中出现一次以上时,其在每一种情况下的定义都是独立的。因此,例如,如果一个基团被0-2个R所取代,则所述基团可以任选地至多被两个R所取代,并且每种情况下的R都有独立的选项。此外,取代基和/或其变体的组合只有在这样的组合会产生稳定的化合物的情况下才是被允许的。
当一个连接基团的数量为0时,比如-(CRR)
0-,表示该连接基团为单键。
当其中一个变量选自单键时,表示其连接的两个基团直接相连,比如A-L-Z中L代表单键时表示该结构实际上是A-Z。
当一个取代基为空缺时,表示该取代基是不存在的,比如A-X中X为空缺时表示该结构实际上是A。当一个取代基的键可以交叉连接到一个环上的两个原子时,这种取代基可以与这个环上的任意原子相键合。当所列举的取代基中没有指明其通过哪一个原子连接到化学结构通式中包括但未具体提及的化合物 时,这种取代基可以通过其任何原子相键合。取代基和/或其变体的组合只有在这样的组合会产生稳定的化合物的情况下才是被允许的。例如,结构单元
 表示其可在环己基或者环己二烯上的任意一个位置发生取代。
表示其可在环己基或者环己二烯上的任意一个位置发生取代。
术语“药学上可接受的”,是针对那些化合物、材料、组合物和/或剂型而言,它们在可靠的医学判断的范围之内适用于与人类和动物的组织接触使用,而没有过多的毒性、刺激性、过敏性反应或其它问题或并发症,与合理的利益/风险比相称。
术语“药学上可接受的盐”是指本发明化合物的盐,作为通式Ⅰ、通式II及通式III的化合物的药学上可接受的盐,例如,可以提及金属盐、铵盐、与有机碱形成的盐、与无机酸形成的盐、与有机酸形成的盐、与碱性或者酸性氨基酸形成的盐等。金属盐的非限制性实例包括但不限于碱金属的盐,例如钠盐、钾盐等;碱土金属的盐,例如钙盐、镁盐、钡盐等;铝盐等。
本发明的药学上可接受的盐可由含有酸根或碱基的母体化合物通过常规化学方法合成。一般情况下,这样的盐的制备方法是:在水或有机溶剂或两者的混合物中,经由游离酸或碱形式的化合物与化学计量的适当的碱或酸反应来制备。一般地,优选醚、乙酸乙酯、乙醇、异丙醇或乙腈等非水介质。
本发明的通式Ⅰ、通式II及通式III的化合物可以以非溶剂化形式或者溶剂化形式存在,包括水合物形式。一般而言,溶剂化形式与非溶剂化的形式相当,都包含在本发明的范围之内。本发明的通式Ⅰ的化合物可以以多晶型物或无定形形式存在。
本发明的通式Ⅰ、通式II及通式III的化合物可以具有不对称碳原子(光学中心)或双键。外消旋体、非对映异构体、几何异构体和单个的异构体都包括在本发明的范围之内。
本发明中的消旋体、ambiscalemic和scalemic或者对映体纯的化合物的图示法来自Maehr,J.Chem.Ed.1985,62:114-120。除非另有说明,用楔形键和虚线键表示一个立体中心的绝对构型。当本发明所述的通式Ⅰ、通式II及通式III的化合物含有烯双键或其它几何不对称中心,除非另有规定,它们包括E和Z几何异构体。同样地,所有的互变异构形式均包括在本发明的范围之内。
本发明的通式Ⅰ、通式II及通式III的化合物可以存在特定的几何或立体异构体形式。本发明设想所有的这类化合物,包括顺式和反式异构体、(-)-和(+)-对映体、(R)-和(S)-对映体、非对映异构体、(D)-异构体、(L)-异构体,及其外消旋混合物和其他混合物,例如对映异构体或非对映体富集的混合物,所有这些混合物都属于本发明的范围之内。烷基等取代基中可存在另外的不对称碳原子。所有这些异构体以及它们的混合物也均包括在本发明的范围之内。
可以通过手性合成或手性试剂或者其他常规技术制备光学活性的(R)-和(S)-异构体以及D和L异构体。如果想得到本发明某一化合物的一种对映体,可以通过不对称合成或者具有手性助剂的衍生作用来制备,其中将所得非对映体混合物分离,并且辅助基团裂开以提供纯的所需对映异构体。或者,当分子中含有碱性官能团(如氨基)或酸性官能团(如羧基)时,与适当的光学活性的酸或碱形成非对映异构体的盐,然后通过本领域技术人员已知的分步结晶法或色谱法进行非对映异构体拆分,然后回收得到纯的对映体。此外,对映异构体和非对映异构体的分离通常是通过使用色谱法完成的,所述色谱法采用手性固定相,并任选地与化学衍生法相结合(例如由胺生成氨基甲酸盐)。
本发明的通式Ⅰ、通式II及通式III的化合物可以在一个或多个构成该化合物的原子上包含非天然比例的原子同位素。例如,可用放射性同位素标记化合物,比如氚(
3H)、碘-125(
125I)或C-14(
14C)。本发明的通式Ⅰ、通式II及通式III的化合物所有同位素组成的变换,无论放射性与否,都包括在本发明的范围之内。
术语“药学上可接受的载体”是指能够递送本发明有效量活性物质、不干扰活性物质的生物活性并且对宿主或者患者无毒副作用的任何制剂或载体介质代表性的载体包括水、油、蔬菜和矿物质、膏基、洗剂基质、软膏基质等。这些基质包括悬浮剂、增粘剂、透皮促进剂等。它们的制剂为化妆品领域或局部药物领域的技术人员所周知。关于载体的其他信息,可以参考Remington:The Science and Practice of Pharmacy,21st Ed.,Lippincott,Williams&Wilkins(2005),该文献的内容通过引用的方式并入本文。
术语“赋形剂”通常是指配制有效的药物组合物所需要载体、稀释剂和/或介质。
针对药物或药理学活性剂而言,术语“有效量”或“治疗有效量”是指无毒的但能达到预期效果的药物或药剂的足够用量。对于本发明中的口服剂型,组合物中一种活性物质的“有效量”是指与该组合物中另一种活性物质联用时为了达到预期效果所需要的用量。有效量的确定因人而异,取决于受体的年龄和一般情况,也取决于具体的活性物质,个案中合适的有效量可以由本领域技术人员根据常规试验确定。
术语“活性成分”、“治疗剂”、“活性物质”或“活性剂”是指一种化学实体,它可以有效地治疗目标紊乱、疾病或病症。
词语“包括(comprise)”或“包含(comprise)”及其英文变体例如comprises或comprising应理解为开放的、非排他性的意义,即“包括但不限于”。
术语“药物组合物”是指一种或多种本发明的化合物或其盐与药学上可接受的辅料组成的混合物。药物组合物的目的是有利于对有机体给予本发明的化合物。
除非另有规定,术语“卤代”或“卤素”本身或作为另一取代基的一部分表示氟、氯、溴或碘原子。此外,术语“卤代烷基”意在包括单卤代烷基和多卤代烷基;例如,术语“卤代C
1-3烷基”意在包括但不仅限于三氟甲基、2,2,2-三氟乙基和3-溴丙基等等。卤代烷基的实例包括但不仅限于:三氟甲基、三氯甲基、五氟乙基和五氯乙基。
术语“羟基”指-OH。
术语“氰基”指-CN。
术语“硝基”指-NO
2。
术语“氨基”是指-NH
2、-NH(烷基)和-N(烷基)
2,氨基的具体例子包括但不限于-NH
2、-NHCH
3、-NHCH(CH
3)
2、-N(CH
3)
2、-NHC
2H
5、-N(CH
3)C
2H
5等。
术语“烷基”是指由碳原子和氢原子组成的直链或支链的饱和脂肪烃基团,例如甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基、壬基、癸基等。所述特定烷基包括其所有同分异构体形式,例如丙基包括-CH
2CH
2CH
3、-CH(CH
3)
2,例如丁基包括-CH
2CH
2CH
2CH
3、-CH(CH
3)(CH
2CH
3)、-C(CH
3)
3、-CH
2CH(CH
3)
2。术语“C
1-8烷基”指具有1-8个碳原子的烷基。术语“C
1-6烷基”指具有1-6个碳原子的烷基。术语“C
1-4烷基”指具有1-4个碳原子的烷基。术语“C
1-3烷基”指具有1-3个碳原子的烷基。所述“烷基”、“C
1-8烷基”、“C
1-6烷基”、“C
1-4烷基”或“C
1-3烷基”可以是非取代的或是被一个或多个选自羟基、卤素或氨基的取代基取代。
术语“环烷基”是指仅由碳原子和氢原子组成的单环的饱和的脂肪烃基团,如C
3-20环烷基,优选为C
3-6环烷基,例如环丙基、环丁基、环戊基、环己基等。所述环烷基可以是非取代的或是被取代的,所述的取代基包括但不限于烷基、烷基氧基、氰基、羧基、芳基、杂芳基、氨基、卤素、磺酰基、亚磺酰基、磷酰基和羟基等。
除非另有规定,术语“杂”表示杂原子或杂原子团(即含有杂原子的原子团),包括碳(C)和氢(H)以外的原子以及含有这些杂原子的原子团,例如包括氧(O)、氮(N)、硫(S)、硅(Si)、锗(Ge)、铝(Al)、硼(B)、-O-、-S-、=O、=S、-C(=O)O-、-C(=O)-、-C(=S)-、-S(=O)、-S(=O)
2-,以及任选被取代的-C(=O)N(H)-、-N(H)-、-C(=NH)-、-S(=O)
2N(H)-或-S(=O)N(H)-。
除非另有规定,“环”表示被取代或未被取代的环烷基、杂环烷基、环烯基、杂环烯基、环炔基、杂环炔基、芳基或杂芳基。所谓的环包括单环、联环、螺环、并环或桥环。环上原子的数目通常被定义为环的元数,例如,“5-7元环”是指环绕排列5-7个原子。除非另有规定,该环任选地包含1-3个杂原子。因此,“5-7元环”包括例如苯基、吡啶和哌啶基;另一方面,术语“5-7元杂环烷基环”包括吡啶基和哌啶基,但不包括苯基。术语“环”还包括含有至少一个环的环系,其中的每一个“环”均独立地符合上述定义。
术语“杂环烷基”是指完全饱和的并且可以以单环、双环或螺环存在的环状基团。除非另有指示,该杂环通常为含有1至3个独立地选自硫、氧和/或氮的杂原子(优选1或2个杂原子)的3至7元环。3元杂环烷基的实例包括但不限于环氧乙烷基、环硫乙烷基、环氮乙烷基,4元杂环烷基的非限制性实例包括但不限于吖丁啶基、噁丁环基、噻丁环基,5元杂环烷基的实例包括但不限于四氢呋喃基、四氢噻吩基、吡咯烷基、异噁唑烷基、噁唑烷基、异噻唑烷基、噻唑烷基、咪唑烷基、四氢吡唑基、吡咯啉基、二氢呋喃基、二氢噻吩基,6元杂环烷基的实例包括但不限于哌啶基、四氢吡喃基、四氢噻喃基、吗啉基、哌嗪基、1,4-噻噁烷基、1,4-二氧六环基、硫代吗啉基、1,2-二噻烷基、1,4-二噻烷基、二氢吡啶基、四氢吡啶基、二氢吡喃基、四氢吡喃基、二氢噻喃基,7元杂环烷基的实例包括但不限于氮杂环庚烷基、氧杂环庚烷基、硫杂环庚烷基。优选为具有5或6个环原子的单环杂环烷基。
术语“芳基”是指具有共轭的π电子体系的全碳单环或稠合多环的芳香环基团。例如,芳基可以具有6-20个碳原子、6-14个碳原子或6-12个碳原子。芳基的非限制性实例包括但不限于苯基、萘基、蒽基和1,2,3,4-四氢化萘等。
术语“杂芳基”是指单环或稠合多环体系,其中含有至少一个选自N、O、S的环原子,其余环原子为C,并且具有至少一个芳香环。优选的杂芳基具有单个4至8元环、尤其是5至8元环,或包含6至14个、尤其是6至10个环原子的多个稠合环。杂芳基的非限制性实例包括但不限于吡咯基、呋喃基、噻吩基、咪唑基、噁唑基、吡唑基、吡啶基、嘧啶基、吡嗪基、喹啉基、异喹啉基、四唑基、三唑基、三嗪基、苯并呋喃基、苯并噻吩基、吲哚基、异吲哚基等。
本发明的药物组合物可通过将本发明的化合物与适宜的药学上可接受的辅料组合而制备,例如可配制成固态、半固态、液态或气态制剂,如片剂、丸剂、胶囊剂、粉剂、颗粒剂、膏剂、乳剂、悬浮剂、栓剂、注射剂、吸入剂、凝胶剂、微球及气溶胶等。
给予本发明化合物或其药学上可接受的盐或其药物组合物的典型途径包括但不限于口服、直肠、局部、吸入、肠胃外、舌下、阴道内、鼻内、眼内、腹膜内、肌内、皮下、静脉内给药。
本发明的药物组合物可以采用本领域众所周知的方法制造,如常规的混合法、溶解法、制粒法、制糖衣药丸法、磨细法、乳化法、冷冻干燥法等。
在一些实施方案中,药物组合物是口服形式。对于口服给药,可以通过将活性化合物与本领域熟知的药学上可接受的辅料混合来配制该药物组合物。这些辅料能使本发明的化合物被配制成片剂、丸剂、锭剂、糖衣剂、胶囊剂、液体、凝胶剂、浆剂、悬浮剂等,用于对患者的口服给药。
可以通过常规的混合、填充或压片方法来制备固体口服组合物。例如,可通过下述方法获得:将所述的活性化合物与固体辅料混合,任选地碾磨所得的混合物,如果需要则加入其它合适的辅料,然后将该混合物加工成颗粒,得到了片剂或糖衣剂的核心。适合的辅料包括但不限于:粘合剂、稀释剂、崩解剂、润滑剂、助流剂、甜味剂或矫味剂等。
药物组合物还可适用于肠胃外给药,如合适的单位剂型的无菌溶液剂、混悬剂或冻干产品。
本文所述的通式Ⅰ、通式II及通式III化合物的所有施用方法中,每天给药的剂量为0.01到200mg/kg体重、优选为0.01到20mg/kg体重、更优选0.01到10mg/kg体重,以单独或分开剂量的形式。
本发明的化合物可以通过本领域技术人员所熟知的多种合成方法来制备,包括下面列举的具体实施方式、其与其他化学合成方法的结合所形成的实施方式以及本领域技术上人员所熟知的等同替换方式,优选的实施方式包括但不限于本发明的实施例。
本发明具体实施方式的化学反应是在合适的溶剂中完成的,所述的溶剂须适合于本发明的化学变化及其所需的试剂和物料。为了获得本发明的化合物,有时需要本领域技术人员在已有实施方式的基础上对合成步骤或者反应流程进行修改或选择。
本领域合成路线规划中的一个重要考量因素是为反应性官能团(如本发明中的氨基)选择合适的保护基,例如,可参考Greene's Protective Groups in Organic Synthesis(4th Ed).Hoboken,New Jersey:John Wiley&Sons,Inc.,本发明引用的所有参考文献整体上并入本发明。
合成路线通式A:

其中,基团和原子定义如通式I化合物所述。
通式化合物1在二氯甲烷中与三光气反应得到通式化合物2;通式化合物2在吡啶中与R
2-L-NH
2反应得到通式化合物3。
合成路线通式B:

其中,基团和原子定义如通式I化合物所述。
通式化合物1在二氯甲烷中与硫光气反应得到通式化合物4;通式化合物4在吡啶中与R
2-L-NH
2反应得到通式化合物5。
下面的具体实施例,其目的是使本领域的技术人员能更清楚地理解和实施本申请。它们不应该被认为是对本申请范围的限制,而只是本申请的示例性说明和典型代表。本领域技术人员应该理解:还有形成本申请化合物的其它合成途径,下面提供的是非限制性的实施例。
除非另有说明,温度是摄氏温度。试剂购自国药集团化学试剂北京有限公司,阿法埃莎(Alfa Aesar),或北京百灵威科技有限公司等商业供应商,并且这些试剂可直接使用无需进一步纯化,除非另有说明。除 非另有说明,下列反应在无水溶剂中、氮气或氩气的正压下或使用干燥管进行;反应瓶上装有橡胶隔膜,以便通过注射器加入底物和试剂;玻璃器皿烘干和/或加热干燥。
除非另有说明,柱色谱纯化使用青岛海洋化工厂的200-300目硅胶;制备薄层色谱分离使用烟台市化学工业研究所生产的薄层色谱硅胶预制板(HSGF254);MS的测定用Thermo LCQ Fleet型(ESI)液相色谱-质谱联用仪。
除非另有说明,核磁数据(
1H-NMR)使用Varian设备于400MHz运行。核磁数据使用的溶剂有CDCl
3、CD
3OD、D
2O、DMSO-d
6等,以四甲基硅烷(0.00ppm)为基准或以残留溶剂为基准(CDCl
3:7.26ppm;CD
3OD:3.31ppm;D
2O:4.79ppm;DMSO-d
6:2.50ppm)。当标明峰形多样性时,以下简写表示不同峰形:s(单峰)、d(双重峰)、t(三重峰)、q(四重峰)、m(多重峰)、br(宽峰)、dd(双双重峰)、dt(双三重峰)。如果给出了耦合常数,则以Hertz(Hz)为单位。
除非另有说明,eq.代表摩尔当量;NBS代表N-溴代丁二酰亚胺;m-CPBA代表间氯过氧化苯甲酸;NCS代表N-氯代琥珀酰亚胺;DPPA代表叠氮磷酸二苯酯;Boc-代表叔丁氧羰基;室温代表20-30℃。
合成通式A

步骤1:通式化合物2的合成
0℃下,向通式化合物1(1.0eq.)的二氯甲烷溶液中加入三光气(0.6eq.),反应液升至室温搅拌2小时。反应结束后,减压浓缩除去溶剂,残留固体通式化合物2直接用于步骤2。
步骤2:通式化合物3的合成
将通式化合物2分批加到R
2-L-NH
2(0.5eq.)的吡啶溶液中,室温搅拌12小时。反应结束后,将反应液倒入水中,用乙酸乙酯萃取,萃取液用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液浓缩,残留物用硅胶色谱柱纯化得到通式化合物3。
合成通式B:

步骤1:通式化合物4的合成
0℃下,向通式化合物1(1.0eq.)的二氯甲烷溶液中加入硫光气(1.2eq.),升到室温搅拌2小时。减压浓缩除去溶剂,残留物用硅胶色谱柱纯化得到通式化合物4。
步骤2:通式化合物5的合成
将通式化合物4分批加到R
2-L-NH
2(1.0eq.)的吡啶溶液中,室温搅拌12小时。反应液倒入水中,用乙酸乙酯萃取,萃取液用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液浓缩,残留物用硅胶柱色谱纯化得到通式化合物5。
中间体1:咪唑并[5,1-a]异喹啉-5-胺的合成

步骤1:3-氯-1-甲基异喹啉的合成
氮气保护下,向1,3-二氯异喹啉(49.6g)的四氢呋喃(500mL)溶液中慢慢滴加三甲基铝的四氢呋喃溶液(139mL,2M),然后加入四三苯基膦钯(2.9g),将混合物加热到85℃搅拌过夜。将反应夜冷却到室温后倒入水(200mL)中,用乙酸乙酯萃取。将有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残留物用硅胶色谱柱纯化(石油醚:乙酸乙酯=20:1)得到3-氯-1-甲基异喹啉(43.0g)。
步骤2:1-溴甲基-3-氯异喹啉的合成
向3-氯-1-甲基异喹啉(43.0g)的四氯化碳(300mL)溶液中依次加入N-溴代丁二酰亚胺(44.0g)和过氧化苯甲酰(6.0g),将混合物加热到80℃搅拌过夜。冷却到室温,过滤,滤液减压浓缩,残留物用硅胶色谱柱纯化(石油醚:乙酸乙酯=20:1)得到1-溴甲基-3-氯异喹啉(50.5g)。
步骤3:1-叠氮甲基-3-氯异喹啉的合成
向1-溴甲基-3-氯异喹啉(50.0g)的丙酮(500mL)和水(50mL)的溶液中加入叠氮化纳(18.2g),加热到50℃搅拌2小时。冷却到室温,倒入水中(100mL),用乙酸乙酯萃取。将有机相用无水硫酸钠干燥,过滤,滤液减压浓缩后得到1-叠氮甲基-3-氯异喹啉(35.6g)。
步骤4:1-氨基甲基-3-氯异喹啉的合成
向1-叠氮甲基-3-氯异喹啉(35.6g)的四氢呋喃(300mL)和水(30mL)的溶液中加入三苯基膦(49.8g),将混合物室温搅拌过夜。将反应混合物倒入水(100mL)中,用乙酸乙酯萃取。将有机相用无水硫酸钠干燥,过滤,将滤液减压浓缩,残留物用硅胶色谱柱纯化(石油醚:乙酸乙酯=4:1)得到1-氨基甲基-3-氯异喹啉(27.7g)。
步骤5:N-((3-氯异喹啉-1-基)甲基)甲酰胺的合成
将1-氨基甲基-3-氯异喹啉(27.7g)和甲酸乙酯(300mL)的混合液加热到65℃搅拌8小时。冷却到室温,减压浓缩得到N-((3-氯异喹啉-1-基)甲基)甲酰胺(30.0g)。
步骤6:5-氯咪唑并[5,1-a]异喹啉的合成
向N-((3-氯异喹啉-1-基)甲基)甲酰胺(30.0g)的二氯甲烷(500mL)溶液中加入三氯氧磷(30.7g),然后慢慢滴加三乙胺(82.0g),将混合物室温搅拌2小时。将反应混合物慢慢倒入碎冰中,用饱和碳酸氢钠水溶液调pH到8,过滤,滤液分液,将有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残留物用硅胶色谱柱纯化(石油醚:乙酸乙酯=2:1)得到5-氯咪唑并[5,1-a]异喹啉(25.0g)。
步骤7:N-(咪唑并[5,1-a]异喹啉-5-基)-1,1-二苯酮缩亚胺的合成
氮气保护下,向5-氯咪唑并[5,1-a]异喹啉(23.0g)的甲苯(300mL)溶液中加入二苯甲酮亚胺(21.7g)、三(二亚苄基丙酮)二钯(4.6g)、1,1'-联萘-2,2'-双二苯膦(6.2g)和碳酸铯(71.7g),将混合物加热到110℃搅拌12小时。冷却到室温,过滤,滤液减压浓缩,残留物用硅胶色谱柱纯化(二氯甲烷:甲醇=20:1)得到N-(咪唑并[5,1-a]异喹啉-5-基)-1,1-二苯酮缩亚胺(25.6g)。
步骤8:咪唑并[5,1-a]异喹啉-5-胺的合成
将N-(咪唑并[5,1-a]异喹啉-5-基)-1,1-二苯酮缩亚胺(15.0g)溶于四氢呋喃(200mL)中,向所得溶液中加入盐酸(30mL,2N),室温搅拌2小时。将反应混合物倒入水中(100mL),用饱和碳酸氢钠水溶液调节pH到8,用乙酸乙酯萃取,将有机相用无水硫酸钠干燥,过滤,将滤液减压浓缩得到咪唑并[5,1-a]异喹啉-5-胺(6.3g)。
1H-NMR(400M,DMSO-d
6):δ9.38(s,1H),8.55(s,1H),8.20(d,J=8.0Hz,1H),7.60(d,J=8.0Hz,1H),7.49(t,J=7.2Hz,1H),7.37(t,J=7.2Hz,1H),6.70(brs,2H),6.29(s,1H)。
中间体2:四氮唑并[5,1-a]异喹啉-5-胺的合成

步骤1:5-氯四氮唑并[5,1-a]异喹啉的合成
向1,3-二氯异喹啉(1.5g)的N,N-二甲基甲酰胺(50mL)溶液中依次加入叠氮基三甲基硅烷(1.8g)和三水合四丁基氟化铵(2.4mg),加热到80℃搅拌12小时。冷却到室温,将反应液倒入水(200mL)中,用乙酸乙酯萃取。将有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残留物用硅胶色谱柱纯化(石油醚:乙酸乙酯=40:1)得到5-氯四氮唑并[5,1-a]异喹啉(0.8g)。
步骤2:N-(四氮唑并[5,1-a]异喹啉-5-基)-1,1-二苯酮缩亚胺的合成
参考中间体1合成中的步骤7。
步骤3:四氮唑并[5,1-a]异喹啉-5-胺的合成
参考中间体1合成中的步骤8。LC-MS[M+1]
+=186。
中间体3:8-氟咪唑并[5,1-a]异喹啉-5-胺的合成

步骤1:5-氟-2-肟基-2,3-二氢-1H-茚-1-酮的合成
将5-氟-2,3-二氢-1H-茚-1-酮(8.0g)溶于乙醚(150mL)中,冷却到0℃,向该溶液中通干燥的氯化氢3小时,然后缓慢滴加亚硝酸异戊酯(12.5g),滴加完毕后升至室温搅拌3小时,出现固体,过滤,滤饼用乙醚洗涤,将滤饼干燥得到5-氟-2-肟基-2,3-二氢-1H-茚-1-酮(7.0g)。
步骤2:1,3-二氯-6-氟异喹啉的合成
0℃下,将5-氟-2-肟基-2,3-二氢-1H-茚-1-酮(7.0g)溶于三氯氧磷(120mL)中,然后分批向其中加入三氯氧磷(1.5g),加毕向溶液中通干燥的氯化氢气体,3小时后停止通气,将反应液加热到60℃搅拌16小时。冷却到室温,减压浓缩除去三氯氧磷,将得到的固体用水洗涤,真空干燥得到1,3-二氯-6-氟异喹啉(6.0g)。
步骤3、4、5、6、7、8、9分别参考中间体1合成中的步骤1、2、3、4、5、6、7
步骤10:8-氟咪唑并[5,1-a]异喹啉-5-胺的合成
参考中间体1合成中的步骤8。
1H-NMR(400MHz,DMSO-d
6):δ8.586(s,1H),8.036(s,1H),7.945-7.924(m,1H),7.257-7.204(m,2H),6.746(s,2H),6.093(s,1H)。
中间体4:7-氟咪唑并[5,1-a]异喹啉-5-胺的合成

步骤1:4-氟-2-肟基-2,3-二氢-1H-茚-1-酮的合成
参考中间体3合成中的步骤1,用4-氟-2,3-二氢-1H-茚-1-酮代替5-氟-2,3-二氢-1H-茚-1-酮。
步骤2、3、4、5、6、7、8、9分别参考中间体3合成中的步骤2、3、4、5、6、7、8、9
步骤10:7-氟咪唑并[5,1-a]异喹啉-5-胺的合成
参考中间体3合成中的步骤10。
1H-NMR(400MHz,DMSO-d
6):δ8.464(s,1H),8.086-8.049(m,1H),7.883(s,1H),7.276-7.243(m,1H),7.036-7.030(m,1H),6.608(s,2H),5.939(s,1H)。
中间体5:7-氟咪唑并[5,1-a]异喹啉-5-胺的合成

步骤1:3-氯-1-甲基-5-硝基异喹啉的合成
0℃下,将3-氯-1-甲基异喹啉(8.3g)缓慢加入到浓硫酸(100mL)中,然后分批加入硝酸钾(5.7g),将混合物在0℃下搅拌1小时,将反应液倒入到碎冰中,用氨水调节pH到9,过滤,滤饼用水洗涤,将滤饼真空干燥后得到3-氯-1-甲基-5-硝基异喹啉(6.2g)。
1H-NMR(400MHz,DMSO-d
6):δ8.713-8.692(m,1H),8.662-8.640(m,1H),8.222(s,1H),7.885-7.845(m,1H),2.968(s,3H)。
步骤2:3-氯-1-甲基-5-氨基异喹啉的合成
将3-氯-1-甲基-5-硝基异喹啉(6.0g)溶于甲醇和水(1:1,200mL)的混合溶剂中,加入氯化铵(14.4g),加热至60℃,溶液变澄清后,加入还原铁粉(15.1g),于60℃下搅拌30分钟,冷却至室温,过滤,滤液减压浓缩除去甲醇,用乙酸乙酯萃取(150mL×2),有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,将滤液减压浓缩,残留物用硅胶柱色谱纯化(石油醚:乙酸乙酯=20:1)得到3-氯-1-甲基-5-氨基异喹啉(3.9g)。
步骤3:3,5-二氯-1-甲基异喹啉的合成
将3-氯-1-甲基-5-氨基异喹啉(3.9g)溶于浓盐酸(50mL)中,冷却到0℃,缓慢滴加亚硝酸钠(1.5 g)的水(10mL)溶液,滴加速度使反应液温度保持低于5℃,20分钟后,加入氯化亚铜(2.4g)的浓盐酸(10mL)溶液,0℃下搅拌1小时。将反应液倒入水中,用乙酸乙酯萃取(150mL×2),有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,将滤液减压浓缩,残留物用硅胶柱色谱纯化(石油醚:乙酸乙酯=20:1)得到3,5-二氯-1-甲基异喹啉(4.2g)。
步骤4、5、6、7、8、9分别参考中间体1合成中的步骤2、3、4、5、6、7。
步骤10:7-氟咪唑并[5,1-a]异喹啉-5-胺的合成
参考中间体1合成中的步骤8。
1H-NMR(400MHz,DMSO-d
6):δ8.543(s,1H),8.053-8.033(m,1H),8.006(s,1H),7.460-7.440(m,1H),7.208-7.169(m,1H),6.819(s,2H),6.231(s,1H)。
中间体6:[1,2,3]三氮唑并[5,1-a]异喹啉-5-胺的合成

步骤1:2-((三甲基硅基)乙炔基)苯甲醛的合成
氮气保护下,向2-溴苯甲醛(18.5g)的四氢呋喃(500mL)溶液中依次加入三甲基硅乙炔(19.6g)、碘化亚铜(0.95g)、三乙胺(20.2g)和双三苯基磷二氯化钯(3.5g),将反应液加热到50℃搅拌过夜。冷却至室温,倒入水中,用乙酸乙酯萃取(300mL×2),有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,将滤液减压浓缩,残留物用硅胶柱色谱纯化(石油醚:乙酸乙酯=20:1)得到2-((三甲基硅基)乙炔基)苯甲醛(19.0g)。
步骤2:三甲基((2(2-硝基乙烯基)苯基)乙炔基)硅烷的合成
将2-((三甲基硅基)乙炔基)苯甲醛(19.0g)、醋酸铵(7.2g)加入到硝基甲烷(50mL)中,反应液加热到110℃后继续搅拌1小时,冷却至室温,减压浓缩,固体残留物用乙酸乙酯(500mL)溶解,有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,将滤液减压浓缩,残留物用硅胶柱色谱纯化(石油醚:乙酸乙酯=20:1)得到三甲基((2(2-硝基乙烯基)苯基)乙炔基)硅烷(15.3g)。
1H-NMR(400MHz,CDCl
3):δ8.460-8.426(d,1H),7.835-7.801(d,1H),7.580-7.535(m,2H),7.441-7.361(m,2H),0.295(s,9H)。
步骤3:[1,2,3]三氮唑并[5,1-a]异喹啉的合成
将三甲基((2(2-硝基乙烯基)苯基)乙炔基)硅烷(15.3g)和叠氮化钠(6.0g)溶于二甲基亚砜(150mL)中,反应液加热到110℃搅拌40分钟。冷却至室温,倒入水中,用乙酸乙酯萃取(300mL×2),有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,将滤液减压浓缩,残留物用硅胶柱色谱纯化(二氯甲烷:甲醇=20:1)得到[1,2,3]三氮唑并[5,1-a]异喹啉(6.8g)。
步骤4:5-溴-[1,2,3]三氮唑并[5,1-a]异喹啉的合成
氮气保护下,将[1,2,3]三氮唑并[5,1-a]异喹啉(2.0g)溶于干燥的甲苯(100mL)中,冷却到-40℃,滴加正丁基锂正己烷溶液(2.4M,13.9mmol)。滴加完毕,反应液在-40℃搅拌2小时,加入1,2-二溴四氯乙烷(4.6g),移去冷却,继续搅拌2小时后倒入水中,用乙酸乙酯萃取(100mL×2),有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,将滤液减压浓缩,残留物用硅胶柱色谱纯化(二氯甲烷:甲醇=20:1)得到5-溴-[1,2,3]三氮唑并[5,1-a]异喹啉(0.54g)。
步骤5和步骤6:[1,2,3]三氮唑并[5,1-a]异喹啉-5-胺的合成
分别参考中间体1合成中的步骤7和步骤8,最终得到[1,2,3]三氮唑并[5,1-a]异喹啉-5-胺。
1H-NMR(400MHz,CDCl
3):δ8.459(s,1H),8.070-8.050(d,1H),7.603-7.583(d,1H),7.542-7.501(m,1H),7.424-7.383(m,1H),6.354(s,1H),5.262(s,2H)。
中间体7:[1,2,4]三氮唑并[4,3-a]喹啉-4-胺的合成

步骤1:3-溴喹啉-1-氮氧化物的合成
向3-溴喹啉(10.0g)的二氯甲烷(300mL)溶液中,分批加入间氯过氧化苯甲酸(17.3g),然后室温搅拌12小时。反应液倒入饱和碳酸氢钠水溶液中,有机相分离,用饱和食盐水洗涤,无水硫酸钠干燥,过滤,将滤液减压浓缩,残留物用硅胶柱色谱纯化(二氯甲烷:甲醇=50:1)得到3-溴喹啉-1-氮氧化物(9.4g)。
步骤2:3-溴-2-氯喹啉的合成
将3-溴喹啉-1-氮氧化物(4.0g)和三氯乙腈(5.2g)溶于甲苯(200mL)中,然后加入三苯基膦(9.4g),将混合液加热到115℃搅拌4小时。冷却到室温,将反应液浓缩,残留物用硅胶色谱柱纯化(二氯甲烷:甲醇=50:1)得到3-溴-2-氯喹啉(0.84g)。
步骤3:3-溴-2-肼基喹啉的合成
将3-溴-2-氯喹啉(0.5g)加入到水合肼(25mL)中,将反应液加热到140℃搅拌1小时。冷却至室温,倒入水中,用乙酸乙酯萃取(100mL×2),有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,将滤液减压浓缩,残留物用硅胶柱色谱纯化(二氯甲烷:甲醇=20:1)得3-溴-2-肼基喹啉(0.4g)。
步骤4:4-溴-[1,2,4]三氮唑并[4,3-a]喹啉的合成
将3-溴-2-肼基喹啉(0.4g)加入到甲酸(25mL)中,然后室温搅拌过夜,将反应液倒入水中,用乙酸乙酯萃取(100mL×2),有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,将滤液减压浓缩,残留物用硅胶柱色谱纯化(二氯甲烷:甲醇=20:1)得到4-溴-[1,2,4]三氮唑并[4,3-a]喹啉(0.32g)。
步骤5和步骤6:[1,2,4]三氮唑并[4,3-a]喹啉-4-胺的合成
分别参考中间体1合成中的步骤7和步骤8,最终得到[1,2,4]三氮唑并[4,3-a]喹啉-4-胺。
1H-NMR(400MHz,DMSO-d
6):δ9.916(s,1H),8.244-8.224(d,1H),7.659-7.637(d,1H),7.424-7.365(m,2H),6.573(s,1H),6.085(s,2H)。
中间体8:咪唑并[1,5-a]吡啶-5-胺的合成

步骤1、2、3、4、5、6分别参考中间体1合成中的步骤2、3、4、5、6、7
步骤7:咪唑并[1,5-a]吡啶-5-胺的合成
参照中间体1合成中的步骤8,得到咪唑并[1,5-a]吡啶-5-胺。
1H-NMR(400MHz,DMSO-d
6):δ8.29(s,1H),7.22(s,1H),6.85(d,J=8.8Hz,1H),6.71(dd,J=8.8Hz,J=6.8Hz,1H),6.44(brs,2H),5.71(dd,J=6.8Hz,J=1.2Hz,1H)。
中间体9:咪唑并[1,5-c]喹唑啉-5-胺的合成

步骤1:4-甲基喹唑啉-2-胺的合成
向2-氟苯乙酮(1.4g)的N,N-二甲基乙酰胺(10mL)溶液中加入胍碳酸盐(2.7g),将混合液加热到130℃搅拌4小时。反应液冷却到室温后加入冷水(20mL)中,过滤,滤饼用石油醚洗涤,干燥后得到4-甲基喹唑啉-2-胺(1.1g)。
步骤2:咪唑并[1,5-c]喹唑啉-5-胺的合成
氮气保护下向4-甲基喹啉-2-胺(1.1g)、甘氨酸(1.0g)、叔丁基过氧化氢(3.60g,70%水溶液)、四丁基碘化铵(0.5g)和醋酸(1.3g)的混合物中加入二甲基亚砜(10mL),将反应液加热到95℃搅拌10小时。冷却到室温,加入水(30ml)和乙酸乙酯(30ml),有机相分离后用饱和盐水洗涤,硫酸钠干燥,过滤,滤液减压浓缩,残留物经硅胶柱层析分离(二氯甲烷:甲醇=20:1)得到咪唑并[1,5-c]喹唑啉-5-胺(0.26g)。
1H-NMR(400MHz,DMSO-d
6):δ8.58(s,1H),7.96(d,J=8.0Hz,1H),7.83(s,1H),7.58(s,2H),7.37(d,J=3.6Hz,2H),7.19-7.23(m,1H)。
中间体10:咪唑并[5,1-f][1,6]萘啶-5-胺的合成

步骤1:2-(2-乙氧基-2-氧代乙基)烟酸的合成
氮气保护下向叔丁醇钾(33.7g)和异丙醇(500ml)的混合物中缓慢滴加乙酰乙酸乙酯(26.0g),滴加完毕后搅拌2小时。向反应液中加入无水醋酸铜(5.5g,0.03mol)和2-氯烟酸(15.8g),加热至80℃搅拌4小时。冷却到室温,用水(500mL)猝灭,接着用2N稀盐酸将pH调节至4。加入乙酸乙酯(500mL),搅拌20分钟后过滤,滤液分液,有机相减压浓缩,向残留物加入甲苯(300mL),搅拌1小时,过滤后得到2-(2-乙氧基-2-氧代乙基)烟酸(15.5g)。
步骤2:1,6-萘啶-5,7(6H,8H)-二酮的合成
0℃下,向2-(2-乙氧基-2-氧代乙基)烟酸(15.5g)的四氢呋喃(200mL)溶液中加入三乙胺(7.5g)和氯甲酸乙酯(9.6g),反应液升至室温搅拌1小时。缓慢地加入氨水(5mL),继续搅拌1小时。加入水(100mL),用稀盐酸将pH调节至7。过滤后得到1,6-萘啶-5,7(6H,8H)-二酮(6.5g)。
步骤3:5,7-二氯-1,6-萘啶的合成
将1,6-萘啶-5,7(6H,8H)-二酮(6.5g)溶于三氯氧磷(150mL)中,冷却到0℃,加入N,N-二异丙基乙胺(10.4g),然后加热到100℃搅拌16小时。冷却到室温,减压除去三氯氧磷,加入冷水(100mL),过滤,固体真空干燥后得到5,7-二氯-1,6-萘啶(4.0g)。
步骤4、5、6、7、8、9、10分别参考中间体1合成中的步骤1、2、3、4、5、6、7。
步骤11:咪唑并[5,1-f][1,6]萘啶-5-胺的合成
参考中间体1合成中的步骤8。LC-MS[M+1]
+=185。
中间体11:6-氯咪唑并[5,1-a]异喹啉-5-胺的合成

步骤1:4-氯-1-甲基异喹啉-3-胺的合成
向1-甲基异喹啉-3-胺(3.2g)的二氯甲烷(50ml)溶液中缓慢加入N-氯代琥珀酰亚胺(2.7g),加毕后室温搅拌1小时,加入冷水(50ml)淬灭反应,分液,将有机相用无水硫酸钠干燥,过滤,减压浓缩,残留物用硅胶色谱柱纯化(二氯甲烷:甲醇=20:1)得到4-氯-1-甲基异喹啉-3-胺(3.5g)。
步骤2:2-(4-氯-1-甲基异喹啉-3-基)异吲哚-1,3-二酮的合成
室温下向醋酸(50ml)中依次加入4-氯-1-甲基异喹啉-3-胺(3.5g)和邻苯二甲酸酐(4.0g),将该混合物加热至120℃搅拌10小时,然后减压浓缩。向残留物加入100ml饱和碳酸氢钠水溶液并搅拌1小时,过滤后得到2-(4-氯-1-甲基异喹啉-3-基)异吲哚-1,3-二酮(4.8g)。
步骤3:2-(1-(溴甲基)-4-氯喹啉-3-基)异吲哚-1,3-二酮的合成
氮气保护下向2-(4-氯-1-甲基异喹啉-3-基)异吲哚-1,3-二酮(4.8g)和N-溴代丁二酰亚胺(3.2g)的混合物中加入过氧化苯甲酰(0.36g)和四氯化碳(200ml),加热到80℃搅拌过夜。冷却到室温,过滤,滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=50:1)得到2-(1-(溴甲基)-4-氯喹啉-3-基)异吲哚-1,3-二酮(3.6g)。
步骤4:2-(1-(叠氮基甲基)-4-氯异喹啉-3-基)异吲哚-1,3-二酮的合成
将2-(1-(溴甲基)-4-氯喹啉-3-基)异吲哚-1,3-二酮(3.6g)和叠氮化钠(0.88g)溶于丙酮和水(10:1,100mL)的混合溶剂中,加热到65℃搅拌3小时。冷却后倒入水(200ml)中,用乙酸乙酯萃取(100mL×2),合并有机相,并用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=20:1)得到2到-(1-(叠氮基甲基)-4-氯异喹啉-3-基)异吲哚-1,3-二酮(2.2g)。
步骤5:2-(1-(氨基甲基)-4-氯异喹啉-3-基)异吲哚-1,3-二酮的合成
将2-(1-(叠氮基甲基)-4-氯异喹啉-3-基)异吲哚-1,3-二酮(2.2g)和三苯基膦(2.3g)溶于四氢呋喃和水(10:1,50mL)的混合溶剂中,室温搅拌过夜。反应混合物倒入水(100mL)中,用乙酸乙酯萃取(100mL×2),有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=20:1)得到2-(1-(氨基甲基)-4-氯异喹啉-3-基)异吲哚-1,3-二酮(1.3g)。
步骤6:2-(6-氯咪唑并[5,1-a]异喹啉-5-基)异吲哚-1,3-二酮的合成
将2-(1-(氨基甲基)-4-氯异喹啉-3-基)异吲哚-1,3-二酮(1.3g)与甲酸乙酯(100mL)混合,加热回流过夜。减压浓缩,得到灰色固体,用四氢呋喃(50mL)溶解,0℃下缓慢加入三氯氧磷(1.2g)和三乙胺(2.3g),然后室温搅拌2小时。减压浓缩,向残留物加入乙酸乙酯(100ml)溶解,所得溶液用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=20:1)得到2-(6-氯咪唑并[5,1-a]异喹啉-5-基)异吲哚-1,3-二酮(0.26g)。
步骤7:6-氯-H-咪唑并[5,1-a]异喹啉-5-胺的合成
将2-(6-氯咪唑并[5,1-a]异喹啉-5-基)异吲哚-1,3-二酮(0.26g)加入到50ml无水乙醇中,搅拌下加入5滴85%水合肼。然后将反应液加热到50℃搅拌10分钟。减压浓缩除去有机溶剂,加入50ml冷水,用乙酸乙酯萃取(50mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=20:1)得到6-氯-H-咪唑并[5,1-a]异喹啉-5-胺(90.5mg)。LC-MS[M+1]
+=218。
中间体12:H-咪唑并[1,5-a]喹啉-4-胺的合成

步骤1:2-甲基喹啉-3-甲酸乙酯的合成
向2-硝基苯甲醛(11.8g)和乙酰乙酸乙酯(12.2g)的醋酸(150mL)溶液中分批加入还原铁粉(21.9g),然后将反应液加热到50℃搅拌2小时。将反应液冷却,过滤,将滤液倒入水(200mL)中,用饱和碳酸氢钠水溶液调节pH到8,用乙酸乙酯萃取(250mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=100:1)得到2-甲基喹啉-3-甲酸乙酯(13.6g)。
步骤2:2-甲基喹啉-3-甲酸的合成
将2-甲基喹啉-3-甲酸乙酯(13.6g)加入四氢呋喃(50mL)、甲醇(50mL)和水(50mL)的混合溶剂中,向该混合物中加入固体一水合氢氧化锂(5.3g),室温搅拌4小时,用稀盐酸(2M)调节pH到5,减压浓缩除去有机溶剂,所得固体过滤,水洗,干燥后得到2-甲基喹啉-3-甲酸(8.1g)。
步骤3:(2-甲基喹啉-3-基)氨基甲酸叔丁酯的合成
氮气保护下将干燥的叔丁醇(10mL)、三乙胺(21.6g)和叠氮磷酸二苯酯(13.1g)加入2-甲基喹啉-3-甲酸(8.1g)的1,4-二氧六环(100mL)溶液中,将反应液加热到100℃搅拌5小时。冷却到室温,过滤,滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=50:1)得到(2-甲基喹啉-3-基)氨基甲酸叔丁酯(3.5g)。
步骤4:咪唑并[1,5-a]喹啉-4-基氨基甲酸叔丁酯的合成
氮气保护下向(2-甲基喹啉-3-基)氨基甲酸叔丁酯(1.8g)、甘氨酸(1.0g)、叔丁基过氧化氢(3.60g,70%水溶液)、四丁基碘化铵(0.5g)和醋酸(1.3g)的混合物中加入二甲基亚砜(10mL),加热到95℃搅拌10小时。冷却到室温,加入水(30ml)和乙酸乙酯(30ml),有机相分离后用饱和盐水洗涤,硫酸钠干燥,过滤,滤液减压浓缩,残留物经硅胶柱层析分离(二氯甲烷:甲醇=20:1)得到咪唑并[1,5-a]喹啉-4-基氨基甲酸叔丁酯(0.66g)。
步骤5:咪唑并[1,5-a]喹啉-4-胺的合成
将咪唑并[1,5-a]喹啉-4-基氨基甲酸叔丁酯(0.66g)加到盐酸乙醇溶液(20mL,4N)中,然后室温搅拌4小时,用饱和碳酸氢钠调节pH到7,用乙酸乙酯萃取(100mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩后得到咪唑并[1,5-a]喹啉-4-胺(0.35g)。
1H-NMR(400MHz,DMSO-d
6):δ9.01(s,1H),8.16(d,J=8.0Hz,1H),7.61(s,1H),7.45(m,1H),7.23(m,2H),6.09(s,1H),5.87(s,2H)。
中间体13:7-氯咪唑并[1,5-a]-5-胺的合成

步骤1:4-氯-6-(乙氧基羰基)-吡啶甲酸的合成
室温下向4-氯吡啶-2,6-二甲酸二乙酯(50.0g)的乙醇(330mL)溶液中慢慢滴加氢氧化钠(7.76g)的水(800mL)溶液,8小时滴加完毕,然后室温搅拌过夜。减压浓缩,将残留物溶于水中,用浓盐酸调节pH到3,用乙酸乙酯萃取(300mL×2),有机相用无水硫酸钠干燥,过滤,减压浓缩后得到4-氯-6-(乙氧基羰基)-吡啶甲酸(21.3g)。
步骤2:6-(叔丁氧基羰基氨基)-4-氯吡啶甲酸乙酯的合成
氮气保护下,将干燥的叔丁醇(10mL)、三乙胺(21.6g)和叠氮磷酸二苯酯(13.1g)加到4-氯-6-(乙氧基羰基)-吡啶甲酸(9.94g)的1,4-二氧六环(100mL)溶液中,将反应液加热到100℃搅拌5小时。冷却到室温,过滤,滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=50:1)得到6-(叔丁氧基羰基氨基)-4-氯吡啶甲酸乙酯(6.1g)。
步骤3:(4-氯-6-(羟甲基)吡啶-2-基)氨基甲酸叔丁酯的合成
向6-(叔丁氧基羰基氨基)-4-氯吡啶甲酸乙酯(11.5g)的乙醇(150mL)溶液中分批加入硼氢化钠(4.4g),反应混合物室温搅拌过夜。向反应混合物慢慢滴加10mL水淬灭反应,减压浓缩除去乙醇,然后加入饱和碳酸氢钠水溶液调节pH到8,用乙酸乙酯萃取(250mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=50:1)得到(4-氯-6-(羟甲基)吡啶-2-基)氨基甲酸叔丁酯(9.6g)。
步骤4:(4-氯-6-(氯代甲基)吡啶-2-基)氨基甲酸叔丁酯的合成
向(4-氯-6-(羟甲基)吡啶-2-基)氨基甲酸叔丁酯(9.6g)的二氯甲烷(200mL)溶液中加入吡啶(3.8g)和二氯亚砜(5.4g),反应混合物室温搅拌过夜。将反应混合物倒入水中,然后加入饱和碳酸氢钠水溶液调节pH到8,用二氯甲烷萃取(250mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=20:1)得到(4-氯-6-(氯代甲基)吡啶-2-基)氨基甲酸叔丁酯(4.8g)。
步骤5:(4-氯-6-((1,3-二氧代异吲哚啉-2-基)甲基)吡啶-2-基)氨基甲酸叔丁酯的合成
向(4-氯-6-(氯代甲基)吡啶-2-基)氨基甲酸叔丁酯(2.8g)的N,N-二甲基甲酰胺(100mL)溶液中加入邻苯二甲酰亚胺钾盐(2.0g),反应混合物室温搅拌过夜。将反应混合物倒入水中,用乙酸乙酯萃取(150mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=20:1)得到(4-氯-6-((1,3-二氧代异吲哚啉-2-基)甲基)吡啶-2-基)氨基甲酸叔丁酯(2.3g)。
步骤6:(6-(氨基甲基)-4-氯吡啶-2-基)氨基甲酸叔丁酯的合成
向(4-氯-6-((1,3-二氧代异吲哚啉-2-基)甲基)吡啶-2-基)氨基甲酸叔丁酯(2.3g)的乙醇(100mL)溶液中加入水合联氨(0.3g),将反应混合物加热到80℃搅拌2小时。将反应混合物冷却到室温,过滤,滤液减压浓缩,残留物用硅胶色谱柱纯化(二氯甲烷:甲醇=20:1),得到(6-(氨基甲基)-4-氯吡啶-2-基)氨基甲酸叔丁酯(1.0g)。
步骤7、8分别参考中间体1合成中的步骤5、6。
步骤9:7-氯咪唑并[1,5-a]-5-胺的合成
参考中间体12合成中的步骤5。
1H-NMR(400MHz,DMSO-d
6):δ8.46(s,1H),7.64(d,J=2.0Hz 1H),7.39(s,1H),6.55(d,J=2.0Hz,1H),6.44(brs,2H)。
中间体14:6-氯咪唑并[1,5-a]-8-胺的合成

步骤1:5-氯-2-甲基-3-硝基吡啶的合成
将1,3-二氯异喹啉替换成2,5-二氯-3-硝基吡啶,参考中间体1合成中的步骤1得到5-氯-2-甲基-3-硝基吡啶。
步骤2:5-氯-2-甲基吡啶-3-胺
参考中间体5合成中的步骤2,得到标题化合物。
步骤3:(5-氯-2-甲基吡啶-3-基)-叔丁氧酰胺
参考中间体15合成中的步骤3,得到标题化合物。
步骤4:(2-溴甲基-5-氯吡啶-3-基)-叔丁氧酰胺
参考中间体1合成中的步骤2,得到标题化合物。
步骤5:(5-氯-2-((1,3-二氧异吲哚啉-2-基)甲基)吡啶-3-基)-叔丁氧酰胺
参考中间体13合成中的步骤5,得到标题化合物。
步骤6:(2-氨基甲基-5-氯吡啶-3-基)-叔丁氧酰胺
参考中间体13合成中的步骤6,得到标题化合物。
步骤7、8分别参考中间体1合成中的步骤5、6。
步骤9:6-氯咪唑并[1,5-a]-8-胺的合成
参考中间体12合成中的步骤5。
1H-NMR(400MHz,DMSO-d6):δ9.09(s,1H),8.45(s,1H),7.96(s,1H),7.00(s,1H),6.87(brs,2H)。
中间体15:6-三氟甲基咪唑并[1,5-a]-8-胺的合成

步骤1:2-氯-5-(三氟甲基)吡啶-3-胺的合成
向5-三氟甲基-3-硝基-2-氯吡啶(10.0g)的乙酸乙酯(250mL)溶液中加入氯化亚锡二水合物(43.9g),将反应混合物加热到80℃搅拌2小时。将反应混合物冷却到室温后倒入水中,用饱和碳酸氢钠水溶液调节pH到8,过滤,将滤液分液,有机相用无水硫酸钠干燥,过滤,减压浓缩后得到2-氯-5-(三氟甲基)吡啶-3-胺(8.3g)。
步骤2:3-氨基-5-(三氟甲基)吡啶甲腈的合成
氮气下向2-氯-5-(三氟甲基)吡啶-3-胺(5.1g)、氰化锌(3.9g)、三(二亚苄基丙酮)二钯(1.2g)和1,1'-联萘-2,2'-双二苯膦(1.42g)的混合物中加入N,N-二甲基甲酰胺(50mL),将混合物加热回流过夜。反应混合物冷却到室温后,倒入水中,用乙酸乙酯萃取,有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,将滤液减压浓缩,残留物用硅胶色谱柱纯化(二氯甲烷:甲醇=20:1)得到3-氨基-5-(三氟甲基)吡啶甲腈(2.5g)。
步骤3:(2-氰基-5-(三氟甲基)吡啶-3-基)氨基甲酸叔丁酯的合成
向3-氨基-5-(三氟甲基)吡啶甲腈(2.0g)的四氢呋喃(100mL)溶液中加入二碳酸二叔丁酯(3.5g)、三乙胺(2.16g)和4-二甲氨基吡啶(0.13g),反应混合物室温搅拌4小时。加入水淬灭反应,用乙酸乙酯萃取(150mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=50:1)得到(2-氰基-5-(三氟甲基)吡啶-3-基)氨基甲酸叔丁酯(2.3g)。
步骤4:(2-(氨基甲基)-5-(三氟甲基)吡啶-3-基)氨基甲酸叔丁酯的合成
向(2-氰基-5-(三氟甲基)吡啶-3-基)氨基甲酸叔丁酯(1.3g)的乙酸乙酯(50mL)溶液中加入雷尼镍(1.0g),混合物在一个大气压氢气下室温搅拌过夜。过滤,滤液减压浓缩,残留物用硅胶色谱柱纯化(二氯甲烷:甲醇=20:1)得到(2-(氨基甲基)-5-(三氟甲基)吡啶-3-基)氨基甲酸叔丁酯(0.3g)。
步骤5、6参考中间体1合成中的步骤5、6。
步骤7:6-三氟甲基咪唑并[1,5-a]-8-胺的合成
参考中间体12合成中的步骤5。
1H-NMR(400MHz,DMSO-d6):δ8.43(1H,s),8.30(1H,s),7.65(1H,s),6.35(2H,brs),5.87(1H,s)。
中间体16:8-氯咪唑并[5,1-a]异喹啉-5-胺的合成

步骤1、2、3、4、5、6、7、8、9、10分别参考中间3的合成步骤1、2、3、4、5、6、7、8、9、10得到标题化合物。
LC-MS[M+1]
+=218。
实施例1-49化合物的合成参考合成通式A得到各标题化合物,实施例50-63的合成参考合成通式B得到各标题化合物,如表1所示:
表1:实施例1-49









试验例1:生物活性测定
1.IDO和TDO蛋白诱导表达及纯化方法
首先,根据试剂盒说明书,PCR扩增IDO(源于HeLa细胞的RNA)和TDO(源于U87MG细胞的RNA)基因,扩增的PCR产物回收,然后将pET28a质粒和IDO(TDO)胶回收产物用NotI和EcoRI(对于TDO质粒构建用BamH1和Xhol)两种限制性内切酶进行酶切(37℃,酶切2h)跑胶并胶回收,将T4快速连接酶连接产物加入到大肠杆菌DH5α(E.coli DH5α)感受态细胞中,冰上放置30min,在42℃下热休克90s,在LB培养基中摇菌并涂板,挑取单克隆过夜摇菌,提取质粒酶切鉴定,再送测序鉴定,拿到阳性克隆,即His-IDO/pET28a和His-TDO/pET28a质粒构建成功。
将构建好的含His-IDO/pET28a或者His-TDO/pET28a质粒的300ml的大肠杆菌BL21菌在37℃下于LB培养基中摇菌至OD
600=0.6-0.8,加入终浓度为0.5mM的IPTG,在30℃下诱导3.5h,诱导后在6000rpm下离心45min收集菌体。
将收集的菌体用裂解液(40mM的Tris-HCl,pH 8.0;110mM的NaCl;2.2mM的KCl;10%的甘油;0.5%的Tween-20;20mM的咪唑;1mM的DTT)重新悬起,超声裂解(功率为30%,冰上裂解15min),裂解后的菌液在12000rpm、4℃下离心60min,将所得上清液用0.22μM的滤器过滤,准备好样品待用。将镍柱用裂解液平衡5个柱体积,然后将准备好的上清液样品上到镍柱上,上样之后,用洗脱液(40mM的Tris-HCl,pH 8.0;110mM的NaCl;2.2mM的KCl;10%的甘油;20-250mM的咪唑)梯度洗脱蛋白,将洗脱的蛋白溶液在4℃下过夜透析,透析之后浓缩分装,于-80℃保存备用。
2.IDO和TDO酶学活性测试
采用光吸收的方法建立了IDO和TDO的酶学活性检测平台,将化合物从10mM开始用100%DMSO进行10倍的梯度稀释(共5个浓度),每个浓度取4μL加入到96μL的反应缓冲液中(50mM K
2HPO
4-KH
2PO
4磷酸盐缓冲液,pH 6.5)混匀,作为4*化合物待用。使用反应缓冲液配制2*IDO酶,终浓度为75nM(TDO酶终浓度为300nM),4*底物(终浓度分别为20μM的L-(+)-Absorbate,购自Alfa Aesar,货号A15613;10μM的Methylene blue,购自Sigma,货号M9140-25G;0.2mg/ml的Catalase购自Sigma,货号C1345-1G和20μM的L-Trp,购自Sigma,货号T0254-25G(TDO为300μM L-Trp))待用。取10μL的4*化合物加入到384孔板(Assay plate,购买于Corning,货号3701),然后加入20μL的2*IDO酶(或者TDO酶),离心混匀,再加入10μL的4*底物混合物启动反应(总反应体积为40μL)。将384孔板放于孵育箱中23℃反应180分钟(TDO反应120分钟),然后每孔加入4μL的6M TCA于55℃下孵育30min,2500rpm下离心10min,每孔取40μL上清液,并加入2%4-二甲基氨基苯甲醛(购自TCI,货号D495,用冰乙酸溶解),5min后在FlexStation 3上读取光吸收值(480nM光吸收)。每个化合物分别在5个浓度下测定酶的活性,数据使用GraphtPad Prism5.0软件计算得到该化合物的IC
50值。
表2:实施例对IDO和TDO酶抑制活性的测定结果

试验例2:药代动力学数据
雄性SD大鼠来源于北京维通利华实验动物技术有限公司,将大鼠分组,每组3只,分别口服单次灌胃给予待测样品混悬液(5mg/kg)。动物在实验前禁食过夜,禁食时间从给药前10小时至给药后4小时。给药后0.25、0.5、1、2、4、6、8、和24小时采血。使用小动物麻醉机经异氟烷麻醉后通过眼底静脉丛采取0.3mL全血,放于肝素抗凝管中,样品于4℃、4000rpm离心5min,血浆转移至离心管中,并放于-80℃保存直到分析。血浆中样品使用蛋白质沉淀法萃取,萃取液通过LC/MS/MS分析。
表3:实施例化合物的药代动力学数据
| 实施例 | 51 | 40 | 52 |
| 剂量(mg/kg) | 5 | 5 | 5 |
| T 1/2(hr) | 3.28 | 4.31 | 3.56 |
| T max(hr) | 0.25 | 0.58 | 0.33 |
| C max(ng/mL) | 721 | 243 | 507 |
| AUC 0-inf(hr*ng/mL) | 1520 | 1018 | 2180 |
Claims (44)
- 式I的化合物或其药学上可接受的盐,
 其中,环A不存在或者环A为苯环或含有1、2或3个选自N、O或S原子的5-6元杂芳环;环B为五元芳香环,其中,W、V、X、Y和Z中的1个、2个、3个或4个选自N或NH,其它选自C或CH,具体的,i)Y和V是N,Z和W是CH,X是C;ii)Y、Z、V和W是N,X是C;iii)X和V是N,Z和W是CH,Y是C;iv)Y、Z和V是N,W是CH,X是C;v)X、Z和V是N,W是CH,Y是C;vi)X是N,Z、V和W是CH,Y是C;vii)Y是N,Z、V和W是CH,X是C;viii)Z是NH,V和W是CH,X和Y是C;ix)V是NH,Z和W是CH,X和Y是C;x)W是NH,V和Z是CH,X和Y是C;xi)V和W是N或NH,Z是CH,X和Y是C;xii)V和Z是N或NH,W是CH,X和Y是C;xiii)Z和W是N或NH,V是CH,X和Y是C;xiv)Y和W是N,V和Z是CH,X是C;xv)X和W是N,V和Z是CH,Y是C;xvi)X和Z是N或NH,V和W是CH,Y是C;xvii)Y和Z是N,V和W是CH,X是C;xviii)Y、V和W是N,Z是CH,X是C;xix)Z、V和W是N或NH,X和Y是C;xx)X、W和V是N,Z是CH,Y是C;xxi)Y、Z和W是N,V是CH,X是C;或xxii)X、Z、V和W是N,Y是C;P为C(R 6)或N;Q为O或S;D选自NH、O、S或CH 2;E选自NH、O或CH 2;L选自单键、S、SO、SO 2、CO、C(O)O、S(O)O或S(O) 2O;每个R 1独立地选自卤素、氨基、羟基、氰基、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基或C 3-6杂环烷基;R 2选自C 3-12环烷基、C 3-12杂环烷基、6-12元芳基或者含有1、2或3个选自N、O或S原子的5-12元杂芳基,其可任选地被一个或多个独立地选自R 3的基团取代;R 3选自卤素、氰基、硝基、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6环烷基CH 2-、C 3-6杂环烷基CH 2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH 2-、-OR 4、-SR 4、-N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-C(O)R 4、-S(O)R 4、-S(O)OR 4、-S(O)N(R 4) 2、-S(O) 2R 4、-S(O) 2OR 4、-S(O) 2N(R 4) 2、-OC(O)R 4、-OC(O)OR 4、-OC(O)N(R 4) 2、-N(R 4)C(O)R 4、-N(R 4)C(O)OR 4、-N(R 4)C(O)N(R 4) 2、-N(R 4)S(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2或-P(O)(OR 4) 2,其中C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6环烷基CH 2-、C 3-6杂环烷基CH 2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH 2-可任选地被一个或多个独立地选自R 5的基团取代;每个R 4独立地选自H、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、苯基或者含有1、2或 3个选自N、O或S原子的5-6元杂芳基;R 5选自卤素、氨基、氰基、羟基、-COOH、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基或C 3-6杂环烷基;R 6选自H、卤素、氨基、氰基、羟基、-COOH或卤代C 1-3烷基;m为0、1或2。
其中,环A不存在或者环A为苯环或含有1、2或3个选自N、O或S原子的5-6元杂芳环;环B为五元芳香环,其中,W、V、X、Y和Z中的1个、2个、3个或4个选自N或NH,其它选自C或CH,具体的,i)Y和V是N,Z和W是CH,X是C;ii)Y、Z、V和W是N,X是C;iii)X和V是N,Z和W是CH,Y是C;iv)Y、Z和V是N,W是CH,X是C;v)X、Z和V是N,W是CH,Y是C;vi)X是N,Z、V和W是CH,Y是C;vii)Y是N,Z、V和W是CH,X是C;viii)Z是NH,V和W是CH,X和Y是C;ix)V是NH,Z和W是CH,X和Y是C;x)W是NH,V和Z是CH,X和Y是C;xi)V和W是N或NH,Z是CH,X和Y是C;xii)V和Z是N或NH,W是CH,X和Y是C;xiii)Z和W是N或NH,V是CH,X和Y是C;xiv)Y和W是N,V和Z是CH,X是C;xv)X和W是N,V和Z是CH,Y是C;xvi)X和Z是N或NH,V和W是CH,Y是C;xvii)Y和Z是N,V和W是CH,X是C;xviii)Y、V和W是N,Z是CH,X是C;xix)Z、V和W是N或NH,X和Y是C;xx)X、W和V是N,Z是CH,Y是C;xxi)Y、Z和W是N,V是CH,X是C;或xxii)X、Z、V和W是N,Y是C;P为C(R 6)或N;Q为O或S;D选自NH、O、S或CH 2;E选自NH、O或CH 2;L选自单键、S、SO、SO 2、CO、C(O)O、S(O)O或S(O) 2O;每个R 1独立地选自卤素、氨基、羟基、氰基、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基或C 3-6杂环烷基;R 2选自C 3-12环烷基、C 3-12杂环烷基、6-12元芳基或者含有1、2或3个选自N、O或S原子的5-12元杂芳基,其可任选地被一个或多个独立地选自R 3的基团取代;R 3选自卤素、氰基、硝基、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6环烷基CH 2-、C 3-6杂环烷基CH 2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH 2-、-OR 4、-SR 4、-N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-C(O)R 4、-S(O)R 4、-S(O)OR 4、-S(O)N(R 4) 2、-S(O) 2R 4、-S(O) 2OR 4、-S(O) 2N(R 4) 2、-OC(O)R 4、-OC(O)OR 4、-OC(O)N(R 4) 2、-N(R 4)C(O)R 4、-N(R 4)C(O)OR 4、-N(R 4)C(O)N(R 4) 2、-N(R 4)S(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2或-P(O)(OR 4) 2,其中C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6环烷基CH 2-、C 3-6杂环烷基CH 2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH 2-可任选地被一个或多个独立地选自R 5的基团取代;每个R 4独立地选自H、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、苯基或者含有1、2或 3个选自N、O或S原子的5-6元杂芳基;R 5选自卤素、氨基、氰基、羟基、-COOH、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基或C 3-6杂环烷基;R 6选自H、卤素、氨基、氰基、羟基、-COOH或卤代C 1-3烷基;m为0、1或2。 - 权利要求1所述的化合物或其药学上可接受的盐,环A不存在或者环A为苯环、呋喃环、噻吩环、吡咯环、吡唑环、咪唑环、吡啶环、嘧啶环、哒嗪环、吡嗪环、噻唑环、异噻唑环、噁唑环、异噁唑环、四唑环或三嗪环;优选,环A不存在或者环A为苯环或吡啶环;进一步优选,环A不存在或者环A为


- 权利要求1或2所述的化合物或其药学上可接受的盐,每个R 1独立地选自卤素、卤代C 1-3烷基或C 1-6烷基;优选,每个R 1独立地选自氟、氯、溴、甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基或三氯甲基;进一步优选,每个R 1独立地选自氟、氯或三氟甲基。
- 权利要求1-3中任一项所述的化合物或其药学上可接受的盐,m为0或1。
- 权利要求1-4中任一项所述的化合物或其药学上可接受的盐,环B为芳香环,其中,i)Y和V是N,Z和W是CH,X是C;ii)Y、Z、V和W是N,X是C;iii)X和V是N,Z和W是CH,Y是C;iv)Y、Z和V是N,W是CH,X是C;v)X、Z和V是N,W是CH,Y是C。
- 权利要求1-5中任一项所述的化合物或其药学上可接受的盐,结构单元选自


- 权利要求1-6中任一项所述的化合物或其药学上可接受的盐,结构单元选自



- 权利要求1-7中任一项所述的化合物或其药学上可接受的盐,D选自NH、O或CH 2;E选自NH、O、或CH 2。
- 权利要求1-8中任一项所述的化合物或其药学上可接受的盐,-D-C(Q)-E-选自-NHC(O)NH-、-NHC(S)NH-、-OC(O)NH-、-NHC(O)CH 2-或-CH 2C(O)NH-。
- 权利要求1-9中任一项所述的化合物或其药学上可接受的盐,L选自单键或SO 2。
- 权利要求1-10中任一项所述的化合物或其药学上可接受的盐,R 2选自C 3-6环烷基、C 3-6杂环烷基、苯基或者含有1、2或3个选自N、O或S原子的5-6元杂芳基,其可任选地被一个或多个独立地选自R 3的基团取代;优选,R 2选自环丙基、环丁基、环戊基、环己基,任意位置失去一个氢原子的

 苯基、呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基,其可任选地被一个或多个独立地选自R 3的基团取代;进一步优选,R 2选自环己基、任意位置失去一个氢原子的
苯基、呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基,其可任选地被一个或多个独立地选自R 3的基团取代;进一步优选,R 2选自环己基、任意位置失去一个氢原子的 苯基、吡啶基或嘧啶基,其可任选地被一个或多个独立地选自R 3的基团取代;更进一步优选,R 2选自环己基、任意位置失去一个氢原子的
苯基、吡啶基或嘧啶基,其可任选地被一个或多个独立地选自R 3的基团取代;更进一步优选,R 2选自环己基、任意位置失去一个氢原子的 苯基、吡啶基或嘧啶基,其可任选地被一个或多个独立地选自R 3的基团取代;更进一步优选,R 2选自
苯基、吡啶基或嘧啶基,其可任选地被一个或多个独立地选自R 3的基团取代;更进一步优选,R 2选自



- 权利要求1-11中任一项所述的化合物或其药学上可接受的盐,R 3选自卤素、氰基、硝基、卤代C 1-3烷基、C 1-6烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6杂环烷基CH 2-、-OR 4、-SR 4、-S(O) 2R 4、-S(O) 2N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2或-P(O)(OR 4) 2,其中C 1-6烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基或C 3-6杂环烷基CH 2-可任选地被一个或多个独立地选自R 5的基团取代;优选,R 3选自氟、氯、溴、氰基、硝基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基、三氯甲基、-OR 4、-SR 4、-S(O) 2R 4、-S(O) 2N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2、-P(O)(OR 4) 2、甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基,任意位置失去一个H原子的

 呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、
呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、

 其中甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基,任意位置失去一个H原子的
其中甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基,任意位置失去一个H原子的

 呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、 哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、
呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、 哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、

 可任选地被一个或多个独立地选自R 5的基团取代;进一步优选,R 3选自氟、氯、溴、氰基、硝基、三氟甲基、-OR 4、-SR 4、-S(O) 2R 4、-S(O) 2N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2、-P(O)(OR 4) 2、甲基、任意位置失去一个氢原子的
可任选地被一个或多个独立地选自R 5的基团取代;进一步优选,R 3选自氟、氯、溴、氰基、硝基、三氟甲基、-OR 4、-SR 4、-S(O) 2R 4、-S(O) 2N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2、-P(O)(OR 4) 2、甲基、任意位置失去一个氢原子的 四唑基或
四唑基或 其中甲基、任意位置失去一个氢原子的
其中甲基、任意位置失去一个氢原子的 四唑基或
四唑基或 可任选地被一个或多个独立地选自R 5的基团取代;更进一步优选,R 3选自氟、氯、溴、氰基、硝基、三氟甲基、-OCF 3、-SCH 3、-S(O) 2CH 3、-S(O) 2NH 2、-C(O)OCH 3、-COOH、-C(O)NHCH 3、-C(O)NH 2、-NHS(O) 2NH 2、-P(O)(CH 3) 2、-P(O)(OCH 2CH 3) 2、-C(OH)(CF 3) 2、
可任选地被一个或多个独立地选自R 5的基团取代;更进一步优选,R 3选自氟、氯、溴、氰基、硝基、三氟甲基、-OCF 3、-SCH 3、-S(O) 2CH 3、-S(O) 2NH 2、-C(O)OCH 3、-COOH、-C(O)NHCH 3、-C(O)NH 2、-NHS(O) 2NH 2、-P(O)(CH 3) 2、-P(O)(OCH 2CH 3) 2、-C(OH)(CF 3) 2、

- 权利要求1-12中任一项所述的化合物或其药学上可接受的盐,每个R 4独立地选自H、卤代C 1-3烷基或C 1-6烷基;优选,每个R 4独立地选自H、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基、三氯甲基、甲基、乙基、丙基、异丙基、丁基、异丁基或叔丁基;进一步优选,每个R 4独立地选自H、三氟甲基、甲基或乙基。
- 权利要求1-13中任一项所述的化合物或其药学上可接受的盐,R 5选自羟基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基或三氯甲基;优选,R 5选自羟基或三氟甲基。
- 权利要求1-14中任一项所述的化合物或其药学上可接受的盐,R 6选自H或卤素,优选R 6选自H或氯。
- 式II的化合物或其药学上可接受的盐,
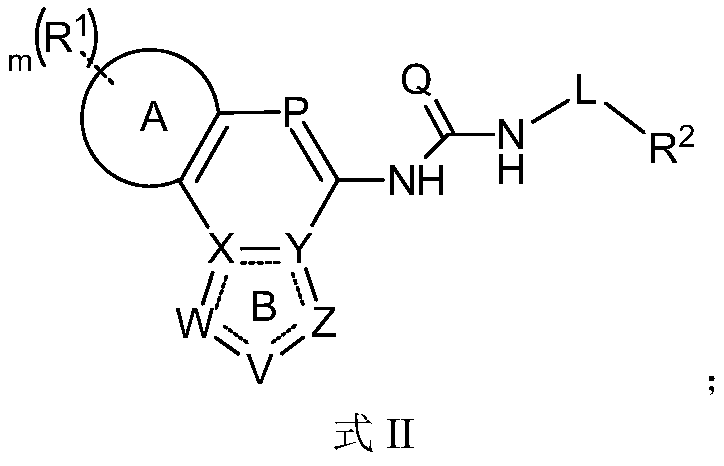 其中,环A为苯环或者含有1、2或3个选自N、O或S原子的5-6元杂芳环;环B为芳香环,其中,W、V、X、Y和Z中的1个、2个、3个或4个选自N或NH,其它选自C或CH,具体的,i)Y和V是N,Z和W是CH,X是C;ii)Y、Z、V和W是N,X是C;iii)X和V是N,Z和W是CH,Y是C;iv)Y、Z和V是N,W是CH,X是C;v)X、Z和V是N,W是CH,Y是C;vi)X是N,Z、V和W是CH,Y是C;vii)Y是N,Z、V和W是CH,X是C;viii)Z是NH,V和W是CH,X和Y是C;ix)V是NH,Z和W是CH,X和Y是C;x)W是NH,V和Z是CH,X和Y是C;xi)V和W是N或NH,Z是CH,X和Y是C;xii)V和Z是N或NH,W是CH,X和Y是C;xiii)Z和W是N或NH,V是CH,X和Y是C;xiv)Y和W是N,V和Z是CH,X是C;xv)X和W是N,V和Z是CH,Y是C;xvi)X和Z是N或NH,V和W是CH,Y是C;xvii)Y和Z是N,V和W是CH,X是C;xviii)Y、V和W是N,Z是CH,X是C;xix)Z、V和W是N或NH,X和Y是C;xx)X、W和V是N,Z是CH,Y是C;xxi)Y、Z和W是N,V是CH,X是C;或xxii)X、Z、V和W是N,Y是C;P为C(R 6)或N;Q为O或S;L选自单键、S、SO、SO 2、CO、C(O)O、S(O)O或S(O) 2O;每个R 1独立地选自卤素、氨基、羟基、氰基、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基或C 3-6杂环烷基;R 2选自C 3-12环烷基、C 3-12杂环烷基、6-12元芳基或者含有1、2或3个选自N、O或S原子的5-12元杂芳基,其可任选地被一个或多个独立地选自R 3的基团取代;R 3选自卤素、氰基、硝基、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6环烷基CH 2-、C 3-6杂环烷基CH 2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH 2-、-OR 4、-SR 4、-N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-C(O)R 4、-S(O)R 4、-S(O)OR 4、-S(O)N(R 4) 2、-S(O) 2R 4、-S(O) 2OR 4、-S(O) 2N(R 4) 2、-OC(O)R 4、-OC(O)OR 4、-OC(O)N(R 4) 2、-N(R 4)C(O)R 4、-N(R 4)C(O)OR 4、-N(R 4)C(O)N(R 4) 2、-N(R 4)S(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2或-P(O)(OR 4) 2,其中C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6环烷基CH 2-、C 3-6杂环烷基CH 2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH 2-可任选地被一个或多个独立地选自R 5的基团取代;每个R 4独立地选自H、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、苯基或者含有1、2或3个选自N、O或S原子的5-6元杂芳基;R 5选自卤素、氨基、氰基、羟基、-COOH、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基或C 3-6杂环烷基;R 6选自H、卤素、氨基、氰基、羟基、-COOH或卤代C 1-3烷基;m为0、1或2。
其中,环A为苯环或者含有1、2或3个选自N、O或S原子的5-6元杂芳环;环B为芳香环,其中,W、V、X、Y和Z中的1个、2个、3个或4个选自N或NH,其它选自C或CH,具体的,i)Y和V是N,Z和W是CH,X是C;ii)Y、Z、V和W是N,X是C;iii)X和V是N,Z和W是CH,Y是C;iv)Y、Z和V是N,W是CH,X是C;v)X、Z和V是N,W是CH,Y是C;vi)X是N,Z、V和W是CH,Y是C;vii)Y是N,Z、V和W是CH,X是C;viii)Z是NH,V和W是CH,X和Y是C;ix)V是NH,Z和W是CH,X和Y是C;x)W是NH,V和Z是CH,X和Y是C;xi)V和W是N或NH,Z是CH,X和Y是C;xii)V和Z是N或NH,W是CH,X和Y是C;xiii)Z和W是N或NH,V是CH,X和Y是C;xiv)Y和W是N,V和Z是CH,X是C;xv)X和W是N,V和Z是CH,Y是C;xvi)X和Z是N或NH,V和W是CH,Y是C;xvii)Y和Z是N,V和W是CH,X是C;xviii)Y、V和W是N,Z是CH,X是C;xix)Z、V和W是N或NH,X和Y是C;xx)X、W和V是N,Z是CH,Y是C;xxi)Y、Z和W是N,V是CH,X是C;或xxii)X、Z、V和W是N,Y是C;P为C(R 6)或N;Q为O或S;L选自单键、S、SO、SO 2、CO、C(O)O、S(O)O或S(O) 2O;每个R 1独立地选自卤素、氨基、羟基、氰基、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基或C 3-6杂环烷基;R 2选自C 3-12环烷基、C 3-12杂环烷基、6-12元芳基或者含有1、2或3个选自N、O或S原子的5-12元杂芳基,其可任选地被一个或多个独立地选自R 3的基团取代;R 3选自卤素、氰基、硝基、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6环烷基CH 2-、C 3-6杂环烷基CH 2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH 2-、-OR 4、-SR 4、-N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-C(O)R 4、-S(O)R 4、-S(O)OR 4、-S(O)N(R 4) 2、-S(O) 2R 4、-S(O) 2OR 4、-S(O) 2N(R 4) 2、-OC(O)R 4、-OC(O)OR 4、-OC(O)N(R 4) 2、-N(R 4)C(O)R 4、-N(R 4)C(O)OR 4、-N(R 4)C(O)N(R 4) 2、-N(R 4)S(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2或-P(O)(OR 4) 2,其中C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6环烷基CH 2-、C 3-6杂环烷基CH 2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH 2-可任选地被一个或多个独立地选自R 5的基团取代;每个R 4独立地选自H、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、苯基或者含有1、2或3个选自N、O或S原子的5-6元杂芳基;R 5选自卤素、氨基、氰基、羟基、-COOH、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基或C 3-6杂环烷基;R 6选自H、卤素、氨基、氰基、羟基、-COOH或卤代C 1-3烷基;m为0、1或2。 - 权利要求16所述的化合物或其药学上可接受的盐,环A为苯环、呋喃环、噻吩环、吡咯环、吡唑环、咪唑环、吡啶环、嘧啶环、哒嗪环、吡嗪环、噻唑环、异噻唑环、噁唑环、异噁唑环、四唑环或三嗪环;优选,环A为苯环或吡啶环;进一步优选,环A不存在或者环A为

- 权利要求16或17所述的化合物或其药学上可接受的盐,每个R 1独立地选自卤素、卤代C 1-3烷基或C 1-6烷基;优选,每个R 1独立地选自氟、氯、溴、甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基或三氯甲基;进一步优选,每个R 1独立地选自氟、氯或三氟甲基。
- 权利要求16-18中任一项所述的化合物或其药学上可接受的盐,m为0或1。
- 权利要求16-19中任一项所述的化合物或其药学上可接受的盐,环B为芳香环,其中,i)Y和V是N,Z和W是CH,X是C;ii)Y、Z、V和W是N,X是C;iii)X和V是N,Z和W是CH,Y是C;iv)Y、Z和V是N,W是CH,X是C;v)X、Z和V是N,W是CH,Y是C。
- 权利要求16-20中任一项所述的化合物或其药学上可接受的盐,结构单元选自


- 权利要求16-21中任一项所述的化合物或其药学上可接受的盐,结构单元选自


- 权利要求16-22中任一项所述的化合物或其药学上可接受的盐,L选自单键或SO 2。
- 权利要求16-23中任一项所述的化合物或其药学上可接受的盐,R 2选自C 3-6环烷基、C 3-6杂环烷基、苯基或者含有1、2或3个选自N、O或S原子的5-6元杂芳基,其可任选地被一个或多个独立地选自R 3的基团取代;优选,R 2选自环丙基、环丁基、环戊基、环己基,任意位置失去一个氢原子的

 苯基、呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基,其可任选地被一个或多个独立地选自R 3的基团取代;进一步优选,R 2选自环己基、任意 位置失去一个氢原子的
苯基、呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基,其可任选地被一个或多个独立地选自R 3的基团取代;进一步优选,R 2选自环己基、任意 位置失去一个氢原子的 苯基、吡啶基或嘧啶基,其可任选地被一个或多个独立地选自R 3的基团取代;更进一步优选,R 2选自
苯基、吡啶基或嘧啶基,其可任选地被一个或多个独立地选自R 3的基团取代;更进一步优选,R 2选自 其可任选地被一个或多个独立地选自R 3的基团取代;更进一步优选,R 2选自
其可任选地被一个或多个独立地选自R 3的基团取代;更进一步优选,R 2选自


- 权利要求16-24中任一项所述的化合物或其药学上可接受的盐,R 3选自卤素、氰基、硝基、卤代C 1-3烷基、C 1-6烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6杂环烷基CH 2-、-OR 4、-SR 4、-S(O) 2R 4、-S(O) 2N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2或-P(O)(OR 4) 2,其中C 1-6烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基或C 3-6杂环烷基CH 2-可任选地被一个或多个独立地选自R 5的基团取代;优选,R 3选自氟、氯、溴、氰基、硝基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基、三氯甲基、-OR 4、-SR 4、-S(O) 2R 4、-S(O) 2N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2、-P(O)(OR 4) 2、甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基,任意位置失去一个H原子的

 呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、 三嗪基、
呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、 三嗪基、

 其中甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基,任意位置失去一个H原子的
其中甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基,任意位置失去一个H原子的

 呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、
呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基、

 可任选地被一个或多个独立地选自R 5的基团取代;进一步优选,R 3选自氟、氯、溴、氰基、硝基、三氟甲基、-OR 4、-SR 4、-S(O) 2R 4、-S(O) 2N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2、-P(O)(OR 4) 2、甲基、任意位置失去一个氢原子的
可任选地被一个或多个独立地选自R 5的基团取代;进一步优选,R 3选自氟、氯、溴、氰基、硝基、三氟甲基、-OR 4、-SR 4、-S(O) 2R 4、-S(O) 2N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2、-P(O)(OR 4) 2、甲基、任意位置失去一个氢原子的 四唑基或
四唑基或 其中甲基、任意位置失去一个氢原子的
其中甲基、任意位置失去一个氢原子的 四唑基或
四唑基或 可任选地被一个或多个独立地选自R 5的基团取代;更进一步优选,R 3选自氟、氯、溴、氰基、硝基、三氟甲基、-OCF 3、-SCH 3、-S(O) 2CH 3、-S(O) 2NH 2、-C(O)OCH 3、-COOH、-C(O)NHCH 3、-C(O)NH 2、-NHS(O) 2NH 2、-P(O)(CH 3) 2、-P(O)(OCH 2CH 3) 2、-C(OH)(CF 3) 2、
可任选地被一个或多个独立地选自R 5的基团取代;更进一步优选,R 3选自氟、氯、溴、氰基、硝基、三氟甲基、-OCF 3、-SCH 3、-S(O) 2CH 3、-S(O) 2NH 2、-C(O)OCH 3、-COOH、-C(O)NHCH 3、-C(O)NH 2、-NHS(O) 2NH 2、-P(O)(CH 3) 2、-P(O)(OCH 2CH 3) 2、-C(OH)(CF 3) 2、

- 权利要求16-25中任一项所述的化合物或其药学上可接受的盐,每个R 4独立地选自H、卤代C 1-3烷基或C 1-6烷基;优选,每个R 4独立地选自H、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基、三氯甲基、甲基、乙基、丙基、异丙基、丁基、异丁基或叔丁基;进一步优选,每个R 4独立地选自H、三氟甲基、甲基或乙基。
- 权利要求16-26中任一项所述的化合物或其药学上可接受的盐,R 5选自羟基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基或三氯甲基;优选,R 5选自羟基或三氟甲基。
- 权利要求16-27中任一项所述的化合物或其药学上可接受的盐,R 6选自H或卤素;优选,R 6选自H或氯。
- 式III的化合物或其药学上可接受的盐,
 其中,环B为芳香环,其中,W、V、X、Y和Z中的1个、2个、3个或4个选自N或NH,其它选自C或CH,具体的,i)Y和V是N,Z和W是CH,X是C;ii)Y、Z、V和W是N,X是C;iii)X和V是N,Z和W是CH,Y是C;iv)Y、Z和V是N,W是CH,X是C;v)X、Z和V是N,W是CH,Y是C;vi)X是N,Z、V和W是CH,Y是C;vii)Y是N,Z、V和W是CH,X是C;viii)Z是NH,V和W是CH,X和Y是C;ix)V是NH,Z和W是CH,X和Y是C;x)W是NH,V和Z是CH,X和Y是C;xi)V和W是N或NH,Z是CH,X和Y是C;xii)V和Z是N或NH,W是CH,X和Y是C;xiii)Z和W是N或NH,V是CH,X和Y是C;xiv)Y和W是N,V和Z是CH,X是C;xv)X和W是N,V和Z是CH,Y是C;xvi)X和Z是N或NH,V和W是CH,Y是C;xvii)Y和Z是N,V和W是CH,X是C;xviii)Y、V和W是N,Z是CH,X是C;xix)Z、V和W是N或NH,X和Y是C;xx)X、W和V是N,Z是CH,Y是C;xxi)Y、Z和W是N,V是CH,X是C;或xxii)X、Z、V和W是N,Y是C;Q为O或S;D选自NH、O、S或CH 2;E选自NH、O或CH 2;L选自单键、S、SO、SO 2、CO、C(O)O、S(O)O或S(O) 2O;每个R 1独立地选自卤素、氨基、羟基、氰基、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基或C 3-6杂环烷基;R 2选自C 3-12环烷基、C 3-12杂环烷基、6-12元芳基或含有1、2或3个选自N、O或S原子的5-12元杂芳基,其可任选地被一个或多个独立地选自R 3的基团取代;R 3选自卤素、氰基、硝基、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6环烷基CH 2-、C 3-6杂环烷基CH 2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH 2-、-OR 4、-SR 4、-N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-C(O)R 4、-S(O)R 4、-S(O)OR 4、-S(O)N(R 4) 2、-S(O) 2R 4、-S(O) 2OR 4、-S(O) 2N(R 4) 2、-OC(O)R 4、-OC(O)OR 4、-OC(O)N(R 4) 2、-N(R 4)C(O)R 4、 -N(R 4)C(O)OR 4、-N(R 4)C(O)N(R 4) 2、-N(R 4)S(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2或-P(O)(OR 4) 2,其中C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6环烷基CH 2-、C 3-6杂环烷基CH 2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH 2-可任选地被一个或多个独立地选自R 5的基团取代;每个R 4独立地选自H、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、苯基或者含有1、2或3个选自N、O或S原子的5-6元杂芳基;R 5选自卤素、氨基、氰基、羟基、-COOH、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基或C 3-6杂环烷基;m为0、1或2。
其中,环B为芳香环,其中,W、V、X、Y和Z中的1个、2个、3个或4个选自N或NH,其它选自C或CH,具体的,i)Y和V是N,Z和W是CH,X是C;ii)Y、Z、V和W是N,X是C;iii)X和V是N,Z和W是CH,Y是C;iv)Y、Z和V是N,W是CH,X是C;v)X、Z和V是N,W是CH,Y是C;vi)X是N,Z、V和W是CH,Y是C;vii)Y是N,Z、V和W是CH,X是C;viii)Z是NH,V和W是CH,X和Y是C;ix)V是NH,Z和W是CH,X和Y是C;x)W是NH,V和Z是CH,X和Y是C;xi)V和W是N或NH,Z是CH,X和Y是C;xii)V和Z是N或NH,W是CH,X和Y是C;xiii)Z和W是N或NH,V是CH,X和Y是C;xiv)Y和W是N,V和Z是CH,X是C;xv)X和W是N,V和Z是CH,Y是C;xvi)X和Z是N或NH,V和W是CH,Y是C;xvii)Y和Z是N,V和W是CH,X是C;xviii)Y、V和W是N,Z是CH,X是C;xix)Z、V和W是N或NH,X和Y是C;xx)X、W和V是N,Z是CH,Y是C;xxi)Y、Z和W是N,V是CH,X是C;或xxii)X、Z、V和W是N,Y是C;Q为O或S;D选自NH、O、S或CH 2;E选自NH、O或CH 2;L选自单键、S、SO、SO 2、CO、C(O)O、S(O)O或S(O) 2O;每个R 1独立地选自卤素、氨基、羟基、氰基、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基或C 3-6杂环烷基;R 2选自C 3-12环烷基、C 3-12杂环烷基、6-12元芳基或含有1、2或3个选自N、O或S原子的5-12元杂芳基,其可任选地被一个或多个独立地选自R 3的基团取代;R 3选自卤素、氰基、硝基、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6环烷基CH 2-、C 3-6杂环烷基CH 2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH 2-、-OR 4、-SR 4、-N(R 4) 2、-C(O)OR 4、-C(O)N(R 4) 2、-C(O)R 4、-S(O)R 4、-S(O)OR 4、-S(O)N(R 4) 2、-S(O) 2R 4、-S(O) 2OR 4、-S(O) 2N(R 4) 2、-OC(O)R 4、-OC(O)OR 4、-OC(O)N(R 4) 2、-N(R 4)C(O)R 4、 -N(R 4)C(O)OR 4、-N(R 4)C(O)N(R 4) 2、-N(R 4)S(O)N(R 4) 2、-N(R 4)S(O) 2N(R 4) 2、-P(O)(R 4) 2或-P(O)(OR 4) 2,其中C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、含有1、2或3个选自N、O或S原子的5-6元杂芳基、C 3-6环烷基CH 2-、C 3-6杂环烷基CH 2-、含有1、2或3个选自N、O或S原子的5-6元杂芳基CH 2-可任选地被一个或多个独立地选自R 5的基团取代;每个R 4独立地选自H、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基、C 3-6杂环烷基、苯基或者含有1、2或3个选自N、O或S原子的5-6元杂芳基;R 5选自卤素、氨基、氰基、羟基、-COOH、卤代C 1-3烷基、C 1-6烷基、C 3-6环烷基或C 3-6杂环烷基;m为0、1或2。 - 权利要求29所述的化合物或其药学上可接受的盐,每个R 1独立地选自卤素、卤代C 1-3烷基或C 1-6烷基;优选,每个R 1独立地选自氟、氯、溴、甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基或三氯甲基;进一步优选,每个R 1独立地选自氯或三氟甲基。
- 权利要求29或30所述的化合物或其药学上可接受的盐,m为0或1。
- 权利要求29-31中任一项所述的化合物或其药学上可接受的盐,环B为芳香环,其中,i)Y和V是N,Z和W是CH,X是C;ii)X和V是N,Z和W是CH,Y是C。
- 权利要求29-32中任一项所述的化合物或其药学上可接受的盐,结构单元选自



- 权利要求29-33中任一项所述的化合物或其药学上可接受的盐,结构单元选自


- 权利要求29-34中任一项所述的化合物或其药学上可接受的盐,D选自NH、O或CH 2;E选自NH、O或CH 2。
- 权利要求29-35中任一项所述的化合物或其药学上可接受的盐,-D-C(Q)-E-选自-NHC(O)NH-、-NHC(S)NH-、-OC(O)NH-、-NHC(O)CH 2-或-CH 2C(O)NH-。
- 权利要求29-36中任一项所述的化合物或其药学上可接受的盐,L选自单键。
- 权利要求29-37中任一项所述的化合物或其药学上可接受的盐,R 2选自苯基或者含有1、2或3个选自N、O或S原子的5-6元杂芳基,其可任选地被一个或多个独立地选自R 3的基团取代;优选,R 2选自苯基、呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基、三嗪基,其可任选地被一个或多个独立地选自R 3的基团取代;进一步优选,R 2选自苯基、吡啶基或嘧啶基,其可任选地被一个或多个独立地选自R 3的基团取代;更进一步优选,R 2选自其可任选地被一个或多个独立地选自R 3的基团取代;更进一步优选,R 2选自



- 权利要求29-38中任一项所述的化合物或其药学上可接受的盐,R 3选自卤素或卤代C 1-3烷基;优选,R 3选自氟、氯、溴或三氟甲基。
- 权利要求1-39中任一项所述的化合物或其药学上可接受的盐,其中,所述化合物优选为:




- 药物组合物,其包含治疗有效量的式I、式II或式III化合物或其药学上可接受的盐和一种或多种药学上可接受的载体或赋形剂。
- 权利要求1-40中任一项所述的化合物或其药学上可接受的盐治疗由吲哚胺2,3-双加氧酶(IDO)和色氨酸2,3-加氧酶(TDO)介导的免疫抑制疾病的方法,包括对需要该治疗的哺乳动物、优选人类给予治疗有效量的式I、式II或式III化合物或其药学上可接受的盐、或权利要求41所述的药物组合物。
- 权利要求1-40中任一项所述的化合物或其药学上可接受的盐或权利要求41所述的药物组合物在制备预防或者治疗由吲哚胺2,3-双加氧酶(IDO)和色氨酸2,3-加氧酶(TDO)介导的免疫抑制疾病的药物中的用途。
- 权利要求43所述的用途,所述免疫抑制疾病与传染性疾病或癌症相关,所述传染性疾病选自下列病毒感染:流感、丙型肝炎病毒(HCV)、人乳头状瘤病毒(HPV)、巨细胞病毒(CMV)、脊髓灰质炎病毒、带状疱疹病毒、人类免疫缺陷病毒(HIV)、爱泼斯坦-巴尔二氏病毒(EBV)或柯萨奇病毒;所述癌症选自结肠癌、胰腺癌、乳腺癌、前列腺癌、肺癌、脑癌、卵巢癌、子宫颈癌、睾丸癌、肾癌、头或颈癌、淋巴瘤、白血病或黑素瘤。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201880015782.2A CN110382500B (zh) | 2017-03-21 | 2018-03-21 | 用于ido和tdo双重抑制剂的脲类化合物 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710168579.6 | 2017-03-21 | ||
| CN201710168579 | 2017-03-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018171602A1 true WO2018171602A1 (zh) | 2018-09-27 |
Family
ID=63585911
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2018/079718 Ceased WO2018171602A1 (zh) | 2017-03-21 | 2018-03-21 | 用于ido和tdo双重抑制剂的脲类化合物 |
Country Status (2)
| Country | Link |
|---|---|
| CN (2) | CN110382500B (zh) |
| WO (1) | WO2018171602A1 (zh) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019138107A1 (en) | 2018-01-15 | 2019-07-18 | Idorsia Pharmaceuticals Ltd | Inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan 2,3-dioxygenase |
| EP3505517A4 (en) * | 2016-08-23 | 2020-01-15 | Beijing InnoCare Pharma Tech Co., Ltd. | Fused heterocyclic derivative, preparation method therefor and medical use thereof |
| WO2021005222A1 (en) | 2019-07-11 | 2021-01-14 | Idorsia Pharmaceuticals Ltd | Inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan 2,3-dioxygenase |
| WO2022178437A1 (en) * | 2021-02-22 | 2022-08-25 | Regents Of The University Of Minnesota | Immunomodulators and immunomodulator conjugates |
| WO2023102800A1 (en) * | 2021-12-09 | 2023-06-15 | Theravance Biopharma R&D Ip, Llc | Synthesis of 5, 7-dichloro-1, 6-naphthyridine |
| JP2024072283A (ja) * | 2022-11-15 | 2024-05-27 | イーライ リリー アンド カンパニー | Ahrアゴニスト |
| WO2024193464A1 (zh) * | 2023-03-17 | 2024-09-26 | 西藏海思科制药有限公司 | 一种含氮三并环衍生物及其在医药上的应用 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115572262B (zh) * | 2022-10-27 | 2024-08-27 | 厦门沃克沃德医药科技有限公司 | 一种异喹啉衍生物及其制备方法 |
| CN119528827A (zh) * | 2023-08-31 | 2025-02-28 | 北京五和博澳药业股份有限公司 | 一种酞嗪酮类化合物及其制备方法、药物组合物及用途 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105884780A (zh) * | 2015-02-16 | 2016-08-24 | 上海迪诺医药科技有限公司 | 多环化合物、其药物组合物及应用 |
| CN106478634A (zh) * | 2015-09-01 | 2017-03-08 | 上海璎黎药业有限公司 | 稠合咪唑化合物、其制备方法、药物组合物和用途 |
| CN107033087A (zh) * | 2016-02-04 | 2017-08-11 | 西华大学 | 1h-吲唑-4-胺类化合物及其作为ido抑制剂的用途 |
| WO2018036414A1 (zh) * | 2016-08-23 | 2018-03-01 | 北京诺诚健华医药科技有限公司 | 稠杂环类衍生物、其制备方法及其在医学上的应用 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0322016D0 (en) * | 2003-09-19 | 2003-10-22 | Merck Sharp & Dohme | New compounds |
-
2018
- 2018-03-21 CN CN201880015782.2A patent/CN110382500B/zh active Active
- 2018-03-21 CN CN202110936214.XA patent/CN113651820A/zh not_active Withdrawn
- 2018-03-21 WO PCT/CN2018/079718 patent/WO2018171602A1/zh not_active Ceased
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105884780A (zh) * | 2015-02-16 | 2016-08-24 | 上海迪诺医药科技有限公司 | 多环化合物、其药物组合物及应用 |
| CN106478634A (zh) * | 2015-09-01 | 2017-03-08 | 上海璎黎药业有限公司 | 稠合咪唑化合物、其制备方法、药物组合物和用途 |
| CN107033087A (zh) * | 2016-02-04 | 2017-08-11 | 西华大学 | 1h-吲唑-4-胺类化合物及其作为ido抑制剂的用途 |
| WO2018036414A1 (zh) * | 2016-08-23 | 2018-03-01 | 北京诺诚健华医药科技有限公司 | 稠杂环类衍生物、其制备方法及其在医学上的应用 |
Non-Patent Citations (1)
| Title |
|---|
| PAWAR C.D.: "Synthesis and Antimicrobial Evaluation of Novel Substituted Acetamido-4-subtituted-thiazole-5-indazole Derivatives", RESEARCH & REVIEWS: JOURNAL OF CHEMISTRY, vol. 5, 2 September 2016 (2016-09-02), pages 3, XP055540615, ISSN: 2319-9849 * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3505517A4 (en) * | 2016-08-23 | 2020-01-15 | Beijing InnoCare Pharma Tech Co., Ltd. | Fused heterocyclic derivative, preparation method therefor and medical use thereof |
| US11046682B2 (en) | 2016-08-23 | 2021-06-29 | Beijing Innocare Pharma Tech Co., Ltd. | Fused heterocyclic derivatives, their preparation methods thereof and medical uses thereof |
| AU2017315572B2 (en) * | 2016-08-23 | 2021-07-15 | Beijing Innocare Pharma Tech Co., Ltd. | Fused heterocyclic derivative, preparation method therefor and medical use thereof |
| WO2019138107A1 (en) | 2018-01-15 | 2019-07-18 | Idorsia Pharmaceuticals Ltd | Inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan 2,3-dioxygenase |
| WO2021005222A1 (en) | 2019-07-11 | 2021-01-14 | Idorsia Pharmaceuticals Ltd | Inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan 2,3-dioxygenase |
| WO2022178437A1 (en) * | 2021-02-22 | 2022-08-25 | Regents Of The University Of Minnesota | Immunomodulators and immunomodulator conjugates |
| WO2023102800A1 (en) * | 2021-12-09 | 2023-06-15 | Theravance Biopharma R&D Ip, Llc | Synthesis of 5, 7-dichloro-1, 6-naphthyridine |
| JP2024072283A (ja) * | 2022-11-15 | 2024-05-27 | イーライ リリー アンド カンパニー | Ahrアゴニスト |
| JP7577819B2 (ja) | 2022-11-15 | 2024-11-05 | イーライ リリー アンド カンパニー | Ahrアゴニスト |
| WO2024193464A1 (zh) * | 2023-03-17 | 2024-09-26 | 西藏海思科制药有限公司 | 一种含氮三并环衍生物及其在医药上的应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN110382500B (zh) | 2021-08-10 |
| CN113651820A (zh) | 2021-11-16 |
| CN110382500A (zh) | 2019-10-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN110382500B (zh) | 用于ido和tdo双重抑制剂的脲类化合物 | |
| AU2020203967B2 (en) | Amide-substituted heterocyclic compounds useful as modulators of il-12, il-23 and/or ifn alpha responses | |
| CA2948587C (en) | Pyrazolopyridines and pyrazolopyrimidines | |
| EP1987030B1 (en) | 3 -deazapurine derivatives as tlr7 modulators | |
| CA3142902A1 (en) | 2,3-dihydroquinazolin compounds as nav1.8 inhibitors | |
| US8557830B2 (en) | RAF kinase modulators and methods of use | |
| AP1164A (en) | Substituted 6,6-hetero-bicyclic derivatives. | |
| KR102318401B1 (ko) | 신규한 이소인돌린 유도체, 이의 약학 조성물 및 용도 | |
| JP2022502476A (ja) | インドールahr阻害剤およびその使用 | |
| KR20150092279A (ko) | 암의 치료를 위한 신규한 2-고리 페닐-피리딘/피라진 | |
| EP4331585A2 (en) | Inhibitors of ret | |
| US10112928B2 (en) | Inhibitors of SYK | |
| KR20250016234A (ko) | 삼환 화합물 및 이의 용도 | |
| CN103502239B (zh) | 可用作脾酪氨酸激酶(syk)抑制剂的吡啶基-和吡嗪基-甲基氧基-芳基衍生物 | |
| AU2023345563A1 (en) | Pcsk9 inhibitors and methods of use thereof | |
| US20240109915A1 (en) | Novel acc inhibitors | |
| AU2022233180B2 (en) | Heteroaryl compounds as inhibitors of rip2 kinase, composition and application thereof | |
| EP4167982A1 (en) | Methods and compounds for restoring mutant p53 function | |
| HK40105122A (zh) | Ret的抑制剂 | |
| EP4568965A1 (en) | Heteroaryl compounds for the treatment of cancer | |
| CN120699000A (zh) | 一种杂双环类化合物及其制备方法与应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18772261 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 18772261 Country of ref document: EP Kind code of ref document: A1 |