WO2016039466A1 - New production method for azodicarbonamide - Google Patents
New production method for azodicarbonamide Download PDFInfo
- Publication number
- WO2016039466A1 WO2016039466A1 PCT/JP2015/075926 JP2015075926W WO2016039466A1 WO 2016039466 A1 WO2016039466 A1 WO 2016039466A1 JP 2015075926 W JP2015075926 W JP 2015075926W WO 2016039466 A1 WO2016039466 A1 WO 2016039466A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- urea
- water
- organic solvent
- reaction
- Prior art date
Links
- 0 C=C=*C(*(N)=C=C)=O Chemical compound C=C=*C(*(N)=C=C)=O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C281/00—Derivatives of carbonic acid containing functional groups covered by groups C07C269/00 - C07C279/00 in which at least one nitrogen atom of these functional groups is further bound to another nitrogen atom not being part of a nitro or nitroso group
- C07C281/20—Derivatives of carbonic acid containing functional groups covered by groups C07C269/00 - C07C279/00 in which at least one nitrogen atom of these functional groups is further bound to another nitrogen atom not being part of a nitro or nitroso group the two nitrogen atoms of the functional groups being doubly-bound to each other, e.g. azoformamide
Definitions
- the present invention relates to a production method for obtaining azodicarbonamide using a urea active derivative and a structure.
- Azodicarbonamide is a useful compound widely used as a foaming agent from the viewpoint of its decomposition behavior, physical properties, and chemical properties.
- the most commonly practiced conventional production method involves reacting hydrazine hydrate produced from urea or ammonia with 2 moles of urea.
- This is a method for producing azodicarbonamide by several stages of reaction including the above (Patent Document 1).
- This conventional production method causes wasteful consumption of raw materials that do not contribute to the molecular structure of the target product azodicarbonamide, which causes an increase in cost.
- Another problem is the generation of a large amount of ammonia as a by-product and carbon dioxide that causes global warming.
- this method uses a chlorinating agent, an oxidizing agent, a strong acid, a heavy metal catalyst, or the like, a large amount of waste water containing a large amount of salts, acids, heavy metals, ammonia nitrogen, and the like is generated as waste. Since disposal is required to dispose of these, it costs a large amount of processing costs, increases product costs, and may cause pollution. Sometimes it is possible.
- Patent Document 4 the structural formula of urea and the structural formula of azodicarbonamide (ADCA) are completely different by using a special apparatus in a special method called organic electrolytic synthesis from urea. Nevertheless, it is described that ADCA is suddenly generated from urea. It is illogical if it reacts with other raw materials from urea to produce various reaction intermediates and the desired ADCA is not obtained. Also in the energy yield, if the electric energy required for the direct reaction is calculated, there is a problem that a large amount of electric energy having a very low electric energy yield is wasted. Details will be given in later sections.
- Patent Document 3 The production method of chlorourea and bromourea used as raw materials or intermediates used in the present invention is described in (Patent Document 3) in addition to (Patent Document 1).
- Patent Document 4 In a recently published document (Patent Document 4), the actual state of the reaction has not been elucidated at all using a special method and a special apparatus called an organic electrolytic synthesis method from urea.
- Various general problems and drawbacks have already been pointed out in the background section. It is a patent that the object that is completely different from the raw material can be unknown at once, special equipment, special technology is necessary, and if you do not perform continuous recycling reaction with raw materials with complicated composition, you can obtain the object I can't. Furthermore, since the raw material is used in a large excess, it is essential to recycle the raw material. Prediction of side reactions and impurities from the side reactions can be predicted, but there is no explanation for these exclusions and purification.
- An object of the present invention is to provide an epoch-making production method of azodicarbonamide that is simple, safe and has a reduced environmental burden.
- the present inventors have conducted intensive research on a simple and efficient method of reacting urea with an active urea derivative and an active structure of urea.
- active urea under normal pressure is analyzed.
- Urea represented by formula (1) is represented by formula (10) in a homogeneous system or a heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water. Since the compound represented by the formula (10) is chemically very active, a de-HBr reaction is immediately generated and converted into the compound of the formula (7). A novel process for producing azodicarbonamide formula (7).
- Urea represented by the formula (1) is converted into water or an organic solvent or ionic liquid and an organic solvent or a homogeneous system or a heterogeneous mixed system of ionic liquid and water under the acidic condition (9)
- a compound of formula (10) is obtained by reacting a urea molecule with a bromourea active transition intermediate represented by the formula:
- Urea represented by the formula (1) can be converted from urea to acidic conditions in a homogeneous or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water.
- N-bromourea represented by the formula (8) is generated by an agent or a bromination method, and then brominated by electrophilic oxidation with an active bromo compound of NH group to which a bromo atom is bonded or a hydrogen peroxide-derived activator.
- a urea active transition intermediate formula (9) is obtained and at the same time urea molecules react to obtain a compound of formula (10).
- Urea represented by formula (1) is subjected to chlorination reaction under acidic conditions in a homogeneous or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water.
- the compound of formula (11) is obtained.
- Monochlorourea of formula (11) (including N-chlorourea Na salt) is subjected to bromination or bromination to obtain N-bromourea represented by formula (8).
- the bromourea active transition intermediate formula (9) is obtained by electrophilic oxidation with the active bromo compound of NH group to which the bromo atom is bonded or the hydrogen peroxide-derived activator, and at the same time, the urea molecule reacts and the formula (10) A compound is obtained.
- Urea represented by the formula (1) can be converted from urea to acidic bromine under acidic conditions in a homogeneous or heterogeneous mixed system of water or organic solvent or ionic liquid and organic solvent or ionic liquid and water.
- N-bromourea represented by the formula (8) is produced by an agent or bromination method, and then electrophilic oxidation of the NH group to which the bromine atom is bonded is electrolyzed and desorbed as a proton by electrolysis, Bromourea active transition intermediate Formula (9).
- urea molecules react to obtain a compound of formula (10).
- Item 6 When an electrolytic reaction is used for urea represented by formula (1) in water or an organic solvent or ionic liquid, and an organic solvent or a homogeneous system or a heterogeneous mixed system of ionic liquid and water.
- a mixture of hydrogen bromide, bromo salt and hydrogen chloride, chloride salt as a supporting electrolyte, two-electron oxidation under acidic conditions to generate a bromo cation to produce N-bromourea represented by formula (8)
- electrophilic oxidation of the NH group to which the bromo atom is bonded is subjected to two-electron oxidation by an electrolytic reaction and eliminated as a proton to obtain a bromourea active transition intermediate formula (9).
- urea molecules react to obtain a compound of formula (10).
- Item 7 The urea represented by the formula (1) is converted from urea to a bromine under acidic conditions in a homogeneous or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water.
- An N-bromourea represented by the formula (8) is produced by a chlorinating agent or a bromination method, or, if necessary, it is represented by the formula (11) from urea by a chlorinating agent or a chlorination method under acidic conditions.
- Chlorourea (including N-chlorourea Na salt) is produced, then N-bromourea represented by the formula (8) is produced by a brominating agent or bromination method, and then supported using an electrolytic reaction.
- Urea represented by the formula (1) is hydrogen bromide under acidic conditions in water or an organic solvent or ionic liquid, and an organic solvent or a homogeneous or heterogeneous mixture of ionic liquid and water. Then, bromine is generated by adding a peroxide such as hydrogen peroxide dropwise to a mixture of bromo salt, hydrogen chloride and chloride salt.
- the urea represented by the formula (1) is generated by the generated bromine, hypobromite, and hypobromite, and the N-bromourea represented by the formula (8) is generated by the two-electron oxidation by the brom cation.
- the bromourea active transition intermediate formula (9) is obtained by electrophilic oxidation with an active bromo compound of NH group to which a bromo atom is bonded or an active substance derived from hydrogen peroxide, and at the same time, a urea molecule reacts with the formula (10 ) Is obtained.
- Item 9 The urea represented by the formula (1) is converted into an organic bromide under acidic conditions in a homogeneous system or a heterogeneous mixed system of water, an organic solvent or an ionic liquid, and an organic solvent or an ionic liquid and water.
- urea is two-electron-oxidized by a brom cation to produce N-bromourea represented by the formula (8), and then a bromine atom is bonded.
- Bromourea active transition intermediate formula (9) is obtained by electrophilic oxidation with an active bromine compound of NH group or an active substance derived from hydrogen peroxide, and at the same time, urea molecules react to obtain a compound of formula (10).
- Item 10 In a homogeneous system or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water, a halogen acid salt such as hydrogen bromide or hydrogen chloride of urea is used as a raw material Then, chlorine or bromine is generated by dropping a peroxide such as hydrogen peroxide under acidic conditions. Generated and converted chlorine, hypochlorous acid, hypochlorite, bromine, hypobromite, hypobromite, chlorinated and brominated urea represented by formula (1) A novel process for producing an azodicarbonamide according to any one of Items 1 to 9
- Item 11 A novel method for producing azodicarbonamide formula (7), wherein the method of the corresponding item is taken from the method of Item 1 to Item 10 using a mixture of urea and urea salt as a raw material.
- Item 13 When a halogen or a halogen compound is used with a peroxide such as hydrogen peroxide as an oxidizing agent, it is stable to water such as cerium compound, vanadium compound, selenium compound, tellurium compound, transition metal compound and aluminum fluoride. 13. A novel process for producing an azodicarbonamide formula (7) according to items 1 to 12, wherein a strong Lewis acid is used as a catalyst and an acid such as formic acid or acetic acid is used.
- a peroxide such as hydrogen peroxide as an oxidizing agent
- M represents hydrogen and a monovalent to tetravalent metal selected from the group consisting of Li, Na, K, Mg, Ca, Mn, Fe, Ni, Cu, Ag, Zn, and Sn.
- X represents a chlorine atom and a bromine atom.
- m represents an integer of 1 to 4.
- M is hydrogen, Li, Na, K, Mg, Ca, Ti, Zr, Mn, Fe, Ni, Cu, Ag, Zn, Al, Si, Sn, or other metal ions, in formula (13)
- the cation portion of the represented compound represents H and ammonium ion, primary ammonium ion, secondary ammonium ion, tertiary ammonium ion, and X is a halogen anion and an organic brominating agent anion.
- iodine Molecule bromine molecule, chlorine molecule, chlorobromine.
- the present invention is an efficient production method for directly obtaining the azodicarbonamide represented by the formula (7) from the urea represented by the formula (1) in one step. Since it is carried out in a homogeneous or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water, it is a simple, safe and inexpensive production method with a low environmental load. Moreover, since the produced azodicarbonamide is precipitated as a solid, separation and purification are easy. Further, since the aqueous solution containing the compounds represented by formulas (12) to (13), hydrogen halide, or halogen and unreacted urea used in the reaction can be circulated and reused as it is, almost no waste is removed from the system.
- the urea of formula (1) may be urea hydrochloride, bromate or other salts, and the hydrogen halide or halogen salt used for the halogenation is in an amount relative to the urea to be reacted. is there.
- the peroxide etc. which are used as an oxidizing agent can be used, since hydrogen peroxide becomes final water, there is no generation of by-products and waste.
- an electrolysis method if a gas diffusion electrode is used, hydrogen is not generated and power consumption can be suppressed.
- the present invention relates to a homogeneous or heterogeneous mixture of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water, with or under active conditions of urea or urea salts under acidic conditions.
- oxidation reaction of bromide compounds and hydrogen peroxide-derived active substances, or by electrolytically oxidizing intermediates of chlorourea and bromourea, by-produced hydrogen halide and halogen salts are recycled and discarded.
- This is a method for producing an azodicarbonamide having no product, and is represented by the following reaction formula.
- the chemical reaction is a homogeneous or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water.
- Urea or a salt thereof represented by formula (1) A compound represented by formula (12) under acidic conditions, a chlorinating agent or a brominating agent prepared from the compound represented by formula (13), and replicated hydrogen chloride or chloride salt and hydrogen bromide Alternatively, the hydrogen bromide salt is recycled using an oxidizing agent such as hydrogen peroxide that does not produce by-products.
- the compound (11) or compound (8) proceeds with an active bromide compound and a hydrogen peroxide-derived activator or by electrolyzing two molecules of urea theoretically by four-electron oxidation.
- the active bromide compound and the hydrogen peroxide-derived activator include compounds and chemical species known in the art, but are not limited to specific compounds and chemical species.
- hydrogen chloride, hydrogen bromide, chlorine, bromine, hypochlorous acid, hypochlorous acid and hydrogen peroxide undergo a correlation reaction with each other, resulting in a competitive reaction. These compounds also coordinate with urea to form complexes, complicating the reaction.
- an active bromine compound is produced.
- the halogen compound acts on hydrogen peroxide and its active substance under acidic conditions, an active substance derived from hydrogen peroxide is generated.
- the active bromide compound and / or the hydrogen peroxide-derived activator may be any compound or chemical species that oxidizes bromourea to form a bromourea active transition intermediate, and there are many compounds and chemical species belonging to them. However, it is not particularly limited to a specific compound or chemical species.
- M represents hydrogen and a monovalent to tetravalent metal selected from the group consisting of Li, Na, K, Mg, Ca, Mn, Fe, Ni, Cu, Ag, Zn, and Sn.
- X represents a chlorine atom, a bromine atom, or an iodine atom.
- m represents an integer of 1 to 4.
- Examples of the compound represented by the formula (12) include hypochlorous acid, lithium hypochlorite, chlorous acid, lithium chlorite, chloric acid, lithium chlorate, perchloric acid, lithium perchlorate, Sodium hypochlorite, sodium chlorite, sodium chlorate, sodium perchlorate, potassium hypochlorite, potassium chlorite, potassium chlorate, potassium perchlorate, calcium hypochlorite, calcium chlorite , Calcium chlorate, calcium perchlorate, magnesium hypochlorite, magnesium chlorite, magnesium chlorate, magnesium perchlorate, hypobromite, lithium hypobromite, bromite, lithium bromite, Bromate, lithium bromate, perbromate, lithium perbromate, sodium hypobromite, sodium bromate, sodium bromate, sodium perbromate , Potassium hypobromite, potassium bromite, potassium bromate, potassium perbromate, calcium hypobromite, calcium bromate, calcium bromate, calcium perbromate, magnesium hypobromite, hypoiodide Acid, lithium
- the compound represented by the formula (12) can be used for generating an active bromide compound and / or an active form derived from hydrogen peroxide.
- the active bromide compound includes a compound generated in the reaction system, and examples thereof include a brom cation, a brom cation radical, a brom radical, bromine chloride, a bromine complex (for example, dioxane complex) and the like.
- the hydrogen peroxide-derived active substance includes compounds generated in the reaction system, such as OH cation, OH radical, OOH radical, singlet oxygen, OOH anion, OOH cation, superoxide, superoxide anion. , Superoxide anion radical, peroxy radical, hydroperoxyl radical and the like.
- the compound represented by the formula (12) is preferably hypochlorous acid, hypobromite, sodium hypochlorite, sodium chlorate, potassium hypochlorite, potassium chlorate, calcium hypochlorite.
- M represents a monovalent to tetravalent metal selected from the group consisting of hydrogen, Li, Na, K, Mg, Ca, Mn, Fe, Ni, Cu, Ag, Zn, and Sn. And H And an ammonium ion, a primary ammonium ion, a secondary ammonium ion, a tertiary ammonium ion and a halogen cation, X is a halogen anion and an organic brominating agent anion component, and a bromine molecule, a chlorine molecule, a chloro molecule. Indicates bromine.
- halogen in X in the formula (13) examples include chlorine, bromine and iodine.
- Examples of the compound represented by the formula (13) include hydrogen chloride, hydrogen bromide, hydrogen iodide, lithium chloride, sodium chloride, potassium chloride, calcium chloride, copper chloride, magnesium chloride, iron chloride, zinc chloride, and chloride.
- the compound represented by the formula (13) can be used to generate an active bromide compound and / or an active form derived from hydrogen peroxide. It can be used to generate active bromide compounds and / or hydrogen peroxide derived activators.
- the active bromide compound includes a compound generated in the reaction system, and examples thereof include a brom cation, a brom cation radical, a brom radical, bromine chloride, a bromine complex (for example, dioxane complex) and the like.
- the hydrogen peroxide-derived active substance includes compounds generated in the reaction system, such as OH cation, OH radical, OOH radical, singlet oxygen, OOH anion, OOH cation, superoxide anion, superoxide. Anion radical, peroxy radical, hydroperoxyl radical and the like can be mentioned.
- the compound represented by the formula (13) is preferably hydrogen chloride, hydrogen bromide, lithium chloride, sodium chloride, potassium chloride, calcium chloride, magnesium chloride, zinc chloride, iron chloride, copper chloride, lithium bromide, Sodium bromide, potassium bromide, calcium bromide, magnesium bromide, zinc bromide, copper bromide, lithium iodide, sodium iodide, potassium iodide, copper iodide, ammonium chloride, ammonium bromide, chlorine, bromine , Iodine, chlorobromine, dibromoisocyanuric acid, N-bromosuccinimide and the like, but are not particularly limited to these compounds.
- halogen in X in the formula (13) examples include fluorine, chlorine, bromine and iodine.
- examples of the hydrogen halide include hydrogen chloride, hydrogen bromide, and hydrogen iodide.
- examples of the halogen include chlorine, bromine, and iodine.
- highly stable strong Lewis acids as catalysts and the use of acids such as formic acid and acetic acid are effective.
- the invention is not particularly limited to this.
- urea as a raw material is contained at a concentration of 0.5 to 50 mol / L in a homogeneous system or a heterogeneous mixed system with water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid.
- concentration is preferably 2 to 30 mol / L.
- the invention is not particularly limited to this.
- the concentration of urea is influenced by the solubility with respect to temperature and the ratio of bromine-based oxidant, but basically, the presence of a high concentration and a large amount of urea is preferable.
- the molar ratio of a compound such as a halogen-based oxidant to urea is preferably such that when the active intermediate transition compound bromourea cation (compound 9) is formed, its surroundings are enveloped with urea.
- the compound such as halogen-based oxidizing agent is 0.01 to 0.45 molar ratio, but preferably 0.05 to 0.3 molar ratio. Hydrogen peroxide, a substantial oxidant, corresponds to these numbers. However, it is not particularly limited to this.
- the series of reactions of the present invention does not require separation of intermediates. If necessary, the compounds represented by the formulas (8) and (11) can be separated and stored, and can be used as raw materials for other derivatives.
- the temperature of the series of reactions of the present invention is too low for converting halogen anions to halogen molecules, halogen cations, active halogen species with an oxidizing agent such as hydrogen peroxide, and for the production of hydrogen peroxide active substances.
- an oxidizing agent such as hydrogen peroxide
- the reaction is carried out at ⁇ 200 to 100 ° C., preferably ⁇ 10 to 70 ° C. It is desirable to adjust the temperature according to the reaction conditions in view of the stability and reactivity of the reaction intermediate and reaction active intermediate.
- an aqueous solution of urea has a significantly reduced freezing point.
- the electrolytic method When the electrolytic method is applied to the intermediate, it is desirable to react at a low temperature in order to decompose the active intermediate and suppress side reactions from the heat generated during electrolysis.
- the reaction is carried out at ⁇ 50 to 100 ° C., preferably ⁇ 20 to 40 ° C. It is also possible to carry out the reaction at a low temperature.
- the reaction can be performed at a low temperature even in a homogeneous mixed system with an organic solvent or an ionic liquid.
- the invention is not particularly limited to this.
- organic solvent examples include halogen solvents such as dichloroethane, carbon tetrachloride, and chlorobenzene, and chain or cyclic alkanes such as n-pentane, n-hexane, cyclopentane, and cyclohexane, Ethers such as tetrahydrofuran, 1,2-dimethoxyethane, dioxane, methyl-t-butyl ether and methylcyclopentyl ether, nitriles such as acetonitrile and propionitrile, ketones such as acetone, methyl ethyl ketone, cyclopentanone and cyclohexanone, methyl
- halogen solvents such as dichloroethane, carbon tetrachloride, and chlorobenzene
- chain or cyclic alkanes such as n-pentane, n-hexane, cyclopentane, and cycl
- an ionic liquid you may have imidazolium which may have a substituent, quaternary ammonium, pyridinium which may have a substituent, phosphonium, and a substituent as a cation component.
- Pyrrolidiniums, sulfoniums, anion components such as boron tetrafluoride, phosphorus hexafluoride, trifluoroacetic acid, trifluoromethanesulfonic acid, bistrifluoromethanesulfonimide, alkylsulfonic acid, acetic acid, nitric acid, chlorine, bromine
- the ionic liquid include salts of these cationic components and anionic components.
- the use of water, an organic solvent, or an ionic liquid alone may be used. A combination of these may also be used.
- the solution containing urea may be a homogeneous phase or a heterogeneous phase (liquid phase separation state).
- a mixed solvent of water and an organic solvent in the case of a heterogeneous system, the raw materials and various reagents are mainly dissolved in the aqueous layer. In addition, various reagents are mainly dissolved.
- urea and salts do not dissolve at all, it is important that urea and salts are dispersed in the solvent as fine particles as possible.
- the halogenating agent is a hydrogen halide or halogen salt after the reaction, but it is regenerated to a halogen with a peroxide such as hydrogen peroxide, so when using hydrogen peroxide in particular,
- a peroxide such as hydrogen peroxide
- the oxidation by-product is a small amount of water, and it is not necessary to adjust the liquid composition from the accumulation of by-products generated in minute amounts to the limit of recycling.
- this electrolytic reaction may be constant voltage electrolysis or constant current electrolysis.
- any method of a non-diaphragm, a diaphragm, and an ion exchange membrane is possible as an electrolysis method.
- a metal electrode, a carbon electrode, and a composite electrode thereof can be used as the anode in the electrolysis of the present invention.
- noble metals such as gold, silver, platinum and ruthenium and minor metals such as titanium, chromium, nickel and manganese, and electrodes coated with noble metals other than noble metals such as titanium, stainless steel, iron and hastelloy And electrodes coated with precious metals on substrates other than metals such as olefin resins, engineering resins, carbon-based substrates, composite coated electrodes of metal oxides such as iridium oxide and ruthenium oxide and platinum, and coatings similar to the above with minor metals An electrode etc. are mentioned.
- examples of the carbon-based electrode include carbon-based electrodes such as carbon, glassy carbon, graphite, graphene, carbon sheet, carbon fiber sheet, carbon fiber cloth, diamond-like coated electrode, and composite electrodes thereof.
- the anode is an electrode in which platinum is coated on a metal substrate other than a noble metal such as platinum, titanium, titanium, stainless steel, iron, or hastelloy, and a substrate other than a metal such as an olefin resin, an engineering resin, or a carbon-based substrate.
- Electrode coated with platinum composite electrode of platinum with metal oxide such as iridium oxide and ruthenium oxide, and carbon, glassy carbon, graphite, graphene, carbon sheet, carbon fiber sheet, carbon fiber cloth, diamond-like coated electrode Carbon-based electrodes such as these, and composite electrodes thereof, but are not particularly limited thereto.
- the cathode is not particularly limited, and the materials exemplified as the anode electrode and general-purpose metals such as iron, copper, and aluminum, and stainless steel, hastelloy, various alloys, and composite electrodes thereof can be used.
- Preferred as the cathode are platinum, stainless steel, titanium, stainless steel, hastelloy, iron, and electrodes in which platinum is coated on a metal base other than noble metal such as titanium, stainless steel, iron, hastelloy, olefin resin, engineering resin, carbon-based group Electrode with platinum coated on base material other than metal such as metal, and composite coated electrode of metal oxide such as iridium oxide and ruthenium oxide and platinum and carbon, glassy carbon, graphite, carbon sheet, carbon fiber sheet, carbon fiber Although it is carbon-type electrodes, such as cloth, these composite electrodes, etc., it is not specifically limited to these.
- the shape of the electrode a plate shape, a cloth shape, a comb shape, a pipe shape, a nonwoven fabric shape, a shape obtained by a paper-making method, a felt shape, etc. can be used. Processed products can be used.
- the anode has a shape without a gap and the cathode has a shape with a hole or a gap.
- the shape of the cathode is not limited. In particular, it is not limited to these.
- the electrolysis of the present invention is carried out in a state where the current density is kept constant at 1 to 20,000 mA / cm 2 , preferably 10 to 5000 mA / cm 2 .
- the electrode potential is set to 0.5 to 100 V vs. Ag / AgCl, preferably 1 to 70 V vs. Although it can hold
- the reaction temperature is preferably ⁇ 40 to 100 ° C.
- the temperature is preferably ⁇ 20 to 70 ° C., but is not particularly limited thereto. It is desirable to adjust the temperature according to the reaction conditions in view of the stability and reactivity of the reaction intermediate and reaction active intermediate. In a homogeneous mixed system with an organic solvent or ionic liquid, the reaction can be performed at a lower temperature.
- Reactivity in this reaction ease of formation of active intermediate, promotion of reactivity of active intermediate with urea, suppression of decomposition of active intermediate, generation of oxidized active species for formation of intermediate, 2 electrons Reactions under acidic conditions are desirable for promoting oxidation and suppressing side reactions such as Hoffman transition.
- the acid added for the acidic condition may be mineral products such as hydrochloric acid, sulfuric acid, phosphoric acid, boric acid, and organic acids such as acetic acid, propionic acid, citric acid.
- the buffer solution which is a mixture with those salts may be sufficient.
- Reference example 1 In a reaction vessel equipped with a stirrer and a thermometer, 75 g (1.25 mol) of urea is dissolved in 150 ml of water, and 321 ml (1.25 mol) of a 29% sodium hypochlorite aqueous solution is added dropwise at 10 ° C. over 1 hour with stirring. A sample is taken, potassium iodide and acetic acid (1: 1) are added, and 0.1N-sodium thiosulfate titration is immediately performed. As a result of titration analysis, it was confirmed that the yield of monochlorourea was 98.4%. When ultraviolet analysis was performed, absorption at 244 nm peculiar to monochlorourea was observed.
- Reference example 3 Monochlorourea produced in Reference Example 1 is separated by crystals when concentrated under reduced pressure. Dissolve 31.5 g (0.33 mol) of monochlorourea in 100 ml of water, add 80 ml of 8% hydrochloric acid, add 34 g (0.33 mol) of sodium bromide, and add 30 ml of 30% hydrogen peroxide at 50 ° C. with stirring. (0.33 mol) is added dropwise over 1 hour. After completion of the dropwise addition, the reaction is terminated by stirring for another 30 minutes. When the sample was collected and subjected to ultraviolet analysis, the absorption at 275 nm characteristic of monobromourea was shown.
- Example 1-1 In a 500 ml round bottom flask equipped with a magnetic stir bar, dropping funnel and thermometer, 90 g (1.5 mol) of urea was dissolved in 100 ml of water, followed by cooling to 5 ° C. under cooling at 5 ° C. 17 .1 g of 32% HCl (0.15 mol) was added with stirring. Adjust the addition amount of 32% HCl so that the pH of the solution is 1. To the solution was added 133 g of 10.7% NaOCl aqueous solution (wt% as active Cl 2 , 0.2 mol) over 14 minutes upon cooling. After stirring for 1 hour after completion of the addition, 20.5 g of NaBr (0.2 mol) was added to the solution.
- the temperature of the reaction solution is raised to 35 ° C. and stirred for 1 hour, then 79.3 g (0.7 mol) of 30% hydrogen peroxide is divided into two, and the first half is added dropwise over 30 minutes. Stir for 1 hour, then drop the remaining half in 30 minutes.
- the reaction is terminated by stirring for 5 hours. As the reaction progresses, a yellow solid is formed and becomes suspended in the reaction solution. After completion of the reaction, the solid is separated by filtration, washed with water and dried.
- the obtained azodicarbonamide was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR).
- Example 1-2 In a 500 ml round bottom flask equipped with a magnetic stir bar, dropping funnel and thermometer, 90 g (1.5 mol) of urea was dissolved in 300 ml of water, followed by cooling to 0 ° C. under cooling at 0 ° C. 22 0.5 g of 35% hydrochloric acid (0.22 mol) was added with stirring. Further, the solution was adjusted with 35% hydrochloric acid so that the pH was 1. To the solution was added 19.6 g ammonium bromide (0.2 mol) and 23.5 g NaCl (0.4 mol).
- Example 1-5 In a 500 ml round bottom flask equipped with a magnetic stir bar, dropping funnel and thermometer, 90 g (1.5 mol) of urea was dissolved in 300 ml of water, followed by cooling to 0 ° C. under cooling at 0 ° C. 22 0.5 g of 35% hydrochloric acid (0.22 mol) was added with stirring. Further, the solution was adjusted with 35% hydrochloric acid so that the pH was 1. To the solution were added 1.96 g ammonium bromide (0.02 mol) and 23.5 g NaCl (0.4 mol).
- azodicarbonamide was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR). The yield of azodicarbonamide from the consumed urea was 94.1%.
- IR KBr
- 3337,3187cm -1 N-H stretching
- 1637cm -1 N-H bending
- 1 H-NMR (D6-DMSO) ⁇ 8.02 (s, 2H, NH), 7.97 (s, 2H, NH).
- Example 15-26 The first oxidant is converted from sodium hypochlorite to bromine, bromide and bromate in amounts considerably smaller than the theoretical amount, and reacted first, and then hydrogen peroxide is added to form hydrogen bromide. Except for using 2 times the amount of hydrogen peroxide corresponding to recycling reaction of bromine, bromide or bromate after converting the product to bromine or bromic acid or bromate, the same as Example 1-3 The reaction and treatment were carried out.
- Example 1-1 an oxidizing agent such as sodium hypochlorite that is the first oxidizing agent is not used, but a small amount of NaBr is added first, and the amount is equivalent to twice the number of moles of dissolved total chloranion.
- the hydrogen peroxide to be added is reacted and treated to separate the produced azodicarbonamide, the mother liquor and the washing solution of the azodicarbonamide isolate are combined, the consumed urea is added, and NaBr is
- the recycling reaction is performed while adjusting the composition by adding these.
- water is removed using a membrane or the like. Other reactions and treatments are carried out in the same manner as in Example 1-1.
- Example 32 Urea (1.2 g, 20 mmol), sodium bromide (41 mg, 0.4 mmol), cooled to 5 ° C. in a beaker-type electrolytic cell equipped with two platinum plate electrodes (1.5 ⁇ 1.0 cm 2 ). 0.34 mg of 32% HCl (0.3 mmol) and water (2.0 g) were weighed and stirred to obtain a homogeneous solution. While the electrolytic cell was immersed in an ice-water bath and cooled, electrolysis was performed for 10.7 hours while keeping the current constant at 100 mA, and 5778 coulombs of electricity was applied.
- Example 33 Electrolysis was carried out in the same manner as described in Example 32 using the monobromourea produced in Reference Example 3 as a raw material instead of the urea in Example 32.
- 213 mg of azodicarbonamide was obtained.
- the yield is 92.0%.
- the electrolysis reaction is carried out with half the energizing time and the energizing electricity.
- a solid form of azodicarbonamide is formed.
- the suspension was filtered, washed with water and dried under reduced pressure.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Provided is an economical, safe, and more environmentally-friendly production method for azodicarbonamide. According to the present invention, in a homogeneous system or heterogeneous miscible system of water or an organic solvent or an ionic solution, and an organic solvent and/or an ionic solution and water, azodicarbonamide is produced by using hydrogen peroxide in urea and reacting active chlorine or active chlorine ions and an active bromine compound such as active bromine or active bromine ions and/or an active form derived from hydrogen peroxide, or by electrolytically oxidizing an intermediate as desired to obtain a target, and recycling and using post-reaction byproducts.
Description
本発明は、尿素活性誘導体及び構造を利用して、アゾジカルボンアミドを得る製造方法に関する。
The present invention relates to a production method for obtaining azodicarbonamide using a urea active derivative and a structure.
アゾジカルボンアミドは、その分解挙動や、物理的性質、化学的性質に優れるという観点から、発泡剤として広く用いられている有用な化合物である。
Azodicarbonamide is a useful compound widely used as a foaming agent from the viewpoint of its decomposition behavior, physical properties, and chemical properties.
最も一般的に実施されている従来の製造方法は、下記反応式(1)で表されるように、尿素又はアンモニアを原料にして製造されるヒドラジンヒドラートと、2モルの尿素との反応を含む数段階の反応でアゾジカルボンアミドを製造する方法である(特許文献1)。この従来の製造法は、目的物アゾジカルボンアミドの分子構造に貢献しない、原料の無駄な消費が生じ、コストアップの原因になっている。また、副生する大量のアンモニアや、地球温暖化の原因となる炭酸ガスの発生も問題である。さらに、この方法では、塩素化剤や酸化剤、あるいは強酸や重金属触媒などを使用するため、廃棄物として多量の塩類や酸、重金属、アンモニア性窒素などを含有する大量の排水が発生する。これらを廃棄する為には処理が必要であることから、大きな処理費用が掛かり、製品コストのアップとなる上、公害の原因となるおそれがあることから、処理を行っても廃棄することが不可能な場合もある。
As shown in the following reaction formula (1), the most commonly practiced conventional production method involves reacting hydrazine hydrate produced from urea or ammonia with 2 moles of urea. This is a method for producing azodicarbonamide by several stages of reaction including the above (Patent Document 1). This conventional production method causes wasteful consumption of raw materials that do not contribute to the molecular structure of the target product azodicarbonamide, which causes an increase in cost. Another problem is the generation of a large amount of ammonia as a by-product and carbon dioxide that causes global warming. Furthermore, since this method uses a chlorinating agent, an oxidizing agent, a strong acid, a heavy metal catalyst, or the like, a large amount of waste water containing a large amount of salts, acids, heavy metals, ammonia nitrogen, and the like is generated as waste. Since disposal is required to dispose of these, it costs a large amount of processing costs, increases product costs, and may cause pollution. Sometimes it is possible.
最近発表された文献では、(特許文献4)尿素から有機電解合成法と言う特殊な方法で特殊な装置を使用して、尿素の構造式とアゾジカルボンアミド(ADCA)の構造式とは全く異なるにも関わらず尿素からいきなりADCAが生成すると記載されている。尿素から他の原料と反応して各種反応中間体生成し、目的物のADCAを得なければ非論理的である。エネルギー収率においても、直接の反応に要する電気エネルギーを計算すると非常に電気エネルギー収率の悪い大量電気エネルギーを無駄に消費する問題点を有する。詳細については、後の項目で示す。
In a recently published document (Patent Document 4), the structural formula of urea and the structural formula of azodicarbonamide (ADCA) are completely different by using a special apparatus in a special method called organic electrolytic synthesis from urea. Nevertheless, it is described that ADCA is suddenly generated from urea. It is illogical if it reacts with other raw materials from urea to produce various reaction intermediates and the desired ADCA is not obtained. Also in the energy yield, if the electric energy required for the direct reaction is calculated, there is a problem that a large amount of electric energy having a very low electric energy yield is wasted. Details will be given in later sections.
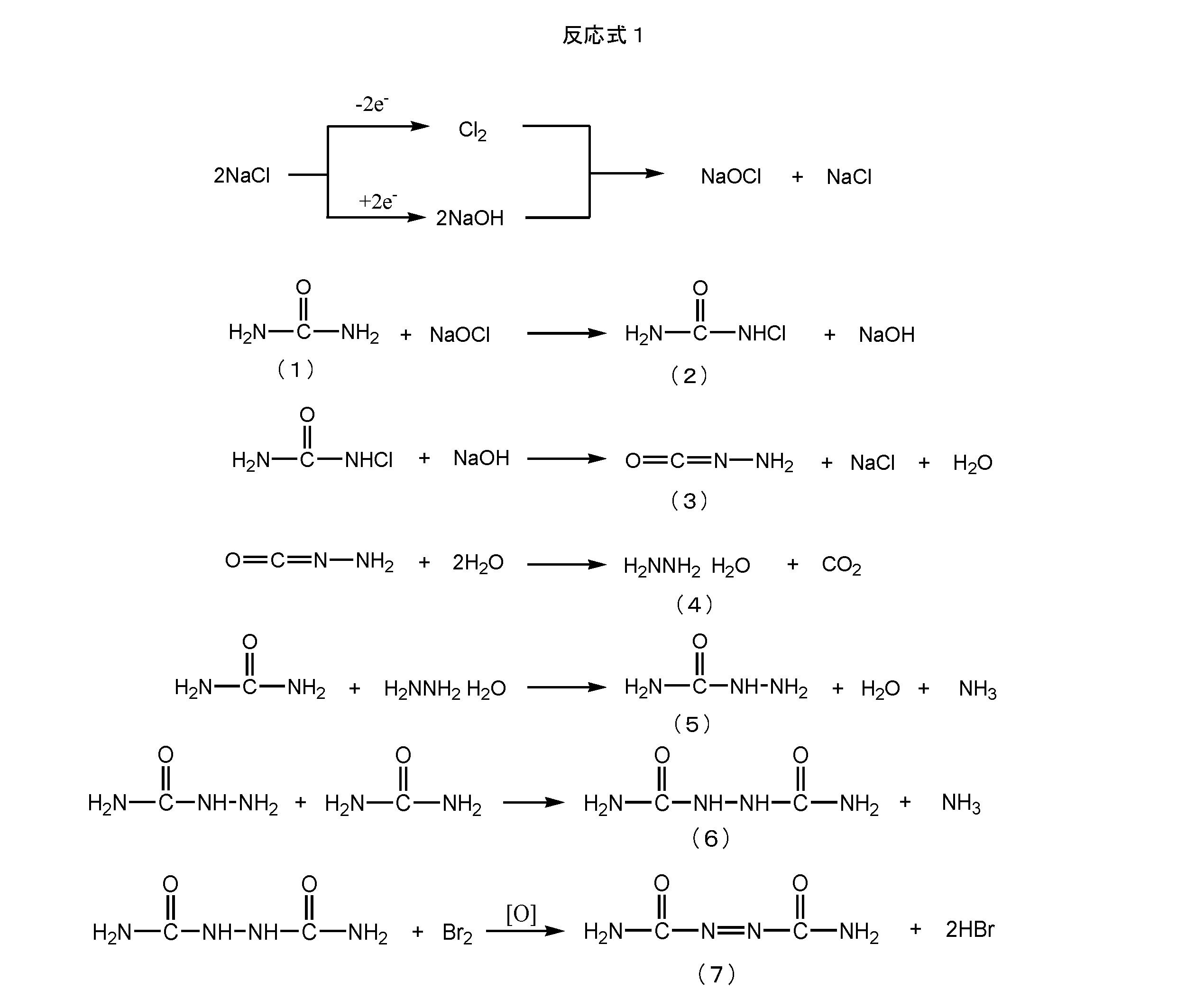
現在実用化されているアゾジカルボンアミドの製造法は、40年以上前に開発された技術であり、その後大きな技術革新が行われていない。比較的最近特許で開示された技術として、上記反応式1のアミノイソシアネート(式(3)で表される化合物)を加圧アンモニア分解してセミカルバジッド(式(5)で表される)を得る方法が報告されている(特許文献2)。しかし、1工程が短縮されるだけであり、その為に加圧容器中での大過剰の液体アンモニアを使用する必要があり、高圧反応の危険性がある。また、高圧反応容器を必要とする。さらに、過剰の液体アンモニアの回収、等量の塩化アンモニウムの副生等の問題もあるため、生産の合理化にも、経済性の革新にもならない。総合的には逆効果である。
[The azodicarbonamide production method currently in practical use is a technology developed more than 40 years ago, and no major technological innovation has been made since then. As a technique disclosed in a relatively recent patent, a semicarbazide (represented by the formula (5)) is obtained by subjecting the amino isocyanate of the above reaction formula 1 (a compound represented by the formula (3)) to ammonia decomposition under pressure. An obtaining method has been reported (Patent Document 2). However, only one step is shortened. For this reason, it is necessary to use a large excess of liquid ammonia in a pressurized container, and there is a risk of a high-pressure reaction. Moreover, a high pressure reaction vessel is required. Furthermore, there are problems such as recovery of excess liquid ammonia and by-production of an equal amount of ammonium chloride, so that neither production rationalization nor economic innovation occurs. Overall, it is counterproductive.
本発明において使用される原料又は中間体として使用されるクロル尿素、ブロム尿素の製造法については、(特許文献1)の他に(特許文献3)に記載されている。
The production method of chlorourea and bromourea used as raw materials or intermediates used in the present invention is described in (Patent Document 3) in addition to (Patent Document 1).
最近発表された文献では、(特許文献4)尿素から有機電解合成法と言う特殊な方法と特殊な装置を使用して、反応の実態が全く解明されていない内容である。一般の種々の問題点、欠点については、既に背景技術の項で指摘されている。原料と全く異なる目的物が一挙に中身不明で出来るという特許である事、特殊装置、特殊技術が必要である事、複雑な組成の原材料で連続のリサイクル反応を行わないと目的物を得る事が出来ない。更に原料を大過剰に使用する為原料のリサイクル使用が必須である。副反応、それからの不純物等の予測が付くが、これ等の除外、精製の説明も無い。製造法全体の評価は別として、本特許文献で示す純粋な化学反応のみの消費エネルギーの評価結果を以下に示す。最大の問題は、製造に必要な他のエネルギーは除外して検討すると、電解法であるから電解反応のみに必要なエネルギーは、電気エネルギーである。大量の電気エネルギーを反応の為消費するので、化学原料と同時に電気エネルギーも原料と見なされる。今日使用される電気エネルギーの単位は、ファラデーではなく、クーロンで示される。特許文献(4)の実施例1の内容を検証すると、実施例(1)で消費された(反応に使用された)電気量は、電流×時間(秒単位)(クーロン)で表される。1.5cm2(電極面積)×66.7×10-3(A/cm2)(電流密度)×5.4×3600秒(5.4時間)=1944.97クーロンである。目的物(ADCA)の収量は純度100%と仮において、0.32gである。アゾジカルボンアミド(ADCA)の分子量は、116である。故に得られたADCAは、0.32÷116=0.00276molである。特許文献中の化学式(8)で1molのADCAを得る為、4電子molが必要と記載されている。クーロンの法則から1mol=96500クーロンで、実質目的物の獲得の為使用された対応電気量は、純度100%と仮定しても266.34クーロン×4=1065.36クーロンである。よって電気収率は、1065.36クーロン(目的物の生成に消費された理論電気量)÷1944.97クーロン(実質消費された電気量)=54.77%である。100%純度のADCAが得られた訳ではなく、分離時におけるロス等も勘案すると、この収率から相当低下する。約50%の非常に電力効率の悪い反応であり、電気エネルギーの塊と言える。電気エネルギーの浪費と言える。社会の流れとして再生可能エネルギーからの発電の要請で電気コストが上がって行く時代である。この特許文献の方法は、製造に高価な電気を大量に浪費する大きな問題点を有していて、経済的に有利な方法、時代の要請に応える方法とは言えない。
In a recently published document (Patent Document 4), the actual state of the reaction has not been elucidated at all using a special method and a special apparatus called an organic electrolytic synthesis method from urea. Various general problems and drawbacks have already been pointed out in the background section. It is a patent that the object that is completely different from the raw material can be unknown at once, special equipment, special technology is necessary, and if you do not perform continuous recycling reaction with raw materials with complicated composition, you can obtain the object I can't. Furthermore, since the raw material is used in a large excess, it is essential to recycle the raw material. Prediction of side reactions and impurities from the side reactions can be predicted, but there is no explanation for these exclusions and purification. Apart from the evaluation of the entire production method, the evaluation results of the energy consumption of only pure chemical reactions shown in this patent document are shown below. The biggest problem is that when the other energy necessary for the production is excluded and examined, since it is an electrolytic method, the energy required only for the electrolytic reaction is electric energy. Since a large amount of electric energy is consumed for the reaction, electric energy is regarded as a raw material as well as a chemical raw material. The unit of electrical energy used today is indicated in coulombs, not Faraday. When the contents of Example 1 of Patent Document (4) are verified, the amount of electricity consumed (used for the reaction) in Example (1) is represented by current × time (second unit) (coulomb). 1.5 cm 2 (electrode area) × 66.7 × 10 −3 (A / cm 2 ) (current density) × 5.4 × 3600 seconds (5.4 hours) = 1944.97 coulombs. The yield of the target product (ADCA) is tentatively 100% and is 0.32 g. The molecular weight of azodicarbonamide (ADCA) is 116. The ADCA thus obtained is 0.32 ÷ 116 = 0.00276 mol. In order to obtain 1 mol ADCA by Chemical formula (8) in patent document, it is described that 4 electron mol is required. According to Coulomb's law, 1 mol = 96500 coulomb, and the corresponding quantity of electricity used to acquire the actual object is 266.34 coulomb × 4 = 1065.36 coulomb even if the purity is assumed to be 100%. Therefore, the electricity yield is 1065.36 coulombs (theoretical electricity consumed for the production of the target product) ÷ 1944.97 coulombs (actually consumed electricity) = 54.77%. The ADCA with 100% purity was not obtained, and when considering the loss at the time of separation, etc., the yield is considerably reduced. It is a very inefficient reaction of about 50% and can be said to be a mass of electric energy. It can be said that electric energy is wasted. It is an era when electricity costs are rising due to demand for power generation from renewable energy as a social trend. The method of this patent document has a big problem of wasting a large amount of expensive electricity for manufacturing, and cannot be said to be an economically advantageous method or a method that meets the demands of the times.
本発明は、アゾジカルボンアミドの、簡便で安全かつ環境負荷の軽減された画期的な製造方法を提供することを課題とする。
An object of the present invention is to provide an epoch-making production method of azodicarbonamide that is simple, safe and has a reduced environmental burden.
本発明者等は、上記の課題を解決するために、簡便かつ効率の良い、活性尿素誘導体及び尿素の活性構造と尿素を反応する方法について、鋭意研究を行った。その結果、水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、反応機構の解析から、論理的な製法として、常圧下で活性尿素誘導体及び尿素の活性構造と尿素の反応を行うことにより、特殊な有機電解装置や微妙なるその運転条件、反応系を常時細かな調整を行う必要がなく、又爆発の危険がある水素の発生がなく、電極間の短絡ショート発火、爆発の危険性の無い、大量、安価に生産されている汎用の工業薬品の過酸化水素や過酸化物、オゾン等の酸化剤とハロゲン酸又はハロゲン塩との組み合わせ使用した、又特殊な装置を新たに製作する事無く、通常の化学製品の生産に係わっている誰でもが容易に運転が出来、現在使用されているプラントをそのまま使用が出来る、実用的で工業的に有利な、環境負荷軽減効果の大きく、省エネルギーなアゾジカルボンアミドの製造方法を見出し、本発明を完成するに至った。
In order to solve the above-mentioned problems, the present inventors have conducted intensive research on a simple and efficient method of reacting urea with an active urea derivative and an active structure of urea. As a result, in a homogeneous or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water, as a logical production method, active urea under normal pressure is analyzed. By reacting the active structure of the derivative and urea with urea, there is no need to make fine adjustments to the special organic electrolyzer, delicate operating conditions and reaction system at all times, and there is a risk of hydrogen that can explode. There is no short circuit between electrodes, there is no risk of explosion, and there is no risk of explosion. Large-scale, inexpensive industrial chemicals such as hydrogen peroxide, peroxide, ozone and other oxidizing agents and halogen acids or halogen salts. Anyone who is involved in the production of normal chemical products can easily operate without using a new device or a special device, and the plant currently in use can be used as it is. Target in industrially advantageous, large environmental load reduction effect, they found a method for producing energy-saving azodicarbonamide, and have completed the present invention.
課題を解決するための手段の詳細な説明
本発明は、下記項1~13に係るアゾジカルボンアミドの製造方法を提供する。
項1:式(1)で表される尿素を、水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において式(10)で表される化合物を使用して、式(10)で表される化合物が、化学的に非常に活性である事から、即時に脱HBr反応を生じて式(7)の化合物に変換することを特徴とするアゾジカルボンアミド式(7)の新規製造方法。 Detailed Description of Means for Solving the Problems The present invention provides a method for producing azodicarbonamide according to the following items 1 to 13.
Item 1: Urea represented by formula (1) is represented by formula (10) in a homogeneous system or a heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water. Since the compound represented by the formula (10) is chemically very active, a de-HBr reaction is immediately generated and converted into the compound of the formula (7). A novel process for producing azodicarbonamide formula (7).
本発明は、下記項1~13に係るアゾジカルボンアミドの製造方法を提供する。
項1:式(1)で表される尿素を、水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において式(10)で表される化合物を使用して、式(10)で表される化合物が、化学的に非常に活性である事から、即時に脱HBr反応を生じて式(7)の化合物に変換することを特徴とするアゾジカルボンアミド式(7)の新規製造方法。 Detailed Description of Means for Solving the Problems The present invention provides a method for producing azodicarbonamide according to the following items 1 to 13.
Item 1: Urea represented by formula (1) is represented by formula (10) in a homogeneous system or a heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water. Since the compound represented by the formula (10) is chemically very active, a de-HBr reaction is immediately generated and converted into the compound of the formula (7). A novel process for producing azodicarbonamide formula (7).
項2:式(1)で表される尿素を、水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において酸性条件下式(9)で表されるブロム尿素活性遷移中間体に尿素分子を反応させる事によって式(10)の化合物を得る。式(10)の化合物は、化学的に活性であり即時に脱HBr反応を生じて式(7)の化合物に変換することを特徴とするアゾジカルボンアミド式(7)の新規製造方法。
Item 2: Urea represented by the formula (1) is converted into water or an organic solvent or ionic liquid and an organic solvent or a homogeneous system or a heterogeneous mixed system of ionic liquid and water under the acidic condition (9) A compound of formula (10) is obtained by reacting a urea molecule with a bromourea active transition intermediate represented by the formula: A novel process for the preparation of azodicarbonamide formula (7), wherein the compound of formula (10) is chemically active and immediately undergoes de-HBr reaction to be converted to the compound of formula (7).
項3:式(1)で表される尿素を、水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、尿素から酸性条件下ブロム化剤又はブロム化方法により式(8)で表されるN-ブロム尿素を生成し、次いでブロム原子が結合するNH基の活性ブロム化合物又は、及び過酸化水素由来活性体による求電子酸化によりブロム尿素活性遷移中間体式(9)を得て、同時に尿素分子が反応し式(10)の化合物を得る。式(10)の化合物は、化学的に活性であり、即時に脱HBr反応を生じて式(7)の化合物に変換することを特徴とするアゾジカルボンアミド式(7)の新規製造方法。
Item 3: Urea represented by the formula (1) can be converted from urea to acidic conditions in a homogeneous or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water. N-bromourea represented by the formula (8) is generated by an agent or a bromination method, and then brominated by electrophilic oxidation with an active bromo compound of NH group to which a bromo atom is bonded or a hydrogen peroxide-derived activator. A urea active transition intermediate formula (9) is obtained and at the same time urea molecules react to obtain a compound of formula (10). A novel process for the preparation of azodicarbonamide formula (7), wherein the compound of formula (10) is chemically active and immediately undergoes de-HBr reaction and is converted to the compound of formula (7).
項4:式(1)で表される尿素を、水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において酸性条件下塩素化反応を行い式(11)の化合物を得る。式(11)のモノクロル尿素(N-クロル尿素Na塩を含む)をブロム化剤又はブロム化方法により式(8)で表されるN-ブロム尿素を得る。次いでブロム原子が結合するNH基の活性ブロム化合物又は、及び過酸化水素由来活性体による求電子酸化によりブロム尿素活性遷移中間体式(9)を得て、同時に尿素分子が反応し式(10)の化合物を得る。式(10)の化合物は、化学的に活性であり、即時に脱HBr反応を生じて式(7)の化合物に変換することを特徴とするアゾジカルボンアミド式(7)の新規製造方法。
Item 4: Urea represented by formula (1) is subjected to chlorination reaction under acidic conditions in a homogeneous or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water. The compound of formula (11) is obtained. Monochlorourea of formula (11) (including N-chlorourea Na salt) is subjected to bromination or bromination to obtain N-bromourea represented by formula (8). Next, the bromourea active transition intermediate formula (9) is obtained by electrophilic oxidation with the active bromo compound of NH group to which the bromo atom is bonded or the hydrogen peroxide-derived activator, and at the same time, the urea molecule reacts and the formula (10) A compound is obtained. A novel process for the preparation of azodicarbonamide formula (7), wherein the compound of formula (10) is chemically active and immediately undergoes de-HBr reaction and is converted to the compound of formula (7).
項5:式(1)で表される尿素を、水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、尿素から酸性条件下ブロム化剤又はブロム化方法により式(8)で表されるN-ブロム尿素を生成し、次いでブロム原子が結合するNH基の求電子酸化を電解反応により2電子酸化してプロトンとして脱離して、ブロム尿素活性遷移中間体式(9)とする。同時に尿素分子が反応し式(10)の化合物を得る。式(10)の化合物は、化学的に活性であり、即時に脱HBr反応を生じて式(7)の化合物に変換することを特徴とするアゾジカルボンアミド式(7)の新規製造方法。
Item 5: Urea represented by the formula (1) can be converted from urea to acidic bromine under acidic conditions in a homogeneous or heterogeneous mixed system of water or organic solvent or ionic liquid and organic solvent or ionic liquid and water. N-bromourea represented by the formula (8) is produced by an agent or bromination method, and then electrophilic oxidation of the NH group to which the bromine atom is bonded is electrolyzed and desorbed as a proton by electrolysis, Bromourea active transition intermediate Formula (9). At the same time, urea molecules react to obtain a compound of formula (10). A novel process for the preparation of azodicarbonamide formula (7), wherein the compound of formula (10) is chemically active and immediately undergoes de-HBr reaction and is converted to the compound of formula (7).
項6:式(1)で表される尿素を、水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、電解反応を利用する場合、支持電解質として臭化水素、ブロム塩と塩化水素、塩化物塩の混合物を使用して酸性条件下2電子酸化してブロムカチオンを生成させて式(8)で表されるN-ブロム尿素を生成し、次いでブロム原子が結合するNH基の求電子酸化を電解反応により2電子酸化してプロトンとして脱離して、ブロム尿素活性遷移中間体式(9)とする。同時に尿素分子が反応し式(10)の化合物を得る。式(10)の化合物は、化学的に活性であり、即時に脱HBr反応を生じて式(7)の化合物に変換することを特徴とするアゾジカルボンアミド式(7)の新規製造方法。
Item 6: When an electrolytic reaction is used for urea represented by formula (1) in water or an organic solvent or ionic liquid, and an organic solvent or a homogeneous system or a heterogeneous mixed system of ionic liquid and water. Using a mixture of hydrogen bromide, bromo salt and hydrogen chloride, chloride salt as a supporting electrolyte, two-electron oxidation under acidic conditions to generate a bromo cation to produce N-bromourea represented by formula (8) Then, electrophilic oxidation of the NH group to which the bromo atom is bonded is subjected to two-electron oxidation by an electrolytic reaction and eliminated as a proton to obtain a bromourea active transition intermediate formula (9). At the same time, urea molecules react to obtain a compound of formula (10). A novel process for the preparation of azodicarbonamide formula (7), wherein the compound of formula (10) is chemically active and immediately undergoes de-HBr reaction and is converted to the compound of formula (7).
項7:式(1)で表される尿素を、水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、尿素から酸性条件下ブロム化剤又はブロム化方法により式(8)で表されるN-ブロム尿素を生成し、又は必要に寄っては、尿素から酸性条件下塩素化剤又は塩素化方法により、式(11)で示されるクロル尿素(N-クロル尿素Na塩を含む)を生成し、次いでブロム化剤又はブロム化方法により式(8)で表されるN-ブロム尿素を生成し、次いで電解反応を利用して支持電解質として臭化水素、ブロム塩と塩化水素、塩化物塩の混合物を使用して2電子酸化してブロムカチオンを生成させて式(8)で表されるN-ブロム尿素を生成し、次いでブロム原子が結合するNH基の求電子酸化を電解反応により2電子酸化してプロトンとして脱離して、ブロム尿素活性遷移中間体式(9)とする。同時に尿素分子が反応し式(10)の化合物を得る。式(10)の化合物は、化学的に活性であり、即時に脱HBr反応を生じて式(7)の化合物に変換することを特徴とするアゾジカルボンアミド式(7)の新規製造方法。
Item 7: The urea represented by the formula (1) is converted from urea to a bromine under acidic conditions in a homogeneous or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water. An N-bromourea represented by the formula (8) is produced by a chlorinating agent or a bromination method, or, if necessary, it is represented by the formula (11) from urea by a chlorinating agent or a chlorination method under acidic conditions. Chlorourea (including N-chlorourea Na salt) is produced, then N-bromourea represented by the formula (8) is produced by a brominating agent or bromination method, and then supported using an electrolytic reaction. Using a mixture of hydrogen bromide, bromo salt and hydrogen chloride, chloride salt as an electrolyte, two-electron oxidation produces a bromo cation to produce N-bromourea represented by formula (8), followed by bromine Electrophilic acid of NH group to which atom is bonded The desorbed as protons and two-electron oxidized by electrolytic reaction, the bromine urea active transition intermediate (9). At the same time, urea molecules react to obtain a compound of formula (10). A novel process for the preparation of azodicarbonamide formula (7), wherein the compound of formula (10) is chemically active and immediately undergoes de-HBr reaction and is converted to the compound of formula (7).
項8:式(1)で表される尿素を、水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、酸性条件下臭化水素、ブロム塩と塩化水素、塩化物塩の混合物を使用してこれに過酸化水素等の過酸化物を滴下する事によって臭素を発生させる。発生、変換された臭素、次亜臭素酸、次亜臭素酸塩によって式(1)で表される尿素がブロムカチオンによって2電子酸化されて式(8)で表されるN-ブロム尿素を生成し、次いでブロム原子が結合するNH基の活性ブロム化合物又は、及び過酸化水素由来活性体による求電子酸化によりブロム尿素活性遷移中間体式(9)を得て、同時に尿素分子が反応し式(10)の化合物を得る。式(10)の化合物は、化学的に活性であり、即時に脱HBr反応を生じて式(7)の化合物に変換することを特徴とするアゾジカルボンアミド式(7)の新規製造方法。
Item 8: Urea represented by the formula (1) is hydrogen bromide under acidic conditions in water or an organic solvent or ionic liquid, and an organic solvent or a homogeneous or heterogeneous mixture of ionic liquid and water. Then, bromine is generated by adding a peroxide such as hydrogen peroxide dropwise to a mixture of bromo salt, hydrogen chloride and chloride salt. The urea represented by the formula (1) is generated by the generated bromine, hypobromite, and hypobromite, and the N-bromourea represented by the formula (8) is generated by the two-electron oxidation by the brom cation. Then, the bromourea active transition intermediate formula (9) is obtained by electrophilic oxidation with an active bromo compound of NH group to which a bromo atom is bonded or an active substance derived from hydrogen peroxide, and at the same time, a urea molecule reacts with the formula (10 ) Is obtained. A novel process for the preparation of azodicarbonamide formula (7), wherein the compound of formula (10) is chemically active and immediately undergoes de-HBr reaction and is converted to the compound of formula (7).
項9:式(1)で表される尿素を、水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、酸性条件下有機ブロム化剤及びブロム‐ジオキサン錯体を酸化剤、活性臭素化化合物発生剤として、尿素がブロムカチオンによって2電子酸化されて式(8)で表されるN-ブロム尿素を生成し、次いでブロム原子が結合するNH基の活性ブロム化合物又は、及び過酸化水素由来活性体による求電子酸化によりブロム尿素活性遷移中間体式(9)を得て、同時に尿素分子が反応し式(10)の化合物を得る。式(10)の化合物は、化学的に活性であり、即時に脱HBr反応を生じて式(7)の化合物に変換することを特徴とするアゾジカルボンアミド式(7)の新規製造方法。
Item 9: The urea represented by the formula (1) is converted into an organic bromide under acidic conditions in a homogeneous system or a heterogeneous mixed system of water, an organic solvent or an ionic liquid, and an organic solvent or an ionic liquid and water. Using an agent and a bromine-dioxane complex as an oxidizing agent and an active brominated compound generator, urea is two-electron-oxidized by a brom cation to produce N-bromourea represented by the formula (8), and then a bromine atom is bonded. Bromourea active transition intermediate formula (9) is obtained by electrophilic oxidation with an active bromine compound of NH group or an active substance derived from hydrogen peroxide, and at the same time, urea molecules react to obtain a compound of formula (10). A novel process for the preparation of azodicarbonamide formula (7), wherein the compound of formula (10) is chemically active and immediately undergoes de-HBr reaction and is converted to the compound of formula (7).
項10:水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、原料として尿素の臭化水素、塩化水素等のハロゲン酸塩を用いて、酸性条件下これに過酸化水素等の過酸化物を滴下する事によって塩素又は、及び臭素を発生させる。発生、変換された塩素、次亜塩素酸、次亜塩素酸塩、臭素、次亜臭素酸、次亜臭素酸塩によって式(1)で表される尿素がクロル化、ブロム化される事を特徴とする項1から項9までのアゾジカルボンアミドの新規製造方法
Item 10: In a homogeneous system or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water, a halogen acid salt such as hydrogen bromide or hydrogen chloride of urea is used as a raw material Then, chlorine or bromine is generated by dropping a peroxide such as hydrogen peroxide under acidic conditions. Generated and converted chlorine, hypochlorous acid, hypochlorite, bromine, hypobromite, hypobromite, chlorinated and brominated urea represented by formula (1) A novel process for producing an azodicarbonamide according to any one of Items 1 to 9
項11:原料に尿素及び尿素の塩の混合物を使用する項1から項10の方法から対応する項の方法を取る事を特徴とするアゾジカルボンアミド式(7)の新規製造方法。
Item 11: A novel method for producing azodicarbonamide formula (7), wherein the method of the corresponding item is taken from the method of Item 1 to Item 10 using a mixture of urea and urea salt as a raw material.
項12:酸性条件下、大過剰の尿素を使用し、過酸化水素等の過酸化物を使用する場合、PH=3以下の酸性条件で、20~80℃の温度で反応を行う。中間体を電解酸化する場合、電解反応時は-25~60℃の間で反応を行う、反応方法、条件に合わせて酸性度、温度選択、過酸化物選択を行う事を特徴とする項1から項11までのアゾジカルボンアミドの新規製造方法。
Item 12: When a large excess of urea is used under acidic conditions and a peroxide such as hydrogen peroxide is used, the reaction is performed at a temperature of 20 to 80 ° C. under acidic conditions of PH = 3 or less. Item 1 is characterized in that when the intermediate is subjected to electrolytic oxidation, the reaction is carried out at a temperature between -25 to 60 ° C. during the electrolytic reaction, and the acidity, temperature and peroxide are selected according to the reaction method and conditions. 12. A novel process for producing azodicarbonamide according to items 11 to 11.
項13:過酸化水素等の過酸化物を酸化剤にしてハロゲン及びハロゲン化合物を使用する場合、セリウム化合物、バナジウム化合物、セレン化合物、テルル化合物、遷移金属化合物及びフッ化アルミニウム等の水に安定な強ルイス酸を触媒とする事、またギ酸、酢酸等の酸を使用する事を特徴とする項1から項12までのアゾジカルボンアミド式(7)の新規製造方法。
Item 13: When a halogen or a halogen compound is used with a peroxide such as hydrogen peroxide as an oxidizing agent, it is stable to water such as cerium compound, vanadium compound, selenium compound, tellurium compound, transition metal compound and aluminum fluoride. 13. A novel process for producing an azodicarbonamide formula (7) according to items 1 to 12, wherein a strong Lewis acid is used as a catalyst and an acid such as formic acid or acetic acid is used.






[式中、Mは、水素及びLi、Na、K、Mg、Ca、Mn、Fe、Ni、Cu、Ag、Zn、Snよりなる群から選択される1~4価の金属を示す。Xは塩素原子及び臭素原子を示す。mは1~4の整数を示す。]及び過酸化水素、オゾン、過酸化物を示す。
[Wherein M represents hydrogen and a monovalent to tetravalent metal selected from the group consisting of Li, Na, K, Mg, Ca, Mn, Fe, Ni, Cu, Ag, Zn, and Sn. X represents a chlorine atom and a bromine atom. m represents an integer of 1 to 4. ], Hydrogen peroxide, ozone, and peroxide.
[式中、Mは、水素、Li、Na、K、Mg、Ca、Ti、Zr、Mn、Fe、Ni、Cu、Ag、Zn、Al、Si、Sn等の金属イオン、式(13)で表される化合物のカチオン部分はH及びアンモニウムイオン、第1級アンモニウムイオン、第2級アンモニウムイオン、第3級アンモニウムイオンを示し、Xは、ハロゲンアニオン及び有機ブロム化剤アニオン成である]及びヨウ素分子、臭素分子、塩素分子、クロル臭素を示す。
[In the formula, M is hydrogen, Li, Na, K, Mg, Ca, Ti, Zr, Mn, Fe, Ni, Cu, Ag, Zn, Al, Si, Sn, or other metal ions, in formula (13) The cation portion of the represented compound represents H and ammonium ion, primary ammonium ion, secondary ammonium ion, tertiary ammonium ion, and X is a halogen anion and an organic brominating agent anion.] And iodine Molecule, bromine molecule, chlorine molecule, chlorobromine.
本発明は、式(1)で表される尿素から、式(7)で表されるアゾジカルボンアミドを1工程で直接得る、効率的な製造方法である。水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系で行われることから、簡便で安全且つ安価で環境負荷の小さい製造方法である。また、生成するアゾジカルボンアミドが固体として析出するため、分離・精製も容易である。さらに、反応で使用する式(12)~(13)で表される化合物、ハロゲン化水素又はハロゲンと未反応の尿素とを含む水溶液を、そのまま循環・再利用できることから、ほとんど廃棄物を系外に排出することがなく、環境負荷の少ない製造方法である。式(1)の尿素は尿素の塩酸塩、臭素酸塩、その他の塩であっても良く、ハロゲン化を行う為使用するハロゲン化水素又はハロゲン塩は、反応を行う尿素に対しての量である。酸化剤として使用する過酸化物等が使用出来るが、過酸化水素は、最終水となる為、副生物、廃棄物の生成が無い。また、電解方法を取る場合、ガス拡散電極を使用すれば、水素の発生もなく、消費電力の抑制が可能となる。
The present invention is an efficient production method for directly obtaining the azodicarbonamide represented by the formula (7) from the urea represented by the formula (1) in one step. Since it is carried out in a homogeneous or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water, it is a simple, safe and inexpensive production method with a low environmental load. Moreover, since the produced azodicarbonamide is precipitated as a solid, separation and purification are easy. Further, since the aqueous solution containing the compounds represented by formulas (12) to (13), hydrogen halide, or halogen and unreacted urea used in the reaction can be circulated and reused as it is, almost no waste is removed from the system. It is a manufacturing method with less environmental impact. The urea of formula (1) may be urea hydrochloride, bromate or other salts, and the hydrogen halide or halogen salt used for the halogenation is in an amount relative to the urea to be reacted. is there. Although the peroxide etc. which are used as an oxidizing agent can be used, since hydrogen peroxide becomes final water, there is no generation of by-products and waste. In addition, when an electrolysis method is used, if a gas diffusion electrode is used, hydrogen is not generated and power consumption can be suppressed.
以下、本発明を詳細に説明する。
本発明は、水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系で、酸性条件下尿素又は尿素の塩の活性ハロゲンによる又は、及び活性ブロム化合物又は、及び過酸化水素由来活性体酸化反応を行うことにより、又はクロル尿素やブロム尿素の中間体を電解酸化を行う事により、副生したハロゲン化水素及びハロゲン塩は循環使用され、廃棄物の出ないアゾジカルボンアミドを製造する方法であり、下記の反応式で表される。 Hereinafter, the present invention will be described in detail.
The present invention relates to a homogeneous or heterogeneous mixture of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water, with or under active conditions of urea or urea salts under acidic conditions. By performing oxidation reaction of bromide compounds and hydrogen peroxide-derived active substances, or by electrolytically oxidizing intermediates of chlorourea and bromourea, by-produced hydrogen halide and halogen salts are recycled and discarded. This is a method for producing an azodicarbonamide having no product, and is represented by the following reaction formula.
本発明は、水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系で、酸性条件下尿素又は尿素の塩の活性ハロゲンによる又は、及び活性ブロム化合物又は、及び過酸化水素由来活性体酸化反応を行うことにより、又はクロル尿素やブロム尿素の中間体を電解酸化を行う事により、副生したハロゲン化水素及びハロゲン塩は循環使用され、廃棄物の出ないアゾジカルボンアミドを製造する方法であり、下記の反応式で表される。 Hereinafter, the present invention will be described in detail.
The present invention relates to a homogeneous or heterogeneous mixture of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water, with or under active conditions of urea or urea salts under acidic conditions. By performing oxidation reaction of bromide compounds and hydrogen peroxide-derived active substances, or by electrolytically oxidizing intermediates of chlorourea and bromourea, by-produced hydrogen halide and halogen salts are recycled and discarded. This is a method for producing an azodicarbonamide having no product, and is represented by the following reaction formula.

前記化学反応は、水又は有機溶媒又は、及びイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系で、式(1)で表される尿素又はその塩を、酸性条件下式(12)で表される化合物、式(13)で表される化合物から調整された塩素化剤又はブロム化剤と反応させ、複製した塩化水素又は塩化物塩及び臭化水素又は臭化水素塩を副生物が生じない過酸化水素等の酸化剤を使用してリサイクル使用する。化合物(11)又は化合物(8)を活性ブロム化合物又は、及び過酸化水素由来活性体によるか又は電解で2分子の尿素を理論的には四電子酸化することにより進行する。活性ブロム化合物及び過酸化水素由来活性体とは、当該技術分野において公知である化合物、化学種を含むが、特定の化合物、化学種に限定されるものではない。本反応において、酸性条件下では、塩化水素、臭化水素、塩素、臭素、次亜塩素酸、次亜臭素酸と過酸化水素とは相互に相関反応を生じ、競争反応が生じる。又これ等化合物は、尿素に配位し錯体を生じて反応を複雑にする。上記のハロゲン系化合物を酸性条件下、過酸化水素又はその活性体が作用した場合、活性ブロム化合物が生じる。逆に酸性条件下、過酸化水素及びその活性体に上記ハロゲン化合物が作用した場合、過酸化水素由来活性体が生じる。それぞれの活性体の生成を助けるのが、テルル化合物、バナジウム化合物、セリウム化合物、セレン化合物、遷移金属化合物及びフッ化アルミニウム等を担持した水に安定な強ルイス酸およびギ酸、酢酸等の化合物である。活性ブロム化合物及び/又は過酸化水素由来活性体は、ブロム尿素を酸化してブロム尿素活性遷移中間体を形成する化合物、化学種であれば良く、それらに属する多々の化合物、化学種が存在するが、特に特定の化合物、化学種に限定されるものでは無い。
The chemical reaction is a homogeneous or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water. Urea or a salt thereof represented by formula (1) A compound represented by formula (12) under acidic conditions, a chlorinating agent or a brominating agent prepared from the compound represented by formula (13), and replicated hydrogen chloride or chloride salt and hydrogen bromide Alternatively, the hydrogen bromide salt is recycled using an oxidizing agent such as hydrogen peroxide that does not produce by-products. The compound (11) or compound (8) proceeds with an active bromide compound and a hydrogen peroxide-derived activator or by electrolyzing two molecules of urea theoretically by four-electron oxidation. The active bromide compound and the hydrogen peroxide-derived activator include compounds and chemical species known in the art, but are not limited to specific compounds and chemical species. In this reaction, under acidic conditions, hydrogen chloride, hydrogen bromide, chlorine, bromine, hypochlorous acid, hypochlorous acid and hydrogen peroxide undergo a correlation reaction with each other, resulting in a competitive reaction. These compounds also coordinate with urea to form complexes, complicating the reaction. When hydrogen peroxide or its active substance acts on the above halogen compounds under acidic conditions, an active bromine compound is produced. On the other hand, when the halogen compound acts on hydrogen peroxide and its active substance under acidic conditions, an active substance derived from hydrogen peroxide is generated. It is the compounds such as strong Lewis acid, formic acid, and acetic acid that are stable in water carrying tellurium compounds, vanadium compounds, cerium compounds, selenium compounds, transition metal compounds, aluminum fluoride, and the like that help generate each active form. . The active bromide compound and / or the hydrogen peroxide-derived activator may be any compound or chemical species that oxidizes bromourea to form a bromourea active transition intermediate, and there are many compounds and chemical species belonging to them. However, it is not particularly limited to a specific compound or chemical species.
[式中、Mは、水素及びLi、Na、K、Mg、Ca、Mn、Fe、Ni、Cu、Ag、Zn、Snよりなる群から選択される1~4価の金属を示す。Xは塩素原子及び臭素原子、ヨウ素原子を示す。mは1~4の整数を示す。]及び過酸化水素、オゾン、過酸化物を示す。
[Wherein M represents hydrogen and a monovalent to tetravalent metal selected from the group consisting of Li, Na, K, Mg, Ca, Mn, Fe, Ni, Cu, Ag, Zn, and Sn. X represents a chlorine atom, a bromine atom, or an iodine atom. m represents an integer of 1 to 4. ], Hydrogen peroxide, ozone, and peroxide.
式(12)で表される化合物としては、例えば、次亜塩素酸、次亜塩素酸リチウム、亜塩素酸、亜塩素酸リチウム、塩素酸、塩素酸リチウム、過塩素酸、過塩素酸リチウム、次亜塩素酸ナトリウム、亜塩素酸ナトリウム、塩素酸ナトリウム、過塩素酸ナトリウム、次亜塩素酸カリウム、亜塩素酸カリウム、塩素酸カリウム、過塩素酸カリウム、次亜塩素酸カルシウム、亜塩素酸カルシウム、塩素酸カルシウム、過塩素酸カルシウム、次亜塩素酸マグネシウム、亜塩素酸マグネシウム、塩素酸マグネシウム、過塩素酸マグネシウム、次亜臭素酸、次亜臭素酸リチウム、亜臭素酸、亜臭素酸リチウム、臭素酸、臭素酸リチウム、過臭素酸、過臭素酸リチウム、次亜臭素酸ナトリウム、亜臭素酸ナトリウム、臭素酸ナトリウム、過臭素酸ナトリウム、次亜臭素酸カリウム、亜臭素酸カリウム、臭素酸カリウム、過臭素酸カリウム、次亜臭素酸カルシウム、亜臭素酸カルシウム、臭素酸カルシウム、過臭素酸カルシウム、次亜臭素酸マグネシウム、次亜沃素酸、次亜沃素酸リチウム、亜沃素酸、亜沃素酸リチウム、沃素酸、沃素酸リチウム、過沃素酸、過沃素酸リチウム、次亜沃素酸ナトリウム、亜沃素酸ナトリウム、沃素酸ナトリウム、過沃素酸ナトリウム、次亜沃素酸カリウム、亜沃素酸カリウム、過沃素酸カリウム、次亜沃素酸カルシウム、亜沃素酸カルシウム、沃素酸カルシウム、過沃素酸カルシム、次亜沃素酸マグネシウム、亜沃素酸マグネシウム、沃素酸マグネシウム、過沃素酸マグネシウム等の次亜ハロゲン酸、亜ハロゲン酸、ハロゲン酸、過ハロゲン酸、及びこれらの塩及び過酸化水素、オゾン、過酸化物を挙げることができる。式(12)で表される化合物は、活性ブロム化合物及び/又は過酸化水素由来活性体を発生させるために使用することができる。活性ブロム化合物は、反応系中で発生する化合物等を含み、例えば、ブロムカチオン、ブロムカチオンラジカル、ブロムラジカル、塩化臭素、臭素錯体(例えば、ジオキサン錯体)等が挙げられる。また、過酸化水素由来活性体としては、反応系中で発生する化合物等を含み、例えば、OHカチオン、OHラジカル、OOHラジカル、1重項酸素、OOHアニオン、OOHカチオン、スーパーオキサイド、スーパーオキサイドアニオン、スーパーオキサイドアニオンラジカル、ペルオキシラジカル、ヒドロペルオキシルラジカル等挙げられる。
Examples of the compound represented by the formula (12) include hypochlorous acid, lithium hypochlorite, chlorous acid, lithium chlorite, chloric acid, lithium chlorate, perchloric acid, lithium perchlorate, Sodium hypochlorite, sodium chlorite, sodium chlorate, sodium perchlorate, potassium hypochlorite, potassium chlorite, potassium chlorate, potassium perchlorate, calcium hypochlorite, calcium chlorite , Calcium chlorate, calcium perchlorate, magnesium hypochlorite, magnesium chlorite, magnesium chlorate, magnesium perchlorate, hypobromite, lithium hypobromite, bromite, lithium bromite, Bromate, lithium bromate, perbromate, lithium perbromate, sodium hypobromite, sodium bromate, sodium bromate, sodium perbromate , Potassium hypobromite, potassium bromite, potassium bromate, potassium perbromate, calcium hypobromite, calcium bromate, calcium bromate, calcium perbromate, magnesium hypobromite, hypoiodide Acid, lithium hypoiodite, iodic acid, lithium iodate, iodic acid, lithium iodate, periodic acid, lithium periodate, sodium hypoiodite, sodium iodate, sodium iodate, periodate Sodium phosphate, potassium hypoiodite, potassium iodate, potassium periodate, calcium hypoiodite, calcium iodate, calcium iodate, calcium periodate, magnesium hypoiodite, magnesium iodate, Hypohalous acid such as magnesium iodate and magnesium periodate, halous acid, halogen acid, perhalogen acid, and salts thereof Fine hydrogen peroxide, mention may be made of ozone, a peroxide. The compound represented by the formula (12) can be used for generating an active bromide compound and / or an active form derived from hydrogen peroxide. The active bromide compound includes a compound generated in the reaction system, and examples thereof include a brom cation, a brom cation radical, a brom radical, bromine chloride, a bromine complex (for example, dioxane complex) and the like. The hydrogen peroxide-derived active substance includes compounds generated in the reaction system, such as OH cation, OH radical, OOH radical, singlet oxygen, OOH anion, OOH cation, superoxide, superoxide anion. , Superoxide anion radical, peroxy radical, hydroperoxyl radical and the like.
式(12)で表される化合物としては、好ましくは、次亜塩素酸、次亜臭素酸、次亜塩素酸ナトリウム、塩素酸ナトリウム、次亜塩素酸カリウム、塩素酸カリウム、次亜塩素酸カルシウム、塩素酸カルシウム、次亜塩素酸マグネシウム、塩素酸マグネシウム、次亜臭素酸、次亜臭素酸ナトリウム、臭素酸ナトリウム、次亜臭素酸カリウム、臭素酸カリウム、次亜臭素酸カルシウム、臭素酸カルシウム、次亜臭素酸マグネシウム、臭素酸マグネシウム、次亜沃素酸、次亜沃素酸ナトリウム、沃素酸ナトリウム、次亜沃素酸カリウム、沃素酸カリウム、次亜沃素酸カルシウム、沃素酸カルシウム、次亜沃素酸マグネシウム、沃素酸マグネシウム等及び過酸化水素、オゾン、過酸化物挙げることができるが、これ等の化合物に限定されるものではない。
The compound represented by the formula (12) is preferably hypochlorous acid, hypobromite, sodium hypochlorite, sodium chlorate, potassium hypochlorite, potassium chlorate, calcium hypochlorite. , Calcium chlorate, magnesium hypochlorite, magnesium chlorate, hypobromite, sodium hypobromite, sodium bromate, potassium hypobromite, potassium bromate, calcium hypobromite, calcium bromate, Magnesium hypobromite, magnesium bromate, hypoiodic acid, sodium hypoiodite, sodium iodate, potassium hypoiodite, potassium iodate, calcium hypoiodite, calcium iodate, magnesium hypoiodite , Magnesium iodate and the like, and hydrogen peroxide, ozone, and peroxide, but are not limited to these compounds. There.
(式中、Mは、水素、Li、Na、K、Mg、Ca、Mn、Fe、Ni、Cu、Ag、Zn、Snよりなる群から選択される1~4価の金属を示す。及びH及びアンモニウムイオン、第1級アンモニウムイオン、第2級アンモニウムイオン、第3級アンモニウムイオン及びハロゲンカチオンを示し、Xは、ハロゲンアニオン及び有機ブロム化剤アニオン成分である。及び臭素分子、塩素分子、クロル臭素を示す。)
(In the formula, M represents a monovalent to tetravalent metal selected from the group consisting of hydrogen, Li, Na, K, Mg, Ca, Mn, Fe, Ni, Cu, Ag, Zn, and Sn. And H And an ammonium ion, a primary ammonium ion, a secondary ammonium ion, a tertiary ammonium ion and a halogen cation, X is a halogen anion and an organic brominating agent anion component, and a bromine molecule, a chlorine molecule, a chloro molecule. Indicates bromine.)
式(13)のXにおけるハロゲンとしては、塩素、臭素、ヨウ素が挙げられる。
Examples of the halogen in X in the formula (13) include chlorine, bromine and iodine.
式(13)で表される化合物としては、例えば、塩化水素、臭化水素、ヨウ化水素、塩化リチウム、塩化ナトリウム、塩化カリウム、塩化カルシウム、塩化銅、塩化マグネシウム、塩化鉄、塩化亜鉛、塩化ニッケル、塩化錫、塩化銀、臭化リチウム、臭化ナトリウム、臭化カリウム、臭化カルシウム、臭化銅、臭化マグネシウム、臭化鉄、臭化マンガン、臭化亜鉛、臭化ニッケル、臭化錫、臭化銀、沃化リチウム、沃化ナトリウム、沃化カリウム、沃化カルシウム、沃化銅、沃化マグネシウム、沃化鉄、沃化マンガン、沃化亜鉛、沃化ニッケル、沃化錫、沃化銀、塩化アンモニウム、モノメチルアンモニウムクロライド、ジメチルアンモニウムクロライド、トリメチルアンモニウムクロライド、テトラメチルアンモニウムクロライド、臭化アンモニウム、モノメチルアンモニウムブロマイド、ジメチルアンモニウムブロマイド、トリメチルアンモニウムブロマイド、テトラメチルアンモニウムブロマイド等の置換基を有しても良いアルキル基、アリール基、アラルキル基、アルケニル基等を有する第1級、第2級、第3級、第4級アンモニウム塩等、含窒素化合物、含窒素環状化合物のハロゲン化水素塩及び塩素、臭素、クロル臭素及び有機ハロゲン化剤であるジブロモイソシアヌル酸、N-ブロモスクシンイミド、ベンジルトリメチルアンモニウムトリブロマイド、ボロントリブロマイド、ピリヂニウムブロマイドパーブロマイド、1-ブチル-3-メチルイミダゾールトリブロマイド、トリフェニルホスフィンヂブロマイド、ベンジルトリメチルテトラクロロよう素酸塩、ベンゼンスルホンクロラミドナトリウム、N-クロロサクシンイミド、シヌル酸クロリド、N-クロロフタルイミド、オギザリルクロリド、メタンスルホニルクロリド等が挙げられる。式(13)で表される化合物は、活性ブロム化合物及び/又は過酸化水素由来活性体を発生させるために使用することができる。活性ブロム化合物及び/又は過酸化水素由来活性体を発生させるために使用することができる。活性ブロム化合物は、反応系中で発生する化合物等を含み、例えば、ブロムカチオン、ブロムカチオンラジカル、ブロムラジカル、塩化臭素、臭素錯体(例えば、ジオキサン錯体)等が挙げられる。また、過酸化水素由来活性体としては、反応系中で発生する化合物等を含み、例えば、OHカチオン、OHラジカル、OOHラジカル、1重項酸素、OOHアニオン、OOHカチオン、スーパーオキサイドアニオン、スーパーオキサイドアニオンラジカル、ペルオキシラジカル、ヒドロペルオキシルラジカル等挙げられる。
Examples of the compound represented by the formula (13) include hydrogen chloride, hydrogen bromide, hydrogen iodide, lithium chloride, sodium chloride, potassium chloride, calcium chloride, copper chloride, magnesium chloride, iron chloride, zinc chloride, and chloride. Nickel, tin chloride, silver chloride, lithium bromide, sodium bromide, potassium bromide, calcium bromide, copper bromide, magnesium bromide, iron bromide, manganese bromide, zinc bromide, nickel bromide, bromide Tin, silver bromide, lithium iodide, sodium iodide, potassium iodide, calcium iodide, copper iodide, magnesium iodide, iron iodide, manganese iodide, zinc iodide, nickel iodide, tin iodide, Silver iodide, ammonium chloride, monomethylammonium chloride, dimethylammonium chloride, trimethylammonium chloride, tetramethylammonium chloride, bromide Primary, secondary, alkyl groups, aryl groups, aralkyl groups, alkenyl groups, etc., which may have substituents such as ammonium, monomethylammonium bromide, dimethylammonium bromide, trimethylammonium bromide, tetramethylammonium bromide, Tertiary and quaternary ammonium salts, such as nitrogen-containing compounds, nitrogen-containing cyclic hydrogen halide salts and chlorine, bromine, chlorobromine and organic halogenating agents dibromoisocyanuric acid, N-bromosuccinimide, benzyltrimethylammonium Tribromide, boron tribromide, pyridinium bromide perbromide, 1-butyl-3-methylimidazole tribromide, triphenylphosphine dibromide, benzyltrimethyltetrachloroiodate, Zen sulfonic Crawlers bromide sodium, N- chlorosuccinimide, cash in acid chloride, N- chloro phthalimide, oxalyl chloride, methanesulfonyl chloride, and the like. The compound represented by the formula (13) can be used to generate an active bromide compound and / or an active form derived from hydrogen peroxide. It can be used to generate active bromide compounds and / or hydrogen peroxide derived activators. The active bromide compound includes a compound generated in the reaction system, and examples thereof include a brom cation, a brom cation radical, a brom radical, bromine chloride, a bromine complex (for example, dioxane complex) and the like. The hydrogen peroxide-derived active substance includes compounds generated in the reaction system, such as OH cation, OH radical, OOH radical, singlet oxygen, OOH anion, OOH cation, superoxide anion, superoxide. Anion radical, peroxy radical, hydroperoxyl radical and the like can be mentioned.
式(13)で表される化合物としては、好ましくは、塩化水素、臭化水素、塩化リチウム、塩化ナトリウム、塩化カリウム、塩化カルシウム、塩化マグネシウム、塩化亜鉛、塩化鉄、塩化銅、臭化リチウム、臭化ナトリウム、臭化カリウム、臭化カルシウム、臭化マグネシウム、臭化亜鉛、臭化銅、沃化リチウム、沃化ナトリウム、沃化カリウム、沃化銅、塩化アンモニウム、臭化アンモニウム、塩素、臭素、ヨウ素、クロル臭素、ジブロモイソシアヌル酸、N-ブロモスクシンイミド等を挙げることができるが、特にこれらの化合物に限定されるものではない。
The compound represented by the formula (13) is preferably hydrogen chloride, hydrogen bromide, lithium chloride, sodium chloride, potassium chloride, calcium chloride, magnesium chloride, zinc chloride, iron chloride, copper chloride, lithium bromide, Sodium bromide, potassium bromide, calcium bromide, magnesium bromide, zinc bromide, copper bromide, lithium iodide, sodium iodide, potassium iodide, copper iodide, ammonium chloride, ammonium bromide, chlorine, bromine , Iodine, chlorobromine, dibromoisocyanuric acid, N-bromosuccinimide and the like, but are not particularly limited to these compounds.
式(13)のXにおけるハロゲンとしては、例えば、フッ素、塩素、臭素、ヨウ素が挙げられる。
Examples of the halogen in X in the formula (13) include fluorine, chlorine, bromine and iodine.
また、上記ハロゲン化水素としては、例えば、塩化水素、臭化水素、沃化水素が挙げられる。
In addition, examples of the hydrogen halide include hydrogen chloride, hydrogen bromide, and hydrogen iodide.
また、上記ハロゲンとしては、例えば、塩素、臭素、ヨウ素が挙げられる。塩素、臭素、ヨウ素、塩素系化合物、臭素系化合物、ヨウ素系化合物を使用して酸化を行う場合、テルル化合物、バナジウム化合物、セリウム化合物、セレン化合物、遷移金属化合物及びフッ化アルミニウム等を担持した水に安定な強ルイス酸を触媒としての使用およびギ酸、酢酸等の酸の使用は効果的である。特にこれに限定されるものではない。
In addition, examples of the halogen include chlorine, bromine, and iodine. When oxidizing using chlorine, bromine, iodine, chlorine compounds, bromine compounds, iodine compounds, water carrying tellurium compounds, vanadium compounds, cerium compounds, selenium compounds, transition metal compounds, aluminum fluoride, etc. The use of highly stable strong Lewis acids as catalysts and the use of acids such as formic acid and acetic acid are effective. The invention is not particularly limited to this.
水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体との均一系又は不均一な混合系中に、原料である尿素を0.5~50mol/Lの濃度で含むことが好ましい。好ましくは、2~30mol/Lの濃度である。特にこれに限定されるものではない。
It is preferable that urea as a raw material is contained at a concentration of 0.5 to 50 mol / L in a homogeneous system or a heterogeneous mixed system with water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid. The concentration is preferably 2 to 30 mol / L. The invention is not particularly limited to this.
尿素の濃度は、温度に対する溶解度とブロム系酸化剤との比率に影響されるが、基本的には、高濃度、大量の尿素の存在が好ましい。ハロゲン系酸化剤等の化合物の尿素に対するモル比率では、活性中間遷移化合物ブロム尿素カチオン(化合物9)が生成した時に、その周囲を尿素で包まれた状態が好ましく、その為に尿素1モルに対してハロゲン系酸化剤等の化合物は、0.01~0.45モル比であるが、好ましくは、0.05~0.3モル比である。実質の酸化剤である過酸化水素は、これ等数値に対応する。しかし特にこれに限定されるものではない。
The concentration of urea is influenced by the solubility with respect to temperature and the ratio of bromine-based oxidant, but basically, the presence of a high concentration and a large amount of urea is preferable. The molar ratio of a compound such as a halogen-based oxidant to urea is preferably such that when the active intermediate transition compound bromourea cation (compound 9) is formed, its surroundings are enveloped with urea. The compound such as halogen-based oxidizing agent is 0.01 to 0.45 molar ratio, but preferably 0.05 to 0.3 molar ratio. Hydrogen peroxide, a substantial oxidant, corresponds to these numbers. However, it is not particularly limited to this.
本発明の一連の反応は、中間体を分離する必要がない。必要があれば式(8)、式(11)で表される化合物は、分離、貯蔵が可能であり、他の誘導体への原料として利用する事も可能である。
The series of reactions of the present invention does not require separation of intermediates. If necessary, the compounds represented by the formulas (8) and (11) can be separated and stored, and can be used as raw materials for other derivatives.
本発明の一連の反応の温度は、ハロゲンアニオンを過酸化水素等の酸化剤でハロゲン分子、ハロゲンカチオン、活性ハロゲン種に変換する場合、及び過酸化水素活性物質の生成の為には、余り低温では、塩の溶解度が低くなる問題及び過酸化水素等の酸化剤の活性の低下となる。-200~100℃で行われるが、好ましくは、-10~70℃である。反応中間体、反応活性中間体の安定性、反応性の兼ね合わせから反応条件に合わせて温度調整を行う事が望ましい。しかし尿素の水溶液は、凝固点が大きく低下する。中間体に電解法を適用する場合、電解時の発熱から活性中間体の分解、副反応の抑制の為、低温で反応する事が望ましい。-50~100℃で行われるが、好ましくは、-20~40℃である。更に低温で反応を行う事も可能である。又有機溶剤又はイオン液体との均一混合系においても低温で反応を行う事が出来る。特にこれに限定されるものではない。
The temperature of the series of reactions of the present invention is too low for converting halogen anions to halogen molecules, halogen cations, active halogen species with an oxidizing agent such as hydrogen peroxide, and for the production of hydrogen peroxide active substances. In this case, the solubility of the salt is lowered and the activity of an oxidizing agent such as hydrogen peroxide is reduced. The reaction is carried out at −200 to 100 ° C., preferably −10 to 70 ° C. It is desirable to adjust the temperature according to the reaction conditions in view of the stability and reactivity of the reaction intermediate and reaction active intermediate. However, an aqueous solution of urea has a significantly reduced freezing point. When the electrolytic method is applied to the intermediate, it is desirable to react at a low temperature in order to decompose the active intermediate and suppress side reactions from the heat generated during electrolysis. The reaction is carried out at −50 to 100 ° C., preferably −20 to 40 ° C. It is also possible to carry out the reaction at a low temperature. In addition, the reaction can be performed at a low temperature even in a homogeneous mixed system with an organic solvent or an ionic liquid. The invention is not particularly limited to this.
また、本発明において使用され得る有機溶剤としては、ジクロルエタン、四塩化炭素、クロルベンゼン等のハロゲン系溶剤類、n-ペンタン、n-ヘキサン、シクロペンタン、シクロヘキサン等の鎖状又は環状のアルカン類、テトラヒドロフラン、1,2-ジメトキシエタン、ジオキサン、メチル-t-ブチルエーテル、メチルシクロペンチルエーテル等のエーテル類、アセトニトリル、プロピオニトリルなどのニトリル類、アセトン、メチルエチルケトン、シクロペンタノン、シクロヘキサノン等のケトン類、メチルアルコール、エチルアルコール、イソプロピルアルコール等のアルコール類、DMF、DMSO、HEMPA、リン酸トリブチル、1,3-ジメチルイミダゾリン等の極性溶媒類等を挙げる事が出来る。またイオン液体については、カチオン成分として、置換基を有しても良いイミダゾールニウム類、第4級アンモニウム類、置換基を有しても良いピリジニウム類、ホスフォニウム類、置換基を有しても良いピロリジニウム類、スルホニウム類、アニオン成分としては、4フッ化ホウ素、6フッ化リン、トリフルオロ酢酸、トリフルオロメタンスルホン酸、ビストリフルオロメタンスルホンイミド、アルキルスルホン酸、酢酸、硝酸、塩素、臭素等のアニオンを挙げる事が出来る。これ等カチオン成分とアニオン成分との塩をイオン液体の例として挙げる事が出来る。特にこれ等に限定されるものではない。水、有機溶媒、イオン液体単独の使用であっても良い。又これ等の組み合わせであっても良い。上記溶媒および各溶媒の組合せ溶剤系において、尿素を含む溶液は、均一相であっても不均一相(液相分離状況)であってもよい。水と有機溶媒との混合溶媒を使用する場合、不均一系の場合は、原料および各種試薬は、主に水層に溶解しており、均一系の場合は、有機溶剤に含まれる水に原料及び各種試薬が主に溶解している。尿素及び塩類が全く溶解しない溶媒系においては、尿素及び塩類を極力微粒子にして溶剤に分散している事が重要である。
Examples of the organic solvent that can be used in the present invention include halogen solvents such as dichloroethane, carbon tetrachloride, and chlorobenzene, and chain or cyclic alkanes such as n-pentane, n-hexane, cyclopentane, and cyclohexane, Ethers such as tetrahydrofuran, 1,2-dimethoxyethane, dioxane, methyl-t-butyl ether and methylcyclopentyl ether, nitriles such as acetonitrile and propionitrile, ketones such as acetone, methyl ethyl ketone, cyclopentanone and cyclohexanone, methyl Examples include alcohols such as alcohol, ethyl alcohol, and isopropyl alcohol, and polar solvents such as DMF, DMSO, HEMPA, tributyl phosphate, and 1,3-dimethylimidazoline. Moreover, about an ionic liquid, you may have imidazolium which may have a substituent, quaternary ammonium, pyridinium which may have a substituent, phosphonium, and a substituent as a cation component. Pyrrolidiniums, sulfoniums, anion components such as boron tetrafluoride, phosphorus hexafluoride, trifluoroacetic acid, trifluoromethanesulfonic acid, bistrifluoromethanesulfonimide, alkylsulfonic acid, acetic acid, nitric acid, chlorine, bromine Can be mentioned. Examples of the ionic liquid include salts of these cationic components and anionic components. In particular, it is not limited to these. The use of water, an organic solvent, or an ionic liquid alone may be used. A combination of these may also be used. In the combined solvent system of the above solvent and each solvent, the solution containing urea may be a homogeneous phase or a heterogeneous phase (liquid phase separation state). When using a mixed solvent of water and an organic solvent, in the case of a heterogeneous system, the raw materials and various reagents are mainly dissolved in the aqueous layer. In addition, various reagents are mainly dissolved. In a solvent system in which urea and salts do not dissolve at all, it is important that urea and salts are dispersed in the solvent as fine particles as possible.
本合成反応においては、添加した原料の尿素を、反応時間内に全て反応させる必要はなく、むしろ大過剰の尿素が活性中間体の周囲に存在する方が式(10)で示される化合物が生じやすく、収率を上げる効果がある。反応で直接生成するアゾジカルボンアミドの固体を連続的に分離し、尿素、ハロゲン化剤、反応促進剤を含有する溶液をリサイクルし、必要に応じて、尿素、ハロゲン化剤、反応促進剤を添加して液組成を調整しながら循環させる製造方法とすることもできる。基本的には、ハロゲン化剤は、反応後ハロゲン化水素又はハロゲン塩になっているが、過酸化水素等の過酸化物でハロゲンに再生されるので、特に過酸化水素を使用する場合は、酸化副生物が少量の水であり、微量に生成する副生物の蓄積からリサイクルの限界まで液組成の調整は必要がない。
In this synthesis reaction, it is not necessary to react all of the added raw material urea within the reaction time. Rather, a compound represented by the formula (10) is produced when a large excess of urea is present around the active intermediate. It is easy to increase the yield. The azodicarbonamide solid produced directly in the reaction is continuously separated, and the solution containing urea, halogenating agent, and reaction accelerator is recycled, and urea, halogenating agent, and reaction accelerator are added as necessary. And it can also be set as the manufacturing method circulated, adjusting a liquid composition. Basically, the halogenating agent is a hydrogen halide or halogen salt after the reaction, but it is regenerated to a halogen with a peroxide such as hydrogen peroxide, so when using hydrogen peroxide in particular, The oxidation by-product is a small amount of water, and it is not necessary to adjust the liquid composition from the accumulation of by-products generated in minute amounts to the limit of recycling.
すなわち、ろ液として回収される未反応の尿素および式(12)~(13)で表される化合物、副生のハロゲン化水素又はハロゲン化塩より選択される化合物と、消費した尿素及び前記化合物を適宜追加して濃度を調整した水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系を、再び反応溶液として用いることができる。これらの処理により、最も高い尿素の反応転換率が達成できる、尿素又は尿素塩の濃度、反応混合液の反応槽での撹拌速度、反応温度、過酸化水素等の酸化剤の添加速度、反応促進剤濃度等の条件で、反応系をリサイクルすることが可能である。
That is, unreacted urea recovered as a filtrate, a compound selected from the compounds represented by formulas (12) to (13), a by-product hydrogen halide or a halide salt, consumed urea and the compound Water, an organic solvent or an ionic liquid, and an organic solvent or a homogeneous system or a heterogeneous mixed system of ionic liquid and water can be used again as a reaction solution. By these treatments, the highest conversion rate of urea can be achieved, the concentration of urea or urea salt, the stirring speed of the reaction mixture in the reaction tank, the reaction temperature, the addition rate of oxidizing agents such as hydrogen peroxide, and the reaction acceleration It is possible to recycle the reaction system under conditions such as the agent concentration.
電解反応の利用又は組み合わせ利用する場合、本電解反応は、定電圧電解でも定電流電解でもよい。また、電解方法として、無隔膜、隔膜、イオン交換膜の何れの方法も可能である。さらに、これら電解装置の陰極にガス拡散電極を設置することも可能である。
When using an electrolytic reaction or a combination, this electrolytic reaction may be constant voltage electrolysis or constant current electrolysis. Moreover, any method of a non-diaphragm, a diaphragm, and an ion exchange membrane is possible as an electrolysis method. Further, it is possible to install a gas diffusion electrode on the cathode of these electrolysis devices.
本発明の電解における陽極としては、金属系電極、炭素系電極およびこれらの複合化電極を用いることができる。
As the anode in the electrolysis of the present invention, a metal electrode, a carbon electrode, and a composite electrode thereof can be used.
陽極材料としては、金、銀、白金、ルテニウム等の貴金属及びチタン、クロム、ニッケル、マンガン等のマイナーメタル、及びチタン、ステンレス、鉄、ハステロイ等の貴金属以外の金属基材に貴金属で被覆した電極及びオレフィン樹脂、エンジニアリング樹脂、炭素系基材等の金属以外の基材に貴金属被覆した電極、酸化イリジウムや酸化ルテニウムなどの金属酸化物と白金との複合被覆電極、マイナーメタルによる上記と同様の被覆電極等が挙げられる。
As anode materials, noble metals such as gold, silver, platinum and ruthenium and minor metals such as titanium, chromium, nickel and manganese, and electrodes coated with noble metals other than noble metals such as titanium, stainless steel, iron and hastelloy And electrodes coated with precious metals on substrates other than metals such as olefin resins, engineering resins, carbon-based substrates, composite coated electrodes of metal oxides such as iridium oxide and ruthenium oxide and platinum, and coatings similar to the above with minor metals An electrode etc. are mentioned.
また、炭素系電極としては、例えば、カーボン、グラッシーカーボン、グラファイト、グラフェン、カーボンシート、炭素繊維シート、炭素繊維布、ダイアモンドライク被覆電極等の炭素系電極及びこれ等の複合化電極が挙げられる。
Also, examples of the carbon-based electrode include carbon-based electrodes such as carbon, glassy carbon, graphite, graphene, carbon sheet, carbon fiber sheet, carbon fiber cloth, diamond-like coated electrode, and composite electrodes thereof.
陽極として好ましいのは、白金、チタン、及びチタン、ステンレス、鉄、ハステロイ等の貴金属以外の金属基材に白金を被覆した電極及びオレフィン樹脂、エンジニアリング樹脂、炭素系基材等の金属以外の基材に白金を被覆した電極、及び酸化イリジウムや酸化ルテニウムなどの金属酸化物と白金との複合被覆電極及びカーボン、グラッシーカーボン、グラファイト、グラフェン、カーボンシート、炭素繊維シート、炭素繊維布、ダイアモンドライク被覆電極等の炭素系電極、及びこれらの複合化電極等であるが、特にこれらに限定されるものではない。
Preferred as the anode is an electrode in which platinum is coated on a metal substrate other than a noble metal such as platinum, titanium, titanium, stainless steel, iron, or hastelloy, and a substrate other than a metal such as an olefin resin, an engineering resin, or a carbon-based substrate. Electrode coated with platinum, composite electrode of platinum with metal oxide such as iridium oxide and ruthenium oxide, and carbon, glassy carbon, graphite, graphene, carbon sheet, carbon fiber sheet, carbon fiber cloth, diamond-like coated electrode Carbon-based electrodes such as these, and composite electrodes thereof, but are not particularly limited thereto.
陰極としては、特に制限されず、陽極電極として例示した材料および鉄、銅、アルミニウム等の汎用金属、及びステンレス、ハステロイ、各種合金及びこれ等の複合化電極が全て使用可能である。
The cathode is not particularly limited, and the materials exemplified as the anode electrode and general-purpose metals such as iron, copper, and aluminum, and stainless steel, hastelloy, various alloys, and composite electrodes thereof can be used.
陰極として好ましいのは、白金、ステンレス、チタン、ステンレス、ハステロイ、鉄、及びチタン、ステンレス、鉄、ハステロイ等の貴金属以外の金属基材に白金を被覆した電極及びオレフィン樹脂、エンジニアリング樹脂、炭素系基材等の金属以外の基材に白金を被覆した電極、及び酸化イリジウムや酸化ルテニウムなどの金属酸化物と白金との複合被覆電極及びカーボン、グラッシーカーボン、グラファイト、カーボンシート、炭素繊維シート、炭素繊維布等の炭素系電極、及びこれらの複合化電極等であるが、特にこれらに限定されるものではない。
Preferred as the cathode are platinum, stainless steel, titanium, stainless steel, hastelloy, iron, and electrodes in which platinum is coated on a metal base other than noble metal such as titanium, stainless steel, iron, hastelloy, olefin resin, engineering resin, carbon-based group Electrode with platinum coated on base material other than metal such as metal, and composite coated electrode of metal oxide such as iridium oxide and ruthenium oxide and platinum and carbon, glassy carbon, graphite, carbon sheet, carbon fiber sheet, carbon fiber Although it is carbon-type electrodes, such as cloth, these composite electrodes, etc., it is not specifically limited to these.
電極の形状としては、板状、布状、すだれ状、パイプ状、不織布状、紙漉き方法で得られる形状物、フェルト状等の形状が使用でき、更にこれら材料を網状、パンチング状、メッシュ状に加工された物が使用できる。好ましくは、陽極は、隙間の無い状態の形状で陰極は穴や隙間がある形状物であるが、ガス拡散電極を使用する場合陰極での水素発生がないため、陰極の形状は限定されない。特にこれらに限定されるものではない。
As the shape of the electrode, a plate shape, a cloth shape, a comb shape, a pipe shape, a nonwoven fabric shape, a shape obtained by a paper-making method, a felt shape, etc. can be used. Processed products can be used. Preferably, the anode has a shape without a gap and the cathode has a shape with a hole or a gap. However, when a gas diffusion electrode is used, there is no hydrogen generation at the cathode, so the shape of the cathode is not limited. In particular, it is not limited to these.
本発明の電気分解は、電流密度を1~20,000mA/cm2、好ましくは、10~5000mA/cm2で一定に保持した状態で行う。あるいは、電極電位を0.5~100V vs.Ag/AgCl、好ましくは、1~70V vs.Ag/AgClで一定に保持し、電解することができるが、特にこれらに限定されるものではない。
The electrolysis of the present invention is carried out in a state where the current density is kept constant at 1 to 20,000 mA / cm 2 , preferably 10 to 5000 mA / cm 2 . Alternatively, the electrode potential is set to 0.5 to 100 V vs. Ag / AgCl, preferably 1 to 70 V vs. Although it can hold | maintain with Ag / AgCl and can be electrolyzed, it is not specifically limited to these.
また、反応温度は-40~100℃であるとよい。好ましくは、-20~70℃であるが、特にこれに限定されるものではない。反応中間体、反応活性中間体の安定性、反応性の兼ね合わせから反応条件に応じて温度調整する事が望ましい。有機溶剤やイオン液体との均一混合系においては、更に低温で反応させる事が出来る。
Also, the reaction temperature is preferably −40 to 100 ° C. The temperature is preferably −20 to 70 ° C., but is not particularly limited thereto. It is desirable to adjust the temperature according to the reaction conditions in view of the stability and reactivity of the reaction intermediate and reaction active intermediate. In a homogeneous mixed system with an organic solvent or ionic liquid, the reaction can be performed at a lower temperature.
本反応における反応性、活性中間体の生成のし易さ、活性中間体の尿素との反応性の促進、活性中間体の分解抑制、中間体の生成の為の酸化活性種の生成、2電子酸化の促進、ホフマン転移等の副反応の抑制等の為には、酸性条件下の反応が望ましい。酸性条件とする為に添加される酸は、塩酸、硫酸、リン酸、ホウ酸等の鉱産及び酢酸、プロピオン酸、クエン酸等の有機酸でも良い。またそれらの塩との混合物である緩衝液でも良い。過酸化水素を酸化剤に使用する場合、過酸化水素の活性化の為にPH=3以下で、好ましくはPH=1で行う。反応工程に対応して過酸化水素の反応温度、添加速度、過酸化水素濃度、反応系の酸性度を調整する事が反応を目的通り進行させるのに重要である。特にこれらに限定されるものではない。
Reactivity in this reaction, ease of formation of active intermediate, promotion of reactivity of active intermediate with urea, suppression of decomposition of active intermediate, generation of oxidized active species for formation of intermediate, 2 electrons Reactions under acidic conditions are desirable for promoting oxidation and suppressing side reactions such as Hoffman transition. The acid added for the acidic condition may be mineral products such as hydrochloric acid, sulfuric acid, phosphoric acid, boric acid, and organic acids such as acetic acid, propionic acid, citric acid. Moreover, the buffer solution which is a mixture with those salts may be sufficient. When hydrogen peroxide is used as the oxidizing agent, PH = 3 or less, preferably PH = 1, for the activation of hydrogen peroxide. It is important to adjust the reaction temperature of hydrogen peroxide, the addition rate, the hydrogen peroxide concentration, and the acidity of the reaction system in accordance with the reaction process in order to proceed the reaction as intended. In particular, it is not limited to these.
以下、実施例を用いて本発明をより詳細に説明するが、本発明はこれら実施例に限定されるものではない。
Hereinafter, the present invention will be described in more detail using examples, but the present invention is not limited to these examples.
以下の各実施例で得られたアゾジカルボンアミドは、赤外吸収スペクトル(IR)及びプロトン核磁気共鳴スペクトル(1H-NMR)により同定した。
IR(KBr):3337、3187cm-1(N-H伸縮)、1743,1731cm-1(C=O伸縮)、1637cm-1(N-H変角)、1369,1331cm-1(C-N伸縮).
1H-NMR(D6-DMSO):δ8.02(s,2H,NH),7.97(s,2H,NH).
以下の各実施例での未反応の尿素の定量は、高速液体クロマトグラフィーにより行った。
高速液体クロマトグラフィーによる分析条件は、以下のとおりである。
カラム:ODS-3 (4.6mmφ ×250mm)
移動相:10mM 酢酸水溶液
流速:1 ml/min
検出波長:205nm The azodicarbonamide obtained in each of the following examples was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR).
IR (KBr): 3337,3187cm -1 ( N-H stretching), 1743,1731cm -1 (C = O stretch), 1637cm -1 (N-H bending), 1369,1331cm -1 (C-N stretch ).
1 H-NMR (D6-DMSO): δ 8.02 (s, 2H, NH), 7.97 (s, 2H, NH).
In the following examples, unreacted urea was quantified by high performance liquid chromatography.
The analysis conditions by high performance liquid chromatography are as follows.
Column: ODS-3 (4.6 mmφ x 250 mm)
Mobile phase: 10 mM aqueous acetic acid flow rate: 1 ml / min
Detection wavelength: 205 nm
IR(KBr):3337、3187cm-1(N-H伸縮)、1743,1731cm-1(C=O伸縮)、1637cm-1(N-H変角)、1369,1331cm-1(C-N伸縮).
1H-NMR(D6-DMSO):δ8.02(s,2H,NH),7.97(s,2H,NH).
以下の各実施例での未反応の尿素の定量は、高速液体クロマトグラフィーにより行った。
高速液体クロマトグラフィーによる分析条件は、以下のとおりである。
カラム:ODS-3 (4.6mmφ ×250mm)
移動相:10mM 酢酸水溶液
流速:1 ml/min
検出波長:205nm The azodicarbonamide obtained in each of the following examples was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR).
IR (KBr): 3337,3187cm -1 ( N-H stretching), 1743,1731cm -1 (C = O stretch), 1637cm -1 (N-H bending), 1369,1331cm -1 (C-N stretch ).
1 H-NMR (D6-DMSO): δ 8.02 (s, 2H, NH), 7.97 (s, 2H, NH).
In the following examples, unreacted urea was quantified by high performance liquid chromatography.
The analysis conditions by high performance liquid chromatography are as follows.
Column: ODS-3 (4.6 mmφ x 250 mm)
Mobile phase: 10 mM aqueous acetic acid flow rate: 1 ml / min
Detection wavelength: 205 nm
下記実施例における変換率とは、消費尿素量を示し、下記の式によって算出したものである。
変換率(%)=[(原料尿素の量-回収尿素の量)/(原料尿素の量)]×100
また、収率は、下記の式によって算出した。
収率(%)=[(得られたアゾジカルボンアミドのモル数)×2/(原料尿素のモル数-回収尿素のモル数)]×100 The conversion rate in the following examples indicates the amount of urea consumed and is calculated by the following formula.
Conversion rate (%) = [(amount of raw urea−amount of recovered urea) / (amount of raw urea)] × 100
The yield was calculated by the following formula.
Yield (%) = [(number of moles of azodicarbonamide obtained) × 2 / (number of moles of raw urea−number of moles of recovered urea)] × 100
変換率(%)=[(原料尿素の量-回収尿素の量)/(原料尿素の量)]×100
また、収率は、下記の式によって算出した。
収率(%)=[(得られたアゾジカルボンアミドのモル数)×2/(原料尿素のモル数-回収尿素のモル数)]×100 The conversion rate in the following examples indicates the amount of urea consumed and is calculated by the following formula.
Conversion rate (%) = [(amount of raw urea−amount of recovered urea) / (amount of raw urea)] × 100
The yield was calculated by the following formula.
Yield (%) = [(number of moles of azodicarbonamide obtained) × 2 / (number of moles of raw urea−number of moles of recovered urea)] × 100
参考例1
攪拌機と温度計を備えた反応容器に尿素75g(1.25mol)を水150mlに溶解し、10℃で29%次亜塩素酸ソーダ水溶液321ml(1.25mol)を撹拌下1時間で滴下する。サンプルを採取してヨウ化カリウム及び酢酸(1:1)を加えて速やかに0.1N-チオ硫酸ナトリウム滴定を行う。滴定分析の結果モノクロル尿素の収率は、98.4%である事を確認した。紫外分析を行うとモノクロロ尿素特有の244nmの吸収を認めた。 Reference example 1
In a reaction vessel equipped with a stirrer and a thermometer, 75 g (1.25 mol) of urea is dissolved in 150 ml of water, and 321 ml (1.25 mol) of a 29% sodium hypochlorite aqueous solution is added dropwise at 10 ° C. over 1 hour with stirring. A sample is taken, potassium iodide and acetic acid (1: 1) are added, and 0.1N-sodium thiosulfate titration is immediately performed. As a result of titration analysis, it was confirmed that the yield of monochlorourea was 98.4%. When ultraviolet analysis was performed, absorption at 244 nm peculiar to monochlorourea was observed.
攪拌機と温度計を備えた反応容器に尿素75g(1.25mol)を水150mlに溶解し、10℃で29%次亜塩素酸ソーダ水溶液321ml(1.25mol)を撹拌下1時間で滴下する。サンプルを採取してヨウ化カリウム及び酢酸(1:1)を加えて速やかに0.1N-チオ硫酸ナトリウム滴定を行う。滴定分析の結果モノクロル尿素の収率は、98.4%である事を確認した。紫外分析を行うとモノクロロ尿素特有の244nmの吸収を認めた。 Reference example 1
In a reaction vessel equipped with a stirrer and a thermometer, 75 g (1.25 mol) of urea is dissolved in 150 ml of water, and 321 ml (1.25 mol) of a 29% sodium hypochlorite aqueous solution is added dropwise at 10 ° C. over 1 hour with stirring. A sample is taken, potassium iodide and acetic acid (1: 1) are added, and 0.1N-sodium thiosulfate titration is immediately performed. As a result of titration analysis, it was confirmed that the yield of monochlorourea was 98.4%. When ultraviolet analysis was performed, absorption at 244 nm peculiar to monochlorourea was observed.
参考例2
尿素塩酸塩の溶液に10.7%NaOCl水溶液(Cl2としての重量%)およびNaBrの38%水溶液(重量%)を添加することによるブロモ尿素溶液の調製(NaOCl:HCl:尿素:NaBrのモル比 1:2.2:37.7:0.8)
撹拌棒と滴下漏斗と温度計とを備えた250mlの丸底フラスコ内で、46.04gの尿素(767mmol)を30.6gのH2Oに溶かし、続いて、5.4gの32%HCl(47.2mmol)を添加した(冷却時、発熱を伴う)。尿素塩酸塩の溶液が得られた。13.5gの10.7%NaOCl水溶液(Cl2としての重量%、20.3mmol)を添加漏斗に入れ、4.84gの38%NaBr溶液を別の添加漏斗に入れた。NaOCl溶液を尿素塩酸塩溶液に添加し、1分遅れで、38%NaBr水溶液を当該溶液に添加した。紫外分析を行うとブロモ尿素に特有の275nmにおける吸収を示した。サンプルを採取してヨウ化カリウム及び酢酸(1:1)を加えて速やかに0.1N-チオ硫酸ナトリウム滴定を行う。滴定分析の結果モノブロモ尿素の収率は、98.9%である事を確認した。 Reference example 2
Preparation of bromo urea solution by adding a solution in 10.7% NaOCl aqueous solution of urea hydrochloride and 38% aqueous solution of NaBr (wt%) (wt% as Cl 2) (NaOCl: HCl: Urea: NaBr molar (Ratio 1: 2.2: 37.7: 0.8)
In a 250 ml round bottom flask equipped with a stir bar, dropping funnel and thermometer, 46.04 g urea (767 mmol) was dissolved in 30.6 g H 2 O followed by 5.4 g 32% HCl ( 47.2 mmol) was added (with exotherm on cooling). A solution of urea hydrochloride was obtained. 13.5 g of 10.7% aqueous NaOCl (wt% as Cl 2 , 20.3 mmol) was placed in the addition funnel and 4.84 g of 38% NaBr solution was placed in another addition funnel. The NaOCl solution was added to the urea hydrochloride solution, and with a delay of 1 minute, 38% NaBr aqueous solution was added to the solution. Ultraviolet analysis showed absorption at 275 nm which is characteristic of bromourea. A sample is taken, potassium iodide and acetic acid (1: 1) are added, and 0.1N-sodium thiosulfate titration is immediately performed. As a result of titration analysis, it was confirmed that the yield of monobromourea was 98.9%.
尿素塩酸塩の溶液に10.7%NaOCl水溶液(Cl2としての重量%)およびNaBrの38%水溶液(重量%)を添加することによるブロモ尿素溶液の調製(NaOCl:HCl:尿素:NaBrのモル比 1:2.2:37.7:0.8)
撹拌棒と滴下漏斗と温度計とを備えた250mlの丸底フラスコ内で、46.04gの尿素(767mmol)を30.6gのH2Oに溶かし、続いて、5.4gの32%HCl(47.2mmol)を添加した(冷却時、発熱を伴う)。尿素塩酸塩の溶液が得られた。13.5gの10.7%NaOCl水溶液(Cl2としての重量%、20.3mmol)を添加漏斗に入れ、4.84gの38%NaBr溶液を別の添加漏斗に入れた。NaOCl溶液を尿素塩酸塩溶液に添加し、1分遅れで、38%NaBr水溶液を当該溶液に添加した。紫外分析を行うとブロモ尿素に特有の275nmにおける吸収を示した。サンプルを採取してヨウ化カリウム及び酢酸(1:1)を加えて速やかに0.1N-チオ硫酸ナトリウム滴定を行う。滴定分析の結果モノブロモ尿素の収率は、98.9%である事を確認した。 Reference example 2
Preparation of bromo urea solution by adding a solution in 10.7% NaOCl aqueous solution of urea hydrochloride and 38% aqueous solution of NaBr (wt%) (wt% as Cl 2) (NaOCl: HCl: Urea: NaBr molar (Ratio 1: 2.2: 37.7: 0.8)
In a 250 ml round bottom flask equipped with a stir bar, dropping funnel and thermometer, 46.04 g urea (767 mmol) was dissolved in 30.6 g H 2 O followed by 5.4 g 32% HCl ( 47.2 mmol) was added (with exotherm on cooling). A solution of urea hydrochloride was obtained. 13.5 g of 10.7% aqueous NaOCl (wt% as Cl 2 , 20.3 mmol) was placed in the addition funnel and 4.84 g of 38% NaBr solution was placed in another addition funnel. The NaOCl solution was added to the urea hydrochloride solution, and with a delay of 1 minute, 38% NaBr aqueous solution was added to the solution. Ultraviolet analysis showed absorption at 275 nm which is characteristic of bromourea. A sample is taken, potassium iodide and acetic acid (1: 1) are added, and 0.1N-sodium thiosulfate titration is immediately performed. As a result of titration analysis, it was confirmed that the yield of monobromourea was 98.9%.
参考例3
参考例1において生成したモノクロロ尿素を減圧化濃縮すると結晶で分離される。100mlの水にモノクロル尿素31.5g(0.33mol)を溶解し、8%の塩酸80mlを加え、臭化ナトリウムを34g(0.33mol)加えて、撹拌下50℃で30%過酸化水素38ml(0.33mol)を1時間で滴下する。滴下終了後更に30分撹拌して反応を終了させる。サンプル採取して紫外分析を行うとモノブロモ尿素に特有の275nmにおける吸収を示した。サンプルを採取してヨウ化カリウム及び酢酸(1:1)を加えて速やかに0.1N-チオ硫酸ナトリウム滴定を行う。滴定分析の結果モノブロモ尿素の収率は、98.7%である事を確認した。紫外分析を行うとブロモ尿素に特有の275nmにおける吸収を示した。モノブロム尿素は、不安定物質であるが、低温で減圧濃縮するか凍結乾燥すると結晶で得る事が出来る。 Reference example 3
Monochlorourea produced in Reference Example 1 is separated by crystals when concentrated under reduced pressure. Dissolve 31.5 g (0.33 mol) of monochlorourea in 100 ml of water, add 80 ml of 8% hydrochloric acid, add 34 g (0.33 mol) of sodium bromide, and add 30 ml of 30% hydrogen peroxide at 50 ° C. with stirring. (0.33 mol) is added dropwise over 1 hour. After completion of the dropwise addition, the reaction is terminated by stirring for another 30 minutes. When the sample was collected and subjected to ultraviolet analysis, the absorption at 275 nm characteristic of monobromourea was shown. A sample is taken, potassium iodide and acetic acid (1: 1) are added, and 0.1N-sodium thiosulfate titration is immediately performed. As a result of titration analysis, it was confirmed that the yield of monobromourea was 98.7%. Ultraviolet analysis showed absorption at 275 nm which is characteristic of bromourea. Monobromourea is an unstable substance, but can be obtained as crystals when concentrated under reduced pressure at low temperature or freeze-dried.
参考例1において生成したモノクロロ尿素を減圧化濃縮すると結晶で分離される。100mlの水にモノクロル尿素31.5g(0.33mol)を溶解し、8%の塩酸80mlを加え、臭化ナトリウムを34g(0.33mol)加えて、撹拌下50℃で30%過酸化水素38ml(0.33mol)を1時間で滴下する。滴下終了後更に30分撹拌して反応を終了させる。サンプル採取して紫外分析を行うとモノブロモ尿素に特有の275nmにおける吸収を示した。サンプルを採取してヨウ化カリウム及び酢酸(1:1)を加えて速やかに0.1N-チオ硫酸ナトリウム滴定を行う。滴定分析の結果モノブロモ尿素の収率は、98.7%である事を確認した。紫外分析を行うとブロモ尿素に特有の275nmにおける吸収を示した。モノブロム尿素は、不安定物質であるが、低温で減圧濃縮するか凍結乾燥すると結晶で得る事が出来る。 Reference example 3
Monochlorourea produced in Reference Example 1 is separated by crystals when concentrated under reduced pressure. Dissolve 31.5 g (0.33 mol) of monochlorourea in 100 ml of water, add 80 ml of 8% hydrochloric acid, add 34 g (0.33 mol) of sodium bromide, and add 30 ml of 30% hydrogen peroxide at 50 ° C. with stirring. (0.33 mol) is added dropwise over 1 hour. After completion of the dropwise addition, the reaction is terminated by stirring for another 30 minutes. When the sample was collected and subjected to ultraviolet analysis, the absorption at 275 nm characteristic of monobromourea was shown. A sample is taken, potassium iodide and acetic acid (1: 1) are added, and 0.1N-sodium thiosulfate titration is immediately performed. As a result of titration analysis, it was confirmed that the yield of monobromourea was 98.7%. Ultraviolet analysis showed absorption at 275 nm which is characteristic of bromourea. Monobromourea is an unstable substance, but can be obtained as crystals when concentrated under reduced pressure at low temperature or freeze-dried.
実施例1-1
磁気撹拌棒と滴下漏斗と温度計とを備えた500mlの丸底フラスコ内で、水100mlに尿素90g(1.5mol)を溶解し、続いて、5℃の冷却下、5℃に冷却した17.1gの32%HCl(0.15mol)を撹拌下添加した。溶液のPH=1になる様に32%HClの添加量で調整する。当該溶液に、14分間にわたって、133gの10.7%NaOCl水溶液(活性Cl2としての重量%、0.2mol)を冷却時に添加した。添加終了後1時間撹拌した後、当該溶液に20.5gのNaBr(0.2mol)を添加した。添加終了後反応液の温度を35℃に上げて1時間撹拌後、30%過酸化水素79.3g(0.7mol)を2分割し、最初の半量を30分間で滴下する。1時間撹拌を行い、その後残りの半量を30分間で滴下する。滴下終了後5時間撹拌を行い反応を終了させる。反応の進行と共に黄色の個体が生成し、反応液に縣濁して来る。反応終了後固形物のろ過分離を行い、水で洗浄した後乾燥させる。得られたアゾジカルボンアミドは、赤外吸収スペクトル(IR)及びプロトン核磁気共鳴スペクトル(1H-NMR)により同定した。消費された尿素からのアゾジカルボンアミドの収率は、95.3%であった。
IR(KBr):3337、3187cm-1(N-H伸縮)、1743,1731cm-1(C=O伸縮)、1637cm-1(N-H変角)、1369,1331cm-1(C-N伸縮).
1H-NMR(D6-DMSO):δ8.02(s,2H,NH),7.97(s,2H,NH). Example 1-1
In a 500 ml round bottom flask equipped with a magnetic stir bar, dropping funnel and thermometer, 90 g (1.5 mol) of urea was dissolved in 100 ml of water, followed by cooling to 5 ° C. under cooling at 5 ° C. 17 .1 g of 32% HCl (0.15 mol) was added with stirring. Adjust the addition amount of 32% HCl so that the pH of the solution is 1. To the solution was added 133 g of 10.7% NaOCl aqueous solution (wt% as active Cl 2 , 0.2 mol) over 14 minutes upon cooling. After stirring for 1 hour after completion of the addition, 20.5 g of NaBr (0.2 mol) was added to the solution. After completion of the addition, the temperature of the reaction solution is raised to 35 ° C. and stirred for 1 hour, then 79.3 g (0.7 mol) of 30% hydrogen peroxide is divided into two, and the first half is added dropwise over 30 minutes. Stir for 1 hour, then drop the remaining half in 30 minutes. After completion of the dropwise addition, the reaction is terminated by stirring for 5 hours. As the reaction progresses, a yellow solid is formed and becomes suspended in the reaction solution. After completion of the reaction, the solid is separated by filtration, washed with water and dried. The obtained azodicarbonamide was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR). The yield of azodicarbonamide from the consumed urea was 95.3%.
IR (KBr): 3337,3187cm -1 ( N-H stretching), 1743,1731cm -1 (C = O stretch), 1637cm -1 (N-H bending), 1369,1331cm -1 (C-N stretch ).
1 H-NMR (D6-DMSO): δ 8.02 (s, 2H, NH), 7.97 (s, 2H, NH).
磁気撹拌棒と滴下漏斗と温度計とを備えた500mlの丸底フラスコ内で、水100mlに尿素90g(1.5mol)を溶解し、続いて、5℃の冷却下、5℃に冷却した17.1gの32%HCl(0.15mol)を撹拌下添加した。溶液のPH=1になる様に32%HClの添加量で調整する。当該溶液に、14分間にわたって、133gの10.7%NaOCl水溶液(活性Cl2としての重量%、0.2mol)を冷却時に添加した。添加終了後1時間撹拌した後、当該溶液に20.5gのNaBr(0.2mol)を添加した。添加終了後反応液の温度を35℃に上げて1時間撹拌後、30%過酸化水素79.3g(0.7mol)を2分割し、最初の半量を30分間で滴下する。1時間撹拌を行い、その後残りの半量を30分間で滴下する。滴下終了後5時間撹拌を行い反応を終了させる。反応の進行と共に黄色の個体が生成し、反応液に縣濁して来る。反応終了後固形物のろ過分離を行い、水で洗浄した後乾燥させる。得られたアゾジカルボンアミドは、赤外吸収スペクトル(IR)及びプロトン核磁気共鳴スペクトル(1H-NMR)により同定した。消費された尿素からのアゾジカルボンアミドの収率は、95.3%であった。
IR(KBr):3337、3187cm-1(N-H伸縮)、1743,1731cm-1(C=O伸縮)、1637cm-1(N-H変角)、1369,1331cm-1(C-N伸縮).
1H-NMR(D6-DMSO):δ8.02(s,2H,NH),7.97(s,2H,NH). Example 1-1
In a 500 ml round bottom flask equipped with a magnetic stir bar, dropping funnel and thermometer, 90 g (1.5 mol) of urea was dissolved in 100 ml of water, followed by cooling to 5 ° C. under cooling at 5 ° C. 17 .1 g of 32% HCl (0.15 mol) was added with stirring. Adjust the addition amount of 32% HCl so that the pH of the solution is 1. To the solution was added 133 g of 10.7% NaOCl aqueous solution (wt% as active Cl 2 , 0.2 mol) over 14 minutes upon cooling. After stirring for 1 hour after completion of the addition, 20.5 g of NaBr (0.2 mol) was added to the solution. After completion of the addition, the temperature of the reaction solution is raised to 35 ° C. and stirred for 1 hour, then 79.3 g (0.7 mol) of 30% hydrogen peroxide is divided into two, and the first half is added dropwise over 30 minutes. Stir for 1 hour, then drop the remaining half in 30 minutes. After completion of the dropwise addition, the reaction is terminated by stirring for 5 hours. As the reaction progresses, a yellow solid is formed and becomes suspended in the reaction solution. After completion of the reaction, the solid is separated by filtration, washed with water and dried. The obtained azodicarbonamide was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR). The yield of azodicarbonamide from the consumed urea was 95.3%.
IR (KBr): 3337,3187cm -1 ( N-H stretching), 1743,1731cm -1 (C = O stretch), 1637cm -1 (N-H bending), 1369,1331cm -1 (C-N stretch ).
1 H-NMR (D6-DMSO): δ 8.02 (s, 2H, NH), 7.97 (s, 2H, NH).
実施例1-2
磁気撹拌棒と滴下漏斗と温度計とを備えた500mlの丸底フラスコ内で、水300mlに尿素90g(1.5mol)を溶解し、続いて、0℃の冷却下、0℃に冷却した22.5gの35%塩酸(0.22mol)を撹拌下添加した。更に溶液がPH=1になる様に35%塩酸で調整を行った。当該溶液に19.6gの臭化アンモニウム(0.2mol)と23.5gのNaCl(0.4mol)を添加した。添加終了後反応液の温度を40℃に上げて、30%過酸化水素140.5g(1.24mol)を2分割し、その半分を30分間で滴下し、1時間撹拌を行った後、残りの過酸化水素を30分間で滴下し、5時間撹拌を行い反応を終了させる。反応の進行と共に黄色の個体が生成し、反応液に縣濁して来る。反応終了後固形物のろ過分離を行い、水で洗浄した後乾燥させる。得られたアゾジカルボンアミドは、赤外吸収スペクトル(IR)及びプロトン核磁気共鳴スペクトル(1H-NMR)により同定した。消費された尿素からのアゾジカルボンアミドの収率は、93.8%であった。
IR(KBr):3337、3187cm-1(N-H伸縮)、1743,1731cm-1(C=O伸縮)、1637cm-1(N-H変角)、1369,1331cm-1(C-N伸縮).
1H-NMR(D6-DMSO):δ8.02(s,2H,NH),7.97(s,2H,NH). Example 1-2
In a 500 ml round bottom flask equipped with a magnetic stir bar, dropping funnel and thermometer, 90 g (1.5 mol) of urea was dissolved in 300 ml of water, followed by cooling to 0 ° C. under cooling at 0 ° C. 22 0.5 g of 35% hydrochloric acid (0.22 mol) was added with stirring. Further, the solution was adjusted with 35% hydrochloric acid so that the pH was 1. To the solution was added 19.6 g ammonium bromide (0.2 mol) and 23.5 g NaCl (0.4 mol). After completion of the addition, the temperature of the reaction solution was raised to 40 ° C., and 140.5 g (1.24 mol) of 30% hydrogen peroxide was divided into two parts, half of which was added dropwise over 30 minutes, and stirred for 1 hour. Of hydrogen peroxide was added dropwise over 30 minutes, and the reaction was terminated by stirring for 5 hours. As the reaction progresses, a yellow solid is formed and becomes suspended in the reaction solution. After completion of the reaction, the solid is separated by filtration, washed with water and dried. The obtained azodicarbonamide was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR). The yield of azodicarbonamide from the consumed urea was 93.8%.
IR (KBr): 3337,3187cm -1 ( N-H stretching), 1743,1731cm -1 (C = O stretch), 1637cm -1 (N-H bending), 1369,1331cm -1 (C-N stretch ).
1 H-NMR (D6-DMSO): δ 8.02 (s, 2H, NH), 7.97 (s, 2H, NH).
磁気撹拌棒と滴下漏斗と温度計とを備えた500mlの丸底フラスコ内で、水300mlに尿素90g(1.5mol)を溶解し、続いて、0℃の冷却下、0℃に冷却した22.5gの35%塩酸(0.22mol)を撹拌下添加した。更に溶液がPH=1になる様に35%塩酸で調整を行った。当該溶液に19.6gの臭化アンモニウム(0.2mol)と23.5gのNaCl(0.4mol)を添加した。添加終了後反応液の温度を40℃に上げて、30%過酸化水素140.5g(1.24mol)を2分割し、その半分を30分間で滴下し、1時間撹拌を行った後、残りの過酸化水素を30分間で滴下し、5時間撹拌を行い反応を終了させる。反応の進行と共に黄色の個体が生成し、反応液に縣濁して来る。反応終了後固形物のろ過分離を行い、水で洗浄した後乾燥させる。得られたアゾジカルボンアミドは、赤外吸収スペクトル(IR)及びプロトン核磁気共鳴スペクトル(1H-NMR)により同定した。消費された尿素からのアゾジカルボンアミドの収率は、93.8%であった。
IR(KBr):3337、3187cm-1(N-H伸縮)、1743,1731cm-1(C=O伸縮)、1637cm-1(N-H変角)、1369,1331cm-1(C-N伸縮).
1H-NMR(D6-DMSO):δ8.02(s,2H,NH),7.97(s,2H,NH). Example 1-2
In a 500 ml round bottom flask equipped with a magnetic stir bar, dropping funnel and thermometer, 90 g (1.5 mol) of urea was dissolved in 300 ml of water, followed by cooling to 0 ° C. under cooling at 0 ° C. 22 0.5 g of 35% hydrochloric acid (0.22 mol) was added with stirring. Further, the solution was adjusted with 35% hydrochloric acid so that the pH was 1. To the solution was added 19.6 g ammonium bromide (0.2 mol) and 23.5 g NaCl (0.4 mol). After completion of the addition, the temperature of the reaction solution was raised to 40 ° C., and 140.5 g (1.24 mol) of 30% hydrogen peroxide was divided into two parts, half of which was added dropwise over 30 minutes, and stirred for 1 hour. Of hydrogen peroxide was added dropwise over 30 minutes, and the reaction was terminated by stirring for 5 hours. As the reaction progresses, a yellow solid is formed and becomes suspended in the reaction solution. After completion of the reaction, the solid is separated by filtration, washed with water and dried. The obtained azodicarbonamide was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR). The yield of azodicarbonamide from the consumed urea was 93.8%.
IR (KBr): 3337,3187cm -1 ( N-H stretching), 1743,1731cm -1 (C = O stretch), 1637cm -1 (N-H bending), 1369,1331cm -1 (C-N stretch ).
1 H-NMR (D6-DMSO): δ 8.02 (s, 2H, NH), 7.97 (s, 2H, NH).
実施例1-3
磁気撹拌棒と滴下漏斗と温度計とを備えた500mlの丸底フラスコ内で、水883gに尿素788g(13.1mol)を溶解し、続いて、0℃の冷却下、0℃に冷却した197gの35%HCl(1.891mol)を撹拌下添加した。当該溶液に44.7gのNH4Cl(836mmol)を添加した。更に40gのNH4Br(408mmol)の添加終了後、反応液をPH=1に調整する。反応液を35℃に上げて30%過酸化水素619g(5.46mol)を3分割し、3分の1量の30%過酸化水素を30分間で滴下した後1時間撹拌を行い、次いで3分の1の30%過酸化水素を30分間で滴下した後1時間撹拌を行い、残りの3分の1の30%過酸化水素を30分間で滴下した後5時間撹拌を行い反応を終了させる。反応の進行と共に黄色の個体が生成し、反応液に縣濁して来る。反応終了後固形物のろ過分離を行い、水で洗浄した後乾燥させる。得られたアゾジカルボンアミドは、赤外吸収スペクトル(IR)及びプロトン核磁気共鳴スペクトル(1H-NMR)により同定した。消費された尿素からのアゾジカルボンアミドの収率は、93.6%であった。
IR(KBr):3337、3187cm-1(N-H伸縮)、1743,1731cm-1(C=O伸縮)、1637cm-1(N-H変角)、1369,1331cm-1(C-N伸縮).
1H-NMR(D6-DMSO):δ8.02(s,2H,NH),7.97(s,2H,NH). Example 1-3
In a 500 ml round bottom flask equipped with a magnetic stir bar, dropping funnel and thermometer, 788 g (13.1 mol) of urea was dissolved in 883 g of water, followed by 197 g of 0 ° C. cooled to 0 ° C. Of 35% HCl (1.891 mol) was added with stirring. To the solution was added 44.7 g NH 4 Cl (836 mmol). Further, after the addition of 40 g of NH 4 Br (408 mmol) is completed, the reaction solution is adjusted to PH = 1. The reaction solution was raised to 35 ° C., and 619 g (5.46 mol) of 30% hydrogen peroxide was divided into three parts. A third amount of 30% hydrogen peroxide was added dropwise over 30 minutes, followed by stirring for 1 hour. 1/3 of 30% hydrogen peroxide was added dropwise over 30 minutes and stirred for 1 hour. The remaining 1/3 of 30% hydrogen peroxide was added dropwise over 30 minutes and stirred for 5 hours to complete the reaction. . As the reaction progresses, a yellow solid is formed and becomes suspended in the reaction solution. After completion of the reaction, the solid is separated by filtration, washed with water and dried. The obtained azodicarbonamide was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR). The yield of azodicarbonamide from the consumed urea was 93.6%.
IR (KBr): 3337,3187cm -1 ( N-H stretching), 1743,1731cm -1 (C = O stretch), 1637cm -1 (N-H bending), 1369,1331cm -1 (C-N stretch ).
1 H-NMR (D6-DMSO): δ 8.02 (s, 2H, NH), 7.97 (s, 2H, NH).
磁気撹拌棒と滴下漏斗と温度計とを備えた500mlの丸底フラスコ内で、水883gに尿素788g(13.1mol)を溶解し、続いて、0℃の冷却下、0℃に冷却した197gの35%HCl(1.891mol)を撹拌下添加した。当該溶液に44.7gのNH4Cl(836mmol)を添加した。更に40gのNH4Br(408mmol)の添加終了後、反応液をPH=1に調整する。反応液を35℃に上げて30%過酸化水素619g(5.46mol)を3分割し、3分の1量の30%過酸化水素を30分間で滴下した後1時間撹拌を行い、次いで3分の1の30%過酸化水素を30分間で滴下した後1時間撹拌を行い、残りの3分の1の30%過酸化水素を30分間で滴下した後5時間撹拌を行い反応を終了させる。反応の進行と共に黄色の個体が生成し、反応液に縣濁して来る。反応終了後固形物のろ過分離を行い、水で洗浄した後乾燥させる。得られたアゾジカルボンアミドは、赤外吸収スペクトル(IR)及びプロトン核磁気共鳴スペクトル(1H-NMR)により同定した。消費された尿素からのアゾジカルボンアミドの収率は、93.6%であった。
IR(KBr):3337、3187cm-1(N-H伸縮)、1743,1731cm-1(C=O伸縮)、1637cm-1(N-H変角)、1369,1331cm-1(C-N伸縮).
1H-NMR(D6-DMSO):δ8.02(s,2H,NH),7.97(s,2H,NH). Example 1-3
In a 500 ml round bottom flask equipped with a magnetic stir bar, dropping funnel and thermometer, 788 g (13.1 mol) of urea was dissolved in 883 g of water, followed by 197 g of 0 ° C. cooled to 0 ° C. Of 35% HCl (1.891 mol) was added with stirring. To the solution was added 44.7 g NH 4 Cl (836 mmol). Further, after the addition of 40 g of NH 4 Br (408 mmol) is completed, the reaction solution is adjusted to PH = 1. The reaction solution was raised to 35 ° C., and 619 g (5.46 mol) of 30% hydrogen peroxide was divided into three parts. A third amount of 30% hydrogen peroxide was added dropwise over 30 minutes, followed by stirring for 1 hour. 1/3 of 30% hydrogen peroxide was added dropwise over 30 minutes and stirred for 1 hour. The remaining 1/3 of 30% hydrogen peroxide was added dropwise over 30 minutes and stirred for 5 hours to complete the reaction. . As the reaction progresses, a yellow solid is formed and becomes suspended in the reaction solution. After completion of the reaction, the solid is separated by filtration, washed with water and dried. The obtained azodicarbonamide was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR). The yield of azodicarbonamide from the consumed urea was 93.6%.
IR (KBr): 3337,3187cm -1 ( N-H stretching), 1743,1731cm -1 (C = O stretch), 1637cm -1 (N-H bending), 1369,1331cm -1 (C-N stretch ).
1 H-NMR (D6-DMSO): δ 8.02 (s, 2H, NH), 7.97 (s, 2H, NH).
実施例1-4
磁気撹拌棒と滴下漏斗と温度計とを備えた500mlの丸底フラスコ内で、水300mlに尿素90g(1.5mol)を溶解し、続いて、0℃の冷却下、0℃に冷却した22.5gの35%塩酸(0.22mol)を撹拌下添加した。更に溶液がPH=2になる様に35%塩酸で調整を行った。当該溶液に19.6gの臭化アンモニウム(0.2mol)と47gのNaCl(0.8mol)を添加した。これにギ酸2.3g(0.05mol)を添加する。全ての添加終了後反応液の温度を40℃に上げて、30%過酸化水素231.2g(2.04mol)を2分割し、その半分を30分間で滴下し、1時間撹拌を行った後、残りの過酸化水素を30分間で滴下し、5時間撹拌を行い反応を終了させた。反応の進行と共に黄色の個体が生成し、反応液に縣濁して来る。反応終了後固形物のろ過分離を行い、水で洗浄した後乾燥させる。得られたアゾジカルボンアミドは、赤外吸収スペクトル(IR)及びプロトン核磁気共鳴スペクトル(1H-NMR)により同定した。消費された尿素からのアゾジカルボンアミドの収率は、94.1%であった。
IR(KBr):3337、3187cm-1(N-H伸縮)、1743,1731cm-1(C=O伸縮)、1637cm-1(N-H変角)、1369,1331cm-1(C-N伸縮).
1H-NMR(D6-DMSO):δ8.02(s,2H,NH),7.97(s,2H,NH). Example 1-4
In a 500 ml round bottom flask equipped with a magnetic stir bar, dropping funnel and thermometer, 90 g (1.5 mol) of urea was dissolved in 300 ml of water, followed by cooling to 0 ° C. under cooling at 0 ° C. 22 0.5 g of 35% hydrochloric acid (0.22 mol) was added with stirring. Furthermore, adjustment was performed with 35% hydrochloric acid so that the solution had a pH = 2. To the solution was added 19.6 g ammonium bromide (0.2 mol) and 47 g NaCl (0.8 mol). To this is added 2.3 g (0.05 mol) of formic acid. After all the additions were completed, the temperature of the reaction solution was raised to 40 ° C., 231.2 g (2.04 mol) of 30% hydrogen peroxide was divided into two portions, half of which was added dropwise over 30 minutes, and stirred for 1 hour. The remaining hydrogen peroxide was added dropwise over 30 minutes, and the reaction was terminated by stirring for 5 hours. As the reaction progresses, a yellow solid is formed and becomes suspended in the reaction solution. After completion of the reaction, the solid is separated by filtration, washed with water and dried. The obtained azodicarbonamide was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR). The yield of azodicarbonamide from the consumed urea was 94.1%.
IR (KBr): 3337,3187cm -1 ( N-H stretching), 1743,1731cm -1 (C = O stretch), 1637cm -1 (N-H bending), 1369,1331cm -1 (C-N stretch ).
1 H-NMR (D6-DMSO): δ 8.02 (s, 2H, NH), 7.97 (s, 2H, NH).
磁気撹拌棒と滴下漏斗と温度計とを備えた500mlの丸底フラスコ内で、水300mlに尿素90g(1.5mol)を溶解し、続いて、0℃の冷却下、0℃に冷却した22.5gの35%塩酸(0.22mol)を撹拌下添加した。更に溶液がPH=2になる様に35%塩酸で調整を行った。当該溶液に19.6gの臭化アンモニウム(0.2mol)と47gのNaCl(0.8mol)を添加した。これにギ酸2.3g(0.05mol)を添加する。全ての添加終了後反応液の温度を40℃に上げて、30%過酸化水素231.2g(2.04mol)を2分割し、その半分を30分間で滴下し、1時間撹拌を行った後、残りの過酸化水素を30分間で滴下し、5時間撹拌を行い反応を終了させた。反応の進行と共に黄色の個体が生成し、反応液に縣濁して来る。反応終了後固形物のろ過分離を行い、水で洗浄した後乾燥させる。得られたアゾジカルボンアミドは、赤外吸収スペクトル(IR)及びプロトン核磁気共鳴スペクトル(1H-NMR)により同定した。消費された尿素からのアゾジカルボンアミドの収率は、94.1%であった。
IR(KBr):3337、3187cm-1(N-H伸縮)、1743,1731cm-1(C=O伸縮)、1637cm-1(N-H変角)、1369,1331cm-1(C-N伸縮).
1H-NMR(D6-DMSO):δ8.02(s,2H,NH),7.97(s,2H,NH). Example 1-4
In a 500 ml round bottom flask equipped with a magnetic stir bar, dropping funnel and thermometer, 90 g (1.5 mol) of urea was dissolved in 300 ml of water, followed by cooling to 0 ° C. under cooling at 0 ° C. 22 0.5 g of 35% hydrochloric acid (0.22 mol) was added with stirring. Furthermore, adjustment was performed with 35% hydrochloric acid so that the solution had a pH = 2. To the solution was added 19.6 g ammonium bromide (0.2 mol) and 47 g NaCl (0.8 mol). To this is added 2.3 g (0.05 mol) of formic acid. After all the additions were completed, the temperature of the reaction solution was raised to 40 ° C., 231.2 g (2.04 mol) of 30% hydrogen peroxide was divided into two portions, half of which was added dropwise over 30 minutes, and stirred for 1 hour. The remaining hydrogen peroxide was added dropwise over 30 minutes, and the reaction was terminated by stirring for 5 hours. As the reaction progresses, a yellow solid is formed and becomes suspended in the reaction solution. After completion of the reaction, the solid is separated by filtration, washed with water and dried. The obtained azodicarbonamide was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR). The yield of azodicarbonamide from the consumed urea was 94.1%.
IR (KBr): 3337,3187cm -1 ( N-H stretching), 1743,1731cm -1 (C = O stretch), 1637cm -1 (N-H bending), 1369,1331cm -1 (C-N stretch ).
1 H-NMR (D6-DMSO): δ 8.02 (s, 2H, NH), 7.97 (s, 2H, NH).
実施例1-5
磁気撹拌棒と滴下漏斗と温度計とを備えた500mlの丸底フラスコ内で、水300mlに尿素90g(1.5mol)を溶解し、続いて、0℃の冷却下、0℃に冷却した22.5gの35%塩酸(0.22mol)を撹拌下添加した。更に溶液がPH=1になる様に35%塩酸で調整を行った。当該溶液に1.96gの臭化アンモニウム(0.02mol)と23.5gのNaCl(0.4mol)を添加した。これにSiC多孔質粒子にフッ化アルミニウム微粒子を焼結担持した触媒をフッ化アルミニウムとして0.84g(10mmol)添加分散して、全ての添加終了後反応液の温度を40℃に上げて、30%過酸化水素140.5g(1.24mol)を2分割し、その半分を30分間で滴下し、1時間撹拌を行った後、残りの過酸化水素を30分間で滴下し、5時間撹拌を行い反応を終了させた。反応の進行と共に黄色の個体が生成し、反応液に縣濁して来る。反応終了後触媒を濾別し、次いで固形物のADCAのろ過分離を行い、水で洗浄した後乾燥させる。得られたアゾジカルボンアミドは、赤外吸収スペクトル(IR)及びプロトン核磁気共鳴スペクトル(1H-NMR)により同定した。消費された尿素からのアゾジカルボンアミドの収率は、94.1%であった。
IR(KBr):3337、3187cm-1(N-H伸縮)、1743,1731cm-1(C=O伸縮)、1637cm-1(N-H変角)、1369,1331cm-1(C-N伸縮).
1H-NMR(D6-DMSO):δ8.02(s,2H,NH),7.97(s,2H,NH). Example 1-5
In a 500 ml round bottom flask equipped with a magnetic stir bar, dropping funnel and thermometer, 90 g (1.5 mol) of urea was dissolved in 300 ml of water, followed by cooling to 0 ° C. under cooling at 0 ° C. 22 0.5 g of 35% hydrochloric acid (0.22 mol) was added with stirring. Further, the solution was adjusted with 35% hydrochloric acid so that the pH was 1. To the solution were added 1.96 g ammonium bromide (0.02 mol) and 23.5 g NaCl (0.4 mol). To this, 0.84 g (10 mmol) of aluminum fluoride fine particles sintered and supported on SiC porous particles was added and dispersed as aluminum fluoride, and the temperature of the reaction solution was raised to 40 ° C. after the completion of all additions. 140.5 g (1.24 mol) of% hydrogen peroxide was divided into two parts, half of which was added dropwise over 30 minutes and stirred for 1 hour, and then the remaining hydrogen peroxide was added dropwise over 30 minutes and stirred for 5 hours. The reaction was terminated. As the reaction progresses, a yellow solid is formed and becomes suspended in the reaction solution. After completion of the reaction, the catalyst is filtered off, then solid ADCA is separated by filtration, washed with water and dried. The obtained azodicarbonamide was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR). The yield of azodicarbonamide from the consumed urea was 94.1%.
IR (KBr): 3337,3187cm -1 ( N-H stretching), 1743,1731cm -1 (C = O stretch), 1637cm -1 (N-H bending), 1369,1331cm -1 (C-N stretch ).
1 H-NMR (D6-DMSO): δ 8.02 (s, 2H, NH), 7.97 (s, 2H, NH).
磁気撹拌棒と滴下漏斗と温度計とを備えた500mlの丸底フラスコ内で、水300mlに尿素90g(1.5mol)を溶解し、続いて、0℃の冷却下、0℃に冷却した22.5gの35%塩酸(0.22mol)を撹拌下添加した。更に溶液がPH=1になる様に35%塩酸で調整を行った。当該溶液に1.96gの臭化アンモニウム(0.02mol)と23.5gのNaCl(0.4mol)を添加した。これにSiC多孔質粒子にフッ化アルミニウム微粒子を焼結担持した触媒をフッ化アルミニウムとして0.84g(10mmol)添加分散して、全ての添加終了後反応液の温度を40℃に上げて、30%過酸化水素140.5g(1.24mol)を2分割し、その半分を30分間で滴下し、1時間撹拌を行った後、残りの過酸化水素を30分間で滴下し、5時間撹拌を行い反応を終了させた。反応の進行と共に黄色の個体が生成し、反応液に縣濁して来る。反応終了後触媒を濾別し、次いで固形物のADCAのろ過分離を行い、水で洗浄した後乾燥させる。得られたアゾジカルボンアミドは、赤外吸収スペクトル(IR)及びプロトン核磁気共鳴スペクトル(1H-NMR)により同定した。消費された尿素からのアゾジカルボンアミドの収率は、94.1%であった。
IR(KBr):3337、3187cm-1(N-H伸縮)、1743,1731cm-1(C=O伸縮)、1637cm-1(N-H変角)、1369,1331cm-1(C-N伸縮).
1H-NMR(D6-DMSO):δ8.02(s,2H,NH),7.97(s,2H,NH). Example 1-5
In a 500 ml round bottom flask equipped with a magnetic stir bar, dropping funnel and thermometer, 90 g (1.5 mol) of urea was dissolved in 300 ml of water, followed by cooling to 0 ° C. under cooling at 0 ° C. 22 0.5 g of 35% hydrochloric acid (0.22 mol) was added with stirring. Further, the solution was adjusted with 35% hydrochloric acid so that the pH was 1. To the solution were added 1.96 g ammonium bromide (0.02 mol) and 23.5 g NaCl (0.4 mol). To this, 0.84 g (10 mmol) of aluminum fluoride fine particles sintered and supported on SiC porous particles was added and dispersed as aluminum fluoride, and the temperature of the reaction solution was raised to 40 ° C. after the completion of all additions. 140.5 g (1.24 mol) of% hydrogen peroxide was divided into two parts, half of which was added dropwise over 30 minutes and stirred for 1 hour, and then the remaining hydrogen peroxide was added dropwise over 30 minutes and stirred for 5 hours. The reaction was terminated. As the reaction progresses, a yellow solid is formed and becomes suspended in the reaction solution. After completion of the reaction, the catalyst is filtered off, then solid ADCA is separated by filtration, washed with water and dried. The obtained azodicarbonamide was identified by infrared absorption spectrum (IR) and proton nuclear magnetic resonance spectrum ( 1 H-NMR). The yield of azodicarbonamide from the consumed urea was 94.1%.
IR (KBr): 3337,3187cm -1 ( N-H stretching), 1743,1731cm -1 (C = O stretch), 1637cm -1 (N-H bending), 1369,1331cm -1 (C-N stretch ).
1 H-NMR (D6-DMSO): δ 8.02 (s, 2H, NH), 7.97 (s, 2H, NH).
実施例2~14
臭化ナトリウムに替えて他のハロゲン化剤を使用した以外は、実施例1-3に記載の方法と同様に反応及び処理を行った。

Examples 2-14
The reaction and treatment were carried out in the same manner as in Example 1-3 except that another halogenating agent was used instead of sodium bromide.
臭化ナトリウムに替えて他のハロゲン化剤を使用した以外は、実施例1-3に記載の方法と同様に反応及び処理を行った。
The reaction and treatment were carried out in the same manner as in Example 1-3 except that another halogenating agent was used instead of sodium bromide.
実施例15~26
最初の酸化剤を次亜塩素酸ソーダから理論量より相当少ない量の臭素、臭化塩、臭素酸塩に変換して最初に反応させ、その後過酸化水素を添加して臭化水素になっている物を臭素又は臭素酸又は臭素酸塩に変換して臭素、臭化塩、臭素酸塩をリサイクル反応させるに対応する過酸化水素の2倍量使用する以外は、実施例1-3と同様の反応及び処理を行った。

Examples 15-26
The first oxidant is converted from sodium hypochlorite to bromine, bromide and bromate in amounts considerably smaller than the theoretical amount, and reacted first, and then hydrogen peroxide is added to form hydrogen bromide. Except for using 2 times the amount of hydrogen peroxide corresponding to recycling reaction of bromine, bromide or bromate after converting the product to bromine or bromic acid or bromate, the same as Example 1-3 The reaction and treatment were carried out.
最初の酸化剤を次亜塩素酸ソーダから理論量より相当少ない量の臭素、臭化塩、臭素酸塩に変換して最初に反応させ、その後過酸化水素を添加して臭化水素になっている物を臭素又は臭素酸又は臭素酸塩に変換して臭素、臭化塩、臭素酸塩をリサイクル反応させるに対応する過酸化水素の2倍量使用する以外は、実施例1-3と同様の反応及び処理を行った。
The first oxidant is converted from sodium hypochlorite to bromine, bromide and bromate in amounts considerably smaller than the theoretical amount, and reacted first, and then hydrogen peroxide is added to form hydrogen bromide. Except for using 2 times the amount of hydrogen peroxide corresponding to recycling reaction of bromine, bromide or bromate after converting the product to bromine or bromic acid or bromate, the same as Example 1-3 The reaction and treatment were carried out.
実施例27~31
実施例1-1において最初の酸化剤である次亜塩素酸ソーダ等の酸化剤を使用せず最初に少量のNaBrを添加して、溶解した総クロルアニオンのモル数の2倍量に相当量する過酸化水素を添加して反応及び処理を行って、生成したアゾジカルボンアミドを分離して、その母液及びアゾジカルボンアミド分離物の洗浄液を併せて、消費した尿素を添加し、またNaBrが、固形物に付着して減少量が問題になった場合、これ等を添加して組成を調整しながらリサイクル反応を行う。また水の量が増加して来るので適当に膜等を使用して水の除去を行う。それ以外の反応と処理は、実施例1-1と同様に行う。

Examples 27-31
In Example 1-1, an oxidizing agent such as sodium hypochlorite that is the first oxidizing agent is not used, but a small amount of NaBr is added first, and the amount is equivalent to twice the number of moles of dissolved total chloranion. The hydrogen peroxide to be added is reacted and treated to separate the produced azodicarbonamide, the mother liquor and the washing solution of the azodicarbonamide isolate are combined, the consumed urea is added, and NaBr is When the amount of reduction becomes a problem due to adhesion to solids, the recycling reaction is performed while adjusting the composition by adding these. Moreover, since the amount of water increases, water is removed using a membrane or the like. Other reactions and treatments are carried out in the same manner as in Example 1-1.
実施例1-1において最初の酸化剤である次亜塩素酸ソーダ等の酸化剤を使用せず最初に少量のNaBrを添加して、溶解した総クロルアニオンのモル数の2倍量に相当量する過酸化水素を添加して反応及び処理を行って、生成したアゾジカルボンアミドを分離して、その母液及びアゾジカルボンアミド分離物の洗浄液を併せて、消費した尿素を添加し、またNaBrが、固形物に付着して減少量が問題になった場合、これ等を添加して組成を調整しながらリサイクル反応を行う。また水の量が増加して来るので適当に膜等を使用して水の除去を行う。それ以外の反応と処理は、実施例1-1と同様に行う。
In Example 1-1, an oxidizing agent such as sodium hypochlorite that is the first oxidizing agent is not used, but a small amount of NaBr is added first, and the amount is equivalent to twice the number of moles of dissolved total chloranion. The hydrogen peroxide to be added is reacted and treated to separate the produced azodicarbonamide, the mother liquor and the washing solution of the azodicarbonamide isolate are combined, the consumed urea is added, and NaBr is When the amount of reduction becomes a problem due to adhesion to solids, the recycling reaction is performed while adjusting the composition by adding these. Moreover, since the amount of water increases, water is removed using a membrane or the like. Other reactions and treatments are carried out in the same manner as in Example 1-1.
実施例32
2枚の白金板電極(1.5×1.0cm2)を取り付けたビーカー型電解セルに、尿素(1.2g、20mmol)、臭化ナトリウム(41mg、0.4mmol)、5℃に冷却した0.34mgの32%HCl(0.3mmol)、水(2.0g)を秤りとり、かき混ぜて均一溶液とした。電解セルを氷水浴に浸して冷却しつつ、100mAに一定に保ちながら10.7時間電解を行い、5778クーロンの電気量を通電した。反応終了後、生成する沈殿物をろ過により分取し、水洗、乾燥後、目的のアゾジカルボンアミド(0.32g、変換率37%、収率74%) が、淡橙色の結晶として得られた。高速液体クロマトグラフィーにより、ろ液の分析を行ったところ、ろ液中に未反応の尿素(0.746g)が確認された。 Example 32
Urea (1.2 g, 20 mmol), sodium bromide (41 mg, 0.4 mmol), cooled to 5 ° C. in a beaker-type electrolytic cell equipped with two platinum plate electrodes (1.5 × 1.0 cm 2 ). 0.34 mg of 32% HCl (0.3 mmol) and water (2.0 g) were weighed and stirred to obtain a homogeneous solution. While the electrolytic cell was immersed in an ice-water bath and cooled, electrolysis was performed for 10.7 hours while keeping the current constant at 100 mA, and 5778 coulombs of electricity was applied. After completion of the reaction, the resulting precipitate was collected by filtration, washed with water and dried, and the target azodicarbonamide (0.32 g, conversion rate 37%, yield 74%) was obtained as pale orange crystals. . When the filtrate was analyzed by high performance liquid chromatography, unreacted urea (0.746 g) was confirmed in the filtrate.
2枚の白金板電極(1.5×1.0cm2)を取り付けたビーカー型電解セルに、尿素(1.2g、20mmol)、臭化ナトリウム(41mg、0.4mmol)、5℃に冷却した0.34mgの32%HCl(0.3mmol)、水(2.0g)を秤りとり、かき混ぜて均一溶液とした。電解セルを氷水浴に浸して冷却しつつ、100mAに一定に保ちながら10.7時間電解を行い、5778クーロンの電気量を通電した。反応終了後、生成する沈殿物をろ過により分取し、水洗、乾燥後、目的のアゾジカルボンアミド(0.32g、変換率37%、収率74%) が、淡橙色の結晶として得られた。高速液体クロマトグラフィーにより、ろ液の分析を行ったところ、ろ液中に未反応の尿素(0.746g)が確認された。 Example 32
Urea (1.2 g, 20 mmol), sodium bromide (41 mg, 0.4 mmol), cooled to 5 ° C. in a beaker-type electrolytic cell equipped with two platinum plate electrodes (1.5 × 1.0 cm 2 ). 0.34 mg of 32% HCl (0.3 mmol) and water (2.0 g) were weighed and stirred to obtain a homogeneous solution. While the electrolytic cell was immersed in an ice-water bath and cooled, electrolysis was performed for 10.7 hours while keeping the current constant at 100 mA, and 5778 coulombs of electricity was applied. After completion of the reaction, the resulting precipitate was collected by filtration, washed with water and dried, and the target azodicarbonamide (0.32 g, conversion rate 37%, yield 74%) was obtained as pale orange crystals. . When the filtrate was analyzed by high performance liquid chromatography, unreacted urea (0.746 g) was confirmed in the filtrate.
実施例33
参考例3で生成したモノブロム尿素を実施例32の尿素に替えて原料にして、実施例32に記載した方法と同様に電解を行った。モノブロム尿素は282mg(2mmol)を使用した所213mgのアゾジカルボンアミドを得た。収率92.0%である。但し通電時間、通電電気量は、半分で電解反応を行う。アゾジカルボンアミドの固形物が生成する。懸濁液をろ過して、水洗して減圧乾燥した。 Example 33
Electrolysis was carried out in the same manner as described in Example 32 using the monobromourea produced in Reference Example 3 as a raw material instead of the urea in Example 32. When 282 mg (2 mmol) of monobromourea was used, 213 mg of azodicarbonamide was obtained. The yield is 92.0%. However, the electrolysis reaction is carried out with half the energizing time and the energizing electricity. A solid form of azodicarbonamide is formed. The suspension was filtered, washed with water and dried under reduced pressure.
参考例3で生成したモノブロム尿素を実施例32の尿素に替えて原料にして、実施例32に記載した方法と同様に電解を行った。モノブロム尿素は282mg(2mmol)を使用した所213mgのアゾジカルボンアミドを得た。収率92.0%である。但し通電時間、通電電気量は、半分で電解反応を行う。アゾジカルボンアミドの固形物が生成する。懸濁液をろ過して、水洗して減圧乾燥した。 Example 33
Electrolysis was carried out in the same manner as described in Example 32 using the monobromourea produced in Reference Example 3 as a raw material instead of the urea in Example 32. When 282 mg (2 mmol) of monobromourea was used, 213 mg of azodicarbonamide was obtained. The yield is 92.0%. However, the electrolysis reaction is carried out with half the energizing time and the energizing electricity. A solid form of azodicarbonamide is formed. The suspension was filtered, washed with water and dried under reduced pressure.
実施例34~52

Examples 34-52
Claims (12)
- 水、有機溶媒、及びイオン液体からなる群より選択される少なくとも1つの溶媒の均一系又は不均一な混合系において、尿素から誘導される式(10)で表される化合物から脱臭化水素反応を特徴とする式(7)で表されるアゾジカルボンアミドの製造方法。

- 水、有機溶媒、及びイオン液体からなる群より選択される少なくとも1つの溶媒の均一系又は不均一な混合系において、尿素から誘導される式(9)ブロム尿素活性遷移中間体と尿素を反応させて得られる式(10)で表される化合物を得る事を特徴とする、請求項1のアゾジカルボンアミドの製造方法。

- 水、有機溶媒、及びイオン液体からなる群より選択される少なくとも1つの溶媒の均一系又は不均一な混合系において、尿素から誘導される式(8)で表されるN-ブロム尿素を活性臭素化合物及び/又は過酸化水素由来活性体によるか、或いは電解で2電子酸化を行って式(9)ブロム尿素活性遷移中間体を得る事を特徴とする、請求項1又は2記載のアゾジカルボンアミドの製造方法。

- 水、有機溶媒、及びイオン液体からなる群より選択される少なくとも1つの溶媒の均一系又は不均一な混合系において、尿素から誘導される式(11)で表されるN-クロル尿素(N-クロル尿素Na塩を含む)をブロム化剤又はブロム化方法によるハロゲン置換反応により式(8)を得る事を特徴とする、請求項1~3のいずれか一項記載のアゾジカルボンアミドの製造方法。

- 水、有機溶媒、及びイオン液体からなる群より選択される少なくとも1つの溶媒の均一系又は不均一な混合系において、尿素から誘導される式(11)で表されるN-クロル尿素(N-クロル尿素Na塩を含む)をブロム化剤又はブロム化方法により式(8)で表されるN-ブロム尿素を生成し、次いで活性臭素化合物又は、及び過酸化水素由来活性体によるか又は電解でブロム原子が結合するNH基の求電子酸化により式(9)で表されるブロム尿素活性遷移中間体を得て、同時に尿素分子が反応し式(10)の化合物を得て、式(10)の化合物は、化学的に活性であり、即時に脱HBr反応を生じて式(7)の化合物に変換することを特徴とする、請求項1~4のいずれか一項記載のアゾジカルボンアミド式の新規製造方法。

- 水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、電解酸化反応を利用する場合、支持電解質及びブロム化剤及び活性臭素化化合物の原料として、臭化水素、ブロム塩を使用して2電子酸化を行い、ブロムカチオンを生成させて式(8)で表されるN-ブロム尿素を生成し、次いでブロム原子が結合するNH基の求電子酸化を電解反応により2電子酸化してH:をプロトン化し、式(9)ブロム尿素活性遷移中間体とし、同時に尿素分子が反応し式(10)で表される化合物が生成する。式(10)の化合物は、化学的に活性であり、即時に脱HBr反応を生じて式(7)の化合物に変換する事を特徴とするアゾジカルボンアミド式の新規製造方法。 When using an electrolytic oxidation reaction in water or an organic solvent or ionic liquid, and an organic solvent or a homogeneous or heterogeneous mixed system of ionic liquid and water, the supporting electrolyte, brominating agent, and active brominated compound Two-electron oxidation is performed using hydrogen bromide and bromo salt as raw materials to generate a bromo cation to generate N-bromourea represented by the formula (8), and then the NH group to which the bromo atom is bonded. Electrophilic oxidation is two-electron-oxidized by an electrolytic reaction to protonate H: to form a formula (9) bromourea active transition intermediate, and at the same time, urea molecules react to produce a compound represented by formula (10). A novel process for producing an azodicarbonamide formula, wherein the compound of the formula (10) is chemically active and immediately undergoes a de-HBr reaction to be converted into the compound of the formula (7).
- 水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、尿素からブロム化剤又はブロム化方法により式(8)で表されるN-ブロム尿素を生成し、又は必要に寄っては、尿素から塩素化剤又は塩素化方法により、式(11)で示されるN-クロル尿素(N-クロル尿素Na塩を含む)を生成し、次いでブロム化剤又はブロム化方法により式(8)で表されるN-ブロム尿素を生成し、次いでN-ブロム尿素を電解反応を利用して支持電解質として臭化水素、ブロム塩を使用して、ブロム原子が結合するNH基の2電子酸化を行いH:をプロトンとして脱離して、式(9)ブロム尿素活性遷移中間体とし、同時に尿素分子が反応し式(10)の化合物を得て、式(10)の化合物は、化学的に活性であり、即時に脱HBr反応を生じて式(7)の化合物に変換することを特徴とするアゾジカルボンアミド式(7)の新規製造方法。 In a homogeneous or heterogeneous mixed system of water or organic solvent or ionic liquid, and organic solvent or ionic liquid and water, N— represented by the formula (8) from urea by a brominating agent or bromination method Producing bromourea or, if necessary, producing N-chlorourea (including N-chlorourea Na salt) of formula (11) from urea by chlorinating agent or chlorination method, N-bromourea represented by the formula (8) is produced by a brominating agent or bromination method, and then N-bromourea is used as a supporting electrolyte using an electrolysis reaction, using hydrogen bromide and bromide salt, Two-electron oxidation of the NH group to which the bromo atom is bonded is performed, and H: is eliminated as a proton to obtain a formula (9) bromourea active transition intermediate, and at the same time a urea molecule reacts to obtain a compound of the formula (10), The compound of formula (10) is Is chemically active, novel method for producing azodicarbonamide type, characterized in that occur de HBr reaction immediately converted to compounds of formula (7) (7).
- 水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、臭化水素、ブロム塩を使用してこれに過酸化水素等の過酸化物を滴下する事によって臭素を発生させ、発生、変換された臭素、次亜臭素酸、次亜臭素酸塩によって式(1)で表される尿素がブロムカチオンによって2電子酸化されて式(8)で表されるN-ブロム尿素を生成する事を特徴とする、請求項1~6のいずれか一項記載のアゾジカルボンアミドの新規製造方法。 In water or organic solvent or ionic liquid, and in organic solvent or homogeneous or heterogeneous mixed system of ionic liquid and water, hydrogen bromide, bromide salt is used and peroxide such as hydrogen peroxide is used. Bromine is generated by dropwise addition of urea, and the urea represented by the formula (1) is generated by the generated, converted bromine, hypobromite, and hypobromite, and the two-electron oxidation of the urea represented by the formula (8) The novel process for producing azodicarbonamide according to any one of claims 1 to 6, wherein N-bromourea represented by the formula:
- 水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、有機ブロム化剤及びブロム‐ジオキサン錯体及びクロル臭素を酸化剤、活性臭素化化合物発生剤として、尿素がブロムカチオンによって2電子酸化されて式(8)で表されるN-ブロム尿素を生成する事を特徴とする、請求項1~7のいずれか一項記載のアゾジカルボンアミドの新規製造方法。 Water or organic solvent or ionic liquid, and organic bromide, bromine-dioxane complex and chlorobromine as oxidizing agent, active brominated compound in homogeneous or heterogeneous mixed system of organic solvent or ionic liquid and water The azodicarbonamide according to any one of claims 1 to 7, wherein urea is two-electron-oxidized with a bromo cation to form N-bromourea represented by the formula (8) as a generator. New manufacturing method.
- 水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、原料として尿素の臭化水素、塩化水素等のハロゲン酸塩を用いて、これに過酸化水素等の過酸化物を滴下する事によって臭素を発生させ、発生、変換された塩素、次亜塩素酸、次亜塩素酸塩、臭素、次亜臭素酸、次亜臭素酸塩によって式(1)で表される尿素がクロル化、次いでブロム化される事を特徴とする、請求項1~8のいずれか一項記載のアゾジカルボンアミドの新規製造方法。 In a homogeneous system or heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water, a halogen salt such as hydrogen bromide or hydrogen chloride of urea is used as a raw material. Bromine is generated by dropping a peroxide such as hydrogen peroxide on the surface, and generated, converted chlorine, hypochlorous acid, hypochlorite, bromine, hypobromite, hypobromite The novel process for producing azodicarbonamide according to any one of claims 1 to 8, wherein the urea represented by the formula (1) is chlorinated and then brominated.
- 水又は有機溶媒又はイオン液体、及び有機溶媒又は、及びイオン液体と水との均一系又は不均一な混合系において、原料として尿素及び尿素の塩の混合物を使用する事を特徴とする、請求項1~10のいずれか一項記載のアゾジカルボンアミドの新規製造方法。 A mixture of urea and a salt of urea is used as a raw material in a homogeneous system or a heterogeneous mixed system of water or an organic solvent or ionic liquid, and an organic solvent or ionic liquid and water. 11. A novel process for producing azodicarbonamide according to any one of 1 to 10.
- 酸性条件下、大過剰の尿素を使用し、80℃以下の低温更に-10~60℃の間で反応条件に合わせて温度選択を行う事及び中間体の酸化に電解反応が入る場合、電解工程は、-20~40℃で反応を行う事を特徴とする、請求項1~11のいずれか一項記載のアゾジカルボンアミドの新規製造方法。 Under acidic conditions, use a large excess of urea, select a temperature according to the reaction conditions at a low temperature of 80 ° C or lower, and between -10 and 60 ° C. The novel process for producing azodicarbonamide according to any one of claims 1 to 11, wherein the reaction is carried out at -20 to 40 ° C.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016547809A JP6165349B2 (en) | 2014-09-12 | 2015-09-11 | New production method of azodicarbonamide |
| CN201580049311.XA CN106795105B (en) | 2014-09-12 | 2015-09-11 | The novel manufacturing method of azodicarbonamide |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JPPCT/JP2014/074315 | 2014-09-12 | ||
| JP2014074315 | 2014-09-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016039466A1 true WO2016039466A1 (en) | 2016-03-17 |
Family
ID=55459209
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/075926 WO2016039466A1 (en) | 2014-09-12 | 2015-09-11 | New production method for azodicarbonamide |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JP6165349B2 (en) |
| CN (1) | CN106795105B (en) |
| WO (1) | WO2016039466A1 (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102019111060A1 (en) * | 2019-04-29 | 2020-10-29 | Rheinisch-Westfälische Technische Hochschule (Rwth) Aachen | Catalysts for the catalytic synthesis of urea |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4886822A (en) * | 1972-02-23 | 1973-11-15 | ||
| JPS5217424A (en) * | 1975-07-31 | 1977-02-09 | Ugine Kuhlmann | Process for manufacturing azodicarbonamides from hydrazines |
| JPS52133924A (en) * | 1976-04-28 | 1977-11-09 | Otsuka Chem Co Ltd | Preparation of azodicarbonamides |
| JPS5398926A (en) * | 1977-02-09 | 1978-08-29 | Otsuka Chem Co Ltd | Preparation of azodicarbonamides |
| JP2002080449A (en) * | 2000-07-19 | 2002-03-19 | Bayer Ag | Method for producing hydrazodicarbonamide(hdc) via ketimine |
| JP2002080448A (en) * | 2000-08-31 | 2002-03-19 | Eiwa Kasei Kogyo Kk | Method for purifying hydrazodicarbonamide |
| WO2012147953A1 (en) * | 2011-04-28 | 2012-11-01 | 大塚化学株式会社 | Novel method for producing azodicarbonamide |
| JP2013540743A (en) * | 2010-09-21 | 2013-11-07 | ブロミン・コンパウンズ・リミテツド | Method for preparing bromourea |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3746300B2 (en) * | 1997-08-11 | 2006-02-15 | 株式会社荏原製作所 | Hydrothermal electrolysis method and apparatus |
| JP2004043900A (en) * | 2002-07-12 | 2004-02-12 | Permelec Electrode Ltd | Electrolytic synthesis method of peracetic acid |
| GB0918616D0 (en) * | 2009-10-23 | 2009-12-09 | 3M Innovative Properties Co | Method of preparing highly fluorinated carboxylic acids and their salts |
-
2015
- 2015-09-11 CN CN201580049311.XA patent/CN106795105B/en active Active
- 2015-09-11 JP JP2016547809A patent/JP6165349B2/en active Active
- 2015-09-11 WO PCT/JP2015/075926 patent/WO2016039466A1/en active Application Filing
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4886822A (en) * | 1972-02-23 | 1973-11-15 | ||
| JPS5217424A (en) * | 1975-07-31 | 1977-02-09 | Ugine Kuhlmann | Process for manufacturing azodicarbonamides from hydrazines |
| JPS52133924A (en) * | 1976-04-28 | 1977-11-09 | Otsuka Chem Co Ltd | Preparation of azodicarbonamides |
| JPS5398926A (en) * | 1977-02-09 | 1978-08-29 | Otsuka Chem Co Ltd | Preparation of azodicarbonamides |
| JP2002080449A (en) * | 2000-07-19 | 2002-03-19 | Bayer Ag | Method for producing hydrazodicarbonamide(hdc) via ketimine |
| JP2002080448A (en) * | 2000-08-31 | 2002-03-19 | Eiwa Kasei Kogyo Kk | Method for purifying hydrazodicarbonamide |
| JP2013540743A (en) * | 2010-09-21 | 2013-11-07 | ブロミン・コンパウンズ・リミテツド | Method for preparing bromourea |
| WO2012147953A1 (en) * | 2011-04-28 | 2012-11-01 | 大塚化学株式会社 | Novel method for producing azodicarbonamide |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2016039466A1 (en) | 2017-04-27 |
| JP6165349B2 (en) | 2017-07-19 |
| CN106795105B (en) | 2019-11-15 |
| CN106795105A (en) | 2017-05-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR20150056627A (en) | Integrated process for producing carboxylic acids from carbon dioxide | |
| JP2010501338A (en) | Process for oxidizing bromide to form bromine and catalyst useful therefor | |
| WO2007125975A1 (en) | Method for producing 1,2,3,4-tetrachlorohexafluorobutane | |
| CN111732520B (en) | Preparation method of 3-methyl-2-aminobenzoic acid | |
| US20220127144A1 (en) | Method for manufacturing sulfur tetrafluoride | |
| CN103360316A (en) | Preparation method of fipronil | |
| CN110753679A (en) | Method and system for forming chloropropanol and propylene oxide | |
| JP6165349B2 (en) | New production method of azodicarbonamide | |
| US6956142B2 (en) | Process for eco-friendly synthesis of bromobenzene | |
| JPH0238573B2 (en) | ||
| JP5378205B2 (en) | Electrochemical preparation method of halogenated carbonyl group-containing compound | |
| Raju et al. | Electrochemical chlorination of toluene by two-phase electrolysis | |
| JP3564979B2 (en) | Method for producing perfluoroalkylcarboxylic acid | |
| Deunf et al. | Direct electrochemical reduction of organic halide droplets dispersed in water | |
| CN107793384B (en) | Method for preparing 2, 5-dimethoxy-2, 5-dihydrofuran | |
| JP2604826B2 (en) | Highly selective oxidation of primary alcohols to aldehydes | |
| CN110565110B (en) | Synthetic method of 2,4, 5-trimethylchlorobenzene | |
| Sharma et al. | NH4Br–Br2 Catalysed Oxidative Bromination of Aromatic Compounds | |
| IL217393A (en) | Process for the preparation of an iodinating agent | |
| JPS6342711B2 (en) | ||
| Inês et al. | Electroepoxidation of natural and synthetic alkenes mediated by sodium bromide | |
| RU2601752C1 (en) | Method for bromination of xylenes into ring | |
| JPH0583533B2 (en) | ||
| JP4121497B2 (en) | Method for producing hydrazodicarbonamide using biuret as starting material | |
| JPH02107791A (en) | Oxidation of secondary alcohol to ketone |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15840196 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2016547809 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15840196 Country of ref document: EP Kind code of ref document: A1 |