WO2016190374A1 - Poly-substituted aromatic compound and method for producing same - Google Patents
Poly-substituted aromatic compound and method for producing same Download PDFInfo
- Publication number
- WO2016190374A1 WO2016190374A1 PCT/JP2016/065516 JP2016065516W WO2016190374A1 WO 2016190374 A1 WO2016190374 A1 WO 2016190374A1 JP 2016065516 W JP2016065516 W JP 2016065516W WO 2016190374 A1 WO2016190374 A1 WO 2016190374A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- group
- general formula
- synthesis
- mmol
- Prior art date
Links
- 0 *C(C(*)=C1I)=C(*)S1=O Chemical compound *C(C(*)=C1I)=C(*)S1=O 0.000 description 6
- NHOFFTFPLIXYRF-UHFFFAOYSA-N Cc(cc1)ccc1-c1c(-c(cc2)ccc2Cl)[s]c(-c2cccc(C)c2)c1OC Chemical compound Cc(cc1)ccc1-c1c(-c(cc2)ccc2Cl)[s]c(-c2cccc(C)c2)c1OC NHOFFTFPLIXYRF-UHFFFAOYSA-N 0.000 description 2
- XIQOLBCCYRYEJJ-UHFFFAOYSA-N CC(C)(C)c(cc1)ccc1-c1c(-c(cc2)ccc2OC)c(-c2ccccc2)c(-c2ccc(C(F)(F)F)cc2)nc1-c1ccccn1 Chemical compound CC(C)(C)c(cc1)ccc1-c1c(-c(cc2)ccc2OC)c(-c2ccccc2)c(-c2ccc(C(F)(F)F)cc2)nc1-c1ccccn1 XIQOLBCCYRYEJJ-UHFFFAOYSA-N 0.000 description 1
- QXORMLLSDGMTQN-UHFFFAOYSA-N CC(C)(C)c(cc1)ccc1-c1nc(-c2ccccn2)c(-c2ccc(C(F)(F)F)cc2)c(-c2ccccc2)c1-c(cc1)ccc1OC Chemical compound CC(C)(C)c(cc1)ccc1-c1nc(-c2ccccn2)c(-c2ccc(C(F)(F)F)cc2)c(-c2ccccc2)c1-c(cc1)ccc1OC QXORMLLSDGMTQN-UHFFFAOYSA-N 0.000 description 1
- HKGIVOCWAZGGLM-UHFFFAOYSA-N Cc(cc1)ccc1-c1c[s]c(-c2cccc(C)c2)c1OC Chemical compound Cc(cc1)ccc1-c1c[s]c(-c2cccc(C)c2)c1OC HKGIVOCWAZGGLM-UHFFFAOYSA-N 0.000 description 1
- VUMTYEMOCRUIQJ-UHFFFAOYSA-N Cc1cccc(-c([s]c(-c(cc2)ccc2Cl)c2-c3ccccc3)c2OC)c1 Chemical compound Cc1cccc(-c([s]c(-c(cc2)ccc2Cl)c2-c3ccccc3)c2OC)c1 VUMTYEMOCRUIQJ-UHFFFAOYSA-N 0.000 description 1
- GWQSENYKCGJTRI-UHFFFAOYSA-N Clc(cc1)ccc1I Chemical compound Clc(cc1)ccc1I GWQSENYKCGJTRI-UHFFFAOYSA-N 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/01—Preparation of ethers
- C07C41/18—Preparation of ethers by reactions not forming ether-oxygen bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C43/00—Ethers; Compounds having groups, groups or groups
- C07C43/02—Ethers
- C07C43/235—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring and to a carbon atom of a ring other than a six-membered aromatic ring
- C07C43/247—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring and to a carbon atom of a ring other than a six-membered aromatic ring containing halogen
Definitions
- the present invention relates to a polysubstituted aromatic compound and a method for producing the same.
- the present invention relates to a fully asymmetric polysubstituted aromatic compound and a method for producing the same.
- Naphthalene and anthracene are organic molecules in which two or three benzene rings are condensed, and are used in fluorescent materials, organic electronics materials, etc. because of their various functions and high stability.
- Benzene on the other hand, is a hexagonal organic molecule with the molecular formula C 6 H 6 , and has been said to be a symbol of organic chemistry because of its simplicity and beauty (tortoise shell).
- Benzene is also the most frequently used structural unit for medicines, agricultural chemicals, fragrances, dyes, plastics, liquid crystals, and electronic materials because of its versatile functions and high stability. For this reason, attempts have been made to impart various functions to naphthalene, anthracene and benzene.
- the structural diversity of polysubstituted naphthalene, polysubstituted anthracene and polysubstituted benzene is remarkably large.
- N (2n + 2n 2 + 4n 3 + 3n 4 + n 6 ) / 12.
- the polysubstituted benzene is not only a hexa (hetero) arylbenzene in which all six hydrogen atoms of benzene are substituted with an aromatic substituent (aryl group or heteroaryl group), but can be a variety of optoelectronic functional materials, In recent years, it is a molecular group that has been attracting attention as a precursor of nanographene.
- An object of the present invention is to provide a polysubstituted aromatic compound substituted with various aryl groups or heteroaryl groups.
- the present inventors obtained a tetra-substituted thiophene S-oxide compound by oxidizing a predetermined tetra-substituted thiophene compound for which program synthesis has already been established. It has been found that various polysubstituted aromatic compounds can be synthesized by reacting with an aromatic compound. In addition, by oxidizing a predetermined tetrasubstituted thiophene compound that has already been established for program synthesis to obtain a tetrasubstituted thiophene S-oxide compound, it is reacted with a compound having a predetermined triple bond to obtain various polysubstituted aromatic compounds. It has been found that group compounds can be synthesized and that all substituents can be different types of substituents. The present invention has been completed as a result of further research based on such knowledge. That is, the present invention includes the following configurations.
- R 1 to R 8 represent an optionally substituted aryl group or an optionally substituted heteroaryl group. Five or more of R 1 to R 8 are different groups.
- R 9 to R 16 each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- R 17 to R 18 each represents a hydrogen atom, an optionally substituted aryl group or an optionally substituted heteroaryl group. 5 or more of R 9 to R 18 are different groups.
- Item 2. The polysubstituted aromatic compound according to Item 1, wherein in the general formula (1), R 1 to R 8 are all different groups.
- Item 3 The polysubstituted aromatic compound according to Item 1, wherein in the general formula (2), R 17 to R 18 are hydrogen atoms, and R 9 to R 16 are all different groups.
- R 17 to R 18 are an optionally substituted aryl group or an optionally substituted heteroaryl group, and R 9 to R 18 are all different groups.
- Item 5 The method for producing a polysubstituted aromatic compound according to any one of Items 1 to 4, General formula (3):
- R 5 to R 8 are the same as defined above.
- One of R 19 and R 20 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group.
- R 13 to R 18 are the same as defined above.
- One of R 21 and R 22 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group.
- the manufacturing method provided with the process with which the compound represented by these is made to react.
- Item 6 The production method according to Item 5, which is obtained by a method comprising a step of reacting a compound represented by formula (I) and a step of substituting a hydroxyl group of the compound obtained in the step with a trifluoromethanesulfonate group.
- R represents a nitrogen atom or a group represented by — (C—R 36 ) ⁇ .
- R 31 to R 36 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- R 35 and R 36 may combine to form a ring.
- R 31b to R 34b are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- A represents a ring.
- Item 9 The polysubstituted aromatic compound according to Item 8, represented by:
- Item 12. The polysubstituted aromatic compound according to any one of Items 8 to 11, wherein each of R 31 to R 36 is different and represents an optionally substituted phenyl group or an optionally substituted monocyclic heteroaryl group.
- Item 13 A method for producing a polysubstituted aromatic compound according to any one of Items 8 to 12, General formula (32):
- R 31 to R 34 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- the compound represented by the general formula (33) is General formula (33A): R 36a -C ⁇ C-R 35a (33A) [Wherein, R 35a and R 36a are different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35a and R 36a are different from any of R 31 to R 34 . ]
- Item 15 The method according to Item 13 or 14, which is obtained by oxidizing the tetrasubstituted thiophene compound represented by:
- polysubstituted aromatic compounds substituted with various aryl groups or heteroaryl groups that have been impossible in the past.
- Such a polysubstituted aromatic compound of the present invention is expected to be applied to various uses such as fluorescent materials and organic electronics materials.
- FIG. 2 is a drawing showing an X-ray crystal structure of the compound 30a obtained in Example 3-2 by thermal vibration ellipsoid drawing software (ORTEP) (the ellipse is 50% of atoms). For clarity, hydrogen atoms are omitted. It is a figure which shows the X-ray crystal structure of the compound 34b obtained in Example 5-1 by thermal vibration ellipsoid drawing software (ORTEP) (the ellipse is 50% of the atoms). For clarity, hydrogen atoms are omitted.
- ORTEP thermal vibration ellipsoid drawing software
- Multi-substituted aromatic compound (first embodiment)
- the polysubstituted aromatic compound of the present invention according to the first aspect is represented by the general formula (1):
- R 1 to R 8 represent an optionally substituted aryl group or an optionally substituted heteroaryl group. Five or more of R 1 to R 8 are different groups.
- a polysubstituted aromatic compound (polysubstituted naphthalene compound) represented by formula (2):
- R 9 to R 16 each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- R 17 to R 18 each represents a hydrogen atom, an optionally substituted aryl group or an optionally substituted heteroaryl group. 5 or more of R 9 to R 18 are different groups.
- R 1 to R 8 are an aryl group or a heteroaryl group. Five or more of these R 1 to R 8 are different groups, but according to the production method of the present invention described later, six or more of R 1 to R 8 can be different groups, or R to seven or more of 1 ⁇ R 8 may be different groups, it may be different based on all the R 1 ⁇ R 8.
- R 9 to R 16 are an aryl group or a heteroaryl group.
- R 17 to R 18 are a hydrogen atom, an aryl group or a heteroaryl group. 5 or more of these R 9 to R 18 are different groups, but according to the production method of the present invention described later, when R 17 to R 18 are hydrogen atoms, 6 or more of R 9 to R 16 are used. it may be a different group, can either be different based on seven more of R 9 ⁇ R 16, may be different based on all R 9 ⁇ R 16.
- R 17 ⁇ R 18 when R 17 ⁇ R 18 is an aryl or heteroaryl group, it can either be different based on six or more of R 9 ⁇ R 18, different groups of seven or more of R 9 ⁇ R 18 it may be a, it may be employed a different group of eight or more of R 9 ⁇ R 18, may be employed a 9 or more different groups of R 9 ⁇ R 18, R 9 ⁇ All of R 18 can be different groups.
- the polysubstituted aromatic compound of the present invention includes the general formula (1A):
- R 1 to R 8 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- R 9 to R 16 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- R 9 to R 18 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- the aryl group represented by R 1 to R 18 is not particularly limited and may be a monocyclic aryl group (phenyl group) or a polycyclic aryl group (fused ring aryl group, polycyclic ring).
- Non-condensed ring aryl group and the like may be used, and examples thereof include a phenyl group, a naphthyl group, an anthracenyl group, and a biphenyl group.
- a monocyclic or condensed ring aryl group is preferable, and a phenyl group, a naphthyl group, and the like are more preferable.
- the aryl group represented by R 1 to R 18 may be substituted.
- the substituent that the aryl group represented by R 1 to R 18 may have is not particularly limited, and examples thereof include a halogen atom (fluorine atom, chlorine atom, bromine atom, etc.), alkyl group (methyl group, ethyl group, C1-6 alkyl group such as t-butyl group), haloalkyl group (C1-6 haloalkyl group such as trifluoromethyl group), alkoxy group (C1-6 alkoxy group such as methoxy group), silyl group (t- Tri (C1-6 alkyl) silyl group such as butyldimethylsilyl group), acyl group (C2-7 acyl group such as acetyl group, propionyl group), alkoxycarbonyl group (methoxycarbonyl group, ethoxycarbonyl group etc.) C1-6 alkoxy) carbonyl group and the like, and amino groups (di (
- the heteroaryl group represented by R 1 to R 18 is not particularly limited, and may be a monocyclic heteroaryl group or a polycyclic heteroaryl group (such as a condensed ring heteroaryl group).
- Examples include isothiazolyl group, furanyl group, thiophenyl group, indolyl group, quinolyl group, isoquinolyl group, benzoimidazolyl group, quinazolyl group, phthalazyl group, purinyl group, pteridyl
- the heteroaryl group represented by R 1 to R 18 may be substituted.
- the substituent that the heteroaryl group represented by R 1 to R 18 may have is not particularly limited, but examples thereof include a halogen atom (fluorine atom, chlorine atom, bromine atom, etc.), alkyl group (methyl group, ethyl group).
- C1-6 alkyl group such as t-butyl group
- haloalkyl group C1-6 haloalkyl group such as trifluoromethyl group
- alkoxy group C1-6 alkoxy group such as methoxy group
- silyl group t -Tri (C1-6 alkyl) silyl group such as butyldimethylsilyl group
- acyl group C2-7 acyl group such as acetyl group, propionyl group
- alkoxycarbonyl group methoxycarbonyl group, ethoxycarbonyl group, etc.
- amino groups di (C1-6 alkyl) amino groups such as diethylamino group) and the like.
- the number of these substituents is preferably 0-6, more preferably 0-3.
- polysubstituted aromatic compound of the present invention that satisfies the above conditions include:
- polysubstituted aromatic compound of the present invention according to the first embodiment is not limited to these. According to the production method of the present invention described later, a desired aryl group and heteroaryl group can be freely introduced into a desired location, and a huge number of polysubstituted aromatic compounds can be freely synthesized. Is possible.
- the method for producing a polysubstituted aromatic compound of the present invention according to the first aspect is a method for producing the polysubstituted aromatic compound of the present invention according to the first aspect described above, and is represented by the general formula (3):
- R 5 to R 8 are the same as defined above.
- One of R 19 and R 20 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group.
- R 13 to R 18 are the same as defined above.
- One of R 21 and R 22 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group.
- the tetrasubstituted thiophene S-oxide compound has the general formula (9):
- R 1 to R 4 are the same as defined above.
- It can obtain by the process (process (1-I)) which oxidizes the tetrasubstituted thiophene compound shown by these.
- R 1 to R 4 can be all different groups, or can be all the same group depending on the type of R 5 to R 8 .
- examples of the aryl group and heteroaryl group represented by R 1 to R 4 include those described above, and the types and numbers of substituents that can be used can also be used.
- Such a tetra-substituted thiophene compound can be synthesized, for example, according to the synthesis method described in Non-Patent Document 1 or by slightly improving the synthesis method. Specifically, the following reaction formula 1:
- R 1 to R 4 are the same as defined above.
- R 24 represents an alkyl group.
- X 1 represents a halogen atom.
- Y 1 is the same or different and represents boronic acid or an ester group thereof.
- Tf represents a trifluoromethanesulfonyl group.
- Examples of the alkyl group represented by R 24 include C1-6 alkyl groups such as a methyl group, an ethyl group, and a t-butyl group, particularly a C1-4 alkyl group.
- Examples of the halogen atom represented by X 1 include a chlorine atom, a bromine atom, and an iodine atom.
- boronic acid represented by Y 1 or an ester group thereof examples include, for example, the general formula (11):
- R 25 are the same or different and each represents a hydrogen atom or an alkyl group. R 25 may be bonded to each other to form a ring. ] The group represented by these is preferable.
- Examples of the alkyl group represented by R 25 include a C1-6 alkyl group such as a methyl group, an ethyl group, and a t-butyl group, particularly a C1-4 alkyl group.
- boronic acid or ester groups thereof examples include:
- R 26 to R 27 are the same or different and each represents an alkyl group. ] The group represented by these is mentioned.
- Examples of the alkyl group represented by R 26 to R 27 include a C1-6 alkyl group such as a methyl group, an ethyl group, and a t-butyl group, particularly a C1-4 alkyl group.
- the halogenating agent that can be used in this step is not particularly limited, and is chlorine (Cl 2 ), bromine (Br 2 ), iodine (I 2 ), 1,2-dibromoethane, N-chlorosuccinimide, N-bromosuccinimide. (NBS), hydrogen bromide and the like.
- the amount of the halogenating agent to be used can be appropriately set depending on the kind of the halogenating agent to be used, but is usually preferably 0.2 to 5 mol, more preferably 0.5 to 2 mol, relative to 1 mol of the compound (10a). .
- the amount of the compound represented by R 4 Y that can be used in this step is usually preferably 0.2 to 5 mol, more preferably 0.5 to 2 mol, per 1 mol of compound (10a).
- Examples of the palladium compound that can be used in this step include palladium acetate (Pd (OCOCH 3 ) 2 ; Pd (OAc) 2 ), tetrakis (triphenylphosphine) palladium (0) (Pd (PPh 3 ) 4 ), trifluoro Palladium acetate (Pd (OCOCF 3 ) 2 ), Palladium chloride (PdCl 2 ), Palladium bromide (PdBr 2 ), Palladium iodide (PdI 2 ), Pd (CH 2 COCH 2 COCH 3 ) 2 , K 2 PdCl 4 , K 2 PdCl 6 , K 2 Pd (NO 3 ) 4 , tris (dibenzylideneacetone) dipalladium (0) (Pd 2 (dba) 3 ), bis (dibenzylideneacetone) palladium (0), dichloro (1,5 -Cyclooctadiene)
- These palladium compounds may be solvates. These can be used alone or in combination of two or more. Among these, in this step, tris (dibenzylideneacetone) dipalladium (0) (Pd 2 (dba) 3 ) is preferable from the viewpoints of yield and ease of synthesis.
- the amount of the palladium compound used is usually preferably from 0.002 to 0.1 mol, more preferably from 0.005 to 0.05 mol, based on 1 mol of the compound (10a).
- a ligand compound can be used as necessary.
- ligand compounds that can be used include triphenylphosphine, trimethoxyphosphine, triethylphosphine, triisopropylphosphine, tri (t-butyl) phosphine, tri (n-butyl) phosphine, triisopropoxyphosphine, and tricyclopentylphosphine.
- Tricyclohexylphosphine Trimesitylphosphine, triphenoxyphosphine, di (t-butyl) methylphosphine, methyldiphenylphosphine, dimethylphenylphosphine, triethylamine, pyridine, 2,2'-bipyridyl, 4,4 '-(t-butyl ) Bipyridyl, 1,1'-bis (diphenylphosphino) ferrocene, 1,1'-bis (t-butyl) ferrocene, diphenylphosphinomethane, 1,2-bis (diphenylphosphino) ethane, 1,3- Bis (Gife Ruphosphino) propane, 1,5-bis (diphenylphosphino) pentane, 1,2-bis (dipentafluorophenylphosphino) ethane, 1,2-bis (dicyclohexylphosphino) ethanethan
- ligand compounds may be solvates. These can be used alone or in combination of two or more. Of these, tri (t-butyl) phosphine is preferable in this step from the viewpoint of yield and ease of synthesis.
- the amount of the ligand compound used is usually preferably 1 to 10 moles, more preferably 3 to 5 moles per mole of palladium compound.
- a base can be used as necessary.
- the base that can be used in this step include metal alkoxides such as potassium t-butoxide, sodium t-butoxide and lithium t-butoxide; alkali metal phosphates such as lithium phosphate, sodium phosphate and potassium phosphate; water Metal hydroxides such as lithium oxide, sodium hydroxide, potassium hydroxide, cesium hydroxide, barium hydroxide; metal carbonates such as lithium carbonate, sodium carbonate, potassium carbonate, cesium carbonate; amines such as triethylamine, diisopropylethylamine; Examples include piperidine, N-methylpiperidine, 2,2,6,6-tetramethylpiperidine (TEMPO) and the like.
- TEMPO 2,2,6,6-tetramethylpiperidine
- the amount of the base to be used is generally preferably 0.5-5 mol, more preferably 1-3 mol, per 1 mol of compound (10a).
- This step can usually be carried out in a solvent.
- the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether, and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran (THF) and dioxane; pentane, hexane, heptane, Aliphatic hydrocarbons such as cyclohexane; Aliphatic halogenated hydrocarbons such as dichloromethane, dichloroethane, chloroform, and carbon tetrachloride; Aromatic hydrocarbons such as benzene, toluene, xylene, and mesitylene; Aromatic halogens such as chlorobenzene and trifluorotoluene Hydrocarbons; alcohols such as methanol, ethanol, isopropyl alcohol and the like.
- This step is preferably performed under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably about ⁇ 50 to 150 ° C., more preferably about 0 to 100 ° C.
- the reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 72 hours, and more preferably about 2 to 48 hours.
- the target compound (10b) can be obtained through ordinary isolation and purification steps as necessary.
- Compound (10b) ⁇ Compound (10c)
- the amount of the compound represented by R 2 Y that can be used in this step is usually preferably 1 to 10 mol, more preferably 3 to 5 mol, per 1 mol of compound (10b).
- palladium acetate Pd (OCOCH 3 ) 2 ; Pd (OAc) 2
- the amount of the palladium compound used is usually preferably from 0.02 to 0.5 mol, more preferably from 0.05 to 0.2 mol, per 1 mol of compound (10b).
- a ligand compound can be used as necessary.
- the ligand compound that can be used is not particularly limited, and examples thereof include those described above.
- 2,2′-bipyridyl, 4,4 ′-(t-butyl) bipyridyl and the like are preferable and 2,2′-bipyridyl is more preferable from the viewpoint of yield and ease of synthesis.
- the amount of the ligand compound used is usually preferably 0.2 to 5 mol, more preferably 0.5 to 2 mol, relative to 1 mol of the palladium compound.
- a base can be used as necessary. It does not specifically limit as a base which can be used at this process, What was mentioned above is mentioned.
- piperidine, N-methylpiperidine, 2,2,6,6-tetramethylpiperidine (TEMPO) and the like are preferable from the viewpoint of yield and ease of synthesis, and 2,2,6,6-tetramethyl is preferable.
- Piperidine (TEMPO) is more preferred.
- the amount of base used is usually preferably 1 to 10 mol, more preferably 3 to 5 mol, per 1 mol of compound (10b).
- This step can usually be carried out in a solvent.
- the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane.
- Aliphatic hydrocarbons such as dichloromethane, dichloroethane, chloroform and carbon tetrachloride; Aromatic hydrocarbons such as benzene, toluene, xylene and mesitylene; Aromatic halogenated hydrocarbons such as chlorobenzene and trifluorotoluene Alcohols such as methanol, ethanol, isopropyl alcohol and the like. These solvents can be used alone or in combination of two or more. In this step, an aromatic halogenated hydrocarbon is preferred and trifluorotoluene is more preferred from the viewpoint of yield and ease of synthesis.
- reaction temperature is usually preferably about 0 to 150 ° C, more preferably about 50 to 100 ° C.
- reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 96 hours, more preferably about 2 to 72 hours.
- the target compound (10c) can be obtained through normal isolation and purification steps as necessary.
- R 1 can be introduced into the compound (10c) using a compound represented by R 1 X in the presence of a palladium compound such as palladium chloride (PdCl 2 ).
- a ligand compound such as bipyridyl and a silver compound such as silver carbonate (Ag 2 CO 3 ) can be used as necessary.
- Compound (10d) ⁇ Compound (10e) This step can be performed according to the method disclosed in Non-Patent Document 1. For example, after dealkylating the compound (10d) using tribromoborane or the like, trifluoromethanesulfonylation can be performed using trifluoromethanesulfonic anhydride or the like. Trifluoromethanesulfonylation can also be performed in the presence of a base such as diisopropylethylamine as necessary.
- a base such as diisopropylethylamine
- the tetrasubstituted thiophene compound (9) it is preferable to use the tetrasubstituted thiophene S-oxide compound (3) as a raw material.
- the tetrasubstituted thiophene S-oxide compound (3) can be obtained by oxidizing the tetrasubstituted thiophene compound (9) (step 1-I).
- the oxidizing agent used for the oxidation is not particularly limited.
- peracid is preferable and m-chloroperbenzoic acid (m-CPBA) is more preferable from the viewpoint of yield and ease of synthesis.
- the amount of the oxidizing agent used is usually preferably 1 to 10 moles, more preferably 3 to 5 moles per mole of the tetrasubstituted thiophene compound (9).
- a boron trifluoride compound as an acid catalyst in addition to the oxidizing agent.
- the boron trifluoride compound that can be used include boron trifluoride diethyl ether complex (BF 3 ⁇ OEt 2 ), boron trifluoride tetrahydrofuran complex, and boron trifluoride methanol complex. These can be used alone or in combination of two or more.
- boron trifluoride diethyl ether complex (BF 3 ⁇ OEt 2 ) is preferable in this step from the viewpoint of yield and ease of synthesis.
- the amount of boron trifluoride compound used is usually preferably 2 to 30 moles, more preferably 5 to 20 moles per mole of tetrasubstituted thiophene compound (9).
- This step can usually be carried out in a solvent.
- the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane.
- Aliphatic hydrocarbons such as dichloromethane, dichloroethane, chloroform and carbon tetrachloride; Aromatic hydrocarbons such as benzene, toluene, xylene and mesitylene; Aromatic halogenated hydrocarbons such as chlorobenzene and trifluorotoluene Alcohols such as methanol, ethanol, isopropyl alcohol and the like. These solvents can be used alone or in combination of two or more. In this step, from the viewpoints of yield and ease of synthesis, aliphatic halogenated hydrocarbons are preferable, and dichloromethane is more preferable.
- This step is preferably performed under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably about ⁇ 100 to 50 ° C., more preferably about ⁇ 50 to 0 ° C.
- the reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 24 hours, and more preferably about 2 to 12 hours.
- the target compound tetrasubstituted thiophene S-oxide compound (3)
- the target compound tetrasubstituted thiophene S-oxide compound (3)
- R 5 to R 8 are the same as defined above.
- One of R 19 and R 20 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group.
- the compound represented by these can be used.
- one of R 19 and R 20 is a carboxy group and the other is an amino group, or one is a silyl group (trimethylsilyl group, triethylsilyl group, etc.) and the other is a trifluoromethanesulfonate group.
- the compound represented by the general formula (4) is, for example,
- OTf represents a trifluoromethanesulfonate group.
- TMS represents a trimethylsilyl group. The same applies hereinafter.
- Etc
- R 5 to R 8 are the same as defined above. Obtained by reacting the compound represented by the formula with maleimide, and reacting the compound obtained in the previous step with the oxidizing agent and the first base, and further reacting with the second base. be able to.
- the amount of maleimide used is usually preferably from 1 to 10 mol, more preferably from 2 to 5 mol, per 1 mol of compound (6).
- This step can usually be carried out in a solvent.
- the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane.
- Aliphatic hydrocarbons such as dichloromethane, dichloroethane, chloroform, carbon tetrachloride; Aromatic compounds such as benzene, toluene, xylene, mesitylene, chlorobenzene, nitrobenzene, trifluorotoluene; Methanol, ethanol, isopropyl alcohol And alcohol. These solvents can be used alone or in combination of two or more. In this step, an aromatic compound is preferable and nitrobenzene is more preferable from the viewpoints of yield and ease of synthesis.
- This step is preferably performed under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably about 100 to 300 ° C, more preferably about 150 to 250 ° C.
- the reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 96 hours, more preferably about 2 to 72 hours.
- the compound represented by the general formula (11) is reacted with the oxidizing agent and the first base, and then further reacted with the second base, whereby the compound represented by the general formula (4) is obtained. Can be obtained.
- the oxidizing agent used include chlorine; hydrogen peroxide; peracids such as peracetic acid, perbenzoic acid and m-chloroperbenzoic acid (m-CPBA); dimethyldioxirane, methyltrifluoromethyldioxirane, Examples thereof include peroxides such as t-butyl peroxide; perhalogenates such as sodium metaperiodate; hypochlorous acid such as sodium hypochlorite. These can be used alone or in combination of two or more. Among these, in this step, hypochlorite is preferable and sodium hypochlorite is more preferable from the viewpoint of yield and ease of synthesis.
- the amount of the oxidizing agent to be used is generally preferably 0.5 to 5 mol, more preferably 1 to 2 mol, relative to 1 mol of compound (11).
- Examples of the first base used include metal alkoxides such as potassium t-butoxide, sodium t-butoxide and lithium t-butoxide; alkali metal phosphates such as lithium phosphate, sodium phosphate and potassium phosphate; water Metal hydroxides such as lithium oxide, sodium hydroxide, potassium hydroxide, cesium hydroxide, barium hydroxide; metal carbonates such as lithium carbonate, sodium carbonate, potassium carbonate, cesium carbonate; amines such as triethylamine, diisopropylethylamine; Examples include piperidine, N-methylpiperidine, 2,2,6,6-tetramethylpiperidine (TEMPO) and the like. These can be used alone or in combination of two or more.
- metal alkoxides such as potassium t-butoxide, sodium t-butoxide and lithium t-butoxide
- alkali metal phosphates such as lithium phosphate, sodium phosphate and potassium phosphate
- water Metal hydroxides such
- the amount of the first base used is usually preferably 2 to 20 mol, more preferably 3 to 10 mol, per 1 mol of compound (11).
- Examples of the second base used include metal alkoxides such as potassium t-butoxide, sodium t-butoxide, and lithium t-butoxide; alkali metal phosphates such as lithium phosphate, sodium phosphate, and potassium phosphate; water Metal hydroxides such as lithium oxide, sodium hydroxide, potassium hydroxide, cesium hydroxide, barium hydroxide; metal carbonates such as lithium carbonate, sodium carbonate, potassium carbonate, cesium carbonate; amines such as triethylamine, diisopropylethylamine; Examples include piperidine, N-methylpiperidine, 2,2,6,6-tetramethylpiperidine (TEMPO) and the like. These can be used alone or in combination of two or more.
- metal alkoxides such as potassium t-butoxide, sodium t-butoxide, and lithium t-butoxide
- alkali metal phosphates such as lithium phosphate, sodium phosphate, and potassium phosphate
- the amount of the second base used is usually preferably 10 to 100 mol, more preferably 20 to 50 mol, per 1 mol of compound (11).
- This step can usually be carried out in a solvent.
- the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane.
- Aliphatic hydrocarbons such as dichloromethane, dichloroethane, chloroform, carbon tetrachloride; Aromatic compounds such as benzene, toluene, xylene, mesitylene, chlorobenzene, nitrobenzene, trifluorotoluene; Methanol, ethanol, isopropyl alcohol And alcohol.
- solvents can be used alone or in combination of two or more. In this step, alcohol is preferable from the viewpoints of yield and ease of synthesis, and methanol, isopropyl alcohol, and the like are more preferable.
- This step is preferably carried out under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably carried out under heating, particularly under reflux.
- the reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 48 hours, and more preferably about 2 to 24 hours.
- R 13 to R 18 are the same as defined above.
- One of R 21 and R 22 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group.
- the compound represented by these can be used.
- one of R 21 and R 22 is a carboxy group and the other is an amino group, or one is a silyl group (trimethylsilyl group, triethylsilyl group, etc.) and the other is a trifluoromethanesulfonate group.
- the compound represented by the general formula (5) is, for example,
- R 17 to R 18 and R 21 to R 22 are the same as defined above.
- R 23 is a silyl group.
- examples of the silyl group represented by R 23 include a trimethylsilyl group and a triethylsilyl group.
- the amount of compound (8) used is usually preferably 1 to 10 mol, more preferably 2 to 5 mol, per 1 mol of compound (7).
- R 21 and R 22 are silyl groups and the other is a trifluoromethanesulfonate group
- the reaction proceeds while deprotecting It is preferable to use a deprotecting agent in combination.
- the deprotecting agent include tetrabutylammonium fluoride.
- the amount of the deprotecting agent used is usually preferably 3 to 20 mol, more preferably 5 to 10 mol, relative to 1 mol of the compound (7).
- This step can usually be carried out in a solvent.
- the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane.
- Aliphatic hydrocarbons such as dichloromethane, dichloroethane, chloroform, carbon tetrachloride; Aromatic compounds such as benzene, toluene, xylene, mesitylene, chlorobenzene, nitrobenzene, trifluorotoluene; Methanol, ethanol, isopropyl alcohol And alcohol.
- solvents can be used alone or in combination of two or more.
- a cyclic ether is preferable, and tetrahydrofuran is more preferable.
- This step is preferably performed under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably ⁇ 50 to 100 ° C., more preferably 0 to 50 ° C.
- the reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 48 hours, more preferably about 2 to 36 hours.
- the hydroxyl group in the compound represented by the general formula (12) is substituted with a trifluoromethanesulfonate group.
- This step can be carried out by a method usually used for protecting a hydroxyl group with a trifluoromethanesulfonate group.
- the compound represented by the general formula (5) can be obtained through ordinary isolation and purification steps as necessary.
- the tetrasubstituted thiophene S-oxide compound (3) described above exhibits reactivity as a diene. Have.
- the compound (4) has R 19 and R 20 (carboxy group and amino group, or silyl group and trifluoromethanesulfonate group) removed, and the general formula (4 ′):
- the compound (5) has R 21 and R 22 (carboxy group and amino group, or silyl group and trifluoromethanesulfonate group) eliminated, and the compound represented by the general formula (5 ′):
- the amount of compound (4) or compound (5) used is usually preferably 0.1 to 2 mol, more preferably 0.2 to 1 mol, per 1 mol of tetrasubstituted thiophene S-oxide compound (3). .
- This step can usually be carried out in a solvent.
- the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane.
- Aliphatic hydrocarbons such as dichloromethane, dichloroethane, chloroform and carbon tetrachloride; Aromatic hydrocarbons such as benzene, toluene, xylene and mesitylene; Aromatic halogenated hydrocarbons such as chlorobenzene and trifluorotoluene Alcohols such as methanol, ethanol, isopropyl alcohol and the like. These solvents can be used alone or in combination of two or more. From the viewpoints of yield and ease of synthesis, cyclic ethers and aliphatic halogenated hydrocarbons are preferable, and tetrahydrofuran and dichloroethane are more preferable.
- This step is preferably performed under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably about ⁇ 50 to 150 ° C., more preferably about 0 to 100 ° C.
- the reaction time can be a time during which the reaction proceeds, and is usually preferably about 0.5 to 48 hours, more preferably about 1 to 36 hours.
- the polysubstituted aromatic compound of the present invention can be obtained through ordinary isolation and purification steps as necessary.
- the target polysubstituted aromatic compound of the present invention is obtained as a mixture of a plurality of isotopes, but can be easily isolated by ordinary isolation and purification means such as thin layer chromatography. Is possible.
- Multi-substituted aromatic compound (second embodiment) According to the second aspect of the present invention, it is possible to freely synthesize fully asymmetric hexa (hetero) arylbenzene, which has heretofore been impossible. In addition, according to the second aspect of the present invention, not only fully asymmetric hexa (hetero) arylbenzene, but also pyridine substituted with 5 types of aromatic substituents and 4 types of aromatic substituents. It is possible to synthesize a wide variety of polysubstituted aromatic compounds such as fused cyclic compounds. Such a polysubstituted aromatic compound of the present invention is expected to be applied to various uses such as medical and agricultural chemicals, fragrances, dyes, plastics, liquid crystals, and electronic materials.
- the polysubstituted aromatic compound of the present invention according to the second aspect has the general formula (31):
- R represents a nitrogen atom or a group represented by — (C—R 36 ) ⁇ .
- R 31 to R 36 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- R 35 and R 36 may combine to form a ring.
- R 31 to R 36 are all different and each represents an aryl group or a heteroaryl group.
- the aryl group represented by R 31 to R 36 is not particularly limited, and may be a monocyclic aryl group (phenyl group) or a polycyclic aryl group (fused ring aryl group, polycyclic non-fused ring aryl group, etc.),
- a phenyl group, a naphthyl group, an anthracenyl group, a biphenyl group, etc. are mentioned.
- a monocyclic or condensed ring aryl group is preferable, and a phenyl group, a naphthyl group, and the like are more preferable.
- the aryl group represented by R 31 to R 36 may be substituted.
- substituent that the aryl group represented by R 31 to R 36 may have include, for example, a halogen atom (a fluorine atom, a chlorine atom, a bromine atom, etc.), an alkyl group (a C1-6 alkyl group such as a methyl group, an ethyl group, etc.) ), Haloalkyl group (C1-6 haloalkyl group such as trifluoromethyl group), alkoxy group (C1-6 alkoxy group such as methoxy group), silyl group (t-butyldimethylsilyl group and the like (C1-6 Alkyl) silyl group), acyl group (C2-7 acyl group such as acetyl group, propionyl group), alkoxycarbonyl group ((C1-6 alkoxy) carbonyl group such as methoxycarbonyl group, ethoxycarbonyl group), amino Group (di (C1-6
- the heteroaryl group represented by R 31 to R 36 is not particularly limited and may be a monocyclic heteroaryl group or a polycyclic heteroaryl group (such as a condensed ring heteroaryl group).
- the heteroaryl group represented by R 31 to R 36 may be substituted.
- substituent that the heteroaryl group represented by R 31 to R 36 may have include, for example, halogen atoms (fluorine atom, chlorine atom, bromine atom, etc.), alkyl groups (C1-6 alkyl such as methyl group, ethyl group, etc.) Group), a haloalkyl group (C1-6 haloalkyl group such as trifluoromethyl group), an alkoxy group (C1-6 alkoxy group such as methoxy group), a silyl group (t-butyldimethylsilyl group and the like (C1- 6alkyl) silyl group), acyl group (C2-7 acyl group such as acetyl group, propionyl group), alkoxycarbonyl group ((C1-6 alkoxy) carbonyl group such as methoxycarbonyl group, ethoxycarbonyl group), An amino group (di (C1-6 alkyl
- R is a nitrogen atom or a group represented by — (C—R 36 ) ⁇ .
- R 35 and R 36 may be bonded to form a ring. That is, in the polysubstituted aromatic compound of the present invention, the benzene ring substituted with six different aryl groups or heteroaryl groups (fully asymmetric hexa (hetero) arylbenzene), and four different aryl groups or heteroaryl groups have one benzene ring. Any of the condensed ring aromatic compounds substituted with pyridine and pyridine substituted with five different aryl groups or heteroaryl groups are included.
- polysubstituted aromatic compound of the present invention has the general formula (31A):
- R 31a to R 36a are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- R 31b to R 34b are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- A represents a ring.
- R 31c to R 35c are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- examples of the aryl group and heteroaryl group represented by R 31a to R 36a include those described above, and the types and numbers of substituents that may be included may be employed.
- examples of the aryl group and heteroaryl group represented by R 31b to R 34b include those described above, and the types and numbers of substituents that may be included may be employed.
- examples of the ring represented by A include:
- examples of the aryl group and heteroaryl group represented by R 31c to R 35c include those described above, and the types and numbers of substituents that may be included may be employed.
- examples of the polysubstituted aromatic compound of the present invention include:
- the polysubstituted aromatic compound of the present invention according to the second embodiment is not limited to these. According to the production method of the present invention to be described later, a desired aryl group and heteroaryl group can be freely introduced at a desired position, and by a combination of about 50 kinds of substituents usually used in organic chemistry. It is possible to freely synthesize a huge number of polysubstituted aromatic compounds of 1.3 billion or more.
- Process for producing polysubstituted aromatic compound (second embodiment)
- the method for producing a polysubstituted aromatic compound according to the second aspect of the present invention is a method for producing the polysubstituted aromatic compound according to the second aspect of the present invention, and is represented by the general formula (32):
- R 31 to R 34 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- the tetrasubstituted thiophene S-oxide compound used as a raw material is represented by the general formula (34):
- R 31 to R 34 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group.
- process (2-I) which oxidizes the tetrasubstituted thiophene compound represented by these.
- R 31 to R 34 are the same as defined above.
- R 37 represents an alkyl group.
- X 2 represents a halogen atom.
- Y 2 is the same or different and represents boronic acid or an ester group thereof.
- Tf represents a trifluoromethanesulfonyl group.
- the cyclization reaction does not proceed even when the above tetrasubstituted thiophene compound (34) is reacted with compound (33) described below.
- the polysubstituted aromatic compound of the present invention according to the second embodiment cannot be obtained.
- the tetrasubstituted thiophene S-oxide compound (33) can be obtained by oxidizing the tetrasubstituted thiophene compound (34).
- Oxidation can be carried out in the same manner as in the first aspect. Therefore, the above method (2-2) can be adopted for the oxidation method and conditions.
- the tetrasubstituted thiophene S-oxide compound (32) described above exhibits reactivity as a diene. Therefore, the polysubstituted aromatic compound of the present invention according to the second aspect can be obtained by reacting with the compound (33).
- examples of the aryl group and heteroaryl group represented by R 35 to R 36 include those described above, and the types and numbers of substituents that can be used may be used.
- R is a nitrogen atom or a group represented by — (C—R 36 ) ⁇ .
- R 35 and R 36 may be bonded to form a ring. That is, the compound (33) has the general formula (33A): R 36a -C ⁇ C-R 35a (33A) [Wherein, R 35a and R 36a are different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35a and R 36a are different from any of R 31 to R 34 . ]
- a ′ represents a ring having a triple bond.
- examples of the aryl group and heteroaryl group represented by R 35a to R 36a include those described above, and the types and numbers of substituents that may be included may be employed.
- examples of the aryl group and heteroaryl group represented by R 35c include those described above, and the types and numbers of substituents that may be included may be employed.
- examples of usable compounds (33) include:
- the amount of compound (33) used is usually preferably 0.5 to 5 moles, more preferably 1 to 3 moles per mole of tetrasubstituted thiophene S-oxide compound (32). Since compound (33C) is usually a liquid and functions as a reaction solvent, when compound (33C) is used, the amount used is excessive with respect to the tetrasubstituted thiophene S-oxide compound (32). It is preferable.
- This step can usually be carried out in a solvent.
- the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane.
- Aliphatic hydrocarbons such as dichloromethane, dichloroethane, chloroform and carbon tetrachloride; Aromatic hydrocarbons such as benzene, toluene, xylene and mesitylene; Aromatic halogenated hydrocarbons such as chlorobenzene and trifluorotoluene ; Alcohol such as methanol, ethanol, isopropyl alcohol and the like.
- solvents can be used alone or in combination of two or more. From the viewpoints of yield and ease of synthesis, cyclic ethers and aromatic hydrocarbons are preferable, and tetrahydrofuran, xylene, and mesitylene are more preferable. Note that since the compound (33C) is usually a liquid, the reaction solvent need not be used when the compound (33C) is used.
- This step is preferably performed under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably about ⁇ 100 to 50 ° C., more preferably about ⁇ 50 to 0 ° C.
- the reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 24 hours, and more preferably about 2 to 12 hours.
- R 31a to R 36a are all different and are the same as defined above. ] It can obtain as a mixture with the compound represented by these. For this reason, the target compound can be isolated through normal isolation and purification steps.
- the compound (31A) and the compound (31A ′) can be easily isolated by substituting R 35a or R 36a with a bulky substituent such as t-butyldimethylsilyloxy group by a conventional method. Is possible.
- the target compound can be obtained through normal isolation and purification steps as necessary.
- R 31c to R 35c are all different and are the same as defined above. ] It can obtain as a mixture with the compound represented by these. For this reason, the target compound can be isolated through normal isolation and purification steps.
- Analytical thin layer chromatography was performed using E. Merck silica gel 60 F 254 precoated plates (0.25 mm). The developed chromatogram was analyzed with a UV lamp (254 nm). Flash column chromatography was performed using E. Merck silica gel 60 (230-400 mesh).
- Preparative recycle gel permeation chromatography was performed using a JAI LC-9204 equipped with a JAIGEL-1H / JAIGEL-2H column with chloroform as the eluent.
- Preparative thin layer chromatography was performed using a Wakogel B5-F silica-coated plate (0.75 mm) prepared in advance.
- HMDS 1,1,1,3,3,3-hexamethyldisilazane.
- THF tetrahydrofuran. The same applies hereinafter.
- a 50 mL two-necked flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen.
- the reaction mixture was refluxed for 16 hours.
- TMSCl represents chlorotrimethylsilane.
- TMSCl represents chlorotrimethylsilane.
- a 100 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen.
- ((2,5-dibromo-1,4-phenylene) bis (oxy)) bis (trimethylsilane) obtained in Synthesis Example 1-1 (Compound 2; 2.06 g, 5 mmol, 1.0 equivalent)
- Sodium (460 mg, 20 mmol, 4.0 eq) and toluene (20 mL) were added under a nitrogen stream.
- Tf 2 O represents trifluoromethanesulfonic anhydride.
- a magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen.
- 2,5-bis (trimethylsilyl) benzene-1,4-diol obtained in Synthesis Example 1-3
- trifluoromethanesulfonic anhydride Tf 2 O; 164 ⁇ L, 1 mmol, 1.0 equivalent
- pyridine 5 mL
- Me represents a methyl group.
- Pd (OAc) 2 represents palladium acetate.
- 2,2'-bipy represents 2,2'-bipyridyl.
- TEMPO represents 2,2,6,6-tetramethylpiperidine 1-oxyl. The same applies hereinafter.
- a 50 mL two-necked flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen.
- the vessel was heated at 80 ° C. for 48 hours.
- the reaction mixture was filtered through silica gel (eluent: CHCl 3 , 100 mL) and volatiles were removed under reduced pressure.
- the vessel was heated at 80 ° C. for 48 hours.
- the reaction mixture was filtered through silica gel (eluent: ethyl acetate, 100 mL) and volatiles were removed under reduced pressure.
- Synthesis was performed in the same manner as in Synthesis Example 3-2 except that Compound 21b obtained in Synthesis Example 2-2 was used instead of Compound 21a obtained in Synthesis Example 2-1.
- the vessel was sealed with an O-ring tap and heated at 120 ° C. for 48 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a short silica gel pad (ethyl acetate). The filtrate was concentrated under vacuum and purified by gel permeation chromatography (GPC) to give compound 3acd as a white solid (1.77 g, 84%).
- GPC gel permeation chromatography
- i-Pr2NEt represents N, N-diisopropylethylamine.
- DMAP represents N, N-dimethylaminopyridine. The same applies hereinafter.
- a 100 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen.
- 2- (4-chlorophenyl) -4-methoxy-5- (m-tolyl) -3- (p-tolyl) thiophene obtained in Synthesis Example 4-1 (Compound 8; 1.76 g, 4.4 mmol, 1.0 equivalent), and dry CH 2 Cl 2 (44 mL) were added under a stream of nitrogen.
- a 100 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen.
- the material obtained in the above step and dry CH 2 Cl 2 (22 mL) were added under a stream of nitrogen.
- the contents were cooled to 0 ° C., and N, N-diisopropylethylamine (i-Pr 2 NEt; 1.14 mL, 6.52 mmol, 1.5 eq), N, N-dimethylaminopyridine (DMAP; 5.3 mg, 43.5 ⁇ mol) was added to the flask.
- i-Pr 2 NEt 1.14 mL, 6.52 mmol, 1.5 eq
- DMAP N-dimethylaminopyridine
- Synthesis was performed in the same manner as in Synthesis Example 4-2, except that Compound 23bce obtained in Synthesis Example 4-3 was used instead of Compound 23acd obtained in Synthesis Example 4-2, and Compound 24bce was obtained as a white solid (136.8 mg, 54%).
- Pd (PPh 3 ) 4 represents tetrakis (triphenylphosphine) palladium (0). The same applies hereinafter.
- a 200 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen.
- a magnetic stirring bar was placed in a 50 mL Schlenk tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen.
- tetrakis (triphenylphosphine) palladium (0) Pd (PPh 3 ) 4 ; 10.4 mg, 9.0 ⁇ mol, 10 mol%), Ba (OH) 2 (30.8 mg, 0.18 mmol, 2.0 equivalents), 4-chlorophenylboronic acid (0.270 mmol, 3.0 equivalents), compound 24acd (0.09 mmol, 1.0 equivalent) obtained in Synthesis Example 5-2, dry 1-butanol (3.6 mL), and H 2 O (3.0 mL) were nitrogenated. Added under air flow.
- Synthesis Examples 6-3 to 6-5 Synthesis of tetrasubstituted thiophene compound 25acdf, tetrasubstituted thiophene compound 25bceg, tetrasubstituted thiophene compound 25bceh
- the tetra-substituted thiophene compound 25acdf was obtained as a white solid (33.7 mg, 69%) in the same manner as in Synthesis Example 6-2 except that 4-methoxyphenylboronic acid was used instead of 4-chlorophenylboronic acid.
- m-CPBA represents m-chloroperbenzoic acid.
- BF 3 ⁇ OEt 2 represents a boron trifluoride diethyl ether complex. The same applies hereinafter.
- reaction time is 6 hours in total.
- Saturated aqueous Na 2 S 2 O 3 and saturated aqueous NaHCO 3 were added to quench the reaction.
- the mixture was extracted with CH 2 Cl 2 , dried over Na 2 SO 4 and concentrated under reduced pressure.
- the crude product was purified by flash column chromatography (CHCl 3 ) to give 2- (4-chlorophenyl) -4- (3,5-dimethoxyphenyl) -5- (m-tolyl) -3- (p-tolyl) Thiophene 1-oxide (Compound 11) was obtained as a yellow solid (98.1 mg, 52%).
- a magnetic stir bar was placed in a 20 mL Schlenk tube, flame dried under vacuum, cooled to room temperature, and then filled with nitrogen.
- the tetrasubstituted thiophene compound 25acde (0.1 mmol, 1.0 equivalent) obtained in Synthesis Example 6-2 and dry CH 2 Cl 2 (400 ⁇ L) were added.
- boron trifluoride diethyl ether complex (BF 3 ⁇ OEt 2 ; 120 ⁇ L, 1.0 mmol, 10 equivalents) was added. The mixture was stirred at ⁇ 20 ° C.
- m-CPBA m-chloroperbenzoic acid
- CH 2 Cl 2 200 ⁇ L
- Saturated aqueous Na 2 S 2 O 3 and saturated aqueous NaHCO 3 were added to quench the reaction.
- the mixture was extracted with CH 2 Cl 2 , dried over Na 2 SO 4 and concentrated under reduced pressure.
- Synthesis Examples 7-3 to 7-5 Synthesis of tetrasubstituted thiophene S-oxide compound 26acdf, tetrasubstituted thiophene S-oxide compound 26bceg, tetrasubstituted thiophene S-oxide compound 26bceh
- Synthesis was performed in the same manner as in Synthesis Example 7-2 except that the tetrasubstituted thiophene compound 25acdf obtained in Synthesis Example 6-3 was used instead of the tetrasubstituted thiophene compound 25acde obtained in Synthesis Example 6-2.
- the oxide compound 26acdf was obtained as a yellow solid (30.0 mg, 54%).
- reaction mixture was stirred at room temperature for 1 hour and then heated at 210 ° C. for 48 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a short silica gel pad (CHCl 3 ) to remove nitrobenzene and filtered through a short silica gel pad (ethyl acetate) to drain compound 13.
- i-PrOH represents isopropyl alcohol.
- a magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen.
- 4- (4- (t-butyl) phenyl) -5- (4-methoxyphenyl) -6-phenyl-7- (4- (trifluoromethyl) phenyl) obtained in Synthesis Example 8-1 Isoindoline-1,3-dione (compound 13; 59.9 mg, 0.1 mmol, 1.0 eq), aqueous NaOCl (ab.
- a magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen.
- KOH 200 mg, 3.6 mmol, 36 equivalents
- dry isopropyl alcohol dry i-PrOH; 2.3 mL
- the contents were refluxed for 16 hours.
- water (10 mL) and CH 2 Cl 2 (10 mL) were added to the mixture.
- the mixture was neutralized with 1M HCl and concentrated with CH 2 Cl 2 .
- the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure.
- TBAF is tetrabutylammonium fluoride; the same shall apply hereinafter.
- a magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen.
- tetrabutylammonium fluoride (TBAF; 480 ⁇ L, 0.48 mmol, 6.0 eq, 1M in THF) was slowly added to the mixture.
- the reaction mixture was stirred at room temperature for 24 hours.
- water (10 mL) and CH 2 Cl 2 (10 mL) was added to the mixture.
- the resulting mixture was extracted with CH 2 Cl 2 .
- the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure.
- a magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, a mixture of compound 16a and compound 16b obtained in Synthesis Example 10-1 (35.4 mg, 0.052 mmol, 1.0 equivalent) and CH 2 Cl 2 (500 ⁇ L) were added under a nitrogen stream.
- a magnetic stirring bar was placed in a 50 mL Schlenk tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen.
- 4-ethynylacetophenone (288 mg, 2.0 mmol, 1.0 equivalent)
- 4-iodopyridine 410 mg, 2.0 mmol, 1.0 equivalent
- bis (triphenylphosphine) palladium (II) dichloride 140 mg, 0.2 mmol, 20 mol%)
- copper (I) iodide 38 mg, 0.2 mmol, 20 mol%)
- triethylamine (836 ⁇ L, 6.0 mmol, 3.0 eq)
- dry THF (10 mL)
- Example 1 (first aspect): Synthesis of compound (1)]
- Example 1-1 Synthesis of Compound 15a and Compound 15b
- a magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen.
- a mixture of Compound 14a and Compound 14b obtained in Synthesis Example 4-1 (8.1 mg, 13.6 ⁇ mol, 1.0 equivalent), 2- (4-chlorophenyl) -4- (3 , 5-Dimethoxyphenyl) -5- (m-tolyl) -3- (p-tolyl) thiophene 1-oxide (Compound 11; 14.3 mg, 27.1 ⁇ mol, 2.0 eq) and dichloroethane (140 ⁇ L) under nitrogen flow Added.
- Example 2 (first aspect): Synthesis of compound (2)]
- Example 2-1 Synthesis of compound 18a and compound 18b
- a magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen.
- a mixture of compound 17a and compound 17b obtained in Synthesis Example 5-2 (11.8 mg, 0.015 mmol, 1.0 equivalent), 2- (4-chlorophenyl) -4- (3 , 5-Dimethoxyphenyl) -5- (m-tolyl) -3- (p-tolyl) thiophene 1-oxide (compound 11; 23.1 mg, 0.44 mmol, 3.0 eq) and THF (150 ⁇ L) under nitrogen flow Added.
- tetrabutylammonium fluoride (TBAF; 66 ⁇ L, 0.066 mmol, 4.5 eq, 1M in THF) was slowly added to the mixture.
- the reaction mixture was stirred at 0 ° C. for 3 hours.
- water (10 mL) and CH 2 Cl 2 (10 mL) was added to the mixture, the resulting mixture was extracted with CH 2 Cl 2 .
- the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure.
- the low polarity compound is Compound 18a and the high polarity compound is Compound 18b.
- Example 3 Synthesis of compound (31A)]
- Example 3-1 Synthesis of polysubstituted aromatic compound 29a and polysubstituted aromatic compound 29b
- Example 3-2 Synthesis of polysubstituted aromatic compound 30a and polysubstituted aromatic compound 30b
- a magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen.
- a mixture (21.9 mg, 0.03 mmol, 1.0 equivalent) of the compound 29a and compound 29b obtained in Example 3-1 and methanol (1.2 mL) were added to the screw cap tube under a nitrogen stream.
- the contents were cooled to 0 ° C. and a solution of sodium borohydride in methanol (300 ⁇ L, 0.3 M, 0.09 mmol) was added slowly.
- the reaction was quenched with aqueous NaHCO 3 solution.
- the mixture was extracted with CH 2 Cl 2 , dried over Na 2 SO 4 and concentrated under reduced pressure. The resulting mixture was used in the next step without further purification.
- a magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen.
- the material obtained in the above step and dry CH 2 Cl 2 (600 ⁇ L) were added under a nitrogen stream.
- the contents were cooled to 0 ° C. and 2,6-lutidine (17.5 ⁇ L, 0.15 mmol) and t-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf: 34.5 ⁇ L, 0.15 mmol) were slowly added to the screw cap tube. .
- TBSOTf t-butyldimethylsilyl trifluoromethanesulfonate
- the more polar compound is the polysubstituted aromatic compound 30a, and the less polar compound is the polysubstituted aromatic compound 30b.
- the structure of the polysubstituted aromatic compound 30a was determined by X-ray crystal structure analysis. The X-ray crystal structure of the polysubstituted aromatic compound 30a is shown in FIG.
- Example 3-3 Synthesis of polysubstituted aromatic compound 31a and polysubstituted aromatic compound 31b
- Example 4 (second aspect): Synthesis of compound (31B)]
- Example 4-1 Synthesis of polysubstituted aromatic compound 32
- a magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen.
- tetrasubstituted thiophene S-oxide compound 26acdf obtained in Synthesis Example 7-3 (25.5 mg, 0.045 mmol, 1.0 equivalent)
- compound 28 obtained in Synthesis Example 12 (dibenzo [a, e] cyclooctyne ) (18.0 mg, 0.09 mmol, 2.0 equivalents) and m-xylene (450 ⁇ L) were added under a stream of nitrogen.
- the flask was heated at 100 ° C. for 16 hours.
- Example 5 Synthesis of compound (31C)
- Example 5-1 Synthesis of polysubstituted aromatic compound 34a and polysubstituted aromatic compound 34b
- a magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen.
- the tetra-substituted thiophene S-oxide compound 26acdf (0.03 mmol, 1.0 equivalent) obtained in Synthesis Example 7-3 and 3-cyanopyridine (300 ⁇ L, 3.0 mmol) were added under a nitrogen stream.
- the flask was heated at 160 ° C. for 24 hours. After the reaction mixture was cooled to room temperature, the mixture was purified by preparative thin layer chromatography to obtain polysubstituted aromatic compound 34a (1.6 mg) and polysubstituted aromatic compound 34b (1.6 mg) in a total yield of 17%.
- the more polar compound is the polysubstituted aromatic compound 34a, and the less polar compound is the polysubstituted aromatic compound 34b.
- the structure of polysubstituted aromatic compound 34b was determined by X-ray crystal structure analysis. The X-ray crystal structure of the polysubstituted aromatic compound 34b is shown in FIG.
- Example 5-2 Synthesis of polysubstituted aromatic compound 35a and polysubstituted aromatic compound 35b
- the amount of tetrasubstituted thiophene S-oxide compound 26acdf was 5.6 mg, 0.01 mmol, 4-methylbenzonitrile (145 mg, 1 mmol) was used instead of 2-cyanopyridine, and the heating temperature was 160 ° C to 230 ° C. Except that described above, synthesis was performed in the same manner as in Example 5-1, and a mixture of the polysubstituted aromatic compound 35a (0.5 mg) and the polysubstituted aromatic compound 35b (0.5 mg) was obtained with a total yield of 16%.
- Example 5-3 Synthesis of polysubstituted aromatic compound 36a and polysubstituted aromatic compound 36b
- Example 5 except that the amount of tetrasubstituted thiophene S-oxide compound 26acdf was 5.6 mg, 0.01 mmol, and 4- (trifluoromethyl) benzonitrile (171 mg, 1 mmol) was used instead of 2-cyanopyridine. Synthesis was carried out in the same manner as for -1, and a polysubstituted aromatic compound 36a and a polysubstituted aromatic compound 36b were obtained as a mixture (0.7 mg, 10% (mixture)).
Abstract
Provided is a poly-substituted aromatic compound represented by general formula (1), (In the formula, R1 to R8 are an aryl group which may be substituted or a heteroaryl group which may be substituted. Five or more of R1 to R8 are different groups.), or general formula (2), (In the formula, R9 to R16 are an aryl group which may be substituted or a heteroaryl group which may be substituted. R17 and R18 are a hydrogen atom, an aryl group which may be substituted, or a heteroaryl group which may be substituted. Five or more of R9 to R18 are different groups.)
Description
本発明は、多置換芳香族化合物及びその製造方法に関する。特に、本発明は、完全非対称多置換芳香族化合物及びその製造方法に関する。
The present invention relates to a polysubstituted aromatic compound and a method for producing the same. In particular, the present invention relates to a fully asymmetric polysubstituted aromatic compound and a method for producing the same.
ナフタレン及びアントラセンは、それぞれベンゼン環2個又は3個が縮合した有機分子であり、その多彩な機能と高い安定性のために、蛍光材料、有機エレクトロニクス材料等に使用されている。一方、ベンゼンは分子式がC6H6である六角形の有機分子であり、その構造の単純さと美しさ(亀の甲)から有機化学のシンボルと言われてきた。また、ベンゼンは、その多彩な機能と高い安定性のために、医農薬、香料、染料、プラスチック、液晶、エレクトロニクス材料に最もよく用いられる構造単位にもなっている。このため、ナフタレン、アントラセン及びベンゼンにさらに様々な機能を付与することが試みられている。
Naphthalene and anthracene are organic molecules in which two or three benzene rings are condensed, and are used in fluorescent materials, organic electronics materials, etc. because of their various functions and high stability. Benzene, on the other hand, is a hexagonal organic molecule with the molecular formula C 6 H 6 , and has been said to be a symbol of organic chemistry because of its simplicity and beauty (tortoise shell). Benzene is also the most frequently used structural unit for medicines, agricultural chemicals, fragrances, dyes, plastics, liquid crystals, and electronic materials because of its versatile functions and high stability. For this reason, attempts have been made to impart various functions to naphthalene, anthracene and benzene.
ナフタレン及びアントラセンに様々な機能を付与するためには、ナフタレン及びアントラセンが有するベンゼン環に結合している水素原子を様々な置換基に置き換えることが求められる。一方、ベンゼンに様々な機能を付与する鍵は、ベンゼン環に結合している6つの水素原子を様々な置換基に置き換えることにある。どのような置換基をどのような配置で導入するかによって、置換芳香族化合物の性質は大きく異なる。このため、所望の置換基をナフタレン、アントラセン及びベンゼンに導入する手法の開発は化学の発展を支える最重要課題の1つとなってきた。しかしながら、多置換ナフタレン、多置換アントラセン及び多置換ベンゼンの破格の構造多様性のために、多置換ナフタレン、多置換アントラセン及び多置換ベンゼンを意のままに合成することは困難であった。この点は「多置換ベンゼン問題」等として、長年化学の未解決問題とされてきた。
In order to impart various functions to naphthalene and anthracene, it is required to replace the hydrogen atom bonded to the benzene ring of naphthalene and anthracene with various substituents. On the other hand, the key to imparting various functions to benzene is to replace the six hydrogen atoms bonded to the benzene ring with various substituents. The nature of the substituted aromatic compound varies greatly depending on what substituent is introduced and in what arrangement. For this reason, the development of techniques for introducing the desired substituents into naphthalene, anthracene and benzene has become one of the most important issues that support the development of chemistry. However, due to the exceptional structural diversity of polysubstituted naphthalene, polysubstituted anthracene and polysubstituted benzene, it was difficult to synthesize polysubstituted naphthalene, polysubstituted anthracene and polysubstituted benzene at will. This point has been regarded as an unsolved problem in chemistry for many years, such as the “multi-substituted benzene problem”.
多置換ナフタレン、多置換アントラセン及び多置換ベンゼンの構造多様性は著しく多い。例えば、多置換ベンゼンの構造多様性は、n種類の置換基の組み合わせから考えられる置換ベンゼンの分子数Nは、N=(2n+2n2+4n3+3n4+n6)/12で表され(バーンサイドの定理)、その構造多様性は有機分子の中でも突出している。具体的には、多置換ベンゼンを意のままに合成することができれば、理論上は、10種類の置換基の組み合わせからは8万以上の、50種類の置換基の組み合わせからは13億以上の多置換ベンゼンが生成可能となる。このため、多置換ナフタレン、多置換アントラセン及び多置換ベンゼンを作り分けすることができれば、その汎用性は非常に高く、インパクトも絶大である。
The structural diversity of polysubstituted naphthalene, polysubstituted anthracene and polysubstituted benzene is remarkably large. For example, the structural diversity of poly-substituted benzene is represented by the formula N = (2n + 2n 2 + 4n 3 + 3n 4 + n 6 ) / 12. (Burnside's theorem), its structural diversity stands out among organic molecules. Specifically, if a polysubstituted benzene can be synthesized at will, theoretically, more than 80,000 from a combination of 10 types of substituents and over 1.3 billion from a combination of 50 types of substituents Multi-substituted benzene can be produced. For this reason, if multi-substituted naphthalene, poly-substituted anthracene and poly-substituted benzene can be made separately, their versatility is very high and the impact is great.
しかしながら、前述した合成の難しさから、多くの異なる置換基を導入した置換ナフタレン及び置換アントラセンはこれまで合成及び単離されたことがなく、その物性等は依然として未知のままである。一方、多置換ベンゼンは、ベンゼンの6つの水素原子を全て芳香族置換基(アリール基又はヘテロアリール基)で置換したヘキサ(ヘテロ)アリールベンゼンは、様々な光電子機能性材料となるばかりでなく、近年ではナノグラフェンの前駆体としても注目を集めている分子群である。しかしながら、前述した合成の難しさから、これまで研究されてきたヘキサ(ヘテロ)アリールベンゼンは1~2種類のアリール基で置換された対称性の高いものばかりであった(例えば、特許文献1等)。特に、6種類の異なるアリール基又はヘテロアリール基で置換された完全非対称ヘキサ(ヘテロ)アリールベンゼンはこれまで合成及び単離されたことがなく、その物性等は依然として未知のままである。
However, due to the difficulty of the synthesis described above, substituted naphthalene and substituted anthracene into which many different substituents have been introduced have not been synthesized and isolated so far, and their physical properties remain unknown. On the other hand, the polysubstituted benzene is not only a hexa (hetero) arylbenzene in which all six hydrogen atoms of benzene are substituted with an aromatic substituent (aryl group or heteroaryl group), but can be a variety of optoelectronic functional materials, In recent years, it is a molecular group that has been attracting attention as a precursor of nanographene. However, because of the difficulty in the synthesis described above, the hexa (hetero) arylbenzenes that have been studied so far have only been highly symmetrical with one or two kinds of aryl groups (for example, Patent Document 1) ). In particular, fully asymmetric hexa (hetero) arylbenzenes substituted with six different aryl groups or heteroaryl groups have never been synthesized and isolated so far, and their physical properties remain unknown.
こうしたなか、様々な多置換有機分子のプログラム合成法の開発研究が盛んに行われている。プログラム合成とは、合成標的とする有機分子において「全ての対象分子構造を意のままにプログラムされた様式で作り分ける」ことを可能にする方法論である。こうした目標設定の中、多置換チオフェン等の様々な多置換有機分子のプログラム合成は確立されている(例えば、非特許文献1等)。しかしながら、その最終目標とも言える多置換ナフタレン、多置換アントラセン及び多置換ベンゼンのプログラム合成は困難を極め、その手がかりすらほとんど得られなかったのが現状である。
Under such circumstances, research and development of program synthesis methods for various polysubstituted organic molecules has been actively conducted. Programmed synthesis is a methodology that allows “creating all target molecular structures in a programmed manner at will” in an organic molecule to be synthesized. In such target setting, program synthesis of various polysubstituted organic molecules such as polysubstituted thiophene has been established (for example, Non-Patent Document 1). However, the program synthesis of polysubstituted naphthalene, polysubstituted anthracene, and polysubstituted benzene, which can be said to be the final goal, is extremely difficult, and even the clues have hardly been obtained.
本発明は、種々のアリール基又はヘテロアリール基で置換された多置換芳香族化合物を提供することを目的とする。
An object of the present invention is to provide a polysubstituted aromatic compound substituted with various aryl groups or heteroaryl groups.
上記の課題に鑑み鋭意研究を重ねた結果、本発明者らは、既にプログラム合成が確立している所定の四置換チオフェン化合物を酸化して四置換チオフェンS-オキシド化合物を得た後に、所定の芳香族化合物と反応させることで、種々の多置換芳香族化合物を合成することができることを見出した。また、既にプログラム合成が確立している所定の四置換チオフェン化合物を酸化して四置換チオフェンS-オキシド化合物を得た後に、所定の三重結合を有する化合物と反応させることで、種々の多置換芳香族化合物を合成することができ、全ての置換基を異なる種類の置換基とすることも可能であることを見出した。本発明は、このような知見に基づき、さらに研究を重ねた結果、完成されたものである。すなわち、本発明は、以下の構成を包含する。
As a result of intensive studies in view of the above problems, the present inventors obtained a tetra-substituted thiophene S-oxide compound by oxidizing a predetermined tetra-substituted thiophene compound for which program synthesis has already been established. It has been found that various polysubstituted aromatic compounds can be synthesized by reacting with an aromatic compound. In addition, by oxidizing a predetermined tetrasubstituted thiophene compound that has already been established for program synthesis to obtain a tetrasubstituted thiophene S-oxide compound, it is reacted with a compound having a predetermined triple bond to obtain various polysubstituted aromatic compounds. It has been found that group compounds can be synthesized and that all substituents can be different types of substituents. The present invention has been completed as a result of further research based on such knowledge. That is, the present invention includes the following configurations.
項1.一般式(1):
Item 1. General formula (1):

[式中、R1~R8は置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R1~R8のうち5個以上は異なる基である。]
、又は一般式(2): [Wherein R 1 to R 8 represent an optionally substituted aryl group or an optionally substituted heteroaryl group. Five or more of R 1 to R 8 are different groups. ]
Or general formula (2):
、又は一般式(2): [Wherein R 1 to R 8 represent an optionally substituted aryl group or an optionally substituted heteroaryl group. Five or more of R 1 to R 8 are different groups. ]
Or general formula (2):

[式中、R9~R16は置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R17~R18は、水素原子、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R9~R18のうち5個以上は異なる基である。]
で表される多置換芳香族化合物。 [Wherein, R 9 to R 16 each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 17 to R 18 each represents a hydrogen atom, an optionally substituted aryl group or an optionally substituted heteroaryl group. 5 or more of R 9 to R 18 are different groups. ]
The polysubstituted aromatic compound represented by these.
で表される多置換芳香族化合物。 [Wherein, R 9 to R 16 each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 17 to R 18 each represents a hydrogen atom, an optionally substituted aryl group or an optionally substituted heteroaryl group. 5 or more of R 9 to R 18 are different groups. ]
The polysubstituted aromatic compound represented by these.
項2.前記一般式(1)において、R1~R8が全て異なる基である、項1に記載の多置換芳香族化合物。
Item 2. Item 4. The polysubstituted aromatic compound according to Item 1, wherein in the general formula (1), R 1 to R 8 are all different groups.
項3.前記一般式(2)において、R17~R18が水素原子であり、R9~R16が全て異なる基である、項1に記載の多置換芳香族化合物。
Item 3. Item 2. The polysubstituted aromatic compound according to Item 1, wherein in the general formula (2), R 17 to R 18 are hydrogen atoms, and R 9 to R 16 are all different groups.
項4.前記一般式(2)において、R17~R18が置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基であり、R9~R18が全て異なる基である、項1に記載の多置換芳香族化合物。
Item 4. In item 1, in the general formula (2), R 17 to R 18 are an optionally substituted aryl group or an optionally substituted heteroaryl group, and R 9 to R 18 are all different groups. The polysubstituted aromatic compound described.
項5.項1~4のいずれかに記載の多置換芳香族化合物の製造方法であって、
一般式(3): Item 5. Item 5. The method for producing a polysubstituted aromatic compound according to any one of Items 1 to 4,
General formula (3):
一般式(3): Item 5. Item 5. The method for producing a polysubstituted aromatic compound according to any one of Items 1 to 4,
General formula (3):

[式中、R1~R4は前記に同じである。]
で表される四置換チオフェンS-オキシド化合物と、
一般式(4): [Wherein R 1 to R 4 are the same as defined above. ]
A tetrasubstituted thiophene S-oxide compound represented by:
General formula (4):
で表される四置換チオフェンS-オキシド化合物と、
一般式(4): [Wherein R 1 to R 4 are the same as defined above. ]
A tetrasubstituted thiophene S-oxide compound represented by:
General formula (4):

[式中、R5~R8は前記に同じである。R19及びR20は片方はカルボキシ基で他方がアミノ基であるか、片方がシリル基で他方がトリフルオロメタンスルホナート基である。]
で表される化合物、又は一般式(5): [Wherein R 5 to R 8 are the same as defined above. One of R 19 and R 20 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group. ]
Or a compound represented by the general formula (5):
で表される化合物、又は一般式(5): [Wherein R 5 to R 8 are the same as defined above. One of R 19 and R 20 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group. ]
Or a compound represented by the general formula (5):

[式中、R13~R18は前記に同じである。R21及びR22は片方はカルボキシ基で他方がアミノ基であるか、片方がシリル基で他方がトリフルオロメタンスルホナート基である。]
で表される化合物とを反応させる工程
を備える、製造方法。 [Wherein R 13 to R 18 are the same as defined above. One of R 21 and R 22 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group. ]
The manufacturing method provided with the process with which the compound represented by these is made to react.
で表される化合物とを反応させる工程
を備える、製造方法。 [Wherein R 13 to R 18 are the same as defined above. One of R 21 and R 22 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group. ]
The manufacturing method provided with the process with which the compound represented by these is made to react.
項6.前記一般式(4)で表される化合物は、一般式(6):
Item 6. The compound represented by the general formula (4) is represented by the general formula (6):

[式中、R5~R8は前記に同じである。]
で示される化合物と、マレイミドとを反応させる工程、及び
前記工程で得られた化合物と、酸化剤及び第1塩基とを反応させた後に、さらに第2塩基と反応させる工程
を備える方法で得られる、項5に記載の製造方法。 [Wherein R 5 to R 8 are the same as defined above. ]
Obtained by reacting the compound represented by the formula with maleimide, and reacting the compound obtained in the previous step with the oxidizing agent and the first base, and further reacting with the second base. Item 6. The production method according to Item 5.
で示される化合物と、マレイミドとを反応させる工程、及び
前記工程で得られた化合物と、酸化剤及び第1塩基とを反応させた後に、さらに第2塩基と反応させる工程
を備える方法で得られる、項5に記載の製造方法。 [Wherein R 5 to R 8 are the same as defined above. ]
Obtained by reacting the compound represented by the formula with maleimide, and reacting the compound obtained in the previous step with the oxidizing agent and the first base, and further reacting with the second base. Item 6. The production method according to Item 5.
項7.前記一般式(5)で表される化合物は、一般式(7):
Item 7. The compound represented by the general formula (5) is represented by the general formula (7):

[式中、R13~R16は前記に同じである。]
で表される化合物と、一般式(8) [Wherein R 13 to R 16 are the same as defined above. ]
And a compound of the general formula (8)
で表される化合物と、一般式(8) [Wherein R 13 to R 16 are the same as defined above. ]
And a compound of the general formula (8)

[式中、R17~R18及びR21~R22は前記に同じである。R23はシリル基である。]
で表される化合物とを反応させる工程、及び
前記工程で得られた化合物の水酸基をトリフルオロメタンスルホナート基に置換する工程
を備える方法で得られる、項5に記載の製造方法。 [Wherein R 17 to R 18 and R 21 to R 22 are the same as defined above. R 23 is a silyl group. ]
Item 6. The production method according to Item 5, which is obtained by a method comprising a step of reacting a compound represented by formula (I) and a step of substituting a hydroxyl group of the compound obtained in the step with a trifluoromethanesulfonate group.
で表される化合物とを反応させる工程、及び
前記工程で得られた化合物の水酸基をトリフルオロメタンスルホナート基に置換する工程
を備える方法で得られる、項5に記載の製造方法。 [Wherein R 17 to R 18 and R 21 to R 22 are the same as defined above. R 23 is a silyl group. ]
Item 6. The production method according to Item 5, which is obtained by a method comprising a step of reacting a compound represented by formula (I) and a step of substituting a hydroxyl group of the compound obtained in the step with a trifluoromethanesulfonate group.
項8.一般式(31):
Item 8. General formula (31):

[式中、Rは窒素原子又は-(C-R36)=で表される基を示す。R31~R36はいずれも異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35とR36は結合して環を形成してもよい。]
で示される多置換芳香族化合物。 [Wherein R represents a nitrogen atom or a group represented by — (C—R 36 ) ═. R 31 to R 36 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35 and R 36 may combine to form a ring. ]
A polysubstituted aromatic compound represented by:
で示される多置換芳香族化合物。 [Wherein R represents a nitrogen atom or a group represented by — (C—R 36 ) ═. R 31 to R 36 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35 and R 36 may combine to form a ring. ]
A polysubstituted aromatic compound represented by:
項9.一般式(31A):
Item 9. General formula (31A):

[式中、R31a~R36aはいずれも異なり、それぞれ置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。]
で表される、項8に記載の多置換芳香族化合物。 [Wherein, R 31a to R 36a are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
Item 9. The polysubstituted aromatic compound according to Item 8, represented by:
で表される、項8に記載の多置換芳香族化合物。 [Wherein, R 31a to R 36a are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
Item 9. The polysubstituted aromatic compound according to Item 8, represented by:
項10.一般式(31B):
Item 10. General formula (31B):

[式中、R31b~R34bはいずれも異なり、それぞれ置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。Aは環を示す。]
で表される、項8に記載の多置換芳香族化合物。 [Wherein, R 31b to R 34b are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. A represents a ring. ]
Item 9. The polysubstituted aromatic compound according to Item 8, represented by:
で表される、項8に記載の多置換芳香族化合物。 [Wherein, R 31b to R 34b are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. A represents a ring. ]
Item 9. The polysubstituted aromatic compound according to Item 8, represented by:
項11.一般式(1C):
Item 11. General formula (1C):

[式中、R31c~R35cはいずれも異なり、それぞれ置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。]
で表される、項8に記載の多置換芳香族化合物。 [Wherein, R 31c to R 35c are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
Item 9. The polysubstituted aromatic compound according to Item 8, represented by:
で表される、項8に記載の多置換芳香族化合物。 [Wherein, R 31c to R 35c are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
Item 9. The polysubstituted aromatic compound according to Item 8, represented by:
項12.前記R31~R36はいずれも異なり、置換されていてもよいフェニル基又は置換されていてもよい単環ヘテロアリール基を示す、項8~11のいずれかに記載の多置換芳香族化合物。
Item 12. Item 12. The polysubstituted aromatic compound according to any one of Items 8 to 11, wherein each of R 31 to R 36 is different and represents an optionally substituted phenyl group or an optionally substituted monocyclic heteroaryl group.
項13.項8~12のいずれかに記載の多置換芳香族化合物の製造方法であって、
一般式(32): Item 13. Item 13. A method for producing a polysubstituted aromatic compound according to any one of Items 8 to 12,
General formula (32):
一般式(32): Item 13. Item 13. A method for producing a polysubstituted aromatic compound according to any one of Items 8 to 12,
General formula (32):

[式中、R31~R34はいずれも異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。]
で表される四置換チオフェンS-オキシド化合物と、
一般式(33):
R≡C-R35 (33)
[式中、Rは窒素原子又は≡(C-R36)で表される基を示す。R35及びR36は異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35及びR36はR31~R34のいずれとも異なる。R35とR36は結合して環を形成してもよい。]
で表される化合物とを反応させる工程
を備える、製造方法。 [Wherein, R 31 to R 34 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
A tetrasubstituted thiophene S-oxide compound represented by:
General formula (33):
R≡C-R 35 (33)
[Wherein R represents a nitrogen atom or a group represented by ≡ (C—R 36 ). R 35 and R 36 are different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35 and R 36 are different from any of R 31 to R 34 . R 35 and R 36 may combine to form a ring. ]
The manufacturing method provided with the process with which the compound represented by these is made to react.
で表される四置換チオフェンS-オキシド化合物と、
一般式(33):
R≡C-R35 (33)
[式中、Rは窒素原子又は≡(C-R36)で表される基を示す。R35及びR36は異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35及びR36はR31~R34のいずれとも異なる。R35とR36は結合して環を形成してもよい。]
で表される化合物とを反応させる工程
を備える、製造方法。 [Wherein, R 31 to R 34 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
A tetrasubstituted thiophene S-oxide compound represented by:
General formula (33):
R≡C-R 35 (33)
[Wherein R represents a nitrogen atom or a group represented by ≡ (C—R 36 ). R 35 and R 36 are different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35 and R 36 are different from any of R 31 to R 34 . R 35 and R 36 may combine to form a ring. ]
The manufacturing method provided with the process with which the compound represented by these is made to react.
項14.前記一般式(33)で表される化合物が、
一般式(33A):
R36a-C≡C-R35a (33A)
[式中、R35a及びR36aは異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35a及びR36aはR31~R34のいずれとも異なる。]
で表される化合物、
一般式(33B): Item 14. The compound represented by the general formula (33) is
General formula (33A):
R 36a -C≡C-R 35a (33A)
[Wherein, R 35a and R 36a are different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35a and R 36a are different from any of R 31 to R 34 . ]
A compound represented by
General formula (33B):
一般式(33A):
R36a-C≡C-R35a (33A)
[式中、R35a及びR36aは異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35a及びR36aはR31~R34のいずれとも異なる。]
で表される化合物、
一般式(33B): Item 14. The compound represented by the general formula (33) is
General formula (33A):
R 36a -C≡C-R 35a (33A)
[Wherein, R 35a and R 36a are different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35a and R 36a are different from any of R 31 to R 34 . ]
A compound represented by
General formula (33B):

[式中、A’は三重結合を有する環を示す。]
で表される化合物、又は
一般式(33C):
N≡C-R35c
[式中、R35cは置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35cはR1~R4のいずれとも異なる。]
で表される化合物である、項13に記載の製造方法。 [Wherein A ′ represents a ring having a triple bond. ]
Or a compound represented by the general formula (33C):
N≡C-R 35c
[Wherein, R 35c represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35c is different from any of R 1 to R 4 . ]
Item 14. The production method according to Item 13, which is a compound represented by:
で表される化合物、又は
一般式(33C):
N≡C-R35c
[式中、R35cは置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35cはR1~R4のいずれとも異なる。]
で表される化合物である、項13に記載の製造方法。 [Wherein A ′ represents a ring having a triple bond. ]
Or a compound represented by the general formula (33C):
N≡C-R 35c
[Wherein, R 35c represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35c is different from any of R 1 to R 4 . ]
Item 14. The production method according to Item 13, which is a compound represented by:
項15.前記一般式(32)で表される四置換チオフェンS-オキシド化合物が、一般式(34):
Item 15. The tetrasubstituted thiophene S-oxide compound represented by the general formula (32) is represented by the general formula (34):

[式中、R31~R34はいずれも異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。]
で表される四置換チオフェン化合物を酸化させる工程により得られる、項13又は14に記載の製造方法。 [Wherein, R 31 to R 34 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
Item 15. The method according to Item 13 or 14, which is obtained by oxidizing the tetrasubstituted thiophene compound represented by:
で表される四置換チオフェン化合物を酸化させる工程により得られる、項13又は14に記載の製造方法。 [Wherein, R 31 to R 34 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
Item 15. The method according to Item 13 or 14, which is obtained by oxidizing the tetrasubstituted thiophene compound represented by:
本発明によれば、従来は不可能とされてきた種々のアリール基又はヘテロアリール基で置換された多置換芳香族化合物を自在に合成することが可能である。このような本発明の多置換芳香族化合物は、蛍光材料、有機エレクトロニクス材料等、種々様々な用途への応用が期待される。
According to the present invention, it is possible to freely synthesize polysubstituted aromatic compounds substituted with various aryl groups or heteroaryl groups that have been impossible in the past. Such a polysubstituted aromatic compound of the present invention is expected to be applied to various uses such as fluorescent materials and organic electronics materials.
1.多置換芳香族化合物(第1の態様)
第1の態様に係る本発明の多置換芳香族化合物は、一般式(1): 1. Multi-substituted aromatic compound (first embodiment)
The polysubstituted aromatic compound of the present invention according to the first aspect is represented by the general formula (1):
第1の態様に係る本発明の多置換芳香族化合物は、一般式(1): 1. Multi-substituted aromatic compound (first embodiment)
The polysubstituted aromatic compound of the present invention according to the first aspect is represented by the general formula (1):

[式中、R1~R8は置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R1~R8のうち5個以上は異なる基である。]
で表される多置換芳香族化合物(多置換ナフタレン化合物)、及び一般式(2): [Wherein R 1 to R 8 represent an optionally substituted aryl group or an optionally substituted heteroaryl group. Five or more of R 1 to R 8 are different groups. ]
And a polysubstituted aromatic compound (polysubstituted naphthalene compound) represented by formula (2):
で表される多置換芳香族化合物(多置換ナフタレン化合物)、及び一般式(2): [Wherein R 1 to R 8 represent an optionally substituted aryl group or an optionally substituted heteroaryl group. Five or more of R 1 to R 8 are different groups. ]
And a polysubstituted aromatic compound (polysubstituted naphthalene compound) represented by formula (2):

[式中、R9~R16は置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R17~R18は、水素原子、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R9~R18のうち5個以上は異なる基である。]
で表される多置換芳香族化合物(多置換アントラセン化合物)である。 [Wherein, R 9 to R 16 each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 17 to R 18 each represents a hydrogen atom, an optionally substituted aryl group or an optionally substituted heteroaryl group. 5 or more of R 9 to R 18 are different groups. ]
Is a polysubstituted aromatic compound (polysubstituted anthracene compound).
で表される多置換芳香族化合物(多置換アントラセン化合物)である。 [Wherein, R 9 to R 16 each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 17 to R 18 each represents a hydrogen atom, an optionally substituted aryl group or an optionally substituted heteroaryl group. 5 or more of R 9 to R 18 are different groups. ]
Is a polysubstituted aromatic compound (polysubstituted anthracene compound).
一般式(1)において、R1~R8はアリール基又はヘテロアリール基である。これらR1~R8のうち5個以上は異なる基であるが、後述する本発明の製造方法によれば、R1~R8のうち6個以上を異なる基とすることもできるし、R1~R8のうち7個以上を異なる基とすることもできるし、R1~R8の全てを異なる基とすることもできる。
In the general formula (1), R 1 to R 8 are an aryl group or a heteroaryl group. Five or more of these R 1 to R 8 are different groups, but according to the production method of the present invention described later, six or more of R 1 to R 8 can be different groups, or R to seven or more of 1 ~ R 8 may be different groups, it may be different based on all the R 1 ~ R 8.
一般式(2)において、R9~R16はアリール基又はヘテロアリール基である。また、R17~R18は水素原子、アリール基又はヘテロアリール基である。これらR9~R18のうち5個以上は異なる基であるが、後述する本発明の製造方法によれば、R17~R18が水素原子の場合、R9~R16のうち6個以上を異なる基とすることもできるし、R9~R16のうち7個以上を異なる基とすることもできるし、R9~R16の全てを異なる基とすることもできる。また、R17~R18がアリール基又はヘテロアリール基の場合、R9~R18のうち6個以上を異なる基とすることもできるし、R9~R18のうち7個以上を異なる基とすることもできるし、R9~R18のうち8個以上を異なる基とすることもできるし、R9~R18のうち9個以上を異なる基とすることもできるし、R9~R18の全てを異なる基とすることもできる。
In the general formula (2), R 9 to R 16 are an aryl group or a heteroaryl group. R 17 to R 18 are a hydrogen atom, an aryl group or a heteroaryl group. 5 or more of these R 9 to R 18 are different groups, but according to the production method of the present invention described later, when R 17 to R 18 are hydrogen atoms, 6 or more of R 9 to R 16 are used. it may be a different group, can either be different based on seven more of R 9 ~ R 16, may be different based on all R 9 ~ R 16. Further, when R 17 ~ R 18 is an aryl or heteroaryl group, it can either be different based on six or more of R 9 ~ R 18, different groups of seven or more of R 9 ~ R 18 it may be a, it may be employed a different group of eight or more of R 9 ~ R 18, may be employed a 9 or more different groups of R 9 ~ R 18, R 9 ~ All of R 18 can be different groups.
つまり、第1の態様に係る本発明の多置換芳香族化合物には、一般式(1A):
That is, the polysubstituted aromatic compound of the present invention according to the first aspect includes the general formula (1A):

[式中、R1~R8は全て異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。]
で表される多置換芳香族化合物、一般式(2A): [Wherein, R 1 to R 8 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
A polysubstituted aromatic compound represented by the general formula (2A):
で表される多置換芳香族化合物、一般式(2A): [Wherein, R 1 to R 8 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
A polysubstituted aromatic compound represented by the general formula (2A):

[式中、R9~R16は全て異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。]
で表される多置換芳香族化合物、一般式(2B): [Wherein, R 9 to R 16 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
A polysubstituted aromatic compound represented by the general formula (2B):
で表される多置換芳香族化合物、一般式(2B): [Wherein, R 9 to R 16 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
A polysubstituted aromatic compound represented by the general formula (2B):

[式中、R9~R18は全て異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。]
で表される多置換芳香族化合物をいずれも包含する。 [Wherein R 9 to R 18 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
Any of the polysubstituted aromatic compounds represented by
で表される多置換芳香族化合物をいずれも包含する。 [Wherein R 9 to R 18 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
Any of the polysubstituted aromatic compounds represented by
一般式(1)及び(2)において、R1~R18で示されるアリール基としては、特に制限はなく、単環アリール基(フェニル基)でも多環アリール基(縮合環アリール基、多環非縮合環アリール基等)でもよいが、例えば、フェニル基、ナフチル基、アントラセニル基、ビフェニル基等が挙げられる。これらのなかでも、合成の容易さ、収率等の観点から、単環若しくは縮合環アリール基が好ましく、フェニル基、ナフチル基等がより好ましい。
In the general formulas (1) and (2), the aryl group represented by R 1 to R 18 is not particularly limited and may be a monocyclic aryl group (phenyl group) or a polycyclic aryl group (fused ring aryl group, polycyclic ring). Non-condensed ring aryl group and the like may be used, and examples thereof include a phenyl group, a naphthyl group, an anthracenyl group, and a biphenyl group. Among these, from the viewpoints of ease of synthesis, yield, and the like, a monocyclic or condensed ring aryl group is preferable, and a phenyl group, a naphthyl group, and the like are more preferable.
R1~R18で示されるアリール基は置換されていてもよい。R1~R18で示されるアリール基が有し得る置換基としては、特に制限はないが、例えば、ハロゲン原子(フッ素原子、塩素原子、臭素原子等)、アルキル基(メチル基、エチル基、t-ブチル基等のC1-6アルキル基等)、ハロアルキル基(トリフルオロメチル基等のC1-6ハロアルキル基等)、アルコキシ基(メトキシ基等のC1-6アルコキシ基)、シリル基(t-ブチルジメチルシリル基等のトリ(C1-6アルキル)シリル基等)、アシル基(アセチル基、プロピオニル基等のC2-7アシル基等)、アルコキシカルボニル基(メトキシカルボニル基、エトキシカルボニル基等の(C1-6アルコキシ)カルボニル基等)、アミノ基(ジエチルアミノ基等のジ(C1-6アルキル)アミノ基等)等が挙げられる。これらの置換基の数は、0~6個が好ましく、0~3個がより好ましい。
The aryl group represented by R 1 to R 18 may be substituted. The substituent that the aryl group represented by R 1 to R 18 may have is not particularly limited, and examples thereof include a halogen atom (fluorine atom, chlorine atom, bromine atom, etc.), alkyl group (methyl group, ethyl group, C1-6 alkyl group such as t-butyl group), haloalkyl group (C1-6 haloalkyl group such as trifluoromethyl group), alkoxy group (C1-6 alkoxy group such as methoxy group), silyl group (t- Tri (C1-6 alkyl) silyl group such as butyldimethylsilyl group), acyl group (C2-7 acyl group such as acetyl group, propionyl group), alkoxycarbonyl group (methoxycarbonyl group, ethoxycarbonyl group etc.) C1-6 alkoxy) carbonyl group and the like, and amino groups (di (C1-6 alkyl) amino groups such as diethylamino group) and the like. The number of these substituents is preferably 0-6, more preferably 0-3.
一般式(1)及び(2)において、R1~R18で示されるヘテロアリール基としては、特に制限はなく、単環ヘテロアリール基でも多環ヘテロアリール基(縮合環ヘテロアリール基等)でもよいが、例えば、ピロリル基、ピリジル基、ピロリジル基、ピペリジル基、イミダゾリル基、ピラゾリル基、ピラジル基、ピリミジル基、ピリダジル基、ピペラジル基、トリアジニル基、オキサゾリル基、イソオキサゾリル基、モルホリル基、チアゾリル基、イソチアゾリル基、フラニル基、チオフェニル基、インドリル基、キノリル基、イソキノリル基、ベンゾイミダゾリル基、キナゾリル基、フタラジル基、プリニル基、プテリジル基、ベンゾフラニル基、クマリル基、クロモニル基、ベンゾチオフェニル基等が挙げられる。これらのなかでも、合成の容易さ、収率等の観点から、単環ヘテロアリール基が好ましい。
In general formulas (1) and (2), the heteroaryl group represented by R 1 to R 18 is not particularly limited, and may be a monocyclic heteroaryl group or a polycyclic heteroaryl group (such as a condensed ring heteroaryl group). For example, pyrrolyl group, pyridyl group, pyrrolidyl group, piperidyl group, imidazolyl group, pyrazolyl group, pyrazyl group, pyrimidyl group, pyridazyl group, piperazyl group, triazinyl group, oxazolyl group, isoxazolyl group, morpholyl group, thiazolyl group, Examples include isothiazolyl group, furanyl group, thiophenyl group, indolyl group, quinolyl group, isoquinolyl group, benzoimidazolyl group, quinazolyl group, phthalazyl group, purinyl group, pteridyl group, benzofuranyl group, coumaryl group, chromonyl group, benzothiophenyl group, etc. . Among these, a monocyclic heteroaryl group is preferable from the viewpoint of ease of synthesis, yield, and the like.
R1~R18で示されるヘテロアリール基は置換されていてもよい。R1~R18で示されるヘテロアリール基が有し得る置換基としては、特に制限はないが、例えば、ハロゲン原子(フッ素原子、塩素原子、臭素原子等)、アルキル基(メチル基、エチル基、t-ブチル基等のC1-6アルキル基等)、ハロアルキル基(トリフルオロメチル基等のC1-6ハロアルキル基等)、アルコキシ基(メトキシ基等のC1-6アルコキシ基)、シリル基(t-ブチルジメチルシリル基等のトリ(C1-6アルキル)シリル基等)、アシル基(アセチル基、プロピオニル基等のC2-7アシル基等)、アルコキシカルボニル基(メトキシカルボニル基、エトキシカルボニル基等の(C1-6アルコキシ)カルボニル基等)、アミノ基(ジエチルアミノ基等のジ(C1-6アルキル)アミノ基等)等が挙げられる。これらの置換基の数は、0~6個が好ましく、0~3個がより好ましい。
The heteroaryl group represented by R 1 to R 18 may be substituted. The substituent that the heteroaryl group represented by R 1 to R 18 may have is not particularly limited, but examples thereof include a halogen atom (fluorine atom, chlorine atom, bromine atom, etc.), alkyl group (methyl group, ethyl group). C1-6 alkyl group such as t-butyl group), haloalkyl group (C1-6 haloalkyl group such as trifluoromethyl group), alkoxy group (C1-6 alkoxy group such as methoxy group), silyl group (t -Tri (C1-6 alkyl) silyl group such as butyldimethylsilyl group), acyl group (C2-7 acyl group such as acetyl group, propionyl group), alkoxycarbonyl group (methoxycarbonyl group, ethoxycarbonyl group, etc.) (C1-6 alkoxy) carbonyl group and the like), amino groups (di (C1-6 alkyl) amino groups such as diethylamino group) and the like. The number of these substituents is preferably 0-6, more preferably 0-3.
以上の条件を満たす本発明の多置換芳香族化合物としては、例えば、
Examples of the polysubstituted aromatic compound of the present invention that satisfies the above conditions include:

[式中、Meはメチル基を示す。t-Buはt-ブチル基を示す。以下同様である。]
等が挙げられる。 [Wherein, Me represents a methyl group. t-Bu represents a t-butyl group. The same applies hereinafter. ]
Etc.
等が挙げられる。 [Wherein, Me represents a methyl group. t-Bu represents a t-butyl group. The same applies hereinafter. ]
Etc.
なお、第1の態様に係る本発明の多置換芳香族化合物は、これらのみに限定されることはない。後述の本発明の製造方法によれば、所望のアリール基及びヘテロアリール基を所望の箇所に自在に導入することが可能であり、膨大な数の多置換芳香族化合物を自在に合成することが可能である。
Note that the polysubstituted aromatic compound of the present invention according to the first embodiment is not limited to these. According to the production method of the present invention described later, a desired aryl group and heteroaryl group can be freely introduced into a desired location, and a huge number of polysubstituted aromatic compounds can be freely synthesized. Is possible.
2.多置換芳香族化合物(第1の態様)の製造方法
第1の態様に係る本発明の多置換芳香族化合物の製造方法は、上記した第1の態様に係る本発明の多置換芳香族化合物を製造する方法であり、一般式(3): 2. Process for producing polysubstituted aromatic compound (first embodiment)
The method for producing a polysubstituted aromatic compound of the present invention according to the first aspect is a method for producing the polysubstituted aromatic compound of the present invention according to the first aspect described above, and is represented by the general formula (3):
第1の態様に係る本発明の多置換芳香族化合物の製造方法は、上記した第1の態様に係る本発明の多置換芳香族化合物を製造する方法であり、一般式(3): 2. Process for producing polysubstituted aromatic compound (first embodiment)
The method for producing a polysubstituted aromatic compound of the present invention according to the first aspect is a method for producing the polysubstituted aromatic compound of the present invention according to the first aspect described above, and is represented by the general formula (3):

[式中、R1~R4は前記に同じである。]
で表される四置換チオフェンS-オキシド化合物と、
一般式(4): [Wherein R 1 to R 4 are the same as defined above. ]
A tetrasubstituted thiophene S-oxide compound represented by:
General formula (4):
で表される四置換チオフェンS-オキシド化合物と、
一般式(4): [Wherein R 1 to R 4 are the same as defined above. ]
A tetrasubstituted thiophene S-oxide compound represented by:
General formula (4):

[式中、R5~R8は前記に同じである。R19及びR20は片方はカルボキシ基で他方がアミノ基であるか、片方がシリル基で他方がトリフルオロメタンスルホナート基である。]
で表される化合物、又は一般式(5): [Wherein R 5 to R 8 are the same as defined above. One of R 19 and R 20 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group. ]
Or a compound represented by the general formula (5):
で表される化合物、又は一般式(5): [Wherein R 5 to R 8 are the same as defined above. One of R 19 and R 20 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group. ]
Or a compound represented by the general formula (5):

[式中、R13~R18は前記に同じである。R21及びR22は片方はカルボキシ基で他方がアミノ基であるか、片方がシリル基で他方がトリフルオロメタンスルホナート基である。]
で表される化合物とを反応させる工程(工程(1-IV))
を備える。 [Wherein R 13 to R 18 are the same as defined above. One of R 21 and R 22 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group. ]
The step of reacting with the compound represented by (Step (1-IV))
Is provided.
で表される化合物とを反応させる工程(工程(1-IV))
を備える。 [Wherein R 13 to R 18 are the same as defined above. One of R 21 and R 22 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group. ]
The step of reacting with the compound represented by (Step (1-IV))
Is provided.
(2-1)工程(1-I):四置換チオフェンS-オキシド化合物及びその製造方法
四置換チオフェンS-オキシド化合物は、一般式(9): (2-1) Step (1-I): Tetrasubstituted thiophene S-oxide compound and method for producing the same
The tetrasubstituted thiophene S-oxide compound has the general formula (9):
四置換チオフェンS-オキシド化合物は、一般式(9): (2-1) Step (1-I): Tetrasubstituted thiophene S-oxide compound and method for producing the same
The tetrasubstituted thiophene S-oxide compound has the general formula (9):

[式中、R1~R4は前記に同じである。]
で示される四置換チオフェン化合物を酸化させる工程(工程(1-I))により得ることができる。この四置換チオフェン化合物におけるR1~R4は、全て異なる基とすることもできるし、R5~R8の種類によっては全て同じ基とすることもできる。 [Wherein R 1 to R 4 are the same as defined above. ]
It can obtain by the process (process (1-I)) which oxidizes the tetrasubstituted thiophene compound shown by these. In the tetrasubstituted thiophene compound, R 1 to R 4 can be all different groups, or can be all the same group depending on the type of R 5 to R 8 .
で示される四置換チオフェン化合物を酸化させる工程(工程(1-I))により得ることができる。この四置換チオフェン化合物におけるR1~R4は、全て異なる基とすることもできるし、R5~R8の種類によっては全て同じ基とすることもできる。 [Wherein R 1 to R 4 are the same as defined above. ]
It can obtain by the process (process (1-I)) which oxidizes the tetrasubstituted thiophene compound shown by these. In the tetrasubstituted thiophene compound, R 1 to R 4 can be all different groups, or can be all the same group depending on the type of R 5 to R 8 .
一般式(9)において、R1~R4で示されるアリール基及びヘテロアリール基は前記したものが挙げられ、有し得る置換基の種類及び数としても上記したものを採用し得る。このような四置換チオフェン化合物は、例えば、非特許文献1に記載の合成方法に準じて、又は該合成方法を若干改良して合成することができる。具体的には、以下の反応式1:
In the general formula (9), examples of the aryl group and heteroaryl group represented by R 1 to R 4 include those described above, and the types and numbers of substituents that can be used can also be used. Such a tetra-substituted thiophene compound can be synthesized, for example, according to the synthesis method described in Non-Patent Document 1 or by slightly improving the synthesis method. Specifically, the following reaction formula 1:

[式中、R1~R4は前記に同じである。R24はアルキル基を示す。X1はハロゲン原子を示す。Y1は同一又は異なって、ボロン酸又はそのエステル基を示す。Tfはトリフルオロメタンスルホニル基を示す。]
にしたがって合成することができる。 [Wherein R 1 to R 4 are the same as defined above. R 24 represents an alkyl group. X 1 represents a halogen atom. Y 1 is the same or different and represents boronic acid or an ester group thereof. Tf represents a trifluoromethanesulfonyl group. ]
Can be synthesized according to
にしたがって合成することができる。 [Wherein R 1 to R 4 are the same as defined above. R 24 represents an alkyl group. X 1 represents a halogen atom. Y 1 is the same or different and represents boronic acid or an ester group thereof. Tf represents a trifluoromethanesulfonyl group. ]
Can be synthesized according to
R24で示されるアルキル基としては、メチル基、エチル基、t-ブチル基等のC1-6アルキル基、特にC1-4アルキル基が挙げられる。
Examples of the alkyl group represented by R 24 include C1-6 alkyl groups such as a methyl group, an ethyl group, and a t-butyl group, particularly a C1-4 alkyl group.
X1で示されるハロゲン原子としては、塩素原子、臭素原子、ヨウ素原子等が挙げられる。
Examples of the halogen atom represented by X 1 include a chlorine atom, a bromine atom, and an iodine atom.
Y1で示されるボロン酸又はそのエステル基としては、例えば、一般式(11):
Examples of the boronic acid represented by Y 1 or an ester group thereof include, for example, the general formula (11):

[式中、R25は同一又は異なって、水素原子又はアルキル基を示す。R25は互いに結合して環を形成してもよい。]
で表される基が好ましい。 [Wherein R 25 are the same or different and each represents a hydrogen atom or an alkyl group. R 25 may be bonded to each other to form a ring. ]
The group represented by these is preferable.
で表される基が好ましい。 [Wherein R 25 are the same or different and each represents a hydrogen atom or an alkyl group. R 25 may be bonded to each other to form a ring. ]
The group represented by these is preferable.
R25で示されるアルキル基としては、メチル基、エチル基、t-ブチル基等のC1-6アルキル基、特にC1-4アルキル基が挙げられる。
Examples of the alkyl group represented by R 25 include a C1-6 alkyl group such as a methyl group, an ethyl group, and a t-butyl group, particularly a C1-4 alkyl group.
このようなボロン酸又はそのエステル基としては、例えば、
Examples of such boronic acid or ester groups thereof include:
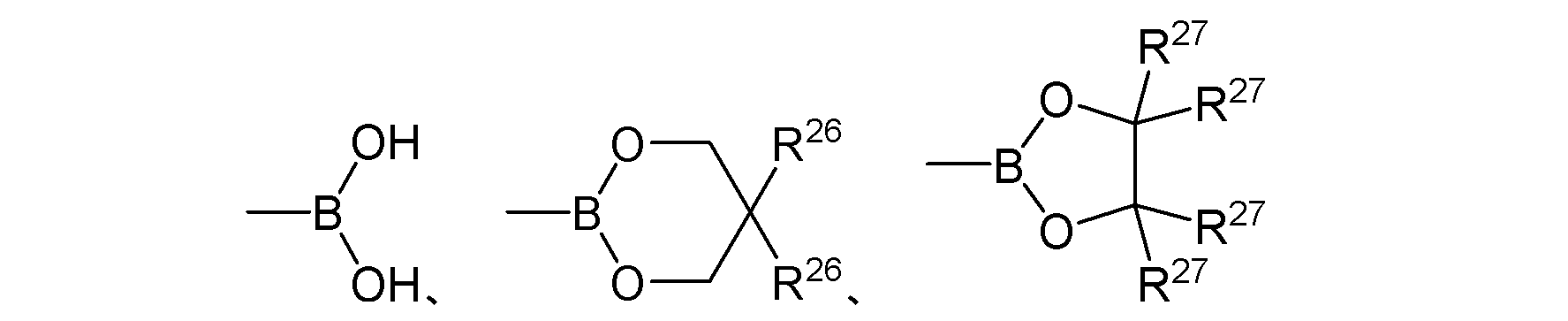
[式中、R26~R27は同一又は異なって、アルキル基を示す。]
で表される基が挙げられる。 [Wherein R 26 to R 27 are the same or different and each represents an alkyl group. ]
The group represented by these is mentioned.
で表される基が挙げられる。 [Wherein R 26 to R 27 are the same or different and each represents an alkyl group. ]
The group represented by these is mentioned.
R26~R27で示されるアルキル基としては、メチル基、エチル基、t-ブチル基等のC1-6アルキル基、特にC1-4アルキル基が挙げられる。
Examples of the alkyl group represented by R 26 to R 27 include a C1-6 alkyl group such as a methyl group, an ethyl group, and a t-butyl group, particularly a C1-4 alkyl group.
化合物(10a)→化合物(10b)
本工程で使用できるハロゲン化剤としては、特に制限されず、塩素(Cl2)、臭素(Br2)、ヨウ素(I2)、1,2-ジブロモエタン、N-クロロスクシンイミド、N-ブロモスクシンイミド(NBS)、臭化水素等が挙げられる。ハロゲン化剤の使用量は、使用するハロゲン化剤の種類に応じて適宜設定され得るが、通常、化合物(10a)1モルに対して、0.2~5モルが好ましく、0.5~2モルがより好ましい。 Compound (10a) → Compound (10b)
The halogenating agent that can be used in this step is not particularly limited, and is chlorine (Cl 2 ), bromine (Br 2 ), iodine (I 2 ), 1,2-dibromoethane, N-chlorosuccinimide, N-bromosuccinimide. (NBS), hydrogen bromide and the like. The amount of the halogenating agent to be used can be appropriately set depending on the kind of the halogenating agent to be used, but is usually preferably 0.2 to 5 mol, more preferably 0.5 to 2 mol, relative to 1 mol of the compound (10a). .
本工程で使用できるハロゲン化剤としては、特に制限されず、塩素(Cl2)、臭素(Br2)、ヨウ素(I2)、1,2-ジブロモエタン、N-クロロスクシンイミド、N-ブロモスクシンイミド(NBS)、臭化水素等が挙げられる。ハロゲン化剤の使用量は、使用するハロゲン化剤の種類に応じて適宜設定され得るが、通常、化合物(10a)1モルに対して、0.2~5モルが好ましく、0.5~2モルがより好ましい。 Compound (10a) → Compound (10b)
The halogenating agent that can be used in this step is not particularly limited, and is chlorine (Cl 2 ), bromine (Br 2 ), iodine (I 2 ), 1,2-dibromoethane, N-chlorosuccinimide, N-bromosuccinimide. (NBS), hydrogen bromide and the like. The amount of the halogenating agent to be used can be appropriately set depending on the kind of the halogenating agent to be used, but is usually preferably 0.2 to 5 mol, more preferably 0.5 to 2 mol, relative to 1 mol of the compound (10a). .
本工程で使用できるR4Yで表される化合物の使用量は、通常、化合物(10a)1モルに対して、0.2~5モルが好ましく、0.5~2モルがより好ましい。
The amount of the compound represented by R 4 Y that can be used in this step is usually preferably 0.2 to 5 mol, more preferably 0.5 to 2 mol, per 1 mol of compound (10a).
本工程で使用できるパラジウム化合物としては、例えば、酢酸パラジウム(Pd(OCOCH3)2;Pd(OAc)2)、テトラキス(トリフェニルホスフィン)パラジウム(0)(Pd(PPh3)4)、トリフルオロ酢酸パラジウム(Pd(OCOCF3)2)、塩化パラジウム(PdCl2)、臭化パラジウム(PdBr2)、ヨウ化パラジウム(PdI2)、Pd(CH2COCH2COCH3)2、K2PdCl4、K2PdCl6、K2Pd(NO3)4、トリス(ジベンジリデンアセトン)ジパラジウム(0)(Pd2(dba)3)、ビス(ジベンジリデンアセトン)パラジウム(0)、ジクロロ(1,5-シクロオクタジエン)パラジウム(II)、2,5-ノルボルナジエンパラジウムジクロリド等が挙げられる。これらのパラジウム化合物は、溶媒和物であってもよい。これらは単独で用いることもでき、2種以上を組合せて用いることもできる。なかでも、本工程では、収率及び合成の容易さの観点から、トリス(ジベンジリデンアセトン)ジパラジウム(0)(Pd2(dba)3)が好ましい。パラジウム化合物の使用量は、通常、化合物(10a)1モルに対して、0.002~0.1モルが好ましく、0.005~0.05モルがより好ましい。
Examples of the palladium compound that can be used in this step include palladium acetate (Pd (OCOCH 3 ) 2 ; Pd (OAc) 2 ), tetrakis (triphenylphosphine) palladium (0) (Pd (PPh 3 ) 4 ), trifluoro Palladium acetate (Pd (OCOCF 3 ) 2 ), Palladium chloride (PdCl 2 ), Palladium bromide (PdBr 2 ), Palladium iodide (PdI 2 ), Pd (CH 2 COCH 2 COCH 3 ) 2 , K 2 PdCl 4 , K 2 PdCl 6 , K 2 Pd (NO 3 ) 4 , tris (dibenzylideneacetone) dipalladium (0) (Pd 2 (dba) 3 ), bis (dibenzylideneacetone) palladium (0), dichloro (1,5 -Cyclooctadiene) palladium (II), 2,5-norbornadiene palladium dichloride, and the like. These palladium compounds may be solvates. These can be used alone or in combination of two or more. Among these, in this step, tris (dibenzylideneacetone) dipalladium (0) (Pd 2 (dba) 3 ) is preferable from the viewpoints of yield and ease of synthesis. The amount of the palladium compound used is usually preferably from 0.002 to 0.1 mol, more preferably from 0.005 to 0.05 mol, based on 1 mol of the compound (10a).
本工程では、必要に応じて配位子化合物を使用することもできる。使用できる配位子化合物としては、例えば、トリフェニルホスフィン、トリメトキシホスフィン、トリエチルホスフィン、トリイソプロピルホスフィン、トリ(t-ブチル)ホスフィン、トリ(n-ブチル)ホスフィン、トリイソプロポキシホスフィン、トリシクロペンチルホスフィン、トリシクロヘキシルホスフィン、トリメシチルホスフィン、トリフェノキシホスフィン、ジ(t-ブチル)メチルホスフィン、メチルジフェニルホスフィン、ジメチルフェニルホスフィン、トリエチルアミン、ピリジン、2,2’-ビピリジル、4,4’-(t-ブチル)ビピリジル、1,1’-ビス(ジフェニルホスフィノ)フェロセン、1,1’-ビス(t-ブチル)フェロセン、ジフェニルホスフィノメタン、1,2-ビス(ジフェニルホスフィノ)エタン、1,3-ビス(ジフェニルホスフィノ)プロパン、1,5-ビス(ジフェニルホスフィノ)ペンタン、1,2-ビス(ジペンタフルオロフェニルホスフィノ)エタン、1,2-ビス(ジシクロヘキシルホスフィノ)エタン、1,3-(ジシクロヘキシルホスフィノ)プロパン、1,2-ビス(ジ-t-ブチルホスフィノ)エタン、1,3-ビス(ジ-t-ブチルホスフィノ)プロパン、1,2-ビス(ジフェニルホスフィノ)ベンゼン、1,5-シクロオクタジエン等が挙げられる。これらの配位子化合物は、溶媒和物であってもよい。これらは単独で用いることもでき、2種以上を組合せて用いることもできる。なかでも、本工程では、収率及び合成の容易さの観点から、トリ(t-ブチル)ホスフィンが好ましい。配位子化合物の使用量は、通常、パラジウム化合物1モルに対して、1~10モルが好ましく、3~5モルがより好ましい。
In this step, a ligand compound can be used as necessary. Examples of ligand compounds that can be used include triphenylphosphine, trimethoxyphosphine, triethylphosphine, triisopropylphosphine, tri (t-butyl) phosphine, tri (n-butyl) phosphine, triisopropoxyphosphine, and tricyclopentylphosphine. , Tricyclohexylphosphine, trimesitylphosphine, triphenoxyphosphine, di (t-butyl) methylphosphine, methyldiphenylphosphine, dimethylphenylphosphine, triethylamine, pyridine, 2,2'-bipyridyl, 4,4 '-(t-butyl ) Bipyridyl, 1,1'-bis (diphenylphosphino) ferrocene, 1,1'-bis (t-butyl) ferrocene, diphenylphosphinomethane, 1,2-bis (diphenylphosphino) ethane, 1,3- Bis (Gife Ruphosphino) propane, 1,5-bis (diphenylphosphino) pentane, 1,2-bis (dipentafluorophenylphosphino) ethane, 1,2-bis (dicyclohexylphosphino) ethane, 1,3- (dicyclohexylphos Fino) propane, 1,2-bis (di-t-butylphosphino) ethane, 1,3-bis (di-t-butylphosphino) propane, 1,2-bis (diphenylphosphino) benzene, 1, And 5-cyclooctadiene. These ligand compounds may be solvates. These can be used alone or in combination of two or more. Of these, tri (t-butyl) phosphine is preferable in this step from the viewpoint of yield and ease of synthesis. The amount of the ligand compound used is usually preferably 1 to 10 moles, more preferably 3 to 5 moles per mole of palladium compound.
本工程では、必要に応じて塩基を使用することもできる。本工程で使用できる塩基としては、例えば、カリウムt-ブトキシド、ナトリウムt-ブトキシド、リチウムt-ブトキシド等の金属アルコキシド;リン酸リチウム、リン酸ナトリウム、リン酸カリウム等のリン酸アルカリ金属塩;水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化セシウム、水酸化バリウム等の金属水酸化物;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の炭酸金属塩;トリエチルアミン、ジイソプロピルエチルアミン等のアミン;ピペリジン、N-メチルピペリジン、2,2,6,6-テトラメチルピペリジン(TEMPO)等が挙げられる。これらは単独で用いることもでき、2種以上を組合せて用いることもできる。なかでも、本工程では、収率及び合成の容易さの観点から、金属水酸化物が好ましく、水酸化ナトリウムがより好ましい。塩基の使用量は、通常、化合物(10a)1モルに対して、0.5~5モルが好ましく、1~3モルがより好ましい。
In this step, a base can be used as necessary. Examples of the base that can be used in this step include metal alkoxides such as potassium t-butoxide, sodium t-butoxide and lithium t-butoxide; alkali metal phosphates such as lithium phosphate, sodium phosphate and potassium phosphate; water Metal hydroxides such as lithium oxide, sodium hydroxide, potassium hydroxide, cesium hydroxide, barium hydroxide; metal carbonates such as lithium carbonate, sodium carbonate, potassium carbonate, cesium carbonate; amines such as triethylamine, diisopropylethylamine; Examples include piperidine, N-methylpiperidine, 2,2,6,6-tetramethylpiperidine (TEMPO) and the like. These can be used alone or in combination of two or more. Among these, in this step, metal hydroxide is preferable and sodium hydroxide is more preferable from the viewpoint of yield and ease of synthesis. The amount of the base to be used is generally preferably 0.5-5 mol, more preferably 1-3 mol, per 1 mol of compound (10a).
本工程は、通常溶媒中で実施することができる。溶媒としては、例えば、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、ジメトキシエタン、シクロペンチルメチルエーテル、t-ブチルメチルエーテル等の鎖状エーテル;テトラヒドロフラン(THF)、ジオキサン等の環状エーテル;ペンタン、ヘキサン、ヘプタン、シクロヘキサン等の脂肪族炭化水素;ジクロロメタン、ジクロロエタン、クロロホルム、四塩化炭素等の脂肪族ハロゲン化炭化水素;ベンゼン、トルエン、キシレン、メシチレン等の芳香族炭化水素;クロロベンゼン、トリフルオロトルエン等の芳香族ハロゲン化炭化水素;メタノール、エタノール、イソプロピルアルコール等のアルコール等が挙げられる。これらの溶媒は単独で用いることもでき、2種以上を組合せて用いることもできる。本工程では、収率及び合成の容易さの観点から、環状エーテルが好ましく、テトラヒドロフランがより好ましい。
This step can usually be carried out in a solvent. Examples of the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether, and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran (THF) and dioxane; pentane, hexane, heptane, Aliphatic hydrocarbons such as cyclohexane; Aliphatic halogenated hydrocarbons such as dichloromethane, dichloroethane, chloroform, and carbon tetrachloride; Aromatic hydrocarbons such as benzene, toluene, xylene, and mesitylene; Aromatic halogens such as chlorobenzene and trifluorotoluene Hydrocarbons; alcohols such as methanol, ethanol, isopropyl alcohol and the like. These solvents can be used alone or in combination of two or more. In this step, from the viewpoint of yield and ease of synthesis, a cyclic ether is preferable, and tetrahydrofuran is more preferable.
本工程は、不活性ガス雰囲気(窒素ガス、アルゴンガス等)下で行うことが好ましく、反応温度は、通常、-50~150℃程度が好ましく、0~100℃程度がより好ましい。反応時間は、反応が進行する時間とすることができ、通常、1~72時間程度が好ましく、2~48時間程度がより好ましい。
This step is preferably performed under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably about −50 to 150 ° C., more preferably about 0 to 100 ° C. The reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 72 hours, and more preferably about 2 to 48 hours.
反応終了後は、必要に応じて通常の単離及び精製工程を経て、目的化合物である化合物(10b)を得ることができる。
After completion of the reaction, the target compound (10b) can be obtained through ordinary isolation and purification steps as necessary.
化合物(10b)→化合物(10c)
本工程で使用できるR2Yで表される化合物の使用量は、通常、化合物(10b)1モルに対して、1~10モルが好ましく、3~5モルがより好ましい。 Compound (10b) → Compound (10c)
The amount of the compound represented by R 2 Y that can be used in this step is usually preferably 1 to 10 mol, more preferably 3 to 5 mol, per 1 mol of compound (10b).
本工程で使用できるR2Yで表される化合物の使用量は、通常、化合物(10b)1モルに対して、1~10モルが好ましく、3~5モルがより好ましい。 Compound (10b) → Compound (10c)
The amount of the compound represented by R 2 Y that can be used in this step is usually preferably 1 to 10 mol, more preferably 3 to 5 mol, per 1 mol of compound (10b).
本工程で使用できるパラジウム化合物としては、特に限定されず、前記したものが挙げられる。本工程では、収率及び合成の容易さの観点から、酢酸パラジウム(Pd(OCOCH3)2;Pd(OAc)2)が好ましい。パラジウム化合物の使用量は、通常、化合物(10b)1モルに対して、0.02~0.5モルが好ましく、0.05~0.2モルがより好ましい。
It does not specifically limit as a palladium compound which can be used at this process, What was mentioned above is mentioned. In this step, palladium acetate (Pd (OCOCH 3 ) 2 ; Pd (OAc) 2 ) is preferable from the viewpoints of yield and ease of synthesis. The amount of the palladium compound used is usually preferably from 0.02 to 0.5 mol, more preferably from 0.05 to 0.2 mol, per 1 mol of compound (10b).
本工程では、必要に応じて配位子化合物を使用することもできる。使用できる配位子化合物としては、特に限定されず、前記したものが挙げられる。本工程では、収率及び合成の容易さの観点から、2,2’-ビピリジル、4,4’-(t-ブチル)ビピリジル等が好ましく、2,2’-ビピリジルがより好ましい。配位子化合物の使用量は、通常、パラジウム化合物1モルに対して、0.2~5モルが好ましく、0.5~2モルがより好ましい。
In this step, a ligand compound can be used as necessary. The ligand compound that can be used is not particularly limited, and examples thereof include those described above. In this step, 2,2′-bipyridyl, 4,4 ′-(t-butyl) bipyridyl and the like are preferable and 2,2′-bipyridyl is more preferable from the viewpoint of yield and ease of synthesis. The amount of the ligand compound used is usually preferably 0.2 to 5 mol, more preferably 0.5 to 2 mol, relative to 1 mol of the palladium compound.
本工程では、必要に応じて塩基を使用することもできる。本工程で使用できる塩基としては、特に限定されず、前記したものが挙げられる。本工程では、収率及び合成の容易さの観点から、ピペリジン、N-メチルピペリジン、2,2,6,6-テトラメチルピペリジン(TEMPO)等が好ましく、2,2,6,6-テトラメチルピペリジン(TEMPO)がより好ましい。塩基の使用量は、通常、化合物(10b)1モルに対して、1~10モルが好ましく、3~5モルがより好ましい。
In this step, a base can be used as necessary. It does not specifically limit as a base which can be used at this process, What was mentioned above is mentioned. In this step, piperidine, N-methylpiperidine, 2,2,6,6-tetramethylpiperidine (TEMPO) and the like are preferable from the viewpoint of yield and ease of synthesis, and 2,2,6,6-tetramethyl is preferable. Piperidine (TEMPO) is more preferred. The amount of base used is usually preferably 1 to 10 mol, more preferably 3 to 5 mol, per 1 mol of compound (10b).
本工程は、通常溶媒中で実施することができる。溶媒としては、例えば、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、ジメトキシエタン、シクロペンチルメチルエーテル、t-ブチルメチルエーテル等の鎖状エーテル;テトラヒドロフラン、ジオキサン等の環状エーテル;ペンタン、ヘキサン、ヘプタン、シクロヘキサン等の脂肪族炭化水素;ジクロロメタン、ジクロロエタン、クロロホルム、四塩化炭素等の脂肪族ハロゲン化炭化水素;ベンゼン、トルエン、キシレン、メシチレン等の芳香族炭化水素;クロロベンゼン、トリフルオロトルエン等の芳香族ハロゲン化炭化水素;メタノール、エタノール、イソプロピルアルコール等のアルコール等が挙げられる。これらの溶媒は単独で用いることもでき、2種以上を組合せて用いることもできる。本工程では、収率及び合成の容易さの観点から、芳香族ハロゲン化炭化水素が好ましく、トリフルオロトルエンがより好ましい。
This step can usually be carried out in a solvent. Examples of the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane. Aliphatic hydrocarbons; Aliphatic halogenated hydrocarbons such as dichloromethane, dichloroethane, chloroform and carbon tetrachloride; Aromatic hydrocarbons such as benzene, toluene, xylene and mesitylene; Aromatic halogenated hydrocarbons such as chlorobenzene and trifluorotoluene Alcohols such as methanol, ethanol, isopropyl alcohol and the like. These solvents can be used alone or in combination of two or more. In this step, an aromatic halogenated hydrocarbon is preferred and trifluorotoluene is more preferred from the viewpoint of yield and ease of synthesis.
本工程は、不活性ガス雰囲気(窒素ガス、アルゴンガス等)下で行うことが好ましいが、脱気しなくてもよいし、水分を除去しなくてもよいため大量合成を容易に行うことができる。反応温度は、通常、0~150℃程度が好ましく、50~100℃程度がより好ましい。反応時間は、反応が進行する時間とすることができ、通常、1~96時間程度が好ましく、2~72時間程度がより好ましい。
Although this step is preferably performed under an inert gas atmosphere (nitrogen gas, argon gas, etc.), it is not necessary to deaerate and it is not necessary to remove moisture, so that mass synthesis can be easily performed. it can. The reaction temperature is usually preferably about 0 to 150 ° C, more preferably about 50 to 100 ° C. The reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 96 hours, more preferably about 2 to 72 hours.
反応終了後は、必要に応じて通常の単離及び精製工程を経て、目的化合物である化合物(10c)を得ることができる。
After completion of the reaction, the target compound (10c) can be obtained through normal isolation and purification steps as necessary.
化合物(10c)→化合物(10d)
本工程は、非特許文献1に開示された方法にしたがって行うことができる。例えば、塩化パラジウム(PdCl2)等のパラジウム化合物の存在下、R1Xで表される化合物を用いて、化合物(10c)に対してR1を導入することができる。この際、必要に応じて、ビピリジル等の配位子化合物、炭酸銀(Ag2CO3)等の銀化合物を使用することもできる。 Compound (10c) → Compound (10d)
This step can be performed according to the method disclosed in Non-Patent Document 1. For example, R 1 can be introduced into the compound (10c) using a compound represented by R 1 X in the presence of a palladium compound such as palladium chloride (PdCl 2 ). At this time, a ligand compound such as bipyridyl and a silver compound such as silver carbonate (Ag 2 CO 3 ) can be used as necessary.
本工程は、非特許文献1に開示された方法にしたがって行うことができる。例えば、塩化パラジウム(PdCl2)等のパラジウム化合物の存在下、R1Xで表される化合物を用いて、化合物(10c)に対してR1を導入することができる。この際、必要に応じて、ビピリジル等の配位子化合物、炭酸銀(Ag2CO3)等の銀化合物を使用することもできる。 Compound (10c) → Compound (10d)
This step can be performed according to the method disclosed in Non-Patent Document 1. For example, R 1 can be introduced into the compound (10c) using a compound represented by R 1 X in the presence of a palladium compound such as palladium chloride (PdCl 2 ). At this time, a ligand compound such as bipyridyl and a silver compound such as silver carbonate (Ag 2 CO 3 ) can be used as necessary.
化合物(10d)→化合物(10e)
本工程は、非特許文献1に開示された方法にしたがって行うことができる。例えば、トリブロモボラン等を用いて化合物(10d)を脱アルキル化した後に、トリフルオロメタンスルホン酸無水物等を用いてトリフルオロメタンスルホニル化を行うことができる。トリフルオロメタンスルホニル化は、必要に応じてジイソプロプルエチルアミン等の塩基の存在下に行うこともできる。 Compound (10d) → Compound (10e)
This step can be performed according to the method disclosed in Non-Patent Document 1. For example, after dealkylating the compound (10d) using tribromoborane or the like, trifluoromethanesulfonylation can be performed using trifluoromethanesulfonic anhydride or the like. Trifluoromethanesulfonylation can also be performed in the presence of a base such as diisopropylethylamine as necessary.
本工程は、非特許文献1に開示された方法にしたがって行うことができる。例えば、トリブロモボラン等を用いて化合物(10d)を脱アルキル化した後に、トリフルオロメタンスルホン酸無水物等を用いてトリフルオロメタンスルホニル化を行うことができる。トリフルオロメタンスルホニル化は、必要に応じてジイソプロプルエチルアミン等の塩基の存在下に行うこともできる。 Compound (10d) → Compound (10e)
This step can be performed according to the method disclosed in Non-Patent Document 1. For example, after dealkylating the compound (10d) using tribromoborane or the like, trifluoromethanesulfonylation can be performed using trifluoromethanesulfonic anhydride or the like. Trifluoromethanesulfonylation can also be performed in the presence of a base such as diisopropylethylamine as necessary.
化合物(10e)→四置換チオフェン化合物(9)
本工程は、非特許文献1に開示された方法にしたがって行うことができる。例えば、テトラキス(トリフェニルホスフィン)パラジウム(0)(Pd(PPh3)4)等のパラジウム化合物の存在下、R3Yで表される化合物を用いて、化合物(10e)に対してR3を導入することができる。この際、必要に応じて、水酸化バリウム等の塩基を使用することもできる。 Compound (10e) → Tetrasubstituted thiophene compound (9)
This step can be performed according to the method disclosed in Non-Patent Document 1. For example, the presence of tetrakis (triphenylphosphine) palladium (0) (Pd (PPh 3) 4) a palladium compound, such as, by using a compound represented by R 3 Y, the R 3 for the compound (10e) Can be introduced. At this time, a base such as barium hydroxide may be used as necessary.
本工程は、非特許文献1に開示された方法にしたがって行うことができる。例えば、テトラキス(トリフェニルホスフィン)パラジウム(0)(Pd(PPh3)4)等のパラジウム化合物の存在下、R3Yで表される化合物を用いて、化合物(10e)に対してR3を導入することができる。この際、必要に応じて、水酸化バリウム等の塩基を使用することもできる。 Compound (10e) → Tetrasubstituted thiophene compound (9)
This step can be performed according to the method disclosed in Non-Patent Document 1. For example, the presence of tetrakis (triphenylphosphine) palladium (0) (Pd (PPh 3) 4) a palladium compound, such as, by using a compound represented by R 3 Y, the R 3 for the compound (10e) Can be introduced. At this time, a base such as barium hydroxide may be used as necessary.
(2-2)工程(1-I):四置換チオフェン化合物(9)の酸化
上記した四置換チオフェン化合物(9)と後述の化合物(4)又は化合物(5)とを反応させても環化反応は進行せず、本発明の多置換芳香族化合物は得られない。本発明では、四置換チオフェン化合物(9)の反応性を向上させるため、原料として四置換チオフェンS-オキシド化合物(3)を用いることが好ましい。 (2-2) Step (1-I): Oxidation of tetrasubstituted thiophene compound (9) Cyclization can be achieved by reacting tetrasubstituted thiophene compound (9) with compound (4) or compound (5) described below. The reaction does not proceed and the polysubstituted aromatic compound of the present invention cannot be obtained. In the present invention, in order to improve the reactivity of the tetrasubstituted thiophene compound (9), it is preferable to use the tetrasubstituted thiophene S-oxide compound (3) as a raw material.
上記した四置換チオフェン化合物(9)と後述の化合物(4)又は化合物(5)とを反応させても環化反応は進行せず、本発明の多置換芳香族化合物は得られない。本発明では、四置換チオフェン化合物(9)の反応性を向上させるため、原料として四置換チオフェンS-オキシド化合物(3)を用いることが好ましい。 (2-2) Step (1-I): Oxidation of tetrasubstituted thiophene compound (9) Cyclization can be achieved by reacting tetrasubstituted thiophene compound (9) with compound (4) or compound (5) described below. The reaction does not proceed and the polysubstituted aromatic compound of the present invention cannot be obtained. In the present invention, in order to improve the reactivity of the tetrasubstituted thiophene compound (9), it is preferable to use the tetrasubstituted thiophene S-oxide compound (3) as a raw material.
本工程では、上記した四置換チオフェン化合物(9)を酸化させる(工程1-I)ことで、四置換チオフェンS-オキシド化合物(3)を得ることができる。
In this step, the tetrasubstituted thiophene S-oxide compound (3) can be obtained by oxidizing the tetrasubstituted thiophene compound (9) (step 1-I).
本工程において、酸化に使用される酸化剤としては、特に制限されないが、例えば、塩素;過酸化水素;過酢酸、過安息香酸、m-クロロ過安息香酸(m-CPBA)等の過酸;t-ブチルペルオキシド等のペルオキシド;メタ過ヨウ素酸ナトリウム等の過ハロゲン酸塩等が挙げられる。これらは単独で用いることもでき、2種以上を組合せて用いることもできる。なかでも、本工程では、収率及び合成の容易さの観点から、過酸が好ましく、m-クロロ過安息香酸(m-CPBA)がより好ましい。酸化剤の使用量は、通常、四置換チオフェン化合物(9)1モルに対して、1~10モルが好ましく、3~5モルがより好ましい。
In this step, the oxidizing agent used for the oxidation is not particularly limited. For example, chlorine; hydrogen peroxide; peracids such as peracetic acid, perbenzoic acid, m-chloroperbenzoic acid (m-CPBA); Examples thereof include peroxides such as t-butyl peroxide; perhalogenates such as sodium metaperiodate. These can be used alone or in combination of two or more. Of these, in this step, peracid is preferable and m-chloroperbenzoic acid (m-CPBA) is more preferable from the viewpoint of yield and ease of synthesis. The amount of the oxidizing agent used is usually preferably 1 to 10 moles, more preferably 3 to 5 moles per mole of the tetrasubstituted thiophene compound (9).
本工程では、上記酸化剤の他、酸触媒として三フッ化ホウ素化合物を使用することが好ましい。使用できる三フッ化ホウ素化合物としては、例えば、三フッ化ホウ素ジエチルエーテル錯体(BF3・OEt2)、三フッ化ホウ素テトラヒドロフラン錯体、三フッ化ホウ素メタノール錯体等が挙げられる。これらは単独で用いることもでき、2種以上を組合せて用いることもできる。なかでも、本工程では、収率及び合成の容易さの観点から、三フッ化ホウ素ジエチルエーテル錯体(BF3・OEt2)が好ましい。三フッ化ホウ素化合物の使用量は、通常、四置換チオフェン化合物(9)1モルに対して、2~30モルが好ましく、5~20モルがより好ましい。
In this step, it is preferable to use a boron trifluoride compound as an acid catalyst in addition to the oxidizing agent. Examples of the boron trifluoride compound that can be used include boron trifluoride diethyl ether complex (BF 3 · OEt 2 ), boron trifluoride tetrahydrofuran complex, and boron trifluoride methanol complex. These can be used alone or in combination of two or more. Among these, boron trifluoride diethyl ether complex (BF 3 · OEt 2 ) is preferable in this step from the viewpoint of yield and ease of synthesis. The amount of boron trifluoride compound used is usually preferably 2 to 30 moles, more preferably 5 to 20 moles per mole of tetrasubstituted thiophene compound (9).
本工程は、通常溶媒中で実施することができる。溶媒としては、例えば、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、ジメトキシエタン、シクロペンチルメチルエーテル、t-ブチルメチルエーテル等の鎖状エーテル;テトラヒドロフラン、ジオキサン等の環状エーテル;ペンタン、ヘキサン、ヘプタン、シクロヘキサン等の脂肪族炭化水素;ジクロロメタン、ジクロロエタン、クロロホルム、四塩化炭素等の脂肪族ハロゲン化炭化水素;ベンゼン、トルエン、キシレン、メシチレン等の芳香族炭化水素;クロロベンゼン、トリフルオロトルエン等の芳香族ハロゲン化炭化水素;メタノール、エタノール、イソプロピルアルコール等のアルコール等が挙げられる。これらの溶媒は単独で用いることもでき、2種以上を組合せて用いることもできる。本工程では、収率及び合成の容易さの観点から、脂肪族ハロゲン化炭化水素が好ましく、ジクロロメタンがより好ましい。
This step can usually be carried out in a solvent. Examples of the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane. Aliphatic hydrocarbons; Aliphatic halogenated hydrocarbons such as dichloromethane, dichloroethane, chloroform and carbon tetrachloride; Aromatic hydrocarbons such as benzene, toluene, xylene and mesitylene; Aromatic halogenated hydrocarbons such as chlorobenzene and trifluorotoluene Alcohols such as methanol, ethanol, isopropyl alcohol and the like. These solvents can be used alone or in combination of two or more. In this step, from the viewpoints of yield and ease of synthesis, aliphatic halogenated hydrocarbons are preferable, and dichloromethane is more preferable.
本工程は、不活性ガス雰囲気(窒素ガス、アルゴンガス等)下で行うことが好ましく、反応温度は、通常、-100~50℃程度が好ましく、-50~0℃程度がより好ましい。反応時間は、反応が進行する時間とすることができ、通常、1~24時間程度が好ましく、2~12時間程度がより好ましい。
This step is preferably performed under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably about −100 to 50 ° C., more preferably about −50 to 0 ° C. The reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 24 hours, and more preferably about 2 to 12 hours.
反応終了後は、必要に応じて通常の単離及び精製工程を経て、目的化合物である四置換チオフェンS-オキシド化合物(3)を得ることができる。
After completion of the reaction, the target compound, tetrasubstituted thiophene S-oxide compound (3), can be obtained through ordinary isolation and purification steps as necessary.
(2-3)工程(1-II)及び(1-III):化合物(4)及び化合物(5)
本発明において、四置換チオフェンS-オキシド化合物(3)と反応させる原料として、上記一般式(4): (2-3) Steps (1-II) and (1-III): Compound (4) and Compound (5)
In the present invention, as a raw material to be reacted with the tetrasubstituted thiophene S-oxide compound (3), the above general formula (4):
本発明において、四置換チオフェンS-オキシド化合物(3)と反応させる原料として、上記一般式(4): (2-3) Steps (1-II) and (1-III): Compound (4) and Compound (5)
In the present invention, as a raw material to be reacted with the tetrasubstituted thiophene S-oxide compound (3), the above general formula (4):

[式中、R5~R8は前記に同じである。R19及びR20は片方はカルボキシ基で他方がアミノ基であるか、片方がシリル基で他方がトリフルオロメタンスルホナート基である。]
で表される化合物を使用することができる。 [Wherein R 5 to R 8 are the same as defined above. One of R 19 and R 20 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group. ]
The compound represented by these can be used.
で表される化合物を使用することができる。 [Wherein R 5 to R 8 are the same as defined above. One of R 19 and R 20 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group. ]
The compound represented by these can be used.
一般式(4)において、R19及びR20は片方はカルボキシ基で他方がアミノ基であるか、片方がシリル基(トリメチルシリル基、トリエチルシリル基等)で他方がトリフルオロメタンスルホナート基である。
In the general formula (4), one of R 19 and R 20 is a carboxy group and the other is an amino group, or one is a silyl group (trimethylsilyl group, triethylsilyl group, etc.) and the other is a trifluoromethanesulfonate group.
このような一般式(4)で表される化合物は、例えば、
The compound represented by the general formula (4) is, for example,

[式中、OTfはトリフルオロメタンスルホナート基を示す。TMSはトリメチルシリル基を示す。以下同様である。]
等が挙げられる。 [Wherein, OTf represents a trifluoromethanesulfonate group. TMS represents a trimethylsilyl group. The same applies hereinafter. ]
Etc.
等が挙げられる。 [Wherein, OTf represents a trifluoromethanesulfonate group. TMS represents a trimethylsilyl group. The same applies hereinafter. ]
Etc.
このような一般式(4)で表される化合物は、一般式(6):
The compound represented by the general formula (4) is represented by the general formula (6):

[式中、R5~R8は前記に同じである。]
で表される化合物と、マレイミドとを反応させる工程、及び
前記工程で得られた化合物と、酸化剤及び第1塩基とを反応させた後に、さらに第2塩基と反応させる工程
を備える方法で得ることができる。 [Wherein R 5 to R 8 are the same as defined above. ]
Obtained by reacting the compound represented by the formula with maleimide, and reacting the compound obtained in the previous step with the oxidizing agent and the first base, and further reacting with the second base. be able to.
で表される化合物と、マレイミドとを反応させる工程、及び
前記工程で得られた化合物と、酸化剤及び第1塩基とを反応させた後に、さらに第2塩基と反応させる工程
を備える方法で得ることができる。 [Wherein R 5 to R 8 are the same as defined above. ]
Obtained by reacting the compound represented by the formula with maleimide, and reacting the compound obtained in the previous step with the oxidizing agent and the first base, and further reacting with the second base. be able to.
一般式(6)で表される化合物は、上記した一般式(3)で表される四置換チオフェンS-オキシド化合物と同様のものを採用することができる。
As the compound represented by the general formula (6), a compound similar to the tetrasubstituted thiophene S-oxide compound represented by the general formula (3) can be employed.
本発明では、まず、一般式(6)で表される化合物とマレイミドとを反応させる。
In the present invention, first, the compound represented by the general formula (6) is reacted with maleimide.
マレイミドの使用量は、通常、化合物(6)1モルに対して、1~10モルが好ましく、2~5モルがより好ましい。
The amount of maleimide used is usually preferably from 1 to 10 mol, more preferably from 2 to 5 mol, per 1 mol of compound (6).
本工程は、通常溶媒中で実施することができる。溶媒としては、例えば、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、ジメトキシエタン、シクロペンチルメチルエーテル、t-ブチルメチルエーテル等の鎖状エーテル;テトラヒドロフラン、ジオキサン等の環状エーテル;ペンタン、ヘキサン、ヘプタン、シクロヘキサン等の脂肪族炭化水素;ジクロロメタン、ジクロロエタン、クロロホルム、四塩化炭素等の脂肪族ハロゲン化炭化水素;ベンゼン、トルエン、キシレン、メシチレン、クロロベンゼン、ニトロベンゼン、トリフルオロトルエン等の芳香族化合物;メタノール、エタノール、イソプロピルアルコール等のアルコール等が挙げられる。これらの溶媒は単独で用いることもでき、2種以上を組合せて用いることもできる。本工程では、収率及び合成の容易さの観点から、芳香族化合物が好ましく、ニトロベンゼンがより好ましい。
This step can usually be carried out in a solvent. Examples of the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane. Aliphatic hydrocarbons; Aliphatic halogenated hydrocarbons such as dichloromethane, dichloroethane, chloroform, carbon tetrachloride; Aromatic compounds such as benzene, toluene, xylene, mesitylene, chlorobenzene, nitrobenzene, trifluorotoluene; Methanol, ethanol, isopropyl alcohol And alcohol. These solvents can be used alone or in combination of two or more. In this step, an aromatic compound is preferable and nitrobenzene is more preferable from the viewpoints of yield and ease of synthesis.
本工程は、不活性ガス雰囲気(窒素ガス、アルゴンガス等)下で行うことが好ましく、反応温度は、通常、100~300℃程度が好ましく、150~250℃程度がより好ましい。反応時間は、反応が進行する時間とすることができ、通常、1~96時間程度が好ましく、2~72時間程度がより好ましい。
This step is preferably performed under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably about 100 to 300 ° C, more preferably about 150 to 250 ° C. The reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 96 hours, more preferably about 2 to 72 hours.
反応終了後は、必要に応じて通常の単離及び精製工程を経て、一般式(11):
After completion of the reaction, the general formula (11):

[式中、R5~R8は前記に同じである。]
で表される化合物を得て、次の工程に用いることができる。 [Wherein R 5 to R 8 are the same as defined above. ]
Can be used in the next step.
で表される化合物を得て、次の工程に用いることができる。 [Wherein R 5 to R 8 are the same as defined above. ]
Can be used in the next step.
次の工程では、得られた一般式(11)で表される化合物と、酸化剤及び第1塩基とを反応させた後に、さらに第2塩基と反応させることにより、一般式(4)で表される化合物を得ることができる。
In the next step, the compound represented by the general formula (11) is reacted with the oxidizing agent and the first base, and then further reacted with the second base, whereby the compound represented by the general formula (4) is obtained. Can be obtained.
使用される酸化剤としては、例えば、塩素;過酸化水素;過酢酸、過安息香酸、m-クロロ過安息香酸(m-CPBA)等の過酸;ジメチルジオキシラン、メチルトリフルオロメチルジオキシラン、t-ブチルペルオキシド等のペルオキシド;メタ過ヨウ素酸ナトリウム等の過ハロゲン酸塩;次亜塩素酸ナトリウム等の次亜塩素酸等が挙げられる。これらは単独で用いることもでき、2種以上を組合せて用いることもできる。なかでも、本工程では、収率及び合成の容易さの観点から、次亜塩素酸塩が好ましく、次亜塩素酸ナトリウムがより好ましい。酸化剤の使用量は、通常、化合物(11)1モルに対して、0.5~5モルが好ましく、1~2モルがより好ましい。
Examples of the oxidizing agent used include chlorine; hydrogen peroxide; peracids such as peracetic acid, perbenzoic acid and m-chloroperbenzoic acid (m-CPBA); dimethyldioxirane, methyltrifluoromethyldioxirane, Examples thereof include peroxides such as t-butyl peroxide; perhalogenates such as sodium metaperiodate; hypochlorous acid such as sodium hypochlorite. These can be used alone or in combination of two or more. Among these, in this step, hypochlorite is preferable and sodium hypochlorite is more preferable from the viewpoint of yield and ease of synthesis. The amount of the oxidizing agent to be used is generally preferably 0.5 to 5 mol, more preferably 1 to 2 mol, relative to 1 mol of compound (11).
使用される第1塩基としては、例えば、カリウムt-ブトキシド、ナトリウムt-ブトキシド、リチウムt-ブトキシド等の金属アルコキシド;リン酸リチウム、リン酸ナトリウム、リン酸カリウム等のリン酸アルカリ金属塩;水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化セシウム、水酸化バリウム等の金属水酸化物;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の炭酸金属塩;トリエチルアミン、ジイソプロピルエチルアミン等のアミン;ピペリジン、N-メチルピペリジン、2,2,6,6-テトラメチルピペリジン(TEMPO)等が挙げられる。これらは単独で用いることもでき、2種以上を組合せて用いることもできる。なかでも、本工程では、収率及び合成の容易さの観点から、金属水酸化物が好ましく、水酸化ナトリウムがより好ましい。第1塩基の使用量は、通常、化合物(11)1モルに対して、2~20モルが好ましく、3~10モルがより好ましい。
Examples of the first base used include metal alkoxides such as potassium t-butoxide, sodium t-butoxide and lithium t-butoxide; alkali metal phosphates such as lithium phosphate, sodium phosphate and potassium phosphate; water Metal hydroxides such as lithium oxide, sodium hydroxide, potassium hydroxide, cesium hydroxide, barium hydroxide; metal carbonates such as lithium carbonate, sodium carbonate, potassium carbonate, cesium carbonate; amines such as triethylamine, diisopropylethylamine; Examples include piperidine, N-methylpiperidine, 2,2,6,6-tetramethylpiperidine (TEMPO) and the like. These can be used alone or in combination of two or more. Among these, in this step, metal hydroxide is preferable and sodium hydroxide is more preferable from the viewpoint of yield and ease of synthesis. The amount of the first base used is usually preferably 2 to 20 mol, more preferably 3 to 10 mol, per 1 mol of compound (11).
使用される第2塩基としては、例えば、カリウムt-ブトキシド、ナトリウムt-ブトキシド、リチウムt-ブトキシド等の金属アルコキシド;リン酸リチウム、リン酸ナトリウム、リン酸カリウム等のリン酸アルカリ金属塩;水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化セシウム、水酸化バリウム等の金属水酸化物;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の炭酸金属塩;トリエチルアミン、ジイソプロピルエチルアミン等のアミン;ピペリジン、N-メチルピペリジン、2,2,6,6-テトラメチルピペリジン(TEMPO)等が挙げられる。これらは単独で用いることもでき、2種以上を組合せて用いることもできる。なかでも、本工程では、収率及び合成の容易さの観点から、金属水酸化物が好ましく、水酸化カリウムがより好ましい。第2塩基の使用量は、通常、化合物(11)1モルに対して、10~100モルが好ましく、20~50モルがより好ましい。
Examples of the second base used include metal alkoxides such as potassium t-butoxide, sodium t-butoxide, and lithium t-butoxide; alkali metal phosphates such as lithium phosphate, sodium phosphate, and potassium phosphate; water Metal hydroxides such as lithium oxide, sodium hydroxide, potassium hydroxide, cesium hydroxide, barium hydroxide; metal carbonates such as lithium carbonate, sodium carbonate, potassium carbonate, cesium carbonate; amines such as triethylamine, diisopropylethylamine; Examples include piperidine, N-methylpiperidine, 2,2,6,6-tetramethylpiperidine (TEMPO) and the like. These can be used alone or in combination of two or more. Among these, in this step, metal hydroxide is preferable and potassium hydroxide is more preferable from the viewpoint of yield and ease of synthesis. The amount of the second base used is usually preferably 10 to 100 mol, more preferably 20 to 50 mol, per 1 mol of compound (11).
本工程は、通常溶媒中で実施することができる。溶媒としては、例えば、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、ジメトキシエタン、シクロペンチルメチルエーテル、t-ブチルメチルエーテル等の鎖状エーテル;テトラヒドロフラン、ジオキサン等の環状エーテル;ペンタン、ヘキサン、ヘプタン、シクロヘキサン等の脂肪族炭化水素;ジクロロメタン、ジクロロエタン、クロロホルム、四塩化炭素等の脂肪族ハロゲン化炭化水素;ベンゼン、トルエン、キシレン、メシチレン、クロロベンゼン、ニトロベンゼン、トリフルオロトルエン等の芳香族化合物;メタノール、エタノール、イソプロピルアルコール等のアルコール等が挙げられる。これらの溶媒は単独で用いることもでき、2種以上を組合せて用いることもできる。本工程では、収率及び合成の容易さの観点から、アルコールが好ましく、メタノール、イソプロピルアルコール等がより好ましい。
This step can usually be carried out in a solvent. Examples of the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane. Aliphatic hydrocarbons; Aliphatic halogenated hydrocarbons such as dichloromethane, dichloroethane, chloroform, carbon tetrachloride; Aromatic compounds such as benzene, toluene, xylene, mesitylene, chlorobenzene, nitrobenzene, trifluorotoluene; Methanol, ethanol, isopropyl alcohol And alcohol. These solvents can be used alone or in combination of two or more. In this step, alcohol is preferable from the viewpoints of yield and ease of synthesis, and methanol, isopropyl alcohol, and the like are more preferable.
本工程は、不活性ガス雰囲気(窒素ガス、アルゴンガス等)下で行うことが好ましく、反応温度は、通常、加熱下、特に還流下に行うことが好ましい。反応時間は、反応が進行する時間とすることができ、通常、1~48時間程度が好ましく、2~24時間程度がより好ましい。
This step is preferably carried out under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably carried out under heating, particularly under reflux. The reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 48 hours, and more preferably about 2 to 24 hours.
反応終了後は、必要に応じて通常の単離及び精製工程を経て、上記一般式(4)で表される化合物を得ることができる。
After completion of the reaction, the compound represented by the above general formula (4) can be obtained through normal isolation and purification steps as necessary.
本発明において、四置換チオフェンS-オキシド化合物(3)と反応させる原料として、上記一般式(5):
In the present invention, as a raw material to be reacted with the tetrasubstituted thiophene S-oxide compound (3), the above general formula (5):

[式中、R13~R18は前記に同じである。R21及びR22は片方はカルボキシ基で他方がアミノ基であるか、片方がシリル基で他方がトリフルオロメタンスルホナート基である。]
で表される化合物を使用することができる。 [Wherein R 13 to R 18 are the same as defined above. One of R 21 and R 22 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group. ]
The compound represented by these can be used.
で表される化合物を使用することができる。 [Wherein R 13 to R 18 are the same as defined above. One of R 21 and R 22 is a carboxy group and the other is an amino group, or one is a silyl group and the other is a trifluoromethanesulfonate group. ]
The compound represented by these can be used.
一般式(5)において、R21及びR22は片方はカルボキシ基で他方がアミノ基であるか、片方がシリル基(トリメチルシリル基、トリエチルシリル基等)で他方がトリフルオロメタンスルホナート基である。
In the general formula (5), one of R 21 and R 22 is a carboxy group and the other is an amino group, or one is a silyl group (trimethylsilyl group, triethylsilyl group, etc.) and the other is a trifluoromethanesulfonate group.
このような一般式(5)で表される化合物は、例えば、
The compound represented by the general formula (5) is, for example,

等が挙げられる。
Etc. *
このような一般式(5)で表される化合物は、一般式(7):
The compound represented by the general formula (5) is represented by the general formula (7):

[式中、R13~R16は前記に同じである。]
で表される化合物と、一般式(8): [Wherein R 13 to R 16 are the same as defined above. ]
And a compound represented by the general formula (8):
で表される化合物と、一般式(8): [Wherein R 13 to R 16 are the same as defined above. ]
And a compound represented by the general formula (8):

[式中、R17~R18及びR21~R22は前記に同じである。R23はシリル基である。]
で表される化合物とを反応させる工程、及び
前記工程で得られた化合物の水酸基をトリフルオロメタンスルホナート基に置換する工程を備える方法で得ることができる。 [Wherein R 17 to R 18 and R 21 to R 22 are the same as defined above. R 23 is a silyl group. ]
And a step of substituting a trifluoromethanesulfonate group for the hydroxyl group of the compound obtained in the above step.
で表される化合物とを反応させる工程、及び
前記工程で得られた化合物の水酸基をトリフルオロメタンスルホナート基に置換する工程を備える方法で得ることができる。 [Wherein R 17 to R 18 and R 21 to R 22 are the same as defined above. R 23 is a silyl group. ]
And a step of substituting a trifluoromethanesulfonate group for the hydroxyl group of the compound obtained in the above step.
一般式(7)で表される化合物は、上記した一般式(3)で表される四置換チオフェンS-オキシド化合物と同様のものを採用することができる。
As the compound represented by the general formula (7), a compound similar to the tetrasubstituted thiophene S-oxide compound represented by the general formula (3) can be employed.
一般式(8)において、R23で示されるシリル基としては、トリメチルシリル基、トリエチルシリル基等が挙げられる。
In the general formula (8), examples of the silyl group represented by R 23 include a trimethylsilyl group and a triethylsilyl group.
化合物(8)の使用量は、通常、化合物(7)1モルに対して、1~10モルが好ましく、2~5モルがより好ましい。
The amount of compound (8) used is usually preferably 1 to 10 mol, more preferably 2 to 5 mol, per 1 mol of compound (7). *
化合物(7)と化合物(8)とを反応させる際には、R21及びR22の片方がシリル基で他方がトリフルオロメタンスルホナート基である場合は、脱保護しつつ反応を進行させるために、脱保護剤を併用することが好ましい。脱保護剤としては、例えば、テトラブチルアンモニウムフルオライド等が挙げられる。脱保護剤の使用量は、通常、化合物(7)1モルに対して、3~20モルが好ましく、5~10モルがより好ましい。
When reacting compound (7) and compound (8), if one of R 21 and R 22 is a silyl group and the other is a trifluoromethanesulfonate group, the reaction proceeds while deprotecting It is preferable to use a deprotecting agent in combination. Examples of the deprotecting agent include tetrabutylammonium fluoride. The amount of the deprotecting agent used is usually preferably 3 to 20 mol, more preferably 5 to 10 mol, relative to 1 mol of the compound (7).
本工程は、通常溶媒中で実施することができる。溶媒としては、例えば、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、ジメトキシエタン、シクロペンチルメチルエーテル、t-ブチルメチルエーテル等の鎖状エーテル;テトラヒドロフラン、ジオキサン等の環状エーテル;ペンタン、ヘキサン、ヘプタン、シクロヘキサン等の脂肪族炭化水素;ジクロロメタン、ジクロロエタン、クロロホルム、四塩化炭素等の脂肪族ハロゲン化炭化水素;ベンゼン、トルエン、キシレン、メシチレン、クロロベンゼン、ニトロベンゼン、トリフルオロトルエン等の芳香族化合物;メタノール、エタノール、イソプロピルアルコール等のアルコール等が挙げられる。これらの溶媒は単独で用いることもでき、2種以上を組合せて用いることもできる。本工程では、収率及び合成の容易さの観点から、環状エーテルが好ましく、テトラヒドロフランがより好ましい。
This step can usually be carried out in a solvent. Examples of the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane. Aliphatic hydrocarbons; Aliphatic halogenated hydrocarbons such as dichloromethane, dichloroethane, chloroform, carbon tetrachloride; Aromatic compounds such as benzene, toluene, xylene, mesitylene, chlorobenzene, nitrobenzene, trifluorotoluene; Methanol, ethanol, isopropyl alcohol And alcohol. These solvents can be used alone or in combination of two or more. In this step, from the viewpoint of yield and ease of synthesis, a cyclic ether is preferable, and tetrahydrofuran is more preferable.
本工程は、不活性ガス雰囲気(窒素ガス、アルゴンガス等)下で行うことが好ましく、反応温度は、通常、-50~100℃が好ましく、0~50℃がより好ましい。反応時間は、反応が進行する時間とすることができ、通常、1~48時間程度が好ましく、2~36時間程度がより好ましい。
This step is preferably performed under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably −50 to 100 ° C., more preferably 0 to 50 ° C. The reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 48 hours, more preferably about 2 to 36 hours.
反応終了後は、必要に応じて通常の単離及び精製工程を経て、一般式(12):
After completion of the reaction, the general formula (12):

[式中、R13~R18及びR23は前記に同じである。]
で表される化合物を得て、次の工程に用いることができる。 [Wherein R 13 to R 18 and R 23 are the same as defined above. ]
Can be used in the next step.
で表される化合物を得て、次の工程に用いることができる。 [Wherein R 13 to R 18 and R 23 are the same as defined above. ]
Can be used in the next step.
次に、一般式(12)で表される化合物における水酸基をトリフルオロメタンスルホナート基に置換する。この工程は、水酸基をトリフルオロメタンスルホナート基で保護する場合に通常使用される方法で行うことができる。その後、必要に応じて通常の単離及び精製工程を経て、上記一般式(5)で表される化合物を得ることができる。
Next, the hydroxyl group in the compound represented by the general formula (12) is substituted with a trifluoromethanesulfonate group. This step can be carried out by a method usually used for protecting a hydroxyl group with a trifluoromethanesulfonate group. Thereafter, the compound represented by the general formula (5) can be obtained through ordinary isolation and purification steps as necessary.
(2-4)工程(1-IV):化合物(3)と、化合物(4)又は化合物(5)との反応
上記した四置換チオフェンS-オキシド化合物(3)は、ジエンとしての反応性を有する。 (2-4) Step (1-IV): Reaction of Compound (3) with Compound (4) or Compound (5) The tetrasubstituted thiophene S-oxide compound (3) described above exhibits reactivity as a diene. Have.
上記した四置換チオフェンS-オキシド化合物(3)は、ジエンとしての反応性を有する。 (2-4) Step (1-IV): Reaction of Compound (3) with Compound (4) or Compound (5) The tetrasubstituted thiophene S-oxide compound (3) described above exhibits reactivity as a diene. Have.
また、化合物(4)は、反応系中でR19及びR20(カルボキシ基及びアミノ基、又はシリル基及びトロフルオロメタンスルホナート基)が抜けて一般式(4’):
In addition, in the reaction system, the compound (4) has R 19 and R 20 (carboxy group and amino group, or silyl group and trifluoromethanesulfonate group) removed, and the general formula (4 ′):

[式中、R5~R8は前記に同じである。]
で表されるようなベンザイン骨格を得ることができ、四置換チオフェンS-オキシド化合物(3)と縮合反応を起こすことができ、本発明の多置換芳香族化合物を得ることができる。 [Wherein R 5 to R 8 are the same as defined above. ]
And a condensation reaction with the tetrasubstituted thiophene S-oxide compound (3) can be obtained, and the polysubstituted aromatic compound of the present invention can be obtained.
で表されるようなベンザイン骨格を得ることができ、四置換チオフェンS-オキシド化合物(3)と縮合反応を起こすことができ、本発明の多置換芳香族化合物を得ることができる。 [Wherein R 5 to R 8 are the same as defined above. ]
And a condensation reaction with the tetrasubstituted thiophene S-oxide compound (3) can be obtained, and the polysubstituted aromatic compound of the present invention can be obtained.
さらに、化合物(5)は、反応系中でR21及びR22(カルボキシ基及びアミノ基、又はシリル基及びトロフルオロメタンスルホナート基)が抜けて一般式(5’):
Further, in the reaction system, the compound (5) has R 21 and R 22 (carboxy group and amino group, or silyl group and trifluoromethanesulfonate group) eliminated, and the compound represented by the general formula (5 ′):

[式中、R13~R18は前記に同じである。]
で表されるようなベンザイン骨格を得ることができ、四置換チオフェンS-オキシド化合物(3)と縮合反応を起こすことができ、本発明の多置換芳香族化合物を得ることができる。 [Wherein R 13 to R 18 are the same as defined above. ]
And a condensation reaction with the tetrasubstituted thiophene S-oxide compound (3) can be obtained, and the polysubstituted aromatic compound of the present invention can be obtained.
で表されるようなベンザイン骨格を得ることができ、四置換チオフェンS-オキシド化合物(3)と縮合反応を起こすことができ、本発明の多置換芳香族化合物を得ることができる。 [Wherein R 13 to R 18 are the same as defined above. ]
And a condensation reaction with the tetrasubstituted thiophene S-oxide compound (3) can be obtained, and the polysubstituted aromatic compound of the present invention can be obtained.
本工程において、化合物(4)又は化合物(5)の使用量は、通常、四置換チオフェンS-オキシド化合物(3)1モルに対して、0.1~2モルが好ましく、0.2~1モルがより好ましい。
In this step, the amount of compound (4) or compound (5) used is usually preferably 0.1 to 2 mol, more preferably 0.2 to 1 mol, per 1 mol of tetrasubstituted thiophene S-oxide compound (3). .
本工程は、通常溶媒中で実施することができる。溶媒としては、例えば、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、ジメトキシエタン、シクロペンチルメチルエーテル、t-ブチルメチルエーテル等の鎖状エーテル;テトラヒドロフラン、ジオキサン等の環状エーテル;ペンタン、ヘキサン、ヘプタン、シクロヘキサン等の脂肪族炭化水素;ジクロロメタン、ジクロロエタン、クロロホルム、四塩化炭素等の脂肪族ハロゲン化炭化水素;ベンゼン、トルエン、キシレン、メシチレン等の芳香族炭化水素;クロロベンゼン、トリフルオロトルエン等の芳香族ハロゲン化炭化水素;メタノール、エタノール、イソプロピルアルコール等のアルコール等が挙げられる。これらの溶媒は単独で用いることもでき、2種以上を組合せて用いることもできる。収率及び合成の容易さの観点から、環状エーテル及び脂肪族ハロゲン化炭化水素が好ましく、テトラヒドロフラン、ジクロロエタンがより好ましい。
This step can usually be carried out in a solvent. Examples of the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane. Aliphatic hydrocarbons; Aliphatic halogenated hydrocarbons such as dichloromethane, dichloroethane, chloroform and carbon tetrachloride; Aromatic hydrocarbons such as benzene, toluene, xylene and mesitylene; Aromatic halogenated hydrocarbons such as chlorobenzene and trifluorotoluene Alcohols such as methanol, ethanol, isopropyl alcohol and the like. These solvents can be used alone or in combination of two or more. From the viewpoints of yield and ease of synthesis, cyclic ethers and aliphatic halogenated hydrocarbons are preferable, and tetrahydrofuran and dichloroethane are more preferable.
本工程は、不活性ガス雰囲気(窒素ガス、アルゴンガス等)下で行うことが好ましく、反応温度は、通常、-50~150℃程度が好ましく、0~100℃程度がより好ましい。反応時間は、反応が進行する時間とすることができ、通常、0.5~48時間程度が好ましく、1~36時間程度がより好ましい。
This step is preferably performed under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably about −50 to 150 ° C., more preferably about 0 to 100 ° C. The reaction time can be a time during which the reaction proceeds, and is usually preferably about 0.5 to 48 hours, more preferably about 1 to 36 hours.
反応終了後は、必要に応じて通常の単離及び精製工程を経て、本発明の多置換芳香族化合物を得ることができる。
After completion of the reaction, the polysubstituted aromatic compound of the present invention can be obtained through ordinary isolation and purification steps as necessary.
本発明においては、目的とする本発明の多置換芳香族化合物は、複数の同位体の混合物として得られるが、薄層クロマトグラフィー等の通常の単離及び精製手段によって容易に単離することが可能である。
In the present invention, the target polysubstituted aromatic compound of the present invention is obtained as a mixture of a plurality of isotopes, but can be easily isolated by ordinary isolation and purification means such as thin layer chromatography. Is possible.
3.多置換芳香族化合物(第2の態様)
本発明の第2の態様によれば、従来は不可能とされてきた完全非対称ヘキサ(ヘテロ)アリールベンゼンを自在に合成することが可能である。また、本発明の第2の態様によれば、完全非対称ヘキサ(ヘテロ)アリールベンゼンのみならず、5種類の芳香族置換基で置換されたピリジンや、4種類の芳香族置換基で置換された縮合環式化合物等、種々様々な多置換芳香族化合物を合成することが可能である。このような本発明の多置換芳香族化合物は、医農薬、香料、染料、プラスチック、液晶、エレクトロニクス材料等、種々様々な用途への応用が期待される。 3. Multi-substituted aromatic compound (second embodiment)
According to the second aspect of the present invention, it is possible to freely synthesize fully asymmetric hexa (hetero) arylbenzene, which has heretofore been impossible. In addition, according to the second aspect of the present invention, not only fully asymmetric hexa (hetero) arylbenzene, but also pyridine substituted with 5 types of aromatic substituents and 4 types of aromatic substituents. It is possible to synthesize a wide variety of polysubstituted aromatic compounds such as fused cyclic compounds. Such a polysubstituted aromatic compound of the present invention is expected to be applied to various uses such as medical and agricultural chemicals, fragrances, dyes, plastics, liquid crystals, and electronic materials.
本発明の第2の態様によれば、従来は不可能とされてきた完全非対称ヘキサ(ヘテロ)アリールベンゼンを自在に合成することが可能である。また、本発明の第2の態様によれば、完全非対称ヘキサ(ヘテロ)アリールベンゼンのみならず、5種類の芳香族置換基で置換されたピリジンや、4種類の芳香族置換基で置換された縮合環式化合物等、種々様々な多置換芳香族化合物を合成することが可能である。このような本発明の多置換芳香族化合物は、医農薬、香料、染料、プラスチック、液晶、エレクトロニクス材料等、種々様々な用途への応用が期待される。 3. Multi-substituted aromatic compound (second embodiment)
According to the second aspect of the present invention, it is possible to freely synthesize fully asymmetric hexa (hetero) arylbenzene, which has heretofore been impossible. In addition, according to the second aspect of the present invention, not only fully asymmetric hexa (hetero) arylbenzene, but also pyridine substituted with 5 types of aromatic substituents and 4 types of aromatic substituents. It is possible to synthesize a wide variety of polysubstituted aromatic compounds such as fused cyclic compounds. Such a polysubstituted aromatic compound of the present invention is expected to be applied to various uses such as medical and agricultural chemicals, fragrances, dyes, plastics, liquid crystals, and electronic materials.
第2の態様に係る本発明の多置換芳香族化合物は、一般式(31):
The polysubstituted aromatic compound of the present invention according to the second aspect has the general formula (31):

[式中、Rは窒素原子又は-(C-R36)=で表される基を示す。R31~R36はいずれも異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35とR36は結合して環を形成してもよい。]
で表される多置換芳香族化合物である。 [Wherein R represents a nitrogen atom or a group represented by — (C—R 36 ) ═. R 31 to R 36 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35 and R 36 may combine to form a ring. ]
It is the polysubstituted aromatic compound represented by these.
で表される多置換芳香族化合物である。 [Wherein R represents a nitrogen atom or a group represented by — (C—R 36 ) ═. R 31 to R 36 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35 and R 36 may combine to form a ring. ]
It is the polysubstituted aromatic compound represented by these.
一般式(31)において、R31~R36はいずれも異なり、それぞれアリール基又はヘテロアリール基である。
In the general formula (31), R 31 to R 36 are all different and each represents an aryl group or a heteroaryl group.
R31~R36で示されるアリール基としては、特に制限はなく、単環アリール基(フェニル基)でも多環アリール基(縮合環アリール基、多環非縮合環アリール基等)でもよいが、例えば、フェニル基、ナフチル基、アントラセニル基、ビフェニル基等が挙げられる。これらのなかでも、合成の容易さ、収率等の観点から、単環若しくは縮合環アリール基が好ましく、フェニル基、ナフチル基等がより好ましい。
The aryl group represented by R 31 to R 36 is not particularly limited, and may be a monocyclic aryl group (phenyl group) or a polycyclic aryl group (fused ring aryl group, polycyclic non-fused ring aryl group, etc.), For example, a phenyl group, a naphthyl group, an anthracenyl group, a biphenyl group, etc. are mentioned. Among these, from the viewpoints of ease of synthesis, yield, and the like, a monocyclic or condensed ring aryl group is preferable, and a phenyl group, a naphthyl group, and the like are more preferable.
R31~R36で示されるアリール基は置換されていてもよい。R31~R36で示されるアリール基が有し得る置換基としては、例えば、ハロゲン原子(フッ素原子、塩素原子、臭素原子等)、アルキル基(メチル基、エチル基等のC1-6アルキル基等)、ハロアルキル基(トリフルオロメチル基等のC1-6ハロアルキル基等)、アルコキシ基(メトキシ基等のC1-6アルコキシ基)、シリル基(t-ブチルジメチルシリル基等のトリ(C1-6アルキル)シリル基等)、アシル基(アセチル基、プロピオニル基等のC2-7アシル基等)、アルコキシカルボニル基(メトキシカルボニル基、エトキシカルボニル基等の(C1-6アルコキシ)カルボニル基等)、アミノ基(ジエチルアミノ基等のジ(C1-6アルキル)アミノ基等)等が挙げられる。これらの置換基の数は、0~6個が好ましく、0~3個がより好ましい。
The aryl group represented by R 31 to R 36 may be substituted. Examples of the substituent that the aryl group represented by R 31 to R 36 may have include, for example, a halogen atom (a fluorine atom, a chlorine atom, a bromine atom, etc.), an alkyl group (a C1-6 alkyl group such as a methyl group, an ethyl group, etc.) ), Haloalkyl group (C1-6 haloalkyl group such as trifluoromethyl group), alkoxy group (C1-6 alkoxy group such as methoxy group), silyl group (t-butyldimethylsilyl group and the like (C1-6 Alkyl) silyl group), acyl group (C2-7 acyl group such as acetyl group, propionyl group), alkoxycarbonyl group ((C1-6 alkoxy) carbonyl group such as methoxycarbonyl group, ethoxycarbonyl group), amino Group (di (C1-6 alkyl) amino group such as diethylamino group) and the like. The number of these substituents is preferably 0-6, more preferably 0-3.
R31~R36で示されるヘテロアリール基としては、特に制限はなく、単環ヘテロアリール基でも多環ヘテロアリール基(縮合環ヘテロアリール基等)でもよいが、例えば、ピロリル基、ピリジル基、ピロリジル基、ピペリジル基、イミダゾリル基、ピラゾリル基、ピラジル基、ピリミジル基、ピリダジル基、ピペラジル基、トリアジニル基、オキサゾリル基、イソオキサゾリル基、モルホリル基、チアゾリル基、イソチアゾリル基、フラニル基、チオフェニル基、インドリル基、キノリル基、イソキノリル基、ベンゾイミダゾリル基、キナゾリル基、フタラジル基、プリニル基、プテリジル基、ベンゾフラニル基、クマリル基、クロモニル基、ベンゾチオフェニル基等が挙げられる。これらのなかでも、合成の容易さ、収率等の観点から、単環ヘテロアリール基が好ましい。
The heteroaryl group represented by R 31 to R 36 is not particularly limited and may be a monocyclic heteroaryl group or a polycyclic heteroaryl group (such as a condensed ring heteroaryl group). For example, a pyrrolyl group, a pyridyl group, Pyrrolidyl group, piperidyl group, imidazolyl group, pyrazolyl group, pyrazyl group, pyrimidyl group, pyridazyl group, piperazyl group, triazinyl group, oxazolyl group, isoxazolyl group, morpholyl group, thiazolyl group, isothiazolyl group, furanyl group, thiophenyl group, indolyl group Quinolyl group, isoquinolyl group, benzimidazolyl group, quinazolyl group, phthalazyl group, purinyl group, pteridyl group, benzofuranyl group, coumaryl group, chromonyl group, benzothiophenyl group and the like. Among these, a monocyclic heteroaryl group is preferable from the viewpoint of ease of synthesis, yield, and the like.
R31~R36で示されるヘテロアリール基は置換されていてもよい。R31~R36で示されるヘテロアリール基が有し得る置換基としては、例えば、ハロゲン原子(フッ素原子、塩素原子、臭素原子等)、アルキル基(メチル基、エチル基等のC1-6アルキル基等)、ハロアルキル基(トリフルオロメチル基等のC1-6ハロアルキル基等)、アルコキシ基(メトキシ基等のC1-6アルコキシ基)、シリル基(t-ブチルジメチルシリル基等のトリ(C1-6アルキル)シリル基等)、アシル基(アセチル基、プロピオニル基等のC2-7アシル基等)、アルコキシカルボニル基(メトキシカルボニル基、エトキシカルボニル基等の(C1-6アルコキシ)カルボニル基等)、アミノ基(ジエチルアミノ基等のジ(C1-6アルキル)アミノ基等)等が挙げられる。これらの置換基の数は、0~6個が好ましく、0~3個がより好ましい。
The heteroaryl group represented by R 31 to R 36 may be substituted. Examples of the substituent that the heteroaryl group represented by R 31 to R 36 may have include, for example, halogen atoms (fluorine atom, chlorine atom, bromine atom, etc.), alkyl groups (C1-6 alkyl such as methyl group, ethyl group, etc.) Group), a haloalkyl group (C1-6 haloalkyl group such as trifluoromethyl group), an alkoxy group (C1-6 alkoxy group such as methoxy group), a silyl group (t-butyldimethylsilyl group and the like (C1- 6alkyl) silyl group), acyl group (C2-7 acyl group such as acetyl group, propionyl group), alkoxycarbonyl group ((C1-6 alkoxy) carbonyl group such as methoxycarbonyl group, ethoxycarbonyl group), An amino group (di (C1-6 alkyl) amino group such as diethylamino group) and the like can be mentioned. The number of these substituents is preferably 0-6, more preferably 0-3.
一般式(31)において、Rは窒素原子又は-(C-R36)=で表される基である。また、R35とR36は結合して環を形成してもよい。つまり、本発明の多置換芳香族化合物は、6つの異なるアリール基又はヘテロアリール基が置換したベンゼン(完全非対称ヘキサ(ヘテロ)アリールベンゼン)、4つの異なるアリール基又はヘテロアリール基が1つのベンゼン環に置換した縮合環芳香族化合物、5つの異なるアリール基又はヘテロアリール基が置換したピリジンのいずれも包含する。
In the general formula (31), R is a nitrogen atom or a group represented by — (C—R 36 ) ═. R 35 and R 36 may be bonded to form a ring. That is, in the polysubstituted aromatic compound of the present invention, the benzene ring substituted with six different aryl groups or heteroaryl groups (fully asymmetric hexa (hetero) arylbenzene), and four different aryl groups or heteroaryl groups have one benzene ring. Any of the condensed ring aromatic compounds substituted with pyridine and pyridine substituted with five different aryl groups or heteroaryl groups are included.
つまり、本発明の多置換芳香族化合物は、一般式(31A):
That is, the polysubstituted aromatic compound of the present invention has the general formula (31A):

[式中、R31a~R36aはいずれも異なり、それぞれ置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。]
で表される化合物、一般式(31B): [Wherein, R 31a to R 36a are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
A compound represented by the general formula (31B):
で表される化合物、一般式(31B): [Wherein, R 31a to R 36a are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
A compound represented by the general formula (31B):

[式中、R31b~R34bはいずれも異なり、それぞれ置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。Aは環を示す。]
で表される化合物、一般式(31C): [Wherein, R 31b to R 34b are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. A represents a ring. ]
A compound represented by the general formula (31C):
で表される化合物、一般式(31C): [Wherein, R 31b to R 34b are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. A represents a ring. ]
A compound represented by the general formula (31C):

[式中、R31c~R35cはいずれも異なり、それぞれ置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。]
で表される化合物のいずれも包含する。 [Wherein, R 31c to R 35c are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
Any of the compounds represented by
で表される化合物のいずれも包含する。 [Wherein, R 31c to R 35c are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
Any of the compounds represented by
一般式(31A)において、R31a~R36aで示されるアリール基及びヘテロアリール基は前記したものが挙げられ、有し得る置換基の種類及び数としても上記したものを採用し得る。
In the general formula (31A), examples of the aryl group and heteroaryl group represented by R 31a to R 36a include those described above, and the types and numbers of substituents that may be included may be employed.
一般式(31B)において、R31b~R34bで示されるアリール基及びヘテロアリール基は前記したものが挙げられ、有し得る置換基の種類及び数としても上記したものを採用し得る。
In the general formula (31B), examples of the aryl group and heteroaryl group represented by R 31b to R 34b include those described above, and the types and numbers of substituents that may be included may be employed.
一般式(31B)において、Aで示される環としては、例えば、
In the general formula (31B), examples of the ring represented by A include:

等が挙げられる。
Etc.
一般式(31C)において、R31c~R35cで示されるアリール基及びヘテロアリール基は前記したものが挙げられ、有し得る置換基の種類及び数としても上記したものを採用し得る。
In the general formula (31C), examples of the aryl group and heteroaryl group represented by R 31c to R 35c include those described above, and the types and numbers of substituents that may be included may be employed.
このため、本発明の多置換芳香族化合物としては、例えば、
For this reason, examples of the polysubstituted aromatic compound of the present invention include:


[式中、TBSはt-ブチルジメチルシリル基を示す。以下同様である。]
等が挙げられる。 [Wherein TBS represents a t-butyldimethylsilyl group. The same applies hereinafter. ]
Etc.
等が挙げられる。 [Wherein TBS represents a t-butyldimethylsilyl group. The same applies hereinafter. ]
Etc.
なお、第2の態様に係る本発明の多置換芳香族化合物は、これらのみに限定されることはない。後述の本発明の製造方法によれば、所望のアリール基及びヘテロアリール基を所望の箇所に自在に導入することが可能であり、有機化学で通常使用される約50種類の置換基の組合せによって13億以上という膨大な数の多置換芳香族化合物を自在に合成することが可能である。
Note that the polysubstituted aromatic compound of the present invention according to the second embodiment is not limited to these. According to the production method of the present invention to be described later, a desired aryl group and heteroaryl group can be freely introduced at a desired position, and by a combination of about 50 kinds of substituents usually used in organic chemistry. It is possible to freely synthesize a huge number of polysubstituted aromatic compounds of 1.3 billion or more.
4.多置換芳香族化合物(第2の態様)の製造方法
第2の態様に係る本発明の多置換芳香族化合物の製造方法は、上記した第2の態様に係る本発明の多置換芳香族化合物を製造する方法であり、一般式(32): 4). Process for producing polysubstituted aromatic compound (second embodiment)
The method for producing a polysubstituted aromatic compound according to the second aspect of the present invention is a method for producing the polysubstituted aromatic compound according to the second aspect of the present invention, and is represented by the general formula (32):
第2の態様に係る本発明の多置換芳香族化合物の製造方法は、上記した第2の態様に係る本発明の多置換芳香族化合物を製造する方法であり、一般式(32): 4). Process for producing polysubstituted aromatic compound (second embodiment)
The method for producing a polysubstituted aromatic compound according to the second aspect of the present invention is a method for producing the polysubstituted aromatic compound according to the second aspect of the present invention, and is represented by the general formula (32):

[式中、R31~R34はいずれも異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。]
で表される四置換チオフェンS-オキシド化合物と、
一般式(33):
R≡C-R35 (33)
[式中、Rは窒素原子又は≡(C-R36)で表される基を示す。R35及びR36は異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35及びR36はR31~R34のいずれとも異なる。R35とR36は結合して環を形成してもよい。]
で表される化合物とを反応させる工程(工程(2-II))を備える。 [Wherein, R 31 to R 34 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
A tetrasubstituted thiophene S-oxide compound represented by:
General formula (33):
R≡C-R 35 (33)
[Wherein R represents a nitrogen atom or a group represented by ≡ (C—R 36 ). R 35 and R 36 are different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35 and R 36 are different from any of R 31 to R 34 . R 35 and R 36 may combine to form a ring. ]
The process with which the compound represented by (process (2-II)) is made to react is provided.
で表される四置換チオフェンS-オキシド化合物と、
一般式(33):
R≡C-R35 (33)
[式中、Rは窒素原子又は≡(C-R36)で表される基を示す。R35及びR36は異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35及びR36はR31~R34のいずれとも異なる。R35とR36は結合して環を形成してもよい。]
で表される化合物とを反応させる工程(工程(2-II))を備える。 [Wherein, R 31 to R 34 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
A tetrasubstituted thiophene S-oxide compound represented by:
General formula (33):
R≡C-R 35 (33)
[Wherein R represents a nitrogen atom or a group represented by ≡ (C—R 36 ). R 35 and R 36 are different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35 and R 36 are different from any of R 31 to R 34 . R 35 and R 36 may combine to form a ring. ]
The process with which the compound represented by (process (2-II)) is made to react is provided.
また、本発明の製造方法において、原料として使用する四置換チオフェンS-オキシド化合物は、一般式(34):
In the production method of the present invention, the tetrasubstituted thiophene S-oxide compound used as a raw material is represented by the general formula (34):

[式中、R31~R34はいずれも異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。]
で表される四置換チオフェン化合物を酸化させる工程(工程(2-I))により得ることができる。 [Wherein, R 31 to R 34 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
It can obtain by the process (process (2-I)) which oxidizes the tetrasubstituted thiophene compound represented by these.
で表される四置換チオフェン化合物を酸化させる工程(工程(2-I))により得ることができる。 [Wherein, R 31 to R 34 are all different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. ]
It can obtain by the process (process (2-I)) which oxidizes the tetrasubstituted thiophene compound represented by these.
(4-1)四置換チオフェン化合物及びその製造方法
一般式(34)において、R31~R34で示されるアリール基及びヘテロアリール基は前記したものが挙げられ、有し得る置換基の種類及び数としても上記したものを採用し得る。このような四置換チオフェン化合物は、例えば、非特許文献1に記載の合成方法に準じて、又は該合成方法を若干改良して合成することができる。具体的には、反応式2: (4-1) Tetra-substituted thiophene compound and method for producing the same In general formula (34), examples of the aryl group and heteroaryl group represented by R 31 to R 34 include those described above. The number described above can be adopted as the number. Such a tetra-substituted thiophene compound can be synthesized, for example, according to the synthesis method described in Non-Patent Document 1 or by slightly improving the synthesis method. Specifically, reaction formula 2:
一般式(34)において、R31~R34で示されるアリール基及びヘテロアリール基は前記したものが挙げられ、有し得る置換基の種類及び数としても上記したものを採用し得る。このような四置換チオフェン化合物は、例えば、非特許文献1に記載の合成方法に準じて、又は該合成方法を若干改良して合成することができる。具体的には、反応式2: (4-1) Tetra-substituted thiophene compound and method for producing the same In general formula (34), examples of the aryl group and heteroaryl group represented by R 31 to R 34 include those described above. The number described above can be adopted as the number. Such a tetra-substituted thiophene compound can be synthesized, for example, according to the synthesis method described in Non-Patent Document 1 or by slightly improving the synthesis method. Specifically, reaction formula 2:

[式中、R31~R34は前記に同じである。R37はアルキル基を示す。X2はハロゲン原子を示す。Y2は同一又は異なって、ボロン酸又はそのエステル基を示す。Tfはトリフルオロメタンスルホニル基を示す。]
にしたがって、上記の反応式1と同じ方法で四置換チオフェン化合物を合成することができる。このため、具体的な方法や条件については、上記(2-1)を採用することができる。 [Wherein R 31 to R 34 are the same as defined above. R 37 represents an alkyl group. X 2 represents a halogen atom. Y 2 is the same or different and represents boronic acid or an ester group thereof. Tf represents a trifluoromethanesulfonyl group. ]
Thus, a tetrasubstituted thiophene compound can be synthesized by the same method as in Reaction Scheme 1 above. Therefore, the above (2-1) can be adopted as a specific method and conditions.
にしたがって、上記の反応式1と同じ方法で四置換チオフェン化合物を合成することができる。このため、具体的な方法や条件については、上記(2-1)を採用することができる。 [Wherein R 31 to R 34 are the same as defined above. R 37 represents an alkyl group. X 2 represents a halogen atom. Y 2 is the same or different and represents boronic acid or an ester group thereof. Tf represents a trifluoromethanesulfonyl group. ]
Thus, a tetrasubstituted thiophene compound can be synthesized by the same method as in Reaction Scheme 1 above. Therefore, the above (2-1) can be adopted as a specific method and conditions.
(4-2)工程(1-I):四置換チオフェン化合物(34)の酸化
上記した四置換チオフェン化合物(34)と後述の化合物(33)とを反応させても環化反応は進行せず、第2の態様に係る本発明の多置換芳香族化合物は得られない。本発明では、四置換チオフェン化合物(34)の反応性を向上させるため、原料として四置換チオフェンS-オキシド化合物(32)を用いることが好ましい。 (4-2) Step (1-I): Oxidation of tetrasubstituted thiophene compound (34) The cyclization reaction does not proceed even when the above tetrasubstituted thiophene compound (34) is reacted with compound (33) described below. Thus, the polysubstituted aromatic compound of the present invention according to the second embodiment cannot be obtained. In the present invention, in order to improve the reactivity of the tetrasubstituted thiophene compound (34), it is preferable to use the tetrasubstituted thiophene S-oxide compound (32) as a raw material.
上記した四置換チオフェン化合物(34)と後述の化合物(33)とを反応させても環化反応は進行せず、第2の態様に係る本発明の多置換芳香族化合物は得られない。本発明では、四置換チオフェン化合物(34)の反応性を向上させるため、原料として四置換チオフェンS-オキシド化合物(32)を用いることが好ましい。 (4-2) Step (1-I): Oxidation of tetrasubstituted thiophene compound (34) The cyclization reaction does not proceed even when the above tetrasubstituted thiophene compound (34) is reacted with compound (33) described below. Thus, the polysubstituted aromatic compound of the present invention according to the second embodiment cannot be obtained. In the present invention, in order to improve the reactivity of the tetrasubstituted thiophene compound (34), it is preferable to use the tetrasubstituted thiophene S-oxide compound (32) as a raw material.
本工程では、上記した四置換チオフェン化合物(34)を酸化させることで、四置換チオフェンS-オキシド化合物(33)を得ることができる。
In this step, the tetrasubstituted thiophene S-oxide compound (33) can be obtained by oxidizing the tetrasubstituted thiophene compound (34).
酸化は、上記の第1の態様と同様の方法で行うことができる。このため、酸化の方法及び条件については、上記(2-2)を採用することができる。
Oxidation can be carried out in the same manner as in the first aspect. Therefore, the above method (2-2) can be adopted for the oxidation method and conditions.
(4-3)工程(2-II):四置換チオフェンS-オキシド化合物(32)と化合物(33)との反応
上記した四置換チオフェンS-オキシド化合物(32)は、ジエンとしての反応性を有するため、化合物(33)と反応させることで、第2の態様に係る本発明の多置換芳香族化合物を得ることができる。 (4-3) Step (2-II): Reaction of Tetrasubstituted Thiophene S-Oxide Compound (32) with Compound (33) The tetrasubstituted thiophene S-oxide compound (32) described above exhibits reactivity as a diene. Therefore, the polysubstituted aromatic compound of the present invention according to the second aspect can be obtained by reacting with the compound (33).
上記した四置換チオフェンS-オキシド化合物(32)は、ジエンとしての反応性を有するため、化合物(33)と反応させることで、第2の態様に係る本発明の多置換芳香族化合物を得ることができる。 (4-3) Step (2-II): Reaction of Tetrasubstituted Thiophene S-Oxide Compound (32) with Compound (33) The tetrasubstituted thiophene S-oxide compound (32) described above exhibits reactivity as a diene. Therefore, the polysubstituted aromatic compound of the present invention according to the second aspect can be obtained by reacting with the compound (33).
一般式(33)において、R35~R36で示されるアリール基及びヘテロアリール基は前記したものが挙げられ、有し得る置換基の種類及び数としても上記したものを採用し得る。
In the general formula (33), examples of the aryl group and heteroaryl group represented by R 35 to R 36 include those described above, and the types and numbers of substituents that can be used may be used.
一般式(33)において、Rは窒素原子又は-(C-R36)=で示される基である。また、R35とR36は結合して環を形成してもよい。つまり、化合物(33)は、一般式(33A):
R36a-C≡C-R35a (33A)
[式中、R35a及びR36aは異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35a及びR36aはR31~R34のいずれとも異なる。]
で表される化合物、
一般式(33B): In the general formula (33), R is a nitrogen atom or a group represented by — (C—R 36 ) ═. R 35 and R 36 may be bonded to form a ring. That is, the compound (33) has the general formula (33A):
R 36a -C≡C-R 35a (33A)
[Wherein, R 35a and R 36a are different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35a and R 36a are different from any of R 31 to R 34 . ]
A compound represented by
General formula (33B):
R36a-C≡C-R35a (33A)
[式中、R35a及びR36aは異なり、置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35a及びR36aはR31~R34のいずれとも異なる。]
で表される化合物、
一般式(33B): In the general formula (33), R is a nitrogen atom or a group represented by — (C—R 36 ) ═. R 35 and R 36 may be bonded to form a ring. That is, the compound (33) has the general formula (33A):
R 36a -C≡C-R 35a (33A)
[Wherein, R 35a and R 36a are different and each represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35a and R 36a are different from any of R 31 to R 34 . ]
A compound represented by
General formula (33B):

[式中、A’は三重結合を有する環を示す。]
で表される化合物、及び
一般式(33C):
N≡C-R35c
[式中、R35cは置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35cはR1~R4のいずれとも異なる。]
で表される化合物のいずれも包含する。 [Wherein A ′ represents a ring having a triple bond. ]
And a compound represented by the general formula (33C):
N≡C-R 35c
[Wherein, R 35c represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35c is different from any of R 1 to R 4 . ]
Any of the compounds represented by
で表される化合物、及び
一般式(33C):
N≡C-R35c
[式中、R35cは置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基を示す。R35cはR1~R4のいずれとも異なる。]
で表される化合物のいずれも包含する。 [Wherein A ′ represents a ring having a triple bond. ]
And a compound represented by the general formula (33C):
N≡C-R 35c
[Wherein, R 35c represents an optionally substituted aryl group or an optionally substituted heteroaryl group. R 35c is different from any of R 1 to R 4 . ]
Any of the compounds represented by
一般式(33A)において、R35a~R36aで示されるアリール基及びヘテロアリール基は前記したものが挙げられ、有し得る置換基の種類及び数としても上記したものを採用し得る。
In the general formula (33A), examples of the aryl group and heteroaryl group represented by R 35a to R 36a include those described above, and the types and numbers of substituents that may be included may be employed.
一般式(33B)において、A'で示される環としては、例えば、
In the general formula (33B), as the ring represented by A ′, for example,

等が挙げられる。
Etc.
一般式(33C)において、R35cで示されるアリール基及びヘテロアリール基は前記したものが挙げられ、有し得る置換基の種類及び数としても上記したものを採用し得る。
In the general formula (33C), examples of the aryl group and heteroaryl group represented by R 35c include those described above, and the types and numbers of substituents that may be included may be employed.
このため、使用できる化合物(33)としては、例えば、
For this reason, examples of usable compounds (33) include:
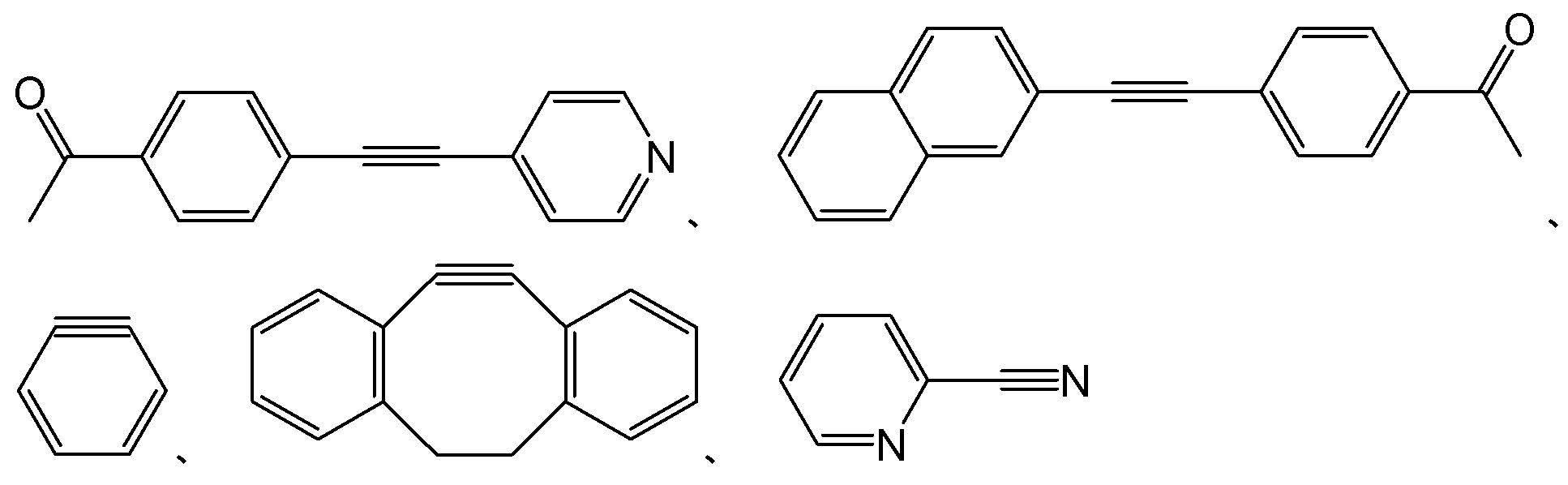
等が挙げられる。
Etc.
本工程において、化合物(33)の使用量は、通常、四置換チオフェンS-オキシド化合物(32)1モルに対して、0.5~5モルが好ましく、1~3モルがより好ましい。なお、化合物(33C)は通常液体であり、反応溶媒としても機能するため、化合物(33C)を用いる場合は、その使用量は四置換チオフェンS-オキシド化合物(32)に対して過剰量とすることが好ましい。
In this step, the amount of compound (33) used is usually preferably 0.5 to 5 moles, more preferably 1 to 3 moles per mole of tetrasubstituted thiophene S-oxide compound (32). Since compound (33C) is usually a liquid and functions as a reaction solvent, when compound (33C) is used, the amount used is excessive with respect to the tetrasubstituted thiophene S-oxide compound (32). It is preferable.
本工程は、通常溶媒中で実施することができる。溶媒としては、例えば、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、ジメトキシエタン、シクロペンチルメチルエーテル、t-ブチルメチルエーテル等の鎖状エーテル;テトラヒドロフラン、ジオキサン等の環状エーテル;ペンタン、ヘキサン、ヘプタン、シクロヘキサン等の脂肪族炭化水素;ジクロロメタン、ジクロロエタン、クロロホルム、四塩化炭素等の脂肪族ハロゲン化炭化水素;ベンゼン、トルエン、キシレン、メシチレン等の芳香族炭化水素;クロロベンゼン、トリフルオロトルエン等の芳香族ハロゲン化炭化水素; メタノール、エタノール、イソプロピルアルコール等のアルコール等が挙げられる。これらの溶媒は単独で用いることもでき、2種以上を組合せて用いることもできる。収率及び合成の容易さの観点から、環状エーテル及び芳香族炭化水素が好ましく、テトラヒドロフラン、キシレン、メシチレンがより好ましい。なお、化合物(33C)は通常液体であるため、化合物(33C)を用いる場合は、反応溶媒を使用しなくてもよい。
This step can usually be carried out in a solvent. Examples of the solvent include chain ethers such as diethyl ether, diisopropyl ether, dibutyl ether, dimethoxyethane, cyclopentyl methyl ether and t-butyl methyl ether; cyclic ethers such as tetrahydrofuran and dioxane; pentane, hexane, heptane and cyclohexane. Aliphatic hydrocarbons; Aliphatic halogenated hydrocarbons such as dichloromethane, dichloroethane, chloroform and carbon tetrachloride; Aromatic hydrocarbons such as benzene, toluene, xylene and mesitylene; Aromatic halogenated hydrocarbons such as chlorobenzene and trifluorotoluene ; Alcohol such as methanol, ethanol, isopropyl alcohol and the like. These solvents can be used alone or in combination of two or more. From the viewpoints of yield and ease of synthesis, cyclic ethers and aromatic hydrocarbons are preferable, and tetrahydrofuran, xylene, and mesitylene are more preferable. Note that since the compound (33C) is usually a liquid, the reaction solvent need not be used when the compound (33C) is used.
本工程は、不活性ガス雰囲気(窒素ガス、アルゴンガス等)下で行うことが好ましく、反応温度は、通常、-100~50℃程度が好ましく、-50~0℃程度がより好ましい。反応時間は、反応が進行する時間とすることができ、通常、1~24時間程度が好ましく、2~12時間程度がより好ましい。
This step is preferably performed under an inert gas atmosphere (nitrogen gas, argon gas, etc.), and the reaction temperature is usually preferably about −100 to 50 ° C., more preferably about −50 to 0 ° C. The reaction time can be a time during which the reaction proceeds, and is usually preferably about 1 to 24 hours, and more preferably about 2 to 12 hours.
このようにして、本発明の多置換芳香族化合物を得ることができる。
In this way, the polysubstituted aromatic compound of the present invention can be obtained. *
化合物(33)として化合物(33A)を用いた場合は、この時点では一般式(31A):
When the compound (33A) is used as the compound (33), the general formula (31A):

[式中、R31a~R36aはいずれも異なり、それぞれ前記に同じである。]
で表される化合物と、一般式(31A’): [Wherein R 31a to R 36a are all different and are the same as defined above. ]
And a compound represented by the general formula (31A ′):
で表される化合物と、一般式(31A’): [Wherein R 31a to R 36a are all different and are the same as defined above. ]
And a compound represented by the general formula (31A ′):

[式中、R31a~R36aはいずれも異なり、それぞれ前記に同じである。]
で表される化合物との混合物として得ることができる。このため、通常の単離及び精製工程を経て、目的化合物を単離することができる。 [Wherein R 31a to R 36a are all different and are the same as defined above. ]
It can obtain as a mixture with the compound represented by these. For this reason, the target compound can be isolated through normal isolation and purification steps.
で表される化合物との混合物として得ることができる。このため、通常の単離及び精製工程を経て、目的化合物を単離することができる。 [Wherein R 31a to R 36a are all different and are the same as defined above. ]
It can obtain as a mixture with the compound represented by these. For this reason, the target compound can be isolated through normal isolation and purification steps.
ただし、R31a~R36aの組合せによっては、上記化合物(31A)と化合物(31A’)とを単離することが困難な場合もある。この場合には、常法により、R35a又はR36aをt-ブチルジメチルシリルオキシ基等のような嵩高い置換基に置換すれば上記化合物(31A)と化合物(31A’)とを単離しやすくすることが可能である。
However, depending on the combination of R 31a to R 36a , it may be difficult to isolate the compound (31A) and the compound (31A ′). In this case, the compound (31A) and the compound (31A ′) can be easily isolated by substituting R 35a or R 36a with a bulky substituent such as t-butyldimethylsilyloxy group by a conventional method. Is possible.
化合物(33)として化合物(33B)を用いた場合は、必要に応じて通常の単離及び精製工程を経て、目的化合物を得ることができる。
When the compound (33B) is used as the compound (33), the target compound can be obtained through normal isolation and purification steps as necessary. *
化合物(33)として化合物(33C)を用いた場合は、この時点では一般式(31C):
When compound (33C) is used as compound (33), general formula (31C):

[式中、R31c~R35cはいずれも異なり、それぞれ前記に同じである。]
で表される化合物と、一般式(31C’): [Wherein R 31c to R 35c are all different and are the same as defined above. ]
And a compound represented by the general formula (31C ′):
で表される化合物と、一般式(31C’): [Wherein R 31c to R 35c are all different and are the same as defined above. ]
And a compound represented by the general formula (31C ′):

[式中、R31c~R35cはいずれも異なり、それぞれ前記に同じである。]
で表される化合物との混合物として得ることができる。このため、通常の単離及び精製工程を経て、目的化合物を単離することができる。 [Wherein R 31c to R 35c are all different and are the same as defined above. ]
It can obtain as a mixture with the compound represented by these. For this reason, the target compound can be isolated through normal isolation and purification steps.
で表される化合物との混合物として得ることができる。このため、通常の単離及び精製工程を経て、目的化合物を単離することができる。 [Wherein R 31c to R 35c are all different and are the same as defined above. ]
It can obtain as a mixture with the compound represented by these. For this reason, the target compound can be isolated through normal isolation and purification steps.
以下、本発明について、実施例を挙げて具体的に説明するが、本発明は、これらの実施例に何ら制約されるものではない。
Hereinafter, the present invention will be specifically described with reference to examples, but the present invention is not limited to these examples.
特に制約しない限り、乾燥溶媒を含む全ての材料は、市販品をそのまま使用した。テトラキス(トリフェニルホスフィン)パラジウム(0)は、既報(J. Chem. Soc. 1186 (1957))にしたがって合成した。N-ブロモスクシンイミドは、水カップリング反応により新たに再結晶した。三フッ化ホウ素ジエチルエーテル錯体(BF3・OEt2)は、酸化反応の前にCaH2で蒸留した。特に断りのない限り、すべての反応は、標準的な真空ライン技法を用いて、フレームドライしたガラス容器中で、窒素(N2)ガス雰囲気下に乾燥溶媒を用いて行った。すべての後処理及び精製手順は、空気中で試薬グレードの溶媒を用いて行った。
Unless otherwise specified, all the materials including the dry solvent were used as they were on the market. Tetrakis (triphenylphosphine) palladium (0) was synthesized according to a previous report (J. Chem. Soc. 1186 (1957)). N-bromosuccinimide was newly recrystallized by a water coupling reaction. Boron trifluoride diethyl ether complex (BF 3 · OEt 2 ) was distilled with CaH 2 before the oxidation reaction. Unless otherwise noted, all reactions were performed in a flame-dried glass container using a dry solvent under a nitrogen (N2) gas atmosphere using standard vacuum line techniques. All work-up and purification procedures were performed in air with reagent grade solvents.
分析用薄層クロマトグラフィー(TLC)は、E. Merckシリカゲル60 F254プレコートプレート(0.25 mm)を用いて行った。開発したクロマトグラムは、UVランプ(254 nm)で分析した。フラッシュカラムクロマトグラフィーは、E. Merckシリカゲル60(230-400メッシュ)を用いて行った。分取リサイクルゲルパーミエーションクロマトグラフィー(GPC)は、溶離液としてクロロホルムを用いてJAIGEL-1H/JAIGEL-2Hカラムを備えたJAI LC-9204を用いて行った。分取薄層クロマトグラフィー(PTLC)はあらかじめ準備したWakogel B5-Fのシリカコートプレート(0.75 mm)を用いて行った。ガスクロマトグラフ質量分析(GC/MS)は、HP-5カラム(30 m×0.25 mm、ヒューレット-パッカード社)を備えた島津GCMS-QP2010で行った。高分解能質量スペクトル(HRMS)は、JMS-T100TD器具(DART)及びThermo Fisher Scientific Exactive(APCI)により得た。核磁気共鳴(NMR)スペクトルは、JEOL JNM-ECA-600分光計(1H 600 MHz、13C 150MHz)、JEOL JNM-ECA-400分光計(1H 400 MHz、13C 100MHz)、及び極低温プローブを備えたJEOL JNM-ECA-600II(1H 600 MHz、13C 150MHz)で記録した。1H NMRの化学シフトはテトラメチルシラン(δ0.00 ppm)又はC2D2Cl4(δ5.98 ppm)の相対的な百万分率(ppm)で表した。13C NMRの化学シフトはCDCl3(δ77.0 ppm)又はC2D2Cl4(δ73.79 ppm)の相対的な百万分率(ppm)で表した。データは、化学シフト、多重度(s =シングレット、d =ダブレット、dd =ダブレットのダブレット、t =トリプレット、m=マルチプレット)、結合定数(Hz)、及び統合の順に報告する。
Analytical thin layer chromatography (TLC) was performed using E. Merck silica gel 60 F 254 precoated plates (0.25 mm). The developed chromatogram was analyzed with a UV lamp (254 nm). Flash column chromatography was performed using E. Merck silica gel 60 (230-400 mesh). Preparative recycle gel permeation chromatography (GPC) was performed using a JAI LC-9204 equipped with a JAIGEL-1H / JAIGEL-2H column with chloroform as the eluent. Preparative thin layer chromatography (PTLC) was performed using a Wakogel B5-F silica-coated plate (0.75 mm) prepared in advance. Gas chromatograph mass spectrometry (GC / MS) was performed on a Shimadzu GCMS-QP2010 equipped with an HP-5 column (30 m × 0.25 mm, Hewlett-Packard). High resolution mass spectra (HRMS) were obtained with a JMS-T100TD instrument (DART) and Thermo Fisher Scientific Exactive (APCI). Nuclear magnetic resonance (NMR) spectra were measured using a JEOL JNM-ECA-600 spectrometer ( 1 H 600 MHz, 13 C 150 MHz), a JEOL JNM-ECA-400 spectrometer ( 1 H 400 MHz, 13 C 100 MHz), and a cryogenic temperature. Recorded on a JEOL JNM-ECA-600II ( 1 H 600 MHz, 13 C 150 MHz) equipped with a probe. 1 H NMR chemical shifts were expressed as relative parts per million (ppm) of tetramethylsilane (δ0.00 ppm) or C 2 D 2 Cl 4 (δ5.98 ppm). 13 C NMR chemical shifts were expressed in relative parts per million (ppm) of CDCl 3 (δ 77.0 ppm) or C 2 D 2 Cl 4 (δ 73.79 ppm). Data are reported in the order of chemical shift, multiplicity (s = singlet, d = doublet, dd = doublet doublet, t = triplet, m = multiplet), binding constant (Hz), and integration.
[合成例1:化合物(8)の合成]
合成例1-1:化合物2の合成 [Synthesis Example 1: Synthesis of Compound (8)]
Synthesis Example 1-1: Synthesis of Compound 2
合成例1-1:化合物2の合成 [Synthesis Example 1: Synthesis of Compound (8)]
Synthesis Example 1-1: Synthesis of Compound 2

[式中、HMDSは1,1,1,3,3,3-ヘキサメチルジシラザンを示す。THFはテトラヒドロフランを示す。以下同様である。]
50 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、2,5-ジブロモヒドロキノン(化合物1; 2.68 g, 10 mmol, 1.0当量)、1,1,1,3,3,3-ヘキサメチルジシラザン(HMDS; 8.38 mL, 40 mmol, 4.0当量)、及びTHF(10 mL)を窒素気流下に添加した。反応混合物を16時間還流した。揮発性物質を減圧下に除去し、((2,5-ジブロモ-1,4-フェニレン)ビス(オキシ))ビス(トリメチルシラン)(化合物2)を白色固体として得た(4.12 g, quant)。
1H NMR (600 MHz, CDCl3): δ 7.04 (s, 2H), 0.29 (s, 18H)。 [Wherein, HMDS represents 1,1,1,3,3,3-hexamethyldisilazane. THF represents tetrahydrofuran. The same applies hereinafter. ]
A 50 mL two-necked flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask was added 2,5-dibromohydroquinone (compound 1; 2.68 g, 10 mmol, 1.0 equiv), 1,1,1,3,3,3-hexamethyldisilazane (HMDS; 8.38 mL, 40 mmol, 4.0 Eq), and THF (10 mL) were added under a stream of nitrogen. The reaction mixture was refluxed for 16 hours. Volatiles were removed under reduced pressure to give ((2,5-dibromo-1,4-phenylene) bis (oxy)) bis (trimethylsilane) (compound 2) as a white solid (4.12 g, quant) .
1 H NMR (600 MHz, CDCl 3 ): δ 7.04 (s, 2H), 0.29 (s, 18H).
50 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、2,5-ジブロモヒドロキノン(化合物1; 2.68 g, 10 mmol, 1.0当量)、1,1,1,3,3,3-ヘキサメチルジシラザン(HMDS; 8.38 mL, 40 mmol, 4.0当量)、及びTHF(10 mL)を窒素気流下に添加した。反応混合物を16時間還流した。揮発性物質を減圧下に除去し、((2,5-ジブロモ-1,4-フェニレン)ビス(オキシ))ビス(トリメチルシラン)(化合物2)を白色固体として得た(4.12 g, quant)。
1H NMR (600 MHz, CDCl3): δ 7.04 (s, 2H), 0.29 (s, 18H)。 [Wherein, HMDS represents 1,1,1,3,3,3-hexamethyldisilazane. THF represents tetrahydrofuran. The same applies hereinafter. ]
A 50 mL two-necked flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask was added 2,5-dibromohydroquinone (compound 1; 2.68 g, 10 mmol, 1.0 equiv), 1,1,1,3,3,3-hexamethyldisilazane (HMDS; 8.38 mL, 40 mmol, 4.0 Eq), and THF (10 mL) were added under a stream of nitrogen. The reaction mixture was refluxed for 16 hours. Volatiles were removed under reduced pressure to give ((2,5-dibromo-1,4-phenylene) bis (oxy)) bis (trimethylsilane) (compound 2) as a white solid (4.12 g, quant) .
1 H NMR (600 MHz, CDCl 3 ): δ 7.04 (s, 2H), 0.29 (s, 18H).
合成例1-2:化合物3の合成
Synthesis Example 1-2: Synthesis of Compound 3

[式中、TMSClはクロロトリメチルシランを示す。以下同様である。]
100 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、合成例1-1で得た((2,5-ジブロモ-1,4-フェニレン)ビス(オキシ))ビス(トリメチルシラン)(化合物2; 2.06 g, 5 mmol, 1.0当量)、ナトリウム(460 mg, 20 mmol, 4.0当量)、及びトルエン(20 mL)を窒素気流下に添加した。混合物を1時間還流した後、クロロトリメチルシラン(TMSCl; 1.4 mL, 11 mmol, 2.2当量)のトルエン(5 mL)溶液をゆっくりと添加した。得られた混合物をさらに16時間還流した。反応混合物を室温まで冷却した後、混合物をガラスフィルター(溶離液:トルエン, 50 mL)でろ過した。揮発性物質を減圧下に除去し、(2,5-ビス((トリメチルシリル)オキシ)-1,4-フェニレン)ビス(トリメチルシラン)(化合物3)を白色固体として得た(1.81 g, 91 %)。
1H NMR (600 MHz, CDCl3): δ 6.76 (s, 2H), 0.29 (s, 18H), 0.24 (s, 18H)。 [Wherein TMSCl represents chlorotrimethylsilane. The same applies hereinafter. ]
A 100 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, ((2,5-dibromo-1,4-phenylene) bis (oxy)) bis (trimethylsilane) obtained in Synthesis Example 1-1 (Compound 2; 2.06 g, 5 mmol, 1.0 equivalent), Sodium (460 mg, 20 mmol, 4.0 eq) and toluene (20 mL) were added under a nitrogen stream. After the mixture was refluxed for 1 hour, a solution of chlorotrimethylsilane (TMSCl; 1.4 mL, 11 mmol, 2.2 eq) in toluene (5 mL) was slowly added. The resulting mixture was refluxed for an additional 16 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a glass filter (eluent: toluene, 50 mL). Volatiles were removed under reduced pressure to give (2,5-bis ((trimethylsilyl) oxy) -1,4-phenylene) bis (trimethylsilane) (compound 3) as a white solid (1.81 g, 91% ).
1 H NMR (600 MHz, CDCl 3 ): δ 6.76 (s, 2H), 0.29 (s, 18H), 0.24 (s, 18H).
100 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、合成例1-1で得た((2,5-ジブロモ-1,4-フェニレン)ビス(オキシ))ビス(トリメチルシラン)(化合物2; 2.06 g, 5 mmol, 1.0当量)、ナトリウム(460 mg, 20 mmol, 4.0当量)、及びトルエン(20 mL)を窒素気流下に添加した。混合物を1時間還流した後、クロロトリメチルシラン(TMSCl; 1.4 mL, 11 mmol, 2.2当量)のトルエン(5 mL)溶液をゆっくりと添加した。得られた混合物をさらに16時間還流した。反応混合物を室温まで冷却した後、混合物をガラスフィルター(溶離液:トルエン, 50 mL)でろ過した。揮発性物質を減圧下に除去し、(2,5-ビス((トリメチルシリル)オキシ)-1,4-フェニレン)ビス(トリメチルシラン)(化合物3)を白色固体として得た(1.81 g, 91 %)。
1H NMR (600 MHz, CDCl3): δ 6.76 (s, 2H), 0.29 (s, 18H), 0.24 (s, 18H)。 [Wherein TMSCl represents chlorotrimethylsilane. The same applies hereinafter. ]
A 100 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, ((2,5-dibromo-1,4-phenylene) bis (oxy)) bis (trimethylsilane) obtained in Synthesis Example 1-1 (Compound 2; 2.06 g, 5 mmol, 1.0 equivalent), Sodium (460 mg, 20 mmol, 4.0 eq) and toluene (20 mL) were added under a nitrogen stream. After the mixture was refluxed for 1 hour, a solution of chlorotrimethylsilane (TMSCl; 1.4 mL, 11 mmol, 2.2 eq) in toluene (5 mL) was slowly added. The resulting mixture was refluxed for an additional 16 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a glass filter (eluent: toluene, 50 mL). Volatiles were removed under reduced pressure to give (2,5-bis ((trimethylsilyl) oxy) -1,4-phenylene) bis (trimethylsilane) (compound 3) as a white solid (1.81 g, 91% ).
1 H NMR (600 MHz, CDCl 3 ): δ 6.76 (s, 2H), 0.29 (s, 18H), 0.24 (s, 18H).
合成例1-3:化合物4の合成
Synthesis Example 1-3: Synthesis of Compound 4

100 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、合成例1-2で得た(2,5-ビス((トリメチルシリル)オキシ)-1,4-フェニレン)ビス(トリメチルシラン)(化合物3; 1.20 g, 3 mmol, 1.0当量)、6N HNO3水溶液(3 mL)、及びジオキサン(48 mL)を窒素気流下に添加した。室温で3時間撹拌した後、混合物に、水(30 mL)及びCH2Cl2(50 mL)を添加した。CH2Cl2で抽出した後、有機層をNa2SO4で乾燥し、揮発性物質を減圧下に除去し、2,5-ビス(トリメチルシリル)ベンゼン-1,4-ジオール(化合物4)を白色固体として得た(710 mg, 93 %)。
1H NMR (600 MHz, CDCl3): δ 7.04 (s, 2H), 2.17 (s, 2H), 0.29 (s, 18H)。 A 100 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, (2,5-bis ((trimethylsilyl) oxy) -1,4-phenylene) bis (trimethylsilane) obtained in Synthesis Example 1-2 (Compound 3; 1.20 g, 3 mmol, 1.0 equivalent), 6N HNO 3 aqueous solution (3 mL) and dioxane (48 mL) were added under a nitrogen stream. After stirring at room temperature for 3 hours, water (30 mL) and CH 2 Cl 2 (50 mL) were added to the mixture. After extraction with CH 2 Cl 2 , the organic layer is dried over Na 2 SO 4 , volatiles are removed under reduced pressure, and 2,5-bis (trimethylsilyl) benzene-1,4-diol (compound 4) is removed. Obtained as a white solid (710 mg, 93%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.04 (s, 2H), 2.17 (s, 2H), 0.29 (s, 18H).
1H NMR (600 MHz, CDCl3): δ 7.04 (s, 2H), 2.17 (s, 2H), 0.29 (s, 18H)。 A 100 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, (2,5-bis ((trimethylsilyl) oxy) -1,4-phenylene) bis (trimethylsilane) obtained in Synthesis Example 1-2 (Compound 3; 1.20 g, 3 mmol, 1.0 equivalent), 6N HNO 3 aqueous solution (3 mL) and dioxane (48 mL) were added under a nitrogen stream. After stirring at room temperature for 3 hours, water (30 mL) and CH 2 Cl 2 (50 mL) were added to the mixture. After extraction with CH 2 Cl 2 , the organic layer is dried over Na 2 SO 4 , volatiles are removed under reduced pressure, and 2,5-bis (trimethylsilyl) benzene-1,4-diol (compound 4) is removed. Obtained as a white solid (710 mg, 93%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.04 (s, 2H), 2.17 (s, 2H), 0.29 (s, 18H).
合成例1-4:化合物5の合成
Synthesis Example 1-4: Synthesis of Compound 5

[式中、Tf2Oはトリフルオロメタンスルホン酸無水物を示す。以下同様である。]
25 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このチューブに、合成例1-3で得た2,5-ビス(トリメチルシリル)ベンゼン-1,4-ジオール(化合物4; 254 mg, 1 mmol, 1.0当量)、トリフルオロメタンスルホン酸無水物(Tf2O; 164μL, 1 mmol, 1.0当量)、及びピリジン(5 mL)を窒素気流下に添加した。得られた混合物を50℃で1時間加熱した。反応混合物を室温まで冷却した後、混合物に水(10 mL)及びCH2Cl2(10 mL)を添加した。CH2Cl2で抽出した後、有機層をNa2SO4で乾燥し、揮発性物質を減圧下に除去した。粗生成物をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル= 30: 1)及びGPCで精製し、4-ヒドロキシ-2,5-ビス(トリメチルシリル)フェニルトリフルオロメタンスルホネート(化合物5)を白色固体として得た(226 mg, 59 %)。
1H NMR (600 MHz, CDCl3): δ 7.23 (s, 1H), 6.75 (s, 1H), 4.92 (s, 1H), 0.35 (s, 9H), 0.31 (s, 9H)。 [In the formula, Tf 2 O represents trifluoromethanesulfonic anhydride. The same applies hereinafter. ]
A magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, 2,5-bis (trimethylsilyl) benzene-1,4-diol (Compound 4; 254 mg, 1 mmol, 1.0 equivalent) obtained in Synthesis Example 1-3, trifluoromethanesulfonic anhydride (Tf 2 O; 164 μL, 1 mmol, 1.0 equivalent) and pyridine (5 mL) were added under a stream of nitrogen. The resulting mixture was heated at 50 ° C. for 1 hour. After the reaction mixture was cooled to room temperature, water (10 mL) and CH 2 Cl 2 (10 mL) were added to the mixture. After extraction with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was purified by flash column chromatography (hexane / ethyl acetate = 30: 1) and GPC to give 4-hydroxy-2,5-bis (trimethylsilyl) phenyl trifluoromethanesulfonate (compound 5) as a white solid. (226 mg, 59%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.23 (s, 1H), 6.75 (s, 1H), 4.92 (s, 1H), 0.35 (s, 9H), 0.31 (s, 9H).
25 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このチューブに、合成例1-3で得た2,5-ビス(トリメチルシリル)ベンゼン-1,4-ジオール(化合物4; 254 mg, 1 mmol, 1.0当量)、トリフルオロメタンスルホン酸無水物(Tf2O; 164μL, 1 mmol, 1.0当量)、及びピリジン(5 mL)を窒素気流下に添加した。得られた混合物を50℃で1時間加熱した。反応混合物を室温まで冷却した後、混合物に水(10 mL)及びCH2Cl2(10 mL)を添加した。CH2Cl2で抽出した後、有機層をNa2SO4で乾燥し、揮発性物質を減圧下に除去した。粗生成物をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル= 30: 1)及びGPCで精製し、4-ヒドロキシ-2,5-ビス(トリメチルシリル)フェニルトリフルオロメタンスルホネート(化合物5)を白色固体として得た(226 mg, 59 %)。
1H NMR (600 MHz, CDCl3): δ 7.23 (s, 1H), 6.75 (s, 1H), 4.92 (s, 1H), 0.35 (s, 9H), 0.31 (s, 9H)。 [In the formula, Tf 2 O represents trifluoromethanesulfonic anhydride. The same applies hereinafter. ]
A magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, 2,5-bis (trimethylsilyl) benzene-1,4-diol (Compound 4; 254 mg, 1 mmol, 1.0 equivalent) obtained in Synthesis Example 1-3, trifluoromethanesulfonic anhydride (Tf 2 O; 164 μL, 1 mmol, 1.0 equivalent) and pyridine (5 mL) were added under a stream of nitrogen. The resulting mixture was heated at 50 ° C. for 1 hour. After the reaction mixture was cooled to room temperature, water (10 mL) and CH 2 Cl 2 (10 mL) were added to the mixture. After extraction with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was purified by flash column chromatography (hexane / ethyl acetate = 30: 1) and GPC to give 4-hydroxy-2,5-bis (trimethylsilyl) phenyl trifluoromethanesulfonate (compound 5) as a white solid. (226 mg, 59%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.23 (s, 1H), 6.75 (s, 1H), 4.92 (s, 1H), 0.35 (s, 9H), 0.31 (s, 9H).
[合成例2:化合物(10b)及び化合物(35b)の合成]
合成例2-1:化合物21aの合成 [Synthesis Example 2: Synthesis of Compound (10b) and Compound (35b)]
Synthesis Example 2-1: Synthesis of Compound 21a
合成例2-1:化合物21aの合成 [Synthesis Example 2: Synthesis of Compound (10b) and Compound (35b)]
Synthesis Example 2-1: Synthesis of Compound 21a

[式中、Meはメチル基を示す。以下同様である。]
500 mLの三つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。0℃で、このフラスコに、3-メトキシチオフェン(3.0 mL, 30 mmol, 1.0当量)のTHF(300 mL)溶液とN-ブロモスクシンイミド(NBS; 5.34 g, 30 mmol, 1.0当量)とを添加した。混合物を0℃で1時間撹拌した後、トリス(ジベンジリデンアセトン)ジパラジウム(0)・クロロホルム付加物(Pd2(dba)3・CHCl3; 465.8 mg, 0.45 mmol, 1.5 mol%)、トリ-t-ブチルホスホニウムテトラフルオロボレート(P(t-Bu)3・HBF4; 522.2 mg, 1.8 mmol, 6 mol%)、4-t-ブチルフェニルボロン酸(31.5 mmol, 1.05当量)、及びNaOH水溶液(3 M, 20 mL, 60 mmol, 2.0当量)を添加し、さらに混合物を60℃で20時間撹拌した。溶媒を蒸発させた後、混合物に水(500 mL)及びCH2Cl2(200 mL)を添加した。CH2Cl2で抽出した後、有機層をNa2SO4で乾燥させ、揮発性物質を減圧下に除去した。フラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=40: 1)により精製し、クーゲルロール(Kugelrohr)により蒸留し、化合物21aを無色油として得た(7.4 g, quant)。
1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 5.6 Hz, 1H), 6.92 (d, J = 5.6 Hz, 1H), 3.90 (s, 3H), 1.33 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 153.3, 149.1, 130.5, 126.5, 125.3, 121.5, 120.1, 117.3, 58.5, 34.4, 31.2; HRMS (DART) m/z calcd for C15H19OS [MH]+: 247.11566, found 247.11528。 [Wherein, Me represents a methyl group. The same applies hereinafter. ]
A 500 mL three-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen. At 0 ° C., a solution of 3-methoxythiophene (3.0 mL, 30 mmol, 1.0 equiv) in THF (300 mL) and N-bromosuccinimide (NBS; 5.34 g, 30 mmol, 1.0 equiv) was added to the flask. . After the mixture was stirred at 0 ° C. for 1 hour, tris (dibenzylideneacetone) dipalladium (0) / chloroform adduct (Pd 2 (dba) 3 .CHCl 3 ; 465.8 mg, 0.45 mmol, 1.5 mol%), tri- t-butylphosphonium tetrafluoroborate (P (t-Bu) 3 · HBF 4 ; 522.2 mg, 1.8 mmol, 6 mol%), 4-t-butylphenylboronic acid (31.5 mmol, 1.05 equivalents), and aqueous NaOH solution ( 3 M, 20 mL, 60 mmol, 2.0 eq) was added and the mixture was further stirred at 60 ° C. for 20 h. After evaporation of the solvent, water (500 mL) and CH 2 Cl 2 (200 mL) were added to the mixture. After extraction with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. Purification by flash column chromatography (hexane / ethyl acetate = 40: 1) and distillation by Kugelrohr gave Compound 21a as a colorless oil (7.4 g, quant).
1 H NMR (400 MHz, CDCl 3 ): δ 7.65 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 5.6 Hz, 1H), 6.92 ( d, J = 5.6 Hz, 1H), 3.90 (s, 3H), 1.33 (s, 9H); 13 C NMR (100 MHz, CDCl 3 ): δ 153.3, 149.1, 130.5, 126.5, 125.3, 121.5, 120.1, 117.3, 58.5, 34.4, 31.2; HRMS (DART) m / z calcd for C 15 H 19 OS [MH] + : 247.11566, found 247.11528.
500 mLの三つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。0℃で、このフラスコに、3-メトキシチオフェン(3.0 mL, 30 mmol, 1.0当量)のTHF(300 mL)溶液とN-ブロモスクシンイミド(NBS; 5.34 g, 30 mmol, 1.0当量)とを添加した。混合物を0℃で1時間撹拌した後、トリス(ジベンジリデンアセトン)ジパラジウム(0)・クロロホルム付加物(Pd2(dba)3・CHCl3; 465.8 mg, 0.45 mmol, 1.5 mol%)、トリ-t-ブチルホスホニウムテトラフルオロボレート(P(t-Bu)3・HBF4; 522.2 mg, 1.8 mmol, 6 mol%)、4-t-ブチルフェニルボロン酸(31.5 mmol, 1.05当量)、及びNaOH水溶液(3 M, 20 mL, 60 mmol, 2.0当量)を添加し、さらに混合物を60℃で20時間撹拌した。溶媒を蒸発させた後、混合物に水(500 mL)及びCH2Cl2(200 mL)を添加した。CH2Cl2で抽出した後、有機層をNa2SO4で乾燥させ、揮発性物質を減圧下に除去した。フラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=40: 1)により精製し、クーゲルロール(Kugelrohr)により蒸留し、化合物21aを無色油として得た(7.4 g, quant)。
1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 5.6 Hz, 1H), 6.92 (d, J = 5.6 Hz, 1H), 3.90 (s, 3H), 1.33 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 153.3, 149.1, 130.5, 126.5, 125.3, 121.5, 120.1, 117.3, 58.5, 34.4, 31.2; HRMS (DART) m/z calcd for C15H19OS [MH]+: 247.11566, found 247.11528。 [Wherein, Me represents a methyl group. The same applies hereinafter. ]
A 500 mL three-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen. At 0 ° C., a solution of 3-methoxythiophene (3.0 mL, 30 mmol, 1.0 equiv) in THF (300 mL) and N-bromosuccinimide (NBS; 5.34 g, 30 mmol, 1.0 equiv) was added to the flask. . After the mixture was stirred at 0 ° C. for 1 hour, tris (dibenzylideneacetone) dipalladium (0) / chloroform adduct (Pd 2 (dba) 3 .CHCl 3 ; 465.8 mg, 0.45 mmol, 1.5 mol%), tri- t-butylphosphonium tetrafluoroborate (P (t-Bu) 3 · HBF 4 ; 522.2 mg, 1.8 mmol, 6 mol%), 4-t-butylphenylboronic acid (31.5 mmol, 1.05 equivalents), and aqueous NaOH solution ( 3 M, 20 mL, 60 mmol, 2.0 eq) was added and the mixture was further stirred at 60 ° C. for 20 h. After evaporation of the solvent, water (500 mL) and CH 2 Cl 2 (200 mL) were added to the mixture. After extraction with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. Purification by flash column chromatography (hexane / ethyl acetate = 40: 1) and distillation by Kugelrohr gave Compound 21a as a colorless oil (7.4 g, quant).
1 H NMR (400 MHz, CDCl 3 ): δ 7.65 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 5.6 Hz, 1H), 6.92 ( d, J = 5.6 Hz, 1H), 3.90 (s, 3H), 1.33 (s, 9H); 13 C NMR (100 MHz, CDCl 3 ): δ 153.3, 149.1, 130.5, 126.5, 125.3, 121.5, 120.1, 117.3, 58.5, 34.4, 31.2; HRMS (DART) m / z calcd for C 15 H 19 OS [MH] + : 247.11566, found 247.11528.
合成例2-2:化合物21bの合成
Synthesis Example 2-2: Synthesis of Compound 21b

アリールボロン酸化合物としてm-トリルボロン酸を使用すること以外は合成例2-1と同様に合成し、化合物21bを無色油として得た(5.5 g, 90 %)。
1H NMR (400 MHz, CDCl3): δ 7.57-7.50 (m, 2H), 7.25 (t, J = 8.0 Hz, 1H), 7.14 (d, J = 5.6 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 6.92 (d, J = 5.6 Hz, 1H), 3.91 (s, 3H), 2.38 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 153.5, 137.9, 133.2, 128.3, 127.5, 127.1, 124.0, 121.9, 120.2, 117.4, 58.6, 21.5; HRMS (DART) m/z calcd for C12H13OS [MH]+: 205.06871, found 205.06863。 Synthesis was carried out in the same manner as in Synthesis Example 2-1 except that m-tolylboronic acid was used as the aryl boronic acid compound to obtain compound 21b as a colorless oil (5.5 g, 90%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.57-7.50 (m, 2H), 7.25 (t, J = 8.0 Hz, 1H), 7.14 (d, J = 5.6 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 6.92 (d, J = 5.6 Hz, 1H), 3.91 (s, 3H), 2.38 (s, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 153.5, 137.9, 133.2 , 128.3, 127.5, 127.1, 124.0, 121.9, 120.2, 117.4, 58.6, 21.5; HRMS (DART) m / z calcd for C 12 H 13 OS [MH] + : 205.06871, found 205.06863.
1H NMR (400 MHz, CDCl3): δ 7.57-7.50 (m, 2H), 7.25 (t, J = 8.0 Hz, 1H), 7.14 (d, J = 5.6 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 6.92 (d, J = 5.6 Hz, 1H), 3.91 (s, 3H), 2.38 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 153.5, 137.9, 133.2, 128.3, 127.5, 127.1, 124.0, 121.9, 120.2, 117.4, 58.6, 21.5; HRMS (DART) m/z calcd for C12H13OS [MH]+: 205.06871, found 205.06863。 Synthesis was carried out in the same manner as in Synthesis Example 2-1 except that m-tolylboronic acid was used as the aryl boronic acid compound to obtain compound 21b as a colorless oil (5.5 g, 90%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.57-7.50 (m, 2H), 7.25 (t, J = 8.0 Hz, 1H), 7.14 (d, J = 5.6 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 6.92 (d, J = 5.6 Hz, 1H), 3.91 (s, 3H), 2.38 (s, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 153.5, 137.9, 133.2 , 128.3, 127.5, 127.1, 124.0, 121.9, 120.2, 117.4, 58.6, 21.5; HRMS (DART) m / z calcd for C 12 H 13 OS [MH] + : 205.06871, found 205.06863.
[合成例3:化合物(10c)及び化合物(35c)の合成]
合成例3-1:化合物7の合成 [Synthesis Example 3: Synthesis of Compound (10c) and Compound (35c)]
Synthesis Example 3-1: Synthesis of Compound 7
合成例3-1:化合物7の合成 [Synthesis Example 3: Synthesis of Compound (10c) and Compound (35c)]
Synthesis Example 3-1: Synthesis of Compound 7

[式中、Meはメチル基を示す。Pd(OAc)2は酢酸パラジウムを示す。2,2’-bipyは2,2’-ビピリジルを示す。TEMPOは2,2,6,6-テトラメチルピペリジン1-オキシルを示す。以下同様である。]
50 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、酢酸パラジウム(Pd(OAc)2; 225.5 mg, 1.0 mmol, 10 mol%)、2,2’-ビピリジル(2,2’-bipy; 155.1 mg, 1.0 mmol, 10 mol%)、p-メチルフェニルボロン酸(5.47 g, 40 mmol, 4.0当量)、2,2,6,6-テトラメチルピペリジン1-オキシル(TEMPO; 4.69 g, 30 mmol, 3.0当量)、合成例2-2で得た3-メトキシ-2-(m-トリル)チオフェン(化合物21b; 2.04 g, 10 mmol, 1.0当量)、及びα,α,α-トリフルオロトルエン(3.0 mL)を窒素気流下に添加した。容器を80℃で48時間加熱した。反応混合物をシリカゲル(溶離液:CHCl3, 100 mL)でろ過し、揮発性物質を減圧下に除去した。粗生成物をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=50: 1)及びゲルパーミエーションクロマトグラフィー(GPC)により精製し、3-メトキシ-2-(m-トリル)-4-(p-トリル)チオフェン(化合物7)を黄色油として得た(2.26 g, 77 %)。
1H NMR (600 MHz, CDCl3): δ 7.59-7.53 (m, 4H), 7.29 (t, J = 7.2 Hz, 1H), 7.23 (d, J = 8.4 Hz, 2H), 7.14 (s, 1H), 7.11 (d, J = 7.2 Hz, 1H), 3.50 (s, 3H), 2.40 (s, 3H), 2.39 (s, 3H)。 [Wherein, Me represents a methyl group. Pd (OAc) 2 represents palladium acetate. 2,2'-bipy represents 2,2'-bipyridyl. TEMPO represents 2,2,6,6-tetramethylpiperidine 1-oxyl. The same applies hereinafter. ]
A 50 mL two-necked flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, palladium acetate (Pd (OAc) 2 ; 225.5 mg, 1.0 mmol, 10 mol%), 2,2'-bipyridyl (2,2'-bipy; 155.1 mg, 1.0 mmol, 10 mol%), p -Methylphenylboronic acid (5.47 g, 40 mmol, 4.0 eq), 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO; 4.69 g, 30 mmol, 3.0 eq), obtained in Synthesis Example 2-2 3-methoxy-2- (m-tolyl) thiophene (Compound 21b; 2.04 g, 10 mmol, 1.0 equivalent) and α, α, α-trifluorotoluene (3.0 mL) were added under a nitrogen stream. The vessel was heated at 80 ° C. for 48 hours. The reaction mixture was filtered through silica gel (eluent: CHCl 3 , 100 mL) and volatiles were removed under reduced pressure. The crude product was purified by flash column chromatography (hexane / ethyl acetate = 50: 1) and gel permeation chromatography (GPC) to give 3-methoxy-2- (m-tolyl) -4- (p-tolyl) Thiophene (compound 7) was obtained as a yellow oil (2.26 g, 77%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.59-7.53 (m, 4H), 7.29 (t, J = 7.2 Hz, 1H), 7.23 (d, J = 8.4 Hz, 2H), 7.14 (s, 1H ), 7.11 (d, J = 7.2 Hz, 1H), 3.50 (s, 3H), 2.40 (s, 3H), 2.39 (s, 3H).
50 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、酢酸パラジウム(Pd(OAc)2; 225.5 mg, 1.0 mmol, 10 mol%)、2,2’-ビピリジル(2,2’-bipy; 155.1 mg, 1.0 mmol, 10 mol%)、p-メチルフェニルボロン酸(5.47 g, 40 mmol, 4.0当量)、2,2,6,6-テトラメチルピペリジン1-オキシル(TEMPO; 4.69 g, 30 mmol, 3.0当量)、合成例2-2で得た3-メトキシ-2-(m-トリル)チオフェン(化合物21b; 2.04 g, 10 mmol, 1.0当量)、及びα,α,α-トリフルオロトルエン(3.0 mL)を窒素気流下に添加した。容器を80℃で48時間加熱した。反応混合物をシリカゲル(溶離液:CHCl3, 100 mL)でろ過し、揮発性物質を減圧下に除去した。粗生成物をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=50: 1)及びゲルパーミエーションクロマトグラフィー(GPC)により精製し、3-メトキシ-2-(m-トリル)-4-(p-トリル)チオフェン(化合物7)を黄色油として得た(2.26 g, 77 %)。
1H NMR (600 MHz, CDCl3): δ 7.59-7.53 (m, 4H), 7.29 (t, J = 7.2 Hz, 1H), 7.23 (d, J = 8.4 Hz, 2H), 7.14 (s, 1H), 7.11 (d, J = 7.2 Hz, 1H), 3.50 (s, 3H), 2.40 (s, 3H), 2.39 (s, 3H)。 [Wherein, Me represents a methyl group. Pd (OAc) 2 represents palladium acetate. 2,2'-bipy represents 2,2'-bipyridyl. TEMPO represents 2,2,6,6-tetramethylpiperidine 1-oxyl. The same applies hereinafter. ]
A 50 mL two-necked flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, palladium acetate (Pd (OAc) 2 ; 225.5 mg, 1.0 mmol, 10 mol%), 2,2'-bipyridyl (2,2'-bipy; 155.1 mg, 1.0 mmol, 10 mol%), p -Methylphenylboronic acid (5.47 g, 40 mmol, 4.0 eq), 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO; 4.69 g, 30 mmol, 3.0 eq), obtained in Synthesis Example 2-2 3-methoxy-2- (m-tolyl) thiophene (Compound 21b; 2.04 g, 10 mmol, 1.0 equivalent) and α, α, α-trifluorotoluene (3.0 mL) were added under a nitrogen stream. The vessel was heated at 80 ° C. for 48 hours. The reaction mixture was filtered through silica gel (eluent: CHCl 3 , 100 mL) and volatiles were removed under reduced pressure. The crude product was purified by flash column chromatography (hexane / ethyl acetate = 50: 1) and gel permeation chromatography (GPC) to give 3-methoxy-2- (m-tolyl) -4- (p-tolyl) Thiophene (compound 7) was obtained as a yellow oil (2.26 g, 77%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.59-7.53 (m, 4H), 7.29 (t, J = 7.2 Hz, 1H), 7.23 (d, J = 8.4 Hz, 2H), 7.14 (s, 1H ), 7.11 (d, J = 7.2 Hz, 1H), 3.50 (s, 3H), 2.40 (s, 3H), 2.39 (s, 3H).
合成例3-2:化合物22acの合成
Synthesis Example 3-2: Synthesis of Compound 22ac

50 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、酢酸パラジウム(Pd(OAc)2; 224.5 mg, 1.0 mmol, 10 mol%)、2,2’-ビピリジル(bipy: 156.8 mg, 1.0 mmol, 10 mol%)、フェニルボロン酸(40 mmol, 4.0当量)、2,2,6,6-テトラメチルピペリジン1-オキシル(TEMPO; 4.69 g, 30 mmol, 3.0当量)、合成例2-1で得た化合物21a(10 mmol, 1.0当量)、及びα,α,α-トリフルオロトルエン(3.3 mL)を窒素雰囲気下に添加した。容器を80℃で48時間加熱した。反応混合物をシリカゲル(溶離液: 酢酸エチル, 100 mL)でろ過し、揮発性物質を減圧下に除去した。フラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=20: 1)及びゲルパーミエーションクロマトグラフィー(GPC)により精製し、化合物22acを白色固体として得た(2.43 g, 75 % (β), β/α= 98: 2)。
1H NMR (400 MHz, CDCl3): δ 7.72-7.64 (m, 4H), 7.45-7.39 (m, 4H), 7.33 (t, J = 8.0 Hz, 1H), 7.16 (s, 1H), 3.51 (s, 3H), 1.36 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 151.3, 150.2, 136.7, 135.1, 130.2, 128.5, 128.2, 127.5, 127.2, 126.9, 125.6, 119.1, 60.7, 34.6, 31.3; HRMS (DART) m/z calcd for C21H23OS [MH]+: 323.14696, found 323.14673。 A 50 mL two-necked flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, palladium acetate (Pd (OAc) 2 ; 224.5 mg, 1.0 mmol, 10 mol%), 2,2'-bipyridyl (bipy: 156.8 mg, 1.0 mmol, 10 mol%), phenylboronic acid (40 mmol) , 4.0 equivalents), 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO; 4.69 g, 30 mmol, 3.0 equivalents), compound 21a obtained in Synthesis Example 2-1 (10 mmol, 1.0 equivalents), And α, α, α-trifluorotoluene (3.3 mL) were added under a nitrogen atmosphere. The vessel was heated at 80 ° C. for 48 hours. The reaction mixture was filtered through silica gel (eluent: ethyl acetate, 100 mL) and volatiles were removed under reduced pressure. Purification by flash column chromatography (hexane / ethyl acetate = 20: 1) and gel permeation chromatography (GPC) gave compound 22ac as a white solid (2.43 g, 75% (β), β / α = 98. : 2).
1 H NMR (400 MHz, CDCl 3 ): δ 7.72-7.64 (m, 4H), 7.45-7.39 (m, 4H), 7.33 (t, J = 8.0 Hz, 1H), 7.16 (s, 1H), 3.51 (s, 3H), 1.36 (s, 9H); 13 C NMR (100 MHz, CDCl 3 ): δ 151.3, 150.2, 136.7, 135.1, 130.2, 128.5, 128.2, 127.5, 127.2, 126.9, 125.6, 119.1, 60.7 , 34.6, 31.3; HRMS (DART) m / z calcd for C 21 H 23 OS [MH] + : 323.14696, found 323.14673.
1H NMR (400 MHz, CDCl3): δ 7.72-7.64 (m, 4H), 7.45-7.39 (m, 4H), 7.33 (t, J = 8.0 Hz, 1H), 7.16 (s, 1H), 3.51 (s, 3H), 1.36 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 151.3, 150.2, 136.7, 135.1, 130.2, 128.5, 128.2, 127.5, 127.2, 126.9, 125.6, 119.1, 60.7, 34.6, 31.3; HRMS (DART) m/z calcd for C21H23OS [MH]+: 323.14696, found 323.14673。 A 50 mL two-necked flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, palladium acetate (Pd (OAc) 2 ; 224.5 mg, 1.0 mmol, 10 mol%), 2,2'-bipyridyl (bipy: 156.8 mg, 1.0 mmol, 10 mol%), phenylboronic acid (40 mmol) , 4.0 equivalents), 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO; 4.69 g, 30 mmol, 3.0 equivalents), compound 21a obtained in Synthesis Example 2-1 (10 mmol, 1.0 equivalents), And α, α, α-trifluorotoluene (3.3 mL) were added under a nitrogen atmosphere. The vessel was heated at 80 ° C. for 48 hours. The reaction mixture was filtered through silica gel (eluent: ethyl acetate, 100 mL) and volatiles were removed under reduced pressure. Purification by flash column chromatography (hexane / ethyl acetate = 20: 1) and gel permeation chromatography (GPC) gave compound 22ac as a white solid (2.43 g, 75% (β), β / α = 98. : 2).
1 H NMR (400 MHz, CDCl 3 ): δ 7.72-7.64 (m, 4H), 7.45-7.39 (m, 4H), 7.33 (t, J = 8.0 Hz, 1H), 7.16 (s, 1H), 3.51 (s, 3H), 1.36 (s, 9H); 13 C NMR (100 MHz, CDCl 3 ): δ 151.3, 150.2, 136.7, 135.1, 130.2, 128.5, 128.2, 127.5, 127.2, 126.9, 125.6, 119.1, 60.7 , 34.6, 31.3; HRMS (DART) m / z calcd for C 21 H 23 OS [MH] + : 323.14696, found 323.14673.
合成例3-3:化合物22bcの合成
Synthesis Example 3-3: Synthesis of Compound 22bc

合成例2-1で得た化合物21aの代わりに合成例2-2で得た化合物21bを使用すること以外は合成例3-2と同様に合成し、化合物22bcを無色油として得た(2.13 g, 76 % (β), β/α= 91: 9)。
1H NMR (400 MHz, CDCl3): δ 7.68-7.64 (m, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.42 (t, J = 7.6 Hz, 2H), 7.36-7.27 (m, 2H), 7.18 (s, 1H), 7.11 (d, J = 7.6 Hz, 1H), 3.50 (s, 3H), 2.40 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 151.5, 138.3, 136.8, 135.0, 133.0, 128.6, 128.5, 128.3, 128.03, 127.97, 127.5, 127.3, 124.4, 119.4, 60.7, 21.5; HRMS (DART) m/z calcd for C18H17OS [MH]+: 281.10001, found 281.10046。 Synthesis was performed in the same manner as in Synthesis Example 3-2 except that Compound 21b obtained in Synthesis Example 2-2 was used instead of Compound 21a obtained in Synthesis Example 2-1. Thus, Compound 22bc was obtained as a colorless oil (2.13 g, 76% (β), β / α = 91: 9).
1 H NMR (400 MHz, CDCl 3 ): δ 7.68-7.64 (m, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.42 (t, J = 7.6 Hz, 2H), 7.36-7.27 (m , 2H), 7.18 (s, 1H), 7.11 (d, J = 7.6 Hz, 1H), 3.50 (s, 3H), 2.40 (s, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 151.5 , 138.3, 136.8, 135.0, 133.0, 128.6, 128.5, 128.3, 128.03, 127.97, 127.5, 127.3, 124.4, 119.4, 60.7, 21.5; HRMS (DART) m / z calcd for C 18 H 17 OS [MH] + : 281.10001, found 281.10046.
1H NMR (400 MHz, CDCl3): δ 7.68-7.64 (m, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.42 (t, J = 7.6 Hz, 2H), 7.36-7.27 (m, 2H), 7.18 (s, 1H), 7.11 (d, J = 7.6 Hz, 1H), 3.50 (s, 3H), 2.40 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 151.5, 138.3, 136.8, 135.0, 133.0, 128.6, 128.5, 128.3, 128.03, 127.97, 127.5, 127.3, 124.4, 119.4, 60.7, 21.5; HRMS (DART) m/z calcd for C18H17OS [MH]+: 281.10001, found 281.10046。 Synthesis was performed in the same manner as in Synthesis Example 3-2 except that Compound 21b obtained in Synthesis Example 2-2 was used instead of Compound 21a obtained in Synthesis Example 2-1. Thus, Compound 22bc was obtained as a colorless oil (2.13 g, 76% (β), β / α = 91: 9).
1 H NMR (400 MHz, CDCl 3 ): δ 7.68-7.64 (m, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.42 (t, J = 7.6 Hz, 2H), 7.36-7.27 (m , 2H), 7.18 (s, 1H), 7.11 (d, J = 7.6 Hz, 1H), 3.50 (s, 3H), 2.40 (s, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 151.5 , 138.3, 136.8, 135.0, 133.0, 128.6, 128.5, 128.3, 128.03, 127.97, 127.5, 127.3, 124.4, 119.4, 60.7, 21.5; HRMS (DART) m / z calcd for C 18 H 17 OS [MH] + : 281.10001, found 281.10046.
[合成例4:化合物(10d)及び化合物(35d)の合成]
合成例4-1:化合物8の合成 [Synthesis Example 4: Synthesis of Compound (10d) and Compound (35d)]
Synthesis Example 4-1: Synthesis of Compound 8
合成例4-1:化合物8の合成 [Synthesis Example 4: Synthesis of Compound (10d) and Compound (35d)]
Synthesis Example 4-1: Synthesis of Compound 8

100 mLのガラス容器にJ. Young Oリングタップを取りつけ、磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。この容器に、塩化パラジウム(PdCl2; 124 mg, 0.66 mmol, 10 mol%)、2,2’-ビピリジル(bipy; 110 mg, 0.66 mmol, 10 mol%)、Ag2CO3(1.93 g, 6.6 mmol, 1.0当量)、合成例3-1で得た化合物7(1.95 g, 6.6 mmol, 1.0当量)、1-クロロ-4-ヨードベンゼン(5.03 g, 19.9 mmol, 3.0当量)、及び乾燥m-キシレン(33 mL)を窒素雰囲気下に添加した。容器をOリングタップで密封し、120℃で36時間加熱した。反応混合物を室温まで冷却した後、混合物を短いシリカゲルパッド(酢酸エチル)でろ過した。ろ液を真空下に濃縮し、粗生成物をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル= 50: 1)及びゲルパーミエーションクロマトグラフィー(GPC)により精製し、2-(4-クロロフェニル)-4-メトキシ-5-(m-トリル)-3-(p-トリル)チオフェン(化合物8)を黄色固体として得た(1.75 g, 66 %)。
1H NMR (600 MHz, CDCl3): δ 7.62-7.58 (m, 2H), 7.30 (t, J = 7.2 Hz, 1H), 7.23-7.19 (m, 4H), 7.19-7.14 (m, 4H), 7.12 (d, J = 7.2 Hz, 1H), 3.38 (s, 3H), 2.41 (s, 3H), 2.38 (s, 3H)。 A J. Young O-ring tap was attached to a 100 mL glass container, a magnetic stirrer was placed, the frame was dried under vacuum, cooled to room temperature, and then filled with nitrogen. In this container, palladium chloride (PdCl 2 ; 124 mg, 0.66 mmol, 10 mol%), 2,2'-bipyridyl (bipy; 110 mg, 0.66 mmol, 10 mol%), Ag 2 CO 3 (1.93 g, 6.6 mmol, 1.0 equivalent), compound 7 obtained in Synthesis Example 3-1 (1.95 g, 6.6 mmol, 1.0 equivalent), 1-chloro-4-iodobenzene (5.03 g, 19.9 mmol, 3.0 equivalent), and dry m- Xylene (33 mL) was added under a nitrogen atmosphere. The vessel was sealed with an O-ring tap and heated at 120 ° C. for 36 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a short silica gel pad (ethyl acetate). The filtrate was concentrated under vacuum and the crude product was purified by flash column chromatography (hexane / ethyl acetate = 50: 1) and gel permeation chromatography (GPC) to give 2- (4-chlorophenyl) -4- Methoxy-5- (m-tolyl) -3- (p-tolyl) thiophene (Compound 8) was obtained as a yellow solid (1.75 g, 66%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.62-7.58 (m, 2H), 7.30 (t, J = 7.2 Hz, 1H), 7.23-7.19 (m, 4H), 7.19-7.14 (m, 4H) , 7.12 (d, J = 7.2 Hz, 1H), 3.38 (s, 3H), 2.41 (s, 3H), 2.38 (s, 3H).
1H NMR (600 MHz, CDCl3): δ 7.62-7.58 (m, 2H), 7.30 (t, J = 7.2 Hz, 1H), 7.23-7.19 (m, 4H), 7.19-7.14 (m, 4H), 7.12 (d, J = 7.2 Hz, 1H), 3.38 (s, 3H), 2.41 (s, 3H), 2.38 (s, 3H)。 A J. Young O-ring tap was attached to a 100 mL glass container, a magnetic stirrer was placed, the frame was dried under vacuum, cooled to room temperature, and then filled with nitrogen. In this container, palladium chloride (PdCl 2 ; 124 mg, 0.66 mmol, 10 mol%), 2,2'-bipyridyl (bipy; 110 mg, 0.66 mmol, 10 mol%), Ag 2 CO 3 (1.93 g, 6.6 mmol, 1.0 equivalent), compound 7 obtained in Synthesis Example 3-1 (1.95 g, 6.6 mmol, 1.0 equivalent), 1-chloro-4-iodobenzene (5.03 g, 19.9 mmol, 3.0 equivalent), and dry m- Xylene (33 mL) was added under a nitrogen atmosphere. The vessel was sealed with an O-ring tap and heated at 120 ° C. for 36 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a short silica gel pad (ethyl acetate). The filtrate was concentrated under vacuum and the crude product was purified by flash column chromatography (hexane / ethyl acetate = 50: 1) and gel permeation chromatography (GPC) to give 2- (4-chlorophenyl) -4- Methoxy-5- (m-tolyl) -3- (p-tolyl) thiophene (Compound 8) was obtained as a yellow solid (1.75 g, 66%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.62-7.58 (m, 2H), 7.30 (t, J = 7.2 Hz, 1H), 7.23-7.19 (m, 4H), 7.19-7.14 (m, 4H) , 7.12 (d, J = 7.2 Hz, 1H), 3.38 (s, 3H), 2.41 (s, 3H), 2.38 (s, 3H).
合成例4-2:化合物23acdの合成
Synthesis Example 4-2: Synthesis of Compound 23acd

100 mLのガラス容器にJ. Young Oリングタップを取りつけ、磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。この容器に、塩化パラジウム(PdCl2; 76.6 mg, 0.45 mmol, 10 mol%)、2,2’-ビピリジル(bipy: 70.2 mg, 0.45 mmol, 10 mol%)、炭酸銀(Ag2CO3; 1.24 g, 4.5 mmol, 1.0当量)、合成例3-2で得た化合物22ac(4.5 mmol, 1.0当量)、4-トリフルオロメチルフェニルヨージド(13.5 mmol, 3.0当量)、及び乾燥m-キシレン(24 mL)を窒素雰囲気下に添加した。容器をOリングタップで密封し、120℃で48時間加熱した。反応混合物を室温まで冷却した後、混合物を短いシリカゲルパッド(酢酸エチル)でろ過した。ろ液を真空下に濃縮し、ゲルパーミエーションクロマトグラフィー(GPC)により精製し、化合物3acdを白色固体として得た(1.77 g, 84 %)。
1H NMR (400 MHz, CDCl3): δ 7.73 (d, J = 8.8 Hz, 2H), 7.49-7.42 (m, 4H), 7.40-7.30 (m, 7H), 3.39 (s, 3H), 1.36 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 152.1, 150.6, 138.1, 134.8, 134.2, 133.6, 130.0, 129.7, 129.0 (q, 2JCF = 32.6 Hz), 128.7, 128.6, 128.0, 127.6, 125.7, 125.3 (q, 3JCF = 3.8 Hz), 124.1 (q, 1JCF = 273 Hz), 60.6, 34.6, 31.3; HRMS (DART) m/z calcd for C28H26F3OS [MH]+: 467.16564, found 467.16622。 A J. Young O-ring tap was attached to a 100 mL glass container, a magnetic stirrer was placed, the frame was dried under vacuum, cooled to room temperature, and then filled with nitrogen. In this container, palladium chloride (PdCl 2 ; 76.6 mg, 0.45 mmol, 10 mol%), 2,2'-bipyridyl (bipy: 70.2 mg, 0.45 mmol, 10 mol%), silver carbonate (Ag 2 CO 3 ; 1.24 g, 4.5 mmol, 1.0 equivalent), compound 22ac obtained in Synthesis Example 3-2 (4.5 mmol, 1.0 equivalent), 4-trifluoromethylphenyl iodide (13.5 mmol, 3.0 equivalent), and dry m-xylene (24 mL) was added under a nitrogen atmosphere. The vessel was sealed with an O-ring tap and heated at 120 ° C. for 48 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a short silica gel pad (ethyl acetate). The filtrate was concentrated under vacuum and purified by gel permeation chromatography (GPC) to give compound 3acd as a white solid (1.77 g, 84%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.73 (d, J = 8.8 Hz, 2H), 7.49-7.42 (m, 4H), 7.40-7.30 (m, 7H), 3.39 (s, 3H), 1.36 (s, 9H); 13 C NMR (100 MHz, CDCl 3 ): δ 152.1, 150.6, 138.1, 134.8, 134.2, 133.6, 130.0, 129.7, 129.0 (q, 2 J CF = 32.6 Hz), 128.7, 128.6, 128.0, 127.6, 125.7, 125.3 (q, 3 J CF = 3.8 Hz), 124.1 (q, 1 J CF = 273 Hz), 60.6, 34.6, 31.3; HRMS (DART) m / z calcd for C 28 H 26 F 3 OS [MH] + : 467.16564, found 467.16622.
1H NMR (400 MHz, CDCl3): δ 7.73 (d, J = 8.8 Hz, 2H), 7.49-7.42 (m, 4H), 7.40-7.30 (m, 7H), 3.39 (s, 3H), 1.36 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 152.1, 150.6, 138.1, 134.8, 134.2, 133.6, 130.0, 129.7, 129.0 (q, 2JCF = 32.6 Hz), 128.7, 128.6, 128.0, 127.6, 125.7, 125.3 (q, 3JCF = 3.8 Hz), 124.1 (q, 1JCF = 273 Hz), 60.6, 34.6, 31.3; HRMS (DART) m/z calcd for C28H26F3OS [MH]+: 467.16564, found 467.16622。 A J. Young O-ring tap was attached to a 100 mL glass container, a magnetic stirrer was placed, the frame was dried under vacuum, cooled to room temperature, and then filled with nitrogen. In this container, palladium chloride (PdCl 2 ; 76.6 mg, 0.45 mmol, 10 mol%), 2,2'-bipyridyl (bipy: 70.2 mg, 0.45 mmol, 10 mol%), silver carbonate (Ag 2 CO 3 ; 1.24 g, 4.5 mmol, 1.0 equivalent), compound 22ac obtained in Synthesis Example 3-2 (4.5 mmol, 1.0 equivalent), 4-trifluoromethylphenyl iodide (13.5 mmol, 3.0 equivalent), and dry m-xylene (24 mL) was added under a nitrogen atmosphere. The vessel was sealed with an O-ring tap and heated at 120 ° C. for 48 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a short silica gel pad (ethyl acetate). The filtrate was concentrated under vacuum and purified by gel permeation chromatography (GPC) to give compound 3acd as a white solid (1.77 g, 84%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.73 (d, J = 8.8 Hz, 2H), 7.49-7.42 (m, 4H), 7.40-7.30 (m, 7H), 3.39 (s, 3H), 1.36 (s, 9H); 13 C NMR (100 MHz, CDCl 3 ): δ 152.1, 150.6, 138.1, 134.8, 134.2, 133.6, 130.0, 129.7, 129.0 (q, 2 J CF = 32.6 Hz), 128.7, 128.6, 128.0, 127.6, 125.7, 125.3 (q, 3 J CF = 3.8 Hz), 124.1 (q, 1 J CF = 273 Hz), 60.6, 34.6, 31.3; HRMS (DART) m / z calcd for C 28 H 26 F 3 OS [MH] + : 467.16564, found 467.16622.
合成例4-3:化合物23bceの合成
Synthesis Example 4-3: Synthesis of Compound 23bce

合成例3-2で得た化合物22acの代わりに合成例3-3で得た化合物22bcを使用し、アリールヨージドとして4-クロロヨードベンゼンを使用すること以外は合成例4-2と同様に合成し、化合物23bceを白色固体として得た(1.69 g, 72 %)。
1H NMR (400 MHz, CDCl3): δ 7.63-7.57 (m, 2H), 7.39-7.28 (m, 6H), 7.23-7.10 (m, 5H), 3.38 (s, 3H), 2.41 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 152.1, 138.3, 134.5, 134.3, 133.9, 133.3, 132.9, 132.6, 130.0, 129.9, 128.7, 128.6, 128.4, 128.1, 127.9, 127.4, 127.1, 124.4, 60.6, 21.5; HRMS (DART) m/z calcd for C24H20ClOS [MH]+: 391.09234, found 391.09257。 Similar to Synthesis Example 4-2, except that Compound 22bc obtained in Synthesis Example 3-3 was used instead of Compound 22ac obtained in Synthesis Example 3-2, and 4-chloroiodobenzene was used as the aryl iodide. Synthesis gave compound 23bce as a white solid (1.69 g, 72%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.63-7.57 (m, 2H), 7.39-7.28 (m, 6H), 7.23-7.10 (m, 5H), 3.38 (s, 3H), 2.41 (s, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 152.1, 138.3, 134.5, 134.3, 133.9, 133.3, 132.9, 132.6, 130.0, 129.9, 128.7, 128.6, 128.4, 128.1, 127.9, 127.4, 127.1, 124.4 HRMS (DART) m / z calcd for C 24 H 20 ClOS [MH] + : 391.09234, found 391.09257.
1H NMR (400 MHz, CDCl3): δ 7.63-7.57 (m, 2H), 7.39-7.28 (m, 6H), 7.23-7.10 (m, 5H), 3.38 (s, 3H), 2.41 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 152.1, 138.3, 134.5, 134.3, 133.9, 133.3, 132.9, 132.6, 130.0, 129.9, 128.7, 128.6, 128.4, 128.1, 127.9, 127.4, 127.1, 124.4, 60.6, 21.5; HRMS (DART) m/z calcd for C24H20ClOS [MH]+: 391.09234, found 391.09257。 Similar to Synthesis Example 4-2, except that Compound 22bc obtained in Synthesis Example 3-3 was used instead of Compound 22ac obtained in Synthesis Example 3-2, and 4-chloroiodobenzene was used as the aryl iodide. Synthesis gave compound 23bce as a white solid (1.69 g, 72%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.63-7.57 (m, 2H), 7.39-7.28 (m, 6H), 7.23-7.10 (m, 5H), 3.38 (s, 3H), 2.41 (s, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 152.1, 138.3, 134.5, 134.3, 133.9, 133.3, 132.9, 132.6, 130.0, 129.9, 128.7, 128.6, 128.4, 128.1, 127.9, 127.4, 127.1, 124.4 HRMS (DART) m / z calcd for C 24 H 20 ClOS [MH] + : 391.09234, found 391.09257.
[合成例5:化合物(10e)及び化合物(35e)の合成]
合成例5-1:化合物9の合成 [Synthesis Example 5: Synthesis of Compound (10e) and Compound (35e)]
Synthesis Example 5-1: Synthesis of Compound 9
合成例5-1:化合物9の合成 [Synthesis Example 5: Synthesis of Compound (10e) and Compound (35e)]
Synthesis Example 5-1: Synthesis of Compound 9

[式中、i-Pr2NEtはN,N-ジイソプロピルエチルアミンを示す。DMAPはN,N-ジメチルアミノピリジンを示す。以下同様である。]
100 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、合成例4-1で得た2-(4-クロロフェニル)-4-メトキシ-5-(m-トリル)-3-(p-トリル)チオフェン(化合物8; 1.76 g, 4.4 mmol, 1.0当量)、及び乾燥CH2Cl2(44 mL)を窒素気流下に添加した。内容物を-78℃まで冷却し、ボロントリブロマイド(5.65 mL, 1 M in CH2Cl2, 6.5 mmol, 1.3当量)をゆっくりと添加した。得られた混合物を-78℃で0.5時間撹拌した。反応混合物を室温まで昇温した後、反応はTLCでモニターした。飽和Na2S2O3水溶液及び飽和NaHCO3水溶液を添加して反応をクエンチした後、混合物に水及びCH2Cl2(20 mL)を添加した。混合物をCH2Cl2で抽出した後、有機層をNa2SO4で乾燥させ、揮発性物質を減圧下に除去した。粗生成物をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル= 50: 1~10: 1)に供して出発材料を除去した。ろ液を真空下に濃縮し、さらなる精製をせずに次の工程に使用した。 [Wherein, i-Pr2NEt represents N, N-diisopropylethylamine. DMAP represents N, N-dimethylaminopyridine. The same applies hereinafter. ]
A 100 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, 2- (4-chlorophenyl) -4-methoxy-5- (m-tolyl) -3- (p-tolyl) thiophene obtained in Synthesis Example 4-1 (Compound 8; 1.76 g, 4.4 mmol, 1.0 equivalent), and dry CH 2 Cl 2 (44 mL) were added under a stream of nitrogen. The contents were cooled to −78 ° C. and boron tribromide (5.65 mL, 1 M in CH 2 Cl 2 , 6.5 mmol, 1.3 eq) was added slowly. The resulting mixture was stirred at −78 ° C. for 0.5 hour. After the reaction mixture was warmed to room temperature, the reaction was monitored by TLC. After quenching the reaction by adding saturated aqueous Na 2 S 2 O 3 and saturated aqueous NaHCO 3 , water and CH 2 Cl 2 (20 mL) were added to the mixture. After the mixture was extracted with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was subjected to flash column chromatography (hexane / ethyl acetate = 50: 1 to 10: 1) to remove starting material. The filtrate was concentrated under vacuum and used in the next step without further purification.
100 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、合成例4-1で得た2-(4-クロロフェニル)-4-メトキシ-5-(m-トリル)-3-(p-トリル)チオフェン(化合物8; 1.76 g, 4.4 mmol, 1.0当量)、及び乾燥CH2Cl2(44 mL)を窒素気流下に添加した。内容物を-78℃まで冷却し、ボロントリブロマイド(5.65 mL, 1 M in CH2Cl2, 6.5 mmol, 1.3当量)をゆっくりと添加した。得られた混合物を-78℃で0.5時間撹拌した。反応混合物を室温まで昇温した後、反応はTLCでモニターした。飽和Na2S2O3水溶液及び飽和NaHCO3水溶液を添加して反応をクエンチした後、混合物に水及びCH2Cl2(20 mL)を添加した。混合物をCH2Cl2で抽出した後、有機層をNa2SO4で乾燥させ、揮発性物質を減圧下に除去した。粗生成物をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル= 50: 1~10: 1)に供して出発材料を除去した。ろ液を真空下に濃縮し、さらなる精製をせずに次の工程に使用した。 [Wherein, i-Pr2NEt represents N, N-diisopropylethylamine. DMAP represents N, N-dimethylaminopyridine. The same applies hereinafter. ]
A 100 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, 2- (4-chlorophenyl) -4-methoxy-5- (m-tolyl) -3- (p-tolyl) thiophene obtained in Synthesis Example 4-1 (Compound 8; 1.76 g, 4.4 mmol, 1.0 equivalent), and dry CH 2 Cl 2 (44 mL) were added under a stream of nitrogen. The contents were cooled to −78 ° C. and boron tribromide (5.65 mL, 1 M in CH 2 Cl 2 , 6.5 mmol, 1.3 eq) was added slowly. The resulting mixture was stirred at −78 ° C. for 0.5 hour. After the reaction mixture was warmed to room temperature, the reaction was monitored by TLC. After quenching the reaction by adding saturated aqueous Na 2 S 2 O 3 and saturated aqueous NaHCO 3 , water and CH 2 Cl 2 (20 mL) were added to the mixture. After the mixture was extracted with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was subjected to flash column chromatography (hexane / ethyl acetate = 50: 1 to 10: 1) to remove starting material. The filtrate was concentrated under vacuum and used in the next step without further purification.
100 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、上記工程で得た物質、及び乾燥CH2Cl2(22 mL)を窒素気流下に添加した。内容物を0℃まで冷却し、このフラスコにN,N-ジイソプロピルエチルアミン(i-Pr2NEt; 1.14 mL, 6.52 mmol, 1.5当量)、N,N-ジメチルアミノピリジン(DMAP; 5.3 mg, 43.5μmol, 1 mol%)、及びトリフルオロメタンスルホン酸無水物(Tf2O; 1.07 mL, 6.52 mmol, 1.5当量)を添加した。得られた混合物を0℃で0.5時間撹拌した。反応混合物を室温まで昇温して12時間撹拌した後、混合物にNaHCO3水溶液、水及びCH2Cl2を添加した。混合物をCH2Cl2で抽出した後、有機層をNa2SO4で乾燥させ、揮発性物質を減圧下に除去した。粗生成物をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル= 50: 1)に供し、5-(4-クロロフェニル)-2-(m-トリル)-4-(p-トリル)チオフェン-3-イルトリフルオロメタンスルホネート(化合物9)を白色固体として得た(1.02 g, 45 %)。
1H NMR (600 MHz, CDCl3): δ 7.45 (s, 1H), 7.42 (d, J = 8.4 Hz, 2H), 7.36 (t, J = 7.8 Hz, 1H), 7.25-7.22 (m, 3H), 7.20-7.14 (m, 6H), 2.42 (s, 3H), 2.38 (s, 3H)。 A 100 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen. To the flask, the material obtained in the above step and dry CH 2 Cl 2 (22 mL) were added under a stream of nitrogen. The contents were cooled to 0 ° C., and N, N-diisopropylethylamine (i-Pr 2 NEt; 1.14 mL, 6.52 mmol, 1.5 eq), N, N-dimethylaminopyridine (DMAP; 5.3 mg, 43.5 μmol) was added to the flask. , 1 mol%), and trifluoromethanesulfonic anhydride (Tf 2 O; 1.07 mL, 6.52 mmol, 1.5 eq). The resulting mixture was stirred at 0 ° C. for 0.5 hour. The reaction mixture was warmed to room temperature and stirred for 12 hours, and then an aqueous NaHCO 3 solution, water and CH 2 Cl 2 were added to the mixture. After the mixture was extracted with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was subjected to flash column chromatography (hexane / ethyl acetate = 50: 1) to give 5- (4-chlorophenyl) -2- (m-tolyl) -4- (p-tolyl) thiophen-3-yltrifluoro. Lomomethanesulfonate (Compound 9) was obtained as a white solid (1.02 g, 45%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.45 (s, 1H), 7.42 (d, J = 8.4 Hz, 2H), 7.36 (t, J = 7.8 Hz, 1H), 7.25-7.22 (m, 3H ), 7.20-7.14 (m, 6H), 2.42 (s, 3H), 2.38 (s, 3H).
1H NMR (600 MHz, CDCl3): δ 7.45 (s, 1H), 7.42 (d, J = 8.4 Hz, 2H), 7.36 (t, J = 7.8 Hz, 1H), 7.25-7.22 (m, 3H), 7.20-7.14 (m, 6H), 2.42 (s, 3H), 2.38 (s, 3H)。 A 100 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen. To the flask, the material obtained in the above step and dry CH 2 Cl 2 (22 mL) were added under a stream of nitrogen. The contents were cooled to 0 ° C., and N, N-diisopropylethylamine (i-Pr 2 NEt; 1.14 mL, 6.52 mmol, 1.5 eq), N, N-dimethylaminopyridine (DMAP; 5.3 mg, 43.5 μmol) was added to the flask. , 1 mol%), and trifluoromethanesulfonic anhydride (Tf 2 O; 1.07 mL, 6.52 mmol, 1.5 eq). The resulting mixture was stirred at 0 ° C. for 0.5 hour. The reaction mixture was warmed to room temperature and stirred for 12 hours, and then an aqueous NaHCO 3 solution, water and CH 2 Cl 2 were added to the mixture. After the mixture was extracted with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was subjected to flash column chromatography (hexane / ethyl acetate = 50: 1) to give 5- (4-chlorophenyl) -2- (m-tolyl) -4- (p-tolyl) thiophen-3-yltrifluoro. Lomomethanesulfonate (Compound 9) was obtained as a white solid (1.02 g, 45%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.45 (s, 1H), 7.42 (d, J = 8.4 Hz, 2H), 7.36 (t, J = 7.8 Hz, 1H), 7.25-7.22 (m, 3H ), 7.20-7.14 (m, 6H), 2.42 (s, 3H), 2.38 (s, 3H).
合成例5-2:化合物24acdの合成
Synthesis Example 5-2: Synthesis of Compound 24acd

50 mLのシュレンクフラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、合成例4-2で得た化合物23acd(0.5 mmol, 1.0当量)、及び乾燥CH2Cl2(5 mL)を窒素気流下に添加した。内容物を-78℃まで冷却し、ボロントリブロマイド(650μL, 1 M in CH2Cl2, 0.65 mmol, 1.3当量)を添加した。得られた混合物を-78℃で0.5時間撹拌した。反応混合物を室温まで昇温してTLCをモニターした後、混合物に水(20 mL)及びCH2Cl2(20 mL)を添加した。CH2Cl2で抽出した後、有機層をNa2SO4で乾燥させ、揮発性物質を減圧下に除去した。粗生成物を短いシリカゲルパッド(ヘキサン/酢酸エチル=1: 1)でろ過した。ろ液を真空下に濃縮し、さらなる精製をせずに次の工程にそのまま使用した。
A 50 mL Schlenk flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, the compound 23acd (0.5 mmol, 1.0 equivalent) obtained in Synthesis Example 4-2 and dry CH 2 Cl 2 (5 mL) were added under a nitrogen stream. The contents were cooled to −78 ° C. and boron tribromide (650 μL, 1 M in CH 2 Cl 2 , 0.65 mmol, 1.3 eq) was added. The resulting mixture was stirred at −78 ° C. for 0.5 hour. After the reaction mixture was warmed to room temperature and TLC was monitored, water (20 mL) and CH 2 Cl 2 (20 mL) were added to the mixture. After extraction with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was filtered through a short silica gel pad (hexane / ethyl acetate = 1: 1). The filtrate was concentrated in vacuo and used directly in the next step without further purification.
20 mLのシュレンクフラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、上記工程で得た物質、及び乾燥CH2Cl2(2.5 mL)を窒素気流下に添加した。内容物を0℃まで冷却し、このフラスコにN,N-ジイソプロピルエチルアミン(i-Pr2NEt; 130.6μL, 0.75 mmol, 1.5当量)、N,N-ジメチルアミノピリジン(5.5 mg, 50μmol, 10 mol%)、及びトリフルオロメタンスルホン酸無水物(Tf2O; 126.2μL, 0.75 mmol, 1.5当量)を添加した。得られた混合物を0℃で0.5時間撹拌した。反応混合物を室温まで昇温して12時間撹拌した後、混合物を短いシリカゲルパッド(ヘキサン/酢酸エチル=1: 1)でろ過した。ろ液を真空下に濃縮し粗生成物を、分取薄層クロマトグラフィー(ヘキサン/酢酸エチル=10: 1)により精製し、化合物24acdを白色固体として得た(130.1 mg, 45 %)。
1H NMR (600 MHz, CDCl3): δ 7.57 (dd, J = 8.4, 1.8 Hz, 2H), 7.52-7.49 (m, 4H), 7.41-7.38 (m, 3H), 7.34-7.29 (m, 4H), 1.37 (s, 9H); 13C NMR (150 MHz, CDCl3): δ 152.6, 137.2, 136.9, 135.2, 133.9, 133.6, 131.8, 130.4, 129.9 (q, 2JCF = 33.2 Hz), 128.9, 128.8, 128.6, 128.3, 127.0, 126.0, 125.6 (q, 3JCF = 2.9 Hz), 123.9 (q, 1JCF = 274 Hz), 117.8 (q, 1JCF = 323 Hz), 34.8, 31.2; HRMS (DART) m/z calcd for C28H23F6O3S2 [MH]+: 585.09928, found 585.10085。 A 20 mL Schlenk flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen. To the flask, the material obtained in the above step and dry CH 2 Cl 2 (2.5 mL) were added under a stream of nitrogen. The contents were cooled to 0 ° C., and N, N-diisopropylethylamine (i-Pr 2 NEt; 130.6 μL, 0.75 mmol, 1.5 eq), N, N-dimethylaminopyridine (5.5 mg, 50 μmol, 10 mol) was cooled to 0 ° C. %), And trifluoromethanesulfonic anhydride (Tf 2 O; 126.2 μL, 0.75 mmol, 1.5 equivalents). The resulting mixture was stirred at 0 ° C. for 0.5 hour. After the reaction mixture was warmed to room temperature and stirred for 12 hours, the mixture was filtered through a short silica gel pad (hexane / ethyl acetate = 1: 1). The filtrate was concentrated in vacuo and the crude product was purified by preparative thin layer chromatography (hexane / ethyl acetate = 10: 1) to give compound 24acd as a white solid (130.1 mg, 45%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.57 (dd, J = 8.4, 1.8 Hz, 2H), 7.52-7.49 (m, 4H), 7.41-7.38 (m, 3H), 7.34-7.29 (m, 4H), 1.37 (s, 9H); 13 C NMR (150 MHz, CDCl 3 ): δ 152.6, 137.2, 136.9, 135.2, 133.9, 133.6, 131.8, 130.4, 129.9 (q, 2 J CF = 33.2 Hz), 128.9, 128.8, 128.6, 128.3, 127.0, 126.0, 125.6 (q, 3 J CF = 2.9 Hz), 123.9 (q, 1 J CF = 274 Hz), 117.8 (q, 1 J CF = 323 Hz), 34.8, 31.2; HRMS (DART) m / z calcd for C 28 H 23 F 6 O 3 S 2 [MH] + : 585.09928, found 585.10085.
1H NMR (600 MHz, CDCl3): δ 7.57 (dd, J = 8.4, 1.8 Hz, 2H), 7.52-7.49 (m, 4H), 7.41-7.38 (m, 3H), 7.34-7.29 (m, 4H), 1.37 (s, 9H); 13C NMR (150 MHz, CDCl3): δ 152.6, 137.2, 136.9, 135.2, 133.9, 133.6, 131.8, 130.4, 129.9 (q, 2JCF = 33.2 Hz), 128.9, 128.8, 128.6, 128.3, 127.0, 126.0, 125.6 (q, 3JCF = 2.9 Hz), 123.9 (q, 1JCF = 274 Hz), 117.8 (q, 1JCF = 323 Hz), 34.8, 31.2; HRMS (DART) m/z calcd for C28H23F6O3S2 [MH]+: 585.09928, found 585.10085。 A 20 mL Schlenk flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen. To the flask, the material obtained in the above step and dry CH 2 Cl 2 (2.5 mL) were added under a stream of nitrogen. The contents were cooled to 0 ° C., and N, N-diisopropylethylamine (i-Pr 2 NEt; 130.6 μL, 0.75 mmol, 1.5 eq), N, N-dimethylaminopyridine (5.5 mg, 50 μmol, 10 mol) was cooled to 0 ° C. %), And trifluoromethanesulfonic anhydride (Tf 2 O; 126.2 μL, 0.75 mmol, 1.5 equivalents). The resulting mixture was stirred at 0 ° C. for 0.5 hour. After the reaction mixture was warmed to room temperature and stirred for 12 hours, the mixture was filtered through a short silica gel pad (hexane / ethyl acetate = 1: 1). The filtrate was concentrated in vacuo and the crude product was purified by preparative thin layer chromatography (hexane / ethyl acetate = 10: 1) to give compound 24acd as a white solid (130.1 mg, 45%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.57 (dd, J = 8.4, 1.8 Hz, 2H), 7.52-7.49 (m, 4H), 7.41-7.38 (m, 3H), 7.34-7.29 (m, 4H), 1.37 (s, 9H); 13 C NMR (150 MHz, CDCl 3 ): δ 152.6, 137.2, 136.9, 135.2, 133.9, 133.6, 131.8, 130.4, 129.9 (q, 2 J CF = 33.2 Hz), 128.9, 128.8, 128.6, 128.3, 127.0, 126.0, 125.6 (q, 3 J CF = 2.9 Hz), 123.9 (q, 1 J CF = 274 Hz), 117.8 (q, 1 J CF = 323 Hz), 34.8, 31.2; HRMS (DART) m / z calcd for C 28 H 23 F 6 O 3 S 2 [MH] + : 585.09928, found 585.10085.
合成例5-3:化合物24bceの合成
Synthesis Example 5-3: Synthesis of Compound 24bce

合成例4-2で得た化合物23acdの代わりに合成例4-3で得た化合物23bceを使用したこと以外は合成例4-2と同様に合成し、化合物24bceを白色固体として得た(136.8 mg, 54 %)。
1H NMR (400 MHz, CDCl3): δ 7.48-7.34 (m, 6H), 7.31-7.21 (m, 5H), 7.15 (d, J = 8.8 Hz, 2H), 2.43 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 138.8, 137.2, 136.1, 134.2, 133.0, 132.8, 131.9, 131.7, 130.4, 130.01, 129.95, 129.8, 129.2, 128.94, 128.88, 128.7, 128.4, 125.7, 117.9 (q, JCF = 323 Hz), 21.3; HRMS (DART) m/z calcd for C24H17ClF3O3S2 [MH]+: 509.02597, found 509.02611。 Synthesis was performed in the same manner as in Synthesis Example 4-2, except that Compound 23bce obtained in Synthesis Example 4-3 was used instead of Compound 23acd obtained in Synthesis Example 4-2, and Compound 24bce was obtained as a white solid (136.8 mg, 54%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.48-7.34 (m, 6H), 7.31-7.21 (m, 5H), 7.15 (d, J = 8.8 Hz, 2H), 2.43 (s, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 138.8, 137.2, 136.1, 134.2, 133.0, 132.8, 131.9, 131.7, 130.4, 130.01, 129.95, 129.8, 129.2, 128.94, 128.88, 128.7, 128.4, 125.7, 117.9 (q , J CF = 323 Hz), 21.3; HRMS (DART) m / z calcd for C 24 H 17 ClF 3 O 3 S 2 [MH] + : 509.02597, found 509.02611.
1H NMR (400 MHz, CDCl3): δ 7.48-7.34 (m, 6H), 7.31-7.21 (m, 5H), 7.15 (d, J = 8.8 Hz, 2H), 2.43 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 138.8, 137.2, 136.1, 134.2, 133.0, 132.8, 131.9, 131.7, 130.4, 130.01, 129.95, 129.8, 129.2, 128.94, 128.88, 128.7, 128.4, 125.7, 117.9 (q, JCF = 323 Hz), 21.3; HRMS (DART) m/z calcd for C24H17ClF3O3S2 [MH]+: 509.02597, found 509.02611。 Synthesis was performed in the same manner as in Synthesis Example 4-2, except that Compound 23bce obtained in Synthesis Example 4-3 was used instead of Compound 23acd obtained in Synthesis Example 4-2, and Compound 24bce was obtained as a white solid (136.8 mg, 54%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.48-7.34 (m, 6H), 7.31-7.21 (m, 5H), 7.15 (d, J = 8.8 Hz, 2H), 2.43 (s, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 138.8, 137.2, 136.1, 134.2, 133.0, 132.8, 131.9, 131.7, 130.4, 130.01, 129.95, 129.8, 129.2, 128.94, 128.88, 128.7, 128.4, 125.7, 117.9 (q , J CF = 323 Hz), 21.3; HRMS (DART) m / z calcd for C 24 H 17 ClF 3 O 3 S 2 [MH] + : 509.02597, found 509.02611.
[合成例6:四置換チオフェン化合物(9)及び四置換チオフェン化合物(34)の合成]
合成例6-1:化合物10の合成 [Synthesis Example 6: Synthesis of tetrasubstituted thiophene compound (9) and tetrasubstituted thiophene compound (34)]
Synthesis Example 6-1: Synthesis of Compound 10
合成例6-1:化合物10の合成 [Synthesis Example 6: Synthesis of tetrasubstituted thiophene compound (9) and tetrasubstituted thiophene compound (34)]
Synthesis Example 6-1: Synthesis of Compound 10

[式中、Pd(PPh3)4はテトラキス(トリフェニルホスフィン)パラジウム(0)を示す。以下同様である。]
200 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、テトラキス(トリフェニルホスフィン)パラジウム(0)(Pd(PPh3)4; 201 mg, 0.74 mmol, 10 mol%)、Ba(OH)2(594 mg, 3.48 mmol, 2.0当量)、(3,5-ジメトキシフェニル)ボロン酸(950 mg, 5.22 mmol, 3.0当量)、合成例5-1で得た5-(4-クロロフェニル)-2-(m-トリル)-4-(p-トリル)チオフェン-3-イルトリフルオロメタンスルホネート(化合物9; 910 mg, 1.74 mmol, 1.0当量)、乾燥1-ブタノール(70 mL)、及びH2O(58 mL)を窒素気流下に添加した。フラスコを65℃で16時間加熱した。反応混合物を室温まで冷却した後、混合物を短いシリカゲルパッド(酢酸エチル)でろ過した。ろ液を真空下に濃縮し、粗生成物をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=20: 1)により精製し、2-(4-クロロフェニル)-4-(3,5-ジメトキシフェニル)-5-(m-トリル)-3-(p-トリル)チオフェン(化合物10)を白色固体として得た(798 mg, 90 %)。
1H NMR (600 MHz, CDCl3): δ 7.20-7.14 (m, 5H), 7.10 (t, J = 7.8 Hz, 1H), 7.05-7.01 (m, 2H), 6.96 (d, J = 8.4 Hz, 2H), 6.86 (d. J = 8.4 Hz, 2H), 6.25 (t, J = 2.4 Hz, 1H), 6.10 (d, J = 2.4 Hz, 2H), 3.49 (s, 3H), 2.28 (s, 3H), 2.27 (s, 3H)。 [Wherein Pd (PPh 3 ) 4 represents tetrakis (triphenylphosphine) palladium (0). The same applies hereinafter. ]
A 200 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, tetrakis (triphenylphosphine) palladium (0) (Pd (PPh 3 ) 4 ; 201 mg, 0.74 mmol, 10 mol%), Ba (OH) 2 (594 mg, 3.48 mmol, 2.0 eq), ( 3,5-Dimethoxyphenyl) boronic acid (950 mg, 5.22 mmol, 3.0 equivalents), 5- (4-chlorophenyl) -2- (m-tolyl) -4- (p-tolyl) obtained in Synthesis Example 5-1. ) Thiophen-3-yl trifluoromethanesulfonate (Compound 9; 910 mg, 1.74 mmol, 1.0 eq), dry 1-butanol (70 mL), and H 2 O (58 mL) were added under a stream of nitrogen. The flask was heated at 65 ° C. for 16 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a short silica gel pad (ethyl acetate). The filtrate was concentrated under vacuum and the crude product was purified by flash column chromatography (hexane / ethyl acetate = 20: 1) to give 2- (4-chlorophenyl) -4- (3,5-dimethoxyphenyl)- 5- (m-Tolyl) -3- (p-tolyl) thiophene (Compound 10) was obtained as a white solid (798 mg, 90%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.20-7.14 (m, 5H), 7.10 (t, J = 7.8 Hz, 1H), 7.05-7.01 (m, 2H), 6.96 (d, J = 8.4 Hz , 2H), 6.86 (d.J = 8.4 Hz, 2H), 6.25 (t, J = 2.4 Hz, 1H), 6.10 (d, J = 2.4 Hz, 2H), 3.49 (s, 3H), 2.28 (s , 3H), 2.27 (s, 3H).
200 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、テトラキス(トリフェニルホスフィン)パラジウム(0)(Pd(PPh3)4; 201 mg, 0.74 mmol, 10 mol%)、Ba(OH)2(594 mg, 3.48 mmol, 2.0当量)、(3,5-ジメトキシフェニル)ボロン酸(950 mg, 5.22 mmol, 3.0当量)、合成例5-1で得た5-(4-クロロフェニル)-2-(m-トリル)-4-(p-トリル)チオフェン-3-イルトリフルオロメタンスルホネート(化合物9; 910 mg, 1.74 mmol, 1.0当量)、乾燥1-ブタノール(70 mL)、及びH2O(58 mL)を窒素気流下に添加した。フラスコを65℃で16時間加熱した。反応混合物を室温まで冷却した後、混合物を短いシリカゲルパッド(酢酸エチル)でろ過した。ろ液を真空下に濃縮し、粗生成物をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=20: 1)により精製し、2-(4-クロロフェニル)-4-(3,5-ジメトキシフェニル)-5-(m-トリル)-3-(p-トリル)チオフェン(化合物10)を白色固体として得た(798 mg, 90 %)。
1H NMR (600 MHz, CDCl3): δ 7.20-7.14 (m, 5H), 7.10 (t, J = 7.8 Hz, 1H), 7.05-7.01 (m, 2H), 6.96 (d, J = 8.4 Hz, 2H), 6.86 (d. J = 8.4 Hz, 2H), 6.25 (t, J = 2.4 Hz, 1H), 6.10 (d, J = 2.4 Hz, 2H), 3.49 (s, 3H), 2.28 (s, 3H), 2.27 (s, 3H)。 [Wherein Pd (PPh 3 ) 4 represents tetrakis (triphenylphosphine) palladium (0). The same applies hereinafter. ]
A 200 mL two-necked flask was charged with a magnetic stir bar, flame-dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, tetrakis (triphenylphosphine) palladium (0) (Pd (PPh 3 ) 4 ; 201 mg, 0.74 mmol, 10 mol%), Ba (OH) 2 (594 mg, 3.48 mmol, 2.0 eq), ( 3,5-Dimethoxyphenyl) boronic acid (950 mg, 5.22 mmol, 3.0 equivalents), 5- (4-chlorophenyl) -2- (m-tolyl) -4- (p-tolyl) obtained in Synthesis Example 5-1. ) Thiophen-3-yl trifluoromethanesulfonate (Compound 9; 910 mg, 1.74 mmol, 1.0 eq), dry 1-butanol (70 mL), and H 2 O (58 mL) were added under a stream of nitrogen. The flask was heated at 65 ° C. for 16 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a short silica gel pad (ethyl acetate). The filtrate was concentrated under vacuum and the crude product was purified by flash column chromatography (hexane / ethyl acetate = 20: 1) to give 2- (4-chlorophenyl) -4- (3,5-dimethoxyphenyl)- 5- (m-Tolyl) -3- (p-tolyl) thiophene (Compound 10) was obtained as a white solid (798 mg, 90%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.20-7.14 (m, 5H), 7.10 (t, J = 7.8 Hz, 1H), 7.05-7.01 (m, 2H), 6.96 (d, J = 8.4 Hz , 2H), 6.86 (d.J = 8.4 Hz, 2H), 6.25 (t, J = 2.4 Hz, 1H), 6.10 (d, J = 2.4 Hz, 2H), 3.49 (s, 3H), 2.28 (s , 3H), 2.27 (s, 3H).
合成例6-2:四置換チオフェン化合物25acdeの合成
Synthesis Example 6-2: Synthesis of tetrasubstituted thiophene compound 25acde

50 mLのシュレンク管に磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このシュレンク管に、テトラキス(トリフェニルホスフィン)パラジウム(0)(Pd(PPh3)4; 10.4 mg, 9.0μmol, 10 mol%)、Ba(OH)2(30.8 mg, 0.18 mmol, 2.0当量)、4-クロロフェニルボロン酸(0.270 mmol, 3.0当量)、合成例5-2で得た化合物24acd(0.09 mmol, 1.0当量)、乾燥1-ブタノール(3.6 mL)、及びH2O(3.0 mL)を窒素気流下に添加した。シュレンク管を65℃で16時間加熱した。反応混合物を室温まで冷却した後、混合物を短いシリカゲルパッド(酢酸エチル)でろ過した。ろ液を真空下に濃縮し、粗生成物を分取薄層クロマトグラフィー(ヘキサン/酢酸エチル=10: 1)により精製し、四置換チオフェン化合物25acdeを白色固体として得た(34.9 mg, 71 %)。
1H NMR (400 MHz, CDCl3): δ 7.45 (d, J = 8.0 Hz, 2H), 7.33-7.23 (m, 4H), 7.21-7.12 (m, 5H), 7.10 (d, J = 8.4 Hz, 2H), 6.95 (dd, J = 7.8, 2.0 Hz, 2H), 6.90 (d, J = 8.4 Hz, 2H), 1.30 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 150.8, 140.4, 140.2, 137.9, 137.7, 136.4, 135.8, 134.7, 132.7, 132.1, 130.6, 130.5, 129.1, 128.9 (q, 2JCF = 32.4 Hz), 128.7, 128.24, 128.18, 127.1, 125.5, 125.3 (q, 3JCF = 3.9 Hz), 124.1 (q, 1JCF = 277 Hz), 34.6, 31.2; HRMS (DART) m/z calcd for C33H27ClF3S [MH]+: 547.14741, found 547.14724。 A magnetic stirring bar was placed in a 50 mL Schlenk tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this Schlenk tube, tetrakis (triphenylphosphine) palladium (0) (Pd (PPh 3 ) 4 ; 10.4 mg, 9.0 μmol, 10 mol%), Ba (OH) 2 (30.8 mg, 0.18 mmol, 2.0 equivalents), 4-chlorophenylboronic acid (0.270 mmol, 3.0 equivalents), compound 24acd (0.09 mmol, 1.0 equivalent) obtained in Synthesis Example 5-2, dry 1-butanol (3.6 mL), and H 2 O (3.0 mL) were nitrogenated. Added under air flow. The Schlenk tube was heated at 65 ° C. for 16 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a short silica gel pad (ethyl acetate). The filtrate was concentrated under vacuum and the crude product was purified by preparative thin layer chromatography (hexane / ethyl acetate = 10: 1) to give the tetrasubstituted thiophene compound 25acde as a white solid (34.9 mg, 71% ).
1 H NMR (400 MHz, CDCl 3 ): δ 7.45 (d, J = 8.0 Hz, 2H), 7.33-7.23 (m, 4H), 7.21-7.12 (m, 5H), 7.10 (d, J = 8.4 Hz , 2H), 6.95 (dd, J = 7.8, 2.0 Hz, 2H), 6.90 (d, J = 8.4 Hz, 2H), 1.30 (s, 9H); 13 C NMR (100 MHz, CDCl 3 ): δ 150.8 , 140.4, 140.2, 137.9, 137.7, 136.4, 135.8, 134.7, 132.7, 132.1, 130.6, 130.5, 129.1, 128.9 (q, 2 J CF = 32.4 Hz), 128.7, 128.24, 128.18, 127.1, 125.5, 125.3 (q , 3 J CF = 3.9 Hz), 124.1 (q, 1 J CF = 277 Hz), 34.6, 31.2; HRMS (DART) m / z calcd for C 33 H 27 ClF 3 S [MH] + : 547.14741, found 547.14724 .
1H NMR (400 MHz, CDCl3): δ 7.45 (d, J = 8.0 Hz, 2H), 7.33-7.23 (m, 4H), 7.21-7.12 (m, 5H), 7.10 (d, J = 8.4 Hz, 2H), 6.95 (dd, J = 7.8, 2.0 Hz, 2H), 6.90 (d, J = 8.4 Hz, 2H), 1.30 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 150.8, 140.4, 140.2, 137.9, 137.7, 136.4, 135.8, 134.7, 132.7, 132.1, 130.6, 130.5, 129.1, 128.9 (q, 2JCF = 32.4 Hz), 128.7, 128.24, 128.18, 127.1, 125.5, 125.3 (q, 3JCF = 3.9 Hz), 124.1 (q, 1JCF = 277 Hz), 34.6, 31.2; HRMS (DART) m/z calcd for C33H27ClF3S [MH]+: 547.14741, found 547.14724。 A magnetic stirring bar was placed in a 50 mL Schlenk tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this Schlenk tube, tetrakis (triphenylphosphine) palladium (0) (Pd (PPh 3 ) 4 ; 10.4 mg, 9.0 μmol, 10 mol%), Ba (OH) 2 (30.8 mg, 0.18 mmol, 2.0 equivalents), 4-chlorophenylboronic acid (0.270 mmol, 3.0 equivalents), compound 24acd (0.09 mmol, 1.0 equivalent) obtained in Synthesis Example 5-2, dry 1-butanol (3.6 mL), and H 2 O (3.0 mL) were nitrogenated. Added under air flow. The Schlenk tube was heated at 65 ° C. for 16 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a short silica gel pad (ethyl acetate). The filtrate was concentrated under vacuum and the crude product was purified by preparative thin layer chromatography (hexane / ethyl acetate = 10: 1) to give the tetrasubstituted thiophene compound 25acde as a white solid (34.9 mg, 71% ).
1 H NMR (400 MHz, CDCl 3 ): δ 7.45 (d, J = 8.0 Hz, 2H), 7.33-7.23 (m, 4H), 7.21-7.12 (m, 5H), 7.10 (d, J = 8.4 Hz , 2H), 6.95 (dd, J = 7.8, 2.0 Hz, 2H), 6.90 (d, J = 8.4 Hz, 2H), 1.30 (s, 9H); 13 C NMR (100 MHz, CDCl 3 ): δ 150.8 , 140.4, 140.2, 137.9, 137.7, 136.4, 135.8, 134.7, 132.7, 132.1, 130.6, 130.5, 129.1, 128.9 (q, 2 J CF = 32.4 Hz), 128.7, 128.24, 128.18, 127.1, 125.5, 125.3 (q , 3 J CF = 3.9 Hz), 124.1 (q, 1 J CF = 277 Hz), 34.6, 31.2; HRMS (DART) m / z calcd for C 33 H 27 ClF 3 S [MH] + : 547.14741, found 547.14724 .
合成例6-3~6-5:四置換チオフェン化合物25acdf、四置換チオフェン化合物25bceg、四置換チオフェン化合物25bcehの合成
Synthesis Examples 6-3 to 6-5: Synthesis of tetrasubstituted thiophene compound 25acdf, tetrasubstituted thiophene compound 25bceg, tetrasubstituted thiophene compound 25bceh

4-クロロフェニルボロン酸の代わりに4-メトキシフェニルボロン酸を使用したこと以外は合成例6-2と同様に合成し、四置換チオフェン化合物25acdfを白色固体として得た(33.7 mg, 69 %)。
1H NMR (400 MHz, CDCl3): δ 7.45 (d, J = 8.8 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 7.28-7.22 (m, 2H), 7.20-7.13 (m, 5H), 6.98-6.94 (m, 2H), 6.88 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 3.75 (s, 3H), 1.29 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 158.3, 150.4, 140.8, 139.5, 139.0, 138.0, 136.2, 135.9, 131.9, 131.0, 130.7, 129.1, 128.8 (q, 2JCF = 32.4 Hz), 128.6, 128.1, 126.9, 125.3, 125.2 (q, 3JCF = 3.8 Hz), 124.1 (q, 1JCF = 277 Hz), 113.3, 55.0, 34.5, 31.2; HRMS (DART) m/z calcd for C34H30F3OS [MH]+: 543.19694, found 543.19636。 The tetra-substituted thiophene compound 25acdf was obtained as a white solid (33.7 mg, 69%) in the same manner as in Synthesis Example 6-2 except that 4-methoxyphenylboronic acid was used instead of 4-chlorophenylboronic acid.
1 H NMR (400 MHz, CDCl 3 ): δ 7.45 (d, J = 8.8 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 7.28-7.22 (m, 2H), 7.20-7.13 (m , 5H), 6.98-6.94 (m, 2H), 6.88 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 3.75 (s, 3H), 1.29 (s, 9H) ; 13 C NMR (100 MHz, CDCl 3 ): δ 158.3, 150.4, 140.8, 139.5, 139.0, 138.0, 136.2, 135.9, 131.9, 131.0, 130.7, 129.1, 128.8 (q, 2 J CF = 32.4 Hz), 128.6 , 128.1, 126.9, 125.3, 125.2 (q, 3 J CF = 3.8 Hz), 124.1 (q, 1 J CF = 277 Hz), 113.3, 55.0, 34.5, 31.2; HRMS (DART) m / z calcd for C 34 H 30 F 3 OS [MH] + : 543.19694, found 543.19636.
1H NMR (400 MHz, CDCl3): δ 7.45 (d, J = 8.8 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 7.28-7.22 (m, 2H), 7.20-7.13 (m, 5H), 6.98-6.94 (m, 2H), 6.88 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 3.75 (s, 3H), 1.29 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 158.3, 150.4, 140.8, 139.5, 139.0, 138.0, 136.2, 135.9, 131.9, 131.0, 130.7, 129.1, 128.8 (q, 2JCF = 32.4 Hz), 128.6, 128.1, 126.9, 125.3, 125.2 (q, 3JCF = 3.8 Hz), 124.1 (q, 1JCF = 277 Hz), 113.3, 55.0, 34.5, 31.2; HRMS (DART) m/z calcd for C34H30F3OS [MH]+: 543.19694, found 543.19636。 The tetra-substituted thiophene compound 25acdf was obtained as a white solid (33.7 mg, 69%) in the same manner as in Synthesis Example 6-2 except that 4-methoxyphenylboronic acid was used instead of 4-chlorophenylboronic acid.
1 H NMR (400 MHz, CDCl 3 ): δ 7.45 (d, J = 8.8 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 7.28-7.22 (m, 2H), 7.20-7.13 (m , 5H), 6.98-6.94 (m, 2H), 6.88 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 3.75 (s, 3H), 1.29 (s, 9H) ; 13 C NMR (100 MHz, CDCl 3 ): δ 158.3, 150.4, 140.8, 139.5, 139.0, 138.0, 136.2, 135.9, 131.9, 131.0, 130.7, 129.1, 128.8 (q, 2 J CF = 32.4 Hz), 128.6 , 128.1, 126.9, 125.3, 125.2 (q, 3 J CF = 3.8 Hz), 124.1 (q, 1 J CF = 277 Hz), 113.3, 55.0, 34.5, 31.2; HRMS (DART) m / z calcd for C 34 H 30 F 3 OS [MH] + : 543.19694, found 543.19636.
合成例5-2で得た化合物24acdの代わりに合成例5-3で得た化合物24cdeを使用し、4-クロロフェニルボロン酸の代わりに3,5-ジメトキシフェニルボロン酸を使用したこと以外は合成例6-2と同様に合成し、四置換チオフェン化合物25bcegを白色固体として得た(42.3 mg, 95 %)。
1H NMR (400 MHz, CDCl3): δ 7.21-7.08 (m, 9H), 7.06-6.96 (m, 4H), 6.42 (t, J = 2.4 Hz, 1H), 6.09 (s, 2H), 3.49 (s, 6H), 2.28 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 160.0, 139.7, 139.2, 139.1, 137.92, 137.89, 136.8, 136.3, 133.8, 133.1, 132.7, 130.6, 130.3, 129.8, 128.5, 128.22, 128.17, 128.0, 126.8, 126.2, 108.8, 99.7, 55.1, 21.4; HRMS (DART) m/z calcd for C31H26ClO2S [MH]+: 497.13420, found 497.13378。 Synthesis except that compound 24cde obtained in Synthesis Example 5-3 was used in place of compound 24acd obtained in Synthesis Example 5-2, and 3,5-dimethoxyphenylboronic acid was used in place of 4-chlorophenylboronic acid Synthesis was carried out in the same manner as in Example 6-2 to obtain 25bceg of a tetrasubstituted thiophene compound as a white solid (42.3 mg, 95%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.21-7.08 (m, 9H), 7.06-6.96 (m, 4H), 6.42 (t, J = 2.4 Hz, 1H), 6.09 (s, 2H), 3.49 (s, 6H), 2.28 (s, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 160.0, 139.7, 139.2, 139.1, 137.92, 137.89, 136.8, 136.3, 133.8, 133.1, 132.7, 130.6, 130.3 , 129.8, 128.5, 128.22, 128.17, 128.0, 126.8, 126.2, 108.8, 99.7, 55.1, 21.4; HRMS (DART) m / z calcd for C 31 H 26 ClO 2 S [MH] + : 497.13420, found 497.13378.
1H NMR (400 MHz, CDCl3): δ 7.21-7.08 (m, 9H), 7.06-6.96 (m, 4H), 6.42 (t, J = 2.4 Hz, 1H), 6.09 (s, 2H), 3.49 (s, 6H), 2.28 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 160.0, 139.7, 139.2, 139.1, 137.92, 137.89, 136.8, 136.3, 133.8, 133.1, 132.7, 130.6, 130.3, 129.8, 128.5, 128.22, 128.17, 128.0, 126.8, 126.2, 108.8, 99.7, 55.1, 21.4; HRMS (DART) m/z calcd for C31H26ClO2S [MH]+: 497.13420, found 497.13378。 Synthesis except that compound 24cde obtained in Synthesis Example 5-3 was used in place of compound 24acd obtained in Synthesis Example 5-2, and 3,5-dimethoxyphenylboronic acid was used in place of 4-chlorophenylboronic acid Synthesis was carried out in the same manner as in Example 6-2 to obtain 25bceg of a tetrasubstituted thiophene compound as a white solid (42.3 mg, 95%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.21-7.08 (m, 9H), 7.06-6.96 (m, 4H), 6.42 (t, J = 2.4 Hz, 1H), 6.09 (s, 2H), 3.49 (s, 6H), 2.28 (s, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 160.0, 139.7, 139.2, 139.1, 137.92, 137.89, 136.8, 136.3, 133.8, 133.1, 132.7, 130.6, 130.3 , 129.8, 128.5, 128.22, 128.17, 128.0, 126.8, 126.2, 108.8, 99.7, 55.1, 21.4; HRMS (DART) m / z calcd for C 31 H 26 ClO 2 S [MH] + : 497.13420, found 497.13378.
合成例5-2で得た化合物24acdの代わりに合成例5-3で得た化合物24bceを使用し、4-クロロフェニルボロン酸の代わりに4-n-ブチルフェニルボロン酸を使用したこと以外は合成例6-2と同様に合成し、四置換チオフェン化合物25bcehを白色固体として得た(42.9 mg, 97 %)。
1H NMR (400 MHz, CDCl3): δ 7.20-6.98 (m, 11H), 6.98-6.89 (m, 4H), 6.84 (d, J = 8.0 Hz, 2H), 2.51 (t, J = 7.6 Hz, 2H), 2.22 (s, 3H), 1.53 (quin, J = 7.8 Hz, 2H), 1.27 (sext, J = 7.6 Hz, 2H), 0.89 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 141.2, 139.9, 139.6, 138.8, 137.8, 136.7, 136.3, 134.0, 133.5, 133.0, 132.9, 130.8, 130.5, 130.3, 129.8, 128.5, 128.1, 128.0, 127.9, 126.7, 126.2, 35.2, 33.4, 22.1, 21.3, 14.0; HRMS (DART) m/z calcd for C33H30ClS [MH]+: 493.17567, found 493.17618。 Synthesis except that compound 24bce obtained in Synthesis Example 5-3 was used in place of compound 24acd obtained in Synthesis Example 5-2 and 4-n-butylphenylboronic acid was used in place of 4-chlorophenylboronic acid Synthesis was conducted in the same manner as in Example 6-2 to obtain the tetrasubstituted thiophene compound 25bceh as a white solid (42.9 mg, 97%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.20-6.98 (m, 11H), 6.98-6.89 (m, 4H), 6.84 (d, J = 8.0 Hz, 2H), 2.51 (t, J = 7.6 Hz , 2H), 2.22 (s, 3H), 1.53 (quin, J = 7.8 Hz, 2H), 1.27 (sext, J = 7.6 Hz, 2H), 0.89 (t, J = 7.6 Hz, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 141.2, 139.9, 139.6, 138.8, 137.8, 136.7, 136.3, 134.0, 133.5, 133.0, 132.9, 130.8, 130.5, 130.3, 129.8, 128.5, 128.1, 128.0, 127.9, 126.7, 126.2 , 35.2, 33.4, 22.1, 21.3, 14.0; HRMS (DART) m / z calcd for C 33 H 30 ClS [MH] + : 493.17567, found 493.17618.
1H NMR (400 MHz, CDCl3): δ 7.20-6.98 (m, 11H), 6.98-6.89 (m, 4H), 6.84 (d, J = 8.0 Hz, 2H), 2.51 (t, J = 7.6 Hz, 2H), 2.22 (s, 3H), 1.53 (quin, J = 7.8 Hz, 2H), 1.27 (sext, J = 7.6 Hz, 2H), 0.89 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 141.2, 139.9, 139.6, 138.8, 137.8, 136.7, 136.3, 134.0, 133.5, 133.0, 132.9, 130.8, 130.5, 130.3, 129.8, 128.5, 128.1, 128.0, 127.9, 126.7, 126.2, 35.2, 33.4, 22.1, 21.3, 14.0; HRMS (DART) m/z calcd for C33H30ClS [MH]+: 493.17567, found 493.17618。 Synthesis except that compound 24bce obtained in Synthesis Example 5-3 was used in place of compound 24acd obtained in Synthesis Example 5-2 and 4-n-butylphenylboronic acid was used in place of 4-chlorophenylboronic acid Synthesis was conducted in the same manner as in Example 6-2 to obtain the tetrasubstituted thiophene compound 25bceh as a white solid (42.9 mg, 97%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.20-6.98 (m, 11H), 6.98-6.89 (m, 4H), 6.84 (d, J = 8.0 Hz, 2H), 2.51 (t, J = 7.6 Hz , 2H), 2.22 (s, 3H), 1.53 (quin, J = 7.8 Hz, 2H), 1.27 (sext, J = 7.6 Hz, 2H), 0.89 (t, J = 7.6 Hz, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 141.2, 139.9, 139.6, 138.8, 137.8, 136.7, 136.3, 134.0, 133.5, 133.0, 132.9, 130.8, 130.5, 130.3, 129.8, 128.5, 128.1, 128.0, 127.9, 126.7, 126.2 , 35.2, 33.4, 22.1, 21.3, 14.0; HRMS (DART) m / z calcd for C 33 H 30 ClS [MH] + : 493.17567, found 493.17618.
[合成例7:四置換チオフェンS-オキシド化合物(3)及び四置換チオフェンS-オキシド化合物(32)の合成]
合成例7-1:化合物11の合成 [Synthesis Example 7: Synthesis of tetrasubstituted thiophene S-oxide compound (3) and tetrasubstituted thiophene S-oxide compound (32)]
Synthesis Example 7-1: Synthesis of Compound 11
合成例7-1:化合物11の合成 [Synthesis Example 7: Synthesis of tetrasubstituted thiophene S-oxide compound (3) and tetrasubstituted thiophene S-oxide compound (32)]
Synthesis Example 7-1: Synthesis of Compound 11

[式中、m-CPBAはm-クロロ過安息香酸を示す。BF3・OEt2は三フッ化ホウ素ジエチルエーテル錯体を示す。以下同様である。]
50 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、合成例6-1で得た2-(4-クロロフェニル)-4-(3,5-ジメトキシフェニル)-5-(m-トリル)-3-(p-トリル)チオフェン(化合物10; 184.5 mg, 0.36 mmol, 1.0当量)、及び乾燥CH2Cl2(1.5 mL)を添加した。-20℃まで冷却した後、三フッ化ホウ素ジエチルエーテル錯体(BF3・OEt2; 452μL, 3.6 mmol, 10当量)を添加した。混合物を-20℃で1時間撹拌した後、m-クロロ過安息香酸(m-CPBA; 0.36 mmol, 1.0当量)のCH2Cl2(720μL)溶液をゆっくりと(1時間ごとに4回、合計4.0当量)添加し、さらに得られた混合物を-20℃で1時間撹拌した。つまり、反応時間は合計6時間である。飽和Na2S2O3水溶液及び飽和NaHCO3水溶液を添加して反応をクエンチした。混合物をCH2Cl2で抽出し、Na2SO4で乾燥し、減圧下に濃縮した。粗生成物をフラッシュカラムクロマトグラフィー(CHCl3)により精製し、2-(4-クロロフェニル)-4-(3,5-ジメトキシフェニル)-5-(m-トリル)-3-(p-トリル)チオフェン1-オキシド(化合物11)を黄色固体として得た(98.1 mg, 52 %)。
1H NMR (500 MHz, CDCl3): δ 7.34-7.27 (m, 3H), 7.27-7.23 (m, 2H), 7.20-7.09 (m, 4H), 6.99 (d, J = 8.4 Hz, 2H), 6.84 (d. J = 8.4 Hz, 2H), 6.25 (t, J = 2.4 Hz, 1H), 6.10 (d, J = 2.4 Hz, 2H), 3.49 (s, 3H), 2.28 (s, 3H), 2.27 (s, 3H)。 [Wherein, m-CPBA represents m-chloroperbenzoic acid. BF 3 · OEt 2 represents a boron trifluoride diethyl ether complex. The same applies hereinafter. ]
A 50 mL two-necked flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, 2- (4-chlorophenyl) -4- (3,5-dimethoxyphenyl) -5- (m-tolyl) -3- (p-tolyl) thiophene (Compound 10) obtained in Synthesis Example 6-1 was added. 184.5 mg, 0.36 mmol, 1.0 eq), and dry CH 2 Cl 2 (1.5 mL) were added. After cooling to −20 ° C., boron trifluoride diethyl ether complex (BF 3 · OEt 2 ; 452 μL, 3.6 mmol, 10 equivalents) was added. After the mixture was stirred at −20 ° C. for 1 hour, a solution of m-chloroperbenzoic acid (m-CPBA; 0.36 mmol, 1.0 equiv) in CH 2 Cl 2 (720 μL) was slowly added (4 times per hour, totaling 4 times). 4.0 equivalents) was added and the resulting mixture was stirred at −20 ° C. for 1 hour. That is, the reaction time is 6 hours in total. Saturated aqueous Na 2 S 2 O 3 and saturated aqueous NaHCO 3 were added to quench the reaction. The mixture was extracted with CH 2 Cl 2 , dried over Na 2 SO 4 and concentrated under reduced pressure. The crude product was purified by flash column chromatography (CHCl 3 ) to give 2- (4-chlorophenyl) -4- (3,5-dimethoxyphenyl) -5- (m-tolyl) -3- (p-tolyl) Thiophene 1-oxide (Compound 11) was obtained as a yellow solid (98.1 mg, 52%).
1 H NMR (500 MHz, CDCl 3 ): δ 7.34-7.27 (m, 3H), 7.27-7.23 (m, 2H), 7.20-7.09 (m, 4H), 6.99 (d, J = 8.4 Hz, 2H) , 6.84 (d.J = 8.4 Hz, 2H), 6.25 (t, J = 2.4 Hz, 1H), 6.10 (d, J = 2.4 Hz, 2H), 3.49 (s, 3H), 2.28 (s, 3H) , 2.27 (s, 3H).
50 mLの二つ首フラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このフラスコに、合成例6-1で得た2-(4-クロロフェニル)-4-(3,5-ジメトキシフェニル)-5-(m-トリル)-3-(p-トリル)チオフェン(化合物10; 184.5 mg, 0.36 mmol, 1.0当量)、及び乾燥CH2Cl2(1.5 mL)を添加した。-20℃まで冷却した後、三フッ化ホウ素ジエチルエーテル錯体(BF3・OEt2; 452μL, 3.6 mmol, 10当量)を添加した。混合物を-20℃で1時間撹拌した後、m-クロロ過安息香酸(m-CPBA; 0.36 mmol, 1.0当量)のCH2Cl2(720μL)溶液をゆっくりと(1時間ごとに4回、合計4.0当量)添加し、さらに得られた混合物を-20℃で1時間撹拌した。つまり、反応時間は合計6時間である。飽和Na2S2O3水溶液及び飽和NaHCO3水溶液を添加して反応をクエンチした。混合物をCH2Cl2で抽出し、Na2SO4で乾燥し、減圧下に濃縮した。粗生成物をフラッシュカラムクロマトグラフィー(CHCl3)により精製し、2-(4-クロロフェニル)-4-(3,5-ジメトキシフェニル)-5-(m-トリル)-3-(p-トリル)チオフェン1-オキシド(化合物11)を黄色固体として得た(98.1 mg, 52 %)。
1H NMR (500 MHz, CDCl3): δ 7.34-7.27 (m, 3H), 7.27-7.23 (m, 2H), 7.20-7.09 (m, 4H), 6.99 (d, J = 8.4 Hz, 2H), 6.84 (d. J = 8.4 Hz, 2H), 6.25 (t, J = 2.4 Hz, 1H), 6.10 (d, J = 2.4 Hz, 2H), 3.49 (s, 3H), 2.28 (s, 3H), 2.27 (s, 3H)。 [Wherein, m-CPBA represents m-chloroperbenzoic acid. BF 3 · OEt 2 represents a boron trifluoride diethyl ether complex. The same applies hereinafter. ]
A 50 mL two-necked flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen. To this flask, 2- (4-chlorophenyl) -4- (3,5-dimethoxyphenyl) -5- (m-tolyl) -3- (p-tolyl) thiophene (Compound 10) obtained in Synthesis Example 6-1 was added. 184.5 mg, 0.36 mmol, 1.0 eq), and dry CH 2 Cl 2 (1.5 mL) were added. After cooling to −20 ° C., boron trifluoride diethyl ether complex (BF 3 · OEt 2 ; 452 μL, 3.6 mmol, 10 equivalents) was added. After the mixture was stirred at −20 ° C. for 1 hour, a solution of m-chloroperbenzoic acid (m-CPBA; 0.36 mmol, 1.0 equiv) in CH 2 Cl 2 (720 μL) was slowly added (4 times per hour, totaling 4 times). 4.0 equivalents) was added and the resulting mixture was stirred at −20 ° C. for 1 hour. That is, the reaction time is 6 hours in total. Saturated aqueous Na 2 S 2 O 3 and saturated aqueous NaHCO 3 were added to quench the reaction. The mixture was extracted with CH 2 Cl 2 , dried over Na 2 SO 4 and concentrated under reduced pressure. The crude product was purified by flash column chromatography (CHCl 3 ) to give 2- (4-chlorophenyl) -4- (3,5-dimethoxyphenyl) -5- (m-tolyl) -3- (p-tolyl) Thiophene 1-oxide (Compound 11) was obtained as a yellow solid (98.1 mg, 52%).
1 H NMR (500 MHz, CDCl 3 ): δ 7.34-7.27 (m, 3H), 7.27-7.23 (m, 2H), 7.20-7.09 (m, 4H), 6.99 (d, J = 8.4 Hz, 2H) , 6.84 (d.J = 8.4 Hz, 2H), 6.25 (t, J = 2.4 Hz, 1H), 6.10 (d, J = 2.4 Hz, 2H), 3.49 (s, 3H), 2.28 (s, 3H) , 2.27 (s, 3H).
合成例7-2:四置換チオフェンS-オキシド化合物26acdeの合成
Synthesis Example 7-2: Synthesis of tetrasubstituted thiophene S-oxide compound 26acde

20 mLのシュレンク管に磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このシュレンク管に、合成例6-2で得た四置換チオフェン化合物25acde(0.1 mmol, 1.0当量)、及び乾燥CH2Cl2(400μL)を添加した。-20℃まで冷却した後、三フッ化ホウ素ジエチルエーテル錯体(BF3・OEt2; 120μL, 1.0 mmol, 10当量)を添加した。混合物を-20℃で1時間撹拌した後、m-クロロ過安息香酸(m-CPBA; 0.1 mmol, 1.0当量)のCH2Cl2(200μL)溶液をゆっくりと(1時間ごとに4回)添加し、さらに得られた混合物を-20℃で1時間撹拌した。飽和Na2S2O3水溶液及び飽和NaHCO3水溶液を添加して反応をクエンチした。混合物をCH2Cl2で抽出し、Na2SO4で乾燥し、減圧下に濃縮した。粗生成物を分取薄層クロマトグラフィー(ヘキサン/CHCl3=2: 3)により精製し、四置換チオフェンS-オキシド化合物26acdeを黄色固体として得た(22.0 mg, 39 %)。
1H NMR (400 MHz, CDCl3): δ 7.52 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.4 Hz, 2H), 7.36-7.24 (m, 5H), 7.23-7.17 (m, 2H), 7.14 (d, J = 8.4 Hz, 2H), 6.95-6.87 (m, 4H), 1.30 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 152.5, 147.9, 144.1, 142.6, 138.7, 134.4, 134.1, 132.6, 131.6, 131.1, 130.3 (q, 2JCF = 33.5 Hz), 129.9, 129.5, 129.3, 128.77, 128.70, 128.66, 126.9, 125.9, 125.6 (q, 3JCF = 3.9 Hz), 123.8 (q, 1JCF = 270 Hz), 34.8, 31.1; HRMS (DART) m/z calcd for C33H27ClF3OS [MH]+: 563.14232, found 563.14329。 A magnetic stir bar was placed in a 20 mL Schlenk tube, flame dried under vacuum, cooled to room temperature, and then filled with nitrogen. To the Schlenk tube, the tetrasubstituted thiophene compound 25acde (0.1 mmol, 1.0 equivalent) obtained in Synthesis Example 6-2 and dry CH 2 Cl 2 (400 μL) were added. After cooling to −20 ° C., boron trifluoride diethyl ether complex (BF 3 · OEt 2 ; 120 μL, 1.0 mmol, 10 equivalents) was added. The mixture was stirred at −20 ° C. for 1 hour, then m-chloroperbenzoic acid (m-CPBA; 0.1 mmol, 1.0 equiv) in CH 2 Cl 2 (200 μL) was added slowly (4 times per hour) The resulting mixture was stirred at -20 ° C for 1 hour. Saturated aqueous Na 2 S 2 O 3 and saturated aqueous NaHCO 3 were added to quench the reaction. The mixture was extracted with CH 2 Cl 2 , dried over Na 2 SO 4 and concentrated under reduced pressure. The crude product was purified by preparative thin layer chromatography (hexane / CHCl 3 = 2: 3) to give the tetrasubstituted thiophene S-oxide compound 26acde as a yellow solid (22.0 mg, 39%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.52 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.4 Hz, 2H), 7.36-7.24 (m, 5H), 7.23-7.17 (m , 2H), 7.14 (d, J = 8.4 Hz, 2H), 6.95-6.87 (m, 4H), 1.30 (s, 9H); 13 C NMR (100 MHz, CDCl 3 ): δ 152.5, 147.9, 144.1, 142.6, 138.7, 134.4, 134.1, 132.6, 131.6, 131.1, 130.3 (q, 2 J CF = 33.5 Hz), 129.9, 129.5, 129.3, 128.77, 128.70, 128.66, 126.9, 125.9, 125.6 (q, 3 J CF = 3.9 Hz), 123.8 (q, 1 J CF = 270 Hz), 34.8, 31.1; HRMS (DART) m / z calcd for C 33 H 27 ClF 3 OS [MH] + : 563.14232, found 563.14329.
1H NMR (400 MHz, CDCl3): δ 7.52 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.4 Hz, 2H), 7.36-7.24 (m, 5H), 7.23-7.17 (m, 2H), 7.14 (d, J = 8.4 Hz, 2H), 6.95-6.87 (m, 4H), 1.30 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 152.5, 147.9, 144.1, 142.6, 138.7, 134.4, 134.1, 132.6, 131.6, 131.1, 130.3 (q, 2JCF = 33.5 Hz), 129.9, 129.5, 129.3, 128.77, 128.70, 128.66, 126.9, 125.9, 125.6 (q, 3JCF = 3.9 Hz), 123.8 (q, 1JCF = 270 Hz), 34.8, 31.1; HRMS (DART) m/z calcd for C33H27ClF3OS [MH]+: 563.14232, found 563.14329。 A magnetic stir bar was placed in a 20 mL Schlenk tube, flame dried under vacuum, cooled to room temperature, and then filled with nitrogen. To the Schlenk tube, the tetrasubstituted thiophene compound 25acde (0.1 mmol, 1.0 equivalent) obtained in Synthesis Example 6-2 and dry CH 2 Cl 2 (400 μL) were added. After cooling to −20 ° C., boron trifluoride diethyl ether complex (BF 3 · OEt 2 ; 120 μL, 1.0 mmol, 10 equivalents) was added. The mixture was stirred at −20 ° C. for 1 hour, then m-chloroperbenzoic acid (m-CPBA; 0.1 mmol, 1.0 equiv) in CH 2 Cl 2 (200 μL) was added slowly (4 times per hour) The resulting mixture was stirred at -20 ° C for 1 hour. Saturated aqueous Na 2 S 2 O 3 and saturated aqueous NaHCO 3 were added to quench the reaction. The mixture was extracted with CH 2 Cl 2 , dried over Na 2 SO 4 and concentrated under reduced pressure. The crude product was purified by preparative thin layer chromatography (hexane / CHCl 3 = 2: 3) to give the tetrasubstituted thiophene S-oxide compound 26acde as a yellow solid (22.0 mg, 39%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.52 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.4 Hz, 2H), 7.36-7.24 (m, 5H), 7.23-7.17 (m , 2H), 7.14 (d, J = 8.4 Hz, 2H), 6.95-6.87 (m, 4H), 1.30 (s, 9H); 13 C NMR (100 MHz, CDCl 3 ): δ 152.5, 147.9, 144.1, 142.6, 138.7, 134.4, 134.1, 132.6, 131.6, 131.1, 130.3 (q, 2 J CF = 33.5 Hz), 129.9, 129.5, 129.3, 128.77, 128.70, 128.66, 126.9, 125.9, 125.6 (q, 3 J CF = 3.9 Hz), 123.8 (q, 1 J CF = 270 Hz), 34.8, 31.1; HRMS (DART) m / z calcd for C 33 H 27 ClF 3 OS [MH] + : 563.14232, found 563.14329.
合成例7-3~7-5:四置換チオフェンS-オキシド化合物26acdf、四置換チオフェンS-オキシド化合物26bceg、四置換チオフェンS-オキシド化合物26bcehの合成
Synthesis Examples 7-3 to 7-5: Synthesis of tetrasubstituted thiophene S-oxide compound 26acdf, tetrasubstituted thiophene S-oxide compound 26bceg, tetrasubstituted thiophene S-oxide compound 26bceh

合成例6-2で得た四置換チオフェン化合物25acdeの代わりに合成例6-3で得た四置換チオフェン化合物25acdfを使用したこと以外は合成例7-2と同様に合成し、四置換チオフェンS-オキシド化合物26acdfを黄色固体として得た(30.0 mg, 54 %)。
1H NMR (400 MHz, CDCl3): δ 7.52 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.8 Hz, 2H), 7.34-7.30 (m, 4H), 7.25-7.16 (m, 3H), 6.94 (d, J = 6.8 Hz, 2H), 6.86 (d, J = 9.2 Hz, 2H), 6.68 (d, J = 9.2 Hz, 2H), 3.75 (s, 3H), 1.29 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 159.4, 152.0, 146.6, 143.7, 143.3, 139.9, 134.4, 133.0, 131.2, 130.1 (q, 2JCF = 37.2 Hz), 129.9, 129.6, 129.3, 128.53, 128.46, 127.5, 125.7, 125.5 (q, 3JCF = 3.8 Hz), 125.2, 123.9 (q, 1JCF = 276 Hz), 113.8, 55.1, 34.7, 31.1; HRMS (DART) m/z calcd for C34H30F3O2S [MH]+: 559.19186, found 559.19277。 Synthesis was performed in the same manner as in Synthesis Example 7-2 except that the tetrasubstituted thiophene compound 25acdf obtained in Synthesis Example 6-3 was used instead of the tetrasubstituted thiophene compound 25acde obtained in Synthesis Example 6-2. The oxide compound 26acdf was obtained as a yellow solid (30.0 mg, 54%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.52 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.8 Hz, 2H), 7.34-7.30 (m, 4H), 7.25-7.16 (m , 3H), 6.94 (d, J = 6.8 Hz, 2H), 6.86 (d, J = 9.2 Hz, 2H), 6.68 (d, J = 9.2 Hz, 2H), 3.75 (s, 3H), 1.29 (s , 9H); 13 C NMR (100 MHz, CDCl 3 ): δ 159.4, 152.0, 146.6, 143.7, 143.3, 139.9, 134.4, 133.0, 131.2, 130.1 (q, 2 J CF = 37.2 Hz), 129.9, 129.6, 129.3, 128.53, 128.46, 127.5, 125.7, 125.5 (q, 3 J CF = 3.8 Hz), 125.2, 123.9 (q, 1 J CF = 276 Hz), 113.8, 55.1, 34.7, 31.1; HRMS (DART) m / z calcd for C 34 H 30 F 3 O 2 S [MH] + : 559.19186, found 559.19277.
1H NMR (400 MHz, CDCl3): δ 7.52 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.8 Hz, 2H), 7.34-7.30 (m, 4H), 7.25-7.16 (m, 3H), 6.94 (d, J = 6.8 Hz, 2H), 6.86 (d, J = 9.2 Hz, 2H), 6.68 (d, J = 9.2 Hz, 2H), 3.75 (s, 3H), 1.29 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 159.4, 152.0, 146.6, 143.7, 143.3, 139.9, 134.4, 133.0, 131.2, 130.1 (q, 2JCF = 37.2 Hz), 129.9, 129.6, 129.3, 128.53, 128.46, 127.5, 125.7, 125.5 (q, 3JCF = 3.8 Hz), 125.2, 123.9 (q, 1JCF = 276 Hz), 113.8, 55.1, 34.7, 31.1; HRMS (DART) m/z calcd for C34H30F3O2S [MH]+: 559.19186, found 559.19277。 Synthesis was performed in the same manner as in Synthesis Example 7-2 except that the tetrasubstituted thiophene compound 25acdf obtained in Synthesis Example 6-3 was used instead of the tetrasubstituted thiophene compound 25acde obtained in Synthesis Example 6-2. The oxide compound 26acdf was obtained as a yellow solid (30.0 mg, 54%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.52 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.8 Hz, 2H), 7.34-7.30 (m, 4H), 7.25-7.16 (m , 3H), 6.94 (d, J = 6.8 Hz, 2H), 6.86 (d, J = 9.2 Hz, 2H), 6.68 (d, J = 9.2 Hz, 2H), 3.75 (s, 3H), 1.29 (s , 9H); 13 C NMR (100 MHz, CDCl 3 ): δ 159.4, 152.0, 146.6, 143.7, 143.3, 139.9, 134.4, 133.0, 131.2, 130.1 (q, 2 J CF = 37.2 Hz), 129.9, 129.6, 129.3, 128.53, 128.46, 127.5, 125.7, 125.5 (q, 3 J CF = 3.8 Hz), 125.2, 123.9 (q, 1 J CF = 276 Hz), 113.8, 55.1, 34.7, 31.1; HRMS (DART) m / z calcd for C 34 H 30 F 3 O 2 S [MH] + : 559.19186, found 559.19277.
合成例6-2で得た四置換チオフェン化合物25acdeの代わりに合成例6-4で得た四置換チオフェン化合物25bcegを使用し、精製処理を分取薄層クロマトグラフィー(CHCl3)としたこと以外は合成例7-2と同様に合成し、四置換チオフェンS-オキシド化合物26bcegを黄色固体として得た(22.4 mg, 44 %)。
1H NMR (400 MHz, CDCl3): δ 7.34-7.28 (m, 3H), 7.28-7.08 (m, 4H), 6.97 (d, J = 7.2 Hz, 2H), 6.28 (t, J = 2.4 Hz, 1H), 6.07-6.03 (m, 2H), 3.49 (s, 6H), 2.29 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 160.4, 146.7, 144.4, 141.7, 140.5, 138.3, 134.75, 134.73, 133.3, 130.9, 130.2, 130.1, 129.7, 129.4, 129.1, 128.9, 128.5, 128.45, 128.41, 126.8, 107.7, 101.0, 55.2, 21.4; HRMS (DART) m/z calcd for C31H26ClO3S [MH]+: 513.12912, found 513.12807。 Except that the tetrasubstituted thiophene compound 25bceg obtained in Synthesis Example 6-4 was used in place of the tetrasubstituted thiophene compound 25acde obtained in Synthesis Example 6-2, and the purification process was preparative thin layer chromatography (CHCl 3 ). Was synthesized in the same manner as in Synthesis Example 7-2 to obtain 26bceg of a tetrasubstituted thiophene S-oxide compound as a yellow solid (22.4 mg, 44%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.34-7.28 (m, 3H), 7.28-7.08 (m, 4H), 6.97 (d, J = 7.2 Hz, 2H), 6.28 (t, J = 2.4 Hz , 1H), 6.07-6.03 (m, 2H), 3.49 (s, 6H), 2.29 (s, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 160.4, 146.7, 144.4, 141.7, 140.5, 138.3 , 134.75, 134.73, 133.3, 130.9, 130.2, 130.1, 129.7, 129.4, 129.1, 128.9, 128.5, 128.45, 128.41, 126.8, 107.7, 101.0, 55.2, 21.4; HRMS (DART) m / z calcd for C 31 H 26 ClO 3 S [MH] + : 513.12912, found 513.12807.
1H NMR (400 MHz, CDCl3): δ 7.34-7.28 (m, 3H), 7.28-7.08 (m, 4H), 6.97 (d, J = 7.2 Hz, 2H), 6.28 (t, J = 2.4 Hz, 1H), 6.07-6.03 (m, 2H), 3.49 (s, 6H), 2.29 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 160.4, 146.7, 144.4, 141.7, 140.5, 138.3, 134.75, 134.73, 133.3, 130.9, 130.2, 130.1, 129.7, 129.4, 129.1, 128.9, 128.5, 128.45, 128.41, 126.8, 107.7, 101.0, 55.2, 21.4; HRMS (DART) m/z calcd for C31H26ClO3S [MH]+: 513.12912, found 513.12807。 Except that the tetrasubstituted thiophene compound 25bceg obtained in Synthesis Example 6-4 was used in place of the tetrasubstituted thiophene compound 25acde obtained in Synthesis Example 6-2, and the purification process was preparative thin layer chromatography (CHCl 3 ). Was synthesized in the same manner as in Synthesis Example 7-2 to obtain 26bceg of a tetrasubstituted thiophene S-oxide compound as a yellow solid (22.4 mg, 44%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.34-7.28 (m, 3H), 7.28-7.08 (m, 4H), 6.97 (d, J = 7.2 Hz, 2H), 6.28 (t, J = 2.4 Hz , 1H), 6.07-6.03 (m, 2H), 3.49 (s, 6H), 2.29 (s, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 160.4, 146.7, 144.4, 141.7, 140.5, 138.3 , 134.75, 134.73, 133.3, 130.9, 130.2, 130.1, 129.7, 129.4, 129.1, 128.9, 128.5, 128.45, 128.41, 126.8, 107.7, 101.0, 55.2, 21.4; HRMS (DART) m / z calcd for C 31 H 26 ClO 3 S [MH] + : 513.12912, found 513.12807.
合成例6-2で得た四置換チオフェン化合物25acdeの代わりに合成例6-5で得た四置換チオフェン化合物25bcehを使用し、精製処理を分取薄層クロマトグラフィー(ヘキサン/酢酸エチル=5: 1)としたこと以外は合成例7-2と同様に合成し、四置換チオフェンS-オキシド化合物26bcehを黄色固体として得た(23.8 mg, 47 %)。
1H NMR (400 MHz, CDCl3): δ 7.31 (d, J = 8.8 Hz, 2H), 7.27-7.06 (m, 9H), 6.97-6.89 (m, 4H), 6.81 (d, J = 8.0 Hz, 2H), 2.52 (t, J = 7.6 Hz, 2H), 2.25 (s, 3H), 1.52 (quin, J = 7.6 Hz, 2H), 1.26 (sext, J = 7.6 Hz, 2H), 0.89 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 146.2, 144.3, 143.1, 142.0, 141.0, 138.2, 134.7, 133.2, 130.9, 130.33, 130.27, 129.7, 129.6, 129.5, 129.2, 128.9, 128.4, 128.30, 128.27, 126.9, 35.3, 33.2, 22.1, 21.4, 13.9; HRMS (APCI) m/z calcd for C33H30ClOS [MH]+: 509.1700, found 509.1688。 Using the tetrasubstituted thiophene compound 25bceh obtained in Synthesis Example 6-5 instead of the tetrasubstituted thiophene compound 25acde obtained in Synthesis Example 6-2, purification treatment was performed by preparative thin layer chromatography (hexane / ethyl acetate = 5: The compound was synthesized in the same manner as in Synthesis Example 7-2 except that 1) was obtained to obtain a tetrasubstituted thiophene S-oxide compound 26bceh as a yellow solid (23.8 mg, 47%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.31 (d, J = 8.8 Hz, 2H), 7.27-7.06 (m, 9H), 6.97-6.89 (m, 4H), 6.81 (d, J = 8.0 Hz , 2H), 2.52 (t, J = 7.6 Hz, 2H), 2.25 (s, 3H), 1.52 (quin, J = 7.6 Hz, 2H), 1.26 (sext, J = 7.6 Hz, 2H), 0.89 (t , J = 7.6 Hz, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 146.2, 144.3, 143.1, 142.0, 141.0, 138.2, 134.7, 133.2, 130.9, 130.33, 130.27, 129.7, 129.6, 129.5, 129.2 , 128.9, 128.4, 128.30, 128.27, 126.9, 35.3, 33.2, 22.1, 21.4, 13.9; HRMS (APCI) m / z calcd for C 33 H 30 ClOS [MH] + : 509.1700, found 509.1688.
1H NMR (400 MHz, CDCl3): δ 7.31 (d, J = 8.8 Hz, 2H), 7.27-7.06 (m, 9H), 6.97-6.89 (m, 4H), 6.81 (d, J = 8.0 Hz, 2H), 2.52 (t, J = 7.6 Hz, 2H), 2.25 (s, 3H), 1.52 (quin, J = 7.6 Hz, 2H), 1.26 (sext, J = 7.6 Hz, 2H), 0.89 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 146.2, 144.3, 143.1, 142.0, 141.0, 138.2, 134.7, 133.2, 130.9, 130.33, 130.27, 129.7, 129.6, 129.5, 129.2, 128.9, 128.4, 128.30, 128.27, 126.9, 35.3, 33.2, 22.1, 21.4, 13.9; HRMS (APCI) m/z calcd for C33H30ClOS [MH]+: 509.1700, found 509.1688。 Using the tetrasubstituted thiophene compound 25bceh obtained in Synthesis Example 6-5 instead of the tetrasubstituted thiophene compound 25acde obtained in Synthesis Example 6-2, purification treatment was performed by preparative thin layer chromatography (hexane / ethyl acetate = 5: The compound was synthesized in the same manner as in Synthesis Example 7-2 except that 1) was obtained to obtain a tetrasubstituted thiophene S-oxide compound 26bceh as a yellow solid (23.8 mg, 47%).
1 H NMR (400 MHz, CDCl 3 ): δ 7.31 (d, J = 8.8 Hz, 2H), 7.27-7.06 (m, 9H), 6.97-6.89 (m, 4H), 6.81 (d, J = 8.0 Hz , 2H), 2.52 (t, J = 7.6 Hz, 2H), 2.25 (s, 3H), 1.52 (quin, J = 7.6 Hz, 2H), 1.26 (sext, J = 7.6 Hz, 2H), 0.89 (t , J = 7.6 Hz, 3H); 13 C NMR (100 MHz, CDCl 3 ): δ 146.2, 144.3, 143.1, 142.0, 141.0, 138.2, 134.7, 133.2, 130.9, 130.33, 130.27, 129.7, 129.6, 129.5, 129.2 , 128.9, 128.4, 128.30, 128.27, 126.9, 35.3, 33.2, 22.1, 21.4, 13.9; HRMS (APCI) m / z calcd for C 33 H 30 ClOS [MH] + : 509.1700, found 509.1688.
[合成例8:化合物(11)の合成]
合成例8-1:化合物13の合成 [Synthesis Example 8: Synthesis of Compound (11)]
Synthesis Example 8-1: Synthesis of Compound 13
合成例8-1:化合物13の合成 [Synthesis Example 8: Synthesis of Compound (11)]
Synthesis Example 8-1: Synthesis of Compound 13

[式中、PhNO2はニトロベンゼンを示す。以下同様である。]
25 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このチューブに、合成例7-3で得た5-(4-(t-ブチル)フェニル)- 4-(4-メトキシフェニル)-3-フェニル-2-(4-(トリフルオロメチル)フェニル)チオフェン1-オキシド(化合物26acdf; 111.7 mg, 0.2 mmol, 1.0当量)、マレイミド(59.5 mg, 0.6 mmol, 3.0当量)、及びニトロベンゼン(PhNO2; 1 mL)を窒素気流下に添加した。反応混合物を室温で1時間撹拌し、その後、210℃で48時間加熱した。反応混合物を室温まで冷却した後、混合物を短いシリカゲルパッド(CHCl3)でろ過してニトロベンゼンを除去し、短いシリカゲルパッド(酢酸エチル)でろ過して化合物13を流出させた。ろ液を濃縮し、粗生成物をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル= 5: 1)で精製し、4-(4-(t-ブチル)フェニル)-5-(4-メトキシフェニル)-6-フェニル-7-(4-(トリフルオロメチル)フェニル)イソインドリン-1,3-ジオン(化合物13)を白色固体として得た(103.3 mg, 85 %)。
1H NMR (600 MHz, CDCl3): δ 7.57 (s, 1H), 7.44 (d, J = 7.8 Hz, 2H), 7.24-7.19 (m, 4H), 7.01 (d, J = 7.2 Hz, 2.4 Hz, 2H), 6.92-6.90 (m, 3H), 6.71-6.69 (m, 2H), 6.59 (dd, J = 6.6 Hz, 2.4 Hz, 2H), 6.41 (dd, J = 6.6 Hz, 2.4 Hz, 2H) 3.60 (s, 3H), 1.27 (s, 9H)。 [Wherein PhNO 2 represents nitrobenzene. The same applies hereinafter. ]
A magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, 5- (4- (t-butyl) phenyl) -4- (4-methoxyphenyl) -3-phenyl-2- (4- (trifluoromethyl) phenyl) obtained in Synthesis Example 7-3 Thiophene 1-oxide (Compound 26acdf; 111.7 mg, 0.2 mmol, 1.0 eq), maleimide (59.5 mg, 0.6 mmol, 3.0 eq), and nitrobenzene (PhNO 2 ; 1 mL) were added under a stream of nitrogen. The reaction mixture was stirred at room temperature for 1 hour and then heated at 210 ° C. for 48 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a short silica gel pad (CHCl 3 ) to remove nitrobenzene and filtered through a short silica gel pad (ethyl acetate) to drain compound 13. The filtrate was concentrated and the crude product was purified by flash column chromatography (hexane / ethyl acetate = 5: 1) to give 4- (4- (t-butyl) phenyl) -5- (4-methoxyphenyl)- 6-phenyl-7- (4- (trifluoromethyl) phenyl) isoindoline-1,3-dione (compound 13) was obtained as a white solid (103.3 mg, 85%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.57 (s, 1H), 7.44 (d, J = 7.8 Hz, 2H), 7.24-7.19 (m, 4H), 7.01 (d, J = 7.2 Hz, 2.4 Hz, 2H), 6.92-6.90 (m, 3H), 6.71-6.69 (m, 2H), 6.59 (dd, J = 6.6 Hz, 2.4 Hz, 2H), 6.41 (dd, J = 6.6 Hz, 2.4 Hz, 2H) 3.60 (s, 3H), 1.27 (s, 9H).
25 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このチューブに、合成例7-3で得た5-(4-(t-ブチル)フェニル)- 4-(4-メトキシフェニル)-3-フェニル-2-(4-(トリフルオロメチル)フェニル)チオフェン1-オキシド(化合物26acdf; 111.7 mg, 0.2 mmol, 1.0当量)、マレイミド(59.5 mg, 0.6 mmol, 3.0当量)、及びニトロベンゼン(PhNO2; 1 mL)を窒素気流下に添加した。反応混合物を室温で1時間撹拌し、その後、210℃で48時間加熱した。反応混合物を室温まで冷却した後、混合物を短いシリカゲルパッド(CHCl3)でろ過してニトロベンゼンを除去し、短いシリカゲルパッド(酢酸エチル)でろ過して化合物13を流出させた。ろ液を濃縮し、粗生成物をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル= 5: 1)で精製し、4-(4-(t-ブチル)フェニル)-5-(4-メトキシフェニル)-6-フェニル-7-(4-(トリフルオロメチル)フェニル)イソインドリン-1,3-ジオン(化合物13)を白色固体として得た(103.3 mg, 85 %)。
1H NMR (600 MHz, CDCl3): δ 7.57 (s, 1H), 7.44 (d, J = 7.8 Hz, 2H), 7.24-7.19 (m, 4H), 7.01 (d, J = 7.2 Hz, 2.4 Hz, 2H), 6.92-6.90 (m, 3H), 6.71-6.69 (m, 2H), 6.59 (dd, J = 6.6 Hz, 2.4 Hz, 2H), 6.41 (dd, J = 6.6 Hz, 2.4 Hz, 2H) 3.60 (s, 3H), 1.27 (s, 9H)。 [Wherein PhNO 2 represents nitrobenzene. The same applies hereinafter. ]
A magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, 5- (4- (t-butyl) phenyl) -4- (4-methoxyphenyl) -3-phenyl-2- (4- (trifluoromethyl) phenyl) obtained in Synthesis Example 7-3 Thiophene 1-oxide (Compound 26acdf; 111.7 mg, 0.2 mmol, 1.0 eq), maleimide (59.5 mg, 0.6 mmol, 3.0 eq), and nitrobenzene (PhNO 2 ; 1 mL) were added under a stream of nitrogen. The reaction mixture was stirred at room temperature for 1 hour and then heated at 210 ° C. for 48 hours. After the reaction mixture was cooled to room temperature, the mixture was filtered through a short silica gel pad (CHCl 3 ) to remove nitrobenzene and filtered through a short silica gel pad (ethyl acetate) to drain compound 13. The filtrate was concentrated and the crude product was purified by flash column chromatography (hexane / ethyl acetate = 5: 1) to give 4- (4- (t-butyl) phenyl) -5- (4-methoxyphenyl)- 6-phenyl-7- (4- (trifluoromethyl) phenyl) isoindoline-1,3-dione (compound 13) was obtained as a white solid (103.3 mg, 85%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.57 (s, 1H), 7.44 (d, J = 7.8 Hz, 2H), 7.24-7.19 (m, 4H), 7.01 (d, J = 7.2 Hz, 2.4 Hz, 2H), 6.92-6.90 (m, 3H), 6.71-6.69 (m, 2H), 6.59 (dd, J = 6.6 Hz, 2.4 Hz, 2H), 6.41 (dd, J = 6.6 Hz, 2.4 Hz, 2H) 3.60 (s, 3H), 1.27 (s, 9H).
[合成例9:化合物(4)の合成]
合成例9-1:化合物14a及び化合物14bの合成 [Synthesis Example 9: Synthesis of Compound (4)]
Synthesis Example 9-1: Synthesis of Compound 14a and Compound 14b
合成例9-1:化合物14a及び化合物14bの合成 [Synthesis Example 9: Synthesis of Compound (4)]
Synthesis Example 9-1: Synthesis of Compound 14a and Compound 14b

[式中、i-PrOHはイソプロピルアルコールを示す。以下同様である。]
25 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このチューブに、合成例8-1で得た4-(4-(t-ブチル)フェニル)-5-(4-メトキシフェニル)-6-フェニル-7-(4-(トリフルオロメチル)フェニル)イソインドリン-1,3-ジオン(化合物13; 59.9 mg, 0.1 mmol, 1.0当量)、NaOCl水溶液(ab. 6%, 200μL, 1.5当量)、NaOH水溶液(3N, 170μL, 5.0当量)、及びメタノール(MeOH; 5.6 mL)を窒素気流下に添加した。得られた混合物を10分間還流した。反応混合物を室温まで冷却した後、混合物に水(10 mL)及びCH2Cl2(10 mL)を添加した。混合物を1M HClで中和し、CH2Cl2で濃縮した。有機層をNa2SO4で乾燥し、揮発性物質を減圧下に除去した。粗生成物を精製せずにそのまま次のステップに使用した。 [Wherein, i-PrOH represents isopropyl alcohol. The same applies hereinafter. ]
A magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, 4- (4- (t-butyl) phenyl) -5- (4-methoxyphenyl) -6-phenyl-7- (4- (trifluoromethyl) phenyl) obtained in Synthesis Example 8-1 Isoindoline-1,3-dione (compound 13; 59.9 mg, 0.1 mmol, 1.0 eq), aqueous NaOCl (ab. 6%, 200 μL, 1.5 eq), aqueous NaOH (3N, 170 μL, 5.0 eq), and methanol ( MeOH; 5.6 mL) was added under a stream of nitrogen. The resulting mixture was refluxed for 10 minutes. After the reaction mixture was cooled to room temperature, water (10 mL) and CH 2 Cl 2 (10 mL) were added to the mixture. The mixture was neutralized with 1M HCl and concentrated with CH 2 Cl 2 . The organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was used directly in the next step without purification.
25 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このチューブに、合成例8-1で得た4-(4-(t-ブチル)フェニル)-5-(4-メトキシフェニル)-6-フェニル-7-(4-(トリフルオロメチル)フェニル)イソインドリン-1,3-ジオン(化合物13; 59.9 mg, 0.1 mmol, 1.0当量)、NaOCl水溶液(ab. 6%, 200μL, 1.5当量)、NaOH水溶液(3N, 170μL, 5.0当量)、及びメタノール(MeOH; 5.6 mL)を窒素気流下に添加した。得られた混合物を10分間還流した。反応混合物を室温まで冷却した後、混合物に水(10 mL)及びCH2Cl2(10 mL)を添加した。混合物を1M HClで中和し、CH2Cl2で濃縮した。有機層をNa2SO4で乾燥し、揮発性物質を減圧下に除去した。粗生成物を精製せずにそのまま次のステップに使用した。 [Wherein, i-PrOH represents isopropyl alcohol. The same applies hereinafter. ]
A magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, 4- (4- (t-butyl) phenyl) -5- (4-methoxyphenyl) -6-phenyl-7- (4- (trifluoromethyl) phenyl) obtained in Synthesis Example 8-1 Isoindoline-1,3-dione (compound 13; 59.9 mg, 0.1 mmol, 1.0 eq), aqueous NaOCl (ab. 6%, 200 μL, 1.5 eq), aqueous NaOH (3N, 170 μL, 5.0 eq), and methanol ( MeOH; 5.6 mL) was added under a stream of nitrogen. The resulting mixture was refluxed for 10 minutes. After the reaction mixture was cooled to room temperature, water (10 mL) and CH 2 Cl 2 (10 mL) were added to the mixture. The mixture was neutralized with 1M HCl and concentrated with CH 2 Cl 2 . The organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was used directly in the next step without purification.
25 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このチューブに、上記工程で得た物質、KOH(200 mg, 3.6 mmol, 36当量)、及び乾燥イソプロピルアルコール(乾燥i-PrOH; 2.3 mL)を窒素気流下に添加した。内容物を16時間還流した。室温まで冷却した後、混合物に水(10 mL)及びCH2Cl2(10 mL)を添加した。混合物を1M HClで中和し、CH2Cl2で濃縮した。有機層をNa2SO4で乾燥し、揮発性物質を減圧下に除去した。粗生成物をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル= 1: 1~0: 1)で精製し、化合物14a及び化合物14bの混合物(14a/ 14b = 2: 3~3: 2)を白色固体として得た(54.2 mg, 91 %)。
A magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen. To the tube, the material obtained in the above step, KOH (200 mg, 3.6 mmol, 36 equivalents), and dry isopropyl alcohol (dry i-PrOH; 2.3 mL) were added under a nitrogen stream. The contents were refluxed for 16 hours. After cooling to room temperature, water (10 mL) and CH 2 Cl 2 (10 mL) were added to the mixture. The mixture was neutralized with 1M HCl and concentrated with CH 2 Cl 2 . The organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was purified by flash column chromatography (hexane / ethyl acetate = 1: 1 to 0: 1), and a mixture of compound 14a and compound 14b (14a / 14b = 2: 3 to 3: 2) as a white solid Obtained (54.2 mg, 91%).
[合成例10:化合物(5)の合成]
合成例10-1:化合物16a及び化合物16bの合成 [Synthesis Example 10: Synthesis of Compound (5)]
Synthesis Example 10-1: Synthesis of Compound 16a and Compound 16b
合成例10-1:化合物16a及び化合物16bの合成 [Synthesis Example 10: Synthesis of Compound (5)]
Synthesis Example 10-1: Synthesis of Compound 16a and Compound 16b

[式中、TBAFはテトラブチルアンモニウムフルオライド;以下同様である。]
25 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このチューブに、合成例7-3で得た5-(4-(t-ブチル)フェニル)- 4-(4-メトキシフェニル)-3-フェニル-2-(4-(トリフルオロメチル)フェニル)チオフェン1-オキシド(化合物26acdf; 44.8 mg, 0.08 mmol, 1.0当量)、合成例1-4で得た4-ヒドロキシ-2,5-ビス(トリメチルシリル)フェニルトリフルオロメタンスルホネート(化合物5; 94.0 mg, 0.24 mmol, 3.0当量)、及びTHF(400μL)を窒素気流下に添加した。その後、混合物に、テトラブチルアンモニウムフルオライド(TBAF; 480μL, 0.48 mmol, 6.0当量, 1M in THF)をゆっくりと添加した。反応混合物を室温で24時間撹拌した。混合物に水(10 mL)及びCH2Cl2(10 mL)を添加した後、得られた混合物をCH2Cl2で抽出した。有機層をNa2SO4で乾燥し、揮発性物質を減圧下に除去した。粗生成物を分取薄層クロマトグラフィー(ヘキサン/酢酸エチル= 10: 1)及びGPCで精製し、化合物16a及び化合物16bの混合物(16a/ 16b = 5: 4~4: 5)を白色固体として得た(45.3 mg, 84 %)。 [Wherein TBAF is tetrabutylammonium fluoride; the same shall apply hereinafter. ]
A magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, 5- (4- (t-butyl) phenyl) -4- (4-methoxyphenyl) -3-phenyl-2- (4- (trifluoromethyl) phenyl) obtained in Synthesis Example 7-3 Thiophene 1-oxide (Compound 26acdf; 44.8 mg, 0.08 mmol, 1.0 equivalent), 4-hydroxy-2,5-bis (trimethylsilyl) phenyl trifluoromethanesulfonate obtained in Synthesis Example 1-4 (Compound 5; 94.0 mg, 0.24 mmol, 3.0 eq), and THF (400 μL) were added under a stream of nitrogen. Then, tetrabutylammonium fluoride (TBAF; 480 μL, 0.48 mmol, 6.0 eq, 1M in THF) was slowly added to the mixture. The reaction mixture was stirred at room temperature for 24 hours. After adding water (10 mL) and CH 2 Cl 2 (10 mL) to the mixture, the resulting mixture was extracted with CH 2 Cl 2 . The organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was purified by preparative thin layer chromatography (hexane / ethyl acetate = 10: 1) and GPC, and a mixture of compound 16a and compound 16b (16a / 16b = 5: 4 to 4: 5) was obtained as a white solid. Obtained (45.3 mg, 84%).
25 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このチューブに、合成例7-3で得た5-(4-(t-ブチル)フェニル)- 4-(4-メトキシフェニル)-3-フェニル-2-(4-(トリフルオロメチル)フェニル)チオフェン1-オキシド(化合物26acdf; 44.8 mg, 0.08 mmol, 1.0当量)、合成例1-4で得た4-ヒドロキシ-2,5-ビス(トリメチルシリル)フェニルトリフルオロメタンスルホネート(化合物5; 94.0 mg, 0.24 mmol, 3.0当量)、及びTHF(400μL)を窒素気流下に添加した。その後、混合物に、テトラブチルアンモニウムフルオライド(TBAF; 480μL, 0.48 mmol, 6.0当量, 1M in THF)をゆっくりと添加した。反応混合物を室温で24時間撹拌した。混合物に水(10 mL)及びCH2Cl2(10 mL)を添加した後、得られた混合物をCH2Cl2で抽出した。有機層をNa2SO4で乾燥し、揮発性物質を減圧下に除去した。粗生成物を分取薄層クロマトグラフィー(ヘキサン/酢酸エチル= 10: 1)及びGPCで精製し、化合物16a及び化合物16bの混合物(16a/ 16b = 5: 4~4: 5)を白色固体として得た(45.3 mg, 84 %)。 [Wherein TBAF is tetrabutylammonium fluoride; the same shall apply hereinafter. ]
A magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, 5- (4- (t-butyl) phenyl) -4- (4-methoxyphenyl) -3-phenyl-2- (4- (trifluoromethyl) phenyl) obtained in Synthesis Example 7-3 Thiophene 1-oxide (Compound 26acdf; 44.8 mg, 0.08 mmol, 1.0 equivalent), 4-hydroxy-2,5-bis (trimethylsilyl) phenyl trifluoromethanesulfonate obtained in Synthesis Example 1-4 (Compound 5; 94.0 mg, 0.24 mmol, 3.0 eq), and THF (400 μL) were added under a stream of nitrogen. Then, tetrabutylammonium fluoride (TBAF; 480 μL, 0.48 mmol, 6.0 eq, 1M in THF) was slowly added to the mixture. The reaction mixture was stirred at room temperature for 24 hours. After adding water (10 mL) and CH 2 Cl 2 (10 mL) to the mixture, the resulting mixture was extracted with CH 2 Cl 2 . The organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was purified by preparative thin layer chromatography (hexane / ethyl acetate = 10: 1) and GPC, and a mixture of compound 16a and compound 16b (16a / 16b = 5: 4 to 4: 5) was obtained as a white solid. Obtained (45.3 mg, 84%).
合成例10-2:化合物17a及び化合物17bの合成
Synthesis Example 10-2: Synthesis of Compound 17a and Compound 17b

25 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このチューブに、合成例10-1で得た化合物16a及び化合物16bの混合物(35.4 mg, 0.052 mmol, 1.0当量)、及びCH2Cl2(500μL)を窒素気流下に添加した。内容物を0℃まで冷却し、このチューブにN,N-ジイソプロピルエチルアミン(i-Pr2NEt; 27.2μL, 0.156 mmol, 3.0当量)、N,N-ジメチルアミノピリジン(DMAP; 1.3 mg, 10μmol, 20 mol%)、及びトリフルオロメタンスルホン酸無水物(Tf2O; 26.2μL, 0.156 mmol, 3.0当量)を添加した。得られた混合物を0℃で0.5時間撹拌した。反応混合物を室温まで昇温して12時間撹拌した後、混合物に水(10 mL)及びCH2Cl2(10 mL)を添加した。混合物をCH2Cl2で抽出した後、有機層をNa2SO4で乾燥させ、揮発性物質を減圧下に除去した。粗生成物を分取薄層クロマトグラフィー(ヘキサン/酢酸エチル= 10: 1)及びGPCで精製し、化合物17a及び化合物17bの混合物(17a/ 17b = 5: 4~4: 5)を白色固体として得た(45.3 mg, 84 %)。
A magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, a mixture of compound 16a and compound 16b obtained in Synthesis Example 10-1 (35.4 mg, 0.052 mmol, 1.0 equivalent) and CH 2 Cl 2 (500 μL) were added under a nitrogen stream. The contents were cooled to 0 ° C., and N, N-diisopropylethylamine (i-Pr 2 NEt; 27.2 μL, 0.156 mmol, 3.0 eq), N, N-dimethylaminopyridine (DMAP; 1.3 mg, 10 μmol, 20 mol%) and trifluoromethanesulfonic anhydride (Tf 2 O; 26.2 μL, 0.156 mmol, 3.0 eq) were added. The resulting mixture was stirred at 0 ° C. for 0.5 hour. The reaction mixture was warmed to room temperature and stirred for 12 hours, and then water (10 mL) and CH 2 Cl 2 (10 mL) were added to the mixture. After the mixture was extracted with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was purified by preparative thin layer chromatography (hexane / ethyl acetate = 10: 1) and GPC, and a mixture of compound 17a and compound 17b (17a / 17b = 5: 4 to 4: 5) was obtained as a white solid. Obtained (45.3 mg, 84%).
[合成例11:化合物(33)の合成]
合成例11-1:化合物27aの合成 [Synthesis Example 11: Synthesis of Compound (33)]
Synthesis Example 11-1: Synthesis of Compound 27a
合成例11-1:化合物27aの合成 [Synthesis Example 11: Synthesis of Compound (33)]
Synthesis Example 11-1: Synthesis of Compound 27a

既報(Org. Lett. 15, 936 (2013))に報告された方法にしたがって4-エチニルアセトフェノンを合成した。
4-Ethynylacetophenone was synthesized according to the method reported in the previous report (Org. Lett. 15, 936 (2013)).
50 mLのシュレンク管に磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このシュレンク管に、4-エチニルアセトフェノン(288 mg, 2.0 mmol, 1.0当量)、4-ヨードピリジン(410 mg, 2.0 mmol, 1.0当量)、ビス(トリフェニルホスフィン)パラジウム(II)ジクロライド(140 mg, 0.2 mmol, 20 mol%)、銅(I)ヨージド(38 mg, 0.2 mmol, 20 mol%)、トリエチルアミン(836μL, 6.0 mmol, 3.0当量)、及び乾燥THF(10 mL)を窒素気流下に添加した。混合物を室温で3時間撹拌した後、水(30 mL)及びCH2Cl2(20 mL)を添加した。CH2Cl2で抽出した後、有機層をNa2SO4で乾燥させ、揮発性物質を減圧下に除去した。フラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=1: 1)及び再結晶(CHCl3/ヘキサン)により精製し、化合物27aを橙色結晶として得た(196 mg, 44 %)。
1H NMR (600 MHz, CDCl3): δ 8.64 (d, J = 5.4 Hz, 2H), 7.97 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.4 Hz, 1H), 7.41 (d, J = 5.4 Hz, 1H), 2.63 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 197.1, 149.9, 136.9, 132.0, 130.7, 128.3, 126.8, 125.5, 92.7, 89.5, 26.6; HRMS (APCI) m/z calcd for C15H12NO [MH]+: 222.09134, found 222.09074。 A magnetic stirring bar was placed in a 50 mL Schlenk tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this Schlenk tube, 4-ethynylacetophenone (288 mg, 2.0 mmol, 1.0 equivalent), 4-iodopyridine (410 mg, 2.0 mmol, 1.0 equivalent), bis (triphenylphosphine) palladium (II) dichloride (140 mg, 0.2 mmol, 20 mol%), copper (I) iodide (38 mg, 0.2 mmol, 20 mol%), triethylamine (836 μL, 6.0 mmol, 3.0 eq), and dry THF (10 mL) were added under a nitrogen stream. . After the mixture was stirred at room temperature for 3 hours, water (30 mL) and CH 2 Cl 2 (20 mL) were added. After extraction with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. Purification by flash column chromatography (hexane / ethyl acetate = 1: 1) and recrystallization (CHCl 3 / hexane) gave compound 27a as orange crystals (196 mg, 44%).
1 H NMR (600 MHz, CDCl 3 ): δ 8.64 (d, J = 5.4 Hz, 2H), 7.97 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.4 Hz, 1H), 7.41 ( d, J = 5.4 Hz, 1H), 2.63 (s, 3H); 13 C NMR (150 MHz, CDCl 3 ): δ 197.1, 149.9, 136.9, 132.0, 130.7, 128.3, 126.8, 125.5, 92.7, 89.5, 26.6 HRMS (APCI) m / z calcd for C 15 H 12 NO [MH] + : 222.09134, found 222.09074.
1H NMR (600 MHz, CDCl3): δ 8.64 (d, J = 5.4 Hz, 2H), 7.97 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.4 Hz, 1H), 7.41 (d, J = 5.4 Hz, 1H), 2.63 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 197.1, 149.9, 136.9, 132.0, 130.7, 128.3, 126.8, 125.5, 92.7, 89.5, 26.6; HRMS (APCI) m/z calcd for C15H12NO [MH]+: 222.09134, found 222.09074。 A magnetic stirring bar was placed in a 50 mL Schlenk tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this Schlenk tube, 4-ethynylacetophenone (288 mg, 2.0 mmol, 1.0 equivalent), 4-iodopyridine (410 mg, 2.0 mmol, 1.0 equivalent), bis (triphenylphosphine) palladium (II) dichloride (140 mg, 0.2 mmol, 20 mol%), copper (I) iodide (38 mg, 0.2 mmol, 20 mol%), triethylamine (836 μL, 6.0 mmol, 3.0 eq), and dry THF (10 mL) were added under a nitrogen stream. . After the mixture was stirred at room temperature for 3 hours, water (30 mL) and CH 2 Cl 2 (20 mL) were added. After extraction with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. Purification by flash column chromatography (hexane / ethyl acetate = 1: 1) and recrystallization (CHCl 3 / hexane) gave compound 27a as orange crystals (196 mg, 44%).
1 H NMR (600 MHz, CDCl 3 ): δ 8.64 (d, J = 5.4 Hz, 2H), 7.97 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.4 Hz, 1H), 7.41 ( d, J = 5.4 Hz, 1H), 2.63 (s, 3H); 13 C NMR (150 MHz, CDCl 3 ): δ 197.1, 149.9, 136.9, 132.0, 130.7, 128.3, 126.8, 125.5, 92.7, 89.5, 26.6 HRMS (APCI) m / z calcd for C 15 H 12 NO [MH] + : 222.09134, found 222.09074.
合成例11-2:化合物27bの合成
Synthesis Example 11-2: Synthesis of Compound 27b

[式中、Acはアセチル基を示す。以下同様である。]
50 mLのシュレンク管に磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このシュレンク管に、4-エチニルアセトフェノン(158.3 mg, 1.1 mmol, 1.1当量)、2-ヨードナフタレン(254.6 mg, 1.0 mmol, 1.0当量)、ビス(トリフェニルホスフィン)パラジウム(II)ジクロライド(35.1 mg, 0.05 mmol, 5 mol%)、銅(I)ヨージド(9.5 mg, 0.05 mmol, 5 mol%)、トリエチルアミン(420μL, 3.0 mmol, 3.0当量)、及び乾燥THF(7 mL)を窒素気流下に添加した。反応混合物を室温で9時間撹拌した後、混合物に水(15 mL)及びCH2Cl2(10 mL)を添加した。CH2Cl2で抽出した後、有機層をNa2SO4で乾燥させ、揮発性物質を減圧下に除去した。フラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=10: 1)及び再結晶(CHCl3/ヘキサン)により精製し、化合物7bを白色結晶として得た(196 mg, 73 %)。
1H NMR (600 MHz, CDCl3): δ 8.08 (s, 1H), 7.95 (d, J = 6.6 Hz, 2H), 7.85-7.81 (m, 3H), 7.65 (d, J = 6.6 Hz, 2H), 7.59 (dd, J = 8.4 Hz, 1.8 Hz, 1H), 7.53-7.49 (m, 2H), 2.62 (s, 3H)。 [In the formula, Ac represents an acetyl group. The same applies hereinafter. ]
A magnetic stirring bar was placed in a 50 mL Schlenk tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this Schlenk tube, 4-ethynylacetophenone (158.3 mg, 1.1 mmol, 1.1 eq), 2-iodonaphthalene (254.6 mg, 1.0 mmol, 1.0 eq), bis (triphenylphosphine) palladium (II) dichloride (35.1 mg, 0.05 mmol, 5 mol%), copper (I) iodide (9.5 mg, 0.05 mmol, 5 mol%), triethylamine (420 μL, 3.0 mmol, 3.0 eq), and dry THF (7 mL) were added under a nitrogen stream. . After the reaction mixture was stirred at room temperature for 9 hours, water (15 mL) and CH 2 Cl 2 (10 mL) were added to the mixture. After extraction with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. Purification by flash column chromatography (hexane / ethyl acetate = 10: 1) and recrystallization (CHCl 3 / hexane) gave compound 7b as white crystals (196 mg, 73%).
1 H NMR (600 MHz, CDCl 3 ): δ 8.08 (s, 1H), 7.95 (d, J = 6.6 Hz, 2H), 7.85-7.81 (m, 3H), 7.65 (d, J = 6.6 Hz, 2H ), 7.59 (dd, J = 8.4 Hz, 1.8 Hz, 1H), 7.53-7.49 (m, 2H), 2.62 (s, 3H).
50 mLのシュレンク管に磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このシュレンク管に、4-エチニルアセトフェノン(158.3 mg, 1.1 mmol, 1.1当量)、2-ヨードナフタレン(254.6 mg, 1.0 mmol, 1.0当量)、ビス(トリフェニルホスフィン)パラジウム(II)ジクロライド(35.1 mg, 0.05 mmol, 5 mol%)、銅(I)ヨージド(9.5 mg, 0.05 mmol, 5 mol%)、トリエチルアミン(420μL, 3.0 mmol, 3.0当量)、及び乾燥THF(7 mL)を窒素気流下に添加した。反応混合物を室温で9時間撹拌した後、混合物に水(15 mL)及びCH2Cl2(10 mL)を添加した。CH2Cl2で抽出した後、有機層をNa2SO4で乾燥させ、揮発性物質を減圧下に除去した。フラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=10: 1)及び再結晶(CHCl3/ヘキサン)により精製し、化合物7bを白色結晶として得た(196 mg, 73 %)。
1H NMR (600 MHz, CDCl3): δ 8.08 (s, 1H), 7.95 (d, J = 6.6 Hz, 2H), 7.85-7.81 (m, 3H), 7.65 (d, J = 6.6 Hz, 2H), 7.59 (dd, J = 8.4 Hz, 1.8 Hz, 1H), 7.53-7.49 (m, 2H), 2.62 (s, 3H)。 [In the formula, Ac represents an acetyl group. The same applies hereinafter. ]
A magnetic stirring bar was placed in a 50 mL Schlenk tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this Schlenk tube, 4-ethynylacetophenone (158.3 mg, 1.1 mmol, 1.1 eq), 2-iodonaphthalene (254.6 mg, 1.0 mmol, 1.0 eq), bis (triphenylphosphine) palladium (II) dichloride (35.1 mg, 0.05 mmol, 5 mol%), copper (I) iodide (9.5 mg, 0.05 mmol, 5 mol%), triethylamine (420 μL, 3.0 mmol, 3.0 eq), and dry THF (7 mL) were added under a nitrogen stream. . After the reaction mixture was stirred at room temperature for 9 hours, water (15 mL) and CH 2 Cl 2 (10 mL) were added to the mixture. After extraction with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. Purification by flash column chromatography (hexane / ethyl acetate = 10: 1) and recrystallization (CHCl 3 / hexane) gave compound 7b as white crystals (196 mg, 73%).
1 H NMR (600 MHz, CDCl 3 ): δ 8.08 (s, 1H), 7.95 (d, J = 6.6 Hz, 2H), 7.85-7.81 (m, 3H), 7.65 (d, J = 6.6 Hz, 2H ), 7.59 (dd, J = 8.4 Hz, 1.8 Hz, 1H), 7.53-7.49 (m, 2H), 2.62 (s, 3H).
[合成例12:化合物(33B)の合成]
合成例12-1:化合物28の合成 [Synthesis Example 12: Synthesis of Compound (33B)]
Synthesis Example 12-1: Synthesis of Compound 28
合成例12-1:化合物28の合成 [Synthesis Example 12: Synthesis of Compound (33B)]
Synthesis Example 12-1: Synthesis of Compound 28

既報(Chem. Commun. 46, 931 (2010))に報告された方法にしたがって化合物28を合成した。
Compound 28 was synthesized according to the method reported in the previous report (Chem. Commun. 46, 931 (2010)).
[実施例1(第1の態様):化合物(1)の合成]
実施例1-1:化合物15a及び化合物15bの合成 [Example 1 (first aspect): Synthesis of compound (1)]
Example 1-1: Synthesis of Compound 15a and Compound 15b
実施例1-1:化合物15a及び化合物15bの合成 [Example 1 (first aspect): Synthesis of compound (1)]
Example 1-1: Synthesis of Compound 15a and Compound 15b

7 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このチューブに、合成例4-1で得た化合物14a及び化合物14bの混合物(8.1 mg, 13.6μmol, 1.0当量)、合成例2-5で得た2-(4-クロロフェニル)-4-(3,5-ジメトキシフェニル)-5-(m-トリル)-3-(p-トリル)チオフェン1-オキシド(化合物11; 14.3 mg, 27.1μmol, 2.0当量)、及びジクロロエタン(140μL)を窒素気流下に添加した。80℃に昇温し、このチューブに、t-ブチルニトリル(2.4μL, 20.4μmol, 1.5当量)のジクロロエタン(140μL)溶液を1時間ゆっくりと添加した。得られた混合物をさらに80℃で1時間加熱した。反応混合物を室温まで冷却した後、混合物に水(5 mL)及びCH2Cl2(5 mL)を添加した。混合物をCH2Cl2で抽出した後、有機層をNa2SO4で乾燥し、揮発性物質を減圧下に除去した。粗生成物を分取薄層クロマトグラフィー(ヘキサン/酢酸エチル= 10: 1)で精製し、化合物15a及び化合物15bの混合物(15a/ 15b = 2: 1)を白色固体として得た(3.0 mg, 22 %)
1-(4-(t-ブチル)フェニル)-5-(4-クロロフェニル)-7-(3,5-ジメトキシフェニル)-2-(4-メトキシフェニル)-3-フェニル-8-(m-トリル)-6-(p-トリル)-4-(4-(トリフルオロメチル)フェニル)ナフタレン(化合物15a):
1H NMR (600 MHz, CDCl3): δ 6.94-6.25 (m, 30H), 5.89 (t, J = 2.4 Hz, 1H), 5.80 (s, 1H), 5.75 (s, 1H), 3.55 (s, 3H) 3.36 (s, 3H), 3.35 (s, 3H), 2.07 (s, 3H), 1.87 (s, 3H), 1.15 (s, 9H)。 A magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, a mixture of Compound 14a and Compound 14b obtained in Synthesis Example 4-1 (8.1 mg, 13.6 μmol, 1.0 equivalent), 2- (4-chlorophenyl) -4- (3 , 5-Dimethoxyphenyl) -5- (m-tolyl) -3- (p-tolyl) thiophene 1-oxide (Compound 11; 14.3 mg, 27.1 μmol, 2.0 eq) and dichloroethane (140 μL) under nitrogen flow Added. The temperature was raised to 80 ° C., and a solution of t-butylnitrile (2.4 μL, 20.4 μmol, 1.5 equivalents) in dichloroethane (140 μL) was slowly added to this tube for 1 hour. The resulting mixture was further heated at 80 ° C. for 1 hour. After the reaction mixture was cooled to room temperature, water (5 mL) and CH 2 Cl 2 (5 mL) were added to the mixture. After the mixture was extracted with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was purified by preparative thin layer chromatography (hexane / ethyl acetate = 10: 1) to obtain a mixture of compound 15a and compound 15b (15a / 15b = 2: 1) as a white solid (3.0 mg, twenty two %)
1- (4- (t-butyl) phenyl) -5- (4-chlorophenyl) -7- (3,5-dimethoxyphenyl) -2- (4-methoxyphenyl) -3-phenyl-8- (m- Tolyl) -6- (p-tolyl) -4- (4- (trifluoromethyl) phenyl) naphthalene (compound 15a):
1 H NMR (600 MHz, CDCl 3 ): δ 6.94-6.25 (m, 30H), 5.89 (t, J = 2.4 Hz, 1H), 5.80 (s, 1H), 5.75 (s, 1H), 3.55 (s , 3H) 3.36 (s, 3H), 3.35 (s, 3H), 2.07 (s, 3H), 1.87 (s, 3H), 1.15 (s, 9H).
1-(4-(t-ブチル)フェニル)-5-(4-クロロフェニル)-7-(3,5-ジメトキシフェニル)-2-(4-メトキシフェニル)-3-フェニル-8-(m-トリル)-6-(p-トリル)-4-(4-(トリフルオロメチル)フェニル)ナフタレン(化合物15a):
1H NMR (600 MHz, CDCl3): δ 6.94-6.25 (m, 30H), 5.89 (t, J = 2.4 Hz, 1H), 5.80 (s, 1H), 5.75 (s, 1H), 3.55 (s, 3H) 3.36 (s, 3H), 3.35 (s, 3H), 2.07 (s, 3H), 1.87 (s, 3H), 1.15 (s, 9H)。 A magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, a mixture of Compound 14a and Compound 14b obtained in Synthesis Example 4-1 (8.1 mg, 13.6 μmol, 1.0 equivalent), 2- (4-chlorophenyl) -4- (3 , 5-Dimethoxyphenyl) -5- (m-tolyl) -3- (p-tolyl) thiophene 1-oxide (Compound 11; 14.3 mg, 27.1 μmol, 2.0 eq) and dichloroethane (140 μL) under nitrogen flow Added. The temperature was raised to 80 ° C., and a solution of t-butylnitrile (2.4 μL, 20.4 μmol, 1.5 equivalents) in dichloroethane (140 μL) was slowly added to this tube for 1 hour. The resulting mixture was further heated at 80 ° C. for 1 hour. After the reaction mixture was cooled to room temperature, water (5 mL) and CH 2 Cl 2 (5 mL) were added to the mixture. After the mixture was extracted with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was purified by preparative thin layer chromatography (hexane / ethyl acetate = 10: 1) to obtain a mixture of compound 15a and compound 15b (15a / 15b = 2: 1) as a white solid (3.0 mg, twenty two %)
1- (4- (t-butyl) phenyl) -5- (4-chlorophenyl) -7- (3,5-dimethoxyphenyl) -2- (4-methoxyphenyl) -3-phenyl-8- (m- Tolyl) -6- (p-tolyl) -4- (4- (trifluoromethyl) phenyl) naphthalene (compound 15a):
1 H NMR (600 MHz, CDCl 3 ): δ 6.94-6.25 (m, 30H), 5.89 (t, J = 2.4 Hz, 1H), 5.80 (s, 1H), 5.75 (s, 1H), 3.55 (s , 3H) 3.36 (s, 3H), 3.35 (s, 3H), 2.07 (s, 3H), 1.87 (s, 3H), 1.15 (s, 9H).
[実施例2(第1の態様):化合物(2)の合成]
実施例2-1:化合物18a及び化合物18bの合成 [Example 2 (first aspect): Synthesis of compound (2)]
Example 2-1: Synthesis of compound 18a and compound 18b
実施例2-1:化合物18a及び化合物18bの合成 [Example 2 (first aspect): Synthesis of compound (2)]
Example 2-1: Synthesis of compound 18a and compound 18b

25 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このチューブに、合成例5-2で得た化合物17a及び化合物17bの混合物(11.8 mg, 0.015 mmol, 1.0当量)、合成例2-5で得た2-(4-クロロフェニル)-4-(3,5-ジメトキシフェニル)-5-(m-トリル)-3-(p-トリル)チオフェン1-オキシド(化合物11; 23.1 mg, 0.44 mmol, 3.0当量)、及びTHF(150μL)を窒素気流下に添加した。0℃まで冷却し、混合物にテトラブチルアンモニウムフルオライド(TBAF; 66μL, 0.066 mmol, 4.5当量, 1M in THF)をゆっくりと添加した。反応混合物を0℃で3時間撹拌した。混合物に水(10 mL)及びCH2Cl2(10 mL)を添加した後、得られた混合物をCH2Cl2で抽出した。有機層をNa2SO4で乾燥し、揮発性物質を減圧下に除去した。粗生成物を分取薄層クロマトグラフィー(ヘキサン/酢酸エチル= 20: 1)で精製し、化合物18a(2.3 mg)及び化合物18b(2.2 mg)を黄色固体として単離することができた(29 %)。低極性化合物が化合物18aであり、高極性化合物が化合物18bである。化合物18aの構造は、X線結晶構造解析により決定した。
1-(4-(t-ブチル)フェニル)-5-(4-クロロフェニル)-7-(3,5-ジメトキシフェニル)-2-(4-メトキシフェニル)-3-フェニル-8-(m-トリル)-6-(p-トリル)-4-(4-(トリフルオロメチル)フェニル)アントラセン(化合物18a):
1H NMR (600 MHz, CDCl3): δ 8.02 (s, 1H), 7.71 (s, 1H), 7.34-6.65 (m, 27H), 6.41-6.38 (m, 2H), 6.01-5.99 (m, 2H), 5.97 (t, J = 2.4 Hz, 1H), 3.60 (s, 3H), 3.43 (s, 6H), 2.15 (s, 3H), 2.12 (s, 3H), 1.28 (s, 9H)。
1-(4-(t-ブチル)フェニル)-8-(4-クロロフェニル)-6-(3,5-ジメトキシフェニル)-2-(4-メトキシフェニル)-3-フェニル-5-(m-トリル)-7-(p-トリル)-4-(4-(トリフルオロメチル)フェニル)アントラセン(化合物18b):
1H NMR (600 MHz, CDCl3): δ 8.05 (s, 1H), 7.65 (s, 1H), 7.34-6.65 (m, 27H), 6.41-6.37 (m, 2H), 6.04-6.01 (m, 2H), 5.97 (t, J = 2.4 Hz, 1H), 3.60 (s, 3H), 3.43 (s, 6H), 2.15 (s, 3H), 2.12 (s, 3H), 1.28 (s, 9H)。 A magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, a mixture of compound 17a and compound 17b obtained in Synthesis Example 5-2 (11.8 mg, 0.015 mmol, 1.0 equivalent), 2- (4-chlorophenyl) -4- (3 , 5-Dimethoxyphenyl) -5- (m-tolyl) -3- (p-tolyl) thiophene 1-oxide (compound 11; 23.1 mg, 0.44 mmol, 3.0 eq) and THF (150 μL) under nitrogen flow Added. After cooling to 0 ° C., tetrabutylammonium fluoride (TBAF; 66 μL, 0.066 mmol, 4.5 eq, 1M in THF) was slowly added to the mixture. The reaction mixture was stirred at 0 ° C. for 3 hours. After adding water (10 mL) and CH 2 Cl 2 (10 mL) to the mixture, the resulting mixture was extracted with CH 2 Cl 2 . The organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was purified by preparative thin layer chromatography (hexane / ethyl acetate = 20: 1) to isolate compound 18a (2.3 mg) and compound 18b (2.2 mg) as a yellow solid (29 %). The low polarity compound is Compound 18a and the high polarity compound is Compound 18b. The structure of compound 18a was determined by X-ray crystal structure analysis.
1- (4- (t-butyl) phenyl) -5- (4-chlorophenyl) -7- (3,5-dimethoxyphenyl) -2- (4-methoxyphenyl) -3-phenyl-8- (m- Tolyl) -6- (p-tolyl) -4- (4- (trifluoromethyl) phenyl) anthracene (compound 18a):
1 H NMR (600 MHz, CDCl 3 ): δ 8.02 (s, 1H), 7.71 (s, 1H), 7.34-6.65 (m, 27H), 6.41-6.38 (m, 2H), 6.01-5.99 (m, 2H), 5.97 (t, J = 2.4 Hz, 1H), 3.60 (s, 3H), 3.43 (s, 6H), 2.15 (s, 3H), 2.12 (s, 3H), 1.28 (s, 9H).
1- (4- (t-butyl) phenyl) -8- (4-chlorophenyl) -6- (3,5-dimethoxyphenyl) -2- (4-methoxyphenyl) -3-phenyl-5- (m- Tolyl) -7- (p-tolyl) -4- (4- (trifluoromethyl) phenyl) anthracene (compound 18b):
1 H NMR (600 MHz, CDCl 3 ): δ 8.05 (s, 1H), 7.65 (s, 1H), 7.34-6.65 (m, 27H), 6.41-6.37 (m, 2H), 6.04-6.01 (m, 2H), 5.97 (t, J = 2.4 Hz, 1H), 3.60 (s, 3H), 3.43 (s, 6H), 2.15 (s, 3H), 2.12 (s, 3H), 1.28 (s, 9H).
1-(4-(t-ブチル)フェニル)-5-(4-クロロフェニル)-7-(3,5-ジメトキシフェニル)-2-(4-メトキシフェニル)-3-フェニル-8-(m-トリル)-6-(p-トリル)-4-(4-(トリフルオロメチル)フェニル)アントラセン(化合物18a):
1H NMR (600 MHz, CDCl3): δ 8.02 (s, 1H), 7.71 (s, 1H), 7.34-6.65 (m, 27H), 6.41-6.38 (m, 2H), 6.01-5.99 (m, 2H), 5.97 (t, J = 2.4 Hz, 1H), 3.60 (s, 3H), 3.43 (s, 6H), 2.15 (s, 3H), 2.12 (s, 3H), 1.28 (s, 9H)。
1-(4-(t-ブチル)フェニル)-8-(4-クロロフェニル)-6-(3,5-ジメトキシフェニル)-2-(4-メトキシフェニル)-3-フェニル-5-(m-トリル)-7-(p-トリル)-4-(4-(トリフルオロメチル)フェニル)アントラセン(化合物18b):
1H NMR (600 MHz, CDCl3): δ 8.05 (s, 1H), 7.65 (s, 1H), 7.34-6.65 (m, 27H), 6.41-6.37 (m, 2H), 6.04-6.01 (m, 2H), 5.97 (t, J = 2.4 Hz, 1H), 3.60 (s, 3H), 3.43 (s, 6H), 2.15 (s, 3H), 2.12 (s, 3H), 1.28 (s, 9H)。 A magnetic stir bar was placed in a 25 mL screw cap tube, flame dried under vacuum, cooled to room temperature, and filled with nitrogen. To this tube, a mixture of compound 17a and compound 17b obtained in Synthesis Example 5-2 (11.8 mg, 0.015 mmol, 1.0 equivalent), 2- (4-chlorophenyl) -4- (3 , 5-Dimethoxyphenyl) -5- (m-tolyl) -3- (p-tolyl) thiophene 1-oxide (compound 11; 23.1 mg, 0.44 mmol, 3.0 eq) and THF (150 μL) under nitrogen flow Added. After cooling to 0 ° C., tetrabutylammonium fluoride (TBAF; 66 μL, 0.066 mmol, 4.5 eq, 1M in THF) was slowly added to the mixture. The reaction mixture was stirred at 0 ° C. for 3 hours. After adding water (10 mL) and CH 2 Cl 2 (10 mL) to the mixture, the resulting mixture was extracted with CH 2 Cl 2 . The organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was purified by preparative thin layer chromatography (hexane / ethyl acetate = 20: 1) to isolate compound 18a (2.3 mg) and compound 18b (2.2 mg) as a yellow solid (29 %). The low polarity compound is Compound 18a and the high polarity compound is Compound 18b. The structure of compound 18a was determined by X-ray crystal structure analysis.
1- (4- (t-butyl) phenyl) -5- (4-chlorophenyl) -7- (3,5-dimethoxyphenyl) -2- (4-methoxyphenyl) -3-phenyl-8- (m- Tolyl) -6- (p-tolyl) -4- (4- (trifluoromethyl) phenyl) anthracene (compound 18a):
1 H NMR (600 MHz, CDCl 3 ): δ 8.02 (s, 1H), 7.71 (s, 1H), 7.34-6.65 (m, 27H), 6.41-6.38 (m, 2H), 6.01-5.99 (m, 2H), 5.97 (t, J = 2.4 Hz, 1H), 3.60 (s, 3H), 3.43 (s, 6H), 2.15 (s, 3H), 2.12 (s, 3H), 1.28 (s, 9H).
1- (4- (t-butyl) phenyl) -8- (4-chlorophenyl) -6- (3,5-dimethoxyphenyl) -2- (4-methoxyphenyl) -3-phenyl-5- (m- Tolyl) -7- (p-tolyl) -4- (4- (trifluoromethyl) phenyl) anthracene (compound 18b):
1 H NMR (600 MHz, CDCl 3 ): δ 8.05 (s, 1H), 7.65 (s, 1H), 7.34-6.65 (m, 27H), 6.41-6.37 (m, 2H), 6.04-6.01 (m, 2H), 5.97 (t, J = 2.4 Hz, 1H), 3.60 (s, 3H), 3.43 (s, 6H), 2.15 (s, 3H), 2.12 (s, 3H), 1.28 (s, 9H).
[実施例3(第2の態様):化合物(31A)の合成]
実施例3-1:多置換芳香族化合物29a及び多置換芳香族化合物29bの合成 [Example 3 (second aspect): Synthesis of compound (31A)]
Example 3-1: Synthesis of polysubstituted aromatic compound 29a and polysubstituted aromatic compound 29b
実施例3-1:多置換芳香族化合物29a及び多置換芳香族化合物29bの合成 [Example 3 (second aspect): Synthesis of compound (31A)]
Example 3-1: Synthesis of polysubstituted aromatic compound 29a and polysubstituted aromatic compound 29b

[式中、t-Buはt-ブチル基を示す。以下同様である。]
7 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このスクリューキャップチューブに、合成例7-3で得た四置換チオフェンS-オキシド化合物26acdf(25.5 mg, 0.045 mmol, 1.0当量)、合成例11-1で得た化合物27a(19.9 mg, 0.090 mmol, 2.0当量)、及びメシチレン(300μL)を窒素気流下に添加した。スクリューキャップチューブを160℃で48時間加熱した。反応混合物を室温まで冷却した後、混合物を真空下に濃縮し、粗生成物を分取薄層クロマトグラフィー(ヘキサン/酢酸エチル=2: 1及びヘキサン/CHCl3=1: 2)により精製し、多置換芳香族化合物29aと多置換芳香族化合物29bとの混合物を白色固体として得た(10.5 mg, 32 % (混合物), 29a/29b = 5:4)。多置換芳香族化合物29aと多置換芳香族化合物29bとの混合物の再結晶を6回繰り返した後、多置換芳香族化合物29aを単独の異性体として得た。
1H NMR (600 MHz, CDCl3): δ 8.08 (dd, J = 4.2, 1.8 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H), 6.94-6.87 (m, 9H), 6.79-6.76 (m, 2H), 6.72 (d, J = 6.0 Hz, 1H), 6.69-6.66 (m, 4H), 6.41 (d, J = 8.4 Hz, 2H), 3.61 (s, 3H), 2.44 (s, 3H), 1.13 (s, 9H); 13C NMR (150 MHz, CDCl3): δ 197.6, 157.3, 148.9, 148.7, 148.3, 144.8, 143.8, 141.5, 141.2, 141.1, 139.6, 138.8, 138.2, 137.7, 136.2, 134.6, 132.2, 132.0, 131.4, 131.3, 131.1, 130.7, 127.7 (q, 2JCF = 31.7 Hz), 127.2, 127.0, 126.2, 125.8, 124.0 (q, 1JCF = 270 Hz), 123.9, 123.8 (q, 3JCF = 2.85 Hz), 112.3, 55.0, 34.2, 31.1, 26.4; HRMS (APCI) m/z calcd for C49H41F3NO2 [MH]+: 732.30839, found 732.30566。 [Wherein t-Bu represents a t-butyl group. The same applies hereinafter. ]
A magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this screw cap tube, tetrasubstituted thiophene S-oxide compound 26acdf obtained in Synthesis Example 7-3 (25.5 mg, 0.045 mmol, 1.0 equivalent), compound 27a obtained in Synthesis Example 11-1 (19.9 mg, 0.090 mmol, 2.0 equivalents) and mesitylene (300 μL) were added under a stream of nitrogen. The screw cap tube was heated at 160 ° C. for 48 hours. After cooling the reaction mixture to room temperature, the mixture is concentrated in vacuo and the crude product is purified by preparative thin layer chromatography (hexane / ethyl acetate = 2: 1 and hexane / CHCl 3 = 1: 2), A mixture of the polysubstituted aromatic compound 29a and the polysubstituted aromatic compound 29b was obtained as a white solid (10.5 mg, 32% (mixture), 29a / 29b = 5: 4). After recrystallization of the mixture of the polysubstituted aromatic compound 29a and the polysubstituted aromatic compound 29b was repeated 6 times, the polysubstituted aromatic compound 29a was obtained as a single isomer.
1 H NMR (600 MHz, CDCl 3 ): δ 8.08 (dd, J = 4.2, 1.8 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H), 6.94-6.87 (m, 9H), 6.79-6.76 (m, 2H), 6.72 (d, J = 6.0 Hz, 1H), 6.69-6.66 (m, 4H), 6.41 (d, J = 8.4 Hz, 2H) , 3.61 (s, 3H), 2.44 (s, 3H), 1.13 (s, 9H); 13 C NMR (150 MHz, CDCl 3 ): δ 197.6, 157.3, 148.9, 148.7, 148.3, 144.8, 143.8, 141.5, 141.2, 141.1, 139.6, 138.8, 138.2, 137.7, 136.2, 134.6, 132.2, 132.0, 131.4, 131.3, 131.1, 130.7, 127.7 (q, 2 J CF = 31.7 Hz), 127.2, 127.0, 126.2, 125.8, 124.0 ( q, 1 J CF = 270 Hz), 123.9, 123.8 (q, 3 J CF = 2.85 Hz), 112.3, 55.0, 34.2, 31.1, 26.4; HRMS (APCI) m / z calcd for C 49 H 41 F 3 NO 2 [MH] + : 732.30839, found 732.30566.
7 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このスクリューキャップチューブに、合成例7-3で得た四置換チオフェンS-オキシド化合物26acdf(25.5 mg, 0.045 mmol, 1.0当量)、合成例11-1で得た化合物27a(19.9 mg, 0.090 mmol, 2.0当量)、及びメシチレン(300μL)を窒素気流下に添加した。スクリューキャップチューブを160℃で48時間加熱した。反応混合物を室温まで冷却した後、混合物を真空下に濃縮し、粗生成物を分取薄層クロマトグラフィー(ヘキサン/酢酸エチル=2: 1及びヘキサン/CHCl3=1: 2)により精製し、多置換芳香族化合物29aと多置換芳香族化合物29bとの混合物を白色固体として得た(10.5 mg, 32 % (混合物), 29a/29b = 5:4)。多置換芳香族化合物29aと多置換芳香族化合物29bとの混合物の再結晶を6回繰り返した後、多置換芳香族化合物29aを単独の異性体として得た。
1H NMR (600 MHz, CDCl3): δ 8.08 (dd, J = 4.2, 1.8 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H), 6.94-6.87 (m, 9H), 6.79-6.76 (m, 2H), 6.72 (d, J = 6.0 Hz, 1H), 6.69-6.66 (m, 4H), 6.41 (d, J = 8.4 Hz, 2H), 3.61 (s, 3H), 2.44 (s, 3H), 1.13 (s, 9H); 13C NMR (150 MHz, CDCl3): δ 197.6, 157.3, 148.9, 148.7, 148.3, 144.8, 143.8, 141.5, 141.2, 141.1, 139.6, 138.8, 138.2, 137.7, 136.2, 134.6, 132.2, 132.0, 131.4, 131.3, 131.1, 130.7, 127.7 (q, 2JCF = 31.7 Hz), 127.2, 127.0, 126.2, 125.8, 124.0 (q, 1JCF = 270 Hz), 123.9, 123.8 (q, 3JCF = 2.85 Hz), 112.3, 55.0, 34.2, 31.1, 26.4; HRMS (APCI) m/z calcd for C49H41F3NO2 [MH]+: 732.30839, found 732.30566。 [Wherein t-Bu represents a t-butyl group. The same applies hereinafter. ]
A magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this screw cap tube, tetrasubstituted thiophene S-oxide compound 26acdf obtained in Synthesis Example 7-3 (25.5 mg, 0.045 mmol, 1.0 equivalent), compound 27a obtained in Synthesis Example 11-1 (19.9 mg, 0.090 mmol, 2.0 equivalents) and mesitylene (300 μL) were added under a stream of nitrogen. The screw cap tube was heated at 160 ° C. for 48 hours. After cooling the reaction mixture to room temperature, the mixture is concentrated in vacuo and the crude product is purified by preparative thin layer chromatography (hexane / ethyl acetate = 2: 1 and hexane / CHCl 3 = 1: 2), A mixture of the polysubstituted aromatic compound 29a and the polysubstituted aromatic compound 29b was obtained as a white solid (10.5 mg, 32% (mixture), 29a / 29b = 5: 4). After recrystallization of the mixture of the polysubstituted aromatic compound 29a and the polysubstituted aromatic compound 29b was repeated 6 times, the polysubstituted aromatic compound 29a was obtained as a single isomer.
1 H NMR (600 MHz, CDCl 3 ): δ 8.08 (dd, J = 4.2, 1.8 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H), 6.94-6.87 (m, 9H), 6.79-6.76 (m, 2H), 6.72 (d, J = 6.0 Hz, 1H), 6.69-6.66 (m, 4H), 6.41 (d, J = 8.4 Hz, 2H) , 3.61 (s, 3H), 2.44 (s, 3H), 1.13 (s, 9H); 13 C NMR (150 MHz, CDCl 3 ): δ 197.6, 157.3, 148.9, 148.7, 148.3, 144.8, 143.8, 141.5, 141.2, 141.1, 139.6, 138.8, 138.2, 137.7, 136.2, 134.6, 132.2, 132.0, 131.4, 131.3, 131.1, 130.7, 127.7 (q, 2 J CF = 31.7 Hz), 127.2, 127.0, 126.2, 125.8, 124.0 ( q, 1 J CF = 270 Hz), 123.9, 123.8 (q, 3 J CF = 2.85 Hz), 112.3, 55.0, 34.2, 31.1, 26.4; HRMS (APCI) m / z calcd for C 49 H 41 F 3 NO 2 [MH] + : 732.30839, found 732.30566.
実施例3-2:多置換芳香族化合物30a及び多置換芳香族化合物30bの合成
Example 3-2: Synthesis of polysubstituted aromatic compound 30a and polysubstituted aromatic compound 30b

7 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このスクリューキャップチューブに、実施例3-1で得た化合物29aと化合物29bとの混合物(21.9 mg, 0.03 mmol, 1.0当量)、及びメタノール(1.2 mL)を窒素気流下に添加した。内容物を0℃まで冷却し、水素化ホウ素ナトリウムのメタノール溶液(300μL, 0.3 M, 0.09 mmol)をゆっくりと添加した。混合物を0.5時間撹拌した後、反応をNaHCO3水溶液でクエンチした。混合物をCH2Cl2で抽出し、Na2SO4で乾燥し、減圧下に濃縮した。得られた混合物はさらに精製せずに次の工程に使用した。
A magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. A mixture (21.9 mg, 0.03 mmol, 1.0 equivalent) of the compound 29a and compound 29b obtained in Example 3-1 and methanol (1.2 mL) were added to the screw cap tube under a nitrogen stream. The contents were cooled to 0 ° C. and a solution of sodium borohydride in methanol (300 μL, 0.3 M, 0.09 mmol) was added slowly. After the mixture was stirred for 0.5 h, the reaction was quenched with aqueous NaHCO 3 solution. The mixture was extracted with CH 2 Cl 2 , dried over Na 2 SO 4 and concentrated under reduced pressure. The resulting mixture was used in the next step without further purification.
7 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このスクリューキャップチューブに、上記工程で得た物質、及び乾燥CH2Cl2(600μL)を窒素気流下に添加した。内容物を0℃まで冷却し、このスクリューキャップチューブに2,6-ルチジン(17.5μL, 0.15 mmol)、及びt-ブチルジメチルシリルトリフルオロメタンスルホネート(TBSOTf: 34.5μL, 0.15 mmol)をゆっくりと添加した。混合物を0.5時間撹拌した後、反応をメタノール(3 mL)でクエンチし、混合物を減圧下に濃縮した。粗生成物を分取薄層クロマトグラフィー(ヘキサン/酢酸エチル=10: 1)により精製し、多置換芳香族化合物30a(9.0 mg)と多置換芳香族化合物30b(8.8 mg)とを白色固体として得た(70 %)。より極性の高い化合物が多置換芳香族化合物30aであり、より極性の低い化合物が多置換芳香族化合物30bである。多置換芳香族化合物30aの構造はX線結晶構造解析で決定した。なお、多置換芳香族化合物30aのX線結晶構造を図1に示す。
多置換芳香族化合物30a:
1H NMR (600 MHz, CDCl3): δ 8.05 (d, J = 6.0 Hz, 2H), 7.08 (d, J = 9.0 Hz, 2H), 6.92-6.86 (m, 8H), 6.80-6.77 (m, 3H), 6.73-6.67 (m, 8H), 6.40 (d, J = 9.0 Hz, 2H), 4.58 (q, J = 6.0 Hz, 1H), 3.60 (s, 3H), 1.19 (d, J = 6.0 Hz, 3H), 1.13 (s, 9H), 0.80 (s, 9H), -0.12 (s, 3H), -0.25 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 157.2, 149.2, 148.7, 147.9, 144.9, 144.3, 141.1, 140.8, 140.5, 140.0, 139.5, 139.2, 137.9, 137.8, 136.5, 132.3, 132.2, 131.6, 131.5, 131.2, 130.9, 130.8, 130.7, 127.3 (q, 2JCF = 33.0 Hz), 126.9, 126.5, 125.6, 124.2, 124.1 (q, 1JCF = 271 Hz), 124.0, 123.8, 123.5, 112.2, 70.6, 55.0, 34.2, 31.2, 27.0, 25.8, 18.2, -4.79, -5.16 (3JCF: not detected); HRMS (APCI) m/z calcd for C55H57F3NO2Si [MH]+: 848.41052 found 848.40628。
多置換芳香族化合物30b:
1H NMR (600 MHz, CDCl3): δ 8.07 (d, J = 5.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.4 Hz, 2H), 6.91-6.76 (m, 9H), 6.73-6.64 (m, 8H), 6.40 (d, J = 8.4 Hz, 2H), 4.59 (q, J = 6.0 Hz, 1H), 3.61 (s, 3H), 1.17 (d, J = 6.0 Hz, 3H), 1.10 (s, 9H), 0.81 (s, 9H), -0.11 (s, 3H), -0.22 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 157.2, 149.0, 148.13, 148.08, 144.5, 144.0, 141.8, 141.1, 140.5, 140.3, 139.8, 138.11, 138.06, 137.1, 136.8, 132.4, 132.3, 131.5, 131.2, 130.84, 130.79, 127.8 (q, 2JCF = 33.0 Hz), 127.0, 126.4, 125.6, 124.0 (q, 1JCF = 270 Hz), 123.84 (3JCF: not detected), 123.78, 123.5, 123.4, 112.20, 112.17, 70.7, 55.0, 34.1, 31.1, 27.1, 25.8, 18.2, -4.6, -5.2; HRMS (APCI) m/z calcd for C55H57F3NO2Si [MH]+: 848.41052 found 848.40868。 A magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To the screw cap tube, the material obtained in the above step and dry CH 2 Cl 2 (600 μL) were added under a nitrogen stream. The contents were cooled to 0 ° C. and 2,6-lutidine (17.5 μL, 0.15 mmol) and t-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf: 34.5 μL, 0.15 mmol) were slowly added to the screw cap tube. . After the mixture was stirred for 0.5 h, the reaction was quenched with methanol (3 mL) and the mixture was concentrated under reduced pressure. The crude product was purified by preparative thin layer chromatography (hexane / ethyl acetate = 10: 1) to give polysubstituted aromatic compound 30a (9.0 mg) and polysubstituted aromatic compound 30b (8.8 mg) as a white solid. Obtained (70%). The more polar compound is the polysubstituted aromatic compound 30a, and the less polar compound is the polysubstituted aromatic compound 30b. The structure of the polysubstituted aromatic compound 30a was determined by X-ray crystal structure analysis. The X-ray crystal structure of the polysubstituted aromatic compound 30a is shown in FIG.
Polysubstituted aromatic compound 30a:
1 H NMR (600 MHz, CDCl 3 ): δ 8.05 (d, J = 6.0 Hz, 2H), 7.08 (d, J = 9.0 Hz, 2H), 6.92-6.86 (m, 8H), 6.80-6.77 (m , 3H), 6.73-6.67 (m, 8H), 6.40 (d, J = 9.0 Hz, 2H), 4.58 (q, J = 6.0 Hz, 1H), 3.60 (s, 3H), 1.19 (d, J = 6.0 Hz, 3H), 1.13 (s, 9H), 0.80 (s, 9H), -0.12 (s, 3H), -0.25 (s, 3H); 13 C NMR (150 MHz, CDCl 3 ): δ 157.2, 149.2, 148.7, 147.9, 144.9, 144.3, 141.1, 140.8, 140.5, 140.0, 139.5, 139.2, 137.9, 137.8, 136.5, 132.3, 132.2, 131.6, 131.5, 131.2, 130.9, 130.8, 130.7, 127.3 (q, 2 J CF = 33.0 Hz), 126.9, 126.5, 125.6, 124.2, 124.1 (q, 1 J CF = 271 Hz), 124.0, 123.8, 123.5, 112.2, 70.6, 55.0, 34.2, 31.2, 27.0, 25.8, 18.2, -4.79 , -5.16 ( 3 J CF : not detected); HRMS (APCI) m / z calcd for C 55 H 57 F 3 NO 2 Si [MH] + : 848.41052 found 848.40628.
Polysubstituted aromatic compound 30b:
1 H NMR (600 MHz, CDCl 3 ): δ 8.07 (d, J = 5.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.4 Hz, 2H), 6.91- 6.76 (m, 9H), 6.73-6.64 (m, 8H), 6.40 (d, J = 8.4 Hz, 2H), 4.59 (q, J = 6.0 Hz, 1H), 3.61 (s, 3H), 1.17 (d , J = 6.0 Hz, 3H), 1.10 (s, 9H), 0.81 (s, 9H), -0.11 (s, 3H), -0.22 (s, 3H); 13 C NMR (150 MHz, CDCl 3 ): δ 157.2, 149.0, 148.13, 148.08, 144.5, 144.0, 141.8, 141.1, 140.5, 140.3, 139.8, 138.11, 138.06, 137.1, 136.8, 132.4, 132.3, 131.5, 131.2, 130.84, 130.79, 127.8 (q, 2 J CF = 33.0 Hz), 127.0, 126.4, 125.6, 124.0 (q, 1 J CF = 270 Hz), 123.84 ( 3 J CF : not detected), 123.78, 123.5, 123.4, 112.20, 112.17, 70.7, 55.0, 34.1, 31.1 , 27.1, 25.8, 18.2, -4.6 , -5.2; HRMS (APCI) m / z calcd for C 55 H 57 F 3 NO 2 Si [MH] +: 848.41052 found 848.40868.
多置換芳香族化合物30a:
1H NMR (600 MHz, CDCl3): δ 8.05 (d, J = 6.0 Hz, 2H), 7.08 (d, J = 9.0 Hz, 2H), 6.92-6.86 (m, 8H), 6.80-6.77 (m, 3H), 6.73-6.67 (m, 8H), 6.40 (d, J = 9.0 Hz, 2H), 4.58 (q, J = 6.0 Hz, 1H), 3.60 (s, 3H), 1.19 (d, J = 6.0 Hz, 3H), 1.13 (s, 9H), 0.80 (s, 9H), -0.12 (s, 3H), -0.25 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 157.2, 149.2, 148.7, 147.9, 144.9, 144.3, 141.1, 140.8, 140.5, 140.0, 139.5, 139.2, 137.9, 137.8, 136.5, 132.3, 132.2, 131.6, 131.5, 131.2, 130.9, 130.8, 130.7, 127.3 (q, 2JCF = 33.0 Hz), 126.9, 126.5, 125.6, 124.2, 124.1 (q, 1JCF = 271 Hz), 124.0, 123.8, 123.5, 112.2, 70.6, 55.0, 34.2, 31.2, 27.0, 25.8, 18.2, -4.79, -5.16 (3JCF: not detected); HRMS (APCI) m/z calcd for C55H57F3NO2Si [MH]+: 848.41052 found 848.40628。
多置換芳香族化合物30b:
1H NMR (600 MHz, CDCl3): δ 8.07 (d, J = 5.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.4 Hz, 2H), 6.91-6.76 (m, 9H), 6.73-6.64 (m, 8H), 6.40 (d, J = 8.4 Hz, 2H), 4.59 (q, J = 6.0 Hz, 1H), 3.61 (s, 3H), 1.17 (d, J = 6.0 Hz, 3H), 1.10 (s, 9H), 0.81 (s, 9H), -0.11 (s, 3H), -0.22 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 157.2, 149.0, 148.13, 148.08, 144.5, 144.0, 141.8, 141.1, 140.5, 140.3, 139.8, 138.11, 138.06, 137.1, 136.8, 132.4, 132.3, 131.5, 131.2, 130.84, 130.79, 127.8 (q, 2JCF = 33.0 Hz), 127.0, 126.4, 125.6, 124.0 (q, 1JCF = 270 Hz), 123.84 (3JCF: not detected), 123.78, 123.5, 123.4, 112.20, 112.17, 70.7, 55.0, 34.1, 31.1, 27.1, 25.8, 18.2, -4.6, -5.2; HRMS (APCI) m/z calcd for C55H57F3NO2Si [MH]+: 848.41052 found 848.40868。 A magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To the screw cap tube, the material obtained in the above step and dry CH 2 Cl 2 (600 μL) were added under a nitrogen stream. The contents were cooled to 0 ° C. and 2,6-lutidine (17.5 μL, 0.15 mmol) and t-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf: 34.5 μL, 0.15 mmol) were slowly added to the screw cap tube. . After the mixture was stirred for 0.5 h, the reaction was quenched with methanol (3 mL) and the mixture was concentrated under reduced pressure. The crude product was purified by preparative thin layer chromatography (hexane / ethyl acetate = 10: 1) to give polysubstituted aromatic compound 30a (9.0 mg) and polysubstituted aromatic compound 30b (8.8 mg) as a white solid. Obtained (70%). The more polar compound is the polysubstituted aromatic compound 30a, and the less polar compound is the polysubstituted aromatic compound 30b. The structure of the polysubstituted aromatic compound 30a was determined by X-ray crystal structure analysis. The X-ray crystal structure of the polysubstituted aromatic compound 30a is shown in FIG.
Polysubstituted aromatic compound 30a:
1 H NMR (600 MHz, CDCl 3 ): δ 8.05 (d, J = 6.0 Hz, 2H), 7.08 (d, J = 9.0 Hz, 2H), 6.92-6.86 (m, 8H), 6.80-6.77 (m , 3H), 6.73-6.67 (m, 8H), 6.40 (d, J = 9.0 Hz, 2H), 4.58 (q, J = 6.0 Hz, 1H), 3.60 (s, 3H), 1.19 (d, J = 6.0 Hz, 3H), 1.13 (s, 9H), 0.80 (s, 9H), -0.12 (s, 3H), -0.25 (s, 3H); 13 C NMR (150 MHz, CDCl 3 ): δ 157.2, 149.2, 148.7, 147.9, 144.9, 144.3, 141.1, 140.8, 140.5, 140.0, 139.5, 139.2, 137.9, 137.8, 136.5, 132.3, 132.2, 131.6, 131.5, 131.2, 130.9, 130.8, 130.7, 127.3 (q, 2 J CF = 33.0 Hz), 126.9, 126.5, 125.6, 124.2, 124.1 (q, 1 J CF = 271 Hz), 124.0, 123.8, 123.5, 112.2, 70.6, 55.0, 34.2, 31.2, 27.0, 25.8, 18.2, -4.79 , -5.16 ( 3 J CF : not detected); HRMS (APCI) m / z calcd for C 55 H 57 F 3 NO 2 Si [MH] + : 848.41052 found 848.40628.
Polysubstituted aromatic compound 30b:
1 H NMR (600 MHz, CDCl 3 ): δ 8.07 (d, J = 5.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.4 Hz, 2H), 6.91- 6.76 (m, 9H), 6.73-6.64 (m, 8H), 6.40 (d, J = 8.4 Hz, 2H), 4.59 (q, J = 6.0 Hz, 1H), 3.61 (s, 3H), 1.17 (d , J = 6.0 Hz, 3H), 1.10 (s, 9H), 0.81 (s, 9H), -0.11 (s, 3H), -0.22 (s, 3H); 13 C NMR (150 MHz, CDCl 3 ): δ 157.2, 149.0, 148.13, 148.08, 144.5, 144.0, 141.8, 141.1, 140.5, 140.3, 139.8, 138.11, 138.06, 137.1, 136.8, 132.4, 132.3, 131.5, 131.2, 130.84, 130.79, 127.8 (q, 2 J CF = 33.0 Hz), 127.0, 126.4, 125.6, 124.0 (q, 1 J CF = 270 Hz), 123.84 ( 3 J CF : not detected), 123.78, 123.5, 123.4, 112.20, 112.17, 70.7, 55.0, 34.1, 31.1 , 27.1, 25.8, 18.2, -4.6 , -5.2; HRMS (APCI) m / z calcd for C 55 H 57 F 3 NO 2 Si [MH] +: 848.41052 found 848.40868.
実施例3-3:多置換芳香族化合物31a及び多置換芳香族化合物31bの合成
Example 3-3: Synthesis of polysubstituted aromatic compound 31a and polysubstituted aromatic compound 31b

合成例7-3で得た四置換チオフェンS-オキシド化合物6acdfの代わりに合成例7-2で得た四置換チオフェンS-オキシド化合物26acde(15.6 mg, 0.028 mmol)を使用し、合成例11-1で得た化合物27aの代わりに合成例11-2で得た化合物27bを使用し、メシチレンの量を190μLとし、精製処理を分取薄層クロマトグラフィー(ヘキサン/酢酸エチル=3: 1)としたこと以外は実施例3-1と同様に合成し、多置換芳香族化合物31a及び多置換芳香族化合物31bを混合物として得た(6.8 mg, 31 %)。
Instead of the tetrasubstituted thiophene S-oxide compound 6acdf obtained in Synthesis Example 7-3, the tetrasubstituted thiophene S-oxide compound 26acde (15.6 mg, 10.028 mmol) obtained in Synthesis Example 7-2 was used, and Synthesis Example 11- The compound 27b obtained in Synthesis Example 11-2 was used in place of the compound 27a obtained in 1, and the amount of mesitylene was 190 μL, and the purification treatment was performed by preparative thin layer chromatography (hexane / ethyl acetate = 3: 1). Except that, the synthesis was performed in the same manner as in Example 3-1, to obtain a polysubstituted aromatic compound 31a and a polysubstituted aromatic compound 31b as a mixture (6.8 mg, 31%).
[実施例4(第2の態様):化合物(31B)の合成]
実施例4-1:多置換芳香族化合物32の合成 [Example 4 (second aspect): Synthesis of compound (31B)]
Example 4-1: Synthesis of polysubstituted aromatic compound 32
実施例4-1:多置換芳香族化合物32の合成 [Example 4 (second aspect): Synthesis of compound (31B)]
Example 4-1: Synthesis of polysubstituted aromatic compound 32

20 mLのシュレンクフラスコに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。この容器に、合成例7-3で得た四置換チオフェンS-オキシド化合物26acdf(25.5 mg, 0.045 mmol, 1.0当量)、2-(トリメチルシリル)フェニルトリフルオロメタンスルホネート(32.7μL, 0.135 mmol, 3.0当量)、及びTHF(450μL)を窒素気流下に添加した。内容物を0℃まで冷却し、このフラスコにテトラブチルアンモニウムフルオライドのTHF溶液(200μL, 1.0 M, 0.2 mmol)を添加した。得られた混合物を室温まで昇温し、1時間撹拌した後、混合物に水(1 mL)を添加した。CH2Cl2で抽出した後、有機層をNa2SO4で乾燥させ、揮発性物質を減圧下に除去した。粗生成物を分取薄層クロマトグラフィー(ヘキサン/CH2Cl2=3: 2)により精製し、多置換芳香族化合物32を白色固体として得た(22.5 mg, 85 %)。
1H NMR (600 MHz, CDCl3): δ 7.72-7.70 (m, 1H), 7.52-7.48 (m, 2H), 7.42-7.38 (m, 2H), 7.33 (d, J = 7.8 Hz, 2H), 7.26 (dd, J = 6.6, 2.4 Hz, 2H, overlapping with the peak of CHCl3), 7.10 (dd, J = 6.6, 1.8 Hz, 2H), 6.90-6.84 (m, 3H), 6.82-6.78 (m, 2H), 6.70 (dd, J = 6.6, 2.4 Hz, 2H), 6.40 (dd, J = 6.9, 2.4 Hz, 2H), 3.60 (s, 3H), 1.30 (s, 9H); 13C NMR (150 MHz, CDCl3): δ 157.1, 149.2, 143.8, 140.2, 139.4, 139.2, 138.5, 136.6, 136.3, 132.7, 132.3, 132.2, 131.6, 131.5, 131.2, 130.8, 128.5 (q, 2JCF = 33.2 Hz), 127.3, 126.8, 126.3, 126.0, 125.9, 125.6, 124.5 (q, 3JCF = 2.9 Hz), 124.4, 124.3 (q, 1JCF = 273 Hz), 112.1, 55.0, 34.4, 31.3; HRMS (APCI) m/z calcd for C40H34F3O [MH]+: 587.25563, found 587.25334。 A 20 mL Schlenk flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen. In this container, the tetrasubstituted thiophene S-oxide compound 26acdf obtained in Synthesis Example 7-3 (25.5 mg, 0.045 mmol, 1.0 equivalent), 2- (trimethylsilyl) phenyl trifluoromethanesulfonate (32.7 μL, 0.135 mmol, 3.0 equivalent) , And THF (450 μL) were added under a stream of nitrogen. The contents were cooled to 0 ° C., and a THF solution of tetrabutylammonium fluoride (200 μL, 1.0 M, 0.2 mmol) was added to the flask. The resulting mixture was warmed to room temperature and stirred for 1 hour, and then water (1 mL) was added to the mixture. After extraction with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was purified by preparative thin layer chromatography (hexane / CH 2 Cl 2 = 3: 2) to give the polysubstituted aromatic compound 32 as a white solid (22.5 mg, 85%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.72-7.70 (m, 1H), 7.52-7.48 (m, 2H), 7.42-7.38 (m, 2H), 7.33 (d, J = 7.8 Hz, 2H) , 7.26 (dd, J = 6.6, 2.4 Hz, 2H, overlapping with the peak of CHCl 3 ), 7.10 (dd, J = 6.6, 1.8 Hz, 2H), 6.90-6.84 (m, 3H), 6.82-6.78 ( m, 2H), 6.70 (dd, J = 6.6, 2.4 Hz, 2H), 6.40 (dd, J = 6.9, 2.4 Hz, 2H), 3.60 (s, 3H), 1.30 (s, 9H); 13 C NMR (150 MHz, CDCl 3 ): δ 157.1, 149.2, 143.8, 140.2, 139.4, 139.2, 138.5, 136.6, 136.3, 132.7, 132.3, 132.2, 131.6, 131.5, 131.2, 130.8, 128.5 (q, 2JCF = 33.2 Hz) , 127.3, 126.8, 126.3, 126.0, 125.9, 125.6, 124.5 (q, 3 J CF = 2.9 Hz), 124.4, 124.3 (q, 1 J CF = 273 Hz), 112.1, 55.0, 34.4, 31.3; HRMS (APCI ) m / z calcd for C 40 H 34 F 3 O [MH] + : 587.25563, found 587.25334.
1H NMR (600 MHz, CDCl3): δ 7.72-7.70 (m, 1H), 7.52-7.48 (m, 2H), 7.42-7.38 (m, 2H), 7.33 (d, J = 7.8 Hz, 2H), 7.26 (dd, J = 6.6, 2.4 Hz, 2H, overlapping with the peak of CHCl3), 7.10 (dd, J = 6.6, 1.8 Hz, 2H), 6.90-6.84 (m, 3H), 6.82-6.78 (m, 2H), 6.70 (dd, J = 6.6, 2.4 Hz, 2H), 6.40 (dd, J = 6.9, 2.4 Hz, 2H), 3.60 (s, 3H), 1.30 (s, 9H); 13C NMR (150 MHz, CDCl3): δ 157.1, 149.2, 143.8, 140.2, 139.4, 139.2, 138.5, 136.6, 136.3, 132.7, 132.3, 132.2, 131.6, 131.5, 131.2, 130.8, 128.5 (q, 2JCF = 33.2 Hz), 127.3, 126.8, 126.3, 126.0, 125.9, 125.6, 124.5 (q, 3JCF = 2.9 Hz), 124.4, 124.3 (q, 1JCF = 273 Hz), 112.1, 55.0, 34.4, 31.3; HRMS (APCI) m/z calcd for C40H34F3O [MH]+: 587.25563, found 587.25334。 A 20 mL Schlenk flask was charged with a magnetic stir bar, flame dried under vacuum, cooled to room temperature and filled with nitrogen. In this container, the tetrasubstituted thiophene S-oxide compound 26acdf obtained in Synthesis Example 7-3 (25.5 mg, 0.045 mmol, 1.0 equivalent), 2- (trimethylsilyl) phenyl trifluoromethanesulfonate (32.7 μL, 0.135 mmol, 3.0 equivalent) , And THF (450 μL) were added under a stream of nitrogen. The contents were cooled to 0 ° C., and a THF solution of tetrabutylammonium fluoride (200 μL, 1.0 M, 0.2 mmol) was added to the flask. The resulting mixture was warmed to room temperature and stirred for 1 hour, and then water (1 mL) was added to the mixture. After extraction with CH 2 Cl 2 , the organic layer was dried over Na 2 SO 4 and volatiles were removed under reduced pressure. The crude product was purified by preparative thin layer chromatography (hexane / CH 2 Cl 2 = 3: 2) to give the polysubstituted aromatic compound 32 as a white solid (22.5 mg, 85%).
1 H NMR (600 MHz, CDCl 3 ): δ 7.72-7.70 (m, 1H), 7.52-7.48 (m, 2H), 7.42-7.38 (m, 2H), 7.33 (d, J = 7.8 Hz, 2H) , 7.26 (dd, J = 6.6, 2.4 Hz, 2H, overlapping with the peak of CHCl 3 ), 7.10 (dd, J = 6.6, 1.8 Hz, 2H), 6.90-6.84 (m, 3H), 6.82-6.78 ( m, 2H), 6.70 (dd, J = 6.6, 2.4 Hz, 2H), 6.40 (dd, J = 6.9, 2.4 Hz, 2H), 3.60 (s, 3H), 1.30 (s, 9H); 13 C NMR (150 MHz, CDCl 3 ): δ 157.1, 149.2, 143.8, 140.2, 139.4, 139.2, 138.5, 136.6, 136.3, 132.7, 132.3, 132.2, 131.6, 131.5, 131.2, 130.8, 128.5 (q, 2JCF = 33.2 Hz) , 127.3, 126.8, 126.3, 126.0, 125.9, 125.6, 124.5 (q, 3 J CF = 2.9 Hz), 124.4, 124.3 (q, 1 J CF = 273 Hz), 112.1, 55.0, 34.4, 31.3; HRMS (APCI ) m / z calcd for C 40 H 34 F 3 O [MH] + : 587.25563, found 587.25334.
実施例4-2:多置換芳香族化合物33の合成
Example 4-2: Synthesis of polysubstituted aromatic compound 33

7 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このスクリューキャップチューブに、合成例7-3で得た四置換チオフェンS-オキシド化合物26acdf(25.5 mg, 0.045 mmol, 1.0当量)、合成例12で得た化合物28(ジベンゾ[a, e]シクロオクチン)(18.0 mg, 0.09 mmol, 2.0当量)、及びm-キシレン(450μL)を窒素気流下に添加した。このフラスコを100℃で16時間加熱した。反応混合物を室温まで冷却した後、混合物を真空下に濃縮し、粗生成物を分取薄層クロマトグラフィー(ヘキサン/CHCl3=2: 1)により精製し、多置換芳香族化合物33を白色固体として得た(14.1 mg, 44 %)。
1H NMR (600 MHz, C2D2Cl4, 146℃): δ 7.14 (d, J = 7.8 Hz, 2H), 7.06 (d, J = 7.8 Hz, 2H), 7.01-6.75 (m, 15H), 6.72-6.63 (m, 4H), 6.45 (d, J = 8.4 Hz, 2H), 3.62 (s, 3H), 3.35-3.23 (m, 2H), 3.03-2.94 (m, 2H), 1.17 (s, 9H); 13C NMR (150 MHz, C2D2Cl4, 146℃): δ 157.4, 148.2, 144.6, 141.0, 140.9, 140.8, 140.7, 140.62, 140.59, 140.3, 140.2, 139.3, 139.0, 137.8, 137.2, 133.1, 132.3, 131.3, 130.9, 130.3, 130.2, 128.5, 128.3, 127.5 (q, 2JCF = 31.7 Hz), 126.4, 126.3, 126.0, 125.2, 124.6, 124.5, 124.1 (q, 1JCF = 274 Hz), 123.1, 123.0, 112.4, 55.1, 33.78, 33.75, 33.73, 30.9 (3JCF: not detected); HRMS (APCI) m/z calcd for C46H34F3O [M-C4H8]+: 659.25563, found 659.25314。 A magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this screw cap tube, tetrasubstituted thiophene S-oxide compound 26acdf obtained in Synthesis Example 7-3 (25.5 mg, 0.045 mmol, 1.0 equivalent), compound 28 obtained in Synthesis Example 12 (dibenzo [a, e] cyclooctyne ) (18.0 mg, 0.09 mmol, 2.0 equivalents) and m-xylene (450 μL) were added under a stream of nitrogen. The flask was heated at 100 ° C. for 16 hours. After cooling the reaction mixture to room temperature, the mixture is concentrated in vacuo and the crude product is purified by preparative thin layer chromatography (hexane / CHCl 3 = 2: 1) to give the polysubstituted aromatic compound 33 as a white solid (14.1 mg, 44%).
1 H NMR (600 MHz, C 2 D 2 Cl 4 , 146 ° C): δ 7.14 (d, J = 7.8 Hz, 2H), 7.06 (d, J = 7.8 Hz, 2H), 7.01-6.75 (m, 15H ), 6.72-6.63 (m, 4H), 6.45 (d, J = 8.4 Hz, 2H), 3.62 (s, 3H), 3.35-3.23 (m, 2H), 3.03-2.94 (m, 2H), 1.17 ( s, 9H); 13 C NMR (150 MHz, C 2 D 2 Cl 4 , 146 ° C): δ 157.4, 148.2, 144.6, 141.0, 140.9, 140.8, 140.7, 140.62, 140.59, 140.3, 140.2, 139.3, 139.0, 137.8, 137.2, 133.1, 132.3, 131.3, 130.9, 130.3, 130.2, 128.5, 128.3, 127.5 (q, 2 J CF = 31.7 Hz), 126.4, 126.3, 126.0, 125.2, 124.6, 124.5, 124.1 (q, 1 J CF = 274 Hz), 123.1, 123.0, 112.4, 55.1, 33.78, 33.75, 33.73, 30.9 ( 3 J CF : not detected); HRMS (APCI) m / z calcd for C 46 H 34 F 3 O [MC 4 H 8 ] + : 659.25563, found 659.25314.
1H NMR (600 MHz, C2D2Cl4, 146℃): δ 7.14 (d, J = 7.8 Hz, 2H), 7.06 (d, J = 7.8 Hz, 2H), 7.01-6.75 (m, 15H), 6.72-6.63 (m, 4H), 6.45 (d, J = 8.4 Hz, 2H), 3.62 (s, 3H), 3.35-3.23 (m, 2H), 3.03-2.94 (m, 2H), 1.17 (s, 9H); 13C NMR (150 MHz, C2D2Cl4, 146℃): δ 157.4, 148.2, 144.6, 141.0, 140.9, 140.8, 140.7, 140.62, 140.59, 140.3, 140.2, 139.3, 139.0, 137.8, 137.2, 133.1, 132.3, 131.3, 130.9, 130.3, 130.2, 128.5, 128.3, 127.5 (q, 2JCF = 31.7 Hz), 126.4, 126.3, 126.0, 125.2, 124.6, 124.5, 124.1 (q, 1JCF = 274 Hz), 123.1, 123.0, 112.4, 55.1, 33.78, 33.75, 33.73, 30.9 (3JCF: not detected); HRMS (APCI) m/z calcd for C46H34F3O [M-C4H8]+: 659.25563, found 659.25314。 A magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this screw cap tube, tetrasubstituted thiophene S-oxide compound 26acdf obtained in Synthesis Example 7-3 (25.5 mg, 0.045 mmol, 1.0 equivalent), compound 28 obtained in Synthesis Example 12 (dibenzo [a, e] cyclooctyne ) (18.0 mg, 0.09 mmol, 2.0 equivalents) and m-xylene (450 μL) were added under a stream of nitrogen. The flask was heated at 100 ° C. for 16 hours. After cooling the reaction mixture to room temperature, the mixture is concentrated in vacuo and the crude product is purified by preparative thin layer chromatography (hexane / CHCl 3 = 2: 1) to give the polysubstituted aromatic compound 33 as a white solid (14.1 mg, 44%).
1 H NMR (600 MHz, C 2 D 2 Cl 4 , 146 ° C): δ 7.14 (d, J = 7.8 Hz, 2H), 7.06 (d, J = 7.8 Hz, 2H), 7.01-6.75 (m, 15H ), 6.72-6.63 (m, 4H), 6.45 (d, J = 8.4 Hz, 2H), 3.62 (s, 3H), 3.35-3.23 (m, 2H), 3.03-2.94 (m, 2H), 1.17 ( s, 9H); 13 C NMR (150 MHz, C 2 D 2 Cl 4 , 146 ° C): δ 157.4, 148.2, 144.6, 141.0, 140.9, 140.8, 140.7, 140.62, 140.59, 140.3, 140.2, 139.3, 139.0, 137.8, 137.2, 133.1, 132.3, 131.3, 130.9, 130.3, 130.2, 128.5, 128.3, 127.5 (q, 2 J CF = 31.7 Hz), 126.4, 126.3, 126.0, 125.2, 124.6, 124.5, 124.1 (q, 1 J CF = 274 Hz), 123.1, 123.0, 112.4, 55.1, 33.78, 33.75, 33.73, 30.9 ( 3 J CF : not detected); HRMS (APCI) m / z calcd for C 46 H 34 F 3 O [MC 4 H 8 ] + : 659.25563, found 659.25314.
[実施例5(第2の態様):化合物(31C)の合成]
実施例5-1:多置換芳香族化合物34a及び多置換芳香族化合物34bの合成 [Example 5 (second aspect): Synthesis of compound (31C)]
Example 5-1: Synthesis of polysubstituted aromatic compound 34a and polysubstituted aromatic compound 34b
実施例5-1:多置換芳香族化合物34a及び多置換芳香族化合物34bの合成 [Example 5 (second aspect): Synthesis of compound (31C)]
Example 5-1: Synthesis of polysubstituted aromatic compound 34a and polysubstituted aromatic compound 34b

7 mLのスクリューキャップチューブに磁気撹拌子を入れ、真空下にフレームドライし、室温まで冷却した後に窒素を充填した。このスクリューキャップチューブに、合成例7-3で得た四置換チオフェンS-オキシド化合物26acdf(0.03 mmol, 1.0当量)、及び3-シアノピリジン(300μL, 3.0 mmol)を窒素気流下に添加した。このフラスコを160℃で24時間加熱した。反応混合物を室温まで冷却した後、混合物を分取薄層クロマトグラフィーにより精製し、多置換芳香族化合物34a(1.6 mg)と多置換芳香族化合物34b(1.6 mg)とを合計収率17 %で得た。より極性の高い化合物が多置換芳香族化合物34aであり、より極性の低い化合物が多置換芳香族化合物34bである。多置換芳香族化合物34bの構造はX線結晶構造解析で決定した。なお、多置換芳香族化合物34bのX線結晶構造を図2に示す。
多置換芳香族化合物34a:
1H NMR (600 MHz, CDCl3): δ 8.50 (d, J = 4.8 Hz, 1H), 7.50-7.45 (m, 3H), 7.41 (d, J = 9.0 Hz, 2H), 7.28 (d, J = 8.4 Hz, 1H), 7.10 (dd, J = 7.8 Hz, 4.8 Hz, 1H), 7.08-7.01 (m, 3H), 6.98 (d, J = 7.8 Hz, 2H), 6.89-6.85 (m, 2H), 6.78 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 6.47 (d, J = 9.0 Hz, 2H), 3.64 (s, 3H), 1.18 (s, 9H); 13C NMR (150 MHz, CDCl3): δ 158.8, 157.9, 156.2, 155.1, 150.2, 149.1, 148.9, 144.4, 137.9, 135.4, 135.3, 134.6, 131.6, 131.2, 130.6, 130.5, 129.8, 129.1 (q, 2JCF = 31.7 Hz), 127.7, 126.6, 124.8, 124.4, 124.2 (q, 1JCF = 271 Hz), 124.1, 122.0, 112.5, 55.0, 34.3, 31.2 (3JCF: not detected); HRMS (APCI) m/z calcd for C40H34F3N2O [MH]+: 615.26177 found 615.25989。
多置換芳香族化合物34b:
1H NMR (600 MHz, CDCl3): δ 8.35 (d, J = 4.8 Hz, 1H), 7.63-7.58 (m, 2H), 7.32 (d, J = 7.8 Hz, 2H), 7.23-7.18 (m, 4H), 7.12-7.08 (m, 1H), 7.02 (d, J = 7.8 Hz, 2H), 6.98-6.93 (m, 3H), 6.79 (d, J = 9.0 Hz, 2H), 6.77-6.72 (m, 2H), 6.56 (d, J = 9.0 Hz, 2H), 3.69 (s, 3H), 1.26 (s, 9H); 13C NMR (150 MHz, CDCl3): δ 158.6, 158.0, 157.2, 155.0, 150.5, 150.3, 148.5, 142.4, 137.65, 137.56, 135.9, 134.4, 132.7, 132.3, 131.3, 130.3, 129.8, 128.0 (q, 2JCF = 31.7 Hz), 127.2, 126.5, 124.8, 124.6, 124.1 (q, 1JCF = 270 Hz), 123.9 (q, 3JCF = 2.85 Hz), 122.2, 113.0, 55.0, 34.5, 31.2; HRMS (APCI) m/z calcd for C40H34F3N2O [MH]+: 615.26177 found 615.26088。 A magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this screw cap tube, the tetra-substituted thiophene S-oxide compound 26acdf (0.03 mmol, 1.0 equivalent) obtained in Synthesis Example 7-3 and 3-cyanopyridine (300 μL, 3.0 mmol) were added under a nitrogen stream. The flask was heated at 160 ° C. for 24 hours. After the reaction mixture was cooled to room temperature, the mixture was purified by preparative thin layer chromatography to obtain polysubstituted aromatic compound 34a (1.6 mg) and polysubstituted aromatic compound 34b (1.6 mg) in a total yield of 17%. Obtained. The more polar compound is the polysubstituted aromatic compound 34a, and the less polar compound is the polysubstituted aromatic compound 34b. The structure of polysubstituted aromatic compound 34b was determined by X-ray crystal structure analysis. The X-ray crystal structure of the polysubstituted aromatic compound 34b is shown in FIG.
Polysubstituted aromatic compound 34a:
1 H NMR (600 MHz, CDCl 3 ): δ 8.50 (d, J = 4.8 Hz, 1H), 7.50-7.45 (m, 3H), 7.41 (d, J = 9.0 Hz, 2H), 7.28 (d, J = 8.4 Hz, 1H), 7.10 (dd, J = 7.8 Hz, 4.8 Hz, 1H), 7.08-7.01 (m, 3H), 6.98 (d, J = 7.8 Hz, 2H), 6.89-6.85 (m, 2H ), 6.78 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 6.47 (d, J = 9.0 Hz, 2H), 3.64 (s, 3H), 1.18 (s, 9H ); 13 C NMR (150 MHz, CDCl 3 ): δ 158.8, 157.9, 156.2, 155.1, 150.2, 149.1, 148.9, 144.4, 137.9, 135.4, 135.3, 134.6, 131.6, 131.2, 130.6, 130.5, 129.8, 129.1 ( q, 2 J CF = 31.7 Hz), 127.7, 126.6, 124.8, 124.4, 124.2 (q, 1 J CF = 271 Hz), 124.1, 122.0, 112.5, 55.0, 34.3, 31.2 ( 3 J CF : not detected); HRMS (APCI) m / z calcd for C 40 H 34 F 3 N 2 O [MH] + : 615.26177 found 615.25989.
Polysubstituted aromatic compound 34b:
1 H NMR (600 MHz, CDCl 3 ): δ 8.35 (d, J = 4.8 Hz, 1H), 7.63-7.58 (m, 2H), 7.32 (d, J = 7.8 Hz, 2H), 7.23-7.18 (m , 4H), 7.12-7.08 (m, 1H), 7.02 (d, J = 7.8 Hz, 2H), 6.98-6.93 (m, 3H), 6.79 (d, J = 9.0 Hz, 2H), 6.77-6.72 ( m, 2H), 6.56 (d, J = 9.0 Hz, 2H), 3.69 (s, 3H), 1.26 (s, 9H); 13 C NMR (150 MHz, CDCl 3 ): δ 158.6, 158.0, 157.2, 155.0 , 150.5, 150.3, 148.5, 142.4, 137.65, 137.56, 135.9, 134.4, 132.7, 132.3, 131.3, 130.3, 129.8, 128.0 (q, 2 J CF = 31.7 Hz), 127.2, 126.5, 124.8, 124.6, 124.1 (q , 1 J CF = 270 Hz), 123.9 (q, 3 J CF = 2.85 Hz), 122.2, 113.0, 55.0, 34.5, 31.2; HRMS (APCI) m / z calcd for C 40 H 34 F 3 N 2 O [ MH] + : 615.26177 found 615.26088.
多置換芳香族化合物34a:
1H NMR (600 MHz, CDCl3): δ 8.50 (d, J = 4.8 Hz, 1H), 7.50-7.45 (m, 3H), 7.41 (d, J = 9.0 Hz, 2H), 7.28 (d, J = 8.4 Hz, 1H), 7.10 (dd, J = 7.8 Hz, 4.8 Hz, 1H), 7.08-7.01 (m, 3H), 6.98 (d, J = 7.8 Hz, 2H), 6.89-6.85 (m, 2H), 6.78 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 6.47 (d, J = 9.0 Hz, 2H), 3.64 (s, 3H), 1.18 (s, 9H); 13C NMR (150 MHz, CDCl3): δ 158.8, 157.9, 156.2, 155.1, 150.2, 149.1, 148.9, 144.4, 137.9, 135.4, 135.3, 134.6, 131.6, 131.2, 130.6, 130.5, 129.8, 129.1 (q, 2JCF = 31.7 Hz), 127.7, 126.6, 124.8, 124.4, 124.2 (q, 1JCF = 271 Hz), 124.1, 122.0, 112.5, 55.0, 34.3, 31.2 (3JCF: not detected); HRMS (APCI) m/z calcd for C40H34F3N2O [MH]+: 615.26177 found 615.25989。
多置換芳香族化合物34b:
1H NMR (600 MHz, CDCl3): δ 8.35 (d, J = 4.8 Hz, 1H), 7.63-7.58 (m, 2H), 7.32 (d, J = 7.8 Hz, 2H), 7.23-7.18 (m, 4H), 7.12-7.08 (m, 1H), 7.02 (d, J = 7.8 Hz, 2H), 6.98-6.93 (m, 3H), 6.79 (d, J = 9.0 Hz, 2H), 6.77-6.72 (m, 2H), 6.56 (d, J = 9.0 Hz, 2H), 3.69 (s, 3H), 1.26 (s, 9H); 13C NMR (150 MHz, CDCl3): δ 158.6, 158.0, 157.2, 155.0, 150.5, 150.3, 148.5, 142.4, 137.65, 137.56, 135.9, 134.4, 132.7, 132.3, 131.3, 130.3, 129.8, 128.0 (q, 2JCF = 31.7 Hz), 127.2, 126.5, 124.8, 124.6, 124.1 (q, 1JCF = 270 Hz), 123.9 (q, 3JCF = 2.85 Hz), 122.2, 113.0, 55.0, 34.5, 31.2; HRMS (APCI) m/z calcd for C40H34F3N2O [MH]+: 615.26177 found 615.26088。 A magnetic stirring bar was placed in a 7 mL screw cap tube, flame-dried under vacuum, cooled to room temperature, and filled with nitrogen. To this screw cap tube, the tetra-substituted thiophene S-oxide compound 26acdf (0.03 mmol, 1.0 equivalent) obtained in Synthesis Example 7-3 and 3-cyanopyridine (300 μL, 3.0 mmol) were added under a nitrogen stream. The flask was heated at 160 ° C. for 24 hours. After the reaction mixture was cooled to room temperature, the mixture was purified by preparative thin layer chromatography to obtain polysubstituted aromatic compound 34a (1.6 mg) and polysubstituted aromatic compound 34b (1.6 mg) in a total yield of 17%. Obtained. The more polar compound is the polysubstituted aromatic compound 34a, and the less polar compound is the polysubstituted aromatic compound 34b. The structure of polysubstituted aromatic compound 34b was determined by X-ray crystal structure analysis. The X-ray crystal structure of the polysubstituted aromatic compound 34b is shown in FIG.
Polysubstituted aromatic compound 34a:
1 H NMR (600 MHz, CDCl 3 ): δ 8.50 (d, J = 4.8 Hz, 1H), 7.50-7.45 (m, 3H), 7.41 (d, J = 9.0 Hz, 2H), 7.28 (d, J = 8.4 Hz, 1H), 7.10 (dd, J = 7.8 Hz, 4.8 Hz, 1H), 7.08-7.01 (m, 3H), 6.98 (d, J = 7.8 Hz, 2H), 6.89-6.85 (m, 2H ), 6.78 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 6.47 (d, J = 9.0 Hz, 2H), 3.64 (s, 3H), 1.18 (s, 9H ); 13 C NMR (150 MHz, CDCl 3 ): δ 158.8, 157.9, 156.2, 155.1, 150.2, 149.1, 148.9, 144.4, 137.9, 135.4, 135.3, 134.6, 131.6, 131.2, 130.6, 130.5, 129.8, 129.1 ( q, 2 J CF = 31.7 Hz), 127.7, 126.6, 124.8, 124.4, 124.2 (q, 1 J CF = 271 Hz), 124.1, 122.0, 112.5, 55.0, 34.3, 31.2 ( 3 J CF : not detected); HRMS (APCI) m / z calcd for C 40 H 34 F 3 N 2 O [MH] + : 615.26177 found 615.25989.
Polysubstituted aromatic compound 34b:
1 H NMR (600 MHz, CDCl 3 ): δ 8.35 (d, J = 4.8 Hz, 1H), 7.63-7.58 (m, 2H), 7.32 (d, J = 7.8 Hz, 2H), 7.23-7.18 (m , 4H), 7.12-7.08 (m, 1H), 7.02 (d, J = 7.8 Hz, 2H), 6.98-6.93 (m, 3H), 6.79 (d, J = 9.0 Hz, 2H), 6.77-6.72 ( m, 2H), 6.56 (d, J = 9.0 Hz, 2H), 3.69 (s, 3H), 1.26 (s, 9H); 13 C NMR (150 MHz, CDCl 3 ): δ 158.6, 158.0, 157.2, 155.0 , 150.5, 150.3, 148.5, 142.4, 137.65, 137.56, 135.9, 134.4, 132.7, 132.3, 131.3, 130.3, 129.8, 128.0 (q, 2 J CF = 31.7 Hz), 127.2, 126.5, 124.8, 124.6, 124.1 (q , 1 J CF = 270 Hz), 123.9 (q, 3 J CF = 2.85 Hz), 122.2, 113.0, 55.0, 34.5, 31.2; HRMS (APCI) m / z calcd for C 40 H 34 F 3 N 2 O [ MH] + : 615.26177 found 615.26088.
実施例5-2:多置換芳香族化合物35a及び多置換芳香族化合物35bの合成
Example 5-2: Synthesis of polysubstituted aromatic compound 35a and polysubstituted aromatic compound 35b

四置換チオフェンS-オキシド化合物26acdfの量を5.6 mg, 0.01 mmolとし、2-シアノピリジンの代わりに4-メチルベンゾニトリル(145 mg, 1 mmol)を使用し、加熱温度を160℃から230℃としたこと以外は実施例5-1と同様に合成し、多置換芳香族化合物35a(0.5 mg)及び多置換芳香族化合物35b(0.5 mg)の混合物を合計収率16 %として得た。
The amount of tetrasubstituted thiophene S-oxide compound 26acdf was 5.6 mg, 0.01 mmol, 4-methylbenzonitrile (145 mg, 1 mmol) was used instead of 2-cyanopyridine, and the heating temperature was 160 ° C to 230 ° C. Except that described above, synthesis was performed in the same manner as in Example 5-1, and a mixture of the polysubstituted aromatic compound 35a (0.5 mg) and the polysubstituted aromatic compound 35b (0.5 mg) was obtained with a total yield of 16%.
実施例5-3:多置換芳香族化合物36a及び多置換芳香族化合物36bの合成
Example 5-3: Synthesis of polysubstituted aromatic compound 36a and polysubstituted aromatic compound 36b

四置換チオフェンS-オキシド化合物26acdfの量を5.6 mg, 0.01 mmolとし、2-シアノピリジンの代わりに4-(トリフルオロメチル)ベンゾニトリル(171 mg, 1 mmol)を使用したこと以外は実施例5-1と同様に合成し、多置換芳香族化合物36a及び多置換芳香族化合物36bを混合物として得た(0.7 mg, 10 %(混合物))。
Example 5 except that the amount of tetrasubstituted thiophene S-oxide compound 26acdf was 5.6 mg, 0.01 mmol, and 4- (trifluoromethyl) benzonitrile (171 mg, 1 mmol) was used instead of 2-cyanopyridine. Synthesis was carried out in the same manner as for -1, and a polysubstituted aromatic compound 36a and a polysubstituted aromatic compound 36b were obtained as a mixture (0.7 mg, 10% (mixture)).
Example 5 except that the amount of tetrasubstituted thiophene S-oxide compound 26acdf was 5.6 mg, 0.01 mmol, and 4- (trifluoromethyl) benzonitrile (171 mg, 1 mmol) was used instead of 2-cyanopyridine. Synthesis was carried out in the same manner as for -1, and a polysubstituted aromatic compound 36a and a polysubstituted aromatic compound 36b were obtained as a mixture (0.7 mg, 10% (mixture)).
Claims (7)
- 一般式(1):

、又は一般式(2):

で表される多置換芳香族化合物。 General formula (1):
Or general formula (2):
The polysubstituted aromatic compound represented by these. - 前記一般式(1)において、R1~R8が全て異なる基である、請求項1に記載の多置換芳香族化合物。 The polysubstituted aromatic compound according to claim 1, wherein in the general formula (1), R 1 to R 8 are all different groups.
- 前記一般式(2)において、R17~R18が水素原子であり、R9~R16が全て異なる基である、請求項1に記載の多置換芳香族化合物。 The polysubstituted aromatic compound according to claim 1, wherein, in the general formula (2), R 17 to R 18 are hydrogen atoms and R 9 to R 16 are all different groups.
- 前記一般式(2)において、R17~R18が置換されていてもよいアリール基又は置換されていてもよいヘテロアリール基であり、R9~R18が全て異なる基である、請求項1に記載の多置換芳香族化合物。 The general formula (2), wherein R 17 to R 18 are an optionally substituted aryl group or an optionally substituted heteroaryl group, and R 9 to R 18 are all different groups. The polysubstituted aromatic compound described in 1.
- 請求項1~4のいずれかに記載の多置換芳香族化合物の製造方法であって、
一般式(3):

で表される四置換チオフェンS-オキシド化合物と、
一般式(4):

で表される化合物、又は一般式(5):

で表される化合物とを反応させる工程
を備える、製造方法。 A method for producing a polysubstituted aromatic compound according to any one of claims 1 to 4,
General formula (3):
A tetrasubstituted thiophene S-oxide compound represented by:
General formula (4):
Or a compound represented by the general formula (5):
The manufacturing method provided with the process with which the compound represented by these is made to react. - 前記一般式(4)で表される化合物は、一般式(6):

で示される化合物と、マレイミドとを反応させる工程、及び
前記工程で得られた化合物と、酸化剤及び第1塩基とを反応させた後に、さらに第2塩基と反応させる工程
を備える方法で得られる、請求項5に記載の製造方法。 The compound represented by the general formula (4) is represented by the general formula (6):
Obtained by reacting the compound represented by the formula with maleimide, and reacting the compound obtained in the previous step with the oxidizing agent and the first base, and further reacting with the second base. The manufacturing method according to claim 5. - 前記一般式(5)で表される化合物は、一般式(7):

で表される化合物と、一般式(8)

で表される化合物とを反応させる工程、及び
前記工程で得られた化合物の水酸基をトリフルオロメタンスルホナート基に置換する工程
を備える方法で得られる、請求項5に記載の製造方法。 The compound represented by the general formula (5) is represented by the general formula (7):
And a compound of the general formula (8)
The manufacturing method of Claim 5 obtained by the method of providing the process with which the compound represented by these are made to react and the process of substituting the hydroxyl group of the compound obtained at the said process by the trifluoromethanesulfonate group.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017520793A JPWO2016190374A1 (en) | 2015-06-26 | 2016-05-25 | Polysubstituted aromatic compound and method for producing the same |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015-105885 | 2015-05-25 | ||
| JP2015105885A JP6709889B2 (en) | 2015-05-25 | 2015-05-25 | Polysubstituted aromatic compound and method for producing the same |
| JP2015129027 | 2015-06-26 | ||
| JP2015-129027 | 2015-06-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016190374A1 true WO2016190374A1 (en) | 2016-12-01 |
Family
ID=57394112
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/065516 WO2016190374A1 (en) | 2015-05-25 | 2016-05-25 | Poly-substituted aromatic compound and method for producing same |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2016190374A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107325120A (en) * | 2017-07-06 | 2017-11-07 | 上海阿拉丁生化科技股份有限公司 | A kind of synthetic method of 2 (trimethyl silicon substrate) phenyl trifluoromethanesulfonate methane sulfonates |
| CN107698616A (en) * | 2017-12-05 | 2018-02-16 | 扬州三友合成化工有限公司 | A kind of Isosorbide-5-Nitrae is double(Dimethylsilyl)The preparation method of benzene |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004535051A (en) * | 2001-07-11 | 2004-11-18 | 富士写真フイルム株式会社 | Light emitting device and aromatic compound |
| CN101279890A (en) * | 2007-04-02 | 2008-10-08 | 中国科学院化学研究所 | Polysubstituted acene derivative and preparation thereof |
| WO2010073864A1 (en) * | 2008-12-26 | 2010-07-01 | パイオニア株式会社 | Organic electroluminescent element |
-
2016
- 2016-05-25 WO PCT/JP2016/065516 patent/WO2016190374A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004535051A (en) * | 2001-07-11 | 2004-11-18 | 富士写真フイルム株式会社 | Light emitting device and aromatic compound |
| CN101279890A (en) * | 2007-04-02 | 2008-10-08 | 中国科学院化学研究所 | Polysubstituted acene derivative and preparation thereof |
| WO2010073864A1 (en) * | 2008-12-26 | 2010-07-01 | パイオニア株式会社 | Organic electroluminescent element |
Non-Patent Citations (2)
| Title |
|---|
| SHIN SUZUKI ET AL.: "Synthesis and characterization of hexaarylbenzenes with five or six different substituents enabled by programmed synthesis", NATURE CHEMISTRY, vol. 7, 26 January 2015 (2015-01-26), pages 227 - 233, XP055335448 * |
| XIN ZHOU ET AL.: "Novel synthetic route to octasubstituted naphthalenes from four alkynes and one olefin unit via zirconacyclopentadienes and 1,2-diiodo-3,4,5,6-tetraalkylbenzene", JOURNAL OF ORGANIC CHEMISTRY, vol. 69, no. 13, 29 May 2004 (2004-05-29), pages 4559 - 4562, XP055335451 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107325120A (en) * | 2017-07-06 | 2017-11-07 | 上海阿拉丁生化科技股份有限公司 | A kind of synthetic method of 2 (trimethyl silicon substrate) phenyl trifluoromethanesulfonate methane sulfonates |
| CN107698616A (en) * | 2017-12-05 | 2018-02-16 | 扬州三友合成化工有限公司 | A kind of Isosorbide-5-Nitrae is double(Dimethylsilyl)The preparation method of benzene |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI823864B (en) | Binaphthyl compounds | |
| JP6855083B2 (en) | Intermediates of deuterated aromatic compounds and methods for preparing deuterated aromatic compounds using them | |
| JP5867583B2 (en) | Novel chalcogen-containing organic compounds and uses thereof | |
| KR20150045507A (en) | Method for producing aromatic compound | |
| Chan et al. | 5, 6-Bis (trimethylsilyl) benzo [c] furan: an isolable versatile building block for linear polycyclic aromatic compounds | |
| WO2016190374A1 (en) | Poly-substituted aromatic compound and method for producing same | |
| Yamada et al. | Synthesis of 5-organostibano-1H-1, 2, 3-triazoles by Cu-catalyzed azide-alkyne cycloaddition and their application in the acyl-induced deantimonation for the preparation of fully substituted 5-acyl-1, 2, 3-triazoles | |
| JP4929468B2 (en) | Synthesis method of oligomeric compounds using cross-coupling reaction | |
| JP6709889B2 (en) | Polysubstituted aromatic compound and method for producing the same | |
| JPWO2016190374A1 (en) | Polysubstituted aromatic compound and method for producing the same | |
| KR102060527B1 (en) | 4 Ring-fused N-heterocycles having luminescent properties and Preparation method thereof | |
| JP6367121B2 (en) | Coupling method and method for producing aromatic group-substituted heterocyclic compound using the coupling method | |
| JP2017171603A (en) | Manufacturing method of condensed polycyclic compound | |
| JP2019085385A (en) | Method for preparing indenoisoquinoline derivative | |
| WO2013183642A1 (en) | Axially chiral pyridine derivative or salt thereof, and method for producing same and chiral catalyst comprising same | |
| Bosiak et al. | Synthesis and photoluminescence properties of star-shaped 2, 3, 6, 7-tetrasubstituted benzo [1, 2-b: 4, 5-b′] difurans | |
| CN115210245A (en) | Preparation method of naphthyl silole, naphthyl silole with heterocyclic group and graphene nanoribbon with heterocyclic group | |
| JP6957084B2 (en) | Method for producing condensed polycyclic compound | |
| JP4362683B2 (en) | Asymmetric dihydrophenazine derivative and method for producing the same | |
| JP2008143857A (en) | Method for producing benzofluorene derivative and its intermediate | |
| Fehér et al. | Palladium-catalysed reactions of 6-halogeno-1, 1′-binaphthyl derivatives. A detailed investigation of structure/reactivity and structure/selectivity relationships | |
| TWI836465B (en) | Precursor for preparing cinnoline derivative, method for preparing cinnoline derivative and cinnoline derivative | |
| TW201514141A (en) | Bis(aryl)acetal compounds | |
| RU2788650C2 (en) | Asymmetrical luminescent donor-acceptor molecules based on triphenylamine-thiophene block with different electron-acceptor groups and their production method | |
| JP2013052384A (en) | Heterogeneous catalyst and method for producing triarylamine compound using the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16800078 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017520793 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16800078 Country of ref document: EP Kind code of ref document: A1 |