WO2015152254A1 - Five-membered ring heteroaryl derivative - Google Patents
Five-membered ring heteroaryl derivative Download PDFInfo
- Publication number
- WO2015152254A1 WO2015152254A1 PCT/JP2015/060155 JP2015060155W WO2015152254A1 WO 2015152254 A1 WO2015152254 A1 WO 2015152254A1 JP 2015060155 W JP2015060155 W JP 2015060155W WO 2015152254 A1 WO2015152254 A1 WO 2015152254A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- pharmaceutically acceptable
- acceptable salt
- optionally substituted
- Prior art date
Links
- VPLRWXUKFRAIBC-UHFFFAOYSA-N Fc(cc1)ccc1-c1ncc(C2CCNCC2)[o]1 Chemical compound Fc(cc1)ccc1-c1ncc(C2CCNCC2)[o]1 VPLRWXUKFRAIBC-UHFFFAOYSA-N 0.000 description 1
- IGIMUFMSZFHQBK-UHFFFAOYSA-N O=C(NC(CC1)CCC1(F)F)N(CC1)CCC1c1cnc(-c(cc2)ccc2F)[o]1 Chemical compound O=C(NC(CC1)CCC1(F)F)N(CC1)CCC1c1cnc(-c(cc2)ccc2F)[o]1 IGIMUFMSZFHQBK-UHFFFAOYSA-N 0.000 description 1
- PYMAQIDIKQXMSN-UHFFFAOYSA-N O=C(NC(CC1)CCC1(F)F)Oc1ccccc1 Chemical compound O=C(NC(CC1)CCC1(F)F)Oc1ccccc1 PYMAQIDIKQXMSN-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/422—Oxazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/08—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/10—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D261/14—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/08—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing alicyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention relates to a novel 5-membered heteroaryl derivative that is a modulator of ⁇ 7 nicotinic acetylcholine receptor ( ⁇ 7 nAChR). Due to their pharmacological properties, the compounds of the present invention can be used for diseases related to cholinergic activity of the central nervous system (CNS) and / or peripheral nervous system (PNS), diseases related to smooth muscle contraction, endocrine diseases, diseases related to neurodegeneration, It may be useful for the treatment of diseases such as inflammation or pain and diseases related to withdrawal symptoms caused by addictive drug abuse.
- CNS central nervous system
- PNS peripheral nervous system
- diseases related to smooth muscle contraction endocrine diseases
- diseases related to neurodegeneration It may be useful for the treatment of diseases such as inflammation or pain and diseases related to withdrawal symptoms caused by addictive drug abuse.
- ⁇ 7 nicotinic acetylcholine receptor represents a valid molecular target for neuroprotection.
- neuroprotection can be achieved by developing an active agonist / positive modulator of the receptor (positive allosteric modulator: PAM).
- PAM positive allosteric modulator
- ⁇ 7 nicotinic receptor agonists have already been identified and evaluated as potential clues for the development of neuroprotective drugs.
- the involvement of ⁇ 7 nicotinic acetylcholine receptors in inflammation has also been reported. From the above, it is assumed that the development of novel modulators of the receptor will lead to novel treatments for nervous system diseases, psychiatric diseases and inflammatory diseases.
- Patent Document 1 Patent Document 2, Patent Document 3, Patent Document 4
- Patent Document 5 Patent Document 6
- the problem to be solved by the present invention is a novel having a positive modulator action of a potent ⁇ 7 nicotinic acetylcholine receptor ( ⁇ 7 nAChR) and useful as a novel therapeutic agent for nervous system diseases, mental diseases and inflammatory diseases. It is to provide a compound.
- ⁇ 7 nAChR potent ⁇ 7 nicotinic acetylcholine receptor
- a novel compound represented by the following formula (I) has a positive modulator action of a potent ⁇ 7 nicotinic acetylcholine receptor ( ⁇ 7 nAChR).
- ⁇ 7 nAChR potent ⁇ 7 nicotinic acetylcholine receptor
- XYZ is N—COR 4A
- R 4A is not C 3 alkyl substituted with 1 to 5 fluorines
- R 3A and R 3B are both C 1-6 When alkyl, together with the nitrogen atom to which they are attached, is substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkyl and C 1-6 alkoxy. It may form a 4- to 10-membered nitrogen-containing saturated heterocyclic ring. Or a pharmaceutically acceptable salt thereof.
- R 3A , R 3B , R 4A , R 4B and R 6 are the same or different and are independently selected from the group consisting of fluorine, C 1-6 alkoxy, and C 3-10 cycloalkyl.
- C 1-6 alkyl optionally substituted with 1-5 substituents; 1-5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl in an optionally substituted C 3-10 cycloalkyl; saturated heterocyclic 4-10 membered; a or a hydrogen atom, wherein, (1) R 4A and R 4B are not hydrogen atom, (2) R 3A and R 3B are not simultaneously hydrogen atoms; (3) when B is B-3, W is an oxygen atom, and XYZ is N-COR 4A , R 4A is not methyl (4) A is unsubstituted phenyl and B is B- In it, W is an oxygen atom, when X-Y-Z is N-COR 4A
- R 3A , R 3B and R 4A are the same or different and are each 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl An optionally substituted C 3-10 cycloalkyl; a 4-10 membered saturated heterocycle; or a hydrogen atom, wherein R 3A and R 3B are not simultaneously a hydrogen atom and R 4A is not a hydrogen atom , Item 3.
- R 4B is good fluorine, C 1-6 alkoxy, and C 3-10 optionally substituted with one to five substituents independently selected from the group consisting of cycloalkyl C 1- 6 alkyl, Item 4.
- A is phenyl or pyridyl (the phenyl and the pyridyl are each substituted with fluorine, C 1-6 alkyl optionally substituted with 1 to 5 fluorine, and 1 to 5 fluorine. And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy).
- Item 5 The compound according to any one of Items 1 to 4 or a pharmaceutically acceptable salt thereof.
- A is phenyl (the phenyl is each fluorine, C 1-6 alkyl optionally substituted with 1 to 5 fluorines, and C 1 optionally substituted with 1 to 5 fluorines) Optionally substituted with 1 to 5 substituents independently selected from the group consisting of -6 alkoxy.) Item 5.
- A is pyridyl (the pyridyl is each fluorine, C 1-6 alkyl optionally substituted with 1 to 5 fluorines, and C 1 optionally substituted with 1 to 5 fluorines) Optionally substituted with 1 to 5 substituents independently selected from the group consisting of -6 alkoxy.) Item 5.
- R 1 is a hydrogen atom; halogen; cyano; C 1-6 alkyl; or C 3-10 cycloalkyl.
- Item 8 The compound according to any one of Items 1 to 7, or a pharmaceutically acceptable salt thereof.
- R 1 is a hydrogen atom, chlorine or cyano.
- Item 8 The compound according to any one of Items 1 to 7, or a pharmaceutically acceptable salt thereof.
- R 1 is a hydrogen atom.
- Item 8 The compound according to any one of Items 1 to 7, or a pharmaceutically acceptable salt thereof.
- R 1 is chlorine.
- Item 8 The compound according to any one of Items 1 to 7, or a pharmaceutically acceptable salt thereof.
- W is an oxygen atom.
- Item 12 The compound according to any one of Items 1 to 11 or a pharmaceutically acceptable salt thereof.
- W is a sulfur atom.
- Item 12 The compound according to any one of Items 1 to 11 or a pharmaceutically acceptable salt thereof.
- R 2A , R 2B , R 2C , R 2D and R 5 are the same or different and are a hydrogen atom or C 1-6 alkyl, wherein R 2A , R 2B , R 2C , R When any two of 2D and R 5 are C 1-6 alkyl, the two alkyls may be combined together to form another ring with the ring to which the alkyl is attached, Item 14.
- R 2A , R 2B , R 2C , R 2D and R 5 are all hydrogen atoms.
- Item 14 The compound according to any one of Items 1 to 13, or a pharmaceutically acceptable salt thereof.
- R 6 is optionally fluorine, C 1-6 alkoxy, and C 3-10 optionally substituted with one to five substituents independently selected from the group consisting of cycloalkyl C 1- 6 alkyl; or a hydrogen atom, Item 16.
- R 6 is a hydrogen atom.
- Item 16 The compound according to any one of Items 1 to 15, or a pharmaceutically acceptable salt thereof.
- R 3B is a hydrogen atom
- R 3A and R 4A are the same or different and are independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl optionally substituted with 5 substituents; or a 4-10 membered saturated heterocycle, Item 18.
- XYZ is N—CO—NR 3A R 3B or N—COR 4A .
- Item 19 The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
- XYZ is N—CO—NR 3A R 3B .
- Item 19 The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
- XYZ is N-COR 4A .
- Item 19 The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
- XYZ is CR 5 —CO—NR 3A R 3B .
- Item 19 The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
- XYZ is CR 5 —NR 6 —COR 4B .
- Item 19 The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
- B is B-1.
- Item 24 The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
- B is B-2.
- Item 24 The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
- [Section 26] B is B-3.
- Item 24 The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
- B is B-4.
- Item 24 The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
- [Item 28] B is B-5.
- Item 24 The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
- a pharmaceutical composition comprising the compound according to any one of items 1 to 29 or a pharmaceutically acceptable salt thereof.
- CIAS cognitive impairment associated with schizophrenia
- cognitive impairment mild cognitive impairment
- memory impairment in schizophrenia Alzheimer's disease, Down's syndrome
- attention deficit / hyperactivity disorder or cerebrovascular angiopathy or Item 31 A pharmaceutical composition according to Item 30, for treating and / or preventing learning disorders.
- a pharmaceutical comprising the compound according to any one of items 1 to 29 or a pharmaceutically acceptable salt thereof and at least one drug selected from atypical antipsychotics .
- [Item 35] Acetylcholine characterized by administering a therapeutically effective amount of the compound according to any one of Items 1 to 29 or a pharmaceutically acceptable salt thereof to a patient in need of treatment.
- the compound of the present invention is useful as a therapeutic and / or prophylactic agent for diseases caused by abnormalities in intracellular signal transduction involving acetylcholine.
- diseases caused by abnormal intracellular signal transduction involving acetylcholine include a neurological disease, a mental disease or an inflammatory disease.
- neurological diseases, psychiatric diseases or inflammatory diseases include schizophrenia, Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder, or cerebrovascular angiopathy.
- the compound of the present invention is (1) CIAS (cognitive dysfunction associated with schizophrenia), or (2) schizophrenia, Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder or cerebrovascular angiopathy, Useful for treating and / or preventing cognitive impairment, mild cognitive impairment, memory impairment or learning impairment.
- CIAS cognitive dysfunction associated with schizophrenia
- schizophrenia Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder or cerebrovascular angiopathy, Useful for treating and / or preventing cognitive impairment, mild cognitive impairment, memory impairment or learning impairment.
- the compound of the present invention can be used in combination with an atypical antipsychotic agent for the purpose of the treatment and / or prevention.
- the compound of the present invention may exist in the form of a hydrate and / or a solvate, a hydrate of the compound represented by formula (I) or a pharmaceutically acceptable salt thereof and / or Alternatively, solvates are also encompassed by the compounds of the present invention.
- the compound of formula (I) may have one or more asymmetric carbon atoms and may cause geometric isomerism and axial chirality, and therefore exist as several stereoisomers. There is. In the present invention, these stereoisomers, mixtures thereof and racemates are included in the compound represented by the formula (I) of the present invention.
- a deuterium converter obtained by converting any one or two or more 1 H of the compound represented by the general formula (I) into 2 H (D) is also included in the compound represented by the general formula (I). Is done.
- Crystalline polymorphisms may exist in the compound represented by the general formula (I) obtained as crystals and pharmaceutically acceptable salts thereof, and the compounds of the present invention may be in any crystalline form. included.
- Alkyl means a linear or branched saturated hydrocarbon group.
- C 1-4 alkyl or “C 1-6 alkyl” has 1 to 4 carbon atoms or Means 1-6 alkyl.
- C 1-6 alkyl “C 1-4 alkyl” is preferable. Specific examples thereof include “C 1-4 alkyl” such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl and the like.
- pentyl, isopentyl, neopentyl, hexyl and the like can be mentioned.
- Cycloalkyl means a group consisting of monocyclic or polycyclic saturated hydrocarbons.
- C 3-10 cycloalkyl means a cyclic alkyl having 3 to 10 carbon atoms.
- crosslinking is mentioned. Specific examples thereof include “C 3-10 cycloalkyl” such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, adamantyl and the like.
- Alkoxy means a group in which a linear or branched saturated hydrocarbon group is bonded to an oxygen atom, and is bonded to another moiety via the oxygen atom.
- C 1-6 alkoxy means alkoxy having 1 to 6 carbon atoms. Specific examples of “C 1-6 alkoxy” include methoxy, ethoxy, propoxy, isopropoxy, butyloxy, pentyloxy, isopentyloxy, neopentyloxy, hexyloxy and the like.
- Halogen means a fluorine atom, a chlorine atom, a bromine atom or an iodine atom. Among them, preferred is a fluorine atom or a chlorine atom.
- the “4- to 10-membered saturated heterocyclic ring” means 4 to 10 atoms including 1 to 2 atoms independently selected from the group consisting of a nitrogen atom, an oxygen atom and a sulfur atom in addition to a carbon atom.
- Preferred 4- to 10-membered saturated heterocycles include tetrahydrofuran, tetrahydropyran, and oxetane. More preferred is tetrahydropyran.
- the “4- to 10-membered nitrogen-containing saturated heterocycle” means 0 to 2 atoms independently selected from the group consisting of 1 to 2 nitrogen atoms, oxygen atoms and sulfur atoms in addition to carbon atoms.
- a saturated heterocyclic ring composed of 4 to 10 atoms is included.
- azetidine, pyrrolidine, piperidine, piperazine, homopiperidine and the like can be mentioned.
- Preferred examples of the 4 to 10-membered nitrogen-containing saturated heterocyclic ring include saturated heterocyclic rings composed of 4 to 10 atoms containing one nitrogen atom in addition to the carbon atom.
- azetidine, pyrrolidine, piperidine and the like can be mentioned.
- A, B, W, XYZ, R 1 , R 2A , R 2B , R 2C , R 2D , R 3A , R 3B , R 4A , R 4B , R 5 and R 6 are preferably as follows, but the technical scope of the present invention is not limited to the scope of the compounds listed below.
- X—Y—Z is preferably N—CO—NR 3A R 3B , N—COR 4A , CR 5 —CO—NR 3A R 3B or CR 5 —NR 6 —COR 4B . More preferably, N—CO—NR 3A R 3B , N—COR 4A or CR 5 —NR 6 —COR 4B may be mentioned. More preferred is N—CO—NR 3A R 3B or N—COR 4A .
- A is preferably phenyl or pyridyl (the phenyl and pyridyl are each substituted with halogen, hydroxyl, C 1-6 alkyl optionally substituted with 1 to 5 fluorine, and 1 to 5 fluorine. And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy. More preferably, phenyl or pyridyl (the phenyl and the pyridyl are each substituted with fluorine, chlorine, C 1-6 alkyl optionally substituted with 1 to 5 fluorine, and 1 to 5 fluorine. And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy.
- it is 1 to 5 substituents independently selected from the group consisting of fluorine, chlorine, C 1-6 alkyl optionally substituted with 1 to 3 fluorine and C 1-6 alkoxy.
- substituents independently selected from the group consisting of fluorine, chlorine, C 1-6 alkyl optionally substituted with 1 to 3 fluorine and C 1-6 alkoxy.
- Examples include optionally substituted phenyl.
- phenyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of fluorine and C 1-6 alkoxy.
- B is preferably B-1, B-2, B-3, B-4 or B-5. More preferred is B-1, B-4 or B-5. More preferred is B-1 or B-4.
- W is preferably an oxygen atom or a sulfur atom. More preferably, an oxygen atom is mentioned.
- R 1 is preferably a hydrogen atom; halogen; cyano; or halogen, hydroxyl, C 3-10 cycloalkyl optionally substituted with 1 to 5 fluorines, C 1-6 alkoxy, and 4 to 10 membered saturation And C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of heterocycles. More preferably, a hydrogen atom, halogen, or cyano is mentioned. More preferably, a hydrogen atom, chlorine, or cyano is mentioned. Most preferably, a hydrogen atom or chlorine is mentioned.
- R 2A , R 2B , R 2C , R 2D and R 5 are preferably the same or different and include a hydrogen atom, fluorine, a hydroxyl group or C 1-6 alkyl.
- R 2A , R 2B , R 2C , R 2D and R 5 are C 1-6 alkyl optionally substituted with 1 to 5 fluorines, the two alkyls together To form a ring different from the ring to which the alkyl is bonded.
- R 2A , R 2B , R 2C , R 2D and R 5 are more preferably the same or different and include a hydrogen atom or C 1-6 alkyl.
- R 2A , R 2B , R 2C , R 2D and R 5 are C 1-6 alkyl
- the two alkyls are joined together to form a ring to which the alkyl is bonded Another ring may be formed.
- R 2A , R 2B , R 2C , R 2D and R 5 are more preferably a hydrogen atom.
- R 3A , R 3B and R 4A are preferably the same or different and are halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4- to 10-membered saturated heterocyclic ring (provided that a carbonyl group is present on the ring).
- C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of: a halogen, a hydroxyl group, C 1-6 alkoxy and C 1-6 alkyl C 3-10 cycloalkyl optionally substituted with 1-5 substituents independently selected from the group consisting of: 4-10 membered saturated heterocycle optionally substituted with C 1-6 alkyl Or a hydrogen atom, wherein (1) R 4A is not a hydrogen atom, (2) R 3A and R 3B are not simultaneously a hydrogen atom, (3) B is B-3, and W is An oxygen atom, X- When -Z is N-COR 4A is, R 4A is not methyl, (4) A a is unsubstituted phenyl, B is B-4, W is an oxygen atom, X-Y- When Z is N—COR 4A , R 4A is not C 3 alkyl substituted with 1 to 5 fluorines.
- R 3A , R 3B and R 4A are the same or different and are 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 3-10 cycloalkyl C 1-6 alkyl optionally substituted with 1-6 alkyls independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl C 3-10 cycloalkyl; a 4-10 membered saturated heterocycle; or a hydrogen atom, where (1) R 4A is not a hydrogen atom and (2) R 3A and R 3B are simultaneously a hydrogen atom And (3) when B is B-3, W is an oxygen atom, and XYZ is N—COR 4A , R 4A is not methyl and (4) A is unsubstituted Phenyl, B is B-4, W is An atom, X-Y-Z is and when is N-COR 4A, R 4A is not C 3 alkyl substituted with 1-5 fluorine.
- R 3A , R 3B and R 4A are the same or different and substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl which may be substituted ; a 4 to 10 membered saturated heterocyclic ring; or a hydrogen atom, wherein R 3A and R 3B are not simultaneously a hydrogen atom and R 4A is not a hydrogen atom .
- R 3A and R 4A the same or different, fluorine and C 1-6 optionally substituted with 1-5 substituents independently selected from the group consisting of alkoxy C 3-10 Examples include cycloalkyl or a 4- to 10-membered saturated heterocyclic ring.
- R 3B includes a hydrogen atom.
- R 4A is not C 3 alkyl substituted with 1 to 5 fluorines means that “R 4A is substituted with 1 to 5 fluorines, It is not C 3 alkyl not substituted with a substituent ”.
- R 4B is preferably selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4-10 membered saturated heterocyclic ring not substituted with a carbonyl group.
- C 1-6 alkyl optionally substituted with 5 substituents; 1-5 substituents independently selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy and C 1-6 alkyl C 3-10 cycloalkyl which may be substituted with a 4-10 membered saturated heterocyclic ring which may be substituted with C 1-6 alkyl.
- fluorine, C 1-6 alkoxy, and C 3-10 1 ⁇ 5 amino optionally substituted with a substituent C 1-6 alkyl independently selected from the group consisting of cycloalkyl; fluorine C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; or 4-10 membered saturation Heterocycles are mentioned. More preferable examples include C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy, and C 3-10 cycloalkyl. It is done. Most preferred is C 1-6 alkyl optionally substituted with 1 to 5 fluorines.
- R 6 is preferably selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4-10 membered saturated heterocyclic ring not substituted with a carbonyl group.
- C 1-6 alkyl optionally substituted with 5 substituents; 1-5 substituents independently selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy and C 1-6 alkyl
- fluorine, C 1-6 alkoxy, and C 3-10 1 ⁇ 5 amino optionally substituted with a substituent C 1-6 alkyl independently selected from the group consisting of cycloalkyl; fluorine C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; A ring; or a hydrogen atom. More preferably, fluorine, C 1-6 alkoxy, and C 3-10 1 ⁇ 5 amino C 1-6 alkyl optionally substituted with a substituent selected independently from the group consisting of cycloalkyl; or A hydrogen atom is mentioned. Most preferably, a hydrogen atom is mentioned.
- preferred compounds include the following compounds or pharmaceutically acceptable salts thereof.
- (A) is mentioned as a preferable aspect.
- (A) XYZ is N—CO—NR 3A R 3B , N—COR 4A , CR 5 —CO—NR 3A R 3B or CR 5 —NR 6 —COR 4B ;
- A is phenyl or pyridyl (the phenyl and the pyridyl may each be substituted with halogen, hydroxyl group, C 1-6 alkyl optionally substituted with 1 to 5 fluorine, and 1 to 5 fluorine; And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy),
- B is B-1, B-2, B-3, B-4 or B-5,
- W is an oxygen atom or a sulfur atom
- R 1 is a hydrogen atom; halogen; cyano; or halogen, hydroxyl, C 3-10 cycloalkyl optionally substituted with 1 to 5 fluorines, C 1-6 al
- R 3A , R 3B and R 4A are the same or different and are halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4- to 10-membered saturated heterocyclic ring (provided that a carbonyl group is present on the ring).
- C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of: a group consisting of halogen, hydroxyl, C 1-6 alkoxy and C 1-6 alkyl
- a C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from: a 4-10 membered saturated heterocycle optionally substituted with C 1-6 alkyl; or (1) R 4A is not a hydrogen atom, (2) R 3A and R 3B are not simultaneously hydrogen atoms, (3) B is B-3, and W is an oxygen atom.
- X-Y-Z is N-COR
- R 4A is not C 3 alkyl substituted with 1 to 5 fluorines
- 1 to 5 R 4B independently selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4-10 membered saturated heterocyclic ring not substituted with a carbonyl group substituted halogen, hydroxyl, with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; the optionally substituted with a substituent C 1-6 alkyl An optionally substituted C 3-10 cycloalkyl; or a 4-10 membered saturated heterocycle optionally substituted with C 1-6 alkyl; 1 to 5 R 6 independently selected from the group
- (B) is mentioned as a more preferable aspect.
- (B) XYZ is N—CO—NR 3A R 3B , N—COR 4A or CR 5 —NR 6 —COR 4B ;
- A is phenyl or pyridyl (each said phenyl and said pyridyl fluorine, chlorine, from one to five fluorines optionally substituted C 1-6 alkyl, optionally substituted with 1-5 fluorine Optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy),
- B is B-1, B-2, B-3, B-4 or B-5, W is an oxygen atom or a sulfur atom,
- R 1 is a hydrogen atom, halogen or cyano
- R 2A , R 2B , R 2C , R 2D and R 5 are the same or different and are a hydrogen atom or C 1-6 alkyl, wherein R 2A , R 2B , R
- R 4A is not a hydrogen atom
- R 3A and R 3B are not simultaneously a hydrogen atom
- B is B-3, W is an oxygen atom and XYZ is N-COR 4A , R 4A is not methyl and (4) A is unsubstituted phenyl; B is B-4, W is an oxygen atom, XY
- Z is N-COR 4A is, R 4A is not C 3 alkyl substituted with 1-5 fluorine, R 4B is selected from the group consisting of fluorine, C 1-6 alkoxy, and C 3-10 1 ⁇ 5 amino optionally substituted with a substituent C 1-6 alkyl independently selected from the group consisting
- (C) is N—CO—NR 3A R 3B or N—COR 4A ;
- A is substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, chlorine, C 1-6 alkyl optionally substituted with 1 to 3 fluorine and C 1-6 alkoxy Which may be phenyl, B is B-1, B-4 or B-5, W is an oxygen atom,
- R 1 is a hydrogen atom, chlorine or cyano
- R 2A , R 2B , R 2C and R 2D are hydrogen atoms,
- R 3A , R 3B and R 4A are the same or different and are substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl; a 4-10 membered saturated heterocyclic ring; or a hydrogen atom, wherein R 3A and R 3B are not simultaneously a hydrogen
- the most preferable embodiment includes the following (D).
- (D) XYZ is N—CO—NR 3A R 3B or N—COR 4A ;
- A is phenyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of fluorine and C 1-6 alkoxy;
- B is B-1 or B-4, W is an oxygen atom,
- R 1 is a hydrogen atom or chlorine;
- R 2A , R 2B , R 2C and R 2D are hydrogen atoms,
- R 3A and R 4A are the same or different and each is a C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of fluorine and C 1-6 alkoxy;
- a 4-10 membered saturated heterocycle A compound of formula (I) or a pharmaceutically acceptable salt thereof, wherein R 3B is a hydrogen atom.
- the pharmaceutically acceptable salt of the compound represented by the formula (I) means a salt formed by adding a pharmaceutically acceptable acid or base to the compound of the formula (I).
- a pharmaceutically acceptable acid or base When the compound of the present invention represented by the formula (I) has a basic functional group such as an amino group, it can form salts with various acids.
- acid addition salts include hydrochloride, hydrobromide, hydroiodide, sulfate, perchlorate, phosphate, and other inorganic acid salts, oxalate, and malonic acid.
- Salt maleate, fumarate, lactate, malate, citrate, tartrate, benzoate, trifluoroacetate, acetate, methanesulfonate, p-toluenesulfonate, trifluoromethane
- organic acid salts such as sulfonate
- amino acid salts such as glutamate and aspartate.
- salts When the compound of the present invention represented by the formula (I) has an acidic functional group such as a carboxyl group, it can form salts with various bases.
- pharmaceutically acceptable salts include alkali metal salts such as sodium salt or potassium salt, alkaline earth metal salts such as calcium salt, or triethylammonium salt, triethanolammonium salt, pyridinium salt, diisopropylammonium salt. Examples thereof include organic base salts such as salts. These salts can be obtained by a conventional method such as recrystallization after mixing the compound of the present invention represented by the formula (I) with a base.
- the method for producing the compound of the present invention is described below.
- the compound of the present invention represented by the formula (I) can be produced, for example, by the following production methods A to M.
- Production method A production method of synthetic intermediate
- synthetic intermediates a6, a8 and a10 of a compound in which B is B-1 can be produced, for example, by the following production method.
- P X represents a protecting group of amino
- R X represents a hydrogen atom, alkyl or phenyl
- R 1X represents halogen
- R 1Y represents the term 1 R 1 as defined is not halogen
- the protective group P X Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
- Compound a1 can be synthesized by a known method such as a halogenation reaction of imidazole, or can be purchased as a commercial product.
- Step A-1 This step is a step of obtaining compound a2 by reacting compound a1 with an appropriate boronic acid derivative in the presence of an appropriate transition metal reagent.
- An example of the transition metal reagent used in this step is copper acetate.
- Examples of the boronic acid derivative used in this step include boronic acid, boronic acid ester, and boronic acid pinacolate.
- the solvent used in this step is selected from the solvents exemplified below, but is preferably dichloroethane or methanol.
- a similar reaction for example, a method described in Journal of Medicinal Chemistry, 2009, 52 (11), 3441-3444 is known and can be synthesized in the same manner.
- reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C. to 200 ° C., preferably ⁇ 20 ° C. to 150 ° C., more preferably room temperature to 100 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step A-2 This step is a step of obtaining compound a4 by reacting compound a2 obtained in step A-1 with compound a3 in the presence of an appropriate transition metal reagent.
- the transition metal reagent used in this step include tris (dibenzylideneacetone) dipalladium and tetrakis (triphenylphosphine) palladium.
- the solvent used in this step is selected from the solvents exemplified below, and is preferably DMF or dioxane.
- a similar reaction for example, a method described in Journal of Organic Chemistry, 2010, 75 (5), 1733-1739 is known and can be synthesized in the same manner.
- reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C. to 200 ° C., preferably ⁇ 20 ° C. to 150 ° C., more preferably room temperature to 100 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step A-3 This step is a step of obtaining compound a5 by reacting compound a4 obtained in step A-2 with a suitable transition metal reagent in a hydrogen atmosphere.
- the transition metal reagent used in this step include palladium / carbon and platinum (IV) oxide.
- the solvent used in this step is selected from the solvents exemplified below, but is preferably methanol or ethanol.
- a similar reaction for example, a method described in Journal of Medicinal Chemistry, 2012, 55 (1), 115-125, etc. is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 20 ° C. to 150 ° C., preferably 0 ° C. to 100 ° C., more preferably room temperature to 60 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step A-4 This step is a protecting group P X of the amino compound a5 prepared in the above Preparation Method A-3, by deprotection, to give compound compound a6.
- This step can be carried out according to the method described in Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts, John Wiley & Sons, Inc., 1999).
- Step A-5 This step is a step of obtaining compound a7 by reacting compound a5 obtained in step A-3 with various halogenating agents in a suitable solvent in the presence of a suitable acid.
- the halogenating agent used in this step is, for example, N-chlorosuccinimide, N-bromosuccinimide, N-iodosuccinimide.
- the solvent used in this step is selected from the solvents exemplified below, and is preferably methylene chloride or dichloroethane.
- the acid used in this step is selected from the acids exemplified below, and preferably trifluoroacetic acid or hydrochloric acid.
- a similar reaction for example, a method described in Bioorg. Med. Chem.
- reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C. to 200 ° C., preferably 0 ° C. to 100 ° C., more preferably room temperature to 70 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step A-6 This step is a step of obtaining compound a8 from compound a7 obtained in step A-5, under the same conditions as in step A-4.
- Step A-7 This step is a step of obtaining compound a9 by reacting compound a7 obtained in step A-5 with a suitable boronic acid derivative in a suitable solvent in the presence of a suitable transition metal reagent.
- a suitable transition metal reagent examples include tris (dibenzylideneacetone) dipalladium and tetrakis (triphenylphosphine) palladium.
- the boronic acid derivative used in this step include boronic acid, boronic acid ester, and boronic acid pinacolate.
- the solvent used in this step is selected from the solvents exemplified below, and is preferably DMF or dioxane. Similar reactions include, for example, Tetrahedron Lett.
- the reaction temperature varies depending on the type of raw material compound used, reagents and the like, but is usually ⁇ 78 ° C. to 200 ° C., preferably ⁇ 20 ° C. to 180 ° C., more preferably room temperature to 150 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step A-8 This step is a step of obtaining compound a10 from compound a9 obtained in step A-7, under the same conditions as in step A-4.
- Production method B (Production method of synthetic intermediate) Among the compounds represented by formula (I), a synthetic intermediate b7 of a compound in which B is B-2 and W is an oxygen atom can be produced, for example, by the following production method. (Wherein, A, R 2A, R 2B , R 2C and R 2D are as defined in claim 1, P X denotes a protective group for amino)
- the protective group P X Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
- Compound b1 can be synthesized by a known method such as oxidation of the corresponding alcohol, or can be purchased as a commercial product.
- Step B-1 This step is a step of obtaining compound b2 by reacting compound b1 with nitromethane in the presence of a suitable base.
- the base used in this step is selected from the bases exemplified below, and preferably tert-butoxy potassium.
- the solvent used in this step is selected from the solvents exemplified below, but is preferably a mixed solvent of tetrahydrofuran and tert-butanol.
- a similar reaction for example, a method described in International Publication No. 2007/41061 pamphlet or the like is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C.
- reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step B-2 This step is a step of obtaining compound b3 by reacting compound b2 obtained in B-1 above in a hydrogen atmosphere using an appropriate transition metal catalyst.
- the transition metal reagent used in this step include palladium / carbon and platinum (IV) oxide.
- the solvent used in this step is selected from the solvents exemplified below, but is preferably methanol or ethanol. As similar reactions, for example, the methods described in Bioorganic and Medicinal Chemistry Letters, 2004, 14 (13), 3419-3424, International Publication No. 2006/19768, etc. are known and can be synthesized similarly. .
- the reaction temperature varies depending on the type of raw material compound used, reagents and the like, but is usually ⁇ 20 ° C. to 200 ° C., preferably 0 ° C. to 100 ° C., more preferably room temperature to 70 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step B-3 In this step, compound b3 obtained in B-2 above is reacted with carboxylic acid b8 or acid chloride b9 in a suitable solvent in the presence or absence of a suitable condensing agent and in the presence of a suitable base.
- compound b4 is obtained.
- the condensing agent used in this step include EDCI (including hydrochloride) or HBTU.
- the base used in this step is selected from the bases exemplified below, and preferably diisopropylethylamine or triethylamine.
- the solvent used in this step is selected from the solvents exemplified below, and preferably dimethylformamide, tetrahydrofuran or methylene chloride.
- reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C. to 200 ° C., preferably 0 ° C. to 150 ° C., more preferably room temperature to 100 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step B-4 This step is a step of obtaining compound b5 by reacting compound b4 obtained in B-3 with an appropriate oxidizing agent.
- the oxidizing agent used in this step include a desmartin reagent.
- the solvent used in this step is selected from the solvents exemplified below, and is preferably methylene chloride or DMSO.
- a similar reaction for example, a method described in International Publication No. 2010/138589 pamphlet is known and can be synthesized in the same manner.
- reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally -78 ° C to 200 ° C, preferably -20 ° C to 150 ° C, more preferably 0 ° C to 100 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step B-5 This step is a step of obtaining compound b6 by reacting compound b5 obtained in B-4 with an appropriate dehydrating reagent.
- the solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran.
- Examples of the dehydrating reagent used in this step include Burgess reagent.
- a similar reaction for example, a method described in Synlett, 1999, 10, 1642-1644 is known, and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C. to 200 ° C., preferably ⁇ 20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step B-6 This step is a step of obtaining the compound b7 from the compound b6 obtained in the step B-5 under the conditions according to the step A-4.
- c2 which is a synthetic intermediate of a compound in which B is B-2 and W is a sulfur atom can be produced, for example, by the following production method.
- A, R 2A, R 2B , R 2C and R 2D are as defined in claim 1, P X denotes a protective group for amino
- the protective group P X Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
- Step C-1 This step is a step of obtaining compound c1 by reacting compound b5 obtained in step B-4 with an appropriate thiocarbonylating reagent.
- the thiocarbonylation reagent used in this step include Lawson's reagent.
- the solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran.
- a similar reaction for example, a method described in US Publication No. 2010/261617, etc. is known, and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C.
- reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step C-2 In this step, compound c2 is obtained from compound c1 obtained in step C-1 under the same conditions as in step A-4.
- d4 that is a synthetic intermediate of a compound in which B is B-3 and W is an oxygen atom can be produced, for example, by the following production method.
- A, R 2A, R 2B , R 2C and R 2D are as defined in claim 1, P X denotes a protective group for amino
- the protective group P X Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
- Compound d1 can be synthesized by a known method such as oxidation of the corresponding alcohol or aldehyde or corresponding ester hydrolysis, or can be purchased as a commercial product.
- Step D-1 This step is a step in which compound d1 and compound d5 are reacted under the same conditions as in step B-3 to obtain compound d2.
- Step D-2 This step is a step of obtaining a compound d3 by reacting the compound d2 obtained in the step D-1 with the conditions according to the step B-5.
- Step D-3 This step is a step of obtaining a compound d4 by reacting the compound d3 obtained in the step D-2 with the conditions according to the step A-4.
- Production method E (Production method of synthetic intermediate) Among the compounds represented by the formula (I), e2 which is a synthetic intermediate of a compound in which B is B-3 and W is a sulfur atom can be produced by, for example, the following production method. (Wherein, A, R 2A, R 2B , R 2C and R 2D are as defined in claim 1, P X denotes a protective group for amino)
- the protective group P X Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
- Step E-1 This step is a step of obtaining a compound e1 by reacting the compound d2 obtained in the step D-1 with the conditions according to the step C-1.
- Step E-2 This step is a step of obtaining a compound e2 by reacting the compound e1 obtained in the step E-1 with the conditions according to the step A-4.
- the protective group P X Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
- Compound f1 can be purchased as a commercial product.
- Compound f4 can be synthesized by known methods such as oxidation of the corresponding alcohol, or can be purchased as a commercial product.
- Step F-1 compound f2 is obtained by reacting compound f1 with a diazotizing reagent using an appropriate base.
- the diazotizing reagent used in this step include 4-toluenesulfonyl azide.
- the base used in this step is selected from the bases exemplified below, and is preferably sodium hydride.
- the solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran.
- the methods described in Bioorganic and Medicinal Chemistry Letters, 2013, 23 (19), 5267-5269, etc. are known, and can be synthesized in the same manner.
- reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C. to 200 ° C., preferably ⁇ 20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step F-2 This step is a step of obtaining compound f3 by reacting compound f2 obtained in step F-1 with compound b1 in the presence of a suitable base.
- the base used in this step is selected from the bases exemplified below, and is preferably potassium carbonate.
- the solvent used in this step is selected from the solvents exemplified below, but is preferably methanol.
- a similar reaction for example, a method described in Journal of the American Chemical Society, 2003, 125 (13), 3714-3715 is known and can be synthesized in the same manner.
- reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally -78 ° C to 200 ° C, preferably -20 ° C to 150 ° C, more preferably 0 ° C to 100 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step F-3 This step is a step of reacting compound f4 and hydroxylamine to obtain compound f5.
- the solvent used in this step is selected from the solvents exemplified below, but is preferably methanol.
- a similar reaction for example, a method described in Organic Letters, 2001, 3 (26), 4209-4211, etc. is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally -78 ° C to 200 ° C, preferably -20 ° C to 150 ° C, more preferably 0 ° C to 100 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step F-4 This step is a step of reacting the compound f5 obtained in the step F-3 with a chlorinating reagent to obtain the compound f6.
- chlorinating reagent used in this step include N-chlorosuccinimide.
- the solvent used in this step is selected from the solvents exemplified below, but is preferably DMF. Similar reactions are described in, for example, Bioorganic and Medicinal Chemistry Letters, 2006, 16 (21), 5576-5579, Journal of Heterocyclic Chemistry, 1996, 33 (6), 1583-1592, and the like. It can be synthesized similarly. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C.
- reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step F-5 In this step, compound f3 obtained in step F-2 and compound f6 obtained in step F-4 are reacted in the presence of a suitable base to obtain compound f7.
- the base used in this step is selected from the bases exemplified below, but is preferably triethylamine.
- the solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran.
- the methods described in Journal of Medicinal Chemistry, 2003, 46 (2), 284-302, European Journal of Medicinal Chemistry, 2012, 54, 324-342, and the like are known. can do.
- reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C. to 200 ° C., preferably ⁇ 20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step F-6 This step is a step of obtaining compound f8 by reacting compound f7 obtained in step F-5 with the conditions according to step A-4.
- Production method G (Production method of synthetic intermediate) Among the compounds represented by the formula (I), g5 which is a synthetic intermediate of a compound in which B is B-5 and W is an oxygen atom can be produced, for example, by the following production method. (Wherein, A, R 2A, R 2B , R 2C and R 2D are as defined in claim 1, P X denotes a protective group for amino)
- the protective group P X Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
- Compound f4 can be synthesized by a known method such as oxidation of the corresponding alcohol, or can be purchased as a commercial product.
- Compound b1 can be synthesized by a known method such as oxidation of the corresponding alcohol, or can be purchased as a commercial product.
- Step G-1 This step is a step of obtaining compound g1 by reacting compound f4 with compound f2 obtained in Step F-1 under the same conditions as in Step F-2.
- Step G-2 This step is a step of obtaining compound g2 by reacting compound b1 under the conditions according to the above F-3 step.
- Step G-3 This step is a step of obtaining compound g3 by reacting compound g2 obtained in the above step G-2 under the same conditions as in the above step F-4.
- Step G-4 This step is a step of obtaining compound g4 by reacting compound g3 obtained in step G-3 and g1 obtained in step G-1 under the same conditions as in step F-5.
- Step G-5 This step is a step of obtaining compound g5 by reacting compound g4 obtained in the above step G-4 with the conditions according to the above step A-4.
- Manufacturing method H Of the compounds represented by the formula (I), the compounds represented by the formulas [H1], [H2] and [H3], wherein B is B-1 and XYZ is N—CO—NR 3A R 3B
- the compounds to be prepared (hereinafter also referred to as compounds H1, H2, and H3) can be produced, for example, by the following production method. Further, using the same production method, compounds represented by the formulas [H2] and [H3] can be produced using the intermediates a8 and a10 obtained by the intermediate production method A as starting materials. Further, using the same production method, the intermediates B7, c2, d4, e2, f8 and g5 obtained by the above intermediate production methods B, C, D, E, F and G are used as starting materials.
- XYZ is N-CO-NR 3A R 3B
- W is an oxygen atom or a sulfur atom.
- A, R 2A , R 2B , R 2C , R 2D , R 3A and R 3B are as defined in item 1; R Y represents hydrogen, nitro or cyano; and R 1X represents halogen. And R 1Y means a non-halogen among R 1 defined in item 1)
- Step H-1 This step is a step of obtaining Compound H1 by reacting Compound a6 obtained in Step A-4 with h1 or h2 which is a urea agent in a suitable solvent in the presence of a suitable base.
- the base used in this step is selected from the bases exemplified below, and is preferably diisopropylethylamine or triethylamine.
- the solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran or methylene chloride. Similar reactions include, for example, J. Org. Chem. 1995, 60 (25), 8262-8266, Bioorg. Med. Chem. Lett.
- reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C. to 200 ° C., preferably ⁇ 20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step H-2 This step is a step for obtaining compound H2 by reacting compound H1 obtained in step H-1 with various halogenating agents in a suitable solvent in the presence of a suitable acid.
- the halogenating agent used in this step is preferably N-chlorosuccinimide, N-bromosuccinimide, or N-iodosuccinimide.
- the solvent used in this step is selected from the solvents exemplified below, and is preferably methylene chloride or dichloroethane.
- the acid used in this step is selected from the acids exemplified below, and preferably trifluoroacetic acid or hydrochloric acid.
- a similar reaction for example, methods described in Bioorg. Med. Chem. Lett.
- reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C. to 200 ° C., preferably ⁇ 20 ° C. to 150 ° C., more preferably 0 ° C. to 70 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step H-3 This step is a step of obtaining compound H3 by reacting compound H2 obtained in step H-2 with an appropriate boronic acid derivative in an appropriate solvent in the presence of an appropriate transition metal reagent.
- the transition metal reagent used in this step include tris (dibenzylideneacetone) dipalladium and tetrakis (triphenylphosphine) palladium.
- the boronic acid derivative used in this step include boronic acid, boronic acid ester, and boronic acid pinacolate.
- the solvent used in this step is selected from the solvents exemplified below, and is preferably DMF or dioxane. Similar reactions include, for example, Tetrahedron Lett.
- the reaction temperature varies depending on the type of raw material compound used, reagents and the like, but is usually ⁇ 78 ° C. to 200 ° C., preferably ⁇ 20 ° C. to 180 ° C., more preferably room temperature to 150 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- XYZ is N-COR 4A
- W is an oxygen atom or a sulfur atom. it can.
- R 1Y represents a non-halogen of R 1 defined in item 1.
- R 1X means halogen
- Step I-1 In this step, compound a6 obtained in step A-4 is reacted with carboxylic acid i1 or acid chloride i2 in the presence of a suitable base in the presence or absence of a suitable condensing agent and in the presence of a suitable base.
- the condensing agent used in this step is, for example, EDCI (including hydrochloride) or HBTU.
- the base used in this step is selected from the bases exemplified below, and is preferably diisopropylethylamine or triethylamine.
- the solvent used in this step is selected from the solvents exemplified below, and preferably dimethylformamide, tetrahydrofuran or methylene chloride.
- reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C. to 200 ° C., preferably ⁇ 20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step I-2 This step is a step of obtaining compound I2 by reacting compound I1 obtained in the above step I-1 under the same conditions as in the above step H-2.
- Step I-3 This step is a step of obtaining compound I3 by reacting compound I2 obtained in the above step I-2 with the conditions according to the above step H-3.
- Manufacturing method J Of the compounds represented by formula (I), the formulas [J1] [J2] [J3] and [J4] wherein B is B-4 and XYZ is CR 5 —NR 6 —COR 4B (Hereinafter also referred to as compounds J1, J2, J3, and J4) can be produced, for example, by the following production method according to the intermediate production method F. Further, by combining the following production method J and the production methods according to the intermediate production methods A, B, C, D, E and G, XYZ is CR 5 —NR 6 —COR 4B ; Is a corresponding compound group in which is B-1, B-2, B-3 and B-5, and W is an oxygen atom or a sulfur atom.
- R 1X represents halogen
- R 1Y represents item 1
- R 1 which is not halogen among the defined R 1 represents R Z represents a leaving group such as halogen
- P X represents an amino protecting group
- the protective group P X Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
- Compound j1 can be synthesized by a known method such as oxidation of the corresponding alcohol, or can be purchased as a commercial product.
- Step J-1 This step is a step wherein compound j2 is obtained by reacting compound j1 under the same conditions as in Step F-2.
- Step J-2 This step is a step of obtaining compound j3 by reacting compound j2 obtained in step J-1 under the same conditions as in step F-5.
- Step J-3 This step is a protecting group P X of the amino compound j3 prepared in the above Preparation Method J-2, is deprotected, to give compound compound j4.
- This step can be carried out according to the method described in Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts, John Wiley & Sons, Inc., 1999).
- Step J-4 In this step, compound j4 obtained in J-3 above is reacted with carboxylic acid j5 or acid chloride j6 in a suitable solvent in the presence or absence of a suitable condensing agent and in the presence of a suitable base.
- compound J1 is obtained.
- the condensing agent used in this step include EDCI (including hydrochloride) or HBTU.
- the base used in this step is selected from the bases exemplified below, and preferably diisopropylethylamine or triethylamine.
- the solvent used in this step is selected from the solvents exemplified below, and preferably dimethylformamide, tetrahydrofuran or methylene chloride.
- reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C. to 200 ° C., preferably 0 ° C. to 150 ° C., more preferably room temperature to 100 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step J-5 This step is a step of obtaining compound J2 by reacting compound J1 obtained in the above step J-4 under the conditions according to the above step H-2.
- Step J-6 In this step, compound J2 obtained in step J-5 is reacted under the same conditions as in step H-3 to obtain compound J3.
- Step J-7 This step is a step of obtaining compound J4 by reacting compound j7 obtained in step J-6 with compound j7 in the presence of various bases in an appropriate solvent.
- the base used in this step is selected from the bases exemplified below, and preferably sodium hydride or diisopropylamine.
- the solvent used in this step is selected from the solvents exemplified below and preferably dimethylformamide or tetrahydrofuran.
- the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 30 ° C. to 200 ° C., preferably 0 ° C. to 150 ° C., more preferably 0 ° C. to 80 ° C.
- the reaction time is usually about 1 to 48 hours, preferably 1 to 24 hours, and more preferably 1 to 16 hours.
- Manufacturing method K Of the compounds represented by formula (I), the compounds of formula [K1] [K2] [K3] and [K3] wherein B is B-4 and XYZ is CR 5 —NR 6 —CONR 3A R 3B
- the compound represented by K4] (hereinafter also referred to as compounds K1, K2, K3 and K4) can be produced, for example, by the following production method.
- XYZ is CR 5 —NR 6 —CONR 3A R 3B by combining the following production method K and production methods according to the above intermediate production methods A, B, C, D, E and G.
- B is B-1, B-2, B-3 and B-5, and corresponding compounds can be produced wherein W is an oxygen atom or a sulfur atom.
- R 1X represents halogen
- R 1Y represents R 1 defined in Item 1 is not halogen
- R Y represents hydrogen, nitro or cyano
- R Z represents a leaving group such as halogen
- Step K-1 This step is a step of obtaining compound K1 by reacting compound j4 obtained in the above step J-3 under the conditions according to the above step H-1.
- Step K-2 This step is a step of obtaining compound K2 by reacting compound K1 obtained in the above step K-1 under the same conditions as in the above step J-7.
- Step K-3 This step is a step of obtaining a compound K3 by reacting the compound K2 obtained in the step K-2 with the conditions according to the step H-2.
- Step K-4 This step is a step of obtaining a compound K4 by reacting the compound K3 obtained in the step K-3 with the conditions according to the step H-3.
- Manufacturing method M Of the compounds represented by the formula (I), the compounds represented by the formulas [M1] [M2] and [M3], wherein B is B-4 and XYZ is CR 5 —CO—NR 3A R 3B
- the compounds to be produced (hereinafter also referred to as compounds M1, M2 and M3) can be produced, for example, by the following production method according to the intermediate production method F. Further, by combining the following production method M and the production methods according to the intermediate production methods A, B, C, D, E and G, XYZ is CR 5 —CO—NR 3A R 3B , Corresponding compound groups in which B is B-1, B-2, B-3 and B-5 and W is an oxygen atom or a sulfur atom can be produced.
- R 1X represents halogen
- R 1Y represents item 1 R 1 which is not halogen among the defined R 1 represents P Y represents a protecting group for a carboxyl group
- the protective group P Y Protective Groups in Organic Synthesis (Theodora W. Greene, Peter GM Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group of a carboxylic acid .
- Compound m1 can be synthesized by a known method such as oxidation of the corresponding alcohol, or can be purchased as a commercial product.
- Step M-1 This step is a step of obtaining compound m2 by reacting compound m1 under the same conditions as in Step F-2.
- Step M-2 This step is a step of obtaining a compound m3 by reacting the compound m2 obtained in the step M-1 with the conditions according to the step F-5.
- This step is a step of obtaining the compound m4 by deprotecting the protecting group P Y of the carboxylic acid of the compound m3 obtained in the step M-2.
- This step can be carried out according to the method described in Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts, John Wiley & Sons, Inc., 1999).
- Step M-4 This step is a step of obtaining compound M1 by reacting compound m4 obtained in step M-3 with amine m5 in the presence of a suitable condensing agent and in the presence of a suitable base in a suitable solvent.
- a suitable condensing agent used in this step include EDCI (including hydrochloride) or HBTU.
- the base used in this step is selected from the bases exemplified below, and preferably diisopropylethylamine or triethylamine.
- the solvent used in this step is selected from the solvents exemplified below, and preferably dimethylformamide, tetrahydrofuran or methylene chloride.
- reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally ⁇ 78 ° C. to 200 ° C., preferably 0 ° C. to 150 ° C., more preferably room temperature to 100 ° C.
- the reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
- Step M-5 In this step, compound M1 obtained in step M-4 is reacted under the same conditions as in step H-2 to obtain compound M2.
- Step M-6 In this step, compound M2 obtained in step M-5 is reacted under the same conditions as in step H-3 to obtain compound M3.
- alkali bicarbonates such as sodium bicarbonate and potassium bicarbonate, sodium carbonate and potassium carbonate
- Alkali carbonates such as sodium hydride, metal hydrides such as sodium hydride and potassium hydride, alkali metal hydroxides such as sodium hydroxide and potassium hydroxide, alkali metals such as sodium methoxide and sodium t-butoxide Alkoxides, organometallic bases such as butyllithium and lithium diisopropylamide, triethylamine, diisopropylethylamine, pyridine, 4-dimethylaminopyridine (DMAP), 1,8-diazabicyclo [5.4.0] -7-undecene ( Organic bases such as DBU) That.
- DMAP 4-dimethylaminopyridine
- DBU 1,8-diazabicyclo [5.4.0] -7-undecene
- the solvent used in each of the above steps should be appropriately selected depending on the reaction and the type of raw material compound.
- alcohols such as methanol, ethanol and isopropanol
- ketones such as acetone and ethyl methyl ketone.
- Halogenated hydrocarbons such as methylene chloride and chloroform, ethers such as tetrahydrofuran (THF) and dioxane, aromatic hydrocarbons such as toluene and benzene, aliphatic hydrocarbons such as hexane and heptane, Esters such as ethyl acetate, propyl acetate, amides such as N, N-dimethylformamide (DMF), N-methyl-2-pyrrolidone, sulfoxides such as dimethyl sulfoxide (DMSO), nitriles such as acetonitrile
- solvents can be used alone There can be used as a mixture of two or more kinds. Depending on the type of reaction, organic bases may be used as a solvent.
- the compound of the present invention represented by the formula (I) or an intermediate thereof can be separated and purified by methods known to those skilled in the art.
- separation or purification methods include extraction, distribution, reprecipitation, column chromatography (for example, silica gel column chromatography, ion exchange column chromatography or preparative liquid chromatography) or recrystallization.
- recrystallization solvent include alcohol solvents such as methanol, ethanol and 2-propanol, ether solvents such as diethyl ether, ester solvents such as ethyl acetate, aromatic hydrocarbon solvents such as benzene and toluene, acetone and the like.
- a spectroscopic method such as nuclear magnetic resonance, infrared absorption, circular dichroism spectrum analysis, And mass spectrometry.
- the compound of the present invention represented by the formula (I) or a pharmaceutically acceptable salt thereof may have asymmetry or may have a substituent having an asymmetric carbon.
- optical isomers exist.
- the compounds of the present invention include mixtures of these isomers and isolated ones, and can be produced according to ordinary methods. Examples of the production method include a method using a raw material having an asymmetric point, or a method of introducing asymmetry at an intermediate stage. For example, by using an optically active raw material or performing optical resolution or the like at an appropriate stage of the production process, the optical properties of the compound of the present invention represented by formula (I) or a pharmaceutically acceptable salt thereof Isomers can be obtained.
- the solvent is an inert solvent (for example, an alcohol solvent such as methanol, ethanol or 2-propanol).
- An ether solvent such as diethyl ether, an ester solvent such as ethyl acetate, a hydrocarbon solvent such as toluene, an aprotic solvent such as acetonitrile, or a mixed solvent thereof), an optically active acid (for example, mandelic acid, N-benzyloxyalanine or monocarboxylic acid such as lactic acid, tartaric acid, dicarboxylic acid such as o-diisopropylidene tartaric acid or malic acid, sulfonic acid such as camphorsulfonic acid or bromocamphorsulfonic acid) Stereomer method can be mentioned.
- an optically active amine for example, 1-phenylethylamine, quinine, quinidine, cinchonidine, cinchonine or strychnine
- the optical resolution can also be carried out by forming a salt using an organic amine).
- the temperature at which the salt is formed is selected from the range from room temperature to the boiling point of the solvent. In order to improve the optical purity, it is desirable to raise the temperature once to near the boiling point of the solvent. When the precipitated salt is collected by filtration, it can be cooled as necessary to improve the yield.
- the amount of the optically active acid or amine used is suitably in the range of about 0.5 to about 2.0 equivalents, preferably in the range of about 1 equivalent, relative to the substrate.
- Crystals in an inert solvent as necessary for example, alcohol solvents such as methanol, ethanol or 2-propanol, ether solvents such as diethyl ether, ester solvents such as ethyl acetate, hydrocarbon solvents such as toluene, acetonitrile, etc. And a high purity optically active salt can be obtained. Further, if necessary, an optically resolved salt can be treated with an acid or a base by a conventional method to obtain a free form.
- an inert solvent for example, alcohol solvents such as methanol, ethanol or 2-propanol, ether solvents such as diethyl ether, ester solvents such as ethyl acetate, hydrocarbon solvents such as toluene, acetonitrile, etc.
- an optically resolved salt can be treated with an acid or a base by a conventional method to obtain a free form.
- an optically active amine for example, 1-phenylethylamine etc.
- an optically active amine for example, 1-phenylethylamine etc.
- the compound of the present invention is useful as a therapeutic and / or prophylactic agent for diseases caused by abnormalities in intracellular signal transduction involving acetylcholine.
- diseases caused by abnormal intracellular signal transduction involving acetylcholine include a neurological disease, a mental disease or an inflammatory disease.
- neurological diseases, psychiatric diseases or inflammatory diseases include schizophrenia, Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder, or cerebrovascular angiopathy.
- the compound of the present invention is (1) CIAS (cognitive dysfunction associated with schizophrenia), or (2) schizophrenia, Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder or cerebrovascular angiopathy, Useful for treating and / or preventing cognitive impairment, mild cognitive impairment, memory impairment or learning impairment.
- CIAS cognitive dysfunction associated with schizophrenia
- schizophrenia Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder or cerebrovascular angiopathy, Useful for treating and / or preventing cognitive impairment, mild cognitive impairment, memory impairment or learning impairment.
- the compounds of the present invention are also useful for treating and / or preventing negative and / or positive symptoms associated with schizophrenia.
- the compound of the present invention can be used in combination with an atypical antipsychotic agent for the purpose of the treatment and / or prevention.
- the pharmaceutical agent in the case where the compound of the present invention or a pharmaceutically acceptable salt thereof and an atypical antipsychotic drug are used in combination may be a single compounding agent, or simultaneously, separately and continuously. Or multiple formulations administered at regular intervals.
- the administration route of the compound of the present invention may be any of oral administration, parenteral administration and rectal administration, and the daily dose varies depending on the type of compound, administration method, patient symptom / age and the like.
- oral administration usually about 0.01 to 1000 mg, more preferably about 0.1 to 500 mg per kg body weight of a human or mammal can be administered in 1 to several divided doses.
- parenteral administration such as intravenous injection, usually, for example, about 0.01 mg to 300 mg, more preferably about 1 mg to 100 mg per kg body weight of a human or mammal can be administered.
- Examples of the dosage form include tablets, capsules, granules, powders, syrups, suspensions, injections, suppositories, eye drops, ointments, coating agents, patches, inhalants and the like.
- These preparations can be prepared according to a conventional method. In the case of a liquid preparation, it may be dissolved or suspended in water, an appropriate aqueous solution or other appropriate medium at the time of use. Tablets and granules may be coated by a known method. In addition, these formulations may contain pharmaceutically acceptable additives.
- Additives are excipients, disintegrants, binders, fluidizers, lubricants, coating agents, solubilizers, solubilizers, thickeners, dispersants, stabilizers, sweeteners depending on the purpose. Perfumes and the like can be used.
- the compound of the present invention can be used in combination with an atypical antipsychotic drug.
- atypical antipsychotics include olanzapine, risperidone, paliperidone, quetiapine, ziprasidone, aripiprazole, asenapine, iloperidone, clozapine, sertindole, blonanserin and lurasidone.
- Me means a methyl group
- Et means an ethyl group
- Ph means a phenyl group
- Ts means a tosyl group.
- TFA means trifluoroacetic acid.
- s is a single line
- d is a double line
- dd is a double double line
- t is a triple line
- td is a triple double line
- q is a quadruple line
- m is Multiple lines
- br means broad
- brs means broad single line
- brd means broad double line
- brt means broad triple line
- J means coupling constant.
- High-performance liquid chromatographic mass spectrometer The measurement conditions of LCMS are as follows, and the observed mass spectrometry value [MS (m / z)] is represented by MH +, and the retention time is represented by Rt (min, min). In each actual measurement value, any one of A to H is added as a measurement condition used for the measurement.
- Reference example 1 4- [5-Chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine dihydrochloride a) Preparation of 4-bromo-1- (3-fluorophenyl) -1H-imidazole (compound Q1) 4-Fluoroimidazole (50 g) in methylene chloride solution (1500 mL) at room temperature with 3-fluorophenylboronic acid ( 94.0 g), copper (II) acetate (91.0 g) and pyridine (50 mL) were added, and the mixture was stirred at 30 ° C. for 48 hours.
- Reference example 2 4- [5-Cyano-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine dihydrochloride a) Preparation of tert-butyl 4- [1- (3-fluorophenyl) -5-iodo-1H-imidazol-4-yl] piperidine-1-carboxylate (Compound Q5) Compound Q3 (5.0 g) in acetonitrile N-iodosuccinimide (3.6 g) and trifluoroacetic acid (0.05 mL) were added to the solution (30 mL) at room temperature, and the mixture was stirred at room temperature for 72 hours under light shielding.
- Reference example 3 4- ⁇ 3- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-5-yl ⁇ piperidine dihydrochloride a) Preparation of tert-butyl 4-ethiylpiperidine-1-carboxylate (Compound Q7) To a methanol solution (50 mL) of tert-butyl 4-formylpiperidine-1-carboxylate (7.8 g) at 0 ° C. Potassium carbonate (11 g) and dimethyl (acetyldiazomethyl) phosphonate (8.6 g) were added and stirred at 0 ° C. for 2 hours.
- Reference example 4 4- ⁇ 5- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-3-yl ⁇ piperidine dihydrochloride a) Preparation of tert-butyl 4-[(hydroxyimino) methyl] piperidine-1-carboxylate (Compound Q11) Methanol / water solution of tert-butyl 4-formylpiperidine-1-carboxylate (11 g) (80 mL / 80 mL) To the mixture, sodium carbonate (2.7 g) and hydroxylamine hydrochloride (4.3 g) were added at room temperature, and the mixture was stirred at room temperature for 16 hours.
- Tetrahydrofuran 40 mL was added to the reaction solution, and then a tetrahydrofuran solution (20 mL) of compound Q12 (2.9 g) and triethylamine (5.6 g) was added at room temperature.
- the reaction solution was heated to 60 ° C. and stirred for 2 hours, and then the solvent was distilled off under reduced pressure.
- the residue was extracted with ethyl acetate (20 mL) and saturated aqueous sodium hydrogen carbonate (30 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure.
- Reference Example 8 4- [5- (4-Fluorophenyl) -1,3-thiazol-2-yl] piperidine hydrochloride (Reference Example 8) a) Preparation of tert-butyl 4- [5- (4-fluorophenyl) -1,3-thiazol-2-yl] piperidine-1-carboxylate (Compound Q22) Compound Q19 (2 g) in tetrahydrofuran (50 mL) After adding Lawson's reagent (1.7 g) at room temperature, the mixture was heated to 70 ° C. and stirred for 4 hours.
- Example 1 4- [5-Chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] -N- (4,4-difluorocyclohexyl) piperidine-1-carboxamide Phenyl 4,4-difluorocyclohexylcarbamate (272 mg) and triethylamine (0.50 mL) were added to a methylene chloride solution (8 mL) of the compound of Reference Example 1 (198 mg) at room temperature, and the temperature was raised to 38 ° C. Stir for 48 hours. After distilling off the reaction solvent under reduced pressure, the compound of Example 1 (200 mg) was obtained by purification by preparative HPLC.
- Example 2 ⁇ 4- [5-Chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidin-1-yl ⁇ (4,4-difluorocyclohexyl) methanone
- a methylene chloride solution (4 mL) of the compound of Reference Example 1 (198 mg), 4,4-difluorocyclohexanecarboxylic acid (175 mg), HBTU (540 mg) and triethylamine (0.50 mL) were added at room temperature. Stir for 24 hours. After distilling off the reaction solvent under reduced pressure, the compound of Example 2 (100 mg) was obtained by purification by preparative HPLC.
- Example 3 4- [5-Cyano-1- (3-fluorophenyl) -1H-imidazol-4-yl] -N- (4,4-difluorocyclohexyl) piperidine-1-carboxamide
- the compound of Example 3 (26 mg) was obtained by the method according to Example 1 using the compound of Reference Example 2 (259 mg).
- Example 4 N- (Tetrahydro-2H-pyran-4-yl) -4- ⁇ 3- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-5-yl ⁇ piperidine-1-carboxamide
- the compound of Example 4 (90 mg) was obtained by the method according to Example 1 using the compound of Reference Example 3 (327 mg).
- Example 5 N- (Tetrahydro-2H-pyran-4-yl) -4- ⁇ 5- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-3-yl ⁇ piperidine-1-carboxamide
- the compound of Example 5 (85 mg) was obtained by the method according to Example 1 using the compound of Reference Example 4 (247 mg).
- Example 6 N- (4,4-difluorocyclohexyl) -4- [2- (4-fluorophenyl) -1,3-oxazol-5-yl] piperidine-1-carboxamide
- the compound of Example 6 (38 mg) was obtained by the method according to Example 1 using the compound of Reference Example 5 (290 mg).
- Example 7 N- (4,4-difluorocyclohexyl) -4- [5- (4-fluorophenyl) -1,3-oxazol-2-yl] piperidine-1-carboxamide
- the compound of Example 7 (200 mg) was obtained by the method according to Example 1 using the compound of Reference Example 6 (300 mg).
- Example 8 N- (4,4-difluorocyclohexyl) -4- [2- (4-fluorophenyl) -1,3-thiazol-5-yl] piperidine-1-carboxamide
- the compound of Example 8 (35 mg) was obtained by the method according to Example 1 using the compound of Reference Example 7 (120 mg).
- Example 9 N- (4,4-difluorocyclohexyl) -4- [5- (4-fluorophenyl) -1,3-thiazol-2-yl] piperidine-1-carboxamide Triethylamine (0.46 mL) was added to a solution of the compound of Reference Example 8 (340 mg) and phenyl 4,4-diphenylcyclohexanecarbamate (434 mg) in acetonitrile (5 mL) at room temperature, and the temperature was raised to 55 ° C. Stir for hours.
- Example 10 4- [4-Chloro-5- (4-fluorophenyl) -1,3-thiazol-2-yl] -N- (4,4-difluorocyclohexyl) piperidine-1-carboxamide N-chlorosuccinimide (38 mg) and trifluoroacetic acid (0.05 mL) were added to an acetonitrile solution (5 mL) of the compound of Example 9 (100 mg) at room temperature, and then the mixture was heated to 55 ° C. and stirred for 4 hours. did. A saturated sodium bicarbonate solution was added to the reaction solution, followed by extraction with ethyl acetate, and the organic layer was washed with water and saturated brine.
- Examples 11 to 39 The compounds shown in Table 1 were obtained by the method according to Example 1 or 3 using the corresponding raw material compounds.
- Examples 40-50 The compounds shown in Table 2 were obtained by the method according to Example 2 using the corresponding raw material compounds.
- Examples 51-91 The compounds shown in Table 3 were obtained by the method according to Example 4 using the corresponding starting compounds.
- Examples 92-98 The compounds shown in Table 4 were obtained by the method according to Reference Example 3 and Example 2 using the corresponding starting compounds.
- Examples 101-142 The compounds shown in Table 6 were obtained by the method according to Example 5 using the corresponding starting compounds.
- Examples 143-148 The compounds shown in Table 7 were obtained by the method according to Reference Example 4 and Example 2 using the corresponding starting compounds.
- Example 149 The compounds shown in Table 8 were obtained by the method according to Example 6 using the corresponding starting compounds.
- Examples 150-151 The compounds shown in Table 9 were obtained by the method according to Reference Example 5 and Example 2 using the corresponding starting compounds.
- Examples 152-154 The compounds shown in Table 10 were obtained by the method according to Example 7 using the corresponding starting compounds.
- Examples 155 to 156 The compounds shown in Table 11 were obtained by the method according to Reference Example 6 and Example 2 using the corresponding starting compounds.
- Examples 157 to 159 The compounds shown in Table 12 were obtained by the method according to Example 8 using the corresponding starting compounds.
- Examples 160-161 The compounds shown in Table 13 were obtained by the method according to Reference Example 7 and Example 2 using the corresponding starting compounds.
- Examples 162-163 The compounds shown in Table 14 were obtained by the method according to Examples 9 and 10 using the corresponding starting compounds.
- Examples 164 to 165 The compounds shown in Table 15 were obtained by the method according to Reference Example 8 and Example 2 using the corresponding starting compounds.
- Examples 166-175 The compounds shown in Table 16 were obtained by the method according to Example 6 using the corresponding starting compounds.
- Examples 176-184 The compounds shown in Table 17 were obtained by the method according to Reference Example 5 and Example 2 using the corresponding starting compounds.
- Examples 185-191 The compounds shown in Table 18 were obtained by the method according to Example 7 using the corresponding starting compounds.
- Example 192 The compounds shown in Table 19 were obtained by the method according to Reference Example 6 and Example 2 using the corresponding starting compounds.
- Examples 193-198 The compounds shown in Table 20 were obtained by the method according to Example 8 using the corresponding starting compounds.
- Examples 199-210 The compounds shown in Table 21 were obtained by the method according to Reference Example 7 and Example 2 using the corresponding starting compounds.
- Examples 211-214 The compounds shown in Table 22 were obtained by the method according to Examples 9 and 10 using the corresponding starting compounds.
- Examples 215 to 219 The compounds shown in Table 23 were obtained by the method according to Reference Example 8 and Example 2 using the corresponding starting compounds.
- Test Example 1 PAM activity evaluation using human ⁇ 7 nACh receptor stable expression cells
- Human ⁇ 7 nAChR stable expression cells Human ⁇ 7 nAChR stable expression cells were prepared and subjected to culture. Specifically, rat pituitary-derived GH4C1 cells (cat. No. CCL-82.2, ATCC, USA) were used as host cells.
- Introduction of pcDNA3.1Zeo vector into which the nucleotide sequence encoding the protein of GenBank BAC81731 was inserted, and introduction of pcDNA3.1 vector into which the human ⁇ 7 nAChR gene was inserted (cat. No.
- V790-20, invitrogen, Carlsbad, CA, USA Thus, aequorin and human ⁇ 7 nAChR stably expressing cells were obtained.
- Zeocin catalog. No. R25001, invitrogen, Carlsbad, CA, USA
- Geneticin catalog. No. 10131-027, invitrogen, Carlsbad, CA, USA
- Medium includes 2.5% fetal bovine serum (cat. No. 2917354, ICN Biomedicals, Inc, USA), 15% inactivated horse serum (cat. No. 26050-088, invitrogen, Carlsbad, CA, USA), 1 ⁇ g / ML Geneticin, F-10 Nutrient Mixture (Ham) medium (cat. No.
- test compound A DMSO solution having a concentration 1000 times the final concentration was prepared as a test compound, and this solution was added to Hanks / 20 mmol / L HEPES / 0.2% BSA (cat. No. A3803, Sigma, St. Louis, MO, USA) and adjusted to 6 times the final concentration.
- PAM activity evaluation FDSS7000 (Hamamatsu Photonics) was used for the detection of the luminescent signal by ⁇ 7 nAChR stimulation. Test compounds were added to the plates to which cells and luminescent substrate had been added, and ACh at a concentration indicating EC 20 was added alone after 150 seconds. The luminescence signal (center wavelength: 465 nm) was measured for 138 seconds after the addition of ACh to calculate RLU (Max-Min), and the ratio of RLU (Max-Min) between the control well and the test compound added well was defined as PAM activity. . Data of ⁇ 7 PAM activity of representative compounds is shown in Table 24, Table 25 and Table 26.
- Test Example 2 Evaluation of cognitive function using mouse novel object recognition test (hereinafter referred to as mORT) Decreased memory for known objects in a novel object recognition test using Slc: ddY mice (male, SLC Japan) weighing 25-30 g depending on the time interval between the first trial (training) and the second trial (test) When a second trial is performed 24 hours later, significant forgetting is observed. Therefore, the compound of the present invention was administered before the first trial, and the memory enhancing action in the second trial was evaluated. As a result, the compounds of Examples 1, 45, 51, 79 and 124 showed a significant memory enhancing action at 3 or 10 mg / kg (oral).
- mORT mouse novel object recognition test
- the compound represented by the formula (I), or a pharmaceutically acceptable salt thereof has a strong ⁇ 7 nicotinic acetylcholine receptor ( ⁇ 7hnAChR) modulating action, and the central nervous system ( CNS) and / or peripheral nervous system (PNS) cholinergic diseases, smooth muscle contraction diseases, endocrine diseases, neurodegenerative diseases, diseases such as inflammation or pain, and withdrawal symptoms caused by addictive drug abuse It is useful for treatment and / or prevention of diseases and the like.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Epidemiology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Hospice & Palliative Care (AREA)
- Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
The present invention provides a compound that has a strong α7 nicotinic acetylcholine receptor (α7 nAChR)-regulating effect and is useful as a therapeutic drug for diseases associated with cholinergicity of the central nervous system (CNS) and/or peripheral nervous system (PNS), diseases associated with smooth muscle contraction, endocrine diseases, diseases associated with neurodegeneration, etc. Provided is a compound of formula (I) or a pharmaceutically acceptable salt of said compound [In the formula, X-Y-Z represents N-CO-NR3AR3B or the like, A represents an optionally substituted phenyl or the like, B represents optionally substituted imidazole or the like, R2A, R2B, R2C, and R2D may be the same or different and represent hydrogen atoms or the like, and R3A and R3B may be the same or different and represent optionally substituted C3-10 cycloalkyls or the like].
Description
本発明は、α7ニコチン性アセチルコリン受容体(α7 nAChR)の調節物質である新規な5員環ヘテロアリール誘導体に関する。それらの薬理学的特性から、本発明の化合物は、中枢神経系(CNS)及び/又は末梢神経系(PNS)のコリン作動性に関する疾患、平滑筋収縮に関する疾患、内分泌疾患、神経変性に関する疾患、炎症又は痛み等の疾患及び常習性の薬物乱用から引き起こされる禁断症状に関する疾患等の治療に有用であり得る。
The present invention relates to a novel 5-membered heteroaryl derivative that is a modulator of α7 nicotinic acetylcholine receptor (α7 nAChR). Due to their pharmacological properties, the compounds of the present invention can be used for diseases related to cholinergic activity of the central nervous system (CNS) and / or peripheral nervous system (PNS), diseases related to smooth muscle contraction, endocrine diseases, diseases related to neurodegeneration, It may be useful for the treatment of diseases such as inflammation or pain and diseases related to withdrawal symptoms caused by addictive drug abuse.
近年、ニコチンの潜在的な神経保護効果が示されており、興奮毒性傷害、栄養欠乏、虚血、外傷、アミロイドベータ(Aβ)媒介神経細胞死及びタンパク質凝集媒介神経変性を伴う動物及び培養細胞の様々な神経変性モデルが提唱されている。ニコチンが神経保護効果を呈する多くの例において、α7サブタイプ含有ニコチン性アセチルコリン受容体が活性化されていることが明らかになっている。これらの報告により、ニコチンが神経保護効果を媒介するために役立つことが示唆され、α7サブタイプを含む受容体の直接的関与が想起されてきた。これらデータから、α7ニコチン性アセチルコリン受容体が、神経保護として妥当な分子標的の代表であることが示唆される。つまり、該受容体の活性なアゴニスト/正のモジュレーター(ポジティブアロステリックモジュレーター:PAM)を開発することにより、神経保護が達成され得る。実際に、α7ニコチン性受容体アゴニストはすでに同定され、神経保護薬の開発のために可能性ある手がかりとして評価されている。また、近年α7ニコチン性アセチルコリン受容体の炎症への関与も、報告されている。以上のことから、該受容体の新規モジュレーターの開発は、神経系疾患、精神疾患及び炎症性疾患の新規な治療に結びつくことが想定される。
In recent years, the potential neuroprotective effects of nicotine have been shown, in animals and cultured cells with excitotoxic injury, nutrient deficiency, ischemia, trauma, amyloid beta (Aβ) mediated neuronal cell death and protein aggregation mediated neurodegeneration. Various neurodegenerative models have been proposed. In many instances where nicotine exhibits a neuroprotective effect, it has been shown that the α7 subtype-containing nicotinic acetylcholine receptor is activated. These reports suggest that nicotine serves to mediate neuroprotective effects and have been recalled for direct involvement of receptors including the α7 subtype. These data suggest that the α7 nicotinic acetylcholine receptor represents a valid molecular target for neuroprotection. Thus, neuroprotection can be achieved by developing an active agonist / positive modulator of the receptor (positive allosteric modulator: PAM). In fact, α7 nicotinic receptor agonists have already been identified and evaluated as potential clues for the development of neuroprotective drugs. In recent years, the involvement of α7 nicotinic acetylcholine receptors in inflammation has also been reported. From the above, it is assumed that the development of novel modulators of the receptor will lead to novel treatments for nervous system diseases, psychiatric diseases and inflammatory diseases.
これまでに、α7ニコチン性アセチルコリン受容体(α7 nAChR)のアゴニスト及び正のモジュレーターに関する開示はあるが、本願発明の化合物とは構造が異なる(特許文献1、特許文献2、特許文献3、特許文献4、特許文献5及び特許文献6)。
So far, there are disclosures regarding agonists and positive modulators of α7 nicotinic acetylcholine receptor (α7 nAChR), but the structure is different from the compounds of the present invention (Patent Document 1, Patent Document 2, Patent Document 3, Patent Document) 4, Patent Document 5 and Patent Document 6).
本発明が解決しようとする課題は、強力なα7ニコチン性アセチルコリン受容体(α7 nAChR)の正のモジュレーター作用を有し、神経系疾患、精神疾患及び炎症性疾患の新規な治療剤として有用な新規化合物を提供することにある。
The problem to be solved by the present invention is a novel having a positive modulator action of a potent α7 nicotinic acetylcholine receptor (α7 nAChR) and useful as a novel therapeutic agent for nervous system diseases, mental diseases and inflammatory diseases. It is to provide a compound.
本発明者らは、鋭意研究を行った結果、下記式(I)で表される新規化合物が強力なα7ニコチン性アセチルコリン受容体(α7 nAChR)の正のモジュレーター作用を有することを見出し、本発明を完成させた。本発明によれば、下記式(I)で表される5員環ヘテロアリール化合物又はその製薬学的に許容される塩(以下、「本発明の化合物」と称することもある)が提供される。
As a result of intensive studies, the present inventors have found that a novel compound represented by the following formula (I) has a positive modulator action of a potent α7 nicotinic acetylcholine receptor (α7 nAChR). Was completed. According to the present invention, a 5-membered heteroaryl compound represented by the following formula (I) or a pharmaceutically acceptable salt thereof (hereinafter sometimes referred to as “the compound of the present invention”) is provided. .
[項1]式(I):
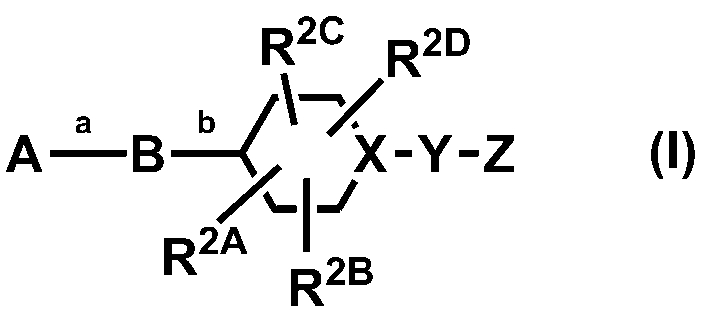

[式中、
X-Y-Zは、N-CO-NR3AR3B、N-COR4A、CR5-CO-NR3AR3B、CR5-NR6-COR4B又はCR5-NR6-CONR3AR3Bを表し、
Aは、フェニル又はピリジル(該フェニル及び該ピリジルは、それぞれハロゲン、水酸基、1から5個のフッ素で置換されていてもよいC1-6アルキル、1から5個のフッ素で置換されていてもよいC1-6アルコキシ及びシアノからなる群から独立して選択される1~5個の置換基で置換されていてもよい)を表し、
Bは、B-1、B-2、B-3、B-4又はB-5を表し、
式(I)の化合物におけるaおよびbは、Bに結合する2つの結合手が、B-1からB-5における2つの結合手のいずれに該当する結合かを示す記号であり、
Wは、酸素原子又は硫黄原子を表し、
R1は、水素原子;ハロゲン;シアノ;C1-6アルキル(該アルキルは、ハロゲン、水酸基、1から5個のフッ素で置換されていてもよいC3-10シクロアルキル、1から5個のフッ素で置換されていてもよいC1-6アルコキシ及び4~10員の飽和複素環からなる群から独立して選択される1~5個の置換基で置換されていてもよい);又はハロゲン、水酸基、C1-6アルキル及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキルを表し、
R2A、R2B、R2C、R2D及びR5は、同一又は異なって、水素原子;フッ素;水酸基;1から5個のフッ素で置換されていてもよいC1-6アルコキシ;又は1から5個のフッ素で置換されていてもよいC1-6アルキルを表し、ここにおいて、R2A、R2B、R2C、R2D及びR5のいずれかの2つが1から5個のフッ素で置換されていてもよいC1-6アルキルのとき、2個のアルキルが一緒になって該アルキルが結合している環と別の環を形成していてもよく、
R3A、R3B、R4A、R4B及びR6は、同一又は異なって、ハロゲン、水酸基、C1-6アルコキシ、C3-10シクロアルキル及び4~10員の飽和複素環(ただし、環上にカルボニル基を有しない)からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;ハロゲン、水酸基、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;C1-6アルキルで置換されていてもよい4~10員の飽和複素環;又は水素原子を表し、ここにおいて、(1)R4A及びR4Bは水素原子ではなく、(2)R3A及びR3Bは同時に水素原子ではなく、(3)BがB-3であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aはメチルではなく、(4)Aが無置換のフェニルであり、BがB-4であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aは1~5個のフッ素で置換されたC3アルキルではなく、かつ(5)R3A及びR3Bが共にC1-6アルキルのとき、それらが結合する窒素原子と一緒になって、フッ素、C1-6アルキル及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい4~10員の含窒素飽和複素環を形成していてもよい]
で表される化合物又はその製薬学的に許容される塩。 [Item 1] Formula (I):
[Where:
XYZ represents N-CO-NR 3A R 3B , N-COR 4A , CR 5 -CO-NR 3A R 3B , CR 5 -NR 6 -COR 4B or CR 5 -NR 6 -CONR 3A R 3B Represents
A is phenyl or pyridyl (said phenyl and said pyridyl, halogen, respectively, hydroxyl, 1 to 5 fluorine optionally substituted by C 1-6 alkyl, optionally substituted with 1-5 fluorine And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and cyano,
B represents B-1, B-2, B-3, B-4 or B-5,
A and b in the compound of the formula (I) are symbols indicating which of the two bonds in B-1 to B-5 the two bonds that bind to B correspond to;
W represents an oxygen atom or a sulfur atom,
R 1 is a hydrogen atom; halogen; cyano; C 1-6 alkyl (wherein the alkyl is halogen, hydroxyl, C 3-10 cycloalkyl optionally substituted with 1 to 5 fluorines, 1 to 5 Halogen optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy optionally substituted with fluorine and a 4- to 10-membered saturated heterocyclic ring; or halogen Represents a C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of hydroxyl, C 1-6 alkyl and C 1-6 alkoxy,
R 2A , R 2B , R 2C , R 2D and R 5 are the same or different and represent a hydrogen atom; fluorine; a hydroxyl group; C 1-6 alkoxy optionally substituted with 1 to 5 fluorines; Represents C 1-6 alkyl optionally substituted with 5 fluorines, wherein two of R 2A , R 2B , R 2C , R 2D and R 5 are substituted with 1 to 5 fluorines In the case of C 1-6 alkyl which may be substituted , two alkyls may be combined to form another ring with the ring to which the alkyl is bonded,
R 3A , R 3B , R 4A , R 4B and R 6 are the same or different and are halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4- to 10-membered saturated heterocyclic ring (provided that the ring C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of (having no carbonyl group above); halogen, hydroxyl, C 1-6 alkoxy and C 1 C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of -6 alkyl; 4 to 10 members optionally substituted with C 1-6 alkyl Or a hydrogen atom, wherein (1) R 4A and R 4B are not hydrogen atoms, (2) R 3A and R 3B are not simultaneously hydrogen atoms, and (3) B is B— 3 and W is an oxygen atom When XYZ is N—COR 4A , R 4A is not methyl, (4) A is unsubstituted phenyl, B is B-4, and W is an oxygen atom. , XYZ is N—COR 4A , R 4A is not C 3 alkyl substituted with 1 to 5 fluorines, and (5) R 3A and R 3B are both C 1-6 When alkyl, together with the nitrogen atom to which they are attached, is substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkyl and C 1-6 alkoxy. It may form a 4- to 10-membered nitrogen-containing saturated heterocyclic ring.
Or a pharmaceutically acceptable salt thereof.
X-Y-Zは、N-CO-NR3AR3B、N-COR4A、CR5-CO-NR3AR3B、CR5-NR6-COR4B又はCR5-NR6-CONR3AR3Bを表し、
Aは、フェニル又はピリジル(該フェニル及び該ピリジルは、それぞれハロゲン、水酸基、1から5個のフッ素で置換されていてもよいC1-6アルキル、1から5個のフッ素で置換されていてもよいC1-6アルコキシ及びシアノからなる群から独立して選択される1~5個の置換基で置換されていてもよい)を表し、
Bは、B-1、B-2、B-3、B-4又はB-5を表し、
式(I)の化合物におけるaおよびbは、Bに結合する2つの結合手が、B-1からB-5における2つの結合手のいずれに該当する結合かを示す記号であり、
Wは、酸素原子又は硫黄原子を表し、
R1は、水素原子;ハロゲン;シアノ;C1-6アルキル(該アルキルは、ハロゲン、水酸基、1から5個のフッ素で置換されていてもよいC3-10シクロアルキル、1から5個のフッ素で置換されていてもよいC1-6アルコキシ及び4~10員の飽和複素環からなる群から独立して選択される1~5個の置換基で置換されていてもよい);又はハロゲン、水酸基、C1-6アルキル及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキルを表し、
R2A、R2B、R2C、R2D及びR5は、同一又は異なって、水素原子;フッ素;水酸基;1から5個のフッ素で置換されていてもよいC1-6アルコキシ;又は1から5個のフッ素で置換されていてもよいC1-6アルキルを表し、ここにおいて、R2A、R2B、R2C、R2D及びR5のいずれかの2つが1から5個のフッ素で置換されていてもよいC1-6アルキルのとき、2個のアルキルが一緒になって該アルキルが結合している環と別の環を形成していてもよく、
R3A、R3B、R4A、R4B及びR6は、同一又は異なって、ハロゲン、水酸基、C1-6アルコキシ、C3-10シクロアルキル及び4~10員の飽和複素環(ただし、環上にカルボニル基を有しない)からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;ハロゲン、水酸基、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;C1-6アルキルで置換されていてもよい4~10員の飽和複素環;又は水素原子を表し、ここにおいて、(1)R4A及びR4Bは水素原子ではなく、(2)R3A及びR3Bは同時に水素原子ではなく、(3)BがB-3であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aはメチルではなく、(4)Aが無置換のフェニルであり、BがB-4であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aは1~5個のフッ素で置換されたC3アルキルではなく、かつ(5)R3A及びR3Bが共にC1-6アルキルのとき、それらが結合する窒素原子と一緒になって、フッ素、C1-6アルキル及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい4~10員の含窒素飽和複素環を形成していてもよい]
で表される化合物又はその製薬学的に許容される塩。 [Item 1] Formula (I):
XYZ represents N-CO-NR 3A R 3B , N-COR 4A , CR 5 -CO-NR 3A R 3B , CR 5 -NR 6 -COR 4B or CR 5 -NR 6 -CONR 3A R 3B Represents
A is phenyl or pyridyl (said phenyl and said pyridyl, halogen, respectively, hydroxyl, 1 to 5 fluorine optionally substituted by C 1-6 alkyl, optionally substituted with 1-5 fluorine And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and cyano,
B represents B-1, B-2, B-3, B-4 or B-5,
A and b in the compound of the formula (I) are symbols indicating which of the two bonds in B-1 to B-5 the two bonds that bind to B correspond to;
W represents an oxygen atom or a sulfur atom,
R 1 is a hydrogen atom; halogen; cyano; C 1-6 alkyl (wherein the alkyl is halogen, hydroxyl, C 3-10 cycloalkyl optionally substituted with 1 to 5 fluorines, 1 to 5 Halogen optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy optionally substituted with fluorine and a 4- to 10-membered saturated heterocyclic ring; or halogen Represents a C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of hydroxyl, C 1-6 alkyl and C 1-6 alkoxy,
R 2A , R 2B , R 2C , R 2D and R 5 are the same or different and represent a hydrogen atom; fluorine; a hydroxyl group; C 1-6 alkoxy optionally substituted with 1 to 5 fluorines; Represents C 1-6 alkyl optionally substituted with 5 fluorines, wherein two of R 2A , R 2B , R 2C , R 2D and R 5 are substituted with 1 to 5 fluorines In the case of C 1-6 alkyl which may be substituted , two alkyls may be combined to form another ring with the ring to which the alkyl is bonded,
R 3A , R 3B , R 4A , R 4B and R 6 are the same or different and are halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4- to 10-membered saturated heterocyclic ring (provided that the ring C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of (having no carbonyl group above); halogen, hydroxyl, C 1-6 alkoxy and C 1 C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of -6 alkyl; 4 to 10 members optionally substituted with C 1-6 alkyl Or a hydrogen atom, wherein (1) R 4A and R 4B are not hydrogen atoms, (2) R 3A and R 3B are not simultaneously hydrogen atoms, and (3) B is B— 3 and W is an oxygen atom When XYZ is N—COR 4A , R 4A is not methyl, (4) A is unsubstituted phenyl, B is B-4, and W is an oxygen atom. , XYZ is N—COR 4A , R 4A is not C 3 alkyl substituted with 1 to 5 fluorines, and (5) R 3A and R 3B are both C 1-6 When alkyl, together with the nitrogen atom to which they are attached, is substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkyl and C 1-6 alkoxy. It may form a 4- to 10-membered nitrogen-containing saturated heterocyclic ring.
Or a pharmaceutically acceptable salt thereof.
[項2]R3A、R3B、R4A、R4B及びR6が、同一又は異なって、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子であり、ここにおいて、(1)R4A及びR4Bは水素原子ではなく、(2)R3A及びR3Bは同時に水素原子ではなく、(3)BがB-3であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aはメチルではなく、(4)Aが無置換のフェニルであり、BがB-4であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aは1~5個のフッ素で置換されたC3アルキルではない、
項1に記載の化合物又はその製薬学的に許容される塩。 [Item 2] R 3A , R 3B , R 4A , R 4B and R 6 are the same or different and are independently selected from the group consisting of fluorine, C 1-6 alkoxy, and C 3-10 cycloalkyl. C 1-6 alkyl optionally substituted with 1-5 substituents; 1-5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl in an optionally substituted C 3-10 cycloalkyl; saturated heterocyclic 4-10 membered; a or a hydrogen atom, wherein, (1) R 4A and R 4B are not hydrogen atom, (2) R 3A and R 3B are not simultaneously hydrogen atoms; (3) when B is B-3, W is an oxygen atom, and XYZ is N-COR 4A , R 4A is not methyl (4) A is unsubstituted phenyl and B is B- In it, W is an oxygen atom, when X-Y-Z is N-COR 4A is, R 4A is not a C 3 alkyl substituted with 1-5 fluorine,
Item 12. The compound according to Item 1 or a pharmaceutically acceptable salt thereof.
項1に記載の化合物又はその製薬学的に許容される塩。 [Item 2] R 3A , R 3B , R 4A , R 4B and R 6 are the same or different and are independently selected from the group consisting of fluorine, C 1-6 alkoxy, and C 3-10 cycloalkyl. C 1-6 alkyl optionally substituted with 1-5 substituents; 1-5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl in an optionally substituted C 3-10 cycloalkyl; saturated heterocyclic 4-10 membered; a or a hydrogen atom, wherein, (1) R 4A and R 4B are not hydrogen atom, (2) R 3A and R 3B are not simultaneously hydrogen atoms; (3) when B is B-3, W is an oxygen atom, and XYZ is N-COR 4A , R 4A is not methyl (4) A is unsubstituted phenyl and B is B- In it, W is an oxygen atom, when X-Y-Z is N-COR 4A is, R 4A is not a C 3 alkyl substituted with 1-5 fluorine,
Item 12. The compound according to Item 1 or a pharmaceutically acceptable salt thereof.
[項3]R3A、R3B及びR4Aが、同一又は異なって、フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子であり、ここにおいて、R3A及びR3Bは同時に水素原子ではなく、R4Aは水素原子ではない、
項1又は2に記載の化合物又はその製薬学的に許容される塩。 [Claim 3] R 3A , R 3B and R 4A are the same or different and are each 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl An optionally substituted C 3-10 cycloalkyl; a 4-10 membered saturated heterocycle; or a hydrogen atom, wherein R 3A and R 3B are not simultaneously a hydrogen atom and R 4A is not a hydrogen atom ,
Item 3. The compound according to Item 1 or 2, or a pharmaceutically acceptable salt thereof.
項1又は2に記載の化合物又はその製薬学的に許容される塩。 [Claim 3] R 3A , R 3B and R 4A are the same or different and are each 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl An optionally substituted C 3-10 cycloalkyl; a 4-10 membered saturated heterocycle; or a hydrogen atom, wherein R 3A and R 3B are not simultaneously a hydrogen atom and R 4A is not a hydrogen atom ,
Item 3. The compound according to Item 1 or 2, or a pharmaceutically acceptable salt thereof.
[項4]R4Bが、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキルである、
項1~3のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Claim 4] R 4B is good fluorine, C 1-6 alkoxy, and C 3-10 optionally substituted with one to five substituents independently selected from the group consisting of cycloalkyl C 1- 6 alkyl,
Item 4. The compound according to any one of Items 1 to 3, or a pharmaceutically acceptable salt thereof.
項1~3のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Claim 4] R 4B is good fluorine, C 1-6 alkoxy, and C 3-10 optionally substituted with one to five substituents independently selected from the group consisting of cycloalkyl C 1- 6 alkyl,
Item 4. The compound according to any one of Items 1 to 3, or a pharmaceutically acceptable salt thereof.
[項5]Aが、フェニル又はピリジル(該フェニル及び該ピリジルは、それぞれフッ素、1から5個のフッ素で置換されていてもよいC1-6アルキル及び1から5個のフッ素で置換されていてもよいC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)である、
項1~4のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 5] A is phenyl or pyridyl (the phenyl and the pyridyl are each substituted with fluorine, C 1-6 alkyl optionally substituted with 1 to 5 fluorine, and 1 to 5 fluorine. And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy).
Item 5. The compound according to any one of Items 1 to 4 or a pharmaceutically acceptable salt thereof.
項1~4のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 5] A is phenyl or pyridyl (the phenyl and the pyridyl are each substituted with fluorine, C 1-6 alkyl optionally substituted with 1 to 5 fluorine, and 1 to 5 fluorine. And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy).
Item 5. The compound according to any one of Items 1 to 4 or a pharmaceutically acceptable salt thereof.
[項6]Aが、フェニル(該フェニルは、それぞれフッ素、1から5個のフッ素で置換されていてもよいC1-6アルキル及び1から5個のフッ素で置換されていてもよいC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)である、
項1~4のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 6] A is phenyl (the phenyl is each fluorine, C 1-6 alkyl optionally substituted with 1 to 5 fluorines, and C 1 optionally substituted with 1 to 5 fluorines) Optionally substituted with 1 to 5 substituents independently selected from the group consisting of -6 alkoxy.)
Item 5. The compound according to any one of Items 1 to 4 or a pharmaceutically acceptable salt thereof.
項1~4のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 6] A is phenyl (the phenyl is each fluorine, C 1-6 alkyl optionally substituted with 1 to 5 fluorines, and C 1 optionally substituted with 1 to 5 fluorines) Optionally substituted with 1 to 5 substituents independently selected from the group consisting of -6 alkoxy.)
Item 5. The compound according to any one of Items 1 to 4 or a pharmaceutically acceptable salt thereof.
[項7]Aが、ピリジル(該ピリジルは、それぞれフッ素、1から5個のフッ素で置換されていてもよいC1-6アルキル及び1から5個のフッ素で置換されていてもよいC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)である、
項1~4のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 7] A is pyridyl (the pyridyl is each fluorine, C 1-6 alkyl optionally substituted with 1 to 5 fluorines, and C 1 optionally substituted with 1 to 5 fluorines) Optionally substituted with 1 to 5 substituents independently selected from the group consisting of -6 alkoxy.)
Item 5. The compound according to any one of Items 1 to 4 or a pharmaceutically acceptable salt thereof.
項1~4のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 7] A is pyridyl (the pyridyl is each fluorine, C 1-6 alkyl optionally substituted with 1 to 5 fluorines, and C 1 optionally substituted with 1 to 5 fluorines) Optionally substituted with 1 to 5 substituents independently selected from the group consisting of -6 alkoxy.)
Item 5. The compound according to any one of Items 1 to 4 or a pharmaceutically acceptable salt thereof.
[項8]R1が、水素原子;ハロゲン;シアノ;C1-6アルキル;又はC3-10シクロアルキルである、
項1~7のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 8] R 1 is a hydrogen atom; halogen; cyano; C 1-6 alkyl; or C 3-10 cycloalkyl.
Item 8. The compound according to any one of Items 1 to 7, or a pharmaceutically acceptable salt thereof.
項1~7のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 8] R 1 is a hydrogen atom; halogen; cyano; C 1-6 alkyl; or C 3-10 cycloalkyl.
Item 8. The compound according to any one of Items 1 to 7, or a pharmaceutically acceptable salt thereof.
[項9]R1が、水素原子、塩素又はシアノである、
項1~7のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 9] R 1 is a hydrogen atom, chlorine or cyano.
Item 8. The compound according to any one of Items 1 to 7, or a pharmaceutically acceptable salt thereof.
項1~7のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 9] R 1 is a hydrogen atom, chlorine or cyano.
Item 8. The compound according to any one of Items 1 to 7, or a pharmaceutically acceptable salt thereof.
[項10]R1が、水素原子である、
項1~7のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 10] R 1 is a hydrogen atom.
Item 8. The compound according to any one of Items 1 to 7, or a pharmaceutically acceptable salt thereof.
項1~7のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 10] R 1 is a hydrogen atom.
Item 8. The compound according to any one of Items 1 to 7, or a pharmaceutically acceptable salt thereof.
[項11]R1が、塩素である、
項1~7のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 11] R 1 is chlorine.
Item 8. The compound according to any one of Items 1 to 7, or a pharmaceutically acceptable salt thereof.
項1~7のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 11] R 1 is chlorine.
Item 8. The compound according to any one of Items 1 to 7, or a pharmaceutically acceptable salt thereof.
[項12]Wが、酸素原子である、
項1~11のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 12] W is an oxygen atom.
Item 12. The compound according to any one of Items 1 to 11 or a pharmaceutically acceptable salt thereof.
項1~11のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 12] W is an oxygen atom.
Item 12. The compound according to any one of Items 1 to 11 or a pharmaceutically acceptable salt thereof.
[項13]Wが、硫黄原子である、
項1~11のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 13] W is a sulfur atom.
Item 12. The compound according to any one of Items 1 to 11 or a pharmaceutically acceptable salt thereof.
項1~11のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 13] W is a sulfur atom.
Item 12. The compound according to any one of Items 1 to 11 or a pharmaceutically acceptable salt thereof.
[項14]R2A、R2B、R2C、R2D及びR5が、同一又は異なって、水素原子、又はC1-6アルキルであり、ここにおいて、R2A、R2B、R2C、R2D及びR5のいずれかの2つがC1-6アルキルのとき、2個のアルキルが一緒になって該アルキルが結合している環と別の環を形成していてもよい、
項1~13のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 14] R 2A , R 2B , R 2C , R 2D and R 5 are the same or different and are a hydrogen atom or C 1-6 alkyl, wherein R 2A , R 2B , R 2C , R When any two of 2D and R 5 are C 1-6 alkyl, the two alkyls may be combined together to form another ring with the ring to which the alkyl is attached,
Item 14. The compound according to any one of Items 1 to 13, or a pharmaceutically acceptable salt thereof.
項1~13のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 14] R 2A , R 2B , R 2C , R 2D and R 5 are the same or different and are a hydrogen atom or C 1-6 alkyl, wherein R 2A , R 2B , R 2C , R When any two of 2D and R 5 are C 1-6 alkyl, the two alkyls may be combined together to form another ring with the ring to which the alkyl is attached,
Item 14. The compound according to any one of Items 1 to 13, or a pharmaceutically acceptable salt thereof.
[項15]R2A、R2B、R2C、R2D及びR5が、すべて水素原子である、
項1~13のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 15] R 2A , R 2B , R 2C , R 2D and R 5 are all hydrogen atoms.
Item 14. The compound according to any one of Items 1 to 13, or a pharmaceutically acceptable salt thereof.
項1~13のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 15] R 2A , R 2B , R 2C , R 2D and R 5 are all hydrogen atoms.
Item 14. The compound according to any one of Items 1 to 13, or a pharmaceutically acceptable salt thereof.
[項16]R6が、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;又は水素原子である、
項1~15のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Claim 16] R 6 is optionally fluorine, C 1-6 alkoxy, and C 3-10 optionally substituted with one to five substituents independently selected from the group consisting of cycloalkyl C 1- 6 alkyl; or a hydrogen atom,
Item 16. The compound according to any one of Items 1 to 15, or a pharmaceutically acceptable salt thereof.
項1~15のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Claim 16] R 6 is optionally fluorine, C 1-6 alkoxy, and C 3-10 optionally substituted with one to five substituents independently selected from the group consisting of cycloalkyl C 1- 6 alkyl; or a hydrogen atom,
Item 16. The compound according to any one of Items 1 to 15, or a pharmaceutically acceptable salt thereof.
[項17]R6が、水素原子である、
項1~15のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Section 17] R 6 is a hydrogen atom.
Item 16. The compound according to any one of Items 1 to 15, or a pharmaceutically acceptable salt thereof.
項1~15のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Section 17] R 6 is a hydrogen atom.
Item 16. The compound according to any one of Items 1 to 15, or a pharmaceutically acceptable salt thereof.
[項18]R3Bが、水素原子であり、R3A及びR4Aが、同一又は異なって、フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;又は4~10員の飽和複素環である、
項1~17のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 18] R 3B is a hydrogen atom, and R 3A and R 4A are the same or different and are independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl optionally substituted with 5 substituents; or a 4-10 membered saturated heterocycle,
Item 18. The compound according to any one of Items 1 to 17, or a pharmaceutically acceptable salt thereof.
項1~17のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 18] R 3B is a hydrogen atom, and R 3A and R 4A are the same or different and are independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl optionally substituted with 5 substituents; or a 4-10 membered saturated heterocycle,
Item 18. The compound according to any one of Items 1 to 17, or a pharmaceutically acceptable salt thereof.
[項19]X-Y-Zが、N-CO-NR3AR3B又はN-COR4Aである、
項1~18のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 19] XYZ is N—CO—NR 3A R 3B or N—COR 4A .
Item 19. The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
項1~18のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 19] XYZ is N—CO—NR 3A R 3B or N—COR 4A .
Item 19. The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
[項20]X-Y-Zが、N-CO-NR3AR3Bである、
項1~18のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 20] XYZ is N—CO—NR 3A R 3B .
Item 19. The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
項1~18のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 20] XYZ is N—CO—NR 3A R 3B .
Item 19. The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
[項21]X-Y-Zが、N-COR4Aである、
項1~18のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 21] XYZ is N-COR 4A .
Item 19. The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
項1~18のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 21] XYZ is N-COR 4A .
Item 19. The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
[項22]X-Y-Zが、CR5-CO-NR3AR3Bである、
項1~18のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 22] XYZ is CR 5 —CO—NR 3A R 3B .
Item 19. The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
項1~18のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 22] XYZ is CR 5 —CO—NR 3A R 3B .
Item 19. The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
[項23]X-Y-Zが、CR5-NR6-COR4Bである、
項1~18のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 23] XYZ is CR 5 —NR 6 —COR 4B .
Item 19. The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
項1~18のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 23] XYZ is CR 5 —NR 6 —COR 4B .
Item 19. The compound according to any one of Items 1 to 18, or a pharmaceutically acceptable salt thereof.
[項24]Bが、B-1である、
項1~23のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Claim 24] B is B-1.
Item 24. The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
項1~23のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Claim 24] B is B-1.
Item 24. The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
[項25]Bが、B-2である、
項1~23のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Claim 25] B is B-2.
Item 24. The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
項1~23のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Claim 25] B is B-2.
Item 24. The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
[項26]Bが、B-3である、
項1~23のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Section 26] B is B-3.
Item 24. The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
項1~23のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Section 26] B is B-3.
Item 24. The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
[項27]Bが、B-4である、
項1~23のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Section 27] B is B-4.
Item 24. The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
項1~23のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Section 27] B is B-4.
Item 24. The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
[項28]Bが、B-5である、
項1~23のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 28] B is B-5.
Item 24. The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
項1~23のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 [Item 28] B is B-5.
Item 24. The compound according to any one of Items 1 to 23 or a pharmaceutically acceptable salt thereof.
[項29]以下の化合物から選択される、項1に記載の化合物又はその製薬学的に許容される塩:
4-[5-クロロ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]-N-(4,4-ジフルオロシクロヘキシル)ピペリジン-1-カルボキサミド(実施例1)、
{4-[5-クロロ-1-(4-フルオロフェニル)-1H-イミダゾール-4-イル]ピペリジン-1-イル}(4,4-ジフルオロシクロヘキシル)メタノン(実施例45)、
N-シクロヘキシル-4-[3-(4-フルオロフェニル)-1,2-オキサゾール-5-イル]ピペリジン-1-カルボキサミド(実施例51)、
N-(4,4-ジフルオロシクロヘキシル)-4-[3-(2,5-ジフルオロフェニル)-1,2-オキサゾール-5-イル]ピペリジン-1-カルボキサミド(実施例77)、
4-[4-クロロ-3-(4-フルオロフェニル)-1,2-オキサゾール-5-イル]-N-(テトラヒドロ-2H-ピラン-4-イル)ピペリジン-1-カルボキサミド(実施例79)、
4-[4-クロロ-5-(4-フルオロフェニル)-1,2-オキサゾール-3-イル]-N-(テトラヒドロ-2H-ピラン-4-イル)ピペリジン-1-カルボキサミド(実施例124)、
4-[4-クロロ-5-(3,4-ジフルオロフェニル)-1,2-オキサゾール-3-イル]-N-(テトラヒドロ-2H-ピラン-4-イル)ピペリジン-1-カルボキサミド(実施例134)、及び
4-[5-(4-クロロフェニル)-1,3-オキサゾール-2-イル]-N-(テトラヒドロ-2H-ピラン-4-イル)ピペリジン-1-カルボキサミド(実施例189)。 [Item 29] The compound according to item 1 or a pharmaceutically acceptable salt thereof selected from the following compounds:
4- [5-Chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] -N- (4,4-difluorocyclohexyl) piperidine-1-carboxamide (Example 1),
{4- [5-Chloro-1- (4-fluorophenyl) -1H-imidazol-4-yl] piperidin-1-yl} (4,4-difluorocyclohexyl) methanone (Example 45),
N-cyclohexyl-4- [3- (4-fluorophenyl) -1,2-oxazol-5-yl] piperidine-1-carboxamide (Example 51),
N- (4,4-difluorocyclohexyl) -4- [3- (2,5-difluorophenyl) -1,2-oxazol-5-yl] piperidine-1-carboxamide (Example 77),
4- [4-Chloro-3- (4-fluorophenyl) -1,2-oxazol-5-yl] -N- (tetrahydro-2H-pyran-4-yl) piperidine-1-carboxamide (Example 79) ,
4- [4-Chloro-5- (4-fluorophenyl) -1,2-oxazol-3-yl] -N- (tetrahydro-2H-pyran-4-yl) piperidine-1-carboxamide (Example 124) ,
4- [4-Chloro-5- (3,4-difluorophenyl) -1,2-oxazol-3-yl] -N- (tetrahydro-2H-pyran-4-yl) piperidine-1-carboxamide (Examples) 134), and 4- [5- (4-Chlorophenyl) -1,3-oxazol-2-yl] -N- (tetrahydro-2H-pyran-4-yl) piperidine-1-carboxamide (Example 189).
4-[5-クロロ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]-N-(4,4-ジフルオロシクロヘキシル)ピペリジン-1-カルボキサミド(実施例1)、
{4-[5-クロロ-1-(4-フルオロフェニル)-1H-イミダゾール-4-イル]ピペリジン-1-イル}(4,4-ジフルオロシクロヘキシル)メタノン(実施例45)、
N-シクロヘキシル-4-[3-(4-フルオロフェニル)-1,2-オキサゾール-5-イル]ピペリジン-1-カルボキサミド(実施例51)、
N-(4,4-ジフルオロシクロヘキシル)-4-[3-(2,5-ジフルオロフェニル)-1,2-オキサゾール-5-イル]ピペリジン-1-カルボキサミド(実施例77)、
4-[4-クロロ-3-(4-フルオロフェニル)-1,2-オキサゾール-5-イル]-N-(テトラヒドロ-2H-ピラン-4-イル)ピペリジン-1-カルボキサミド(実施例79)、
4-[4-クロロ-5-(4-フルオロフェニル)-1,2-オキサゾール-3-イル]-N-(テトラヒドロ-2H-ピラン-4-イル)ピペリジン-1-カルボキサミド(実施例124)、
4-[4-クロロ-5-(3,4-ジフルオロフェニル)-1,2-オキサゾール-3-イル]-N-(テトラヒドロ-2H-ピラン-4-イル)ピペリジン-1-カルボキサミド(実施例134)、及び
4-[5-(4-クロロフェニル)-1,3-オキサゾール-2-イル]-N-(テトラヒドロ-2H-ピラン-4-イル)ピペリジン-1-カルボキサミド(実施例189)。 [Item 29] The compound according to item 1 or a pharmaceutically acceptable salt thereof selected from the following compounds:
4- [5-Chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] -N- (4,4-difluorocyclohexyl) piperidine-1-carboxamide (Example 1),
{4- [5-Chloro-1- (4-fluorophenyl) -1H-imidazol-4-yl] piperidin-1-yl} (4,4-difluorocyclohexyl) methanone (Example 45),
N-cyclohexyl-4- [3- (4-fluorophenyl) -1,2-oxazol-5-yl] piperidine-1-carboxamide (Example 51),
N- (4,4-difluorocyclohexyl) -4- [3- (2,5-difluorophenyl) -1,2-oxazol-5-yl] piperidine-1-carboxamide (Example 77),
4- [4-Chloro-3- (4-fluorophenyl) -1,2-oxazol-5-yl] -N- (tetrahydro-2H-pyran-4-yl) piperidine-1-carboxamide (Example 79) ,
4- [4-Chloro-5- (4-fluorophenyl) -1,2-oxazol-3-yl] -N- (tetrahydro-2H-pyran-4-yl) piperidine-1-carboxamide (Example 124) ,
4- [4-Chloro-5- (3,4-difluorophenyl) -1,2-oxazol-3-yl] -N- (tetrahydro-2H-pyran-4-yl) piperidine-1-carboxamide (Examples) 134), and 4- [5- (4-Chlorophenyl) -1,3-oxazol-2-yl] -N- (tetrahydro-2H-pyran-4-yl) piperidine-1-carboxamide (Example 189).
[項30]項1~29のいずれか一項に記載の化合物又はそれらの製薬学的に許容される塩を含有する医薬組成物。
[Item 30] A pharmaceutical composition comprising the compound according to any one of items 1 to 29 or a pharmaceutically acceptable salt thereof.
[項31]項1~29のいずれか一項に記載の化合物又はそれらの製薬学的に許容される塩を有効成分とする、アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患の治療剤及び/又は予防剤。
[Item 31] Treatment of a disease caused by an abnormality in intracellular signal transduction involving acetylcholine, comprising the compound according to any one of items 1 to 29 or a pharmaceutically acceptable salt thereof as an active ingredient Agent and / or preventive agent.
[項32]アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患が、神経疾患、精神疾患又は炎症性疾患である、項31に記載の治療剤及び/又は予防剤。
[Item 32] The therapeutic and / or prophylactic agent according to item 31, wherein the disease caused by abnormality in intracellular signal transduction involving acetylcholine is a neurological disease, a mental disease or an inflammatory disease.
[項32]神経疾患、精神疾患又は炎症性疾患が、統合失調症、アルツハイマー病、ダウン症、注意欠陥・多動性障害又は脳血管アンギオパチーである、項32に記載の治療剤及び/又は予防剤。
[Item 32] The therapeutic and / or prophylactic agent according to item 32, wherein the neurological disease, mental disease or inflammatory disease is schizophrenia, Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder or cerebrovascular angiopathy. .
[項33]CIAS(統合失調症に伴う認知機能障害)、或いは、統合失調症、アルツハイマー病、ダウン症、注意欠陥・多動性障害又は脳血管アンギオパチーにおける、認知障害、軽度認知障害、記憶障害又は学習障害を治療及び/又は予防するための、項30に記載の医薬組成物。
[Claim 33] CIAS (cognitive impairment associated with schizophrenia) or cognitive impairment, mild cognitive impairment, memory impairment in schizophrenia, Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder or cerebrovascular angiopathy or Item 31. A pharmaceutical composition according to Item 30, for treating and / or preventing learning disorders.
[項34]項1~29のいずれか一項に記載の化合物又はそれらの製薬学的に許容される塩と、非定型抗精神病薬から選択される少なくとも1種以上の薬剤とを含有する医薬。
[Item 34] A pharmaceutical comprising the compound according to any one of items 1 to 29 or a pharmaceutically acceptable salt thereof and at least one drug selected from atypical antipsychotics .
[項35]治療が必要な患者に、治療上の有効量の項1~29のいずれか一項に記載の化合物又はその製薬学的に許容される塩を投与することを特徴とする、アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患を治療及び/又は予防するための方法。
[Item 35] Acetylcholine characterized by administering a therapeutically effective amount of the compound according to any one of Items 1 to 29 or a pharmaceutically acceptable salt thereof to a patient in need of treatment. A method for treating and / or preventing a disease caused by an abnormality in intracellular signal transduction associated with a cell.
[項36]アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患の治療剤及び/又は予防剤を製造するための、項1~29のいずれか一項に記載の化合物又はその製薬学的に許容される塩の使用。
[Item 36] The compound according to any one of items 1 to 29 or a pharmacological agent thereof for producing a therapeutic and / or prophylactic agent for a disease caused by an abnormality in intracellular signal transduction involving acetylcholine Use of acceptable salts.
[項37]アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患の治療及び/又は予防に使用するための、項1~29のいずれか一項に記載の化合物又はその製薬学的に許容される塩を含有する医薬組成物。
[Item 37] The compound according to any one of items 1 to 29 or a pharmaceutically acceptable salt thereof for use in the treatment and / or prevention of a disease caused by an abnormality in intracellular signal transduction involving acetylcholine The pharmaceutical composition containing the salt made.
本発明の化合物は、アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患の治療剤及び/又は予防剤として有用である。アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患としては、神経疾患、精神疾患又は炎症性疾患が挙げられる。神経疾患、精神疾患又は炎症性疾患の具体例としては、統合失調症、アルツハイマー病、ダウン症、注意欠陥・多動性障害又は脳血管アンギオパチーが挙げられる。また、本発明の化合物は、(1)CIAS(統合失調症に伴う認知機能障害)、或いは、(2)統合失調症、アルツハイマー病、ダウン症、注意欠陥・多動性障害又は脳血管アンギオパチーにおける、認知障害、軽度認知障害、記憶障害又は学習障害を治療及び/又は予防するために有用である。
The compound of the present invention is useful as a therapeutic and / or prophylactic agent for diseases caused by abnormalities in intracellular signal transduction involving acetylcholine. Examples of the disease caused by abnormal intracellular signal transduction involving acetylcholine include a neurological disease, a mental disease or an inflammatory disease. Specific examples of neurological diseases, psychiatric diseases or inflammatory diseases include schizophrenia, Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder, or cerebrovascular angiopathy. In addition, the compound of the present invention is (1) CIAS (cognitive dysfunction associated with schizophrenia), or (2) schizophrenia, Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder or cerebrovascular angiopathy, Useful for treating and / or preventing cognitive impairment, mild cognitive impairment, memory impairment or learning impairment. *
さらに、本発明の化合物は、前記治療及び/又は予防の目的で、非定型抗精神病薬と併用して用いることもできる。
Furthermore, the compound of the present invention can be used in combination with an atypical antipsychotic agent for the purpose of the treatment and / or prevention.
本発明の化合物は、水和物及び/又は溶媒和物の形で存在することもあるので、式(I)で表される化合物又はその製薬学的に許容される塩の水和物及び/又は溶媒和物もまた本発明の化合物に包含される。
Since the compound of the present invention may exist in the form of a hydrate and / or a solvate, a hydrate of the compound represented by formula (I) or a pharmaceutically acceptable salt thereof and / or Alternatively, solvates are also encompassed by the compounds of the present invention.
式(I)の化合物は、1個又は場合によりそれ以上の不斉炭素原子を有する場合があり、また幾何異性や軸性キラリティを生じることがあるので、数種の立体異性体として存在することがある。本発明においては、これらの立体異性体、それらの混合物及びラセミ体は本発明の式(I)で表される化合物に包含される。
The compound of formula (I) may have one or more asymmetric carbon atoms and may cause geometric isomerism and axial chirality, and therefore exist as several stereoisomers. There is. In the present invention, these stereoisomers, mixtures thereof and racemates are included in the compound represented by the formula (I) of the present invention.
また、一般式(I)で表される化合物のいずれか1つ又は2つ以上の1Hを2H(D)に変換した重水素変換体も一般式(I)で表される化合物に包含される。
In addition, a deuterium converter obtained by converting any one or two or more 1 H of the compound represented by the general formula (I) into 2 H (D) is also included in the compound represented by the general formula (I). Is done.
結晶として得られる一般式(I)で表される化合物及びその製薬学的に許容される塩には、結晶多形が存在する場合があり、本発明の化合物には、あらゆる結晶形のものが含まれる。
Crystalline polymorphisms may exist in the compound represented by the general formula (I) obtained as crystals and pharmaceutically acceptable salts thereof, and the compounds of the present invention may be in any crystalline form. included.
つぎに、本明細書における用語について以下に説明する。
Next, terms used in this specification will be described below.
「アルキル」とは、直鎖状又は分枝鎖状の飽和炭化水素基を意味し、例えば、「C1-4アルキル」又は「C1-6アルキル」とは炭素原子数が1~4又は1~6のアルキルを意味する。「C1-6アルキル」の中で好ましくは、「C1-4アルキル」が挙げられる。その具体例として、「C1-4アルキル」の場合には、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル等が挙げられる。「C1-6アルキル」の場合には、前記に加えて、ぺンチル、イソペンチル、ネオペンチル、ヘキシル等が挙げられる。
“Alkyl” means a linear or branched saturated hydrocarbon group. For example, “C 1-4 alkyl” or “C 1-6 alkyl” has 1 to 4 carbon atoms or Means 1-6 alkyl. Among “C 1-6 alkyl”, “C 1-4 alkyl” is preferable. Specific examples thereof include “C 1-4 alkyl” such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl and the like. In the case of “C 1-6 alkyl”, in addition to the above, pentyl, isopentyl, neopentyl, hexyl and the like can be mentioned.
「シクロアルキル」とは、単環又は多環式飽和炭化水素からなる基を意味し、例えば「C3-10シクロアルキル」とは炭素原子数が3~10の環状アルキルを意味し、一部架橋された構造のもの等が挙げられる。その具体例として、「C3-10シクロアルキル」の場合には、シクロプロピル、シクロブチル、シクロペンチル、シクロへキシル、シクロヘプチル、シクロオクチル、アダマンチル等が挙げられる。
“Cycloalkyl” means a group consisting of monocyclic or polycyclic saturated hydrocarbons. For example, “C 3-10 cycloalkyl” means a cyclic alkyl having 3 to 10 carbon atoms. The thing of the structure of bridge | crosslinking is mentioned. Specific examples thereof include “C 3-10 cycloalkyl” such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, adamantyl and the like.
「アルコキシ」とは、直鎖状又は分枝鎖状の飽和炭化水素基が酸素原子と結合している基であって、当該酸素原子を介して他の部分に結合する基を意味し、例えば、「C1-6アルコキシ」とは炭素原子数が1~6のアルコキシを意味する。その具体例として、「C1-6アルコキシ」の場合には、メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブチルオキシ、ペンチルオキシ、イソペンチルオキシ、ネオペンチルオキシ、ヘキシルオキシ等が挙げられる。
“Alkoxy” means a group in which a linear or branched saturated hydrocarbon group is bonded to an oxygen atom, and is bonded to another moiety via the oxygen atom. “C 1-6 alkoxy” means alkoxy having 1 to 6 carbon atoms. Specific examples of “C 1-6 alkoxy” include methoxy, ethoxy, propoxy, isopropoxy, butyloxy, pentyloxy, isopentyloxy, neopentyloxy, hexyloxy and the like.
「ハロゲン」とは、フッ素原子、塩素原子、臭素原子又はヨウ素原子を意味する。中でも好ましくは、フッ素原子又は塩素原子である。
“Halogen” means a fluorine atom, a chlorine atom, a bromine atom or an iodine atom. Among them, preferred is a fluorine atom or a chlorine atom.
「4~10員の飽和複素環」とは、炭素原子以外に窒素原子、酸素原子及び硫黄原子からなる群から独立して選択される1~2個の原子を含む4~10個の原子で構成される飽和複素環を意味し、好ましくは、単環の4~10員の飽和複素環を意味する。例えば、アゼチジン、ピロリジン、ピペリジン、ピペラジン、モルホリン、ホモピペリジン、テトラヒドロフラン、テトラヒドロピラン、オキセタン等が挙げられる。好ましい4~10員の飽和複素環としては、テトラヒドロフラン、テトラヒドロピラン、オキセタンが挙げられる。さらに好ましくは、テトラヒドロピランである。
The “4- to 10-membered saturated heterocyclic ring” means 4 to 10 atoms including 1 to 2 atoms independently selected from the group consisting of a nitrogen atom, an oxygen atom and a sulfur atom in addition to a carbon atom. Means a saturated heterocyclic ring, preferably a monocyclic 4- to 10-membered saturated heterocyclic ring. Examples thereof include azetidine, pyrrolidine, piperidine, piperazine, morpholine, homopiperidine, tetrahydrofuran, tetrahydropyran, oxetane and the like. Preferred 4- to 10-membered saturated heterocycles include tetrahydrofuran, tetrahydropyran, and oxetane. More preferred is tetrahydropyran.
「4~10員の含窒素飽和複素環」とは、炭素原子以外に1~2個の窒素原子及び、酸素原子及び硫黄原子からなる群から独立して選択される0~2個の原子を含む4~10個の原子で構成される飽和複素環を意味する。例えば、アゼチジン、ピロリジン、ピペリジン、ピペラジン、ホモピペリジン等が挙げられる。好ましい4~10員の含窒素飽和複素環としては、炭素原子以外に1個の窒素原子を含む4~10個の原子で構成される飽和複素環が挙げられる。例えば、アゼチジン、ピロリジン、ピペリジン等が挙げられる。
The “4- to 10-membered nitrogen-containing saturated heterocycle” means 0 to 2 atoms independently selected from the group consisting of 1 to 2 nitrogen atoms, oxygen atoms and sulfur atoms in addition to carbon atoms. A saturated heterocyclic ring composed of 4 to 10 atoms is included. For example, azetidine, pyrrolidine, piperidine, piperazine, homopiperidine and the like can be mentioned. Preferred examples of the 4 to 10-membered nitrogen-containing saturated heterocyclic ring include saturated heterocyclic rings composed of 4 to 10 atoms containing one nitrogen atom in addition to the carbon atom. For example, azetidine, pyrrolidine, piperidine and the like can be mentioned.
式(I)で表される本発明の化合物の中でも、A、B、W、X-Y-Z、R1、R2A、R2B、R2C、R2D、R3A、R3B、R4A、R4B、R5及びR6で、好ましいものは以下のとおりであるが、本発明の技術的範囲は下記に挙げる化合物の範囲に限定されるものではない。
Among the compounds of the present invention represented by the formula (I), A, B, W, XYZ, R 1 , R 2A , R 2B , R 2C , R 2D , R 3A , R 3B , R 4A , R 4B , R 5 and R 6 are preferably as follows, but the technical scope of the present invention is not limited to the scope of the compounds listed below.
X-Y-Zとして好ましくは、N-CO-NR3AR3B、N-COR4A、CR5-CO-NR3AR3B又はCR5-NR6-COR4Bが挙げられる。
より好ましくは、N-CO-NR3AR3B、N-COR4A又はCR5-NR6-COR4Bが挙げられる。
さらに好ましくは、N-CO-NR3AR3B又はN-COR4Aが挙げられる。 X—Y—Z is preferably N—CO—NR 3A R 3B , N—COR 4A , CR 5 —CO—NR 3A R 3B or CR 5 —NR 6 —COR 4B .
More preferably, N—CO—NR 3A R 3B , N—COR 4A or CR 5 —NR 6 —COR 4B may be mentioned.
More preferred is N—CO—NR 3A R 3B or N—COR 4A .
より好ましくは、N-CO-NR3AR3B、N-COR4A又はCR5-NR6-COR4Bが挙げられる。
さらに好ましくは、N-CO-NR3AR3B又はN-COR4Aが挙げられる。 X—Y—Z is preferably N—CO—NR 3A R 3B , N—COR 4A , CR 5 —CO—NR 3A R 3B or CR 5 —NR 6 —COR 4B .
More preferably, N—CO—NR 3A R 3B , N—COR 4A or CR 5 —NR 6 —COR 4B may be mentioned.
More preferred is N—CO—NR 3A R 3B or N—COR 4A .
Aとして好ましくは、フェニル又はピリジル(該フェニル及び該ピリジルは、それぞれハロゲン、水酸基、1から5個のフッ素で置換されていてもよいC1-6アルキル、及び1から5個のフッ素で置換されていてもよいC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)が挙げられる。
より好ましくは、フェニル又はピリジル(該フェニル及び該ピリジルは、それぞれフッ素、塩素、1から5個のフッ素で置換されていてもよいC1-6アルキル、及び1から5個のフッ素で置換されていてもよいC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)が挙げられる。
さらに好ましくは、フッ素、塩素、1~3個のフッ素で置換されていてもよいC1-6アルキル及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいフェニルが挙げられる。
もっとも好ましくは、フッ素及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいフェニルが挙げられる。 A is preferably phenyl or pyridyl (the phenyl and pyridyl are each substituted with halogen, hydroxyl, C 1-6 alkyl optionally substituted with 1 to 5 fluorine, and 1 to 5 fluorine. And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy.
More preferably, phenyl or pyridyl (the phenyl and the pyridyl are each substituted with fluorine, chlorine, C 1-6 alkyl optionally substituted with 1 to 5 fluorine, and 1 to 5 fluorine. And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy.
More preferably, it is 1 to 5 substituents independently selected from the group consisting of fluorine, chlorine, C 1-6 alkyl optionally substituted with 1 to 3 fluorine and C 1-6 alkoxy. Examples include optionally substituted phenyl.
Most preferably, mention may be made of phenyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of fluorine and C 1-6 alkoxy.
より好ましくは、フェニル又はピリジル(該フェニル及び該ピリジルは、それぞれフッ素、塩素、1から5個のフッ素で置換されていてもよいC1-6アルキル、及び1から5個のフッ素で置換されていてもよいC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)が挙げられる。
さらに好ましくは、フッ素、塩素、1~3個のフッ素で置換されていてもよいC1-6アルキル及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいフェニルが挙げられる。
もっとも好ましくは、フッ素及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいフェニルが挙げられる。 A is preferably phenyl or pyridyl (the phenyl and pyridyl are each substituted with halogen, hydroxyl, C 1-6 alkyl optionally substituted with 1 to 5 fluorine, and 1 to 5 fluorine. And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy.
More preferably, phenyl or pyridyl (the phenyl and the pyridyl are each substituted with fluorine, chlorine, C 1-6 alkyl optionally substituted with 1 to 5 fluorine, and 1 to 5 fluorine. And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy.
More preferably, it is 1 to 5 substituents independently selected from the group consisting of fluorine, chlorine, C 1-6 alkyl optionally substituted with 1 to 3 fluorine and C 1-6 alkoxy. Examples include optionally substituted phenyl.
Most preferably, mention may be made of phenyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of fluorine and C 1-6 alkoxy.
Bとして好ましくは、B-1、B-2、B-3、B-4又はB-5が挙げられる。より好ましくは、B-1、B-4又はB-5が挙げられる。さらに好ましくは、B-1又はB-4が挙げられる。
B is preferably B-1, B-2, B-3, B-4 or B-5. More preferred is B-1, B-4 or B-5. More preferred is B-1 or B-4.
Wとして好ましくは、酸素原子又は硫黄原子が挙げられる。より好ましくは、酸素原子が挙げられる。
W is preferably an oxygen atom or a sulfur atom. More preferably, an oxygen atom is mentioned.
R1として好ましくは、水素原子;ハロゲン;シアノ;又はハロゲン、水酸基、1から5個のフッ素で置換されていてもよいC3-10シクロアルキル、C1-6アルコキシ及び4~10員の飽和複素環からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキルが挙げられる。より好ましくは、水素原子、ハロゲン又はシアノが挙げられる。さらに好ましくは、水素原子、塩素又はシアノが挙げられる。もっとも好ましくは、水素原子又は塩素が挙げられる。
R 1 is preferably a hydrogen atom; halogen; cyano; or halogen, hydroxyl, C 3-10 cycloalkyl optionally substituted with 1 to 5 fluorines, C 1-6 alkoxy, and 4 to 10 membered saturation And C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of heterocycles. More preferably, a hydrogen atom, halogen, or cyano is mentioned. More preferably, a hydrogen atom, chlorine, or cyano is mentioned. Most preferably, a hydrogen atom or chlorine is mentioned.
R2A、R2B、R2C、R2D及びR5として好ましくは、同一又は異なって、水素原子、フッ素、水酸基又はC1-6アルキルが挙げられる。ここにおいて、R2A、R2B、R2C、R2D及びR5のいずれかの2つが1から5個のフッ素で置換されていてもよいC1-6アルキルのとき、2個のアルキルが一緒になって該アルキルが結合している環と別の環を形成していてもよい。
R2A、R2B、R2C、R2D及びR5としてより好ましくは、同一又は異なって、水素原子、又はC1-6アルキルが挙げられる。ここにおいて、R2A、R2B、R2C、R2D及びR5のいずれかの2つが、C1-6アルキルのとき、2個のアルキルが一緒になって該アルキルが結合している環と別の環を形成していてもよい。
R2A、R2B、R2C、R2D及びR5としてさらに好ましくは、水素原子が挙げられる。 R 2A , R 2B , R 2C , R 2D and R 5 are preferably the same or different and include a hydrogen atom, fluorine, a hydroxyl group or C 1-6 alkyl. Here, when any two of R 2A , R 2B , R 2C , R 2D and R 5 are C 1-6 alkyl optionally substituted with 1 to 5 fluorines, the two alkyls together To form a ring different from the ring to which the alkyl is bonded.
R 2A , R 2B , R 2C , R 2D and R 5 are more preferably the same or different and include a hydrogen atom or C 1-6 alkyl. Here, when any two of R 2A , R 2B , R 2C , R 2D and R 5 are C 1-6 alkyl, the two alkyls are joined together to form a ring to which the alkyl is bonded Another ring may be formed.
R 2A , R 2B , R 2C , R 2D and R 5 are more preferably a hydrogen atom.
R2A、R2B、R2C、R2D及びR5としてより好ましくは、同一又は異なって、水素原子、又はC1-6アルキルが挙げられる。ここにおいて、R2A、R2B、R2C、R2D及びR5のいずれかの2つが、C1-6アルキルのとき、2個のアルキルが一緒になって該アルキルが結合している環と別の環を形成していてもよい。
R2A、R2B、R2C、R2D及びR5としてさらに好ましくは、水素原子が挙げられる。 R 2A , R 2B , R 2C , R 2D and R 5 are preferably the same or different and include a hydrogen atom, fluorine, a hydroxyl group or C 1-6 alkyl. Here, when any two of R 2A , R 2B , R 2C , R 2D and R 5 are C 1-6 alkyl optionally substituted with 1 to 5 fluorines, the two alkyls together To form a ring different from the ring to which the alkyl is bonded.
R 2A , R 2B , R 2C , R 2D and R 5 are more preferably the same or different and include a hydrogen atom or C 1-6 alkyl. Here, when any two of R 2A , R 2B , R 2C , R 2D and R 5 are C 1-6 alkyl, the two alkyls are joined together to form a ring to which the alkyl is bonded Another ring may be formed.
R 2A , R 2B , R 2C , R 2D and R 5 are more preferably a hydrogen atom.
R3A、R3B及びR4Aとして好ましくは、同一又は異なって、ハロゲン、水酸基、C1-6アルコキシ、C3-10シクロアルキル及び4~10員の飽和複素環(ただし、環上にカルボニル基を有しない)からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;ハロゲン、水酸基、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;C1-6アルキルで置換されていてもよい4~10員の飽和複素環;又は水素原子が挙げられ、ここにおいて、(1)R4Aは水素原子ではなく、(2)R3A及びR3Bは同時に水素原子ではなく、(3)BがB-3であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aはメチルではなく、(4)Aが無置換のフェニルであり、BがB-4であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aは1~5個のフッ素で置換されたC3アルキルではない。
R3A、R3B及びR4Aとしてより好ましくは、同一又は異なって、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子が挙げられ、ここにおいて、(1)R4Aは水素原子ではなく、(2)R3A及びR3Bは同時に水素原子ではなく、(3)BがB-3であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aはメチルではなく、(4)Aが無置換のフェニルであり、BがB-4であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aは1~5個のフッ素で置換されたC3アルキルではない。
R3A、R3B及びR4Aとしてさらに好ましくは、同一又は異なって、フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子が挙げられ、ここにおいて、R3A及びR3Bは同時に水素原子ではなく、R4Aは水素原子ではない。
R3A及びR4Aとしてもっとも好ましくは、同一又は異なって、フッ素及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル又は4~10員の飽和複素環が挙げられる。
R3Bとしてもっとも好ましくは、水素原子が挙げられる。
なお、本明細書において、「R4Aは1~5個のフッ素で置換されたC3アルキルではない」とは、「R4Aは1~5個のフッ素で置換され、当該フッ素以外の他の置換基で置換されていないC3アルキルではない」ことを意味する。 R 3A , R 3B and R 4A are preferably the same or different and are halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4- to 10-membered saturated heterocyclic ring (provided that a carbonyl group is present on the ring). C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of: a halogen, a hydroxyl group, C 1-6 alkoxy and C 1-6 alkyl C 3-10 cycloalkyl optionally substituted with 1-5 substituents independently selected from the group consisting of: 4-10 membered saturated heterocycle optionally substituted with C 1-6 alkyl Or a hydrogen atom, wherein (1) R 4A is not a hydrogen atom, (2) R 3A and R 3B are not simultaneously a hydrogen atom, (3) B is B-3, and W is An oxygen atom, X- When -Z is N-COR 4A is, R 4A is not methyl, (4) A a is unsubstituted phenyl, B is B-4, W is an oxygen atom, X-Y- When Z is N—COR 4A , R 4A is not C 3 alkyl substituted with 1 to 5 fluorines.
More preferably, R 3A , R 3B and R 4A are the same or different and are 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 3-10 cycloalkyl C 1-6 alkyl optionally substituted with 1-6 alkyls independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl C 3-10 cycloalkyl; a 4-10 membered saturated heterocycle; or a hydrogen atom, where (1) R 4A is not a hydrogen atom and (2) R 3A and R 3B are simultaneously a hydrogen atom And (3) when B is B-3, W is an oxygen atom, and XYZ is N—COR 4A , R 4A is not methyl and (4) A is unsubstituted Phenyl, B is B-4, W is An atom, X-Y-Z is and when is N-COR 4A, R 4A is not C 3 alkyl substituted with 1-5 fluorine.
More preferably, R 3A , R 3B and R 4A are the same or different and substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl which may be substituted ; a 4 to 10 membered saturated heterocyclic ring; or a hydrogen atom, wherein R 3A and R 3B are not simultaneously a hydrogen atom and R 4A is not a hydrogen atom .
Most preferably the R 3A and R 4A, the same or different, fluorine and C 1-6 optionally substituted with 1-5 substituents independently selected from the group consisting of alkoxy C 3-10 Examples include cycloalkyl or a 4- to 10-membered saturated heterocyclic ring.
Most preferably, R 3B includes a hydrogen atom.
In the present specification, “R 4A is not C 3 alkyl substituted with 1 to 5 fluorines” means that “R 4A is substituted with 1 to 5 fluorines, It is not C 3 alkyl not substituted with a substituent ”.
R3A、R3B及びR4Aとしてより好ましくは、同一又は異なって、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子が挙げられ、ここにおいて、(1)R4Aは水素原子ではなく、(2)R3A及びR3Bは同時に水素原子ではなく、(3)BがB-3であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aはメチルではなく、(4)Aが無置換のフェニルであり、BがB-4であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aは1~5個のフッ素で置換されたC3アルキルではない。
R3A、R3B及びR4Aとしてさらに好ましくは、同一又は異なって、フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子が挙げられ、ここにおいて、R3A及びR3Bは同時に水素原子ではなく、R4Aは水素原子ではない。
R3A及びR4Aとしてもっとも好ましくは、同一又は異なって、フッ素及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル又は4~10員の飽和複素環が挙げられる。
R3Bとしてもっとも好ましくは、水素原子が挙げられる。
なお、本明細書において、「R4Aは1~5個のフッ素で置換されたC3アルキルではない」とは、「R4Aは1~5個のフッ素で置換され、当該フッ素以外の他の置換基で置換されていないC3アルキルではない」ことを意味する。 R 3A , R 3B and R 4A are preferably the same or different and are halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4- to 10-membered saturated heterocyclic ring (provided that a carbonyl group is present on the ring). C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of: a halogen, a hydroxyl group, C 1-6 alkoxy and C 1-6 alkyl C 3-10 cycloalkyl optionally substituted with 1-5 substituents independently selected from the group consisting of: 4-10 membered saturated heterocycle optionally substituted with C 1-6 alkyl Or a hydrogen atom, wherein (1) R 4A is not a hydrogen atom, (2) R 3A and R 3B are not simultaneously a hydrogen atom, (3) B is B-3, and W is An oxygen atom, X- When -Z is N-COR 4A is, R 4A is not methyl, (4) A a is unsubstituted phenyl, B is B-4, W is an oxygen atom, X-Y- When Z is N—COR 4A , R 4A is not C 3 alkyl substituted with 1 to 5 fluorines.
More preferably, R 3A , R 3B and R 4A are the same or different and are 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 3-10 cycloalkyl C 1-6 alkyl optionally substituted with 1-6 alkyls independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl C 3-10 cycloalkyl; a 4-10 membered saturated heterocycle; or a hydrogen atom, where (1) R 4A is not a hydrogen atom and (2) R 3A and R 3B are simultaneously a hydrogen atom And (3) when B is B-3, W is an oxygen atom, and XYZ is N—COR 4A , R 4A is not methyl and (4) A is unsubstituted Phenyl, B is B-4, W is An atom, X-Y-Z is and when is N-COR 4A, R 4A is not C 3 alkyl substituted with 1-5 fluorine.
More preferably, R 3A , R 3B and R 4A are the same or different and substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl which may be substituted ; a 4 to 10 membered saturated heterocyclic ring; or a hydrogen atom, wherein R 3A and R 3B are not simultaneously a hydrogen atom and R 4A is not a hydrogen atom .
Most preferably the R 3A and R 4A, the same or different, fluorine and C 1-6 optionally substituted with 1-5 substituents independently selected from the group consisting of alkoxy C 3-10 Examples include cycloalkyl or a 4- to 10-membered saturated heterocyclic ring.
Most preferably, R 3B includes a hydrogen atom.
In the present specification, “R 4A is not C 3 alkyl substituted with 1 to 5 fluorines” means that “R 4A is substituted with 1 to 5 fluorines, It is not C 3 alkyl not substituted with a substituent ”.
R4Bとして好ましくは、ハロゲン、水酸基、C1-6アルコキシ、C3-10シクロアルキル及びカルボニル基で置換されていない4~10員の飽和複素環からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;ハロゲン、水酸基、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;C1-6アルキルで置換されていてもよい4~10員の飽和複素環が挙げられる。
より好ましくは、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;又は4~10員の飽和複素環が挙げられる。
さらに好ましくは、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキルが挙げられる。
もっとも好ましくは、1~5個のフッ素で置換されていてもよいC1-6アルキルが挙げられる。 R 4B is preferably selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4-10 membered saturated heterocyclic ring not substituted with a carbonyl group. C 1-6 alkyl optionally substituted with 5 substituents; 1-5 substituents independently selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy and C 1-6 alkyl C 3-10 cycloalkyl which may be substituted with a 4-10 membered saturated heterocyclic ring which may be substituted with C 1-6 alkyl.
More preferably fluorine, C 1-6 alkoxy, and C 3-10 1 ~ 5 amino optionally substituted with a substituent C 1-6 alkyl independently selected from the group consisting of cycloalkyl; fluorine C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; or 4-10 membered saturation Heterocycles are mentioned.
More preferable examples include C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy, and C 3-10 cycloalkyl. It is done.
Most preferred is C 1-6 alkyl optionally substituted with 1 to 5 fluorines.
より好ましくは、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;又は4~10員の飽和複素環が挙げられる。
さらに好ましくは、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキルが挙げられる。
もっとも好ましくは、1~5個のフッ素で置換されていてもよいC1-6アルキルが挙げられる。 R 4B is preferably selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4-10 membered saturated heterocyclic ring not substituted with a carbonyl group. C 1-6 alkyl optionally substituted with 5 substituents; 1-5 substituents independently selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy and C 1-6 alkyl C 3-10 cycloalkyl which may be substituted with a 4-10 membered saturated heterocyclic ring which may be substituted with C 1-6 alkyl.
More preferably fluorine, C 1-6 alkoxy, and C 3-10 1 ~ 5 amino optionally substituted with a substituent C 1-6 alkyl independently selected from the group consisting of cycloalkyl; fluorine C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; or 4-10 membered saturation Heterocycles are mentioned.
More preferable examples include C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy, and C 3-10 cycloalkyl. It is done.
Most preferred is C 1-6 alkyl optionally substituted with 1 to 5 fluorines.
R6として好ましくは、ハロゲン、水酸基、C1-6アルコキシ、C3-10シクロアルキル及びカルボニル基で置換されていない4~10員の飽和複素環からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;ハロゲン、水酸基、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;C1-6アルキルで置換されていてもよい4~10員の飽和複素環;又は水素原子が挙げられる。
より好ましくは、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子が挙げられる。
さらに好ましくは、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;又は水素原子が挙げられる。
もっとも好ましくは、水素原子が挙げられる。 R 6 is preferably selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4-10 membered saturated heterocyclic ring not substituted with a carbonyl group. C 1-6 alkyl optionally substituted with 5 substituents; 1-5 substituents independently selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl which may be substituted with a 4 to 10-membered saturated heterocyclic ring which may be substituted with a C 1-6 alkyl; or a hydrogen atom.
More preferably fluorine, C 1-6 alkoxy, and C 3-10 1 ~ 5 amino optionally substituted with a substituent C 1-6 alkyl independently selected from the group consisting of cycloalkyl; fluorine C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; A ring; or a hydrogen atom.
More preferably, fluorine, C 1-6 alkoxy, and C 3-10 1 ~ 5 amino C 1-6 alkyl optionally substituted with a substituent selected independently from the group consisting of cycloalkyl; or A hydrogen atom is mentioned.
Most preferably, a hydrogen atom is mentioned.
より好ましくは、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子が挙げられる。
さらに好ましくは、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;又は水素原子が挙げられる。
もっとも好ましくは、水素原子が挙げられる。 R 6 is preferably selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4-10 membered saturated heterocyclic ring not substituted with a carbonyl group. C 1-6 alkyl optionally substituted with 5 substituents; 1-5 substituents independently selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl which may be substituted with a 4 to 10-membered saturated heterocyclic ring which may be substituted with a C 1-6 alkyl; or a hydrogen atom.
More preferably fluorine, C 1-6 alkoxy, and C 3-10 1 ~ 5 amino optionally substituted with a substituent C 1-6 alkyl independently selected from the group consisting of cycloalkyl; fluorine C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; A ring; or a hydrogen atom.
More preferably, fluorine, C 1-6 alkoxy, and C 3-10 1 ~ 5 amino C 1-6 alkyl optionally substituted with a substituent selected independently from the group consisting of cycloalkyl; or A hydrogen atom is mentioned.
Most preferably, a hydrogen atom is mentioned.
式(I)で表される化合物のうちで、好ましい化合物としては、以下のような化合物またはその製薬学的に許容される塩が挙げられる。
Among the compounds represented by the formula (I), preferred compounds include the following compounds or pharmaceutically acceptable salts thereof.
好ましい態様としては、以下の(A)が挙げられる。
(A)
X-Y-Zが、N-CO-NR3AR3B、N-COR4A、CR5-CO-NR3AR3B又はCR5-NR6-COR4Bであり、
Aが、フェニル又はピリジル(該フェニル及び該ピリジルはそれぞれハロゲン、水酸基、1から5個のフッ素で置換されていてもよいC1-6アルキル、及び1から5個のフッ素で置換されていてもよいC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)であり、
Bが、B-1、B-2、B-3、B-4又はB-5であり、
Wが、酸素原子又は硫黄原子であり、
R1が、水素原子;ハロゲン;シアノ;又はハロゲン、水酸基、1から5個のフッ素で置換されていてもよいC3-10シクロアルキル、C1-6アルコキシ及び4~10員の飽和複素環からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキルであり、
R2A、R2B、R2C、R2D及びR5が、同一又は異なって、水素原子、フッ素、水酸基又はC1-6アルキルであり、ここにおいて、R2A、R2B、R2C、R2D及びR5のいずれかの2つが1から5個のフッ素で置換されていてもよいC1-6アルキルのとき、2個のアルキルが一緒になって該アルキルが結合している環と別の環を形成していてもよく、
R3A、R3B及びR4Aが、同一又は異なって、ハロゲン、水酸基、C1-6アルコキシ、C3-10シクロアルキル及び4~10員の飽和複素環(ただし、環上にカルボニル基を有しない)からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;ハロゲン、水酸基、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;C1-6アルキルで置換されていてもよい4~10員の飽和複素環;又は水素原子であり、ここにおいて、(1)R4Aは水素原子ではなく、(2)R3A及びR3Bは同時に水素原子ではなく、(3)BがB-3であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aはメチルではなく、(4)Aが無置換のフェニルであり、BがB-4であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aは1~5個のフッ素で置換されたC3アルキルではなく、
R4Bが、ハロゲン、水酸基、C1-6アルコキシ、C3-10シクロアルキル及びカルボニル基で置換されていない4~10員の飽和複素環からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;ハロゲン、水酸基、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;又はC1-6アルキルで置換されていてもよい4~10員の飽和複素環であり、
R6が、ハロゲン、水酸基、C1-6アルコキシ、C3-10シクロアルキル及びカルボニル基で置換されていない4~10員の飽和複素環からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;ハロゲン、水酸基、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;C1-6アルキルで置換されていてもよい4~10員の飽和複素環;又は水素原子である、
式(I)の化合物又はその製薬学的に許容される塩。 The following (A) is mentioned as a preferable aspect.
(A)
XYZ is N—CO—NR 3A R 3B , N—COR 4A , CR 5 —CO—NR 3A R 3B or CR 5 —NR 6 —COR 4B ;
A is phenyl or pyridyl (the phenyl and the pyridyl may each be substituted with halogen, hydroxyl group, C 1-6 alkyl optionally substituted with 1 to 5 fluorine, and 1 to 5 fluorine; And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy),
B is B-1, B-2, B-3, B-4 or B-5,
W is an oxygen atom or a sulfur atom,
R 1 is a hydrogen atom; halogen; cyano; or halogen, hydroxyl, C 3-10 cycloalkyl optionally substituted with 1 to 5 fluorines, C 1-6 alkoxy, and a 4- to 10-membered saturated heterocyclic ring C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of:
R 2A , R 2B , R 2C , R 2D and R 5 are the same or different and are a hydrogen atom, fluorine, hydroxyl group or C 1-6 alkyl, wherein R 2A , R 2B , R 2C , R 2D And any two of R 5 are C 1-6 alkyl optionally substituted with 1 to 5 fluorines, and the two alkyls together are different from the ring to which the alkyl is bonded. May form a ring,
R 3A , R 3B and R 4A are the same or different and are halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4- to 10-membered saturated heterocyclic ring (provided that a carbonyl group is present on the ring). C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of: a group consisting of halogen, hydroxyl, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from: a 4-10 membered saturated heterocycle optionally substituted with C 1-6 alkyl; or (1) R 4A is not a hydrogen atom, (2) R 3A and R 3B are not simultaneously hydrogen atoms, (3) B is B-3, and W is an oxygen atom. There, X-Y-Z is N-COR When an A is, R 4A is not methyl, (4) A a is unsubstituted phenyl, B is B-4, W is an oxygen atom, X-Y-Z is N-COR 4A When R 4A is not C 3 alkyl substituted with 1 to 5 fluorines,
1 to 5 R 4B independently selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4-10 membered saturated heterocyclic ring not substituted with a carbonyl group substituted halogen, hydroxyl, with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; the optionally substituted with a substituent C 1-6 alkyl An optionally substituted C 3-10 cycloalkyl; or a 4-10 membered saturated heterocycle optionally substituted with C 1-6 alkyl;
1 to 5 R 6 independently selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4-10 membered saturated heterocyclic ring not substituted with a carbonyl group substituted halogen, hydroxyl, with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; the optionally substituted with a substituent C 1-6 alkyl A C 3-10 cycloalkyl which may be substituted; a 4-10 membered saturated heterocyclic ring optionally substituted with C 1-6 alkyl; or a hydrogen atom,
A compound of formula (I) or a pharmaceutically acceptable salt thereof.
(A)
X-Y-Zが、N-CO-NR3AR3B、N-COR4A、CR5-CO-NR3AR3B又はCR5-NR6-COR4Bであり、
Aが、フェニル又はピリジル(該フェニル及び該ピリジルはそれぞれハロゲン、水酸基、1から5個のフッ素で置換されていてもよいC1-6アルキル、及び1から5個のフッ素で置換されていてもよいC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)であり、
Bが、B-1、B-2、B-3、B-4又はB-5であり、
Wが、酸素原子又は硫黄原子であり、
R1が、水素原子;ハロゲン;シアノ;又はハロゲン、水酸基、1から5個のフッ素で置換されていてもよいC3-10シクロアルキル、C1-6アルコキシ及び4~10員の飽和複素環からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキルであり、
R2A、R2B、R2C、R2D及びR5が、同一又は異なって、水素原子、フッ素、水酸基又はC1-6アルキルであり、ここにおいて、R2A、R2B、R2C、R2D及びR5のいずれかの2つが1から5個のフッ素で置換されていてもよいC1-6アルキルのとき、2個のアルキルが一緒になって該アルキルが結合している環と別の環を形成していてもよく、
R3A、R3B及びR4Aが、同一又は異なって、ハロゲン、水酸基、C1-6アルコキシ、C3-10シクロアルキル及び4~10員の飽和複素環(ただし、環上にカルボニル基を有しない)からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;ハロゲン、水酸基、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;C1-6アルキルで置換されていてもよい4~10員の飽和複素環;又は水素原子であり、ここにおいて、(1)R4Aは水素原子ではなく、(2)R3A及びR3Bは同時に水素原子ではなく、(3)BがB-3であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aはメチルではなく、(4)Aが無置換のフェニルであり、BがB-4であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aは1~5個のフッ素で置換されたC3アルキルではなく、
R4Bが、ハロゲン、水酸基、C1-6アルコキシ、C3-10シクロアルキル及びカルボニル基で置換されていない4~10員の飽和複素環からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;ハロゲン、水酸基、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;又はC1-6アルキルで置換されていてもよい4~10員の飽和複素環であり、
R6が、ハロゲン、水酸基、C1-6アルコキシ、C3-10シクロアルキル及びカルボニル基で置換されていない4~10員の飽和複素環からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;ハロゲン、水酸基、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;C1-6アルキルで置換されていてもよい4~10員の飽和複素環;又は水素原子である、
式(I)の化合物又はその製薬学的に許容される塩。 The following (A) is mentioned as a preferable aspect.
(A)
XYZ is N—CO—NR 3A R 3B , N—COR 4A , CR 5 —CO—NR 3A R 3B or CR 5 —NR 6 —COR 4B ;
A is phenyl or pyridyl (the phenyl and the pyridyl may each be substituted with halogen, hydroxyl group, C 1-6 alkyl optionally substituted with 1 to 5 fluorine, and 1 to 5 fluorine; And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy),
B is B-1, B-2, B-3, B-4 or B-5,
W is an oxygen atom or a sulfur atom,
R 1 is a hydrogen atom; halogen; cyano; or halogen, hydroxyl, C 3-10 cycloalkyl optionally substituted with 1 to 5 fluorines, C 1-6 alkoxy, and a 4- to 10-membered saturated heterocyclic ring C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of:
R 2A , R 2B , R 2C , R 2D and R 5 are the same or different and are a hydrogen atom, fluorine, hydroxyl group or C 1-6 alkyl, wherein R 2A , R 2B , R 2C , R 2D And any two of R 5 are C 1-6 alkyl optionally substituted with 1 to 5 fluorines, and the two alkyls together are different from the ring to which the alkyl is bonded. May form a ring,
R 3A , R 3B and R 4A are the same or different and are halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4- to 10-membered saturated heterocyclic ring (provided that a carbonyl group is present on the ring). C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of: a group consisting of halogen, hydroxyl, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from: a 4-10 membered saturated heterocycle optionally substituted with C 1-6 alkyl; or (1) R 4A is not a hydrogen atom, (2) R 3A and R 3B are not simultaneously hydrogen atoms, (3) B is B-3, and W is an oxygen atom. There, X-Y-Z is N-COR When an A is, R 4A is not methyl, (4) A a is unsubstituted phenyl, B is B-4, W is an oxygen atom, X-Y-Z is N-COR 4A When R 4A is not C 3 alkyl substituted with 1 to 5 fluorines,
1 to 5 R 4B independently selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4-10 membered saturated heterocyclic ring not substituted with a carbonyl group substituted halogen, hydroxyl, with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; the optionally substituted with a substituent C 1-6 alkyl An optionally substituted C 3-10 cycloalkyl; or a 4-10 membered saturated heterocycle optionally substituted with C 1-6 alkyl;
1 to 5 R 6 independently selected from the group consisting of halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4-10 membered saturated heterocyclic ring not substituted with a carbonyl group substituted halogen, hydroxyl, with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; the optionally substituted with a substituent C 1-6 alkyl A C 3-10 cycloalkyl which may be substituted; a 4-10 membered saturated heterocyclic ring optionally substituted with C 1-6 alkyl; or a hydrogen atom,
A compound of formula (I) or a pharmaceutically acceptable salt thereof.
より好ましい態様としては、以下の(B)が挙げられる。
(B)
X-Y-Zが、N-CO-NR3AR3B、N-COR4A又はCR5-NR6-COR4Bであり、
Aが、フェニル又はピリジル(該フェニル及び該ピリジルはそれぞれフッ素、塩素、1から5個のフッ素で置換されていてもよいC1-6アルキル、1から5個のフッ素で置換されていてもよいC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)であり、
Bが、B-1、B-2、B-3、B-4又はB-5であり、
Wが、酸素原子又は硫黄原子であり、
R1が、水素原子、ハロゲン又はシアノであり、
R2A、R2B、R2C、R2D及びR5が、同一又は異なって、水素原子、又はC1-6アルキルであり、ここにおいて、R2A、R2B、R2C、R2D及びR5のいずれかの2つがC1-6アルキルのとき、2個のアルキルが一緒になって該アルキルが結合している環と別の環を形成していてもよく、
R3A、R3B及びR4Aが、同一又は異なって、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子であり、ここにおいて、(1)R4Aは水素原子ではなく、(2)R3A及びR3Bは同時に水素原子ではなく、(3)BがB-3であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aはメチルではなく、(4)Aが無置換のフェニルであり、BがB-4であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aは1~5個のフッ素で置換されたC3アルキルではなく、
R4Bが、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;又は4~10員の飽和複素環であり、
R6が、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子である、
式(I)の化合物又はその製薬学的に許容される塩。 The following (B) is mentioned as a more preferable aspect.
(B)
XYZ is N—CO—NR 3A R 3B , N—COR 4A or CR 5 —NR 6 —COR 4B ;
A is phenyl or pyridyl (each said phenyl and said pyridyl fluorine, chlorine, from one to five fluorines optionally substituted C 1-6 alkyl, optionally substituted with 1-5 fluorine Optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy),
B is B-1, B-2, B-3, B-4 or B-5,
W is an oxygen atom or a sulfur atom,
R 1 is a hydrogen atom, halogen or cyano,
R 2A , R 2B , R 2C , R 2D and R 5 are the same or different and are a hydrogen atom or C 1-6 alkyl, wherein R 2A , R 2B , R 2C , R 2D and R 5 Any two of the above are C 1-6 alkyl, the two alkyls may be combined to form a ring with the ring to which the alkyl is attached;
R 3A , R 3B and R 4A are the same or different and are substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy, and C 3-10 cycloalkyl. which may be C 1-6 alkyl; fluorine, C 1-6 alkoxy and C 1-6 independently from the group consisting of alkyl optionally substituted with 1 to 5 substituents selected C 3- 10 cycloalkyl; 4 to 10 membered saturated heterocycle; or a hydrogen atom, wherein (1) R 4A is not a hydrogen atom, and (2) R 3A and R 3B are not simultaneously a hydrogen atom, (3 ) When B is B-3, W is an oxygen atom and XYZ is N-COR 4A , R 4A is not methyl and (4) A is unsubstituted phenyl; B is B-4, W is an oxygen atom, XY When Z is N-COR 4A is, R 4A is not C 3 alkyl substituted with 1-5 fluorine,
R 4B is selected from the group consisting of fluorine, C 1-6 alkoxy, and C 3-10 1 ~ 5 amino optionally substituted with a substituent C 1-6 alkyl independently selected from the group consisting of cycloalkyl; fluorine C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; or 4-10 membered saturation A heterocycle,
R 6 is fluorine, C 1-6 alkoxy, and C 3-10 1 ~ 5 amino optionally substituted with a substituent C 1-6 alkyl independently selected from the group consisting of cycloalkyl; fluorine C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; A ring; or a hydrogen atom,
A compound of formula (I) or a pharmaceutically acceptable salt thereof.
(B)
X-Y-Zが、N-CO-NR3AR3B、N-COR4A又はCR5-NR6-COR4Bであり、
Aが、フェニル又はピリジル(該フェニル及び該ピリジルはそれぞれフッ素、塩素、1から5個のフッ素で置換されていてもよいC1-6アルキル、1から5個のフッ素で置換されていてもよいC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)であり、
Bが、B-1、B-2、B-3、B-4又はB-5であり、
Wが、酸素原子又は硫黄原子であり、
R1が、水素原子、ハロゲン又はシアノであり、
R2A、R2B、R2C、R2D及びR5が、同一又は異なって、水素原子、又はC1-6アルキルであり、ここにおいて、R2A、R2B、R2C、R2D及びR5のいずれかの2つがC1-6アルキルのとき、2個のアルキルが一緒になって該アルキルが結合している環と別の環を形成していてもよく、
R3A、R3B及びR4Aが、同一又は異なって、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子であり、ここにおいて、(1)R4Aは水素原子ではなく、(2)R3A及びR3Bは同時に水素原子ではなく、(3)BがB-3であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aはメチルではなく、(4)Aが無置換のフェニルであり、BがB-4であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aは1~5個のフッ素で置換されたC3アルキルではなく、
R4Bが、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;又は4~10員の飽和複素環であり、
R6が、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子である、
式(I)の化合物又はその製薬学的に許容される塩。 The following (B) is mentioned as a more preferable aspect.
(B)
XYZ is N—CO—NR 3A R 3B , N—COR 4A or CR 5 —NR 6 —COR 4B ;
A is phenyl or pyridyl (each said phenyl and said pyridyl fluorine, chlorine, from one to five fluorines optionally substituted C 1-6 alkyl, optionally substituted with 1-5 fluorine Optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy),
B is B-1, B-2, B-3, B-4 or B-5,
W is an oxygen atom or a sulfur atom,
R 1 is a hydrogen atom, halogen or cyano,
R 2A , R 2B , R 2C , R 2D and R 5 are the same or different and are a hydrogen atom or C 1-6 alkyl, wherein R 2A , R 2B , R 2C , R 2D and R 5 Any two of the above are C 1-6 alkyl, the two alkyls may be combined to form a ring with the ring to which the alkyl is attached;
R 3A , R 3B and R 4A are the same or different and are substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy, and C 3-10 cycloalkyl. which may be C 1-6 alkyl; fluorine, C 1-6 alkoxy and C 1-6 independently from the group consisting of alkyl optionally substituted with 1 to 5 substituents selected C 3- 10 cycloalkyl; 4 to 10 membered saturated heterocycle; or a hydrogen atom, wherein (1) R 4A is not a hydrogen atom, and (2) R 3A and R 3B are not simultaneously a hydrogen atom, (3 ) When B is B-3, W is an oxygen atom and XYZ is N-COR 4A , R 4A is not methyl and (4) A is unsubstituted phenyl; B is B-4, W is an oxygen atom, XY When Z is N-COR 4A is, R 4A is not C 3 alkyl substituted with 1-5 fluorine,
R 4B is selected from the group consisting of fluorine, C 1-6 alkoxy, and C 3-10 1 ~ 5 amino optionally substituted with a substituent C 1-6 alkyl independently selected from the group consisting of cycloalkyl; fluorine C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; or 4-10 membered saturation A heterocycle,
R 6 is fluorine, C 1-6 alkoxy, and C 3-10 1 ~ 5 amino optionally substituted with a substituent C 1-6 alkyl independently selected from the group consisting of cycloalkyl; fluorine C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl; A ring; or a hydrogen atom,
A compound of formula (I) or a pharmaceutically acceptable salt thereof.
さらに好ましい態様としては、以下の(C)が挙げられる。
(C)
X-Y-Zが、N-CO-NR3AR3B又はN-COR4Aであり、
Aが、フッ素、塩素、1~3個のフッ素で置換されていてもよいC1-6アルキル及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいフェニルであり、
Bが、B-1、B-4又はB-5であり、
Wが、酸素原子であり、
R1が、水素原子、塩素又はシアノであり、
R2A、R2B、R2C及びR2Dが、水素原子であり、
R3A、R3B及びR4Aが、同一又は異なって、フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子であり、ここにおいて、R3A及びR3Bは同時に水素原子ではなく、R4Aは水素原子ではない、
式(I)の化合物又はその製薬学的に許容される塩。 The following (C) is mentioned as a more preferable aspect.
(C)
XYZ is N—CO—NR 3A R 3B or N—COR 4A ;
A is substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, chlorine, C 1-6 alkyl optionally substituted with 1 to 3 fluorine and C 1-6 alkoxy Which may be phenyl,
B is B-1, B-4 or B-5,
W is an oxygen atom,
R 1 is a hydrogen atom, chlorine or cyano,
R 2A , R 2B , R 2C and R 2D are hydrogen atoms,
R 3A , R 3B and R 4A are the same or different and are substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl; a 4-10 membered saturated heterocyclic ring; or a hydrogen atom, wherein R 3A and R 3B are not simultaneously a hydrogen atom, and R 4A is not a hydrogen atom,
A compound of formula (I) or a pharmaceutically acceptable salt thereof.
(C)
X-Y-Zが、N-CO-NR3AR3B又はN-COR4Aであり、
Aが、フッ素、塩素、1~3個のフッ素で置換されていてもよいC1-6アルキル及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいフェニルであり、
Bが、B-1、B-4又はB-5であり、
Wが、酸素原子であり、
R1が、水素原子、塩素又はシアノであり、
R2A、R2B、R2C及びR2Dが、水素原子であり、
R3A、R3B及びR4Aが、同一又は異なって、フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子であり、ここにおいて、R3A及びR3Bは同時に水素原子ではなく、R4Aは水素原子ではない、
式(I)の化合物又はその製薬学的に許容される塩。 The following (C) is mentioned as a more preferable aspect.
(C)
XYZ is N—CO—NR 3A R 3B or N—COR 4A ;
A is substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, chlorine, C 1-6 alkyl optionally substituted with 1 to 3 fluorine and C 1-6 alkoxy Which may be phenyl,
B is B-1, B-4 or B-5,
W is an oxygen atom,
R 1 is a hydrogen atom, chlorine or cyano,
R 2A , R 2B , R 2C and R 2D are hydrogen atoms,
R 3A , R 3B and R 4A are the same or different and are substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl; a 4-10 membered saturated heterocyclic ring; or a hydrogen atom, wherein R 3A and R 3B are not simultaneously a hydrogen atom, and R 4A is not a hydrogen atom,
A compound of formula (I) or a pharmaceutically acceptable salt thereof.
もっとも好ましい態様としては、以下の(D)が挙げられる。
(D)
X-Y-Zが、N-CO-NR3AR3B又はN-COR4Aであり、
Aが、フッ素及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいフェニルであり、
Bが、B-1又はB-4であり、
Wが、酸素原子であり、
R1が、水素原子又は塩素であり、
R2A、R2B、R2C及びR2Dが、水素原子であり、
R3A及びR4Aが、同一又は異なって、フッ素及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル又は4~10員の飽和複素環であり、
R3Bが、水素原子である、式(I)の化合物又はその製薬学的に許容される塩。 The most preferable embodiment includes the following (D).
(D)
XYZ is N—CO—NR 3A R 3B or N—COR 4A ;
A is phenyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of fluorine and C 1-6 alkoxy;
B is B-1 or B-4,
W is an oxygen atom,
R 1 is a hydrogen atom or chlorine;
R 2A , R 2B , R 2C and R 2D are hydrogen atoms,
R 3A and R 4A are the same or different and each is a C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of fluorine and C 1-6 alkoxy; A 4-10 membered saturated heterocycle,
A compound of formula (I) or a pharmaceutically acceptable salt thereof, wherein R 3B is a hydrogen atom.
(D)
X-Y-Zが、N-CO-NR3AR3B又はN-COR4Aであり、
Aが、フッ素及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいフェニルであり、
Bが、B-1又はB-4であり、
Wが、酸素原子であり、
R1が、水素原子又は塩素であり、
R2A、R2B、R2C及びR2Dが、水素原子であり、
R3A及びR4Aが、同一又は異なって、フッ素及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル又は4~10員の飽和複素環であり、
R3Bが、水素原子である、式(I)の化合物又はその製薬学的に許容される塩。 The most preferable embodiment includes the following (D).
(D)
XYZ is N—CO—NR 3A R 3B or N—COR 4A ;
A is phenyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of fluorine and C 1-6 alkoxy;
B is B-1 or B-4,
W is an oxygen atom,
R 1 is a hydrogen atom or chlorine;
R 2A , R 2B , R 2C and R 2D are hydrogen atoms,
R 3A and R 4A are the same or different and each is a C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of fluorine and C 1-6 alkoxy; A 4-10 membered saturated heterocycle,
A compound of formula (I) or a pharmaceutically acceptable salt thereof, wherein R 3B is a hydrogen atom.
式(I)で表される化合物の製薬学的に許容される塩とは、式(I)の化合物に製薬学的に許容される酸又は塩基を加えて形成された塩を意味する。
式(I)で表される本発明の化合物がアミノ基などの塩基性官能基を有する場合、各種の酸と塩を形成しうる。このような酸付加塩の具体例としては、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硫酸塩、過塩素酸塩、リン酸塩等の無機酸塩、シュウ酸塩、マロン酸塩、マレイン酸塩、フマル酸塩、乳酸塩、リンゴ酸塩、クエン酸塩、酒石酸塩、安息香酸塩、トリフルオロ酢酸塩、酢酸塩、メタンスルホン酸塩、p-トルエンスルホン酸塩、トリフルオロメタンスルホン酸塩等の有機酸塩、又はグルタミン酸塩、アスパラギン酸塩等のアミノ酸塩が挙げられる。これらの塩は、式(I)で表される本発明の化合物を酸と混合した後、再結晶などの常法により得ることができる。 The pharmaceutically acceptable salt of the compound represented by the formula (I) means a salt formed by adding a pharmaceutically acceptable acid or base to the compound of the formula (I).
When the compound of the present invention represented by the formula (I) has a basic functional group such as an amino group, it can form salts with various acids. Specific examples of such acid addition salts include hydrochloride, hydrobromide, hydroiodide, sulfate, perchlorate, phosphate, and other inorganic acid salts, oxalate, and malonic acid. Salt, maleate, fumarate, lactate, malate, citrate, tartrate, benzoate, trifluoroacetate, acetate, methanesulfonate, p-toluenesulfonate, trifluoromethane Examples thereof include organic acid salts such as sulfonate, and amino acid salts such as glutamate and aspartate. These salts can be obtained by a conventional method such as recrystallization after mixing the compound of the present invention represented by the formula (I) with an acid.
式(I)で表される本発明の化合物がアミノ基などの塩基性官能基を有する場合、各種の酸と塩を形成しうる。このような酸付加塩の具体例としては、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硫酸塩、過塩素酸塩、リン酸塩等の無機酸塩、シュウ酸塩、マロン酸塩、マレイン酸塩、フマル酸塩、乳酸塩、リンゴ酸塩、クエン酸塩、酒石酸塩、安息香酸塩、トリフルオロ酢酸塩、酢酸塩、メタンスルホン酸塩、p-トルエンスルホン酸塩、トリフルオロメタンスルホン酸塩等の有機酸塩、又はグルタミン酸塩、アスパラギン酸塩等のアミノ酸塩が挙げられる。これらの塩は、式(I)で表される本発明の化合物を酸と混合した後、再結晶などの常法により得ることができる。 The pharmaceutically acceptable salt of the compound represented by the formula (I) means a salt formed by adding a pharmaceutically acceptable acid or base to the compound of the formula (I).
When the compound of the present invention represented by the formula (I) has a basic functional group such as an amino group, it can form salts with various acids. Specific examples of such acid addition salts include hydrochloride, hydrobromide, hydroiodide, sulfate, perchlorate, phosphate, and other inorganic acid salts, oxalate, and malonic acid. Salt, maleate, fumarate, lactate, malate, citrate, tartrate, benzoate, trifluoroacetate, acetate, methanesulfonate, p-toluenesulfonate, trifluoromethane Examples thereof include organic acid salts such as sulfonate, and amino acid salts such as glutamate and aspartate. These salts can be obtained by a conventional method such as recrystallization after mixing the compound of the present invention represented by the formula (I) with an acid.
式(I)で表される本発明の化合物がカルボキシル基などの酸性官能基を有する場合、各種の塩基と塩を形成しうる。この場合の製薬学的に許容される塩としては、ナトリウム塩もしくはカリウム塩などのアルカリ金属塩、カルシウム塩などのアルカリ土類金属塩、又はトリエチルアンモニウム塩、トリエタノールアンモニウム塩、ピリジニウム塩、ジイソプロピルアンモニウム塩等の有機塩基塩等などが挙げられる。これらの塩は、式(I)で表される本発明の化合物を塩基と混合した後、再結晶などの常法により得ることができる。
When the compound of the present invention represented by the formula (I) has an acidic functional group such as a carboxyl group, it can form salts with various bases. In this case, pharmaceutically acceptable salts include alkali metal salts such as sodium salt or potassium salt, alkaline earth metal salts such as calcium salt, or triethylammonium salt, triethanolammonium salt, pyridinium salt, diisopropylammonium salt. Examples thereof include organic base salts such as salts. These salts can be obtained by a conventional method such as recrystallization after mixing the compound of the present invention represented by the formula (I) with a base.
なお、本明細書において記載の簡略化のために、次に挙げる略号を用いることもある。o-:ortho-、m-:meta-、p-:para-、t-:tert-、s-:sec-、THF:テトラヒドロフラン、DMF:N,N-ジメチルホルムアミド、DMSO:ジメチルスルホキシド、d6-DMSO:重ジメチルスルホキシド、HEPES:N-2-ヒドロキシエチルピペラジン-N’-2-エタンスルホン酸、BSA:bovine serum albumin、牛血清アルブミン、FDSS:Functional Drug Screening System、ACh:アセチルコリン、PAM:ポジティブアロステリックモジュレーター、nAChR:ニコチン性アセチルコリン受容体、EC20:20%効果濃度、RLU:Relative Light Unit、Boc:tert-ブトキシカルボニル、EDCI:1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド、HOBt:1-ヒドロキシベンゾトリアゾール、HBTU:2-(1H-7-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロリン酸塩。
Note that the following abbreviations may be used to simplify the description in this specification. o-: ortho-, m-: meta-, p-: para-, t-: tert-, s-: sec-, THF: tetrahydrofuran, DMF: N, N-dimethylformamide, DMSO: dimethyl sulfoxide, d 6 -DMSO: heavy dimethyl sulfoxide, HEPES: N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid, BSA: bovine serum albumin, bovine serum albumin, FDSS: Functional Drug Screening System, ACh: acetylcholine, PAM: positive Allosteric modulator, nAChR: nicotinic acetylcholine receptor, EC20: 20% effective concentration, RLU: Relative Light Unit, Boc: tert-butoxycarbonyl, DCI: 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide, HOBt: 1-hydroxybenzotriazole, HBTU: 2- (1H-7-benzotriazol-1-yl) -1,1,3,3- Tetramethyluronium hexafluorophosphate.
本発明の化合物の製造方法について以下に述べる。式(I)で表される本発明の化合物は、例えば下記の製造法A~Mにより製造することができる。
The method for producing the compound of the present invention is described below. The compound of the present invention represented by the formula (I) can be produced, for example, by the following production methods A to M.
製造法A(合成中間体の製法)
式(I)で表される化合物のうち、BがB-1である化合物の合成中間体a6、a8及びa10は、例えば下記の製法により製造することができる。
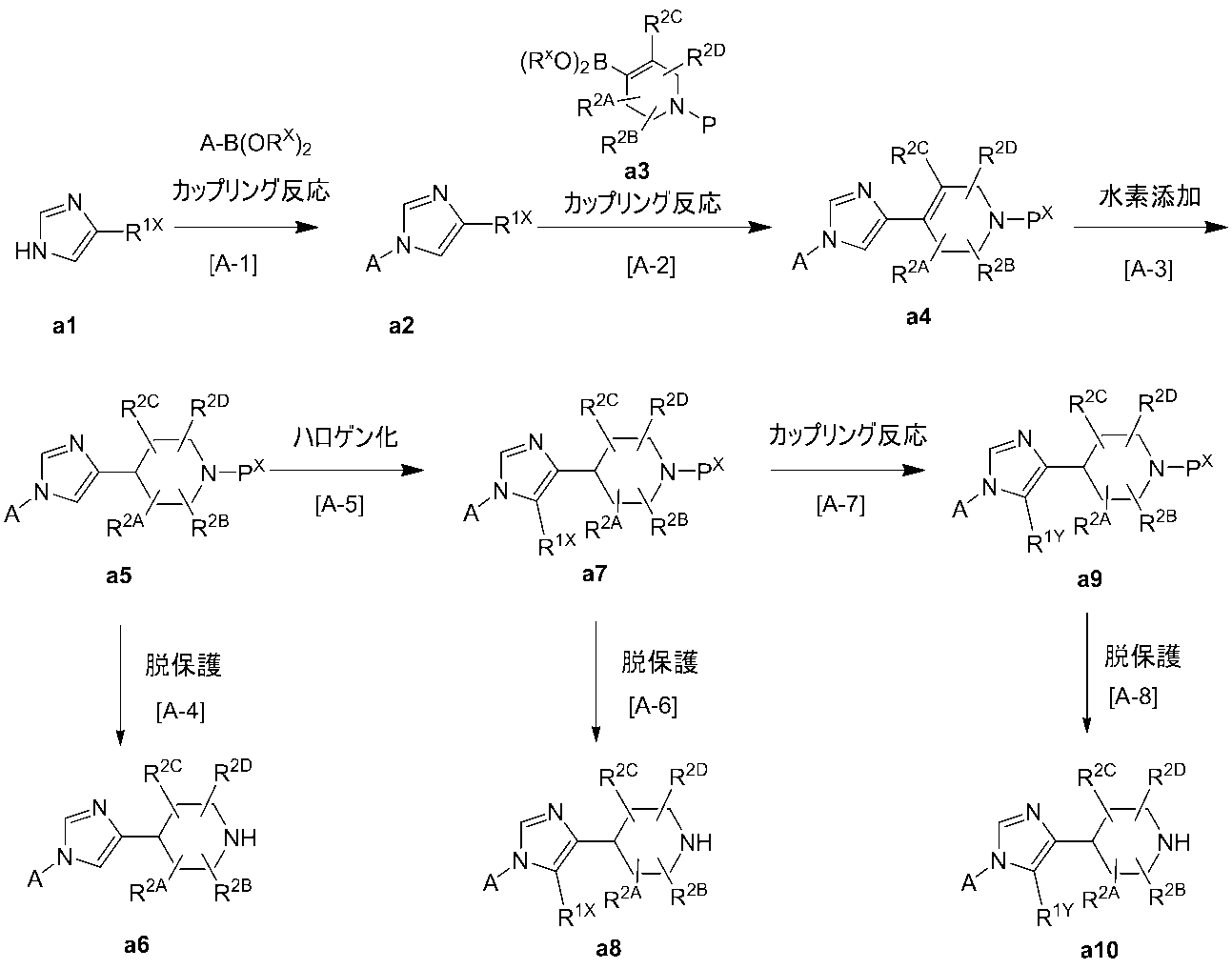
(式中、A、R2A、R2B、R2C及びR2Dは項1に定義されるとおりであり、PXはアミノの保護基を表し、RXは水素原子、アルキル又はフェニルを表し、ここにおいてRXがアルキルのとき、2個の当該アルキルが一緒になって4~10員の含酸素飽和複素環を形成していてもよく、R1Xはハロゲンを表し、R1Yは項1に定義されるR1のうちハロゲンではないものを意味する)
Production method A (production method of synthetic intermediate)
Among the compounds represented by the formula (I), synthetic intermediates a6, a8 and a10 of a compound in which B is B-1 can be produced, for example, by the following production method.
(Wherein, A, R 2A, R 2B , R 2C and R 2D are as defined in claim 1, P X represents a protecting group of amino, R X represents a hydrogen atom, alkyl or phenyl, In this case, when R X is alkyl, the two alkyls together may form a 4- to 10-membered oxygen-containing saturated heterocyclic ring, R 1X represents halogen, R 1Y represents the term 1 R 1 as defined is not halogen)
式(I)で表される化合物のうち、BがB-1である化合物の合成中間体a6、a8及びa10は、例えば下記の製法により製造することができる。
Among the compounds represented by the formula (I), synthetic intermediates a6, a8 and a10 of a compound in which B is B-1 can be produced, for example, by the following production method.
保護基PXとしては、Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年) にアミノの保護基として記載されているものが挙げられる。
The protective group P X, Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
化合物a1はイミダゾールのハロゲン化反応などの既知の方法により合成できるか、又は市販品として購入できる。
Compound a1 can be synthesized by a known method such as a halogenation reaction of imidazole, or can be purchased as a commercial product.
[A-1工程]
本工程は化合物a1に、適当な遷移金属試薬の存在下、適当なボロン酸誘導体と反応させることにより、化合物a2を得る工程である。本工程において使用される遷移金属試薬は、例えば酢酸銅が挙げられる。本工程において使用されるボロン酸誘導体は、例えばボロン酸、ボロン酸エステル、ボロン酸ピナコラートが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはジクロロエタン又はメタノールである。類似反応として、例えば、Journal of Medicinal Chemistry, 2009, 52(11), 3441-3444などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは室温~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step A-1]
This step is a step of obtaining compound a2 by reacting compound a1 with an appropriate boronic acid derivative in the presence of an appropriate transition metal reagent. An example of the transition metal reagent used in this step is copper acetate. Examples of the boronic acid derivative used in this step include boronic acid, boronic acid ester, and boronic acid pinacolate. The solvent used in this step is selected from the solvents exemplified below, but is preferably dichloroethane or methanol. As a similar reaction, for example, a method described in Journal of Medicinal Chemistry, 2009, 52 (11), 3441-3444 is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably room temperature to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は化合物a1に、適当な遷移金属試薬の存在下、適当なボロン酸誘導体と反応させることにより、化合物a2を得る工程である。本工程において使用される遷移金属試薬は、例えば酢酸銅が挙げられる。本工程において使用されるボロン酸誘導体は、例えばボロン酸、ボロン酸エステル、ボロン酸ピナコラートが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはジクロロエタン又はメタノールである。類似反応として、例えば、Journal of Medicinal Chemistry, 2009, 52(11), 3441-3444などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは室温~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step A-1]
This step is a step of obtaining compound a2 by reacting compound a1 with an appropriate boronic acid derivative in the presence of an appropriate transition metal reagent. An example of the transition metal reagent used in this step is copper acetate. Examples of the boronic acid derivative used in this step include boronic acid, boronic acid ester, and boronic acid pinacolate. The solvent used in this step is selected from the solvents exemplified below, but is preferably dichloroethane or methanol. As a similar reaction, for example, a method described in Journal of Medicinal Chemistry, 2009, 52 (11), 3441-3444 is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably room temperature to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[A-2工程]
本工程は上記A-1工程で得られた化合物a2と化合物a3を、適当な遷移金属試薬の存在下で反応させることにより、化合物a4を得る工程である。本工程において使用される遷移金属試薬は、例えばトリス(ジベンジリデンアセトン)ジパラジウムやテトラキス(トリフェニルフォスフィン)パラジウムが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはDMF又はジオキサンである。類似反応として、例えば、Journal of Organic Chemistry, 2010, 75(5), 1733-1739などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは室温~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step A-2]
This step is a step of obtaining compound a4 by reacting compound a2 obtained in step A-1 with compound a3 in the presence of an appropriate transition metal reagent. Examples of the transition metal reagent used in this step include tris (dibenzylideneacetone) dipalladium and tetrakis (triphenylphosphine) palladium. The solvent used in this step is selected from the solvents exemplified below, and is preferably DMF or dioxane. As a similar reaction, for example, a method described in Journal of Organic Chemistry, 2010, 75 (5), 1733-1739 is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably room temperature to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記A-1工程で得られた化合物a2と化合物a3を、適当な遷移金属試薬の存在下で反応させることにより、化合物a4を得る工程である。本工程において使用される遷移金属試薬は、例えばトリス(ジベンジリデンアセトン)ジパラジウムやテトラキス(トリフェニルフォスフィン)パラジウムが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはDMF又はジオキサンである。類似反応として、例えば、Journal of Organic Chemistry, 2010, 75(5), 1733-1739などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは室温~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step A-2]
This step is a step of obtaining compound a4 by reacting compound a2 obtained in step A-1 with compound a3 in the presence of an appropriate transition metal reagent. Examples of the transition metal reagent used in this step include tris (dibenzylideneacetone) dipalladium and tetrakis (triphenylphosphine) palladium. The solvent used in this step is selected from the solvents exemplified below, and is preferably DMF or dioxane. As a similar reaction, for example, a method described in Journal of Organic Chemistry, 2010, 75 (5), 1733-1739 is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably room temperature to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[A-3工程]
本工程は上記A-2工程で得られた化合物a4を、適当な遷移金属試薬を用いて水素雰囲気下で反応させることにより化合物a5を得る工程である。本工程において使用される遷移金属試薬は、例えばパラジウム/炭素や酸化プラチナ(IV)が挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはメタノール又はエタノールである。類似反応として、例えば、Journal of Medicinal Chemistry, 2012, 55(1), 115-125などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-20℃~150℃、好ましくは0℃~100℃であり、より好ましくは室温~60℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step A-3]
This step is a step of obtaining compound a5 by reacting compound a4 obtained in step A-2 with a suitable transition metal reagent in a hydrogen atmosphere. Examples of the transition metal reagent used in this step include palladium / carbon and platinum (IV) oxide. The solvent used in this step is selected from the solvents exemplified below, but is preferably methanol or ethanol. As a similar reaction, for example, a method described in Journal of Medicinal Chemistry, 2012, 55 (1), 115-125, etc. is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −20 ° C. to 150 ° C., preferably 0 ° C. to 100 ° C., more preferably room temperature to 60 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記A-2工程で得られた化合物a4を、適当な遷移金属試薬を用いて水素雰囲気下で反応させることにより化合物a5を得る工程である。本工程において使用される遷移金属試薬は、例えばパラジウム/炭素や酸化プラチナ(IV)が挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはメタノール又はエタノールである。類似反応として、例えば、Journal of Medicinal Chemistry, 2012, 55(1), 115-125などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-20℃~150℃、好ましくは0℃~100℃であり、より好ましくは室温~60℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step A-3]
This step is a step of obtaining compound a5 by reacting compound a4 obtained in step A-2 with a suitable transition metal reagent in a hydrogen atmosphere. Examples of the transition metal reagent used in this step include palladium / carbon and platinum (IV) oxide. The solvent used in this step is selected from the solvents exemplified below, but is preferably methanol or ethanol. As a similar reaction, for example, a method described in Journal of Medicinal Chemistry, 2012, 55 (1), 115-125, etc. is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −20 ° C. to 150 ° C., preferably 0 ° C. to 100 ° C., more preferably room temperature to 60 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[A-4工程]
本工程は上記製造法A-3で得られた化合物a5のアミノの保護基PXを、脱保護することにより、化合物a6を得る工程である。本工程はProtective Groups in Organic Synthesis(Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年)に記載されている方法等に準じて行うことができる。 [Step A-4]
This step is a protecting group P X of the amino compound a5 prepared in the above Preparation Method A-3, by deprotection, to give compound compound a6. This step can be carried out according to the method described in Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts, John Wiley & Sons, Inc., 1999).
本工程は上記製造法A-3で得られた化合物a5のアミノの保護基PXを、脱保護することにより、化合物a6を得る工程である。本工程はProtective Groups in Organic Synthesis(Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年)に記載されている方法等に準じて行うことができる。 [Step A-4]
This step is a protecting group P X of the amino compound a5 prepared in the above Preparation Method A-3, by deprotection, to give compound compound a6. This step can be carried out according to the method described in Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts, John Wiley & Sons, Inc., 1999).
[A-5工程]
本工程は上記A-3工程で得られた化合物a5に、適当な溶媒中、適当な酸の存在下で、種々のハロゲン化剤を反応させることにより、化合物a7を得る工程である。本工程において使用されるハロゲン化剤は、例えばN-クロロスクシンイミド、N-ブロモスクシンイミド、N-ヨードスクシンイミドである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくは塩化メチレン又はジクロロエタンである。本工程において使用される酸は、後記に例示する酸等から選択されるが、好ましくはトリフルオロ酢酸又は塩酸である。類似反応として、例えば、Bioorg. Med. Chem. Lett. 2008, 18(5), 1702-1707、J. Org. Chem. 2002, 67(17), 5913-5918などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは0℃~100℃であり、より好ましくは室温~70℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step A-5]
This step is a step of obtaining compound a7 by reacting compound a5 obtained in step A-3 with various halogenating agents in a suitable solvent in the presence of a suitable acid. The halogenating agent used in this step is, for example, N-chlorosuccinimide, N-bromosuccinimide, N-iodosuccinimide. The solvent used in this step is selected from the solvents exemplified below, and is preferably methylene chloride or dichloroethane. The acid used in this step is selected from the acids exemplified below, and preferably trifluoroacetic acid or hydrochloric acid. As a similar reaction, for example, a method described in Bioorg. Med. Chem. Lett. 2008, 18 (5), 1702-1707, J. Org. Chem. 2002, 67 (17), 5913-5918, etc. is known. And can be synthesized similarly. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably 0 ° C. to 100 ° C., more preferably room temperature to 70 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記A-3工程で得られた化合物a5に、適当な溶媒中、適当な酸の存在下で、種々のハロゲン化剤を反応させることにより、化合物a7を得る工程である。本工程において使用されるハロゲン化剤は、例えばN-クロロスクシンイミド、N-ブロモスクシンイミド、N-ヨードスクシンイミドである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくは塩化メチレン又はジクロロエタンである。本工程において使用される酸は、後記に例示する酸等から選択されるが、好ましくはトリフルオロ酢酸又は塩酸である。類似反応として、例えば、Bioorg. Med. Chem. Lett. 2008, 18(5), 1702-1707、J. Org. Chem. 2002, 67(17), 5913-5918などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは0℃~100℃であり、より好ましくは室温~70℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step A-5]
This step is a step of obtaining compound a7 by reacting compound a5 obtained in step A-3 with various halogenating agents in a suitable solvent in the presence of a suitable acid. The halogenating agent used in this step is, for example, N-chlorosuccinimide, N-bromosuccinimide, N-iodosuccinimide. The solvent used in this step is selected from the solvents exemplified below, and is preferably methylene chloride or dichloroethane. The acid used in this step is selected from the acids exemplified below, and preferably trifluoroacetic acid or hydrochloric acid. As a similar reaction, for example, a method described in Bioorg. Med. Chem. Lett. 2008, 18 (5), 1702-1707, J. Org. Chem. 2002, 67 (17), 5913-5918, etc. is known. And can be synthesized similarly. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably 0 ° C. to 100 ° C., more preferably room temperature to 70 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[A-6工程]
本工程は上記A-5工程で得られた化合物a7に、上記A-4工程に準じた条件で、化合物a8を得る工程である。 [Step A-6]
This step is a step of obtaining compound a8 from compound a7 obtained in step A-5, under the same conditions as in step A-4.
本工程は上記A-5工程で得られた化合物a7に、上記A-4工程に準じた条件で、化合物a8を得る工程である。 [Step A-6]
This step is a step of obtaining compound a8 from compound a7 obtained in step A-5, under the same conditions as in step A-4.
[A-7工程]
本工程は上記A-5工程で得られた化合物a7に、適当な溶媒中、適当な遷移金属試薬の存在下、適当なボロン酸誘導体と反応させることにより、化合物a9を得る工程である。本工程において使用される遷移金属試薬は、例えばトリス(ジベンジリデンアセトン)ジパラジウムやテトラキス(トリフェニルフォスフィン)パラジウムが挙げられる。本工程において使用されるボロン酸誘導体は、例えばボロン酸、ボロン酸エステル、ボロン酸ピナコラートが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはDMF又はジオキサンである。類似反応として、例えば、Tetrahedron Lett. 2003, 44(7), 1379-1382、J. Med. Chem. 2009, 52(14), 4370-4379、Bioorg. Med. Chem. Lett. 2012, 20(9), 3009-3015、J. Org. Chem. 2002, 67(10), 3365-3373、Tetrahedron Lett. 2007, 48(13), 2339-2343などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~180℃であり、より好ましくは室温~150℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step A-7]
This step is a step of obtaining compound a9 by reacting compound a7 obtained in step A-5 with a suitable boronic acid derivative in a suitable solvent in the presence of a suitable transition metal reagent. Examples of the transition metal reagent used in this step include tris (dibenzylideneacetone) dipalladium and tetrakis (triphenylphosphine) palladium. Examples of the boronic acid derivative used in this step include boronic acid, boronic acid ester, and boronic acid pinacolate. The solvent used in this step is selected from the solvents exemplified below, and is preferably DMF or dioxane. Similar reactions include, for example, Tetrahedron Lett. 2003, 44 (7), 1379-1382, J. Med. Chem. 2009, 52 (14), 4370-4379, Bioorg. Med. Chem. Lett. 2012, 20 (9 ), 3009-3015, J. Org. Chem. 2002, 67 (10), 3365-3373, Tetrahedron Lett. 2007, 48 (13), 2339-2343, etc. Can be synthesized. The reaction temperature varies depending on the type of raw material compound used, reagents and the like, but is usually −78 ° C. to 200 ° C., preferably −20 ° C. to 180 ° C., more preferably room temperature to 150 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記A-5工程で得られた化合物a7に、適当な溶媒中、適当な遷移金属試薬の存在下、適当なボロン酸誘導体と反応させることにより、化合物a9を得る工程である。本工程において使用される遷移金属試薬は、例えばトリス(ジベンジリデンアセトン)ジパラジウムやテトラキス(トリフェニルフォスフィン)パラジウムが挙げられる。本工程において使用されるボロン酸誘導体は、例えばボロン酸、ボロン酸エステル、ボロン酸ピナコラートが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはDMF又はジオキサンである。類似反応として、例えば、Tetrahedron Lett. 2003, 44(7), 1379-1382、J. Med. Chem. 2009, 52(14), 4370-4379、Bioorg. Med. Chem. Lett. 2012, 20(9), 3009-3015、J. Org. Chem. 2002, 67(10), 3365-3373、Tetrahedron Lett. 2007, 48(13), 2339-2343などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~180℃であり、より好ましくは室温~150℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step A-7]
This step is a step of obtaining compound a9 by reacting compound a7 obtained in step A-5 with a suitable boronic acid derivative in a suitable solvent in the presence of a suitable transition metal reagent. Examples of the transition metal reagent used in this step include tris (dibenzylideneacetone) dipalladium and tetrakis (triphenylphosphine) palladium. Examples of the boronic acid derivative used in this step include boronic acid, boronic acid ester, and boronic acid pinacolate. The solvent used in this step is selected from the solvents exemplified below, and is preferably DMF or dioxane. Similar reactions include, for example, Tetrahedron Lett. 2003, 44 (7), 1379-1382, J. Med. Chem. 2009, 52 (14), 4370-4379, Bioorg. Med. Chem. Lett. 2012, 20 (9 ), 3009-3015, J. Org. Chem. 2002, 67 (10), 3365-3373, Tetrahedron Lett. 2007, 48 (13), 2339-2343, etc. Can be synthesized. The reaction temperature varies depending on the type of raw material compound used, reagents and the like, but is usually −78 ° C. to 200 ° C., preferably −20 ° C. to 180 ° C., more preferably room temperature to 150 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[A-8工程]
本工程は上記A-7工程で得られた化合物a9に、上記A-4工程に準じた条件で、化合物a10を得る工程である。 [Step A-8]
This step is a step of obtaining compound a10 from compound a9 obtained in step A-7, under the same conditions as in step A-4.
本工程は上記A-7工程で得られた化合物a9に、上記A-4工程に準じた条件で、化合物a10を得る工程である。 [Step A-8]
This step is a step of obtaining compound a10 from compound a9 obtained in step A-7, under the same conditions as in step A-4.
製造法B(合成中間体の製法)
式(I)で表される化合物のうち、BがB-2であり、Wが酸素原子である化合物の合成中間体b7は、例えば下記の製法により製造することができる。
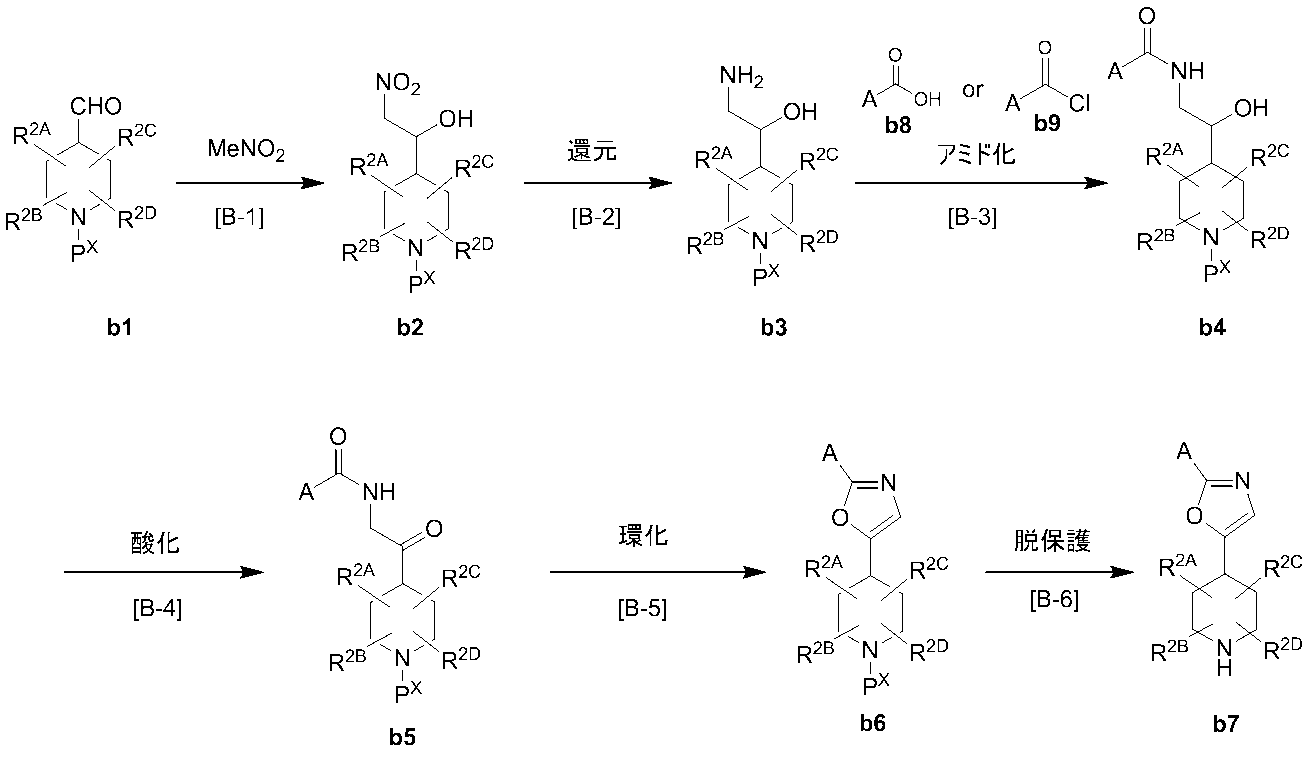
(式中、A、R2A、R2B、R2C及びR2Dは項1に定義されるとおりであり、PXはアミノの保護基を意味する)
Production method B (Production method of synthetic intermediate)
Among the compounds represented by formula (I), a synthetic intermediate b7 of a compound in which B is B-2 and W is an oxygen atom can be produced, for example, by the following production method.
(Wherein, A, R 2A, R 2B , R 2C and R 2D are as defined in claim 1, P X denotes a protective group for amino)
式(I)で表される化合物のうち、BがB-2であり、Wが酸素原子である化合物の合成中間体b7は、例えば下記の製法により製造することができる。
Among the compounds represented by formula (I), a synthetic intermediate b7 of a compound in which B is B-2 and W is an oxygen atom can be produced, for example, by the following production method.
保護基PXとしては、Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年) にアミノの保護基として記載されているものが挙げられる。
The protective group P X, Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
化合物b1は対応するアルコールの酸化などの既知の方法により合成できるか、又は市販品として購入できる。
Compound b1 can be synthesized by a known method such as oxidation of the corresponding alcohol, or can be purchased as a commercial product.
[B-1工程]
本工程は化合物b1に、適当な塩基存在下でニトロメタンを反応させることにより、化合物b2を得る工程である。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはtert-ブトキシカリウムである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはテトラヒドロフランとtert-ブタノールの混合溶媒である。類似反応として、例えば、国際公開第2007/41061号パンフレットなどに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step B-1]
This step is a step of obtaining compound b2 by reacting compound b1 with nitromethane in the presence of a suitable base. The base used in this step is selected from the bases exemplified below, and preferably tert-butoxy potassium. The solvent used in this step is selected from the solvents exemplified below, but is preferably a mixed solvent of tetrahydrofuran and tert-butanol. As a similar reaction, for example, a method described in International Publication No. 2007/41061 pamphlet or the like is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は化合物b1に、適当な塩基存在下でニトロメタンを反応させることにより、化合物b2を得る工程である。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはtert-ブトキシカリウムである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはテトラヒドロフランとtert-ブタノールの混合溶媒である。類似反応として、例えば、国際公開第2007/41061号パンフレットなどに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step B-1]
This step is a step of obtaining compound b2 by reacting compound b1 with nitromethane in the presence of a suitable base. The base used in this step is selected from the bases exemplified below, and preferably tert-butoxy potassium. The solvent used in this step is selected from the solvents exemplified below, but is preferably a mixed solvent of tetrahydrofuran and tert-butanol. As a similar reaction, for example, a method described in International Publication No. 2007/41061 pamphlet or the like is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[B-2工程]
本工程は上記B-1で得られた化合物b2を、適当な遷移金属触媒を用いて水素雰囲気下で反応させることにより、化合物b3を得る工程である。本工程において使用される遷移金属試薬は、例えばパラジウム/炭素や酸化プラチナ(IV)が挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはメタノール又はエタノールである。類似反応として、例えば、Bioorganic and Medicinal Chemistry Letters, 2004, 14(13), 3419-3424、国際公開第2006/19768号パンフレットなどに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-20℃~200℃、好ましくは0℃~100℃であり、より好ましくは室温~70℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step B-2]
This step is a step of obtaining compound b3 by reacting compound b2 obtained in B-1 above in a hydrogen atmosphere using an appropriate transition metal catalyst. Examples of the transition metal reagent used in this step include palladium / carbon and platinum (IV) oxide. The solvent used in this step is selected from the solvents exemplified below, but is preferably methanol or ethanol. As similar reactions, for example, the methods described in Bioorganic and Medicinal Chemistry Letters, 2004, 14 (13), 3419-3424, International Publication No. 2006/19768, etc. are known and can be synthesized similarly. . The reaction temperature varies depending on the type of raw material compound used, reagents and the like, but is usually −20 ° C. to 200 ° C., preferably 0 ° C. to 100 ° C., more preferably room temperature to 70 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記B-1で得られた化合物b2を、適当な遷移金属触媒を用いて水素雰囲気下で反応させることにより、化合物b3を得る工程である。本工程において使用される遷移金属試薬は、例えばパラジウム/炭素や酸化プラチナ(IV)が挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはメタノール又はエタノールである。類似反応として、例えば、Bioorganic and Medicinal Chemistry Letters, 2004, 14(13), 3419-3424、国際公開第2006/19768号パンフレットなどに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-20℃~200℃、好ましくは0℃~100℃であり、より好ましくは室温~70℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step B-2]
This step is a step of obtaining compound b3 by reacting compound b2 obtained in B-1 above in a hydrogen atmosphere using an appropriate transition metal catalyst. Examples of the transition metal reagent used in this step include palladium / carbon and platinum (IV) oxide. The solvent used in this step is selected from the solvents exemplified below, but is preferably methanol or ethanol. As similar reactions, for example, the methods described in Bioorganic and Medicinal Chemistry Letters, 2004, 14 (13), 3419-3424, International Publication No. 2006/19768, etc. are known and can be synthesized similarly. . The reaction temperature varies depending on the type of raw material compound used, reagents and the like, but is usually −20 ° C. to 200 ° C., preferably 0 ° C. to 100 ° C., more preferably room temperature to 70 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[B-3工程]
本工程は上記B-2で得られた化合物b3に、適当な縮合剤存在下又は非存在下、適当な塩基存在下、適当な溶媒中でカルボン酸b8又は酸クロライドb9と反応させることにより、化合物b4を得る工程である。本工程において使用される縮合剤は、例えばEDCI(塩酸塩を含む)又はHBTUが挙げられる。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはジイソプロピルエチルアミン又はトリエチルアミンが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはジメチルホルムアミド、テトラヒドロフラン又は塩化メチレンが挙げられる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは0℃~150℃であり、より好ましくは室温~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step B-3]
In this step, compound b3 obtained in B-2 above is reacted with carboxylic acid b8 or acid chloride b9 in a suitable solvent in the presence or absence of a suitable condensing agent and in the presence of a suitable base. In this step, compound b4 is obtained. Examples of the condensing agent used in this step include EDCI (including hydrochloride) or HBTU. The base used in this step is selected from the bases exemplified below, and preferably diisopropylethylamine or triethylamine. The solvent used in this step is selected from the solvents exemplified below, and preferably dimethylformamide, tetrahydrofuran or methylene chloride. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably 0 ° C. to 150 ° C., more preferably room temperature to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記B-2で得られた化合物b3に、適当な縮合剤存在下又は非存在下、適当な塩基存在下、適当な溶媒中でカルボン酸b8又は酸クロライドb9と反応させることにより、化合物b4を得る工程である。本工程において使用される縮合剤は、例えばEDCI(塩酸塩を含む)又はHBTUが挙げられる。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはジイソプロピルエチルアミン又はトリエチルアミンが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはジメチルホルムアミド、テトラヒドロフラン又は塩化メチレンが挙げられる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは0℃~150℃であり、より好ましくは室温~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step B-3]
In this step, compound b3 obtained in B-2 above is reacted with carboxylic acid b8 or acid chloride b9 in a suitable solvent in the presence or absence of a suitable condensing agent and in the presence of a suitable base. In this step, compound b4 is obtained. Examples of the condensing agent used in this step include EDCI (including hydrochloride) or HBTU. The base used in this step is selected from the bases exemplified below, and preferably diisopropylethylamine or triethylamine. The solvent used in this step is selected from the solvents exemplified below, and preferably dimethylformamide, tetrahydrofuran or methylene chloride. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably 0 ° C. to 150 ° C., more preferably room temperature to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[B-4工程]
本工程は上記B-3で得られた化合物b4を、適当な酸化剤と反応させることにより、化合物b5を得る工程である。本工程において使用される酸化剤は、例えばデスマーチン試薬が挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくは塩化メチレン又はDMSOである。類似反応として、例えば、国際公開第2010/138589号パンフレットなどに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step B-4]
This step is a step of obtaining compound b5 by reacting compound b4 obtained in B-3 with an appropriate oxidizing agent. Examples of the oxidizing agent used in this step include a desmartin reagent. The solvent used in this step is selected from the solvents exemplified below, and is preferably methylene chloride or DMSO. As a similar reaction, for example, a method described in International Publication No. 2010/138589 pamphlet is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally -78 ° C to 200 ° C, preferably -20 ° C to 150 ° C, more preferably 0 ° C to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記B-3で得られた化合物b4を、適当な酸化剤と反応させることにより、化合物b5を得る工程である。本工程において使用される酸化剤は、例えばデスマーチン試薬が挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくは塩化メチレン又はDMSOである。類似反応として、例えば、国際公開第2010/138589号パンフレットなどに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step B-4]
This step is a step of obtaining compound b5 by reacting compound b4 obtained in B-3 with an appropriate oxidizing agent. Examples of the oxidizing agent used in this step include a desmartin reagent. The solvent used in this step is selected from the solvents exemplified below, and is preferably methylene chloride or DMSO. As a similar reaction, for example, a method described in International Publication No. 2010/138589 pamphlet is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally -78 ° C to 200 ° C, preferably -20 ° C to 150 ° C, more preferably 0 ° C to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[B-5工程]
本工程は上記B-4で得られた化合物b5を、適当な脱水化試薬と反応させることにより、化合物b6を得る工程である。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはテトラヒドロフランである。本工程において使用される脱水化試薬は、例えばバージェス試薬が挙げられる。類似反応として、例えば、Synlett, 1999, 10, 1642-1644などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step B-5]
This step is a step of obtaining compound b6 by reacting compound b5 obtained in B-4 with an appropriate dehydrating reagent. The solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran. Examples of the dehydrating reagent used in this step include Burgess reagent. As a similar reaction, for example, a method described in Synlett, 1999, 10, 1642-1644 is known, and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記B-4で得られた化合物b5を、適当な脱水化試薬と反応させることにより、化合物b6を得る工程である。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはテトラヒドロフランである。本工程において使用される脱水化試薬は、例えばバージェス試薬が挙げられる。類似反応として、例えば、Synlett, 1999, 10, 1642-1644などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step B-5]
This step is a step of obtaining compound b6 by reacting compound b5 obtained in B-4 with an appropriate dehydrating reagent. The solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran. Examples of the dehydrating reagent used in this step include Burgess reagent. As a similar reaction, for example, a method described in Synlett, 1999, 10, 1642-1644 is known, and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[B-6工程]
本工程は上記B-5工程で得られた化合物b6に、上記A-4工程に準じた条件で、化合物b7を得る工程である。 [Step B-6]
This step is a step of obtaining the compound b7 from the compound b6 obtained in the step B-5 under the conditions according to the step A-4.
本工程は上記B-5工程で得られた化合物b6に、上記A-4工程に準じた条件で、化合物b7を得る工程である。 [Step B-6]
This step is a step of obtaining the compound b7 from the compound b6 obtained in the step B-5 under the conditions according to the step A-4.
製造法C(合成中間体の製法)
式(I)で表される化合物のうち、BがB-2であり、Wが硫黄原子である化合物の合成中間体となるc2は、例えば下記の製法により製造することができる。

(式中、A、R2A、R2B、R2C及びR2Dは項1に定義されるとおりであり、PXはアミノの保護基を意味する)
Production Method C (Production of synthetic intermediate)
Among the compounds represented by the formula (I), c2 which is a synthetic intermediate of a compound in which B is B-2 and W is a sulfur atom can be produced, for example, by the following production method.
(Wherein, A, R 2A, R 2B , R 2C and R 2D are as defined in claim 1, P X denotes a protective group for amino)
式(I)で表される化合物のうち、BがB-2であり、Wが硫黄原子である化合物の合成中間体となるc2は、例えば下記の製法により製造することができる。
Among the compounds represented by the formula (I), c2 which is a synthetic intermediate of a compound in which B is B-2 and W is a sulfur atom can be produced, for example, by the following production method.
保護基PXとしては、Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年) にアミノの保護基として記載されているものが挙げられる。
The protective group P X, Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
[C-1工程]
本工程は上記B-4工程で得られた化合物b5に、適当なチオカルボニル化試薬を反応させることにより、化合物c1を得る工程である。本工程において使用されるチオカルボニル化試薬は、例えばローソンズ試薬が挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはテトラヒドロフランである。類似反応として、例えば、米国公開第2010/261697号パンフレットなどに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step C-1]
This step is a step of obtaining compound c1 by reacting compound b5 obtained in step B-4 with an appropriate thiocarbonylating reagent. Examples of the thiocarbonylation reagent used in this step include Lawson's reagent. The solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran. As a similar reaction, for example, a method described in US Publication No. 2010/261617, etc. is known, and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記B-4工程で得られた化合物b5に、適当なチオカルボニル化試薬を反応させることにより、化合物c1を得る工程である。本工程において使用されるチオカルボニル化試薬は、例えばローソンズ試薬が挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはテトラヒドロフランである。類似反応として、例えば、米国公開第2010/261697号パンフレットなどに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step C-1]
This step is a step of obtaining compound c1 by reacting compound b5 obtained in step B-4 with an appropriate thiocarbonylating reagent. Examples of the thiocarbonylation reagent used in this step include Lawson's reagent. The solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran. As a similar reaction, for example, a method described in US Publication No. 2010/261617, etc. is known, and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[C-2工程]
本工程は上記C-1工程で得られた化合物c1に、上記A-4工程に準じた条件で、化合物c2を得る工程である。 [Step C-2]
In this step, compound c2 is obtained from compound c1 obtained in step C-1 under the same conditions as in step A-4.
本工程は上記C-1工程で得られた化合物c1に、上記A-4工程に準じた条件で、化合物c2を得る工程である。 [Step C-2]
In this step, compound c2 is obtained from compound c1 obtained in step C-1 under the same conditions as in step A-4.
製造法D(合成中間体の製法)
式(I)で表される化合物のうち、BがB-3であり、Wが酸素原子である化合物の合成中間体となるd4は、例えば下記の製法により製造することができる。

(式中、A、R2A、R2B、R2C及びR2Dは項1に定義されるとおりであり、PXはアミノの保護基を意味する)
Production Method D (Production Method of Synthetic Intermediate)
Among the compounds represented by the formula (I), d4 that is a synthetic intermediate of a compound in which B is B-3 and W is an oxygen atom can be produced, for example, by the following production method.
(Wherein, A, R 2A, R 2B , R 2C and R 2D are as defined in claim 1, P X denotes a protective group for amino)
式(I)で表される化合物のうち、BがB-3であり、Wが酸素原子である化合物の合成中間体となるd4は、例えば下記の製法により製造することができる。
Among the compounds represented by the formula (I), d4 that is a synthetic intermediate of a compound in which B is B-3 and W is an oxygen atom can be produced, for example, by the following production method.
保護基PXとしては、Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年) にアミノの保護基として記載されているものが挙げられる。
The protective group P X, Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
化合物d1は対応するアルコール又はアルデヒドの酸化や対応するエステル加水分解などの既知の方法により合成できるか、又は市販品として購入できる。
Compound d1 can be synthesized by a known method such as oxidation of the corresponding alcohol or aldehyde or corresponding ester hydrolysis, or can be purchased as a commercial product.
[D-1工程]
本工程は化合物d1と化合物d5を、上記B-3工程に準じた条件で反応させて、化合物d2を得る工程である。 [Step D-1]
This step is a step in which compound d1 and compound d5 are reacted under the same conditions as in step B-3 to obtain compound d2.
本工程は化合物d1と化合物d5を、上記B-3工程に準じた条件で反応させて、化合物d2を得る工程である。 [Step D-1]
This step is a step in which compound d1 and compound d5 are reacted under the same conditions as in step B-3 to obtain compound d2.
[D-2工程]
本工程は上記D-1工程で得られた化合物d2を、上記B-5工程に準じた条件で反応させて、化合物d3を得る工程である。 [Step D-2]
This step is a step of obtaining a compound d3 by reacting the compound d2 obtained in the step D-1 with the conditions according to the step B-5.
本工程は上記D-1工程で得られた化合物d2を、上記B-5工程に準じた条件で反応させて、化合物d3を得る工程である。 [Step D-2]
This step is a step of obtaining a compound d3 by reacting the compound d2 obtained in the step D-1 with the conditions according to the step B-5.
[D-3工程]
本工程は上記D-2工程で得られた化合物d3を、上記A-4工程に準じた条件で反応させて、化合物d4を得る工程である。 [Step D-3]
This step is a step of obtaining a compound d4 by reacting the compound d3 obtained in the step D-2 with the conditions according to the step A-4.
本工程は上記D-2工程で得られた化合物d3を、上記A-4工程に準じた条件で反応させて、化合物d4を得る工程である。 [Step D-3]
This step is a step of obtaining a compound d4 by reacting the compound d3 obtained in the step D-2 with the conditions according to the step A-4.
製造法E(合成中間体の製法)
式(I)で表される化合物のうち、BがB-3であり、Wが硫黄原子である化合物の合成中間体となるe2は、例えば下記の製法により製造することができる。

(式中、A、R2A、R2B、R2C及びR2Dは項1に定義されるとおりであり、PXはアミノの保護基を意味する)
Production method E (Production method of synthetic intermediate)
Among the compounds represented by the formula (I), e2 which is a synthetic intermediate of a compound in which B is B-3 and W is a sulfur atom can be produced by, for example, the following production method.
(Wherein, A, R 2A, R 2B , R 2C and R 2D are as defined in claim 1, P X denotes a protective group for amino)
式(I)で表される化合物のうち、BがB-3であり、Wが硫黄原子である化合物の合成中間体となるe2は、例えば下記の製法により製造することができる。
Among the compounds represented by the formula (I), e2 which is a synthetic intermediate of a compound in which B is B-3 and W is a sulfur atom can be produced by, for example, the following production method.
保護基PXとしては、Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年) にアミノの保護基として記載されているものが挙げられる。
The protective group P X, Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
[E-1工程]
本工程は上記D-1工程で得られた化合物d2を、上記C-1工程に準じた条件で反応させて、化合物e1得る工程である。 [Step E-1]
This step is a step of obtaining a compound e1 by reacting the compound d2 obtained in the step D-1 with the conditions according to the step C-1.
本工程は上記D-1工程で得られた化合物d2を、上記C-1工程に準じた条件で反応させて、化合物e1得る工程である。 [Step E-1]
This step is a step of obtaining a compound e1 by reacting the compound d2 obtained in the step D-1 with the conditions according to the step C-1.
[E-2工程]
本工程は上記E-1工程で得られた化合物e1を、上記A-4工程に準じた条件で反応させて、化合物e2得る工程である。 [Step E-2]
This step is a step of obtaining a compound e2 by reacting the compound e1 obtained in the step E-1 with the conditions according to the step A-4.
本工程は上記E-1工程で得られた化合物e1を、上記A-4工程に準じた条件で反応させて、化合物e2得る工程である。 [Step E-2]
This step is a step of obtaining a compound e2 by reacting the compound e1 obtained in the step E-1 with the conditions according to the step A-4.
製造法F(合成中間体の製法)
式(I)で表される化合物のうち、BがB-4であり、Wが酸素原子である化合物の合成中間体となるf8は、例えば下記の製法により製造することができる。

(式中、A、R2A、R2B、R2C及びR2Dは項1に定義されるとおりであり、PXはアミノの保護基を意味する)
Production Method F (Production of synthetic intermediate)
Of the compounds represented by formula (I), f8, which is a synthetic intermediate of a compound in which B is B-4 and W is an oxygen atom, can be produced, for example, by the following production method.
(Wherein, A, R 2A, R 2B , R 2C and R 2D are as defined in claim 1, P X denotes a protective group for amino)
式(I)で表される化合物のうち、BがB-4であり、Wが酸素原子である化合物の合成中間体となるf8は、例えば下記の製法により製造することができる。
Of the compounds represented by formula (I), f8, which is a synthetic intermediate of a compound in which B is B-4 and W is an oxygen atom, can be produced, for example, by the following production method.
保護基PXとしては、Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年) にアミノの保護基として記載されているものが挙げられる。
The protective group P X, Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
化合物f1は市販品として購入できる。化合物f4は、対応するアルコールの酸化などの既知の方法により合成できるか、又は市販品として購入できる。
Compound f1 can be purchased as a commercial product. Compound f4 can be synthesized by known methods such as oxidation of the corresponding alcohol, or can be purchased as a commercial product.
[F-1工程]
本工程は化合物f1と適当な塩基を用いて、ジアゾ化試薬と反応させることにより、化合物f2を得る工程である。本工程において使用されるジアゾ化試薬は、例えば4-トルエンスルフォニルアジドが挙げられる。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくは水素化ナトリウムである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはテトラヒドロフランである。類似反応として、例えば、Bioorganic and Medicinal Chemistry Letters, 2013, 23(19), 5267-5269などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step F-1]
In this step, compound f2 is obtained by reacting compound f1 with a diazotizing reagent using an appropriate base. Examples of the diazotizing reagent used in this step include 4-toluenesulfonyl azide. The base used in this step is selected from the bases exemplified below, and is preferably sodium hydride. The solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran. As a similar reaction, for example, the methods described in Bioorganic and Medicinal Chemistry Letters, 2013, 23 (19), 5267-5269, etc. are known, and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は化合物f1と適当な塩基を用いて、ジアゾ化試薬と反応させることにより、化合物f2を得る工程である。本工程において使用されるジアゾ化試薬は、例えば4-トルエンスルフォニルアジドが挙げられる。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくは水素化ナトリウムである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはテトラヒドロフランである。類似反応として、例えば、Bioorganic and Medicinal Chemistry Letters, 2013, 23(19), 5267-5269などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step F-1]
In this step, compound f2 is obtained by reacting compound f1 with a diazotizing reagent using an appropriate base. Examples of the diazotizing reagent used in this step include 4-toluenesulfonyl azide. The base used in this step is selected from the bases exemplified below, and is preferably sodium hydride. The solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran. As a similar reaction, for example, the methods described in Bioorganic and Medicinal Chemistry Letters, 2013, 23 (19), 5267-5269, etc. are known, and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[F-2工程]
本工程は上記F-1工程で得られる化合物f2と化合物b1を、適当な塩基存在下で反応させることにより、化合物f3を得る工程である。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくは炭酸カリウムである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはメタノールである。類似反応として、例えば、Journal of the American Chemical Society, 2003, 125(13), 3714-3715などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step F-2]
This step is a step of obtaining compound f3 by reacting compound f2 obtained in step F-1 with compound b1 in the presence of a suitable base. The base used in this step is selected from the bases exemplified below, and is preferably potassium carbonate. The solvent used in this step is selected from the solvents exemplified below, but is preferably methanol. As a similar reaction, for example, a method described in Journal of the American Chemical Society, 2003, 125 (13), 3714-3715 is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally -78 ° C to 200 ° C, preferably -20 ° C to 150 ° C, more preferably 0 ° C to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記F-1工程で得られる化合物f2と化合物b1を、適当な塩基存在下で反応させることにより、化合物f3を得る工程である。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくは炭酸カリウムである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはメタノールである。類似反応として、例えば、Journal of the American Chemical Society, 2003, 125(13), 3714-3715などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step F-2]
This step is a step of obtaining compound f3 by reacting compound f2 obtained in step F-1 with compound b1 in the presence of a suitable base. The base used in this step is selected from the bases exemplified below, and is preferably potassium carbonate. The solvent used in this step is selected from the solvents exemplified below, but is preferably methanol. As a similar reaction, for example, a method described in Journal of the American Chemical Society, 2003, 125 (13), 3714-3715 is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally -78 ° C to 200 ° C, preferably -20 ° C to 150 ° C, more preferably 0 ° C to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[F-3工程]
本工程は化合物f4とヒドロキシルアミンを反応させて、化合物f5を得る工程である。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはメタノールである。類似反応として、例えば、Organic Letters, 2001, 3(26), 4209-4211などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step F-3]
This step is a step of reacting compound f4 and hydroxylamine to obtain compound f5. The solvent used in this step is selected from the solvents exemplified below, but is preferably methanol. As a similar reaction, for example, a method described in Organic Letters, 2001, 3 (26), 4209-4211, etc. is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally -78 ° C to 200 ° C, preferably -20 ° C to 150 ° C, more preferably 0 ° C to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は化合物f4とヒドロキシルアミンを反応させて、化合物f5を得る工程である。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはメタノールである。類似反応として、例えば、Organic Letters, 2001, 3(26), 4209-4211などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step F-3]
This step is a step of reacting compound f4 and hydroxylamine to obtain compound f5. The solvent used in this step is selected from the solvents exemplified below, but is preferably methanol. As a similar reaction, for example, a method described in Organic Letters, 2001, 3 (26), 4209-4211, etc. is known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally -78 ° C to 200 ° C, preferably -20 ° C to 150 ° C, more preferably 0 ° C to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[F-4工程]
本工程は上記F-3工程で得られる化合物f5とクロロ化試薬を反応させて、化合物f6を得る工程である。本工程において使用されるクロロ化試薬は、例えばN-クロロスクシンイミドが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはDMFである。類似反応として、例えば、Bioorganic and Medicinal Chemistry Letters, 2006, 16(21), 5576-5579、Journal of Heterocyclic Chemistry, 1996, 33(6), 1583-1592などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step F-4]
This step is a step of reacting the compound f5 obtained in the step F-3 with a chlorinating reagent to obtain the compound f6. Examples of the chlorinating reagent used in this step include N-chlorosuccinimide. The solvent used in this step is selected from the solvents exemplified below, but is preferably DMF. Similar reactions are described in, for example, Bioorganic and Medicinal Chemistry Letters, 2006, 16 (21), 5576-5579, Journal of Heterocyclic Chemistry, 1996, 33 (6), 1583-1592, and the like. It can be synthesized similarly. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記F-3工程で得られる化合物f5とクロロ化試薬を反応させて、化合物f6を得る工程である。本工程において使用されるクロロ化試薬は、例えばN-クロロスクシンイミドが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはDMFである。類似反応として、例えば、Bioorganic and Medicinal Chemistry Letters, 2006, 16(21), 5576-5579、Journal of Heterocyclic Chemistry, 1996, 33(6), 1583-1592などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step F-4]
This step is a step of reacting the compound f5 obtained in the step F-3 with a chlorinating reagent to obtain the compound f6. Examples of the chlorinating reagent used in this step include N-chlorosuccinimide. The solvent used in this step is selected from the solvents exemplified below, but is preferably DMF. Similar reactions are described in, for example, Bioorganic and Medicinal Chemistry Letters, 2006, 16 (21), 5576-5579, Journal of Heterocyclic Chemistry, 1996, 33 (6), 1583-1592, and the like. It can be synthesized similarly. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[F-5工程]
本工程は上記F-2工程で得られる化合物f3と上記F-4工程で得られる化合物f6を、適当な塩基存在下で反応させて、化合物f7を得る工程である。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはトリエチルアミンである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはテトラヒドロフランである。類似反応として、例えば、Journal of Medicinal Chemistry, 2003, 46(2), 284-302、European Journal of Medicinal Chemistry, 2012, 54, 324-342などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step F-5]
In this step, compound f3 obtained in step F-2 and compound f6 obtained in step F-4 are reacted in the presence of a suitable base to obtain compound f7. The base used in this step is selected from the bases exemplified below, but is preferably triethylamine. The solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran. As similar reactions, for example, the methods described in Journal of Medicinal Chemistry, 2003, 46 (2), 284-302, European Journal of Medicinal Chemistry, 2012, 54, 324-342, and the like are known. can do. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記F-2工程で得られる化合物f3と上記F-4工程で得られる化合物f6を、適当な塩基存在下で反応させて、化合物f7を得る工程である。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはトリエチルアミンである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはテトラヒドロフランである。類似反応として、例えば、Journal of Medicinal Chemistry, 2003, 46(2), 284-302、European Journal of Medicinal Chemistry, 2012, 54, 324-342などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step F-5]
In this step, compound f3 obtained in step F-2 and compound f6 obtained in step F-4 are reacted in the presence of a suitable base to obtain compound f7. The base used in this step is selected from the bases exemplified below, but is preferably triethylamine. The solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran. As similar reactions, for example, the methods described in Journal of Medicinal Chemistry, 2003, 46 (2), 284-302, European Journal of Medicinal Chemistry, 2012, 54, 324-342, and the like are known. can do. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[F-6工程]
本工程は上記F-5工程で得られた化合物f7を、上記A-4工程に準じた条件で反応させて、化合物f8を得る工程である。 [Step F-6]
This step is a step of obtaining compound f8 by reacting compound f7 obtained in step F-5 with the conditions according to step A-4.
本工程は上記F-5工程で得られた化合物f7を、上記A-4工程に準じた条件で反応させて、化合物f8を得る工程である。 [Step F-6]
This step is a step of obtaining compound f8 by reacting compound f7 obtained in step F-5 with the conditions according to step A-4.
製造法G(合成中間体の製法)
式(I)で表される化合物のうち、BがB-5であり、Wが酸素原子である化合物の合成中間体となるg5は、例えば下記の製法により製造することができる。
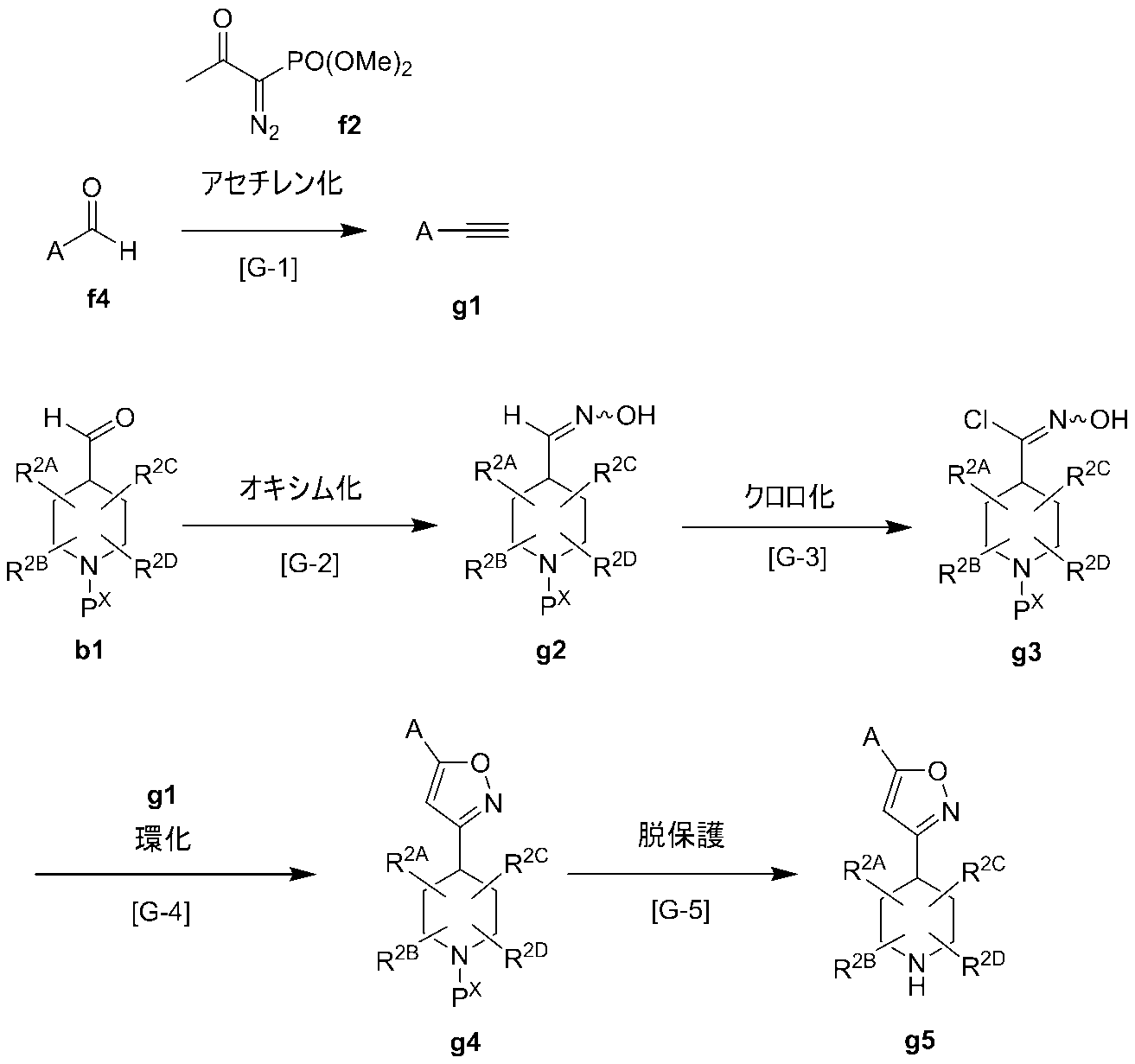
(式中、A、R2A、R2B、R2C及びR2Dは項1に定義されるとおりであり、PXはアミノの保護基を意味する)
Production method G (Production method of synthetic intermediate)
Among the compounds represented by the formula (I), g5 which is a synthetic intermediate of a compound in which B is B-5 and W is an oxygen atom can be produced, for example, by the following production method.
(Wherein, A, R 2A, R 2B , R 2C and R 2D are as defined in claim 1, P X denotes a protective group for amino)
式(I)で表される化合物のうち、BがB-5であり、Wが酸素原子である化合物の合成中間体となるg5は、例えば下記の製法により製造することができる。
Among the compounds represented by the formula (I), g5 which is a synthetic intermediate of a compound in which B is B-5 and W is an oxygen atom can be produced, for example, by the following production method.
保護基PXとしては、Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年) にアミノの保護基として記載されているものが挙げられる。
The protective group P X, Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
化合物f4は、対応するアルコールの酸化などの既知の方法により合成できるか、又は市販品として購入できる。
Compound f4 can be synthesized by a known method such as oxidation of the corresponding alcohol, or can be purchased as a commercial product.
化合物b1は対応するアルコールの酸化などの既知の方法により合成できるか、又は市販品として購入できる。
Compound b1 can be synthesized by a known method such as oxidation of the corresponding alcohol, or can be purchased as a commercial product.
[G-1工程]
本工程は化合物f4と上記F-1工程で得られる化合物f2を、上記F-2工程に準じた条件で反応させて、化合物g1を得る工程である。 [Step G-1]
This step is a step of obtaining compound g1 by reacting compound f4 with compound f2 obtained in Step F-1 under the same conditions as in Step F-2.
本工程は化合物f4と上記F-1工程で得られる化合物f2を、上記F-2工程に準じた条件で反応させて、化合物g1を得る工程である。 [Step G-1]
This step is a step of obtaining compound g1 by reacting compound f4 with compound f2 obtained in Step F-1 under the same conditions as in Step F-2.
[G-2工程]
本工程は化合物b1を、上記F-3工程に準じた条件で反応させて、化合物g2を得る工程である。 [Step G-2]
This step is a step of obtaining compound g2 by reacting compound b1 under the conditions according to the above F-3 step.
本工程は化合物b1を、上記F-3工程に準じた条件で反応させて、化合物g2を得る工程である。 [Step G-2]
This step is a step of obtaining compound g2 by reacting compound b1 under the conditions according to the above F-3 step.
[G-3工程]
本工程は上記G-2工程で得られる化合物g2を、上記F-4工程に準じた条件で反応させて、化合物g3を得る工程である。 [Step G-3]
This step is a step of obtaining compound g3 by reacting compound g2 obtained in the above step G-2 under the same conditions as in the above step F-4.
本工程は上記G-2工程で得られる化合物g2を、上記F-4工程に準じた条件で反応させて、化合物g3を得る工程である。 [Step G-3]
This step is a step of obtaining compound g3 by reacting compound g2 obtained in the above step G-2 under the same conditions as in the above step F-4.
[G-4工程]
本工程は上記G-3工程で得られる化合物g3と上記G-1工程で得られるg1を、上記工程F-5に準じた条件で反応させて、化合物g4を得る工程である。 [Step G-4]
This step is a step of obtaining compound g4 by reacting compound g3 obtained in step G-3 and g1 obtained in step G-1 under the same conditions as in step F-5.
本工程は上記G-3工程で得られる化合物g3と上記G-1工程で得られるg1を、上記工程F-5に準じた条件で反応させて、化合物g4を得る工程である。 [Step G-4]
This step is a step of obtaining compound g4 by reacting compound g3 obtained in step G-3 and g1 obtained in step G-1 under the same conditions as in step F-5.
[G-5工程]
本工程は上記G-4工程で得られる化合物g4を、上記A-4工程に準じた条件で反応させて、化合物g5を得る工程である。 [Step G-5]
This step is a step of obtaining compound g5 by reacting compound g4 obtained in the above step G-4 with the conditions according to the above step A-4.
本工程は上記G-4工程で得られる化合物g4を、上記A-4工程に準じた条件で反応させて、化合物g5を得る工程である。 [Step G-5]
This step is a step of obtaining compound g5 by reacting compound g4 obtained in the above step G-4 with the conditions according to the above step A-4.
製造法H
式(I)で表される化合物のうち、BがB-1であり、X-Y-ZがN-CO-NR3AR3Bである式[H1]、[H2]及び[H3]で表される化合物(以下、化合物H1、H2、H3とも称する)は、例えば下記の製法により製造することができる。また、同様の製造法を用いて、上記中間体製造法Aで得られる中間体a8及びa10を出発原料として式[H2]及び[H3]で表される化合物を製造することができる。さらに、同様の製造法を用いて、上記中間体製造法B、C、D、E、F及びGで得られる中間体b7、c2、d4、e2、f8及びg5を出発物質として、BがB-2、B-3、B-4及びB-5であり、X-Y-ZがN-CO-NR3AR3Bであり、Wが酸素原子又は硫黄原子で表される対応の化合物群を製造することができる。

(式中、A、R2A、R2B、R2C、R2D、R3A及びR3Bは項1に定義されるとおりであり、RYは水素、ニトロ又はシアノを表し、R1Xはハロゲンを表し、R1Yは項1に定義されるR1のうちハロゲンではないものを意味する)
Manufacturing method H
Of the compounds represented by the formula (I), the compounds represented by the formulas [H1], [H2] and [H3], wherein B is B-1 and XYZ is N—CO—NR 3A R 3B The compounds to be prepared (hereinafter also referred to as compounds H1, H2, and H3) can be produced, for example, by the following production method. Further, using the same production method, compounds represented by the formulas [H2] and [H3] can be produced using the intermediates a8 and a10 obtained by the intermediate production method A as starting materials. Further, using the same production method, the intermediates B7, c2, d4, e2, f8 and g5 obtained by the above intermediate production methods B, C, D, E, F and G are used as starting materials. -2, B-3, B-4 and B-5, XYZ is N-CO-NR 3A R 3B , and W is an oxygen atom or a sulfur atom. Can be manufactured.
(In the formula, A, R 2A , R 2B , R 2C , R 2D , R 3A and R 3B are as defined in item 1; R Y represents hydrogen, nitro or cyano; and R 1X represents halogen. And R 1Y means a non-halogen among R 1 defined in item 1)
式(I)で表される化合物のうち、BがB-1であり、X-Y-ZがN-CO-NR3AR3Bである式[H1]、[H2]及び[H3]で表される化合物(以下、化合物H1、H2、H3とも称する)は、例えば下記の製法により製造することができる。また、同様の製造法を用いて、上記中間体製造法Aで得られる中間体a8及びa10を出発原料として式[H2]及び[H3]で表される化合物を製造することができる。さらに、同様の製造法を用いて、上記中間体製造法B、C、D、E、F及びGで得られる中間体b7、c2、d4、e2、f8及びg5を出発物質として、BがB-2、B-3、B-4及びB-5であり、X-Y-ZがN-CO-NR3AR3Bであり、Wが酸素原子又は硫黄原子で表される対応の化合物群を製造することができる。
Of the compounds represented by the formula (I), the compounds represented by the formulas [H1], [H2] and [H3], wherein B is B-1 and XYZ is N—CO—NR 3A R 3B The compounds to be prepared (hereinafter also referred to as compounds H1, H2, and H3) can be produced, for example, by the following production method. Further, using the same production method, compounds represented by the formulas [H2] and [H3] can be produced using the intermediates a8 and a10 obtained by the intermediate production method A as starting materials. Further, using the same production method, the intermediates B7, c2, d4, e2, f8 and g5 obtained by the above intermediate production methods B, C, D, E, F and G are used as starting materials. -2, B-3, B-4 and B-5, XYZ is N-CO-NR 3A R 3B , and W is an oxygen atom or a sulfur atom. Can be manufactured.
[H-1工程]
本工程は上記A-4工程で得られた化合物a6に、適当な塩基存在下、適当な溶媒中、ウレア化剤であるh1又はh2を反応させることにより、化合物H1を得る工程である。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはジイソプロピルエチルアミン又はトリエチルアミンである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはテトラヒドロフラン又は塩化メチレンである。類似反応として、例えば、J. Org. Chem. 1995, 60(25), 8262-8266、Bioorg. Med. Chem. Lett. 2004, 14(3), 727-779、Tetrahedron Lett. 2001, 42(8), 1445-1447などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step H-1]
This step is a step of obtaining Compound H1 by reacting Compound a6 obtained in Step A-4 with h1 or h2 which is a urea agent in a suitable solvent in the presence of a suitable base. The base used in this step is selected from the bases exemplified below, and is preferably diisopropylethylamine or triethylamine. The solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran or methylene chloride. Similar reactions include, for example, J. Org. Chem. 1995, 60 (25), 8262-8266, Bioorg. Med. Chem. Lett. 2004, 14 (3), 727-779, Tetrahedron Lett. 2001, 42 (8 ), 1445-1447, etc. are known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記A-4工程で得られた化合物a6に、適当な塩基存在下、適当な溶媒中、ウレア化剤であるh1又はh2を反応させることにより、化合物H1を得る工程である。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはジイソプロピルエチルアミン又はトリエチルアミンである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはテトラヒドロフラン又は塩化メチレンである。類似反応として、例えば、J. Org. Chem. 1995, 60(25), 8262-8266、Bioorg. Med. Chem. Lett. 2004, 14(3), 727-779、Tetrahedron Lett. 2001, 42(8), 1445-1447などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step H-1]
This step is a step of obtaining Compound H1 by reacting Compound a6 obtained in Step A-4 with h1 or h2 which is a urea agent in a suitable solvent in the presence of a suitable base. The base used in this step is selected from the bases exemplified below, and is preferably diisopropylethylamine or triethylamine. The solvent used in this step is selected from the solvents exemplified below, and is preferably tetrahydrofuran or methylene chloride. Similar reactions include, for example, J. Org. Chem. 1995, 60 (25), 8262-8266, Bioorg. Med. Chem. Lett. 2004, 14 (3), 727-779, Tetrahedron Lett. 2001, 42 (8 ), 1445-1447, etc. are known and can be synthesized in the same manner. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[H-2工程]
本工程は上記H-1工程で得られた化合物H1に、適当な溶媒中、適当な酸の存在下で、種々のハロゲン化剤を反応させることにより、化合物H2を得る工程である。本工程において使用されるハロゲン化剤は、好ましくはN-クロロスクシンイミド、N-ブロモスクシンイミド、N-ヨードスクシンイミドである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくは塩化メチレン又はジクロロエタンである。本工程において使用される酸は、後記に例示する酸等から選択されるが、好ましくはトリフルオロ酢酸又は塩酸である。類似反応として、例えば、Bioorg. Med. Chem. Lett. 2008, 18(5), 1702-1707、J. Org. Chem. 2002, 67(17), 5913-5918などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~70℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step H-2]
This step is a step for obtaining compound H2 by reacting compound H1 obtained in step H-1 with various halogenating agents in a suitable solvent in the presence of a suitable acid. The halogenating agent used in this step is preferably N-chlorosuccinimide, N-bromosuccinimide, or N-iodosuccinimide. The solvent used in this step is selected from the solvents exemplified below, and is preferably methylene chloride or dichloroethane. The acid used in this step is selected from the acids exemplified below, and preferably trifluoroacetic acid or hydrochloric acid. As a similar reaction, for example, methods described in Bioorg. Med. Chem. Lett. 2008, 18 (5), 1702-1707, J. Org. Chem. 2002, 67 (17), 5913-5918 and the like are known. And can be synthesized similarly. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 70 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記H-1工程で得られた化合物H1に、適当な溶媒中、適当な酸の存在下で、種々のハロゲン化剤を反応させることにより、化合物H2を得る工程である。本工程において使用されるハロゲン化剤は、好ましくはN-クロロスクシンイミド、N-ブロモスクシンイミド、N-ヨードスクシンイミドである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくは塩化メチレン又はジクロロエタンである。本工程において使用される酸は、後記に例示する酸等から選択されるが、好ましくはトリフルオロ酢酸又は塩酸である。類似反応として、例えば、Bioorg. Med. Chem. Lett. 2008, 18(5), 1702-1707、J. Org. Chem. 2002, 67(17), 5913-5918などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~70℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step H-2]
This step is a step for obtaining compound H2 by reacting compound H1 obtained in step H-1 with various halogenating agents in a suitable solvent in the presence of a suitable acid. The halogenating agent used in this step is preferably N-chlorosuccinimide, N-bromosuccinimide, or N-iodosuccinimide. The solvent used in this step is selected from the solvents exemplified below, and is preferably methylene chloride or dichloroethane. The acid used in this step is selected from the acids exemplified below, and preferably trifluoroacetic acid or hydrochloric acid. As a similar reaction, for example, methods described in Bioorg. Med. Chem. Lett. 2008, 18 (5), 1702-1707, J. Org. Chem. 2002, 67 (17), 5913-5918 and the like are known. And can be synthesized similarly. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 70 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[H-3工程]
本工程は上記H-2工程で得られた化合物H2に、適当な溶媒中、適当な遷移金属試薬の存在下で、適当なボロン酸誘導体と反応させることにより、化合物H3を得る工程である。本工程において使用される遷移金属試薬は、例えばトリス(ジベンジリデンアセトン)ジパラジウムやテトラキス(トリフェニルフォスフィン)パラジウムが挙げられる。本工程において使用されるボロン酸誘導体は、例えばボロン酸、ボロン酸エステル、ボロン酸ピナコラートが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはDMF又はジオキサンである。類似反応として、例えば、Tetrahedron Lett. 2003, 44(7), 1379-1382、J. Med. Chem. 2009, 52(14), 4370-4379、Bioorg. Med. Chem. Lett. 2012, 20(9), 3009-3015、J. Org. Chem. 2002, 67(10), 3365-3373、Tetrahedron Lett. 2007, 48(13), 2339-2343などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~180℃であり、より好ましくは室温~150℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step H-3]
This step is a step of obtaining compound H3 by reacting compound H2 obtained in step H-2 with an appropriate boronic acid derivative in an appropriate solvent in the presence of an appropriate transition metal reagent. Examples of the transition metal reagent used in this step include tris (dibenzylideneacetone) dipalladium and tetrakis (triphenylphosphine) palladium. Examples of the boronic acid derivative used in this step include boronic acid, boronic acid ester, and boronic acid pinacolate. The solvent used in this step is selected from the solvents exemplified below, and is preferably DMF or dioxane. Similar reactions include, for example, Tetrahedron Lett. 2003, 44 (7), 1379-1382, J. Med. Chem. 2009, 52 (14), 4370-4379, Bioorg. Med. Chem. Lett. 2012, 20 (9 ), 3009-3015, J. Org. Chem. 2002, 67 (10), 3365-3373, Tetrahedron Lett. 2007, 48 (13), 2339-2343, and the like. Can be synthesized. The reaction temperature varies depending on the type of raw material compound used, reagents and the like, but is usually −78 ° C. to 200 ° C., preferably −20 ° C. to 180 ° C., more preferably room temperature to 150 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記H-2工程で得られた化合物H2に、適当な溶媒中、適当な遷移金属試薬の存在下で、適当なボロン酸誘導体と反応させることにより、化合物H3を得る工程である。本工程において使用される遷移金属試薬は、例えばトリス(ジベンジリデンアセトン)ジパラジウムやテトラキス(トリフェニルフォスフィン)パラジウムが挙げられる。本工程において使用されるボロン酸誘導体は、例えばボロン酸、ボロン酸エステル、ボロン酸ピナコラートが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはDMF又はジオキサンである。類似反応として、例えば、Tetrahedron Lett. 2003, 44(7), 1379-1382、J. Med. Chem. 2009, 52(14), 4370-4379、Bioorg. Med. Chem. Lett. 2012, 20(9), 3009-3015、J. Org. Chem. 2002, 67(10), 3365-3373、Tetrahedron Lett. 2007, 48(13), 2339-2343などに記載されている方法が既知であり、同様に合成することができる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~180℃であり、より好ましくは室温~150℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step H-3]
This step is a step of obtaining compound H3 by reacting compound H2 obtained in step H-2 with an appropriate boronic acid derivative in an appropriate solvent in the presence of an appropriate transition metal reagent. Examples of the transition metal reagent used in this step include tris (dibenzylideneacetone) dipalladium and tetrakis (triphenylphosphine) palladium. Examples of the boronic acid derivative used in this step include boronic acid, boronic acid ester, and boronic acid pinacolate. The solvent used in this step is selected from the solvents exemplified below, and is preferably DMF or dioxane. Similar reactions include, for example, Tetrahedron Lett. 2003, 44 (7), 1379-1382, J. Med. Chem. 2009, 52 (14), 4370-4379, Bioorg. Med. Chem. Lett. 2012, 20 (9 ), 3009-3015, J. Org. Chem. 2002, 67 (10), 3365-3373, Tetrahedron Lett. 2007, 48 (13), 2339-2343, and the like. Can be synthesized. The reaction temperature varies depending on the type of raw material compound used, reagents and the like, but is usually −78 ° C. to 200 ° C., preferably −20 ° C. to 180 ° C., more preferably room temperature to 150 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
製造法I
式(I)で表される化合物のうち、BがB-1であり、X-Y-ZがN-COR4Aである式[I1][I2][I3]で表される化合物(以下、化合物I1、I2、I3とも称する)は、例えば下記の製法により製造することができる。
また、同様の製造法を用いて、上記中間体製造法Aで得られる中間体a8及びa10を出発原料として式[I2]及び[I3]で表される化合物を製造することができる。
さらに、同様の製造法を用いて、上記中間体製造法B、C、D、E、F及びGで得られる中間体b7、c2、d4、e2、f8及びg5を出発物質として、BがB-2、B-3、B-4及びB-5であり、X-Y-ZがN-COR4Aであり、Wが酸素原子又は硫黄原子で表される対応の化合物群を製造することができる。

(式中、A、R2A、R2B、R2C、R2D及びR4Aは項1に定義されるとおりであり、R1Yは項1に定義されるR1のうちハロゲンではないものを表し、R1Xはハロゲンを意味する)
Manufacturing method I
Of the compounds represented by the formula (I), compounds represented by the formula [I1] [I2] [I3] wherein B is B-1 and XYZ is N—COR 4A (hereinafter, Compounds I1, I2, and I3) can be produced, for example, by the following production method.
Further, using the same production method, compounds represented by the formulas [I2] and [I3] can be produced using the intermediates a8 and a10 obtained by the intermediate production method A as starting materials.
Further, using the same production method, the intermediates B7, c2, d4, e2, f8 and g5 obtained by the above intermediate production methods B, C, D, E, F and G are used as starting materials. -2, B-3, B-4, and B-5, XYZ is N-COR 4A , and W is an oxygen atom or a sulfur atom. it can.
(In the formula, A, R 2A , R 2B , R 2C , R 2D and R 4A are as defined in item 1, and R 1Y represents a non-halogen of R 1 defined in item 1. , R 1X means halogen)
式(I)で表される化合物のうち、BがB-1であり、X-Y-ZがN-COR4Aである式[I1][I2][I3]で表される化合物(以下、化合物I1、I2、I3とも称する)は、例えば下記の製法により製造することができる。
また、同様の製造法を用いて、上記中間体製造法Aで得られる中間体a8及びa10を出発原料として式[I2]及び[I3]で表される化合物を製造することができる。
さらに、同様の製造法を用いて、上記中間体製造法B、C、D、E、F及びGで得られる中間体b7、c2、d4、e2、f8及びg5を出発物質として、BがB-2、B-3、B-4及びB-5であり、X-Y-ZがN-COR4Aであり、Wが酸素原子又は硫黄原子で表される対応の化合物群を製造することができる。
Of the compounds represented by the formula (I), compounds represented by the formula [I1] [I2] [I3] wherein B is B-1 and XYZ is N—COR 4A (hereinafter, Compounds I1, I2, and I3) can be produced, for example, by the following production method.
Further, using the same production method, compounds represented by the formulas [I2] and [I3] can be produced using the intermediates a8 and a10 obtained by the intermediate production method A as starting materials.
Further, using the same production method, the intermediates B7, c2, d4, e2, f8 and g5 obtained by the above intermediate production methods B, C, D, E, F and G are used as starting materials. -2, B-3, B-4, and B-5, XYZ is N-COR 4A , and W is an oxygen atom or a sulfur atom. it can.
[I-1工程]
本工程は上記A-4工程で得られた化合物a6に、適当な縮合剤存在下または非存在下、適当な塩基存在下、適当な溶媒中で、カルボン酸i1又は酸クロライドi2と反応させることにより、化合物I1を得る工程である。本工程において使用される縮合剤は、例えばEDCI(塩酸塩を含む)又はHBTUである。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはジイソプロピルエチルアミン又はトリエチルアミンである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはジメチルホルムアミド、テトラヒドロフラン又は塩化メチレンである。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step I-1]
In this step, compound a6 obtained in step A-4 is reacted with carboxylic acid i1 or acid chloride i2 in the presence of a suitable base in the presence or absence of a suitable condensing agent and in the presence of a suitable base. To obtain compound I1. The condensing agent used in this step is, for example, EDCI (including hydrochloride) or HBTU. The base used in this step is selected from the bases exemplified below, and is preferably diisopropylethylamine or triethylamine. The solvent used in this step is selected from the solvents exemplified below, and preferably dimethylformamide, tetrahydrofuran or methylene chloride. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記A-4工程で得られた化合物a6に、適当な縮合剤存在下または非存在下、適当な塩基存在下、適当な溶媒中で、カルボン酸i1又は酸クロライドi2と反応させることにより、化合物I1を得る工程である。本工程において使用される縮合剤は、例えばEDCI(塩酸塩を含む)又はHBTUである。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはジイソプロピルエチルアミン又はトリエチルアミンである。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはジメチルホルムアミド、テトラヒドロフラン又は塩化メチレンである。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは-20℃~150℃であり、より好ましくは0℃~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step I-1]
In this step, compound a6 obtained in step A-4 is reacted with carboxylic acid i1 or acid chloride i2 in the presence of a suitable base in the presence or absence of a suitable condensing agent and in the presence of a suitable base. To obtain compound I1. The condensing agent used in this step is, for example, EDCI (including hydrochloride) or HBTU. The base used in this step is selected from the bases exemplified below, and is preferably diisopropylethylamine or triethylamine. The solvent used in this step is selected from the solvents exemplified below, and preferably dimethylformamide, tetrahydrofuran or methylene chloride. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably −20 ° C. to 150 ° C., more preferably 0 ° C. to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[I-2工程]
本工程は上記I-1工程で得られる化合物I1を、上記H-2工程に準じた条件で反応させて、化合物I2を得る工程である。 [Step I-2]
This step is a step of obtaining compound I2 by reacting compound I1 obtained in the above step I-1 under the same conditions as in the above step H-2.
本工程は上記I-1工程で得られる化合物I1を、上記H-2工程に準じた条件で反応させて、化合物I2を得る工程である。 [Step I-2]
This step is a step of obtaining compound I2 by reacting compound I1 obtained in the above step I-1 under the same conditions as in the above step H-2.
[I-3工程]
本工程は上記I-2工程で得られる化合物I2を、上記H-3工程に準じた条件で反応させて、化合物I3を得る工程である。 [Step I-3]
This step is a step of obtaining compound I3 by reacting compound I2 obtained in the above step I-2 with the conditions according to the above step H-3.
本工程は上記I-2工程で得られる化合物I2を、上記H-3工程に準じた条件で反応させて、化合物I3を得る工程である。 [Step I-3]
This step is a step of obtaining compound I3 by reacting compound I2 obtained in the above step I-2 with the conditions according to the above step H-3.
製造法J
式(I)で表される化合物のうち、BがB-4であり、X-Y-ZがCR5-NR6-COR4Bである式[J1][J2][J3]及び[J4]で表される化合物(以下、化合物J1、J2、J3及びJ4とも称する)は、例えば上記中間体製造方法Fに準じた下記の製法により製造することができる。
また、下記製造法Jと上記中間体製造法A、B、C、D、E及びGに準じた製法を組み合わせることにより、X-Y-ZがCR5-NR6-COR4Bであり、BがB-1、B-2、B-3及びB-5であり、Wが酸素原子又は硫黄原子で表される対応の化合物群を製造することができる。

(式中、A、R2A、R2B、R2C、R2D、R4B、R5及びR6は項1に定義されるとおりであり、R1Xはハロゲンを表し、R1Yは項1に定義されるR1のうちハロゲンではないものを表し、RZはハロゲン等の脱離基を表し、PXはアミノの保護基を意味する)
Manufacturing method J
Of the compounds represented by formula (I), the formulas [J1] [J2] [J3] and [J4] wherein B is B-4 and XYZ is CR 5 —NR 6 —COR 4B (Hereinafter also referred to as compounds J1, J2, J3, and J4) can be produced, for example, by the following production method according to the intermediate production method F.
Further, by combining the following production method J and the production methods according to the intermediate production methods A, B, C, D, E and G, XYZ is CR 5 —NR 6 —COR 4B ; Is a corresponding compound group in which is B-1, B-2, B-3 and B-5, and W is an oxygen atom or a sulfur atom.
(In the formula, A, R 2A , R 2B , R 2C , R 2D , R 4B , R 5 and R 6 are as defined in item 1; R 1X represents halogen; R 1Y represents item 1; R 1 which is not halogen among the defined R 1 represents R Z represents a leaving group such as halogen, and P X represents an amino protecting group)
式(I)で表される化合物のうち、BがB-4であり、X-Y-ZがCR5-NR6-COR4Bである式[J1][J2][J3]及び[J4]で表される化合物(以下、化合物J1、J2、J3及びJ4とも称する)は、例えば上記中間体製造方法Fに準じた下記の製法により製造することができる。
また、下記製造法Jと上記中間体製造法A、B、C、D、E及びGに準じた製法を組み合わせることにより、X-Y-ZがCR5-NR6-COR4Bであり、BがB-1、B-2、B-3及びB-5であり、Wが酸素原子又は硫黄原子で表される対応の化合物群を製造することができる。
Of the compounds represented by formula (I), the formulas [J1] [J2] [J3] and [J4] wherein B is B-4 and XYZ is CR 5 —NR 6 —COR 4B (Hereinafter also referred to as compounds J1, J2, J3, and J4) can be produced, for example, by the following production method according to the intermediate production method F.
Further, by combining the following production method J and the production methods according to the intermediate production methods A, B, C, D, E and G, XYZ is CR 5 —NR 6 —COR 4B ; Is a corresponding compound group in which is B-1, B-2, B-3 and B-5, and W is an oxygen atom or a sulfur atom.
保護基PXとしては、Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年) にアミノの保護基として記載されているものが挙げられる。
The protective group P X, Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group for amino.
化合物j1は、対応するアルコールの酸化などの既知の方法により合成できるか、又は市販品として購入できる。
Compound j1 can be synthesized by a known method such as oxidation of the corresponding alcohol, or can be purchased as a commercial product.
[J-1工程]
本工程は化合物j1を、上記F-2工程に準じた条件で反応させて、化合物j2を得る工程である。 [Step J-1]
This step is a step wherein compound j2 is obtained by reacting compound j1 under the same conditions as in Step F-2.
本工程は化合物j1を、上記F-2工程に準じた条件で反応させて、化合物j2を得る工程である。 [Step J-1]
This step is a step wherein compound j2 is obtained by reacting compound j1 under the same conditions as in Step F-2.
[J-2工程]
本工程は上記J-1工程で得られる化合物j2を、上記F-5工程に準じた条件で反応させて、化合物j3を得る工程である。 [Step J-2]
This step is a step of obtaining compound j3 by reacting compound j2 obtained in step J-1 under the same conditions as in step F-5.
本工程は上記J-1工程で得られる化合物j2を、上記F-5工程に準じた条件で反応させて、化合物j3を得る工程である。 [Step J-2]
This step is a step of obtaining compound j3 by reacting compound j2 obtained in step J-1 under the same conditions as in step F-5.
[J-3工程]
本工程は上記製造法J-2で得られた化合物j3のアミノの保護基PXを、脱保護することにより、化合物j4を得る工程である。本工程はProtective Groups in Organic Synthesis(Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年)に記載されている方法等に準じて行うことができる。 [Step J-3]
This step is a protecting group P X of the amino compound j3 prepared in the above Preparation Method J-2, is deprotected, to give compound compound j4. This step can be carried out according to the method described in Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts, John Wiley & Sons, Inc., 1999).
本工程は上記製造法J-2で得られた化合物j3のアミノの保護基PXを、脱保護することにより、化合物j4を得る工程である。本工程はProtective Groups in Organic Synthesis(Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年)に記載されている方法等に準じて行うことができる。 [Step J-3]
This step is a protecting group P X of the amino compound j3 prepared in the above Preparation Method J-2, is deprotected, to give compound compound j4. This step can be carried out according to the method described in Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts, John Wiley & Sons, Inc., 1999).
[J-4工程]
本工程は上記J-3で得られた化合物j4に、適当な縮合剤存在下又は非存在下、適当な塩基存在下、適当な溶媒中でカルボン酸j5又は酸クロライドj6と反応させることにより、化合物J1を得る工程である。本工程において使用される縮合剤は、例えばEDCI(塩酸塩を含む)又はHBTUが挙げられる。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはジイソプロピルエチルアミン又はトリエチルアミンが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはジメチルホルムアミド、テトラヒドロフラン又は塩化メチレンが挙げられる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは0℃~150℃であり、より好ましくは室温~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step J-4]
In this step, compound j4 obtained in J-3 above is reacted with carboxylic acid j5 or acid chloride j6 in a suitable solvent in the presence or absence of a suitable condensing agent and in the presence of a suitable base. In this step, compound J1 is obtained. Examples of the condensing agent used in this step include EDCI (including hydrochloride) or HBTU. The base used in this step is selected from the bases exemplified below, and preferably diisopropylethylamine or triethylamine. The solvent used in this step is selected from the solvents exemplified below, and preferably dimethylformamide, tetrahydrofuran or methylene chloride. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably 0 ° C. to 150 ° C., more preferably room temperature to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記J-3で得られた化合物j4に、適当な縮合剤存在下又は非存在下、適当な塩基存在下、適当な溶媒中でカルボン酸j5又は酸クロライドj6と反応させることにより、化合物J1を得る工程である。本工程において使用される縮合剤は、例えばEDCI(塩酸塩を含む)又はHBTUが挙げられる。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはジイソプロピルエチルアミン又はトリエチルアミンが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはジメチルホルムアミド、テトラヒドロフラン又は塩化メチレンが挙げられる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは0℃~150℃であり、より好ましくは室温~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step J-4]
In this step, compound j4 obtained in J-3 above is reacted with carboxylic acid j5 or acid chloride j6 in a suitable solvent in the presence or absence of a suitable condensing agent and in the presence of a suitable base. In this step, compound J1 is obtained. Examples of the condensing agent used in this step include EDCI (including hydrochloride) or HBTU. The base used in this step is selected from the bases exemplified below, and preferably diisopropylethylamine or triethylamine. The solvent used in this step is selected from the solvents exemplified below, and preferably dimethylformamide, tetrahydrofuran or methylene chloride. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably 0 ° C. to 150 ° C., more preferably room temperature to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[J-5工程]
本工程は上記J-4工程で得られる化合物J1を、上記H-2工程に準じた条件で反応させて、化合物J2を得る工程である。 [Step J-5]
This step is a step of obtaining compound J2 by reacting compound J1 obtained in the above step J-4 under the conditions according to the above step H-2.
本工程は上記J-4工程で得られる化合物J1を、上記H-2工程に準じた条件で反応させて、化合物J2を得る工程である。 [Step J-5]
This step is a step of obtaining compound J2 by reacting compound J1 obtained in the above step J-4 under the conditions according to the above step H-2.
[J-6工程]
本工程は上記J-5工程で得られる化合物J2を、上記H-3工程に準じた条件で反応させて、化合物J3を得る工程である。 [Step J-6]
In this step, compound J2 obtained in step J-5 is reacted under the same conditions as in step H-3 to obtain compound J3.
本工程は上記J-5工程で得られる化合物J2を、上記H-3工程に準じた条件で反応させて、化合物J3を得る工程である。 [Step J-6]
In this step, compound J2 obtained in step J-5 is reacted under the same conditions as in step H-3 to obtain compound J3.
[J-7工程]
本工程は上記J-6工程で得られた化合物J3に、種々の塩基存在下、適当な溶媒中、化合物j7を反応させることにより、化合物J4を得る工程である。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくは水素化ナトリウム又はジイソプロピルアミンが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはジメチルホルムアミド又はテトラヒドロフランが挙げられる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-30℃~200℃、好ましくは0℃~150℃であり、より好ましくは0℃~80℃である。反応時間は通常1時間~48時間程度であり、好ましくは1~24時間、さらに好ましくは1~16時間である。 [Step J-7]
This step is a step of obtaining compound J4 by reacting compound j7 obtained in step J-6 with compound j7 in the presence of various bases in an appropriate solvent. The base used in this step is selected from the bases exemplified below, and preferably sodium hydride or diisopropylamine. The solvent used in this step is selected from the solvents exemplified below and preferably dimethylformamide or tetrahydrofuran. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −30 ° C. to 200 ° C., preferably 0 ° C. to 150 ° C., more preferably 0 ° C. to 80 ° C. The reaction time is usually about 1 to 48 hours, preferably 1 to 24 hours, and more preferably 1 to 16 hours.
本工程は上記J-6工程で得られた化合物J3に、種々の塩基存在下、適当な溶媒中、化合物j7を反応させることにより、化合物J4を得る工程である。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくは水素化ナトリウム又はジイソプロピルアミンが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはジメチルホルムアミド又はテトラヒドロフランが挙げられる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-30℃~200℃、好ましくは0℃~150℃であり、より好ましくは0℃~80℃である。反応時間は通常1時間~48時間程度であり、好ましくは1~24時間、さらに好ましくは1~16時間である。 [Step J-7]
This step is a step of obtaining compound J4 by reacting compound j7 obtained in step J-6 with compound j7 in the presence of various bases in an appropriate solvent. The base used in this step is selected from the bases exemplified below, and preferably sodium hydride or diisopropylamine. The solvent used in this step is selected from the solvents exemplified below and preferably dimethylformamide or tetrahydrofuran. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −30 ° C. to 200 ° C., preferably 0 ° C. to 150 ° C., more preferably 0 ° C. to 80 ° C. The reaction time is usually about 1 to 48 hours, preferably 1 to 24 hours, and more preferably 1 to 16 hours.
製造法K
式(I)で表される化合物のうち、BがB-4であり、X-Y-ZがCR5-NR6-CONR3AR3Bである式[K1][K2][K3]及び[K4]で表される化合物(以下、化合物K1、K2、K3及びK4とも称する)は、例えば下記の製法により製造することができる。
また、下記製造法K及び上記中間体製造法A、B、C、D、E及びGに準じた製法を組み合わせることにより、X-Y-ZがCR5-NR6-CONR3AR3Bであり、BがB-1、B-2、B-3及びB-5であり、Wが酸素原子又は硫黄原子で表される対応の化合物群を製造することができる。

(式中、A、R2A、R2B、R2C、R2D、R3A、R3B、R5及びR6は項1に定義されるとおりであり、R1Xはハロゲンを表し、R1Yは項1に定義されるR1のうちハロゲンではないものを表し、RYは水素、ニトロ又はシアノを表し、RZはハロゲン等の脱離基を意味する)
Manufacturing method K
Of the compounds represented by formula (I), the compounds of formula [K1] [K2] [K3] and [K3] wherein B is B-4 and XYZ is CR 5 —NR 6 —CONR 3A R 3B The compound represented by K4] (hereinafter also referred to as compounds K1, K2, K3 and K4) can be produced, for example, by the following production method.
Further, XYZ is CR 5 —NR 6 —CONR 3A R 3B by combining the following production method K and production methods according to the above intermediate production methods A, B, C, D, E and G. , B is B-1, B-2, B-3 and B-5, and corresponding compounds can be produced wherein W is an oxygen atom or a sulfur atom.
(In the formula, A, R 2A , R 2B , R 2C , R 2D , R 3A , R 3B , R 5 and R 6 are as defined in Item 1, R 1X represents halogen, and R 1Y represents R 1 defined in Item 1 is not halogen, R Y represents hydrogen, nitro or cyano, and R Z represents a leaving group such as halogen)
式(I)で表される化合物のうち、BがB-4であり、X-Y-ZがCR5-NR6-CONR3AR3Bである式[K1][K2][K3]及び[K4]で表される化合物(以下、化合物K1、K2、K3及びK4とも称する)は、例えば下記の製法により製造することができる。
また、下記製造法K及び上記中間体製造法A、B、C、D、E及びGに準じた製法を組み合わせることにより、X-Y-ZがCR5-NR6-CONR3AR3Bであり、BがB-1、B-2、B-3及びB-5であり、Wが酸素原子又は硫黄原子で表される対応の化合物群を製造することができる。
Of the compounds represented by formula (I), the compounds of formula [K1] [K2] [K3] and [K3] wherein B is B-4 and XYZ is CR 5 —NR 6 —CONR 3A R 3B The compound represented by K4] (hereinafter also referred to as compounds K1, K2, K3 and K4) can be produced, for example, by the following production method.
Further, XYZ is CR 5 —NR 6 —CONR 3A R 3B by combining the following production method K and production methods according to the above intermediate production methods A, B, C, D, E and G. , B is B-1, B-2, B-3 and B-5, and corresponding compounds can be produced wherein W is an oxygen atom or a sulfur atom.
[K-1工程]
本工程は上記J-3工程で得られる化合物j4を、上記H-1工程に準じた条件で反応させて、化合物K1を得る工程である。 [Step K-1]
This step is a step of obtaining compound K1 by reacting compound j4 obtained in the above step J-3 under the conditions according to the above step H-1.
本工程は上記J-3工程で得られる化合物j4を、上記H-1工程に準じた条件で反応させて、化合物K1を得る工程である。 [Step K-1]
This step is a step of obtaining compound K1 by reacting compound j4 obtained in the above step J-3 under the conditions according to the above step H-1.
[K-2工程]
本工程は上記K-1工程で得られる化合物K1を、上記J-7工程に準じた条件で反応させて、化合物K2を得る工程である。 [Step K-2]
This step is a step of obtaining compound K2 by reacting compound K1 obtained in the above step K-1 under the same conditions as in the above step J-7.
本工程は上記K-1工程で得られる化合物K1を、上記J-7工程に準じた条件で反応させて、化合物K2を得る工程である。 [Step K-2]
This step is a step of obtaining compound K2 by reacting compound K1 obtained in the above step K-1 under the same conditions as in the above step J-7.
[K-3工程]
本工程は上記K-2工程で得られる化合物K2を、上記H-2工程に準じた条件で反応させて、化合物K3を得る工程である。 [Step K-3]
This step is a step of obtaining a compound K3 by reacting the compound K2 obtained in the step K-2 with the conditions according to the step H-2.
本工程は上記K-2工程で得られる化合物K2を、上記H-2工程に準じた条件で反応させて、化合物K3を得る工程である。 [Step K-3]
This step is a step of obtaining a compound K3 by reacting the compound K2 obtained in the step K-2 with the conditions according to the step H-2.
[K-4工程]
本工程は上記K-3工程で得られる化合物K3を、上記H-3工程に準じた条件で反応させて、化合物K4を得る工程である。 [Step K-4]
This step is a step of obtaining a compound K4 by reacting the compound K3 obtained in the step K-3 with the conditions according to the step H-3.
本工程は上記K-3工程で得られる化合物K3を、上記H-3工程に準じた条件で反応させて、化合物K4を得る工程である。 [Step K-4]
This step is a step of obtaining a compound K4 by reacting the compound K3 obtained in the step K-3 with the conditions according to the step H-3.
製造法M
式(I)で表される化合物のうち、BがB-4であり、X-Y-ZがCR5-CO-NR3AR3Bである式[M1][M2]及び[M3]で表される化合物(以下、化合物M1、M2及びM3とも称する)は、例えば上記中間体製造方法Fに準じた下記の製法により製造することができる。
また、下記製造法Mと上記中間体製造法A、B、C、D、E及びGに準じた製法を組み合わせることにより、X-Y-ZがCR5-CO-NR3AR3Bであり、BがB-1、B-2、B-3及びB-5であり、Wが酸素原子又は硫黄原子で表される対応の化合物群を製造することができる。

(式中、A、R2A、R2B、R2C、R2D、R3A、R3B及びR5は項1に定義されるとおりであり、R1Xはハロゲンを表し、R1Yは項1に定義されるR1のうちハロゲンではないものを表し、PYはカルボキシル基の保護基を意味する)
Manufacturing method M
Of the compounds represented by the formula (I), the compounds represented by the formulas [M1] [M2] and [M3], wherein B is B-4 and XYZ is CR 5 —CO—NR 3A R 3B The compounds to be produced (hereinafter also referred to as compounds M1, M2 and M3) can be produced, for example, by the following production method according to the intermediate production method F.
Further, by combining the following production method M and the production methods according to the intermediate production methods A, B, C, D, E and G, XYZ is CR 5 —CO—NR 3A R 3B , Corresponding compound groups in which B is B-1, B-2, B-3 and B-5 and W is an oxygen atom or a sulfur atom can be produced.
(In the formula, A, R 2A , R 2B , R 2C , R 2D , R 3A , R 3B and R 5 are as defined in item 1, R 1X represents halogen, and R 1Y represents item 1 R 1 which is not halogen among the defined R 1 represents P Y represents a protecting group for a carboxyl group)
式(I)で表される化合物のうち、BがB-4であり、X-Y-ZがCR5-CO-NR3AR3Bである式[M1][M2]及び[M3]で表される化合物(以下、化合物M1、M2及びM3とも称する)は、例えば上記中間体製造方法Fに準じた下記の製法により製造することができる。
また、下記製造法Mと上記中間体製造法A、B、C、D、E及びGに準じた製法を組み合わせることにより、X-Y-ZがCR5-CO-NR3AR3Bであり、BがB-1、B-2、B-3及びB-5であり、Wが酸素原子又は硫黄原子で表される対応の化合物群を製造することができる。
Of the compounds represented by the formula (I), the compounds represented by the formulas [M1] [M2] and [M3], wherein B is B-4 and XYZ is CR 5 —CO—NR 3A R 3B The compounds to be produced (hereinafter also referred to as compounds M1, M2 and M3) can be produced, for example, by the following production method according to the intermediate production method F.
Further, by combining the following production method M and the production methods according to the intermediate production methods A, B, C, D, E and G, XYZ is CR 5 —CO—NR 3A R 3B , Corresponding compound groups in which B is B-1, B-2, B-3 and B-5 and W is an oxygen atom or a sulfur atom can be produced.
保護基PYとしては、Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年) にカルボン酸の保護基として記載されているものが挙げられる。
The protective group P Y, Protective Groups in Organic Synthesis (Theodora W. Greene, Peter GM Wuts al, John Wiley & Sons, Inc. published in 1999), and those described in as a protecting group of a carboxylic acid .
化合物m1は、対応するアルコールの酸化などの既知の方法により合成できるか、又は市販品として購入できる。
Compound m1 can be synthesized by a known method such as oxidation of the corresponding alcohol, or can be purchased as a commercial product.
[M-1工程]
本工程は化合物m1を、上記F-2工程に準じた条件で反応させて、化合物m2を得る工程である。 [Step M-1]
This step is a step of obtaining compound m2 by reacting compound m1 under the same conditions as in Step F-2.
本工程は化合物m1を、上記F-2工程に準じた条件で反応させて、化合物m2を得る工程である。 [Step M-1]
This step is a step of obtaining compound m2 by reacting compound m1 under the same conditions as in Step F-2.
[M-2工程]
本工程は上記M-1工程で得られる化合物m2を、上記F-5工程に準じた条件で反応させて、化合物m3を得る工程である。 [Step M-2]
This step is a step of obtaining a compound m3 by reacting the compound m2 obtained in the step M-1 with the conditions according to the step F-5.
本工程は上記M-1工程で得られる化合物m2を、上記F-5工程に準じた条件で反応させて、化合物m3を得る工程である。 [Step M-2]
This step is a step of obtaining a compound m3 by reacting the compound m2 obtained in the step M-1 with the conditions according to the step F-5.
[M-3工程]
本工程は上記M-2工程で得られた化合物m3のカルボン酸の保護基PYを、脱保護することにより、化合物m4を得る工程である。本工程はProtective Groups in Organic Synthesis(Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年)に記載されている方法等に準じて行うことができる。 [M-3 process]
This step is a step of obtaining the compound m4 by deprotecting the protecting group P Y of the carboxylic acid of the compound m3 obtained in the step M-2. This step can be carried out according to the method described in Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts, John Wiley & Sons, Inc., 1999).
本工程は上記M-2工程で得られた化合物m3のカルボン酸の保護基PYを、脱保護することにより、化合物m4を得る工程である。本工程はProtective Groups in Organic Synthesis(Theodora W. Greene, Peter G. M. Wuts著、John Wiley & Sons, Inc.発行、1999年)に記載されている方法等に準じて行うことができる。 [M-3 process]
This step is a step of obtaining the compound m4 by deprotecting the protecting group P Y of the carboxylic acid of the compound m3 obtained in the step M-2. This step can be carried out according to the method described in Protective Groups in Organic Synthesis (Theodora W. Greene, Peter G. M. Wuts, John Wiley & Sons, Inc., 1999).
[M-4工程]
本工程は上記M-3工程で得られた化合物m4に、適当な縮合剤存在下、適当な塩基存在下、適当な溶媒中でアミンm5と反応させることにより、化合物M1を得る工程である。本工程において使用される縮合剤は、例えばEDCI(塩酸塩を含む)又はHBTUが挙げられる。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはジイソプロピルエチルアミン又はトリエチルアミンが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはジメチルホルムアミド、テトラヒドロフラン又は塩化メチレンが挙げられる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは0℃~150℃であり、より好ましくは室温~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step M-4]
This step is a step of obtaining compound M1 by reacting compound m4 obtained in step M-3 with amine m5 in the presence of a suitable condensing agent and in the presence of a suitable base in a suitable solvent. Examples of the condensing agent used in this step include EDCI (including hydrochloride) or HBTU. The base used in this step is selected from the bases exemplified below, and preferably diisopropylethylamine or triethylamine. The solvent used in this step is selected from the solvents exemplified below, and preferably dimethylformamide, tetrahydrofuran or methylene chloride. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably 0 ° C. to 150 ° C., more preferably room temperature to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
本工程は上記M-3工程で得られた化合物m4に、適当な縮合剤存在下、適当な塩基存在下、適当な溶媒中でアミンm5と反応させることにより、化合物M1を得る工程である。本工程において使用される縮合剤は、例えばEDCI(塩酸塩を含む)又はHBTUが挙げられる。本工程において使用される塩基は、後記に例示する塩基等から選択されるが、好ましくはジイソプロピルエチルアミン又はトリエチルアミンが挙げられる。本工程において使用される溶媒は、後記に例示する溶媒等から選択されるが、好ましくはジメチルホルムアミド、テトラヒドロフラン又は塩化メチレンが挙げられる。反応温度は、用いられる原料化合物の種類、試薬等により異なるが、通常、-78℃~200℃、好ましくは0℃~150℃であり、より好ましくは室温~100℃である。反応時間は通常5分~72時間程度であり、好ましくは30分~48時間、さらに好ましくは1時間~24時間である。 [Step M-4]
This step is a step of obtaining compound M1 by reacting compound m4 obtained in step M-3 with amine m5 in the presence of a suitable condensing agent and in the presence of a suitable base in a suitable solvent. Examples of the condensing agent used in this step include EDCI (including hydrochloride) or HBTU. The base used in this step is selected from the bases exemplified below, and preferably diisopropylethylamine or triethylamine. The solvent used in this step is selected from the solvents exemplified below, and preferably dimethylformamide, tetrahydrofuran or methylene chloride. While the reaction temperature varies depending on the type of raw material compound used, reagents and the like, it is generally −78 ° C. to 200 ° C., preferably 0 ° C. to 150 ° C., more preferably room temperature to 100 ° C. The reaction time is usually about 5 minutes to 72 hours, preferably 30 minutes to 48 hours, more preferably 1 hour to 24 hours.
[M-5工程]
本工程は上記M-4工程で得られる化合物M1を、上記H-2工程に準じた条件で反応させて、化合物M2を得る工程である。 [Step M-5]
In this step, compound M1 obtained in step M-4 is reacted under the same conditions as in step H-2 to obtain compound M2.
本工程は上記M-4工程で得られる化合物M1を、上記H-2工程に準じた条件で反応させて、化合物M2を得る工程である。 [Step M-5]
In this step, compound M1 obtained in step M-4 is reacted under the same conditions as in step H-2 to obtain compound M2.
[M-6工程]
本工程は上記M-5工程で得られる化合物M2を、上記H-3工程に準じた条件で反応させて、化合物M3を得る工程である。 [Step M-6]
In this step, compound M2 obtained in step M-5 is reacted under the same conditions as in step H-3 to obtain compound M3.
本工程は上記M-5工程で得られる化合物M2を、上記H-3工程に準じた条件で反応させて、化合物M3を得る工程である。 [Step M-6]
In this step, compound M2 obtained in step M-5 is reacted under the same conditions as in step H-3 to obtain compound M3.
上記の各工程において使用される塩基は、反応や原料化合物の種類等によって適時選択されるべきであるが、例えば重炭酸ナトリウム、重炭酸カリウムのような重炭酸アルカリ類、炭酸ナトリウム、炭酸カリウムのような炭酸アルカリ類、水素化ナトリウム、水素化カリウムのような金属水素化類、水酸化ナトリウム、水酸化カリウムのようなアルカリ金属水酸化物、ナトリウムメトキシド、ナトリウムt-ブトキシドのようなアルカリ金属アルコキシド類、ブチルリチウム、リチウムジイソプロピルアミドのような有機金属塩基類、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、4-ジメチルアミノピリジン(DMAP)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(DBU)のような有機塩基類が挙げられる。
The base used in each of the above steps should be selected in a timely manner depending on the reaction and the type of raw material compound. For example, alkali bicarbonates such as sodium bicarbonate and potassium bicarbonate, sodium carbonate and potassium carbonate Alkali carbonates such as sodium hydride, metal hydrides such as sodium hydride and potassium hydride, alkali metal hydroxides such as sodium hydroxide and potassium hydroxide, alkali metals such as sodium methoxide and sodium t-butoxide Alkoxides, organometallic bases such as butyllithium and lithium diisopropylamide, triethylamine, diisopropylethylamine, pyridine, 4-dimethylaminopyridine (DMAP), 1,8-diazabicyclo [5.4.0] -7-undecene ( Organic bases such as DBU) That.
上記の各工程において使用される溶媒は、反応や原料化合物の種類等によって適宜選択されるべきであるが、例えばメタノール、エタノール、イソプロパノールのようなアルコール類、アセトン、エチルメチルケトンのようなケトン類、塩化メチレン、クロロホルムのようなハロゲン化炭化水素類、テトラヒドロフラン(THF)、ジオキサンのようなエーテル類、トルエン、ベンゼンのような芳香族炭化水素類、ヘキサン、ヘプタンのような脂肪族炭化水素類、酢酸エチル、酢酸プロピルのようなエステル類、N,N-ジメチルホルムアミド(DMF)、N-メチル-2-ピロリドンのようなアミド類、ジメチルスルホキシド(DMSO)のようなスルホキシド類、アセトニトリルのようなニトリル類が挙げられ、これらの溶媒は単独あるいは2種類以上混合して用いることができる。また反応の種類によっては、有機塩基類を溶媒として用いてもよい。
The solvent used in each of the above steps should be appropriately selected depending on the reaction and the type of raw material compound. For example, alcohols such as methanol, ethanol and isopropanol, and ketones such as acetone and ethyl methyl ketone. Halogenated hydrocarbons such as methylene chloride and chloroform, ethers such as tetrahydrofuran (THF) and dioxane, aromatic hydrocarbons such as toluene and benzene, aliphatic hydrocarbons such as hexane and heptane, Esters such as ethyl acetate, propyl acetate, amides such as N, N-dimethylformamide (DMF), N-methyl-2-pyrrolidone, sulfoxides such as dimethyl sulfoxide (DMSO), nitriles such as acetonitrile These solvents can be used alone There can be used as a mixture of two or more kinds. Depending on the type of reaction, organic bases may be used as a solvent.
式(I)で表される本発明の化合物又はその中間体は、当業者に公知の方法で分離、精製することができる。そのような分離または精製方法としては、例えば、抽出、分配、再沈殿、カラムクロマトグラフィー(例えば、シリカゲルカラムクロマトグラフィー、イオン交換カラムクロマトグラフィーもしくは分取液体クロマトグラフィー)又は再結晶などが挙げられる。再結晶溶媒としては例えば、メタノール、エタノールもしくは2-プロパノールなどのアルコール系溶媒、ジエチルエーテルなどのエーテル系溶媒、酢酸エチルなどのエステル系溶媒、ベンゼンもしくはトルエンなどの芳香族炭化水素系溶媒、アセトンなどのケトン系溶媒、ジクロロメタンもしくはクロロホルムなどのハロゲン系溶媒、ヘキサンなどの炭化水素系溶媒、ジメチルホルムアミドもしくはアセトニトリルなどの非プロトン系溶媒、水、又はこれらの混合溶媒などを用いることができる。その他の精製方法としては、実験化学講座(日本化学会編、丸善)1巻などに記載された方法などを用いることができる。また、本発明の化合物の分子構造の決定は、それぞれの原料化合物に由来する構造を参照して、核磁気共鳴法、赤外吸収法、円二色性スペクトル分析法などの分光学的手法、及び質量分析法により容易に行える。
The compound of the present invention represented by the formula (I) or an intermediate thereof can be separated and purified by methods known to those skilled in the art. Examples of such separation or purification methods include extraction, distribution, reprecipitation, column chromatography (for example, silica gel column chromatography, ion exchange column chromatography or preparative liquid chromatography) or recrystallization. Examples of the recrystallization solvent include alcohol solvents such as methanol, ethanol and 2-propanol, ether solvents such as diethyl ether, ester solvents such as ethyl acetate, aromatic hydrocarbon solvents such as benzene and toluene, acetone and the like. A ketone solvent, a halogen solvent such as dichloromethane or chloroform, a hydrocarbon solvent such as hexane, an aprotic solvent such as dimethylformamide or acetonitrile, water, or a mixed solvent thereof. As other purification methods, the methods described in Experimental Chemistry Course (The Chemical Society of Japan, Maruzen) vol. 1 can be used. Further, the molecular structure of the compound of the present invention is determined by referring to the structure derived from each raw material compound, a spectroscopic method such as nuclear magnetic resonance, infrared absorption, circular dichroism spectrum analysis, And mass spectrometry.
式(I)で表される本発明の化合物またはそれらの製薬学的に許容される塩には、不斉が生じる場合又は不斉炭素を有する置換基を有する場合があり、そのような化合物にあっては光学異性体が存在する。本発明の化合物にはこれらの各異性体の混合物や単離されたものも含まれ、通常の方法に従って製造することができる。製造方法としては例えば、不斉点を有する原料を用いる方法か、又は途中の段階で不斉を導入する方法が挙げられる。例えば、光学活性な原料を用いるか、製造工程の適当な段階で光学分割などを行うことで、式(I)で表される本発明の化合物またはそれらの製薬学的に許容される塩の光学異性体を得ることができる。光学分割法としては例えば、式(I)で表される化合物又はその中間体が、塩基性官能基を有する場合には、不活性溶媒中(例えばメタノール、エタノールもしくは2-プロパノールなどのアルコール系溶媒、ジエチルエーテルなどのエーテル系溶媒、酢酸エチルなどのエステル系溶媒、トルエンなどの炭化水素系溶媒、アセトニトリルなどの非プロトン系溶媒、又はこれらの混合溶媒)、光学活性な酸(例えば、マンデル酸、N-ベンジルオキシアラニンもしくは乳酸などのモノカルボン酸、酒石酸、o-ジイソプロピリデン酒石酸もしくはリンゴ酸などのジカルボン酸、カンファースルフォン酸もしくはブロモカンファースルホン酸などのスルホン酸)を用いて塩を形成させるジアステレオマー法が挙げられる。式(I)で表される本発明の化合物の中間体が、カルボキシル基などの酸性官能基を有する場合には、光学活性なアミン(例えば1-フェニルエチルアミン、キニン、キニジン、シンコニジン、シンコニンもしくはストリキニーネなどの有機アミン)を用いて、塩を形成させることにより、光学分割を行うこともできる。
The compound of the present invention represented by the formula (I) or a pharmaceutically acceptable salt thereof may have asymmetry or may have a substituent having an asymmetric carbon. In that case, optical isomers exist. The compounds of the present invention include mixtures of these isomers and isolated ones, and can be produced according to ordinary methods. Examples of the production method include a method using a raw material having an asymmetric point, or a method of introducing asymmetry at an intermediate stage. For example, by using an optically active raw material or performing optical resolution or the like at an appropriate stage of the production process, the optical properties of the compound of the present invention represented by formula (I) or a pharmaceutically acceptable salt thereof Isomers can be obtained. As the optical resolution method, for example, when the compound represented by the formula (I) or an intermediate thereof has a basic functional group, the solvent is an inert solvent (for example, an alcohol solvent such as methanol, ethanol or 2-propanol). An ether solvent such as diethyl ether, an ester solvent such as ethyl acetate, a hydrocarbon solvent such as toluene, an aprotic solvent such as acetonitrile, or a mixed solvent thereof), an optically active acid (for example, mandelic acid, N-benzyloxyalanine or monocarboxylic acid such as lactic acid, tartaric acid, dicarboxylic acid such as o-diisopropylidene tartaric acid or malic acid, sulfonic acid such as camphorsulfonic acid or bromocamphorsulfonic acid) Stereomer method can be mentioned. When the intermediate of the compound of the present invention represented by the formula (I) has an acidic functional group such as a carboxyl group, an optically active amine (for example, 1-phenylethylamine, quinine, quinidine, cinchonidine, cinchonine or strychnine) The optical resolution can also be carried out by forming a salt using an organic amine).
塩を形成させる温度としては、室温から溶媒の沸点までの範囲から選択される。光学純度を向上させるためには、一旦、溶媒の沸点付近まで温度を上げることが望ましい。析出した塩を濾取する際、必要に応じて冷却し、収率を向上させることができる。光学活性な酸又はアミンの使用量は、基質に対し約0.5~約2.0当量の範囲、好ましくは1当量前後の範囲が適当である。必要に応じ結晶を不活性溶媒中(例えばメタノール、エタノールもしくは2-プロパノールなどのアルコール系溶媒、ジエチルエーテルなどのエーテル系溶媒、酢酸エチルなどのエステル系溶媒、トルエンなどの炭化水素系溶媒、アセトニトリルなどの非プロトン系溶媒、又はこれらの混合溶媒)で再結晶し、高純度の光学活性な塩を得ることもできる。また、必要に応じて光学分割した塩を通常の方法で酸又は塩基で処理し、フリー体として得ることもできる。
The temperature at which the salt is formed is selected from the range from room temperature to the boiling point of the solvent. In order to improve the optical purity, it is desirable to raise the temperature once to near the boiling point of the solvent. When the precipitated salt is collected by filtration, it can be cooled as necessary to improve the yield. The amount of the optically active acid or amine used is suitably in the range of about 0.5 to about 2.0 equivalents, preferably in the range of about 1 equivalent, relative to the substrate. Crystals in an inert solvent as necessary (for example, alcohol solvents such as methanol, ethanol or 2-propanol, ether solvents such as diethyl ether, ester solvents such as ethyl acetate, hydrocarbon solvents such as toluene, acetonitrile, etc. And a high purity optically active salt can be obtained. Further, if necessary, an optically resolved salt can be treated with an acid or a base by a conventional method to obtain a free form.
あるいは式(I)で表される本発明の化合物の中間体が、カルボキシル基を有する場合には、光学活性なアミン(例えば、1-フェニルエチルアミンなど)を用いてアミドを形成させることにより、光学分割を行うこともできる。
Alternatively, when the intermediate of the compound of the present invention represented by the formula (I) has a carboxyl group, an optically active amine (for example, 1-phenylethylamine etc.) is used to form an amide. Division can also be performed.
本発明の化合物は、アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患の治療剤及び/又は予防剤として有用である。アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患としては、神経疾患、精神疾患又は炎症性疾患が挙げられる。神経疾患、精神疾患又は炎症性疾患の具体例としては、統合失調症、アルツハイマー病、ダウン症、注意欠陥・多動性障害又は脳血管アンギオパチーが挙げられる。また、本発明の化合物は、(1)CIAS(統合失調症に伴う認知機能障害)、或いは、(2)統合失調症、アルツハイマー病、ダウン症、注意欠陥・多動性障害又は脳血管アンギオパチーにおける、認知障害、軽度認知障害、記憶障害又は学習障害を治療及び/又は予防するために有用である。また、本発明の化合物は、統合失調症に伴う陰性症状及び/又は陽性症状を治療及び/又は予防するために有用である。
The compound of the present invention is useful as a therapeutic and / or prophylactic agent for diseases caused by abnormalities in intracellular signal transduction involving acetylcholine. Examples of the disease caused by abnormal intracellular signal transduction involving acetylcholine include a neurological disease, a mental disease or an inflammatory disease. Specific examples of neurological diseases, psychiatric diseases or inflammatory diseases include schizophrenia, Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder, or cerebrovascular angiopathy. In addition, the compound of the present invention is (1) CIAS (cognitive dysfunction associated with schizophrenia), or (2) schizophrenia, Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder or cerebrovascular angiopathy, Useful for treating and / or preventing cognitive impairment, mild cognitive impairment, memory impairment or learning impairment. The compounds of the present invention are also useful for treating and / or preventing negative and / or positive symptoms associated with schizophrenia.
さらに、本発明の化合物は、前記治療及び/又は予防の目的で、非定型抗精神病薬と併用して用いることもできる。本発明の化合物又はその製薬学的に許容される塩と、非定型抗精神病薬とを併用する場合における医薬は、単一の配合剤であってもよく、あるいは、同時に、別々に、連続してまたは一定間隔を空けて投与される複数製剤であってもよい。
Furthermore, the compound of the present invention can be used in combination with an atypical antipsychotic agent for the purpose of the treatment and / or prevention. The pharmaceutical agent in the case where the compound of the present invention or a pharmaceutically acceptable salt thereof and an atypical antipsychotic drug are used in combination may be a single compounding agent, or simultaneously, separately and continuously. Or multiple formulations administered at regular intervals.
本発明の化合物の投与経路としては、経口投与、非経口投与又は直腸内投与のいずれでもよく、その一日投与量は、化合物の種類、投与方法、患者の症状・年齢等により異なる。例えば、経口投与の場合は、通常、ヒト又は哺乳動物1kg体重当たり約0.01~1000mg、更に好ましくは約0.1~500mgを1~数回に分けて投与することができる。静注等の非経口投与の場合は、通常、例えば、ヒト又は哺乳動物1kg体重当たり約0.01mg~300mg、更に好ましくは約1mg~100mgを投与することができる。
The administration route of the compound of the present invention may be any of oral administration, parenteral administration and rectal administration, and the daily dose varies depending on the type of compound, administration method, patient symptom / age and the like. For example, in the case of oral administration, usually about 0.01 to 1000 mg, more preferably about 0.1 to 500 mg per kg body weight of a human or mammal can be administered in 1 to several divided doses. In the case of parenteral administration such as intravenous injection, usually, for example, about 0.01 mg to 300 mg, more preferably about 1 mg to 100 mg per kg body weight of a human or mammal can be administered.
剤形としては、錠剤、カプセル剤、顆粒剤、散剤、シロップ剤、懸濁剤、注射剤、坐剤、点眼剤、軟膏剤、塗布剤、貼付剤、吸入剤等が挙げられる。これらの製剤は常法に従って調製することができる。なお、液体製剤にあっては、用時、水、適当な水溶液又は他の適当な媒体に溶解又は懸濁する形であってもよい。また、錠剤及び顆粒剤は周知の方法でコーティングしてもよい。更に、これらの製剤は製薬学的に許容される添加剤を含有してもよい。添加剤は、目的に応じて、賦形剤、崩壊剤、結合剤、流動化剤、滑沢剤、コーティング剤、溶解剤、溶解補助剤、増粘剤、分散剤、安定化剤、甘味剤、香料等を用いることができる。
Examples of the dosage form include tablets, capsules, granules, powders, syrups, suspensions, injections, suppositories, eye drops, ointments, coating agents, patches, inhalants and the like. These preparations can be prepared according to a conventional method. In the case of a liquid preparation, it may be dissolved or suspended in water, an appropriate aqueous solution or other appropriate medium at the time of use. Tablets and granules may be coated by a known method. In addition, these formulations may contain pharmaceutically acceptable additives. Additives are excipients, disintegrants, binders, fluidizers, lubricants, coating agents, solubilizers, solubilizers, thickeners, dispersants, stabilizers, sweeteners depending on the purpose. Perfumes and the like can be used.
本発明の化合物は、非定型抗精神病薬と併用することができる。非定型抗精神病薬としては、例えば、オランザピン、リスペリドン、パリペリドン、ケチアピン、ジプラシドン、アリピプラゾール、アセナピン、イロペリドン、クロザピン、セルティンドール、ブロナンセリン及びルラシドンが挙げられる。
The compound of the present invention can be used in combination with an atypical antipsychotic drug. Examples of atypical antipsychotics include olanzapine, risperidone, paliperidone, quetiapine, ziprasidone, aripiprazole, asenapine, iloperidone, clozapine, sertindole, blonanserin and lurasidone.
以下に参考例、実施例及び試験例を挙げて本発明を更に具体的に説明するが、これらは本発明を限定するものではない。なお、化合物の同定は元素分析値、マス・スペクトル、高速液体クロマト質量分析計;LCMS、IRスペクトル、NMRスペクトル、高速液体クロマトグラフィー(HPLC)等により行った。
Hereinafter, the present invention will be described more specifically with reference to reference examples, examples and test examples, but these examples do not limit the present invention. The compound was identified by elemental analysis, mass spectrum, high performance liquid chromatography / mass spectrometer; LCMS, IR spectrum, NMR spectrum, high performance liquid chromatography (HPLC) and the like.
明細書の記載を簡略化するために参考例、実施例及び実施例中の表において以下に示すような略号を用いることもある。置換基として用いられる略号としては、Meはメチル基、Etはエチル基、Phはフェニル基、Tsはトシル基を意味する。TFAはトリフルオロ酢酸を意味する。NMRに用いられる記号としては、sは一重線、dは二重線、ddは二重の二重線、tは三重線、tdは三重線の二重線、qは四重線、mは多重線、brは幅広い、brsは幅広い一重線、brdは幅広い二重線、brtは幅広い三重線及びJは結合定数を意味する。
In order to simplify the description of the specification, the following abbreviations may be used in the reference examples, examples, and tables in the examples. As abbreviations used as substituents, Me means a methyl group, Et means an ethyl group, Ph means a phenyl group, and Ts means a tosyl group. TFA means trifluoroacetic acid. The symbols used in NMR are as follows: s is a single line, d is a double line, dd is a double double line, t is a triple line, td is a triple double line, q is a quadruple line, m is Multiple lines, br means broad, brs means broad single line, brd means broad double line, brt means broad triple line, and J means coupling constant.
高速液体クロマト質量分析計;LCMSの測定条件は、以下の通りであり、観察された質量分析の値[MS(m/z)]をMH+で、保持時間をRt(分、min)で示す。なお、各実測値においては、測定に用いた測定条件としてA~Hのいずれかを付記する。
High-performance liquid chromatographic mass spectrometer: The measurement conditions of LCMS are as follows, and the observed mass spectrometry value [MS (m / z)] is represented by MH +, and the retention time is represented by Rt (min, min). In each actual measurement value, any one of A to H is added as a measurement condition used for the measurement.
測定条件A
検出機器:Waters ACQUITY UPLC
column:ACQUITY UPLC BEH C18 1.7μm 2.1×50 mm column
Solvent:A液:0.05% HCOOH/H2O、B液:CH3CN
Gradient Condition:
0.0-1.3分;A/B=90:10~1:99(linear gradient)
1.35-1.5分;A/B=1:99
1.5-2分;A/B=90:10
Flow Rate:0.75 mL/min.
UV:220 nm, 254 nm
カラム温度:50℃ Measurement condition A
Detection equipment: Waters ACQUITY UPLC
column: ACQUITY UPLC BEH C18 1.7μm 2.1 × 50 mm column
Solvent: Liquid A: 0.05% HCOOH / H 2 O, Liquid B: CH 3 CN
Gradient Condition:
0.0-1.3 min; A / B = 90: 10 to 1:99 (linear gradient)
1.35-1.5 minutes; A / B = 1: 99
1.5-2 minutes; A / B = 90: 10
Flow Rate: 0.75 mL / min.
UV: 220 nm, 254 nm
Column temperature: 50 ° C
検出機器:Waters ACQUITY UPLC
column:ACQUITY UPLC BEH C18 1.7μm 2.1×50 mm column
Solvent:A液:0.05% HCOOH/H2O、B液:CH3CN
Gradient Condition:
0.0-1.3分;A/B=90:10~1:99(linear gradient)
1.35-1.5分;A/B=1:99
1.5-2分;A/B=90:10
Flow Rate:0.75 mL/min.
UV:220 nm, 254 nm
カラム温度:50℃ Measurement condition A
Detection equipment: Waters ACQUITY UPLC
column: ACQUITY UPLC BEH C18 1.7μm 2.1 × 50 mm column
Solvent: Liquid A: 0.05% HCOOH / H 2 O, Liquid B: CH 3 CN
Gradient Condition:
0.0-1.3 min; A / B = 90: 10 to 1:99 (linear gradient)
1.35-1.5 minutes; A / B = 1: 99
1.5-2 minutes; A / B = 90: 10
Flow Rate: 0.75 mL / min.
UV: 220 nm, 254 nm
Column temperature: 50 ° C
測定条件B
検出機器:Shimadzu LCMS-2020
Column:Phenomenex Kinetex 1.7μm C18 2.1 mm×50 mm
Solvent:A液:MeCN, B液 : 0.05% TFA/H2O
Gradient condition:
0 min: A/B= 10:90
0-1.70 min: A/B= 10:90-99:1 (linear gradient)
1.71-1.90 min: A/B=99:1
1.91-3.00 min: A/B=10:90
Flow Rate:0.5 mL/min.
UV:220 nm
カラム温度:40℃ Measurement condition B
Detection equipment: Shimadzu LCMS-2020
Column: Phenomenex Kinetex 1.7μm C18 2.1 mm x 50 mm
Solvent: A solution: MeCN, B solution: 0.05% TFA / H 2 O
Gradient condition:
0 min: A / B = 10: 90
0-1.70 min: A / B = 10: 90-99: 1 (linear gradient)
1.71-1.90 min: A / B = 99: 1
1.91-3.00 min: A / B = 10: 90
Flow Rate: 0.5 mL / min.
UV: 220 nm
Column temperature: 40 ° C
検出機器:Shimadzu LCMS-2020
Column:Phenomenex Kinetex 1.7μm C18 2.1 mm×50 mm
Solvent:A液:MeCN, B液 : 0.05% TFA/H2O
Gradient condition:
0 min: A/B= 10:90
0-1.70 min: A/B= 10:90-99:1 (linear gradient)
1.71-1.90 min: A/B=99:1
1.91-3.00 min: A/B=10:90
Flow Rate:0.5 mL/min.
UV:220 nm
カラム温度:40℃ Measurement condition B
Detection equipment: Shimadzu LCMS-2020
Column: Phenomenex Kinetex 1.7μm C18 2.1 mm x 50 mm
Solvent: A solution: MeCN, B solution: 0.05% TFA / H 2 O
Gradient condition:
0 min: A / B = 10: 90
0-1.70 min: A / B = 10: 90-99: 1 (linear gradient)
1.71-1.90 min: A / B = 99: 1
1.91-3.00 min: A / B = 10: 90
Flow Rate: 0.5 mL / min.
UV: 220 nm
Column temperature: 40 ° C
測定条件C
検出機器:Shimadzu LC:20A, MS:2010
Column:Xtimate 3μm C18 2.1 mm×30 mm
Solvent:A液:1.5mL/4L TFA/H2O, B液 : 0.75mL/4L TFA/MeCN
Gradient condition:
0 min: A/B= 90:10
0-2.2 min: A/B= 90:10-20:80 (linear gradient)
2.21-2.5 min: A/B=20:80
Flow Rate:0.8 mL/min.
UV:220 nm
カラム温度:50℃ Measurement condition C
Detection equipment: Shimadzu LC: 20A, MS: 2010
Column: Xtimate 3μm C18 2.1 mm x 30 mm
Solvent: Liquid A: 1.5mL / 4L TFA / H 2 O, Liquid B: 0.75mL / 4L TFA / MeCN
Gradient condition:
0 min: A / B = 90:10
0-2.2 min: A / B = 90: 10-20: 80 (linear gradient)
2.21-2.5 min: A / B = 20: 80
Flow Rate: 0.8 mL / min.
UV: 220 nm
Column temperature: 50 ° C
検出機器:Shimadzu LC:20A, MS:2010
Column:Xtimate 3μm C18 2.1 mm×30 mm
Solvent:A液:1.5mL/4L TFA/H2O, B液 : 0.75mL/4L TFA/MeCN
Gradient condition:
0 min: A/B= 90:10
0-2.2 min: A/B= 90:10-20:80 (linear gradient)
2.21-2.5 min: A/B=20:80
Flow Rate:0.8 mL/min.
UV:220 nm
カラム温度:50℃ Measurement condition C
Detection equipment: Shimadzu LC: 20A, MS: 2010
Column: Xtimate 3μm C18 2.1 mm x 30 mm
Solvent: Liquid A: 1.5mL / 4L TFA / H 2 O, Liquid B: 0.75mL / 4L TFA / MeCN
Gradient condition:
0 min: A / B = 90:10
0-2.2 min: A / B = 90: 10-20: 80 (linear gradient)
2.21-2.5 min: A / B = 20: 80
Flow Rate: 0.8 mL / min.
UV: 220 nm
Column temperature: 50 ° C
測定条件D
検出機器:Shimadzu LC:20A, MS:2010
Column:Xtimate 3μm C18 2.1 mm×30 mm
Solvent:A液:1.5mL/4L TFA/H2O, B液 : 0.75mL/4L TFA/MeCN
Gradient condition:
0 min: A/B= 70:30
0-2.2 min: A/B= 70:30-10:90 (linear gradient)
2.21-2.5 min: A/B=10:90
Flow Rate:0.8 mL/min.
UV:220 nm
カラム温度:50℃ Measurement condition D
Detection equipment: Shimadzu LC: 20A, MS: 2010
Column: Xtimate 3μm C18 2.1 mm x 30 mm
Solvent: Liquid A: 1.5mL / 4L TFA / H 2 O, Liquid B: 0.75mL / 4L TFA / MeCN
Gradient condition:
0 min: A / B = 70:30
0-2.2 min: A / B = 70: 30-10: 90 (linear gradient)
2.21-2.5 min: A / B = 10: 90
Flow Rate: 0.8 mL / min.
UV: 220 nm
Column temperature: 50 ° C
検出機器:Shimadzu LC:20A, MS:2010
Column:Xtimate 3μm C18 2.1 mm×30 mm
Solvent:A液:1.5mL/4L TFA/H2O, B液 : 0.75mL/4L TFA/MeCN
Gradient condition:
0 min: A/B= 70:30
0-2.2 min: A/B= 70:30-10:90 (linear gradient)
2.21-2.5 min: A/B=10:90
Flow Rate:0.8 mL/min.
UV:220 nm
カラム温度:50℃ Measurement condition D
Detection equipment: Shimadzu LC: 20A, MS: 2010
Column: Xtimate 3μm C18 2.1 mm x 30 mm
Solvent: Liquid A: 1.5mL / 4L TFA / H 2 O, Liquid B: 0.75mL / 4L TFA / MeCN
Gradient condition:
0 min: A / B = 70:30
0-2.2 min: A / B = 70: 30-10: 90 (linear gradient)
2.21-2.5 min: A / B = 10: 90
Flow Rate: 0.8 mL / min.
UV: 220 nm
Column temperature: 50 ° C
測定条件E
検出機器:Shimadzu LC:20A, MS:2010
Column:Xtimate 3μm C18 2.1 mm×30 mm
Solvent:A液:1.5mL/4L TFA/H2O, B液 : 0.75mL/4L TFA/MeCN
Gradient condition:
0 min: A/B= 100:0
0-2.2 min: A/B= 100:0-40:60 (linear gradient)
2.21-2.5 min: A/B=40:60
Flow Rate:0.8 mL/min.
UV:220 nm
カラム温度:50℃ Measurement condition E
Detection equipment: Shimadzu LC: 20A, MS: 2010
Column: Xtimate 3μm C18 2.1 mm x 30 mm
Solvent: Liquid A: 1.5mL / 4L TFA / H 2 O, Liquid B: 0.75mL / 4L TFA / MeCN
Gradient condition:
0 min: A / B = 100: 0
0-2.2 min: A / B = 100: 0-40: 60 (linear gradient)
2.21-2.5 min: A / B = 40: 60
Flow Rate: 0.8 mL / min.
UV: 220 nm
Column temperature: 50 ° C
検出機器:Shimadzu LC:20A, MS:2010
Column:Xtimate 3μm C18 2.1 mm×30 mm
Solvent:A液:1.5mL/4L TFA/H2O, B液 : 0.75mL/4L TFA/MeCN
Gradient condition:
0 min: A/B= 100:0
0-2.2 min: A/B= 100:0-40:60 (linear gradient)
2.21-2.5 min: A/B=40:60
Flow Rate:0.8 mL/min.
UV:220 nm
カラム温度:50℃ Measurement condition E
Detection equipment: Shimadzu LC: 20A, MS: 2010
Column: Xtimate 3μm C18 2.1 mm x 30 mm
Solvent: Liquid A: 1.5mL / 4L TFA / H 2 O, Liquid B: 0.75mL / 4L TFA / MeCN
Gradient condition:
0 min: A / B = 100: 0
0-2.2 min: A / B = 100: 0-40: 60 (linear gradient)
2.21-2.5 min: A / B = 40: 60
Flow Rate: 0.8 mL / min.
UV: 220 nm
Column temperature: 50 ° C
測定条件F
検出機器:Agilent LC:1200, MS:6110
Column:Xbrige RP-18.5μm 2.1 mm×50 mm
Solvent:A液:0.5mL/1L NH3・H2O/H2O, B液 : MeCN
Gradient condition:
0 min: A/B= 10:90
0-2.0 min: A/B= 10:90-20:80 (linear gradient)
2.01-2.5 min: A/B=20:80
Flow Rate:1.0 mL/min.
UV:220 nm
カラム温度:30℃ Measurement condition F
Detection equipment: Agilent LC: 1200, MS: 6110
Column: Xbrige RP-18.5μm 2.1 mm x 50 mm
Solvent: A solution: 0.5mL / 1L NH3 · H2O / H 2 O, B solution: MeCN
Gradient condition:
0 min: A / B = 10: 90
0-2.0 min: A / B = 10: 90-20: 80 (linear gradient)
2.01-2.5 min: A / B = 20: 80
Flow Rate: 1.0 mL / min.
UV: 220 nm
Column temperature: 30 ° C
検出機器:Agilent LC:1200, MS:6110
Column:Xbrige RP-18.5μm 2.1 mm×50 mm
Solvent:A液:0.5mL/1L NH3・H2O/H2O, B液 : MeCN
Gradient condition:
0 min: A/B= 10:90
0-2.0 min: A/B= 10:90-20:80 (linear gradient)
2.01-2.5 min: A/B=20:80
Flow Rate:1.0 mL/min.
UV:220 nm
カラム温度:30℃ Measurement condition F
Detection equipment: Agilent LC: 1200, MS: 6110
Column: Xbrige RP-18.5μm 2.1 mm x 50 mm
Solvent: A solution: 0.5mL / 1L NH3 · H2O / H 2 O, B solution: MeCN
Gradient condition:
0 min: A / B = 10: 90
0-2.0 min: A / B = 10: 90-20: 80 (linear gradient)
2.01-2.5 min: A / B = 20: 80
Flow Rate: 1.0 mL / min.
UV: 220 nm
Column temperature: 30 ° C
測定条件G
検出機器:Agilent LC:1200, MS:6110
Column:Durashell 3.0μm C18 2.1 mm×30 mm
Solvent:A液:1.5mL/4L TFA/H2O, B液 : 0.75mL/4L TFA/MeCN
Gradient condition:
0 min: A/B= 10:90
0-2.2 min: A/B= 10:90-20:80 (linear gradient)
2.21-2.5 min: A/B=20:80
Flow Rate:0.8 mL/min.
UV:220 nm
カラム温度:50℃ Measurement condition G
Detection equipment: Agilent LC: 1200, MS: 6110
Column: Durashell 3.0μm C18 2.1 mm x 30 mm
Solvent: Liquid A: 1.5mL / 4L TFA / H 2 O, Liquid B: 0.75mL / 4L TFA / MeCN
Gradient condition:
0 min: A / B = 10: 90
0-2.2 min: A / B = 10: 90-20: 80 (linear gradient)
2.21-2.5 min: A / B = 20: 80
Flow Rate: 0.8 mL / min.
UV: 220 nm
Column temperature: 50 ° C
検出機器:Agilent LC:1200, MS:6110
Column:Durashell 3.0μm C18 2.1 mm×30 mm
Solvent:A液:1.5mL/4L TFA/H2O, B液 : 0.75mL/4L TFA/MeCN
Gradient condition:
0 min: A/B= 10:90
0-2.2 min: A/B= 10:90-20:80 (linear gradient)
2.21-2.5 min: A/B=20:80
Flow Rate:0.8 mL/min.
UV:220 nm
カラム温度:50℃ Measurement condition G
Detection equipment: Agilent LC: 1200, MS: 6110
Column: Durashell 3.0μm C18 2.1 mm x 30 mm
Solvent: Liquid A: 1.5mL / 4L TFA / H 2 O, Liquid B: 0.75mL / 4L TFA / MeCN
Gradient condition:
0 min: A / B = 10: 90
0-2.2 min: A / B = 10: 90-20: 80 (linear gradient)
2.21-2.5 min: A / B = 20: 80
Flow Rate: 0.8 mL / min.
UV: 220 nm
Column temperature: 50 ° C
測定条件H
検出機器:APIシリーズ用Agilent 1100シリーズ (applied Biosystems社製)
HPLC:API 150EX LC/MS system (applied Biosystems社製)
Column:YMC CombiScreen Hydrosphere C18 (S-5, 12 nm, 4.6×50 mm)
Solvent:A液:0.05 % TFA/H2O、B液:0.05 % TFA/MeOH
Gradient Condition:
0.0-6.0分;A/B=75:25-1:99(linear gradient)
Flow rate:3.5 mL/分
UV:254 nm Measurement condition H
Detection equipment: Agilent 1100 series for API series (Applied Biosystems)
HPLC: API 150EX LC / MS system (Applied Biosystems)
Column: YMC CombiScreen Hydrosphere C18 (S-5, 12 nm, 4.6 × 50 mm)
Solvent: Liquid A: 0.05% TFA / H 2 O, Liquid B: 0.05% TFA / MeOH
Gradient Condition:
0.0-6.0 min; A / B = 75: 25-1: 99 (linear gradient)
Flow rate: 3.5 mL / min UV: 254 nm
検出機器:APIシリーズ用Agilent 1100シリーズ (applied Biosystems社製)
HPLC:API 150EX LC/MS system (applied Biosystems社製)
Column:YMC CombiScreen Hydrosphere C18 (S-5, 12 nm, 4.6×50 mm)
Solvent:A液:0.05 % TFA/H2O、B液:0.05 % TFA/MeOH
Gradient Condition:
0.0-6.0分;A/B=75:25-1:99(linear gradient)
Flow rate:3.5 mL/分
UV:254 nm Measurement condition H
Detection equipment: Agilent 1100 series for API series (Applied Biosystems)
HPLC: API 150EX LC / MS system (Applied Biosystems)
Column: YMC CombiScreen Hydrosphere C18 (S-5, 12 nm, 4.6 × 50 mm)
Solvent: Liquid A: 0.05% TFA / H 2 O, Liquid B: 0.05% TFA / MeOH
Gradient Condition:
0.0-6.0 min; A / B = 75: 25-1: 99 (linear gradient)
Flow rate: 3.5 mL / min UV: 254 nm
参考例1
4-[5-クロロ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]ピペリジン・2塩酸塩

a)4-ブロモ-1-(3-フルオロフェニル)-1H-イミダゾール(化合物Q1)の製造
4-ブロモイミダゾール (50g)の塩化メチレン溶液(1500mL)に、室温にて3-フルオロフェニルボロン酸(94.0g)、酢酸銅(II)(91.0g)及びピリジン(50mL)を加えた後に、30℃にて48時間撹拌した。反応溶液をセライト濾過した後に、得られたろ液の溶媒を減圧留去した。シリカゲルカラムクロマトグラフィーにて精製することで、化合物Q1(35g)を得た。
1H-NMR (400 MHz, CDCl3): δ7.75 (s, 1H), 7.50-7.47 (m, 1H), 7.29 (s, 1H), 7.21-7.11 (m, 3H). Reference example 1
4- [5-Chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine dihydrochloride
a) Preparation of 4-bromo-1- (3-fluorophenyl) -1H-imidazole (compound Q1) 4-Fluoroimidazole (50 g) in methylene chloride solution (1500 mL) at room temperature with 3-fluorophenylboronic acid ( 94.0 g), copper (II) acetate (91.0 g) and pyridine (50 mL) were added, and the mixture was stirred at 30 ° C. for 48 hours. The reaction solution was filtered through Celite, and then the solvent of the obtained filtrate was distilled off under reduced pressure. Purification by silica gel column chromatography gave Compound Q1 (35 g).
1 H-NMR (400 MHz, CDCl3): δ 7.75 (s, 1H), 7.50-7.47 (m, 1H), 7.29 (s, 1H), 7.21-7.11 (m, 3H).
4-[5-クロロ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]ピペリジン・2塩酸塩
4-ブロモイミダゾール (50g)の塩化メチレン溶液(1500mL)に、室温にて3-フルオロフェニルボロン酸(94.0g)、酢酸銅(II)(91.0g)及びピリジン(50mL)を加えた後に、30℃にて48時間撹拌した。反応溶液をセライト濾過した後に、得られたろ液の溶媒を減圧留去した。シリカゲルカラムクロマトグラフィーにて精製することで、化合物Q1(35g)を得た。
1H-NMR (400 MHz, CDCl3): δ7.75 (s, 1H), 7.50-7.47 (m, 1H), 7.29 (s, 1H), 7.21-7.11 (m, 3H). Reference example 1
4- [5-Chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine dihydrochloride
1 H-NMR (400 MHz, CDCl3): δ 7.75 (s, 1H), 7.50-7.47 (m, 1H), 7.29 (s, 1H), 7.21-7.11 (m, 3H).
b)tert-ブチル 4-[1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(化合物Q2)の製造
化合物Q1(50g)のDMF(350mL)及び水(35mL)の混合溶液に、室温にてtert-ブチル 4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(57.0g)、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウムジクロライド(5.6g)及び炭酸カリウム(64.0g)を加えた後に、窒素雰囲気下、100℃にて24時間撹拌した。反応溶液をセライト濾過した。得られたろ液を酢酸エチルで抽出、硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィーで精製することにより、化合物Q2(32g)を取得した。
1H-NMR (400 MHz, DMSO-d6): δ8.31 (s, 1H), 7.82 (s, 1H), 7.66-7.62 (m, 1H), 7.54-7.52 (m, 2H), 7.18-7.16 (m, 1H), 6.32 (brs, 1H), 3.98 (brs, 2H), 3.52-3.49 (m, 2H), 2.39 (brs, 2H), 1.05 (s, 9H). b) Preparation of tert-butyl 4- [1- (3-fluorophenyl) -1H-imidazol-4-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (compound Q2) Compound Q1 (50 g) Tert-butyl 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -3, was added to a mixed solution of DMF (350 mL) and water (35 mL) at room temperature. After adding 6-dihydropyridine-1 (2H) -carboxylate (57.0 g), [1,1′-bis (diphenylphosphino) ferrocene] palladium dichloride (5.6 g) and potassium carbonate (64.0 g). The mixture was stirred at 100 ° C. for 24 hours under a nitrogen atmosphere. The reaction solution was filtered through celite. The obtained filtrate was extracted with ethyl acetate and dried over sodium sulfate, and then the solvent was distilled off under reduced pressure. Compound Q2 (32 g) was obtained by purification by silica gel column chromatography.
1 H-NMR (400 MHz, DMSO-d6): δ8.31 (s, 1H), 7.82 (s, 1H), 7.66-7.62 (m, 1H), 7.54-7.52 (m, 2H), 7.18-7.16 (m, 1H), 6.32 (brs, 1H), 3.98 (brs, 2H), 3.52-3.49 (m, 2H), 2.39 (brs, 2H), 1.05 (s, 9H).
化合物Q1(50g)のDMF(350mL)及び水(35mL)の混合溶液に、室温にてtert-ブチル 4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(57.0g)、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウムジクロライド(5.6g)及び炭酸カリウム(64.0g)を加えた後に、窒素雰囲気下、100℃にて24時間撹拌した。反応溶液をセライト濾過した。得られたろ液を酢酸エチルで抽出、硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィーで精製することにより、化合物Q2(32g)を取得した。
1H-NMR (400 MHz, DMSO-d6): δ8.31 (s, 1H), 7.82 (s, 1H), 7.66-7.62 (m, 1H), 7.54-7.52 (m, 2H), 7.18-7.16 (m, 1H), 6.32 (brs, 1H), 3.98 (brs, 2H), 3.52-3.49 (m, 2H), 2.39 (brs, 2H), 1.05 (s, 9H). b) Preparation of tert-butyl 4- [1- (3-fluorophenyl) -1H-imidazol-4-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (compound Q2) Compound Q1 (50 g) Tert-butyl 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -3, was added to a mixed solution of DMF (350 mL) and water (35 mL) at room temperature. After adding 6-dihydropyridine-1 (2H) -carboxylate (57.0 g), [1,1′-bis (diphenylphosphino) ferrocene] palladium dichloride (5.6 g) and potassium carbonate (64.0 g). The mixture was stirred at 100 ° C. for 24 hours under a nitrogen atmosphere. The reaction solution was filtered through celite. The obtained filtrate was extracted with ethyl acetate and dried over sodium sulfate, and then the solvent was distilled off under reduced pressure. Compound Q2 (32 g) was obtained by purification by silica gel column chromatography.
1 H-NMR (400 MHz, DMSO-d6): δ8.31 (s, 1H), 7.82 (s, 1H), 7.66-7.62 (m, 1H), 7.54-7.52 (m, 2H), 7.18-7.16 (m, 1H), 6.32 (brs, 1H), 3.98 (brs, 2H), 3.52-3.49 (m, 2H), 2.39 (brs, 2H), 1.05 (s, 9H).
c)tert-ブチル 4-[1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]ピペリジン-1-カルボキシレート(化合物Q3)の製造
化合物Q2(32.0g)のメタノール溶液(200mL)に、室温にて含水の10% パラジウムカーボン(5.0g)を加えた後に、2気圧の水素雰囲気下、30℃で24時間撹拌した。反応液をセライト濾過した。得られたろ液の溶媒を減圧留去することで、化合物Q3(33g)を粗生成物として取得した。
1H-NMR (400 MHz, CDCl3): δ7.80 (s, 1H), 7.64-7.44 (m, 1H), 7.20-7.18 (m, 1H), 7.13-7.10 (m, 2H), 6.99 (s, 1H), 4.20 (brs, 2H), 2.91-2.78 (m, 3H), 2.07-2.03 (m, 2H), 1.63-1.55 (m, 2H), 1.49 (s, 9H). c) Preparation of tert-butyl 4- [1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine-1-carboxylate (Compound Q3) Compound Q2 (32.0 g) in methanol (200 mL) After adding 10% palladium carbon (5.0 g) containing water at room temperature, the mixture was stirred at 30 ° C. for 24 hours under a hydrogen atmosphere of 2 atm. The reaction solution was filtered through celite. The solvent of the obtained filtrate was distilled off under reduced pressure to obtain Compound Q3 (33 g) as a crude product.
1 H-NMR (400 MHz, CDCl3): δ7.80 (s, 1H), 7.64-7.44 (m, 1H), 7.20-7.18 (m, 1H), 7.13-7.10 (m, 2H), 6.99 (s , 1H), 4.20 (brs, 2H), 2.91-2.78 (m, 3H), 2.07-2.03 (m, 2H), 1.63-1.55 (m, 2H), 1.49 (s, 9H).
化合物Q2(32.0g)のメタノール溶液(200mL)に、室温にて含水の10% パラジウムカーボン(5.0g)を加えた後に、2気圧の水素雰囲気下、30℃で24時間撹拌した。反応液をセライト濾過した。得られたろ液の溶媒を減圧留去することで、化合物Q3(33g)を粗生成物として取得した。
1H-NMR (400 MHz, CDCl3): δ7.80 (s, 1H), 7.64-7.44 (m, 1H), 7.20-7.18 (m, 1H), 7.13-7.10 (m, 2H), 6.99 (s, 1H), 4.20 (brs, 2H), 2.91-2.78 (m, 3H), 2.07-2.03 (m, 2H), 1.63-1.55 (m, 2H), 1.49 (s, 9H). c) Preparation of tert-butyl 4- [1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine-1-carboxylate (Compound Q3) Compound Q2 (32.0 g) in methanol (200 mL) After adding 10% palladium carbon (5.0 g) containing water at room temperature, the mixture was stirred at 30 ° C. for 24 hours under a hydrogen atmosphere of 2 atm. The reaction solution was filtered through celite. The solvent of the obtained filtrate was distilled off under reduced pressure to obtain Compound Q3 (33 g) as a crude product.
1 H-NMR (400 MHz, CDCl3): δ7.80 (s, 1H), 7.64-7.44 (m, 1H), 7.20-7.18 (m, 1H), 7.13-7.10 (m, 2H), 6.99 (s , 1H), 4.20 (brs, 2H), 2.91-2.78 (m, 3H), 2.07-2.03 (m, 2H), 1.63-1.55 (m, 2H), 1.49 (s, 9H).
d)tert-ブチル 4-[5-クロロ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]ピペリジン-1-カルボキシレート(化合物Q4)の製造
化合物Q3(5.1g)のアセトニトリル溶液(10mL)に、室温にてN-クロロスクシンイミド(2.2g)及びトリフルオロ酢酸(0.05mL)を加えた後に、30℃にて48時間撹拌した。反応液に水を加えた後に、酢酸エチルで抽出、水と飽和食塩水で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィーで精製することにより、化合物Q4(4.0g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.60 (s, 1H), 7.51-7.47 (m, 1H), 7.20-7.13 (m, 3H), 4.25 (brs, 2H), 2.88-2.81 (m, 3H), 1.89-1.83 (m, 4H), 1.49 (s, 9H). d) Preparation of tert-butyl 4- [5-chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine-1-carboxylate (Compound Q4) Acetonitrile of Compound Q3 (5.1 g) N-chlorosuccinimide (2.2 g) and trifluoroacetic acid (0.05 mL) were added to the solution (10 mL) at room temperature, and the mixture was stirred at 30 ° C. for 48 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate and washed with water and saturated brine. The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Compound Q4 (4.0 g) was obtained by purification by silica gel column chromatography.
1 H-NMR (400 MHz, CDCl3): δ7.60 (s, 1H), 7.51-7.47 (m, 1H), 7.20-7.13 (m, 3H), 4.25 (brs, 2H), 2.88-2.81 (m , 3H), 1.89-1.83 (m, 4H), 1.49 (s, 9H).
化合物Q3(5.1g)のアセトニトリル溶液(10mL)に、室温にてN-クロロスクシンイミド(2.2g)及びトリフルオロ酢酸(0.05mL)を加えた後に、30℃にて48時間撹拌した。反応液に水を加えた後に、酢酸エチルで抽出、水と飽和食塩水で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィーで精製することにより、化合物Q4(4.0g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.60 (s, 1H), 7.51-7.47 (m, 1H), 7.20-7.13 (m, 3H), 4.25 (brs, 2H), 2.88-2.81 (m, 3H), 1.89-1.83 (m, 4H), 1.49 (s, 9H). d) Preparation of tert-butyl 4- [5-chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine-1-carboxylate (Compound Q4) Acetonitrile of Compound Q3 (5.1 g) N-chlorosuccinimide (2.2 g) and trifluoroacetic acid (0.05 mL) were added to the solution (10 mL) at room temperature, and the mixture was stirred at 30 ° C. for 48 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate and washed with water and saturated brine. The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Compound Q4 (4.0 g) was obtained by purification by silica gel column chromatography.
1 H-NMR (400 MHz, CDCl3): δ7.60 (s, 1H), 7.51-7.47 (m, 1H), 7.20-7.13 (m, 3H), 4.25 (brs, 2H), 2.88-2.81 (m , 3H), 1.89-1.83 (m, 4H), 1.49 (s, 9H).
e)4-[5-クロロ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]ピペリジン・2塩酸塩(参考例1)の製造
化合物Q4(3.0g)のジオキサン溶液(5mL)に、室温にて4規定の塩化水素/ジオキサン溶液(10mL)を加えた後に、室温にて3時間撹拌した。反応溶媒を減圧留去して、参考例1の化合物(4.6g)を取得した。 e) Preparation of 4- [5-chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine dihydrochloride (Reference Example 1) Compound Q4 (3.0 g) in dioxane (5 mL) ), 4N hydrogen chloride / dioxane solution (10 mL) was added at room temperature, and the mixture was stirred at room temperature for 3 hours. The reaction solvent was distilled off under reduced pressure to obtain the compound of Reference Example 1 (4.6 g).
化合物Q4(3.0g)のジオキサン溶液(5mL)に、室温にて4規定の塩化水素/ジオキサン溶液(10mL)を加えた後に、室温にて3時間撹拌した。反応溶媒を減圧留去して、参考例1の化合物(4.6g)を取得した。 e) Preparation of 4- [5-chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine dihydrochloride (Reference Example 1) Compound Q4 (3.0 g) in dioxane (5 mL) ), 4N hydrogen chloride / dioxane solution (10 mL) was added at room temperature, and the mixture was stirred at room temperature for 3 hours. The reaction solvent was distilled off under reduced pressure to obtain the compound of Reference Example 1 (4.6 g).
参考例2
4-[5-シアノ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]ピペリジン・2塩酸塩

a)tert-ブチル 4-[1-(3-フルオロフェニル)-5-ヨード-1H-イミダゾール-4-イル]ピペリジン-1-カルボキシレート(化合物Q5)の製造
化合物Q3(5.0g) のアセトニトリル溶液(30mL)に、室温にてN-ヨードスクシンイミド(3.6g)及びトリフルオロ酢酸(0.05mL)を加えた後に、室温で遮光下72時間攪拌した。反応液にチオ硫酸ナトリウム水溶液を加えた後に、酢酸エチルで抽出、飽和食塩水で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィーで精製することにより、化合物Q5(4.3g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.78 (s, 1H), 7.49-7.48 (m, 1H), 7.21-7.08 (m, 3H), 4.24 (brs, 2H), 2.84-2.75 (m, 3H), 1.88-1.75 (m, 4H), 1.48 (s, 9H). Reference example 2
4- [5-Cyano-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine dihydrochloride
a) Preparation of tert-butyl 4- [1- (3-fluorophenyl) -5-iodo-1H-imidazol-4-yl] piperidine-1-carboxylate (Compound Q5) Compound Q3 (5.0 g) in acetonitrile N-iodosuccinimide (3.6 g) and trifluoroacetic acid (0.05 mL) were added to the solution (30 mL) at room temperature, and the mixture was stirred at room temperature for 72 hours under light shielding. A sodium thiosulfate aqueous solution was added to the reaction solution, followed by extraction with ethyl acetate and washing with saturated brine. The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Compound Q5 (4.3 g) was obtained by purification by silica gel column chromatography.
1 H-NMR (400 MHz, CDCl3): δ7.78 (s, 1H), 7.49-7.48 (m, 1H), 7.21-7.08 (m, 3H), 4.24 (brs, 2H), 2.84-2.75 (m , 3H), 1.88-1.75 (m, 4H), 1.48 (s, 9H).
4-[5-シアノ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]ピペリジン・2塩酸塩
化合物Q3(5.0g) のアセトニトリル溶液(30mL)に、室温にてN-ヨードスクシンイミド(3.6g)及びトリフルオロ酢酸(0.05mL)を加えた後に、室温で遮光下72時間攪拌した。反応液にチオ硫酸ナトリウム水溶液を加えた後に、酢酸エチルで抽出、飽和食塩水で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィーで精製することにより、化合物Q5(4.3g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.78 (s, 1H), 7.49-7.48 (m, 1H), 7.21-7.08 (m, 3H), 4.24 (brs, 2H), 2.84-2.75 (m, 3H), 1.88-1.75 (m, 4H), 1.48 (s, 9H). Reference example 2
4- [5-Cyano-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine dihydrochloride
1 H-NMR (400 MHz, CDCl3): δ7.78 (s, 1H), 7.49-7.48 (m, 1H), 7.21-7.08 (m, 3H), 4.24 (brs, 2H), 2.84-2.75 (m , 3H), 1.88-1.75 (m, 4H), 1.48 (s, 9H).
b)tert-ブチル 4-[5-シアノ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]ピペリジン-1-カルボキシレート(化合物Q6)の製造
化合物Q5(4.3g)のジメチルホルムアミド(30mL)溶液に、室温にてシアン化銅(2.6g)を加えて、120℃で18時間加熱攪拌した。反応液をセライト濾過した後に、得られたろ液の溶媒を減圧留去した。シリカゲルカラムクロマトグラフィーで精製することにより、化合物Q6(1.2g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.79 (s, 1H), 7.57-7.55 (m, 1H), 7.30-7.21 (m, 3H), 4.27 (brs, 2H), 3.08-3.04 (m, 1H), 2.87 (brs, 2H), 1.97-1.86 (m, 4H), 1.48 (s, 9H). b) Preparation of tert-butyl 4- [5-cyano-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine-1-carboxylate (Compound Q6) Dimethyl of Compound Q5 (4.3 g) Copper cyanide (2.6 g) was added to a formamide (30 mL) solution at room temperature, and the mixture was heated with stirring at 120 ° C. for 18 hours. After the reaction solution was filtered through Celite, the solvent of the obtained filtrate was distilled off under reduced pressure. Compound Q6 (1.2 g) was obtained by purification by silica gel column chromatography.
1 H-NMR (400 MHz, CDCl3): δ 7.79 (s, 1H), 7.57-7.55 (m, 1H), 7.30-7.21 (m, 3H), 4.27 (brs, 2H), 3.08-3.04 (m , 1H), 2.87 (brs, 2H), 1.97-1.86 (m, 4H), 1.48 (s, 9H).
化合物Q5(4.3g)のジメチルホルムアミド(30mL)溶液に、室温にてシアン化銅(2.6g)を加えて、120℃で18時間加熱攪拌した。反応液をセライト濾過した後に、得られたろ液の溶媒を減圧留去した。シリカゲルカラムクロマトグラフィーで精製することにより、化合物Q6(1.2g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.79 (s, 1H), 7.57-7.55 (m, 1H), 7.30-7.21 (m, 3H), 4.27 (brs, 2H), 3.08-3.04 (m, 1H), 2.87 (brs, 2H), 1.97-1.86 (m, 4H), 1.48 (s, 9H). b) Preparation of tert-butyl 4- [5-cyano-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine-1-carboxylate (Compound Q6) Dimethyl of Compound Q5 (4.3 g) Copper cyanide (2.6 g) was added to a formamide (30 mL) solution at room temperature, and the mixture was heated with stirring at 120 ° C. for 18 hours. After the reaction solution was filtered through Celite, the solvent of the obtained filtrate was distilled off under reduced pressure. Compound Q6 (1.2 g) was obtained by purification by silica gel column chromatography.
1 H-NMR (400 MHz, CDCl3): δ 7.79 (s, 1H), 7.57-7.55 (m, 1H), 7.30-7.21 (m, 3H), 4.27 (brs, 2H), 3.08-3.04 (m , 1H), 2.87 (brs, 2H), 1.97-1.86 (m, 4H), 1.48 (s, 9H).
c)4-[5-シアノ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]ピペリジン・2塩酸塩(参考例2)の製造
化合物Q6(1.2g)を用いて、参考例1の工程e)に準ずる方法により参考例2の化合物(1.1g)を取得した。 c) Preparation of 4- [5-cyano-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine dihydrochloride (Reference Example 2) Compound Q6 (1.2 g) was used as a reference. The compound of Reference Example 2 (1.1 g) was obtained by the method according to Step e) of Example 1.
化合物Q6(1.2g)を用いて、参考例1の工程e)に準ずる方法により参考例2の化合物(1.1g)を取得した。 c) Preparation of 4- [5-cyano-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidine dihydrochloride (Reference Example 2) Compound Q6 (1.2 g) was used as a reference. The compound of Reference Example 2 (1.1 g) was obtained by the method according to Step e) of Example 1.
参考例3
4-{3-[5-(トリフルオロメチル)ピリジン-2-イル]-1,2-オキサゾール-5-イル}ピペリジン・2塩酸塩

a)tert-ブチル 4-エチイルピペリジン-1-カルボキシレート(化合物Q7)の製造
tert-ブチル 4-ホルミルピペリジン-1-カルボキシレート(7.8g)のメタノール溶液(50mL)に、0℃にて炭酸カリウム(11g)及びジメチル(アセチルジアゾメチル)ホスホネート(8.6g)を加えて、0℃で2時間攪拌した。反応液に飽和塩化アンモニウム水溶液(50mL)を加えた後に、溶媒を減圧留去し、酢酸エチルで抽出した。得られた有機層を硫酸ナトリウムで乾燥、溶媒を減圧留去した後に、シリカゲルカラムクロマトグラフィーで精製することにより、化合物Q7(3.7g)を取得した。
1H-NMR (400 MHz, CDCl3): δ3.71-3.68 (m, 2H), 3.21-3.15 (m, 2H), 2.63-2.53 (m, 1H), 2.10-2.11 (m, 1H), 1.80-1.75 (m, 2H), 1.60-1.55 (m, 2H). Reference example 3
4- {3- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-5-yl} piperidine dihydrochloride
a) Preparation of tert-butyl 4-ethiylpiperidine-1-carboxylate (Compound Q7) To a methanol solution (50 mL) of tert-butyl 4-formylpiperidine-1-carboxylate (7.8 g) at 0 ° C. Potassium carbonate (11 g) and dimethyl (acetyldiazomethyl) phosphonate (8.6 g) were added and stirred at 0 ° C. for 2 hours. Saturated aqueous ammonium chloride solution (50 mL) was added to the reaction mixture, and the solvent was evaporated under reduced pressure and extracted with ethyl acetate. The obtained organic layer was dried over sodium sulfate, the solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography to obtain compound Q7 (3.7 g).
1 H-NMR (400 MHz, CDCl3): δ3.71-3.68 (m, 2H), 3.21-3.15 (m, 2H), 2.63-2.53 (m, 1H), 2.10-2.11 (m, 1H), 1.80 -1.75 (m, 2H), 1.60-1.55 (m, 2H).
4-{3-[5-(トリフルオロメチル)ピリジン-2-イル]-1,2-オキサゾール-5-イル}ピペリジン・2塩酸塩
tert-ブチル 4-ホルミルピペリジン-1-カルボキシレート(7.8g)のメタノール溶液(50mL)に、0℃にて炭酸カリウム(11g)及びジメチル(アセチルジアゾメチル)ホスホネート(8.6g)を加えて、0℃で2時間攪拌した。反応液に飽和塩化アンモニウム水溶液(50mL)を加えた後に、溶媒を減圧留去し、酢酸エチルで抽出した。得られた有機層を硫酸ナトリウムで乾燥、溶媒を減圧留去した後に、シリカゲルカラムクロマトグラフィーで精製することにより、化合物Q7(3.7g)を取得した。
1H-NMR (400 MHz, CDCl3): δ3.71-3.68 (m, 2H), 3.21-3.15 (m, 2H), 2.63-2.53 (m, 1H), 2.10-2.11 (m, 1H), 1.80-1.75 (m, 2H), 1.60-1.55 (m, 2H). Reference example 3
4- {3- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-5-yl} piperidine dihydrochloride
1 H-NMR (400 MHz, CDCl3): δ3.71-3.68 (m, 2H), 3.21-3.15 (m, 2H), 2.63-2.53 (m, 1H), 2.10-2.11 (m, 1H), 1.80 -1.75 (m, 2H), 1.60-1.55 (m, 2H).
b)N-ヒドロキシ-1-[5-(トリフルオロメチル)ピリジン-2-イル]メタンイミン(化合物Q8)の製造
5-(トリフルオロメチル)ピリジン-2-カルバアルデヒド(2.0g)のメタノール溶液(60mL)に、0℃にてトリエチルアミン(10mL)及びヒドロキシルアミンの塩酸塩(3.0g)を加えた後に、室温まで昇温して16時間攪拌した。反応液を減圧留去した後に、酢酸エチル(20mL)で抽出、飽和炭酸水素ナトリウム水溶液(30mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルクロマトグラフィー (石油エーテル/酢酸エチル = 90/10)で精製することにより、化合物Q8(0.9g)を取得した。
1H-NMR (400 MHz, DMSO-d6): δ12.10 (s, 1H), 8.97 (s, 1H), 8.22 (d, J=8.4Hz, 1H), 8.17 (s, 1H), 7.98 (d, J=8.4Hz, 1H). b) Preparation of N-hydroxy-1- [5- (trifluoromethyl) pyridin-2-yl] methanimine (Compound Q8) 5- (Trifluoromethyl) pyridine-2-carbaldehyde (2.0 g) in methanol (60 mL), triethylamine (10 mL) and hydroxylamine hydrochloride (3.0 g) were added at 0 ° C., and then the mixture was warmed to room temperature and stirred for 16 hours. The reaction mixture was evaporated under reduced pressure, extracted with ethyl acetate (20 mL), and washed with saturated aqueous sodium hydrogen carbonate solution (30 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel chromatography (petroleum ether / ethyl acetate = 90/10) gave Compound Q8 (0.9 g).
1 H-NMR (400 MHz, DMSO-d6): δ12.10 (s, 1H), 8.97 (s, 1H), 8.22 (d, J = 8.4Hz, 1H), 8.17 (s, 1H), 7.98 ( d, J = 8.4Hz, 1H).
5-(トリフルオロメチル)ピリジン-2-カルバアルデヒド(2.0g)のメタノール溶液(60mL)に、0℃にてトリエチルアミン(10mL)及びヒドロキシルアミンの塩酸塩(3.0g)を加えた後に、室温まで昇温して16時間攪拌した。反応液を減圧留去した後に、酢酸エチル(20mL)で抽出、飽和炭酸水素ナトリウム水溶液(30mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルクロマトグラフィー (石油エーテル/酢酸エチル = 90/10)で精製することにより、化合物Q8(0.9g)を取得した。
1H-NMR (400 MHz, DMSO-d6): δ12.10 (s, 1H), 8.97 (s, 1H), 8.22 (d, J=8.4Hz, 1H), 8.17 (s, 1H), 7.98 (d, J=8.4Hz, 1H). b) Preparation of N-hydroxy-1- [5- (trifluoromethyl) pyridin-2-yl] methanimine (Compound Q8) 5- (Trifluoromethyl) pyridine-2-carbaldehyde (2.0 g) in methanol (60 mL), triethylamine (10 mL) and hydroxylamine hydrochloride (3.0 g) were added at 0 ° C., and then the mixture was warmed to room temperature and stirred for 16 hours. The reaction mixture was evaporated under reduced pressure, extracted with ethyl acetate (20 mL), and washed with saturated aqueous sodium hydrogen carbonate solution (30 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel chromatography (petroleum ether / ethyl acetate = 90/10) gave Compound Q8 (0.9 g).
1 H-NMR (400 MHz, DMSO-d6): δ12.10 (s, 1H), 8.97 (s, 1H), 8.22 (d, J = 8.4Hz, 1H), 8.17 (s, 1H), 7.98 ( d, J = 8.4Hz, 1H).
c)N-ヒドロキシ-5-(トリフルオロメチル)ピリジン-2-カルボキシイミドイル クロライド(化合物Q9)の製造
化合物Q8(0.9g)のDMF溶液(7mL)に、0℃にてN-クロロスクシンイミド(770mg)を加えた後に、室温まで昇温して16時間撹拌した。反応液に水(25mL)を加えた後に、酢酸エチル(15mL)で抽出、水(50mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去することにより、化合物Q9(930mg)を取得した。
1H-NMR (400 MHz, DMSO-d6): δ13.07 (s, 1H), 9.08 (s, 1H), 8.31-8.29 (m, 1H), 8.10-8.08 (m, 1H). c) Preparation of N-hydroxy-5- (trifluoromethyl) pyridine-2-carboximidoyl chloride (Compound Q9) N-chlorosuccinimide at 0 ° C. in DMF solution (7 mL) of Compound Q8 (0.9 g) (770 mg) was added, and the mixture was warmed to room temperature and stirred for 16 hours. Water (25 mL) was added to the reaction solution, followed by extraction with ethyl acetate (15 mL) and washing with water (50 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was evaporated under reduced pressure to obtain Compound Q9 (930 mg).
1 H-NMR (400 MHz, DMSO-d6): δ13.07 (s, 1H), 9.08 (s, 1H), 8.31-8.29 (m, 1H), 8.10-8.08 (m, 1H).
化合物Q8(0.9g)のDMF溶液(7mL)に、0℃にてN-クロロスクシンイミド(770mg)を加えた後に、室温まで昇温して16時間撹拌した。反応液に水(25mL)を加えた後に、酢酸エチル(15mL)で抽出、水(50mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去することにより、化合物Q9(930mg)を取得した。
1H-NMR (400 MHz, DMSO-d6): δ13.07 (s, 1H), 9.08 (s, 1H), 8.31-8.29 (m, 1H), 8.10-8.08 (m, 1H). c) Preparation of N-hydroxy-5- (trifluoromethyl) pyridine-2-carboximidoyl chloride (Compound Q9) N-chlorosuccinimide at 0 ° C. in DMF solution (7 mL) of Compound Q8 (0.9 g) (770 mg) was added, and the mixture was warmed to room temperature and stirred for 16 hours. Water (25 mL) was added to the reaction solution, followed by extraction with ethyl acetate (15 mL) and washing with water (50 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was evaporated under reduced pressure to obtain Compound Q9 (930 mg).
1 H-NMR (400 MHz, DMSO-d6): δ13.07 (s, 1H), 9.08 (s, 1H), 8.31-8.29 (m, 1H), 8.10-8.08 (m, 1H).
d)tert-ブチル 4-{3-[5-(トリフルオロメチル)ピリジン-2-イル]-1,2-オキサゾール-5-イル}ピペリジン-1-カルボキシレート(化合物Q10)の製造
化合物Q9(930mg)及び化合物Q7(690mg)のテトラヒドロフラン溶液(10mL)に、室温にてトリエチルアミン(1.7mL)のテトラヒドロフラン溶液(5mL)を加えた後に、60℃まで昇温して2時間撹拌した。反応液を減圧留去した後に、飽和炭酸水素ナトリウム水溶液(10mL)を加えて、酢酸エチル(5mL)で抽出した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルクロマトグラフィー (石油エーテル/酢酸エチル = 90/10)で精製することにより、化合物Q10(500mg)を取得した。
1H-NMR (400 MHz, CDCl3): δ8.93 (s, 1H), 8.23-8.20 (m, 1H), 8.05-8.03 (m, 1H), 6.70 (s, 1H), 4.18 (brs, 2H), 3.05-2.90 (m, 3H), 2.11-2.08 (m, 2H), 1.74-1.70 (m, 2H), 1.48 (s, 9H). d) Preparation of tert-butyl 4- {3- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-5-yl} piperidine-1-carboxylate (Compound Q10) Compound Q9 ( To a tetrahydrofuran solution (10 mL) of 930 mg) and compound Q7 (690 mg) was added a tetrahydrofuran solution (5 mL) of triethylamine (1.7 mL) at room temperature, and then the mixture was heated to 60 ° C. and stirred for 2 hours. The reaction mixture was evaporated under reduced pressure, saturated aqueous sodium hydrogen carbonate solution (10 mL) was added, and the mixture was extracted with ethyl acetate (5 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel chromatography (petroleum ether / ethyl acetate = 90/10) gave Compound Q10 (500 mg).
1 H-NMR (400 MHz, CDCl3): δ8.93 (s, 1H), 8.23-8.20 (m, 1H), 8.05-8.03 (m, 1H), 6.70 (s, 1H), 4.18 (brs, 2H ), 3.05-2.90 (m, 3H), 2.11-2.08 (m, 2H), 1.74-1.70 (m, 2H), 1.48 (s, 9H).
化合物Q9(930mg)及び化合物Q7(690mg)のテトラヒドロフラン溶液(10mL)に、室温にてトリエチルアミン(1.7mL)のテトラヒドロフラン溶液(5mL)を加えた後に、60℃まで昇温して2時間撹拌した。反応液を減圧留去した後に、飽和炭酸水素ナトリウム水溶液(10mL)を加えて、酢酸エチル(5mL)で抽出した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルクロマトグラフィー (石油エーテル/酢酸エチル = 90/10)で精製することにより、化合物Q10(500mg)を取得した。
1H-NMR (400 MHz, CDCl3): δ8.93 (s, 1H), 8.23-8.20 (m, 1H), 8.05-8.03 (m, 1H), 6.70 (s, 1H), 4.18 (brs, 2H), 3.05-2.90 (m, 3H), 2.11-2.08 (m, 2H), 1.74-1.70 (m, 2H), 1.48 (s, 9H). d) Preparation of tert-butyl 4- {3- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-5-yl} piperidine-1-carboxylate (Compound Q10) Compound Q9 ( To a tetrahydrofuran solution (10 mL) of 930 mg) and compound Q7 (690 mg) was added a tetrahydrofuran solution (5 mL) of triethylamine (1.7 mL) at room temperature, and then the mixture was heated to 60 ° C. and stirred for 2 hours. The reaction mixture was evaporated under reduced pressure, saturated aqueous sodium hydrogen carbonate solution (10 mL) was added, and the mixture was extracted with ethyl acetate (5 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel chromatography (petroleum ether / ethyl acetate = 90/10) gave Compound Q10 (500 mg).
1 H-NMR (400 MHz, CDCl3): δ8.93 (s, 1H), 8.23-8.20 (m, 1H), 8.05-8.03 (m, 1H), 6.70 (s, 1H), 4.18 (brs, 2H ), 3.05-2.90 (m, 3H), 2.11-2.08 (m, 2H), 1.74-1.70 (m, 2H), 1.48 (s, 9H).
e)4-{3-[5-(トリフルオロメチル)ピリジン-2-イル]-1,2-オキサゾール-5-イル}ピペリジン・2塩酸塩(参考例3)の製造
化合物Q10(500mg)を用いて、参考例1の工程e)に準ずる方法により参考例3の化合物(360mg)を取得した。 e) Preparation of 4- {3- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-5-yl} piperidine dihydrochloride (Reference Example 3) Compound Q10 (500 mg) And the compound of Reference Example 3 (360 mg) was obtained by the method according to Step e) of Reference Example 1.
化合物Q10(500mg)を用いて、参考例1の工程e)に準ずる方法により参考例3の化合物(360mg)を取得した。 e) Preparation of 4- {3- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-5-yl} piperidine dihydrochloride (Reference Example 3) Compound Q10 (500 mg) And the compound of Reference Example 3 (360 mg) was obtained by the method according to Step e) of Reference Example 1.
参考例4
4-{5-[5-(トリフルオロメチル)ピリジン-2-イル]-1,2-オキサゾール-3-イル}ピペリジン・2塩酸塩

a)tert-ブチル 4-[(ヒドロキシイミノ)メチル]ピペリジン-1-カルボキシレート(化合物Q11)の製造
tert-ブチル 4-ホルミルピペリジン-1-カルボキシレート(11g) のメタノール/水溶液(80mL/80mL)に、室温にて炭酸ナトリウム(2.7g)及びとヒドロキシルアミンの塩酸塩(4.3g)を加えて、室温で16時間撹拌した。反応液を減圧留去した後に、塩化メチレン(50mL)で抽出した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去することにより、化合物Q11(8.5g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.40 (brs, 1H), 7.34 (d, J=5.6Hz, 1H), 4.08 (brs, 2H), 2.82-2.77 (m, 2H), 2.41-2.34 (m, 1H), 1.77-1.75 (m, 2H), 1.49-1.42 (m, 11H). Reference example 4
4- {5- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-3-yl} piperidine dihydrochloride
a) Preparation of tert-butyl 4-[(hydroxyimino) methyl] piperidine-1-carboxylate (Compound Q11) Methanol / water solution of tert-butyl 4-formylpiperidine-1-carboxylate (11 g) (80 mL / 80 mL) To the mixture, sodium carbonate (2.7 g) and hydroxylamine hydrochloride (4.3 g) were added at room temperature, and the mixture was stirred at room temperature for 16 hours. The reaction solution was distilled off under reduced pressure and then extracted with methylene chloride (50 mL). The obtained organic layer was dried over sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain Compound Q11 (8.5 g).
1 H-NMR (400 MHz, CDCl3): δ7.40 (brs, 1H), 7.34 (d, J = 5.6Hz, 1H), 4.08 (brs, 2H), 2.82-2.77 (m, 2H), 2.41- 2.34 (m, 1H), 1.77-1.75 (m, 2H), 1.49-1.42 (m, 11H).
4-{5-[5-(トリフルオロメチル)ピリジン-2-イル]-1,2-オキサゾール-3-イル}ピペリジン・2塩酸塩
tert-ブチル 4-ホルミルピペリジン-1-カルボキシレート(11g) のメタノール/水溶液(80mL/80mL)に、室温にて炭酸ナトリウム(2.7g)及びとヒドロキシルアミンの塩酸塩(4.3g)を加えて、室温で16時間撹拌した。反応液を減圧留去した後に、塩化メチレン(50mL)で抽出した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去することにより、化合物Q11(8.5g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.40 (brs, 1H), 7.34 (d, J=5.6Hz, 1H), 4.08 (brs, 2H), 2.82-2.77 (m, 2H), 2.41-2.34 (m, 1H), 1.77-1.75 (m, 2H), 1.49-1.42 (m, 11H). Reference example 4
4- {5- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-3-yl} piperidine dihydrochloride
1 H-NMR (400 MHz, CDCl3): δ7.40 (brs, 1H), 7.34 (d, J = 5.6Hz, 1H), 4.08 (brs, 2H), 2.82-2.77 (m, 2H), 2.41- 2.34 (m, 1H), 1.77-1.75 (m, 2H), 1.49-1.42 (m, 11H).
b)tert-ブチル 4-[クロロ(ヒドロキシイミノ)メチル]ピペリジン-1-カルボキシレート(化合物Q12)の製造
化合物Q11(8.5g)のDMF溶液(7mL)に、0℃にてN-クロロスクシンイミド(6.0g)を加えた後に、室温まで昇温して4時間撹拌した。反応液に水(50mL)を加えて後に、析出した固体をろ取することで、化合物Q12(2.9g)を取得した。
1H-NMR (400 MHz, CDCl3): δ8.87 (s, 1H), 4.14 (brs, 2H), 2.82-2.78 (m, 2H), 2.61-2.55 (m, 1H), 1.94-1.91 (m, 2H), 1.66-1.62 (m, 2H), 1.46 (s, 9H). b) Preparation of tert-butyl 4- [chloro (hydroxyimino) methyl] piperidine-1-carboxylate (compound Q12) N-chlorosuccinimide in a DMF solution (7 mL) of compound Q11 (8.5 g) at 0 ° C (6.0 g) was added, and the mixture was warmed to room temperature and stirred for 4 hours. After adding water (50 mL) to the reaction solution, the precipitated solid was collected by filtration to obtain Compound Q12 (2.9 g).
1 H-NMR (400 MHz, CDCl3): δ8.87 (s, 1H), 4.14 (brs, 2H), 2.82-2.78 (m, 2H), 2.61-2.55 (m, 1H), 1.94-1.91 (m , 2H), 1.66-1.62 (m, 2H), 1.46 (s, 9H).
化合物Q11(8.5g)のDMF溶液(7mL)に、0℃にてN-クロロスクシンイミド(6.0g)を加えた後に、室温まで昇温して4時間撹拌した。反応液に水(50mL)を加えて後に、析出した固体をろ取することで、化合物Q12(2.9g)を取得した。
1H-NMR (400 MHz, CDCl3): δ8.87 (s, 1H), 4.14 (brs, 2H), 2.82-2.78 (m, 2H), 2.61-2.55 (m, 1H), 1.94-1.91 (m, 2H), 1.66-1.62 (m, 2H), 1.46 (s, 9H). b) Preparation of tert-butyl 4- [chloro (hydroxyimino) methyl] piperidine-1-carboxylate (compound Q12) N-chlorosuccinimide in a DMF solution (7 mL) of compound Q11 (8.5 g) at 0 ° C (6.0 g) was added, and the mixture was warmed to room temperature and stirred for 4 hours. After adding water (50 mL) to the reaction solution, the precipitated solid was collected by filtration to obtain Compound Q12 (2.9 g).
1 H-NMR (400 MHz, CDCl3): δ8.87 (s, 1H), 4.14 (brs, 2H), 2.82-2.78 (m, 2H), 2.61-2.55 (m, 1H), 1.94-1.91 (m , 2H), 1.66-1.62 (m, 2H), 1.46 (s, 9H).
c)tert-ブチル 4-{5-[5-(トリフルオロメチル)ピリジン-2-イル]-1,2-オキサゾール-3-イル}ピペリジン-1-カルボキシレート(化合物Q13)の製造
5-(トリフルオロメチル)ピリジン-2-カルバルデヒド(1.0 g)のメタノール溶液(10mL)に、0℃にてジメチル(アセチルジアゾメチル)ホスホネート(1.1g)及び炭酸カリウム(1.6g)を加えた後に、室温まで昇温して窒素雰囲気下で16時間撹拌した。反応液にテトラヒドロフラン(40mL)を加えた後に、室温にて化合物Q12(2.9g)及びトリエチルアミン(5.6g)のテトラヒドロフラン溶液(20mL)を加えた。反応液を60℃に昇温して2時間撹拌した後に、溶媒を減圧留去した。残渣に酢酸エチル(20mL)及び飽和炭酸水素ナトリウム水溶液(30mL)を加えて抽出した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去。シリカゲルクロマトグラフィー (石油エーテル/酢酸エチル = 90/10)で精製することにより、化合物Q13(270mg)を取得した。
1H-NMR (400 MHz, CDCl3): δ8.92 (s, 1H), 8.08-8.00 (m, 2H), 6.91 (s, 1H), 4.20 (brs, 2H), 3.04-2.89 (m, 3H), 2.02-1.99 (m, 2H), 1.77-1.67 (m, 2H), 1.48 (s, 9H). c) Preparation of tert-butyl 4- {5- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-3-yl} piperidine-1-carboxylate (Compound Q13) 5- ( Dimethyl (acetyldiazomethyl) phosphonate (1.1 g) and potassium carbonate (1.6 g) were added to a methanol solution (10 mL) of trifluoromethyl) pyridine-2-carbaldehyde (1.0 g) at 0 ° C. Then, the temperature was raised to room temperature and stirred for 16 hours under a nitrogen atmosphere. Tetrahydrofuran (40 mL) was added to the reaction solution, and then a tetrahydrofuran solution (20 mL) of compound Q12 (2.9 g) and triethylamine (5.6 g) was added at room temperature. The reaction solution was heated to 60 ° C. and stirred for 2 hours, and then the solvent was distilled off under reduced pressure. The residue was extracted with ethyl acetate (20 mL) and saturated aqueous sodium hydrogen carbonate (30 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. The product was purified by silica gel chromatography (petroleum ether / ethyl acetate = 90/10) to obtain Compound Q13 (270 mg).
1 H-NMR (400 MHz, CDCl3): δ8.92 (s, 1H), 8.08-8.00 (m, 2H), 6.91 (s, 1H), 4.20 (brs, 2H), 3.04-2.89 (m, 3H ), 2.02-1.99 (m, 2H), 1.77-1.67 (m, 2H), 1.48 (s, 9H).
5-(トリフルオロメチル)ピリジン-2-カルバルデヒド(1.0 g)のメタノール溶液(10mL)に、0℃にてジメチル(アセチルジアゾメチル)ホスホネート(1.1g)及び炭酸カリウム(1.6g)を加えた後に、室温まで昇温して窒素雰囲気下で16時間撹拌した。反応液にテトラヒドロフラン(40mL)を加えた後に、室温にて化合物Q12(2.9g)及びトリエチルアミン(5.6g)のテトラヒドロフラン溶液(20mL)を加えた。反応液を60℃に昇温して2時間撹拌した後に、溶媒を減圧留去した。残渣に酢酸エチル(20mL)及び飽和炭酸水素ナトリウム水溶液(30mL)を加えて抽出した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去。シリカゲルクロマトグラフィー (石油エーテル/酢酸エチル = 90/10)で精製することにより、化合物Q13(270mg)を取得した。
1H-NMR (400 MHz, CDCl3): δ8.92 (s, 1H), 8.08-8.00 (m, 2H), 6.91 (s, 1H), 4.20 (brs, 2H), 3.04-2.89 (m, 3H), 2.02-1.99 (m, 2H), 1.77-1.67 (m, 2H), 1.48 (s, 9H). c) Preparation of tert-butyl 4- {5- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-3-yl} piperidine-1-carboxylate (Compound Q13) 5- ( Dimethyl (acetyldiazomethyl) phosphonate (1.1 g) and potassium carbonate (1.6 g) were added to a methanol solution (10 mL) of trifluoromethyl) pyridine-2-carbaldehyde (1.0 g) at 0 ° C. Then, the temperature was raised to room temperature and stirred for 16 hours under a nitrogen atmosphere. Tetrahydrofuran (40 mL) was added to the reaction solution, and then a tetrahydrofuran solution (20 mL) of compound Q12 (2.9 g) and triethylamine (5.6 g) was added at room temperature. The reaction solution was heated to 60 ° C. and stirred for 2 hours, and then the solvent was distilled off under reduced pressure. The residue was extracted with ethyl acetate (20 mL) and saturated aqueous sodium hydrogen carbonate (30 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. The product was purified by silica gel chromatography (petroleum ether / ethyl acetate = 90/10) to obtain Compound Q13 (270 mg).
1 H-NMR (400 MHz, CDCl3): δ8.92 (s, 1H), 8.08-8.00 (m, 2H), 6.91 (s, 1H), 4.20 (brs, 2H), 3.04-2.89 (m, 3H ), 2.02-1.99 (m, 2H), 1.77-1.67 (m, 2H), 1.48 (s, 9H).
d)4-{5-[5-(トリフルオロメチル)ピリジン-2-イル]-1,2-オキサゾール-3-イル}ピペリジン・2塩酸塩(参考例4)の製造
化合物Q13(270mg)を用いて、参考例1の工程e)に準ずる方法により参考例4の化合物(210mg)を取得した。 d) Preparation of 4- {5- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-3-yl} piperidine dihydrochloride (Reference Example 4) Compound Q13 (270 mg) And the compound of Reference Example 4 (210 mg) was obtained by the method according to Step e) of Reference Example 1.
化合物Q13(270mg)を用いて、参考例1の工程e)に準ずる方法により参考例4の化合物(210mg)を取得した。 d) Preparation of 4- {5- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-3-yl} piperidine dihydrochloride (Reference Example 4) Compound Q13 (270 mg) And the compound of Reference Example 4 (210 mg) was obtained by the method according to Step e) of Reference Example 1.
参考例5
4-[2-(4-フルオロフェニル)-1,3-オキサゾール-5-イル]ピペリジン・塩酸塩

a)tert-ブチル 4-(1-ヒドロキシ-2-ニトロエチル)ピペリジン-1-カルボキシレート(化合物Q14)の製造
tert-ブチル 4-ホルミルピペリジン-1-カルボキシレート(5.0g)のテトラヒドロフラン/tert-ブタノール溶液(40mL/40mL)に、室温にてtert-ブトキシカリウム(2.63g)及びニトロメタン(2.51mL)を加えた後に、室温で2時間撹拌した。反応液に室温にて酢酸(1.6mL)加えた後に、酢酸エチル(200mL)を加えて抽出、飽和食塩水で洗浄をおこなった。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、化合物Q14(6.8g)を取得した。
1H-NMR (400 MHz, CDCl3): δ4.47-4.40 (m, 1H), 4.18-4.12 (m, 3H), 2.70-2.63 (m, 2H), 2.07-2.04 (m, 1H), 1.61-1.57 (m, 2H), 1.44 (s, 9H), 1.30-1.25 (m, 4H). Reference Example 5
4- [2- (4-Fluorophenyl) -1,3-oxazol-5-yl] piperidine hydrochloride
a) Preparation of tert-butyl 4- (1-hydroxy-2-nitroethyl) piperidine-1-carboxylate (compound Q14) tert-butyl 4-formylpiperidine-1-carboxylate (5.0 g) in tetrahydrofuran / tert- To a butanol solution (40 mL / 40 mL) were added tert-butoxypotassium (2.63 g) and nitromethane (2.51 mL) at room temperature, and the mixture was stirred at room temperature for 2 hours. Acetic acid (1.6 mL) was added to the reaction solution at room temperature, followed by extraction with ethyl acetate (200 mL) and washing with saturated brine. The obtained organic layer was dried over sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain Compound Q14 (6.8 g).
1 H-NMR (400 MHz, CDCl3): δ4.47-4.40 (m, 1H), 4.18-4.12 (m, 3H), 2.70-2.63 (m, 2H), 2.07-2.04 (m, 1H), 1.61 -1.57 (m, 2H), 1.44 (s, 9H), 1.30-1.25 (m, 4H).
4-[2-(4-フルオロフェニル)-1,3-オキサゾール-5-イル]ピペリジン・塩酸塩
tert-ブチル 4-ホルミルピペリジン-1-カルボキシレート(5.0g)のテトラヒドロフラン/tert-ブタノール溶液(40mL/40mL)に、室温にてtert-ブトキシカリウム(2.63g)及びニトロメタン(2.51mL)を加えた後に、室温で2時間撹拌した。反応液に室温にて酢酸(1.6mL)加えた後に、酢酸エチル(200mL)を加えて抽出、飽和食塩水で洗浄をおこなった。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去することで、化合物Q14(6.8g)を取得した。
1H-NMR (400 MHz, CDCl3): δ4.47-4.40 (m, 1H), 4.18-4.12 (m, 3H), 2.70-2.63 (m, 2H), 2.07-2.04 (m, 1H), 1.61-1.57 (m, 2H), 1.44 (s, 9H), 1.30-1.25 (m, 4H). Reference Example 5
4- [2- (4-Fluorophenyl) -1,3-oxazol-5-yl] piperidine hydrochloride
1 H-NMR (400 MHz, CDCl3): δ4.47-4.40 (m, 1H), 4.18-4.12 (m, 3H), 2.70-2.63 (m, 2H), 2.07-2.04 (m, 1H), 1.61 -1.57 (m, 2H), 1.44 (s, 9H), 1.30-1.25 (m, 4H).
b)tert-ブチル 4-(2-アミノ-1-ヒドロキシエチル)ピペリジン-1-カルボキシレート(化合物Q15)の製造
化合物Q14(6.8g)のメタノール溶液(65mL)に、室温にて10%パラジウムカーボン(0.68g)を加えた後に、水素雰囲気下で2時間撹拌した。反応液をセライト濾過した後に、ろ液の溶媒を減圧留去することで、化合物Q15(6.0g)を取得した。
1H-NMR (400 MHz, CDCl3): δ4.13 (brs, 2H), 3.36 (brs, 1H), 2.93 (brs, 5H), 2.65 (brs, 2H), 1.84-1.81 (m, 1H), 1.60-1.54 (m, 2H), 1.45 (s, 9H), 1.27-1.16 (m, 2H). b) Preparation of tert-butyl 4- (2-amino-1-hydroxyethyl) piperidine-1-carboxylate (Compound Q15) To a methanol solution (65 mL) of Compound Q14 (6.8 g) at room temperature with 10% palladium After adding carbon (0.68 g), the mixture was stirred for 2 hours under a hydrogen atmosphere. The reaction solution was filtered through Celite, and then the solvent of the filtrate was distilled off under reduced pressure to obtain Compound Q15 (6.0 g).
1 H-NMR (400 MHz, CDCl3): δ4.13 (brs, 2H), 3.36 (brs, 1H), 2.93 (brs, 5H), 2.65 (brs, 2H), 1.84-1.81 (m, 1H), 1.60-1.54 (m, 2H), 1.45 (s, 9H), 1.27-1.16 (m, 2H).
化合物Q14(6.8g)のメタノール溶液(65mL)に、室温にて10%パラジウムカーボン(0.68g)を加えた後に、水素雰囲気下で2時間撹拌した。反応液をセライト濾過した後に、ろ液の溶媒を減圧留去することで、化合物Q15(6.0g)を取得した。
1H-NMR (400 MHz, CDCl3): δ4.13 (brs, 2H), 3.36 (brs, 1H), 2.93 (brs, 5H), 2.65 (brs, 2H), 1.84-1.81 (m, 1H), 1.60-1.54 (m, 2H), 1.45 (s, 9H), 1.27-1.16 (m, 2H). b) Preparation of tert-butyl 4- (2-amino-1-hydroxyethyl) piperidine-1-carboxylate (Compound Q15) To a methanol solution (65 mL) of Compound Q14 (6.8 g) at room temperature with 10% palladium After adding carbon (0.68 g), the mixture was stirred for 2 hours under a hydrogen atmosphere. The reaction solution was filtered through Celite, and then the solvent of the filtrate was distilled off under reduced pressure to obtain Compound Q15 (6.0 g).
1 H-NMR (400 MHz, CDCl3): δ4.13 (brs, 2H), 3.36 (brs, 1H), 2.93 (brs, 5H), 2.65 (brs, 2H), 1.84-1.81 (m, 1H), 1.60-1.54 (m, 2H), 1.45 (s, 9H), 1.27-1.16 (m, 2H).
c)tert-ブチル 4-{2-[(4-フルオロベンゾイル)アミノ]-1-ヒドロキシエチル}ピペリジン-1-カルボキシレート(化合物Q16)の製造
化合物Q15(6.0g)のDMF溶液(50mL)に、室温にて4-フルオロ安息香酸(2.5g)、EDCI(4.4g)、HOBt(3.1g)及びトリエチルアミン(3.5g)を加えた後に、室温にて18時間撹拌した。反応液に酢酸エチル(300mL)を加えた後に、1mol/Lの塩酸水溶液(200mL)、水(200mL)及び、1mol/Lの炭酸水素ナトリウム水溶液(200mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(石油エーテル/酢酸エチル = 2/1)で精製することにより、化合物Q16(3.8g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.81-7.78 (m, 2H), 7.13-7.09 (m, 2H), 6.67-6.66 (m, 1H), 4.14-4.09 (m, 1H), 3.73-3.68 (m, 1H), 3.57 (brs, 1H), 3.41-3.35 (m, 1H), 2.93 (brs, 2H), 2.67 (brs, 2H), 1.68-1.65 (m, 1H), 1.56-1.24 (m, 13H). c) Preparation of tert-butyl 4- {2-[(4-fluorobenzoyl) amino] -1-hydroxyethyl} piperidine-1-carboxylate (Compound Q16) Compound D15 (6.0 g) in DMF (50 mL) 4-fluorobenzoic acid (2.5 g), EDCI (4.4 g), HOBt (3.1 g) and triethylamine (3.5 g) were added thereto at room temperature, followed by stirring at room temperature for 18 hours. Ethyl acetate (300 mL) was added to the reaction solution, and then washed with a 1 mol / L hydrochloric acid aqueous solution (200 mL), water (200 mL), and a 1 mol / L sodium hydrogen carbonate aqueous solution (200 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (petroleum ether / ethyl acetate = 2/1) gave Compound Q16 (3.8 g).
1 H-NMR (400 MHz, CDCl3): δ7.81-7.78 (m, 2H), 7.13-7.09 (m, 2H), 6.67-6.66 (m, 1H), 4.14-4.09 (m, 1H), 3.73 -3.68 (m, 1H), 3.57 (brs, 1H), 3.41-3.35 (m, 1H), 2.93 (brs, 2H), 2.67 (brs, 2H), 1.68-1.65 (m, 1H), 1.56-1.24 (m, 13H).
化合物Q15(6.0g)のDMF溶液(50mL)に、室温にて4-フルオロ安息香酸(2.5g)、EDCI(4.4g)、HOBt(3.1g)及びトリエチルアミン(3.5g)を加えた後に、室温にて18時間撹拌した。反応液に酢酸エチル(300mL)を加えた後に、1mol/Lの塩酸水溶液(200mL)、水(200mL)及び、1mol/Lの炭酸水素ナトリウム水溶液(200mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(石油エーテル/酢酸エチル = 2/1)で精製することにより、化合物Q16(3.8g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.81-7.78 (m, 2H), 7.13-7.09 (m, 2H), 6.67-6.66 (m, 1H), 4.14-4.09 (m, 1H), 3.73-3.68 (m, 1H), 3.57 (brs, 1H), 3.41-3.35 (m, 1H), 2.93 (brs, 2H), 2.67 (brs, 2H), 1.68-1.65 (m, 1H), 1.56-1.24 (m, 13H). c) Preparation of tert-butyl 4- {2-[(4-fluorobenzoyl) amino] -1-hydroxyethyl} piperidine-1-carboxylate (Compound Q16) Compound D15 (6.0 g) in DMF (50 mL) 4-fluorobenzoic acid (2.5 g), EDCI (4.4 g), HOBt (3.1 g) and triethylamine (3.5 g) were added thereto at room temperature, followed by stirring at room temperature for 18 hours. Ethyl acetate (300 mL) was added to the reaction solution, and then washed with a 1 mol / L hydrochloric acid aqueous solution (200 mL), water (200 mL), and a 1 mol / L sodium hydrogen carbonate aqueous solution (200 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (petroleum ether / ethyl acetate = 2/1) gave Compound Q16 (3.8 g).
1 H-NMR (400 MHz, CDCl3): δ7.81-7.78 (m, 2H), 7.13-7.09 (m, 2H), 6.67-6.66 (m, 1H), 4.14-4.09 (m, 1H), 3.73 -3.68 (m, 1H), 3.57 (brs, 1H), 3.41-3.35 (m, 1H), 2.93 (brs, 2H), 2.67 (brs, 2H), 1.68-1.65 (m, 1H), 1.56-1.24 (m, 13H).
d)tert-ブチル 4-[N-(4-フルオロベンゾイル)グリシル]ピペリジン-1-カルボキシレート(化合物Q17)の製造
化合物Q16(3.3g)の塩化メチレン溶液(40mL)に、氷冷下にてデスマーチン試薬(7.6g)を加えた後に、室温まで昇温して4時間撹拌した。反応液に塩化メチレン(60mL)を加えて抽出した後に、飽和炭酸水素ナトリウム水溶液(80mL)及び水(100mL)で洗浄をおこなった。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(石油エーテル/酢酸エチル = 2/1)で精製することにより、化合物Q17(2.1g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.85-7.81 (m, 2H), 7.14-7.10 (m, 2H), 6.91 (brs, 1H), 4.40-4.39 (m, 2H), 4.14-4.09 (m, 2H), 2.84-2.78 (m, 2H), 2.62-2.61 (m, 1H), 1.89-1.86 (m, 2H), 1.65-1.60 (m, 2H), 1.46 (s, 9H). d) Preparation of tert-butyl 4- [N- (4-fluorobenzoyl) glycyl] piperidine-1-carboxylate (Compound Q17) To a solution of Compound Q16 (3.3 g) in methylene chloride (40 mL) under ice-cooling After adding Dess-Martin reagent (7.6 g), the mixture was warmed to room temperature and stirred for 4 hours. The reaction solution was extracted with methylene chloride (60 mL), and then washed with a saturated aqueous sodium hydrogen carbonate solution (80 mL) and water (100 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (petroleum ether / ethyl acetate = 2/1) gave Compound Q17 (2.1 g).
1 H-NMR (400 MHz, CDCl3): δ7.85-7.81 (m, 2H), 7.14-7.10 (m, 2H), 6.91 (brs, 1H), 4.40-4.39 (m, 2H), 4.14-4.09 (m, 2H), 2.84-2.78 (m, 2H), 2.62-2.61 (m, 1H), 1.89-1.86 (m, 2H), 1.65-1.60 (m, 2H), 1.46 (s, 9H).
化合物Q16(3.3g)の塩化メチレン溶液(40mL)に、氷冷下にてデスマーチン試薬(7.6g)を加えた後に、室温まで昇温して4時間撹拌した。反応液に塩化メチレン(60mL)を加えて抽出した後に、飽和炭酸水素ナトリウム水溶液(80mL)及び水(100mL)で洗浄をおこなった。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(石油エーテル/酢酸エチル = 2/1)で精製することにより、化合物Q17(2.1g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.85-7.81 (m, 2H), 7.14-7.10 (m, 2H), 6.91 (brs, 1H), 4.40-4.39 (m, 2H), 4.14-4.09 (m, 2H), 2.84-2.78 (m, 2H), 2.62-2.61 (m, 1H), 1.89-1.86 (m, 2H), 1.65-1.60 (m, 2H), 1.46 (s, 9H). d) Preparation of tert-butyl 4- [N- (4-fluorobenzoyl) glycyl] piperidine-1-carboxylate (Compound Q17) To a solution of Compound Q16 (3.3 g) in methylene chloride (40 mL) under ice-cooling After adding Dess-Martin reagent (7.6 g), the mixture was warmed to room temperature and stirred for 4 hours. The reaction solution was extracted with methylene chloride (60 mL), and then washed with a saturated aqueous sodium hydrogen carbonate solution (80 mL) and water (100 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (petroleum ether / ethyl acetate = 2/1) gave Compound Q17 (2.1 g).
1 H-NMR (400 MHz, CDCl3): δ7.85-7.81 (m, 2H), 7.14-7.10 (m, 2H), 6.91 (brs, 1H), 4.40-4.39 (m, 2H), 4.14-4.09 (m, 2H), 2.84-2.78 (m, 2H), 2.62-2.61 (m, 1H), 1.89-1.86 (m, 2H), 1.65-1.60 (m, 2H), 1.46 (s, 9H).
e)tert-ブチル 4-[2-(4-フルオロフェニル)-1,3-オキサゾール-5-イル]ピペリジン-1-カルボキシレート(化合物Q18)の製造
化合物Q17(1.2g)のテトラヒドロフラン溶液(13mL)に、室温にてバージェス試薬(1.6g)を加えた後に、70℃まで昇温して12時間撹拌した。反応液を室温まで冷却した後に、酢酸エチル(50mL)を加えた。有機層を飽和炭酸水素ナトリウム水溶液(30mL)及び水(40mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(石油エーテル/酢酸エチル = 2/1)で精製することにより、化合物Q18(0.9g)を取得した。
1H-NMR (400 MHz, CDCl3): δ8.00-7.97 (m, 2H), 7.16-7.11 (m, 2H), 6.83 (s, 1H), 4.17 (brs, 2H), 2.93-2.88 (m, 3H), 2.05-2.01 (m, 2H), 1.68-1.64 (m, 2H), 1.48 (s, 9H). e) Preparation of tert-butyl 4- [2- (4-fluorophenyl) -1,3-oxazol-5-yl] piperidine-1-carboxylate (Compound Q18) Compound Q17 (1.2 g) in tetrahydrofuran solution ( 13 mL) was added Burgess reagent (1.6 g) at room temperature, and then the mixture was heated to 70 ° C. and stirred for 12 hours. After cooling the reaction solution to room temperature, ethyl acetate (50 mL) was added. The organic layer was washed with saturated aqueous sodium hydrogen carbonate solution (30 mL) and water (40 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (petroleum ether / ethyl acetate = 2/1) gave Compound Q18 (0.9 g).
1 H-NMR (400 MHz, CDCl3): δ8.00-7.97 (m, 2H), 7.16-7.11 (m, 2H), 6.83 (s, 1H), 4.17 (brs, 2H), 2.93-2.88 (m , 3H), 2.05-2.01 (m, 2H), 1.68-1.64 (m, 2H), 1.48 (s, 9H).
化合物Q17(1.2g)のテトラヒドロフラン溶液(13mL)に、室温にてバージェス試薬(1.6g)を加えた後に、70℃まで昇温して12時間撹拌した。反応液を室温まで冷却した後に、酢酸エチル(50mL)を加えた。有機層を飽和炭酸水素ナトリウム水溶液(30mL)及び水(40mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(石油エーテル/酢酸エチル = 2/1)で精製することにより、化合物Q18(0.9g)を取得した。
1H-NMR (400 MHz, CDCl3): δ8.00-7.97 (m, 2H), 7.16-7.11 (m, 2H), 6.83 (s, 1H), 4.17 (brs, 2H), 2.93-2.88 (m, 3H), 2.05-2.01 (m, 2H), 1.68-1.64 (m, 2H), 1.48 (s, 9H). e) Preparation of tert-butyl 4- [2- (4-fluorophenyl) -1,3-oxazol-5-yl] piperidine-1-carboxylate (Compound Q18) Compound Q17 (1.2 g) in tetrahydrofuran solution ( 13 mL) was added Burgess reagent (1.6 g) at room temperature, and then the mixture was heated to 70 ° C. and stirred for 12 hours. After cooling the reaction solution to room temperature, ethyl acetate (50 mL) was added. The organic layer was washed with saturated aqueous sodium hydrogen carbonate solution (30 mL) and water (40 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (petroleum ether / ethyl acetate = 2/1) gave Compound Q18 (0.9 g).
1 H-NMR (400 MHz, CDCl3): δ8.00-7.97 (m, 2H), 7.16-7.11 (m, 2H), 6.83 (s, 1H), 4.17 (brs, 2H), 2.93-2.88 (m , 3H), 2.05-2.01 (m, 2H), 1.68-1.64 (m, 2H), 1.48 (s, 9H).
f)4-[2-(4-フルオロフェニル)-1,3-オキサゾール-5-イル]ピペリジン・塩酸塩(参考例5)の製造
化合物Q18(500mg)を用いて、参考例1の工程e)に準ずる方法により参考例5の化合物(580mg)を取得した。 f) Preparation of 4- [2- (4-fluorophenyl) -1,3-oxazol-5-yl] piperidine hydrochloride (Reference Example 5) Step e of Reference Example 1 using Compound Q18 (500 mg) ) To obtain the compound of Reference Example 5 (580 mg).
化合物Q18(500mg)を用いて、参考例1の工程e)に準ずる方法により参考例5の化合物(580mg)を取得した。 f) Preparation of 4- [2- (4-fluorophenyl) -1,3-oxazol-5-yl] piperidine hydrochloride (Reference Example 5) Step e of Reference Example 1 using Compound Q18 (500 mg) ) To obtain the compound of Reference Example 5 (580 mg).
参考例6
4-[5-(4-フルオロフェニル)-1,3-オキサゾール-2-イル]ピペリジン・塩酸塩

a)tert-ブチル 4-{[2-(4-フルオロフェニル)-2-オキソエチル]カルバモイル}ピペリジン-1-カルボキシレート(化合物Q19)の製造
1-(tert-ブトキシカルボニル)ピペリジン-4-カルボン酸(8.0g)及び2-アミノ-1-(4-フルオロフェニル)エタノン(7.0g)のピリジン溶液(160mL)に、10℃にてEDCI(13.3g)を加えた後に、10℃にて3時間撹拌した。反応液に水(1.5L)を加えて5分間撹拌した後に、析出した固体をろ取した。得られた固体を減圧乾燥することで、化合物Q19(11g)得た。
1H-NMR (400 MHz, CDCl3): δ8.03-7.99 (m, 2H), 7.20-7.16 (m, 2H), 6.60 (brs, 1H), 4.73-4.72 (m, 2H), 4.16 (brs, 2H), 2.80-2.75 (m, 2H), 2.40-2.36 (m, 1H), 1.88-1.85 (m, 2H), 1.72-1.66 (m, 2H), 1.46 (s, 9H). Reference Example 6
4- [5- (4-Fluorophenyl) -1,3-oxazol-2-yl] piperidine hydrochloride
a) Preparation of tert-butyl 4-{[2- (4-fluorophenyl) -2-oxoethyl] carbamoyl} piperidine-1-carboxylate (Compound Q19) 1- (tert-Butoxycarbonyl) piperidine-4-carboxylic acid EDCI (13.3 g) was added at 10 ° C. to a pyridine solution (160 mL) of (8.0 g) and 2-amino-1- (4-fluorophenyl) ethanone (7.0 g). And stirred for 3 hours. Water (1.5 L) was added to the reaction solution and stirred for 5 minutes, and then the precipitated solid was collected by filtration. The obtained solid was dried under reduced pressure to obtain Compound Q19 (11 g).
1 H-NMR (400 MHz, CDCl3): δ8.03-7.99 (m, 2H), 7.20-7.16 (m, 2H), 6.60 (brs, 1H), 4.73-4.72 (m, 2H), 4.16 (brs) , 2H), 2.80-2.75 (m, 2H), 2.40-2.36 (m, 1H), 1.88-1.85 (m, 2H), 1.72-1.66 (m, 2H), 1.46 (s, 9H).
4-[5-(4-フルオロフェニル)-1,3-オキサゾール-2-イル]ピペリジン・塩酸塩
1-(tert-ブトキシカルボニル)ピペリジン-4-カルボン酸(8.0g)及び2-アミノ-1-(4-フルオロフェニル)エタノン(7.0g)のピリジン溶液(160mL)に、10℃にてEDCI(13.3g)を加えた後に、10℃にて3時間撹拌した。反応液に水(1.5L)を加えて5分間撹拌した後に、析出した固体をろ取した。得られた固体を減圧乾燥することで、化合物Q19(11g)得た。
1H-NMR (400 MHz, CDCl3): δ8.03-7.99 (m, 2H), 7.20-7.16 (m, 2H), 6.60 (brs, 1H), 4.73-4.72 (m, 2H), 4.16 (brs, 2H), 2.80-2.75 (m, 2H), 2.40-2.36 (m, 1H), 1.88-1.85 (m, 2H), 1.72-1.66 (m, 2H), 1.46 (s, 9H). Reference Example 6
4- [5- (4-Fluorophenyl) -1,3-oxazol-2-yl] piperidine hydrochloride
1 H-NMR (400 MHz, CDCl3): δ8.03-7.99 (m, 2H), 7.20-7.16 (m, 2H), 6.60 (brs, 1H), 4.73-4.72 (m, 2H), 4.16 (brs) , 2H), 2.80-2.75 (m, 2H), 2.40-2.36 (m, 1H), 1.88-1.85 (m, 2H), 1.72-1.66 (m, 2H), 1.46 (s, 9H).
b)tert-ブチル 4-[5-(4-フルオロフェニル)-1,3-オキサゾール-2-イル]ピペリジン-1-カルボキシレート(化合物Q20)の製造
化合物Q19(3.0g)のテトラヒドロフラン溶液(33mL)に、室温にてバージェス試薬(1.6g)を加えた後に、65℃まで昇温して14時間撹拌した。反応液を減圧留去した後に、酢酸エチル(80mL)を加え、飽和炭酸水素ナトリウム水溶液(40mL)及び、飽和食塩水(40mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(石油エーテル/酢酸エチル = 100/1から3/1)で精製することにより、化合物Q20(2.2g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.59-7.56 (m, 2H), 7.17 (s, 1H), 7.13-7.08 (m, 2H), 4.12 (brs, 2H), 3.04-2.95 (m, 3H), 2.10-2.06 (m, 2H), 1.85-1.81 (m, 2H), 1.47 (s, 9H). b) Preparation of tert-butyl 4- [5- (4-fluorophenyl) -1,3-oxazol-2-yl] piperidine-1-carboxylate (Compound Q20) Compound Q19 (3.0 g) in tetrahydrofuran solution ( (33 mL), Burgess reagent (1.6 g) was added at room temperature, and the mixture was heated to 65 ° C. and stirred for 14 hours. The reaction solution was evaporated under reduced pressure, ethyl acetate (80 mL) was added, and the mixture was washed with a saturated aqueous sodium hydrogen carbonate solution (40 mL) and saturated brine (40 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (petroleum ether / ethyl acetate = 100/1 to 3/1) gave Compound Q20 (2.2 g).
1 H-NMR (400 MHz, CDCl3): δ7.59-7.56 (m, 2H), 7.17 (s, 1H), 7.13-7.08 (m, 2H), 4.12 (brs, 2H), 3.04-2.95 (m , 3H), 2.10-2.06 (m, 2H), 1.85-1.81 (m, 2H), 1.47 (s, 9H).
化合物Q19(3.0g)のテトラヒドロフラン溶液(33mL)に、室温にてバージェス試薬(1.6g)を加えた後に、65℃まで昇温して14時間撹拌した。反応液を減圧留去した後に、酢酸エチル(80mL)を加え、飽和炭酸水素ナトリウム水溶液(40mL)及び、飽和食塩水(40mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(石油エーテル/酢酸エチル = 100/1から3/1)で精製することにより、化合物Q20(2.2g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.59-7.56 (m, 2H), 7.17 (s, 1H), 7.13-7.08 (m, 2H), 4.12 (brs, 2H), 3.04-2.95 (m, 3H), 2.10-2.06 (m, 2H), 1.85-1.81 (m, 2H), 1.47 (s, 9H). b) Preparation of tert-butyl 4- [5- (4-fluorophenyl) -1,3-oxazol-2-yl] piperidine-1-carboxylate (Compound Q20) Compound Q19 (3.0 g) in tetrahydrofuran solution ( (33 mL), Burgess reagent (1.6 g) was added at room temperature, and the mixture was heated to 65 ° C. and stirred for 14 hours. The reaction solution was evaporated under reduced pressure, ethyl acetate (80 mL) was added, and the mixture was washed with a saturated aqueous sodium hydrogen carbonate solution (40 mL) and saturated brine (40 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (petroleum ether / ethyl acetate = 100/1 to 3/1) gave Compound Q20 (2.2 g).
1 H-NMR (400 MHz, CDCl3): δ7.59-7.56 (m, 2H), 7.17 (s, 1H), 7.13-7.08 (m, 2H), 4.12 (brs, 2H), 3.04-2.95 (m , 3H), 2.10-2.06 (m, 2H), 1.85-1.81 (m, 2H), 1.47 (s, 9H).
c)4-[5-(4-フルオロフェニル)-1,3-オキサゾール-2-イル]ピペリジン・塩酸塩(参考例6)の製造
化合物Q20(800mg)を用いて、参考例1の工程e)に準ずる方法により参考例6の化合物(700mg)を取得した。 c) Preparation of 4- [5- (4-fluorophenyl) -1,3-oxazol-2-yl] piperidine hydrochloride (Reference Example 6) Step e of Reference Example 1 using Compound Q20 (800 mg) ) To obtain the compound of Reference Example 6 (700 mg).
化合物Q20(800mg)を用いて、参考例1の工程e)に準ずる方法により参考例6の化合物(700mg)を取得した。 c) Preparation of 4- [5- (4-fluorophenyl) -1,3-oxazol-2-yl] piperidine hydrochloride (Reference Example 6) Step e of Reference Example 1 using Compound Q20 (800 mg) ) To obtain the compound of Reference Example 6 (700 mg).
参考例7
4-[2-(4-フルオロフェニル)-1,3-チアゾール-5-イル]ピペリジン・塩酸塩

a) tert-ブチル 4-[2-(4-フルオロフェニル)-1,3-チアゾール-5-イル]ピペリジン-1-カルボキシレート(化合物Q21)
化合物Q17(1.3g)のテトラヒドロフラン溶液(50mL)に、室温にてローソン試薬(1.7g)を加えた後に、70℃まで昇温して12時間撹拌した。反応液を室温まで冷却した後に、酢酸エチル(200mL)を加えて抽出後、有機層を水(100mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(石油エーテル/酢酸エチル = 2/1)で精製することにより、化合物Q21(650mg)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.92-7.88 (m, 2H), 7.56 (s, 1H), 7.16-7.12 (m, 2H), 4.24 (brs, 2H), 3.08-3.02 (m, 1H), 2.91-2.85 (m, 2H), 2.06-2.02 (m, 2H), 1.73-1.63 (m, 2H), 1.50 (s, 9H). Reference Example 7
4- [2- (4-Fluorophenyl) -1,3-thiazol-5-yl] piperidine hydrochloride
a) tert-butyl 4- [2- (4-fluorophenyl) -1,3-thiazol-5-yl] piperidine-1-carboxylate (compound Q21)
To a tetrahydrofuran solution (50 mL) of compound Q17 (1.3 g), Lawesson's reagent (1.7 g) was added at room temperature, and then the mixture was heated to 70 ° C. and stirred for 12 hours. The reaction solution was cooled to room temperature, extracted with ethyl acetate (200 mL), and the organic layer was washed with water (100 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (petroleum ether / ethyl acetate = 2/1) gave Compound Q21 (650 mg).
1 H-NMR (400 MHz, CDCl3): δ7.92-7.88 (m, 2H), 7.56 (s, 1H), 7.16-7.12 (m, 2H), 4.24 (brs, 2H), 3.08-3.02 (m , 1H), 2.91-2.85 (m, 2H), 2.06-2.02 (m, 2H), 1.73-1.63 (m, 2H), 1.50 (s, 9H).
4-[2-(4-フルオロフェニル)-1,3-チアゾール-5-イル]ピペリジン・塩酸塩
化合物Q17(1.3g)のテトラヒドロフラン溶液(50mL)に、室温にてローソン試薬(1.7g)を加えた後に、70℃まで昇温して12時間撹拌した。反応液を室温まで冷却した後に、酢酸エチル(200mL)を加えて抽出後、有機層を水(100mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(石油エーテル/酢酸エチル = 2/1)で精製することにより、化合物Q21(650mg)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.92-7.88 (m, 2H), 7.56 (s, 1H), 7.16-7.12 (m, 2H), 4.24 (brs, 2H), 3.08-3.02 (m, 1H), 2.91-2.85 (m, 2H), 2.06-2.02 (m, 2H), 1.73-1.63 (m, 2H), 1.50 (s, 9H). Reference Example 7
4- [2- (4-Fluorophenyl) -1,3-thiazol-5-yl] piperidine hydrochloride
To a tetrahydrofuran solution (50 mL) of compound Q17 (1.3 g), Lawesson's reagent (1.7 g) was added at room temperature, and then the mixture was heated to 70 ° C. and stirred for 12 hours. The reaction solution was cooled to room temperature, extracted with ethyl acetate (200 mL), and the organic layer was washed with water (100 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (petroleum ether / ethyl acetate = 2/1) gave Compound Q21 (650 mg).
1 H-NMR (400 MHz, CDCl3): δ7.92-7.88 (m, 2H), 7.56 (s, 1H), 7.16-7.12 (m, 2H), 4.24 (brs, 2H), 3.08-3.02 (m , 1H), 2.91-2.85 (m, 2H), 2.06-2.02 (m, 2H), 1.73-1.63 (m, 2H), 1.50 (s, 9H).
b) 4-[2-(4-フルオロフェニル)-1,3-チアゾール-5-イル]ピペリジン・塩酸塩(参考例7)
化合物Q21(600mg)を用いて、参考例1の工程e)に準ずる方法により参考例7の化合物(600mg)を取得した。 b) 4- [2- (4-Fluorophenyl) -1,3-thiazol-5-yl] piperidine hydrochloride (Reference Example 7)
The compound (600 mg) of Reference Example 7 was obtained by the method according to Step e) of Reference Example 1 using Compound Q21 (600 mg).
化合物Q21(600mg)を用いて、参考例1の工程e)に準ずる方法により参考例7の化合物(600mg)を取得した。 b) 4- [2- (4-Fluorophenyl) -1,3-thiazol-5-yl] piperidine hydrochloride (Reference Example 7)
The compound (600 mg) of Reference Example 7 was obtained by the method according to Step e) of Reference Example 1 using Compound Q21 (600 mg).
参考例8
4-[5-(4-フルオロフェニル)-1,3-チアゾール-2-イル]ピペリジン・塩酸塩(参考例8)

a)tert-ブチル 4-[5-(4-フルオロフェニル)-1,3-チアゾール-2-イル]ピペリジン-1-カルボキシレート(化合物Q22)の製造
化合物Q19(2g)のテトラヒドロフラン溶液(50mL)に、室温にてローソン試薬(1.7g)を加えた後に、70℃まで昇温して4時間撹拌した。反応液を減圧留去した後に、塩化メチレン(100mL)を加え、飽和炭酸水素ナトリウム水溶液(30mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(石油エーテル/酢酸エチル = 90/10から50/50)で精製することにより、化合物Q22(1.3g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.76 (s, 1H), 7.51-7.48 (m, 2H), 7.11-7.09 (m, 2H), 4.21 (brs, 2H), 3.15-3.10 (m, 1H), 2.93-2.90 (m, 2H), 2.14-2.05 (m, 2H), 1.78-1.74 (m, 2H), 1.48 (s, 9H). Reference Example 8
4- [5- (4-Fluorophenyl) -1,3-thiazol-2-yl] piperidine hydrochloride (Reference Example 8)
a) Preparation of tert-butyl 4- [5- (4-fluorophenyl) -1,3-thiazol-2-yl] piperidine-1-carboxylate (Compound Q22) Compound Q19 (2 g) in tetrahydrofuran (50 mL) After adding Lawson's reagent (1.7 g) at room temperature, the mixture was heated to 70 ° C. and stirred for 4 hours. The reaction solution was evaporated under reduced pressure, methylene chloride (100 mL) was added, and the mixture was washed with a saturated aqueous sodium hydrogen carbonate solution (30 mL). The obtained organic layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (petroleum ether / ethyl acetate = 90/10 to 50/50) gave Compound Q22 (1.3 g).
1 H-NMR (400 MHz, CDCl3): δ7.76 (s, 1H), 7.51-7.48 (m, 2H), 7.11-7.09 (m, 2H), 4.21 (brs, 2H), 3.15-3.10 (m , 1H), 2.93-2.90 (m, 2H), 2.14-2.05 (m, 2H), 1.78-1.74 (m, 2H), 1.48 (s, 9H).
4-[5-(4-フルオロフェニル)-1,3-チアゾール-2-イル]ピペリジン・塩酸塩(参考例8)
化合物Q19(2g)のテトラヒドロフラン溶液(50mL)に、室温にてローソン試薬(1.7g)を加えた後に、70℃まで昇温して4時間撹拌した。反応液を減圧留去した後に、塩化メチレン(100mL)を加え、飽和炭酸水素ナトリウム水溶液(30mL)で洗浄した。得られた有機層を硫酸ナトリウムで乾燥後、溶媒を減圧留去した。シリカゲルカラムクロマトグラフィー(石油エーテル/酢酸エチル = 90/10から50/50)で精製することにより、化合物Q22(1.3g)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.76 (s, 1H), 7.51-7.48 (m, 2H), 7.11-7.09 (m, 2H), 4.21 (brs, 2H), 3.15-3.10 (m, 1H), 2.93-2.90 (m, 2H), 2.14-2.05 (m, 2H), 1.78-1.74 (m, 2H), 1.48 (s, 9H). Reference Example 8
4- [5- (4-Fluorophenyl) -1,3-thiazol-2-yl] piperidine hydrochloride (Reference Example 8)
1 H-NMR (400 MHz, CDCl3): δ7.76 (s, 1H), 7.51-7.48 (m, 2H), 7.11-7.09 (m, 2H), 4.21 (brs, 2H), 3.15-3.10 (m , 1H), 2.93-2.90 (m, 2H), 2.14-2.05 (m, 2H), 1.78-1.74 (m, 2H), 1.48 (s, 9H).
b)4-[5-(4-フルオロフェニル)-1,3-チアゾール-2-イル]ピペリジン・塩酸塩(参考例8)の製造
化合物Q22(840mg)を用いて、参考例1の工程e)に準ずる方法により参考例8の化合物(680mg)を取得した。 b) Preparation of 4- [5- (4-fluorophenyl) -1,3-thiazol-2-yl] piperidine hydrochloride (Reference Example 8) Step e of Reference Example 1 using Compound Q22 (840 mg) ) To obtain the compound of Reference Example 8 (680 mg).
化合物Q22(840mg)を用いて、参考例1の工程e)に準ずる方法により参考例8の化合物(680mg)を取得した。 b) Preparation of 4- [5- (4-fluorophenyl) -1,3-thiazol-2-yl] piperidine hydrochloride (Reference Example 8) Step e of Reference Example 1 using Compound Q22 (840 mg) ) To obtain the compound of Reference Example 8 (680 mg).
実施例1
4-[5-クロロ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]-N-(4,4-ジフルオロシクロヘキシル)ピペリジン-1-カルボキサミド

参考例1の化合物(198mg)の塩化メチレン溶液(8mL)に、室温にてフェニル 4,4-ジフルオロシクロヘキシルカルバメート(272mg)及びトリエチルアミン(0.50mL)を加えた後に、38℃まで昇温して48時間撹拌した。反応溶媒を減圧留去した後に、分取HPLCで精製することにより、実施例1の化合物(200mg)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.60 (s, 1H), 7.52-7.45 (m, 1H), 7.21-7.13 (m, 3H), 4.33-4.31 (m, 1H), 4.08-4.04 (m, 2H), 3.84-3.83 (m, 1H), 2.98-2.86 (m, 3H), 2.09-1.88 (m, 10H), 1.55-1.45 (m, 2H).
LCMS ; [M+H]+ / Rt (min) : 測定条件 (441 / 1.94 : F) Example 1
4- [5-Chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] -N- (4,4-difluorocyclohexyl) piperidine-1-carboxamide
Phenyl 4,4-difluorocyclohexylcarbamate (272 mg) and triethylamine (0.50 mL) were added to a methylene chloride solution (8 mL) of the compound of Reference Example 1 (198 mg) at room temperature, and the temperature was raised to 38 ° C. Stir for 48 hours. After distilling off the reaction solvent under reduced pressure, the compound of Example 1 (200 mg) was obtained by purification by preparative HPLC.
1 H-NMR (400 MHz, CDCl3): δ7.60 (s, 1H), 7.52-7.45 (m, 1H), 7.21-7.13 (m, 3H), 4.33-4.31 (m, 1H), 4.08-4.04 (m, 2H), 3.84-3.83 (m, 1H), 2.98-2.86 (m, 3H), 2.09-1.88 (m, 10H), 1.55-1.45 (m, 2H).
LCMS; [M + H] + / Rt (min): Measurement conditions (441 / 1.94: F)
4-[5-クロロ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]-N-(4,4-ジフルオロシクロヘキシル)ピペリジン-1-カルボキサミド
1H-NMR (400 MHz, CDCl3): δ7.60 (s, 1H), 7.52-7.45 (m, 1H), 7.21-7.13 (m, 3H), 4.33-4.31 (m, 1H), 4.08-4.04 (m, 2H), 3.84-3.83 (m, 1H), 2.98-2.86 (m, 3H), 2.09-1.88 (m, 10H), 1.55-1.45 (m, 2H).
LCMS ; [M+H]+ / Rt (min) : 測定条件 (441 / 1.94 : F) Example 1
4- [5-Chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] -N- (4,4-difluorocyclohexyl) piperidine-1-carboxamide
1 H-NMR (400 MHz, CDCl3): δ7.60 (s, 1H), 7.52-7.45 (m, 1H), 7.21-7.13 (m, 3H), 4.33-4.31 (m, 1H), 4.08-4.04 (m, 2H), 3.84-3.83 (m, 1H), 2.98-2.86 (m, 3H), 2.09-1.88 (m, 10H), 1.55-1.45 (m, 2H).
LCMS; [M + H] + / Rt (min): Measurement conditions (441 / 1.94: F)
実施例2
{4-[5-クロロ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]ピペリジン-1-イル}(4,4-ジフルオロシクロヘキシル)メタノン
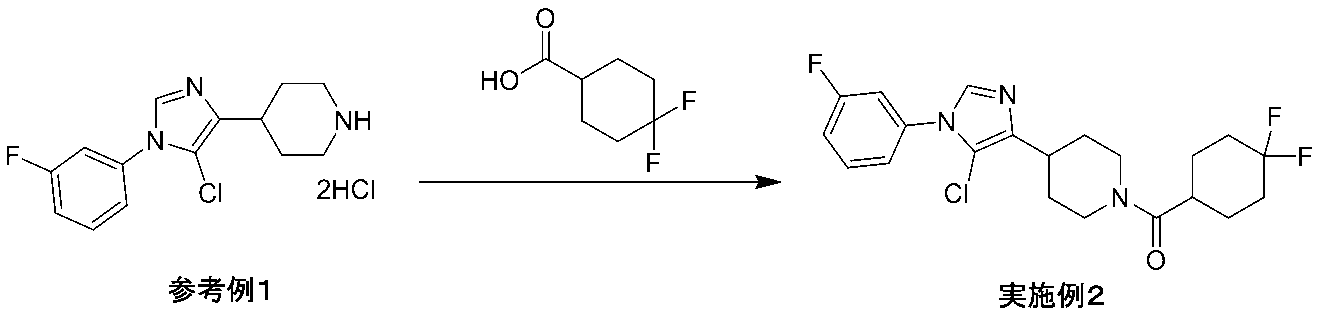
参考例1の化合物(198mg)の塩化メチレン溶液(4mL)に、室温にて4,4-ジフルオロシクロヘキサンカルボン酸(175mg)、HBTU(540mg)及びトリエチルアミン(0.50mL)を加えた後に、室温で24時間撹拌した。反応溶媒を減圧留去した後に、分取HPLCで精製することにより、実施例2の化合物(100mg)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.61 (s, 1H), 7.52-7.49 (m, 1H), 7.22-7.14 (m, 3H), 4.78-4.75 (m, 1H), 4.05-4.02 (m, 1H), 3.24-3.20 (m, 1H), 3.00-2.95 (m, 1H), 2.73-2.64 (m, 2H), 2.25-2.22 (m, 2H), 1.95-1.78 (m, 10H).
LCMS ; [M+H]+ / Rt (min) : 測定条件 (426 / 1.96 : F) Example 2
{4- [5-Chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidin-1-yl} (4,4-difluorocyclohexyl) methanone
To a methylene chloride solution (4 mL) of the compound of Reference Example 1 (198 mg), 4,4-difluorocyclohexanecarboxylic acid (175 mg), HBTU (540 mg) and triethylamine (0.50 mL) were added at room temperature. Stir for 24 hours. After distilling off the reaction solvent under reduced pressure, the compound of Example 2 (100 mg) was obtained by purification by preparative HPLC.
1 H-NMR (400 MHz, CDCl3): δ7.61 (s, 1H), 7.52-7.49 (m, 1H), 7.22-7.14 (m, 3H), 4.78-4.75 (m, 1H), 4.05-4.02 (m, 1H), 3.24-3.20 (m, 1H), 3.00-2.95 (m, 1H), 2.73-2.64 (m, 2H), 2.25-2.22 (m, 2H), 1.95-1.78 (m, 10H) .
LCMS; [M + H] + / Rt (min): Measurement conditions (426 / 1.96: F)
{4-[5-クロロ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]ピペリジン-1-イル}(4,4-ジフルオロシクロヘキシル)メタノン
1H-NMR (400 MHz, CDCl3): δ7.61 (s, 1H), 7.52-7.49 (m, 1H), 7.22-7.14 (m, 3H), 4.78-4.75 (m, 1H), 4.05-4.02 (m, 1H), 3.24-3.20 (m, 1H), 3.00-2.95 (m, 1H), 2.73-2.64 (m, 2H), 2.25-2.22 (m, 2H), 1.95-1.78 (m, 10H).
LCMS ; [M+H]+ / Rt (min) : 測定条件 (426 / 1.96 : F) Example 2
{4- [5-Chloro-1- (3-fluorophenyl) -1H-imidazol-4-yl] piperidin-1-yl} (4,4-difluorocyclohexyl) methanone
1 H-NMR (400 MHz, CDCl3): δ7.61 (s, 1H), 7.52-7.49 (m, 1H), 7.22-7.14 (m, 3H), 4.78-4.75 (m, 1H), 4.05-4.02 (m, 1H), 3.24-3.20 (m, 1H), 3.00-2.95 (m, 1H), 2.73-2.64 (m, 2H), 2.25-2.22 (m, 2H), 1.95-1.78 (m, 10H) .
LCMS; [M + H] + / Rt (min): Measurement conditions (426 / 1.96: F)
実施例3
4-[5-シアノ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]-N-(4,4-ジフルオロシクロヘキシル)ピペリジン-1-カルボキサミド
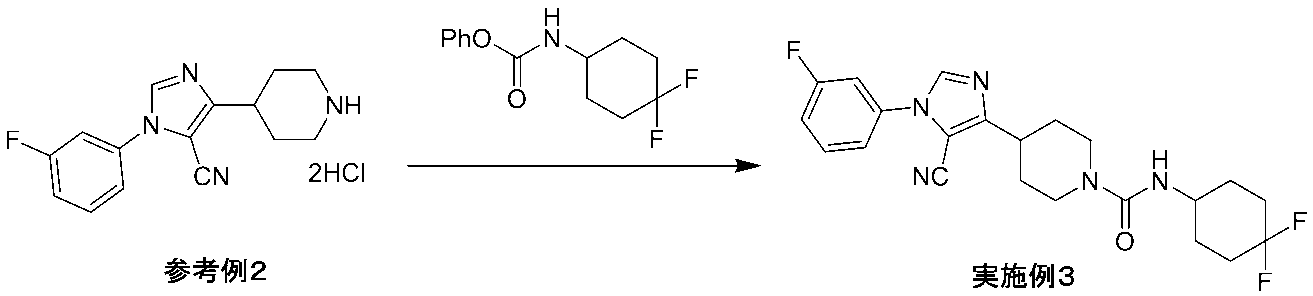
参考例2の化合物(259mg)を用いて、実施例1に準ずる方法により、実施例3の化合物(26mg)を取得した。
LCMS ; [M+H]+ / Rt (min) : 測定条件 (432 / 2.01 : C) Example 3
4- [5-Cyano-1- (3-fluorophenyl) -1H-imidazol-4-yl] -N- (4,4-difluorocyclohexyl) piperidine-1-carboxamide
The compound of Example 3 (26 mg) was obtained by the method according to Example 1 using the compound of Reference Example 2 (259 mg).
LCMS; [M + H] + / Rt (min): Measurement conditions (432 / 2.01: C)
4-[5-シアノ-1-(3-フルオロフェニル)-1H-イミダゾール-4-イル]-N-(4,4-ジフルオロシクロヘキシル)ピペリジン-1-カルボキサミド
LCMS ; [M+H]+ / Rt (min) : 測定条件 (432 / 2.01 : C) Example 3
4- [5-Cyano-1- (3-fluorophenyl) -1H-imidazol-4-yl] -N- (4,4-difluorocyclohexyl) piperidine-1-carboxamide
LCMS; [M + H] + / Rt (min): Measurement conditions (432 / 2.01: C)
実施例4
N-(テトラヒドロ-2H-ピラン-4-イル)-4-{3-[5-(トリフルオロメチル)ピリジン-2-イル]-1,2-オキサゾール-5-イル}ピペリジン-1-カルボキサミド

参考例3の化合物(327mg)を用いて、実施例1に準ずる方法により、実施例4の化合物(90mg)を取得した。
LCMS ; [M+H]+ / Rt (min) : 測定条件 (425 / 1.68 : F) Example 4
N- (Tetrahydro-2H-pyran-4-yl) -4- {3- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-5-yl} piperidine-1-carboxamide
The compound of Example 4 (90 mg) was obtained by the method according to Example 1 using the compound of Reference Example 3 (327 mg).
LCMS; [M + H] + / Rt (min): Measurement conditions (425 / 1.68: F)
N-(テトラヒドロ-2H-ピラン-4-イル)-4-{3-[5-(トリフルオロメチル)ピリジン-2-イル]-1,2-オキサゾール-5-イル}ピペリジン-1-カルボキサミド
LCMS ; [M+H]+ / Rt (min) : 測定条件 (425 / 1.68 : F) Example 4
N- (Tetrahydro-2H-pyran-4-yl) -4- {3- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-5-yl} piperidine-1-carboxamide
LCMS; [M + H] + / Rt (min): Measurement conditions (425 / 1.68: F)
実施例5
N-(テトラヒドロ-2H-ピラン-4-イル)-4-{5-[5-(トリフルオロメチル)ピリジン-2-イル]-1,2-オキサゾール-3-イル}ピペリジン-1-カルボキサミド
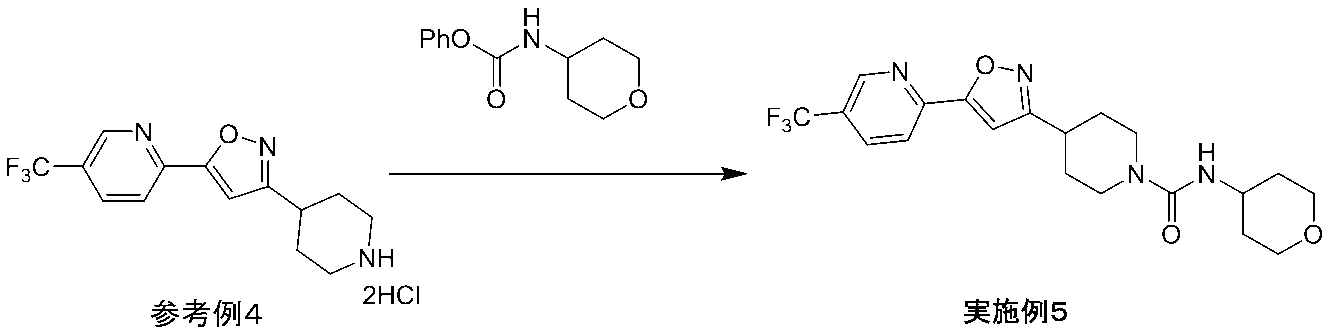
参考例4の化合物(247mg)を用いて、実施例1に準ずる方法により、実施例5の化合物(85mg)を取得した。
LCMS ; [M+H]+ / Rt (min) : 測定条件 (425 / 1.85 : C) Example 5
N- (Tetrahydro-2H-pyran-4-yl) -4- {5- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-3-yl} piperidine-1-carboxamide
The compound of Example 5 (85 mg) was obtained by the method according to Example 1 using the compound of Reference Example 4 (247 mg).
LCMS; [M + H] + / Rt (min): Measurement conditions (425 / 1.85: C)
N-(テトラヒドロ-2H-ピラン-4-イル)-4-{5-[5-(トリフルオロメチル)ピリジン-2-イル]-1,2-オキサゾール-3-イル}ピペリジン-1-カルボキサミド
LCMS ; [M+H]+ / Rt (min) : 測定条件 (425 / 1.85 : C) Example 5
N- (Tetrahydro-2H-pyran-4-yl) -4- {5- [5- (trifluoromethyl) pyridin-2-yl] -1,2-oxazol-3-yl} piperidine-1-carboxamide
LCMS; [M + H] + / Rt (min): Measurement conditions (425 / 1.85: C)
実施例6
N-(4,4-ジフルオロシクロヘキシル)-4-[2-(4-フルオロフェニル)-1,3-オキサゾール-5-イル]ピペリジン-1-カルボキサミド

参考例5の化合物(290mg)を用いて、実施例1に準ずる方法により、実施例6の化合物(38mg)を取得した。
LCMS ; [M+H]+ / Rt (min) : 測定条件 (408 / 2.53 : C) Example 6
N- (4,4-difluorocyclohexyl) -4- [2- (4-fluorophenyl) -1,3-oxazol-5-yl] piperidine-1-carboxamide
The compound of Example 6 (38 mg) was obtained by the method according to Example 1 using the compound of Reference Example 5 (290 mg).
LCMS; [M + H] + / Rt (min): Measurement conditions (408 / 2.53: C)
N-(4,4-ジフルオロシクロヘキシル)-4-[2-(4-フルオロフェニル)-1,3-オキサゾール-5-イル]ピペリジン-1-カルボキサミド
LCMS ; [M+H]+ / Rt (min) : 測定条件 (408 / 2.53 : C) Example 6
N- (4,4-difluorocyclohexyl) -4- [2- (4-fluorophenyl) -1,3-oxazol-5-yl] piperidine-1-carboxamide
LCMS; [M + H] + / Rt (min): Measurement conditions (408 / 2.53: C)
実施例7
N-(4,4-ジフルオロシクロヘキシル)-4-[5-(4-フルオロフェニル)-1,3-オキサゾール-2-イル]ピペリジン-1-カルボキサミド

参考例6の化合物(300mg)を用いて、実施例1に準ずる方法により、実施例7の化合物(200mg)を取得した。
LCMS ; [M+H]+ / Rt (min) : 測定条件 (408 / 2.04 : C) Example 7
N- (4,4-difluorocyclohexyl) -4- [5- (4-fluorophenyl) -1,3-oxazol-2-yl] piperidine-1-carboxamide
The compound of Example 7 (200 mg) was obtained by the method according to Example 1 using the compound of Reference Example 6 (300 mg).
LCMS; [M + H] + / Rt (min): Measurement conditions (408 / 2.04: C)
N-(4,4-ジフルオロシクロヘキシル)-4-[5-(4-フルオロフェニル)-1,3-オキサゾール-2-イル]ピペリジン-1-カルボキサミド
LCMS ; [M+H]+ / Rt (min) : 測定条件 (408 / 2.04 : C) Example 7
N- (4,4-difluorocyclohexyl) -4- [5- (4-fluorophenyl) -1,3-oxazol-2-yl] piperidine-1-carboxamide
LCMS; [M + H] + / Rt (min): Measurement conditions (408 / 2.04: C)
実施例8
N-(4,4-ジフルオロシクロヘキシル)-4-[2-(4-フルオロフェニル)-1,3-チアゾール-5-イル]ピペリジン-1-カルボキサミド

参考例7の化合物(120mg)を用いて、実施例1に準ずる方法により、実施例8の化合物(35mg)を取得した。
LCMS ; [M+H]+ / Rt (min) : 測定条件 (424 / 2.65 : C) Example 8
N- (4,4-difluorocyclohexyl) -4- [2- (4-fluorophenyl) -1,3-thiazol-5-yl] piperidine-1-carboxamide
The compound of Example 8 (35 mg) was obtained by the method according to Example 1 using the compound of Reference Example 7 (120 mg).
LCMS; [M + H] + / Rt (min): Measurement conditions (424 / 2.65: C)
N-(4,4-ジフルオロシクロヘキシル)-4-[2-(4-フルオロフェニル)-1,3-チアゾール-5-イル]ピペリジン-1-カルボキサミド
LCMS ; [M+H]+ / Rt (min) : 測定条件 (424 / 2.65 : C) Example 8
N- (4,4-difluorocyclohexyl) -4- [2- (4-fluorophenyl) -1,3-thiazol-5-yl] piperidine-1-carboxamide
LCMS; [M + H] + / Rt (min): Measurement conditions (424 / 2.65: C)
実施例9
N-(4,4-ジフルオロシクロヘキシル)-4-[5-(4-フルオロフェニル)-1,3-チアゾール-2-イル]ピペリジン-1-カルボキサミド

参考例8の化合物(340mg)及びフェニル 4,4-ジフェニルシクロヘキサンカルバメート(434mg)のアセトニトリル溶液(5mL)に、室温にてトリエチルアミン(0.46mL)を加えた後に、55℃まで昇温して4時間撹拌した。反応溶媒を減圧留去した後に、シリカゲルカラムクロマトグラフィー(酢酸エチル/メタノール = 100/0から92/8)で精製することにより、実施例9の化合物(57mg)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.76 (s, 1H), 7.50-7.47 (m, 2H), 7.11-7.07 (m, 2H), 4.37-4.30 (m, 1H), 4.05-3.98 (m, 2H), 3.80-3.73 (m, 1H), 3.18-3.10 (m, 1H), 3.00-2.93 (m, 2H), 2.20-2.05 (m, 6H), 1.93-1.75 (m, 4H), 1.53-1.40 (m, 2H).
LCMS ; [M+H]+ / Rt (min) : 測定条件 (424 / 1.10 : C) Example 9
N- (4,4-difluorocyclohexyl) -4- [5- (4-fluorophenyl) -1,3-thiazol-2-yl] piperidine-1-carboxamide
Triethylamine (0.46 mL) was added to a solution of the compound of Reference Example 8 (340 mg) and phenyl 4,4-diphenylcyclohexanecarbamate (434 mg) in acetonitrile (5 mL) at room temperature, and the temperature was raised to 55 ° C. Stir for hours. After distilling off the reaction solvent under reduced pressure, the compound of Example 9 (57 mg) was obtained by purification by silica gel column chromatography (ethyl acetate / methanol = 100/0 to 92/8).
1 H-NMR (400 MHz, CDCl3): δ7.76 (s, 1H), 7.50-7.47 (m, 2H), 7.11-7.07 (m, 2H), 4.37-4.30 (m, 1H), 4.05-3.98 (m, 2H), 3.80-3.73 (m, 1H), 3.18-3.10 (m, 1H), 3.00-2.93 (m, 2H), 2.20-2.05 (m, 6H), 1.93-1.75 (m, 4H) , 1.53-1.40 (m, 2H).
LCMS; [M + H] + / Rt (min): Measurement conditions (424 / 1.10: C)
N-(4,4-ジフルオロシクロヘキシル)-4-[5-(4-フルオロフェニル)-1,3-チアゾール-2-イル]ピペリジン-1-カルボキサミド
1H-NMR (400 MHz, CDCl3): δ7.76 (s, 1H), 7.50-7.47 (m, 2H), 7.11-7.07 (m, 2H), 4.37-4.30 (m, 1H), 4.05-3.98 (m, 2H), 3.80-3.73 (m, 1H), 3.18-3.10 (m, 1H), 3.00-2.93 (m, 2H), 2.20-2.05 (m, 6H), 1.93-1.75 (m, 4H), 1.53-1.40 (m, 2H).
LCMS ; [M+H]+ / Rt (min) : 測定条件 (424 / 1.10 : C) Example 9
N- (4,4-difluorocyclohexyl) -4- [5- (4-fluorophenyl) -1,3-thiazol-2-yl] piperidine-1-carboxamide
1 H-NMR (400 MHz, CDCl3): δ7.76 (s, 1H), 7.50-7.47 (m, 2H), 7.11-7.07 (m, 2H), 4.37-4.30 (m, 1H), 4.05-3.98 (m, 2H), 3.80-3.73 (m, 1H), 3.18-3.10 (m, 1H), 3.00-2.93 (m, 2H), 2.20-2.05 (m, 6H), 1.93-1.75 (m, 4H) , 1.53-1.40 (m, 2H).
LCMS; [M + H] + / Rt (min): Measurement conditions (424 / 1.10: C)
実施例10
4-[4-クロロ-5-(4-フルオロフェニル)-1,3-チアゾール-2-イル]-N-(4,4-ジフルオロシクロヘキシル)ピペリジン-1-カルボキサミド

実施例9の化合物(100mg)のアセトニトリル溶液(5mL)に、室温にてN-クロロスクシンイミド(38mg)及びトリフルオロ酢酸(0.05mL)を加えた後に、55℃まで昇温して4時間撹拌した。反応液に飽和炭酸水素ナトリウム溶液を加えた後に、酢酸エチルで抽出し、有機層を水及び飽和食塩水で洗浄した。得られた有機層を硫酸ナトリウムで乾燥、溶媒を減圧留去した後に、分取HPLCで精製することにより、実施例10の化合物(64mg)を取得した。
1H-NMR (400 MHz, CDCl3): δ7.61-7.57 (m, 2H), 7.16-7.11 (m, 2H), 4.30-4.28 (m, 1H), 4.05-4.01 (m, 2H), 3.78-3.73 (m, 1H), 3.16-3.08 (m, 1H), 2.99-2.92 (m, 2H), 2.17-2.02 (m, 6H), 1.83-1.75 (m, 4H), 1.51-1.42 (m, 2H).
LCMS ; [M+H]+ / Rt (min) : 測定条件 (458 / 1.45 : C) Example 10
4- [4-Chloro-5- (4-fluorophenyl) -1,3-thiazol-2-yl] -N- (4,4-difluorocyclohexyl) piperidine-1-carboxamide
N-chlorosuccinimide (38 mg) and trifluoroacetic acid (0.05 mL) were added to an acetonitrile solution (5 mL) of the compound of Example 9 (100 mg) at room temperature, and then the mixture was heated to 55 ° C. and stirred for 4 hours. did. A saturated sodium bicarbonate solution was added to the reaction solution, followed by extraction with ethyl acetate, and the organic layer was washed with water and saturated brine. The obtained organic layer was dried over sodium sulfate, the solvent was distilled off under reduced pressure, and the residue was purified by preparative HPLC to obtain the compound of Example 10 (64 mg).
1 H-NMR (400 MHz, CDCl3): δ7.61-7.57 (m, 2H), 7.16-7.11 (m, 2H), 4.30-4.28 (m, 1H), 4.05-4.01 (m, 2H), 3.78 -3.73 (m, 1H), 3.16-3.08 (m, 1H), 2.99-2.92 (m, 2H), 2.17-2.02 (m, 6H), 1.83-1.75 (m, 4H), 1.51-1.42 (m, 2H).
LCMS; [M + H] + / Rt (min): Measurement conditions (458 / 1.45: C)
4-[4-クロロ-5-(4-フルオロフェニル)-1,3-チアゾール-2-イル]-N-(4,4-ジフルオロシクロヘキシル)ピペリジン-1-カルボキサミド
1H-NMR (400 MHz, CDCl3): δ7.61-7.57 (m, 2H), 7.16-7.11 (m, 2H), 4.30-4.28 (m, 1H), 4.05-4.01 (m, 2H), 3.78-3.73 (m, 1H), 3.16-3.08 (m, 1H), 2.99-2.92 (m, 2H), 2.17-2.02 (m, 6H), 1.83-1.75 (m, 4H), 1.51-1.42 (m, 2H).
LCMS ; [M+H]+ / Rt (min) : 測定条件 (458 / 1.45 : C) Example 10
4- [4-Chloro-5- (4-fluorophenyl) -1,3-thiazol-2-yl] -N- (4,4-difluorocyclohexyl) piperidine-1-carboxamide
1 H-NMR (400 MHz, CDCl3): δ7.61-7.57 (m, 2H), 7.16-7.11 (m, 2H), 4.30-4.28 (m, 1H), 4.05-4.01 (m, 2H), 3.78 -3.73 (m, 1H), 3.16-3.08 (m, 1H), 2.99-2.92 (m, 2H), 2.17-2.02 (m, 6H), 1.83-1.75 (m, 4H), 1.51-1.42 (m, 2H).
LCMS; [M + H] + / Rt (min): Measurement conditions (458 / 1.45: C)
実施例11~39
対応する原料化合物を用いて、実施例1又は3に準ずる方法により、表1に示す化合物を得た。
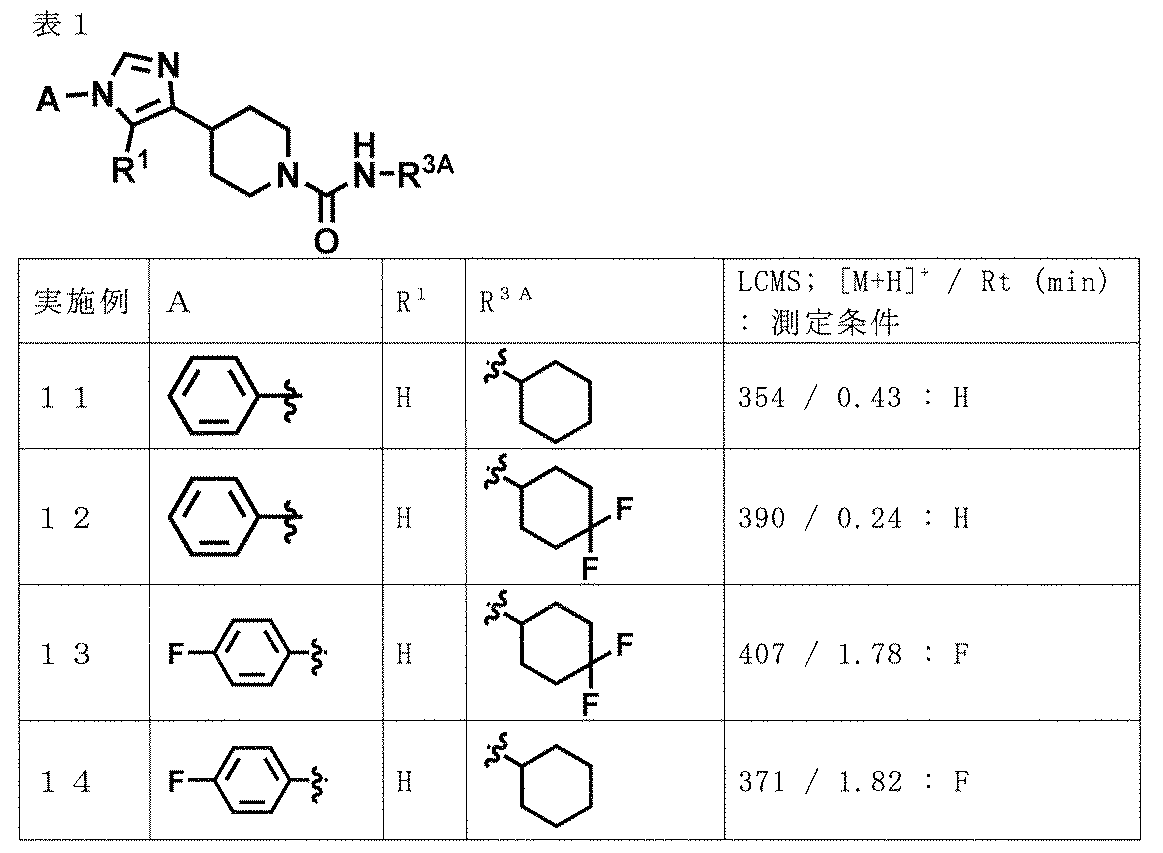


Examples 11 to 39
The compounds shown in Table 1 were obtained by the method according to Example 1 or 3 using the corresponding raw material compounds.
対応する原料化合物を用いて、実施例1又は3に準ずる方法により、表1に示す化合物を得た。
The compounds shown in Table 1 were obtained by the method according to Example 1 or 3 using the corresponding raw material compounds.
実施例40~50
対応する原料化合物を用いて、実施例2に準ずる方法により、表2に示す化合物を得た。

Examples 40-50
The compounds shown in Table 2 were obtained by the method according to Example 2 using the corresponding raw material compounds.
対応する原料化合物を用いて、実施例2に準ずる方法により、表2に示す化合物を得た。
The compounds shown in Table 2 were obtained by the method according to Example 2 using the corresponding raw material compounds.
実施例51~91
対応する原料化合物を用いて、実施例4に準ずる方法により、表3に示す化合物を得た。
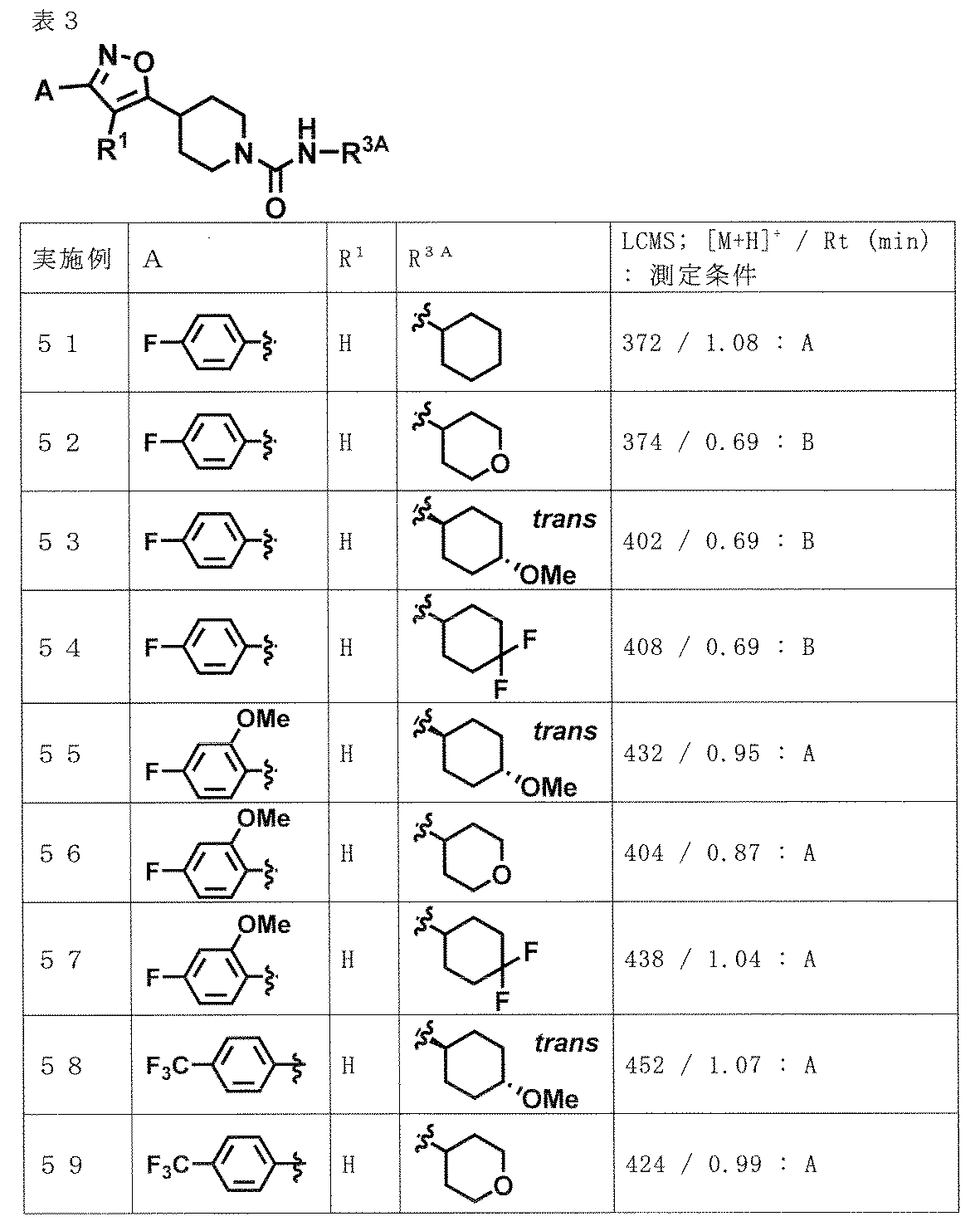

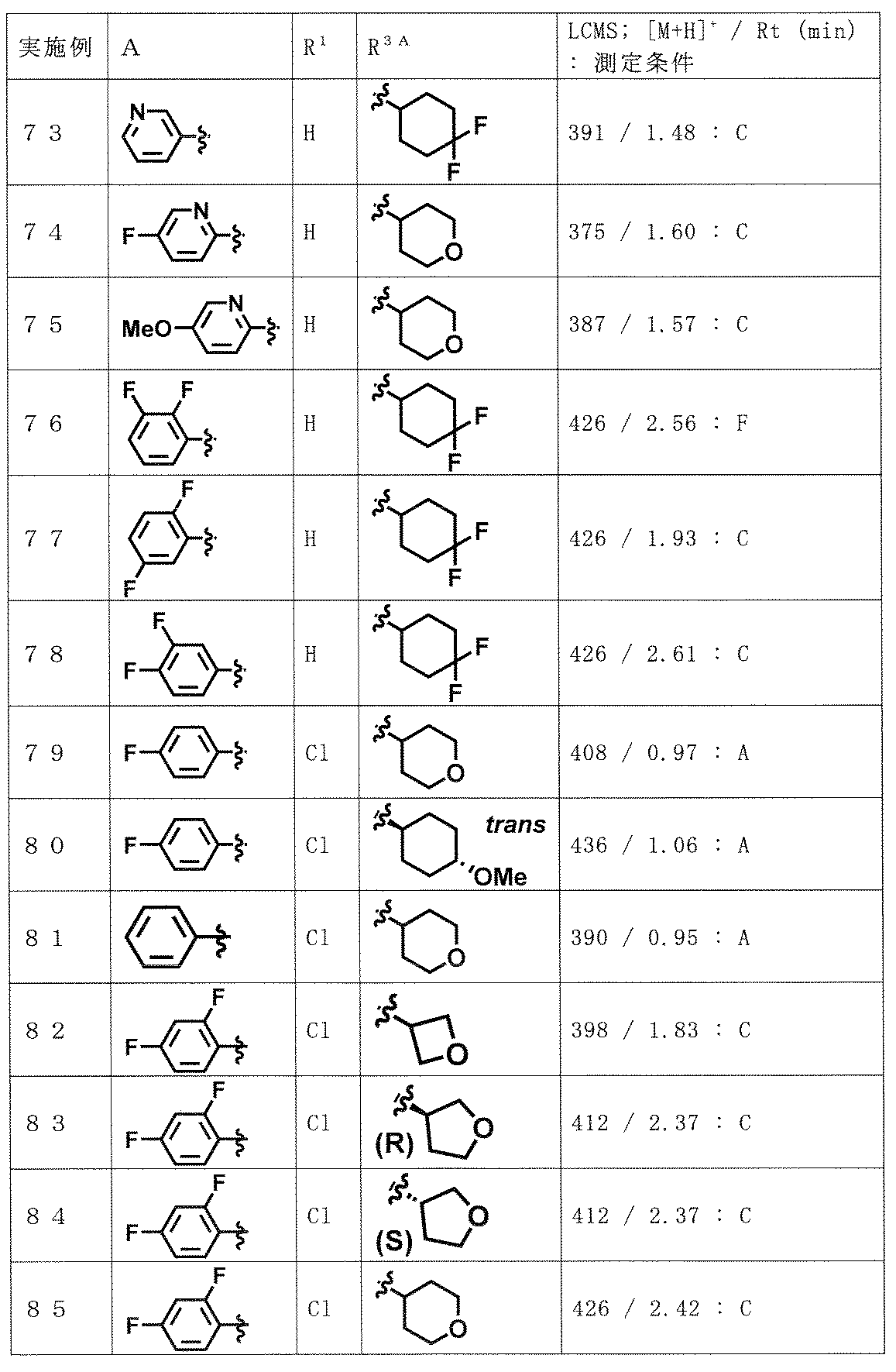
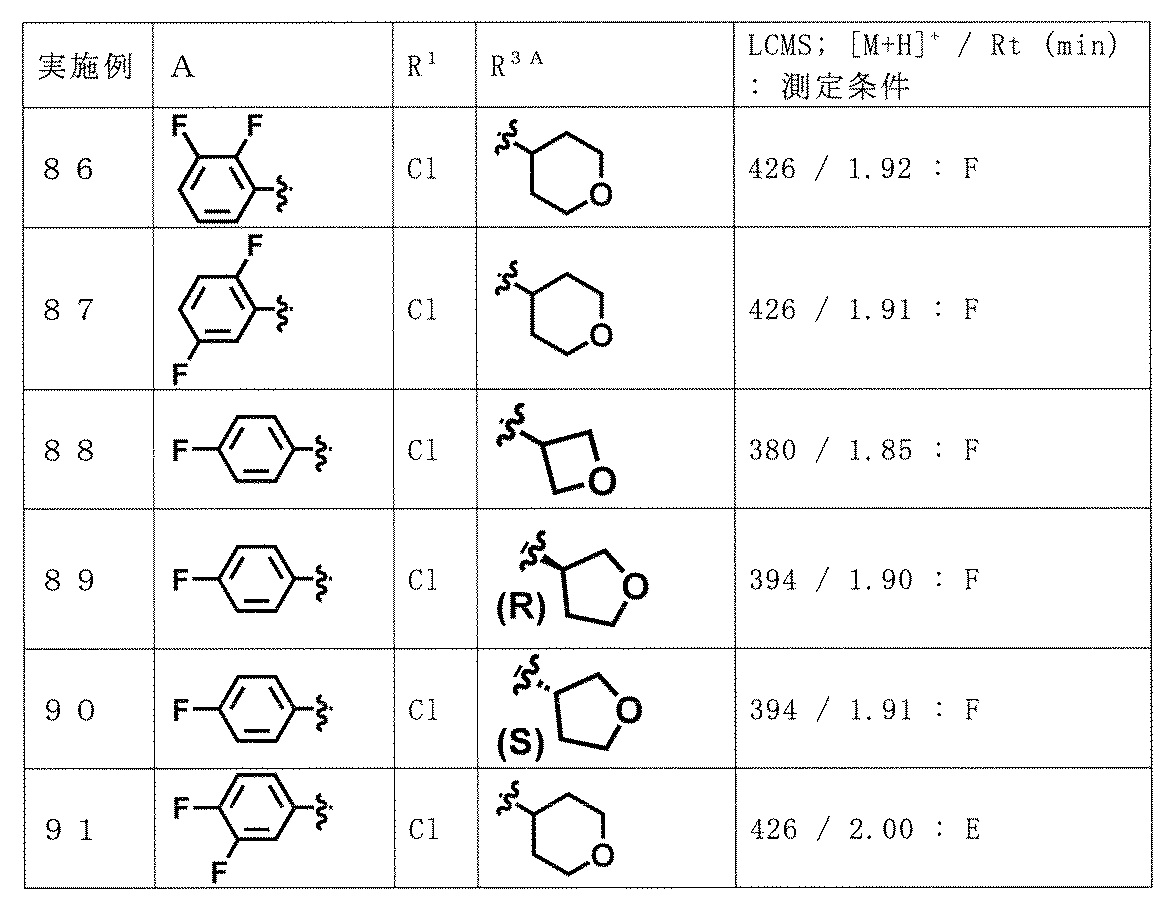
Examples 51-91
The compounds shown in Table 3 were obtained by the method according to Example 4 using the corresponding starting compounds.
対応する原料化合物を用いて、実施例4に準ずる方法により、表3に示す化合物を得た。
The compounds shown in Table 3 were obtained by the method according to Example 4 using the corresponding starting compounds.
実施例92~98
対応する原料化合物を用いて、参考例3及び実施例2に準ずる方法により、表4に示す化合物を得た。
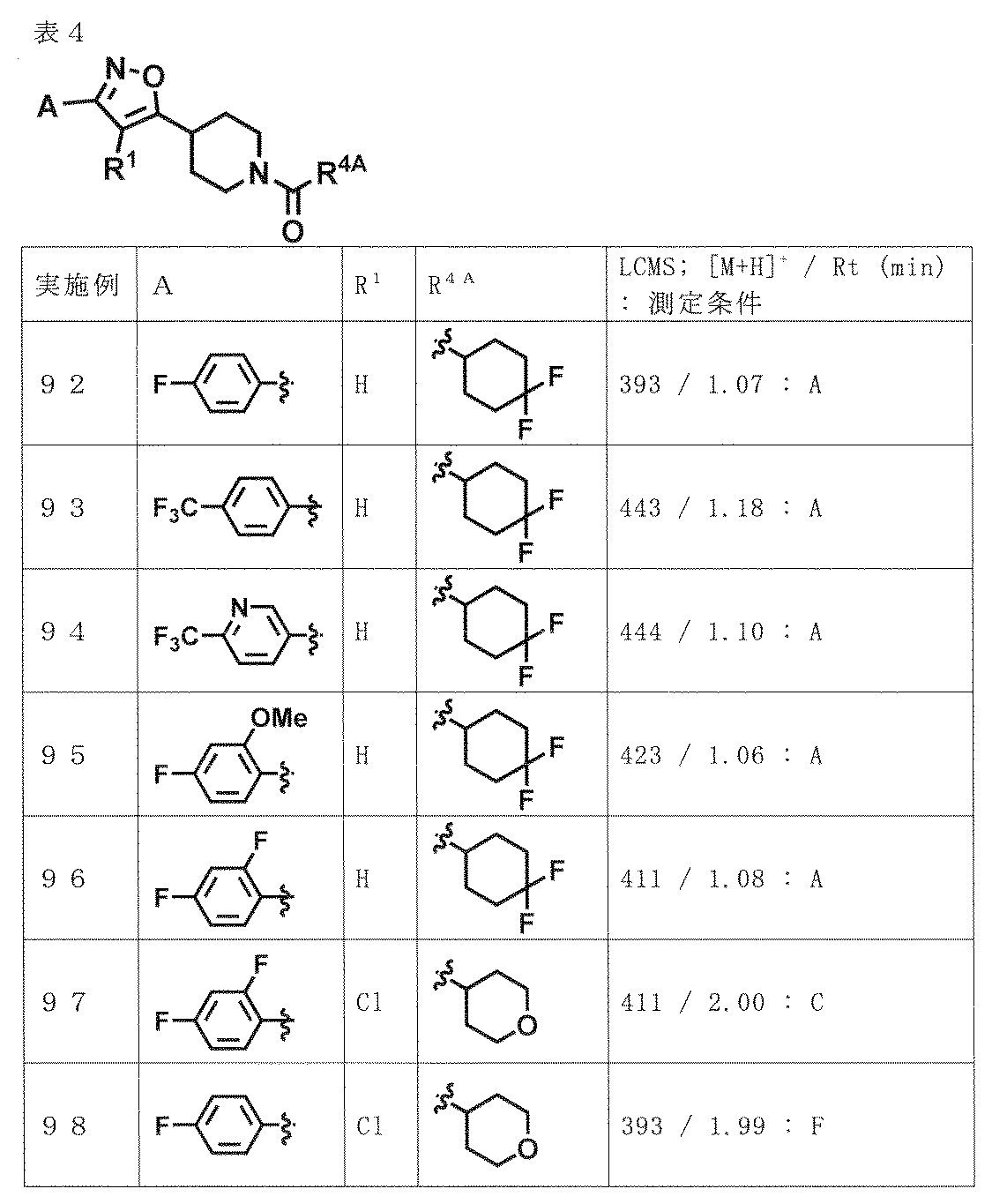
Examples 92-98
The compounds shown in Table 4 were obtained by the method according to Reference Example 3 and Example 2 using the corresponding starting compounds.
対応する原料化合物を用いて、参考例3及び実施例2に準ずる方法により、表4に示す化合物を得た。
The compounds shown in Table 4 were obtained by the method according to Reference Example 3 and Example 2 using the corresponding starting compounds.
実施例99~100
対応する原料化合物を用いて、参考例3及び実施例2に準ずる方法により、表5に示す化合物を得た。
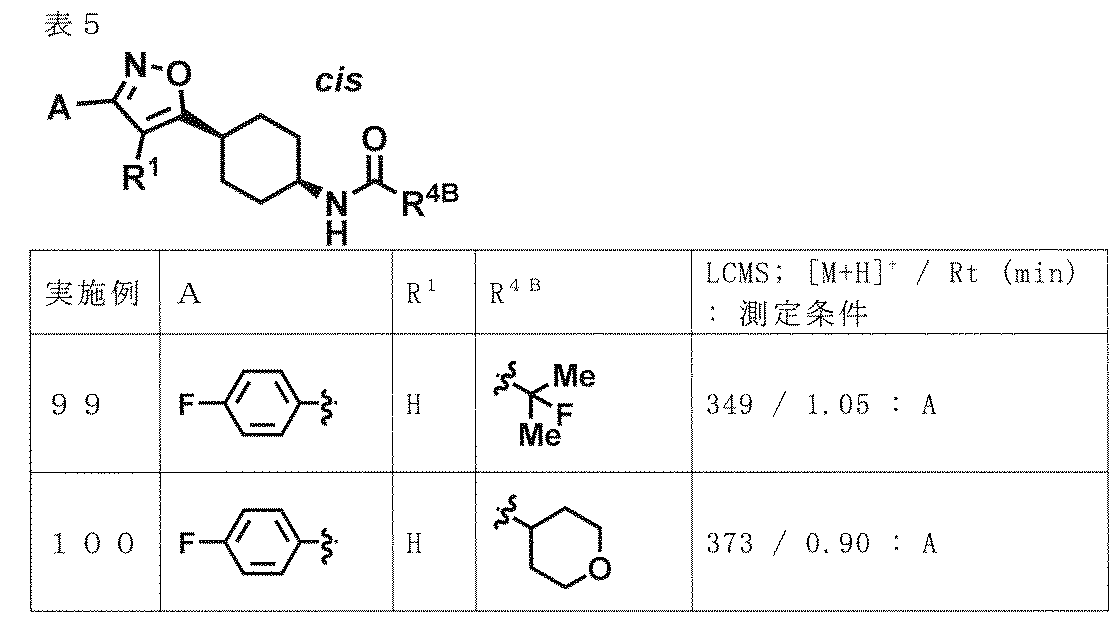
Examples 99-100
The compounds shown in Table 5 were obtained by the method according to Reference Example 3 and Example 2 using the corresponding starting compounds.
対応する原料化合物を用いて、参考例3及び実施例2に準ずる方法により、表5に示す化合物を得た。
The compounds shown in Table 5 were obtained by the method according to Reference Example 3 and Example 2 using the corresponding starting compounds.
実施例101~142
対応する原料化合物を用いて、実施例5に準ずる方法により、表6に示す化合物を得た。



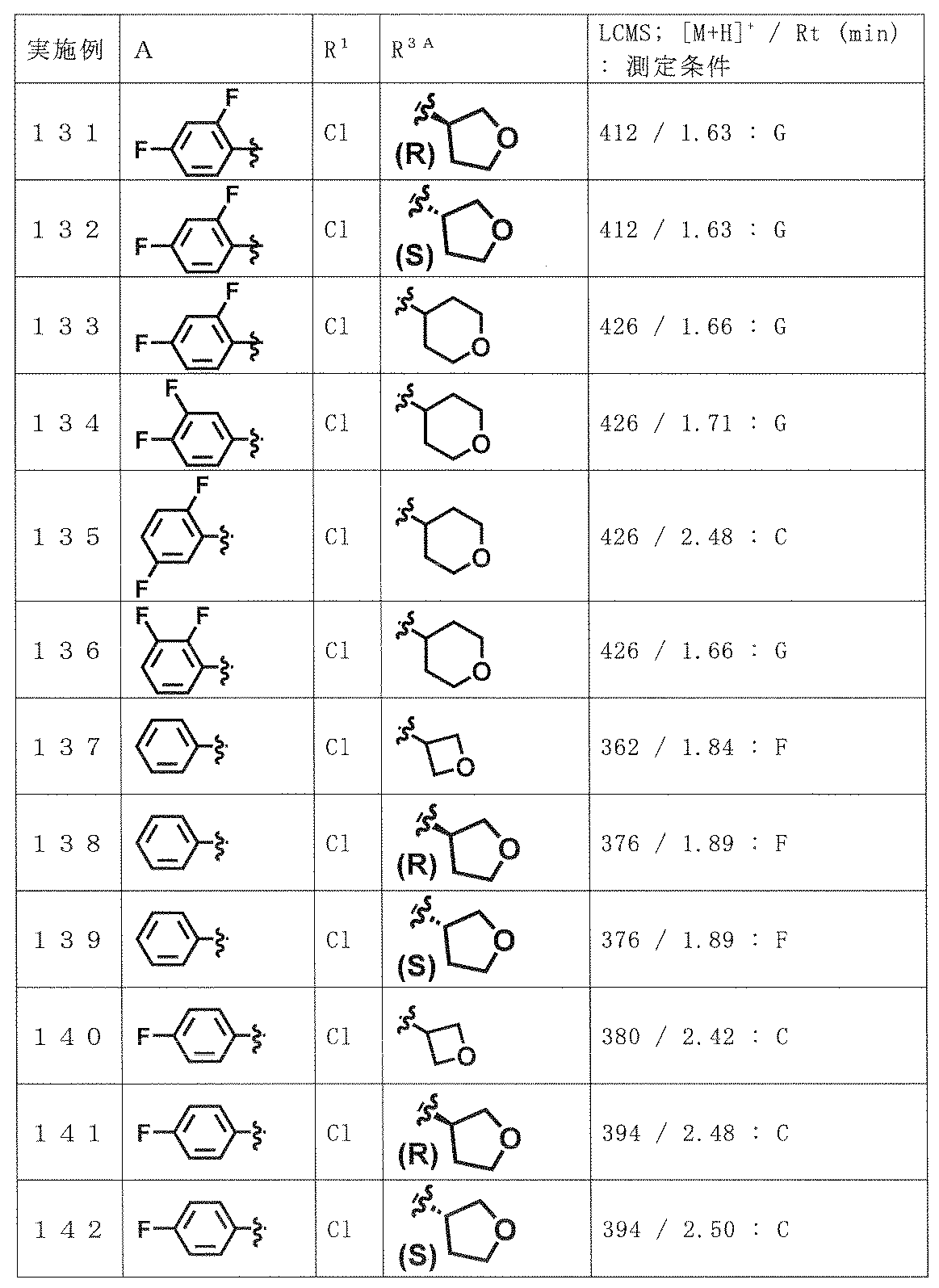
Examples 101-142
The compounds shown in Table 6 were obtained by the method according to Example 5 using the corresponding starting compounds.
対応する原料化合物を用いて、実施例5に準ずる方法により、表6に示す化合物を得た。
The compounds shown in Table 6 were obtained by the method according to Example 5 using the corresponding starting compounds.
実施例143~148
対応する原料化合物を用いて、参考例4及び実施例2に準ずる方法により、表7に示す化合物を得た。

Examples 143-148
The compounds shown in Table 7 were obtained by the method according to Reference Example 4 and Example 2 using the corresponding starting compounds.
対応する原料化合物を用いて、参考例4及び実施例2に準ずる方法により、表7に示す化合物を得た。
The compounds shown in Table 7 were obtained by the method according to Reference Example 4 and Example 2 using the corresponding starting compounds.
実施例149
対応する原料化合物を用いて、実施例6に準ずる方法により、表8に示す化合物を得た。

Example 149
The compounds shown in Table 8 were obtained by the method according to Example 6 using the corresponding starting compounds.
対応する原料化合物を用いて、実施例6に準ずる方法により、表8に示す化合物を得た。
The compounds shown in Table 8 were obtained by the method according to Example 6 using the corresponding starting compounds.
実施例150~151
対応する原料化合物を用いて、参考例5及び実施例2に準ずる方法により、表9に示す化合物を得た。

Examples 150-151
The compounds shown in Table 9 were obtained by the method according to Reference Example 5 and Example 2 using the corresponding starting compounds.
対応する原料化合物を用いて、参考例5及び実施例2に準ずる方法により、表9に示す化合物を得た。
The compounds shown in Table 9 were obtained by the method according to Reference Example 5 and Example 2 using the corresponding starting compounds.
実施例152~154
対応する原料化合物を用いて、実施例7に準ずる方法により、表10に示す化合物を得た。

Examples 152-154
The compounds shown in Table 10 were obtained by the method according to Example 7 using the corresponding starting compounds.
対応する原料化合物を用いて、実施例7に準ずる方法により、表10に示す化合物を得た。
The compounds shown in Table 10 were obtained by the method according to Example 7 using the corresponding starting compounds.
実施例155~156
対応する原料化合物を用いて、参考例6及び実施例2に準ずる方法により、表11に示す化合物を得た。

Examples 155 to 156
The compounds shown in Table 11 were obtained by the method according to Reference Example 6 and Example 2 using the corresponding starting compounds.
対応する原料化合物を用いて、参考例6及び実施例2に準ずる方法により、表11に示す化合物を得た。
The compounds shown in Table 11 were obtained by the method according to Reference Example 6 and Example 2 using the corresponding starting compounds.
実施例157~159
対応する原料化合物を用いて、実施例8に準ずる方法により、表12に示す化合物を得た。
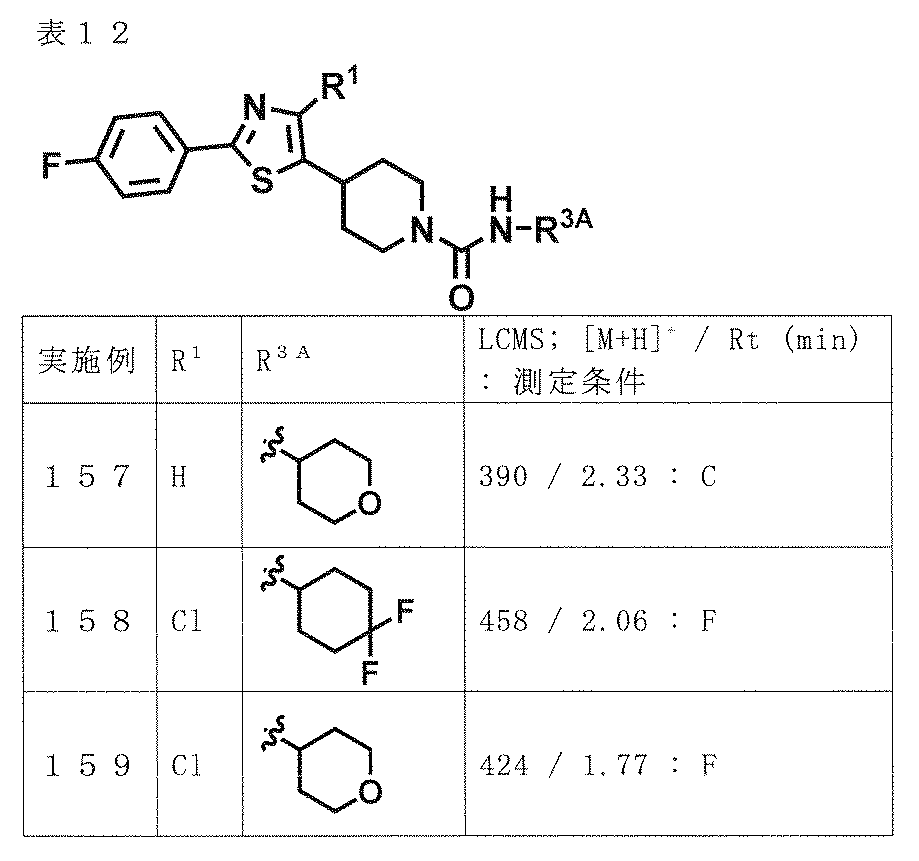
Examples 157 to 159
The compounds shown in Table 12 were obtained by the method according to Example 8 using the corresponding starting compounds.
対応する原料化合物を用いて、実施例8に準ずる方法により、表12に示す化合物を得た。
The compounds shown in Table 12 were obtained by the method according to Example 8 using the corresponding starting compounds.
実施例160~161
対応する原料化合物を用いて、参考例7及び実施例2に準ずる方法により、表13に示す化合物を得た。

Examples 160-161
The compounds shown in Table 13 were obtained by the method according to Reference Example 7 and Example 2 using the corresponding starting compounds.
対応する原料化合物を用いて、参考例7及び実施例2に準ずる方法により、表13に示す化合物を得た。
The compounds shown in Table 13 were obtained by the method according to Reference Example 7 and Example 2 using the corresponding starting compounds.
実施例162~163
対応する原料化合物を用いて、実施例9及び10に準ずる方法により、表14に示す化合物を得た。
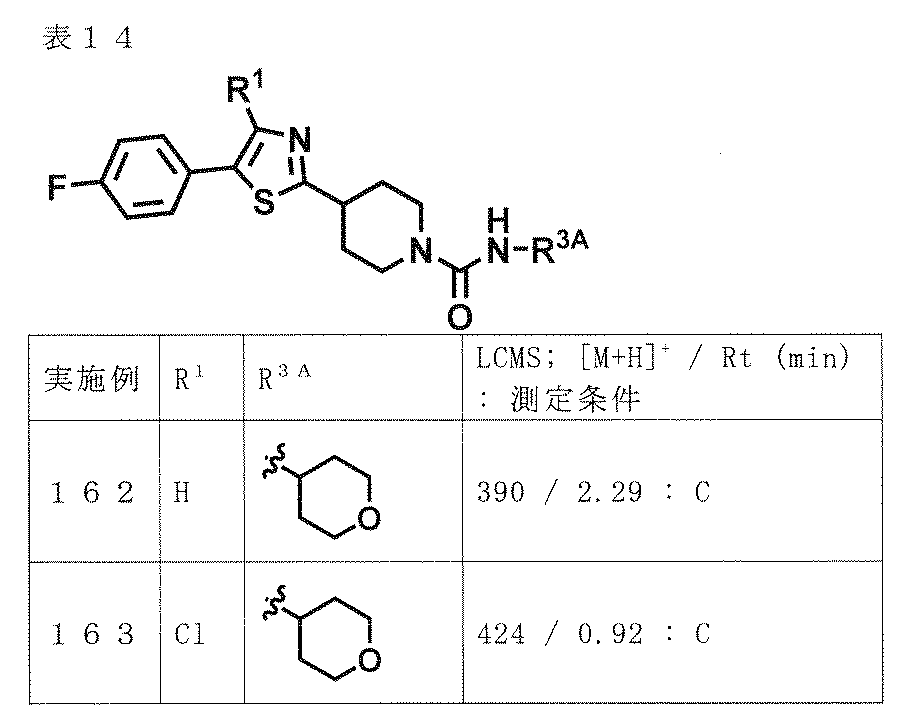
Examples 162-163
The compounds shown in Table 14 were obtained by the method according to Examples 9 and 10 using the corresponding starting compounds.
対応する原料化合物を用いて、実施例9及び10に準ずる方法により、表14に示す化合物を得た。
The compounds shown in Table 14 were obtained by the method according to Examples 9 and 10 using the corresponding starting compounds.
実施例164~165
対応する原料化合物を用いて、参考例8及び実施例2に準ずる方法により、表15に示す化合物を得た。

Examples 164 to 165
The compounds shown in Table 15 were obtained by the method according to Reference Example 8 and Example 2 using the corresponding starting compounds.
対応する原料化合物を用いて、参考例8及び実施例2に準ずる方法により、表15に示す化合物を得た。
The compounds shown in Table 15 were obtained by the method according to Reference Example 8 and Example 2 using the corresponding starting compounds.
実施例166~175
対応する原料化合物を用いて、実施例6に準ずる方法により、表16に示す化合物を得た。

Examples 166-175
The compounds shown in Table 16 were obtained by the method according to Example 6 using the corresponding starting compounds.
対応する原料化合物を用いて、実施例6に準ずる方法により、表16に示す化合物を得た。
The compounds shown in Table 16 were obtained by the method according to Example 6 using the corresponding starting compounds.
実施例176~184
対応する原料化合物を用いて、参考例5及び実施例2に準ずる方法により、表17に示す化合物を得た。

Examples 176-184
The compounds shown in Table 17 were obtained by the method according to Reference Example 5 and Example 2 using the corresponding starting compounds.
対応する原料化合物を用いて、参考例5及び実施例2に準ずる方法により、表17に示す化合物を得た。
The compounds shown in Table 17 were obtained by the method according to Reference Example 5 and Example 2 using the corresponding starting compounds.
実施例185~191
対応する原料化合物を用いて、実施例7に準ずる方法により、表18に示す化合物を得た。

Examples 185-191
The compounds shown in Table 18 were obtained by the method according to Example 7 using the corresponding starting compounds.
対応する原料化合物を用いて、実施例7に準ずる方法により、表18に示す化合物を得た。
The compounds shown in Table 18 were obtained by the method according to Example 7 using the corresponding starting compounds.
実施例192
対応する原料化合物を用いて、参考例6及び実施例2に準ずる方法により、表19に示す化合物を得た。

Example 192
The compounds shown in Table 19 were obtained by the method according to Reference Example 6 and Example 2 using the corresponding starting compounds.
対応する原料化合物を用いて、参考例6及び実施例2に準ずる方法により、表19に示す化合物を得た。
The compounds shown in Table 19 were obtained by the method according to Reference Example 6 and Example 2 using the corresponding starting compounds.
実施例193~198
対応する原料化合物を用いて、実施例8に準ずる方法により、表20に示す化合物を得た。

Examples 193-198
The compounds shown in Table 20 were obtained by the method according to Example 8 using the corresponding starting compounds.
対応する原料化合物を用いて、実施例8に準ずる方法により、表20に示す化合物を得た。
The compounds shown in Table 20 were obtained by the method according to Example 8 using the corresponding starting compounds.
実施例199~210
対応する原料化合物を用いて、参考例7及び実施例2に準ずる方法により、表21に示す化合物を得た。


Examples 199-210
The compounds shown in Table 21 were obtained by the method according to Reference Example 7 and Example 2 using the corresponding starting compounds.
対応する原料化合物を用いて、参考例7及び実施例2に準ずる方法により、表21に示す化合物を得た。
The compounds shown in Table 21 were obtained by the method according to Reference Example 7 and Example 2 using the corresponding starting compounds.
実施例211~214
対応する原料化合物を用いて、実施例9及び10に準ずる方法により、表22に示す化合物を得た。
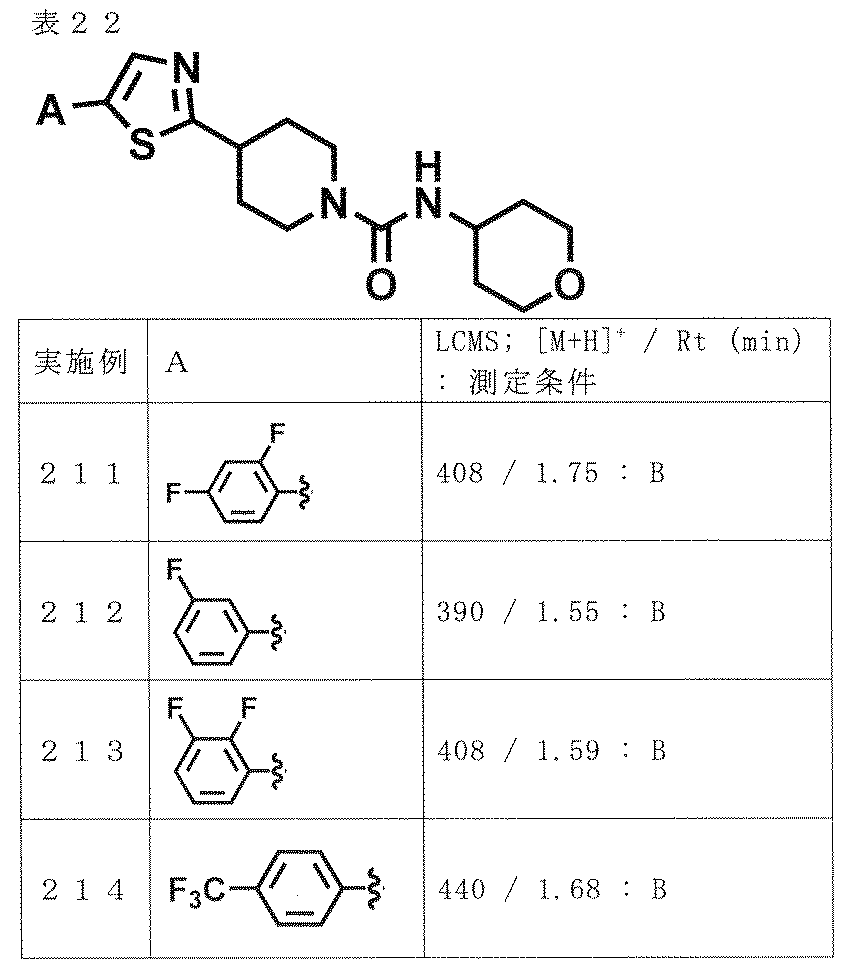
Examples 211-214
The compounds shown in Table 22 were obtained by the method according to Examples 9 and 10 using the corresponding starting compounds.
対応する原料化合物を用いて、実施例9及び10に準ずる方法により、表22に示す化合物を得た。
The compounds shown in Table 22 were obtained by the method according to Examples 9 and 10 using the corresponding starting compounds.
実施例215~219
対応する原料化合物を用いて、参考例8及び実施例2に準ずる方法により、表23に示す化合物を得た。

Examples 215 to 219
The compounds shown in Table 23 were obtained by the method according to Reference Example 8 and Example 2 using the corresponding starting compounds.
対応する原料化合物を用いて、参考例8及び実施例2に準ずる方法により、表23に示す化合物を得た。
The compounds shown in Table 23 were obtained by the method according to Reference Example 8 and Example 2 using the corresponding starting compounds.
試験例
以下に、本発明の代表的化合物の薬理試験結果を示し、該化合物についての薬理作用を説明するが、本発明はこれらの試験例に限定されるものではない。 Test Examples Hereinafter, pharmacological test results of representative compounds of the present invention will be shown and the pharmacological action of the compounds will be described. However, the present invention is not limited to these test examples.
以下に、本発明の代表的化合物の薬理試験結果を示し、該化合物についての薬理作用を説明するが、本発明はこれらの試験例に限定されるものではない。 Test Examples Hereinafter, pharmacological test results of representative compounds of the present invention will be shown and the pharmacological action of the compounds will be described. However, the present invention is not limited to these test examples.
試験例1.ヒトα7 nACh受容体安定発現細胞を用いたPAM活性評価
(1)ヒトα7 nAChR安定発現細胞
ヒトα7 nAChR安定発現細胞を作製し、培養に供した。具体的には、宿主細胞としてラット下垂体由来GH4C1細胞(cat. no. CCL-82.2, ATCC, USA)を用いた。GenBank BAC81731の蛋白をコードする塩基配列を挿入したpcDNA3.1Zeoベクターの導入、及びヒトα7 nAChR遺伝子を挿入したpcDNA3.1ベクター(cat. no. V790-20, invitrogen, Carlsbad,CA,USA)の導入によりエクオリン及びヒトα7 nAChR安定発現細胞を得た。選別にはそれぞれZeocin (cat. no. R25001, invitrogen, Carlsbad,CA,USA)及びGeneticin(cat. no. 10131-027, invitrogen, Carlsbad,CA,USA)を用いた。
培地には2.5%ウシ胎児血清(cat. no. 2917354, ICN Biomedicals, Inc, USA)、15%非働化ウマ血清(cat. no. 26050-088, invitrogen, Carlsbad,CA,USA)、1μg/mL Geneticin、5μg/mL Puromycin(cat. no. 14861-84, invitrogen, Carlsbad,CA,USA)を含むF-10 Nutrient Mixture(Ham)培地(cat. no. 11550-043, invitrogen, Carlsbad,CA,USA)を用い、コラーゲンType1コートディッシュ(cat. no. 4030-010, iwaki, Tokyo, Japan)にて培養を行った。培養中、2-3日毎に培地交換を行い、7日毎にTrypLE Express(cat. no. 45604-021, invitrogen, Carlsbad,CA,USA)処理にて細胞を回収し、継代培養を行った。
継代から7日後、約80%コンフルエントな状態でTrypLE Express処理にて細胞を回収し、Hanks (cat. no. 14065-056, invitrogen, Carlsbad,CA,USA)/20mmol/L Hepes(cat. no. 15630-080, invitrogen, Carlsbad,CA,USA) Buffer (pH7.4)、F-10 Nutrient Mixture (Ham)、0.1mg/mL Geneticinからなる反応培地にて20000 cells/25μL/wellとなるように懸濁し、384 wellプレート(cat. no. 781090, Greiner,Germany)に播種した。
播種翌日、Viviren(cat. no. E649X, Promega, Madison,WI,USA)を終濃度4μmol/Lとなるように添加し(15μL/well)、遠心後4時間室温、遮光下で静置した。 Test Example 1 PAM activity evaluation using human α7 nACh receptor stable expression cells (1) Human α7 nAChR stable expression cells Human α7 nAChR stable expression cells were prepared and subjected to culture. Specifically, rat pituitary-derived GH4C1 cells (cat. No. CCL-82.2, ATCC, USA) were used as host cells. Introduction of pcDNA3.1Zeo vector into which the nucleotide sequence encoding the protein of GenBank BAC81731 was inserted, and introduction of pcDNA3.1 vector into which the human α7 nAChR gene was inserted (cat. No. V790-20, invitrogen, Carlsbad, CA, USA) Thus, aequorin and human α7 nAChR stably expressing cells were obtained. For selection, Zeocin (cat. No. R25001, invitrogen, Carlsbad, CA, USA) and Geneticin (cat. No. 10131-027, invitrogen, Carlsbad, CA, USA) were used.
Medium includes 2.5% fetal bovine serum (cat. No. 2917354, ICN Biomedicals, Inc, USA), 15% inactivated horse serum (cat. No. 26050-088, invitrogen, Carlsbad, CA, USA), 1 μg / ML Geneticin, F-10 Nutrient Mixture (Ham) medium (cat. No. 11550-043, invitrogen, Carlsbad, CA) containing 5 μg / mL Puromycin (cat. No. 14861-84, invitrogen, Carlsbad, CA, USA) , USA) and cultured in a collagen Type 1 coat dish (cat. No. 4030-010, iwaki, Tokyo, Japan). During the culture, the medium was changed every 2-3 days, and the cells were collected by TrypLE Express (cat. No. 45604-021, invitrogen, Carlsbad, CA, USA) treatment every 7 days and subcultured.
Seven days after passage, cells were collected by TrypLE Express treatment in a state of about 80% confluence, Hanks (cat. No. 14065-056, invitrogen, Carlsbad, CA, USA) / 20 mmol / L Hepes (cat. No 15630-080, invitrogen, Carlsbad, CA, USA) To reach 20000 cells / 25μL / well in a reaction medium consisting of Buffer (pH7.4), F-10 Nutrient Mixture (Ham), and 0.1mg / mL Geneticin And seeded on 384 well plates (cat. No. 781090, Greiner, Germany).
The day after sowing, Viviren (cat. No. E649X, Promega, Madison, Wis., USA) was added to a final concentration of 4 μmol / L (15 μL / well), and the mixture was allowed to stand at room temperature for 4 hours after light centrifugation at room temperature.
(1)ヒトα7 nAChR安定発現細胞
ヒトα7 nAChR安定発現細胞を作製し、培養に供した。具体的には、宿主細胞としてラット下垂体由来GH4C1細胞(cat. no. CCL-82.2, ATCC, USA)を用いた。GenBank BAC81731の蛋白をコードする塩基配列を挿入したpcDNA3.1Zeoベクターの導入、及びヒトα7 nAChR遺伝子を挿入したpcDNA3.1ベクター(cat. no. V790-20, invitrogen, Carlsbad,CA,USA)の導入によりエクオリン及びヒトα7 nAChR安定発現細胞を得た。選別にはそれぞれZeocin (cat. no. R25001, invitrogen, Carlsbad,CA,USA)及びGeneticin(cat. no. 10131-027, invitrogen, Carlsbad,CA,USA)を用いた。
培地には2.5%ウシ胎児血清(cat. no. 2917354, ICN Biomedicals, Inc, USA)、15%非働化ウマ血清(cat. no. 26050-088, invitrogen, Carlsbad,CA,USA)、1μg/mL Geneticin、5μg/mL Puromycin(cat. no. 14861-84, invitrogen, Carlsbad,CA,USA)を含むF-10 Nutrient Mixture(Ham)培地(cat. no. 11550-043, invitrogen, Carlsbad,CA,USA)を用い、コラーゲンType1コートディッシュ(cat. no. 4030-010, iwaki, Tokyo, Japan)にて培養を行った。培養中、2-3日毎に培地交換を行い、7日毎にTrypLE Express(cat. no. 45604-021, invitrogen, Carlsbad,CA,USA)処理にて細胞を回収し、継代培養を行った。
継代から7日後、約80%コンフルエントな状態でTrypLE Express処理にて細胞を回収し、Hanks (cat. no. 14065-056, invitrogen, Carlsbad,CA,USA)/20mmol/L Hepes(cat. no. 15630-080, invitrogen, Carlsbad,CA,USA) Buffer (pH7.4)、F-10 Nutrient Mixture (Ham)、0.1mg/mL Geneticinからなる反応培地にて20000 cells/25μL/wellとなるように懸濁し、384 wellプレート(cat. no. 781090, Greiner,Germany)に播種した。
播種翌日、Viviren(cat. no. E649X, Promega, Madison,WI,USA)を終濃度4μmol/Lとなるように添加し(15μL/well)、遠心後4時間室温、遮光下で静置した。 Test Example 1 PAM activity evaluation using human α7 nACh receptor stable expression cells (1) Human α7 nAChR stable expression cells Human α7 nAChR stable expression cells were prepared and subjected to culture. Specifically, rat pituitary-derived GH4C1 cells (cat. No. CCL-82.2, ATCC, USA) were used as host cells. Introduction of pcDNA3.1Zeo vector into which the nucleotide sequence encoding the protein of GenBank BAC81731 was inserted, and introduction of pcDNA3.1 vector into which the human α7 nAChR gene was inserted (cat. No. V790-20, invitrogen, Carlsbad, CA, USA) Thus, aequorin and human α7 nAChR stably expressing cells were obtained. For selection, Zeocin (cat. No. R25001, invitrogen, Carlsbad, CA, USA) and Geneticin (cat. No. 10131-027, invitrogen, Carlsbad, CA, USA) were used.
Medium includes 2.5% fetal bovine serum (cat. No. 2917354, ICN Biomedicals, Inc, USA), 15% inactivated horse serum (cat. No. 26050-088, invitrogen, Carlsbad, CA, USA), 1 μg / ML Geneticin, F-10 Nutrient Mixture (Ham) medium (cat. No. 11550-043, invitrogen, Carlsbad, CA) containing 5 μg / mL Puromycin (cat. No. 14861-84, invitrogen, Carlsbad, CA, USA) , USA) and cultured in a collagen Type 1 coat dish (cat. No. 4030-010, iwaki, Tokyo, Japan). During the culture, the medium was changed every 2-3 days, and the cells were collected by TrypLE Express (cat. No. 45604-021, invitrogen, Carlsbad, CA, USA) treatment every 7 days and subcultured.
Seven days after passage, cells were collected by TrypLE Express treatment in a state of about 80% confluence, Hanks (cat. No. 14065-056, invitrogen, Carlsbad, CA, USA) / 20 mmol / L Hepes (cat. No 15630-080, invitrogen, Carlsbad, CA, USA) To reach 20000 cells / 25μL / well in a reaction medium consisting of Buffer (pH7.4), F-10 Nutrient Mixture (Ham), and 0.1mg / mL Geneticin And seeded on 384 well plates (cat. No. 781090, Greiner, Germany).
The day after sowing, Viviren (cat. No. E649X, Promega, Madison, Wis., USA) was added to a final concentration of 4 μmol / L (15 μL / well), and the mixture was allowed to stand at room temperature for 4 hours after light centrifugation at room temperature.
(2)試験化合物の調製
試験化合物は最終濃度の1000倍濃度のDMSO溶液を作製し、この溶液をHanks/20 mmol/L HEPES/0.2% BSA(cat. no. A3803, Sigma,St.Louis, MO, USA)にて最終濃度の6倍濃度に調整した。 (2) Preparation of test compound A DMSO solution having a concentration 1000 times the final concentration was prepared as a test compound, and this solution was added to Hanks / 20 mmol / L HEPES / 0.2% BSA (cat. No. A3803, Sigma, St. Louis, MO, USA) and adjusted to 6 times the final concentration.
試験化合物は最終濃度の1000倍濃度のDMSO溶液を作製し、この溶液をHanks/20 mmol/L HEPES/0.2% BSA(cat. no. A3803, Sigma,St.Louis, MO, USA)にて最終濃度の6倍濃度に調整した。 (2) Preparation of test compound A DMSO solution having a concentration 1000 times the final concentration was prepared as a test compound, and this solution was added to Hanks / 20 mmol / L HEPES / 0.2% BSA (cat. No. A3803, Sigma, St. Louis, MO, USA) and adjusted to 6 times the final concentration.
(3)PAM活性評価
α7 nAChR刺激による発光シグナルの検出にはFDSS7000(浜松ホトニクス)を用いた。細胞及び発光基質を添加したプレートに試験化合物を添加し、150秒後に単独処置でEC20を示す濃度のAChを添加した。ACh添加後138秒間発光シグナル(中心波長:465 nm)を測定してRLU(Max-Min)を算出し、コントロールwellと試験化合物添加wellとのRLU(Max-Min)の比をPAM活性とした。
代表的化合物のα7 PAM活性のデータを表24、表25及び表26に示す。

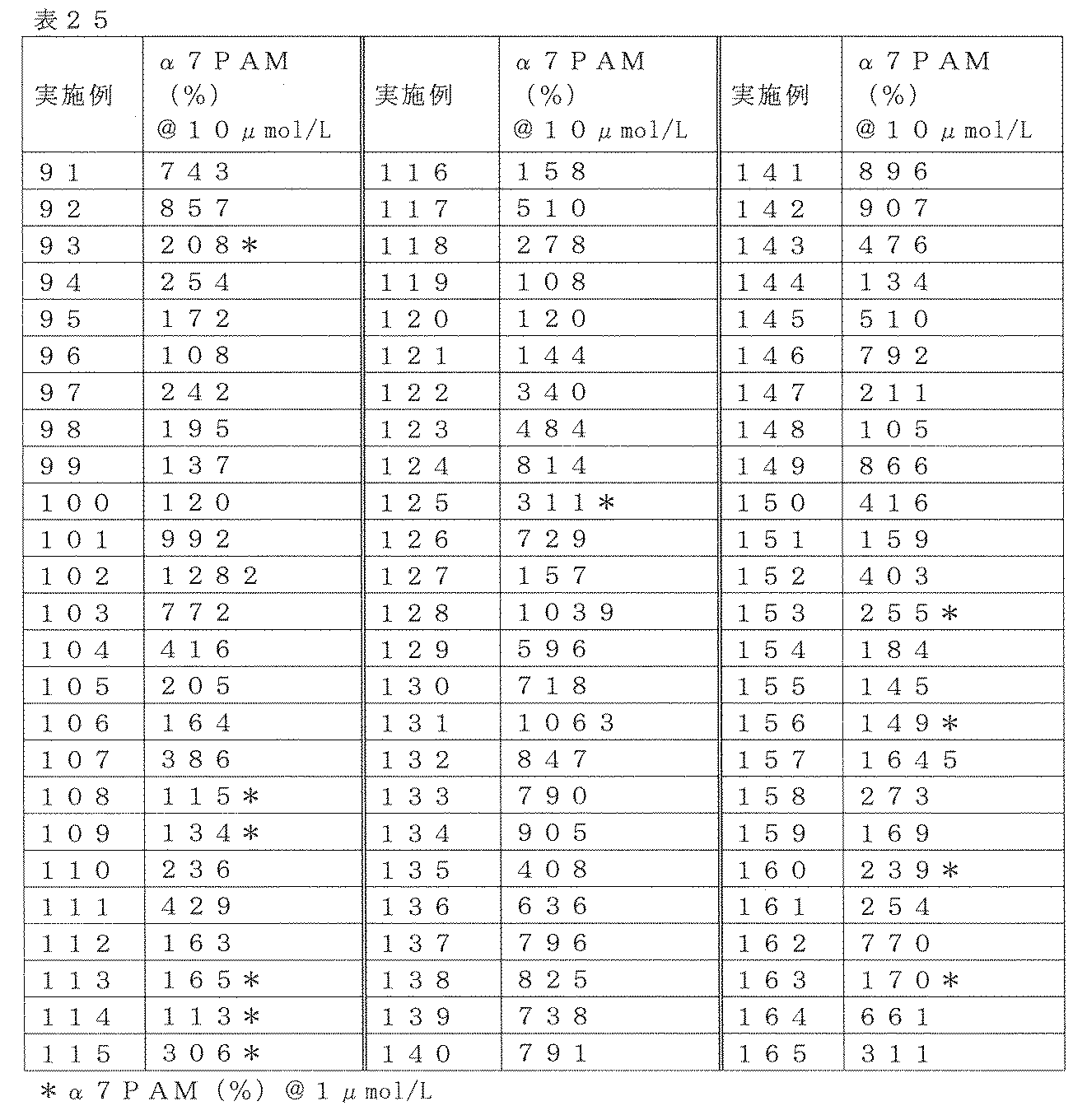

(3) PAM activity evaluation FDSS7000 (Hamamatsu Photonics) was used for the detection of the luminescent signal by α7 nAChR stimulation. Test compounds were added to the plates to which cells and luminescent substrate had been added, and ACh at a concentration indicating EC 20 was added alone after 150 seconds. The luminescence signal (center wavelength: 465 nm) was measured for 138 seconds after the addition of ACh to calculate RLU (Max-Min), and the ratio of RLU (Max-Min) between the control well and the test compound added well was defined as PAM activity. .
Data of α7 PAM activity of representative compounds is shown in Table 24, Table 25 and Table 26.
α7 nAChR刺激による発光シグナルの検出にはFDSS7000(浜松ホトニクス)を用いた。細胞及び発光基質を添加したプレートに試験化合物を添加し、150秒後に単独処置でEC20を示す濃度のAChを添加した。ACh添加後138秒間発光シグナル(中心波長:465 nm)を測定してRLU(Max-Min)を算出し、コントロールwellと試験化合物添加wellとのRLU(Max-Min)の比をPAM活性とした。
代表的化合物のα7 PAM活性のデータを表24、表25及び表26に示す。
Data of α7 PAM activity of representative compounds is shown in Table 24, Table 25 and Table 26.
試験例2.マウス新奇物体認識試験を用いた認知機能評価(以下、mORTという)
体重25-30gのSlc:ddYマウス(雄性、日本エスエルシー)を用いた新奇物体認識試験において、第一試行(トレーニング)と第二試行(テスト)の間隔時間依存的に、既知物体に対する記憶低下が認められ、24時間後に第二試行を行った場合、顕著な忘却が認められる。そこで本発明化合物を第一試行前に投与し、第二試行における記憶増強作用を評価した。その結果、実施例1、45、51、79及び124の化合物は、3または10mg/kg(経口)において有意な記憶増強作用を示した。
Test Example 2 Evaluation of cognitive function using mouse novel object recognition test (hereinafter referred to as mORT)
Decreased memory for known objects in a novel object recognition test using Slc: ddY mice (male, SLC Japan) weighing 25-30 g depending on the time interval between the first trial (training) and the second trial (test) When a second trial is performed 24 hours later, significant forgetting is observed. Therefore, the compound of the present invention was administered before the first trial, and the memory enhancing action in the second trial was evaluated. As a result, the compounds of Examples 1, 45, 51, 79 and 124 showed a significant memory enhancing action at 3 or 10 mg / kg (oral).
体重25-30gのSlc:ddYマウス(雄性、日本エスエルシー)を用いた新奇物体認識試験において、第一試行(トレーニング)と第二試行(テスト)の間隔時間依存的に、既知物体に対する記憶低下が認められ、24時間後に第二試行を行った場合、顕著な忘却が認められる。そこで本発明化合物を第一試行前に投与し、第二試行における記憶増強作用を評価した。その結果、実施例1、45、51、79及び124の化合物は、3または10mg/kg(経口)において有意な記憶増強作用を示した。
Test Example 2 Evaluation of cognitive function using mouse novel object recognition test (hereinafter referred to as mORT)
Decreased memory for known objects in a novel object recognition test using Slc: ddY mice (male, SLC Japan) weighing 25-30 g depending on the time interval between the first trial (training) and the second trial (test) When a second trial is performed 24 hours later, significant forgetting is observed. Therefore, the compound of the present invention was administered before the first trial, and the memory enhancing action in the second trial was evaluated. As a result, the compounds of Examples 1, 45, 51, 79 and 124 showed a significant memory enhancing action at 3 or 10 mg / kg (oral).
以上で説明したように、式(I)で表される化合物、またはその製薬学上許容される塩は、強いα7ニコチン性アセチルコリン受容体(α7 nAChR)の調節作用を有し、中枢神経系(CNS)及び/又は末梢神経系(PNS)のコリン作動性に関する疾患、平滑筋収縮に関する疾患、内分泌疾患、神経変性に関する疾患、炎症又は痛み等の疾患及び常習性の薬物乱用から引き起こされる禁断症状に関する疾患等の治療及び/又は予防に有用である。
As described above, the compound represented by the formula (I), or a pharmaceutically acceptable salt thereof, has a strong α7 nicotinic acetylcholine receptor (α7hnAChR) modulating action, and the central nervous system ( CNS) and / or peripheral nervous system (PNS) cholinergic diseases, smooth muscle contraction diseases, endocrine diseases, neurodegenerative diseases, diseases such as inflammation or pain, and withdrawal symptoms caused by addictive drug abuse It is useful for treatment and / or prevention of diseases and the like.
Claims (28)
- 式(I):


X-Y-Zは、N-CO-NR3AR3B、N-COR4A、CR5-CO-NR3AR3B、CR5-NR6-COR4B又はCR5-NR6-CONR3AR3Bを表し、
Aは、フェニル又はピリジル(該フェニル及び該ピリジルは、それぞれハロゲン、水酸基、1から5個のフッ素で置換されていてもよいC1-6アルキル、1から5個のフッ素で置換されていてもよいC1-6アルコキシ及びシアノからなる群から独立して選択される1~5個の置換基で置換されていてもよい)を表し、
Bは、B-1、B-2、B-3、B-4又はB-5を表し、
式(I)の化合物におけるaおよびbは、Bに結合する2つの結合手が、B-1からB-5における2つの結合手のいずれに該当する結合かを示す記号であり、
Wは、酸素原子又は硫黄原子を表し、
R1は、水素原子;ハロゲン;シアノ;C1-6アルキル(該アルキルは、ハロゲン、水酸基、1から5個のフッ素で置換されていてもよいC3-10シクロアルキル、1から5個のフッ素で置換されていてもよいC1-6アルコキシ及び4~10員の飽和複素環からなる群から独立して選択される1~5個の置換基で置換されていてもよい);又はハロゲン、水酸基、C1-6アルキル及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキルを表し、
R2A、R2B、R2C、R2D及びR5は、同一又は異なって、水素原子;フッ素;水酸基;1から5個のフッ素で置換されていてもよいC1-6アルコキシ;又は1から5個のフッ素で置換されていてもよいC1-6アルキルを表し、ここにおいて、R2A、R2B、R2C、R2D及びR5のいずれかの2つが1から5個のフッ素で置換されていてもよいC1-6アルキルのとき、2個のアルキルが一緒になって該アルキルが結合している環とは別の環を形成していてもよく、
R3A、R3B、R4A、R4B及びR6は、同一又は異なって、ハロゲン、水酸基、C1-6アルコキシ、C3-10シクロアルキル及び4~10員の飽和複素環(ただし、環上にカルボニル基を有しない)からなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;ハロゲン、水酸基、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;C1-6アルキルで置換されていてもよい4~10員の飽和複素環;又は水素原子を表し、ここにおいて、(1)R4A及びR4Bは水素原子ではなく、(2)R3A及びR3Bは同時に水素原子ではなく、(3)BがB-3であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aはメチルではなく、(4)Aが無置換のフェニルであり、BがB-4であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aは1~5個のフッ素で置換されたC3アルキルではなく、かつ(5)R3A及びR3Bが共にC1-6アルキルのとき、それらが結合する窒素原子と一緒になって、フッ素、C1-6アルキル及びC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい4~10員の含窒素飽和複素環を形成していてもよい]
で表される化合物又はその製薬学的に許容される塩。 Formula (I):
XYZ represents N-CO-NR 3A R 3B , N-COR 4A , CR 5 -CO-NR 3A R 3B , CR 5 -NR 6 -COR 4B or CR 5 -NR 6 -CONR 3A R 3B Represents
A is phenyl or pyridyl (said phenyl and said pyridyl, halogen, respectively, hydroxyl, 1 to 5 fluorine optionally substituted by C 1-6 alkyl, optionally substituted with 1-5 fluorine And optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy and cyano,
B represents B-1, B-2, B-3, B-4 or B-5,
A and b in the compound of the formula (I) are symbols indicating which of the two bonds in B-1 to B-5 the two bonds that bind to B correspond to;
W represents an oxygen atom or a sulfur atom,
R 1 is a hydrogen atom; halogen; cyano; C 1-6 alkyl (wherein the alkyl is halogen, hydroxyl, C 3-10 cycloalkyl optionally substituted with 1 to 5 fluorines, 1 to 5 Halogen optionally substituted with 1 to 5 substituents independently selected from the group consisting of C 1-6 alkoxy optionally substituted with fluorine and a 4- to 10-membered saturated heterocyclic ring; or halogen Represents a C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of hydroxyl, C 1-6 alkyl and C 1-6 alkoxy,
R 2A , R 2B , R 2C , R 2D and R 5 are the same or different and represent a hydrogen atom; fluorine; a hydroxyl group; C 1-6 alkoxy optionally substituted with 1 to 5 fluorines; Represents C 1-6 alkyl optionally substituted with 5 fluorines, wherein two of R 2A , R 2B , R 2C , R 2D and R 5 are substituted with 1 to 5 fluorines In the case of C 1-6 alkyl, which may be substituted , two alkyls may be combined to form a ring different from the ring to which the alkyl is bonded;
R 3A , R 3B , R 4A , R 4B and R 6 are the same or different and are halogen, hydroxyl group, C 1-6 alkoxy, C 3-10 cycloalkyl and a 4- to 10-membered saturated heterocyclic ring (provided that the ring C 1-6 alkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of (having no carbonyl group above); halogen, hydroxyl, C 1-6 alkoxy and C 1 C 3-10 cycloalkyl optionally substituted with 1 to 5 substituents independently selected from the group consisting of -6 alkyl; 4 to 10 members optionally substituted with C 1-6 alkyl Or a hydrogen atom, wherein (1) R 4A and R 4B are not hydrogen atoms, (2) R 3A and R 3B are not simultaneously hydrogen atoms, and (3) B is B— 3 and W is an oxygen atom When XYZ is N—COR 4A , R 4A is not methyl, (4) A is unsubstituted phenyl, B is B-4, and W is an oxygen atom. , XYZ is N—COR 4A , R 4A is not C 3 alkyl substituted with 1 to 5 fluorines, and (5) R 3A and R 3B are both C 1-6 When alkyl, together with the nitrogen atom to which they are attached, is substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkyl and C 1-6 alkoxy. It may form a 4- to 10-membered nitrogen-containing saturated heterocyclic ring.
Or a pharmaceutically acceptable salt thereof. - R3A、R3B、R4A、R4B及びR6が、同一又は異なって、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキル;フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子であり、ここにおいて、(1)R4A及びR4Bは水素原子ではなく、(2)R3A及びR3Bは同時に水素原子ではなく、(3)BがB-3であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aはメチルではなく、(4)Aが無置換のフェニルであり、BがB-4であり、Wが酸素原子であり、X-Y-ZがN-COR4Aであるときは、R4Aは1~5個のフッ素で置換されたC3アルキルではない、
請求項1に記載の化合物又はその製薬学的に許容される塩。 1 to 5 R 3A , R 3B , R 4A , R 4B and R 6 are the same or different and are independently selected from the group consisting of fluorine, C 1-6 alkoxy, and C 3-10 cycloalkyl of which may be substituted with a substituent C 1-6 alkyl; fluorine, optionally substituted with one to five substituents independently selected from the group consisting of C 1-6 alkoxy and C 1-6 alkyl An optionally substituted C 3-10 cycloalkyl; a 4-10 membered saturated heterocycle; or a hydrogen atom, wherein (1) R 4A and R 4B are not hydrogen atoms, but (2) R 3A and R 3B Are not hydrogen atoms at the same time, (3) when B is B-3, W is an oxygen atom, and XYZ is N—COR 4A , R 4A is not methyl and (4) A is unsubstituted phenyl, B is B-4 W is an oxygen atom, when X-Y-Z is N-COR 4A is, R 4A is not a C 3 alkyl substituted with 1-5 fluorine,
The compound according to claim 1 or a pharmaceutically acceptable salt thereof. - R3A、R3B及びR4Aが、同一又は異なって、フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;4~10員の飽和複素環;又は水素原子であり、ここにおいて、R3A及びR3Bは同時に水素原子ではなく、R4Aは水素原子ではない、
請求項1又は2に記載の化合物又はその製薬学的に許容される塩。 R 3A , R 3B and R 4A are the same or different and are substituted with 1 to 5 substituents independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl; a 4-10 membered saturated heterocyclic ring; or a hydrogen atom, wherein R 3A and R 3B are not simultaneously a hydrogen atom, and R 4A is not a hydrogen atom,
The compound according to claim 1 or 2, or a pharmaceutically acceptable salt thereof. - R4Bが、フッ素、C1-6アルコキシ、及びC3-10シクロアルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC1-6アルキルである、
請求項1~3のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 R 4B is selected from the group consisting of fluorine, is C 1-6 alkoxy, and C 3-10 optionally substituted with 1 to 5 substituents selected independently from the group consisting of cycloalkyl C 1-6 alkyl ,
The compound according to any one of claims 1 to 3 or a pharmaceutically acceptable salt thereof. - Aが、フェニル又はピリジル(該フェニル及び該ピリジルは、それぞれフッ素、1から5個のフッ素で置換されていてもよいC1-6アルキル及び1から5個のフッ素で置換されていてもよいC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)である、
請求項1~4のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 A is phenyl or pyridyl (the phenyl and the pyridyl are each fluorine, C 1-6 alkyl optionally substituted with 1 to 5 fluorines, and C optionally substituted with 1 to 5 fluorines) Which may be substituted with 1 to 5 substituents independently selected from the group consisting of 1-6 alkoxy).
The compound according to any one of claims 1 to 4 or a pharmaceutically acceptable salt thereof. - R1が、水素原子;ハロゲン;シアノ;C1-6アルキル;又はC3-10シクロアルキルである、
請求項1~5のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 R 1 is a hydrogen atom; halogen; cyano; C 1-6 alkyl; or C 3-10 cycloalkyl.
The compound according to any one of claims 1 to 5 or a pharmaceutically acceptable salt thereof. - Wが酸素原子である、
請求項1~6のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 W is an oxygen atom,
The compound according to any one of claims 1 to 6, or a pharmaceutically acceptable salt thereof. - Wが硫黄原子である、
請求項1~6のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 W is a sulfur atom,
The compound according to any one of claims 1 to 6, or a pharmaceutically acceptable salt thereof. - R2A、R2B、R2C、R2D及びR5が、すべて水素原子である、
請求項1~8のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 R 2A , R 2B , R 2C , R 2D and R 5 are all hydrogen atoms,
The compound according to any one of claims 1 to 8, or a pharmaceutically acceptable salt thereof. - R6が、水素原子である、
請求項1~9のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 R 6 is a hydrogen atom,
The compound according to any one of claims 1 to 9, or a pharmaceutically acceptable salt thereof. - R3Bが、水素原子であり、R3A及びR4Aが、同一又は異なって、フッ素、C1-6アルコキシ及びC1-6アルキルからなる群から独立して選択される1~5個の置換基で置換されていてもよいC3-10シクロアルキル;又は4~10員の飽和複素環である、
請求項1~10のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 1 to 5 substituents in which R 3B is a hydrogen atom, and R 3A and R 4A are the same or different and are independently selected from the group consisting of fluorine, C 1-6 alkoxy and C 1-6 alkyl A C 3-10 cycloalkyl optionally substituted with a group; or a 4-10 membered saturated heterocycle,
The compound according to any one of claims 1 to 10, or a pharmaceutically acceptable salt thereof. - X-Y-Zが、N-CO-NR3AR3B又はN-COR4Aである、
請求項1~11のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 XYZ is N—CO—NR 3A R 3B or N—COR 4A ;
The compound according to any one of claims 1 to 11, or a pharmaceutically acceptable salt thereof. - X-Y-Zが、N-CO-NR3AR3Bである、
請求項1~11のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 XYZ is N—CO—NR 3A R 3B ;
The compound according to any one of claims 1 to 11, or a pharmaceutically acceptable salt thereof. - X-Y-Zが、N-COR4Aである、
請求項1~11のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 XYZ is N-COR 4A ;
The compound according to any one of claims 1 to 11, or a pharmaceutically acceptable salt thereof. - Bが、B-1である、
請求項1~14のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 B is B-1.
The compound according to any one of claims 1 to 14, or a pharmaceutically acceptable salt thereof. - Bが、B-2である、
請求項1~14のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 B is B-2,
The compound according to any one of claims 1 to 14, or a pharmaceutically acceptable salt thereof. - Bが、B-3である、
請求項1~14のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 B is B-3,
The compound according to any one of claims 1 to 14, or a pharmaceutically acceptable salt thereof. - Bが、B-4である、
請求項1~14のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 B is B-4,
The compound according to any one of claims 1 to 14, or a pharmaceutically acceptable salt thereof. - Bが、B-5である、
請求項1~14のいずれか一項に記載の化合物又はその製薬学的に許容される塩。 B is B-5,
The compound according to any one of claims 1 to 14, or a pharmaceutically acceptable salt thereof. - 請求項1~19のいずれか一項に記載の化合物又はそれらの製薬学的に許容される塩を含有する医薬組成物。 A pharmaceutical composition comprising the compound according to any one of claims 1 to 19 or a pharmaceutically acceptable salt thereof.
- 請求項1~19のいずれか一項に記載の化合物又はそれらの製薬学的に許容される塩を有効成分とする、アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患の治療剤及び/又は予防剤。 A therapeutic agent for a disease caused by an abnormality in intracellular signal transduction involving acetylcholine, comprising the compound according to any one of claims 1 to 19 or a pharmaceutically acceptable salt thereof as an active ingredient, and / or Or a preventive agent.
- アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患が、神経疾患、精神疾患又は炎症性疾患である、請求項21に記載の治療剤及び/又は予防剤。 The therapeutic and / or prophylactic agent according to claim 21, wherein the disease caused by abnormality in intracellular signal transduction involving acetylcholine is a neurological disease, a mental disease or an inflammatory disease.
- 神経疾患、精神疾患又は炎症性疾患が、統合失調症、アルツハイマー病、ダウン症、注意欠陥・多動性障害又は脳血管アンギオパチーである、請求項22に記載の治療剤及び/又は予防剤。 The therapeutic agent and / or preventive agent according to claim 22, wherein the neurological disease, psychiatric disease or inflammatory disease is schizophrenia, Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder or cerebrovascular angiopathy.
- CIAS(統合失調症に伴う認知機能障害)、或いは、統合失調症、アルツハイマー病、ダウン症、注意欠陥・多動性障害又は脳血管アンギオパチーにおける、認知障害、軽度認知障害、記憶障害又は学習障害を治療及び/又は予防するための、請求項20に記載の医薬組成物。 Treat cognitive impairment, mild cognitive impairment, memory impairment or learning impairment in CIAS (cognitive impairment associated with schizophrenia) or schizophrenia, Alzheimer's disease, Down's syndrome, attention deficit / hyperactivity disorder or cerebrovascular angiopathy 21. A pharmaceutical composition according to claim 20, for and / or prevention.
- 請求項1~19のいずれか一項に記載の化合物又はそれらの製薬学的に許容される塩と、非定型抗精神病薬から選択される少なくとも1種以上の薬剤とを含有する医薬。 A medicament comprising the compound according to any one of claims 1 to 19, or a pharmaceutically acceptable salt thereof, and at least one drug selected from atypical antipsychotic drugs.
- 治療が必要な患者に、治療上の有効量の請求項1~19のいずれか一項に記載の化合物又はその製薬学的に許容される塩を投与することを特徴とする、アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患を治療及び/又は予防するための方法。 Involves acetylcholine, characterized in that a patient in need of treatment is administered a therapeutically effective amount of a compound according to any one of claims 1 to 19 or a pharmaceutically acceptable salt thereof. A method for treating and / or preventing a disease caused by abnormal intracellular signaling.
- アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患の治療剤及び/又は予防剤を製造するための、請求項1~19のいずれか一項に記載の化合物又はその製薬学的に許容される塩の使用。 The compound according to any one of claims 1 to 19 or a pharmaceutically acceptable salt thereof for the manufacture of a therapeutic and / or prophylactic agent for a disease caused by abnormal intracellular signaling involving acetylcholine. Use of salt.
- アセチルコリンが関与する細胞内シグナル伝達の異常に起因する疾患の治療に使用するための、請求項1~19のいずれか一項に記載の化合物又はその製薬学的に許容される塩を含む医薬組成物。 A pharmaceutical composition comprising the compound according to any one of claims 1 to 19 or a pharmaceutically acceptable salt thereof for use in the treatment of a disease caused by abnormal intracellular signaling involving acetylcholine. object.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-075265 | 2014-04-01 | ||
| JP2014075265A JP2017100949A (en) | 2014-04-01 | 2014-04-01 | 5-membered heteroaryl derivative |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015152254A1 true WO2015152254A1 (en) | 2015-10-08 |
Family
ID=54240573
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/060155 WO2015152254A1 (en) | 2014-04-01 | 2015-03-31 | Five-membered ring heteroaryl derivative |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2017100949A (en) |
| WO (1) | WO2015152254A1 (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7460649B2 (en) | 2019-03-12 | 2024-04-02 | サイティアー セラピューティクス,インコーポレイティド | RAD51 inhibitor |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993020061A1 (en) * | 1992-04-01 | 1993-10-14 | The University Of Toledo | 4-[4'-piperidinyl or 3'-pirrolidinyl] substituted imidazoles as h3-receptor antagonists and therapeutic uses thereof |
| WO1996016049A1 (en) * | 1994-11-23 | 1996-05-30 | Glaxo Wellcome Inc. | OXAZOLE DERIVATIVES AS ANTAGONISTS OF α-1C ADRENERGIC RECEPTORS |
| WO2007070433A2 (en) * | 2005-12-12 | 2007-06-21 | Merck & Co., Inc. | 2-arylthiazole derivatives as cxcr3 receptor modulators |
| WO2013083741A1 (en) * | 2011-12-08 | 2013-06-13 | Boehringer Ingelheim International Gmbh | 1,2,4 -triazoles as allosteric modulators of mglu5 receptor activity for the treatment of schizophrenia of dementia |
| WO2014054635A1 (en) * | 2012-10-02 | 2014-04-10 | 大日本住友製薬株式会社 | Imidazole derivative |
-
2014
- 2014-04-01 JP JP2014075265A patent/JP2017100949A/en active Pending
-
2015
- 2015-03-31 WO PCT/JP2015/060155 patent/WO2015152254A1/en active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993020061A1 (en) * | 1992-04-01 | 1993-10-14 | The University Of Toledo | 4-[4'-piperidinyl or 3'-pirrolidinyl] substituted imidazoles as h3-receptor antagonists and therapeutic uses thereof |
| WO1996016049A1 (en) * | 1994-11-23 | 1996-05-30 | Glaxo Wellcome Inc. | OXAZOLE DERIVATIVES AS ANTAGONISTS OF α-1C ADRENERGIC RECEPTORS |
| WO2007070433A2 (en) * | 2005-12-12 | 2007-06-21 | Merck & Co., Inc. | 2-arylthiazole derivatives as cxcr3 receptor modulators |
| WO2013083741A1 (en) * | 2011-12-08 | 2013-06-13 | Boehringer Ingelheim International Gmbh | 1,2,4 -triazoles as allosteric modulators of mglu5 receptor activity for the treatment of schizophrenia of dementia |
| WO2014054635A1 (en) * | 2012-10-02 | 2014-04-10 | 大日本住友製薬株式会社 | Imidazole derivative |
Non-Patent Citations (4)
| Title |
|---|
| CARROLL F. I.: "Epibatidine structure-activity relationships", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 14, no. 8, 2004, pages 1889 - 96, XP009082279 * |
| DATABASE REGISTRY [o] 25 March 2010 (2010-03-25), retrieved from STN Database accession no. 1214439-84-5 * |
| DATABASE REGISTRY 5 July 2012 (2012-07-05), Database accession no. 1381644-38-7 * |
| FLIPO M. ET AL.: "Ethionamide Boosters. 2. Combining Bioisosteric Replacement and Structure-Based Drug Design To Solve Pharmacokinetic Issues in a Series of Potent 1,2,4-Oxadiazole EthR Inhibitors", JOURNAL OF MEDICINAL CHEMISTRY, vol. 55, no. 1, 2012, pages 68 - 83, XP055068481 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2017100949A (en) | 2017-06-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10160753B2 (en) | Indazole compounds as IRAK4 inhibitors | |
| WO2012176763A1 (en) | Novel indazole derivative | |
| JP5586632B2 (en) | Azaspiranyl-alkylcarbamate derivatives of 5-membered heterocyclic compounds, their preparation and therapeutic use | |
| JP6088491B2 (en) | New 1-substituted indazole derivatives | |
| WO2001058900A1 (en) | 1h-imidazopyridine derivatives | |
| WO2014054635A1 (en) | Imidazole derivative | |
| CA2730793A1 (en) | Azole compound | |
| WO2012133509A1 (en) | Novel benzimidazole derivative | |
| JP5560287B2 (en) | Alkyl-heterocyclic carbamate derivatives, their preparation and their use | |
| JP2016028016A (en) | Oxadiazole derivatives and pharmaceutical uses thereof | |
| WO2017073743A1 (en) | Tricyclic compound | |
| WO2017018475A1 (en) | Condensed pyrazole derivative having new linker site and medicinal use of same | |
| WO2014054634A1 (en) | Pyrimidine derivative | |
| WO2015152254A1 (en) | Five-membered ring heteroaryl derivative | |
| JP2015199722A (en) | Medicine comprising imidazole derivative | |
| KR20240017785A (en) | Novel (homo)piperidinyl heterocycle as a sigma ligand | |
| JP2014073982A (en) | Pharmaceutical containing novel benzimidazole derivative | |
| KR101337163B1 (en) | Quinazoline derivatives as nk3 receptor antagonists | |
| WO2014038623A1 (en) | 3-(4-piperidyl)-indazole derivative | |
| JP6088476B2 (en) | A pharmaceutical comprising a novel 1-substituted indazole derivative | |
| WO2015152253A1 (en) | 4-aryl imidazole derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15772675 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15772675 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |