WO2016207656A1 - Methods of diagnosing bladder cancer - Google Patents
Methods of diagnosing bladder cancer Download PDFInfo
- Publication number
- WO2016207656A1 WO2016207656A1 PCT/GB2016/051903 GB2016051903W WO2016207656A1 WO 2016207656 A1 WO2016207656 A1 WO 2016207656A1 GB 2016051903 W GB2016051903 W GB 2016051903W WO 2016207656 A1 WO2016207656 A1 WO 2016207656A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mvps
- seq
- nos
- methylated
- identified
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/686—Polymerase chain reaction [PCR]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6827—Hybridisation assays for detection of mutation or polymorphism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6858—Allele-specific amplification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2523/00—Reactions characterised by treatment of reaction samples
- C12Q2523/10—Characterised by chemical treatment
- C12Q2523/125—Bisulfite(s)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/118—Prognosis of disease development
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/154—Methylation markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/16—Primer sets for multiplex assays
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/574—Immunoassay; Biospecific binding assay; Materials therefor for cancer
- G01N33/57407—Specifically defined cancers
Definitions
- the present invention relates to methods of diagnosing bladder cancer in a patient, involving determining the methylation status of Methylation Variable Positions (MVPs) in DNA from the patient and providing a diagnosis based on methylation status data.
- the invention also relates to methods of treating bladder cancer comprising providing a diagnosis of bladder cancer by the diagnostic methods defined herein followed by administering one or more anti-cancer agents to a patient.
- the invention also relates to methylation-discriminatory arrays comprising probes directed to the MVPs defined herein and kits comprising the arrays.
- Bladder cancer represents one of the most common malignancies in the western world, ranking as the 5th most common cancer in the United States and causing approximately 3% of all cancer-related deaths [1, 2].
- the foremost clinical sign at presentation is hematuria, and bladder cancer is detected in about 10% of all such cases investigated [3].
- Bladder cancer is more likely in older male patients, current or past smokers and patients exposed to industrial carcinogens [4]. Younger females with nonvisible hematuria are less likely to harbor bladder cancer and for these patients delay in detection of bladder cancer, following misdiagnosis of haematuria, is a frequent event [5]. Cystoscopy is the current gold standard for detecting bladder cancer and is an invasive, uncomfortable procedure requiring clinic or hospital attendance and posing a small but significant risk of infection [6-9].
- bladder cancer Each year in the UK, approximately 10,300 people are diagnosed with bladder cancer and 5,000 die from the disease. However, more than 100,000 cases per year are referred from primary care to urology haematuria clinics for cystoscopy and imaging. Bladder cancer is detected in only 10% of patients referred.
- NMIBC muscle invasive bladder cancer
- DNA methylation biomarkers in body fluids, including urine [15-23], plasma/serum [24-26], and sputum [27, 28], for the non-invasive detection of cancer.
- Changes in DNA methylation play a key role in malignant transformation, leading to the silencing of tumor-suppressor genes and overexpression of oncogenes [29].
- the ontogenic plasticity and relative stability of DNA methylation makes epigenetic changes ideal biomarkers for diagnosis.
- Detected protein biomarkers include human complement factor H-related protein,
- CEA carcinoembryonic antigen
- NMP22 nuclear mitotic apparatus protein 22
- WO2014042763 describes a nine -biomarker panel consisting of IL-8, MMP9, SDC1, CCL18,
- SERPINEl CD44, VEGF-A, CA9, and ANG for detection of protein in urine samples; a further nine-biomarker panel consisting of CA9, CCL18, MMP12, TMEM45A, MMP9, SEMA3D, ERBB2, CRH, and MXRA8; as well as a three-biomarker panel consisting of CCL18, CD44, and VEGF-A.
- DNA methylation biomarker assays for the detection of bladder cancer have been centered on the analysis of only a small number of loci, in part due to technological limitations and derivation of targets with cancer specificity [1 1-19].
- reported sensitivities and specificities are high relative to established assays, but would fail to attain performance characteristics achieved by cystoscopy.
- Methylation markers for bladder cancer previously studied include DAPK, BCL2, TERT, TWIST 1, NID2, RARbeta, E-cadherin and pi 6.
- International patent application publication WO2013/144362 describes a diagnostic assay for bladder cancer involving detecting methylation of the promoter of the ECRG4 and/or the ITIH5 gene.
- US patent application publication US2013224738 describes a diagnostic assay for bladder cancer involving assessing the methylation status of genes consisting of BCL2, CDKN2A and NID2.
- Diagnostic methods which can detect bladder cancer from a biological sample, particularly a voided urine sample, with robust and high sensitivity and specificity, and which have the potential to reduce the need for cystoscopy in patients referred with haematuria and in patients undergoing surveillance for disease recurrence. Avoiding cystoscopy will reduce the cost of bladder cancer management and positively impact on patient wellbeing, reducing both the number of hospital visits and the inconvenience of an invasive investigation.
- the invention provides the following:
- the invention provides a method of diagnosing bladder cancer in an individual comprising:
- the group of MVPs may comprises at least 40 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG], and wherein bladder cancer is diagnosed when at least 25 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] are methylated.
- the group of MVPs may comprise at least 50 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG], or may comprise at least 100 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG].
- the group of MVPs may comprise all 150 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG].
- cancer may be diagnosed in the individual when at least 40 of the MVPs selected from the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] are methylated, or when at least 50 of the MVPs selected from the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] are methylated, or when at least 100 of the MVPs are methylated, or when all 150 MVPs are methylated.
- the MVPs determined to be methylated may include the MVPs identified in SEQ ID NOS 1 to 3 and denoted by [CG], or may include the MVPs identified in SEQ ID NOS 1 to 5 and denoted by [CG], or may include the MVPs identified in SEQ ID NOS 1 to 10 and denoted by [CG], or may include the MVPs identified in SEQ ID NOS 1 to 40 and denoted by [CG].
- the group of MVPs may comprise all 150 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG], wherein bladder cancer is diagnosed in the individual when at least 40 of the MVPs selected from the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] are methylated, and wherein the MVPs determined to be methylated include the MVPs identified in SEQ ID NOS 1 to 10 and denoted by [CG].
- the step of determining whether each one the MVPs is methylated may comprise bisulphite converting the DNA.
- the step of determining whether each one the MVPs is methylated may comprise:
- an amplification step may be performed, wherein loci comprising each MVP are amplified.
- Amplification may be performed by PCR.
- a capturing step may be performed before the sequencing or hybridization steps.
- the capturing step may involve binding polynucleotides comprising the MVP loci to binding molecules specific to the MVP loci and collecting complexes comprising MVP loci and binding molecules; and wherein:
- the capturing step occurs before the step of bisulphite converting the DNA
- the capturing step occurs after the step of bisulphite converting the DNA but before the amplification or hybridization steps;
- the capturing step occurs after the step of bisulphite converting the DNA and after the amplification step.
- the binding molecules may be oligonucleotides specific for each MVP, preferably DNA or RNA molecules each comprising a sequence which is
- the binding molecule may be coupled to a purification moiety.
- the purification moiety may comprise a first purification moiety and the step of collecting complexes comprising MVP loci and binding molecules may comprise binding the first purification moiety to substrates comprising a second purification moiety, wherein first and second purification moieties form an interaction complex.
- the first purification moiety may be biotin and the second purification moiety may be streptavidin; or the first purification moiety may be streptavidin and the second purification moiety may be biotin.
- the step of amplifying loci comprising MVPs may comprise the use of primers which are independent of the methylation status of the MVP.
- the step of amplifying loci comprising MVPs may be performed by
- the biological sample obtained from the individual may be a sample of urine, blood, serum, plasma or cell-free DNA.
- the method may achieve a ROC sensitivity of 95% or greater and a ROC specificity of 90% or greater; preferably a ROC sensitivity of 96% and a ROC specificity of 97%, preferably a ROC AUC of 95% or greater, preferably 98%o.
- the method may achieve a negative predictive value (NPV) of 95% or greater, preferably 97%.
- NPV negative predictive value
- the step of diagnosing bladder cancer in the individual may further comprise:
- the invention additionally provides a method of treating bladder cancer in an individual comprising:
- the invention additionally provides a method of treating bladder cancer in an individual comprising:
- the invention additionally provides a method of treating bladder cancer in an individual comprising:
- the invention additionally provides a method of treating bladder cancer in an individual comprising administering one or more bladder cancer treatments to the individual, wherein the individual has been diagnosed with bladder cancer by steps comprising:
- the invention additionally provides a method of diagnosing bladder cancer in an individual comprising:
- the cancer may be a non-muscle invasive bladder cancer (NMIBC).
- NMIBC non-muscle invasive bladder cancer
- MIBC muscle invasive bladder cancer
- the invention additionally provides an array capable of discriminating between methylated and non-methylated forms of MVPs; the array comprising oligonucleotide probes specific for a methylated form of each MVP in a MVP panel and oligonucleotide probes specific for a non-methylated form of each MVP in the panel; wherein the panel consists of at least 25 MVPs selected from the MVPs identified in SEQ ID NOS 1 to 150.
- the array is not an Infinium HumanMethylation450 BeadChip array.
- the number of MVP-specific oligonucleotide probes of the array is less than 482,421, preferably 482,000 or less, 480,000 or less, 450,000 or less, 440,000 or less, 430,000 or less, 420,000 or less, 410,000 or less, or 400,000 or less.
- the panel may consist of at least 40 MVPs selected from the MVPs identified in SEQ ID NOS 1 to 150; preferably at least 50 MVPs, at least 60 MVPs, at least 70 MVPs, at least 80 MVPs, at least 90 MVPs, at least 100 MVPs, at least 110 MVPs, at least 120 MVPs, at least 130 MVPs, at least 140 MVPs, at least 145 MVPs, or all 150 MVPs identified in SEQ ID NOS 1 to 150.
- the panel may include the MVPs defined by SEQ ID NOS 1 to 3, or the MVPs defined by SEQ ID NOS 1 to 5, or the MVPs defined by SEQ ID NOS 1 to 10, or the MVPs defined by SEQ ID NOS 1 to 20, or the MVPs defined by SEQ ID NOS 1 to 30, or the MVPs defined by SEQ ID NOS 1 to 40, or the MVPs defined by SEQ ID NOS 1 to 50, or the MVPs defined by SEQ ID NOS 1 to 60, or the MVPs defined by SEQ ID NOS 1 to 70, or the MVPs defined by SEQ ID NOS 1 to 80, or the MVPs defined by SEQ ID NOS 1 to 90, or the MVPs defined by SEQ ID NOS 1 to 100, or the MVPs defined by SEQ ID NOS 1 to 100, or the MVPs defined by SEQ ID NOS 1 to 120, or the MVPs defined by SEQ ID NOS 1 to 130, or the MVPs defined by SEQ ID NOS 1 to 140, or the MVPs defined by SEQ ID NOS 1 to 3
- the panel may include all MVPs defined by SEQ ID NOS 1 to 150.
- the array may further comprise one or more oligonucleotides comprising a MVP selected from any of the MVPs defined in SEQ ID NOS 1 to 150, wherein the one or more oligonucleotides are hybridized to
- the one or more oligonucleotides may comprise at least 20 MVPs selected from the MVPs identified in SEQ ID NOS 1 to 150; preferably at least 50 MVPs, at least 60 MVPs, at least 70 MVPs, at least 80 MVPs, at least 90 MVPs, at least 100 MVPs, at least 110 MVPs, at least 120 MVPs, at least 130 MVPs, at least 140 MVPs, at least 145 MVPs, or all 150 MVPs identified in SEQ ID NOS 1 to 150.
- the one or more oligonucleotides may comprise the MVPs defined by SEQ ID NOS 1 to 10, or the MVPs defined by SEQ ID NOS 1 to 20, or the MVPs defined by SEQ ID NOS 1 to 30, or the MVPs defined by SEQ ID NOS 1 to 40, or the MVPs defined by SEQ ID NOS 1 to 50, or the MVPs defined by SEQ ID NOS 1 to 60, or the MVPs defined by SEQ ID NOS 1 to 70, or the MVPs defined by SEQ ID NOS 1 to 80, or the MVPs defined by SEQ ID NOS 1 to 90, or the MVPs defined by SEQ ID NOS 1 to 100, or the MVPs defined by SEQ ID NOS 1 to 110, or the MVPs defined by SEQ ID NOS 1 to 120, or the MVPs defined by SEQ ID NOS 1 to 130, or the MVPs defined by SEQ ID NOS 1 to 140, or the MVPs defined by SEQ ID NOS 1 to 150.
- the one or more oligonucleotides may comprise all MVPs defined by SEQ ID NOS 1 to 150.
- Arrays as described above may be obtainable by hybridizing to an array as described above a group of oligonucleotides each comprising a different MVP selected from any of the MVPs defined in SEQ ID NOS 1 to 150, and wherein the group comprises at least 40 oligonucleotides.
- the group may comprise at least 50 oligonucleotides.
- the group may comprise at least 60, at least 70, at least 80, at least 90, at least 100, at least 110, at least 120, at least 130, at least 140, at least 145, or at least 150
- the group may comprise at least 40 oligonucleotides comprising the MVPs defined by SEQ ID NOS 1 to 20, or wherein the group may comprise at least 50 oligonucleotides comprising the MVPs defined by SEQ ID NOS 1 to 50, or wherein the group may comprise at least 60 oligonucleotides comprising the MVPs defined by SEQ ID NOS 1 to 60, or wherein the group may comprise at least 70 oligonucleotides comprising the MVPs defined by SEQ ID NOS 1 to 70, or wherein the group may comprise at least 80 oligonucleotides comprising the MVPs defined by SEQ ID NOS 1 to 80, or wherein the group may comprise at least 90 oligonucleotides comprising the MVPs defined by SEQ ID NOS 1 to 90, or wherein the group may comprise at least 100 oligonucleotides comprising the MVPs defined by SEQ ID NOS 1 to 100, or wherein the group may comprise at least 110 oligonu
- the invention also provides a process for making the hybridized array as defined above, comprising contacting an array as defined above with a group of
- oligonucleotides each comprising a different MVP selected from any of the MVPs defined in SEQ ID NOS 1 to 150, and wherein the group comprises at least 25 oligonucleotides.
- the invention also provides a process for making a hybridized array as defined above, comprising contacting an array as defined above with a group of
- the invention also provides a kit comprising any of the arrays described above.
- the kit may further comprise a DNA modifying regent that is capable of modifying a non-methylated cytosine in a MVP dinucleotide but is not capable of modifying a methylated cytosine in a MVP dinucleotide, optionally wherein the dinucleotide is CpG.
- the DNA modifying regent may be a bisulphite reagent.
- FIG. 1 Heatmap of 9786 MVPs (1746 hypermethylated MVPs, 8040 hypomethylated MVPs) between bladder cancer (red) and 30 normal urothelium (blue).
- Figure 3. Heat map of the 150 loci involved in the UroMark assay for normal urothelium (Blue) bladder cancer (Red) compared with the predicted (Light blue/light red) and actual (Blue/Red) status of bladder cancer.
- FIG. 4 MDS plots for bladder tumour and normal urothelium based on the methylation state of 150 loci in bladder cancer samples from UCL and TCGA Bladder Cancer.
- the MDS (Multidimensional scaling) plot represents the dissimilarly of phenotypes based on the methylation state of the 150 loci with the panel, and clearly shows that the 150 marker can accurately separate tumour from normal bladder.
- Axis represent the Euclidean distance between samples.
- Figure 5. ROC plot for UroMark model for the detection of bladder cancer in urine.
- FIG. 6 ROC plots for top performing (A) 3, (B) 5 and (C) 10 marker panels.
- Top 3 MVPs are listed as SEQ ID NOs: 1 to 3
- Top 5 MVPs are listed as SEQ ID NOs: 1 to 5
- Top 10 MVPs are listed as SEQ ID NOs: 1 to 10, all in rank order (see Table 1).
- FIG. 7 ROC plot for UroMark model for the detection of bladder cancer from 176 unique urine samples (98 non-cancer urines and 78 cancer urines).
- FIG. 8 Comparison of DNA quality (concentration, purity and integrity) from patients' urine.
- FIG. 9 ROC plot for UroMark model for the detection of bladder cancer from Validation Cohort 2 - 96 unique urine samples (64 non-cancer urines and 32 cancer urines).
- Figure 10. ROC plot for UroMark model for the detection of bladder cancer from Validation Cohort 3 - 92 urine samples (65 non-cancer urines and 27 cancer urines).
- bladder cancers represent one of the most prevalent groups of cancers in the western world.
- Transitional cell carcinoma is the most common type, and accounts for approximately 90% of bladder cancers.
- Transitional cell carcinomas arise from the transitional epithelium, which is a tissue lining the inner surface of the bladder.
- the remaining 10% of bladder cancers are mainly comprised of squamous cell carcinoma, adenocarcinoma, sarcoma, and small cell carcinoma.
- Squamous cell carcinoma also arises from epithelial tissue, from squamous cells. These are thin, flat cells found in the most superficial epithelial layer.
- Adenocarcinomas form from epithelial cells having glandular characteristics and/or origin.
- Sarcomas derive from cells of mesenchymal origin, such as the cells of the fat and muscle layers of the bladder.
- Small cell carcinomas have a rapid doubling time and are capable of earlier metastases, making them particularly aggressive.
- Bladder cancers may also be classified as non-muscle invasive bladder cancer (NMIBC) and muscle invasive bladder cancer (MIBC).
- NMIBC non-muscle invasive bladder cancer
- MIBC muscle invasive bladder cancer
- the diagnostic and treatment methods described herein are capable of positively identifying malignant cells of all classifications of bladder cancer.
- any of the methods described herein may be used to diagnose transitional cell carcinoma of the bladder, squamous cell carcinoma of the bladder, adenocarcinoma of the bladder, sarcoma of the bladder, small cell carcinoma of the bladder, metastatic bladder cancer, leiomyosarcoma (a tumor arising from smooth muscle), lymphoma (a tumor that usually arises in the lymph nodes), malignant melanoma (a tumor that usually arises from the skin) and large cell neuroendocrine carcinoma.
- Primary forms and recurrent forms of bladder cancer are included.
- the cancer to be diagnosed or treated as described herein may be a urothelial cell cancer.
- the cancer may be cancer of the ureter, urethra or renal pelvis.
- the most preferred patient type to which the diagnostic assays described herein are applicable are humans.
- the diagnostic assays described herein may also be used to identify bladder cancer in a non-human animal.
- non-human animals may contain tissue derived from humans, e.g. xenografts.
- diagnostic assays may be used to diagnose human bladder cancer in an animal model of human bladder cancer.
- Typical non-human animals to which the diagnostic assays described herein are applicable are rodents such as rats or mice.
- MVPs Methylation Variable Positions
- Methylation of DNA is a recognised form of epigenetic modification which has the capability of altering the expression of genes and other elements such as
- methylation may have the effect of e.g. silencing tumor suppressor genes and/or increasing the expression of oncogenes. Other forms of dysregulation may occur as a result of methylation.
- Methylation of DNA occurs at discrete loci which are predominately dinucleotide consisting of a CpG motif, but may also occur at CHH motifs (where H is A, C, or T).
- a methyl group is added to the fifth carbon of cytosine bases to create methylcytosine.
- Methylation can occur throughout the genome and is not limited to regions with respect to an expressed sequence such as a gene. Methylation typically, but not always, occurs in a promoter or other regulatory region of an expressed sequence.
- a Methylation Variable Position as defined herein is any dinucleotide locus which may show a variation in its methylation status between phenotypes, i.e. between tumour and normal tissue.
- An MVP is preferably a CpG or a CHH
- An MPV as defined herein is not limited to the position of the locus with respect to a corresponding expressed sequence.
- an assessment of DNA methylation status involves analysing the presence or absence of methyl groups in DNA, for example methyl groups on the 5 th position of one or more cytosine nucleotides.
- the methylation status of one or more cytosine nucleotides present as a CpG dinucleotide (where C stands for
- Cytosine, G for Guanine and p for the phosphate group linking the two is assessed.
- assessing the methylation status of an MVP or determining whether an MVP is methylated it is meant that a determination is made as to whether an MVP was methylated or unmethylated in the starting sample of DNA obtained from the individual prior to subsequent processing.
- An MVP is herein defined as methylated if one or more alleles of that MVP in a sample of genomic DNA from the patient is determined to possess one or more methylated CpG dinucleotide loci.
- the MVPs determined to be methylated are methylated relative to normal urothelium control and/or whole blood control.
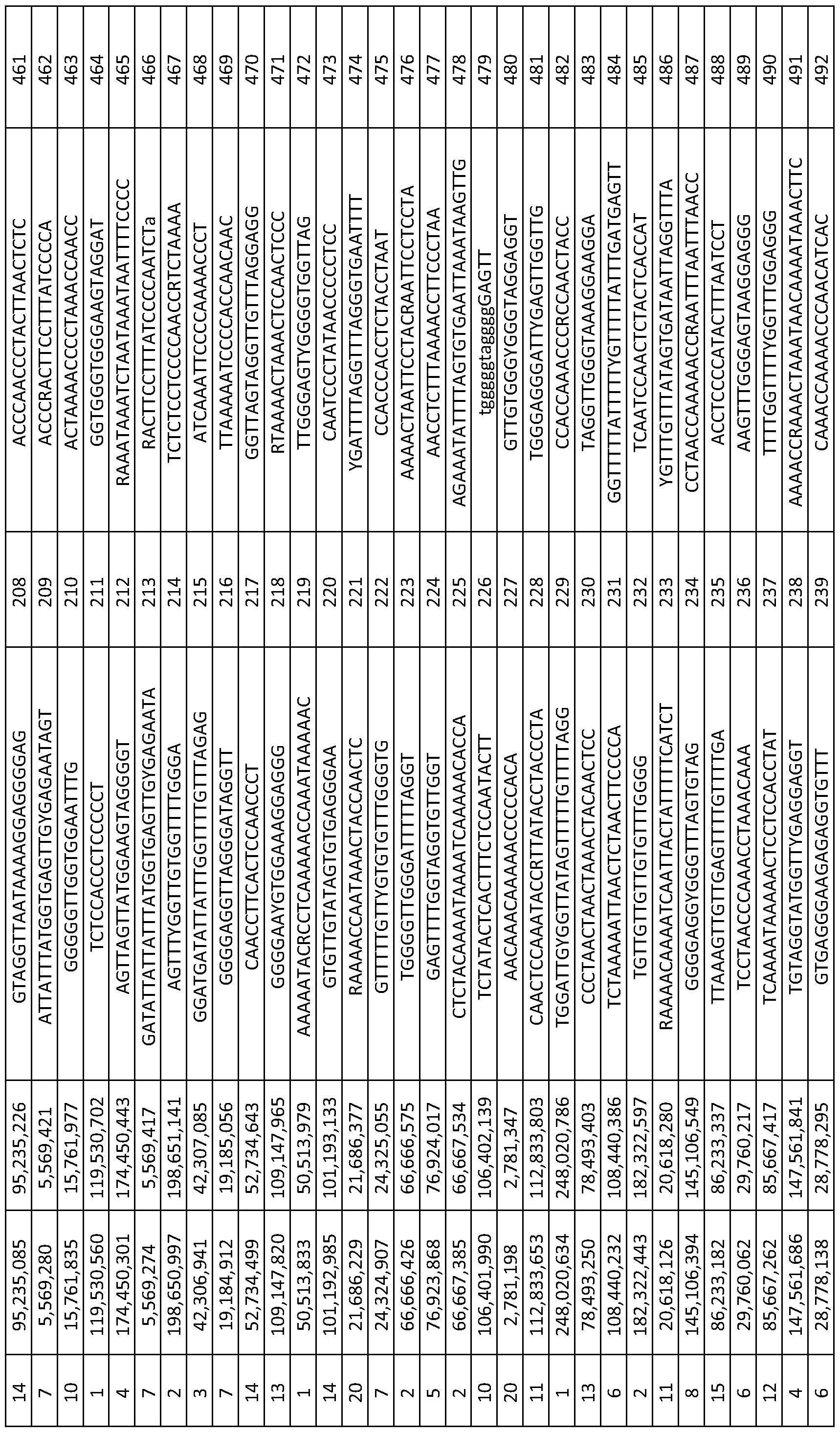
- MVPs useful for diagnostic purposes are set forth in Table 1 and are identified by SEQ ID number, as well as Illumina ID number (limn ID).
- Exemplary primers for amplifying the defined MVPs are set forth in Table 2 and are also identified by SEQ ID number. Identification and assessment of Methylation Variable Position (MVP) status
- MVPs Methylation Variable Positions
- Methyl groups are lost from a starting DNA molecule during conventional in vitro handling steps such as PCR.
- techniques for the detection of methyl groups commonly involve the preliminary treatment of DNA prior to subsequent processing, in a way that preserves the methylation status information of the original DNA molecule.
- Such preliminary techniques involve three main categories of processing, i.e. bisulphite modification, restriction enzyme digestion and affinity-based analysis. Products of these techniques can then be coupled with sequencing or array- based platforms for subsequent identification or qualitative assessment of MVP methylation status.
- cytosine bases can be detected by a variety of techniques. For example, primers specific for unmethylated versus methylated DNA can be generated and used for PCR- based identification of methylated CpG dinucleotides. A separation/capture step may be performed, e.g. using binding molecules such as complementary oligonucleotide sequences. Standard and next-generation DNA sequencing protocols can also be used.
- methylation-sensitive enzymes can be employed which digest or cut only in the presence of methylated DNA. Analysis of resulting fragments is commonly carried out using microarrays.
- binding molecules such as anti-5- methylcytosine antibodies are commonly employed prior to subsequent processing steps such as PCR and sequencing.
- any suitable method can be employed.
- Preferred methods involve bisulphite treatment of DNA, including amplification of the identified MVP loci for methylation specific PCR and/or sequencing and/or assessment of the methylation status of target loci using methylation-discriminatory microarrays.
- Amplification of MVP loci can be achieved by a variety of approaches.
- MVP loci are amplified using PCR.
- MVP may also be amplified by other techniques such as multiplex ligation-dependent probe amplification (MLPA).
- MLPA multiplex ligation-dependent probe amplification
- PCR-based approaches may be used.
- methylation-specific primers may be hybridized to DNA containing the MVP sequence of interest. Such primers may be designed to anneal to a sequence derived from either a methylated or non-methylated MVP locus.
- a PCR reaction is performed and the presence of a subsequent PCR product indicates the presence of an annealed MVP of identifiable sequence.
- DNA is bisulphite converted prior to amplification.
- MSP methylation specific PCR
- PCR primers may anneal to the MVP sequence of interest independently of the methylation status, and further processing steps may be used to determine the status of the MVP.
- Assays are designed so that the MVP site(s) are located between primer annealing sites. This method scheme is used in techniques such as bisulphite genomic sequencing [54], COBRA [55], Ms-SNuPE [56]. In such methods, DNA can be bisulphite converted before or after amplification.
- small-scale PCR approaches are used. Such approaches commonly involve mass partitioning of samples (e.g. digital PCR). These techniques offer robust accuracy and sensitivity in the context of a highly miniaturised system (pico-liter sized droplets), ideal for the subsequent handling of small quantities of DNA obtainable from the potentially small volume of cellular material present in biological samples, particularly urine samples.
- a variety of such small-scale PCR techniques are widely available. For example, microdroplet-based PCR instruments are available from a variety of suppliers, including RainDance Technologies, Inc. (Billerica, MA;
- Microarray platforms may also be used to carry out small-scale PCR. Such platforms may include microfiuidic network-based arrays e.g. available from Fluidigm Corp.
- amplified PCR products may be coupled to subsequent analytical platforms in order to determine the methylation status of the MVPs of interest.
- the PCR products may be directly sequenced to determine the presence or absence of a methyl cytosine at the target MVP or analysed by array-based techniques.
- any suitable sequencing techniques may be employed to determine the sequence of target DNA.
- second generation large numbers of DNA molecules are sequenced in parallel. Typically, tens of thousands of molecules are anchored to a given location at high density and sequences are determined in a process dependent upon DNA synthesis. Reactions generally consist of successive reagent delivery and washing steps, e.g. to allow the incorporation of reversible labelled terminator bases, and scanning steps to determine the order of base incorporation.
- Array-based systems of this type are available commercially e.g. from Illumina, Inc. (San Diego, CA;
- Third generation techniques are typically defined by the absence of a requirement to halt the sequencing process between detection steps and can therefore be viewed as real-time systems.
- the base-specific release of hydrogen ions which occurs during the incorporation process, can be detected in the context of microwell systems (e.g. see the Ion Torrent system available from Life Technologies; http://www.lifetechnologies.com/).
- PPi pyrophosphate
- nanopore technologies DNA molecules are passed through or positioned next to nanopores, and the identities of individual bases are determined following movement of the DNA molecule relative to the nanopore. Systems of this type are available commercially e.g.
- a DNA polymerase enzyme is confined in a "zero-mode waveguide" and the identity of incorporated bases are determined with florescence detection of gamma-labeled phosphonucleotides (see e.g. Pacific Biosciences; http://www.pacificbiosciences.com/).
- sequencing steps may be omitted.
- amplified PCR products may be applied directly to hybridization arrays based on the principle of the annealing of two complementary nucleic acid strands to form a double-stranded molecule.
- Hybridization arrays may be designed to include probes which are able to hybridize to amplification products of an MVP and allow discrimination between methylated and non-methylated loci.
- probes may be designed which are able to selectively hybridize to an MVP locus containing thymine, indicating the generation of uracil following bisulphite conversion of an unmethylated cytosine in the starting template DNA.
- probes may be designed which are able to selectively hybridize to an MVP locus containing cytosine, indicating the absence of uracil conversion following bisulphite treatment. This corresponds with a methylated MVP locus in the starting template DNA.
- Detection systems may include, e.g. the addition of fluorescent molecules following a methylation status-specific probe extension reaction. Such techniques allow MVP status determination without the specific need for the sequencing of MVP amplification products.
- array-based discriminatory probes may be termed methylation-specific probes.
- Any suitable methylation-discriminatory microarrays may be employed to assess the methylation status of the MVPs described herein.
- a preferred methylation- discriminatory microarray system is provided by Illumina, Inc. (San Diego, CA;
- BeadChip array system may be used to assess the methylation status of diagnostic MVPs for bladder cancer as described herein.
- the array comprises beads to which are coupled oligonucleotide probes specific for DNA sequences corresponding to the unmethylated form of an MVP, as well as separate beads to which are coupled oligonucleotide probes specific for DNA sequences corresponding to the methylated form of an MVP.
- Candidate DNA molecules are applied to the array and selectively hybridize, under appropriate conditions, to the oligonucleotide probe corresponding to the relevant epigenetic form.
- corresponding genomic DNA will selectively attach to the bead comprising the methylation-specific oligonucleotide probe, but will fail to attach to the bead
- the bladder cancer-specific diagnostic MVP biomarkers defined herein were initially identified using the Illumina Infinium HumanMethylation450 BeadChip array system, the same chip system can be used to interrogate those same MVPs in the diagnostic assays described herein.
- Alternative or customised arrays could, however, be employed to interrogate the bladder cancer-specific diagnostic MVP biomarkers defined herein, provided that they comprise means for interrogating all MVPs for a given method, as defined herein.
- DNA containing MVP sequences of interest may be hybridized to microarrays and then subjected to DNA sequencing to determine the status of the MVP as described above.
- sequences corresponding to MVP loci may also be subjected to an enrichment process.
- DNA containing MVP sequences of interest may be captured by binding molecules such as oligonucleotide probes complementary to the MVP target sequence of interest.
- Sequences corresponding to MVP loci may be captured before or after bisulphite conversion or before or after amplification. Probes may be designed to be complementary to bisulphite converted DNA. Captured DNA may then be subjected to further processing steps to determine the status of the MVP, such as DNA sequencing steps.
- Capture/separation steps may be custom designed. Alternatively a variety of such techniques are available commercially, e.g. the SureSelect target enrichment system available from Agilent Technologies (http://www.agilent.com/home). In this system biotinylated "bait” or “probe” sequences (e.g. RNA) complementary to the DNA containing MVP sequences of interest are hybridized to sample nucleic acids.
- Streptavidin-coated magnetic beads are then used to capture sequences of interest hybridized to bait sequences. Unbound fractions are discarded. Bait sequences are then removed (e.g. by digestion of RNA) thus providing an enriched pool of MVP target sequences separated from non-MVP sequences.
- template DNA is subjected to bisulphite conversion and target loci are then amplified by small-scale PCR such as microdroplet PCR using primers which are independent of the methylation status of the MVP.
- small-scale PCR such as microdroplet PCR using primers which are independent of the methylation status of the MVP.
- samples are subjected to a capture step to enrich for PCR products containing the target MVP, e.g. captured and purified using magnetic beads, as described above.
- PCR reaction is carried out to incorporate DNA sequencing barcodes into MVP-containing amplicons. PCR products are again purified and then subjected to DNA sequencing and analysis to determine the presence or absence of a methylcytosine at the target genomic MVP [31].
- the MVP biomarker loci defined herein are identified e.g. by Illumina® identifiers (IlmnID). These MVP loci identifiers refer to individual MVP sites used in the commercially available Illumina® Infinium Human Methylation450 BeadChip kit. The identity of each MVP site represented by each MVP loci identifier is publicly available from the Illumina, Inc. website under reference to the MVP sites used in the
- Illumina® has developed a method to consistently designate MVP/CpG loci based on the actual or contextual sequence of each individual MVP/CpG locus.
- Illumina® has developed a consistent and deterministic MVP loci database to ensure uniformity in the reporting of methylation data.
- the Illumina® method takes advantage of sequences flanking a MVP locus to generate a unique MVP locus cluster ID. This number is based on sequence information only and is unaffected by genome version.
- Illumina's standardized nomenclature also parallels the TOP/BOT strand nomenclature (which indicates the strand orientation) commonly used for single nucleotide polymorphism (SNP) designation.
- SNP single nucleotide polymorphism
- Illumina® Identifiers for the Infinium Human Methylation450 BeadChip system are also available from public repositories such as Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/).
- GEO Gene Expression Omnibus
- An MVP as defined herein thus refers to the CG dinucleotide motif identified in relation to each SEQ ID NO. and Illumina Identifier (limn ID) as listed in Table 1, wherein the cytosine base of the dinucleotide (noted in bold and square brackets in the sequences listed at Table 1) may (or may not) be modified.
- determining the methylation status of a CpG defined by or identified in a given SEQ ID NO., or determining whether such a CpG is methylated it is meant that a determination is made as to whether the cytosine of the CG dinucleotide motif identified in bold and in square brackets in a sequence shown in Table 1 is methylated or not at one or more loci in the sample of DNA from the individual, accepting that variation in the sequence upstream and downstream of any given CpG may exist due to sequencing errors or variation between individuals.
- the invention provides a method of diagnosing bladder cancer in an individual comprising:
- the group of MVPs may comprise 26 or more of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG]; or the group may comprise 27 or more, 28 or more, 29 or more, 30 or more, 31 or more, 32 or more, 33 or more, 34 or more, 35 or more, 36 or more, 37 or more, 38 or more, 39 or more, 40 or more, 41 or more, 42 or more, 43 or more, 44 or more, 45 or more, 46 or more, 47 or more, 48 or more, 49 or more, 50 or more, 51 or more, 52 or more, 53 or more, 54 or more, 55 or more, 56 or more, 57 or more, 58 or more, 59 or more, 60 or more, 61 or more, 62 or more, 63 or more, 64 or more, 65
- bladder cancer may be diagnosed when at least 25 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] are methylated.
- Bladder cancer may be diagnosed when 26 or more of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] are methylated; or when 27 or more, 28 or more, 29 or more, 30 or more, 31 or more, 32 or more, 33 or more, 34 or more, 35 or more, 36 or more, 37 or more, 38 or more, 39 or more, 40 or more, 41 or more, 42 or more, 43 or more, 44 or more, 45 or more, 46 or more, 47 or more, 48 or more, 49 or more, 50 or more, 51 or more, 52 or more, 53 or more, 54 or more, 55 or more, 56 or more, 57 or more, 58 or more, 59 or more, 60 or more, 61 or more, 62 or more, 63 or more, 64 or more, 65 or more, 66 or more, 67 or more
- bladder cancer may be diagnosed when 40 or more of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] are methylated.
- Bladder cancer may also be diagnosed when 50 or more, 60 or more, 70 or more or 80 or more, 90 or more or 100 or more of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] are methylated.
- the MVPs determined to be methylated may include the MVPs identified in SEQ ID NOS 1 to 3 and denoted by [CG], or may include the MVPs identified in SEQ ID NOS 1 to 5 and denoted by [CG], or may include the MVPs identified in SEQ ID NOS 1 to 10 and denoted by [CG], or may include the MVPs identified in SEQ ID NOS 1 to 20 and denoted by [CG], or may include the MVPs identified in SEQ ID NOS 1 to 30 and denoted by [CG], or may include the MVPs identified in SEQ ID NOS 1 to 40 and denoted by [CG], or may include the MVPs identified in SEQ ID NOS 1 to 50 and denoted by [CG], or may include the MVPs identified in SEQ ID NOS 1 to 60 and denoted by [CG], or may include the MVPs identified in SEQ ID NOS 1 to 70 and denoted by [CG], or may include the MVPs identified in SEQ ID NOS 1 to 80 and denoted by [CG],
- the group of MVPs (i.e. those MVPs the methylation status of which are to be determined) may comprises all 150 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG], and in this method bladder cancer is diagnosed in the individual when at least 40 of the MVPs selected from the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] are methylated, and in this method the MVPs determined to be methylated include the MVPs identified in SEQ ID NOS 1 to 10 and denoted by [CG].
- DNA sequences and in primer design for the purposes of methylation-specific analyses are generally available and have been described previously [57, 58, 59].
- MVP methylation status assays described herein may be defined using standard receiver operating characteristic (ROC) statistical analysis [52].
- ROC receiver operating characteristic
- a bladder cancer diagnostic assay in accordance with the invention described herein can achieve a ROC sensitivity of 90%> or greater, 91%> or greater, 92% or greater, 93%> or greater, 94%> or greater, 95% or greater, 96%> or greater, 97% or greater, 98%> or greater or 99%.
- ROC sensitivity may be 100%.
- Diagnostic assays in accordance with the invention can achieve a ROC specificity of 90% or greater, 91% or greater, 92% or greater, 93% or greater, 94% or greater, 95% or greater, 96% or greater, 97% or greater, 98% or greater or 99%.
- ROC specificity may be 100%. Diagnostic assays in accordance with the invention may have an associated combination of ROC sensitivity and ROC specificity values wherein the combination is any one of the above-listed sensitivity values and any one of the above-listed specificity values, provided that the sensitivity value is equal to or less than the specificity value.
- the ROC specificity may be 100% and the ROC sensitivity may be 90% or greater, 91%> or greater, 92% or greater, 93% or greater, 94%> or greater, 95% or greater, 96% or greater, 97% or greater, 98% or greater, 99% or 100%.
- the ROC specificity may be 99% and the ROC sensitivity may be 90% > or greater, 91%o or greater, 92% or greater, 93% or greater, 94% or greater, 95% or greater, 96% or greater, 97% or greater, 98% or 99%.
- the ROC specificity may be 98% and the ROC sensitivity may be 90% or greater, 91% or greater, 92% or greater, 93% or greater, 94% or greater, 95% or greater, 96% or greater, 97% or 98%.
- the ROC specificity may be 97% and the ROC sensitivity may be 90% or greater, 91% or greater, 92% or greater, 93% or greater, 94% or greater, 95% or greater, 96% or 97%.
- the ROC specificity may be 96% and the ROC sensitivity may be 90% or greater, 91% or greater, 92% or greater, 93% or greater, 94% or greater, 95% or 96%.
- the ROC specificity may be 95% and the ROC sensitivity may be 90% or greater, 91% or greater, 92% or greater, 93% or greater, 94%o or 95%.
- the ROC specificity may be 94% and the ROC sensitivity may be 90% or greater, 91% or greater, 92% or greater, 93% or 94%.
- the ROC specificity may be 93% and the ROC sensitivity may be 90% or greater, 91% or greater, 92% or 93%.
- the ROC specificity may be 92% and the ROC sensitivity may be 90% or greater, 91% or 92%.
- the ROC specificity may be 91% and the ROC sensitivity may be 90% or 91%.
- the ROC specificity may be 90% and the ROC sensitivity may be 90%.
- the assay may achieve a ROC sensitivity of 95% or greater and a ROC specificity of 90%o or greater; preferably a ROC sensitivity of 96% and a ROC specificity of 97%.
- ROC plots corresponding to example methods representative of the diagnostic methods defined herein are presented at Figures 5 and 7, demonstrating the extraordinar sensitivity and selectivity of the MVP -based assays. This contrasts with assays conducted with smaller panels of biomarkers, which have been described previously. Thus, comparative data demonstrate the superior predictive power of the assays defined herein.
- a further metric which can be employed to classify the accuracy of the MVP- based assays is ROC AUC.
- AUC area under the curve of a ROC plot
- the AUC score for the ROC plot will be 0.5.
- the number of true positives will be 100% and the number of false positives will be 0%.
- the AUC score for the ROC plot will be 1.
- a bladder cancer diagnostic assay in accordance with the invention can achieve a ROC AUC of 0.90 or greater, 0.91 or greater, 0.92 or greater, 0.93 or greater, 0.94 or greater, 0.95 or greater, 0.96 or greater, 0.97 or greater, 0.98 or greater, 0.99 or 1.
- the diagnostic assay can achieve a ROC AUC of 0.98 or greater.
- Bladder cancer diagnostic tests based on the MVP methylation status assays described herein may also be characterised using a Negative Predictive Value (NPV) metric.
- NPV Negative Predictive Value
- a bladder cancer diagnostic assay in accordance with the invention described herein can achieve an NPV of 90% or greater, 91 > or greater, 92% or greater, 93% or greater, 94% or greater, 95% or greater, 96% or greater, 97% or greater, 98% or greater or 99% or 100%.
- the bladder cancer diagnostic assays described herein may be performed on any suitable biological material obtained from the patient.
- Preferred biological material is urine.
- samples of bladder tissue e.g. obtained via biopsy or aspirates, or obtained from preserved samples ⁇ e.g. cryopreserved material, tissue sections etc.
- Samples of biological material may also include solid tissue samples, aspirates, samples of biological fluids, blood, serum, plasma, ascitic fluid, lymph, peripheral blood, cerebrospinal fluid, fine needle aspirate, saliva, sputum, bone marrow, skin, epithelial samples (including buccal, cervical or vaginal epithelia) or other tissue derived from the ectoderm, vaginal fluid, semen etc.
- Tissue scrapes may include biological material from e.g. buccal, oesophageal, bladder, vaginal, urethral or cervical scrapes.
- the cells of the sample may comprise inflammatory cells, such as lymphocytes.
- any of the assays and methods described herein may involve providing a biological sample from the patient as the source of patient DNA for methylation analysis.
- any of the assays and methods described herein may involve obtaining patient DNA from a biological sample which has previously been obtained from the patient.
- any of the assays and methods described herein may involve obtaining a biological sample from the patient as the source of patient DNA for methylation analysis.
- Procedures for obtaining a biological sample from the patient may be noninvasive, such as collecting cells from urine. Alternatively, invasive procedures such as biopsies may be used.
- the level of detection is such that 2 tumor cells may be detected in a sample comprising 150,000 cells or more.
- the sample may comprise 160,000 cells or more, 170,000 cells or more, 180,000 cells or more, 190,000 cells or more, 200,000 cells or more, 210,000 cells or more, 220,000 cells or more, 230,000 cells or more, 240,000 cells or more, 250,000 cells or more, 260,000 cells or more, 270,000 cells or more, 280,000 cells or more, 280,000 cells or more, or 300,000 cells or more.
- the number of tumor cells that can be detected is 10 or more, 20 or more, 30 or more, 40 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more, 200 or more, 300 or more, 400 or more, 500 or more,
- the invention also encompasses the performance of one or more treatment steps following a positive diagnosis of bladder cancer by the diagnostic methods described herein.
- the invention also encompasses a method of treating bladder cancer in an individual comprising:
- the invention also encompasses a method of treating bladder cancer in an individual comprising:
- the invention also encompasses a method of treating bladder cancer in an individual comprising: (a) determining whether each one of a group of MVPs selected from a panel comprising the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] is methylated in DNA from a sample from the individual, wherein the group comprises at least 25 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG];
- the invention also encompasses a method of treating bladder cancer in an individual comprising administering one or more bladder cancer treatments to the individual, wherein the individual has been diagnosed with bladder cancer by steps comprising:
- the group of MVPs which are selected from a panel comprising the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] may comprise any number of MVPs as described and defined herein, provided that the group comprises at least 25 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG].
- bladder cancer may be diagnosed in the individual when the number of MVPs of the group which are determined to be methylated is any number of MVPs as described and defined herein, provided that at least 25 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] are determined to be methylated.
- the invention encompasses administration of one or more surgical procedures, one or more chemotherapeutic agents, one or more immunotherapeutic agents, one or more radiotherapeutic agents, one or more hormonal therapeutic agents or any combination of the above following a positive diagnosis of bladder cancer.
- Surgical procedures include transurethral resection of bladder tumor (TURBT), cystectomy, open radical cystectomy (ORC), laparoscopic radical cystectomy (LRC) and robot-assisted radical cystectomy (RARC).
- TURBT transurethral resection of bladder tumor
- ORC open radical cystectomy
- LRC laparoscopic radical cystectomy
- RARC robot-assisted radical cystectomy
- Chemotherapeutic agents include the following. Alkylating agents, which include the nitrogen mustards, nitrosoureas, tetrazines, aziridines, cisplatin and platinum based derivatives, as well as the non-classical alkylating agents. Antimetabolites, which include the anti-folates, fluoropyrimidines, deoxynucleoside analogues and thiopurines. Microtubule disrupting agents, which include the vinca alkaloids and taxanes, as well as dolastatin 10 and derivatives thereof. Topoisomerase inhibitors, which include camptothecin, irinotecan and topotecan.
- Topoisomerase II poisons which include etoposide, doxorubicin, mitoxantrone and teniposide.
- Topoisomerase II catalytic inhibitors which include novobiocin, merbarone, and aclarubicin.
- Cytotoxic antibiotics which include anthracyclines, actinomycin, bleomycin, plicamycin, and mitomycin.
- Combinations of agents include but are not limited to MVAC (Methotrexate, Vinblastine, Vinblastine and Vinblastine), Gem-Cis (GC) (Gemcitabine and Cisplatin), Lapatinib and gemcitabine.
- MVAC Metalhotrexate, Vinblastine, Vinblastine and Vinblastine
- GC Gem-Cis
- Lapatinib and gemcitabine include but are not limited to MVAC (Methotrexate, Vinblastine, Vinblastine and Vinblastine), Gem-Cis (GC) (Gemcitabine and Cisplatin), Lapatinib and gemcitabine.
- Immunotherapeutics include bacilli Calmette-Guerin (BCG) immunotherapy as well as monoclonal antibodies and antibody-drug conjugates.
- Antibody-drug conjugates include antibodies conjugated to microtubule disrupting agents and DNA modifying agents as described above.
- Combination therapies include intravesical, sequential BCG, followed by electromotive administration (EMDA) of MMC (EMDA-MMC) as well as microwave- induced bladder wall hyperthermia (HT) and intravesical MMC.
- EMDA electromotive administration
- HT microwave- induced bladder wall hyperthermia
- Cancer therapeutic agents are administered to a subject already suffering from a disorder or condition, in an amount sufficient to cure, alleviate or partially arrest the condition or one or more of its symptoms. Such therapeutic treatment may result in a decrease in severity of disease symptoms, or an increase in frequency or duration of symptom-free periods. An amount adequate to accomplish this is defined as "therapeutically effective amount”. Effective amounts for a given purpose will depend on the severity of the disease as well as the weight and general state of the subject. As used herein, the term "subject" includes any human. Arrays
- the invention also encompasses arrays capable of discriminating between methylated and non-methylated forms of MVPs as defined herein; the arrays may comprise oligonucleotide probes specific for methylated forms of MVPs as defined herein and oligonucleotide probes specific for non-methylated forms of MVPs as defined herein.
- probes comprise sequences which are complementary to those of the oligonucleotides comprising the MVP such they may hybridize, particularly under stringent conditions.
- HumanMethylation450 BeadChip array Infinium HumanMethylation450 BeadChip array.
- the number of MVP-specific oligonucleotide probes of the array is less than 482,421, preferably 482,000 or less, 480,000 or less, 450,000 or less, 440,000 or less, 430,000 or less, 420,000 or less, 410,000 or less, or 400,000 or less, 375,000 or less, 350,000 or less, 325,000 or less, 300,000 or less, 275,000 or less, 250,000 or less, 225,000 or less, 200,000 or less, 175,000 or less, 150,000 or less, 125,000 or less, 100,000 or less, 75,000 or less, 50,000 or less, 45,000 or less, 40,000 or less, 35,000 or less, 30,000 or less, 25,000 or less, 20,000 or less, 15,000 or less, 10,000 or less, 5,000 or less, 4,000 or less, 3,000 or less or 2,000 or less.
- the invention further encompasses the use of any of the arrays as defined herein in any of the methods which require determining the methylation status of MVPs for the purposes of diagnosing bladder cancer cells in an individual. Kits
- any of the arrays as defined herein may be comprised in a kit.
- the kit may comprise any array as defined herein.
- the kit may comprise any array as defined herein together with instructions for use.
- the kit may additionally comprise a DNA modifying regent, such as a bisulphite reagent.
- a DNA modifying regent such as a bisulphite reagent.
- the kit may additionally comprise reagents for amplifying DNA, such as primers directed to any of the MVPs as defined herein as identified in SEQ ID NOS 1 to 150 (see Table 2).
- the invention further encompasses a method of determining a methylation profile of a sample from an individual, the method comprising:
- the group of MVPs which are selected from a panel comprising the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] may comprise any number of MVPs as described and defined herein, provided that the group comprises at least 25 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG].
- the methylation status of MVPs may be determined using any of the arrays described herein. Further methods
- the step of diagnosing bladder cancer in the individual may further comprise:
- the invention also encompasses a method of determining the risk of the development of bladder cancer in an individual, the method comprising:
- the group of MVPs which are selected from a panel comprising the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG] may comprise any number of MVPs as described and defined herein, provided that the group comprises at least 25 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG].
- the methylation status of MVPs may be determined using any of the arrays described herein.
- the invention also encompasses the use of a group of MVPs in the diagnosis of bladder cancer in an individual or in determining the risk of the development of bladder cancer in an individual.
- the group of MVPs are selected from a panel comprising the
- the group of MVPs may comprise any number of MVPs as described and defined herein, provided that the group comprises at least 25 of the MVPs identified in SEQ ID NOS 1 to 150 and denoted by [CG].
- the diagnosis of bladder cancer in an individual or the determination of the risk of the development of bladder cancer in an individual may be performed by any of the respective methods described and defined herein. Furthermore, in any such methods, the methylation status of MVPs may be determined using any of the arrays described herein.
- next-generation DNA sequencing platforms hold particular promise for the development of highly sensitive epigenetic biomarker panels.
- the microdroplet-based PCR amplification of bisulphite converted DNA, followed by next-generation sequencing of the amplified target loci developed by RainDance Technologies [30] enables the sensitive, specific and simultaneous amplification of up to 20,000 bisulfite-converted target loci.
- Highly parallel microdroplet-based PCR amplification of bisulphite-converted DNA has shown utility in the validation of epigenetic alterations in a range of tissues [31 -33].
- it has not been applied to the development of non-invasive diagnostic biomarkers for the detection of bladder cancer.
- bladder-specific epigenetic biomarkers have been defined and the sensitivity and specificity of a 150 loci panel using
- Genome-wide DNA methylation profiling was performed on DNA from 81 bladder cancers and 30 age-matched normal urothelium samples collected from UCLH (London) Department of Urology and CIEMAT (Madrid). The cohort included 35 low- grade non-muscle-invasive cancers. Pathological review of representative H&E sections was conducted to include specimens with tumor cellularity > 80%. Blood methylome data was download from the MARMAL-aid database (http:/marmal-aid.org, [34]).
- er%20Urofheiiaj%2QCarciaoma consisting of MIBC bladder cancer and 20 normal urothelium samples.
- Urine samples were obtained from patients attending haematuria clinics or undergoing cystoscopy for recurrent bladder cancer at University College Hospital. For comparison of clinic versus home urine samples, patients were asked to supply three samples: a clinic sample and two home samples, one of which should be a first void. 40- 100ml were obtained per sample.
- the home urine kit for one sample comprised up to four 25ml sterile tubes, mailing tubes with absorbent pads and a pre-addressed padded envelope, designed to fit through a Royal Mail post box.
- Urinary DNA was extracted using a DNeasy blood and tissue kit (Qiagen, UK) in accordance with the manufacturer's instructions. DNA was quantified by
- the bisulfite-treated genomic DNA template mix was then applied to a fully automated Thunderstorm system (RainDance Technologies) following the
- primer panel droplets (MethylSeq Solution, RainDance Technologies) were dispensed to a microfiuidic chip.
- the DNA template mix was converted into droplets within the microfiuidic chip.
- the primer pair droplets and template droplets were then paired together in a 1 : 1 ratio. Paired droplets passed through an electric field inducing the discrete droplets to coalesce into a single PCR droplet (26 pi); approximately 1 million PCR droplets are collected per sample.
- PCR droplets were processed in a PTC-225 thermocycler (MJ Research) as follows: 94°C for 2 min; 55 cycles of 94°C for 30 s, 54°C for 45 s, 68°C for 80 s;
- the pooled sequencing library (12 pM) and custom sequencing primers (0.5 ⁇ ) were applied to a MiSeq-cycle PE consumable cartridge (Illumina) according to the manufacturer's protocol.
- the DNA sequences of the custom sequencing primers are provided in Table 2 below. Sequencing was performed on a MiSeq DNA sequencer
- the RainDance Thunderstorm® System was also used for the sequencing of nucleic acids (http://raindancetech.com/targeted-dna-sequencing/thunderstorm/).
- Sequencing adapters were trimmed from the raw sequencing reads using the fastq-mcf tool of ea-utils vl .1.2-537 [60]. Trimmed sequencing data were mapped to an in silico bisulfite-converted human reference genome (GRCh37) using Bismark vO.7.12
- the epigenetic alterations associated with bladder cancer were initially defined by performing genome-wide DNA methylation profiling on DNA from 81 high-grade and 30 normal urothelium.
- MVPs Methods (Methylation Variable Positions) between bladder cancer and normal tissue. MVPs were selected on the basis of statistical significance
- methylated ( ⁇ >50%) as discussed herein in relation to the development of this initial DNA methylation biomarker panel, it is meant that for any given locus, >50% of cells in a patient sample are determined to be methylated with respect to that MVP.
- alterations which are bladder cancer specific, whole blood and urine from 10 patients attending hematuria clinics and who had a confirmed non- cancer diagnosis was also profiled.
- Random Forest framework which resulted in a classification signature consisting of 150 CpG loci ( Figure 2, see Table 1 below).
- Using this core set of 150 markers we performed an internal cross validation of the classifier with predicted likelihood values i.e. likelihood of a sample being cancer or not for each sample independent of its relationship to the group of samples. This resulted in a cross validated sensitivity of 100% and specificity of 100% for the detection of cancer, showing that 150 epigenetic loci can clearly stratify normal urothelium from bladder cancer ( Figure 3, Figure 4).
- Example 3 Validation of detection panel
- Applying the epi-signature to this validation cohort of give a sensitivity of 95% and specificity of 96% and an AUC of 97% for the detection of bladder cancer in this independent validation cohort (Figure 5).
- the large marker panel was also compared to the best performing single markers from the training cohort, this includes CpG loci from regions previously published as potential urinary biomarkers in genes including OTX1, COD I and MEIS1. For each CpG a methylation threshold was defined, based on the highest ⁇ -value obtained from non-bladder cancer controls in the training cohort. This value was then used to predict the likely presence of bladder cancer in the validation cohort.
- the best performing single markers were combined and a predictive classifier developed to explore the potential for an "oligo panel" based on 3, 5 or 10 markers. Although sensitivities improve over single markers (best single marker 72%, best combined marker 70%), they still do not reach the required level to replace cystoscopy.
- RainDrop BS-seq [31] allows large scale targeted bisulphite sequencing of a large number of regions (up to 20,00 unique amplicons) in parallel. This technology has been validated previously and shown to be highly correlated with the 45 OK methylation array, and its utility with low template input has also been validated [31 , 32].
- a bisulphite converted sequencing primer panel was designed to measure the methylation state of the 150 selected genomic loci (see Table 2 below). Primers were designed to interrogate both Watson and Crick strands independently where possible.
- Validation of the urinary epi-signature was conducted using RainDrop BS-seq in a second independent cohort of 96 cases. DNA from urinary sediment cells was obtained from 26 patients with bladder cancer and 64 non-cancer patient controls. Methylation score for each of the 150 loci was generated using the Bismark algorithm, using this data the urinary epi-signature predicted the likely presence of bladder cancer with a sensitivity of 96%, specificity of 97% and an AUC of 0.96.
- Biomarker-driven early non-invasive detection of bladder cancer has the potential to radically improve the management of this disease.
- Highly sensitive and specific assays have the potential utility in both the detection of de novo disease in patients attending haematuria clinic and also in the screening for recurrence in existing bladder cancer populations.
- DNA methylation patterns are highly cell-specific and the ontogenic stability of these epigenetic events make DNA methylation an ideal biomarker for the detection and diagnosis of disease. Changes in global DNA methylation patterns are a common feature of neoplastic transformation and is a frequent event in bladder cancer. Previous studies have shown that methylation changes between bladder cancer, both non-muscle invasive bladder cancer (NMIBC) and muscle invasive bladder cancer (MIBC) and normal urothelium are reflected in urinary sediment cells from bladder cancer patients, and as such could be a useful diagnostic marker. Several studies now have shown the utility of urinary epigenetic markers in the diagnosis of bladder cancer.
- NMIBC non-muscle invasive bladder cancer
- MIBC muscle invasive bladder cancer
- DNA methylation biomarker assays (along with other DNA based markers, e.g. mutations) have been limited by the low resolution of primary analysis in identifying putative biomarkers, using either a candidate approach or a low-resolution microarray based platforms, and also by the limitations in the technology available for analysing candidate markers in urine. This has resulted in single/small biomarker panels being interrogated in the final biomarker panel [15-23]. However, in order for small biomarker panels to show the sensitivity and specificity to match those of cystoscopy they rely heavily on a low intra and inter tumour heterogeneity across a wide spectrum of disease states [42].
- the inventors have shown that by applying a large scale highly multiplexed next generation assay, which is both highly sensitive and quantitative, the presence of bladder cancer in urine can be detected with a higher sensitivity and specificity than previously published methylation assays, and has a PPV comparable to cystoscopy.
- the invention demonstrates that the combination of novel technologies, which allow the interrogation of larger panels, and bladder cancer-specific epigenetic biomarkers can be utilized to detect bladder cancer, allowing a reduction in the number of cystoscopies and consequently improve the quality of life for the patients as well as decrease health care expenditure. Furthermore, the utility of large panel assays allows for the potential of multiple clinical parameters to be evaluated from within the same data. For example, the stratification of tumour grade, recurrence or progression of non- muscular invasive disease, the likely response to therapy for muscular invasive disease or the differential diagnosis of multiple conditions.
- the standard UCL urine collection tubes contain 70mg/ml of StabilurTM urinary
- Table 1 below provides a list of 150 MVPs (CpGs) as used in the methods described herein.
- CpGs the Illumina Identifier 5
- CH chromosome number
- MAP INFO chromosome position
- the cytosine of the CG dinucleotide motif subject to modification is identified in bold and in square brackets in each sequence.
- the CpGs are listed in rank order from 1 to 150. The rank order is in respect to the number of tumours in which a given locus is methylated.
- the MVP corresponding to SEQ ID NO: 1 is methylated in the largest number of tumours
- the MVP corresponding to SEQ ID NO: 150 is methylated in the lowest 10 number of tumours.
- Table 2 below provides exemplary primers which may be used to amplify or sequence MVPs defined in Table 1 above.
- Table 3 below provides statistical information of exemplary assays involving all 150 MVPs as defined in Table 1 above, the top 3 ranked MVPs (SEQ ID NOs: 1-3), the top 5 ranked MVPs (SEQ ID NOs: 1-5) and the top 10 ranked MVPs (SEQ ID NOs: 1-10).
- Belinsky SA Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer 2004, 4:707-717.
- Hajdinjak T UroVysion FISH test for detecting urothelial cancers: metaanalysis of diagnostic accuracy and comparison with urinary cytology testing. Urol Oncol 2008, 26:646-651.
- Methylation-specific PCR a novel PCR assay for methylation status of CpG islands. Proc. Natl Acad. Sci. USA 1996, 93: 9821-9826.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Immunology (AREA)
- Analytical Chemistry (AREA)
- Genetics & Genomics (AREA)
- Pathology (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Hospice & Palliative Care (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Apparatus Associated With Microorganisms And Enzymes (AREA)
Abstract
Description


















Claims
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201680048957.0A CN108350504B (en) | 2015-06-24 | 2016-06-24 | How to diagnose bladder cancer |
| EP16732729.5A EP3314016B1 (en) | 2015-06-24 | 2016-06-24 | Methods of diagnosing bladder cancer |
| AU2016283063A AU2016283063B2 (en) | 2015-06-24 | 2016-06-24 | Methods of diagnosing bladder cancer |
| US15/738,539 US11473148B2 (en) | 2015-06-24 | 2016-06-24 | Methods of diagnosing bladder cancer |
| JP2017567208A JP7002334B2 (en) | 2015-06-24 | 2016-06-24 | How to Diagnose Bladder Cancer |
| JP2021169917A JP2022025090A (en) | 2015-06-24 | 2021-10-15 | Methods of diagnosing bladder cancer |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB1511152.9 | 2015-06-24 | ||
| GBGB1511152.9A GB201511152D0 (en) | 2015-06-24 | 2015-06-24 | Method of diagnosing bladder cancer |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016207656A1 true WO2016207656A1 (en) | 2016-12-29 |
Family
ID=53784470
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2016/051903 Ceased WO2016207656A1 (en) | 2015-06-24 | 2016-06-24 | Methods of diagnosing bladder cancer |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US11473148B2 (en) |
| EP (1) | EP3314016B1 (en) |
| JP (2) | JP7002334B2 (en) |
| CN (1) | CN108350504B (en) |
| AU (1) | AU2016283063B2 (en) |
| GB (1) | GB201511152D0 (en) |
| MA (1) | MA42238A (en) |
| WO (1) | WO2016207656A1 (en) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108588219A (en) * | 2018-04-19 | 2018-09-28 | 安徽达健医学科技有限公司 | A kind of kit and its application method for the detection of early stage carcinoma of urinary bladder |
| WO2018213550A1 (en) * | 2017-05-18 | 2018-11-22 | Genomic Health, Inc. | Dna methylation and mutational analysis methods for bladder cancer surveillance |
| WO2019068082A1 (en) * | 2017-09-29 | 2019-04-04 | Arizona Board Of Regents On Behalf Of The University Of Arizona | DNA METHYLATION BIOMARKERS FOR THE DIAGNOSIS OF CANCER |
| EP3880847A2 (en) * | 2018-11-16 | 2021-09-22 | Oslo Universitetssykehus HF | Methods and compositions for characterizing bladder cancer |
| EP3856903A4 (en) * | 2018-09-27 | 2022-07-27 | Grail, LLC | METHYLATION MARKERS AND TARGETED METHYLATION PROBE PANELS |
| EP3963095A4 (en) * | 2019-05-03 | 2023-01-25 | Cornell University | MARKERS FOR THE IDENTIFICATION AND QUANTIFICATION OF A MUTATION, EXPRESSION, SPLICING VARIANT, TRANSLOCATION, COPY NUMBER OR NUCLEIC ACID SEQUENCE METHYLATION CHANGES |
| EP3964578A4 (en) * | 2019-04-30 | 2023-06-07 | Shanghai Epiprobe Biotechnology Co., Ltd. | Methylation-based modified tumor marker stamp-ep8 and application thereof |
| JP2023100848A (en) * | 2017-11-30 | 2023-07-19 | マヨ ファウンデーション フォア メディカル エデュケーション アンド リサーチ | breast cancer detection |
| CN116987787A (en) * | 2023-06-09 | 2023-11-03 | 北京泛生子基因科技有限公司 | Apparatus for detecting recurrence of bladder cancer and computer readable storage medium |
| EP4083232A4 (en) * | 2019-12-26 | 2024-03-20 | Anchordx Medical Co., Ltd. | COMBINATION OF DNA METHYLATION BIOMARKERS AND DETECTION METHOD THEREOF AND KIT THEREOF |
| EP4144859A4 (en) * | 2020-04-30 | 2024-07-24 | Creative Biosciences (Guangzhou) Co., Ltd. | REAGENT AND KIT FOR THE DETECTION OF TUMORS |
| US12435375B2 (en) | 2018-04-02 | 2025-10-07 | Grail, Inc. | Methylation markers and targeted methylation probe panel |
| US12454726B2 (en) | 2019-12-26 | 2025-10-28 | Anchordx Medical Co., Ltd. | Combinations, detection methods and kits of DNA methylation biomarker |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20210172029A1 (en) * | 2018-07-11 | 2021-06-10 | Stichting Vumc | Urine dna methylation markers for bladder cancer |
| CN111206093A (en) * | 2018-11-21 | 2020-05-29 | 立森印迹诊断技术(无锡)有限公司 | Marker for detecting invasive bladder cancer and application thereof |
| CN113728115A (en) * | 2019-01-25 | 2021-11-30 | 格里尔公司 | Detecting cancer, cancer-derived tissue and/or cancer cell types |
| CA3127894A1 (en) * | 2019-02-05 | 2020-08-13 | Grail, Inc. | Detecting cancer, cancer tissue of origin, and/or a cancer cell type |
| US11396679B2 (en) | 2019-05-31 | 2022-07-26 | Universal Diagnostics, S.L. | Detection of colorectal cancer |
| US20220251663A1 (en) * | 2019-06-14 | 2022-08-11 | Arizona Board Of Regents On Behalf Of The University Of Arizona | Dna methylation biomarkers for cancer diagnosing and treatment |
| US11898199B2 (en) | 2019-11-11 | 2024-02-13 | Universal Diagnostics, S.A. | Detection of colorectal cancer and/or advanced adenomas |
| CN110872631B (en) * | 2019-12-26 | 2021-08-10 | 广州市基准医疗有限责任公司 | DNA methylation biomarker combination, detection method and kit |
| CN117265121A (en) * | 2019-12-31 | 2023-12-22 | 上海奕谱生物科技有限公司 | Novel tumor diagnosis marker and application thereof |
| KR102637032B1 (en) * | 2020-01-28 | 2024-02-15 | 주식회사 젠큐릭스 | Composition for diagnosing bladder cancer using CpG methylation status of specific gene and uses thereof |
| US12467096B2 (en) | 2020-05-15 | 2025-11-11 | Universal Diagnostics, S.A. | Methods and systems for identifying methylation biomarkers |
| CN111560436A (en) * | 2020-05-28 | 2020-08-21 | 郑成云 | TERT mutation detection kit and application thereof in noninvasive tumor diagnosis |
| CN115997035A (en) * | 2020-06-30 | 2023-04-21 | 通用诊断股份公司 | Systems and methods for detecting multiple cancer types |
| WO2022255472A1 (en) | 2021-06-03 | 2022-12-08 | 日本精工株式会社 | Grease deterioration detecting method, and lubricant deterioration detecting method |
| CN113652490B (en) * | 2021-09-27 | 2022-07-22 | 广州凯普医药科技有限公司 | Primer probe combination and kit for early screening and/or prognosis monitoring of bladder cancer |
| CN114107513B (en) * | 2022-01-27 | 2022-05-03 | 北京优乐复生科技有限责任公司 | Detection method and kit for bladder urothelial cancer diagnosis |
| CN116042820B (en) * | 2022-09-07 | 2023-09-29 | 浙江大学 | Colon cancer DNA methylation molecular markers and application thereof in preparation of early diagnosis kit for colon cancer |
| WO2024176212A1 (en) * | 2023-02-21 | 2024-08-29 | Nucleix Ltd. | Assessment of hematuria and other urinary tract symptoms |
| CN116555432B (en) * | 2023-07-05 | 2023-09-05 | 广州凯普医药科技有限公司 | Rapid detection kit for bladder cancer |
| CN118006781B (en) * | 2024-02-27 | 2025-01-07 | 广州中鑫基因医学科技有限公司 | Markers, primer sets, high-sensitivity and high-specificity kits and detection methods for detecting urothelial carcinoma |
| CN118222710A (en) * | 2024-03-25 | 2024-06-21 | 江苏阔然生物医药科技有限公司 | Application of reagent for detecting methylation levels of DMRTA, RUNX3 and TWIST1 genes in preparation of bladder cancer diagnosis products |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010123354A2 (en) * | 2009-04-20 | 2010-10-28 | Erasmus University Medical Center Rotterdam | Method of diagnosing bladder cancer |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010149782A1 (en) | 2009-06-26 | 2010-12-29 | Epigenomics Ag | Methods and nucleic acids for analysis of bladder cell proliferative disorders |
| US9249467B2 (en) | 2011-09-16 | 2016-02-02 | Steven Goodison | Bladder cancer detection composition, kit and associated methods |
| US9096905B2 (en) | 2012-02-23 | 2015-08-04 | Medical Diagnostic Laboratories, Llc | Detecting DNA methylation of BCL2, CDKN2A and NID2 genes to predict bladder cancer in humans |
-
2015
- 2015-06-24 GB GBGB1511152.9A patent/GB201511152D0/en not_active Ceased
-
2016
- 2016-06-24 US US15/738,539 patent/US11473148B2/en active Active
- 2016-06-24 JP JP2017567208A patent/JP7002334B2/en active Active
- 2016-06-24 EP EP16732729.5A patent/EP3314016B1/en active Active
- 2016-06-24 MA MA042238A patent/MA42238A/en unknown
- 2016-06-24 WO PCT/GB2016/051903 patent/WO2016207656A1/en not_active Ceased
- 2016-06-24 CN CN201680048957.0A patent/CN108350504B/en active Active
- 2016-06-24 AU AU2016283063A patent/AU2016283063B2/en active Active
-
2021
- 2021-10-15 JP JP2021169917A patent/JP2022025090A/en active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010123354A2 (en) * | 2009-04-20 | 2010-10-28 | Erasmus University Medical Center Rotterdam | Method of diagnosing bladder cancer |
Non-Patent Citations (3)
| Title |
|---|
| ANDREW FEBER ET AL: "Using high-density DNA methylation arrays to profile copy number alterations", GENOME BIOLOGY, BIOMED CENTRAL LTD., LONDON, GB, vol. 15, no. 2, 3 February 2014 (2014-02-03), pages R30, XP021185518, ISSN: 1465-6906, DOI: 10.1186/GB-2014-15-2-R30 * |
| T. REINERT ET AL: "Comprehensive Genome Methylation Analysis in Bladder Cancer: Identification and Validation of Novel Methylated Genes and Application of These as Urinary Tumor Markers", CLINICAL CANCER RESEARCH, vol. 17, no. 17, 25 July 2011 (2011-07-25), US, pages 5582 - 5592, XP055299578, ISSN: 1078-0432, DOI: 10.1158/1078-0432.CCR-10-2659 * |
| YOSHITOMO CHIHARA ET AL: "Diagnostic markers of urothelial cancer based on DNA methylation analysis", BMC CANCER, BIOMED CENTRAL, LONDON, GB, vol. 13, no. 1, 4 June 2013 (2013-06-04), pages 275, XP021153060, ISSN: 1471-2407, DOI: 10.1186/1471-2407-13-275 * |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018213550A1 (en) * | 2017-05-18 | 2018-11-22 | Genomic Health, Inc. | Dna methylation and mutational analysis methods for bladder cancer surveillance |
| CN110621788A (en) * | 2017-05-18 | 2019-12-27 | 基美健有限公司 | DNA methylation and mutation analysis methods for bladder cancer monitoring |
| US11649506B2 (en) | 2017-05-18 | 2023-05-16 | Genomic Health, Inc. | DNA methylation and mutational analysis methods for bladder cancer surveillance |
| WO2019068082A1 (en) * | 2017-09-29 | 2019-04-04 | Arizona Board Of Regents On Behalf Of The University Of Arizona | DNA METHYLATION BIOMARKERS FOR THE DIAGNOSIS OF CANCER |
| US11851711B2 (en) | 2017-09-29 | 2023-12-26 | Arizona Board Of Regents On Behalf Of The University Of Arizona | DNA methylation biomarkers for cancer diagnosing |
| JP7757338B2 (en) | 2017-11-30 | 2025-10-21 | マヨ ファウンデーション フォア メディカル エデュケーション アンド リサーチ | Breast cancer detection |
| USRE50621E1 (en) | 2017-11-30 | 2025-10-07 | Mayo Foundation For Medical Education And Research | Detecting breast cancer |
| US12325878B2 (en) | 2017-11-30 | 2025-06-10 | Mayo Foundation For Medical Education And Research | Detecting breast cancer |
| JP2023100848A (en) * | 2017-11-30 | 2023-07-19 | マヨ ファウンデーション フォア メディカル エデュケーション アンド リサーチ | breast cancer detection |
| US12435375B2 (en) | 2018-04-02 | 2025-10-07 | Grail, Inc. | Methylation markers and targeted methylation probe panel |
| CN108588219A (en) * | 2018-04-19 | 2018-09-28 | 安徽达健医学科技有限公司 | A kind of kit and its application method for the detection of early stage carcinoma of urinary bladder |
| CN108588219B (en) * | 2018-04-19 | 2021-10-26 | 安徽达健医学科技有限公司 | Kit for early bladder cancer detection and use method thereof |
| US11725251B2 (en) | 2018-09-27 | 2023-08-15 | Grail, Llc | Methylation markers and targeted methylation probe panel |
| US11685958B2 (en) | 2018-09-27 | 2023-06-27 | Grail, Llc | Methylation markers and targeted methylation probe panel |
| EP3856903A4 (en) * | 2018-09-27 | 2022-07-27 | Grail, LLC | METHYLATION MARKERS AND TARGETED METHYLATION PROBE PANELS |
| US11795513B2 (en) | 2018-09-27 | 2023-10-24 | Grail, Llc | Methylation markers and targeted methylation probe panel |
| US12410482B2 (en) | 2018-09-27 | 2025-09-09 | Grail, Inc. | Methylation markers and targeted methylation probe panel |
| EP3880847B1 (en) * | 2018-11-16 | 2025-09-03 | Oslo Universitetssykehus HF | Methods and compositions for characterizing bladder cancer |
| EP3880847A2 (en) * | 2018-11-16 | 2021-09-22 | Oslo Universitetssykehus HF | Methods and compositions for characterizing bladder cancer |
| EP3964578A4 (en) * | 2019-04-30 | 2023-06-07 | Shanghai Epiprobe Biotechnology Co., Ltd. | Methylation-based modified tumor marker stamp-ep8 and application thereof |
| EP3963095A4 (en) * | 2019-05-03 | 2023-01-25 | Cornell University | MARKERS FOR THE IDENTIFICATION AND QUANTIFICATION OF A MUTATION, EXPRESSION, SPLICING VARIANT, TRANSLOCATION, COPY NUMBER OR NUCLEIC ACID SEQUENCE METHYLATION CHANGES |
| EP4083232A4 (en) * | 2019-12-26 | 2024-03-20 | Anchordx Medical Co., Ltd. | COMBINATION OF DNA METHYLATION BIOMARKERS AND DETECTION METHOD THEREOF AND KIT THEREOF |
| US12454726B2 (en) | 2019-12-26 | 2025-10-28 | Anchordx Medical Co., Ltd. | Combinations, detection methods and kits of DNA methylation biomarker |
| AU2020445677B2 (en) * | 2020-04-30 | 2025-05-29 | Creative Biosciences (Guangzhou) Co., Ltd. | Tumor detection reagent and kit |
| EP4144859A4 (en) * | 2020-04-30 | 2024-07-24 | Creative Biosciences (Guangzhou) Co., Ltd. | REAGENT AND KIT FOR THE DETECTION OF TUMORS |
| CN116987787A (en) * | 2023-06-09 | 2023-11-03 | 北京泛生子基因科技有限公司 | Apparatus for detecting recurrence of bladder cancer and computer readable storage medium |
Also Published As
| Publication number | Publication date |
|---|---|
| MA42238A (en) | 2018-05-02 |
| JP7002334B2 (en) | 2022-02-04 |
| JP2022025090A (en) | 2022-02-09 |
| EP3314016A1 (en) | 2018-05-02 |
| JP2018520672A (en) | 2018-08-02 |
| CN108350504A (en) | 2018-07-31 |
| AU2016283063B2 (en) | 2022-01-20 |
| CN108350504B (en) | 2023-09-29 |
| US20180305765A1 (en) | 2018-10-25 |
| US11473148B2 (en) | 2022-10-18 |
| AU2016283063A1 (en) | 2018-02-08 |
| GB201511152D0 (en) | 2015-08-05 |
| EP3314016B1 (en) | 2024-05-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3314016B1 (en) | Methods of diagnosing bladder cancer | |
| JP7289264B2 (en) | Detection of Colon Tumors by Analysis of Methylated DNA | |
| US20230028856A1 (en) | Detecting gastrointestinal neoplasms | |
| EP3889611A1 (en) | Compositions and methods for performing methylation detection assays | |
| CN109072310A (en) | Cancer is detected in urine | |
| EP3904515A1 (en) | Tumor marker stamp-ep3 based on methylation modification | |
| JP2024020392A (en) | Composition for diagnosing liver cancer using CPG methylation changes in specific genes and its use | |
| EP3828273A1 (en) | Methylation modification-based tumor marker stamp-ep2 | |
| US20130071842A1 (en) | Hypermethylation Biomarkers for Detection of Head and Neck Squamous Cell Cancer | |
| US20220025466A1 (en) | Differential methylation | |
| EP3904516A1 (en) | Methylation modification-based tumor marker stamp-ep6 | |
| US20210062268A1 (en) | Method of Identifying Metastatic Breast Cancer by Differentially Methylated Regions | |
| WO2018158589A1 (en) | Diagnostic and prognostic methods | |
| EP3964578A1 (en) | Methylation-based modified tumor marker stamp-ep8 and application thereof | |
| WO2017119510A1 (en) | Test method, gene marker, and test agent for diagnosing breast cancer | |
| JP2023500923A (en) | Colorectal cancer detection method | |
| EP3052660A1 (en) | Kits and methods for diagnosis, screening, treatment and disease monitoring | |
| JP7447155B2 (en) | Method for detecting methylation of SDC2 gene | |
| JP2023530984A (en) | Methods for detecting and predicting grade 3 cervical epithelial neoplasia (CIN3) and/or cancer | |
| AU2021291134A1 (en) | Methods for detecting and predicting cancer | |
| EP3964580A1 (en) | Tumor marker stamp-ep9 based on methylation modification and application thereof | |
| EP3964581A1 (en) | Tumor marker stamp-ep7 based on methylated modification and use thereof | |
| CN116783309A (en) | Methods for detecting and predicting cancer and/or CIN3 | |
| JP2015177745A (en) | Method of examining lung cancer | |
| EP3887549A1 (en) | Molecular signature |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16732729 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15738539 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2017567208 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016732729 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2016283063 Country of ref document: AU Date of ref document: 20160624 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201680048957.0 Country of ref document: CN |