WO2018042305A1 - Improved processes for preparation of bilastine using novel intermediates - Google Patents
Improved processes for preparation of bilastine using novel intermediates Download PDFInfo
- Publication number
- WO2018042305A1 WO2018042305A1 PCT/IB2017/055146 IB2017055146W WO2018042305A1 WO 2018042305 A1 WO2018042305 A1 WO 2018042305A1 IB 2017055146 W IB2017055146 W IB 2017055146W WO 2018042305 A1 WO2018042305 A1 WO 2018042305A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- chloride
- methyl
- potassium
- formula
- sodium
- Prior art date
Links
- ACCMWZWAEFYUGZ-UHFFFAOYSA-N bilastine Chemical compound N=1C2=CC=CC=C2N(CCOCC)C=1C(CC1)CCN1CCC1=CC=C(C(C)(C)C(O)=O)C=C1 ACCMWZWAEFYUGZ-UHFFFAOYSA-N 0.000 title claims abstract description 68
- 229960004314 bilastine Drugs 0.000 title claims abstract description 66
- 238000000034 method Methods 0.000 title claims abstract description 58
- 238000002360 preparation method Methods 0.000 title claims abstract description 32
- 239000000543 intermediate Substances 0.000 title abstract description 11
- 150000003839 salts Chemical class 0.000 claims abstract description 75
- 239000002904 solvent Substances 0.000 claims description 65
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 54
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 45
- 238000006243 chemical reaction Methods 0.000 claims description 44
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 37
- 150000001875 compounds Chemical class 0.000 claims description 32
- 239000000203 mixture Substances 0.000 claims description 32
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 26
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 24
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 24
- 239000002253 acid Substances 0.000 claims description 22
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 claims description 21
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 21
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 claims description 20
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 18
- 239000002585 base Substances 0.000 claims description 18
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 15
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 claims description 14
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 claims description 14
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 claims description 14
- -1 2-[4-(2-chloroethyl)phenyl]-2-methyl- propanoic acid compound Chemical class 0.000 claims description 12
- NFSUTBPTKCWLLP-UHFFFAOYSA-N 2-[4-(2-chloroethyl)phenyl]-2-methylpropanoic acid Chemical compound OC(=O)C(C)(C)C1=CC=C(CCCl)C=C1 NFSUTBPTKCWLLP-UHFFFAOYSA-N 0.000 claims description 12
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 12
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 claims description 12
- 239000003444 phase transfer catalyst Substances 0.000 claims description 12
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 12
- 239000002841 Lewis acid Substances 0.000 claims description 11
- DKPFZGUDAPQIHT-UHFFFAOYSA-N butyl acetate Chemical compound CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 claims description 11
- 239000003153 chemical reaction reagent Substances 0.000 claims description 11
- 150000007517 lewis acids Chemical class 0.000 claims description 11
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 claims description 10
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 9
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 claims description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims description 8
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 claims description 8
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 8
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims description 8
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 claims description 8
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 claims description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 8
- HTZCNXWZYVXIMZ-UHFFFAOYSA-M benzyl(triethyl)azanium;chloride Chemical compound [Cl-].CC[N+](CC)(CC)CC1=CC=CC=C1 HTZCNXWZYVXIMZ-UHFFFAOYSA-M 0.000 claims description 8
- 150000007529 inorganic bases Chemical class 0.000 claims description 8
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 claims description 8
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 claims description 8
- NHGXDBSUJJNIRV-UHFFFAOYSA-M tetrabutylammonium chloride Chemical compound [Cl-].CCCC[N+](CCCC)(CCCC)CCCC NHGXDBSUJJNIRV-UHFFFAOYSA-M 0.000 claims description 8
- 239000008096 xylene Substances 0.000 claims description 8
- TXUICONDJPYNPY-UHFFFAOYSA-N (1,10,13-trimethyl-3-oxo-4,5,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl) heptanoate Chemical compound C1CC2CC(=O)C=C(C)C2(C)C2C1C1CCC(OC(=O)CCCCCC)C1(C)CC2 TXUICONDJPYNPY-UHFFFAOYSA-N 0.000 claims description 7
- 229910021578 Iron(III) chloride Inorganic materials 0.000 claims description 7
- 229910021626 Tin(II) chloride Inorganic materials 0.000 claims description 7
- 229910021623 Tin(IV) bromide Inorganic materials 0.000 claims description 7
- 229910021627 Tin(IV) chloride Inorganic materials 0.000 claims description 7
- 239000003638 chemical reducing agent Substances 0.000 claims description 7
- RBTARNINKXHZNM-UHFFFAOYSA-K iron trichloride Chemical compound Cl[Fe](Cl)Cl RBTARNINKXHZNM-UHFFFAOYSA-K 0.000 claims description 7
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 7
- 239000001119 stannous chloride Substances 0.000 claims description 7
- 235000011150 stannous chloride Nutrition 0.000 claims description 7
- PUGUQINMNYINPK-UHFFFAOYSA-N tert-butyl 4-(2-chloroacetyl)piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCN(C(=O)CCl)CC1 PUGUQINMNYINPK-UHFFFAOYSA-N 0.000 claims description 7
- LTSUHJWLSNQKIP-UHFFFAOYSA-J tin(iv) bromide Chemical compound Br[Sn](Br)(Br)Br LTSUHJWLSNQKIP-UHFFFAOYSA-J 0.000 claims description 7
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 claims description 7
- 239000011592 zinc chloride Substances 0.000 claims description 7
- 235000005074 zinc chloride Nutrition 0.000 claims description 7
- PEQBOAIOIXHJTL-UHFFFAOYSA-N 2-[4-(2-chloroacetyl)phenyl]-2-methylpropanoic acid Chemical compound ClCC(=O)C1=CC=C(C=C1)C(C(=O)O)(C)C PEQBOAIOIXHJTL-UHFFFAOYSA-N 0.000 claims description 6
- YYEROYLAYAVZNW-UHFFFAOYSA-N 2-methyl-2-phenylpropanoic acid Chemical compound OC(=O)C(C)(C)C1=CC=CC=C1 YYEROYLAYAVZNW-UHFFFAOYSA-N 0.000 claims description 6
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 claims description 6
- WTEOIRVLGSZEPR-UHFFFAOYSA-N boron trifluoride Chemical compound FB(F)F WTEOIRVLGSZEPR-UHFFFAOYSA-N 0.000 claims description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 6
- 235000011181 potassium carbonates Nutrition 0.000 claims description 6
- YKYONYBAUNKHLG-UHFFFAOYSA-N propyl acetate Chemical compound CCCOC(C)=O YKYONYBAUNKHLG-UHFFFAOYSA-N 0.000 claims description 6
- VGCXGMAHQTYDJK-UHFFFAOYSA-N Chloroacetyl chloride Chemical compound ClCC(Cl)=O VGCXGMAHQTYDJK-UHFFFAOYSA-N 0.000 claims description 5
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 claims description 5
- ZFFBIQMNKOJDJE-UHFFFAOYSA-N 2-bromo-1,2-diphenylethanone Chemical compound C=1C=CC=CC=1C(Br)C(=O)C1=CC=CC=C1 ZFFBIQMNKOJDJE-UHFFFAOYSA-N 0.000 claims description 4
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 claims description 4
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 claims description 4
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 claims description 4
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 claims description 4
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 claims description 4
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 claims description 4
- 229910021529 ammonia Inorganic materials 0.000 claims description 4
- 229960000510 ammonia Drugs 0.000 claims description 4
- KXHPPCXNWTUNSB-UHFFFAOYSA-M benzyl(trimethyl)azanium;chloride Chemical compound [Cl-].C[N+](C)(C)CC1=CC=CC=C1 KXHPPCXNWTUNSB-UHFFFAOYSA-M 0.000 claims description 4
- ZMCUDHNSHCRDBT-UHFFFAOYSA-M caesium bicarbonate Chemical compound [Cs+].OC([O-])=O ZMCUDHNSHCRDBT-UHFFFAOYSA-M 0.000 claims description 4
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 4
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 4
- VSGNNIFQASZAOI-UHFFFAOYSA-L calcium acetate Chemical compound [Ca+2].CC([O-])=O.CC([O-])=O VSGNNIFQASZAOI-UHFFFAOYSA-L 0.000 claims description 4
- 239000001639 calcium acetate Substances 0.000 claims description 4
- 235000011092 calcium acetate Nutrition 0.000 claims description 4
- 229960005147 calcium acetate Drugs 0.000 claims description 4
- 150000003983 crown ethers Chemical class 0.000 claims description 4
- OIKHZBFJHONJJB-UHFFFAOYSA-N dimethyl(phenyl)silicon Chemical compound C[Si](C)C1=CC=CC=C1 OIKHZBFJHONJJB-UHFFFAOYSA-N 0.000 claims description 4
- AFRJJFRNGGLMDW-UHFFFAOYSA-N lithium amide Chemical compound [Li+].[NH2-] AFRJJFRNGGLMDW-UHFFFAOYSA-N 0.000 claims description 4
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 claims description 4
- 229910052808 lithium carbonate Inorganic materials 0.000 claims description 4
- HQRPHMAXFVUBJX-UHFFFAOYSA-M lithium;hydrogen carbonate Chemical compound [Li+].OC([O-])=O HQRPHMAXFVUBJX-UHFFFAOYSA-M 0.000 claims description 4
- UEGPKNKPLBYCNK-UHFFFAOYSA-L magnesium acetate Chemical compound [Mg+2].CC([O-])=O.CC([O-])=O UEGPKNKPLBYCNK-UHFFFAOYSA-L 0.000 claims description 4
- 239000011654 magnesium acetate Substances 0.000 claims description 4
- 235000011285 magnesium acetate Nutrition 0.000 claims description 4
- 229940069446 magnesium acetate Drugs 0.000 claims description 4
- 229940098779 methanesulfonic acid Drugs 0.000 claims description 4
- PARWUHTVGZSQPD-UHFFFAOYSA-N phenylsilane Chemical compound [SiH3]C1=CC=CC=C1 PARWUHTVGZSQPD-UHFFFAOYSA-N 0.000 claims description 4
- 235000011056 potassium acetate Nutrition 0.000 claims description 4
- 229960004109 potassium acetate Drugs 0.000 claims description 4
- 235000015497 potassium bicarbonate Nutrition 0.000 claims description 4
- 229910000028 potassium bicarbonate Inorganic materials 0.000 claims description 4
- 239000011736 potassium bicarbonate Substances 0.000 claims description 4
- RPDAUEIUDPHABB-UHFFFAOYSA-N potassium ethoxide Chemical compound [K+].CC[O-] RPDAUEIUDPHABB-UHFFFAOYSA-N 0.000 claims description 4
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 claims description 4
- BDAWXSQJJCIFIK-UHFFFAOYSA-N potassium methoxide Chemical compound [K+].[O-]C BDAWXSQJJCIFIK-UHFFFAOYSA-N 0.000 claims description 4
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 claims description 4
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 claims description 4
- 239000001632 sodium acetate Substances 0.000 claims description 4
- 235000017281 sodium acetate Nutrition 0.000 claims description 4
- 229960004249 sodium acetate Drugs 0.000 claims description 4
- ODZPKZBBUMBTMG-UHFFFAOYSA-N sodium amide Chemical compound [NH2-].[Na+] ODZPKZBBUMBTMG-UHFFFAOYSA-N 0.000 claims description 4
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 claims description 4
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 claims description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 4
- IPILPUZVTYHGIL-UHFFFAOYSA-M tributyl(methyl)azanium;chloride Chemical compound [Cl-].CCCC[N+](C)(CCCC)CCCC IPILPUZVTYHGIL-UHFFFAOYSA-M 0.000 claims description 4
- ZDHXKXAHOVTTAH-UHFFFAOYSA-N trichlorosilane Chemical compound Cl[SiH](Cl)Cl ZDHXKXAHOVTTAH-UHFFFAOYSA-N 0.000 claims description 4
- 239000005052 trichlorosilane Substances 0.000 claims description 4
- PQDJYEQOELDLCP-UHFFFAOYSA-N trimethylsilane Chemical compound C[SiH](C)C PQDJYEQOELDLCP-UHFFFAOYSA-N 0.000 claims description 4
- AKQNYQDSIDKVJZ-UHFFFAOYSA-N triphenylsilane Chemical compound C1=CC=CC=C1[SiH](C=1C=CC=CC=1)C1=CC=CC=C1 AKQNYQDSIDKVJZ-UHFFFAOYSA-N 0.000 claims description 4
- 229910015900 BF3 Inorganic materials 0.000 claims description 3
- 150000004677 hydrates Chemical class 0.000 claims description 3
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 claims description 3
- 229940011051 isopropyl acetate Drugs 0.000 claims description 3
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 claims description 3
- 239000012453 solvate Substances 0.000 claims description 3
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 claims description 3
- JYZIHLWOWKMNNX-UHFFFAOYSA-N benzimidazole Chemical compound C1=C[CH]C2=NC=NC2=C1 JYZIHLWOWKMNNX-UHFFFAOYSA-N 0.000 claims 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 21
- 239000010410 layer Substances 0.000 description 18
- 238000010992 reflux Methods 0.000 description 17
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 14
- 229910052799 carbon Inorganic materials 0.000 description 10
- 238000003756 stirring Methods 0.000 description 10
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 8
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 7
- 238000010626 work up procedure Methods 0.000 description 7
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 238000001035 drying Methods 0.000 description 5
- 238000001704 evaporation Methods 0.000 description 5
- 230000008020 evaporation Effects 0.000 description 5
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 5
- 238000002955 isolation Methods 0.000 description 5
- 230000035484 reaction time Effects 0.000 description 5
- HBOGHPAOOWUTLB-UHFFFAOYSA-N 2-piperidin-4-yl-1h-benzimidazole Chemical compound C1CNCCC1C1=NC2=CC=CC=C2N1 HBOGHPAOOWUTLB-UHFFFAOYSA-N 0.000 description 4
- 239000004215 Carbon black (E152) Substances 0.000 description 4
- 208000024780 Urticaria Diseases 0.000 description 4
- 238000004821 distillation Methods 0.000 description 4
- KZTGKKWHYBIJER-UHFFFAOYSA-N ethyl 4-(1h-benzimidazol-2-yl)piperidine-1-carboxylate Chemical compound C1CN(C(=O)OCC)CCC1C1=NC2=CC=CC=C2N1 KZTGKKWHYBIJER-UHFFFAOYSA-N 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 229930195733 hydrocarbon Natural products 0.000 description 4
- 150000002430 hydrocarbons Chemical class 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 238000001953 recrystallisation Methods 0.000 description 4
- GPTVQTPMFOLLOA-UHFFFAOYSA-N 1-chloro-2-ethoxyethane Chemical compound CCOCCCl GPTVQTPMFOLLOA-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 206010010744 Conjunctivitis allergic Diseases 0.000 description 3
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 3
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 3
- 201000010435 allergic urticaria Diseases 0.000 description 3
- 239000012296 anti-solvent Substances 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- 238000004042 decolorization Methods 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- 239000012153 distilled water Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- 150000007530 organic bases Chemical class 0.000 description 3
- 238000010979 pH adjustment Methods 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 3
- 238000010791 quenching Methods 0.000 description 3
- 230000000171 quenching effect Effects 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000012312 sodium hydride Substances 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 229910000102 alkali metal hydride Inorganic materials 0.000 description 2
- 150000008046 alkali metal hydrides Chemical class 0.000 description 2
- 239000000739 antihistaminic agent Substances 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 239000003610 charcoal Substances 0.000 description 2
- 150000008280 chlorinated hydrocarbons Chemical class 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- JQVDAXLFBXTEQA-UHFFFAOYSA-N dibutylamine Chemical compound CCCCNCCCC JQVDAXLFBXTEQA-UHFFFAOYSA-N 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 239000002360 explosive Substances 0.000 description 2
- 150000008282 halocarbons Chemical class 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 239000013557 residual solvent Substances 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 238000002636 symptomatic treatment Methods 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 0 *C(c(cc1)ccc1C(CN)=O)(N)O Chemical compound *C(c(cc1)ccc1C(CN)=O)(N)O 0.000 description 1
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 1
- YBJXRWANRTYCJE-UHFFFAOYSA-N 1-(2-ethoxyethyl)-2-piperidin-4-ylbenzimidazole Chemical compound N=1C2=CC=CC=C2N(CCOCC)C=1C1CCNCC1 YBJXRWANRTYCJE-UHFFFAOYSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- BEQDKWKSUMQVMX-UHFFFAOYSA-N 2,4-dimethyl-4,5-dihydro-1,3-oxazole Chemical compound CC1COC(C)=N1 BEQDKWKSUMQVMX-UHFFFAOYSA-N 0.000 description 1
- NJBCRXCAPCODGX-UHFFFAOYSA-N 2-methyl-n-(2-methylpropyl)propan-1-amine Chemical compound CC(C)CNCC(C)C NJBCRXCAPCODGX-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 125000003504 2-oxazolinyl group Chemical group O1C(=NCC1)* 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical group N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 230000003266 anti-allergic effect Effects 0.000 description 1
- 230000001387 anti-histamine Effects 0.000 description 1
- 229940058303 antinematodal benzimidazole derivative Drugs 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 150000001556 benzimidazoles Chemical class 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 210000000748 cardiovascular system Anatomy 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- SRJOCJYGOFTFLH-UHFFFAOYSA-N isonipecotic acid Chemical compound OC(=O)C1CCNCC1 SRJOCJYGOFTFLH-UHFFFAOYSA-N 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000003880 polar aprotic solvent Substances 0.000 description 1
- 229920000137 polyphosphoric acid Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 229910021653 sulphate ion Inorganic materials 0.000 description 1
- FIAFUQMPZJWCLV-UHFFFAOYSA-N suramin Chemical compound OS(=O)(=O)C1=CC(S(O)(=O)=O)=C2C(NC(=O)C3=CC=C(C(=C3)NC(=O)C=3C=C(NC(=O)NC=4C=C(C=CC=4)C(=O)NC=4C(=CC=C(C=4)C(=O)NC=4C5=C(C=C(C=C5C(=CC=4)S(O)(=O)=O)S(O)(=O)=O)S(O)(=O)=O)C)C=CC=3)C)=CC=C(S(O)(=O)=O)C2=C1 FIAFUQMPZJWCLV-UHFFFAOYSA-N 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- YBRBMKDOPFTVDT-UHFFFAOYSA-N tert-butylamine Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/347—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups
- C07C51/373—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups by introduction of functional groups containing oxygen only in doubly bound form
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/347—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups
- C07C51/377—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups by splitting-off hydrogen or functional groups; by hydrogenolysis of functional groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C57/00—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms
- C07C57/52—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms containing halogen
- C07C57/58—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms containing halogen containing six-membered aromatic rings
Definitions
- the present invention relates to improved, commercially viable and industrially advantageous processes for the preparation of Bilastine or a pharmaceutically acceptable salt thereof using novel intermediates, in high yield and purity.
- U.S. Patent No. 5,877,187 discloses a variety of benzimidazole derivatives, processes for their preparation, pharmaceutical compositions comprising the derivatives, and methods of use thereof. These compounds have high Hi antihistaminic and antiallergic activity and are devoid of effects on the central nervous and cardiovascular systems.
- Bilastine chemically named 2- [4-[2-[4-[l-(2-ethoxyethyl)-benzimidazol-2-yl]piperidin-l-yl]ethyl]phenyl]-2-methyl propanoic acid, is a selective histamine Hi receptor antagonist used for treatment of allergic rhinoconjunctivitis and urticaria (hives).
- Bilastine is represented by the following structural formula I:
- Bilastine a novel second-generation Hi-antihistamine, is approved for the symptomatic treatment of allergic rhinoconjunctivitis and urticaria in adults and children over 12 years of age.
- Bilastine has a favourable pharmacokinetic profile, being rapidly absorbed resulting in an onset of clinical effect within one hour of administration, and has a long duration of action, exceeding 24 hours, which allows for once-daily dosing.
- Bilastine was developed by FAES Farma and approved in the European Union for the symptomatic treatment of allergic rhinoconjunctivitis and urticaria. Bilastine is marketed under the trade names Bilaxten ® (in Spain, Colombia, Australia, and several other countries), Ilaxten ® (in United Kingdom), and BlextenTM(in Canada).
- Bilastine According to the synthetic route described in the US' 187 patent, Bilastine is prepared by the following main reaction steps: a) 2-(4-(l-(4,4-dimethyl-A -oxazoline-2- yl)-l-methylethyl)phenyl)ethylp-toluenesulphonate is reacted with 2-(4-piperidinyl)-lH- benzimidazole in the presence of sodium carbonate to produce 2-[l-(2-(4-(l-(4,4-dimethyl- ⁇ -oxazoline-2-yl)-l-methylethyl)phenyl)ethyl)piperidine-4-yl]-lH-benzimidazole; b) the resulting dimethyl-oxazoline intermediate is reacted with 2-chloroethyl ethylether in dimethylformamide in the presence of sodium hydride at a temperature of 80°C, followed by tedious work- up and isolation
- the object of the present invention is to provide novel, commercially viable and industrially advantageous processes for the preparation of Bilastine and its intermediates in high yields and purity.
- Bilastine or a pharmaceutically acceptable salt thereof can be prepared, in high purity and with high yield, by reacting 2-methyl-2- phenyl-propanoic acid with an acylating agent in the presence of a suitable Lewis acid to produce 2- [4-(2-chloroacetyl)phenyl]-2-methyl- propanoic acid, followed by reduction with a suitable reducing agent in the presence of a Lewis acid to produce 2-[4-(2- chloroethyl)phenyl]-2-methyl-propanoic acid, which is then condensed with l-(2- ethoxyethyl)-2-(piperidin-4-yl)-benzimidazole in the presence of a suitable base to produce Bilastine or a pharmaceutically acceptable salt thereof.
- the overall process involves a reduced number of process steps, shorter reaction times and less expensive reagents, thereby making the process cost effective;
- the process avoids the use of the explosive and difficult to handle reagents like Sodium hydride;
- the process avoids the use of tedious and cumbersome procedures like prolonged reaction time periods, multiple process steps, column chromatographic purifications and additional purifications or isolations.
- step-(a) reducing the compound of formula III obtained in step-(a) with a hydrosilane reagent in the presence of an acid to produce 2-[4-(2-chloroethyl)phenyl]-2-methyl-propanoic acid compound of formula IIIA:
- the solvent used for isolating, purifying and/or recrystallizing the compounds of formula I, III and IIIA obtained by the processes described in the present invention is selected from the group consisting of water, an alcohol, a ketone, an ether, an ester, a hydrocarbon, a halogenated hydrocarbon, and mixtures thereof.
- the solvent is selected from the group consisting of water, methanol, ethanol, 1-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl- tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, butyl acetate, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
- 'base' as used herein includes, but is not limited to, organic bases and inorganic bases such as carbonates, bicarbonates, hydroxides, alkoxides, acetates and amides of alkali or alkali earth metals.
- the inorganic base is selected from the group consisting of sodium carbonate, potassium carbonate, lithium carbonate, cesium carbonate, sodium bicarbonate, potassium bicarbonate, lithium bicarbonate, cesium bicarbonate, sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium methoxide, sodium ethoxide, potassium methoxide, potassium ethoxide, sodium tertbutoxide, potassium tert.butoxide, sodium amide, potassium amide, lithium amide, ammonia, sodium acetate, potassium acetate, magnesium acetate, calcium acetate, and mixtures thereof.
- the organic base is selected from the group consisting of dimethylamine, diethylamine, diisopropyl amine, diisopropylethylamine, di n-butylamine, diisobutylamine, triethylamine, tributylamine, tert-butyl amine, pyridine, 4- dimethylaminopyridine (DMAP), and mixtures thereof.
- phase transfer catalysts' as used herein include, but are not limited to, tetrabutylammonium bromide, tetrabutylammonium chloride, tetrabutylammonium iodide, benzyltrimethyl ammonium chloride, benzyltriethyl ammonium chloride, methyltributyl ammonium chloride, crown ethers and the like.
- salts may include acid addition salts and base addition salts.
- Acid addition salts may be derived from organic and inorganic acids.
- Exemplary acid addition salts include, but are not limited to, hydrochloride, hydrobromide, sulphate, nitrate, phosphate, acetate, propionate, oxalate, succinate, maleate, fumarate, benzenesulfonate, toluenesulfonate, citrate, tartrate, and the like.
- a most specific acid addition salt is hydrochloride salt.
- Base addition salts may be derived from an organic or an inorganic base.
- the base addition salts are derived from alkali or alkaline earth metals such as sodium, calcium, potassium and magnesium, ammonium salt and the like.
- the highly pure Bilastine or a pharmaceutically acceptable salt thereof obtained by the process disclosed herein has a purity of greater than about 99.5%, specifically greater than about 99.8%, more specifically greater than about 99.9% as measured by HPLC.
- the purity of the highly pure Bilastine or a pharmaceutically acceptable salt thereof obtained by the processes disclosed herein is about 99.5% to about 99.99% as measured by HPLC.
- reflux temperature means the temperature at which the solvent or solvent system refluxes or boils at atmospheric pressure.
- room temperature refers to a temperature of about 20°C to about 35°C.
- room temperature can refer to a temperature of about 25°C to about 30°C.
- Exemplary Lewis acids used in step-(a) include, but are not limited to, aluminum chloride, aluminum bromide, boron trifluoride, boron tribromide, boron trichloride, tin tetrachloride, tin tetrabromide, stannous chloride, ferric chloride, zinc chloride, titanium tetrachloride, and hydrates or solvates thereof.
- a most specific Lewis acid used in step-(a) is aluminum chloride.
- step-(a) is carried out in a suitable solvent.
- suitable solvents used in step-(a) include, but are not limited to, a halogenated hydrocarbon, a ketone, an ether, an ester, a hydrocarbon, and mixtures thereof.
- the solvent used in step-(a) is selected from the group consisting of dichloromethane, dichloroethane, chloroform, acetone, methyl ethyl ketone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-propyl acetate, isopropyl acetate, n-butyl acetate, cyclohexane, toluene, xylene, and mixtures thereof.
- a most specific solvent is dichloromethane.
- the reaction in step-(a) is carried out at a temperature of about -10°C to about 50°C, and more specifically at a temperature of about -5°C to about 35°C.
- the reaction time may vary between about 30 minutes to about 5 hours, and specifically about 1 hour to about 3 hours.
- the reaction mass containing the compound of formula III or a salt thereof obtained in step-(a) may be subjected to usual work up methods such as a washing, a quenching, an extraction, a pH adjustment, an evaporation, a layer separation, decolorization, a carbon treatment, or a combination thereof.
- the reaction mass may be used directly in the next step to produce the compound of formula IIIA, or the compound of formula III or a salt thereof may be isolated and/or recrystallized and then used in the next step.
- the carbon treatment is carried out by methods known in the art, for example, by stirring the reaction mass/solution with finely powdered carbon at a temperature of about 40°C to the reflux temperature for at least 5 minutes, specifically at the reflux temperature; and filtering the resulting mixture through charcoal bed to obtain a filtrate containing compound by removing charcoal.
- finely powdered carbon is a special carbon or an active carbon.
- the compound of formula III or a salt thereof may be isolated and/or re-crystallized from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
- the solvent used for work up, isolation and/or recrystallization of the compound of formula III obtained by the process described herein is selected from the group as described hereinabove.
- the hydrosilane reducing agent used in step-(b) is selected from the group consisting of triethylsilane, trimethylsilane, dimethyl phenyl silane, phenyl silane, triphenylsilane, trichloro silane, and the like; and a most specific reducing agent is triethylsilane.
- the acid used in step-(b) is selected from the group consisting of boron trifluoride diethyl etherate, titanium tetrachloride, aluminum chloride, aluminum bromide, boron tribromide, tin tetrachloride, tin tetrabromide, stannous chloride, ferric chloride, zinc chloride, trifluoro acetic acid and methanesulfonic acid.
- a most specific acid used is titanium tetrachloride.
- Exemplary solvents used in step-(b) include, but are not limited to, a hydrocarbon solvent, a chlorinated hydrocarbon solvent, and mixtures thereof.
- the solvent used in step-(b) is selected from the group consisting of toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof; and a most specific solvent is dichloromethane.
- the reaction in step-(b) is carried out at a temperature of about -10°C to 50°C; and specifically at a temperature of about 10°C to about 40°C.
- the reaction time may vary between about 2 hours to 8 hours, and more specifically about 4 hours to 6 hours.
- the reaction mass containing the compound of formula IIIA or a salt thereof obtained in step-(b) may be subjected to usual work up methods such as a washing, a quenching, an extraction, a pH adjustment, an evaporation, a layer separation, decolorization, a carbon treatment, or a combination thereof.
- the reaction mass may be used directly in the next step to produce the compound of formula I, or the compound of formula IIIA or a salt thereof may be isolated and/or recrystallized and then used in the next step.
- the compound of formula IIIA or a salt thereof may be isolated and/or re-crystallized from a suitable solvent by conventional methods as described hereinabove.
- the solvent used for work up, isolation and/or recrystallization of the compound of formula IIIA obtained by the process described herein is selected from the group as described hereinabove.
- the base used in step-(c) is an organic base or an inorganic base selected from the group as described hereinabove.
- the base used in step- (c) is an inorganic base.
- a most specific base used in step-(c) is sodium carbonate or potassium carbonate.
- reaction in step-(c) is carried out in the presence of a phase transfer catalyst.
- the phase transfer catalyst can be selected from the group as described hereinabove.
- Exemplary solvents used in step-(c) include, but are not limited, water, acetone, methyl ethyl ketone, methyl isobutyl ketone, acetonitrile, propionitrile and mixtures thereof.
- a most specific solvent used in step-(c) is water.
- the reaction in step-(c) is carried out at a temperature of about 10°C to the reflux temperature of the solvent used, specifically at a temperature of about 30°C to the reflux temperature of the solvent used, and more specifically at the reflux temperature of the solvent used.
- the reaction time may vary from about 15 hours to about 25 hours.
- the reaction mass containing the Bilastine of formula I or a salt thereof obtained in step-(c) may be subjected to usual work up methods such as a washing, a quenching, an extraction, a pH adjustment, an evaporation, a layer separation, decolorization, a carbon treatment, or a combination thereof.
- the Bilastine of formula I or a salt thereof may be isolated, purified and/or re-crystallized from a suitable solvent by conventional methods as described hereinabove.
- the solvent used for work up, isolation, recrystallization and/or purification of the Bilastine of formula I or a salt thereof obtained by the process described herein is selected from the group as described hereinabove.
- the crude Bilastine obtained in step-(c) is, optionally subjected to carbon treatment or silica gel treatment.
- the carbon treatment or silica gel treatment is carried out by methods known in the art, for example, as per the methods described hereinabove.
- the solvent used for purification of Bilastine obtained in step- (c) is selected from the group consisting of water, acetone, methanol, ethanol, isopropyl alcohol, ethyl acetate, butyl acetate, and mixtures thereof.
- anti- solvent refers to a solvent which when added to an existing solution of a substance reduces the solubility of the substance.
- exemplary anti-solvents include, but are not limited to, water, an alcohol, a ketone, a chlorinated hydrocarbon, a hydrocarbon, an ester, a nitrile, an ether, a polar aprotic solvent, and mixtures thereof.
- Removal of solvent is accomplished, for example, by substantially complete evaporation of the solvent, concentrating the solution or distillation of solvent, under inert atmosphere to obtain highly pure Bilastine or a salt thereof.
- step-(a) reducing the compound of formula III obtained in step-(a) with a hydrosilane reagent in the presence of an acid to produce 2-[4-(2-chloroethyl)phenyl]-2-methyl-propanoic acid of formula IIIA or a salt thereof.
- the preparation of the Bilastine of formula I or a pharmaceutically acceptable salt thereof as described in the above process steps-(a) and (b) can be carried out by using the suitable solvents, reagents, methods, parameters and conditions as described hereinabove.
- Bilastine of formula I or a pharmaceutically acceptable salt thereof in the presence of a base, optionally in the presence of a phase transfer catalyst, in a suitable solvent to produce Bilastine of formula I or a salt thereof, and optionally purifying the Bilastine obtained with a suitable solvent to produce highly pure Bilastine or a pharmaceutically acceptable salt thereof.
- the preparation of the Bilastine of formula I or a pharmaceutically acceptable salt thereof can be carried out by using the suitable solvents, reagents, methods, parameters and conditions as described hereinabove.
- the highly pure Bilastine or a salt thereof obtained by the above processes may be further dried in, for example, a Vacuum Tray Dryer, a Rotocon Vacuum Dryer, a Vacuum Paddle Dryer or a pilot plant Rota vapor, to further lower residual solvents. Drying can be carried out under reduced pressure until the residual solvent content reduces to the desired amount such as an amount that is within the limits given by the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (“ICH”) guidelines.
- ICH International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use
- the drying is carried out at atmospheric pressure or reduced pressures, such as below about 200 mm Hg, or below about 50 mm Hg, at temperatures such as about 35°C to about 90°C, and specifically at about 75°C to about 85°C.
- the drying can be carried out for any desired time period that achieves the desired result, such as times about 1 to 20 hours.
- Drying may also be carried out for shorter or longer periods of time depending on the product specifications. Temperatures and pressures will be chosen based on the volatility of the solvent being used and the foregoing should be considered as only a general guidance. Drying can be suitably carried out in a tray dryer, vacuum oven, air oven, or using a fluidized bed drier, spin flash dryer, flash dryer, and the like.
- Dichloro methane 700 ml was added to 2-[4-(2-chloroacetyl)phenyl]-2-methyl-propanoic acid (35 g) and the mixture was cooled to 0-5°C, followed by slow addition of titanium tetrachloride (140 g) at the same temperature. The temperature of the resulting mass was raised to 20-25°C, followed by the addition of triethylsilane (64.4 g) and then stirring the reaction mixture at 25-30°C for 4 hours. The reaction mass was cooled to below 10°C and then water (980 ml) was added at the same temperature. The organic layer was separated and the aqueous layer was extracted with dichloromethane (500 ml x 2).
- the resulting organic layers were combined, followed by removal of the solvent completely by distillation under vacuum to produce a crude compound.
- Aqueous NaOH solution was added to the resulting crude compound while adjusting the pH to 9-10, and then washed with toluene (75 ml x 2).
- the layers were separated, followed adjusting the pH of the aqueous layer to 1-2 with dilute hydrochloric acid at 10-15°C.
- the resulting acidic aqueous layer was extracted thrice with ethyl acetate (100 ml x 3).
- the combined organic layers were washed with water (100 ml), and then distilled-off the solvent completely under vacuum to produce 31 g of 2-[4-(2-chloroethyl)phenyl]-2-methyl-propanoic acid.
- Orthophenylenediamine (20 g), polyphosphoric acid (120 g) and isonipecotic acid (26.5 g) were taken into a reaction flask and the resulting mixture was heated to 115-120°C, followed by stirring for 20 hours at the same temperature. After completion of the reaction, the reaction mass was cooled to 90°C, quenched with distilled water (260 ml) and then cooled to room temperature (25-30°C). The resulting mass was further cooled to 10-15°C, followed by adjusting the pH of the reaction mass to 9-10 with dilute sodium hydroxide solution, and then stirring for 30 minutes at 10-15°C. The separated solid was filtered and washed with distilled water. The wet material was dried at 40-45°C.
- reaction mass was cooled to room temperature, followed by the addition of water (210 ml) and then stirring for 10 minutes at room temperature.
- the resulting mass was neutralized with dilute hydrochloric acid.
- the layers were separated and the aqueous layer was extracted twice with ethyl acetate (200 ml x 2).
- the toluene layer and ethyl acetate layers were combined and washed with distilled water (250 ml).
- the solvents were distilled off completely under reduced pressure to give 51 g of ethyl 4-[l-(2-ethoxyethyl)- benzimidazol-2-yl] -piperidine- 1 -carboxylate.
- reaction mass was cooled to 25-30°C, filtered the material and then washed with acetone (30 ml) to give 32.5 g of pure l-(2-ethoxyethyl)-2- (piperidin-4-yl)-benzimidazole.
- the aqueous layer was separated and then neutralized with acetic acid, followed by extracting thrice with dichloromethane (800 ml x 3).
- Activated carbon (12 g) was added to the resulting organic layers and then stirred for 10 minutes.
- the resulting mixture was filtered through hyflo-bed and then washed the bed with dichloro methane (50 ml).
- the resulting filtrate was distilled-off under vacuum to remove the solvent completely.
- Acetone (110 ml) was added to the resulting crude compound and then stirred for 10-15 minutes at room temperature.
- the solvent was distilled-off completely from the resulting mass, acetone (85 ml) was added again and then stirred for 1 hour at room temperature.
- the separated solid was filtered and washed with acetone (30 ml) to produce 19 g of crude Bilastine.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Provided herein are improved, commercially viable and industrially advantageous processes for the preparation of Bilastine or a pharmaceutically acceptable salt thereof using novel intermediates, in high yield and purity.
Description
IMPROVED PROCESSES FOR PREPARATION OF BILASTINE USING NOVEL
INTERMEDIATES
CROSS REFERENCE TO RELATED APPLICATION
This patent application claims the benefit of priority to Indian Provisional Patent
Application No. 201641029306, filed on August 29, 2016, which is incorporated herein by reference in its entirety.
FIELD OF THE INVENTION
The present invention relates to improved, commercially viable and industrially advantageous processes for the preparation of Bilastine or a pharmaceutically acceptable salt thereof using novel intermediates, in high yield and purity.
BACKGROUND OF THE INVENTION
U.S. Patent No. 5,877,187 (hereinafter referred to as the US '187 patent) discloses a variety of benzimidazole derivatives, processes for their preparation, pharmaceutical compositions comprising the derivatives, and methods of use thereof. These compounds have high Hi antihistaminic and antiallergic activity and are devoid of effects on the central nervous and cardiovascular systems. Among them, Bilastine, chemically named 2- [4-[2-[4-[l-(2-ethoxyethyl)-benzimidazol-2-yl]piperidin-l-yl]ethyl]phenyl]-2-methyl propanoic acid, is a selective histamine Hi receptor antagonist used for treatment of allergic rhinoconjunctivitis and urticaria (hives). Bilastine is represented by the following structural formula I:

(I) Bilastine, a novel second-generation Hi-antihistamine, is approved for the symptomatic treatment of allergic rhinoconjunctivitis and urticaria in adults and children over 12 years of age. Bilastine has a favourable pharmacokinetic profile, being rapidly absorbed resulting in an onset of clinical effect within one hour of administration, and has a long duration of action, exceeding 24 hours, which allows for once-daily dosing.
Bilastine was developed by FAES Farma and approved in the European Union for the symptomatic treatment of allergic rhinoconjunctivitis and urticaria. Bilastine is marketed under the trade names Bilaxten® (in Spain, Colombia, Australia, and several other countries), Ilaxten® (in United Kingdom), and Blexten™(in Canada).
Various processes for the preparation of Bilastine, its intermediates, and related compounds are described in U.S. Patent Nos. US 5,877,187 and US 8,367,704; PCT Publication Nos. WO 2014/188453, WO2014/026657; Chinese Patent Application Publication No. CN 102675101; and Journal Articles: Syn. Comm., 41(9), 1394-1402, 2011; and Drugs of future 35(2), 98-105, 2010.
The synthesis of Bilastine was first described in the US' 187 patent. According to the US' 187 patent, Bilastine is prepared by a process as depicted in scheme 1:
Scheme- 1:
Sodium carbonate
Dimethylformamide
ethyl ethyl ether
ydride
formamide

2-[l-(2-(4-(l-(4,4-Dimethyl-delta2-oxazoline-2-yl)- l-methylethyl)phenyl)ethyl)piperidine-4-yl]-lH- benzimidazole

-oxazoline-2-vl)-l-methvlethvl)phenvl)ethvl)piperid

Bilastine
According to the synthetic route described in the US' 187 patent, Bilastine is prepared by the following main reaction steps: a) 2-(4-(l-(4,4-dimethyl-A -oxazoline-2- yl)-l-methylethyl)phenyl)ethylp-toluenesulphonate is reacted with 2-(4-piperidinyl)-lH- benzimidazole in the presence of sodium carbonate to produce 2-[l-(2-(4-(l-(4,4-dimethyl- Δ -oxazoline-2-yl)-l-methylethyl)phenyl)ethyl)piperidine-4-yl]-lH-benzimidazole; b) the resulting dimethyl-oxazoline intermediate is reacted with 2-chloroethyl ethylether in dimethylformamide in the presence of sodium hydride at a temperature of 80°C, followed by tedious work- up and isolation methods to produce the l-(2-ethoxyethyl)-2-l-(2-(4-(l-
(4,4-dimethyl-A -oxazoline-2-yl)- l-methylethyl)phenyl)ethyl)piperidine-4-yl- 1H- benzimidazole; and c) the resulting 2-ethoxyethyl compound is reacted with 3N
Hydrochloric acid to produce 2-4-(2-(4-(l-(2-ethoxyethyl)benzimidazole-2-yl)piperidine- l-yl)ethyl)phenyl-2-methylpropanoic acid (Bilastine).
The process for the preparation of Bilastine as described in the aforementioned prior art suffers from the following major disadvantages and shortcomings: (a) the introduction of the oxazoline group and its subsequent hydrolysis inevitably comprised in the process leads to the formation of several by-products, thereby resulting in a poor product yields and quality and making the whole process lengthy and cumbersome; b) the reaction between 2-[l-(2-(4-(l-(4,4-dimethyl-A -oxazoline-2-yl)-l-methylethyl)phenyl) ethyl)piperidine-4-yl]-lH-benzimidazole and 2-chloroethyl ethylether is performed under very stringent reaction condition and involves the use of dangerous and explosive alkali metal hydrides such as sodium hydride; c) use of alkali metal hydrides is not advisable for commercial scale operations from safety point of view.
A need remains for novel, commercially viable and environmentally friendly processes for the preparation of Bilastine and its intermediates with high yields and purity, to resolve the problems associated with the processes described in the prior art, and that will be suitable for large-scale preparation.
SUMMARY OF THE INVENTION
The object of the present invention is to provide novel, commercially viable and industrially advantageous processes for the preparation of Bilastine and its intermediates in high yields and purity.
The present inventors have found that Bilastine or a pharmaceutically acceptable salt thereof can be prepared, in high purity and with high yield, by reacting 2-methyl-2- phenyl-propanoic acid with an acylating agent in the presence of a suitable Lewis acid to
produce 2- [4-(2-chloroacetyl)phenyl]-2-methyl- propanoic acid, followed by reduction with a suitable reducing agent in the presence of a Lewis acid to produce 2-[4-(2- chloroethyl)phenyl]-2-methyl-propanoic acid, which is then condensed with l-(2- ethoxyethyl)-2-(piperidin-4-yl)-benzimidazole in the presence of a suitable base to produce Bilastine or a pharmaceutically acceptable salt thereof.
In another aspect, provided herein is a novel intermediate compound, 2-[4-(2- chloroethyl)phenyl]-2-methyl-propanoic acid, of formula IIIA:

In another aspect, provided also herein is a process for the preparation of the novel intermediate compound, 2-[4-(2-chloroethyl)phenyl]-2-methyl-propanoic acid, of formula
IIIA.
The novel process for the preparation of Bilastine disclosed in the present invention is represented by a schematic diagram as depicted in scheme-2:
Scheme 2:

(I)
Bilastine
The process for the preparation of Bilastine described herein has the following advantages over the processes described in the prior art:
i) the process involves the use of novel intermediate compounds;
ii) the overall process involves a reduced number of process steps, shorter reaction times and less expensive reagents, thereby making the process cost effective;
iii) the process avoids the use of the explosive and difficult to handle reagents like Sodium hydride;
iv) the process avoids the use of tedious and cumbersome procedures like prolonged reaction time periods, multiple process steps, column chromatographic purifications and additional purifications or isolations.
DETAILED DESCRIPTION OF THE INVENTION
According to one aspect, there is provided a novel and industrially advantageous process for the preparation of highly pure Bilastine of formula I:

a) reacting 2-methyl-2-phenyl-propanoic acid of formula II:

or a salt thereof, with chloroacetyl chloride of formula VI:
 optionally in the presence of a Lewis acid, to produce 2-[4-(2-chloroacetyl)phenyl]-2- methyl-propanoic acid of formula III:
optionally in the presence of a Lewis acid, to produce 2-[4-(2-chloroacetyl)phenyl]-2- methyl-propanoic acid of formula III:
in
 or a salt thereof;
or a salt thereof;
b) reducing the compound of formula III obtained in step-(a) with a hydrosilane reagent in the presence of an acid to produce 2-[4-(2-chloroethyl)phenyl]-2-methyl-propanoic acid compound of formula IIIA:

c) condensing the compound of formula IIIA obtained in step-(b) with l-(2-ethoxyethyl)- 2-(piperidin-4-yl)benzimidazole of formula IV:

Unless otherwise specified, the solvent used for isolating, purifying and/or recrystallizing the compounds of formula I, III and IIIA obtained by the processes described in the present invention is selected from the group consisting of water, an alcohol, a ketone, an ether, an ester, a hydrocarbon, a halogenated hydrocarbon, and mixtures thereof. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, 1-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl- tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, butyl acetate, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
Unless otherwise specified, the term 'base' as used herein includes, but is not limited to, organic bases and inorganic bases such as carbonates, bicarbonates, hydroxides, alkoxides, acetates and amides of alkali or alkali earth metals.
Specifically, the inorganic base is selected from the group consisting of sodium carbonate, potassium carbonate, lithium carbonate, cesium carbonate, sodium bicarbonate, potassium bicarbonate, lithium bicarbonate, cesium bicarbonate, sodium hydroxide,
potassium hydroxide, lithium hydroxide, sodium methoxide, sodium ethoxide, potassium methoxide, potassium ethoxide, sodium tertbutoxide, potassium tert.butoxide, sodium amide, potassium amide, lithium amide, ammonia, sodium acetate, potassium acetate, magnesium acetate, calcium acetate, and mixtures thereof.
Specifically, the organic base is selected from the group consisting of dimethylamine, diethylamine, diisopropyl amine, diisopropylethylamine, di n-butylamine, diisobutylamine, triethylamine, tributylamine, tert-butyl amine, pyridine, 4- dimethylaminopyridine (DMAP), and mixtures thereof.
Unless otherwise specified, the term 'phase transfer catalysts' as used herein include, but are not limited to, tetrabutylammonium bromide, tetrabutylammonium chloride, tetrabutylammonium iodide, benzyltrimethyl ammonium chloride, benzyltriethyl ammonium chloride, methyltributyl ammonium chloride, crown ethers and the like.
Unless otherwise specified, the term 'salt' as used herein may include acid addition salts and base addition salts.
Acid addition salts may be derived from organic and inorganic acids. Exemplary acid addition salts include, but are not limited to, hydrochloride, hydrobromide, sulphate, nitrate, phosphate, acetate, propionate, oxalate, succinate, maleate, fumarate, benzenesulfonate, toluenesulfonate, citrate, tartrate, and the like. A most specific acid addition salt is hydrochloride salt.
Base addition salts may be derived from an organic or an inorganic base. For example, the base addition salts are derived from alkali or alkaline earth metals such as sodium, calcium, potassium and magnesium, ammonium salt and the like.
The highly pure Bilastine or a pharmaceutically acceptable salt thereof obtained by the process disclosed herein has a purity of greater than about 99.5%, specifically greater than about 99.8%, more specifically greater than about 99.9% as measured by HPLC. For example, the purity of the highly pure Bilastine or a pharmaceutically acceptable salt thereof obtained by the processes disclosed herein is about 99.5% to about 99.99% as measured by HPLC.
As used herein, the term "reflux temperature" means the temperature at which the solvent or solvent system refluxes or boils at atmospheric pressure.
As used herein, the term "room temperature" refers to a temperature of about 20°C to about 35°C. For example, "room temperature" can refer to a temperature of about 25°C to about 30°C.
Exemplary Lewis acids used in step-(a) include, but are not limited to, aluminum chloride, aluminum bromide, boron trifluoride, boron tribromide, boron trichloride, tin tetrachloride, tin tetrabromide, stannous chloride, ferric chloride, zinc chloride, titanium tetrachloride, and hydrates or solvates thereof. A most specific Lewis acid used in step-(a) is aluminum chloride.
The reaction in step-(a) is carried out in a suitable solvent. Exemplary solvents used in step-(a) include, but are not limited to, a halogenated hydrocarbon, a ketone, an ether, an ester, a hydrocarbon, and mixtures thereof.
Specifically, the solvent used in step-(a) is selected from the group consisting of dichloromethane, dichloroethane, chloroform, acetone, methyl ethyl ketone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-propyl acetate, isopropyl acetate, n-butyl acetate, cyclohexane, toluene, xylene, and mixtures thereof. A most specific solvent is dichloromethane.
Specifically, the reaction in step-(a) is carried out at a temperature of about -10°C to about 50°C, and more specifically at a temperature of about -5°C to about 35°C. The reaction time may vary between about 30 minutes to about 5 hours, and specifically about 1 hour to about 3 hours.
The reaction mass containing the compound of formula III or a salt thereof obtained in step-(a) may be subjected to usual work up methods such as a washing, a quenching, an extraction, a pH adjustment, an evaporation, a layer separation, decolorization, a carbon treatment, or a combination thereof. The reaction mass may be used directly in the next step to produce the compound of formula IIIA, or the compound of formula III or a salt thereof may be isolated and/or recrystallized and then used in the next step.
Unless otherwise specified, the carbon treatment is carried out by methods known in the art, for example, by stirring the reaction mass/solution with finely powdered carbon at a temperature of about 40°C to the reflux temperature for at least 5 minutes, specifically at the reflux temperature; and filtering the resulting mixture through charcoal bed to obtain a filtrate containing compound by removing charcoal. Specifically, finely powdered carbon is a special carbon or an active carbon.
In one embodiment, the compound of formula III or a salt thereof may be isolated and/or re-crystallized from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
The solvent used for work up, isolation and/or recrystallization of the compound of formula III obtained by the process described herein is selected from the group as described hereinabove.
In one embodiment, the hydrosilane reducing agent used in step-(b) is selected from the group consisting of triethylsilane, trimethylsilane, dimethyl phenyl silane, phenyl silane, triphenylsilane, trichloro silane, and the like; and a most specific reducing agent is triethylsilane.
In another embodiment, the acid used in step-(b) is selected from the group consisting of boron trifluoride diethyl etherate, titanium tetrachloride, aluminum chloride, aluminum bromide, boron tribromide, tin tetrachloride, tin tetrabromide, stannous chloride, ferric chloride, zinc chloride, trifluoro acetic acid and methanesulfonic acid. A most specific acid used is titanium tetrachloride.
Exemplary solvents used in step-(b) include, but are not limited to, a hydrocarbon solvent, a chlorinated hydrocarbon solvent, and mixtures thereof.
Specifically, the solvent used in step-(b) is selected from the group consisting of toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof; and a most specific solvent is dichloromethane.
In another embodiment, the reaction in step-(b) is carried out at a temperature of about -10°C to 50°C; and specifically at a temperature of about 10°C to about 40°C. The reaction time may vary between about 2 hours to 8 hours, and more specifically about 4 hours to 6 hours.
The reaction mass containing the compound of formula IIIA or a salt thereof obtained in step-(b) may be subjected to usual work up methods such as a washing, a quenching, an extraction, a pH adjustment, an evaporation, a layer separation, decolorization, a carbon treatment, or a combination thereof. The reaction mass may be used directly in the next step to produce the compound of formula I, or the compound of formula IIIA or a salt thereof may be isolated and/or recrystallized and then used in the next step.
In one embodiment, the compound of formula IIIA or a salt thereof may be isolated and/or re-crystallized from a suitable solvent by conventional methods as described hereinabove.
The solvent used for work up, isolation and/or recrystallization of the compound of formula IIIA obtained by the process described herein is selected from the group as described hereinabove.
In one embodiment, the base used in step-(c) is an organic base or an inorganic base selected from the group as described hereinabove. Specifically, the base used in step- (c) is an inorganic base. A most specific base used in step-(c) is sodium carbonate or potassium carbonate.
In another embodiment, the reaction in step-(c) is carried out in the presence of a phase transfer catalyst. The phase transfer catalyst can be selected from the group as described hereinabove.
Exemplary solvents used in step-(c) include, but are not limited, water, acetone, methyl ethyl ketone, methyl isobutyl ketone, acetonitrile, propionitrile and mixtures thereof. A most specific solvent used in step-(c) is water.
In one embodiment, the reaction in step-(c) is carried out at a temperature of about 10°C to the reflux temperature of the solvent used, specifically at a temperature of about 30°C to the reflux temperature of the solvent used, and more specifically at the reflux temperature of the solvent used. The reaction time may vary from about 15 hours to about 25 hours.
The reaction mass containing the Bilastine of formula I or a salt thereof obtained in step-(c) may be subjected to usual work up methods such as a washing, a quenching, an extraction, a pH adjustment, an evaporation, a layer separation, decolorization, a carbon treatment, or a combination thereof.
In one embodiment, the Bilastine of formula I or a salt thereof may be isolated, purified and/or re-crystallized from a suitable solvent by conventional methods as described hereinabove.
The solvent used for work up, isolation, recrystallization and/or purification of the Bilastine of formula I or a salt thereof obtained by the process described herein is selected from the group as described hereinabove.
The crude Bilastine obtained in step-(c) is, optionally subjected to carbon treatment or silica gel treatment. The carbon treatment or silica gel treatment is carried out by methods known in the art, for example, as per the methods described hereinabove.
In one embodiment, the solvent used for purification of Bilastine obtained in step- (c) is selected from the group consisting of water, acetone, methanol, ethanol, isopropyl alcohol, ethyl acetate, butyl acetate, and mixtures thereof.
The term "anti- solvent" refers to a solvent which when added to an existing solution of a substance reduces the solubility of the substance.
Exemplary anti-solvents include, but are not limited to, water, an alcohol, a ketone, a chlorinated hydrocarbon, a hydrocarbon, an ester, a nitrile, an ether, a polar aprotic solvent, and mixtures thereof.
Removal of solvent is accomplished, for example, by substantially complete evaporation of the solvent, concentrating the solution or distillation of solvent, under inert atmosphere to obtain highly pure Bilastine or a salt thereof.
According to another aspect, there is provided a novel compound, 2-[4-(2- chloroethyl)phenyl]-2-methyl-propanoic acid, of formula IIIA:

According to another aspect, there is provided a process for the preparation of 2- [4- (2-chloroethyl)phenyl]-2-methyl-propanoic acid of formula IIIA:

or a salt thereof, comprising:
a) reacting the 2-methyl-2-phenyl-propanoic acid of formula II:



b) reducing the compound of formula III obtained in step-(a) with a hydrosilane reagent in the presence of an acid to produce 2-[4-(2-chloroethyl)phenyl]-2-methyl-propanoic acid of formula IIIA or a salt thereof.
The preparation of the 2-[4-(2-chloroethyl)phenyl]-2-methyl-propanoic acid compound of formula IIIA or a salt thereof as described in the above process steps-(a) and (b) can be carried out by using the suitable solvents, reagents, methods, parameters and conditions as described hereinabove.
According to another aspect, there is provided a process for the preparation of highly pure Bilastine of formula I:

a) reducing the 2-[4-(2-chloroacetyl)phenyl]-2-methyl-propanoic acid compound of formula III:



The preparation of the Bilastine of formula I or a pharmaceutically acceptable salt thereof as described in the above process steps-(a) and (b) can be carried out by using the suitable solvents, reagents, methods, parameters and conditions as described hereinabove.
According to another aspect, there is provided a process for the preparation of highly pure Bilastine of formula I:
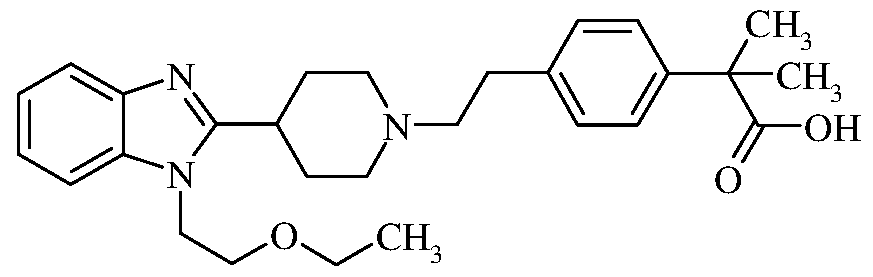


The highly pure Bilastine or a salt thereof obtained by the above processes may be further dried in, for example, a Vacuum Tray Dryer, a Rotocon Vacuum Dryer, a Vacuum Paddle Dryer or a pilot plant Rota vapor, to further lower residual solvents. Drying can be carried out under reduced pressure until the residual solvent content reduces to the desired amount such as an amount that is within the limits given by the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use ("ICH") guidelines.
In one embodiment, the drying is carried out at atmospheric pressure or reduced pressures, such as below about 200 mm Hg, or below about 50 mm Hg, at temperatures such as about 35°C to about 90°C, and specifically at about 75°C to about 85°C. The drying can be carried out for any desired time period that achieves the desired result, such as times about 1 to 20 hours.
Drying may also be carried out for shorter or longer periods of time depending on the product specifications. Temperatures and pressures will be chosen based on the volatility of the solvent being used and the foregoing should be considered as only a general guidance. Drying can be suitably carried out in a tray dryer, vacuum oven, air oven, or using a fluidized bed drier, spin flash dryer, flash dryer, and the like.
The following examples are given only to illustrate the present invention. However, they should not be considered as limitation on the scope or spirit of the invention.
EXAMPLES
Example 1
Preparation of 2-[4-(2-Chloroacetyl)phenyl]-2-methyl-propanoic acid
Aluminum chloride (122 g) was slowly added to a mixture of 2-methyl-2-phenyl- propanoic acid (25 g) and dichloro methane (250 ml) at room temperature. The resulting mixture was cooled to -5 to -10°C, followed by drop wise addition of chloroacetyl chloride (34.5 g) at the same temperature. The temperature of the reaction mass was raised to 25-30°C and then stirred for 2 hours at the same temperature. After completion of the reaction, the reaction mass was poured into water (1300 ml), ice (160 g) and hydrochloric acid (220 ml) at 10-15°C. The layers were separated and the aqueous layer was extracted twice with dichloro methane (250 ml x 2). The organic layers were combined and then
washed with 1 N HC1 solution (250 ml x 2), followed by water (250 ml x 2). The solvent was removed from the organic layer by distillation under vacuum to produce 35 g of 2-[4- (2-chloroacetyl)phenyl]-2-methyl-propanoic acid. Example 2
Preparation of 2-[4-(2-Chloroethyl)phenyl]-2-methyl-propanoic acid
Dichloro methane (700 ml) was added to 2-[4-(2-chloroacetyl)phenyl]-2-methyl-propanoic acid (35 g) and the mixture was cooled to 0-5°C, followed by slow addition of titanium tetrachloride (140 g) at the same temperature. The temperature of the resulting mass was raised to 20-25°C, followed by the addition of triethylsilane (64.4 g) and then stirring the reaction mixture at 25-30°C for 4 hours. The reaction mass was cooled to below 10°C and then water (980 ml) was added at the same temperature. The organic layer was separated and the aqueous layer was extracted with dichloromethane (500 ml x 2). The resulting organic layers were combined, followed by removal of the solvent completely by distillation under vacuum to produce a crude compound. Aqueous NaOH solution was added to the resulting crude compound while adjusting the pH to 9-10, and then washed with toluene (75 ml x 2). The layers were separated, followed adjusting the pH of the aqueous layer to 1-2 with dilute hydrochloric acid at 10-15°C. The resulting acidic aqueous layer was extracted thrice with ethyl acetate (100 ml x 3). The combined organic layers were washed with water (100 ml), and then distilled-off the solvent completely under vacuum to produce 31 g of 2-[4-(2-chloroethyl)phenyl]-2-methyl-propanoic acid.
Example 3
Preparation of 2-(4-Piperidinyl)-lH-benzimidazole
Orthophenylenediamine (20 g), polyphosphoric acid (120 g) and isonipecotic acid (26.5 g) were taken into a reaction flask and the resulting mixture was heated to 115-120°C, followed by stirring for 20 hours at the same temperature. After completion of the reaction, the reaction mass was cooled to 90°C, quenched with distilled water (260 ml) and then cooled to room temperature (25-30°C). The resulting mass was further cooled to 10-15°C, followed by adjusting the pH of the reaction mass to 9-10 with dilute sodium hydroxide solution, and then stirring for 30 minutes at 10-15°C. The separated solid was filtered and washed with distilled water. The wet material was dried at 40-45°C. Methanol (720 ml) was added to the resulting material and stirred for 1 hour at room temperature. The
resulting mass was filtered and washed with methanol (250 ml). The resulting filtrate was concentrated under reduced pressure to give 36 g of 2-(4-Piperidinyl)-lH-benzimidazole.
Example 4
Preparation of Ethyl 4-(lH-Benzimidazol-2-yl)-piperidine-l-carboxylate
2-(4-Piperidinyl)-lH-benzimidazole (36 g) and chloroform (720 ml) were taken into a reaction flask and then triethylamine (45 g) was added at room temperature. The resulting mass was cooled to 0-5°C, followed by drop-wise addition of a solution of ethyl chloroformate (21.5 g) in chloroform (80 ml) at the same temperature for 30-45 minutes. After completion of the reaction, water (360 ml) was added to the reaction mass at the same temperature and then stirred for 10 minutes. The organic layer was separated and the aqueous layer was extracted with chloroform (250 ml). The chloroform layers were combined and then washed twice with water (250 ml x 2). The chloroform was distilled off under reduced pressure from the resulting organic layer to give 46 g of crude 4-(lH- benzimidazol-2-yl)-piperidine-l-carboxylic acid ethyl ester. The crude compound was added to cyclohexane (157.5 ml) and then heated to reflux temperature. The resulting mass was stirred for 30-45 minutes at reflux temperature for 30-45 minutes and then cooled the reaction mass to room temperature, followed by stirring for 1 hour at the same temperature. The separated solid was filtered and washed with cyclohexane (45 ml) to give 44 g of pure ethyl 4-(lH-benzimidazol-2-yl)-piperidine-l-carboxylate.
Example 5
Preparation of Ethyl 4-[l-(2-Ethoxyethyl)-benzimidazol-2-yl]-piperidine-l- carboxylate
Ethyl 4-(lH-benzimidazol-2-yl)-piperidine-l-carboxylate (42 g) and toluene (210 ml) were taken into a reaction flask and then heated to 40-45°C. To the reaction mass, sodium hydroxide (18.5 g), potassium iodide (4.6 g), 2-chloroethyl ethyl ether (25 ml) and tetrabutylammonium bromide (10 g) were added. The resulting mixture was heated to 95- 100°C and then stirred for 16 hours at the same temperature. After completion of the reaction, the reaction mass was cooled to room temperature, followed by the addition of water (210 ml) and then stirring for 10 minutes at room temperature. The resulting mass was neutralized with dilute hydrochloric acid. The layers were separated and the aqueous layer was extracted twice with ethyl acetate (200 ml x 2). The toluene layer and ethyl acetate layers were combined and washed with distilled water (250 ml). The solvents were
distilled off completely under reduced pressure to give 51 g of ethyl 4-[l-(2-ethoxyethyl)- benzimidazol-2-yl] -piperidine- 1 -carboxylate.
Example 6
Preparation of l-(2-Ethoxyethyl)-2-(piperidin-4-yl)-benzimidazole
Ethyl 4- [l-(2-ethoxyethyl)-benzimidazol-2-yl] -piperidine- 1 -carboxylate (50 g) and isopropyl alcohol (350 ml) were taken into a reaction flask and then potassium hydroxide (100 g) was added at room temperature. The resulting mixture was heated to reflux and then maintained for 10 hours at reflux temperature. The solvent was distilled off completely from the reaction mass under reduced pressure to obtain crude product. Water (850 ml) was added to the crude product and then neutralized with dilute hydrochloric acid. The resulting mass was extracted with 1-butanol (600 ml) and again extracted with 1- butanol (300 ml x 2) twice. The solvent was distilled off under reduced pressure, followed by co-distillation with toluene (100 ml). The resulting crude was cooled to room temperature and then toluene (135 ml) was added, followed by stirring for 20-30 minutes. The separated solid was filtered and washed with toluene (40 ml) to give 41 g of crude 1- (2-ethoxyethyl)-2-(piperidin-4-yl)-benzimidazole. The crude compound was added to acetone (410 ml) and then heated to reflux, followed by stirring the reaction mass at reflux temperature for 30 minutes. The reaction mass was cooled to 25-30°C, filtered the material and then washed with acetone (30 ml) to give 32.5 g of pure l-(2-ethoxyethyl)-2- (piperidin-4-yl)-benzimidazole.
Example 7
Preparation of Pure Bilastine
Step-(a): Preparation of Crude Bilastine
2-[4-(2-Chloroethyl)phenyl]-2-methyl-propanoic acid (20 g), water (200 ml), l-(2- ethoxyethyl)-2-(piperidin-4-yl)-benzimidazole (26 g) and sodium carbonate (28 g) were taken into a reaction flask at room temperature. The resulting mixture was heated to reflux temperature and maintained for 22 hours at the same temperature. After completion of reaction, the resulting mass was cooled to room temperature, followed by addition of water (960 ml) at the same temperature. The layers were separated and the aqueous layer was washed with toluene (640 ml x 2). The aqueous layer was separated and then neutralized with acetic acid, followed by extracting thrice with dichloromethane (800 ml x 3). Activated carbon (12 g) was added to the resulting organic layers and then stirred for 10
minutes. The resulting mixture was filtered through hyflo-bed and then washed the bed with dichloro methane (50 ml). The resulting filtrate was distilled-off under vacuum to remove the solvent completely. Acetone (110 ml) was added to the resulting crude compound and then stirred for 10-15 minutes at room temperature. The solvent was distilled-off completely from the resulting mass, acetone (85 ml) was added again and then stirred for 1 hour at room temperature. The separated solid was filtered and washed with acetone (30 ml) to produce 19 g of crude Bilastine.
Step-(b): Purification of crude Bilastine:
Crude Bilastine (19 g) was added to butyl acetate (570 ml) and then heated to reflux temperature while stirring, followed by maintaining the reaction mass for 15 minutes at the same temperature. The resulting mass was cooled to room temperature and then stirred for 30 minutes at the same temperature. The separated solid was filtered and washed with butyl acetate (30 ml). Butyl acetate (540 ml) was added to the resulting wet material and then repeated the above re-crystallization process. The resulting wet material was taken in methanol (320 ml) and then heated to reflux, followed by stirring the reaction mass for 30 minutes at reflux temperature. Activated carbon (1.6 g) was added to the reaction mass and then stirred for 10 minutes. The resulting mixture was filtered through hyflo-bed and then washed the bed with hot methanol (16 ml). The resulting filtrate was cooled to 20-22°C and then stirred for 40 minutes at the same temperature. The separated solid was filtered, washed with cold methanol (16 ml) and then dried the material at 60-70°C to give 15 g of pure Bilastine (Purity by HPLC: 99.6%).
Claims
1. A process for the preparation of highly pure Bilastine of formula I:

a) reacting 2-methyl-2-phenyl-propanoic acid of formula II:



b) reducing the compound of formula III obtained in step-(a) with a hydrosilane reagent in the presence of an acid to produce 2-[4-(2-chloroethyl)phenyl]-2-methyl- propanoic acid compound of formula IIIA:


The process of claim 1, wherein the Lewis acid used in step-(a) is selected from the group consisting of aluminum chloride, aluminum bromide, boron trifluoride, boron tribromide, boron trichloride, tin tetrachloride, tin tetrabromide, stannous chloride, ferric chloride, zinc chloride, titanium tetrachloride, and hydrates or solvates thereof; wherein the reaction in step-(a) is carried out in a suitable solvent selected from the group consisting of dichloromethane, dichloroethane, chloroform, acetone, methyl ethyl ketone, tetrahydrofuran,
2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert- butyl ether, ethyl acetate, n-propyl acetate, isopropyl acetate, n-butyl acetate, cyclohexane, toluene, xylene, and mixtures thereof; wherein the hydrosilane reducing agent used in step-(b) is selected from the group consisting of triethylsilane, trimethylsilane, dimethyl phenyl silane, phenyl silane, triphenylsilane, trichlorosilane; wherein the acid used in step-(b) is selected from the group consisting of boron trifluoride diethyl etherate, titanium tetrachloride, aluminum chloride, aluminum bromide, boron tribromide, tin tetrachloride, tin tetrabromide, stannous chloride, ferric chloride, zinc chloride, trifluoro acetic acid and methanesulfonic acid; wherein the solvent used in step-(b) is selected from the group consisting of toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof; wherein the base used in step-(c) is an inorganic base selected from the group consisting of sodium carbonate, potassium carbonate, lithium carbonate, cesium carbonate, sodium bicarbonate, potassium bicarbonate, lithium bicarbonate, cesium bicarbonate, sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium methoxide, sodium ethoxide, potassium methoxide, potassium ethoxide, sodium tertbutoxide, potassium
tert.butoxide, sodium amide, potassium amide, lithium amide, ammonia, sodium acetate, potassium acetate, magnesium acetate, calcium acetate, and mixtures thereof; wherein the phase transfer catalyst used in step-(c) is selected from the group consisting of tetrabutylammonium bromide, tetrabutylammonium chloride, tetrabutylammonium iodide, benzyltrimethyl ammonium chloride, benzyltriethyl ammonium chloride, methyltributyl ammonium chloride, and crown ethers; and wherein the solvent used in step-(c) is selected from the group consisting of water, acetone, methyl ethyl ketone, methyl isobutyl ketone, acetonitrile, propionitrile and mixtures thereof.
3. The process of claim 2, wherein the Lewis acid used in step-(a) is aluminum chloride; wherein the solvent used in step-(a) is dichloro methane; wherein the hydrosilane reducing agent used in step-(b) is triethylsilane; wherein the acid used in step-(b) is titanium tetrachloride; wherein the solvent used in step-(b) is dichloro methane; wherein the base used in step-(c) is sodium carbonate or potassium carbonate; and wherein the solvent used in step-(c) is water.
4. A compound, 2-[4-(2-chloroethyl)phenyl]-2-methyl-propanoic acid, of formula IIIA:

A process for the preparation of 2-[4-(2-chloroethyl)phenyl]-2-methyl-propano of formula IIIA:

or a salt thereof, comprising:
a) reacting the 2-methyl-2-phenyl-propanoic acid of formula II:
 or a salt thereof, with chloroacetyl chloride of formula VI:
or a salt thereof, with chloroacetyl chloride of formula VI:
 optionally in the presence of a Lewis acid, to produce 2-[4-(2-chloroacetyl)phenyl]- 2-methyl-propanoic acid compound of formula III:
optionally in the presence of a Lewis acid, to produce 2-[4-(2-chloroacetyl)phenyl]- 2-methyl-propanoic acid compound of formula III:

b) reducing the compound of formula III obtained in step-(a) with a hydrosilane reagent in the presence of an acid to produce 2-[4-(2-chloroethyl)phenyl]-2-methyl- propanoic acid of formula IIIA or a salt thereof.
6. The process of claim 5, wherein the Lewis acid used in step-(a) is selected from the group consisting of aluminum chloride, aluminum bromide, boron trifluoride, boron tribromide, boron trichloride, tin tetrachloride, tin tetrabromide, stannous chloride, ferric chloride, zinc chloride, titanium tetrachloride, and hydrates or solvates thereof; wherein the reaction in step-(a) is carried out in a suitable solvent selected from the group consisting of dichloromethane, dichloroethane, chloroform, acetone, methyl ethyl ketone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert- butyl ether, ethyl acetate, n-propyl acetate, isopropyl acetate, n-butyl acetate, cyclohexane, toluene, xylene, and mixtures thereof; wherein the hydrosilane reducing agent used in step-(b) is selected from the group consisting of triethylsilane, trimethylsilane, dimethyl phenyl silane, phenyl silane, triphenylsilane, trichlorosilane; wherein the acid used in step-(b) is selected from the group consisting of boron trifluoride diethyl etherate, titanium tetrachloride, aluminum chloride, aluminum bromide, boron tribromide, tin tetrachloride, tin tetrabromide, stannous chloride, ferric
chloride, zinc chloride, trifluoroacetic acid and methanesulfonic acid; and wherein the solvent used in step-(b) is selected from the group consisting of toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
A process for the preparation of highly pure Bilastine of formula I:

a) reducing the 2-[4-(2-chloroacetyl)phenyl]-2-methyl-propanoic acid compound of formula III:
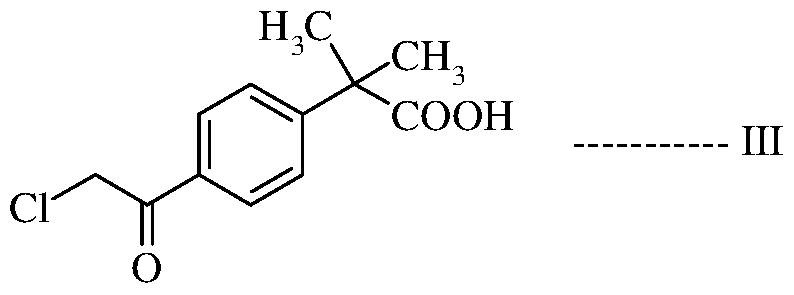

b) condensing the compound of formula IIIA obtained in step-(a) with l-(2- ethoxyethyl)-2-(piperidin-4-yl)benzimidazole of formula IV:

The process of claim 7, wherein the hydrosilane reducing agent used in step-(a) is selected from the group consisting of triethylsilane, trimethylsilane, dimethyl phenyl silane, phenyl silane, triphenylsilane, trichlorosilane; wherein the acid used in step-(a) is selected from the group consisting of boron trifluoride diethyl etherate, titanium tetrachloride, aluminum chloride, aluminum bromide, boron tribromide, tin tetrachloride, tin tetrabromide, stannous chloride, ferric chloride, zinc chloride, trifluoro acetic acid and methanesulfonic acid; wherein the solvent used in step-(a) is selected from the group consisting of toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof; wherein the base used in step-(b) is an inorganic base selected from the group consisting of sodium carbonate, potassium carbonate, lithium carbonate, cesium carbonate, sodium bicarbonate, potassium bicarbonate, lithium bicarbonate, cesium bicarbonate, sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium methoxide, sodium ethoxide, potassium methoxide, potassium ethoxide, sodium tertbutoxide, potassium tert.butoxide, sodium amide, potassium amide, lithium amide, ammonia, sodium acetate, potassium acetate, magnesium acetate, calcium acetate, and mixtures thereof; wherein the phase transfer catalyst used in step-(b) is selected from the group consisting of tetrabutylammonium bromide, tetrabutylammonium chloride, tetrabutylammonium iodide, benzyltrimethyl ammonium chloride, benzyltriethyl ammonium chloride, methyltributyl ammonium chloride, and crown ethers; and wherein the solvent used in step-(b) is selected from the group consisting of water, acetone, methyl ethyl ketone, methyl isobutyl ketone, acetonitrile, propionitrile and mixtures thereof.
9. A process for the preparation of highly pure Bilastine of formula I:
or a pharmaceutically acceptable salt thereof, comprising condensing the 2-[4-(2- chloroethyl)phenyl]-2-methyl-propanoic acid compound of formula IIIA:
ΠΙΑ

IV:

The process of claim 9, wherein the base used in the reaction is an inorganic base selected from the group consisting of sodium carbonate, potassium carbonate, lithium carbonate, cesium carbonate, sodium bicarbonate, potassium bicarbonate, lithium bicarbonate, cesium bicarbonate, sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium methoxide, sodium ethoxide, potassium methoxide, potassium ethoxide, sodium tertbutoxide, potassium tert.butoxide, sodium amide, potassium amide, lithium amide, ammonia, sodium acetate, potassium acetate, magnesium acetate, calcium acetate, and mixtures thereof; wherein the phase transfer catalyst used in the reaction is selected from the group consisting of tetrabutylammonium bromide, tetrabutylammonium chloride, tetrabutylammonium iodide, benzyltrimethyl ammonium chloride, benzyltriethyl ammonium chloride, methyltributyl ammonium chloride, and crown ethers; and wherein the solvent used in the reaction is selected from the group consisting of water, acetone, methyl ethyl ketone, methyl isobutyl ketone, acetonitrile, propionitrile and mixtures thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN201641029306 | 2016-08-29 | ||
| IN201641029306 | 2016-08-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018042305A1 true WO2018042305A1 (en) | 2018-03-08 |
Family
ID=61300223
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2017/055146 WO2018042305A1 (en) | 2016-08-29 | 2017-08-28 | Improved processes for preparation of bilastine using novel intermediates |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2018042305A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110105204A (en) * | 2019-06-04 | 2019-08-09 | 荆楚理工学院 | A kind of 4- (the halogenated isobutyryl of 2-) benzyl carbinol derivative and preparation method thereof |
| CN111689953A (en) * | 2019-03-14 | 2020-09-22 | 北京万全德众医药生物技术有限公司 | Preparation method of bilastine intermediate |
| CN114591291A (en) * | 2020-12-05 | 2022-06-07 | 鲁南制药集团股份有限公司 | A kind of bilastine intermediate compound and preparation method thereof |
| CN114671802A (en) * | 2022-04-14 | 2022-06-28 | 江苏联环药业股份有限公司 | Preparation method of high-purity ebastine |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009102155A2 (en) * | 2008-02-12 | 2009-08-20 | Yuhan Corporation | Process for preparation of 2-methyl-2´-phenylpropionic acid derivatives and novel intermediate compounds |
| WO2014188453A2 (en) * | 2013-05-24 | 2014-11-27 | Msn Laboratories Private Limited | Novel process for the preparation of 2-[4-(2-{4-[1-(2-ethoxyethyl)-1h-benzimidazol-2-yl]-1-piperidinyl}ethyl) phenyl]-2-methylpropanoic acid |
-
2017
- 2017-08-28 WO PCT/IB2017/055146 patent/WO2018042305A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009102155A2 (en) * | 2008-02-12 | 2009-08-20 | Yuhan Corporation | Process for preparation of 2-methyl-2´-phenylpropionic acid derivatives and novel intermediate compounds |
| WO2014188453A2 (en) * | 2013-05-24 | 2014-11-27 | Msn Laboratories Private Limited | Novel process for the preparation of 2-[4-(2-{4-[1-(2-ethoxyethyl)-1h-benzimidazol-2-yl]-1-piperidinyl}ethyl) phenyl]-2-methylpropanoic acid |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111689953A (en) * | 2019-03-14 | 2020-09-22 | 北京万全德众医药生物技术有限公司 | Preparation method of bilastine intermediate |
| CN110105204A (en) * | 2019-06-04 | 2019-08-09 | 荆楚理工学院 | A kind of 4- (the halogenated isobutyryl of 2-) benzyl carbinol derivative and preparation method thereof |
| CN114591291A (en) * | 2020-12-05 | 2022-06-07 | 鲁南制药集团股份有限公司 | A kind of bilastine intermediate compound and preparation method thereof |
| CN114591291B (en) * | 2020-12-05 | 2025-06-17 | 鲁南制药集团股份有限公司 | A bilastine intermediate compound and preparation method thereof |
| CN114671802A (en) * | 2022-04-14 | 2022-06-28 | 江苏联环药业股份有限公司 | Preparation method of high-purity ebastine |
| CN114671802B (en) * | 2022-04-14 | 2024-05-17 | 江苏联环药业股份有限公司 | Preparation method of high-purity ebastine |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2010220204B2 (en) | Improved process | |
| WO2018042305A1 (en) | Improved processes for preparation of bilastine using novel intermediates | |
| US8481730B2 (en) | Method of synthesis of Bosentan, its polymorphic forms and its salts | |
| EP2768806B1 (en) | Method for preparing silodosin | |
| WO2008005423A1 (en) | Improved method of making sufentanil | |
| US20100311969A1 (en) | Process For Preparation of Paliperidone | |
| WO2020194115A1 (en) | Process for the preparation of elagolix sodium and intermediates thereof | |
| CN111630049A (en) | Process for the preparation of 2- (5-methoxyisochroman-1-yl) -4, 5-dihydro-1H-imidazole and its bisulphate | |
| JP5819284B2 (en) | Method for producing olopatadine | |
| EP3413891B1 (en) | Processes for the preparation of highly pure prucalopride succinate | |
| CA2890961A1 (en) | Novel polymorphs of azilsartan medoxomil | |
| WO2016199020A1 (en) | Process for preparation of ceritinib | |
| US7777037B2 (en) | Ziprasidone process | |
| US20070129549A1 (en) | Stable lamotrigine pharmaceutical compositions and processes for their preparation | |
| CN112062763B (en) | Hydroxypyrimido [1,2-b ] [1,2,5] triazepine derivative, preparation and application | |
| US11401269B2 (en) | Intermediates and processes for the preparation of Linagliptin and its salts | |
| WO2011051957A2 (en) | A process for the preparation of donepezil hydrochloride | |
| WO2012134445A1 (en) | An improved process for the preparation of paliperidone | |
| WO2009084030A2 (en) | Improved process for the preparation of (1-benzyl-4-(5,6,- dimethoxyind anone-2-yl)methylpiperidine) hydrochloride-form iii | |
| US20120259116A1 (en) | Novel Process for the Preparation of Paliperidone | |
| WO2014049609A2 (en) | Novel salts of vilazodone | |
| WO2012001357A1 (en) | Crystalline form of prulifloxacin and processes for its preparation | |
| EP2765131B1 (en) | Process for the production of Moxonidine | |
| WO2012035554A1 (en) | An improved process for the preparation of highly pure paliperidone | |
| WO2018011676A1 (en) | Novel processes for the preparation of 2-oxy-benzoxazinone derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17845618 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 17845618 Country of ref document: EP Kind code of ref document: A1 |