WO2017064627A2 - Process for preparation of eribulin and intermediates thereof - Google Patents
Process for preparation of eribulin and intermediates thereof Download PDFInfo
- Publication number
- WO2017064627A2 WO2017064627A2 PCT/IB2016/056097 IB2016056097W WO2017064627A2 WO 2017064627 A2 WO2017064627 A2 WO 2017064627A2 IB 2016056097 W IB2016056097 W IB 2016056097W WO 2017064627 A2 WO2017064627 A2 WO 2017064627A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- protecting group
- alcohol
- silyl
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 84
- 238000002360 preparation method Methods 0.000 title claims abstract description 44
- UFNVPOGXISZXJD-XJPMSQCNSA-N eribulin Chemical compound C([C@H]1CC[C@@H]2O[C@@H]3[C@H]4O[C@H]5C[C@](O[C@H]4[C@H]2O1)(O[C@@H]53)CC[C@@H]1O[C@H](C(C1)=C)CC1)C(=O)C[C@@H]2[C@@H](OC)[C@@H](C[C@H](O)CN)O[C@H]2C[C@@H]2C(=C)[C@H](C)C[C@H]1O2 UFNVPOGXISZXJD-XJPMSQCNSA-N 0.000 title claims abstract description 14
- 229960003649 eribulin Drugs 0.000 title claims abstract description 14
- 239000000543 intermediate Substances 0.000 title description 10
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims abstract description 13
- 150000001875 compounds Chemical class 0.000 claims description 214
- -1 acetylene compound Chemical class 0.000 claims description 76
- 125000006241 alcohol protecting group Chemical group 0.000 claims description 69
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 claims description 31
- 229910052736 halogen Inorganic materials 0.000 claims description 28
- 150000002367 halogens Chemical group 0.000 claims description 28
- 125000004665 trialkylsilyl group Chemical group 0.000 claims description 21
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 claims description 17
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 13
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 13
- 229910052794 bromium Inorganic materials 0.000 claims description 13
- 125000006239 protecting group Chemical group 0.000 claims description 10
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 9
- 239000001257 hydrogen Substances 0.000 claims description 9
- 229910052739 hydrogen Inorganic materials 0.000 claims description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 8
- 125000003118 aryl group Chemical group 0.000 claims description 7
- 150000002440 hydroxy compounds Chemical class 0.000 claims description 7
- 150000003839 salts Chemical class 0.000 claims description 6
- 229920002554 vinyl polymer Polymers 0.000 claims description 6
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 claims description 5
- MLUCVPSAIODCQM-NSCUHMNNSA-N crotonaldehyde Chemical compound C\C=C\C=O MLUCVPSAIODCQM-NSCUHMNNSA-N 0.000 claims description 5
- MLUCVPSAIODCQM-UHFFFAOYSA-N crotonaldehyde Natural products CC=CC=O MLUCVPSAIODCQM-UHFFFAOYSA-N 0.000 claims description 5
- 239000011630 iodine Substances 0.000 claims description 5
- 229910052740 iodine Inorganic materials 0.000 claims description 5
- 150000002118 epoxides Chemical class 0.000 claims description 3
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 claims 1
- TXBFKXDFIPFHAV-UHFFFAOYSA-N 3-methylideneoxolane Chemical compound C=C1CCOC1 TXBFKXDFIPFHAV-UHFFFAOYSA-N 0.000 abstract description 6
- FXNFULJVOQMBCW-VZBLNRDYSA-N halichondrin b Chemical class O([C@@H]1[C@@H](C)[C@@H]2O[C@@H]3C[C@@]4(O[C@H]5[C@@H](C)C[C@@]6(C[C@@H]([C@@H]7O[C@@H](C[C@@H]7O6)[C@@H](O)C[C@@H](O)CO)C)O[C@H]5C4)O[C@@H]3C[C@@H]2O[C@H]1C[C@@H]1C(=C)[C@H](C)C[C@@H](O1)CC[C@H]1C(=C)C[C@@H](O1)CC1)C(=O)C[C@H](O2)CC[C@H]3[C@H]2[C@H](O2)[C@@H]4O[C@@H]5C[C@@]21O[C@@H]5[C@@H]4O3 FXNFULJVOQMBCW-VZBLNRDYSA-N 0.000 abstract description 4
- 238000006243 chemical reaction Methods 0.000 description 96
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 65
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 59
- 239000000243 solution Substances 0.000 description 48
- 239000011541 reaction mixture Substances 0.000 description 45
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 42
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 42
- 239000007787 solid Substances 0.000 description 41
- 239000000047 product Substances 0.000 description 40
- 239000003153 chemical reaction reagent Substances 0.000 description 39
- 238000001914 filtration Methods 0.000 description 38
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 34
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 33
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 30
- 239000000203 mixture Substances 0.000 description 29
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 27
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 27
- 235000019439 ethyl acetate Nutrition 0.000 description 27
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 25
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 24
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 24
- 239000010410 layer Substances 0.000 description 24
- 150000002170 ethers Chemical class 0.000 description 23
- 239000012044 organic layer Substances 0.000 description 23
- 239000002904 solvent Substances 0.000 description 23
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 22
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 22
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 21
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 20
- 125000001931 aliphatic group Chemical group 0.000 description 20
- 229930195733 hydrocarbon Natural products 0.000 description 20
- 239000003880 polar aprotic solvent Substances 0.000 description 20
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 18
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 18
- 238000005119 centrifugation Methods 0.000 description 18
- 238000010908 decantation Methods 0.000 description 18
- 238000000605 extraction Methods 0.000 description 18
- 230000005484 gravity Effects 0.000 description 18
- 239000002245 particle Substances 0.000 description 18
- 238000010791 quenching Methods 0.000 description 18
- 230000000171 quenching effect Effects 0.000 description 18
- 238000000967 suction filtration Methods 0.000 description 18
- 238000010626 work up procedure Methods 0.000 description 18
- 150000008282 halocarbons Chemical class 0.000 description 16
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 15
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 15
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 15
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 14
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 14
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 14
- 239000002585 base Substances 0.000 description 14
- 239000003054 catalyst Substances 0.000 description 14
- 150000002825 nitriles Chemical class 0.000 description 14
- 239000010948 rhodium Substances 0.000 description 14
- 150000002576 ketones Chemical class 0.000 description 13
- XLSZMDLNRCVEIJ-UHFFFAOYSA-N 4-methylimidazole Chemical compound CC1=CNC=N1 XLSZMDLNRCVEIJ-UHFFFAOYSA-N 0.000 description 12
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 12
- ZMXDDKWLCZADIW-UHFFFAOYSA-N dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 12
- 239000003921 oil Substances 0.000 description 11
- 229910000027 potassium carbonate Inorganic materials 0.000 description 11
- 235000011181 potassium carbonates Nutrition 0.000 description 11
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 10
- 239000012043 crude product Substances 0.000 description 10
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 10
- 239000011736 potassium bicarbonate Substances 0.000 description 10
- 235000015497 potassium bicarbonate Nutrition 0.000 description 10
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 10
- 239000011734 sodium Substances 0.000 description 10
- 229910000029 sodium carbonate Inorganic materials 0.000 description 10
- 235000017550 sodium carbonate Nutrition 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 9
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000012267 brine Substances 0.000 description 9
- 239000013058 crude material Substances 0.000 description 9
- 150000002148 esters Chemical class 0.000 description 9
- 238000003818 flash chromatography Methods 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 9
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 9
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 9
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 8
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 8
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 8
- LEHBURLTIWGHEM-UHFFFAOYSA-N pyridinium chlorochromate Chemical compound [O-][Cr](Cl)(=O)=O.C1=CC=[NH+]C=C1 LEHBURLTIWGHEM-UHFFFAOYSA-N 0.000 description 8
- 239000011347 resin Substances 0.000 description 8
- 229920005989 resin Polymers 0.000 description 8
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 8
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 7
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 7
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-diisopropylethylamine Substances CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 7
- 150000001298 alcohols Chemical class 0.000 description 7
- 238000002955 isolation Methods 0.000 description 7
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 7
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 7
- 150000007530 organic bases Chemical class 0.000 description 7
- 229910052708 sodium Inorganic materials 0.000 description 7
- LXBGSDVWAMZHDD-UHFFFAOYSA-N 2-methyl-1h-imidazole Chemical compound CC1=NC=CN1 LXBGSDVWAMZHDD-UHFFFAOYSA-N 0.000 description 6
- 0 CC=*CC#CCCC(C1)C(CC*2CC2)CC1*1CCC1 Chemical compound CC=*CC#CCCC(C1)C(CC*2CC2)CC1*1CCC1 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- 150000001340 alkali metals Chemical class 0.000 description 6
- 150000004703 alkoxides Chemical class 0.000 description 6
- HOPRXXXSABQWAV-UHFFFAOYSA-N anhydrous collidine Natural products CC1=CC=NC(C)=C1C HOPRXXXSABQWAV-UHFFFAOYSA-N 0.000 description 6
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 6
- 229910000024 caesium carbonate Inorganic materials 0.000 description 6
- 229910000019 calcium carbonate Inorganic materials 0.000 description 6
- 235000010216 calcium carbonate Nutrition 0.000 description 6
- 125000004432 carbon atom Chemical group C* 0.000 description 6
- UTBIMNXEDGNJFE-UHFFFAOYSA-N collidine Natural products CC1=CC=C(C)C(C)=N1 UTBIMNXEDGNJFE-UHFFFAOYSA-N 0.000 description 6
- 239000003446 ligand Substances 0.000 description 6
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 6
- 239000001095 magnesium carbonate Substances 0.000 description 6
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 6
- 235000014380 magnesium carbonate Nutrition 0.000 description 6
- 150000002894 organic compounds Chemical class 0.000 description 6
- BDAWXSQJJCIFIK-UHFFFAOYSA-N potassium methoxide Chemical compound [K+].[O-]C BDAWXSQJJCIFIK-UHFFFAOYSA-N 0.000 description 6
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 6
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 6
- GFYHSKONPJXCDE-UHFFFAOYSA-N sym-collidine Natural products CC1=CN=C(C)C(C)=C1 GFYHSKONPJXCDE-UHFFFAOYSA-N 0.000 description 6
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 6
- VVDJVCJVVHHCIB-UHFFFAOYSA-N 1-bromoethenyl(trimethyl)silane Chemical compound C[Si](C)(C)C(Br)=C VVDJVCJVVHHCIB-UHFFFAOYSA-N 0.000 description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 5
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 5
- 229910000272 alkali metal oxide Inorganic materials 0.000 description 5
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 5
- 125000000217 alkyl group Chemical group 0.000 description 5
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 5
- 239000000920 calcium hydroxide Substances 0.000 description 5
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 5
- 229910052799 carbon Inorganic materials 0.000 description 5
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 5
- 239000011591 potassium Substances 0.000 description 5
- 229910052700 potassium Inorganic materials 0.000 description 5
- 150000003333 secondary alcohols Chemical class 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- AVFZOVWCLRSYKC-UHFFFAOYSA-N 1-methylpyrrolidine Chemical compound CN1CCCC1 AVFZOVWCLRSYKC-UHFFFAOYSA-N 0.000 description 4
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 4
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 4
- FEJUGLKDZJDVFY-UHFFFAOYSA-N 9-borabicyclo(3.3.1)nonane Chemical compound C1CCC2CCCC1B2 FEJUGLKDZJDVFY-UHFFFAOYSA-N 0.000 description 4
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 4
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 4
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 4
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- AHVYPIQETPWLSZ-UHFFFAOYSA-N N-methyl-pyrrolidine Natural products CN1CC=CC1 AHVYPIQETPWLSZ-UHFFFAOYSA-N 0.000 description 4
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 150000007513 acids Chemical group 0.000 description 4
- 150000001299 aldehydes Chemical class 0.000 description 4
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 4
- 229910021529 ammonia Inorganic materials 0.000 description 4
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 4
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 4
- SIPUZPBQZHNSDW-UHFFFAOYSA-N diisobutylaluminium hydride Substances CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 4
- 235000019253 formic acid Nutrition 0.000 description 4
- AQTUHJABKZECGA-UHFFFAOYSA-N hept-1-en-4-ol Chemical compound CCCC(O)CC=C AQTUHJABKZECGA-UHFFFAOYSA-N 0.000 description 4
- 229940071870 hydroiodic acid Drugs 0.000 description 4
- 238000011065 in-situ storage Methods 0.000 description 4
- 239000003456 ion exchange resin Substances 0.000 description 4
- 229920003303 ion-exchange polymer Polymers 0.000 description 4
- 229910052744 lithium Inorganic materials 0.000 description 4
- 229910052749 magnesium Inorganic materials 0.000 description 4
- 239000011777 magnesium Substances 0.000 description 4
- 229910052751 metal Inorganic materials 0.000 description 4
- 239000002184 metal Substances 0.000 description 4
- 229910021645 metal ion Inorganic materials 0.000 description 4
- 229940098779 methanesulfonic acid Drugs 0.000 description 4
- GDOPTJXRTPNYNR-UHFFFAOYSA-N methyl-cyclopentane Natural products CC1CCCC1 GDOPTJXRTPNYNR-UHFFFAOYSA-N 0.000 description 4
- 229910017604 nitric acid Inorganic materials 0.000 description 4
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 4
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 4
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 4
- 150000003138 primary alcohols Chemical class 0.000 description 4
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 4
- IARSSOVWSJAVSZ-UHFFFAOYSA-N tris(dimethylamino)sulfanium Chemical compound CN(C)[S+](N(C)C)N(C)C IARSSOVWSJAVSZ-UHFFFAOYSA-N 0.000 description 4
- WGECXQBGLLYSFP-UHFFFAOYSA-N 2,3-dimethylpentane Chemical compound CCC(C)C(C)C WGECXQBGLLYSFP-UHFFFAOYSA-N 0.000 description 3
- BZHMBWZPUJHVEE-UHFFFAOYSA-N 2,3-dimethylpentane Natural products CC(C)CC(C)C BZHMBWZPUJHVEE-UHFFFAOYSA-N 0.000 description 3
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 3
- ZNORAFJUESSLTM-UHFFFAOYSA-N [4-[5-bis(3,5-ditert-butyl-4-methoxyphenyl)phosphanyl-1,3-benzodioxol-4-yl]-1,3-benzodioxol-5-yl]-bis(3,5-ditert-butyl-4-methoxyphenyl)phosphane Chemical compound C1=C(C(C)(C)C)C(OC)=C(C(C)(C)C)C=C1P(C=1C(=C2OCOC2=CC=1)C=1C(=CC=C2OCOC2=1)P(C=1C=C(C(OC)=C(C=1)C(C)(C)C)C(C)(C)C)C=1C=C(C(OC)=C(C=1)C(C)(C)C)C(C)(C)C)C1=CC(C(C)(C)C)=C(OC)C(C(C)(C)C)=C1 ZNORAFJUESSLTM-UHFFFAOYSA-N 0.000 description 3
- 230000003213 activating effect Effects 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 3
- 239000010779 crude oil Substances 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- AFABGHUZZDYHJO-UHFFFAOYSA-N dimethyl butane Natural products CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 3
- 150000002430 hydrocarbons Chemical class 0.000 description 3
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 239000012280 lithium aluminium hydride Substances 0.000 description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 3
- 239000012266 salt solution Substances 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 3
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 2
- DRZBLHZZDMCPGX-FGZHOGPDSA-N (s)-tert-butyl-[3-[tert-butyl(methyl)phosphanyl]quinoxalin-2-yl]-methylphosphane Chemical compound C1=CC=C2N=C([P@@](C)C(C)(C)C)C([P@@](C)C(C)(C)C)=NC2=C1 DRZBLHZZDMCPGX-FGZHOGPDSA-N 0.000 description 2
- GETTZEONDQJALK-UHFFFAOYSA-N (trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC=CC=C1 GETTZEONDQJALK-UHFFFAOYSA-N 0.000 description 2
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 2
- UBOXGVDOUJQMTN-UHFFFAOYSA-N 1,1,2-trichloroethane Chemical compound ClCC(Cl)Cl UBOXGVDOUJQMTN-UHFFFAOYSA-N 0.000 description 2
- FYGHSUNMUKGBRK-UHFFFAOYSA-N 1,2,3-trimethylbenzene Chemical compound CC1=CC=CC(C)=C1C FYGHSUNMUKGBRK-UHFFFAOYSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- GJFNRSDCSTVPCJ-UHFFFAOYSA-N 1,8-bis(dimethylamino)naphthalene Chemical compound C1=CC(N(C)C)=C2C(N(C)C)=CC=CC2=C1 GJFNRSDCSTVPCJ-UHFFFAOYSA-N 0.000 description 2
- HNRMPXKDFBEGFZ-UHFFFAOYSA-N 2,2-dimethylbutane Chemical compound CCC(C)(C)C HNRMPXKDFBEGFZ-UHFFFAOYSA-N 0.000 description 2
- ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 2,3-dimethylbutane Chemical compound CC(C)C(C)C ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 0.000 description 2
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 2
- GXDHCNNESPLIKD-UHFFFAOYSA-N 2-methylhexane Natural products CCCCC(C)C GXDHCNNESPLIKD-UHFFFAOYSA-N 0.000 description 2
- AEXMKKGTQYQZCS-UHFFFAOYSA-N 3,3-dimethylpentane Chemical compound CCC(C)(C)CC AEXMKKGTQYQZCS-UHFFFAOYSA-N 0.000 description 2
- LAIUFBWHERIJIH-UHFFFAOYSA-N 3-Methylheptane Chemical compound CCCCC(C)CC LAIUFBWHERIJIH-UHFFFAOYSA-N 0.000 description 2
- QCHPKSFMDHPSNR-UHFFFAOYSA-N 3-aminoisobutyric acid Chemical compound NCC(C)C(O)=O QCHPKSFMDHPSNR-UHFFFAOYSA-N 0.000 description 2
- AORMDLNPRGXHHL-UHFFFAOYSA-N 3-ethylpentane Chemical compound CCC(CC)CC AORMDLNPRGXHHL-UHFFFAOYSA-N 0.000 description 2
- VLJXXKKOSFGPHI-UHFFFAOYSA-N 3-methylhexane Chemical compound CCCC(C)CC VLJXXKKOSFGPHI-UHFFFAOYSA-N 0.000 description 2
- PFEOZHBOMNWTJB-UHFFFAOYSA-N 3-methylpentane Chemical compound CCC(C)CC PFEOZHBOMNWTJB-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N Ethylbenzene Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- UUIQMZJEGPQKFD-UHFFFAOYSA-N Methyl butyrate Chemical compound CCCC(=O)OC UUIQMZJEGPQKFD-UHFFFAOYSA-N 0.000 description 2
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 2
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 238000006405 Nozaki-Hiyama-Kishi reaction Methods 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical group CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- URLKBWYHVLBVBO-UHFFFAOYSA-N Para-Xylene Chemical group CC1=CC=C(C)C=C1 URLKBWYHVLBVBO-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- QYTDEUPAUMOIOP-UHFFFAOYSA-N TEMPO Chemical group CC1(C)CCCC(C)(C)N1[O] QYTDEUPAUMOIOP-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- ZBIKORITPGTTGI-UHFFFAOYSA-N [acetyloxy(phenyl)-$l^{3}-iodanyl] acetate Chemical compound CC(=O)OI(OC(C)=O)C1=CC=CC=C1 ZBIKORITPGTTGI-UHFFFAOYSA-N 0.000 description 2
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 2
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 2
- INLLPKCGLOXCIV-UHFFFAOYSA-N bromoethene Chemical class BrC=C INLLPKCGLOXCIV-UHFFFAOYSA-N 0.000 description 2
- DKPFZGUDAPQIHT-UHFFFAOYSA-N butyl acetate Chemical compound CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- DMEGYFMYUHOHGS-UHFFFAOYSA-N cycloheptane Chemical compound C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- VURFVHCLMJOLKN-UHFFFAOYSA-N diphosphane Chemical compound PP VURFVHCLMJOLKN-UHFFFAOYSA-N 0.000 description 2
- OBNCKNCVKJNDBV-UHFFFAOYSA-N ethyl butyrate Chemical compound CCCC(=O)OCC OBNCKNCVKJNDBV-UHFFFAOYSA-N 0.000 description 2
- FKRCODPIKNYEAC-UHFFFAOYSA-N ethyl propionate Chemical compound CCOC(=O)CC FKRCODPIKNYEAC-UHFFFAOYSA-N 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- 230000026030 halogenation Effects 0.000 description 2
- 238000005658 halogenation reaction Methods 0.000 description 2
- 238000005647 hydrohalogenation reaction Methods 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N indane Chemical compound C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 2
- QWTDNUCVQCZILF-UHFFFAOYSA-N isopentane Chemical compound CCC(C)C QWTDNUCVQCZILF-UHFFFAOYSA-N 0.000 description 2
- 229960004592 isopropanol Drugs 0.000 description 2
- IVSZLXZYQVIEFR-UHFFFAOYSA-N m-xylene Chemical group CC1=CC=CC(C)=C1 IVSZLXZYQVIEFR-UHFFFAOYSA-N 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 2
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 2
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- CRSOQBOWXPBRES-UHFFFAOYSA-N neopentane Chemical compound CC(C)(C)C CRSOQBOWXPBRES-UHFFFAOYSA-N 0.000 description 2
- KPSSIOMAKSHJJG-UHFFFAOYSA-N neopentyl alcohol Chemical compound CC(C)(C)CO KPSSIOMAKSHJJG-UHFFFAOYSA-N 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- AQIXEPGDORPWBJ-UHFFFAOYSA-N pentan-3-ol Chemical compound CCC(O)CC AQIXEPGDORPWBJ-UHFFFAOYSA-N 0.000 description 2
- FDPIMTJIUBPUKL-UHFFFAOYSA-N pentan-3-one Chemical compound CCC(=O)CC FDPIMTJIUBPUKL-UHFFFAOYSA-N 0.000 description 2
- 229960005235 piperonyl butoxide Drugs 0.000 description 2
- 238000000634 powder X-ray diffraction Methods 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- YKYONYBAUNKHLG-UHFFFAOYSA-N propyl acetate Chemical compound CCCOC(C)=O YKYONYBAUNKHLG-UHFFFAOYSA-N 0.000 description 2
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 2
- 229910000077 silane Inorganic materials 0.000 description 2
- 238000004808 supercritical fluid chromatography Methods 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- BWIVUQBSKOJUJI-VTFAUUCOSA-N tert-butyl-[3-[(2S,5S)-5-[(3R,5R)-3-[tert-butyl(dimethyl)silyl]oxy-5-methyl-7-tri(propan-2-yl)silylhept-6-ynyl]-4-methylideneoxolan-2-yl]propoxy]-diphenylsilane Chemical compound C(C)(C)(C)[Si](C1=CC=CC=C1)(C1=CC=CC=C1)OCCC[C@@H]1O[C@H](C(C1)=C)CC[C@H](C[C@H](C#C[Si](C(C)C)(C(C)C)C(C)C)C)O[Si](C)(C)C(C)(C)C BWIVUQBSKOJUJI-VTFAUUCOSA-N 0.000 description 2
- BLHYCNPXNDAJTO-MNPUUQGPSA-N tert-butyl-[3-[(2S,5S)-5-[(3R,5R)-3-[tert-butyl(dimethyl)silyl]oxy-5-methylhept-6-ynyl]-4-methylideneoxolan-2-yl]propoxy]-dimethylsilane Chemical compound C(C)(C)(C)[Si](C)(C)OCCC[C@@H]1O[C@H](C(C1)=C)CC[C@H](C[C@H](C#C)C)O[Si](C)(C)C(C)(C)C BLHYCNPXNDAJTO-MNPUUQGPSA-N 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 2
- DBGVGMSCBYYSLD-UHFFFAOYSA-N tributylstannane Chemical compound CCCC[SnH](CCCC)CCCC DBGVGMSCBYYSLD-UHFFFAOYSA-N 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- ZISSAWUMDACLOM-UHFFFAOYSA-N triptane Chemical compound CC(C)C(C)(C)C ZISSAWUMDACLOM-UHFFFAOYSA-N 0.000 description 2
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 2
- UBKCMQCDHVYUAF-UHFFFAOYSA-N (2,2-dichloro-3-diphenylphosphanylpropyl)-diphenylphosphane Chemical compound ClC(CP(C1=CC=CC=C1)C1=CC=CC=C1)(CP(C1=CC=CC=C1)C1=CC=CC=C1)Cl UBKCMQCDHVYUAF-UHFFFAOYSA-N 0.000 description 1
- BIIBYWQGRFWQKM-JVVROLKMSA-N (2S)-N-[4-(cyclopropylamino)-3,4-dioxo-1-[(3S)-2-oxopyrrolidin-3-yl]butan-2-yl]-2-[[(E)-3-(2,4-dichlorophenyl)prop-2-enoyl]amino]-4,4-dimethylpentanamide Chemical compound CC(C)(C)C[C@@H](C(NC(C[C@H](CCN1)C1=O)C(C(NC1CC1)=O)=O)=O)NC(/C=C/C(C=CC(Cl)=C1)=C1Cl)=O BIIBYWQGRFWQKM-JVVROLKMSA-N 0.000 description 1
- RBVGOQHQBUPSGX-ZJZGAYNASA-N (2s,5s)-1-[2-[(2s,5s)-2,5-di(propan-2-yl)phospholan-1-yl]phenyl]-2,5-di(propan-2-yl)phospholane Chemical compound CC(C)[C@@H]1CC[C@@H](C(C)C)P1C1=CC=CC=C1P1[C@H](C(C)C)CC[C@H]1C(C)C RBVGOQHQBUPSGX-ZJZGAYNASA-N 0.000 description 1
- IRCDUOCGSIGEAI-XUXIUFHCSA-N (2s,5s)-1-[2-[(2s,5s)-2,5-dimethylphospholan-1-yl]ethyl]-2,5-dimethylphospholane Chemical compound C[C@H]1CC[C@H](C)P1CCP1[C@@H](C)CC[C@@H]1C IRCDUOCGSIGEAI-XUXIUFHCSA-N 0.000 description 1
- ZZDPPKWLRNLKEU-UKMLZYKCSA-N (3R,5R)-1-[(2S,5S)-5-(3-hydroxypropyl)-3-methylideneoxolan-2-yl]-5-methylhept-6-yn-3-ol Chemical compound OCCC[C@H]1CC([C@@H](O1)CC[C@H](C[C@H](C#C)C)O)=C ZZDPPKWLRNLKEU-UKMLZYKCSA-N 0.000 description 1
- DKJMCYPXFCHYCK-FWYOQMDTSA-N (3R,5R)-1-[(2S,5S)-5-(3-hydroxypropyl)-3-methylideneoxolan-2-yl]-6-iodo-5-methylhept-6-en-3-ol Chemical compound OCCC[C@H]1CC([C@@H](O1)CC[C@H](C[C@H](C(=C)I)C)O)=C DKJMCYPXFCHYCK-FWYOQMDTSA-N 0.000 description 1
- QIVUCLWGARAQIO-OLIXTKCUSA-N (3s)-n-[(3s,5s,6r)-6-methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-yl]-2-oxospiro[1h-pyrrolo[2,3-b]pyridine-3,6'-5,7-dihydrocyclopenta[b]pyridine]-3'-carboxamide Chemical compound C1([C@H]2[C@H](N(C(=O)[C@@H](NC(=O)C=3C=C4C[C@]5(CC4=NC=3)C3=CC=CN=C3NC5=O)C2)CC(F)(F)F)C)=C(F)C=CC(F)=C1F QIVUCLWGARAQIO-OLIXTKCUSA-N 0.000 description 1
- YCBUVNQBVZVSFY-OAHLLOKOSA-N (4R)-7-[tert-butyl(dimethyl)silyl]oxy-2-trimethylsilylhept-1-en-4-ol Chemical compound [Si](C)(C)(C(C)(C)C)OCCC[C@H](CC(=C)[Si](C)(C)C)O YCBUVNQBVZVSFY-OAHLLOKOSA-N 0.000 description 1
- ONGNQLQQVGZJDZ-XZOQPEGZSA-N (4R,6R)-4-[tert-butyl(dimethyl)silyl]oxy-6-methyl-8-tri(propan-2-yl)silyloct-7-yn-1-ol Chemical compound [Si](C)(C)(C(C)(C)C)O[C@H](CCCO)C[C@H](C#C[Si](C(C)C)(C(C)C)C(C)C)C ONGNQLQQVGZJDZ-XZOQPEGZSA-N 0.000 description 1
- USBXQGGSAFINOC-XZOQPEGZSA-N (4R,6R)-4-[tert-butyl(dimethyl)silyl]oxy-6-methyl-8-tri(propan-2-yl)silyloct-7-ynal Chemical compound [Si](C)(C)(C(C)(C)C)O[C@H](CCC=O)C[C@H](C#C[Si](C(C)C)(C(C)C)C(C)C)C USBXQGGSAFINOC-XZOQPEGZSA-N 0.000 description 1
- VQECHDYCJMBWSQ-ZWKOTPCHSA-N (4R,6R)-6-methyl-8-tri(propan-2-yl)silyloct-1-en-7-yn-4-ol Chemical compound C[C@H](C[C@@H](CC=C)O)C#C[Si](C(C)C)(C(C)C)C(C)C VQECHDYCJMBWSQ-ZWKOTPCHSA-N 0.000 description 1
- BYEAHWXPCBROCE-UHFFFAOYSA-N 1,1,1,3,3,3-hexafluoropropan-2-ol Chemical compound FC(F)(F)C(O)C(F)(F)F BYEAHWXPCBROCE-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 1
- LVEYOSJUKRVCCF-UHFFFAOYSA-N 1,3-Bis(diphenylphosphino)propane Substances C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCP(C=1C=CC=CC=1)C1=CC=CC=C1 LVEYOSJUKRVCCF-UHFFFAOYSA-N 0.000 description 1
- RDZHCKRAHUPIFK-UHFFFAOYSA-N 1,3-diiodo-5,5-dimethylimidazolidine-2,4-dione Chemical compound CC1(C)N(I)C(=O)N(I)C1=O RDZHCKRAHUPIFK-UHFFFAOYSA-N 0.000 description 1
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 1
- RRQYJINTUHWNHW-UHFFFAOYSA-N 1-ethoxy-2-(2-ethoxyethoxy)ethane Chemical compound CCOCCOCCOCC RRQYJINTUHWNHW-UHFFFAOYSA-N 0.000 description 1
- GPAAEZIXSQCCES-UHFFFAOYSA-N 1-methoxy-2-(2-methoxyethoxymethoxymethoxy)ethane Chemical compound COCCOCOCOCCOC GPAAEZIXSQCCES-UHFFFAOYSA-N 0.000 description 1
- SDTORDSXCYSNTD-UHFFFAOYSA-N 1-methoxy-4-[(4-methoxyphenyl)methoxymethyl]benzene Chemical compound C1=CC(OC)=CC=C1COCC1=CC=C(OC)C=C1 SDTORDSXCYSNTD-UHFFFAOYSA-N 0.000 description 1
- 229940044613 1-propanol Drugs 0.000 description 1
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 1
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 1
- FJSKXQVRKZTKSI-UHFFFAOYSA-N 2,3-dimethylfuran Chemical compound CC=1C=COC=1C FJSKXQVRKZTKSI-UHFFFAOYSA-N 0.000 description 1
- YHGKEORTCHVBQH-UHFFFAOYSA-M 2,4,6-tri(propan-2-yl)benzenesulfonate Chemical compound CC(C)C1=CC(C(C)C)=C(S([O-])(=O)=O)C(C(C)C)=C1 YHGKEORTCHVBQH-UHFFFAOYSA-M 0.000 description 1
- JAPYIBBSTJFDAK-UHFFFAOYSA-N 2,4,6-tri(propan-2-yl)benzenesulfonyl chloride Chemical compound CC(C)C1=CC(C(C)C)=C(S(Cl)(=O)=O)C(C(C)C)=C1 JAPYIBBSTJFDAK-UHFFFAOYSA-N 0.000 description 1
- SBASXUCJHJRPEV-UHFFFAOYSA-N 2-(2-methoxyethoxy)ethanol Chemical compound COCCOCCO SBASXUCJHJRPEV-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- ZSDGHWLLLGYAJV-AHEHSYJASA-N 2-[(E)-[(E)-3-[1-(2-nitrophenyl)pyrrol-2-yl]prop-2-enylidene]amino]guanidine Chemical compound NC(N)=N\N=C\C=C\C1=CC=CN1C1=CC=CC=C1[N+]([O-])=O ZSDGHWLLLGYAJV-AHEHSYJASA-N 0.000 description 1
- SSORSZACHCNXSJ-UHFFFAOYSA-N 2-[2-(3,4-dichlorophenyl)-3-[2-(2-hydroxypropylamino)pyrimidin-4-yl]imidazol-4-yl]acetonitrile Chemical compound ClC=1C=C(C=CC=1Cl)C=1N(C(=CN=1)CC#N)C1=NC(=NC=C1)NCC(C)O SSORSZACHCNXSJ-UHFFFAOYSA-N 0.000 description 1
- DILISPNYIVRDBP-UHFFFAOYSA-N 2-[3-[2-(2-hydroxypropylamino)pyrimidin-4-yl]-2-naphthalen-2-ylimidazol-4-yl]acetonitrile Chemical compound OC(CNC1=NC=CC(=N1)N1C(=NC=C1CC#N)C1=CC2=CC=CC=C2C=C1)C DILISPNYIVRDBP-UHFFFAOYSA-N 0.000 description 1
- DWKNOLCXIFYNFV-HSZRJFAPSA-N 2-[[(2r)-1-[1-[(4-chloro-3-methylphenyl)methyl]piperidin-4-yl]-5-oxopyrrolidine-2-carbonyl]amino]-n,n,6-trimethylpyridine-4-carboxamide Chemical compound CN(C)C(=O)C1=CC(C)=NC(NC(=O)[C@@H]2N(C(=O)CC2)C2CCN(CC=3C=C(C)C(Cl)=CC=3)CC2)=C1 DWKNOLCXIFYNFV-HSZRJFAPSA-N 0.000 description 1
- NAMYKGVDVNBCFQ-UHFFFAOYSA-N 2-bromopropane Chemical compound CC(C)Br NAMYKGVDVNBCFQ-UHFFFAOYSA-N 0.000 description 1
- NCKJIJSEWKIXAT-UHFFFAOYSA-N 2-diphenylphosphanylethenyl(diphenyl)phosphane Chemical group C=1C=CC=CC=1P(C=1C=CC=CC=1)C=CP(C=1C=CC=CC=1)C1=CC=CC=C1 NCKJIJSEWKIXAT-UHFFFAOYSA-N 0.000 description 1
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 1
- 229940093475 2-ethoxyethanol Drugs 0.000 description 1
- GGDYAKVUZMZKRV-UHFFFAOYSA-N 2-fluoroethanol Chemical compound OCCF GGDYAKVUZMZKRV-UHFFFAOYSA-N 0.000 description 1
- MSXVEPNJUHWQHW-UHFFFAOYSA-N 2-methylbutan-2-ol Chemical compound CCC(C)(C)O MSXVEPNJUHWQHW-UHFFFAOYSA-N 0.000 description 1
- KIPMDPDAFINLIV-UHFFFAOYSA-N 2-nitroethanol Chemical compound OCC[N+]([O-])=O KIPMDPDAFINLIV-UHFFFAOYSA-N 0.000 description 1
- RCLQNICOARASSR-SECBINFHSA-N 3-[(2r)-2,3-dihydroxypropyl]-6-fluoro-5-(2-fluoro-4-iodoanilino)-8-methylpyrido[2,3-d]pyrimidine-4,7-dione Chemical compound FC=1C(=O)N(C)C=2N=CN(C[C@@H](O)CO)C(=O)C=2C=1NC1=CC=C(I)C=C1F RCLQNICOARASSR-SECBINFHSA-N 0.000 description 1
- ATVJXMYDOSMEPO-UHFFFAOYSA-N 3-prop-2-enoxyprop-1-ene Chemical compound C=CCOCC=C ATVJXMYDOSMEPO-UHFFFAOYSA-N 0.000 description 1
- LZPWAYBEOJRFAX-UHFFFAOYSA-N 4,4,5,5-tetramethyl-1,3,2$l^{2}-dioxaborolane Chemical compound CC1(C)O[B]OC1(C)C LZPWAYBEOJRFAX-UHFFFAOYSA-N 0.000 description 1
- UXHQLGLGLZKHTC-CUNXSJBXSA-N 4-[(3s,3ar)-3-cyclopentyl-7-(4-hydroxypiperidine-1-carbonyl)-3,3a,4,5-tetrahydropyrazolo[3,4-f]quinolin-2-yl]-2-chlorobenzonitrile Chemical compound C1CC(O)CCN1C(=O)C1=CC=C(C=2[C@@H]([C@H](C3CCCC3)N(N=2)C=2C=C(Cl)C(C#N)=CC=2)CC2)C2=N1 UXHQLGLGLZKHTC-CUNXSJBXSA-N 0.000 description 1
- HFGHRUCCKVYFKL-UHFFFAOYSA-N 4-ethoxy-2-piperazin-1-yl-7-pyridin-4-yl-5h-pyrimido[5,4-b]indole Chemical compound C1=C2NC=3C(OCC)=NC(N4CCNCC4)=NC=3C2=CC=C1C1=CC=NC=C1 HFGHRUCCKVYFKL-UHFFFAOYSA-N 0.000 description 1
- RSIWALKZYXPAGW-NSHDSACASA-N 6-(3-fluorophenyl)-3-methyl-7-[(1s)-1-(7h-purin-6-ylamino)ethyl]-[1,3]thiazolo[3,2-a]pyrimidin-5-one Chemical compound C=1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)N=C2SC=C(C)N2C(=O)C=1C1=CC=CC(F)=C1 RSIWALKZYXPAGW-NSHDSACASA-N 0.000 description 1
- SJVGFKBLUYAEOK-SFHVURJKSA-N 6-[4-[(3S)-3-(3,5-difluorophenyl)-3,4-dihydropyrazole-2-carbonyl]piperidin-1-yl]pyrimidine-4-carbonitrile Chemical compound FC=1C=C(C=C(C=1)F)[C@@H]1CC=NN1C(=O)C1CCN(CC1)C1=CC(=NC=N1)C#N SJVGFKBLUYAEOK-SFHVURJKSA-N 0.000 description 1
- AMKGKYQBASDDJB-UHFFFAOYSA-N 9$l^{2}-borabicyclo[3.3.1]nonane Chemical compound C1CCC2CCCC1[B]2 AMKGKYQBASDDJB-UHFFFAOYSA-N 0.000 description 1
- 108090000531 Amidohydrolases Proteins 0.000 description 1
- 102000004092 Amidohydrolases Human genes 0.000 description 1
- 206010055113 Breast cancer metastatic Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- FLKWZFLWBMNKEA-OMFQWQAWSA-O C[C@@H]([C@@H](C1)OC2[C@H]3O[C@]1(CC[C@@H](C1)O[C@@H](CC[C@@H](C[C@H]4C)O[C@H](C[C@@H]([C@@H]5C6)O[C@H](C[C@@H](CN)O)[C@@H]5OC)/C4=N4)C1=C)[OH2+])C2OC(CC1)[C@]34O[C@H]1CC6=O Chemical compound C[C@@H]([C@@H](C1)OC2[C@H]3O[C@]1(CC[C@@H](C1)O[C@@H](CC[C@@H](C[C@H]4C)O[C@H](C[C@@H]([C@@H]5C6)O[C@H](C[C@@H](CN)O)[C@@H]5OC)/C4=N4)C1=C)[OH2+])C2OC(CC1)[C@]34O[C@H]1CC6=O FLKWZFLWBMNKEA-OMFQWQAWSA-O 0.000 description 1
- NXNAVZJYVCATJP-OAHLLOKOSA-N C[C@H](CC=O)C#C[Si](C(C)C)(C(C)C)C(C)C Chemical compound C[C@H](CC=O)C#C[Si](C(C)C)(C(C)C)C(C)C NXNAVZJYVCATJP-OAHLLOKOSA-N 0.000 description 1
- LUDPPJVPATXCNJ-VHSXEESVSA-N C[C@H](C[C@@H](CCC=O)N=O)C#CC#[I]=N Chemical compound C[C@H](C[C@@H](CCC=O)N=O)C#CC#[I]=N LUDPPJVPATXCNJ-VHSXEESVSA-N 0.000 description 1
- DCERHCFNWRGHLK-UHFFFAOYSA-N C[Si](C)C Chemical compound C[Si](C)C DCERHCFNWRGHLK-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 229910021555 Chromium Chloride Inorganic materials 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 1
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 238000003747 Grignard reaction Methods 0.000 description 1
- NHTMVDHEPJAVLT-UHFFFAOYSA-N Isooctane Chemical compound CC(C)CC(C)(C)C NHTMVDHEPJAVLT-UHFFFAOYSA-N 0.000 description 1
- HETCEOQFVDFGSY-UHFFFAOYSA-N Isopropenyl acetate Chemical compound CC(=C)OC(C)=O HETCEOQFVDFGSY-UHFFFAOYSA-N 0.000 description 1
- 108090001060 Lipase Proteins 0.000 description 1
- 239000004367 Lipase Substances 0.000 description 1
- 102000004882 Lipase Human genes 0.000 description 1
- 239000012448 Lithium borohydride Substances 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 1
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 1
- 229940122255 Microtubule inhibitor Drugs 0.000 description 1
- 238000006751 Mitsunobu reaction Methods 0.000 description 1
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 1
- BBVOPOMPARWKIS-LBPRGKRZSA-N N-[2-[(4R)-4-propan-2-yl-4,5-dihydro-1,3-oxazol-2-yl]phenyl]methanesulfonamide Chemical compound C(C)(C)[C@H]1N=C(OC1)C1=C(C=CC=C1)NS(=O)(=O)C BBVOPOMPARWKIS-LBPRGKRZSA-N 0.000 description 1
- PHSPJQZRQAJPPF-UHFFFAOYSA-N N-alpha-Methylhistamine Chemical compound CNCCC1=CN=CN1 PHSPJQZRQAJPPF-UHFFFAOYSA-N 0.000 description 1
- 229910021586 Nickel(II) chloride Inorganic materials 0.000 description 1
- 108010084311 Novozyme 435 Proteins 0.000 description 1
- GZMYLSJUNSCMTD-MOPGFXCFSA-N OC[C@@H](C)NC1=NC(=CC(=C1)C=1C=C(C=CC=1C)NC(=O)N1C[C@@H](CC1)CC(F)(F)F)N1CCOCC1 Chemical compound OC[C@@H](C)NC1=NC(=CC(=C1)C=1C=C(C=CC=1C)NC(=O)N1C[C@@H](CC1)CC(F)(F)F)N1CCOCC1 GZMYLSJUNSCMTD-MOPGFXCFSA-N 0.000 description 1
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 238000003477 Sonogashira cross-coupling reaction Methods 0.000 description 1
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 1
- MCRWZBYTLVCCJJ-DKALBXGISA-N [(1s,3r)-3-[[(3s,4s)-3-methoxyoxan-4-yl]amino]-1-propan-2-ylcyclopentyl]-[(1s,4s)-5-[6-(trifluoromethyl)pyrimidin-4-yl]-2,5-diazabicyclo[2.2.1]heptan-2-yl]methanone Chemical compound C([C@]1(N(C[C@]2([H])C1)C(=O)[C@@]1(C[C@@H](CC1)N[C@@H]1[C@@H](COCC1)OC)C(C)C)[H])N2C1=CC(C(F)(F)F)=NC=N1 MCRWZBYTLVCCJJ-DKALBXGISA-N 0.000 description 1
- WRJSSAQXXHKDRP-JGCGQSQUSA-N [(4R)-2-bromo-7-[tert-butyl(diphenyl)silyl]oxyhept-1-en-4-yl] 2,4,6-tri(propan-2-yl)benzenesulfonate Chemical compound C(C)(C)C1=C(C(=CC(=C1)C(C)C)C(C)C)S(=O)(=O)O[C@@H](CC(=C)Br)CCCO[Si](C1=CC=CC=C1)(C1=CC=CC=C1)C(C)(C)C WRJSSAQXXHKDRP-JGCGQSQUSA-N 0.000 description 1
- JEDZLBFUGJTJGQ-UHFFFAOYSA-N [Na].COCCO[AlH]OCCOC Chemical compound [Na].COCCO[AlH]OCCOC JEDZLBFUGJTJGQ-UHFFFAOYSA-N 0.000 description 1
- KGEMRSFKCAFWTB-UHFFFAOYSA-N [SiH4].C1(CCCCC1)B Chemical class [SiH4].C1(CCCCC1)B KGEMRSFKCAFWTB-UHFFFAOYSA-N 0.000 description 1
- YVIMHTIMVIIXBQ-UHFFFAOYSA-N [SnH3][Al] Chemical compound [SnH3][Al] YVIMHTIMVIIXBQ-UHFFFAOYSA-N 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 150000004645 aluminates Chemical class 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- DALDUXIBIKGWTK-UHFFFAOYSA-N benzene;toluene Chemical compound C1=CC=CC=C1.CC1=CC=CC=C1 DALDUXIBIKGWTK-UHFFFAOYSA-N 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- AZWXAPCAJCYGIA-UHFFFAOYSA-N bis(2-methylpropyl)alumane Chemical compound CC(C)C[AlH]CC(C)C AZWXAPCAJCYGIA-UHFFFAOYSA-N 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- KVNRLNFWIYMESJ-UHFFFAOYSA-N butyronitrile Chemical compound CCCC#N KVNRLNFWIYMESJ-UHFFFAOYSA-N 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000012018 catalyst precursor Substances 0.000 description 1
- ZDQWVKDDJDIVAL-UHFFFAOYSA-N catecholborane Chemical compound C1=CC=C2O[B]OC2=C1 ZDQWVKDDJDIVAL-UHFFFAOYSA-N 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- QSWDMMVNRMROPK-UHFFFAOYSA-K chromium(3+) trichloride Chemical compound [Cl-].[Cl-].[Cl-].[Cr+3] QSWDMMVNRMROPK-UHFFFAOYSA-K 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 1
- NKNDPYCGAZPOFS-UHFFFAOYSA-M copper(i) bromide Chemical compound Br[Cu] NKNDPYCGAZPOFS-UHFFFAOYSA-M 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- 229940045803 cuprous chloride Drugs 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- VRLDVERQJMEPIF-UHFFFAOYSA-N dbdmh Chemical compound CC1(C)N(Br)C(=O)N(Br)C1=O VRLDVERQJMEPIF-UHFFFAOYSA-N 0.000 description 1
- XNYOSXARXANYPB-UHFFFAOYSA-N dicyclohexylborane Chemical compound C1CCCCC1BC1CCCCC1 XNYOSXARXANYPB-UHFFFAOYSA-N 0.000 description 1
- WHYBPBROKHGRNU-UHFFFAOYSA-N diethylaluminum;tributyltin Chemical compound CC[Al]CC.CCCC[Sn](CCCC)CCCC WHYBPBROKHGRNU-UHFFFAOYSA-N 0.000 description 1
- XXJWXESWEXIICW-UHFFFAOYSA-N diethylene glycol monoethyl ether Chemical compound CCOCCOCCO XXJWXESWEXIICW-UHFFFAOYSA-N 0.000 description 1
- 229940075557 diethylene glycol monoethyl ether Drugs 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- JVSWJIKNEAIKJW-UHFFFAOYSA-N dimethyl-hexane Natural products CCCCCC(C)C JVSWJIKNEAIKJW-UHFFFAOYSA-N 0.000 description 1
- UBHZUDXTHNMNLD-UHFFFAOYSA-N dimethylsilane Chemical compound C[SiH2]C UBHZUDXTHNMNLD-UHFFFAOYSA-N 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 229940093476 ethylene glycol Drugs 0.000 description 1
- KZGWPHUWNWRTEP-UHFFFAOYSA-N ethynyl-tri(propan-2-yl)silane Chemical group CC(C)[Si](C#C)(C(C)C)C(C)C KZGWPHUWNWRTEP-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 230000002140 halogenating effect Effects 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- 229910000043 hydrogen iodide Inorganic materials 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 229910052741 iridium Inorganic materials 0.000 description 1
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 1
- GJRQTCIYDGXPES-UHFFFAOYSA-N iso-butyl acetate Natural products CC(C)COC(C)=O GJRQTCIYDGXPES-UHFFFAOYSA-N 0.000 description 1
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-M isobutyrate Chemical compound CC(C)C([O-])=O KQNPFQTWMSNSAP-UHFFFAOYSA-M 0.000 description 1
- FGKJLKRYENPLQH-UHFFFAOYSA-M isocaproate Chemical compound CC(C)CCC([O-])=O FGKJLKRYENPLQH-UHFFFAOYSA-M 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- OQAGVSWESNCJJT-UHFFFAOYSA-N isovaleric acid methyl ester Natural products COC(=O)CC(C)C OQAGVSWESNCJJT-UHFFFAOYSA-N 0.000 description 1
- 235000019421 lipase Nutrition 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- HNQIVZYLYMDVSB-UHFFFAOYSA-N methanesulfonimidic acid Chemical compound CS(N)(=O)=O HNQIVZYLYMDVSB-UHFFFAOYSA-N 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- NSPJNIDYTSSIIY-UHFFFAOYSA-N methoxy(methoxymethoxy)methane Chemical compound COCOCOC NSPJNIDYTSSIIY-UHFFFAOYSA-N 0.000 description 1
- SKTCDJAMAYNROS-UHFFFAOYSA-N methoxycyclopentane Chemical compound COC1CCCC1 SKTCDJAMAYNROS-UHFFFAOYSA-N 0.000 description 1
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 1
- CPZBTYRIGVOOMI-UHFFFAOYSA-N methylsulfanyl(methylsulfanylmethoxy)methane Chemical compound CSCOCSC CPZBTYRIGVOOMI-UHFFFAOYSA-N 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 231100000782 microtubule inhibitor Toxicity 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- 239000011733 molybdenum Substances 0.000 description 1
- PYLWMHQQBFSUBP-UHFFFAOYSA-N monofluorobenzene Chemical compound FC1=CC=CC=C1 PYLWMHQQBFSUBP-UHFFFAOYSA-N 0.000 description 1
- VZUGBLTVBZJZOE-KRWDZBQOSA-N n-[3-[(4s)-2-amino-1,4-dimethyl-6-oxo-5h-pyrimidin-4-yl]phenyl]-5-chloropyrimidine-2-carboxamide Chemical compound N1=C(N)N(C)C(=O)C[C@@]1(C)C1=CC=CC(NC(=O)C=2N=CC(Cl)=CN=2)=C1 VZUGBLTVBZJZOE-KRWDZBQOSA-N 0.000 description 1
- VOVZXURTCKPRDQ-CQSZACIVSA-N n-[4-[chloro(difluoro)methoxy]phenyl]-6-[(3r)-3-hydroxypyrrolidin-1-yl]-5-(1h-pyrazol-5-yl)pyridine-3-carboxamide Chemical compound C1[C@H](O)CCN1C1=NC=C(C(=O)NC=2C=CC(OC(F)(F)Cl)=CC=2)C=C1C1=CC=NN1 VOVZXURTCKPRDQ-CQSZACIVSA-N 0.000 description 1
- IYRGXJIJGHOCFS-UHFFFAOYSA-N neocuproine Chemical compound C1=C(C)N=C2C3=NC(C)=CC=C3C=CC2=C1 IYRGXJIJGHOCFS-UHFFFAOYSA-N 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- QMMRZOWCJAIUJA-UHFFFAOYSA-L nickel dichloride Chemical compound Cl[Ni]Cl QMMRZOWCJAIUJA-UHFFFAOYSA-L 0.000 description 1
- 229940078552 o-xylene Drugs 0.000 description 1
- KSWNZTXYXRDYAM-UHFFFAOYSA-N oct-1-en-7-yn-4-ol Chemical compound C=CCC(O)CCC#C KSWNZTXYXRDYAM-UHFFFAOYSA-N 0.000 description 1
- ATCNYMVVGBLQMQ-UHFFFAOYSA-N oct-7-yn-1-ol Chemical compound OCCCCCCC#C ATCNYMVVGBLQMQ-UHFFFAOYSA-N 0.000 description 1
- RIWDMMJFVMXOQU-UHFFFAOYSA-N oct-7-yn-4-ol Chemical compound CCCC(O)CCC#C RIWDMMJFVMXOQU-UHFFFAOYSA-N 0.000 description 1
- XULSCZPZVQIMFM-IPZQJPLYSA-N odevixibat Chemical compound C12=CC(SC)=C(OCC(=O)N[C@@H](C(=O)N[C@@H](CC)C(O)=O)C=3C=CC(O)=CC=3)C=C2S(=O)(=O)NC(CCCC)(CCCC)CN1C1=CC=CC=C1 XULSCZPZVQIMFM-IPZQJPLYSA-N 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- VUYVXCJTTQJVKJ-UHFFFAOYSA-L palladium(2+);tricyclohexylphosphane;dichloride Chemical compound Cl[Pd]Cl.C1CCCCC1P(C1CCCCC1)C1CCCCC1.C1CCCCC1P(C1CCCCC1)C1CCCCC1 VUYVXCJTTQJVKJ-UHFFFAOYSA-L 0.000 description 1
- QJPQVXSHYBGQGM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 QJPQVXSHYBGQGM-UHFFFAOYSA-N 0.000 description 1
- VWYVHZFRBJJWSM-UHFFFAOYSA-N pent-4-ynal Chemical compound O=CCCC#C VWYVHZFRBJJWSM-UHFFFAOYSA-N 0.000 description 1
- JYVLIDXNZAXMDK-UHFFFAOYSA-N pentan-2-ol Chemical compound CCCC(C)O JYVLIDXNZAXMDK-UHFFFAOYSA-N 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 1
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- QMKUYPGVVVLYSR-UHFFFAOYSA-N propyl 2,2-dimethylpropanoate Chemical compound CCCOC(=O)C(C)(C)C QMKUYPGVVVLYSR-UHFFFAOYSA-N 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 229910052707 ruthenium Inorganic materials 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- ODZPKZBBUMBTMG-UHFFFAOYSA-N sodium amide Chemical compound [NH2-].[Na+] ODZPKZBBUMBTMG-UHFFFAOYSA-N 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 239000012419 sodium bis(2-methoxyethoxy)aluminum hydride Substances 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 150000003461 sulfonyl halides Chemical class 0.000 description 1
- WMOVHXAZOJBABW-UHFFFAOYSA-N tert-butyl acetate Chemical compound CC(=O)OC(C)(C)C WMOVHXAZOJBABW-UHFFFAOYSA-N 0.000 description 1
- KEFUQHPJWHCFJF-XZOQPEGZSA-N tert-butyl-dimethyl-[(4R,6R)-6-methyl-8-tri(propan-2-yl)silyloct-1-en-7-yn-4-yl]oxysilane Chemical compound C(C)(C)(C)[Si](O[C@H](CC=C)C[C@H](C#C[Si](C(C)C)(C(C)C)C(C)C)C)(C)C KEFUQHPJWHCFJF-XZOQPEGZSA-N 0.000 description 1
- 238000005809 transesterification reaction Methods 0.000 description 1
- LGSAOJLQTXCYHF-UHFFFAOYSA-N tri(propan-2-yl)-tri(propan-2-yl)silyloxysilane Chemical compound CC(C)[Si](C(C)C)(C(C)C)O[Si](C(C)C)(C(C)C)C(C)C LGSAOJLQTXCYHF-UHFFFAOYSA-N 0.000 description 1
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- AKQNYQDSIDKVJZ-UHFFFAOYSA-N triphenylsilane Chemical compound C1=CC=CC=C1[SiH](C=1C=CC=CC=1)C1=CC=CC=C1 AKQNYQDSIDKVJZ-UHFFFAOYSA-N 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- KMIOJWCYOHBUJS-HAKPAVFJSA-N vorolanib Chemical compound C1N(C(=O)N(C)C)CC[C@@H]1NC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C KMIOJWCYOHBUJS-HAKPAVFJSA-N 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic System
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/26—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D307/28—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic System
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic System
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
Definitions
- aspects of the present application relate to a process for preparation of 4- Methylene tetrahydrofuran compound of formula II, which is useful as an intermediate for the preparation of halichondrin B analogues such as Eribulin.
- the drug compound having the adopted name Eribulin is a synthetic analogue of halichondrin B, and is represented by structure of formula I.
- Eribulin is a microtubule inhibitor indicated for the treatment of patients with metastatic breast cancer who have previously received at least two chemotherapeutic regimens for the treatment of metastatic disease.
- U.S. Patent No. 6,214,865 discloses eribulin and its pharmaceutically acceptable salts.
- a 4-methylene tetrahydrofuran compound of formula I I is used as an intermediate for the preparation of halichondrin B analogues such as Eribulin.
- R is selected from straight or branched C Cio alkyl or optionally substituted C 5 -Ci 2 aryl or optionally substituted aralkyl; X is halogen.
- the present application provides a process for preparation of 4-Methylene tetrahydrofuran compound of formula II,
- Pi is H or an alcohol protecting group
- P2 is H or an alcohol protecting group or -S0 2 (R); wherein R is selected from straight or branched C1 -C10 alkyl or optionally substituted C5-C 12 aryl or optionally substituted aralkyl
- X is halogen
- Ri is H or trialkyi silyl protecting group
- LG is -OS02(R); wherein R is selected from straight or branched C1 -C10 alkyl or optionally substituted C5-C12 aryl or optionally substituted aralkyl;
- Pi is an alcohol-protecting group
- P-i is H or an alcohol protecting group and X is halogen, preferably Iodine;
- the present application provides a process for preparation of acetylene compoun wherein is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group; which includes one or more of the following steps:
- R ! is H or trialkyi silyl protecting group
- Ri is H or trialkyi silyl protecting group
- P-i is an alcohol-protecting group
- Ri is H or trialkyi silyl protecting group
- P-i is an alcohol-protecting group
- Ri is H or trialkyi silyl protecting group
- the present application provides a process for preparation of an acetylene compound of formula III, wherein Pi is an alcohol-protecting group; Ri is H or trialkyi silyl; which includes one or more of the following steps:
- Ri is H or trialkyi silyl
- Pi is H or an alcohol-protecting group
- X is halogen
- Ri is H or trialkyi silyl
- Ri is H or trialkyi silyl
- Ri is H or trialkyi silyl
- Pi is an alcohol-protecting group
- P 2 is hydrogen or an alcohol-protecting group
- the present application provides a process for preparation of a compound of formula IV;
- P ⁇ is an alcohol-protecting group
- LG is -OS0 2 (R); wherein R is selected from straight or branched C Cio alkyl or optionally substituted C5-C15 aryl or optionally substituted aralkyl
- X is halogen, preferably bromine
- R 2 is trialkyl silyl; P ⁇ is an alcohol-protecting group; X is halogen, preferably bromine;
- P ⁇ is an alcohol-protecting group
- X is halogen, preferably bromine or iodine
- P-i is an alcohol-protecting group
- Ri is H or trialkyi silyl protecting group
- the present application provides a compound formula VI or isomers thereof.
- the present application provides a compound of formula VII or isomers thereof.
- P-i is an alcohol-protecting group
- the present application provides a compound of formula VII I or isomers thereof.
- Pi is an alcohol-protecting group
- Ri is H or trialkyi silyl protecting group
- the present application provides a compound of formula IVa or isomers thereof.
- the present application provides a compound having the following formula or isomers thereof.
- the present application provides a process for preparation of eribulin or a pharmaceutically acceptable salt thereof comprising synthesizing eribulin or its pharmaceutically acceptable salt from one or more compounds of first embodiment to eleventh embodiment.
- Figure 1 is powder X-ray diffraction ("PXRD”) pattern of compound of formula
- the present application provides a process for preparation of 4-methylene tetrahydrofuran compound of formula II,
- R is selected from straight or branched C Ci 0 alkyl or optionally substituted C 5 -Ci 2 aryl or optionally substituted aralkyl;
- X is halogen
- Pi is an alcohol-protecting group
- Ri is H or trialkyi silyl protecting group
- LG is -OS0 2 (R); wherein R is selected from straight or branched C Cio alkyl or optionally substituted C 5 -d 2 aryl or optionally substituted aralkyl;
- Pi is an alcohol-protecting group
- Step (a) involves reacting acetylene compound of formula II I with vinyl halide compound of formula IV to provide compound of formula V;
- Suitable reagents that may be used in step (a) include, chromium chloride and optionally a ligand such as '(R)-N-(2-(4-isopropyl-4,5-dihydrooxazol-2- yl)phenyl)methanesulfonamide and the like, nickel chloride and optionally a ligand such as 2,9-dimethyl-1 ,10-phenanthroline and the like or any other suitable catalyst or ligands known in the art used in Nozaki-Hiyama-Kishi (NHK) reaction.
- a ligand such as '(R)-N-(2-(4-isopropyl-4,5-dihydrooxazol-2- yl)phenyl)methanesulfonamide and the like
- nickel chloride and optionally a ligand such as 2,9-dimethyl-1 ,10-phenanthroline and the like or any other suitable catalyst or ligands known in the art used
- Suitable bases that may be used in step (a) include, sodium hydride, potassium tert-butoxide, sodium methoxide, lithium hexamethyldisilazide, sodium amide, 1 ,8- bis(dimethylamino)naphthalene (Proton-sponge) and the like; other organic bases, such as for example, N-methylmorpholine, N-methylpyrrolidine, pyridine, 4-(N,N- dimethylamino)pyridine, morpholine, imidazole and the like or any other suitable base known in the art.
- Suitable solvents that may be used in step (a) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents or mixtures thereof.
- the reaction mixture obtained from step (a) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (a) may be isolated directly from the reaction mixture itself after the reaction is complete in step (a), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- an obtained crude product may be directly used for step (b) or it may be isolated and further purified.
- Step (b) involves optionally deprotecting compound of formula V to provide dihydroxy compound of formula V
- Suitable reagents that may be used in step (b) include, hydrochloric acid, hydrobromic acid, hydroiodic acid, nitric acid, sulfuric acid, acetic acid, formic acid, phosphoric acid, methanesulfonic acid, p-toluenesulfonic acid, tetra-n-butylammoniu m fluoride (TBAF), tris(dimethylamino)sulfonium difluorotrimethylsilicate, ammonia, sodium hydroxide, potassium hydroxide, sodium methoxide, potassium t-butoxide, sodium t-butoxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, and the like; ion exchange resins, such as: resins bound to metal ions, including lithium, sodium, potassium, and the like; and resins bound to acids, including phosphoric, sulfonic, methanesulfonic, p-toluenesulfonic,
- Suitable solvents that may be used in step (b) include water, alcohols, ketones, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane or mixtures thereof.
- Suitable temperature that may be used in step (b) may be less than about 100°C, less than about 70°C, less than about 40°C, less than about 30°C, less than about 10°C, less than about 0°C, less than about -10°C, less than about -20°C, or any other suitable temperature.
- the reaction mixture obtained from step (b) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (b) may be isolated directly from the reaction mixture itself after the reaction is complete in step (b), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- an obtained crude product may be directly used for step (c) or it may be isolated and further purified.
- Step (c) involves optionally protecting dihydroxy compound of formula VI to provide compound of formula VII;
- Suitable bases that may be used in step (c) include, alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide; carbonates such as sodium carbonate, potassium carbonate, cesium carbonate, calcium carbonate, magnesium carbonate, sodium bicarbonate, potassium bicarbonate, alkoxides such as sodium methoxide, potassium methoxide; organic bases, such as for example, triethylamine, tributylamine, N- methylmorpholine, ⁇ , ⁇ -diisopropylethylamine, N-methylpyrrolidine, pyridine, collidine, 4-(N,N-dimethylamino)pyridine, morpholine, imidazole, 2-methylimidazole, 4- methylimidazole and the like or any other suitable base known in the art.
- alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide
- Suitable solvents that may be used in step (c) include ketones, esters, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane or mixtures thereof.
- the reaction mixture obtained from step (c) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (c) may be isolated directly from the reaction mixture itself after the reaction is complete in step (c), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- an obtained crude product may be directly used for step (d) or it may be isolated and further purified.
- Step (d) involves converting compound of formula VI or formula VI I to provide compound of formula VIII;
- step (d) may involve reduction followed by halogenation, hydrohalogenation or any other suitable method known in the art.
- Suitable reducing agents that may be used in case of reduction in step (d) include, sodium borohydride, lithium aluminum hydride, sodium trimethoxy borohydride, Lithium borohydride, acetoxyborohydride, cyanoborohydride, sodium dihydro-bis-(2-methoxyethoxy) aluminate solution (VITRIDE®), diisobutyl aluminium hydride, 9-borabicyclo(3.3.1 )nonane (9-BBN), catecholborane, pinacolborane, diiasamylboirane, cyclohexylborane silanes such as triethylsilane, triphenylsilane and tributyltin hydride, tin-aluminium reagents such as diethyl(tributylstannyl)aluminum and the like or any other suitable reductants known in the art.
- Suitable catalysts that may be used in said reduction in
- Suitable halogenating agents that may be used in step (d) include, N- halosuccinimides such as N-Chlorosuccinimide, N-Bromosuccinimide, N- lodosuccinimide or any other suitable agents known in the art; 1 ,3-Dihalo-5,5- dimethylhydantoin such as 1 ,3-Diiodo-5,5-dimethylhydantoin and the like; halides such as iodine, bromine or any other suitable agents known in the art.
- Suitable agents for halogenation or hydrohalogenation include, hydrogen halides such as hydrogen chloride, hydrogen bromide, hydrogen iodide or any other suitable agents known in the art.
- Suitable solvents that may be used in step (d) include alcohols, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents or mixtures thereof.
- the reaction mixture obtained from step (d) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other suitable techniques for the removal of solids.
- the product of step (d) may be isolated directly from the reaction mixture itself after the reaction is complete in step (d), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- an obtained crude product may be directly used for step (e) or it may be isolated and further purified.
- Step (e) involves converting compound of formula VII I to compound of formula
- Conversion of compound of formula VIII to compound of formula II may involve (i) deprotecting both primary and secondary alcohol protected compound of formula VIII, (ii) optionally protecting primary alcohol with suitable alcohol protecting group preferably using pivaloyl chloride and protecting or activating secondary alcohol with a suitable protecting group or leaving group preferably using mesyl chloride or tosyl chloride.
- Suitable reagents that may be used in step (e) for deprotecting both primary and secondary alcohol protected compound of formula VIII, include, hydrochloric acid, hydrobromic acid, hydroiodic acid, nitric acid, sulfuric acid, acetic acid, formic acid, phosphoric acid, methanesulfonic acid, p-toluenesulfonic acid, tetra-n-butylammoniu m fluoride (TBAF), tris(dimethylamino)sulfonium difluorotrimethylsilicate, ammonia, sodium hydroxide, potassium hydroxide, sodium methoxide, potassium t-butoxide, sodium t-butoxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, and the like; ion exchange resins, such as: resins bound to metal ions, including lithium, sodium, potassium, and the like; and resins bound to acids, including phosphoric, sulfonic, methanesulf
- Suitable solvents that may be used in step (e) include water, alcohols, ketones, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane and mixtures thereof.
- Suitable bases that may be used in step (e) for protecting the primary alcohol, for protecting or activating secondary alcohol include, alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide; carbonates such as sodium carbonate, potassium carbonate, cesium carbonate, calcium carbonate, magnesium carbonate, sodium bicarbonate, potassium bicarbonate, alkoxides such as sodium methoxide, potassium methoxide; organic bases, such as for example, triethylamine, tributylamine, N-methylmorpholine, ⁇ , ⁇ -diisopropylethylamine, N-methylpyrrolidine, pyridine, collidine, 4-(N,N- dimethylamino)pyridine, morpholine, imidazole, 2-methylimidazole, 4-methylimidazole and the like or any other suitable base known in the art.
- alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide,
- Suitable solvents that may be used in step (e) include ketones, esters, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane or mixtures thereof.
- steps (a) to (e) or any two or more steps may be carried out in-situ i.e. without isolating the intermediates in each stage.
- the present application provides a process for preparation of acetylene compoun wherein is an alcohol-protecting group; Ri is H or trialkyl silyl protecting group; which includes one or more of the following steps:
- Ri is H or trialkyl silyl protecting group
- Ri is H or trialkyi silyl protecting group
- P-i is an alcohol-protecting group
- Ri is H or trialkyi silyl protecting group
- Pi is an alcohol-protecting group
- Ri is H or trialkyi silyl protecting group
- Step (a) involves treating crotonaldehyde IX with acetylene compound of formula X to provide compound of formula XI;
- Suitable reagents that may be used in step (a) may include rhodium(l) catalyst precursors such as hydroxy(cyclooctadiene)rhodium(l) dimer and the like; bulky diphosphine ligands such as (S)-(+)-5,5'-bis[di(3,5-di-tert-butyl-4- methoxyphenyl)phosphino]-4,4'-bi-1 ,3-benzodioxole ((S)-DTBM-SEGPHOS) and the like or any other suitable catalyst or ligands known in the art.
- Suitable solvents that may be used in step (a) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, polar aprotic solvents or mixtures thereof.
- the reaction mixture obtained from step (a) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (a) may be isolated directly from the reaction mixture itself after the reaction is complete in step (a), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- an obtained crude product may be directly used for step (b) or it may be isolated and further purified.
- Step (b) involves reacting compound of formula XI with (+)-B-allyldiisopino campheylborane to provide hydroxy compound of formula XII;
- Ri is H or trialkyl silyl protecting group
- Suitable solvents that may be used in step (b) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, polar aprotic solvents or mixtures thereof.
- the reaction mixture obtained from step (b) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (b) may be isolated directly from the reaction mixture itself after the reaction is complete in step (b), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- an obtained crude product may be directly used for step (c) or it may be isolated and further purified.
- Step (c) involves optionally protecting hydroxy compound of formula XII to provide compound of formula XIII;
- Pi is an alcohol-protecting group
- Ri is H or trialkyl silyl protecting group
- Suitable base that may be used in step (c) include, alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide; carbonates such as sodium carbonate, potassium carbonate, cesium carbonate, calcium carbonate, magnesium carbonate, sodium bicarbonate, potassium bicarbonate, alkoxides such as sodium methoxide, potassium methoxide; organic bases, such as for example, triethylamine, tributylamine, N-methylmorpholine, ⁇ , ⁇ -diisopropylethylamine, N-methylpyrrolidine, pyridine, collidine, 4-(N,N- dimethylamino)pyridine, morpholine, imidazole, 2-methylimidazole, 4-methylimidazole and the like or any other suitable bases known in the art.
- alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide
- carbonates
- Suitable solvents that may be used in step (c) include ketones, esters, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane or mixtures thereof.
- the reaction mixture obtained from step (c) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (c) may be isolated directly from the reaction mixture itself after the reaction is complete in step (c), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- an obtained crude product may be directly used for step (d) or it may be isolated and further purified.
- Step (d) involves converting compound of formula XII I to provide compound of formula XIV;
- Pi is an alcohol-protecting group
- Ri is H or trialkyl silyl protecting group
- Suitable reagents that may be used in step (d) include, 9- borabicyclo(3.3.1 )nonane (9-BBN), borane, bis-3-methyl-2-butylborane, dicyclohexylborane, boron trifluoride diethyl etherate, or any other suitable reagent that are known in the art.
- Suitable solvents that may be used in step (d) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, polar aprotic solvents or mixtures thereof.
- the reaction mixture obtained from step (d) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (d) may be isolated directly from the reaction mixture itself after the reaction is complete in step (d), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- an obtained crude product may be directly used for step (e) or it may be isolated and further purified.
- Step (e) involves converting compound of formula XIV to compound of formula
- Suitable reagents that may be used in step (e) include, (2,2,6,6-tetramethyl- piperidin-1 -yl)oxyl (TEMPO), pyridinium chlorochromate (PCC), oxalyl chloride & dimethyl sulfoxide (DMSO), dicyclohexylcarbodiimide & DMSO, Dess-Martin periodinane, [bis(acetoxy)iodo]-benzene (BAIB) and the like or any other oxidizing agent known in the art.
- TEMPO (2,2,6,6-tetramethyl- piperidin-1 -yl)oxyl
- PCC pyridinium chlorochromate
- DMSO oxalyl chloride & dimethyl sulfoxide
- BAIB Dess-Martin periodinane
- BAIB [bis(acetoxy)iodo]-benzene
- Suitable solvents that may be used in step (e) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, halogenated hydrocarbons, polar aprotic solvents or mixtures thereof.
- the reaction mixture obtained from step (e) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (e) may be isolated directly from the reaction mixture itself after the reaction is complete in step (e), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- steps (a) to (e) or any two or more steps may be carried out in-situ i.e. without isolating the intermediates in each stage.
- the present application provides a process for preparation of an acetylene compound of formula III
- Pi is an alcohol-protecting group
- Ri is H or trialkyi silyl; which includes one or more of the following steps:
- Ri is H or trialkylsilyl
- Pi is H or an alcohol-protecting group
- X is halogen
- Ri is H or trialkyi silyl
- Ri is H or trialkyi silyl
- Ri is H or trialkyl silyl
- Pi is an alcohol-protecting group
- P 2 is hydrogen or an alcohol-protecting group
- Ri is H or trialkyl silyl
- Step (a) involves treating a compound of formula XV with an acetylene compound of formula X to provide a compound of formula
- P ⁇ is H or trialkyl silyl
- P1 is an alcohol-protecting group
- X is halogen
- Suitable catalysts that may be used in step (a) include, dichloro[1 ,3- bis(diphenylphosphino)propane]palladium(ll) ((dppp)PdCI 2 ), [1 ,1 '-
- Suitable bases that may be used in step (a) include, alkali metal or alkaline earth metal carbonates such as sodium carbonate, potassium carbonate, cesium carbonate, calcium carbonate, magnesium carbonate, sodium bicarbonate, potassium bicarbonate, alkoxides such as sodium methoxide, potassium methoxide; organic bases, such as for example, triethylamine, tributylamine, A/-methylmorpholine, N,N- diisopropylethylamine, A/-methylpyrrolidine, pyridine, piperidine, collidine, 4-(N,N- dimethylamino)pyridine, morpholine, imidazole, 2-methylimidazole, 4-methylimidazole and the like or any other suitable base known in the art.
- alkali metal or alkaline earth metal carbonates such as sodium carbonate, potassium carbonate, cesium carbonate, calcium carbonate, magnesium carbonate, sodium bicarbonate, potassium bicarbonate, alkoxides such as sodium methoxide, potassium
- Suitable solvents that may be used in step (a) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, nitriles, ethers, polar aprotic solvents or mixtures thereof.
- the reaction mixture obtained from step (a) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (a) may be isolated directly from the reaction mixture itself after the reaction is complete in step (a), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- the resulting product may be directly used for step (b) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
- Step (b) involves optionally deprotecting a compound of formula XVI to provide a compound of formula XVII;
- Suitable reagents that may be used in step (b) include, hydrochloric acid, hydrobromic acid, hydroiodic acid, nitric acid, sulfuric acid, acetic acid, formic acid, phosphoric acid, methanesulfonic acid, p-toluenesulfonic acid, tetra-n-butylammoniu m fluoride (TBAF), tris(dimethylamino)sulfonium difluorotrimethylsilicate, ammonia, sodium hydroxide, potassium hydroxide, sodium methoxide, potassium t-butoxide, sodium t-butoxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, and the like; ion exchange resins, such as: resins bound to metal ions, including lithium, sodium, potassium, and the like; and resins bound to acids, including phosphoric, sulfonic, methanesulfonic, p-toluenesulfonic,
- Suitable solvents that may be used in step (b) include water, alcohols, ketones, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane and mixtures thereof.
- Suitable temperatures that may be used in step (b) may be less than about 1 00°C, less than about 70°C, less than about 40°C, less than about 30°C, less than about 10°C, less than about 0°C, less than about -10°C, less than about -20°C, or any other suitable temperature.
- the reaction mixture obtained from step (b) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration , suction filtration, or any other suitable techniques for the removal of solids.
- the product of step (b) may be isolated directly from the reaction mixture itself after the reaction is complete in step (b), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- an obtained crude product may be directly used for step (c) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
- Step (c) involves converting a compound of formula XVI or compound of formula XVI I to a compound of for
- Conversion of a compound of formula XVI or compound of formula XVI I to a compound of formula XVI II may be carried out using hydrogen and a catalyst;
- Suitable catalysts that may be used in step (c) include, [((S,S)- QuinoxP*)Rh(cod)]BF 4 , [((fl,fl)-Norphos)Rh(cod)]BF 4 , [((S,S)-Me-BPE)Rh(cod)]BF 4 , [((fl,fl)-Et-BPE)Rh(cod)]BF 4 , [((S,S)-Ph-BPP)Rh(cod)]BF 4 , [((S,S)-tBu- ferrotane)Rh(cod)]BF 4 , [((fl,fl)-Me-DuPhos)Rh(cod)]BF 4 , [((fi)-BINAP)Rh(cod)]BF 4 , [((S,S)-iPr-DuPhos)Rh(cod)]BF 4 , [((fl,f
- Suitable solvents that may be used in step (c) include alcohols, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, polar aprotic solvents or mixtures thereof.
- the reaction mixture obtained from step (c) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other suitable techniques for the removal of solids.
- the product of step (c) may be isolated directly from the reaction mixture itself after the reaction is complete in step (c), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- the resulting product may be directly used for step (d) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
- Step (d) involves protecting a diol compound of formula XVIII to provide a compound of formula XIX;
- Ri is H or trialkyl silyl
- Pi is an alcohol-protecting group
- P 2 is hydrogen or an alcohol-protecting group
- diol compound of formula XVIII may be protected sequentially to provide a compound of formula XIX or mono protected compound of formula XIV or compound of formula XXII I may be converted to compound of formula XIX or two or more compounds mixture of compound of formula XVIII, compound of formula XIV and compound of formula XXII I may be converted to compound of formula XIX
- ⁇ - ⁇ and P 2 is alcohol protecting group; Ri is H or trialkyl silyl;
- Suitable bases that may be used in step (d) include, alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide; carbonates such as sodium carbonate, potassium carbonate, cesium carbonate, calcium carbonate, magnesium carbonate, sodium bicarbonate, potassium bicarbonate, alkoxides such as sodium methoxide, potassium methoxide; organic bases, such as for example, triethylamine, tributylamine, N- methylmorpholine, A/,A/-diisopropylethylamine, A/-methylpyrrolidine, pyridine, collidine 4-(A/,A/-dimethylamino)pyridine, morpholine, imidazole, piperidine, 2-methylimidazole, 4-methylimidazole and the like or any other suitable bases known in the art.
- alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide
- Suitable catalysts that may be used in step (d) include enzymes such as lipases, esterases, amidases and any other suitable trans esterification enzymes known in the art, other catalysts such as oxo[hexa(trifluoroacetato)]tetrazinc and the like or any other suitable catalysts known in the art that are used for selective alcohol acylation or protection..
- Suitable solvents that may be used in step (d) include ketones, esters, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane or mixtures thereof.
- the reaction mixture obtained from step (d) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (d) may be isolated directly from the reaction mixture itself after the reaction is complete in step (d), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- the resulting product may be directly used for step (e) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
- Step (e) involves optionally deprotecting a compound of formula XIX to provide a compound of formula XIV;
- Suitable reagents that may be used in step (e) include, hydrochloric acid, hydrobromic acid, hydroiodic acid, nitric acid, sulfuric acid, acetic acid, formic acid, phosphoric acid, methanesulfonic acid, p-toluenesulfonic acid, tetra-n-butylammoniu m fluoride (TBAF), tris(dimethylamino)sulfonium difluorotrimethylsilicate, ammonia, sodium hydroxide, potassium hydroxide, sodium methoxide, potassium f-butoxide, sodium f-butoxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, and the like; ion exchange resins, such as: resins bound to metal ions, including lithium, sodium, potassium, and the like; and resins bound to acids, including phosphoric, sulfonic, methanesulfonic, p-toluenesulfonic,
- Suitable solvents that may be used in step (e) include water, alcohols, ketones, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane and mixtures thereof.
- Suitable temperatures that may be used in step (e) may be less than about 100°C, less than about 70°C, less than about 40°C, less than about 30°C, less than about 10°C, less than about 0°C, less than about -10°C, less than about -20°C, or any other suitable temperature.
- the reaction mixture obtained from step (e) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (e) may be isolated directly from the reaction mixture itself after the reaction is complete in step (e, or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- the resulting product may be directly used for step (f) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
- Step (f) involves converting a compound of formula XIV to a compound of formula II I.
- Suitable reagents that may be used in step (f) include, (2,2,6,6-tetramethyl- piperidin-1 -yl)oxyl (TEMPO), pyridinium chlorochromate (PCC), oxalyl chloride & dimethyl sulfoxide (DMSO), dicyclohexylcarbodiimide & DMSO, Dess-Martin periodinane, [bis(acetoxy)iodo]-benzene (BAIB), manganese dioxide and the like or any other suitable oxidizing agent known in the art.
- TEMPO (2,2,6,6-tetramethyl- piperidin-1 -yl)oxyl
- PCC pyridinium chlorochromate
- DMSO oxalyl chloride & dimethyl sulfoxide
- BAIB dicyclohexylcarbodiimide & DMSO
- BAIB [bis(acetoxy)iodo]-benzene
- Suitable solvents that may be used in step (f) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, halogenated hydrocarbons, polar aprotic solvents or mixtures thereof.
- the reaction mixture obtained from step (f) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (f) may be isolated directly from the reaction mixture itself after the reaction is complete in step (f), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- steps (a) to (f) or any two or more steps may be carried out in-situ i.e. without isolating the intermediates in each stage.
- a compound of formula XIV can also be prepared by using the (S)-enantiomer or enantiomerically enriched compound of formula XV.
- Inversion of the stereocentre at the secondary alcohol ⁇ (S)-enantiomer of a compound of formula XVI II ⁇ may be carried out by employing the Mitsunobu reaction conditions or any other suitable chiral inversion reaction conditions known in the art.
- the present application provides a process for preparation of a compound of formula IV;
- Pi is an alcohol-protecting group
- LG is -OSC>2(R); wherein R is selected from straight or branched C Cio alkyl or optionally substituted C5-C15 aryl or optionally substituted aralkyl
- X is halogen, preferably bromine
- R 2 is trialkyl silyl
- Pi is an alcohol-protecting group
- X is halogen, preferably bromine
- Step (a) involves reacting an epoxy compound of formula XX with a compound of formula XXI to provide a compound of formula XXII;
- R 2 is trialkyl silyl
- Pi is an alcohol-protecting group
- X is halogen, preferably bromine
- Suitable forms of magnesium metal that may be used in step (a) include, any form of magnesium metal such as magnesium turnings, magnesium beads, magnesium powder, magnesium chips and the like.
- Suitable reagents that may be used in step (a) to activate magnesium include, alkyl halides such as methyl iodide, methyl ethyl bromide, 1 , 2-dibromoethane and the like, iodine, diisobutylaluminium hydride (DIBAL-H), sodium bis(2- methoxyethoxy)aluminumhydride (Red-AI), lithium aluminium hydride (LAH) and the like or any other suitable magnesium activating reagents known in the art.
- alkyl halides such as methyl iodide, methyl ethyl bromide, 1 , 2-dibromoethane and the like
- iodine diisobutylaluminium hydride (DIBAL-H)
- DIBAL-H diisobutylaluminium hydride
- Red-AI sodium bis(2- methoxyethoxy)aluminumhydride
- Suitable solvents that may be used in step (a) include ethers, aromatic hydrocarbons, or mixtures thereof.
- step (a) may be carried out in presence of a metal catalyst such as cuprous chloride, cuprous bromide, cuprous iodide, zinc chloride and the like or any other suitable metal catalyst known in the art employed for the Grignard reaction.
- the reaction mixture obtained from step (a) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (a) may be isolated directly from the reaction mixture itself after the reaction is complete in step (a), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- the resulting product may be directly used for step (b) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
- Step (b) involves converting a compound of formula XXII to a compound of formula XV;
- Pi is an alcohol-protecting group
- X is halogen, preferably bromine
- Suitable reagents that may be used in step (b) include, bromine, N- bromosuccinimide, 1 ,3-dibromo-5,5-dimethylhydantoin, pyridinium perbromide phosphorus tribromide, iodine, A/-iodosuccinimide and the like or any other suitable reagents known in the art.
- Suitable solvents that may be used in step (b) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, halogenated hydrocarbons, esters, ketones, polar aprotic solvents or mixtures thereof.
- the reaction mixture obtained from step (b) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (b) may be isolated directly from the reaction mixture itself after the reaction is complete in step (b), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- the resulting product may be directly used for step (c) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
- Step (c) involves converting a compound of formula XV to a compound of formula IV.
- the compound of formula IV is prepared by treating a compound of formula XV with sulfonyl halides or esters and the like.
- Suitable sulfonyl reagents that may be used in step (c) include, methanesulfonyl, p-toluenesulfonyl, 2,4,6- triisopropylbenzenesulfonyl, 4-propoxybenzene-1 -sulfonyl, 4-methoxybenzenesulfonyl, phenylmethanesulfonyl, 2,4-dichloropyrimidine-5-sulfonyl, benzenesulfonyl and the like.
- Suitable base that may be used in step (c) include, alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide; carbonates such as sodium carbonate, potassium carbonate, cesium carbonate, calcium carbonate, magnesium carbonate, sodium bicarbonate, potassium bicarbonate, alkoxides such as sodium methoxide, potassium methoxide; organic bases, such as for example, triethylamine, tributylamine, A/-methylmorpholine, A/,A/-diisopropylethylamine, A/-methylpyrrolidine, pyridine, collidine, 4-(N,N- dimethylamino)pyridine, morpholine, imidazole, 2-methylimidazole, 4-methylimidazole and the like or any other suitable bases known in the art.
- alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide
- Suitable solvents that may be used in step (c) include ketones, esters, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane or mixtures thereof.
- the reaction mixture obtained from step (c) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids.
- the product of step (c) may be isolated directly from the reaction mixture itself after the reaction is complete in step (c), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
- steps (a) to (c) may be carried out in-situ i.e. without isolating the intermediates in each stage.
- the present application provides a compound of formula V or isomers thereof.
- P-i is an alcohol-protecting group
- Ri is H or trialkyi silyl protecting group preferably triisopropylsilyl ether
- the present application provides a compound formula VI or isomers thereof.
- the present application provides a compound of formula VII or isomers thereof.
- P-i is an alcohol-protecting group
- the present application provides a compound of formula VII I or isomers thereof.
- Pi is an alcohol-protecting group
- Ri is H or trialkyi silyl protecting group
- the present application provides a compound of formula IVa or isomers thereof.
- the present application provides a compound having the following formula or isomers thereof.
- the present application provides a process for preparation of eribulin or a pharmaceutically acceptable salt thereof comprising synthesizing eribulin or its pharmaceutically acceptable salt from one or more compounds of first embodiment to eleventh embodiment.
- One or more compounds of first embodiment to eleventh embodiment include compound of formula III, formula IV, formula IVa, formula V, formula VI, formula VII, formula VIII, formula XI, formula XII, formula XIII, formula XIV, formula XV, formula XVI, formula XVII, formula XVII I, formula XIX, formula XXII and compounds in eleventh embodiment.
- Isomers of one or more compounds of first embodiment to twelfth embodiment isomers include stereoisomers of formula III, formula IV, formula IVa, formula V, formula VI, formula VII, formula VIII, formula XI, formula XII, formula XIII, formula XIV, formula XV, formula XVI, formula XVII, formula XVIII, formula XIX, formula XXI I and compounds in eleventh embodiment.
- the number of carbon atoms present in a given group or compound is designated “C x -C y ", where x and y are the lower and upper limits, respectively.
- a group designated as “CrC 6 " contains from 1 to 6 carbon atoms.
- the carbon number as used in the definitions herein refers to carbon backbone and carbon branching, but does not include carbon atoms of the substituents, such as alkoxy substitutions or the like.
- an alcohol protecting group is a functional group that protects the alcohol group from participating in reactions that are occurring in other parts of the molecule.
- Suitable alcohol protecting groups that are used in step (a) include, acetyl, benzoyl, benzyl, ⁇ -methoxyethoxymethyl ether, methoxymethyl ether, dimethoxytrityl, p-methoxybenzyl ether, methylthiomethyl ether, allyl ether, f-butyl ether, pivaloyl, trityl, silyl ether (e.g., trimethylsilyl (TMS), f-butyldimethylsilyl (TBMDS), f-butyldiphenylsilyl (TBDPS), f-butyldimethylsilyloxymethyl (TOM) or triisopropylsilyl (TIPS) ether), tetrahydropyranyl (THP), methyl ether and ethyl,
- Alcohol is an organic compound containing a carbon bound to a hydroxyl group.
- Ci-C 6 alcohols include methanol, ethanol, 2-nitroethanol, 2-fluoroethanol, 2,2,2-trifluoroethanol, hexafluoroisopropyl alcohol, ethylene glycol, 1 -propanol, 2- propanol (isopropyl alcohol), 2-methoxyethanol, 1 -butanol, 2-butanol, i-butyl alcohol, t- butyl alcohol, 2-ethoxyethanol, diethylene glycol, 1 -, 2-, or 3-pentanol, neo-pentyl alcohol, t-pentyl alcohol, cyclohexanol, phenol, glycerol and the like.
- aliphatic hydrocarbon is a liquid hydrocarbon compound, which may be linear, branched, or cyclic and may be saturated or have as many as two double bonds.
- a liquid hydrocarbon compound that contains a six-carbon group having three double bonds in a ring is called “aromatic.”
- C 5 -C 8 aliphatic include n- pentane, isopentane, neopentane, n-hexane, isohexane, 3-methylpentane, 2,3- dimethylbutane, neohexane, n-heptane, isoheptane, 3-methylhexane, neoheptane, 2,3-dimethylpentane, 2,4-dimethylpentane, 3,3-dimethylpentane, 3-ethylpentane, 2,2,3-trimethylbutane, n-octane, isooctane, 3-methylheptane
- aromatic hydrocarbon solvent refers to a liquid, unsaturated, cyclic, hydrocarbon containing one or more rings which has delocalized conjugated ⁇ system.
- aromatic hydrocarbon solvent include benzene toluene, ethylbenzene, m-xylene, o-xylene, p-xylene, indane, naphthalene, tetralin, trimethylbenzene, chlorobenzene, fluorobenzene, trifluorotoluene, anisole, C 6 -Ci 2 aromatic hydrocarbons and the like.
- C 3 -C 6 esters include ethyl acetate, n-propyl acetate, n-butyl acetate, isobutyl acetate, t-butyl acetate, ethyl formate, methyl acetate, methyl propanoate, ethyl propanoate, methyl butanoate, ethyl butanoate and the like.
- ether is an organic compound containing an oxygen atom -O- bonded to two other carbon atoms.
- C 2 -C 6 ethers include diethyl ether, diisopropyl ether, methyl t-butyl ether, glyme, diglyme, tetrahydrofuran, 2-methyltetrahydrofuran, 1 , 4-dioxane, dibutyl ether, dimethylfuran, anisole, diethylene glycol monomethyl ether, diethylene glycol monoethyl ether, cyclopentyl methyl ether and the like.
- halogenated hydrocarbon is an organic compound containing a carbon bound to a halogen.
- Halogenated hydrocarbons include dichloromethane, 1 ,2- dichloroethane, trichloroethylene, perchloroethylene, 1 ,1 ,1 -trichloroethane, 1 ,1 ,2- trichloroethane, chloroform, carbon tetrachloride and the like.
- C 3 -C 6 ketones include acetone, ethyl methyl ketone, diethyl ketone, methyl isobutyl ketone, ketones and the like.
- a “nitrile” is an organic compound containing a cyano -(C ⁇ N) bonded to another carbon atom.
- C 2 -C 6 Nitriles include acetonitrile, propionitrile, butanenitrile and the like.
- a "polar aprotic solvents” include ⁇ , ⁇ -dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, sulfolane, N-methylpyrrolidone and the like;
- Example-1 Preparation of (fi)-3-Methyl-5-(triisopropylsilyl)pent-4-ynal.
- Example-2 Preparation of (4ff,6fi)-6-methyl-8-(triisopropylsilyl)oct-1 -en-7-yn-4- ol.
- Example-3 Preparation of ieri-Butyldimethyl(((4R,6R)-6-methyl-8- (triisopropylsilyl)oct-1-en-7-yn-4-yl)oxy)silane.
- Example-4 Preparation of (4ff,6fi)-4-((tert-butyldimethylsilyl)oxy)-6-methyl-8- (triisopropylsilyl)oct-7-yn-1-ol.
- Example-5 Preparation of (4ff,6fi)-4-((ieri-Butyldimethylsilyl)oxy)-6-methyl-8- (triisopropylsilyl)oct-7-ynal.
- Example-6 Preparation of tert-butyl(3-((2S,5S)-5-((3R,5R)-3-((tert- butyldimethylsilyl)oxy)-5-methyl-7-(triisopropylsilyl)hept-6-yn-1-yl)-4-methylen tetrahydrofuran-2-yl)propoxy)diphenylsilane.
- Aldehyde and vinyl bromide compounds solution preparation In two separate 500 ml flasks, (4R,6R)-4-((tert-Butyldimethylsilyl)oxy)-6-methyl-8-(triisopropylsilyl)oct- 7-ynal (61 .9 g) and (R)-2-bromo-7-((tert-butyldiphenylsilyl) oxy)hept-1 -en-4-yl 2,4,6- triisopropylbenzenesulfonate (124.7 g) were evaporated with toluene (30 mL) three times to remove any water.
- the flasks were placed under a flow of nitrogen and acetonitrile (200 mL) was added to the flask containing bromo compound. After dissolution of bromo compound in acetonitrile, transferred to the flask containing aldehyde and stirred for 15 minutes to get clear solution.
- the reaction mass was warmed to 40 °C and stirred at 40 °C for 4 hours.
- Water (600 mL) and 2-Me-THF (600 mL) was added to the reaction mass and stirred for 15 minutes.
- Example-7 (3ff,5fi)-1 -((2S,5S)-5-(3-Hydroxypropyl)-3-methylenetetrahydrofuran- 2-yl)-5-methylhept-6-yn-3-ol
- Example-8 Preparation of ieri-Butyl(3-((2S,5S)-5-((3ff,5fi)-3-((tert- butyldimethylsilyl)oxy)-5-methylhept-6-yn-1-yl)-4-methylenetetrahydrofuran-2- yl)propoxy)dimethylsilane.
- DIBAL-H (1 M in THF, 1 .62 mL) was slowly added to a suspension of NiCI 2 (bis(diphenylphosphino)ethylene) (42.7 mg) in THF (0.5 mL) and stirred for 15 minutes.
- Example-10 Preparation of 3-((2S,5S)-5-((3ff,5fl)-3-Hydroxy-6-iodo-5- methylhept-6-en-1-yl)-4-methylenetetrahydrofuran-2-yl)propyl pivalate.
- Example-11 Preparation of (fi)-7-((ieri-butyldimethylsilyl)oxy)-2- (trimethylsilyl)hept-1-en-4-ol.
- 1 -bromovinyltrimethylsilane solution preparation 62.1 g of 1 -bromovinyltrimethylsilane was dissolved in 1 00 mL THF. Iodine (1 .17 g) was added to the magnesium turnings (9.55 g) at 23 °C and allowed to stand for 10 minutes. Anhydrous tetrahydrofuran (50 mL) followed by 1 - bromovinyltrimethylsilane solution in THF (10 mL) was added to the reaction mass at 24 °C and stirred for 30 minutes. Another lot of 1 -bromovinyltrimethylsilane solution in THF (10 mL) was added to the reaction mass at 24 °C and stirred for 20 minutes.
- Triethylamine (0.320 g) was added to the reaction mass containing (R)-7-((tert- butyldimethylsilyl)oxy)-2-(trimethylsilyl)hept-1 -en-4-ol (1 .0 g) and toluene (15 mL) at - 30 °C.
- bromine solution (0.606 g of bromine in 10 mL of toluene) was added slowly to the reaction mass at -58 °C.
- Sodium methanolate solution (2 mL; 25 wt. % in methanol) was added at -58 °C and the resultant reaction mixture was stirred at -48 °C for 5 hours.
- Example-13 Preparation of (fi)-2-bromo-7-((ieri-butyldimethylsilyl)oxy)hept-1- en-4-ol
- Example-14 Preparation of (fi)-2-bromo-7-((ieri-butyldimethylsilyl)oxy)hept-1- en-4-yl 2,4,6-triisopropylbenzenesulfonate.
- Example-15 Preparation of (fi)-1 -((ieri-butyldimethylsilyl)oxy)-6-methylene-8- (triisopropylsilyl)oct-7-yn-4-ol.
- the precipitate was removed by filtration through a pad of silica gel (100 g), the filter cake was washed with MTBE (250 mL). The combined filtrates were washed with water (500 mL), sat. NH 4 CI sol. (500 mL), water (500 mL) and brine solution (500 mL). The washed filtrates were dried over MgS0 4 and then concentrated under reduced pressure to afford title compound (157.47 g).
- Example-16 Preparation of (fi)-6-methylene-8-(triisopropylsilyl)oct-7-yne-1 ,4- diol.
- Example-17 Preparation of (4ff, 6fi)-6-Methyl-8-(triisopropylsilyl)oct-7-yne-1 ,4- diol.
- Example-19 preparation of (4ff, 6fi)-4-((ieri-Butyldimethylsilyl)oxy)-6-methyl-8- (triisopropylsilyl)oct-7-yn-1-ol.
- BusSn-AIEt? solution preparation 1 .6 M nBuLi in hexanes (8.8 mL) was added dropwise to the solution containing isopropylamine (1 .98 mL) and THF (34 mL) at -10 °C under nitrogen and the resulting solution was stirred at -10 °C for 30 minutes. Bu 3 SnH (3.8 mL) was added to the solution dropwise at below -30 °C and stirred for 30 minutes. 1 .8 M Et 2 AICI in PhMe (7.9 mL) was added dropwise at -40 °C and the reaction mixture was stirred for 10 minutes at -30 °C.
Abstract
The present application provides process for preparation of 4-Methylene tetrahydrofuran compound of formula II, which is useful as an intermediate for the preparation of halichondrin B analogues such as Eribulin.
Description
PROCESS FOR PREPARATION OF ERIBULIN AND INTERMEDIATES THEREOF
INTRODUCTION
Aspects of the present application relate to a process for preparation of 4- Methylene tetrahydrofuran compound of formula II, which is useful as an intermediate for the preparation of halichondrin B analogues such as Eribulin.
The drug compound having the adopted name Eribulin, is a synthetic analogue of halichondrin B, and is represented by structure of formula I.

I
Eribulin is a microtubule inhibitor indicated for the treatment of patients with metastatic breast cancer who have previously received at least two chemotherapeutic regimens for the treatment of metastatic disease. U.S. Patent No. 6,214,865 discloses eribulin and its pharmaceutically acceptable salts. A 4-methylene tetrahydrofuran compound of formula I I is used as an intermediate for the preparation of halichondrin B analogues such as Eribulin.

Processes for the preparation of a 4-methylene tetrahydrofuran compound of formula II have been disclosed in PCT application No. 2005/1 18565A1 , J. Am. Chem. Soc, 1992, 114, 3162 and Org. Lett., 2002, 4, 341 1-3414. The reported processes
suffer from major disadvantages, including use of highly expensive reagents, large amounts of catalysts, low temperature and longer reaction time.
Hence, there remains a need to provide an alternative process for the preparation of 4-Methylene tetrahydrofuran compound of formula II which is simple, economic and industrially viable, which in turn can be converted to Eribulin.
SUMMARY
In the first embodiment, the present application provides a process for preparation of 4-Methylene tetrahydrofuran compound of formula II,

I I
wherein Pi is H or an alcohol protecting group; P2 is H or an alcohol protecting group or -S02(R); wherein R is selected from straight or branched C1 -C10 alkyl or optionally substituted C5-C12 aryl or optionally substituted aralkyl; X is halogen;
which includes one or more of the following steps:
(a) reacting acetylene compound of formula III with vinyl halide compound of formula IV to provide compound of formula V;

V
wherein is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group; LG is -OS02(R); wherein R is selected from straight or branched C1 -C10 alkyl or optionally substituted C5-C12 aryl or optionally substituted aralkyl;
(b) optionally deprotecting compound of formula V to provide dihydroxy compound of formula VI;

VI
(c) optionally protecting dihydroxy compound of formula VI to provide compound of formula VII;

VI I
wherein Pi is an alcohol-protecting group;
(d) converting compound of formula VI or formula VII to provide compound of formula VIII;

VII I
wherein P-i is H or an alcohol protecting group and X is halogen, preferably Iodine;
(e) converting compound of formula VIII to a compound of formula II.
In the second embodiment, the present application provides a process for preparation of acetylene compoun
 wherein is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group; which includes one or more of the following steps:
wherein is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group; which includes one or more of the following steps:
(a) treating crotonaldehyde IX with acetylene compound of formula X to provide compound of formula XI;

IX X XI
wherein R! is H or trialkyi silyl protecting group;
(b) reacting compound of formula XI with (+)-B-allyldiisopinocampheylborane to provide hydroxy compound of formula XII;

XII
wherein Ri is H or trialkyi silyl protecting group;
(c) optionally protecting hydroxy compound of formula XII to provide a compound of formula XIII;

XII I
wherein P-i is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group;
(d) converting compound of formula XIII to provide compound of formula XIV; and

XIV
wherein P-i is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group
(e) converting compound of formula XIV to compound of formula I II.
In the third embodiment, the present application provides a process for preparation of an acetylene compound of formula III,
 wherein Pi is an alcohol-protecting group; Ri is H or trialkyi silyl; which includes one or more of the following steps:
wherein Pi is an alcohol-protecting group; Ri is H or trialkyi silyl; which includes one or more of the following steps:
(a) treating a compound of formula XV with an acetylene compound of formula X to provide a compound of formula XVI;

XV X XVI
wherein Ri is H or trialkyi silyl; Pi is H or an alcohol-protecting group; X is halogen;
(b) optionally deprotecting a compound of formula XVI to provide a compound of formula XVII;

XVII
wherein Ri is H or trialkyi silyl;
(c) converting a compound of formula XVI or compound of formula XVII to a compound of formula XVIII;

XVI I I
wherein Ri is H or trialkyi silyl;
(d) protecting a diol compound of formula XVIII to provide a compound of formula XIX;

XIX
wherein Ri is H or trialkyi silyl; Pi is an alcohol-protecting group; P2 is hydrogen or an alcohol-protecting group;
(e) optionally deprotecting a compound of formula XIX to provide a compound of formula XIV;

XIV
wherein is H or trialkyl silyl;
(f) converting a compound of formula XIV to a compound of formula III.
In the fourth embodiment, the present application provides a process for preparation of a compound of formula IV;

wherein P^ is an alcohol-protecting group; LG is -OS02(R); wherein R is selected from straight or branched C Cio alkyl or optionally substituted C5-C15 aryl or optionally substituted aralkyl; X is halogen, preferably bromine;
which includes one or more of the following steps:
(a) reacting an epoxide of formula XX with a compound of formula XXI to provide a compound of formula XXII;

wherein R2 is trialkyl silyl; P^ is an alcohol-protecting group; X is halogen, preferably bromine;
(b) converting a compound of formula XXII to a vinyl halide compound of formula XV;

wherein P^ is an alcohol-protecting group; X is halogen, preferably bromine or iodine;
(c) converting a compound of formula XV to a compound of formula IV.
In the fifth embodiment, the present application provides a compound of formula V or isomers thereof.

In the sixth embodiment, the present application provides a compound formula VI or isomers thereof.

In the seventh embodiment, the present application provides a compound of formula VII or isomers thereof.

VI I
wherein P-i is an alcohol-protecting group;
In the eighth embodiment, the present application provides a compound of formula VII I or isomers thereof.

VI I I
wherein Pi is H or an alcohol protecting group and X is halogen.
In the ninth embodiment, the present application provides a compound of formula II I or isomers thereof;

I I I
wherein Pi is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group;
In the tenth embodiment, the present application provides a compound of formula IVa or isomers thereof.

IVa
wherein P is an alcohol protecting group and X is halogen;
In the eleventh embodiment, the present application provides a compound having the following formula or isomers thereof.


In the twelfth embodiment, the present application provides a process for preparation of eribulin or a pharmaceutically acceptable salt thereof comprising synthesizing eribulin or its pharmaceutically acceptable salt from one or more compounds of first embodiment to eleventh embodiment.
DRAWINGS
Figure 1 is powder X-ray diffraction ("PXRD") pattern of compound of formula
VI.
DETAILED DESCRIPTION
In the first embodiment, the present application provides a process for preparation of 4-methylene tetrahydrofuran compound of formula II,

I I
wherein is H or an alcohol protecting group; P2 is H or an alcohol protecting group or -S02(R); wherein R is selected from straight or branched C Ci0 alkyl or optionally substituted C5-Ci2 aryl or optionally substituted aralkyl; X is halogen
which includes one or more of the following steps:
(a) reacting acetylene compound of formula I II with vinyl halide compound of formula IV to provide compound of formula V;

V
wherein Pi is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group; LG is -OS02(R); wherein R is selected from straight or branched C Cio alkyl or optionally substituted C5-d2 aryl or optionally substituted aralkyl;
(b) optionally deprotecting compound of formula V to provide dihydroxy compound of formula VI;

(c) optionally protecting dihydroxy compound of formula VI to provide compound of formula VII;

VI I
wherein Pi is an alcohol-protecting group;
(d) converting compound of formula VI or formula VII to provide compound of formula VII I;

VII I
wherein P-i is H or an alcohol protected group and X is halogen, preferably Iodine; and (e) converting compound of formula VII I to compound of formula II.
Step (a) involves reacting acetylene compound of formula II I with vinyl halide compound of formula IV to provide compound of formula V;

v
Suitable reagents that may be used in step (a) include, chromium chloride and optionally a ligand such as '(R)-N-(2-(4-isopropyl-4,5-dihydrooxazol-2-
yl)phenyl)methanesulfonamide and the like, nickel chloride and optionally a ligand such as 2,9-dimethyl-1 ,10-phenanthroline and the like or any other suitable catalyst or ligands known in the art used in Nozaki-Hiyama-Kishi (NHK) reaction.
Suitable bases that may be used in step (a) include, sodium hydride, potassium tert-butoxide, sodium methoxide, lithium hexamethyldisilazide, sodium amide, 1 ,8- bis(dimethylamino)naphthalene (Proton-sponge) and the like; other organic bases, such as for example, N-methylmorpholine, N-methylpyrrolidine, pyridine, 4-(N,N- dimethylamino)pyridine, morpholine, imidazole and the like or any other suitable base known in the art.
Suitable solvents that may be used in step (a) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents or mixtures thereof.
The reaction mixture obtained from step (a) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (a) may be isolated directly from the reaction mixture itself after the reaction is complete in step (a), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, an obtained crude product may be directly used for step (b) or it may be isolated and further purified.
Step (b) involves optionally deprotecting compound of formula V to provide dihydroxy compound of formula V

VI
Suitable reagents that may be used in step (b) include, hydrochloric acid, hydrobromic acid, hydroiodic acid, nitric acid, sulfuric acid, acetic acid, formic acid, phosphoric acid, methanesulfonic acid, p-toluenesulfonic acid, tetra-n-butylammoniu m fluoride (TBAF), tris(dimethylamino)sulfonium difluorotrimethylsilicate, ammonia, sodium hydroxide, potassium hydroxide, sodium methoxide, potassium t-butoxide, sodium t-butoxide, sodium carbonate, potassium carbonate, sodium bicarbonate,
potassium bicarbonate, and the like; ion exchange resins, such as: resins bound to metal ions, including lithium, sodium, potassium, and the like; and resins bound to acids, including phosphoric, sulfonic, methanesulfonic, p-toluenesulfonic, and the like or any other suitable reagents and mixtures thereof.
Suitable solvents that may be used in step (b) include water, alcohols, ketones, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane or mixtures thereof.
Suitable temperature that may be used in step (b) may be less than about 100°C, less than about 70°C, less than about 40°C, less than about 30°C, less than about 10°C, less than about 0°C, less than about -10°C, less than about -20°C, or any other suitable temperature.
The reaction mixture obtained from step (b) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (b) may be isolated directly from the reaction mixture itself after the reaction is complete in step (b), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, an obtained crude product may be directly used for step (c) or it may be isolated and further purified.
Step (c) involves optionally protecting dihydroxy compound of formula VI to provide compound of formula VII;

VI I
wherein is an alcohol-protecting group;
Suitable bases that may be used in step (c) include, alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide; carbonates such as sodium carbonate, potassium carbonate, cesium carbonate, calcium carbonate, magnesium carbonate, sodium bicarbonate, potassium bicarbonate, alkoxides such as sodium methoxide, potassium methoxide; organic bases, such as for example, triethylamine, tributylamine, N-
methylmorpholine, Ν,Ν-diisopropylethylamine, N-methylpyrrolidine, pyridine, collidine, 4-(N,N-dimethylamino)pyridine, morpholine, imidazole, 2-methylimidazole, 4- methylimidazole and the like or any other suitable base known in the art.
Suitable solvents that may be used in step (c) include ketones, esters, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane or mixtures thereof.
The reaction mixture obtained from step (c) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (c) may be isolated directly from the reaction mixture itself after the reaction is complete in step (c), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, an obtained crude product may be directly used for step (d) or it may be isolated and further purified.
Step (d) involves converting compound of formula VI or formula VI I to provide compound of formula VIII;
Suitably, step (d) may involve reduction followed by halogenation, hydrohalogenation or any other suitable method known in the art.
Suitable reducing agents that may be used in case of reduction in step (d) include, sodium borohydride, lithium aluminum hydride, sodium trimethoxy borohydride, Lithium borohydride, acetoxyborohydride, cyanoborohydride, sodium dihydro-bis-(2-methoxyethoxy) aluminate solution (VITRIDE®), diisobutyl aluminium hydride, 9-borabicyclo(3.3.1 )nonane (9-BBN), catecholborane, pinacolborane, diiasamylboirane, cyclohexylborane silanes such as triethylsilane, triphenylsilane and tributyltin hydride, tin-aluminium reagents such as diethyl(tributylstannyl)aluminum and the like or any other suitable reductants known in the art. Suitable catalysts that may be used in said reduction in step (d) includes, compounds of or complexes of nickel, ruthenium, palladium, copper, molybdenum and the like or any other suitable metal catalysts known in the art.
Suitable halogenating agents that may be used in step (d) include, N- halosuccinimides such as N-Chlorosuccinimide, N-Bromosuccinimide, N- lodosuccinimide or any other suitable agents known in the art; 1 ,3-Dihalo-5,5- dimethylhydantoin such as 1 ,3-Diiodo-5,5-dimethylhydantoin and the like; halides such as iodine, bromine or any other suitable agents known in the art. Suitable agents for
halogenation or hydrohalogenation include, hydrogen halides such as hydrogen chloride, hydrogen bromide, hydrogen iodide or any other suitable agents known in the art.
Suitable solvents that may be used in step (d) include alcohols, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents or mixtures thereof.
The reaction mixture obtained from step (d) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other suitable techniques for the removal of solids. The product of step (d) may be isolated directly from the reaction mixture itself after the reaction is complete in step (d), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, an obtained crude product may be directly used for step (e) or it may be isolated and further purified.
Step (e) involves converting compound of formula VII I to compound of formula
II.
Conversion of compound of formula VIII to compound of formula II may involve (i) deprotecting both primary and secondary alcohol protected compound of formula VIII, (ii) optionally protecting primary alcohol with suitable alcohol protecting group preferably using pivaloyl chloride and protecting or activating secondary alcohol with a suitable protecting group or leaving group preferably using mesyl chloride or tosyl chloride.
Suitable reagents that may be used in step (e) for deprotecting both primary and secondary alcohol protected compound of formula VIII, include, hydrochloric acid, hydrobromic acid, hydroiodic acid, nitric acid, sulfuric acid, acetic acid, formic acid, phosphoric acid, methanesulfonic acid, p-toluenesulfonic acid, tetra-n-butylammoniu m fluoride (TBAF), tris(dimethylamino)sulfonium difluorotrimethylsilicate, ammonia, sodium hydroxide, potassium hydroxide, sodium methoxide, potassium t-butoxide, sodium t-butoxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, and the like; ion exchange resins, such as: resins bound to metal ions, including lithium, sodium, potassium, and the like; and resins bound to acids, including phosphoric, sulfonic, methanesulfonic, p-toluenesulfonic, and the like or any other suitable reagents and mixtures thereof.
Suitable solvents that may be used in step (e) include water, alcohols, ketones, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane and mixtures thereof.
Suitable bases that may be used in step (e) for protecting the primary alcohol, for protecting or activating secondary alcohol include, alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide; carbonates such as sodium carbonate, potassium carbonate, cesium carbonate, calcium carbonate, magnesium carbonate, sodium bicarbonate, potassium bicarbonate, alkoxides such as sodium methoxide, potassium methoxide; organic bases, such as for example, triethylamine, tributylamine, N-methylmorpholine, Ν,Ν-diisopropylethylamine, N-methylpyrrolidine, pyridine, collidine, 4-(N,N- dimethylamino)pyridine, morpholine, imidazole, 2-methylimidazole, 4-methylimidazole and the like or any other suitable base known in the art.
Suitable solvents that may be used in step (e) include ketones, esters, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane or mixtures thereof.
Optionally steps (a) to (e) or any two or more steps may be carried out in-situ i.e. without isolating the intermediates in each stage.
In the second embodiment, the present application provides a process for preparation of acetylene compoun
 wherein is an alcohol-protecting group; Ri is H or trialkyl silyl protecting group; which includes one or more of the following steps:
wherein is an alcohol-protecting group; Ri is H or trialkyl silyl protecting group; which includes one or more of the following steps:
(a) treating crotonaldehyde IX with acetylene compound of formula X to provide compound of formul

wherein Ri is H or trialkyl silyl protecting group;
(b) reacting compound of formula XI with (+)-B-Allyldiisopinocampheylborane to provide hydroxy compound of form

XII
wherein Ri is H or trialkyi silyl protecting group
(c) optionally protecting hydroxyl compound of formula XII to provide compound of formula XII I;

XII I
wherein P-i is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group
(d) converting compound of formula XIII to provide compound of formula XIV; and

XIV
wherein Pi is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group;
(e) converting compound of formula XIV to compound of formula I II.
Step (a) involves treating crotonaldehyde IX with acetylene compound of formula X to provide compound of formula XI;

Suitable reagents that may be used in step (a) may include rhodium(l) catalyst precursors such as hydroxy(cyclooctadiene)rhodium(l) dimer and the like; bulky diphosphine ligands such as (S)-(+)-5,5'-bis[di(3,5-di-tert-butyl-4- methoxyphenyl)phosphino]-4,4'-bi-1 ,3-benzodioxole ((S)-DTBM-SEGPHOS) and the like or any other suitable catalyst or ligands known in the art.
Suitable solvents that may be used in step (a) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, polar aprotic solvents or mixtures thereof.
The reaction mixture obtained from step (a) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (a) may be isolated directly from the reaction mixture itself after the reaction is complete in step (a), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, an obtained crude product may be directly used for step (b) or it may be isolated and further purified.
Step (b) involves reacting compound of formula XI with (+)-B-allyldiisopino campheylborane to provide hydroxy compound of formula XII;

XII
wherein Ri is H or trialkyl silyl protecting group
Suitable solvents that may be used in step (b) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, polar aprotic solvents or mixtures thereof.
The reaction mixture obtained from step (b) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (b) may be isolated directly from the reaction mixture itself after the reaction is complete in step (b), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, an obtained crude product may be directly used for step (c) or it may be isolated and further purified.
Step (c) involves optionally protecting hydroxy compound of formula XII to provide compound of formula XIII;

XII I
wherein Pi is an alcohol-protecting group; Ri is H or trialkyl silyl protecting group
Suitable base that may be used in step (c) include, alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide; carbonates such as sodium carbonate, potassium carbonate, cesium carbonate, calcium carbonate, magnesium carbonate, sodium bicarbonate, potassium bicarbonate, alkoxides such as sodium methoxide, potassium methoxide; organic bases, such as for example, triethylamine, tributylamine, N-methylmorpholine, Ν,Ν-diisopropylethylamine, N-methylpyrrolidine, pyridine, collidine, 4-(N,N- dimethylamino)pyridine, morpholine, imidazole, 2-methylimidazole, 4-methylimidazole and the like or any other suitable bases known in the art.
Suitable solvents that may be used in step (c) include ketones, esters, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane or mixtures thereof.
The reaction mixture obtained from step (c) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (c) may be isolated directly from the reaction mixture itself after the reaction is complete in step (c), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, an obtained crude product may be directly used for step (d) or it may be isolated and further purified.
Step (d) involves converting compound of formula XII I to provide compound of formula XIV; and

XIV
wherein Pi is an alcohol-protecting group; Ri is H or trialkyl silyl protecting group;
Suitable reagents that may be used in step (d) include, 9- borabicyclo(3.3.1 )nonane (9-BBN), borane, bis-3-methyl-2-butylborane, dicyclohexylborane, boron trifluoride diethyl etherate, or any other suitable reagent that are known in the art.
Suitable solvents that may be used in step (d) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, polar aprotic solvents or mixtures thereof.
The reaction mixture obtained from step (d) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (d) may be isolated directly from the reaction mixture itself after the reaction is complete in step (d), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, an obtained crude product may be directly used for step (e) or it may be isolated and further purified.
Step (e) involves converting compound of formula XIV to compound of formula
III.
Suitable reagents that may be used in step (e) include, (2,2,6,6-tetramethyl- piperidin-1 -yl)oxyl (TEMPO), pyridinium chlorochromate (PCC), oxalyl chloride & dimethyl sulfoxide (DMSO), dicyclohexylcarbodiimide & DMSO, Dess-Martin periodinane, [bis(acetoxy)iodo]-benzene (BAIB) and the like or any other oxidizing agent known in the art.
Suitable solvents that may be used in step (e) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, halogenated hydrocarbons, polar aprotic solvents or mixtures thereof.
The reaction mixture obtained from step (e) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (e) may be isolated directly from the reaction mixture itself after the reaction is complete in step (e), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
Optionally steps (a) to (e) or any two or more steps may be carried out in-situ i.e. without isolating the intermediates in each stage.
In the third embodiment, the present application provides a process for preparation of an acetylene compound of formula III

I I I
wherein Pi is an alcohol-protecting group; Ri is H or trialkyi silyl; which includes one or more of the following steps:
(a) treating a compound of formula XV with an acetylene compound of formula X to provide a compound of formula XVI;

XV X XVI
wherein Ri is H or trialkylsilyl; Pi is H or an alcohol-protecting group; X is halogen;
(b) optionally deprotecting a compound of formula XVI to provide a compound of formula XVII;

XVII
wherein Ri is H or trialkyi silyl;
(c) converting a compound of formula XVI or compound of formula XVII to a compound of formula XVIII;
Ri
I
HON '
XVI I I
wherein Ri is H or trialkyi silyl;
(d) protecting a diol compound of formula XVIII to provide a compound of formula XIX;

XIX
wherein Ri is H or trialkyl silyl; Pi is an alcohol-protecting group; P2 is hydrogen or an alcohol-protecting group;
(e) optionally deprotecting a compound of formula XIX to provide a compound of formula XIV;

XIV
wherein Ri is H or trialkyl silyl;
(f) converting a compound of formula XIV to a compound of formula III.
Step (a) involves treating a compound of formula XV with an acetylene compound of formula X to provide a compound of formula

XV XVI
wherein P^ is H or trialkyl silyl; P1 is an alcohol-protecting group; X is halogen;
Suitable catalysts that may be used in step (a) include, dichloro[1 ,3- bis(diphenylphosphino)propane]palladium(ll) ((dppp)PdCI2), [1 ,1 '-
Bis(diphenylphosphino)ferrocene]dichloropalladium(ll) (Pd(dppf)CI2), bis(triphenylphosphine)palladium(ll) dichloride (PdCI2(PPh3)2), palladium- tetrakis(triphenylphosphine) (Pd(PPh3)4), tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3), dichlorobis(tricyclohexylphosphine)palladium(ll) (PdCI2[P(Cy)3]2) and the like or any other suitable catalysts or ligands known in the art used in said Sonogashira reaction in step (a).
Suitable bases that may be used in step (a) include, alkali metal or alkaline earth metal carbonates such as sodium carbonate, potassium carbonate, cesium
carbonate, calcium carbonate, magnesium carbonate, sodium bicarbonate, potassium bicarbonate, alkoxides such as sodium methoxide, potassium methoxide; organic bases, such as for example, triethylamine, tributylamine, A/-methylmorpholine, N,N- diisopropylethylamine, A/-methylpyrrolidine, pyridine, piperidine, collidine, 4-(N,N- dimethylamino)pyridine, morpholine, imidazole, 2-methylimidazole, 4-methylimidazole and the like or any other suitable base known in the art.
Suitable solvents that may be used in step (a) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, nitriles, ethers, polar aprotic solvents or mixtures thereof.
The reaction mixture obtained from step (a) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (a) may be isolated directly from the reaction mixture itself after the reaction is complete in step (a), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, the resulting product may be directly used for step (b) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
Step (b) involves optionally deprotecting a compound of formula XVI to provide a compound of formula XVII;

XVII
wherein is H or trialkyl silyl;
Suitable reagents that may be used in step (b) include, hydrochloric acid, hydrobromic acid, hydroiodic acid, nitric acid, sulfuric acid, acetic acid, formic acid, phosphoric acid, methanesulfonic acid, p-toluenesulfonic acid, tetra-n-butylammoniu m fluoride (TBAF), tris(dimethylamino)sulfonium difluorotrimethylsilicate, ammonia, sodium hydroxide, potassium hydroxide, sodium methoxide, potassium t-butoxide, sodium t-butoxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, and the like; ion exchange resins, such as: resins bound to metal ions, including lithium, sodium, potassium, and the like; and resins bound to
acids, including phosphoric, sulfonic, methanesulfonic, p-toluenesulfonic, and the like or any other suitable reagents and mixtures thereof.
Suitable solvents that may be used in step (b) include water, alcohols, ketones, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane and mixtures thereof.
Suitable temperatures that may be used in step (b) may be less than about 1 00°C, less than about 70°C, less than about 40°C, less than about 30°C, less than about 10°C, less than about 0°C, less than about -10°C, less than about -20°C, or any other suitable temperature.
The reaction mixture obtained from step (b) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration , suction filtration, or any other suitable techniques for the removal of solids. The product of step (b) may be isolated directly from the reaction mixture itself after the reaction is complete in step (b), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, an obtained crude product may be directly used for step (c) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
Step (c) involves converting a compound of formula XVI or compound of formula XVI I to a compound of for

XVI I I
wherein is H or trialkyl silyl ;
Conversion of a compound of formula XVI or compound of formula XVI I to a compound of formula XVI II may be carried out using hydrogen and a catalyst;
Suitable catalysts that may be used in step (c) include, [((S,S)- QuinoxP*)Rh(cod)]BF4, [((fl,fl)-Norphos)Rh(cod)]BF4, [((S,S)-Me-BPE)Rh(cod)]BF4, [((fl,fl)-Et-BPE)Rh(cod)]BF4, [((S,S)-Ph-BPP)Rh(cod)]BF4, [((S,S)-tBu- ferrotane)Rh(cod)]BF4, [((fl,fl)-Me-DuPhos)Rh(cod)]BF4, [((fi)-BINAP)Rh(cod)]BF4, [((S,S)-iPr-DuPhos)Rh(cod)]BF4, [((fl,fl)-iPr-BPE)Rh(cod)]BF4, [((R,R,S,S)- DuanPhos)Rh(cod)]BF4, [RuCI((S)-BINAP)2^-CI)3][NH2Me2], [(S)-
SEGPHOS)Ru(OAc)2], [(S)-BINAP)Ru(OAc)2] and the like or any iridium (Ir) complexes known in the art used for hydrogenation or any other suitable metal complexes known in the art.
Suitable solvents that may be used in step (c) include alcohols, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, polar aprotic solvents or mixtures thereof.
The reaction mixture obtained from step (c) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other suitable techniques for the removal of solids. The product of step (c) may be isolated directly from the reaction mixture itself after the reaction is complete in step (c), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, the resulting product may be directly used for step (d) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
Step (d) involves protecting a diol compound of formula XVIII to provide a compound of formula XIX;

XIX
wherein Ri is H or trialkyl silyl; Pi is an alcohol-protecting group; P2 is hydrogen or an alcohol-protecting group;
Optionally diol compound of formula XVIII may be protected sequentially to provide a compound of formula XIX or mono protected compound of formula XIV or compound of formula XXII I may be converted to compound of formula XIX or two or more compounds mixture of compound of formula XVIII, compound of formula XIV and compound of formula XXII I may be converted to compound of formula XIX

xviii xiv xxiii
Wherein Ρ-ι and P2 is alcohol protecting group; Ri is H or trialkyl silyl;
Suitable bases that may be used in step (d) include, alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide; carbonates such as sodium carbonate, potassium carbonate, cesium carbonate, calcium carbonate, magnesium carbonate, sodium bicarbonate, potassium bicarbonate, alkoxides such as sodium methoxide, potassium methoxide; organic bases, such as for example, triethylamine, tributylamine, N- methylmorpholine, A/,A/-diisopropylethylamine, A/-methylpyrrolidine, pyridine, collidine 4-(A/,A/-dimethylamino)pyridine, morpholine, imidazole, piperidine, 2-methylimidazole, 4-methylimidazole and the like or any other suitable bases known in the art.
Suitable catalysts that may be used in step (d) include enzymes such as lipases, esterases, amidases and any other suitable trans esterification enzymes known in the art, other catalysts such as oxo[hexa(trifluoroacetato)]tetrazinc and the like or any other suitable catalysts known in the art that are used for selective alcohol acylation or protection..
Suitable solvents that may be used in step (d) include ketones, esters, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane or mixtures thereof.
The reaction mixture obtained from step (d) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (d) may be isolated directly from the reaction mixture itself after the reaction is complete in step (d), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, the resulting product may be directly used for step (e) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
Step (e) involves optionally deprotecting a compound of formula XIX to provide a compound of formula XIV;

XIV
Suitable reagents that may be used in step (e) include, hydrochloric acid, hydrobromic acid, hydroiodic acid, nitric acid, sulfuric acid, acetic acid, formic acid, phosphoric acid, methanesulfonic acid, p-toluenesulfonic acid, tetra-n-butylammoniu m fluoride (TBAF), tris(dimethylamino)sulfonium difluorotrimethylsilicate, ammonia, sodium hydroxide, potassium hydroxide, sodium methoxide, potassium f-butoxide, sodium f-butoxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, and the like; ion exchange resins, such as: resins bound to metal ions, including lithium, sodium, potassium, and the like; and resins bound to acids, including phosphoric, sulfonic, methanesulfonic, p-toluenesulfonic, pyridinium p- toluenesulfonate and the like or any other suitable reagents and mixtures thereof.
Suitable solvents that may be used in step (e) include water, alcohols, ketones, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane and mixtures thereof.
Suitable temperatures that may be used in step (e) may be less than about 100°C, less than about 70°C, less than about 40°C, less than about 30°C, less than about 10°C, less than about 0°C, less than about -10°C, less than about -20°C, or any other suitable temperature.
The reaction mixture obtained from step (e) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (e) may be isolated directly from the reaction mixture itself after the reaction is complete in step (e, or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, the resulting product may be directly used for step (f) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
Step (f) involves converting a compound of formula XIV to a compound of formula II I.
Suitable reagents that may be used in step (f) include, (2,2,6,6-tetramethyl- piperidin-1 -yl)oxyl (TEMPO), pyridinium chlorochromate (PCC), oxalyl chloride &
dimethyl sulfoxide (DMSO), dicyclohexylcarbodiimide & DMSO, Dess-Martin periodinane, [bis(acetoxy)iodo]-benzene (BAIB), manganese dioxide and the like or any other suitable oxidizing agent known in the art.
Suitable solvents that may be used in step (f) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, halogenated hydrocarbons, polar aprotic solvents or mixtures thereof.
The reaction mixture obtained from step (f) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (f) may be isolated directly from the reaction mixture itself after the reaction is complete in step (f), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
Optionally steps (a) to (f) or any two or more steps may be carried out in-situ i.e. without isolating the intermediates in each stage.
A compound of formula XIV can also be prepared by using the (S)-enantiomer or enantiomerically enriched compound of formula XV. Inversion of the stereocentre at the secondary alcohol {(S)-enantiomer of a compound of formula XVI II} may be carried out by employing the Mitsunobu reaction conditions or any other suitable chiral inversion reaction conditions known in the art.
In the fourth embodiment, the present application provides a process for preparation of a compound of formula IV;

wherein Pi is an alcohol-protecting group; LG is -OSC>2(R); wherein R is selected from straight or branched C Cio alkyl or optionally substituted C5-C15 aryl or optionally substituted aralkyl; X is halogen, preferably bromine;
which includes one or more of the following steps:
(a) reacting an epoxide of formula XX with a compound of formula XXI to provide a compound of formula XXI I;

** ™ XXII
wherein R2 is trialkyl silyl; Pi is an alcohol-protecting group; X is halogen, preferably bromine;
(b) converting a compound of formula XXII to a compound of formula XV;
X OH XV
wherein is an alcohol-protecting group; X is halogen;
(c) converting a compound of formula XV to a compound of formula IV.
Step (a) involves reacting an epoxy compound of formula XX with a compound of formula XXI to provide a compound of formula XXII;

** ™ XXII
wherein R2 is trialkyl silyl; Pi is an alcohol-protecting group; X is halogen, preferably bromine;
Suitable forms of magnesium metal that may be used in step (a) include, any form of magnesium metal such as magnesium turnings, magnesium beads, magnesium powder, magnesium chips and the like.
Suitable reagents that may be used in step (a) to activate magnesium include, alkyl halides such as methyl iodide, methyl ethyl bromide, 1 , 2-dibromoethane and the like, iodine, diisobutylaluminium hydride (DIBAL-H), sodium bis(2- methoxyethoxy)aluminumhydride (Red-AI), lithium aluminium hydride (LAH) and the like or any other suitable magnesium activating reagents known in the art.
Suitable solvents that may be used in step (a) include ethers, aromatic hydrocarbons, or mixtures thereof.
Optionally, step (a) may be carried out in presence of a metal catalyst such as cuprous chloride, cuprous bromide, cuprous iodide, zinc chloride and the like or any other suitable metal catalyst known in the art employed for the Grignard reaction.
The reaction mixture obtained from step (a) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (a) may be isolated directly from the reaction mixture itself after the reaction is complete in step (a), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, the resulting product may be directly used for step (b) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
Step (b) involves converting a compound of formula XXII to a compound of formula XV;

wherein Pi is an alcohol-protecting group; X is halogen, preferably bromine;
Suitable reagents that may be used in step (b) include, bromine, N- bromosuccinimide, 1 ,3-dibromo-5,5-dimethylhydantoin, pyridinium perbromide phosphorus tribromide, iodine, A/-iodosuccinimide and the like or any other suitable reagents known in the art.
Suitable solvents that may be used in step (b) include, ethers, aliphatic and alicyclic hydrocarbons, aromatic hydrocarbons, halogenated hydrocarbons, esters, ketones, polar aprotic solvents or mixtures thereof.
The reaction mixture obtained from step (b) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (b) may be isolated directly from the reaction mixture itself after the reaction is complete in step (b), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like. Optionally, the resulting product may be directly used for step (c) with or without isolation or it may be further purified, if isolated, to improve the purity of the product.
Step (c) involves converting a compound of formula XV to a compound of formula IV.
The compound of formula IV is prepared by treating a compound of formula XV with sulfonyl halides or esters and the like. Suitable sulfonyl reagents that may be used in step (c) include, methanesulfonyl, p-toluenesulfonyl, 2,4,6- triisopropylbenzenesulfonyl, 4-propoxybenzene-1 -sulfonyl, 4-methoxybenzenesulfonyl, phenylmethanesulfonyl, 2,4-dichloropyrimidine-5-sulfonyl, benzenesulfonyl and the like.
Suitable base that may be used in step (c) include, alkali metal or alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide; carbonates such as sodium carbonate, potassium carbonate, cesium carbonate, calcium carbonate, magnesium carbonate, sodium bicarbonate, potassium bicarbonate, alkoxides such as sodium methoxide, potassium methoxide; organic bases, such as for example, triethylamine, tributylamine, A/-methylmorpholine, A/,A/-diisopropylethylamine, A/-methylpyrrolidine, pyridine, collidine, 4-(N,N- dimethylamino)pyridine, morpholine, imidazole, 2-methylimidazole, 4-methylimidazole and the like or any other suitable bases known in the art.
Suitable solvents that may be used in step (c) include ketones, esters, ethers, aliphatic and alicyclic hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, nitriles, polar aprotic solvents, nitromethane or mixtures thereof.
The reaction mixture obtained from step (c) may be optionally processed to remove any insoluble solids, and particles may be removed by methods such as decantation, centrifugation, gravity filtration, suction filtration, or any other techniques for the removal of solids. The product of step (c) may be isolated directly from the reaction mixture itself after the reaction is complete in step (c), or after conventional work up with techniques such as filtration, quenching with a suitable reagent, extraction, or the like.
Optionally steps (a) to (c) may be carried out in-situ i.e. without isolating the intermediates in each stage.
In the fifth embodiment, the present application provides a compound of formula V or isomers thereof.
 wherein P-i is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group preferably triisopropylsilyl ether;
wherein P-i is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group preferably triisopropylsilyl ether;
In the sixth embodiment, the present application provides a compound formula VI or isomers thereof.

In the seventh embodiment, the present application provides a compound of formula VII or isomers thereof.

VI I
wherein P-i is an alcohol-protecting group;
In the eighth embodiment, the present application provides a compound of formula VII I or isomers thereof.

VII I
wherein Pi is H or an alcohol protecting group and X is halogen.
In the ninth embodiment, the present application provides a compound formula II I or isomers thereof;

I I I
wherein Pi is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group;
In the tenth embodiment, the present application provides a compound of formula IVa or isomers thereof.

IVa
wherein Pi is an alcohol protecting group and X is halogen;
In the eleventh embodiment, the present application provides a compound having the following formula or isomers thereof.


In the twelfth embodiment, the present application provides a process for preparation of eribulin or a pharmaceutically acceptable salt thereof comprising synthesizing eribulin or its pharmaceutically acceptable salt from one or more compounds of first embodiment to eleventh embodiment.
One or more compounds of first embodiment to eleventh embodiment include compound of formula III, formula IV, formula IVa, formula V, formula VI, formula VII, formula VIII, formula XI, formula XII, formula XIII, formula XIV, formula XV, formula XVI, formula XVII, formula XVII I, formula XIX, formula XXII and compounds in eleventh embodiment.
Isomers of one or more compounds of first embodiment to twelfth embodiment isomers include stereoisomers of formula III, formula IV, formula IVa, formula V, formula VI, formula VII, formula VIII, formula XI, formula XII, formula XIII, formula XIV, formula XV, formula XVI, formula XVII, formula XVIII, formula XIX, formula XXI I and compounds in eleventh embodiment.
DEFINITIONS
The following definitions are used in connection with the present application unless the context indicates otherwise. In general, the number of carbon atoms present in a given group or compound is designated "Cx-Cy", where x and y are the lower and upper limits, respectively. For example, a group designated as "CrC6" contains from 1 to 6 carbon atoms. The carbon number as used in the definitions herein refers to carbon backbone and carbon branching, but does not include carbon atoms of the substituents, such as alkoxy substitutions or the like.
As used herein, "an alcohol protecting group" is a functional group that protects the alcohol group from participating in reactions that are occurring in other parts of the molecule. Suitable alcohol protecting groups that are used in step (a) include, acetyl, benzoyl, benzyl, β-methoxyethoxymethyl ether, methoxymethyl ether, dimethoxytrityl, p-methoxybenzyl ether, methylthiomethyl ether, allyl ether, f-butyl ether, pivaloyl, trityl, silyl ether (e.g., trimethylsilyl (TMS), f-butyldimethylsilyl (TBMDS), f-butyldiphenylsilyl (TBDPS), f-butyldimethylsilyloxymethyl (TOM) or triisopropylsilyl (TIPS) ether), tetrahydropyranyl (THP), methyl ether and ethoxyethyl ether (EE) or any suitable alcohol protecting group known in the art.
An "alcohol" is an organic compound containing a carbon bound to a hydroxyl group. "Ci-C6 alcohols" include methanol, ethanol, 2-nitroethanol, 2-fluoroethanol, 2,2,2-trifluoroethanol, hexafluoroisopropyl alcohol, ethylene glycol, 1 -propanol, 2- propanol (isopropyl alcohol), 2-methoxyethanol, 1 -butanol, 2-butanol, i-butyl alcohol, t- butyl alcohol, 2-ethoxyethanol, diethylene glycol, 1 -, 2-, or 3-pentanol, neo-pentyl alcohol, t-pentyl alcohol, cyclohexanol, phenol, glycerol and the like.
An "aliphatic hydrocarbon" is a liquid hydrocarbon compound, which may be linear, branched, or cyclic and may be saturated or have as many as two double bonds. A liquid hydrocarbon compound that contains a six-carbon group having three double bonds in a ring is called "aromatic." Examples of "C5-C8 aliphatic" include n- pentane, isopentane, neopentane, n-hexane, isohexane, 3-methylpentane, 2,3- dimethylbutane, neohexane, n-heptane, isoheptane, 3-methylhexane, neoheptane, 2,3-dimethylpentane, 2,4-dimethylpentane, 3,3-dimethylpentane, 3-ethylpentane, 2,2,3-trimethylbutane, n-octane, isooctane, 3-methylheptane, neooctane, cyclohexane, methylcyclohexane, cycloheptane, petroleum ethers and the like.
An "aromatic hydrocarbon solvent" refers to a liquid, unsaturated, cyclic, hydrocarbon containing one or more rings which has delocalized conjugated π system. Examples of an aromatic hydrocarbon solvent include benzene toluene, ethylbenzene, m-xylene, o-xylene, p-xylene, indane, naphthalene, tetralin, trimethylbenzene, chlorobenzene, fluorobenzene, trifluorotoluene, anisole, C6-Ci2 aromatic hydrocarbons and the like.
An "ester" is an organic compound containing a carboxyl group -(C=0)-0- bonded to two other carbon atoms. "C3-C6 esters" include ethyl acetate, n-propyl acetate, n-butyl acetate, isobutyl acetate, t-butyl acetate, ethyl formate, methyl acetate, methyl propanoate, ethyl propanoate, methyl butanoate, ethyl butanoate and the like.
An "ether" is an organic compound containing an oxygen atom -O- bonded to two other carbon atoms. "C2-C6 ethers" include diethyl ether, diisopropyl ether, methyl t-butyl ether, glyme, diglyme, tetrahydrofuran, 2-methyltetrahydrofuran, 1 , 4-dioxane, dibutyl ether, dimethylfuran, anisole, diethylene glycol monomethyl ether, diethylene glycol monoethyl ether, cyclopentyl methyl ether and the like.
A "halogenated hydrocarbon" is an organic compound containing a carbon bound to a halogen. Halogenated hydrocarbons include dichloromethane, 1 ,2- dichloroethane, trichloroethylene, perchloroethylene, 1 ,1 ,1 -trichloroethane, 1 ,1 ,2- trichloroethane, chloroform, carbon tetrachloride and the like.
A "ketone" is an organic compound containing a carbonyl group -(C=0)- bonded to two other carbon atoms. "C3-C6 ketones" include acetone, ethyl methyl ketone, diethyl ketone, methyl isobutyl ketone, ketones and the like.
A "nitrile" is an organic compound containing a cyano -(C≡N) bonded to another carbon atom. "C2-C6 Nitriles" include acetonitrile, propionitrile, butanenitrile and the like.
A "polar aprotic solvents" include Ν,Ν-dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, sulfolane, N-methylpyrrolidone and the like;
Certain specific aspects and embodiments of the present application will be explained in greater detail with reference to the following examples, which are provided only for purposes of illustration and should not be construed as limiting the scope of the application in any manner. Reasonable variations of the described procedures are intended to be within the scope of the present application. While particular aspects of the present application have been illustrated and described, it would be obvious to those skilled in the art that various other changes and modifications can be made without departing from the spirit and scope of the invention. It is therefore intended to cover in the appended claims all such changes and modifications that are within the scope of this application.
EXAMPLES
Example-1 : Preparation of (fi)-3-Methyl-5-(triisopropylsilyl)pent-4-ynal.

[Rh(OH)(cod)]2 (5.87 g) and (S)-DTBM-SEGPHOS (36.45, 6 mol%) were partially dissolved in dioxane (732 mL) and warmed to 53 °C. After 1 .25 h, the resulting orange solution was allowed to cool to room temperature. TIPS acetylene (188 g) was charged to the reaction vessel over 10 min followed by crotonaldehyde (42.7 mL) was added to the reaction mass via pump syringe over 3 h and the resultant reaction mass was stirred at 43 °C for 16 h. Reaction mass was concentrated in vacuo and the obtained compound was purified by wipe-film-evaporation to afford the titled aldehyde (42.9 g, 170 mmol, 33%) as a pale yellow oil.
Example-2: Preparation of (4ff,6fi)-6-methyl-8-(triisopropylsilyl)oct-1 -en-7-yn-4- ol.

(R)-3-Methyl-5-(triisopropylsilyl)pent-4-ynal (10.77 g) in diethyl ether (50 mL) was added slowly to the solution of (+)-lpc2B-allyl (1 M in pentane, 51 .2 mL) in diethyl ether (50 mL) at -70 °C. The resultant reaction mass was stirred at -70 °C for 1 hour 30 minutes, warmed to 0 °C and stirred at 0 °C for 1 hour 30 minutes. 3M NaOH (80.2 mL) followed by hydrogen peroxide (30% aq. solution, 33 mL) was added slowly to the reaction mass at 27 °C and stirred at 27 °C for 17 hours. Reaction mass was passed through a pad of Celite, washed with MTBE (50 mL). Layers were separated, aqueous layer was extracted with MTBE (3 x 50 mL) and the combined organic extracts were dried over MgSC¼ and concentrated in vacuo. The obtained compound was purified by flash chromatography (Si02) using 0 to 5 to 10% EtOAc/heptane to afford titled compound (10.6 g).
Example-3: Preparation of ieri-Butyldimethyl(((4R,6R)-6-methyl-8- (triisopropylsilyl)oct-1-en-7-yn-4-yl)oxy)silane.

To a solution of (4R,6R)-6-methyl-8-(triisopropylsilyl)oct-1 -en-7-yn-4-ol (126 g) in DMF (250 mL) was added imidazole (87.6 g) followed by TBSCI (96.9 g). The solution was warmed to 44 °C and stirred at 44 °C for 1 8 hours. Methanol (10 mL), water (250 mL) and heptane (350 mL) was added slowly to the reaction mixture at 27 °C and stirred for 15 minutes. Layers were separated and the aqueous layer was extracted with heptane (250 mL). The combined organic layers were dried over MgSC¼ and then concentrated in vacuo. The obtained crude material was purified by flash chromatography (Si02) using (1 ) heptane (1 .5 L); (2) 1 % heptane/EtOAc (2 L); (3) 2% EtOAc/heptane (10 L) sequentially to afford the titled product (159.7 g).
Example-4: Preparation of (4ff,6fi)-4-((tert-butyldimethylsilyl)oxy)-6-methyl-8- (triisopropylsilyl)oct-7-yn-1-ol.
T

9-BBN-H (0.5 M in THF, 41 .9 mL) was added slowly to the solution of tert- Butyldimethyl(((4R,6R)-6-methyl-8-(triisopropylsilyl)oct-1 -en-7-yn-4-yl)oxy)silane (5.7 g) and THF (30 mL) at 0 °C. The solution was allowed to warm to 27 °C and stirred at 27 °C for 15 hours. Water (103 mL) followed by NaB03.4H20 (10.3 g) was added portion wise to the reaction mass at 20 °C and the resultant reaction mass was stirred at 27 °C for 22 hours. Reaction mass was filtered through a pad of Celite, washed with MTBE (250 mL). Layers were separated, aqueous layer was extracted with MTBE (250 mL). The combined organic layers were dried over MgSC and then concentrated in vacuo. The obtained crude material was purified by flash chromatography (Si02) using 0%, 10% and 20% EtOAc/heptane sequentially to afford titled product (5.45 g).
Example-5: Preparation of (4ff,6fi)-4-((ieri-Butyldimethylsilyl)oxy)-6-methyl-8- (triisopropylsilyl)oct-7-ynal.

To a solution of (4R,6R)-4-((tert-butyldimethylsilyl)oxy)-6-methyl-8- (triisopropylsilyl)oct-7-yn-1 -ol (122 g) in dichloromethane (500 mL) was added TEMPO (4.47 g) followed by Phl(OAc)2 (1 .2 g) at 27 °C and stirred at 27 °C for 18 hours. Dichloromethane (500 mL) was added to the reaction mass and the resultant reaction mass was washed with sat. aq. Na2S03 (500 mL), sat. aq. NaHC03 (500 mL) then brine (500 mL). The organic layer was concentrated in vacuo and the obtained crude material was purified by flash chromatography (Si02, 1 .4 kg) with (1 ) heptane (1 L), (2) 2% EtOAc/heptane (1 L), (3) 3% EtOAc/heptane (3 L) and (4) 4% EtOAc/heptane (3 L) sequentially to afford titled product (98.8 g).
Example-6: Preparation of tert-butyl(3-((2S,5S)-5-((3R,5R)-3-((tert- butyldimethylsilyl)oxy)-5-methyl-7-(triisopropylsilyl)hept-6-yn-1-yl)-4-methylen tetrahydrofuran-2-yl)propoxy)diphenylsilane.

Aldehyde and vinyl bromide compounds solution preparation: In two separate 500 ml flasks, (4R,6R)-4-((tert-Butyldimethylsilyl)oxy)-6-methyl-8-(triisopropylsilyl)oct- 7-ynal (61 .9 g) and (R)-2-bromo-7-((tert-butyldiphenylsilyl) oxy)hept-1 -en-4-yl 2,4,6- triisopropylbenzenesulfonate (124.7 g) were evaporated with toluene (30 mL) three times to remove any water. The flasks were placed under a flow of nitrogen and acetonitrile (200 mL) was added to the flask containing bromo compound. After dissolution of bromo compound in acetonitrile, transferred to the flask containing aldehyde and stirred for 15 minutes to get clear solution.
A 3L, 3-neck flask equipped with a mechanical stirrer and a nitrogen inlet and a gas outlet, was dried with a heatgun under a flow of nitrogen for 20 min. It was left under a nitrogen flow overnight. 1 ,8-bis(dimethylamino)naphthalene (Proton sponge) (96.6 g), (R)-N-(2-(4-isopropyl-4,5-dihydrooxazol-2-yl)phenyl)methanesulfonamide (133.8 g), and CrCI2 (55.4 g) were added to the flask. Acetonitrile (600 mL) was slowly added to the flask and the resultant reaction mass was stirred at 27 °C for 1 hour 30 minutes. NiCI2-2,9-dimethyl-1 ,10-phenanthroline (2.46 g, 5%), followed by the solution of aldehyde and vinyl bromide compounds was added to the reaction mass and the resultant reaction mass was stirred at 27 °C for 20 hours. Ethylenediamine (97 mL) was slowly added to the reaction mass at below 20 °C and stirred for 10 minutes. The reaction mass was warmed to 40 °C and stirred at 40 °C for 4 hours. Water (600 mL) and 2-Me-THF (600 mL) was added to the reaction mass and stirred for 15 minutes. The layers were separated, the aqueous layer was extracted twice with 2-Me-THF (600 mL), combined organic layers washed with brine (100 mL) and dried over MgSC¼. The obtained organic layer was filtered, concentrated in vacuo. THF (300 mL) was added to the crude compound, separated solid was filtered and wet cake was
washed with THF (300 mL). Acetonitrile (700 mL) and heptane (700 mL) was added to the wet compound, stirred for 30 minutes and the resultant reaction mass was filtered. Separated filtrate layers, extracted acetonitrile layer with heptane (3x700 mL). Evaporated all the heptane layer and the obtained crude material was purified using column chromatography (Heptane/MTBE (95/5, 2.5 L, then Heptane/MTBE (90/10, 4.5I)) to afford title compound (55 g).
Example-7: (3ff,5fi)-1 -((2S,5S)-5-(3-Hydroxypropyl)-3-methylenetetrahydrofuran- 2-yl)-5-methylhept-6-yn-3-ol

To a solution of tert-butyl(3-((2S,5S)-5-((3R,5R)-3-((tert-butyldimethylsilyl)oxy)- 5-methyl-7-(triisopropylsilyl)hept-6-yn-1 -yl)-4-methylene tetrahydrofuran-2-yl)propoxy) diphenylsilane (27.0 g) in THF (135 mL) was added TBAF (1 M in THF, 209 mL) over 25 min and the resulting brown solution was stirred at 27 °C for 16 hours. MTBE (270 mL) was added to the reaction mass, and the resultant reaction mass was washed with water/sat aq. NH4CI (1 :1 , 270 mL). Aqueous layer was extracted with MTBE (270 mL), combined organic layers were concentrated in vacuo. The obtained crude oil was dissolved in methanol (270 mL) and cone. HCI (8.1 mL) was added slowly. The solution was stirred at 27 °C for 2 hours and quenched with solid NaHC03 (7 g). The resultant suspension was filtered and the filtrate concentrated in vacuo. The obtained oil was dissolved in MTBE (270 mL) and washed with sat aq. NaHC03 (270 mL). Aqueous layer was extracted with MTBE (2 x 270 mL), the combined organic layers were concentrated in vacuo and the obtained crude material was purified by flash chromatography (Si02) with: (1 ) 20% EtOAc/heptane (0.5 L), (2) 60% EtOAc/heptane (1 L), (3) 80% EtOAc/heptane (2 L) and (4) EtOAc (0.5 L) sequentially to afford crystalline titled product (7.41 g; dr A ).
Example-8: Preparation of ieri-Butyl(3-((2S,5S)-5-((3ff,5fi)-3-((tert- butyldimethylsilyl)oxy)-5-methylhept-6-yn-1-yl)-4-methylenetetrahydrofuran-2- yl)propoxy)dimethylsilane.

To a solution of (3R,5R)-1 -((2S,5S)-5-(3-Hydroxypropyl)-3- methylenetetrahydrofuran-2-yl)-5-methylhept-6-yn-3-ol (9.85 g) and imidazole (12.6 g) in DMF (39 mL) was added TBSCI (13.9 g). The resulting reaction mass was warmed to 47°C and stirred at 47 °C for 16 hours. Methanol (4 mL) was added to the reaction mass then cooled to 27 °C. Heptane (100 mL) was added to the reaction mass and washed with water (2 x 200 mL) then sat aq. NH4CI (100 mL). The organic layer was concentrated in vacuo, the obtained crude oil was purified by passing through a silica plug using (1 ) heptane (200 mL) and (2) 5% EtOAc/heptane (600 mL) to afford the titled product (17.4 g; drA'A ).
Example-9: Preparation of (3ff,5fl)-1-((2S,5S)-5-(3-hydroxypropyl)-3- methylenetetrahydrofuran-2-yl)-6-iodo-5-methylhept-6-en-3-ol

DIBAL-H (1 M in THF, 1 .62 mL) was slowly added to a suspension of NiCI2 (bis(diphenylphosphino)ethylene) (42.7 mg) in THF (0.5 mL) and stirred for 15 minutes. A solution of tert-Butyl(3-((2S,5S)-5-((3R,5R)-3-((tert-butyldimethylsilyl)oxy)- 5-methylhept-6-yn-1 -yl)-4-methylenetetrahydrofuran-2-yl)propoxy)dimethylsilane (400 mg) in THF (1 .5 mL) was added to the reaction mass at 0 °C and stirred at 0 °C for 30 minutes. Reaction mass was warmed to 27 °C and stirred at 27 °C for 4 hours. A solution of N-iodosuccinimide (364 mg) in THF (1 .5 mL) was added to the reaction mass at -78 °C and stirred at -78 °C for 1 hour. Reaction mass was quenched into sat aq. Rochelle's salt (15 mL), extracted with MTBE (2 x 15 mL) and the combined organic layers were concentrated in vacuo. The resulting crude oil was dissolved in methanol (4 mL). 37% HCI (0.04 mL) was added to the reaction mass at 27 °C and stirred at 27 °C for 2 hours. Solid NaHC03 was added to the reaction mass, the suspension was filtered and the filtrate concentrated in vacuo. To the obtained crude
material, EtOAc (15 mL) and water (15 mL) was added. The layers were separated and the aqueous was extracted again with EtOAc (15 mL). The combined organic layers were dried over MgS04 and concentrated in vacuo. The resulting oil was purified by fluid chromatography using (1 ) 5% acetone/heptane (70 mL); (2) 40% EtOAc/acetone (80 mL) followed by supercritical fluid chromatography (SFC) to afford the titled product (178 mg).
SFC Method: Column: YMC amylose-C 150 x 4.6, 5 μ; Flow: 1 mL/min (Isocratic); Run-time: 15 minutes; Diluent: acetonitrile; Sample Concentration: 1 mg/mL in diluent; Mobile phase: acetonitrile and water in 95:5 ratio (v/v)
Example-10: Preparation of 3-((2S,5S)-5-((3ff,5fl)-3-Hydroxy-6-iodo-5- methylhept-6-en-1-yl)-4-methylenetetrahydrofuran-2-yl)propyl pivalate.

A solution of (3R,5R)-1 -((2S,5S)-5-(3-hydroxypropyl)-3-methylenetetrahydro furan-2-yl)-6-iodo-5-methylhept-6-en-3-ol (480 mg) in dichloromethane (4.8 mL) was cooled to 0 °C. Pyridine (296 μΙ_), 4-DMAP (9.6 mg) and then PivCI (157 μΙ_) was added to the reaction mass. The resultant reaction mass was stirred at 0 °C for 30 minutes then at 27 °C for 2 hours. Dichloromethane (5 mL) was added to the reaction mass and the obtained reaction mass was washed with sat. aq. NH4CI (10 mL). The organic layer was concentrated in vacuo and the resultant crude material was purified by FC (Si02) with (1 ) 20% EtOAc/heptane (60 mL) and 40% EtOAc/heptane (60 mL) sequentially to afford the titled product (480 mg).
Example-11 : Preparation of (fi)-7-((ieri-butyldimethylsilyl)oxy)-2- (trimethylsilyl)hept-1-en-4-ol.

1 -bromovinyltrimethylsilane solution preparation: 62.1 g of 1 -bromovinyltrimethylsilane was dissolved in 1 00 mL THF.
Iodine (1 .17 g) was added to the magnesium turnings (9.55 g) at 23 °C and allowed to stand for 10 minutes. Anhydrous tetrahydrofuran (50 mL) followed by 1 - bromovinyltrimethylsilane solution in THF (10 mL) was added to the reaction mass at 24 °C and stirred for 30 minutes. Another lot of 1 -bromovinyltrimethylsilane solution in THF (10 mL) was added to the reaction mass at 24 °C and stirred for 20 minutes. Anhydrous tetrahydrofuran (200 mL) followed by remaining 1 - bromovinyltrimethylsilane solution in THF (-130 mL) was added slowly to the reaction mass and stirred for 30 minutes. The resultant reaction mixture slowly cooled to 25 °C and further cooled to -25 °C. Copper (I) iodide (2.2 g) was added to the reaction mass at -25 °C under argon and stirred at -25 °C for 45 minutes. (fl)-ferf-butyldimethyl(3- (oxiran-2-yl)propoxy)silane solution in THF (50 g in 150 mL THF) was slowly added to the reaction mass at -25 °C over a period of 1 hour and stirred at -25 °C for 1 hour. Saturated ammonium chloride solution (200 mL) was added slowly to the reaction mass at 2 °C and stirred for 10 minutes. Water (200 mL) was added and stirred for 30 minutes, layers were separated. Aqueous layer was extracted with ethyl acetate (2X100 mL), combined organic layer was washed with water (200 mL), brine solution (200 mL) and dried with anhydrous sodium sulfate (20 g). The organic layer was concentrated in vacuo, the obtained crude product was purified by using column chromatography (eluent: EtOAc/hexane) to afford the titled product (68 g).
Example-12: Preparation of (fi)-2-bromo-7-((ieri-butyldimethylsilyl)oxy)hept-1 - en-4-ol

Triethylamine (0.320 g) was added to the reaction mass containing (R)-7-((tert- butyldimethylsilyl)oxy)-2-(trimethylsilyl)hept-1 -en-4-ol (1 .0 g) and toluene (15 mL) at - 30 °C. Pre cooled (-30 °C) bromine solution (0.606 g of bromine in 10 mL of toluene) was added slowly to the reaction mass at -58 °C. Sodium methanolate solution (2 mL; 25 wt. % in methanol) was added at -58 °C and the resultant reaction mixture was stirred at -48 °C for 5 hours. Methanol (10 mL) was added to the reaction mass at -42 °C and the reaction mass slowly warmed to 28 °C. Solvent was removed under reduced pressure at 48 °C. The obtained crude compound was dissolved in ethyl acetate (50 mL), washed with saturated aqueous ammonium chloride solution (10 mL)
and water (5 mL). The resultant organic layer was concentrated in vacuo at below 48 °C to afford the title compound.
Example-13: Preparation of (fi)-2-bromo-7-((ieri-butyldimethylsilyl)oxy)hept-1- en-4-ol

1 3-dibromo-5 5-dimethylimidazolidine-2 4-dione (10.84 g) was slowly added to the reaction mass containing (/^)-7-{(fe/ -butyldimethylsilyl)oxy)-2-(trimethylsilyl)hept- 1 -en-4-ol (10 g) and dichloromethane (150 mL) at -10 °C and the resultant reaction mass was stirred at -10 °C for 2 hours. Sodium methanolate solution (5.12 g; 30 wt. % in methanol) was added at -52 °C and the resultant reaction mixture was stirred at -52 °C for 2 hours 20 minutes. Methanol (100 mL) was added to the reaction mass at 0 °C and the reaction mass pH was slowly adjusted to 6.5 with aqueous ammonium chloride solution. Reaction mass concentrated under reduced pressure, the obtained crude compound was dissolved in ethyl acetate (100 mL), washed with saturated aqueous ammonium chloride solution (100 mL) and water (150 mL). The resultant organic layer was concentrated in vacuo at below 47 °C. The obtained crude compound was purified by flash chromatography to afford the titled compound (4.88 g)-
Example-14: Preparation of (fi)-2-bromo-7-((ieri-butyldimethylsilyl)oxy)hept-1- en-4-yl 2,4,6-triisopropylbenzenesulfonate.

4-(Dimethylamino)pyridine (286 mg) and triethylamine (316 mg) was added to the reaction mass containing (/:?)-2-bromo-7-((ferf-butyldimethylsilyl)oxy)hept-1 -en-4-ol (505 mg) and dichloromethane (5 mL) at 0 °C. Solution of 2,4,6-triisopropylbenzene-1 - sulfonyl chloride (710 mg in 1 .5 mL dichloromethane) was added to the reaction mass
at 0 °C and stirred at 25 °C for 22 hours. Reaction mass was washed with water (2x20 mL) and the resultant organic layer was concentrated in vacuo. The obtained crude compound was purified by flash chromatography to afford the titled compound (780 mg).
Example-15: Preparation of (fi)-1 -((ieri-butyldimethylsilyl)oxy)-6-methylene-8- (triisopropylsilyl)oct-7-yn-4-ol.

A solution of the bromide (100.31 g) in dry DMF (600 mL), dry triethylamine (130 mL) and (triisopropylsilyl)acetylene (104 mL) was added to the reaction mass containing [1 ,1 '-Bis(diphenylphosphino)ethane]dichloropalladium(ll) (1 .793 g) and copper iodide (1 .187 g) under N2 . The resultant reaction mass was stirred at 55 °C for 16 hours. MTBE (750 mL) was added to the reaction mass at 25 °C and stirred at 25 °C for 1 hour. The precipitate was removed by filtration through a pad of silica gel (100 g), the filter cake was washed with MTBE (250 mL). The combined filtrates were washed with water (500 mL), sat. NH4CI sol. (500 mL), water (500 mL) and brine solution (500 mL). The washed filtrates were dried over MgS04 and then concentrated under reduced pressure to afford title compound (157.47 g).
Example-16: Preparation of (fi)-6-methylene-8-(triisopropylsilyl)oct-7-yne-1 ,4- diol.

12 M HCI (1 .5 mL) was added to the reaction mass containing (fl)-1 -((ferf- butyldimethylsilyl)oxy)-6-methylene-8-(triisopropylsilyl)oct-7-yn-4-ol (157.47 g) and methanol (750 ml) and the resultant reaction mixture was stirred at 25 °C for 2 hours. Solid sodium hydrogen carbonate (2.27 g) was added to the reaction and stirred for 1 hour at 25 °C. The solids were removed by filtration and the filtrates concentrated under reduced pressure. The concentrated residue was dissolved in ethyl acetate (750 mL), washed with water (500 ml) and brine (500 ml). The resultant organic layer
was dried over magnesium sulfate and concentrated in vacuo to afford the title compound (108.7 g).
Example-17: Preparation of (4ff, 6fi)-6-Methyl-8-(triisopropylsilyl)oct-7-yne-1 ,4- diol.

(fl)-6-methylene-8-(triisopropylsilyl)oct-7-yne-1 ,4-diol (1 .0 g) and MeOH (10 mL) was charged into 50 mL Parr pressure vessel under a nitrogen atmosphere containing [((S,S)-QuinoxP*)Rh(cod)]BF4 (6.8 mg). The vessel was topped up to 10 bar pressure with hydrogen. Pressure was monitored and when it dropped to 9 bar or below, the pressure was increased to 1 0 bar by topping up with more hydrogen. Reaction mass was stirred until no further hydrogen uptake was observed. Pressure was released and the resulting solution was concentrated in vacuo to afford the titled compound (0.92 g).
Example-18: Preparation of (4ff, 6fi)-4-((tert-Butyldimethylsilyl)oxy)-6-methyl-8- (triisopropylsilyl)oct-7-yn-1-ol.

To a solution of (4R, 6fl)-6-Methyl-8-(triisopropylsilyl)oct-7-yne-1 ,4-diol (274 mg) in DMF (1 mL) was added imidazole (298 mg) followed by ferf-Butyldimethylsilyl chloride (330 mg) and the resultant reaction mass was stirred for 4 hours at 47 °C. MeOH (150 μΙ_) was added to the reaction mass at 25 °C and stirred at 25 °C for 2 hours. MTBE (20 mL) and water (20 mL) was added to the reaction mass and layers were separated. Organic layer washed with water (20 mL), dried over MgS04 and concentrated in vacuo to afford the crude di-TBS protected intermediate (467 mg) as a pale yellow oil.
To a solution of the crude di-TBS protected intermediate in EtOH (2.8 mL) was added pyridinium p-toluenesulfonate (21 .7 mg) and the resultant reaction mass was
stirred at 25 °C for 19 hours. Reaction mass was diluted with MTBE (20 mL) and washed with brine (20 mL). The organic layer was dried over MgSC¼ and concentrated in vacuo. The obtained crude compound was purified by flash chromatography (Si02) (0:100 to 20:80, EtOAc/heptane) to afforded the titled compound (214 mg).
Example-19: preparation of (4ff, 6fi)-4-((ieri-Butyldimethylsilyl)oxy)-6-methyl-8- (triisopropylsilyl)oct-7-yn-1-ol.
T

A mixture of (4R, 6/¾-6-Methyl-8-(triisopropylsilyl)oct-7-yne-1 ,4-diol and (4^,6^)-4-((ferf-butyldimethylsilyl)oxy)-6-methyl-8-(triisopropylsilyl)oct-7-yn-1 -ol (1 .36 g), isopropenyl acetate (0.8 mL) and Novozyme 435 (1 00 mg) in ferf-Butyl Methyl ether (1 5 mL) was stirred at 21 °C under nitrogen for 18 hours. Reaction mass filtered through a pad of Celite, washed with MTBE and the resultant filtrate was concentrated. The obtained crude compound was dissolved in dimethylformamide (4 mL).7erf-butyldimethylsilyl chloride (1 .2 g) and imidazole (1 .3 g) were added to the reaction mass and the resulting solution was stirred at 21 °C under nitrogen for 16 hours. Methanol (0.5 mL) was added to the reaction mass and the mixture was stirred at 21 °C for 3 hours. MTBE (30 mL) and water (20 mL) was added to the reaction mass at 21 °C and stirred for 1 5 minutes. The layers were separated and the aqueous layer was extracted with MTBE (30 mL). The combined layer was washed with water (10 mL) and then concentrated to yield a slightly hazy oil (1 .17 g). The oil was dissolved in methanol (15 mL) and potassium carbonate (100 mg) was added to the reaction mass. The resulting solution was stirred at 21 °C under nitrogen for 5 hours. The reaction mass was poured into a mixture of MTBE (20 mL) and water (30 mL) and layers were separated. The aqueous layer was extracted with MTBE (2 x 20 mL). The combined organic layer was filtered through a pad of magnesium sulfate and concentrated to afford title compound (750 mg).
Example-20: Preparation of (4ff,6fi)-4-((ieri-Butyldimethylsilyl)oxy)-6-methyl-8- (triisopropylsilyl)oct-7-ynal.

To a solution of (4/:?,6/:?)-4-((ferf-butyldimethylsilyl)oxy)-6-methyl-8- (triisopropylsilyl)oct-7-yn-1 -ol (122 g) in dichloromethane (500 mL) was added TEMPO (4.47 g) followed by Phl(OAc)2 (1 .2 g) at 27 °C and stirred at 27 °C for 18 hours. Dichloromethane (500 mL) was added to the reaction mass and the resultant reaction mass was washed with sat. aq. Na2S03 (500 mL), sat. aq. NaHC03 (500 mL) then brine (500 mL). The organic layer was concentrated in vacuo and the obtained crude material was purified by flash chromatography (Si02, 1 .4 kg) with (1 ) heptane (1 L), (2) 2% EtOAc/heptane (1 L), (3) 3% EtOAc/heptane (3 L) and (4) 4% EtOAc/heptane (3 L) sequentially to afford titled product (98.8 g).
Example-21 : Preparation of (3ff,5fl)-1-((2S,5S)-5-(3-hydroxypropyl)-3- methylenetetrahydrofuran-2-yl)-6-iodo-5-methylhept-6-en-3-ol

BusSn-AIEt? solution preparation: 1 .6 M nBuLi in hexanes (8.8 mL) was added dropwise to the solution containing isopropylamine (1 .98 mL) and THF (34 mL) at -10 °C under nitrogen and the resulting solution was stirred at -10 °C for 30 minutes. Bu3SnH (3.8 mL) was added to the solution dropwise at below -30 °C and stirred for 30 minutes. 1 .8 M Et2AICI in PhMe (7.9 mL) was added dropwise at -40 °C and the reaction mixture was stirred for 10 minutes at -30 °C.
tert-Butyl(3-((2S,5S)-5-((3R,5R)-3-((tert-butyldimethylsilyl)oxy)-5-methylhept-6- yn-1 -yl)-4-methylenetetrahydrofuran-2-yl)propoxy)dimethylsilane (3.3 g) in THF (17.5 mL) was added to a reaction mass that contained [Ph3PCul]4 (1 .28 g) under nitrogen atmosphere and the resulting suspension was stirred for 5 minutes at 0 °C. The Bu3Sn-AIEt2 solution was added to the reaction mixture and the resultant reaction
mixture stirred for 1 hour 30 minutes at 0 °C. Sat. aq. NH4CI solution (35 mL) was added slowly and the resulting mixture was diluted with 2-MeTHF (70 mL) and 20% w/w aq. Rochelle's salt solution (70 mL), warmed to room temperature, and stirred rapidly for 1 hour. The biphasic mixture was transferred into a separating funnel, further portions of 2-MeTHF (70 mL) and 20% w/w aq. Rochelle's salt solution (70 mL) were added and the phases were mixed, separated and the organic layer was washed with 20% w/w aq. Rochelle's salt solution (70 mL), brine (150 mL), dried with MgS04, filtered and concentrated to give a colourless oil containing an off-white precipitate. The oil was solubilised in heptane (150 mL) and stirred for 1 h, the resulting suspension filtered and the filtrate concentrated to give colourless oil.
The crude material (colourless oil) was dissolved in DCM (170 mL) and cooled to -40 °C under nitrogen. A/-lodosuccinimide (3.18 g) was added and the reaction stirred for 1 hour. The reaction mixture was quenched with 20% w/w aq. Na2S203 (1 50 mL) and warmed to room temperature. The layers were separated and the aqueous layer was washed with DCM (2x 150 mL). The combined organic layers were washed with brine solution (150 mL), dried with MgS04, filtered and concentrated to give 8.02 g pale yellow oil; potency 35.7 wt% for desired a-regioisomer, 2.86 g of title compound.
Claims
1. A process for preparation of compound of formula I I comprising one or more of the following steps:

I I
wherein Pi is H or an alcohol protecting group; P2 is H or an alcohol protecting group or -S02(R); wherein R is selected from straight or branched C1-C10 alkyl or optionally substituted C5-Ci2 aryl or optionally substituted aralkyl ; X is halogen;
(a) reacting acetylene compound of formula I II with vinyl halide compound of formula IV to provide compound of formula V;

V
wherein is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group; LG is -OS02(R); wherein R is selected from straight or branched C1 -C10 alkyl or optionally substituted C5-C12 aryl or optionally substituted aralkyl ;
(b) optionally deprotecting compound of formula V to provide dihydroxy compound of formula VI;

(c) optionally protecting dihydroxy compound of formula VI to provide compound of formula VII;

VI I
wherein Pi is an alcohol-protecting group;
(d) converting compound of formula VI or formula VII to provide compound of formula VII I;

VII I
wherein P-i is H or an alcohol protecting group and X is halogen;
(e) converting compound of formula VII I to a compound of formula II.
2. The process according to claim 1 , wherein the compound of formula II I is

3. The process according to claim 1 , wherein the compound of formula IV is

4. The process according to claim 1 , wherein the compound of formula I I I is prepared by a process comprising one or more of the following steps:
(a) treating crotonaldehyde IX with acetylene compound of formula X to provide compound of formula XI;
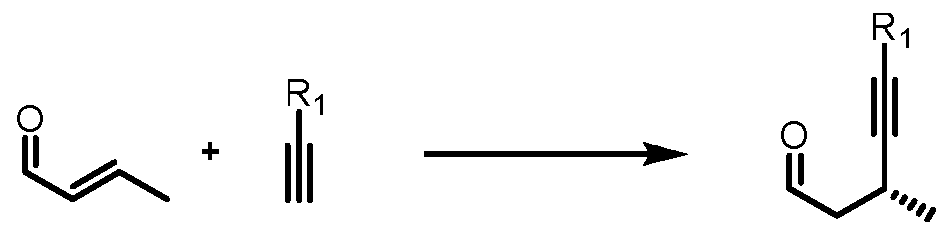
IX X XI
wherein is H or trialkyl silyl protecting group;
(b) reacting compound of formula XI with (+)-B-Allyldiisopinocampheylborane to provide hydroxy compound of formula XII;

XII
wherein Ri is H or trialkyl silyl protecting group;
(c) optionally protecting hydroxy compound of formula XII to provide a compound of formula XIII;

XII I
wherein P-i is an alcohol-protecting group; Ri is H or trialkyl silyl protecting group;
(d) converting compound of formula XIII to provide compound of formula XIV; and

XIV
wherein is an alcohol-protecting group; Ri is H or trialkyl silyl protecting group (e) converting compound of formula XIV to compound of formula I II.
5. The process according to claim 1 , wherein the compound of formula III is prepared by process comprising one or more of the following steps:
(a) treating a compound of formula XV with an acetylene compound of formula X to provide a compound of formula XVI;

XV X XVI
wherein is H or trialkyi silyl; Pi is H or an alcohol-protecting group; X is halogen;
(b) optionally deprotecting a compound of formula XVI to provide a compound of formula XVII;

XVII
wherein is H or trialkyi silyl;
(c) converting a compound of formula XVI or a compound of formula XVII to a compound of formula XVIII;
Ri
OH II
HO„
XVI I I
wherein P^ is H or trialkyi silyl;
(d) protecting a diol compound of formula XVIII to provide a compound of formula XIX;

XIX
wherein is H or trialkyi silyl; Pi is an alcohol-protecting group; P2 is hydrogen or an alcohol-protecting group;
(e) optionally deprotecting a compound of formula XIX to provide a compound of formula XIV;
XIV
wherein Ri is H or trialkyi silyl;
(f) converting a compound of formula XIV to a compound of formula III.
6. The process according to claim 1 , wherein the compound of formula IV is prepared by a process comprising one or more of the following steps:
(a) reacting an epoxide of formula XX with a compound of formula XXI to provide a compound of formula XXII;

XX XXI
XXII
wherein R2 is trialkyi silyl; Pi is an alcohol-protecting group; X is halogen, preferably bromine;
(b) converting a compound of formula XXII to a vinyl halide compound of formula XV;

XV
wherein P-i is an alcohol-protecting group; X is halogen, preferably bromine or iodine;
(c) converting a compound of formula XV to a compound of formula IV.
7. A compound of formula V or isomers thereof.

8. A compound of formula VI or isomers thereof.

9. The compound of claim 8 in crystalline form.
10. A compound of formula VII or isomers thereof.

VI I
wherein P-i is an alcohol-protecting group;
11. A compound of formula VII I or isomers thereof.

VII I
wherein P-i is H or an alcohol protecting group and X is halogen.
12. A compound of formula IVa or isomers thereof.

wherein P is an alcohol protecting group and X is halogen.
13. A compound of claim 1 2 having the formula.

14. A compound of formula III or isomers thereof;

I I I
wherein P-i is an alcohol-protecting group; Ri is H or trialkyi silyl protecting group; 15. A compound of claim 14 having the formula.

16. A compound having the following formula or isomers thereof.


17. A process for preparation of eribulin or a pharmaceutically acceptable salt thereof from a compound of any one of claims 1 -16.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN5501CH2015 | 2015-10-14 | ||
| IN5501/CHE/2015 | 2015-10-14 | ||
| IN7087/CHE/2015 | 2015-12-30 | ||
| IN7087CH2015 | 2015-12-30 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2017064627A2 true WO2017064627A2 (en) | 2017-04-20 |
| WO2017064627A3 WO2017064627A3 (en) | 2017-07-06 |
Family
ID=58517094
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2016/056097 WO2017064627A2 (en) | 2015-10-14 | 2016-10-12 | Process for preparation of eribulin and intermediates thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2017064627A2 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018096478A2 (en) | 2016-11-23 | 2018-05-31 | Dr. Reddy’S Laboratories Limited | Process for preparation of eribulin and intermediates thereof |
| US20190177291A1 (en) * | 2016-05-26 | 2019-06-13 | Dr. Reddy's Laboratories Limited | Process for preparation of eribulin and intermediates thereof |
| CN110922423A (en) * | 2019-10-16 | 2020-03-27 | 杭州励德生物科技有限公司 | Synthetic method of eribulin intermediate |
| CN111285894A (en) * | 2018-12-10 | 2020-06-16 | 北京天一绿甫医药科技有限公司 | Intermediate for preparing halichondrin compound and preparation method thereof |
| CN111793047A (en) * | 2019-06-13 | 2020-10-20 | 南通诺泰生物医药技术有限公司 | Preparation method of eribulin intermediate |
| CN113166096A (en) * | 2018-10-09 | 2021-07-23 | 雷迪博士实验室有限公司 | Method for preparing eribulin and intermediate thereof |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102013104007A1 (en) * | 2013-04-19 | 2014-10-23 | Schenck Rotec Gmbh | reinflator |
| EP3066102B1 (en) * | 2013-11-04 | 2020-02-26 | Eisai R&D Management Co., Ltd. | Macrocyclization reactions and intermediates useful in the synthesis of analogs of halichondrin b |
-
2016
- 2016-10-12 WO PCT/IB2016/056097 patent/WO2017064627A2/en active Application Filing
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20190177291A1 (en) * | 2016-05-26 | 2019-06-13 | Dr. Reddy's Laboratories Limited | Process for preparation of eribulin and intermediates thereof |
| JP2019521969A (en) * | 2016-05-26 | 2019-08-08 | ドクター レディズ ラボラトリーズ リミテッド | Method for producing eribulin and its intermediate |
| US11059799B2 (en) | 2016-05-26 | 2021-07-13 | Dr. Reddy's Laboratories Limited. | Process for preparation of eribulin and intermediates thereof |
| WO2018096478A2 (en) | 2016-11-23 | 2018-05-31 | Dr. Reddy’S Laboratories Limited | Process for preparation of eribulin and intermediates thereof |
| US10836776B2 (en) | 2016-11-23 | 2020-11-17 | Dr. Reddy's Laboratories Limited | Process for preparation of eribulin and intermediates thereof |
| CN113166096A (en) * | 2018-10-09 | 2021-07-23 | 雷迪博士实验室有限公司 | Method for preparing eribulin and intermediate thereof |
| CN111285894A (en) * | 2018-12-10 | 2020-06-16 | 北京天一绿甫医药科技有限公司 | Intermediate for preparing halichondrin compound and preparation method thereof |
| CN111285894B (en) * | 2018-12-10 | 2021-03-05 | 北京天一绿甫医药科技有限公司 | Intermediate for preparing halichondrin compound and preparation method thereof |
| CN111793047A (en) * | 2019-06-13 | 2020-10-20 | 南通诺泰生物医药技术有限公司 | Preparation method of eribulin intermediate |
| CN111793047B (en) * | 2019-06-13 | 2023-08-22 | 南通诺泰生物医药技术有限公司 | Preparation method of eribulin intermediate |
| CN110922423A (en) * | 2019-10-16 | 2020-03-27 | 杭州励德生物科技有限公司 | Synthetic method of eribulin intermediate |
| CN110922423B (en) * | 2019-10-16 | 2020-10-20 | 杭州励德生物科技有限公司 | Synthetic method of eribulin intermediate |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2017064627A3 (en) | 2017-07-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2017064627A2 (en) | Process for preparation of eribulin and intermediates thereof | |
| Fuwa et al. | Synthetic studies on a marine polyether toxin, gambierol: stereoselective synthesis of the EFGH ring system via B-alkyl Suzuki coupling | |
| CA2868611C (en) | Alpha2 adrenoceptor agonists | |
| Ueda et al. | Reaction of cyclopropenes with a trichloromethyl radical: unprecedented ring-opening reaction of cyclopropanes with migration | |
| EP1613646A2 (en) | Process for preparation of cyclosporin "a" analogues having a terminal diene group | |
| EP2495235A1 (en) | Process for the synthesis of prostaglandins and intermediates thereof | |
| EP2555625A1 (en) | Process for preparing benzoxaboroles | |
| JP2836556B2 (en) | Method for producing intermediate of 5,6,7-trinor-4,8-inter-m-phenylene PG12 derivative | |
| EP3334714A1 (en) | Process for making beraprost | |
| IL111851A (en) | Intermediates useful in the synthesis of bisindolylmaleimides and process for their preparation | |
| EP3464297B1 (en) | Process for preparation of eribulin and intermediates thereof | |
| CN101790509A (en) | Process for preparing estrogen-antagonistic 11 ss-fluoro-17alpha-alkylestra-1,3,5(10)-triene-3,17-diols having a 7alpha-(xi-alkylamino-omega-perfluoroalkyl)alkyl side chain and a-alkyl(amino)-omega-perfluoro(alkyl)alkanes and processes for their preparation | |
| Fukuda et al. | Synthetic studies on macrolactin A by using a (diene) Fe (CO) 3 complex | |
| CN110713477B (en) | Preparation method of LUBIPROSTONE (LUBIPROSTONE) and intermediate thereof | |
| EP2655373A1 (en) | Preparation process of an antiviral drug (entecavir) and intermediates thereof | |
| Matsumura et al. | Synthesis of 7-fluoro-2, 4-methylene-17, 20-dimethylprostacyclins. Novel stable prostacyclin analogs as potent anti-anginal agents | |
| KR20170028990A (en) | Metal-catalyzed asymmetric 1,4-conjugate addition of vinylboron compounds to 2-substituted-4-oxy-cyclopent-2-en-1-ones yielding prostaglandins and prostaglandin analogs | |
| WO2017168309A1 (en) | Process for preparation of eribulin and intermediates thereof | |
| CN109232637B (en) | Preparation method of entecavir intermediate | |
| JPWO2004065346A1 (en) | (-)-Tetrahydrolipstatin and method for producing the intermediate | |
| US10836776B2 (en) | Process for preparation of eribulin and intermediates thereof | |
| Tang et al. | The study of intramolecular tandem radical cyclizations of acylsilanes with radicalphiles attached on silicon | |
| Prasad et al. | Synthetic studies on cytotoxic macrolides cruentarens A and B: stereoselective synthesis of the C8–C19 segment of cruentarens A and B | |
| RO129083B1 (en) | Polyfunctional cyclopentanic chloroester and ()-lactone oxabicyclo [3.3.0] octane key compounds prepared by stereoselective transformation of ()-lactone intermediates and processes for preparing the same | |
| EP2861572A1 (en) | Improved process for the preparation of 2-substituted-2-(6-(substituted)-7-methylbenzo[d][1,3]dioxol-4-yl)acetic acid derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16855036 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16855036 Country of ref document: EP Kind code of ref document: A2 |