WO2015163211A1 - Biological substance quantitation method, image processing device, pathological diagnosis support system, and image processing program - Google Patents
Biological substance quantitation method, image processing device, pathological diagnosis support system, and image processing program Download PDFInfo
- Publication number
- WO2015163211A1 WO2015163211A1 PCT/JP2015/061578 JP2015061578W WO2015163211A1 WO 2015163211 A1 WO2015163211 A1 WO 2015163211A1 JP 2015061578 W JP2015061578 W JP 2015061578W WO 2015163211 A1 WO2015163211 A1 WO 2015163211A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- image
- fluorescent
- bright spot
- biological material
- profile
- Prior art date
Links
Images




















Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N21/00—Investigating or analysing materials by the use of optical means, i.e. using sub-millimetre waves, infrared, visible or ultraviolet light
- G01N21/62—Systems in which the material investigated is excited whereby it emits light or causes a change in wavelength of the incident light
- G01N21/63—Systems in which the material investigated is excited whereby it emits light or causes a change in wavelength of the incident light optically excited
- G01N21/64—Fluorescence; Phosphorescence
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/483—Physical analysis of biological material
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/531—Production of immunochemical test materials
- G01N33/532—Production of labelled immunochemicals
- G01N33/533—Production of labelled immunochemicals with fluorescent label
Definitions
- the present invention relates to a biological material quantification method, an image processing device, a pathological diagnosis support system, and an image processing program.
- Patent Document 1 describes a method in which a biological material is stained with a fluorescent material such as an organic fluorescent dye or a quantum dot and measured using a three-dimensional image analysis apparatus. According to the method described in Patent Document 1, by reconstructing and analyzing a fluorescence image in three dimensions, a plurality of fluorescence signals that appear to overlap in two dimensions can be separated and counted one by one. .
- a fluorescent material such as an organic fluorescent dye or a quantum dot
- Patent Document 2 an average luminance value per particle is obtained by staining a tissue section using fluorescent dye-integrated particles to which a biological substance recognition site is bound, and analyzing the luminance distribution of fluorescent emission points.
- a method for calculating the number of particles in each bright spot has been proposed.
- fluorescent dye-aggregated particles have high luminance per particle. Therefore, even when the fluorescently stained biological material is a protein, the fluorescent signal is observed in the form of dots and the number of particles is calculated. can do.
- the main object of the present invention is to provide a biological material quantification method for accurately quantifying the amount of a specific biological material in a specimen using a simple microscope.
- a biological material quantification method for quantifying the biological material from a specimen stained with a specific biological material using fluorescent particles in which a plurality of fluorescent materials are accumulated as a staining reagent An input step of continuously changing a depth of focus at a predetermined interval, and inputting a fluorescence image representing the expression of the biological material in the specimen as a fluorescent luminescent spot at each depth of focus; A profile creation step of creating a bright spot image in which a bright spot region is extracted from the fluorescent image at each focal depth, and creating a brightness profile for each bright spot image; The brightness profile of the fluorescent particles measured in advance as a reference profile is created, and the brightness profile of the bright spot image at each focal depth is analyzed based on the reference profile, whereby the number of fluorescent particles included in the fluorescence image is determined.
- a calculation step to calculate, A method for quantifying a biological material is provided.
- the reference profile includes information on a relative distance from a fluorescent particle serving as a fluorescent bright spot source and luminance information.
- the position of the fluorescent particles included in the fluorescent image is specified,
- a method for quantifying a biological material is provided.
- a biological material quantification method in the biological material quantification method according to any one of claims 1 to 3, is provided, wherein the fluorescent particles have an average particle size of 20 to 200 nm.
- a biological material quantification method is provided, wherein the coefficient of variation of the particle size of the fluorescent particles is 15% or less.
- a biological material quantification method is provided, wherein the biological material is a protein.
- a biological material quantification method for quantifying the biological material from a specimen stained with a specific biological material using fluorescent particles in which a plurality of fluorescent materials are accumulated as a staining reagent
- Input means for continuously changing the depth of focus at a predetermined interval, and inputting a fluorescence image representing the expression of the biological material in the specimen as a fluorescence bright spot at each depth of focus
- a profile creating means for generating a bright spot image in which a bright spot region is extracted from the fluorescent image at each focal depth and creating a brightness profile for each bright spot image;
- the brightness profile of the fluorescent particles measured in advance as a reference profile is created, and the brightness profile of the bright spot image at each focal depth is analyzed based on the reference profile, whereby the number of fluorescent particles included in the fluorescence image is determined.
- An image processing apparatus is provided.
- An image processing apparatus according to claim 7; An image acquisition device for acquiring the fluorescent image used in the image processing device; A pathological diagnosis support system is provided.
- a computer for quantifying the biological material from a specimen stained with a specific biological material using fluorescent particles in which a plurality of fluorescent materials are accumulated as a staining reagent, Input means for continuously changing a focal depth at a predetermined interval, and inputting a fluorescence image representing the expression of the biological material in the specimen as a fluorescent luminescent spot at each focal depth; Profile creation means for generating a bright spot image from which a bright spot region is extracted from the fluorescent image at each focal depth, and creating a brightness profile for each bright spot image, The brightness profile of the fluorescent particles measured in advance as a reference profile is created, and the brightness profile of the bright spot image at each focal depth is analyzed based on the reference profile, whereby the number of fluorescent particles included in the fluorescence image is determined.
- a sample stained with fluorescent particles in which a plurality of fluorescent substances are accumulated is analyzed using images of fluorescence intensities using images obtained at a plurality of focal depths, and a simple microscope is used.
- the amount of a specific biological substance in the observation target cell can be accurately quantified.
- FIG. 5 S4 It is a figure which shows the system configuration
- FIG. 10B is an enlarged view of one bright spot region surrounded by a square in FIG. 10A. It is a fluorescence image of the site
- FIG. 10B is a second fluorescence image generated by masking the image of FIG. 10C with FIG. 10B. It is an example of the brightness
- luminance profile which shows the brightness
- FIG. 1 shows an example of the overall configuration of a pathological diagnosis support system 100 using the biological material quantification method of the present invention.
- the pathological diagnosis support system 100 acquires a microscopic image of a human tissue sample stained with a predetermined staining reagent, and analyzes the acquired microscopic image, thereby expressing the expression of a specific biological material in the tissue sample to be observed. This is a system that outputs feature quantities quantitatively.
- the pathological diagnosis support system 100 is configured by connecting a microscope image acquisition device 1A and an image processing device 2A through a cable 3A or the like so as to be able to transmit and receive data.
- the connection method between the microscope image acquisition device 1A and the image processing device 2A is not particularly limited.
- the microscope image acquisition device 1A and the image processing device 2A may be connected via a LAN (Local Area Network) or may be configured to be connected wirelessly.
- LAN Local Area Network
- the microscope image acquisition apparatus 1A is a known optical microscope with a camera, and acquires a microscope image of a tissue specimen on a slide placed on a slide fixing stage, and transmits the microscope image to the image processing apparatus 2A.
- the microscope image acquisition apparatus 1A includes an irradiation unit, an imaging unit, an imaging unit, a communication I / F, and the like.
- the irradiation means includes a light source, a filter, and the like, and irradiates the tissue specimen on the slide placed on the slide fixing stage with light.
- the imaging means is composed of an eyepiece lens, an objective lens, and the like, and forms an image of transmitted light, reflected light, or fluorescence emitted from the tissue specimen on the slide by the irradiated light.
- the imaging means is a microscope-installed camera that includes a CCD (Charge Coupled Device) sensor and the like, captures an image formed on the imaging surface by the imaging means, and generates digital image data of the microscope image.
- the communication I / F transmits image data of the generated microscope image to the image processing apparatus 2A.
- the microscope image acquisition apparatus 1A includes a bright field unit that combines an irradiation unit and an imaging unit suitable for bright field observation, and a fluorescence unit that combines an irradiation unit and an imaging unit suitable for fluorescence observation. It is possible to switch between bright field / fluorescence by switching units.
- a light source for fluorescence observation any lamp such as a mercury lamp, a xenon lamp, an LED, or a laser beam can be used.
- the microscope image acquisition apparatus 1A is not limited to a microscope with a camera.
- a virtual microscope slide creation apparatus for example, a special microscope microscope
- Table 2002-514319 may be used.
- the virtual microscope slide creation device it is possible to acquire image data that allows the entire tissue specimen image on the slide to be viewed on the display unit at a time.
- the image processing apparatus 2A calculates the expression distribution of a specific biological substance in the tissue specimen to be observed by analyzing the microscope image transmitted from the microscope image acquisition apparatus 1A.
- FIG. 2 shows a functional configuration example of the image processing apparatus 2A.
- the image processing apparatus 2 ⁇ / b> A includes a control unit 21, an operation unit 22, a display unit 23, a communication I / F 24, a storage unit 25, and the like, and each unit is connected via a bus 26. ing.
- the control unit 21 includes a CPU (Central Processing Unit), a RAM (Random Access Memory), and the like.
- the control unit 21 executes various processes in cooperation with various programs stored in the storage unit 25, and performs image processing 2A. Overall control of the operation.
- the control unit 21 executes an image analysis process (see FIG. 5) in cooperation with a program stored in the storage unit 25, and executes a profile creation process, a calculation process, an extraction process, and a generation process.
- the function as a means is realized.
- the operation unit 22 includes a keyboard having character input keys, numeric input keys, various function keys, and the like, and a pointing device such as a mouse, and a key pressing signal pressed by the keyboard and an operation signal by the mouse. Are output to the control unit 21 as an input signal.
- the display unit 23 includes, for example, a monitor such as a CRT (Cathode Ray Tube) or an LCD (Liquid Crystal Display), and displays various screens in accordance with display signal instructions input from the control unit 21.
- the display unit 23 functions as an output unit for outputting an image analysis result.
- the communication I / F 24 is an interface for transmitting and receiving data to and from external devices such as the microscope image acquisition device 1A.
- the communication I / F 24 functions as a means for executing a bright-field image and fluorescent image input process.
- the storage unit 25 includes, for example, an HDD (Hard Disk Drive), a semiconductor nonvolatile memory, or the like. As described above, the storage unit 25 stores various programs, various data, and the like.
- the image processing apparatus 2A may include a LAN adapter, a router, and the like and be connected to an external device via a communication network such as a LAN.
- the image processing apparatus 2A in the present embodiment preferably performs analysis using the bright field image and the fluorescence image transmitted from the microscope image acquisition apparatus 1A.
- the bright field image is obtained by enlarging and photographing a tissue specimen stained with H (hematoxylin) staining reagent and HE (hematoxylin-eosin) staining reagent in a bright field in the microscope image acquisition apparatus 1A. It is an image and is a cell form image showing the form of the cell in the tissue specimen.
- Hematoxylin is a blue-violet pigment that stains cell nuclei, bone tissue, part of cartilage tissue, serous components, etc. (basophilic tissue, etc.).
- Eosin is a red to pink pigment that stains the cytoplasm, connective tissue of soft tissues, red blood cells, fibrin, endocrine granules, etc. (eosinophilic tissues, etc.).
- FIG. 3 shows an example of a bright field image obtained by photographing a tissue specimen subjected to HE staining.
- a fluorescent image includes a staining reagent including nanoparticles encapsulating a fluorescent substance bound with a biological substance recognition site that specifically binds and / or reacts with a specific biological substance (hereinafter referred to as a fluorescent substance-encapsulated nanoparticle or fluorescent particle).
- the fluorescence appearing in the fluorescence image indicates the expression of a specific biological material corresponding to the biological material recognition site in the tissue specimen.
- FIG. 4 shows an example of the fluorescence image.
- a method for acquiring a fluorescent image including a staining reagent (fluorescent substance-containing nanoparticles) used when acquiring the fluorescent image, a method of staining a tissue specimen with the staining reagent, and the like.
- a staining reagent fluorescent substance-containing nanoparticles
- fluorescent substance examples include fluorescent organic dyes and quantum dots (semiconductor particles). When excited by ultraviolet to near infrared light having a wavelength in the range of 200 to 700 nm, it preferably emits visible to near infrared light having a wavelength in the range of 400 to 1100 nm.
- fluorescent organic dyes examples include fluorescein dye molecules, rhodamine dye molecules, Alexa Fluor (Invitrogen) dye molecules, BODIPY (Invitrogen) dye molecules, cascade dye molecules, coumarin dye molecules, and eosin dyes. And a molecule, an NBD dye molecule, a pyrene dye molecule, a cyanine dye molecule, and an aromatic hydrocarbon molecule.
- quantum dots containing II-VI group compounds, III-V group compounds, or group IV elements as components ("II-VI group quantum dots”, "III-V group quantum dots”, " Or “Group IV quantum dots”). You may use individually or what mixed multiple types.
- CdSe CdS, CdS, CdTe, ZnSe, ZnS, ZnTe, InP, InN, InAs, InGaP, GaP, GaAs, Si, and Ge, but are not limited thereto.
- a quantum dot having the above quantum dot as a core and a shell provided thereon.
- CdSe / ZnS when the core is CdSe and the shell is ZnS, it is expressed as CdSe / ZnS.
- CdSe / ZnS, CdS / ZnS, InP / ZnS, InGaP / ZnS, Si / SiO 2 , Si / ZnS, Ge / GeO 2 , Ge / ZnS, and the like can be used, but are not limited thereto.
- the quantum dots those subjected to surface treatment with an organic polymer or the like may be used as necessary. Examples thereof include CdSe / ZnS having a surface carboxy group (manufactured by Invitrogen), CdSe / ZnS having a surface amino group (manufactured by Invitrogen), and the like.
- the fluorescent substance-encapsulating nanoparticles are those in which the fluorescent substance is dispersed inside the nanoparticles, whether the fluorescent substance and the nanoparticles themselves are chemically bonded or not. Good.
- the material constituting the nanoparticles is not particularly limited, and examples thereof include polystyrene, polylactic acid, silica, and melamine.
- Fluorescent substance-containing nanoparticles used in the present embodiment can be produced by a known method.
- silica nanoparticles encapsulating a fluorescent organic dye can be synthesized with reference to the synthesis of FITC-encapsulated silica particles described in Langmuir 8, Vol. 2921 (1992).
- Various fluorescent organic dye-containing silica nanoparticles can be synthesized by using a desired fluorescent organic dye in place of FITC.
- Silica nanoparticles encapsulating quantum dots can be synthesized with reference to the synthesis of CdTe-encapsulated silica nanoparticles described in New Journal of Chemistry, Vol. 33, p. 561 (2009).
- Polystyrene nanoparticles encapsulating a fluorescent organic dye may be copolymerized using an organic dye having a polymerizable functional group described in US Pat. No. 4,326,008 (1982) or polystyrene described in US Pat. No. 5,326,692 (1992). It can be produced using a method of impregnating nanoparticles with a fluorescent organic dye.
- Polymer nanoparticles encapsulating quantum dots can be prepared using the method of impregnating polystyrene nanoparticles with quantum dots described in Nature Biotechnology, Vol. 19, page 631 (2001).
- the average particle size of the fluorescent substance-containing nanoparticles used in this embodiment is not particularly limited, but those having a large particle size are difficult to access the antigen, and those having a small particle size and low luminance are signals of the fluorescent substance-containing nanoparticles. Are buried in the background noise (camera noise and cell autofluorescence), and those of about 20 to 200 nm are preferable.
- the average particle diameter is obtained by taking an electron micrograph using a scanning electron microscope (SEM), measuring the cross-sectional area of a sufficient number of particles, and taking each measured value as the area of the circle. As sought.
- SEM scanning electron microscope
- the arithmetic average of the particle sizes of 1000 particles is defined as the average particle size.
- the coefficient of variation was calculated from the particle size of 1000 particles in the same manner as the average particle size.
- the biological material recognition site is a site that specifically binds and / or reacts with the target biological material.
- the target biological substance is not particularly limited as long as a substance that specifically binds to the target biological substance exists, but typically, protein (peptide), nucleic acid (oligonucleotide, polynucleotide), antibody, etc. Is mentioned. Accordingly, substances that bind to the target biological substance include antibodies that recognize the protein as an antigen, other proteins that specifically bind to the protein, and nucleic acids having a base sequence that hybridizes to the nucleic acid. Is mentioned.
- an anti-HER2 antibody that specifically binds to HER2 which is a protein present on the cell surface
- an anti-ER antibody that specifically binds to an estrogen receptor (ER) present in the cell nucleus and actin that forms a cytoskeleton
- an anti-actin antibody that specifically binds to are preferable.
- antigens examples include M. actin, MS actin, SM actin, ACTH, Alk-1, ⁇ 1-antichymotrypsin, ⁇ 1-antitrypsin, AFP, bcl-2, bcl-6, ⁇ -catenin, BCA 225, CA19-9, CA125 , Calcitonin, calretinin, CD1a, CD3, CD4, CD5, CD8, CD10, CD15, CD20, CD21, CD23, CD30, CD31, CD34, CD43, CD45, CD45R, CD56, CD57, CD61, CD68, CD79a, "CD99, MIC2 ", CD138, chromogranin, c-KIT, c-MET, collagen type IV, Cox-2, cyclin D1, keratin, cytokeratin (high molecular weight), pankeratin, pankeratin
- examples of the specific nucleic acid gene that has been pointed out to be associated with a disease can include the following, and probes that recognize each specific nucleic acid gene are available as BAC probes: Can be created based on general knowledge. Specific examples of specific nucleic acid genes are as follows. HER2, TOP2A, HER3, EGFR, P53, MET, etc. are mentioned as genes related to cancer growth and molecular target drug response. Furthermore, the following genes are known as various cancer-related genes. Can be mentioned.
- Tyrosine kinase-related genes include ALK, FLT3, AXL, FLT4 (VEGFR3, DDR1, FMS (CSF1R), DDR2, EGFR (ERBB1), HER4 (ERBB4), EML4-ALK, IGF1R, EPHA1, INSR, EPHA2, IRR (INSRR) ), EPHA3, KIT, EPHA4, LTK, EPHA5, MER (MERTK), EPHA6, MET, EPHA7, MUSK, EPHA8, NPM1-ALK, EPHB1, PDGFR ⁇ (PDGFRA), EPHB2, PDGFR ⁇ (PDGFRB), EPHEP3, T RON (MST1R), FGFR1, ROS (ROS1), FGFR2, TIE2 (TEK), FGFR3, TRKA (NTRK1), FGFR4, TRKB (NT RK2), FLT1 (VEGFR1), TRKC (NTRK3), and breast cancer-related genes are ATM, BRCA1, BRCA2, BRCA3, CC
- Cancer-related genes include APC, MSH6, AXIN2, MYH, BMPR1A, p53, DCC, PMS2, KRAS2 (or Ki-ras), PTEN, MLH1, and SMA.
- MSH2, STK11, MSH6 Lung cancer-related genes include ALK, PTEN, CCND1, RASSF1A, CDKN2A, RB1, EGFR, RET, EML4, ROS1, KRAS2, TP53, MYC.
- genes include Axin1, MALAT1, b-catenin, p16 INK4A, c-ERBB-2, p53, CTNNB1, RB1, Cyclin D1, SMAD2, EGFR, SMAD4, IGFR2, TCF1, and KRAS.
- Related genes include Alpha, PRCC, ASPSCR1, PSF, CLTC, TFE3, p54nrb / NONO, and TFEB As thyroid cancer-related genes, AKAP10, NTRK1, and AKA 9, RET, BRAF, TFG, ELE1, TPM3, H4 / D10S170, TPR and the like.
- Examples of ovarian cancer-related genes include AKT2, MDM2, BCL2, MYC, BRCA1, NCOA4, CDKN2A, p53, ERBB2, PIK3CA, GATA4, RB, HRAS, RET, KRAS, and RNASET2.
- Examples of prostate cancer-related genes include AR, KLK3, BRCA2, MYC, CDKN1B, NKX3.1, EZH2, p53, GSTP1, and PTEN.
- Examples of bone tumor-related genes include CDH11, COL12A1, CNBP, OMD, COL1A1, THRAP3, COL4A5, and USP6.
- the mode of binding between the biological substance recognition site and the fluorescent substance-encapsulating nanoparticles is not particularly limited, and examples thereof include covalent bonding, ionic bonding, hydrogen bonding, coordination bonding, physical adsorption, and chemical adsorption.
- a bond having a strong bonding force such as a covalent bond is preferred from the viewpoint of bond stability.
- an organic molecule that connects between the biological substance recognition site and the fluorescent substance-containing nanoparticle.
- a polyethylene glycol chain can be used, and SM (PEG) 12 manufactured by Thermo Scientific can be used.
- a silane coupling agent that is a compound widely used for bonding an inorganic substance and an organic substance can be used.
- This silane coupling agent is a compound having an alkoxysilyl group that gives a silanol group by hydrolysis at one end of the molecule and a functional group such as a carboxyl group, an amino group, an epoxy group, an aldehyde group at the other end, Bonding with an inorganic substance through an oxygen atom of the silanol group.
- silane coupling agent having a polyethylene glycol chain (for example, PEG-silane no. SIM6492.7 manufactured by Gelest), etc. Is mentioned.
- silane coupling agent you may use 2 or more types together.
- a publicly known method can be used for the reaction procedure of the fluorescent organic dye-encapsulated silica nanoparticles and the silane coupling agent.
- the obtained fluorescent organic dye-encapsulated silica nanoparticles are dispersed in pure water, aminopropyltriethoxysilane is added, and the mixture is reacted at room temperature for 12 hours.
- fluorescent organic dye-encapsulated silica nanoparticles whose surface is modified with an aminopropyl group can be obtained by centrifugation or filtration.
- the antibody can be bound to the fluorescent organic dye-encapsulated silica nanoparticles via an amide bond.
- a condensing agent such as EDC (1-Ethyl-3- [3-Dimethylaminopropyl] carbodiimide Hydrochloride: manufactured by Pierce) can also be used.
- a linker compound having a site that can be directly bonded to the fluorescent organic dye-encapsulated silica nanoparticles modified with organic molecules and a site that can be bonded to the molecular target substance can be used.
- sulfo-SMCC Sulfosuccinimidyl 4 [N-maleimidomethyl] -cyclohexane-1-carboxylate: manufactured by Pierce
- sulfo-SMCC Sulfosuccinimidyl 4 [N-maleimidomethyl] -cyclohexane-1-carboxylate: manufactured by Pierce
- the same procedure can be applied regardless of whether the fluorescent substance is a fluorescent organic dye or a quantum dot. That is, by impregnating a polystyrene nanoparticle having a functional group such as an amino group with a fluorescent organic dye or a quantum dot, a fluorescent substance-containing polystyrene nanoparticle having a functional group can be obtained, and thereafter EDC or sulfo-SMCC is used. Thus, antibody-bound fluorescent substance-encapsulated polystyrene nanoparticles can be produced.
- the same procedure as that for the fluorescent substance-encapsulated silica nanoparticles can be applied.
- the number of surface amino groups may be increased by reacting melamine nanoparticles with a polyfunctional amine compound in advance.
- the operator immerses the tissue specimen in a container containing xylene to remove paraffin.
- the temperature is not particularly limited, but can be performed at room temperature.
- the immersion time is preferably 3 minutes or longer and 30 minutes or shorter. If necessary, xylene may be exchanged during the immersion.
- the tissue specimen is immersed in a container containing ethanol to remove xylene.
- the temperature is not particularly limited, but can be performed at room temperature.
- the immersion time is preferably 3 minutes or longer and 30 minutes or shorter. Further, if necessary, ethanol may be exchanged during the immersion.
- the tissue specimen is immersed in a container containing water to remove ethanol.
- the temperature is not particularly limited, but can be performed at room temperature.
- the immersion time is preferably 3 minutes or longer and 30 minutes or shorter. Moreover, you may exchange water in the middle of immersion as needed.
- the activation conditions are not particularly defined, but as the activation liquid, 0.01 M citrate buffer (pH 6.0), 1 mM EDTA solution (pH 8.0), 5% urea, 0.1 M Tris-HCl buffer, etc. Can be used.
- the heating device an autoclave, a microwave, a pressure cooker, a water bath, or the like can be used.
- the temperature is not particularly limited, but can be performed at room temperature. The temperature can be 50 to 130 ° C. and the time can be 5 to 30 minutes.
- the tissue specimen after the activation treatment is immersed in a container containing PBS (Phosphate Buffered Saline) and washed.
- PBS Phosphate Buffered Saline
- the temperature is not particularly limited, but can be performed at room temperature.
- the immersion time is preferably 3 minutes or longer and 30 minutes or shorter. If necessary, the PBS may be replaced during the immersion.
- each fluorescent substance-encapsulated nanoparticle PBS dispersion may be mixed in advance or separately placed on a tissue specimen separately. Also good.
- the temperature is not particularly limited, but can be performed at room temperature.
- the reaction time is preferably 30 minutes or more and 24 hours or less.
- a known blocking agent such as BSA-containing PBS
- the stained tissue specimen is immersed in a container containing PBS to remove unreacted fluorescent substance-containing nanoparticles.
- the temperature is not particularly limited, but can be performed at room temperature.
- the immersion time is preferably 3 minutes or longer and 30 minutes or shorter. If necessary, the PBS may be replaced during the immersion. Place the cover glass on the tissue specimen and enclose it. A commercially available encapsulant may be used as necessary.
- staining using a HE dyeing reagent HE dyeing is performed before enclosure with a cover glass.
- a microscope image (fluorescence image) with a wide field of view is acquired from the stained tissue specimen using the microscope image acquisition device 1A.
- an excitation light source and a fluorescence detection optical filter corresponding to the absorption maximum wavelength and fluorescence wavelength of the fluorescent material used for the staining reagent are selected.
- the magnification of the objective lens is preferably 4 to 100 times
- the numerical aperture (NA) is preferably 0.6 or more, more preferably 0.8 or more. It is.
- the sampling pitch of the camera for imaging is preferably 400 nm or less, and more preferably 150 nm or less.
- the operator uses two types of staining reagents, a HE staining reagent and a staining reagent using fluorescent substance-encapsulated nanoparticles bound with a biological substance recognition site that recognizes a specific protein as a fluorescent labeling material. Stain. Thereafter, in the microscope image acquisition apparatus 1A, a bright field image and a fluorescence image are acquired by the following procedures (a1) to (a6). (A1) The operator places the tissue specimen stained with the HE staining reagent and the staining reagent containing the fluorescent substance-containing nanoparticles on the slide, and places the slide on the slide fixing stage of the microscope image acquisition apparatus 1A.
- (A2) Set in the bright field unit, adjust the imaging magnification and focus, place the area to be observed on the tissue specimen in the field of view, and start imaging in the direction of focus depth movement (here, the vertical direction) Set the position, shooting end position, and pitch.
- the slide fixing stage is moved upward or downward to a predetermined photographing start position.
- (A3) Shooting is performed by the imaging unit to generate bright field image data, and the image data is transmitted to the image processing apparatus 2A.
- (A4) Change the unit to a fluorescent unit.
- Photographing is performed by the imaging means without changing the position of the slide fixing stage and the photographing magnification to generate image data of a fluorescent image, and the image data is transmitted to the image processing apparatus 2A.
- FIG. 5 shows a flowchart of image analysis processing in the image processing apparatus 2A.
- the image analysis processing shown in FIG. 5 is executed in cooperation with the control unit 21 and the program stored in the storage unit 25.
- step S1 when a bright field image is input from the microscope image acquisition device 1A through the communication I / F 24 (step S1), the control unit 21 extracts a cell region from the bright field image (step S2).
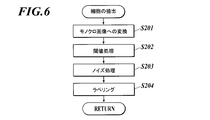
- FIG. 6 shows a detailed flow of the process in step S2. The process of step S2 is executed in cooperation with the control unit 21 and the program stored in the storage unit 25.
- step S2 first, a bright-field image is converted into a monochrome image (step S201).
- FIG. 7A shows an example of a bright field image.
- threshold processing is performed on the monochrome image using a predetermined threshold, and the value of each pixel is binarized (step S202).
- noise processing is performed (step S203).
- the noise process can be performed by performing a closing process on the binary image.
- the closing process is a process in which the contraction process is performed the same number of times after the expansion process is performed.
- the expansion process is a process of replacing a target pixel with white when at least one pixel in the range of n ⁇ n pixels (n is an integer of 2 or more) from the target pixel is white.
- the contraction process is a process of replacing a target pixel with black when at least one pixel in the range of n ⁇ n pixels from the target pixel contains black.
- FIG. 7B shows an example of an image after noise processing. As shown in FIG. 7B, after noise processing, an image (cell image) from which cells are extracted is generated.
- the labeling process is a process for identifying an object in an image by assigning the same label (number) to connected pixels. By the labeling process, each cell can be identified from the image after the noise process and a label can be applied.
- step S3 when a fluorescent image is input from the microscope image acquisition device 1A through the communication I / F 24 (step S3), the control unit 21 extracts fluorescent particles from the fluorescent image (step S4).
- FIG. 8 shows a detailed flow of the process in step S4. The process of step S4 is executed in cooperation with the control unit 21 and the program stored in the storage unit 25.
- step S4 first, a color component corresponding to the wavelength of the fluorescent bright spot is extracted from the fluorescent image (step S401).
- FIG. 9A shows an example of a fluorescence image.
- step S401 for example, when the emission wavelength of the fluorescent particles is 550 nm, only the fluorescent bright spot having the wavelength component is extracted as an image.
- threshold processing is performed on the extracted image to generate a binary image, and a bright spot region is extracted (step S402).
- noise removal processing such as cell autofluorescence and other unnecessary signal components may be performed before the threshold processing, and a low-pass filter such as a Gaussian filter or a high-pass filter such as a second derivative is preferably used.
- FIG. 9B shows an example of an image from which the bright spot region is extracted. As shown in FIG. 9B, a bright spot region centered on the fluorescent bright spot is extracted from such an image.
- step S403 profile creation process
- step S404 the number of fluorescent particles in each bright spot region and the position of each fluorescent particle are calculated.
- the “luminance profile” is luminance value distribution information created based on the image extracted from the fluorescence image using the image from which the luminescent spot region is extracted as a mask. The luminance value in the luminescent spot region and its range (luminance distribution) Spread).
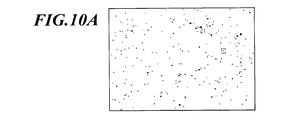
- FIG. 10A is an example of an image in which a bright spot region is extracted from a fluorescence image. Based on the image from which the bright spot area has been extracted, for each bright spot area, the image from which the bright spot area has been extracted (FIG. 10B) and the fluorescence image of the portion corresponding to the bright spot area (FIG. 10C) are superimposed.
- FIG. 10B is an enlarged view of a region surrounded by a square in FIG. 10A and shows one bright spot region.
- FIG. 10C is a fluorescence image of a portion corresponding to the bright spot region of FIG. 10B.
- a bright spot image corresponding to the bright spot area is generated from the fluorescent image (eg, FIG.
- FIG. 10C uses the image from which the bright spot area is extracted (eg, FIG. 10B) as a mask (FIG. 10D).
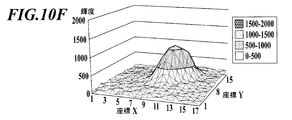
- the brightness value of the bright spot image measured for each pixel and displayed at the X coordinate position and the Y coordinate position is the distribution of brightness values created as a brightness profile in step S403 (FIG. 10E).
- the luminance profile may be one in which the luminance at the X coordinate position and the Y coordinate position is expressed two-dimensionally as shown in FIG. 10E, or as shown in FIG. 10F. ) And the luminance (height) at the Y coordinate position (vertical) may be expressed three-dimensionally.
- one bright spot region includes one or a plurality of fluorescent particles
- the luminance profile includes a luminance value and a range (luminance distribution) corresponding to the number of fluorescent particles and the position of each fluorescent particle.
- a luminance profile of one fluorescent particle is created in advance as a reference profile from an image of a single fluorescent particle taken under the same image capturing conditions as the fluorescent image input in step S3.
- a reference profile created from a fluorescent image in which one fluorescent particle is in focus has a normal distribution shape having one sharp peak at the center as shown in FIG. 11B.
- a reference profile created from a fluorescent image whose focus is shifted downward from the fluorescent particles has a large spread in the vertical and horizontal directions and a low peak as shown in FIG. 11A, for example.
- the reference profile created from the fluorescence image in which the focal point is shifted upward from the fluorescent particles for example, as shown in FIG.
- the center has a slightly low brightness and a concave shape.
- FIGS. 12A to 12C are schematic diagrams showing cross sections of a three-dimensional luminance profile created from the same region of fluorescent images (left diagrams of FIGS. 12A to 12C) acquired at focal depths Z1, Z2, and Z3 at predetermined intervals. It is a figure (each right figure of FIG. 12A-FIG. 12C).
- the luminance profile shown here includes three bright spot regions.
- the leftmost luminance profile shows a normal distribution shape having a sharp peak at the focal depth Z1 (FIG. 12A), but the center is at the focal depth Z2 (FIG. 12B).
- the dent and the spread in the X-axis direction become large. Furthermore, at the focal depth Z3 (FIG.
- the rightmost luminance profile has a concave center at the focal depth Z1 (FIG. 12A) and a very large spread in the X-axis direction, and has a concave center at the focal depth Z2 (FIG. 12B).
- the spread of the direction is slightly narrowed, and a normal distribution shape having a sharp peak at the center is shown at the focal depth Z3 (FIG. 12C).
- the position of the fluorescent particle included in each bright spot region is calculated as a position on the three-dimensional coordinate obtained by adding the focal depth to the position on the two-dimensional coordinate on each fluorescent image.
- the fluorescent image is subjected to a labeling process, and a label is given to the bright spot image at the position of the fluorescent particle calculated in the step S404 (step S405).
- a label for identifying the fluorescent particles in the bright spot image (fluorescent particle image) most focused on the fluorescent particles Will be granted.
- 12A to 12C show an example in which one fluorescent particle is included in each bright spot region. For example, even when a plurality of fluorescent particles are included in one bright spot region, By analyzing a plurality of fluorescent images with different focal depths based on the reference profile, it is possible to separate and measure a plurality of fluorescent particles that appear to overlap on a two-dimensional fluorescent image. Then, for each of the plurality of fluorescent particles, a label is given to the bright spot image (fluorescent particle image) that is most focused.
- the number and position of the fluorescent particles are determined using the luminance profile for one fluorescent particle as a reference profile.
- a luminance profile composed of a plurality of fluorescent particles is prepared in advance as a reference profile. Then, the number and position of the fluorescent particles may be determined.
- the luminance profile itself composed of a plurality of fluorescent particles may be subjected to known arbitrary image processing such as two-dimensional Fourier transform to decompose the waveform to determine the number and position of the fluorescent particles.
- step S2 and step S4 After the process of step S2 and step S4 is completed, the process returns to the process of FIG. 5, the addition process of the cell image (see FIG. 7B) and the fluorescence image is performed (step S5), and one cell given in step S204 is removed. The number of fluorescent particles per cell is calculated from the label shown and the label of the fluorescent particles given in step S405 (calculation step).
- one reconstructed image (focused image) in which a plurality of fluorescent images with different depths of focus are reconstructed is generated (generation process).
- the fluorescent particle image to which the fluorescent particle label is assigned in step S405 is extracted and used for reconstruction of the focused image (extraction process).
- the extracted plurality of fluorescent particle images is, for example, an average value or addition of luminance values for each pixel. Reconstructed into a single image whose value has been calculated. The bright spot image without the label is not used for reconstruction because the focus is shifted from the fluorescent particles.
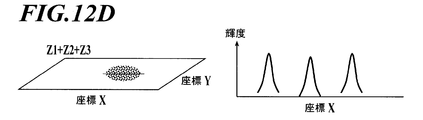
- FIG. 12D is a schematic diagram showing a cross-section of the focused image reconstructed from the three fluorescent images of FIGS. 12A to 12C and the luminance profile of the focused image.
- FIG. 12A A bright spot image with depth Z1 (FIG. 12A), a bright spot image with focal depth Z2 (FIG. 12B) in the central bright spot area, and a bright spot image with focal depth Z3 (FIG. 12C) in the right bright spot area.
- An example extracted and reconstructed as a fluorescent particle image is shown.
- the luminance profile created from the focused image (right diagram in FIG. 12D) has a normal distribution shape having sharp peaks in all the bright spot regions, and an image in which each fluorescent particle is focused is obtained.
- step S6 the reconstructed image and the cell image are superimposed, and an image showing the distribution of the fluorescent particles on the cell is displayed.
- cells are extracted by the processing of steps S1 to S2, the bright spot region is extracted by the processing of steps S3 to S402, and then the fluorescence on the cells is processed by the processing of steps S403 to S404.
- the particle distribution is specifically grasped on the three-dimensional coordinates.
- steps S5 to S6 a bright spot image focused on the fluorescent particles is extracted and reconstructed into a single fluorescent image, and the distribution of the fluorescent particles on the cells is displayed. In this way, it is possible to accurately quantify the expression (number of expression and position of expression) of a specific protein in the observation target cell using a simple microscope, and the bright spot blur focused on each fluorescent particle. No fluorescence image can be obtained.
- the HER2 protein in breast cancer is mentioned as an example of the specific protein, but it is not limited to this.
- the feature quantity that quantitatively indicates the expression level of the specific protein corresponding to the lesion type It can be provided to a doctor.
- each color component is extracted using filter work or the like in step S401, and the processing of steps S402 to S405 is executed for each extracted color component (wavelength component).
- the cell region image is extracted.
- the fluorescent particle image created for each color component may be added.
- the fluorescent particles may be directly bound to a biological substance recognition site that binds to a specific protein as in the above embodiment, but indirectly through another substance, as in a known indirect method in immunostaining. May be combined.
- a secondary antibody having the primary antibody as an antigen may be reacted with a fluorescent particle bound thereto and stained.
- a fluorescent particle modified with streptavidin is reacted with streptavidin. You may dye
- an HDD or a semiconductor non-volatile memory is used as a computer-readable medium of the program according to the present invention, but the present invention is not limited to this example.
- a portable recording medium such as a CD-ROM can be applied.
- a carrier wave carrier wave is also applied as a medium for providing program data according to the present invention via a communication line.
- the mixture was centrifuged at 20000 G for 15 minutes in a centrifuge (Kubota Micro Cooling Centrifuge 3740), and after removing the supernatant, ultrapure water was added and ultrasonically irradiated to redisperse. Centrifugation, supernatant removal, and washing by redispersion in ultrapure water were repeated 5 times.
- the obtained melamine particles were positively charged because the melamine resin itself contains many amino groups in the skeleton. The charge of the particles was evaluated by resin component analysis by NMR, IR, etc. and zeta potential measurement.
- the obtained fluorescent particles were observed with a scanning electron microscope (SEM; Model S-800 manufactured by Hitachi (registered trademark)), and the average particle size and coefficient of variation were calculated.
- fluorescent particles having an average particle diameter of 200, 170, 150, 100, 80, 60, 40, and 20 nm and a variation coefficient of 12% were used.
- Step (1) 1 mg of fluorescent particles was dispersed in 5 mL of pure water. Next, 100 ⁇ L of aminopropyltriethoxysilane aqueous dispersion (LS-3150, manufactured by Shin-Etsu Chemical Co., Ltd.) was added and stirred at room temperature for 12 hours. Step (2): The reaction mixture was centrifuged at 10,000 G for 60 minutes, and the supernatant was removed. Step (3): Ethanol was added to disperse the sediment, followed by centrifugation again. Washing with ethanol and pure water was performed once by the same procedure. When the FT-IR measurement was performed on the resulting amino group-modified fluorescent particles, absorption derived from the amino group could be observed, confirming that the amino group was modified.
- Step (2) The reaction mixture was centrifuged at 10,000 G for 60 minutes, and the supernatant was removed.
- Step (3) Ethanol was added to disperse the sediment, followed by centrifugation again. Washing with ethanol and pure water was performed once by the same procedure
- Step (4) The amino group-modified fluorescent particles obtained in step (3) were adjusted to 3 nM using PBS containing 2 mM of EDTA (ethylenediaminetetraacetic acid).
- Step (6) The reaction mixture was centrifuged at 10,000 G for 60 minutes, and the supernatant was removed.
- Step (7) PBS containing 2 mM of EDTA was added, the precipitate was dispersed, and centrifuged again. The washing
- Step (8) When 100 ⁇ g of the anti-HER2 antibody was dissolved in 100 ⁇ L of PBS, 1 M dithiothreitol (DTT) was added and reacted for 30 minutes.
- Step (11) 4 ⁇ L of 10 mM mercaptoethanol was added to stop the reaction.
- reagent for FISH staining As a probe for FISH staining, a biotinylated HER-2 DNA probe in which biotin was introduced into the HER-2 DNA probe by Nick translation was used.
- the staining reagent b for visualizing the probe includes streptavidin-modified fluorescent particles prepared by using streptavidin instead of anti-HER2 antibody in step (8) of (A1-2), and (A1-3 ) And the same fluorescent dye and quantum dot used in combination with streptavidin.
- B Tissue staining
- IHC Immunohistochemistry
- Step (1) The tissue specimen was immersed in a container containing xylene for 30 minutes. The xylene was changed three times during the process.
- Step (2) The tissue specimen was immersed in a container containing ethanol for 30 minutes. The ethanol was changed three times during the process.
- Step (3) The tissue specimen was immersed in a container containing water for 30 minutes. The water was changed three times along the way.
- Step (4) The tissue specimen was immersed in 10 mM citrate buffer (pH 6.0) for 30 minutes.
- Step (6) The tissue specimen after the autoclave treatment was immersed in a container containing PBS for 30 minutes.
- Step (7) PBS containing 1% BSA was placed on the tissue specimen and allowed to stand for 1 hour.
- Step (11) After adding Aquatex made by Merck Chemicals, a cover glass was placed and sealed.
- B2 Fluorescence in situ hybridization (FISH) method
- FISH Fluorescence in situ hybridization
- HER2 genes are usually present in the cell and may increase to four during cell proliferation.
- the number of genes is 1 to 4.
- C2 Conventional image analysis processing method
- the luminance is obtained by using only one image (single image) in which the outer edge of the cell is in focus among the obtained 20 microscope images.
- the area where the value exceeded a predetermined threshold was measured as a bright spot, and the number of fluorescent particles per cell (the number of bright spots) was calculated.
- Table 1 shows the number of bright spots per cell calculated from the tissue specimen stained with the HER2 protein by the IHC method described in (B1) using the staining reagent a described in (A1).
- Example 1 the number of bright spots was calculated from the tissue specimen stained with the fluorescent particle-derived staining reagent a using the image analysis processing method (C1) of the present invention.
- Comparative Example 1 the number of bright spots was calculated from a tissue specimen stained with a fluorescent particle-derived staining reagent a using a conventional image analysis processing method (C2).
- Comparative Examples 2 and 3 the number of bright spots was calculated from the tissue specimen stained with the fluorescent dye and the staining reagent a derived from quantum dots using the image analysis processing method (C1) of the present invention.
- the IHC method compares the average particle diameter of fluorescent substances and the lot of tissue specimens in the same combination. According to the method (C1) using the reconstructed image and the luminance profile, the number of bright spots was always calculated larger than in the conventional method (C2), and a result close to the true value was obtained.
- the HER2 + tissue sample is always 2.5 times as large as the HER2 ⁇ tissue sample.
- the number of bright spots above (average: about 3.7 times) was calculated.
- the number of bright spots was calculated to be 4.6 to 5.8 times (average: about 5.2 times) that of the HER2 + tissue specimen.
- Comparative Example 1 in the HER2 + tissue sample, 1.3 to 2.3 times (average: about 1.7 times) the number of bright spots were calculated as compared to the HER2 ⁇ tissue sample.
- the number of bright spots was calculated to be 4.6 to 6 times (average: about 5.2 times) that of the HER2 + tissue specimen.
- the fluorescently stained protein can be measured as a fluorescent bright spot. Furthermore, according to the biological material quantification method of the present invention, since analysis is performed using a plurality of fluorescent images with different depths of focus, it is possible to detect fluorescent particles from the whole cell in consideration of the spread in the direction of the depth of focus. it can. Furthermore, since it is possible to separate and measure the adjacent fluorescent particles by the analysis based on the luminance profile, the number of bright spots can be measured more accurately.
- Example 1 and Comparative Example 1 the number of bright spots measured from the tissue specimens with the tissue specimen lots HER2 3+ and HER2 + were compared with the same average particle diameter of the fluorescent particles. The number of bright spots about 2.5 times that of the example was calculated. As described above, according to the present invention, since a large number of bright spots across the depth of focus can be accurately measured, it is easy to detect a slight difference in the expression level, and the difference in the expression level can be detected even when the expression of HER2 is small. It can be clearly distinguished and diagnosed.
- Table 2 shows the number of bright spots per cell calculated from the tissue specimen in which the HER2 gene was stained by the FISH method described in (B2) using the staining reagent b described in (A2).
- Example 2 the number of bright spots was calculated from the tissue specimen stained with the fluorescent particle-derived staining reagent b using the image analysis processing method (C1) of the present invention.
- Comparative Example 4 the number of bright spots was calculated from the tissue specimen stained with the fluorescent particle-derived staining reagent b using the conventional image analysis processing method (C2).
- Comparative Examples 5 and 6 from the tissue specimen stained with the fluorescent dye and the staining reagent b derived from quantum dots, the image analysis processing method (C1) of the present invention and the conventional image analysis processing method (C2) are used. The number of bright spots was calculated.
- the fluorescent dyes and quantum dots have low luminance per molecule, and it has been difficult to obtain a reference luminance profile by changing the depth of focus. Therefore, in Comparative Example 5-1 and Comparative Example 6-1, image analysis processing was performed using the luminance profile of the fluorescent particles.
- Example 2 Focusing on the average particle diameter of fluorescent particles and the relationship between the image analysis method and the number of bright spots, from Example 2 and Comparative Example 4, the average particle diameter of fluorescent particles and the tissue sample in the tissue sample stained with the HER2 gene by the FISH method
- the number of bright spots is always larger than that of the conventional method (Comparative Example 4). Points were calculated (20-55% increase) and results close to true values were obtained.
- the HER2 small amplification tissue specimen calculates the number of bright spots more than about 4 times that of the tissue specimen without HER2 amplification, and HER2 large amplification.
- the number of bright spots about 2.3 times that of the tissue sample with a small HER2 amplification was calculated.
- the degree of increase was not particularly different in any of the particle sizes and the image analysis processing methods shown in Table 2.
- fluorescent particles as the fluorescent material because the number of calculated bright spots is increased even in the case of staining of genes by the FISH method in which fluorescent dyes and quantum dots can be observed as bright spots.
- the change rate of the number of bright spots when the FISH method changes from “no amplification” to “small amplification” is There was almost no difference between the method of the present invention and the conventional method, and all were in the range of 4 to 5 times.
- the change rate of the number of bright spots when “HER2 ⁇ ” is changed to “HER2 +” is the method of the present invention (about 3.7 times) compared to the conventional method (about 1.7 times). Then, it can be said that the rate of change is extremely increased and the sensitivity is improved. From the above results, the method of the present invention is effective for quantification of both genes and proteins, and can calculate the number of bright spots closer to the true value than the conventional method. It has a remarkable effect in protein detection.
- the present invention is characterized in that the number of specific biological substances in a tissue specimen can be accurately quantified, and can be particularly suitably used for generating highly accurate pathological diagnosis information.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Biomedical Technology (AREA)
- Molecular Biology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Physics & Mathematics (AREA)
- General Health & Medical Sciences (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Biochemistry (AREA)
- Food Science & Technology (AREA)
- Medicinal Chemistry (AREA)
- Microbiology (AREA)
- Cell Biology (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Investigating, Analyzing Materials By Fluorescence Or Luminescence (AREA)
- Microscoopes, Condenser (AREA)
- Image Processing (AREA)
- Investigating Or Analysing Biological Materials (AREA)
Abstract
A biological substance quantitation method for quantitating a specific biological substance from a sample in which the biological substance has been stained using, as a staining reagent, fluorescent particles in which a plurality of fluorescent substances have been agglomerated, said method being characterized by having an input step for consecutively varying a depth of focus at a prescribed interval and inputting, for each depth of focus, a fluorescence image indicating the appearance of the biological substance in the sample as fluorescence bright spots; a profile creation step for creating, from the fluorescence image at each depth of focus, a bright spot image of an extracted bright spot area and creating a brightness profile for each bright spot image; and a calculation step for creating, as a reference profile, a brightness profile for fluorescent particles measured beforehand, analyzing the brightness profile for each bright spot image at each depth of focus on the basis of the reference profile, and thereby calculating the number of fluorescent particles included in the bright spot area.
Description
本発明は、生体物質定量方法、画像処理装置、病理診断支援システム及び画像処理プログラムに関する。
The present invention relates to a biological material quantification method, an image processing device, a pathological diagnosis support system, and an image processing program.
近年、抗体医薬を中心とした分子標的薬治療の広がりに伴い、分子標的薬をより効果的に設計するため、観察対象細胞上の生体物質の定量が求められている。生体物質の存在を確認する方法として、特定の生体物質に結合可能な蛍光物質を用いた組織染色に基づく、組織分析方法が知られている。
In recent years, with the spread of molecular targeted drug treatment centering on antibody drugs, in order to design molecular targeted drugs more effectively, the quantification of biological substances on cells to be observed has been demanded. As a method for confirming the presence of a biological material, a tissue analysis method based on tissue staining using a fluorescent material that can bind to a specific biological material is known.
例えば、特許文献1では、生体物質を有機蛍光色素、量子ドット等の蛍光物質で染色し、三次元画像解析装置を用いて計測する方法が記載されている。
特許文献1に記載の方法によれば、蛍光画像を三次元に再構築して解析することにより、二次元では重なって見える複数個の蛍光シグナルを、一つ一つ分離して数えることができる。 For example,Patent Document 1 describes a method in which a biological material is stained with a fluorescent material such as an organic fluorescent dye or a quantum dot and measured using a three-dimensional image analysis apparatus.
According to the method described inPatent Document 1, by reconstructing and analyzing a fluorescence image in three dimensions, a plurality of fluorescence signals that appear to overlap in two dimensions can be separated and counted one by one. .
特許文献1に記載の方法によれば、蛍光画像を三次元に再構築して解析することにより、二次元では重なって見える複数個の蛍光シグナルを、一つ一つ分離して数えることができる。 For example,
According to the method described in
特許文献1に記載の方法では、蛍光色素を用いた一般的な蛍光in situ ハイブリダイゼーション(FISH)法により染色される遺伝子は、一細胞当たりの発現数が比較的少ないため、撮影条件を適宜調整することによって蛍光色素による蛍光シグナルがドット状に観察され、数を数えることができる(特許文献1の図1(B)、(C))。しかし、一細胞当たりの発現数が多いタンパク質を免疫組織化学法(IHC)により蛍光色素標識した場合は、蛍光シグナルはドット状に観察されにくく、細胞全体が光って見える(特許文献1の図1(D))。そのため、タンパク質の定量は蛍光輝度の計測によって行うこととなり、発現数の正確な定量や、撮影条件が異なる画像間での比較が困難であるという問題がある。
また、特許文献1の実施例においては、画像の取得は共焦点顕微鏡を用いて行っている。発明者らが、有機蛍光色素又は量子ドットを用いてFISH法又はIHC法により染色した組織標本を通常の蛍光顕微鏡(BZ-9000、キーエンス社製)で撮像したところ、いずれの場合も、得られた蛍光画像では細胞全体が蛍光を発しており、蛍光シグナルをドット状に観察することは困難であった。つまり、1分子当たりの輝度値が小さい蛍光色素又は量子ドットの微小な蛍光シグナルをドット状に撮像するためには、FISH法により遺伝子を染色された標本であっても、背景ノイズが少なくコントラストの高い画像が得られる共焦点顕微鏡の使用が必要であった。
しかし、共焦点顕微鏡は、一般的に励起光としてレーザー光を用いる必要があることから、使用できる波長が限られ、また、撮像の手間がかかって簡便性が低いことが知られる。 In the method described inPatent Document 1, since the gene stained by a general fluorescence in situ hybridization (FISH) method using a fluorescent dye has a relatively small number of expression per cell, the imaging conditions are appropriately adjusted. By doing so, the fluorescent signal by the fluorescent dye is observed in the form of dots, and the number can be counted (FIGS. 1B and 1C of Patent Document 1). However, when a protein having a high expression number per cell is labeled with a fluorescent dye by immunohistochemistry (IHC), the fluorescent signal is hardly observed in a dot shape, and the whole cell appears to be shining (FIG. 1 of Patent Document 1). (D)). Therefore, protein quantification is performed by measuring fluorescence luminance, and there is a problem that accurate quantification of the expression number and comparison between images with different imaging conditions are difficult.
Moreover, in the Example ofpatent document 1, acquisition of the image is performed using the confocal microscope. When the inventors imaged a tissue specimen stained by an FISH method or an IHC method using an organic fluorescent dye or quantum dots with a normal fluorescence microscope (BZ-9000, manufactured by Keyence Corporation), in either case, it was obtained. In the fluorescent image, the whole cell was fluorescent, and it was difficult to observe the fluorescent signal in the form of dots. In other words, in order to image a fluorescent signal with a small luminance value per molecule or a minute fluorescent signal of a quantum dot in a dot shape, even a sample stained with a gene by the FISH method has little background noise and contrast. It was necessary to use a confocal microscope that could provide a high image.
However, it is known that the confocal microscope generally needs to use laser light as excitation light, so that the wavelength that can be used is limited, and it is troublesome to take an image and is not convenient.
また、特許文献1の実施例においては、画像の取得は共焦点顕微鏡を用いて行っている。発明者らが、有機蛍光色素又は量子ドットを用いてFISH法又はIHC法により染色した組織標本を通常の蛍光顕微鏡(BZ-9000、キーエンス社製)で撮像したところ、いずれの場合も、得られた蛍光画像では細胞全体が蛍光を発しており、蛍光シグナルをドット状に観察することは困難であった。つまり、1分子当たりの輝度値が小さい蛍光色素又は量子ドットの微小な蛍光シグナルをドット状に撮像するためには、FISH法により遺伝子を染色された標本であっても、背景ノイズが少なくコントラストの高い画像が得られる共焦点顕微鏡の使用が必要であった。
しかし、共焦点顕微鏡は、一般的に励起光としてレーザー光を用いる必要があることから、使用できる波長が限られ、また、撮像の手間がかかって簡便性が低いことが知られる。 In the method described in
Moreover, in the Example of
However, it is known that the confocal microscope generally needs to use laser light as excitation light, so that the wavelength that can be used is limited, and it is troublesome to take an image and is not convenient.
一方、特許文献2では、生体物質認識部位が結合された蛍光色素集積粒子を用いて組織切片を染色し、蛍光発光輝点の輝度分布を解析することで、一粒子当たりの平均輝度値を求め、各輝点内の粒子数を算出する方法が提案されている。
特許文献2に記載の方法によれば、蛍光色素集積粒子は一粒子当たりの輝度が高いため、蛍光染色された生体物質がタンパク質の場合でも、蛍光シグナルをドット状に観察し、粒子数を算出することができる。 On the other hand, in Patent Document 2, an average luminance value per particle is obtained by staining a tissue section using fluorescent dye-integrated particles to which a biological substance recognition site is bound, and analyzing the luminance distribution of fluorescent emission points. A method for calculating the number of particles in each bright spot has been proposed.
According to the method described in Patent Document 2, fluorescent dye-aggregated particles have high luminance per particle. Therefore, even when the fluorescently stained biological material is a protein, the fluorescent signal is observed in the form of dots and the number of particles is calculated. can do.
特許文献2に記載の方法によれば、蛍光色素集積粒子は一粒子当たりの輝度が高いため、蛍光染色された生体物質がタンパク質の場合でも、蛍光シグナルをドット状に観察し、粒子数を算出することができる。 On the other hand, in Patent Document 2, an average luminance value per particle is obtained by staining a tissue section using fluorescent dye-integrated particles to which a biological substance recognition site is bound, and analyzing the luminance distribution of fluorescent emission points. A method for calculating the number of particles in each bright spot has been proposed.
According to the method described in Patent Document 2, fluorescent dye-aggregated particles have high luminance per particle. Therefore, even when the fluorescently stained biological material is a protein, the fluorescent signal is observed in the form of dots and the number of particles is calculated. can do.
しかし、特許文献2に記載の手法においても、蛍光色素集積粒子の粒径のばらつきの程度や、蛍光染色された生体物質の発現量によっては、蛍光色素集積粒子のクラスタを一つの輝点と換算して粒子数を低く見積もったり、計測された輝度分布から一粒子当りの平均輝度値を読み取る際に誤差が生じやすく、正確な粒子数を算出できない場合があるという問題があった。
また、特許文献2においても、実施例では共焦点顕微鏡を用いて画像を取得しており、通常の蛍光顕微鏡を用いた場合には、焦点が大きくずれた蛍光色素集積粒子のシグナルは背景ノイズに埋もれて識別し難いこと等の影響により、上述したような誤差はさらに大きくなり得る。 However, even in the technique described in Patent Document 2, a cluster of fluorescent dye integrated particles is converted into one bright spot depending on the degree of variation in the particle diameter of the fluorescent dye integrated particles and the expression level of the fluorescently stained biological material. Thus, there is a problem in that the number of particles is estimated to be low, or an error is likely to occur when reading the average luminance value per particle from the measured luminance distribution, and the accurate number of particles may not be calculated.
Also in Patent Document 2, images are acquired using a confocal microscope in the examples, and when a normal fluorescent microscope is used, the signal of the fluorescent dye-integrated particles whose focus is greatly deviated becomes background noise. Due to the influence of being buried and difficult to identify, the error as described above can be further increased.
また、特許文献2においても、実施例では共焦点顕微鏡を用いて画像を取得しており、通常の蛍光顕微鏡を用いた場合には、焦点が大きくずれた蛍光色素集積粒子のシグナルは背景ノイズに埋もれて識別し難いこと等の影響により、上述したような誤差はさらに大きくなり得る。 However, even in the technique described in Patent Document 2, a cluster of fluorescent dye integrated particles is converted into one bright spot depending on the degree of variation in the particle diameter of the fluorescent dye integrated particles and the expression level of the fluorescently stained biological material. Thus, there is a problem in that the number of particles is estimated to be low, or an error is likely to occur when reading the average luminance value per particle from the measured luminance distribution, and the accurate number of particles may not be calculated.
Also in Patent Document 2, images are acquired using a confocal microscope in the examples, and when a normal fluorescent microscope is used, the signal of the fluorescent dye-integrated particles whose focus is greatly deviated becomes background noise. Due to the influence of being buried and difficult to identify, the error as described above can be further increased.
本発明の主な目的は、簡易な顕微鏡を用いて、標本内の特定の生体物質の量を正確に定量する生体物質定量方法を提供することにある。
The main object of the present invention is to provide a biological material quantification method for accurately quantifying the amount of a specific biological material in a specimen using a simple microscope.
上記課題を解決するため、本発明の第1の態様によれば、
蛍光物質を複数集積した蛍光粒子を染色試薬として用いて特定の生体物質が染色された標本から、前記生体物質を定量する生体物質定量方法において、
焦点深度を所定の間隔で連続的に変え、各焦点深度において、前記標本における前記生体物質の発現を蛍光輝点で表す蛍光画像を入力する入力工程と、
各焦点深度における前記蛍光画像から、輝点領域が抽出された輝点画像を生成し、当該輝点画像ごとに輝度プロファイルを作成するプロファイル作成工程と、
基準プロファイルとして予め計測された蛍光粒子の輝度プロファイルを作成し、各焦点深度における輝点画像の輝度プロファイルを、前記基準プロファイルに基づいて解析することにより、前記蛍光画像に含まれる蛍光粒子の数を算出する算出工程と、
を有することを特徴とする生体物質定量方法が提供される。 In order to solve the above problems, according to the first aspect of the present invention,
In a biological material quantification method for quantifying the biological material from a specimen stained with a specific biological material using fluorescent particles in which a plurality of fluorescent materials are accumulated as a staining reagent,
An input step of continuously changing a depth of focus at a predetermined interval, and inputting a fluorescence image representing the expression of the biological material in the specimen as a fluorescent luminescent spot at each depth of focus;
A profile creation step of creating a bright spot image in which a bright spot region is extracted from the fluorescent image at each focal depth, and creating a brightness profile for each bright spot image;
The brightness profile of the fluorescent particles measured in advance as a reference profile is created, and the brightness profile of the bright spot image at each focal depth is analyzed based on the reference profile, whereby the number of fluorescent particles included in the fluorescence image is determined. A calculation step to calculate,
A method for quantifying a biological material is provided.
蛍光物質を複数集積した蛍光粒子を染色試薬として用いて特定の生体物質が染色された標本から、前記生体物質を定量する生体物質定量方法において、
焦点深度を所定の間隔で連続的に変え、各焦点深度において、前記標本における前記生体物質の発現を蛍光輝点で表す蛍光画像を入力する入力工程と、
各焦点深度における前記蛍光画像から、輝点領域が抽出された輝点画像を生成し、当該輝点画像ごとに輝度プロファイルを作成するプロファイル作成工程と、
基準プロファイルとして予め計測された蛍光粒子の輝度プロファイルを作成し、各焦点深度における輝点画像の輝度プロファイルを、前記基準プロファイルに基づいて解析することにより、前記蛍光画像に含まれる蛍光粒子の数を算出する算出工程と、
を有することを特徴とする生体物質定量方法が提供される。 In order to solve the above problems, according to the first aspect of the present invention,
In a biological material quantification method for quantifying the biological material from a specimen stained with a specific biological material using fluorescent particles in which a plurality of fluorescent materials are accumulated as a staining reagent,
An input step of continuously changing a depth of focus at a predetermined interval, and inputting a fluorescence image representing the expression of the biological material in the specimen as a fluorescent luminescent spot at each depth of focus;
A profile creation step of creating a bright spot image in which a bright spot region is extracted from the fluorescent image at each focal depth, and creating a brightness profile for each bright spot image;
The brightness profile of the fluorescent particles measured in advance as a reference profile is created, and the brightness profile of the bright spot image at each focal depth is analyzed based on the reference profile, whereby the number of fluorescent particles included in the fluorescence image is determined. A calculation step to calculate,
A method for quantifying a biological material is provided.
本発明の第2の態様によれば、請求項1に記載の生体物質定量方法において、
前記基準プロファイルは、蛍光輝点源となる蛍光粒子からの相対距離と輝度の情報を備えることを特徴とする生体物質定量方法が提供される。 According to a second aspect of the present invention, in the biological material quantification method according toclaim 1,
The reference profile includes information on a relative distance from a fluorescent particle serving as a fluorescent bright spot source and luminance information.
前記基準プロファイルは、蛍光輝点源となる蛍光粒子からの相対距離と輝度の情報を備えることを特徴とする生体物質定量方法が提供される。 According to a second aspect of the present invention, in the biological material quantification method according to
The reference profile includes information on a relative distance from a fluorescent particle serving as a fluorescent bright spot source and luminance information.
本発明の第3の態様によれば、請求項1又は2に記載の生体物質定量方法において、
前記算出工程において、前記蛍光画像に含まれる蛍光粒子の位置を特定し、
各焦点深度における前記輝点画像の中から、算出された蛍光粒子の位置に最も近い位置の輝点画像を抽出する抽出工程と、
前記抽出された輝点画像を合成して一枚の画像に再構成した再構成画像を生成する生成工程と、
を有することを特徴とする生体物質定量方法が提供される。 According to a third aspect of the present invention, in the biological material quantification method according toclaim 1 or 2,
In the calculation step, the position of the fluorescent particles included in the fluorescent image is specified,
An extraction step for extracting a bright spot image at a position closest to the calculated fluorescent particle position from the bright spot images at each focal depth;
A generating step of generating a reconstructed image obtained by synthesizing the extracted bright spot images and reconstructing into one image;
A method for quantifying a biological material is provided.
前記算出工程において、前記蛍光画像に含まれる蛍光粒子の位置を特定し、
各焦点深度における前記輝点画像の中から、算出された蛍光粒子の位置に最も近い位置の輝点画像を抽出する抽出工程と、
前記抽出された輝点画像を合成して一枚の画像に再構成した再構成画像を生成する生成工程と、
を有することを特徴とする生体物質定量方法が提供される。 According to a third aspect of the present invention, in the biological material quantification method according to
In the calculation step, the position of the fluorescent particles included in the fluorescent image is specified,
An extraction step for extracting a bright spot image at a position closest to the calculated fluorescent particle position from the bright spot images at each focal depth;
A generating step of generating a reconstructed image obtained by synthesizing the extracted bright spot images and reconstructing into one image;
A method for quantifying a biological material is provided.
本発明の第4の態様によれば、請求項1~3の何れか一項に記載の生体物質定量方法において、
前記蛍光粒子の平均粒径が20~200nmであることを特徴とする生体物質定量方法が提供される。 According to a fourth aspect of the present invention, in the biological material quantification method according to any one ofclaims 1 to 3,
A biological material quantification method is provided, wherein the fluorescent particles have an average particle size of 20 to 200 nm.
前記蛍光粒子の平均粒径が20~200nmであることを特徴とする生体物質定量方法が提供される。 According to a fourth aspect of the present invention, in the biological material quantification method according to any one of
A biological material quantification method is provided, wherein the fluorescent particles have an average particle size of 20 to 200 nm.
本発明の第5の態様によれば、請求項1~4の何れか一項に記載の生体物質定量方法において、
前記蛍光粒子の粒径の変動係数が15%以下であることを特徴とする生体物質定量方法が提供される。 According to a fifth aspect of the present invention, in the biological material quantification method according to any one ofclaims 1 to 4,
A biological material quantification method is provided, wherein the coefficient of variation of the particle size of the fluorescent particles is 15% or less.
前記蛍光粒子の粒径の変動係数が15%以下であることを特徴とする生体物質定量方法が提供される。 According to a fifth aspect of the present invention, in the biological material quantification method according to any one of
A biological material quantification method is provided, wherein the coefficient of variation of the particle size of the fluorescent particles is 15% or less.
本発明の第6の態様によれば、請求項1~5の何れか一項に記載の生体物質定量方法において、
前記生体物質がタンパク質であることを特徴とする生体物質定量方法が提供される。 According to a sixth aspect of the present invention, in the biological material quantification method according to any one ofclaims 1 to 5,
A biological material quantification method is provided, wherein the biological material is a protein.
前記生体物質がタンパク質であることを特徴とする生体物質定量方法が提供される。 According to a sixth aspect of the present invention, in the biological material quantification method according to any one of
A biological material quantification method is provided, wherein the biological material is a protein.
本発明の第7の態様によれば、
蛍光物質を複数集積した蛍光粒子を染色試薬として用いて特定の生体物質が染色された標本から、前記生体物質を定量する生体物質定量方法において、
焦点深度を所定の間隔で連続的に変え、各焦点深度において、前記標本における前記生体物質の発現を蛍光輝点で表す蛍光画像を入力する入力手段と、
各焦点深度における前記蛍光画像から、輝点領域が抽出された輝点画像を生成し、当該輝点画像ごとに輝度プロファイルを作成するプロファイル作成手段と、
基準プロファイルとして予め計測された蛍光粒子の輝度プロファイルを作成し、各焦点深度における輝点画像の輝度プロファイルを、前記基準プロファイルに基づいて解析することにより、前記蛍光画像に含まれる蛍光粒子の数を算出する算出手段と、
を有することを特徴とする画像処理装置が提供される。 According to a seventh aspect of the present invention,
In a biological material quantification method for quantifying the biological material from a specimen stained with a specific biological material using fluorescent particles in which a plurality of fluorescent materials are accumulated as a staining reagent,
Input means for continuously changing the depth of focus at a predetermined interval, and inputting a fluorescence image representing the expression of the biological material in the specimen as a fluorescence bright spot at each depth of focus;
A profile creating means for generating a bright spot image in which a bright spot region is extracted from the fluorescent image at each focal depth and creating a brightness profile for each bright spot image;
The brightness profile of the fluorescent particles measured in advance as a reference profile is created, and the brightness profile of the bright spot image at each focal depth is analyzed based on the reference profile, whereby the number of fluorescent particles included in the fluorescence image is determined. A calculating means for calculating;
An image processing apparatus is provided.
蛍光物質を複数集積した蛍光粒子を染色試薬として用いて特定の生体物質が染色された標本から、前記生体物質を定量する生体物質定量方法において、
焦点深度を所定の間隔で連続的に変え、各焦点深度において、前記標本における前記生体物質の発現を蛍光輝点で表す蛍光画像を入力する入力手段と、
各焦点深度における前記蛍光画像から、輝点領域が抽出された輝点画像を生成し、当該輝点画像ごとに輝度プロファイルを作成するプロファイル作成手段と、
基準プロファイルとして予め計測された蛍光粒子の輝度プロファイルを作成し、各焦点深度における輝点画像の輝度プロファイルを、前記基準プロファイルに基づいて解析することにより、前記蛍光画像に含まれる蛍光粒子の数を算出する算出手段と、
を有することを特徴とする画像処理装置が提供される。 According to a seventh aspect of the present invention,
In a biological material quantification method for quantifying the biological material from a specimen stained with a specific biological material using fluorescent particles in which a plurality of fluorescent materials are accumulated as a staining reagent,
Input means for continuously changing the depth of focus at a predetermined interval, and inputting a fluorescence image representing the expression of the biological material in the specimen as a fluorescence bright spot at each depth of focus;
A profile creating means for generating a bright spot image in which a bright spot region is extracted from the fluorescent image at each focal depth and creating a brightness profile for each bright spot image;
The brightness profile of the fluorescent particles measured in advance as a reference profile is created, and the brightness profile of the bright spot image at each focal depth is analyzed based on the reference profile, whereby the number of fluorescent particles included in the fluorescence image is determined. A calculating means for calculating;
An image processing apparatus is provided.
本発明の第8の態様によれば、
請求項7に記載の画像処理装置と、
前記画像処理装置で使用される前記蛍光画像を取得する画像取得装置と、
を備えることを特徴とする病理診断支援システムが提供される。 According to an eighth aspect of the present invention,
An image processing apparatus according toclaim 7;
An image acquisition device for acquiring the fluorescent image used in the image processing device;
A pathological diagnosis support system is provided.
請求項7に記載の画像処理装置と、
前記画像処理装置で使用される前記蛍光画像を取得する画像取得装置と、
を備えることを特徴とする病理診断支援システムが提供される。 According to an eighth aspect of the present invention,
An image processing apparatus according to
An image acquisition device for acquiring the fluorescent image used in the image processing device;
A pathological diagnosis support system is provided.
本発明の第9の態様によれば、
蛍光物質を複数集積した蛍光粒子を染色試薬として用いて特定の生体物質が染色された標本から、前記生体物質を定量するコンピュータを、
焦点深度を所定の間隔で連続的に変え、各焦点深度において、前記標本における前記生体物質の発現を蛍光輝点で表す蛍光画像を入力する入力手段、
各焦点深度における前記蛍光画像から、輝点領域が抽出された輝点画像を生成し、当該輝点画像ごとに輝度プロファイルを作成するプロファイル作成手段、
基準プロファイルとして予め計測された蛍光粒子の輝度プロファイルを作成し、各焦点深度における輝点画像の輝度プロファイルを、前記基準プロファイルに基づいて解析することにより、前記蛍光画像に含まれる蛍光粒子の数を算出する算出手段、
として機能させるための画像処理プログラムが提供される。 According to a ninth aspect of the present invention,
A computer for quantifying the biological material from a specimen stained with a specific biological material using fluorescent particles in which a plurality of fluorescent materials are accumulated as a staining reagent,
Input means for continuously changing a focal depth at a predetermined interval, and inputting a fluorescence image representing the expression of the biological material in the specimen as a fluorescent luminescent spot at each focal depth;
Profile creation means for generating a bright spot image from which a bright spot region is extracted from the fluorescent image at each focal depth, and creating a brightness profile for each bright spot image,
The brightness profile of the fluorescent particles measured in advance as a reference profile is created, and the brightness profile of the bright spot image at each focal depth is analyzed based on the reference profile, whereby the number of fluorescent particles included in the fluorescence image is determined. Calculating means for calculating,
An image processing program for functioning as
蛍光物質を複数集積した蛍光粒子を染色試薬として用いて特定の生体物質が染色された標本から、前記生体物質を定量するコンピュータを、
焦点深度を所定の間隔で連続的に変え、各焦点深度において、前記標本における前記生体物質の発現を蛍光輝点で表す蛍光画像を入力する入力手段、
各焦点深度における前記蛍光画像から、輝点領域が抽出された輝点画像を生成し、当該輝点画像ごとに輝度プロファイルを作成するプロファイル作成手段、
基準プロファイルとして予め計測された蛍光粒子の輝度プロファイルを作成し、各焦点深度における輝点画像の輝度プロファイルを、前記基準プロファイルに基づいて解析することにより、前記蛍光画像に含まれる蛍光粒子の数を算出する算出手段、
として機能させるための画像処理プログラムが提供される。 According to a ninth aspect of the present invention,
A computer for quantifying the biological material from a specimen stained with a specific biological material using fluorescent particles in which a plurality of fluorescent materials are accumulated as a staining reagent,
Input means for continuously changing a focal depth at a predetermined interval, and inputting a fluorescence image representing the expression of the biological material in the specimen as a fluorescent luminescent spot at each focal depth;
Profile creation means for generating a bright spot image from which a bright spot region is extracted from the fluorescent image at each focal depth, and creating a brightness profile for each bright spot image,
The brightness profile of the fluorescent particles measured in advance as a reference profile is created, and the brightness profile of the bright spot image at each focal depth is analyzed based on the reference profile, whereby the number of fluorescent particles included in the fluorescence image is determined. Calculating means for calculating,
An image processing program for functioning as
本発明によれば、蛍光物質を複数集積した蛍光粒子を用いて染色した標本を、複数の焦点深度で得られた画像を蛍光輝度のプロファイルを用いて解析することにより、簡易な顕微鏡を用いて、観察対象細胞内の特定の生体物質の量を正確に定量することができる。
According to the present invention, a sample stained with fluorescent particles in which a plurality of fluorescent substances are accumulated is analyzed using images of fluorescence intensities using images obtained at a plurality of focal depths, and a simple microscope is used. The amount of a specific biological substance in the observation target cell can be accurately quantified.
以下、図を参照して本発明を実施するための形態について説明するが、本発明はこれらに限定されない。
Hereinafter, modes for carrying out the present invention will be described with reference to the drawings, but the present invention is not limited thereto.
<病理診断支援システム100の構成>
図1に、本発明の生体物質定量方法を用いた病理診断支援システム100の全体構成例を示す。病理診断支援システム100は、所定の染色試薬で染色された人体の組織標本の顕微鏡画像を取得し、取得された顕微鏡画像を解析することにより、観察対象の組織標本における特定の生体物質の発現を定量的に表す特徴量を出力するシステムである。 <Configuration of PathologicalDiagnosis Support System 100>
FIG. 1 shows an example of the overall configuration of a pathologicaldiagnosis support system 100 using the biological material quantification method of the present invention. The pathological diagnosis support system 100 acquires a microscopic image of a human tissue sample stained with a predetermined staining reagent, and analyzes the acquired microscopic image, thereby expressing the expression of a specific biological material in the tissue sample to be observed. This is a system that outputs feature quantities quantitatively.
図1に、本発明の生体物質定量方法を用いた病理診断支援システム100の全体構成例を示す。病理診断支援システム100は、所定の染色試薬で染色された人体の組織標本の顕微鏡画像を取得し、取得された顕微鏡画像を解析することにより、観察対象の組織標本における特定の生体物質の発現を定量的に表す特徴量を出力するシステムである。 <Configuration of Pathological
FIG. 1 shows an example of the overall configuration of a pathological
図1に示すように、病理診断支援システム100は、顕微鏡画像取得装置1Aと、画像処理装置2Aとがケーブル3A等のインターフェースを介してデータ送受信可能に接続されて構成されている。なお、顕微鏡画像取得装置1Aと画像処理装置2Aとの接続方式は特に限定されない。例えば、顕微鏡画像取得装置1Aと画像処理装置2AはLAN(Local Area Network)により接続されることとしてもよいし、無線により接続される構成としてもよい。
As shown in FIG. 1, the pathological diagnosis support system 100 is configured by connecting a microscope image acquisition device 1A and an image processing device 2A through a cable 3A or the like so as to be able to transmit and receive data. The connection method between the microscope image acquisition device 1A and the image processing device 2A is not particularly limited. For example, the microscope image acquisition device 1A and the image processing device 2A may be connected via a LAN (Local Area Network) or may be configured to be connected wirelessly.
顕微鏡画像取得装置1Aは、公知のカメラ付き光学顕微鏡であり、スライド固定ステージ上に載置されたスライド上の組織標本の顕微鏡画像を取得し、画像処理装置2Aに送信するものである。
顕微鏡画像取得装置1Aは、照射手段、結像手段、撮像手段、及び通信I/F等を備えて構成されている。照射手段は、光源及びフィルター等により構成され、スライド固定ステージに載置されたスライド上の組織標本に光を照射する。結像手段は、接眼レンズ、対物レンズ等により構成され、照射した光によりスライド上の組織標本から発せられる透過光、反射光、又は蛍光を結像する。撮像手段は、CCD(Charge Coupled Device)センサー等を備え、結像手段により結像面に結像される像を撮像して顕微鏡画像のデジタル画像データを生成する顕微鏡設置カメラである。通信I/Fは、生成された顕微鏡画像の画像データを画像処理装置2Aに送信する。本実施の形態において、顕微鏡画像取得装置1Aは、明視野観察に適した照射手段及び結像手段を組み合わせた明視野ユニット、蛍光観察に適した照射手段及び結像手段を組み合わせた蛍光ユニットが備えられており、ユニットを切り替えることにより明視野/蛍光を切り替えることが可能である。蛍光観察時の光源としては、水銀ランプ、キセノンランプ、LED、又はレーザー光など、任意のものが使用できる。 The microscopeimage acquisition apparatus 1A is a known optical microscope with a camera, and acquires a microscope image of a tissue specimen on a slide placed on a slide fixing stage, and transmits the microscope image to the image processing apparatus 2A.
The microscopeimage acquisition apparatus 1A includes an irradiation unit, an imaging unit, an imaging unit, a communication I / F, and the like. The irradiation means includes a light source, a filter, and the like, and irradiates the tissue specimen on the slide placed on the slide fixing stage with light. The imaging means is composed of an eyepiece lens, an objective lens, and the like, and forms an image of transmitted light, reflected light, or fluorescence emitted from the tissue specimen on the slide by the irradiated light. The imaging means is a microscope-installed camera that includes a CCD (Charge Coupled Device) sensor and the like, captures an image formed on the imaging surface by the imaging means, and generates digital image data of the microscope image. The communication I / F transmits image data of the generated microscope image to the image processing apparatus 2A. In the present embodiment, the microscope image acquisition apparatus 1A includes a bright field unit that combines an irradiation unit and an imaging unit suitable for bright field observation, and a fluorescence unit that combines an irradiation unit and an imaging unit suitable for fluorescence observation. It is possible to switch between bright field / fluorescence by switching units. As a light source for fluorescence observation, any lamp such as a mercury lamp, a xenon lamp, an LED, or a laser beam can be used.
顕微鏡画像取得装置1Aは、照射手段、結像手段、撮像手段、及び通信I/F等を備えて構成されている。照射手段は、光源及びフィルター等により構成され、スライド固定ステージに載置されたスライド上の組織標本に光を照射する。結像手段は、接眼レンズ、対物レンズ等により構成され、照射した光によりスライド上の組織標本から発せられる透過光、反射光、又は蛍光を結像する。撮像手段は、CCD(Charge Coupled Device)センサー等を備え、結像手段により結像面に結像される像を撮像して顕微鏡画像のデジタル画像データを生成する顕微鏡設置カメラである。通信I/Fは、生成された顕微鏡画像の画像データを画像処理装置2Aに送信する。本実施の形態において、顕微鏡画像取得装置1Aは、明視野観察に適した照射手段及び結像手段を組み合わせた明視野ユニット、蛍光観察に適した照射手段及び結像手段を組み合わせた蛍光ユニットが備えられており、ユニットを切り替えることにより明視野/蛍光を切り替えることが可能である。蛍光観察時の光源としては、水銀ランプ、キセノンランプ、LED、又はレーザー光など、任意のものが使用できる。 The microscope
The microscope
なお、顕微鏡画像取得装置1Aとしては、カメラ付き顕微鏡に限定されず、例えば、顕微鏡のスライド固定ステージ上のスライドをスキャンして組織標本全体の顕微鏡画像を取得するバーチャル顕微鏡スライド作成装置(例えば、特表2002-514319号公報参照)等を用いてもよい。バーチャル顕微鏡スライド作成装置によれば、スライド上の組織標本全体像を表示部で一度に閲覧可能な画像データを取得することができる。
Note that the microscope image acquisition apparatus 1A is not limited to a microscope with a camera. For example, a virtual microscope slide creation apparatus (for example, a special microscope microscope) that acquires a microscope image of an entire tissue specimen by scanning a slide on a microscope slide fixing stage. Table 2002-514319) may be used. According to the virtual microscope slide creation device, it is possible to acquire image data that allows the entire tissue specimen image on the slide to be viewed on the display unit at a time.
画像処理装置2Aは、顕微鏡画像取得装置1Aから送信された顕微鏡画像を解析することにより、観察対象の組織標本における特定の生体物質の発現分布を算出する。
図2に、画像処理装置2Aの機能構成例を示す。図2に示すように、画像処理装置2Aは、制御部21、操作部22、表示部23、通信I/F24、及び記憶部25等を備えて構成され、各部はバス26を介して接続されている。 Theimage processing apparatus 2A calculates the expression distribution of a specific biological substance in the tissue specimen to be observed by analyzing the microscope image transmitted from the microscope image acquisition apparatus 1A.
FIG. 2 shows a functional configuration example of theimage processing apparatus 2A. As shown in FIG. 2, the image processing apparatus 2 </ b> A includes a control unit 21, an operation unit 22, a display unit 23, a communication I / F 24, a storage unit 25, and the like, and each unit is connected via a bus 26. ing.
図2に、画像処理装置2Aの機能構成例を示す。図2に示すように、画像処理装置2Aは、制御部21、操作部22、表示部23、通信I/F24、及び記憶部25等を備えて構成され、各部はバス26を介して接続されている。 The
FIG. 2 shows a functional configuration example of the
制御部21は、CPU(Central Processing Unit)、RAM(Random Access Memory)等を備えて構成され、記憶部25に記憶されている各種プログラムとの協働により各種処理を実行し、画像処理装置2Aの動作を統括的に制御する。例えば、制御部21は、記憶部25に記憶されているプログラムとの協働により画像解析処理(図5参照)を実行し、プロファイル作成工程、算出工程、抽出工程、及び生成工程を実行するための手段としての機能を実現する。
The control unit 21 includes a CPU (Central Processing Unit), a RAM (Random Access Memory), and the like. The control unit 21 executes various processes in cooperation with various programs stored in the storage unit 25, and performs image processing 2A. Overall control of the operation. For example, the control unit 21 executes an image analysis process (see FIG. 5) in cooperation with a program stored in the storage unit 25, and executes a profile creation process, a calculation process, an extraction process, and a generation process. The function as a means is realized.
操作部22は、文字入力キー、数字入力キー、及び各種機能キー等を備えたキーボードと、マウス等のポインティングデバイスを備えて構成され、キーボードで押下操作されたキーの押下信号とマウスによる操作信号とを、入力信号として制御部21に出力する。
The operation unit 22 includes a keyboard having character input keys, numeric input keys, various function keys, and the like, and a pointing device such as a mouse, and a key pressing signal pressed by the keyboard and an operation signal by the mouse. Are output to the control unit 21 as an input signal.
表示部23は、例えば、CRT(Cathode Ray Tube)やLCD(Liquid Crystal Display)等のモニタを備えて構成されており、制御部21から入力される表示信号の指示に従って、各種画面を表示する。本実施の形態において、表示部23は、画像解析結果を出力するための出力手段として機能する。
The display unit 23 includes, for example, a monitor such as a CRT (Cathode Ray Tube) or an LCD (Liquid Crystal Display), and displays various screens in accordance with display signal instructions input from the control unit 21. In the present embodiment, the display unit 23 functions as an output unit for outputting an image analysis result.
通信I/F24は、顕微鏡画像取得装置1Aをはじめとする外部機器との間でデータ送受信を行なうためのインターフェースである。通信I/F24は、明視野画像と蛍光画像の入力工程を実行するための手段として機能する。
The communication I / F 24 is an interface for transmitting and receiving data to and from external devices such as the microscope image acquisition device 1A. The communication I / F 24 functions as a means for executing a bright-field image and fluorescent image input process.
記憶部25は、例えばHDD(Hard Disk Drive)や半導体の不揮発性メモリー等で構成されている。記憶部25には、前述のように各種プログラムや各種データ等が記憶されている。
その他、画像処理装置2Aは、LANアダプターやルーター等を備え、LAN等の通信ネットワークを介して外部機器と接続される構成としてもよい。 Thestorage unit 25 includes, for example, an HDD (Hard Disk Drive), a semiconductor nonvolatile memory, or the like. As described above, the storage unit 25 stores various programs, various data, and the like.
In addition, theimage processing apparatus 2A may include a LAN adapter, a router, and the like and be connected to an external device via a communication network such as a LAN.
その他、画像処理装置2Aは、LANアダプターやルーター等を備え、LAN等の通信ネットワークを介して外部機器と接続される構成としてもよい。 The
In addition, the
本実施の形態における画像処理装置2Aは、顕微鏡画像取得装置1Aから送信された明視野画像及び蛍光画像を用いて解析を行うことが好ましい。
明視野画像は、H(ヘマトキシリン)染色試薬、HE(ヘマトキシリン-エオジン)染色試薬を用いて染色された組織標本を、顕微鏡画像取得装置1Aにおいて明視野で拡大結像及び撮影することにより得られる顕微鏡画像であって、当該組織標本における細胞の形態を表す細胞形態画像である。ヘマトキシリンは青紫色の色素であり、細胞核、骨組織、軟骨組織の一部、漿液成分など(好塩基性の組織等)を染色する。エオジンは赤~ピンク色の色素であり、細胞質、軟部組織の結合組織、赤血球、線維素、内分泌顆粒など(好酸性の組織等)を染色する。図3に、HE染色を行った組織標本を撮影した明視野画像の一例を示す。
蛍光画像は、特定の生体物質と特異的に結合及び/又は反応する生体物質認識部位が結合した蛍光物質を内包したナノ粒子(以下、蛍光物質内包ナノ粒子又は蛍光粒子と呼ぶ)を含む染色試薬を用いて染色された組織標本に対し、顕微鏡画像取得装置1Aにおいて所定波長の励起光を照射して蛍光物質内包ナノ粒子を発光(蛍光)させ、この蛍光を拡大結像及び撮影することにより得られる顕微鏡画像である。即ち、蛍光画像に現れる蛍光は、組織標本における、生体物質認識部位に対応する特定の生体物質の発現を示すものである。図4に、蛍光画像の一例を示す。 Theimage processing apparatus 2A in the present embodiment preferably performs analysis using the bright field image and the fluorescence image transmitted from the microscope image acquisition apparatus 1A.
The bright field image is obtained by enlarging and photographing a tissue specimen stained with H (hematoxylin) staining reagent and HE (hematoxylin-eosin) staining reagent in a bright field in the microscopeimage acquisition apparatus 1A. It is an image and is a cell form image showing the form of the cell in the tissue specimen. Hematoxylin is a blue-violet pigment that stains cell nuclei, bone tissue, part of cartilage tissue, serous components, etc. (basophilic tissue, etc.). Eosin is a red to pink pigment that stains the cytoplasm, connective tissue of soft tissues, red blood cells, fibrin, endocrine granules, etc. (eosinophilic tissues, etc.). FIG. 3 shows an example of a bright field image obtained by photographing a tissue specimen subjected to HE staining.
A fluorescent image includes a staining reagent including nanoparticles encapsulating a fluorescent substance bound with a biological substance recognition site that specifically binds and / or reacts with a specific biological substance (hereinafter referred to as a fluorescent substance-encapsulated nanoparticle or fluorescent particle). It is obtained by irradiating a fluorescent material-containing nanoparticle by emitting excitation light of a predetermined wavelength in the microscopeimage acquisition device 1A to a tissue specimen stained with a fluorescent material, and enlarging and photographing this fluorescence. It is a microscope image obtained. That is, the fluorescence appearing in the fluorescence image indicates the expression of a specific biological material corresponding to the biological material recognition site in the tissue specimen. FIG. 4 shows an example of the fluorescence image.
明視野画像は、H(ヘマトキシリン)染色試薬、HE(ヘマトキシリン-エオジン)染色試薬を用いて染色された組織標本を、顕微鏡画像取得装置1Aにおいて明視野で拡大結像及び撮影することにより得られる顕微鏡画像であって、当該組織標本における細胞の形態を表す細胞形態画像である。ヘマトキシリンは青紫色の色素であり、細胞核、骨組織、軟骨組織の一部、漿液成分など(好塩基性の組織等)を染色する。エオジンは赤~ピンク色の色素であり、細胞質、軟部組織の結合組織、赤血球、線維素、内分泌顆粒など(好酸性の組織等)を染色する。図3に、HE染色を行った組織標本を撮影した明視野画像の一例を示す。
蛍光画像は、特定の生体物質と特異的に結合及び/又は反応する生体物質認識部位が結合した蛍光物質を内包したナノ粒子(以下、蛍光物質内包ナノ粒子又は蛍光粒子と呼ぶ)を含む染色試薬を用いて染色された組織標本に対し、顕微鏡画像取得装置1Aにおいて所定波長の励起光を照射して蛍光物質内包ナノ粒子を発光(蛍光)させ、この蛍光を拡大結像及び撮影することにより得られる顕微鏡画像である。即ち、蛍光画像に現れる蛍光は、組織標本における、生体物質認識部位に対応する特定の生体物質の発現を示すものである。図4に、蛍光画像の一例を示す。 The
The bright field image is obtained by enlarging and photographing a tissue specimen stained with H (hematoxylin) staining reagent and HE (hematoxylin-eosin) staining reagent in a bright field in the microscope
A fluorescent image includes a staining reagent including nanoparticles encapsulating a fluorescent substance bound with a biological substance recognition site that specifically binds and / or reacts with a specific biological substance (hereinafter referred to as a fluorescent substance-encapsulated nanoparticle or fluorescent particle). It is obtained by irradiating a fluorescent material-containing nanoparticle by emitting excitation light of a predetermined wavelength in the microscope
<蛍光画像の取得>
ここで、蛍光画像の取得方法について、この蛍光画像の取得に際して用いられる染色試薬(蛍光物質内包ナノ粒子)、及び染色試薬による組織標本の染色方法等も含めて詳細に説明する。 <Acquisition of fluorescence image>
Here, a method for acquiring a fluorescent image will be described in detail, including a staining reagent (fluorescent substance-containing nanoparticles) used when acquiring the fluorescent image, a method of staining a tissue specimen with the staining reagent, and the like.
ここで、蛍光画像の取得方法について、この蛍光画像の取得に際して用いられる染色試薬(蛍光物質内包ナノ粒子)、及び染色試薬による組織標本の染色方法等も含めて詳細に説明する。 <Acquisition of fluorescence image>
Here, a method for acquiring a fluorescent image will be described in detail, including a staining reagent (fluorescent substance-containing nanoparticles) used when acquiring the fluorescent image, a method of staining a tissue specimen with the staining reagent, and the like.
〔蛍光物質〕
蛍光画像の取得のための染色試薬に用いられる蛍光物質としては、蛍光有機色素及び量子ドット(半導体粒子)を挙げることができる。200~700nmの範囲内の波長の紫外~近赤外光により励起されたときに、400~1100nmの範囲内の波長の可視~近赤外光の発光を示すことが好ましい。 [Fluorescent substance]
Examples of the fluorescent substance used in the staining reagent for obtaining a fluorescent image include fluorescent organic dyes and quantum dots (semiconductor particles). When excited by ultraviolet to near infrared light having a wavelength in the range of 200 to 700 nm, it preferably emits visible to near infrared light having a wavelength in the range of 400 to 1100 nm.
蛍光画像の取得のための染色試薬に用いられる蛍光物質としては、蛍光有機色素及び量子ドット(半導体粒子)を挙げることができる。200~700nmの範囲内の波長の紫外~近赤外光により励起されたときに、400~1100nmの範囲内の波長の可視~近赤外光の発光を示すことが好ましい。 [Fluorescent substance]
Examples of the fluorescent substance used in the staining reagent for obtaining a fluorescent image include fluorescent organic dyes and quantum dots (semiconductor particles). When excited by ultraviolet to near infrared light having a wavelength in the range of 200 to 700 nm, it preferably emits visible to near infrared light having a wavelength in the range of 400 to 1100 nm.
蛍光有機色素としては、フルオレセイン系色素分子、ローダミン系色素分子、Alexa Fluor(インビトロジェン社製)系色素分子、BODIPY(インビトロジェン社製)系色素分子、カスケード系色素分子、クマリン系色素分子、エオジン系色素分子、NBD系色素分子、ピレン系色素分子、シアニン系色素分子、芳香族炭化水素系分子等を挙げることができる。
Examples of fluorescent organic dyes include fluorescein dye molecules, rhodamine dye molecules, Alexa Fluor (Invitrogen) dye molecules, BODIPY (Invitrogen) dye molecules, cascade dye molecules, coumarin dye molecules, and eosin dyes. And a molecule, an NBD dye molecule, a pyrene dye molecule, a cyanine dye molecule, and an aromatic hydrocarbon molecule.
具体的には、5-カルボキシ-フルオレセイン、6-カルボキシ-フルオレセイン、5,6-ジカルボキシ-フルオレセイン、6-カルボキシ-2’,4,4’,5’,7,7’-ヘキサクロロフルオレセイン、6-カルボキシ-2’,4,7,7’-テトラクロロフルオレセイン、6-カルボキシ-4’,5’-ジクロロ-2’,7’-ジメトキシフルオレセイン、ナフトフルオレセイン、5-カルボキシ-ローダミン、6-カルボキシ-ローダミン、5,6-ジカルボキシ-ローダミン、ローダミン 6G、テトラメチルローダミン、X-ローダミン、スルホローダミンB、スルホローダミン101、及びAlexa Fluor 350、Alexa Fluor 405、Alexa Fluor 430、Alexa Fluor 488、Alexa Fluor 500、Alexa Fluor 514、Alexa Fluor 532、Alexa Fluor 546、Alexa Fluor 555、Alexa Fluor 568、Alexa Fluor 594、Alexa Fluor 610、Alexa Fluor 633、Alexa Fluor 635、Alexa Fluor 647、Alexa Fluor 660、Alexa Fluor 680、Alexa Fluor 700、Alexa Fluor 750、BODIPY FL、BODIPY TMR、BODIPY 493/503、BODIPY 530/550、BODIPY 558/568、BODIPY 564/570、BODIPY 576/589、BODIPY 581/591、BODIPY 630/650、BODIPY 650/665(以上インビトロジェン社製)、メトキシクマリン、エオジン、NBD、ピレン、Cy5、Cy5.5、Cy7、HiLyte Fluor 594(登録商標、アナスペック社製)、DyLight 594(登録商標、サーモサイエンティフィック社製)系色素分子、ATTO 594(登録商標、ATTO-TEC社製)、MFP 594(登録商標、Mobitec社製)、5,10,15,20-テトラフェニルポルフィンテトラスルホン酸、亜鉛5,10,15,20-テトラフェニルポルフィンテトラスルホン酸、フタロシアニンテトラスルホン酸、亜鉛フタロシアニンテトラスルホン酸、N,N-Bis-(2,6-diisopropylphenyl)-1,6,7,12-(4-tert-butylphenoxy)-perylen-3,4,9,10-tetracarbonacid diimide、N,N’-Bis(2,6-diisopropylphenyl)-1,6,7,12-tetraphenoxyperylene-3,4:9,10-tetracarboxdiimide、Benzenesulfonic acid, 4,4',4'',4'''-[(1,3,8,10-tetrahydro-1,3,8,10-tetraoxoperylo[3,4-cd:9,10-c'd']dipyran-5,6,12,13-tetrayl)tetralis(oxy)]tetrakis-等を挙げることができる。単独でも複数種を混合したものを用いてもよい。
Specifically, 5-carboxy-fluorescein, 6-carboxy-fluorescein, 5,6-dicarboxy-fluorescein, 6-carboxy-2 ′, 4,4 ′, 5 ′, 7,7′-hexachlorofluorescein, 6 -Carboxy-2 ', 4,7,7'-tetrachlorofluorescein, 6-carboxy-4', 5'-dichloro-2 ', 7'-dimethoxyfluorescein, naphthofluorescein, 5-carboxy-rhodamine, 6-carboxy -Rhodamine, 5,6-dicarboxy-rhodamine, rhodamine 6G, tetramethylrhodamine, X-rhodamine, sulforhodamine B, sulforhodamine 101, and Alexa Fluor 350, Alexa Fluor 405, Alexa Fluor 430, Alexa Fluor 488, Alexa Fluor 500, Alexa Fluor 514, Alexa Fluor 532, Alexa Fluor 546, A lexa Fluor 555, Alexa Fluor 568, Alexa Fluor 594, Alexa Fluor 610, Alexa Fluor 633, Alexa Fluor 635, Alexa Fluor 647, Alexa Fluor 660, Alexa Fluor 680, Alexa Fluor 700, Alexa Fluor 750, BODIPY FLROD BODIPY 493/503, BODIPY 530/550, BODIPY 558/568, BODIPY 564/570, BODIPY 576/589, BODIPY 581/591, BODIPY 630/650, BODIPY 650/665 (more from Invitrogen), methoxycoumarin, eosin , NBD, pyrene, Cy5, Cy5.5, Cy7, HiLyte Fluor 594 (registered trademark, manufactured by Anaspec), DyLight 594 (registered trademark, manufactured by Thermo Scientific), dye molecule, ATTO 594 (registered trademark, ATTO) -TEC), MFP 594 (registered trademark, manufactured by Mobitec), 5,10,15,20-tetraphenylporphine tetrasulfonic acid, zinc 5,10,15,20-tetraphenylporphine tetrasulfonic acid, phthalocyanine Nine tetrasulfonic acid, zinc phthalocyanine tetrasulfonic acid, N, N-Bis- (2,6-diisopropylphenyl) -1,6,7,12- (4-tert-butylphenoxy) -perylen-3,4,9,10 -tetracarbonacid diimide, N, N'-Bis (2,6-diisopropylphenyl) -1,6,7,12-tetraphenoxyperylene-3,4: 9,10-tetracarboxdiimide, Benzenesulfonic acid, 4,4 ', 4' ', 4 '' '-[(1,3,8,10-tetrahydro-1,3,8,10-tetraoxoperylo [3,4-cd: 9,10-c'd'] dipyran-5,6,12, 13-tetrayl) tetralis (oxy)] tetrakis- and the like. You may use individually or what mixed multiple types.
量子ドットとしては、II-VI族化合物、III-V族化合物、又はIV族元素を成分として含有する量子ドット(それぞれ、「II-VI族量子ドット」、「III-V族量子ドット」、「IV族量子ドット」ともいう。)のいずれかを用いることができる。単独でも複数種を混合したものを用いてもよい。
As quantum dots, quantum dots containing II-VI group compounds, III-V group compounds, or group IV elements as components ("II-VI group quantum dots", "III-V group quantum dots", " Or “Group IV quantum dots”). You may use individually or what mixed multiple types.
具体的には、CdSe、CdS、CdTe、ZnSe、ZnS、ZnTe、InP、InN、InAs、InGaP、GaP、GaAs、Si、Geが挙げられるが、これらに限定されない。
Specific examples include CdSe, CdS, CdTe, ZnSe, ZnS, ZnTe, InP, InN, InAs, InGaP, GaP, GaAs, Si, and Ge, but are not limited thereto.
上記量子ドットをコアとし、その上にシェルを設けた量子ドットを用いることもできる。以下、本明細書中シェルを有する量子ドットの表記法として、コアがCdSe、シェルがZnSの場合、CdSe/ZnSと表記する。例えば、CdSe/ZnS、CdS/ZnS、InP/ZnS、InGaP/ZnS、Si/SiO2、Si/ZnS、Ge/GeO2、Ge/ZnS等を用いることができるが、これらに限定されない。
量子ドットは必要に応じて、有機ポリマー等により表面処理が施されているものを用いてもよい。例えば、表面カルボキシ基を有するCdSe/ZnS(インビトロジェン社製)、表面アミノ基を有するCdSe/ZnS(インビトロジェン社製)等が挙げられる。 It is also possible to use a quantum dot having the above quantum dot as a core and a shell provided thereon. Hereinafter, as a notation of a quantum dot having a shell in this specification, when the core is CdSe and the shell is ZnS, it is expressed as CdSe / ZnS. For example, CdSe / ZnS, CdS / ZnS, InP / ZnS, InGaP / ZnS, Si / SiO 2 , Si / ZnS, Ge / GeO 2 , Ge / ZnS, and the like can be used, but are not limited thereto.
As the quantum dots, those subjected to surface treatment with an organic polymer or the like may be used as necessary. Examples thereof include CdSe / ZnS having a surface carboxy group (manufactured by Invitrogen), CdSe / ZnS having a surface amino group (manufactured by Invitrogen), and the like.
量子ドットは必要に応じて、有機ポリマー等により表面処理が施されているものを用いてもよい。例えば、表面カルボキシ基を有するCdSe/ZnS(インビトロジェン社製)、表面アミノ基を有するCdSe/ZnS(インビトロジェン社製)等が挙げられる。 It is also possible to use a quantum dot having the above quantum dot as a core and a shell provided thereon. Hereinafter, as a notation of a quantum dot having a shell in this specification, when the core is CdSe and the shell is ZnS, it is expressed as CdSe / ZnS. For example, CdSe / ZnS, CdS / ZnS, InP / ZnS, InGaP / ZnS, Si / SiO 2 , Si / ZnS, Ge / GeO 2 , Ge / ZnS, and the like can be used, but are not limited thereto.
As the quantum dots, those subjected to surface treatment with an organic polymer or the like may be used as necessary. Examples thereof include CdSe / ZnS having a surface carboxy group (manufactured by Invitrogen), CdSe / ZnS having a surface amino group (manufactured by Invitrogen), and the like.
〔蛍光物質内包ナノ粒子〕
本実施の形態において蛍光物質内包ナノ粒子とは、蛍光物質がナノ粒子内部に分散されたものをいい、蛍光物質とナノ粒子自体とが化学的に結合していても、結合していなくてもよい。
ナノ粒子を構成する素材は特に限定されるものではなく、ポリスチレン、ポリ乳酸、シリカ、メラミン等を挙げることができる。 [Fluorescent substance-containing nanoparticles]
In the present embodiment, the fluorescent substance-encapsulating nanoparticles are those in which the fluorescent substance is dispersed inside the nanoparticles, whether the fluorescent substance and the nanoparticles themselves are chemically bonded or not. Good.
The material constituting the nanoparticles is not particularly limited, and examples thereof include polystyrene, polylactic acid, silica, and melamine.
本実施の形態において蛍光物質内包ナノ粒子とは、蛍光物質がナノ粒子内部に分散されたものをいい、蛍光物質とナノ粒子自体とが化学的に結合していても、結合していなくてもよい。
ナノ粒子を構成する素材は特に限定されるものではなく、ポリスチレン、ポリ乳酸、シリカ、メラミン等を挙げることができる。 [Fluorescent substance-containing nanoparticles]
In the present embodiment, the fluorescent substance-encapsulating nanoparticles are those in which the fluorescent substance is dispersed inside the nanoparticles, whether the fluorescent substance and the nanoparticles themselves are chemically bonded or not. Good.
The material constituting the nanoparticles is not particularly limited, and examples thereof include polystyrene, polylactic acid, silica, and melamine.
本実施の形態で用いられる蛍光物質内包ナノ粒子は、公知の方法により作製することが可能である。例えば、蛍光有機色素を内包したシリカナノ粒子は、ラングミュア 8巻 2921ページ(1992)に記載されているFITC内包シリカ粒子の合成を参考に合成することができる。FITCの代わりに所望の蛍光有機色素を用いることで種々の蛍光有機色素内包シリカナノ粒子を合成することができる。
Fluorescent substance-containing nanoparticles used in the present embodiment can be produced by a known method. For example, silica nanoparticles encapsulating a fluorescent organic dye can be synthesized with reference to the synthesis of FITC-encapsulated silica particles described in Langmuir 8, Vol. 2921 (1992). Various fluorescent organic dye-containing silica nanoparticles can be synthesized by using a desired fluorescent organic dye in place of FITC.
量子ドットを内包したシリカナノ粒子は、ニュー・ジャーナル・オブ・ケミストリー 33巻 561ページ(2009)に記載されているCdTe内包シリカナノ粒子の合成を参考に合成することができる。
Silica nanoparticles encapsulating quantum dots can be synthesized with reference to the synthesis of CdTe-encapsulated silica nanoparticles described in New Journal of Chemistry, Vol. 33, p. 561 (2009).
蛍光有機色素を内包したポリスチレンナノ粒子は、米国特許4326008(1982)に記載されている重合性官能基をもつ有機色素を用いた共重合法や、米国特許5326692(1992)に記載されているポリスチレンナノ粒子への蛍光有機色素の含浸法を用いて作製することができる。
Polystyrene nanoparticles encapsulating a fluorescent organic dye may be copolymerized using an organic dye having a polymerizable functional group described in US Pat. No. 4,326,008 (1982) or polystyrene described in US Pat. No. 5,326,692 (1992). It can be produced using a method of impregnating nanoparticles with a fluorescent organic dye.
量子ドットを内包したポリマーナノ粒子は、ネイチャー・バイオテクノロジー19巻631ページ(2001)に記載されているポリスチレンナノ粒子への量子ドットの含浸法を用いて作製することができる。
Polymer nanoparticles encapsulating quantum dots can be prepared using the method of impregnating polystyrene nanoparticles with quantum dots described in Nature Biotechnology, Vol. 19, page 631 (2001).
本実施の形態で用いられる蛍光物質内包ナノ粒子の平均粒径は特に限定されないが、粒径が大きいものは抗原にアクセスしにくく、粒径が小さく輝度の低いものは蛍光物質内包ナノ粒子の信号がバックグラウンドノイズ(カメラのノイズや細胞の自家蛍光)に埋もれてしまうことから、20~200nm程度のものが好適である。
また、粒径のばらつきを示す変動係数(=(標準偏差/平均値)×100%)は、粒径のばらつきが大きい場合は、抗原へのアクセスがばらつくという観点から、15%以下のものを用いることが好ましい。
平均粒径は、走査型電子顕微鏡(SEM)を用いて電子顕微鏡写真を撮影し十分な数の粒子について断面積を計測し、各計測値を円の面積としたときの円の直径を粒径として求めた。本願においては、1000個の粒子の粒径の算術平均を平均粒径とした。変動係数も、平均粒径と同様に1000個の粒子の粒径から算出した。 The average particle size of the fluorescent substance-containing nanoparticles used in this embodiment is not particularly limited, but those having a large particle size are difficult to access the antigen, and those having a small particle size and low luminance are signals of the fluorescent substance-containing nanoparticles. Are buried in the background noise (camera noise and cell autofluorescence), and those of about 20 to 200 nm are preferable.
In addition, the coefficient of variation (= (standard deviation / average value) × 100%) indicating the variation in particle size is 15% or less from the viewpoint that when the particle size variation is large, access to the antigen varies. It is preferable to use it.
The average particle diameter is obtained by taking an electron micrograph using a scanning electron microscope (SEM), measuring the cross-sectional area of a sufficient number of particles, and taking each measured value as the area of the circle. As sought. In the present application, the arithmetic average of the particle sizes of 1000 particles is defined as the average particle size. The coefficient of variation was calculated from the particle size of 1000 particles in the same manner as the average particle size.
また、粒径のばらつきを示す変動係数(=(標準偏差/平均値)×100%)は、粒径のばらつきが大きい場合は、抗原へのアクセスがばらつくという観点から、15%以下のものを用いることが好ましい。
平均粒径は、走査型電子顕微鏡(SEM)を用いて電子顕微鏡写真を撮影し十分な数の粒子について断面積を計測し、各計測値を円の面積としたときの円の直径を粒径として求めた。本願においては、1000個の粒子の粒径の算術平均を平均粒径とした。変動係数も、平均粒径と同様に1000個の粒子の粒径から算出した。 The average particle size of the fluorescent substance-containing nanoparticles used in this embodiment is not particularly limited, but those having a large particle size are difficult to access the antigen, and those having a small particle size and low luminance are signals of the fluorescent substance-containing nanoparticles. Are buried in the background noise (camera noise and cell autofluorescence), and those of about 20 to 200 nm are preferable.
In addition, the coefficient of variation (= (standard deviation / average value) × 100%) indicating the variation in particle size is 15% or less from the viewpoint that when the particle size variation is large, access to the antigen varies. It is preferable to use it.
The average particle diameter is obtained by taking an electron micrograph using a scanning electron microscope (SEM), measuring the cross-sectional area of a sufficient number of particles, and taking each measured value as the area of the circle. As sought. In the present application, the arithmetic average of the particle sizes of 1000 particles is defined as the average particle size. The coefficient of variation was calculated from the particle size of 1000 particles in the same manner as the average particle size.
〔生体物質認識部位と蛍光物質内包ナノ粒子との結合〕
本実施の形態に係る生体物質認識部位とは、目的とする生体物質と特異的に結合及び/又は反応する部位である。目的とする生体物質は、それと特異的に結合する物質が存在するものであれば特に限定されるものではないが、代表的にはタンパク質(ペプチド)および核酸(オリゴヌクレオチド、ポリヌクレオチド)、抗体等が挙げられる。したがって、そのような目的とする生体物質に結合する物質としては、前記タンパク質を抗原として認識する抗体やそれに特異的に結合する他のタンパク質等、および前記核酸にハイブリタイズする塩基配列を有する核酸等が挙げられる。具体的には、細胞表面に存在するタンパク質であるHER2に特異的に結合する抗HER2抗体、細胞核に存在するエストロゲン受容体(ER)に特異的に結合する抗ER抗体、細胞骨格を形成するアクチンに特異的に結合する抗アクチン抗体等があげられる。中でも抗HER2抗体及び抗ER抗体を蛍光物質内包ナノ粒子に結合させたものは、乳癌の投薬選定に用いることができ、好ましい。 [Bonding of biological substance recognition sites and fluorescent substance-containing nanoparticles]
The biological material recognition site according to the present embodiment is a site that specifically binds and / or reacts with the target biological material. The target biological substance is not particularly limited as long as a substance that specifically binds to the target biological substance exists, but typically, protein (peptide), nucleic acid (oligonucleotide, polynucleotide), antibody, etc. Is mentioned. Accordingly, substances that bind to the target biological substance include antibodies that recognize the protein as an antigen, other proteins that specifically bind to the protein, and nucleic acids having a base sequence that hybridizes to the nucleic acid. Is mentioned. Specifically, an anti-HER2 antibody that specifically binds to HER2, which is a protein present on the cell surface, an anti-ER antibody that specifically binds to an estrogen receptor (ER) present in the cell nucleus, and actin that forms a cytoskeleton And an anti-actin antibody that specifically binds to. Among them, those in which anti-HER2 antibody and anti-ER antibody are bound to fluorescent substance-encapsulating nanoparticles can be used for selection of breast cancer medication, and are preferable.
本実施の形態に係る生体物質認識部位とは、目的とする生体物質と特異的に結合及び/又は反応する部位である。目的とする生体物質は、それと特異的に結合する物質が存在するものであれば特に限定されるものではないが、代表的にはタンパク質(ペプチド)および核酸(オリゴヌクレオチド、ポリヌクレオチド)、抗体等が挙げられる。したがって、そのような目的とする生体物質に結合する物質としては、前記タンパク質を抗原として認識する抗体やそれに特異的に結合する他のタンパク質等、および前記核酸にハイブリタイズする塩基配列を有する核酸等が挙げられる。具体的には、細胞表面に存在するタンパク質であるHER2に特異的に結合する抗HER2抗体、細胞核に存在するエストロゲン受容体(ER)に特異的に結合する抗ER抗体、細胞骨格を形成するアクチンに特異的に結合する抗アクチン抗体等があげられる。中でも抗HER2抗体及び抗ER抗体を蛍光物質内包ナノ粒子に結合させたものは、乳癌の投薬選定に用いることができ、好ましい。 [Bonding of biological substance recognition sites and fluorescent substance-containing nanoparticles]
The biological material recognition site according to the present embodiment is a site that specifically binds and / or reacts with the target biological material. The target biological substance is not particularly limited as long as a substance that specifically binds to the target biological substance exists, but typically, protein (peptide), nucleic acid (oligonucleotide, polynucleotide), antibody, etc. Is mentioned. Accordingly, substances that bind to the target biological substance include antibodies that recognize the protein as an antigen, other proteins that specifically bind to the protein, and nucleic acids having a base sequence that hybridizes to the nucleic acid. Is mentioned. Specifically, an anti-HER2 antibody that specifically binds to HER2, which is a protein present on the cell surface, an anti-ER antibody that specifically binds to an estrogen receptor (ER) present in the cell nucleus, and actin that forms a cytoskeleton And an anti-actin antibody that specifically binds to. Among them, those in which anti-HER2 antibody and anti-ER antibody are bound to fluorescent substance-encapsulating nanoparticles can be used for selection of breast cancer medication, and are preferable.
特定抗原としては以下を例示することができ、各抗原を認識する抗体はさまざまな抗体メーカーから入手可能であるとともに一般的な知識に基づいて作成可能である。例示としてM.アクチン、M.S.アクチン、S.M.アクチン、ACTH、Alk-1、α1-アンチキモトリプシン、α1-アンチトリプシン、AFP、bcl-2、bcl-6、β-カテニン、BCA 225、CA19-9、CA125、カルシトニン、カルレチニン、CD1a、CD3、CD4、CD5、CD8、CD10、CD15、CD20、CD21、CD23、CD30、CD31、CD34、CD43、CD45、CD45R、CD56、CD57、CD61、CD68、CD79a、"CD99、MIC2"、CD138、クロモグラニン、c-KIT、c-MET、コラーゲン タイプIV、Cox-2、サイクリンD1、ケラチン、サイトケラチン(高分子量)、パンケラチン、パンケラチン、サイトケラチン5/6、サイトケラチン7、サイトケラチン8、サイトケラチン8/18、サイトケラチン14、サイトケラチン19、サイトケラチン20、CMV、E-カドヘリン、EGFR、ER、EMA、EBV、第VIII因子関連抗原、ファッシン、FSH、ガレクチン-3、ガストリン、GFAP、グルカゴン、グリコフォリンA、グランザイムB、hCG、hGH、ヘリコバクターピロリ、HBc抗原、HBs抗原、ヘパトサイト特異抗原、HER2、HSV-I、HSV-II、HHV-8、IgA、IgG、IgM、IGF-1R、インヒビン、インスリン、カッパL鎖、Ki67、ラムダL鎖、LH、リゾチーム、マクロファージ、メランA、MLH-1、MSH-2、ミエロパーオキシダーゼ、ミオゲニン、ミオグロビン、ミオシン、ニューロフィラメント、NSE、p27(Kip1)、p53、p53、P63、PAX5、PLAP、ニューモシスティス カリニ、ポドプラニン(D2-40)、PGR、プロラクチン、PSA、前立腺酸性フォスファターゼ、Renal Cell Carcinoma、S100、ソマトスタチン、スペクトリン、シナプトフィジン、TAG-72、TdT、サイログロブリン、TSH、TTF-1、TRAcP、トリプターゼ、ビリン、ビメンチン、WT1、Zap-70が挙げられる。
The following are examples of specific antigens, and antibodies that recognize each antigen are available from various antibody manufacturers and can be prepared based on general knowledge. Examples include M. actin, MS actin, SM actin, ACTH, Alk-1, α1-antichymotrypsin, α1-antitrypsin, AFP, bcl-2, bcl-6, β-catenin, BCA 225, CA19-9, CA125 , Calcitonin, calretinin, CD1a, CD3, CD4, CD5, CD8, CD10, CD15, CD20, CD21, CD23, CD30, CD31, CD34, CD43, CD45, CD45R, CD56, CD57, CD61, CD68, CD79a, "CD99, MIC2 ", CD138, chromogranin, c-KIT, c-MET, collagen type IV, Cox-2, cyclin D1, keratin, cytokeratin (high molecular weight), pankeratin, pankeratin, cytokeratin 5/6, cytokeratin 7 , Cytokeratin 8, cytokeratin 8/18, cytokeratin 14, cytokeratin 19, cytokeratin 20, CMV, E-cadherin, EGFR, ER, EMA, EBV, factor VIII related antigen, fascin, FSH, galectin-3 , Gastrin, GFAP, glucagon, glycofoli A, Granzyme B, hCG, hGH, Helicobacter pylori, HBc antigen, HBs antigen, hepatocyte specific antigen, HER2, HSV-I, HSV-II, HHV-8, IgA, IgG, IgM, IGF-1R, inhibin, insulin, Kappa light chain, Ki67, lambda light chain, LH, lysozyme, macrophage, Melan A, MLH-1, MSH-2, myeloperoxidase, myogenin, myoglobin, myosin, neurofilament, NSE, p27 (Kip1), p53, p53 , P63, PAX5, PLAP, Pneumocystis carini, podoplanin (D2-40), PGR, prolactin, PSA, prostate acid phosphatase, Renal Cell Carcinoma, S100, somatostatin, spectrin, synaptophysin, TAG-72, TdT, thyroglobulin, Examples include TSH, TTF-1, TRAcP, tryptase, villin, vimentin, WT1, and Zap-70.
目的とする生体物質が核酸の場合、病気との関連が指摘されている特定核酸遺伝子としては以下を例示することができ、各特定核酸遺伝子を認識するプローブは、BACプローブとして入手可能であるとともに一般的な知識に基づいて作成可能である。具体的な特定核酸遺伝子の例示は以下の通り。癌の増殖や分子標的薬の奏効率に関係する遺伝子として、HER2、TOP2A、HER3、EGFR、P53、METなどが挙げられ、さらに、各種癌関連遺伝子として知られている遺伝子として、以下のものが挙げられる。チロシンキナーゼ関連遺伝子として、ALK、FLT3、AXL、FLT4(VEGFR3、DDR1、FMS(CSF1R)、DDR2、EGFR(ERBB1)、HER4(ERBB4)、EML4-ALK、IGF1R、EPHA1、INSR、EPHA2、IRR(INSRR)、EPHA3、KIT、EPHA4、LTK、EPHA5、MER(MERTK)、EPHA6、MET、EPHA7、MUSK、EPHA8、NPM1-ALK、EPHB1、PDGFRα(PDGFRA)、EPHB2、PDGFRβ(PDGFRB)EPHB3、RET、EPHB4、RON(MST1R)、FGFR1、ROS(ROS1)、FGFR2、TIE2(TEK)、FGFR3、TRKA(NTRK1)、FGFR4、TRKB(NTRK2)、FLT1(VEGFR1)、TRKC(NTRK3)が挙げられる。また、乳がん関連の遺伝子としてATM、BRCA1、BRCA2、BRCA3、CCND1、E-Cadherin、ERBB2、ETV6、FGFR1、HRAS、KRAS、NRAS、NTRK3、p53、PTENが挙げられる。カルチノイド腫瘍に関連する遺伝子として、BCL2、BRD4、CCND1、CDKN1A、CDKN2A、CTNNB1、HES1、MAP2、MEN1、NF1、NOTCH1、NUT、RAF、SDHD、VEGFAが挙げられる。大腸がん関連遺伝子として、APC、MSH6、AXIN2、MYH、BMPR1A、p53、DCC、PMS2、KRAS2(or Ki-ras)、PTEN、MLH1、SMAD4、MSH2、STK11、MSH6が挙げられる。肺がん関連の遺伝子としては、ALK、PTEN、CCND1、RASSF1A、CDKN2A、RB1、EGFR、RET、EML4、ROS1、KRAS2、TP53、MYCが挙げられる。肝臓がん関連の遺伝子としては、Axin1、MALAT1、b-catenin、p16 INK4A、c-ERBB-2、p53、CTNNB1、RB1、Cyclin D1、SMAD2、EGFR、SMAD4、IGFR2、TCF1、KRASが挙げられる。腎臓がん関連遺伝子として、Alpha、PRCC、ASPSCR1、PSF、CLTC、TFE3、p54nrb/NONO、TFEBが挙げられる。甲状腺がん関連遺伝子として、AKAP10、NTRK1、AKAP9、RET、BRAF、TFG、ELE1、TPM3、H4/D10S170、TPRが挙げられる。卵巣がん関連遺伝子として、AKT2、MDM2、BCL2、MYC、BRCA1、NCOA4、CDKN2A、p53、ERBB2、PIK3CA、GATA4、RB、HRAS、RET、KRAS、RNASET2が挙げられる。前立腺がん関連遺伝子として、AR、KLK3、BRCA2、MYC、CDKN1B、NKX3.1、EZH2、p53、GSTP1、PTENが挙げられる。骨腫瘍関連遺伝子として、CDH11、COL12A1、CNBP、OMD、COL1A1、THRAP3、COL4A5、USP6が挙げられる。
When the target biological material is a nucleic acid, examples of the specific nucleic acid gene that has been pointed out to be associated with a disease can include the following, and probes that recognize each specific nucleic acid gene are available as BAC probes: Can be created based on general knowledge. Specific examples of specific nucleic acid genes are as follows. HER2, TOP2A, HER3, EGFR, P53, MET, etc. are mentioned as genes related to cancer growth and molecular target drug response. Furthermore, the following genes are known as various cancer-related genes. Can be mentioned. Tyrosine kinase-related genes include ALK, FLT3, AXL, FLT4 (VEGFR3, DDR1, FMS (CSF1R), DDR2, EGFR (ERBB1), HER4 (ERBB4), EML4-ALK, IGF1R, EPHA1, INSR, EPHA2, IRR (INSRR) ), EPHA3, KIT, EPHA4, LTK, EPHA5, MER (MERTK), EPHA6, MET, EPHA7, MUSK, EPHA8, NPM1-ALK, EPHB1, PDGFRα (PDGFRA), EPHB2, PDGFRβ (PDGFRB), EPHEP3, T RON (MST1R), FGFR1, ROS (ROS1), FGFR2, TIE2 (TEK), FGFR3, TRKA (NTRK1), FGFR4, TRKB (NT RK2), FLT1 (VEGFR1), TRKC (NTRK3), and breast cancer-related genes are ATM, BRCA1, BRCA2, BRCA3, CCND1, E-cadherin, ERBB2, ETV6, FGFR1, HRAS, KRAS, NRAS, NTR, NTR Genes associated with carcinoid tumors include BCL2, BRD4, CCND1, CDKN1A, CDKN2A, CTNNB1, HES1, MAP2, MEN1, NF1, NOTCH1, NUT, RAF, SDHD, and VEGFA. Cancer-related genes include APC, MSH6, AXIN2, MYH, BMPR1A, p53, DCC, PMS2, KRAS2 (or Ki-ras), PTEN, MLH1, and SMA. 4, MSH2, STK11, MSH6 Lung cancer-related genes include ALK, PTEN, CCND1, RASSF1A, CDKN2A, RB1, EGFR, RET, EML4, ROS1, KRAS2, TP53, MYC. Related genes include Axin1, MALAT1, b-catenin, p16 INK4A, c-ERBB-2, p53, CTNNB1, RB1, Cyclin D1, SMAD2, EGFR, SMAD4, IGFR2, TCF1, and KRAS. Related genes include Alpha, PRCC, ASPSCR1, PSF, CLTC, TFE3, p54nrb / NONO, and TFEB As thyroid cancer-related genes, AKAP10, NTRK1, and AKA 9, RET, BRAF, TFG, ELE1, TPM3, H4 / D10S170, TPR and the like. Examples of ovarian cancer-related genes include AKT2, MDM2, BCL2, MYC, BRCA1, NCOA4, CDKN2A, p53, ERBB2, PIK3CA, GATA4, RB, HRAS, RET, KRAS, and RNASET2. Examples of prostate cancer-related genes include AR, KLK3, BRCA2, MYC, CDKN1B, NKX3.1, EZH2, p53, GSTP1, and PTEN. Examples of bone tumor-related genes include CDH11, COL12A1, CNBP, OMD, COL1A1, THRAP3, COL4A5, and USP6.
生体物質認識部位と蛍光物質内包ナノ粒子の結合の態様としては特に限定されず、共有結合、イオン結合、水素結合、配位結合、物理吸着及び化学吸着等が挙げられる。結合の安定性から共有結合等の結合力の強い結合が好ましい。
The mode of binding between the biological substance recognition site and the fluorescent substance-encapsulating nanoparticles is not particularly limited, and examples thereof include covalent bonding, ionic bonding, hydrogen bonding, coordination bonding, physical adsorption, and chemical adsorption. A bond having a strong bonding force such as a covalent bond is preferred from the viewpoint of bond stability.
また、生体物質認識部位と蛍光物質内包ナノ粒子の間を連結する有機分子があってもよい。例えば、生体物質との非特異的吸着を抑制するため、ポリエチレングリコール鎖を用いることができ、Thermo Scientific社製SM(PEG)12を用いることができる。
In addition, there may be an organic molecule that connects between the biological substance recognition site and the fluorescent substance-containing nanoparticle. For example, in order to suppress non-specific adsorption with a biological substance, a polyethylene glycol chain can be used, and SM (PEG) 12 manufactured by Thermo Scientific can be used.
蛍光物質内包シリカナノ粒子へ生体物質認識部位を結合させる場合、蛍光物質が蛍光有機色素の場合でも、量子ドットの場合でも同様の手順を適用することができる。例えば、無機物と有機物を結合させるために広く用いられている化合物であるシランカップリング剤を用いることができる。このシランカップリング剤は、分子の一端に加水分解でシラノール基を与えるアルコキシシリル基を有し、他端に、カルボキシル基、アミノ基、エポキシ基、アルデヒド基等の官能基を有する化合物であり、上記シラノール基の酸素原子を介して無機物と結合する。具体的には、メルカプトプロピルトリエトキシシラン、グリシドキシプロピルトリエトキシシラン、アミノプロピルトリエトキシシラン、ポリエチレングリコール鎖をもつシランカップリング剤(例えば、Gelest社製PEG-silane no.SIM6492.7)等が挙げられる。シランカップリング剤を用いる場合、二種以上を併用してもよい。
When binding a biological substance recognition site to a fluorescent substance-encapsulating silica nanoparticle, the same procedure can be applied regardless of whether the fluorescent substance is a fluorescent organic dye or a quantum dot. For example, a silane coupling agent that is a compound widely used for bonding an inorganic substance and an organic substance can be used. This silane coupling agent is a compound having an alkoxysilyl group that gives a silanol group by hydrolysis at one end of the molecule and a functional group such as a carboxyl group, an amino group, an epoxy group, an aldehyde group at the other end, Bonding with an inorganic substance through an oxygen atom of the silanol group. Specifically, mercaptopropyltriethoxysilane, glycidoxypropyltriethoxysilane, aminopropyltriethoxysilane, a silane coupling agent having a polyethylene glycol chain (for example, PEG-silane no. SIM6492.7 manufactured by Gelest), etc. Is mentioned. When using a silane coupling agent, you may use 2 or more types together.
蛍光有機色素内包シリカナノ粒子とシランカップリング剤との反応手順は、公知の手法を用いることができる。例えば、得られた蛍光有機色素内包シリカナノ粒子を純水中に分散させ、アミノプロピルトリエトキシシランを添加し、室温で12時間反応させる。反応終了後、遠心分離又はろ過により表面がアミノプロピル基で修飾された蛍光有機色素内包シリカナノ粒子を得ることができる。続いてアミノ基と抗体中のカルボキシル基とを反応させることで、アミド結合を介し抗体を蛍光有機色素内包シリカナノ粒子と結合させることができる。必要に応じて、EDC(1-Ethyl-3-[3-Dimethylaminopropyl]carbodiimide Hydrochloride:Pierce社製、登録商標)のような縮合剤を用いることもできる。
A publicly known method can be used for the reaction procedure of the fluorescent organic dye-encapsulated silica nanoparticles and the silane coupling agent. For example, the obtained fluorescent organic dye-encapsulated silica nanoparticles are dispersed in pure water, aminopropyltriethoxysilane is added, and the mixture is reacted at room temperature for 12 hours. After completion of the reaction, fluorescent organic dye-encapsulated silica nanoparticles whose surface is modified with an aminopropyl group can be obtained by centrifugation or filtration. Subsequently, by reacting an amino group with a carboxyl group in the antibody, the antibody can be bound to the fluorescent organic dye-encapsulated silica nanoparticles via an amide bond. If necessary, a condensing agent such as EDC (1-Ethyl-3- [3-Dimethylaminopropyl] carbodiimide Hydrochloride: manufactured by Pierce) can also be used.
必要により、有機分子で修飾された蛍光有機色素内包シリカナノ粒子と直接結合しうる部位と、分子標的物質と結合しうる部位とを有するリンカー化合物を用いることができる。具体例として、アミノ基と選択的に反応する部位とメルカプト基と選択的に反応する部位の両方をもつsulfo-SMCC(Sulfosuccinimidyl 4[N-maleimidomethyl]-cyclohexane-1-carboxylate:Pierce社製)を用いると、アミノプロピルトリエトキシシランで修飾した蛍光有機色素内包シリカナノ粒子のアミノ基と、抗体中のメルカプト基を結合させることで、抗体結合した蛍光有機色素内包シリカナノ粒子ができる。
If necessary, a linker compound having a site that can be directly bonded to the fluorescent organic dye-encapsulated silica nanoparticles modified with organic molecules and a site that can be bonded to the molecular target substance can be used. As a specific example, sulfo-SMCC (Sulfosuccinimidyl 4 [N-maleimidomethyl] -cyclohexane-1-carboxylate: manufactured by Pierce) having both a site that selectively reacts with an amino group and a site that reacts selectively with a mercapto group When used, by binding the amino group of the fluorescent organic dye-encapsulated silica nanoparticles modified with aminopropyltriethoxysilane and the mercapto group in the antibody, antibody-bound fluorescent organic dye-encapsulated silica nanoparticles can be produced.
蛍光物質内包ポリスチレンナノ粒子へ生体物質認識部位を結合させる場合、蛍光物質が蛍光有機色素の場合でも、量子ドットの場合でも同様の手順を適用することができる。すなわち、アミノ基等の官能基をもつポリスチレンナノ粒子へ蛍光有機色素、量子ドットを含浸することにより、官能基もつ蛍光物質内包ポリスチレンナノ粒子を得ることができ、以降EDC又はsulfo-SMCCを用いることで、抗体結合した蛍光物質内包ポリスチレンナノ粒子ができる。
蛍光物質内包メラミンナノ粒子へ生体物質認識部位を結合させる場合、蛍光物質内包シリカナノ粒子と同様の手順を適用することができる。また、より反応性を向上させるため、メラミンナノ粒子と多官能性アミン化合物をあらかじめ反応させて表面アミノ基数を増やしても良い。 When the biological substance recognition site is bound to the fluorescent substance-encapsulated polystyrene nanoparticles, the same procedure can be applied regardless of whether the fluorescent substance is a fluorescent organic dye or a quantum dot. That is, by impregnating a polystyrene nanoparticle having a functional group such as an amino group with a fluorescent organic dye or a quantum dot, a fluorescent substance-containing polystyrene nanoparticle having a functional group can be obtained, and thereafter EDC or sulfo-SMCC is used. Thus, antibody-bound fluorescent substance-encapsulated polystyrene nanoparticles can be produced.
When the biological substance recognition site is bound to the fluorescent substance-encapsulated melamine nanoparticles, the same procedure as that for the fluorescent substance-encapsulated silica nanoparticles can be applied. In order to further improve the reactivity, the number of surface amino groups may be increased by reacting melamine nanoparticles with a polyfunctional amine compound in advance.
蛍光物質内包メラミンナノ粒子へ生体物質認識部位を結合させる場合、蛍光物質内包シリカナノ粒子と同様の手順を適用することができる。また、より反応性を向上させるため、メラミンナノ粒子と多官能性アミン化合物をあらかじめ反応させて表面アミノ基数を増やしても良い。 When the biological substance recognition site is bound to the fluorescent substance-encapsulated polystyrene nanoparticles, the same procedure can be applied regardless of whether the fluorescent substance is a fluorescent organic dye or a quantum dot. That is, by impregnating a polystyrene nanoparticle having a functional group such as an amino group with a fluorescent organic dye or a quantum dot, a fluorescent substance-containing polystyrene nanoparticle having a functional group can be obtained, and thereafter EDC or sulfo-SMCC is used. Thus, antibody-bound fluorescent substance-encapsulated polystyrene nanoparticles can be produced.
When the biological substance recognition site is bound to the fluorescent substance-encapsulated melamine nanoparticles, the same procedure as that for the fluorescent substance-encapsulated silica nanoparticles can be applied. In order to further improve the reactivity, the number of surface amino groups may be increased by reacting melamine nanoparticles with a polyfunctional amine compound in advance.
〔染色方法〕
以下、組織標本の染色方法について述べるが、本発明は組織標本に限定されるものではなく、基板上に固定した細胞等の標本にも適用可能である。
また、以下に説明する染色方法が適用できる組織標本の作製法は特に限定されず、公知の方法により作製されたものを用いることができる。 [Dyeing method]
Hereinafter, a method for staining a tissue specimen will be described, but the present invention is not limited to a tissue specimen, and can also be applied to a specimen such as a cell fixed on a substrate.
Moreover, the preparation method of the tissue specimen which can apply the dyeing | staining method demonstrated below is not specifically limited, What was produced by the well-known method can be used.
以下、組織標本の染色方法について述べるが、本発明は組織標本に限定されるものではなく、基板上に固定した細胞等の標本にも適用可能である。
また、以下に説明する染色方法が適用できる組織標本の作製法は特に限定されず、公知の方法により作製されたものを用いることができる。 [Dyeing method]
Hereinafter, a method for staining a tissue specimen will be described, but the present invention is not limited to a tissue specimen, and can also be applied to a specimen such as a cell fixed on a substrate.
Moreover, the preparation method of the tissue specimen which can apply the dyeing | staining method demonstrated below is not specifically limited, What was produced by the well-known method can be used.
1)脱パラフィン工程
まず、操作者は、キシレンを入れた容器に組織標本を浸漬させ、パラフィンを除去する。温度は特に限定されるものではないが、室温で行うことができる。浸漬時間は、3分以上30分以下であることが好ましい。また、必要により浸漬途中でキシレンを交換してもよい。
次いで、エタノールを入れた容器に組織標本を浸漬させ、キシレンを除去する。温度は特に限定されるものではないが、室温で行うことができる。浸漬時間は、3分以上30分以下であることが好ましい。また、必要により浸漬途中でエタノールを交換してもよい。
次いで、水を入れた容器に組織標本を浸漬させ、エタノールを除去する。温度は特に限定されるものではないが、室温で行うことができる。浸漬時間は、3分以上30分以下であることが好ましい。また、必要により浸漬途中で水を交換してもよい。 1) Deparaffinization process First, the operator immerses the tissue specimen in a container containing xylene to remove paraffin. The temperature is not particularly limited, but can be performed at room temperature. The immersion time is preferably 3 minutes or longer and 30 minutes or shorter. If necessary, xylene may be exchanged during the immersion.
Next, the tissue specimen is immersed in a container containing ethanol to remove xylene. The temperature is not particularly limited, but can be performed at room temperature. The immersion time is preferably 3 minutes or longer and 30 minutes or shorter. Further, if necessary, ethanol may be exchanged during the immersion.
Next, the tissue specimen is immersed in a container containing water to remove ethanol. The temperature is not particularly limited, but can be performed at room temperature. The immersion time is preferably 3 minutes or longer and 30 minutes or shorter. Moreover, you may exchange water in the middle of immersion as needed.
まず、操作者は、キシレンを入れた容器に組織標本を浸漬させ、パラフィンを除去する。温度は特に限定されるものではないが、室温で行うことができる。浸漬時間は、3分以上30分以下であることが好ましい。また、必要により浸漬途中でキシレンを交換してもよい。
次いで、エタノールを入れた容器に組織標本を浸漬させ、キシレンを除去する。温度は特に限定されるものではないが、室温で行うことができる。浸漬時間は、3分以上30分以下であることが好ましい。また、必要により浸漬途中でエタノールを交換してもよい。
次いで、水を入れた容器に組織標本を浸漬させ、エタノールを除去する。温度は特に限定されるものではないが、室温で行うことができる。浸漬時間は、3分以上30分以下であることが好ましい。また、必要により浸漬途中で水を交換してもよい。 1) Deparaffinization process First, the operator immerses the tissue specimen in a container containing xylene to remove paraffin. The temperature is not particularly limited, but can be performed at room temperature. The immersion time is preferably 3 minutes or longer and 30 minutes or shorter. If necessary, xylene may be exchanged during the immersion.
Next, the tissue specimen is immersed in a container containing ethanol to remove xylene. The temperature is not particularly limited, but can be performed at room temperature. The immersion time is preferably 3 minutes or longer and 30 minutes or shorter. Further, if necessary, ethanol may be exchanged during the immersion.
Next, the tissue specimen is immersed in a container containing water to remove ethanol. The temperature is not particularly limited, but can be performed at room temperature. The immersion time is preferably 3 minutes or longer and 30 minutes or shorter. Moreover, you may exchange water in the middle of immersion as needed.
2)賦活化処理
操作者は、公知の方法にならい、目的とする生体物質の賦活化処理を行う。賦活化条件に特に定めはないが、賦活液としては、0.01M クエン酸緩衝液(pH6.0)、1mM EDTA溶液(pH8.0)、5% 尿素、0.1M トリス塩酸緩衝液等を用いることができる。加熱機器は、オートクレーブ、マイクロウェーブ、圧力鍋、ウォーターバス等を用いることができる。温度は特に限定されるものではないが、室温で行うことができる。温度は50~130℃、時間は5~30分で行うことができる。
次いで、PBS(Phosphate Buffered Saline:リン酸緩衝生理食塩水)を入れた容器に、賦活化処理後の組織標本を浸漬させ、洗浄を行う。温度は特に限定されるものではないが、室温で行うことができる。浸漬時間は、3分以上30分以下であることが好ましい。また、必要により浸漬途中でPBSを交換してもよい。 2) Activation process An operator performs the activation process of the target biological substance according to a well-known method. The activation conditions are not particularly defined, but as the activation liquid, 0.01 M citrate buffer (pH 6.0), 1 mM EDTA solution (pH 8.0), 5% urea, 0.1 M Tris-HCl buffer, etc. Can be used. As the heating device, an autoclave, a microwave, a pressure cooker, a water bath, or the like can be used. The temperature is not particularly limited, but can be performed at room temperature. The temperature can be 50 to 130 ° C. and the time can be 5 to 30 minutes.
Next, the tissue specimen after the activation treatment is immersed in a container containing PBS (Phosphate Buffered Saline) and washed. The temperature is not particularly limited, but can be performed at room temperature. The immersion time is preferably 3 minutes or longer and 30 minutes or shorter. If necessary, the PBS may be replaced during the immersion.
操作者は、公知の方法にならい、目的とする生体物質の賦活化処理を行う。賦活化条件に特に定めはないが、賦活液としては、0.01M クエン酸緩衝液(pH6.0)、1mM EDTA溶液(pH8.0)、5% 尿素、0.1M トリス塩酸緩衝液等を用いることができる。加熱機器は、オートクレーブ、マイクロウェーブ、圧力鍋、ウォーターバス等を用いることができる。温度は特に限定されるものではないが、室温で行うことができる。温度は50~130℃、時間は5~30分で行うことができる。
次いで、PBS(Phosphate Buffered Saline:リン酸緩衝生理食塩水)を入れた容器に、賦活化処理後の組織標本を浸漬させ、洗浄を行う。温度は特に限定されるものではないが、室温で行うことができる。浸漬時間は、3分以上30分以下であることが好ましい。また、必要により浸漬途中でPBSを交換してもよい。 2) Activation process An operator performs the activation process of the target biological substance according to a well-known method. The activation conditions are not particularly defined, but as the activation liquid, 0.01 M citrate buffer (pH 6.0), 1 mM EDTA solution (pH 8.0), 5% urea, 0.1 M Tris-HCl buffer, etc. Can be used. As the heating device, an autoclave, a microwave, a pressure cooker, a water bath, or the like can be used. The temperature is not particularly limited, but can be performed at room temperature. The temperature can be 50 to 130 ° C. and the time can be 5 to 30 minutes.
Next, the tissue specimen after the activation treatment is immersed in a container containing PBS (Phosphate Buffered Saline) and washed. The temperature is not particularly limited, but can be performed at room temperature. The immersion time is preferably 3 minutes or longer and 30 minutes or shorter. If necessary, the PBS may be replaced during the immersion.
3)生体物質認識部位が結合された蛍光物質内包ナノ粒子を用いた染色
操作者は、生体物質認識部位が結合された蛍光物質内包ナノ粒子のPBS分散液を組織標本に載せ、目的とする生体物質と反応させる。蛍光物質内包ナノ粒子と結合させる生体物質認識部位を変えることにより、さまざまな生体物質に対応した染色が可能となる。数種類の生体物質認識部位が結合された蛍光物質内包ナノ粒子を用いる場合には、それぞれの蛍光物質内包ナノ粒子PBS分散液を予め混合しておいてもよいし、別々に順次組織標本に載せてもよい。
温度は特に限定されるものではないが、室温で行うことができる。反応時間は、30分以上24時間以下であることが好ましい。
蛍光物質内包ナノ粒子による染色を行う前に、BSA含有PBS等、公知のブロッキング剤を滴下することが好ましい。
次いで、PBSを入れた容器に、染色後の組織標本を浸漬させ、未反応蛍光物質内包ナノ粒子の除去を行う。温度は特に限定されるものではないが、室温で行うことができる。浸漬時間は、3分以上30分以下であることが好ましい。また、必要により浸漬途中でPBSを交換してもよい。カバーガラスを組織標本に載せ、封入する。必要に応じて市販の封入剤を使用してもよい。
なお、HE染色試薬を用いて染色を行う場合、カバーガラスによる封入前にHE染色を行う。 3) Staining using fluorescent substance-encapsulating nanoparticles with a biological substance recognition site bonded The operator places a PBS dispersion of fluorescent substance-encapsulating nanoparticles with a biological substance recognition site on a tissue specimen, React with substance. By changing the biological material recognition site to be combined with the fluorescent substance-containing nanoparticles, staining corresponding to various biological materials becomes possible. When using fluorescent substance-encapsulated nanoparticles to which several kinds of biological substance recognition sites are bound, each fluorescent substance-encapsulated nanoparticle PBS dispersion may be mixed in advance or separately placed on a tissue specimen separately. Also good.
The temperature is not particularly limited, but can be performed at room temperature. The reaction time is preferably 30 minutes or more and 24 hours or less.
It is preferable to drop a known blocking agent such as BSA-containing PBS before staining with fluorescent substance-encapsulating nanoparticles.
Next, the stained tissue specimen is immersed in a container containing PBS to remove unreacted fluorescent substance-containing nanoparticles. The temperature is not particularly limited, but can be performed at room temperature. The immersion time is preferably 3 minutes or longer and 30 minutes or shorter. If necessary, the PBS may be replaced during the immersion. Place the cover glass on the tissue specimen and enclose it. A commercially available encapsulant may be used as necessary.
In addition, when dyeing | staining using a HE dyeing reagent, HE dyeing is performed before enclosure with a cover glass.
操作者は、生体物質認識部位が結合された蛍光物質内包ナノ粒子のPBS分散液を組織標本に載せ、目的とする生体物質と反応させる。蛍光物質内包ナノ粒子と結合させる生体物質認識部位を変えることにより、さまざまな生体物質に対応した染色が可能となる。数種類の生体物質認識部位が結合された蛍光物質内包ナノ粒子を用いる場合には、それぞれの蛍光物質内包ナノ粒子PBS分散液を予め混合しておいてもよいし、別々に順次組織標本に載せてもよい。
温度は特に限定されるものではないが、室温で行うことができる。反応時間は、30分以上24時間以下であることが好ましい。
蛍光物質内包ナノ粒子による染色を行う前に、BSA含有PBS等、公知のブロッキング剤を滴下することが好ましい。
次いで、PBSを入れた容器に、染色後の組織標本を浸漬させ、未反応蛍光物質内包ナノ粒子の除去を行う。温度は特に限定されるものではないが、室温で行うことができる。浸漬時間は、3分以上30分以下であることが好ましい。また、必要により浸漬途中でPBSを交換してもよい。カバーガラスを組織標本に載せ、封入する。必要に応じて市販の封入剤を使用してもよい。
なお、HE染色試薬を用いて染色を行う場合、カバーガラスによる封入前にHE染色を行う。 3) Staining using fluorescent substance-encapsulating nanoparticles with a biological substance recognition site bonded The operator places a PBS dispersion of fluorescent substance-encapsulating nanoparticles with a biological substance recognition site on a tissue specimen, React with substance. By changing the biological material recognition site to be combined with the fluorescent substance-containing nanoparticles, staining corresponding to various biological materials becomes possible. When using fluorescent substance-encapsulated nanoparticles to which several kinds of biological substance recognition sites are bound, each fluorescent substance-encapsulated nanoparticle PBS dispersion may be mixed in advance or separately placed on a tissue specimen separately. Also good.
The temperature is not particularly limited, but can be performed at room temperature. The reaction time is preferably 30 minutes or more and 24 hours or less.
It is preferable to drop a known blocking agent such as BSA-containing PBS before staining with fluorescent substance-encapsulating nanoparticles.
Next, the stained tissue specimen is immersed in a container containing PBS to remove unreacted fluorescent substance-containing nanoparticles. The temperature is not particularly limited, but can be performed at room temperature. The immersion time is preferably 3 minutes or longer and 30 minutes or shorter. If necessary, the PBS may be replaced during the immersion. Place the cover glass on the tissue specimen and enclose it. A commercially available encapsulant may be used as necessary.
In addition, when dyeing | staining using a HE dyeing reagent, HE dyeing is performed before enclosure with a cover glass.
〔蛍光画像の取得〕
染色した組織標本に対し顕微鏡画像取得装置1Aを用いて、広視野の顕微鏡画像(蛍光画像)を取得する。顕微鏡画像取得装置1Aにおいて、染色試薬に用いた蛍光物質の吸収極大波長及び蛍光波長に対応した励起光源及び蛍光検出用光学フィルターを選択する。
解析に用いられる蛍光顕微鏡画像(蛍光画像)の取得条件には特に制限はないが、対物レンズの倍率は4~100倍が好ましく、開口数(NA)は0.6以上が好ましく、さらに好ましくは0.8以上である。また、撮像するカメラのサンプリングピッチは、400nm以下が好ましく、さらに好ましくは150nm以下である。 (Fluorescence image acquisition)
A microscope image (fluorescence image) with a wide field of view is acquired from the stained tissue specimen using the microscopeimage acquisition device 1A. In the microscope image acquisition apparatus 1A, an excitation light source and a fluorescence detection optical filter corresponding to the absorption maximum wavelength and fluorescence wavelength of the fluorescent material used for the staining reagent are selected.
There are no particular restrictions on the acquisition conditions of the fluorescence microscope image (fluorescence image) used for the analysis, but the magnification of the objective lens is preferably 4 to 100 times, and the numerical aperture (NA) is preferably 0.6 or more, more preferably 0.8 or more. It is. Further, the sampling pitch of the camera for imaging is preferably 400 nm or less, and more preferably 150 nm or less.
染色した組織標本に対し顕微鏡画像取得装置1Aを用いて、広視野の顕微鏡画像(蛍光画像)を取得する。顕微鏡画像取得装置1Aにおいて、染色試薬に用いた蛍光物質の吸収極大波長及び蛍光波長に対応した励起光源及び蛍光検出用光学フィルターを選択する。
解析に用いられる蛍光顕微鏡画像(蛍光画像)の取得条件には特に制限はないが、対物レンズの倍率は4~100倍が好ましく、開口数(NA)は0.6以上が好ましく、さらに好ましくは0.8以上である。また、撮像するカメラのサンプリングピッチは、400nm以下が好ましく、さらに好ましくは150nm以下である。 (Fluorescence image acquisition)
A microscope image (fluorescence image) with a wide field of view is acquired from the stained tissue specimen using the microscope
There are no particular restrictions on the acquisition conditions of the fluorescence microscope image (fluorescence image) used for the analysis, but the magnification of the objective lens is preferably 4 to 100 times, and the numerical aperture (NA) is preferably 0.6 or more, more preferably 0.8 or more. It is. Further, the sampling pitch of the camera for imaging is preferably 400 nm or less, and more preferably 150 nm or less.
<病理診断支援システム100の動作(画像処理方法を含む。)>
以下、病理診断支援システム100において、上記説明した蛍光画像及び明視野画像を取得して解析を行う動作について説明する。ここでは、乳癌組織におけるHER2タンパク(以下、特定タンパクと呼ぶ。)を認識する生体物質認識部位が結合した蛍光物質内包ナノ粒子を含む染色試薬を用いて染色された組織標本を観察対象とする場合を例にとり説明するが、これに限定されるものではない。 <Operation of Pathological Diagnosis Support System 100 (Including Image Processing Method)>
Hereinafter, an operation of acquiring and analyzing the above-described fluorescence image and bright field image in the pathologicaldiagnosis support system 100 will be described. Here, when a tissue specimen stained with a staining reagent containing fluorescent substance-containing nanoparticles bound to a biological substance recognition site that recognizes HER2 protein (hereinafter referred to as a specific protein) in breast cancer tissue is to be observed. However, the present invention is not limited to this.
以下、病理診断支援システム100において、上記説明した蛍光画像及び明視野画像を取得して解析を行う動作について説明する。ここでは、乳癌組織におけるHER2タンパク(以下、特定タンパクと呼ぶ。)を認識する生体物質認識部位が結合した蛍光物質内包ナノ粒子を含む染色試薬を用いて染色された組織標本を観察対象とする場合を例にとり説明するが、これに限定されるものではない。 <Operation of Pathological Diagnosis Support System 100 (Including Image Processing Method)>
Hereinafter, an operation of acquiring and analyzing the above-described fluorescence image and bright field image in the pathological
まず、操作者は、HE染色試薬と、特定タンパクを認識する生体物質認識部位が結合した蛍光物質内包ナノ粒子を蛍光標識材料とした染色試薬との、2種の染色試薬を用いて組織標本を染色する。
その後、顕微鏡画像取得装置1Aにおいて、以下の(a1)~(a6)の手順により明視野画像及び蛍光画像を取得する。
(a1)操作者は、HE染色試薬と蛍光物質内包ナノ粒子を含む染色試薬とにより染色された組織標本をスライドに載置し、そのスライドを顕微鏡画像取得装置1Aのスライド固定ステージに設置する。
(a2)明視野ユニットに設定し、撮影倍率及びピントの調整を行って、組織標本上の観察対象の領域を視野に納め、焦点深度の移動方向(ここでは、上下方向とする)の撮影開始位置、撮影終了位置、及びピッチを設定する。スライド固定ステージを所定の撮影開始位置まで上方又は下方に移動する。
(a3)撮像手段で撮影を行って明視野画像の画像データを生成し、画像処理装置2Aに画像データを送信する。
(a4)ユニットを蛍光ユニットに変更する。
(a5)スライド固定ステージの位置及び撮影倍率を変えずに撮像手段で撮影を行って蛍光画像の画像データを生成し、画像処理装置2Aに画像データを送信する。
(a6)スライド固定ステージを上方又は下方に所定のピッチ移動させて焦点深度を変えた後、再び(a3)~(a5)の手順を行う。
操作者は、スライド固定ステージが所定の撮影終了位置に到達するまで、(a6)の手順を繰り返す。 First, the operator uses two types of staining reagents, a HE staining reagent and a staining reagent using fluorescent substance-encapsulated nanoparticles bound with a biological substance recognition site that recognizes a specific protein as a fluorescent labeling material. Stain.
Thereafter, in the microscopeimage acquisition apparatus 1A, a bright field image and a fluorescence image are acquired by the following procedures (a1) to (a6).
(A1) The operator places the tissue specimen stained with the HE staining reagent and the staining reagent containing the fluorescent substance-containing nanoparticles on the slide, and places the slide on the slide fixing stage of the microscopeimage acquisition apparatus 1A.
(A2) Set in the bright field unit, adjust the imaging magnification and focus, place the area to be observed on the tissue specimen in the field of view, and start imaging in the direction of focus depth movement (here, the vertical direction) Set the position, shooting end position, and pitch. The slide fixing stage is moved upward or downward to a predetermined photographing start position.
(A3) Shooting is performed by the imaging unit to generate bright field image data, and the image data is transmitted to theimage processing apparatus 2A.
(A4) Change the unit to a fluorescent unit.
(A5) Photographing is performed by the imaging means without changing the position of the slide fixing stage and the photographing magnification to generate image data of a fluorescent image, and the image data is transmitted to theimage processing apparatus 2A.
(A6) The slide fixing stage is moved upward or downward by a predetermined pitch to change the depth of focus, and then the procedures (a3) to (a5) are performed again.
The operator repeats the procedure of (a6) until the slide fixing stage reaches a predetermined photographing end position.
その後、顕微鏡画像取得装置1Aにおいて、以下の(a1)~(a6)の手順により明視野画像及び蛍光画像を取得する。
(a1)操作者は、HE染色試薬と蛍光物質内包ナノ粒子を含む染色試薬とにより染色された組織標本をスライドに載置し、そのスライドを顕微鏡画像取得装置1Aのスライド固定ステージに設置する。
(a2)明視野ユニットに設定し、撮影倍率及びピントの調整を行って、組織標本上の観察対象の領域を視野に納め、焦点深度の移動方向(ここでは、上下方向とする)の撮影開始位置、撮影終了位置、及びピッチを設定する。スライド固定ステージを所定の撮影開始位置まで上方又は下方に移動する。
(a3)撮像手段で撮影を行って明視野画像の画像データを生成し、画像処理装置2Aに画像データを送信する。
(a4)ユニットを蛍光ユニットに変更する。
(a5)スライド固定ステージの位置及び撮影倍率を変えずに撮像手段で撮影を行って蛍光画像の画像データを生成し、画像処理装置2Aに画像データを送信する。
(a6)スライド固定ステージを上方又は下方に所定のピッチ移動させて焦点深度を変えた後、再び(a3)~(a5)の手順を行う。
操作者は、スライド固定ステージが所定の撮影終了位置に到達するまで、(a6)の手順を繰り返す。 First, the operator uses two types of staining reagents, a HE staining reagent and a staining reagent using fluorescent substance-encapsulated nanoparticles bound with a biological substance recognition site that recognizes a specific protein as a fluorescent labeling material. Stain.
Thereafter, in the microscope
(A1) The operator places the tissue specimen stained with the HE staining reagent and the staining reagent containing the fluorescent substance-containing nanoparticles on the slide, and places the slide on the slide fixing stage of the microscope
(A2) Set in the bright field unit, adjust the imaging magnification and focus, place the area to be observed on the tissue specimen in the field of view, and start imaging in the direction of focus depth movement (here, the vertical direction) Set the position, shooting end position, and pitch. The slide fixing stage is moved upward or downward to a predetermined photographing start position.
(A3) Shooting is performed by the imaging unit to generate bright field image data, and the image data is transmitted to the
(A4) Change the unit to a fluorescent unit.
(A5) Photographing is performed by the imaging means without changing the position of the slide fixing stage and the photographing magnification to generate image data of a fluorescent image, and the image data is transmitted to the
(A6) The slide fixing stage is moved upward or downward by a predetermined pitch to change the depth of focus, and then the procedures (a3) to (a5) are performed again.
The operator repeats the procedure of (a6) until the slide fixing stage reaches a predetermined photographing end position.
画像処理装置2Aにおいては、明視野画像及び蛍光画像に基づき画像解析処理が実行される。
図5に、画像処理装置2Aにおける画像解析処理のフローチャートを示す。図5に示す画像解析処理は、制御部21と記憶部25に記憶されているプログラムとの協働により実行される。 In theimage processing apparatus 2A, image analysis processing is executed based on the bright field image and the fluorescence image.
FIG. 5 shows a flowchart of image analysis processing in theimage processing apparatus 2A. The image analysis processing shown in FIG. 5 is executed in cooperation with the control unit 21 and the program stored in the storage unit 25.
図5に、画像処理装置2Aにおける画像解析処理のフローチャートを示す。図5に示す画像解析処理は、制御部21と記憶部25に記憶されているプログラムとの協働により実行される。 In the
FIG. 5 shows a flowchart of image analysis processing in the
まず、通信I/F24により顕微鏡画像取得装置1Aからの明視野画像が入力されると(ステップS1)、制御部21により、明視野画像から細胞領域の抽出が行われる(ステップS2)。
図6に、ステップS2における処理の詳細フローを示す。ステップS2の処理は、制御部21と記憶部25に記憶されているプログラムとの協働により実行される。 First, when a bright field image is input from the microscopeimage acquisition device 1A through the communication I / F 24 (step S1), the control unit 21 extracts a cell region from the bright field image (step S2).
FIG. 6 shows a detailed flow of the process in step S2. The process of step S2 is executed in cooperation with thecontrol unit 21 and the program stored in the storage unit 25.
図6に、ステップS2における処理の詳細フローを示す。ステップS2の処理は、制御部21と記憶部25に記憶されているプログラムとの協働により実行される。 First, when a bright field image is input from the microscope
FIG. 6 shows a detailed flow of the process in step S2. The process of step S2 is executed in cooperation with the
ステップS2においては、まず、明視野画像のモノクロ画像への変換が行われる(ステップS201)。図7Aに、明視野画像の一例を示す。
次いで、モノクロ画像に対し予め定められた閾値を用いて閾値処理が施され、各画素の値が二値化される(ステップS202)。 In step S2, first, a bright-field image is converted into a monochrome image (step S201). FIG. 7A shows an example of a bright field image.
Next, threshold processing is performed on the monochrome image using a predetermined threshold, and the value of each pixel is binarized (step S202).
次いで、モノクロ画像に対し予め定められた閾値を用いて閾値処理が施され、各画素の値が二値化される(ステップS202)。 In step S2, first, a bright-field image is converted into a monochrome image (step S201). FIG. 7A shows an example of a bright field image.
Next, threshold processing is performed on the monochrome image using a predetermined threshold, and the value of each pixel is binarized (step S202).
次いで、ノイズ処理が行われる(ステップS203)。ノイズ処理は、具体的には、二値画像にクロージング処理が施されることにより行うことができる。クロージング処理は、膨張処理を行ってから同じ回数分だけ収縮処理を行う処理である。膨張処理は、注目画素からn×n画素(nは2以上の整数)の範囲内にある画素に1つでも白が含まれている場合に注目画素を白に置き換える処理である。収縮処理は、注目画素からn×n画素の範囲内にある画素に1つでも黒が含まれている場合に注目画素を黒に置き換える処理である。クロージング処理により、ノイズ等の小さい領域を除去することができる。図7Bに、ノイズ処理後の画像の一例を示す。図7Bに示すように、ノイズ処理後には、細胞が抽出された画像(細胞画像)が生成される。
Next, noise processing is performed (step S203). Specifically, the noise process can be performed by performing a closing process on the binary image. The closing process is a process in which the contraction process is performed the same number of times after the expansion process is performed. The expansion process is a process of replacing a target pixel with white when at least one pixel in the range of n × n pixels (n is an integer of 2 or more) from the target pixel is white. The contraction process is a process of replacing a target pixel with black when at least one pixel in the range of n × n pixels from the target pixel contains black. By the closing process, a small area such as noise can be removed. FIG. 7B shows an example of an image after noise processing. As shown in FIG. 7B, after noise processing, an image (cell image) from which cells are extracted is generated.
次いで、ノイズ処理後の画像にラベリング処理が施され、抽出された細胞のそれぞれにラベルが付与される(ステップS204)。ラベリング処理とは、連結している画素に同じラベル(番号)を付与していくことで画像内のオブジェクトを識別する処理である。ラベリング処理により、ノイズ処理後の画像から各細胞を識別してラベルを付与することができる。
Next, a labeling process is performed on the image after the noise process, and a label is assigned to each of the extracted cells (step S204). The labeling process is a process for identifying an object in an image by assigning the same label (number) to connected pixels. By the labeling process, each cell can be identified from the image after the noise process and a label can be applied.
一方、通信I/F24により顕微鏡画像取得装置1Aからの蛍光画像が入力されると(ステップS3)、制御部21により、蛍光画像から蛍光粒子が抽出される(ステップS4)。
図8に、ステップS4における処理の詳細フローを示す。ステップS4の処理は、制御部21と記憶部25に記憶されているプログラムとの協働により実行される。 On the other hand, when a fluorescent image is input from the microscopeimage acquisition device 1A through the communication I / F 24 (step S3), the control unit 21 extracts fluorescent particles from the fluorescent image (step S4).
FIG. 8 shows a detailed flow of the process in step S4. The process of step S4 is executed in cooperation with thecontrol unit 21 and the program stored in the storage unit 25.
図8に、ステップS4における処理の詳細フローを示す。ステップS4の処理は、制御部21と記憶部25に記憶されているプログラムとの協働により実行される。 On the other hand, when a fluorescent image is input from the microscope
FIG. 8 shows a detailed flow of the process in step S4. The process of step S4 is executed in cooperation with the
ステップS4においては、まず、蛍光画像から蛍光輝点の波長に応じた色成分の抽出が行われる(ステップS401)。図9Aに、蛍光画像の一例を示す。ステップS401では、たとえば、蛍光粒子の発光波長が550nmである場合には、その波長成分を有する蛍光輝点のみが画像として抽出される。
In step S4, first, a color component corresponding to the wavelength of the fluorescent bright spot is extracted from the fluorescent image (step S401). FIG. 9A shows an example of a fluorescence image. In step S401, for example, when the emission wavelength of the fluorescent particles is 550 nm, only the fluorescent bright spot having the wavelength component is extracted as an image.
次いで、抽出された画像に閾値処理が施されて二値画像が生成され、輝点領域が抽出される(ステップS402)。
なお、閾値処理の前に細胞自家蛍光や他の不要信号成分等のノイズ除去処理が施されてもよく、ガウシアンフィルタ等のローパスフィルタや二次微分等のハイパスフィルタが好ましく用いられる。
図9Bに、輝点領域が抽出された画像の一例を示す。図9Bに示すように、かかる画像では蛍光輝点を中心とした輝点領域が抽出されている。 Next, threshold processing is performed on the extracted image to generate a binary image, and a bright spot region is extracted (step S402).
Note that noise removal processing such as cell autofluorescence and other unnecessary signal components may be performed before the threshold processing, and a low-pass filter such as a Gaussian filter or a high-pass filter such as a second derivative is preferably used.
FIG. 9B shows an example of an image from which the bright spot region is extracted. As shown in FIG. 9B, a bright spot region centered on the fluorescent bright spot is extracted from such an image.
なお、閾値処理の前に細胞自家蛍光や他の不要信号成分等のノイズ除去処理が施されてもよく、ガウシアンフィルタ等のローパスフィルタや二次微分等のハイパスフィルタが好ましく用いられる。
図9Bに、輝点領域が抽出された画像の一例を示す。図9Bに示すように、かかる画像では蛍光輝点を中心とした輝点領域が抽出されている。 Next, threshold processing is performed on the extracted image to generate a binary image, and a bright spot region is extracted (step S402).
Note that noise removal processing such as cell autofluorescence and other unnecessary signal components may be performed before the threshold processing, and a low-pass filter such as a Gaussian filter or a high-pass filter such as a second derivative is preferably used.
FIG. 9B shows an example of an image from which the bright spot region is extracted. As shown in FIG. 9B, a bright spot region centered on the fluorescent bright spot is extracted from such an image.
次いで、輝点領域が抽出された画像と蛍光画像とが重ね合わせられ、輝点領域内の輝度信号情報がマップ化されることで輝度プロファイルが作成され(ステップS403:プロファイル作成工程)、その輝度プロファイルから、各輝点領域における蛍光粒子の数と各蛍光粒子の位置とが算出される(ステップS404)。
「輝度プロファイル」とは、輝点領域が抽出された画像をマスクとして蛍光画像から抽出された画像に基づき作成される輝度値の分布情報であり、輝点領域における輝度値とその範囲(輝度分布の広がり)とを示すものである。 Next, the image from which the bright spot area is extracted and the fluorescence image are superimposed, and the brightness signal information in the bright spot area is mapped to create a brightness profile (step S403: profile creation process), and the brightness From the profile, the number of fluorescent particles in each bright spot region and the position of each fluorescent particle are calculated (step S404).
The “luminance profile” is luminance value distribution information created based on the image extracted from the fluorescence image using the image from which the luminescent spot region is extracted as a mask. The luminance value in the luminescent spot region and its range (luminance distribution) Spread).
「輝度プロファイル」とは、輝点領域が抽出された画像をマスクとして蛍光画像から抽出された画像に基づき作成される輝度値の分布情報であり、輝点領域における輝度値とその範囲(輝度分布の広がり)とを示すものである。 Next, the image from which the bright spot area is extracted and the fluorescence image are superimposed, and the brightness signal information in the bright spot area is mapped to create a brightness profile (step S403: profile creation process), and the brightness From the profile, the number of fluorescent particles in each bright spot region and the position of each fluorescent particle are calculated (step S404).
The “luminance profile” is luminance value distribution information created based on the image extracted from the fluorescence image using the image from which the luminescent spot region is extracted as a mask. The luminance value in the luminescent spot region and its range (luminance distribution) Spread).
すなわち、図10Aは、蛍光画像から輝点領域が抽出された画像の一例である。輝点領域が抽出された画像に基づいて、輝点領域ごとに、輝点領域が抽出された画像(図10B)と、その輝点領域に対応する部位の蛍光画像(図10C)とが重ね合わせられる。図10Bは、図10A内の□で囲まれた領域の拡大図であり、1つの輝点領域を示す。図10Cは、図10Bの輝点領域に対応する部位の蛍光画像である。
次いで、輝点領域が抽出された画像(例えば、図10B)をマスクとして、蛍光画像(例えば、図10C)から、輝点領域に対応する輝点画像が生成される(図10D)。輝点画像の輝度値を画素ごとに計測し、X座標位置及びY座標位置に表示したものが、ステップS403において輝度プロファイルとして作成される輝度値の分布である(図10E)。 That is, FIG. 10A is an example of an image in which a bright spot region is extracted from a fluorescence image. Based on the image from which the bright spot area has been extracted, for each bright spot area, the image from which the bright spot area has been extracted (FIG. 10B) and the fluorescence image of the portion corresponding to the bright spot area (FIG. 10C) are superimposed. Adapted. FIG. 10B is an enlarged view of a region surrounded by a square in FIG. 10A and shows one bright spot region. FIG. 10C is a fluorescence image of a portion corresponding to the bright spot region of FIG. 10B.
Next, a bright spot image corresponding to the bright spot area is generated from the fluorescent image (eg, FIG. 10C) using the image from which the bright spot area is extracted (eg, FIG. 10B) as a mask (FIG. 10D). The brightness value of the bright spot image measured for each pixel and displayed at the X coordinate position and the Y coordinate position is the distribution of brightness values created as a brightness profile in step S403 (FIG. 10E).
次いで、輝点領域が抽出された画像(例えば、図10B)をマスクとして、蛍光画像(例えば、図10C)から、輝点領域に対応する輝点画像が生成される(図10D)。輝点画像の輝度値を画素ごとに計測し、X座標位置及びY座標位置に表示したものが、ステップS403において輝度プロファイルとして作成される輝度値の分布である(図10E)。 That is, FIG. 10A is an example of an image in which a bright spot region is extracted from a fluorescence image. Based on the image from which the bright spot area has been extracted, for each bright spot area, the image from which the bright spot area has been extracted (FIG. 10B) and the fluorescence image of the portion corresponding to the bright spot area (FIG. 10C) are superimposed. Adapted. FIG. 10B is an enlarged view of a region surrounded by a square in FIG. 10A and shows one bright spot region. FIG. 10C is a fluorescence image of a portion corresponding to the bright spot region of FIG. 10B.
Next, a bright spot image corresponding to the bright spot area is generated from the fluorescent image (eg, FIG. 10C) using the image from which the bright spot area is extracted (eg, FIG. 10B) as a mask (FIG. 10D). The brightness value of the bright spot image measured for each pixel and displayed at the X coordinate position and the Y coordinate position is the distribution of brightness values created as a brightness profile in step S403 (FIG. 10E).
なお、輝度プロファイルは、図10Eに示すように、X座標位置及びY座標位置における輝度が2次元的に表現されたものであってもよいし、図10Fに示すように、X座標位置(横)及びY座標位置(縦)における輝度(高さ)が3次元的に表現されたものであってもよい。
The luminance profile may be one in which the luminance at the X coordinate position and the Y coordinate position is expressed two-dimensionally as shown in FIG. 10E, or as shown in FIG. 10F. ) And the luminance (height) at the Y coordinate position (vertical) may be expressed three-dimensionally.
そして実際のところ、1つの輝点領域には1個または複数個の蛍光粒子が含まれ、かかる輝度プロファイルには蛍光粒子の数と各蛍光粒子の位置とに応じた輝度値と範囲(輝度分布の広がり)とが示される。
本実施形態では、ステップS3において入力された蛍光画像と同一の画像取り込み条件で撮影された単独の蛍光粒子の画像から、1つの蛍光粒子の輝度プロファイルが、基準プロファイルとして予め作成される。輝点画像から作成された輝度プロファイルを、基準プロファイルに基づいて解析することで、焦点深度の異なる複数の輝点画像の中から、焦点の合った画像のみを抽出でき、また、各輝点領域における蛍光粒子の数と各蛍光粒子の位置とが算出できる。
例えば、1つの蛍光粒子に焦点が合っている蛍光画像から作成された基準プロファイルは、図11Bのように中心に一つの鋭いピークを持つ正規分布形状となる。焦点が蛍光粒子よりも下方にずれている蛍光画像から作成された基準プロファイルは、例えば図11Aのように、縦横方向の広がりが大きく、ピークが低くなる。一方、焦点が蛍光粒子よりも上方にずれている蛍光画像から作成された基準プロファイルは、例えば図11Cのように、中心から一定距離離れた位置に最も輝度が高い部位がリング状に見られ、中心はやや輝度が低く凹んだ形となる。 Actually, one bright spot region includes one or a plurality of fluorescent particles, and the luminance profile includes a luminance value and a range (luminance distribution) corresponding to the number of fluorescent particles and the position of each fluorescent particle. Spread).
In the present embodiment, a luminance profile of one fluorescent particle is created in advance as a reference profile from an image of a single fluorescent particle taken under the same image capturing conditions as the fluorescent image input in step S3. By analyzing the brightness profile created from the bright spot image based on the reference profile, it is possible to extract only the focused image from multiple bright spot images with different depths of focus. The number of fluorescent particles and the position of each fluorescent particle can be calculated.
For example, a reference profile created from a fluorescent image in which one fluorescent particle is in focus has a normal distribution shape having one sharp peak at the center as shown in FIG. 11B. A reference profile created from a fluorescent image whose focus is shifted downward from the fluorescent particles has a large spread in the vertical and horizontal directions and a low peak as shown in FIG. 11A, for example. On the other hand, in the reference profile created from the fluorescence image in which the focal point is shifted upward from the fluorescent particles, for example, as shown in FIG. The center has a slightly low brightness and a concave shape.
本実施形態では、ステップS3において入力された蛍光画像と同一の画像取り込み条件で撮影された単独の蛍光粒子の画像から、1つの蛍光粒子の輝度プロファイルが、基準プロファイルとして予め作成される。輝点画像から作成された輝度プロファイルを、基準プロファイルに基づいて解析することで、焦点深度の異なる複数の輝点画像の中から、焦点の合った画像のみを抽出でき、また、各輝点領域における蛍光粒子の数と各蛍光粒子の位置とが算出できる。
例えば、1つの蛍光粒子に焦点が合っている蛍光画像から作成された基準プロファイルは、図11Bのように中心に一つの鋭いピークを持つ正規分布形状となる。焦点が蛍光粒子よりも下方にずれている蛍光画像から作成された基準プロファイルは、例えば図11Aのように、縦横方向の広がりが大きく、ピークが低くなる。一方、焦点が蛍光粒子よりも上方にずれている蛍光画像から作成された基準プロファイルは、例えば図11Cのように、中心から一定距離離れた位置に最も輝度が高い部位がリング状に見られ、中心はやや輝度が低く凹んだ形となる。 Actually, one bright spot region includes one or a plurality of fluorescent particles, and the luminance profile includes a luminance value and a range (luminance distribution) corresponding to the number of fluorescent particles and the position of each fluorescent particle. Spread).
In the present embodiment, a luminance profile of one fluorescent particle is created in advance as a reference profile from an image of a single fluorescent particle taken under the same image capturing conditions as the fluorescent image input in step S3. By analyzing the brightness profile created from the bright spot image based on the reference profile, it is possible to extract only the focused image from multiple bright spot images with different depths of focus. The number of fluorescent particles and the position of each fluorescent particle can be calculated.
For example, a reference profile created from a fluorescent image in which one fluorescent particle is in focus has a normal distribution shape having one sharp peak at the center as shown in FIG. 11B. A reference profile created from a fluorescent image whose focus is shifted downward from the fluorescent particles has a large spread in the vertical and horizontal directions and a low peak as shown in FIG. 11A, for example. On the other hand, in the reference profile created from the fluorescence image in which the focal point is shifted upward from the fluorescent particles, for example, as shown in FIG. The center has a slightly low brightness and a concave shape.
図12A~図12Cは、所定間隔の焦点深度Z1、Z2、Z3において取得された蛍光画像(図12A~図12Cのそれぞれ左図)の同一領域から作成された3次元輝度プロファイルの断面を示す模式図(図12A~図12Cのそれぞれ右図)である。ここで図示される輝度プロファイルには、3つの輝点領域が含まれている。
図12A~図12Cに示される輝度プロファイルのうち、左端の輝度プロファイルは、焦点深度Z1(図12A)において鋭いピークを持つ正規分布形状を示すが、焦点深度Z2(図12B)においては、中心が凹み、X軸方向の広がりが大きくなる。さらに、焦点深度Z3(図12C)においては、中心が凹んだ形状のまま、X軸方向の広がりがさらに大きくなる。これらの輝度プロファイルを基準プロファイルに基づいて解析すると、例えば焦点深度Z1(図12A)において最も焦点が合っている蛍光粒子が1つ存在していると判断される。
また、中央の輝度プロファイルは、焦点深度Z2(図12B)において中心に鋭いピークを持つ正規分布形状を示すが、焦点深度Z1及びZ3(図12A及び図12C)においては、中心が凹み、X軸方向の広がりが大きいほぼ同一形状を示している。基準プロファイルに基づく解析によれば、この輝点領域には、例えば、焦点深度Z2(図12B)において最も焦点が合っている蛍光粒子が1つ存在していると判断される。
また、右端の輝度プロファイルは、焦点深度Z1(図12A)においては、中心が凹んでX軸方向の広がりが非常に大きく、焦点深度Z2(図12B)においては、中心は凹んでいるがX軸方向の広がりがやや狭まり、焦点深度Z3(図12C)においては中心に鋭いピークを持つ正規分布形状を示している。基準プロファイルに基づく解析によれば、この輝点領域には、例えば、焦点深度Z3(図12C)において最も焦点が合っている蛍光粒子が1つ存在していると判断される。 FIGS. 12A to 12C are schematic diagrams showing cross sections of a three-dimensional luminance profile created from the same region of fluorescent images (left diagrams of FIGS. 12A to 12C) acquired at focal depths Z1, Z2, and Z3 at predetermined intervals. It is a figure (each right figure of FIG. 12A-FIG. 12C). The luminance profile shown here includes three bright spot regions.
Of the luminance profiles shown in FIGS. 12A to 12C, the leftmost luminance profile shows a normal distribution shape having a sharp peak at the focal depth Z1 (FIG. 12A), but the center is at the focal depth Z2 (FIG. 12B). The dent and the spread in the X-axis direction become large. Furthermore, at the focal depth Z3 (FIG. 12C), the spread in the X-axis direction is further increased while the center is recessed. When these luminance profiles are analyzed based on the reference profile, for example, it is determined that there is one fluorescent particle that is most focused at the focal depth Z1 (FIG. 12A).
The central luminance profile shows a normal distribution shape having a sharp peak at the center at the focal depth Z2 (FIG. 12B). However, at the focal depths Z1 and Z3 (FIGS. 12A and 12C), the center is depressed, and the X axis It shows substantially the same shape with a large extent of direction. According to the analysis based on the reference profile, it is determined that, for example, one fluorescent particle that is most focused at the focal depth Z2 (FIG. 12B) exists in this bright spot region.
Further, the rightmost luminance profile has a concave center at the focal depth Z1 (FIG. 12A) and a very large spread in the X-axis direction, and has a concave center at the focal depth Z2 (FIG. 12B). The spread of the direction is slightly narrowed, and a normal distribution shape having a sharp peak at the center is shown at the focal depth Z3 (FIG. 12C). According to the analysis based on the reference profile, it is determined that, for example, one fluorescent particle that is most focused at the focal depth Z3 (FIG. 12C) exists in this bright spot region.
図12A~図12Cに示される輝度プロファイルのうち、左端の輝度プロファイルは、焦点深度Z1(図12A)において鋭いピークを持つ正規分布形状を示すが、焦点深度Z2(図12B)においては、中心が凹み、X軸方向の広がりが大きくなる。さらに、焦点深度Z3(図12C)においては、中心が凹んだ形状のまま、X軸方向の広がりがさらに大きくなる。これらの輝度プロファイルを基準プロファイルに基づいて解析すると、例えば焦点深度Z1(図12A)において最も焦点が合っている蛍光粒子が1つ存在していると判断される。
また、中央の輝度プロファイルは、焦点深度Z2(図12B)において中心に鋭いピークを持つ正規分布形状を示すが、焦点深度Z1及びZ3(図12A及び図12C)においては、中心が凹み、X軸方向の広がりが大きいほぼ同一形状を示している。基準プロファイルに基づく解析によれば、この輝点領域には、例えば、焦点深度Z2(図12B)において最も焦点が合っている蛍光粒子が1つ存在していると判断される。
また、右端の輝度プロファイルは、焦点深度Z1(図12A)においては、中心が凹んでX軸方向の広がりが非常に大きく、焦点深度Z2(図12B)においては、中心は凹んでいるがX軸方向の広がりがやや狭まり、焦点深度Z3(図12C)においては中心に鋭いピークを持つ正規分布形状を示している。基準プロファイルに基づく解析によれば、この輝点領域には、例えば、焦点深度Z3(図12C)において最も焦点が合っている蛍光粒子が1つ存在していると判断される。 FIGS. 12A to 12C are schematic diagrams showing cross sections of a three-dimensional luminance profile created from the same region of fluorescent images (left diagrams of FIGS. 12A to 12C) acquired at focal depths Z1, Z2, and Z3 at predetermined intervals. It is a figure (each right figure of FIG. 12A-FIG. 12C). The luminance profile shown here includes three bright spot regions.
Of the luminance profiles shown in FIGS. 12A to 12C, the leftmost luminance profile shows a normal distribution shape having a sharp peak at the focal depth Z1 (FIG. 12A), but the center is at the focal depth Z2 (FIG. 12B). The dent and the spread in the X-axis direction become large. Furthermore, at the focal depth Z3 (FIG. 12C), the spread in the X-axis direction is further increased while the center is recessed. When these luminance profiles are analyzed based on the reference profile, for example, it is determined that there is one fluorescent particle that is most focused at the focal depth Z1 (FIG. 12A).
The central luminance profile shows a normal distribution shape having a sharp peak at the center at the focal depth Z2 (FIG. 12B). However, at the focal depths Z1 and Z3 (FIGS. 12A and 12C), the center is depressed, and the X axis It shows substantially the same shape with a large extent of direction. According to the analysis based on the reference profile, it is determined that, for example, one fluorescent particle that is most focused at the focal depth Z2 (FIG. 12B) exists in this bright spot region.
Further, the rightmost luminance profile has a concave center at the focal depth Z1 (FIG. 12A) and a very large spread in the X-axis direction, and has a concave center at the focal depth Z2 (FIG. 12B). The spread of the direction is slightly narrowed, and a normal distribution shape having a sharp peak at the center is shown at the focal depth Z3 (FIG. 12C). According to the analysis based on the reference profile, it is determined that, for example, one fluorescent particle that is most focused at the focal depth Z3 (FIG. 12C) exists in this bright spot region.
以上のステップS404の工程により、各輝点領域に含まれる蛍光粒子の位置が、各蛍光画像上の2次元座標上の位置に焦点深度を加えた3次元座標上の位置として算出される。
次いで、蛍光画像にラベリング処理が施され、ステップS404の工程において算出された蛍光粒子の位置の輝点画像にラベルが付与される(ステップS405)。ここでは、各輝点領域に対応する焦点深度の異なる複数の輝点画像の中で、最も蛍光粒子に焦点が合っている輝点画像(蛍光粒子画像)に、蛍光粒子を識別するためのラベルが付与されることとなる。 Through the above-described step S404, the position of the fluorescent particle included in each bright spot region is calculated as a position on the three-dimensional coordinate obtained by adding the focal depth to the position on the two-dimensional coordinate on each fluorescent image.
Next, the fluorescent image is subjected to a labeling process, and a label is given to the bright spot image at the position of the fluorescent particle calculated in the step S404 (step S405). Here, among a plurality of bright spot images with different focal depths corresponding to each bright spot region, a label for identifying the fluorescent particles in the bright spot image (fluorescent particle image) most focused on the fluorescent particles Will be granted.
次いで、蛍光画像にラベリング処理が施され、ステップS404の工程において算出された蛍光粒子の位置の輝点画像にラベルが付与される(ステップS405)。ここでは、各輝点領域に対応する焦点深度の異なる複数の輝点画像の中で、最も蛍光粒子に焦点が合っている輝点画像(蛍光粒子画像)に、蛍光粒子を識別するためのラベルが付与されることとなる。 Through the above-described step S404, the position of the fluorescent particle included in each bright spot region is calculated as a position on the three-dimensional coordinate obtained by adding the focal depth to the position on the two-dimensional coordinate on each fluorescent image.
Next, the fluorescent image is subjected to a labeling process, and a label is given to the bright spot image at the position of the fluorescent particle calculated in the step S404 (step S405). Here, among a plurality of bright spot images with different focal depths corresponding to each bright spot region, a label for identifying the fluorescent particles in the bright spot image (fluorescent particle image) most focused on the fluorescent particles Will be granted.
なお、図12A~図12Cにおいては、各輝点領域に1つの蛍光粒子が含まれている例を示したが、例えば、1つの輝点領域に複数の蛍光粒子が含まれている場合でも、焦点深度をずらした複数の蛍光画像を基準プロファイルに基づいて解析することにより、2次元画像である蛍光画像上では重なって見える複数の蛍光粒子を分離して計測することができる。そして、複数の蛍光粒子のそれぞれについて、最も焦点が合った輝点画像(蛍光粒子画像)にラベルが付与される。
12A to 12C show an example in which one fluorescent particle is included in each bright spot region. For example, even when a plurality of fluorescent particles are included in one bright spot region, By analyzing a plurality of fluorescent images with different focal depths based on the reference profile, it is possible to separate and measure a plurality of fluorescent particles that appear to overlap on a two-dimensional fluorescent image. Then, for each of the plurality of fluorescent particles, a label is given to the bright spot image (fluorescent particle image) that is most focused.
また、上記実施形態では蛍光粒子1個分の輝度プロファイルを基準プロファイルとして蛍光粒子の数と位置とを判別することとしたが、例えば、あらかじめ複数個の蛍光粒子からなる輝度プロファイルを基準プロファイルとして準備して、蛍光粒子の数と位置とを判別してもよい。また、複数個の蛍光粒子からなる輝度プロファイルそのものに対して2次元フーリエ変換等の公知の任意の画像処理を施して波形を分解し、蛍光粒子の数と位置とを判別してもよい。
In the above embodiment, the number and position of the fluorescent particles are determined using the luminance profile for one fluorescent particle as a reference profile. For example, a luminance profile composed of a plurality of fluorescent particles is prepared in advance as a reference profile. Then, the number and position of the fluorescent particles may be determined. Alternatively, the luminance profile itself composed of a plurality of fluorescent particles may be subjected to known arbitrary image processing such as two-dimensional Fourier transform to decompose the waveform to determine the number and position of the fluorescent particles.
ステップS2とステップS4の処理の終了後、図5の処理に戻り、細胞画像(図7B参照)と蛍光画像との加算処理が行われ(ステップS5)、ステップS204で付与された1つの細胞を示すラベルとステップS405で付与された蛍光粒子のラベルから、一細胞当たりの蛍光粒子数が算出される(算出工程)。
After the process of step S2 and step S4 is completed, the process returns to the process of FIG. 5, the addition process of the cell image (see FIG. 7B) and the fluorescence image is performed (step S5), and one cell given in step S204 is removed. The number of fluorescent particles per cell is calculated from the label shown and the label of the fluorescent particles given in step S405 (calculation step).
さらに、焦点深度の異なる複数の蛍光画像が再構成された一枚の再構成画像(合焦点画像)が生成される(生成工程)。このとき、各輝点領域においては、ステップS405で蛍光粒子のラベルが付与された蛍光粒子画像が抽出されて合焦点画像の再構成に用いられる(抽出工程)。1つの輝点領域に複数の蛍光粒子が含まれ、それらが異なる焦点深度に存在している場合には、抽出された複数の蛍光粒子画像は、例えば、画素ごとに輝度値の平均値又は加算値が算出された一枚の画像に再構成される。ラベルが付与されていない輝点画像は、蛍光粒子から焦点がずれているため再構成には使用されない。
図12Dは、図12A~図12Cの3枚の蛍光画像から再構成された合焦点画像及び合焦点画像の輝度プロファイルの断面を示す模式図であり、輝度プロファイルの左の輝点領域においては焦点深度Z1(図12A)の輝点画像、中央の輝点領域においては焦点深度Z2(図12B)の輝点画像、右の輝点領域においては焦点深度Z3(図12C)の輝点画像が、蛍光粒子画像として抽出され、再構成された例を示す。合焦点画像から作成された輝度プロファイル(図12Dの右図)は、全ての輝点領域において鋭いピークを持つ正規分布形状となり、各蛍光粒子に焦点が合った画像が得られる。
次いで、ステップS6では、再構成画像と細胞画像が重ね合わせられて、細胞上での蛍光粒子の分布を示す画像が表示される。 Furthermore, one reconstructed image (focused image) in which a plurality of fluorescent images with different depths of focus are reconstructed is generated (generation process). At this time, in each bright spot region, the fluorescent particle image to which the fluorescent particle label is assigned in step S405 is extracted and used for reconstruction of the focused image (extraction process). When a plurality of fluorescent particles are included in one bright spot region and they are present at different depths of focus, the extracted plurality of fluorescent particle images is, for example, an average value or addition of luminance values for each pixel. Reconstructed into a single image whose value has been calculated. The bright spot image without the label is not used for reconstruction because the focus is shifted from the fluorescent particles.
FIG. 12D is a schematic diagram showing a cross-section of the focused image reconstructed from the three fluorescent images of FIGS. 12A to 12C and the luminance profile of the focused image. In the bright spot region on the left side of the luminance profile, FIG. A bright spot image with depth Z1 (FIG. 12A), a bright spot image with focal depth Z2 (FIG. 12B) in the central bright spot area, and a bright spot image with focal depth Z3 (FIG. 12C) in the right bright spot area, An example extracted and reconstructed as a fluorescent particle image is shown. The luminance profile created from the focused image (right diagram in FIG. 12D) has a normal distribution shape having sharp peaks in all the bright spot regions, and an image in which each fluorescent particle is focused is obtained.
Next, in step S6, the reconstructed image and the cell image are superimposed, and an image showing the distribution of the fluorescent particles on the cell is displayed.
図12Dは、図12A~図12Cの3枚の蛍光画像から再構成された合焦点画像及び合焦点画像の輝度プロファイルの断面を示す模式図であり、輝度プロファイルの左の輝点領域においては焦点深度Z1(図12A)の輝点画像、中央の輝点領域においては焦点深度Z2(図12B)の輝点画像、右の輝点領域においては焦点深度Z3(図12C)の輝点画像が、蛍光粒子画像として抽出され、再構成された例を示す。合焦点画像から作成された輝度プロファイル(図12Dの右図)は、全ての輝点領域において鋭いピークを持つ正規分布形状となり、各蛍光粒子に焦点が合った画像が得られる。
次いで、ステップS6では、再構成画像と細胞画像が重ね合わせられて、細胞上での蛍光粒子の分布を示す画像が表示される。 Furthermore, one reconstructed image (focused image) in which a plurality of fluorescent images with different depths of focus are reconstructed is generated (generation process). At this time, in each bright spot region, the fluorescent particle image to which the fluorescent particle label is assigned in step S405 is extracted and used for reconstruction of the focused image (extraction process). When a plurality of fluorescent particles are included in one bright spot region and they are present at different depths of focus, the extracted plurality of fluorescent particle images is, for example, an average value or addition of luminance values for each pixel. Reconstructed into a single image whose value has been calculated. The bright spot image without the label is not used for reconstruction because the focus is shifted from the fluorescent particles.
FIG. 12D is a schematic diagram showing a cross-section of the focused image reconstructed from the three fluorescent images of FIGS. 12A to 12C and the luminance profile of the focused image. In the bright spot region on the left side of the luminance profile, FIG. A bright spot image with depth Z1 (FIG. 12A), a bright spot image with focal depth Z2 (FIG. 12B) in the central bright spot area, and a bright spot image with focal depth Z3 (FIG. 12C) in the right bright spot area, An example extracted and reconstructed as a fluorescent particle image is shown. The luminance profile created from the focused image (right diagram in FIG. 12D) has a normal distribution shape having sharp peaks in all the bright spot regions, and an image in which each fluorescent particle is focused is obtained.
Next, in step S6, the reconstructed image and the cell image are superimposed, and an image showing the distribution of the fluorescent particles on the cell is displayed.
以上の本実施形態によれば、ステップS1~S2の処理により細胞が抽出され、ステップS3~S402の処理により輝点領域が抽出され、その後、ステップS403~S404の処理により、細胞上での蛍光粒子の分布が3次元座標上で具体的に把握されるようになっている。さらに、ステップS5~S6の処理により、蛍光粒子に焦点が合った輝点画像が抽出されて一枚の蛍光画像に再構成され、細胞上での蛍光粒子の分布が表示される。
こうして、簡易な顕微鏡を用いて観察対象細胞内での特定タンパク質の発現(発現数とその発現位置)を正確に定量することができ、それぞれの蛍光粒子に焦点が合った、輝点のボケがない蛍光画像を得ることができる。 According to the present embodiment described above, cells are extracted by the processing of steps S1 to S2, the bright spot region is extracted by the processing of steps S3 to S402, and then the fluorescence on the cells is processed by the processing of steps S403 to S404. The particle distribution is specifically grasped on the three-dimensional coordinates. Further, by the processing in steps S5 to S6, a bright spot image focused on the fluorescent particles is extracted and reconstructed into a single fluorescent image, and the distribution of the fluorescent particles on the cells is displayed.
In this way, it is possible to accurately quantify the expression (number of expression and position of expression) of a specific protein in the observation target cell using a simple microscope, and the bright spot blur focused on each fluorescent particle. No fluorescence image can be obtained.
こうして、簡易な顕微鏡を用いて観察対象細胞内での特定タンパク質の発現(発現数とその発現位置)を正確に定量することができ、それぞれの蛍光粒子に焦点が合った、輝点のボケがない蛍光画像を得ることができる。 According to the present embodiment described above, cells are extracted by the processing of steps S1 to S2, the bright spot region is extracted by the processing of steps S3 to S402, and then the fluorescence on the cells is processed by the processing of steps S403 to S404. The particle distribution is specifically grasped on the three-dimensional coordinates. Further, by the processing in steps S5 to S6, a bright spot image focused on the fluorescent particles is extracted and reconstructed into a single fluorescent image, and the distribution of the fluorescent particles on the cells is displayed.
In this way, it is possible to accurately quantify the expression (number of expression and position of expression) of a specific protein in the observation target cell using a simple microscope, and the bright spot blur focused on each fluorescent particle. No fluorescence image can be obtained.
なお、上記実施形態における記述内容は、本発明の好適な一例であり、これに限定されるものではない。
In addition, the description content in the said embodiment is a suitable example of this invention, and is not limited to this.
上記実施形態では、特定タンパクの例として乳癌におけるHER2タンパクを挙げたが、これに限定されない。診断対象となる病変(がん)種に応じて、蛍光画像を取得する際の生体物質認識部位を異なるものとすれば、病変種に応じた特定タンパクの発現量を定量的に示す特徴量を医師に提供することが可能となる。
In the above embodiment, the HER2 protein in breast cancer is mentioned as an example of the specific protein, but it is not limited to this. Depending on the type of lesion (cancer) to be diagnosed, if the biological material recognition site when acquiring a fluorescence image is different, the feature quantity that quantitatively indicates the expression level of the specific protein corresponding to the lesion type It can be provided to a doctor.
また、上記実施形態では、1種の特定タンパクのみを対象としたが、複数の特定タンパクに対し、発光波長が互いに異なる2種以上の蛍光粒子を用いてもよい。
かかる場合、ステップS401においてフィルターワーク等を用いてそれぞれの色成分を抽出し、その抽出した色成分(波長成分)ごとにステップS402~S405の処理を実行し、ステップS5-S6において、細胞領域画像と色成分ごと作成された蛍光粒子画像とを加算すればよい。 Moreover, in the said embodiment, although only 1 type of specific protein was made into object, you may use 2 or more types of fluorescent particles from which a light emission wavelength mutually differs with respect to several specific protein.
In such a case, each color component is extracted using filter work or the like in step S401, and the processing of steps S402 to S405 is executed for each extracted color component (wavelength component). In step S5-S6, the cell region image is extracted. And the fluorescent particle image created for each color component may be added.
かかる場合、ステップS401においてフィルターワーク等を用いてそれぞれの色成分を抽出し、その抽出した色成分(波長成分)ごとにステップS402~S405の処理を実行し、ステップS5-S6において、細胞領域画像と色成分ごと作成された蛍光粒子画像とを加算すればよい。 Moreover, in the said embodiment, although only 1 type of specific protein was made into object, you may use 2 or more types of fluorescent particles from which a light emission wavelength mutually differs with respect to several specific protein.
In such a case, each color component is extracted using filter work or the like in step S401, and the processing of steps S402 to S405 is executed for each extracted color component (wavelength component). In step S5-S6, the cell region image is extracted. And the fluorescent particle image created for each color component may be added.
また、蛍光粒子は、上記実施形態のように特定タンパクに結合する生体物質認識部位に直接結合されても良いが、免疫染色における公知の間接法のように、別の物質を介して間接的に結合されても良い。例えば、組織標本に特定タンパクを抗原とする一次抗体を反応させた後、一次抗体を抗原とする二次抗体に蛍光粒子を結合したものを反応させて染色しても良い。また、例えば、組織標本に特定タンパクを抗原とする一次抗体及び、一次抗体を抗原とするビオチン化二次抗体を反応させた後、ストレプトアビジンにより修飾された蛍光粒子を反応させて、ストレプトアビジンとビオチンが特異的に結合して複合体を形成することを利用して染色しても良い。
In addition, the fluorescent particles may be directly bound to a biological substance recognition site that binds to a specific protein as in the above embodiment, but indirectly through another substance, as in a known indirect method in immunostaining. May be combined. For example, after reacting a tissue specimen with a primary antibody having a specific protein as an antigen, a secondary antibody having the primary antibody as an antigen may be reacted with a fluorescent particle bound thereto and stained. In addition, for example, after reacting a tissue sample with a primary antibody having a specific protein as an antigen and a biotinylated secondary antibody having a primary antibody as an antigen, a fluorescent particle modified with streptavidin is reacted with streptavidin. You may dye | stain using biotin specifically couple | bonding and forming a complex.
また、上記の説明では、本発明に係るプログラムのコンピュータ読み取り可能な媒体としてHDDや半導体の不揮発性メモリー等を使用した例を開示したが、この例に限定されない。その他のコンピュータ読み取り可能な媒体として、CD-ROM等の可搬型記録媒体を適用することが可能である。また、本発明に係るプログラムのデータを、通信回線を介して提供する媒体として、キャリアウエーブ(搬送波)も適用される。
In the above description, an example in which an HDD or a semiconductor non-volatile memory is used as a computer-readable medium of the program according to the present invention is disclosed, but the present invention is not limited to this example. As another computer-readable medium, a portable recording medium such as a CD-ROM can be applied. Further, a carrier wave (carrier wave) is also applied as a medium for providing program data according to the present invention via a communication line.
その他、病理診断支援システム100を構成する各装置の細部構成及び細部動作に関しても、発明の趣旨を逸脱することのない範囲で適宜変更可能である。
In addition, the detailed configuration and detailed operation of each device constituting the pathological diagnosis support system 100 can be changed as appropriate without departing from the spirit of the invention.
(A1)IHC染色用試薬の作製
蛍光物質に抗HER2抗体を結合させたIHC染色用の染色試薬aを、以下の方法で作成した。 (A1) Preparation of IHC staining reagent A staining reagent a for IHC staining in which an anti-HER2 antibody was bound to a fluorescent substance was prepared by the following method.
蛍光物質に抗HER2抗体を結合させたIHC染色用の染色試薬aを、以下の方法で作成した。 (A1) Preparation of IHC staining reagent A staining reagent a for IHC staining in which an anti-HER2 antibody was bound to a fluorescent substance was prepared by the following method.
(A1-1)蛍光物質内包ナノ粒子の作製
蛍光色素として赤色発光色素であるSulforhodamine101(シグマアルドリッチ社製)14.4mgを水22mLに加えて溶解させた。その後、この溶液に乳化重合用乳化剤のエマルゲン(登録商標)430(ポリオキシエチレンオレイルエーテル、花王社製)の5%水溶液を2mL加えた。この溶液をホットスターラー上で撹拌しながら70℃まで昇温させた後、この溶液にメラミン樹脂原料ニカラックMX-035(日本カーバイド工業社製)を0.65g加えた。
さらに、この溶液に界面活性剤としてドデシルベンゼンスルホン酸(関東化学社製)の10%水溶液を1000μL加え、70℃で50分間加熱撹拌した。その後、90℃に昇温して20分間加熱撹拌した。得られた色素樹脂粒子の分散液から、余剰の樹脂原料や蛍光色素等の不純物を除くため、純水による洗浄を行った。
具体的には、遠心分離機(クボタ社製マイクロ冷却遠心機3740)にて20000Gで15分間、遠心分離し、上澄み除去後、超純水を加えて超音波照射して再分散した。遠心分離、上澄み除去および超純水への再分散による洗浄を5回繰り返した。得られたメラミン粒子はメラミン樹脂自体が骨格に多くのアミノ基を含むことから、プラス電荷となった。粒子の電荷の評価は、NMRやIR等による樹脂成分分析と、ゼータ電位測定により行なった。 (A1-1) Preparation of fluorescent substance-encapsulated nanoparticles 14.4 mg of Sulforhodamine 101 (manufactured by Sigma-Aldrich), which is a red luminescent dye, was added to 22 mL of water and dissolved. Thereafter, 2 mL of a 5% aqueous solution of Emulgen (registered trademark) 430 (polyoxyethylene oleyl ether, manufactured by Kao Corporation), an emulsifier for emulsion polymerization, was added to this solution. This solution was heated to 70 ° C. while stirring on a hot stirrer, and then 0.65 g of melamine resin raw material Nicalak MX-035 (manufactured by Nippon Carbide Industries Co., Ltd.) was added to this solution.
Further, 1000 μL of a 10% aqueous solution of dodecylbenzenesulfonic acid (manufactured by Kanto Chemical Co., Inc.) as a surfactant was added to this solution, and the mixture was heated and stirred at 70 ° C. for 50 minutes. Then, it heated up at 90 degreeC and heat-stirred for 20 minutes. In order to remove impurities such as excess resin raw materials and fluorescent dyes from the obtained dispersion of the dye resin particles, washing with pure water was performed.
Specifically, the mixture was centrifuged at 20000 G for 15 minutes in a centrifuge (Kubota Micro Cooling Centrifuge 3740), and after removing the supernatant, ultrapure water was added and ultrasonically irradiated to redisperse. Centrifugation, supernatant removal, and washing by redispersion in ultrapure water were repeated 5 times. The obtained melamine particles were positively charged because the melamine resin itself contains many amino groups in the skeleton. The charge of the particles was evaluated by resin component analysis by NMR, IR, etc. and zeta potential measurement.
蛍光色素として赤色発光色素であるSulforhodamine101(シグマアルドリッチ社製)14.4mgを水22mLに加えて溶解させた。その後、この溶液に乳化重合用乳化剤のエマルゲン(登録商標)430(ポリオキシエチレンオレイルエーテル、花王社製)の5%水溶液を2mL加えた。この溶液をホットスターラー上で撹拌しながら70℃まで昇温させた後、この溶液にメラミン樹脂原料ニカラックMX-035(日本カーバイド工業社製)を0.65g加えた。
さらに、この溶液に界面活性剤としてドデシルベンゼンスルホン酸(関東化学社製)の10%水溶液を1000μL加え、70℃で50分間加熱撹拌した。その後、90℃に昇温して20分間加熱撹拌した。得られた色素樹脂粒子の分散液から、余剰の樹脂原料や蛍光色素等の不純物を除くため、純水による洗浄を行った。
具体的には、遠心分離機(クボタ社製マイクロ冷却遠心機3740)にて20000Gで15分間、遠心分離し、上澄み除去後、超純水を加えて超音波照射して再分散した。遠心分離、上澄み除去および超純水への再分散による洗浄を5回繰り返した。得られたメラミン粒子はメラミン樹脂自体が骨格に多くのアミノ基を含むことから、プラス電荷となった。粒子の電荷の評価は、NMRやIR等による樹脂成分分析と、ゼータ電位測定により行なった。 (A1-1) Preparation of fluorescent substance-encapsulated nanoparticles 14.4 mg of Sulforhodamine 101 (manufactured by Sigma-Aldrich), which is a red luminescent dye, was added to 22 mL of water and dissolved. Thereafter, 2 mL of a 5% aqueous solution of Emulgen (registered trademark) 430 (polyoxyethylene oleyl ether, manufactured by Kao Corporation), an emulsifier for emulsion polymerization, was added to this solution. This solution was heated to 70 ° C. while stirring on a hot stirrer, and then 0.65 g of melamine resin raw material Nicalak MX-035 (manufactured by Nippon Carbide Industries Co., Ltd.) was added to this solution.
Further, 1000 μL of a 10% aqueous solution of dodecylbenzenesulfonic acid (manufactured by Kanto Chemical Co., Inc.) as a surfactant was added to this solution, and the mixture was heated and stirred at 70 ° C. for 50 minutes. Then, it heated up at 90 degreeC and heat-stirred for 20 minutes. In order to remove impurities such as excess resin raw materials and fluorescent dyes from the obtained dispersion of the dye resin particles, washing with pure water was performed.
Specifically, the mixture was centrifuged at 20000 G for 15 minutes in a centrifuge (Kubota Micro Cooling Centrifuge 3740), and after removing the supernatant, ultrapure water was added and ultrasonically irradiated to redisperse. Centrifugation, supernatant removal, and washing by redispersion in ultrapure water were repeated 5 times. The obtained melamine particles were positively charged because the melamine resin itself contains many amino groups in the skeleton. The charge of the particles was evaluated by resin component analysis by NMR, IR, etc. and zeta potential measurement.
得られた蛍光粒子を走査型電子顕微鏡(SEM;日立(登録商標)社製S-800型)で観察し、平均粒径及び変動係数を算出した。本実施例及び比較例においては、平均粒径が200、170、150、100、80、60、40、20nmであり、変動係数が12%の蛍光粒子を用いた。
The obtained fluorescent particles were observed with a scanning electron microscope (SEM; Model S-800 manufactured by Hitachi (registered trademark)), and the average particle size and coefficient of variation were calculated. In this example and comparative example, fluorescent particles having an average particle diameter of 200, 170, 150, 100, 80, 60, 40, and 20 nm and a variation coefficient of 12% were used.
(A1-2)蛍光粒子への抗体の結合
下記の工程(1)~(12)の方法により、蛍光粒子に対して抗HER2抗体を結合させた。
工程(1):1mgの蛍光粒子を純水5mLに分散させた。次いで、アミノプロピルトリエトキシシラン水分散液(LS-3150、信越化学工業社製)100μLを添加し、室温で12時間撹拌した。
工程(2):反応混合物を10000Gで60分遠心分離を行い、上澄みを除去した。
工程(3):エタノールを加え、沈降物を分散させ、再度遠心分離を行った。同様の手順でエタノールと純水による洗浄を1回ずつ行った。
得られたアミノ基修飾した蛍光粒子のFT-IR測定を行ったところ、アミノ基に由来する吸収が観測でき、アミノ基修飾されたことが確認できた。 (A1-2) Binding of antibody to fluorescent particles Anti-HER2 antibody was bound to the fluorescent particles by the following steps (1) to (12).
Step (1): 1 mg of fluorescent particles was dispersed in 5 mL of pure water. Next, 100 μL of aminopropyltriethoxysilane aqueous dispersion (LS-3150, manufactured by Shin-Etsu Chemical Co., Ltd.) was added and stirred at room temperature for 12 hours.
Step (2): The reaction mixture was centrifuged at 10,000 G for 60 minutes, and the supernatant was removed.
Step (3): Ethanol was added to disperse the sediment, followed by centrifugation again. Washing with ethanol and pure water was performed once by the same procedure.
When the FT-IR measurement was performed on the resulting amino group-modified fluorescent particles, absorption derived from the amino group could be observed, confirming that the amino group was modified.
下記の工程(1)~(12)の方法により、蛍光粒子に対して抗HER2抗体を結合させた。
工程(1):1mgの蛍光粒子を純水5mLに分散させた。次いで、アミノプロピルトリエトキシシラン水分散液(LS-3150、信越化学工業社製)100μLを添加し、室温で12時間撹拌した。
工程(2):反応混合物を10000Gで60分遠心分離を行い、上澄みを除去した。
工程(3):エタノールを加え、沈降物を分散させ、再度遠心分離を行った。同様の手順でエタノールと純水による洗浄を1回ずつ行った。
得られたアミノ基修飾した蛍光粒子のFT-IR測定を行ったところ、アミノ基に由来する吸収が観測でき、アミノ基修飾されたことが確認できた。 (A1-2) Binding of antibody to fluorescent particles Anti-HER2 antibody was bound to the fluorescent particles by the following steps (1) to (12).
Step (1): 1 mg of fluorescent particles was dispersed in 5 mL of pure water. Next, 100 μL of aminopropyltriethoxysilane aqueous dispersion (LS-3150, manufactured by Shin-Etsu Chemical Co., Ltd.) was added and stirred at room temperature for 12 hours.
Step (2): The reaction mixture was centrifuged at 10,000 G for 60 minutes, and the supernatant was removed.
Step (3): Ethanol was added to disperse the sediment, followed by centrifugation again. Washing with ethanol and pure water was performed once by the same procedure.
When the FT-IR measurement was performed on the resulting amino group-modified fluorescent particles, absorption derived from the amino group could be observed, confirming that the amino group was modified.
工程(4):工程(3)で得られたアミノ基修飾した蛍光粒子を、EDTA(エチレンジアミン四酢酸)を2mM含有したPBSを用いて3nMに調整した。
工程(5):工程(4)で調整した溶液に、最終濃度10mMとなるようSM(PEG)12(サーモサイエンティフィック社製、succinimidyl-[(N-maleomidopropionamid)-dodecaethyleneglycol]ester)を混合し、1時間反応させた。
工程(6):反応混合液を10000Gで60分遠心分離を行い、上澄みを除去した。
工程(7):EDTAを2mM含有したPBSを加え、沈降物を分散させ、再度遠心分離を行った。同様の手順による洗浄を3回行った。最後に500μLのPBSを用いて再分散させ、抗体結合用マレイミド結合蛍光粒子を得た。 Step (4): The amino group-modified fluorescent particles obtained in step (3) were adjusted to 3 nM using PBS containing 2 mM of EDTA (ethylenediaminetetraacetic acid).
Step (5): SM (PEG) 12 (manufactured by Thermo Scientific, succinimidyl-[(N-maleomidopropionamid) -dodecaethyleneglycol] ester) is mixed with the solution prepared in step (4) to a final concentration of 10 mM. The reaction was performed for 1 hour.
Step (6): The reaction mixture was centrifuged at 10,000 G for 60 minutes, and the supernatant was removed.
Step (7): PBS containing 2 mM of EDTA was added, the precipitate was dispersed, and centrifuged again. The washing | cleaning by the same procedure was performed 3 times. Finally, 500 μL of PBS was redispersed to obtain maleimide-bound fluorescent particles for antibody binding.
工程(5):工程(4)で調整した溶液に、最終濃度10mMとなるようSM(PEG)12(サーモサイエンティフィック社製、succinimidyl-[(N-maleomidopropionamid)-dodecaethyleneglycol]ester)を混合し、1時間反応させた。
工程(6):反応混合液を10000Gで60分遠心分離を行い、上澄みを除去した。
工程(7):EDTAを2mM含有したPBSを加え、沈降物を分散させ、再度遠心分離を行った。同様の手順による洗浄を3回行った。最後に500μLのPBSを用いて再分散させ、抗体結合用マレイミド結合蛍光粒子を得た。 Step (4): The amino group-modified fluorescent particles obtained in step (3) were adjusted to 3 nM using PBS containing 2 mM of EDTA (ethylenediaminetetraacetic acid).
Step (5): SM (PEG) 12 (manufactured by Thermo Scientific, succinimidyl-[(N-maleomidopropionamid) -dodecaethyleneglycol] ester) is mixed with the solution prepared in step (4) to a final concentration of 10 mM. The reaction was performed for 1 hour.
Step (6): The reaction mixture was centrifuged at 10,000 G for 60 minutes, and the supernatant was removed.
Step (7): PBS containing 2 mM of EDTA was added, the precipitate was dispersed, and centrifuged again. The washing | cleaning by the same procedure was performed 3 times. Finally, 500 μL of PBS was redispersed to obtain maleimide-bound fluorescent particles for antibody binding.
工程(8):抗HER2抗体100μgを100μLのPBSに溶解させたところに、1Mジチオスレイトール(DTT)を添加し、30分反応させた。
工程(9):反応混合物についてゲルろ過カラムにより過剰のDTTを除去し、蛍光粒子に結合可能な還元化抗HER2抗体溶液を得た。
工程(10):蛍光粒子を出発原料として工程(7)で得られた蛍光粒子分散液と工程(9)で得られた還元化抗HER2抗体溶液とをPBS中で混合し、1時間反応させた。
工程(11):10mMメルカプトエタノール4μLを添加し、反応を停止させた。
工程(12):反応混合物を10000Gで60分遠心分離を行い、上澄みを除去した後、EDTAを2mM含有したPBSを加え、沈降物を分散させ、再度遠心分離を行った。同様の手順による洗浄を3回行った。最後に500μLのPBSを用いて再分散させ、抗HER2抗体が結合された蛍光粒子を得た。 Step (8): When 100 μg of the anti-HER2 antibody was dissolved in 100 μL of PBS, 1 M dithiothreitol (DTT) was added and reacted for 30 minutes.
Step (9): Excess DTT was removed from the reaction mixture using a gel filtration column to obtain a reduced anti-HER2 antibody solution capable of binding to fluorescent particles.
Step (10): Using fluorescent particles as a starting material, the fluorescent particle dispersion obtained in step (7) and the reduced anti-HER2 antibody solution obtained in step (9) are mixed in PBS and allowed to react for 1 hour. It was.
Step (11): 4 μL of 10 mM mercaptoethanol was added to stop the reaction.
Step (12): The reaction mixture was centrifuged at 10,000 G for 60 minutes, and the supernatant was removed. Then, PBS containing 2 mM of EDTA was added, the precipitate was dispersed, and centrifuged again. The washing | cleaning by the same procedure was performed 3 times. Finally, 500 μL of PBS was redispersed to obtain fluorescent particles bound with anti-HER2 antibody.
工程(9):反応混合物についてゲルろ過カラムにより過剰のDTTを除去し、蛍光粒子に結合可能な還元化抗HER2抗体溶液を得た。
工程(10):蛍光粒子を出発原料として工程(7)で得られた蛍光粒子分散液と工程(9)で得られた還元化抗HER2抗体溶液とをPBS中で混合し、1時間反応させた。
工程(11):10mMメルカプトエタノール4μLを添加し、反応を停止させた。
工程(12):反応混合物を10000Gで60分遠心分離を行い、上澄みを除去した後、EDTAを2mM含有したPBSを加え、沈降物を分散させ、再度遠心分離を行った。同様の手順による洗浄を3回行った。最後に500μLのPBSを用いて再分散させ、抗HER2抗体が結合された蛍光粒子を得た。 Step (8): When 100 μg of the anti-HER2 antibody was dissolved in 100 μL of PBS, 1 M dithiothreitol (DTT) was added and reacted for 30 minutes.
Step (9): Excess DTT was removed from the reaction mixture using a gel filtration column to obtain a reduced anti-HER2 antibody solution capable of binding to fluorescent particles.
Step (10): Using fluorescent particles as a starting material, the fluorescent particle dispersion obtained in step (7) and the reduced anti-HER2 antibody solution obtained in step (9) are mixed in PBS and allowed to react for 1 hour. It was.
Step (11): 4 μL of 10 mM mercaptoethanol was added to stop the reaction.
Step (12): The reaction mixture was centrifuged at 10,000 G for 60 minutes, and the supernatant was removed. Then, PBS containing 2 mM of EDTA was added, the precipitate was dispersed, and centrifuged again. The washing | cleaning by the same procedure was performed 3 times. Finally, 500 μL of PBS was redispersed to obtain fluorescent particles bound with anti-HER2 antibody.
(A1-3)蛍光色素及び量子ドットへの抗体の結合
蛍光色素(平均粒径:1nm以下)及び量子ドット(平均粒径:約5nm)に対して抗HER2抗体を結合させた染色試薬aを公知の方法により準備した。 (A1-3) Binding of antibody to fluorescent dye and quantum dot Staining reagent a in which anti-HER2 antibody is bound to fluorescent dye (average particle size: 1 nm or less) and quantum dot (average particle size: about 5 nm) Prepared by a known method.
蛍光色素(平均粒径:1nm以下)及び量子ドット(平均粒径:約5nm)に対して抗HER2抗体を結合させた染色試薬aを公知の方法により準備した。 (A1-3) Binding of antibody to fluorescent dye and quantum dot Staining reagent a in which anti-HER2 antibody is bound to fluorescent dye (average particle size: 1 nm or less) and quantum dot (average particle size: about 5 nm) Prepared by a known method.
(A2)FISH染色用試薬の作製
FISH染色用プローブは、HER-2 DNAプローブにNickトランスレーションによりビオチンを導入した、ビオチン化HER-2 DNAプローブを用いた。
また、プローブを可視化するための染色試薬bとしては、(A1-2)の工程(8)において抗HER2抗体の代わりにストレプトアビジンを用いて作成したストレプトアビジン修飾蛍光粒子、及び、(A1-3)と同様の蛍光色素及び量子ドットに対してストレプトアビジンを結合させたものを用いた。 (A2) Preparation of reagent for FISH staining As a probe for FISH staining, a biotinylated HER-2 DNA probe in which biotin was introduced into the HER-2 DNA probe by Nick translation was used.
The staining reagent b for visualizing the probe includes streptavidin-modified fluorescent particles prepared by using streptavidin instead of anti-HER2 antibody in step (8) of (A1-2), and (A1-3 ) And the same fluorescent dye and quantum dot used in combination with streptavidin.
FISH染色用プローブは、HER-2 DNAプローブにNickトランスレーションによりビオチンを導入した、ビオチン化HER-2 DNAプローブを用いた。
また、プローブを可視化するための染色試薬bとしては、(A1-2)の工程(8)において抗HER2抗体の代わりにストレプトアビジンを用いて作成したストレプトアビジン修飾蛍光粒子、及び、(A1-3)と同様の蛍光色素及び量子ドットに対してストレプトアビジンを結合させたものを用いた。 (A2) Preparation of reagent for FISH staining As a probe for FISH staining, a biotinylated HER-2 DNA probe in which biotin was introduced into the HER-2 DNA probe by Nick translation was used.
The staining reagent b for visualizing the probe includes streptavidin-modified fluorescent particles prepared by using streptavidin instead of anti-HER2 antibody in step (8) of (A1-2), and (A1-3 ) And the same fluorescent dye and quantum dot used in combination with streptavidin.
(B)組織染色
(B1)免疫組織化学(IHC)法
下記工程(1)~(10)の方法により、染色試薬aを用い、ヒト乳房組織標本の免疫染色を行った。染色標本はコスモバイオ社製の組織アレイスライド(CB-A712)を用い、あらかじめDAB染色によりHER2染色濃度を観察し、HER2高発現(HER2 3+)、HER2低発現(HER2 +)、HER2陰性(HER2 -)、の3種のロットを用意し、それぞれ染色を行った。 (B) Tissue staining (B1) Immunohistochemistry (IHC) method Human breast tissue specimens were immunostained using staining reagent a by the methods of the following steps (1) to (10). As a stained specimen, a tissue array slide (CB-A712) manufactured by Cosmo Bio Co., Ltd. was used, and the HER2 staining concentration was observed in advance by DAB staining. -), Three types of lots were prepared and each was stained.
(B1)免疫組織化学(IHC)法
下記工程(1)~(10)の方法により、染色試薬aを用い、ヒト乳房組織標本の免疫染色を行った。染色標本はコスモバイオ社製の組織アレイスライド(CB-A712)を用い、あらかじめDAB染色によりHER2染色濃度を観察し、HER2高発現(HER2 3+)、HER2低発現(HER2 +)、HER2陰性(HER2 -)、の3種のロットを用意し、それぞれ染色を行った。 (B) Tissue staining (B1) Immunohistochemistry (IHC) method Human breast tissue specimens were immunostained using staining reagent a by the methods of the following steps (1) to (10). As a stained specimen, a tissue array slide (CB-A712) manufactured by Cosmo Bio Co., Ltd. was used, and the HER2 staining concentration was observed in advance by DAB staining. -), Three types of lots were prepared and each was stained.
工程(1):キシレンを入れた容器に組織標本を30分浸漬させた。途中3回キシレンを交換した。
工程(2):エタノールを入れた容器に組織標本を30分浸漬させた。途中3回エタノールを交換した。
工程(3):水を入れた容器に組織標本を30分浸漬させた。途中3回水を交換した。
工程(4):10mMクエン酸緩衝液(pH6.0)に組織標本を30分浸漬させた。
工程(5):121度で10分オートクレーブ処理を行った。
工程(6):PBSを入れた容器に、オートクレーブ処理後の組織標本を30分浸漬させた。
工程(7):1%BSA含有PBSを組織標本に載せて、1時間放置した。
工程(8):1%BSA含有PBSで0.05nMに希釈した抗HER2抗体が結合された染色試薬aを、それぞれ組織標本に載せて3時間放置した。
工程(9):PBSを入れた容器に、染色後の組織標本をそれぞれ30分浸漬させた。
工程(10)4%中性パラホルムアルデヒド溶液で10分間固定処理した後、HE染色を行った。
工程(11):Merck Chemicals社製Aquatexを滴下後、カバーガラスを載せ封入した。 Step (1): The tissue specimen was immersed in a container containing xylene for 30 minutes. The xylene was changed three times during the process.
Step (2): The tissue specimen was immersed in a container containing ethanol for 30 minutes. The ethanol was changed three times during the process.
Step (3): The tissue specimen was immersed in a container containing water for 30 minutes. The water was changed three times along the way.
Step (4): The tissue specimen was immersed in 10 mM citrate buffer (pH 6.0) for 30 minutes.
Step (5): Autoclaving was performed at 121 degrees for 10 minutes.
Step (6): The tissue specimen after the autoclave treatment was immersed in a container containing PBS for 30 minutes.
Step (7): PBS containing 1% BSA was placed on the tissue specimen and allowed to stand for 1 hour.
Step (8): The staining reagent a to which the anti-HER2 antibody diluted to 0.05 nM with PBS containing 1% BSA was bound was placed on each tissue specimen and allowed to stand for 3 hours.
Step (9): Each stained tissue specimen was immersed in a container containing PBS for 30 minutes.
Step (10) After fixing with a 4% neutral paraformaldehyde solution for 10 minutes, HE staining was performed.
Step (11): After adding Aquatex made by Merck Chemicals, a cover glass was placed and sealed.
工程(2):エタノールを入れた容器に組織標本を30分浸漬させた。途中3回エタノールを交換した。
工程(3):水を入れた容器に組織標本を30分浸漬させた。途中3回水を交換した。
工程(4):10mMクエン酸緩衝液(pH6.0)に組織標本を30分浸漬させた。
工程(5):121度で10分オートクレーブ処理を行った。
工程(6):PBSを入れた容器に、オートクレーブ処理後の組織標本を30分浸漬させた。
工程(7):1%BSA含有PBSを組織標本に載せて、1時間放置した。
工程(8):1%BSA含有PBSで0.05nMに希釈した抗HER2抗体が結合された染色試薬aを、それぞれ組織標本に載せて3時間放置した。
工程(9):PBSを入れた容器に、染色後の組織標本をそれぞれ30分浸漬させた。
工程(10)4%中性パラホルムアルデヒド溶液で10分間固定処理した後、HE染色を行った。
工程(11):Merck Chemicals社製Aquatexを滴下後、カバーガラスを載せ封入した。 Step (1): The tissue specimen was immersed in a container containing xylene for 30 minutes. The xylene was changed three times during the process.
Step (2): The tissue specimen was immersed in a container containing ethanol for 30 minutes. The ethanol was changed three times during the process.
Step (3): The tissue specimen was immersed in a container containing water for 30 minutes. The water was changed three times along the way.
Step (4): The tissue specimen was immersed in 10 mM citrate buffer (pH 6.0) for 30 minutes.
Step (5): Autoclaving was performed at 121 degrees for 10 minutes.
Step (6): The tissue specimen after the autoclave treatment was immersed in a container containing PBS for 30 minutes.
Step (7): PBS containing 1% BSA was placed on the tissue specimen and allowed to stand for 1 hour.
Step (8): The staining reagent a to which the anti-HER2 antibody diluted to 0.05 nM with PBS containing 1% BSA was bound was placed on each tissue specimen and allowed to stand for 3 hours.
Step (9): Each stained tissue specimen was immersed in a container containing PBS for 30 minutes.
Step (10) After fixing with a 4% neutral paraformaldehyde solution for 10 minutes, HE staining was performed.
Step (11): After adding Aquatex made by Merck Chemicals, a cover glass was placed and sealed.
(B2)蛍光in situ ハイブリダイゼーション(FISH)法
染色試薬bを用いて、FISH法によるヒト乳房組織標本の染色を行った。ビオチン化HER-2 DNAプローブのハイブリダイゼーションは、公知の方法により行なった。また、ハイブリダイゼーションしたビオチン化HER-2 DNAプローブに対して、染色試薬bを公知の方法により結合して可視化した。
染色標本はコスモバイオ社製の組織アレイスライド(CB-A712)を用い、あらかじめパスビジョン(登録商標)HER-2 DNAプローブキット(アボット社製)を用いて各スポット当りのFISHスコアを算出した。算出されたFISHスコアに基づいて、HER2遺伝子量の異なる3種の組織標本のロットを用意し、それぞれ染色を行った。HER2遺伝子は通常、細胞内に2つ存在し、細胞増殖時には4個に増える場合もある。正常な細胞である「増幅無し」の組織標本のロットにおいては、遺伝子数が1~4個となる。一方、HER2「大増幅」の組織標本のロットにおいては、細胞あたり10個以上のHER2遺伝子がある。また、HER2「小増幅」の組織標本のロットにおいては、細胞あたり6~10個のHER2遺伝子がある。 (B2) Fluorescence in situ hybridization (FISH) method Using a staining reagent b, a human breast tissue specimen was stained by the FISH method. Hybridization of the biotinylated HER-2 DNA probe was performed by a known method. Further, staining reagent b was bound to the hybridized biotinylated HER-2 DNA probe by a known method and visualized.
As a stained specimen, a tissue array slide (CB-A712) manufactured by Cosmo Bio Co., Ltd. was used, and a FISH score for each spot was calculated in advance using Pathvision (registered trademark) HER-2 DNA probe kit (manufactured by Abbott). Based on the calculated FISH score, lots of three kinds of tissue specimens having different HER2 gene amounts were prepared and stained. Two HER2 genes are usually present in the cell and may increase to four during cell proliferation. In a lot of “unamplified” tissue specimens that are normal cells, the number of genes is 1 to 4. On the other hand, there are more than 10 HER2 genes per cell in a lot of HER2 “large amplification” tissue specimens. In addition, there are 6-10 HER2 genes per cell in a HER2 “small amplification” tissue specimen lot.
染色試薬bを用いて、FISH法によるヒト乳房組織標本の染色を行った。ビオチン化HER-2 DNAプローブのハイブリダイゼーションは、公知の方法により行なった。また、ハイブリダイゼーションしたビオチン化HER-2 DNAプローブに対して、染色試薬bを公知の方法により結合して可視化した。
染色標本はコスモバイオ社製の組織アレイスライド(CB-A712)を用い、あらかじめパスビジョン(登録商標)HER-2 DNAプローブキット(アボット社製)を用いて各スポット当りのFISHスコアを算出した。算出されたFISHスコアに基づいて、HER2遺伝子量の異なる3種の組織標本のロットを用意し、それぞれ染色を行った。HER2遺伝子は通常、細胞内に2つ存在し、細胞増殖時には4個に増える場合もある。正常な細胞である「増幅無し」の組織標本のロットにおいては、遺伝子数が1~4個となる。一方、HER2「大増幅」の組織標本のロットにおいては、細胞あたり10個以上のHER2遺伝子がある。また、HER2「小増幅」の組織標本のロットにおいては、細胞あたり6~10個のHER2遺伝子がある。 (B2) Fluorescence in situ hybridization (FISH) method Using a staining reagent b, a human breast tissue specimen was stained by the FISH method. Hybridization of the biotinylated HER-2 DNA probe was performed by a known method. Further, staining reagent b was bound to the hybridized biotinylated HER-2 DNA probe by a known method and visualized.
As a stained specimen, a tissue array slide (CB-A712) manufactured by Cosmo Bio Co., Ltd. was used, and a FISH score for each spot was calculated in advance using Pathvision (registered trademark) HER-2 DNA probe kit (manufactured by Abbott). Based on the calculated FISH score, lots of three kinds of tissue specimens having different HER2 gene amounts were prepared and stained. Two HER2 genes are usually present in the cell and may increase to four during cell proliferation. In a lot of “unamplified” tissue specimens that are normal cells, the number of genes is 1 to 4. On the other hand, there are more than 10 HER2 genes per cell in a lot of HER2 “large amplification” tissue specimens. In addition, there are 6-10 HER2 genes per cell in a HER2 “small amplification” tissue specimen lot.
(C)画像解析処理
染色試薬a及びbを用いて染色した組織標本について、顕微鏡画像(明視野画像及び蛍光画像)を取得した。
顕微鏡は、蛍光顕微鏡(BZ-9000、キーエンス社製)を用い、対物レンズ倍率を40倍に設定した。蛍光画像の取得にあたっては、中心波長560nmの励起光を照射して、組織標本から発せられる630nmの中心波長を有する蛍光を結像し、顕微鏡設置カメラ(モノクロ)により顕微鏡画像(画像データ)を取得した。
顕微鏡画像の取得は、まず、細胞の外縁にピントを合わせてから、ステージを下方に2μm下げた位置で顕微鏡画像を取得し、ステージを0.2μm上方向にずらして再度顕微鏡画像を取得することを繰り返し、計20枚の蛍光顕微鏡画像を取得した。 (C) Image analysis processing Microscopic images (bright-field images and fluorescent images) were obtained for tissue specimens stained using staining reagents a and b.
The microscope used was a fluorescence microscope (BZ-9000, manufactured by Keyence Corporation), and the objective lens magnification was set to 40 times. In acquiring the fluorescence image, the excitation light having the center wavelength of 560 nm is irradiated to form an image of the fluorescence having the center wavelength of 630 nm emitted from the tissue specimen, and the microscope image (image data) is acquired by the microscope installation camera (monochrome). did.
To obtain a microscopic image, first focus on the outer edge of the cell, then acquire the microscopic image at a position where the stage is lowered 2 μm downward, and then acquire the microscopic image again by shifting the stage upward 0.2 μm. And a total of 20 fluorescence microscope images were acquired.
染色試薬a及びbを用いて染色した組織標本について、顕微鏡画像(明視野画像及び蛍光画像)を取得した。
顕微鏡は、蛍光顕微鏡(BZ-9000、キーエンス社製)を用い、対物レンズ倍率を40倍に設定した。蛍光画像の取得にあたっては、中心波長560nmの励起光を照射して、組織標本から発せられる630nmの中心波長を有する蛍光を結像し、顕微鏡設置カメラ(モノクロ)により顕微鏡画像(画像データ)を取得した。
顕微鏡画像の取得は、まず、細胞の外縁にピントを合わせてから、ステージを下方に2μm下げた位置で顕微鏡画像を取得し、ステージを0.2μm上方向にずらして再度顕微鏡画像を取得することを繰り返し、計20枚の蛍光顕微鏡画像を取得した。 (C) Image analysis processing Microscopic images (bright-field images and fluorescent images) were obtained for tissue specimens stained using staining reagents a and b.
The microscope used was a fluorescence microscope (BZ-9000, manufactured by Keyence Corporation), and the objective lens magnification was set to 40 times. In acquiring the fluorescence image, the excitation light having the center wavelength of 560 nm is irradiated to form an image of the fluorescence having the center wavelength of 630 nm emitted from the tissue specimen, and the microscope image (image data) is acquired by the microscope installation camera (monochrome). did.
To obtain a microscopic image, first focus on the outer edge of the cell, then acquire the microscopic image at a position where the stage is lowered 2 μm downward, and then acquire the microscopic image again by shifting the stage upward 0.2 μm. And a total of 20 fluorescence microscope images were acquired.
(C1)本発明の画像解析処理方法
本発明の合焦点画像を用いた計測として、得られた20枚の顕微鏡画像に図5の画像解析処理を実行し、輝度プロファイルに基づいて、1細胞当たりの蛍光粒子数(輝点数)を算出した。 (C1) Image Analysis Processing Method of the Present Invention As measurement using the focused image of the present invention, the image analysis processing of FIG. 5 is performed on the obtained 20 microscope images, and per cell based on the luminance profile. The number of fluorescent particles (number of bright spots) was calculated.
本発明の合焦点画像を用いた計測として、得られた20枚の顕微鏡画像に図5の画像解析処理を実行し、輝度プロファイルに基づいて、1細胞当たりの蛍光粒子数(輝点数)を算出した。 (C1) Image Analysis Processing Method of the Present Invention As measurement using the focused image of the present invention, the image analysis processing of FIG. 5 is performed on the obtained 20 microscope images, and per cell based on the luminance profile. The number of fluorescent particles (number of bright spots) was calculated.
(C2)従来の画像解析処理方法
従来の計測手法による比較として、取得した20枚の顕微鏡画像のうち、細胞の外縁にピントを合わせた一枚の画像(単一画像)のみを用いて、輝度が所定の閾値を超えた領域を輝点として計測し、1細胞当たりの蛍光粒子数(輝点数)を算出した。 (C2) Conventional image analysis processing method As a comparison by the conventional measurement method, the luminance is obtained by using only one image (single image) in which the outer edge of the cell is in focus among the obtained 20 microscope images. The area where the value exceeded a predetermined threshold was measured as a bright spot, and the number of fluorescent particles per cell (the number of bright spots) was calculated.
従来の計測手法による比較として、取得した20枚の顕微鏡画像のうち、細胞の外縁にピントを合わせた一枚の画像(単一画像)のみを用いて、輝度が所定の閾値を超えた領域を輝点として計測し、1細胞当たりの蛍光粒子数(輝点数)を算出した。 (C2) Conventional image analysis processing method As a comparison by the conventional measurement method, the luminance is obtained by using only one image (single image) in which the outer edge of the cell is in focus among the obtained 20 microscope images. The area where the value exceeded a predetermined threshold was measured as a bright spot, and the number of fluorescent particles per cell (the number of bright spots) was calculated.
<実験結果1>
(A1)に記載の染色試薬aを用いて、(B1)に記載のIHC法によってHER2タンパク質が染色された組織標本から算出された一細胞当たりの輝点数を、表1に示す。
実施例1においては、蛍光粒子由来の染色試薬aを用いて染色した組織標本から、本発明の画像解析処理方法(C1)を用いて輝点数を算出した。
比較例1においては、蛍光粒子由来の染色試薬aを用いて染色した組織標本から、従来の画像解析処理方法(C2)を用いて輝点数を算出した。
比較例2、3においては、蛍光色素及び量子ドット由来の染色試薬aを用いて染色した組織標本から、本発明の画像解析処理方法(C1)を用いて輝点数を算出した。 <Experimental result 1>
Table 1 shows the number of bright spots per cell calculated from the tissue specimen stained with the HER2 protein by the IHC method described in (B1) using the staining reagent a described in (A1).
In Example 1, the number of bright spots was calculated from the tissue specimen stained with the fluorescent particle-derived staining reagent a using the image analysis processing method (C1) of the present invention.
In Comparative Example 1, the number of bright spots was calculated from a tissue specimen stained with a fluorescent particle-derived staining reagent a using a conventional image analysis processing method (C2).
In Comparative Examples 2 and 3, the number of bright spots was calculated from the tissue specimen stained with the fluorescent dye and the staining reagent a derived from quantum dots using the image analysis processing method (C1) of the present invention.
(A1)に記載の染色試薬aを用いて、(B1)に記載のIHC法によってHER2タンパク質が染色された組織標本から算出された一細胞当たりの輝点数を、表1に示す。
実施例1においては、蛍光粒子由来の染色試薬aを用いて染色した組織標本から、本発明の画像解析処理方法(C1)を用いて輝点数を算出した。
比較例1においては、蛍光粒子由来の染色試薬aを用いて染色した組織標本から、従来の画像解析処理方法(C2)を用いて輝点数を算出した。
比較例2、3においては、蛍光色素及び量子ドット由来の染色試薬aを用いて染色した組織標本から、本発明の画像解析処理方法(C1)を用いて輝点数を算出した。 <
Table 1 shows the number of bright spots per cell calculated from the tissue specimen stained with the HER2 protein by the IHC method described in (B1) using the staining reagent a described in (A1).
In Example 1, the number of bright spots was calculated from the tissue specimen stained with the fluorescent particle-derived staining reagent a using the image analysis processing method (C1) of the present invention.
In Comparative Example 1, the number of bright spots was calculated from a tissue specimen stained with a fluorescent particle-derived staining reagent a using a conventional image analysis processing method (C2).
In Comparative Examples 2 and 3, the number of bright spots was calculated from the tissue specimen stained with the fluorescent dye and the staining reagent a derived from quantum dots using the image analysis processing method (C1) of the present invention.

蛍光粒子の平均粒径及び画像解析方法と輝点数の関係に着目すると、表1より、IHC法において、蛍光物質の平均粒径及び組織標本のロットが同一の組み合わせの場合で比較すると、本発明の再構成画像と輝度プロファイルを用いた方法(C1)によれば、従来の方法(C2)と比べて、常に輝点数が多く算出され、真値に近い結果が得られた。
Focusing on the average particle diameter of fluorescent particles and the relationship between the image analysis method and the number of bright spots, it can be seen from Table 1 that the IHC method compares the average particle diameter of fluorescent substances and the lot of tissue specimens in the same combination. According to the method (C1) using the reconstructed image and the luminance profile, the number of bright spots was always calculated larger than in the conventional method (C2), and a result close to the true value was obtained.
また、事前にDAB染色により計測されたHER2染色濃度と輝点数の関係に着目すると、実施例1によれば、HER2 +の組織標本においては、HER2 -の組織標本と比べて常に2.5倍以上(平均:約3.7倍)の数の輝点が算出された。また、HER2 3+の組織標本においては、HER2 +の組織標本の4.6~5.8倍(平均:約5.2倍)の輝点数が算出された。
一方、比較例1によれば、HER2 +の組織標本においては、HER2 -の組織標本の1.3~2.3倍(平均:約1.7倍)の数の輝点が算出された。また、HER2 3+の組織標本においては、HER2 +の組織標本の4.6~6倍(平均:約5.2倍)の輝点数が算出された。 Further, focusing on the relationship between the HER2 staining density and the number of bright spots measured in advance by DAB staining, according to Example 1, the HER2 + tissue sample is always 2.5 times as large as the HER2− tissue sample. The number of bright spots above (average: about 3.7 times) was calculated. In theHER2 3+ tissue specimen, the number of bright spots was calculated to be 4.6 to 5.8 times (average: about 5.2 times) that of the HER2 + tissue specimen.
On the other hand, according to Comparative Example 1, in the HER2 + tissue sample, 1.3 to 2.3 times (average: about 1.7 times) the number of bright spots were calculated as compared to the HER2 − tissue sample. Further, in theHER2 3+ tissue specimen, the number of bright spots was calculated to be 4.6 to 6 times (average: about 5.2 times) that of the HER2 + tissue specimen.
一方、比較例1によれば、HER2 +の組織標本においては、HER2 -の組織標本の1.3~2.3倍(平均:約1.7倍)の数の輝点が算出された。また、HER2 3+の組織標本においては、HER2 +の組織標本の4.6~6倍(平均:約5.2倍)の輝点数が算出された。 Further, focusing on the relationship between the HER2 staining density and the number of bright spots measured in advance by DAB staining, according to Example 1, the HER2 + tissue sample is always 2.5 times as large as the HER2− tissue sample. The number of bright spots above (average: about 3.7 times) was calculated. In the
On the other hand, according to Comparative Example 1, in the HER2 + tissue sample, 1.3 to 2.3 times (average: about 1.7 times) the number of bright spots were calculated as compared to the HER2 − tissue sample. Further, in the
また、比較例2及び3で得られた蛍光画像においては、細胞全体が均一に蛍光を発しており、蛍光色素及び量子ドットの一つ一つをドット状に観察することが不可能であったため、輝点数を算出することができなかった。
In addition, in the fluorescence images obtained in Comparative Examples 2 and 3, the entire cell was uniformly fluorescent, and it was impossible to observe each of the fluorescent dye and the quantum dots in a dot shape. The number of bright spots could not be calculated.
実験結果1より、平均粒径20~200nmの蛍光粒子を用いて染色することによって、蛍光染色されたタンパク質を蛍光輝点として計測可能であることがわかる。
さらに、本発明の生体物質定量方法によれば、焦点深度を変えた複数の蛍光画像を用いて解析を行うので、焦点深度の方向の広がりも加味した細胞の全体から蛍光粒子を検出することができる。さらに、輝度プロファイルに基づいた解析により、近接した蛍光粒子を分離して計測可能であるため、より正確な輝点数を計測できる。実際、実施例1及び比較例1において、組織標本のロットがHER2 3+及びHER2 +の組織標本から計測された輝点数を、蛍光粒子の平均粒径が同一のもので比較すると、実施例では比較例の約2.5倍の輝点数が算出されていた。
このように、本発明によれば焦点深度の方向にも渡る多数の輝点を正確に計測できるため、発現量のわずかな差を検出しやすく、HER2の発現が少ない場合でも発現量の差を明確に区別して診断することができる。 From theexperimental result 1, it can be seen that by staining with fluorescent particles having an average particle diameter of 20 to 200 nm, the fluorescently stained protein can be measured as a fluorescent bright spot.
Furthermore, according to the biological material quantification method of the present invention, since analysis is performed using a plurality of fluorescent images with different depths of focus, it is possible to detect fluorescent particles from the whole cell in consideration of the spread in the direction of the depth of focus. it can. Furthermore, since it is possible to separate and measure the adjacent fluorescent particles by the analysis based on the luminance profile, the number of bright spots can be measured more accurately. In fact, in Example 1 and Comparative Example 1, the number of bright spots measured from the tissue specimens with the tissuespecimen lots HER2 3+ and HER2 + were compared with the same average particle diameter of the fluorescent particles. The number of bright spots about 2.5 times that of the example was calculated.
As described above, according to the present invention, since a large number of bright spots across the depth of focus can be accurately measured, it is easy to detect a slight difference in the expression level, and the difference in the expression level can be detected even when the expression of HER2 is small. It can be clearly distinguished and diagnosed.
さらに、本発明の生体物質定量方法によれば、焦点深度を変えた複数の蛍光画像を用いて解析を行うので、焦点深度の方向の広がりも加味した細胞の全体から蛍光粒子を検出することができる。さらに、輝度プロファイルに基づいた解析により、近接した蛍光粒子を分離して計測可能であるため、より正確な輝点数を計測できる。実際、実施例1及び比較例1において、組織標本のロットがHER2 3+及びHER2 +の組織標本から計測された輝点数を、蛍光粒子の平均粒径が同一のもので比較すると、実施例では比較例の約2.5倍の輝点数が算出されていた。
このように、本発明によれば焦点深度の方向にも渡る多数の輝点を正確に計測できるため、発現量のわずかな差を検出しやすく、HER2の発現が少ない場合でも発現量の差を明確に区別して診断することができる。 From the
Furthermore, according to the biological material quantification method of the present invention, since analysis is performed using a plurality of fluorescent images with different depths of focus, it is possible to detect fluorescent particles from the whole cell in consideration of the spread in the direction of the depth of focus. it can. Furthermore, since it is possible to separate and measure the adjacent fluorescent particles by the analysis based on the luminance profile, the number of bright spots can be measured more accurately. In fact, in Example 1 and Comparative Example 1, the number of bright spots measured from the tissue specimens with the tissue
As described above, according to the present invention, since a large number of bright spots across the depth of focus can be accurately measured, it is easy to detect a slight difference in the expression level, and the difference in the expression level can be detected even when the expression of HER2 is small. It can be clearly distinguished and diagnosed.
<実験結果2>
(A2)に記載の染色試薬bを用いて、(B2)に記載のFISH法によってHER2遺伝子が染色された組織標本から算出された一細胞当たりの輝点数を、表2に示す。
実施例2においては、蛍光粒子由来の染色試薬bを用いて染色した組織標本から、本発明の画像解析処理方法(C1)を用いて輝点数を算出した。
比較例4においては、蛍光粒子由来の染色試薬bを用いて染色した組織標本から、従来の画像解析処理方法(C2)を用いて輝点数を算出した。
比較例5、6においては、蛍光色素及び量子ドット由来の染色試薬bを用いて染色した組織標本から、本発明の画像解析処理方法(C1)及び従来の画像解析処理方法(C2)を用いて輝点数を算出した。なお、蛍光色素及び量子ドットは1分子当たりの輝度が小さく、基準となる輝度プロファイルを焦点深度を変えて得ることが困難であった。そのため、比較例5-1及び比較例6-1においては、蛍光粒子の輝度プロファイルを用いて画像解析処理を行った。 <Experimental result 2>
Table 2 shows the number of bright spots per cell calculated from the tissue specimen in which the HER2 gene was stained by the FISH method described in (B2) using the staining reagent b described in (A2).
In Example 2, the number of bright spots was calculated from the tissue specimen stained with the fluorescent particle-derived staining reagent b using the image analysis processing method (C1) of the present invention.
In Comparative Example 4, the number of bright spots was calculated from the tissue specimen stained with the fluorescent particle-derived staining reagent b using the conventional image analysis processing method (C2).
In Comparative Examples 5 and 6, from the tissue specimen stained with the fluorescent dye and the staining reagent b derived from quantum dots, the image analysis processing method (C1) of the present invention and the conventional image analysis processing method (C2) are used. The number of bright spots was calculated. The fluorescent dyes and quantum dots have low luminance per molecule, and it has been difficult to obtain a reference luminance profile by changing the depth of focus. Therefore, in Comparative Example 5-1 and Comparative Example 6-1, image analysis processing was performed using the luminance profile of the fluorescent particles.
(A2)に記載の染色試薬bを用いて、(B2)に記載のFISH法によってHER2遺伝子が染色された組織標本から算出された一細胞当たりの輝点数を、表2に示す。
実施例2においては、蛍光粒子由来の染色試薬bを用いて染色した組織標本から、本発明の画像解析処理方法(C1)を用いて輝点数を算出した。
比較例4においては、蛍光粒子由来の染色試薬bを用いて染色した組織標本から、従来の画像解析処理方法(C2)を用いて輝点数を算出した。
比較例5、6においては、蛍光色素及び量子ドット由来の染色試薬bを用いて染色した組織標本から、本発明の画像解析処理方法(C1)及び従来の画像解析処理方法(C2)を用いて輝点数を算出した。なお、蛍光色素及び量子ドットは1分子当たりの輝度が小さく、基準となる輝度プロファイルを焦点深度を変えて得ることが困難であった。そのため、比較例5-1及び比較例6-1においては、蛍光粒子の輝度プロファイルを用いて画像解析処理を行った。 <Experimental result 2>
Table 2 shows the number of bright spots per cell calculated from the tissue specimen in which the HER2 gene was stained by the FISH method described in (B2) using the staining reagent b described in (A2).
In Example 2, the number of bright spots was calculated from the tissue specimen stained with the fluorescent particle-derived staining reagent b using the image analysis processing method (C1) of the present invention.
In Comparative Example 4, the number of bright spots was calculated from the tissue specimen stained with the fluorescent particle-derived staining reagent b using the conventional image analysis processing method (C2).
In Comparative Examples 5 and 6, from the tissue specimen stained with the fluorescent dye and the staining reagent b derived from quantum dots, the image analysis processing method (C1) of the present invention and the conventional image analysis processing method (C2) are used. The number of bright spots was calculated. The fluorescent dyes and quantum dots have low luminance per molecule, and it has been difficult to obtain a reference luminance profile by changing the depth of focus. Therefore, in Comparative Example 5-1 and Comparative Example 6-1, image analysis processing was performed using the luminance profile of the fluorescent particles.

蛍光粒子の平均粒径及び画像解析方法と輝点数の関係に着目すると、実施例2及び比較例4より、FISH法によってHER2遺伝子が染色された組織標本において、蛍光粒子の平均粒径及び組織標本のロットが同一の組み合わせの場合で比較すると、本発明の再構成画像と輝度プロファイルを用いた方法(実施例2)によれば、従来の方法(比較例4)と比べて、常に多数の輝点が算出され(20~55%増加)、真値に近い結果が得られた。
Focusing on the average particle diameter of fluorescent particles and the relationship between the image analysis method and the number of bright spots, from Example 2 and Comparative Example 4, the average particle diameter of fluorescent particles and the tissue sample in the tissue sample stained with the HER2 gene by the FISH method When the lots of the same combination are compared, according to the method (Example 2) using the reconstructed image and the luminance profile of the present invention, the number of bright spots is always larger than that of the conventional method (Comparative Example 4). Points were calculated (20-55% increase) and results close to true values were obtained.
比較例5~6においては、染色に用いた蛍光色素及び量子ドットではなく蛍光粒子の輝度プロファイルを用いて画像解析を行ったため、本発明の方法で算出された輝点数は、従来の方法で算出された輝点数よりは増加するものの、増加の程度は14~18%と小さく、蛍光粒子を用いた実施例2と比べると定量精度は低かった。
In Comparative Examples 5 to 6, since the image analysis was performed using the luminance profile of the fluorescent particles instead of the fluorescent dye and quantum dots used for staining, the number of bright spots calculated by the method of the present invention was calculated by the conventional method. Although the number of bright spots increased, the degree of increase was as small as 14 to 18%, and the quantitative accuracy was lower than that in Example 2 using fluorescent particles.
また、事前に計測されたFISHスコアと輝点数の関係に着目すると、HER2小増幅の組織標本においては、HER2増幅無しの組織標本と比べて約4倍以上の輝点数が算出され、HER2大増幅の組織標本においては、HER2小増幅の組織標本の約2.3倍の輝点数が算出された。これらの増加の程度は、表2に記載の何れの粒径、何れの画像解析処理方法の場合でも特に差はなかった。
Focusing on the relationship between the pre-measured FISH score and the number of bright spots, the HER2 small amplification tissue specimen calculates the number of bright spots more than about 4 times that of the tissue specimen without HER2 amplification, and HER2 large amplification. The number of bright spots about 2.3 times that of the tissue sample with a small HER2 amplification was calculated. The degree of increase was not particularly different in any of the particle sizes and the image analysis processing methods shown in Table 2.
実験結果2より、蛍光色素及び量子ドットを輝点として観察可能なFISH法による遺伝子の染色の場合でも、蛍光物質として蛍光粒子を用いれば、算出される輝点数がより増加するため、好適である。
From the experimental result 2, it is preferable to use fluorescent particles as the fluorescent material because the number of calculated bright spots is increased even in the case of staining of genes by the FISH method in which fluorescent dyes and quantum dots can be observed as bright spots. .
実験結果1及び2より、HER2タンパク又は遺伝子が正常状態からわずかに増加した場合の検出感度として考えると、FISH法では「増幅無し」から「小増幅」となった場合の輝点数の変化率は、本発明の方法と従来の方法による差はほとんどなく、いずれも4~5倍の範囲であった。一方、IHC法では「HER2 -」から「HER2 +」となった場合の輝点数の変化率は、従来の方法(約1.7倍)に対して本発明の方法(約3.7倍)では極めて変化率が増加し、感度が向上したと言える。以上の結果から、本発明の方法は、遺伝子及びタンパク質のいずれの定量にも有効であり、従来の方法より真値に近い輝点数を算出可能であるが、検出感度の向上という点では、特にタンパク質の検出において著しい効果を発揮する。
From the experimental results 1 and 2, when considering the detection sensitivity when the HER2 protein or gene is slightly increased from the normal state, the change rate of the number of bright spots when the FISH method changes from “no amplification” to “small amplification” is There was almost no difference between the method of the present invention and the conventional method, and all were in the range of 4 to 5 times. On the other hand, in the IHC method, the change rate of the number of bright spots when “HER2 −” is changed to “HER2 +” is the method of the present invention (about 3.7 times) compared to the conventional method (about 1.7 times). Then, it can be said that the rate of change is extremely increased and the sensitivity is improved. From the above results, the method of the present invention is effective for quantification of both genes and proteins, and can calculate the number of bright spots closer to the true value than the conventional method. It has a remarkable effect in protein detection.
本発明は、組織標本内での特定の生体物質の数を正確に定量できることを特徴とし、高精度な病理診断情報の生成に特に好適に利用することができる。
The present invention is characterized in that the number of specific biological substances in a tissue specimen can be accurately quantified, and can be particularly suitably used for generating highly accurate pathological diagnosis information.
1A 顕微鏡画像取得装置
2A 画像処理装置
3A ケーブル
21 制御部
22 操作部
23 表示部
24 通信I/F
25 記憶部
26 バス
100 病理診断支援システム DESCRIPTION OFSYMBOLS 1A Microscope image acquisition apparatus 2A Image processing apparatus 3A Cable 21 Control part 22 Operation part 23 Display part 24 Communication I / F
25storage unit 26 bus 100 pathological diagnosis support system
2A 画像処理装置
3A ケーブル
21 制御部
22 操作部
23 表示部
24 通信I/F
25 記憶部
26 バス
100 病理診断支援システム DESCRIPTION OF
25
Claims (9)
- 蛍光物質を複数集積した蛍光粒子を染色試薬として用いて特定の生体物質が染色された標本から、前記生体物質を定量する生体物質定量方法において、
焦点深度を所定の間隔で連続的に変え、各焦点深度において、前記標本における前記生体物質の発現を蛍光輝点で表す蛍光画像を入力する入力工程と、
各焦点深度における前記蛍光画像から、輝点領域が抽出された輝点画像を生成し、当該輝点画像ごとに輝度プロファイルを作成するプロファイル作成工程と、
基準プロファイルとして予め計測された蛍光粒子の輝度プロファイルを作成し、各焦点深度における輝点画像の輝度プロファイルを、前記基準プロファイルに基づいて解析することにより、前記蛍光画像に含まれる蛍光粒子の数を算出する算出工程と、
を有することを特徴とする生体物質定量方法。 In a biological material quantification method for quantifying the biological material from a specimen stained with a specific biological material using fluorescent particles in which a plurality of fluorescent materials are accumulated as a staining reagent,
An input step of continuously changing a depth of focus at a predetermined interval, and inputting a fluorescence image representing the expression of the biological material in the specimen as a fluorescent luminescent spot at each depth of focus;
A profile creation step of creating a bright spot image in which a bright spot region is extracted from the fluorescent image at each focal depth, and creating a brightness profile for each bright spot image;
The brightness profile of the fluorescent particles measured in advance as a reference profile is created, and the brightness profile of the bright spot image at each focal depth is analyzed based on the reference profile, whereby the number of fluorescent particles included in the fluorescence image is determined. A calculation step to calculate,
A biological substance quantification method characterized by comprising: - 請求項1に記載の生体物質定量方法において、
前記基準プロファイルは、蛍光輝点源となる蛍光粒子からの相対距離と輝度の情報を備えることを特徴とする生体物質定量方法。 The biological material quantification method according to claim 1,
The biological material quantification method, wherein the reference profile includes information on a relative distance and luminance from a fluorescent particle serving as a fluorescent bright spot source. - 請求項1又は2に記載の生体物質定量方法において、
前記算出工程において、前記蛍光画像に含まれる蛍光粒子の位置を特定し、
各焦点深度における前記輝点画像の中から、算出された蛍光粒子の位置に最も近い位置の輝点画像を抽出する抽出工程と、
前記抽出された輝点画像を合成して一枚の画像に再構成した再構成画像を生成する生成工程と、
を有することを特徴とする生体物質定量方法。 The biological substance quantification method according to claim 1 or 2,
In the calculation step, the position of the fluorescent particles included in the fluorescent image is specified,
An extraction step for extracting a bright spot image at a position closest to the calculated fluorescent particle position from the bright spot images at each focal depth;
A generating step of generating a reconstructed image obtained by synthesizing the extracted bright spot images and reconstructing into one image;
A biological substance quantification method characterized by comprising: - 請求項1~3の何れか一項に記載の生体物質定量方法において、
前記蛍光粒子の平均粒径が20~200nmであることを特徴とする生体物質定量方法。 The biological material quantification method according to any one of claims 1 to 3,
A biological material quantification method, wherein the fluorescent particles have an average particle diameter of 20 to 200 nm. - 請求項1~4の何れか一項に記載の生体物質定量方法において、
前記蛍光粒子の粒径の変動係数が15%以下であることを特徴とする生体物質定量方法。 The biological material quantification method according to any one of claims 1 to 4,
A biological material quantification method, wherein a coefficient of variation in particle diameter of the fluorescent particles is 15% or less. - 請求項1~5の何れか一項に記載の生体物質定量方法において、
前記生体物質がタンパク質であることを特徴とする生体物質定量方法。 The biological material quantification method according to any one of claims 1 to 5,
A biological material quantification method, wherein the biological material is a protein. - 蛍光物質を複数集積した蛍光粒子を染色試薬として用いて特定の生体物質が染色された標本から、前記生体物質を定量する画像処理装置において、
焦点深度を所定の間隔で連続的に変え、各焦点深度において、前記標本における前記生体物質の発現を蛍光輝点で表す蛍光画像を入力する入力手段と、
各焦点深度における前記蛍光画像から、輝点領域が抽出された輝点画像を生成し、当該輝点画像ごとに輝度プロファイルを作成するプロファイル作成手段と、
基準プロファイルとして予め計測された蛍光粒子の輝度プロファイルを作成し、各焦点深度における輝点画像の輝度プロファイルを、前記基準プロファイルに基づいて解析することにより、前記蛍光画像に含まれる蛍光粒子の数を算出する算出手段と、
を有することを特徴とする画像処理装置。 In an image processing apparatus for quantifying the biological material from a specimen in which a specific biological material is stained using fluorescent particles in which a plurality of fluorescent materials are accumulated as a staining reagent,
Input means for continuously changing the depth of focus at a predetermined interval, and inputting a fluorescence image representing the expression of the biological material in the specimen as a fluorescence bright spot at each depth of focus;
A profile creating means for generating a bright spot image in which a bright spot region is extracted from the fluorescent image at each focal depth and creating a brightness profile for each bright spot image;
The brightness profile of the fluorescent particles measured in advance as a reference profile is created, and the brightness profile of the bright spot image at each focal depth is analyzed based on the reference profile, whereby the number of fluorescent particles included in the fluorescence image is determined. A calculating means for calculating;
An image processing apparatus comprising: - 請求項7に記載の画像処理装置と、
前記画像処理装置で使用される前記蛍光画像を取得する画像取得装置と、
を備えることを特徴とする病理診断支援システム。 An image processing apparatus according to claim 7;
An image acquisition device for acquiring the fluorescent image used in the image processing device;
A pathological diagnosis support system comprising: - 蛍光物質を複数集積した蛍光粒子を染色試薬として用いて特定の生体物質が染色された標本から、前記生体物質を定量するコンピュータを、
焦点深度を所定の間隔で連続的に変え、各焦点深度において、前記標本における前記生体物質の発現を蛍光輝点で表す蛍光画像を入力する入力手段、
各焦点深度における前記蛍光画像から、輝点領域が抽出された輝点画像を生成し、当該輝点画像ごとに輝度プロファイルを作成するプロファイル作成手段、
基準プロファイルとして予め計測された蛍光粒子の輝度プロファイルを作成し、各焦点深度における輝点画像の輝度プロファイルを、前記基準プロファイルに基づいて解析することにより、前記蛍光画像に含まれる蛍光粒子の数を算出する算出手段、
として機能させるための画像処理プログラム。 A computer for quantifying the biological material from a specimen stained with a specific biological material using fluorescent particles in which a plurality of fluorescent materials are accumulated as a staining reagent,
Input means for continuously changing a focal depth at a predetermined interval, and inputting a fluorescence image representing the expression of the biological material in the specimen as a fluorescent luminescent spot at each focal depth;
Profile creation means for generating a bright spot image from which a bright spot region is extracted from the fluorescent image at each focal depth, and creating a brightness profile for each bright spot image,
The brightness profile of the fluorescent particles measured in advance as a reference profile is created, and the brightness profile of the bright spot image at each focal depth is analyzed based on the reference profile, whereby the number of fluorescent particles included in the fluorescence image is determined. Calculating means for calculating,
Image processing program to function as
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016514882A JP6504160B2 (en) | 2014-04-21 | 2015-04-15 | Method for quantifying biological material, image processing apparatus, pathological diagnosis support system, and image processing program |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-087155 | 2014-04-21 | ||
| JP2014087155 | 2014-04-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015163211A1 true WO2015163211A1 (en) | 2015-10-29 |
Family
ID=54332378
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/061578 WO2015163211A1 (en) | 2014-04-21 | 2015-04-15 | Biological substance quantitation method, image processing device, pathological diagnosis support system, and image processing program |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP6504160B2 (en) |
| WO (1) | WO2015163211A1 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017110986A (en) * | 2015-12-16 | 2017-06-22 | コニカミノルタ株式会社 | Focus position specification system for fluorescent image, focus position specification method, and focus position specification program |
| JP2017140006A (en) * | 2016-02-12 | 2017-08-17 | 大日本印刷株式会社 | Culture state analysis system, culture state analysis method, and program |
| WO2018008309A1 (en) * | 2016-07-05 | 2018-01-11 | コニカミノルタ株式会社 | Biological material quantifying method, image processing device, pathological diagnosis support system and program |
| WO2019087853A1 (en) * | 2017-11-06 | 2019-05-09 | コニカミノルタ株式会社 | Biological material quantification method, image processing device, and program |
| JP2020165765A (en) * | 2019-03-29 | 2020-10-08 | シスメックス株式会社 | Fluorescence image analyzer and fluorescence image analysis method |
| WO2020209217A1 (en) * | 2019-04-12 | 2020-10-15 | コニカミノルタ株式会社 | Image processing system, image processing method, and program |
| JPWO2019082788A1 (en) * | 2017-10-26 | 2020-12-17 | コニカミノルタ株式会社 | Image processing device, focusing position identification method and focusing position identification program |
| JPWO2019078230A1 (en) * | 2017-10-19 | 2020-12-17 | コニカミノルタ株式会社 | Biomaterial quantification method, image processing device, pathological diagnosis support system and program |
| WO2021117153A1 (en) * | 2019-12-11 | 2021-06-17 | 株式会社日立ハイテク | Fluorescence detection device and fluorescence detection method |
| WO2021124866A1 (en) * | 2019-12-19 | 2021-06-24 | コニカミノルタ株式会社 | Image processing method, image processing system, and program |
| WO2021149356A1 (en) * | 2020-01-21 | 2021-07-29 | コニカミノルタ株式会社 | Malaria test method and malaria test device |
| WO2022059312A1 (en) * | 2020-09-17 | 2022-03-24 | コニカミノルタ株式会社 | Focused image selection method, focused image selection device, and focused image selection program |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008542826A (en) * | 2005-05-23 | 2008-11-27 | エフ. ヘスス ハラルド | Optical microscopy using optically convertible optical labels |
| JP2010540939A (en) * | 2007-09-26 | 2010-12-24 | マサチューセッツ インスティテュート オブ テクノロジー | High resolution 3D imaging of single semiconductor nanocrystals |
| WO2012029342A1 (en) * | 2010-08-30 | 2012-03-08 | コニカミノルタエムジー株式会社 | Tissue staining method, tissue evaluation method and biosubstance detection method |
-
2015
- 2015-04-15 JP JP2016514882A patent/JP6504160B2/en active Active
- 2015-04-15 WO PCT/JP2015/061578 patent/WO2015163211A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008542826A (en) * | 2005-05-23 | 2008-11-27 | エフ. ヘスス ハラルド | Optical microscopy using optically convertible optical labels |
| JP2010540939A (en) * | 2007-09-26 | 2010-12-24 | マサチューセッツ インスティテュート オブ テクノロジー | High resolution 3D imaging of single semiconductor nanocrystals |
| WO2012029342A1 (en) * | 2010-08-30 | 2012-03-08 | コニカミノルタエムジー株式会社 | Tissue staining method, tissue evaluation method and biosubstance detection method |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017110986A (en) * | 2015-12-16 | 2017-06-22 | コニカミノルタ株式会社 | Focus position specification system for fluorescent image, focus position specification method, and focus position specification program |
| JP2017140006A (en) * | 2016-02-12 | 2017-08-17 | 大日本印刷株式会社 | Culture state analysis system, culture state analysis method, and program |
| WO2018008309A1 (en) * | 2016-07-05 | 2018-01-11 | コニカミノルタ株式会社 | Biological material quantifying method, image processing device, pathological diagnosis support system and program |
| JPWO2018008309A1 (en) * | 2016-07-05 | 2019-04-25 | コニカミノルタ株式会社 | Method for quantifying biological material, image processing apparatus, pathological diagnosis support system and program |
| US11600020B2 (en) * | 2017-10-19 | 2023-03-07 | Konica Minolta, Inc. | Biological substance quantification method, image processing device, pathological diagnosis support system, and recording medium storing computer readable program |
| JP7184046B2 (en) | 2017-10-19 | 2022-12-06 | コニカミノルタ株式会社 | Biological substance quantification method, image processing device, pathological diagnosis support system and program |
| JPWO2019078230A1 (en) * | 2017-10-19 | 2020-12-17 | コニカミノルタ株式会社 | Biomaterial quantification method, image processing device, pathological diagnosis support system and program |
| EP3702779A4 (en) * | 2017-10-26 | 2021-01-06 | Konica Minolta, Inc. | Image processing device, in-focus position specifying method, and in-focus position specifying program |
| JP7173034B2 (en) | 2017-10-26 | 2022-11-16 | コニカミノルタ株式会社 | Image processing device, focus position specifying method and focus position specifying program |
| JPWO2019082788A1 (en) * | 2017-10-26 | 2020-12-17 | コニカミノルタ株式会社 | Image processing device, focusing position identification method and focusing position identification program |
| JP7160047B2 (en) | 2017-11-06 | 2022-10-25 | コニカミノルタ株式会社 | Biological substance quantification method, image processing device and program |
| JPWO2019087853A1 (en) * | 2017-11-06 | 2020-12-24 | コニカミノルタ株式会社 | Biomaterial quantification method, image processing device and program |
| WO2019087853A1 (en) * | 2017-11-06 | 2019-05-09 | コニカミノルタ株式会社 | Biological material quantification method, image processing device, and program |
| JP2020165765A (en) * | 2019-03-29 | 2020-10-08 | シスメックス株式会社 | Fluorescence image analyzer and fluorescence image analysis method |
| US11781987B2 (en) | 2019-03-29 | 2023-10-10 | Sysmex Corporation | Fluorescence image analyzer and fluorescence image analyzing method |
| JP7403965B2 (en) | 2019-03-29 | 2023-12-25 | シスメックス株式会社 | Fluorescence image analysis device and fluorescence image analysis method |
| WO2020209217A1 (en) * | 2019-04-12 | 2020-10-15 | コニカミノルタ株式会社 | Image processing system, image processing method, and program |
| WO2021117153A1 (en) * | 2019-12-11 | 2021-06-17 | 株式会社日立ハイテク | Fluorescence detection device and fluorescence detection method |
| WO2021124866A1 (en) * | 2019-12-19 | 2021-06-24 | コニカミノルタ株式会社 | Image processing method, image processing system, and program |
| WO2021149356A1 (en) * | 2020-01-21 | 2021-07-29 | コニカミノルタ株式会社 | Malaria test method and malaria test device |
| WO2022059312A1 (en) * | 2020-09-17 | 2022-03-24 | コニカミノルタ株式会社 | Focused image selection method, focused image selection device, and focused image selection program |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6504160B2 (en) | 2019-04-24 |
| JPWO2015163211A1 (en) | 2017-04-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6504160B2 (en) | Method for quantifying biological material, image processing apparatus, pathological diagnosis support system, and image processing program | |
| JP5768945B1 (en) | Biological substance determination method, image processing apparatus, pathological diagnosis support system, and program | |
| US11035844B2 (en) | Image processing device, pathological diagnosis support system, storage medium for image processing, and image processing method | |
| JP5892238B2 (en) | Medical image processing apparatus and program | |
| JP6350527B2 (en) | Image processing apparatus, pathological diagnosis support system, image processing program, and pathological diagnosis support method | |
| US10309900B1 (en) | Biological substance quantitation method, pathological diagnosis support system and recording medium storing computer readable program | |
| JP6717751B2 (en) | Image processing method, image generation method, image processing device, and program | |
| WO2017126420A1 (en) | Image processing device and program | |
| JPWO2015145644A1 (en) | Image processing apparatus and image processing program | |
| JP5835536B1 (en) | Tissue evaluation method, image processing apparatus, pathological diagnosis support system, and program | |
| JP6493398B2 (en) | Diagnosis support information generation method, image processing apparatus, diagnosis support information generation system, and image processing program | |
| JP6372370B2 (en) | Biological substance quantification method, image processing apparatus, and program | |
| JP6337629B2 (en) | Diagnosis support information generation method, image processing apparatus, diagnosis support information generation system, and image processing program | |
| JP6375925B2 (en) | Image processing apparatus, image processing system, image processing program, and image processing method | |
| JP6405985B2 (en) | Image processing apparatus, image processing system, image processing program, and image processing method |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15782524 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016514882 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15782524 Country of ref document: EP Kind code of ref document: A1 |