WO2016150902A1 - Use of 4-(4-fluoro-2-methoxyphenyl)-n-{3-[(s-methylsulfonimidoyl)methyl]phenyl}-1,3,5-triazin-2-amine for treating gastric cancers - Google Patents
Use of 4-(4-fluoro-2-methoxyphenyl)-n-{3-[(s-methylsulfonimidoyl)methyl]phenyl}-1,3,5-triazin-2-amine for treating gastric cancers Download PDFInfo
- Publication number
- WO2016150902A1 WO2016150902A1 PCT/EP2016/056108 EP2016056108W WO2016150902A1 WO 2016150902 A1 WO2016150902 A1 WO 2016150902A1 EP 2016056108 W EP2016056108 W EP 2016056108W WO 2016150902 A1 WO2016150902 A1 WO 2016150902A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methoxyphenyl
- amine
- amplification
- methylsulfonimidoyl
- fluoro
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- the present invention relates to the use of 4-(4-Fluoro-2-methoxyphenyl)-N- ⁇ 3-[(S- methylsulfonimidoyl)methyl]phenyl ⁇ -l,3,5-triazin-2-amine (compound A), more particularly (+)-4-(4- Fluoro-2-methoxyphenyl)-N- ⁇ 3-[(S-methylsulfonimidoyl)methyl]phenyl ⁇ -l,3,5-triazin-2-amine (compound A'), for treating gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene.
- CDK cyclin-dependent kinase
- the family of cyclin-dependent kinase (CDK) proteins consists of members that are key regulators of the cell division cycle (cell cycle CDK's), that are involved in regulation of gene transcription (transcriptional CDK's), and of members with other functions. CDKs require for activation the association with a regulatory cyclin subunit.
- the cell cycle CDKs CDKl/cyclin B, CDK2/cyclin A, CDK2/cyclinE, CDK4/cyclinD, and CDK6/cyclinD get activated in a sequential order to drive a cell into and through the cell division cycle.
- Positive transcription factor b P-TEFb
- CDK9 NCBI GenBank Gene ID 1025
- CDK7 in addition participates in cell cycle regulation as CDK-activating kinase (CAK).
- RNA polymerase II Transcription of genes by RNA polymerase II is initiated by assembly of the pre-initiation complex at the promoter region and phosphorylation of Ser 5 and Ser 7 of the CTD by CDK7/cyclin H. For a major fraction of genes RNA polymerase II stops mRNA transcription after it moved 20-40 nucleotides along the DNA template. This promoter-proximal pausing of RNA polymerase ⁇ is mediated by negative elongation factors and is recognized as a major control mechanism to regulate expression of rapidly induced genes in response to a variety of stimuli (Cho et al., Cell Cycle 2010, 9, 1697).
- P-TEFb is crucially involved in overcoming promoter-proximal pausing of RNA polymerase ⁇ and transition into a productive elongation state by phosphorylation of Ser 2 of the CTD as well as by phosphorylation and inactivation of negative elongation factors.
- P-TEFb activity is regulated by several mechanisms. About half of cellular P-TEFb exists in an inactive complex with 7SK small nuclear RNA (7SK snRNA), La-related protein 7 (LARP7/PIP7S) and hexamethylene bis-acetamide inducible proteins 1/2 (HEXIM1/2, He et al., Mol. Cell 2008, 29, 588). The remaining half of P-TEFb exists in an active complex containing the bromodomain protein Brd4 (Yang et al., Mol. Cell 2005, 19, 535). Brd4 recruits P-TEFb through interaction with acetylated histones to chromatin areas primed for gene transcription.
- 7SK snRNA 7SK small nuclear RNA
- LRP7/PIP7S La-related protein 7
- HEXIM1/2 hexamethylene bis-acetamide inducible proteins 1/2
- Brd4 recruits P-TEFb through interaction with acetylated histones to chromatin areas primed
- P-TEFb is maintained in a functional equilibrium: P-TEFb bound to the 7SK snRNA complex represents a reservoir from which active P-TEFb can be released on demand of cellular transcription and cell proliferation (Zhou & Yik, Microbiol. Mol. Biol. Rev. 2006, 70, 646). Furthermore, the activity of P-TEFb is regulated by posttranslational modifications including phosphorylation/de-phosphorylation, ubiquitination, and acetylation (reviewed in Cho et al., Cell Cycle 2010, 9, 1697).
- Deregulated CDK9 kinase activity of the P-TEFb heterodimer is associated with a variety of human pathological settings such as hyper-proliferative diseases (e.g. cancer), virally induced infectious diseases or cardiovascular diseases.
- hyper-proliferative diseases e.g. cancer
- virally induced infectious diseases e.g. cancer
- cardiovascular diseases e.g. cancer
- Cancer is regarded as a hyper-proliferative disorder mediated by a disbalance of proliferation and cell death (apoptosis).
- High levels of anti-apoptotic Bcl-2-family proteins are found in various human tumours and account for prolonged survival of tumour cells and therapy resistance.
- Inhibition of P- TEFb kinase activity was shown to reduce transcriptional activity of RNA polymerase ⁇ leading to a decline of short-lived anti-apoptotic proteins, especially Mcl-l and XIAP, reinstalling the ability of tumour cells to undergo apoptosis.
- a number of other proteins associated with the transformed tumour phenotype are either short-lived proteins or are encoded by short-lived transcripts which are sensitive to reduced RNA polymerase II activity mediated by P-TEFb inhibition (reviewed in Wang & Fischer, Trends Pharmacol. Sci. 2008, 29, 302).
- Tat viral transcription activator
- Cardiac hypertrophy the heart's adaptive response to mechanical overload and pressure (hemodynamic stress e.g. hypertension, myocardial infarction), can lead, on a long term, to heart failure and death. Cardiac hypertrophy was shown to be associated with increased transcriptional activity and RNA polymerase ⁇ CTD phosphorylation in cardiac muscle cells. P-TEFb was found to be activated by dissociation from the inactive 7SK snRNA/HEXEMl/2 complex. These findings suggest pharmacological inhibition of P-TEFb kinase activity as a therapeutic approach to treat cardiac hypertrophy (reviewed in Dey et al., Cell Cycle 2007, 6, 1856).
- CDK9 belongs to a family of at least 13 closely related kinases of which the subgroup of the cell cycle CDK's fulfils multiple roles in regulation of cell proliferation.
- co- inhibition of cell cycle CDK's e.g.
- CDKl/cyclin B, CDK2/cyclin A, CDK2/cyclinE, CDK4/cyclinD, CDK6/cyclinD) and of CDK9 is expected to impact normal proliferating tissues such as intestinal mucosa, lymphatic and hematopoietic organs, and reproductive organs.
- CDK9 kinase inhibitors molecules with high selectivity towards CDK9 are therefore required.
- CDK inhibitors in general as well as CDK9 inhibitors are described in a number of different publications: WO2008129070 and WO2008129071 both describe 2,4 substituted aminopyrimidines as CDK inhibitors in general. It is also asserted that some of these compounds may act as selective CDK9 inhibitors (WO2008129070) and as CDK5 inhibitors (WO2008129071), respectively, but no specific CDK9 IC50 (WO2008129070) or CDK5 IC50 (WO200812971) data is presented.
- WO2008129080 discloses 4,6 substituted aminopyrimidines and demonstrates that these compounds show an inhibitory effect on the protein kinase activity of various protein kinases, such as CDK1, CDK2, CDK4, CDK5, CDK6 and CDK9, with a preference for CDK9 inhibition (example 80).
- EP1218360 Bl describes triazin derivatives as kinase inhibitors, but does not disclose potent or selective CDK9 inhibitors.
- WO2008079933 discloses aminopyridine and aminopyrimidine derivatives and their use as CDK1, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8 or CDK9 inhibitors.
- WO2011012661 describes aminopyridine derivatives useful as CDK inhibitors. Wang et al. (Chemistry & Biology 2010, 17, 1111-1121) describe 2-anilino-4-(thiazol-5-yl)pyrimidine transcriptional CDK inhibitors, which show anticancer activity in animal models. WO2004009562 discloses substituted triazine kinase inhibitors. For selected compounds CDKl and CDK 4 test data, but no CDK9 data is presented.
- WO2004072063 describes heteroaryl (pyrimidine, triazine) substituted pyrroles as inhibitors of protein kinases such as ERK2, GSK3, PKA or CDK2.
- WO2010009155 discloses triazine and pyrimidine derivatives as inhibitors of histone deacetylase and/or cyclin dependent kinases (CDKs). For selected compounds CDK2 test data is described.
- WO2003037346 (corresponding to US7618968B2, US7291616B2, US2008064700A1, US2003153570A1) relates to aryl triazines and uses thereof, including to inhibit lysophosphatidic acid acyltransferase beta (LPAAT-beta) activity and/or proliferation of cells such as tumour cells.
- LPAAT-beta lysophosphatidic acid acyltransferase beta
- WO2008025556 describes carbamoyl sulfoximides having a pyrimidine core, which are useful as kinase inhibitors. No CDK9 data is presented.
- WO2002066481 describes pyrimidine derivatives as cyclin dependent kinase inhibitors CDK9 is not mentioned and no CDK9 data is presented.
- WO2008109943 concerns phenyl aminopyri(mi)dine compounds and their use as kinase inhibitors, in particular as JAK2 kinase inhibitors.
- the specific examples focus on compounds having a pyrimidine core.
- WO2009032861 describes substituted pyrimidinyl amines as JNK kinase inhibitors.
- the specific examples focus on compounds having a pyrimidine core.
- WO2011046970 concerns amino-pyrimidine compounds as inhibitors of TBKL and/or ⁇ epsilon.
- the specific examples focus on compounds having a pyrimidine core.
- WO2012160034 the compounds of the present invention. It is disclosed the compounds inhibit the cell proliferation of HeLa cells (cervical cancer), HeLa/MaTu/ADR cells (cervical cancer), NCI-H460 cells (non-small cell lung cancer), DU145 cells (hormone-independent human prostate cancer), Caco-2 cells (colorectal cancer) and B 16F10 cells (melanoma).
- the object of the present invention is to improve the treatment of gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene. Treatment of gastric cancer
- Gastric cancer is an aggressive disease and the second leading cause of cancer-related mortality worldwide (Jemal A et al. Global cancer statistics. CA Cancer J Clin 2011; 61(2): 69-90). Dietary improvements and reduction in H. pylori infections, due to the use of antibiotics, have resulted in a steady fall in incidence and mortality rates, however, the prognosis for gastric cancer patients remains poor in Western countries (Lordick F. Unmet needs and challenges in gastric cancer: The way forward. Can Treat Rev 2014; 40: 692-700). This is in stark contrast to the overall five-year survival rate of gastric cancer patients in Japan, where regular screening leading to early stage diagnosis has resulted in 70% patient survival (European Union Network of Excellence (EUNE) for Gastric Cancer Steering Group. Gastric cancer in Europe. Br J Surg 2008; 95:406 ⁇ 108). Unfortunately, early stage gastric cancer rarely presents symptoms that cannot be explained by other factors and thus remains undiagnosed until it has progressed to an advanced metastatic phase.
- EUNE European Union
- the single curative therapy for localised late stage gastric cancer is surgical resection where tumour removal coupled with extensive lymph node dissection and long-term follow-up results in increased efficacy.
- Preoperative chemotherapy consisting of epirubicin, cisplatin and capecitabine (or other platinum/fluoropyrimidine combinations) has also proven beneficial in improving overall survival rates and has been widely adopted as the standard of care in Europe.
- ESMO-ESSO-ESTRO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Radiotherapy and Oncology 2014;110: 189-194 In the case of inoperable, recurring or metastatic cancers, chemotherapy and best supportive care provide enhanced quality of life and improved patient prognosis. However, response to first-line chemotherapeutic agents still remains poor.
- trastuzumab an anti-HER2 (epidermal growth factor receptor kinase 2) antibody, in combination with cisplatin and fluoropyrimidine, leading to enhanced overall survival was granted FDA approval in 2010 for the treatment of patients with HER2-overexpressing metastatic gastric cancer.
- HER2 epidermal growth factor receptor kinase 2
- trastuzumab is a great example of how advances in molecular characterisation of tumours can be translated into successful and targetable therapeutics (Bang YJ et al., Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2 -positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial, Lancet 2010; 376 (9742): 687-697).
- MYC Molecular pathways and ongoing investigations. Biochimica et Biophysica Acta 2014;1846: 232-7). Deregulated MYC expression underlies the pathogenesis of numerous human neoplasms, and seems to be at the crossroad of many important pathways and processes involved in carcinogenesis (Fletcher S et al. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 2014).
- the C-MYC gene is an important member of the MYC proto-oncogene family and several studies have demonstrated an association between C-MYC expression and gastric cancer.
- C-MYC overexpression has been described in over 40% of gastric cancers (Milne AN et al. Early onset gastric cancer: on the road to unravelling gastric carcinogenesis. Curr Mol Med 2007;7(1): 15-28) and in both intestinal- and diffuse-type gastric adenocarcinomas (Calcagno DQ et al. Interrelationship between chromosome 8 aneuploidy, C-MYC amplification and increased expression in individuals from northern Brazil with gastric adenocarcinoma.
- C-MYC amplification may be an important instigator of gastric cancer and disease progression potentially due to the fundamental role of this transcription factor in the regulation of cell proliferation and apoptosis.
- Gene amplification also known as gene duplication or chromosomal duplication, is a cellular process in which multiple copies of a gene are produced. The genes on each of the copies can be transcribed and translated, leading to an overproduction of the mRNA and protein corresponding to the amplified genes. Gene amplifications are important features of many advanced cancers and have prognostic as well as therapeutic significance in clinical cancer treatment (Myllykangas S, Knuutila S. manifestation, mechanisms and mystery of gene amplifications. Cancer Lett 2006; 232(l):79-89). Various methods can be used to detect gene amplifications in clinical samples.
- cytogenetics chromosome banding
- CGH chromosomal comparative genomic hybridization
- FISH fluorescent in situ hybridization
- NGS next-generation sequencing
- Double -hit mantle cell lymphoma with MYC gene rearrangement or amplification a report of four cases and review of the literature, hit J Clin Exp Pathol 2013;6(2): 155- 67; Balko JM et al. Molecular profiling of the residual disease of triple -negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets.
- compound A is a selected sulphoximine-substituted anilinopyrimidine derivative which can be separated into two stereoisomers, viz.:
- Compound A' is preferred and in clinical development as BAY1143572.
- the present invention is directed to the use of
- gastric cancers for the treatment and/or prophylaxis of gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene.
- Preferred is the use of compound A' for the treatment and/or prophylaxis of gastric cancers in which cells have an amplification of the C-MYC gene.
- the methods for detecting this amplification include, but are not limited to conventional cytogenetics (chromosome banding), chromosomal comparative genomic hybridization (CGH), fluorescent in situ hybridization (FISH) next-generation sequencing (NGS), multiplex ligation-dependent probe amplification (MLPA), PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
- CGH chromosomal comparative genomic hybridization
- FISH fluorescent in situ hybridization
- NGS next-generation sequencing
- MLPA multiplex ligation-dependent probe amplification
- PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
- the present application is further directed to the use of
- Another aspect of the present invention is the use of
- Preferred is the use of compound A' in the manufacture of a medicament for treating gastric cancers in which cells have an amplification of the C-MYC gene.
- the methods for detecting this amplification include, but are not limited to
- cytogenetics chromosome banding
- CGH chromosomal comparative genomic hybridization
- FISH fluorescent in situ hybridization
- NGS next-generation sequencing
- MLPA multiplex ligation-dependent probe amplification
- qPCR quantitative PCR
- SNP single nucleotide polymorphism
- treating gastric cancer preferably gastric cancers in which cells have an amplification of the C-MYC gene.
- gastric cancers in which cells have an amplification of the C-MYC gene preferably gastric cancers in which cells have an amplification of the C-MYC gene.
- compound A' for the use of treating gastric cancers in which cells have an amplification of the C-MYC gene.
- the methods for detecting this amplification include, but are not limited to
- cytogenetics chromosome banding
- CGH chromosomal comparative genomic hybridization
- FISH fluorescent in situ hybridization
- NGS next-generation sequencing
- MLPA multiplex ligation-dependent probe amplification
- PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
- the present invention is also directed to
- gastric cancers in which cells have an amplification of the C-MYC gene preferably gastric cancers in which cells have an amplification of the C-MYC gene.
- compound A' for the use in a method of treatment and/or prophylaxis of gastric cancers in which cells have an amplification of the C-MYC gene.
- the methods for detecting this amplification include, but are not limited to
- cytogenetics chromosome banding
- CGH chromosomal comparative genomic hybridization
- FISH fluorescent in situ hybridization
- NGS next-generation sequencing
- MLPA multiplex ligation-dependent probe amplification
- PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
- Another aspect of the present invention is
- said method comprising the steps:
- Another aspect of the present invention is a
- Another aspect of the present invention is a method of treatment and/or prophylaxis of gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene using an effective amount of 4-(4-Fluoro-2-methoxyphenyl)-N- ⁇ 3-[(S-methylsulfonimidoyl)methyl]phenyl ⁇ - 1 ,3,5- triazin-2-amine (compound A) of formula I or one of its physiologically acceptable salts or enantiomers,
- a preferred method of treatment is a method of treatment and/or prophylaxis of gastric cancers in which cells have an amplification of the C-MYC gene using an effective amount of compound A'.
- the methods for detecting this amplification include, but are not limited to
- cytogenetics chromosome banding
- CGH chromosomal comparative genomic hybridization
- FISH fluorescent in situ hybridization
- NGS next-generation sequencing
- MLPA multiplex ligation-dependent probe amplification
- PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
- the present application further provides pharmaceutical compositions containing
- gastric cancer preferably gastric cancers in which cells have an amplification of the C- MYC gene.
- the present invention is also directed to pharmaceutical compositions comprising 4-(4-Fluoro-2-methoxyphenyl)-N- ⁇ 3-[(S-methylsulfonimidoyl)methyl]phenyl ⁇ -l,3,5-triazin-2-amine (compound A) of formula I or one of its hysiologically acceptable salts or enantiomers,
- a preferred pharmaceutical composition is a pharmaceutical composition comprising compound A' for the treatment of gastric cancers in which cells have an amplification of the C-MYC gene.
- the methods for detecting this amplification include, but are not limited to
- cytogenetics chromosome banding
- CGH chromosomal comparative genomic hybridization
- FISH fluorescent in situ hybridization
- NGS next-generation sequencing
- MLPA multiplex ligation-dependent probe amplification
- qPCR quantitative PCR
- SNP single nucleotide polymorphism arrays
- Southern blotting and slot blot methods PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
- qPCR quantitative PCR
- SNP single nucleotide polymorphism
- gastric cancer preferably gastric cancers in which cells have an amplification of the C-MYC gene.
- the present invention is also directed to
- gastric cancer preferably gastric cancers in which cells have an amplification of the C-MYC gene.
- a preferred pharmaceutical combination is a pharmaceutical combination comprising compound A' for the treatment of gastric cancers in which cells have an amplification of the C-MYC gene.
- the methods for detecting this amplification include, but are not limited to conventional cytogenetics (chromosome banding), chromosomal comparative genomic hybridization (CGH), fluorescent in situ hybridization (FISH), next-generation sequencing (NGS), multiplex ligation-dependent probe amplification (MLPA), PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
- CGH chromosomal comparative genomic hybridization
- FISH fluorescent in situ hybridization
- NGS next-generation sequencing
- MLPA multiplex ligation-dependent probe amplification
- PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
- Another aspect of the present invention is a method for identifying a patient disposed to respond favorably to a CDK9-inhibitor for treating gastric cancer
- CDK9-inhibitor is compound A and
- the method comprises the detection of an amplification of the C-MYC gene in tumour cells in a tissue sample from the patient.
- Preferred is a method for identifying a patient disposed to respond favorably to a CDK9-inhibitor for treating gastric cancer
- CDK9-inhibitor is compound A' and
- the method comprises the detection of an amplification of the C-MYC gene in tumour cells in a tissue sample from the patient and
- Another aspect of the present invention is a method for identifying a patient disposed to respond favorably to
- the method comprises the detection of C-MYC amplification in tumor cells in a tissue sample from the patient and
- the methods for detecting this amplification include, but are not limited to
- cytogenetics chromosome banding
- CGH chromosomal comparative genomic hybridization
- FISH fluorescent in situ hybridization
- NGS next-generation sequencing
- MLPA multiplex ligation-dependent probe amplification
- PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
- Another aspect of the present invention is a method of predicting whether a patient will be respond to the treatment with
- the method comprises the detection of C-MYC amplification in tumour cells in a tissue sample from the patient.
- the methods for detecting this amplification include, but are not limited to
- cytogenetics chromosome banding
- CGH chromosomal comparative genomic hybridization
- FISH fluorescent in situ hybridization
- NGS next-generation sequencing
- MLPA multiplex ligation- dependent probe amplification
- PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
- Physiologically safe salts of compound A encompass acid addition salts of mineral acids, carboxylic acids and sulphonic acids, for example salts of hydrochloric acid, hydrobromic acid, sulphuric acid, phosphoric acid, methanesulphonic acid, ethanesulphonic acid, toluenesulphonic acid, benzenesulphonic acid, naphthalenedisulphonic acid, acetic acid, trifluoroacetic acid, propionic acid, lactic acid, tartaric acid, malic acid, citric acid, fumaric acid, maleic acid and benzoic acid.
- Physiologically safe salts of compound A also encompass salts of customary bases, such as, by way of example and preferably, alkali metal salts (e.g. sodium and potassium salts), alkaline earth metal salts (e.g. calcium and magnesium salts) and ammonium salts derived from ammonia or organic amines having from 1 to 16 C atoms, such as, by way of example and preferably, ethylamine, diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, arginine, lysine, ethylenediamine and N-methylpiperidine.
- alkali metal salts e.g. sodium and potassium salts
- alkaline earth metal salts e.g. calcium and magnesium salts
- the present invention further provides drugs containing compound A and at least one or more further active ingredients for treating gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene.
- Compound A may have systemic and/or local activity.
- it can be administered in a suitable manner, such as, for example, orally, parenterally, via the pulmonary route, nasal, sublingually, lingually, buccally, rectally, vaginally, dermally, transdermally, conjuntivally, otically or as an implant or stent.
- compound A according to the invention may be administered in suitable administration forms.
- Suitable for oral administration forms which function according to the prior art and deliver compound A of the invention rapidly and/or in a modified manner and which comprise compound A according to the invention in crystalline and/or amorphised and/or dissolved form, such as, for example, tablets (uncoated or coated tablets, for example with coatings which are resistant to gastric juice or dissolve with a delay or are insoluble and control the release of the compound of the invention), tablets which disintegrate rapidly in the oral cavity, or films/wafers, films/lyophilisates, capsules (for example hard or soft gelatine capsules), sugar-coated tablets, granules, pellets, powders, emulsions, suspensions, aerosols or solutions.
- tablets uncoated or coated tablets, for example with coatings which are resistant to gastric juice or dissolve with a delay or are insoluble and control the release of the compound of the invention
- tablets which disintegrate rapidly in the oral cavity or films/wafers, films/lyophilisates
- capsules for example hard or soft ge
- Parenteral administration can be effected with avoidance of an absorption step (for example intravenous, intraarterial, intracardial, intraspinal or intralumbal) or with inclusion of absorption (for example intramuscular, subcutaneous, intracutaneous, percutaneous or intraperidoneal).
- absorption step for example intravenous, intraarterial, intracardial, intraspinal or intralumbal
- absorption for example intramuscular, subcutaneous, intracutaneous, percutaneous or intraperidoneal.
- Administration forms which are suitable for parenteral administration are, inter alia, preparations for injection and infusion in the form of solutions, suspensions, emulsions, lyophilisates or sterile powders.
- Examples which are suitable for other administration routes are pharmaceutical forms for inhalation [inter alia power inhalers, nebulizers], nasal drops, solutions, sprays; tablets, films/wafers or capsules, to be administered lingually, sublingually or buccaly, suppositories, preparations for the eyes and the ears, eye baths, ocular insert, ear drops, ear powders, ear-rinses, ear tampons, vaginal capsules, aqueous suspensions (lotions, mixturae agitandae), lipophilic suspensions, ointments, creams, transdermal therapeutic systems (such as, for example, patches), milk, pastes, foams, dusting powders, implants or stents.
- Compound A can be converted into the stated administration forms. This can be effected in a manner known per se by mixing with inert, non-toxic, pharmaceutically suitable adjuvants.
- adjuvants include, inter alia,
- fillers and excipients for example cellulose, microcrystalline cellulose, such as, for example, Avicel®, lactose, mannitol, starch, calcium phosphate such as, for example, Di-Cafos®),
- ointment bases for example petroleum jelly, paraffins, triglycerides, waxes, wool wax, wool wax alcohols, lanolin, hydrophilic ointment, polyethylene glycols
- ointment bases for example petroleum jelly, paraffins, triglycerides, waxes, wool wax, wool wax alcohols, lanolin, hydrophilic ointment, polyethylene glycols
- bases for suppositories for example polyethylene glycols, cacao butter, hard fat
- solvents for example water, ethanol, Isopropanol, glycerol, propylene glycol, medium chain- length triglycerides fatty oils, liquid polyethylene glycols, paraffins
- surfactants for example sodium dodecyle sulphate, lecithin, phospholipids, fatty alcohols such as, for example, Lanette®, sorbitan fatty acid esters such as, for example, Span®, polyoxyethylene sorbitan fatty acid esters such as, for example, Tween®, polyoxyethylene fatty acid glycerides such as, for example, Cremophor®, polyoxethylene fatty acid esters, polyoxyethylene fatty alcohol ethers, glycerol fatty acid esters, poloxamers such as, for example, Pluronic®),
- surfactants for example sodium dodecyle sulphate, lecithin, phospholipids, fatty alcohols such as, for example, Lanette®, sorbitan fatty acid esters such as, for example, Span®, polyoxyethylene sorbitan fatty acid esters such as, for example, Tween®, polyoxyethylene fatty acid glycerides such as, for example, Crem
- buffers and also acids and bases for example phosphates, carbonates, citric acid, acetic acid, hydrochloric acid, sodium hydroxide solution, ammonium carbonate, trometamol, triethanolamine
- isotonicity agents for example glucose, sodium chloride
- adsorbents for example highly-disperse silicas
- viscosity-increasing agents for example polyvinylpyrrolidon, methylcellulose, hydroxypropylmethylcellulose, hydroxypropylcellulose, carboxymethylcellulose-sodium, starch, carbomers, polyacrylic acids such as, for example, Carbopol®, alginates, gelatine),
- disintegrants for example modified starch, carboxymethylcellulose-sodium, sodium starch glycolate such as, for example, Explotab®, cross- linked polyvinylpyrrolidon, croscarmellose- sodium such as, for example, AcDiSol®
- modified starch carboxymethylcellulose-sodium, sodium starch glycolate such as, for example, Explotab®, cross- linked polyvinylpyrrolidon, croscarmellose- sodium such as, for example, AcDiSol®
- disintegrants for example modified starch, carboxymethylcellulose-sodium, sodium starch glycolate such as, for example, Explotab®, cross- linked polyvinylpyrrolidon, croscarmellose- sodium such as, for example, AcDiSol®
- flow regulators for example magnesium stearate, stearic acid, talc, highly-disperse silicas such as, for example, Aerosil®
- lubricants for example magnesium stearate, stearic acid, talc, highly-disperse silicas such as, for example, Aerosil®
- coating materials for example sugar, shellac
- film formers for films or diffusion membranes which dissolve rapidly or in a modified manner for example polyvinylpyrrolidones such as, for example, Kollidon®, polyvinyl alcohol, hydroxypropylmethylcellulose, hydroxypropylcellulose, ethylcellulose, hydroxypropylmethylcellulose phthalate, cellulose acetate, cellulose acetate phthalate, polyacrylates, polymethacrylates such as, for example, Eudragit®),
- capsule materials for example gelatine, hydroxypropylmethylcellulose
- synthetic polymers for example polylactides, polyglycolides, polyacrylates, polymethacrylates such as, for example, Eudragit®, polyvinylpyrrolidones such as, for example, Kollidon®, polyvinyl alcohols, polyvinyl acetates, polyethylene oxides, polyethylene glycols and their copolymers and blockcopolymers
- synthetic polymers for example polylactides, polyglycolides, polyacrylates, polymethacrylates such as, for example, Eudragit®, polyvinylpyrrolidones such as, for example, Kollidon®, polyvinyl alcohols, polyvinyl acetates, polyethylene oxides, polyethylene glycols and their copolymers and blockcopolymers
- plasticisers for example polyethylene glycols, propylene glycol, glycerol, triacetine, triacetyl citrate, dibutyl phthalate
- plasticisers for example polyethylene glycols, propylene glycol, glycerol, triacetine, triacetyl citrate, dibutyl phthalate
- stabilisers for example antioxidants such as, for example, ascorbic acid, ascorbyl palmitate, sodium ascorbate, butylhydroxyanisole, butylhydroxytoluene, propyl gallate
- antioxidants such as, for example, ascorbic acid, ascorbyl palmitate, sodium ascorbate, butylhydroxyanisole, butylhydroxytoluene, propyl gallate
- preservatives for example parabens, sorbic acid, thiomersal, benzalkonium chloride, chlorhexidine acetate, sodium benzoate
- preservatives for example parabens, sorbic acid, thiomersal, benzalkonium chloride, chlorhexidine acetate, sodium benzoate
- colourants for example inorganic pigments such as, for example, iron oxides, titanium dioxide
- flavourings sweeteners, flavour- and/or odour-masking agents.
- the present invention furthermore relates to medicaments which comprise at least one compound according to the invention, conventionally together with one or more inert, non-toxic, pharmaceutically suitable adjuvants, and to their use for the above mentioned purposes.
- the dosage and the treatment regimen can and must be varied depending on the carcinoma type and the treatment goal.
- the daily dose is generally between 20 mg and 850 mg and can be divided into a plurality of identical or different dosage units, preferably 2 which can be taken simultaneously or according to a certain time schedule.
- the daily dose is between 30 mg and 500 mg and can be divided into a plurality of identical or different dosage units, preferably 2 which can be taken simultaneously or according to a certain time schedule.
- a preferred daily dose is between 20 mg and 400 mg and can be divided into a plurality of identical or different dosage units, preferably 2 which can be taken simultaneously or according to a certain time schedule.
- the daily dose is between 40 mg and 300 mg and can be divided into a plurality of identical or different dosage units, preferably 2 which can be taken simultaneously or according to a certain time schedule.
- a more preferred daily dose is between 20 mg and 200 mg and can be divided into a plurality of identical or different dosage units, preferably 2 which can be taken simultaneously or according to a certain time schedule.
- An even more preferred daily dose is between 50 mg and 180 mg and can be divided into a plurality of identical or different dosage units, preferably 2 which can be taken simultaneously or according to a certain time schedule.
- Treatment can be carried out in regularly repeated cycles. Treatment cycles may have varying duration, such as 21 days or 28 days, whereby dosing is given continuously, or intermittently. Preferred is a cycle length of 28 days, whereby dosing is given continuously, or intermittently.
- Continuous schedules involve daily dosing, for example, 21 daily doses in a 21 -day cycle, or 28 daily doses in a 28-day cycle.
- a preferred continuous schedule is 28 daily doses in a 28 daily cycle.
- Intermittent schedules involve a period of treatment followed by a period of non-treatment, for example in a cycle of 21 days, or a cycle of 28 days.
- a preferred cycle duration for an intermittent schedule is 28 days.
- the period of treatment may be repeated more than once in a given treatment cycle.
- the period of treatment may be for example 1 to 21 days, more preferably 3 to 14 days.
- An even more preferred intermittent schedule involves treatment for 3 days followed by non-treatment for 4 days, repeated every week in such a way that a 28-day treatment cycle is completed.
- Treatment is successful when there is at least disease stabilization and the adverse effects occur to an extent which is easily treatable, but at least easily acceptable.
- the number of cycles of treatment applied may vary from patient to patient, according to treatment response and tolerability.
- Treatment is successful when there is at least disease stabilization and the adverse effects occur to an extent which is easily treatable, but at least easily acceptable.
- Compound A can be used on its own or, if required, in combination with one or more other pharmacologically effective substances, provided said combination does not lead to undesired and unacceptable adverse effects.
- the present invention therefore further provides drugs containing compound A according to the invention and one or more further active ingredients, in particular for treating and/or preventing the above-mentioned diseases.
- compound A can be combined with known anti-hyperproliferative, cytostatic or cytotoxic substances for treating cancers.
- the combination compound A according to the invention with other substances in use for cancer therapy or else with radiotherapy is especially advisable.
- Suitable active ingredients for combination purposes include:
- compound A of the present invention can be combined with the following active ingredients:
- Compound A can also achieve positive effects in combination with other therapies directed against angiogenesis, such as, for example, with avastin, axitinib, regorafenib, recentin, sorafenib or sunitinib.
- Combinations with inhibitors of the proteasome and of mTOR and also antihormones and steroidal metabolic enzyme inhibitors are especially useful because of their favourable profile of adverse effects.
- compound A according to the invention can also be used in connection with radiotherapy and/or a surgical intervention.
- Table 1 List of the cell lines investigated and results of the proliferation assays.
- the aim of the present experiments was to assess the in vivo efficacy and tolerability of Compound A' in monotherapy and in combination with paclitaxel or cisplatin in two gastric cancer models subcutaneously implanted in NMRI nu/nu mice.
- Compound A' was assessed at one dose level in mono- and in combination therapy with paclitaxel and cisplatin.
- Anti-tumour activity and tolerability of all groups were assessed using the vehicle control group as a reference.
- Cisplatin 2 1,4,7,10,13,16,19,22,25, 28 i.p. 3.3 Experimental procedures
- the animals were housed in individually ventilated cages. The animals were monitored twice daily. All materials were autoclaved prior to use. Food and water were provided ad libitum.
- the patient-derived GXF 251 tumour model used in this study was obtained from surgical specimens from cancer patients.
- GXA SCH LX was a cell line-derived cancer model (Oncotest, Freiburg, Germany). Both of these gastric cancer xenografts exhibit C-MYC amplification.
- Gastric cancer tumour fragments were obtained from xenografts in serial passage in nude mice and placed in PBS containing 10% penicillin/streptomycin. Tumour fragments (one fragment per animal; 3- 4 mm edge length) were then subcutaneously implanted in the flank of NMRI nu/nu recipient mice under isofluorane anaesthesia.
- Vehicle Compound A' 80% (m/V) PEG400 in water for injection
- Compound A' preparation of a dosing solution (2.5 mg/ml) once weekly by diluting the Compound A' powder at 0.25% (w/v) in vehicle; storage of the dosing solution at 4°C; dosing volume 10 ml/kg
- Paclitaxel 1.6 ml of stock solution was mixed with 6.4 ml of vehicle to obtain the dosing solution.
- Cisplatin 1.6 mg of cisplatin was resuspended in 8 ml of vehicle to obtain the dosing solution.
- mice were weighed twice a week. Relative body weights of individual mice in % were calculated by dividing the individual body weight on day X (BWx) by the individual body weight on day 0 (BWo) multiplied by 100 according to the formula:
- tumour volumes were determined by two-dimensional measurement with a caliper on the day of randomisation (day 0) and then twice weekly (i.e. on the same days on which mice were weighed). Tumour volumes were calculated according to the formulas:
- Relative volumes of individual tumours (RTVs) for Day x were calculated by dividing the absolute individual tumour volume on Day x (T x ) by the absolute individual tumour volume of the same tumour on Day 0 (T 0 ) multiplied by 100%:
- Anti-tumour activity was evaluated as maximum tumour volume inhibition versus the vehicle control group.
- Tumour inhibition for a particular day was calculated from the ratio of the median RTV values of test versus control groups multiplied by 100. Median relative tumour volume of the test group on Day x
- T/C% The minimum (or optimum) T/C% value recorded for a particular test group during an experiment represents the maximum anti-tumour activity for the respective treatment. T/C values were calculated if at least four of the randomised animals in a group were alive on the day in question. 3.3.5.6 Efficacy Criteria
- Compound A' was assessed at one dose level, in mono- and in combination therapy with paclitaxel or cisplatin, in two gastric cancer models subcutaneously implanted in NMRI nu/nu mice.
- Compound A', paclitaxel or cisplatin monotherapy displayed moderate anti-tumour activity with minimum T/C values of 44.6%, 36.2% and 39.7%, respectively.
- the combination of Compound A' with paclitaxel or cisplatin increased the anti-tumour efficacy of the respective monotherapies leading to optimal T/C values of 22.0% (high activity) and 27.7% (moderate activity), respectively.
- Tumour growth of GXA SCH LX was significantly reduced by Compound A' in combination treatments as compared to the respective vehicle control groups, as determined by the non-parametric Kruskal- Wallis test, followed by Dunn's post-test.
- Tumour growth of GFX 251 was significantly reduced by Compound A' combination treatments as compared to the respective vehicle control groups and to paclitaxel or cisplatin monotherapies alone, as determined by the Kruskal-Wallis test, followed by Dunn's post-test.
- Vehicle Compound A' 80% PEG400 in water for injection
- tumour growth was significantly attenuated by Compound A' in combination with either paclitaxel or cisplatin as compared to the respective vehicle control group (Kruskal-Wallis test, followed by Dunn's post-test). No or moderate group median BWLs of up to 7.9% were observed in GXA SCH LX and GXF 251 tumour models. In conclusion, these data indicate significant and meaningful anti- tumour activity of Compound A' in combination with either paclitaxel or cisplatin in patients with gastric cancer, preferably with C-MYC amplification.
Abstract
The present invention relates to the use of 4-(4-Fluoro-2-methoxyphenyl)-N-{3-[(S- methylsulfonimidoyl)methyl]phenyl}-1,3,5-triazin-2-amine (compound A), more particularly (+)-4-(4- Fluoro-2-methoxyphenyl)-N-{3-[(S-methylsulfonimidoyl)methyl]phenyl}-1,3,5-triazin-2-amine (compound A´), for treating gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene.
Description
Use of 4-(4-Fluoro-2-methoxyphenyl)-N-{3 (S-methylsulfonimidoyl)methyl]phenyl}-l,3,5- triazin-2-amine for treating gastric cancers
The present invention relates to the use of 4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S- methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine (compound A), more particularly (+)-4-(4- Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine (compound A'), for treating gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene. The family of cyclin-dependent kinase (CDK) proteins consists of members that are key regulators of the cell division cycle (cell cycle CDK's), that are involved in regulation of gene transcription (transcriptional CDK's), and of members with other functions. CDKs require for activation the association with a regulatory cyclin subunit. The cell cycle CDKs CDKl/cyclin B, CDK2/cyclin A, CDK2/cyclinE, CDK4/cyclinD, and CDK6/cyclinD get activated in a sequential order to drive a cell into and through the cell division cycle. The transcriptional CDKs CDK9/cyclin T and CDK7/cyclin H regulate the activity of RNA polymerase II via phosphorylation of the carboxy-terminal domain (CTD). Positive transcription factor b (P-TEFb) is a heterodimer of CDK9 and one of four cyclin partners, cyclin Tl, cyclin K, cyclin T2a or T2b. Whereas CDK9 (NCBI GenBank Gene ID 1025) is exclusively involved in transcriptional regulation, CDK7 in addition participates in cell cycle regulation as CDK-activating kinase (CAK).
Transcription of genes by RNA polymerase II is initiated by assembly of the pre-initiation complex at the promoter region and phosphorylation of Ser 5 and Ser 7 of the CTD by CDK7/cyclin H. For a major fraction of genes RNA polymerase II stops mRNA transcription after it moved 20-40 nucleotides along the DNA template. This promoter-proximal pausing of RNA polymerase Π is mediated by negative elongation factors and is recognized as a major control mechanism to regulate expression of rapidly induced genes in response to a variety of stimuli (Cho et al., Cell Cycle 2010, 9, 1697). P-TEFb is crucially involved in overcoming promoter-proximal pausing of RNA polymerase Π and transition into a productive elongation state by phosphorylation of Ser 2 of the CTD as well as by phosphorylation and inactivation of negative elongation factors.
Activity of P-TEFb itself is regulated by several mechanisms. About half of cellular P-TEFb exists in an inactive complex with 7SK small nuclear RNA (7SK snRNA), La-related protein 7 (LARP7/PIP7S) and hexamethylene bis-acetamide inducible proteins 1/2 (HEXIM1/2, He et al., Mol. Cell 2008, 29, 588). The remaining half of P-TEFb exists in an active complex containing the bromodomain protein Brd4 (Yang et al., Mol. Cell 2005, 19, 535). Brd4 recruits P-TEFb through interaction with acetylated
histones to chromatin areas primed for gene transcription. Through alternately interacting with its positive and negative regulators, P-TEFb is maintained in a functional equilibrium: P-TEFb bound to the 7SK snRNA complex represents a reservoir from which active P-TEFb can be released on demand of cellular transcription and cell proliferation (Zhou & Yik, Microbiol. Mol. Biol. Rev. 2006, 70, 646). Furthermore, the activity of P-TEFb is regulated by posttranslational modifications including phosphorylation/de-phosphorylation, ubiquitination, and acetylation (reviewed in Cho et al., Cell Cycle 2010, 9, 1697).
Deregulated CDK9 kinase activity of the P-TEFb heterodimer is associated with a variety of human pathological settings such as hyper-proliferative diseases (e.g. cancer), virally induced infectious diseases or cardiovascular diseases.
Cancer is regarded as a hyper-proliferative disorder mediated by a disbalance of proliferation and cell death (apoptosis). High levels of anti-apoptotic Bcl-2-family proteins are found in various human tumours and account for prolonged survival of tumour cells and therapy resistance. Inhibition of P- TEFb kinase activity was shown to reduce transcriptional activity of RNA polymerase Π leading to a decline of short-lived anti-apoptotic proteins, especially Mcl-l and XIAP, reinstalling the ability of tumour cells to undergo apoptosis. A number of other proteins associated with the transformed tumour phenotype (such as Myc, NF-kB responsive gene transcripts, mitotic kinases) are either short-lived proteins or are encoded by short-lived transcripts which are sensitive to reduced RNA polymerase II activity mediated by P-TEFb inhibition (reviewed in Wang & Fischer, Trends Pharmacol. Sci. 2008, 29, 302).
Many viruses rely on the transcriptional machinery of the host cell for the transcription of their own genome. In case of HIV-1 RNA polymerase Π gets recruited to the promoter region within the viral LTR's. The viral transcription activator (Tat) protein binds to nascent viral transcripts and overcomes promoter-proximal RNA polymerase Π pausing by recruitment of P-TEFb which in turn promotes transcriptional elongation. Furthermore, the Tat protein increases the fraction of active P-TEFb by replacement of the P-TEFb inhibitory proteins HEXIM1/2 within the 7SK snRNA complex. Recent data have shown that inhibition of the kinase activity of P-TEFb is sufficient to block HIV-1 replication at kinase inhibitor concentrations that are not cytotoxic to the host cells (reviewed in Wang & Fischer, Trends Pharmacol. Sci. 2008, 29, 302). Similarly, recruitment of P-TEFb by viral proteins has been reported for other viruses such as B-cell cancer-associated Epstein-Barr virus, where the nuclear antigen EBNA2 protein interacts with P-TEFb (Bark-Jones et al., Oncogene 2006, 25, 1775), and the human T-lympho tropic virus type 1 (HTLV-1), where the transcriptional activator Tax recruits P-TEFb (Zhou et al., J. Virol. 2006, 80, 4781).
Cardiac hypertrophy, the heart's adaptive response to mechanical overload and pressure (hemodynamic stress e.g. hypertension, myocardial infarction), can lead, on a long term, to heart failure and death. Cardiac hypertrophy was shown to be associated with increased transcriptional activity and RNA polymerase Π CTD phosphorylation in cardiac muscle cells. P-TEFb was found to be activated by dissociation from the inactive 7SK snRNA/HEXEMl/2 complex. These findings suggest pharmacological inhibition of P-TEFb kinase activity as a therapeutic approach to treat cardiac hypertrophy (reviewed in Dey et al., Cell Cycle 2007, 6, 1856). In summary, multiple lines of evidence suggest that selective inhibition of the CDK9 kinase activity of the P-TEFb heterodimer (= CDK9 and one of four cyclin partners, cyclin Tl, cyclin K, cyclin T2a or T2b) represents an innovative approach for the treatment of diseases such as cancer, viral diseases, and/or diseases of the heart. CDK9 belongs to a family of at least 13 closely related kinases of which the subgroup of the cell cycle CDK's fulfils multiple roles in regulation of cell proliferation. Thus, co- inhibition of cell cycle CDK's (e.g. CDKl/cyclin B, CDK2/cyclin A, CDK2/cyclinE, CDK4/cyclinD, CDK6/cyclinD) and of CDK9 is expected to impact normal proliferating tissues such as intestinal mucosa, lymphatic and hematopoietic organs, and reproductive organs. To maximize the therapeutic margin of CDK9 kinase inhibitors, molecules with high selectivity towards CDK9 are therefore required.
CDK inhibitors in general as well as CDK9 inhibitors are described in a number of different publications: WO2008129070 and WO2008129071 both describe 2,4 substituted aminopyrimidines as CDK inhibitors in general. It is also asserted that some of these compounds may act as selective CDK9 inhibitors (WO2008129070) and as CDK5 inhibitors (WO2008129071), respectively, but no specific CDK9 IC50 (WO2008129070) or CDK5 IC50 (WO200812971) data is presented.
WO2008129080 discloses 4,6 substituted aminopyrimidines and demonstrates that these compounds show an inhibitory effect on the protein kinase activity of various protein kinases, such as CDK1, CDK2, CDK4, CDK5, CDK6 and CDK9, with a preference for CDK9 inhibition (example 80).
EP1218360 Bl describes triazin derivatives as kinase inhibitors, but does not disclose potent or selective CDK9 inhibitors.
WO2008079933 discloses aminopyridine and aminopyrimidine derivatives and their use as CDK1, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8 or CDK9 inhibitors.
WO2011012661 describes aminopyridine derivatives useful as CDK inhibitors.
Wang et al. (Chemistry & Biology 2010, 17, 1111-1121) describe 2-anilino-4-(thiazol-5-yl)pyrimidine transcriptional CDK inhibitors, which show anticancer activity in animal models. WO2004009562 discloses substituted triazine kinase inhibitors. For selected compounds CDKl and CDK 4 test data, but no CDK9 data is presented.
WO2004072063 describes heteroaryl (pyrimidine, triazine) substituted pyrroles as inhibitors of protein kinases such as ERK2, GSK3, PKA or CDK2. WO2010009155 discloses triazine and pyrimidine derivatives as inhibitors of histone deacetylase and/or cyclin dependent kinases (CDKs). For selected compounds CDK2 test data is described.
WO2003037346 (corresponding to US7618968B2, US7291616B2, US2008064700A1, US2003153570A1) relates to aryl triazines and uses thereof, including to inhibit lysophosphatidic acid acyltransferase beta (LPAAT-beta) activity and/or proliferation of cells such as tumour cells.
WO2008025556 describes carbamoyl sulfoximides having a pyrimidine core, which are useful as kinase inhibitors. No CDK9 data is presented.
WO2002066481 describes pyrimidine derivatives as cyclin dependent kinase inhibitors CDK9 is not mentioned and no CDK9 data is presented.
WO2008109943 concerns phenyl aminopyri(mi)dine compounds and their use as kinase inhibitors, in particular as JAK2 kinase inhibitors. The specific examples focus on compounds having a pyrimidine core.
WO2009032861 describes substituted pyrimidinyl amines as JNK kinase inhibitors. The specific examples focus on compounds having a pyrimidine core.
WO2011046970 concerns amino-pyrimidine compounds as inhibitors of TBKL and/or ΓΚΚ epsilon. The specific examples focus on compounds having a pyrimidine core.
WO2012160034 the compounds of the present invention. It is disclosed the compounds inhibit the cell proliferation of HeLa cells (cervical cancer), HeLa/MaTu/ADR cells (cervical cancer), NCI-H460 cells (non-small cell lung cancer), DU145 cells (hormone-independent human prostate cancer), Caco-2 cells (colorectal cancer) and B 16F10 cells (melanoma).
The object of the present invention is to improve the treatment of gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene.
Treatment of gastric cancer
Gastric cancer is an aggressive disease and the second leading cause of cancer-related mortality worldwide (Jemal A et al. Global cancer statistics. CA Cancer J Clin 2011; 61(2): 69-90). Dietary improvements and reduction in H. pylori infections, due to the use of antibiotics, have resulted in a steady fall in incidence and mortality rates, however, the prognosis for gastric cancer patients remains poor in Western countries (Lordick F. Unmet needs and challenges in gastric cancer: The way forward. Can Treat Rev 2014; 40: 692-700). This is in stark contrast to the overall five-year survival rate of gastric cancer patients in Japan, where regular screening leading to early stage diagnosis has resulted in 70% patient survival (European Union Network of Excellence (EUNE) for Gastric Cancer Steering Group. Gastric cancer in Europe. Br J Surg 2008; 95:406^108). Unfortunately, early stage gastric cancer rarely presents symptoms that cannot be explained by other factors and thus remains undiagnosed until it has progressed to an advanced metastatic phase.
The single curative therapy for localised late stage gastric cancer is surgical resection where tumour removal coupled with extensive lymph node dissection and long-term follow-up results in increased efficacy. Preoperative chemotherapy consisting of epirubicin, cisplatin and capecitabine (or other platinum/fluoropyrimidine combinations) has also proven beneficial in improving overall survival rates and has been widely adopted as the standard of care in Europe. (Waddell et al. Gastric cancer: ESMO-ESSO-ESTRO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Radiotherapy and Oncology 2014;110: 189-194). In the case of inoperable, recurring or metastatic cancers, chemotherapy and best supportive care provide enhanced quality of life and improved patient prognosis. However, response to first-line chemotherapeutic agents still remains poor.
Trastuzumab, an anti-HER2 (epidermal growth factor receptor kinase 2) antibody, in combination with cisplatin and fluoropyrimidine, leading to enhanced overall survival was granted FDA approval in 2010 for the treatment of patients with HER2-overexpressing metastatic gastric cancer. Importantly, trastuzumab is a great example of how advances in molecular characterisation of tumours can be translated into successful and targetable therapeutics (Bang YJ et al., Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2 -positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial, Lancet 2010; 376 (9742): 687-697).
C-MYC amplification in gastric cancer
In gastric cancer, commonly recurring genomic aberrations include TP53, PIK3CA, ErbB2, ErbB3, KRAS, MET and MYC (Yang W et al. Targeted therapy for gastric cancer: Molecular pathways and ongoing investigations. Biochimica et Biophysica Acta 2014;1846: 232-7).
Deregulated MYC expression underlies the pathogenesis of numerous human neoplasms, and seems to be at the crossroad of many important pathways and processes involved in carcinogenesis (Fletcher S et al. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 2014). The C-MYC gene is an important member of the MYC proto-oncogene family and several studies have demonstrated an association between C-MYC expression and gastric cancer. C-MYC overexpression has been described in over 40% of gastric cancers (Milne AN et al. Early onset gastric cancer: on the road to unravelling gastric carcinogenesis. Curr Mol Med 2007;7(1): 15-28) and in both intestinal- and diffuse-type gastric adenocarcinomas (Calcagno DQ et al. Interrelationship between chromosome 8 aneuploidy, C-MYC amplification and increased expression in individuals from northern Brazil with gastric adenocarcinoma. World J Gastroenterol 2006; 12(38):6207— 6211). Higher C-MYC expression has also been associated with metastasis (Kozma L et al. C-MYC amplification and cluster analysis in human gastric carcinoma. Anticancer Res 2001 ; 21(1B):707-710; Onoda N et al. Overexpression of C-MYC messenger RNA in primary and metastatic lesions of carcinoma of the stomach. J Am Coll Surg 1996; 182(1):55— 59) and with poor prognosis (Han S et al. C-MYC expression is related with cell proliferation and associated with poor clinical outcome in human gastric cancer. J Korean Med Sci 1999; 14(5):526-530; De Souza C et al. MYC Deregulation in Gastric Cancer and Its Clinicopathological Implications. Plos One 2013; 8(5):e64420).
Altogether, mounting evidence suggests that C-MYC amplification may be an important instigator of gastric cancer and disease progression potentially due to the fundamental role of this transcription factor in the regulation of cell proliferation and apoptosis.
Detection of C-MYC gene amplification
Gene amplification, also known as gene duplication or chromosomal duplication, is a cellular process in which multiple copies of a gene are produced. The genes on each of the copies can be transcribed and translated, leading to an overproduction of the mRNA and protein corresponding to the amplified genes. Gene amplifications are important features of many advanced cancers and have prognostic as well as therapeutic significance in clinical cancer treatment (Myllykangas S, Knuutila S. Manifestation, mechanisms and mysteries of gene amplifications. Cancer Lett 2006; 232(l):79-89). Various methods can be used to detect gene amplifications in clinical samples. Among these, conventional cytogenetics (chromosome banding), chromosomal comparative genomic hybridization (CGH), fluorescent in situ hybridization (FISH) and next-generation sequencing (NGS) are routinely employed in clinical trials to determine copy number alterations in patient samples (Myllykangas S, Knuutila S. Manifestation, mechanisms and mysteries of gene amplifications. Cancer Lett 2006;232(l):79-89). Although there is no standard method to detect the amplification of C-MYC, all of the above-mentioned techniques as well as multiplex ligation-dependent probe amplification (MLPA),
PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, and the traditional Southern blotting and slot blot methods have been used and are strongly supported by several publications (Deming SL et al. C-myc amplification in breast cancer: a meta-analysis of its occurrence and prognostic relevance. Br J Cancer 2000;83(12): 1688-1695; Setoodeh R et al. Double -hit mantle cell lymphoma with MYC gene rearrangement or amplification: a report of four cases and review of the literature, hit J Clin Exp Pathol 2013;6(2): 155- 67; Balko JM et al. Molecular profiling of the residual disease of triple -negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov 2014;4(2):232-45; Kim S et al. High-throughput sequencing and copy number variation detection using formalin fixed embedded tissue in metastatic gastric cancer. PLoS One 2014;9(l l):el 11693; Minca EC et al. Genomic microarray analysis on formalin-fixed paraffin-embedded material for uveal melanoma prognostication. Cancer Genet 2014;207(7-8):306-15; Poddighe PJ et al. Genomic amplification of MYC as double minutes in a patient with APL-like leukemia, Mol Cytogenet 2014;7(1):67; Vogt N et al. Amplicon rearrangements during the extrachromosomal and intrachromosomal amplification process in a glioma. Nucleic Acids Res 2014;42(21): 13194-205; Baykara O et al. Amplification of chromosome 8 genes in lung cancer. J Cancer 2015;6(3):270-5; Ooi A et al. Semi-comprehensive analysis of gene amplification in gastric cancers using multiplex ligation-dependent probe amplification and fluorescence in situ hybridization. Mod Pathol 2015: doi: 10.1038/modpathol.2015.33; Verbeke SL et al. Array CGH analysis identifies two distinct subgroups of primary angiosarcoma of bone. Genes Chromosomes Cancer 2015;54(2):72-81)..
It has now been found that the compound 4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S- methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine (compound A, formula (I)) or one of its physiologically acceptable salts or enantiomers,

Compound A
more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2- amine (compound A') or one of its physiologically acceptable,
acts in specific tumour types which had previously not yet been contemplated, viz. gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene.
4-(4-Fluoro-2-methoxyphenyl)-N-
(compound A) is a selected sulphoximine-substituted anilinopyrimidine derivative which can be separated into two stereoisomers, viz.:
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2- amine (compound A') and
(-)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2- amine (compound A").
Compound A' is preferred and in clinical development as BAY1143572.
Where compound A is mentioned below, both the pure stereoisomers A' and A", and also any mixture of these two, are meant thereby.
The present invention is directed to the use of
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine (compound A) or one of its physiologically acceptable salts or enantiomers,
more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2- amine (compound A') or one of its physiologically acceptable salts,
for the treatment and/or prophylaxis of gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene.
Preferred is the use of compound A' for the treatment and/or prophylaxis of gastric cancers in which cells have an amplification of the C-MYC gene.
The methods for detecting this amplification include, but are not limited to conventional cytogenetics (chromosome banding), chromosomal comparative genomic hybridization (CGH), fluorescent in situ hybridization (FISH) next-generation sequencing (NGS), multiplex ligation-dependent probe amplification (MLPA), PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
The present application is further directed to the use of
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine or one of its physiologically acceptable salts or enantiomers,
more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2- amine or one of its physiologically acceptable salts,
for preparing a medicament for treating gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene.
Another aspect of the present invention is the use of
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-13,5-triazin-2-amine (compound A) according to formula I) or one of its physiologically acceptable salts or enantiomers,

more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N- { 3- [(S-methylsulfonimidoyl)methyl]phenyl } - 1 ,3,5-triazin-2- amine or one of its physiologically acceptable salts,
in the manufacture of a medicament for treating cancer in a subject, wherein the medicament is manufactured for treating gastric cancers.
Preferred is the use of compound A' in the manufacture of a medicament for treating gastric cancers in which cells have an amplification of the C-MYC gene.
The methods for detecting this amplification include, but are not limited to
conventional cytogenetics (chromosome banding), chromosomal comparative genomic hybridization (CGH), fluorescent in situ hybridization (FISH), next-generation sequencing (NGS), multiplex ligation-dependent probe amplification (MLPA), PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods. The present application further provides
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine of formula I (compound A or one of its physiologically acceptable salts or enantiomers,

more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2- amine (compound A') or one of its physiologically acceptable salts,
for the use of treating gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene.
Preferred is compound A' for the use of treating gastric cancers in which cells have an amplification of the C-MYC gene.
The methods for detecting this amplification include, but are not limited to
conventional cytogenetics (chromosome banding), chromosomal comparative genomic hybridization (CGH), fluorescent in situ hybridization (FISH), next-generation sequencing (NGS), multiplex ligation-dependent probe amplification (MLPA), PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
The present invention is also directed to
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine of formula I (compound A or one of its physiologically acceptable salts or enantiomers,

more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2- amine (compound A') or one of its physiologically acceptable salts,
for the use in a method of treatment and/or prophylaxis of gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene. Preferred is compound A', for the use in a method of treatment and/or prophylaxis of gastric cancers in which cells have an amplification of the C-MYC gene.
The methods for detecting this amplification include, but are not limited to
conventional cytogenetics (chromosome banding), chromosomal comparative genomic hybridization (CGH), fluorescent in situ hybridization (FISH), next-generation sequencing (NGS), multiplex ligation-dependent probe amplification (MLPA), PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
Another aspect of the present invention is
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine according to formula (I) or one of its physiologically acceptable salts or enantiomers
more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl
amine (compound A') or one of its physiologically acceptable salts
for the use in a method for treating a human patient diagnosed with gastric cancer
characterized by a C-MYC amplification
said method comprising the steps:
a) assaying a tumour sample from the patient and
b) determining if C-MYC gene is amplified and
c) administering a therapeutically effective amount of 4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S- methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2-amine according to formula (I) or one of its physiologically acceptable salts or enantiomers,
more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2- amine (compound A') or one of its physiologically acceptable salts,
if C-MYC amplification is detected as defined in step b.
Another aspect of the present invention is a
method of treating gastric cancer comprising the steps
a) assaying a tumour sample from the patient and
b) determining if C-MYC gene is amplified and
c) administering a therapeutically effective amount of 4-(4-Fluoro-2-methoxyphenyl)-N-{3-[(S- methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2-amine (compound A) of formula I or one of its physiologically acceptable salts or enantiomers,
preferably (+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}- l,3,5-triazin-2-amine (compound A') or one of its physiologically acceptable salts if C-MYC gene is amplified as defined in step b.
Another aspect of the present invention is a method of treatment and/or prophylaxis of gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene using an effective amount of 4-(4-Fluoro-2-methoxyphenyl)-N- { 3-[(S-methylsulfonimidoyl)methyl]phenyl } - 1 ,3,5- triazin-2-amine (compound A) of formula I or one of its physiologically acceptable salts or enantiomers,
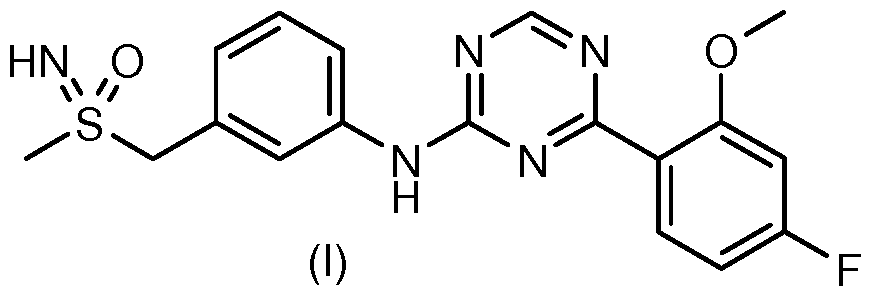
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l^
amine (compound A') or one of its physiologically acceptable salts. A preferred method of treatment is a method of treatment and/or prophylaxis of gastric cancers in which cells have an amplification of the C-MYC gene using an effective amount of compound A'. The methods for detecting this amplification include, but are not limited to
conventional cytogenetics (chromosome banding), chromosomal comparative genomic hybridization (CGH), fluorescent in situ hybridization (FISH), next-generation sequencing (NGS), multiplex ligation-dependent probe amplification (MLPA), PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
The present application further provides pharmaceutical compositions containing
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine or one of its physiologically acceptable salts or enantiomers,
more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2- amine or one of its physiologically acceptable salts,
for treating gastric cancer, preferably gastric cancers in which cells have an amplification of the C- MYC gene.
The present invention is also directed to pharmaceutical compositions comprising 4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine (compound A) of formula I or one of its hysiologically acceptable salts or enantiomers,

more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2- amine (compound A') or one of its physiologically acceptable salts ,
and at least one inert, nontoxic, pharmaceutically suitable adjuvant for the treatment and/or prophylaxis of gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene.
A preferred pharmaceutical composition is a pharmaceutical composition comprising compound A' for the treatment of gastric cancers in which cells have an amplification of the C-MYC gene.
The methods for detecting this amplification include, but are not limited to
conventional cytogenetics (chromosome banding), chromosomal comparative genomic hybridization (CGH), fluorescent in situ hybridization (FISH), next-generation sequencing (NGS), multiplex ligation-dependent probe amplification (MLPA), PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods. The present application further provides combinations of
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine (compound A),
more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2- amine (compound A'),
with at least one further active ingredient for treating gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene.
The present invention is also directed to
pharmaceutical combinations comprising
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine (compound A) of formula I or one of its hysiologically acceptable salts or enantiomers,

more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N- { 3- [(S-methylsulfonimidoyl)methyl]phenyl } - 1 ,3,5-triazin-2- amine (compound A') or one of its physiologically acceptable salts ,
and at least one or more further active ingredients for the treatment and/or prophylaxis of gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene.
A preferred pharmaceutical combination is a pharmaceutical combination comprising compound A' for the treatment of gastric cancers in which cells have an amplification of the C-MYC gene.
The methods for detecting this amplification include, but are not limited to
conventional cytogenetics (chromosome banding), chromosomal comparative genomic hybridization (CGH), fluorescent in situ hybridization (FISH), next-generation sequencing (NGS), multiplex ligation-dependent probe amplification (MLPA), PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
Another aspect of the present invention is a method for identifying a patient disposed to respond favorably to a CDK9-inhibitor for treating gastric cancer,
wherein the CDK9-inhibitor is compound A and
wherein the method comprises the detection of an amplification of the C-MYC gene in tumour cells in a tissue sample from the patient.
Preferred is a method for identifying a patient disposed to respond favorably to a CDK9-inhibitor for treating gastric cancer
wherein the CDK9-inhibitor is compound A' and
wherein the method comprises the detection of an amplification of the C-MYC gene in tumour cells in a tissue sample from the patient and
wherein those patients are identified for a treatment of gastric cancer with a CDK9-inhibitor whose tumour cells have an amplification of the C-MYC gene.
Another aspect of the present invention is a method for identifying a patient disposed to respond favorably to
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine according to formula (I) or one of its physiologically acceptable salts or enantiomers
more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2- amine (compound A') or one of its physiologically acceptable salts,
for treating gastric cancer,
wherein the method comprises the detection of C-MYC amplification in tumor cells in a tissue sample from the patient and
wherein those patients are identified for a treatment of gastric cancer with a CDK9-inhibitor whose tumour cells have an amplification of the C-MYC gene.
The methods for detecting this amplification include, but are not limited to
conventional cytogenetics (chromosome banding), chromosomal comparative genomic hybridization (CGH), fluorescent in situ hybridization (FISH), next-generation sequencing (NGS), multiplex ligation-dependent probe amplification (MLPA), PCR-based methods such as quantitative PCR
(qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods.
Another aspect of the present invention is a method of predicting whether a patient will be respond to the treatment with
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2-amine according to formula (I) or one of its physiologically acceptable salts or enantiomers,
more particularly
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2- amine (compound A') or one of its physiologically acceptable salts,
wherein the method comprises the detection of C-MYC amplification in tumour cells in a tissue sample from the patient.
The methods for detecting this amplification include, but are not limited to
conventional cytogenetics (chromosome banding), chromosomal comparative genomic hybridization (CGH), fluorescent in situ hybridization (FISH), next-generation sequencing (NGS) multiplex ligation- dependent probe amplification (MLPA), PCR-based methods such as quantitative PCR (qPCR) and digital PCR, single nucleotide polymorphism (SNP) arrays, Southern blotting and slot blot methods. The use of the physiologically tolerable salts of compound A should likewise be considered to be covered by the present invention.
Physiologically safe salts of compound A encompass acid addition salts of mineral acids, carboxylic acids and sulphonic acids, for example salts of hydrochloric acid, hydrobromic acid, sulphuric acid, phosphoric acid, methanesulphonic acid, ethanesulphonic acid, toluenesulphonic acid, benzenesulphonic acid, naphthalenedisulphonic acid, acetic acid, trifluoroacetic acid, propionic acid, lactic acid, tartaric acid, malic acid, citric acid, fumaric acid, maleic acid and benzoic acid.
Physiologically safe salts of compound A also encompass salts of customary bases, such as, by way of example and preferably, alkali metal salts (e.g. sodium and potassium salts), alkaline earth metal salts (e.g. calcium and magnesium salts) and ammonium salts derived from ammonia or organic amines having from 1 to 16 C atoms, such as, by way of example and preferably, ethylamine, diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, arginine, lysine, ethylenediamine and N-methylpiperidine.
The present invention further provides drugs containing compound A and at least one or more further active ingredients for treating gastric cancer, preferably gastric cancers in which cells have an amplification of the C-MYC gene. Compound A may have systemic and/or local activity. For this purpose, it can be administered in a suitable manner, such as, for example, orally, parenterally, via the pulmonary route, nasal, sublingually, lingually, buccally, rectally, vaginally, dermally, transdermally, conjuntivally, otically or as an implant or stent. For these administration routes, compound A according to the invention may be administered in suitable administration forms.
Suitable for oral administration forms which function according to the prior art and deliver compound A of the invention rapidly and/or in a modified manner and which comprise compound A according to the invention in crystalline and/or amorphised and/or dissolved form, such as, for example, tablets (uncoated or coated tablets, for example with coatings which are resistant to gastric juice or dissolve with a delay or are insoluble and control the release of the compound of the invention), tablets which disintegrate rapidly in the oral cavity, or films/wafers, films/lyophilisates, capsules (for example hard or soft gelatine capsules), sugar-coated tablets, granules, pellets, powders, emulsions, suspensions, aerosols or solutions.
Parenteral administration can be effected with avoidance of an absorption step (for example intravenous, intraarterial, intracardial, intraspinal or intralumbal) or with inclusion of absorption (for example intramuscular, subcutaneous, intracutaneous, percutaneous or intraperidoneal). Administration forms which are suitable for parenteral administration are, inter alia, preparations for injection and infusion in the form of solutions, suspensions, emulsions, lyophilisates or sterile powders.
Examples which are suitable for other administration routes are pharmaceutical forms for inhalation [inter alia power inhalers, nebulizers], nasal drops, solutions, sprays; tablets, films/wafers or capsules, to be administered lingually, sublingually or buccaly, suppositories, preparations for the eyes and the ears, eye baths, ocular insert, ear drops, ear powders, ear-rinses, ear tampons, vaginal capsules, aqueous suspensions (lotions, mixturae agitandae), lipophilic suspensions, ointments, creams, transdermal therapeutic systems (such as, for example, patches), milk, pastes, foams, dusting powders, implants or stents.
Compound A can be converted into the stated administration forms. This can be effected in a manner known per se by mixing with inert, non-toxic, pharmaceutically suitable adjuvants. These adjuvants include, inter alia,
• fillers and excipients (for example cellulose, microcrystalline cellulose, such as, for example, Avicel®, lactose, mannitol, starch, calcium phosphate such as, for example, Di-Cafos®),
• ointment bases (for example petroleum jelly, paraffins, triglycerides, waxes, wool wax, wool wax alcohols, lanolin, hydrophilic ointment, polyethylene glycols),
• bases for suppositories (for example polyethylene glycols, cacao butter, hard fat)
• solvents (for example water, ethanol, Isopropanol, glycerol, propylene glycol, medium chain- length triglycerides fatty oils, liquid polyethylene glycols, paraffins),
• surfactants, emulsifiers, dispersants or wetters (for example sodium dodecyle sulphate, lecithin, phospholipids, fatty alcohols such as, for example, Lanette®, sorbitan fatty acid esters such as, for example, Span®, polyoxyethylene sorbitan fatty acid esters such as, for example, Tween®, polyoxyethylene fatty acid glycerides such as, for example, Cremophor®, polyoxethylene fatty acid esters, polyoxyethylene fatty alcohol ethers, glycerol fatty acid esters, poloxamers such as, for example, Pluronic®),
• buffers and also acids and bases (for example phosphates, carbonates, citric acid, acetic acid, hydrochloric acid, sodium hydroxide solution, ammonium carbonate, trometamol, triethanolamine)
• isotonicity agents (for example glucose, sodium chloride),
• adsorbents (for example highly-disperse silicas)
• viscosity-increasing agents, gel formers, thickeners and/or binders (for example polyvinylpyrrolidon, methylcellulose, hydroxypropylmethylcellulose, hydroxypropylcellulose, carboxymethylcellulose-sodium, starch, carbomers, polyacrylic acids such as, for example, Carbopol®, alginates, gelatine),
• disintegrants (for example modified starch, carboxymethylcellulose-sodium, sodium starch glycolate such as, for example, Explotab®, cross- linked polyvinylpyrrolidon, croscarmellose- sodium such as, for example, AcDiSol®),
• flow regulators, lubricants, glidant and mould release agents (for example magnesium stearate, stearic acid, talc, highly-disperse silicas such as, for example, Aerosil®),
• coating materials (for example sugar, shellac) and film formers for films or diffusion membranes which dissolve rapidly or in a modified manner (for example polyvinylpyrrolidones such as, for example, Kollidon®, polyvinyl alcohol, hydroxypropylmethylcellulose, hydroxypropylcellulose, ethylcellulose,
hydroxypropylmethylcellulose phthalate, cellulose acetate, cellulose acetate phthalate, polyacrylates, polymethacrylates such as, for example, Eudragit®),
capsule materials (for example gelatine, hydroxypropylmethylcellulose),
synthetic polymers (for example polylactides, polyglycolides, polyacrylates, polymethacrylates such as, for example, Eudragit®, polyvinylpyrrolidones such as, for example, Kollidon®, polyvinyl alcohols, polyvinyl acetates, polyethylene oxides, polyethylene glycols and their copolymers and blockcopolymers),
plasticisers (for example polyethylene glycols, propylene glycol, glycerol, triacetine, triacetyl citrate, dibutyl phthalate),
penetration enhancers,
stabilisers (for example antioxidants such as, for example, ascorbic acid, ascorbyl palmitate, sodium ascorbate, butylhydroxyanisole, butylhydroxytoluene, propyl gallate),
preservatives (for example parabens, sorbic acid, thiomersal, benzalkonium chloride, chlorhexidine acetate, sodium benzoate),
colourants (for example inorganic pigments such as, for example, iron oxides, titanium dioxide),
flavourings, sweeteners, flavour- and/or odour-masking agents.
The present invention furthermore relates to medicaments which comprise at least one compound according to the invention, conventionally together with one or more inert, non-toxic, pharmaceutically suitable adjuvants, and to their use for the above mentioned purposes.
Dosage and treatment regimen
The dosage and the treatment regimen can and must be varied depending on the carcinoma type and the treatment goal.
The daily dose is generally between 20 mg and 850 mg and can be divided into a plurality of identical or different dosage units, preferably 2 which can be taken simultaneously or according to a certain time schedule.
In particular the daily dose is between 30 mg and 500 mg and can be divided into a plurality of identical or different dosage units, preferably 2 which can be taken simultaneously or according to a certain time schedule.
A preferred daily dose is between 20 mg and 400 mg and can be divided into a plurality of identical or different dosage units, preferably 2 which can be taken simultaneously or according to a certain time schedule. More particularly, the daily dose is between 40 mg and 300 mg and can be divided into a plurality of identical or different dosage units, preferably 2 which can be taken simultaneously or according to a certain time schedule.
A more preferred daily dose is between 20 mg and 200 mg and can be divided into a plurality of identical or different dosage units, preferably 2 which can be taken simultaneously or according to a certain time schedule.
An even more preferred daily dose is between 50 mg and 180 mg and can be divided into a plurality of identical or different dosage units, preferably 2 which can be taken simultaneously or according to a certain time schedule.
This applies both to monotherapy and to combination therapy with other anti-hyperproliferative, cytostatic or cytotoxic substances, the combination therapy possibly requiring a reduction in dose. The treatment can be carried out in regularly repeated cycles. Treatment cycles may have varying duration, such as 21 days or 28 days, whereby dosing is given continuously, or intermittently. Preferred is a cycle length of 28 days, whereby dosing is given continuously, or intermittently.
Continuous schedules involve daily dosing, for example, 21 daily doses in a 21 -day cycle, or 28 daily doses in a 28-day cycle. A preferred continuous schedule is 28 daily doses in a 28 daily cycle.
Intermittent schedules involve a period of treatment followed by a period of non-treatment, for example in a cycle of 21 days, or a cycle of 28 days. A preferred cycle duration for an intermittent schedule is 28 days.
The period of treatment may be repeated more than once in a given treatment cycle. The period of treatment may be for example 1 to 21 days, more preferably 3 to 14 days.
An even more preferred intermittent schedule involves treatment for 3 days followed by non-treatment for 4 days, repeated every week in such a way that a 28-day treatment cycle is completed.
Treatment is successful when there is at least disease stabilization and the adverse effects occur to an extent which is easily treatable, but at least easily acceptable. Thus the number of cycles of treatment applied may vary from patient to patient, according to treatment response and tolerability.
Treatment is successful when there is at least disease stabilization and the adverse effects occur to an extent which is easily treatable, but at least easily acceptable.
Compound A can be used on its own or, if required, in combination with one or more other pharmacologically effective substances, provided said combination does not lead to undesired and unacceptable adverse effects. The present invention therefore further provides drugs containing compound A according to the invention and one or more further active ingredients, in particular for treating and/or preventing the above-mentioned diseases.
For example, compound A can be combined with known anti-hyperproliferative, cytostatic or cytotoxic substances for treating cancers. The combination compound A according to the invention with other substances in use for cancer therapy or else with radiotherapy is especially advisable.
Examples of suitable active ingredients for combination purposes include:
abraxane, afinitor, aldesleukin, alendronic acid, alfaferone, alitretinoin, allopurinol, aloprim, aloxi, altretamine, aminoglutethimide, amifostine, amrubicin, amsacrine, anastrozole, anzemet, aranesp, arglabin, arsenic trioxide, aromasin, 5-azacytidine, azathioprine, BCG or tice-BCG, bestatin, betamethasone acetate, betamethasone sodium phosphate, bexarotene, bleomycin sulphate, broxuridine, bortezomib, busulfan, calcitonin, campath, capecitabine, carboplatin, casodex, cefesone, celmoleukin, cerubidine, chlorambucil, cisplatin, cladribine, clodronic acid, cyclophosphamide, cytarabine, dacarbazine, dactinomycin, daunoxome, decadron, decadron phosphate, delestrogen, denileukin diftitox, depo-medrol, deslorelin, dexrazoxane, diethylstilbestrol, diflucan, docetaxel, doxifluridine, doxorubicin, dronabinol, DW-166HC, eligard, elitek, ellence, emend, epirubicin, epoetin alfa, epogen, eptaplatin, ergamisol, estrace, estradiol, estramustine sodium phosphate, ethinyl estradiol, ethyol, etidronic acid, etopophos, etoposide, fadrozole, fareston, filgrastim, finasteride, filgrastim, floxuridine, fluconazole, fludarabine, 5-fluorodeoxyuridine monophosphate, 5-fluorouracil (5-FU), fluoxymesterone, flutamide, formestane, fosteabine, fotemustine, fulvestrant, gammagard, gemcitabine, gemtuzumab, gleevec, gliadel, goserelin, granisetron hydrochloride, histrelin, hycamtin, hydrocortone, erythro-hydroxynonyladenine, hydroxyurea, ibritumomab tiuxetan, idarubicin,
ifosfamide, interferon alpha, interferon alpha 2, interferon alpha 2a, interferon alpha 2β, interferon alpha nl, interferon alpha n3, interferon beta, interferon gamma la, interleukin 2, intron A, iressa, irinotecan, kytril, lapatinib, lentinan sulphate, letrozole, leucovorin, leuprolide, leuprolide acetate, levamisole, levofolinic acid calcium salt, levothroid, levoxyl, lomustine, lonidamine, marinol, mechlorethamine, mecobalamin, medroxyprogesterone acetate, megestrol acetate, melphalan, menest, 6-mercaptopurine, mesna, methotrexate, metvix, miltefosine, minocycline, mitomycin C, mitotane, mitoxantrone, modrenal, myocet, nedaplatin, neulasta, neumega, neupogen, nilutamide, nolvadex, NSC-631570, OCT-43, octreotide, ondansetron hydrochloride, orapred, oxaliplatin, paclitaxel, pediapred, pegaspargase, pegasys, pentostatin, picibanil, pilocarpine hydrochloride, pirarubicin, plicamycin, porfimer sodium, prednimustine, prednisolone, prednisone, premarin, procarbazine, procrit, raltitrexed, RDEA119, rebif, rhenium- 186 etidronate, rituximab, roferon-A, romurtide, salagen, sandostatin, sargramostim, semustine, sizofiran, sobuzoxane, solu-medrol, streptozocin, strontium-89 chloride, synthroid, tamoxifen, tamsulosin, tasonermin, tastolactone, taxotere, teceleukin, temozolomide, teniposide, testosterone propionate, testred, thioguanine, thiotepa, thyrotropin, tiludronic acid, topotecan, toremifene, tositumomab, trastuzumab, treosulfan, tretinoin, trexall, trimethylmelamine, trimetrexate, triptorelin acetate, triptorelin pamoate, UFT, uridine, valrubicin, vesnarinone, vinblastine, vincristine, vindesine, vinorelbine, virulizin, zinecard, zinostatin stimalamer, zofran; ABI-007, acolbifene, actimmune, affinitak, aminopterin, arzoxifene, asoprisnil, atamestane, atrasentan, BAY 43-9006 (sorafenib), avastin, CCI-779, CDC -501, celebrex, cetuximab, crisnatol, cyproterone acetate, decitabine, DN-101, doxorubicin MTC, dSLEM, dutasteride, edotecarin, eflornithine, exatecan, fenretinide, histamine dihydrochloride, histrelin hydrogel implant, holmium- 166 DOTMP, ibandronic acid, interferon gamma, intron-PEG, ixabepilone, keyhole limpet hemocyanin, L-651582, lanreotide, lasofoxifene, libra, lonafarnib, miproxifen, minodronate, MS-209, liposomal MTP-PE, MX-6, nafarelin, nemorubicin, neovastat, nolatrexed, oblimersen, onco-TCS, osidem, paclitaxel polyglutamate, pamidronate disodium, PN-401, QS-21, quazepam, R-1549, raloxifene, ranpirnase, 13-cw-retinoic acid, satraplatin, seocalcitol, T-138067, tarceva, taxoprexin, thymosin alpha 1, tiazofurin, tipifarnib, tirapazamine, TLK-286, toremifene, transMID-107R, valspodar, vapreotide, vatalanib, verteporfin, vinflunine, Z-100, zoledronic acid, and also combinations thereof.
In a preferred embodiment, compound A of the present invention can be combined with the following active ingredients:
1311-chTNT, abarelix, abiraterone, aclarubicin, aldesleukin, alemtuzumab, alitretinoin, altretamine, aminoglutethimide, amrubicin, amsacrine, anastrozole, arglabin, arsenic trioxide, asparaginase, azacitidine, basiliximab, BAY 80-6946, belotecan, bendamustine, bevacizumab, bexarotene,
bicalutamide, bisantrene, bleomycin, bortezomib, buserelin, busulfan, cabazitaxel, calcium folinate, calcium levofolinate, capecitabine, carboplatin, carmofur, carmustine, catumaxomab, celecoxib, celmoleukin, cetuximab, chlorambucil, chlormadinone, chlormethine, cisplatin, cladribine, clodronic acid, clofarabine, crisantaspase, cyclophosphamide, cyproterone, cytarabine, dacarbazine, dactinomycin, darbepoetin alfa, dasatinib, daunorubicin, decitabine, degarelix, denileukin diftitox, denosumab, deslorelin, dibrospidium chloride, docetaxel, doxifluridine, doxorubicin, doxorubicin + estrone, eculizumab, edrecolomab, elliptinium acetate, eltrombopag, endostatin, enocitabine, epirubicin, epitiostanol, epoetin alfa, epoetin beta, eptaplatin, eribulin, erlotinib, estradiol, estramustine, etoposide, everolimus, exemestane, fadrozole, filgrastim, fludarabine, fluorouracil, flutamide, formestane, fotemustine, fulvestrant, gallium nitrate, ganirelix, gefitinib, gemcitabine, gemtuzumab, glutoxim, goserelin, histamine dihydrochloride, histrelin, hydroxycarbamide, 1-125 seeds, ibandronic acid, ibritumomab tiuxetan, idarubicin, ifosfamide, imatinib, imiquimod, improsulfan, interferon alpha, interferon beta, interferon gamma, ipilimumab, irinotecan, ixabepilone, lanreotide, lapatinib, lenalidomide, lenograstim, lentinan, letrozole, leuprorelin, levamisole, lisuride, lobaplatin, lomustine, lonidamine, masoprocol, medroxyprogesterone, megestrol, melphalan, mepitiostane, mercaptopurine, methotrexate, methoxsalen, methyl aminolevulinate, methyltestosterone, mifamurtide, miltefosine, miriplatin, mitobronitol, mitoguazone, mitolactol, mitomycin, mitotane, mitoxantrone, nedaplatin, nelarabine, nilotinib, nilutamide, nimotuzumab, nimustine, nitracrine, ofatumumab, omeprazole, oprelvekin, oxaliplatin, p53 gene therapy, paclitaxel, palifermin, palladium- 103 seed, pamidronic acid, panitumumab, pazopanib, pegaspargase, PEG- epoetin beta (methoxy-PEG-epoetin beta), pegfilgrastim, peginterferon alfa 2b, pemetrexed, pentazocine, pentostatin, peplomycin, perfosfamide, picibanil, pirarubicin, plerixafor, plicamycin, poliglusam, polyestradiol phosphate, polysaccharide-K, porfimer sodium, pralatrexate, prednimustine, procarbazine, quinagolide, radium-223 chloride, raloxifene, raltitrexed, ranimustine, razoxane, refametinib, regorafenib, risedronic acid, rituximab, romidepsin, romiplostim, sargramostim, sipuleucel-T, sizofiran, sobuzoxane, sodium glycididazole, sorafenib, streptozocin, sunitinib, talaporfin, tamibarotene, tamoxifen, tasonermin, teceleukin, tegafur, tegafur + gimeracil + oteracil, temoporfin, temozolomide, temsirolimus, teniposide, testosterone, tetrofosmin, thalidomide, thiotepa, thymalfasin, tioguanine, tocilizumab, topotecan, toremifene, tositumomab, trabectedin, trastuzumab, treosulfan, tretinoin, trilostane, triptorelin, trofosfamide, tryptophan, ubenimex, valrubicin, vandetanib, vapreotide, vemurafenib, vinblastine, vincristine, vindesine, vinflunine, vinorelbine, vorinostat, vorozole, yttrium-90 glass microspheres, zinostatin, zinostatin stimalamer, zoledronic acid, zorubicin.
Promisingly, compound A can also be combined with biological therapeutics such as antibodies (e.g. avastin, rituxan, erbitux, herceptin, cetuximab) and recombinant proteins.
Compound A can also achieve positive effects in combination with other therapies directed against angiogenesis, such as, for example, with avastin, axitinib, regorafenib, recentin, sorafenib or sunitinib. Combinations with inhibitors of the proteasome and of mTOR and also antihormones and steroidal metabolic enzyme inhibitors are especially useful because of their favourable profile of adverse effects.
In general, the combination of compound A with other cytostatic or cytotoxic agents makes it possible to pursue the following goals:
• improved efficacy in slowing the growth of a tumour, in reducing its size or even in completely eliminating it in comparison with treatment using an individual active ingredient;
• the possibility of employing the chemotherapeutics used in a lower dosage than in the case of monotherapy;
• the possibility of a more tolerable therapy with fewer adverse effects in comparison with individual administration;
• the possibility of treating a broader spectrum of tumour diseases;
• achieving a higher response rate to the therapy;
• longer patient survival time in comparison with current standard therapy.
Furthermore, compound A according to the invention can also be used in connection with radiotherapy and/or a surgical intervention.
Examples
1. Preparation of Compound A Compound A' was prepared according to the procedure described in example 4 of WO2012/160034.
2. Proliferation assay
Table 1 : List of the cell lines investigated and results of the proliferation assays.

*After 72 hours of incubation with the substance
3. In vivo Experiments
The aim of the present experiments was to assess the in vivo efficacy and tolerability of Compound A' in monotherapy and in combination with paclitaxel or cisplatin in two gastric cancer models subcutaneously implanted in NMRI nu/nu mice.
Acronyms and Abbreviations
Table 2: Acronyms and abbreviations

The study included two in vivo efficacy experiments with female NMRI nu/nu mice bearing subcutaneous gastric cancer xenografts with C-MYC amplification. Compound A' was assessed at one dose level in mono- and in combination therapy with paclitaxel and cisplatin. Anti-tumour activity and tolerability of all groups were assessed using the vehicle control group as a reference.
Total Daily Dose Schedule Appl. No. of
Group ID Therapy
[mg/kg/day] [Dosing days] Route Animals
1 Vehicle 10 mL/kg/day 1-28 p.o. 10
2 Compound A' 25 1-28 p.o. 10
3 Paclitaxel 12 1, 8, 15, 22 i.v. 10
4 Cisplatin 2 1,4,7,10,13,16,19,22,25, 28 i.p. 10
Compound A' 25 1-28 p.o .
5 II II // II 10
Paclitaxel 12 1,8,15,22 i.v.
Compound A' 25 1-28 p.o.
6 II II // II 10
Cisplatin 2 1,4,7,10,13,16,19,22,25, 28 i.p.
3.3 Experimental procedures
Specific Animal Information
Mouse strain, sex: NMRI nu/nu, female
Animals supplied by: Harlan
Total number of mice
Efficacy test (implanted / randomised): 280 / 120
Approximate age at implantation: 5-7 weeks
Approximate age at randomisation: 7.5-13 weeks
Housing conditions
The animals were housed in individually ventilated cages. The animals were monitored twice daily. All materials were autoclaved prior to use. Food and water were provided ad libitum.
3.3.2 Tumour Information
3.3.2.1 Characterization of Test Tumours
The patient-derived GXF 251 tumour model used in this study was obtained from surgical specimens from cancer patients. GXA SCH LX was a cell line-derived cancer model (Oncotest, Freiburg, Germany). Both of these gastric cancer xenografts exhibit C-MYC amplification.
3.3.2.2 Tumour Implantation
Gastric cancer tumour fragments were obtained from xenografts in serial passage in nude mice and placed in PBS containing 10% penicillin/streptomycin. Tumour fragments (one fragment per animal; 3- 4 mm edge length) were then subcutaneously implanted in the flank of NMRI nu/nu recipient mice under isofluorane anaesthesia.
3.3.3 Randomisation
Animals and tumour implants were monitored daily until the maximum number of implants showed clear signs of beginning solid tumour growth. At randomisation, the volume of growing tumours was initially determined. Animals bearing one tumour of a volume of 50 - 250 mm3, preferably 80 - 200 mm3, were distributed in experimental groups according to the study protocol, considering a comparable median and mean of group tumour volume of approximately 100 - 120 mm3. The result of the randomisation was documented and maintained with the experimental data. Animals not randomised were euthanised. The day of randomisation is designated as day 0 of an experiment.
3.3.4. Test Reagents
Vehicle Compound A': 80% (m/V) PEG400 in water for injection
Vehicle paclitaxel and cisplatin: 0.9% saline
Compound A': preparation of a dosing solution (2.5 mg/ml) once weekly by diluting the Compound A' powder at 0.25% (w/v) in vehicle; storage of the dosing solution at 4°C; dosing volume 10 ml/kg
Paclitaxel: 1.6 ml of stock solution was mixed with 6.4 ml of vehicle to obtain the dosing solution.
Cisplatin: 1.6 mg of cisplatin was resuspended in 8 ml of vehicle to obtain the dosing solution.
3.3.5. Observations and Calculations
3.3.5.1 Mortality
Mortality checks were conducted daily during routine monitoring.
3.3.5.2 Body Weight
Mice were weighed twice a week. Relative body weights of individual mice in % were calculated by dividing the individual body weight on day X (BWx) by the individual body weight on day 0 (BWo) multiplied by 100 according to the formula:
Relative Body Weight (Dayx) [%] = BWX x 100
BWo
Group median relative body weights were calculated as well, considering only the weights of mice that were alive on the day in question.
3.3.5.3 Tumour Volume
The tumour volumes were determined by two-dimensional measurement with a caliper on the day of randomisation (day 0) and then twice weekly (i.e. on the same days on which mice were weighed). Tumour volumes were calculated according to the formulas:
Tumour volume (a x b2) x 0.5
where a represents the largest and b the perpendicular tumour diameter.
Relative volumes of individual tumours (RTVs) for Day x were calculated by dividing the absolute individual tumour volume on Day x (Tx) by the absolute individual tumour volume of the same tumour on Day 0 (T0) multiplied by 100%:

3.3.5.4 Anti-tumour Activity
Anti-tumour activity was evaluated as maximum tumour volume inhibition versus the vehicle control group.
3.3.5.5 Tumour Inhibition, Test/Control Value in %
Tumour inhibition for a particular day (T/C in %) was calculated from the ratio of the median RTV values of test versus control groups multiplied by 100. Median relative tumour volume of the test group on Dayx
1/C (Day x) I % l = , , ,. — : — = 100
Median relative tumour volume of the control group on Dayx
The minimum (or optimum) T/C% value recorded for a particular test group during an experiment represents the maximum anti-tumour activity for the respective treatment. T/C values were calculated if at least four of the randomised animals in a group were alive on the day in question. 3.3.5.6 Efficacy Criteria
Group optimum T/C values (in %) were used for activity rating as follows:
Table 3: Efficacy criteria
- Inactive T/C > 65%
+1- Borderline activity 50% <T/C < 65%
+ Moderate activity 25% <T/C <50%
++ High activity 10% <T/C <25%
+++ Very high activity
5% <T/C <10%
++++ Complete remission
T/C <5%
3.4 Results
3.4.1 Anti-tumour Efficacy of Compound A' in Xenograft-bearing Mice
Compound A' was assessed at one dose level, in mono- and in combination therapy with paclitaxel or cisplatin, in two gastric cancer models subcutaneously implanted in NMRI nu/nu mice. The two gastric cancer xenograft models, GXA SCH LX and GXF 251, exhibit C-MYC amplification.
In the GXA SCH LX tumour model, Compound A', paclitaxel or cisplatin monotherapy displayed moderate anti-tumour activity with minimum T/C values of 44.6%, 36.2% and 39.7%, respectively. The combination of Compound A' with paclitaxel or cisplatin increased the anti-tumour efficacy of the respective monotherapies leading to optimal T/C values of 22.0% (high activity) and 27.7% (moderate activity), respectively.
Tumour growth of GXA SCH LX was significantly reduced by Compound A' in combination treatments as compared to the respective vehicle control groups, as determined by the non-parametric Kruskal- Wallis test, followed by Dunn's post-test.
In the GFX 251 tumour model, Compound A' monotherapy resulted in borderline anti-tumour activity with an optimal T/C value of 59.9%, whereas paclitaxel or cisplatin as single agents displayed no anti- tumour efficacy. However, the combination of Compound A' with paclitaxel or cisplatin increased the anti -tumour efficacy of the respective monotherapies leading to optimal TC values of 18.0% (high activity) and 38.1 % (moderate activity), respectively.
Tumour growth of GFX 251 was significantly reduced by Compound A' combination treatments as compared to the respective vehicle control groups and to paclitaxel or cisplatin monotherapies alone, as determined by the Kruskal-Wallis test, followed by Dunn's post-test.
Table 4 - Summary of anti-tumour efficacy of Compound A"

Vehicle Compound A': 80% PEG400 in water for injection;
Vehicle paclitaxel and cisplatin: 0.9% saline
In conclusion, these data indicate significant and meaningful anti-tumour activity of Compound A' in combination with paclitaxel or cisplatin in patients with gastric cancer, displaying an amplification of C- MYC.
3.4.2. Survival and Body Weight Changes
No or moderate group median BWLs up to 7.9% were observed in tumour models GXA SCH LX and GXF 251. Groups receiving the control vehicle, cisplatin or paclitaxel monotherapies showed survival rates of 100%. Groups receiving Compound A' mono- or combination treatments exhibited survival rates ranging from 80- 90%, with the exception of animals receiving the combination of Compound A' and paclitaxel in model GXA SCH LX, which displayed a survival rate of 70%.
In conclusion, Compound A' showed an acceptable tolerability profile in two C-MYC amplified gastric cancer xenograft bearing mice.
3.5. Summary and Conclusion
The in vivo efficacy and tolerability of Bayer Healthcare's investigational compound Compound A' was assessed in mono- and in combination therapy with paclitaxel or cisplatin in two subcutaneously implanted gastric cancer xenograft models. The two gastric cancer xenografts, GXA SCH LX and GXF 251, which exhibit C-MYC amplification, were subcutaneously implanted into female NMRI nu/nu mice. Compound A' was administered orally at one dose level (25 mg kg/day), once daily, and treatments were initiated once subcutaneous tumours were established. A vehicle-treated control group was included in each experiment. Group sizes were 10 mice per group. Anti-tumour activity (tumour growth inhibition) and tolerability of all groups were assessed using the vehicle control group as a reference.
In the GXA SCH LX tumour model, Compound A', paclitaxel or cisplatin monotherapy displayed moderate anti-tumour activity with minimum T/C values of 44.6%, 36.2% and 39.7%, respectively. Moderate anti-tumour activity was also observed with Compound A' and cisplatin combination in both GXA SCH LX and GFX 251 gastric cancer models, with minimum T/C values of 27.7% and 38.1%, respectively. High anti-tumour activity was observed with Compound A' and paclitaxel in both GXA SCH LX and GFX 251 gastric cancer models, with minimum T/C values of 22% and 18%, respectively. In all these cases, tumour growth was significantly attenuated by Compound A' in combination with either paclitaxel or cisplatin as compared to the respective vehicle control group (Kruskal-Wallis test, followed by Dunn's post-test). No or moderate group median BWLs of up to 7.9% were observed in GXA SCH LX and GXF 251 tumour models. In conclusion, these data indicate significant and meaningful anti- tumour activity of Compound A' in combination with either paclitaxel or cisplatin in patients with gastric cancer, preferably with C-MYC amplification.
Claims
Claims

in the manufacture of a medicament for treating cancer in a subject,
wherein the medicament is manufactured for treating gastric cancer.
Use of a compound of formula (I) according to any one of claims 1,
wherein the enantiomer
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin- 2-amine or one of its physiologically acceptable salts is used.
Use according to claim 1 or 2, wherein
the subject who shall be treated is one for whom a C-MYC amplification has been detected in a tissue sample containing tumour cells from the subject.
Use according to claim 3, wherein the C-MYC amplification is detected by conventional cytogenetics, chromosomal comparative genomic hybridization, fluorescent in situ hybridization, next-generation sequencing, multiplex ligation-dependent probe amplification, quantitative PCR or digital PCR, single nucleotide polymorphism arrays, Southern blotting or slot blot methods.
5. A method for identifying a patient disposed to respond favourably to a CDK9-inhibitor for treating gastric cancer,
wherein the CDK9-inhibitor is a compound of formula I according to claim 1 or claim 2 and wherein the method comprises the detection of C-MYC amplification in tumour cells in a tissue sample from the patient and
wherein those patients are identified for a treatment of gastric cancer with a CDK9-inhibitor whose tumour cells have an amplification of the C-MYC gene.
A method according to claim 5 for identifying a patient disposed to respond favorably to 4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2- amine according to formula (I) or one of its physiologically acceptable salts or enantiomers for treating gastric cancer,
wherein the method comprises the detection of C-MYC amplification in tumour cells in a tissue sample from the patient and
wherein those patients are identified for a treatment of gastric cancer with 4-(4-Fluoro-2- methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2-amine according to formula (I) whose tumour cells have an amplification of the C-MYC gene.
A method according to claim 6,
wherein the C-MYC amplification
is detected by conventional cytogenetics, chromosomal comparative genomic hybridization, fluorescent in situ hybridization, next-generation sequencing, multiplex ligation-dependent probe amplification, quantitative PCR or digital PCR, single nucleotide polymorphism arrays, Southern blotting or slot blot methods.
Compound
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2- amine of formula I or one of its hysiologically acceptable salts or enantiomers

for the use of treating gastric cancer.
Compound according to 8, wherein the enantiomer
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin- 2-amine or one of its physiologically acceptable salts is used.
Compound according to claim 8 or 9,
wherein the subject who shall be treated is one for whom a C-MYC amplification has been detected in a tissue sample containing tumour cells from the subject.
Compound according to claim 10,
wherein the C-MYC amplification is detected by conventional cytogenetics, chromosomal comparative genomic hybridization, fluorescent in situ hybridization, next-generation sequencing, multiplex ligation-dependent probe amplification, quantitative PCR or digital PCR, single nucleotide polymorphism arrays, Southern blotting or slot blot methods.
12. Compound
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2- amine of formula I or one of its hysiologically acceptable salts or enantiomers

for the use in a method of treatment and/or prophylaxis of gastric cancer.
13. Compound according to any one of claim 12, wherein the enantiomer
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin- 2-amine or one of its physiologically acceptable salts is used.
14. Compound according to claim 12 or 13,
wherein the subject who shall be treated is one for whom a C-MYC amplification has been detected in a tissue sample containing tumour cells from the subject.
15. 4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2- amine according to formula (I) or one of its physiologically acceptable salts or enantiomers for the use in a method for treating a human patient diagnosed with gastric cancer
characterized by a C-MYC amplification
said method comprising the steps
a) assaying a tumour sample from the patient and
b) determining if C-MYC gene is amplified and
c) administering a therapeutically effective amount of 4-(4-Fluoro-2-methoxyphenyl)-N-{3- [(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin-2-amine of formula (I) or one of its physiologically acceptable salts or enantiomers
if C-MYC amplification is detected as defined in step b.
Method of treating gastric cancer comprising the steps
a) assaying a tumour sample from the patient and
b) determining if C-MYC is amplified and
c) administering a therapeutically effective amount of
4-(4-Fluoro-2-methoxyphenyl)-N- { 3-[(S-methylsulfonimidoyl)methyl]phenyl } -1,3,5- triazin-2-amine of formula I or one of its hysiologically acceptable salts or enantiomers

if C-MYC is amplified as defined in step b. 17. Method of claim 16, wherein in step c)
(+)-4-(4-Fluoro-2-methoxyphenyl)-N- { 3-[(S-methylsulfonimidoyl)methyl]phenyl } - 1 ,3,5-triazin- 2-amine or one of its physiologically acceptable salts is administered.
18. Use of
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2- amine of formula I or one of its hysiologically acceptable salts or enantiomers

for the treatment and/or prophylaxis of gastric cancers. 19. Use of a compound of formula (I) according to claim 18, wherein the enantiomer
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin- 2-amine or one of its physiologically acceptable salts is used
20. Use according to claim 18 or 19, wherein the subject who shall be treated is one for whom C- MYC amplification has been detected in a tissue sample containing tumour cells from the subject.
Use according to claim 20,
wherein the C-MYC amplification is detected by conventional cytogenetics, chromosomal comparative genomic hybridization, fluorescent in situ hybridization, next-generation sequencing, multiplex ligation-dependent probe amplification, quantitative PCR or digital PCR, single nucleotide polymorphism arrays, Southern blotting or slot blot methods.
22. Pharmaceutical combination comprising
4-(4-Fluoro-2-methoxyphenyl)-N- { 3-[(S-methylsulfonimidoyl)methyl]phenyl } - 1 ,3,5-triazin-2- amine of formula I or one of its hysiologically acceptable salts or enantiomers,

as defined in claim 1 and at least one or more further active ingredients for the treatment and/or prophylaxis of gastric cancers.
23. Pharmaceutical compositions comprising
4-(4-Fluoro-2-methoxyphenyl)-N- { 3-[(S-methylsulfonimidoyl)methyl]phenyl } - 1 ,3,5-triazin-2- amine of formula I or one of its hysiologically acceptable salts or enantiomers,

as defined in claim 1 and at least one inert, nontoxic, pharmaceutically suitable adjuvant for the treatment and/or prophylaxis of gastric cancers.
24. Pharmaceutical combination or pharmaceutical composition according to claim 22 or 23,
wherein (+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}- l,3,5-triazin-2-amine or one of its physiologically acceptable salts is comprised. 25. Pharmaceutical combination or pharmaceutical composition according to any one of claims claim 22 to 24, wherein the subject who shall be treated is one for whom a C-MYC amplification has been detected in a tissue sample containing tumour cells from the subject.
Pharmaceutical combination or pharmaceutical composition according to claim 25,
wherein the C-MYC amplification is detected by conventional cytogenetics, chromosomal comparative genomic hybridization, fluorescent in situ hybridization, next-generation sequencing, multiplex ligation-dependent probe amplification, as quantitative PCR or digital PCR, single nucleotide polymorphism arrays, Southern blotting or slot blot methods.
27 Use of the pharmaceutical combinations or the pharmaceutical compositions according to any one of claim 22 to 26 for the treatment and/or prophylaxis of gastric cancers.
28 Method of treatment and/or prophylaxis of gastric cancers using an effective amount of 4-(4- Fluoro-2-methoxyphenyl)-N- { 3-[(S-methylsulfonimidoyl)methyl]phenyl } - 1 ,3,5-triazin-2-amine of formula I or one of its hysiologically acceptable salts or enantiomers,

Method of treatment according to claim 28, wherein the enantiomer
(+)-4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl}-l,3,5-triazin- 2-amine or one of its physiologically acceptable salts is used.
30. Method of treatment according to claim 28 or 29, wherein the subject who shall be treated is one for whom a C-MYC amplification has been detected in a tissue sample containing tumour cells from the subject.
31. Method of treatment according to claim 30,
wherein the C-MYC amplification is detected by conventional cytogenetics, chromosomal comparative genomic hybridization, fluorescent in situ hybridization, next-generation sequencing, multiplex ligation-dependent probe amplification, quantitative PCR or digital PCR, single nucleotide polymorphism arrays, Southern blotting or slot blot methods.
A method of predicting whether a patient will be respond to the treatment with
4-(4-Fluoro-2-methoxyphenyl)-N-{ 3-[(S-methylsulfonimidoyl)methyl]phenyl }-l,3,5-triazin-2- amine of formula (I) or one of its physiologically acceptable salts or enantiomers,
wherein the method comprises the detection of C-MYC amplification in tumour cells in a tissue sample from the patient.
A method according to claim 32, wherein the C-MYC amplification
is detected by conventional cytogenetics, chromosomal comparative genomic hybridization, fluorescent in situ hybridization, next-generation sequencing, multiplex ligation-dependent probe amplification, quantitative PCR or digital PCR, single nucleotide polymorphism arrays, Southern blotting or slot blot methods.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15160587 | 2015-03-24 | ||
| EP15160587.0 | 2015-03-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016150902A1 true WO2016150902A1 (en) | 2016-09-29 |
Family
ID=52779521
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2016/056108 WO2016150902A1 (en) | 2015-03-24 | 2016-03-21 | Use of 4-(4-fluoro-2-methoxyphenyl)-n-{3-[(s-methylsulfonimidoyl)methyl]phenyl}-1,3,5-triazin-2-amine for treating gastric cancers |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW201642865A (en) |
| WO (1) | WO2016150902A1 (en) |
Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002066481A1 (en) | 2001-02-17 | 2002-08-29 | Astrazeneca Ab | Pyrimidine derivatives for inhibition of cell-proliferation |
| WO2003037346A1 (en) | 2001-10-31 | 2003-05-08 | Cell Therapeutics, Inc. | 6-phenyl-n-phenyl-(1,3,5) -triazine-2,4-diamine derivatives and related compounds with lysophphosphatidic acid acyltransferase beta (lpaat-beta) inhibitory activity for use in the treatment of cancer |
| WO2004009562A1 (en) | 2002-07-18 | 2004-01-29 | Janssen Pharmaceutica, Nv | Substituted triazine kinase inhibitors |
| WO2004072063A1 (en) | 2003-02-07 | 2004-08-26 | Vertex Pharmaceuticals Incorporated | Heteroaryl substituted pyrolls useful as inhibitors of protein kinases |
| WO2008012971A1 (en) | 2006-07-25 | 2008-01-31 | Toyota Jidosha Kabushiki Kaisha | Spark ignition type internal combustion engine |
| WO2008025556A1 (en) | 2006-08-29 | 2008-03-06 | Bayer Schering Pharma Aktiengesellschaft | Sulphoximides as protein kinase inhibitors |
| EP1218360B1 (en) | 1999-10-07 | 2008-05-28 | Amgen Inc., | Triazine kinase inhibitors |
| WO2008079933A2 (en) | 2006-12-22 | 2008-07-03 | Novartis Ag | Heteroaryl-heteroaryl compounds as cdk inhibitors for the treatment of cancer, inflammation and viral infections |
| WO2008109943A1 (en) | 2007-03-12 | 2008-09-18 | Cytopia Research Pty Ltd | Phenyl amino pyrimidine compounds and uses thereof |
| WO2008129080A1 (en) | 2007-04-24 | 2008-10-30 | Ingenium Pharmaceuticals Gmbh | 4, 6-disubstituted aminopyrimidine derivatives as inhibitors of protein kinases |
| WO2008129070A1 (en) | 2007-04-24 | 2008-10-30 | Ingenium Pharmaceuticals Gmbh | Inhibitors of protein kinases |
| WO2008129071A1 (en) | 2007-04-24 | 2008-10-30 | Ingenium Pharmaceuticals Gmbh | Inhibitors of protein kinases |
| WO2009032861A1 (en) | 2007-09-04 | 2009-03-12 | The Scripps Research Institute | Substituted pyrimidinyl-amines as protein kinase inhibitors |
| WO2010009155A2 (en) | 2008-07-14 | 2010-01-21 | Gilead Colorado, Inc. | Fused heterocyclyc inhibitor compounds |
| WO2011012661A1 (en) | 2009-07-30 | 2011-02-03 | Novartis Ag | Pyridine and pyrazine derivatives as protein kinase modulators |
| WO2011046970A1 (en) | 2009-10-12 | 2011-04-21 | Myrexis, Inc. | Amino - pyrimidine compounds as inhibitors of tbkl and/or ikk epsilon |
| EP2527332A1 (en) * | 2011-05-24 | 2012-11-28 | Bayer Intellectual Property GmbH | 4-Aryl-N-phenyl-1,3,5-triazin-2-amines containing a sulfoximine group as CDK9 inhibitors |
| WO2012160034A1 (en) | 2011-05-24 | 2012-11-29 | Bayer Intellectual Property Gmbh | 4-aryl-n-phenyl-1,3,5-triazin-2-amines containing a sulfoximine group |
-
2016
- 2016-03-21 WO PCT/EP2016/056108 patent/WO2016150902A1/en active Application Filing
- 2016-03-24 TW TW105109282A patent/TW201642865A/en unknown
Patent Citations (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1218360B1 (en) | 1999-10-07 | 2008-05-28 | Amgen Inc., | Triazine kinase inhibitors |
| WO2002066481A1 (en) | 2001-02-17 | 2002-08-29 | Astrazeneca Ab | Pyrimidine derivatives for inhibition of cell-proliferation |
| US7618968B2 (en) | 2001-10-31 | 2009-11-17 | Cell Therapeutics, Inc. | Aryl triazines as LPAAT-β inhibitors and uses thereof |
| WO2003037346A1 (en) | 2001-10-31 | 2003-05-08 | Cell Therapeutics, Inc. | 6-phenyl-n-phenyl-(1,3,5) -triazine-2,4-diamine derivatives and related compounds with lysophphosphatidic acid acyltransferase beta (lpaat-beta) inhibitory activity for use in the treatment of cancer |
| US20030153570A1 (en) | 2001-10-31 | 2003-08-14 | Cell Therapeutics, Inc. | Aryl triazines as LPAAT-SS inhibitors and uses thereof |
| US7291616B2 (en) | 2001-10-31 | 2007-11-06 | Cell Therapeutics, Inc. | Aryl triazines as LPAAT-β inhibitors and uses thereof |
| US20080064700A1 (en) | 2001-10-31 | 2008-03-13 | Cell Therapeutics, Inc. | Aryl triazines as LPAAT-beta inhibitors and uses thereof |
| WO2004009562A1 (en) | 2002-07-18 | 2004-01-29 | Janssen Pharmaceutica, Nv | Substituted triazine kinase inhibitors |
| WO2004072063A1 (en) | 2003-02-07 | 2004-08-26 | Vertex Pharmaceuticals Incorporated | Heteroaryl substituted pyrolls useful as inhibitors of protein kinases |
| WO2008012971A1 (en) | 2006-07-25 | 2008-01-31 | Toyota Jidosha Kabushiki Kaisha | Spark ignition type internal combustion engine |
| WO2008025556A1 (en) | 2006-08-29 | 2008-03-06 | Bayer Schering Pharma Aktiengesellschaft | Sulphoximides as protein kinase inhibitors |
| WO2008079933A2 (en) | 2006-12-22 | 2008-07-03 | Novartis Ag | Heteroaryl-heteroaryl compounds as cdk inhibitors for the treatment of cancer, inflammation and viral infections |
| WO2008109943A1 (en) | 2007-03-12 | 2008-09-18 | Cytopia Research Pty Ltd | Phenyl amino pyrimidine compounds and uses thereof |
| WO2008129080A1 (en) | 2007-04-24 | 2008-10-30 | Ingenium Pharmaceuticals Gmbh | 4, 6-disubstituted aminopyrimidine derivatives as inhibitors of protein kinases |
| WO2008129070A1 (en) | 2007-04-24 | 2008-10-30 | Ingenium Pharmaceuticals Gmbh | Inhibitors of protein kinases |
| WO2008129071A1 (en) | 2007-04-24 | 2008-10-30 | Ingenium Pharmaceuticals Gmbh | Inhibitors of protein kinases |
| WO2009032861A1 (en) | 2007-09-04 | 2009-03-12 | The Scripps Research Institute | Substituted pyrimidinyl-amines as protein kinase inhibitors |
| WO2010009155A2 (en) | 2008-07-14 | 2010-01-21 | Gilead Colorado, Inc. | Fused heterocyclyc inhibitor compounds |
| WO2011012661A1 (en) | 2009-07-30 | 2011-02-03 | Novartis Ag | Pyridine and pyrazine derivatives as protein kinase modulators |
| WO2011046970A1 (en) | 2009-10-12 | 2011-04-21 | Myrexis, Inc. | Amino - pyrimidine compounds as inhibitors of tbkl and/or ikk epsilon |
| EP2527332A1 (en) * | 2011-05-24 | 2012-11-28 | Bayer Intellectual Property GmbH | 4-Aryl-N-phenyl-1,3,5-triazin-2-amines containing a sulfoximine group as CDK9 inhibitors |
| WO2012160034A1 (en) | 2011-05-24 | 2012-11-29 | Bayer Intellectual Property Gmbh | 4-aryl-n-phenyl-1,3,5-triazin-2-amines containing a sulfoximine group |
Non-Patent Citations (37)
| Title |
|---|
| "European Union Network of Excellence (EUNE) for Gastric Cancer Steering Group. Gastric cancer in Europe", BR J SURG, vol. 95, 2008, pages 406 - 408 |
| ANONYMOUS: "Open Label Phase I Dose Escalation Study With BAY1143572 in Patients With Advanced Cancer", 5 September 2013 (2013-09-05), XP002757577, Retrieved from the Internet <URL:https://clinicaltrials.gov/ct2/show/NCT01938638?term=bay+1143572&rank=2> * |
| ARNE SCHOLZ ET AL: "Abstract DDT02-02: BAY 1143572: A first-in-class, highly selective, potent and orally available inhibitor of PTEFb/CDK9 currently in Phase I, inhibits MYC and shows convincing anti-tumor activity in multiple xenograft models by the induction of apoptosis", CANCER RESEARCH, 1 August 2015 (2015-08-01), XP055271477, Retrieved from the Internet <URL:http://cancerres.aacrjournals.org/content/75/15_Supplement/DDT02-02> [retrieved on 20160510] * |
| BALKO JM ET AL.: "Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets", CANCER DISCOV, vol. 4, no. 2, 2014, pages 232 - 245 |
| BANG YJ ET AL.: "Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial", LANCET, vol. 376, no. 9742, 2010, pages 687 - 697 |
| BARK-JONES ET AL., ONCOGENE, vol. 25, 2006, pages 1775 |
| BAYKARA 0 ET AL.: "Amplification of chromosome 8 genes in lung cancer", J CANCER, vol. 6, no. 3, 2015, pages 270 - 275 |
| CALCAGNO DQ ET AL.: "Interrelationship between chromosome 8 aneuploidy, C-MYC amplification and increased expression in individuals from northern Brazil with gastric adenocarcinoma", WORLD J GASTROENTEROL, vol. 12, no. 38, 2006, pages 6207 - 6211 |
| CHO ET AL., CELL CYCLE, vol. 9, 2010, pages 1697 |
| DE SOUZA C ET AL.: "MYC Deregulation in Gastric Cancer and Its Clinicopathological Implications", PLOS ONE, vol. 8, no. 5, 2013, pages E64420 |
| DEMING SL ET AL.: "C-myc amplification in breast cancer: a meta-analysis of its occurrence and prognostic relevance", BR J CANCER, vol. 83, no. 12, 2000, pages 1688 - 1695 |
| DEY ET AL., CELL CYCLE, vol. 6, 2007, pages 1856 |
| FLETCHER S ET AL.: "Small-molecule inhibitors of the Myc oncoprotein", BIOCHIM. BIOPHYS. ACTA, 2014 |
| HAN S ET AL.: "C-MYC expression is related with cell proliferation and associated with poor clinical outcome in human gastric cancer", J KOREAN MED SCI, vol. 14, no. 5, 1999, pages 526 - 530 |
| HE ET AL., MOL. CELL, vol. 29, 2008, pages 588 |
| JEMAL A ET AL.: "Global cancer statistics", CA CANCER J CLIN, vol. 61, no. 2, 2011, pages 69 - 90 |
| JONAS CICENAS ET AL: "The CDK inhibitors in cancer research and therapy", JOURNAL OF CANCER RESEARCH AND CLINICAL ONCOLOGY, SPRINGER, BERLIN, DE, vol. 137, no. 10, 30 August 2011 (2011-08-30), pages 1409 - 1418, XP019951971, ISSN: 1432-1335, DOI: 10.1007/S00432-011-1039-4 * |
| KIM S ET AL.: "High-throughput sequencing and copy number variation detection using formalin fixed embedded tissue in metastatic gastric cancer", PLOS ONE, vol. 9, no. LL, 2014, pages EL 11693 |
| KOZMA L ET AL.: "C-MYC amplification and cluster analysis in human gastric carcinoma", ANTICANCER RES, vol. 21, no. 1B, 2001, pages 707 - 710 |
| LORDICK F: "Unmet needs and challenges in gastric cancer: The way forward", CAN TREAT REV, vol. 40, 2014, pages 692 - 700 |
| MILNE AN ET AL.: "Early onset gastric cancer: on the road to unravelling gastric carcinogenesis", CURR MOL MED, vol. 7, no. 1, 2007, pages 15 - 28 |
| MINCA EC ET AL.: "Genomic microarray analysis on formalin-fixed paraffin-embedded material for uveal melanoma prognostication", CANCER GENET, vol. 207, no. 7-8, 2014, pages 306 - 315 |
| MYLLYKANGAS S; KNUUTILA S: "Manifestation, mechanisms and mysteries of gene amplifications", CANCER LETT, vol. 232, no. 1, 2006, pages 79 - 89 |
| ONODA N ET AL.: "Overexpression of C-MYC messenger RNA in primary and metastatic lesions of carcinoma of the stomach", J AM COLL SURG, vol. 182, no. 1, 1996, pages 55 - 59 |
| OOI A ET AL.: "Semi-comprehensive analysis of gene amplification in gastric cancers using multiplex ligation-dependent probe amplification and fluorescence in situ hybridization", MOD PATHOL, 2015 |
| PODDIGHE PJ ET AL.: "Genomic amplification of MYC as double minutes in a patient with APL-like leukemia", MOL CYTOGENET, vol. 7, no. 1, 2014, pages 67 |
| SETOODEH R ET AL.: "Double-hit mantle cell lymphoma with MYC gene rearrangement or amplification: a report of four cases and review of the literature", INT J CLIN EXP PATHOL, vol. 6, no. 2, 2013, pages 155 - 167 |
| ULRICH TJ LUECKING ET AL: "Abstract 2828: Rapid identification of potent and highly selective, oral PTEFb Inhibitor BAY 1143572 with first in class potential", CANCER RESEARCH, 1 August 2015 (2015-08-01), XP055271531, Retrieved from the Internet <URL:http://cancerres.aacrjournals.org/content/75/15_Supplement/2828> [retrieved on 20160510] * |
| VERBEKE SL ET AL.: "Array CGH analysis identifies two distinct subgroups of primary angiosarcoma of bone", GENES CHROMOSOMES CANCER, vol. 54, no. 2, 2015, pages 72 - 81 |
| VOGT N ET AL.: "Amplicon rearrangements during the extrachromosomal and intrachromosomal amplification process in a glioma", NUCLEIC ACIDS RES, vol. 42, no. 21, 2014, pages 13194 - 13205 |
| WADDELL ET AL.: "Gastric cancer: ESMO-ESSO-ESTRO Clinical Practice Guidelines for diagnosis, treatment and follow-up", RADIOTHERAPY AND ONCOLOGY, vol. 110, 2014, pages 189 - 194 |
| WANG ET AL., CHEMISTRY & BIOLOGY, vol. 17, 2010, pages 1111 - 1121 |
| WANG; FISCHER, TRENDS PHARMACOL. SCI., vol. 29, 2008, pages 302 |
| YANG ET AL., MOL. CELL, vol. 19, 2005, pages 535 |
| YANG W ET AL.: "Targeted therapy for gastric cancer: Molecular pathways and ongoing investigations", BIOCHIMICA ET BIOPHYSICA ACTA, vol. 1846, 2014, pages 232 - 237 |
| ZHOU ET AL., J. VIROL., vol. 80, 2006, pages 4781 |
| ZHOU; YIK, MICROBIOL. MOL. BIOL. REV., vol. 70, 2006, pages 646 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW201642865A (en) | 2016-12-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI689307B (en) | Use of substituted 2,3-dihydroimidazo[1,2-c]quinazolines | |
| US10406162B2 (en) | Substituted 2,3-dihydroimidazo[1,2-C]quinazoline-containing combinations | |
| TWI639599B (en) | Use of substituted 2,3-dihydroimidazo[1,2-c]quinazolines | |
| US11701347B2 (en) | Use of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine for treating diffuse large B-cell lymphoma | |
| WO2016113187A1 (en) | Use of 4-(4-fluoro-2-methoxyphenyl)-n-{3-[(s-methylsulfonimidoyl)methyl]phenyl}-1,3,5-triazin-2-amine for treating leukemias | |
| CA2980507A1 (en) | Use of 4-(4-fluoro-2-methoxyphenyl)-n-{3-[(s-methylsulfonimidoyl)methyl]phenyl}-1,3,5-triazin-2-amine for treating lymphomas | |
| EP3274338A1 (en) | Use of 4-(4-fluoro-2-methoxyphenyl)-n-{3-[(s-methylsulfonimidoyl)methyl]phenyl}-1,3,5-triazin-2-amine for treating multiple myeloma | |
| WO2016150902A1 (en) | Use of 4-(4-fluoro-2-methoxyphenyl)-n-{3-[(s-methylsulfonimidoyl)methyl]phenyl}-1,3,5-triazin-2-amine for treating gastric cancers | |
| CA2904149A1 (en) | Use of (rs)-s-cyclopropyl-s-(4-{[4-{[(1r,2r)-2-hydroxy-1-methylpropyl]- oxy}-5-(trifluoromethyl)pyrimidin-2-yl]amino}phenyl)sulfoximide for treatment of specific tumours |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16713785 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16713785 Country of ref document: EP Kind code of ref document: A1 |