WO2016185200A1 - Antibacterial agent - Google Patents
Antibacterial agent Download PDFInfo
- Publication number
- WO2016185200A1 WO2016185200A1 PCT/GB2016/051420 GB2016051420W WO2016185200A1 WO 2016185200 A1 WO2016185200 A1 WO 2016185200A1 GB 2016051420 W GB2016051420 W GB 2016051420W WO 2016185200 A1 WO2016185200 A1 WO 2016185200A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- optionally substituted
- halogen
- membered heterocyclyl
- Prior art date
Links
- 0 CC(*)(C(*C(*)(*)*)N(*)C(*)=O)S Chemical compound CC(*)(C(*C(*)(*)*)N(*)C(*)=O)S 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/0606—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing heteroatoms not provided for by C07K5/06086 - C07K5/06139, e.g. Ser, Met, Cys, Thr
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/34—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/36—Oxygen or sulfur atoms
- C07D207/40—2,5-Pyrrolidine-diones
- C07D207/416—2,5-Pyrrolidine-diones with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to other ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to compounds useful as antibacterial agents.
- the use of such compounds in treating or preventing bacterial infection is also described.
- the potassium efflux system protects bacteria against the detrimental effects of electrophilic compounds via acidification of the bacterial cytoplasm.
- Glutathione (GSH) and glutathione-S-conjugate (GS-X) compounds are known to affect Kef.
- GSH Glutathione
- GS-X glutathione-S-conjugate
- GSH inhibits Kef
- some GS-X compounds activate Kef (Healy et al, Understanding the Structural Requirements for Activators of the Kef Bacterial Potassium Efflux System, Biochemistry, 2014 Apr 1 ; 53(12): 1982-92).
- Activation of the Kef system results in K+ efflux and concomitant H+ influx.
- Kef activation results in a decrease in cellular pH, protonating nucleophiles in DNA and proteins and minimising their reactivity with electrophilic species. Kef activation is thus known to interfere with normal bacterial functioning and therefore has the potential as a therapeutic target for preventing or treating bacterial infections.
- the invention provides a compound which is a peptide derivative of formula (1) or a salt thereof:
- R G is H, -CH 2 -R GA or C 1-12 hydrocarbyl optionally substituted with 1 to 4
- R G and R N1 together form a 5-6 membered heterocyclic ring which is optionally substituted with 1 to 3 substituents selected from R a , -OR' and halogen;
- R GA is Ci-ii hydrocarbyl optionally substituted with 1 to 4 substituents selected from - R'R", -OR', -NO2, -CN, halogen and 5-6 membered heterocyclyl;
- R s is Ci-16 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group,
- R SA is Ci-ie hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group
- R b is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl;
- Z is a group selected from -C(0)-0-R° and -CN 4 R' (tetrazolyl);
- each R C1 , R C2 , R', R", R" and R"" is independently H or C alkyl and is optionally substituted with 1 to 3 substituents selected from halogen, -OH and -O-C1-4 alkyl;
- each R a is independently C1-4 alkyl which is optionally substituted with 1 to 3 halogen atoms.
- the inventors have found surprisingly that the glutamate residue of GS-X compounds makes a negligible contribution to the affinity of the compounds to Kef.
- truncation or replacement of the glutamate residue in GS-X compounds has allowed the development of a new class of antibacterial compounds.
- These compounds not only demonstrate good binding to Kef, but have also been shown to cause activation of the Kef system.
- the compounds have reduced polar groups and a shorter peptide chain, and therefore are expected to demonstrate better ability to cross cell membranes than the known GSH.
- the compounds are thus useful in the treatment and prevention of bacterial infections in animals as well as plants. These compounds can effectively inhibit or damage bacteria through activation of the Kef system and resulting K+ efflux.
- the invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a
- the invention also provides a compound which is a peptide derivative or a salt thereof as defined herein, for use in the treatment or prevention of an infection caused by bacteria, wherein the salt is a pharmaceutically acceptable salt.
- the invention also provides a method of treating or preventing infection caused by bacteria in a subject, wherein the infection is as defined herein, which method comprises administering an effective amount of a peptide derivative or a salt thereof as defined herein to the subject, wherein the salt is a pharmaceutically acceptable salt.
- the invention also provides a combination comprising (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a pharmaceutically acceptable salt, and (b) a further active agent which is an antibiotic compound or a pharmaceutically acceptable salt thereof.
- the invention also provides a combination for use in the treatment or prevention of an infection caused by bacteria as defined herein, which combination comprises (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a pharmaceutically acceptable salt, and (b) a further active agent which is an antibiotic compound.
- the invention also provides a kit comprising (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a pharmaceutically acceptable salt, and (b) a further antibiotic compound or a pharmaceutically acceptable salt thereof.
- the invention also provides a compound which is a peptide derivative or a salt thereof as defined herein for use in a method of activating potassium efflux in bacteria, wherein the bacteria are as defined herein.
- the invention also provides a method for preventing or treating infection of a plant caused by bacteria, which method comprises treating the plant, the seed from which the plant grows, or the place at which the plant grows, with a composition comprising a compound as defined herein.
- the invention also provides an in vitro method for killing or inhibiting the growth of bacteria, which method comprises applying a composition comprising a compound which is a peptide derivative or salt thereof as defined herein to a surface, to an article or to the skin.
- a composition comprising a compound which is a peptide derivative or salt thereof as defined herein to a surface, to an article or to the skin.
- the method is typically not a method of treatment of the human or animal body, but merely a method for killing or inhibiting bacteria present on the skin.
- the invention also provides a soap product, washing product or cleaning product comprising a compound which is a peptide derivative or salt thereof as defined herein.
- the invention also provides a process for producing a compound which is a peptide derivative of formula (la) or a salt thereof:
- R G , R N1 , R C1 , Q, R C2 and Z are as defined herein and the group
- Figures 1 to 3 show the results of competition fluorescence assays for compounds according to the invention and comparative compounds.
- Figures 4 to 10 show Kd curves for various compounds obtained by CPMG MR.
- Figure 11 shows the results of a Kirby-Bauer disk diffusion assay performed for a compound of the invention.
- a hydrocarbyl group is a group comprising only carbon atoms and hydrogen atoms.
- hydrocarbyl groups are those derived by the removal of a hydrogen atom from a hydrocarbon compound.
- hydrocarbyl groups include alkyl groups, cycloalkyl groups, alkenyl groups, cycloalkenyl groups, alkynyl groups, aryl groups, aryl groups substituted with one or more alkyl groups, alkyl groups substituted with one or more aryl groups and alkyl groups substituted with aryl groups which are themselves substituted with one or more alkyl groups.
- a hydrocarbyl group may be defined by the number of carbon atoms it contains.
- a Ci-i6 hydrocarbyl group comprises from 1 to 16 carbon atoms.
- Examples of a Ci- 16 hydrocarbyl group include Ci-i6 alkyl, phenyl, C3-6 cycloalkyl, and Ci-10 alkyl substituted with a phenyl or C3-6 cycloalkyl group, wherein each of these groups may be substituted with 1 to 3 Ci-3 alkyl groups.
- C1-16 hydrocarbyl is typically Ci-12 hydrocarbyl or C 1-11
- Ci-12 hydrocarbyl or C 1-11 hydrocarbyl is Ci -8 alkyl, or a C3-6 cycloalkyl, phenyl, benzyl or ethyl-phenyl group, each of which may optionally be substituted with 1 to 3 C1-4 alkyl groups.
- An alkyl group is a linear or branched chain saturated hydrocarbon group.
- An alkyl group may be a C1-16 alkyl group, a Ci -8 alkyl group, or a C1-4 alkyl group.
- Examples of a C1-16 alkyl group are methyl, ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, sec-butyl, tert-butyl, pentyl, neopentyl, hexyl, heptyl and octyl.
- a cycloalkyl group is typically a C3-6 cycloalkyl group.
- Examples of a C3-6 cycloalkyl group include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- a cycloalkyl group is unsubstituted or substituted. Typically, it is substituted with one or two C1-4 alkyl groups, e.g. 1 or 2 methyl groups, or is unsubstituted.
- a cycloalkyl group is unsubstituted.
- An alkenyl group is a linear or branched chain hydrocarbon radical comprising one or more double bonds.
- An alkenyl group may be a C2-10 alkenyl group.
- Alkenyl groups typically comprise one or two double bonds.
- An alkynyl group is a linear or branched chain hydrocarbon radical comprising one or more triple bonds.
- An alkynyl group may be a C2-10 alkynyl group.
- Examples of C2-10 alkynyl groups include those related to C2-10 alkyl groups by the insertion of one or more triple bonds, e.g. ethynyl (-C ⁇ CH), propynyl (-CH 2 -C ⁇ CH or -C ⁇ C-CH 3 ) and butynyl (-CH2-CH2- C ⁇ CH or -CH2-C ⁇ C-CH3 or -C ⁇ C-CH2-CH3).
- Alkynyl groups typically comprise one or two triple bonds.
- alkyl, alkenyl or alkynyl group may be unsubstituted or substituted with one or more, e.g. from 1 to 3, substituents.
- Preferred substituents are selected from - R'R", -OR', halogen and 5-6 membered heterocyclyl, wherein each R' and R" is independently H or Ci-4 alkyl, most preferably from -NR'R", -OR', halogen and morpholinyl, wherein each R' and R" is independently H or Ci-4 alkyl.
- an alkyl, alkenyl or alkynyl group is unsubstituted or substituted with from 1 to 3 halogen atoms, e.g. it is unsubstituted.
- An aryl group is a monocyclic, bicyclic or polycyclic aromatic ring which contains from 6 to 14 carbon atoms, typically from 6 to 10 carbon atoms, in the ring portion. Examples include phenyl, naphthyl, indenyl, indanyl, anthracenyl and pyrenyl groups. Often an aryl group is phenyl.
- a heterocyclyl group is a monovalent moiety obtained by removing a hydrogen atom from a ring atom of a heterocyclic compound.
- a heterocyclyl group has from 5 to 10 ring atoms (5-10 membered heterocyclyl), more preferably 5 or 6 ring atoms (5-6 membered heterocyclyl), including at least one (e.g. one, two, three or four, preferably one or two) heteroatoms selected from N, O and S.
- a 5-10 membered heterocyclyl group may for instance be a group derived from pyrrolidine, imidazolidine, tetrahydrofuran,
- 5-6 membered heterocyclyl may be derived from pyrrolidine, imidazolidine, tetrahydrofuran, tetrahydropyran, piperidine, piperazine, and morpholine, i.e. pyrrolidinyl, imidazolidinyl, tetrahydrofuranyl, tetrahydropyranyl, piperidinyl, piperazinyl and morpholinyl.
- the heterocyclyl group may be linked to the rest of the molecule via any of its ring atoms, typically a ring C or N atom.
- 5-6 membered heterocyclyl is
- a heteroaryl group is a monocyclic or bicyclic heteroaromatic ring which typically contains from six to ten atoms in the ring portion including one or more heteroatoms.
- a heteroaryl group is generally a 5- or 6-membered ring, containing at least one heteroatom selected from O, S and N. It may contain, for example, one, two, three or four, e.g. one or two,
- heteroaryl groups include pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, furanyl, thienyl, pyrazolidinyl, pyrrolyl, tetrazolyl, oxazolyl, oxadiazolyl, isoxazolyl, thiadiazolyl, thiazolyl, isothiazolyl, imidazolyl, pyrazolyl, quinolyl and isoquinolyl.
- a heteroaryl group is pyridyl, pyrrolyl, tetrazolyl or oxazolyl.
- An aryl, heteroaryl or heterocyclyl group may be unsubstituted or substituted.
- a substituted group typically carries from 1 to 5, e.g. 1, 2 or 3, substituents.
- Halogen is chlorine, fluorine, bromine or iodine (a chloro group, a fluoro group, a bromo group or an iodo group). Halogen is typically chlorine or fluorine.
- Ci-4 alkoxy group is a group of formula -OR wherein R is a C1-4 alkyl group.
- Examples of Ci-4 alkoxy include -OMe, -OEt, -O n Pr, - ⁇ and -0*Bu.
- An alkylene group is a bivalent group derived by removing two hydrogen atoms from a straight chain or branched alkane.
- Examples of a C1-4 alkylene group are -CH 2 - -CH(CH 3 )- , -C(CH 3 ) 2 - -CH 2 CH 2 - -CH(CH 3 )CH 2 - -CH 2 CH(CH 3 )-, -CH 2 C(CH 3 ) 2 - -C(CH 3 ) 2 CH 2 - -CH 2 CH 2 CH 2 - -CH(CH 3 )CH 2 CH 2 -, -CH 2 CH(CH 3 )CH 2 - -CH 2 CH 2 CH(CH 3 )- and - CH 2 CH 2 CH 2 CH 2 -.
- a C alkylene group is -CH 2 - -CH 2 CH 2 - -CH 2 CH 2 CH 2 - or -CH 2 CH 2 CH 2 CH 2 - and a Ci -3 alkylene group is -CH 2 - -CH 2 CH 2 - or -CH 2 CH 2 CH 2 -.
- a reference to carboxylic acid or carboxyl group also includes the anionic (carboxylate) form (-COO " ), a salt or solvate thereof.
- a reference to an amino group includes the protonated form (- ⁇ + ⁇ 3 ⁇ 43 ⁇ 4 2 ), a salt or solvate of the amino group, for example, a hydrochloride salt.
- a reference to a hydroxyl group also includes the anionic form (-0 " ), a salt or solvate thereof.
- Certain compounds may exist in one or more particular geometric, optical, enantiomeric, diastereomeric, epimeric, atropic, stereoisomeric, tautomeric, conformational, or anomeric forms, including but not limited to, cis- and trans-forms; E- and Z-forms; c-, t-, and r- forms; endo- and exo-forms; R-, S-, and meso-forms; D- and L-forms; d- and 1-forms; (+) and (-) forms; keto-, enol-, and enolate-forms; syn- and anti-forms; synclinal- and anticlinal-forms; a- and ⁇ -forms; axial and equatorial forms; boat-, chair-, twist-, envelope-, and halfchair- forms; and combinations thereof, hereinafter collectively referred to as "isomers” (or "isomeric forms").
- a reference to a particular compound includes all such isomeric forms, including (wholly or partially) racemic and other mixtures thereof. Substantially optically pure forms are also included. Methods for the preparation (e.g., asymmetric synthesis) and separation (e.g., fractional crystallisation and chromatographic means) of such isomeric forms are either known in the art or are readily obtained by adapting known methods, in a known manner.
- salt refers to salt comprising a cationic or ionic form of the compound together with a counter ion. Such salts are typically acid addition salts. Often, the salt is a pharmaceutically acceptable salt.
- a pharmaceutically acceptable salt is a salt formed with a pharmaceutically acceptable acid or base.
- Pharmaceutically acceptable acids include both inorganic acids such as hydrochloric, sulphuric, phosphoric, diphosphoric, hydrobromic or nitric acid and organic acids such as citric, fumaric, maleic, malic, ascorbic, succinic, tartaric, benzoic, acetic, methanesulphonic, ethanesulphonic, benzenesulphonic or p- toluenesulphonic acid.
- Pharmaceutically acceptable bases include alkali metal (e.g. sodium or potassium) and alkali earth metal (e.g. calcium or magnesium) hydroxides and organic bases such as alkyl amines, aralkyl amines and heterocyclic amines.
- the invention provides a compound which is a peptide derivative of formula (1) or a salt thereof as defined above.
- Q is typically -C(0)-N(R N2 )-, -C(0)-C(R'R")-, -C(0)-0- -N(R N2 )-C(0)-, -C(R'R")- C(O)- or -O-C(O)-.
- Q is -C(0)-N(R N2 )-, -C(0)-CH 2 - or -C(0)-0-.
- Q is -C(0)-N(R N2 )-, most preferably -C(0)- H-
- Z is -C(0)-0-R° and R° is H, Ci-4 alkyl, phenyl or benzyl, wherein the phenyl and benzyl groups are optionally substituted with 1 to 4 substituents selected from R a , -OR' and halogen.
- Preferred substituents are Ci-4 alkyl, -CF 3 , -0(Ci-4)alkyl and halogen, in particular methyl and halogen.
- R° is unsubstituted.
- R° is H or Ci-4 alkyl optionally substituted with -O-Ci-4 alkyl.
- R° may be H or methyl, e.g. H.
- Z may be -CN4H (tetrazolyl).
- each R a is independently methyl or ethyl which is optionally substituted with 1 to 3 fluorine atoms.
- each R N1 and R N2 is independently H or methyl.
- R N1 and R N2 are each H.
- R N1 and R N2 may be the same or different.
- each R C1 is the same or different and is independently H or methyl.
- each R C2 is the same or different and is
- R C1 and R C2 are all H.
- each R', R", R" and R"" is independently H or C1-4 alkyl, preferably H.
- R G is H, -CH 2 -R GA or Ci-12 hydrocarbyl optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl; and R GA is Ci-n hydrocarbyl optionally substituted with 1 to 4 substituents selected from -NR'R", -OR', halogen and 5-6 membered heterocyclyl.
- R G may be H, -CH 2 -R GA or a Ci -8 alkyl group which is optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl; or R G and R N1 together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from R a , -OR' and halogen.
- R G is H, -CH 2 -R GA or a Ci-8 alkyl group which is optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl.
- R GA is typically a C 1 -7 alkyl group which is optionally substituted with 1 to 4 substituents selected from -NR'R", -OR', halogen and 5-6 membered heterocyclyl.
- R G may be C 1 -8 alkyl, phenyl or benzyl, each of which is optionally substituted with 1 to 4 substituents selected from halogen, -OH and -O-Ci-4 alkyl.
- R G is C 1 -8 alkyl optionally substituted with 1 to 4 substituents selected from halogen, -OH and -O-Ci-4 alkyl.
- R G is methyl or ethyl.
- R° is H or Ci-4 alkyl optionally substituted with -O-Ci-4 alkyl.
- R° is H, methyl or ethyl.
- R G may be C 1 -8 alkyl optionally substituted with 1 to 4 substituents selected from halogen, -OH and -O-Ci-4 alkyl and R° may be H or Ci-4 alkyl optionally substituted with -O-Ci-4 alkyl.
- R s is Ci-i6 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group, -(Ci-4 alkylene)-R SA or -(Ci-4 alkylene)-L-R SA ;
- R SA is Ci-ie hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group;
- L is -SO2-; wherein R s and R SA are optionally substituted with a group R b and/or from 1 to 5 substituents selected from R a , -NR'R", -OR',
- R s and an R C1 together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from R a , -OR' and halogen; wherein Ra, R' and R" are as defined herein, and R b is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl.
- each R' and R" is independently H or C1-4 alkyl which is optionally substituted with 1 to 3 substituents selected from halogen, -OH and -O- Ci-4 alkyl; and each R a is independently Ci-4 alkyl which is optionally substituted with 1 to 3 halogen atoms.
- R a is Ci-4 alkyl or -CF 3 and R' and R" are each independently H or Ci-4 alkyl.
- R s may be:
- R s is a group of formula:
- R x is H, phenyl, benzyl, C 3 - 6 cycloalkyl or Ci-4 alkyl.
- R s may be a group selected from the following groups:
- each R c and R is independently H or Ci-4 alkyl, preferably H.
- the compound is a peptide derivative of formula (2) or a salt thereof:
- R G is H, -CH 2 -R GA or a C 1-8 alkyl group which is optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl; or R G and R N1 together form a 5-6 membered heterocyclic ring which is optionally substituted with 1 to 3 substituents selected from R a , -OR' and halogen;
- R GA is Ci-7 alkyl group which is optionally substituted with 1 to 4 substituents selected from - R'R", -OR', halogen and 5-6 membered heterocyclyl;
- R s is Ci-16 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group, -(Ci-4 alkylene)-R SA or -(Ci-4 alkylene)-L-R SA ;
- R SA is Ci-i6 hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group;
- L is -SO2-; and wherein R s and R SA are optionally substituted with a group R b and/or from 1 to 5 substituents selected from R a , - R'R", -OR', - N0 2 , -CN, -C(0)R', -C(0)OR', -C(0) R'R", -0-C(0)R', -N(R')-C(0)R",
- R b is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl;
- R° is H, Ci-4 alkyl, phenyl or benzyl, wherein the phenyl and benzyl groups are optionally substituted with 1 to 4 substituents selected from R a , -OR' and halogen;
- R N1 and R N2 are each independently H or C1-4 alkyl
- R C1 and R C2 are each independently H or C1-4 alkyl
- each R' and R" is independently H or C1-4 alkyl which is optionally substituted with 1 to 3 substituents selected from halogen, -OH and -O-C1-4 alkyl;
- each R a is independently C1-4 alkyl which is optionally substituted with 1 to 3 halogen atoms.
- the compound is a peptide derivative of formula (3) or a salt thereof:
- R G is H, -CH 2 -R GA or a Ci -8 alkyl group which is optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl;
- R GA is Ci-7 alkyl group which is optionally substituted with 1 to 4 substituents selected from -NR'R", -OR', halogen and 5-6 membered heterocyclyl;
- R s is Ci-16 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group,
- R SA is Ci-ie hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group;
- R b is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl;
- R° is H or Ci-4 alkyl optionally substituted with -O-C1-4 alkyl
- each R' and R" is independently H or C1-4 alkyl.
- the compound of the invention may be a peptide derivative of formula (3) or a salt thereof:
- R G is Ci-8 alkyl optionally substituted with 1 to 4 substituents selected from halogen, -OH and -O-C1-4 alkyl.
- the compound is a peptide derivative of formula (4) or a salt thereof:
- R , R , R , R , R and R u are as defined herein.
- R G is methyl and R° is H or methyl.
- the compound of the invention may for instance be a compound of formula
- R s is a rou selected from
- the compound is , or a salt thereof.
- the compound of the invention is a salt of the peptide derivative, it is typically an acid addition salt.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a pharmaceutically acceptable salt, and (b) one or more pharmaceutically acceptable excipients or diluents.
- the compounds of the invention may be administered as pharmaceutical compositions in a variety of dosage forms. Thus, they can be administered orally, for example as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules.
- the compounds of the invention may also be administered parenterally, whether subcutaneously, intravenously, intramuscularly, intrasternally, transdermally or by infusion techniques.
- the compounds may also be administered as suppositories.
- the one or more pharmaceutically acceptable excipients or diluents may be any suitable excipients or diluents.
- a pharmaceutical composition which is a solid oral form may contain, together with the active compound, diluents, e.g. lactose, dextrose, saccharose, cellulose, corn starch or potato starch; lubricants, e.g. silica, talc, stearic acid, magnesium or calcium stearate, and/or polyethylene glycols; binding agents; e.g. starches, arabic gums, gelatin, methylcellulose, carboxymethylcellulose or polyvinyl pyrrolidone; disaggregating agents, e.g.
- a pharmaceutical composition which is a liquid dispersion for oral administration may be a syrup, emulsion and suspension.
- the syrups may contain as carriers, for example, saccharose or saccharose with glycerine and/or mannitol and/or sorbitol.
- Suspensions and emulsions may contain as carrier, for example a natural gum, agar, sodium alginate, pectin, methylcellulose, carboxymethylcellulose, or polyvinyl alcohol.
- the suspension or solutions for intramuscular injections may contain, together with the active compound, a pharmaceutically acceptable carrier, e.g. sterile water, olive oil, ethyl oleate, glycols, e.g. propylene glycol, and if desired, a suitable amount of lidocaine hydrochloride.
- Solutions for injection or infusion may contain as carrier, for example, sterile water or preferably they may be in the form of sterile, aqueous, isotonic saline solutions.
- antibiotic compounds can be used in combination with further active agents, and in particular in combination with one or more antibiotic compounds.
- antibiotic compounds may be provided in the form of pharmaceutically acceptable salts.
- the invention therefore provides a combination comprising (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a pharmaceutically acceptable salt, and (b) a further active agent which is an antibiotic compound.
- the further active agent may be any suitable antibiotic compound.
- the antibiotic compound may be a compound of the aminoglycoside class, ansamycin class, carbacephem class, carbapenem class, cephalosporin class, glycopeptide class, lincosamide class, lipopeptide class, macrolide class, monobactam class, nitrofuran class, oxazolidinone class, penicillin class, polypeptide class, quinolone class, sulfonamide class or tetracyclines class.
- the further antibiotic may be phenoxymethylpenicillin, flucloxacillin, amoxicillin, cefaclor, cefadroxil, cefalexin, tetracycline, doxycycline, lymecycline, gentamicin, tobramycin, erythromycin, azithromycin, clarithromycin, clindamycin, co-trimoxazole, metronidazole, tinidazole, ciprofloxacin, levofloxacin or norfloxacin.
- the combination typically further comprises (c) one or more pharmaceutically acceptable excipients or diluents.
- the one or more pharmaceutically acceptable excipients or diluents may be as defined above.
- the combination of the invention may for instance be a fixed combination. In a fixed combination, components (a) and (b) are present in the same composition.
- the fixed combination can be used for simultaneous administration of two compounds.
- the fixed combination is typically a solid composition (e.g. a tablet) or a solution (e.g. an injectable composition).
- the two components in a fixed combination are typically intermixed.
- the combination of the invention may be a free combination.
- the active components (a) and (b) are typically separate from each other and packaged in one unit for simultaneous, substantially simultaneous, separate or sequential administration.
- the invention also provides a kit comprising (a) a compound which is a peptide derivative or a salt thereof as herein, wherein the salt is a pharmaceutically acceptable salt, and (b) a further antibiotic compound.
- the kit may further comprise instructions for using or administering components (a) and (b), for instance in a method of treating or preventing an infection caused by bacteria.
- a soap product is a composition or article which is suitable for washing the skin.
- the soap product may be a liquid soap product (for instance a hand wash, body wash, shampoo or face wash) or a solid soap product (for instance a bar of soap or a disposable wipe).
- a soap product according to the invention typically further comprises a soap, which is usually a surfactant. Examples of soaps include salts of fatty acids (i.e.
- linear C 6 -i8 alkyl or alkenyl carboxylic acids for instance a sodium or potassium salt of lauric acid, myristic acid, palmitic acid, stearic acid, oleic acid, linoleic acid or linolenic acid, and salts of long chain sulfonic acids (i.e. linear C 6 -i8 alkyl or alkenyl sulfonic acids), for instance sodium laureth sulfate.
- the soap product may for instance be a liquid soap product comprising one or more solvents, for instance selected from water and alcohols. Often, the soap product is a liquid soap product which further comprises ethanol.
- a cleaning product includes household cleaning products such as surface cleaning products, fabric cleaners, washing powders, and cleaning products suitable for use in a medical environment, for example to clean or disinfect surfaces in a hospital or equipment found in a clinical environment such as medical equipment. Treatment or prevention of bacterial infections
- the compounds of the invention can kill or inhibit the growth of bacteria.
- the invention therefore provides a compound which is a peptide derivative or a salt thereof as defined herein, for use in the treatment or prevention of an infection caused by bacteria, wherein the salt is a pharmaceutically acceptable salt.
- the compounds of the invention can treat a number of different bacterial infections.
- the infection may be caused by bacteria which are gram-negative or gram-positive.
- the bacterial infection is caused by gram-negative bacteria.
- the infection may be caused by bacteria of genus Shewanella, Acinetobacter, Bacillus, Bartonella, Bordetella, Borrelia, Brucella, Campylobacter, Chlamydia, Chlamydophila, Clostridium, Corynebacterium, Enterobacter, Enterococcus, Escherichia, Francisella, Haemophilus, Helicobacter,
- the bacterial infection may be caused by bacteria of genus Shewanella or Escherichia.
- Gram-negative bacteria include bacteria of the genus Shewanella, Escherichia, Salmonella, Shigella, Pseudomonas, Moraxella, Helicobacter, Stenotrophomonas, Bdellovibrio and Legionella.
- the compound of the invention may be used to treat or prevent a bacterial infection caused by Staphylococcus aureus, Escherichia coli, Shewanella denitrificans, Hemophilus influenzae, Klebsiella pneumoniae, Legionella
- pneumophila Pseudomonas aeruginosa, Proteus mirabilis, Enterobacter cloacae, Serratia marcescens, Helicobacter pylori, Salmonella enteritidis, Salmonella typhi ox Acinetobacter baumannii.
- the invention also provides a method of treating or preventing infection caused by bacteria as defined above in a subject, which method comprises administering an effective amount of a peptide derivative or a salt thereof as defined herein to the subject, wherein the salt is a pharmaceutically acceptable salt.
- the invention also provides the use of a peptide derivative or a salt thereof as defined herein in the manufacture of a medicament for use in the treatment or prevention of infection caused by bacteria as defined above in a subject.
- the subject is typically a mammal, more typically a human.
- the bacterial infection may be of any part of the body, for instance part of the skin, the lungs or the gut.
- a therapeutically effective amount of a compound of the invention is administered to a patient.
- a typical dose is from 0.001 to 1000 mg, according to the activity of the specific compound, the age, weight and conditions of the subject to be treated, the type and severity of the disease and the frequency and route of administration.
- daily dosage levels are from 0.001 mg to 4000 mg.
- the compounds of the invention may be used alone, or alternatively they may be used in combination with a second active agent, typically an antibiotic compound.
- the combination may be for use in the treatment or prevention of an infection caused by bacteria as defined above, which combination comprises (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a pharmaceutically acceptable salt, and (b) a further active agent which is an antibiotic compound or a pharmaceutically acceptable salt thereof.
- the invention provides a compound which is a peptide derivative or a salt thereof as defined herein, for use in the treatment or prevention of an infection caused by bacteria as defined herein combination with a further active agent which is an antibiotic compound, wherein the salt is a pharmaceutically acceptable salt.
- a method of treating or preventing infection caused by bacteria as defined herein in a subject is also described, which method comprises administering an effective amount of a peptide derivative or a salt thereof as defined herein to the subject in combination with a further active agent which is an antibiotic compound, wherein the salt is a pharmaceutically acceptable salt.
- the invention also provides the use of a peptide derivative or a salt thereof as defined herein in a the manufacture of a medicament for use in the treatment or prevention of infection caused by bacteria as defined herein in a subject, in combination with a further active agent which is an antibiotic compound.
- the compound of the invention and the further active agent may be used simultaneously or sequentially.
- the invention also provides a method for preventing or treating infection of a plant caused by bacteria, which method comprises treating the plant, the seed from which the plant grows, or the place at which the plant grows, with a composition comprising a compound as defined herein.
- the infection of the plant is typically caused by bacteria of the genus Shewanella, Erwinia, Agrobacterium, Phytoplasma, Burkholderia, Proteobacteria, Xanthomonas or Pseudomonas.
- Treating the plant, seed or place typically comprises applying a composition comprising the compound to the plant, seed or place. This may comprise spraying an aqueous solution of a compound of the invention on the plant, seed or place. The treatment may be repeated daily or weekly.
- the plant to be treated may be a wide range of plants and is typically a crop plant.
- the plant may be a cereal-producing plant, a vegetable-producing plant or a fruit- producing plant.
- the invention also provides an in vitro method for killing or inhibiting the growth of bacteria, which method comprises applying a composition comprising a compound which is a peptide derivative or salt thereof as defined herein to a surface, to an article or to the skin.
- the bacteria may be as defined herein. Said method is typically not a method of treatment.
- Compounds of the invention may be produced by a thiol-ene reaction.
- the invention also provides a process for producing a compound which is a peptide derivative of formula (la) or a salt thereof:
- R G , R N 1 , R C1 , Q, R C2 and Z are as defined herein and the group
- R S1 , R S2 , R S4 are independently H or C1-4 alkyl and
- R S3 is a Ci-16 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group, -(C1-2 alkylene)-R SA or -(C1-2 alkylene)-L-R SA ;
- R SA is C1-16 hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group;
- R b is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl;
- each R' and R" is independently H or C1-4 alkyl which is optionally substituted with 1 to 3 substituents selected from halogen, -OH and -O-C1-4 alkyl;
- each R a is independently C1-4 alkyl which is optionally substituted with 1 to 3 halogen atoms.
- R S1 , R S2 , R S4 may be H and R S3 may be phenyl or -S02-phenyl and R S3 is optionally substituted with a group selected from halogen, -NO2, -OMe and -CF 3 .
- compound (A) is a compound of formula (3 A) or a salt thereof:
- R G is H, -CH 2 -R GA or a C 1-8 alkyl group which is optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl;
- R GA is Ci-7 alkyl group which is optionally substituted with 1 to 4 substituents selected from - R'R", -OR', halogen and 5-6 membered heterocyclyl;
- R° is H or Ci-4 alkyl optionally substituted with -O-Ci-4 alkyl
- each R' and R" is independently H or Ci-4 alkyl.
- R G may be Ci -8 alkyl optionally substituted with 1 to 4 substituents selected from halogen, -OH and -O-Ci-4 alkyl and R° may be H or Ci-4 alkyl optionally substituted with - O-Ci-4 alkyl.
- compound (B) is a compound selected from the formulae:
- n is from 0 to 11.
- the initiator may be a chemical initiator (for instance a free radical) or a physical initiator (for instance UV light or heat). Typically, the initiator is UV light.
- the reaction may use UV light together with a chemical initiator (e.g. 2,2-dimethoxyphenyl acetophenone).
- the temperature of the reaction is typically from 10 to 40°C.
- the alkene (compound (B)) and the thiol (compound (A)) are stirred at from 10 to 40°C in a solvent (for instance THF/H2O (1 :2, 3 mL)) in the presence of light (e.g. at 365 nm) for from 1 to 10 h. After this time the reaction may be filtered, and the solid washed with ethanol and water.
- a solvent for instance THF/H2O (1 :2, 3 mL
- GSH DNGSH N-Allyl-5-(dimethylamino)naphthalene-l -sulfonamide (0.10 g, 0.34 mmol, 1.0 eq.)
- L- glutathione (0.42 g, 1.36 mmol, 4.0 eq.)
- TCEP HCl (0.19 mg, 0.68 mmol, 2.0 eq.
- 2,2- dimethoxyphenyl acetophenone 0.02 g, 0.07 mmol, 0.2 eq.
- reaction solution was lyophilized, purified by RP C-18 silica gel chromatography, eluting with water to afford ⁇ -N-tert-butylsuccinimido glutathione 3 ⁇ 4uSG (mixture of diastereoisomers) as a colourless hygroscopic solid (0.26 g, 87%): R t 0.7 (water (RP)); [ ⁇ ° -13.3 (c 1.0, water) (Lit.
- Glu-Cys (15.3 mg, 0.06 mmol, 1.0 eq.) and NaOH (2.4 mg, 0.06 mmol, 1.0 eq.) were stirred in water (0.4 mL), to this N-tert-butyl-maleimide (9.4 mg, 8.8 ⁇ ., 0.06 mmol, 1.0 eq.) was added and the resulting solution stirred at RT for 4.5 h.
- N-acetyl cysteine (20.0 mg, 0.12 mmol, 1.0 eq.) and NaOH (4.9 mg, 0.12 mmol, 1.0 eq.) were stirred in water (0.9 mL), to this N-tert-butyl-maleimide (18.7 mg, 17.7 L, 0.12 mmol, 1.0 eq.) was added and the resulting solution stirred at RT for 3.5 h.
- Ci 3 H 2 i0 5 N 2 S requires M + 317.11657); m/z (ES + ) 317 ([M+H] + , 100%); Analytical HPLC @ 220 nm (Acclaim ® 120 C18 RP LC
- N-acetyl-L-cysteine methyl ester 12 (0.40 g, 2.26 mmol, 1.0 eq.) was stirred in a mixture of toluene (3 mL) and ammonium hydroxide (3 mL, 25% solution in water) under argon at RT for 19 h. After this time the reaction mixture was concentrated in vacuo at 60 °C to afford a mixture of N-acetyl-L-cysteinamide and oxidised N-acetyl-L-cysteinamide (0.37 g) as a colourless solid, which was used without further purification.
- Ci 4 H 24 N 3 0 5 S requires M + 346.14312); m/z (ES + ) 346 ([M+H] + , 100%); Analytical HPLC @ 220 nm (Acclaim ® 120 C18 RP LC Column; 95:5:0.1
- N-Fmoc-D-Cys(Trt)-OH (0.50 g, 0.85 mmol, 1.05 eq.)
- glycine tert-butyl ester hydrochloride (0.14 g, 0.81 mmol, 1.0 eq.)
- HBTU 0.32 g, 0.85 mmol, 1.05 eq.
- DIPEA 0.21 g, 0.28 mL, 1.62 mmol, 2.0 eq.
- N-Fmoc- ⁇ -trityl-D-cysteinylglycine tert-butyl ester 14 (0.4 g, 0.57 mmol, 1.0 eq.) was stirred under argon at RT in 20% piped dine in DMF (3.2 mL) for 4 h.
- ⁇ -trityl-D-cysteinyl tert-butyl ester 15 (0.11 g, 0.23 mmol, 1.0 eq.) and triethylsilane (0.06 g, 0.08 mL, 0.5 mmol, 2.2 eq.) were stirred under argon at RT in 20% TFA in CH 2 C1 2 (5.8 mL) for 7 h. After this time the reaction solution was concentrated in vacuo and resulting residue was purified by RP C-18 silica gel column chromatography, eluting with water to afford D- cysteinylglycine trifluoroacetic acid salt as an off-white sticky foam (55.2 mg), which was used without further purification.
- Ci 3 H 22 N 3 0 5 S requires M + 332.12747); m/z (ES + ) 332 ([M+H] + , 100%); Analytical HPLC @ 220 nm (Acclaim ® 120 C18 RP LC Column; 95:5:0.1
- Ci 5 H 24 N 3 0 6 S requires M + 374.13803); m/z (ES + ) 374 ([M+H] + , 100%); Analytical HPLC @ 220 nm (Acclaim ® 120 C18 RP LC Column; 95:5:0.1
- Ci 5 H 22 0 6 N 3 S requires M " 372.12348); m z (ES + ) 374 ([M+H] + , 100%); Analytical HPLC @ 220 nm (Acclaim ® 120 CI 8 RP LC
- Boc-J-Cys(Trt)-OH (0.1 g, 0.22 mmol, 1.0 eq.) and fert-butyl-2-hydroxyacetate (55.6 mg, 0.42 mmol, 1.95 eq.) were stirred in anhydrous acetonitrile at 0 °C, to this ⁇ , ⁇ '- dicyclohexylcarbodiimide (86.8 mg, 0.42 mmol, 1.95 eq.) was added and the resulting solution stirred under argon at 0 °C for a further 2 h before being stirred at RT for 15.5 h.
- N- (tert-butoxycarbonyl ⁇ -trityl-J-cysteinate (0.11 g) and triethylsilane (0.08 g, 0.11 mL, 0.69 mmol) were stirred under argon at RT in 20% TFA in CH2CI2 (6.5 mL) for 2 h. After this time the reaction solution was concentrated in vacuo and the resulting residue was purified by RP C-18 silica gel column chromatography, eluting with water to afford 2-((L- cysteinyl)oxy)acetic acid as an impure colourless solid (32.2 mg), which was used without further purification.
- reaction solution was lyophilized and the resulting solid was purified by semi-preparative HPLC (Agilent ZORBAX 300SB-C18 column; 95 :5:0.1 5:95:0.1 ; H 2 0 : MeCN : TFA) to afford (2R)-3-((l-(tert-butyl)-2,5- dioxopyrrolidin-3 -yl)thio)- 1 -(carboxymethoxy)- 1 -oxopropan-2-aminium 2,2,2- trifluoroacetate (mixture of diastereoisomers) as a colourless solid (25.5 mg), which was used without further purification.
- Ci 5 H 23 0 7 N 2 S requires M + 375.12205); m z (ES + ) 375 ([M+H] + , 100%); Analytical HPLC @ 220 nm (Acclaim® 120 C18 RP LC Column; 95:5:0.1
- N-Fmoc-Cys(Trt)-OH 20 (0.75 g, 1.28 mmol, 1.05 eq.), glycine tert-butyl ester hydrochloride (0.20 g, 1.22 mmol, 1.0 eq.) and HBTU (0.49 g, 1.28 mmol, 1.05 eq.) were stirred in anhydrous DMF (3 mL) for 5 minutes. After this time, DIPEA (0.32 g, 0.42 mL, 2.43 mmol, 2.0 eq.) was added and the reaction mixture was stirred under argon at RT for 24 h.
- reaction mixture was partitioned between ethyl acetate and 0.5 M LiCl and the organic layer was collected, dried over Na 2 S04, filtered and concentrated in vacuo.
- the resulting crude material was adsorbed onto Celite ® and purified by silica gel chromatography, eluting with ethyl acetate, petroleum ether and triethylamine (10:89: 1 - 80: 19: 1; ethyl acetate :
- N-Fmoc- ⁇ -trityl-L-cysteinylglycine tert-butyl ester 21 (0.75 g, 1.07 mmol, 1.0 eq.) was stirred under argon at RT in 20% piperidine in DMF (6 mL) for 5 h.
- ⁇ -Trityl-L-cysteinylglycine tert-butyl ester 22 (89.3 mg, 0.19 mmol, 1.0 eq.), N-boc-Y- aminobutyric acid (38.1 mg, 0.19 mmol, 1.0 eq.), EDC DHC1 (41.3 mg, 0.22 mmol, 1.15 eq.), HBTU (7.1 mg, 0.02 mmol, 0.1 eq.) and DIPEA (48.4 mg, 65.3 L, 0.37 mmol, 2.0 eq.) were stirred at RT in anhydrous CH2CI2 (9 mL) for 24 h.
- reaction mixture was concentrated in vacuo and the resulting residue was adsorbed onto Celite and purified by silica gel chromatography, eluting with ethyl acetate, petroleum ether and triethylamine (12:87: 1 - 99:0: 1; ethyl acetate : petroleum ether : triethylamine) to yield N-boc-Y- aminobutyric acid- ⁇ -trityl-L-cysteinylglycine tert-butyl ester 23 (0.10 g) as a colourless oil, which was used without further purification.
- N-Boc- Y-aminobutyric acid- ⁇ -trityl-L-cysteinylglycine tert-butyl ester 23 (95.0 mg) and triethylsilane (50.1 mg, 68.8 L, 0.43 mmol) were stirred in 20% TFA/CH 2 C1 2 (5 mL) at RT for 6.5 h. After this time, the reaction mixture was concentrated in vacuo and the resulting residue was purified by RP C-18 silica gel column chromatography, eluting with water to afford Y-aminobutyric acid-L-cysteinylglycine trifluoroacetic acid salt (33.3 mg) as a colourless residue, which was used without further purification.
- ⁇ -Trityl-L-cysteinylglycine tert-butyl ester (0.21 g, 0.44 mmol, 1.0 eq.) and triethylamine (0.06 g, 0.08 mL, 0.57 mmol, 1.3 eq.), were stirred at RT in anhydrous chloroform (2 mL) for 5 minutes. After this time, glutaric anhydride (0.06 g, 0.48 mmol, 1.1 eq.) was added and the reaction mixture was stirred at RT for 15.5 h.
- reaction solution was concentrated in vacuo and the resulting residue was adsorbed onto Celite ® and purified by silica gel chromatography, eluting with ethyl acetate, petroleum ether and formic acid (60:40: 1) to yield slightly impure glutaric acid- ⁇ -trityl-L-cysteinylglycine tert-butyl ester (21.9 mg) as a colourless oil, which was used without further purification.
- the compounds tested are as follows.
- KD values were determined for certain compounds using 3 ⁇ 4 CPMG NMR.
- the titration, experiments were performed with a fixed concentration of the ligand (typically 10 ⁇ ) and a range of protein concentrations (typically 0-30 ⁇ ; the concentration was increased until complete saturation of the ligand signal was achieved).
- the % decrease in signal intensity of the ligand was calculated as follows:
- IB is the intensity of the ligand signal in the absence of protein and Ip is the intensity of the ligand signal in the presence of the protein at a given concentration.
- y is the % decrease in ligand signal intensity
- A is the maximum loss in the signal intensity
- C is the ligand concentration
- B is the KD
- X is the concentration of the protein.
- test discs were prepared by impregnating 20 iL of different concentrations of test compounds onto Whatman ® antibiotic assay discs of 6 mm diameters (Sigma- Aldrich Co.). After the cell culture had been reached the cell density of 0.5 McFarland turbidity standard, the cell isolate was inoculated onto Muller-Hinton agar (MHA; Sigma-Aldrich Co.) plates by using sterile cotton swab. Subsequently the compound discs were aseptically placed onto the bacterial plates. After 24 h of incubation at 37 °C, inhibition zone diameters associated with the inhibition effect of the compounds were recorded by averaging both vertical and horizontal diameters of each zone. The whole experimental procedures were performed according to the standards set by the Clinical and Laboratory Standards Institute.
- the assay was performed using compound 4 which was used at a number of concentrations.
- the bacteria used was E. coli MJF335 (pTrcSdKefH6) which has the genotype Frag5, lacl, kefB, kefC::Tnl0, gshA::Tnl0(Kan).
- the disk diameter was 6 mm.
- the results of the assay are shown in Figure 11. A dose dependent increase in bacterial inhibition is clearly visible.
Abstract
The invention provides a compound which is a peptide derivative of formula (1) as defined in the claims or a salt thereof: RG is H, -CH2-RGA or C1-12 hydrocarbyl optionally substituted with 1 to 4 substituents selected from -OR', -NO2, -CN, halogen and 5-6 membered heterocyclyl; or RG and RN1 together form a 5-6 membered heterocyclic ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen; RGA is C1-11 hydrocarbyl optionally substituted with 1 to 4 substituents selected from -NR'R", -OR', -NO2, -CN, halogen and 5-6 membered heterocyclyl; RS
is C1-16 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group, -(C1-4 alkylene)-RSA or -(C1-4 alkylene)-L-RSA; RSA is C1-16 hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group; L is -SO2-; and wherein RS and RSA are optionally substituted with a group Rb and/or from 1 to 5 substituents selected from Ra, -NR'R", -OR', -NO2, -CN, -C(O)R', -C(O)OR', -C(O)NR'R", -0-C(0)R', -N(R')-C(0)R", -SR', -S(O)R', -S(O)2R', -S(O)2OR', -P(O)(OR')(OR"), =O, -SiR'R"R" and halogen; or Rs and an RC1 together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen; Rb is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl; Q is -C(O)-N(RN2)-, -C(O)-C(R'R")-, -C(O)-O- -N(RN2)-C(O)-, -C(R'R")- C(O)-, -O-C(O)-, -C(R'R")-C(R"'R"")-, -C(R'R")-O- -O-C(R'R")-, -C(R'R")-N(RN2)-, -N(RN2)-C(R'R")- or -C(R')=C(R")-; Z is a group selected from -C(O)-O-RO and -CN4R' (tetrazolyl); RO is H or C1-12 hydrocarbyl optionally substituted with 1 to 4 substituents selected from -NR'R", -OR', -NO2, -CN, -C(O)R', -C(O)OR', -C(O)NR'R", -O-C(O)R', -N(R')- C(O)R", -SR', -S(O)R', -S(O)2R', -S(O)2OR', -P(O)(OR')(OR"), =O, halogen and 5-6 membered heterocyclyl; or RO and an RC2 together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen; each RN1 and RN2 is independently H or C1-4 alkyl; each RC1, RC2, R', R", R" and R"" is independently H or C1-4 alkyl and is optionally substituted with 1 to 3 substituents selected from halogen, -OH and -O-C1-4 alkyl; and each Ra is independently C1-4 alkyl which is optionally substituted with 1 to 3 halogen atoms.
Description
ANTIBACTERIAL AGENT
FIELD OF THE INVENTION
The present invention relates to compounds useful as antibacterial agents. The use of such compounds in treating or preventing bacterial infection is also described.
BACKGROUND OF THE INVENTION
The potassium efflux system, Kef, protects bacteria against the detrimental effects of electrophilic compounds via acidification of the bacterial cytoplasm. Glutathione (GSH) and glutathione-S-conjugate (GS-X) compounds are known to affect Kef. In particular, GSH inhibits Kef, whereas some GS-X compounds activate Kef (Healy et al, Understanding the Structural Requirements for Activators of the Kef Bacterial Potassium Efflux System, Biochemistry, 2014 Apr 1 ; 53(12): 1982-92). Activation of the Kef system results in K+ efflux and concomitant H+ influx. Consequently, Kef activation results in a decrease in cellular pH, protonating nucleophiles in DNA and proteins and minimising their reactivity with electrophilic species. Kef activation is thus known to interfere with normal bacterial functioning and therefore has the potential as a therapeutic target for preventing or treating bacterial infections.
SUMMARY OF THE INVENTION
The invention provides a compound which is a peptide derivative of formula (1) or a salt thereof:

wherein:
RG is H, -CH2-RGA or C1-12 hydrocarbyl optionally substituted with 1 to 4
substituents selected from -OR', -N02, -CN, halogen and 5-6 membered heterocyclyl; or RG and RN1 together form a 5-6 membered heterocyclic ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen;
RGA is Ci-ii hydrocarbyl optionally substituted with 1 to 4 substituents selected from - R'R", -OR', -NO2, -CN, halogen and 5-6 membered heterocyclyl;
Rs is Ci-16 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group,
-(Ci-4 alkylene)-RSA or -(C1-4 alkylene)-L-RSA; RSA is Ci-ie hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group; L is -SO2-; and wherein Rs and RSA are optionally substituted with a group Rb and/or from 1 to 5 substituents selected from Ra, -NR'R", -OR', -NO2, -CN, -C(0)R', -C(0)OR', -C(0)NR'R", -0-C(0)R', -N(R')-C(0)R", -SR', -S(0)R', -S(0)2R', -S(0)2OR', -P(0)(OR')(OR"), =0, -SiR'R"R" and halogen; or Rs and an RC1 together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen;
Rb is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl;
Q is -C(0)-N(RN2)-, -C(0)-C(R'R")-, -C(0)-0- -N(RN2)-C(0)-, -C(R'R")- C(O)-, -O-C(O)-, -C(R'R")-C(R"'R"")-, -C(R'R")-0- -0-C(R'R")-, -C(R'R")-N(RN2)-, -N(RN2)-C(R'R")- or -C(R')=C(R")-;
Z is a group selected from -C(0)-0-R° and -CN4R' (tetrazolyl);
R° is H or Ci-12 hydrocarbyl optionally substituted with 1 to 4 substituents selected from -NR'R", -OR', -NO2, -CN, -C(0)R', -C(0)OR', -C(0)NR'R", -0-C(0)R', -N(R')- C(0)R", -SR', -S(0)R', -S(0)2R', -S(0)2OR', -P(0)(OR')(OR"), =0, halogen and 5-6 membered heterocyclyl; or R° and an RC2 together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen; each RN1 and RN2 is independently H or C1-4 alkyl;
each RC1, RC2, R', R", R" and R"" is independently H or C alkyl and is optionally substituted with 1 to 3 substituents selected from halogen, -OH and -O-C1-4 alkyl; and
each Ra is independently C1-4 alkyl which is optionally substituted with 1 to 3 halogen atoms.
The inventors have found surprisingly that the glutamate residue of GS-X compounds makes a negligible contribution to the affinity of the compounds to Kef. Thus, truncation or replacement of the glutamate residue in GS-X compounds has allowed the development of a new class of antibacterial compounds. These compounds not only demonstrate good binding
to Kef, but have also been shown to cause activation of the Kef system. Moreover, the compounds have reduced polar groups and a shorter peptide chain, and therefore are expected to demonstrate better ability to cross cell membranes than the known GSH. The compounds are thus useful in the treatment and prevention of bacterial infections in animals as well as plants. These compounds can effectively inhibit or damage bacteria through activation of the Kef system and resulting K+ efflux.
The invention also provides a pharmaceutical composition comprising (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a
pharmaceutically acceptable salt, and (b) one or more pharmaceutically acceptable excipients or diluents.
The invention also provides a compound which is a peptide derivative or a salt thereof as defined herein, for use in the treatment or prevention of an infection caused by bacteria, wherein the salt is a pharmaceutically acceptable salt.
The invention also provides a method of treating or preventing infection caused by bacteria in a subject, wherein the infection is as defined herein, which method comprises administering an effective amount of a peptide derivative or a salt thereof as defined herein to the subject, wherein the salt is a pharmaceutically acceptable salt.
The invention also provides a combination comprising (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a pharmaceutically acceptable salt, and (b) a further active agent which is an antibiotic compound or a pharmaceutically acceptable salt thereof.
The invention also provides a combination for use in the treatment or prevention of an infection caused by bacteria as defined herein, which combination comprises (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a pharmaceutically acceptable salt, and (b) a further active agent which is an antibiotic compound.
The invention also provides a kit comprising (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a pharmaceutically acceptable salt, and (b) a further antibiotic compound or a pharmaceutically acceptable salt thereof.
The invention also provides a compound which is a peptide derivative or a salt thereof as defined herein for use in a method of activating potassium efflux in bacteria, wherein the bacteria are as defined herein.
The invention also provides a method for preventing or treating infection of a plant caused by bacteria, which method comprises treating the plant, the seed from which the plant grows, or the place at which the plant grows, with a composition comprising a compound as defined herein.
The invention also provides an in vitro method for killing or inhibiting the growth of bacteria, which method comprises applying a composition comprising a compound which is a peptide derivative or salt thereof as defined herein to a surface, to an article or to the skin. In the case of application to the skin, the method is typically not a method of treatment of the human or animal body, but merely a method for killing or inhibiting bacteria present on the skin.
The invention also provides a soap product, washing product or cleaning product comprising a compound which is a peptide derivative or salt thereof as defined herein.
The invention also provides a process for producing a compound which is a peptide derivative of formula (la) or a salt thereof:

which process comprises reacting a thiol compound of formula (A) with an alkene of formula (B) in the presence of an initiator,

wherein RG, RN1, RC1, Q, RC2 and Z are as defined herein and the group

BRIEF DESCRIPTION OF THE FIGURES
Figures 1 to 3 show the results of competition fluorescence assays for compounds according to the invention and comparative compounds.
Figures 4 to 10 show Kd curves for various compounds obtained by CPMG MR.
Figure 11 shows the results of a Kirby-Bauer disk diffusion assay performed for a compound of the invention.
DETAILED DESCRIPTION OF THE INVENTION
Definitions
A hydrocarbyl group is a group comprising only carbon atoms and hydrogen atoms. Thus, hydrocarbyl groups are those derived by the removal of a hydrogen atom from a hydrocarbon compound. Examples of hydrocarbyl groups include alkyl groups, cycloalkyl groups, alkenyl groups, cycloalkenyl groups, alkynyl groups, aryl groups, aryl groups substituted with one or more alkyl groups, alkyl groups substituted with one or more aryl groups and alkyl groups substituted with aryl groups which are themselves substituted with one or more alkyl groups. A hydrocarbyl group may be defined by the number of carbon atoms it contains. For instance, a Ci-i6 hydrocarbyl group comprises from 1 to 16 carbon atoms. Examples of a Ci- 16 hydrocarbyl group include Ci-i6 alkyl, phenyl, C3-6 cycloalkyl, and Ci-10 alkyl substituted
with a phenyl or C3-6 cycloalkyl group, wherein each of these groups may be substituted with 1 to 3 Ci-3 alkyl groups. C1-16 hydrocarbyl is typically Ci-12 hydrocarbyl or C 1-11
hydrocarbyl. Typically, Ci-12 hydrocarbyl or C 1-11 hydrocarbyl is Ci-8 alkyl, or a C3-6 cycloalkyl, phenyl, benzyl or ethyl-phenyl group, each of which may optionally be substituted with 1 to 3 C1-4 alkyl groups.
An alkyl group is a linear or branched chain saturated hydrocarbon group. An alkyl group may be a C1-16 alkyl group, a Ci-8 alkyl group, or a C1-4 alkyl group. Examples of a C1-16 alkyl group are methyl, ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, sec-butyl, tert-butyl, pentyl, neopentyl, hexyl, heptyl and octyl.
A cycloalkyl group is typically a C3-6 cycloalkyl group. Examples of a C3-6 cycloalkyl group include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl. A cycloalkyl group is unsubstituted or substituted. Typically, it is substituted with one or two C1-4 alkyl groups, e.g. 1 or 2 methyl groups, or is unsubstituted. Preferably, a cycloalkyl group is unsubstituted.
An alkenyl group is a linear or branched chain hydrocarbon radical comprising one or more double bonds. An alkenyl group may be a C2-10 alkenyl group. Examples of C2-10 alkenyl groups include those related to C2-10 alkyl groups by the insertion of one or more double bonds, e.g. ethenyl (-CH=CH2), propenyl (-CH2-CH=CH2 or -CH=CH-CH3) and butenyl (- CH2-CH2-CH=CH2 or -CH2-CH=CH-CH3 or -CH=CH-CH2-CH3). Alkenyl groups typically comprise one or two double bonds.
An alkynyl group is a linear or branched chain hydrocarbon radical comprising one or more triple bonds. An alkynyl group may be a C2-10 alkynyl group. Examples of C2-10 alkynyl groups include those related to C2-10 alkyl groups by the insertion of one or more triple bonds, e.g. ethynyl (-C≡CH), propynyl (-CH2-C≡CH or -C≡C-CH3) and butynyl (-CH2-CH2- C≡CH or -CH2-C≡C-CH3 or -C≡C-CH2-CH3). Alkynyl groups typically comprise one or two triple bonds.
An alkyl, alkenyl or alkynyl group may be unsubstituted or substituted with one or more, e.g. from 1 to 3, substituents. Typical substituents are selected from - R'R", -OR', halogen, 5-6 membered heterocyclyl, -C(0)R', -C(0)OR', -C(0) R'R", -0-C(0)R', -N(R')-C(0)R", - SR', -S(0)R', -S(0)2R', -S(0)2OR', -P(0)(OR')(OR"), =0 and -SiR'R"R", wherein each R', R" and R" is independently H or C1-4 alkyl and is optionally substituted with 1 to 3 substituents selected from halogen, -OH and -O-C1-4 alkyl. Preferred substituents are
selected from - R'R", -OR', halogen and 5-6 membered heterocyclyl, wherein each R' and R" is independently H or Ci-4 alkyl, most preferably from -NR'R", -OR', halogen and morpholinyl, wherein each R' and R" is independently H or Ci-4 alkyl. Typically, an alkyl, alkenyl or alkynyl group is unsubstituted or substituted with from 1 to 3 halogen atoms, e.g. it is unsubstituted.
An aryl group is a monocyclic, bicyclic or polycyclic aromatic ring which contains from 6 to 14 carbon atoms, typically from 6 to 10 carbon atoms, in the ring portion. Examples include phenyl, naphthyl, indenyl, indanyl, anthracenyl and pyrenyl groups. Often an aryl group is phenyl.
A heterocyclyl group is a monovalent moiety obtained by removing a hydrogen atom from a ring atom of a heterocyclic compound. Preferably, a heterocyclyl group has from 5 to 10 ring atoms (5-10 membered heterocyclyl), more preferably 5 or 6 ring atoms (5-6 membered heterocyclyl), including at least one (e.g. one, two, three or four, preferably one or two) heteroatoms selected from N, O and S. A 5-10 membered heterocyclyl group may for instance be a group derived from pyrrolidine, imidazolidine, tetrahydrofuran,
tetrahydropyran, piperidine, piperazine, morpholine, azepane, oxepane and indoline. 5-6 membered heterocyclyl may be derived from pyrrolidine, imidazolidine, tetrahydrofuran, tetrahydropyran, piperidine, piperazine, and morpholine, i.e. pyrrolidinyl, imidazolidinyl, tetrahydrofuranyl, tetrahydropyranyl, piperidinyl, piperazinyl and morpholinyl. The heterocyclyl group may be linked to the rest of the molecule via any of its ring atoms, typically a ring C or N atom. Typically, 5-6 membered heterocyclyl is

A heteroaryl group is a monocyclic or bicyclic heteroaromatic ring which typically contains from six to ten atoms in the ring portion including one or more heteroatoms. A heteroaryl group is generally a 5- or 6-membered ring, containing at least one heteroatom selected from O, S and N. It may contain, for example, one, two, three or four, e.g. one or two,
heteroatoms. Examples of heteroaryl groups include pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, furanyl, thienyl, pyrazolidinyl, pyrrolyl, tetrazolyl, oxazolyl, oxadiazolyl,
isoxazolyl, thiadiazolyl, thiazolyl, isothiazolyl, imidazolyl, pyrazolyl, quinolyl and isoquinolyl. Typically, a heteroaryl group is pyridyl, pyrrolyl, tetrazolyl or oxazolyl.
An aryl, heteroaryl or heterocyclyl group may be unsubstituted or substituted. A substituted group typically carries from 1 to 5, e.g. 1, 2 or 3, substituents. Substituents are typically selected from Ra, - R'R", -OR', -N02, -CN, -C(0)R', -C(0)OR', -C(0) R'R", -O- C(0)R', -N(R')-C(0)R", -SR', -S(0)R', -S(0)2R', -S(0)2OR', -P(0)(OR')(OR"), =0, - SiR'R"R" and halogen, wherein Ra, R', R" and R" are independently H or Ci-4 alkyl and are optionally substituted with 1 to 3 substituents selected from halogen, -OH and -O-Ci-4 alkyl. Preferred substituents on an aryl, heteroaryl or heterocyclyl group are Ci-4 alkyl, -CF3, - NR'R", -OR', -NO2, -CN, -C(0)R', -C(0)OR', -C(0)NR'R", =0 and halogen, wherein R' and R" are independently H or C1-4 alkyl.
Halogen is chlorine, fluorine, bromine or iodine (a chloro group, a fluoro group, a bromo group or an iodo group). Halogen is typically chlorine or fluorine.
A Ci-4 alkoxy group is a group of formula -OR wherein R is a C1-4 alkyl group. Examples of Ci-4 alkoxy include -OMe, -OEt, -OnPr, -ΟΨτ and -0*Bu.
An alkylene group is a bivalent group derived by removing two hydrogen atoms from a straight chain or branched alkane. Examples of a C1-4 alkylene group are -CH2- -CH(CH3)- , -C(CH3)2- -CH2CH2- -CH(CH3)CH2- -CH2CH(CH3)-, -CH2C(CH3)2- -C(CH3)2CH2- -CH2CH2CH2- -CH(CH3)CH2CH2-, -CH2CH(CH3)CH2- -CH2CH2CH(CH3)- and - CH2CH2CH2CH2-. Typically, a C alkylene group is -CH2- -CH2CH2- -CH2CH2CH2- or -CH2CH2CH2CH2- and a Ci-3 alkylene group is -CH2- -CH2CH2- or -CH2CH2CH2-.
Unless otherwise specified, included in groups and substituents defined herein are the well known ionic, salt, solvate, and protected forms of these groups and substituents. For example, a reference to carboxylic acid or carboxyl group (-COOH) also includes the anionic (carboxylate) form (-COO"), a salt or solvate thereof. Similarly, a reference to an amino group includes the protonated form (-Ν+ϊ¾¾2), a salt or solvate of the amino group, for example, a hydrochloride salt. Similarly, a reference to a hydroxyl group also includes the anionic form (-0"), a salt or solvate thereof.
Certain compounds may exist in one or more particular geometric, optical, enantiomeric, diastereomeric, epimeric, atropic, stereoisomeric, tautomeric, conformational, or anomeric forms, including but not limited to, cis- and trans-forms; E- and Z-forms; c-, t-, and r- forms; endo- and exo-forms; R-, S-, and meso-forms; D- and L-forms; d- and 1-forms; (+) and (-) forms; keto-, enol-, and enolate-forms; syn- and anti-forms; synclinal- and anticlinal-forms; a- and β-forms; axial and equatorial forms; boat-, chair-, twist-, envelope-, and halfchair- forms; and combinations thereof, hereinafter collectively referred to as "isomers" (or "isomeric forms").
Unless otherwise specified, a reference to a particular compound includes all such isomeric forms, including (wholly or partially) racemic and other mixtures thereof. Substantially optically pure forms are also included. Methods for the preparation (e.g., asymmetric synthesis) and separation (e.g., fractional crystallisation and chromatographic means) of such isomeric forms are either known in the art or are readily obtained by adapting known methods, in a known manner.
The term "salt", as used herein, refers to salt comprising a cationic or ionic form of the compound together with a counter ion. Such salts are typically acid addition salts. Often, the salt is a pharmaceutically acceptable salt. A pharmaceutically acceptable salt is a salt formed with a pharmaceutically acceptable acid or base. Pharmaceutically acceptable acids include both inorganic acids such as hydrochloric, sulphuric, phosphoric, diphosphoric, hydrobromic or nitric acid and organic acids such as citric, fumaric, maleic, malic, ascorbic, succinic, tartaric, benzoic, acetic, methanesulphonic, ethanesulphonic, benzenesulphonic or p- toluenesulphonic acid. Pharmaceutically acceptable bases include alkali metal (e.g. sodium or potassium) and alkali earth metal (e.g. calcium or magnesium) hydroxides and organic bases such as alkyl amines, aralkyl amines and heterocyclic amines.
Compound
The invention provides a compound which is a peptide derivative of formula (1) or a salt thereof as defined above.
Q is typically -C(0)-N(RN2)-, -C(0)-C(R'R")-, -C(0)-0- -N(RN2)-C(0)-, -C(R'R")- C(O)- or -O-C(O)-.
More typically, Q is -C(0)-N(RN2)-, -C(0)-CH2- or -C(0)-0-.
Preferably, Q is -C(0)-N(RN2)-, most preferably -C(0)- H-
Typically, Z is -C(0)-0-R° and R° is H, C1-4 alkyl, phenyl or benzyl, and R° is optionally substituted with 1 to 4 substituents selected from Ra, - R'R", -OR', -C(0)R', -C(0)OR', - 0-C(0)R', =0 and halogen.
Preferably, Z is -C(0)-0-R° and R° is H, Ci-4 alkyl, phenyl or benzyl, wherein the phenyl and benzyl groups are optionally substituted with 1 to 4 substituents selected from Ra, -OR' and halogen. Preferred substituents are Ci-4 alkyl, -CF3, -0(Ci-4)alkyl and halogen, in particular methyl and halogen. Typically, R° is unsubstituted.
Preferably, R° is H or Ci-4 alkyl optionally substituted with -O-Ci-4 alkyl. For example, R° may be H or methyl, e.g. H.
Alternatively, Z may be -CN4H (tetrazolyl).
Often, each Ra is independently methyl or ethyl which is optionally substituted with 1 to 3 fluorine atoms.
Typically, in formula (1) or (2), each RN1 and RN2 is independently H or methyl. Preferably, RN1 and RN2 are each H. RN1 and RN2 may be the same or different.
Typically, in formula (1) or (2), each RC1 is the same or different and is independently H or methyl. Typically, in formula (1) or (2), each RC2 is the same or different and is
independently H or methyl. Preferably, RC1 and RC2 are all H.
Typically, each R', R", R" and R"" is independently H or C1-4 alkyl, preferably H.
Typically, RG is H, -CH2-RGA or Ci-12 hydrocarbyl optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl; and RGA is Ci-n hydrocarbyl optionally substituted with 1 to 4 substituents selected from -NR'R", -OR', halogen and 5-6 membered heterocyclyl.
Alternatively, RG may be H, -CH2-RGA or a Ci-8 alkyl group which is optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl; or RG and RN1 together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen. Preferably, RG is H, -CH2-RGA or a
Ci-8 alkyl group which is optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl. RGA is typically a C1 -7 alkyl group which is optionally substituted with 1 to 4 substituents selected from -NR'R", -OR', halogen and 5-6 membered heterocyclyl.
For instance, RG may be C1 -8 alkyl, phenyl or benzyl, each of which is optionally substituted with 1 to 4 substituents selected from halogen, -OH and -O-Ci-4 alkyl.
More typically, RG is C1 -8 alkyl optionally substituted with 1 to 4 substituents selected from halogen, -OH and -O-Ci-4 alkyl.
Preferably, RG is methyl or ethyl.
Typically, R° is H, C1 -8 alkyl, phenyl or benzyl, each of which is optionally substituted with 1 to 4 substituents selected from -NR'R", -OR', -C(0)R', -C(0)OR', -C(0)NR'R", -O- C(0)R', -N(R')-C(0)R", =0, halogen and 5-6 membered heterocyclyl.
More typically, R° is H or Ci-4 alkyl optionally substituted with -O-Ci-4 alkyl.
Preferably, R° is H, methyl or ethyl.
For instance, RG may be C1 -8 alkyl optionally substituted with 1 to 4 substituents selected from halogen, -OH and -O-Ci-4 alkyl and R° may be H or Ci-4 alkyl optionally substituted with -O-Ci-4 alkyl.
Typically, Rs is Ci-i6 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group, -(Ci-4 alkylene)-RSA or -(Ci-4 alkylene)-L-RSA; RSA is Ci-ie hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group; L is -SO2-; wherein Rs and RSA are optionally substituted with a group Rb and/or from 1 to 5 substituents selected from Ra, -NR'R", -OR',
-NO2, -CN, -C(0)R', -C(0)OR', -C(0)NR'R", -0-C(0)R', -N(R')-C(0)R", -SR.',
=0 and halogen; or Rs and an RC1 together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen; wherein Ra, R' and R" are as defined herein, and Rb is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl.
Typically, where Rs is as defined above, each R' and R" is independently H or C1-4 alkyl which is optionally substituted with 1 to 3 substituents selected from halogen, -OH and -O- Ci-4 alkyl; and each Ra is independently Ci-4 alkyl which is optionally substituted with 1 to 3 halogen atoms. Preferably, where Rs is as defined above, Ra is Ci-4 alkyl or -CF3 and R' and R" are each independently H or Ci-4 alkyl.
Typical substituents on Rs are a group Rb and/or from 1 to 5 substituents selected from Ci-4 alkyl, -CF3, - R'R", -OR', -N02, -CN, -C(0)R', -C(0)OR', -C(0) R'R", =0 and halogen. Preferably, Rs is substituted with 0 or 1 groups Rb and 0, 1, 2 or 3 substituents selected from Ci-4 alkyl, -CF3, -OCi-4alkyl, -N02, -CN, =0 and halogen; wherein Rb is phenyl, benzyl or C3-6 cycloalkyl.
More typically, Rs is Ci-io alkyl, -(C1-3 alkylene)-RSA, -(C1-3 alkyl ene)-L-RSA, C2-10 alkenyl, C2-10 alkynyl, C3 -6 cycloalkyl, 5- to 10 membered heterocyclyl, C5-10 aryl or 5- to 10- membered heteroaryl, wherein L is SO2, RSA is C3 -6 cycloalkyl, 5- to 10 membered heterocyclyl, C5-10 aryl or 5- to 10-membered heteroaryl and Rs and RSA are optionally substituted with a group Rb and/or from 1 to 5 substituents selected from C1-4 alkyl, -CF3, - NR'R", -OR', -NO2, -CN, -C(0)R', -C(0)OR', -C(0)NR'R", =0 and halogen; wherein each R' and R" is independently H or C1-4 alkyl; and Rb is phenyl, benzyl or C3 -6 cycloalkyl.
More typically, Rs is Ci-10 alkyl, -(Ci-3 alkylene)-RSA, -(Ci-3 alkyl ene)-L-RSA, C3-6 cycloalkyl, 5-6 membered heterocyclyl or phenyl, wherein L is SO2, RSA is C3 -6 cycloalkyl, 5- 6 membered heterocyclyl or phenyl and Rs and RSA are optionally substituted with a group Rb and/or from 1 to 3 substituents selected from C1-4 alkyl, -CF3, -OCi-4alkyl, -NO2, -CN, =0 and halogen; wherein Rb is phenyl, benzyl or C3 -6 cycloalkyl.
For instance, Rs may be:
(i) Ci-10 alkyl, -(C1-2 alkylene)-(C3 -6 cycloalkyl) or C3 -6 cycloalkyl, wherein the cycloalkyl ring is optionally substituted with from 1 to 3 methyl groups;
(ii) 5-6 membered heterocyclyl or -CH2CH2-(5-6 membered heterocyclyl) wherein the heterocyclyl group in each case is optionally substituted with phenyl, benzyl or C3-6 cycloalkyl and/or with from 1 to 3 substituents selected from C1-4 alkyl, -NO2, halogen, Ci-4 alkoxy, and =0; or
(iii) -CH2CH2-Ph-(Y)„ or -CH2CH2-S02-Ph-(Y)„, wherein n is 0 or 1 and Y is N02, -CF3, -F or -OC¾.
In some embodiments, Rs is a group of formula:

where Rx is H, phenyl, benzyl, C3 -6 cycloalkyl or Ci-4 alkyl.
For instance, Rs may be a group selected from the following groups:

Typically, each Rc and R is independently H or Ci-4 alkyl, preferably H.
Typically, the compound is a peptide derivative of formula (2) or a salt thereof:

wherein:
RG is H, -CH2-RGA or a C1-8 alkyl group which is optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl; or RG and RN1 together form a 5-6 membered heterocyclic ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen;
RGA is Ci-7 alkyl group which is optionally substituted with 1 to 4 substituents selected from - R'R", -OR', halogen and 5-6 membered heterocyclyl;
Rs is Ci-16 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group, -(Ci-4 alkylene)-RSA or -(Ci-4 alkylene)-L-RSA; RSA is Ci-i6 hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group; L is -SO2-; and wherein Rs and RSA are optionally substituted with a group Rb and/or from 1 to 5 substituents selected from Ra, - R'R", -OR', - N02, -CN, -C(0)R', -C(0)OR', -C(0) R'R", -0-C(0)R', -N(R')-C(0)R", -SR', =0 and halogen; or Rs and an RC1 together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen;
Rb is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl;
R° is H, Ci-4 alkyl, phenyl or benzyl, wherein the phenyl and benzyl groups are optionally substituted with 1 to 4 substituents selected from Ra, -OR' and halogen;
RN1 and RN2 are each independently H or C1-4 alkyl;
RC1 and RC2 are each independently H or C1-4 alkyl;
each R' and R" is independently H or C1-4 alkyl which is optionally substituted with 1 to 3 substituents selected from halogen, -OH and -O-C1-4 alkyl; and
each Ra is independently C1-4 alkyl which is optionally substituted with 1 to 3 halogen atoms.
More typically, the compound is a peptide derivative of formula (3) or a salt thereof:

wherein:
RG is H, -CH2-RGA or a Ci-8 alkyl group which is optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl;
RGA is Ci-7 alkyl group which is optionally substituted with 1 to 4 substituents selected from -NR'R", -OR', halogen and 5-6 membered heterocyclyl;
Rs is Ci-16 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group,
-(Ci-4 alkylene)-RSA or -(Ci-4 alkylene)-L-RSA; RSA is Ci-ie hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group; L is -SO2-; and wherein Rs and RSA are optionally
substituted with a group Rb and/or from 1 to 5 substituents selected from Ci-4 alkyl, -CF3, - R'R", -OR', -N02, -CN, -C(0)R', -C(0)OR', -C(0) R'R", =0 and halogen;
Rb is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl;
R° is H or Ci-4 alkyl optionally substituted with -O-C1-4 alkyl; and
each R' and R" is independently H or C1-4 alkyl.
Thus, the compound of the invention may be a peptide derivative of formula (3) or a salt thereof:

wherein:
RG is Ci-8 alkyl optionally substituted with 1 to 4 substituents selected from halogen, -OH and -O-C1-4 alkyl.
R° is H, Ci-8 alkyl, phenyl or benzyl, each of which is optionally substituted with 1 to 4 substituents selected from -NR'R", -OR', -C(0)R', -C(0)OR', -C(0)NR'R", -0-C(0)R', -N(R')-C(0)R", =0, halogen and 5-6 membered heterocyclyl;
Rs is (i) 5-6 membered heterocyclyl or -CH2CH2-(5-6 membered heterocyclyl) wherein the heterocyclyl group in each case is optionally substituted with phenyl, benzyl or C3-6 cycloalkyl and/or with from 1 to 3 substituents selected from C1-4 alkyl, -NO2, halogen, Ci-4 alkoxy, and =0; or (ii) -CH2CH2-Ph-(Y)„ or -CH2CH2-S02-Ph-(Y)„, wherein n is 0 or 1 and Y is -NO2, -CF3, -F or -OCH3.
Preferably, the compound is a peptide derivative of formula (4) or a salt thereof:

Preferably in a compound of the invention RG is methyl and R° is H or methyl.
The compound of the invention may for instance be a compound of formula

or a salt thereof, and where Rs is a rou selected from

Preferably, the compound is
 , or a salt thereof.
, or a salt thereof.
If the compound of the invention is a salt of the peptide derivative, it is typically an acid addition salt.
Compositions and products
The invention provides a pharmaceutical composition comprising (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a pharmaceutically acceptable salt, and (b) one or more pharmaceutically acceptable excipients or diluents.
The compounds of the invention may be administered as pharmaceutical compositions in a variety of dosage forms. Thus, they can be administered orally, for example as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules. The compounds of the invention may also be administered parenterally, whether subcutaneously, intravenously, intramuscularly, intrasternally, transdermally or by infusion techniques. The compounds may also be administered as suppositories.
The one or more pharmaceutically acceptable excipients or diluents may be any suitable excipients or diluents. For example, a pharmaceutical composition which is a solid oral form may contain, together with the active compound, diluents, e.g. lactose, dextrose, saccharose, cellulose, corn starch or potato starch; lubricants, e.g. silica, talc, stearic acid, magnesium or calcium stearate, and/or polyethylene glycols; binding agents; e.g. starches, arabic gums, gelatin, methylcellulose, carboxymethylcellulose or polyvinyl pyrrolidone; disaggregating agents, e.g. starch, alginic acid, alginates or sodium starch glycolate; effervescing mixtures; dyestuffs; sweeteners; wetting agents, such as lecithin, polysorbates, laurylsulphates; and, in general, non toxic and pharmacologically inactive substances used in pharmaceutical formulations. Such pharmaceutical preparations may be manufactured in known manner, for example, by means of mixing, granulating, tableting, sugar coating, or film coating processes.
A pharmaceutical composition which is a liquid dispersion for oral administration may be a syrup, emulsion and suspension. The syrups may contain as carriers, for example, saccharose or saccharose with glycerine and/or mannitol and/or sorbitol.
Suspensions and emulsions may contain as carrier, for example a natural gum, agar, sodium alginate, pectin, methylcellulose, carboxymethylcellulose, or polyvinyl alcohol. The suspension or solutions for intramuscular injections may contain, together with the active compound, a pharmaceutically acceptable carrier, e.g. sterile water, olive oil, ethyl oleate, glycols, e.g. propylene glycol, and if desired, a suitable amount of lidocaine hydrochloride.
Solutions for injection or infusion may contain as carrier, for example, sterile water or preferably they may be in the form of sterile, aqueous, isotonic saline solutions.
The compounds of the invention can be used in combination with further active agents, and in particular in combination with one or more antibiotic compounds. For the avoidance of doubt, antibiotic compounds may be provided in the form of pharmaceutically acceptable salts.
The invention therefore provides a combination comprising (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a pharmaceutically acceptable salt, and (b) a further active agent which is an antibiotic compound.
The further active agent may be any suitable antibiotic compound. The antibiotic compound may be a compound of the aminoglycoside class, ansamycin class, carbacephem class, carbapenem class, cephalosporin class, glycopeptide class, lincosamide class, lipopeptide class, macrolide class, monobactam class, nitrofuran class, oxazolidinone class, penicillin class, polypeptide class, quinolone class, sulfonamide class or tetracyclines class. For instance, the further antibiotic may be phenoxymethylpenicillin, flucloxacillin, amoxicillin, cefaclor, cefadroxil, cefalexin, tetracycline, doxycycline, lymecycline, gentamicin, tobramycin, erythromycin, azithromycin, clarithromycin, clindamycin, co-trimoxazole, metronidazole, tinidazole, ciprofloxacin, levofloxacin or norfloxacin.
The combination typically further comprises (c) one or more pharmaceutically acceptable excipients or diluents. The one or more pharmaceutically acceptable excipients or diluents may be as defined above.
The combination of the invention may for instance be a fixed combination. In a fixed combination, components (a) and (b) are present in the same composition. The fixed combination can be used for simultaneous administration of two compounds. The fixed combination is typically a solid composition (e.g. a tablet) or a solution (e.g. an injectable composition). The two components in a fixed combination are typically intermixed.
Alternatively, the combination of the invention may be a free combination. In a free combination, the active components (a) and (b) are typically separate from each other and packaged in one unit for simultaneous, substantially simultaneous, separate or sequential administration.
The invention also provides a kit comprising (a) a compound which is a peptide derivative or a salt thereof as herein, wherein the salt is a pharmaceutically acceptable salt, and (b) a further antibiotic compound. The kit may further comprise instructions for using or administering components (a) and (b), for instance in a method of treating or preventing an infection caused by bacteria.
The invention provides a soap product, cleaning product or washing product comprising a compound as described herein. A soap product is a composition or article which is suitable for washing the skin. The soap product may be a liquid soap product (for instance a hand wash, body wash, shampoo or face wash) or a solid soap product (for instance a bar of soap or a disposable wipe). A soap product according to the invention typically further comprises a soap, which is usually a surfactant. Examples of soaps include salts of fatty acids (i.e. linear C6-i8 alkyl or alkenyl carboxylic acids), for instance a sodium or potassium salt of lauric acid, myristic acid, palmitic acid, stearic acid, oleic acid, linoleic acid or linolenic acid, and salts of long chain sulfonic acids (i.e. linear C6-i8 alkyl or alkenyl sulfonic acids), for instance sodium laureth sulfate.
The soap product may for instance be a liquid soap product comprising one or more solvents, for instance selected from water and alcohols. Often, the soap product is a liquid soap product which further comprises ethanol.
A cleaning product includes household cleaning products such as surface cleaning products, fabric cleaners, washing powders, and cleaning products suitable for use in a medical environment, for example to clean or disinfect surfaces in a hospital or equipment found in a clinical environment such as medical equipment.
Treatment or prevention of bacterial infections
The compounds of the invention can kill or inhibit the growth of bacteria. The invention therefore provides a compound which is a peptide derivative or a salt thereof as defined herein, for use in the treatment or prevention of an infection caused by bacteria, wherein the salt is a pharmaceutically acceptable salt.
The compounds of the invention can treat a number of different bacterial infections. The infection may be caused by bacteria which are gram-negative or gram-positive. Typically, the bacterial infection is caused by gram-negative bacteria. The infection may be caused by bacteria of genus Shewanella, Acinetobacter, Bacillus, Bartonella, Bordetella, Borrelia, Brucella, Campylobacter, Chlamydia, Chlamydophila, Clostridium, Corynebacterium, Enterobacter, Enterococcus, Escherichia, Francisella, Haemophilus, Helicobacter,
Klebsiella, Legionella, Leptospira, Listeria, Mycobacterium, Mycoplasma, Neisseria, Pseudomonas, Rickettsia, Salmonella, Shigella, Staphylococcus, Streptococcus, Treponema, Vibrio or Yersinia. For example, the bacterial infection may be caused by bacteria of genus Shewanella or Escherichia.
Gram-negative bacteria include bacteria of the genus Shewanella, Escherichia, Salmonella, Shigella, Pseudomonas, Moraxella, Helicobacter, Stenotrophomonas, Bdellovibrio and Legionella. In particular, the compound of the invention may be used to treat or prevent a bacterial infection caused by Staphylococcus aureus, Escherichia coli, Shewanella denitrificans, Hemophilus influenzae, Klebsiella pneumoniae, Legionella
pneumophila, Pseudomonas aeruginosa, Proteus mirabilis, Enterobacter cloacae, Serratia marcescens, Helicobacter pylori, Salmonella enteritidis, Salmonella typhi ox Acinetobacter baumannii.
The invention also provides a method of treating or preventing infection caused by bacteria as defined above in a subject, which method comprises administering an effective amount of a peptide derivative or a salt thereof as defined herein to the subject, wherein the salt is a pharmaceutically acceptable salt. The invention also provides the use of a peptide derivative or a salt thereof as defined herein in the manufacture of a medicament for use in the treatment or prevention of infection caused by bacteria as defined above in a subject.
In the invention, the subject is typically a mammal, more typically a human. The bacterial infection may be of any part of the body, for instance part of the skin, the lungs or the gut.
A therapeutically effective amount of a compound of the invention is administered to a patient. A typical dose is from 0.001 to 1000 mg, according to the activity of the specific compound, the age, weight and conditions of the subject to be treated, the type and severity of the disease and the frequency and route of administration. Preferably, daily dosage levels are from 0.001 mg to 4000 mg.
As discussed above, the compounds of the invention may be used alone, or alternatively they may be used in combination with a second active agent, typically an antibiotic compound. The combination may be for use in the treatment or prevention of an infection caused by bacteria as defined above, which combination comprises (a) a compound which is a peptide derivative or a salt thereof as defined herein, wherein the salt is a pharmaceutically acceptable salt, and (b) a further active agent which is an antibiotic compound or a pharmaceutically acceptable salt thereof.
The invention provides a compound which is a peptide derivative or a salt thereof as defined herein, for use in the treatment or prevention of an infection caused by bacteria as defined herein combination with a further active agent which is an antibiotic compound, wherein the salt is a pharmaceutically acceptable salt. A method of treating or preventing infection caused by bacteria as defined herein in a subject is also described, which method comprises administering an effective amount of a peptide derivative or a salt thereof as defined herein to the subject in combination with a further active agent which is an antibiotic compound, wherein the salt is a pharmaceutically acceptable salt. The invention also provides the use of a peptide derivative or a salt thereof as defined herein in a the manufacture of a medicament for use in the treatment or prevention of infection caused by bacteria as defined herein in a subject, in combination with a further active agent which is an antibiotic compound.
In the uses of the combination according to the invention, the compound of the invention and the further active agent may be used simultaneously or sequentially.
Methods of controlling bacteria
The invention also provides a method for preventing or treating infection of a plant caused by bacteria, which method comprises treating the plant, the seed from which the plant grows, or the place at which the plant grows, with a composition comprising a compound as defined herein.
The infection of the plant is typically caused by bacteria of the genus Shewanella, Erwinia, Agrobacterium, Phytoplasma, Burkholderia, Proteobacteria, Xanthomonas or Pseudomonas.
Treating the plant, seed or place typically comprises applying a composition comprising the compound to the plant, seed or place. This may comprise spraying an aqueous solution of a compound of the invention on the plant, seed or place. The treatment may be repeated daily or weekly.
The plant to be treated may be a wide range of plants and is typically a crop plant. For instance, the plant may be a cereal-producing plant, a vegetable-producing plant or a fruit- producing plant.
The invention also provides an in vitro method for killing or inhibiting the growth of bacteria, which method comprises applying a composition comprising a compound which is a peptide derivative or salt thereof as defined herein to a surface, to an article or to the skin. The bacteria may be as defined herein. Said method is typically not a method of treatment.
Process for producing compounds
Compounds of the invention may be produced by a thiol-ene reaction. Thus, the invention also provides a process for producing a compound which is a peptide derivative of formula (la) or a salt thereof:

which process comprises reacting a thiol compound of formula (A) with an alkene of formula (B) in the presence of an initiator,
wherein RG, RN
 1, RC1, Q, RC2 and Z are as defined herein and the group
1, RC1, Q, RC2 and Z are as defined herein and the group
corresponds to Rs as defined herein.
Typically:
RS1, RS2, RS4 are independently H or C1-4 alkyl and
RS3 is a Ci-16 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group, -(C1-2 alkylene)-RSA or -(C1-2 alkylene)-L-RSA; RSA is C1-16 hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group; L is -SO2-; and wherein RS3 and RSA are optionally substituted with a group Rb and/or from 1 to 5 substituents selected from Ra, - R'R", -OR', - N02, -CN, -C(0)R', -C(0)OR', -C(0) R'R", -0-C(0)R', -N(R')-C(0)R", -SR', =0 and halogen; or
wherein RS1 and RS3 together form a 5-10 membered heterocyclyl group or a C3-6 cycloalkyl group optionally substituted with a group Rb and/or from 1 to 5 substituents selected from Ra, -NR'R", -OR', -NO2, -CN, -C(0)R', -C(0)OR', -C(0)NR'R", -O- C(0)R', -N(R')-C(0)R", -SR', =0 and halogen;
Rb is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl;
each R' and R" is independently H or C1-4 alkyl which is optionally substituted with 1 to 3 substituents selected from halogen, -OH and -O-C1-4 alkyl; and
each Ra is independently C1-4 alkyl which is optionally substituted with 1 to 3 halogen atoms.
For instance, RS1, RS2, RS4 may be H and RS3 may be phenyl or -S02-phenyl and RS3 is optionally substituted with a group selected from halogen, -NO2, -OMe and -CF3.
Typically, compound (A) is a compound of formula (3 A) or a salt thereof:

wherein:
RG is H, -CH2-RGA or a C1-8 alkyl group which is optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl;
RGA is Ci-7 alkyl group which is optionally substituted with 1 to 4 substituents selected from - R'R", -OR', halogen and 5-6 membered heterocyclyl;
R° is H or Ci-4 alkyl optionally substituted with -O-Ci-4 alkyl; and
each R' and R" is independently H or Ci-4 alkyl.
Typically RG may be Ci-8 alkyl optionally substituted with 1 to 4 substituents selected from halogen, -OH and -O-Ci-4 alkyl and R° may be H or Ci-4 alkyl optionally substituted with - O-Ci-4 alkyl.
Typically, compound (B) is a compound selected from the formulae:
C-i alkyl

The initiator may be a chemical initiator (for instance a free radical) or a physical initiator (for instance UV light or heat). Typically, the initiator is UV light. The reaction may use UV light together with a chemical initiator (e.g. 2,2-dimethoxyphenyl acetophenone).
The temperature of the reaction is typically from 10 to 40°C.
Typically, the alkene (compound (B)) and the thiol (compound (A)) are stirred at from 10 to 40°C in a solvent (for instance THF/H2O (1 :2, 3 mL)) in the presence of light (e.g. at 365 nm) for from 1 to 10 h. After this time the reaction may be filtered, and the solid washed with ethanol and water.
The following Examples illustrate the invention. They do not however, limit the invention in any way. In this regard, it is important to understand that the particular assay used in the Examples section is designed only to provide an indication of activity. There are many assays available to determine such activity, and a negative result in any one particular assay is therefore not determinative.
EXAMPLES
Example - synthesis procedure
The following compounds were sy es described below.

BuSG

4


9 10
Synthesis of DNGSH

GSH DNGSH
N-Allyl-5-(dimethylamino)naphthalene-l -sulfonamide (0.10 g, 0.34 mmol, 1.0 eq.), L- glutathione (0.42 g, 1.36 mmol, 4.0 eq.), TCEP HCl (0.19 mg, 0.68 mmol, 2.0 eq.) and 2,2- dimethoxyphenyl acetophenone (0.02 g, 0.07 mmol, 0.2 eq.) were stirred at RT in THF/H2O (1 :2, 3 mL) in the presence of light (365 nm, 4 x 15 W) for 5 h. After this time, the reaction was extracted with CH2CI2. The aqueous layer was lyophilized and the crude material purified by RP C-18 silica gel column chromatography (MeOH : H20 0: 100 50:50), yielding DNGSH (0.02 g, 11%) as a hygroscopic yellow solid: Rt 0.4 (50:50 MeOH : H20 (RP)); [αβ°-19.1 (c 0.25, water) (Lit. Value [αβ° -19.2 (c 0.25, water))2; 1H MR (400 MHz; D20): δ/ppm 8.55 (d, 1H, J7.8 Hz, Ar C8H), 8.44 (d, 1H, J 8.7 Hz, Ar C6H), 8.32 (d, 1H, J 7.4 Hz, Ar C3H), 7.83-7.74 (m, 3H, Ar C2H & Ar C4H & Ar C7H), 4.26 (dd, 1H, J 8.8 Hz, J 5.0 Hz, Cys-a-CH), 3.76-3.68 (m, 3H, Gly-C¾ Glu-a-CH), 3.21 (s, 6H, N(G%)2), 3.00 (t, 2H, J6.8 Hz, Linker-G%), 2.54 (dd, 1H, J 14.2 Hz, J 5.0 Hz, Cys-p-G%), 2.49-2.38 (m, 3H, Cys-p-G¾ Glu-p-G%), 2.21-2.13 (m, 2H, 6.8 Hz, Linker-G%), 2.12-2.03 (m, 2H, Glu-Y-G%), 1.45 (qn, 2H, J 6.8 Hz, Linker-G%), m/z (ES+) 598 ([M+H]+, 100%). The data are in good agreement with the literature values.2
Synthesis of ¾uSG:

GSH
L-Glutathione (0.2 g, 0.65 mmol, 1.0 eq.) and NaOH (0.03 g, 0.65 mmol, 1.0 eq.) were stirred in water (5 mL), to this N-tert-butyl-maleimide (0.01 g, 0.09 mL, 0.65 mmol, 1.0 eq.) was added and the resulting solution stirred at RT for 20 minutes. After this time, the reaction solution was lyophilized, purified by RP C-18 silica gel chromatography, eluting with water to afford ^-N-tert-butylsuccinimido glutathione ¾uSG (mixture of diastereoisomers) as a colourless hygroscopic solid (0.26 g, 87%): Rt 0.7 (water (RP)); [αβ° -13.3 (c 1.0, water) (Lit. Value [αβ° -12.3 (c 1.0, water))2; ¾ MR (400 MHz; D20): δ/ppm 4.68-4.60 (m, 1H, Cys-a-CH), 3.91-3.81 (m, 1H, Succinimide-CH), 3.81-3.67 (m, 3H, Glu-a-CH, Gly-G%), 3.32 (dd, 0.5H, J 14.2 Hz, J 4.9 Hz, Cys-p-G%), 3.26-3.04 (m, 2H, Cys-p-G%, Succinimide- CH2), 2.96 (dd, 0.5H, J 14.2 Hz, J 9.2 Hz, Cys-p-G%), 2.60-2.43 (m, 3H, Glu-y-CH2, Succinimide-G%), 2.20-2.07 (m, 2H, Glu-p-G%), 1.51 (s, 4.5H, NC(G%)3), 1.51 (s, 4.5H,
NC(C%)3); m/z (ES") 459 ([M-H]", 100%). The data are in good agreement with literature values2
Synthesis of compound 1 :

glu-cys
Glu-Cys (15.3 mg, 0.06 mmol, 1.0 eq.) and NaOH (2.4 mg, 0.06 mmol, 1.0 eq.) were stirred in water (0.4 mL), to this N-tert-butyl-maleimide (9.4 mg, 8.8 μΐ., 0.06 mmol, 1.0 eq.) was added and the resulting solution stirred at RT for 4.5 h. After this time, the reaction solution was lyophilized and the resulting solid was purified by semi-preparative HPLC (Agilent ZORBAX 300SB-C 18 column; 95 :5 :0.1 5 :95 :0.1 ; H20 : MeCN : TFA) to afford 1 (mixture of diastereoisomers) as a colourless solid (19.8 mg, 63%): Rf 0.8 (50:50 acetonitrile : water (RP)); [αβ° -6.4 (c 1.1, H20); vm cm l (neat) 1739.3 (s), 1367.7 (w), 1215.6 (w); ¾ MR (500 MHz; D20): δ/ppm 4.71-4.63 (m, 1H, Cys-a-CH), 4.04 (dd, 1H, J 7.0 Hz, J 6.5 Hz, Glu-a-CH), 3.92-3.85 (m, 1H, Succinimide-CH), 3.37 (dd, 0.5H, J 14.1 Hz, J 4.6 Hz, Cys-p-G%), 3.27 (dd, 0.5H, J 14.1 Hz, J 4.6 Hz, Cys-p-G%), 3.23-3.10 (m, 1.5H, Cys-β- CH2, Succinimide-CH2), 3.04 (dd, 0.5H, J 14.1 Hz, J 8.7 Hz, Cys-p-G%), 2.64-2.52 (m, 3H, Glu-Y-G¾ Succinimide-CH2), 2.31-2.15 (m, 2H, Glu-p-G%), 1.54 (s, 4.5H, NC(C%)3), 1.53 (s, 4.5H, NC(C%)3); 19F NMR (decoupled) (376.6 MHz; D20): δ/ppm -75.7
(OCOGF5); 13C NMR (128.5 MHz; D20): δ/ppm 180.1 (Succinimide-CO), 180.0
(Succinimide-CO), 179.1 (Succinimide-CO), 179.0 (Succinimide-CO), 174.3 (Glu-CO), 174.3 (Glu-CO), 173.5 (Cys-COOH), 173.5 (Cys-COOH), 172.0 (Glu-COOH), 163.0 (q, J 35.3 Hz, OCOCF3), 1 16.3 (q, J 291.5 Hz, OCOCF3), 59.2 (NC(C¾)3), 59.2 (NC(C¾)3), 52.6 (Glu-a-CH), 52.2 (Cys-a-CH), 40.9 (Succinimide-CH), 39.9 (Succinimide-CH), 36.3 (Succinimide-CH2), 35.8 (Succinimide-CH2), 32.3 (Cys-p-CH2), 31.8 (Cys-p-CH2), 31.0 (Glu-Y-CH2), 27.3 (NC(CH3)3), 25.7 (Glu-p-CH2); HRMS m/z (ESI+) (Found: [M+H]+ 404.14836. Ci6H2607N3S requires M+ 404.14860); m/z (ES+) 404 ([M+H]+, 100%);
Analytical HPLC @ 220 nm (Acclaim® 120 C 18 RP LC Column; 95 :5 :0.1 5 :95 :0.1 ; H20 : MeCN : TFA) Ret. Time = 9.094 min, Purity: 98.57%.
Synthesis of compound 2:

JV-acetyl cys 2
N-acetyl cysteine (20.0 mg, 0.12 mmol, 1.0 eq.) and NaOH (4.9 mg, 0.12 mmol, 1.0 eq.) were stirred in water (0.9 mL), to this N-tert-butyl-maleimide (18.7 mg, 17.7 L, 0.12 mmol, 1.0 eq.) was added and the resulting solution stirred at RT for 3.5 h. After this time, the reaction solution was lyophilized and the resulting solid was purified by semi-preparative HPLC (Agilent ZORB AX 300SB-C18 column; 95 :5:0.1 5:95:0.1 ; H20 : MeCN : TFA) to afford 2 (mixture of diastereoisomers) as a pale yellow solid (35.6 mg, 67%): Rf 0.8 (50:50 acetonitrile : water (RP)); [αβ°-17.3 (c 1.0, MeOH); Vmax/cm"1 (neat) 3329.1 (w), 2970.4 (w), 1737.9 (s), 1718.6 (s), 1697.4 (s), 1612.5 (m), 1558.5 (m), 1423.5 (w), 1365.6 (m), 1348.2 (m), 1315.5 (w), 1263.4 (m), 1228.7 (m), 1217.1 (s), 1205.5 (m), 1161.2 (m), 1116.8 (w), 1037.7 (w); ¾ NMR (500 MHz; D20): δ/ppm 4.70-4.63 (m, 1H, Cys-a-CH), 3.92-3.85 (m, 1H, Succinimide-CH), 3.36 (dd, 0.2H, J 14.1 Hz, J4.6 Hz, Cys-p-CH2), 3.29-3.10 (m, 2.6H, Cys-p-C¾ Succinimide-CH2), 3.04 (dd, 0.2H, J 14.1 Hz, J 8.6 Hz, Cys-p-CH2), 2.61-2.51 (m, 1H, Succinimide-CH2), 2.06 (s, 0.4H, C%), 2.05 (s, 2.6H, C%), 1.53 (s, 9H, NC(C%)3); 19F NMR (decoupled) (376.6 MHz; D20): δ/ppm -75.7 (OCOCF5); 13C NMR (128.5 MHz; D20): δ/ppm 180.2 (Succinimide-CO), 180.0 (Succinimide-CO), 179.1 (Succinimide-CO), 179.0 (Succinimide-CO), 174.2 (COCH3), 174.2 (COCH3), 173.5 (COOH), 162.9 (q, J35.3 Hz, OCOCF3), 116.3 (q, 291.5 Hz, OCOCF3), 59.2 (NC(CH3)3), 52.6 (Cys-a-CH), 52.1 (Cys- a-CH), 40.9 (Succinimide-CH), 39.9 (Succinimide-CH), 36.2 (Succinimide-CH2), 35.8 (Succinimide-CH2), 32.4 (Cys-p-CH2), 31.8 (Cys-p-CH2), 27.3 (NC(CH3)3), 21.6 (CH3), HRMS m/z (ESI+) (Found: [M+H]+ 317.11603. Ci3H2i05N2S requires M+ 317.11657); m/z (ES+) 317 ([M+H]+, 100%); Analytical HPLC @ 220 nm (Acclaim® 120 C18 RP LC
Column; 95 :5:0.1 5:95:0.1 ; H20 : MeCN : TFA) Ret. Time = 9.817 min, Purity: 97.46%; Chiral HPLC @ 220 nm (ChiralPak® AD-H column (5 μ m, 4.6 x 150 mm); Isocratic method: 75 : 25 Hexane : IP A, 0.3 mL / min) Ret. Time = 20.397 min, 99.99%.
Synthesis of compound 3:

JV-acetyl cys 11 3
Synthesis of 11:
To a solution of N-acetyl-cysteine (1.00 g, 6.13 mmol, 1.0 eq.) stirring in methanol (22 mL) was added thionyl chloride (0.85 g, 0.52 mL, 7.11 mmol, 1.16 eq.) dropwise and the reaction mixture was stirred under argon at RT for 1.5 h. After this time, the reaction mixture was concentrated in vacuo and the resulting residue was partitioned between brine and ethyl acetate. The aqueous layer was extracted with ethyl acetate, and the organic layers were combined, dried over Na2S04, filtered and concentrated in vacuo. The resulting crude material was adsorbed onto Celite® and purified by silica gel chromatography, eluting with methanol and chloroform (1 :99 -> 10:90; methanol : chloroform) to yield N-acetyl-L- cysteine methyl ester 12 as a colourless solid (0.49 g, 45%): Rf 0.6 (5:95 MeOH :
chloroform); mp 79-81 °C (MeOH : chloroform) (Lit. Value: 79-80 °C (ethyl acetate))3; [αβ° +75.5 (c 1.0, chloroform) (Lit. Value [a]D +71.0 (c 1.0, chloroform))4; ¾ NMR (400 MHz; CDCls): δ/ppm 6.68 (d, 1H, J 5.8 Hz, NH), 4.86-4.78 (m, 1H, Cys-a-CH), 3.72 (s, 3H, COOGft), 2.93 (dd, 2H, J 9.0 Hz, J4.4 Hz, Cys-p-G%), 2.01 (s, 3H, COG%), 1.36 (t, 1H, J 8.8 Hz, SH); m/z (ES+) 178 ([M+H]+, 100%). The data are in good agreement with literature values. 3'4
Synthesis of 3:
N-acetyl-L-cysteine methyl ester 12 (0.40 g, 2.26 mmol, 1.0 eq.) was stirred in a mixture of toluene (3 mL) and ammonium hydroxide (3 mL, 25% solution in water) under argon at RT for 19 h. After this time the reaction mixture was concentrated in vacuo at 60 °C to afford a mixture of N-acetyl-L-cysteinamide and oxidised N-acetyl-L-cysteinamide (0.37 g) as a colourless solid, which was used without further purification. 20.0 mg of the colourless solid containing N-acetyl-L-cysteinamide and oxidised N-acetyl-L-cysteinamide was stirred with triphenylphosphine, polymer-bound (79.0 mg, ~3 mmol/g triphenylphosphine loading - Sigma Aldrich) in DMF (0.5 mL) at RT for 4 h. After this time, the reaction mixture was
filtered and washed through with 0.5 mL DMF. N-tert-Butyl-maleimide (9.2 mg, 8.7 [iL) and triethylamine (8.4 μΐ/) were added to the filtrate, which was stirred at RT for 168 h. After this time, the reaction mixture was concentrated in vacuo and the crude reaction mixture was purified by semi-preparative HPLC (Agilent ZORBAX 300SB-C18 column; 95 :5:0.1 5:95:0.1 ; H20 : MeCN : TFA) to afford 3 as a colourless solid (3.3 mg, 6%, over two steps): [αβ° -11.4 (c 0.33, water); Vmax/cm"1 (neat); 3390.9 (w), 3315.6 (w), 2970.4 (w), 1737.9 (w), 1699.3 (s), 1635.6 (s), 1521.8 (m), 1458.2 (w), 1425.4 (m), 1398.4 (w), 1373.3 (m), 1346.3 (m), 1311.6 (w), 1280.7 (w), 1263.4 (m), 1228.7 (w), 1197.8 (m), 1166.9 (m), 1153.4 (m), 1114.9 (w), 1041.6 (w); 1H NMR (500 MHz; D20): δ/ppm 4.61-4.54 (m, 1H, Cys-a-CH), 3.92-3.84 (m, 1H, Succinimide-CH), 3.36-3.10 (m, 2.5H, Cys-p-CH2, Succinimide-C%), 3.00 (dd, 0.5H, J 14.1 Hz, J 8.8 Hz, Cys-p-CH2), 2.58 (dd, 0.5H, J 4.7 Hz, J 4.3 Hz,
Succinimide-C%), 2.54 (dd, 0.5H, J4.7 Hz, J4.3 Hz, Succinimide-C%), 2.07 (s, 1.5H, CH3), 2.07 (s, 1.5H, CH3), 1.54 (s, 9H, NC(C%)3); 19F NMR (decoupled) (376.6 MHz; D20): δ/ppm -75.6 (OCOCF5); 13C NMR (128.5 MHz; D20): δ/ppm 180.2 (Succinimide- CO), 180.2 (Succinimide-CO), 179.1 (Succinimide-CO), 179.0 (Succinimide-CO), 174.6 (CONH2), 174.6 (CONH2), 174.4 (COCH3), 174.3 (COCH3), 59.2 (NC(CH3)3), 59.1
(NC(CH3)3), 53.0 (Cys-a-CH), 52.6 (Cys-a-CH), 40.9 (Succinimide-CH), 40.0 (Succinimide- CH), 36.2 (Succinimide-CH2), 35.9 (Succinimide-CH2), 32.7 (Cys-p-CH2), 32.3 (Cys-β- CH2), 27.3 (NC(CH3)3), 21.7 (CH3); HRMS m/z (ESI+) (Found: [M+H]+ 316.13229.
Ci3H22N304S requires M+ 316.13255); m/z (ES+) 316 ([M+H]+, 100%); Analytical HPLC @ 220 nm (Acclaim® 120 C18 RP LC Column; 95:5:0.1 5:95:0.1 ; H20 : MeCN : TFA) Ret. Time = 9.061 min, Purity: 96.00%; Chiral HPLC @ 220 nm (ChiralPak® AD-H column (5 μ m, 4.6 x 150 mm); Isocratic method: 75 : 25 Hexane : IP A, 0.3 mL / min) Diastereomer 1 : Ret. Time = 24.185 min, 45.97%; Diastereomer 2: Ret. Time = 32.510 min, 53.84%.
Synthesis of compound 4:

cys-gly 12 4
Synthesis of 12:
To a solution of cys-gly (24.6 mg, 0.14 mmol, 1.0 eq.) stirring in methanol (1.1 mL) was added thionyl chloride (19.1 mg, 11.6 [iL, 0.16 mmol, 1.16 eq.) and the reaction mixture was stirred at RT for 2.5 h. After this time, the reaction mixture was concentrated in vacuo and the resulting residue was lyophilized to afford cys-gly methyl ester as a colourless solid
(26.5 mg), which was used without further purification. To cys-gly methyl ester (26.5 mg) stirring in water (1.1 mL) was added N-tert-butyl-maleimide (21.1 mg, 20.0 [iL, 0.14 mmol, 1.0 eq. with respect to the previous step) and the resulting solution was stirred at RT for 8.5 h. After this time, the reaction solution was lyophilized and the resulting solid was purified by semi-preparative HPLC (Agilent ZORB AX 300SB-C18 column; 95 :5:0.1 43 :57:0.1; H20 : MeCN : TFA) to afford 12 (mixture of diastereoisomers) as a colourless solid (35.9 mg, 57%): [αβ° +20.4 (c 1.0, chloroform); Vmax/cm"1 (neat) 2980.0 (w), 1747.5 (w), 1664.6 (s), 1460.1 (w), 1417.7 (w), 1367.5 (w), 1344.4 (m), 1265.3 (m), 1197.8 (s), 1165.0 (s), 1128.4 (s), 1028.1 (w); ¾ NMR (500 MHz; D20): δ/ppm 4.43-4.34 (m, 1H, Cys-a-CH), 4.16-4.05 (m, 2H, Gly-G%), 3.96-3.90 (m, 1H, Succinimide-CH), 3.77 (s, 1.5H, COOCft), 3.76 (s, 1.5H, COOCft), 3.48 (dd, 0.5H, J 14.7 Hz, J 5.9 Hz, Cys-p-CH2), 3.41-3.31 (m, 1H, Cys-β- CH2), 3.24-3.12 (m, 1.5H, Cys-p-CH2, Succinimide-CH2), 2.58 (dd, 0.5H, J 5.2 Hz, J4.6 Hz, Succinimide-CH2), 2.55 (dd, 0.5H, J 5.2 Hz, J4.6 Hz, Succinimide-CH2), 1.54 (s, 9H, NC(C%)3); 19F MR (decoupled) (376.6 MHz; D20): δ/ppm -75.6 (OCOCF5); 13C MR (128.5 MHz; D20): δ/ppm 180.5 (Succinimide-CO), 180.2 (Succinimide-CO), 178.8
(Succinimide-CO), 178.8 (Succinimide-CO), 171.5 (COOCH3), 171.5 (COOCH3), 168.6 (Cys-CO), 168.6 (Cys-CO), 163.0 (q, J 35.1 Hz, OCOCF3), 116.4 (q, J291.5 Hz, OCOCF3), 59.2 (NC(CH3)3), 52.9 (COOCH3), 52.5 (Cys-a-CH), 52.2 (Cys-a-CH), 41.3 (Gly-CH2), 41.2 (Gly-CH2), 41.0 (Succinimide-CH), 40.1 (Succinimide-CH2), 35.9 (Succinimide-CH2), 35.6 (Succinimide-CH2), 32.8 (Cys-p-CH2), 32.2 (Cys-p-CH2), 27.3 (NC(CH3)3); HRMS m/z (ESI+) (Found: [M+H]+ 346.14322. Ci4H24N305S requires M+ 346.14312); m/z (ES+) 346 ([M+H]+, 100%); Analytical HPLC @ 220 nm (Acclaim® 120 C18 RP LC Column; 95:5:0.1
5:95:0.1 ; H20 : MeCN : TFA) Diastereomer 1 : Ret. Time = 8.812 min, Purity: 37.16%; Diastereomer 2: Ret. Time = 8.940 min, Purity: 59.20%;
Synthesis of 4:
To a solution of 12 (36.6 mg, 0.08 mmol, 1.0 eq.) and anhydrous triethylamine (17.0 mg, 23.4 μΐ^, 0.17 mmol 2.0 eq.) stirring in anhydrous THF (0.9 mL) was added acetyl chloride
(7.3 mg, 6.6 [iL, 0.09 mmol, 1.1 eq.). The reaction mixture was stirred under nitrogen at RT for 4 h. After this time the reaction mixture was diluted with water, lyophilized and the resulting solid was purified by semi-preparative HPLC (Agilent ZORBAX 300SB-C18 column; 95 :5 - 5 :95; H20 : MeCN) to afford 4 (mixture of diastereoisomers) as a colourless hygroscopic solid (20.8 mg, 64%): [αβ° -14.5 (c 0.55, water); vm cm l (neat) 3297.8 (w), 2976.6 (w), 1749.4 (m), 1695.7 (s), 1650.1 (m), 1529.5 (m), 1457.9 (m), 1437.4 (m), 141 1.4 (m), 1369.0 (m), 1342.2 (m), 1263.4 (m), 1204.2 (m), 1 163.5 (m), 1039.9 (m); ¾ NMR (500 MHz; D20): δ/ppm 4.68-4.61 (m, 1H, Cys-a-CH), 4.04 (s, 2H, Gly-C%), 3.93-3.85 (m, 1H, Succinimide-CH), 3.75 (s, 3H, COOCft), 3.30 (dd, 0.5H, J 14.1 Hz, J 5.2 Hz, Cys-p-CH2), 3.24-3.10 (m, 2H, Cys-p-CH2, Succinimide-CH2), 3.01 (dd, 0.5H, J 14.1 Hz, J 8.7 Hz, Cys-β- CH2), 2.58 (dd, 0.5H, J 4.8 Hz, J 4.1 Hz, Succinimide-CH2), 2.54 (dd, 0.5H, J4.8 Hz, J4.1 Hz, Succinimide-CH2), 2.08 (s, 1.5H, COC%), 2.07 (s, 1.5H, COC%), 1.54 (s, 9H,
NC(C%)3); 13C NMR (128.5 MHz; D20): δ/ppm 180.2 (Succinimide-CO), 180.1
(Succinimide-CO), 179.1 (Succinimide-CO), 179.0 (Succinimide-CO), 174.3 (COCH3), 174.3 (COCH3), 172.6 (Cys-CO), 172.5 (Cys-CO), 171.7 (COOCH3), 171.7 (COOCH3), 59.2 (NC(CH3)3), 59.1 (NC(CH3)3), 53.2 (Cys-a-CH), 52.8 (COOCH3), 52.7 (Cys-a-CH), 41.2 (Gly-CH2), 40.8 (Succinimide-CH), 40.0 (Succinimide-CH), 36.2 (Succinimide-CH2), 35.9 (Succinimide-CH2), 32.6 (Cys-p-CH2), 32.3 (Cys-p-CH2), 27.3 (NC(CH3)3), 21.7 (COCH3); HRMS m/z (ESI+) (Found: [M+H]+ 388.15286. Ci6H26N306S requires M+ 388.15368); m/z (ES+) 388 ([M+H]+, 100%); Analytical HPLC @ 220 nm (Acclaim® 120 C18 RP LC
Column; 95 :5 :0.1 5 :95 :0.1 ; H20 : MeCN : TFA) Ret. Time = 9.677 min, Purity: 97.16%; Chiral HPLC @ 220 nm (ChiralPak® AD-H column (5 μ m, 4.6 x 150 mm); Isocratic method: 75 : 25 Hexane : IP A, 0.3 mL / min) Diastereomer 1 : Ret. Time = 37.366 min, 38.54%; Diastereomer 2: Ret. Time = 40.515 min, 61.46%.
Synthesis of compound 5:
FmocHN

13

5 16
Synthesis of 14:
N-Fmoc-D-Cys(Trt)-OH (0.50 g, 0.85 mmol, 1.05 eq.), glycine tert-butyl ester hydrochloride (0.14 g, 0.81 mmol, 1.0 eq.) and HBTU (0.32 g, 0.85 mmol, 1.05 eq.) were stirred in anhydrous DMF (2 mL) for 5 minutes. After this time, DIPEA (0.21 g, 0.28 mL, 1.62 mmol, 2.0 eq.) was added and the reaction mixture was stirred under nitrogen at RT for 120 h. The reaction mixture was partitioned over ethyl acetate and 0.5 M LiCl and the organic layer was collected, dried over Na2SC"4, filtered and concentrated in vacuo. The resulting crude material was adsorbed onto Celite® and purified by silica gel chromatography, eluting with ethyl acetate, petroleum ether and triethylamine (10:89: 1 - 80: 19: 1; ethyl acetate : petroleum ether : triethylamine) to yield N-Fmoc-^-trityl-D-cysteinylglycine tert-butyl ester (0.50 g, 84%) as a colourless solid: Rt 0.4 (30:70 ethyl acetate : petroleum ether); mp 85-88 °C (ethyl acetate : petroleum ether); [a]¾° -5.4 (c 1.0, chloroform); Vmax/cm"1 (neat); 3296.4 (w), 3057.2 (w), 1728.2 (m), 1662.6 (m), 1595.1 (m), 1519.9 (m), 1489.1 (m), 1444.7 (m), 1367.5 (m), 1319.3 (m), 1217.1 (m), 1151.5 (m), 1082.1 (m), 1033.9 (m); ¾ NMR (500 MHz; CDC13): δ/ppm 7.82-7.18 (m, 23H, Ar CH), 6.34 (s, 1H, Gly-NH), 5.03 (d, 1H, J 7.3 Hz, Cys-NH), 4.44-4.36 (m, 2H, Fmoc-CH2), 4.22 (t, 1H, J 7.3 Hz, J 6.8 Hz, Fmoc-CH), 3.93-3.77 (m, 3H, Cys-a-CH, Gly-CH2), 2.75-2.61 (m, 2H, Cys-p-Ci¾), 1.46 (s, 9H, OC(CH3)3); 13C NMR (128.5 MHz; CDCI3): δ/ppm 170.1 (Cys-CO), 168.4 (COO¾u), 156.1 (Fmoc-CONH), 144.5 (Ar C), 143.8 (Ar C), 141.4 (Ar C), 129.7 (Ar CH), 128.2 (Ar CH), 127.9 (Ar CH), 127.2 (Ar CH), 127.1 (Ar CH), 125.2 (Ar CH), 120.1 (Ar CH), 82.5 (OC(CH3)3), 67.5 (Fmoc-CH2), 67.2 (SCPh3) 54.0 (Cys-a-CH), 47.2 (Fmoc-CH), 42.2 (Gly-CH2), 33.9 (Cys-p-CH2), 28.1
(OC(CH3)3); HRMS m/z (ESI+) (Found: [M+Na]+ 721.26843. C43H42N2Na05S requires M+ 721.27066); m/z (ES+) 117 (100%), 721 ([M+Na]+, 3%); Analytical HPLC @ 254 nm
(Acclaim® 120 C18 RP LC Column; 95:5:0.1 5:95:0.1; H20 : MeCN : TFA) Ret. Time = 16.693 min, Purity: 97.97%.
Synthesis of 15:
N-Fmoc-^-trityl-D-cysteinylglycine tert-butyl ester 14 (0.4 g, 0.57 mmol, 1.0 eq.) was stirred under argon at RT in 20% piped dine in DMF (3.2 mL) for 4 h. After this time, the reaction mixture was concentrated in vacuo and the resulting residue was adsorbed onto Celite® and purified by silica gel chromatography, eluting with ethyl acetate, petroleum ether and triethylamine (12:87: 1 - 99:0: 1; ethyl acetate : petroleum ether : triethylamine) to yield S- trityl-D-cysteinylglycine tert-butyl ester 15 (0.13 g, 48%) as a yellow oil: Rf 0.3 (50:50 ethyl acetate : petroleum ether); [a]¾° -12.0 (c 0.59, chloroform); Vmax/cm"1 (chloroform); 3373.5 (w), 2980.0 (m), 2929.9 (w), 1739.8 (m), 1672.3 (m), 1595.1 (m), 1514.1 (m), 1491.0 (m), 1444.7 (m), 1392.6 (m), 1367.5 (m), 1244.1 (m), 1155.4 (s), 1082.1 (m), 1033.9 (m); ¾ MR (500 MHz; CDC13): δ/ppm 7.51 (t, 1H, J 5.3 Hz, Gly-NH), 7.48-7.15 (m, 15H, Ar CH), 3.90 (dd, 1H, J 18.4 Hz, J 5.3 Hz, Gly C¾HB), 3.81 (dd, 1H, J 18.4 Hz, J 5.3 Hz, Gly
CHA¾), 3.06-2.96 (m, 1H, Cys-a-CH), 2.82-2.73 (m, 1H, Cys-p-C%), 2.55 (dd, 1H, J 12.8 Hz, J 8.8 Hz, Cys-p-C%), 1.45 (s, 9H, OC(C%)3); 13C NMR (128.5 MHz; CDC13): δ/ppm 173.2 (Cys-CO), 169.0 (COOC(CH3)3), 144.7 (Ar C), 129.8 (Ar CH), 128.1 (Ar CH), 127.0 (Ar CH), 82.3 (OC(CH3)3), 67.2 (SCPh3), 54.1 (Cys-a-CH), 41.9 (Gly-CH2), 37.4 (Cys-β- CH2), 28.2 (OC(CH3)3); HRMS m/z (ESI+) (Found: [M+H]+ 477.22004. C28H33N203S requires M+ 477.22064); m/z (ES+) 477 ([M+H]+, 100%); Analytical HPLC @ 254 nm (Acclaim® 120 C18 RP LC Column; 95:5:0.1 5:95:0.1; H20 : MeCN : TFA) Ret. Time = 11.734 min, Purity: 95.46%.
Synthesis of 16:
^-trityl-D-cysteinyl tert-butyl ester 15 (0.11 g, 0.23 mmol, 1.0 eq.) and triethylsilane (0.06 g, 0.08 mL, 0.5 mmol, 2.2 eq.) were stirred under argon at RT in 20% TFA in CH2C12 (5.8 mL) for 7 h. After this time the reaction solution was concentrated in vacuo and resulting residue was purified by RP C-18 silica gel column chromatography, eluting with water to afford D- cysteinylglycine trifluoroacetic acid salt as an off-white sticky foam (55.2 mg), which was used without further purification. D-cysteinylglycine trifluoroacetic acid salt (35.5 mg) was
stirred in water (1.0 mL), to this N-tert-butyl-maleimide (21.0 mg, 19.8 μΐ., 0.14 mmol) was added and the resulting solution stirred at RT for 16 h. After this time, the reaction solution was lyophilized and the resulting solid was purified by semi-preparative HPLC (Agilent ZORBAX 300SB-C18 column; 95 :5:0.1 5:95:0.1; H20 : MeCN : TFA) to afford 16
(mixture of diastereoisomers) as a colourless solid (47.1 mg, 71% over 2 steps): [a]¾° -26.4 (c 1.0, water); vm cm l (neat); 2970.4 (w), 2941.4 (w), 1737.9 (m), 1681.9 (s), 1460.1 (m), 1417.7 (m), 1348.2 (m), 1265.3 (m), 1228.7 (m), 1197.8 (s), 1166.9 (s), 1134.1 (s), 1024.2 (m); ¾ NMR (500 MHz; D20): δ/ppm 4.39 (dd, 0.5H, J 7.3 Hz, J 5.8 Hz, Cys-a-CH), 4.35 (dd, 0.5H, J 7.3 Hz, J 5.7 Hz, Cys-a-CH), 4.09 (s, 1H, Gly-G%), 4.08 (s, 1H, Gly-G%), 3.94-3.90 (m, 1H, Succinimide-CH), 3.48 (dd, 0.5H, J 14.8 Hz, J 5.9 Hz, Cys-p-C%), 3.41- 3.30 (m, 1H, Cys-p-C%), 3.22-3.11 (m, 1.5H, Cys-p-C¾ Succinimide-CH2), 2.57 (dd, 0.5H, J 5.4 Hz, J 5.0 Hz, Succinimide-CH2), 2.54 (dd, 0.5H, J 5.4 Hz, J 5.0 Hz, Succinimide- CH2), 1.53 (s, 9H, NC(C%)3); 19F NMR (decoupled) (376.6 MHz; D20): δ/ppm -75.6 (OCOCF5); 13C NMR (128.5 MHz; D20): δ/ppm 180.4 (Succinimide-CO), 180.1
(Succinimide-CO), 178.8 (Succinimide-CO), 178.8 (Succinimide-CO), 172.7 (COOH), 172.7 (COOH), 168.5 (Cys-CO), 168.5 (Cys-CO), 162.9 (q, J35.4 Hz, OCOCF3), 116.3 (q, J291.5 Hz, OCOCF3), 59.2 (NC(CH3)3), 52.5 (Cys-a-CH), 52.3 (Cys-a-CH), 41.2 (Gly-CH2), 41.2 (Gly-CH2), 41.0 (Succinimide-CH), 40.1 (Succinimide-CH), 35.9 (Succinimide-CH2), 35.6 (Succinimide-CH2), 32.8 (Cys-p-CH2), 32.2 (Cys-p-CH2), 27.3 (NC(CH3)3); HRMS m/z (ESI+) (Found: [M+H]+ 332.12676. Ci3H22N305S requires M+ 332.12747); m/z (ES+) 332 ([M+H]+, 100%); Analytical HPLC @ 220 nm (Acclaim® 120 C18 RP LC Column; 95:5:0.1
5:95:0.1 ; H20 : MeCN : TFA) Diastereomer 1 : Ret. Time = 8.448 min, Purity: 35.99%; Diastereomer 2: Ret. Time = 8.541 min, Purity: 61.26%.
Synthesis of 5:
To a solution of 16 (27.5 mg, 0.06 mmol, 1.0 eq.) and anhydrous triethylamine (12.5 mg, 17.2 μΐ^, 0.12 mmol, 2.0 eq.) stirring in anhydrous THF (0.6 mL) was added acetyl chloride (5.3 mg, 4.8 [iL, 0.07 mmol, 1.1 eq.). The reaction mixture was stirred at RT for 4 h. After this time the reaction mixture was diluted with water, lyophilized and the resulting solid was purified by semi-preparative HPLC (Agilent ZORBAX 300SB-C18 column; 95 :5:0.1 5:95:0.1 ; H20 : MeCN : TFA) to afford 5 (mixture of diastereoisomers) as a colourless solid (13.9 mg, 46%): [αβ° +15.0 (c 1.0, water); vm cm l (neat); 3304.1 (w), 2980.0 (w), 1768.7 (w), 1693.5 (s), 1658.8 (m), 1531.5 (m), 1460.1 (m), 1402.3 (m), 1369.5 (m), 1342.5 (m),
1263.4 (m), 1232.5 (m), 1197.8 (m), 1163.1 (m), 1041.6 (m); ¾ NMR (500 MHz; D20): δ/ppm 4.68-4.61 (m, 1H, Cys-a-CH), 4.01 (s, 2H, Gly-G%), 3.92-3.84 (m, 1H, Succinimide- CH), 3.30 (dd, 0.5H, J 14.1 Hz, J 5.1 Hz, Cys-p-CH2), 3.24-3.09 (m, 2H, Cys-p-CH2, Succinimide-CH2), 2.99 (dd, 0.5H, J 14.1 Hz, J 8.7 Hz, Cys-p-CH2), 2.57 (dd, 0.5H, J4.4 Hz, J 3.9 Hz, Succinimide-CH2), 2.53 (dd, 0.5H, J4.4 Hz, J 3.9 Hz, Succinimide-CH2), 2.07 (s, 1.5H, COC%), 2.06 (s, 1.5H, COC%), 1.53 (s, 9H, NC(C%)3); 19F NMR (decoupled) (376.6 MHz; D20): δ/ppm -75.7 (OCOCF5); 13C NMR (128.5 MHz; D20): δ/ppm 180.2 (Succinimide-CO), 180.2 (Succinimide-CO), 179.1 (Succinimide-CO), 179.0 (Succinimide- CO), 174.4 (COCH3), 174.3 (COCH3), 172.9 (COOH), 172.9 (COOH), 172.4 (Cys-CO), 172.4 (Cys-CO), 163.0 (q, J35.1 Hz, OCOCF3), 116.3 (q, J290.9 Hz, OCOCF3), 59.2 (NC(CH3)3), 59.1 (NC(CH3)3), 53.1 (Cys-a-CH), 52.7 (Cys-a-CH), 41.1 (Gly-CH2), 40.8 (Succinimide-CH), 40.0 (Succinimide-CH), 36.2 (Succinimide-CH2), 35.9 (Succinimide- CH2), 32.7 (Cys-p-CH2), 32.3 (Cys-p-CH2), 27.3 (NC(CH3)3), 21.7 (COCH3); HRMS m/z (ESI+) (Found: [M+H]+ 374.13761. Ci5H24N306S requires M+ 374.13803); m/z (ES+) 374 ([M+H]+, 100%); Analytical HPLC @ 220 nm (Acclaim® 120 C18 RP LC Column; 95:5:0.1
5:95:0.1 ; H20 : MeCN : TFA) Ret. Time = 9.237 min, Purity: 97.26%; Chiral HPLC @ 220 nm (ChiralPak® AD-H column (5 μ m, 4.6 x 150 mm); Isocratic method: 75 : 25 Hexane : IP A, 0.3 mL / min) Diastereomer 1 : Ret. Time = 40.368 min, 53.55%; Diastereomer 2: Ret. Time = 51.065 min, 44.86%.
Synthesis of compound 6:
cys-gly
Cys-Gly (15.0 mg, 0.08 mmol, 1.0 eq.) and NaOH (3.4 mg, 0.08 mmol, 1.0 eq.) were stirred in water (0.7 mL), to this N-tert-butyl-maleimide (12.9 mg, 12.2 μΐ^, 0.08 mmol, 1.0 eq.) was added and the resulting solution stirred at RT for 1.5 h. After this time, the reaction solution was lyophilized and the resulting solid was purified by semi-preparative HPLC (Agilent ZORBAX 300SB-C18 column; 95 :5:0.1 81 : 19:0.1; H20 : MeCN : TFA) to afford 6
(mixture of diastereoisomers) as a colourless solid (21.9 mg, 58%): Rf0.3 (water (RP));
[αβ°+25.8 (c 0.65, H20); vm cm l (neat); 2970.9 (w), 1675.7 (s), 1349.1 (m), 1264.9 (m),
1200.1 (s); ¾ NMR (500 MHz; D20): δ/ppm 4.39 (dd, 0.5H, J7.5 Hz, J 5.8 Hz, Cys-a-CH), 4.35 (dd, 0.5H, J 7.5 Hz, J 5.8 Hz, Cys-a-CH), 4.08-3.96 (m, 2H, Gly-C%), 3.95-3.89 (m, 1H, Succinimide-CH), 3.49 (dd, 0.5H, J 14.7 Hz, J 5.8 Hz, Cys-p-C%), 3.41-3.30 (m, 1H, Cys-p-G%), 3.22-3.12 (m, 1.5H, Cys-p-C¾ Succinimide-CH2), 2.58 (dd, 0.5H, J 5.6 Hz, J 5.2 Hz, Succinimide-CH2), 2.55 (dd, 0.5H, J 5.6 Hz, J 5.2 Hz, Succinimide-CH2), 1.54 (s, 9H, NC(C%)3); 19F MR (decoupled) (376.6 MHz; D20): δ/ppm -75.6 (OCOCF5);
13C NMR (128.5 MHz; D20): δ/ppm 180.5 (Succinimide-CO), 180.2 (Succinimide-CO), 178.8 (Succinimide-CO), 178.8 (Succinimide-CO), 173.6 (COOH), 173.6 (COOH), 168.4 (Cys-CO), 168.3 (Cys-CO), 163.0 (q, J 35.6 Hz, OCOCF3), 116.4 (q, J290.9 Hz, OCOCF3), 59.2 ((NC(CH3)3), 52.6 (Cys-a-CH), 52.3 (Cys-a-CH), 41.8 (Gly-CH2), 41.8 (Gly-CH2), 41.0 (Succinimide-CH), 40.1 (Succinimide-CH), 35.9 (Succinimide-CH2), 35.6 (Succinimide- CH2), 32.8 (Cys-p-CH2), 32.2 (Cys-p-CH2), 27.3 (NC(CH3)3); HRMS m/z (ESI+) (Found: [M+Na]+ 354.1098. Ci3H2iN3Na05S requires M+ 354.1094); m/z (ES+) 332 ([M+H]+, 100%); Analytical HPLC (Acclaim® 120 C18 RP LC Column; 95:5:0.1 5:95:0.1 ; H20 : MeCN : TFA) Diastereomer 1 : Ret. Time = 8.517 min, Purity: 40.55%; Diastereomer 2: Ret. Time = 8.605 min, Purity: 57.21%.
Synthesis of compound 7:

6
To a solution of 6 (15.0 mg, 0.03 mmol, 1.0 eq.) and anhydrous triethylamine (6.8 mg, 9.4 μί, 0.07 mmol, 2.0 eq.) stirring in anhydrous THF (0.3 mL) was added acetyl chloride (2.9 mg, 2.6 0.04 mmol, 1.1 eq.). The reaction mixture was stirred at RT for 6.5 h. After this time, the reaction mixture was diluted with water, lyophilized and the resulting solid was purified by semi-preparative HPLC (Agilent ZORBAX 300SB-C18 column; 95 :5:0.1 5:95:0.1 ; H20 : MeCN : TFA) to afford 7 (mixture of diastereoisomers) as a colourless solid (9.0 mg, 55%): Rt 1.0 (water (RP)); [αβ° -14.8 (c 0.46, H20); Vmax/cm"1 (neat) 2970.8 (w), 1737.8 (m), 1694.1 (s), 1532.9 (m), 1346.2 (m), 1263.7 (m), 1201.8 (m), 1163.7 (s); ¾ NMR (500 MHz; D20): δ/ppm 4.69-4.61 (m, 1H, Cys-a-CH), 4.01 (s, 2Η, Gly-CH2), 3.92- 3.84 (m, 1Η, Succinimide-CH), 3.31 (dd, 0.5Η, J 14.0 Hz, J 5.1 Hz, Cys-p-CH2) 3.24-3.10
(m, 2H, Cys-p-G¾ Succinimide-CH2), 3.00 (dd, 0.5H, J 14.0 Hz, J 8.7 Hz, Cys-p-C%), 2.57 (dd, 0.5H, J4.3 Hz, J 3.6 Hz, Succinimide-CH2), 2.54 (dd, 0.5H, J4.3 Hz, J3.6 Hz, Succinimide-CH2), 2.08 (s, 1.5H, COC%), 2.07 (s, 1.5H, COC%), 1.54 (s, 4.5H, NC(C%)3), 1.53 (s, 4.5H, NC(G%)3); 19F NMR (decoupled) (376.6 MHz; D20): δ/ppm -75.6 (OCOCF5); 13C NMR (128.5 MHz; D20): δ/ppm 180.2 (Succinimide-CO), 180.2 (Succinimide-CO), 179.1 (Succinimide-CO), 179.1 (Succinimide-CO), 174.4 (COCH3), 174.3 (COC¾), 172.9 (COOH), 172.4 (Cys-CO), 172.4 (Cys-CO), 163.0 (q, J35.4 Hz, OCOCF3), 116.4 (q, J290.5 Hz, OCOCF3), 59.2 (NC(CH3)3), 59.1 (NC(CH3)3), 53.1 (Cys-a-CH), 52.7 (Cys-a-CH), 41.1 (Gly-CH2), 40.8 (Succinimide-CH), 40.0 (Succinimide-CH), 36.2 (Succinimide-CH2), 35.9 (Succinimide-CH2), 32.7 (Cys-p-CH2), 32.3 (Cys-p-CH2), 27.3 (NC(CH3)3), 21.7 (COCH3); HRMS m/z (ESI") (Found: [M-H]" 372.12347. Ci5H2206N3S requires M" 372.12348); m z (ES+) 374 ([M+H]+, 100%); Analytical HPLC @ 220 nm (Acclaim® 120 CI 8 RP LC
Column; 95 :5:0.1 5:95:0.1 ; H20 : MeCN : TFA) Ret. Time = 9.519 min, Purity: 98.82%. Chiral HPLC @ 220 nm (ChiralPak® AD-H column (5 μ m, 4.6 x 150 mm); Isocratic method: 75 : 25 Hexane : IP A, 0.3 mL / min) Diastereomer 1 : Ret. Time = 21.810 min, 32.99%; Diastereomer 2: Ret. Time = 23.640 min, 66.99%.
Synthesis of compound 8:

17 18 19

8
Boc-J-Cys(Trt)-OH (0.1 g, 0.22 mmol, 1.0 eq.) and fert-butyl-2-hydroxyacetate (55.6 mg, 0.42 mmol, 1.95 eq.) were stirred in anhydrous acetonitrile at 0 °C, to this Ν,Ν'-
dicyclohexylcarbodiimide (86.8 mg, 0.42 mmol, 1.95 eq.) was added and the resulting solution stirred under argon at 0 °C for a further 2 h before being stirred at RT for 15.5 h. After this time, the reaction mixture was filtered under reduced pressure, the filtrate was concentrated in vacuo and the resulting residue was adsorbed onto Celite® and purified by silica gel chromatography, eluting with ethyl acetate and petroleum ether (5:95 - 40:60; ethyl acetate : petroleum ether) to yield 2-(tert-butoxy)-2-oxoethyl N-(tert-butoxycarbonyl)- ^-trityl-J-cysteinate as an impure oil (0.11 g), which was used without further purification. N- (tert-butoxycarbonyl^-trityl-J-cysteinate (0.11 g) and triethylsilane (0.08 g, 0.11 mL, 0.69 mmol) were stirred under argon at RT in 20% TFA in CH2CI2 (6.5 mL) for 2 h. After this time the reaction solution was concentrated in vacuo and the resulting residue was purified by RP C-18 silica gel column chromatography, eluting with water to afford 2-((L- cysteinyl)oxy)acetic acid as an impure colourless solid (32.2 mg), which was used without further purification. To 2-((J-Cysteinyl)oxy)acetic acid (32.2 mg) stirring in water (3.6 mL) was added N-tert-butyl-maleimide (27.5 mg, 26.0 μL, 0.18 mmol) and the resulting solution was stirred at RT for 7 h. After this time, the reaction solution was lyophilized and the resulting solid was purified by semi-preparative HPLC (Agilent ZORBAX 300SB-C18 column; 95 :5:0.1 5:95:0.1 ; H20 : MeCN : TFA) to afford (2R)-3-((l-(tert-butyl)-2,5- dioxopyrrolidin-3 -yl)thio)- 1 -(carboxymethoxy)- 1 -oxopropan-2-aminium 2,2,2- trifluoroacetate (mixture of diastereoisomers) as a colourless solid (25.5 mg), which was used without further purification.
To a solution of (2R)-3-((l-(tert-butyl)-2,5-dioxopyrrolidin-3-yl)thio)-l-(carboxymethoxy)-l- oxopropan-2-aminium 2,2,2-trifluoroacetate (25.5 mg) and anhydrous triethylamine (11.5 mg, 15.9 [iL, 0.11 mmol) stirring in anhydrous THF (0.5 mL) was added acetyl chloride (5.0 mg, 4.5 μL, 0.06 mmol). The reaction mixture was stirred at RT for 3.5 h. After this time, the reaction mixture was diluted with water, lyophilized and the resulting solid was purified by semi-preparative HPLC (Agilent ZORBAX 300SB-C18 column; 95 :5:0.1 5:95:0.1; H20 : MeCN : TFA) to afford 8 (mixture of diastereoisomers) as a colourless solid (11.6 mg, 14%, over four steps): [a]D 2° -24.0 (c 0.75, H20); Vmax/cm"1 (neat); 2980.0 (m), 1745.6 (m), 1693.5 (s), 1541.1 (m), 1460.1 (w), 1419.6 (m), 1371.4 (m), 1344.4 (m), 1263.4 (m), 1163.1 (s), 1053.1 (m); ¾ NMR (500 MHz; D20): δ/ppm 4.86-4.73 (m, 3H, Cys-a-CH, OCH2CO2H), 3.94-3.85 (m, 1H, Succinimide-CH), 3.42 (dd, 0.5H, J 14.2 Hz, J4.9 Hz, Cys-p-C%), 3.31 (dd, 0.5H, J 14.2 Hz, J4.9 Hz, Cys-p-C%), 3.25-3.10 (m, 1.5H, Cys-p-C%, Succinimide- CH2), 3.06 (dd, 0.5H, J 14.2 Hz, J 8.9 Hz, Cys-p-C%), 2.58 (dd, 0.5H, J 4.4 Hz, J 3.2 Hz,
Succinimide-CH2) 2.54 (dd, 0.5H, J4.4 Hz, J 3.2 Hz, Succinimide-CH2), 2.07 (s, 1.5H,
COCH5), 2.06 (s, 1.5H, COCH5), 1.53 (s, 4.5H, NC(CH3)3), 1.53 (s, 4.5H, NC(CH5)3); 19F NMR (decoupled) (376.6 MHz; D20): δ/ppm -75.7 (OCOCF5); 13C NMR (128.5 MHz; D20): δ/ppm 180.1 (Succinimide-CO), 180.0 (Succinimide-CO), 179.0 (Succinimide-CO), 179.0 (Succinimide-CO), 174.3 (COCH3), 174.2 (COC¾), 171.2 (Cys-CO), 171.2 (Cys-CO), 171.1 (CO2H), 162.9 (q, J 35.3 Hz, OCOCF3), 116.3 (q, J 290.7 Hz, OCOCF3), 61.9 (OCH2C02H), 59.2 (NC(CH3)3), 59.2 (NC(CH3)3), 52.5 (Cys-a-CH), 52.0 (Cys-a-CH), 40.9 (Succinimide- CH), 39.9 (Succinimide-CH), 36.2 (Succinimide-CH2), 35.8 (Succinimide-CH2), 32.2 (Cys- p-CH2), 31.7 (Cys-p-CH2), 27.3 (NC(CH3)3), 21.6 (COCH3), 21.5 (COCH3); HRMS m/z (ESI+) (Found: [M+H]+ 375.12147. Ci5H2307N2S requires M+ 375.12205); m z (ES+) 375 ([M+H]+, 100%); Analytical HPLC @ 220 nm (Acclaim® 120 C18 RP LC Column; 95:5:0.1
5:95:0.1 ; H20 : MeCN : TFA) Ret. Time = 9.776 min, Purity: 95.47%; Chiral HPLC @ 220 nm (ChiralPak® AD-H column (5 μ m, 4.6 x 150 mm); Isocratic method: 75 : 25 Hexane : IP A, 0.3 mL / min) Diastereomer 1 : Ret. Time = 20.625 min, 50.48%; Diastereomer 2: Ret. Time = 29.359 min, 49.32%.
Synthesis of compound 9:

9
Synthesis of compound 21:
N-Fmoc-Cys(Trt)-OH 20 (0.75 g, 1.28 mmol, 1.05 eq.), glycine tert-butyl ester hydrochloride (0.20 g, 1.22 mmol, 1.0 eq.) and HBTU (0.49 g, 1.28 mmol, 1.05 eq.) were stirred in anhydrous DMF (3 mL) for 5 minutes. After this time, DIPEA (0.32 g, 0.42 mL, 2.43 mmol,
2.0 eq.) was added and the reaction mixture was stirred under argon at RT for 24 h. The reaction mixture was partitioned between ethyl acetate and 0.5 M LiCl and the organic layer was collected, dried over Na2S04, filtered and concentrated in vacuo. The resulting crude material was adsorbed onto Celite® and purified by silica gel chromatography, eluting with ethyl acetate, petroleum ether and triethylamine (10:89: 1 - 80: 19: 1; ethyl acetate :
petroleum ether : triethylamine) to yield N-Fmoc-^-trityl-L-cysteinylglycine tert-butyl ester 21 (0.85 g, 95%) as a colourless solid: Ri 0.8 (40:60 ethyl acetate : petroleum ether); mp 79- 83 °C (ethyl acetate : petroleum ether); [αβ° +5.8 (c 1.0, chloroform) (Lit. Value [αβ5 +2.4 (c 0.25, chloroform))5; ¾ NMR (400 MHz; CDC13): δ/ppm 7.73-7.08 (m, 23H, Ar CH), 6.25 (s, IH, CONH), 4.92 (d, IH, J 7.5 Hz, Cys-NH), 4.36-4.26 (m, 2H, Fmoc-C%), 4.13 (t, IH, J 7.5 Hz, J 6.8 Hz, Fmoc-CH), 3.85-3.67 (m, 3H, Cys-a-CH, Gly-C%), 2.67-2.52 (m, 2H, Cys- p-C%), 1.37 (s, 9H, OC(C%)3); m/z (ES+) 721 ([M+Na]+, 100%). The data are in good agreement with literature values.5
Synthesis of compound 22:
N-Fmoc-^-trityl-L-cysteinylglycine tert-butyl ester 21 (0.75 g, 1.07 mmol, 1.0 eq.) was stirred under argon at RT in 20% piperidine in DMF (6 mL) for 5 h. After this time, the reaction mixture was concentrated in vacuo and the resulting residue was adsorbed onto Celite® and purified by silica gel chromatography, eluting with ethyl acetate, petroleum ether and triethylamine (12:87: 1 - 99:0: 1; ethyl acetate : petroleum ether : triethylamine) to yield S-trityl-L-cysteinylglycine fert-butyl ester 22 (0.38 g, 74%) as a yellow oil: Rt 0.2 (50:50 ethyl acetate : petroleum ether); [αβ° +13.2 (c 1.0, chloroform) (Lit. Value [αβ5 +9.2 (c 0.5, chloroform))5; ¾ MR (400 MHz; CDCI3): δ/ppm 7.45 (t, IH, J 5.3 Hz, Gly-NH), 7.40-7.02 (m, 15H, Ar CH), 3.77 (dd, IH, J 18.3 Hz, J 5.4 Hz, Gly C¾HB), 3.69 (dd, IH, J 18.3 Hz, J 5.4 Hz, Gly CHA¾), 2.91 (dd, IH, J 8.8 Hz, J 3.8 Hz, Cys-a-CH), 2.65 (dd, IH, J 12.8 Hz, J 3.8 Hz, Cys-p-C%), 2.45 (dd, IH, J 12.8 Hz, J 8.8 Hz, Cys-p-C%), 1.33 (s, 9H, OC(C%)3); m/z (ES+) 477 ([M+H]+, 100%). The data are in good agreement with literature values.5
Synthesis of compound 9:
^-Trityl-L-cysteinylglycine tert-butyl ester 22 (89.3 mg, 0.19 mmol, 1.0 eq.), N-boc-Y- aminobutyric acid (38.1 mg, 0.19 mmol, 1.0 eq.), EDC DHC1 (41.3 mg, 0.22 mmol, 1.15 eq.), HBTU (7.1 mg, 0.02 mmol, 0.1 eq.) and DIPEA (48.4 mg, 65.3 L, 0.37 mmol, 2.0 eq.) were stirred at RT in anhydrous CH2CI2 (9 mL) for 24 h. After this time, the reaction mixture was
concentrated in vacuo and the resulting residue was adsorbed onto Celite and purified by silica gel chromatography, eluting with ethyl acetate, petroleum ether and triethylamine (12:87: 1 - 99:0: 1; ethyl acetate : petroleum ether : triethylamine) to yield N-boc-Y- aminobutyric acid-^-trityl-L-cysteinylglycine tert-butyl ester 23 (0.10 g) as a colourless oil, which was used without further purification.
N-Boc- Y-aminobutyric acid-^-trityl-L-cysteinylglycine tert-butyl ester 23 (95.0 mg) and triethylsilane (50.1 mg, 68.8 L, 0.43 mmol) were stirred in 20% TFA/CH2C12 (5 mL) at RT for 6.5 h. After this time, the reaction mixture was concentrated in vacuo and the resulting residue was purified by RP C-18 silica gel column chromatography, eluting with water to afford Y-aminobutyric acid-L-cysteinylglycine trifluoroacetic acid salt (33.3 mg) as a colourless residue, which was used without further purification. To Y-aminobutyric acid-L- cysteinylglycine trifluoroacetic acid salt (25.0 mg) stirring in water (0.5 mL) was added N- tert-butyl-maleimide (10.2 mg, 9.6 μL, 0.07 mmol) and the resulting solution was stirred at RT for 19.5 h. After this time, the reaction solution was lyophilized and the resulting solid was purified by semi-preparative HPLC (Agilent ZORB AX 300SB-C 18 column; 95 :5:0.1 5:95:0.1 ; H20 : MeCN : TFA) to afford 9 (mixture of diastereoisomers) as a colourless solid (25.9 mg, 37% over 3 steps): [αβ° -14.5 (c 1.0, water); Vmax/cm"1 (neat) 2939.5 (w), 1689.6 (m), 1653.0 (s), 1527.6 (m), 1460.1 (m), 1417.7 (m), 1346.3 (m), 1263.4 (m), 1230.6 (m), 1166.9 (s), 1130.3 (s); ¾ NMR (400 MHz; D20): δ/ppm 4.66-4.57 (m, 1H, Cys-a-CH), 3.97 (s, 2H, Gly-G%), 3.90-3.79 (m, 1H, Succinimide-CH), 3.28 (dd, 0.5H, J 14.2 Hz, J 5.1 Hz, Cys-p-G%), 3.22-3.04 (m, 2H, Cys-p-G¾ Succinimide-CH2), 3.04-2.89 (m, 2.5H, CH2, Cys-p-G%), 2.54 (dd, 0.5H, J 8.1 Hz, J 4.3 Hz, Succinimide-CH2), 2.49 (dd, 0.5H, J 8.1 Hz, J4.3 Hz, Succinimide-CH2), 2.46-2.38 (m, 2H, CH2), 1.99-1.87 (m, 2H, CH2), 1.49 (s, 4.5H, NC(C%)3), 1.49 (s, 4.5H, NC(C%)3); 19F NMR (decoupled) (376.6 MHz; D20): δ/ppm -75.6 (OCOGF5); 13C NMR (128.5 MHz; D20): δ/ppm 180.5 (Succinimide-CO), 180.5
(Succinimide-CO), 179.4 (Succinimide-CO), 179.4 (Succinimide-CO), 175.4
(CH2CH2CONH), 175.3 (CH2CH2CONH), 173.2 (Gly-COOH), 173.2 (Gly-COOH), 172.8 (Cys-CO), 172.7 (Cys-CO), 163.3 (q, J 35.2 Hz, OCOCF3), 116.7 (q, J292.0 Hz, OCOCF3), 59.6 (NC(CH3)3), 59.6 (NC(CH3)3), 53.4 (Cys-a-CH), 53.0 (Cys-a-CH), 41.5 (Gly-CH2), 41.2 (Succinimide-CH), 40.4 (Succinimide-CH), 39.2 (CH2), 36.6 (Succinimide-CH2), 36.3 (Succinimide-CH2), 33.0 (Cys-p-CH2), 32.8 (Cys-p-CH2), 32.5 (CH2), 27.7 (NC(CH3)3), 23.1 (CH2); HRMS m/z (ESI+) (Found: [M+H]+ 417.17911 CnH^OeS requires M+ 417.18023); m/z (ES+) 833 ([2M+H]+, 100%); Analytical HPLC @ 220 nm (Acclaim® 120 CI 8 RP LC
Column; 95 :5:0.1 5:95:0.1 ; H20 : MeCN : TFA) Diastereomer 1 : Ret. Time Purity: 28.42%; Diastereomer 2: Ret. Time = 8.553 min, Purity: 70.16%.
Synthesis of compound 10:

10
Compounds 20-22 where synthesis in an equivalent manner to compound 9. Synthesis of compound 10:
^-Trityl-L-cysteinylglycine tert-butyl ester (0.21 g, 0.44 mmol, 1.0 eq.) and triethylamine (0.06 g, 0.08 mL, 0.57 mmol, 1.3 eq.), were stirred at RT in anhydrous chloroform (2 mL) for 5 minutes. After this time, glutaric anhydride (0.06 g, 0.48 mmol, 1.1 eq.) was added and the reaction mixture was stirred at RT for 15.5 h. The reaction solution was concentrated in vacuo and the resulting residue was adsorbed onto Celite® and purified by silica gel chromatography, eluting with ethyl acetate, petroleum ether and formic acid (60:40: 1) to yield slightly impure glutaric acid-^-trityl-L-cysteinylglycine tert-butyl ester (21.9 mg) as a colourless oil, which was used without further purification. Glutaric acid-S-trityl-L- cysteinylglycine tert-butyl ester (15.9 mg) and triethylsilane (94.1 mg, 0.13 mL, 0.81 mmol) were stirred at RT under argon in 20% TFA/CH2C12 (9.4 mL) at RT for 6.5 h. After this time, the reaction mixture was concentrated in vacuo and the resulting residue was purified by RP C-18 silica gel column chromatography, eluting with water to afford slightly impure glutaric acid-L-cysteinylglycine trifluoroacetic acid salt (23.7 mg) as a colourless solid, which was used without further purification.
To glutaric acid-L-cysteinylglycine trifluoroacetic acid salt (22.4 mg) stirring in water (0.4 mL) was added N-tert-butyl-maleimide (8.4 mg, 8.0 μΐ^, 0.06 mmol) and the resulting solution was stirred at RT for 14 h. After this time, the reaction solution was lyophilized and the resulting solid was purified by semi-preparative HPLC (Agilent ZORBAX 300SB-C18 column; 95 :5 :0.1 5 :95 :0.1 ; H20 : MeCN : TFA) to afford 10 (mixture of diastereoisomers) as a colourless solid (20.2 mg, 12% over 3 steps): [a]¾° -20.0 (c 1.0, water); Vmax/cm"1 (neat) 2970.4 (w), 2941.4 (w), 1737.9 (m), 1689.6 (s), 1529.6 (m), 1456.3 (m), 1365.6 (m), 1346.3 (m), 1263.4 (m), 1228.7 (m), 1217.1 (s), 1201.7 (s), 1 159.2 (s), 1041.6 (m); ¾ NMR (500 MHz; D20): δ/ppm 4.70-4.63 (m, 1H, Cys-a-CH), 4.01 (s, 2H, Gly-C%), 3.91-3.83 (m, 1H, Succinimide-CH), 3.33 (dd, 0.5H, J 14.2 Hz, J 5.1 Hz, Cys-p-C%), 3.23 (dd, 0.5H, J 14.2 Hz, J 5.1 Hz, Cys-p-C%), 3.19-3.09 (m, 1.5H, Cys-p-C¾ Succinimide-CH2), 2.98 (dd, 0.5H, J 14.2 Hz, J 9.2 Hz, Cys-p-C%), 2.55 (dd, 0.5H, J 5.2 Hz, J4.6 Hz, Succinimide- CH2), 2.51 (dd, 0.5H, J 5.2 Hz, J 4.6 Hz, Succinimide-CH2), 2.49-2.33 (m, 4H, CH2), 1.96- 1.86 (m, 2H, CH2), 1.53 (s, 4.5H, NC(C%)3), 1.52 (s, 4.5H, NC(C%)3); 19F NMR
(decoupled) (376.6 MHz; D20): δ/ppm -75.7 (OCOCF5); 13C NMR (128.5 MHz; D20): δ/ppm 180.2 (Succinimide-CO), 180.1 (Succinimide-CO), 179.0 (Succinimide-CO), 179.0 (Succinimide-CO), 177.7 (CH2CH2COOH), 176.1 (CH2CH2CONH), 176.0 (CH2CH2CONH), 172.9 (Gly-COOH), 172.4 (Cys-CO), 172.4 (Cys-CO), 162.9 (q, J 35.5 Hz, OCOCF3), 1 16.3 (q, J292.0 Hz, OCOCF3), 59.1 (NC(C¾)3), 59.1 (NC(C¾)3), 53.1 (Cys-a-CH), 52.5 (Cys- a-CH), 41.1 (Gly-CH2), 41.1 (Gly-CH2), 40.8 (Succinimide-CH), 39.9 (Succinimide-CH), 36.1 (Succinimide-CH2), 35.9 (Succinimide- H2), 34.4 (CH2), 32.8 (Cys-p-CH2), 32.7 (CH2), 32.5 (Cys-p-CH2), 27.3 (NC(CH5)3), 20.4 (CH2); HRMS m/z (ESI+) (Found: [M+Na]+ 468.13940. Ci8H27N3Na08S requires M+ 468.141 1 1); m/z (ES+) 468 ([M+Na]+, 100%);
Analytical HPLC @ 220 nm (Acclaim® 120 C 18 RP LC Column; 95 :5 :0.1 5 :95 :0.1 ; H20 : MeCN : TFA) Ret. Time = 9.1 16 min, Purity: 97.61%.
References: 1. Mechanism of ligand-gated potassium efflux in bacterial pathogens. PNAS 107, 19784-19789 (2010); 2. Healy, J. et al. Understanding the structural requirements for activators of the Kef bacterial potassium efflux system. Biochemistry 53, 1982-1992 (2014); 3. Bernardes, G. J. L. et al. From Disulfide- to Thioether-Linked Glycoproteins. Angewandte Chemie 120, 2276-2279 (2008); 4. Aroyan, C. E., Dermenci, A. & Miller, S. J. Development of a cysteine-catalyzed enantioselective Rauhut-Currier reaction. J. Org. Chem. 75, 5784- 5796 (2010); 5. Healy, J. Studies on the bacterial potassium efflux system KefC, and its ancillary protein KefF.
Example - competition fluorescence assay
Competition fluorescence assays were performed on a number compounds to determine binding to Kef relative to GS-X compounds. All measurements were performed by a Perkin Elmer LS-50B Spectrometer. Samples were run with the expressed C-terminal domain of Schewanella denitrificans Kef (6 μΜ) and DNGSH (5 μΜ) in 50 mM phosphate buffer pH 7.4 and 150 mM NaCl. The competing ligand was added to each sample at a final concentration of 1 mM. The samples were excited at 340 nm and the emission spectra measured from 375-670 nm. The data are represented as ratios of the fluorescence intensities before and after the addition of the tested compound at 525.5 nm (FB/FL) - 1.
The compounds tested are as follows.

10
Fluorescence competition assay results are shown in Figures 1 to 3. Significance of changes evaluated by a Student's t-test are shown in the Figures (where ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05). It can be observed that the compounds of the invention competitively bind to Kef.
Example - CPMG NMR and KD
KD values were determined for certain compounds using ¾ CPMG NMR.
All the NMR experiments were performed on a Bruker Avance III 700 MHz spectrometer equipped with a TCI inverse cryoprobe at 298 K and processed and analysed by Bruker Topspin™ 3.2. The PRO JECT-CPMG pulse sequence (90ox-[x-180o y-T-90o y-T-180o y- x]n-acq) as described by Aguilar, et al (Spin echo NMR spectra without J modulation. Chem.
Commun. 2012, 48, 811-813) was used to remove the broad resonances of the protein. All spectra were processed with a Lorentzian line broadening of 0.3 Hz. The CPMG edited ¾ NMR experiments were recorded with a total filter time of 88 ms. Water suppression was achieved by presaturation. Bruker, MATCH (3 mm diameter) NMR tubes were used with a total sample volume of 160 μΐ.. The solutions were buffered in 50 mM Na-phosphate, 150 mM NaCl, pH 7.4. For each sample the pulse tip-angle calibration was carried out using the single pulse nutation method (Wu, P. S. C; Otting, G. Rapid pulse length determination in high-resolution NMR. J. Magn. Reson. 2005, 176, 115-119).
The titration, experiments were performed with a fixed concentration of the ligand (typically 10 μΜ) and a range of protein concentrations (typically 0-30 μΜ; the concentration was increased until complete saturation of the ligand signal was achieved). The % decrease in signal intensity of the ligand was calculated as follows:
Ip - IB
% Decrease in signal intensity = — x 100
IB
Where: IB is the intensity of the ligand signal in the absence of protein and Ip is the intensity of the ligand signal in the presence of the protein at a given concentration.
The titration data (% decrease in ligand signal intensity plotted against protein concentration) was fitted using OriginPro 9.0 (Origin lab, Northampton, MA, USA) and the value of the KD
was extracted using the equation described by Dalvit (Dalvit, C. Theoretical analysis of the competition ligand-based MR experiments and selected applications to fragment screening and binding constant measurements. Concepts in Magnetic Resonance Part A. 2008 32A(5), 341-372): y = A*(l/(2*C))*((B+x+C)-sqrt(((B+x+C)A2)-(4*x*C)))
Where: y is the % decrease in ligand signal intensity; A is the maximum loss in the signal intensity; C is the ligand concentration; B is the KD, X is the concentration of the protein.
The results are shown in Figures 4 to 10.
Example - antibacterial activity of compounds
Antimicrobial susceptibility tests were carried out using a Kirby-Bauer disk diffusion assay. Escherichia coli strains (Table 1) or transformants were inoculated onto 2x TY agar plate [16 g/L Tryptone (Oxoid Ltd.), 10 g/L Yeast extract (Oxoid Ltd.), 5 g/L NaCl, 1.5% (w/v) Oxoid™ Bacto Agar (Thermo Fisher Scientific Inc.)] and incubated for 18-24 h at 37 °C for selection of well-isolated colonies. The full-length Shewanella denitrificans Kef channel (GenBank ID: ABE53663.1) encoded by the pTrcSdKefHe plasmid (J. Healy et al,
Understanding the structural requirements for activators of the Kef bacterial potassium efflux system. Biochemistry. 53, 1982-1992 (2014)) was transformed into the E. coR MJF335 host (Table 1), in which its endogenous E. coli Kef systems were knocked out (S. Miller, R. M. Douglas, P. Carter, I. R. Booth, Mutations in the glutathi one-gated KefC K+ efflux system of Escherichia coli that cause constitutive activation (1997)), as a model strain for testing in this study. For each isolate, three to five morphologically similar colonies were selected from the agar plate from the above step and transferred into a sterile Falcon™ 50-mL conical centrifuge tube (Thermo Fisher Scientific Inc.) containing 10 mL of sterile Mueller-Hinton broth (MUB; Sigma-Aldrich Co.) by using inoculation loop. The bacterial culture was allowed to grow aerobically to 0.5 McFarland turbidity standard (Clinical and Laboratory Standards Institute, M02-A11 : Performance standards for antimicrobial disk susceptibility tests; approved standard— eleventh edition. 32, 1-76 (2012); Clinical and Laboratory Standards Institute, M100-S24: Performance standards for antimicrobial susceptibility testing; twenty- fourth informational supplement (ed. 24, 2014)) at 37 °C with an agitation speed of 200 r.p.m. During this incubation period, test discs were prepared by impregnating 20 iL of different concentrations of test compounds onto Whatman® antibiotic assay discs of
6 mm diameters (Sigma- Aldrich Co.). After the cell culture had been reached the cell density of 0.5 McFarland turbidity standard, the cell isolate was inoculated onto Muller-Hinton agar (MHA; Sigma-Aldrich Co.) plates by using sterile cotton swab. Subsequently the compound discs were aseptically placed onto the bacterial plates. After 24 h of incubation at 37 °C, inhibition zone diameters associated with the inhibition effect of the compounds were recorded by averaging both vertical and horizontal diameters of each zone. The whole experimental procedures were performed according to the standards set by the Clinical and Laboratory Standards Institute.
Table 1. E. coli strains used in this study.
E. coli strain Genotype
Fragl F", lacZ82(Am), λ', rha-4, thiEI, gal-33
Frag5 Fragl, A(kdpC-kdpA)18
Frag56 Frag5, gshA::Tnl0(Kan)
MJF335 Frag5, lad, kejB, kefC::Tnl0, gs/? vTn70(Kan)
See S. Miller, R. M. Douglas, P. Carter, I. R. Booth, Mutations in the glutathione-gated KefC K+ efflux system of Escherichia coli that cause constitutive activation (1997) and W. Epstein, B. S. Kim, Potassium transport loci in Escherichia coli K-12. J. Bacterid. 108, 639-644 (1971).
The assay was performed using compound 4 which was used at a number of concentrations.

4
The bacteria used was E. coli MJF335 (pTrcSdKefH6) which has the genotype Frag5, lacl, kefB, kefC::Tnl0, gshA::Tnl0(Kan). The disk diameter was 6 mm. The results of the assay are shown in Figure 11. A dose dependent increase in bacterial inhibition is clearly visible.
Claims
1. A compound which is a eptide derivative of formula (1) or a salt thereof:

wherein:
RG is H, -CH2-RGA or C1-12 hydrocarbyl optionally substituted with 1 to 4 substituents selected from -OR', -N02, -CN, halogen and 5-6 membered heterocyclyl; or RG and RN1 together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen;
RGA is Ci-ii hydrocarbyl optionally substituted with 1 to 4 substituents selected from - R'R", -OR', -NO2, -CN, halogen and 5-6 membered heterocyclyl;
Rs is Ci-16 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group,
-(Ci-4 alkylene)-RSA or -(C1-4 alkylene)-L-RSA; RSA is Ci-ie hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group; L is -SO2-; and wherein Rs and RSA are optionally substituted with a group Rb and/or from 1 to 5 substituents selected from Ra, -NR'R", -OR', -NO2, -CN, -C(0)R', -C(0)OR', -C(0)NR'R", -0-C(0)R', -N(R')-C(0)R", -SR', -S(0)R', -S(0)2R', -S(0)2OR', -P(0)(OR')(OR"), =0, -SiR'R"R" and halogen; or Rs and an RC1 together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen;
Rb is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl;
Q is -C(0)-N(RN2)-, -C(0)-C(R'R")-, -C(0)-0- -N(RN2)-C(0)-, -C(R'R")- C(O)-, -O-C(O)-, -C(R'R")-C(R"'R"")-, -C(R'R")-0- -0-C(R'R")-, -C(R'R")-N(RN2)-, -N(RN2)-C(R'R")- or -C(R')=C(R")-;
Z is a group selected from -C(0)-0-R° and -CN4R' (tetrazolyl);
R° is H or Ci-12 hydrocarbyl optionally substituted with 1 to 4 substituents selected from -NR'R", -OR', -NO2, -CN, -C(0)R', -C(0)OR', -C(0)NR'R", -0-C(0)R', -N(R')- C(0)R", -SR', -S(0)R', -S(0)2R', -S(0)2OR', -P(0)(OR')(OR"), =0, halogen and 5-6
membered heterocyclyl; or R and an R together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen; each RN1 and RN2 is independently H or Ci-4 alkyl;
each RC1, RC2, R', R", R" and R"" is independently H or C alkyl and is optionally substituted with 1 to 3 substituents selected from halogen, -OH and -O-Ci-4 alkyl; and
each Ra is independently Ci-4 alkyl which is optionally substituted with 1 to 3 halogen atoms.
2. A compound according to claim 1, wherein Q is -C(0)-N(R )-.
3. A compound according to claim 1 or claim 2, wherein
Z is -C(0)-0-R° and R° is H, Ci-4 alkyl, phenyl or benzyl, wherein the phenyl and benzyl groups are optionally substituted with 1 to 4 substituents selected from Ra, -OR' and halogen, or
Z is -CN4H (tetrazolyl).
4. A compound according to any one of the preceding claims, which compound is a peptide derivative of formula 2) or a salt thereof:

wherein:
RG is H, -CH2-RGA or a Ci-8 alkyl group which is optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl; or RG and RN1 together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen;
RGA is Ci-7 alkyl group which is optionally substituted with 1 to 4 substituents selected from - R'R", -OR', halogen and 5-6 membered heterocyclyl;
Rs is Ci-16 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group,
-(Ci-4 alkylene)-RSA or -(C1-4 alkylene)-L-RSA; RSA is Ci-ie hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group; L is -SO2-; and wherein Rs and RSA are optionally substituted with a group Rb and/or from 1 to 5 substituents selected from Ra, - R'R", -OR', -NO2, -CN, -C(0)R', -C(0)OR', -C(0) R'R", -0-C(0)R', -N(R')-C(0)R", -SR',
=0 and halogen; or Rs and an RC1 together form a 5-6 membered heterocyclyl ring which is optionally substituted with 1 to 3 substituents selected from Ra, -OR' and halogen;
Rb is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl;
R° is H, Ci-4 alkyl, phenyl or benzyl, wherein the phenyl and benzyl groups are optionally substituted with 1 to 4 substituents selected from Ra, -OR' and halogen;
RN1 and RN2 are each independently H or C1-4 alkyl;
RC1 and RC2 are each independently H or C1-4 alkyl;
each R' and R" is independently H or C1-4 alkyl which is optionally substituted with 1 to 3 substituents selected from halogen, -OH and -O-C1-4 alkyl; and
each Ra is independently C1-4 alkyl which is optionally substituted with 1 to 3 halogen atoms.
5. A compound according to any one of the preceding claims, wherein each RN1, RN2, RC1 and RC2 is independently H or methyl.
6. A compound according to any one of the preceding claims, which compound is a peptide derivative of formula (3) or a salt thereof:
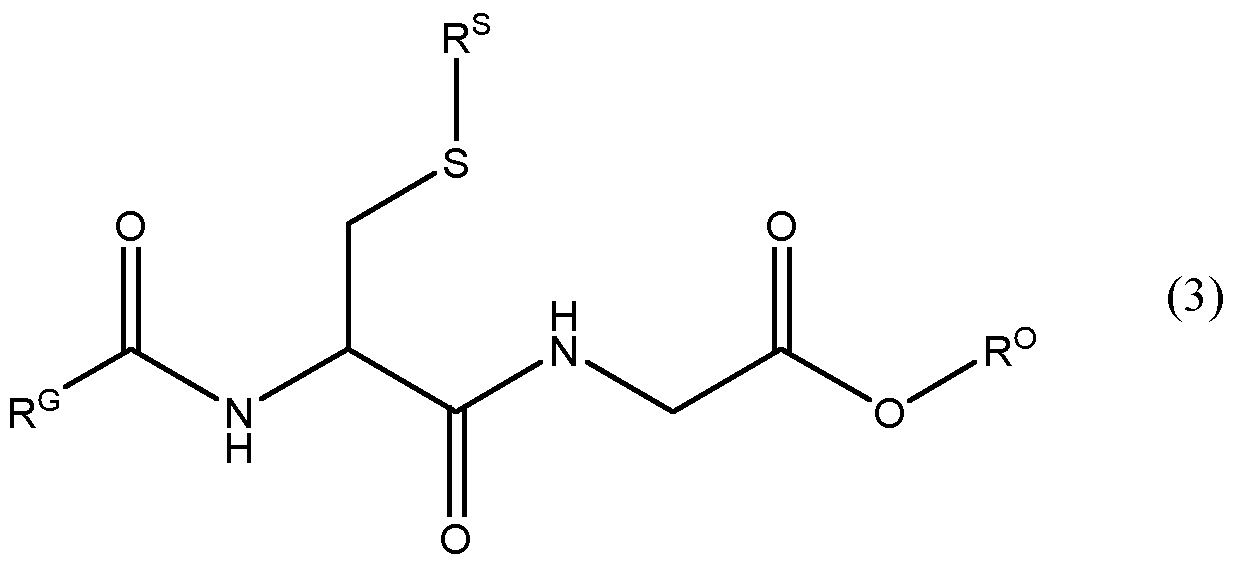
wherein:
RG is H, -CH2-RGA or a Ci-8 alkyl group which is optionally substituted with 1 to 4 substituents selected from -OR', halogen and 5-6 membered heterocyclyl;
RGA is Ci-7 alkyl group which is optionally substituted with 1 to 4 substituents selected from -NR'R", -OR', halogen and 5-6 membered heterocyclyl;
Rs is Ci-16 hydrocarbyl, a 5-10 membered heterocyclyl or heteroaryl group,
-(Ci-4 alkylene)-RSA or -(C1-4 alkylene)-L-RSA; RSA is Ci-ie hydrocarbyl, or a 5-10 membered heterocyclyl or heteroaryl group; L is -SO2-; and wherein Rs and RSA are optionally substituted with a group Rb and/or from 1 to 5 substituents selected from C1-4 alkyl, -CF3, - R'R", -OR', -NO2, -CN, -C(0)R', -C(0)OR', -C(0) R'R", =0 and halogen;
Rb is phenyl, benzyl, C3-6 cycloalkyl or 5-6 membered heterocyclyl;
R° is H or Ci-4 alkyl optionally substituted with -O-C1-4 alkyl; and
each R' and R" is independently H or C1-4 alkyl.
7. A compound according to any one of the preceding claims, wherein RG is Ci-8 alkyl optionally substituted with 1 to 4 substituents selected from halogen, -OH and -O-C1-4 alkyl, preferably wherein RG is methyl or ethyl.
8. A compound according to any one of the preceding claims, wherein R° is H or C1-4 alkyl optionally substituted with -O-C1-4 alkyl, preferably wherein R° is H, methyl or ethyl.
9. A compound according to any one of the preceding claims, wherein RS is Ci-10 alkyl, -(Ci-3 alkylene)-RSA, -(C1-3 alkylene)-L-RSA, C2-10 alkenyl, C2-10 alkynyl, C3-6 cycloalkyl, 5- to 10 membered heterocyclyl, C6-io aryl or 5- to 10-membered heteroaryl, wherein L is SO2, RSA is C3-6 cycloalkyl, 5- to 10 membered heterocyclyl, C6-io aryl or 5- to 10-membered heteroaryl and Rs and RSA are optionally substituted with a group Rb and/or from 1 to 5 substituents selected from CM alkyl, -CF3, -NR'R", -OR', -NO2, -CN, -C(0)R', -C(0)OR', -C(0)NR'R", =0 and halogen; wherein each R' and R" is independently H or C1-4 alkyl; and Rb is phenyl, benzyl or C3-6 cycloalkyl.
10. A compound according to any one of the preceding claims, wherein RS is Ci-10 alkyl, -(Ci-3 alkylene)-RSA, -(C1-3 alkylene)-L-RSA, C3-6 cycloalkyl, 5-6 membered heterocyclyl or phenyl, wherein L is SO2, RSA is C3-6 cycloalkyl, 5-6 membered heterocyclyl or phenyl and Rs and RSA are optionally substituted with a group Rb and/or from 1 to 3 substituents selected from Ci-4 alkyl, -CF3, -OCi-4alkyl, -NO2, -CN, =0 and halogen; wherein Rb is phenyl, benzyl or C3-6 cycloalkyl.
11. A compound according to any one of the preceding claims, wherein each R', R", R" and R"" is independently H or C1-4 alkyl, preferably H.
12. A compound according to any one of the preceding claims, wherein Rs is
(i) Ci-io alkyl, -(C1-2 alkylene)-(C3-6 cycloalkyl) or C3-6 cycloalkyl, wherein the cycloalkyl ring is optionally substituted with from 1 to 3 methyl groups;
(ii) 5-6 membered heterocyclyl or -CH2CH2-(5-6 membered heterocyclyl) wherein the heterocyclyl group in each case is optionally substituted with phenyl, benzyl or C3-6 cycloalkyl and/or with from 1 to 3 substituents selected from C1-4 alkyl, -NO2, halogen, Ci-4 alkoxy, and =0; or
(iii) -CH2CH2-Ph-(Y)„ or -CH2CH2-S02-Ph-(Y)„, wherein n is 0 or 1 and Y is -

13. A compound according to any one of the preceding claims, wherein Rs is a group of formula:

where R is H, phenyl, benzyl, C3-6 cycloalkyl or C1-4 alkyl.
14. A compound according to any one of the preceding claims, wherein Rs is a group

15. A compound according to any one of claims 1 to 4, which compound is a peptide derivative of formula (4) or a salt thereof:

wherein RG, RN1, Rs, RC1, RN2, RC2 and R° are as defined in any one of the preceding claims.
16. A compound according to any one of the preceding claims, wherein RG is methyl and R° is H or methyl.
17. A com ound according to claim 1, which compound is

or a salt thereof, and where Rs is a group selected from

18. A compound according to any one of the preceding claims, which compound is

19. A pharmaceutical composition comprising (a) a compound which is a peptide derivative or a salt thereof as defined in any one of claims 1 to 18, wherein the salt is a pharmaceutically acceptable salt, and (b) one or more pharmaceutically acceptable excipients or diluents.
20. A compound which is a peptide derivative or a salt thereof as defined in any one of claims 1 to 18, for use in a method of treatment or prevention of bacterial infection, wherein the salt is a pharmaceutically acceptable salt.
21. A compound for use according to claim 20, wherein the bacterial infection is caused by bacteria of genus Shewanella, Acinetobacter, Bacillus, Bartonella, Bordetella, Borrelia, Brucella, Campylobacter, Chlamydia, Chlamydophila, Clostridium, Corynebacterium, Enterobacter, Enterococcus, Escherichia, Francisella, Haemophilus, Helicobacter,
Klebsiella, Legionella, Leptospira, Listeria, Mycobacterium, Mycoplasma, Neisseria, Pseudomonas, Rickettsia, Salmonella, Shigella, Staphylococcus, Streptococcus, Treponema, Vibrio or Yersinia. For example, the bacterial infection may be caused by bacteria of genus Shewanella or Escherichia.
22. A compound for use according to claim 20 or 21, wherein the bacterial infection is caused by Staphylococcus aureus, Escherichia coli, Shewanella denitrificans, Hemophilus
influenzae, Klebsiella pneumoniae, Legionella pneumophila, Pseudomonas aeruginosa, Proteus mirabilis, Enterobacter cloacae, Serratia marcescens, Helicobacter pylori,
Salmonella enteritidis, Salmonella typhi or Acinetobacter baumannii.
23. A method of treating or preventing bacterial infection in a subject, wherein the infection is as defined in any one of claims 20 to 22, which method comprises administering an effective amount of a peptide derivative or a salt thereof as defined in any one of claims 1 to 18 to the subject, wherein the salt is a pharmaceutically acceptable salt.
24. Use of a peptide derivative or a salt thereof as defined in any one of claims 1 to 18 in the manufacture of a medicament for use in a method of treatment or prevention of bacterial infection, wherein the infection is as defined in any one of claims 20 to 22, and wherein the salt is a pharmaceutically acceptable salt.
25. A combination comprising (a) a compound which is a peptide derivative or a salt thereof as defined in any one of claims 1 to 18, wherein the salt is a pharmaceutically acceptable salt, and (b) a further active agent which is an antibiotic compound.
26. A compound for use according to any one of claims 20 to 22, wherein the method comprises administering the compound in combination with a further active agent which is an antibiotic compound.
27. A method of treating or preventing infection according to claim 23, wherein the method comprises administering the peptide derivative or salt thereof in combination with a further active agent which is an antibiotic compound.
28. A kit comprising (a) a compound which is a peptide derivative or a salt thereof as defined in any one of claims 1 to 18, wherein the salt is a pharmaceutically acceptable salt, and (b) a further active agent which is an antibiotic compound.
29. Use according to claim 24, wherein the method comprises administering the compound in combination with a further active agent which is an antibiotic compound.
30. A compound which is a peptide derivative or a salt thereof as defined in any one of claims 1 to 18 for use in a method of activating potassium efflux in bacteria, wherein the bacteria are as defined in any one of claims 20 to 22.
31. A method for preventing or treating bacterial infection of a plant, which method comprises treating the plant, the seed from which the plant grows, or the place at which the plant grows, with a composition comprising a compound as defined in any one of claims 1 to
18.
32. A method according to claim 31, wherein the infection is caused by bacteria of the genus Shewanella, Erwinia, Agrobacterium, Phytoplasma, Burkholderia, Proteobacteria, Xanthomonas or Pseudomonas.
33. An in vitro method for killing or inhibiting the growth of bacteria, which method comprises applying a composition comprising a compound which is a peptide derivative or salt thereof as defined in any one of claims 1 to 18 to a surface, to an article or to the skin.
33. A soap product, washing product or cleaning product comprising a compound which is a peptide derivative or salt thereof as defined in any one of claims 1 to 18.
34. A process for producing a compound which is a peptide derivative of formula (la) or a salt thereof:

which process comprises reacting a thiol compound of formula (A) with an alkene of formula (B) in the presence of an initiator,

wherein RG RN1, RC1, Q, RC2 and Z are as defined in any one of claims 1 to 18 and the group
 is a group Rs as defined in any one of claims 1 to 18.
is a group Rs as defined in any one of claims 1 to 18.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1508497.3A GB201508497D0 (en) | 2015-05-18 | 2015-05-18 | Antibacterial agent |
| GB1508497.3 | 2015-05-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016185200A1 true WO2016185200A1 (en) | 2016-11-24 |
Family
ID=53505944
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2016/051420 WO2016185200A1 (en) | 2015-05-18 | 2016-05-17 | Antibacterial agent |
Country Status (2)
| Country | Link |
|---|---|
| GB (1) | GB201508497D0 (en) |
| WO (1) | WO2016185200A1 (en) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014113634A1 (en) * | 2013-01-17 | 2014-07-24 | University Of Kansas | Toll-like receptor 2-agonistic lipopeptides, and method of making the same |
-
2015
- 2015-05-18 GB GBGB1508497.3A patent/GB201508497D0/en not_active Ceased
-
2016
- 2016-05-17 WO PCT/GB2016/051420 patent/WO2016185200A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014113634A1 (en) * | 2013-01-17 | 2014-07-24 | University Of Kansas | Toll-like receptor 2-agonistic lipopeptides, and method of making the same |
Non-Patent Citations (1)
| Title |
|---|
| JESSICA HEALY ET AL: "Understanding the Structural Requirements for Activators of the Kef Bacterial Potassium Efflux System", BIOCHEMISTRY, vol. 53, no. 12, 1 April 2014 (2014-04-01), US, pages 1982 - 1992, XP055286326, ISSN: 0006-2960, DOI: 10.1021/bi5001118 * |
Also Published As
| Publication number | Publication date |
|---|---|
| GB201508497D0 (en) | 2015-07-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2603198T3 (en) | 1,6-diazabicyclo [3,2,1] octan-7-one derivatives and their use in the treatment of bacterial infections | |
| EP3233889B1 (en) | Antimicrobial polymyxins for treatment of bacterial infections | |
| AU2010212183B2 (en) | Actagardine derivatives | |
| WO2012168820A1 (en) | Polymyxin derivatives useful as antibacterial agents | |
| JP7280273B2 (en) | Monocyclic β-Lactam Compounds for Treating Bacterial Infections | |
| JP2017510570A (en) | Polymyxin derivatives and their use in combination therapy with different antibiotics | |
| KR20150107793A (en) | Compositions, methods of making and methods of use | |
| EP2739289B1 (en) | Glycopeptide antibiotic analogs effective against vancomycin-resistant bacterial strains | |
| JP2012501300A (en) | Organic compounds | |
| JP6643484B2 (en) | 7-oxo-6- (sulfoxy) -1,6-diazabicyclo [3.2.1] octane-2-carboxamide containing compounds and their use in the treatment of bacterial infections | |
| EP3111948A1 (en) | New bicyclic lipopeptide, preparation and use as antimicrobial agent | |
| KR100652470B1 (en) | Cycloalkyl-substituted aminomethylpyrrolidine derivatives | |
| JP2021100939A (en) | Novel peptide derivatives and uses thereof | |
| WO2016185200A1 (en) | Antibacterial agent | |
| JP2011511811A (en) | Antibacterial fluoroquinolone analog | |
| CN110938114A (en) | Vancomycin sulfonium derivatives, preparation method, pharmaceutical composition and application thereof | |
| JP2001500853A (en) | Pharmaceutical compounds | |
| DE102004025731A1 (en) | Antibacterial amide macrocycles III | |
| TW201741308A (en) | 6-(buta-1,3-diyn-1-yl)benzo[D]thiazole derivatives | |
| US9981986B2 (en) | Antimicrobial compounds | |
| GB2289274A (en) | Angiogenesis inhibiting sialic acid derivatives and process for their preparation | |
| RU2181361C2 (en) | Novel polycyclic derivatives of phthalazine and their use | |
| WO2017042099A1 (en) | Efflux-pump inhibitors and therapeutic uses thereof | |
| RU2785715C1 (en) | USE OF MONOCYCLIC β-LACTAM COMPOUND IN PHARMACY | |
| US20230339973A1 (en) | Ring-fused 2-pyridone compounds, methods for preparation thereof and their use in the treatment and/or prevention of a disease involving gram-positive bacteria |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16725562 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16725562 Country of ref document: EP Kind code of ref document: A1 |