WO2016164754A1 - Fgfr4 inhibitors - Google Patents
Fgfr4 inhibitors Download PDFInfo
- Publication number
- WO2016164754A1 WO2016164754A1 PCT/US2016/026690 US2016026690W WO2016164754A1 WO 2016164754 A1 WO2016164754 A1 WO 2016164754A1 US 2016026690 W US2016026690 W US 2016026690W WO 2016164754 A1 WO2016164754 A1 WO 2016164754A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mmol
- methyl
- nitro
- pyrazol
- stirred
- Prior art date
Links
- 0 COc1cc(OC)c(*)c(NC(N(*)c2cc(Nc3c(NC(C=C*)=O)[n](*)nc3)ncn2)=O)c1* Chemical compound COc1cc(OC)c(*)c(NC(N(*)c2cc(Nc3c(NC(C=C*)=O)[n](*)nc3)ncn2)=O)c1* 0.000 description 5
- PAPUTLFQKXYDAC-UHFFFAOYSA-N CC(C)(C)OC(N(C(N(C)c1cc(Cl)ncn1)=O)c(c(C)c(cc1OC)OC)c1F)=O Chemical compound CC(C)(C)OC(N(C(N(C)c1cc(Cl)ncn1)=O)c(c(C)c(cc1OC)OC)c1F)=O PAPUTLFQKXYDAC-UHFFFAOYSA-N 0.000 description 1
- ISWMUFWPHKULTA-UHFFFAOYSA-N CC(C)(C)OC(N(C(N(Cc1ccccc1)c1cc(Cl)ncn1)=O)c(c(Cl)c(cc1OC)OC)c1Cl)=O Chemical compound CC(C)(C)OC(N(C(N(Cc1ccccc1)c1cc(Cl)ncn1)=O)c(c(Cl)c(cc1OC)OC)c1Cl)=O ISWMUFWPHKULTA-UHFFFAOYSA-N 0.000 description 1
- XDZYZZXNNWHBBL-UHFFFAOYSA-N CC(C)(C)OC(Nc1n[n](CCN(C)C)cc1[N+]([O-])=O)=O Chemical compound CC(C)(C)OC(Nc1n[n](CCN(C)C)cc1[N+]([O-])=O)=O XDZYZZXNNWHBBL-UHFFFAOYSA-N 0.000 description 1
- ICRWAXJHUWVZFN-ZETCQYMHSA-N CC1(C)O[C@H](C[n](cc2N)nc2[N+](OC)=O)CN1 Chemical compound CC1(C)O[C@H](C[n](cc2N)nc2[N+](OC)=O)CN1 ICRWAXJHUWVZFN-ZETCQYMHSA-N 0.000 description 1
- YIDDGBNJBPDAKQ-UHFFFAOYSA-N CCN(CC1)CCC1[n](cc1N(C(C=C)=O)C2=O)nc1N2c1cc(N(C)C(Nc(c(Cl)c(cc2OC)OC)c2Cl)=O)ncn1 Chemical compound CCN(CC1)CCC1[n](cc1N(C(C=C)=O)C2=O)nc1N2c1cc(N(C)C(Nc(c(Cl)c(cc2OC)OC)c2Cl)=O)ncn1 YIDDGBNJBPDAKQ-UHFFFAOYSA-N 0.000 description 1
- HSSHOHYWEFZQKI-UHFFFAOYSA-N CN(C(Nc(c(Cl)c(cc1OC)OC)c1Cl)=O)c1cc(Nc2n[n](CCO)cc2NC(C=C)=O)ncn1 Chemical compound CN(C(Nc(c(Cl)c(cc1OC)OC)c1Cl)=O)c1cc(Nc2n[n](CCO)cc2NC(C=C)=O)ncn1 HSSHOHYWEFZQKI-UHFFFAOYSA-N 0.000 description 1
- DNMHSAGVWLYNNK-UHFFFAOYSA-N CN(C(Nc(c(Cl)c(cc1OC)OC)c1F)=O)c1cc(Cl)ncn1 Chemical compound CN(C(Nc(c(Cl)c(cc1OC)OC)c1F)=O)c1cc(Cl)ncn1 DNMHSAGVWLYNNK-UHFFFAOYSA-N 0.000 description 1
- BRXIXTDMKVPKGX-UHFFFAOYSA-N CN(C(Nc(c(F)c(cc1OC)OC)c1F)=O)c1cc(Nc2n[n](C)cc2NC(C=C)=O)ncn1 Chemical compound CN(C(Nc(c(F)c(cc1OC)OC)c1F)=O)c1cc(Nc2n[n](C)cc2NC(C=C)=O)ncn1 BRXIXTDMKVPKGX-UHFFFAOYSA-N 0.000 description 1
- OATIUDCZTISRHG-UHFFFAOYSA-N CN(C)CC[n](cc1NC(C=C)=O)nc1Nc1ncnc(N(C)C(Nc(c(Cl)c(cc2OC)OC)c2Cl)=O)c1 Chemical compound CN(C)CC[n](cc1NC(C=C)=O)nc1Nc1ncnc(N(C)C(Nc(c(Cl)c(cc2OC)OC)c2Cl)=O)c1 OATIUDCZTISRHG-UHFFFAOYSA-N 0.000 description 1
- HZSOSCBVILJUDO-UHFFFAOYSA-N COC(c1n[n](CC2CC2)cc1[N+]([O-])=O)=O Chemical compound COC(c1n[n](CC2CC2)cc1[N+]([O-])=O)=O HZSOSCBVILJUDO-UHFFFAOYSA-N 0.000 description 1
- QGRSCDXIVJLNJA-UHFFFAOYSA-N COc(c(Cl)c1NC(N(Cc2ccccc2)c2cc(Cl)ncn2)=O)cc(OC)c1Cl Chemical compound COc(c(Cl)c1NC(N(Cc2ccccc2)c2cc(Cl)ncn2)=O)cc(OC)c1Cl QGRSCDXIVJLNJA-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- Fibroblast growth factors are a family of more than 20 structurally related proteins with a variety of biological activities. Their main receptors, the fibroblast growth factor receptors (FGFR1, FGFR2, FGFR3 and FGFR4), are a family of receptor tyrosine kinases that bind FGF and are involved in processes of cell proliferation and differentiation. Deregulation of FGFR signaling networks is implicated in a number of pathophysiological conditions, including many types of human cancers.
- FGFR4 Fibroblast Growth Factor Receptor 4" or "FGFR4" is known to regulate proliferation and antiapoptosis and is expressed or highly expressed in many cancers. See, e.g., Dieci et al. 2013, Cancer Discovery, 0F1-0F16. Studies have shown that expression of FGFR4 is predictive of a more aggressive phenotype of the cancer, and knockdown or reduction of FGFR4 expression serves to reduce proliferation and promote apoptosis. See, e.g. , Wesche et al. 201 1, Biochem J 437: 199-213.
- FGFR4 expression or overexpression is associated with cancer
- sarcoma such as rhabdomyosarcoma (Taylor VI et al. 2009, J Clin Invest, 119(1 1):3395-3407), skin cancer such as melanoma (Streit et al. 2006, British J Cancer, 94:1879-1886), liver cancer such as cholangiocarcinoma (Sia et al. 2013, Gastroenterology 144:829-840) and hepatocellular carcinoma (French et al. 2012, PLoS ONE 7(5): e367313;
- pancreatic cancer such as pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma (Motoda et al. 201 1, Int'l J Oncol 38:133-143)
- lung cancer such as non-small-cell lung cancer (Fawdar et al. 2013, PNAS 110(30): 12426- 12431), colorectal cancer (Pelaez-Garcia et al. 2013, PLoS ONE 8(5): e63695; Barderas et al.
- a purpose of the present invention is to provide a compound of Formula I:
- X is H or CI
- R 1 is selected from the group consisting of:
- C 1-6 alkyl which C 1-6 alkyl may be unsubstituted or substituted 1-3 times with an independently selected substituent from the group consisting of: hydroxy, methoxy, C3- 6 cycloalkyl, and -NR 3 R 4 , wherein R 3 and R 4 are each independently hydrogen or methyl;
- heterocyclyl which heterocyclyl may be unsubstituted or substituted with C 1-6 alkyl;
- R 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl;
- Y and Z are each independently fluoro or chloro
- a further purpose of the present invention is to provide a compound of Formula 1(a):
- a further purpose is to provide a compound of Formula II:
- X is H or CI
- R 3 is hydrogen or C 1-6 alkyl
- R 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl;
- Y and Z are each independently fluoro or chloro
- a further purpose is to provide a compound of Formula 11(a):
- X is H or CI
- R 3 is hydrogen or C 1-6 alkyl
- R 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl;
- Y and Z are each independently fluoro or chloro
- a further purpose is to provide a compound of Formula III:
- X is H or CI
- R 3 is hydrogen or Ci -6 alkyl
- R 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl;
- Y and Z are each independently fluoro or chloro
- a further purpose is to provide a compound of Formula 111(a):
- X is H or CI
- R 3 is hydrogen or Ci -6 alkyl
- R 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl;
- Y and Z are each independently fluoro or chloro
- a further purpose is to provide a compound of Formula IV:
- X is H or CI
- Ri is selected from the group consisting of
- C 1-6 alkyl which Ci -6 alkyl may be unsubstituted or substituted 1-3 times with an independently selected substituent from the group consisting of: hydroxy, methoxy, C 3 - 6 cycloalkyl, and -NR 3 R 4 , wherein R 3 and R 4 are each independently hydrogen or methyl; C3. 6 cycloalkyl; and
- heterocyclyl which heterocyclyl may be unsubstituted or substituted with C 1-6 alkyl;
- R 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl;
- Y and Z are each independently fluoro or chloro
- a further purpose is to provide a compound of Formula IV(a):
- X is H or CI
- Ri is selected from the group consisting of
- Ci -6 alkyl which Ci -6 alkyl may be unsubstituted or substituted 1-3 times with an independently selected substituent from the group consisting of: hydroxy, methoxy, C 3 - 6 cycloalkyl, and -NR3R4, wherein R 3 and R 4 are each independently hydrogen or methyl; C 3- 6cycloalkyl; and
- R 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl;
- Y and Z are each independently fluoro or chloro
- a further purpose is a pharmaceutical composition
- a pharmaceutical composition comprising a compound as described herein and a pharmaceutically acceptable earner.
- the composition is formulated for oral or parenteral administration.
- a further purpose is a method of treating hepatocellular carcinoma in a subject in need thereof comprising administering to said subject a treatment effective amount of a compound or salt or composition as described herein.
- hepatocellular carcinoma has altered FGFR4 and/or FGF19 status (e.g., increased expression of FGFR4 and/or FGF19).
- a further purpose is a method of treating hepatocellular carcinoma in a subject in need thereof, comprising: detecting an altered FGFR4 and/or FGF19 status (e.g., increased expression of FGFR4 and/or FGF19) in a biological sample containing cells of said hepatocellular carcinoma, and if said hepatocellular carcinoma has said altered FGFR4 and/or FGF19 status, administering a compound or composition described herein to said subject in a treatment- effective amount.
- an altered FGFR4 and/or FGF19 status e.g., increased expression of FGFR4 and/or FGF19
- a further purpose is the use of a compound or salt or a composition as described herein in a method of treatment of hepatocellular carcinoma.
- a further purpose is the use of a compound or salt described herein in the preparation of a medicament for the treatment of hepatocellular carcinoma.
- the compounds are selective FGFR4 inhibitors in that they have a greater binding affinity and/or inhibitory effect of FGFR4 as compared to that of FGFRl and/or FGFR2 and/or FGFR3 (e.g., by 10-fold, 100-fold, or 1000-fold greater or more).
- compounds of the invention may optionally be substituted with one or more substituents, such as those illustrated generally herein, or as exemplified by particular classes, subclasses, and species of the invention.
- substituted refers to the replacement of hydrogen in a given structure with a specified substituent.
- a substituted group may have a substituent at each substitutable position of the group, and when more than one position in any given structure may be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at every position.
- Combinations of substituents envisioned by this invention are preferably those that result in the formation of stable compounds.
- “Stable” as used herein refers to a chemically feasible compound that is not substantially altered when kept at a temperature of 40 °C or less, in the absence of moisture or other chemically reactive conditions, for at least one week.
- H is hydrogen
- C is carbon
- N is nitrogen
- S is sulfur
- O oxygen
- Alkyl or "alkyl group,” as used herein, means a straight-chain (i.e., unbranched), or branched chain that is completely saturated.
- the alkyl has 1, 2, 3, 4, 5 or 6 carbon atoms, i.e., methyl, ethyl, propyl, butyl, etc.
- alkyl groups contain 1-6 carbon atoms (C 1-6 alkyl).
- alkyl groups contain 1-4 carbon atoms (Ci -4 alkyl).
- alkyl groups contain 1-3 carbon atoms (Ci -3 alkyl).
- alkyl groups contain 2-3 carbon atoms (C 2-3 alkyl), and in yet other embodiments alkyl groups contain 1-2 carbon atoms (Ci -2 alkyl).
- alkenyl refers to a straight-chain (i.e., unbranched), or branched hydrocarbon chain that has one or more double bonds.
- the alkenyl has 2, 3, 4, 5 or 6 carbon atoms.
- alkenyl groups contain 2-8 carbon atoms (C 2-8 alkenyl).
- alkenyl groups contain 2-6 carbon atoms (C 2-6 alkenyl).
- alkenyl groups contain 3-4 carbon atoms (C 3-4 alkenyl), and in yet other embodiments alkenyl groups contain 2-3 carbon atoms (C 2-3 alkenyl).
- alkenyl refers to a straight chain hydrocarbon having two double bonds, also referred to as "diene.”
- Alkynyl or “alkynyl group” as used herein refers to a straight-chain (i.e., unbranched), branched, or cyclic hydrocarbon chain that has one or more triple bonds.
- the alkynyl has 2, 3, 4, 5 or 6 carbon atoms.
- alkynyl groups contain 2-8 carbon atoms (C 2- 8alkynyl).
- alkynyl groups contain 2-6 carbon atoms (C2 -6 alkynyl).
- alkynyl groups contain 3-4 carbon atoms (C 3-4 alkynyl), and in yet other embodiments alkynyl groups contain 2-3 carbon atoms (C 2-3 alkynyl).
- Ar or aryl refer to an aromatic carbocyclic moiety having one or more closed rings. Examples include, without limitation, phenyl, naphthyl, anthracenyl, phenanthracenyl, biphenyl, and pyrenyl.
- Benzyl refers to a phenyl group appended to the parent compound through a methyl group (-CH 2 C 6 H 5 ).
- “Pyridylmethyl” refers to a pyridine ring appended to the parent compound through a methyl group (-CH 2 C 5 H 4 N).
- Halo refers to chloro (CI), fluoro (F), bromo (Br) or iodo (I).
- Haloalkyl refers to one or more halo groups appended to the parent molecular moiety through an alkyl group. Examples include, but are not limited to, chloromethyl, fluoromethyl, trifluoromethyl, etc.
- Heteroaryl refers to a cyclic moiety having one or more closed rings, with one or more heteroatoms (oxygen, nitrogen or sulfur) in at least one of the rings, wherein at least one of the rings is aromatic, and wherein the ring or rings may independently be fused, and/or bridged.
- Examples include without limitation quinolinyl, isoquinolinyl, indolyl, furyl, thienyl, pyrazolyl, quinoxalinyl, pyrrolyl, indazolyl, thieno[2,3-c]pyrazolyl, benzofuryl, pyrazolo[l,5-a]pyridyl, thiophenylpyrazolyl, benzothienyl, benzothiazolyl, thiazolyl, 2-phenylthiazolyl, and isoxazolyl.
- Alkoxy or alkylthio refers to an alkyl group, as previously defined, attached to the principal carbon chain through an oxygen (“alkoxy”) or sulfur (“alkylthio”) atom. Examples include, but are not limited to, methoxy, ethoxy, butoxy, etc.
- Hydrophilicity refers to an -OH group.
- “-OR” refers to an R group appended to the parent molecular moiety through an oxy group, wherein R is alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocyclo, or heteroaryl.
- -SR refers to an R group appended to the parent molecular moiety through a sulfur atom, wherein R is alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocyclo, or heteroaryl.
- -SR include, but are not limited to, ethanethiyl, 3 -methyl- 1- butanethiyl, phenylthiyl and the like.
- Cycloalkyl refers to a saturated cyclic hydrocarbon group containing from 3 to 8 carbons or more.
- Representative examples of cycloalkyl include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl.
- Cycloalkenyl refers to an unsaturated cyclic hydrocarbon group containing from 3 to 8 carbons or more having one or more double bonds.
- Electrophile refers to a group having reduced electron density, typically comprising a carbon atom that is directly bonded to a more electronegative atom, such as an oxygen, nitrogen or halo.
- electrophiles include, but are not limited to, diazomethane, trimethylsilyldiazomethane, alkyl halides, such as for example methyl iodide, benzyl bromide and the like, alkyl triflates, such as for example methyl triflate and the like, alkyl sulfonates, such as for example ethyl toluenesulfonate, butyl methanesulfonate and the like, acyl halides, such as for example acetyl chloride, benzoyl bromide and the like, acid anhydrides, such as for example acetic anhydride, succinic anhydride, maleic anhydride and the like, isocyanates, such as for example methyl
- electrophiles are alpha-haloketones, isothiocyanates, epoxides, aziridines, sulfonyl halides, or alpha-beta-unsaturated carbonyls.
- the electrophile is a Michael acceptor. As known in the ait, a
- “Michael acceptor” is an alkene or alkyne of the form ⁇ — Z ; wherein Z comprises an electron withdrawing group, including, but not limited to, CHO, COR, COOR, CONRR', CONROR', CN, N0 2 , SOR, S0 2 R.
- R may be H, alkyl, or aryl ; wherein R is alkyl, alkenyl, alkoxy or aryl.
- azodicarboxamides and quinones are Michael acceptors. See, Santos, M.M.M. and Moreira, R., Mini-Reviews in Medicinal Chemistry, 7: 1040-1050, 2007.
- the Michael acceptors are alpha-beta-unsaturated carbonyl compounds including, but not limited to, alpha-beta-unstaturated amides, alpha-beta-unstaturated ketones, alpha-beta- unstaturated esters, conjugated alkynyl carbonyls and alpha-beta-unsaturated nitriles.
- Alpha-beta-unsaturated amide or “unsaturated amide” as used herein refers to an amide comprising an alkene or alkyne bonded directly to the amide carbonyl group and is represented
- R is hydrogen or alkyl
- Heteroatom refers to O, S or N.
- Heterocycle or “heterocyclyl” as used herein, means a monocyclic heterocycle, a bicyclic heterocycle, or a tricyclic heterocycle containing at least one heteroatom in the ring.
- the monocyclic heterocycle is a 3-, 4-, 5-, 6-, 7, or 8-membered ring containing at least one heteroatom independently selected from the group consisting of O, N, and S.
- the heterocycle is a 3- or 4-membered ring containing one heteroatom selected from the group consisting of O, N and S.
- the heterocycle is a 5-membered ring containing zero or one double bond and one, two or three heteroatoms selected from the group consisting of O, N and S.
- the heterocycle is a 6-, 7-, or 8-membered ring containing zero, one or two double bonds and one, two or three heteroatoms selected from the group consisting of 0, N and S.
- monocyclic heterocycle include, but are not limited to, azetidinyl, azepanyl, aziridinyl, diazepanyl, 1,3-dioxanyl, 1,3-dioxolanyl, dihydropyranyl (including 3,4-dihydro-2H-pyran-6-yl), 1 ,3-dithiolanyl, 1,3-dithianyl, imidazolinyl, imidazolidinyl, isothiazolinyl, isothiazolidinyl, isoxazolinyl, isoxazolidinyl, morpholinyl, oxadiazolinyl, oxadiazolidinyl, oxazolinyl, oxazolidinyl, piperazinyl, piperidinyl, pyranyl, pyrazolinyl, pyrazolidinyl, pyrrolinyl, pyrrolidiny
- the bicyclic heterocycles of the present invention are exemplified by a monocyclic heterocycle fused to an aryl group, or a monocyclic heterocycle fused to a monocyclic cycloalkyl, or a monocyclic heterocycle fused to a monocyclic cycloalkenyl, or a monocyclic heterocycle fused to a monocyclic heterocycle.
- bicyclic heterocycles include, but are not limited to, 3,4-dihydro-2H-pyranyl, 1,3-benzodioxolyl, 1,3-benzodithiolyl, 2,3-dihydro-l,4-benzodioxinyl, 2,3-dihydro-l-benzofuranyl, 2,3-dihydro-l-benzothienyl, 2,3- dihydro-lH-indolyl, 3,4-dihydroquinolin-2(lH)-one and 1,2,3,4- tetrahydroquinolinyl.
- the tricyclic heterocycle is a bicyclic heterocycle fused to an aryl group, or a bicyclic heterocycle fused to a monocyclic cycloalkyl, or a bicyclic heterocycle fused to a monocyclic cycloalkenyl, or a bicyclic heterocycle fused to a monocyclic heterocycle.
- tricyclic heterocycles include, but are not limited to, 2,3,4,4a,9,9a-hexahydro-lH- carbazolyl, 5a,6,7,8,9,9a-hexahydrodibenzo[b,d]furanyl, and 5a,6,7,8,9,9a-hexahydrodibenzo[b, djthienyl.
- the nitrogen or sulfur atoms can be optionally oxidized to various oxidation states.
- the group S(O) 0-2 refers to— S- (sulfide), -S(O)- (sulfoxide), an -SO 2 - (sulfone) respectively.
- nitrogens particularly but not exclusively, those defined as annular aromatic nitrogens, are meant to include those corresponding N-oxide forms.
- the corresponding pyridyl-N-oxide is meant to be included as another compound of the invention.
- “Pharmaceutically acceptable salt” as used herein refer to acid addition salts or base addition salts of the compounds in the present disclosure.
- a pharmaceutically acceptable salt is any salt which retains the activity of the parent compound and does not impart any deleterious or undesirable effect on a subject to whom it is administered and in the context in which it is administered.
- Pharmaceutically acceptable salts include, but are not limited to, metal complexes and salts of both inorganic and carboxylic acids.
- Pharmaceutically acceptable salts also include metal salts such as aluminum, calcium, iron, magnesium, manganese and complex salts.
- salts include, but are not limited to, acid salts such as acetic, aspartic, alkylsulfonic, arylsulfonic, axetil, benzenesulfonic, benzoic, bicarbonic, bisulfuric, bitartaric, butyric, calcium edetate, camsylic, carbonic, chlorobenzoic, citric, edetic, edisylic, estolic, esyl, esylic, formic, fumaric, gluceptic, gluconic, glutamic, glycolic, glycolylarsanilic, hexamic, hexylresorcjnoic, hydrabamic, hydrobromic, hydrochloric, hydroiodic, hydroxynaphthoic, isethionic, lactic, lactobionic, maleic, malic, malonic, mandelic, methanesulfonic, methylnitric, methyls
- Pharmaceutically acceptable salts may be derived from amino acids including, but not limited to, cysteine.
- Methods for producing compounds as salts are known to those of skill in the art ⁇ see, e.g., Stahl et al., Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Wiley-VCH; Verlag Helvetica Chimica Acta, Zurich, 2002; Berge et al., J. Pharm. Sci. 66: 1, 1977).
- structures depicted herein are meant to include all enantiomeric, diastereomeric, and geometric (or conformational) forms of the structure; for example, the R and S configurations for each asymmetric center, (Z) and (E) double bond isomers, and (Z) and (E) conformational isomers. Therefore, single stereochemical isomers as well as enantiomeric, diastereomeric, and geometric (or conformational) mixtures of the present compounds are within the scope of the invention. Unless otherwise stated, all tautomeric forms of the compounds of the invention are within the scope of the invention. In addition, unless otherwise stated, all rotamer forms of the compounds of the invention are within the scope of the invention.
- structures depicted herein are also meant to include compounds that differ only in the presence of one or more isotopically enriched atoms.
- compounds having the present structures except for the replacement of hydrogen by deuterium or tritium, or the replacement of a carbon by a 13 C- or 14 C-enriched carbon are within the scope of this invention.
- Such compounds are useful, for example, as analytical tools or probes in biological assays.
- “Isomers” refer to compounds having the same number and kind of atoms and hence the same molecular weight, but differing with respect to the arrangement or configuration of the atoms. It will be understood, however, that some isomers or racemates or others mixtures of isomers may exhibit more activity than others. "Stereoisomers” refer to isomers that differ only in the arrangement of the atoms in space. "Diastereoisomers” refer to stereoisomers that are not mirror images of each other. “Enantiomers” refers to stereoisomers that are non-superimposable miiTor images of one another.
- enantiomeric compounds taught herein may be "enantiomerically pure" isomers that comprise substantially a single enantiomer, for example, greater than or equal to 90%, 92%, 95%, 98%, or 99%, or equal to 100% of a single enantiomer.
- enantiomeric compounds taught herein may be stereomerically pure.
- “Stereomerically pure” as used herein means a compound or composition thereof that comprises one stereoisomer of a compound and is substantially free of other stereoisomers of that compound.
- a stereomerically pure composition of a compound having one chiral center will be substantially free of the opposite enantiomer of the compound.
- a typical stereomerically pure compound comprises greater than about 80% by weight of one stereoisomer of the compound and less than about 20% by weight of the other stereoisomers of the compound, more preferably greater than about 90% by weight of one stereoisomer of the compound and less than about 10% by weight of the other stereoisomers of the compound, even more preferably greater than about 95% by weight of one stereoisomer of the compound and less than about 5% by weight of the other stereoisomers of the compound, and most preferably greater than about 97% by weight of one stereoisomer of the compound and less than about 3% by weight of the other stereoisomers of the compound. See, e.g., US Patent No. 7,189,715.
- Enantiomeric excess (ee) of an enantiomer is [(the mole fraction of the major enantiomer) minus (the mole fraction of the minor enantiomer)] x 100.
- X is H or CI
- Ri is selected from the group consisting of:
- Ci -6 alkyl which Ci_ 6 alkyl may be unsubstituted or substituted 1-3 times with an independently selected substituent from the group consisting of: hydroxy, methoxy, C 3 - 6 cycloalkyl, and -NR3R 4 , wherein R 3 and R4 are each independently hydrogen or methyl;
- heterocyclyl which heterocyclyl may be unsubstituted or substituted with Ci -6 alkyl;
- R 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl;
- Y and Z are each independently fluoro or chloro
- the compound is a compound of Formula 1(a):
- X is H or CI
- R 3 is hydrogen or C] -6 alkyl
- R 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl;
- Y and Z are each independently fluoro or chloro
- the compound is a compound of Formula 11(a):
- X is H or CI
- R 3 is hydrogen or C] -6 alkyl
- R 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl;
- Y and Z are each independently fluoro or chloro
- X is H or CI
- R 3 is hydrogen or Ci -6 alkyl
- R 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl;
- Y and Z are each independently fluoro or chloro
- the compound is a compound of Formula 111(a):
- X is H or CI
- R 3 is hydrogen or Ci -6 alkyl
- R 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl;
- Y and Z are each independently fluoro or chloro
- X is H or CI
- Ri is selected from the group consisting of
- Ci -6 alkyl which Ci -6 alkyl may be unsubstituted or substituted 1-3 times with an independently selected substituent from the group consisting of: hydroxy, methoxy, C3- 6 cycloalkyl, and -NRsR ⁇ wherein R 3 and R 4 are each independently hydrogen or methyl; C 1-6 cycloalkyl; and
- heterocyclyl which heterocyclyl may be unsubstituted or substituted with Ci-6 alkyl;
- R 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl;
- Y and Z are each independently fluoro or chloro
- the compound is a compound of Formula IV(a):
- X is H or CI
- Ri is selected from the group consisting of
- Ci -6 alkyl which Ci -6 alkyl may be unsubstituted or substituted 1-3 times with an independently selected substituent from the group consisting of: hydroxy, methoxy, C 3 - 6 cycloalkyl, and -NR 3 R 4 , wherein R 3 and R 4 are each independently hydrogen or methyl; C 3-6 cycloalkyl; and
- heterocyclyl which heterocyclyl may be unsubstituted or substituted with C 1-6 alkyl; 2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
- Y and Z are each independently fluoro or chloro
- Active compounds of the present invention can be combined with a pharmaceutically acceptable carrier to provide pharmaceutical formulations thereof.
- a pharmaceutically acceptable carrier to provide pharmaceutical formulations thereof.
- the particular choice of carrier and formulation will depend upon the particular route of administration for which the composition is intended.
- “Pharmaceutically acceptable carrier” refers to a nontoxic carrier, adjuvant, or vehicle that does not destroy the pharmacological activity of the compound with which it is formulated.
- Pharmaceutically acceptable carriers, adjuvants or vehicles that may be used in the compositions of this invention include, but are not limited to, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene glycol and wool fat.
- compositions of the present invention may be suitable for parenteral, oral, inhalation spray, topical, rectal, nasal, buccal, vaginal or implanted reservoir administration, etc.
- parenteral as used herein includes subcutaneous, intravenous, intramuscular, intra-articular, intra-synovial, intrasternal, intrathecal, intrahepatic, intralesional and intracranial injection or infusion techniques.
- the compounds are administered intravenously, orally, subcutaneously, or via intramuscular administration.
- Sterile injectable forms of the compositions of this invention may be aqueous or oleaginous suspension. These suspensions may be formulated according to techniques known in the art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a nontoxic parenterally acceptable diluent or solvent.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or di- glycerides.
- Fatty acids and their glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions.
- These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant, such as carboxymethyl cellulose or similar dispersing agents that are commonly used in the formulation of pharmaceutically acceptable dosage forms including emulsions and suspensions.
- Other commonly used surfactants, such as Tweens, Spans and other emulsifying agents that are commonly used in the manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms may also be used for the purposes of formulation.
- the active compounds may be provided in an acceptable oral dosage form, including, but not limited to, capsules, tablets, aqueous suspensions or solutions.
- carriers commonly used include lactose and corn starch.
- Lubricating agents such as magnesium stearate, may also be added.
- useful diluents include lactose and dried cornstarch.
- the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening, flavoring or coloring agents may also be added. In addition preservatives may also be added.
- Suitable examples of pharmaceutically acceptable preservatives include, but are not limited to, various antibacterial and antifungal agents such as solvents, for example ethanol, propylene glycol, benzyl alcohol, chlorobutanol, quaternary ammonium salts, and parabens (such as methyl paraben, ethyl paraben, propyl paraben, etc.).
- solvents for example ethanol, propylene glycol, benzyl alcohol, chlorobutanol, quaternary ammonium salts, and parabens (such as methyl paraben, ethyl paraben, propyl paraben, etc.).
- Compounds of the present invention may be used to treat hepatocellular carcinoma.
- Treatment refers to reversing, alleviating, delaying the onset of, inhibiting the progress of, or otherwise ameliorating a disease or disorder as described herein.
- treatment may be administered after one or more symptoms have developed.
- treatment may be administered in the absence of symptoms.
- treatment may be administered to a susceptible individual prior to the onset of symptoms (e.g., in light of a history of symptoms and/or in light of genetic or other susceptibility factors).
- Treatment may also be continued after symptoms have resolved, for example to prevent or delay their recurrence.
- "Patient” or “subject”, as used herein, means an animal subject, preferably a mammalian subject (e.g., dog, cat, horse, cow, sheep, goat, monkey, etc.), and particularly human subjects (including both male and female subjects, and including neonatal, infant, juvenile, adolescent, adult and geriatric subjects).
- treatment is provided to a subject having a cancer with altered FGFR4 and/or FGF19 (fibroblast growth factor 19) status, including hepatocellular carcinoma.
- FGF19 fibroblast growth factor 19
- treatment may include analyzing FGFR4 and/or FGF19 status in a biological sample containing cells of said cancer, and if said cancer exhibits an FGFR4 and/or FGF19 alteration, treating a subject with a treatment effective amount of a compound or composition as described herein.
- Altered status as used herein with reference to FGFR4 and/or FGF19 includes an increased expression thereof (e.g., increased levels of the niRNA or increased levels of the protein), increased copy number in the genome, increased activity of the encoded protein as a result of mutation, etc., as compared to a corresponding non-cancerous tissue.
- Altered status of FGFR4 and/or FGF19 includes gene and/or encoded protein mutations that result in an increase in activity or are otherwise associated with a more aggressive form of cancer.
- “Expression” of FGFR4 and/or FGF19 means that a gene encoding the same is transcribed, and preferably, translated. Typically, expression of a coding region will result in production of the encoded polypeptide.
- FGFR and FGF19 proteins are known, and their altered status and/or expression may be measured using techniques standard in the art, e.g., genomic analysis of mutations or copy number aberrations such as by nucleic acid amplification, sequencing analysis, and/or hybridization-based techniques, RNA expression analysis such as northern blot or qRT-PCR, western blot or other imniunoblot or immunoassay, fluorescent activated cell sorting (FACS), etc.
- genomic analysis of mutations or copy number aberrations such as by nucleic acid amplification, sequencing analysis, and/or hybridization-based techniques
- RNA expression analysis such as northern blot or qRT-PCR, western blot or other imniunoblot or immunoassay, fluorescent activated cell sorting (FACS), etc.
- FACS fluorescent activated cell sorting
- Microwave heating was done using a Biotage Emrys Liberator or Initiator microwave. Column chiOmatography was carried out using an Isco Rf200d. Solvent removal was earned out using either a Biichi rotary evaporator or a Genevac centrifugal evaporator. Preparative LC/MS was conducted using a Waters autopurifier and 19 x 100mm XTerra 5 micron MS CI 8 column under acidic mobile phase conditions. NMR spectra were recorded using a Varian 400MHz spectrometer.
- inerted e.g., a reaction vessel, flask, glass reactor, and the like
- inert gas such as nitrogen, argon, and the like
- DIPEA N,N-diisopropylethylamine
- HATU 1 -[Bis(dimethylamino)methylene]- 1 H- 1 ,2,3 -trizolo [4,5-b]pyridinium 3 -oxid hexafluorophosphate
- NaOAc Sodium acetate
- NMO N-Methylmorpholine-N-oxide
- TBSOTf tert-Butyldimethylsilyl trifluoromethanesulfonate
- TESC1 Chlorotriethylsilane
- PPTS Pyridinium p-toluenesulfonate
- PE Petroleum ether
- Ti(0'Pr) 4 Titanium isopropoxide
- CDCI 3 CDCI 3 ) ⁇ 2.16 (s, 3H), 3.77 23 (s, 1H), 6.75 (s, 2H), 7.20 (s, 1H).
- tert-Butyl 4-iodopiperidine-l-carboxylate (11.26g, 36.23 mmol) was slowly added to a stirred solution of methyl 4-nitro-lH-pyrazole-3-carboxylate (5g, 24.15 mmol) and K 2 C0 3 (9.99g, 72.46 mmol) in DMF (50mL) at 0 °C.
- the reaction mass was stirred at room temperature for 18hr. Quenched the reaction with ice cold water (lOOmL) and ethyl acetate (25mL). The aqueous layer was separated and extracted with ethyl acetate (3x25mL).
- TEA tert-butyl (4-amino-l-(l- ethylpiperidin-4-yl)-lH-pyrazol-3-yl)(6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methyl-3-((2- (trimethylsilyl)ethoxy)methyl)ureido)pyrimidin-4-yl)carbamate (0.25g, 0.3 mmol, Preparation from Example 610) in anhydrous DCM (6mL) under argon atmosphere at 0 °C.
- Triethylamine (0.072g, 0.71 mmol) was added to a stirred solution of diboc protected l-(6-((4- amino-l-methyl-lH-pyrazol-3-yl)amino)pyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)- 1-methylurea (0.2g, 0.28 mol) in anhydrous DCM (l OmL) under argon atmosphere at 0 °C. The resulting mixture was stirred for 10 min and slowly added propanoyl chloride (0.039g, 0.43 mmol) at 0 °C. The resulting reaction mixture stirred at room temperature for lh.
- Lithium hydroxide (0.9g, 22.61mmol) in water (5mL) was added to a stirred solution of methyl l-ethyl-4-nitro-lH-pyrazole-3-carboxylate (1.8g, 9.04 mmol) in a mixture of methanol
- reaction mass was quenched with saturated NH 4 C1 solution. Extracted the compound to ethyl acetate and the ethyl acetate layer was washed with brine, dried over Na 2 S0 4 , filtered and concentrated under vacuum. The residue was purified by basic alumina column by eluting with 2.5% methanol-DCM to afford the title compound as the free base (0.048g, 30%).
- tert-Butylchlorodimethylsilane (1.09g, 7.2 mmol) was slowly added to a stirred solution of 3-(3- amino-4-nitro-lH-pyrazol-l-yl)propan-l-ol (0.9g, 4.8 mmol) and imidazole (0.65g, 9.6 mmol) in DCM (lOmL) at 0 °C. Stirred the reaction mass for 5min and DMAP (0.29g, 2.4 mmol) was added to the reaction mass. The reaction mass was stirred at room temperature for lh. Quenched the reaction mass with ice cold water and diluted with ethyl acetate (lOOmL).
- Lithium hydroxide (2.4g, 57.14 mmol) in water (lOmL) was added to a stirred solution of methyl 1 -ethyl -4-nitro-lH-pyrazole-3-carboxylate (3.5g, 14 mmol) in a mixture of methanol
- tert-Butyl(3-chloiOpiOpoxy)dimethylsilane (15g, 72.4 mmol) was slowly added to a stirred solution of methyl 4-nitro-lH-pyrazole-3-carboxylate, (10. Og, 48.3 mmol) in DMF (lOOmL) at 0 °C.
- TEA TEA (0.16g, 1.61 mmol) was added to a stirred solution of Diboc-protected l-(6-((4-amino-l- methyl-lH-pyrazol-3-yl)amino)pyrimidin-4-yl)-l-benzyl-3-(2,6-dichloro-3,5-dimethoxyphenyl) urea (0.8g, 1.07mmol) in anhydrous DCM (15mL) under argon atmosphere at 0 °C. The resulting mixture was stirred for 5 min and then slowly added the acryloyl chloride (0.116g, 1.29 mmol) at 0 °C. The resulting reaction mixture was allowed to warm to room temperature and stirred for 30 min.
- IC50 Profiling of Kinase Activity Inhibition Compounds were profiled for FGFR inhibition activity at Reaction Biology Corporation (Malvern, Pennsylvania) with their Kinase HotSpot assay. See, Anastassiadis et al., 2011, Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol 29, 1039-1045.
- FGFR1 Recombinant FGFR1 (2.5 nM), FGFR2 (1 nM), FGFR3 (5 nM), or FGFR4 (12 nM) (InvitrogenTM) was prepared as a mixture with substrate KKKSPGEYVNIEFG (SEQ ID NO:l) (20 ⁇ , FGFR1 substrate); and Poly [E,Y] 4:1 (0.2 mg/ml, FGFR2,3,4 substrate)] in kinase reaction buffer (20 mM HEPES-HC1, pH 7.5, 10 mM MgCl 2 , 2 mM MnCl 2 , 1 mM EGTA, 0.02% Brij35, 0.1 mM Na 3 V0 4 , 0.02 mg/ml BSA, 2 mM DTT, and 1% DMSO) (reagents from Reaction Biology Corp., Malvern, Pennsylvania).
- kinase reaction buffer (20 mM HEPES-HC1, pH 7.5, 10 mM MgCl
- the IC50 activity of FGFR1 is generally representative of the activity of FGFR1, FGFR2, and FGFR3. See also, Dieci et al., 2013, Fibroblast Growth Factor Receptor Inhibitors as a Cancer Treatment: From a Biologic Rationale to Medical Perspectives. Cancer Discovery, F1-F16.
- Ba/F3 is a murine, interleukin-3 dependent hematopoietic cell line, purchased from Riken Cell Bank (Ibaraki, Japan), and maintained in culture medium (RPMI-1640 (Gibco) supplemented with 10% FBS (Gibco) and 10 ng/mL recombinant mouse IL-3 (R&D Systems)).
- RPMI-1640 Gibco
- FBS FBS
- 10 ng/mL recombinant mouse IL-3 R&D Systems
- pLenti- FGFR4 or pLenti-FGFRl lentivirus were infected to the Ba/F3 parental cell lines (seeded 5E5 cells/mL, lmL/well in 12-well plate) to generate Ba/F3-FGFR4 or Ba/F3-FGFR1 by using 20ug/mL blasticidin selection marker.
- Ba/F3-FGFR4 viability assay 4,000 cells were dispensed into each well of a 96-well plate in 90 ⁇ xL of growth media (RPMI-1640 supplemented with 10%) FBS), and incubated for 24 hours at 37°C and 5% C0 2 . 10 of serially diluted compound were added to appropriate wells in triplicates, and cells were incubated for 72 hours at 37°C and 5% C0 2 . Viability was measured by adding 50 ⁇ xL of the CellTiter-Glo® (Promega) reagent and measuring luminescence, reported as relative light units (RLU). GI 50 values were calculated by producing nine-point dose-response curves, with normalization to media control, DMSO control and day 0 RLU measurements taken at the time of compound addition. Best-fit curves were generated with Hill equation.
- Ba/F3-FGFR1 viability assay Assay was performed as described above for Ba/F3- FGFR4.
- Ba/F3-Parental viability assay Assay to test general cytotoxic effects was performed as described above for Ba/F3-FGFR4, except for a difference in the growth media (RPMI-1640 supplemented with 10% FBS and lOng/ml IL-3).
Abstract
We provide FGFR inhibitors, their salts, methods of manufacture, and methods of use.
Description
FGFR4 INHIBITORS
BACKGROUND
Fibroblast growth factors (FGF) are a family of more than 20 structurally related proteins with a variety of biological activities. Their main receptors, the fibroblast growth factor receptors (FGFR1, FGFR2, FGFR3 and FGFR4), are a family of receptor tyrosine kinases that bind FGF and are involved in processes of cell proliferation and differentiation. Deregulation of FGFR signaling networks is implicated in a number of pathophysiological conditions, including many types of human cancers.
"Fibroblast Growth Factor Receptor 4" or "FGFR4" is known to regulate proliferation and antiapoptosis and is expressed or highly expressed in many cancers. See, e.g., Dieci et al. 2013, Cancer Discovery, 0F1-0F16. Studies have shown that expression of FGFR4 is predictive of a more aggressive phenotype of the cancer, and knockdown or reduction of FGFR4 expression serves to reduce proliferation and promote apoptosis. See, e.g. , Wesche et al. 201 1, Biochem J 437: 199-213.
For example, FGFR4 expression or overexpression is associated with cancer
aggressiveness in gastric cancer (Ye et al. 2011, Cancer, 5304-5313), prostate cancer (Xu et al.
2011, BMC Cancer, 11 ;84), sarcoma such as rhabdomyosarcoma (Taylor VI et al. 2009, J Clin Invest, 119(1 1):3395-3407), skin cancer such as melanoma (Streit et al. 2006, British J Cancer, 94:1879-1886), liver cancer such as cholangiocarcinoma (Sia et al. 2013, Gastroenterology 144:829-840) and hepatocellular carcinoma (French et al. 2012, PLoS ONE 7(5): e367313;
Miura et al. 2012, BMC Cancer 12:56; Chiang et al. 2008, Cancer Res 68(16):6779-6788; Sawey et al. 2011, Cancer Cell 19:347-358), pancreatic cancer such as pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma (Motoda et al. 201 1, Int'l J Oncol 38:133-143), lung cancer such as non-small-cell lung cancer (Fawdar et al. 2013, PNAS 110(30): 12426- 12431), colorectal cancer (Pelaez-Garcia et al. 2013, PLoS ONE 8(5): e63695; Barderas et al.
2012, J Proteomics 75:4647-4655), and ovarian cancer (Zaid et al. 2013, Clin Cancer Res 19:809-820).
Clinical development of several FGFR inhibitors have confirmed their utility as antitumor agents. Dieci et al. 2013, Cancer Discovery, 0F1-0F16. However, new agents are needed that are useful to target FGFR, and FGFR4, in particular.
SUMMARY
A purpose of the present invention is to provide a compound of Formula I:

I
wherein:
X is H or CI;
R1 is selected from the group consisting of:
hydrogen;
C1-6alkyl, which C1-6alkyl may be unsubstituted or substituted 1-3 times with an independently selected substituent from the group consisting of: hydroxy, methoxy, C3-6cycloalkyl, and -NR3R4, wherein R3 and R4 are each independently hydrogen or methyl;
C3-6cycloalkyl; and
heterocyclyl, which heterocyclyl may be unsubstituted or substituted with C1-6 alkyl;
R2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
A further purpose of the present invention is to provide a compound of Formula 1(a):

wherein X, Ri, R2, Y and Z are as defined above, or a pharmaceutically acceptable salt thereof.
A further purpose is to provide a compound of Formula II:

II
wherein
X is H or CI;
R3 is hydrogen or C1-6alkyl;
R2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
A further purpose is to provide a compound of Formula 11(a):

11(a)
wherein
X is H or CI;
R3 is hydrogen or C1-6alkyl;
R2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
A further purpose is to provide a compound of Formula III:

III wherein:
X is H or CI;
R3 is hydrogen or Ci-6alkyl;
R2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
A further purpose is to provide a compound of Formula 111(a):

111(a) wherein:
X is H or CI;
R3 is hydrogen or Ci-6alkyl;
R2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
A further purpose is to provide a compound of Formula IV:

IV
wherein:
X is H or CI;
Ri is selected from the group consisting of
hydrogen;
C1-6alkyl, which Ci-6alkyl may be unsubstituted or substituted 1-3 times with an independently selected substituent from the group consisting of: hydroxy, methoxy, C3-6cycloalkyl, and -NR3R4, wherein R3 and R4 are each independently hydrogen or methyl; C3.6cycloalkyl; and
heterocyclyl, which heterocyclyl may be unsubstituted or substituted with C1-6 alkyl;
R2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
A further purpose is to provide a compound of Formula IV(a):

IV(a)
wherein:
X is H or CI;
Ri is selected from the group consisting of
hydrogen;
C1-6alkyl, which Ci-6alkyl may be unsubstituted or substituted 1-3 times with an independently selected substituent from the group consisting of: hydroxy, methoxy, C3-6cycloalkyl, and -NR3R4, wherein R3 and R4 are each independently hydrogen or methyl; C3-6cycloalkyl; and
heterocyclyl, which heterocyclyl may be unsubstituted or substituted with Ci_6 alkyl;
R2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
Also provided is a compound selected from those presented in Table 1 below, or a pharmaceutically acceptable salt thereof.
A further purpose is a pharmaceutical composition comprising a compound as described herein and a pharmaceutically acceptable earner. In some embodiments, the composition is formulated for oral or parenteral administration.
A further purpose is a method of treating hepatocellular carcinoma in a subject in need thereof comprising administering to said subject a treatment effective amount of a compound or salt or composition as described herein. In some embodiments, hepatocellular carcinoma has altered FGFR4 and/or FGF19 status (e.g., increased expression of FGFR4 and/or FGF19).
A further purpose is a method of treating hepatocellular carcinoma in a subject in need thereof, comprising: detecting an altered FGFR4 and/or FGF19 status (e.g., increased expression of FGFR4 and/or FGF19) in a biological sample containing cells of said hepatocellular carcinoma, and if said hepatocellular carcinoma has said altered FGFR4 and/or FGF19 status, administering a compound or composition described herein to said subject in a treatment- effective amount.
A further purpose is the use of a compound or salt or a composition as described herein in a method of treatment of hepatocellular carcinoma.
A further purpose is the use of a compound or salt described herein in the preparation of a medicament for the treatment of hepatocellular carcinoma.
DETAILED DESCRIPTION OF EMBODIMENTS
Provided herein are compounds useful as FGFR4 inhibitors. In some embodiments, the compounds are selective FGFR4 inhibitors in that they have a greater binding affinity and/or inhibitory effect of FGFR4 as compared to that of FGFRl and/or FGFR2 and/or FGFR3 (e.g., by 10-fold, 100-fold, or 1000-fold greater or more).
A. Definitions
Compounds useful as active agents in accordance with the present disclosure include those described generally above and below, and are further illustrated by the embodiments, sub-
embodiments, and species disclosed herein. As used herein, the following definitions shall apply unless otherwise indicated.
As described herein, compounds of the invention may optionally be substituted with one or more substituents, such as those illustrated generally herein, or as exemplified by particular classes, subclasses, and species of the invention. In general, the term "substituted" refers to the replacement of hydrogen in a given structure with a specified substituent. Unless otherwise indicated, a substituted group may have a substituent at each substitutable position of the group, and when more than one position in any given structure may be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at every position. Combinations of substituents envisioned by this invention are preferably those that result in the formation of stable compounds. "Stable" as used herein refers to a chemically feasible compound that is not substantially altered when kept at a temperature of 40 °C or less, in the absence of moisture or other chemically reactive conditions, for at least one week.
As would be understood by those of skill in the art, as used herein "H" is hydrogen, "C" is carbon, "N" is nitrogen, "S" is sulfur, and "O" is oxygen.
"Alkyl" or "alkyl group," as used herein, means a straight-chain (i.e., unbranched), or branched chain that is completely saturated. In some embodiments, the alkyl has 1, 2, 3, 4, 5 or 6 carbon atoms, i.e., methyl, ethyl, propyl, butyl, etc. In certain embodiments, alkyl groups contain 1-6 carbon atoms (C1-6alkyl). In certain embodiments, alkyl groups contain 1-4 carbon atoms (Ci-4alkyl). In certain embodiments, alkyl groups contain 1-3 carbon atoms (Ci-3alkyl). In still other embodiments, alkyl groups contain 2-3 carbon atoms (C2-3alkyl), and in yet other embodiments alkyl groups contain 1-2 carbon atoms (Ci-2alkyl).
"Alkenyl" or "alkenyl group," as used herein, refers to a straight-chain (i.e., unbranched), or branched hydrocarbon chain that has one or more double bonds. In some embodiments, the alkenyl has 2, 3, 4, 5 or 6 carbon atoms. In certain embodiments, alkenyl groups contain 2-8 carbon atoms (C2-8alkenyl). In certain embodiments, alkenyl groups contain 2-6 carbon atoms (C2-6alkenyl). In still other embodiments, alkenyl groups contain 3-4 carbon atoms (C3-4alkenyl), and in yet other embodiments alkenyl groups contain 2-3 carbon atoms (C2-3alkenyl). According to another aspect, the term alkenyl refers to a straight chain hydrocarbon having two double bonds, also referred to as "diene." Non-limiting examples of exemplary alkenyl groups include— CH=CH2, -CH2CH=CH2, -CH=CHCH3, -C¾CH2CH=CH2,
-CH2CH=CHCH3, -CH=CHCH2CH3, and -CH=CHCH=CH2.
"Alkynyl" or "alkynyl group" as used herein refers to a straight-chain (i.e., unbranched), branched, or cyclic hydrocarbon chain that has one or more triple bonds. In some embodiments,
the alkynyl has 2, 3, 4, 5 or 6 carbon atoms. In certain embodiments, alkynyl groups contain 2-8 carbon atoms (C2-8alkynyl). In certain embodiments, alkynyl groups contain 2-6 carbon atoms (C2-6alkynyl). In still other embodiments, alkynyl groups contain 3-4 carbon atoms (C3-4alkynyl), and in yet other embodiments alkynyl groups contain 2-3 carbon atoms (C2-3 alkynyl).
"Ar" or "aryl" refer to an aromatic carbocyclic moiety having one or more closed rings. Examples include, without limitation, phenyl, naphthyl, anthracenyl, phenanthracenyl, biphenyl, and pyrenyl.
"Benzyl" refers to a phenyl group appended to the parent compound through a methyl group (-CH2C6H5).
"Pyridylmethyl" refers to a pyridine ring appended to the parent compound through a methyl group (-CH2C5H4N).
"Halo" refers to chloro (CI), fluoro (F), bromo (Br) or iodo (I).
"Haloalkyl" refers to one or more halo groups appended to the parent molecular moiety through an alkyl group. Examples include, but are not limited to, chloromethyl, fluoromethyl, trifluoromethyl, etc.
"Heteroaryl" refers to a cyclic moiety having one or more closed rings, with one or more heteroatoms (oxygen, nitrogen or sulfur) in at least one of the rings, wherein at least one of the rings is aromatic, and wherein the ring or rings may independently be fused, and/or bridged. Examples include without limitation quinolinyl, isoquinolinyl, indolyl, furyl, thienyl, pyrazolyl, quinoxalinyl, pyrrolyl, indazolyl, thieno[2,3-c]pyrazolyl, benzofuryl, pyrazolo[l,5-a]pyridyl, thiophenylpyrazolyl, benzothienyl, benzothiazolyl, thiazolyl, 2-phenylthiazolyl, and isoxazolyl.
"Alkoxy" or "alkylthio" as used herein refers to an alkyl group, as previously defined, attached to the principal carbon chain through an oxygen ("alkoxy") or sulfur ("alkylthio") atom. Examples include, but are not limited to, methoxy, ethoxy, butoxy, etc.
"Hydroxy" refers to an -OH group.
"-OR" refers to an R group appended to the parent molecular moiety through an oxy group, wherein R is alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocyclo, or heteroaryl.
Representative examples of "-OR" include, but are not limited to, methoxy, ethoxy, propoxy, phenoxy, 2-propoxy, butoxy, tert-butoxy, pentyloxy, hexyloxy and the like.
"-SR" refers to an R group appended to the parent molecular moiety through a sulfur atom, wherein R is alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocyclo, or heteroaryl.
Representative examples of "-SR" include, but are not limited to, ethanethiyl, 3 -methyl- 1- butanethiyl, phenylthiyl and the like.
"Cycloalkyl" as used herein, refers to a saturated cyclic hydrocarbon group containing from 3 to 8 carbons or more. Representative examples of cycloalkyl include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl.
"Cycloalkenyl" as used herein, refers to an unsaturated cyclic hydrocarbon group containing from 3 to 8 carbons or more having one or more double bonds.
"Electrophile" as used herein refers to a group having reduced electron density, typically comprising a carbon atom that is directly bonded to a more electronegative atom, such as an oxygen, nitrogen or halo. Exemplary electrophiles include, but are not limited to, diazomethane, trimethylsilyldiazomethane, alkyl halides, such as for example methyl iodide, benzyl bromide and the like, alkyl triflates, such as for example methyl triflate and the like, alkyl sulfonates, such as for example ethyl toluenesulfonate, butyl methanesulfonate and the like, acyl halides, such as for example acetyl chloride, benzoyl bromide and the like, acid anhydrides, such as for example acetic anhydride, succinic anhydride, maleic anhydride and the like, isocyanates, such as for example methyl isocyanate, phenyl isocyanate and the like, isothiocyanates, such as for example methyl isothiocyanate, phenyl isothiocyanate and the like, chloroformates, such as for example methyl chloroformate, ethyl chloroformate, benzyl chloroformate and the like, sulfonyl halides, such as for example methanesulfonyl chloride, methanesulfonyl fluoride, p-toluenesulfonyl chloride and the like, silyl halides, such as for example trimethylsilyl chloride, tert- butyldimethylsilyl chloride and the like, phosphoryl halides such as for example dimethyl chlorophosphate and the like, epoxides such as for example 2-methyloxirane, aziridines such as for example 2-methylaziridine, alpha-haloketone such as for example l-chloro-2-propanone, alpha-beta-unsaturated carbonyl compounds such as for example acrolein, methyl vinyl ketone, cinnamaldehyde, Ν,Ν-dimethylacrylamide and the like, and gamma-halo-alpha-beta-unsaturated carbonyl compounds such as for example (E)-6-chlorohex-4-en-3-one. In some embodiments, electrophiles are alpha-haloketones, isothiocyanates, epoxides, aziridines, sulfonyl halides, or alpha-beta-unsaturated carbonyls.
In some embodiments, the electrophile is a Michael acceptor. As known in the ait, a
"Michael acceptor" is an alkene or alkyne of the form■— Z ; wherein Z comprises an electron withdrawing group, including, but not limited to, CHO, COR, COOR, CONRR', CONROR', CN, N02, SOR, S02R. R may be H, alkyl, or aryl; wherein R is alkyl, alkenyl, alkoxy or aryl. In another embodiment, azodicarboxamides and quinones are Michael acceptors. See, Santos, M.M.M. and Moreira, R., Mini-Reviews in Medicinal Chemistry, 7: 1040-1050, 2007.
An example of the Michael Reaction is depicted in the scheme below:
Z-CH2-Z' + =— Ζ' base H H2 H2
C— C -C -Z"
Z'
Michael donor Michael acceptor wherein electron withdrawing groups Z, Z' and Z" are as described above. In some
embodiments, the Michael acceptors are alpha-beta-unsaturated carbonyl compounds including, but not limited to, alpha-beta-unstaturated amides, alpha-beta-unstaturated ketones, alpha-beta- unstaturated esters, conjugated alkynyl carbonyls and alpha-beta-unsaturated nitriles.
"Alpha-beta-unsaturated amide" or "unsaturated amide" as used herein refers to an amide comprising an alkene or alkyne bonded directly to the amide carbonyl group and is represented
by the structure
 wherein R is hydrogen or alkyl.
wherein R is hydrogen or alkyl.
"Heteroatom" refers to O, S or N.
"Heterocycle" or "heterocyclyl" as used herein, means a monocyclic heterocycle, a bicyclic heterocycle, or a tricyclic heterocycle containing at least one heteroatom in the ring.
The monocyclic heterocycle is a 3-, 4-, 5-, 6-, 7, or 8-membered ring containing at least one heteroatom independently selected from the group consisting of O, N, and S. In some embodiments, the heterocycle is a 3- or 4-membered ring containing one heteroatom selected from the group consisting of O, N and S. In some embodiments, the heterocycle is a 5-membered ring containing zero or one double bond and one, two or three heteroatoms selected from the group consisting of O, N and S. In some embodiments, the heterocycle is a 6-, 7-, or 8-membered ring containing zero, one or two double bonds and one, two or three heteroatoms selected from the group consisting of 0, N and S. Representative examples of monocyclic heterocycle include, but are not limited to, azetidinyl, azepanyl, aziridinyl, diazepanyl, 1,3-dioxanyl, 1,3-dioxolanyl, dihydropyranyl (including 3,4-dihydro-2H-pyran-6-yl), 1 ,3-dithiolanyl, 1,3-dithianyl, imidazolinyl, imidazolidinyl, isothiazolinyl, isothiazolidinyl, isoxazolinyl, isoxazolidinyl, morpholinyl, oxadiazolinyl, oxadiazolidinyl, oxazolinyl, oxazolidinyl, piperazinyl, piperidinyl, pyranyl, pyrazolinyl, pyrazolidinyl, pyrrolinyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl (including tetrahydro-2H-pyran-4-yl), tetrahydrothienyl, thiadiazolinyl, thiadiazolidinyl, thiazolinyl, thiazolidinyl, thiomorpholinyl, 1,1-dioxidotHomorpholinyl (thiomorpholine sulfone), thiopyranyl, and trithianyl.
The bicyclic heterocycles of the present invention are exemplified by a monocyclic heterocycle fused to an aryl group, or a monocyclic heterocycle fused to a monocyclic
cycloalkyl, or a monocyclic heterocycle fused to a monocyclic cycloalkenyl, or a monocyclic heterocycle fused to a monocyclic heterocycle. Representative examples of bicyclic heterocycles include, but are not limited to, 3,4-dihydro-2H-pyranyl, 1,3-benzodioxolyl, 1,3-benzodithiolyl, 2,3-dihydro-l,4-benzodioxinyl, 2,3-dihydro-l-benzofuranyl, 2,3-dihydro-l-benzothienyl, 2,3- dihydro-lH-indolyl, 3,4-dihydroquinolin-2(lH)-one and 1,2,3,4- tetrahydroquinolinyl.
The tricyclic heterocycle is a bicyclic heterocycle fused to an aryl group, or a bicyclic heterocycle fused to a monocyclic cycloalkyl, or a bicyclic heterocycle fused to a monocyclic cycloalkenyl, or a bicyclic heterocycle fused to a monocyclic heterocycle. Representative examples of tricyclic heterocycles include, but are not limited to, 2,3,4,4a,9,9a-hexahydro-lH- carbazolyl, 5a,6,7,8,9,9a-hexahydrodibenzo[b,d]furanyl, and 5a,6,7,8,9,9a-hexahydrodibenzo[b, djthienyl.
In the above heteroaryl and heterocycles the nitrogen or sulfur atoms can be optionally oxidized to various oxidation states. In a specific example, the group S(O)0-2 refers to— S- (sulfide), -S(O)- (sulfoxide), an -SO2- (sulfone) respectively. For convenience, nitrogens, particularly but not exclusively, those defined as annular aromatic nitrogens, are meant to include those corresponding N-oxide forms. Thus, for a compound of the invention having, for example, a pyridyl ring; the corresponding pyridyl-N-oxide is meant to be included as another compound of the invention.
"Pharmaceutically acceptable salt" as used herein refer to acid addition salts or base addition salts of the compounds in the present disclosure. A pharmaceutically acceptable salt is any salt which retains the activity of the parent compound and does not impart any deleterious or undesirable effect on a subject to whom it is administered and in the context in which it is administered. Pharmaceutically acceptable salts include, but are not limited to, metal complexes and salts of both inorganic and carboxylic acids. Pharmaceutically acceptable salts also include metal salts such as aluminum, calcium, iron, magnesium, manganese and complex salts. In addition, pharmaceutically acceptable salts include, but are not limited to, acid salts such as acetic, aspartic, alkylsulfonic, arylsulfonic, axetil, benzenesulfonic, benzoic, bicarbonic, bisulfuric, bitartaric, butyric, calcium edetate, camsylic, carbonic, chlorobenzoic, citric, edetic, edisylic, estolic, esyl, esylic, formic, fumaric, gluceptic, gluconic, glutamic, glycolic, glycolylarsanilic, hexamic, hexylresorcjnoic, hydrabamic, hydrobromic, hydrochloric, hydroiodic, hydroxynaphthoic, isethionic, lactic, lactobionic, maleic, malic, malonic, mandelic, methanesulfonic, methylnitric, methylsulfuric, mucic, muconic, napsylic, nitric, oxalic, p- nitrophenylsulfonic, pamoic, pantothenic, phosphoric, monohydrogen phosphoric, dihydrogen phosphoric, phthalic, polygalactouronic, propionic, salicylic, stearic, succinic, sulfamic,
sulfanlic, sulfonic, sulfuric, tannic, tartaric, teoclic, toluenesulfonic, and the like.
Pharmaceutically acceptable salts may be derived from amino acids including, but not limited to, cysteine. Methods for producing compounds as salts are known to those of skill in the art {see, e.g., Stahl et al., Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Wiley-VCH; Verlag Helvetica Chimica Acta, Zurich, 2002; Berge et al., J. Pharm. Sci. 66: 1, 1977).
Unless indicated otherwise, nomenclature used to describe chemical groups or moieties as used herein follow the convention where, reading the name from left to right, the point of attachment to the rest of the molecule is at the right-hand side of the name. For example, the group "arylCi-6alkyl," is attached to the rest of the molecule at the alkyl end.
Unless indicated otherwise, where a chemical group is described by its chemical formula, including a terminal bond moiety indicated by it will be understood that the attachment is read from left to right.
Unless otherwise stated, structures depicted herein are meant to include all enantiomeric, diastereomeric, and geometric (or conformational) forms of the structure; for example, the R and S configurations for each asymmetric center, (Z) and (E) double bond isomers, and (Z) and (E) conformational isomers. Therefore, single stereochemical isomers as well as enantiomeric, diastereomeric, and geometric (or conformational) mixtures of the present compounds are within the scope of the invention. Unless otherwise stated, all tautomeric forms of the compounds of the invention are within the scope of the invention. In addition, unless otherwise stated, all rotamer forms of the compounds of the invention are within the scope of the invention.
Inclusion of stereoisomers in a formula as taught herein may be depicted as wavy line:
X , which indicates a compound having any of the possible stereoisomers {e.g. , enantiomers, cis and trans isomers, etc.) at that position.
Unless otherwise stated, structures depicted herein are also meant to include compounds that differ only in the presence of one or more isotopically enriched atoms. For example, compounds having the present structures except for the replacement of hydrogen by deuterium or tritium, or the replacement of a carbon by a 13C- or 14C-enriched carbon are within the scope of this invention. Such compounds are useful, for example, as analytical tools or probes in biological assays.
"Isomers" refer to compounds having the same number and kind of atoms and hence the same molecular weight, but differing with respect to the arrangement or configuration of the atoms. It will be understood, however, that some isomers or racemates or others mixtures of isomers may exhibit more activity than others. "Stereoisomers" refer to isomers that differ only
in the arrangement of the atoms in space. "Diastereoisomers" refer to stereoisomers that are not mirror images of each other. "Enantiomers" refers to stereoisomers that are non-superimposable miiTor images of one another.
In some embodiments, enantiomeric compounds taught herein may be "enantiomerically pure" isomers that comprise substantially a single enantiomer, for example, greater than or equal to 90%, 92%, 95%, 98%, or 99%, or equal to 100% of a single enantiomer.
In some embodiments, enantiomeric compounds taught herein may be stereomerically pure. "Stereomerically pure" as used herein means a compound or composition thereof that comprises one stereoisomer of a compound and is substantially free of other stereoisomers of that compound. For example, a stereomerically pure composition of a compound having one chiral center will be substantially free of the opposite enantiomer of the compound. A
stereomerically pure composition of a compound having two chiral centers will be substantially free of diastereomers, and substantially free of the opposite enantiomer, of the compound. A typical stereomerically pure compound comprises greater than about 80% by weight of one stereoisomer of the compound and less than about 20% by weight of the other stereoisomers of the compound, more preferably greater than about 90% by weight of one stereoisomer of the compound and less than about 10% by weight of the other stereoisomers of the compound, even more preferably greater than about 95% by weight of one stereoisomer of the compound and less than about 5% by weight of the other stereoisomers of the compound, and most preferably greater than about 97% by weight of one stereoisomer of the compound and less than about 3% by weight of the other stereoisomers of the compound. See, e.g., US Patent No. 7,189,715.
"R" and "S" as terms describing isomers are descriptors of the stereochemical configuration at an asymmetrically substituted carbon atom. The designation of an
asymmetrically substituted carbon atom as "R" or "S" is done by application of the Cahn-Ingold- Prelog priority rules, as are well known to those skilled in the art, and described in the
International Union of Pure and Applied Chemistry (IUPAC) Rules for the Nomenclature of Organic Chemistry. Section E, Stereochemistry.
"Enantiomeric excess" (ee) of an enantiomer is [(the mole fraction of the major enantiomer) minus (the mole fraction of the minor enantiomer)] x 100.
Compounds
Provided herein according to some embodiments is a compound of Formula I:

I
wherein:
X is H or CI;
Ri is selected from the group consisting of:
hydrogen;
Ci-6alkyl, which Ci_6alkyl may be unsubstituted or substituted 1-3 times with an independently selected substituent from the group consisting of: hydroxy, methoxy, C3-6cycloalkyl, and -NR3R4, wherein R3 and R4 are each independently hydrogen or methyl;
C1-6cycloalkyl; and
heterocyclyl, which heterocyclyl may be unsubstituted or substituted with Ci-6 alkyl;
R2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
In some embodiments, the compound is a compound of Formula 1(a):

wherein X, R1} R2, Y and Z are as defined above, or a pharmaceutically acceptable salt thereof.
Provided herein according to some embodiments is a compound of Formula II:

II
wherein
X is H or CI;
R3 is hydrogen or C]-6alkyl;
R2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
In some embodiments, the compound is a compound of Formula 11(a):

11(a)
wherein
X is H or CI;
R3 is hydrogen or C]-6alkyl;
R2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
Provided herein according to some embodiments is a compound of Formula III:

III wherein:
X is H or CI;
R3 is hydrogen or Ci-6alkyl;
R2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
In some embodiments, the compound is a compound of Formula 111(a):

111(a) wherein:
X is H or CI;
R3 is hydrogen or Ci-6alkyl;
R2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
Provided herein according to some embodiments is a compound of Formula IV:

IV
wherein:
X is H or CI;
Ri is selected from the group consisting of
hydrogen;
Ci-6alkyl, which Ci-6alkyl may be unsubstituted or substituted 1-3 times with an independently selected substituent from the group consisting of: hydroxy, methoxy, C3-6cycloalkyl, and -NRsR^ wherein R3 and R4 are each independently hydrogen or methyl; C1-6cycloalkyl; and
heterocyclyl, which heterocyclyl may be unsubstituted or substituted with Ci-6 alkyl;
R2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
In some embodiments, the compound is a compound of Formula IV(a):

IV(a)
wherein:
X is H or CI;
Ri is selected from the group consisting of
hydrogen;
Ci-6alkyl, which Ci-6alkyl may be unsubstituted or substituted 1-3 times with an independently selected substituent from the group consisting of: hydroxy, methoxy, C3-6cycloalkyl, and -NR3R4, wherein R3 and R4 are each independently hydrogen or methyl; C3-6cycloalkyl; and
heterocyclyl, which heterocyclyl may be unsubstituted or substituted with C1-6 alkyl;
2 is selected from the group consisting of methyl, ethyl, benzyl, and 2- pyridylmethyl; and
Y and Z are each independently fluoro or chloro,
or a pharmaceutically acceptable salt thereof.
Also provided is a compound selected from those presented in Table 1 below, or a pharmaceutically acceptable salt thereof.
C. Pharmaceutical formulations
Active compounds of the present invention can be combined with a pharmaceutically acceptable carrier to provide pharmaceutical formulations thereof. The particular choice of carrier and formulation will depend upon the particular route of administration for which the composition is intended.
"Pharmaceutically acceptable carrier" as used herein refers to a nontoxic carrier, adjuvant, or vehicle that does not destroy the pharmacological activity of the compound with which it is formulated. Pharmaceutically acceptable carriers, adjuvants or vehicles that may be used in the compositions of this invention include, but are not limited to, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene glycol and wool fat.
The compositions of the present invention may be suitable for parenteral, oral, inhalation spray, topical, rectal, nasal, buccal, vaginal or implanted reservoir administration, etc. The term "parenteral" as used herein includes subcutaneous, intravenous, intramuscular, intra-articular, intra-synovial, intrasternal, intrathecal, intrahepatic, intralesional and intracranial injection or infusion techniques. In particular embodiments, the compounds are administered intravenously, orally, subcutaneously, or via intramuscular administration. Sterile injectable forms of the compositions of this invention may be aqueous or oleaginous suspension. These suspensions may be formulated according to techniques known in the art using suitable dispersing or wetting agents and suspending agents. The sterile injectable preparation may also be a sterile injectable solution or suspension in a nontoxic parenterally acceptable diluent or solvent. Among the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic
sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium.
For this purpose, any bland fixed oil may be employed including synthetic mono- or di- glycerides. Fatty acids and their glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions. These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant, such as carboxymethyl cellulose or similar dispersing agents that are commonly used in the formulation of pharmaceutically acceptable dosage forms including emulsions and suspensions. Other commonly used surfactants, such as Tweens, Spans and other emulsifying agents that are commonly used in the manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms may also be used for the purposes of formulation.
For oral administration, the active compounds may be provided in an acceptable oral dosage form, including, but not limited to, capsules, tablets, aqueous suspensions or solutions. In the case of tablets for oral use, carriers commonly used include lactose and corn starch.
Lubricating agents, such as magnesium stearate, may also be added. For oral administration in a capsule form, useful diluents include lactose and dried cornstarch. When aqueous suspensions are required for oral use, the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening, flavoring or coloring agents may also be added. In addition preservatives may also be added. Suitable examples of pharmaceutically acceptable preservatives include, but are not limited to, various antibacterial and antifungal agents such as solvents, for example ethanol, propylene glycol, benzyl alcohol, chlorobutanol, quaternary ammonium salts, and parabens (such as methyl paraben, ethyl paraben, propyl paraben, etc.).
D. Subjects and methods of use
Compounds of the present invention may be used to treat hepatocellular carcinoma.
"Treatment," "treat," and "treating" refer to reversing, alleviating, delaying the onset of, inhibiting the progress of, or otherwise ameliorating a disease or disorder as described herein. In some embodiments, treatment may be administered after one or more symptoms have developed. In other embodiments, treatment may be administered in the absence of symptoms. For example, treatment may be administered to a susceptible individual prior to the onset of symptoms (e.g., in light of a history of symptoms and/or in light of genetic or other susceptibility factors).
Treatment may also be continued after symptoms have resolved, for example to prevent or delay their recurrence.
"Patient" or "subject", as used herein, means an animal subject, preferably a mammalian subject (e.g., dog, cat, horse, cow, sheep, goat, monkey, etc.), and particularly human subjects (including both male and female subjects, and including neonatal, infant, juvenile, adolescent, adult and geriatric subjects).
In some embodiments, treatment is provided to a subject having a cancer with altered FGFR4 and/or FGF19 (fibroblast growth factor 19) status, including hepatocellular carcinoma.
In some embodiments, treatment may include analyzing FGFR4 and/or FGF19 status in a biological sample containing cells of said cancer, and if said cancer exhibits an FGFR4 and/or FGF19 alteration, treating a subject with a treatment effective amount of a compound or composition as described herein.
"Altered status" as used herein with reference to FGFR4 and/or FGF19 includes an increased expression thereof (e.g., increased levels of the niRNA or increased levels of the protein), increased copy number in the genome, increased activity of the encoded protein as a result of mutation, etc., as compared to a corresponding non-cancerous tissue. Altered status of FGFR4 and/or FGF19 includes gene and/or encoded protein mutations that result in an increase in activity or are otherwise associated with a more aggressive form of cancer.
"Expression" of FGFR4 and/or FGF19 means that a gene encoding the same is transcribed, and preferably, translated. Typically, expression of a coding region will result in production of the encoded polypeptide.
The FGFR and FGF19 proteins are known, and their altered status and/or expression may be measured using techniques standard in the art, e.g., genomic analysis of mutations or copy number aberrations such as by nucleic acid amplification, sequencing analysis, and/or hybridization-based techniques, RNA expression analysis such as northern blot or qRT-PCR, western blot or other imniunoblot or immunoassay, fluorescent activated cell sorting (FACS), etc.
In order that the invention described herein may be more fully understood, the following examples are set forth. It should be understood that these examples are for illustrative purposes only and are not to be construed as limiting.
EXAMPLES
General:
Microwave heating was done using a Biotage Emrys Liberator or Initiator microwave. Column chiOmatography was carried out using an Isco Rf200d. Solvent removal was earned out
using either a Biichi rotary evaporator or a Genevac centrifugal evaporator. Preparative LC/MS was conducted using a Waters autopurifier and 19 x 100mm XTerra 5 micron MS CI 8 column under acidic mobile phase conditions. NMR spectra were recorded using a Varian 400MHz spectrometer.
When the term "inerted" is used to describe a reactor (e.g., a reaction vessel, flask, glass reactor, and the like) it is meant that the air in the reactor has been replaced with an essentially moisture-free or dry, inert gas (such as nitrogen, argon, and the like).
General methods and experimentals for preparing compounds of the present invention are set forth below. In certain cases, a particular compound is described by way of example.
However, it will be appreciated that in each case a series of compounds of the present invention were prepared in accordance with the schemes and experimentals described below.
Preparative HPLC Conditions for the Purification of Target Compounds
Chromatography Conditions:
Instrument: Waters 2767-SQD Mass trigger Prep System
Column : Waters Xbridge CI 8 150mm* 19mm* 5um
Detector: VWD SQD
Flow Rate : 15 mL/min
Gradient Time:
Time(min) B%
0 5
7.5 70
8 95
1 1 95
Representative Mobile Phase:
1)
Mobile Phase: A: 0.1%TFA in water
Mobile Phase: B: ACN
2)
Mobile Phase: A: 0.1%NH4HCO3 in water
Mobile Phase: B: ACN
3)
Mobile Phase: A: 0.1%NH OAc in water
Mobile Phase: B: ACN
4)
Mobile Phase: A: 0.1%NH4OH in water
Mobile Phase: B: ACN
The following abbreviations are used herein:
Definitions: The following abbreviations have the indicated meanings:
ACN: Acetonitrile
Boc20: Di-tert-butyl dicarbonate
Brettphos: 2-(Dicyclohexylphosphino)-3,6-dimethoxy-2',4',6'-triisopi pyl-l, -biphenyl tBuONa: Sodium tert-butoxide
CH3I: Iodomethane
Cs2C03: Cesium carbonate
DCC: N,N'-dicyclohexylcarbodiimide
DCM: Dichloromethane
DIPEA: N,N-diisopropylethylamine
DMAP: 4-(Dimethylamino)pyridine
DME: Dimethyl ether
DMF: Dimethylformamide
DPPA: Diphenylphosphoryl azide
EDCI.HCl: N-(3-Dimethylaminopropyl)-N' -ethylcarbodiimide hydrochloride
ESI-MS: Electrospray ionization - mass spectrometry
EtOH: Ethanol
HATU: 1 -[Bis(dimethylamino)methylene]- 1 H- 1 ,2,3 -trizolo [4,5-b]pyridinium 3 -oxid hexafluorophosphate
H2SO4: Sulfuric acid
HOBt: Hydroxybenzotriazole
iPrOH: Isopropanol
K2C03: Potassium carbonate
KHMDS: Potassium bis(trimethylsilyl)amide
KOH: Potassium hydroxide
LCMS: Liquid chromatography - mass spectrometry
MeOH: Methanol
MsCl: Methansulfonyl chloride
NaBH3CN: Sodium cyanoborohydride
NaBH(OAc)3: Sodium triacetoxyborohydride
NH4CI: Ammonium chloride
NH4HC03: Ammonium bicarbonate
Nal: Sodium iodide
NaN(¾: Sodium nitrate
NaOAc: Sodium acetate
nBuOH: n-Butanol
NMO: N-Methylmorpholine-N-oxide
NMP: N-Methyl-2-pyrrolidone
prep-HPLC: Preparative high-performance liquid chromatography
prep-TLC: Preparative thin layer chromatography
Selectfluor®: l-Chloromethyl-4-fluoro-l,4-diazoniabicyclo [2,2,2] octane bis(tetrafluoroborate)
SEMC1: 2-(trimethylsilyl)ethoxymethyl chloride
TBAF: Tetrabutylammonium fluoride
TBSC1: tert-Butyldimethylsilyl chloride
TBSOTf: tert-Butyldimethylsilyl trifluoromethanesulfonate
TEA: Triethylamine
TESC1: Chlorotriethylsilane
THF: Tetrahydrofuran
TLC: Thin-layer chromatography
PPTS: Pyridinium p-toluenesulfonate
PE: Petroleum ether
Pt02: platinum dioxide
EtOAc: Ethyl acetate
Pd/C: Palladium (0) on carbon
Pd2(dba)3: Tris(dibenzylideneacetone) dipalladium(O)
Pd(dppf)2Cl2: [1,1 '-Bis(diphenylphosphino)feiTOcene]dichloropalladium(II)
Ti(0'Pr)4: Titanium isopropoxide
Xantphos: 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene
Materials: The following compounds are commercially available and/or can be prepared in a number of ways well known to one skilled in the art of organic synthesis. More specifically, disclosed compounds can be prepared using the reactions and techniques described herein. In the description of the synthetic methods described below, it is to be understood that all proposed reaction conditions, including choice of solvent, reaction atmosphere, reaction temperature, duration of the experiment, and workup procedures, can be chosen to be the conditions standard for that reaction, unless otherwise indicated. It is understood by one skilled in the art of organic synthesis that the functionality present on various portions of the molecule should be compatible with the reagents and reactions proposed. Substituents not compatible with the reaction
conditions will be apparent to one skilled in the art, and alternate methods are therefore indicated. The starting materials for the examples are either commercially available or are readily prepared by standard methods from known materials.
SYNTHESIS OF EXAMPLE COMPOUNDS
Compounds of Table 1 were prepared by the procedure of Scheme I and modifications thereof described below.






Procedure 6A: Example - 601

N-(3-{6-[3-(2,6-DICHLORO-3,5-DIMETHOXY-PHENYL)-l-METHYL-UREIDO]- PYRIMIDIN-4-YLAMINO}-l-METHYL-lH-PYRAZOL-4-YL)-ACRYL AMIDE
Preparation of l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimetho
((2- (trimethylsilyl) eth oxy) methyl) urea

To a solution of 3,5-dimethoxy-phenylamine (20 g, 0.131 mol) in toluene (110 mL) was added acetic anhydride (14 g, 0.137 mmol) at room temperature. The resulting mixture was stirred for 18 hours at room temperature. PE (55 mL) was added, the precipitate was filtered and washed with PE (100 mL) to obtain the title compound (24.2 g, yield: 95%). 1H-NMR (400 MHz,
CDCI3) δ 2.16 (s, 3H), 3.77 23 (s, 1H), 6.75 (s, 2H), 7.20 (s, 1H).

b. N-(2,6-Dichloro-3,5-dimethoxy-phenyl)-acetamide
To a solution of N-(3,5-dimethoxy-phenyl)-acetamide (5 g, 25.6 mmol) in ACN (75 mL) was added sulfuryl chloride (6.9 g, 51.2 mmol) at 0 °C under nitrogen atmosphere. The resulting mixture was stirred for 30 minutes at this temperature and quenched with saturated aqueous NaHC03 (40 mL). The precipitate was filtered, washed with water and dried to obtain the title compound (2.3 g, yield: 34%). ]H-NMR (400 MHz, CDC13) δ 2.25 (s, 3H), 3.86 (s, 6H), 6.54 (s, 1H), 6.90 (s, 1H).

c. 2,6-Dichloro-3,5-dimethoxy-phenylamine
A solution of N-(2,6-dichloro-3,5-dimethoxy-phenyl)-acetamide (3.6 g, 13.7 mmol) in EtOH (130 mL) and KOH (2M, 75 mL) was heated to reflux for 24 hours. The reaction was cooled to 0 °C and stirred for 1 hour at this temperature. The precipitate was filtered and dried to obtain the title compound (2.3 g, yield: 76%). 1H-NMR (400 MHz, CDC13) δ 3.90 (s, 6H), 4.57 (bs, 2H), 6.05 (s, 1H).

d. 2,4-Dichloro-3-isocyanato-l,5~dimethoxy-benzene
A mixture of 2,6-dichloro- 3,5-dimethoxy-phenylamine (500 mg, 2.25 mmol), triphosgene (335 mg, 1.12 mmol) and TEA (342 mg, 3.38 mmol) in dioxane (15 mL) was heated to 130 °C for 2 hours under microwave. The reaction was concentrated and the residue was purified by flash
chromatography on silica eluting with DCM to obtain the title compound (450 mg, yield: 80%). 1H-NMR (400 MHz, CDC13) δ 3.92 (s, 6H), 6.42 (s, 1H).

e. ( 6-Chloro-pyrimidin-4-yl)-methyl-amine
To a solution of 4,6-dichloro-pyrimidine (7.45 g, 50 mmol) in iPrOH (50 mL) was added a solution of methyl amine in THF (2M, 30 mL, 60 mmol) at room temperature. The resulting mixture was stirred for 18 hours. The mixture was concentrated and the residue was purified by flash chromatography on silica eluting with DCM:EtOAc = 6: 1—1 : 1 to obtain the title compound
(4.4 g, yield: 62%) as a white solid. 1H-NMR (400 MHz, CDC13) δ 2.96 (d, 3H), 5.22-5.36 (bs, 1H), 6.35 (s, 1H), 8.35 s, 1H); MS (ESI): 144 [M+H]+.

l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-m
(trimethylsilyl) eth oxy) methyl) urea
To a solution of 6-chloro-N-methylpyrimidin-4-amine (Procedure 2A , step e; 460 mg, 3.21 mmol) in DMF (15 mL) was added NaH (60%, 193 mg, 4.81 mmol) at 0 °C, and the mixture was stirred for 30 minutes at room temperature. A solution of 2,4-dichloro-3-isocyanato- 1,5- dimethoxy-benzene (Procedure 2A , steps a-d; 1.03 g, 4.17 mmol) in DMF (5 mL) was added dropwise at room temperature. The resulting mixture was stirred for 0.5 hour. SEMC1 (804 mg, 4.81 mmol) in DMF (2 mL) was added. The reaction mixture was stirred at room temperature for 1 hour. Saturated aqueous NH4C1 was added to quench the reaction. The mixture was diluted with water and extracted with EtOAc. The combined extracts were washed with water and brine, dried over anhydrous Na2S04 and filtered. The filtrate was evaporated under vacuum to give crude product, which was purified by flash chromatography on silica to obtain the title compound (470 mg, yield: 28%). MS (ESI): 521 [M+H]+.
Preparation of l-methyl-4-nitro-lH-pyrazol-3-amine

g. N-(l-Methyl-lH-pyrazol-3-yl)-acetamide HCI salt
To a solution of 1 -methyl- lH-pyrazol-3-ylamine (5 g, 50 mmol) in THF (50 mL), was added dropwise acetyl chloride (7 mL, 100 mmol) at 0°C. The reaction mixture was stirred at room temperature for 2 hours. The resulting solid was filtered and washed with THF twice, dried under vacuum to afford the title compound (9 g, quant.).

h. l-Methyl-4-nitro-lH-pyrazol-3-ylamine
To a cooled (0°C) solution of N-(l -methyl- lH-pyrazol-3-yl)-acetamide HCI salt (3.5 g, 20 mmol) in con. H2S04 (11 mL), was added fuming HN03 (1.65 g, 28 mmol) in 2 min. The solution was stirred at 0°C for 2 hours and poured into ice-water. The reaction mixture was stirred overnight, then neutralized with 4 N NaOH and extracted with EtOAc twice. The combined extracts were washed with water and brine, dried over anhydrous sodium sulfate, and concentrated to give a crude product, which was purified by flash chromatography on silica to obtain the title product (970 mg, yield: 30%). MS (ESI): 143 [M+H]+

/. l-(2,6-Dichloro-3,5-dimethoxy-phenyl)-3-methyl-3-[6-(l-methyl-4-nitro^
ylamino)-pyrimidin-4-yl]A-(2-tnmethylsilanyl-ethoxym A degassed mixture of 1- methyl-4-nitro-lH-pyrazol-3-ylamine (200 mg, 1.4 mmol), l-(6-chloro-pyrimidin-4-yl)-3-(2,6- dichloro-3 ,5-dimethoxy-phenyl)- 1 -pyridin-2-ylmethyl-3 -(2-trimethylsilanyl-ethoxymethyl)-urea (886 mg, 1.7 mmol), Pd2(dba)3 (162 mg, 0.17 mmol), Xantphos (196 mg, 0.34 mmol) and
Cs2C03 (913 mg, 2.8 mmol) in toluene (10 mL) was heated at 100°C for 4 hours. The reaction was concentrated, and the residue was purified by flash chromatography on silica to obtain the title compound 310 mg, yield: 35%) as a red solid. MS (ESI): 627 [M+H]+.

j. {6-[3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-l-methyl-3-(2-trimethylsU^
ureido]-pyrimidin-4-yl}-(l-methyl-4-nitro-lH-pyrazol-3-yl)-carbam acid tert-butyl ester A mixture of 1 -(2,6-dichloro-3,5-dimethoxy-phenyl)-3-methyl-3-[6-(l-methyl-4-nitro-lH-pyrazol- 3-ylamino)-pyrimidin-4-yl]-l-(2-trimethylsilanyl-ethoxymethyl)-urea (310 mg, 0.5 mmol), (Boc)20 (130 mg, 0.6 mmol) and catalytic amount of DMAP in THF (10 mL) was heated under reflux for 1 hour. The mixture was concentrated and the residue was used for the next step without further urification. MS (ESI): 727 [M+H]+

k. (4-Amino-l-methyl-lH-pyrazol-3-yl)-{6-[3-(2,6-dichloro-3,5-dimethoxy-phen^
(2-trimethylsilanyl-ethoxymethyl)-ureido]-pyrimidin-4-yl}-carba acid tert-butyl ester.
To a solution of {6-[3-(2,6-dichloro-3,5-dimethoxy-phenyl)-l-methyl-3-(2-trimethylsilanyl- ethoxymethyl)-ureido]-pyrimidin-4-yl}-(l-methyl-4-nitro-lH-pyrazol-3-yl)-carbamic acid tert- butyl ester (crude, prepared above) in THF (10 mL) and MeOH (5 mL) was added Pt02 (50 mg) at room temperature, the resulting mixture was stirred under hydrogen atmosphere (1 atm) overnight. The reaction was filtered and concentrated. The residue was purified by flash chromatography on silica to obtain the title compound (188 mg, yield: 55% in two steps). MS (ESI): 697 [M+H]+.

/. (4-Acryloylamino-l-methyl-lH-pyrazol-3-yl)-{6-[3-(2,6-dichloro-3,5-d
methyl-3-(2-trimethylsilanyl-ethoxymethyl)~ureido]-pyrimidin^ acid tert-butyl ester.
To a solution of (4-amino-l -methyl- lH-pyrazol-3-yl^
phenyl)-l-methyl-3-(2-trimethylsilanyl-ethoxymethyl)-ureido]-pyrimidin-4-yl}-carbamic acid tert-butyl ester (188 mg, 0.27 mmol) in DCM (10 mL) was added a solution of TEA (10 mg/mL, 3 mL, 0.3 mmol) and a solution of acryloyl chloride (10 mg/mL, 2.7 mL, 0.3 mmol) in DCM dropwise at 0 °C, and the resulting mixture was stirred at room temperature for 1 hour. ESI-MS showed that the reaction was complete. Water (5 mL) was added to quench the reaction, and the reaction mixture was extracted with DCM. The combined extracts were washed with brine, dried over anhydrous sodium sulfate and concentrated to give a crude product, which was purified by flash chromatography on silica to obtain the title compound (145 mg, yield: 72%). MS (ESI): 751 [M+H]+.

m. N-(3-{6-[3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-l-methyl-urete^
methyl-lH-pyrazol-4-yl)-acrylamide.
To a solution of (4-acryloylamino-l-methyl-lH-pyrazol-3-yl)-{6-[3-(2,6-dichloro-3,5- dimethoxy-phenyl)- 1 -methyl-3-(2-trimethylsilanyl-ethoxymethyl)-ureido]-pyrimidin-4-yl} - carbamic acid tert-butyl ester (145 mg, 0.193 mmol) in DCM (15 mL) was added TFA (5 mL) at 0 °C, the resulting mixture was stirred for 3 hours at room temperature. After removal of all volatiles in vacuo, the residue was re-dissolved in DCM, neutralized with ammonia hydroxide to
give the crude compound, which was purified by Prep-HPLC to obtain title compound (32 mg, yield: 32%).1H NMR (300 MHz, DMSO-d6) δ 12.07 (s, 1H), 9.82 (s, 1H), 9.48 (s, 1H), 8.42 (s, 1H), 8.08 (s, 1H), 7.05 (s, 1H), 6.90 (s, 1H), 6.40 (dd, 1H), 6.21 (d, 1H), 5.71 (d, 1H), 3.93 (s, 6H), 3.76 (s, 3H), 3.31 (s, 3H); MS (ESI): 521 [M+H]+
Example 602

Preparation of N-(5-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidin- 4-yl)amino)-l-methyl-lH-pyrazol-4-yl)acrylamide TFA salt
The compound was synthesized following the approach outlined in Procedure 6 A substituting 1- methyl-4-nitro-lH-pyrazol-5-amine (Procedure shown below) in step (i) to afford the title compound (36 mg, yield: 38% 4 steps) 1H-NMR (400 MHz, DMSO-d6) δ 3.60 (s, 3 H) 3.94 (s, 6 H) 5.62 - 5.71 (m, 1 H) 6.14 - 6.19 (m, 2 H) 6.29 - 6.52 (m, 1 H) 6.90 (s, 1 H) 7.80 (d, 1 H) 8.39 (s, 1 H) 9.21 (s, 1 H) 9.56 (s, 1 H) 11.85 (s, 1 H); ESI-MS: 521 [M+H] +
Preparation of l-methyI-4-nitro-lH-pyrazol-5-amine
yield: 40%
a. ethyl l-methyl-4-nitro-lH-pyrazole-5-carboxylate
Ethyl 4-nitro-lH-pyrazole-3-carboxylate (1.5 g, 8.10 mmol) and K2C03 (2.2 g, 16.20 mmol) were stirred in DMF (15.0 ml). To the mixture was added iodomethane (1.201 g, 8.51 mmol). The reaction mixture was stirred at room temperature for 12 hours. The reaction mixture was poured into EtO Ac/brine. The organic layer was washed with brine three times, dried over MgS04 and evaporated. The resulting material was purified by flash chromatography on silica eluting with 5% to 80% EtOAc/Hexane to afford the title compound (non-polar fractions) (638 vmg, yield: 40%). The polar fractions afforded ethyl l-methyl-4-nitro-lH-pyrazole-3- carboxylate. The regioisomers were assigned based on the reported NMR shift
(WO2012073143(Al)). 1H-NMR (400 MHz, CDC13) δ 1.42 (t, 3 H) 1.55 (s, 1 H) 4.01 (s, 3 H) 4.47 (q, 2 H) 8.13 (s, 1 H)

yield: 82%
b. l-methyl-4-nitro-lH-pyrazole-5-carboxylic acid
Ethyl l-methyl-4-nitro-lH-pyrazole-5-carboxylate (638 mg, 3.20 mmol) was stirred in THF (8.0 ml) and Methanol (4.0 ml). 1M LiOH aq (4.8 ml, 4.801 mmol) was added and the reaction mixture was stirred at room temperature for 12 hours. The solvent was evaporated. The remaining material was acidified with IN HC1 and extracted with EtOAc. The aqueous layer was extracted with EtOAc again and the combined org layer was dried over MgS04 and evaporated to afford the title com ound (451 mg, yield: 82 %). ESI-MS: 171 [M+H]+

tBuOH
100°C, 12h
yield: 25%
c. tert-butyl (l-methyl-4-nitro-lH-pyrazol-5-yl)carbamate
l-methyl-4-mtro-lH-pyrazole-5-carboxylic acid (370 mg, 2.16 mmol), Diphenylphosphoryl azide (490 μΐ, 2.27 mmol) and TEA (316 μΐ, 2.27 mmol) were stirred in tert-butanol (12ml) under N2. The reaction mixture was stirred at room temperature for 30 minutes and then heated at 100 °C for 12 hours. Solvent was evaporated and the remaining material was purified by flash chromatography on silica eluting with 0% to 30% EtOAc/Hexane to afford the title compound
(130 mg, yield: 25 %). H-NMR (400 MHz, CDC13) δ 1.55 (s, 9 H) 3.86 (s, 3 H) 7.66 (brs., 1 H) 8.03 (s, 1 H)

yield: 97%
d. l-methyl-4-nitro-lH-pyrazol-5-amine
tert-butyl (l-methyl-4-nitro-lH-pyrazol-5-yl)carbamate (130 mg, 0.537 mmol) was stirred in DCM (3.0 ml). TFA (2.0 ml, 25.96 mmol) was added and stirred at room temperature for 1 hour.
Solvent was evaporated and remaining material was dried under high vacuum to afford the title compound (74 mg, yield: 97 %). ESI-MS: 143 [M+H]+
Example 603

Preparation of N-(4-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidin- 4-yl)amino)-l-methyl-lH-pyrazol-3-yl)acrylamide 2,2,2-trifluoroacetate
The compound was synthesized following the approach outlined in Procedure 6A substituting 1 - methyl-3-nitro-lH-pyrazol-4-amine 2,2,2-trifluoroacetate (procedure shown below) and sodium tert-butoxide in step (i) to afford the title compound (21 mg, yield: 10% 5 steps) 'H-NMR (400 MHz, MeOH-d4) δ 3.41 (s, 3 H) 3.86 (s, 3 H) 3.95 (s, 6 H) 5.80 - 5.83 (m, 1 H) 6.37 - 6.51 (m, 3 H) 6.81 (s, 1 H) 8.00 - 8.07 (m, 1 H) 8.39 (s, 1 H); ESI-MS: 521 [M+H] +

90°C, 12h yield: 96%
yield: 38%
Preparation of l-methyl-3-nitro-lH-pyrazol-4-amine 2,2,2-trifluoroacetate
The compound was synthesized following the approach outlined in Example 602, substituting ethyl 3-nitro-lH-pyrazole-4-carboxylate to afford the title compound (181 mg, yield: 24% 4 steps) ESI-MS: 143 [M+H]+
Example 604

Preparation of N-(4-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methyIureido)pyrimidin- 4-yl)amino)-lH-pyrazoI-5-yl)acryIamide 2,2,2-trifIuoroacetate
The compound was synthesized following the approach outlined in Procedure 6 A substituting 3- nitro-l-((2-(trimethylsilyl)ethoxy)methyl)-lH-pyrazol-4-amine (procedure shown below) in step (i) to afford the title compound (19 mg, yield: 41% 5 steps) H-NMR (400 MHz, MeOH-d4) δ 3.42 (s, 3 H) 3.94 (s, 6 H) 5.82 (dd, 1 H) 6.34 - 6.53 (m, 3 H) 6.81 (s, 1 H) 8.01 (br. s., 1 H) 8.40 (s, 1 H); ESI-MS: 507 [M+H] +
Preparation of 3-nitro-l-((2-(trimethylsiIyl)ethoxy)methyl)-lH-pyrazol-4-

yield: 52%
a. ethyl 3-nitro-l-((2-(trimethylsilyl)ethoxy)methyl)-lH-pyrazole-4-carboxylate
Ethyl 3-nitro-lH-pyrazole-4-carboxylate (1.0 g, 5.40 mmol) was stirred in DMF (5.0 ml) under nitrogen over ice bath. NaH (60% dispersion in oil, 0.324 g, 8.10 mmol) was added and the mixture was stirred over ice bath for 60 minutes. A solution of (2-
(chloromethoxy)ethyl)trimethylsilane (0.964 g, 5.78 mmol) in DMF (1 ml) was added and the mixture was stirred at room temperature for 1 hour. The reaction was quenched by addition of saturated NH4C1 and extracted with EtOAc. The org. layer was washed with brine, dried over MgS04 and evaporated. The remaining material was purified by flash chromatography on silica eluting with 0% to 70% EtOAc/Hexane to afford the title compound (polar fractions) (890 mg, yield: 52%>). The non-polar fractions afforded ethyl 5-nitro-l-((2-(trimethylsilyl)ethoxy)methyl)- lH-pyrazole-4-carboxylate. ^-NMR (400 MHz, CDC13) δ 0.02 (s, 9 H) 0.91 - 0.99 (m, 2 H) 3.61 - 3.70 (m, 2 H) 5.47 (s, 2 H) 8.07 (s, 1 H)

b. 3-nitro-l-((2-(trimethylsilyl)ethoxy)methyl)-lH-pyrazole-4-carboxylic acid
Ethyl 3-nitiO-l-((2-(trimethylsilyl)ethoxy)methyl)-lH-pyrazole-4-carboxylate (890 mg, 2.82 mmol) was stirred in THF (10.0 ml) and Methanol (5.0 ml). 1M LiOH aq (4.5 ml, 4.50 mmol) was added and the reaction mixture was stirred at room temperature for 2 hours. The solvent was evaporated. The remaining material was acidified with IN HC1 and extracted with EtOAc. The aqueous layer was extracted with EtOAc again and the combined org layer was dried over MgS04 and evaporated to afford the title compound (673 mg, yield: 83 %). ESI-MS: 286 [M-H]~

80°C, 12h
yield: 60%
c. ethyl (3-nitro-l-((2-(trimethylsilyl)ethoxy)methyl)-lH-pyrazol-4-yl)carbamate
3-nitro-l-((2-(trimethylsilyl)ethoxy)methyl)-lH-pyrazole-4-carboxylic acid (646 mg, 2.25 mmol), Diphenylphosphoryl azide (510 μΐ, 2.36 mmol) and TEA (329 μΐ, 2.36 mmol) were stirred in EtOH (5.0 ml) and 1,4-dioxane (5.0 ml) under N2. The reaction mixture was stirred at room temperature for 1 hour and then heated at 80 °C for 12 hours. Solvent was evaporated and the remaining material was purified by flash chromatography on silica eluting with 0% to 30% EtOAc/Hexane to afford the title compound (449 mg, yield: 60 %). 1H-NMR (400 MHz, CDC13) δ 0.01 (s, 9 H) 0.90 - 0.98 (m, 2 H) 1.35 (t, 3 H) 3.58 - 3.64 (m, 2 H) 4.28 (d, 2 H) 5.45 (s, 2 H) 8.18 (br. s., 1 H) 8

d. 3-nitro-l-((2-(trimethylsilyl)ethoxy)methyl)-lH-pyrazol-4-amine
Ethyl (3-nitro-l-((2-(trimethylsilyl)ethoxy)methyl)-lH-pyrazol-4-yl)carbamate (449 mg, 1.34 mmol) was dissolved in EtOH (10.0 ml) and 3M NaOH aq (4.5 ml, 13.50 mmol) was added. The reaction mixture was then heated at 120 °C using microwave (Biotage Initiator) for 10 minutes. The reaction mixture was poured into EtO Ac/Water. The organic layer was washed with brine, dried over MgS04 and evaporated to give afford the title compound (295 mg, yield: 84 %>) which was used in the next reaction without further purification. ESI-MS: 259 [M+H]+
Example 605

N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidin-4-yl)amino)-l- (2~methoxyethyl)-lH-pyrazoI-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A by substituting l-(2-Methoxyethyl)-4-nitro-lH-pyrazol-3 -amine (procedure shown below) in step (i) to afford the title compound (80 mg, yield: 3.3%). 1H NMR (DMSO-d6, 400MHz): δ 12.4 (s, 1H), 9.2 (brs, 1H), 8.43 (s, 1H), 8.03 (s, 1H), 7.61 (s, 1H), 6.52 (s, 2H), 6.38 (d, 1H), 6.23-6.17 (m, 1H), 5.73 (d, 1H), 4.19-4.17 (m, 2H), 3.92 (s, 6H), 3.75-3.72 (m, 2H), 3.42 (s, 3H), 3.34 (s, 3H); ESI- MS: m/z = 565.1 (M+H)+; HPLC: 98.68%, rt: 4.52 min.
Preparation of l-(2-Methox ethyl)-4-nitro-lH-pyrazol-3-amine

a. Methyl l-(2-methoxyethyl)-4-nitro-lH-pyrazole-3-carboxylate
l-Bromo-2-methoxyethane (4.3g, 30.9mmol) was slowly added to a stirred solution of methyl 4- nitro-lH-pyrazole-3-carboxylate (6g, 28.9 mmol) in DMF (120mL) at 0 °C. The reaction mass was stirred at room temperature for 18h. Quenched the reaction mass with ice cold water and extracted with ethyl acetate (3x25mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to obtain crude residue. The residue was purified by silica gel column chromatography to afford the desired title compound (4g, 60%).
1HNMR (CDCI3, 300MHz): δ 8.25 (s, 1H), 4.35-4.32 (m, 2H), 3.98 (s, 3H), 3.75-3.71 (m,
3.35 (s, 3H).

b. l-(2-Methoxyethyl)-4-nitro-lH-pyrazole-3-carboxylic acid
Lithium hydroxide (2.5g, 591 mmol) was added to a stirred solution of methyl l-(2- methoxyethyl)-4-nitro-lH-pyrazole-3-carboxylate (4g, 17.4 mmol) in a mixture of 40:60 of methanohTHF (50mL) at room temperature and stirred the reaction mass for 4h. Concentrated the reaction mass under vacuum and diluted with ice water, acidified with 2N HC1 to pH=2. Extracted with ethyl acetate (3x50mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude title compound (3.5g, 94%). ESI-MS: m/z = 216.1 (M+H)+.

c. tert-Butyl (l-(2-methoxyethyl)-4-nitro-lH-pyrazol-3-yl)carbamate
Added TEA (2.5g, 25 mmol) at 0 °C to a solution of l-(2-methoxyethyl)-4-nitro-lH-pyrazole-3- carboxylic acid (3.5g, 16.4 mmol) in a mixture of DMF (lOmL) and tert-butanol (20mL) followed by the addition of DPP A (5g, 18mrnol). Heated the reaction mass to 80 °C slowly and maintained for 2h. Concentrated the reaction mass under vacuum and diluted with ethyl acetate (lOOmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by silica gel column chromatography by eluting with 5% methanol-DCM to afford title compound (3.15g, 68%). 1HNMR (CDCI3, 300MHz): δ 8.41(s, 1H), 8.16 (s, 1H), 4.28-4.25 (m, 2H), 3.73-3.70 (m, 2H),
3.34 (s, 3H), 1.53 (s, 9H).

d. l-(2-Methoxyethyl)-4-nitro-lH-pyrazol-3-amine
TFA (5mL, 65 mmol) was added drop wise to a solution of tert-butyl (l-(2-methoxyethyl)-4- nitro-lH-pyrazol-3-yl)carbamate (3.15g, 11 mmol) in DCM (15mL) at 0 °C under nitrogen atmosphere. Stirred the reaction mass at room temperature for 4h. Concentrated the reaction
mass under vacuum and triturated with diethyl ether. Solid obtained dried under vacuum to yield the title compound (2g, 60.6%). ESI-MS: mix = 187.1 (M+H)+.
Example 606

N-(3-((6-(3-(2,6-dichIoro-3,5-dimethoxyphenyl)-l-methyIureido)pyrimidin-4-yl)amino)-l- (2-hydroxyethyl)-lH-pyrazol-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A by substituting l-(2-((tert-butyldimethylsilyl)oxy)ethyl)-4-nitro-lH-pyrazol-3 -amine (procedure shown below) in step (i) to afford the title compound (93 mg, yield: 5.9%). 1H NMR (DMSO-d6, 400MHz): δ 12.18 (s, 1H), 9.86 (s, 1H), 9.49 (s, 1H), 8.42 (s, 1H), 8.10 (s, 1H), 7.12 (brs, 1H), 6.90 (s, 1H), 6.44-6.37 (m, 1H), 6.22 (dd, 1H), 5.72 (d, 1H), 4.89 (t, 1H), 4.07 (t, 2H), 3.93 (s, 6H), 3.75-3.71 (m, 2H), 3.32 (s, 3H); ESI-MS: mlz = 551.1 (M+H)+; HPLC: 99.18%, rt: 3.82 min.
Preparation of l- 2-( (tert-butyldimethylsilyl)oxy)ethyl)-4-nitro-lH-pyrazol-3-amine

a. methyl l-(2-((tert-butyldimethylsilyl)oxy)ethyl)-4-nitro-lH-pyrazole-3-carboxylate
(2-Bromoethoxy)(tert-butyl)dimethylsilane (7.7g, 32 mmol) was slowly added to a stirred solution of methyl 4-nitro-lH-pyrazole-3-carboxylate, (6.0g, 28.9 mmol) in DMF (70mL) at 0 °C. The reaction mass was stirred at room temperature for 18hr. Quenched the reaction mass with ice cold water and diluted with ethyl acetate. The aqueous layer was separated and extracted with ethyl acetate (3x5 OmL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum. The residue was purified by silica gel column chromatography afforded title compound (Polar spot in TLC) (4.3g, 47%). ESI-MS: m/z = 330.2 (M+H)+.

b. l-(2-((tert-butyldimethyIsilyl)oxy)ethyl)-4-nitro-lH-pyrazole-3-carboxy acid
Lithium hydroxide (1.47g, 35 mmol) was added to a stirred solution of methyl l-(2-((tert- butyldimethylsilyl)oxy)ethyl)-4-nitro-lH-pyrazole-3-carboxylate (5.5g, 17.5 mmol) in a mixture of 60:40 methanol :THF (80mL) at room temperature and stirred the reaction mass for 4h. Concentrated the reaction mass under vacuum and diluted with ice water, acidified with 2N HC1 to pH=2. Extracted with ethyl acetate (3x50mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude title compound (4.1g, 78%). ESI-MS: m/z = 316.0 (M+H)+.

c tert-butyl (l-(2-((teH-butyldimethylsilyl)oxy)ethyl)-4-nitro-lH-py zol-3-yl)carbamate
Added TEA (2.5g, 25mmol) at 0 °C to a solution of l-(2-((tert-butyldimethylsilyl)oxy)ethyl)-4- nitro-lH-pyrazole-3-carboxylic acid (4g, 12.6 mmol) in a mixture of 20mL DMF and 80mL tert- butanol followed by the addition of DPP A (4g, 14.5mmol). Heated the reaction mass to 80 °C slowly and maintained for 5h. Concentrated the reaction mass under vacuum and diluted with ethyl acetate (lOOmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by silica gel column chromatography to afford title compound (2.3g, 47%). ESI-MS: m/z = 287.3 (M- Boc+H)+.

d. l-(2-((tert-butyldimethylsilyl)oxy)ethyl)-4-nitro-lH-pymzol-3-amine. TFA salt
TFA (4mL, 3.54 mmol) was added drop wise to a solution of tert-butyl (l-(2-((tert- butyldimethylsilyl)oxy)ethyl)-4-nitro-lH-pyrazol-3-yl)carbamate (2.3g, 0.59 mmol) in DCM (20mL) at 0 °C under nitrogen atmosphere. Stirred the reaction mass at room temperature for 2h. Concentrated the reaction mass under vacuum and triturated with diethyl ether. Solid obtained dried under vacuum to yield the title compound (1.35g, 79%). ESI-MS: m/z = 287.1 (M+H)+.
Example 607

Preparation of (R)-N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureido) pyrimidin-4- - - - l - - z l-4- lami

4-nitro-lH-pyrazol-3-yl)amino)pyrimidin-4-yl)-3-methyl-l-((2- (trimethylsttyl)ethoxy) methyl) urea
The compound was synthesized following the approach outlined in Procedure 6A step (i) substituting (R)- 1 -((2,2-dimethyl- 1 ,3 -dioxolan-4-yl)methyl)-3 -nitro- 1 H-pyrazol-4-amine (Procedure shown below) and heating for 12 hours to afford the title compound (206 mg, yield: 91% -MS: 727 [M+H] +

b. (R)-l-( 6-( (4-amino-l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-lH-pyrazol-3- yl)amino)pyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-me
(trimethyIsilyl)ethoxy)methyl)urea
The compound was synthesized following the approach outlined in Procedure 6A step (k) substituting (R)-l-(2,6-dichloro-3,5-dimethoxyphenyl)-3-(6-((l-((2,2-dimethyl-l,3-dioxolan-4- yl)methyl)-4-nitro-lH-pyrazol-3-yl)amino)pyrimidin-4-yl)-3-methyl-l-((2- (trimethylsilyl)ethoxy)methyl)urea and purification by flash chromatography on silica eluting with 0% to 10% MeOH/DCM to afford the title compound (23 mg, yield: 52%). ESI-MS: 697 [M+H] +

c. (R)-N-(3-((6-(3-(2,6-dichloro-3,S-dimethoxyphenyl)-l-methyl-3-((2-
(trimethylsilyl)ethoxy)methyl)ureido)pyrimidin-4-yl)amino)-l-((2,2-dim
yl)methyl)-lH-pyrazol-4-yl)acrylamide
(R)-l-(6-((4-amino-l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-lH-pyrazol-3- yl)amino)pyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methyl-3-((2- (trimethylsilyl)ethoxy)methyl)urea (23 mg, 0.033 mmol) was dissolved in DCM (1.5 ml) and stirred under nitrogen. Acrylic acid (27 μΐ, 0.039 mmol) (as 10% solution in DCM) and DIEA (12 μΐ, 0.069 mmol) were added followed by 1-Propanephosphonic acid cyclic anhydride (29 μΐ, 0.049 mmol). The reaction mixture was stirred at rt for 6 hours. The reaction mixture was charged directly onto a column and eluted at gradient 0% to 10%o MeOH in DCM to afford the title -MS: 751 [M+H] +

d. (R)-N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureid
l-(2,3-dihydroxypropyl)-lH-pyrazol-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A step (n) substituting (R)-N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methyl-3-((2- (trimethylsilyl)ethoxy)methyl)ureido)pyrimidin-4-yl)amino)-l-((2,2-dimethyl-l,3-dioxolan-4-
yl)methyl)-lH-pyrazol-4-yl)acrylamide and purification by flash chromatography on silica eluting with 0% to 10% MeOH/DCM to afford the title compound (18 mg, yield: 63%). !H- NMR (400 MHz, DMSO-d6) δ 3.29 (br. s., 2 H) 3.32 - 3.40 (m, 4 H) 3.75 - 3.87 (m, 2 H) 3.89 - 3.99 (m, 15 H) 4.14 (dd, 3 H) 4.63 - 4.75 (m, 1 H) 4.96 (d, 1 H) 5.69 - 5.75 (m, 2 H) 6.19 (d, 1 H) 6.24 (d, 1 H) 6.36 - 6.45 (m, 2 H) 6.90 (s, 2 H) 7.05 - 7.22 (m, 2 H) 8.08 (s, 2 H) 8.42 (s, 2 H) 9.48 (br. s., 1 H) 9.87 (br. s., 1 H) 12.20 (s, 1 H); ESI-MS: 581 [M+H] +
Preparation of (R)-l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-3-nitro-lH-pyrazol-4-amine

a. (R)-ethyl l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-4-nitro-lH-pyrazole-3-carboxylate
Ethyl 4-nitro-lH-pyrazole-3-carboxylate (620 mg, 3.349 mmol), (S)-(2,2-dimethyl-l,3-dioxolan- 4-yl)methyl 4-methylbenzenesulfonate (1055 mg, 3.684 mmol) and K2C03 (926 mg, 6.698 mmol) were stirred in DMF (5.0 ml). The reaction mixture was stirred at 80 °C for 12 hours. The reaction mixture was poured into EtO Ac/brine. The organic layer was washed with brine three times, dried over MgS04 and evaporated. The remaining material was purified by flash chromatography on silica eluting with 0% to 40% EtOAc/Hexane to afford the title compound (polar fractions) (503 mg, yield: 52%). The non-polar fractions afforded (R)-ethyl l-((2,2- dimethyl- 1 ,3 -dioxolan-4-yl)methyl)-4-nitro- 1 H-pyrazole-5-carboxylate. 1H-NMR (400 MHz, CDC13) δ 1.35 - 1.45 (m, 9 H) 3.76 (dd, 1 H) 4.10 - 4.27 (m, 2 H) 4.33 - 4.41 (m, 1 H) 4.44 - 4.52 (m, 3 H) 8.28 (s, 1 H)

b. (R)-l-( (2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-4-nitro-lH-pyrazole-3-carboxylic acid
The compound was synthesized following the approach outlined in Example 602, step (b) substituting (R)-ethyl 1 -((2,2-dimethyl- 1 ,3 -dioxolan-4-yl)methyl)-4-nitro- 1 H-pyrazole-3 - carboxylate to afford the title compound (328 mg, yield: 72%). ESI-MS: 272 [M+H] +

yield: 28%
c. (R)-ethyl (l-((2,2-dimethylA,3-dioxolan-4-yl)methyl)-3-nitroAH-pyraz
The compound was synthesized following the approach outlined in Example 602, step (c) substituting (R)-l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-4-nitro-lH-pyrazole-3-carboxylic acid to afford the title compound (108 mg, yield: 28%). ESI-MS: 315 [M+H] +

(R)-l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-3-nitro-lH-pyrazol-4-amine
The compound was synthesized following the approach outlined in Example 603, step (d) substituting (R)-ethyl ( 1 -((2,2-dimethyl- 1 ,3 -dioxolan-4-yl)methyl)-3 -nitro- 1 H-pyrazol-4- yl)carbamate to afford the title compound (75 mg, yield: 90%). ESI-MS: 243 [M+H] +
Example 608

Preparation of (S)-N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyI)-l- methylureido)pyrimidin-4-yl)amino)-l-(2 -dihydroxypropyl)-lH-pyrazoI-4-yl)acrylamide
The compound was synthesized following the approach outlined in Example 607 substituting (S)-l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-3-nitro-lH-pyrazol-4-amine (Procedure shown below) to afford the title compound (12 mg, yield: 18% 4 steps) 1H-NMR (400 MHz, DMSO-d6) δ 3.40 (s, 3 H) 3.55 (d, 2 H) 3.94 (s, 6 H) 3.99 - 4.10 (m, 2 H) 4.21 - 4.27 (m, 1 H) 5.73 - 5.78
(m, 1 H) 6.31 - 6.45 (m, 2 H) 6.79 (s, 1 H) 7.00 (s, 1 H) 8.00 (s, 1 H) 8.39 (d, 1 H); ESI-MS: 581 [M+H] +
Preparation of (S)-l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-3-nitro-lH-pyrazol-4-amine
The compound was synthesized following the approach outlined in Example 607 substituting (S)-ethyl l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-4-nitro-lH-pyrazole-3-carboxylate to afford the title compound (97 mg, yield: 4% over four steps). ESI-MS: 243 [M+H]+.
Example 609

N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidin-4-yI)amino)-l- (2-(dimethylamino)ethyl)-lH-pyrazol-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A by substituting l-(2-(dimethylamino)ethyl)-4-nitro-lH-pyrazol-3 -amine (procedure shown below) in step (i) to afford the title compound (33 mg, yield: 4.6%). 1HNMR (CDC13, 400MHz): δ 12.40 (s, 1H), 8.43 (s, 1H), 8.05 (s, 1H), 7.37 (s, 1H), 6.53 (s, 1H), 6.44 (s, 1H), 6.40-6.36 (m, 1H), 6.24-6.17 (m, 2H), 5.73 (dd, 1H), 4.13-4.09 (m, 2H), 3.93 (s, 6H), 3.42 (s, 3H), 2.76-2.73 (m, 2H), 2.28 (m, 6H); ESI-MS: m/z = 578.5 (M+H)+; HPLC: 97.39%, rt: 6.26 min.
Preparation of l-(2-(dimethylamino)ethyl)-4-nitro-lH-pyrazol-3-amine

a. tert-Butyl (l-(2-hydroxyethyl)-4-nitro-lH-pyrazol-3-yl)carbamate
Added 1M TBAF solution in THF (3.65g, 13.9 mmol) at 0 °C to a solution of tert-butyl (l-(2- ((tert-butyldimethylsilyl)oxy)ethyl)-4-nitro- 1 H-pyrazol-3 -yl)carbamate (2.7g, 6.98mmol, Procedure shown in Example 606) in 50mL THF. Stirred the reaction mass at room temperature for 3-4h. Concentrated the reaction mass under vacuum and triturated with n-hexane to afford the title compound (1.6g, 84%). !HNMR (CDC13, 400MHz): δ 8.41 (s, 1H), 8.19 (s, 1H), 4.25-4.23 (m, 2H), 4.05-4.03 (m, 2H), 1.53 (s, 9H).

b. 2-(3-((tert-Butoxycarbonyl)amino)-4-nitro-lH-pyrazol-l-yl)ethyl methanesulfonate
To a solution of tert-butyl (l-(2-hydroxyethyl)-4-nitro-lH-pyrazol-3-yl)carbamate (1.2g, 4.4 mmol) in dry DCM (25mL) was added methane sulfonyl chloride (0.6g, 5.2mmol) drop wise at 0 °C and the reaction mixture was stirred for 2h. The reaction mixture was quenched with ice water and DCM (25mL each). The aqueous layer was separated and extracted with DCM (3x25mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and
concentrated under vacuum to obtain crude residue. The residue was purified by silica gel column chromatography to afford the desired title compound (1.4g, 90%). 1HNMR (DMSO-£¾,
400MHz): δ 9.42 (s, 1H), 8.85 (s, 1H), 4.58-4.55 (m, 2H), 4.45-4.43 (m, 2H), 3.14 (s, 3H), 1.40
(s, 9H).

c. tert-Butyl (l-(2-(dimethylamino)ethyl)-4-nitro-lH-pyrazol-3-yl)carbamate
To a solution of 2-(3-((tert-butoxycarbonyl)amino)-4-nitro-lH-pyrazol-l-yl)ethyl
methanesulfonate (1.4g, 3.9mmol) in dry THF (l OmL) was added potassium carbonate (0.8g, 5.8mmol) followed by the addition of 2M methyl amine solution in THF (9.99mL, 19 mmol) in a sealed tube at 10 °C and the reaction mixture was heated to 80 °C for lOh. The reaction mixture was quenched with ice water and ethyl acetate (50mL each). The aqueous layer was separated and extracted with ethyl acetate (3x25mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to obtain crude residue. The residue was purified by silica gel column chromatography to afford the desired title compound (1.12g, 94%). ESI-MS: m/z = 300.5 M+H)+.

d. l-(2-(Dimethylamino)ethyl)-4-nitro-lH-pyrazol-3-amine. TFA salt
TFA (4mL) was added drop wise to a solution of tert-butyl (l-(2-(dimethylamino)ethyl)-4-nitro- lH-pyrazol-3-yi)carbamate (1.12g, 3.7 mmol) in DCM (15mL) at 0 °C under nitrogen
atmosphere. Stirred the reaction mass at room temperature for 3h. Concentrated the reaction
mass under vacuum and triturated with n-pentane. Solid obtained dried under vacuum to yield the title compound (1.15g, 98%). ESI-MS: m/z = 200.3 (M+H)+.
Example 610

N-(3-((6-(3-(2,6-dichIoro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidin-4-yl)amino)-l- (l-ethylpiperidin-4-yl)-lH-pyrazol-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6 A by substituting l-(l-ethylpiperidin-4-yl)-4-nitro-lH-pyrazol-3 -amine (procedure shown below) in step (i) to afford the title compound (13 mg, yield: 5.9%). 'HNMR (CDC13, 400MHZ): δ 12.4 (s, 1H), 8.43 (s, 1H), 8.04 (s, 1H), 7.49 (s, 1H), 6.53-6.49 (m, 2H), 6.39-6.35 (d, 1H), 6.24-6.17 (m, 1H), 5.74-5.72 (m, 1H), 3.99-3.97 (m, 1H), 3.92 (s, 6H), 3.48 (s, 1H), 3.42 (s, 3H), 3.07-3.04 (m, 2H), 2.49-2.43 (m, 2H), 2.17-2.03 (m, 6H), 1.13 (t, 3H); ESI-MS : m/z = 618.3 (M+H)+; HPLC:
98.89%, rt: 6.22 min.
Preparation of l-(l-eth lpiperidin-4-yl)-4-nitro-lH-pyrazol-3-amine

a. tert-butyl 4-(3-(methoxycarbonyl)-4-nitro-lH-pyrazol-l-yl)piperidine-l-carboxylate
tert-Butyl 4-iodopiperidine-l-carboxylate (11.26g, 36.23 mmol) was slowly added to a stirred solution of methyl 4-nitro-lH-pyrazole-3-carboxylate (5g, 24.15 mmol) and K2C03 (9.99g, 72.46 mmol) in DMF (50mL) at 0 °C. The reaction mass was stirred at room temperature for 18hr. Quenched the reaction with ice cold water (lOOmL) and ethyl acetate (25mL). The aqueous layer was separated and extracted with ethyl acetate (3x25mL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum. The crude residue was purified by column chromatography by eluting with 10% ethyl acetate -hexane on silica gel to afford the desired title compound (Polar spot in TLC, 1.6g, 18.7%). ESI-MS: m/z = 255(M- Boc+H)+.

b. l-(l-(tert-butoxycarbonyl)piperidin-4-yl)-4-niiro-lH-pyrazole-3-carboxylic acid
Lithium hydroxide (0.57g, 13.55 mmol) in water (lOmL) was added to a stirred solution of tert- butyl 4-(3-(methoxycarbonyl)-4-nitro-lH-pyrazol-l-yl)piperidine-l-carboxylate (1.6g, 4.51 mmol) in a mixture of methanohTHF (20mL:10mL) at room temperature and stirred the reaction mass for lh. Concentrated the reaction mass under vacuum and diluted with ice water, acidified with citric acid to pH=2. Extracted with ethyl acetate (3x25mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to afford the title compound (1.33g, 88%). ESI-MS m/z = 241.1 (M-Boc+H)+.

c. tert-butyl 4-(3-((ethoxycarbonyl)amino)-4-nitro-lH-pyrazol-l-yl)piperidin^
Added TEA (0.59g, 5.86mmol) at 0 °C to a solution of l-(l-(tert-butoxycarbonyl)piperidin-4-yl)- 4-nitro-lH-pyrazole-3-carboxylic acid (1.33g, 3.91mmol) in 1,4-dioxane (20mL) followed by the addition of DPPA (1.0 lmL, 4.69 mmol). Stirred the reaction mass at room temperature for lh. and added ethanol (3mL). Heated the reaction mass slowly to 80 °C and maintained for 18h. Concentrated the reaction mass under vacuum and the crude residue was purified by silica gel column chromatography by eluting with 0-35% ethyl acetate -hexane system to afford the title compound (0.65g, 43%). ESI-MS: m/z = 284 (M-Boc+H)+.

d. ethyl (4-nitw-l-(piperidin~4-yl)-lH-pyrazol-3-yl)carbamate
TFA (1.8mL, 2.7v) was added drop wise to a solution of tert-butyl 4-(3- ((ethoxycarbonyl)amino)-4-nitro-lH-pyrazol-l-yl)piperidine-l-carboxylate (0.65g, 1.69 mmol) in DCM (7mL) at 0 °C under nitrogen atmosphere. Stirred the reaction mass at room temperature
for lh. Concentrated the reaction mass under vacuum and triturated with diethyl ether. Solid obtained dried under vacuum to yield the title compound (0.65g crude). ESI-MS: m/z = 284
(M+H)+.

e. ethyl (l-(l-ethylpiperidin-4-yl)-4-nitro-lH-pyrazol-3-yl)carbamate
Bromoethane (0.2g, 1.9mmol) was added to a stirred solution of ethyl (4-nitro-l-(piperidin-4-yl)- lH-pyrazol-3-yl)carbamate (0.65g, 1.61 mmol) and TEA (0.56g, 5.7 mmol) in anhydrous DCM (8mL) at 0 °C under argon atmosphere. The reaction mixture was stirred at room temperature for 18h. After completion of the reaction by TLC, diluted the reaction mass with DCM and water. The aqueous layer was separated and extracted with DCM (3x25mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum. The residue was triturated with 5% mixture of methanol-diethyl ether, and the precipitate was filtered and concentrated under vacuum to afford the desired title compound (0.4g, 56.3%). ESI-MS: m/z = 312.3 (M+H +.

f. l-(l-ethylpiperidin-4-yl)-4-nitro-lH-pyrazol-3-amine
3M NaOH (6mL) was added to a stirred solution of ethyl (l-(l-ethylpiperidin-4-yl)-4-nitro-lH- pyrazol-3-yl)carbamate (0.4g, 1.2mmol) in ethanol (5mL) and the reaction mixture was stirred at reflux condition for 6-7h. After completion of the reaction by TLC, concentrated the reaction mass under vacuum. Diluted the reaction mass with DCM (25mL) and the organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum. The residue triturated with 5% mixture of methanol-diethyl ether to afford the desired title compound (0.3g, 69%). ESI-MS: m/z = 240.3 (M+H)+.
Example 611

l-(6-(4-acryloyl-2-(l-ethylpiperidin-4-yl)-5-oxo-4,5-dihydroimidazo[4,5-c]pyrazol-6(2H)- yl)pyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylurea

aJ-(6-(4-acryloyl-2-(l-ethylpiperidin-4-yl)-5-oxo-4,S-dihydroimidazo[4
yl)pyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methyl-3
(trimethylsilyl) eth oxy) methyl) urea
TEA (0.09g, 0.9 mmol) was added to a stirred solution of tert-butyl (4-amino-l-(l- ethylpiperidin-4-yl)-lH-pyrazol-3-yl)(6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methyl-3-((2- (trimethylsilyl)ethoxy)methyl)ureido)pyrimidin-4-yl)carbamate (0.25g, 0.3 mmol, Preparation from Example 610) in anhydrous DCM (6mL) under argon atmosphere at 0 °C. The resulting mixture was stirred for 15 min and then slowly added the acryloyl chloride (0.03mL, 0.3 mmol) at 0 °C. The resulting reaction mixture was allowed to warm to room temperature and stirred for 30min. The reaction mixture was quenched with ice cold water (25mL) and diluted with DCM (25mL). The aqueous layer was separated and extracted with DCM (3x20mL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated. The residue was purified by 60-120 silica gel column chromatography by eluting with 5% methanol-DCM to afford desired title compound (0.2g, crude). ESI-MS: m/z = 776.3 (M+3).

J-(6-(4-acryloyl-2-(l-ethylpiperidin-4-yl)-5-oxo-4,5-dihydroi^
ytypyrimidin^-ylj-S-ft e-dichloro-SfS-dimethoxyphenyi i-methyhirea
TFA (0.6mL) was slowly added to a stirred solution of tert-butyl (4-acrylamido-l-(l- ethylpiperidin-4-yl)-lH-pyrazol-3-yl)(6-(3-(2^
(trimethylsilyl)ethoxy)methyl)ureido)pyrimidin-4-yl)carbamate (0.2g, 0.2 nimol) in dry DCM (5mL) under argon atmosphere at 0 °C. The resulting reaction mixture was allowed to warm to room temperature and stirred for 12h. Reaction progress was monitored by ESI-MS. After completion of the reaction, excess solvents were removed under reduced pressure. The resulting residue was diluted with DCM and quenched with a saturated aqueous solution of NaHC03. The aqueous layer was separated and extracted with DCM (3x1 OmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by silica gel column chromatography which was then purified by preparative HPLC (Conditions: Column: Zorbax CI 8 (21.2mm x 150mm particle size 5μπι); (Mobile Phase: A; 0.05% NH4OH in Water, B; ACN) to afford the desired compound (0.005g,
3%) as the free base. 1HNMR (CDC13, 400MHz): δ 12.6 (s, 1H), 8.40 (s, 1H), 7.64 (s, 1H), 7.36 (s, 1H), 6.91 (s, 1H), 6.53 (s, 1H), 6.41 (d, 1H), 6.20-6.17 (m, 1H), 5.59 (d, 1H), 4.12-4.05 (m, 1H), 3.93 (s, 6H), 3.72-3.68 (m, 2H), 3.51 (s, 3H), 3.13-3.10 (m, 2H), 2.26-2.17 (m, 2H), 2.09- 2.06 (m, 2H), 1.17-1.12 (m, 4H); ESI-MS: m/z = 645.7 (M+H)+; HPLC: 97.11%, rt: 6.26 min.
Example 612

N-(3-((6-(3-(2,6-dichIoro-3,5-dimethoxyphenyl)-l-(pyridin-2-ylmethyl)ureido)pyrimidin-4- yl)amino)-l-methyI-lH-pyrazol-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A by substituting l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloiO-3,5-dimethoxyphenyl)-l-(pyridin-2-ylmethyl)-3-((2- (trimethylsilyl)ethoxy)methyl)urea (procedure shown below) in step (i) to afford the title compound (1 1 mg, yield: 3.3%). 1HNMR (CDC13, 400MHz): δ 12.36 (s, 1H), 8.52 (d, IK), 8.44 (s, 1H), 8.01 (s, 1H), 7.62-7.69 (m, 1H), 7.49-7.52 (m, 1H), 7.23-7.22 (m, 2H), 7.05 (s, 1H), 6.69 (s, 1H), 6.55 (s, 1H), 6.32 (d, 1H), 6.1-6.2 (m, 1H) 5.71(d, 1H), 5.35 (s, 2H), 3.95 (s, 6H), 3.77 (s, 3H); ESI-MS: m/z = 598.2 (M+H)+; HPLC: 96.17%, rt: 3.74 min.
Preparation of l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l- (pyridin-2-ylmethy -3-((2-(trimethylsilyl)ethoxy)methyl)urea

a. ( 6-Chloro-pyrimidin-4-yl)-pyridin-2-ylmethyl-amine
To a solution of 4,6-dichloro-pyrimidine (1 g, 7 mmol) in iPrOH (40 mL) and DIPEA (1.16 g, 9 mmol) was added a solution of 2-pyridinylmethanamine (970 mg, 9 mmol) at room temperature. The resulting mixture was stirred at room temperature for 2.5 hours. Water was added and the mixture was extracted with DCM. The combined extracts were washed with brine, dried over anhydrous sodium sulfate and concentrated to give a crude product, which was purified by flash chromatography on silica to obtain the title compound (1.2 g, yield: 78%). MS (ESI): 221
[M+H]+.

b. l-(6-Chloro-pynmidin-4-yl)-3-(2,6-dichlow-3,5-dimethoxy-phenyl)-l-p^
(2-trimethylsilanyl-ethoxymethyl)-urea
To a solution of (6-chloro-pyrimidin-4-yl)-pyridin-2-ylmethyl-amine (200 mg, 0.91 mmol) in DMF (5 mL) was added NaH (60%, 55 mg, 1.37 mmol) at 0 °C, and the mixture was stirred for
10 minutes at room temperature. A solution of l-isocyanato-3,5-dimethoxy-benzene (Procedure 2 A , a-d; 337 mg, 1.37 mmol) in DMF (2 mL) was added dropwise at 0 °C. The resulting mixture was stirred for 30 minutes. SEMC1 (230 mg, 1.37 mmol) in DMF (2 mL) was added and the reaction mixture was stirred at room temperature for 1 hour. Saturated aqueous NH4C1 was added to quench the reaction. The mixture was diluted with water and extracted with EtOAc. The combined extracts were washed with water and brine, dried over anhydrous Na2S04 and filtered. The filtrate was concentrated to give a crude product, which was purified by flash
chromatography on silica to obtain the title product (420 mg, yield: 78%). 1H NMR (400 MHz,
CHLOROFORM-^) δ ppm 0.00 (s, 9 H) 0.86 - 0.97 (m, 2 H) 1.25 (t, J=7.15 Hz, 2 H) 3.76 (s, 6 H) 3.80 - 3.95 (m, 3 H) 4.97 (s, 2 H) 5.20 (s, 2 H) 6.09 (s, 1 H) 6.57 (d, J=7.91 Hz, 1 H) 6.95 (dd, J=6.90, 5.14 Hz, 1 H) 7.25 - 7.35 (m, 2 H) 7.60 (d, J=0.75 Hz, 1 H) 8.32 (d, J=4.14 Hz, 1 H) 8.42 (s, 1 H). MS (ESI): 598 [M+H]+.
Example 613

N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-(2-(isopropylamino)ethyl)ureido) pyrimidin-4-yl)ammo)-l-methyI-lH-pyrazoI-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A by substituting tert-butyl (2-(l-(6-chloiOpyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-3-tert-butyl carbonate ureido)ethyl)(isopropyl)carbamate (procedure shown below) in step (i) to afford the title compound (13 mg, yield: 5.9%). 1HNMR (CDC13, 400MHz): δ 10.05 (brs, 1H), 8.41 (s, 1H), 8.0 (s, 1H), 7.1 (s, 1H), 6.5 (s, 1H), 6.3-6.38 (dd, 1H), 6.18-6.22 (m, 1H), 5.7 (d, 1H), 4.25 (s, 1H), 3.92 (s, 6H), 3.77 (s, 3H), 3.05 (d, 2H), 2.9-2.95 (m, 1H), 1.07(s, 6H). ESI-MS: m/z = 592.1 (M+H)+; HPLC: 94.53%, rt: 5.97 min.
Preparation of tert-butyl (2-(l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5- dimethoxyphenyr)-3-fert-butyl carbonate ureido)ethyl)(isopropyl)carbamate

a. tert-butyl isopropyl(2-((2-nitrophenyl)sulfonamido)ethyl)carbamate
To a solution of N-isopropylethylenediamine (2g, 19.6 mmol) and triethyl amine (4.16g, 29.4 mmol) in DCM (40mL) and 2-nitrobenzene sulfonyl chloride (4.3g, 19.6mmol) was gradually added at 0 °C. The reaction mass was stirred at room temperature for 4h and cooled again to 0 °C and added Boc anhydride ((Boc)20) (5.12g, 23 mmol). The resultant reaction mass was stirred at room temperature for overnight. The reaction mixture was diluted with water and DCM. DCM layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum. The crude residue was purified by silica column chromatography by eluting with 0-30% ethyl acetate-hexane system to afford the title compound (6.4g, 85%). ESI-MS: m/z = 288.1 (M- Boc+H)+.

b. tert-butyl (2-aminoethyl)(isopropyl) carbamate
Cesium carbonate (67.47g, 206 mmol) was added to a solution of tert-butyl isopropyl(2-((2- nitrophenyl)sulfonamido)ethyl)carbamate (40g, 103 mmol) in acetonitrile (400mL) followed by the addition of benzenethiol (17.14g, 154 mmol) and the resultant reaction mixture was stirred at room temperature for one day. The reaction mass was diluted with DCM and filtered. The filtrate was concentrated and the residue was washed with saturated NaHC03 and DCM. The organic phase was washed with brine, dried over Na2S04, filtered and concentrated. The crude oil was diluted with hexane and the separated solid was filtered off and the filtrate was concentrated under vacuum to afford the title compound (18g). ESI-MS: m/z = 203.3 (M+H)+.


d. tert-but l (2-(l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxy
ethyl) (isopropyl) carbamate
Cesium carbonate (7.7g, 23.8 mmol) was added to a stirred solution of tert-butyl (2-((6- chloropyrimidin-4-yl)amino)ethyl)(isopropyl)carbamate (5g, 15.9 mmol) in DMF (50mL) under argon atmosphere at 0 °C. Added 2,4-dichloro-3-isocyanato-l,5-dimethoxybenzene (5.9g, 23.8 mmol) portionwise at 0 °C The reaction mixture was stirred at room temperature for 2h and quenched with ice-water. The separated solid was filtered and washed with water, dried under vacuum to afford the title compound (5.8g )+.

e. tert-butyl (2-(l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyI)-3-tert- butyl carbonate ureido)ethyI)(isopropyl)carbamate
DMAP (0.5g) and di-tert-butyl dicarbonate (2.9g, 13.3 mmol) were added to a stirred solution of tert-butyl (2-(l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)ureido)ethyl) (isopropyl)carbamate (5g, 8.8 mmol) in anhydrous DCM (lOOmL) in TEA (2.69mL, 26.4 mmol) under an argon atmosphere at 0 °C. The resulting mixture was then stirred at room temperature for 3h. After completion of the reaction by TLC, reaction mass was diluted with DCM and water. The organic layer was washed with brine, dried over Na2S04, filtered and concentrated to obtain crude residue. The crude residue was purified by column chromatography on silica gel by
eluting with a 20% ethyl acetate-hexane system to afford the desired title compound (3.1 g, 53%). ESI-MS: m/z = 661.6 (M+H)+. - 614

N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidin-4-yI)ammo)-l- methyI- -pyrazol-4-yl)propionamide.

1. Diboc protected N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureido) pyrimidin- 4-yl)amino)-l-methyl-lH-pyrazol-4-yl)propionamide.
Triethylamine (0.072g, 0.71 mmol) was added to a stirred solution of diboc protected l-(6-((4- amino-l-methyl-lH-pyrazol-3-yl)amino)pyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)- 1-methylurea (0.2g, 0.28 mol) in anhydrous DCM (l OmL) under argon atmosphere at 0 °C. The resulting mixture was stirred for 10 min and slowly added propanoyl chloride (0.039g, 0.43 mmol) at 0 °C. The resulting reaction mixture stirred at room temperature for lh. Then the reaction mixture was quenched with water, wanned to room temperature and diluted with DCM and water. The aqueous layer was separated and extracted with DCM (3x30mL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to afford desired title compound (0.17g, 82%). 'HNMR (DMS0-i 400MHz): δ 9.6 (s, 1H), 8.38 (s, 1H), 8.11 (s, 1H), 8.02 (s, 1H), 6.94 (s, 1H), 3.93 (s, 6H), 3.8 (q, 2H), 3.73 (s, 3H), 2.83 (s, 3H), 1.36 (s, 18H), 1.02 (t, 3H).

a. N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidm^
methyl-lH-pyrazol-4-yl)propionamide.
TFA (0.2mL) was slowly added to a stirred solution of diboc protected N-(3-((6-(3-(2,6- dichloro-3 ,5-dimethoxyphenyl)- 1 -methylureido)pyrimidin-4-yl)amino)- 1 -methyl- lH-pyrazol-4- yl)propionamide (0.17g, 0.23 mmol) in dry DCM (5mL) under argon atmosphere at 0 °C. The resulting reaction mixture was allowed to warm to room temperature and stirred for 16h. After completion of the reaction, excess solvents were removed under reduced pressure. The resulting residue was triturated with diethyl ether and the solid separated is filtered and dissolved in DCM and a saturated aqueous solution of NaHC03 (25mL). The aqueous layer was separated and extracted with DCM (3x1 OmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum. The residue was purified by preparative TLC by eluting with 50% ethyl acetate-hexane to afford the title compound as the free base (0.055g, 45%).
1HNMR (DMSO-ffc, 400MHz): δ 12.08 (s, 1H), 9.5-9.4 (m, 2H), 8.41 (s, 1H), 7.96 (s, 1H), 7.04 (brs, 1H), 6.89 (s, 1H), 3.93 (s, 6H), 3.76 (s, 3H), 3.30 (s, 3H), 2.26 (q, 2H), 1.04 (t, 3H). ESI- MS: m/z = 523.0 (M+H)+; HPLC: 96.16%, rt: 6.84min. - 615

N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidin-4-yl)amino)-l- ethyI-lH-pyrazoI-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A by substituting l-ethyl-4-nitro-lH-pyrazol-3 -amine (preparation shown below) and Boc-protected l-(6- chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylurea (procedure shown
below) in step (i) to afford the title compound (45 mg, yield: 4.6%). HNMR (DMSO-<i6, 400MHz): δ 12.2 (s, 1H), 9.8 (s, 1H), 9.49 (s, 1H), 8.42 (s, 1H), 8.11 (s, 1H), 7.1 (brs, 1H), 6.9 (s, 1H), 6.37-6.41 (m, 1H), 6.21 (dd, 1H), 5.76-5.70 (m, 1H), 4.09 (q, 2H), 3.94 (s, 6H), 3.31 (s, 3H), 1.37 (t, 3H); ESI-MS: m/z = 535.3 (M+H)+; HPLC: 99.71%, rt: 4.64 min.
Preparation of l-EthyI-4-nitro-lH-pyrazol-3-amine

a. Methyl l-ethyl-4-nitro-lH-pyrazole-3-carboxylate
Ethyl bromide (2.4g, 22 mmol) was slowly added to a stirred solution of methyl 4-nitro-lH- pyrazole-3-carboxylate (3.5g, 18.3 mmol) in DMF (30mL) at 0 °C. The reaction mass was stirred at room temperature for 4h. Quenched the reaction mass with ice cold water and diluted with ethyl acetate (lOOmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by silica gel column chromatography by eluting with 0-30% ethyl acetate-hexane (Lower spot on TLC) to afford the title compound (1.8g, 59%). ESI-MS: m/z = 199.7 (M+H)+.

b. l-Ethyl-4-nitro-lH-pyrazole-3-carboxylic acid
Lithium hydroxide (0.9g, 22.61mmol) in water (5mL) was added to a stirred solution of methyl l-ethyl-4-nitro-lH-pyrazole-3-carboxylate (1.8g, 9.04 mmol) in a mixture of methanol
(10mL):THF (l OmL) at room temperature and stirred the reaction mass for 2h. Concentrated the reaction mass under vacuum and diluted with ice water, acidified with 2N HC1 to pH=2.
Extracted with ethyl acetate (3x50mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get desired title compound (1.6g, 90%). ESI-MS: m/z = 141 0 (M-COOH)+.

c. tert-Butyl (l-ethyl-4-nitro-lH-pyrazol-3-yl)carbamate
Added TEA (2.5mL, 17.3 mmol) at 0 °C to a solution of l-ethyl-4-nitro-lH-pyrazole-3- carboxylic acid (1.6g, 8.64 mmol) in a mixture of DMF (15mL) and tert-butanol (5mL) followed
by the addition of DPP A (3.6g, 12.97 mmol). Heated the reaction mass to 80 °C slowly and maintained for 4h. Concentrated the reaction mass under vacuum and diluted with ethyl acetate (l OOmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by silica gel column chromatography by eluting with 0-40% ethyl acetate-hexane to afford the title compound (2g, 90%). 1HNM (CDC13, 300MHz): δ 8.39 (brs, 1H), 8.06 (s, 1H), 4.2 (q, 2H), 1.57-1.51 (m, 12H).

d. l-Ethyl-4-nitro-lH-pyrazol-3-amine
TFA (6mL) was added dropwise to a solution of tert-butyl (l -ethyl-4-nitro-lH-pyrazol-3- yl)carbamate (2g, 7.8 mmol) in DCM (l OmL) at 0 °C under nitrogen atmosphere. Stirred the reaction mass at room temperature for 4-5h. Concentrated the reaction mass under vacuum and triturated with diethyl ether. Solid obtained dried under vacuum to yield the desired title compound (l g, 80%). ESI-MS: m/z = 157 (M+H)+.
Preparation of l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methyl- 3-tert-butyl carbonate urea

DMAP (0.080g, 0.655 mmol) and Di-te/t-butyl dicarbonate (2.9mL, 12.6 mmol) was added to a stirred solution of l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l- methylurea (Procedure 2E, step b; 2.6g, 6.632 mmol) in anhydrous THF (20mL) under an argon atmosphere at 0 °C. The resulting mixture was then refluxed for 2h. After completion of the reaction by TLC (EtOAc:Hexane 3 :7), reaction mixture was cooled to room temperature and concentrated under vacuum to obtain a crude residue. The residue was purified by silica gel column chromatography to afford the title compound (2.5g, yield: 69.4%) as a solid. 1H-NMR (CDCI3, 300MHz): δ 8.73 (d, 1H), 7.95 (d, 1H), 6.61 (s, 1H), 3.95 (s, 6H), 3.63 (s, 3H), 1.35 (s, 9H).
Example 616

N-(3-((6-(3-(2,6-dichIoro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidin-4-yl)amino)-l- isopropyl- 1 H-py razol-4-yl)acry lamide
The compound was synthesized following the approach outlined in Procedure 6A by substituting l-isopropyl-4-nitro-lH-pyrazol-3 -amine (preparation shown below) and Boc-protected l-(6- chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylurea (Example 615) in step (i) to afford the title compound (0.043g, 7.7%). IHNMR (CDC13, 400MHz): δ 12.4 (s, IH), 9.2 (brs, IH), 8.42 (s, IH), 8.02 (s, IH), 7.56 (s, IH), 6.58-6.51 (m, 2H), 6.39-6.35 (m, IH), 6.23-6.16 (m, IH), 5.73 (d, IH), 4.37-4.34 (m, IH), 3.91 (s, 6H), 3.42 (s, 3H), 1.49 (d, 6H). ESI- MS: m/z = 549.3 (M+H)+; HPLC: 99.08%, rt: 4.57 min.
Preparation of l-isopropyl-4-nitro-lH-pyrazol-3-amine

a. Methyl-l-isopropyl-4-n itro-1 H-py razole-3 -car boxy late
Isopropyl bromide (0.35g, 2.89 mmol) was slowly added to a stirred solution of methyl 4-nitro- lH-pyrazole-3-carboxylate (0.5g, 2.4 mmol) in DMF (lOmL) at 0 °C. The reaction mass was stirred at room temperature for 5h. Quenched the reaction mass with ice cold water and diluted with ethyl acetate (lOOmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by silica gel column chromatography by eluting with 0 - 40% ethyl acetate-hexane to afford the title compound (0.3g, 58%). ESI-MS: m/z = 214.0 (M+H)+.

a. l-Isopropyl-4-nitro~lH-pyrazole-3-carboxylic acid
Lithium hydroxide (2.1g, 52 mmol) in water (5mL) was added to a stirred solution of methyl 1- isopropyl-4-nitro-lH-pyrazole-3-carboxylate (2.8g, 13.3 mmol) in a mixture of methanol (10mL):THF (20mL) at room temperature. Stirred the reaction mass for 3h. Concentrated the reaction mass under vacuum and diluted with ice water, acidified with 2N HC1 to pH=2.
Extracted with ethyl acetate (3x50mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get desired title compound (2.1g, 80%). ESI-MS: m/z = 200.2 M+H)+.

c. tert-Butyl (l-isopropyl~4-nitro-lH-pyrazol-3-yl)carbamate
Added TEA (3.5g, 35.4 mmol) at 0 °C to a solution of l-isopropyl-4-nitro-lZ7-pyrazole-3- carboxylic acid (3g, 17.7 mmol) in a mixture of DMF (30mL) and tert-butanol (30mL) followed by the addition of DPPA (5.8g, 21.3mmol). Heated the reaction mass to 80 °C slowly and maintained for 4h. Concentrated the reaction mass under vacuum and diluted with ethyl acetate (lOOmL). The organic phase was washed with brine, dried over Na2S04, filtered and
concentrated under vacuum to get a crude residue. The residue was purified by silica gel column chromatography by eluting with 0-30% ethyl acetate-hexane to afford the title compound (2.5g, 52%). ESI-MS: m/z = 271.3 (M+H)+.

d. l-isopropyl-4-nitro-lH-pyrazol-3-amine
TFA (3mL) was added drop wise to a solution of tert-butyl (l-isopropyl-4-nitro-lH-pyrazol-3- yl)carbamate (2.5g, 9.25 mmol) in DCM (25mL) at 0 °C under nitrogen atmosphere. Stirred the reaction mass at room temperature for 12h. Concentrated the reaction mass under vacuum and triturated with diethyl ether. Solid obtained dried under vacuum to yield the desired title compound (1.3g, 82%). ESI-MS: m/z = 171.1 (M+H)+.
Example 617

(Z)-3-Chloro-N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidin-4- yl)amino -l-methyl-lH-pyrazol-4-yl)acrylamide

a. tert-But l (Z)-(4-(3-chloroacrylamido)-l-methyl-lH-pyrazol-3-yl) ( 6-(3-(2, 6-dichloro-3,5- dimethoxyphenyl)-l-methyl-3-((2-(trimethylsilyl)ethoxy)methyl)ureido
carbamate
To a stirred solution of tert-hutyl (4-amino-l-methyl-lH-pyrazol-3-yl)(6-(3-(2,6-dichloro-3,5- dimethoxyphenyl)- 1 -methyl-3 -((2-(trimethylsilyl)ethoxy)methyl)ureido)pyrimidin-4- yl)carbamate (Procedure 6 A, step (k)) (O.lg, 0.14 mmol) was added cis-3-chloro acrylic acid (89mg, 0.84 mmol) in anhydrous pyridine (lmL) under argon atmosphere at -40 °C. The resulting mixture was stirred for 15min. and added the EDCI.HCl (161mg, 0.84 mmol) at -40 °C. The resulting reaction mixture was stirred at -20 °C to 0 °C and further stirred at room
temperature for 2h. Then the reaction mixture was quenched with water, warmed to room temperature and diluted with ethyl acetate and water. The aqueous layer was separated and extracted with ethyl acetate (3x30mL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum. The residue was purified by prep TLC by eluting with 5% Methanol-DCM to afford the desired title compound (0.02g, 16%) as a solid. ESI-MS: m/z = 785.0 (M+H)+.

b.(Z)-3-chloro-N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureM
yl)amino)-l-methyl-lH-pyrazol-4-yl)acrylamide
TFA (0.5mL) was slowly added to a stirred solution of tert-butyl (Z)-(4-(3-chloroacrylamido)-l- methyl-lH-pyrazol-3-yl)(6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methyl-3-((2- (trimethylsilyl)ethoxy)methyl)ureido)pyrimidin-4-yl)carbamate (0.02g, 0.025 mmol) in dry DCM (lmL) under argon atmosphere at 0 °C. The resulting reaction mixture was allowed to warm to room temperature and stirred for 2h. After completion of the reaction, excess solvents were removed under reduced pressure. The resulting residue was stirred with aq. ammonia (5mL) and stirred for 2h. The aqueous layer was extracted with DCM (3x1 OmL). The organic phase was washed with brine, dried over a2S04, filtered and concentrated under vacuum. The residue was purified by preparative TLC by eluting with a 5% Methanol-DCM system to afford the title compound as the free base (0.008g, 57%). 1HNMR (CD3OD, 400MHz): δ 12.4 (s, 1H), 8.42 (s, 1H), 8.02 (s, 1H), 7.19 (s, 1H), 6.59 (d, 1H), 6.53 (s, 1H), 6.36 (s, 1H), 6.29 (d, 1H), 5.30 (s, 1H), 3.93 (s, 6H), 3.83 (s, 3H), 3.42 (s, 3H). ESI-MS: m/z = 555.0 (M+H)+; HPLC: 97.05%, rt: 3.84 min.
Example 618

N-(l-(cyclopropylmethyl)-3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureido) pyrimidin-4-yl)amino)-lH~pyrazol-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A by substituting l-(cyclopropylmethyl)-4-nitro-lH-pyrazol-3 -amine (preparation shown below) and Boc-
protected l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloiO-3,5-dimethoxyphenyl)-l -methylurea (Example 615) in step (i) to afford the title compound (0.035g, 3.2%) as a white solid. 'HNMR (DMSO-i 6, 400MHz): δ 12.2 (s, 1H), 9.81 (s, 1H), 9.44 (s, 1H), 8.43 (s, 1H), 7.16 (brs, 1H), 6.90 (s, 1H), 6.44-6.37 (m, 1H), 6.22 (d, 1H), 5.72 (d, 1H), 3.94-3.90 (s, 8H), 3.32 (s, 3H), 1.23 (brs, 2H), 0.55-0.51 (m, 2H), 0.37(m, 2H); ESI-MS: m/z = 561.1 (M+H)+; HPLC: 95.44%, rt: 4.63 min.
Preparation of l-(cyclo ropylmethyl)-4-nitro-lH-pyrazol-3-amine

a. Methyl l-(cyclopropylmethyl)-4-nitro-lH-pyrazole-3-carboxylate
Cyclopropylmethyl bromide (2.34g, 17.34 mmol) was slowly added to a stirred solution of methyl 4-nitro-lH-pyrazole-3-carboxylate (3g, 14.45 mmol) and potassium carbonate (2.36g, 17.34 mmol) in DMF (40mL) at 0 °C. The reaction mass was stirred at room temperature for 4h. Quenched the reaction mass with ice cold water, diluted with ethyl acetate (50mL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by silica gel column chromatography by eluting with 0-30% ethyl acetate-hexane to afford the title compound (2.7g, 84%). ESI-MS: m/z = 226.1
(M+H)+.

b. l-(cyclopropylmethyl)-4-nitro-lH-pyrazole-3-carboxylic acid
Lithium hydroxide (2.9g, 70.2 mmol) in water (6mL) was added to a stirred solution of methyl 1 - (cyclopropylmethyl)-4-nitro-lH-pyrazole-3-carboxylate (3.16g, 14 mmol) in a mixture of methanol (6mL):THF (15mL) at room temperature and stirred the reaction mass for 3h. Concentrated the reaction mass under vacuum and diluted with ice water, acidified with 2N HC1 to pH=2. Extracted with ethyl acetate (3x50mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get desired title compound
(2.1g, 70%).

c. tert-Butyl (l-(cyclopropylmethyl)-4-nitro-lH-pyrazol-3-yl)carbamate
Added TEA (2g, 19.8mmol) at 0 °C to a solution of l-(cyclopropylmethyl)-4-nitro-lH-pyrazole- 3-carboxylic acid (2.1g, 9.9 mmol) in a mixture of DMF (20mL) and tert-butanol (20mL) followed by the addition of DPP A (3.28g, 11.93 mmol). Heated the reaction mass to 80 °C slowly and maintained for overnight. Concentrated the reaction mass under vacuum and diluted with ethyl acetate (lOOmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by silica gel column chromatography by eluting with 0-20% ethyl acetate-hexane to afford the title compound (l . lg, 39% . ESI-MS: m/z = 283.1 (M+H)+.

d. l~(cyclopropylmethyl)-4-nitro-lH-pyrazol-3-amine
TFA (2mL) was added drop wise to a solution of tert-butyl (l-(cyclopropylmethyl)-4-nitro-lH- pyrazol-3-yl)carbamate (l .lg, 4 mmol) in DCM (20mL) at 0 °C under nitrogen atmosphere. Stirred the reaction mass at room temperature for overnight. Concentrated the reaction mass under vacuum and triturated with diethyl ether. Solid obtained dried under vacuum to yield the desired title compound (0.6g, 85%). ESI-MS: m/z = 183.1 (M+H)+.
Example 619

The compound was synthesized following the approach outlined in Procedure 6A by substituting 1 -(3-((tert-Butyldimethylsilyl)oxy)propyl)-4-nitro- 1 H-pyrazol-3 -amine (preparation shown below) and Boc-protected l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l- methylurea (Example 615) in step (i), followed by the additional step:

1M TBAF solution in THF (0.9ml) was slowly added to a stirred solution of N-(l-(3-((tert- butyldimethylsilyl)oxy)propyl)-3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l- methylureido)pyrimidin-4-yl)amino)-lH-pyrazol-4-yl)acrylamide (0.2g, 0.2 rnmol) in dry THF (5mL) under argon atmosphere at 0 °C. The resulting reaction mixture was allowed to warm to room temperature and stirred for 3h at ambient temperature. After completion of the reaction, the reaction mass was quenched with saturated NH4C1 solution. Extracted the compound to ethyl acetate and the ethyl acetate layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum. The residue was purified by basic alumina column by eluting with 2.5% methanol-DCM to afford the title compound as the free base (0.048g, 30%). 'HNMR (DMSO- , 400MHz): δ 12.08 (s, 1H), 9.86 (s, 1H), 9.49 (s, 1H), 8.42 (s, 1H), 8.10 (s, 1H), 7.09 (brs, 1H), 6.90 (s, 1H), 6.37-6.44 (m, 1H), 6.1-6.24 (m, 1H), 5.71 (d, 1H), 4.59 (t, 1H), 4.10 (t, 2H), 3.94 (s, 6H), 3.40 (dd, 2H), 3.30 (s, 3H), 1.88-1.94 (m, 2H). ESI-MS: m/z = 565.1 (M+H)+; HPLC: 98.46%, it: 3.92min.
Preparation of l-(3-((tert-ButyIdimethyIsiIyl)oxy)propyl)-4-nitro-lH-pyrazol-3-amine

a. Methyl l-(3-( (iert-butyldimethylsilyl)oxy)propyl)-4-nitro-lH-pyrazole-3-carboxylate tert-Butyl(3-chloropropoxy)dimethylsilane (15g, 72.1 mmol) was slowly added to a stirred solution of methyl 4-nitro-lH-pyrazole-3-carboxylate (l Og, 48.3 mmol) and K2C03 (13.3g, 97 mmol) in DMF (lOOmL) at 0 °C. The reaction mass was stirred at room temperature for 16h.
Quenched the reaction mass with ice cold water and diluted with ethyl acetate (lOOmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by 60-120 silica gel column
chromatography by eluting with 0 - 40% ethyl acetate-hexane to afford the title compound (3.2g, 19%) (Lower spot on TLC). 1HNMR (CDC13, 400MHz): δ 8.16 (s, 1H), 4.32 (t, 2H), 3.99 (s, 3H), 3.6 (t, 2H), 2.1 (t, 2H), 0.9 (s, 9H), 0.2 (s, 6H).

b. l-(3-( (tert-Butyldimethylsilyl)oxy)propyl)-4-nitro-lH-pyrazole-3-carboxylic acid
Lithium hydroxide (1.56g, 37.3 mmol) in water (lOmL) was added to a stirred solution of methyl l-(3-((tert-butyldimethylsilyl)oxy)propyl)-4-nitro-lH-pyrazole-3-carboxylate (3.2g, 9.3 mmol) in a mixture of methanol (20mL):THF (20mL) at room temperature and stirred the reaction mass for 3h. Concentrated the reaction mass under vacuum and diluted with ice water, acidified with 2N HC1 to pH=2. Extracted with ethyl acetate (3x50mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get desired title compound (2.7g, 90%).

c. tert-Butyl (l-(3-( (tert-butyldimethylsilyl)oxy)propyl)-4-nitro-lH-pyrazol-3-yl)carbamate
Added TEA (1.6g, 15.8 mmol) at 0 °C to a solution of l-(3-((tert-butyldimethylsilyl)oxy)propyl)- 4-nitro-lH-pyrazole-3-carboxylic acid (2.7g, 8.2 mmol) in a mixture of DMF (20mL) and tert- butanol (lOmL) followed by the addition of DPPA (3.38g, 12.3mmol). Heated the reaction mass to 95 °C slowly and maintained for 3h. Concentrated the reaction mass under vacuum and diluted with ethyl acetate (lOOmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by 60- 120 silica gel column chromatography by eluting with 0-30% ethyl acetate-hexane to afford the title compound (l.lg, 34%). ESI-MS: m/z = 301.2 M-Boc+H)+.

d. 3-(3-Amino-4-nitro-lH-pyrazol-l-yl)propan-l-ol
TFA (1.5mL) was added drop wise to a solution of tert-butyl (l-(3-((tert- butyldimethylsilyl)oxy)propyl)-4-nitro-lH-pyrazol-3-yl)carbamate (l .lg, 2.75 mmol) in DCM (lOmL) at 0 °C under nitrogen atmosphere. Stirred the reaction mass at room temperature for 2h. Concentrated the reaction mass under vacuum and triturated with diethyl ether. Solid obtained dried under vacuum to yield the desired title compound (0.9g crude). ESI-MS: m/z = 187.1 (M+H)+.

e. l-(3-((tert-Butyldimethylsilyl)oxy)propyl)-4-nitro-lH^ymzol-3-amine
tert-Butylchlorodimethylsilane (1.09g, 7.2 mmol) was slowly added to a stirred solution of 3-(3- amino-4-nitro-lH-pyrazol-l-yl)propan-l-ol (0.9g, 4.8 mmol) and imidazole (0.65g, 9.6 mmol) in DCM (lOmL) at 0 °C. Stirred the reaction mass for 5min and DMAP (0.29g, 2.4 mmol) was added to the reaction mass. The reaction mass was stirred at room temperature for lh. Quenched the reaction mass with ice cold water and diluted with ethyl acetate (lOOmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by basic alumina column chromatography by eluting with 0 - 20% ethyl acetate-hexane to afford the title compound (0.2g, 13%). ESI-MS: m/z = 301.2 (M+H)+.
Example 620

N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidin-4-yl)amino)-l- (3-methoxypropyl)-lH-pyrazol-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A by substituting l-(3-Methoxypropyl)-4-nitro-lH-pyrazol-3-amine (preparation shown below) and Boc-protected 1 -(6-chloropyrimidin-4-yl)-3-(2,6-dichloiO-3,5-dimethoxyphenyl)- 1 -methylurea (Example 615) in step (i) to afford the title compound (0.268g, 43.8% over four steps). 1HNMR (CDC13,
300MHz): δ 12.4 (s, 1H), 8.43 (s, 1H), 8.0 (s, 1H), 7.49 (s, 1H), 6.52 (s, 1H), 6.35-6.42 (m, 2H),
6.21-6.24 (m, 1H), 5.7 (dd, 1H), 4.09-4.14 (m, 2H), 3.92 (s, 6H), 3.42 (s, 3H), 3.33-3.37 (m, 5H), 2.07-2.11 (m, 2H). ESI-MS: m/z = 579.1 (M+H)+; HPLC: 96.37%, rt: 4.69min.
Preparation of l- 3-Methoxypropyl)-4-nitro-lH-pyrazol-3-amine

a. Methyl l-(3-methoxypropyl)-4-nitro-lH-pyrazole-3-carboxylate
l-Bromo-3-methoxypropane (4.43g, 28 mmol) was slowly added to a stirred solution of methyl 4-nitro-lH-pyrazole-3-carboxylate hydrochloride (4g, 19 mmol) and 2C03 (5.35g, 38.8 mmol) in DMF (40mL) at 0 °C. The reaction mass was stirred at room temperature for 12h. Quenched the reaction mass with ice cold water, diluted with ethyl acetate (lOOmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by 60-120 silica gel column chromatography by eluting with 0- 30% ethyl acetate-hexane (Lower spot on TLC) to afford the title compound (3.5g, 74%). ESI- MS: m/z = 244.0 (M+H +.

b. l-(3-Methoxypropyl)-4-nitro-lH-pyrazole-3-carboxylic acid
Lithium hydroxide (2.4g, 57.14 mmol) in water (lOmL) was added to a stirred solution of methyl 1 -ethyl -4-nitro-lH-pyrazole-3-carboxylate (3.5g, 14 mmol) in a mixture of methanol
(30mL):THF (5mL) at room temperature and stirred the reaction mass for lh. Concentrated the reaction mass under vacuum and diluted with ice water, acidified with 2N HC1 to pH=2.
Extracted with ethyl acetate (3x50mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get desired title compound (3.6g crude). ESI-MS: m/z = 230.0 (M+H)+.
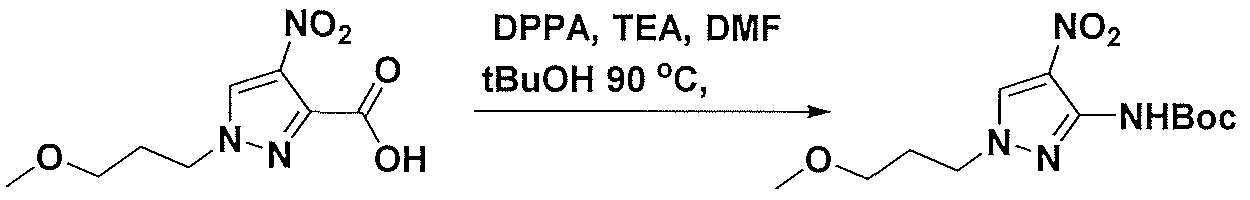
c. tert-Butyl (l-(3-methoxypropyl)-4-nitro-lH-pyrazol-3-yl)carbamate
Added TEA (4.36mL, 31 mmol) at 0 °C to a solution of l~(3-methoxypropyl)-4-nitro-lH- pyrazole-3-carboxylic acid (3.6g, 15.6 mmol) in DMF (30mL) followed by the addition of DPP A
(6g, 23 mmol) and stirred the reaction mass for 30 min at room temperature. Added tert-butanol (5mL) and heated the reaction mass to 90 °C slowly and maintained for 3h. Diluted the reaction mass with ice cold water and ethyl acetate (50mL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by 60-120 silica gel column chromatography by eluting with 0-30% ethyl acetate- hexane to afford the title compound (3.5g, 74%). ESI-MS: m/z = 301 (M+H)+.

d.l-(3-Methoxypropyl)-4-nitro-lH-pyrazol-3-amine
TFA (7mL) was slowly added to a stirred solution of tert-butyl (l-(3-methoxypropyl)-4-nitro- lH-pyrazol-3-yl)carbamate (3.5g, 11.6 mmol) in dry DCM (20mL) under argon atmosphere at 0 °C. The resulting reaction mixture was allowed to warm to room temperature and stirred for 2h. After completion of the reaction, excess solvents were removed under reduced pressure, diluted the reaction mass with DCM and basified with NaHC03 solution. The separated organic layer was washed with brine solution followed by water. Dried the organic layer over anhydrous sodium sulfate and concentrated under vacuum to afford the title compound (2g, 86%). ESI-MS: m/z = 201.1 (M+H)+.
Example 621

N-(3-((6-(3-(2,6-Dichloro-3,5-dimethoxyphenyl)-l-methyIureido)pyrimidin-4-yl)amino)-l- (2-(dimethylamino)propyl)-lH-pyrazol-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A by substituting l-(2-(Dimethylamino) propyl)-4-niti -lH-pyrazol-3 -amine (preparation shown below) and Boc- protected l-(6-chloi pyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylurea (Example 615) in step (i) and zinc/ammonium chloride in step (k) to afford the title compound compound (0.018 g) as the free base. 1HNMR (CDC13, 400MHz): δ 12.40 (s, 1H), 8.45 (s, 1H), 8.02 (s, 1H), 7.38 (s, 1H), 6.54 (s, 1H), 6.41 (s, 1H), 6.36 (s, 1H), 6.22-6.15 (m, 2H), 5.73 (dd,
1H), 4.06-4.12 (m, 2H), 3.93 (s, 6H), 3.43 (s, 3H), 2.25-2.27 (m, 2H), 2.22 (s, 6H), 2.00-2.02 (m, 2H); LCMS: m/z = 592.0 (M+H)+; HPLC: 92.61%, rt: 3.56 min.
Preparation of l-(2-(Dimethylamino) propyl)-4-nitro-lH-pyrazol-3-amine.TFA salt

a. Methyl l-(2-((tert-butyldimethylsilyl)oxy)propyl)-4-nitro-lH-pyrazole-3-carboxy
tert-Butyl(3-chloiOpiOpoxy)dimethylsilane (15g, 72.4 mmol) was slowly added to a stirred solution of methyl 4-nitro-lH-pyrazole-3-carboxylate, (10. Og, 48.3 mmol) in DMF (lOOmL) at 0 °C. The reaction mass was stirred at room temperature for 16h. Quenched the reaction mass with ice cold water and diluted with ethyl acetate. The aqueous layer was separated and extracted with ethyl acetate (3x50mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum. The residue was purified by silica gel column chromatography afforded title compound (polar spot in TLC) (3.2g, 19%). LCMS: m/z = 344.1 (M+H)+.

b. l-(2-((tert-Butyldimethylsilyl)oxy) propyl)-4-nitro-lH-pyrazole-3-carboxylic acid
Lithium hydroxide (4.89g, 116 mmol) was added to a stirred solution of methyl l-(2-((tert- butyldimethylsilyl)oxy) propyl)-4-nitro-lH-pyrazole-3-carboxylate (lOg, 29 mmol) in a mixture of 60:40 methanol:THF (lOOmL) at room temperature. Stirred the reaction mass for 2h. Concentrated the reaction mass under vacuum and diluted with ice water, acidified with 2N HCl to pH=2. Extracted with ethyl acetate (3x50mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get the crude title compound (3.2g, 33%).

c. tert-Butyl (l-(2-( (tert-butyldimethylsilyl)oxy)propyl)-4-nitro-lH-pyrazol-3-yl)carbamate
Added TEA (2.28g, 16.4 mmol) at 0 °C to a solution of l-(2-((tert-butyldimethylsilyl)oxy) propyl)-4-nitro-lH-pyrazole-3-carboxylic acid (2.7g, 8.2 mmol) in a mixture of DMF (20mL)
and tert-butanol (lOmL) followed by the addition of DPPA (2.65g, 12.3 mmol). Heated the reaction mass to 90 °C slowly and maintained for 3h. Concentrated the reaction mass under vacuum and diluted with ethyl acetate (lOOmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by 60-120 silica gel column chromatography to afford the title compound (l .lg, 33%). LCMS: m/z = 301.1 (M-Boc+H)+.

d. tert-Butyl (l-(2-hydroxy propyl)-4-nitro-lH-pyrazol-3-yl)carbamate
Added 1M TBAF solution (8.7mL, 8.7 mmol) at 0 °C to a solution of tert-butyl (l-(2-((tert- butyldimethylsilyl)oxy)propyl)-4-nitro-lH-pyrazol-3-yl)carbamate (1.4g, 3.5 mmol) in lOmL THF. Stirred the reaction mass at room temperature for 2h. Concentrated the reaction mass under vacuum and triturated with n-hexane to afford the title compound (lg crude). LCMS: m/z = 186 (M-Boc+H)+.

e. 2-(3-((tert-Butoxycarbonyl)amino)-4-nitro-lH-pyrazol-l-yl) propyl methanes ulfonate
To a solution of tert-butyl (l-(2-hydroxypropyl)-4-nitro-lH-pyrazol-3-yl)carbamate (lg, 3.4 mmol) in dry DCM (lOmL) was added methanesulfonyl chloride (0.4mL, 5.2 mmol) dropwise at 0 °C and the reaction mixture was stirred for 2h. The reaction mixture was quenched with ice water and DCM (25mL each). The aqueous layer was separated and extracted with DCM (3x25mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to afford the desired title compound (1.4g crude). LCMS: m/z = 265.0 (M-Boc+H)+.
2M Dimethyl amine

f. tert-Butyl (l-(2-(dimethylamino) propyl)-4-nitro-lH-pyrazol-3-yl)carbamate
To a solution of 2-(3-((tert-butoxycarbonyl)amino)-4-nitro-lH-pyrazol-l-yl)propyl methanesulfonate (1.2g, 3.2 mmol) in dry THF (lOmL) was added potassium carbonate (0.68g, 4 mmol) followed by the addition of 2M methyl amine solution in THF (8.2mL, 16 mmol) in a sealed tube at 10 °C and the reaction mixture was heated to 65 °C for 16h. The reaction mixture
was quenched with ice water and ethyl acetate (50mL each). The aqueous layer was separated and extracted with ethyl acetate (3x25mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to obtain the crude residue. The residue was purified by silica gel column chromatography to afford the desired title compound (0.85g, 82%). LCMS: m/z = 314.1 M+H)+.

g. l-(2-(Dimethylamino)propyl)-4-nitro-lH-pyrazol-3-amine. TFA salt
TFA (3.2mL) was added drop wise to a solution of tert-butyl (l-(2-(dimethylamino)propyi)-4- nitro-lH-pyrazol-3-yl)carbamate (0.85g, 2.7 mmol) in DCM (lOmL) at 0 °C under nitrogen atmosphere. Stirred the reaction mass at room temperature for 16h. Concentrated the reaction mass under vacuum and triturated with n-pentane to yield the title compound (0.95g crude). LCMS: m/z = 214.1 (M+H)+. - Example 622

N-(3-((6-(l-BenzyI-3-(2,6-dichIoro-3,5-dimethoxyphenyI)ureido)pyrimidin-4-yl)amino)-l- methyl-lH-pyrazol-4-yl)acrylamide

a. l-Benzyl-l-(6-chloropyrimidin-4-yl)-3-(3,5-dimethoxyphenyl)urea
To a stirred solution of 3,5-dimethoxyaniline (3g, 19.6 mmol) in dioxane (30mL) was added 20% phosgene (38.4g, 78.43 mmol) in toluene under argon atmosphere at 0 °C. The resulting mixture was heated to 70 °C, stirred for 4h, and allowed to cool to room temperature. The resulting reaction mixture was concentrated under vacuum and co-distilled with toluene to afford the isocyanate intermediate and was taken in toluene (30mL) and added N-benzyl-6- chloiOpyrimidin-4-amine (4.3g, 19.6 mmol) under argon at 0 °C. The resulting reaction mixture was refluxed for 12h. After completion of the reaction by TLC, reaction mixture was concentrated under vacuum. The residue was purified by 60-120 silica gel column chromatography t 3 (M+H)+

b. l-Benzyl-l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimeth^
Sulfuryl chloride (1.62g, 12.06 mmol) was added dropwise to a solution of 1 -benzyl- 1 -(6- chloropyrimidin-4-yl)-3-(3,5-dimethoxyphenyl)urea (3.2g, 8.04 mmol) in acetonitrile (30mL) at -20 °C. The resulting reaction mixture was stirred at -10 °C to -5 °C slowly and stirred for lh. The reaction mixture was quenched with ice-cold water. The precipitated solid was filtered and washed with ether and pentane to afford desired title compound (2.8g, 75%). ESI-MS: m/z = 467.0 (M+H)+.

c. Boc-protected l-benzyl-l-(6-chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl) urea
DMAP (0.26g, 2.22 mmol) and Boc20 (1.7g, 8.35 mmol) was added to 1 -benzyl- 1 -(6- chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)urea (2.6g, 5.56 mmol) in THF (30mL) at 0 °C. Allowed the reaction mass to stir at room temperature for 15min. and then further heated to 70 °C for 2h. Cooled and diluted the reaction mass with water (50mL) and DCM. Separated the DCM layer and washed it with water followed by brine, dried over Na2S04, filtered and concentrated. The residue was purified by 60-120 silica gel column chromatography to afford de red title compound (1.9g ESI-MS: m/z = 567.0 (M+H)+.

d. Boc-protected l-benzyl-3-(2, 6-dichloro-3,5-dimethoxyphenyl)-l-(6-( (l-methyl-4-nitro-lH- pyrazol-3-yl)amino)pyrimidin-4-yl)urea
l-Methyl-4-nitro-lH-pyrazol-3-amine (0.5g, 3.52 mmol) and boc protected 1 -benzyl- 1 -(6- chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)urea (1.9g, 4.22 mmol) were taken in a mixture of toluene (lOmL) and DMF (lOmL) in a seal tube at room temperature and argon gas was purged for 5-10 min. Then Cs C03 (2.2g, 7.04 mmol) and Xantphos (0.20g, 0.35 mmol) were added and the resulting reaction mixture was purged with argon gas for 5min, followed by the addition of Pd2(dba)3 (0.32g, 0.35mmol). The argon gas purging was continued for an additional 15min. before sealing the reaction vial. Then the reaction mixture was heated at 120 °C for 3h. After completion of the reaction by TLC, the reaction mass was filtered through Celite® and the filtrate was evaporated and the residue was purified by combiflash by eluting with 40% ethyl acetate-hexane to afford the desired title compound (lg, 42%). ESI-MS: m/z = 573.0 (M-Boc+H)+.

e. Diboc-protected l-benzyl~3-(2,6-dichloro-3,5-dimethoxyphenyl)~l-(6-((l-methyl-4-nti^ pyrazol-3-yl)amino)pyrimidin-4-yl)urea
DMAP (0.07g, 0.59 mmol) and Boc20 (0.48g, 4.6 mmol) was added to Boc protected 1-benzyl- 3-(2,6-dic loro-3,5-dimethoxyphenyl)-l-(6-((l-methyl-4-nitro-lH-pyrazol-3- yl)amino)pyrimidin-4-yl)urea (lg, 1.48 mmol) in THF (lOmL) at 0 °C. Allowed the reaction mass to stir at room temperature for 15 min and further heated to 70 °C for 2h. Cooled and diluted the reaction mass with water (50mL) and DCM. Separated DCM layer and washed with water followed by brine, dried over Na2S04, filtered and concentrated. The residue was purified by 60-120 silica gel column chromatography to afford desired title compound (lg, 90%). ESI- MS: m/z = 775.2 M+3).

f. Diboc-protected l-(6-( (4-amino-l-methyl-lH-pyrazol-3-yl)amino)pyrimidin-4~yl)-l-ben^ (2, 6-dichloro-3,5-dimethoxyphenyl) urea
To a solution of Diboc protected l-benzyl-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-(6-((l- methyl-4-nitro-lH-pyrazol-3-yl)amino)pyrimidin-4-yl)urea (lg, 1.29 mmol) in THF (lOmL) was added a solution of NH4C1 (1.04g, 19.37 mmol) in H20 (2mL) and stirred at room temperature for 10 min. Zinc powder (1.24g, 19.37 mmol) was added to the resulting reaction mixture and stirring was continued for 2h at room temperature. The reaction mixture was diluted with ethyl acetate (20mL) and filtered over a Celite® bed. The organic layer was washed with brine, dried over Na2S04, filtered and concentrated to obtain the title compound (0.80g crude). ESI-MS: m/z = 743.1 (M+H)+.

g. Diboc-protected N-(3-((6-(l-benzyl-3-(2,6-dichloro-3,5-dimethoxyphenyl)ureido)pyrimidin- 4-yl)amino)-l-methyl-lH~pyrazol-4-yl)acrylamide
TEA (0.16g, 1.61 mmol) was added to a stirred solution of Diboc-protected l-(6-((4-amino-l- methyl-lH-pyrazol-3-yl)amino)pyrimidin-4-yl)-l-benzyl-3-(2,6-dichloro-3,5-dimethoxyphenyl)
urea (0.8g, 1.07mmol) in anhydrous DCM (15mL) under argon atmosphere at 0 °C. The resulting mixture was stirred for 5 min and then slowly added the acryloyl chloride (0.116g, 1.29 mmol) at 0 °C. The resulting reaction mixture was allowed to warm to room temperature and stirred for 30 min. The reaction mixture was quenched with a saturated sodium bicarbonate solution (25mL) and diluted with DCM (50mL). The aqueous layer was separated and extracted with DCM (3x30mL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated. The residue was purified by silica gel flash chromatography to afford desired title compou -MS: m/z = 797.1 (M+H)+.

h. N-(3-((6-(l-Benzyl-3-(2,6-dichloro-3,5-dimethoxyphenyl)ureido)pyrimidin^
methyl-lH-pyrazol-4-yl)acrylamide
TFA (1.5mL) was slowly added to a stirred solution of Diboc protected N-(3-((6-(l-benzyl-3- (2,6-dichloro-3,5-dimethoxyphenyl)ureido)pyrimidin-4-yl)amino)-l-methyl-lH-pyrazol-4- yl)acrylamide (0.6g, 0.75 mmol) in dry DCM (2mL) under argon atmosphere at 0 °C. The resulting reaction mixture was allowed to warm to room temperature and stirred for 16h at ambient temperature. After completion of the reaction, excess solvents were removed under reduced pressure. The resulting residue was triturated with diethyl ether and the solid separated is filtered and dissolved in DCM and a saturated aqueous solution of NaHC03 (25mL). The aqueous layer was separated and extracted with DCM (3x1 OmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum. The residue was purified by silica gel flash chromatography by eluting with 5% methanol-DCM to afford the title compound as the free base (0.06g, 13.6%). 1HNMR (CDC13, 400MHz): δ 12.58 (s, 1H), 8.42 (s, 1H), 7.93 (s, 1H), 7.30 (s, 5H), 7.24 (brs, 1H), 6.54 (s, 1H), 6.32-6.36 (m, 2H), 6.10-6.17 (m, 2H), 5.70 (d, 1H), 5.2 (s, 2H), 3.93 (s, 6H), 3.75 (s, 3H). ESI-MS: m/z = 597.1 (M+H)+; HPLC: 95.21%, rt: 4.53min.
Example 623

N-(3-((6-(3-(2,6-Dichloro-3,5-dimethoxyphenyl)-l-ethylureido)pyrimidin-4-yl)amino)-l- methyl-lH-pyrazol-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6B by substituting added N-ethyl-6-chloropyrimidin-4-amine in step (a) to afford the title compound (0.12 g, 9.6% over seven steps). 1HNMR (CDC13, 400MHz): δ 11.78 (s, 1H), 9.2 (brs, 2H), 8.41 (s, 1H), 8.12 (s, 1H), 6.58 (t, 1H), 6.39-6.44 (m, 1H), 6.26-6.32 (m, 1H), 5.74-5.77 (m, 1H), 3.89 (s, 6H), 3.85 (s, 3H), 3.51 (s, 3H). ESI-MS: m/z = 489.2 (M+H)+; HPLC: 97.09%, rt: 4.16min.
Example 624

N-(3-((6-(3-(2,6-Difluoro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidin-4-yl)amino)-l- methyl-lH-pyrazol-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A by substituting Selectfluor® fluorinating reagent in step (b) and zinc/NH4Cl in step (k) to afford the title compound (0.12 g, 9.6% over seven steps). 1HNMR (CDC13, 400MHz): δ 11.78 (s, 1H), 9.2 (brs, 2H), 8.41 (s, 1H), 8.12 (s, 1H), 6.58 (t, 1H), 6.39-6.44 (m, 1H), 6.26-6.32 (m, 1H), 5.74- 5.77 (m, 1H), 3.89 (s, 6H), 3.85 (s, 3H), 3.51 (s, 3H). ESI-MS: m/z = 489.2 (M+H)+; HPLC: 97.09%, rt: 4.16min.
Example 625

N-(3-((6-(3-(2-Chloro-6-fluoro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidin-4- yl)amino) -1 -methyl- lH-pyrazol-4-yl)acryIamide
The compound was synthesized following the approach outlined in Procedure 6A by substituting Boc protected 3-(2-chloro-6-fluoro-3,5-dimethoxyphenyl)- 1 -(6-chloropyrimidin-4-yl)-l - methylurea in step (b) and zinc NH4Cl in step (k) to afford the title compound (0.014g). 1HNMR (CDC13, 400MHz): δ 12.4 (s, 1H), 9.3 (brs, 1H), 8.43 (s, 1H), 7.99 (s, 1H), 7.65 (brs, 1H), 6.47- 6.53 (m, 2H), 6.35-6.40 (m, 1H), 6.17-6.24 (m, 1H), 5.73 (d, 1H), 3.9 (d, 6H), 3.82 (s, 3H), 3.41 (s, 3H). LCMS: m/z = 505.2 (M+H)+; HPLC: 98.78%, it: 4.29min.
Preparation of Boc protected 3-(2-chloro-6-fluoro-3,5-dimethoxyphenyl)-l-(6-chIoro pyrimidin-4-yl)-l-methyl rea

a. Methyl 2-chloro-6-fluoro-3,5-dimethoxybenzoate
S02C12 (4.13g, 30.84 mmol) was added dropwise to a solution of methyl 2-fluoro-3,5-dimethoxy benzoate (6g, 28.03 mmol) in acetonitrile (lOOmL) at 0 °C under nitrogen atmosphere. The resulting reaction mixture was wanned to room temperature slowly and stined for lh. The reaction mixture was quenched with saturated sodium bicarbonate solution, and extracted with ethyl acetate (3x30mL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated. The residue was purified by triturating with 30% ether-pentane to afford the desired title compound (5g, 71%) as a solid. 'HNMR (CDCI3, 400MHz): δ 6.63 (d, 1H), 3.96 (s, 3H), 3.9 (d, 6H).

b. 2-Chloro-6-fluoro-3,5-dimethoxybenzoic acid
A suspension of methyl 2-chloro-6-fluoro-3,5-dimethoxybenzoate (5g, 20.1mmol) and sodium hydroxide (3.2g, 80 mmol) in anhydrous ethanol (50mL) was refluxed for 24h. The resulting reaction mixture was cooled to room temperature and concentrated under vacuum to get a crude residue. The crude residue was dissolved in water and extracted with ether (2X30mL). The aqueous layer was acidified with cone. HC1 and the precipitated solid was filtered, washed with cold water and dried in vacuum to afford the desired title compound (4.4g, 81%) as a solid. LCMS: m/z = 234.8 (M+H +.

c. 2-Chloro-6-fluoro-3,5-dimethoxyaniline
A suspension of 2-chloro-6-fluoro-3,5-dimethoxybenzoic acid (4.4g, 18.8 mmol) and triethyl amine (2.8g, 28.2 mmol) in tert-BuOH (60mL) was stirred for 5 min. To the resulting reaction mixture was added diphenyl phosphoryl azide (6.2g, 22.5 mmol) and the mixture was heated to 100 °C and maintained for overnight. The reaction mixture was then concentrated in vacuum to obtain a crude residue. The crude residue was dissolved in dichloromethane (20mL) and cooled to 0 °C. TFA (4mL) was added to the reaction mixture and the resultant reaction mixture was then stirred at room temperature for 2h. The solvents were removed under vacuum and the crude residue was diluted with dichloromethane and saturated sodium carbonate solution. The aqueous layer was separated and extracted with dichloromethane (3x30mL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated. The residue was purified by 60- 120 silica gel column chromatography using 0^-0% ethyl acetate-hexane to afford the pure product (2.6g, 67.5% as a solid. LCMS: m/z = 206.1 (M+H)+.

d. 3-(2-Chloro-6-fluoro-3,5-dimethoxyphenyl)-l-(6-chloropynmidin-4-yl)^
To a stirred solution of 2-chloro-6-fluoro-3,5-dimethoxyaniline (2g, 9.85 mmol) in dioxane (30 mL) was added 20% phosgene in toluene (20mL, 39.0 mmol) under argon atmosphere at 0 °C. The resulting mixture was stirred for 6h at 90 °C, and then allowed to cool to room temperature. The solvents were removed and the residue was dissolved in toluene (50mL). To this, was added 6-chloro-N-methylpyrimidin-4-amine (0.82g, 5.8mmol). The resultant reaction mixture was then
refluxed for 6h. After completion of the reaction by TLC, the reaction mixture was cooled to room temperature and concentrated under vacuum to obtain a crude reaction mixture. The solid precipitated on addition of ethyl acetate to the crude reaction mixture and was filtered, washed with ether to afford the title com ound (l.lg, 30%). LCMS: m/z = 375.0 (M+H)+.

e. Boc protected 3-(2-chloro-6-fluoro-3,5-dimethoxyphenyl)-l-(6-chloropyrimidin-4-yl)-l- methylurea
DMAP (0.17g, 1.4 mmol) and Boc20 (0.95g, 4.3 mmol) was added to 3-(2-chloro-6-fluoro-3,5- dimethoxyphenyl)-l-(6-chloropyrimidin-4-yl)-l -methylurea (l .lg, 2.9 mmol) in THF (20mL) at 0 °C. Allowed the reaction mass to stir at 70 °C for 4h. Diluted the reaction mass with water and ethyl acetate. Separated ethyl acetate layer was washed with water followed by brine, dried over Na2S04, filtered and concentrated. The residue was purified by 100-200 silica gel column chromatography to afford the desired title compound (1.15g, 82%). LCMS: m/z = 475.1 (M+H)+.
Example 626

N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylureido)pyrimidin-4-yI)amino)-l- propyl-lH-pyrazol-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A substituting 1- propyl-4-nitro-lH-pyrazol-3 -amine (procedure shown below) and boc-protected l-(6- chloropyrimidin-4-yl)-3-(2,6-dichloiO-3,5-dimethoxyphenyl)-l-methylurea in step (i) and zinc/ammonium chloride in step (k) to afford the title compound (0.07g, 50%). 'HNMR (DMSO- d6, 400MHz): δ 12.2 (s, 1H), 9.86 (s, 1H), 9.489 (s, 1H), 8.43 (s, 1H), 8.1 1 (s, 1H), 7.1 (brs, 1H),
6.91 (s, 1H), 6.37-6.44 (m, 1H), 6.21 (dd, 1H), 5.70-5.76 (m, 1H), 4.01 (t, 2H), 3.94 (s, 6H), 3.30
(s, 3H), 1.78 (q, 2H), 0.84 (t, 3H). ; MS (ESI): 549.3 [M+H]+.
Preparation of 1- Propyl -4-nitro-lH- razol-3-amine

a. Methyl 1-propyl -4-nitro-lH-pyrazole-3-carboxylate
Propyl bromide (3.2g, 26 mmol) was slowly added to a stirred solution of methyl 4-nitro-lH- pyrazole-3-carboxylate (5g, 25 mmol) in DMF (50mL) at 0 °C. The reaction mass was stirred at room temperature for 2h. Quenched the reaction mass with ice cold water and diluted with ethyl acetate (lOOmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by silica gel column chromatography by eluting with 0-30% ethyl acetate-hexane (Lower spot on TLC) to afford the title compound (3.7g, 70%). MS (ESI): 214.1 [M+H]+.

b. l-Propyl-4-nitro-lH-pyrazole-3-carboxylic acid
Lithium hydroxide (2.6g, 65 mmol) in water (lOmL) was added to a stirred solution of methyl 1- propyl-4-nitro-lH-pyrazole-3-carboxylate (3.5g, 16.4 mmol) in a mixture of methanol (20mL):THF (20mL) at room temperature and stirred the reaction mass for 2h. Concentrated the reaction mass under vacuum and diluted with ice water, acidified with 2N HC1 to pH=2. Extracted with ethyl acetate (3 50mL). The combined organic layer was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get desired title compound (2.3g, 71%). MS (ESI): 200.1 [M+H]+.

c. tert-Butyl (l-propyl-4-nitro-lH-pyrazol-3-yl)carbamate
Added TEA (2.3g, 23 mmol) at 0 °C to a solution of l-propyl-4-nitro-lH-pyrazole-3-carboxylic acid (2.3g, 11.5 mmol) in a mixture of DMF (18.4mL) and DPPA (4.76g, 17.2 mmol), stirred the reaction mass for 30min at room temperature followed by the addition of tert-butanol (6mL).
Heated the reaction mass to 80 °C slowly and maintained for 5-6h. Concentrated the reaction mass under vacuum and diluted with ethyl acetate (lOOmL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated under vacuum to get a crude residue. The residue was purified by silica gel column chromatography by eluting with 0-40% ethyl acetate- hexane to afford the title com ound (2.7g, 87%). MS (ESI): 170.15 (M-Boc+H)+.

d. 1-Propyl -4-nitro-lH-pyrazol-3-amine
TFA (lmL) was added dropwise to a solution of tert-butyl (l-propyl-4-nitro-lH-pyrazol-3- yl)carbamate (lg, 3.7 mmol) in DCM (3mL) at 0 °C under nitrogen atmosphere. Stirred the reaction mass at room temperature for 2h. Concentrated the reaction mass under vacuum and triturated with diethyl ether. Solid obtained dried under vacuum to yield the desired title compound (0.5g crude). MS (ESI): 171.2 [M+H]+.
Example 627

( )-3-chIoro-N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyI)-l-methylureido)pyrimidin-4- yl)amino)-l-ethyl-lH-pyrazol-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A by substituting l-ethyl-4-nitro-lH-pyrazol-3 -amine (preparation shown in Example 615) and Boc-protected 1- (6-chloropyrimidin-4-yl)-3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-methylurea (preparation shown in Example 615) in step (i) and cis-chloro acrylic acid, EDC1.HC1, and HOBt in step (k) to afford the title compound as free base (0.02g, 33%); 'HNMR (DMSO-C 400MHz): δ 12.08 (s, 1H), 9.87 (s, 1H), 9.48 (s, 1H), 8.42 (s, 1H), 8.15 (s, 1H), 7.05 (brs, 1H), 6.87-6.91 (m, 2H), 6.52 (d, 1H), 4.08 (q, 2H), 3.94 (s, 6H), 3.31 (s, 3H), 1.37 (t, 3H). MS (ESI): 571.3 (M+3).
Example 632

N-(3-((6-(3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-((tetrahydrofuran-3- yl)methyl)ureido)pyrimidin-4-yl)amino)-l-methyl-lH-pyrazol-4-yl)acrylamide
The compound was synthesized following the approach outlined in Procedure 6A substituting (tetrahydrofuran-3-yl)methanamine in step (e) and zinc/ammonium chloride in step (k) to afford the title compound (50 mg, yield: 19%). 1HNMR (CDC13, 400MHz): δ 12.4 (s, 1H), 8.45 (s, 1H), 7.92 (s, 1H), 7.56 (s, 1H), 6.65 (brs, 1H), 6.53 (s, 1H), 6.37 (d, 1H), 6.18-6.25 (m, 1H), 5.75 (dd, 1H), 4.15-4.20 (m, 1H), 3.93 (s, 6H), 3.76-3.88 (m, 5H), 3.66-3.69 (m, 1H), 2.75-2.79 (m, 2H), 2.01-2.06 (m, 2H), 1.81-1.85 (m, 2H); MS (ESI): 593.3 (M+3); HPLC: 97.43%, rt: 4.2 min.
Assays of Biological Activity
Assay of Binding to FGFR4. Purified, recombinant FGFR4 was pre-incubated with 10μΜ of either compound 601 or compound 609 for 1 hour at room temperature. FGFR4 was then concentrated and buffer exchanged on an OPTI-TRAP protein concentrating and desalting C4 column (Optimize Technologies). Protein was eluted in acetonitrile containing 0.1 % formic acid and run by direct injection on a Thermo Scientific Q Exactive LCMS to identify modified, intact FGFR4. Results are shown in Table 2.
TABLE 2

IC50 Profiling of Kinase Activity Inhibition. Compounds were profiled for FGFR inhibition activity at Reaction Biology Corporation (Malvern, Pennsylvania) with their Kinase
HotSpot assay. See, Anastassiadis et al., 2011, Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol 29, 1039-1045.
Recombinant FGFR1 (2.5 nM), FGFR2 (1 nM), FGFR3 (5 nM), or FGFR4 (12 nM) (Invitrogen™) was prepared as a mixture with substrate KKKSPGEYVNIEFG (SEQ ID NO:l) (20 μΜ, FGFR1 substrate); and Poly [E,Y] 4:1 (0.2 mg/ml, FGFR2,3,4 substrate)] in kinase reaction buffer (20 mM HEPES-HC1, pH 7.5, 10 mM MgCl2, 2 mM MnCl2, 1 mM EGTA, 0.02% Brij35, 0.1 mM Na3V04, 0.02 mg/ml BSA, 2 mM DTT, and 1% DMSO) (reagents from Reaction Biology Corp., Malvern, Pennsylvania). Compound was added to the enzyme/substrate mixture using acoustic technology (Labcyte Echo550) (Olechno et al., 2006, Improving IC50 results with acoustic droplet ejection. JALA 11, 240-246) and pre-incubated for 0, 15, or 60 minutes at room temperature. After compound pre-incubation, a mixture of ATP (Sigma- Aldrich®) and 33Ρ-γ-ΑΤΡ (PerkinElmer) was added to a final concentration of 10 μΜ to initiate kinase reactions. Reactions were incubated for 120 minutes at room temperature and then spotted onto P81 ion exchange filter paper (Whatman). Unbound phosphate was removed by extensively washing filters in 0.75% phosphoric acid. See, Anastassiadis et al., 2011, Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol 29, 1039-1045.
Results for FGFR4 and FGFR1 are shown next to individual compounds listed in Table 1 above. (The absence of a value in these tables indicates that it was untested.) Many of the compounds showed selective inhibition of FGFR4, with a higher IC50 for FGFR1.
Without wishing to be bound by theory, the IC50 activity of FGFR1 is generally representative of the activity of FGFR1, FGFR2, and FGFR3. See also, Dieci et al., 2013, Fibroblast Growth Factor Receptor Inhibitors as a Cancer Treatment: From a Biologic Rationale to Medical Perspectives. Cancer Discovery, F1-F16.
To confirm, compound 601 and compound 606 were also tested for FGFR2 and FGFR3 inhibition. These results shown below in Table 3 are consistent with the IC50 activity of FGFR1 being generally representative of the activity of FGFR1, FGFR2, and FGFR3, and further demonstrates the selectivity of these FGFR4 inhibitors.
TABLE 3
 Ba F3 Cellular Assays
Ba F3 Cellular Assays
Cell-based activity assays were performed with Ba/F3 cells. Ba/F3 is a murine, interleukin-3 dependent hematopoietic cell line, purchased from Riken Cell Bank (Ibaraki, Japan), and maintained in culture medium (RPMI-1640 (Gibco) supplemented with 10% FBS (Gibco) and 10 ng/mL recombinant mouse IL-3 (R&D Systems)). Ba/F3 is a model system for assessing the ability of kinase inhibitors to block kinase activity. See Warmuth et al., 2007, Ba/F3 cells and their use in kinase drug discovery, Curr Opin Oncolo, Jan;19(l):55-60.
Generation of Ba/F3-FGFR4 and Ba/F3-FGFR1 cell lines. Fusion proteins of Tel (1 to 149 aa) and FGFR4 (442-802 aa) or Tel (1 to 149 aa) and FGFR1 (470-822 aa) were generated by PCR based methods and cloned into pLenti vectors (Life technologies). Lentivirus was produced by transfection of pLenti vectors into 293T cells (Clontech laboratories). pLenti- FGFR4 or pLenti-FGFRl lentivirus were infected to the Ba/F3 parental cell lines (seeded 5E5 cells/mL, lmL/well in 12-well plate) to generate Ba/F3-FGFR4 or Ba/F3-FGFR1 by using 20ug/mL blasticidin selection marker.
Ba/F3-FGFR4 viability assay. 4,000 cells were dispensed into each well of a 96-well plate in 90 \xL of growth media (RPMI-1640 supplemented with 10%) FBS), and incubated for 24 hours at 37°C and 5% C02. 10 of serially diluted compound were added to appropriate wells in triplicates, and cells were incubated for 72 hours at 37°C and 5% C02. Viability was measured by adding 50 \xL of the CellTiter-Glo® (Promega) reagent and measuring luminescence, reported as relative light units (RLU). GI50 values were calculated by producing nine-point dose-response curves, with normalization to media control, DMSO control and day 0 RLU measurements taken at the time of compound addition. Best-fit curves were generated with Hill equation.
Ba/F3-FGFR1 viability assay. Assay was performed as described above for Ba/F3- FGFR4.
Ba/F3-Parental viability assay. Assay to test general cytotoxic effects was performed as described above for Ba/F3-FGFR4, except for a difference in the growth media (RPMI-1640 supplemented with 10% FBS and lOng/ml IL-3).
Results of these assays for example compounds are shown in Table 1 above.
The foregoing is illustrative of the present invention, and is not to be construed as limiting thereof. The invention is defined by the following claims, with equivalents of the claims to be included therein.
Claims
1. A compound of the following formula:

or a pharmaceutically acceptable salt thereof
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562145143P | 2015-04-09 | 2015-04-09 | |
| US62/145,143 | 2015-04-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016164754A1 true WO2016164754A1 (en) | 2016-10-13 |
Family
ID=55759934
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2016/026690 WO2016164754A1 (en) | 2015-04-09 | 2016-04-08 | Fgfr4 inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2016164754A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108239069A (en) * | 2016-12-26 | 2018-07-03 | 南京药捷安康生物科技有限公司 | It is a kind of novel for inhibitor of fibroblast growth factor acceptor and application thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7189715B2 (en) | 2002-10-24 | 2007-03-13 | Sepracor Inc. | Compositions comprising zopiclone derivatives and methods of making and using the same |
| WO2008042639A1 (en) * | 2006-10-02 | 2008-04-10 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| WO2008157131A1 (en) * | 2007-06-15 | 2008-12-24 | Irm Llc | Protein kinase inhibitors and methods for using thereof |
| WO2012073143A1 (en) | 2010-12-01 | 2012-06-07 | Pfizer Inc. | Kat ii inhibitors |
-
2016
- 2016-04-08 WO PCT/US2016/026690 patent/WO2016164754A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7189715B2 (en) | 2002-10-24 | 2007-03-13 | Sepracor Inc. | Compositions comprising zopiclone derivatives and methods of making and using the same |
| WO2008042639A1 (en) * | 2006-10-02 | 2008-04-10 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| WO2008157131A1 (en) * | 2007-06-15 | 2008-12-24 | Irm Llc | Protein kinase inhibitors and methods for using thereof |
| WO2012073143A1 (en) | 2010-12-01 | 2012-06-07 | Pfizer Inc. | Kat ii inhibitors |
Non-Patent Citations (24)
| Title |
|---|
| ANASTASSIADIS ET AL.: "Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity", NAT BIOTECHNOL, vol. 29, 2011, pages 1039 - 1045 |
| BARDERAS ET AL., J PROTEOMICS, vol. 75, 2012, pages 4647 - 4655 |
| BERGE ET AL., J. PHARM. SCI., vol. 66, 1977, pages 1 |
| CHIANG ET AL., CANCER RES, vol. 68, no. 16, 2008, pages 6779 - 6788 |
| DIECI ET AL., CANCER DISCOVERY, 2013, pages 0F I - 0F 16 |
| DIECI ET AL., CANCER DISCOVERY, 2013, pages 0F1 - 0F16 |
| DIECI ET AL.: "Fibroblast Growth Factor Receptor Inhibitors as a Cancer Treatment: From a Biologic Rationale to Medical Perspectives", CANCER DISCOVERY, 2013, pages F1 - F16 |
| FAWDAR ET AL., PNAS, vol. 110, no. 30, 2013, pages 12426 - 12431 |
| FRENCH ET AL., PLOS ONE, vol. 7, no. 5, 2012, pages E367313 |
| MIURA ET AL., BMC CANCER, vol. 12, 2012, pages 56 |
| MOTODA ET AL., INT'L J ONCOL, vol. 38, 2011, pages 133 - 143 |
| OLECHNO ET AL.: "Improving IC50 results with acoustic droplet ejection", JALA, vol. 11, 2006, pages 240 - 246 |
| PELAEZ-GARCIA ET AL., PLOS ONE, vol. 8, no. 5, 2013, pages E63695 |
| SANTOS, M.M.M.; MOREIRA, R., MINI-REVIEWS IN MEDICINAL CHEMISTRY, vol. 7, 2007, pages 1040 - 1050 |
| SAWEY ET AL., CANCER CELL, vol. 19, 2011, pages 347 - 358 |
| SIA ET AL., GASTROENTEROLOGY, vol. 144, 2013, pages 829 - 840 |
| STAHL ET AL.: "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", 2002, WILEY-VCH; VERLAG HELVETICA CHIMICA ACTA |
| STREIT ET AL., BRITISH J CANCER, vol. 94, 2006, pages 1879 - 1886 |
| TAYLOR VI ET AL., J CLIN INVEST, vol. 119, no. 11, 2009, pages 3395 - 3407 |
| WARMUTH ET AL.: "Ba/F3 cells and their use in kinase drug discovery", CURR OPIN ONCOLO, vol. 19, no. L, January 2007 (2007-01-01), pages 55 - 60 |
| WESCHE ET AL., BIOCHEM J, vol. 437, 2011, pages 199 - 213 |
| XU ET AL., BMC CANCER, vol. 11, 2011, pages 84 |
| YE ET AL., CANCER, 2011, pages 5304 - 5313 |
| ZAID ET AL., CLIN CANCER RES, vol. 19, 2013, pages 809 - 820 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108239069A (en) * | 2016-12-26 | 2018-07-03 | 南京药捷安康生物科技有限公司 | It is a kind of novel for inhibitor of fibroblast growth factor acceptor and application thereof |
| CN108239069B (en) * | 2016-12-26 | 2021-01-05 | 南京药捷安康生物科技有限公司 | Inhibitor for fibroblast growth factor receptor and application thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2018271284B2 (en) | Pyrimidine FGFR4 inhibitors | |
| KR102073797B1 (en) | Aminopyridazinone compounds as protein kinase inhibitors | |
| WO2016164703A1 (en) | Fgfr4 inhibitors | |
| EP2857404B1 (en) | IMIDAZO[1,2-b]PYRIDAZINE DERIVATIVES AS KINASE INHIBITORS | |
| RU2629194C2 (en) | Derivatives of 1,5- and 1,7-naphthyridine useful in treatment of fgfr-mediated diseases | |
| EP3173412B1 (en) | 2,4-disubstituted 7h-pyrrolo[2,3-d]pyrimidine derivative, preparation method and medicinal use thereof | |
| ES2899937T3 (en) | Pyrazolylquinoxaline kinase inhibitors | |
| WO2005080330A1 (en) | Heteroarylphenylurea derivative | |
| WO2016054987A1 (en) | Egfr inhibitor, and preparation and application thereof | |
| SG174964A1 (en) | 1-heterocyclyl-1, 5-dihydro-pyrazolo [3, 4-d] pyrimidin-4-one derivatives and their use as pde9a modulators | |
| CN109867676B (en) | Pyrrolopyrimidine derivative compound, pharmaceutical composition and application thereof | |
| BR112014017749B1 (en) | ALKNYLBENZENE COMPOSITE 3,5-DISPLACED AND SALT OF THE SAME | |
| WO2005082854A1 (en) | Novel pyridine derivative and pyrimidine derivative (1) | |
| KR20150001734A (en) | 3-aryl-5-substituted-isoquinolin-1-one compounds and their therapeutic use | |
| JP2013534221A (en) | 4- (1H-Indol-3-yl) -pyrimidine as an ALK inhibitor | |
| JP2020512315A (en) | Heteroaryl compounds useful as MK2 inhibitors | |
| WO2013118817A1 (en) | Quinolyl pyrrolopyrimidine compound or salt thereof | |
| JP2024505732A (en) | Pyridopyrimidinone derivatives and their production methods and uses | |
| WO2023067546A1 (en) | Novel bicyclic heteroaryl derivatives as sos1:kras proteinprotein interaction inhibitors | |
| WO2020207476A1 (en) | Pyrazolopyrazine derived compounds, pharmaceutical composition and use thereof | |
| JP2023145548A (en) | Cd73 inhibitor, preparation method therefor and application thereof | |
| WO2020207260A1 (en) | Cdk inhibitor and application thereof | |
| CN111655690B (en) | Pyrazolopyridinone compounds | |
| KR20200090828A (en) | Pyrazolopyridinone compounds | |
| WO2016164754A1 (en) | Fgfr4 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16717230 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16717230 Country of ref document: EP Kind code of ref document: A1 |