WO2015083571A1 - Carbon catalyst and production method therefor as well as electrode and cell using such carbon catalyst - Google Patents
Carbon catalyst and production method therefor as well as electrode and cell using such carbon catalyst Download PDFInfo
- Publication number
- WO2015083571A1 WO2015083571A1 PCT/JP2014/081010 JP2014081010W WO2015083571A1 WO 2015083571 A1 WO2015083571 A1 WO 2015083571A1 JP 2014081010 W JP2014081010 W JP 2014081010W WO 2015083571 A1 WO2015083571 A1 WO 2015083571A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- carbon catalyst
- electrode
- metal
- carbonized material
- carbon
- Prior art date
Links
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 title claims abstract description 92
- 239000003054 catalyst Substances 0.000 title claims abstract description 71
- 229910052799 carbon Inorganic materials 0.000 title claims abstract description 54
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 16
- 239000005539 carbonized material Substances 0.000 claims abstract description 74
- 229910052751 metal Inorganic materials 0.000 claims abstract description 54
- 239000002184 metal Substances 0.000 claims abstract description 54
- 238000002441 X-ray diffraction Methods 0.000 claims abstract description 23
- 239000002994 raw material Substances 0.000 claims description 51
- 238000000034 method Methods 0.000 claims description 44
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 34
- 239000001301 oxygen Substances 0.000 claims description 34
- 229910052760 oxygen Inorganic materials 0.000 claims description 34
- 238000010000 carbonizing Methods 0.000 claims description 18
- 239000004071 soot Substances 0.000 claims description 14
- 238000003763 carbonization Methods 0.000 abstract description 28
- 230000003197 catalytic effect Effects 0.000 abstract description 10
- 150000001721 carbon Chemical class 0.000 abstract 1
- 239000007858 starting material Substances 0.000 abstract 1
- XMWRBQBLMFGWIX-UHFFFAOYSA-N C60 fullerene Chemical compound C12=C3C(C4=C56)=C7C8=C5C5=C9C%10=C6C6=C4C1=C1C4=C6C6=C%10C%10=C9C9=C%11C5=C8C5=C8C7=C3C3=C7C2=C1C1=C2C4=C6C4=C%10C6=C9C9=C%11C5=C5C8=C3C3=C7C1=C1C2=C4C6=C2C9=C5C3=C12 XMWRBQBLMFGWIX-UHFFFAOYSA-N 0.000 description 28
- 229910003472 fullerene Inorganic materials 0.000 description 28
- 230000000052 comparative effect Effects 0.000 description 25
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 16
- 239000000126 substance Substances 0.000 description 15
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 13
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 12
- 239000000463 material Substances 0.000 description 12
- 239000002904 solvent Substances 0.000 description 10
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- 230000010757 Reduction Activity Effects 0.000 description 8
- 229910052697 platinum Inorganic materials 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 7
- 230000004913 activation Effects 0.000 description 7
- 239000000446 fuel Substances 0.000 description 7
- 238000010438 heat treatment Methods 0.000 description 7
- 239000012299 nitrogen atmosphere Substances 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical group [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 6
- 229910021529 ammonia Inorganic materials 0.000 description 6
- 239000006227 byproduct Substances 0.000 description 6
- 229910052742 iron Inorganic materials 0.000 description 6
- 238000002156 mixing Methods 0.000 description 6
- 125000004433 nitrogen atom Chemical group N* 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- 238000001228 spectrum Methods 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 4
- 238000004140 cleaning Methods 0.000 description 4
- 239000001257 hydrogen Substances 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- IEQIEDJGQAUEQZ-UHFFFAOYSA-N phthalocyanine Chemical compound N1C(N=C2C3=CC=CC=C3C(N=C3C4=CC=CC=C4C(=N4)N3)=N2)=C(C=CC=C2)C2=C1N=C1C2=CC=CC=C2C4=N1 IEQIEDJGQAUEQZ-UHFFFAOYSA-N 0.000 description 4
- 229920005989 resin Polymers 0.000 description 4
- 239000011347 resin Substances 0.000 description 4
- 230000002441 reversible effect Effects 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 3
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 3
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- 229910003481 amorphous carbon Inorganic materials 0.000 description 3
- 239000003575 carbonaceous material Substances 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 239000011261 inert gas Substances 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 239000007791 liquid phase Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 229910017604 nitric acid Inorganic materials 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 230000000737 periodic effect Effects 0.000 description 3
- 239000005518 polymer electrolyte Substances 0.000 description 3
- 238000010298 pulverizing process Methods 0.000 description 3
- 239000002002 slurry Substances 0.000 description 3
- 229910021642 ultra pure water Inorganic materials 0.000 description 3
- 239000012498 ultrapure water Substances 0.000 description 3
- 239000008096 xylene Substances 0.000 description 3
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 229920000877 Melamine resin Polymers 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 229920002845 Poly(methacrylic acid) Polymers 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- 229910052796 boron Inorganic materials 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 238000005229 chemical vapour deposition Methods 0.000 description 2
- 239000011651 chromium Substances 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 239000008151 electrolyte solution Substances 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 239000011572 manganese Substances 0.000 description 2
- 229910044991 metal oxide Inorganic materials 0.000 description 2
- 150000004706 metal oxides Chemical class 0.000 description 2
- 229910052976 metal sulfide Inorganic materials 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- 239000010955 niobium Substances 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- -1 phosphino group Chemical group 0.000 description 2
- 125000004437 phosphorous atom Chemical group 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 239000011148 porous material Substances 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- XSCHRSMBECNVNS-UHFFFAOYSA-N quinoxaline Chemical compound N1=CC=NC2=CC=CC=C21 XSCHRSMBECNVNS-UHFFFAOYSA-N 0.000 description 2
- 239000010948 rhodium Substances 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 239000010936 titanium Substances 0.000 description 2
- 229910052723 transition metal Inorganic materials 0.000 description 2
- 150000003624 transition metals Chemical class 0.000 description 2
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 1
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- VEUMANXWQDHAJV-UHFFFAOYSA-N 2-[2-[(2-hydroxyphenyl)methylideneamino]ethyliminomethyl]phenol Chemical compound OC1=CC=CC=C1C=NCCN=CC1=CC=CC=C1O VEUMANXWQDHAJV-UHFFFAOYSA-N 0.000 description 1
- KXGFMDJXCMQABM-UHFFFAOYSA-N 2-methoxy-6-methylphenol Chemical compound [CH]OC1=CC=CC([CH])=C1O KXGFMDJXCMQABM-UHFFFAOYSA-N 0.000 description 1
- LXBGSDVWAMZHDD-UHFFFAOYSA-N 2-methyl-1h-imidazole Chemical compound CC1=NC=CN1 LXBGSDVWAMZHDD-UHFFFAOYSA-N 0.000 description 1
- KGIGUEBEKRSTEW-UHFFFAOYSA-N 2-vinylpyridine Chemical compound C=CC1=CC=CC=N1 KGIGUEBEKRSTEW-UHFFFAOYSA-N 0.000 description 1
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 1
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 1
- 239000002028 Biomass Substances 0.000 description 1
- UJOBWOGCFQCDNV-UHFFFAOYSA-N Carbazole Natural products C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 229910052684 Cerium Inorganic materials 0.000 description 1
- 229920002101 Chitin Polymers 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 1
- 239000004640 Melamine resin Substances 0.000 description 1
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 1
- 229920000557 Nafion® Polymers 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 239000004696 Poly ether ether ketone Substances 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 239000004962 Polyamide-imide Substances 0.000 description 1
- 239000004693 Polybenzimidazole Substances 0.000 description 1
- 239000004642 Polyimide Substances 0.000 description 1
- 239000004721 Polyphenylene oxide Substances 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 229920001328 Polyvinylidene chloride Polymers 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 241000234314 Zingiber Species 0.000 description 1
- 235000006886 Zingiber officinale Nutrition 0.000 description 1
- 229910052768 actinide Inorganic materials 0.000 description 1
- 150000001255 actinides Chemical class 0.000 description 1
- IBVAQQYNSHJXBV-UHFFFAOYSA-N adipic acid dihydrazide Chemical compound NNC(=O)CCCCC(=O)NN IBVAQQYNSHJXBV-UHFFFAOYSA-N 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 238000001241 arc-discharge method Methods 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 239000003849 aromatic solvent Substances 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- HCOMFAYPHBFMKU-UHFFFAOYSA-N butanedihydrazide Chemical compound NNC(=O)CCC(=O)NN HCOMFAYPHBFMKU-UHFFFAOYSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000006229 carbon black Substances 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- GWXLDORMOJMVQZ-UHFFFAOYSA-N cerium Chemical compound [Ce] GWXLDORMOJMVQZ-UHFFFAOYSA-N 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 238000009841 combustion method Methods 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 150000004696 coordination complex Chemical class 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003822 epoxy resin Substances 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000007849 furan resin Substances 0.000 description 1
- 235000008397 ginger Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229910021397 glassy carbon Inorganic materials 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 229910052747 lanthanoid Inorganic materials 0.000 description 1
- 150000002602 lanthanoids Chemical class 0.000 description 1
- 229920005610 lignin Polymers 0.000 description 1
- 239000003077 lignite Substances 0.000 description 1
- 238000004502 linear sweep voltammetry Methods 0.000 description 1
- 229910052748 manganese Inorganic materials 0.000 description 1
- JDSHMPZPIAZGSV-UHFFFAOYSA-N melamine Chemical compound NC1=NC(N)=NC(N)=N1 JDSHMPZPIAZGSV-UHFFFAOYSA-N 0.000 description 1
- 150000001247 metal acetylides Chemical class 0.000 description 1
- 150000002736 metal compounds Chemical class 0.000 description 1
- 229910000000 metal hydroxide Inorganic materials 0.000 description 1
- 150000004692 metal hydroxides Chemical class 0.000 description 1
- 229940117841 methacrylic acid copolymer Drugs 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- 239000011733 molybdenum Substances 0.000 description 1
- 239000004570 mortar (masonry) Substances 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229910052758 niobium Inorganic materials 0.000 description 1
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 1
- 150000004767 nitrides Chemical class 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 229920000368 omega-hydroxypoly(furan-2,5-diylmethylene) polymer Polymers 0.000 description 1
- 239000011368 organic material Substances 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 229920001568 phenolic resin Polymers 0.000 description 1
- 239000005011 phenolic resin Substances 0.000 description 1
- 230000010287 polarization Effects 0.000 description 1
- 229920000962 poly(amidoamine) Polymers 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920002492 poly(sulfone) Polymers 0.000 description 1
- 229920002037 poly(vinyl butyral) polymer Polymers 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 229920002239 polyacrylonitrile Polymers 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 229920002312 polyamide-imide Polymers 0.000 description 1
- 229920000767 polyaniline Polymers 0.000 description 1
- 229920002480 polybenzimidazole Polymers 0.000 description 1
- 229920001088 polycarbazole Polymers 0.000 description 1
- 229920000647 polyepoxide Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 229920002530 polyetherether ketone Polymers 0.000 description 1
- 229920001721 polyimide Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920000128 polypyrrole Polymers 0.000 description 1
- 239000004814 polyurethane Substances 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 239000005033 polyvinylidene chloride Substances 0.000 description 1
- 229920002717 polyvinylpyridine Polymers 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- 229910052707 ruthenium Inorganic materials 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 229910052706 scandium Inorganic materials 0.000 description 1
- SIXSYDAISGFNSX-UHFFFAOYSA-N scandium atom Chemical compound [Sc] SIXSYDAISGFNSX-UHFFFAOYSA-N 0.000 description 1
- VSZWPYCFIRKVQL-UHFFFAOYSA-N selanylidenegallium;selenium Chemical compound [Se].[Se]=[Ga].[Se]=[Ga] VSZWPYCFIRKVQL-UHFFFAOYSA-N 0.000 description 1
- 238000000967 suction filtration Methods 0.000 description 1
- JBQYATWDVHIOAR-UHFFFAOYSA-N tellanylidenegermanium Chemical compound [Te]=[Ge] JBQYATWDVHIOAR-UHFFFAOYSA-N 0.000 description 1
- 229920005992 thermoplastic resin Polymers 0.000 description 1
- 229920001187 thermosetting polymer Polymers 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 1
- LEONUFNNVUYDNQ-UHFFFAOYSA-N vanadium atom Chemical compound [V] LEONUFNNVUYDNQ-UHFFFAOYSA-N 0.000 description 1
- 239000012808 vapor phase Substances 0.000 description 1
- 238000001075 voltammogram Methods 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 description 1
Images


Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/18—Carbon
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/74—Iron group metals
- B01J23/745—Iron
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
- B01J37/082—Decomposition and pyrolysis
- B01J37/084—Decomposition of carbon-containing compounds into carbon
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/86—Inert electrodes with catalytic activity, e.g. for fuel cells
- H01M4/90—Selection of catalytic material
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M2008/1095—Fuel cells with polymeric electrolytes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/30—Hydrogen technology
- Y02E60/50—Fuel cells
Definitions
- the present invention relates to a carbon catalyst and a method for producing the same, and an electrode and a battery using the carbon catalyst.
- the catalytic activity for example, oxygen reduction activity
- a conventional carbon catalyst not using platinum is not always sufficient.
- the present invention has been made in view of the above problems, and an object of the present invention is to provide a carbon catalyst exhibiting improved catalytic activity, a method for producing the same, and an electrode and a battery using the carbon catalyst.
- a carbon catalyst according to an embodiment of the present invention for solving the above-described problems is obtained by using a diffraction angle of 18 with respect to a peak maximum intensity (I F ) at a diffraction angle of 11.5 ° to 15.0 ° in an X-ray diffraction method. It has a carbon structure in which the ratio (I G / I F ) of the peak maximum intensity (I G ) at 0.0 ° to 26.5 ° is 0.8 or more and 11 or less.
- the carbon catalyst may include a carbonized material obtained by carbonizing a raw material containing fullerene soot and a metal.
- a carbon catalyst according to an embodiment of the present invention for solving the above problems includes a carbonized material obtained by carbonizing a raw material containing fullerene soot and a metal.
- Any of the above carbon catalysts may be a carbon catalyst having an oxygen reduction starting potential of 0.60 V (vs. RHE) or more.
- An electrode according to an embodiment of the present invention for solving the above problems includes any one of the above carbon catalysts.
- a battery according to an embodiment of the present invention for solving the above problems includes the electrode.
- a method for producing a catalyst according to an embodiment of the present invention for solving the above-mentioned problem is to obtain a carbonized material by carbonizing a raw material containing fullerene soot and a metal, and a carbon catalyst containing the carbonized material. Including.
- FIG. 3 is an X-ray diffraction pattern of a carbonized material according to Comparative Example 1. It is a figure after correcting a background about the X-ray diffraction pattern of the carbonization material concerning one embodiment of the present invention. It is the figure after correcting a background about the X-ray-diffraction pattern of the carbonization material which concerns on the comparative example 1.
- FIG. It is a table
- This method is a method including carbonizing a raw material containing a fullerene soot and a metal to obtain a carbonized material, and producing a carbon catalyst containing the carbonized material.
- a raw material containing fullerene soot and metal is prepared. That is, at least the fuller soot and the metal are mixed.
- the mixing method is not particularly limited, and for example, one or more mixing methods such as powder mixing using a mortar or stirring device and powder mixing for adding a solvent and solvent mixing for adding a solvent can be used.
- Fullerene soot is a component other than the extracted fullerene (fullerene dissolved in the solvent) obtained when fullerene is extracted from the fullerene-containing soot obtained in the production of fullerene using a solvent (hereinafter referred to as “fullerene” in the present invention).
- a carbon material comprising a molecule derived from "byproduct” and having a structure in which five-membered and six-membered rings of carbon atoms are arranged three-dimensionally and does not form a closed space It is.
- the fullerene soot is a carbon material containing an amorphous carbon structure, and the amorphous carbon structure may contain the molecule derived from fullerene byproduct.
- the fullerene soot may be a carbon material including, for example, an amorphous carbon structure including the molecule derived from the fullerene byproduct ginger and a random gradual carbon network structure. .
- the above-mentioned fullerene byproduct koji is, for example, the solvent other than the extracted fullerene (fullerene dissolved in the solvent) obtained when fullerene is extracted from the fullerene-containing koji obtained in the production of fullerene using a solvent. It is good also as a component which did not melt
- the method for producing the fullerene is not particularly limited as long as the fullerene-containing soot is obtained.
- the fullerene-containing method is at least one selected from the group consisting of an arc discharge method, a resistance heating method, a laser evaporation method, and a combustion method. It is good as well.
- the solvent used when extracting fullerene from fullerene-containing soot is not particularly limited as long as it can extract fullerene from fullerene-containing soot to obtain fullerene by-product soot.
- it is an organic solvent.
- the organic solvent may be, for example, an aromatic solvent, and more specifically, for example, toluene and / or xylene.
- the fullerene by-product is an extracted fullerene (fullerene dissolved in toluene and / or xylene) obtained when fullerene is extracted from the fullerene-containing soot obtained in the production of fullerene using toluene and / or xylene. It is good also as being components other than.
- the amount of fullerene soot contained in the raw material is not particularly limited.
- the weight ratio of the fullerene soot to the raw material may be 40 wt% or more, or may be 50 wt% or more. Moreover, it is good also as being 99.5 wt% or less. That is, the weight ratio of the fullerene soot to the raw material may be 40 to 99.5 wt%, 50 to 99.5 wt%, or 50 to 80 wt%.
- the metal contained in the raw material is not particularly limited as long as it does not inhibit the activity of the carbon catalyst obtained by this method. That is, for example, one or more metals selected from the group consisting of Groups 3 to 16 of the periodic table may be used.
- Group 3A Group 3) element, Group 4A (Group 4) element, Group 5A (Group 5) element, Group 6A (Group 6) element, Group 7A (Group 7) element, Group 8 (8 Group, Group 9 and 10) element, Group 1B (Group 11) element, Group 2B (Group 12) element, Group 3B (Group 13) element, Group 4B (Group 14) element, Group 5B (Group 15) element and
- 6B group (16 group) elements can be used, transition metals (groups 3 to 12 of the periodic table) can be preferably used, from group 3 of the periodic table Transition metals belonging to the fourth period of Group 12 can be used more preferably.
- the metal can be used as a simple substance of the metal or a compound of the metal.
- the metal compound for example, one or more selected from the group consisting of metal salts, metal oxides, metal hydroxides, metal nitrides, metal sulfides, metal carbides and metal complexes can be used.
- One or more selected from the group consisting of salts, metal oxides, metal sulfides, and metal complexes can be preferably used.
- the raw material contains a ligand capable of coordinating to a metal
- a metal complex formed by the metal and the ligand is formed in the raw material.
- the amount of metal contained in the raw material is not particularly limited, for example, the weight ratio of the metal with respect to the raw material may be 0.5 to 10 wt% or 2 to 5 wt%.
- the amount of fullerene soot and metal contained in the raw material is not particularly limited.
- the raw material is a raw material containing 50-80 wt% fullerene soot and 2-5 wt% metal. Also good.
- the raw material may further contain other components. That is, the raw material may include, for example, an organic material.
- the present method includes a carbonized material obtained by carbonizing a raw material containing fullerene soot, a metal, and an organic substance.
- the organic substance contained in the raw material is not particularly limited as long as it can be carbonized. That is, for example, one or both of a high molecular weight organic substance (for example, a resin such as a thermosetting resin or a thermoplastic resin) and a low molecular weight organic substance can be used. Biomass can also be used.
- the weight ratio of fullerene soot, metal, and organic substance contained in the raw material is not particularly limited.
- the raw material may be a raw material containing 50-80 wt% fuller soot, 2-5 wt% of the metal, and 18-45 wt% of the organic substance.
- a ligand capable of coordinating with a metal can be preferably used. That is, in this case, an organic substance containing one or more coordination atoms in the molecule is used. More specifically, for example, as a coordination atom, an organic substance containing one or more selected from the group consisting of a nitrogen atom, a phosphorus atom, an oxygen atom and a sulfur atom in the molecule can be used. In addition, for example, an organic substance containing one or more selected from the group consisting of an amino group, a phosphino group, a carboxyl group, and a thiol group in the molecule can be used as a coordination group.
- the organic substance contains, for example, at least one selected from the group consisting of a nitrogen atom, a boron atom, a phosphorus atom, an oxygen atom, and a sulfur atom as a component that improves the activity of the carbon catalyst produced by this method. You can also.
- examples of the organic substance include acrylonitrile, polyacrylonitrile, melamine, melamine resin, pyrrole, polypyrrole, 3-methylpolypyrrole, polyvinylpyrrole, thiazole, pyrazole, vinylpyridine, polyvinylpyridine, pyridazine, pyrimidine, piperazine, imidazole, 1-methylimidazole, 2-methylimidazole, quinoxaline, aniline, polyaniline, benzimidazole, polybenzimidazole, hydrazine, polycarbazole, triazine, polycarbodiimide, chelate resin, polyamideimide resin, polyacrylonitrile-poly Methacrylic acid copolymer, oxazole, morpholine, succinic dihydrazide, adipic dihydrazide, polybismaleimide, polyaminobismale , Polyimide, polyacrylamide, polyamide, chitin, chito-vin
- This method includes carbonizing the above-described raw material to obtain a carbonized material.
- Carbonization of a raw material is performed by heating the above-mentioned raw material and holding at a predetermined temperature (carbonization temperature) at which the raw material can be carbonized.
- the carbonization temperature is not particularly limited as long as the raw material can be carbonized, and may be, for example, 300 ° C. or higher. More specifically, the carbonization temperature may be, for example, 300 ° C. or higher and 1500 ° C. or lower.
- the heating rate when heating the raw material to the carbonization temperature is not particularly limited, and may be, for example, 0.5 ° C./min or more and 300 ° C./min or less.
- the time for holding the raw material at the carbonization temperature is not particularly limited as long as the raw material can be carbonized, and may be, for example, 5 minutes or more, or 5 minutes or more and 240 minutes or less.
- Carbonization is preferably performed under an inert gas such as nitrogen (for example, under the flow of an inert gas).
- a carbonized material generated by carbonizing the above-described raw material is obtained.
- the obtained carbonized material may be pulverized.
- the method for pulverizing the carbonized material is not particularly limited, and for example, a pulverizing apparatus such as a ball mill or a bead mill can be used.
- the average particle size of the carbonized material after pulverization can be, for example, 150 ⁇ m or less, and preferably 100 ⁇ m or less.
- the carbonized material obtained by carbonizing the above raw materials has catalytic activity (for example, oxygen reduction activity). That is, the carbon catalyst manufactured by this method should just contain the carbonization material obtained by carbonizing the above-mentioned raw material, for example, it is good also as obtaining the said carbonization material itself as a carbon catalyst.
- the present method may include carbonizing a raw material containing fullerene soot and a metal to obtain a carbonized material, and obtaining the carbonized material as a carbon catalyst.
- the carbonized material generated by carbonization may be further treated to obtain the carbonized material subjected to the treatment as a carbon catalyst.
- a pulverized carbonized material that has been treated may be obtained as a carbon catalyst.
- nitrogen atoms and / or boron atoms may be introduced (doped) into the carbonized material in an arbitrary step. That is, in this method, for example, nitrogen atoms and / or boron atoms are introduced into one or more of a carbonized material obtained by carbonizing a raw material and a carbonized material after metal removal treatment described later, It is good also as manufacturing the carbon catalyst containing a carbonization material.
- a method for introducing nitrogen atoms and / or boron atoms for example, a vapor phase doping method such as an ammoxidation method or a CVD (Chemical Vapor Deposition) method, a liquid phase doping method, or a gas phase-liquid phase doping method is used. be able to.
- the carbonized material is heated to 400 ° C. or more and 1200 ° C. or less in an inert gas atmosphere such as nitrogen, argon, or helium, and then a nitrogen source such as ammonia or a boron source such as boron chloride.
- nitrogen atoms and / or boron atoms can be introduced into the carbonized material by holding at a temperature of 400 ° C. or higher and 1200 ° C. or lower for 5 minutes to 180 minutes. Further, the obtained carbonized material is subjected to carbon dioxide activation, phosphoric acid activation, alkali activation, ammonia activation, activation with nitric oxide, activation treatment such as electrolytic activation, and / or liquid phase oxidation such as mixed acid oxidation and hydrogen peroxide oxidation. Can also be applied.
- the carbonization material obtained by the above-described carbonization may be subjected to metal removal treatment.
- the metal removal process is a process for removing the metal contained in the carbonized material.
- the metal removal treatment is not particularly limited as long as it can remove the metal contained in the carbonized material or reduce the amount of the metal, and for example, an acid cleaning treatment or an electrolytic treatment can be performed.
- the acid used for the acid cleaning treatment is not particularly limited as long as the effect of the metal removal treatment can be obtained, and any one or more of them can be used. That is, for example, it is possible to use one or more selected from the group consisting of hydrochloric acid (eg, dilute hydrochloric acid and concentrated hydrochloric acid), nitric acid (eg, dilute nitric acid and concentrated nitric acid) and sulfuric acid (eg, dilute sulfuric acid and concentrated sulfuric acid). it can.
- the method of the acid cleaning treatment is not particularly limited, and for example, a method of immersing and holding the carbonized material in an acid-containing solution and / or stirring can be preferably used.
- the present catalyst In the X-ray diffraction method, the present catalyst has a peak maximum intensity (I F ) at a diffraction angle of 18.0 ° to 26.5 ° with respect to a peak maximum intensity (I F ) at a diffraction angle of 11.5 ° to 15.0 °.
- the above-mentioned I G / IF ratio is not particularly limited as long as it is 0.8 or more and 11 or less, but may be 0.8 or more and 7 or less, for example, 0.8 or more and 5 or less. It may be, may be 0.9 or more and 11 or less, may be 0.9 or more and 7 or less, and may be 0.9 or more and 5 or less. Also in Examples 1 to 7 (I G / I F of 1 or more and 3 or less) described later, improved catalytic activity (oxygen reduction activity) was obtained.
- This catalyst has, for example, an oxygen reduction activity as one of the catalytic activities. That is, the present catalyst may be a carbon catalyst having a carbon structure in which the above-mentioned IG / IF ratio is 0.8 or more and 11 or less and having oxygen reduction activity in the X-ray diffraction method. Good.
- the oxygen reduction activity of the catalyst described above can be evaluated by the oxygen reduction start potential.
- the oxygen reduction start potential is, for example, data indicating the relationship between the voltage and current density obtained when the potential is swept and applied using a rotating ring disk electrode device having a working electrode coated with this catalyst (oxygen reduction voltammogram).
- the voltage (E O2 ) when a reduction current of ⁇ 10 ⁇ A / cm 2 flows is obtained.
- the oxygen reduction starting potential of the present catalyst may be 0.60 V (vs. RHE) or higher, or 0.80 V (vs. RHE) or higher.
- RHE is an abbreviation for Reversible Hydrogen Electrode and represents a reversible hydrogen electrode. That is, vs. RHE represents a potential measured with respect to a reversible hydrogen electrode.
- this catalyst has the carbon structure which shows the above-mentioned I G / IF ratio, a manufacturing method will not be restricted especially, For example, it can manufacture efficiently by the above-mentioned this method. That is, the present catalyst may contain a carbonized material obtained by carbonizing a raw material containing fullerene soot and a metal.
- the carbonized material contained in the present catalyst may contain a metal.
- the metal may be contained in the carbonized material. That is, the present catalyst includes a carbonized material obtained by carbonizing a raw material containing fullerene soot and a metal, and the carbonized material may include at least a metal inside.
- the carbonized material contained in the present catalyst may contain, for example, 0.01 to 10 wt% of metal or 0.01 to 5 wt% of metal.
- the content of the metal can be confirmed by a method such as elemental analysis.
- the present catalyst may be a carbon catalyst obtained, for example, by subjecting a carbonized material obtained by carbonizing a raw material to the above-described metal removal treatment.
- the carbonized material may be substantially free of metal, but the remaining metal may be included in the carbonized material.
- the electrode according to the present embodiment (hereinafter referred to as “main electrode”) is an electrode including the above-described catalyst. That is, this electrode is, for example, an electrode on which the present catalyst is supported. Specifically, the present electrode is, for example, an electrode having a predetermined electrode base material and the present catalyst supported on the electrode base material.
- the electrode can be, for example, a fuel cell electrode, and preferably a polymer electrolyte fuel cell (PEFC) electrode. Moreover, this electrode can be used as the electrode for air batteries, for example.
- the main electrode is a fuel cell electrode or an air cell electrode, the main electrode is used as a cathode (oxygen electrode).
- the battery according to the present embodiment is a battery including the present electrode.
- the battery can be, for example, a fuel cell, preferably PEFC.
- the battery can be an air battery, for example.
- the battery can be, for example, a fuel cell or an air battery including the electrode as one or both of a cathode and an anode.
- the battery includes the electrode as at least a cathode (oxygen electrode). Is preferred.
- a polymer electrolyte membrane and a cathode (oxygen electrode) and an anode (fuel electrode) formed on one side and the other side of the polymer electrolyte membrane are integrated, for example.
- a PEFC may be provided that includes a membrane / electrode assembly and includes the electrode on one or both of the cathode and the anode.
- the battery preferably includes the electrode at least on the cathode.
- a carbonized material was obtained in the same manner as in Example 1 except that fullerene soot and phthalocyanine iron were used so that the weight ratio of iron contained in the phthalocyanine iron to the fullerene soot was 10 wt%.
- Example 4 The carbonized material obtained in Example 1 was subjected to metal removal treatment by acid cleaning to obtain a carbonized material. That is, the carbonized material obtained in Example 1 was added to 1M hydrochloric acid and stirred at 70 ° C. using a stirrer for 2 hours. Next, the solution containing the carbonized material was suction filtered using a filter having a pore size of 0.1 ⁇ m and washed with distilled water. This operation was repeated three times. The collected carbonized material was dried under reduced pressure at 60 ° C. for 12 hours to obtain a carbonized material. [Example 4]
- Example 5 The raw material used in Example 1 was subjected to ammoxidation treatment to obtain a carbonized material. That is, 0.3 g of the raw material used in Example 1 was heated at a heating rate of 30 ° C./min in an image furnace in a nitrogen atmosphere. Then, after reaching 800 ° C., a mixed gas of ammonia and air (ammonia concentration 70%) was introduced into the image furnace and held for 2 hours. Thereafter, the inside of the furnace was again switched to a nitrogen atmosphere and held for 10 minutes to obtain a carbonized material. [Example 5]
- Example 6 A carbonized material was obtained in the same manner as in Example 1 except that the carbonization temperature was 700 ° C.
- a carbonized material was obtained in the same manner as in Example 1 except that the carbonization temperature was 900 ° C. [Example 7]
- a carbonized material was obtained in the same manner as in Example 1 except that the carbonization temperature was 1000 ° C. [Comparative Example 1]
- Example 2 The fuller soot used in Example 1 (nanom black ST, Frontier Carbon Co.) itself was used as the sample of Comparative Example 1. [Comparative Example 2]
- Fullerene soot was carbonized at 400 ° C. in a nitrogen atmosphere to obtain a carbonized material. That is, 0.3 g of fullerene soot was heated in an image furnace in a nitrogen atmosphere at a heating rate of 30 ° C./min. And the carbonization was performed by hold
- the fullerence soot was subjected to ammoxidation treatment at 400 ° C. to obtain a carbonization catalyst. That is, 0.3 g of fuller soot (nanom black ST, Frontier Carbon) was heated in an image furnace in a nitrogen atmosphere at a heating rate of 30 ° C./min. Then, after reaching 400 ° C., a mixed gas of ammonia and air (ammonia concentration 70%) was introduced into the image furnace and held for 2 hours. Thereafter, the inside of the furnace was again switched to a nitrogen atmosphere and held for 10 minutes to obtain a carbonized material. [Comparative Example 6]
- a carbonized material was obtained in the same manner as in Comparative Example 5 except that the ammoxidation treatment was performed at 600 ° C.
- a catalyst slurry was prepared. Specifically, 5 mg of the carbonized material obtained as described above was mixed with 50 ⁇ L of a binder solution (Nafion (registered trademark), DuPont Co., Ltd.), 150 ⁇ L of ethanol, 150 ⁇ L of ultrapure water, and 2 cups (about 15 grains) of spatula. Glass beads (diameter 1 mm) were added and sonicated for 20 minutes to obtain a catalyst slurry.
- a binder solution Nafion (registered trademark), DuPont Co., Ltd.
- 150 ⁇ L of ethanol 150 ⁇ L of ultrapure water
- 2 cups about 15 grains
- the catalyst slurry was sucked with a pipette and applied to the disk electrode (area 0.1256 cm 2 ) of the rotating ring disk electrode device (RRDE-3A Ver. 1.2S, manufactured by BAS Co., Ltd.).
- the working electrode was prepared by drying.
- a platinum electrode was used as the ring electrode
- glassy carbon was used as the counter electrode
- a reversible hydrogen electrode was used as the reference electrode.
- As the electrolyte solution a 0.5 M sulfuric acid aqueous solution was bubbled with oxygen at room temperature and saturated with oxygen.
- linear sweep voltammetry was performed using an electrochemical analyzer (CHI700E, ALS / DY2323, manufactured by BAS Co., Ltd.).
- the measurement was started.
- the working electrode was rotated at a rotation speed of 1500 rpm, and the potential was swept from 1 V (vs. RHE) to 0 V (vs. RHE) at a sweep speed of 1 mV / sec at 25 ° C. The value of the current flowing through the working electrode was measured.
- FIG. 1A, FIG. 1B, with reference to FIGS. 2A and 2B I G, illustrating a method of determining the I F from X-ray diffraction pattern obtained in the manner described above.
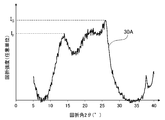
- FIG. 1A is a diagram showing the results of X-ray diffraction of the carbonized material obtained in Example 1, wherein the horizontal axis represents the diffraction angle 2 ⁇ (°) and the vertical axis represents the diffraction intensity (arbitrary unit).
- the horizontal axis represents the diffraction angle 2 ⁇ (°)
- the vertical axis represents the diffraction intensity (arbitrary unit).
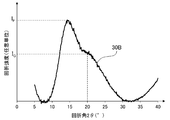
- FIG. 2A is a diagram showing a peak spectrum 30A obtained from the X-ray diffraction pattern of FIG. 1A.
- the maximum intensity of a peak definitive range diffraction angle 2 ⁇ of 11.5 ⁇ 15.0 ° and I F, the maximum peak in the range of 18.0 ⁇ 26.5 ° strength was I G.
- FIG. 1B is a diagram showing the results of X-ray diffraction of Comparative Example 1.
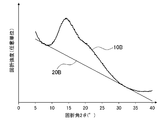
- the X-ray diffraction pattern 10B obtained by the X-ray diffraction method has a downwardly convex portion with a diffraction angle 2 ⁇ in the range of 5.5 to 12.5 °, and 30.5.
- a straight line 20B in contact with both of the downward convex portions in a range of ⁇ 37 ° was drawn, and the straight line was used as the background 20B of the X-ray diffraction pattern 10B.
- a peak spectrum 30B was created by subtracting the value of the background 20B from the X-ray diffraction pattern 10B.
- the peak spectrum 30B since the peak of the I G had become shoulder, the diffraction intensity at a diffraction angle 20 ° was I G.
- FIG. 3 shows the results of evaluating the carbon catalysts obtained in Examples 1 to 7 and Comparative Examples 1 to 6 by the above-described method. That is, in FIG. 3, each of the embodiments, the carbon catalyst obtained in Comparative Example, the oxygen reduction onset potential of the carbon catalyst (E O2) (V (vs.RHE )), of the carbon catalyst I G / IF ratio (-) is shown.
- E O2 oxygen reduction onset potential of the carbon catalyst
- V vs.RHE
- the carbon catalysts according to Examples 1 to 7 have an I G / IF ratio in the range of 1.0 to 3.0, and the oxygen reduction starting potential (E O2 ) is 0.80 V ( vs. RHE) or higher.
- the carbon catalyst according to Comparative Example 1 is I G / I F ratio is as low as 0.6, the oxygen reduction onset potential (E O2) includes a 0.27V (Vs.RHE) It was lower than Examples 1-7.
- the I G / IF ratio is 0.5 to 0.00. 7
- the oxygen reduction starting potential (E O2 ) was also as low as 0.39 to 0.47 V (vs. RHE).
- carbon catalyst I G / I F ratio is 1.0 or more, compared to the case I G / I F ratio is less than 1.0, and had a high oxygen reduction activity.
- carbonized material according to Comparative Example 6 is an I G / I F ratio 12, an oxygen reduction onset potential (E O2) was 0.52V (vs.RHE). Furthermore, carbonized material according to Comparative Example 5 is an I G / I F ratio 18.9, oxygen reduction onset potential (E O2), the carbon of Comparative Example 6 and 0.49V (vs.RHE) It was even lower than the material. Thus, the carbonized material having an I G / IF ratio of 12 or more had a lower oxygen reduction initiation potential (E O2 ) than that when the I G / IF ratio was less than 12.
- the carbonized material according to Example 3 has an oxygen reduction start potential (E O2 ) of 0.83 V (vs. RHE), and the carbonized material according to Example 1 (oxygen reduction start potential of 0.84 V ( v. RHE)), it had high oxygen reduction activity.
- the carbonized material according to Example 3 is obtained by subjecting the carbonized material used in Example 1 to metal removal treatment. That is, the carbonized material according to Example 1 had high catalytic activity before and after metal removal. Thus, it was considered that the high catalytic activity of the carbonized material according to the example was contributed by the carbon structure represented by the above-mentioned I G / IF ratio.
- the I G / I F ratio was also obtained for the carbonized material produced in the same manner as in Example 1 except that carbon black (Vulcan: XC) was used instead of fullerene soot. I tried to evaluate, but the peak of the I F has not been confirmed.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Electrochemistry (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Carbon And Carbon Compounds (AREA)
- Catalysts (AREA)
- Inert Electrodes (AREA)
- Fuel Cell (AREA)
Abstract
Provided are a carbon catalyst showing improved catalytic activity, a production method therefor, and an electrode and cell using the carbon catalyst. This carbon catalyst is characterized in having a carbon structure which indicates that, in X-ray diffraction, a ratio (IG/IF) of the maximum intensity (IG) of the peak at a diffraction angle of 18.0 to 26.5˚ to the maximum intensity (IF) of the peak at a diffraction angle of 11.5 to 15.0˚ is 0.8 to 11. The carbon catalyst may comprise a carbonized material obtained by carbonization of a starting material containing fullerene soot and metal.
Description
本発明は、炭素触媒及びその製造方法、並びに当該炭素触媒を用いた電極及び電池に関する。
The present invention relates to a carbon catalyst and a method for producing the same, and an electrode and a battery using the carbon catalyst.
現在、多くの化学反応や次世代電池において、白金触媒が使用されている。しかしながら、例えば、白金の埋蔵量が限られていること、固体高分子形燃料電池(PEFC)においては、白金の使用によってコストが高くなること、空気電池においては白金の使用によってコストが高くなるだけでなく白金による電解質溶液の分解等の化学反応が起こること等、解決すべき問題が多い。このため、白金を使用しない代替技術の開発が進められている。上述の代替技術としては、従来、例えば、炭素触媒を用いたものが提案されている(例えば特許文献1)。
Currently, platinum catalysts are used in many chemical reactions and next-generation batteries. However, for example, platinum reserves are limited, solid polymer fuel cells (PEFC) are expensive due to the use of platinum, and air batteries are only expensive due to the use of platinum. In addition, there are many problems to be solved, such as chemical reactions such as decomposition of the electrolyte solution by platinum. For this reason, the development of alternative technologies that do not use platinum is underway. Conventionally, for example, a technique using a carbon catalyst has been proposed as an alternative technique described above (for example, Patent Document 1).
しかしながら、例えば、従来の白金を使用しない炭素触媒の触媒活性(例えば、酸素還元活性)は、必ずしも十分ではなかった。
However, for example, the catalytic activity (for example, oxygen reduction activity) of a conventional carbon catalyst not using platinum is not always sufficient.
本発明は、上記課題に鑑みて為されたものであり、向上した触媒活性を示す炭素触媒及びその製造方法、並びに当該炭素触媒を用いた電極及び電池を提供することを目的の一つとする。
The present invention has been made in view of the above problems, and an object of the present invention is to provide a carbon catalyst exhibiting improved catalytic activity, a method for producing the same, and an electrode and a battery using the carbon catalyst.
上記課題を解決するための本発明の一実施形態に係る炭素触媒は、X線回折法において、回折角11.5°~15.0°におけるピークの最大強度(IF)に対する、回折角18.0°~26.5°におけるピークの最大強度(IG)の比(IG/IF)が、0.8以上、11以下を示す炭素構造、を有することを特徴とする。
A carbon catalyst according to an embodiment of the present invention for solving the above-described problems is obtained by using a diffraction angle of 18 with respect to a peak maximum intensity (I F ) at a diffraction angle of 11.5 ° to 15.0 ° in an X-ray diffraction method. It has a carbon structure in which the ratio (I G / I F ) of the peak maximum intensity (I G ) at 0.0 ° to 26.5 ° is 0.8 or more and 11 or less.
上記炭素触媒は、フラーレンスートと、金属と、を含む原料を炭素化して得られた炭素化材料、を含むこととしてもよい。
The carbon catalyst may include a carbonized material obtained by carbonizing a raw material containing fullerene soot and a metal.
上記課題を解決するための本発明の一実施形態に係る炭素触媒は、フラーレンスートと、金属と、を含む原料を炭素化して得られた炭素化材料、を含む。
A carbon catalyst according to an embodiment of the present invention for solving the above problems includes a carbonized material obtained by carbonizing a raw material containing fullerene soot and a metal.
上記いずれかの炭素触媒は、酸素還元開始電位が0.60V(vs.RHE)以上である炭素触媒であることとしてもよい。
Any of the above carbon catalysts may be a carbon catalyst having an oxygen reduction starting potential of 0.60 V (vs. RHE) or more.
上記課題を解決するための本発明の一実施形態に係る電極は、上記いずれかの炭素触媒を含む。
An electrode according to an embodiment of the present invention for solving the above problems includes any one of the above carbon catalysts.
上記課題を解決するための本発明の一実施形態に係る電池は、前記電極を含む。
A battery according to an embodiment of the present invention for solving the above problems includes the electrode.
上記課題を解決するための本発明の一実施形態に係る触媒の製造方法は、フラーレンスートと、金属と、を含む原料を炭素化して炭素化材料を得ること、前記炭素化材料を含む炭素触媒を得ること、を含む。
A method for producing a catalyst according to an embodiment of the present invention for solving the above-mentioned problem is to obtain a carbonized material by carbonizing a raw material containing fullerene soot and a metal, and a carbon catalyst containing the carbonized material. Including.
本発明によれば、向上した触媒活性を持った炭素触媒及びその製造方法、並びにこれを用いた電極、及び電池を提供することができる。
According to the present invention, it is possible to provide a carbon catalyst having improved catalytic activity, a method for producing the same, an electrode using the same, and a battery.
以下に、本発明の一実施形態について説明する。なお、本発明は本実施形態で示す例に限られない。
Hereinafter, an embodiment of the present invention will be described. The present invention is not limited to the example shown in the present embodiment.
まず、本実施形態に係る炭素触媒の製造方法(以下、「本方法」という)について説明する。本方法は、フラーレンスートと、金属と、を含む原料を炭素化して炭素化材料を得ること、前記炭素化材料を含む炭素触媒を製造すること、を含む、方法である。
First, a method for producing a carbon catalyst according to this embodiment (hereinafter referred to as “the present method”) will be described. This method is a method including carbonizing a raw material containing a fullerene soot and a metal to obtain a carbonized material, and producing a carbon catalyst containing the carbonized material.
本方法では、フラーレンスートと、金属と、を含む原料を調製する。すなわち、少なくともフラーレンスートと、金属と、を混合する。混合する方法は特に限られず、例えば、乳鉢や撹拌装置を使用し、粉末状で混合する粉末混合、溶媒を添加して混合する溶媒混合等、1種以上の混合方法を使用することができる。
In this method, a raw material containing fullerene soot and metal is prepared. That is, at least the fuller soot and the metal are mixed. The mixing method is not particularly limited, and for example, one or more mixing methods such as powder mixing using a mortar or stirring device and powder mixing for adding a solvent and solvent mixing for adding a solvent can be used.
フラーレンスートは、フラーレンの製造において得られるフラーレン含有煤からフラーレンを溶媒を使用して抽出した場合に得られる、抽出されたフラーレン(当該溶媒に溶解したフラーレン)以外の成分(以下本発明において「フラーレン副生煤」という)に由来する分子であって、炭素原子の五員環及び六員環が三次元的に配置された構造を有し閉じた空間を形成していない分子、を含む炭素材料である。
Fullerene soot is a component other than the extracted fullerene (fullerene dissolved in the solvent) obtained when fullerene is extracted from the fullerene-containing soot obtained in the production of fullerene using a solvent (hereinafter referred to as “fullerene” in the present invention). A carbon material comprising a molecule derived from "byproduct" and having a structure in which five-membered and six-membered rings of carbon atoms are arranged three-dimensionally and does not form a closed space It is.
また、フラーレンスートは、非晶質な炭素構造を含む炭素材料であって、前記非晶質な炭素構造はフラーレン副生煤に由来する上記分子を含むこととしてもよい。具体的には、フラーレンスートは、例えば、フラーレン副生煤に由来する上記分子を含む非晶質な炭素構造と、ランダムな緩やかな炭素網面構造と、を含む炭素材料であることとしてもよい。
Also, the fullerene soot is a carbon material containing an amorphous carbon structure, and the amorphous carbon structure may contain the molecule derived from fullerene byproduct. Specifically, the fullerene soot may be a carbon material including, for example, an amorphous carbon structure including the molecule derived from the fullerene byproduct ginger and a random gradual carbon network structure. .
上述のフラーレン副生煤は、例えば、フラーレンの製造において得られるフラーレン含有煤からフラーレンを溶媒を使用して抽出した場合に得られる、抽出されたフラーレン(当該溶媒に溶解したフラーレン)以外の当該溶媒に溶解せず固形分として残った成分であることとしてもよい。
The above-mentioned fullerene byproduct koji is, for example, the solvent other than the extracted fullerene (fullerene dissolved in the solvent) obtained when fullerene is extracted from the fullerene-containing koji obtained in the production of fullerene using a solvent. It is good also as a component which did not melt | dissolve in and remained as solid content.
上述のフラーレンの製造の方法は、フラーレン含有煤が得られれば特に限られないが、例えば、アーク放電法、抵抗加熱法、レーザー蒸発法及び燃焼法からなる群より選択される1種以上であることとしてもよい。
The method for producing the fullerene is not particularly limited as long as the fullerene-containing soot is obtained. For example, the fullerene-containing method is at least one selected from the group consisting of an arc discharge method, a resistance heating method, a laser evaporation method, and a combustion method. It is good as well.
また、フラーレン含有煤からフラーレンを抽出する場合に使用される上記溶媒は、フラーレン含有煤からフラーレンを抽出しフラーレン副生煤が得られるものであれば特に限られないが、例えば、有機溶媒であることとしてもよい。当該有機溶媒は、具体的には、例えば、芳香族溶媒であることとしてもよく、より具体的には、例えば、トルエン及び/又はキシレンであることとしてもよい。すなわち、フラーレン副生煤は、フラーレンの製造において得られるフラーレン含有煤からフラーレンをトルエン及び/又はキシレンを使用して抽出した場合に得られる、抽出されたフラーレン(トルエン及び/又はキシレンに溶解したフラーレン)以外の成分であることとしてもよい。
Further, the solvent used when extracting fullerene from fullerene-containing soot is not particularly limited as long as it can extract fullerene from fullerene-containing soot to obtain fullerene by-product soot. For example, it is an organic solvent. It is good as well. Specifically, the organic solvent may be, for example, an aromatic solvent, and more specifically, for example, toluene and / or xylene. That is, the fullerene by-product is an extracted fullerene (fullerene dissolved in toluene and / or xylene) obtained when fullerene is extracted from the fullerene-containing soot obtained in the production of fullerene using toluene and / or xylene. It is good also as being components other than.
原料に含まれるフラーレンスートの量は特に限られないが、例えば、当該原料に対する当該フラーレンスートの重量割合が、40wt%以上であることとしてもよく、50wt%以上であることとしてもよい。また、99.5wt%以下であることとしてもよい。すなわち、当該原料に対する当該フラーレンスートの重量割合は、40~99.5wt%であることとしてもよく、50~99.5wt%であることとしてもよく、50~80wt%であることとしてもよい。
The amount of fullerene soot contained in the raw material is not particularly limited. For example, the weight ratio of the fullerene soot to the raw material may be 40 wt% or more, or may be 50 wt% or more. Moreover, it is good also as being 99.5 wt% or less. That is, the weight ratio of the fullerene soot to the raw material may be 40 to 99.5 wt%, 50 to 99.5 wt%, or 50 to 80 wt%.
原料に含まれる金属は、本方法で得られる炭素触媒の活性を阻害しないものであれば特に限られない。すなわち、例えば、周期表の3族~16族からなる群より選択される1種以上の金属を使用することとしてもよい。
The metal contained in the raw material is not particularly limited as long as it does not inhibit the activity of the carbon catalyst obtained by this method. That is, for example, one or more metals selected from the group consisting of Groups 3 to 16 of the periodic table may be used.
この場合、周期表の3A族(3族)元素、4A族(4族)元素、5A族(5族)元素、6A族(6族)元素、7A族(7族)元素、8族(8族、9族及び10族)元素、1B族(11族)元素、2B族(12族)元素、3B族(13族)元素、4B族(14族)元素、5B族(15族)元素及び6B族(16族)元素からなる群より選択される1種以上を使用することができ、遷移金属(周期表の3族から12族)を好ましく使用することができ、周期表の3族から12族の第4周期に属する遷移金属をさらに好ましく使用することができる。
In this case, Group 3A (Group 3) element, Group 4A (Group 4) element, Group 5A (Group 5) element, Group 6A (Group 6) element, Group 7A (Group 7) element, Group 8 (8 Group, Group 9 and 10) element, Group 1B (Group 11) element, Group 2B (Group 12) element, Group 3B (Group 13) element, Group 4B (Group 14) element, Group 5B (Group 15) element and One or more selected from the group consisting of 6B group (16 group) elements can be used, transition metals (groups 3 to 12 of the periodic table) can be preferably used, from group 3 of the periodic table Transition metals belonging to the fourth period of Group 12 can be used more preferably.
具体的に、例えば、スカンジウム(Sc)、チタン(Ti)、バナジウム(V)、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)、ニッケル(Ni)、銅(Cu)、亜鉛(Zn)、イットリウム(Y)、ジルコニウム(Zr)、ニオブ(Nb)、モリブデン(Mo)、ルテニウム(Ru)、ロジウム(Rh)、パラジウム(Pd)、ランタノイド(セリウム(Ce)等)及びアクチノイドからなる群より選択される1種以上の金属を好ましく使用することができる。
Specifically, for example, scandium (Sc), titanium (Ti), vanadium (V), chromium (Cr), manganese (Mn), iron (Fe), cobalt (Co), nickel (Ni), copper (Cu) Zinc (Zn), yttrium (Y), zirconium (Zr), niobium (Nb), molybdenum (Mo), ruthenium (Ru), rhodium (Rh), palladium (Pd), lanthanoid (cerium (Ce), etc.) and One or more metals selected from the group consisting of actinides can be preferably used.
金属は、当該金属の単体又は当該金属の化合物として使用することができる。金属化合物としては、例えば、金属塩、金属酸化物、金属水酸化物、金属窒化物、金属硫化物、金属炭化物及び金属錯体からなる群より選択される1種以上を使用することができ、金属塩、金属酸化物、金属硫化物及び金属錯体からなる群より選択される1種以上を好ましく使用することができる。なお、後述するように、原料が金属に配位可能な配位子を含む場合には、原料中において当該金属と当該配位子とで形成される金属錯体が形成されることとなる。
The metal can be used as a simple substance of the metal or a compound of the metal. As the metal compound, for example, one or more selected from the group consisting of metal salts, metal oxides, metal hydroxides, metal nitrides, metal sulfides, metal carbides and metal complexes can be used. One or more selected from the group consisting of salts, metal oxides, metal sulfides, and metal complexes can be preferably used. As will be described later, when the raw material contains a ligand capable of coordinating to a metal, a metal complex formed by the metal and the ligand is formed in the raw material.
原料に含まれる金属の量は特に限られないが、例えば、当該原料に対する当該金属の重量割合は、0.5~10wt%であることとしてもよく、2~5wt%であることとしてもよい。
Although the amount of metal contained in the raw material is not particularly limited, for example, the weight ratio of the metal with respect to the raw material may be 0.5 to 10 wt% or 2 to 5 wt%.
また、本方法は、原料に含まれるフラーレンスート及び金属の量は特に限られないが、例えば、当該原料は、フラーレンスートを50~80wt%、当該金属を2~5wt%含む原料であることとしてもよい。
In this method, the amount of fullerene soot and metal contained in the raw material is not particularly limited. For example, the raw material is a raw material containing 50-80 wt% fullerene soot and 2-5 wt% metal. Also good.
また、原料は、さらに他の成分を含むこととしてもよい。すなわち、原料は、例えば、有機物を含むこととしてもよい。この場合、本方法は、フラーレンスートと、金属と、有機物と、を含む原料を炭素化して得られた炭素化材料、を含むこととなる。原料に含まれる有機物は、炭素化できるものであれば特に限られない。すなわち、例えば、高分子量の有機物(例えば、熱硬化性樹脂や熱可塑性樹脂等の樹脂)及び低分子量の有機物の一方又は両方を使用することができる。また、バイオマスを使用することもできる。
The raw material may further contain other components. That is, the raw material may include, for example, an organic material. In this case, the present method includes a carbonized material obtained by carbonizing a raw material containing fullerene soot, a metal, and an organic substance. The organic substance contained in the raw material is not particularly limited as long as it can be carbonized. That is, for example, one or both of a high molecular weight organic substance (for example, a resin such as a thermosetting resin or a thermoplastic resin) and a low molecular weight organic substance can be used. Biomass can also be used.
原料が有機物を含む場合、原料に含まれるフラーレンスート、金属、及び有機物の重量割合は、特に限られない。例えば、当該原料は、フラーレンスートを50~80wt%、当該金属を2~5wt%、当該有機物を18~45wt%、含む原料あることとしてもよい。
When the raw material contains an organic substance, the weight ratio of fullerene soot, metal, and organic substance contained in the raw material is not particularly limited. For example, the raw material may be a raw material containing 50-80 wt% fuller soot, 2-5 wt% of the metal, and 18-45 wt% of the organic substance.
有機物としては、例えば、金属に配位可能な配位子を好ましく使用することができる。すなわち、この場合、その分子内に1又は複数個の配位原子を含む有機物を使用する。より具体的に、例えば、配位原子として、その分子内に窒素原子、リン原子、酸素原子及び硫黄原子からなる群より選択される1種以上を含む有機物を使用することができる。また、例えば、配位基として、その分子内にアミノ基、フォスフィノ基、カルボキシル基及びチオール基からなる群より選択される1種以上を含む有機物を使用することもできる。
As the organic substance, for example, a ligand capable of coordinating with a metal can be preferably used. That is, in this case, an organic substance containing one or more coordination atoms in the molecule is used. More specifically, for example, as a coordination atom, an organic substance containing one or more selected from the group consisting of a nitrogen atom, a phosphorus atom, an oxygen atom and a sulfur atom in the molecule can be used. In addition, for example, an organic substance containing one or more selected from the group consisting of an amino group, a phosphino group, a carboxyl group, and a thiol group in the molecule can be used as a coordination group.
また、有機物は、例えば、本方法により製造される炭素触媒の活性を向上させる成分として、窒素原子、ホウ素原子、リン原子、酸素原子及び硫黄原子からなる群より選択される1種以上を含むこともできる。
The organic substance contains, for example, at least one selected from the group consisting of a nitrogen atom, a boron atom, a phosphorus atom, an oxygen atom, and a sulfur atom as a component that improves the activity of the carbon catalyst produced by this method. You can also.
具体的に、有機物としては、例えば、アクリロニトリル、ポリアクリロニトリル、メラミン、メラミン樹脂、ピロール、ポリピロール、3-メチルポリピロール、ポリビニルピロール、チアゾール、ピラゾール、ビニルピリジン、ポリビニルピリジン、ピリダジン、ピリミジン、ピペラジン、イミダゾール、1-メチルイミダゾール、2-メチルイミダゾ-ル、キノキサリン、アニリン、ポリアニリン、ベンゾイミダゾ-ル、ポリベンゾイミダゾ-ル、ヒドラジン、ポリカルバゾール、トリアジン、ポリカルボジイミド、キレート樹脂、ポリアミドイミド樹脂、ポリアクリロニトリル-ポリメタクリル酸共重合体、オキサゾール、モルホリン、コハク酸ジヒドラジド、アジピン酸ジヒドラジド、ポリビスマレイミド、ポリアミノビスマレイミド、ポリイミド、ポリアクリルアミド、ポリアミド、キチン、キトサン、タンパク質、ペプチド、アミノ酸、ポリアミノ酸、核酸、ヒドラジド、尿素、サレン、ポリウレタン、ポリアミドアミン、フェノール樹脂、フェノールホルムアルデヒド樹脂、ポリフルフリルアルコール、フラン、フラン樹脂、エポキシ樹脂、ピラン、ポリスルフォン、ポリビニルアルコール、ポリビニルブチラール、ポリエステル、ポリエ-テル、ポリ乳酸、ポリエ-テルエ-テルケトン、セルロ-ス、カルボキシメチルセルロース、リグニン、ポリアクリル酸、ポリアクリル酸エステル、ポリメタクリル酸エステル、ポリメタクリル酸、ポリ塩化ビニリデン、チオフェン、ピッチ及び褐炭からなる群より選択される1種以上を使用することができる。
Specifically, examples of the organic substance include acrylonitrile, polyacrylonitrile, melamine, melamine resin, pyrrole, polypyrrole, 3-methylpolypyrrole, polyvinylpyrrole, thiazole, pyrazole, vinylpyridine, polyvinylpyridine, pyridazine, pyrimidine, piperazine, imidazole, 1-methylimidazole, 2-methylimidazole, quinoxaline, aniline, polyaniline, benzimidazole, polybenzimidazole, hydrazine, polycarbazole, triazine, polycarbodiimide, chelate resin, polyamideimide resin, polyacrylonitrile-poly Methacrylic acid copolymer, oxazole, morpholine, succinic dihydrazide, adipic dihydrazide, polybismaleimide, polyaminobismale , Polyimide, polyacrylamide, polyamide, chitin, chitosan, protein, peptide, amino acid, polyamino acid, nucleic acid, hydrazide, urea, salen, polyurethane, polyamidoamine, phenol resin, phenol formaldehyde resin, polyfurfuryl alcohol, furan, furan resin , Epoxy resin, pyran, polysulfone, polyvinyl alcohol, polyvinyl butyral, polyester, polyether, polylactic acid, polyetheretherketone, cellulose, carboxymethylcellulose, lignin, polyacrylic acid, polyacrylic ester, polymethacrylic One or more selected from the group consisting of acid esters, polymethacrylic acid, polyvinylidene chloride, thiophene, pitch and lignite can be used.
本方法は、上述の原料を炭素化して炭素化材料を得ることを含む。原料の炭素化は、上述の原料を加熱して、当該原料を炭素化できる所定温度(炭素化温度)で保持することにより行う。炭素化温度は、原料を炭素化できる温度であれば特に限られず、例えば、300℃以上であることとしてもよい。より具体的に、炭素化温度は、例えば、300℃以上、1500℃以下であることとしてもよい。
This method includes carbonizing the above-described raw material to obtain a carbonized material. Carbonization of a raw material is performed by heating the above-mentioned raw material and holding at a predetermined temperature (carbonization temperature) at which the raw material can be carbonized. The carbonization temperature is not particularly limited as long as the raw material can be carbonized, and may be, for example, 300 ° C. or higher. More specifically, the carbonization temperature may be, for example, 300 ° C. or higher and 1500 ° C. or lower.
原料を炭素化温度まで加熱する際の昇温速度は、特に限られず、例えば、0.5℃/分以上、300℃/分以下であることとしてもよい。原料を炭素化温度で保持する時間は、原料を炭素化できる時間であれば特に限られず、例えば、5分以上であることとしてもよく、5分以上、240分以下であることとしてもよい。また、炭素化は、窒素等の不活性ガス下(例えば、不活性ガスの流通下)で行うことが好ましい。
The heating rate when heating the raw material to the carbonization temperature is not particularly limited, and may be, for example, 0.5 ° C./min or more and 300 ° C./min or less. The time for holding the raw material at the carbonization temperature is not particularly limited as long as the raw material can be carbonized, and may be, for example, 5 minutes or more, or 5 minutes or more and 240 minutes or less. Carbonization is preferably performed under an inert gas such as nitrogen (for example, under the flow of an inert gas).
本方法においては、上述の原料を炭素化して生成された炭素化材料を得る。得られた炭素化材料は、粉砕することとしてもよい。炭素化材料を粉砕する方法は、特に限られず、例えば、ボールミルやビーズミル等の粉砕装置を使用することができる。粉砕後の炭素化材料の平均粒径は、例えば、150μm以下とすることができ、好ましくは100μm以下とすることができる。
In this method, a carbonized material generated by carbonizing the above-described raw material is obtained. The obtained carbonized material may be pulverized. The method for pulverizing the carbonized material is not particularly limited, and for example, a pulverizing apparatus such as a ball mill or a bead mill can be used. The average particle size of the carbonized material after pulverization can be, for example, 150 μm or less, and preferably 100 μm or less.
上述の原料を炭素化して得られる炭素化材料は触媒活性(例えば、酸素還元活性)を有する。すなわち、本方法により製造される炭素触媒は、上述の原料を炭素化して得られる炭素化材料を含んでいればよく、例えば、当該炭素化材料自体を炭素触媒として得ることとしてもよい。つまり、本方法は、フラーレンスートと、金属と、を含む原料を炭素化して炭素化材料を得ること、前記炭素化材料を炭素触媒として得ること、を含むこととしてもよい。
The carbonized material obtained by carbonizing the above raw materials has catalytic activity (for example, oxygen reduction activity). That is, the carbon catalyst manufactured by this method should just contain the carbonization material obtained by carbonizing the above-mentioned raw material, for example, it is good also as obtaining the said carbonization material itself as a carbon catalyst. In other words, the present method may include carbonizing a raw material containing fullerene soot and a metal to obtain a carbonized material, and obtaining the carbonized material as a carbon catalyst.
また、本方法においては、炭素化により生成された炭素化材料に、さらなる処理を施し、当該処理が施された炭素化材料を炭素触媒として得ることとしてもよい。この場合、処理が施された炭素化材料を粉砕したものを炭素触媒として得ることとしてもよい。
Further, in this method, the carbonized material generated by carbonization may be further treated to obtain the carbonized material subjected to the treatment as a carbon catalyst. In this case, a pulverized carbonized material that has been treated may be obtained as a carbon catalyst.
また、本方法においては、任意の工程で炭素化材料に窒素原子及び/又はホウ素原子を導入(ドープ)することとしてもよい。すなわち、本方法は、例えば、原料を炭素化して得られた炭素化材料、後述の金属除去処理後の炭素化材料の一つ以上に対して、窒素原子及び/又はホウ素原子を導入し、当該炭素化材料を含む炭素触媒を製造することとしてもよい。窒素原子及び/又はホウ素原子を導入する方法としては、例えば、アンモオキシデーション法やCVD(Chemical Vapor Deposition)法などの気相ドープ法、液相ドープ法、気相―液相ドープ法を使用することができる。具体的には、例えば、炭素化材料を窒素、アルゴン、ヘリウム等の不活性ガス雰囲気下で400℃以上、1200℃以下まで昇温し、次いで、アンモニア等の窒素源又は塩化ホウ素等のホウ素源を導入し、400℃以上、1200℃以下の温度で5分以上、180分以下の時間保持することにより、当該炭素化材料に窒素原子及び/又はホウ素原子を導入することができる。また、得られた炭素化材料に、二酸化炭素賦活、リン酸賦活、アルカリ賦活、アンモニア賦活、酸化窒素による賦活、電解賦活等の賦活処理及び/又は混酸酸化、過酸化水素酸化等の液相酸化を施すこともできる。
Moreover, in this method, nitrogen atoms and / or boron atoms may be introduced (doped) into the carbonized material in an arbitrary step. That is, in this method, for example, nitrogen atoms and / or boron atoms are introduced into one or more of a carbonized material obtained by carbonizing a raw material and a carbonized material after metal removal treatment described later, It is good also as manufacturing the carbon catalyst containing a carbonization material. As a method for introducing nitrogen atoms and / or boron atoms, for example, a vapor phase doping method such as an ammoxidation method or a CVD (Chemical Vapor Deposition) method, a liquid phase doping method, or a gas phase-liquid phase doping method is used. be able to. Specifically, for example, the carbonized material is heated to 400 ° C. or more and 1200 ° C. or less in an inert gas atmosphere such as nitrogen, argon, or helium, and then a nitrogen source such as ammonia or a boron source such as boron chloride. Then, nitrogen atoms and / or boron atoms can be introduced into the carbonized material by holding at a temperature of 400 ° C. or higher and 1200 ° C. or lower for 5 minutes to 180 minutes. Further, the obtained carbonized material is subjected to carbon dioxide activation, phosphoric acid activation, alkali activation, ammonia activation, activation with nitric oxide, activation treatment such as electrolytic activation, and / or liquid phase oxidation such as mixed acid oxidation and hydrogen peroxide oxidation. Can also be applied.
また、本方法においては、例えば、上述の炭素化によって得られた炭素化材料に、金属除去処理を施すこととしてもよい。金属除去処理は、炭素化材料に含まれる金属を除去する処理である。金属除去処理は、炭素化材料に含まれる金属を除去し、又は当該金属の量を低減できる処理であれば特に限られず、例えば、酸による洗浄処理や電解処理を実施することができる。
In this method, for example, the carbonization material obtained by the above-described carbonization may be subjected to metal removal treatment. The metal removal process is a process for removing the metal contained in the carbonized material. The metal removal treatment is not particularly limited as long as it can remove the metal contained in the carbonized material or reduce the amount of the metal, and for example, an acid cleaning treatment or an electrolytic treatment can be performed.
酸による洗浄処理に使用する酸は、金属除去処理の効果が得られるものであれば特に限られず、任意の1種以上を使用することができる。すなわち、例えば、塩酸(例えば、希塩酸及び濃塩酸)、硝酸(例えば、希硝酸及び濃硝酸)及び硫酸(例えば、希硫酸及び濃硫酸)からなる群より選択される1種以上を使用することができる。酸による洗浄処理の方法は、特に限られず、例えば、酸を含有する溶液中に炭素化材料を浸漬して保持する及び/又は撹拌する方法を好ましく使用することができる。
The acid used for the acid cleaning treatment is not particularly limited as long as the effect of the metal removal treatment can be obtained, and any one or more of them can be used. That is, for example, it is possible to use one or more selected from the group consisting of hydrochloric acid (eg, dilute hydrochloric acid and concentrated hydrochloric acid), nitric acid (eg, dilute nitric acid and concentrated nitric acid) and sulfuric acid (eg, dilute sulfuric acid and concentrated sulfuric acid). it can. The method of the acid cleaning treatment is not particularly limited, and for example, a method of immersing and holding the carbonized material in an acid-containing solution and / or stirring can be preferably used.
次に、本実施形態に係る炭素触媒(以下、「本触媒」)を説明する。本触媒は、X線回折法において、回折角11.5°~15.0°におけるピークの最大強度(IF)に対する、回折角18.0°~26.5°におけるピークの最大強度(IG)の比(IG/IF)が0.8以上、11以下を示す炭素構造、を有する炭素化材料、を含む。発明者らは、鋭意検討の結果、炭素化材料が、IGのピークに対応する炭素構造と、IFのピークに対応する炭素構造とを、特定のバランスで有することにより、優れた触媒活性を示すことを見出した。
Next, the carbon catalyst according to the present embodiment (hereinafter “the present catalyst”) will be described. In the X-ray diffraction method, the present catalyst has a peak maximum intensity (I F ) at a diffraction angle of 18.0 ° to 26.5 ° with respect to a peak maximum intensity (I F ) at a diffraction angle of 11.5 ° to 15.0 °. A carbonized material having a carbon structure in which the ratio of G 1 ) (I G / I F ) is 0.8 or more and 11 or less. We result of intensive studies, carbonized material, and carbon structure corresponding to the peak of I G, and a carbon structure corresponding to the peak of the I F, by having a particular balance, excellent catalytic activity It was found to show.
上述のIG/IF比は、0.8以上、11以下であれば特に限られないが、例えば、0.8以上、7以下であることとしてもよく、0.8以上、5以下であることとしてもよく、0.9以上、11以下であることとしてもよく、0.9以上、7以下であることとしてもよく、0.9以上、5以下であることとしてもよい。後述する実施例1~7(IG/IFが1以上、3以下)においても、向上した触媒活性(酸素還元活性)が得られている。
The above-mentioned I G / IF ratio is not particularly limited as long as it is 0.8 or more and 11 or less, but may be 0.8 or more and 7 or less, for example, 0.8 or more and 5 or less. It may be, may be 0.9 or more and 11 or less, may be 0.9 or more and 7 or less, and may be 0.9 or more and 5 or less. Also in Examples 1 to 7 (I G / I F of 1 or more and 3 or less) described later, improved catalytic activity (oxygen reduction activity) was obtained.
本触媒は、例えば、触媒活性の一つとして酸素還元活性を有する。すなわち、本触媒は、例えば、X線回折法において、上述のIG/IF比が、0.8以上、11以下を示す炭素構造を含み、酸素還元活性を有する炭素触媒であることとしてもよい。上述の本触媒の酸素還元活性は、酸素還元開始電位により評価することができる。酸素還元開始電位は、例えば、本触媒を塗布した作用電極を有する回転リングディスク電極装置を用いて、電位を掃引印加した場合に得られる電圧と電流密度との関係を示すデータ(酸素還元ボルタモグラム)における、-10μA/cm2の還元電流が流れた時の電圧(EO2)として求められる。本触媒の、酸素還元開始電位は、0.60V(vs.RHE)以上であることとしてもよく、0.80V(vs.RHE)以上であることとしてもよい。ここで、RHEはReversible Hydrogen Electrodeの略であり、可逆水素電極を表す。すなわち、vs.RHEは、可逆水素電極を基準にして測定された電位であることを表す。
This catalyst has, for example, an oxygen reduction activity as one of the catalytic activities. That is, the present catalyst may be a carbon catalyst having a carbon structure in which the above-mentioned IG / IF ratio is 0.8 or more and 11 or less and having oxygen reduction activity in the X-ray diffraction method. Good. The oxygen reduction activity of the catalyst described above can be evaluated by the oxygen reduction start potential. The oxygen reduction start potential is, for example, data indicating the relationship between the voltage and current density obtained when the potential is swept and applied using a rotating ring disk electrode device having a working electrode coated with this catalyst (oxygen reduction voltammogram). The voltage (E O2 ) when a reduction current of −10 μA / cm 2 flows is obtained. The oxygen reduction starting potential of the present catalyst may be 0.60 V (vs. RHE) or higher, or 0.80 V (vs. RHE) or higher. Here, RHE is an abbreviation for Reversible Hydrogen Electrode and represents a reversible hydrogen electrode. That is, vs. RHE represents a potential measured with respect to a reversible hydrogen electrode.
本触媒は、上述のIG/IF比を示す炭素構造を有していれば、製造方法は特に限られないが、例えば、上述の本方法によって、効率的に製造することができる。すなわち、本触媒は、フラーレンスートと、金属と、を含む原料を炭素化して得られた炭素化材料、を含むこととしてもよい。
If this catalyst has the carbon structure which shows the above-mentioned I G / IF ratio, a manufacturing method will not be restricted especially, For example, it can manufacture efficiently by the above-mentioned this method. That is, the present catalyst may contain a carbonized material obtained by carbonizing a raw material containing fullerene soot and a metal.
本触媒に含まれる炭素化材料は、金属を含むこととしてもよい。この場合、例えば、当該金属は炭素化材料の内部に含まれることとしてもよい。すなわち、本触媒は、フラーレンスートと、金属と、を含む原料を炭素化して得られた炭素化材料、を含み、当該炭素化材料は、少なくとも内部に金属を含むこととしてもよい。
The carbonized material contained in the present catalyst may contain a metal. In this case, for example, the metal may be contained in the carbonized material. That is, the present catalyst includes a carbonized material obtained by carbonizing a raw material containing fullerene soot and a metal, and the carbonized material may include at least a metal inside.
本触媒に含まれる炭素化材料は、例えば、金属を、0.01~10wt%含むこととしてもよく、0.01~5wt%含むこととしてもよい。金属の当該含有量は、元素分析等の方法により確認することができる。
The carbonized material contained in the present catalyst may contain, for example, 0.01 to 10 wt% of metal or 0.01 to 5 wt% of metal. The content of the metal can be confirmed by a method such as elemental analysis.
また、本触媒は、例えば、原料を炭素化して得られる炭素化材料に、上述の金属除去処理を施すことにより得られる炭素触媒であることとしてもよい。この場合、当該炭素化材料は、実質的に金属を含有しないこととしてもよいが、残存した金属を炭素化材料の内部に含むこととしてもよい。
The present catalyst may be a carbon catalyst obtained, for example, by subjecting a carbonized material obtained by carbonizing a raw material to the above-described metal removal treatment. In this case, the carbonized material may be substantially free of metal, but the remaining metal may be included in the carbonized material.
本実施形態に係る電極(以下、「本電極」)は、上述した本触媒を含む電極である。すなわち、本電極は、例えば、本触媒が担持された電極である。具体的に、本電極は、例えば、所定の電極基材と、当該電極基材に担持された本触媒と、を有する電極である。
The electrode according to the present embodiment (hereinafter referred to as “main electrode”) is an electrode including the above-described catalyst. That is, this electrode is, for example, an electrode on which the present catalyst is supported. Specifically, the present electrode is, for example, an electrode having a predetermined electrode base material and the present catalyst supported on the electrode base material.
本電極は、例えば、燃料電池用電極とすることができ、好ましくは、固体高分子形燃料電池(PEFC)用電極とすることができる。また、本電極は、例えば、空気電池用電極とすることができる。本電極が燃料電池用電極又は空気電池電極である場合、当該本電極は、カソード(酸素極)として使用される。
The electrode can be, for example, a fuel cell electrode, and preferably a polymer electrolyte fuel cell (PEFC) electrode. Moreover, this electrode can be used as the electrode for air batteries, for example. When the main electrode is a fuel cell electrode or an air cell electrode, the main electrode is used as a cathode (oxygen electrode).
本実施形態に係る電池(以下、「本電池」)は、本電極を含む電池である。本電池は、例えば、燃料電池とすることができ、好ましくはPEFCとすることができる。また、本電池は、例えば、空気電池とすることができる。
The battery according to the present embodiment (hereinafter “the present battery”) is a battery including the present electrode. The battery can be, for example, a fuel cell, preferably PEFC. The battery can be an air battery, for example.
すなわち、本電池は、例えば、カソード及びアノードの一方又は両方として本電極を含む燃料電池又は空気電池とすることができ、この場合、本電池は、少なくともカソード(酸素極)として本電極を含むことが好ましい。
That is, the battery can be, for example, a fuel cell or an air battery including the electrode as one or both of a cathode and an anode. In this case, the battery includes the electrode as at least a cathode (oxygen electrode). Is preferred.
具体的に、本電池は、例えば、高分子電解質膜と、当該高分子電解質膜の一方側及び他方側にそれぞれ形成されたカソード(酸素極)及びアノード(燃料極)と、が一体化された膜/電極接合体を備え、当該カソード及び当該アノードの一方又は両方に本電極を含むPEFCとすることができる。この場合、本電池は、少なくともカソードに本電極を含むことが好ましい。
Specifically, in this battery, for example, a polymer electrolyte membrane and a cathode (oxygen electrode) and an anode (fuel electrode) formed on one side and the other side of the polymer electrolyte membrane are integrated, for example. A PEFC may be provided that includes a membrane / electrode assembly and includes the electrode on one or both of the cathode and the anode. In this case, the battery preferably includes the electrode at least on the cathode.
次に、本実施形態に係る具体的な実施例について説明する。
[実施例1] Next, specific examples according to the present embodiment will be described.
[Example 1]
[実施例1] Next, specific examples according to the present embodiment will be described.
[Example 1]
[原料の調製]
フラーレンスート(nanom black ST、フロンティアカーボン社製)と、フタロシアニン鉄(純度95%、東京化成工業株式会社)と、を当該フラーレンスートに対する当該フタロシアニン鉄に含まれる鉄の重量割合が3wt%になるように超純水中、ボールミルを用いて湿式混合を行った。得られた混合物を、孔径0.1μmのフィルターを用いて吸引ろ過し、溶媒である超純水を除去した。その後、混合物を60℃で一晩減圧乾燥することにより原料を得た。 [Preparation of raw materials]
Fullerence soot (nanom black ST, manufactured by Frontier Carbon Co., Ltd.) and phthalocyanine iron (purity 95%, Tokyo Chemical Industry Co., Ltd.) so that the weight ratio of iron contained in the phthalocyanine iron to the fullerence soot is 3 wt%. Wet mixing was performed in ultrapure water using a ball mill. The obtained mixture was subjected to suction filtration using a filter having a pore size of 0.1 μm to remove ultrapure water as a solvent. Then, the raw material was obtained by drying a mixture under reduced pressure at 60 degreeC overnight.
フラーレンスート(nanom black ST、フロンティアカーボン社製)と、フタロシアニン鉄(純度95%、東京化成工業株式会社)と、を当該フラーレンスートに対する当該フタロシアニン鉄に含まれる鉄の重量割合が3wt%になるように超純水中、ボールミルを用いて湿式混合を行った。得られた混合物を、孔径0.1μmのフィルターを用いて吸引ろ過し、溶媒である超純水を除去した。その後、混合物を60℃で一晩減圧乾燥することにより原料を得た。 [Preparation of raw materials]
Fullerence soot (nanom black ST, manufactured by Frontier Carbon Co., Ltd.) and phthalocyanine iron (purity 95%, Tokyo Chemical Industry Co., Ltd.) so that the weight ratio of iron contained in the phthalocyanine iron to the fullerence soot is 3 wt%. Wet mixing was performed in ultrapure water using a ball mill. The obtained mixture was subjected to suction filtration using a filter having a pore size of 0.1 μm to remove ultrapure water as a solvent. Then, the raw material was obtained by drying a mixture under reduced pressure at 60 degreeC overnight.
[原料の炭素化]
次に、得られた原料の炭素化を行った。すなわち、原料0.3gをイメージ炉内、窒素雰囲気下で、昇温速度10℃/minにて昇温した。そして、原料を800℃で1時間保持することにより炭素化を行い、炭素化材料を得た。
[実施例2] [Carbonization of raw materials]
Next, the obtained raw material was carbonized. That is, 0.3 g of the raw material was heated at a temperature rising rate of 10 ° C./min in an image furnace in a nitrogen atmosphere. And carbonization was performed by hold | maintaining a raw material at 800 degreeC for 1 hour, and the carbonization material was obtained.
[Example 2]
次に、得られた原料の炭素化を行った。すなわち、原料0.3gをイメージ炉内、窒素雰囲気下で、昇温速度10℃/minにて昇温した。そして、原料を800℃で1時間保持することにより炭素化を行い、炭素化材料を得た。
[実施例2] [Carbonization of raw materials]
Next, the obtained raw material was carbonized. That is, 0.3 g of the raw material was heated at a temperature rising rate of 10 ° C./min in an image furnace in a nitrogen atmosphere. And carbonization was performed by hold | maintaining a raw material at 800 degreeC for 1 hour, and the carbonization material was obtained.
[Example 2]
フラーレンスート及びフタロシアニン鉄を、当該フラーレンスートに対する当該フタロシアニン鉄に含まれる鉄の重量割合が10wt%になるように用いたこと以外は、実施例1と同様にして、炭素化材料を得た。
[実施例3] A carbonized material was obtained in the same manner as in Example 1 except that fullerene soot and phthalocyanine iron were used so that the weight ratio of iron contained in the phthalocyanine iron to the fullerene soot was 10 wt%.
[Example 3]
[実施例3] A carbonized material was obtained in the same manner as in Example 1 except that fullerene soot and phthalocyanine iron were used so that the weight ratio of iron contained in the phthalocyanine iron to the fullerene soot was 10 wt%.
[Example 3]
実施例1で得られた炭素化材料に、酸洗浄による金属除去処理を施し、炭素化材料を得た。すなわち、実施例1で得られた炭素化材料を、1Mの塩酸中に添加し、70℃でスターラーを用いて2時間撹拌した。次いで、当該炭素化材料を含む当該溶液を、孔径0.1μmのフィルターを用いて吸引ろ過し、蒸留水で洗浄した。この操作を3回繰り返した。回収した炭素化材料を60℃で12時間減圧乾燥し、炭素化材料を得た。
[実施例4] The carbonized material obtained in Example 1 was subjected to metal removal treatment by acid cleaning to obtain a carbonized material. That is, the carbonized material obtained in Example 1 was added to 1M hydrochloric acid and stirred at 70 ° C. using a stirrer for 2 hours. Next, the solution containing the carbonized material was suction filtered using a filter having a pore size of 0.1 μm and washed with distilled water. This operation was repeated three times. The collected carbonized material was dried under reduced pressure at 60 ° C. for 12 hours to obtain a carbonized material.
[Example 4]
[実施例4] The carbonized material obtained in Example 1 was subjected to metal removal treatment by acid cleaning to obtain a carbonized material. That is, the carbonized material obtained in Example 1 was added to 1M hydrochloric acid and stirred at 70 ° C. using a stirrer for 2 hours. Next, the solution containing the carbonized material was suction filtered using a filter having a pore size of 0.1 μm and washed with distilled water. This operation was repeated three times. The collected carbonized material was dried under reduced pressure at 60 ° C. for 12 hours to obtain a carbonized material.
[Example 4]
実施例1で用いた原料に、アンモオキシデーション処理を施し、炭素化材料を得た。すなわち、実施例1で用いた原料0.3gをイメージ炉内、窒素雰囲気下で、昇温速度30℃/minにて昇温した。そして、800℃に到達した後、アンモニアと空気との混合ガス(アンモニア濃度70%)をイメージ炉内に導入し、2時間保持した。その後再び炉内を窒素雰囲気に切り替えて10分保持することにより炭素化材料を得た。
[実施例5] The raw material used in Example 1 was subjected to ammoxidation treatment to obtain a carbonized material. That is, 0.3 g of the raw material used in Example 1 was heated at a heating rate of 30 ° C./min in an image furnace in a nitrogen atmosphere. Then, after reaching 800 ° C., a mixed gas of ammonia and air (ammonia concentration 70%) was introduced into the image furnace and held for 2 hours. Thereafter, the inside of the furnace was again switched to a nitrogen atmosphere and held for 10 minutes to obtain a carbonized material.
[Example 5]
[実施例5] The raw material used in Example 1 was subjected to ammoxidation treatment to obtain a carbonized material. That is, 0.3 g of the raw material used in Example 1 was heated at a heating rate of 30 ° C./min in an image furnace in a nitrogen atmosphere. Then, after reaching 800 ° C., a mixed gas of ammonia and air (ammonia concentration 70%) was introduced into the image furnace and held for 2 hours. Thereafter, the inside of the furnace was again switched to a nitrogen atmosphere and held for 10 minutes to obtain a carbonized material.
[Example 5]
炭素化温度が700℃であること以外は、上述の実施例1と同様にして、炭素化材料を得た。
[実施例6] A carbonized material was obtained in the same manner as in Example 1 except that the carbonization temperature was 700 ° C.
[Example 6]
[実施例6] A carbonized material was obtained in the same manner as in Example 1 except that the carbonization temperature was 700 ° C.
[Example 6]
炭素化温度が900℃であること以外は、上述の実施例1と同様にして、炭素化材料を得た。
[実施例7] A carbonized material was obtained in the same manner as in Example 1 except that the carbonization temperature was 900 ° C.
[Example 7]
[実施例7] A carbonized material was obtained in the same manner as in Example 1 except that the carbonization temperature was 900 ° C.
[Example 7]
炭素化温度が1000℃であること以外は、上述の実施例1と同様にして、炭素化材料を得た。
[比較例1] A carbonized material was obtained in the same manner as in Example 1 except that the carbonization temperature was 1000 ° C.
[Comparative Example 1]
[比較例1] A carbonized material was obtained in the same manner as in Example 1 except that the carbonization temperature was 1000 ° C.
[Comparative Example 1]
実施例1で用いたフラーレンスート(nanom black ST、フロンティアカーボン社)そのものを比較例1の試料として用いた。
[比較例2] The fuller soot used in Example 1 (nanom black ST, Frontier Carbon Co.) itself was used as the sample of Comparative Example 1.
[Comparative Example 2]
[比較例2] The fuller soot used in Example 1 (nanom black ST, Frontier Carbon Co.) itself was used as the sample of Comparative Example 1.
[Comparative Example 2]
フラーレンスートを窒素雰囲気下、400℃で炭素化して炭素化材料を得た。すなわち、フラーレンスート0.3gをイメージ炉内、窒素雰囲気下で、昇温速度30℃/minにて昇温した。そして、当該フラーレンスートを400℃で2時間保持することにより炭素化を行い、炭素化材料を得た。
[比較例3] Fullerene soot was carbonized at 400 ° C. in a nitrogen atmosphere to obtain a carbonized material. That is, 0.3 g of fullerene soot was heated in an image furnace in a nitrogen atmosphere at a heating rate of 30 ° C./min. And the carbonization was performed by hold | maintaining the said fullerene soot for 2 hours at 400 degreeC, and the carbonization material was obtained.
[Comparative Example 3]
[比較例3] Fullerene soot was carbonized at 400 ° C. in a nitrogen atmosphere to obtain a carbonized material. That is, 0.3 g of fullerene soot was heated in an image furnace in a nitrogen atmosphere at a heating rate of 30 ° C./min. And the carbonization was performed by hold | maintaining the said fullerene soot for 2 hours at 400 degreeC, and the carbonization material was obtained.
[Comparative Example 3]
炭素化の温度が600℃である以外は、上述の比較例2と同様にして、炭素化材料を得た。
[比較例4] A carbonized material was obtained in the same manner as Comparative Example 2 except that the carbonization temperature was 600 ° C.
[Comparative Example 4]
[比較例4] A carbonized material was obtained in the same manner as Comparative Example 2 except that the carbonization temperature was 600 ° C.
[Comparative Example 4]
炭素化の温度が800℃である以外は、上述の比較例2と同様にして、炭素化材料を得た。
[比較例5] A carbonized material was obtained in the same manner as in Comparative Example 2 except that the carbonization temperature was 800 ° C.
[Comparative Example 5]
[比較例5] A carbonized material was obtained in the same manner as in Comparative Example 2 except that the carbonization temperature was 800 ° C.
[Comparative Example 5]
フラーレンスートに400℃でアンモオキシデーション処理を施し、炭素化触媒を得た。すなわち、フラーレンスート(nanom black ST、フロンティアカーボン社)0.3gをイメージ炉内、窒素雰囲気下で、昇温速度30℃/minにて昇温した。そして、400℃に到達した後、アンモニアと空気との混合ガス(アンモニア濃度70%)をイメージ炉内に導入し、2時間保持した。その後再び炉内を窒素雰囲気に切り替えて10分保持することにより炭素化材料を得た。
[比較例6] The fullerence soot was subjected to ammoxidation treatment at 400 ° C. to obtain a carbonization catalyst. That is, 0.3 g of fuller soot (nanom black ST, Frontier Carbon) was heated in an image furnace in a nitrogen atmosphere at a heating rate of 30 ° C./min. Then, after reaching 400 ° C., a mixed gas of ammonia and air (ammonia concentration 70%) was introduced into the image furnace and held for 2 hours. Thereafter, the inside of the furnace was again switched to a nitrogen atmosphere and held for 10 minutes to obtain a carbonized material.
[Comparative Example 6]
[比較例6] The fullerence soot was subjected to ammoxidation treatment at 400 ° C. to obtain a carbonization catalyst. That is, 0.3 g of fuller soot (nanom black ST, Frontier Carbon) was heated in an image furnace in a nitrogen atmosphere at a heating rate of 30 ° C./min. Then, after reaching 400 ° C., a mixed gas of ammonia and air (ammonia concentration 70%) was introduced into the image furnace and held for 2 hours. Thereafter, the inside of the furnace was again switched to a nitrogen atmosphere and held for 10 minutes to obtain a carbonized material.
[Comparative Example 6]
600℃でアンモオキシデーション処理を施したこと以外は、比較例5と同様にして、炭素化材料を得た。
A carbonized material was obtained in the same manner as in Comparative Example 5 except that the ammoxidation treatment was performed at 600 ° C.
[酸素還元開始電位の測定]
上述の実施例1~7、比較例1~6で得られた炭素化材料各々について、酸素還元開始電位を測定した。 [Measurement of oxygen reduction starting potential]
For each of the carbonized materials obtained in Examples 1 to 7 and Comparative Examples 1 to 6, the oxygen reduction starting potential was measured.
上述の実施例1~7、比較例1~6で得られた炭素化材料各々について、酸素還元開始電位を測定した。 [Measurement of oxygen reduction starting potential]
For each of the carbonized materials obtained in Examples 1 to 7 and Comparative Examples 1 to 6, the oxygen reduction starting potential was measured.
まず、触媒スラリーを調製した。具体的に、上述のようにして得られた炭素化材料5mgにバインダー溶液(ナフィオン(商標登録)、デュポン株式会社)50μL、エタノール150μL、超純水150μL、スパチュラで2杯(約15粒)のガラスビーズ(直径1mm)を加え、20分間超音波処理することにより、触媒スラリーを得た。
First, a catalyst slurry was prepared. Specifically, 5 mg of the carbonized material obtained as described above was mixed with 50 μL of a binder solution (Nafion (registered trademark), DuPont Co., Ltd.), 150 μL of ethanol, 150 μL of ultrapure water, and 2 cups (about 15 grains) of spatula. Glass beads (diameter 1 mm) were added and sonicated for 20 minutes to obtain a catalyst slurry.
次に、触媒スラリーをピペットにより1.8μL吸い取り、回転リングディスク電極装置(RRDE-3A Ver.1.2S、ビー・エー・エス株式会社製)のディスク電極(面積0.1256cm2)に塗布し、乾燥させることにより、作用電極を作製した。また、リング電極としては白金電極を、対極としてはガラス状炭素を、参照電極としては可逆水素電極を用いた。電解質溶液としては、0.5M硫酸水溶液に酸素を常温でバブリングさせ、酸素飽和させたものを用いた。
Next, 1.8 μL of the catalyst slurry was sucked with a pipette and applied to the disk electrode (area 0.1256 cm 2 ) of the rotating ring disk electrode device (RRDE-3A Ver. 1.2S, manufactured by BAS Co., Ltd.). The working electrode was prepared by drying. In addition, a platinum electrode was used as the ring electrode, glassy carbon was used as the counter electrode, and a reversible hydrogen electrode was used as the reference electrode. As the electrolyte solution, a 0.5 M sulfuric acid aqueous solution was bubbled with oxygen at room temperature and saturated with oxygen.
そして、電気化学アナライザー(CHI700E、ALS/DY2323、ビー・エー・エス株式会社製)を用いてリニアスイープボルタンメトリーを行った。
Then, linear sweep voltammetry was performed using an electrochemical analyzer (CHI700E, ALS / DY2323, manufactured by BAS Co., Ltd.).
まず、25℃で酸素を20分間バブリングすることにより電解質溶液を酸素飽和させた後、測定を開始した。次いで、初期電位を600秒保持した後に、作用電極を回転速度1500rpmで回転させ、25℃にて、掃引速度1mV/秒で1V(vs.RHE)から0V(vs.RHE)まで電位を掃引し、作用電極に流れる電流値を測定した。
First, after the oxygen solution was saturated with oxygen by bubbling oxygen at 25 ° C. for 20 minutes, the measurement was started. Next, after maintaining the initial potential for 600 seconds, the working electrode was rotated at a rotation speed of 1500 rpm, and the potential was swept from 1 V (vs. RHE) to 0 V (vs. RHE) at a sweep speed of 1 mV / sec at 25 ° C. The value of the current flowing through the working electrode was measured.
このときの電流を電位の関数として記録した。そして、得られた分極曲線から、-10μA/cm2の還元電流が流れた電圧を酸素還元開始電位(EO2)(vs.RHE)として記録した。
The current at this time was recorded as a function of potential. Then, from the obtained polarization curve, a voltage at which a reduction current of −10 μA / cm 2 flowed was recorded as an oxygen reduction starting potential (E O2 ) (vs. RHE).
[X線回折]
上述の実施例1~7、比較例1~6で得られた炭素触媒各々について、X線回折法による解析を行い、IFに対するIGの比を評価した。すなわち、X線回折装置(XRD-6100、SHIMADZU)を用いてX線回折測定を行った。X線管球への印加電圧及び電流はそれぞれ40kV及び30mAとし、サンプリング間隔は0.1°、走査速度は2.0°/分、測定角度範囲(2θ)は5~40°とした。入射X線はCuKα線を用いた。 [X-ray diffraction]
Examples 1-7 described above, the carbon catalyst each obtained in Comparative Examples 1-6, analyzed by X-ray diffraction, to evaluate the ratio of I G for I F. That is, X-ray diffraction measurement was performed using an X-ray diffractometer (XRD-6100, SHIMADZU). The applied voltage and current to the X-ray tube were 40 kV and 30 mA, the sampling interval was 0.1 °, the scanning speed was 2.0 ° / min, and the measurement angle range (2θ) was 5 to 40 °. The incident X-ray was a CuKα ray.
上述の実施例1~7、比較例1~6で得られた炭素触媒各々について、X線回折法による解析を行い、IFに対するIGの比を評価した。すなわち、X線回折装置(XRD-6100、SHIMADZU)を用いてX線回折測定を行った。X線管球への印加電圧及び電流はそれぞれ40kV及び30mAとし、サンプリング間隔は0.1°、走査速度は2.0°/分、測定角度範囲(2θ)は5~40°とした。入射X線はCuKα線を用いた。 [X-ray diffraction]
Examples 1-7 described above, the carbon catalyst each obtained in Comparative Examples 1-6, analyzed by X-ray diffraction, to evaluate the ratio of I G for I F. That is, X-ray diffraction measurement was performed using an X-ray diffractometer (XRD-6100, SHIMADZU). The applied voltage and current to the X-ray tube were 40 kV and 30 mA, the sampling interval was 0.1 °, the scanning speed was 2.0 ° / min, and the measurement angle range (2θ) was 5 to 40 °. The incident X-ray was a CuKα ray.
次に、図1A、図1B、図2A及び図2Bを用いて、上述の方法で得られたX線回折パターンからIG、IFを決定する方法を説明する。
Next, FIG. 1A, FIG. 1B, with reference to FIGS. 2A and 2B, I G, illustrating a method of determining the I F from X-ray diffraction pattern obtained in the manner described above.
図1Aは、実施例1で得られた炭素化材料のX線回折の結果を示す図であり、横軸は回折角2θ(°)、縦軸は回折強度(任意単位)である。まず、得られたX線回折パターン10Aに、回折角2θが5.5~12.5°の範囲で下に凸の部分、及び30.5~37°の範囲で下に凸の部分、の両方に接する直線を引き、当該直線を当該X線回折パターンのバックグラウンド20Aとした。次に、当該X線回折パターンから、前述のバックグランド20Aの値を指し引いたグラフ(以下、「ピークスペクトル」と示す)を作成した。図2Aは、図1AのX線回折パターンから得られたピークスペクトル30Aを表す図である。上述の方法によって得られたピークスペクトル30Aにおいて、回折角2θが11.5~15.0°の範囲おけるピークの最大強度をIFとし、18.0~26.5°の範囲におけるピークの最大強度をIGとした。実施例1においては、IGがIFに対して相対的に大きかった(具体的には、IG/IF=1.2)。
FIG. 1A is a diagram showing the results of X-ray diffraction of the carbonized material obtained in Example 1, wherein the horizontal axis represents the diffraction angle 2θ (°) and the vertical axis represents the diffraction intensity (arbitrary unit). First, in the obtained X-ray diffraction pattern 10A, a downward convex portion with a diffraction angle 2θ in the range of 5.5 to 12.5 °, and a downward convex portion in the range of 30.5 to 37 °, A straight line in contact with both was drawn, and the straight line was used as the background 20A of the X-ray diffraction pattern. Next, a graph (hereinafter referred to as “peak spectrum”) in which the value of the background 20A described above was subtracted from the X-ray diffraction pattern was created. FIG. 2A is a diagram showing a peak spectrum 30A obtained from the X-ray diffraction pattern of FIG. 1A. In the peak spectrum 30A obtained by the method described above, the maximum intensity of a peak definitive range diffraction angle 2θ of 11.5 ~ 15.0 ° and I F, the maximum peak in the range of 18.0 ~ 26.5 ° strength was I G. In Example 1, I G is greater relative to I F (specifically, I G / I F = 1.2 ).
図1Bは、比較例1のX線回折の結果を示す図である。上述の図1Aの場合と同様に、X線回折法により得られたX線回折パターン10Bに、回折角2θが5.5~12.5°の範囲で下に凸の部分、及び30.5~37°の範囲で下に凸の部分、の両方において接する直線20Bを引き、当該直線を当該X線回折パターン10Bのバックグラウンド20Bとした。次に、当該X線回折パターン10Bから、前述のバックグランド20Bの値を指し引いたピークスペクトル30Bを作成した。ピークスペクトル30Bにおいては、IGのピークがショルダーになっていたため、回折角20°における回折強度をIGとした。比較例1においては、IGがIFに対して相対的に小さかった(IG/IF=0.6)。
FIG. 1B is a diagram showing the results of X-ray diffraction of Comparative Example 1. Similarly to the case of FIG. 1A described above, the X-ray diffraction pattern 10B obtained by the X-ray diffraction method has a downwardly convex portion with a diffraction angle 2θ in the range of 5.5 to 12.5 °, and 30.5. A straight line 20B in contact with both of the downward convex portions in a range of ˜37 ° was drawn, and the straight line was used as the background 20B of the X-ray diffraction pattern 10B. Next, a peak spectrum 30B was created by subtracting the value of the background 20B from the X-ray diffraction pattern 10B. In the peak spectrum 30B, since the peak of the I G had become shoulder, the diffraction intensity at a diffraction angle 20 ° was I G. In Comparative Example 1, I G is smaller relative to I F (I G / I F = 0.6).
なお、上述の比較例1のように、IG、IFのピーク形状が不明瞭な場合は、回折角15°における回折強度をIFと、回折角20°における回折強度をIGとした。
Incidentally, as in Comparative Example 1 described above, when I G, is unclear peak shape I F is a diffraction intensity at a diffraction angle 15 ° was and I F, the diffraction intensity at a diffraction angle 20 ° and I G .
実施例2~7、比較例2~6においても、上述同様の方法でIG、IFを決定し、IFに対するIGの比(IG/IF)を求めた。
Examples 2-7, in Comparative Examples 2 to 6, I G in the above same method to determine the I F, to determine the ratio of I G for I F (I G / I F ).
[評価結果]
図3には、実施例1~7、比較例1~6において得られた炭素触媒を上述の方法で評価した結果を示す。すなわち、図3には、各実施例、各比較例で得られた炭素触媒について、当該炭素触媒の酸素還元開始電位(EO2)(V(vs.RHE))、当該炭素触媒のIG/IF比(-)を示す。 [Evaluation results]
FIG. 3 shows the results of evaluating the carbon catalysts obtained in Examples 1 to 7 and Comparative Examples 1 to 6 by the above-described method. That is, in FIG. 3, each of the embodiments, the carbon catalyst obtained in Comparative Example, the oxygen reduction onset potential of the carbon catalyst (E O2) (V (vs.RHE )), of the carbon catalyst I G / IF ratio (-) is shown.
図3には、実施例1~7、比較例1~6において得られた炭素触媒を上述の方法で評価した結果を示す。すなわち、図3には、各実施例、各比較例で得られた炭素触媒について、当該炭素触媒の酸素還元開始電位(EO2)(V(vs.RHE))、当該炭素触媒のIG/IF比(-)を示す。 [Evaluation results]
FIG. 3 shows the results of evaluating the carbon catalysts obtained in Examples 1 to 7 and Comparative Examples 1 to 6 by the above-described method. That is, in FIG. 3, each of the embodiments, the carbon catalyst obtained in Comparative Example, the oxygen reduction onset potential of the carbon catalyst (E O2) (V (vs.RHE )), of the carbon catalyst I G / IF ratio (-) is shown.
図3で示すように、実施例1~7に係る炭素触媒は、IG/IF比が1.0~3.0の範囲であり、酸素還元開始電位(EO2)は0.80V(vs.RHE)以上であった。
As shown in FIG. 3, the carbon catalysts according to Examples 1 to 7 have an I G / IF ratio in the range of 1.0 to 3.0, and the oxygen reduction starting potential (E O2 ) is 0.80 V ( vs. RHE) or higher.
これに対し、比較例1に係る炭素触媒(フラーレンスートそのもの)は、IG/IF比が0.6と低く、酸素還元開始電位(EO2)は、0.27V(vs.RHE)と、実施例1~7に比べ低かった。
In contrast, the carbon catalyst according to Comparative Example 1 (fullerene soot itself) is I G / I F ratio is as low as 0.6, the oxygen reduction onset potential (E O2) includes a 0.27V (Vs.RHE) It was lower than Examples 1-7.
また、フラーレンスートを400℃、600℃、800℃と温度を変化させて炭素化して得た、比較例2~4に係る炭素触媒については、IG/IF比は0.5~0.7と実施例1~7に比べ低く、酸素還元開始電位(EO2)についても、0.39~0,47V(vs.RHE)と低かった。
For the carbon catalysts according to Comparative Examples 2 to 4 obtained by carbonizing the fullerene soot at 400 ° C., 600 ° C., and 800 ° C., the I G / IF ratio is 0.5 to 0.00. 7, the oxygen reduction starting potential (E O2 ) was also as low as 0.39 to 0.47 V (vs. RHE).
このように、IG/IF比が1.0以上である炭素触媒は、IG/IF比が1.0未満の場合に比べ、高い酸素還元活性を有していた。
Thus, carbon catalyst I G / I F ratio is 1.0 or more, compared to the case I G / I F ratio is less than 1.0, and had a high oxygen reduction activity.
一方、比較例6に係る炭素化材料は、IG/IF比が12であり、酸素還元開始電位(EO2)は、0.52V(vs.RHE)であった。また、比較例5に係る炭素化材料は、IG/IF比が18.9であり、酸素還元開始電位(EO2)は、0.49V(vs.RHE)と比較例6の炭素化材料に対して、さらに低かった。このように、IG/IF比が12以上である炭素化材料は、酸素還元開始電位(EO2)がIG/IF比が12未満である場合に比べて低くかった。
On the other hand, carbonized material according to Comparative Example 6 is an I G / I F ratio 12, an oxygen reduction onset potential (E O2) was 0.52V (vs.RHE). Furthermore, carbonized material according to Comparative Example 5 is an I G / I F ratio 18.9, oxygen reduction onset potential (E O2), the carbon of Comparative Example 6 and 0.49V (vs.RHE) It was even lower than the material. Thus, the carbonized material having an I G / IF ratio of 12 or more had a lower oxygen reduction initiation potential (E O2 ) than that when the I G / IF ratio was less than 12.
また、実施例3に係る炭素化材料は、酸素還元開始電位(EO2)が0.83V(vs.RHE)であり、実施例1に係る炭素化材料(酸素還元開始電位が0.84V(vs.RHE))と同様に、高い酸素還元活性を有していた。実施例3に係る炭素化材料は、実施例1で用いた炭素化材料に、金属除去処理を施したものである。すなわち、実施例1に係る炭素化材料は、金属除去前後を通じて、高い触媒活性を有していたことになる。このように、実施例に係る炭素化材料の高い触媒活性は、上述のIG/IF比で示される炭素構造が寄与しているものと考えられた。
The carbonized material according to Example 3 has an oxygen reduction start potential (E O2 ) of 0.83 V (vs. RHE), and the carbonized material according to Example 1 (oxygen reduction start potential of 0.84 V ( v. RHE)), it had high oxygen reduction activity. The carbonized material according to Example 3 is obtained by subjecting the carbonized material used in Example 1 to metal removal treatment. That is, the carbonized material according to Example 1 had high catalytic activity before and after metal removal. Thus, it was considered that the high catalytic activity of the carbonized material according to the example was contributed by the carbon structure represented by the above-mentioned I G / IF ratio.
また、図3には記載していないが、フラーレンスートに替えてカーボンブラック(バルカン:XC)を使用した以外は実施例1と同様にして製造した炭素化材料についても、IG/IF比を評価しようとしたが、IFのピークが確認されなかった。
Further, although not shown in FIG. 3, the I G / I F ratio was also obtained for the carbonized material produced in the same manner as in Example 1 except that carbon black (Vulcan: XC) was used instead of fullerene soot. I tried to evaluate, but the peak of the I F has not been confirmed.
Further, although not shown in FIG. 3, the I G / I F ratio was also obtained for the carbonized material produced in the same manner as in Example 1 except that carbon black (Vulcan: XC) was used instead of fullerene soot. I tried to evaluate, but the peak of the I F has not been confirmed.
Claims (7)
- X線回折法において、回折角11.5°~15.0°におけるピークの最大強度(IF)に対する、回折角18.0°~26.5°におけるピークの最大強度(IG)の比(IG/IF)が、0.8以上、11以下を示す炭素構造、を有する
ことを特徴とする炭素触媒。 In the X-ray diffraction method, the ratio of the peak maximum intensity (I G ) at a diffraction angle of 18.0 ° to 26.5 ° to the peak maximum intensity (I F ) at a diffraction angle of 11.5 ° to 15.0 ° (I G / IF ) has a carbon structure showing 0.8 or more and 11 or less. - フラーレンスートと、金属と、を含む原料を炭素化して得られた炭素化材料、を含む
ことを特徴とする請求項1に記載の炭素触媒。 The carbon catalyst according to claim 1, comprising a carbonized material obtained by carbonizing a raw material containing fullerene soot and a metal. - フラーレンスートと、金属と、を含む原料を炭素化して得られた炭素化材料、を含む
ことを特徴とする炭素触媒。 A carbon catalyst comprising: a carbonized material obtained by carbonizing a raw material containing fullerene soot and a metal. - 酸素還元開始電位が0.60V(vs.RHE)以上である
ことを特徴とする請求項1乃至3のいずれかに記載の炭素触媒。 The carbon catalyst according to any one of claims 1 to 3, wherein an oxygen reduction starting potential is 0.60 V (vs. RHE) or more. - 請求項1乃至4のいずれかに記載された炭素触媒を含む
ことを特徴とする電極。 An electrode comprising the carbon catalyst according to any one of claims 1 to 4. - 請求項5に記載された電極を含む
ことを特徴とする電池。 A battery comprising the electrode according to claim 5. - フラーレンスートと、金属と、を含む原料を炭素化して炭素化材料を得ること、及び
前記炭素化材料を含む炭素触媒を得ること、
を含む、
ことを特徴とする炭素触媒の製造方法。
Carbonizing a raw material containing fullerence soot and a metal to obtain a carbonized material, and obtaining a carbon catalyst containing the carbonized material,
including,
A method for producing a carbon catalyst.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-249263 | 2013-12-02 | ||
| JP2013249263A JP6189197B2 (en) | 2013-12-02 | 2013-12-02 | Carbon catalyst, method for producing the same, electrode and battery using the same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015083571A1 true WO2015083571A1 (en) | 2015-06-11 |
Family
ID=53273336
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/081010 WO2015083571A1 (en) | 2013-12-02 | 2014-11-25 | Carbon catalyst and production method therefor as well as electrode and cell using such carbon catalyst |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP6189197B2 (en) |
| WO (1) | WO2015083571A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3570353A1 (en) * | 2018-05-14 | 2019-11-20 | Commissariat à l'Energie Atomique et aux Energies Alternatives | Catalytic layers comprising a fullerene |
| CN113368899A (en) * | 2021-07-03 | 2021-09-10 | 太原理工大学 | Preparation method of high-acid-density pseudo-cellulase resin solid acid catalyst |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010275140A (en) * | 2009-05-27 | 2010-12-09 | Hokkaido Univ | Hydrogen storage carbon material and manufacturing method thereof |
| JP2012200643A (en) * | 2011-03-24 | 2012-10-22 | Nec Corp | Oxygen reduction catalyst and method for preparing the same |
| JP2013111496A (en) * | 2011-11-25 | 2013-06-10 | Gunma Univ | Support for supporting metal, metal-supported catalyst, methanation reaction apparatus, and method relating to these |
-
2013
- 2013-12-02 JP JP2013249263A patent/JP6189197B2/en active Active
-
2014
- 2014-11-25 WO PCT/JP2014/081010 patent/WO2015083571A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010275140A (en) * | 2009-05-27 | 2010-12-09 | Hokkaido Univ | Hydrogen storage carbon material and manufacturing method thereof |
| JP2012200643A (en) * | 2011-03-24 | 2012-10-22 | Nec Corp | Oxygen reduction catalyst and method for preparing the same |
| JP2013111496A (en) * | 2011-11-25 | 2013-06-10 | Gunma Univ | Support for supporting metal, metal-supported catalyst, methanation reaction apparatus, and method relating to these |
Non-Patent Citations (2)
| Title |
|---|
| JUN'ICHI OZAKI: "Preparation of cathode catalysts for PEMFC by carbon-alloying techniques", TANSO, vol. 2005, no. 218, 15 June 2005 (2005-06-15), pages 178 - 184 * |
| SHIGERU SHUTO ET AL.: "Fullerene Soot-kei Denkyoku no Denki Kagaku Yoryo to Nano Kozo tono Sokan", ABSTRACTS OF ANNUAL MEETING OF THE CARBON SOCIETY OF JAPAN, 7 December 2005 (2005-12-07), pages 324 - 325 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3570353A1 (en) * | 2018-05-14 | 2019-11-20 | Commissariat à l'Energie Atomique et aux Energies Alternatives | Catalytic layers comprising a fullerene |
| CN113368899A (en) * | 2021-07-03 | 2021-09-10 | 太原理工大学 | Preparation method of high-acid-density pseudo-cellulase resin solid acid catalyst |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6189197B2 (en) | 2017-08-30 |
| JP2015104717A (en) | 2015-06-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4964292B2 (en) | Electrode and battery | |
| JP5149364B2 (en) | Carbon catalyst, method for producing the same, electrode and battery using the same | |
| US11398629B2 (en) | Carbon catalyst, electrode, and battery | |
| US11014074B2 (en) | Cell electrode, composition for cell electrode catalyst layer, and cell | |
| Sharma et al. | Effects of structural disorder and nitrogen content on the oxygen reduction activity of polyvinylpyrrolidone-derived multi-doped carbon | |
| US12080896B2 (en) | Carbon catalyst, battery electrode, and battery | |
| JP5689379B2 (en) | Catalyst support carrier, catalyst support, electrode and battery | |
| CN110891682A (en) | Carbon catalyst, battery electrode and battery | |
| JP6189197B2 (en) | Carbon catalyst, method for producing the same, electrode and battery using the same | |
| JP5732667B2 (en) | Method for producing carbon catalyst |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14868070 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14868070 Country of ref document: EP Kind code of ref document: A1 |