WO2017004073A1 - Bisnitroxides - Google Patents
Bisnitroxides Download PDFInfo
- Publication number
- WO2017004073A1 WO2017004073A1 PCT/US2016/039877 US2016039877W WO2017004073A1 WO 2017004073 A1 WO2017004073 A1 WO 2017004073A1 US 2016039877 W US2016039877 W US 2016039877W WO 2017004073 A1 WO2017004073 A1 WO 2017004073A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- optionally substituted
- alkyl
- compounds
- group
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/92—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with a hetero atom directly attached to the ring nitrogen atom
- C07D211/94—Oxygen atom, e.g. piperidine N-oxide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- Microdialysis is a powerful analytical technique used to recover analytes from the extracellular space of the brain (neurotransmitters, metabolites, amino acids, neuropeptides).
- Microdialysis probes consist of inlet tubing that leads to a semi-permeable membrane at which analytes diffuse across the membrane and are collected through outlet tubing. This simple technique can be coupled to various analytical methods (high-performance liquid chromatography, mass spectrometry, capillary electrophoresis) for simultaneous detection of small molecules. Due to the simplicity and versatility of the method, brain microdialysis has significantly impacted our understanding of brain function, neurological diseases, drug addiction, and traumatic brain injury.
- probes are 200-300 ⁇ in diameter. Implantation of probes into the brain damages the surrounding tissue decreasing blood flow, neurons and increasing microglia and astrocytes. This penetration injury causes a progressive decline in dopamine (DA) an important neurotransmitter often studied using microdialysis, specifically, probe implantation significantly decreases evoked DA release in the surrounding tissue. Although, DA terminals survive probe implantation, they do not seem to function on a normal level in tissue surrounding the probe.
- DA dopamine
- Retrodialysis of dexamethasone an anti-inflammatory steroid increases blood flow to the area surrounding the probe and reduces gliosis for at least 5 days.
- DEX dexamethasone
- Pharmacological mitigation of probe induced tissue damage through the use of an anti-inflammatory provides a platform for improving long-term microdialysis.
- a concern with DEX is that steroids are can affect neurotransmission, some steroids acting specifically on the central dopaminergic systems. Therefore it is of important to investigate other non-steroidal anti-inflammatory agents and their ability to prevent tissue damage and loss of evoked DA release near probes.
- a compound, or a pharmaceutically acceptable salt thereof having a structure of:
- Ri, Ria R2, R2a, R 4 , R20 and R21 are each independently hydrogen, halo, or an optionally substituted alkyl;
- R 31 - N(R 33 ) - R 32 Formula 2 wherein R 31 and R 32 are each independently
- R 33 is H or an optionally substituted alkyl.
- Y is -NH-, -0-, -N(alkyl)-, -N(aryl)-, -N(cycloalkyl)-, -N(heteroaryl)-, or -CH(R 41 )-;
- R 41 is H, optionally substituted aliphatic, optionally substituted cycloalkyl, optionally substituted aryl, or optionally heteroaryl;
- each of R 42 - R 45 is H, optionally substituted aliphatic, optionally substituted cycloalkyl, optionally substituted aryl, optionally heteroaryl, or fluorine;
- each Z is independently -0-, -NH- or -C3 ⁇ 4-;
- each R 40 is independently
- FIG. 1 Top panel: Chemical structures of anti-inflammatory agents.
- Bottom panel :
- FIGS. 2A-2C Average (+SEM) DA responses to a 25 s stimulation of the MFB recorded in the striatum premicrodialysis probe implantation (blue), post-probe implantation (red), and post- probe, post-nomifensine (green).
- FIGS. 3A and 3B Effects of anti-inflammatory retrodialysis of agents on maximum evoked DA (FIG. 3A) post-probe implantation and (FIG. 3B) post-nomifensine.
- FIGS. 4A-4C Average (+SEM) DA responses to a 25 s stimulation of recorded in the striatum pre-microdialysis probe implantation (blue), post-probe implantation (red), and post-probe, post-nomifensine (green).
- FIGS. 5A and 5B Effects ROS scavengers retrodialysis of maximum evoked DA (FIG. 5A) post-probe implantation and (FIG. 5B) postnomifensine.
- FIGS. 6 A Average (+SEM) current responses to a 25 s stimulation of the recorded in the striatum postprobe implantation with aCSF (red), and post- probe with aCSF plus nomifensine (green).
- FIG. 6B Average (+SEM) DA responses to a 25 s stimulation of the MFB recorded in the striatum post-probe implantation with DEX (red), and post- probe with DEX plus nomifensine (green).
- Acyl refers to a group having the structure -C(0)R, where R may be, for example, optionally substituted alkyl, optionally substituted aryl, or optionally substituted heteroaryl.
- “Lower acyl” groups are those that contain one to six carbon atoms.
- administering is inclusive of administration by another person to the subject or self-administration by the subject.
- aliphatic is defined as including alkyl, alkenyl, alkynyl, halogenated alkyl and cycloalkyl groups.
- a "lower aliphatic” group is a branched or unbranched aliphatic group having from 1 to 10 carbon atoms.
- Alkanediyl refers to a divalent radical derived from aliphatic, cycloaliphatic, aryl, and alkanearyl hydrocarbons.
- Alkenyl refers to a cyclic, branched or straight chain group containing only carbon and hydrogen, and contains one or more double bonds that may or may not be conjugated. Alkenyl groups may be unsubstituted or substituted. "Lower alkenyl” groups contain one to six carbon atoms.
- alkoxy refers to a straight, branched or cyclic hydrocarbon configuration and combinations thereof, including from 1 to 20 carbon atoms, preferably from 1 to 8 carbon atoms (referred to as a "lower alkoxy”), more preferably from 1 to 4 carbon atoms, that include an oxygen atom at the point of attachment.
- alkoxy group is represented by the formula - OR, where R can be an alkyl group, optionally substituted with an alkenyl, alkynyl, aryl, aralkyl, cycloalkyl, halogenated alkyl, alkoxy or heterocycloalkyl group.
- Suitable alkoxy groups include methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, i-butoxy, sec-butoxy, tert-butoxy cyclopropoxy, cyclohexyloxy, and the like.
- Alkoxycarbonyl refers to an alkoxy substituted carbonyl radical, -C(0)OR, wherein R represents an optionally substituted alkyl, aryl, aralkyl, cycloalkyl, cycloalkylalkyl or similar moiety.
- alkyl refers to a branched or unbranched saturated hydrocarbon group of 1 to 24 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, i-butyl, pentyl, hexyl, heptyl, octyl, decyl, tetradecyl, hexadecyl, eicosyl, tetracosyl and the like.
- a “lower alkyl” group is a saturated branched or unbranched hydrocarbon having from 1 to 6 carbon atoms. Preferred alkyl groups have 1 to 4 carbon atoms.
- Alkyl groups may be "substituted alkyls" wherein one or more hydrogen atoms are substituted with a substituent such as halogen, cycloalkyl, alkoxy, amino, hydroxyl, aryl, alkenyl, or carboxyl.
- a lower alkyl or (Ci-Ce)alkyl can be methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, sec-butyl, pentyl, 3-pentyl, or hexyl;
- (C3-Ce)cycloalkyl can be cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl;
- C3-C6)cycloalkyl(Ci-C6)alkyl can be cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclohexylmethyl, 2-cyclopropylethyl, 2-cyclobutylethyl, 2-cyclopentylethyl, or 2-cyclohexylethyl;
- (Ci-Ce)alkoxy can be methoxy, ethoxy, propoxy, isopropoxy, butoxy, iso-
- amine refers to a group of the formula -NRR', where R and R' can be, independently, hydrogen or an alkyl, alkenyl, alkynyl, acyl, aryl, aralkyl, cycloalkyl, halogenated alkyl, or heterocycloalkyl group.
- R and R' can be, independently, hydrogen or an alkyl, alkenyl, alkynyl, acyl, aryl, aralkyl, cycloalkyl, halogenated alkyl, or heterocycloalkyl group.
- R and R' can be, independently, hydrogen or an alkyl, alkenyl, alkynyl, acyl, aryl, aralkyl, cycloalkyl, halogenated alkyl, or heterocycloalkyl group.
- an “alkylamino” or “alkylated amino” refers to -NRR', wherein at least one of R or R'
- aminoalkyl refers to alkyl groups as defined above where at least one hydrogen atom is replaced with an amino group (e.g, -CH2-NH2).
- aminocarbonyl alone or in combination, means an amino substituted carbonyl
- (carbamoyl) radical wherein the amino radical may optionally be mono- or di-substituted, such as, for example, with alkyl, aryl, acyl, aralkyl, cycloalkyl, cycloalkylalkyl, alkanoyl, alkoxycarbonyl, aralkoxycarbonyl and the like.
- amide or “amido” is represented by the formula -C(0)NRR', where R and R' independently can be, for example, a hydrogen, alkyl, alkenyl, alkynyl, acyl, aryl, aralkyl, cycloalkyl, halogenated alkyl, or heterocycloalkyl group.
- an "animal” refers to living multi-cellular vertebrate organisms, a category that includes, for example, mammals and birds.
- the term mammal includes both human and non-human mammals.
- the term “subject” includes both human and non-human subjects, including birds and non-human mammals, such as non-human primates, companion animals (such as dogs and cats), livestock (such as pigs, sheep, cows), as well as non-domesticated animals, such as the big cats.
- the term subject applies regardless of the stage in the organism's life-cycle. Thus, the term subject applies to an organism in utero or in ovo, depending on the organism (that is, whether the organism is a mammal or a bird, such as a domesticated or wild fowl).
- aralkyl refers to an alkyl group wherein an aryl group is substituted for a hydrogen of the alkyl group.
- An example of an aralkyl group is a benzyl group.
- Aryl refers to a monovalent unsaturated aromatic carbocyclic group having a single ring (e.g., phenyl) or multiple condensed rings (e.g., naphthyl or anthryl), which can optionally be unsubstituted or substituted.
- a "heteroaryl group,” is defined as an aromatic group that has at least one heteroatom incorporated within the ring of the aromatic group. Examples of heteroatoms include, but are not limited to, nitrogen, oxygen, sulfur, and phosphorous.
- Heteroaryl includes, but is not limited to, pyridinyl, pyrazinyl, pyrimidinyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isooxazolyl, thiadiazolyl, oxadiazolyl, thiophenyl, furanyl, quinolinyl, isoquinolinyl, benzimidazolyl, benzooxazolyl, quinoxalinyl, and the like.
- the aryl or heteroaryl group can be substituted with one or more groups including, but not limited to, alkyl, alkynyl, alkenyl, aryl, halide, nitro, amino, ester, ketone, aldehyde, hydroxy, carboxylic acid, or alkoxy, or the aryl or heteroaryl group can be unsubstituted.
- Aryloxy or “heteroaryloxy” refers to a group of the formula -OAr, wherein Ar is an aryl group or a heteroaryl group, respectively.
- cycloalkyl refers to a non-aromatic carbon-based ring composed of at least three carbon atoms.
- examples of cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and the like.
- heterocycloalkyl group is a cycloalkyl group as defined above where at least one of the carbon atoms of the ring is substituted with a heteroatom such as, but not limited to, nitrogen, oxygen, sulfur, or phosphorous.
- esters refers to a carboxyl group-containing moiety having the hydrogen replaced with, for example, a Ci-6alkyl group ("carboxylCi-6alkyl” or “alkylester”), an aryl or aralkyl group (“arylester” or “aralkylester”) and so on.
- CC Ci-salkyl groups are preferred, such as for example, methylester (CO 2 Me), ethylester (CC Et) and propylester (CC Pr) and includes reverse esters thereof (e.g. -OCOMe, -OCOEt and -OCOPr).
- halogenated alkyl or “haloalkyl group” refer to an alkyl group with one or more hydrogen atoms present on these groups substituted with a halogen (F, CI, Br, I).
- hydroxyl is represented by the formula -OH.
- hydroxy alkyl refers to an alkyl group that has at least one hydrogen atom substituted with a hydroxyl group.
- alkoxyalkyl group is defined as an alkyl group that has at least one hydrogen atom substituted with an alkoxy group described above.
- Inhibiting refers to inhibiting the full development of a disease or condition. “Inhibiting” also refers to any quantitative or qualitative reduction in biological activity or binding, relative to a control.
- ROS reactive oxygen species
- subject includes both human and non-human subjects, including birds and non-human mammals, such as non-human primates, companion animals (such as dogs and cats), livestock (such as pigs, sheep, cows), as well as non-domesticated animals, such as the big cats.
- non-human mammals such as non-human primates, companion animals (such as dogs and cats), livestock (such as pigs, sheep, cows), as well as non-domesticated animals, such as the big cats.
- subject applies regardless of the stage in the organism's life-cycle. Thus, the term subject applies to an organism in utero or in ovo, depending on the organism (that is, whether the organism is a mammal or a bird, such as a domesticated or wild fowl).
- substituted or “substitution” refers to replacement of a hydrogen atom of a molecule or an R-group with one or more additional R-groups.
- optionally- substituted” or “optional substituent” as used herein refers to a group which may or may not be further substituted with 1, 2, 3, 4 or more groups, preferably 1, 2 or 3, more preferably 1 or 2 groups.
- the substituents may be selected, for example, from Ci-6alkyl, C2-6alkenyl, C2- 6 alkynyl, C3- 8cycloalkyl, hydroxyl, oxo, Ci-6alkoxy, aryloxy, Ci-6alkoxyaryl, halo, Ci-6alkylhalo (such as CF3 and CHF2), Ci-6alkoxyhalo (such as OCF3 and OCHF2), carboxyl, esters, cyano, nitro, amino, substituted amino, disubstituted amino, acyl, ketones, amides, aminoacyl, substituted amides, disubstituted amides, thiol, alkylthio, thioxo, sulfates, sulfonates, sulfinyl, substituted sulfinyl, sulfonyl, substituted sulfonyl, sulfonylamides, substituted sul
- N-heterocycles may also include but are not limited to Ci-6alkyl i.e. N-Ci-3alkyl, more preferably methyl particularly N-methyl.
- a “therapeutically effective amount” refers to a quantity of a specified agent sufficient to achieve a desired effect in a subject being treated with that agent. Ideally, a therapeutically effective amount of an agent is an amount sufficient to inhibit or treat the disease or condition without causing a substantial cytotoxic effect in the subject. The therapeutically effective amount of an agent will be dependent on the subject being treated, the severity of the affliction, and the manner of administration of the therapeutic composition.
- Treatment refers to a therapeutic intervention that ameliorates a sign or symptom of a disease or pathological condition after it has begun to develop, or administering a compound or composition to a subject who does not exhibit signs of a disease or exhibits only early signs for the purpose of decreasing the risk of developing a pathology or condition, or diminishing the severity of a pathology or condition.
- the term “ameliorating,” with reference to a disease or pathological condition refers to any observable beneficial effect of the treatment.
- the beneficial effect can be evidenced, for example, by a delayed onset of clinical symptoms of the disease in a susceptible subject, a reduction in severity of some or all clinical symptoms of the disease, a slower progression of the disease, an improvement in the overall health or well-being of the subject, or by other parameters well known in the art that are specific to the particular disease.
- treating a disease refers to inhibiting the full development of a disease, for example, in a subject who is at risk for a disease such as cancer.
- Preventing a disease or condition refers to
- compositions are compositions that include an amount (for example, a unit dosage) of one or more of the disclosed compounds together with one or more non-toxic pharmaceutically acceptable additives, including carriers, diluents, and/or adjuvants, and optionally other biologically active ingredients.
- Such pharmaceutical compositions can be prepared by standard pharmaceutical formulation techniques such as those disclosed in Remington's
- salts or esters refers to salts or esters prepared by conventional means that include salts, e.g., of inorganic and organic acids, including but not limited to hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, malic acid, acetic acid, oxalic acid, tartaric acid, citric acid, lactic acid, fumaric acid, succinic acid, maleic acid, salicylic acid, benzoic acid, phenylacetic acid, mandelic acid and the like.
- inorganic and organic acids including but not limited to hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, malic acid, acetic acid, oxalic acid, tartaric acid, citric acid, lactic acid, fumaric acid, succinic acid, maleic acid, salicylic acid, benzoic acid,
- “Pharmaceutically acceptable salts” of the presently disclosed compounds also include those formed from cations such as sodium, potassium, aluminum, calcium, lithium, magnesium, zinc, and from bases such as ammonia, ethylenediamine, N-methyl-glutamine, lysine, arginine, ornithine, choline, ⁇ , ⁇ '-dibenzylethylenediamine, chloroprocaine, diethanolamine, procaine, N-benzylphenethylamine, diethylamine, piperazine, tris(hydroxymethyl)aminomethane, and tetramethylammonium hydroxide.
- bases such as ammonia, ethylenediamine, N-methyl-glutamine, lysine, arginine, ornithine, choline, ⁇ , ⁇ '-dibenzylethylenediamine, chloroprocaine, diethanolamine, procaine, N-benzylphenethylamine, diethylamine, piperazine
- any chemical compound recited in this specification may alternatively be administered as a pharmaceutically acceptable salt thereof.
- “Pharmaceutically acceptable salts” are also inclusive of the free acid, base, and zwitterionic forms. Descriptions of suitable pharmaceutically acceptable salts can be found in Handbook of Pharmaceutical Salts, Properties, Selection and Use, Wiley VCH (2002). When compounds disclosed herein include an acidic function such as a carboxy group, then suitable pharmaceutically acceptable cation pairs for the carboxy group are well known to those skilled in the art and include alkaline, alkaline earth, ammonium, quaternary ammonium cations and the like. Such salts are known to those of skill in the art. For additional examples of
- “Pharmaceutically acceptable esters” includes those derived from compounds described herein that are modified to include a carboxyl group.
- An in vivo hydrolysable ester is an ester, which is hydrolysed in the human or animal body to produce the parent acid or alcohol.
- esters thus include carboxylic acid esters in which the non-carbonyl moiety of the carboxylic acid portion of the ester grouping is selected from straight or branched chain alkyl (for example, methyl, n-propyl, t-butyl, or n-butyl), cycloalkyl, alkoxyalkyl (for example,
- aralkyl for example benzyl
- aryloxyalkyl for example, phenoxymethyl
- aryl for example, phenyl, optionally substituted by, for example, halogen, C.sub.1-4 alkyl, or C.sub.l- 4 alkoxy) or amino
- sulphonate esters such as alkyl- or aralkylsulphonyl (for example, methanesulphonyl); or amino acid esters (for example, L-valyl or L-isoleucyl).
- esters also includes inorganic esters such as mono-, di-, or triphosphate esters. In such esters, unless otherwise specified, any alkyl moiety present
- esters advantageously contains from 1 to 18 carbon atoms, particularly from 1 to 6 carbon atoms, more particularly from 1 to 4 carbon atoms. Any cycloalkyl moiety present in such esters
- esters advantageously contains from 3 to 6 carbon atoms.
- Any aryl moiety present in such esters advantageously comprises a phenyl group, optionally substituted as shown in the definition of carbocycylyl above.
- Pharmaceutically acceptable esters thus include C1-C22 fatty acid esters, such as acetyl, t-butyl or long chain straight or branched unsaturated or omega-6 monounsaturated fatty acids such as palmoyl, stearoyl and the like.
- Alternative aryl or heteroaryl esters include benzoyl, pyridylmethyloyl and the like any of which may be substituted, as defined in carbocyclyl above.
- Additional pharmaceutically acceptable esters include aliphatic L-amino acid esters such as leucyl, isoleucyl and especially valyl.
- salts of the compounds are those wherein the counter-ion is pharmaceutically acceptable.
- salts of acids and bases which are non-pharmaceutically acceptable may also find use, for example, in the preparation or purification of a pharmaceutically acceptable compound.
- the pharmaceutically acceptable acid and base addition salts as mentioned hereinabove are meant to comprise the therapeutically active non-toxic acid and base addition salt forms which the compounds are able to form.
- the pharmaceutically acceptable acid addition salts can conveniently be obtained by treating the base form with such appropriate acid.
- Appropriate acids comprise, for example, inorganic acids such as hydrohalic acids, e.g. hydrochloric or hydrobromic acid, sulfuric, nitric, phosphoric and the like acids; or organic acids such as, for example, acetic, propanoic, hydroxy acetic, lactic, pyruvic, oxalic (i.e. ethanedioic), malonic, succinic (i.e.
- salt forms can be converted by treatment with an appropriate base into the free base form.
- the compounds containing an acidic proton may also be converted into their non-toxic metal or amine addition salt forms by treatment with appropriate organic and inorganic bases.
- Appropriate base salt forms comprise, for example, the ammonium salts, the alkali and earth alkaline metal salts, e.g. the lithium, sodium, potassium, magnesium, calcium salts and the like, salts with organic bases, e.g. the benzathine, N-methyl-D-glucamine, hydrabamine salts, and salts with amino acids such as, for example, arginine, lysine and the like.
- addition salt as used hereinabove also comprises the solvates which the compounds described herein are able to form.
- solvates are for example hydrates, alcoholates and the like.
- quaternary amine as used hereinbefore defines the quaternary ammonium salts which the compounds are able to form by reaction between a basic nitrogen of a compound and an appropriate quaternizing agent, such as, for example, an optionally substituted alkylhalide, arylhalide or arylalkylhalide, e.g. methyliodide or benzyliodide.
- an appropriate quaternizing agent such as, for example, an optionally substituted alkylhalide, arylhalide or arylalkylhalide, e.g. methyliodide or benzyliodide.
- Other reactants with good leaving groups may also be used, such as alkyl trifluoromethanesulfonates, alkyl methanesulfonates, and alkyl p-toluenesulfonates.
- a quaternary amine has a positively charged nitrogen.
- Pharmaceutically acceptable counterions include chloro, bro
- Prodrugs of the disclosed compounds also are contemplated herein.
- a prodrug is an active or inactive compound that is modified chemically through in vivo physiological action, such as hydrolysis, metabolism and the like, into an active compound following administration of the prodrug to a subject.
- the term "prodrug” as used throughout this text means the pharmacologically acceptable derivatives such as esters, amides and phosphates, such that the resulting in vivo biotransformation product of the derivative is the active drug as defined in the compounds described herein.
- Prodrugs preferably have excellent aqueous solubility, increased bioavailability and are readily metabolized into the active inhibitors in vivo.
- Prodrugs of a compounds described herein may be prepared by modifying functional groups present in the compound in such a way that the modifications are cleaved, either by routine manipulation or in vivo, to the parent compound.
- the suitability and techniques involved in making and using prodrugs are well known by those skilled in the art. F or a general discussion of prodrugs involving esters see Svensson and Tunek, Drug Metabolism Reviews 165 (1988) and Bundgaard, Design of Prodrugs, Elsevier (1985).
- prodrug also is intended to include any covalently bonded carriers that release an active parent drug of the present invention in vivo when the prodrug is administered to a subject. Since prodrugs often have enhanced properties relative to the active agent pharmaceutical, such as, solubility and bioavailability, the compounds disclosed herein can be delivered in prodrug form. Thus, also contemplated are prodrugs of the presently disclosed compounds, methods of delivering prodrugs and compositions containing such prodrugs. Prodrugs of the disclosed compounds typically are prepared by modifying one or more functional groups present in the compound in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to yield the parent compound.
- Prodrugs include compounds having a phosphonate and/or amino group functionalized with any group that is cleaved in vivo to yield the corresponding amino and/or phosphonate group, respectively.
- Examples of prodrugs include, without limitation, compounds having an acylated amino group and/or a phosphonate ester or phosphonate amide group.
- a prodrug is a lower alkyl phosphonate ester, such as an isopropyl phosphonate ester.
- Protected derivatives of the disclosed compounds also are contemplated.
- a variety of suitable protecting groups for use with the disclosed compounds are disclosed in Greene and Wuts, Protective Groups in Organic Synthesis; 3rd Ed.; John Wiley & Sons, New York, 1999.
- protecting groups are removed under conditions that will not affect the remaining portion of the molecule. These methods are well known in the art and include acid hydrolysis, hydrogenolysis and the like.
- One preferred method involves the removal of an ester, such as cleavage of a phosphonate ester using Lewis acidic conditions, such as in TMS-Br mediated ester cleavage to yield the free phosphonate.
- a second preferred method involves removal of a protecting group, such as removal of a benzyl group by hydrogenolysis utilizing palladium on carbon in a suitable solvent system such as an alcohol, acetic acid, and the like or mixtures thereof.
- a t-butoxy-based group, including t-butoxy carbonyl protecting groups can be removed utilizing an inorganic or organic acid, such as HC1 or trifluoroacetic acid, in a suitable solvent system, such as water, dioxane and/or methylene chloride.
- a suitable solvent system such as water, dioxane and/or methylene chloride.
- Another exemplary protecting group, suitable for protecting amino and hydroxy functions amino is trityl.
- Other conventional protecting groups are known and suitable protecting groups can be selected by those of skill in the art in consultation with Greene and Wuts, Protective Groups in Organic Synthesis; 3rd Ed.; John Wiley & Sons, New York, 1999.
- an amine is deprotected, the resulting salt can readily be neutralized to yield the free amine.
- an acid moiety such as a phosphonic acid moiety is unveiled, the compound may be isolated as the acid compound or as a salt thereof.
- compounds and compositions may be provided as individual pure enantiomers or as stereoisomeric mixtures, including racemic mixtures.
- the compounds disclosed herein are synthesized in or are purified to be in substantially enantiopure form, such as in a 90%
- the compounds can be isolated as a single isomer or as mixture of isomers. All tautomers of the compounds are also considered part of the disclosure.
- the presently disclosed compounds also includes all isotopes of atoms present in the compounds, which can include, but are not limited to, deuterium, tritium, 18 F, etc.
- mitochondria targeted nitroxides Enrichment in mitochondria of mitochondria targeted nitroxides has been demonstrated by EPR spectroscopy as well as by MS analysis of their content in mitochondria obtained from cells incubated with mitochondria targeted nitroxides. Delivery of mitochondria targeted-nitroxides into mitochondria does not depend on the mitochondrial membrane potential. Therefore, mitochondria targeted nitroxides can accumulate not only in intact but also in de-energized or damaged mitochondria with low membrane potential. Moreover, mitochondria targeted nitroxide conjugates are delivered into mitochondria without affecting the mitochondrial membrane potential. Hence, they do not impair the major mitochondrial function, the energy production, in cells. In addition, the conjugated nitroxides provide a new important feature, post irradiation protection.
- conjugated mitochondria targeted nitroxides might potentially lower blood pressure and sympathetic nerve activity.
- the dramatically reduced dose of mitochondria targeted nitroxides (about 1,000-fold), compared to non-conjugated parental nitroxides, may be significantly below of those inducing side effects.
- XJB-5-131 XJB
- ROS reactive oxygen species
- This particular ROS scavenger is unique in that it targets the inner mitochondrial membrane and contains a potent nitroxide group responsible for electron and radical scavenging.
- the neuroprotective qualities of XJB make ROS ' s good candidates for pharmacologically enhanced microdialysis.
- ibuprofen IBU
- PPads pyridoxalphosphate-6-azophenyl-2',4'-disulfonic acid tetrasodium salt
- JP4-039 JP4
- JRS527 JRS527
- Ri, Ria R2, R2a, R 4 , R20 and R21 are each independently hydrogen, halo, or an optionally substituted alkyl;
- Rio is H
- R* is H
- Ri a R2, and R2a are each H.
- Ri is alkyl, particularly Ci-C 6 alkyl, most particularly propyl (e.g., isopropyl) or butyl (e.g., isobutyl).
- Ri a and R2a are H, and R2 is aralkyl, particularly benzyl.
- R 6 is alkyl or aralkyl, particularly isopropyl or benzyl.
- At least one of Ri, Ri a R2, R2a, R 4 , R20 or R21 is Ci-C 6 straight or branched-chain alkyl optionally substituted one or more phenyl (-C6H5) groups, and the phenyl group is optionally methyl-, ethyl-, hydroxyl- or fluoro-substituted.
- the - ⁇ -0 ⁇ containing group is selected from:
- both R5 and Rio are identical to each other.
- Rio is H
- R5 and R 6 are both
- R 33 is H or an optionally substituted alkyl.
- Y is -NH-, -0-, -N(alkyl)-, -N(aryl)-, -N(cycloalkyl)-, -N(heteroaryl)-, or -CH(R 41 )-;
- R 41 is H, optionally substituted aliphatic, optionally substituted cycloalkyl, optionally substituted aryl, or optionally heteroaryl;
- each of R 42 - R 45 is H, optionally substituted aliphatic, optionally substituted cycloalkyl, optionally substituted aryl, optionally heteroaryl, or fluorine;
- each Z is independently -0-, -NH- or -C3 ⁇ 4-;
- each R 40 is independently
- ach -ZR 40 is independently
- each of R -R 45 is H or (Ci-C6)alkyl.
- R 41 is propyl or butyl, particularly isobutyl.
- Y is -NH-.
- R 42 -R 45 are each H.
- nitroxides (- ⁇ ⁇ ) disclosed herein may undergo reduction in vivo to their corresponding hydroxylamines (-N-OH) and/or may undergo oxidation in vivo to their corresponding nitroxonium ions. All three forms may be in equilibrium and their ratio depends on the redox environment. Any one of the three forms, or a mixture thereof, could be administered to a subject.
- nitroxide and nitroxide derivatives are stable radicals that can withstand biological environments. Therefore, the presence of the 4-amino-TEMPO, TEMPOL or another nitroxide "payload" within the mitochondria membrane can serve as an effective and efficient
- SUBSTITUTE SHEET (RULE 26) electron scavenger of the ROS being produced within the membrane.
- Non-limiting examples of this include TEMPO (2,2,6,6-Tetramethyl-4-piperidine 1-oxyl) and TEMPOL (4-Hydroxy- TEMPO), in which, when incorporated into the compound described herein, form, for example, when R 3 is -NH-R5, -O-R5:
- a method of scavenging free-radicals in a subject comprising administering to the subject an amount of one or more compound described herein and having a free-radical scavenging group, such as a nitroxide-containing group effective to scavenge free radicals.
- a free-radical scavenging group such as a nitroxide-containing group effective to scavenge free radicals.
- any agent or agents used for prevention, mitigation or treatment in a subject of injury caused by radiation exposure may be administered in an amount effective to prevent, mitigate of treat such injury, namely in an amount and in a dosage regimen effective to prevent injury or to reduce the duration and/or severity of the injury resulting from radiation exposure.
- an effective dose ranges from 0.1 or 1 mg/kg to 100 mg/kg, including any increment or range therebetween, including 1 mg/kg, 5 mg/kg, 10 mg/kg, 20 mg/kg, 25 mg/kg, 50 mg/kg, and 75 mg/kg.
- an effective dose or dose range is expected to vary from that of other compounds described herein for any number of reasons, including the molecular weight of the compound, bioavailability, specific activity, etc.
- the therapeutic window between the minimally-effective dose, and maximum tolerable dose in a subject can be determined empirically by a person of skill in the art, with end points being determinable by in vitro and in vivo assays, such as those described herein and/or are acceptable in the pharmaceutical and medical arts for obtaining such information regarding radioprotective agents.
- Different concentrations of the agents described herein are expected to achieve similar results, with the drug product administered, for example and without limitation, once prior to an expected radiation dose, such as prior to radiation therapy or diagnostic exposure to ionizing radiation, during exposure to radiation, or after exposure in any effective dosage regimen.
- the compounds can be administered continuously, such as intravenously, one or more times daily, once every two, three, four, five or more days, weekly, monthly, etc., including increments therebetween.
- a person of ordinary skill in the pharmaceutical and medical arts will appreciate that it will be a matter of simple design choice and optimization to identify a suitable dosage regimen for prevention, mitigation or treatment of injury due to exposure to radiation.
- the compounds described herein also are useful in preventing, mitigating (to make less severe) and/or treating injury caused by radiation exposure.
- radiation in the context of this disclosure, it is meant types of radiation that result in the generation of free radicals, e.g., reactive oxygen species (ROS), as described herein.
- the free radicals are produced, for example and without limitation, by direct action of the radiation, as a physiological response to the radiation and/or as a consequence of damage/injury caused by the radiation.
- the radiation is ionizing radiation.
- Ionizing radiation consists of highly-energetic particles or waves that can detach (ionize) at least one electron from an atom or molecule. Examples of ionizing radiation are energetic beta particles, neutrons, and alpha particles.
- X-rays and gamma rays can ionize almost any molecule or atom; far ultraviolet light can ionize many atoms and molecules; near ultraviolet and visible light are ionizing to very few molecules.
- Microwaves and radio waves typically are considered to be non-ionizing radiation, though damage caused by, e.g., microwaves, may result in the production of free-radicals as part of the injury and/or physiological response to the injury.
- the compounds typically are administered in an amount and dosage regimen to prevent, mitigate or treat the effects of exposure of a subject to radiation.
- the compounds may be administered in any manner that is effective to treat, mitigate or prevent damage caused by the radiation.
- Examples of delivery routes include, without limitation: topical, for example, epicutaneous, inhalational, enema, ocular, otic and intranasal delivery; enteral, for example, orally, by gastric feeding tube and rectally; and parenteral, such as, intravenous, intraarterial,
- the compounds disclosed herein may be useful for treating neurodegenerative disorders such as, for example, Alzheimer's disease, ataxia telangiectasia, Parkinson's disease, amyotrophic lateral sclerosis, and Huntington's disease.
- one or more of the disclosed compounds are mixed or combined with a suitable pharmaceutically acceptable carrier to prepare a pharmaceutical composition.
- Pharmaceutical carriers or vehicles suitable for administration of the compounds provided herein include any such carriers known to be suitable for the particular mode of administration.
- compositions and formulations suitable for pharmaceutical delivery of the compounds disclosed herein describes exemplary compositions and formulations suitable for pharmaceutical delivery of the compounds disclosed herein.
- the compounds may be formulated as the sole pharmaceutically active ingredient in the composition or may be combined with other active ingredients.
- the resulting mixture may be a solution, suspension, emulsion, or the like.
- Liposomal suspensions may also be suitable as pharmaceutically acceptable carriers. These may be prepared according to methods known to those skilled in the art. The form of the resulting mixture depends upon a number of factors, including the intended mode of administration and the solubility of the compound in the selected carrier or vehicle. Where the compounds exhibit insufficient solubility, methods for solubilizing may be used. Such methods are known and include, but are not limited to, using cosolvents such as dimethylsulfoxide (DMSO), using surfactants such as Tween®, and dissolution in aqueous sodium bicarbonate.
- DMSO dimethylsulfoxide
- surfactants such as Tween®
- Derivatives of the compounds may also be used in formulating effective pharmaceutical compositions.
- the disclosed compounds may also be prepared with carriers that protect them against rapid elimination from the body, such as time-release formulations or coatings.
- Such carriers include controlled release formulations, such as, but not limited to, microencapsulated delivery systems.
- kits for example, including component parts that can be assembled for use.
- one or more of the disclosed compounds may be provided in a lyophilized form and a suitable diluent may be provided as separated components for combination prior to use.
- a kit may include a disclosed compound and a second therapeutic agent for co-administration.
- the compound and second therapeutic agent may be provided as separate component parts.
- a kit may include a plurality of containers, each container holding one or more unit dose of the compound.
- the containers are preferably adapted for the desired mode of administration, including, but not limited to tablets, gel capsules, sustained-release capsules, and the like for oral administration; depot products, pre-filled syringes, ampoules, vials, and the like for parenteral administration; and patches, medipads, creams, and the like for topical administration.
- the active compound is included in the pharmaceutically acceptable carrier in an amount sufficient to exert a therapeutically useful effect in the absence of undesirable side effects on the subject treated.
- a therapeutically effective concentration may be determined empirically by testing the compounds in known in vitro and in vivo model systems for the treated disorder.
- a therapeutically effective amount of the compound is an amount that lessens or ameliorates at least one symptom of the disorder for which the compound is administered.
- compositions are formulated for single dosage administration.
- concentration of active compound in the drug composition will depend on absorption, inactivation, and excretion rates of the active compound, the dosage schedule, and amount administered as well as other factors known to those of skill in the art.
- 0.1 mg to 1000 mg of a disclosed compound, a mixture of such compounds, or a physiologically acceptable salt or ester thereof, is compounded with a
- compositions are formulated in a unit dosage form, each dosage containing from about 1 mg to about 1000 mg (for example, about 2 mg to about 500 mg, about 5 mg to 50 mg, about 10 mg to 100 mg, or about 25 mg to 75 mg) of the one or more compounds.
- the unit dosage form includes about 0.1 mg, about 1 mg, about 5 mg, about 10 mg, about 20 mg, about 30 mg, about 40 mg, about 50 mg, about 60 mg, about 70 mg, about 80 mg, about 90 mg, about 100 mg, about 150 mg, about 200 mg, about 250 mg, about 300 mg, about 400 mg, about 500 mg, about 600 mg, about 700 mg, about 800 mg, about 900 mg, about 1000 mg, or more of the disclosed compound(s).
- the disclosed compounds or compositions may be administered as a single dose, or may be divided into a number of smaller doses to be administered at intervals of time.
- the therapeutic compositions can be administered in a single dose delivery, by continuous delivery over an extended time period, in a repeated administration protocol (for example, by a multi-daily, daily, weekly, or monthly repeated administration protocol). It is understood that the precise dosage, timing, and duration of treatment is a function of the disease being treated and may be determined empirically using known testing protocols or by extrapolation from in vivo or in vitro test data. It is to be noted that concentrations and dosage values may also vary with the severity of the condition to be alleviated. In addition, it is understood that for a specific subject, dosage regimens may be adjusted over time according to the individual need and the professional judgment of the person administering or supervising the administration of the compositions, and that the concentration ranges set forth herein are exemplary only.
- these compositions When administered orally as a suspension, these compositions are prepared according to techniques well known in the art of pharmaceutical formulation and may contain microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners/flavoring agents. As immediate release tablets, these compositions may contain microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants.
- the compound is typically provided in a composition that protects it from the acidic environment of the stomach.
- the composition can be formulated in an enteric coating that maintains its integrity in the stomach and releases the active compound in the intestine.
- the composition may also be formulated in combination with an antacid or other such ingredient.
- Oral compositions will generally include an inert diluent or an edible carrier and may be compressed into tablets or enclosed in gelatin capsules.
- the active compound or compounds can be incorporated with excipients and used in the form of tablets, capsules, or troches.
- Pharmaceutically compatible binding agents and adjuvant materials can be included as part of the composition.
- the tablets, pills, capsules, troches, and the like can contain any of the following ingredients or compounds of a similar nature: a binder such as, but not limited to, gum tragacanth, acacia, corn starch, or gelatin; an excipient such as microcrystalline cellulose, starch, or lactose; a disintegrating agent such as, but not limited to, alginic acid and corn starch; a lubricant such as, but not limited to, magnesium stearate; a gildant, such as, but not limited to, colloidal silicon dioxide; a sweetening agent such as sucrose or saccharin; and a flavoring agent such as peppermint, methyl salicylate, or fruit flavoring.
- a binder such as, but not limited to, gum tragacanth, acacia, corn starch, or gelatin
- an excipient such as microcrystalline cellulose, starch, or lactose
- a disintegrating agent such as, but not limited to, alg
- the dosage unit form When the dosage unit form is a capsule, it can contain, in addition to material of the above type, a liquid carrier such as a fatty oil.
- dosage unit forms can contain various other materials, which modify the physical form of the dosage unit, for example, coatings of sugar and other enteric agents.
- the compounds can also be administered as a component of an elixir, suspension, syrup, wafer, chewing gum or the like.
- a syrup may contain, in addition to the active compounds, sucrose as a sweetening agent and certain preservatives, dyes and colorings, and flavors.
- the compounds When administered orally, the compounds can be administered in usual dosage forms for oral administration.
- dosage forms include the usual solid unit dosage forms of tablets and capsules as well as liquid dosage forms such as solutions, suspensions, and elixirs.
- solid dosage forms When the solid dosage forms are used, it is preferred that they be of the sustained release type so that the compounds need to be administered only once or twice daily.
- an oral dosage form is administered to the subject 1, 2, 3, 4, or more times daily.
- the oral dosage is from about 1 mg/day to about 500 mg/day, about 2 mg/day to about 200 mg/day, or about 5 mg/day to about 50 mg/day. It is understood that while a subject may be started at one dose, that dose may be varied over time as the subject's condition changes.
- the compounds can be administered orally to humans in a dosage range of 1 to 1000 mg/kg body weight in single or divided doses.
- One illustrative dosage range is 0.1 to 200 mg/kg body weight orally (such as 0.5 to 100 mg/kg body weight orally) in single or divided doses.
- the compositions may be provided in the form of tablets containing about 1 to 1000 milligrams of the active ingredient, particularly 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900, or 1000 milligrams of the active ingredient.
- Injectable solutions or suspensions may also be formulated, using suitable non-toxic, parenterally- acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution or isotonic sodium chloride solution, or suitable dispersing or wetting and suspending agents, such as sterile, bland, fixed oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- suitable non-toxic, parenterally- acceptable diluents or solvents such as mannitol, 1,3-butanediol, water, Ringer's solution or isotonic sodium chloride solution, or suitable dispersing or wetting and suspending agents, such as sterile, bland, fixed oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- Solutions or suspensions used for parenteral, intradermal, subcutaneous, or topical application can include any of the following components: a sterile diluent such as water for injection, saline solution, fixed oil, a naturally occurring vegetable oil such as sesame oil, coconut oil, peanut oil, cottonseed oil, and the like, or a synthetic fatty vehicle such as ethyl oleate, and the like, polyethylene glycol, glycerine, propylene glycol, or other synthetic solvent; antimicrobial agents such as benzyl alcohol and methyl parabens; antioxidants such as ascorbic acid and sodium bisulfite; chelating agents such as ethylenediaminetetraacetic acid (EDTA); buffers such as acetates, citrates, and phosphates; and agents for the adjustment of tonicity such as sodium chloride and dextrose.
- Parenteral preparations can be enclosed in ampoules, disposable syringes, or multiple dose vials made of glass,
- suitable carriers include physiological saline, phosphate buffered saline (PBS), and solutions containing thickening and solubilizing agents such as glucose, polyethylene glycol, polypropyleneglycol, and mixtures thereof.
- PBS phosphate buffered saline
- suitable carriers include physiological saline, phosphate buffered saline (PBS), and solutions containing thickening and solubilizing agents such as glucose, polyethylene glycol, polypropyleneglycol, and mixtures thereof.
- Liposomal suspensions including tissue-targeted liposomes may also be suitable as pharmaceutically acceptable carriers.
- the compounds can be administered parenterally, for example, by IV, IM, depo-IM, SC, or depo-SC.
- a therapeutically effective amount of about 0.1 to about 500 mg/day (such as about 1 mg/day to about 100 mg/day, or about 5 mg/day to about 50 mg/day) may be delivered.
- a depot formulation is used for injection once a month or once every two weeks, the dose may be about 0.1 mg/day to about 100 mg/day, or a monthly dose of from about 3 mg to about 3000 mg.
- the compounds can also be administered sublingually. When given sublingually, the compounds should be given one to four times daily in the amounts described above for IM administration.
- the compounds can also be administered intranasally.
- the appropriate dosage forms are a nasal spray or dry powder.
- the dosage of the compounds for intranasal administration is the amount described above for IM administration.
- these compositions may be prepared according to techniques well known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents.
- the compounds can be administered intrathecally.
- the appropriate dosage form can be a parenteral dosage form.
- the dosage of the compounds for intrathecal administration is the amount described above for IM administration.
- the compounds can be administered topically.
- the appropriate dosage form is a cream, ointment, or patch.
- an illustrative dosage is from about 0.5 mg/day to about 200 mg/day. Because the amount that can be delivered by a patch is limited, two or more patches may be used.
- the compounds can be administered rectally by suppository.
- an illustrative therapeutically effective amount may range from about 0.5 mg to about 500 mg.
- these compositions may be prepared by mixing the drug with a suitable non-irritating excipient, such as cocoa butter, synthetic glyceride esters of polyethylene glycols, which are solid at ordinary temperatures, but liquefy and/or dissolve in the rectal cavity to release the drug.
- IBU and Ppads Preserve Evoked DA Release Near Probes.
- cells up-regulate production of pro- inflammatory proteins causing activation of phospholipase A2.
- COX cyclooxygenase
- Both DEX and IBU work to intervene at different stages of the inflammatory cellular mechanism.
- DEX acts by down- regulating proinflammatory proteins in cells and up-regulating anti-inflammatory proteins produced by the cell.
- IBU is considered a COX-1 and COX-2 inhibitor thereby inhibiting COX modification and preventing inflammatory mediator signaling.
- the non-steroidal anti-inflammatory IBU when perfused through microdialysis probes prevented complete loss of evoked DA release near probes ( Figure 2-middle panel, red).
- Administration of nomifensine greatly impacted maximum evoked DA release near probes, increasing it to nearly pre-probe implantation amplitude ( Figure 2-middle panel, green).
- IBU had similar effects as DEX suggesting that DEX's effects on evoked DA release are due to both its anti-inflammatory actions, not steroidal actions on DA terminals.
- PPads is a non-selective P2 purinergic antagonist that blocks P2Y receptors. PPads reduces inflammation in the brain by blocking purine receptors on microglia limiting their response.
- Microglia are immune cells that are the first to respond to injury in the central nervous system.
- PPads significantly decreased the number and motility of the microglia responding to the site of an injury.
- PPads perfused through microdialysis also preserved evoked DA release near probes ( Figure 2-right panel, red).
- Nomifensine further increased evoked DA release but not as dramatically as DEX and IBU ( Figure 2-right panel, green).
- DEX, IBU, and PPads all significantly increased evoked DA release near probes (Figure 3). DEX's effect was more significant than IBU and PPads, and this is most likely due to DEX's duel action to both decrease pro-inflammatory proteins and increase anti-inflammatory proteins. Post- nomifensine maximum evoked DA responses were subject to a one-way ANOVA. DEX, IBU, and PPads all significantly increased maximum evoked DA release, DEX and IBU to above 80% of the pre -probe response. These findings demonstrate the ability of anti-inflammatories to preserve DA release and uptake activity in tissue near microdialysis probes.
- JP4 and JRS Inflammation is known to induce mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system.
- XJB a first generation ROS scavenger, infiltrates mitochondria and has been shown to be effective in neuroprotection. XJB prevents neuronal loss and preserves evoked DA near probes.
- a second generation isostere of XJB, the small molecule JP4 was developed offering an improved pharmacokinetic profile while maintaining its impressive ROS scavenging abilities.
- JP4 specifically targets mitochondria, similar to XJB and catalyzes the dismutation of superoxide radical anions and other reactive oxygen species. JP4 protective qualities mitigate radiation damage in blood cells in vitro and in vivo.
- JRS a modified analog of JP4, represents the first bis-nitroxide in this series and is expected to increase potency (Figure 1).
- JRS Preserve Evoked DA Release Near Probes Similar to anti-inflammatory drugs, probes perfused with JP4 and JRS decreased but did not abolish evoked DA release near probes (Figure 4). JP4 and JRS all substantially increased the post-nomifensine response. Histograms representing ROS effect on evoked DA release are represented in Figure 5. XJB and JRS significantly increased evoked DA release near probes ( Figure 5). All responses post-probe with JP4 gave a measurable DA signal however, this increase was not significantly greater than zero. JRS significantly increased evoked DA release compared to aCSF ( Figure 5).
- Nomifensine Perfusion does not Impact DA Terminals 70-100 ⁇ Away.
- Nomifensine a competitive DA transporter inhibitor was unable to act on DA terminals 70-100 ⁇ away from the probe.
- nomifensine perfusion through microdialysis is known to increase extracellular levels of DA within 10 minutes of administration.
- Nomifensine is a smaller molecule than DEX; therefore it would expect to diffuse through brain tissue more efficiently than DEX.
- DEX's effects on evoked responses are attributable to its anti-inflammatory actions, not direct actions on DA terminals. This evidence further supports our conclusion that DEX is acting on the tissue to improve cellular health during inflammation as opposed to DEX acting on DA terminals, specifically the DA transporter 70 ⁇ away.
- microdialysis probe implantation decreased evoked DA release in tissue near probes.
- DEX anti-inflammatory steroid
- XJB novel ROS scavenger
- IBU, PPads, XJB and JRS significantly attenuate the loss of DA activity in tissue near microdialysis probes.
- long- term protection with DEX preserves evoked DA release measured in tissue near the probe and at the outlet of probes, these drugs present other options to improve chronic microdialysis.
- pharmacological enhanced microdialysis provides new insight into acute mitigation of microdialysis probe penetration injury and has the potential to successfully mitigate chronic implantation for long-term in vivo monitoring of neurochemicals.
- Dexamethasone sodium phosphate (DEX: AAP
- ibuprofen IBU: Sigma Aldrich, St. Louis, MO
- PPads Sigma Aldrich, St. Louis, MO
- XJB-5-131 XJB
- JP4-039 JP4
- JRS527 JRS was prepared as described below. All solutions were prepared with ultrapure water (Nanopure, Barnstead, Dubuque, IA).
- DEX, and PPads were diluted to 10 ⁇ in aCSF.
- IBU, XJB, JP4, and JRS were dissolved in aCSF containing 1% DMSO (Sigma Aldrich, St. Louis, MO).
- Nomifensine (20 mg/kg, i.p., Sigma Aldrich, St. Louis, MO) was used as received.
- Nomifensine was dissolved in phosphate- buffered saline (PBS: 155 mM NaCl, 100 mM Na3 ⁇ 4P04, pH 7.40) for i.p. injections and diluted to 1 ⁇ in aCSF for microdialysis perfusion.
- PBS phosphate- buffered saline
- Electrodes were pre-treated in isopropyl alcohol (Sigma Aldrich, St. Louis, MO) and decolorizing carbon (Fisher, Pittsburgh, PA). Post- calibration of electrodes was performed with DA (Sigma Aldrich, St. Louis, MO) standards prepared in N2-purged aCSF.
- a carbon fiber electrode was implanted at a 5° angle into the striatum of each rat.
- a stimulating electrode was lowered towards the medial forebrain bundle (MFB) until maximum evoked DA was observed at the electrode (4.3 mm below bregma, 1.2 mm lateral to bregma, and 7.2-8.5 mm below dura).
- Electrical stimulation of the MFB was performed for 25 s at 45 Hz (waveform: biphasic, square, constant current, 300 ⁇ ) and evoked DA release was measured in the striatum in twenty minute intervals.
- the microdialysis probe was 70 ⁇ from the tip of the carbon fiber, and 100 ⁇ from the base of the electrode. The probe was left to perfuse in the brain for 2 hours. Following this, three more stimulus responses were recorded. Finally, nomifensine was administered (20 mg/kg i.p.) and one more stimulus response was collected.
Abstract
A compound, or a pharmaceutically acceptable salt thereof, having a structure of: Formula I wherein X is, or R1, R1a R2, R2a, R4, R20 and R21 are each independently hydrogen, halo, or an optionally substituted alkyl; R3 is -N(R5)(R10), wherein R5 and R10 are each independently H or an -Ν-O•, -N-OH or -N=O containing group, provided at least one of R5 is an -Ν-O•, -N-OH or -N=O containing group; and R is -C(O)-R6, -C(O)O-R6, or -P(O)-(R6)2, wherein R6 is an optionally substituted alkyl or an -Ν-O•, -N-OH or -N=O containing group, provided that if only one of R5 or R10 is an -Ν-O•, -N-OH or -N=O containing group then R6 is an optionally substituted alkyl or an -Ν-O•, -N-OH or -N=O containing group.
Description
BISNITROXIDES
CROSS REFERENCE TO RELATED APPLICATIONS
This application claims the benefit of U.S. Provisional Application No. 62/186,131, filed
June 29, 2015; U.S. Provisional Application No. 62/186,268, filed June 29, 2015; and U.S.
Provisional Application No. 62/187,701, filed July 1, 2015, all of which are incorporated by reference in their entirety. ACKNOWLEDGMENT OF GOVERNMENT SUPPORT
This invention was made with government support under grant number NS081744 awarded by the National Institutes of Health. The government has certain rights in the invention.
BACKGROUND
Microdialysis is a powerful analytical technique used to recover analytes from the extracellular space of the brain (neurotransmitters, metabolites, amino acids, neuropeptides). Microdialysis probes consist of inlet tubing that leads to a semi-permeable membrane at which analytes diffuse across the membrane and are collected through outlet tubing. This simple technique can be coupled to various analytical methods (high-performance liquid chromatography, mass spectrometry, capillary electrophoresis) for simultaneous detection of small molecules. Due to the simplicity and versatility of the method, brain microdialysis has significantly impacted our understanding of brain function, neurological diseases, drug addiction, and traumatic brain injury.
Commonly used probes are 200-300 μιη in diameter. Implantation of probes into the brain damages the surrounding tissue decreasing blood flow, neurons and increasing microglia and astrocytes. This penetration injury causes a progressive decline in dopamine (DA) an important neurotransmitter often studied using microdialysis, specifically, probe implantation significantly decreases evoked DA release in the surrounding tissue. Although, DA terminals survive probe implantation, they do not seem to function on a normal level in tissue surrounding the probe.
Retrodialysis of dexamethasone (DEX), an anti-inflammatory steroid increases blood flow to the area surrounding the probe and reduces gliosis for at least 5 days. By improving the health of the tissue DEX preserves neurons, DA terminals, and subsequently evoked DA release near probes at 4 and 24 hours. Pharmacological mitigation of probe induced tissue
damage through the use of an anti-inflammatory provides a platform for improving long-term microdialysis. A concern with DEX is that steroids are can affect neurotransmission, some steroids acting specifically on the central dopaminergic systems. Therefore it is of important to investigate other non-steroidal anti-inflammatory agents and their ability to prevent tissue damage and loss of evoked DA release near probes.
SUMMARY
Disclosed herein are novel bisnitroxides or higher nitroxides.
In one embodiment, there is disclosed a compound, or a pharmaceutically acceptable salt thereof, having a structure of:

Formula 1 wherein X is

Ri, Ria R2, R2a, R4, R20 and R21 are each independently hydrogen, halo, or an optionally substituted alkyl;
R3 is -N(R5)(Rio), wherein R5 and Rio are each independently H or an -Ν-0·, -N-OH or -N=0 containing group, provided at least one of R5 is an -Ν-0·, -N-OH or -N=0 containing group; and
R is -C(0)-R6, -C(0)0-R6, or -P(0)-(Re)2, wherein R6 is an optionally substituted alkyl or an -N- 0·, -N-OH or -N=0 containing group, provided that if only one of R5 or Rio is an -Ν-0·, -N-OH or -N=0 containing group then R6 is an optionally substituted alkyl or an -Ν-0·, -N-OH or -N=0 containing group.
In another embodiment, there is disclosed a compound, or a pharmaceutically acceptable salt thereof, having a structure of:
R31 - N(R33) - R32 Formula 2 wherein R31 and R32 are each independently

R33 is H or an optionally substituted alkyl.
Further disclosed herein are compounds, or pharmaceutically acceptable salts thereof, having a structure of:

Formula 3 A ; or

Formula 3B
wherein Y is -NH-, -0-, -N(alkyl)-, -N(aryl)-, -N(cycloalkyl)-, -N(heteroaryl)-, or -CH(R41)-; R41 is H, optionally substituted aliphatic, optionally substituted cycloalkyl, optionally substituted aryl, or optionally heteroaryl;
each of R42 - R45 is H, optionally substituted aliphatic, optionally substituted cycloalkyl, optionally substituted aryl, optionally heteroaryl, or fluorine;
each Z is independently -0-, -NH- or -C¾-; and
each R40 is independently

Further disclosed herein is a method for mitigating probe-induced tissue damage in a subject, comprising administering to a subject in need thereof, at least one compound disclosed herein.
The foregoing will become more apparent from the following detailed description, which proceeds with reference to the accompanying figures.
BRIEF DESCRIPTION OF THE DRAWINGS FIG. 1. Top panel: Chemical structures of anti-inflammatory agents. Bottom panel:
Chemical structures of reactive oxygen species scavengers.
FIGS. 2A-2C. Average (+SEM) DA responses to a 25 s stimulation of the MFB recorded in the striatum premicrodialysis probe implantation (blue), post-probe implantation (red), and post- probe, post-nomifensine (green). Microdialysis probes were perfused with either DEX (Fig. 2A), IBU (Fig. 2B), or PPads (Fig. 2C) (n=6 rats per agent). Solid lines represent the mean response and dotted lines the SEM. Black diamond indicate the beginning and end of stimulation. DEX data previously published.
FIGS. 3A and 3B. Effects of anti-inflammatory retrodialysis of agents on maximum evoked DA (FIG. 3A) post-probe implantation and (FIG. 3B) post-nomifensine. (FIG. 3A) As DA was non-detectable in the case of aCSF a one-sample t-tests was performed to compare each post-probe response to zero. DEX, IBU and PPads significantly increased maximum evoked DA compared to aCSF: One-sample, one-tailed t-test: DEX t(5)=8.21, ***p<0.0005, IBU t(5)=4.11,**p<0.005, PPads t(5)=3.46, *p<0.05. DEX is significantly different from IBU and PPads: one-way ANOVA F(2,15)=5.20, p<0.05, Post-hoc tukey DEX is significantly different from IBU p<0.05, PPads p<0.05. (FIG. 3B) Post-nomifensine, DEX, IBU, and Ppads increased evoked DA compared to aCSF: One-way ANOVA F(3,20)=7.28, p<0.005, Post-hoc tukey DEX: **p<0.005, IBU:
**p<0.005, PPads: *p<0.05.
FIGS. 4A-4C. Average (+SEM) DA responses to a 25 s stimulation of recorded in the striatum pre-microdialysis probe implantation (blue), post-probe implantation (red), and post-probe, post-nomifensine (green). Microdialysis probes were perfused with either XJB (Fig. 4A), JP4 (FIG. 4B), or JRS (FIG. 4C) (n=6 rats per agent). Solid lines represent the mean response and dotted lines the SEM. Black diamond indicate the beginning and end of stimulation. XJB data previously published.
FIGS. 5A and 5B. Effects ROS scavengers retrodialysis of maximum evoked DA (FIG. 5A) post-probe implantation and (FIG. 5B) postnomifensine. (FIG. 5A) As DA was non-detectable in the case of aCSF a one-sample t-tests was performed to compare each post-probe response to zero. XJB, and JRS significantly increased maximum evoked DA compared to aCSF: One sample, one- tailed t-test XJB t(5)=2.02, *p<0.05, JRS t(5)=2.24, *p<0.05. (FIG. 5B) Post-nomifensine, JRS
increased evoked DA compared to aCSF: One-way ANOVA F(3,20)=3.98, p<0.05, post-hoc tukey test: JRS: *p<0.05.
FIGS. 6A-6D. Effects of nomifensine added to perfusion fluid of microdialysis probes on evoked DA release near probes (n=3 rats/group). (FIG. 6 A) Average (+SEM) current responses to a 25 s stimulation of the recorded in the striatum postprobe implantation with aCSF (red), and post- probe with aCSF plus nomifensine (green). (FIG. 6B) Average (+SEM) DA responses to a 25 s stimulation of the MFB recorded in the striatum post-probe implantation with DEX (red), and post- probe with DEX plus nomifensine (green). (FIG. 6C) There was no difference in maximum current post-probe implantation with aCSF (red), and post-probe with aCSF plus nomifensine (green), paired t-test (p>0.05). (FIG. 6D) There was no difference in maximum DA release post-probe implantation with DEX (red), and post-probe with DEX plus nomifensine (green), paired t-test (p>0.05).
DETAILED DESCRIPTION
Terminology
The following explanations of terms and methods are provided to better describe the present compounds, compositions and methods, and to guide those of ordinary skill in the art in the practice of the present disclosure. It is also to be understood that the terminology used in the disclosure is for the purpose of describing particular embodiments and examples only and is not intended to be limiting.
"Acyl" refers to a group having the structure -C(0)R, where R may be, for example, optionally substituted alkyl, optionally substituted aryl, or optionally substituted heteroaryl.
"Lower acyl" groups are those that contain one to six carbon atoms.
"Administration" as used herein is inclusive of administration by another person to the subject or self-administration by the subject.
The term "aliphatic" is defined as including alkyl, alkenyl, alkynyl, halogenated alkyl and cycloalkyl groups. A "lower aliphatic" group is a branched or unbranched aliphatic group having from 1 to 10 carbon atoms.
Alkanediyl," "cycloalkanediyl," "aryldiyl," "alkanearyldiyl" refers to a divalent radical derived from aliphatic, cycloaliphatic, aryl, and alkanearyl hydrocarbons.
"Alkenyl" refers to a cyclic, branched or straight chain group containing only carbon and hydrogen, and contains one or more double bonds that may or may not be conjugated. Alkenyl groups may be unsubstituted or substituted. "Lower alkenyl" groups contain one to six carbon atoms.
The term "alkoxy" refers to a straight, branched or cyclic hydrocarbon configuration and combinations thereof, including from 1 to 20 carbon atoms, preferably from 1 to 8 carbon atoms (referred to as a "lower alkoxy"), more preferably from 1 to 4 carbon atoms, that include an oxygen atom at the point of attachment. An example of an "alkoxy group" is represented by the formula - OR, where R can be an alkyl group, optionally substituted with an alkenyl, alkynyl, aryl, aralkyl, cycloalkyl, halogenated alkyl, alkoxy or heterocycloalkyl group. Suitable alkoxy groups include methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, i-butoxy, sec-butoxy, tert-butoxy cyclopropoxy, cyclohexyloxy, and the like.
"Alkoxycarbonyl" refers to an alkoxy substituted carbonyl radical, -C(0)OR, wherein R represents an optionally substituted alkyl, aryl, aralkyl, cycloalkyl, cycloalkylalkyl or similar moiety.
The term "alkyl" refers to a branched or unbranched saturated hydrocarbon group of 1 to 24 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, i-butyl, pentyl, hexyl, heptyl, octyl, decyl, tetradecyl, hexadecyl, eicosyl, tetracosyl and the like. A "lower alkyl" group is a saturated branched or unbranched hydrocarbon having from 1 to 6 carbon atoms. Preferred alkyl groups have 1 to 4 carbon atoms. Alkyl groups may be "substituted alkyls" wherein one or more hydrogen atoms are substituted with a substituent such as halogen, cycloalkyl, alkoxy, amino, hydroxyl, aryl, alkenyl, or carboxyl. For example, a lower alkyl or (Ci-Ce)alkyl can be methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, sec-butyl, pentyl, 3-pentyl, or hexyl; (C3-Ce)cycloalkyl can be cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl; (C3-C6)cycloalkyl(Ci-C6)alkyl can be cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclohexylmethyl, 2-cyclopropylethyl, 2-cyclobutylethyl, 2-cyclopentylethyl, or 2-cyclohexylethyl; (Ci-Ce)alkoxy can be methoxy, ethoxy, propoxy, isopropoxy, butoxy, iso-butoxy, sec-butoxy, pentoxy, 3-pentoxy, or hexyloxy; (C2-C6)alkenyl can be vinyl, allyl, 1-propenyl, 2-propenyl, 1-butenyl, 2-butenyl, 3-butenyl, 1,- pentenyl, 2-pentenyl, 3-pentenyl, 4-pentenyl, 1- hexenyl, 2-hexenyl, 3-hexenyl, 4-hexenyl, or 5- hexenyl; (C2-C6)alkynyl can be ethynyl, 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-butynyl, 1-pentynyl, 2-pentynyl, 3-pentynyl, 4-pentynyl, 1- hexynyl, 2-hexynyl, 3-hexynyl, 4-hexynyl, or 5- hexynyl; (Ci-Ce)alkanoyl can be acetyl, propanoyl or butanoyl; halo(Ci-Ce)alkyl can be iodomethyl, bromomethyl, chloromethyl, fluoromethyl, trifluoromethyl, 2-chloroethyl, 2- fluoroethyl, 2,2,2-trifluoroethyl, or pentafluoroethyl; hydroxy(Ci-Ce)alkyl can be hydroxymethyl, 1 -hydroxy ethyl, 2-hydroxyethyl, 1-hydroxypropyl, 2-hydroxypropyl, 3-hydroxypropyl, 1- hydroxybutyl, 4-hydroxybutyl, 1-hydroxypentyl, 5-hydroxypentyl, 1 -hydroxyhexyl, or 6- hydroxyhexyl; (Ci-Ce)alkoxycarbonyl can be methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, isopropoxycarbonyl, butoxycarbonyl, pentoxycarbonyl, or hexyloxycarbonyl; (Ci-Ce)alkylthio can
be methylthio, ethylthio, propylthio, isopropylthio, butylthio, isobutylthio, pentylthio, or hexylthio; (C2-Ce)alkanoyloxy can be acetoxy, propanoyloxy, butanoyloxy, isobutanoyloxy, pentanoyloxy, or hexanoyloxy.
The term "amine" or "amino" refers to a group of the formula -NRR', where R and R' can be, independently, hydrogen or an alkyl, alkenyl, alkynyl, acyl, aryl, aralkyl, cycloalkyl, halogenated alkyl, or heterocycloalkyl group. For example, an "alkylamino" or "alkylated amino" refers to -NRR', wherein at least one of R or R' is an alkyl. A suitable amine or amino group is acetamido.
The term "aminoalkyl" refers to alkyl groups as defined above where at least one hydrogen atom is replaced with an amino group (e.g, -CH2-NH2).
"Aminocarbonyl" alone or in combination, means an amino substituted carbonyl
(carbamoyl) radical, wherein the amino radical may optionally be mono- or di-substituted, such as, for example, with alkyl, aryl, acyl, aralkyl, cycloalkyl, cycloalkylalkyl, alkanoyl, alkoxycarbonyl, aralkoxycarbonyl and the like.
The term "amide" or "amido" is represented by the formula -C(0)NRR', where R and R' independently can be, for example, a hydrogen, alkyl, alkenyl, alkynyl, acyl, aryl, aralkyl, cycloalkyl, halogenated alkyl, or heterocycloalkyl group.
An "animal" refers to living multi-cellular vertebrate organisms, a category that includes, for example, mammals and birds. The term mammal includes both human and non-human mammals. Similarly, the term "subject" includes both human and non-human subjects, including birds and non-human mammals, such as non-human primates, companion animals (such as dogs and cats), livestock (such as pigs, sheep, cows), as well as non-domesticated animals, such as the big cats. The term subject applies regardless of the stage in the organism's life-cycle. Thus, the term subject applies to an organism in utero or in ovo, depending on the organism (that is, whether the organism is a mammal or a bird, such as a domesticated or wild fowl).
The term "aralkyl" refers to an alkyl group wherein an aryl group is substituted for a hydrogen of the alkyl group. An example of an aralkyl group is a benzyl group.
"Aryl" refers to a monovalent unsaturated aromatic carbocyclic group having a single ring (e.g., phenyl) or multiple condensed rings (e.g., naphthyl or anthryl), which can optionally be unsubstituted or substituted. A "heteroaryl group," is defined as an aromatic group that has at least one heteroatom incorporated within the ring of the aromatic group. Examples of heteroatoms include, but are not limited to, nitrogen, oxygen, sulfur, and phosphorous. Heteroaryl includes, but is not limited to, pyridinyl, pyrazinyl, pyrimidinyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isooxazolyl, thiadiazolyl, oxadiazolyl, thiophenyl, furanyl, quinolinyl, isoquinolinyl,
benzimidazolyl, benzooxazolyl, quinoxalinyl, and the like. The aryl or heteroaryl group can be substituted with one or more groups including, but not limited to, alkyl, alkynyl, alkenyl, aryl, halide, nitro, amino, ester, ketone, aldehyde, hydroxy, carboxylic acid, or alkoxy, or the aryl or heteroaryl group can be unsubstituted.
"Aryloxy" or "heteroaryloxy" refers to a group of the formula -OAr, wherein Ar is an aryl group or a heteroaryl group, respectively.
The term "cycloalkyl" refers to a non-aromatic carbon-based ring composed of at least three carbon atoms. Examples of cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and the like. The term "heterocycloalkyl group" is a cycloalkyl group as defined above where at least one of the carbon atoms of the ring is substituted with a heteroatom such as, but not limited to, nitrogen, oxygen, sulfur, or phosphorous.
The term "ester" refers to a carboxyl group-containing moiety having the hydrogen replaced with, for example, a Ci-6alkyl group ("carboxylCi-6alkyl" or "alkylester"), an aryl or aralkyl group ("arylester" or "aralkylester") and so on. CC Ci-salkyl groups are preferred, such as for example, methylester (CO 2Me), ethylester (CC Et) and propylester (CC Pr) and includes reverse esters thereof (e.g. -OCOMe, -OCOEt and -OCOPr).
The terms "halogenated alkyl" or "haloalkyl group" refer to an alkyl group with one or more hydrogen atoms present on these groups substituted with a halogen (F, CI, Br, I).
The term "hydroxyl" is represented by the formula -OH.
The term "hydroxy alkyl" refers to an alkyl group that has at least one hydrogen atom substituted with a hydroxyl group. The term "alkoxyalkyl group" is defined as an alkyl group that has at least one hydrogen atom substituted with an alkoxy group described above.
"Inhibiting" refers to inhibiting the full development of a disease or condition. "Inhibiting" also refers to any quantitative or qualitative reduction in biological activity or binding, relative to a control.
The term "reactive oxygen species" ("ROS") includes, but is not limited to, superoxide anion, hydroxyl, and hydroperoxide radicals.
The term "subject" includes both human and non-human subjects, including birds and non- human mammals, such as non-human primates, companion animals (such as dogs and cats), livestock (such as pigs, sheep, cows), as well as non-domesticated animals, such as the big cats.
The term subject applies regardless of the stage in the organism's life-cycle. Thus, the term subject applies to an organism in utero or in ovo, depending on the organism (that is, whether the organism is a mammal or a bird, such as a domesticated or wild fowl).
"Substituted" or "substitution" refers to replacement of a hydrogen atom of a molecule or an R-group with one or more additional R-groups. Unless otherwise defined, the term "optionally- substituted" or "optional substituent" as used herein refers to a group which may or may not be further substituted with 1, 2, 3, 4 or more groups, preferably 1, 2 or 3, more preferably 1 or 2 groups. The substituents may be selected, for example, from Ci-6alkyl, C2-6alkenyl, C2-6alkynyl, C3- 8cycloalkyl, hydroxyl, oxo, Ci-6alkoxy, aryloxy, Ci-6alkoxyaryl, halo, Ci-6alkylhalo (such as CF3 and CHF2), Ci-6alkoxyhalo (such as OCF3 and OCHF2), carboxyl, esters, cyano, nitro, amino, substituted amino, disubstituted amino, acyl, ketones, amides, aminoacyl, substituted amides, disubstituted amides, thiol, alkylthio, thioxo, sulfates, sulfonates, sulfinyl, substituted sulfinyl, sulfonyl, substituted sulfonyl, sulfonylamides, substituted sulfonamides, disubstituted
sulfonamides, aryl, arCi-6alkyl, heterocyclyl and heteroaryl wherein each alkyl, alkenyl, alkynyl, cycloalkyl, aryl and heterocyclyl and groups containing them may be further optionally substituted. Optional substituents in the case N-heterocycles may also include but are not limited to Ci-6alkyl i.e. N-Ci-3alkyl, more preferably methyl particularly N-methyl.
A "therapeutically effective amount" refers to a quantity of a specified agent sufficient to achieve a desired effect in a subject being treated with that agent. Ideally, a therapeutically effective amount of an agent is an amount sufficient to inhibit or treat the disease or condition without causing a substantial cytotoxic effect in the subject. The therapeutically effective amount of an agent will be dependent on the subject being treated, the severity of the affliction, and the manner of administration of the therapeutic composition.
"Treatment" refers to a therapeutic intervention that ameliorates a sign or symptom of a disease or pathological condition after it has begun to develop, or administering a compound or composition to a subject who does not exhibit signs of a disease or exhibits only early signs for the purpose of decreasing the risk of developing a pathology or condition, or diminishing the severity of a pathology or condition. As used herein, the term "ameliorating," with reference to a disease or pathological condition, refers to any observable beneficial effect of the treatment. The beneficial effect can be evidenced, for example, by a delayed onset of clinical symptoms of the disease in a susceptible subject, a reduction in severity of some or all clinical symptoms of the disease, a slower progression of the disease, an improvement in the overall health or well-being of the subject, or by other parameters well known in the art that are specific to the particular disease. The phrase
"treating a disease" refers to inhibiting the full development of a disease, for example, in a subject who is at risk for a disease such as cancer. "Preventing" a disease or condition refers to
prophylactic administering a composition to a subject who does not exhibit signs of a disease or
exhibits only early signs for the purpose of decreasing the risk of developing a pathology or condition, or diminishing the severity of a pathology or condition.
"Pharmaceutical compositions" are compositions that include an amount (for example, a unit dosage) of one or more of the disclosed compounds together with one or more non-toxic pharmaceutically acceptable additives, including carriers, diluents, and/or adjuvants, and optionally other biologically active ingredients. Such pharmaceutical compositions can be prepared by standard pharmaceutical formulation techniques such as those disclosed in Remington's
Pharmaceutical Sciences, Mack Publishing Co., Easton, PA (19th Edition).
The terms "pharmaceutically acceptable salt or ester" refers to salts or esters prepared by conventional means that include salts, e.g., of inorganic and organic acids, including but not limited to hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, malic acid, acetic acid, oxalic acid, tartaric acid, citric acid, lactic acid, fumaric acid, succinic acid, maleic acid, salicylic acid, benzoic acid, phenylacetic acid, mandelic acid and the like. "Pharmaceutically acceptable salts" of the presently disclosed compounds also include those formed from cations such as sodium, potassium, aluminum, calcium, lithium, magnesium, zinc, and from bases such as ammonia, ethylenediamine, N-methyl-glutamine, lysine, arginine, ornithine, choline, Ν,Ν'-dibenzylethylenediamine, chloroprocaine, diethanolamine, procaine, N-benzylphenethylamine, diethylamine, piperazine, tris(hydroxymethyl)aminomethane, and tetramethylammonium hydroxide. These salts may be prepared by standard procedures, for example by reacting the free acid with a suitable organic or inorganic base. Any chemical compound recited in this specification may alternatively be administered as a pharmaceutically acceptable salt thereof. "Pharmaceutically acceptable salts" are also inclusive of the free acid, base, and zwitterionic forms. Descriptions of suitable pharmaceutically acceptable salts can be found in Handbook of Pharmaceutical Salts, Properties, Selection and Use, Wiley VCH (2002). When compounds disclosed herein include an acidic function such as a carboxy group, then suitable pharmaceutically acceptable cation pairs for the carboxy group are well known to those skilled in the art and include alkaline, alkaline earth, ammonium, quaternary ammonium cations and the like. Such salts are known to those of skill in the art. For additional examples of
"pharmacologically acceptable salts," see Berge et al., /. Pharm. Sci. 66: 1 (1977).
"Pharmaceutically acceptable esters" includes those derived from compounds described herein that are modified to include a carboxyl group. An in vivo hydrolysable ester is an ester, which is hydrolysed in the human or animal body to produce the parent acid or alcohol.
Representative esters thus include carboxylic acid esters in which the non-carbonyl moiety of the carboxylic acid portion of the ester grouping is selected from straight or branched chain alkyl (for
example, methyl, n-propyl, t-butyl, or n-butyl), cycloalkyl, alkoxyalkyl (for example,
methoxymethyl), aralkyl (for example benzyl), aryloxyalkyl (for example, phenoxymethyl), aryl (for example, phenyl, optionally substituted by, for example, halogen, C.sub.1-4 alkyl, or C.sub.l- 4 alkoxy) or amino); sulphonate esters, such as alkyl- or aralkylsulphonyl (for example, methanesulphonyl); or amino acid esters (for example, L-valyl or L-isoleucyl). A
"pharmaceutically acceptable ester" also includes inorganic esters such as mono-, di-, or triphosphate esters. In such esters, unless otherwise specified, any alkyl moiety present
advantageously contains from 1 to 18 carbon atoms, particularly from 1 to 6 carbon atoms, more particularly from 1 to 4 carbon atoms. Any cycloalkyl moiety present in such esters
advantageously contains from 3 to 6 carbon atoms. Any aryl moiety present in such esters advantageously comprises a phenyl group, optionally substituted as shown in the definition of carbocycylyl above. Pharmaceutically acceptable esters thus include C1-C22 fatty acid esters, such as acetyl, t-butyl or long chain straight or branched unsaturated or omega-6 monounsaturated fatty acids such as palmoyl, stearoyl and the like. Alternative aryl or heteroaryl esters include benzoyl, pyridylmethyloyl and the like any of which may be substituted, as defined in carbocyclyl above.
Additional pharmaceutically acceptable esters include aliphatic L-amino acid esters such as leucyl, isoleucyl and especially valyl.
For therapeutic use, salts of the compounds are those wherein the counter-ion is pharmaceutically acceptable. However, salts of acids and bases which are non-pharmaceutically acceptable may also find use, for example, in the preparation or purification of a pharmaceutically acceptable compound.
The pharmaceutically acceptable acid and base addition salts as mentioned hereinabove are meant to comprise the therapeutically active non-toxic acid and base addition salt forms which the compounds are able to form. The pharmaceutically acceptable acid addition salts can conveniently be obtained by treating the base form with such appropriate acid. Appropriate acids comprise, for example, inorganic acids such as hydrohalic acids, e.g. hydrochloric or hydrobromic acid, sulfuric, nitric, phosphoric and the like acids; or organic acids such as, for example, acetic, propanoic, hydroxy acetic, lactic, pyruvic, oxalic (i.e. ethanedioic), malonic, succinic (i.e. butanedioic acid), maleic, fumaric, malic (i.e. hydroxybutanedioic acid), tartaric, citric, methanesulfonic, ethanesulfonic, benzenesulfonic, p-toluenesulfonic, cyclamic, salicylic, p-aminosalicylic, pamoic and the like acids. Conversely said salt forms can be converted by treatment with an appropriate base into the free base form.
The compounds containing an acidic proton may also be converted into their non-toxic metal or amine addition salt forms by treatment with appropriate organic and inorganic bases.
Appropriate base salt forms comprise, for example, the ammonium salts, the alkali and earth alkaline metal salts, e.g. the lithium, sodium, potassium, magnesium, calcium salts and the like, salts with organic bases, e.g. the benzathine, N-methyl-D-glucamine, hydrabamine salts, and salts with amino acids such as, for example, arginine, lysine and the like.
The term "addition salt" as used hereinabove also comprises the solvates which the compounds described herein are able to form. Such solvates are for example hydrates, alcoholates and the like.
The term "quaternary amine" as used hereinbefore defines the quaternary ammonium salts which the compounds are able to form by reaction between a basic nitrogen of a compound and an appropriate quaternizing agent, such as, for example, an optionally substituted alkylhalide, arylhalide or arylalkylhalide, e.g. methyliodide or benzyliodide. Other reactants with good leaving groups may also be used, such as alkyl trifluoromethanesulfonates, alkyl methanesulfonates, and alkyl p-toluenesulfonates. A quaternary amine has a positively charged nitrogen. Pharmaceutically acceptable counterions include chloro, bromo, iodo, trifluoroacetate and acetate. The counterion of choice can be introduced using ion exchange resins.
Prodrugs of the disclosed compounds also are contemplated herein. A prodrug is an active or inactive compound that is modified chemically through in vivo physiological action, such as hydrolysis, metabolism and the like, into an active compound following administration of the prodrug to a subject. The term "prodrug" as used throughout this text means the pharmacologically acceptable derivatives such as esters, amides and phosphates, such that the resulting in vivo biotransformation product of the derivative is the active drug as defined in the compounds described herein. Prodrugs preferably have excellent aqueous solubility, increased bioavailability and are readily metabolized into the active inhibitors in vivo. Prodrugs of a compounds described herein may be prepared by modifying functional groups present in the compound in such a way that the modifications are cleaved, either by routine manipulation or in vivo, to the parent compound. The suitability and techniques involved in making and using prodrugs are well known by those skilled in the art. F or a general discussion of prodrugs involving esters see Svensson and Tunek, Drug Metabolism Reviews 165 (1988) and Bundgaard, Design of Prodrugs, Elsevier (1985).
The term "prodrug" also is intended to include any covalently bonded carriers that release an active parent drug of the present invention in vivo when the prodrug is administered to a subject. Since prodrugs often have enhanced properties relative to the active agent pharmaceutical, such as, solubility and bioavailability, the compounds disclosed herein can be delivered in prodrug form. Thus, also contemplated are prodrugs of the presently disclosed compounds, methods of delivering prodrugs and compositions containing such prodrugs. Prodrugs of the disclosed compounds
typically are prepared by modifying one or more functional groups present in the compound in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to yield the parent compound. Prodrugs include compounds having a phosphonate and/or amino group functionalized with any group that is cleaved in vivo to yield the corresponding amino and/or phosphonate group, respectively. Examples of prodrugs include, without limitation, compounds having an acylated amino group and/or a phosphonate ester or phosphonate amide group. In particular examples, a prodrug is a lower alkyl phosphonate ester, such as an isopropyl phosphonate ester.
Protected derivatives of the disclosed compounds also are contemplated. A variety of suitable protecting groups for use with the disclosed compounds are disclosed in Greene and Wuts, Protective Groups in Organic Synthesis; 3rd Ed.; John Wiley & Sons, New York, 1999.
In general, protecting groups are removed under conditions that will not affect the remaining portion of the molecule. These methods are well known in the art and include acid hydrolysis, hydrogenolysis and the like. One preferred method involves the removal of an ester, such as cleavage of a phosphonate ester using Lewis acidic conditions, such as in TMS-Br mediated ester cleavage to yield the free phosphonate. A second preferred method involves removal of a protecting group, such as removal of a benzyl group by hydrogenolysis utilizing palladium on carbon in a suitable solvent system such as an alcohol, acetic acid, and the like or mixtures thereof. A t-butoxy-based group, including t-butoxy carbonyl protecting groups can be removed utilizing an inorganic or organic acid, such as HC1 or trifluoroacetic acid, in a suitable solvent system, such as water, dioxane and/or methylene chloride. Another exemplary protecting group, suitable for protecting amino and hydroxy functions amino is trityl. Other conventional protecting groups are known and suitable protecting groups can be selected by those of skill in the art in consultation with Greene and Wuts, Protective Groups in Organic Synthesis; 3rd Ed.; John Wiley & Sons, New York, 1999. When an amine is deprotected, the resulting salt can readily be neutralized to yield the free amine. Similarly, when an acid moiety, such as a phosphonic acid moiety is unveiled, the compound may be isolated as the acid compound or as a salt thereof.
Particular examples of the presently disclosed agents may include one or more asymmetric centers; thus these compounds can exist in different stereoisomeric forms. Accordingly, compounds and compositions may be provided as individual pure enantiomers or as stereoisomeric mixtures, including racemic mixtures. In certain embodiments the compounds disclosed herein are synthesized in or are purified to be in substantially enantiopure form, such as in a 90%
enantiomeric excess, a 95% enantiomeric excess, a 97% enantiomeric excess or even in greater than a 99% enantiomeric excess, such as in enantiopure form.
The presently disclosed agents can have at least one asymmetric center or geometric center, cis-trans center(C=C, C=N). All chiral, diasteromeric, racemic, meso, rotational and geometric isomers of the structures are intended unless otherwise specified. The compounds can be isolated as a single isomer or as mixture of isomers. All tautomers of the compounds are also considered part of the disclosure. The presently disclosed compounds also includes all isotopes of atoms present in the compounds, which can include, but are not limited to, deuterium, tritium, 18F, etc.
While the generation of ROS in small amounts is a typical byproduct of the cellular respiration pathway, certain conditions, including a disease or other medical condition, may occur in the patient when the amount of ROS is excessive to the point where natural enzyme mechanisms cannot scavenge the amount of ROS being produced. Therefore, compounds, compositions and methods that scavenge reactive oxygen species that are present within the mitochondrial membrane of the cell are useful and are provided herein. These compounds, compositions and methods have the utility of being able to scavenge an excess amount of ROS being produced that naturally occurring enzymes SOD and catalase, among others, cannot cope with.
Compounds and compositions described herein have use in the prophylaxis and treatment of exposure to ionizing radiation, in anti-ageing therapies and, generally, in treating conditions that benefit from antioxidant treatment. Examples of these compounds are provided below and in the claims. For example, the effective mitochondrial concentration of mitochondria targeted conjugated nitroxides (-N-0, -N-OH or N=0 containing compounds and groups) against γ-irradiation could be increased up to 1,000 times (and their required tissues concentrations can be reduced 1,000 times from 10 mM to 10 μΜ) compared with parent non-conjugated nitroxides. Enrichment in mitochondria of mitochondria targeted nitroxides has been demonstrated by EPR spectroscopy as well as by MS analysis of their content in mitochondria obtained from cells incubated with mitochondria targeted nitroxides. Delivery of mitochondria targeted-nitroxides into mitochondria does not depend on the mitochondrial membrane potential. Therefore, mitochondria targeted nitroxides can accumulate not only in intact but also in de-energized or damaged mitochondria with low membrane potential. Moreover, mitochondria targeted nitroxide conjugates are delivered into mitochondria without affecting the mitochondrial membrane potential. Hence, they do not impair the major mitochondrial function, the energy production, in cells. In addition, the conjugated nitroxides provide a new important feature, post irradiation protection.
Like other nitroxides, conjugated mitochondria targeted nitroxides might potentially lower blood pressure and sympathetic nerve activity. However, the dramatically reduced dose of mitochondria targeted nitroxides (about 1,000-fold), compared to non-conjugated parental nitroxides, may be significantly below of those inducing side effects.
XJB-5-131 (XJB), a reactive oxygen species (ROS) scavenger also partially prevented evoked DA loss near probes for 4 hours by improving overall tissue health. This particular ROS scavenger is unique in that it targets the inner mitochondrial membrane and contains a potent nitroxide group responsible for electron and radical scavenging. The neuroprotective qualities of XJB make ROS ' s good candidates for pharmacologically enhanced microdialysis.
Disclosed herein are the effects of two non-steroidal anti-inflammatories, ibuprofen (IBU) and pyridoxalphosphate-6-azophenyl-2',4'-disulfonic acid tetrasodium salt (PPads) and two ROS scavengers, JP4-039 (JP4) and JRS527 (JRS) on the preservation of evoked DA responses near microdialysis probes. As the effects of both DEX and XJB were profound after only 4 hours, this timeframe was used for the present study. Carbon fiber microelectrodes were coupled with fast scan cyclic voltammetry (FSCV) for detection of evoked DA release in tissue near probes perfused separately with each pharmacological agent.
Disclosed herein are compounds, or pharmaceutically acceptable salts thereof, having a

Formula 1 wherein X is

R3 is -N(R5)(Rio), wherein R5 and Rio are each independently H or an -Ν-0·, -N-OH or -N=0 containing group, provided at least one of R5 is an -Ν-0·, -N-OH or -N=0 containing group; and R is -C(0)-R6, -C(0)0-R6, or -P(0)-(R6)2, wherein R6 is an optionally substituted alkyl or an -N- Ο·, -N-OH or -N=0 containing group, provided that if only one of R5 or Rio is an -Ν-0·, -N-OH or -N=0 containing group then R6 is an optionally substituted alkyl or an -Ν-0·, -N-OH or -N=0 containing group.
In certain embodiments, both R5 and Rio are each an -Ν-0·, -N-OH or -N=0 containing group.
In certain embodiments, R5 is an -Ν-0·, -N-OH or -N=0 containing group, Rio is H, and R6 is an -Ν-0·, -N-OH or -N=0
In certain embodiment
In certain embodiment
 , and R* is H.
, and R* is H.
In certain embodiments, Ria R2, and R2a are each H.
In certain embodiments, Ri is alkyl, particularly Ci-C6 alkyl, most particularly propyl (e.g., isopropyl) or butyl (e.g., isobutyl).
In certain embodiments, Ria and R2a are H, and R2 is aralkyl, particularly benzyl.
In certain embodiments, R6 is alkyl or aralkyl, particularly isopropyl or benzyl.
In certain embodiments, at least one of Ri, Ria R2, R2a, R4, R20 or R21 is Ci-C6 straight or branched-chain alkyl optionally substituted one or more phenyl (-C6H5) groups, and the phenyl group is optionally methyl-, ethyl-, hydroxyl- or fluoro-substituted.
In certain embodiments, the -Ν-0· containing group is selected from:


In certain embodiments, both R5 and Rio are

In certain embodiments, Rio is H, and R5 and R6 are both

Also disclosed herein are compounds, or pharmaceutically acceptable salts thereof, having ; structure of:
R31 - N(R33) - R32
Formula 2 wherein R31 and R32 are each independently

R33 is H or an optionally substituted alkyl.
Further disclosed herein are compounds, or pharmaceutically acceptable salts thereof, having a structure of:


Formula 3B
wherein Y is -NH-, -0-, -N(alkyl)-, -N(aryl)-, -N(cycloalkyl)-, -N(heteroaryl)-, or -CH(R41)-; R41 is H, optionally substituted aliphatic, optionally substituted cycloalkyl, optionally substituted aryl, or optionally heteroaryl;
each of R42 - R45 is H, optionally substituted aliphatic, optionally substituted cycloalkyl, optionally substituted aryl, optionally heteroaryl, or fluorine;
each Z is independently -0-, -NH- or -C¾-; and
each R40 is independently


In certain embodiments, each of R -R45 is H or (Ci-C6)alkyl.
In certain embodiments, R41 is propyl or butyl, particularly isobutyl.
In certain embodiments, Y is -NH-.
In certain embodiments, R42-R45 are each H.
Illustrative compounds are shown below:

SUBSTITUTE SHEET (RULE 26)


JiR:SS27-S3

RS527*3S
As indicated above, R5 is an -Ν-0·, -N-OH or -N=0 containing group (not -Ν-0·, -N-OH or -N=0, but groups containing those moieties, such as TEMPO, etc, as described herein). It should be recognized that the nitroxides (-Ν ·) disclosed herein may undergo reduction in vivo to their corresponding hydroxylamines (-N-OH) and/or may undergo oxidation in vivo to their corresponding nitroxonium ions. All three forms may be in equilibrium and their ratio depends on the redox environment. Any one of the three forms, or a mixture thereof, could be administered to a subject.
As is known to one ordinarily skilled in the art, nitroxide and nitroxide derivatives, including TEMPOL and associated TEMPO derivatives are stable radicals that can withstand biological environments. Therefore, the presence of the 4-amino-TEMPO, TEMPOL or another nitroxide "payload" within the mitochondria membrane can serve as an effective and efficient
SUBSTITUTE SHEET (RULE 26)
electron scavenger of the ROS being produced within the membrane. Non-limiting examples of this include TEMPO (2,2,6,6-Tetramethyl-4-piperidine 1-oxyl) and TEMPOL (4-Hydroxy- TEMPO), in which, when incorporated into the compound described herein, form, for example, when R3 is -NH-R5, -O-R5:

Additional non-limiting examples of -Ν-0·, -N-OH or N=0 containing group are provided in Table 1 (from Jiang, J., et al. "Structural Requirements for Optimized Delivery, Inhibition of Oxidative Stress, and Antiapoptotic Activity of Targeted Nitroxides", J Pharmacol Exp Therap. 2007, 320(3): 1050-60). A person of ordinary skill in the art would be able to conjugate (covalently attach) any of these compounds to the rest of the compound using common linkers and/or conjugation chemistries, such as the chemistries described herein. Table 1 provides a non-limiting excerpt from a list of over 300 identified commercially-available -Ν-0·, -N-OH or N=0 containing compounds that may be useful in preparation of the compounds or compositions described herein.
Table 1 - Commercially-available -Ν-0·, -N-OH or N=0 containing groups

SUBSTITUTE SHEET (RULE 26)

SUBSTITUTE SHEET (RULE 26)

ide
SUBSTITUTE SHEET (RULE 26)

SUBSTITUTE SHEET (RULE 26)

SUBSTITUTE SHEET (RULE 26)

SUBSTITUTE SHEET (RULE 26)

SUBSTITUTE SHEET (RULE 26)
 1-
1-
Triisopropylsilanyl
-lh-Pyrrolo[2,3-
B]Pyridine-5-
Carboxylic Acid
Methoxy+
-Methyl-Amide
The compounds described above can be synthesized by any useful method.
Methods of Use and Pharmaceutical Compositions
In a therapeutic embodiment, a method of scavenging free-radicals in a subject (e.g., a patient in need of treatment with a free-radical scavenger) is provided, comprising administering to the subject an amount of one or more compound described herein and having a free-radical scavenging group, such as a nitroxide-containing group effective to scavenge free radicals. As described above, a number of diseases, conditions or injuries can be ameliorated or otherwise treated or prevented by administration of free radical scavenging compounds, such as those described herein.
Any agent or agents used for prevention, mitigation or treatment in a subject of injury caused by radiation exposure may be administered in an amount effective to prevent, mitigate of treat such injury, namely in an amount and in a dosage regimen effective to prevent injury or to reduce the duration and/or severity of the injury resulting from radiation exposure. According to one non-limiting embodiment, an effective dose ranges from 0.1 or 1 mg/kg to 100 mg/kg, including any increment or range therebetween, including 1 mg/kg, 5 mg/kg, 10 mg/kg, 20 mg/kg, 25 mg/kg, 50 mg/kg, and 75 mg/kg. However, for each compound described herein, an effective dose or dose range is expected to vary from that of other compounds described herein for any number of reasons, including the molecular weight of the compound, bioavailability, specific activity, etc. The therapeutic window between the minimally-effective dose, and maximum tolerable dose in a subject can be determined empirically by a person of skill in the art, with end points being determinable by in vitro and in vivo assays, such as those described herein and/or are acceptable in the pharmaceutical and medical arts for obtaining such information regarding radioprotective agents. Different concentrations of the agents described herein are expected to achieve similar results, with the drug product administered, for example and without limitation,
once prior to an expected radiation dose, such as prior to radiation therapy or diagnostic exposure to ionizing radiation, during exposure to radiation, or after exposure in any effective dosage regimen. The compounds can be administered continuously, such as intravenously, one or more times daily, once every two, three, four, five or more days, weekly, monthly, etc., including increments therebetween. A person of ordinary skill in the pharmaceutical and medical arts will appreciate that it will be a matter of simple design choice and optimization to identify a suitable dosage regimen for prevention, mitigation or treatment of injury due to exposure to radiation.
The compounds described herein also are useful in preventing, mitigating (to make less severe) and/or treating injury caused by radiation exposure. By radiation, in the context of this disclosure, it is meant types of radiation that result in the generation of free radicals, e.g., reactive oxygen species (ROS), as described herein. The free radicals are produced, for example and without limitation, by direct action of the radiation, as a physiological response to the radiation and/or as a consequence of damage/injury caused by the radiation. In one embodiment, the radiation is ionizing radiation. Ionizing radiation consists of highly-energetic particles or waves that can detach (ionize) at least one electron from an atom or molecule. Examples of ionizing radiation are energetic beta particles, neutrons, and alpha particles. The ability of light waves (photons) to ionize an atom or molecule varies across the electromagnetic spectrum. X-rays and gamma rays can ionize almost any molecule or atom; far ultraviolet light can ionize many atoms and molecules; near ultraviolet and visible light are ionizing to very few molecules. Microwaves and radio waves typically are considered to be non-ionizing radiation, though damage caused by, e.g., microwaves, may result in the production of free-radicals as part of the injury and/or physiological response to the injury.
The compounds typically are administered in an amount and dosage regimen to prevent, mitigate or treat the effects of exposure of a subject to radiation. The compounds may be administered in any manner that is effective to treat, mitigate or prevent damage caused by the radiation. Examples of delivery routes include, without limitation: topical, for example, epicutaneous, inhalational, enema, ocular, otic and intranasal delivery; enteral, for example, orally, by gastric feeding tube and rectally; and parenteral, such as, intravenous, intraarterial,
intramuscular, intracardiac, subcutaneous, intraosseous, intradermal, intrathecal, intraperitoneal, transdermal, iontophoretic, transmucosal, epidural and intravitreal, with oral, intravenous, intramuscular and transdermal approaches being preferred in many instances.
In certain embodiments, the compounds disclosed herein may be useful for treating neurodegenerative disorders such as, for example, Alzheimer's disease, ataxia telangiectasia, Parkinson's disease, amyotrophic lateral sclerosis, and Huntington's disease.
In some embodiments, one or more of the disclosed compounds are mixed or combined with a suitable pharmaceutically acceptable carrier to prepare a pharmaceutical composition.
Pharmaceutical carriers or vehicles suitable for administration of the compounds provided herein include any such carriers known to be suitable for the particular mode of administration.
Remington: The Science and Practice of Pharmacy, The University of the Sciences in Philadelphia, Editor, Lippincott, Williams, & Wilkins, Philadelphia, PA, 21st Edition (2005), describes exemplary compositions and formulations suitable for pharmaceutical delivery of the compounds disclosed herein. In addition, the compounds may be formulated as the sole pharmaceutically active ingredient in the composition or may be combined with other active ingredients.
Upon mixing or addition of the compound(s) to a pharmaceutically acceptable carrier, the resulting mixture may be a solution, suspension, emulsion, or the like. Liposomal suspensions may also be suitable as pharmaceutically acceptable carriers. These may be prepared according to methods known to those skilled in the art. The form of the resulting mixture depends upon a number of factors, including the intended mode of administration and the solubility of the compound in the selected carrier or vehicle. Where the compounds exhibit insufficient solubility, methods for solubilizing may be used. Such methods are known and include, but are not limited to, using cosolvents such as dimethylsulfoxide (DMSO), using surfactants such as Tween®, and dissolution in aqueous sodium bicarbonate. Derivatives of the compounds, such as salts or prodrugs may also be used in formulating effective pharmaceutical compositions. The disclosed compounds may also be prepared with carriers that protect them against rapid elimination from the body, such as time-release formulations or coatings. Such carriers include controlled release formulations, such as, but not limited to, microencapsulated delivery systems.
The disclosed compounds and/or compositions can be enclosed in multiple or single dose containers. The compounds and/or compositions can also be provided in kits, for example, including component parts that can be assembled for use. For example, one or more of the disclosed compounds may be provided in a lyophilized form and a suitable diluent may be provided as separated components for combination prior to use. In some examples, a kit may include a disclosed compound and a second therapeutic agent for co-administration. The compound and second therapeutic agent may be provided as separate component parts. A kit may include a plurality of containers, each container holding one or more unit dose of the compound. The containers are preferably adapted for the desired mode of administration, including, but not limited to tablets, gel capsules, sustained-release capsules, and the like for oral administration; depot products, pre-filled syringes, ampoules, vials, and the like for parenteral administration; and patches, medipads, creams, and the like for topical administration.
The active compound is included in the pharmaceutically acceptable carrier in an amount sufficient to exert a therapeutically useful effect in the absence of undesirable side effects on the subject treated. A therapeutically effective concentration may be determined empirically by testing the compounds in known in vitro and in vivo model systems for the treated disorder. In some examples, a therapeutically effective amount of the compound is an amount that lessens or ameliorates at least one symptom of the disorder for which the compound is administered.
Typically, the compositions are formulated for single dosage administration. The concentration of active compound in the drug composition will depend on absorption, inactivation, and excretion rates of the active compound, the dosage schedule, and amount administered as well as other factors known to those of skill in the art.
In some examples, about 0.1 mg to 1000 mg of a disclosed compound, a mixture of such compounds, or a physiologically acceptable salt or ester thereof, is compounded with a
physiologically acceptable vehicle, carrier, excipient, binder, preservative, stabilizer, flavor, etc., in a unit dosage form. The amount of active substance in those compositions or preparations is such that a suitable dosage in the range indicated is obtained. The term "unit dosage form" refers to physically discrete units suitable as unitary dosages for human subjects and other mammals, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect, in association with a suitable pharmaceutical excipient. In some examples, the compositions are formulated in a unit dosage form, each dosage containing from about 1 mg to about 1000 mg (for example, about 2 mg to about 500 mg, about 5 mg to 50 mg, about 10 mg to 100 mg, or about 25 mg to 75 mg) of the one or more compounds. In other examples, the unit dosage form includes about 0.1 mg, about 1 mg, about 5 mg, about 10 mg, about 20 mg, about 30 mg, about 40 mg, about 50 mg, about 60 mg, about 70 mg, about 80 mg, about 90 mg, about 100 mg, about 150 mg, about 200 mg, about 250 mg, about 300 mg, about 400 mg, about 500 mg, about 600 mg, about 700 mg, about 800 mg, about 900 mg, about 1000 mg, or more of the disclosed compound(s).
The disclosed compounds or compositions may be administered as a single dose, or may be divided into a number of smaller doses to be administered at intervals of time. The therapeutic compositions can be administered in a single dose delivery, by continuous delivery over an extended time period, in a repeated administration protocol (for example, by a multi-daily, daily, weekly, or monthly repeated administration protocol). It is understood that the precise dosage, timing, and duration of treatment is a function of the disease being treated and may be determined empirically using known testing protocols or by extrapolation from in vivo or in vitro test data. It is to be noted that concentrations and dosage values may also vary with the severity of the condition
to be alleviated. In addition, it is understood that for a specific subject, dosage regimens may be adjusted over time according to the individual need and the professional judgment of the person administering or supervising the administration of the compositions, and that the concentration ranges set forth herein are exemplary only.
When administered orally as a suspension, these compositions are prepared according to techniques well known in the art of pharmaceutical formulation and may contain microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners/flavoring agents. As immediate release tablets, these compositions may contain microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants. If oral administration is desired, the compound is typically provided in a composition that protects it from the acidic environment of the stomach. For example, the composition can be formulated in an enteric coating that maintains its integrity in the stomach and releases the active compound in the intestine. The composition may also be formulated in combination with an antacid or other such ingredient.
Oral compositions will generally include an inert diluent or an edible carrier and may be compressed into tablets or enclosed in gelatin capsules. For the purpose of oral therapeutic administration, the active compound or compounds can be incorporated with excipients and used in the form of tablets, capsules, or troches. Pharmaceutically compatible binding agents and adjuvant materials can be included as part of the composition. The tablets, pills, capsules, troches, and the like can contain any of the following ingredients or compounds of a similar nature: a binder such as, but not limited to, gum tragacanth, acacia, corn starch, or gelatin; an excipient such as microcrystalline cellulose, starch, or lactose; a disintegrating agent such as, but not limited to, alginic acid and corn starch; a lubricant such as, but not limited to, magnesium stearate; a gildant, such as, but not limited to, colloidal silicon dioxide; a sweetening agent such as sucrose or saccharin; and a flavoring agent such as peppermint, methyl salicylate, or fruit flavoring.
When the dosage unit form is a capsule, it can contain, in addition to material of the above type, a liquid carrier such as a fatty oil. In addition, dosage unit forms can contain various other materials, which modify the physical form of the dosage unit, for example, coatings of sugar and other enteric agents. The compounds can also be administered as a component of an elixir, suspension, syrup, wafer, chewing gum or the like. A syrup may contain, in addition to the active compounds, sucrose as a sweetening agent and certain preservatives, dyes and colorings, and flavors.
When administered orally, the compounds can be administered in usual dosage forms for oral administration. These dosage forms include the usual solid unit dosage forms of tablets and capsules as well as liquid dosage forms such as solutions, suspensions, and elixirs. When the solid dosage forms are used, it is preferred that they be of the sustained release type so that the compounds need to be administered only once or twice daily. In some examples, an oral dosage form is administered to the subject 1, 2, 3, 4, or more times daily. In certain examples, the oral dosage is from about 1 mg/day to about 500 mg/day, about 2 mg/day to about 200 mg/day, or about 5 mg/day to about 50 mg/day. It is understood that while a subject may be started at one dose, that dose may be varied over time as the subject's condition changes.
In additional examples, the compounds can be administered orally to humans in a dosage range of 1 to 1000 mg/kg body weight in single or divided doses. One illustrative dosage range is 0.1 to 200 mg/kg body weight orally (such as 0.5 to 100 mg/kg body weight orally) in single or divided doses. For oral administration, the compositions may be provided in the form of tablets containing about 1 to 1000 milligrams of the active ingredient, particularly 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900, or 1000 milligrams of the active ingredient. It will be understood, however, that the specific dose level and frequency of dosage for any particular patient may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the host undergoing therapy.
Injectable solutions or suspensions may also be formulated, using suitable non-toxic, parenterally- acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution or isotonic sodium chloride solution, or suitable dispersing or wetting and suspending agents, such as sterile, bland, fixed oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid. Solutions or suspensions used for parenteral, intradermal, subcutaneous, or topical application can include any of the following components: a sterile diluent such as water for injection, saline solution, fixed oil, a naturally occurring vegetable oil such as sesame oil, coconut oil, peanut oil, cottonseed oil, and the like, or a synthetic fatty vehicle such as ethyl oleate, and the like, polyethylene glycol, glycerine, propylene glycol, or other synthetic solvent; antimicrobial agents such as benzyl alcohol and methyl parabens; antioxidants such as ascorbic acid and sodium bisulfite; chelating agents such as ethylenediaminetetraacetic acid (EDTA); buffers such as acetates, citrates, and phosphates; and agents for the adjustment of tonicity such as sodium chloride and dextrose. Parenteral preparations can be enclosed in ampoules, disposable syringes, or multiple
dose vials made of glass, plastic, or other suitable material. Buffers, preservatives, antioxidants, and the like can be incorporated as required.
Where administered intravenously, suitable carriers include physiological saline, phosphate buffered saline (PBS), and solutions containing thickening and solubilizing agents such as glucose, polyethylene glycol, polypropyleneglycol, and mixtures thereof. Liposomal suspensions including tissue-targeted liposomes may also be suitable as pharmaceutically acceptable carriers.
The compounds can be administered parenterally, for example, by IV, IM, depo-IM, SC, or depo-SC. When administered parenterally, a therapeutically effective amount of about 0.1 to about 500 mg/day (such as about 1 mg/day to about 100 mg/day, or about 5 mg/day to about 50 mg/day) may be delivered. When a depot formulation is used for injection once a month or once every two weeks, the dose may be about 0.1 mg/day to about 100 mg/day, or a monthly dose of from about 3 mg to about 3000 mg.
The compounds can also be administered sublingually. When given sublingually, the compounds should be given one to four times daily in the amounts described above for IM administration.
The compounds can also be administered intranasally. When given by this route, the appropriate dosage forms are a nasal spray or dry powder. The dosage of the compounds for intranasal administration is the amount described above for IM administration. When administered by nasal aerosol or inhalation, these compositions may be prepared according to techniques well known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents.
The compounds can be administered intrathecally. When given by this route, the appropriate dosage form can be a parenteral dosage form. The dosage of the compounds for intrathecal administration is the amount described above for IM administration.
The compounds can be administered topically. When given by this route, the appropriate dosage form is a cream, ointment, or patch. When administered topically, an illustrative dosage is from about 0.5 mg/day to about 200 mg/day. Because the amount that can be delivered by a patch is limited, two or more patches may be used.
The compounds can be administered rectally by suppository. When administered by suppository, an illustrative therapeutically effective amount may range from about 0.5 mg to about 500 mg. When rectally administered in the form of suppositories, these compositions may be prepared by mixing the drug with a suitable non-irritating excipient, such as cocoa butter, synthetic
glyceride esters of polyethylene glycols, which are solid at ordinary temperatures, but liquefy and/or dissolve in the rectal cavity to release the drug.
Examples
Control Experiments. In all of the following experiments evoked DA release was measured in the striatum during a 25 s stimulation of the MFB. Responses measured pre-probe implantation showed robust maximum evoked DA release ranging from 12-20 μΜ (blue responses in Figure 2 and 4). As previously reported, microdialysis probes perfused with aCSF (no pharmacological agent) eliminated evoked DA release nearby (70-100 μιη away). To determine if any DA terminals survived probe implantation, a competitive DA transporter inhibitor, nomifensine was administered by i.p. injection. Nomifensine revived evoked DA release near probes. The immediate response to stimulation proves that DA terminals survive directly next to the electrode, no further than 70 μιη from the probe.
Anti-Inflammatory and ROS Scavenger Agents. This study compares the effects of three anti-inflammatory agents and three ROS scavengers on preserving evoked DA release in tissue surrounding microdialysis probes. DEX and XJB have previously been reported on (Nesbitt et al. (2013) Pharmacological mitigation of tissue damage during brain microdialysis, Anal Chem. 85, 8173-8179), and are represented here for comparison purposes. The chemical structures of all pharmacological agents are represented in Figure 1. The top panel i ll u s trate s the inflammatory agents and the bottom panel the ROS scavengers.
IBU and Ppads Preserve Evoked DA Release Near Probes. During an inflammatory event such as microdialysis probe implantation, cells up-regulate production of pro- inflammatory proteins causing activation of phospholipase A2. Through cyclooxygenase (COX) modification a cascade of inflammatory mediators are produced. Both DEX and IBU work to intervene at different stages of the inflammatory cellular mechanism. DEX acts by down- regulating proinflammatory proteins in cells and up-regulating anti-inflammatory proteins produced by the cell. IBU is considered a COX-1 and COX-2 inhibitor thereby inhibiting COX modification and preventing inflammatory mediator signaling. The non-steroidal anti-inflammatory IBU, when perfused through microdialysis probes prevented complete loss of evoked DA release near probes (Figure 2-middle panel, red). Administration of nomifensine greatly impacted maximum evoked DA release near probes, increasing it to nearly pre-probe implantation amplitude (Figure 2-middle panel, green). IBU had similar effects as DEX suggesting that DEX's effects on evoked DA release are due to both its anti-inflammatory actions, not steroidal actions on DA terminals.
PPads is a non-selective P2 purinergic antagonist that blocks P2Y receptors. PPads reduces inflammation in the brain by blocking purine receptors on microglia limiting their response. Microglia are immune cells that are the first to respond to injury in the central nervous system. In a study investigating the migration of microglia, PPads significantly decreased the number and motility of the microglia responding to the site of an injury. PPads perfused through microdialysis also preserved evoked DA release near probes (Figure 2-right panel, red).
Nomifensine further increased evoked DA release but not as dramatically as DEX and IBU (Figure 2-right panel, green).
DEX, IBU, and PPads all significantly increased evoked DA release near probes (Figure 3). DEX's effect was more significant than IBU and PPads, and this is most likely due to DEX's duel action to both decrease pro-inflammatory proteins and increase anti-inflammatory proteins. Post- nomifensine maximum evoked DA responses were subject to a one-way ANOVA. DEX, IBU, and PPads all significantly increased maximum evoked DA release, DEX and IBU to above 80% of the pre -probe response. These findings demonstrate the ability of anti-inflammatories to preserve DA release and uptake activity in tissue near microdialysis probes.
JP4 and JRS. Inflammation is known to induce mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. XJB, a first generation ROS scavenger, infiltrates mitochondria and has been shown to be effective in neuroprotection. XJB prevents neuronal loss and preserves evoked DA near probes. A second generation isostere of XJB, the small molecule JP4, was developed offering an improved pharmacokinetic profile while maintaining its impressive ROS scavenging abilities. JP4 specifically targets mitochondria, similar to XJB and catalyzes the dismutation of superoxide radical anions and other reactive oxygen species. JP4 protective qualities mitigate radiation damage in blood cells in vitro and in vivo. JRS, a modified analog of JP4, represents the first bis-nitroxide in this series and is expected to increase potency (Figure 1).
JRS Preserve Evoked DA Release Near Probes. Similar to anti-inflammatory drugs, probes perfused with JP4 and JRS decreased but did not abolish evoked DA release near probes (Figure 4). JP4 and JRS all substantially increased the post-nomifensine response. Histograms representing ROS effect on evoked DA release are represented in Figure 5. XJB and JRS significantly increased evoked DA release near probes (Figure 5). All responses post-probe with JP4 gave a measurable DA signal however, this increase was not significantly greater than zero. JRS significantly increased evoked DA release compared to aCSF (Figure 5). One-way AVONA comparing post-nomifensine responses with ROS scavengers and aCSF). Nomifensine increase
all ROS DA responses, but it is clear that JRS had the greatest effect on protective evoked DA near probes, which was expected.
Nomifensine Perfusion does not Impact DA Terminals 70-100 μηι Away. In a final study, nomifensine was administrated by perfusion through microdialysis probes instead of an i.p. injection. Voltammetry near probes perfused with nomifensine was performed in two groups of rats (n=3 rats/group), one group with perfusion of aCSF plus 1 μΜ nomifensine, and the other with perfusion of DEX plus 1 μΜ nomifensine.
Post-probe implantation responses were recorded before nomifensine was added to the perfusion fluid and gave similar results to experiments performed previously. At probes perfused with only aCSF DA was not detected (Figure 6a). At probes perfused with DEX, evoked DA was measured (Figure 6b). Note that the aCSF results are reported as current (Figure 6a and 6c) as DA was undetected (aka. results did not produce a corresponding DA cyclic voltammograms). Nomifensine was added to each perfusion fluid and evoked DA responses were monitored near probes every 20 minutes for at least 2 hours. Nomifensine had no effect on evoked DA responses near probes whether DEX was present or not in the perfusion fluid.
Nomifensine, a competitive DA transporter inhibitor was unable to act on DA terminals 70-100 μιη away from the probe. However nomifensine perfusion through microdialysis is known to increase extracellular levels of DA within 10 minutes of administration. When measured by microdialysis probes, Nomifensine is a smaller molecule than DEX; therefore it would expect to diffuse through brain tissue more efficiently than DEX. We have previously shown that DEX's effects on evoked responses are attributable to its anti-inflammatory actions, not direct actions on DA terminals. This evidence further supports our conclusion that DEX is acting on the tissue to improve cellular health during inflammation as opposed to DEX acting on DA terminals, specifically the DA transporter 70 μιη away.
It has previously shown that microdialysis probe implantation decreased evoked DA release in tissue near probes. Through mitigation of the injury site with an anti- inflammatory steroid, DEX, and a novel ROS scavenger, XJB, DA activity is preserved. Herein it is shown that other non-steroidal anti-inflammatory agents and ROS scavengers that protect DA release and uptake near microdialysis probes. IBU, PPads, XJB and JRS significantly attenuate the loss of DA activity in tissue near microdialysis probes. As we have previously shown long- term protection with DEX preserves evoked DA release measured in tissue near the probe and at the outlet of probes, these drugs present other options to improve chronic microdialysis.
The protective effects were only partial, not completely restoring evoked DA release to its pre-probe response amplitude. Combination (e.g., via co-administration) of these
pharmacologically agents may also further promote evoked DA release in probe induced tissue damage. Overall, pharmacological enhanced microdialysis provides new insight into acute mitigation of microdialysis probe penetration injury and has the potential to successfully mitigate chronic implantation for long-term in vivo monitoring of neurochemicals.
METHODS
Reagents and Solutions. Dexamethasone sodium phosphate (DEX: AAP
Pharmaceruticals LLC, Schaumburg, IL), ibuprofen (IBU: Sigma Aldrich, St. Louis, MO) pyridoxalphosphate-6-azophenyl-2',4'-disulfonic acid tetrasodium salt (PPads: Sigma Aldrich, St. Louis, MO), were used as received from their suppliers. XJB-5-131 (XJB), and JP4-039 (JP4), were prepared as described by Wipf and co-workers. JRS527 (JRS) was prepared as described below. All solutions were prepared with ultrapure water (Nanopure, Barnstead, Dubuque, IA). Artificial cerebrospinal fluid (aCSF: 142 mM NaCl, 1.2 mM CaCl2, 2.7 mM KC1, 1.0 mM MgCl2, 2.0 mM Na¾P04, pH 7.4) was used as the perfusion fluid for
microdialysis.
DEX, and PPads were diluted to 10 μΜ in aCSF. IBU, XJB, JP4, and JRS were dissolved in aCSF containing 1% DMSO (Sigma Aldrich, St. Louis, MO). Nomifensine (20 mg/kg, i.p., Sigma Aldrich, St. Louis, MO) was used as received. Nomifensine was dissolved in phosphate- buffered saline (PBS: 155 mM NaCl, 100 mM Na¾P04, pH 7.40) for i.p. injections and diluted to 1 μΜ in aCSF for microdialysis perfusion. Electrodes were pre-treated in isopropyl alcohol (Sigma Aldrich, St. Louis, MO) and decolorizing carbon (Fisher, Pittsburgh, PA). Post- calibration of electrodes was performed with DA (Sigma Aldrich, St. Louis, MO) standards prepared in N2-purged aCSF.
Synthetic Procedure of JRS527. A solution of 4-hydroxy-TEMPO (67 mg, 0.38 mmol) in DMF (700 μ > was treated with N,N'-disuccinimidyl carbonate (115 mg, 0.45 mmol) and pyridine (62 μί, 0.075 mmol) and the resulting mixture was heated at 40 °C for 14 h. The solution was cooled to room temperature and directly used for the next step.
To a solution of JP4-039 (160 mg, 0.377 mmol) in CH2CI2 (3.5 mL) was added trifluoroacetic acid (565 μΐ,, 7.53 mmol) at 0 °C. The reaction was stirred and warmed to room temperature for 3 h and the solvent was removed under reduced pressure. The crude material was dissolved in
EtOAc, washed with 10% aq. Na2C03 and the aqueous layers was extracted with further portions of EtOAc. The combined organic layer was dried (Na2C03), filtered and the solvent was removed under reduced pressure. To the crude product dissolved in DMF (2.1 mL) and H2O (700
μί) was then added K3PO4 (83 mg, 0.38 mmol) and freshly prepared TEMPO-succinimidyl carbonate. After stirring at rt for 16 h the mixture was diluted with water (20 mL) and EtOAc (10 mL). The layers were separated, and the aqueous layer was extracted with EtOAc. The combined organics were washed with 5% aqueous LiCl, dried over MgSOz)., and concentrated to dryness. The residue was purified by flash column chromatography (S1O2, 40-80% EtOAc/hexanes) to provide JRS527-84 (110 mg, 56%) as a pale orange solid. HRMS (ESI) m/z calcd for
C28H51N4O5 [M+H]+ 523.3846, found 523.3854; IR (neat) 3310, 2972, 2934, 2871, 1713, 1649, 1528, 1465, 1363, 1316, 1239, 1178, 1117, 1085, 1049, 971, 776, 742, 685; m.p. 89- 92 °C;
[a]D+0.032 (c=0.5)
Voltammetry and Microdialysis. Carbon fiber microelectrodes (7 μιη in diameter, and 400 μιη in length) were coupled with fast scan cyclic voltammetry for in vivo measurement of evoked DA. Concentric microdialysis probes (300 μιη diameter, 4 mm length) were constructed in house as previously described with hollow fiber 13 kDa MWCO membranes (Specta/Por RC, Spectrum Laboratories Inc., Ranco Dominguez, CA). The probe inlet tubing (PE-20, Becton Dickinson, Franklin Lakes, NJ) was connected to a syringe pump (Harvard Apparatus, Holliston, MA) at a rate of 0.610 μΙ7ιηίη. The outlet made of fused silica capillary was led to waste as the dialysate fluid was not analyzed in this experiment.
Voltammetry Next to Microdialysis Probes. All procedures involving animals were approved by the University of Pittsburgh's Animal Care and Use Committee. Male Sprague- Dawley rats (250-350g; Charles Rivers, Raleigh, NC) were anesthetized with isoflurane (2.5 % by volume) and placed in a stereotaxic frame. Voltammetry next to microdialysis probes was performed in six groups of rats (6 rats per perfusion fluid unless otherwise noted).
A carbon fiber electrode was implanted at a 5° angle into the striatum of each rat. A stimulating electrode was lowered towards the medial forebrain bundle (MFB) until maximum evoked DA was observed at the electrode (4.3 mm below bregma, 1.2 mm lateral to bregma, and 7.2-8.5 mm below dura). Electrical stimulation of the MFB was performed for 25 s at 45 Hz (waveform: biphasic, square, constant current, 300 μΑ) and evoked DA release was measured in the striatum in twenty minute intervals.
Once consistent evoked DA responses were observed, a microdialysis probe was slowly
(over 30 minutes) implanted next to the electrode (0.7 mm above bregma, 2.5 mm lateral from bregma, and 7 mm below dura). In its final position, the microdialysis probe was 70 μιη from the tip of the carbon fiber, and 100 μιη from the base of the electrode. The probe was left to
perfuse in the brain for 2 hours. Following this, three more stimulus responses were recorded. Finally, nomifensine was administered (20 mg/kg i.p.) and one more stimulus response was collected.
Data Analysis. For comparison, evoked DA responses near probes perfused with only aCSF (no pharmacological agent) were used as the control. These results have previously been reported in Nesbitt et al 2013. For statistical analysis, maximum DA release was normalized with respect to the maximum amplitude of the responses recorded before each probe was implanted and presented in histograms (Figure 3 and 4). As in the case of probe perfusion with aCSF, DA was undetected after probe implantation but before nomifensine, a one-sample t-test was performed comparing DEX, IBU, PPads, XJB, JP4, and JRS to zero. Statistical analysis was performed using IBM Statistical Package for the Social Sciences (SPSS) 22 software. Details of statistical test of in the figure captions.
In view of the many possible embodiments to which the principles of the disclosed compounds, compositions, and methods may be applied, it should be recognized that the illustrated embodiments are only preferred examples of the invention and should not be taken as limiting the scope of the invention.
Claims
What is claimed is:
1. A compound, or a pharmaceutically acceptable salt thereof, having a structure of:

Ri, Ria R2, R2a, R4, R20 and R21 are each independently hydrogen, halo, or an optionally substituted alkyl;
R3 is -N(R5)(Rio), wherein R5 and Rio are each independently H or an -Ν-0·, -N-OH or -N=0 containing group, provided at least one of R5 is an -Ν-0·, -N-OH or -N=0 containing group; and R is -C(0)-R6, -C(0)0-R6, or -P(0)-(Re)2, wherein R6 is an optionally substituted alkyl or an -N- Ο·, -N-OH or -N=0 containing group, provided that if only one of R5 or Rio is an -Ν-0·, -N-OH or -N=0 containing group then R6 is an optionally substituted alkyl or an -Ν-0·, -N-OH or -N=0 containing group.
2. The compound of claim 1, wherein both R5 and Rio are each an -Ν-0·, -N-OH or - N=0 containing group.
3. The compound of claim 1, wherein R5 is an -Ν-0·, -N-OH or -N=0 containing group, Rio is H, and R6 is an -Ν-0·, -N-OH or -N=0 containing group.
The compound of any one of claims 1 to 3, wherein X
The compound of any one of claims 1 to 3, wherein X
 , and R4 is H.
, and R4 is H.
6. The compound of any one of claims 1 to 5, wherein Ria R2, and R2a are each H.
7. The compound of any one of claims 1 to 6, wherein Ri is alkyl.
8. The compound of any one of claims 1 to 5 or 7, wherein Ria and R2a are H, and R2 is aralkyl.
9. The compound of any one of claims 1 to 8, wherein R6 is alkyl or aralkyl.
10. The compound of claim 1, wherein at least one of Ri, Ria R2, R2a, R4, R20 or R21 is Ci-C6 straight or branched-chain alkyl optionally substituted one or more phenyl (-C6H5) groups, and the phenyl group is optionally methyl-, ethyl-, hydroxyl- or fluoro-substituted.
11. The compound of any one of claims 1 to 10, wherein the -Ν-0· containing group is selected from:


12. The compound of any one of claims 1 to 11, wherein Rio is H and R5 is

13. The compound of any one of claims 1 to 11, wherein both R5 and Rio are

14. The compound of any one of claims 1 to 11, wherein Rio is H, and R5 and R6 are

15. The compound of claim 1, wherein the compound is

JRS 52:74*
or

«527.Μ
16. A compound, or a pharmaceutically acceptable salt thereof, having a structure of:
R31 - N(R33) - R32
Formula 2
wherein R31 and R32 are each independently
SUBSTITUTE SHEET (RULE 26)


R33 is H or an optionally substituted alkyl.
The compound of claim 16, wherein R31 and R32 are each independently

18. The compound of claim 16, wherein the compound is

or

19. A compound, or a pharmaceutically acceptable salt thereof, having a structure of:
SUBSTITUTE SHEET (RULE 26)

Formula 3 A ; or

Formula 3B
wherein Y is -NH-, -0-, -N(alkyl)-, -N(aryl)-, -N(cycloalkyl)-, -N(heteroaryl)-, or -CH(R41)-; R41 is H, optionally substituted aliphatic, optionally substituted cycloalkyl, optionally substituted aryl, or optionally heteroaryl;
each of R42 - R45 is H, optionally substituted aliphatic, optionally substituted cycloalkyl, optionally substituted aryl, optionally heteroaryl, or fluorine;
each Z is independently -0-, -NH- or -C¾-; and
each R40 is independently


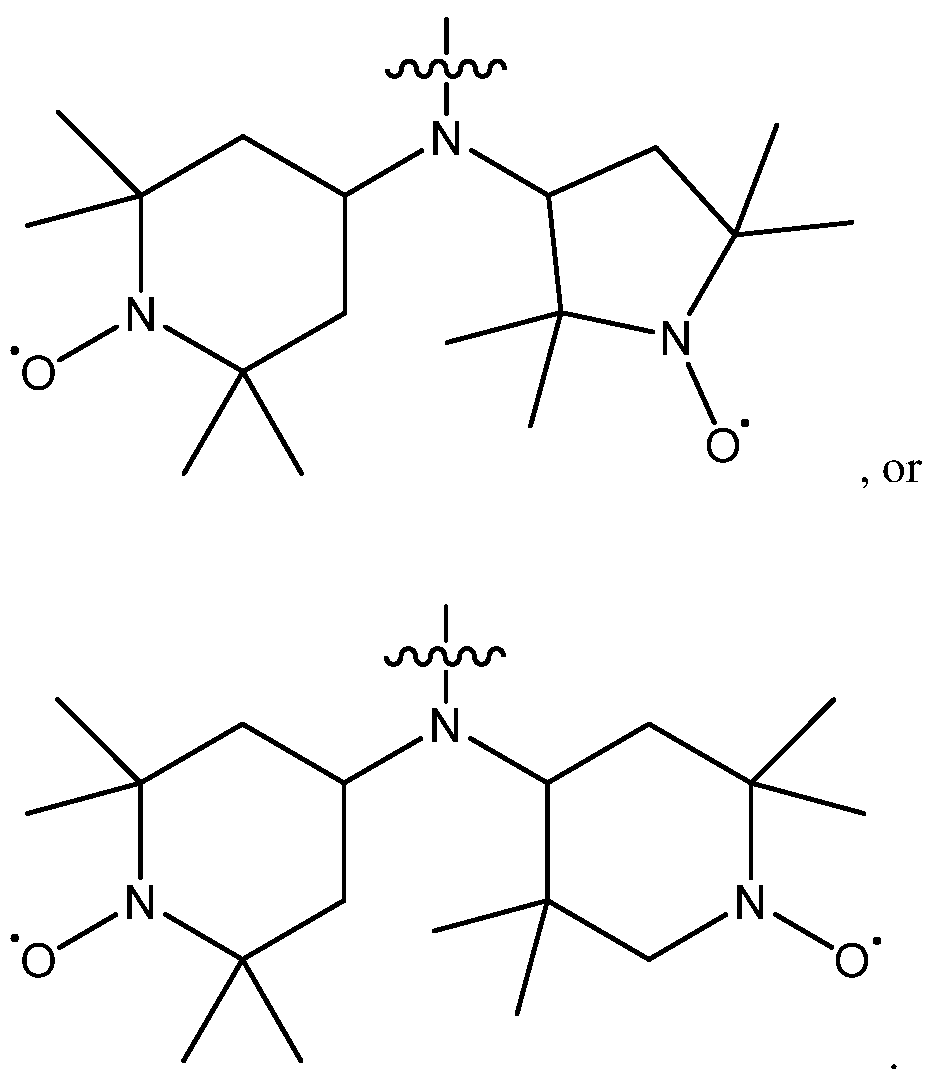
20. A pharmaceutical composition comprising at least one compound of any one of claims 1 to 19, and at least one pharmaceutically acceptable additive.
21. A method for mitigating probe-induced tissue damage in a subject, comprising administering to a subject in need thereof, at least one compound of any one of claims 1 to 19.
22. The method of claim 21, wherein the probe is a microdialysis probe.
23. The method of claim 21 or 22, wherein the method mitigates loss of evoked dopamine release.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562186131P | 2015-06-29 | 2015-06-29 | |
| US201562186268P | 2015-06-29 | 2015-06-29 | |
| US62/186,268 | 2015-06-29 | ||
| US62/186,131 | 2015-06-29 | ||
| US201562187701P | 2015-07-01 | 2015-07-01 | |
| US62/187,701 | 2015-07-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017004073A1 true WO2017004073A1 (en) | 2017-01-05 |
Family
ID=57609090
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2016/039877 WO2017004073A1 (en) | 2015-06-29 | 2016-06-28 | Bisnitroxides |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2017004073A1 (en) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010009405A2 (en) * | 2008-07-17 | 2010-01-21 | University Of Pittsburgh-Of The Commonwealth System Of Higher Education | Targeted nitroxide agents |
| WO2010009327A1 (en) * | 2008-07-17 | 2010-01-21 | University Of Pittsburgh Of The Commonwealth System Of Higher Education | Compounds for the treatment of pathologies associated with aging and degenerative disorders |
| WO2012112851A2 (en) * | 2011-02-18 | 2012-08-23 | University Of Pittsburgh - Of The Commonwealth System Of Higher Education | Targeted nitroxide agents |
-
2016
- 2016-06-28 WO PCT/US2016/039877 patent/WO2017004073A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010009405A2 (en) * | 2008-07-17 | 2010-01-21 | University Of Pittsburgh-Of The Commonwealth System Of Higher Education | Targeted nitroxide agents |
| WO2010009327A1 (en) * | 2008-07-17 | 2010-01-21 | University Of Pittsburgh Of The Commonwealth System Of Higher Education | Compounds for the treatment of pathologies associated with aging and degenerative disorders |
| WO2012112851A2 (en) * | 2011-02-18 | 2012-08-23 | University Of Pittsburgh - Of The Commonwealth System Of Higher Education | Targeted nitroxide agents |
Non-Patent Citations (5)
| Title |
|---|
| FRANTZ MARIE-CELINE ET AL.: "Synthesis of analogs of the radiation mitigator JP 4-039 and Visualization of BODIPY derivatives in mitochondria", ORG BIOMOL CHEM., vol. 11, no. 25, 7 July 2013 (2013-07-07), pages 4147 - 4153, XP055344247, Retrieved from the Internet <URL:https://www.ncbi.nlm.nih.gov/pmc/ articles/PMC3729477> * |
| KAVALA MIROSLAV: "Preparation and Spectroscopic, Magnetic, and Electrochemical Studies of Mono-/Biradical TEMPO Derivatives", JOURNAL OF ORGANIC. CHEMISTRY., vol. 78, no. 13, 3 June 2013 (2013-06-03), pages 6558 - 6569, XP055344254 * |
| NESBITT, KATHRYN M. ET AL.: "Pharmacological Mitigation of Tissue Damage During Brain Microdialysis", ANAL. CHEM., vol. 85, no. 17, 20 August 2013 (2013-08-20), pages 8173 - 8179, XP055344257, Retrieved from the Internet <URL:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3799822> * |
| NESBITT, KATHRYN M.: "Retrodialysis of Pharmacological Agents Mitigates Tissue Damage during Brain Microdialysis and Preserves Dopamine Activity in Surrounding Tissue.", DOCTORAL DISSERTATION, 22 June 2015 (2015-06-22), pages 1 - 149, XP055344263, Retrieved from the Internet <URL:http://d- scholarship.pitt.edu/24621> * |
| WEN-GUANG LI ET AL.: "The relationship between structure and antioxidative activity of piperidine nitroxides", JOURNAL OF PHARMACY AND PHARMACOLOGY, vol. 58, no. 7, 1 July 2006 (2006-07-01), pages 941 - 949, XP009105130, Retrieved from the Internet <URL:https://www.researchgate.net/ publication/6978637> * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11426419B2 (en) | Compositions and methods for the repair of myelin | |
| JP7470151B2 (en) | Use of chloroquine and clemizole compounds to treat inflammatory and cancerous conditions | |
| JP6564380B2 (en) | Compounds for treating prostate cancer | |
| ES2872335T3 (en) | 2,4-Thiazolidinedione derivatives in the treatment of central nervous system disorders | |
| EP2945947B1 (en) | Inhibiting the transient receptor potential a1 ion channel | |
| BRPI0708071A2 (en) | deuterated catecholamine derivatives and medicaments comprising said compounds | |
| US9649300B2 (en) | Inhibition of WNT, TGF beta and Hippo signaling pathways to treat cancer, organ fibrosis and metabolic disorders | |
| WO2010036395A2 (en) | Treatment of organophosphate exposure with tetrahydroindolone arylpiperazine compounds | |
| EP3187485A1 (en) | Dimethylphenylammonium long-chain compound, preparation, self-assembled structure and use thereof | |
| US20160264593A1 (en) | Protein phosphatase inhibitors that cross the blood brain barrier | |
| US8283375B2 (en) | 2, 6 xylidine derivatives for the treatment of pain | |
| JP7346571B2 (en) | Sugar-conjugated L-DOPA and/or DOPA decarboxylase inhibitors for the treatment of dopamine-responsive disorders | |
| WO2017004073A1 (en) | Bisnitroxides | |
| JP2020505388A (en) | Bicyclic compounds as caspase inhibitors | |
| US10441565B2 (en) | Conjugate of memantine and arctigenin, and composition and use thereof | |
| EP4056565A1 (en) | Liver targeting drug, pharmaceutical composition and use thereof | |
| EP2246055A1 (en) | The use of aryl piperazine derivatives in manufacturing medicants for treating pain | |
| US9394308B2 (en) | Inhibiting the transient receptor potential A1 ion channel | |
| EP3085698B1 (en) | Pain-related compound and medicinal composition | |
| US10759822B2 (en) | Brain-targeting prodrug for AMPA receptor synergist, and pharmaceutical applications thereof | |
| WO2021187314A1 (en) | Mitochondrial dysfunction improving agent | |
| US20210171553A1 (en) | Compositions useful in therapy of autophagy-related pathologies, and methods of making and using the same | |
| GR1010438B (en) | Farmaceutical compounds based on the association of an histamine antagonist and a hydrogen sulfide donor for the use in the treatment of pruritus | |
| BR112016022974B1 (en) | USE OF A COMPOUND OR MIXTURE OF 2,4-THIAZOLIDINEDIONE DERIVATIVES IN THE TREATMENT OF DISORDERS OF THE CENTRAL NERVOUS SYSTEM |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16818612 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16818612 Country of ref document: EP Kind code of ref document: A1 |