WO2017168431A1 - Process for the preparation of salmeterol xinafoate - Google Patents
Process for the preparation of salmeterol xinafoate Download PDFInfo
- Publication number
- WO2017168431A1 WO2017168431A1 PCT/IN2016/000136 IN2016000136W WO2017168431A1 WO 2017168431 A1 WO2017168431 A1 WO 2017168431A1 IN 2016000136 W IN2016000136 W IN 2016000136W WO 2017168431 A1 WO2017168431 A1 WO 2017168431A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- salmeterol
- methanol
- solvent
- Prior art date
Links
- GIIZNNXWQWCKIB-UHFFFAOYSA-N Serevent Chemical compound C1=C(O)C(CO)=CC(C(O)CNCCCCCCOCCCCC=2C=CC=CC=2)=C1 GIIZNNXWQWCKIB-UHFFFAOYSA-N 0.000 title claims abstract description 54
- 238000000034 method Methods 0.000 title claims abstract description 28
- 229960005018 salmeterol xinafoate Drugs 0.000 title claims abstract description 28
- 238000002360 preparation method Methods 0.000 title claims abstract description 22
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 90
- 150000001875 compounds Chemical class 0.000 claims description 56
- 239000002904 solvent Substances 0.000 claims description 31
- 229960004017 salmeterol Drugs 0.000 claims description 28
- 239000003638 chemical reducing agent Substances 0.000 claims description 11
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 10
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 10
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 claims description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 7
- 238000009833 condensation Methods 0.000 claims description 7
- 230000005494 condensation Effects 0.000 claims description 7
- 239000012279 sodium borohydride Substances 0.000 claims description 7
- 229910000033 sodium borohydride Inorganic materials 0.000 claims description 7
- 239000001257 hydrogen Substances 0.000 claims description 6
- 229910052739 hydrogen Inorganic materials 0.000 claims description 6
- 239000003054 catalyst Substances 0.000 claims description 5
- 229910052751 metal Inorganic materials 0.000 claims description 5
- 239000002184 metal Substances 0.000 claims description 5
- 238000006264 debenzylation reaction Methods 0.000 claims description 4
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 42
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 33
- 239000011541 reaction mixture Substances 0.000 description 24
- 229940093499 ethyl acetate Drugs 0.000 description 14
- 235000019439 ethyl acetate Nutrition 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 12
- 239000010410 layer Substances 0.000 description 12
- -1 5-(2-amino-l-hydroxyethyl)-2-hydroxybenzyl Chemical group 0.000 description 9
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 9
- 239000002585 base Substances 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 8
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 8
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 8
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000000706 filtrate Substances 0.000 description 6
- 239000012458 free base Substances 0.000 description 6
- 239000000203 mixture Substances 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 150000001298 alcohols Chemical class 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 239000008213 purified water Substances 0.000 description 4
- 238000006722 reduction reaction Methods 0.000 description 4
- 150000003839 salts Chemical class 0.000 description 4
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 239000012299 nitrogen atmosphere Substances 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 229910052938 sodium sulfate Inorganic materials 0.000 description 3
- 235000011152 sodium sulphate Nutrition 0.000 description 3
- 238000004809 thin layer chromatography Methods 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- LDCRTTXIJACKKU-ONEGZZNKSA-N dimethyl fumarate Chemical compound COC(=O)\C=C\C(=O)OC LDCRTTXIJACKKU-ONEGZZNKSA-N 0.000 description 2
- 229960004419 dimethyl fumarate Drugs 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical group [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- SGRHVVLXEBNBDV-UHFFFAOYSA-N 1,6-dibromohexane Chemical compound BrCCCCCCBr SGRHVVLXEBNBDV-UHFFFAOYSA-N 0.000 description 1
- HYRIAERKUPZOCS-UHFFFAOYSA-N 4-(6-bromohexoxy)butylbenzene Chemical compound BrCCCCCCOCCCCC1=CC=CC=C1 HYRIAERKUPZOCS-UHFFFAOYSA-N 0.000 description 1
- PZSMUPGANZGPBF-UHFFFAOYSA-N 4-[5-(dithiolan-3-yl)pentanoylamino]butanoic acid Chemical compound OC(=O)CCCNC(=O)CCCCC1CCSS1 PZSMUPGANZGPBF-UHFFFAOYSA-N 0.000 description 1
- LDZLXQFDGRCELX-UHFFFAOYSA-N 4-phenylbutan-1-ol Chemical compound OCCCCC1=CC=CC=C1 LDZLXQFDGRCELX-UHFFFAOYSA-N 0.000 description 1
- 208000009079 Bronchial Spasm Diseases 0.000 description 1
- 208000014181 Bronchial disease Diseases 0.000 description 1
- 206010006482 Bronchospasm Diseases 0.000 description 1
- 0 COc(c(C=O)c1)ccc1C(C*(CCCCCCOCCCCc1ccccc1)Cc1ccccc1)=O Chemical compound COc(c(C=O)c1)ccc1C(C*(CCCCCCOCCCCc1ccccc1)Cc1ccccc1)=O 0.000 description 1
- SXVCOGWSXNSYQW-UHFFFAOYSA-N Oc(cc1)c(C=O)cc1C(CN(CCCCCCOCCCCc1ccccc1)Cc1ccccc1)=O Chemical compound Oc(cc1)c(C=O)cc1C(CN(CCCCCCOCCCCc1ccccc1)Cc1ccccc1)=O SXVCOGWSXNSYQW-UHFFFAOYSA-N 0.000 description 1
- SXVCOGWSXNSYQW-UHFFFAOYSA-O Oc(cc1)c(C=O)cc1C(C[NH+](CCCCCCOCCCCc1ccccc1)Cc1ccccc1)=O Chemical compound Oc(cc1)c(C=O)cc1C(C[NH+](CCCCCCOCCCCc1ccccc1)Cc1ccccc1)=O SXVCOGWSXNSYQW-UHFFFAOYSA-O 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- JEDZLBFUGJTJGQ-UHFFFAOYSA-N [Na].COCCO[AlH]OCCOC Chemical compound [Na].COCCO[AlH]OCCOC JEDZLBFUGJTJGQ-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 239000000048 adrenergic agonist Substances 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 238000011021 bench scale process Methods 0.000 description 1
- 150000003939 benzylamines Chemical class 0.000 description 1
- 230000002051 biphasic effect Effects 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- 229940125389 long-acting beta agonist Drugs 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229910003446 platinum oxide Inorganic materials 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- 239000010948 rhodium Substances 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- SMQUZDBALVYZAC-UHFFFAOYSA-N salicylaldehyde Chemical class OC1=CC=CC=C1C=O SMQUZDBALVYZAC-UHFFFAOYSA-N 0.000 description 1
- 238000013341 scale-up Methods 0.000 description 1
- 229940090585 serevent Drugs 0.000 description 1
- 239000012419 sodium bis(2-methoxyethoxy)aluminum hydride Substances 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 229950000339 xinafoate Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/41—Preparation of salts of carboxylic acids
- C07C51/412—Preparation of salts of carboxylic acids by conversion of the acids, their salts, esters or anhydrides with the same carboxylic acid part
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/08—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions not involving the formation of amino groups, hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C221/00—Preparation of compounds containing amino groups and doubly-bound oxygen atoms bound to the same carbon skeleton
Definitions
- the present invention relates to an improved process for the preparation of Salmeterol Xinafoate of Formu la (I).
- Salmeterol Xinafoate is the racemic form of the l -hydroxy-2-naphthoic acid salt of Salmeterol. It
- Salmeterol Xinafoate is a Long-acting p 2 -adrenergic agonist (LABA). It is used for the treatment of asthma and in the prevention of bronchospasm. Salmeterol Xinafoate is commercialized under the brand name of SEREVENT ® .
- Salmeterol was first described and claimed in U.S. Pat. No. 4,992,474 A.
- This patent describes a specific method for producing Salmeterol by the reaction of 4-phenyl-l-butanol of Formula 1 with 1 ,6-dibromohexane of Formula 2 in the presence of sodium hydride (NaH) in tetrahydrofuran (THF) to give the ether derivative [4-[(6-bromohexyl)oxy]butyl]benzene of Formula 3, which is then condensed with 5-(2-amino-l-hydroxyethyl)-2-hydroxybenzyl alcohol of Formula 4 in the presence of potassium iodide ( I) and triethylamine in hot dimethylfumarate (DMF) to give Salmeterol of Formula 1-1.
- the process is shown in the scheme I given below:
- ES 2065269 B l describes a process for the preparation of Salmeterol by reacting 6-(4- phenylbutoxy)]hexyl]benzylamine of Formula 6 with salicylaldehyde derivative of Formula 5 followed by, catalytic hydrogenation of the obtained compound of Formula 7 to give Salmeterol of
- the main objective of the present invention is to provide cost effective and commercially viable process for the preparation of Salmeterol Xinafoate.
- Another objective of the present invention is to provide a process for the preparation of Salmeterol Xinafoate which employs less expensive, easily available and environment friendly reagents.
- the present invention provides an improved process for the preparation of Salmeterol Xinafoate of Formula (I)
- the present invention provides an improved process for the preparation of compound of Formula D
- the main embodiment, of the present invention provides an improved process for the preparation of Salmeterol Xinafoate of Formula (I) as shown in the scheme IV given below:
- the solvent used for condensation is selected from alcohols such as methanol, ethanol, n-propanol, isopropyl alcohol, isobutanol, n-butanol, tert-butanol and the like; preferably the solvent used is methanol.
- the base used for condensation is selected from amines such as triethylamine or diisopropylethylamine. Preferably the base used is diisopropylethylamine (DfPEA).
- the condensation reaction temperature may range from 0 °C to 15 °C and preferably at a temperature in the range from 5 °C to 10 °C.
- the duration of the reaction may range from 10 hours to 20 hours, preferably for a period of 15 hours.
- Step 2 After the completion of the reaction, the reaction mass is quenched into water and extracted with n-heptane. The n-heptane layer (which contains the compound of Formula C) is further extracted with methanol before proceeding to step-2 to give compound of Formula C in methanol. Step 2:
- the reducing agent used for reduction of compound of Formula C to compound of Formula D is selected from sodium borohydride, sodium cyanoborohydride, sodium triacetoxy borohydride, sodium bis(2-methoxyethoxy)aluminum hydride.
- the reducing agent used is sodium borohydride.
- the reduction reaction temperature may range from 20 °C to 35 °C and preferably at a temperature in the range from 25 °C to 30 °C.
- the duration of the reaction may range from 7 hours to 9 hours, preferably for a period of 8 hours.
- the debenzylating agent is selected from platinum, platinum oxide, palladium, Raney nickel or rhodium, on a support, such as charcoal or mixtures thereof. Preferably debenzylation is carried out by using palladium on carbon.
- the solvent used is selected from alcohols such as methanol, ethanol, isopropanol, n-propanol, n-butanol, isobutanol, t-butanol and the like. Preferably the solvent used is methanol.
- the reaction temperature may range from 20 °C to 30 °C and preferably at a temperature in the range from 25 °C to 28 °C.
- the duration of the reaction may range from 1 hour to 2 hours, preferably for a period of 1 .5 hours.
- Salmeterol free base of Formula 1-1 is subjected to purification using a solvent selected from esters such as ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate.
- a solvent selected from esters such as ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate.
- the solvent used is ethylacetate.
- the solvent used for salt formation is selected from alcohols such as methanol, ethanol, isopropanol, n-propanol, n-butanol, isobutanol, t-butanol and the like.
- the solvent used is methanol.
- the reaction temperature may range from 20 °C to 30 °C and preferably at a temperature in the range from 25 °C to 30 °C.
- the duration of the reaction may range from 1 hour to 2 hours, preferably for a period of 1 .5 hours.
- Salmeterol Xinafoate of Formula (I) is subjected to purification using a solvent selected from alcohols such as methanol, ethanol, isopropanol, n-propanol, n-butanol, isobutanol, t-butanol and the like.
- a solvent selected from alcohols such as methanol, ethanol, isopropanol, n-propanol, n-butanol, isobutanol, t-butanol and the like.
- the solvent used is methanol.
- the present invention provides an improved process for the preparation of Salmeterol Xinafoate of Formula (I)
- the present invention provides an improved process for the preparation of Salmeterol Xinafoate of Formula (I), which comprises:
- the present invention provides an improved process for the preparation of com ound of Formula D,
- the present invention provides an improved process for the preparation of compound of Formula D,
- Step I Preparation of 2-hydroxy-5-[[[6-(4-phenylbutoxy)hexylbenzyI]amino] acetyl] benzaldehyde (Formula C):
- n-Heptane 1000 mL and vacuum ( 166 grams) were added into reaction mixture at 20 ⁇ 3 °C. Separated the organic layer and washed with purified water (500 mL) and vacuum salt (42.6 grams) at 25-30 °C. Filtered the organic (n-Heptane) layer through (hyflo 25 g with n-Heptane 250 mL) hyflo bed and washed with n-Heptane (333.33 mL). Dried the filtrate over sodium sulphate (83.33 grams) at 25-30 °C. Filtered the sodium sulphate and washed with n-Heptane (125 mL). Extracted the filtrate with methanol (1666.66 mL) at -5 to 0 °C. Separated the n-heptane layer from the methanol ic layer at -5 to 0 °C.
- Step 2 Preparation of 4-hydroxy-a'-[[[6-(4-phenylbutoxy)hexyl]benzyIamino]methyl]-l,3- ben- zenedimethanol (Formula D - Benzyl Salmeterol)
- Salmeterol base (57 grams) and Ethylacetate (456 mL) were taken in round bottom flask. Heated the reaction mixture to 40-45 °C and stirred for 30 min. Cooled the reaction mixture to 0 ⁇ 2 °C and stirred for 1 hour. Filtered the product and washed with chilled ethylacetate (22.8 mL). Wet compound (65 grams) and Ethylacetate (456 mL) were taken in round bottom flask. Heated the reaction mixture to 40-45 °C and stirred for 30 min. Cooled the reaction mixture to 0 ⁇ 2 °C and stirred for 1 hour. Filtered the product and washed with chilled ethylacetate (22.8 mL).
- Step 1 Preparation of Salmeterol Xinafoate
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The present invention relates to an improved process for the preparation of Salmeterol Xinafoate of Formula (I).
Description
PROCESS FOR THE PREPARATION OF SALMETEROL XINAFOATE
Field of the Invention
The present invention relates to an improved process for the preparation of Salmeterol Xinafoate of Formu la (I).

Background of the Invention
Salmeterol Xinafoate is the racemic form of the l -hydroxy-2-naphthoic acid salt of Salmeterol. It
1
has the chemical name 4-hydroxy-a -[[[6-(4-phenylbutoxy)hexyl]amino]methyl]-l,3- benzenedimethanol, l-hydroxy-2-naphthalencarboxylate.
Salmeterol Xinafoate is a Long-acting p2-adrenergic agonist (LABA). It is used for the treatment of asthma and in the prevention of bronchospasm. Salmeterol Xinafoate is commercialized under the brand name of SEREVENT®.
Salmeterol was first described and claimed in U.S. Pat. No. 4,992,474 A. This patent describes a specific method for producing Salmeterol by the reaction of 4-phenyl-l-butanol of Formula 1 with 1 ,6-dibromohexane of Formula 2 in the presence of sodium hydride (NaH) in tetrahydrofuran (THF) to give the ether derivative [4-[(6-bromohexyl)oxy]butyl]benzene of Formula 3, which is then condensed with 5-(2-amino-l-hydroxyethyl)-2-hydroxybenzyl alcohol of Formula 4 in the presence of potassium iodide ( I) and triethylamine in hot dimethylfumarate (DMF) to give Salmeterol of Formula 1-1. The process is shown in the scheme I given below:

Scheme I
ES 2065269 B l describes a process for the preparation of Salmeterol by reacting 6-(4- phenylbutoxy)]hexyl]benzylamine of Formula 6 with salicylaldehyde derivative of Formula 5 followed by, catalytic hydrogenation of the obtained compound of Formula 7 to give Salmeterol of
Formula I-l. The process is shown in the scheme II given below:
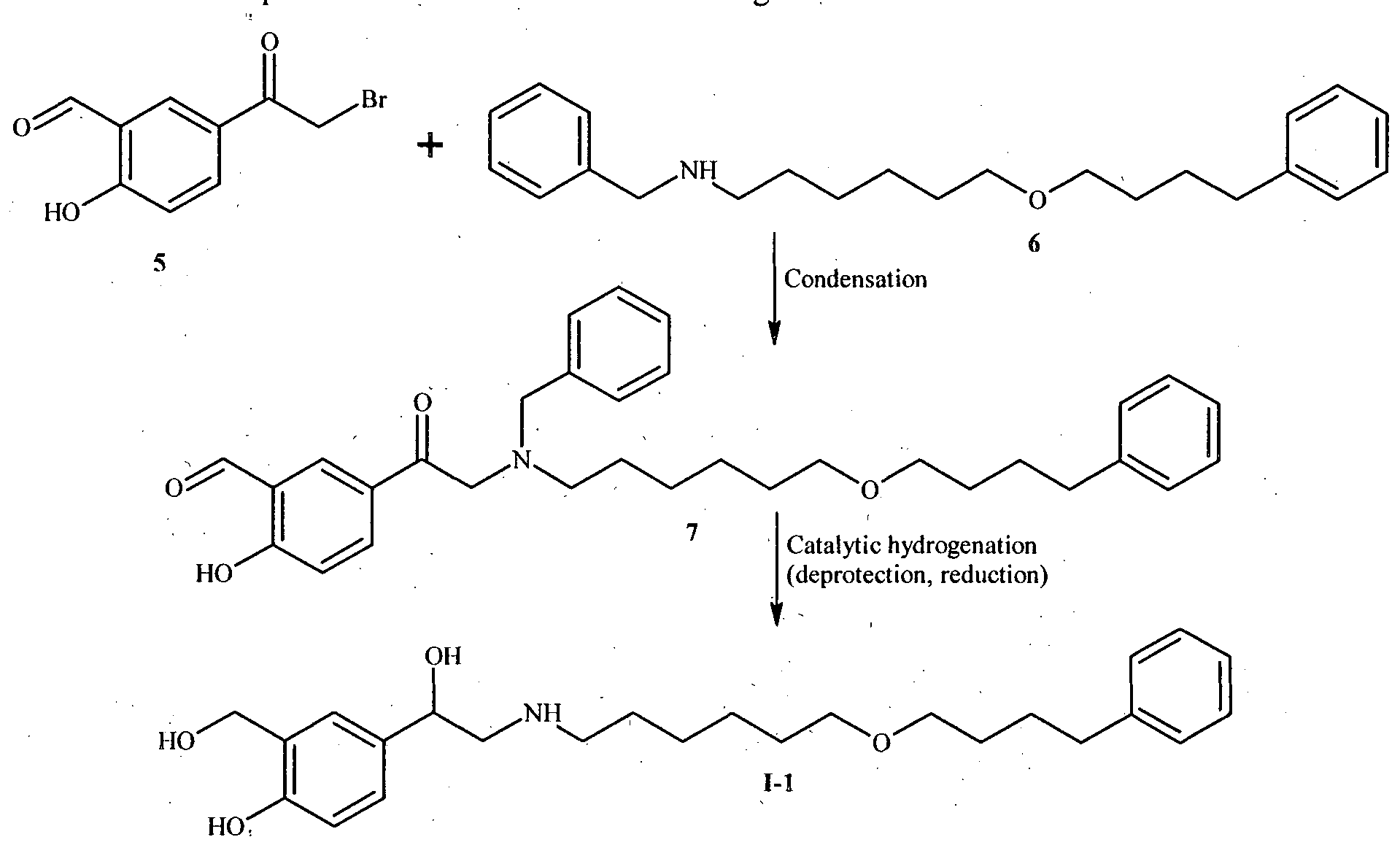
Scheme II
US 8,648,214 B2 discloses a process for preparing Salmeterol or a salt or solvate thereof, comprising the steps of reacting an amine compound of Formula 9 with an aldehyde compound of Formula 8 to obtain compound of Formula 10; followed by reducing the compound of Formula 10 to alcohol compound of Formula 11 in a biphasic solvent system and finally deprotecting the alcohol compound of Formula 11 to obtain Salmeterol of Formula 1-1. The process is shown in the scheme III given below:

wherein X is a leaving group and Pg is a protecting group
Scheme III
The processes described in the above patents are lengthy, cumbersome and results in low yield and purity.
Therefore, there is need of providing an improved process for the preparation of Salmeterol Xinafoate, which is simple, eco-friendly, inexpensive, reproducible and well suited for commercial scale up.
Objective of the Invention
The main objective of the present invention is to provide cost effective and commercially viable process for the preparation of Salmeterol Xinafoate. Another objective of the present invention is to provide a process for the preparation of Salmeterol Xinafoate which employs less expensive, easily available and environment friendly reagents.
Summary of the Invention
Accordingly, the present invention provides an improved process for the preparation of Salmeterol Xinafoate of Formula (I)

which comprises:
i) condensation of com ound of Formula A
 with compound of Formula B,
with compound of Formula B,

in a solvent in the presence of a base to give compound of Formula C;

ii) reducing the compound of Formula C in a monophasic solvent with a reducing agent to obtain compound of Formula D;

iii) debenzylating compound of Formula D with hydrogen and a metal catalyst in a solvent to obtain Salmeterol of Formula I-l and

In another aspect, the present invention provides an improved process for the preparation of compound of Formula D


Detailed Description of the Invention
The main embodiment, of the present invention provides an improved process for the preparation of Salmeterol Xinafoate of Formula (I) as shown in the scheme IV given below:


Scheme IV
Step 1:
Condensation of compound of Formula A with compound of Formula B in a solvent in the presence of a base to give compound of Formula C.
The solvent used for condensation is selected from alcohols such as methanol, ethanol, n-propanol, isopropyl alcohol, isobutanol, n-butanol, tert-butanol and the like; preferably the solvent used is methanol. The base used for condensation is selected from amines such as triethylamine or diisopropylethylamine. Preferably the base used is diisopropylethylamine (DfPEA). The condensation reaction temperature may range from 0 °C to 15 °C and preferably at a temperature in the range from 5 °C to 10 °C. The duration of the reaction may range from 10 hours to 20 hours, preferably for a period of 15 hours.
After the completion of the reaction, the reaction mass is quenched into water and extracted with n-heptane. The n-heptane layer (which contains the compound of Formula C) is further extracted with methanol before proceeding to step-2 to give compound of Formula C in methanol. Step 2:
Reduction of compound of Formula C in methanol with reducing agent to give compound of Formula D.
The reducing agent used for reduction of compound of Formula C to compound of Formula D is selected from sodium borohydride, sodium cyanoborohydride, sodium triacetoxy borohydride, sodium bis(2-methoxyethoxy)aluminum hydride. Preferably the reducing agent used is sodium borohydride. The reduction reaction temperature may range from 20 °C to 35 °C and preferably at a temperature in the range from 25 °C to 30 °C. The duration of the reaction may range from 7 hours to 9 hours, preferably for a period of 8 hours.
Step 3:
Debenzylation of compound of Formula D with hydrogen and a metal catalyst in a solvent to give Salmeterol free base of Formula 1-1.
The debenzylating agent is selected from platinum, platinum oxide, palladium, Raney nickel or rhodium, on a support, such as charcoal or mixtures thereof. Preferably debenzylation is carried out by using palladium on carbon. The solvent used is selected from alcohols such as methanol, ethanol, isopropanol, n-propanol, n-butanol, isobutanol, t-butanol and the like. Preferably the solvent used is methanol. The reaction temperature may range from 20 °C to 30 °C and preferably at a temperature in the range from 25 °C to 28 °C. The duration of the reaction may range from 1 hour to 2 hours, preferably for a period of 1 .5 hours. Salmeterol free base of Formula 1-1 is subjected to purification using a solvent selected from esters such as ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate. Preferably the solvent used is ethylacetate.
Step 4:
Xinafoate salt formation of Salmeterol in a solvent using l -hydroxy-2-napthoic acid to give Salmeterol Xinafoate of Formula (I).
The solvent used for salt formation is selected from alcohols such as methanol, ethanol, isopropanol, n-propanol, n-butanol, isobutanol, t-butanol and the like. Preferably the solvent used is methanol. The reaction temperature may range from 20 °C to 30 °C and preferably at a temperature in the
range from 25 °C to 30 °C. The duration of the reaction may range from 1 hour to 2 hours, preferably for a period of 1 .5 hours.
Salmeterol Xinafoate of Formula (I) is subjected to purification using a solvent selected from alcohols such as methanol, ethanol, isopropanol, n-propanol, n-butanol, isobutanol, t-butanol and the like. Preferably the solvent used is methanol. in a preferred embodiment, the present invention provides an improved process for the preparation of Salmeterol Xinafoate of Formula (I)

i) condensation of com ound of Formula A
 with compound of Formula B,
with compound of Formula B,



iii) debenzylating compound of Formula D with hydrogen and a metal catalyst in a solvent to obtain Salmeterol of Formula 1-1 and

In a most preferred embodiment, the present invention provides an improved process for the preparation of Salmeterol Xinafoate of Formula (I),
 which comprises:
which comprises:
i) condensation of compound of Formula A

with compound of Formula B


ii) reducing the compound of Formula C in methanol solvent with sodium borohydride to obtain compound of Formula D;

iii) debenzylating the compound of Formula D with hydrogen and palladium on carbon in methanol to obtain Salmeterol of Formula I-l and

iv) converting the compound of Formula I-l to Salmeterol Xinafoate of Formula (I).
In another preferred embodiment, the present invention provides an improved process for the preparation of com ound of Formula D,


in a monophasic solvent with a reducing agent to obtain compound of Formula D.
In another most preferred embodiment, the present invention provides an improved process for the preparation of compound of Formula D,

which com rises reducing compound of Formula C

in methanol with sodium borohydride to obtain compound of Formula D.
The following examples describes the nature of the invention and are given only for the purpose of illustrating the present invention in more detail and are not limited and relate to solutions which have been particularly effective on a bench scale.
Examples
Example-1: Preparation of Salmeterol free base of Formula 1-1
Step I: Preparation of 2-hydroxy-5-[[[6-(4-phenylbutoxy)hexylbenzyI]amino] acetyl] benzaldehyde (Formula C):
Mixture of Methylethylketone (1250 mL), benzyl amine derivative (146.66 grams) were taken in round bottom flask and stirred for 1 5 minutes at 25-30 °C. Cooled the reaction mixture to 0 °C and diisopropylethylarnine (DIPEA) (66.66 grams) was added. Bromo acetyl derivative (100 grams) was added lot wise in 2 hours intervals at 0-5 °C. Raised the reaction mixture temperature to 5- 10 °C and stirred for 15 hours. After the completion of the reaction by High Performance Liquid Chromatography (HPLC), the reaction mixture was quenched into purified water (7833 mL) at
20±3 °C. n-Heptane (1000 mL) and vacuum ( 166 grams) were added into reaction mixture at 20±3 °C. Separated the organic layer and washed with purified water (500 mL) and vacuum salt (42.6 grams) at 25-30 °C. Filtered the organic (n-Heptane) layer through (hyflo 25 g with n-Heptane 250 mL) hyflo bed and washed with n-Heptane (333.33 mL). Dried the filtrate over sodium sulphate (83.33 grams) at 25-30 °C. Filtered the sodium sulphate and washed with n-Heptane (125 mL). Extracted the filtrate with methanol (1666.66 mL) at -5 to 0 °C. Separated the n-heptane layer from the methanol ic layer at -5 to 0 °C.
Step 2: Preparation of 4-hydroxy-a'-[[[6-(4-phenylbutoxy)hexyl]benzyIamino]methyl]-l,3- ben- zenedimethanol (Formula D - Benzyl Salmeterol)
Cooled the methanolic layer obtained in Step 1 to -5 °C and sodium borohydride (1 10 grams) was added slowly at -5 °C to 10 °C under nitrogen atmosphere. Raised the temperature of the reaction mixture to 25-30 °C and stirred for 8.0 hours under nitrogen atmosphere. After the completion of the reaction by HPLC, heated the reaction mixture to 30-35 °C. Washed the reaction mixture with n-Heptane (2x833.33 mL) at 30-35 °C. Cooled the methanolic layer to 0-10 °C and purified water (4166.66 mL) was added followed by adjusting the pH to 2-3 with hydrochloric acid at below 10 °C. Distilled the methanol completely under vacuum at below 40 °C. Cooled the reaction mixture to 15-20 °C and Ethylacetate (2333.33 mL) was added followed by adjusting the pH to 7.3-7.7 with saturated sodium bicarbonate solution. Heated the reaction mixture to 25-30 °C, separated the aqueous layer and extracted with Ethylacetate (833.33 mL). The combined ethylacetate layers were washed with purified water (500 mL). Dried the ethylacetate layer over sodium sulphate (83.33 grams), filtered and washed with Ethyl acetate (166.66 mL). Concentrated the filtrate under vacuum at 45 °C to obtain compound of Formula D (Benzyl Salmeterol) as oily residue. Step 3: Preparation of Salmeterol free base (Formula 1-1)
Mixture of methanol (1833 mL) and Benzyl salmeterol compound of Formula D oily residue obtained in Step 2 were taken in hydrogenator at 25 °C under nitrogen atmosphere. Palladium on carbon (33.33 g 20%) was added into the reaction mixture and hydrogen gas was purged at 8-10 psi hydrogen pressure at 25±2 °C. Maintained the reaction mixture for 1 hour 10 minutes at the same temperature. After completion of reaction by Thin layer chromatography (TLC), the reaction mixture was filtered and washed with methanol (166.66 mL). Concentrated the filtrate under
vacumm at below 45 °C and co-distilled with thrice Ethylacetate (3x 1000 mL). Ethylacetate (1000 mL) was added into reaction mixture and stirred for dissolution. Cooled the reaction mixture to - 5±2 °C and stirred for 2 hours. Salmeterol base (83 mgrams) was added into reaction mixture and stirred for 10-30 hours at -5±2 °C. Filtered the product and washed with chilled Ethylacetate (83.33 mL). Dried the product in vacuum tray dryer for 8 hours at 40-45 °C to obtain 57 grams of Salmeterol free base.
Step 4: Purification of Salmeterol free base (Formula 1-1)
Salmeterol base (57 grams) and Ethylacetate (456 mL) were taken in round bottom flask. Heated the reaction mixture to 40-45 °C and stirred for 30 min. Cooled the reaction mixture to 0±2 °C and stirred for 1 hour. Filtered the product and washed with chilled ethylacetate (22.8 mL). Wet compound (65 grams) and Ethylacetate (456 mL) were taken in round bottom flask. Heated the reaction mixture to 40-45 °C and stirred for 30 min. Cooled the reaction mixture to 0±2 °C and stirred for 1 hour. Filtered the product and washed with chilled ethylacetate (22.8 mL). Dried the product in vacuum tray dryer for 12 hours at 40-45 °C to obtain pure Salmeterol base (45 grams). Ί INMR (DMSO, 300 MHz): 1.26-1.62 (m, 12H), 2.49-2.59 (m, 6H), 3.28-3.36 (m 4H), 4.46-4.51 (m, 3H), 5.03, (br s l H), 6.67-6.70 (d I ), 6.96-6.99 (dd l H), 7.13-7.18 (m 3H), 7.24-7.28 (m 3H). MS: m/z = 416 [M+H]+. Example-2: Preparation of Salmeterol Xinafoate
Step 1 : Preparation of Salmeterol Xinafoate
Mixture of Salmeterol base (30 grams) and methanol (150 mL) were taken in round bottom flask and stirred for clear solution at 25-30 °C. Activated carbon (3.0 grams) was added and stirred for 30 minutes at 25-30 °C. Filtered the reaction mixture through hyflo bed and washed with methanol (30 mL). l -hydroxy-2-napthoic acid ( 13.59 grams) was added to the filtrate at 30±2 °C. Stirred the reaction mixture for 1.0 hour at 30±2 °C and cooled to 18-22 °C. Stirred the reaction mixture for 2.0 hours and the product was filtered, slurry washed with chilled methanol (60 mL). Dried the product in vacuum tray dryer for 12 hours at 40-45 °C to obtain of Salmeterol Xinafoate (36 grams).
Step 2: Purification of Salmeterol Xinafoate
Mixture o Salmeterol Xinafoate (25 grams) and methanol (150 mL) were taken in round bottom flask and heated at 45-50 °C to get clear solution. Activated carbon (Norit Darco G60) (1.25 grams) was added and stirred for 15 minutes at 45-50 °C. Filtered the reaction mixture was through hyflow bed and washed with methanol (8.33 mL). The filtrate was taken into round bottom flask and heated at 45-50 °C to get clear solution. Cooled the reaction mixture slowly to 20±2 °C and stirred for 4.0 hours. Filtered the precipitated product and slurry washed with chilled methanol (25 mL). Dried the product in vacuum tray dryer for 12 hours at 40-45 °C to obtain pure Salmeterol Xinafoate (20.75 gram's).
,'HNMR (DMSO, 300 MHz): 1.29-1.64 (m, 12H), 2.50-2.58 (m, 2H), 2.91-3.10 (m 4H), 3.27-3.34 (m, 4H), 4.5 (s 2H), 4.83-4.87 (d 2H), 5.05 (br s I H), 6.13 (br s 1 H), 6.77-6.79 (d 1H), 7.02-7.08 (m 2H), 7.13-7.18 (m 3H), 7.23-7.28 (m 3H), 7.36-7.40 (m 2H), 7.43-7.48 (m I H), 7.70-7.79 (m 2H), 8.19-8.22 (d I H), 8.82 (br s IH), 9.52 (br s IH).
MS: m/z = 416 [M+H]+.
Advantages of the present invention:
1. Extraction of compound of Formula C in n-heptane layer to methanol ic layer improved the purity of the compound of Formula C as the impurities were removed in the n-Heptane layer.
2. Reduction reaction completed with 1st time TLC.
3. Purity of Salmeterol Xinafoate is > 99.5%.
4. Consistency in yields.
Claims
We Claim:


ii. reducing the compound of Formula C in a monophasic solvent with a reducing agent to obtain compound of Formula D;


2. The process as claimed in (step i) of claim 1, wherein the base used in the condensation is diisopropylethylamine and the solvent used is methylethylketone.
3. The process as claimed in (step ii) of claim 1 , where in the reducing agent used for reduction of compound of Formula C to compound of Formula D is sodium borohydride.
4. The process as claimed in (step ii) of claim 1 , where in the solvent used for reduction is methanol.
5. The process as claimed in (step iii) of claim 1 , wherein the metal catalyst used for debenzylation of compound of Formula D to Salmeterol of Formula I-l is palladium on carbon.
6. The process as claimed in (step iii) of claim 1 , wherein the solvent used for debenzylation is methanol.
7. The process as claimed in (step iv) of claim 1 , wherein the solvent used for converting the Salmeterol of Formula 1-1 to Salmeterol Xinafoate of Formula (I) is methanol.
8. An im roved process for the preparation of compound of Formula D,

9. The process as claimed in claim 8, where in the reducing agent used for reduction of compound of Formula C to compound of Formula D is sodium borohydride.
10. The process as claimed in claim 8, where in the solvent used for reduction is methanol.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN20164101105 | 2016-03-30 | ||
| IN20164101105 | 2016-03-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017168431A1 true WO2017168431A1 (en) | 2017-10-05 |
Family
ID=59963627
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2016/000136 WO2017168431A1 (en) | 2016-03-30 | 2016-05-26 | Process for the preparation of salmeterol xinafoate |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2017168431A1 (en) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8648214B2 (en) * | 2005-10-17 | 2014-02-11 | Generics [Uk] Limited | Processes suitable for the preparation of salmeterol |
-
2016
- 2016-05-26 WO PCT/IN2016/000136 patent/WO2017168431A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8648214B2 (en) * | 2005-10-17 | 2014-02-11 | Generics [Uk] Limited | Processes suitable for the preparation of salmeterol |
Non-Patent Citations (1)
| Title |
|---|
| "Contemporary Drug Synthesis", ISBN: 0-47 1 -2 1 480-9, article JIE-JACK LI ET AL., pages: 206 - 207 * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5599994A (en) | Amino acid-derived diaminopropanols | |
| US9035061B2 (en) | Process for preparing a biphenyl-2-ylcarbamic acid | |
| CN101959870B (en) | Preparation of morpholine derivatives | |
| CA2884197C (en) | Methods for the preparation of indacaterol and pharmaceutically acceptable salts thereof | |
| US20110178326A1 (en) | Unsaturated cinacalcet salts and processes for preparing cinacalcet hydrochloride | |
| JP7685292B2 (en) | Edoxaban key intermediate and its synthesis method | |
| WO2013049605A1 (en) | Processes for the preparation of an intermediate in the synthesis of eltrombopag | |
| KR101308258B1 (en) | A novel method of making Endoxifen | |
| JP2006249095A (en) | Amino acid-derived diaminopropanol | |
| WO2005061446A2 (en) | Processes for the preparation of aminoalkyl phenylcarbamates | |
| CA2960473A1 (en) | Processes for the preparation of tadalafil and intermediates thereof | |
| US20180237386A1 (en) | Process For Preparation Of Vortioxetine Hydrobromide | |
| WO2017168431A1 (en) | Process for the preparation of salmeterol xinafoate | |
| EP2178864B1 (en) | Process for the preparation of alfuzosin hydrochloride | |
| WO1999052855A1 (en) | A process for preparing chiral (s)-2,3-disubstituted-1-propylamine derivatives | |
| TW500721B (en) | Process for the preparation of 3-N,N-dicyclobutylamino-8-fluoro-3,4-dihydor-2H-1-benzopyran-5-carboxamide | |
| ES2957319T3 (en) | Methods for producing (6S,15S)-3,8,13,18-tetraazalcosane-6,15-diol | |
| EP0129383B1 (en) | Process for the preparation of 3,4-di-isobutyryloxy-n-methyl-phenethylamine | |
| US8629146B2 (en) | Method for stereoselective synthesis of bicyclic heterocyclic compounds | |
| JPS597709B2 (en) | Novel morpholine derivative and its production method | |
| JP2016511761A (en) | Method for synthesizing 4-piperidin-4-yl-benzene-1,3-diol and salts thereof, and novel compound tert-butyl 4- (2,4-dihydroxy-phenyl) -4-hydroxy-piperidine-1-carboxylate | |
| US20140256963A1 (en) | Process for the preparation of aliskiren | |
| KR101974388B1 (en) | Alkyl diethylene triamine derivatives and a process of the preparation thereof | |
| WO2025047770A1 (en) | Method for producing deoxynyboquinone derivative | |
| CN119176757A (en) | Preparation method of mianserin hydrochloride intermediate and derivative thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16896675 Country of ref document: EP Kind code of ref document: A1 |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16896675 Country of ref document: EP Kind code of ref document: A1 |