WO2017083867A1 - Ion channel inhibitory compounds, pharmaceutical formulations and uses - Google Patents
Ion channel inhibitory compounds, pharmaceutical formulations and uses Download PDFInfo
- Publication number
- WO2017083867A1 WO2017083867A1 PCT/US2016/061918 US2016061918W WO2017083867A1 WO 2017083867 A1 WO2017083867 A1 WO 2017083867A1 US 2016061918 W US2016061918 W US 2016061918W WO 2017083867 A1 WO2017083867 A1 WO 2017083867A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- unsubstituted
- azaspiro
- compound
- pharmaceutically acceptable
- Prior art date
Links
- 0 CCCCc(cc1)ccc1OC*CCCC1CCOCC1 Chemical compound CCCCc(cc1)ccc1OC*CCCC1CCOCC1 0.000 description 18
- BICUCGPTPDFRIK-UHFFFAOYSA-N CNC(c1cc(Cl)cc(Cl)c1)=O Chemical compound CNC(c1cc(Cl)cc(Cl)c1)=O BICUCGPTPDFRIK-UHFFFAOYSA-N 0.000 description 2
- IPUWGHBPSVVPML-UHFFFAOYSA-N O=C(c1ccccc1)ON1CCC2(CC2)CC1 Chemical compound O=C(c1ccccc1)ON1CCC2(CC2)CC1 IPUWGHBPSVVPML-UHFFFAOYSA-N 0.000 description 2
- VAWJZOCSSWEFGT-UHFFFAOYSA-N CC(C)(C)C(CN(CC1)CCC1(C1)C1NC(c1cc(Cl)cc(Cl)c1)=O)O Chemical compound CC(C)(C)C(CN(CC1)CCC1(C1)C1NC(c1cc(Cl)cc(Cl)c1)=O)O VAWJZOCSSWEFGT-UHFFFAOYSA-N 0.000 description 1
- ZGJWDBFSLCWTSL-UHFFFAOYSA-N CC(C)(C)CCN(CC1)CCC1(C1)C1N(C)C(c1cc(Cl)cc(Cl)c1)=O Chemical compound CC(C)(C)CCN(CC1)CCC1(C1)C1N(C)C(c1cc(Cl)cc(Cl)c1)=O ZGJWDBFSLCWTSL-UHFFFAOYSA-N 0.000 description 1
- BCTAIKQJLQIDSY-UHFFFAOYSA-N CC(C)(C)NC(CN(CC1)CCC1(C1)C1NC(c1cc(Cl)cc(Cl)c1)=O)=O Chemical compound CC(C)(C)NC(CN(CC1)CCC1(C1)C1NC(c1cc(Cl)cc(Cl)c1)=O)=O BCTAIKQJLQIDSY-UHFFFAOYSA-N 0.000 description 1
- BCTAIKQJLQIDSY-INIZCTEOSA-N CC(C)(C)NC(CN(CC1)CCC1(C1)[C@H]1NC(c1cc(Cl)cc(Cl)c1)=O)=O Chemical compound CC(C)(C)NC(CN(CC1)CCC1(C1)[C@H]1NC(c1cc(Cl)cc(Cl)c1)=O)=O BCTAIKQJLQIDSY-INIZCTEOSA-N 0.000 description 1
- NQVKUBIPDCUQTC-UHFFFAOYSA-N CC(C)(C)NC(CN1CCC(C)(CNC(c2cccc(OC)c2)=O)CC1)=O Chemical compound CC(C)(C)NC(CN1CCC(C)(CNC(c2cccc(OC)c2)=O)CC1)=O NQVKUBIPDCUQTC-UHFFFAOYSA-N 0.000 description 1
- VFYQSLCBRGDSNO-UHFFFAOYSA-N Fc1ccc(CCS)cc1 Chemical compound Fc1ccc(CCS)cc1 VFYQSLCBRGDSNO-UHFFFAOYSA-N 0.000 description 1
- DLGXOZCUHNFARA-UHFFFAOYSA-N N#CC1(COC(c2ccccc2)=O)CCN(CC(NI)=O)CC1 Chemical compound N#CC1(COC(c2ccccc2)=O)CCN(CC(NI)=O)CC1 DLGXOZCUHNFARA-UHFFFAOYSA-N 0.000 description 1
- DELNZTRPJTUOIP-UHFFFAOYSA-N NC(c1cc(Cl)cc(Cl)c1)=O Chemical compound NC(c1cc(Cl)cc(Cl)c1)=O DELNZTRPJTUOIP-UHFFFAOYSA-N 0.000 description 1
- OHDHZWCGXUKAOW-UHFFFAOYSA-N NCC1(COC(c2ccccc2)=O)CCN(CC(Nc2ccccc2)=O)CC1 Chemical compound NCC1(COC(c2ccccc2)=O)CCN(CC(Nc2ccccc2)=O)CC1 OHDHZWCGXUKAOW-UHFFFAOYSA-N 0.000 description 1
- QLUFMSCHWIQZCV-UHFFFAOYSA-N OC(CN(CC1)CCC1(C1)C1NC(c1cc(Cl)cc(Cl)c1)=O)c1ccc(C(F)(F)F)cc1 Chemical compound OC(CN(CC1)CCC1(C1)C1NC(c1cc(Cl)cc(Cl)c1)=O)c1ccc(C(F)(F)F)cc1 QLUFMSCHWIQZCV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D221/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00
- C07D221/02—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00 condensed with carbocyclic rings or ring systems
- C07D221/20—Spiro-condensed ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4453—Non condensed piperidines, e.g. piperocaine only substituted in position 1, e.g. propipocaine, diperodon
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4747—Quinolines; Isoquinolines spiro-condensed
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/26—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
- C07D211/58—Nitrogen atoms attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the field of the present invention relates to ion channel modulators (inhibitors or antagonists of one type ion channel and/or combination of inhibition of multiple ion channels), compositions including ion channel modulators, and methods of treating conditions and disorders using the compounds and compositions.
- a more particular field involves compounds having selective T-type Ca v3 channel inhibitory effects for mitigating neuropathic and/or inflammatory pain, pharmaceutical formulations including such compounds, and methods for selective treatment of neuropathic and inflammatory pain, other neurological disorders, such epilepsy, essential tremor, migraine and Parkinson disease, and psychiatric disorders, e.g., anxiety, depression and schizophrenia.
- Voltage-gated calcium (Ca 2+ ) channels play an integral role in the regulation of membrane ion conductance, cellular excitability and neurotransmitter release.
- VGCC are composed of the pore-forming al subunit and auxiliary ⁇ 2 ⁇ ppm and ⁇ subunits that modulate channel expression and function.
- the Ca v3 channel subtype which mediates T-type calcium currents that may be targeted for treatment of epilepsy, especially children absence epilepsy and chronic pain (Huguenard, 1998, Cribbs et al., 2000, Perez-Reyes et al., 2009, Perez-Reyes, 2010).
- T-type channels are activated at relatively negative membrane potentials ( ⁇ -60 mV).
- Ca 2+ influx through T channels triggers low-threshold Ca 2+ spikes, which in turn elicit a burst of action potentials mediated by voltage-gated sodium (Na + ) channels.
- T channels can be activated by mild depolarization of the cell membrane (Talley et al., 1999, Perez-Reyes, 2003, Perez-Reyes, 2010, Pexton et al., 2011, Todorovic and Jevtovic-Todorovic, 2011).
- the Ca V 3.2 channel is highly expressed in dorsal root ganglion (DRG) neurons, whereas little Ca V 3 . i and virtually no Ca v3 .3 are expressed in the small diameter DRG neurons (Nelson et al., 1992).
- the Ca v3 .2 channels are also expressed at a lower level in several non-neuronal tissues, including heart, liver, kidney, and pituitary. Both diabetic neuropathy and chronic constriction injury models in rats lead to DRG neuron-specific upregulation of the Ca V 3.2 channel and the T current density.
- T-type channel inhibitors have two known uses in the clinic.
- the anti-absence seizure effects of ethosuximide and lamotrigine are thought to be mediated by the inhibition of T channel activity in the thalamus (Gomora et al., 2001, Huguenard, 2002).
- both drugs are weak and not specific against the T channel (Xie et al., 1995, Zhang et al., 1996).
- the antihypertensive effect of mibefradil is conventionally attributed to its inhibition of the T channel.
- T-type has poor selectivity with about 3-10 times more potent inhibition of the T-type than of the L-type Ca 2+ current or the voltage-gated Na + current (Avdonin et al., 2000). Because there are no selective T channel blockers, it is unclear whether and to what extend the inhibition of T channel activity at therapeutically relevant concentrations contributes to the therapeutic usefulness of a wide range of drugs, such as analgesics, antiepileptics, neuroprotectants, antipsychotics, antidepressants, antiarrhythmics and antihypertensives.
- analgesics such as analgesics, antiepileptics, neuroprotectants, antipsychotics, antidepressants, antiarrhythmics and antihypertensives.
- T-channel particularly the Ca v3 2 isoform
- Ca v3 2 isoform Targeting a T-channel, particularly the Ca v3 2 isoform, would be highly useful in reduction of thermal hyperalgesia and mechanical allodynia under pathological conditions, for example diabetic neuropathy.
- Several efforts to discover potent and selective T-type Ca 2+ channels have been described in the literature, as exemplified below.
- TTA-A2 suppresses active wake, promotes slow-wave sleep (Kraus et al., 2010), and prevents weight gain in mice on a high-fat diet (Uebele et al., 2009).
- ML218 (3,5-Dichloro-N-[[(la,5a,6-exo,6a)- 3-(3,3-dimethylbutyl)-3-azabicyclo[3.1.0]hex-6-yl]methyl]benzamide, CID 45115620) a selective T-Type Ca 2+ inhibitor.
- ML218 possess acceptable in vivo rat PK and was efficacious in a preclinical Parkinson model.
- ML218 is a useful new biologic probe to study T-Type Ca 2+ function in vitro and in vivo (Xie et al., 2010, Xiang et al., 2011).
- lactam acetamides have been described by Abbott and others as Ca v2 2 and Ca v3 2 calcium channel blockers, and ABT-639 has been reported as a Ca v3 2 calcium channel blocker for treatment of diabetic neuropathic pain through peripheral action, because ABT- 639 is presumed to not penetrate the blood brain barrier (Jarvis et al., 2014).
- N-Piperidinyl acetamide derivatives as calcium channel blockers have been described by Zalicus Pharmaceuticals, Ltd. [US8569344 (2013); US8377968 (2013)].
- a piperidine-based compound, Z944 inhibits Ca v3 channels in a voltage-dependent manner and is able to attenuate thalamic burst firing and suppress absence seizures in rats (Tringham et al., 2012).
- Z944 has shown promising results in clinical Phase I studies of pain in humans (Lee, 2014).
- T-type Ca channel inhibitors have been discovered and have advanced to different stages of development, no FDA-approved selective T-type channel inhibitory compounds are available for clinical applications.
- the current invention provides compounds, formulations and methods of useful for the treatment and prevention of neurological and psychiatric disorders and diseases, especially neuropathic pain and inflammatory pain, in which key ion channels, particularly the T-type Ca 2+ channels are involved.
- the compounds and methods can be used in both human and veterinary medicine.
- the compound or formulation contains the components expressly listed, and may contain other components which do not substantially affect the condition being treated. That is, the compound or formulation either does not contain any other components which do substantially affect the condition being treated other than those components expressly listed; or, if the compound or formulation does contain extra components other than those listed which substantially affect the condition being treated, the compound or formulation does not contain a sufficient concentration or amount of those extra components to substantially affect the condition being treated.
- FIG. 1A Examples of compounds that mitigate neuropathic pain induced by Spared Nerve Injury (SNI) in mice.
- SNI Spared Nerve Injury
- Each SNI mouse received either EXAMPLE (EX)-l 1, -14, -38, -62, or 113, or gabapentin, as benchmark (each 30 mg/kg, 0.2 mL I P ), or vehicle (0.2 mL I P. 2% DMSO in 0.5% Hydroxyl Propyl Cellulose, HPC).
- FIG. IB Mechanical pain threshold assessment (using von Frey monofilament test) of the same cohort of SNI mice's left hindpaws.
- Application of EXAMPLE-11, -14, -38, -62, or 113 (or gabapentin) reduced allodynia compared to the SNI-vehicle control group.
- FIG. 2A Other examples of compounds that alleviate neuropathic pain induced by SNI in another cohort of mice.
- Each SNI mouse received either EXAMPLE-12, -16, -19, -39, or -44 (each 30 mg/kg, 0.2 mL I P ), or vehicle (0.2 mL I P. 2% DMSO in 0.5% HPC).
- FIG. 2B Mechanical pain threshold assessment (von Frey monofilament test) of the same cohort of SNI mice's left hindpaws.
- Application of EXAMPLE-12, -16, -19, -39, or - 44 reduced allodynia compared to the SNI-vehicle control group.
- FIG. 3B Mechanical pain threshold assessment (von Frey monofilament test) of SNI rats' left hindpaws.
- Each SNI rat received either EXAMPLE-106, -107, and -115 (each 30 mg/kg, 1 mL I P ), or vehicle (1 mL I P. 2% DMSO in 0.5% HPC). Sham rats also received vehicle LP. injections.
- FIG. 4 Mechanical pain threshold assessment (von Frey monofilament test) of left hindpaws in another cohort of SNI rats.
- Each SNI rat received either EXAMPLE-128, -130, - 132, or Z944 (as a reference compound, each 10 mg/kg, 2 mL oral gavage, P.O.).
- FIG. 5A Mechanical pain threshold assessment (von Frey monofilament test) of spinal nerve ligation (SNL) rats' left hindpaws.
- SNL rat received either EXAMPLE- 128, at 3, 10 or 30 mg/kg (2 mL oral gavage, P.O.), or vehicle (2 mL 2% DMSO in 0.5% HPC, P.O.).
- FIG. 5B Mechanical pain threshold assessment (von Frey monofilament test) of SNL rats' left hindpaws. In the same cohort of animals, each SNL rat received either
- EXAMPLE-128, -130, Z944 (as a reference compound, each at 30 mg/kg, 2 mL oral gavage, P.O.), or vehicle (2 mL 2% DMSO in 0.5% HPC, P.O.).
- the degree of analgesia against allodynia in the order of EXAMPLE-128 > Z944 > EXAMPLE- 130 compared to the SNI- vehicle group. Mechanical pain responses were measured at 1, 2, 4 and 6 hours after treatment (n 8 rats per treatment group).
- FIG. 7 Effects of EXAMPLE-96, 107 or 115 (each 30 mg /kg, I P.) or gabapentin (as a benchmark, 100 mg/kg, LP) on Complete Freund's Adjuvant (CFA)-induced
- Mechanical pain threshold assessment of CFA mice's left hindpaws were measured using von Frey monofilament test at 0.5, 1, 2, 4, and 6 hours after treatment.
- the test compound was LP. administered 30 min prior to local injection of 10% formalin into the left hindpaw (10 ⁇ ). Time spent in grooming, licking or biting the injury paw was manually scored immediately following formalin injection in 5-min time bin for 45 min.
- Mechanical pain threshold assessment von Frey monofilament test of the mice's incision left hindpaws.
- Each mouse received either EXAMPLE-116, -118, or ML218 (as a reference compound, each 30 mg /kg, LP.) or vehicle (2% DMSO in 0.5% HPC, LP.). Mechanical pain responses were measured at 0.5, 1, 2, 4, and 5 hours after treatment.
- PTZ pentylenetetrazol
- mice Sixty minutes (min) prior to receiving PTZ (40 mg/kg, LP.), each mouse was pretreated with either an Example compound, ML218, Z944 or vehicle (2% DMSO in 0.5% HPC). Fatality rate (%) of mice in each group that dies within the 20 minutes observation period - was calculated. Both fatality rate and latency (data not shown) to death were used to evaluate the Example compound's ability to prevent death resulting from PTZ-induced seizures. A cutoff of 20 minutes was set for observational and statistical analysis purposes.
- FIG. 11 Effects of EXAMPLE- 128 on baclofen-induced absence seizures in mice.
- R-baclofen (5 mg/kg, LP.) was administered to mice (C57BL/6, male, 2 months) caused abnormal brain seizure activity so-called “spike wave discharges" (SWD) revealed by the electroencephalogram (EEG, indicated by an arrow), mimicking absence seizures.
- SWD spike wave discharges
- FIG. 12 Effects of EXAMPLE-128 on harmaline-induced essential tremor model in mice. Harmaline was dissolved in saline and administrated at 10 mg/kg (S.C.) mice (C57BL/6, male, 3 - 5 months). Ten minutes after tremors occurred, mice were treated with either EXAMPLE-128 (30 mg/kg, LP.) or vehicle (2% DMSO in 0.5% HPC). Tremor episodes numbers and each episode duration were offline human scored; EXAMPLE-128 significantly reduced the total tremor occurring time in a period of 60 minutes following the LP. drug treatment.
- Compound of the invention refers to the compounds discussed herein and salts (e.g. pharmaceutically acceptable salts) of these compounds.
- Alkyl is intended to embrace a univalent saturated linear or branched
- Alkylene refers to a similar group, which is divalent.
- Optionally substituted alkylene groups can be unsubstituted, or substituted in the same manner as substituted alkyl groups.
- Cycloalkyl is intended to embrace a univalent saturated cyclic hydrocarbon chain having the number of carbon atoms specified, or if no number is specified, having 3 to 10 carbon atoms, or 3 to 8 carbon atoms, preferably 3 to 6 carbon atoms.
- Cycloalkylene refers to a similar group, which is divalent. Cycloalkyl and cycloalkylene groups can be unsubstituted, or substituted in the same manner as substituted alkyl groups.
- Alkenyl is intended to embrace a univalent linear or branched hydrocarbon chain having at least one carbon-carbon double bond, and having the number of carbon atoms specified, or if no number is specified, having 2 to 8 carbon atoms.
- Alkenylene refers to a similar group, which is divalent. Alkenyl and alkenylene groups can be unsubstituted, or substituted in the same manner as substituted alkyl groups where chemically possible.
- Cycloalkenyl is intended to embrace a univalent cyclic hydrocarbon chain having at least one carbon-carbon double bond and having the number of carbon atoms specified, or if no number is specified, having 4 to 10 carbon atoms, or 4 to 8 carbon atoms, or 4 to 6 carbon atoms.
- Cycloalkenylene refers to a similar group, which is divalent. Cycloalkenyl and cycloalkenylene groups can be unsubstituted, or substituted in the same manner as substituted alkyl groups where chemically possible.
- Alkynyl is intended to embrace a univalent linear or branched hydrocarbon chain having at least one carbon-carbon triple bond, and having the number of carbon atoms specified, or if no number is specified, having 2 to 8 carbon atoms.
- Alkynylene refers to a similar group, which is divalent. Alkynyl and alkynylene groups can be unsubstituted, or substituted in the same manner as substituted alkyl groups where chemically possible.
- Aryl is defined as a univalent aromatic ring system.
- Aryl groups include monocyclic aromatic rings and polycyclic aromatic ring systems containing the number of carbon atoms specified, or if no number is specified, containing six to twenty carbon atoms. In other embodiments, aryl groups may contain six to ten carbon atoms.
- Non-limiting examples of aryl and heteroaryl groups include phenyl, 1-naphthyl, 2-naphthyl, 1 -pyrrol yl, 2- pyrrolyl, 3 -pyrrol yl, 3-pyrazolyl, 2-imidazolyl, 4-imidazolyl, pyrazinyl, 2-oxazolyl, 4- oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2-thiazolyl, 4-thiazolyl, 5- thiazolyl, 2-furyl, 3-furyl, 2-thienyl, 3-thienyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl, 4- pyrimidyl, 5-benzothiazolyl, purinyl, 2-benzimidazolyl, 5-indolyl, 1-isoquinolyl, 5- isoquinolyl,
- aryl groups can be unsubstituted. In other embodiments, aryl groups can be substituted with, for example, one, two, three or more substituents
- any of the aryl and heteroaryl groups is optionally substituted, e.g., with one or more groups referred to herein as an "aryl group substituent".
- aryl group substituent refers to a similar group, which is divalent.
- heteroaryl refers to aryl groups (or rings) that contain from one to four heteroatoms selected from N, O, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized.
- a heteroaryl group can be attached to the remainder of the molecule through a heteroatom.
- Hydrocarbyl is defined as a univalent hydrocarbon group, that is, a group comprised of hydrogen and carbon, whether aliphatic or aromatic, acyclic or cyclic, or any combination of, or all of, aliphatic, aromatic, acyclic and cyclic. Hydrocarbyl groups have the number of carbon atoms specified, or if no number is specified, having 1 to 10 carbon atoms. "Hydrocarbylene” refers to a similar group, which is divalent. Hydrocarbyl and hydrocarbylene groups can be unsubstituted, or substituted in the same manner as substituted alkyl groups where chemically possible.
- Heterocycle or a “heterocyclic group” is defined as a ring system which contains the number of carbon atoms specified, and one or more heteroatoms (such as one to six heteroatoms, or one to three heteroatoms, or one heteroatom), where heteroatoms include, but are not limited to, oxygen, nitrogen, sulfur, and phosphorus.
- Heteroaryl is defined as an aromatic ring system which contains the number of carbon atoms specified, and one or more heteroatoms (such as one to six heteroatoms, or one to three heteroatoms, or one heteroatom), where heteroatoms include, but are not limited to, oxygen, nitrogen, sulfur, and phosphorus; heteroaryl groups are a subset of heterocyclic groups.
- heteroatoms for heterocyclyl and heteroaryl groups are selected from the group consisting of oxygen and nitrogen.
- heterocyclic groups may contain two to twenty carbon atoms and one to six heteroatoms, two to twelve carbon atoms and one to four heteroatoms, two to twelve carbon atoms and one to three heteroatoms, two to ten carbon atoms and one to three heteroatoms, two to six carbon atoms and one to three heteroatoms, or two to six carbon atoms and two to four heteroatoms.
- heterocyclic groups can be unsubstituted.
- heterocycles include aziridine, oxirane, oxetane, azetidine, pyrrolidine, pyrrole, tetrahydrofuran, furan, thiolane, thiophene, imidazolidine, imidazole, pyrazolidine, pyrazole, 1,2,3-triazole, 1,2,4-triazole, piperidine, pyridine, pyran, piperazine, and
- a “heteroalkyl” group is defined as a univalent hydrocarbyl group, where one or more of the carbon atoms have been independently replaced by a heteroatom at any chemically possible location, where heteroatoms include, but are not limited to, oxygen, nitrogen, sulfur, and phosphorus. Heteroalkyl groups have the number of carbon atoms specified, or if no number is specified, having 1 to 10 carbon atoms, and also at least one heteroatom, such as 1 to 5 heteroatoms, 1 to 4 heteroatoms, 1 to 3 heteroatoms, 1 to 2 heteroatoms, or one heteroatom. "Heteroalkylene” refers to a similar group, which is divalent.
- Heteroalkyl and heteroalkylene groups can be unsubstituted, or substituted in the same manner as substituted alkyl groups where chemically possible.
- heteroalkyl and heteroalkylene groups include, but are not limited to, ethylene glycol and polyethylene glycol moieties, such as (-CH 2 CH 2 -0) n -H (a monovalent heterohalkyl group) and (-CH 2 CH 2 -0-)n (a divalent heteroalkylene group) where n is an integer from 1 to 12 inclusive, and propylene glycol and polypropylene glycol moieties, such as
- heteroatom(s) O, N and S and Si may be placed at any interior position of the heteroalkyl group or at the position at which the alkyl group is attached to the remainder of the molecule.
- heteroalkylene by itself or as part of another substituent means a divalent radical derived from heteroalkyl, as exemplified, but not limited by, -CH 2 -CH 2 -S-CH 2 -CH 2 - and - CH 2 -S-CH 2 -CH 2 - H-CH 2 -.
- heteroatoms can also occupy either or both of the chain termini (e.g., alkyleneoxy, alkylenedioxy, alkyl eneamino,
- alkylenediamino and the like. Still further, for alkylene and heteroalkylene linking groups, no orientation of the linking group is implied by the direction in which the formula of the linking group is written. For example, the formula -C0 2 R'- represents both -C(0)OR' and - OC(0)R' .
- groups described above can be attached to the remainder of the molecule at any chemically possible location on the fragment, including attachment via a substituent when the group is substituted.
- groups are typically attached by replacement of a hydrogen, hydroxyl, methyl, or methoxy group on a "complete" molecule to generate the appropriate fragment, and a bond is drawn from the open valence on the fragment to the remainder of the molecule.
- attachment of the heteroalkyl group -CH 2 -0-CH 3 proceeds by removal of a hydrogen from one of the methyl groups of CH 3 -0-CH 3 , to generate the heteroalkyl fragment -CH 2 -0-CH 3 , from which a bond is drawn from the open valence to the remainder of the molecule.
- Reference to "about” a value or parameter herein includes (and describes) variations that are directed to that value or parameter per se. For example, description referring to "about X” includes description of "X”.
- the description is intended to embrace all salts of the compounds described herein, as well as methods of using such salts of the compounds.
- the salts of the compounds comprise pharmaceutically acceptable salts.
- Pharmaceutically acceptable salts are those salts which can be administered as drugs or pharmaceuticals to humans and/or animals and which, upon administration, retain at least some of the biological activity of the free compound (neutral compound or non-salt compound).
- the desired salt of a basic compound may be prepared by methods known to those of skill in the art by treating the compound with an acid. Examples of inorganic acids include, but are not limited to, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, and phosphoric acid.
- organic acids include, but are not limited to, formic acid, acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, sulfonic acids, and salicylic acid.
- Salts of basic compounds with amino acids, such as aspartate salts and glutamate salts can also be prepared.
- the desired salt of an acidic compound can be prepared by methods known to those of skill in the art by treating the compound with a base.
- inorganic salts of acid compounds include, but are not limited to, alkali metal and alkaline earth salts, such as sodium salts, potassium salts, magnesium salts, and calcium salts; ammonium salts; and aluminum salts.
- organic salts of acid compounds include, but are not limited to, procaine, dibenzylamine, N-ethylpiperidine, ⁇ , ⁇ '- dibenzylethylenediamine, and triethylamine salts. Salts of acidic compounds with amino acids, such as lysine salts, can also be prepared. For lists of pharmaceutically acceptable salts, see, for example, P. H. Stahl and C. G.
- terapéuticaally effective amount means that amount of a compound, material, or formulation comprising a compound of the present invention which is effective for producing some desired therapeutic effect by inhibition of a T-Channel in at least a sub-population of cells in an animal, thereby blocking or lessening the biological consequences of that pathway in the treated cells, at a reasonable benefit/risk ratio applicable to any medical treatment.
- a T-Channel in at least a sub-population of cells in an animal
- neurodegenerative diseases e.g., Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis
- neuropsychiatric diseases e.g. Schizophrenia and anxieties, such as general anxiety disorder
- exemplary neurological disorders include MLS (cerebellar ataxia), Huntington's disease, Down syndrome, multi-infarct dementia, status epilepticus, contusive injuries (e.g. spinal cord injury and head injury), viral infection induced neurodegeneration, (e.g.
- Neurological disorder also includes any condition associated with the disorder. For instance, a method of treating a
- neurodegenerative disorder includes methods of treating loss of memory and/or loss of cognition associated with a neurodegenerative disorder. Such method would also include treating or preventing loss of neuronal function characteristic of neurodegenerative disorder.
- Pain is an unpleasant sensory and emotional experience. Pain classifications have been based on duration, etiology or pathophysiology, mechanism, intensity, and symptoms.
- the term "pain” as used herein refers to all categories of pain, including pain that is described in terms of stimulus or nerve response, e.g., somatic pain (normal nerve response to a noxious stimulus) and neuropathic pain (abnormal response of a injured or altered sensory pathway, often without clear noxious input); pain that is categorized temporally, e.g., chronic pain and acute pain; pain that is categorized in terms of its severity, e.g., mild, moderate, or severe; and pain that is a symptom or a result of a disease state or syndrome, e.g., inflammatory pain, cancer pain, AIDS pain, arthropathy, migraine, trigeminal neuralgia, cardiac ischaemia, and diabetic peripheral neuropathic pain (see, e.g., Harrison's Principles of Internal Medicine,
- Pain is also meant to include mixed etiology pain, dual mechanism pain, allodynia, causalgia, central pain, hyperesthesia, hyperpathia, dysesthesia, and hyperalgesia.
- Somatic pain refers to a normal nerve response to a noxious stimulus such as injury or illness, e.g., trauma, burn, infection, inflammation, or disease process such as cancer, and includes both cutaneous pain (e.g., skin, muscle or joint derived) and visceral pain (e.g., organ derived).
- a noxious stimulus such as injury or illness, e.g., trauma, burn, infection, inflammation, or disease process such as cancer
- cutaneous pain e.g., skin, muscle or joint derived
- visceral pain e.g., organ derived
- Neuroneuropathic pain is a heterogeneous group of neurological conditions that result from damage to the nervous system.
- Neuroopathic pain refers to pain resulting from injury to or dysfunctions of peripheral and/or central sensory pathways, and from dysfunctions of the nervous system, where the pain often occurs or persists without an obvious noxious input. This includes pain related to peripheral neuropathies as well as central neuropathic pain.
- Common types of peripheral neuropathic pain include diabetic neuropathy (also called diabetic peripheral neuropathic pain, or DN, DPN, or DPNP), postherpetic neuralgia (PHN), and trigeminal neuralgia (TGN).
- Central neuropathic pain involving damage to the brain or spinal cord, can occur following stroke, spinal cord injury, and as a result of multiple sclerosis.
- Other types of pain that are meant to be included in the definition of neuropathic pain include pain from spinal cord injury, neuropathic cancer pain, HIV/ AIDS induced pain, phantom limb pain, and complex regional pain syndrome.
- the compounds of the invention are of use for treating neuropathic pain.
- An exemplary compound of use in this embodiment is a compound according to Formula I in which each of R x -R 3 is hydrogen, and R 4 is selected such that the compound is a free acid or salt thereof.
- neuropathic pain Common clinical features of neuropathic pain include sensory loss, allodynia (non- noxious stimuli produce pain), hyperalgesia and hyperpathia (delayed perception, summation, and painful aftersensation). Pain is often a combination of nociceptive and neuropathic types, for example, mechanical spinal pain and radiculopathy or myelopathy.
- Acute pain is the normal, predicted physiological response to a noxious chemical, thermal or mechanical stimulus typically associated with invasive procedures, trauma and disease. It is generally time-limited, and may be viewed as an appropriate response to a stimulus that threatens and/or produces tissue injury. "Acute pain”, as described above, refers to pain, which is marked by short duration or sudden onset.
- Chronic pain occurs in a wide range of disorders, for example, trauma,
- Chronic pain usually lasts more than about six months.
- intensity of chronic pain may be disproportionate to the intensity of the noxious stimulus or underlying process.
- Chronic pain refers to pain associated with a chronic disorder, or pain that persists beyond resolution of an underlying disorder or healing of an injury, and that is often more intense than the underlying process would predict. It may be subject to frequent recurrence.
- Inflammatory pain is adaptive in that it elicits physiologic responses that promote healing. However, inflammation may also affect neuronal function.
- Inflammatory pain refers to pain, which is produced as a symptom or a result of inflammation or an immune system disorder.
- Visceral pain refers to pain, which is located in an internal organ.
- Mated etiology pain refers to pain that contains both inflammatory and neuropathic components.
- “Dual mechanism” pain refers to pain that is amplified and maintained by both peripheral and central sensitization.
- Ceralgia refers to a syndrome of sustained burning, allodynia, and hyperpathia after a traumatic nerve lesion, often combined with vasomotor and sudomotor dysfunction and later trophic changes.
- Central pain refers to pain initiated by a primary lesion or dysfunction in the central nervous system.
- “Hyperesthesia”, as described above, refers to increased sensitivity to stimulation, excluding the special senses.
- Hyperpathia refers to a painful syndrome characterized by an abnormally painful reaction to a stimulus, especially a repetitive stimulus, as well as an increased threshold. It may occur with allodynia, hyperesthesia, hyperalgesia, or dysesthesia.
- Dysesthesia refers to an unpleasant abnormal sensation, whether spontaneous or evoked. Special cases of dysesthesia include hyperalgesia and allodynia,
- pain includes pain resulting from dysfunction of the nervous system: organic pain states that share clinical features of neuropathic pain and possible common pathophysiology mechanisms, but are not initiated by an identifiable lesion in any part of the nervous system.
- DPNP Diabetic Peripheral Neuropathic Pain
- DN diabetic peripheral neuropathy
- DN diabetic peripheral neuropathy
- DN diabetic peripheral neuropathy
- the classic presentation of DPNP is pain or tingling in the feet that can be described not only as “burning” or “shooting” but also as severe aching pain. Less commonly, patients may describe the pain as itching, tearing, or like a toothache. The pain may be accompanied by allodynia and hyperalgesia and an absence of symptoms, such as numbness.
- Post-Herpetic Neuralgia also called “Postherpetic Neuralgia” (PHN)
- PPN Postherpetic Neuralgia
- VZV varicella zoster virus
- neurodegeneration pain refers to peripheral neuropathic pain as a result of cancer, and can be caused directly by infiltration or compression of a nerve by a tumor, or indirectly by cancer treatments such as radiation therapy and chemotherapy (chemotherapy- induced neuropathy).
- HIV/ AIDS peripheral neuropathy or "HIV/ AIDS -related neuropathy” refers to peripheral neuropathy caused by HIV/ AIDS, such as acute or chronic inflammatory demyelinating neuropathy (AIDP and CIDP, respectively), as well as peripheral neuropathy resulting as a side effect of drugs used to treat HIV/ AIDS.
- HIV/ AIDS peripheral neuropathy or "HIV/ AIDS -related neuropathy” refers to peripheral neuropathy caused by HIV/ AIDS, such as acute or chronic inflammatory demyelinating neuropathy (AIDP and CIDP, respectively), as well as peripheral neuropathy resulting as a side effect of drugs used to treat HIV/ AIDS.
- AIDP and CIDP chronic inflammatory demyelinating neuropathy
- Phantom Limb Pain refers to pain appearing to come from where an amputated limb used to be. Phantom limb pain can also occur in limbs following paralysis. It is usually chronic in nature. It is similar in nature to the limb pain experienced by patients with paralysis following spinal cord injury.
- TN Trigeminal Neuralgia
- TN Trigeminal Neuralgia
- CRPS Complex Regional Pain Syndrome
- RSD Reflex Sympathetic Dystrophy
- CRPS is a chronic pain condition.
- the key symptom of CRPS is continuous, intense pain out of proportion to the severity of the injury, which gets worse rather than better over time.
- CRPS is divided into type 1, which includes conditions caused by tissue injury other than peripheral nerve, and type 2, in which the syndrome is provoked by major nerve injury, and is sometimes called causalgia.
- Fibromyalgia refers to a chronic condition characterized by diffuse or specific muscle, joint, or bone pain, along with fatigue and a range of other symptoms.
- fibromyalgia was known by other names such as fibrositis, chronic muscle pain syndrome, psychogenic rheumatism and tension myalgias.
- convulsion refers to a CNS disorder and is used interchangeably with “seizure,” although there are many types of seizure, some of which have subtle or mild symptoms instead of convulsions. Seizures of all types may be caused by disorganized and sudden electrical activity in the brain. Convulsions are a rapid and uncontrollable shaking. During convulsions, the muscles contract and relax repeatedly.
- the term "method of treating pain” means relief from the symptoms or the prevention of pain, which includes the descriptions of pain provided herein. Additional examples include, but are not limited to, migraine, chronic back pain, phantom limb pain, neuropathic pain such as diabetic neuropathy, and post herpetic neuropathy.
- method of treating anxiety disorders means relief from the symptoms or the prevention of anxiety disorders, which include, but are not limited to, panic disorder with or without agoraphobia, agoraphobia without history of panic disorder, animal or other phobias including social phobias, obsessive-compulsive disorder, stress disorders including post-traumatic and acute stress disorder, situational anxiety, generalized anxiety disorder and substance-induced anxiety disorder.
- the term "method of treating psychotic disorders” as used herein means relief from the symptoms or the prevention of psychotic disorders, which include, but are not limited to, schizophrenia, schizophreniform disorder, schizoaffective disorder, delusional disorder, brief psychotic disorder, shared psychotic disorder, psychotic disorder due to general medical condition, substance-induced psychotic disorder, or psychotic disorder not otherwise specified (Diagnostic and Stastistical Manual of Mental Disorders, (Ed. 4 ) American Psychiatric Association, Washington, D.C. (1994)).
- the term "method of treating convulsive disorders” means relief from the symptoms or the prevention of epilepsy, which include, but are not limited to, altered consciousness, altered motor activity, autonomic responses, inappropriate behavior patterns seizures including tonic or clonic jerking of extremities, emotional stress, sense of terror, uneasiness, nervousness, headache, fatigue, auditory hallucinations, aggressive outbursts, acute skeletal muscle spasm, and spasticity.
- method of treating depressive or bipolar disorders means relief from the symptoms or the prevention of depressive disorders, which include, but are not limited to, single-episode or recurrent major depressive disorder, seasonal affective disorder (SAD), dysthymic disorder, bipolar I and bipolar II manic disorders, and
- the term "method of treating cognitive disorders” means relief from the symptoms or the prevention of cognitive disorders, which includes, but is not limited to delirium, dementia, amnesic disorders, and cognitive deficits, including age-related memory deficits, due to traumatic injury, stroke, Parkinson's disease, attention deficit disorder and Downs Syndrome. Any of these conditions may be attributable to substance abuse or withdrawal.
- dementia include dementia of the Alzheimer's type with early or late onset, and vascular dementia, any of which may be uncomplicated or accompanied by delirium, delusions or depressed mood; and dementia due to HIV virus, head trauma, Parkinson's disease or Creutzfeldt- Jakob disease.
- the term "method of treating sleeping disorders”, as used herein, means relief from the symptoms or the prevention of sleep disorders or states that affect a subject's ability to sleep, which includes, but are not limited to, insomnia, sleep apnea, REM sleep interruptions, parasomnia, jet-lag syndrome, hypersomnia, shift workers' sleep disturbances, dysomnias, night terror, narcolepsy, disturbed sleep patterns, disturbed biological or circadian rhythms, sleep disturbances associated with such diseases as neurological disorders, neuropathic pain and restless leg syndrome, or providing sleep induction before surgical procedures or in disturbed or anxious states.
- the term "method of treating neurodegenerative eye diseases”, as used herein, means relief of symptoms or the prevention of neurodegenerative eye diseases, which includes, but is not limited to retinoschisis, vascular diseases of the retina, diseases caused by venous and/or arterial vascular occlusions, macular degenerations, traumatic retinal changes such as contusion of the eye, perforating eye injuries, siderosis/hemidosis, chalcosis, bums, retinopathia traumatica and/or injury to the retina from light, diseases of the choroid, diseases of the optic nerve, anterior ischemic optic neuropathy, optic atrophy, glaucoma, glaucoma simplex, secondary glaucoma and/or ocular hypertension.
- method of treating emesis means relief from the symptoms or the prevention of emesis, which includes, but is not limited to, acute, delayed and anticipatory emesis, emesis induced by chemotherapy or radiation, as well as motion sickness, and postoperative nausea and vomiting.
- the term "method of treating eating disorders” means relief from the symptoms or the prevention of eating disorders, which include, but are not limited to, anorexia nervosa, bulimia nervosa, obesity, weight-gain after smoking cessation, snacking and binge eating.
- benzodiazepine receptor includes the benzodiazepine receptor/GABA receptor/chloride channel complex (benzodiazepine receptor complex) and benzodiazepine receptor-agonist binding sites at or near the receptor complex. Both central nervous system (“central”) and peripheral benzodiazepine receptors (“peripheral”) are encompassed by the use of this term.
- the invention provides a method for treating a condition described herein in a mammal, the method comprises administering to the mammal a therapeutically effective amount of a compound described herein, or a compound according to a formula described herein, or a pharmaceutically acceptable salt thereof, sufficient to treat the condition, thereby treating the condition.
- the mammal is in need of treatment with the compound.
- the mammal is not otherwise in need of treatment with the compound.
- the mammal is a mouse or a rat.
- the mammal is a human.
- the invention provides a method for treating a condition described herein in a mammal, the method comprises administering to the mammal a therapeutically effective amount of a pharmaceutical formulation described herein, sufficient to treat the condition, thereby treating the condition.
- the mammal is in need of treatment with the pharmaceutical formulation.
- the mammal is not otherwise in need of treatment with the pharmaceutical formulation.
- the mammal is a mouse or a rat.
- the mammal is a human.
- the invention provides a method for treating pain in a mammal, the method comprises administering to the mammal a therapeutically effective amount of a compound described herein, or a compound according to a formula described herein, or a pharmaceutically acceptable salt thereof, sufficient to treat the pain, thereby treating the pain.
- the mammal is in need of treatment with the compound.
- the mammal is not otherwise in need of treatment with the compound.
- the mammal is a mouse or a rat.
- the mammal is a human.
- the invention provides a method for treating pain in a mammal, the method comprises administering to the mammal a therapeutically effective amount of a pharmaceutical formulation described herein, sufficient to treat the pain, thereby treating the pain.
- the mammal is in need of treatment with the pharmaceutical formulation.
- the mammal is not otherwise in need of treatment with the pharmaceutical formulation.
- the mammal is a mouse or a rat.
- the mammal is a human.
- the invention provides a method for treating epilepsy in a mammal, the method comprises administering to the mammal a therapeutically effective amount of a compound described herein, or a compound according to a formula described herein, or a pharmaceutically acceptable salt thereof, sufficient to treat the epilepsy, thereby treating the epilepsy.
- the mammal is in need of treatment with the compound.
- the mammal is not otherwise in need of treatment with the compound.
- the epilepsy is child absence epilepsy.
- the mammal is a mouse or a rat.
- the mammal is a human.
- the invention provides a method for treating epilepsy in a mammal, the method comprises administering to the mammal a therapeutically effective amount of a pharmaceutical formulation described herein, sufficient to treat the epilepsy, thereby treating the epilepsy.
- the mammal is in need of treatment with the pharmaceutical formulation.
- the mammal is not otherwise in need of treatment with the pharmaceutical formulation.
- the epilepsy is child absence epilepsy.
- the mammal is a mouse or a rat.
- the mammal is a human.
- the invention provides a method for treating essential tremor in a mammal, the method comprises administering to the mammal a therapeutically effective amount of a compound described herein, or a compound according to a formula described herein, or a pharmaceutically acceptable salt thereof, sufficient to treat the essential tremor, thereby treating the essential tremor.
- the mammal is in need of treatment with the compound.
- the mammal is not otherwise in need of treatment with the compound.
- the mammal is a mouse or a rat.
- the mammal is a human.
- the invention provides a method for treating essential tremor in a mammal, the method comprises administering to the mammal a therapeutically effective amount of a pharmaceutical formulation described herein, sufficient to treat the essential tremor, thereby treating the essential tremor.
- the mammal is in need of treatment with the pharmaceutical formulation.
- the mammal is not otherwise in need of treatment with the pharmaceutical formulation.
- the mammal is a mouse or a rat.
- the mammal is a human.
- IC 50 refers to the concentration causing a 50% inhibition of the specific binding of the control substance.
- the invention provides a compound of the invention.
- the invention provides a compound described herein, or a salt thereof.
- the salt of a compound described herein is a pharmaceutically acceptable salt.
- the invention provides a compound described herein, or a pharmaceutically acceptable salt thereof.
- the invention provides a compound described in a formula provided herein.
- the invention provides a compound described herein.
- One object of the present invention is to provide a compound, or a pharmaceutically acceptable salt thereof, of the general structure:
- R 1 is substituted or unsubstituted alkyl, substituted or unsubstituted aryl or substituted or unsubstituted heteroaryl;
- R 14 is H or substituted or unsubstituted Ci-C 6 alkyl or substituted or unsubstituted cycloalkyl, or substituted or unsubstituted heteroalkyl
- R 2 is selected from substituted or unsubstituted alkyl and substituted or unsubstituted heteroalkyl
- R 3 , R 4 , R 5 , R 6 are each independently hydrogen, substituted or unsubstituted -Ci -6 alkyl, substituted or unsubstituted -Ci -6 haloalkyl, a 3-, 4-, 5- or 6-membered substituted or unsubstituted cycloalkyl or substituted or unsubstituted heterocycloalkyl group, wherein R 3 and R 4 along with the carbon to which they are attached optionally form a 3 - to 6-membered substituted or
- R 5 and R 6 together with the carbon to which they are attached, optionally form a 3-, 4-, 5- or 6- membered substituted or unsubstituted cycloalkyl or substituted or unsubstituted heterocycloalkyl group, said heterocycloalkyl group optionally containing 1 or 2 heteroatoms independently selected from
- R 7 , R 8 , R 9 , and R 10 are each independently hydrogen, fluorine, substituted or unsubstituted-Ci-6 alkyl, substituted or unsubstituted-Ci -6 haloalkyl, a 3-, 4-, 5- or 6-membered substituted or unsubstituted cycloalkyl or substituted or unsubstituted heterocycloalkyl group, wherein R 7 and R 8 , together with the carbon to which they are attached, optionally form a 3-, 4-, 5-, or 6- membered substituted or unsubstituted cycloalkyl or substituted or unsubstituted
- heterocycloalkyl group said heterocycloalkyl group optionally containing 1 or 2 heteroatoms independently selected from O, N or S, and said substituted cycloalkyl or substituted heterocycloalkyl group is optionally substituted with 1, 2 or 3 substituents independently selected from F, -Ci -6 alkyl, or -CF 3 ; R 9 and R 10 , together with the carbon to which they are attached, optionally form a 3-, 4-, 5- or 6-membered substituted or unsubstituted cycloalkyl or substituted or unsubstituted heterocycloalkyl group, said heterocycloalkyl group containing from 1 or 2 heteroatoms independently selected from O, N or S, and the substituted cycloalkyl or substituted heterocycloalkyl group is optionally substituted with 1,
- R 11 , R 12 and R 13 are each independently hydrogen, fluorine, substituted or unsubstituted-Ci -6 alkyl, substituted or unsubstituted-Ci-6 haloalkyl, a 3-, 4-, 5- or 6-membered substituted or unsubstituted cycloalkyl or substituted or unsubstituted heterocycloalkyl group, wherein R 11 and either R 12 or R 13 , together with the carbons to which they are attached, optionally form a 3-, 4-, 5-, 6- or 7-membered substituted or unsubstituted cycloalkyl or substituted or unsubstituted heterocycloalkyl group.
- R 1 is selected from substituted or unsubstituted benzyl or a substituted or unsubstituted polycyclic cycloalkyl ring, e.g., adamantyl.
- R 1 is substituted adamantyl.
- R 1 is substituted adamant-1- yl.
- R 1 is unsubstituted adamantyl.
- R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , R 11 , R 12 , R 13 , and X are as described herein, R 1 is unsubstituted adamant- 1-yl.
- R 1 is substituted by 1, 2, 3 or 4 non-hydrogen substituents selected from halo, haloalkyl, substituted or unsubstituted alkoxy, and cyano.
- the compounds of the invention have a structure according to Formula I, wherein R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , R 11 , R 12 , R 13 , and X are as described herein, R 1 is of the formula
- A, B, C and D are independently selected from "aryl group substituents"; and the indices a, b, c, and d are independently selected from 0 and 1.
- the compounds of the invention have a structure according to Formula I, wherein R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , R 11 , R 12 , R 13 , and X are as described herein, A, B, C and D are independently selected from CN, CI, Br, F, substituted or unsubstituted Ci-C 6 alkyl, substituted or unsubstituted Ci-C 6 heteroalkyl, substituted or unsubstituted Ci-C 6 haloalkyl, and substituted or unsubstituted Ci-C 6 alkoxy.
- R 2 is selected from Ci-Cio straight chain or branched substituted or unsubstituted Ci-C 6 alkyl, and Ci-Cio substituted or unsubstituted Ci-C 6 alkyl heteroalkyl.
- R 2 includes substituted alkyl or substituted heteroalkyl groups, which are substituted with amide, oxo, substituted or unsubstituted aryl or substituted or unsubstituted heterocycloalkyl.
- exemplary substituted aryl groups include substituted or unsubstituted phenyl, and an exemplary heterocycloalkyl moiety is an oxygen- containing heterocycle.
- a compound of the invention has the formula:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , R lla , R ll , and X are as described herein, wherein q is an integer selected from 1, 2, 3, 4 and 5.
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , and X are as described herein, wherein R lla and R ll are members each independently selected from H, methyl, and fluorine.
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , and X are as described herein, q is 1, R lla is H and R ll is F. In an exemplary embodiment, wherein R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , and X are as described herein, q is 1, R lla is F and R ll is F.
- a compound of the invention has the formula:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , and X are as described herein, q is an integer selected from 1, 2, 3, 4 and 5.
- a compound of the invention has the formula:
- a compound of the invention has the formula:
- a compound of the invention has the formula:
- a compound of the invention has the formula:
- a compound of the invention has the formula:
- R 1 is unsubstituted phenyl.
- R 1 is substituted or unsubstituted adamantyl.
- q, R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , and X are as described herein, wherein R 1 is substituted or unsubstituted adamantyl.
- R 1 is or or , wherein represents the covalent link between R 1 and X; and R la and R l are members each independently selected from halo, haloalkyl, substituted or unsubstituted alkoxy, and cyano.
- R 1 is or or , wherein represents the covalent link between R 1 and X; and R la and R l are members each independently selected from halo, haloalkyl, substituted or unsubstituted alkoxy, and cyano.
- R la and R l are members each independently selected from F, CI,
- R 6 , R 7 , R 8 , R 9 , R 10 , and X are as described herein, wherein R is
- R a and R are members each independently selected from F, CI, and CF 3 .
- R 2 is substituted or unsubstituted alkyl.
- R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , and X are as described herein, wherein R 2 is substituted or unsubstituted heteroalkyl.
- R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , and X are as described herein, wherein R 2 is substituted or unsubstituted heteroalkyl.
- R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , and X are as described herein, wherein R 2 is
- the compound has the following formula:
- the compound has the following formula: , wherein X, R 2 , and R 1 are as described herein.
- the compound is according to a formula described herein, and R 1 is phenyl substituted with one to three substituents, each of which is a member selected from the group consisting of F, CI, and CF 3 .
- the compound is according to a formula described herein, and R 1 is phenyl substituted with one substituent which is a member selected from the group consisting of F, CI, and CF 3 .
- the compound is according to a formula described herein, and R 1 is phenyl substituted with two substituents which are members each individually selected from the group consisting of F, CI, and CF 3 .
- the compound is according to a formula described herein, and R 1 is phenyl substituted with thee substituents which are members each individually selected from the group consisting of F, CI, and CF 3 .
- R 1 is phenyl substituted with thee substituents which are members each individually selected from the group consisting of F, CI, and CF 3 .
- the compound has the following formula: , wherein X,
- R 2 , and R 1 are as described herein.
- the compound has the
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound is wherein X and R 1 are as described herein. In an exemplary embodiment, the compound is
- n X and R 1 are as described herein.
- the compound is
- the compound is , wherein R 1 and R 2 are as described herein. In an exemplary embodiment, the compound is , wherein R 1 and R 2 are as described herein. In an exemplary embodiment, the compound is , wherein R 1 and R 2 are as described herein. In an exemplary
- the compound and R 2 are as described herein.
- the compound is_
- R la and R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- R la and R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is , wherein X and R 2 are as described herein, R 1 is as described herein. In an exemplary embodiment, the
- the compound is wherein X and R 2 are as described herein, R 1 is unsubstituted adamantyl.
- the compound is wherein X and R 2 are as described herein, R 1 is unsubstituted adamant- 1-yl.
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound is according to a formula described herein which contains R la and R l , wherein R la and R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is according to a formula described herein which contains R la and R l , wherein R la and R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is according to a formula described herein which contains R la and R l , wherein R la is as described herein, and R l is F.
- the compound is according to a formula described herein which contains R a and R , wherein R la is as described herein, and R l is CI. In an exemplary embodiment, the compound is according to a formula described herein which contains R la and R l , wherein R la is as described herein, and R l is CF 3 . In an exemplary embodiment, the compound is according to a formula described herein which contains R la and R l , wherein R l is as described herein, and R la is F. In an exemplary embodiment, the compound is according to a formula described herein which contains R la and R l , wherein R l is as described herein, and R la is CI. In an exemplary embodiment, the compound is according to a formula described herein which contains R la and R l , wherein R l is as described herein, and R la is CF 3 . In an exemplary embodiment, the compound is according to a formula described herein which contains R la and R l , wherein R l is
- the compound is , a and R are as described herein. In an exemplary embodiment, the compound is
- R 2 , R la and R l are as described herein.
- R a and R are as described herein.
- the compound is
- the compound is , wherein R 2 , R la and R l are as described herein.
- the compound is , wherein R 2 , R la and R l are as described herein.
- the compound is , wherein R 2 , R la and R l are as described herein.
- the compound is according to a formula described herein which contains R la and R l , wherein R la and R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is according to a formula described herein which contains R la and R l , wherein R la and R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is according to a formula described herein which contains R la and R l , wherein R la is as described herein, and R l is F.
- the compound is according to a formula described herein which contains R la and R l , wherein R la is as described herein, and R l is CI. In an exemplary embodiment, the compound is according to a formula described herein which contains R la and R l , wherein R la is as described herein, and R l is CF 3 . In an exemplary embodiment, the compound is according to a formula described herein which contains R la and R l , wherein R l is as described herein, and R la is F. In an exemplary embodiment, the compound is according to a formula described herein which contains R la and R l , wherein R l is as described herein, and R la is CI. In an exemplary embodiment, the compound is according to a formula described herein which contains R la and l , wherein R l is as described herein, and R la is CF 3 . In an exemplary embodiment, the compound is according to a formula described herein which contains R la and l , wherein R l is as
- R a and R are as described herein.
- the compound is wherein R 2 .
- R la and R l are as described herein.
- the compound is , wherein R a and R are as described herein. In an exemplary embodiment, the compound is
- R 2 , R la and R l are as described herein.
- the compound is wherein R R la and R l are as described herein.
- the compound is , wherein
- R 2 is as described herein, and R 1 is unsubstituted adamantyl.
- R 1 is unsubstituted adamantyl.
- the com rein R 2 is as described herein, and R 1 is unsubstituted adamant-l-yl.
- the compound is wherein R 2 is as described herein, and R 1 is unsubstituted
- the compound is
- R 2 is as described herein, and R 1 is unsubstituted adamant-l-yl.
- the compound is , wherein R z is as described herein, and R 1 is unsubstituted adamantyl.
- the compound is wherein R 2 is as described herein, and R 1 is unsubstituted adamant- 1-yl.
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- R 1 is as described herein.
- the compound is
- erein R 1 is as described herein.
- the compound is wherein R is as described herein. In an exemplary embodiment, the compound is
- the compound is wherein R 1 is as described herein. In an exemplary embodiment, the compound is
- R 1 is as described herein. In an exemplary embodiment, the compound is wherein 1 is as described herein. In an exemplary embodiment, the compound is wherein R 1 is as described herein. In an exemplary embodiment, the compound is wherein R 1 is as described herein. In an exemplary
- the compound wherein R 1 is as described herein is as described herein.
- the compound is , wherein R 1 is as described herein.
- the compound is , wherein R 1 is as described herein.
- the compound is (H 3 C ) 3 C ? wherein R 1 is as described herein.
- the compound is
- R 1 is as described herein.
- the compound is N-(00130]-2-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(00130]-2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound is , wherein X is as described herein, and R 1 is unsubstituted adamantyl. In an exemplary embodiment, the compound is , wherein X is as described herein, and R 1 is unsubstituted adamant-l-yl. In an exemplary embodiment, the compound is , wherein X is as described herein, and R 1 is unsubstituted adamantyl. In an exemplary embodiment, the compound is
- the compound is
- the compound is
- the compound is
- X is as described herein, and R is unsubstituted adamantyl.
- the compound is
- X is as described herein, and R 1 is unsubstituted adamant-l-yl.
- the compound is
- X is as described herein, and R 1 is unsubstituted adamant- 1-yl.
- the compound is wherein X is as described herein, and R 1 is unsubstituted adamantyl.
- the compound is wherein X is as described herein, and R 1 is unsubstituted adamant- 1-yl.
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- R a and R are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is
- R la and R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is
- R a and R are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is R la and R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is
- R a and R are members each independently selected from th an exemplary
- the compound is , wherein R a and
- R are members each independently .
- R la and R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is
- R d R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is wherein R a and
- R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is
- R la and R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is wherein R a and
- R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- R la and R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is R a and R are members each independently selected from th an exemplary
- the compound is wherein R
- R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is wherein R a and R are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is R and R are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is , wherein R a and R are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is , wherein R a and
- R l are members each independently
- R a and R are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is wherein R a and R are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is wherein R la and R are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is
- R a and R are members each independently selected from the group consisting of F, CI, and CF 3 .
- R a and R are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is wherein R la and
- R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is
- R la and R l are members each independently selected from the group consisting of F, CI, and CF 3 .
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound is unsubstituted adamant-l-yl.
- the compound is wherein R 1 is unsubstituted adamantyl.
- the compound rein R 1 is unsubstituted adamant-l-yl.
- the compound is
- the compound is n R 1 is unsubstituted adamant-l-yl. In an exemplary embodiment, the compound is
- R 1 is unsubstituted adamantyl.
- the compound is wherein R is unsubstituted adamant- 1-yl.
- the compound is , wherein 1 is unsubstituted adamantyl. In an exemplary embodiment, the compound is wherei 1 is unsubstituted adamant- 1-yl.
- the compound is wherein 1 is unsubstituted adamantyl. In an exemplary embodiment, the compound is wherei 1 is unsubstituted adamant- 1-yl.
- the compound is wherein R 1 is unsubstituted adamantyl. In an exemplary embodiment, the compound is wherei 1 is unsubstituted adamant- 1-yl. In an
- the compound is wherein R 1 is unsubstituted adamantyl. In an exemplary embodiment, the compound is is unsubstituted adamant- 1-yl. In an exemplary embodiment, the compound is
- R 1 is unsubstituted adamantyl.
- the compound wherein R 1 is unsubstituted adamant- 1- l.
- the compound i in R 1 is unsubstituted adamantyl.
- the compound is
- the compound is wherei 1 is unsubstituted adamant- 1-yl.
- the compound is wherein R 1 is unsubstituted adamantyl. In an exemplary embodiment, the compound is wherein R 1 is unsubstituted adamant-l-yl.
- the compound is N-(00135]-2-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(00135]-2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl
- the compound is or a pharmaceutically acceptable salt thereof. In an exemplary embodiment, the compound is
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound is or a pharmaceutically acceptable salt thereof. In an exemplary embodiment, the compound is
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound is
- the compound is or a pharmaceutically acceptable salt thereof.
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound is
- the compound is or a pharmaceutically acceptable salt thereof.
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound is or a pharmaceutically acceptable salt thereof.
- the compound is
- the compound is
- the compound is
- the compound is or a pharmaceutically acceptable salt thereof.
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound is
- the compound is or a pharmaceutically acceptable salt thereof.
- the compound is or a pharmaceutically acceptable salt thereof. In an exemplary embodiment, the compound is or a pharmaceutically acceptable salt thereof. In an exemplary embodiment, the compound is or a pharmaceutically acceptable salt thereof.
- the compound is H
- R 1 is unsubstituted adamant-l-yl.
- the compound is or a pharmaceutically acceptable salt thereof, wherein R 1 is unsubstituted adamant-l-yl.
- the compound is or a pharmaceutically acceptable salt thereof, wherein R 1 is unsubstituted adamant-l-yl.
- the compound is r a pharmaceutically acceptable salt thereof.
- the compoun or a pharmaceutically acceptable salt thereof In an exemplary embodiment, the compound is or a pharmaceutically acceptable salt thereof. [00145] The invention is of a scope including any compound disclosed herein whether it is or is not covered by Formula I.
- the invention also provides a pharmaceutical formulation comprising a
- the formulation further includes a pharmaceutically acceptable carrier.
- the invention also provides a method for treating pain responsive to selective inhibition of the T Channel known as the Ca v3 2 channel comprising administering to a mammal a therapeutically effective amount of a compound according to Formula I or individually disclosed herein.
- the present invention provides compounds which are selective T-Channel inhibitory compounds useful for relief of neuropathic and/or inflammatory pain, and epilepsy.
- the invention also includes, where chemically possible, all stereoisomers and geometric isomers of the compounds, including diastereomers, enantiomers, and cis/trans (E/Z) isomers.
- the invention also includes mixtures of stereoisomers and/or geometric isomers in any ratio, including, but not limited to, racemic mixtures. Unless stereochemistry is explicitly indicated in a structure, the structure is intended to embrace all possible stereoisomers of the compound depicted. If stereochemistry is explicitly indicated for one portion or portions of a molecule, but not for another portion or portions of a molecule, the structure is intended to embrace all possible stereoisomers for the portion or portions where stereochemistry is not explicitly indicated.
- the compounds of the present invention may also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds.
- the compounds may be radiolabeled with radioactive isotopes, such as for example tritium ( 3 H), iodine-125 ( 125 I) or carbon-14 ( 14 C). All isotopic variations of the compounds of the present invention, whether radioactive or not, are intended to be encompassed within the scope of the present invention. Unless a specific isotope is indicated, the invention includes all isotopologues of the compounds disclosed herein, such as, for example, deuterated derivatives of the compounds (where H can be 2 H, i.e., D).
- compounds that are considered to possess activity as T-Channel inhibitors are those displaying 50% inhibition of the Ca ++ voltage (IC 50 ) at a concentration of not higher than about 100 ⁇ , preferably, not higher than about 10 ⁇ , more preferably not higher than about 1 ⁇ and most preferably not higher than about 100 nM.
- a protecting group refers to a group which is used to mask a functionality during a process step in which it would otherwise react, but in which reaction is undesirable.
- the protecting group prevents reaction at that step, but may be subsequently removed to expose the original functionality. The removal or "deprotection” occurs after the completion of the reaction or reactions in which the functionality would interfere.
- the compounds of the present invention may be prepared by the methods illustrated in the general reaction schemes as, for example, described below, or by modifications thereof, using readily available starting materials, reagents and conventional synthesis procedures. In these reactions, it is also possible to make use of variants that are in themselves known, but are not mentioned here.
- the starting materials for example in the case of suitably substituted benzimidazole ring compounds, are either commercially available, synthesized as described in the examples or may be obtained by the methods well known to persons of skill in the art.
- the present invention further provides pharmaceutical formulations comprising as active agents, the compounds described herein.
- the invention is a pharmaceutical formulation comprising a therapeutically effective amount of a compound described herein, or a pharmaceutically acceptable salt thereof.
- the invention is a pharmaceutical formulation comprising a therapeutically effective amount of a compound of a formula described herein, or a pharmaceutically acceptable salt thereof.
- the invention is a pharmaceutical formulation comprising a therapeutically effective amount of a compound described herein, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the invention is a pharmaceutical formulation comprising a therapeutically effective amount of a compound of a formula described herein, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the invention is a pharmaceutical formulation described herein, wherein the formulation is in a unit dosage form.
- a "pharmaceutical formulation” refers to a preparation of one or more of the compounds described herein, or physiologically acceptable salts or solvates (including hydrates) thereof, with other chemical components such as physiologically suitable carriers and excipients.
- a prophylactic or therapeutic dose typically varies with the nature and severity of the condition to be treated and the route of administration.
- the dose, and perhaps the dose frequency, will also vary according to the age, body weight and response of the individual patient. In general, a dose ranges from about 0.1 mg to about 7000 mg, preferably about 1 mg to about 100 mg, and more preferably, about 25 mg to about 50 mg, in single or divided doses.
- a dose may range from about 50 mg to about 500 mg, and preferably, about 100 mg to about 500 mg. Such doses may be administered 1, 2, 3, 4, 5, 6 or more times in a day. It may be recommended that children, patients over 65 years old, and those with impaired renal or hepatic function, initially receive low doses and that the dosage is titrated based on individual responses and/or blood levels. It may be necessary to use dosages outside these ranges in some cases, as will be apparent to those in the art. Further, it is noted that the clinician or treating physician knows how and when to interrupt, adjust or terminate therapy in conjunction with individual patient's response.
- compositions for use in accordance with the present invention thus may be formulated in a conventional manner using one or more physiologically acceptable carriers comprising excipients and auxiliaries, which facilitate processing of the active compounds into preparations which, can be used pharmaceutically.
- the carriers must be acceptable in the sense of being compatible with the other ingredients of the formulation and not deleterious to the recipient thereof. Proper formulation is dependent upon the route of administration chosen.
- Compounds that inhibit T-Channels can be formulated as pharmaceutical formulations and administered to a mammalian subject, such as a human patient in a variety of forms adapted to the chosen route of administration, i.e., oral, rectal, topical (including dermal, buccal, sublingual, and intraocular), or parenteral, by intravenous, intramuscular, topical, transdermal, intradermal, intraarticular, or subcutaneous routes.
- a mammalian subject such as a human patient in a variety of forms adapted to the chosen route of administration, i.e., oral, rectal, topical (including dermal, buccal, sublingual, and intraocular), or parenteral, by intravenous, intramuscular, topical, transdermal, intradermal, intraarticular, or subcutaneous routes.
- the compounds can be formulated readily by combining the active compounds with pharmaceutically acceptable carriers well known in the art.
- Such carriers enable the compounds of the invention to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions, and the like, for oral ingestion by a patient.
- Pharmacological preparations for oral use can be made using a solid excipient, optionally grinding the resulting mixture, and processing the mixture of granules, after adding suitable auxiliaries if desired, to obtain tablets or dragee cores.
- Suitable excipients are, in particular, fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium
- carbomethylcellulose and/or physiologically acceptable polymers such as
- PVP polyvinylpyrrolidone
- disintegrating agents such as cross- linked polyvinyl pyrrolidone, agar or alginic acid or a salt thereof such as sodium alginate.
- an enteric coating may be useful as it is may be desirable to prevent exposure of the compounds of the invention to the gastric environment.
- compositions which can be used orally, include push-fit capsules made of gelatin as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol.
- the push-fit capsules may contain the active ingredients in admixture with filler such as lactose, binders such as starches, lubricants such as talc or magnesium stearate and, optionally, stabilizers.
- the active compounds may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols.
- stabilizers may be added.
- Formulations for topical administration in the mouth include lozenges comprising the active ingredient in a flavored basis such as sucrose and acacia or tragacanth, and pastilles comprising the active ingredient in a basis such as gelatin and glycerin or sucrose and acacia. All formulations for oral administration should be in dosages suitable for the chosen route of administration.
- the compounds of the invention may be formulated in aqueous solutions, preferably in physiologically compatible buffers such as Hank's or Ringer's solution or physiological saline buffer.
- physiologically compatible buffers such as Hank's or Ringer's solution or physiological saline buffer.
- penetrants appropriate to the barrier to be permeated may be used in the formulation.
- penetrants including for example DMSO or polyethylene glycol, are known in the art.
- the compounds for use according to the present invention are conveniently delivered in the form of an aerosol spray presentation from a pressurized pack or a nebulizer with the use of a suitable propellant, e.g.,
- the dosage unit may be determined by providing a valve to deliver a metered amount.
- Capsules and cartridges of, e.g., gelatin for use in an inhaler or insufflator may be formulated containing a powder mix of the compound and a suitable powder base such as lactose or starch.
- compositions for parenteral administration include aqueous and non-aqueous sterile injection solutions, which may contain anti -oxidants, buffers, bacteriostats and solutes, which render the formulation isotonic with the blood of the intended recipient.
- Formulations also include aqueous and non-aqueous sterile suspensions, which may include suspending agents and thickening agents.
- Pharmaceutical formulations for parenteral administration in an aqueous solution contain the active ingredients in water- soluble form. Additionally, suspensions of the active compounds may be prepared as appropriate oily injection suspensions.
- Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acids esters such as ethyl oleate, triglycerides or liposomes.
- Aqueous injection suspensions may contain substances, which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol or dextran.
- the suspension may also contain suitable stabilizers or agents, which increase the solubility of the compounds, to allow for the preparation of highly concentrated solutions.
- the formulations may be presented in unit-dose of multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of a sterile liquid carrier, for example saline, phosphate- buffered saline (PBS) or the like, immediately prior to use.
- a sterile liquid carrier for example saline, phosphate- buffered saline (PBS) or the like.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
- the compounds of the present invention may also be formulated in rectal formulations such as suppositories or retention enemas, using, e.g., conventional suppository bases such as cocoa butter, polyethylene glycol, or other glycerides.
- dosing can also be a single administration of a slow release formulations, with course of treatment lasting from several days to several weeks or until cure is effected or diminution of the disease state is achieved.
- the amount of a formulation to be administered will, of course, be dependent on many factors including the subject being treated, the severity of the affliction, the manner of administration, the judgment of the prescribing physician.
- the compounds of the invention may be administered orally or via injection at a dose from 0.001 to 250 mg/kg per day.
- the dose range for adult humans is generally from 0.5 mg to 10 g/day.
- Tablets or other forms of presentation provided in discrete units may conveniently contain an amount of compound of the invention which is effective at such dosage or as a multiple of the same, for instance, units containing 5 mg to 500 mg, usually around 10 mg to 200 mg.
- the precise amount of compound administered to a patient will be the responsibility of the attendant physician. However, the dose employed will depend on a number of factors, including the age and sex of the patient, the precise disorder being treated, and its severity. Also, the route of administration may vary depending on the condition and its severity.
- solvate refers to a compound described herein and/or from Formula I in the solid state, wherein molecules of a suitable solvent are incorporated in the crystal lattice.
- a suitable solvent for therapeutic administration is physiologically tolerable at the dosage administered. Examples of suitable solvents for therapeutic administration are ethanol and water. When water is the solvent, the solvate is referred to as a hydrate. In general, solvates are formed by dissolving the compound in the appropriate solvent and isolating the solvate by cooling or using an antisolvent.
- the solvate is typically dried or azeotroped under ambient conditions.
- Inclusion complexes are described in Remington: The Science and Practice of Pharmacy 19th Ed. (1995) volume 1, page 176-177, which is incorporated herein by reference. The most commonly employed inclusion complexes are those with cyclodextrins, and all cyclodextrin complexes, natural and synthetic, are specifically encompassed within the claims.
- salts refers to salts prepared from pharmaceutically acceptable non-toxic acids or bases including inorganic acids and bases and organic acids and bases.
- salts may be prepared from pharmaceutically acceptable non-toxic acids including inorganic and organic acids.
- Suitable pharmaceutically acceptable acid addition salts for the compounds of the present invention include acetic, benzenesulfonic (besylate), benzoic, camphorsulfonic, citric, ethenesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric acid, p-toluenesulfonic, and the like.
- suitable pharmaceutically acceptable base addition salts for the compounds of the present invention include metallic salts made from aluminum, calcium, lithium, magnesium, potassium, sodium and zinc or organic salts made from lysine, ⁇ , ⁇ '-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, meglumine (N-methylglucamine) and procaine.
- preventing refers to administering a medicament beforehand to forestall or obtund an attack.
- the person of ordinary skill in the medical art recognizes that the term “prevent” is not an absolute term. In the medical art it is understood to refer to the prophylactic administration of a drug to substantially diminish the likelihood or seriousness of a condition, and this is the sense intended herein.
- formulations of this invention may include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavoring agents.
- the formulations may be presented in a packaging device or dispenser, which may contain one or more unit dosage forms containing the active ingredient.
- a packaging device include metal or plastic foil, such as a blister pack and a nebulizer for inhalation.
- the packaging device or dispenser may be accompanied by instructions for administration.
- Formulations comprising a compound of the present invention formulated in a compatible pharmaceutical carrier may also be placed in an appropriate container and labeled for treatment of an indicated condition.
- Step A To ethyl 2-(diethoxyphosphoryl)acetate (4.68 mL, 23.58 mmol) in THF (50 mL) at -78 °C was added n-BuLi (2.5M in hexanes, 9.43 mL, 23.58 mmol) and the reaction mixture was stirred for 10 min. To this mixture was added benzyl 4-oxopiperidine- 1-carboxylate (5.00 g, 21.44 mmol) and the mixture was allowed to warm to RT and stirred for 30 min. The solvent was removed and the resulting solids were triturated with 100 mL hexanes. The mixture was filtered through Celite ® (J.T. Baker, Phillipsberg, NJ,
- Step B Potassium tert-butoxide (8.43 g, 75.1 mmol) was added to a solution of trimethylsulfoxonium iodide (17.5 g, 79.5 mmol) in DMSO (100 mL) in one portion. The mixture was stirred at RT for 2 h, after which benzyl 4-(2-ethoxy-2-oxoethylidene)piperidine- 1-carboxylate (13.4 g, 44.2 mmol) in DMSO (50 mL) was added. The mixture was stirred at RT overnight and then slowly added to a sat. aq. H 4 C1 solution at 0 °C with stirring. The mixture was extracted with ether.
- Step C 6-Benzyl 1-ethyl 6-azaspiro[2.5]octane-l,6-dicarboxylate (4.06 mmol, 1.29 g) was dissolved in MeOH (8 mL), THF (8 mL), and H 2 0 (8 mL). Li OH was added (8.13 mmol, 0.33 g), and the reaction was stirred overnight at room temperature. After the reaction was complete, the mixture was acidified to pH 1 with 1 N HC1 and extracted with EtOAc.
- Step D To a solution of 6-(benzyloxycarbonyl)-6-azaspiro[2.5]octane-l-carboxylic acid (1.9g, 6.6 mmol) in t-BuOH (19 mL) was added DPPA (2.7 g, 9.8 mmol) and
- Step F To a solution of 6-benzyloxycarbonyl-l-amino-6-azaspiro[2.5]octane hydrochloride (0.8 g, 2.7 mmol) in DCM was added EDCI (0.78 g, 4.05 mmol), HOBT (0.55 g, 4.05 mmol), DIPEA (1.04 g, 8.1 mmol) and benzoyl chloride (0.57 g, 4.05 mmol). The reaction mixture was stirred at RT for 12 h, then diluted with DCM, and washed successively with sat. H 4 C1 solution, sat. NaHC0 3 solution, water and brine.
- Step G To a solution ofN-(6-benzyloxycarbonyl-6-azaspiro[2.5]octan-l- yl)benzamide (lg, 2.7 mmol) in MeOH was added Pd/C. The flask was purged and backfilled with H 2 3 times, and then stirred at RT under an H 2 atmosphere for 2 h. The mixture was then filtered through a Celite ® (J.T. Baker, Phillipsberg, NJ, diatomaceous earth) pad, and the filtrate was concentrated to give the crude product N-(6-azaspiro[2.5]octan-l- yl)benzamide (0.6 g, 95% yield), which was used in next step without purification.
- Celite ® J.T. Baker, Phillipsberg, NJ, diatomaceous earth
- Step H A solution of N-(6-azaspiro[2.5]octan-l-yl)benzamide (76 mg, 0.33 mmol), l-bromo-2-methylpropane (55 mg, 0.44 mmol) and K 2 C0 3 (91 mg, 0.66 mmol) in DMF/CH 3 CN (1 : 1, 3mL) was stirred at 60 °C for 16 h. Then 10 mL of water was added and the solution was extracted with EtOAc (20 mL ⁇ 3). The combined organic layers were washed with brine, dried over Na 2 S0 4 and concentrated in vacuo.
- Examples 02 to 18 were also prepared by procedures similar to the one described in Example 01, replacing benzoyl chloride in step F and/or l-bromo-2-methylpropane in step H with the designated reagents.
- Examples 19 to 36 can also prepared by procedures similar to the one described in Example 01, replacing benzoyl chloride in step F with the appropriate acyl chloride.
- Examples 51 to 53 were also prepared by procedures similar to the one described in Example 01, replacing benzoyl chloride in step F with 3,5-dichlorobenzenesulfonyl chloride (CAS# 705-21-5) and l-bromo-2-methylpropane in step H with the designated reagents.
- Step A Benzyl l-(3,5-dichlorobenzamido)-6-azaspiro[2.5]octane-6-carboxylate, prepared by a procedure similar to the on described in Example 1, Step F, is alkylated with Mel in DMF in presence of a base, such as NaH to give benzyl l-(3,5-dichloro-N- methylbenzamido)-6-azaspiro[2.5]octane-6-carboxylate.
- a base such as NaH
- Step B To a solution of benzyl l-(3,5-dichloro-N-methylbenzamido)-6- azaspiro[2.5]octane-6-carboxylate from Step A in AcOH (10 mL) was added FIBr in AcOH (33%, 3 mL) at rt. The resulting mixture was stirred at rt for 2 h. Solvent was removed, the residue was diluted with ethyl acetate (20 mL) and washed with sat. aq. NaHC0 3 (20 mL) and extracted with ethyl acetate (2 ⁇ 30 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated to give a product that was used in next step without further purification.
- Step C 3,5-Dichloro-N-methyl-N-(6-azaspiro[2.5]octan-l-yl)benzamide obtained in Step B was converted to 3,5-dichloro-N-(6-(3,3-dimethylbutyl)-6-azaspiro[2.5]octan-l-yl)- N-methylbenzamide by a procedure similar to the one described in Example 1 Step H.
- Example 54 replacing methyliodide in Step A with (bromomethyl)cyclopropane.
- Examples 1 - 55 of the present invention can be prepared as single enantiomers by separating the racemic mixtures by HPLC using a chiral column, such as, for example, CHIRALPAK ® (Chiral Technologies, Inc., West Chester, PA, USA). Alternatively, enantiomerically pure or enriched compounds can be prepared as described in Example 56.
- a chiral column such as, for example, CHIRALPAK ® (Chiral Technologies, Inc., West Chester, PA, USA).
- enantiomerically pure or enriched compounds can be prepared as described in Example 56.
- Step A To a solution of 6-benzyl 1-ethyl 6-azaspiro[2.5]octane-l,6-dicarboxylate (4.06 mmol, 1.29 g; Example 1, Step B) in a mixture of MeOH (8 mL), THF (8 mL) and H 2 0 (8 mL) was added LiOH (8.13 mmol, 0.33 g) and the reaction was stirred at room temperature
- Step B To a suspension of 6-(benzyloxycarbonyl)-6-azaspiro[2.5]octane-l- carboxylic acid (289 mg, 1 mmol) in dichloromethane (5 mL) was added oxalyl chloride (152 mg, 1.2 mmol) followed by the addition of one drop of DMF. After the reaction was stirred at rt for 3h, the solvent was evaporated to give 6-(benzyloxycarbonyl)-6-azaspiro[2.5]octane-
- Step C To a solution of (4R, 5S)-4-methyl-5-phenyloxazolidin-2-one (2.4 g, 13.56 mmol; CAS # 77943-39-6) in THF (12 mL) was added n-BuLi (8.2 mL, 2M in hexane, 1.2 eq) dropwise at -78 °C. After addition, the reaction was warmed to rt and stirred for 1 hour.
- Step D Purification of diastereomeric mixture of benzyl (S)-l-((4R,5S)-4-methyl-
- Step F To a solution of (S)-6-((benzyloxy)carbonyl)-6-azaspiro[2.5]octane-l- carboxylic acid or (R)-6-((benzyloxy)carbonyl)-6-azaspiro[2.5]octane-l-carboxylic acid (850 mg, 2.94 mmol) in t-BuOH (20 mL ) was added DPPA (1.21 g, 4.4 mmol) and
- tetraethylammonium chloride (TEA-C1) (600mg, 5.9 mmol).
- the reaction mixture was refluxed for 6 h, then concentrated to give the crude residue which was purified by silica gel chromatography (hexanes: EtOAc 10: 1 to 5: 1) to afford benzyl (S)-l-((tert- butoxycarbonyl)amino)-6-azaspiro[2.5]octane-6-carboxylate or benzyl (R)-l-((tert- butoxycarbonyl)amino)-6-azaspiro[2.5]octane-6-carboxylate (440 mg, 42% yield).
- Mass Spectrum (ESI) m/z 383 (M+23).
- Step G A solution of benzyl (S)-l-((tert-butoxycarbonyl)amino)-6- azaspiro[2.5]octane-6-carboxylate or benzyl (R)-l-((tert-butoxycarbonyl)amino)-6- azaspiro[2.5]octane-6-carboxylate (440mg, 1.2 mmol) in HC1 in Dioxane (4M, lOmL) was stirred at rt for 1 h. The solvent was removed and the residue (420mg, crude) was used in the next step without further purification.
- Mass Spectrum (ESI) m/z 261 (M+l).
- Step H To a solution of the crude product from Step G (420mg, 1.4 mmol) in DCM (10 mL) was added EDCI (403 mg, 2.1 mmol), HOBT (284 mg, 2.1 mmol) , DIPEA (542 mg, 4.2 mmol) and 3,5-dichlorobenzoic acid (401mg, 2.1 mmol). The reaction mixture was stirred at rt for 12 h, then diluted with DCM (20 mL), washed successively with sat. aq. H 4 C1 solution, sat. aq. NaHC0 3 , water and brine and concentrated.
- Step J A solution of the prodcuct from Step I (60 mg, 0.2 mmol), 2-bromo-N-tert- butylacetamide (35 mg, 0.18 mmol) and K 2 C0 3 (83 mg, 0.6 mmol) in DMF/MeCN (1/1, 3mL) was stirred at 60 °C for 16 h. 10 mL of water was added and the mixture was extracted with EA (20 mL X 3). The combined organic layers were washed with brine, dried over Na 2 S0 4 and concentrated.
- Example 49 To a solution of Example 49 in MeOH was added NaBH 4 at 0 °C, and the resulting mixture was stirred at rt for 1 h. The solution was concentrated and the residue was diluted with ethyl acetate (30 mL) and washed with water (30 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated. The crude product was purified by prep-TLC with DCM/MeOH to obtan the desired product as a white solid.
- Example 59 can be prepared from Example 50 using a procedure similar to the one described in Example 58.
- Step A To a solution of diphenyl sulfide (7.44 g, 40 mmol) and l-chloro-3- iodopropane (24.5 g, 120 mmol) in nitromethane (15 mL) at RT was added silver tetrafluorob orate (7.37 g, 38 mmol) in one portion. A yellow precipitate formed, and the reaction temperature initially rose to around 40-50 °C, then gradually fell to RT. The reaction mixture was stirred at RT for 20 hr then diluted with dichloromethane (15 mL) and filtered through a sintered funnel packed with Celite ® (J.T.
- Step B To a slurry of 3-chloropropyldiphenylsulfonium fluoroborate (13 g, 37.2 mmol) in tetrahydrofuran (150 mL) was added a solution (1.28M) of potassium tert-butoxide (37.2 mol, 4.2 g) in dimethyl sulfoxide (29 mL) at RT over a period of 1 h.
- the reaction mixture turned a deep amber color when the base was added [the colour disappeared quickly after each addition, but at the end of the addition, the color remained amber].
- the reaction mixture was diluted with dichloromethane (80 mL), stirred for 10 min then poured onto a mixture of dichloromethane/water (1 : 1, 100 mL). The phases were separated and the dichloromethane phase collected and evaporated to dryness. The residue was stirred in diethyl ether (1L) for 1 hr and then decanted. The oily residue was crystallized using ethanol/di ethyl ether to give the product (4.1 g, 13.1 mmol, 35%) as a white solid.
- Step C To a solution of cyclopropyldiphenylsulfonium tetrafluorob orate (4 g, 12.74 mmol) in tetrahydrofuran (80 mL) at -40 °C was added potassium
- Step D Dried crude t-butyl 10-oxa-7-azadispiro[2.0.5.1]decane-7-carboxylate (6.2 g, 26 mmol) was dissolved in anhydrous toluene (60 mL). To this solution was added lithium tetrafluorob orate (122 mg) in one lot. The mixture was heated to 80 °C for 2 h. After cooling to RT, about 50 mL of toluene was removed under reduced pressure. The reaction mixture was then quenched with water (40 mL).
- Step E To a solution of tert-butyl l-oxo-7-azaspiro[3,5]-nonane-7-carboxylate (2.96 g, 12.4 mmol) in anhydrous 1,2-dichloroethane (35 mL) was added benzylamine (1.6 g, 14.9 mmol) followed by sodium triacetoxyborohydride (5.3 g, 24.8 mmol). The mixture was stirred at rt for 18 h and then quenched with saturated aqueous sodium bicarbonate and the product extracted with EtOAc (3 x 50 mL).
- Step F To a solution of tert-butyl l-(benzylamino)-7-azaspiro[3.5]nonane-7- carboxylate (2.63 g, 7.96 mmol) in methanol (60 mL) was added Pd/C (260 mg) in one portion. The reaction mixture was stirred at rt under a hydrogen atmosphere (balloon) for 16 h, and then the catalyst was filtered off through a Celite ® (J.T. Baker, Phillipsberg, NJ, diatomaceous earth)-packed funnel. The Celite ® layer was washed with methanol (3 ⁇ 20 mL). The combined filtrate and washings were concentrated to dryness to afford tert-butyl l,8-diazaspiro[5.6]decane-8-carboxylate as a yellow oil (1.9 g, 99%).
- Step G A solution of tert-butyl l,8-diazaspiro[5.6]decane-8-carboxylate (600 mg, 2.5 mmol), benzoic acid (458 mg, 3.75 mmol), HOBT (675 mg, 5 mmol), EDCI (955 mg, 5 mmol) and diisopropylethylamine (968 g, 7.5 mmol) in DMF/DCM (2 mL/8 mL) was stirred at RT for 3 hours. Then 20 mL of water was added and the solution was extracted with DCM (30 mL x 3).
- Step I A solution of N-(7-azaspiro[3.5]nonan-l-yl)benzamide (80 mg, 0.33 mmol), l-Bromo-2-methylpropane (55 mg, 0.44 mmol) and K 2 C0 3 (91 mg, 0.66 mmol) in
- Examples 61 to 71 were also prepared by procedures similar to the one described in Example 60, replacing benzoic acid in step G and/or l-bromo-2-methylpropane in step I with the designated reagents.
- Step A 8-Azaspiro[4.5]decan-l-one (CAS No. 198133-82-3) is commercially available or can be prepared as described in W09737992. 8-Azaspiro[4.5]decan-l-one can be treated with (Boc) 2 0 in presence of a base to give tert-butyl l-oxo-8-azaspiro[4.5]decane- 8-carboxylate.
- Step B tert-butyl l-oxo-8-azaspiro[4.5]decane-8-carboxylate can be converted into tert-butyl l-(benzylamino)-8-azaspiro[4.5]decane-8-carboxylate by a procedure similar to the one described in Example 58 Step E.
- Step C tert-butyl l-(benzylamino)-8-azaspiro[4.5]decane-8-carboxylate can be converted into tert-butyl l-amino-8-azaspiro[4.5]decane-8-carboxylate by a procedure similar to the one described in Example 58 Step F.
- Step D tert-butyl l-amino-8-azaspiro[4.5]decane-8-carboxylate can be converted into tert-butyl l-benzamido-8-azaspiro[4.5]decane-8-carboxylate by a procedure similar to the one described in Example 58 Step G.
- Step E tert-butyl l-benzamido-8-azaspiro[4.5]decane-8-carboxylate can be converted into N-(8-azaspiro[4.5]decan-l-yl)benzamide by a procedure similar to the one described in Example 58 Step H.
- Step F N-(8-azaspiro[4.5]decan-l-yl)benzamide can be converted into N-(8- isobutyl-8-azaspiro[4.5]decan-l-yl)benzamide by a procedure similar to the one described in Example 16 Step I.
- Examples 73 to 86 can also prepared by procedures similar to the one described in Example 70, replacing benzoyl chloride in step D and/or l-bromo-2-methylpropane in step F with the designated reagents.
- Step B To a solution of the product from Step A (1.5 g, 5.5 mmol) in DCM (20 mL) was added DMP (2.8 g, 6.6 mmol) at 0 °C, then warmed to rt and stirred for 2 hours. The mixture was diluted with DCM (80 mL) and washed with 1M aq. Na 2 S 2 0 3 solution (30 mL), saturated aq. NaHC0 3 solution (30 m) and brine (30 mL).
- Step C To a solution of the product from Step B (1.6 g, 5.9 mmol) and 2- methylpropane-2-sulfinamide (860 mg, 7.1 mmol) in THF (25 mL) was added Ti(OEt) 4 (2 g, 8.9 mmol) dropwise at 0 °C. The mixture was refluxed for 16 h. Then the solvent was removed, to give the crude product benzyl l-(((tert-butylsulfinyl)-14-azanylidene)methyl)-6- azaspiro[2.5]octane-6-carboxylate which was used in the next step without further purification.
- Mass Spectrum (ESI) m/z 377.
- Step D To a solution of benzyl l-(((tert-butylsulfinyl)-14-azanylidene)methyl)-6- azaspiro[2.5]octane-6-carboxylate (l . lg, 3 mmol) in THF (10 mL) was added NaBH 4 (2.2 g, 5.9 mmol) at 0 °C, The reaction mixture was stirred at RT for 2 h, then diluted with EA(50 mL) washed with sat. aq. H 4 C1 solution (20 mL), sat. aq. NaHC0 3 solution (20 mL) and brine (20 mL) and concentrated under reduced pressure.
- Step F To a stirred solution of benzyl l-(aminomethyl)-6-azaspiro[2.5]octane-6- carboxylate (930mg, 3 mmol) in DCM (15 mL) was added EDCI (864 mg, 1.5 mmol) , HOBT (486 mg, 3.6 mmol), DIPEA (1.2 g, 9 mmol) and 3,5-dichlorobenzoic acid (688mg, 3.6 mmol), The reaction mixture was stirred at RT for 12 h, then diluted with DCM (30 mL), washed with brine (30 mL) and extracted with DCM (30 mL x 3). The combined extracts were concentrated under reduced pressure. The crude residue was purified by
- Step G To a stirred solution of benzyl l-((3,5-dichlorobenzamido)methyl)-6- azaspiro[2.5]octane-6-carboxylate (900mg, 3 mmol) in AcOH (10 mL) was added HBr in AcOH (33 ), 4 mL) at rt. After the mixture was stirred at rt for 2 h, solvent was removed, the residue was diluted with ethyl acetate (30 mL) and washed with sat. aq NaHC0 3 solution (30 mL) and extracted with ethyl acetate (2 x 30 mL).
- Step H A solution of N-((6-azaspiro[2.5]octan-l-yl)methyl)-3,5- dichlorobenzamide (120mg, 0.39mmol), 2-bromo-N-tert-butylacetamide (67 mg, 0.34 mmol) and K 2 C0 3 (210mg, 1.54mmol) in DMF/MeCN (1/1, 3mL) was stirred at 60 °C for 16 h. 10 mL of water was added and the solution was extracted with EA (20 mL x 3). The combined organic layers were washed with brine, dried over Na 2 S0 4 filtered and the filtrate was concentrated. The residue was purified by silica gel chromatography eluting with 5%
- Examples 88 and 89 can also prepared by procedures similar to the one described in Example 87, replacing 2-brom -N-tert-butylacetamide in step H with the designated reagents.
- Step A A solution of 6-(Benzyloxycarbonyl)-6-azaspiro[2.5]octane-l-carboxylic acid (1 g, 3.46 mmol; Example 1, Step C), 3,5-dichlorobenzenamine (1.1 g, 6.92 mmol), HATU (2.6 g, 6.92 mmol), and DIEA (1.8 g, 13.8 mmol) in DCM/DMF (12 mL / 3 mL ) was stirred at RT for 1 h. 20 mL of water was added and the solution was extracted with DCM (30 mL x 3). The combined organic layers were washed with brine, dried over Na 2 S0 4 and concentrated in vacuo. The residue was purified by chromatography on silica gel
- Step B To a solution of benzyl l-((3,5-dichlorophenyl)carbamoyl)-6- azaspiro[2.5]octane-6-carboxylate (700 mg, 1.62 mmol) in AcOH (10 mL) was added HBr in
- Step C A solution of N-(3,5-dichlorophenyl)-6-azaspiro[2.5]octane-l-carboxamide (100 mg, 0.2 mmol), 2-bromo-N-tert-butylacetamide (80 mg, 0.41 mmol) and K 2 C0 3 (188 mg, 1.36 mmol) in DMF/MeCN (1/1, 3mL) was stirred at 60 °C for 16 h. Water (20 mL) was added and the solution was extracted with EA (30 mL X 3). The combined organic layers were washed with brine, dried over Na 2 S0 4 and concentrated.
- Examples 91 and 92 can also prepared by procedures similar to the one described in Example 90, replacing 2-bromo- -tert-butylacetamide in step C with the designated reagents.
- Step A To a stirred solution of benzyl l-((3,5-dichlorophenyl)carbamoyl)-6- azaspiro[2.5]octane-6-carboxylate (500 mg, 1.23 mmol; Example 90, Step A) in anhydrous THF (10 mL) was added a solution of BH 3 -THF complex (1 M in THF, 5 mL) at 0 °C. The reaction was warmed to rt and stirred for 16 h. Then the reaction was quenched by addition of a saturated aq. solution of H 4 CI (20 mL) until gas evolution ceased, and extracted with EtOAc (30 mL x 3).
- Step B N-((6-Azaspiro[2.5]octan-l-yl)methyl)-3,5-dichloroaniline was prepared from benzyl l-(((3,5-dichlorophenyl)amino)methyl)-6-azaspiro[2.5]octane-6-carboxylate as described in Example 90, Step B and was used in the next step without further purification.
- Mass Spectrum (ESI) m/z 285 (M+H).
- N-((6-Azaspiro[2.5]octan-l-yl)methyl)-3,5-dichloroaniline was alkylated with 2-bromo-N-tert-butylacetamide as described in Example 90, Step C.
- the product was purified by silica gel chromatography eluting with 5% DCM/MeOH to give 6-(2-(tert- butylamino)-2-oxoethyl)-N-(3,5-dichlorophenyl)-6-azaspiro[2.5]octane-l-carboxamide as a yellow solid (30 mg, 22%).
- Examples 94 and 95 can also prepared by procedures similar to the one described in Example 93, replacing 2-bromo-N-tert-butylacetamide in step C with the designated reagents.
- Step A A mixture of 4-cyano piperidine (1 g, 9.1 mmol), N-tert-buty ⁇ -2- chloroacetamide (1.35 g, 9.1 mmol), and potassium carbonate (2.5 g, 18.2 mmol) in acetonitrile (30 mL) was heated to reflux under argon for 9 hours. The resulting mixture was cooled and filtered through a layer of potassium carbonate. The filtrate was concentrated and crystallized from ethyl ether to give N-tert-butyl-2-(4-cyanopiperidin-l-yl)acetamide (1.82 g, 90 % yield) as a white solid.
- Step B To a cold solution of N-tert-butyl-2-(4-cyanopiperidin-lyl)acetamide (Example 43, Step A) (0.7 g, 3.1 mmol) and MeOH (20 mL), cobalt (II) chloride hexahydrate (0.37 g, 1.6 mmol) was added followed by the addition of sodium borohydride (0.53 g, 14.1 mmol). After stirring for 15 hours at 25 °C, the reaction solution was diluted with 25 mL of 5% aqueous ammonium hydroxide and extracted three times with dichloromethane. The combined organic layer was washed with brine, dried over anhydrous Na 2 S0 4 and
- Step C To a cold solution of 0.20 g (0.88 mmol) of 2-(4-(aminomethyl)piperidin- l-yl)-N-(tert-butyl)acetamide in freshly distilled dichloromethane under argon, pyridine (1.3 mmol) was added followed by the addition of 0.12 g (0.88 mmol) of benzoyl chloride. The reaction was stirred at 0 °C for 30 min.
- reaction solution was diluted with 20 mL of aqueous sodium bicarbonate and extracted three times with DCM. The combined organic layer was washed with brine, dried (anhydrous Na 2 S0 4 ), concentrated, and column chromatographed on silica gel using a mixture of dichloromethane and MeOH (10: 1) as eluant to give the title compound, N-((l-(2-(tert-butylamino)-2-oxoethyl)piperidin-4- yl)methyl)benzamide, (0.15g, 51% yield) as a white solid.
- Examples 97 to 106 can also prepared by procedures similar to the one described in Example 96, replacing N-tert-butyl-2-chloroacetamide in step A and/or benzoyl chloride in step C with the designated reagents.
- Step A A solution of 4-cyanopiperdine 0.5 g (4.54 mmol) was dissolved in 25 mL of dry THF and cooled to -78 °C. LDA (0.227 M in 20 mL, 4.54 mmol) was slowly added to the above solution over 30 min at -78 °C under argon. A pale brown solution was obtained. Stirring continued for 2 h at -78 °C and iodomethane (0.64 g, 4.54 mmol) was added dropwise into the reaction while keeping the temperature at -78 °C.
- Step B To a solution of 4-methylpiperidine-4-carbonitrile (0.19 g, 1.50 mmol) in dry CH 3 CN (8 mL), 2-chloro-N-(l,l -dimethyl ethyl)acetamide (0.22 g, 1.50 mmol) and anhydrous K 2 C0 3 (0.41 g, 3.0 mmol) were added and refluxed at 85 °C for 8 h.
- Step C To a solution of N-(tert-butyl)-2-(4-cyano-4-methylpiperidin-l- yl)acetamide (0.23 g, 1.06 mmol) in 8 mL of methanol, CoCl 2 .6H 2 0 (0.13 g, 0.53 mmol) was added and stirred. The reaction mixture was cooled to 0 °C and sodium borohydride (0.18 g, 4.77 mmol) was added in several portions over 30 min. The resulting solution was stirred for 18 h at rt, diluted with 15 mL of cold saturated NH 4 C1 and 15 mL of water, and extracted three times with ethyl acetate.
- Step D To a solution of 2-(4-(aminomethyl)-4-methylpiperidin-l-yl)-N-(tert- butyl)acetamide (0.15 g, 0.625 mmol) in freshly distilled dichloromethane (7.5 mL) dry pyridine (81 ⁇ ⁇ , 0.625 mmol) was added and the resulting solution was stirred at 0 °C. To this mixture benzoyl chloride (88 mg, 0.625 mmol) was added and the reaction mixture was stirred for 3 h at rt. The reaction mixture was partitioned between dichloromethane and water and basified to pH 8 using NaHC0 3 .
- Step A To a solution of piperidine-4-carbonitrile (0.5 g, 4.54 mmol) in 25 mL of distilled dichloromethane under argon, distilled triethylamine (1.3 mL, 9.08 mmol) was added. While stirring, the solution was cooled to 0 °C and di-tert-butyl dicarbonate (1.19 g, 5.4 mmol) was added in several portions over 15 min (bubbling observed).
- Step B A solution of tert-butyl 4-cyanopiperdine-l-carboxylate 0.95 g (4.54 mmol) was dissolved in 25 mL of dry THF and cooled to -78 °C. LDA (0.23 M in 20 mL, 4.54 mmol) was slowly added to the reaction over 30 min at -78 °C under argon. A pale brown solution was obtained. Stirring continued for 2 h at -78 °C, and a solution of paraformaldehyde (0.16 g, 5.4 mmol) in 25 mL of freshly distilled THF was added slowly into the reaction solution while keeping the temperature at -78 °C.
- reaction mixture was allowed to reach room temperature while stirred for 15 h under argon.
- the reaction mixture was diluted in 100 mL of water, saturated NaCl (50 mL) was added, and extracted with DCM three times (250 mL each).
- the combined organic layers were washed with brine (100 mL), dried (Na 2 S0 4 ), concentrated, and column chromatographed on silica gel using a gradient mixture of hexane and ethyl acetate as eluant to give tert-butyl 4-cyano-4- (hydroxymethyl)piperidine-l-carboxylate, 0.78 g (72% yield) as a white solid.
- Step C To a solution of tert-butyl 4-cyano-4-(hydroxym ethyl )piperi dine- 1- carboxylate (0.49 g, 2.0 mmol) in DCM (8 mL), pyridine (0.24 mL, 3.03 mmol) was added and the mixure was stirred at 0 °C. To this mixture, benzoyl chloride (0.25 mL, 2.0 mmol) was added and the reaction mixture was stirred for 8 h at rt. The reaction mixture was partitioned between dichloromethane and water and basified to pH 8 using NaHC0 3 . The organic layer was separated, and the aqueous layer was extracted three times with
- Step D A solution of 4-((benzoyloxy)methyl)-4-cyanopiperidine-l-carboxylate (1.12 mmol) in 2 mL dichloromethane containing 10% trifiuoroacetic acid (TFA) was stirred at room temperature for 1 h. The solvent was removed and the remaining solid was kept under high vacuum to yield 0.27 g (>95% yield) of (4-cyanopiperidin-4-yl)methyl benzoate as a yellow solid.
- TFA trifiuoroacetic acid
- Step E To a solution of (4-cyanopiperidin-4-yl)methyl benzoate (0.10 g, 0.41 mmol) in dry CH 3 CN, N-(chloromethyl)-benzamide (57 mg, 0.41 mmol) and anhydrous K 2 C0 3 (0.17 g, 1.23 mmol) were added and refluxed at 85 °C for 12 h.
- reaction mixture was filtered and evaporated to yield a yellow oil that was purified by column chromatography on silica gel using a mixture of CH 2 C1 2 and MeOH (30: 1) as eluant to give (4-cyano-l-(2- oxo-2-(phenylamino)ethyl)piperidin-4-yl)methyl benzoate, 0.15 g (97% yield).
- Step F To a solution of (4-cyano-l-(2-oxo-2-(phenylamino)ethyl)piperidin-4- yl)methyl benzoate (0.16 g, 0.42 mmol) in 5 mL of methanol, CoCl 2 .6H 2 0 (51 mg 0.21 mmol) was added and stirred. The reaction mixture was cooled to 0 °C and sodium borohydride (86 mg, 2.27 mmol) was added in several portions over 30 min. The resulting solution was stirred for 18 h at room temperature, diluted with 15 mL of cold saturated
- Step G To a solution of (4-(aminomethyl)-l-(2-oxo-2-(phenylamino)ethyl) piperidin-4-yl)methyl benzoate (50 mg, 0.13 mmol) in dichloromethane (5 mL) was added dry pyridine (19 ⁇ ., 0.17 mmol) and the mixture was stirred at 0 °C. To the reaction solution, 2-fluorobenzoyl chloride (27 mg, 017 mmol) was added and the mixture was stirred for 3 h at room temperature. The reaction mixture was partitioned between dichloromethane and water, and basified to pH 8 using NaHC0 3 .
- Examples 60 - 71 of the present invention can be prepared as single enantiomers by separating the racemic mixtures by HPLC using a chiral column, such as, for example, CHIRALPAK ® (Chiral Technologies, Inc., West Chester, PA, USA). Alternatively, enantiomerically pure or enriched compounds can be prepared as described in Example 111.
- a chiral column such as, for example, CHIRALPAK ® (Chiral Technologies, Inc., West Chester, PA, USA).
- enantiomerically pure or enriched compounds can be prepared as described in Example 111.
- Step A The Boc group of tert-butyl l-oxo-7-azaspiro[3,5]-nonane-7-carboxylate was removed as described in Example 60, Step H to give 7-azaspiro[3.5]nonan-l-one.
- Step B 7-Azaspiro[3.5]nonan-l-one was alkylated with phenethylbromide to give 7-phenethyl-7-azaspiro[3.5]nonan-l-one by a procedure similar to the one described in Example 60, Step I.
- Step C 7-Phenethyl-7-azaspiro[3.5]nonan-l-one was converted to (S)-2-methyl-N-
- Step D (S)-2-methyl-N-(7-phenethyl-7-azaspiro[3.5]nonan-l-ylidene)propane-2- sulfinamide was reduced with NaBH 4 in THF to give a mixture of (S)-2-methyl-N-((R)-7- phenethyl-7-azaspiro[3.5]nonan-l-yl)propane-2-sulfinamide and (S)-2-m ethyl -N-((S)-7- phenethyl-7-azaspiro[3.5]nonan-l-yl)propane-2-sulfinamide which was separated by chromatography on silica gel to give (S)-2-methyl-N-((R)-7-phenethyl-7-azaspiro[3.5]nonan- l-yl)propane-2-sulfinamide as the major isomer.
- Step E Treatment of (S)-2-methyl-N-((R)-7-phenethyl-7-azaspiro[3.5]nonan-l- yl)propane-2-sulfinamide with HC1 gave (R)-7-phenethyl-7-azaspiro[3.5]nonan-l-amine.
- Step F (R)-7-phenethyl-7-azaspiro[3.5]nonan-l -amine was acylated with benzoic acid using a procedure similar to the one described in Example 60, Step G to give (R)-N-(7- phenethyl-7-azaspiro[3.5]nonan-l-yl)benzamide.
- Example 103 using the commercially available (R)-2-methyl-2-propanesulfinamide (Sigma- Aldrich, Milwaukee, Order No. : 497401) in Step C.
- Example 112 using 2-bromo-N-(tert-butyl) acetamide in Step B and 3,5-dichlorobenzoic acid in Step F.
- 2-carboxamides can be synthesized by a variety of methods well known to well known to the practitioner in the art.
- an arylacetic acid or heteroaryl acetic acid can be converted to an acid chloride using SOCl 2 , POCl 3 , (COCl) 2 or the like (Step A).
- the acid chloride can be converted to an acylthiosemicarbazide by reaction with hydrazine
- Step B The acylthiosemicarbazide can be cyclized to give the 5-aryl-l,3,4- oxadiazol-2-amine or 5-heteroaryl-l,3,4-oxadiazol-2-amine by a variety of methods well known to those skilled in the art (Step C).
- l,3-dibromo-5,5-dimethylhydantoin is an effective agent for this cyclization reaction (Rivera, N.R.; Balsells, J.; Hansen, K.B., Tetrahedron Lett., ⁇ 7, 4889, (2006)).
- 5-Phenyl-l,3,4-oxadiazol-2-amine (CAS No. 1612-76-6) is commercially available (e.g. Sigma-Aldrich, Milwaukee, US; Catalog No. 663395).
Abstract
Description







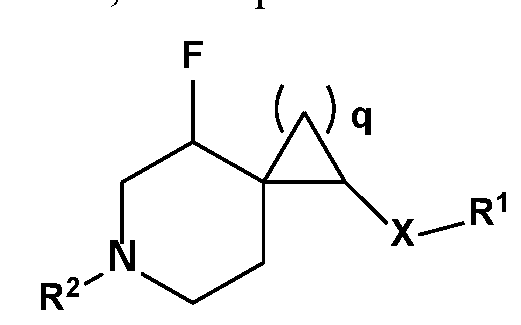
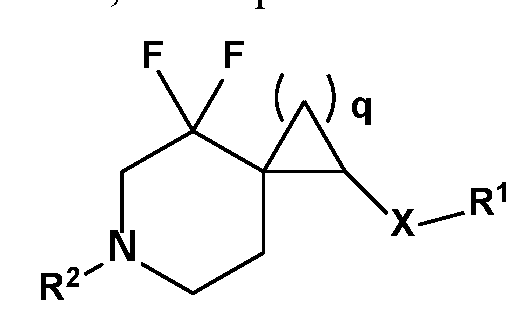

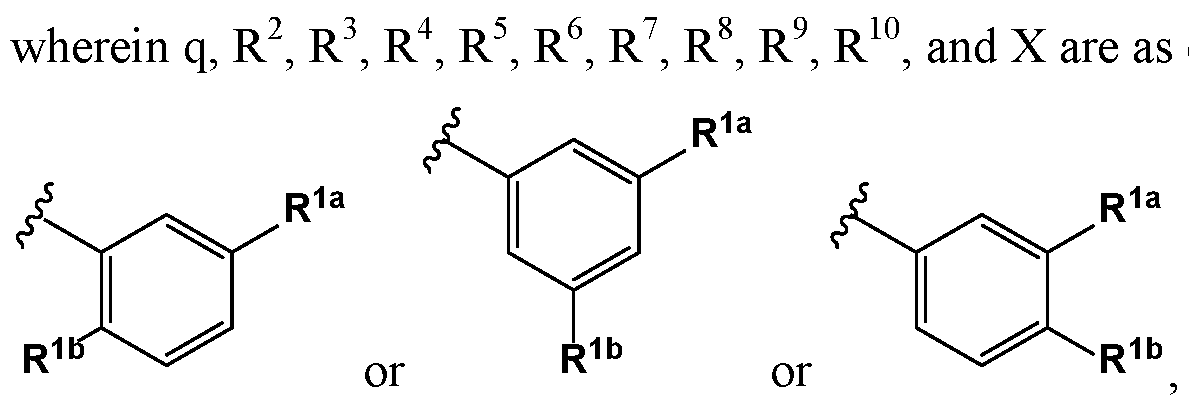

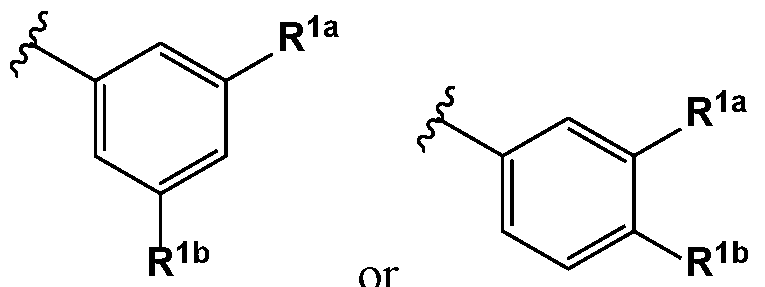



















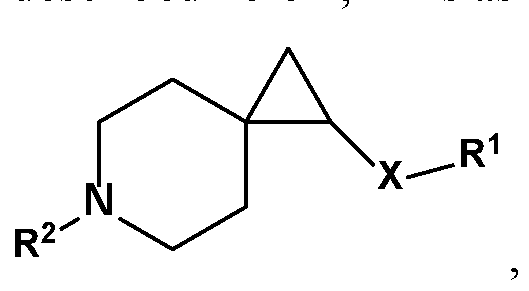



















































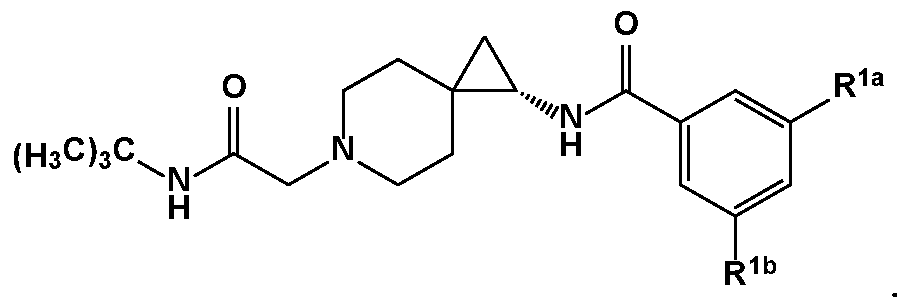













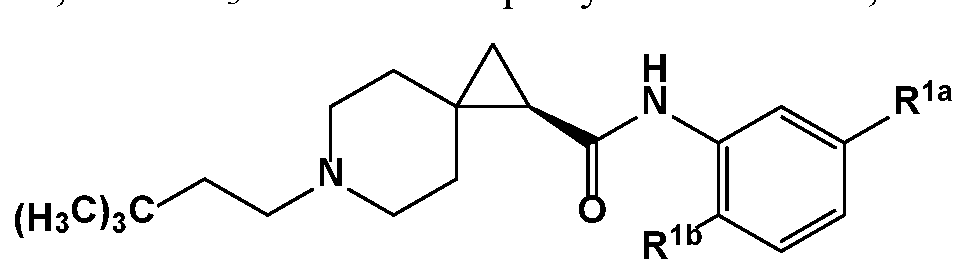

























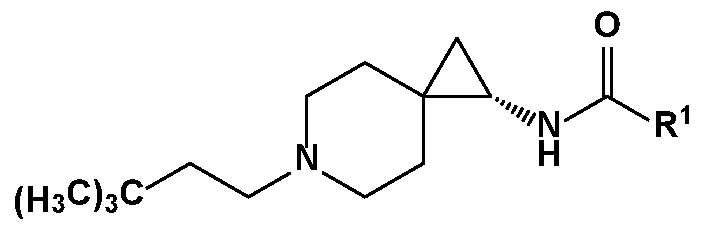


















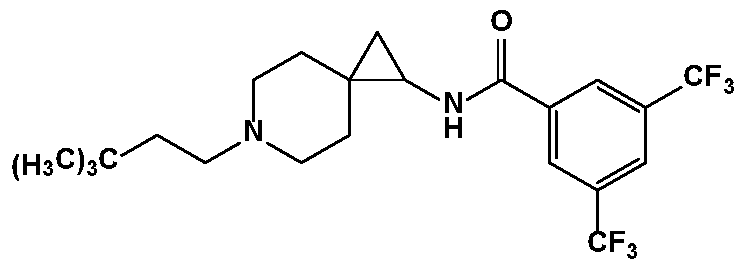


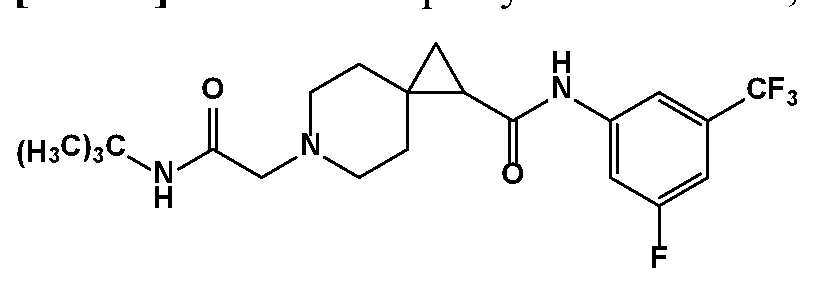



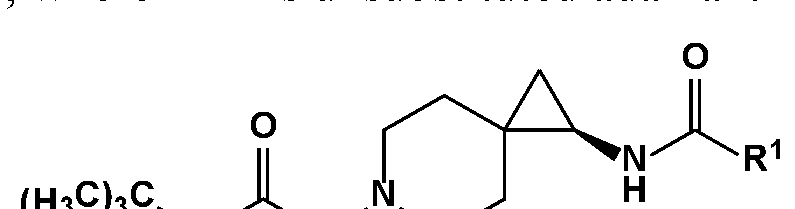





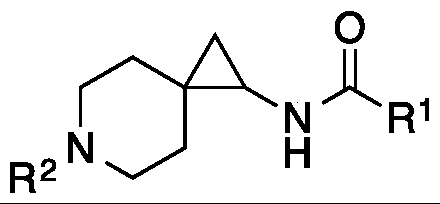











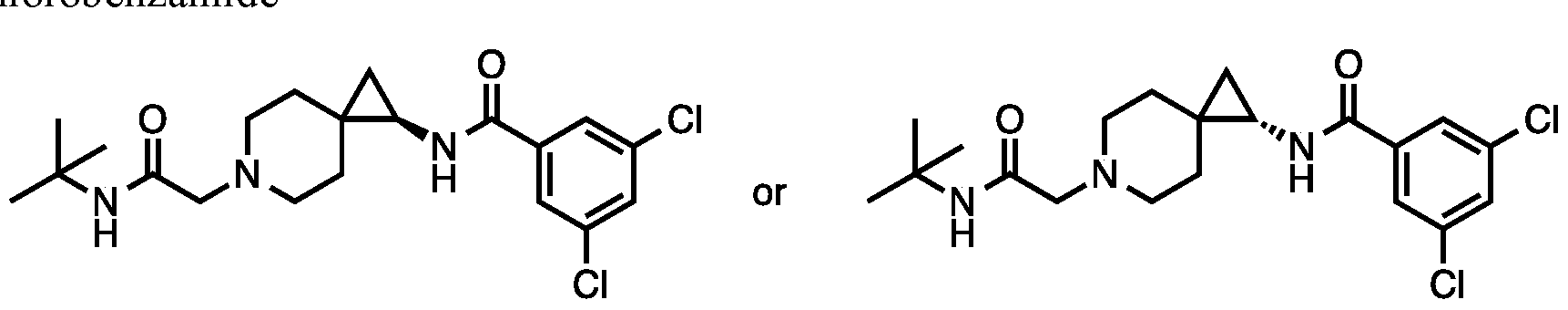
































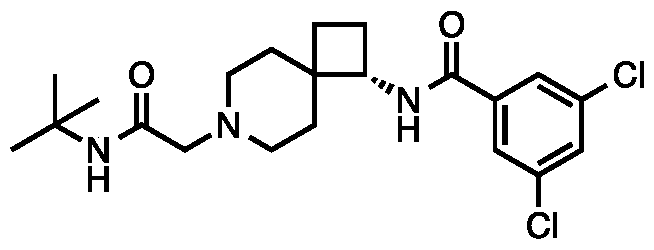

















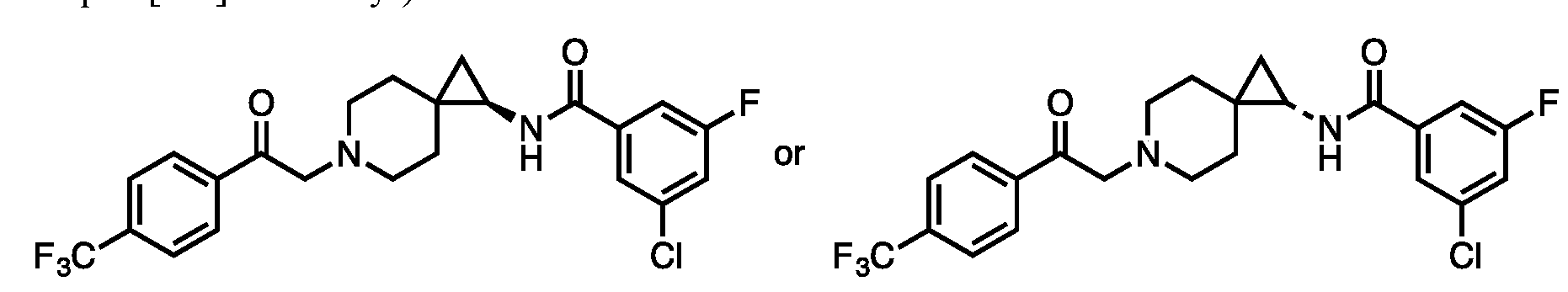

























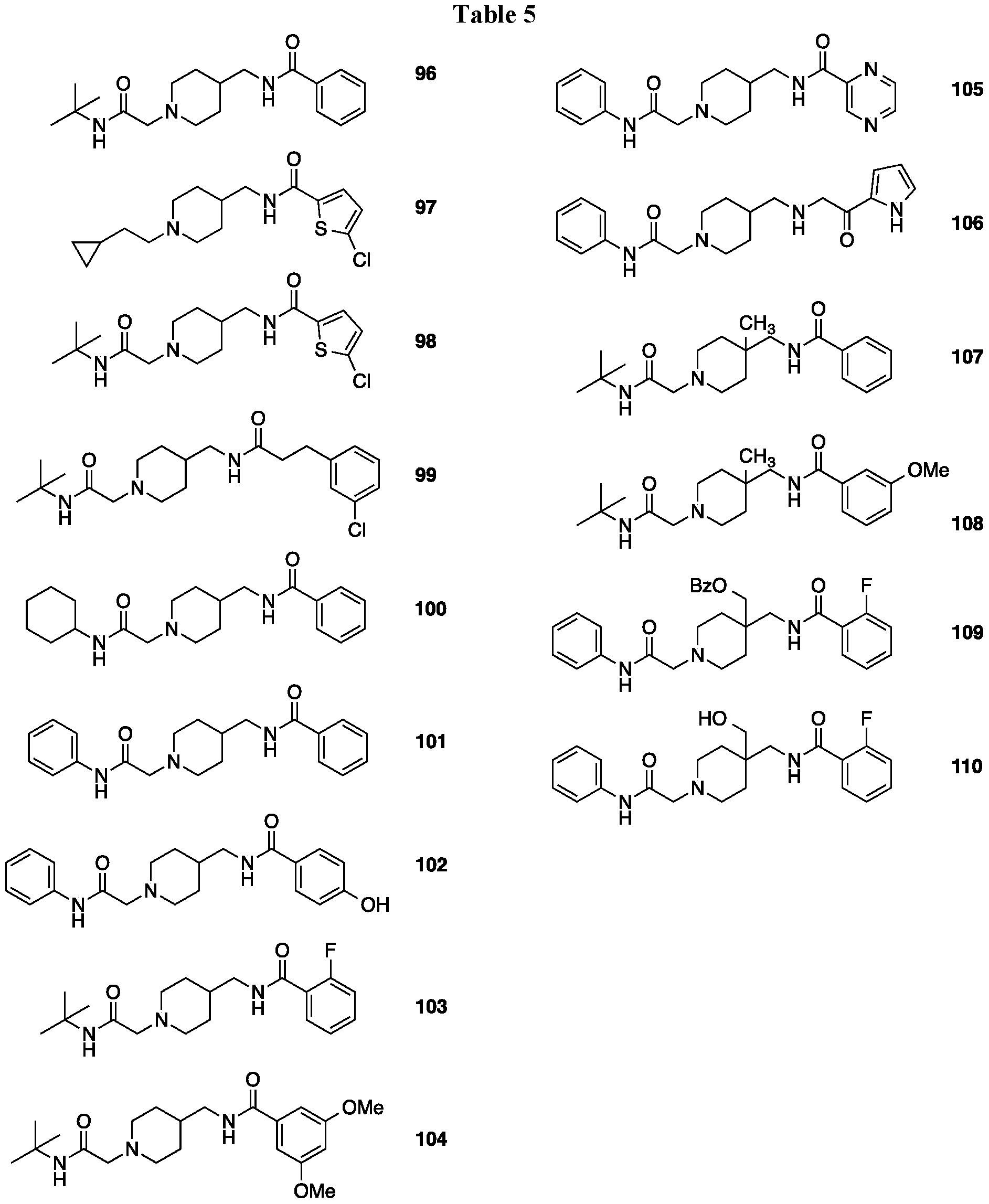

Claims




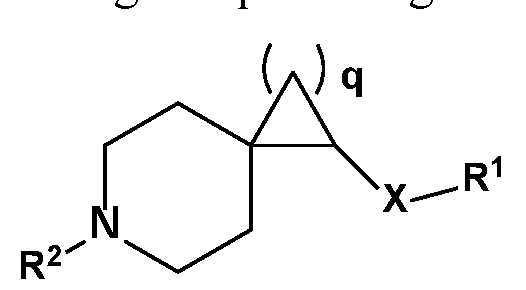





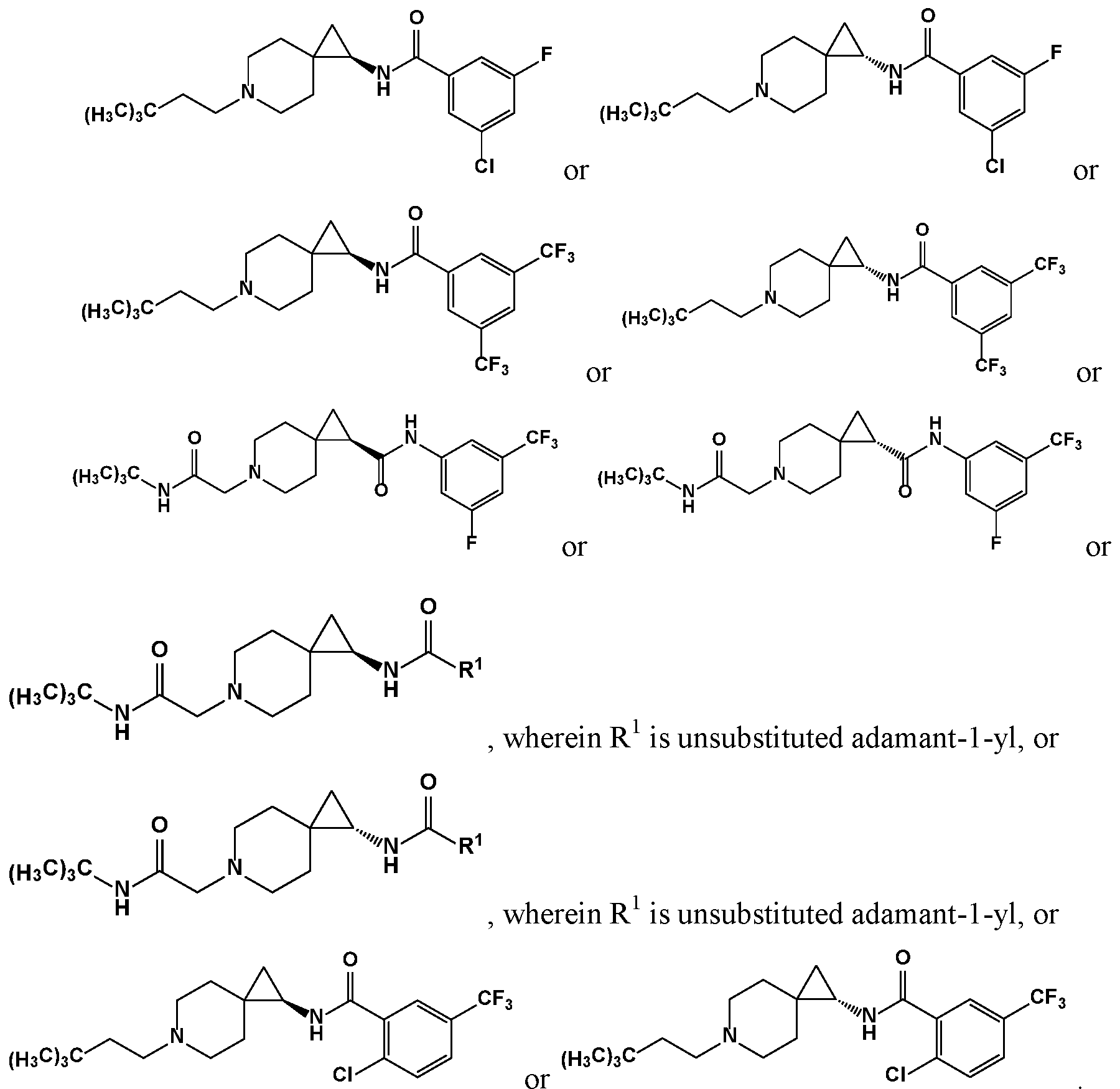

Priority Applications (16)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA3004448A CA3004448A1 (en) | 2015-11-12 | 2016-11-14 | Ion channel inhibitory compounds, pharmaceutical formulations and uses |
| NZ742033A NZ742033A (en) | 2015-11-12 | 2016-11-14 | Ion channel inhibitory compounds, pharmaceutical formulations and uses |
| EP16865245.1A EP3340988A4 (en) | 2015-11-12 | 2016-11-14 | Ion channel inhibitory compounds, pharmaceutical formulations and uses |
| MX2018005242A MX2018005242A (en) | 2015-11-12 | 2016-11-14 | Ion channel inhibitory compounds, pharmaceutical formulations and uses. |
| SG11201803475YA SG11201803475YA (en) | 2015-11-12 | 2016-11-14 | Ion channel inhibitory compounds, pharmaceutical formulations and uses |
| JP2018523749A JP6790094B2 (en) | 2015-11-12 | 2016-11-14 | Ion channel inhibitor compounds, pharmaceutical formulations and uses |
| BR112018009439-0A BR112018009439B1 (en) | 2015-11-12 | 2016-11-14 | ION CHANNEL INHIBITOR COMPOUNDS, PHARMACEUTICAL FORMULATIONS AND USES |
| US15/772,365 US10562857B2 (en) | 2015-11-12 | 2016-11-14 | Ion channel inhibitory compounds, pharmaceutical formulations, and uses |
| CN201680066520.XA CN108289886B (en) | 2015-11-12 | 2016-11-14 | Ion channel inhibiting compounds, pharmaceutical formulations and uses |
| AU2016353446A AU2016353446B2 (en) | 2015-11-12 | 2016-11-14 | Ion channel inhibitory compounds, pharmaceutical formulations and uses |
| KR1020187013376A KR102217524B1 (en) | 2015-11-12 | 2016-11-14 | Ion channel inhibitory compounds, formulations and uses |
| RU2018121288A RU2746188C2 (en) | 2015-11-12 | 2016-11-14 | Ion channel inhibitors, pharmaceutical formulations and applications |
| PH12018501000A PH12018501000A1 (en) | 2015-11-12 | 2018-05-08 | Ion channel inhibitory compounds, pharmaceutical formulations and uses |
| IL259199A IL259199B (en) | 2015-11-12 | 2018-05-08 | Ion channel inhibitory compounds, pharmaceutical formulations and uses |
| ZA2018/03739A ZA201803739B (en) | 2015-11-12 | 2018-06-06 | Ion channel inhibitory compounds, pharmaceutical formulations and uses |
| HK18113486.4A HK1254407A1 (en) | 2015-11-12 | 2018-10-22 | Ion channel inhibitory compounds, pharmaceutical formulations and uses |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562254564P | 2015-11-12 | 2015-11-12 | |
| US62/254,564 | 2015-11-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017083867A1 true WO2017083867A1 (en) | 2017-05-18 |
Family
ID=58696184
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2016/061918 WO2017083867A1 (en) | 2015-11-12 | 2016-11-14 | Ion channel inhibitory compounds, pharmaceutical formulations and uses |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US10562857B2 (en) |
| EP (1) | EP3340988A4 (en) |
| JP (1) | JP6790094B2 (en) |
| KR (1) | KR102217524B1 (en) |
| CN (1) | CN108289886B (en) |
| AU (1) | AU2016353446B2 (en) |
| BR (1) | BR112018009439B1 (en) |
| CA (1) | CA3004448A1 (en) |
| HK (1) | HK1254407A1 (en) |
| IL (1) | IL259199B (en) |
| MX (1) | MX2018005242A (en) |
| NZ (1) | NZ742033A (en) |
| PH (1) | PH12018501000A1 (en) |
| RU (1) | RU2746188C2 (en) |
| SG (1) | SG11201803475YA (en) |
| WO (1) | WO2017083867A1 (en) |
| ZA (1) | ZA201803739B (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108658933A (en) * | 2018-07-13 | 2018-10-16 | 南京大学 | A kind of preparation method of diaryl bithiophene type or diaryl sulfide type deuteroalkyl |
| WO2019014427A1 (en) * | 2017-07-12 | 2019-01-17 | Vanderbilt University | Antagonists of the muscarinic acetylcholine receptor m4 |
| CN110294717A (en) * | 2018-03-21 | 2019-10-01 | 湖南加法检测有限公司 | 2,2- bis- replaces cyclobutanone impurity and its derivative, preparation method and application in pesticide full constituent |
| WO2020047168A1 (en) * | 2018-08-28 | 2020-03-05 | Afasci, Inc. | Methods of treating hypersensitive cough or itching using ion channel inhibitory compounds |
| WO2020223136A1 (en) | 2019-05-02 | 2020-11-05 | Merck Sharp & Dohme Corp. | Spiropiperidine allosteric modulators of nicotinic acetylcholine receptors |
| US11149022B2 (en) | 2017-10-17 | 2021-10-19 | Vanderbilt University | Antagonists of the muscarinic acetylcholine receptor M4 |
| US11325896B2 (en) | 2017-12-20 | 2022-05-10 | Vanderbilt University | Antagonists of the muscarinic acetylcholine receptor M4 |
| US11427540B2 (en) | 2019-07-11 | 2022-08-30 | Praxis Precision Medicines, Inc. | Formulations of T-type calcium channel modulators and methods of use thereof |
| JP7472143B2 (en) | 2018-08-28 | 2024-04-22 | アファスシ,インコーポレイテッド | Methods of Treating Irritable Cough or Itch Using Ion Channel Blocking Compounds - Patent application |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3177277A1 (en) * | 2020-04-29 | 2021-11-04 | Kiran Reddy | Methods of use of t-type calcium channel modulators |
| CN112574085A (en) * | 2020-12-18 | 2021-03-30 | 南通药明康德医药科技有限公司 | Preparation method of 5-amino-2-azaspiro [3.4] octane-2-carboxylic acid tert-butyl ester |
| WO2023150703A2 (en) * | 2022-02-03 | 2023-08-10 | Praxis Precision Medicines, Inc. | Methods of treatment using t-type calcium channel modulators |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997037992A2 (en) | 1996-04-10 | 1997-10-16 | Hoechst Marion Roussel, Inc. | Spiro[cyclopent[b]indole-piperidines] and n-phenyl-hydrazon intermediates for their preparation, both being acetylcholinesterase and mao inhibitors |
| US20100222387A1 (en) | 2005-06-23 | 2010-09-02 | Barrow James C | 3-Fluoro-Piperidine T-Type Calcium Channel Antagonists |
| WO2012047703A2 (en) * | 2010-10-04 | 2012-04-12 | Schering Corporation | Cyclopropyl-spiro-piperidines useful as sodium channel blockers |
| US8377968B2 (en) | 2008-06-02 | 2013-02-19 | Zalicus Pharmaceuticals, Ltd. | N-piperidinyl acetamide derivatives as calcium channel blockers |
| US8501773B2 (en) | 2005-06-29 | 2013-08-06 | Merck Sharp & Dohme Corp. | 4-fluoro-piperidine T-type calcium channel antagonists |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102005030231B4 (en) | 2005-06-29 | 2007-05-31 | Forschungszentrum Karlsruhe Gmbh | Method for applying a high-temperature suitable FeCrAl protective layer, cladding tube with such a protective layer applied and use of such a cladding tube |
| PT2155663T (en) * | 2007-06-15 | 2018-01-16 | Newron Pharm Spa | Substituted 2-[2-(phenyl)ethylamino]alkaneamide derivatives and their use as sodium and/or calcium channel modulators |
| US20140005174A1 (en) | 2011-03-17 | 2014-01-02 | Anilkumar G. Nair | Indole derivatives useful as ccr2 antagonists |
| EP3292110B1 (en) * | 2011-09-22 | 2019-08-21 | Merck Sharp & Dohme B.V. | N-piperidin-4-yl derivatives |
| JP6404717B2 (en) * | 2012-03-02 | 2018-10-17 | ジェネンテック, インコーポレイテッド | Amidospirocyclic amide and sulfonamide derivatives |
| KR101556318B1 (en) * | 2013-05-15 | 2015-10-01 | 한국과학기술연구원 | Novel 6-pyrazolylamido-3-substituted azabicyclo[3.1.0]hexane compounds as calcium channel inhibitors |
| WO2015000949A1 (en) * | 2013-07-03 | 2015-01-08 | F. Hoffmann-La Roche Ag | Heteroaryl pyridone and aza-pyridone amide compounds |
| JP2017001954A (en) | 2013-11-08 | 2017-01-05 | 石原産業株式会社 | Nitrogen-containing saturated heterocyclic compound |
-
2016
- 2016-11-14 CA CA3004448A patent/CA3004448A1/en active Pending
- 2016-11-14 SG SG11201803475YA patent/SG11201803475YA/en unknown
- 2016-11-14 US US15/772,365 patent/US10562857B2/en active Active
- 2016-11-14 NZ NZ742033A patent/NZ742033A/en unknown
- 2016-11-14 BR BR112018009439-0A patent/BR112018009439B1/en active IP Right Grant
- 2016-11-14 RU RU2018121288A patent/RU2746188C2/en active
- 2016-11-14 AU AU2016353446A patent/AU2016353446B2/en active Active
- 2016-11-14 KR KR1020187013376A patent/KR102217524B1/en active IP Right Grant
- 2016-11-14 CN CN201680066520.XA patent/CN108289886B/en active Active
- 2016-11-14 JP JP2018523749A patent/JP6790094B2/en active Active
- 2016-11-14 MX MX2018005242A patent/MX2018005242A/en unknown
- 2016-11-14 WO PCT/US2016/061918 patent/WO2017083867A1/en active Application Filing
- 2016-11-14 EP EP16865245.1A patent/EP3340988A4/en active Pending
-
2018
- 2018-05-08 IL IL259199A patent/IL259199B/en unknown
- 2018-05-08 PH PH12018501000A patent/PH12018501000A1/en unknown
- 2018-06-06 ZA ZA2018/03739A patent/ZA201803739B/en unknown
- 2018-10-22 HK HK18113486.4A patent/HK1254407A1/en unknown
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997037992A2 (en) | 1996-04-10 | 1997-10-16 | Hoechst Marion Roussel, Inc. | Spiro[cyclopent[b]indole-piperidines] and n-phenyl-hydrazon intermediates for their preparation, both being acetylcholinesterase and mao inhibitors |
| US20100222387A1 (en) | 2005-06-23 | 2010-09-02 | Barrow James C | 3-Fluoro-Piperidine T-Type Calcium Channel Antagonists |
| US8501773B2 (en) | 2005-06-29 | 2013-08-06 | Merck Sharp & Dohme Corp. | 4-fluoro-piperidine T-type calcium channel antagonists |
| US8377968B2 (en) | 2008-06-02 | 2013-02-19 | Zalicus Pharmaceuticals, Ltd. | N-piperidinyl acetamide derivatives as calcium channel blockers |
| US20130065926A1 (en) * | 2008-06-02 | 2013-03-14 | Hassan A. Pajouhesh | N-piperidinyl acetamide derivatives as calcium channel blockers |
| US8569344B2 (en) | 2008-06-02 | 2013-10-29 | Zalicus Pharmaceuticals Ltd. | N-piperidinyl acetamide derivatives as calcium channel blockers |
| WO2012047703A2 (en) * | 2010-10-04 | 2012-04-12 | Schering Corporation | Cyclopropyl-spiro-piperidines useful as sodium channel blockers |
Non-Patent Citations (14)
| Title |
|---|
| "Diagnostic and Stastistical Manual of Mental Disorders", 1994, AMERICAN PSYCHIATRIC ASSOCIATION |
| "Handbook of Pharmaceutical Salts, Properties, Selection and Use", 2011, WILEY-VCH |
| "Harrison's Principles of Internal Medicine", 1991, pages: 93 - 98 |
| "Remington: The Science and Practice of Pharmacy", vol. 1, 1995, pages: 176 - 177 |
| BERGE, J. PHARM. SCI., vol. 66, 1977, pages 1 |
| CHEMICAL ABSTRACTS, Columbus, Ohio, US; abstract no. 198133-82-3 |
| J. MED. CHEM., vol. 51, 2008, pages 6471 |
| J. ORG. CHEM., vol. 74, 2009, pages 1304 - 1313 |
| LIU, G.COGAN, D. A.OWENS, T. D.TANG, T. P.ELLMAN, J. A., J. ORG. CHEM., vol. 64, 1999, pages 1278 - 1284 |
| OBACH, R.S. ET AL., J. PHARMACOL. EXP. THER., vol. 283, no. 1, 1997, pages 46 - 58 |
| PETER G. M. WUTS: "Greene's Protective Groups in Organic Synthesis", 2014, JOHN WILEY & SONS |
| See also references of EP3340988A4 |
| TROST, B. M.BOGDANOWICZ, M. J., J. AM. CHEM. SOC., vol. 95, 1973, pages 5298 - 5307 |
| WILLIAMS ET AL., J. OF MED. CHEM., vol. 42, 1999, pages 1481 - 1485 |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019014427A1 (en) * | 2017-07-12 | 2019-01-17 | Vanderbilt University | Antagonists of the muscarinic acetylcholine receptor m4 |
| CN110891569A (en) * | 2017-07-12 | 2020-03-17 | 范德堡大学 | Antagonists of muscarinic acetylcholine receptor M4 |
| US11149022B2 (en) | 2017-10-17 | 2021-10-19 | Vanderbilt University | Antagonists of the muscarinic acetylcholine receptor M4 |
| US11325896B2 (en) | 2017-12-20 | 2022-05-10 | Vanderbilt University | Antagonists of the muscarinic acetylcholine receptor M4 |
| CN110294717B (en) * | 2018-03-21 | 2021-01-19 | 湖南加法检测有限公司 | 2, 2-disubstituted cyclobutanone impurity in pesticide complete component and its derivative, preparation method and application |
| CN110294717A (en) * | 2018-03-21 | 2019-10-01 | 湖南加法检测有限公司 | 2,2- bis- replaces cyclobutanone impurity and its derivative, preparation method and application in pesticide full constituent |
| CN108658933A (en) * | 2018-07-13 | 2018-10-16 | 南京大学 | A kind of preparation method of diaryl bithiophene type or diaryl sulfide type deuteroalkyl |
| US20200069665A1 (en) * | 2018-08-28 | 2020-03-05 | Afasci, Inc. | Methods of treating hypersensitive cough or itching using ion channel inhibitory compounds |
| WO2020047168A1 (en) * | 2018-08-28 | 2020-03-05 | Afasci, Inc. | Methods of treating hypersensitive cough or itching using ion channel inhibitory compounds |
| EP3843731A4 (en) * | 2018-08-28 | 2022-05-11 | Afasci, Inc. | Methods of treating hypersensitive cough or itching using ion channel inhibitory compounds |
| US11744825B2 (en) * | 2018-08-28 | 2023-09-05 | Afasci, Inc. | Methods of treating hypersensitive cough or itching using ion channel inhibitory compounds |
| JP7472143B2 (en) | 2018-08-28 | 2024-04-22 | アファスシ,インコーポレイテッド | Methods of Treating Irritable Cough or Itch Using Ion Channel Blocking Compounds - Patent application |
| WO2020223136A1 (en) | 2019-05-02 | 2020-11-05 | Merck Sharp & Dohme Corp. | Spiropiperidine allosteric modulators of nicotinic acetylcholine receptors |
| EP3962483A4 (en) * | 2019-05-02 | 2023-01-25 | Merck Sharp & Dohme LLC | Spiropiperidine allosteric modulators of nicotinic acetylcholine receptors |
| US11427540B2 (en) | 2019-07-11 | 2022-08-30 | Praxis Precision Medicines, Inc. | Formulations of T-type calcium channel modulators and methods of use thereof |
| US11649207B2 (en) | 2019-07-11 | 2023-05-16 | Praxis Precision Medicines, Inc. | Formulations of T-type calcium channel modulators and methods of use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| IL259199A (en) | 2018-07-31 |
| HK1254407A1 (en) | 2019-07-19 |
| CN108289886B (en) | 2022-01-28 |
| RU2746188C2 (en) | 2021-04-08 |
| RU2018121288A (en) | 2019-12-16 |
| AU2016353446B2 (en) | 2022-05-19 |
| EP3340988A4 (en) | 2019-05-15 |
| IL259199B (en) | 2022-03-01 |
| CN108289886A (en) | 2018-07-17 |
| BR112018009439B1 (en) | 2023-12-12 |
| MX2018005242A (en) | 2019-03-18 |
| BR112018009439A2 (en) | 2018-12-04 |
| RU2018121288A3 (en) | 2020-03-27 |
| CA3004448A1 (en) | 2017-05-18 |
| PH12018501000A1 (en) | 2019-01-28 |
| AU2016353446A1 (en) | 2018-05-24 |
| KR102217524B1 (en) | 2021-02-19 |
| ZA201803739B (en) | 2019-04-24 |
| KR20180074705A (en) | 2018-07-03 |
| US10562857B2 (en) | 2020-02-18 |
| SG11201803475YA (en) | 2018-05-30 |
| EP3340988A1 (en) | 2018-07-04 |
| JP6790094B2 (en) | 2020-11-25 |
| US20180312471A1 (en) | 2018-11-01 |
| NZ742033A (en) | 2022-11-25 |
| JP2018533593A (en) | 2018-11-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2016353446B2 (en) | Ion channel inhibitory compounds, pharmaceutical formulations and uses | |
| KR102479789B1 (en) | Cyclopropyl-amide compounds as dual LSD1/HDAC inhibitors | |
| KR101839137B1 (en) | Oxadiazole Amine Derivative Compounds as Histone Deacetylase 6 Inhibitor, and the Pharmaceutical Composition Comprising the same | |
| JP6830671B2 (en) | Prodrug of γ-hydroxybutyric acid (GHB), its composition and use | |
| JP5989660B2 (en) | Substituted 1-benzylcycloalkyl carboxylic acids and uses thereof | |
| AU2013354189B2 (en) | Cyclopropylamine derivatives useful as inhibitors of histone demethylases KDM1A | |
| TWI655186B (en) | S1P regulator | |
| KR101944559B1 (en) | Branched 3-phenylpropionic acid derivatives and the use thereof | |
| JP5139307B2 (en) | Prolinamide derivatives as sodium channel modulators | |
| CA2721098C (en) | Oxo-heterocyclic substituted carboxylic acid derivates and the use thereof | |
| RU2477721C2 (en) | Substituted n-phenylpyrrolidinyl methylpyrrolidine amides and therapeutic use thereof as modulators of histamine h3 receptor | |
| WO2006001463A1 (en) | Compound having s1p receptor binding potency and use thereof | |
| MX2012004951A (en) | Substituted 3-phenylpropionic acids and the use thereof. | |
| TW202340191A (en) | Tetrahydro-imidazo quinoline compounds as cbp/p300 inhibitors | |
| WO2016109501A1 (en) | Amide compounds as tryptophan hydroxylase inhibitors | |
| CA3217565A1 (en) | Cycloalkyl 3-oxopiperazine carboxamides and cycloheteroalkyl 3-oxopiperazine carboxamides as nav1.8 inhibitors | |
| WO2018140876A1 (en) | Nrf2 activator | |
| CA2979926A1 (en) | Substituted n-bicyclo-2-aryl-quinolin-4-carboxamides and use thereof | |
| TW201821404A (en) | Tricyclic sulfones as ROR gamma modulators | |
| WO2020154431A1 (en) | Inhibitors of spinster homolog 2 (spns2) for use in therapy | |
| KR20180094946A (en) | Alkyl dihydroquinoline sulfonamide compound | |
| TW202233620A (en) | Cftr modulator compounds, compositions, and uses thereof | |
| JP2005510475A6 (en) | Tetrahydroisoquinolines, methods for their production, and their use as anesthetics | |
| JP2024517678A (en) | Modulators of sortilin activity | |
| NZ626745B2 (en) | Cyclic amide derivatives as inhibitors of 11 - beta - hydroxysteroid dehydrogenase and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16865245 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016865245 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11201803475Y Country of ref document: SG Ref document number: MX/A/2018/005242 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15772365 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018523749 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 3004448 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 259199 Country of ref document: IL Ref document number: 12018501000 Country of ref document: PH |
|
| ENP | Entry into the national phase |
Ref document number: 20187013376 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112018009439 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2016353446 Country of ref document: AU Date of ref document: 20161114 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018121288 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: 112018009439 Country of ref document: BR Kind code of ref document: A2 Effective date: 20180509 |