WO2015046405A1 - Analgesic - Google Patents
Analgesic Download PDFInfo
- Publication number
- WO2015046405A1 WO2015046405A1 PCT/JP2014/075571 JP2014075571W WO2015046405A1 WO 2015046405 A1 WO2015046405 A1 WO 2015046405A1 JP 2014075571 W JP2014075571 W JP 2014075571W WO 2015046405 A1 WO2015046405 A1 WO 2015046405A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- mmol
- cyano
- piperidine
- benzyl
- Prior art date
Links
- 0 **(*)C(N(CCC1)C[C@@]1C(NCc1ccccc1)=O)=O Chemical compound **(*)C(N(CCC1)C[C@@]1C(NCc1ccccc1)=O)=O 0.000 description 2
- JEXYLWKDNWOPPB-GFCCVEGCSA-N O=C([C@H]1CNCCC1)NCc1ccccc1 Chemical compound O=C([C@H]1CNCCC1)NCc1ccccc1 JEXYLWKDNWOPPB-GFCCVEGCSA-N 0.000 description 2
- LQLOGZQVKUNBRX-UHFFFAOYSA-N NCc1cc(I)ccc1 Chemical compound NCc1cc(I)ccc1 LQLOGZQVKUNBRX-UHFFFAOYSA-N 0.000 description 1
Images



Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/453—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
- A61P29/02—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID] without antiinflammatory effect
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to an analgesic.
- Pain is an experience with an unpleasant sensation or unpleasant emotion that occurs when tissue damage or potential damage is caused. Pain is mainly classified into nociceptive pain and neuropathic pain, depending on the cause. In the present specification, “pain” and “pain” are synonymous.
- Nociceptive pain refers to pain caused by damage to a living tissue or the addition of a noxious stimulus having such a risk, and refers to pain via a nociceptor. For example, physiological pain and inflammatory pain are applicable.
- Neuropathic pain is pathological pain caused by abnormal functioning of the peripheral or central nervous system itself, and is caused by direct damage or compression of nerve tissue even though nociceptors are not subjected to noxious stimulation. This refers to the pain that occurs.
- Neuropathic pain is defined as “pain caused directly by damage or disease to the somatosensory system” at the International Society of Pain (IASP) (Non-Patent Document 1), along with nociceptive pain and the like. It is positioned as one, and is classified into peripheral and central depending on the causative disease (Non-patent Document 2).
- Neuropathic pain is characterized by persistent spontaneous pain, allodynia (pain response to non-nociceptive stimuli) and hyperalgesia, and in patients with neuropathic pain, severe pain due to these is a long-term of several months to 10 years or more. Since QOL (Quality of life) is significantly reduced (Non-patent Document 3), active treatment is required.
- Non-steroidal anti-inflammatory analgesics NSAIDs
- narcotic analgesics opioid, etc.
- anticonvulsants and antidepressants are used as analgesics for neuropathic pain.
- Antiepileptic agents such as anti-anxiety agents, gabapentin and pregabalin are used.
- EETs epoxyeicosatrienoic acids
- sEH soluble epoxide hydrolase
- DHETs dihydroxyeicosatrienoic acids
- DHETs soluble epoxide hydrolase inhibitors
- sEH inhibitors have been shown to increase the amount of EETs (Non-Patent Document 5).
- compounds having sEH inhibitory activity have been reported, and these compounds have been shown to have analgesic effects on nociceptive pain and neuropathic pain (Non-Patent Documents 6 and 7).
- Patent Document 1 a heteroaryl amide derivative in which a heteroarylamine is condensed with nipecotic acid as a compound having a nipecotic acid diamide structure
- Patent Document 2 Amidine derivatives
- Patent Document 3 hydroxamic acid derivatives
- Non-steroidal anti-inflammatory analgesics used for nociceptive pain are associated with side effects such as gastrointestinal disorders and renal disorders, and narcotic analgesics are known to have side effects such as constipation, sleepiness, nausea and vomiting. And its use is known to be limited.
- Anticonvulsants, antidepressants, anxiolytics and antiepileptics used for neuropathic pain frequently involve central side effects such as dizziness, nausea and vomiting, which are difficult to discontinue for a long time or are discontinued. It has been pointed out that there are cases in which these drugs do not provide sufficient analgesic effects with these drugs (Non-patent Document 4).
- an object of the present invention is to provide a new analgesic effective for pain, particularly nociceptive pain and neuropathic pain.
- a novel nipecotic acid derivative or a pharmacologically acceptable salt thereof exhibits sEH inhibitory activity, and pain based on its action, In particular, it has been found that it exhibits an excellent analgesic effect against nociceptive pain and neuropathic pain, and has completed the present invention.
- the present invention provides an analgesic containing an nipecotic acid derivative represented by the following general formula (I) or a pharmacologically acceptable salt thereof as an active ingredient.
- R 1 represents a hydroxyl group, a cyano group, an alkyl group or alkyloxy group having 1 to 6 carbon atoms, a cycloalkyl group or cycloalkyloxy group having 3 to 6 carbon atoms, or an alkyloxyalkyl group having 2 to 7 carbon atoms.
- a cycloalkylalkyl group having 4 to 7 carbon atoms (the alkyl group, alkyloxy group, cycloalkyl group, cycloalkyloxy group, alkyloxyalkyl group and cycloalkylalkyl group have 1 to 3 hydrogen atoms, Each independently may be substituted with a halogen atom, a hydroxyl group, a cyano group, —SR 6 , —S ( ⁇ O) —R 6 or —S ( ⁇ O) 2 R 6 ), —N (R 6 ) C ( ⁇ O) R 7 , —N (R 6 ) S ( ⁇ O) 2 R 7 , —C ( ⁇ O) N (R 6 ) R 7 or a heteroaryl group having 5 ring atoms, R 2 and R 3 are Independently, a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, or an alkyloxyalkyl group having 2 to 7 carbon atoms (the alkyl group
- R 2 and R 3 each independently represents a hydrogen atom or an alkyl group having 1 to 6 carbon atoms, or together represent — (CH 2 ) 1 —.
- R 4 represents a substituent at the 2-position on the benzene ring
- R 5 represents a substituent at the 4-position on the benzene ring.
- R 1 represents —N (R 6 ) C ( ⁇ O) R 7 or —N (R 6 ) S ( ⁇ O) 2 R 7
- R 4 represents a halogen atom.
- R 5 represents a halogen atom, a cyano group, or an alkyl group or alkyloxy group having 1 to 6 carbon atoms
- R 6 represents a hydrogen atom.
- R 1 represents —N (H) C ( ⁇ O) CH 2 CH 3
- R 2 and R 3 together represent — (CH 2 ) 3 —
- R 4 represents It is particularly preferred that it represents —OCF 3 and R 5 represents a cyano group.
- the above analgesic is preferably an analgesic for nociceptive pain and / or neuropathic pain.
- the analgesic of the present invention exhibits a high therapeutic effect or preventive effect on nociceptive pain and neuropathic pain based on sEH inhibitory activity, so that it can be administered as a medicine for a wide range of pain symptoms. Become.
- the analgesic of the present invention is characterized by containing a nipecotic acid derivative represented by the following general formula (I) or a pharmacologically acceptable salt thereof as an active ingredient.
- R 1 represents a hydroxyl group, a cyano group, an alkyl group or alkyloxy group having 1 to 6 carbon atoms, a cycloalkyl group or cycloalkyloxy group having 3 to 6 carbon atoms, or an alkyloxyalkyl group having 2 to 7 carbon atoms.
- a cycloalkylalkyl group having 4 to 7 carbon atoms (the alkyl group, alkyloxy group, cycloalkyl group, cycloalkyloxy group, alkyloxyalkyl group and cycloalkylalkyl group have 1 to 3 hydrogen atoms, Each independently may be substituted with a halogen atom, a hydroxyl group, a cyano group, —SR 6 , —S ( ⁇ O) —R 6 or —S ( ⁇ O) 2 R 6 ), —N (R 6 ) C ( ⁇ O) R 7 , —N (R 6 ) S ( ⁇ O) 2 R 7 , —C ( ⁇ O) N (R 6 ) R 7 or a heteroaryl group having 5 ring atoms, R 2 and R 3 are Independently, a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, or an alkyloxyalkyl group having 2 to 7 carbon atoms (the alkyl group
- C1-C6 alkyl group means a straight-chain saturated hydrocarbon group having 1 to 6 carbon atoms or a branched saturated hydrocarbon group having 3 to 6 carbon atoms, such as a methyl group, Ethyl group, 1-propyl group, 2-propyl group, 1-butyl group, 2-butyl group, 2-methyl-2-propyl group (tert-butyl group), 2-methyl-1-propyl group, 2,2 -Dimethyl-1-propyl group, 1-pentyl group, 2-pentyl group or 3-pentyl group.
- C 1-6 alkyloxy group means a group in which the above C 1-6 alkyl group is bonded to an oxygen atom, such as a methoxy group, an ethoxy group, a 1-propyloxy group, -Propyloxy group, 1-butyloxy group or 2-butyloxy group.
- C3-C6 cycloalkyl group means a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, or a cyclohexyl group.
- C3-C6 cycloalkyloxy group means a cyclopropyloxy group, a cyclobutyloxy group, a cyclopentyloxy group, or a cyclohexyloxy group.
- C2-C7 alkyloxyalkyl group means a group having 2 to 7 carbon atoms in which one hydrogen atom of the alkyl group is substituted with an alkyloxy group.
- cycloalkyl alkyl group having 4 to 7 carbon atoms means a group having 4 to 7 carbon atoms in which one hydrogen atom of the alkyl group is substituted with a cycloalkyl group. Examples thereof include a methyl group, a cyclopropylethyl group, a cyclopropylpropyl group, a cyclobutylmethyl group, a cyclopentylmethyl group, and a cyclohexylmethyl group.
- Halogen atom means a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- heteroaryl group having 5 ring atoms means that the number of ring atoms is 5 including 1 to 4 identical or different atoms selected from the group consisting of a nitrogen atom, an oxygen atom and a sulfur atom. And includes, for example, pyrrolyl group, imidazolyl group, pyrazolyl group, triazolyl group, oxazolyl group, isoxazolyl group, furanyl group and thiazolyl group.
- R 1 may be —N (R 6 ) C ( ⁇ O) R 7 or —N (R 6 ) S ( ⁇ O) 2 R 7
- R 1 is acetylamidyl, propionamidyl group or methanesulfonylamidyl group.
- R 2 and R 3 are each independently a hydrogen atom or an alkyl group having 1 to 6 carbon atoms, or are preferably taken together as — (CH 2 ) 1 —.
- R 4 is preferably a substituent at the 2-position on the benzene ring.
- R 4 is preferably a halogen atom, an alkyl group having 1 to 6 carbon atoms or an alkyloxy group, more preferably a halogen atom or an alkyloxy group, and further preferably an alkyloxy group.
- R 5 is preferably a substituent at the 4-position on the benzene ring.
- R 5 is preferably a halogen atom, a cyano group, an alkyl group having 1 to 6 carbon atoms, or an alkyloxy group having 1 to 6 carbon atoms, and more preferably a halogen atom or a cyano group.
- R 6 is preferably a hydrogen atom
- R 7 is preferably a methyl group or an ethyl group.
- L is preferably 2 or 3
- m is preferably 2
- n is preferably 2.
- nipecotic acid derivative (I) has at least one asymmetric carbon atom and has optical isomers and diastereomers.
- the nipecotic acid derivative (I) includes not only a single isomer but also a racemate and a mixture of diastereomers.
- all rotamers are included.
- Examples of the pharmacologically acceptable salt of the nipecotic acid derivative (I) include, for example, hydrochloride, trifluoroacetate, sulfate, nitrate, hydrobromide, hydroiodide or methane as an acid addition salt.
- Examples of the sulfonate include hydrochloride, sulfate, hydrobromide, hydroiodide, and methanesulfonate.
- the starting materials and reagents used for the production of the nipecotic acid derivative (I) may be commercially available products or may be synthesized by known methods.
- the nipecotic acid derivative (Ia) can be produced, for example, by a condensation reaction of an amine derivative (II) and a carboxylic acid derivative (III) in the presence of a base and a condensing agent as shown in the following scheme 1.
- Scheme 1 [Wherein R 1 ′ represents a hydroxyl group, a cyano group, an alkyl group or alkyloxy group having 1 to 6 carbon atoms, a cycloalkyl group or cycloalkyloxy group having 3 to 6 carbon atoms, or an alkyloxy group having 2 to 7 carbon atoms.
- alkyl group, a cycloalkylalkyl group having 4 to 7 carbon atoms (the alkyl group, alkyloxy group, cycloalkyl group, cycloalkyloxy group, alkyloxyalkyl group and cycloalkylalkyl group have 1 to 3 hydrogen atoms)
- R 2 to R 6 are the same as defined above. ]
- condensing agent used in the condensation reaction examples include cyclohexylcarbodiimide, N-ethyl-N′-3-dimethylaminopropylcarbodiimide hydrochloride, benzotriazol-1-yloxy-trisdimethylaminophosphonium salt (BOP reagent), 1- [ Bis (dimethylamino) methylene] -1H-benzotriazolium-3-oxide hexafluorophosphate (HBTU) or O- (7-azabenzotriazol-1-yl) tetramethyluronium hexafluorophosphate HATU), and HATU is preferred.
- the equivalent of the condensing agent is preferably 1 to 10 equivalents, and more preferably 1 to 3 equivalents.
- Examples of the solvent used in the condensation reaction include N, N-dimethylformamide (hereinafter referred to as DMF), tetrahydrofuran (hereinafter referred to as THF), dichloromethane, chloroform, diethyl ether or dimethyl ether, with DMF or THF being preferred, and DMF being More preferred.
- DMF N, N-dimethylformamide
- THF tetrahydrofuran
- dichloromethane dichloromethane
- chloroform chloroform
- diethyl ether or dimethyl ether dimethyl ether
- Examples of the base used in the condensation reaction include organic bases such as diisopropylethylamine (hereinafter DIPEA), triethylamine (hereinafter TEA), pyridine or N-methylmorpholine, or organic acid salts such as potassium carbonate, sodium carbonate or sodium bicarbonate. DIPEA or TEA is preferable.
- the equivalent of the base is preferably 1 to 100 equivalents and more preferably 1 to 10 equivalents with respect to the amine derivative (II).
- the equivalent amount of the carboxylic acid derivative (III) used in the condensation reaction is preferably 0.1 to 100 equivalents, more preferably 0.1 to 10 equivalents, and even more preferably 0.8 to 2 equivalents with respect to the amine derivative (II). .
- the reaction temperature of the condensation reaction is preferably ⁇ 50 to 100 ° C., more preferably 0 to 50 ° C., and further preferably 0 to 30 ° C.
- the reaction time for the condensation reaction is preferably 1 minute to 48 hours, more preferably 1 minute to 24 hours, and even more preferably 10 minutes to 24 hours.
- the concentration of the amine derivative (II) at the start of the condensation reaction is preferably 0.01 to 100M, more preferably 0.01 to 10M, and even more preferably 0.1 to 10M.
- nipecotic acid derivative (Ib) in which R 1 is —N (H) C ( ⁇ O) R 7 is represented by, for example, an amine derivative (IV) in the presence of a base as shown in Scheme 2 below.
- an acid chloride derivative (V), or a condensation reaction between an amine derivative (IV) and a carboxylic acid derivative (VI) in the presence of a base and a condensing agent. Scheme 2 [Wherein R 2 to R 5 and R 7 are the same as defined above]. ]
- Examples of the solvent used for the condensation reaction with the acid chloride derivative (V) include dichloromethane, 1,2-dichloroethane, acetonitrile, DMF, THF, dioxane, diethyl ether or 1,2-dimethoxyethane. 1,2-dichloroethane, acetonitrile or THF is preferred, and dichloromethane or 1,2-dichloroethane is more preferred.
- the equivalent amount of the acid chloride (V) used in the condensation reaction with the acid chloride derivative (V) is preferably 0.1 to 10 equivalents, more preferably 1 to 3 equivalents, relative to the amine derivative (IV). 5 equivalents are more preferred.
- Examples of the base used for the condensation reaction with the acid chloride derivative (V) include organic bases such as DIPEA, TEA, pyridine and N-methylmorpholine, with DIPEA or TEA being preferred.
- the equivalent of the base is preferably 1 to 100 equivalents and more preferably 1 to 10 equivalents with respect to the amine derivative (IV).
- the reaction temperature of the condensation reaction with the acid chloride derivative (V) is preferably ⁇ 50 to 100 ° C., more preferably ⁇ 20 to 60 ° C., and further preferably 0 to 40 ° C.
- the reaction time for the condensation reaction with acid chloride (V) is preferably 30 minutes to 24 hours, more preferably 30 minutes to 12 hours, and even more preferably 30 minutes to 8 hours.
- the concentration at the start of the reaction of the amine derivative (IV) in the condensation reaction with the acid chloride derivative (V) is preferably 0.01 to 100M, more preferably 0.01 to 10M, and further preferably 0.1 to 10M.
- nipecotic acid derivative (Ic) in which R 1 is —N (H) S ( ⁇ O) 2 R 7 can be synthesized, for example, with an amine derivative (IV) in the presence of a base as shown in Scheme 3 below. It can be produced by a sulfonamidation reaction with a sulfonic acid chloride derivative (VII). (Scheme 3) [Wherein R 2 to R 5 and R 7 are the same as defined above]. ]
- Examples of the solvent used in the sulfonamidation reaction include dichloromethane, 1,2-dichloroethane, acetonitrile, DMF, THF, dioxane, diethyl ether, or 1,2-dimethoxyethane, but dichloromethane, 1,2-dichloroethane, Acetonitrile or THF is preferred, and dichloromethane or 1,2-dichloroethane is more preferred.
- the equivalent amount of the sulfonic acid chloride derivative (VII) used in the sulfonamidation reaction is preferably 0.1 to 10 equivalents, more preferably 1 to 3 equivalents, and further preferably 1 to 1.5 equivalents with respect to the amine derivative (IV). preferable.
- Examples of the base used in the sulfonamidation reaction include organic bases such as DIPEA, TEA, pyridine and N-methylmorpholine, with DIPEA or TEA being preferred.
- the equivalent of the base is preferably 1 to 100 equivalents and more preferably 1 to 10 equivalents with respect to the amine derivative (IV).
- the reaction temperature of the sulfonamidation reaction is preferably ⁇ 50 to 50 ° C., more preferably ⁇ 30 to 30 ° C., and further preferably ⁇ 20 to 20 ° C.
- the reaction time of the sulfonamidation reaction is preferably 30 minutes to 24 hours, more preferably 30 minutes to 12 hours, and further preferably 30 minutes to 8 hours.
- the concentration of the amine derivative (IV) at the start of the reaction in the sulfonamidation reaction is preferably 0.01 to 100M, more preferably 0.01 to 10M, and further preferably 0.1 to 10M.
- the amine derivative (IV) which is the starting material in the above-mentioned schemes 2 and 3, is, for example, after the condensation reaction between the amine derivative (II) and the carboxylic acid derivative (VIII) in the presence of a base, as shown in the following scheme 4.
- the deprotection reaction following the condensation reaction can be performed by a known method described in, for example, Protective Groups in Organic Synthesis 3rd Edition (Green et al., 1999, John Wiley & Sons, Inc.).
- the protecting group is a tert-butoxycarbonyl group
- the protecting group can be removed by treatment with a strong acid such as trifluoroacetic acid.
- the amine derivative (II) which is the starting material in the above-mentioned schemes 1 and 4 is, for example, as shown in the following scheme 5, in the presence of a base and a condensing agent, benzylamine derivative (IX), nipecotic acid derivative (X) and After the condensation reaction, a deprotection reaction for removing the protecting group can be used.
- Scheme 5 [Wherein R 4 , R 5 and R 8 are the same as defined above. ]
- the deprotection reaction can be carried out under the same conditions as in the method described in Scheme 4.
- the condensation reaction of Scheme 5 can also be performed in the presence of a base by converting the nipecotic acid derivative (X) into an acid chloride.
- Examples of the reagent for converting the nipecotic acid derivative (X) into acid chloride include oxalyl chloride and thionyl chloride, with oxalyl chloride being preferred.
- the equivalent amount of the reagent is preferably 1 to 10 equivalents, more preferably 1 to 1.5 equivalents, relative to the nipecotic acid derivative (X).
- Examples of the solvent used for converting the nipecotic acid derivative (X) into the acid chloride include dichloromethane, chloroform, THF, 1,2-dichloroethane, acetonitrile, 1,4-dioxane, and DMF, and dichloromethane, THF, or DMF.
- a mixed solvent thereof is preferable, and a mixed solvent of dichloromethane and DMF or a mixed solvent of THF and DMF is more preferable.
- the reaction temperature for converting the nipecotic acid derivative (X) to acid chloride is preferably -50 to 100 ° C, more preferably -30 to 30 ° C, and further preferably -20 to 0 ° C.
- the reaction time for converting the nipecotic acid derivative (X) to acid chloride is preferably 30 minutes to 24 hours, more preferably 30 minutes to 12 hours, and even more preferably 30 minutes to 2 hours.
- the concentration of the nipecotic acid derivative (X) at the start of the reaction when converting the nipecotic acid derivative (X) to acid chloride is preferably 0.01 to 100M, more preferably 0.01 to 10M, and more preferably 0.1 to 3M. Is more preferable.
- nipecotic acid derivative (I) obtained as described above, a pharmacologically acceptable salt thereof, or an intermediate, a raw material compound or a reagent used for the production of the nipecotic acid derivative (I) is necessary. Depending on the method, it may be isolated and purified by a method such as extraction, distillation, chromatography or recrystallization.
- the above analgesic is characterized by containing the above nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof as an active ingredient, and is based on sEH inhibitory activity, and in particular, nociceptive pain In addition, it can exert a high analgesic effect on neuropathic pain.
- SEH is an abbreviation for soluble epoxide hydrolase, which is a metabolic enzyme that catalyzes the hydrolysis of epoxide and converts it to the corresponding diol.
- the most known substrate of sEH is EETs, which is one of endothelial cell-derived hyperpolarizing factors, and sEH has an action of metabolizing EETs to DHETs and inactivating them.
- EETs is an abbreviation for Epoxyeicosatrienoic acids
- DHETs is an abbreviation for Dihydroxyeicosatrienoic acids. Examples of the EETs include 14,15-epoxyeicosatrienoic acid (hereinafter, 14,15-EET). Examples of DHETs include 14,15-dihydroxyeicosatrienoic acid (14,15-DHET).
- SEH inhibitory activity means an activity that inhibits the action of sEH. Therefore, the sEH inhibitory activity includes the activity of inhibiting the enzymatic reaction of sEH to catalyze the hydrolysis of EETs, which is one of sEH substrates.
- SEH inhibitor means a compound showing sEH inhibitory activity or a composition containing the compound as an active ingredient.
- the sEH inhibitory activity is, for example, that human sEH and its substrate EETs are reacted in the presence of an sEH inhibitor, and the amount of DHETs produced is compared with the amount of DHETs produced in the absence of the sEH inhibitor. Can be measured.
- a commercially available measurement kit Soluable Epoxide Hydrose Inhibitor Screening Assay Kit; Cayman
- the sEH inhibitory activity of the inhibitor can be measured.
- racemic 4-nitrophenyl-trans-2,3-epoxy-3-phenylpropyl carbonate was used as a substrate for sEH, and 4-nitrophenolate anion was used.
- the appearance of 6-methoxy-2-naphthaldehyde is measured using cyano (6-methoxynaphthalen-2-yl) methyl 2- (3-phenyloxiran-2-yl) acetate as a substrate for sEH.
- the sEH inhibitory activity of the sEH inhibitor can also be measured by measuring the appearance.
- Nociceptive pain refers to pain caused by damage to biological tissue or the addition of a noxious stimulus having such a risk, and refers to pain via nociceptors.
- nociceptive pain examples include pain caused by injuries such as fractures and cuts, postoperative pain, sprain pain, bruise pain, joint pain, low back pain, muscle pain, post-extraction pain, toothache, appendicitis, rheumatoid arthritis. Pain due to inflammatory diseases such as rheumatic fever, osteoarthritis, ankylosing spondylitis, osteoarthritis, cervical shoulder arm syndrome, periarthritis, connective tissue inflammation, acute otitis media, prostatitis, alveolar periosteitis, vaginitis Can be mentioned.
- the nociceptive pain includes deep pain and visceral pain (for example, headache, abdominal pain, back and back pain, chronic pelvic pain syndrome, pain associated with endometriosis, pain associated with urolithiasis and urethral stones, Pain associated with gastrointestinal lesions, pelvic pain, pain associated with urological disorders).
- More preferable target diseases of the above analgesic include rheumatoid arthritis, osteoarthritis, postoperative pain, joint pain, low back pain, muscle pain or toothache.
- Neuroopathic pain is pathological pain caused by abnormalities in the function of the peripheral or central nervous system itself, and direct damage or compression of nerve tissue even though nociceptors are not subjected to noxious stimuli. This refers to pain caused by the like.
- neuropathic pain examples include cancer pain, herpes zoster pain, postherpetic neuralgia, painful diabetic neuropathic pain, AIDS-related neuralgia, neuropathic back pain, phantom limb pain, anticancer Neuropathic pain caused by administration of agents, pain after spinal cord injury, pain due to strangulated neuropathy such as carpal trunk syndrome and spinal canal stenosis, pain due to Guillain-Barre syndrome, or trigeminal neuralgia.
- nipecotic acid derivative (I) which is an active ingredient of the above-mentioned analgesic agent, or a pharmacologically acceptable salt thereof should be evaluated using an appropriate animal model.
- Suitable animal models for nociceptive pain include, for example, the mouse acetate rising model, the rat formalin test, the rat carrageenan-induced inflammation model, the mouse or rat hot plate test for acute pain, or the tail flick test.
- suitable animal models of neuropathic pain include, for example, a mouse or rat partial sciatic nerve ligation model or a mouse or rat spinal nerve ligation model, or a mouse or rat streptozotocin-induced diabetic neuropathy model.
- Acute pain is usually short-term, but includes acute postoperative pain, herpes zoster pain, post-extraction pain, or trigeminal neuralgia.
- Chronic pain is usually defined as pain lasting for 3-6 months and includes somatic and psychogenic pain, such as rheumatoid arthritis, osteoarthritis, painful diabetic neuropathic pain Chronic postoperative neuropathic pain or postherpetic neuralgia.
- analgesic is prepared by using a nipecotic acid derivative (I), which is an active ingredient, or a pharmacologically acceptable salt thereof as it is or as a pharmaceutical composition in an appropriate dosage form for mammals (eg, mice, rats, etc.). , Hamsters, rabbits, dogs, monkeys, cows, sheep or humans) orally or parenterally (eg, transdermal, intravenous, rectal, inhalation, nasal or ophthalmic) Can be administered.
- mammals eg, mice, rats, etc.
- mammals eg, mice, rats, etc.
- parenterally eg, transdermal, intravenous, rectal, inhalation, nasal or ophthalmic
- Examples of the dosage form for administration to mammals include tablets, powders, pills, capsules, granules, syrups, solutions, injections, emulsions, suspensions or suppositories, or known continuous forms.
- a formulation is mentioned.
- These dosage forms can be produced by a known method and contain a carrier generally used in the pharmaceutical field. Examples of such carriers include excipients, lubricants, binders, disintegrants in solid preparations, and solvents, solubilizers, suspending agents, or soothing agents in liquid preparations.
- excipient examples include lactose, D-mannitol, starch, sucrose, corn starch, crystalline cellulose, and light anhydrous silicic acid.
- lubricant examples include magnesium stearate, calcium stearate, talc, and colloidal silica.
- binder examples include crystalline cellulose, D-mannitol, dextrin, hydroxypropylcellulose, hydroxypropylmethylcellulose, polyvinylpyrrolidone, starch, sucrose, gelatin, methylcellulose or sodium carboxymethylcellulose.
- disintegrant examples include starch, carboxymethyl cellulose, carboxymethyl cellulose calcium, croscarmellose sodium, carboxymethyl starch sodium, and L-hydroxypropyl cellulose.
- solvent examples include water for injection, alcohol, propylene glycol, macrogol, sesame oil or corn oil.
- solubilizer examples include polyethylene glycol, propylene glycol, D-mannitol, benzyl benzoate, ethanol, cholesterol, triethanolamine, sodium carbonate, or sodium citrate.
- suspending agent examples include surfactants such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalkonium chloride, benzethonium chloride or glyceryl monostearate, or polyvinyl alcohol, polyvinylpyrrolidone, methylcellulose. , Hydrophilic polymers such as hydroxymethylcellulose, hydroxyethylcellulose or hydroxypropylcellulose.
- surfactants such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalkonium chloride, benzethonium chloride or glyceryl monostearate, or polyvinyl alcohol, polyvinylpyrrolidone, methylcellulose.
- Hydrophilic polymers such as hydroxymethylcellulose, hydroxyethylcellulose or hydroxypropylcellulose.
- Examples of soothing agents include benzyl alcohol.
- Examples of the isotonic agent include glucose, sodium chloride, D-sorbitol, and D-mannitol.
- buffer solutions such as phosphate, acetate, carbonate or citrate.
- preservative examples include p-oxybenzoates, chlorobutanol, benzyl alcohol, phenethyl alcohol, dehydroacetic acid, and sorbic acid.
- antioxidant examples include sulfite and ascorbic acid.
- the analgesic preferably contains 0.001 to 99% by weight, more preferably 0.01 to 99% by weight, of the nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof.
- the effective dosage and frequency of administration of the nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof vary depending on the dosage form, patient age, body weight, or the nature or severity of the symptoms to be treated / prevented. However, usually 1 to 1000 mg, preferably 1 to 300 mg per day for an adult can be administered in one or several divided doses.
- analgesics may be administered alone, but may be combined with other drugs or combined with other drugs to supplement or enhance the preventive or therapeutic effect of the disease or reduce the dose. It can also be used in combination.
- Examples of other drugs that can be combined or used together include antitussives, expectorants, antitussive expectorants, bronchodilators, peptic ulcers, antibiotics, or narcotic analgesics.
- the timing of administration of the analgesic and the concomitant drug is not particularly limited, and these may be administered simultaneously to the administration subject, May be administered.
- the concomitant drug may be a low molecular compound, a polymer such as a protein, polypeptide or antibody, or a vaccine.
- the dose of the concomitant drug can be appropriately selected based on the clinically used dose.
- the mixing ratio of the analgesic and the concomitant drug can be appropriately selected depending on the administration subject, administration route, target disease, symptom, combination of the above analgesic and concomitant drug, and the like. For example, when the administration subject is a human, the concomitant drug may be used in a mixing ratio of 0.01 to 99.99 with respect to the above analgesic.
- Antitussives, expectorants and antitussive expectorants include, for example, dextromethorphan, benproperine, dimemorphan, clofedanol, ephedrine, decoben, minc ), Methylephedrine, acetylcysteine, ambroxol, carbocysteine, bromhexine, epradinone, incode, indehyde Ydrocodeine) or tipepidine (tipepidine) and the like.
- bronchodilators examples include clenbuterol, cromoglycate, salbutamol, salmeterol, tulobuterol, and theophylline.
- peptic ulcer agents examples include azulene, aldioxa, irsogladine, ecabet, omeprazole, ornoprostil, cimetidine, cimetidine, cimetidine. , Sulpiride, cetraxate or famotidine.
- Antibiotics include, for example, amoxicillin, azithromycin, erythromycin, clarithromycin, tetracycline, and doxycycline (doxycycline).
- narcotic analgesics examples include opium alkaloids, ethyl morphine, oxycodone, morphine, cocaine, fentanyl, and pethidine.
- Step 2 Synthesis of (4-bromo-2- (trifluoromethoxy) phenyl) methanol: At ⁇ 10 ° C., sodium borohydride (2.4 g, 63 mmol) was added to a methanol (0.23 L) solution of Reference Example compound 1 (16 g, 59 mmol). After stirring at ⁇ 10 ° C. for 10 minutes, acetone (10 mL) and 1N hydrochloric acid (10 mL) were added to the reaction solution. The reaction solution was concentrated under reduced pressure, water was added to the obtained crude product, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure.
- Step 3 Synthesis of 4-bromo-2- (trifluoromethoxy) benzyl methanesulfonate: Methanesulfonyl chloride (0.93 g, 8.1 mmol) was added to a solution of Reference Example Compound 2 (2.0 g, 7.4 mmol) and TEA (1.2 mL, 8.9 mmol) in dichloromethane (20 mL) under ice cooling. It was. After stirring at room temperature for 3 hours, water was added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. 2.6 g of 4-bromo-2- (trifluoromethoxy) benzyl methanesulfonate (hereinafter referred to as Reference Example Compound 3) ( (Quantitative).
- Step 5 Synthesis of (4-bromo-2- (trifluoromethoxy) phenyl) methanamine: Hydrazine monohydrate (0.98 g, 19 mmol) was added to a methanol (40 mL) solution of Reference Example compound 4 (2.6 g, 6.5 mmol) at room temperature. After stirring at 60 ° C. for 2 hours, the solid precipitated at room temperature was filtered off. The filtrate was concentrated under reduced pressure, and the resulting crude product was dissolved in ethyl acetate and washed with water and a saturated aqueous sodium chloride solution.
- Step 7 Synthesis of (R) -tert-butyl 3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carboxylate: At room temperature, a solution of Reference Compound 6 (0.050 g, 0.10 mmol) and zinc cyanide (0.012 g, 0.10 mmol) in DMF (2.0 mL) was added to tetrakistriphenylphosphine palladium (0) (0. 030 g, 0.026 mmol) was added. After stirring at 150 ° C. for 30 minutes, water was added to the reaction solution at room temperature, and the mixture was extracted with diethyl ether.
- Step 8 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide: Under ice-cooling, trifluoroacetic acid (hereinafter TFA) (35 mL, 0.45 mol) was added to a solution of Reference Example Compound 7 (6.9 g, 16 mmol) in dichloromethane (0.16 L). After stirring at room temperature for 1 hour, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane, neutralized with a saturated aqueous sodium carbonate solution, and extracted with dichloromethane.
- TFA trifluoroacetic acid
- Example Compound 1 (1-propionamidocyclobutanecarbonyl) piperidine-3-carboxamide was obtained in an amount of 6.2 g (71%).
- Example Compound 2 (2-Methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 2) was obtained in an amount of 2.4 g (87%).
- Example Compound 3 (1- (methylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 3) was obtained in an amount of 0.56 g (74%).
- Example 4 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (trifluoromethyl) cyclopropanecarbonyl) piperidine-3-carboxamide: The same reaction as in Example 1 [Step 9] was carried out using 1- (trifluoromethyl) cyclopropanecarboxylic acid (0.054 g, 0.17 mmol) to give (R) -N- (4-cyano- 0.044 g (58%) of 2- (trifluoromethoxy) benzyl) -1- (1- (trifluoromethyl) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 4) was obtained.
- Example 5 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (methylsulfonamido) cyclobutanecarbonyl) piperidine-3-carboxamide: (R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 2 [Step 3] using Reference compound 10 (0.020 g, 0.047 mmol). 0.017 g (71%) of benzyl) -1- (1- (methylsulfonamido) cyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 5) was obtained.
- Example 6 Synthesis of (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-isobutyramidecyclobutanecarbonyl) piperidine-3-carboxamide: (R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by carrying out the same reaction as in Example 1 [Step 11] using isobutyryl chloride (0.0055 g, 0.052 mmol). 0.022 g (95%) of (benzyl) -1- (1-isobutyramidecyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 6) was obtained.
- Example 7 Synthesis of (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-pivalamidocyclobutanecarbonyl) piperidine-3-carboxamide: (R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by carrying out the same reaction as in Example 1 [Step 11] using pivaloyl chloride (0.0063 g, 0.052 mmol). 0.017 g (72%) of (benzyl) -1- (1-pivalamidocyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 7) was obtained.
- Example 9 Synthesis of (R) -1- (1-acetamidocyclobutanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide: (R) -1- (1-acetamidocyclobutanecarbonyl) -N- (4) was prepared by performing the same reaction as in Example 8 [Step 3] using Reference compound 10 (0.020 g, 0.047 mmol). 0.013 g (60%) of -cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 9) was obtained.
- Step 2 Synthesis of (R) -1-((R) -2-acetamido-3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide: (R) -1-((R) -2-acetamido-3-methyl) was prepared by performing the same reaction as in Example 8 [Step 3] using Reference compound 23 (0.020 g, 0.047 mmol). 0.018 g (83%) of butanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 11) was obtained.
- Example 12 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3- Synthesis of carboxamide: (R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by conducting the same reaction as in Example 2 [Step 3] using Reference Example Compound 23 (0.020 g, 0.047 mmol). 0.020 g (83%) of benzyl) -1-((R) -3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 12) was obtained.
- Example Compound 13 was obtained in an amount of 0.0080 g (13%).
- Example Compound 14 (2-Methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide was obtained in an amount of 0.013 g (63%).
- Example 15 (R) -N- (4-carbamoyl-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide Synthesis: (R) -N- (4-carbamoyl-2- (trifluoromethoxy) benzyl) -1 was prepared by carrying out the same reaction as in Example 14 using Example Compound 3 (0.025 g, 0.051 mmol). As a result, 0.020 g (77%) of — (2-methyl-2- (methylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 15) was obtained.
- Example 16 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-hydroxy-2-methylpropanoyl) piperidine-3-carboxamide: The same reaction as in Example 1 [Step 9] was performed using 2-hydroxy-2-methylpropanoic acid (0.15 g, 0.46 mmol) to give (R) -N- (4-cyano-2- 0.12 g (62%) of (trifluoromethoxy) benzyl) -1- (2-hydroxy-2-methylpropanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 16) was obtained.
- Example 17 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-pivaloylpiperidine-3-carboxamide: By performing the same reaction as in Example 1 [Step 11] using Reference Example Compound 8 (0.15 g, 0.46 mmol) and pivaloyl chloride (0.066 g, 0.55 mmol), (R) — 0.19 g (quantitative) of N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-pivaloylpiperidine-3-carboxamide (hereinafter, Example Compound 17) was obtained.
- Step 2 Synthesis of ethyl 1- (N-methylmethylsulfonamido) cyclopropanecarboxylate: Sodium hydride (55 wt%, 0.51 g, 12 mmol) was added to a DMF (10 mL) solution of Reference Example Compound 24 (2.0 g, 9.7 mmol) under ice cooling. The mixture was stirred for 10 minutes under ice cooling, and then stirred for 30 minutes at room temperature. Methyl iodide (0.78 mL, 13 mmol) was added to the reaction solution under ice cooling.
- Step 3 Synthesis of 1- (N-methylmethylsulfonamido) cyclopropanecarboxylic acid: A 1N aqueous sodium hydroxide solution (12 mL, 12 mmol) was added to a methanol (20 mL) solution of Reference Example Compound 25 (1.8 g, 7.9 mmol) at room temperature. After stirring at 50 ° C. for 3 hours, 1N hydrochloric acid was added to the reaction solution at room temperature, and the mixture was extracted with chloroform. The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to obtain 0.88 g (58 %)Obtained.
- HATU (0.35 g, 0.93 mmol) was added to a DMF (1.6 mL) solution of the obtained crude product (0.10 g) and DIPEA (0.30 mL, 1.7 mmol) under ice cooling. After stirring for 15 minutes under ice cooling, Reference Example Compound 8 (0.28 g, 0.85 mmol) was added to the reaction solution. After stirring overnight at room temperature, water was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure.
- Step 3 Synthesis of (R) -1-((R) -2-aminobutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide: (R) -1-((R) -2-aminobutanoyl)-was prepared by carrying out the same reaction as in Example 1 [Step 10] using Reference Example Compound 28 (0.47 g, 0.91 mmol). 0.38 g (quantitative) of N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 29) was obtained.
- Step 4 Synthesis of (R) -1-((R) -2-acetamidobutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide: (R) -1-((R) -2-acetamidobutanoyl)-was prepared by conducting the same reaction as in Example 8 [Step 3] using Reference compound 29 (0.091 g, 0.22 mmol). 0.085 g (85%) of N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 21) was obtained.
- Example 22 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide: (R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 2 [Step 3] using Reference compound 29 (0.096 g, 0.23 mmol). 0.090 g (79%) of (benzyl) -1-((R) -2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 22) was obtained.
- Example 23 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-cyanocyclopropanecarbonyl) piperidine-3-carboxamide: The same reaction as in Example 1 [Step 9] was performed using 1-cyanocyclopropanecarboxylic acid (0.034 g, 0.31 mmol) to give (R) -N- (4-cyano-2- (tri 0.081 g (63%) of fluoromethoxy) benzyl) -1- (1-cyanocyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 23) was obtained.
- Morpholine (0.36 mL, 4.1 mmol) was added to a solution of the obtained crude product (0.53 g) in DMF (4.0 mL) at room temperature. After stirring at room temperature for 6 hours, water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure.
- Example Compound 24 The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure.
- Example 25 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide: (R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 2 [Step 3] using Reference compound 30 (0.10 g, 0.25 mmol). 0.099 g (83%) of benzyl) -1-((R) -2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 25) was obtained.
- Example 26 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-isobutyramidecyclopropanecarbonyl) piperidine-3-carboxamide: By performing the same reaction as in Example 1 [Step 11] using Reference Example Compound 14 (0.020 g, 0.049 mmol) and isobutyryl chloride (0.0062 g, 0.058 mmol), (R) — 0.017 g (71%) of N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-isobutyramidecyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 26) was obtained. It was.
- Example 27 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-pivalamidocyclopropanecarbonyl) piperidine-3-carboxamide: By performing the same reaction as in Example 1 [Step 11] using Reference Example Compound 14 (0.020 g, 0.049 mmol) and pivaloyl chloride (0.0064 g, 0.058 mmol), (R) — 0.018 g (73%) of N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-pivalamidocyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 27) Obtained.
- Example 28 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (4- (methylsulfonamido) tetrahydro-2H-pyran-4-carbonyl) piperidine-3- Synthesis of carboxamide: [Step 1] Synthesis of 8-oxa-1,3-diazaspiro [4.5] decane-2,4-dione: Dihydro-2H-pyran 4 (3H) -one (2.0 g, 20 mmol), ammonium carbonate (9.6 g, 0.10 mol), TEA (2.8 mL, 20 mmol) in water: methanol (water: methanol) at room temperature.
- Example 29 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (cyclopropanecarboxamido) cyclobutanecarbonyl) piperidine-3-carboxamide: (R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by carrying out the same reaction as in Example 1 [Step 11] using cyclopropanecarbonyl chloride (0.0059 g, 0.057 mmol). 0.012 g (52%) of benzyl) -1- (1- (cyclopropanecarboxamide) cyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 29) was obtained.
- Step 2 Synthesis of (S) -tert-butyl (1,3-dihydroxy-3-methylbutan-2-yl) carbamate: Methyl magnesium bromide diethyl ether solution (3N, 30 mL, 91 mmol) was added to a diethyl ether (0.12 L) solution of Reference Example compound 35 (4.0 g, 18 mmol) at ⁇ 78 ° C. After stirring at room temperature for 1 hour, a saturated aqueous ammonium chloride solution and water were added to the reaction solution under ice cooling, and the mixture was extracted with ethyl acetate.
- Example Compound 30 ((R) -1-((R) -2-acetamido-3-hydroxy 0.029 g (66%) of -3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 30) was obtained.
- Example 31 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -3-hydroxy-3-methyl-2-propionamidobutanoyl) piperidine- Synthesis of 3-carboxamide: (R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by conducting the same reaction as in Example 1 [Step 11] using Reference Example Compound 39 (0.0083 g, 0.019 mmol). As a result, 0.0065 g (70%) of benzyl) -1-((R) -3-hydroxy-3-methyl-2-propionamidobutanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 31) was obtained.
- Example Compound 32 0.038 g (81%) of (benzyl) -1-((R) -3-hydroxy-3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 32) was obtained. .
- Example 33 Synthesis of (R) -1- (1-butylamidocyclobutanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide: (R) -1- (1-Butylamidocyclobutanecarbonyl) -N— (4) was prepared by performing the same reaction as in Example 1 [Step 11] using butyryl chloride (0.0060 g, 0.057 mmol). 0.018 g (79%) of -cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 33) was obtained.
- Example 34 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (2-cyclopropylacetamido) cyclobutanecarbonyl) piperidine-3-carboxamide: By performing the same reaction as in Example 1 [Step 9] using Reference Example Compound 10 (0.021 g, 0.049 mmol) and 2-cyclopropylacetic acid (0.0059 g, 0.058 mmol), (R) 0.0065 g of —N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (2-cyclopropylacetamido) cyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 34) (26%) obtained.
- Example 35 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (2-cyclopropylacetamido) cyclopropanecarbonyl) piperidine-3-carboxamide: By performing the same reaction as in Example 1 [Step 9] using Reference Example Compound 14 (0.020 g, 0.049 mmol) and 2-cyclopropylacetic acid (0.0059 g, 0.058 mmol), (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (2-cyclopropylacetamido) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 35) 0083 g (35%) was obtained.
- Step 2 Synthesis of sodium 2-methyl-2- (1H-1,2,4-triazol-1-yl) propanoate: Under ice-cooling, 1N aqueous sodium hydroxide solution (3.9 mL, 3.9 mmol) was added to a solution of Reference Example Compound 40 (0.65 g, 3.6 mmol) in ethanol (18 mL). After stirring at room temperature for 1 hour, the reaction solution was concentrated under reduced pressure, and sodium 2-methyl-2- (1H-1,2,4-triazol-1-yl) propanoate (hereinafter referred to as Reference Example Compound 41) was reduced to 0. Obtained .67 g (quantitative).
- Example 37 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (1H-pyrazol-1-yl) propanoyl) piperidine-3- Synthesis of carboxamide: [Step 1] Synthesis of ethyl 2-methyl-2- (1H-pyrazol-1-yl) propanoate: The same reaction as in Example 36 [Step 1] was performed using 1H-pyrazole (0.42 g, 6.2 mmol) to give ethyl 2-methyl-2- (1H-pyrazol-1-yl) propanoate ( In the following, 0.81 g (87%) of Reference Example Compound 42) was obtained.
- Step 2 Synthesis of sodium 2-methyl-2- (1H-pyrazol-1-yl) propanoate: By performing the same reaction as in Example 36 [Step 2] using Reference Example Compound 42 (0.81 g, 4.5 mmol), sodium 2-methyl-2- (1H-pyrazol-1-yl) propanoate 0.80 g of Reference Example Compound 43 was obtained.
- Step 3 Synthesis of (R) -N- (2,4-dichlorobenzyl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide: (R) -N- (2,4-dichlorobenzyl) -1- (1-hydroxycyclohexane) was prepared by performing the same reaction as in Example 20 using Reference Example Compound 45 (0.10 g, 0.35 mmol). 0.030 g (24%) of carbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 38) was obtained.
- Example Compound 39 ((R) -1-((R) -2-acetamido-3-hydroxy 0.021 g (96%) of -3-methylbutanoyl) -N- (2,4-dichlorobenzyl) piperidine-3-carboxamide (hereinafter, Example Compound 39) was obtained.
- Example 40 Synthesis of (R) -N- (2,4-dichlorobenzyl) -1-((R) -3-hydroxy-3-methyl-2-propionamidobutanoyl) piperidine-3-carboxamide: By performing the same reaction as in Example 1 [Step 11] using Reference Compound 47 (0.020 g, 0.050 mmol), (R) -N- (2,4-dichlorobenzyl) -1- ( 0.018 g (77%) of (R) -3-hydroxy-3-methyl-2-propionamidobutanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 40) was obtained.
- Example 41 (R) -N- (2,4-dichlorobenzyl) -1-((R) -3-hydroxy-3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide Synthesis: The same reaction as in Example 2 [Step 3] was carried out using Reference Compound 47 (0.020 g, 0.050 mmol) to give (R) -N- (2,4-dichlorobenzyl) -1- ( 0.020 g (85%) of (R) -3-hydroxy-3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 41) was obtained.
- Example 42 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2-hydroxypropanoyl) piperidine-3-carboxamide: (R) -N- (4-cyano-2) was obtained by carrying out the same reaction as in Example 1 [Step 9] using (R) -2-hydroxypropanoic acid sodium salt (0.019 g, 0.17 mmol). 0.045 g (74%) of-(trifluoromethoxy) benzyl) -1-((R) -2-hydroxypropanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 42) was obtained.
- Example 43 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2-hydroxybutanoyl) piperidine-3-carboxamide: (R) -2-N- (4-cyano-2-) is prepared by carrying out the same reaction as in Example 1 [Step 9] using (R) -2-hydroxybutanoic acid (0.017 g, 0.17 mmol). 0.056 g (89%) of (trifluoromethoxy) benzyl) -1-((R) -2-hydroxybutanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 43) was obtained.
- Example 44 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2-hydroxy-3-methylbutanoyl) piperidine-3-carboxamide : (R) -2-Hydroxy-3-methylbutanoic acid (0.018 g, 0.15 mmol) was used for the same reaction as in Example 1 [Step 9] to give (R) -N- (4-cyano 0.042 g (64%) of 2- (trifluoromethoxy) benzyl) -1-((R) -2-hydroxy-3-methylbutanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 44) was obtained. It was.
- Tables 1-1 to 1-6 show physical property data of Example compounds 1 to 44, Table 2 shows physical property data of Comparative compounds 1 and 2, and Tables 3-1 to 3-5 The physical property data of Reference Example compounds 1 to 49 are shown in FIG. In the table, N.I. D. Represents “no data”.
- the solvent name in the 1H-NMR data indicates the solvent used for the measurement.
- the 400 MHz NMR spectrum was measured using a JNM-AL400 type nuclear magnetic resonance apparatus (JEOL Ltd.). The chemical shift is represented by ⁇ (unit: ppm) based on tetramethylsilane, and the signals are s (single line), d (double line), t (triple line), q (quadruple line), m ( Multiple line), brs (wide), dd (double double line), dt (double triple line), ddd (double double line), dq (double quadruple line), td (triple double line) (Multiple line) or tt (triple triple line). All solvents were commercially available.
- the ESI-MS spectrum was measured using Agilent Technologies 1200 Series, G6130A (Agilent Technology).
- Example 45 Evaluation test of sEH inhibitory activity in vitro: Inhibition of sEH of nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof using human sEH based on the method described in known literature (Analytical Biochemistry, 2005, 343, p. 66-75) Activity was evaluated.
- Example compounds 1 to 44 showed a very strong inhibitory activity against the enzymatic reaction of human sEH, as compared with Comparative compounds 1 and 2.
- nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof exhibits a very strong inhibitory activity on the enzyme reaction of human sEH.
- Example 46 Drug efficacy evaluation test in mouse acetate rising model: The analgesic action of nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof was examined using a mouse acetate rising method capable of evaluating nociceptive pain.
- mice The mouse acetate rising method was performed according to an existing method (Endoh et al., Life Science, 1999, Vol. 65, p. 1685-1694).
- ddY male mice 5-6 weeks old (Japan SLC) were bred under free drinking, and the test compound solution or its solvent (Vehicle) was orally administered (10 mL / kg).
- Tween 80 methyl cellulose (hereinafter referred to as MC): distilled water (0.5: 0.5: 99) was used as a solvent for the test compound solution.
- the * mark in the figure indicates that it is statistically significant in comparison with the solvent administration group (Student's t test, p ⁇ 0.05).
- Example Compound 1 When the Example Compound 1 was orally administered at 30 mg / kg, a significant decrease in the number of rising reactions was observed as compared with the solvent administration group. On the other hand, when 30 mg / kg of Comparative Example Compound 1 or Comparative Example Compound 2 was orally administered, no significant reduction in the number of rising reactions was observed compared to the solvent administration group. From the above results, Example Compound 1 was shown to have an analgesic effect on nociceptive pain, and nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof was effective against nociceptive pain. It was shown to have analgesic action.
- nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof was effective against nociceptive pain. It was shown to have analgesic action.
- the mouse partial sciatic nerve ligation model was prepared according to the method of Seltzer et al. (Malberg et al., Pain, 1998, Vol. 76, p. 215-222).
- a group that was ligated three times in half-strength was called a “ligation group”, and a group that only exposed the sciatic nerve but did not ligate was called a “sham surgery group”.
- neuropathic pain (hereinafter referred to as von Frey test) was performed by acclimating a mouse for at least 1 hour in a measurement acrylic cage (Natsume Seisakusho) installed on a wire mesh and then applying a filament (0.16 g pressure) Using North Coast Medical, Inc.
- the pre value was measured before oral administration of the test compound 7 days after the sciatic nerve ligation operation, and the mice in the ligation group were divided into the solvent administration group and the test compound administration group so that the total score of the von Frey test was uniform. Divided.
- the test compound solution or its solvent (Vehicle) was orally administered (10 mL / kg) to mice in the ligation group, and von Frey tests were performed 1 hour and 2 hours after oral administration, respectively. The value was used as an index of analgesic action. Tween 80: MC: distilled water (0.5: 0.5: 99) was used as a solvent for the test compound solution.
- the results are shown in FIGS. It should be noted that the group administered with the solvent was the “solvent administered group”, the group administered with Example Compound 1 at a dose of 30 mg / kg was the “Example Compound 1 administered group”, and the comparative compound 1 was administered at a dose of 30 mg / kg. The group administered in 1) was designated as “Comparative Example Compound 1 Administration Group”, and the group administered with Comparative Compound 2 at a dose of 30 mg / kg was designated as “Comparative Example Compound 2 Administration Group”.
- the * mark in the figure indicates that it is statistically significant in comparison with the solvent administration group (Student's t test, p ⁇ 0.05).
- Example Compound 1 was shown to have an analgesic effect on neuropathic pain, and nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof was effective against neuropathic pain. It was shown to have analgesic action.
- the analgesic of the present invention comprises a nipecotic acid derivative or a pharmacologically acceptable salt thereof as an active ingredient, and exhibits a medicinal effect based on sEH inhibitory activity, in particular nociceptive pain and / or neuropathy. It can be used as an analgesic for pain.
Abstract
The purpose of the present invention is to provide a novel analgesic effective against pain, particularly nociceptive pain and neuropathic pain. The present invention provides an analgesic containing, as an active ingredient, a nipecotic acid derivative represented by the chemical formula or a pharmaceutically acceptable salt thereof.
Description
本発明は、鎮痛剤に関する。
The present invention relates to an analgesic.
痛みとは、組織の損傷を引き起こす時又は損傷を引き起こす可能性のある時に生じる不快な感覚や不快な情動を伴う体験のことである。痛みは、その原因により、主に、侵害受容性疼痛と神経障害性疼痛とに分類される。なお、本明細書において、「痛み」と「疼痛」は同義とする。
Pain is an experience with an unpleasant sensation or unpleasant emotion that occurs when tissue damage or potential damage is caused. Pain is mainly classified into nociceptive pain and neuropathic pain, depending on the cause. In the present specification, “pain” and “pain” are synonymous.
侵害受容性疼痛とは、生体組織が損傷を受けたり、そのような危険を有する侵害刺激が加えられたりすることで生じる痛みであり、侵害受容器を介した疼痛のことをいう。例えば、生理的な痛みや炎症性疼痛が該当する。
“Nociceptive pain” refers to pain caused by damage to a living tissue or the addition of a noxious stimulus having such a risk, and refers to pain via a nociceptor. For example, physiological pain and inflammatory pain are applicable.
神経障害性疼痛とは、末梢又は中枢神経系そのものの機能異常による病的な痛みであり、侵害受容器が侵害刺激を受けていないにもかかわらず、神経組織の直接的な損傷や圧迫等によって生じる疼痛のことをいう。神経障害性疼痛は、国際疼痛学会(IASP)において「体性感覚系に対する損傷や疾患によって直接的に引き起こされる疼痛」と定義され(非特許文献1)、侵害受容性疼痛などと並び慢性疼痛の一つと位置づけられており、その原因疾患により末梢性及び中枢性に分類される(非特許文献2)。神経障害性疼痛は、持続的な自発痛、アロディニア(非侵害刺激に対する疼痛反応)及び痛覚過敏が特徴として挙げられ、神経障害性疼痛患者では、これらによる激しい疼痛が数ヵ月から10年以上の長期にわたり継続し、QOL(Quality of life)が著しく低下するため(非特許文献3)、積極的な治療を要する。
Neuropathic pain is pathological pain caused by abnormal functioning of the peripheral or central nervous system itself, and is caused by direct damage or compression of nerve tissue even though nociceptors are not subjected to noxious stimulation. This refers to the pain that occurs. Neuropathic pain is defined as “pain caused directly by damage or disease to the somatosensory system” at the International Society of Pain (IASP) (Non-Patent Document 1), along with nociceptive pain and the like. It is positioned as one, and is classified into peripheral and central depending on the causative disease (Non-patent Document 2). Neuropathic pain is characterized by persistent spontaneous pain, allodynia (pain response to non-nociceptive stimuli) and hyperalgesia, and in patients with neuropathic pain, severe pain due to these is a long-term of several months to 10 years or more. Since QOL (Quality of life) is significantly reduced (Non-patent Document 3), active treatment is required.
侵害受容性疼痛の鎮痛剤としては、非ステロイド性消炎鎮痛剤(NSAIDs)や麻薬性鎮痛剤(オピオイド等)等が使用され、神経障害性疼痛の鎮痛剤としては、抗痙攣剤、抗うつ剤、抗不安剤、ガバペンチン、プレガバリン等の抗てんかん剤が使用されている。
Non-steroidal anti-inflammatory analgesics (NSAIDs), narcotic analgesics (opioids, etc.) are used as analgesics for nociceptive pain, and anticonvulsants and antidepressants are used as analgesics for neuropathic pain. Antiepileptic agents such as anti-anxiety agents, gabapentin and pregabalin are used.
近年、内皮細胞由来の過分極因子の一つであるエポキシエイコサトリエン酸(Epoxyeicosatrienoic acids;以下、EETs)が、侵害受容性疼痛に対する鎮痛効果を示すことが報告された(非特許文献4)。EETsは、可溶性エポキシド加水分解酵素(soluble epoxide hydrolase;以下、sEH)によってジヒドロキシエイコサトリエン酸(dihydroxyeicosatrienoic acids;以下、DHETs)に代謝されて失活するが、可溶性エポキシド加水分解酵素阻害剤(以下、sEH阻害剤)は、EETsの量を増加させることが示されている(非特許文献5)。最近になって、sEH阻害活性を有する化合物が報告され、これら化合物が侵害受容性疼痛及び神経障害性疼痛に対する鎮痛効果を有することが示されている(非特許文献6及び7)。
Recently, it has been reported that epoxyeicosatrienoic acids (hereinafter referred to as EETs), which is one of endothelial cell-derived hyperpolarizing factors, exhibits an analgesic effect on nociceptive pain (Non-patent Document 4). EETs are metabolized and inactivated by a soluble epoxide hydrolase (hereinafter, sEH) to dihydroxyeicosatrienoic acids (hereinafter, DHETs), but are soluble epoxide hydrolase inhibitors (hereinafter, DHETs). sEH inhibitors) have been shown to increase the amount of EETs (Non-Patent Document 5). Recently, compounds having sEH inhibitory activity have been reported, and these compounds have been shown to have analgesic effects on nociceptive pain and neuropathic pain (Non-Patent Documents 6 and 7).
これまでに報告されたsEH阻害活性を有する化合物には、ニペコチン酸ジアミド構造を有するものはなく、ニペコチン酸ジアミド構造を有する化合物としてニペコチン酸にヘテロアリールアミンが縮合したヘテロアリールアミド誘導体(特許文献1)、アミジン誘導体(特許文献2)及びヒドロキサム酸誘導体(特許文献3)が報告されているが、sEH阻害活性との関係については開示も示唆もされていない。
None of the compounds having sEH inhibitory activity reported so far has a nipecotic acid diamide structure, and a heteroaryl amide derivative in which a heteroarylamine is condensed with nipecotic acid as a compound having a nipecotic acid diamide structure (Patent Document 1) ), Amidine derivatives (Patent Document 2) and hydroxamic acid derivatives (Patent Document 3) have been reported, but the relationship with sEH inhibitory activity has not been disclosed or suggested.
しかしながら、侵害受容性疼痛に用いられる非ステロイド性消炎鎮痛剤は、胃腸障害や腎障害等の副作用を伴い、また麻薬性鎮痛剤は、便秘、眠気、悪心、嘔吐等の副作用を伴うことが知られており、その使用が制限されるケースがあることが知られている。また、神経障害性疼痛に用いられる抗痙攣剤、抗うつ剤、抗不安剤及び抗てんかん剤は、めまい、悪心、嘔吐等の中枢性の副作用を高い頻度で伴い、長期投与が困難あるいは投薬中止に至るケースがあること、及びこれらの薬剤では十分な鎮痛効果の得られない患者が存在することが指摘されている(非特許文献4)。
However, non-steroidal anti-inflammatory analgesics used for nociceptive pain are associated with side effects such as gastrointestinal disorders and renal disorders, and narcotic analgesics are known to have side effects such as constipation, sleepiness, nausea and vomiting. And its use is known to be limited. Anticonvulsants, antidepressants, anxiolytics and antiepileptics used for neuropathic pain frequently involve central side effects such as dizziness, nausea and vomiting, which are difficult to discontinue for a long time or are discontinued. It has been pointed out that there are cases in which these drugs do not provide sufficient analgesic effects with these drugs (Non-patent Document 4).
そこで本発明は、疼痛、特に、侵害受容性疼痛及び神経障害性疼痛に対して有効な新たな鎮痛剤を提供することを目的とする。
Therefore, an object of the present invention is to provide a new analgesic effective for pain, particularly nociceptive pain and neuropathic pain.
本発明者らは上記課題を解決するために鋭意研究を重ねた結果、新規なニペコチン酸誘導体又はその薬理学的に許容される塩がsEH阻害活性を示すこと、並びに、その作用に基づき疼痛、特に、侵害受容性疼痛及び神経障害性疼痛に対して優れた鎮痛効果を発揮することを見出し、本発明を完成させた。
As a result of intensive studies to solve the above problems, the present inventors have shown that a novel nipecotic acid derivative or a pharmacologically acceptable salt thereof exhibits sEH inhibitory activity, and pain based on its action, In particular, it has been found that it exhibits an excellent analgesic effect against nociceptive pain and neuropathic pain, and has completed the present invention.
すなわち、本発明は、以下の一般式(I)で示されるニペコチン酸誘導体又はその薬理学的に許容される塩を有効成分として含有する鎮痛剤を提供する。

[式中、R1は、水酸基、シアノ基、炭素数1~6のアルキル基若しくはアルキルオキシ基、炭素数3~6のシクロアルキル基若しくはシクロアルキルオキシ基、炭素数2~7のアルキルオキシアルキル基、炭素数4~7のシクロアルキルアルキル基(該アルキル基、アルキルオキシ基、シクロアルキル基、シクロアルキルオキシ基、アルキルオキシアルキル基及びシクロアルキルアルキル基は、1~3個の水素原子が、それぞれ独立して、ハロゲン原子、水酸基、シアノ基、-SR6、-S(=O)-R6又は-S(=O)2R6で置換されていてもよい)、-N(R6)C(=O)R7、-N(R6)S(=O)2R7、-C(=O)N(R6)R7又は環構成原子数5のヘテロアリール基を表し、R2及びR3は、それぞれ独立して、水素原子、炭素数1~6のアルキル基又は炭素数2~7のアルキルオキシアルキル基(該アルキル基及びアルキルオキシアルキル基は、1~3個の水素原子が、それぞれ独立して、ハロゲン原子、水酸基又はシアノ基で置換されていてもよい)を表すか、又は、一緒になって-(CH2)l-若しくは-(CH2)m-O-(CH2)n-を表すが、同時に水素原子を表すことはなく、R4及びR5は、それぞれ独立して、水素原子、ハロゲン原子、シアノ基、炭素数1~6のアルキル基若しくはアルキルオキシ基、炭素数3~6のシクロアルキル基若しくはシクロアルキルオキシ基(該アルキル基、アルキルオキシ基、シクロアルキル基及びシクロアルキルオキシ基は、1~3個の水素原子が、それぞれ独立して、ハロゲン原子で置換されていてもよい)又は-C(=O)NH2を表すが、同時にアルキルオキシ基を表すことはなく、R6は、水素原子又は炭素数1~6のアルキル基を表し、R7は、炭素数1~6のアルキル基、炭素数3~6のシクロアルキル基、炭素数2~7のアルキルオキシアルキル基又は炭素数4~7のシクロアルキルアルキル基(該アルキル基、シクロアルキル基、アルキルオキシアルキル基及びシクロアルキルアルキル基は、1~3個の水素原子が、それぞれ独立して、ハロゲン原子、水酸基又はシアノ基で置換されていてもよい)を表し、lは、2~5の整数を表し、m及びnは、それぞれ独立して、1又は2を表す。]
That is, the present invention provides an analgesic containing an nipecotic acid derivative represented by the following general formula (I) or a pharmacologically acceptable salt thereof as an active ingredient.
[Wherein R 1 represents a hydroxyl group, a cyano group, an alkyl group or alkyloxy group having 1 to 6 carbon atoms, a cycloalkyl group or cycloalkyloxy group having 3 to 6 carbon atoms, or an alkyloxyalkyl group having 2 to 7 carbon atoms. A cycloalkylalkyl group having 4 to 7 carbon atoms (the alkyl group, alkyloxy group, cycloalkyl group, cycloalkyloxy group, alkyloxyalkyl group and cycloalkylalkyl group have 1 to 3 hydrogen atoms, Each independently may be substituted with a halogen atom, a hydroxyl group, a cyano group, —SR 6 , —S (═O) —R 6 or —S (═O) 2 R 6 ), —N (R 6 ) C (═O) R 7 , —N (R 6 ) S (═O) 2 R 7 , —C (═O) N (R 6 ) R 7 or a heteroaryl group having 5 ring atoms, R 2 and R 3 are Independently, a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, or an alkyloxyalkyl group having 2 to 7 carbon atoms (the alkyl group and the alkyloxyalkyl group each have 1 to 3 hydrogen atoms independently And may be substituted with a halogen atom, a hydroxyl group or a cyano group, or together, — (CH 2 ) 1 — or — (CH 2 ) m —O— (CH 2 ) n -Represents a hydrogen atom, but R 4 and R 5 are each independently a hydrogen atom, a halogen atom, a cyano group, an alkyl group having 1 to 6 carbon atoms or an alkyloxy group, 3 to 6 cycloalkyl groups or cycloalkyloxy groups (the alkyl group, alkyloxy group, cycloalkyl group and cycloalkyloxy group each have 1 to 3 hydrogen atoms independently represented by Halogen may be substituted by atoms) or -C (= O) represents the NH 2, not simultaneously represent an alkyl group, R 6 represents a hydrogen atom or an alkyl group having 1 to 6 carbon atoms , R 7 represents an alkyl group having 1 to 6 carbon atoms, a cycloalkyl group having 3 to 6 carbon atoms, an alkyloxyalkyl group having 2 to 7 carbon atoms, or a cycloalkylalkyl group having 4 to 7 carbon atoms (the alkyl group, A cycloalkyl group, an alkyloxyalkyl group and a cycloalkylalkyl group each represent 1 to 3 hydrogen atoms each independently may be substituted with a halogen atom, a hydroxyl group or a cyano group, Represents an integer of 2 to 5, and m and n each independently represent 1 or 2. ]
上記のニペコチン酸誘導体は、R2及びR3が、それぞれ独立して、水素原子若しくは炭素数1~6のアルキル基を表すか、又は、一緒になって-(CH2)l-を表すが、同時に水素原子を表すことはなく、R4が、ベンゼン環上の2位の置換基を表し、R5が、ベンゼン環上の4位の置換基を表すことが好ましい。
In the above nipecotic acid derivative, R 2 and R 3 each independently represents a hydrogen atom or an alkyl group having 1 to 6 carbon atoms, or together represent — (CH 2 ) 1 —. At the same time, it is preferable that R 4 represents a substituent at the 2-position on the benzene ring and R 5 represents a substituent at the 4-position on the benzene ring.
この場合には、より強いsEH阻害活性を有し、疼痛に対するより優れた鎮痛効果が発揮される。
In this case, it has a stronger sEH inhibitory activity and exhibits a more excellent analgesic effect on pain.
また、上記のニペコチン酸誘導体は、R1が、-N(R6)C(=O)R7又は-N(R6)S(=O)2R7を表し、R4が、ハロゲン原子又は炭素数1~6のアルキル基若しくはアルキルオキシ基を表し、R5が、ハロゲン原子、シアノ基又は炭素数1~6のアルキル基若しくはアルキルオキシ基を表し、R6が、水素原子を表すことがより好ましく、R1が、-N(H)C(=O)CH2CH3を表し、R2及びR3が、一緒になって-(CH2)3-を表し、R4が、-OCF3を表し、R5が、シアノ基を表すことが特に好ましい。
In the above nipecotic acid derivative, R 1 represents —N (R 6 ) C (═O) R 7 or —N (R 6 ) S (═O) 2 R 7 , and R 4 represents a halogen atom. Or an alkyl group or alkyloxy group having 1 to 6 carbon atoms, R 5 represents a halogen atom, a cyano group, or an alkyl group or alkyloxy group having 1 to 6 carbon atoms, and R 6 represents a hydrogen atom. Is more preferred, R 1 represents —N (H) C (═O) CH 2 CH 3 , R 2 and R 3 together represent — (CH 2 ) 3 —, and R 4 represents It is particularly preferred that it represents —OCF 3 and R 5 represents a cyano group.
この場合には、より強いsEH阻害活性を有し、薬物動態も優れていることから、疼痛に対するさらに優れた鎮痛効果が発揮される。
In this case, since it has stronger sEH inhibitory activity and excellent pharmacokinetics, a further excellent analgesic effect on pain is exhibited.
上記の鎮痛剤は、侵害受容性疼痛及び/又は神経障害性疼痛の鎮痛剤であることが好ましい。
The above analgesic is preferably an analgesic for nociceptive pain and / or neuropathic pain.
本発明の鎮痛剤は、sEH阻害活性に基づき、侵害受容性疼痛及び神経障害性疼痛に対して高い治療効果又は予防効果を発揮するため、広い範囲の疼痛症状に対し医薬としての投与が可能となる。
The analgesic of the present invention exhibits a high therapeutic effect or preventive effect on nociceptive pain and neuropathic pain based on sEH inhibitory activity, so that it can be administered as a medicine for a wide range of pain symptoms. Become.
本発明の鎮痛剤は、以下の一般式(I)で示されるニペコチン酸誘導体又はその薬理学的に許容される塩を有効成分として含有することを特徴としている。

[式中、R1は、水酸基、シアノ基、炭素数1~6のアルキル基若しくはアルキルオキシ基、炭素数3~6のシクロアルキル基若しくはシクロアルキルオキシ基、炭素数2~7のアルキルオキシアルキル基、炭素数4~7のシクロアルキルアルキル基(該アルキル基、アルキルオキシ基、シクロアルキル基、シクロアルキルオキシ基、アルキルオキシアルキル基及びシクロアルキルアルキル基は、1~3個の水素原子が、それぞれ独立して、ハロゲン原子、水酸基、シアノ基、-SR6、-S(=O)-R6又は-S(=O)2R6で置換されていてもよい)、-N(R6)C(=O)R7、-N(R6)S(=O)2R7、-C(=O)N(R6)R7又は環構成原子数5のヘテロアリール基を表し、R2及びR3は、それぞれ独立して、水素原子、炭素数1~6のアルキル基又は炭素数2~7のアルキルオキシアルキル基(該アルキル基及びアルキルオキシアルキル基は、1~3個の水素原子が、それぞれ独立して、ハロゲン原子、水酸基又はシアノ基で置換されていてもよい)を表すか、又は、一緒になって-(CH2)l-若しくは-(CH2)m-O-(CH2)n-を表すが、同時に水素原子を表すことはなく、R4及びR5は、それぞれ独立して、水素原子、ハロゲン原子、シアノ基、炭素数1~6のアルキル基若しくはアルキルオキシ基、炭素数3~6のシクロアルキル基若しくはシクロアルキルオキシ基(該アルキル基、アルキルオキシ基、シクロアルキル基及びシクロアルキルオキシ基は、1~3個の水素原子が、それぞれ独立して、ハロゲン原子で置換されていてもよい)又は-C(=O)NH2を表すが、同時にアルキルオキシ基を表すことはなく、R6は、水素原子又は炭素数1~6のアルキル基を表し、R7は、炭素数1~6のアルキル基、炭素数3~6のシクロアルキル基、炭素数2~7のアルキルオキシアルキル基又は炭素数4~7のシクロアルキルアルキル基(該アルキル基、シクロアルキル基、アルキルオキシアルキル基及びシクロアルキルアルキル基は、1~3個の水素原子が、それぞれ独立して、ハロゲン原子、水酸基又はシアノ基で置換されていてもよい)を表し、lは、2~5の整数を表し、m及びnは、それぞれ独立して、1又は2を表す。]
The analgesic of the present invention is characterized by containing a nipecotic acid derivative represented by the following general formula (I) or a pharmacologically acceptable salt thereof as an active ingredient.
[Wherein R 1 represents a hydroxyl group, a cyano group, an alkyl group or alkyloxy group having 1 to 6 carbon atoms, a cycloalkyl group or cycloalkyloxy group having 3 to 6 carbon atoms, or an alkyloxyalkyl group having 2 to 7 carbon atoms. A cycloalkylalkyl group having 4 to 7 carbon atoms (the alkyl group, alkyloxy group, cycloalkyl group, cycloalkyloxy group, alkyloxyalkyl group and cycloalkylalkyl group have 1 to 3 hydrogen atoms, Each independently may be substituted with a halogen atom, a hydroxyl group, a cyano group, —SR 6 , —S (═O) —R 6 or —S (═O) 2 R 6 ), —N (R 6 ) C (═O) R 7 , —N (R 6 ) S (═O) 2 R 7 , —C (═O) N (R 6 ) R 7 or a heteroaryl group having 5 ring atoms, R 2 and R 3 are Independently, a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, or an alkyloxyalkyl group having 2 to 7 carbon atoms (the alkyl group and the alkyloxyalkyl group each have 1 to 3 hydrogen atoms independently And may be substituted with a halogen atom, a hydroxyl group or a cyano group, or together, — (CH 2 ) 1 — or — (CH 2 ) m —O— (CH 2 ) n -Represents a hydrogen atom, but R 4 and R 5 are each independently a hydrogen atom, a halogen atom, a cyano group, an alkyl group having 1 to 6 carbon atoms or an alkyloxy group, 3 to 6 cycloalkyl groups or cycloalkyloxy groups (the alkyl group, alkyloxy group, cycloalkyl group and cycloalkyloxy group each have 1 to 3 hydrogen atoms independently represented by Halogen may be substituted by atoms) or -C (= O) represents the NH 2, not simultaneously represent an alkyl group, R 6 represents a hydrogen atom or an alkyl group having 1 to 6 carbon atoms , R 7 represents an alkyl group having 1 to 6 carbon atoms, a cycloalkyl group having 3 to 6 carbon atoms, an alkyloxyalkyl group having 2 to 7 carbon atoms, or a cycloalkylalkyl group having 4 to 7 carbon atoms (the alkyl group, A cycloalkyl group, an alkyloxyalkyl group and a cycloalkylalkyl group each represent 1 to 3 hydrogen atoms each independently may be substituted with a halogen atom, a hydroxyl group or a cyano group, Represents an integer of 2 to 5, and m and n each independently represent 1 or 2. ]
「炭素数1~6のアルキル基」とは、炭素原子を1~6個有する直鎖状又は炭素原子を3~6個有する分岐鎖状の飽和炭化水素基を意味し、例えば、メチル基、エチル基、1-プロピル基、2-プロピル基、1-ブチル基、2-ブチル基、2-メチル-2-プロピル基(tert-ブチル基)、2-メチル-1-プロピル基、2,2-ジメチル-1-プロピル基、1-ペンチル基、2-ペンチル基又は3-ペンチル基が挙げられる。
“C1-C6 alkyl group” means a straight-chain saturated hydrocarbon group having 1 to 6 carbon atoms or a branched saturated hydrocarbon group having 3 to 6 carbon atoms, such as a methyl group, Ethyl group, 1-propyl group, 2-propyl group, 1-butyl group, 2-butyl group, 2-methyl-2-propyl group (tert-butyl group), 2-methyl-1-propyl group, 2,2 -Dimethyl-1-propyl group, 1-pentyl group, 2-pentyl group or 3-pentyl group.
「炭素数1~6のアルキルオキシ基」とは、上記の炭素数1~6のアルキル基が酸素原子に結合した基を意味し、例えば、メトキシ基、エトキシ基、1-プロピルオキシ基、2-プロピルオキシ基、1-ブチルオキシ基又は2-ブチルオキシ基が挙げられる。
The “C 1-6 alkyloxy group” means a group in which the above C 1-6 alkyl group is bonded to an oxygen atom, such as a methoxy group, an ethoxy group, a 1-propyloxy group, -Propyloxy group, 1-butyloxy group or 2-butyloxy group.
「炭素数3~6のシクロアルキル基」とは、シクロプロピル基、シクロブチル基、シクロペンチル基又はシクロヘキシル基を意味する。
“C3-C6 cycloalkyl group” means a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, or a cyclohexyl group.
「炭素数3~6のシクロアルキルオキシ基」とは、シクロプロピルオキシ基、シクロブチルオキシ基、シクロペンチルオキシ基又はシクロヘキシルオキシ基を意味する。
“C3-C6 cycloalkyloxy group” means a cyclopropyloxy group, a cyclobutyloxy group, a cyclopentyloxy group, or a cyclohexyloxy group.
「炭素数2~7のアルキルオキシアルキル基」とは、炭素原子を2~7個有し、アルキル基の1個の水素原子がアルキルオキシ基で置換された基を意味し、例えば、メトキシメチル基、メトキシエチル基、メトキシプロピル基、エトキシメチル基、プロポキシメチル基又はイソプロポキシメチル基が挙げられる。
“C2-C7 alkyloxyalkyl group” means a group having 2 to 7 carbon atoms in which one hydrogen atom of the alkyl group is substituted with an alkyloxy group. Group, methoxyethyl group, methoxypropyl group, ethoxymethyl group, propoxymethyl group or isopropoxymethyl group.
「炭素数4~7のシクロアルキルアルキル基」とは、炭素原子を4~7個有し、アルキル基の1個の水素原子がシクロアルキル基で置換された基を意味し、例えば、シクロプロピルメチル基、シクロプロピルエチル基、シクロプロピルプロピル基、シクロブチルメチル基、シクロペンチルメチル基又はシクロヘキシルメチル基が挙げられる。
The “cycloalkyl alkyl group having 4 to 7 carbon atoms” means a group having 4 to 7 carbon atoms in which one hydrogen atom of the alkyl group is substituted with a cycloalkyl group. Examples thereof include a methyl group, a cyclopropylethyl group, a cyclopropylpropyl group, a cyclobutylmethyl group, a cyclopentylmethyl group, and a cyclohexylmethyl group.
「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子又はヨウ素原子を意味する。
“Halogen atom” means a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
「環構成原子数5のヘテロアリール基」とは、窒素原子、酸素原子及び硫黄原子からなる群から選択される同一又は異なる原子を環構成原子として1~4個含む、環構成原子数が5の複素芳香族基を意味し、例えば、ピロリル基、イミダゾリル基、ピラゾリル基、トリアゾリル基、オキサゾリル基、イソキサゾリル基、フラニル基又はチアゾリル基が挙げられる。
The term “heteroaryl group having 5 ring atoms” means that the number of ring atoms is 5 including 1 to 4 identical or different atoms selected from the group consisting of a nitrogen atom, an oxygen atom and a sulfur atom. And includes, for example, pyrrolyl group, imidazolyl group, pyrazolyl group, triazolyl group, oxazolyl group, isoxazolyl group, furanyl group and thiazolyl group.
上記のニペコチン酸誘導体は、一般式(I)において、R1は、-N(R6)C(=O)R7又は-N(R6)S(=O)2R7であることが好ましく、アセチルアミジル、プロピオンアミジル基又はメタンスルホニルアミジル基であることがより好ましい。
In the above nipecotic acid derivative, in general formula (I), R 1 may be —N (R 6 ) C (═O) R 7 or —N (R 6 ) S (═O) 2 R 7 Preferably, it is acetylamidyl, propionamidyl group or methanesulfonylamidyl group.
R2及びR3は、それぞれ独立して、水素原子若しくは炭素数1~6のアルキル基であるか、又は、一緒になって-(CH2)l-であることが好ましく、それぞれ独立して、水素原子若しくは炭素数1~3のアルキル基(該アルキル基は、1個の水素原子が、水酸基で置換されていてもよい)であるか、又は、一緒になって-(CH2)2-若しくは-(CH2)3-であることがより好ましく、それぞれ独立して、水素原子、メチル基若しくは2-ヒドロキシ-2-プロピル基であるか、又は、一緒になって-(CH2)2-若しくは-(CH2)3-であることがさらに好ましいが、同時に水素原子となることはない。
R 2 and R 3 are each independently a hydrogen atom or an alkyl group having 1 to 6 carbon atoms, or are preferably taken together as — (CH 2 ) 1 —. , A hydrogen atom or an alkyl group having 1 to 3 carbon atoms (in which the alkyl group, one hydrogen atom may be substituted with a hydroxyl group), or together, — (CH 2 ) 2 -Or-(CH 2 ) 3- is more preferable, and each independently represents a hydrogen atom, a methyl group or a 2-hydroxy-2-propyl group, or together,-(CH 2 ) 2 -or-(CH 2 ) 3- is more preferred, but they are not simultaneously hydrogen atoms.
R4は、ベンゼン環上の2位の置換基であることが好ましい。また、R4は、ハロゲン原子又は炭素数1~6のアルキル基若しくはアルキルオキシ基であることが好ましく、ハロゲン原子又はアルキルオキシ基であることがより好ましく、アルキルオキシ基であることがさらに好ましい。
R 4 is preferably a substituent at the 2-position on the benzene ring. R 4 is preferably a halogen atom, an alkyl group having 1 to 6 carbon atoms or an alkyloxy group, more preferably a halogen atom or an alkyloxy group, and further preferably an alkyloxy group.
R5は、ベンゼン環上の4位の置換基であることが好ましい。また、R5は、ハロゲン原子、シアノ基又は炭素数1~6のアルキル基若しくは炭素数1~6のアルキルオキシ基であることが好ましく、ハロゲン原子又はシアノ基であることがより好ましい。
R 5 is preferably a substituent at the 4-position on the benzene ring. R 5 is preferably a halogen atom, a cyano group, an alkyl group having 1 to 6 carbon atoms, or an alkyloxy group having 1 to 6 carbon atoms, and more preferably a halogen atom or a cyano group.
R6は、水素原子であることが好ましく、R7は、メチル基又はエチル基であることが好ましい。
R 6 is preferably a hydrogen atom, and R 7 is preferably a methyl group or an ethyl group.
また、lは、2又は3であることが好ましく、mは、2であることが好ましく、nは、2であることが好ましい。
L is preferably 2 or 3, m is preferably 2, and n is preferably 2.
上記の一般式(I)で示されるニペコチン酸誘導体(以下、ニペコチン酸誘導体(I))は、少なくとも1個の不斉炭素原子を有しており、光学異性体やジアステレオマーが存在するものであるが、ニペコチン酸誘導体(I)は単一異性体のみならず、ラセミ体及びジアステレオマー混合物も包含するものである。また、回転異性体が存在する場合、全ての回転異性体を包含する。
The nipecotic acid derivative represented by the above general formula (I) (hereinafter referred to as nipecotic acid derivative (I)) has at least one asymmetric carbon atom and has optical isomers and diastereomers. However, the nipecotic acid derivative (I) includes not only a single isomer but also a racemate and a mixture of diastereomers. Moreover, when a rotamer exists, all rotamers are included.
ニペコチン酸誘導体(I)の薬理学的に許容される塩としては、例えば、酸付加塩として塩酸塩、トリフルオロ酢酸塩、硫酸塩、硝酸塩、臭化水素酸塩、ヨウ化水素酸塩又はメタンスルホン酸塩が挙げられるが、塩酸塩、硫酸塩、臭化水素酸塩、ヨウ化水素酸塩又はメタンスルホン酸塩が好ましい。
Examples of the pharmacologically acceptable salt of the nipecotic acid derivative (I) include, for example, hydrochloride, trifluoroacetate, sulfate, nitrate, hydrobromide, hydroiodide or methane as an acid addition salt. Examples of the sulfonate include hydrochloride, sulfate, hydrobromide, hydroiodide, and methanesulfonate.
ニペコチン酸誘導体(I)の製造に使用する出発物質と試薬は、市販品をそのまま利用してもよいし、又は、公知の方法により合成しても構わない。
The starting materials and reagents used for the production of the nipecotic acid derivative (I) may be commercially available products or may be synthesized by known methods.
ニペコチン酸誘導体(I-a)は、例えば、以下のスキーム1に示すように、塩基及び縮合剤存在下、アミン誘導体(II)とカルボン酸誘導体(III)との縮合反応により製造できる。
(スキーム1)

[式中、R1’は、水酸基、シアノ基、炭素数1~6のアルキル基若しくはアルキルオキシ基、炭素数3~6のシクロアルキル基若しくはシクロアルキルオキシ基、炭素数2~7のアルキルオキシアルキル基、炭素数4~7のシクロアルキルアルキル基(該アルキル基、アルキルオキシ基、シクロアルキル基、シクロアルキルオキシ基、アルキルオキシアルキル基及びシクロアルキルアルキル基は、1~3個の水素原子が、それぞれ独立して、ハロゲン原子、水酸基、シアノ基、-SR6、-S(=O)-R6又は-S(=O)2R6で置換されていてもよい)を表す。R2~R6は、上記定義に同じ。]
The nipecotic acid derivative (Ia) can be produced, for example, by a condensation reaction of an amine derivative (II) and a carboxylic acid derivative (III) in the presence of a base and a condensing agent as shown in the following scheme 1.
(Scheme 1)
[Wherein R 1 ′ represents a hydroxyl group, a cyano group, an alkyl group or alkyloxy group having 1 to 6 carbon atoms, a cycloalkyl group or cycloalkyloxy group having 3 to 6 carbon atoms, or an alkyloxy group having 2 to 7 carbon atoms. An alkyl group, a cycloalkylalkyl group having 4 to 7 carbon atoms (the alkyl group, alkyloxy group, cycloalkyl group, cycloalkyloxy group, alkyloxyalkyl group and cycloalkylalkyl group have 1 to 3 hydrogen atoms) Each independently represents a halogen atom, a hydroxyl group, a cyano group, —SR 6 , —S (═O) —R 6 or —S (═O) 2 R 6 . R 2 to R 6 are the same as defined above. ]
(スキーム1)
(Scheme 1)
縮合反応に用いる縮合剤としては、例えば、シクロヘキシルカルボジイミド、N-エチル-N‘-3-ジメチルアミノプロピルカルボジイミド塩酸塩、ベンゾトリアゾール-1-イルオキシ-トリスジメチルアミノホスホニウム塩(BOP試薬)、1-[ビス(ジメチルアミノ)メチレン]-1H-ベンゾトリアゾリウム-3-オキシドヘキサフルオロホスファート(HBTU)又はO-(7-アザベンゾトリアゾール-1-イル)テトラメチルウロニウム ヘキサフルオロホスファート(以下、HATU)が挙げられるが、HATUが好ましい。該縮合剤の当量は、1~10当量が好ましく、1~3当量がより好ましい。
Examples of the condensing agent used in the condensation reaction include cyclohexylcarbodiimide, N-ethyl-N′-3-dimethylaminopropylcarbodiimide hydrochloride, benzotriazol-1-yloxy-trisdimethylaminophosphonium salt (BOP reagent), 1- [ Bis (dimethylamino) methylene] -1H-benzotriazolium-3-oxide hexafluorophosphate (HBTU) or O- (7-azabenzotriazol-1-yl) tetramethyluronium hexafluorophosphate HATU), and HATU is preferred. The equivalent of the condensing agent is preferably 1 to 10 equivalents, and more preferably 1 to 3 equivalents.
縮合反応に用いる溶媒としては、例えば、N,N-ジメチルホルムアミド(以下、DMF)、テトラヒドロフラン(以下、THF)、ジクロロメタン、クロロホルム、ジエチルエーテル又はジメチルエーテルが挙げられるが、DMF又はTHFが好ましく、DMFがより好ましい。
Examples of the solvent used in the condensation reaction include N, N-dimethylformamide (hereinafter referred to as DMF), tetrahydrofuran (hereinafter referred to as THF), dichloromethane, chloroform, diethyl ether or dimethyl ether, with DMF or THF being preferred, and DMF being More preferred.
縮合反応に用いる塩基としては、例えば、ジイソプロピルエチルアミン(以下、DIPEA)、トリエチルアミン(以下、TEA)、ピリジン若しくはN-メチルモルホリン等の有機塩基又は炭酸カリウム、炭酸ナトリウム若しくは炭酸水素ナトリウム等の有機酸塩が挙げられるが、DIPEA又はTEAが好ましい。該塩基の当量は、アミン誘導体(II)に対して1~100当量が好ましく、1~10当量がより好ましい。
Examples of the base used in the condensation reaction include organic bases such as diisopropylethylamine (hereinafter DIPEA), triethylamine (hereinafter TEA), pyridine or N-methylmorpholine, or organic acid salts such as potassium carbonate, sodium carbonate or sodium bicarbonate. DIPEA or TEA is preferable. The equivalent of the base is preferably 1 to 100 equivalents and more preferably 1 to 10 equivalents with respect to the amine derivative (II).
縮合反応に用いるカルボン酸誘導体(III)の当量は、アミン誘導体(II)に対して0.1~100当量が好ましく、0.1~10当量がより好ましく、0.8~2当量がさらに好ましい。
The equivalent amount of the carboxylic acid derivative (III) used in the condensation reaction is preferably 0.1 to 100 equivalents, more preferably 0.1 to 10 equivalents, and even more preferably 0.8 to 2 equivalents with respect to the amine derivative (II). .
縮合反応の反応温度は、-50~100℃が好ましく、0~50℃がより好ましく、0~30℃がさらに好ましい。また、縮合反応の反応時間は、1分間~48時間が好ましく、1分間~24時間がより好ましく、10分間~24時間がさらに好ましい。
The reaction temperature of the condensation reaction is preferably −50 to 100 ° C., more preferably 0 to 50 ° C., and further preferably 0 to 30 ° C. The reaction time for the condensation reaction is preferably 1 minute to 48 hours, more preferably 1 minute to 24 hours, and even more preferably 10 minutes to 24 hours.
縮合反応におけるアミン誘導体(II)の反応開始時の濃度は、0.01~100Mが好ましく、0.01~10Mがより好ましく、0.1~10Mがさらに好ましい。
The concentration of the amine derivative (II) at the start of the condensation reaction is preferably 0.01 to 100M, more preferably 0.01 to 10M, and even more preferably 0.1 to 10M.
また、R1が、-N(H)C(=O)R7であるニペコチン酸誘導体(I-b)は、例えば、以下のスキーム2に示すように、塩基存在下、アミン誘導体(IV)と酸クロリド誘導体(V)との縮合反応、又は、塩基及び縮合剤存在下、アミン誘導体(IV)とカルボン酸誘導体(VI)との縮合反応により製造できる。
(スキーム2)

[式中、R2~R5及びR7は、上記定義に同じ。]
In addition, the nipecotic acid derivative (Ib) in which R 1 is —N (H) C (═O) R 7 is represented by, for example, an amine derivative (IV) in the presence of a base as shown in Scheme 2 below. And an acid chloride derivative (V), or a condensation reaction between an amine derivative (IV) and a carboxylic acid derivative (VI) in the presence of a base and a condensing agent.
(Scheme 2)
[Wherein R 2 to R 5 and R 7 are the same as defined above]. ]
(スキーム2)
(Scheme 2)
酸クロリド誘導体(V)との縮合反応に用いる溶媒としては、例えば、ジクロロメタン、1,2-ジクロロエタン、アセトニトリル、DMF、THF、ジオキサン、ジエチルエーテル又は1,2-ジメトキシエタンが挙げられるが、ジクロロメタン、1,2-ジクロロエタン、アセトニトリル又はTHFが好ましく、ジクロロメタン又は1,2-ジクロロエタンがより好ましい。
Examples of the solvent used for the condensation reaction with the acid chloride derivative (V) include dichloromethane, 1,2-dichloroethane, acetonitrile, DMF, THF, dioxane, diethyl ether or 1,2-dimethoxyethane. 1,2-dichloroethane, acetonitrile or THF is preferred, and dichloromethane or 1,2-dichloroethane is more preferred.
酸クロリド誘導体(V)との縮合反応に用いる酸クロリド(V)の当量は、アミン誘導体(IV)に対して0.1~10当量が好ましく、1~3当量がより好ましく、1~1.5当量がさらに好ましい。
The equivalent amount of the acid chloride (V) used in the condensation reaction with the acid chloride derivative (V) is preferably 0.1 to 10 equivalents, more preferably 1 to 3 equivalents, relative to the amine derivative (IV). 5 equivalents are more preferred.
酸クロリド誘導体(V)との縮合反応に用いる塩基としては、例えば、DIPEA、TEA、ピリジン又はN-メチルモルホリン等の有機塩基が挙げられるが、DIPEA又はTEAが好ましい。該塩基の当量は、アミン誘導体(IV)に対して1~100当量が好ましく、1~10当量がより好ましい。
Examples of the base used for the condensation reaction with the acid chloride derivative (V) include organic bases such as DIPEA, TEA, pyridine and N-methylmorpholine, with DIPEA or TEA being preferred. The equivalent of the base is preferably 1 to 100 equivalents and more preferably 1 to 10 equivalents with respect to the amine derivative (IV).
酸クロリド誘導体(V)との縮合反応の反応温度は、-50~100℃が好ましく、-20~60℃がより好ましく、0~40℃がさらに好ましい。また、酸クロリド(V)との縮合反応の反応時間は、30分間~24時間が好ましく、30分間~12時間がより好ましく、30分間~8時間がさらに好ましい。
The reaction temperature of the condensation reaction with the acid chloride derivative (V) is preferably −50 to 100 ° C., more preferably −20 to 60 ° C., and further preferably 0 to 40 ° C. The reaction time for the condensation reaction with acid chloride (V) is preferably 30 minutes to 24 hours, more preferably 30 minutes to 12 hours, and even more preferably 30 minutes to 8 hours.
酸クロリド誘導体(V)との縮合反応におけるアミン誘導体(IV)の反応開始時の濃度は、0.01~100Mが好ましく、0.01~10Mがより好ましく、0.1~10Mがさらに好ましい。
The concentration at the start of the reaction of the amine derivative (IV) in the condensation reaction with the acid chloride derivative (V) is preferably 0.01 to 100M, more preferably 0.01 to 10M, and further preferably 0.1 to 10M.
一方、アミン誘導体(IV)とカルボン酸誘導体(VI)との縮合反応は、スキーム1と同様の条件により行うことができる。
On the other hand, the condensation reaction between the amine derivative (IV) and the carboxylic acid derivative (VI) can be performed under the same conditions as in Scheme 1.
R1が、-N(H)S(=O)2R7であるニペコチン酸誘導体(I-c)は、例えば、以下のスキーム3に示すように、塩基存在下、アミン誘導体(IV)とスルホン酸クロリド誘導体(VII)とのスルホンアミド化反応により製造できる。
(スキーム3)

[式中、R2~R5及びR7は、上記定義に同じ。]
The nipecotic acid derivative (Ic) in which R 1 is —N (H) S (═O) 2 R 7 can be synthesized, for example, with an amine derivative (IV) in the presence of a base as shown in Scheme 3 below. It can be produced by a sulfonamidation reaction with a sulfonic acid chloride derivative (VII).
(Scheme 3)
[Wherein R 2 to R 5 and R 7 are the same as defined above]. ]
(スキーム3)
(Scheme 3)
スルホンアミド化反応に用いる溶媒としては、例えば、ジクロロメタン、1,2-ジクロロエタン、アセトニトリル、DMF、THF、ジオキサン、ジエチルエーテル又は1,2-ジメトキシエタンが挙げられるが、ジクロロメタン、1,2-ジクロロエタン、アセトニトリル又はTHFが好ましく、ジクロロメタン又は1,2-ジクロロエタンがより好ましい。
Examples of the solvent used in the sulfonamidation reaction include dichloromethane, 1,2-dichloroethane, acetonitrile, DMF, THF, dioxane, diethyl ether, or 1,2-dimethoxyethane, but dichloromethane, 1,2-dichloroethane, Acetonitrile or THF is preferred, and dichloromethane or 1,2-dichloroethane is more preferred.
スルホンアミド化反応に用いるスルホン酸クロリド誘導体(VII)の当量は、アミン誘導体(IV)に対して0.1~10当量が好ましく、1~3当量がより好ましく、1~1.5当量がさらに好ましい。
The equivalent amount of the sulfonic acid chloride derivative (VII) used in the sulfonamidation reaction is preferably 0.1 to 10 equivalents, more preferably 1 to 3 equivalents, and further preferably 1 to 1.5 equivalents with respect to the amine derivative (IV). preferable.
スルホンアミド化反応に用いる塩基としては、例えば、DIPEA、TEA、ピリジン又はN-メチルモルホリン等の有機塩基が挙げられるが、DIPEA又はTEAが好ましい。該塩基の当量は、アミン誘導体(IV)に対して1~100当量が好ましく、1~10当量がより好ましい。
Examples of the base used in the sulfonamidation reaction include organic bases such as DIPEA, TEA, pyridine and N-methylmorpholine, with DIPEA or TEA being preferred. The equivalent of the base is preferably 1 to 100 equivalents and more preferably 1 to 10 equivalents with respect to the amine derivative (IV).
スルホンアミド化反応の反応温度は、-50~50℃が好ましく、-30~30℃がより好ましく、-20~20℃がさらに好ましい。また、スルホンアミド化反応の反応時間は、30分間~24時間が好ましく、30分間~12時間がより好ましく、30分間~8時間がさらに好ましい。
The reaction temperature of the sulfonamidation reaction is preferably −50 to 50 ° C., more preferably −30 to 30 ° C., and further preferably −20 to 20 ° C. The reaction time of the sulfonamidation reaction is preferably 30 minutes to 24 hours, more preferably 30 minutes to 12 hours, and further preferably 30 minutes to 8 hours.
スルホンアミド化反応におけるアミン誘導体(IV)の反応開始時の濃度は、0.01~100Mが好ましく、0.01~10Mがより好ましく、0.1~10Mがさらに好ましい。
The concentration of the amine derivative (IV) at the start of the reaction in the sulfonamidation reaction is preferably 0.01 to 100M, more preferably 0.01 to 10M, and further preferably 0.1 to 10M.
上記のスキーム2及び3における出発物質であるアミン誘導体(IV)は、例えば、以下のスキーム4に示すように、塩基存在下、アミン誘導体(II)とカルボン酸誘導体(VIII)との縮合反応後、保護基を除去する脱保護反応により製造できる。
(スキーム4)

[式中、R2~R5は、上記定義に同じであり、R8は、保護基を表す。]
The amine derivative (IV), which is the starting material in the above-mentioned schemes 2 and 3, is, for example, after the condensation reaction between the amine derivative (II) and the carboxylic acid derivative (VIII) in the presence of a base, as shown in the following scheme 4. Can be produced by a deprotection reaction to remove the protecting group.
(Scheme 4)
[Wherein R 2 to R 5 are the same as defined above, and R 8 represents a protecting group. ]
(スキーム4)
(Scheme 4)
アミン誘導体(II)とカルボン酸誘導体(VIII)との縮合反応は、スキーム1と同様の条件により行うことができる。
The condensation reaction of the amine derivative (II) and the carboxylic acid derivative (VIII) can be performed under the same conditions as in Scheme 1.
縮合反応に続く脱保護反応は、例えば、Protective Groups in Organic Synthesis第3版(Greenら著、1999年、John Wiley & Sons,Inc.)に記されている公知の方法により行うことができる。例えば、保護基が、tert-ブトキシカルボニル基である場合には、トリフルオロ酢酸などの強酸で処理することで保護基を除去することができる。
The deprotection reaction following the condensation reaction can be performed by a known method described in, for example, Protective Groups in Organic Synthesis 3rd Edition (Green et al., 1999, John Wiley & Sons, Inc.). For example, when the protecting group is a tert-butoxycarbonyl group, the protecting group can be removed by treatment with a strong acid such as trifluoroacetic acid.
上記のスキーム4におけるカルボン酸誘導体(VIII)は、市販品をそのまま利用してもよいし、又は、公知の方法により製造しても構わない。
As the carboxylic acid derivative (VIII) in the above scheme 4, a commercially available product may be used as it is, or it may be produced by a known method.
上記のスキーム1及び4における出発物質であるアミン誘導体(II)は、例えば、以下のスキーム5に示すように、塩基及び縮合剤存在下、ベンジルアミン誘導体(IX)とニペコチン酸誘導体(X)との縮合反応後、保護基を除去する脱保護反応により製造できる。
(スキーム5)

[式中、R4、R5及びR8は、上記定義に同じ。]
The amine derivative (II) which is the starting material in the above-mentioned schemes 1 and 4 is, for example, as shown in the following scheme 5, in the presence of a base and a condensing agent, benzylamine derivative (IX), nipecotic acid derivative (X) and After the condensation reaction, a deprotection reaction for removing the protecting group can be used.
(Scheme 5)
[Wherein R 4 , R 5 and R 8 are the same as defined above. ]
(スキーム5)
(Scheme 5)
ベンジルアミン誘導体(IX)とニペコチン酸誘導体(X)との縮合反応は、スキーム1と同様の条件により行うことができる。
The condensation reaction of the benzylamine derivative (IX) and the nipecotic acid derivative (X) can be carried out under the same conditions as in Scheme 1.
脱保護反応は、スキーム4記載の方法と同様の条件にて行うことができる。
The deprotection reaction can be carried out under the same conditions as in the method described in Scheme 4.
スキーム5の縮合反応は、ニペコチン酸誘導体(X)を酸クロリドへ変換し、塩基存在下にて行うこともできる。
The condensation reaction of Scheme 5 can also be performed in the presence of a base by converting the nipecotic acid derivative (X) into an acid chloride.
ニペコチン酸誘導体(X)を酸クロリドへ変換する試薬としては、オキサリルクロリド又は塩化チオニルが挙げられるが、オキサリルクロリドが好ましい。該試薬の当量は、ニペコチン酸誘導体(X)に対して1~10当量が好ましく、1~1.5当量がより好ましい。
Examples of the reagent for converting the nipecotic acid derivative (X) into acid chloride include oxalyl chloride and thionyl chloride, with oxalyl chloride being preferred. The equivalent amount of the reagent is preferably 1 to 10 equivalents, more preferably 1 to 1.5 equivalents, relative to the nipecotic acid derivative (X).
ニペコチン酸誘導体(X)を酸クロリドへ変換する際に用いる溶媒としては、ジクロロメタン、クロロホルム、THF、1,2-ジクロロエタン、アセトニトリル、1,4-ジオキサン又はDMFが挙げられるが、ジクロロメタン、THF若しくはDMF、又は、これらの混合溶媒が好ましく、ジクロロメタンとDMFとの混合溶媒又はTHFとDMFとの混合溶媒がより好ましい。混合溶媒の比率としては、例えば、ジクロロメタンとDMFとの混合溶媒の場合、ジクロロメタン:DMF=1~1000:1が好ましく、1~100:1がより好ましい。
Examples of the solvent used for converting the nipecotic acid derivative (X) into the acid chloride include dichloromethane, chloroform, THF, 1,2-dichloroethane, acetonitrile, 1,4-dioxane, and DMF, and dichloromethane, THF, or DMF. Alternatively, a mixed solvent thereof is preferable, and a mixed solvent of dichloromethane and DMF or a mixed solvent of THF and DMF is more preferable. As a ratio of the mixed solvent, for example, in the case of a mixed solvent of dichloromethane and DMF, dichloromethane: DMF = 1 to 1000: 1 is preferable, and 1 to 100: 1 is more preferable.
ニペコチン酸誘導体(X)を酸クロリドへ変換する際の反応温度は、-50~100℃が好ましく、-30~30℃が好ましく、-20~0℃がさらに好ましい。また、ニペコチン酸誘導体(X)を酸クロリドへ変換する際の反応時間は、30分間~24時間が好ましく、30分間~12時間がより好ましく、30分間~2時間がさらに好ましい。
The reaction temperature for converting the nipecotic acid derivative (X) to acid chloride is preferably -50 to 100 ° C, more preferably -30 to 30 ° C, and further preferably -20 to 0 ° C. The reaction time for converting the nipecotic acid derivative (X) to acid chloride is preferably 30 minutes to 24 hours, more preferably 30 minutes to 12 hours, and even more preferably 30 minutes to 2 hours.
ニペコチン酸誘導体(X)を酸クロリドへ変換する際の反応開始時のニペコチン酸誘導体(X)の濃度は、0.01~100Mが好ましく、0.01~10Mがより好ましく、0.1~3Mがさらに好ましい。
The concentration of the nipecotic acid derivative (X) at the start of the reaction when converting the nipecotic acid derivative (X) to acid chloride is preferably 0.01 to 100M, more preferably 0.01 to 10M, and more preferably 0.1 to 3M. Is more preferable.
以上のようにして得られる上記のニペコチン酸誘導体(I)、その薬理学的に許容される塩、又は、上記のニペコチン酸誘導体(I)の製造に用いる中間体、原料化合物若しくは試薬は、必要に応じて、抽出、蒸留、クロマトグラフィー又は再結晶等の方法で単離精製してもよい。
The nipecotic acid derivative (I) obtained as described above, a pharmacologically acceptable salt thereof, or an intermediate, a raw material compound or a reagent used for the production of the nipecotic acid derivative (I) is necessary. Depending on the method, it may be isolated and purified by a method such as extraction, distillation, chromatography or recrystallization.
上記の鎮痛剤は、上記のニペコチン酸誘導体(I)又はその薬理学的に許容される塩を有効成分として含有することを特徴としており、sEH阻害活性に基づき、疼痛、特に、侵害受容性疼痛及び神経障害性疼痛に対して高い鎮痛効果を発揮することができる。
The above analgesic is characterized by containing the above nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof as an active ingredient, and is based on sEH inhibitory activity, and in particular, nociceptive pain In addition, it can exert a high analgesic effect on neuropathic pain.
「sEH」は、可溶性エポキシド加水分解酵素(soluble epoxide hydrolase)の略であり、エポキシドの加水分解を触媒し、それに対応するジオールに変換する代謝酵素である。sEHの最も知られた基質は、内皮細胞由来の過分極因子の一つであるEETsであり、sEHは、EETsをDHETsに代謝して失活させる作用を有している。「EETs」は、エポキシエイコサトリエン酸(Epoxyeicosatrienoic acids)の略であり、「DHETs」は、ジヒドロキシエイコサトリエン酸(Dihydroxyeicosatrienoic acids)の略である。EETsとして、例えば、14,15―エポキシエイコサトリエン酸(14,15―Epoxyeicosatrienoic acid;以下、14,15―EET)が挙げられる。DHETsとして、例えば、14,15―ジヒドロキシエイコサトリエン酸(14,15―Dihydroxyeicosatrienoic acid;以下、14,15―DHET)が挙げられる。
“SEH” is an abbreviation for soluble epoxide hydrolase, which is a metabolic enzyme that catalyzes the hydrolysis of epoxide and converts it to the corresponding diol. The most known substrate of sEH is EETs, which is one of endothelial cell-derived hyperpolarizing factors, and sEH has an action of metabolizing EETs to DHETs and inactivating them. “EETs” is an abbreviation for Epoxyeicosatrienoic acids, and “DHETs” is an abbreviation for Dihydroxyeicosatrienoic acids. Examples of the EETs include 14,15-epoxyeicosatrienoic acid (hereinafter, 14,15-EET). Examples of DHETs include 14,15-dihydroxyeicosatrienoic acid (14,15-DHET).
「sEH阻害活性」とは、sEHの作用を阻害する活性を意味する。したがって、sEH阻害活性とは、sEHの基質の一つであるEETsの加水分解を触媒するというsEHの酵素反応を阻害する活性を包含する。
“SEH inhibitory activity” means an activity that inhibits the action of sEH. Therefore, the sEH inhibitory activity includes the activity of inhibiting the enzymatic reaction of sEH to catalyze the hydrolysis of EETs, which is one of sEH substrates.
「sEH阻害剤」とは、sEH阻害活性を示す化合物又は該化合物を有効成分として含有する組成物を意味する。
“SEH inhibitor” means a compound showing sEH inhibitory activity or a composition containing the compound as an active ingredient.
sEH阻害活性は、例えば、ヒトsEHと、その基質であるEETsとをsEH阻害剤の存在下で反応させ、産生されるDHETsの量を、sEH阻害剤非存在下におけるDHETs産生量と比較することで測定できる。また、市販の測定キット(Soluble Epoxide Hydrolase Inhibitor Screening Assay Kit;Cayman社)を用いるか、公知文献(Analytical Biochemistry、2005年、第343巻、p.66-75等)に記載の方法によっても、sEH阻害剤のsEH阻害活性を測定できる。
The sEH inhibitory activity is, for example, that human sEH and its substrate EETs are reacted in the presence of an sEH inhibitor, and the amount of DHETs produced is compared with the amount of DHETs produced in the absence of the sEH inhibitor. Can be measured. In addition, a commercially available measurement kit (Soluable Epoxide Hydrose Inhibitor Screening Assay Kit; Cayman) is used, or is described in a publicly known document (Analytical Biochemistry, 2005, Vol. 343, p. 66-75, E, etc.). The sEH inhibitory activity of the inhibitor can be measured.
また、sEH阻害剤存在下及び非存在下で、sEHの基質としてラセミ性4-ニトロフェニル-トランス-2,3-エポキシ-3-フェニルプロピルカルボナートを用いて、4-ニトロフェノラート陰イオンの出現を測定するか、又は、sEHの基質としてシアノ(6-メトキシナフタレン-2-イル)メチル 2-(3-フェニルオキシラン-2-イル)アセタートを用いて、6-メトキシ-2-ナフトアルデヒドの出現を測定することによっても、sEH阻害剤のsEH阻害活性を測定できる。
In addition, in the presence and absence of an sEH inhibitor, racemic 4-nitrophenyl-trans-2,3-epoxy-3-phenylpropyl carbonate was used as a substrate for sEH, and 4-nitrophenolate anion was used. The appearance of 6-methoxy-2-naphthaldehyde is measured using cyano (6-methoxynaphthalen-2-yl) methyl 2- (3-phenyloxiran-2-yl) acetate as a substrate for sEH. The sEH inhibitory activity of the sEH inhibitor can also be measured by measuring the appearance.
「侵害受容性疼痛」とは、生体組織が損傷を受けたり、そのような危険を有する侵害刺激が加えられたりすることで生じる痛みであり、侵害受容器を介した疼痛のことをいう。
“Nociceptive pain” refers to pain caused by damage to biological tissue or the addition of a noxious stimulus having such a risk, and refers to pain via nociceptors.
ここでいう侵害受容性疼痛としては、例えば、骨折や切傷等の受傷による痛み、術後疼痛、ねんざ痛、打撲痛、関節痛、腰痛、筋肉痛、抜歯後疼痛、歯痛、虫垂炎、慢性関節リウマチ、リウマチ熱、変形性関節症、強直性脊椎炎、変形性脊椎症、頸肩腕症候群、関節周囲炎、結合組織炎、急性中耳炎、前立腺炎、歯槽骨膜炎、膣炎等炎症性疾患による痛みが挙げられる。また、上記侵害受容性疼痛には、深部痛及び内臓痛(例えば、頭痛、腹痛、腰背部痛、慢性骨盤痛症候群、子宮内膜症に伴う痛み、尿路結石症や尿道結石に伴う痛み、消化器病変に伴う疝痛、骨盤痛、泌尿器疾患に伴う痛み)が含まれるものとする。上記の鎮痛剤のより好ましい対象疾患としては、慢性関節リウマチ、変形性関節症、術後疼痛、関節痛、腰痛、筋肉痛又は歯痛が挙げられる。
Examples of nociceptive pain include pain caused by injuries such as fractures and cuts, postoperative pain, sprain pain, bruise pain, joint pain, low back pain, muscle pain, post-extraction pain, toothache, appendicitis, rheumatoid arthritis. Pain due to inflammatory diseases such as rheumatic fever, osteoarthritis, ankylosing spondylitis, osteoarthritis, cervical shoulder arm syndrome, periarthritis, connective tissue inflammation, acute otitis media, prostatitis, alveolar periosteitis, vaginitis Can be mentioned. In addition, the nociceptive pain includes deep pain and visceral pain (for example, headache, abdominal pain, back and back pain, chronic pelvic pain syndrome, pain associated with endometriosis, pain associated with urolithiasis and urethral stones, Pain associated with gastrointestinal lesions, pelvic pain, pain associated with urological disorders). More preferable target diseases of the above analgesic include rheumatoid arthritis, osteoarthritis, postoperative pain, joint pain, low back pain, muscle pain or toothache.
「神経障害性疼痛」とは、末梢又は中枢神経系そのものの機能異常による病的な痛みであり、侵害受容器が侵害刺激を受けていないにもかかわらず、神経組織の直接的な損傷や圧迫等によって生じる疼痛のことをいう。
“Neuropathic pain” is pathological pain caused by abnormalities in the function of the peripheral or central nervous system itself, and direct damage or compression of nerve tissue even though nociceptors are not subjected to noxious stimuli. This refers to pain caused by the like.
ここでいう神経障害性疼痛としては、例えば、癌性疼痛、帯状疱疹痛、帯状疱疹後神経痛、有痛性糖尿病性神経障害痛、エイズ関連神経痛、神経障害性腰痛、幻肢痛、抗がん剤投与による神経障害痛、脊髄損傷後疼痛、手根幹症候群や脊柱管狭窄症などの絞扼性神経障害による疼痛、ギランバレー症候群による疼痛又は三叉神経痛が挙げられる。
Examples of neuropathic pain include cancer pain, herpes zoster pain, postherpetic neuralgia, painful diabetic neuropathic pain, AIDS-related neuralgia, neuropathic back pain, phantom limb pain, anticancer Neuropathic pain caused by administration of agents, pain after spinal cord injury, pain due to strangulated neuropathy such as carpal trunk syndrome and spinal canal stenosis, pain due to Guillain-Barre syndrome, or trigeminal neuralgia.
上記の鎮痛剤の有効成分であるニペコチン酸誘導体(I)又はその薬理学的に許容される塩の侵害受容性疼痛及び神経障害性疼痛に対する鎮痛効果は、適切な動物モデルを用いて評価することができる。侵害受容性疼痛の適切な動物モデルとしては、例えば、マウス酢酸ライジングモデル、ラットホルマリンテスト、ラットカラゲニン誘発炎症モデル、急性疼痛のためのマウス若しくはラットホットプレートテスト、又はテールフリックテストが挙げられる。
The analgesic effect on nociceptive pain and neuropathic pain of nipecotic acid derivative (I), which is an active ingredient of the above-mentioned analgesic agent, or a pharmacologically acceptable salt thereof should be evaluated using an appropriate animal model. Can do. Suitable animal models for nociceptive pain include, for example, the mouse acetate rising model, the rat formalin test, the rat carrageenan-induced inflammation model, the mouse or rat hot plate test for acute pain, or the tail flick test.
また、神経障害性疼痛の適切な動物モデルとしては、例えば、マウス若しくはラット坐骨神経部分結紮モデル又はマウス若しくはラット脊髄神経結紮モデル、あるいはマウス若しくはラットストレプトゾトシン誘発糖尿病性神経障害モデルが挙げられる。
Also, suitable animal models of neuropathic pain include, for example, a mouse or rat partial sciatic nerve ligation model or a mouse or rat spinal nerve ligation model, or a mouse or rat streptozotocin-induced diabetic neuropathy model.
上記の鎮痛剤は、急性及び慢性疼痛に対しても有用である。急性疼痛は、通常短期間であるが、例えば、急性術後疼痛、帯状疱疹痛、抜歯後疼痛又は三叉神経痛が挙げられる。慢性疼痛は、通常3~6ヶ月間持続する疼痛と定義され、かつ体因性疼痛及び心因性疼痛を含むが、例えば、慢性関節リウマチ、変形性関節症、有痛性糖尿病性神経障害痛、慢性術後神経障害性疼痛又は帯状疱疹後神経痛が挙げられる。
The above analgesics are also useful for acute and chronic pain. Acute pain is usually short-term, but includes acute postoperative pain, herpes zoster pain, post-extraction pain, or trigeminal neuralgia. Chronic pain is usually defined as pain lasting for 3-6 months and includes somatic and psychogenic pain, such as rheumatoid arthritis, osteoarthritis, painful diabetic neuropathic pain Chronic postoperative neuropathic pain or postherpetic neuralgia.
上記の鎮痛剤は、有効成分であるニペコチン酸誘導体(I)又はその薬理学的に許容される塩を、そのまま、又は、適当な剤形の医薬組成物として、哺乳動物(例えば、マウス、ラット、ハムスター、ウサギ、イヌ、サル、ウシ、ヒツジ又はヒト)に対して、経口的又は非経口的(例えば、経皮投与、静脈投与、直腸内投与、吸入投与、点鼻投与又は点眼投与)に投与することができる。
The above-mentioned analgesic is prepared by using a nipecotic acid derivative (I), which is an active ingredient, or a pharmacologically acceptable salt thereof as it is or as a pharmaceutical composition in an appropriate dosage form for mammals (eg, mice, rats, etc.). , Hamsters, rabbits, dogs, monkeys, cows, sheep or humans) orally or parenterally (eg, transdermal, intravenous, rectal, inhalation, nasal or ophthalmic) Can be administered.
哺乳動物への投与のための剤形としては、例えば、錠剤、散剤、丸剤、カプセル剤、顆粒剤、シロップ剤、液剤、注射剤、乳剤、懸濁剤若しくは坐剤、又は公知の持続型製剤が挙げられる。これら剤形は、公知の方法によって製造でき、製剤分野において一般的に用いられる担体を含有するものである。そのような担体としては、例えば、固形製剤における賦形剤、滑沢剤、結合剤、崩壊剤、あるいは、液状製剤における溶剤、溶解補助剤、懸濁化剤又は無痛化剤が挙げられる。また必要に応じて、等張化剤、緩衝剤、防腐剤、抗酸化剤、着色剤、甘味剤、吸着剤又は湿潤剤等の添加物を用いても構わない。
Examples of the dosage form for administration to mammals include tablets, powders, pills, capsules, granules, syrups, solutions, injections, emulsions, suspensions or suppositories, or known continuous forms. A formulation is mentioned. These dosage forms can be produced by a known method and contain a carrier generally used in the pharmaceutical field. Examples of such carriers include excipients, lubricants, binders, disintegrants in solid preparations, and solvents, solubilizers, suspending agents, or soothing agents in liquid preparations. Moreover, you may use additives, such as a tonicity agent, a buffering agent, antiseptic | preservative, an antioxidant, a coloring agent, a sweetener, an adsorbent, or a wetting agent, as needed.
賦形剤としては、例えば、乳糖、D-マンニトール、澱粉、ショ糖、コーンスターチ、結晶セルロース又は軽質無水ケイ酸が挙げられる。
Examples of the excipient include lactose, D-mannitol, starch, sucrose, corn starch, crystalline cellulose, and light anhydrous silicic acid.
滑沢剤としては、例えば、ステアリン酸マグネシウム、ステアリン酸カルシウム、タルク又はコロイドシリカが挙げられる。
Examples of the lubricant include magnesium stearate, calcium stearate, talc, and colloidal silica.
結合剤としては、例えば、結晶セルロース、D-マンニトール、デキストリン、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、澱粉、ショ糖、ゼラチン、メチルセルロース又はカルボキシメチルセルロースナトリウムが挙げられる。
Examples of the binder include crystalline cellulose, D-mannitol, dextrin, hydroxypropylcellulose, hydroxypropylmethylcellulose, polyvinylpyrrolidone, starch, sucrose, gelatin, methylcellulose or sodium carboxymethylcellulose.
崩壊剤としては、例えば、澱粉、カルボキシメチルセルロース、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、カルボキシメチルスターチナトリウム又はL-ヒドロキシプロピルセルロースが挙げられる。
Examples of the disintegrant include starch, carboxymethyl cellulose, carboxymethyl cellulose calcium, croscarmellose sodium, carboxymethyl starch sodium, and L-hydroxypropyl cellulose.
溶剤としては、例えば、注射用水、アルコール、プロピレングリコール、マクロゴール、ゴマ油又はトウモロコシ油が挙げられる。
Examples of the solvent include water for injection, alcohol, propylene glycol, macrogol, sesame oil or corn oil.
溶解補助剤としては、例えば、ポリエチレングリコール、プロピレングリコール、D-マンニトール、安息香酸ベンジル、エタノール、コレステロール、トリエタノールアミン、炭酸ナトリウム又はクエン酸ナトリウムが挙げられる。
Examples of the solubilizer include polyethylene glycol, propylene glycol, D-mannitol, benzyl benzoate, ethanol, cholesterol, triethanolamine, sodium carbonate, or sodium citrate.
懸濁化剤としては、例えば、ステアリルトリエタノールアミン、ラウリル硫酸ナトリウム、ラウリルアミノプロピオン酸、レシチン、塩化ベンザルコニウム、塩化ベンゼトニウム若しくはモノステアリン酸グリセリン等の界面活性剤又はポリビニルアルコール、ポリビニルピロリドン、メチルセルロース、ヒドロキシメチルセルロース、ヒドロキシエチルセルロース若しくはヒドロキシプロピルセルロース等の親水性高分子が挙げられる。
Examples of the suspending agent include surfactants such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalkonium chloride, benzethonium chloride or glyceryl monostearate, or polyvinyl alcohol, polyvinylpyrrolidone, methylcellulose. , Hydrophilic polymers such as hydroxymethylcellulose, hydroxyethylcellulose or hydroxypropylcellulose.
無痛化剤としては、例えば、ベンジルアルコールが挙げられる。
Examples of soothing agents include benzyl alcohol.
等張化剤としては、例えば、ブドウ糖、塩化ナトリウム、D-ソルビトール又はD-マンニトールが挙げられる。
Examples of the isotonic agent include glucose, sodium chloride, D-sorbitol, and D-mannitol.
緩衝剤としては、例えば、リン酸塩、酢酸塩、炭酸塩又はクエン酸塩等の緩衝液が挙げられる。
Examples of the buffer include buffer solutions such as phosphate, acetate, carbonate or citrate.
防腐剤としては、例えば、パラオキシ安息香酸エステル類、クロロブタノール、ベンジルアルコール、フェネチルアルコール、デヒドロ酢酸又はソルビン酸が挙げられる。
Examples of the preservative include p-oxybenzoates, chlorobutanol, benzyl alcohol, phenethyl alcohol, dehydroacetic acid, and sorbic acid.
抗酸化剤としては、例えば、亜硫酸塩又はアスコルビン酸が挙げられる。
Examples of the antioxidant include sulfite and ascorbic acid.
上記の鎮痛剤は、ニペコチン酸誘導体(I)又はその薬理学的に許容される塩を、0.001~99重量%含有することが好ましく、0.01~99重量%含有することがより好ましい。ニペコチン酸誘導体(I)又はその薬理学的に許容される塩の、有効投与量及び投与回数は、投与形態、患者の年齢、体重又は治療/予防すべき症状の性質若しくは重篤度によっても異なるが、通常成人1日当り1~1000mgを、好ましくは1~300mgを、1回又は数回に分けて投与することができる。
The analgesic preferably contains 0.001 to 99% by weight, more preferably 0.01 to 99% by weight, of the nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof. . The effective dosage and frequency of administration of the nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof vary depending on the dosage form, patient age, body weight, or the nature or severity of the symptoms to be treated / prevented. However, usually 1 to 1000 mg, preferably 1 to 300 mg per day for an adult can be administered in one or several divided doses.
なお、上記の鎮痛剤は、単独で投与してもよいが、疾患の予防効果若しくは治療効果の補完又は増強、あるいは投与量の低減のために、他の薬剤と配合するか、他の薬剤と併用して使用することもできる。
The above analgesics may be administered alone, but may be combined with other drugs or combined with other drugs to supplement or enhance the preventive or therapeutic effect of the disease or reduce the dose. It can also be used in combination.
配合又は併用し得る他の薬剤(以下、併用薬剤)としては、例えば、鎮咳剤、去痰剤、鎮咳去痰剤、気管支拡張剤、消化性潰瘍剤、抗生物質又は麻薬性鎮痛剤が挙げられる。
Examples of other drugs that can be combined or used together (hereinafter referred to as combined drugs) include antitussives, expectorants, antitussive expectorants, bronchodilators, peptic ulcers, antibiotics, or narcotic analgesics.
上記の鎮痛剤を併用薬剤と併用して使用する場合には、上記の鎮痛剤と併用薬剤の投与時期は特に限定されず、これらを投与対象に対して同時に投与してもよいし、時間差をおいて投与しても構わない。また、併用薬剤は低分子化合物であってもよいし、タンパク質、ポリペプチド若しくは抗体等の高分子又はワクチン等であっても構わない。併用薬剤の投与量は、臨床上用いられている投与量を基準として、適宜選択することができる。上記の鎮痛剤と併用薬剤との配合比は、投与対象、投与ルート、対象疾患、症状又は、上記の鎮痛剤と併用薬剤の組み合わせ等により適宜選択することができる。例えば、投与対象がヒトである場合には、上記の鎮痛剤に対し、併用薬剤を0.01~99.99の配合比で用いればよい。
When the analgesic is used in combination with a concomitant drug, the timing of administration of the analgesic and the concomitant drug is not particularly limited, and these may be administered simultaneously to the administration subject, May be administered. The concomitant drug may be a low molecular compound, a polymer such as a protein, polypeptide or antibody, or a vaccine. The dose of the concomitant drug can be appropriately selected based on the clinically used dose. The mixing ratio of the analgesic and the concomitant drug can be appropriately selected depending on the administration subject, administration route, target disease, symptom, combination of the above analgesic and concomitant drug, and the like. For example, when the administration subject is a human, the concomitant drug may be used in a mixing ratio of 0.01 to 99.99 with respect to the above analgesic.
鎮咳剤、去痰剤及び鎮咳去痰剤としては、例えば、デキストロメトルファン(dextromethorphan)、ベンプロペリン(benproperine)、ジメモルファン(dimemorfan)、クロフェダノール(clofedanol)、エフェドリン(ephedrine)、フスコデ(huscode)、ホミノベン(fominoben)、メチルエフェドリン(methylephedrine)、アセチルシステイン(acetylcysteine)、アンブロキソール(ambroxol)、カルボシステイン(carbocisteine)、ブロムヘキシン(bromhexine)、エプラジノン(eprazinone)、オウヒエキス、コデイン(codeine)、ジヒドロコデイン(dihydrocodeine)又はチペピジン(tipepidine)が挙げられる。
Antitussives, expectorants and antitussive expectorants include, for example, dextromethorphan, benproperine, dimemorphan, clofedanol, ephedrine, decoben, minc ), Methylephedrine, acetylcysteine, ambroxol, carbocysteine, bromhexine, epradinone, incode, indehyde Ydrocodeine) or tipepidine (tipepidine) and the like.
気管支拡張剤としては、例えば、クレンブテロール(clenbuterol)、クロモグリク酸(cromoglycate)、サルブタモール(salbutamol)、サルメテロール(salmeterol)、ツロブテロール(tulobuterol)、テオフィリン(theophylline)又はプロカテロール(procaterol)が挙げられる。
Examples of bronchodilators include clenbuterol, cromoglycate, salbutamol, salmeterol, tulobuterol, and theophylline.
消化性潰瘍剤としては、例えば、アズレン(azulene)、アルジオキサ(aldioxa)、イルソグラジン(irsogladine)、エカベト(ecabet)、オメプラゾール(omeprazole)、オルノプロスチル(ornoprostil)、シメチジン(cimetidine)、スクラルファート(sucralfate)、スルピリド(sulpiride)、セトラキサート(cetraxate)又はファモチジン(famotidine)が挙げられる。
Examples of peptic ulcer agents include azulene, aldioxa, irsogladine, ecabet, omeprazole, ornoprostil, cimetidine, cimetidine, cimetidine. , Sulpiride, cetraxate or famotidine.
抗生物質としては、例えば、アモキシシリン(amoxicillin)、アジスロマイシン(azithromycin)、エリスロマイシン(erythromycin)、クラリスロマイシン(clarithromycin)、テトラサイクリン(tetracycline)又はドキシサイクリン(doxycycline)が挙げられる。
Antibiotics include, for example, amoxicillin, azithromycin, erythromycin, clarithromycin, tetracycline, and doxycycline (doxycycline).
麻薬性鎮痛剤としては、例えば、アヘンアルカロイド、エチルモルヒネ(ethylmorphine)、オキシコドン(oxycodone)、モルヒネ(morphine)、コカイン(cocaine)、フェンタニル(fentanyl)又はペチジン(pethidine)が挙げられる。
Examples of narcotic analgesics include opium alkaloids, ethyl morphine, oxycodone, morphine, cocaine, fentanyl, and pethidine.
以下、実施例を挙げて本発明を具体的に説明するが、本発明はこれらに限定されるものではない。シリカゲルカラムクロマトグラフィーによる精製は、特に記述のない場合、「HI-FLASH」カラム(山善社)及びPurif-α2(昭光サイエンティフィックス社)を用いて行った。
Hereinafter, the present invention will be specifically described with reference to examples, but the present invention is not limited thereto. Purification by silica gel column chromatography was performed using a “HI-FLASH” column (Yamazen Co., Ltd.) and Purif-α2 (Shoko Scientific Co., Ltd.) unless otherwise specified.
(実施例1) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-プロピオンアミドシクロブタンカルボニル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
4-ブロモ-2-(トリフルオロメトキシ)ベンズアルデヒドの合成:
-78℃下、4-ブロモ-1-ヨード-2-(トリフルオロメトキシ)ベンゼン(25g、68mmol)のTHF(0.40L)溶液に、n-ブチルリチウムヘキサン溶液(1.6規定、86mL、0.14mol)を1.5時間かけて滴下した。-78℃下で1時間撹拌した後、反応溶液に、DMF(11mL、0.14mmol)を10分間かけて滴下した。-78℃下で2時間撹拌した後、反応溶液に、クエン酸水溶液(0.25M、0.25L、63mmol)を加え、ジエチルエーテルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、4-ブロモ-2-(トリフルオロメトキシ)ベンズアルデヒド(以下、参考例化合物1)を16g(87%)得た。 Example 1 Synthesis of (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-propionamidocyclobutanecarbonyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of 4-bromo-2- (trifluoromethoxy) benzaldehyde:
To a THF (0.40 L) solution of 4-bromo-1-iodo-2- (trifluoromethoxy) benzene (25 g, 68 mmol) at −78 ° C., an n-butyllithium hexane solution (1.6 N, 86 mL, 0.14 mol) was added dropwise over 1.5 hours. After stirring at −78 ° C. for 1 hour, DMF (11 mL, 0.14 mmol) was added dropwise to the reaction solution over 10 minutes. After stirring at −78 ° C. for 2 hours, an aqueous citric acid solution (0.25 M, 0.25 L, 63 mmol) was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain 16 g (87%) of 4-bromo-2- (trifluoromethoxy) benzaldehyde (hereinafter referred to as Reference Example Compound 1).
〔ステップ1〕
4-ブロモ-2-(トリフルオロメトキシ)ベンズアルデヒドの合成:
-78℃下、4-ブロモ-1-ヨード-2-(トリフルオロメトキシ)ベンゼン(25g、68mmol)のTHF(0.40L)溶液に、n-ブチルリチウムヘキサン溶液(1.6規定、86mL、0.14mol)を1.5時間かけて滴下した。-78℃下で1時間撹拌した後、反応溶液に、DMF(11mL、0.14mmol)を10分間かけて滴下した。-78℃下で2時間撹拌した後、反応溶液に、クエン酸水溶液(0.25M、0.25L、63mmol)を加え、ジエチルエーテルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、4-ブロモ-2-(トリフルオロメトキシ)ベンズアルデヒド(以下、参考例化合物1)を16g(87%)得た。 Example 1 Synthesis of (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-propionamidocyclobutanecarbonyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of 4-bromo-2- (trifluoromethoxy) benzaldehyde:
To a THF (0.40 L) solution of 4-bromo-1-iodo-2- (trifluoromethoxy) benzene (25 g, 68 mmol) at −78 ° C., an n-butyllithium hexane solution (1.6 N, 86 mL, 0.14 mol) was added dropwise over 1.5 hours. After stirring at −78 ° C. for 1 hour, DMF (11 mL, 0.14 mmol) was added dropwise to the reaction solution over 10 minutes. After stirring at −78 ° C. for 2 hours, an aqueous citric acid solution (0.25 M, 0.25 L, 63 mmol) was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain 16 g (87%) of 4-bromo-2- (trifluoromethoxy) benzaldehyde (hereinafter referred to as Reference Example Compound 1).
〔ステップ2〕
(4-ブロモ-2-(トリフルオロメトキシ)フェニル)メタノールの合成:
-10℃下、参考例化合物1(16g、59mmol)のメタノール(0.23L)溶液に、水素化ホウ素ナトリウム(2.4g、63mmol)を加えた。-10℃下で10分間撹拌した後、反応溶液に、アセトン(10mL)、1規定塩酸(10mL)を加えた。反応溶液を減圧濃縮し、得られた粗生成物に、水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=50:1→1:1)で精製し、(4-ブロモ-2-(トリフルオロメトキシ)フェニル)メタノール(以下、参考例化合物2)を15g(91%)得た。 [Step 2]
Synthesis of (4-bromo-2- (trifluoromethoxy) phenyl) methanol:
At −10 ° C., sodium borohydride (2.4 g, 63 mmol) was added to a methanol (0.23 L) solution of Reference Example compound 1 (16 g, 59 mmol). After stirring at −10 ° C. for 10 minutes, acetone (10 mL) and 1N hydrochloric acid (10 mL) were added to the reaction solution. The reaction solution was concentrated under reduced pressure, water was added to the obtained crude product, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 50: 1 → 1: 1), and (4-bromo 15 g (91%) of -2- (trifluoromethoxy) phenyl) methanol (hereinafter referred to as Reference Example Compound 2) was obtained.
(4-ブロモ-2-(トリフルオロメトキシ)フェニル)メタノールの合成:
-10℃下、参考例化合物1(16g、59mmol)のメタノール(0.23L)溶液に、水素化ホウ素ナトリウム(2.4g、63mmol)を加えた。-10℃下で10分間撹拌した後、反応溶液に、アセトン(10mL)、1規定塩酸(10mL)を加えた。反応溶液を減圧濃縮し、得られた粗生成物に、水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=50:1→1:1)で精製し、(4-ブロモ-2-(トリフルオロメトキシ)フェニル)メタノール(以下、参考例化合物2)を15g(91%)得た。 [Step 2]
Synthesis of (4-bromo-2- (trifluoromethoxy) phenyl) methanol:
At −10 ° C., sodium borohydride (2.4 g, 63 mmol) was added to a methanol (0.23 L) solution of Reference Example compound 1 (16 g, 59 mmol). After stirring at −10 ° C. for 10 minutes, acetone (10 mL) and 1N hydrochloric acid (10 mL) were added to the reaction solution. The reaction solution was concentrated under reduced pressure, water was added to the obtained crude product, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 50: 1 → 1: 1), and (4-bromo 15 g (91%) of -2- (trifluoromethoxy) phenyl) methanol (hereinafter referred to as Reference Example Compound 2) was obtained.
〔ステップ3〕
4-ブロモ-2-(トリフルオロメトキシ)ベンジル メタンスルホナートの合成:
氷冷下、参考例化合物2(2.0g、7.4mmol)、TEA(1.2mL、8.9mmol)のジクロロメタン(20mL)溶液に、メタンスルホニルクロリド(0.93g、8.1mmol)を加えた。室温下で3時間撹拌した後、反応溶液に、水を加え、ジクロロメタンで抽出した。有機層を飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮し、4-ブロモ-2-(トリフルオロメトキシ)ベンジル メタンスルホナート(以下、参考例化合物3)を2.6g(定量的)得た。 [Step 3]
Synthesis of 4-bromo-2- (trifluoromethoxy) benzyl methanesulfonate:
Methanesulfonyl chloride (0.93 g, 8.1 mmol) was added to a solution of Reference Example Compound 2 (2.0 g, 7.4 mmol) and TEA (1.2 mL, 8.9 mmol) in dichloromethane (20 mL) under ice cooling. It was. After stirring at room temperature for 3 hours, water was added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. 2.6 g of 4-bromo-2- (trifluoromethoxy) benzyl methanesulfonate (hereinafter referred to as Reference Example Compound 3) ( (Quantitative).
4-ブロモ-2-(トリフルオロメトキシ)ベンジル メタンスルホナートの合成:
氷冷下、参考例化合物2(2.0g、7.4mmol)、TEA(1.2mL、8.9mmol)のジクロロメタン(20mL)溶液に、メタンスルホニルクロリド(0.93g、8.1mmol)を加えた。室温下で3時間撹拌した後、反応溶液に、水を加え、ジクロロメタンで抽出した。有機層を飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮し、4-ブロモ-2-(トリフルオロメトキシ)ベンジル メタンスルホナート(以下、参考例化合物3)を2.6g(定量的)得た。 [Step 3]
Synthesis of 4-bromo-2- (trifluoromethoxy) benzyl methanesulfonate:
Methanesulfonyl chloride (0.93 g, 8.1 mmol) was added to a solution of Reference Example Compound 2 (2.0 g, 7.4 mmol) and TEA (1.2 mL, 8.9 mmol) in dichloromethane (20 mL) under ice cooling. It was. After stirring at room temperature for 3 hours, water was added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. 2.6 g of 4-bromo-2- (trifluoromethoxy) benzyl methanesulfonate (hereinafter referred to as Reference Example Compound 3) ( (Quantitative).
〔ステップ4〕
2-(4-ブロモ-2-(トリフルオロメトキシ)ベンジル)イソインドリン-1,3-ジオンの合成:
氷冷下、参考例化合物3(2.6g、7.4mmol)のDMF(20mL)溶液に、フタルイミドカリウム(2.1g、11mmol)を加えた。室温下で14時間撹拌した後、反応溶液に、水を加えた。析出した固体をろ取し、水で洗浄し、乾燥し、2-(4-ブロモ-2-(トリフルオロメトキシ)ベンジル)イソインドリン-1,3-ジオン(以下、参考例化合物4)を2.7g(91%)得た。 [Step 4]
Synthesis of 2- (4-bromo-2- (trifluoromethoxy) benzyl) isoindoline-1,3-dione:
Under ice-cooling, potassium phthalimide (2.1 g, 11 mmol) was added to a DMF (20 mL) solution of Reference Example compound 3 (2.6 g, 7.4 mmol). After stirring at room temperature for 14 hours, water was added to the reaction solution. The precipitated solid was collected by filtration, washed with water, and dried to give 2- (4-bromo-2- (trifluoromethoxy) benzyl) isoindoline-1,3-dione (hereinafter referred to as Reference Example Compound 4). 0.7 g (91%) was obtained.
2-(4-ブロモ-2-(トリフルオロメトキシ)ベンジル)イソインドリン-1,3-ジオンの合成:
氷冷下、参考例化合物3(2.6g、7.4mmol)のDMF(20mL)溶液に、フタルイミドカリウム(2.1g、11mmol)を加えた。室温下で14時間撹拌した後、反応溶液に、水を加えた。析出した固体をろ取し、水で洗浄し、乾燥し、2-(4-ブロモ-2-(トリフルオロメトキシ)ベンジル)イソインドリン-1,3-ジオン(以下、参考例化合物4)を2.7g(91%)得た。 [Step 4]
Synthesis of 2- (4-bromo-2- (trifluoromethoxy) benzyl) isoindoline-1,3-dione:
Under ice-cooling, potassium phthalimide (2.1 g, 11 mmol) was added to a DMF (20 mL) solution of Reference Example compound 3 (2.6 g, 7.4 mmol). After stirring at room temperature for 14 hours, water was added to the reaction solution. The precipitated solid was collected by filtration, washed with water, and dried to give 2- (4-bromo-2- (trifluoromethoxy) benzyl) isoindoline-1,3-dione (hereinafter referred to as Reference Example Compound 4). 0.7 g (91%) was obtained.
〔ステップ5〕
(4-ブロモ-2-(トリフルオロメトキシ)フェニル)メタンアミンの合成:
室温下、参考例化合物4(2.6g、6.5mmol)のメタノール(40mL)溶液に、ヒドラジン一水和物(0.98g、19mmol)を加えた。60℃下で2時間撹拌した後、室温下で析出した固体をろ別した。ろ液を減圧濃縮し、得られた粗生成物を酢酸エチルに溶解させ、水、飽和塩化ナトリウム水溶液で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(4-ブロモ-2-(トリフルオロメトキシ)フェニル)メタンアミン(以下、参考例化合物5)を1.5g(85%)得た。 [Step 5]
Synthesis of (4-bromo-2- (trifluoromethoxy) phenyl) methanamine:
Hydrazine monohydrate (0.98 g, 19 mmol) was added to a methanol (40 mL) solution of Reference Example compound 4 (2.6 g, 6.5 mmol) at room temperature. After stirring at 60 ° C. for 2 hours, the solid precipitated at room temperature was filtered off. The filtrate was concentrated under reduced pressure, and the resulting crude product was dissolved in ethyl acetate and washed with water and a saturated aqueous sodium chloride solution. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain 1.5 g (85%) of (4-bromo-2- (trifluoromethoxy) phenyl) methanamine (hereinafter referred to as Reference Example Compound 5).
(4-ブロモ-2-(トリフルオロメトキシ)フェニル)メタンアミンの合成:
室温下、参考例化合物4(2.6g、6.5mmol)のメタノール(40mL)溶液に、ヒドラジン一水和物(0.98g、19mmol)を加えた。60℃下で2時間撹拌した後、室温下で析出した固体をろ別した。ろ液を減圧濃縮し、得られた粗生成物を酢酸エチルに溶解させ、水、飽和塩化ナトリウム水溶液で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(4-ブロモ-2-(トリフルオロメトキシ)フェニル)メタンアミン(以下、参考例化合物5)を1.5g(85%)得た。 [Step 5]
Synthesis of (4-bromo-2- (trifluoromethoxy) phenyl) methanamine:
Hydrazine monohydrate (0.98 g, 19 mmol) was added to a methanol (40 mL) solution of Reference Example compound 4 (2.6 g, 6.5 mmol) at room temperature. After stirring at 60 ° C. for 2 hours, the solid precipitated at room temperature was filtered off. The filtrate was concentrated under reduced pressure, and the resulting crude product was dissolved in ethyl acetate and washed with water and a saturated aqueous sodium chloride solution. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain 1.5 g (85%) of (4-bromo-2- (trifluoromethoxy) phenyl) methanamine (hereinafter referred to as Reference Example Compound 5).
〔ステップ6〕
(R)-tert-ブチル 3-((4-ブロモ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボキシラートの合成:
室温下、参考例化合物5(0.50g、1.9mmol)、(R)-1-(tert-ブトキシカルボニル)ピペリジン-3-カルボン酸(0.43g、1.9mmol)、DIPEA(0.53g、4.1mmol)のDMF(3.0mL)溶液に、HATU(0.77g、2.0mmol)を加えた。室温下で15時間撹拌した後、反応溶液に、酢酸エチルを加え、有機層を飽和炭酸水素ナトリウム水溶液、水、飽和塩化ナトリウム水溶液で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=9:1→1:1)で精製し、(R)-tert-ブチル 3-((4-ブロモ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボキシラート(以下、参考例化合物6)を0.89g(定量的)得た。 [Step 6]
Synthesis of (R) -tert-butyl 3-((4-bromo-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carboxylate:
Reference temperature compound 5 (0.50 g, 1.9 mmol), (R) -1- (tert-butoxycarbonyl) piperidine-3-carboxylic acid (0.43 g, 1.9 mmol), DIPEA (0.53 g) at room temperature 4.1 mmol) in DMF (3.0 mL) was added HATU (0.77 g, 2.0 mmol). After stirring at room temperature for 15 hours, ethyl acetate was added to the reaction solution, and the organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, water, and a saturated aqueous sodium chloride solution. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 9: 1 → 1: 1), and (R) — As a result, 0.89 g (quantitative) of tert-butyl 3-((4-bromo-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carboxylate (hereinafter referred to as Reference Example Compound 6) was obtained.
(R)-tert-ブチル 3-((4-ブロモ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボキシラートの合成:
室温下、参考例化合物5(0.50g、1.9mmol)、(R)-1-(tert-ブトキシカルボニル)ピペリジン-3-カルボン酸(0.43g、1.9mmol)、DIPEA(0.53g、4.1mmol)のDMF(3.0mL)溶液に、HATU(0.77g、2.0mmol)を加えた。室温下で15時間撹拌した後、反応溶液に、酢酸エチルを加え、有機層を飽和炭酸水素ナトリウム水溶液、水、飽和塩化ナトリウム水溶液で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=9:1→1:1)で精製し、(R)-tert-ブチル 3-((4-ブロモ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボキシラート(以下、参考例化合物6)を0.89g(定量的)得た。 [Step 6]
Synthesis of (R) -tert-butyl 3-((4-bromo-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carboxylate:
Reference temperature compound 5 (0.50 g, 1.9 mmol), (R) -1- (tert-butoxycarbonyl) piperidine-3-carboxylic acid (0.43 g, 1.9 mmol), DIPEA (0.53 g) at room temperature 4.1 mmol) in DMF (3.0 mL) was added HATU (0.77 g, 2.0 mmol). After stirring at room temperature for 15 hours, ethyl acetate was added to the reaction solution, and the organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, water, and a saturated aqueous sodium chloride solution. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 9: 1 → 1: 1), and (R) — As a result, 0.89 g (quantitative) of tert-butyl 3-((4-bromo-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carboxylate (hereinafter referred to as Reference Example Compound 6) was obtained.
〔ステップ7〕
(R)-tert-ブチル 3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボキシラートの合成:
室温下、参考例化合物6(0.050g、0.10mmol)、シアン化亜鉛(0.012g、0.10mmol)のDMF(2.0mL)溶液に、テトラキストリフェニルホスフィンパラジウム(0)(0.030g、0.026mmol)を加えた。150℃下で30分間撹拌した後、室温下で反応溶液に、水を加え、ジエチルエーテルで抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=20:1→1:2)で精製し、(R)-tert-ブチル 3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボキシラート(以下、参考例化合物7)を0.017g(39%)得た。 [Step 7]
Synthesis of (R) -tert-butyl 3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carboxylate:
At room temperature, a solution of Reference Compound 6 (0.050 g, 0.10 mmol) and zinc cyanide (0.012 g, 0.10 mmol) in DMF (2.0 mL) was added to tetrakistriphenylphosphine palladium (0) (0. 030 g, 0.026 mmol) was added. After stirring at 150 ° C. for 30 minutes, water was added to the reaction solution at room temperature, and the mixture was extracted with diethyl ether. The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 20: 1 → 1: 2), and (R) -tert-butyl 3-((4-cyano-2- ( 0.017 g (39%) of trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carboxylate (hereinafter referred to as Reference Example Compound 7) was obtained.
(R)-tert-ブチル 3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボキシラートの合成:
室温下、参考例化合物6(0.050g、0.10mmol)、シアン化亜鉛(0.012g、0.10mmol)のDMF(2.0mL)溶液に、テトラキストリフェニルホスフィンパラジウム(0)(0.030g、0.026mmol)を加えた。150℃下で30分間撹拌した後、室温下で反応溶液に、水を加え、ジエチルエーテルで抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=20:1→1:2)で精製し、(R)-tert-ブチル 3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボキシラート(以下、参考例化合物7)を0.017g(39%)得た。 [Step 7]
Synthesis of (R) -tert-butyl 3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carboxylate:
At room temperature, a solution of Reference Compound 6 (0.050 g, 0.10 mmol) and zinc cyanide (0.012 g, 0.10 mmol) in DMF (2.0 mL) was added to tetrakistriphenylphosphine palladium (0) (0. 030 g, 0.026 mmol) was added. After stirring at 150 ° C. for 30 minutes, water was added to the reaction solution at room temperature, and the mixture was extracted with diethyl ether. The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 20: 1 → 1: 2), and (R) -tert-butyl 3-((4-cyano-2- ( 0.017 g (39%) of trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carboxylate (hereinafter referred to as Reference Example Compound 7) was obtained.
〔ステップ8〕
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物7(6.9g、16mmol)のジクロロメタン(0.16L)溶液に、トリフルオロ酢酸(以下、TFA)(35mL、0.45mol)を加えた。室温下で1時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタンに溶解させ、飽和炭酸ナトリウム水溶液で中和し、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物8)を5.2g(98%)得た。 [Step 8]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
Under ice-cooling, trifluoroacetic acid (hereinafter TFA) (35 mL, 0.45 mol) was added to a solution of Reference Example Compound 7 (6.9 g, 16 mmol) in dichloromethane (0.16 L). After stirring at room temperature for 1 hour, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane, neutralized with a saturated aqueous sodium carbonate solution, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to give (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 8). 2 g (98%) were obtained.
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物7(6.9g、16mmol)のジクロロメタン(0.16L)溶液に、トリフルオロ酢酸(以下、TFA)(35mL、0.45mol)を加えた。室温下で1時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタンに溶解させ、飽和炭酸ナトリウム水溶液で中和し、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物8)を5.2g(98%)得た。 [Step 8]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
Under ice-cooling, trifluoroacetic acid (hereinafter TFA) (35 mL, 0.45 mol) was added to a solution of Reference Example Compound 7 (6.9 g, 16 mmol) in dichloromethane (0.16 L). After stirring at room temperature for 1 hour, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane, neutralized with a saturated aqueous sodium carbonate solution, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to give (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 8). 2 g (98%) were obtained.
〔ステップ9〕
(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)シクロブチル)カルバマートの合成:
氷冷下、参考例化合物8(0.20g、0.67mmol)、1-((tert-ブトキシカルボニル)アミノ)シクロブタンカルボン酸(0.15g、0.67mmol)、DIPEA(0.24mL、1.3mmol)のDMF(0.70mL)溶液に、HATU(0.28g、0.73mmol)を加えた。室温下で86時間撹拌した後、反応溶液に、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;ヘキサン:酢酸エチル=7:3→4:6)で精製し、(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)シクロブチル)カルバマート(以下、参考例化合物9)を0.25g(78%)得た。 [Step 9]
Synthesis of (R) -tert-butyl (1- (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carbonyl) cyclobutyl) carbamate:
Reference Example Compound 8 (0.20 g, 0.67 mmol), 1-((tert-butoxycarbonyl) amino) cyclobutanecarboxylic acid (0.15 g, 0.67 mmol), DIPEA (0.24 mL, 1. To a solution of 3 mmol) in DMF (0.70 mL) was added HATU (0.28 g, 0.73 mmol). After stirring at room temperature for 86 hours, 1N hydrochloric acid was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (amine silica gel DM1020, manufactured by Fuji Silysia Chemical Ltd., eluent; hexane: ethyl acetate = 7: 3 → 4: 6), and (R) -tert-butyl (1 0.25 g (78%) of — (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carbonyl) cyclobutyl) carbamate (hereinafter referred to as Reference Example Compound 9) was obtained.
(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)シクロブチル)カルバマートの合成:
氷冷下、参考例化合物8(0.20g、0.67mmol)、1-((tert-ブトキシカルボニル)アミノ)シクロブタンカルボン酸(0.15g、0.67mmol)、DIPEA(0.24mL、1.3mmol)のDMF(0.70mL)溶液に、HATU(0.28g、0.73mmol)を加えた。室温下で86時間撹拌した後、反応溶液に、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;ヘキサン:酢酸エチル=7:3→4:6)で精製し、(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)シクロブチル)カルバマート(以下、参考例化合物9)を0.25g(78%)得た。 [Step 9]
Synthesis of (R) -tert-butyl (1- (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carbonyl) cyclobutyl) carbamate:
Reference Example Compound 8 (0.20 g, 0.67 mmol), 1-((tert-butoxycarbonyl) amino) cyclobutanecarboxylic acid (0.15 g, 0.67 mmol), DIPEA (0.24 mL, 1. To a solution of 3 mmol) in DMF (0.70 mL) was added HATU (0.28 g, 0.73 mmol). After stirring at room temperature for 86 hours, 1N hydrochloric acid was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (amine silica gel DM1020, manufactured by Fuji Silysia Chemical Ltd., eluent; hexane: ethyl acetate = 7: 3 → 4: 6), and (R) -tert-butyl (1 0.25 g (78%) of — (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carbonyl) cyclobutyl) carbamate (hereinafter referred to as Reference Example Compound 9) was obtained.
〔ステップ10〕
(R)-1-(1-アミノシクロブタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物9(0.25g、0.47mmol)のジクロロメタン(1.4mL)溶液に、TFA(0.70mL、9.1mmol)を加えた。室温下で3時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタンに溶解させ、飽和炭酸ナトリウム水溶液で中和し、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-1-(1-アミノシクロブタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物10)を0.18g(90%)得た。 [Step 10]
Synthesis of (R) -1- (1-aminocyclobutanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
TFA (0.70 mL, 9.1 mmol) was added to a dichloromethane (1.4 mL) solution of Reference Example compound 9 (0.25 g, 0.47 mmol) under ice cooling. After stirring at room temperature for 3 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane, neutralized with a saturated aqueous sodium carbonate solution, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and (R) -1- (1-aminocyclobutanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide ( Hereinafter, 0.18 g (90%) of Reference Example Compound 10) was obtained.
(R)-1-(1-アミノシクロブタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物9(0.25g、0.47mmol)のジクロロメタン(1.4mL)溶液に、TFA(0.70mL、9.1mmol)を加えた。室温下で3時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタンに溶解させ、飽和炭酸ナトリウム水溶液で中和し、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-1-(1-アミノシクロブタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物10)を0.18g(90%)得た。 [Step 10]
Synthesis of (R) -1- (1-aminocyclobutanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
TFA (0.70 mL, 9.1 mmol) was added to a dichloromethane (1.4 mL) solution of Reference Example compound 9 (0.25 g, 0.47 mmol) under ice cooling. After stirring at room temperature for 3 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane, neutralized with a saturated aqueous sodium carbonate solution, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and (R) -1- (1-aminocyclobutanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide ( Hereinafter, 0.18 g (90%) of Reference Example Compound 10) was obtained.
〔ステップ11〕
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-プロピオンアミドシクロブタンカルボニル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物10(7.6g、18mmol)、TEA(5.5mL、40mmol)のジクロロメタン(54mL)溶液に、プロピオニルクロリド(1.8g、20mmol)を加えた。氷冷下で1時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-プロピオンアミドシクロブタンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物1)を6.2g(71%)得た。 [Step 11]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-propionamidocyclobutanecarbonyl) piperidine-3-carboxamide:
Under ice-cooling, propionyl chloride (1.8 g, 20 mmol) was added to a solution of Reference Example Compound 10 (7.6 g, 18 mmol) and TEA (5.5 mL, 40 mmol) in dichloromethane (54 mL). After stirring for 1 hour under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 95: 5), and (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl was obtained. ) -1- (1-propionamidocyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 1) was obtained in an amount of 6.2 g (71%).
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-プロピオンアミドシクロブタンカルボニル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物10(7.6g、18mmol)、TEA(5.5mL、40mmol)のジクロロメタン(54mL)溶液に、プロピオニルクロリド(1.8g、20mmol)を加えた。氷冷下で1時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-プロピオンアミドシクロブタンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物1)を6.2g(71%)得た。 [Step 11]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-propionamidocyclobutanecarbonyl) piperidine-3-carboxamide:
Under ice-cooling, propionyl chloride (1.8 g, 20 mmol) was added to a solution of Reference Example Compound 10 (7.6 g, 18 mmol) and TEA (5.5 mL, 40 mmol) in dichloromethane (54 mL). After stirring for 1 hour under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 95: 5), and (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl was obtained. ) -1- (1-propionamidocyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 1) was obtained in an amount of 6.2 g (71%).
(実施例2) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-2-メチル-1-オキソプロパン-2-イル)カルバマートの合成:
氷冷下、参考例化合物8(3.5g、11mmol)、2-((tert-ブトキシカルボニル)アミノ)-2-メチルプロパン酸(2.6g、13mmol)、DIPEA(4.1mL、24mmol)のDMF(40mL)溶液に、HATU(4.9g、13mmol)を加えた。室温下で1時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;ヘキサン:酢酸エチル=9:1→4:6)で精製し、(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-2-メチル-1-オキソプロパン-2-イル)カルバマート(以下、参考例化合物11)を5.2g(95%)得た。 Example 2 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide:
[Step 1]
(R) -tert-butyl (1- (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-yl) -2-methyl-1-oxopropan-2-yl) Synthesis of carbamate:
Under ice-cooling, Reference Example Compound 8 (3.5 g, 11 mmol), 2-((tert-butoxycarbonyl) amino) -2-methylpropanoic acid (2.6 g, 13 mmol), DIPEA (4.1 mL, 24 mmol) To a DMF (40 mL) solution, HATU (4.9 g, 13 mmol) was added. After stirring at room temperature for 1 hour, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (amine silica gel DM1020, manufactured by Fuji Silysia Chemical Ltd., eluent; hexane: ethyl acetate = 9: 1 → 4: 6), and (R) -tert-butyl (1 -(3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-yl) -2-methyl-1-oxopropan-2-yl) carbamate (hereinafter referred to as Reference Example Compound 11) Of 5.2 g (95%).
〔ステップ1〕
(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-2-メチル-1-オキソプロパン-2-イル)カルバマートの合成:
氷冷下、参考例化合物8(3.5g、11mmol)、2-((tert-ブトキシカルボニル)アミノ)-2-メチルプロパン酸(2.6g、13mmol)、DIPEA(4.1mL、24mmol)のDMF(40mL)溶液に、HATU(4.9g、13mmol)を加えた。室温下で1時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;ヘキサン:酢酸エチル=9:1→4:6)で精製し、(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-2-メチル-1-オキソプロパン-2-イル)カルバマート(以下、参考例化合物11)を5.2g(95%)得た。 Example 2 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide:
[Step 1]
(R) -tert-butyl (1- (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-yl) -2-methyl-1-oxopropan-2-yl) Synthesis of carbamate:
Under ice-cooling, Reference Example Compound 8 (3.5 g, 11 mmol), 2-((tert-butoxycarbonyl) amino) -2-methylpropanoic acid (2.6 g, 13 mmol), DIPEA (4.1 mL, 24 mmol) To a DMF (40 mL) solution, HATU (4.9 g, 13 mmol) was added. After stirring at room temperature for 1 hour, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (amine silica gel DM1020, manufactured by Fuji Silysia Chemical Ltd., eluent; hexane: ethyl acetate = 9: 1 → 4: 6), and (R) -tert-butyl (1 -(3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-yl) -2-methyl-1-oxopropan-2-yl) carbamate (hereinafter referred to as Reference Example Compound 11) Of 5.2 g (95%).
〔ステップ2〕
(R)-1-(2-アミノ-2-メチルプロパノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物11(5.2g、10mmol)のジクロロメタン(0.10L)溶液に、TFA(25mL、0.32mol)を加えた。室温下で1.5時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタンに溶解させ、飽和炭酸ナトリウム水溶液で中和し、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-1-(2-アミノ-2-メチルプロパノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物12)を3.4g(82%)得た。 [Step 2]
Synthesis of (R) -1- (2-amino-2-methylpropanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
TFA (25 mL, 0.32 mol) was added to a solution of Reference Example Compound 11 (5.2 g, 10 mmol) in dichloromethane (0.10 L) under ice cooling. After stirring at room temperature for 1.5 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane, neutralized with a saturated aqueous sodium carbonate solution, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and (R) -1- (2-amino-2-methylpropanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine- 3.4 g (82%) of 3-carboxamide (hereinafter referred to as Reference Example Compound 12) was obtained.
(R)-1-(2-アミノ-2-メチルプロパノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物11(5.2g、10mmol)のジクロロメタン(0.10L)溶液に、TFA(25mL、0.32mol)を加えた。室温下で1.5時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタンに溶解させ、飽和炭酸ナトリウム水溶液で中和し、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-1-(2-アミノ-2-メチルプロパノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物12)を3.4g(82%)得た。 [Step 2]
Synthesis of (R) -1- (2-amino-2-methylpropanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
TFA (25 mL, 0.32 mol) was added to a solution of Reference Example Compound 11 (5.2 g, 10 mmol) in dichloromethane (0.10 L) under ice cooling. After stirring at room temperature for 1.5 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane, neutralized with a saturated aqueous sodium carbonate solution, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and (R) -1- (2-amino-2-methylpropanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine- 3.4 g (82%) of 3-carboxamide (hereinafter referred to as Reference Example Compound 12) was obtained.
〔ステップ3〕
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物12(2.3g、5.6mmol)、TEA(1.6mL、11mmol)のジクロロメタン(15mL)溶液に、メタンスルホニルクロリド(0.97g、8.5mmol)を加えた。氷冷下で5分間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物2)を2.4g(87%)得た。 [Step 3]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide:
Methanesulfonyl chloride (0.97 g, 8.5 mmol) was added to a solution of Reference Example Compound 12 (2.3 g, 5.6 mmol) and TEA (1.6 mL, 11 mmol) in dichloromethane (15 mL) under ice cooling. After stirring for 5 minutes under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 95: 5), and (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl was obtained. ) -1- (2-Methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 2) was obtained in an amount of 2.4 g (87%).
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物12(2.3g、5.6mmol)、TEA(1.6mL、11mmol)のジクロロメタン(15mL)溶液に、メタンスルホニルクロリド(0.97g、8.5mmol)を加えた。氷冷下で5分間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物2)を2.4g(87%)得た。 [Step 3]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide:
Methanesulfonyl chloride (0.97 g, 8.5 mmol) was added to a solution of Reference Example Compound 12 (2.3 g, 5.6 mmol) and TEA (1.6 mL, 11 mmol) in dichloromethane (15 mL) under ice cooling. After stirring for 5 minutes under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 95: 5), and (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl was obtained. ) -1- (2-Methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 2) was obtained in an amount of 2.4 g (87%).
(実施例3) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(メチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)シクロプロピル)カルバマートの合成:
氷冷下、参考例化合物8(0.67g、2.0mmol)、1-((tert-ブトキシカルボニル)アミノ)シクロプロパンカルボン酸(0.49g、2.4mmol)、DIPEA(1.1mL、6.1mmol)のDMF(5.0mL)溶液に、HATU(1.1g、2.5mmol)を加えた。室温下で1時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;ヘキサン:酢酸エチル=9:1→4:6)で精製し、(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)シクロプロピル)カルバマート(以下、参考例化合物13)を0.92g(88%)得た。 Example 3 Synthesis of (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (methylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of (R) -tert-butyl (1- (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carbonyl) cyclopropyl) carbamate:
Under ice-cooling, Reference Compound 8 (0.67 g, 2.0 mmol), 1-((tert-butoxycarbonyl) amino) cyclopropanecarboxylic acid (0.49 g, 2.4 mmol), DIPEA (1.1 mL, 6 mmol) .1 mmol) in DMF (5.0 mL) was added HATU (1.1 g, 2.5 mmol). After stirring at room temperature for 1 hour, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (amine silica gel DM1020, manufactured by Fuji Silysia Chemical Ltd., eluent; hexane: ethyl acetate = 9: 1 → 4: 6), and (R) -tert-butyl (1 0.92 g (88%) of — (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carbonyl) cyclopropyl) carbamate (hereinafter referred to as Reference Example Compound 13) was obtained.
〔ステップ1〕
(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)シクロプロピル)カルバマートの合成:
氷冷下、参考例化合物8(0.67g、2.0mmol)、1-((tert-ブトキシカルボニル)アミノ)シクロプロパンカルボン酸(0.49g、2.4mmol)、DIPEA(1.1mL、6.1mmol)のDMF(5.0mL)溶液に、HATU(1.1g、2.5mmol)を加えた。室温下で1時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;ヘキサン:酢酸エチル=9:1→4:6)で精製し、(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)シクロプロピル)カルバマート(以下、参考例化合物13)を0.92g(88%)得た。 Example 3 Synthesis of (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (methylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of (R) -tert-butyl (1- (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carbonyl) cyclopropyl) carbamate:
Under ice-cooling, Reference Compound 8 (0.67 g, 2.0 mmol), 1-((tert-butoxycarbonyl) amino) cyclopropanecarboxylic acid (0.49 g, 2.4 mmol), DIPEA (1.1 mL, 6 mmol) .1 mmol) in DMF (5.0 mL) was added HATU (1.1 g, 2.5 mmol). After stirring at room temperature for 1 hour, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (amine silica gel DM1020, manufactured by Fuji Silysia Chemical Ltd., eluent; hexane: ethyl acetate = 9: 1 → 4: 6), and (R) -tert-butyl (1 0.92 g (88%) of — (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carbonyl) cyclopropyl) carbamate (hereinafter referred to as Reference Example Compound 13) was obtained.
〔ステップ2〕
(R)-1-(1-アミノシクロプロパンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物13(0.92g、1.8mmol)のジクロロメタン(20mL)溶液に、TFA(4.7mL、61mmol)を加えた。室温下で1.5時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタンに溶解させ、飽和炭酸水素ナトリウム水溶液で中和し、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-1-(1-アミノシクロプロパンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物14)を0.63g(87%)得た。 [Step 2]
Synthesis of (R) -1- (1-aminocyclopropanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
TFA (4.7 mL, 61 mmol) was added to a dichloromethane (20 mL) solution of Reference Example Compound 13 (0.92 g, 1.8 mmol) under ice cooling. After stirring at room temperature for 1.5 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane, neutralized with a saturated aqueous sodium hydrogen carbonate solution, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate, concentrated under reduced pressure, and (R) -1- (1-aminocyclopropanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide. As a result, 0.63 g (87%) of Reference Example Compound 14 was obtained.
(R)-1-(1-アミノシクロプロパンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物13(0.92g、1.8mmol)のジクロロメタン(20mL)溶液に、TFA(4.7mL、61mmol)を加えた。室温下で1.5時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタンに溶解させ、飽和炭酸水素ナトリウム水溶液で中和し、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-1-(1-アミノシクロプロパンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物14)を0.63g(87%)得た。 [Step 2]
Synthesis of (R) -1- (1-aminocyclopropanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
TFA (4.7 mL, 61 mmol) was added to a dichloromethane (20 mL) solution of Reference Example Compound 13 (0.92 g, 1.8 mmol) under ice cooling. After stirring at room temperature for 1.5 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane, neutralized with a saturated aqueous sodium hydrogen carbonate solution, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate, concentrated under reduced pressure, and (R) -1- (1-aminocyclopropanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide. As a result, 0.63 g (87%) of Reference Example Compound 14 was obtained.
〔ステップ3〕
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(メチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物14(0.63g、1.5mmol)、TEA(1.1mL、7.7mmol)のジクロロメタン(5.0mL)溶液に、メタンスルホニルクロリド(0.27g、2.3mmol)を加えた。氷冷下で1.5時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(メチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物3)を0.56g(74%)得た。 [Step 3]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (methylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide:
Under ice cooling, methanesulfonyl chloride (0.27 g, 2.3 mmol) was added to a solution of Reference Compound 14 (0.63 g, 1.5 mmol) and TEA (1.1 mL, 7.7 mmol) in dichloromethane (5.0 mL). Was added. After stirring for 1.5 hours under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 95: 5), and (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl was obtained. ) -1- (1- (methylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 3) was obtained in an amount of 0.56 g (74%).
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(メチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物14(0.63g、1.5mmol)、TEA(1.1mL、7.7mmol)のジクロロメタン(5.0mL)溶液に、メタンスルホニルクロリド(0.27g、2.3mmol)を加えた。氷冷下で1.5時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(メチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物3)を0.56g(74%)得た。 [Step 3]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (methylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide:
Under ice cooling, methanesulfonyl chloride (0.27 g, 2.3 mmol) was added to a solution of Reference Compound 14 (0.63 g, 1.5 mmol) and TEA (1.1 mL, 7.7 mmol) in dichloromethane (5.0 mL). Was added. After stirring for 1.5 hours under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 95: 5), and (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl was obtained. ) -1- (1- (methylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 3) was obtained in an amount of 0.56 g (74%).
(実施例4) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(トリフルオロメチル)シクロプロパンカルボニル)ピペリジン-3-カルボキサミドの合成:
1-(トリフルオロメチル)シクロプロパンカルボン酸(0.054g、0.17mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(トリフルオロメチル)シクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物4)を0.044g(58%)得た。 Example 4 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (trifluoromethyl) cyclopropanecarbonyl) piperidine-3-carboxamide:
The same reaction as in Example 1 [Step 9] was carried out using 1- (trifluoromethyl) cyclopropanecarboxylic acid (0.054 g, 0.17 mmol) to give (R) -N- (4-cyano- 0.044 g (58%) of 2- (trifluoromethoxy) benzyl) -1- (1- (trifluoromethyl) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 4) was obtained.
1-(トリフルオロメチル)シクロプロパンカルボン酸(0.054g、0.17mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(トリフルオロメチル)シクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物4)を0.044g(58%)得た。 Example 4 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (trifluoromethyl) cyclopropanecarbonyl) piperidine-3-carboxamide:
The same reaction as in Example 1 [Step 9] was carried out using 1- (trifluoromethyl) cyclopropanecarboxylic acid (0.054 g, 0.17 mmol) to give (R) -N- (4-cyano- 0.044 g (58%) of 2- (trifluoromethoxy) benzyl) -1- (1- (trifluoromethyl) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 4) was obtained.
(実施例5) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(メチルスルホンアミド)シクロブタンカルボニル)ピペリジン-3-カルボキサミドの合成:
参考例化合物10(0.020g、0.047mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(メチルスルホンアミド)シクロブタンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物5)を0.017g(71%)得た。 Example 5 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (methylsulfonamido) cyclobutanecarbonyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 2 [Step 3] using Reference compound 10 (0.020 g, 0.047 mmol). 0.017 g (71%) of benzyl) -1- (1- (methylsulfonamido) cyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 5) was obtained.
参考例化合物10(0.020g、0.047mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(メチルスルホンアミド)シクロブタンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物5)を0.017g(71%)得た。 Example 5 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (methylsulfonamido) cyclobutanecarbonyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 2 [Step 3] using Reference compound 10 (0.020 g, 0.047 mmol). 0.017 g (71%) of benzyl) -1- (1- (methylsulfonamido) cyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 5) was obtained.
(実施例6) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-イソブチルアミドシクロブタンカルボニル)ピペリジン-3-カルボキサミドの合成:
イソブチリルクロリド(0.0055g、0.052mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-イソブチルアミドシクロブタンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物6)を0.022g(95%)得た。 Example 6 Synthesis of (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-isobutyramidecyclobutanecarbonyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by carrying out the same reaction as in Example 1 [Step 11] using isobutyryl chloride (0.0055 g, 0.052 mmol). 0.022 g (95%) of (benzyl) -1- (1-isobutyramidecyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 6) was obtained.
イソブチリルクロリド(0.0055g、0.052mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-イソブチルアミドシクロブタンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物6)を0.022g(95%)得た。 Example 6 Synthesis of (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-isobutyramidecyclobutanecarbonyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by carrying out the same reaction as in Example 1 [Step 11] using isobutyryl chloride (0.0055 g, 0.052 mmol). 0.022 g (95%) of (benzyl) -1- (1-isobutyramidecyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 6) was obtained.
(実施例7) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-ピバルアミドシクロブタンカルボニル)ピペリジン-3-カルボキサミドの合成:
ピバロイルクロリド(0.0063g、0.052mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-ピバルアミドシクロブタンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物7)を0.017g(72%)得た。 Example 7 Synthesis of (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-pivalamidocyclobutanecarbonyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by carrying out the same reaction as in Example 1 [Step 11] using pivaloyl chloride (0.0063 g, 0.052 mmol). 0.017 g (72%) of (benzyl) -1- (1-pivalamidocyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 7) was obtained.
ピバロイルクロリド(0.0063g、0.052mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-ピバルアミドシクロブタンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物7)を0.017g(72%)得た。 Example 7 Synthesis of (R) —N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-pivalamidocyclobutanecarbonyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by carrying out the same reaction as in Example 1 [Step 11] using pivaloyl chloride (0.0063 g, 0.052 mmol). 0.017 g (72%) of (benzyl) -1- (1-pivalamidocyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 7) was obtained.
(実施例8) (R)-1-(1-アセトアミドシクロペンタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)シクロペンチル)カルバマートの合成:
1-((tert-ブトキシカルボニル)アミノ)シクロペンタンカルボン酸(0.078g、0.34mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)シクロペンチル)カルバマート(以下、参考例化合物15)を0.13g(77%)得た。 Example 8 Synthesis of (R) -1- (1-acetamidocyclopentanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of (R) -tert-butyl (1- (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carbonyl) cyclopentyl) carbamate:
(R) -tert-butyl was prepared by carrying out the same reaction as in Example 1 [Step 9] using 1-((tert-butoxycarbonyl) amino) cyclopentanecarboxylic acid (0.078 g, 0.34 mmol). 0.13 g (77%) of (1- (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carbonyl) cyclopentyl) carbamate (hereinafter referred to as Reference Example Compound 15) was obtained. .
〔ステップ1〕
(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)シクロペンチル)カルバマートの合成:
1-((tert-ブトキシカルボニル)アミノ)シクロペンタンカルボン酸(0.078g、0.34mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-tert-ブチル (1-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)シクロペンチル)カルバマート(以下、参考例化合物15)を0.13g(77%)得た。 Example 8 Synthesis of (R) -1- (1-acetamidocyclopentanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of (R) -tert-butyl (1- (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carbonyl) cyclopentyl) carbamate:
(R) -tert-butyl was prepared by carrying out the same reaction as in Example 1 [Step 9] using 1-((tert-butoxycarbonyl) amino) cyclopentanecarboxylic acid (0.078 g, 0.34 mmol). 0.13 g (77%) of (1- (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidine-1-carbonyl) cyclopentyl) carbamate (hereinafter referred to as Reference Example Compound 15) was obtained. .
〔ステップ2〕
(R)-1-(1-アミノシクロペンタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
参考例化合物15(0.13g、0.24mmol)を用いて実施例1〔ステップ10〕と同様の反応を行うことにより、(R)-1-(1-アミノシクロペンタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物16)を0.051g(49%)得た。 [Step 2]
Synthesis of (R) -1- (1-aminocyclopentanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1- (1-aminocyclopentanecarbonyl) -N— (R) -1- (1-aminocyclopentanecarbonyl) -N— (Reference Example Compound 15 (0.13 g, 0.24 mmol) was used for the same reaction as in Example 1 [Step 10]. 0.051 g (49%) of 4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 16) was obtained.
(R)-1-(1-アミノシクロペンタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
参考例化合物15(0.13g、0.24mmol)を用いて実施例1〔ステップ10〕と同様の反応を行うことにより、(R)-1-(1-アミノシクロペンタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物16)を0.051g(49%)得た。 [Step 2]
Synthesis of (R) -1- (1-aminocyclopentanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1- (1-aminocyclopentanecarbonyl) -N— (R) -1- (1-aminocyclopentanecarbonyl) -N— (Reference Example Compound 15 (0.13 g, 0.24 mmol) was used for the same reaction as in Example 1 [Step 10]. 0.051 g (49%) of 4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 16) was obtained.
〔ステップ3〕
(R)-1-(1-アセトアミドシクロペンタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物16(0.020g、0.046mmol)、TEA(0.019mL、0.14mmol)のジクロロメタン(0.20mL)溶液に、無水酢酸(0.0070g、0.068mmol)を加えた。氷冷下で1時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、(R)-1-(1-アセトアミドシクロペンタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物8)を0.014g(62%)得た。 [Step 3]
Synthesis of (R) -1- (1-acetamidocyclopentanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
Under ice-cooling, acetic anhydride (0.0070 g, 0.068 mmol) was added to a solution of Reference Example Compound 16 (0.020 g, 0.046 mmol) and TEA (0.019 mL, 0.14 mmol) in dichloromethane (0.20 mL). added. After stirring for 1 hour under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 95: 5), and (R) -1- (1-acetamidocyclopentanecarbonyl) -N- (4 0.014 g (62%) of -cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 8) was obtained.
(R)-1-(1-アセトアミドシクロペンタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物16(0.020g、0.046mmol)、TEA(0.019mL、0.14mmol)のジクロロメタン(0.20mL)溶液に、無水酢酸(0.0070g、0.068mmol)を加えた。氷冷下で1時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、(R)-1-(1-アセトアミドシクロペンタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物8)を0.014g(62%)得た。 [Step 3]
Synthesis of (R) -1- (1-acetamidocyclopentanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
Under ice-cooling, acetic anhydride (0.0070 g, 0.068 mmol) was added to a solution of Reference Example Compound 16 (0.020 g, 0.046 mmol) and TEA (0.019 mL, 0.14 mmol) in dichloromethane (0.20 mL). added. After stirring for 1 hour under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 95: 5), and (R) -1- (1-acetamidocyclopentanecarbonyl) -N- (4 0.014 g (62%) of -cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 8) was obtained.
(実施例9) (R)-1-(1-アセトアミドシクロブタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
参考例化合物10(0.020g、0.047mmol)を用いて実施例8〔ステップ3〕と同様の反応を行うことにより、(R)-1-(1-アセトアミドシクロブタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物9)を0.013g(60%)得た。 Example 9 Synthesis of (R) -1- (1-acetamidocyclobutanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1- (1-acetamidocyclobutanecarbonyl) -N- (4) was prepared by performing the same reaction as in Example 8 [Step 3] using Reference compound 10 (0.020 g, 0.047 mmol). 0.013 g (60%) of -cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 9) was obtained.
参考例化合物10(0.020g、0.047mmol)を用いて実施例8〔ステップ3〕と同様の反応を行うことにより、(R)-1-(1-アセトアミドシクロブタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物9)を0.013g(60%)得た。 Example 9 Synthesis of (R) -1- (1-acetamidocyclobutanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1- (1-acetamidocyclobutanecarbonyl) -N- (4) was prepared by performing the same reaction as in Example 8 [Step 3] using Reference compound 10 (0.020 g, 0.047 mmol). 0.013 g (60%) of -cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 9) was obtained.
(実施例10) (R)-N-(4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンゾニトリルの合成:
氷冷下、2,2,2-トリフルオロエタノール(1.5g、15mmol)のTHF(50mL)溶液に、水素化ナトリウム(55重量%、0.67g、15mmol)を加えた。氷冷下で10分間撹拌した後、室温下で30分間撹拌した。氷冷下、反応溶液に、4-クロロ-2-フルオロベンゾニトリル(2.0g、13mmol)を加えた。室温下で1時間撹拌した後、氷冷下、反応溶液に、0.1規定塩酸を加え、酢酸エチルで抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=10:0→3:1)で精製し、4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンゾニトリル(以下、参考例化合物17)を2.6g(86%)得た。 Example 10 (R) -N- (4-Chloro-2- (2,2,2-trifluoroethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) propanoyl) piperidine- Synthesis of 3-carboxamide:
[Step 1]
Synthesis of 4-chloro-2- (2,2,2-trifluoroethoxy) benzonitrile:
Sodium hydride (55 wt%, 0.67 g, 15 mmol) was added to a solution of 2,2,2-trifluoroethanol (1.5 g, 15 mmol) in THF (50 mL) under ice cooling. The mixture was stirred for 10 minutes under ice cooling, and then stirred for 30 minutes at room temperature. 4-Chloro-2-fluorobenzonitrile (2.0 g, 13 mmol) was added to the reaction solution under ice cooling. After stirring at room temperature for 1 hour, 0.1N hydrochloric acid was added to the reaction solution under ice cooling, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 10: 0 → 3: 1), and 4-chloro-2- (2,2,2-trifluoroethoxy) benzo 2.6 g (86%) of nitrile (hereinafter referred to as Reference Example Compound 17) was obtained.
〔ステップ1〕
4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンゾニトリルの合成:
氷冷下、2,2,2-トリフルオロエタノール(1.5g、15mmol)のTHF(50mL)溶液に、水素化ナトリウム(55重量%、0.67g、15mmol)を加えた。氷冷下で10分間撹拌した後、室温下で30分間撹拌した。氷冷下、反応溶液に、4-クロロ-2-フルオロベンゾニトリル(2.0g、13mmol)を加えた。室温下で1時間撹拌した後、氷冷下、反応溶液に、0.1規定塩酸を加え、酢酸エチルで抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=10:0→3:1)で精製し、4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンゾニトリル(以下、参考例化合物17)を2.6g(86%)得た。 Example 10 (R) -N- (4-Chloro-2- (2,2,2-trifluoroethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) propanoyl) piperidine- Synthesis of 3-carboxamide:
[Step 1]
Synthesis of 4-chloro-2- (2,2,2-trifluoroethoxy) benzonitrile:
Sodium hydride (55 wt%, 0.67 g, 15 mmol) was added to a solution of 2,2,2-trifluoroethanol (1.5 g, 15 mmol) in THF (50 mL) under ice cooling. The mixture was stirred for 10 minutes under ice cooling, and then stirred for 30 minutes at room temperature. 4-Chloro-2-fluorobenzonitrile (2.0 g, 13 mmol) was added to the reaction solution under ice cooling. After stirring at room temperature for 1 hour, 0.1N hydrochloric acid was added to the reaction solution under ice cooling, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 10: 0 → 3: 1), and 4-chloro-2- (2,2,2-trifluoroethoxy) benzo 2.6 g (86%) of nitrile (hereinafter referred to as Reference Example Compound 17) was obtained.
〔ステップ2〕
(4-クロロ-2-(2,2,2-トリフルオロエトキシ)フェニル)メタンアミンの合成:
氷冷下、参考例化合物17(2.5g、11mmol)のジエチルエーテル(30mL)溶液に、水素化アルミニウムリチウム(1.0g、27mmol)を加えた。室温下で4時間撹拌した後、氷冷下、反応溶液に、THF(20mL)、水(1.0mL)、1規定水酸化ナトリウム水溶液(1.0mL)、水(3.0mL)を加えた。反応溶液をろ過後、ろ液を減圧濃縮し、(4-クロロ-2-(2,2,2-トリフルオロエトキシ)フェニル)メタンアミン(以下、参考例化合物18)を2.4g(94%)得た。 [Step 2]
Synthesis of (4-chloro-2- (2,2,2-trifluoroethoxy) phenyl) methanamine:
Under ice cooling, lithium aluminum hydride (1.0 g, 27 mmol) was added to a solution of Reference Example Compound 17 (2.5 g, 11 mmol) in diethyl ether (30 mL). After stirring at room temperature for 4 hours, THF (20 mL), water (1.0 mL), 1N aqueous sodium hydroxide solution (1.0 mL), and water (3.0 mL) were added to the reaction solution under ice cooling. . After the reaction solution was filtered, the filtrate was concentrated under reduced pressure to obtain 2.4 g (94%) of (4-chloro-2- (2,2,2-trifluoroethoxy) phenyl) methanamine (hereinafter referred to as Reference Example Compound 18). Obtained.
(4-クロロ-2-(2,2,2-トリフルオロエトキシ)フェニル)メタンアミンの合成:
氷冷下、参考例化合物17(2.5g、11mmol)のジエチルエーテル(30mL)溶液に、水素化アルミニウムリチウム(1.0g、27mmol)を加えた。室温下で4時間撹拌した後、氷冷下、反応溶液に、THF(20mL)、水(1.0mL)、1規定水酸化ナトリウム水溶液(1.0mL)、水(3.0mL)を加えた。反応溶液をろ過後、ろ液を減圧濃縮し、(4-クロロ-2-(2,2,2-トリフルオロエトキシ)フェニル)メタンアミン(以下、参考例化合物18)を2.4g(94%)得た。 [Step 2]
Synthesis of (4-chloro-2- (2,2,2-trifluoroethoxy) phenyl) methanamine:
Under ice cooling, lithium aluminum hydride (1.0 g, 27 mmol) was added to a solution of Reference Example Compound 17 (2.5 g, 11 mmol) in diethyl ether (30 mL). After stirring at room temperature for 4 hours, THF (20 mL), water (1.0 mL), 1N aqueous sodium hydroxide solution (1.0 mL), and water (3.0 mL) were added to the reaction solution under ice cooling. . After the reaction solution was filtered, the filtrate was concentrated under reduced pressure to obtain 2.4 g (94%) of (4-chloro-2- (2,2,2-trifluoroethoxy) phenyl) methanamine (hereinafter referred to as Reference Example Compound 18). Obtained.
〔ステップ3〕
(R)-tert-ブチル 3-((4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボキシラートの合成:
参考例化合物18(2.4g、10mmol)を用いて実施例1〔ステップ6〕と同様の反応を行うことにより、(R)-tert-ブチル 3-((4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボキシラート(以下、参考例化合物19)を4.5g(定量的)得た。 [Step 3]
Synthesis of (R) -tert-butyl 3-((4-chloro-2- (2,2,2-trifluoroethoxy) benzyl) carbamoyl) piperidine-1-carboxylate:
(R) -tert-butyl 3-((4-chloro-2- (2,4 g, 10 mmol) was used in the same manner as in Example 1 [Step 6] using Compound 18 (2.4 g, 10 mmol). 4.5 g (quantitative) of 2,2-trifluoroethoxy) benzyl) carbamoyl) piperidine-1-carboxylate (hereinafter referred to as Reference Example Compound 19) was obtained.
(R)-tert-ブチル 3-((4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボキシラートの合成:
参考例化合物18(2.4g、10mmol)を用いて実施例1〔ステップ6〕と同様の反応を行うことにより、(R)-tert-ブチル 3-((4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボキシラート(以下、参考例化合物19)を4.5g(定量的)得た。 [Step 3]
Synthesis of (R) -tert-butyl 3-((4-chloro-2- (2,2,2-trifluoroethoxy) benzyl) carbamoyl) piperidine-1-carboxylate:
(R) -tert-butyl 3-((4-chloro-2- (2,4 g, 10 mmol) was used in the same manner as in Example 1 [Step 6] using Compound 18 (2.4 g, 10 mmol). 4.5 g (quantitative) of 2,2-trifluoroethoxy) benzyl) carbamoyl) piperidine-1-carboxylate (hereinafter referred to as Reference Example Compound 19) was obtained.
〔ステップ4〕
(R)-N-(4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物19(2.0g、4.4mmol)に、濃塩酸(10mL、0.12mol)を加えた。室温下で3時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタン(10mL)に溶解させ、飽和炭酸水素ナトリウム水溶液(10mL)を加えた。室温下で30分間撹拌した後、反応溶液に、水を加え、ジクロロメタンで抽出した。有機層を水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-N-(4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物20)を1.4g(87%)得た。 [Step 4]
Synthesis of (R) -N- (4-chloro-2- (2,2,2-trifluoroethoxy) benzyl) piperidine-3-carboxamide:
Concentrated hydrochloric acid (10 mL, 0.12 mol) was added to Reference Example Compound 19 (2.0 g, 4.4 mmol) under ice cooling. After stirring at room temperature for 3 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane (10 mL), and saturated aqueous sodium hydrogen carbonate solution (10 mL) was added. After stirring at room temperature for 30 minutes, water was added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with water, dried over anhydrous sodium sulfate, concentrated under reduced pressure, and (R) -N- (4-chloro-2- (2,2,2-trifluoroethoxy) benzyl) piperidine-3-carboxamide. 1.4 g (87%) ofReference Example Compound 20 was obtained.
(R)-N-(4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物19(2.0g、4.4mmol)に、濃塩酸(10mL、0.12mol)を加えた。室温下で3時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタン(10mL)に溶解させ、飽和炭酸水素ナトリウム水溶液(10mL)を加えた。室温下で30分間撹拌した後、反応溶液に、水を加え、ジクロロメタンで抽出した。有機層を水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-N-(4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物20)を1.4g(87%)得た。 [Step 4]
Synthesis of (R) -N- (4-chloro-2- (2,2,2-trifluoroethoxy) benzyl) piperidine-3-carboxamide:
Concentrated hydrochloric acid (10 mL, 0.12 mol) was added to Reference Example Compound 19 (2.0 g, 4.4 mmol) under ice cooling. After stirring at room temperature for 3 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane (10 mL), and saturated aqueous sodium hydrogen carbonate solution (10 mL) was added. After stirring at room temperature for 30 minutes, water was added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with water, dried over anhydrous sodium sulfate, concentrated under reduced pressure, and (R) -N- (4-chloro-2- (2,2,2-trifluoroethoxy) benzyl) piperidine-3-carboxamide. 1.4 g (87%) of
〔ステップ5〕
(R)-tert-ブチル (1-3-((4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-2-メチル-1-オキソプロパン-2-イル)カルバマートの合成:
参考例化合物20(0.60g、1.7mmol)を用いて実施例2〔ステップ1〕と同様の反応を行うことにより、(R)-tert-ブチル (1-3-((4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-2-メチル-1-オキソプロパン-2-イル)カルバマート(以下、参考例化合物21)を0.77g(84%)得た。 [Step 5]
(R) -tert-butyl (1-3-((4-chloro-2- (2,2,2-trifluoroethoxy) benzyl) carbamoyl) piperidin-1-yl) -2-methyl-1-oxopropane Synthesis of -2-yl) carbamate:
(R) -tert-butyl (1-3-((4-chloro-) was obtained by conducting the same reaction as in Example 2 [Step 1] using Reference Example Compound 20 (0.60 g, 1.7 mmol). 0.77 g of 2- (2,2,2-trifluoroethoxy) benzyl) carbamoyl) piperidin-1-yl) -2-methyl-1-oxopropan-2-yl) carbamate (hereinafter referred to as Reference Example Compound 21) (84%) obtained.
(R)-tert-ブチル (1-3-((4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-2-メチル-1-オキソプロパン-2-イル)カルバマートの合成:
参考例化合物20(0.60g、1.7mmol)を用いて実施例2〔ステップ1〕と同様の反応を行うことにより、(R)-tert-ブチル (1-3-((4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-2-メチル-1-オキソプロパン-2-イル)カルバマート(以下、参考例化合物21)を0.77g(84%)得た。 [Step 5]
(R) -tert-butyl (1-3-((4-chloro-2- (2,2,2-trifluoroethoxy) benzyl) carbamoyl) piperidin-1-yl) -2-methyl-1-oxopropane Synthesis of -2-yl) carbamate:
(R) -tert-butyl (1-3-((4-chloro-) was obtained by conducting the same reaction as in Example 2 [Step 1] using Reference Example Compound 20 (0.60 g, 1.7 mmol). 0.77 g of 2- (2,2,2-trifluoroethoxy) benzyl) carbamoyl) piperidin-1-yl) -2-methyl-1-oxopropan-2-yl) carbamate (hereinafter referred to as Reference Example Compound 21) (84%) obtained.
〔ステップ6〕
(R)-1-(2-アミノ-2-メチルプロパノイル)-N-(4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物21(0.83g、1.5mmol)に、TFA(10mL)を加えた。室温下で3時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタン(10mL)に溶解させ、飽和炭酸水素ナトリウム水溶液(10mL)を加えた。室温下で30分間撹拌した後、反応溶液に、水を加え、ジクロロメタンで抽出した。有機層を水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-1-(2-アミノ-2-メチルプロパノイル)-N-(4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物22)を0.65g(96%)得た。 [Step 6]
Synthesis of (R) -1- (2-amino-2-methylpropanoyl) -N- (4-chloro-2- (2,2,2-trifluoroethoxy) benzyl) piperidine-3-carboxamide:
TFA (10 mL) was added to Reference Example Compound 21 (0.83 g, 1.5 mmol) under ice cooling. After stirring at room temperature for 3 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane (10 mL), and saturated aqueous sodium hydrogen carbonate solution (10 mL) was added. After stirring at room temperature for 30 minutes, water was added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with water, dried over anhydrous sodium sulfate, concentrated under reduced pressure, and (R) -1- (2-amino-2-methylpropanoyl) -N- (4-chloro-2- (2,2 , 2-trifluoroethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Reference Example Compound 22) was obtained in an amount of 0.65 g (96%).
(R)-1-(2-アミノ-2-メチルプロパノイル)-N-(4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物21(0.83g、1.5mmol)に、TFA(10mL)を加えた。室温下で3時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタン(10mL)に溶解させ、飽和炭酸水素ナトリウム水溶液(10mL)を加えた。室温下で30分間撹拌した後、反応溶液に、水を加え、ジクロロメタンで抽出した。有機層を水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-1-(2-アミノ-2-メチルプロパノイル)-N-(4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物22)を0.65g(96%)得た。 [Step 6]
Synthesis of (R) -1- (2-amino-2-methylpropanoyl) -N- (4-chloro-2- (2,2,2-trifluoroethoxy) benzyl) piperidine-3-carboxamide:
TFA (10 mL) was added to Reference Example Compound 21 (0.83 g, 1.5 mmol) under ice cooling. After stirring at room temperature for 3 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane (10 mL), and saturated aqueous sodium hydrogen carbonate solution (10 mL) was added. After stirring at room temperature for 30 minutes, water was added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with water, dried over anhydrous sodium sulfate, concentrated under reduced pressure, and (R) -1- (2-amino-2-methylpropanoyl) -N- (4-chloro-2- (2,2 , 2-trifluoroethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Reference Example Compound 22) was obtained in an amount of 0.65 g (96%).
〔ステップ7〕
(R)-N-(4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物22(0.080g、0.18mmol)、ピリジン(0.030mL、0.37mmol)のジクロロメタン(2.0mL)溶液に、メタンスルホニルクロリド(0.023g、0.20mmol)を加えた。室温下で10時間撹拌した後、反応溶液に、水を加え、ジクロロメタンで抽出した。有機層を0.1規定塩酸、水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=100:1→10:1)で精製し、(R)-N-(4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物10)を0.030g(32%)得た。 [Step 7]
Synthesis of (R) -N- (4-chloro-2- (2,2,2-trifluoroethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide :
Under ice cooling, a solution of Reference Compound 22 (0.080 g, 0.18 mmol) and pyridine (0.030 mL, 0.37 mmol) in dichloromethane (2.0 mL) was added to methanesulfonyl chloride (0.023 g, 0.20 mmol). Was added. After stirring at room temperature for 10 hours, water was added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with 0.1N hydrochloric acid, water, and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 100: 1 → 10: 1), and (R) -N- (4-chloro-2- (2,2,2 -Trifluoroethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 10) was obtained in an amount of 0.030 g (32%).
(R)-N-(4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物22(0.080g、0.18mmol)、ピリジン(0.030mL、0.37mmol)のジクロロメタン(2.0mL)溶液に、メタンスルホニルクロリド(0.023g、0.20mmol)を加えた。室温下で10時間撹拌した後、反応溶液に、水を加え、ジクロロメタンで抽出した。有機層を0.1規定塩酸、水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=100:1→10:1)で精製し、(R)-N-(4-クロロ-2-(2,2,2-トリフルオロエトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物10)を0.030g(32%)得た。 [Step 7]
Synthesis of (R) -N- (4-chloro-2- (2,2,2-trifluoroethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide :
Under ice cooling, a solution of Reference Compound 22 (0.080 g, 0.18 mmol) and pyridine (0.030 mL, 0.37 mmol) in dichloromethane (2.0 mL) was added to methanesulfonyl chloride (0.023 g, 0.20 mmol). Was added. After stirring at room temperature for 10 hours, water was added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with 0.1N hydrochloric acid, water, and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 100: 1 → 10: 1), and (R) -N- (4-chloro-2- (2,2,2 -Trifluoroethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 10) was obtained in an amount of 0.030 g (32%).
(実施例11) (R)-1-((R)-2-アセトアミド-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
(R)-1-((R)-2-アミノ-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物8(0.21g、0.65mmol)、(R)-2-((((9H-フルオレン-9-イル)メトキシ)カルボニル)アミノ)-3-メチルブタン酸(0.24g、0.72mmol)、DIPEA(0.14mL、0.78mmol)のDMF(3.5mL)溶液に、HATU(0.30g、0.78mmol)を加えた。室温下で1時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=9:1→1:9)で精製し、粗生成物を0.30g得た。氷冷下、得られた粗生成物(0.30g)のDMF(2.0mL)溶液に、モルホリン(0.20mL、2.3mmol)を加えた。室温下で2.5時間撹拌した後、反応溶液に、水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→90:10)で精製し、(R)-1-((R)-2-アミノ-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物23)を0.18g(2段階収率65%)得た。 Example 11 Synthesis of (R) -1-((R) -2-acetamido-3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide :
[Step 1]
Synthesis of (R) -1-((R) -2-amino-3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
Reference Example Compound 8 (0.21 g, 0.65 mmol), (R) -2-((((9H-fluoren-9-yl) methoxy) carbonyl) amino) -3-methylbutanoic acid (0. HATU (0.30 g, 0.78 mmol) was added to a DMF (3.5 mL) solution of 24 g, 0.72 mmol), DIPEA (0.14 mL, 0.78 mmol). After stirring at room temperature for 1 hour, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 9: 1 → 1: 9) to obtain 0.30 g of a crude product. Under ice-cooling, morpholine (0.20 mL, 2.3 mmol) was added to a solution of the obtained crude product (0.30 g) in DMF (2.0 mL). After stirring at room temperature for 2.5 hours, water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 90: 10) to give (R) -1-((R) -2-amino-3-methylbutaline. Noyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 23) (0.18 g, 2-step yield 65%) was obtained.
〔ステップ1〕
(R)-1-((R)-2-アミノ-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物8(0.21g、0.65mmol)、(R)-2-((((9H-フルオレン-9-イル)メトキシ)カルボニル)アミノ)-3-メチルブタン酸(0.24g、0.72mmol)、DIPEA(0.14mL、0.78mmol)のDMF(3.5mL)溶液に、HATU(0.30g、0.78mmol)を加えた。室温下で1時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=9:1→1:9)で精製し、粗生成物を0.30g得た。氷冷下、得られた粗生成物(0.30g)のDMF(2.0mL)溶液に、モルホリン(0.20mL、2.3mmol)を加えた。室温下で2.5時間撹拌した後、反応溶液に、水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→90:10)で精製し、(R)-1-((R)-2-アミノ-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物23)を0.18g(2段階収率65%)得た。 Example 11 Synthesis of (R) -1-((R) -2-acetamido-3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide :
[Step 1]
Synthesis of (R) -1-((R) -2-amino-3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
Reference Example Compound 8 (0.21 g, 0.65 mmol), (R) -2-((((9H-fluoren-9-yl) methoxy) carbonyl) amino) -3-methylbutanoic acid (0. HATU (0.30 g, 0.78 mmol) was added to a DMF (3.5 mL) solution of 24 g, 0.72 mmol), DIPEA (0.14 mL, 0.78 mmol). After stirring at room temperature for 1 hour, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 9: 1 → 1: 9) to obtain 0.30 g of a crude product. Under ice-cooling, morpholine (0.20 mL, 2.3 mmol) was added to a solution of the obtained crude product (0.30 g) in DMF (2.0 mL). After stirring at room temperature for 2.5 hours, water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 90: 10) to give (R) -1-((R) -2-amino-3-methylbutaline. Noyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 23) (0.18 g, 2-step yield 65%) was obtained.
〔ステップ2〕
(R)-1-((R)-2-アセトアミド-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
参考例化合物23(0.020g、0.047mmol)を用いて実施例8〔ステップ3〕と同様の反応を行うことにより、(R)-1-((R)-2-アセトアミド-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物11)を0.018g(83%)得た。 [Step 2]
Synthesis of (R) -1-((R) -2-acetamido-3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1-((R) -2-acetamido-3-methyl) was prepared by performing the same reaction as in Example 8 [Step 3] using Reference compound 23 (0.020 g, 0.047 mmol). 0.018 g (83%) of butanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 11) was obtained.
(R)-1-((R)-2-アセトアミド-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
参考例化合物23(0.020g、0.047mmol)を用いて実施例8〔ステップ3〕と同様の反応を行うことにより、(R)-1-((R)-2-アセトアミド-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物11)を0.018g(83%)得た。 [Step 2]
Synthesis of (R) -1-((R) -2-acetamido-3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1-((R) -2-acetamido-3-methyl) was prepared by performing the same reaction as in Example 8 [Step 3] using Reference compound 23 (0.020 g, 0.047 mmol). 0.018 g (83%) of butanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 11) was obtained.
(実施例12) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-3-メチル-2-(メチルスルホンアミド)ブタノイル)ピペリジン-3-カルボキサミドの合成:
参考例化合物23(0.020g、0.047mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-3-メチル-2-(メチルスルホンアミド)ブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物12)を0.020g(83%)得た。 Example 12 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3- Synthesis of carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by conducting the same reaction as in Example 2 [Step 3] using Reference Example Compound 23 (0.020 g, 0.047 mmol). 0.020 g (83%) of benzyl) -1-((R) -3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 12) was obtained.
参考例化合物23(0.020g、0.047mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-3-メチル-2-(メチルスルホンアミド)ブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物12)を0.020g(83%)得た。 Example 12 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3- Synthesis of carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by conducting the same reaction as in Example 2 [Step 3] using Reference Example Compound 23 (0.020 g, 0.047 mmol). 0.020 g (83%) of benzyl) -1-((R) -3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 12) was obtained.
(実施例13) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(エチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物14(0.050g、0.12mmol)、DIPEA(0.043mL、0.24mmol)のジクロロメタン(3.0mL)溶液に、エタンスルホニルクロリド(0.017g、0.13mmol)を加えた。室温下で3時間撹拌した後、ピリジン(0.30mL、3.7mmol)を加えた。室温下で3時間撹拌した後、反応溶液に、水を加え、ジクロロメタンで抽出した。有機層を0.1規定塩酸、水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=100:1→10:1)で精製し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(エチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物13)を0.0080g(13%)得た。 Example 13 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (ethylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide:
Under ice-cooling, ethanesulfonyl chloride (0.017 g, 0.13 mmol) was added to a solution of Reference Example Compound 14 (0.050 g, 0.12 mmol) and DIPEA (0.043 mL, 0.24 mmol) in dichloromethane (3.0 mL). Was added. After stirring at room temperature for 3 hours, pyridine (0.30 mL, 3.7 mmol) was added. After stirring at room temperature for 3 hours, water was added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with 0.1N hydrochloric acid, water, and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 100: 1 → 10: 1), and (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl was obtained. ) -1- (1- (ethylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 13) was obtained in an amount of 0.0080 g (13%).
氷冷下、参考例化合物14(0.050g、0.12mmol)、DIPEA(0.043mL、0.24mmol)のジクロロメタン(3.0mL)溶液に、エタンスルホニルクロリド(0.017g、0.13mmol)を加えた。室温下で3時間撹拌した後、ピリジン(0.30mL、3.7mmol)を加えた。室温下で3時間撹拌した後、反応溶液に、水を加え、ジクロロメタンで抽出した。有機層を0.1規定塩酸、水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=100:1→10:1)で精製し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(エチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物13)を0.0080g(13%)得た。 Example 13 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (ethylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide:
Under ice-cooling, ethanesulfonyl chloride (0.017 g, 0.13 mmol) was added to a solution of Reference Example Compound 14 (0.050 g, 0.12 mmol) and DIPEA (0.043 mL, 0.24 mmol) in dichloromethane (3.0 mL). Was added. After stirring at room temperature for 3 hours, pyridine (0.30 mL, 3.7 mmol) was added. After stirring at room temperature for 3 hours, water was added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was washed with 0.1N hydrochloric acid, water, and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 100: 1 → 10: 1), and (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl was obtained. ) -1- (1- (ethylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 13) was obtained in an amount of 0.0080 g (13%).
(実施例14) (R)-N-(4-カルバモイル-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミドの合成:
氷冷下、実施例化合物2(0.020g、0.041mmol)、炭酸カリウム(0.0028g、0.020mmol)のDMF(0.38mL)溶液に、過酸化水素水(30重量%、0.021mL)を加えた。室温下で18時間撹拌した後、反応溶液に、水を加え、酢酸エチルで抽出した。有機層をチオ硫酸ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→85:15)で精製し、(R)-N-(4-カルバモイル-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物14)を0.013g(63%)得た。 Example 14 Synthesis of (R) -N- (4-carbamoyl-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide:
Under ice-cooling, a hydrogen peroxide solution (30% by weight, 0.0. 021 mL) was added. After stirring at room temperature for 18 hours, water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was washed with an aqueous sodium thiosulfate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 85: 15), and (R) —N- (4-carbamoyl-2- (trifluoromethoxy) benzyl was obtained. ) -1- (2-Methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 14) was obtained in an amount of 0.013 g (63%).
氷冷下、実施例化合物2(0.020g、0.041mmol)、炭酸カリウム(0.0028g、0.020mmol)のDMF(0.38mL)溶液に、過酸化水素水(30重量%、0.021mL)を加えた。室温下で18時間撹拌した後、反応溶液に、水を加え、酢酸エチルで抽出した。有機層をチオ硫酸ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→85:15)で精製し、(R)-N-(4-カルバモイル-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物14)を0.013g(63%)得た。 Example 14 Synthesis of (R) -N- (4-carbamoyl-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide:
Under ice-cooling, a hydrogen peroxide solution (30% by weight, 0.0. 021 mL) was added. After stirring at room temperature for 18 hours, water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was washed with an aqueous sodium thiosulfate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 85: 15), and (R) —N- (4-carbamoyl-2- (trifluoromethoxy) benzyl was obtained. ) -1- (2-Methyl-2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 14) was obtained in an amount of 0.013 g (63%).
(実施例15) (R)-N-(4-カルバモイル-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミドの合成:
実施例化合物3(0.025g、0.051mmol)を用いて実施例14と同様の反応を行うことにより、(R)-N-(4-カルバモイル-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物15)を0.020g(77%)得た。 Example 15 (R) -N- (4-carbamoyl-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide Synthesis:
(R) -N- (4-carbamoyl-2- (trifluoromethoxy) benzyl) -1 was prepared by carrying out the same reaction as in Example 14 using Example Compound 3 (0.025 g, 0.051 mmol). As a result, 0.020 g (77%) of — (2-methyl-2- (methylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 15) was obtained.
実施例化合物3(0.025g、0.051mmol)を用いて実施例14と同様の反応を行うことにより、(R)-N-(4-カルバモイル-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(メチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物15)を0.020g(77%)得た。 Example 15 (R) -N- (4-carbamoyl-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (methylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide Synthesis:
(R) -N- (4-carbamoyl-2- (trifluoromethoxy) benzyl) -1 was prepared by carrying out the same reaction as in Example 14 using Example Compound 3 (0.025 g, 0.051 mmol). As a result, 0.020 g (77%) of — (2-methyl-2- (methylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 15) was obtained.
(実施例16) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-ヒドロキシ-2-メチルプロパノイル)ピペリジン-3-カルボキサミドの合成:
2-ヒドロキシ-2-メチルプロパン酸(0.15g、0.46mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-ヒドロキシ-2-メチルプロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物16)を0.12g(62%)得た。 Example 16 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-hydroxy-2-methylpropanoyl) piperidine-3-carboxamide:
The same reaction as in Example 1 [Step 9] was performed using 2-hydroxy-2-methylpropanoic acid (0.15 g, 0.46 mmol) to give (R) -N- (4-cyano-2- 0.12 g (62%) of (trifluoromethoxy) benzyl) -1- (2-hydroxy-2-methylpropanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 16) was obtained.
2-ヒドロキシ-2-メチルプロパン酸(0.15g、0.46mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-ヒドロキシ-2-メチルプロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物16)を0.12g(62%)得た。 Example 16 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-hydroxy-2-methylpropanoyl) piperidine-3-carboxamide:
The same reaction as in Example 1 [Step 9] was performed using 2-hydroxy-2-methylpropanoic acid (0.15 g, 0.46 mmol) to give (R) -N- (4-cyano-2- 0.12 g (62%) of (trifluoromethoxy) benzyl) -1- (2-hydroxy-2-methylpropanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 16) was obtained.
(実施例17) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-ピバロイルピペリジン-3-カルボキサミドの合成:
参考例化合物8(0.15g、0.46mmol)、ピバロイルクロリド(0.066g、0.55mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-ピバロイルピペリジン-3-カルボキサミド(以下、実施例化合物17)を0.19g(定量的)得た。 Example 17 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-pivaloylpiperidine-3-carboxamide:
By performing the same reaction as in Example 1 [Step 11] using Reference Example Compound 8 (0.15 g, 0.46 mmol) and pivaloyl chloride (0.066 g, 0.55 mmol), (R) — 0.19 g (quantitative) of N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-pivaloylpiperidine-3-carboxamide (hereinafter, Example Compound 17) was obtained.
参考例化合物8(0.15g、0.46mmol)、ピバロイルクロリド(0.066g、0.55mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-ピバロイルピペリジン-3-カルボキサミド(以下、実施例化合物17)を0.19g(定量的)得た。 Example 17 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-pivaloylpiperidine-3-carboxamide:
By performing the same reaction as in Example 1 [Step 11] using Reference Example Compound 8 (0.15 g, 0.46 mmol) and pivaloyl chloride (0.066 g, 0.55 mmol), (R) — 0.19 g (quantitative) of N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-pivaloylpiperidine-3-carboxamide (hereinafter, Example Compound 17) was obtained.
(実施例18) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(N-メチルメチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
1-(メチルスルホンアミド)シクロプロパンカルボン酸エチルの合成:
氷冷下、1-アミノシクロプロパンカルボン酸エチル塩酸塩(2.0g、12mmol)、DIPEA(6.3mL、36mmol)のジクロロメタン(35mL)溶液に、メタンスルホニルクロリド(1.4g、12mmol)を加えた。氷冷下で3時間撹拌した後、反応溶液に、1規定塩酸を加え、酸性にした後、酢酸エチルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=10:1→1:2)で精製し、1-(メチルスルホンアミド)シクロプロパンカルボン酸エチル(以下、参考例化合物24)を2.3g(60%)得た。 Example 18 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (N-methylmethylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide :
[Step 1]
Synthesis of ethyl 1- (methylsulfonamido) cyclopropanecarboxylate:
Methanesulfonyl chloride (1.4 g, 12 mmol) was added to a solution of ethyl 1-aminocyclopropanecarboxylic acid hydrochloride (2.0 g, 12 mmol) and DIPEA (6.3 mL, 36 mmol) in dichloromethane (35 mL) under ice cooling. It was. After stirring for 3 hours under ice cooling, 1N hydrochloric acid was added to the reaction solution to make it acidic, followed by extraction with ethyl acetate. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 10: 1 → 1: 2) to give ethyl 1- (methylsulfonamido) cyclopropanecarboxylate (hereinafter referred to as Reference Example Compound). 24) 2.3 g (60%) was obtained.
〔ステップ1〕
1-(メチルスルホンアミド)シクロプロパンカルボン酸エチルの合成:
氷冷下、1-アミノシクロプロパンカルボン酸エチル塩酸塩(2.0g、12mmol)、DIPEA(6.3mL、36mmol)のジクロロメタン(35mL)溶液に、メタンスルホニルクロリド(1.4g、12mmol)を加えた。氷冷下で3時間撹拌した後、反応溶液に、1規定塩酸を加え、酸性にした後、酢酸エチルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=10:1→1:2)で精製し、1-(メチルスルホンアミド)シクロプロパンカルボン酸エチル(以下、参考例化合物24)を2.3g(60%)得た。 Example 18 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (N-methylmethylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide :
[Step 1]
Synthesis of ethyl 1- (methylsulfonamido) cyclopropanecarboxylate:
Methanesulfonyl chloride (1.4 g, 12 mmol) was added to a solution of ethyl 1-aminocyclopropanecarboxylic acid hydrochloride (2.0 g, 12 mmol) and DIPEA (6.3 mL, 36 mmol) in dichloromethane (35 mL) under ice cooling. It was. After stirring for 3 hours under ice cooling, 1N hydrochloric acid was added to the reaction solution to make it acidic, followed by extraction with ethyl acetate. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 10: 1 → 1: 2) to give ethyl 1- (methylsulfonamido) cyclopropanecarboxylate (hereinafter referred to as Reference Example Compound). 24) 2.3 g (60%) was obtained.
〔ステップ2〕
1-(N-メチルメチルスルホンアミド)シクロプロパンカルボン酸エチルの合成:
氷冷下、参考例化合物24(2.0g、9.7mmol)のDMF(10mL)溶液に、水素化ナトリウム(55重量%、0.51g、12mmol)を加えた。氷冷下で10分間撹拌した後、室温下で30分間撹拌した。氷冷下、反応溶液に、ヨウ化メチル(0.78mL、13mmol)を加えた。室温下で14時間撹拌した後、氷冷下、反応溶液に、0.1規定塩酸を加え、ヘキサン:酢酸エチル混合溶媒(ヘキサン:酢酸エチル=1:2)で抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=10:1→1:1)で精製し、1-(N-メチルメチルスルホンアミド)シクロプロパンカルボン酸エチル(以下、参考例化合物25)を1.8g(84%)得た。 [Step 2]
Synthesis of ethyl 1- (N-methylmethylsulfonamido) cyclopropanecarboxylate:
Sodium hydride (55 wt%, 0.51 g, 12 mmol) was added to a DMF (10 mL) solution of Reference Example Compound 24 (2.0 g, 9.7 mmol) under ice cooling. The mixture was stirred for 10 minutes under ice cooling, and then stirred for 30 minutes at room temperature. Methyl iodide (0.78 mL, 13 mmol) was added to the reaction solution under ice cooling. After stirring at room temperature for 14 hours, 0.1N hydrochloric acid was added to the reaction solution under ice cooling, and the mixture was extracted with a mixed solvent of hexane: ethyl acetate (hexane: ethyl acetate = 1: 2). The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 10: 1 → 1: 1), and ethyl 1- (N-methylmethylsulfonamido) cyclopropanecarboxylate (hereinafter referred to as “eluent”). 1.8 g (84%) of Reference Example Compound 25) was obtained.
1-(N-メチルメチルスルホンアミド)シクロプロパンカルボン酸エチルの合成:
氷冷下、参考例化合物24(2.0g、9.7mmol)のDMF(10mL)溶液に、水素化ナトリウム(55重量%、0.51g、12mmol)を加えた。氷冷下で10分間撹拌した後、室温下で30分間撹拌した。氷冷下、反応溶液に、ヨウ化メチル(0.78mL、13mmol)を加えた。室温下で14時間撹拌した後、氷冷下、反応溶液に、0.1規定塩酸を加え、ヘキサン:酢酸エチル混合溶媒(ヘキサン:酢酸エチル=1:2)で抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=10:1→1:1)で精製し、1-(N-メチルメチルスルホンアミド)シクロプロパンカルボン酸エチル(以下、参考例化合物25)を1.8g(84%)得た。 [Step 2]
Synthesis of ethyl 1- (N-methylmethylsulfonamido) cyclopropanecarboxylate:
Sodium hydride (55 wt%, 0.51 g, 12 mmol) was added to a DMF (10 mL) solution of Reference Example Compound 24 (2.0 g, 9.7 mmol) under ice cooling. The mixture was stirred for 10 minutes under ice cooling, and then stirred for 30 minutes at room temperature. Methyl iodide (0.78 mL, 13 mmol) was added to the reaction solution under ice cooling. After stirring at room temperature for 14 hours, 0.1N hydrochloric acid was added to the reaction solution under ice cooling, and the mixture was extracted with a mixed solvent of hexane: ethyl acetate (hexane: ethyl acetate = 1: 2). The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 10: 1 → 1: 1), and ethyl 1- (N-methylmethylsulfonamido) cyclopropanecarboxylate (hereinafter referred to as “eluent”). 1.8 g (84%) of Reference Example Compound 25) was obtained.
〔ステップ3〕
1-(N-メチルメチルスルホンアミド)シクロプロパンカルボン酸の合成:
室温下で、参考例化合物25(1.8g、7.9mmol)のメタノール(20mL)溶液に、1規定水酸化ナトリウム水溶液(12mL、12mmol)を加えた。50℃下で3時間撹拌した後、室温下、反応溶液に、1規定塩酸を加え、クロロホルムで抽出した。有機層を飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮し、1-(N-メチルメチルスルホンアミド)シクロプロパンカルボン酸(以下、参考例化合物26)を0.88g(58%)得た。 [Step 3]
Synthesis of 1- (N-methylmethylsulfonamido) cyclopropanecarboxylic acid:
A 1N aqueous sodium hydroxide solution (12 mL, 12 mmol) was added to a methanol (20 mL) solution of Reference Example Compound 25 (1.8 g, 7.9 mmol) at room temperature. After stirring at 50 ° C. for 3 hours, 1N hydrochloric acid was added to the reaction solution at room temperature, and the mixture was extracted with chloroform. The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to obtain 0.88 g (58 %)Obtained.
1-(N-メチルメチルスルホンアミド)シクロプロパンカルボン酸の合成:
室温下で、参考例化合物25(1.8g、7.9mmol)のメタノール(20mL)溶液に、1規定水酸化ナトリウム水溶液(12mL、12mmol)を加えた。50℃下で3時間撹拌した後、室温下、反応溶液に、1規定塩酸を加え、クロロホルムで抽出した。有機層を飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮し、1-(N-メチルメチルスルホンアミド)シクロプロパンカルボン酸(以下、参考例化合物26)を0.88g(58%)得た。 [Step 3]
Synthesis of 1- (N-methylmethylsulfonamido) cyclopropanecarboxylic acid:
A 1N aqueous sodium hydroxide solution (12 mL, 12 mmol) was added to a methanol (20 mL) solution of Reference Example Compound 25 (1.8 g, 7.9 mmol) at room temperature. After stirring at 50 ° C. for 3 hours, 1N hydrochloric acid was added to the reaction solution at room temperature, and the mixture was extracted with chloroform. The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to obtain 0.88 g (58 %)Obtained.
〔ステップ4〕
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(N-メチルメチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミドの合成:
参考例化合物26(0.13g、0.67mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(N-メチルメチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物18)を0.17g(60%)得た。 [Step 4]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (N-methylmethylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 1 [Step 9] using Reference Example Compound 26 (0.13 g, 0.67 mmol). 0.17 g (60%) of benzyl) -1- (1- (N-methylmethylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 18) was obtained.
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(N-メチルメチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミドの合成:
参考例化合物26(0.13g、0.67mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(N-メチルメチルスルホンアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物18)を0.17g(60%)得た。 [Step 4]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (N-methylmethylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 1 [Step 9] using Reference Example Compound 26 (0.13 g, 0.67 mmol). 0.17 g (60%) of benzyl) -1- (1- (N-methylmethylsulfonamido) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 18) was obtained.
(実施例19) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(3-ヒドロキシ-2,2-ジメチルプロパノイル)ピペリジン-3-カルボキサミドの合成:
室温下、3-ヒドロキシ-2,2-ジメチルプロパン酸メチル(1.0g、7.6mmol)のメタノール(7.5mL)溶液に、1規定水酸化ナトリウム水溶液(9.1mL)を加えた。室温下で4時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物に、1規定塩酸を加え、減圧濃縮した。氷冷下、得られた粗生成物(0.10g)、DIPEA(0.30mL、1.7mmol)のDMF(1.6mL)溶液に、HATU(0.35g、0.93mmol)を加えた。氷冷下で15分間撹拌した後、反応溶液に、参考例化合物8(0.28g、0.85mmol)を加えた。室温下で一晩撹拌した後、反応溶液に、水を加え、ジエチルエーテルで抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=8:2→酢酸エチルのみ)で精製し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(3-ヒドロキシ-2,2-ジメチルプロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物19)を0.24g(2段階収率65%)得た。 Example 19 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (3-hydroxy-2,2-dimethylpropanoyl) piperidine-3-carboxamide:
A 1N aqueous sodium hydroxide solution (9.1 mL) was added to a solution of methyl 3-hydroxy-2,2-dimethylpropanoate (1.0 g, 7.6 mmol) in methanol (7.5 mL) at room temperature. After stirring at room temperature for 4 hours, the reaction solution was concentrated under reduced pressure. 1N Hydrochloric acid was added to the obtained crude product, and the mixture was concentrated under reduced pressure. HATU (0.35 g, 0.93 mmol) was added to a DMF (1.6 mL) solution of the obtained crude product (0.10 g) and DIPEA (0.30 mL, 1.7 mmol) under ice cooling. After stirring for 15 minutes under ice cooling, Reference Example Compound 8 (0.28 g, 0.85 mmol) was added to the reaction solution. After stirring overnight at room temperature, water was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 8: 2 → ethyl acetate only), and (R) -N- (4-cyano-2- (trifluoromethoxy) 0.24 g (2-step yield 65%) of benzyl) -1- (3-hydroxy-2,2-dimethylpropanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 19) was obtained.
室温下、3-ヒドロキシ-2,2-ジメチルプロパン酸メチル(1.0g、7.6mmol)のメタノール(7.5mL)溶液に、1規定水酸化ナトリウム水溶液(9.1mL)を加えた。室温下で4時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物に、1規定塩酸を加え、減圧濃縮した。氷冷下、得られた粗生成物(0.10g)、DIPEA(0.30mL、1.7mmol)のDMF(1.6mL)溶液に、HATU(0.35g、0.93mmol)を加えた。氷冷下で15分間撹拌した後、反応溶液に、参考例化合物8(0.28g、0.85mmol)を加えた。室温下で一晩撹拌した後、反応溶液に、水を加え、ジエチルエーテルで抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=8:2→酢酸エチルのみ)で精製し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(3-ヒドロキシ-2,2-ジメチルプロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物19)を0.24g(2段階収率65%)得た。 Example 19 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (3-hydroxy-2,2-dimethylpropanoyl) piperidine-3-carboxamide:
A 1N aqueous sodium hydroxide solution (9.1 mL) was added to a solution of methyl 3-hydroxy-2,2-dimethylpropanoate (1.0 g, 7.6 mmol) in methanol (7.5 mL) at room temperature. After stirring at room temperature for 4 hours, the reaction solution was concentrated under reduced pressure. 1N Hydrochloric acid was added to the obtained crude product, and the mixture was concentrated under reduced pressure. HATU (0.35 g, 0.93 mmol) was added to a DMF (1.6 mL) solution of the obtained crude product (0.10 g) and DIPEA (0.30 mL, 1.7 mmol) under ice cooling. After stirring for 15 minutes under ice cooling, Reference Example Compound 8 (0.28 g, 0.85 mmol) was added to the reaction solution. After stirring overnight at room temperature, water was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 8: 2 → ethyl acetate only), and (R) -N- (4-cyano-2- (trifluoromethoxy) 0.24 g (2-step yield 65%) of benzyl) -1- (3-hydroxy-2,2-dimethylpropanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 19) was obtained.
(実施例20) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-ヒドロキシシクロヘキサンカルボニル)ピペリジン-3-カルボキサミドの合成:
氷冷下、シクロヘキサノン(3.0g、31mmol)、シアン化カリウム(2.2g、34mmol)の水(5.6mL)溶液に、硫酸水溶液(40重量%、5.6mL)を加えた。室温下で1時間撹拌した後、反応溶液に、水を加え、ジエチルエーテルで抽出した。有機層を飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物に、濃塩酸(60mL)を加えた。80℃下で16時間撹拌した後、反応溶液を減圧濃縮し、粗生成物を4.0g得た。氷冷下、得られた粗生成物(0.16g)、参考例化合物8(0.15g、0.46mmol)、DIPEA(0.24mL、1.4mmol)のDMF(1.0mL)溶液に、HATU(0.45g、0.60mmol)を加えた。室温下で2時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;ヘキサン:酢酸エチル=7:3→4:6)で精製し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-ヒドロキシシクロヘキサンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物20)を0.061g(2段階収率29%)得た。 Example 20 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide:
Under ice-cooling, an aqueous sulfuric acid solution (40 wt%, 5.6 mL) was added to a solution of cyclohexanone (3.0 g, 31 mmol) and potassium cyanide (2.2 g, 34 mmol) in water (5.6 mL). After stirring at room temperature for 1 hour, water was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. Concentrated hydrochloric acid (60 mL) was added to the obtained crude product. After stirring at 80 ° C. for 16 hours, the reaction solution was concentrated under reduced pressure to obtain 4.0 g of a crude product. Under ice cooling, the obtained crude product (0.16 g), Reference Example Compound 8 (0.15 g, 0.46 mmol), DIPEA (0.24 mL, 1.4 mmol) in DMF (1.0 mL) solution, HATU (0.45 g, 0.60 mmol) was added. After stirring at room temperature for 2 hours, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium bicarbonate solution and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (amine silica gel DM1020, manufactured by Fuji Silysia Chemical Ltd., eluent; hexane: ethyl acetate = 7: 3 → 4: 6), and (R) -N- (4- 0.061 g (2-step yield 29%) of cyano-2- (trifluoromethoxy) benzyl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 20) was obtained.
氷冷下、シクロヘキサノン(3.0g、31mmol)、シアン化カリウム(2.2g、34mmol)の水(5.6mL)溶液に、硫酸水溶液(40重量%、5.6mL)を加えた。室温下で1時間撹拌した後、反応溶液に、水を加え、ジエチルエーテルで抽出した。有機層を飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物に、濃塩酸(60mL)を加えた。80℃下で16時間撹拌した後、反応溶液を減圧濃縮し、粗生成物を4.0g得た。氷冷下、得られた粗生成物(0.16g)、参考例化合物8(0.15g、0.46mmol)、DIPEA(0.24mL、1.4mmol)のDMF(1.0mL)溶液に、HATU(0.45g、0.60mmol)を加えた。室温下で2時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;ヘキサン:酢酸エチル=7:3→4:6)で精製し、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-ヒドロキシシクロヘキサンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物20)を0.061g(2段階収率29%)得た。 Example 20 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide:
Under ice-cooling, an aqueous sulfuric acid solution (40 wt%, 5.6 mL) was added to a solution of cyclohexanone (3.0 g, 31 mmol) and potassium cyanide (2.2 g, 34 mmol) in water (5.6 mL). After stirring at room temperature for 1 hour, water was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. Concentrated hydrochloric acid (60 mL) was added to the obtained crude product. After stirring at 80 ° C. for 16 hours, the reaction solution was concentrated under reduced pressure to obtain 4.0 g of a crude product. Under ice cooling, the obtained crude product (0.16 g), Reference Example Compound 8 (0.15 g, 0.46 mmol), DIPEA (0.24 mL, 1.4 mmol) in DMF (1.0 mL) solution, HATU (0.45 g, 0.60 mmol) was added. After stirring at room temperature for 2 hours, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium bicarbonate solution and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (amine silica gel DM1020, manufactured by Fuji Silysia Chemical Ltd., eluent; hexane: ethyl acetate = 7: 3 → 4: 6), and (R) -N- (4- 0.061 g (2-step yield 29%) of cyano-2- (trifluoromethoxy) benzyl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 20) was obtained.
(実施例21) (R)-1-((R)-2-アセトアミドブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
(R)-2-((tert-ブトキシカルボニル)アミノ)ブタン酸の合成:
氷冷下、(R)-2-アミノブタン酸(2.0g、19mmol)、炭酸水素ナトリウム(1.6g、19mmol)の1,4-ジオキサン:水(1,4-ジオキサン:水=3:10、26mL)混合溶液に、ジ-tert-ブチルジカルボナート(4.7g、21mmol)を加えた。室温下で120時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をクロロホルムに溶解させ、1規定塩酸を加え、クロロホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-2-((tert-ブトキシカルボニル)アミノ)ブタン酸(以下、参考例化合物27)を2.0g(定量的)得た。 Example 21 Synthesis of (R) -1-((R) -2-acetamidobutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of (R) -2-((tert-butoxycarbonyl) amino) butanoic acid:
Under ice-cooling, (R) -2-aminobutanoic acid (2.0 g, 19 mmol), sodium bicarbonate (1.6 g, 19 mmol) in 1,4-dioxane: water (1,4-dioxane: water = 3: 10) , 26 mL) di-tert-butyl dicarbonate (4.7 g, 21 mmol) was added to the mixed solution. After stirring at room temperature for 120 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in chloroform, 1N hydrochloric acid was added, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain 2.0 g (quantitative) of (R) -2-((tert-butoxycarbonyl) amino) butanoic acid (hereinafter referred to as Reference Example Compound 27).
〔ステップ1〕
(R)-2-((tert-ブトキシカルボニル)アミノ)ブタン酸の合成:
氷冷下、(R)-2-アミノブタン酸(2.0g、19mmol)、炭酸水素ナトリウム(1.6g、19mmol)の1,4-ジオキサン:水(1,4-ジオキサン:水=3:10、26mL)混合溶液に、ジ-tert-ブチルジカルボナート(4.7g、21mmol)を加えた。室温下で120時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をクロロホルムに溶解させ、1規定塩酸を加え、クロロホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-2-((tert-ブトキシカルボニル)アミノ)ブタン酸(以下、参考例化合物27)を2.0g(定量的)得た。 Example 21 Synthesis of (R) -1-((R) -2-acetamidobutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of (R) -2-((tert-butoxycarbonyl) amino) butanoic acid:
Under ice-cooling, (R) -2-aminobutanoic acid (2.0 g, 19 mmol), sodium bicarbonate (1.6 g, 19 mmol) in 1,4-dioxane: water (1,4-dioxane: water = 3: 10) , 26 mL) di-tert-butyl dicarbonate (4.7 g, 21 mmol) was added to the mixed solution. After stirring at room temperature for 120 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in chloroform, 1N hydrochloric acid was added, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain 2.0 g (quantitative) of (R) -2-((tert-butoxycarbonyl) amino) butanoic acid (hereinafter referred to as Reference Example Compound 27).
〔ステップ2〕
tert-ブチル ((R)-1-((R)-3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-1-オキソブタン-2-イル)カルバマートの合成:
参考例化合物27(0.20g、1.0mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、tert-ブチル ((R)-1-((R)-3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-1-オキソブタン-2-イル)カルバマート(以下、参考例化合物28)を0.47g(定量的)得た。 [Step 2]
tert-Butyl ((R) -1-((R) -3-((4-Cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-yl) -1-oxobutan-2-yl) carbamate Synthesis of:
The same reaction as in Example 1 [Step 9] was carried out using Reference Compound 27 (0.20 g, 1.0 mmol) to give tert-butyl ((R) -1-((R) -3- ( 0.47 g (quantitative) of (4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-yl) -1-oxobutan-2-yl) carbamate (hereinafter referred to as Reference Example Compound 28) was obtained. .
tert-ブチル ((R)-1-((R)-3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-1-オキソブタン-2-イル)カルバマートの合成:
参考例化合物27(0.20g、1.0mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、tert-ブチル ((R)-1-((R)-3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-1-オキソブタン-2-イル)カルバマート(以下、参考例化合物28)を0.47g(定量的)得た。 [Step 2]
tert-Butyl ((R) -1-((R) -3-((4-Cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-yl) -1-oxobutan-2-yl) carbamate Synthesis of:
The same reaction as in Example 1 [Step 9] was carried out using Reference Compound 27 (0.20 g, 1.0 mmol) to give tert-butyl ((R) -1-((R) -3- ( 0.47 g (quantitative) of (4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-yl) -1-oxobutan-2-yl) carbamate (hereinafter referred to as Reference Example Compound 28) was obtained. .
〔ステップ3〕
(R)-1-((R)-2-アミノブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
参考例化合物28(0.47g、0.91mmol)を用いて実施例1〔ステップ10〕と同様の反応を行うことにより、(R)-1-((R)-2-アミノブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物29)を0.38g(定量的)得た。 [Step 3]
Synthesis of (R) -1-((R) -2-aminobutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1-((R) -2-aminobutanoyl)-was prepared by carrying out the same reaction as in Example 1 [Step 10] using Reference Example Compound 28 (0.47 g, 0.91 mmol). 0.38 g (quantitative) of N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 29) was obtained.
(R)-1-((R)-2-アミノブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
参考例化合物28(0.47g、0.91mmol)を用いて実施例1〔ステップ10〕と同様の反応を行うことにより、(R)-1-((R)-2-アミノブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物29)を0.38g(定量的)得た。 [Step 3]
Synthesis of (R) -1-((R) -2-aminobutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1-((R) -2-aminobutanoyl)-was prepared by carrying out the same reaction as in Example 1 [Step 10] using Reference Example Compound 28 (0.47 g, 0.91 mmol). 0.38 g (quantitative) of N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 29) was obtained.
〔ステップ4〕
(R)-1-((R)-2-アセトアミドブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
参考例化合物29(0.091g、0.22mmol)を用いて実施例8〔ステップ3〕と同様の反応を行うことにより、(R)-1-((R)-2-アセトアミドブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物21)を0.085g(85%)得た。 [Step 4]
Synthesis of (R) -1-((R) -2-acetamidobutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1-((R) -2-acetamidobutanoyl)-was prepared by conducting the same reaction as in Example 8 [Step 3] using Reference compound 29 (0.091 g, 0.22 mmol). 0.085 g (85%) of N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 21) was obtained.
(R)-1-((R)-2-アセトアミドブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
参考例化合物29(0.091g、0.22mmol)を用いて実施例8〔ステップ3〕と同様の反応を行うことにより、(R)-1-((R)-2-アセトアミドブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物21)を0.085g(85%)得た。 [Step 4]
Synthesis of (R) -1-((R) -2-acetamidobutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1-((R) -2-acetamidobutanoyl)-was prepared by conducting the same reaction as in Example 8 [Step 3] using Reference compound 29 (0.091 g, 0.22 mmol). 0.085 g (85%) of N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 21) was obtained.
(実施例22) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-(メチルスルホンアミド)ブタノイル)ピペリジン-3-カルボキサミドの合成:
参考例化合物29(0.096g、0.23mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-(メチルスルホンアミド)ブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物22)を0.090g(79%)得た。 Example 22 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 2 [Step 3] using Reference compound 29 (0.096 g, 0.23 mmol). 0.090 g (79%) of (benzyl) -1-((R) -2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 22) was obtained.
参考例化合物29(0.096g、0.23mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-(メチルスルホンアミド)ブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物22)を0.090g(79%)得た。 Example 22 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 2 [Step 3] using Reference compound 29 (0.096 g, 0.23 mmol). 0.090 g (79%) of (benzyl) -1-((R) -2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 22) was obtained.
(実施例23) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-シアノシクロプロパンカルボニル)ピペリジン-3-カルボキサミドの合成:
1-シアノシクロプロパンカルボン酸(0.034g、0.31mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-シアノシクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物23)を0.081g(63%)得た。 Example 23 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-cyanocyclopropanecarbonyl) piperidine-3-carboxamide:
The same reaction as in Example 1 [Step 9] was performed using 1-cyanocyclopropanecarboxylic acid (0.034 g, 0.31 mmol) to give (R) -N- (4-cyano-2- (tri 0.081 g (63%) of fluoromethoxy) benzyl) -1- (1-cyanocyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 23) was obtained.
1-シアノシクロプロパンカルボン酸(0.034g、0.31mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-シアノシクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物23)を0.081g(63%)得た。 Example 23 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-cyanocyclopropanecarbonyl) piperidine-3-carboxamide:
The same reaction as in Example 1 [Step 9] was performed using 1-cyanocyclopropanecarboxylic acid (0.034 g, 0.31 mmol) to give (R) -N- (4-cyano-2- (tri 0.081 g (63%) of fluoromethoxy) benzyl) -1- (1-cyanocyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 23) was obtained.
(実施例24) (R)-1-((R)-2-アセトアミドプロパノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
(R)-1-((R)-2-アミノプロパノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
室温下、参考例化合物8(0.35g、1.1mmol)、(R)-2-((((9H-フルオレン-9-イル)メトキシ)カルボニル)アミノ)プロパン酸(0.37g、1.2mmol)、DIPEA(0.56mL、3.2mmol)のDMF(2.0mL)溶液に、HATU(0.53g、1.4mmol)を加えた。室温下で30分間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;ヘキサン:酢酸エチル=9:1→1:9)で精製し、粗生成物を0.53g得た。室温下、得られた粗生成物(0.53g)のDMF(4.0mL)溶液に、モルホリン(0.36mL、4.1mmol)を加えた。室温下で6時間撹拌した後、反応溶液に、水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、(R)-1-((R)-2-アミノプロパノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物30)を0.29g(2段階収率67%)得た。 Example 24 Synthesis of (R) -1-((R) -2-acetamidopropanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of (R) -1-((R) -2-aminopropanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
Reference Example Compound 8 (0.35 g, 1.1 mmol), (R) -2-((((9H-fluoren-9-yl) methoxy) carbonyl) amino) propanoic acid (0.37 g, 1. 2 mmol), DIPEA (0.56 mL, 3.2 mmol) in DMF (2.0 mL) was added HATU (0.53 g, 1.4 mmol). After stirring at room temperature for 30 minutes, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (Amine silica gel DM1020 manufactured by Fuji Silysia Chemical Ltd., eluent; hexane: ethyl acetate = 9: 1 → 1: 9) to obtain 0.53 g of the crude product. . Morpholine (0.36 mL, 4.1 mmol) was added to a solution of the obtained crude product (0.53 g) in DMF (4.0 mL) at room temperature. After stirring at room temperature for 6 hours, water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (amine silica gel DM1020 manufactured by Fuji Silysia Chemical Ltd., eluent; chloroform: methanol = 99: 1 → 95: 5), and (R) -1-((R) -2-Aminopropanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 30) (0.29 g, 2-step yield 67%) was obtained. It was.
〔ステップ1〕
(R)-1-((R)-2-アミノプロパノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
室温下、参考例化合物8(0.35g、1.1mmol)、(R)-2-((((9H-フルオレン-9-イル)メトキシ)カルボニル)アミノ)プロパン酸(0.37g、1.2mmol)、DIPEA(0.56mL、3.2mmol)のDMF(2.0mL)溶液に、HATU(0.53g、1.4mmol)を加えた。室温下で30分間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;ヘキサン:酢酸エチル=9:1→1:9)で精製し、粗生成物を0.53g得た。室温下、得られた粗生成物(0.53g)のDMF(4.0mL)溶液に、モルホリン(0.36mL、4.1mmol)を加えた。室温下で6時間撹拌した後、反応溶液に、水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、(R)-1-((R)-2-アミノプロパノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物30)を0.29g(2段階収率67%)得た。 Example 24 Synthesis of (R) -1-((R) -2-acetamidopropanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of (R) -1-((R) -2-aminopropanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
Reference Example Compound 8 (0.35 g, 1.1 mmol), (R) -2-((((9H-fluoren-9-yl) methoxy) carbonyl) amino) propanoic acid (0.37 g, 1. 2 mmol), DIPEA (0.56 mL, 3.2 mmol) in DMF (2.0 mL) was added HATU (0.53 g, 1.4 mmol). After stirring at room temperature for 30 minutes, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (Amine silica gel DM1020 manufactured by Fuji Silysia Chemical Ltd., eluent; hexane: ethyl acetate = 9: 1 → 1: 9) to obtain 0.53 g of the crude product. . Morpholine (0.36 mL, 4.1 mmol) was added to a solution of the obtained crude product (0.53 g) in DMF (4.0 mL) at room temperature. After stirring at room temperature for 6 hours, water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (amine silica gel DM1020 manufactured by Fuji Silysia Chemical Ltd., eluent; chloroform: methanol = 99: 1 → 95: 5), and (R) -1-((R) -2-Aminopropanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 30) (0.29 g, 2-step yield 67%) was obtained. It was.
〔ステップ2〕
(R)-1-((R)-2-アセトアミドプロパノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物30(0.10g、0.25mmol)、TEA(0.11mL、0.14mmol)のジクロロメタン(1.0mL)溶液に、無水酢酸(0.032g、0.32mmol)を加えた。氷冷下で5分間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→90:10)で精製し、(R)-1-((R)-2-アセトアミドプロパノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物24)を0.11g(定量的)得た。 [Step 2]
Synthesis of (R) -1-((R) -2-acetamidopropanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
Acetic anhydride (0.032 g, 0.32 mmol) was added to a dichloromethane (1.0 mL) solution of Reference Example Compound 30 (0.10 g, 0.25 mmol) and TEA (0.11 mL, 0.14 mmol) under ice cooling. added. After stirring for 5 minutes under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 90: 10), and (R) -1-((R) -2-acetamidopropanoyl) -N 0.11 g (quantitative) of-(4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 24) was obtained.
(R)-1-((R)-2-アセトアミドプロパノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物30(0.10g、0.25mmol)、TEA(0.11mL、0.14mmol)のジクロロメタン(1.0mL)溶液に、無水酢酸(0.032g、0.32mmol)を加えた。氷冷下で5分間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→90:10)で精製し、(R)-1-((R)-2-アセトアミドプロパノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物24)を0.11g(定量的)得た。 [Step 2]
Synthesis of (R) -1-((R) -2-acetamidopropanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
Acetic anhydride (0.032 g, 0.32 mmol) was added to a dichloromethane (1.0 mL) solution of Reference Example Compound 30 (0.10 g, 0.25 mmol) and TEA (0.11 mL, 0.14 mmol) under ice cooling. added. After stirring for 5 minutes under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 90: 10), and (R) -1-((R) -2-acetamidopropanoyl) -N 0.11 g (quantitative) of-(4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 24) was obtained.
(実施例25) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミドの合成:
参考例化合物30(0.10g、0.25mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物25)を0.099g(83%)得た。 Example 25 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 2 [Step 3] using Reference compound 30 (0.10 g, 0.25 mmol). 0.099 g (83%) of benzyl) -1-((R) -2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 25) was obtained.
参考例化合物30(0.10g、0.25mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-(メチルスルホンアミド)プロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物25)を0.099g(83%)得た。 Example 25 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 2 [Step 3] using Reference compound 30 (0.10 g, 0.25 mmol). 0.099 g (83%) of benzyl) -1-((R) -2- (methylsulfonamido) propanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 25) was obtained.
(実施例26) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-イソブチルアミドシクロプロパンカルボニル)ピペリジン-3-カルボキサミドの合成:
参考例化合物14(0.020g、0.049mmol)、イソブチリルクロリド(0.0062g、0.058mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-イソブチルアミドシクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物26)を0.017g(71%)得た。 Example 26 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-isobutyramidecyclopropanecarbonyl) piperidine-3-carboxamide:
By performing the same reaction as in Example 1 [Step 11] using Reference Example Compound 14 (0.020 g, 0.049 mmol) and isobutyryl chloride (0.0062 g, 0.058 mmol), (R) — 0.017 g (71%) of N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-isobutyramidecyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 26) was obtained. It was.
参考例化合物14(0.020g、0.049mmol)、イソブチリルクロリド(0.0062g、0.058mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-イソブチルアミドシクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物26)を0.017g(71%)得た。 Example 26 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-isobutyramidecyclopropanecarbonyl) piperidine-3-carboxamide:
By performing the same reaction as in Example 1 [Step 11] using Reference Example Compound 14 (0.020 g, 0.049 mmol) and isobutyryl chloride (0.0062 g, 0.058 mmol), (R) — 0.017 g (71%) of N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-isobutyramidecyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 26) was obtained. It was.
(実施例27) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-ピバルアミドシクロプロパンカルボニル)ピペリジン-3-カルボキサミドの合成:
参考例化合物14(0.020g、0.049mmol)、ピバロイルクロリド(0.0064g、0.058mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-ピバルアミドシクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物27)を0.018g(73%)得た。 Example 27 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-pivalamidocyclopropanecarbonyl) piperidine-3-carboxamide:
By performing the same reaction as in Example 1 [Step 11] using Reference Example Compound 14 (0.020 g, 0.049 mmol) and pivaloyl chloride (0.0064 g, 0.058 mmol), (R) — 0.018 g (73%) of N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-pivalamidocyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 27) Obtained.
参考例化合物14(0.020g、0.049mmol)、ピバロイルクロリド(0.0064g、0.058mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-ピバルアミドシクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物27)を0.018g(73%)得た。 Example 27 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-pivalamidocyclopropanecarbonyl) piperidine-3-carboxamide:
By performing the same reaction as in Example 1 [Step 11] using Reference Example Compound 14 (0.020 g, 0.049 mmol) and pivaloyl chloride (0.0064 g, 0.058 mmol), (R) — 0.018 g (73%) of N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1-pivalamidocyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 27) Obtained.
(実施例28) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(4-(メチルスルホンアミド)テトラヒドロ-2H-ピラン-4-カルボニル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
8-オキサ-1,3-ジアザスピロ[4.5]デカン-2,4-ジオンの合成:
室温下、ジヒドロ-2H-ピラン4(3H)-オン(2.0g、20mmol)、炭酸アンモニウム(9.6g、0.10mol)、TEA(2.8mL、20mmol)の水:メタノール(水:メタノール=1:1、60mL)混合溶液に、シアン化カリウム(3.9g、60mmol)を加えた。加熱環流下で48時間撹拌した後、反応溶液を減圧濃縮し、溶媒を半分程度留去し、析出した固体をろ取し、水で洗浄後、乾燥し、8-オキサ-1,3-ジアザスピロ[4.5]デカン-2,4-ジオン(以下、参考例化合物31)を1.4g(41%)得た。また、ろ液に、酸性になるまで濃塩酸を加え、析出した固体をろ取し、水で洗浄後、乾燥し、参考例化合物31を0.80g(24%)得た。 Example 28 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (4- (methylsulfonamido) tetrahydro-2H-pyran-4-carbonyl) piperidine-3- Synthesis of carboxamide:
[Step 1]
Synthesis of 8-oxa-1,3-diazaspiro [4.5] decane-2,4-dione:
Dihydro-2H-pyran 4 (3H) -one (2.0 g, 20 mmol), ammonium carbonate (9.6 g, 0.10 mol), TEA (2.8 mL, 20 mmol) in water: methanol (water: methanol) at room temperature. = 1: 1, 60 mL) Potassium cyanide (3.9 g, 60 mmol) was added to the mixed solution. After stirring for 48 hours under reflux with heating, the reaction solution is concentrated under reduced pressure, the solvent is distilled off by about half, the precipitated solid is collected by filtration, washed with water and dried, and then 8-oxa-1,3-diazaspiro. [4.5] 1.4 g (41%) of decane-2,4-dione (hereinafter referred to as Reference Example Compound 31) was obtained. Concentrated hydrochloric acid was added to the filtrate until acidic, and the precipitated solid was collected by filtration, washed with water and dried to obtain 0.80 g (24%) of Reference Example Compound 31.
〔ステップ1〕
8-オキサ-1,3-ジアザスピロ[4.5]デカン-2,4-ジオンの合成:
室温下、ジヒドロ-2H-ピラン4(3H)-オン(2.0g、20mmol)、炭酸アンモニウム(9.6g、0.10mol)、TEA(2.8mL、20mmol)の水:メタノール(水:メタノール=1:1、60mL)混合溶液に、シアン化カリウム(3.9g、60mmol)を加えた。加熱環流下で48時間撹拌した後、反応溶液を減圧濃縮し、溶媒を半分程度留去し、析出した固体をろ取し、水で洗浄後、乾燥し、8-オキサ-1,3-ジアザスピロ[4.5]デカン-2,4-ジオン(以下、参考例化合物31)を1.4g(41%)得た。また、ろ液に、酸性になるまで濃塩酸を加え、析出した固体をろ取し、水で洗浄後、乾燥し、参考例化合物31を0.80g(24%)得た。 Example 28 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (4- (methylsulfonamido) tetrahydro-2H-pyran-4-carbonyl) piperidine-3- Synthesis of carboxamide:
[Step 1]
Synthesis of 8-oxa-1,3-diazaspiro [4.5] decane-2,4-dione:
Dihydro-2H-pyran 4 (3H) -one (2.0 g, 20 mmol), ammonium carbonate (9.6 g, 0.10 mol), TEA (2.8 mL, 20 mmol) in water: methanol (water: methanol) at room temperature. = 1: 1, 60 mL) Potassium cyanide (3.9 g, 60 mmol) was added to the mixed solution. After stirring for 48 hours under reflux with heating, the reaction solution is concentrated under reduced pressure, the solvent is distilled off by about half, the precipitated solid is collected by filtration, washed with water and dried, and then 8-oxa-1,3-diazaspiro. [4.5] 1.4 g (41%) of decane-2,4-dione (hereinafter referred to as Reference Example Compound 31) was obtained. Concentrated hydrochloric acid was added to the filtrate until acidic, and the precipitated solid was collected by filtration, washed with water and dried to obtain 0.80 g (24%) of Reference Example Compound 31.
〔ステップ2〕
4-((tert-ブトキシカルボニル)アミノ)テトラヒドロ-2H-ピラン-4-カルボン酸の合成:
室温下、参考例化合物31(2.2g、13mmol)の水(30mL)溶液に水酸化カルシウム(3.0g、41mmol)を加えた。加熱環流下で48時間撹拌した後、反応溶液をセライトろ過し、ろ物を熱湯で洗浄した。ろ液を減圧濃縮し、得られた粗生成物を水:1,4-ジオキサン:メタノール(水:1,4-ジオキサン:メタノール=10:10:3、23mL)混合溶液に溶解させた。室温下、反応溶液に、ジ-tert-ブチルジカルボナート(3.4g、16mmol)、水酸化ナトリウム(0.50g、13mmol)を加えた。室温下で15時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物に、希塩酸(15mL)を加え、クロロホルム:メタノール(クロロホルム:メタノール=10:1)混合溶液で抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、4-((tert-ブトキシカルボニル)アミノ)テトラヒドロ-2H-ピラン-4-カルボン酸(以下、参考例化合物32)を2.6g(82%)得た。 [Step 2]
Synthesis of 4-((tert-butoxycarbonyl) amino) tetrahydro-2H-pyran-4-carboxylic acid:
Calcium hydroxide (3.0 g, 41 mmol) was added to a solution of Reference Example Compound 31 (2.2 g, 13 mmol) in water (30 mL) at room temperature. After stirring for 48 hours under heating reflux, the reaction solution was filtered through Celite, and the residue was washed with hot water. The filtrate was concentrated under reduced pressure, and the resulting crude product was dissolved in a mixed solution of water: 1,4-dioxane: methanol (water: 1,4-dioxane: methanol = 10: 10: 3, 23 mL). Di-tert-butyl dicarbonate (3.4 g, 16 mmol) and sodium hydroxide (0.50 g, 13 mmol) were added to the reaction solution at room temperature. After stirring at room temperature for 15 hours, the reaction solution was concentrated under reduced pressure. Dilute hydrochloric acid (15 mL) was added to the resulting crude product, and the mixture was extracted with a mixed solution of chloroform: methanol (chloroform: methanol = 10: 1). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain 2.6 g (82%) of 4-((tert-butoxycarbonyl) amino) tetrahydro-2H-pyran-4-carboxylic acid (hereinafter referred to as Reference Example Compound 32). )Obtained.
4-((tert-ブトキシカルボニル)アミノ)テトラヒドロ-2H-ピラン-4-カルボン酸の合成:
室温下、参考例化合物31(2.2g、13mmol)の水(30mL)溶液に水酸化カルシウム(3.0g、41mmol)を加えた。加熱環流下で48時間撹拌した後、反応溶液をセライトろ過し、ろ物を熱湯で洗浄した。ろ液を減圧濃縮し、得られた粗生成物を水:1,4-ジオキサン:メタノール(水:1,4-ジオキサン:メタノール=10:10:3、23mL)混合溶液に溶解させた。室温下、反応溶液に、ジ-tert-ブチルジカルボナート(3.4g、16mmol)、水酸化ナトリウム(0.50g、13mmol)を加えた。室温下で15時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物に、希塩酸(15mL)を加え、クロロホルム:メタノール(クロロホルム:メタノール=10:1)混合溶液で抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、4-((tert-ブトキシカルボニル)アミノ)テトラヒドロ-2H-ピラン-4-カルボン酸(以下、参考例化合物32)を2.6g(82%)得た。 [Step 2]
Synthesis of 4-((tert-butoxycarbonyl) amino) tetrahydro-2H-pyran-4-carboxylic acid:
Calcium hydroxide (3.0 g, 41 mmol) was added to a solution of Reference Example Compound 31 (2.2 g, 13 mmol) in water (30 mL) at room temperature. After stirring for 48 hours under heating reflux, the reaction solution was filtered through Celite, and the residue was washed with hot water. The filtrate was concentrated under reduced pressure, and the resulting crude product was dissolved in a mixed solution of water: 1,4-dioxane: methanol (water: 1,4-dioxane: methanol = 10: 10: 3, 23 mL). Di-tert-butyl dicarbonate (3.4 g, 16 mmol) and sodium hydroxide (0.50 g, 13 mmol) were added to the reaction solution at room temperature. After stirring at room temperature for 15 hours, the reaction solution was concentrated under reduced pressure. Dilute hydrochloric acid (15 mL) was added to the resulting crude product, and the mixture was extracted with a mixed solution of chloroform: methanol (chloroform: methanol = 10: 1). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain 2.6 g (82%) of 4-((tert-butoxycarbonyl) amino) tetrahydro-2H-pyran-4-carboxylic acid (hereinafter referred to as Reference Example Compound 32). )Obtained.
〔ステップ3〕
(R)-tert-ブチル (4-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)テトラヒドロ-2H-ピラン-4-イル)カルバマートの合成:
参考例化合物32(0.082g、0.34mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-tert-ブチル (4-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)テトラヒドロ-2H-ピラン-4-イル)カルバマート(以下、参考例化合物33)を0.10g(62%)得た。 [Step 3]
Synthesis of (R) -tert-butyl (4- (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-carbonyl) tetrahydro-2H-pyran-4-yl) carbamate:
(R) -tert-butyl (4- (3-((4-cyano) was obtained by conducting the same reaction as in Example 9 [Step 9] using Reference Example Compound 32 (0.082 g, 0.34 mmol). 0.10 g (62%) of -2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-carbonyl) tetrahydro-2H-pyran-4-yl) carbamate (hereinafter referred to as Reference Example Compound 33) was obtained.
(R)-tert-ブチル (4-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)テトラヒドロ-2H-ピラン-4-イル)カルバマートの合成:
参考例化合物32(0.082g、0.34mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-tert-ブチル (4-(3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-カルボニル)テトラヒドロ-2H-ピラン-4-イル)カルバマート(以下、参考例化合物33)を0.10g(62%)得た。 [Step 3]
Synthesis of (R) -tert-butyl (4- (3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-carbonyl) tetrahydro-2H-pyran-4-yl) carbamate:
(R) -tert-butyl (4- (3-((4-cyano) was obtained by conducting the same reaction as in Example 9 [Step 9] using Reference Example Compound 32 (0.082 g, 0.34 mmol). 0.10 g (62%) of -2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-carbonyl) tetrahydro-2H-pyran-4-yl) carbamate (hereinafter referred to as Reference Example Compound 33) was obtained.
〔ステップ4〕
(R)-1-(4-アミノテトラヒドロ-2H-ピランカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
参考例化合物33(0.10g、0.19mmol)を用いて実施例1〔ステップ10〕と同様の反応を行うことにより、(R)-1-(4-アミノテトラヒドロ-2H-ピランカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物34)を0.050g(58%)得た。 [Step 4]
Synthesis of (R) -1- (4-aminotetrahydro-2H-pyrancarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1- (4-Aminotetrahydro-2H-pyrancarbonyl)-was prepared by carrying out the same reaction as in Example 1 [Step 10] using Reference Example Compound 33 (0.10 g, 0.19 mmol). 0.050 g (58%) of N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 34) was obtained.
(R)-1-(4-アミノテトラヒドロ-2H-ピランカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
参考例化合物33(0.10g、0.19mmol)を用いて実施例1〔ステップ10〕と同様の反応を行うことにより、(R)-1-(4-アミノテトラヒドロ-2H-ピランカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物34)を0.050g(58%)得た。 [Step 4]
Synthesis of (R) -1- (4-aminotetrahydro-2H-pyrancarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1- (4-Aminotetrahydro-2H-pyrancarbonyl)-was prepared by carrying out the same reaction as in Example 1 [Step 10] using Reference Example Compound 33 (0.10 g, 0.19 mmol). 0.050 g (58%) of N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 34) was obtained.
〔ステップ5〕
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(4-(メチルスルホンアミド)テトラヒドロ-2H-ピラン-4-カルボニル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物34(0.040g、0.088mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(4-(メチルスルホンアミド)テトラヒドロ-2H-ピラン-4-カルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物28)を0.024g(5.1%)得た。 [Step 5]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (4- (methylsulfonamido) tetrahydro-2H-pyran-4-carbonyl) piperidine-3-carboxamide:
The same reaction as in Example 2 [Step 3] was carried out using Reference Example Compound 34 (0.040 g, 0.088 mmol) under ice cooling to give (R) -N- (4-cyano-2- ( 0.024 g (5.1%) of (trifluoromethoxy) benzyl) -1- (4- (methylsulfonamido) tetrahydro-2H-pyran-4-carbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 28) Obtained.
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(4-(メチルスルホンアミド)テトラヒドロ-2H-ピラン-4-カルボニル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物34(0.040g、0.088mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(4-(メチルスルホンアミド)テトラヒドロ-2H-ピラン-4-カルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物28)を0.024g(5.1%)得た。 [Step 5]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (4- (methylsulfonamido) tetrahydro-2H-pyran-4-carbonyl) piperidine-3-carboxamide:
The same reaction as in Example 2 [Step 3] was carried out using Reference Example Compound 34 (0.040 g, 0.088 mmol) under ice cooling to give (R) -N- (4-cyano-2- ( 0.024 g (5.1%) of (trifluoromethoxy) benzyl) -1- (4- (methylsulfonamido) tetrahydro-2H-pyran-4-carbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 28) Obtained.
(実施例29) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(シクロプロパンカルボキサミド)シクロブタンカルボニル)ピペリジン-3-カルボキサミドの合成:
シクロプロパンカルボニルクロリド(0.0059g、0.057mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(シクロプロパンカルボキサミド)シクロブタンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物29)を0.012g(52%)得た。 Example 29 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (cyclopropanecarboxamido) cyclobutanecarbonyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by carrying out the same reaction as in Example 1 [Step 11] using cyclopropanecarbonyl chloride (0.0059 g, 0.057 mmol). 0.012 g (52%) of benzyl) -1- (1- (cyclopropanecarboxamide) cyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 29) was obtained.
シクロプロパンカルボニルクロリド(0.0059g、0.057mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(シクロプロパンカルボキサミド)シクロブタンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物29)を0.012g(52%)得た。 Example 29 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (cyclopropanecarboxamido) cyclobutanecarbonyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by carrying out the same reaction as in Example 1 [Step 11] using cyclopropanecarbonyl chloride (0.0059 g, 0.057 mmol). 0.012 g (52%) of benzyl) -1- (1- (cyclopropanecarboxamide) cyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 29) was obtained.
(実施例30) (R)-1-((R)-2-アセトアミド-3-ヒドロキシ-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
(R)-2-((tert-ブトキシカルボニル)アミノ)-3-ヒドロキシプロパン酸メチルの合成:
氷冷下、(R)-2-アミノ-3-ヒドロキシプロパン酸メチル(3.0g、19mmol)、TEA(8.1mL、58mmol)のメタノール(50mL)溶液に、ジ-tert-ブチルジカルボナート(4.6g、21mmol)を加えた。室温下で14時間撹拌した後、反応溶液に、希塩酸を加えた。室温下で1時間環撹拌した後、反応溶液に、水を加え、酢酸エチルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液、水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=20:1→2:1)で精製し、(R)-2-((tert-ブトキシカルボニル)アミノ)-3-ヒドロキシプロパン酸メチル(以下、参考例化合物35)を4.1g(97%)得た。 Example 30 (R) -1-((R) -2-Acetamido-3-hydroxy-3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3 -Synthesis of carboxamide:
[Step 1]
Synthesis of methyl (R) -2-((tert-butoxycarbonyl) amino) -3-hydroxypropanoate:
Under ice-cooling, a solution of methyl (R) -2-amino-3-hydroxypropanoate (3.0 g, 19 mmol) and TEA (8.1 mL, 58 mmol) in methanol (50 mL) was added to di-tert-butyl dicarbonate. (4.6 g, 21 mmol) was added. After stirring for 14 hours at room temperature, dilute hydrochloric acid was added to the reaction solution. After stirring at room temperature for 1 hour, water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution and water, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 20: 1 → 2: 1), and (R) -2-((tert-butoxycarbonyl) amino) -3- 4.1 g (97%) of methyl hydroxypropanoate (hereinafter referred to as Reference Example Compound 35) was obtained.
〔ステップ1〕
(R)-2-((tert-ブトキシカルボニル)アミノ)-3-ヒドロキシプロパン酸メチルの合成:
氷冷下、(R)-2-アミノ-3-ヒドロキシプロパン酸メチル(3.0g、19mmol)、TEA(8.1mL、58mmol)のメタノール(50mL)溶液に、ジ-tert-ブチルジカルボナート(4.6g、21mmol)を加えた。室温下で14時間撹拌した後、反応溶液に、希塩酸を加えた。室温下で1時間環撹拌した後、反応溶液に、水を加え、酢酸エチルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液、水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=20:1→2:1)で精製し、(R)-2-((tert-ブトキシカルボニル)アミノ)-3-ヒドロキシプロパン酸メチル(以下、参考例化合物35)を4.1g(97%)得た。 Example 30 (R) -1-((R) -2-Acetamido-3-hydroxy-3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3 -Synthesis of carboxamide:
[Step 1]
Synthesis of methyl (R) -2-((tert-butoxycarbonyl) amino) -3-hydroxypropanoate:
Under ice-cooling, a solution of methyl (R) -2-amino-3-hydroxypropanoate (3.0 g, 19 mmol) and TEA (8.1 mL, 58 mmol) in methanol (50 mL) was added to di-tert-butyl dicarbonate. (4.6 g, 21 mmol) was added. After stirring for 14 hours at room temperature, dilute hydrochloric acid was added to the reaction solution. After stirring at room temperature for 1 hour, water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution and water, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 20: 1 → 2: 1), and (R) -2-((tert-butoxycarbonyl) amino) -3- 4.1 g (97%) of methyl hydroxypropanoate (hereinafter referred to as Reference Example Compound 35) was obtained.
〔ステップ2〕
(S)-tert-ブチル (1,3-ジヒドロキシ-3-メチルブタン-2-イル)カルバメートの合成:
-78℃下、参考例化合物35(4.0g、18mmol)のジエチルエーテル(0.12L)溶液に、メチルマグネシウムブロミドジエチルエーテル溶液(3規定、30mL、91mmol)を加えた。室温下で1時間撹拌した後、反応溶液に、氷冷下、飽和塩化アンモニウム水溶液、水を加え、酢酸エチルで抽出した。有機層を0.1規定希塩酸、水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=10:1→3:1)で精製し、(S)-tert-ブチル (1,3-ジヒドロキシ-3-メチルブタン-2-イル)カルバメート(以下、参考例化合物36)を2.8g(84%)得た。 [Step 2]
Synthesis of (S) -tert-butyl (1,3-dihydroxy-3-methylbutan-2-yl) carbamate:
Methyl magnesium bromide diethyl ether solution (3N, 30 mL, 91 mmol) was added to a diethyl ether (0.12 L) solution of Reference Example compound 35 (4.0 g, 18 mmol) at −78 ° C. After stirring at room temperature for 1 hour, a saturated aqueous ammonium chloride solution and water were added to the reaction solution under ice cooling, and the mixture was extracted with ethyl acetate. The organic layer was washed with 0.1N dilute hydrochloric acid and water, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 10: 1 → 3: 1), and (S) -tert-butyl (1,3-dihydroxy-3-methylbutane- 2.8 g (84%) of 2-yl) carbamate (hereinafter referred to as Reference Example Compound 36) was obtained.
(S)-tert-ブチル (1,3-ジヒドロキシ-3-メチルブタン-2-イル)カルバメートの合成:
-78℃下、参考例化合物35(4.0g、18mmol)のジエチルエーテル(0.12L)溶液に、メチルマグネシウムブロミドジエチルエーテル溶液(3規定、30mL、91mmol)を加えた。室温下で1時間撹拌した後、反応溶液に、氷冷下、飽和塩化アンモニウム水溶液、水を加え、酢酸エチルで抽出した。有機層を0.1規定希塩酸、水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=10:1→3:1)で精製し、(S)-tert-ブチル (1,3-ジヒドロキシ-3-メチルブタン-2-イル)カルバメート(以下、参考例化合物36)を2.8g(84%)得た。 [Step 2]
Synthesis of (S) -tert-butyl (1,3-dihydroxy-3-methylbutan-2-yl) carbamate:
Methyl magnesium bromide diethyl ether solution (3N, 30 mL, 91 mmol) was added to a diethyl ether (0.12 L) solution of Reference Example compound 35 (4.0 g, 18 mmol) at −78 ° C. After stirring at room temperature for 1 hour, a saturated aqueous ammonium chloride solution and water were added to the reaction solution under ice cooling, and the mixture was extracted with ethyl acetate. The organic layer was washed with 0.1N dilute hydrochloric acid and water, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 10: 1 → 3: 1), and (S) -tert-butyl (1,3-dihydroxy-3-methylbutane- 2.8 g (84%) of 2-yl) carbamate (hereinafter referred to as Reference Example Compound 36) was obtained.
〔ステップ3〕
(R)-2-((tert-ブトキシカルボニル)アミノ)-3-ヒドロキシ-3-メチルブタン酸の合成:
35℃下、参考例化合物36(2.8g、13mmol)、2,2,6,6,-テトラメチルピペリジン 1-オキシル(0.40g、2.6mmol)、標準中性リン酸緩衝液(45mL)のアセトニトリル(50mL)溶液に、次亜塩素酸ナトリウム(0.8g、0.64mmol)の水(5.0mL)溶液、亜塩素酸(2.4g、27mmol)の水(10mL)溶液を、同時に別々にゆっくり加えた。35℃下で3時間撹拌した後、反応溶液に、酢酸(1.0mL)を加えた。35℃下で3時間撹拌した後、反応溶液に、水を加え、酢酸エチルで抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物を飽和炭酸水素ナトリウム水溶液に溶解させ、酢酸エチルで洗浄した。水層に、3規定塩酸を加え、酸性にし、酢酸エチルで抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-2-((tert-ブトキシカルボニル)アミノ)-3-ヒドロキシ-3-メチルブタン酸(以下、参考例化合物37)を2.5g(84%)得た。 [Step 3]
Synthesis of (R) -2-((tert-butoxycarbonyl) amino) -3-hydroxy-3-methylbutanoic acid:
Reference compound 36 (2.8 g, 13 mmol), 2,2,6,6, -tetramethylpiperidine 1-oxyl (0.40 g, 2.6 mmol), standard neutral phosphate buffer (45 mL) at 35 ° C. ) In acetonitrile (50 mL), a solution of sodium hypochlorite (0.8 g, 0.64 mmol) in water (5.0 mL), chlorous acid (2.4 g, 27 mmol) in water (10 mL), Slowly added separately at the same time. After stirring at 35 ° C. for 3 hours, acetic acid (1.0 mL) was added to the reaction solution. After stirring at 35 ° C. for 3 hours, water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was dissolved in a saturated aqueous sodium hydrogen carbonate solution and washed with ethyl acetate. The aqueous layer was acidified with 3N hydrochloric acid and extracted with ethyl acetate. The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, concentrated under reduced pressure, and (R) -2-((tert-butoxycarbonyl) amino) -3-hydroxy-3-methylbutanoic acid (hereinafter referred to as “R”). Then, 2.5 g (84%) of Reference Example Compound 37) was obtained.
(R)-2-((tert-ブトキシカルボニル)アミノ)-3-ヒドロキシ-3-メチルブタン酸の合成:
35℃下、参考例化合物36(2.8g、13mmol)、2,2,6,6,-テトラメチルピペリジン 1-オキシル(0.40g、2.6mmol)、標準中性リン酸緩衝液(45mL)のアセトニトリル(50mL)溶液に、次亜塩素酸ナトリウム(0.8g、0.64mmol)の水(5.0mL)溶液、亜塩素酸(2.4g、27mmol)の水(10mL)溶液を、同時に別々にゆっくり加えた。35℃下で3時間撹拌した後、反応溶液に、酢酸(1.0mL)を加えた。35℃下で3時間撹拌した後、反応溶液に、水を加え、酢酸エチルで抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物を飽和炭酸水素ナトリウム水溶液に溶解させ、酢酸エチルで洗浄した。水層に、3規定塩酸を加え、酸性にし、酢酸エチルで抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-2-((tert-ブトキシカルボニル)アミノ)-3-ヒドロキシ-3-メチルブタン酸(以下、参考例化合物37)を2.5g(84%)得た。 [Step 3]
Synthesis of (R) -2-((tert-butoxycarbonyl) amino) -3-hydroxy-3-methylbutanoic acid:
Reference compound 36 (2.8 g, 13 mmol), 2,2,6,6, -tetramethylpiperidine 1-oxyl (0.40 g, 2.6 mmol), standard neutral phosphate buffer (45 mL) at 35 ° C. ) In acetonitrile (50 mL), a solution of sodium hypochlorite (0.8 g, 0.64 mmol) in water (5.0 mL), chlorous acid (2.4 g, 27 mmol) in water (10 mL), Slowly added separately at the same time. After stirring at 35 ° C. for 3 hours, acetic acid (1.0 mL) was added to the reaction solution. After stirring at 35 ° C. for 3 hours, water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was dissolved in a saturated aqueous sodium hydrogen carbonate solution and washed with ethyl acetate. The aqueous layer was acidified with 3N hydrochloric acid and extracted with ethyl acetate. The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, concentrated under reduced pressure, and (R) -2-((tert-butoxycarbonyl) amino) -3-hydroxy-3-methylbutanoic acid (hereinafter referred to as “R”). Then, 2.5 g (84%) of Reference Example Compound 37) was obtained.
〔ステップ4〕
tert-ブチル ((R)-1-((R)-3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-3-ヒドロキシ-3-メチル-1-オキソブタン-2-イル)カルバマートの合成:
室温下、参考例化合物8(0.30g、0.92mmol)、参考例化合物37(0.24g、1.0mmol)、DIPEA(0.35mL、2.0mmol)のDMF(2.0mL)溶液に、HATU(0.42g、1.1mmol)を加えた。室温下で1時間撹拌した後、反応溶液に、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;ヘキサン:酢酸エチル=7:3→4:6)で精製し、tert-ブチル ((R)-1-((R)-3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-3-ヒドロキシ-3-メチル-1-オキソブタン-2-イル)カルバマート(以下、参考例化合物38)を0.44g(89%)得た。 [Step 4]
tert-butyl ((R) -1-((R) -3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-yl) -3-hydroxy-3-methyl-1 Synthesis of -oxobutan-2-yl) carbamate:
To a DMF (2.0 mL) solution of Reference Example Compound 8 (0.30 g, 0.92 mmol), Reference Example Compound 37 (0.24 g, 1.0 mmol) and DIPEA (0.35 mL, 2.0 mmol) at room temperature. , HATU (0.42 g, 1.1 mmol) was added. After stirring at room temperature for 1 hour, 1N hydrochloric acid was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium bicarbonate solution and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (Amine Silica DM1020, manufactured by Fuji Silysia Chemical Ltd., eluent; hexane: ethyl acetate = 7: 3 → 4: 6), and tert-butyl ((R) -1 -((R) -3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-yl) -3-hydroxy-3-methyl-1-oxobutan-2-yl) carbamate ( In the following, 0.44 g (89%) of Reference Example Compound 38) was obtained.
tert-ブチル ((R)-1-((R)-3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-3-ヒドロキシ-3-メチル-1-オキソブタン-2-イル)カルバマートの合成:
室温下、参考例化合物8(0.30g、0.92mmol)、参考例化合物37(0.24g、1.0mmol)、DIPEA(0.35mL、2.0mmol)のDMF(2.0mL)溶液に、HATU(0.42g、1.1mmol)を加えた。室温下で1時間撹拌した後、反応溶液に、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(富士シリシア化学社製アミンシリカゲルDM1020、溶離液;ヘキサン:酢酸エチル=7:3→4:6)で精製し、tert-ブチル ((R)-1-((R)-3-((4-シアノ-2-(トリフルオロメトキシ)ベンジル)カルバモイル)ピペリジン-1-イル)-3-ヒドロキシ-3-メチル-1-オキソブタン-2-イル)カルバマート(以下、参考例化合物38)を0.44g(89%)得た。 [Step 4]
tert-butyl ((R) -1-((R) -3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-yl) -3-hydroxy-3-methyl-1 Synthesis of -oxobutan-2-yl) carbamate:
To a DMF (2.0 mL) solution of Reference Example Compound 8 (0.30 g, 0.92 mmol), Reference Example Compound 37 (0.24 g, 1.0 mmol) and DIPEA (0.35 mL, 2.0 mmol) at room temperature. , HATU (0.42 g, 1.1 mmol) was added. After stirring at room temperature for 1 hour, 1N hydrochloric acid was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium bicarbonate solution and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (Amine Silica DM1020, manufactured by Fuji Silysia Chemical Ltd., eluent; hexane: ethyl acetate = 7: 3 → 4: 6), and tert-butyl ((R) -1 -((R) -3-((4-cyano-2- (trifluoromethoxy) benzyl) carbamoyl) piperidin-1-yl) -3-hydroxy-3-methyl-1-oxobutan-2-yl) carbamate ( In the following, 0.44 g (89%) of Reference Example Compound 38) was obtained.
〔ステップ5〕
(R)-1-((R)-2-アミノ-3-ヒドロキシ-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物38(0.44g、0.82mmol)のジクロロメタン(8.0mL)溶液に、TFA(1.8mL、23mmol)を加えた。室温下で2時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタンに溶解させ、飽和炭酸ナトリウム水溶液で中和し、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-1-((R)-2-アミノ-3-ヒドロキシ-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物39)を0.20g(58%)得た。 [Step 5]
Synthesis of (R) -1-((R) -2-amino-3-hydroxy-3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
TFA (1.8 mL, 23 mmol) was added to a solution of Reference Example Compound 38 (0.44 g, 0.82 mmol) in dichloromethane (8.0 mL) under ice cooling. After stirring at room temperature for 2 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane, neutralized with a saturated aqueous sodium carbonate solution, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and (R) -1-((R) -2-amino-3-hydroxy-3-methylbutanoyl) -N- (4-cyano-2- ( 0.20 g (58%) of (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 39) was obtained.
(R)-1-((R)-2-アミノ-3-ヒドロキシ-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物38(0.44g、0.82mmol)のジクロロメタン(8.0mL)溶液に、TFA(1.8mL、23mmol)を加えた。室温下で2時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタンに溶解させ、飽和炭酸ナトリウム水溶液で中和し、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-1-((R)-2-アミノ-3-ヒドロキシ-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物39)を0.20g(58%)得た。 [Step 5]
Synthesis of (R) -1-((R) -2-amino-3-hydroxy-3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
TFA (1.8 mL, 23 mmol) was added to a solution of Reference Example Compound 38 (0.44 g, 0.82 mmol) in dichloromethane (8.0 mL) under ice cooling. After stirring at room temperature for 2 hours, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane, neutralized with a saturated aqueous sodium carbonate solution, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and (R) -1-((R) -2-amino-3-hydroxy-3-methylbutanoyl) -N- (4-cyano-2- ( 0.20 g (58%) of (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 39) was obtained.
〔ステップ6〕
((R)-1-((R)-2-アセトアミド-3-ヒドロキシ-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物39(0.040g、0.090mmol)、DIPEA(0.035mL、0.20mmol)のジクロロメタン(0.30mL)溶液に、無水酢酸(0.010g、0.10mmol)を加えた。氷冷下で10分間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、((R)-1-((R)-2-アセトアミド-3-ヒドロキシ-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物30)を0.029g(66%)得た。 [Step 6]
Synthesis of ((R) -1-((R) -2-acetamido-3-hydroxy-3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide :
Under ice cooling, acetic anhydride (0.010 g, 0.10 mmol) was added to a solution of Reference Example Compound 39 (0.040 g, 0.090 mmol) and DIPEA (0.035 mL, 0.20 mmol) in dichloromethane (0.30 mL). added. After stirring for 10 minutes under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 95: 5) to obtain ((R) -1-((R) -2-acetamido-3-hydroxy 0.029 g (66%) of -3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 30) was obtained.
((R)-1-((R)-2-アセトアミド-3-ヒドロキシ-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物39(0.040g、0.090mmol)、DIPEA(0.035mL、0.20mmol)のジクロロメタン(0.30mL)溶液に、無水酢酸(0.010g、0.10mmol)を加えた。氷冷下で10分間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、((R)-1-((R)-2-アセトアミド-3-ヒドロキシ-3-メチルブタノイル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物30)を0.029g(66%)得た。 [Step 6]
Synthesis of ((R) -1-((R) -2-acetamido-3-hydroxy-3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide :
Under ice cooling, acetic anhydride (0.010 g, 0.10 mmol) was added to a solution of Reference Example Compound 39 (0.040 g, 0.090 mmol) and DIPEA (0.035 mL, 0.20 mmol) in dichloromethane (0.30 mL). added. After stirring for 10 minutes under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 95: 5) to obtain ((R) -1-((R) -2-acetamido-3-hydroxy 0.029 g (66%) of -3-methylbutanoyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 30) was obtained.
(実施例31) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-3-ヒドロキシ-3-メチル-2-プロピオンアミドブタノイル)ピペリジン-3-カルボキサミドの合成:
参考例化合物39(0.0083g、0.019mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-3-ヒドロキシ-3-メチル-2-プロピオンアミドブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物31)を0.0065g(70%)得た。 Example 31 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -3-hydroxy-3-methyl-2-propionamidobutanoyl) piperidine- Synthesis of 3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by conducting the same reaction as in Example 1 [Step 11] using Reference Example Compound 39 (0.0083 g, 0.019 mmol). As a result, 0.0065 g (70%) of benzyl) -1-((R) -3-hydroxy-3-methyl-2-propionamidobutanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 31) was obtained.
参考例化合物39(0.0083g、0.019mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-3-ヒドロキシ-3-メチル-2-プロピオンアミドブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物31)を0.0065g(70%)得た。 Example 31 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -3-hydroxy-3-methyl-2-propionamidobutanoyl) piperidine- Synthesis of 3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was prepared by conducting the same reaction as in Example 1 [Step 11] using Reference Example Compound 39 (0.0083 g, 0.019 mmol). As a result, 0.0065 g (70%) of benzyl) -1-((R) -3-hydroxy-3-methyl-2-propionamidobutanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 31) was obtained.
(実施例32) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-3-ヒドロキシ-3-メチル-2-(メチルスルホンアミド)ブタノイル)ピペリジン-3-カルボキサミドの合成:
参考例化合物39(0.040g、0.090mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-3-ヒドロキシ-3-メチル-2-(メチルスルホンアミド)ブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物32)を0.038g(81%)得た。 Example 32 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -3-hydroxy-3-methyl-2- (methylsulfonamido) butanoyl) Synthesis of piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 2 [Step 3] using Reference Example Compound 39 (0.040 g, 0.090 mmol). 0.038 g (81%) of (benzyl) -1-((R) -3-hydroxy-3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 32) was obtained. .
参考例化合物39(0.040g、0.090mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-3-ヒドロキシ-3-メチル-2-(メチルスルホンアミド)ブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物32)を0.038g(81%)得た。 Example 32 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -3-hydroxy-3-methyl-2- (methylsulfonamido) butanoyl) Synthesis of piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 2 [Step 3] using Reference Example Compound 39 (0.040 g, 0.090 mmol). 0.038 g (81%) of (benzyl) -1-((R) -3-hydroxy-3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 32) was obtained. .
(実施例33) (R)-1-(1-ブチルアミドシクロブタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
ブチリルクロリド(0.0060g、0.057mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-1-(1-ブチルアミドシクロブタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物33)を0.018g(79%)得た。 Example 33 Synthesis of (R) -1- (1-butylamidocyclobutanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1- (1-Butylamidocyclobutanecarbonyl) -N— (4) was prepared by performing the same reaction as in Example 1 [Step 11] using butyryl chloride (0.0060 g, 0.057 mmol). 0.018 g (79%) of -cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 33) was obtained.
ブチリルクロリド(0.0060g、0.057mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-1-(1-ブチルアミドシクロブタンカルボニル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物33)を0.018g(79%)得た。 Example 33 Synthesis of (R) -1- (1-butylamidocyclobutanecarbonyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
(R) -1- (1-Butylamidocyclobutanecarbonyl) -N— (4) was prepared by performing the same reaction as in Example 1 [Step 11] using butyryl chloride (0.0060 g, 0.057 mmol). 0.018 g (79%) of -cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter, Example Compound 33) was obtained.
(実施例34) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(2-シクロプロピルアセトアミド)シクロブタンカルボニル)ピペリジン-3-カルボキサミドの合成:
参考例化合物10(0.021g、0.049mmol)、2-シクロプロピル酢酸(0.0059g、0.058mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(2-シクロプロピルアセトアミド)シクロブタンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物34)を0.0065g(26%)得た。 Example 34 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (2-cyclopropylacetamido) cyclobutanecarbonyl) piperidine-3-carboxamide:
By performing the same reaction as in Example 1 [Step 9] using Reference Example Compound 10 (0.021 g, 0.049 mmol) and 2-cyclopropylacetic acid (0.0059 g, 0.058 mmol), (R) 0.0065 g of —N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (2-cyclopropylacetamido) cyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 34) (26%) obtained.
参考例化合物10(0.021g、0.049mmol)、2-シクロプロピル酢酸(0.0059g、0.058mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(2-シクロプロピルアセトアミド)シクロブタンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物34)を0.0065g(26%)得た。 Example 34 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (2-cyclopropylacetamido) cyclobutanecarbonyl) piperidine-3-carboxamide:
By performing the same reaction as in Example 1 [Step 9] using Reference Example Compound 10 (0.021 g, 0.049 mmol) and 2-cyclopropylacetic acid (0.0059 g, 0.058 mmol), (R) 0.0065 g of —N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (2-cyclopropylacetamido) cyclobutanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 34) (26%) obtained.
(実施例35) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(2-シクロプロピルアセトアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミドの合成:
参考例化合物14(0.020g、0.049mmol)、2-シクロプロピル酢酸(0.0059g、0.058mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(2-シクロプロピルアセトアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物35)を0.0083g(35%)得た。 Example 35 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (2-cyclopropylacetamido) cyclopropanecarbonyl) piperidine-3-carboxamide:
By performing the same reaction as in Example 1 [Step 9] using Reference Example Compound 14 (0.020 g, 0.049 mmol) and 2-cyclopropylacetic acid (0.0059 g, 0.058 mmol), (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (2-cyclopropylacetamido) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 35) 0083 g (35%) was obtained.
参考例化合物14(0.020g、0.049mmol)、2-シクロプロピル酢酸(0.0059g、0.058mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(1-(2-シクロプロピルアセトアミド)シクロプロパンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物35)を0.0083g(35%)得た。 Example 35 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (2-cyclopropylacetamido) cyclopropanecarbonyl) piperidine-3-carboxamide:
By performing the same reaction as in Example 1 [Step 9] using Reference Example Compound 14 (0.020 g, 0.049 mmol) and 2-cyclopropylacetic acid (0.0059 g, 0.058 mmol), (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (1- (2-cyclopropylacetamido) cyclopropanecarbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 35) 0083 g (35%) was obtained.
(実施例36) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(1H-1,2,4-トリアゾール-1-イル)プロパノイル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
2-メチル-2-(1H-1,2,4-トリアゾール-1-イル)プロパン酸エチルの合成:
室温下、2-ブロモ-2-メチルプロパン酸エチル(1.0g、5.1mmol)のDMF(25mL)溶液に、炭酸セシウム(5.0g、15mmol)、1,2,4-トリアゾール-1-イドナトリウム(0.58g、6.2mmol)を加えた。50℃下で24時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物に、水を加え、クロロホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=7:3→4:6)で精製し、2-メチル-2-(1H-1,2,4-トリアゾール-1-イル)プロパン酸エチル(以下、参考例化合物40)を0.84g(89%)得た。 Example 36 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (1H-1,2,4-triazol-1-yl) Synthesis of propanoyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of ethyl 2-methyl-2- (1H-1,2,4-triazol-1-yl) propanoate:
At room temperature, a solution of ethyl 2-bromo-2-methylpropanoate (1.0 g, 5.1 mmol) in DMF (25 mL) was added to cesium carbonate (5.0 g, 15 mmol), 1,2,4-triazole-1- Id sodium (0.58 g, 6.2 mmol) was added. After stirring at 50 ° C. for 24 hours, the reaction solution was concentrated under reduced pressure. Water was added to the obtained crude product and extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 7: 3 → 4: 6) to give 2-methyl- 0.84 g (89%) of ethyl 2- (1H-1,2,4-triazol-1-yl) propanoate (hereinafter referred to as Reference Example Compound 40) was obtained.
〔ステップ1〕
2-メチル-2-(1H-1,2,4-トリアゾール-1-イル)プロパン酸エチルの合成:
室温下、2-ブロモ-2-メチルプロパン酸エチル(1.0g、5.1mmol)のDMF(25mL)溶液に、炭酸セシウム(5.0g、15mmol)、1,2,4-トリアゾール-1-イドナトリウム(0.58g、6.2mmol)を加えた。50℃下で24時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物に、水を加え、クロロホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=7:3→4:6)で精製し、2-メチル-2-(1H-1,2,4-トリアゾール-1-イル)プロパン酸エチル(以下、参考例化合物40)を0.84g(89%)得た。 Example 36 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (1H-1,2,4-triazol-1-yl) Synthesis of propanoyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of ethyl 2-methyl-2- (1H-1,2,4-triazol-1-yl) propanoate:
At room temperature, a solution of ethyl 2-bromo-2-methylpropanoate (1.0 g, 5.1 mmol) in DMF (25 mL) was added to cesium carbonate (5.0 g, 15 mmol), 1,2,4-triazole-1- Id sodium (0.58 g, 6.2 mmol) was added. After stirring at 50 ° C. for 24 hours, the reaction solution was concentrated under reduced pressure. Water was added to the obtained crude product and extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 7: 3 → 4: 6) to give 2-methyl- 0.84 g (89%) of ethyl 2- (1H-1,2,4-triazol-1-yl) propanoate (hereinafter referred to as Reference Example Compound 40) was obtained.
〔ステップ2〕
2-メチル-2-(1H-1,2,4-トリアゾール-1-イル)プロパン酸ナトリウムの合成:
氷冷下、参考例化合物40(0.65g、3.6mmol)のエタノール(18mL)溶液に、1規定水酸化ナトリウム水溶液(3.9mL、3.9mmol)を加えた。室温下で1時間撹拌した後、反応溶液を減圧濃縮し、2-メチル-2-(1H-1,2,4-トリアゾール-1-イル)プロパン酸ナトリウム(以下、参考例化合物41)を0.67g(定量的)得た。 [Step 2]
Synthesis of sodium 2-methyl-2- (1H-1,2,4-triazol-1-yl) propanoate:
Under ice-cooling, 1N aqueous sodium hydroxide solution (3.9 mL, 3.9 mmol) was added to a solution of Reference Example Compound 40 (0.65 g, 3.6 mmol) in ethanol (18 mL). After stirring at room temperature for 1 hour, the reaction solution was concentrated under reduced pressure, and sodium 2-methyl-2- (1H-1,2,4-triazol-1-yl) propanoate (hereinafter referred to as Reference Example Compound 41) was reduced to 0. Obtained .67 g (quantitative).
2-メチル-2-(1H-1,2,4-トリアゾール-1-イル)プロパン酸ナトリウムの合成:
氷冷下、参考例化合物40(0.65g、3.6mmol)のエタノール(18mL)溶液に、1規定水酸化ナトリウム水溶液(3.9mL、3.9mmol)を加えた。室温下で1時間撹拌した後、反応溶液を減圧濃縮し、2-メチル-2-(1H-1,2,4-トリアゾール-1-イル)プロパン酸ナトリウム(以下、参考例化合物41)を0.67g(定量的)得た。 [Step 2]
Synthesis of sodium 2-methyl-2- (1H-1,2,4-triazol-1-yl) propanoate:
Under ice-cooling, 1N aqueous sodium hydroxide solution (3.9 mL, 3.9 mmol) was added to a solution of Reference Example Compound 40 (0.65 g, 3.6 mmol) in ethanol (18 mL). After stirring at room temperature for 1 hour, the reaction solution was concentrated under reduced pressure, and sodium 2-methyl-2- (1H-1,2,4-triazol-1-yl) propanoate (hereinafter referred to as Reference Example Compound 41) was reduced to 0. Obtained .67 g (quantitative).
〔ステップ3〕
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(1H-1,2,4-トリアゾール-1-イル)プロパノイル)ピペリジン-3-カルボキサミドの合成:
参考例化合物41(0.090g、0.51mmol)を用いて実施例1〔ステップ6〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(1H-1,2,4-トリアゾール-1-イル)プロパノイル)ピペリジン-3(以下、実施例化合物36)を0.12g(54%)得た。 [Step 3]
(R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (1H-1,2,4-triazol-1-yl) propanoyl) piperidine-3 -Synthesis of carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 1 [Step 6] using Reference compound 41 (0.090 g, 0.51 mmol). 0.12 g (54%) of (benzyl) -1- (2-methyl-2- (1H-1,2,4-triazol-1-yl) propanoyl) piperidine-3 (hereinafter, Example Compound 36) was obtained. .
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(1H-1,2,4-トリアゾール-1-イル)プロパノイル)ピペリジン-3-カルボキサミドの合成:
参考例化合物41(0.090g、0.51mmol)を用いて実施例1〔ステップ6〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(1H-1,2,4-トリアゾール-1-イル)プロパノイル)ピペリジン-3(以下、実施例化合物36)を0.12g(54%)得た。 [Step 3]
(R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (1H-1,2,4-triazol-1-yl) propanoyl) piperidine-3 -Synthesis of carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 1 [Step 6] using Reference compound 41 (0.090 g, 0.51 mmol). 0.12 g (54%) of (benzyl) -1- (2-methyl-2- (1H-1,2,4-triazol-1-yl) propanoyl) piperidine-3 (hereinafter, Example Compound 36) was obtained. .
(実施例37) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(1H-ピラゾール-1-イル)プロパノイル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
2-メチル-2-(1H-ピラゾール-1-イル)プロパン酸エチルの合成:
1H-ピラゾール(0.42g、6.2mmol)を用いて実施例36〔ステップ1〕と同様の反応を行うことにより、2-メチル-2-(1H-ピラゾール-1-イル)プロパン酸エチル(以下、参考例化合物42)を0.81g(87%)得た。 Example 37 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (1H-pyrazol-1-yl) propanoyl) piperidine-3- Synthesis of carboxamide:
[Step 1]
Synthesis of ethyl 2-methyl-2- (1H-pyrazol-1-yl) propanoate:
The same reaction as in Example 36 [Step 1] was performed using 1H-pyrazole (0.42 g, 6.2 mmol) to give ethyl 2-methyl-2- (1H-pyrazol-1-yl) propanoate ( In the following, 0.81 g (87%) of Reference Example Compound 42) was obtained.
〔ステップ1〕
2-メチル-2-(1H-ピラゾール-1-イル)プロパン酸エチルの合成:
1H-ピラゾール(0.42g、6.2mmol)を用いて実施例36〔ステップ1〕と同様の反応を行うことにより、2-メチル-2-(1H-ピラゾール-1-イル)プロパン酸エチル(以下、参考例化合物42)を0.81g(87%)得た。 Example 37 (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (1H-pyrazol-1-yl) propanoyl) piperidine-3- Synthesis of carboxamide:
[Step 1]
Synthesis of ethyl 2-methyl-2- (1H-pyrazol-1-yl) propanoate:
The same reaction as in Example 36 [Step 1] was performed using 1H-pyrazole (0.42 g, 6.2 mmol) to give ethyl 2-methyl-2- (1H-pyrazol-1-yl) propanoate ( In the following, 0.81 g (87%) of Reference Example Compound 42) was obtained.
〔ステップ2〕
2-メチル-2-(1H-ピラゾール-1-イル)プロパン酸ナトリウムの合成:
参考例化合物42(0.81g、4.5mmol)を用いて実施例36〔ステップ2〕と同様の反応を行うことにより、2-メチル-2-(1H-ピラゾール-1-イル)プロパン酸ナトリウム(以下、参考例化合物43)を0.80g得た。 [Step 2]
Synthesis of sodium 2-methyl-2- (1H-pyrazol-1-yl) propanoate:
By performing the same reaction as in Example 36 [Step 2] using Reference Example Compound 42 (0.81 g, 4.5 mmol), sodium 2-methyl-2- (1H-pyrazol-1-yl) propanoate 0.80 g of Reference Example Compound 43 was obtained.
2-メチル-2-(1H-ピラゾール-1-イル)プロパン酸ナトリウムの合成:
参考例化合物42(0.81g、4.5mmol)を用いて実施例36〔ステップ2〕と同様の反応を行うことにより、2-メチル-2-(1H-ピラゾール-1-イル)プロパン酸ナトリウム(以下、参考例化合物43)を0.80g得た。 [Step 2]
Synthesis of sodium 2-methyl-2- (1H-pyrazol-1-yl) propanoate:
By performing the same reaction as in Example 36 [Step 2] using Reference Example Compound 42 (0.81 g, 4.5 mmol), sodium 2-methyl-2- (1H-pyrazol-1-yl) propanoate 0.80 g of Reference Example Compound 43 was obtained.
〔ステップ3〕
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(1H-ピラゾール-1-イル)プロパノイル)ピペリジン-3-カルボキサミドの合成:
参考例化合物43(0.090g、0.51mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(1H-ピラゾール-1-イル)プロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物37)を0.16g(68%)得た。 [Step 3]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (1H-pyrazol-1-yl) propanoyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 1 [Step 9] using Reference Example Compound 43 (0.090 g, 0.51 mmol). 0.16 g (68%) of benzyl) -1- (2-methyl-2- (1H-pyrazol-1-yl) propanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 37) was obtained.
(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(1H-ピラゾール-1-イル)プロパノイル)ピペリジン-3-カルボキサミドの合成:
参考例化合物43(0.090g、0.51mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-(2-メチル-2-(1H-ピラゾール-1-イル)プロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物37)を0.16g(68%)得た。 [Step 3]
Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1- (2-methyl-2- (1H-pyrazol-1-yl) propanoyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2- (trifluoromethoxy) was obtained by conducting the same reaction as in Example 1 [Step 9] using Reference Example Compound 43 (0.090 g, 0.51 mmol). 0.16 g (68%) of benzyl) -1- (2-methyl-2- (1H-pyrazol-1-yl) propanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 37) was obtained.
(実施例38) (R)-N-(2,4-ジクロロベンジル)-1-(1-ヒドロキシシクロヘキサンカルボニル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
(R)-tert-ブチル 3-((2,4-ジクロロベンジル)カルバモイル)ピペリジン-1-カルボキシラートの合成:
2,4-ジクロロベンジルアミン(1.5g、8.5mmol)を用いて実施例1〔ステップ6〕と同様の反応を行うことにより、(R)-tert-ブチル 3-((2,4-ジクロロベンジル)カルバモイル)ピペリジン-1-カルボキシラート(以下、参考例化合物44)を2.6g(定量的)得た。 Example 38 Synthesis of (R) -N- (2,4-dichlorobenzyl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of (R) -tert-butyl 3-((2,4-dichlorobenzyl) carbamoyl) piperidine-1-carboxylate:
The same reaction as in Example 1 [Step 6] was performed using 2,4-dichlorobenzylamine (1.5 g, 8.5 mmol) to give (R) -tert-butyl 3-((2,4- 2.6 g (quantitative) of dichlorobenzyl) carbamoyl) piperidine-1-carboxylate (hereinafter referred to as Reference Example Compound 44) was obtained.
〔ステップ1〕
(R)-tert-ブチル 3-((2,4-ジクロロベンジル)カルバモイル)ピペリジン-1-カルボキシラートの合成:
2,4-ジクロロベンジルアミン(1.5g、8.5mmol)を用いて実施例1〔ステップ6〕と同様の反応を行うことにより、(R)-tert-ブチル 3-((2,4-ジクロロベンジル)カルバモイル)ピペリジン-1-カルボキシラート(以下、参考例化合物44)を2.6g(定量的)得た。 Example 38 Synthesis of (R) -N- (2,4-dichlorobenzyl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of (R) -tert-butyl 3-((2,4-dichlorobenzyl) carbamoyl) piperidine-1-carboxylate:
The same reaction as in Example 1 [Step 6] was performed using 2,4-dichlorobenzylamine (1.5 g, 8.5 mmol) to give (R) -tert-butyl 3-((2,4- 2.6 g (quantitative) of dichlorobenzyl) carbamoyl) piperidine-1-carboxylate (hereinafter referred to as Reference Example Compound 44) was obtained.
〔ステップ2〕
(R)-N-(2,4-ジクロロベンジル)ピペリジン-3-カルボキサミドの合成:
参考例化合物44(2.6g、6.7mmol)を用いて実施例1〔ステップ8〕と同様の反応を行うことにより、(R)-N-(2,4-ジクロロベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物45)を1.8g(95%)得た。 [Step 2]
Synthesis of (R) -N- (2,4-dichlorobenzyl) piperidine-3-carboxamide:
(R) -N- (2,4-dichlorobenzyl) piperidine-3-reaction was carried out in the same manner as in Example 1 [Step 8] using Reference Compound 44 (2.6 g, 6.7 mmol). 1.8 g (95%) of carboxamide (hereinafter referred to as Reference Example Compound 45) was obtained.
(R)-N-(2,4-ジクロロベンジル)ピペリジン-3-カルボキサミドの合成:
参考例化合物44(2.6g、6.7mmol)を用いて実施例1〔ステップ8〕と同様の反応を行うことにより、(R)-N-(2,4-ジクロロベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物45)を1.8g(95%)得た。 [Step 2]
Synthesis of (R) -N- (2,4-dichlorobenzyl) piperidine-3-carboxamide:
(R) -N- (2,4-dichlorobenzyl) piperidine-3-reaction was carried out in the same manner as in Example 1 [Step 8] using Reference Compound 44 (2.6 g, 6.7 mmol). 1.8 g (95%) of carboxamide (hereinafter referred to as Reference Example Compound 45) was obtained.
〔ステップ3〕
(R)-N-(2,4-ジクロロベンジル)-1-(1-ヒドロキシシクロヘキサンカルボニル)ピペリジン-3-カルボキサミドの合成:
参考例化合物45(0.10g、0.35mmol)を用いて実施例20と同様の反応を行うことにより、(R)-N-(2,4-ジクロロベンジル)-1-(1-ヒドロキシシクロヘキサンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物38)を0.030g(24%)得た。 [Step 3]
Synthesis of (R) -N- (2,4-dichlorobenzyl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide:
(R) -N- (2,4-dichlorobenzyl) -1- (1-hydroxycyclohexane) was prepared by performing the same reaction as in Example 20 using Reference Example Compound 45 (0.10 g, 0.35 mmol). 0.030 g (24%) of carbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 38) was obtained.
(R)-N-(2,4-ジクロロベンジル)-1-(1-ヒドロキシシクロヘキサンカルボニル)ピペリジン-3-カルボキサミドの合成:
参考例化合物45(0.10g、0.35mmol)を用いて実施例20と同様の反応を行うことにより、(R)-N-(2,4-ジクロロベンジル)-1-(1-ヒドロキシシクロヘキサンカルボニル)ピペリジン-3-カルボキサミド(以下、実施例化合物38)を0.030g(24%)得た。 [Step 3]
Synthesis of (R) -N- (2,4-dichlorobenzyl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide:
(R) -N- (2,4-dichlorobenzyl) -1- (1-hydroxycyclohexane) was prepared by performing the same reaction as in Example 20 using Reference Example Compound 45 (0.10 g, 0.35 mmol). 0.030 g (24%) of carbonyl) piperidine-3-carboxamide (hereinafter, Example Compound 38) was obtained.
(実施例39) ((R)-1-((R)-2-アセトアミド-3-ヒドロキシ-3-メチルブタノイル)-N-(2,4-ジクロロベンジル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
tert-ブチル ((R)-1-((R)-3-(2,4-ジクロロベンジル)カルバモイル)ピペリジン-1-イル)-3-ヒドロキシ-3-メチル-1-オキソブタン-2-イル)カルバマートの合成:
氷冷下、参考例化合物37(0.089g、0.38mmol)、参考例化合物45(0.10g、0.35mmol)、DIPEA(0.20mL、1.1mmol)のDMF(0.70mL)溶液に、HATU(0.16g、0.42mmol)を加えた。室温下で1時間撹拌した後、反応溶液に、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=7:3→4:6)で精製し、tert-ブチル ((R)-1-((R)-3-(2,4-ジクロロベンジル)カルバモイル)ピペリジン-1-イル)-3-ヒドロキシ-3-メチル-1-オキソブタン-2-イル)カルバマート(以下、参考例化合物46)を0.15g(82%)得た。 Example 39 Synthesis of ((R) -1-((R) -2-acetamido-3-hydroxy-3-methylbutanoyl) -N- (2,4-dichlorobenzyl) piperidine-3-carboxamide:
[Step 1]
tert-butyl ((R) -1-((R) -3- (2,4-dichlorobenzyl) carbamoyl) piperidin-1-yl) -3-hydroxy-3-methyl-1-oxobutan-2-yl) Synthesis of carbamate:
Reference Example Compound 37 (0.089 g, 0.38 mmol), Reference Example Compound 45 (0.10 g, 0.35 mmol), DIPEA (0.20 mL, 1.1 mmol) in DMF (0.70 mL) under ice cooling Was added HATU (0.16 g, 0.42 mmol). After stirring at room temperature for 1 hour, 1N hydrochloric acid was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 7: 3 → 4: 6), and tert-butyl ((R) -1-((R) -3- ( 0.15 g (82%) of 2,4-dichlorobenzyl) carbamoyl) piperidin-1-yl) -3-hydroxy-3-methyl-1-oxobutan-2-yl) carbamate (hereinafter referred to as Reference Example Compound 46) was obtained. It was.
〔ステップ1〕
tert-ブチル ((R)-1-((R)-3-(2,4-ジクロロベンジル)カルバモイル)ピペリジン-1-イル)-3-ヒドロキシ-3-メチル-1-オキソブタン-2-イル)カルバマートの合成:
氷冷下、参考例化合物37(0.089g、0.38mmol)、参考例化合物45(0.10g、0.35mmol)、DIPEA(0.20mL、1.1mmol)のDMF(0.70mL)溶液に、HATU(0.16g、0.42mmol)を加えた。室温下で1時間撹拌した後、反応溶液に、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=7:3→4:6)で精製し、tert-ブチル ((R)-1-((R)-3-(2,4-ジクロロベンジル)カルバモイル)ピペリジン-1-イル)-3-ヒドロキシ-3-メチル-1-オキソブタン-2-イル)カルバマート(以下、参考例化合物46)を0.15g(82%)得た。 Example 39 Synthesis of ((R) -1-((R) -2-acetamido-3-hydroxy-3-methylbutanoyl) -N- (2,4-dichlorobenzyl) piperidine-3-carboxamide:
[Step 1]
tert-butyl ((R) -1-((R) -3- (2,4-dichlorobenzyl) carbamoyl) piperidin-1-yl) -3-hydroxy-3-methyl-1-oxobutan-2-yl) Synthesis of carbamate:
Reference Example Compound 37 (0.089 g, 0.38 mmol), Reference Example Compound 45 (0.10 g, 0.35 mmol), DIPEA (0.20 mL, 1.1 mmol) in DMF (0.70 mL) under ice cooling Was added HATU (0.16 g, 0.42 mmol). After stirring at room temperature for 1 hour, 1N hydrochloric acid was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent; hexane: ethyl acetate = 7: 3 → 4: 6), and tert-butyl ((R) -1-((R) -3- ( 0.15 g (82%) of 2,4-dichlorobenzyl) carbamoyl) piperidin-1-yl) -3-hydroxy-3-methyl-1-oxobutan-2-yl) carbamate (hereinafter referred to as Reference Example Compound 46) was obtained. It was.
〔ステップ2〕
氷冷下、参考例化合物46(0.70g、1.7mmol)のジクロロメタン(2.0mL)溶液に、TFA(1.0mL、13mmol)を加えた。室温下で2.5時間撹拌した後、反応溶液に、TFA(1.0mL、13mmol)を加えた。室温下で1時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタンに溶解させ、飽和炭酸ナトリウム水溶液で中和し、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-1-((R)-2-アミノ-3-ヒドロキシ-3-メチルブタノイル)-N-(2,4-ジクロロベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物47)を0.47g(84%)得た。 [Step 2]
TFA (1.0 mL, 13 mmol) was added to a dichloromethane (2.0 mL) solution of Reference Example compound 46 (0.70 g, 1.7 mmol) under ice cooling. After stirring at room temperature for 2.5 hours, TFA (1.0 mL, 13 mmol) was added to the reaction solution. After stirring at room temperature for 1 hour, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane, neutralized with a saturated aqueous sodium carbonate solution, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and (R) -1-((R) -2-amino-3-hydroxy-3-methylbutanoyl) -N- (2,4-dichlorobenzyl) 0.47 g (84%) of piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 47) was obtained.
氷冷下、参考例化合物46(0.70g、1.7mmol)のジクロロメタン(2.0mL)溶液に、TFA(1.0mL、13mmol)を加えた。室温下で2.5時間撹拌した後、反応溶液に、TFA(1.0mL、13mmol)を加えた。室温下で1時間撹拌した後、反応溶液を減圧濃縮した。得られた粗生成物をジクロロメタンに溶解させ、飽和炭酸ナトリウム水溶液で中和し、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、(R)-1-((R)-2-アミノ-3-ヒドロキシ-3-メチルブタノイル)-N-(2,4-ジクロロベンジル)ピペリジン-3-カルボキサミド(以下、参考例化合物47)を0.47g(84%)得た。 [Step 2]
TFA (1.0 mL, 13 mmol) was added to a dichloromethane (2.0 mL) solution of Reference Example compound 46 (0.70 g, 1.7 mmol) under ice cooling. After stirring at room temperature for 2.5 hours, TFA (1.0 mL, 13 mmol) was added to the reaction solution. After stirring at room temperature for 1 hour, the reaction solution was concentrated under reduced pressure. The obtained crude product was dissolved in dichloromethane, neutralized with a saturated aqueous sodium carbonate solution, and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and (R) -1-((R) -2-amino-3-hydroxy-3-methylbutanoyl) -N- (2,4-dichlorobenzyl) 0.47 g (84%) of piperidine-3-carboxamide (hereinafter referred to as Reference Example Compound 47) was obtained.
〔ステップ3〕
((R)-1-((R)-2-アセトアミド-3-ヒドロキシ-3-メチルブタノイル)-N-(2,4-ジクロロベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物47(0.020g、0.050mmol)、TEA(0.014mL、0.099mmol)のジクロロメタン(0.15mL)溶液に、無水酢酸(0.0056g、0.055mmol)を加えた。氷冷下で10分間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、((R)-1-((R)-2-アセトアミド-3-ヒドロキシ-3-メチルブタノイル)-N-(2,4-ジクロロベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物39)を0.021g(96%)得た。 [Step 3]
Synthesis of ((R) -1-((R) -2-acetamido-3-hydroxy-3-methylbutanoyl) -N- (2,4-dichlorobenzyl) piperidine-3-carboxamide:
Acetic anhydride (0.0056 g, 0.055 mmol) was added to a dichloromethane (0.15 mL) solution of Reference Example Compound 47 (0.020 g, 0.050 mmol) and TEA (0.014 mL, 0.099 mmol) under ice cooling. added. After stirring for 10 minutes under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 95: 5) to obtain ((R) -1-((R) -2-acetamido-3-hydroxy 0.021 g (96%) of -3-methylbutanoyl) -N- (2,4-dichlorobenzyl) piperidine-3-carboxamide (hereinafter, Example Compound 39) was obtained.
((R)-1-((R)-2-アセトアミド-3-ヒドロキシ-3-メチルブタノイル)-N-(2,4-ジクロロベンジル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物47(0.020g、0.050mmol)、TEA(0.014mL、0.099mmol)のジクロロメタン(0.15mL)溶液に、無水酢酸(0.0056g、0.055mmol)を加えた。氷冷下で10分間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;クロロホルム:メタノール=99:1→95:5)で精製し、((R)-1-((R)-2-アセトアミド-3-ヒドロキシ-3-メチルブタノイル)-N-(2,4-ジクロロベンジル)ピペリジン-3-カルボキサミド(以下、実施例化合物39)を0.021g(96%)得た。 [Step 3]
Synthesis of ((R) -1-((R) -2-acetamido-3-hydroxy-3-methylbutanoyl) -N- (2,4-dichlorobenzyl) piperidine-3-carboxamide:
Acetic anhydride (0.0056 g, 0.055 mmol) was added to a dichloromethane (0.15 mL) solution of Reference Example Compound 47 (0.020 g, 0.050 mmol) and TEA (0.014 mL, 0.099 mmol) under ice cooling. added. After stirring for 10 minutes under ice cooling, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (eluent: chloroform: methanol = 99: 1 → 95: 5) to obtain ((R) -1-((R) -2-acetamido-3-hydroxy 0.021 g (96%) of -3-methylbutanoyl) -N- (2,4-dichlorobenzyl) piperidine-3-carboxamide (hereinafter, Example Compound 39) was obtained.
(実施例40) (R)-N-(2,4-ジクロロベンジル)-1-((R)-3-ヒドロキシ-3-メチル-2-プロピオンアミドブタノイル)ピペリジン-3-カルボキサミドの合成:
参考例化合物47(0.020g、0.050mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(2,4-ジクロロベンジル)-1-((R)-3-ヒドロキシ-3-メチル-2-プロピオンアミドブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物40)を0.018g(77%)得た。 Example 40 Synthesis of (R) -N- (2,4-dichlorobenzyl) -1-((R) -3-hydroxy-3-methyl-2-propionamidobutanoyl) piperidine-3-carboxamide:
By performing the same reaction as in Example 1 [Step 11] using Reference Compound 47 (0.020 g, 0.050 mmol), (R) -N- (2,4-dichlorobenzyl) -1- ( 0.018 g (77%) of (R) -3-hydroxy-3-methyl-2-propionamidobutanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 40) was obtained.
参考例化合物47(0.020g、0.050mmol)を用いて実施例1〔ステップ11〕と同様の反応を行うことにより、(R)-N-(2,4-ジクロロベンジル)-1-((R)-3-ヒドロキシ-3-メチル-2-プロピオンアミドブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物40)を0.018g(77%)得た。 Example 40 Synthesis of (R) -N- (2,4-dichlorobenzyl) -1-((R) -3-hydroxy-3-methyl-2-propionamidobutanoyl) piperidine-3-carboxamide:
By performing the same reaction as in Example 1 [Step 11] using Reference Compound 47 (0.020 g, 0.050 mmol), (R) -N- (2,4-dichlorobenzyl) -1- ( 0.018 g (77%) of (R) -3-hydroxy-3-methyl-2-propionamidobutanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 40) was obtained.
(実施例41) (R)-N-(2,4-ジクロロベンジル)-1-((R)-3-ヒドロキシ-3-メチル-2-(メチルスルホンアミド)ブタノイル)ピペリジン-3-カルボキサミドの合成:
参考例化合物47(0.020g、0.050mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(2,4-ジクロロベンジル)-1-((R)-3-ヒドロキシ-3-メチル-2-(メチルスルホンアミド)ブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物41)を0.020g(85%)得た。 Example 41 (R) -N- (2,4-dichlorobenzyl) -1-((R) -3-hydroxy-3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide Synthesis:
The same reaction as in Example 2 [Step 3] was carried out using Reference Compound 47 (0.020 g, 0.050 mmol) to give (R) -N- (2,4-dichlorobenzyl) -1- ( 0.020 g (85%) of (R) -3-hydroxy-3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 41) was obtained.
参考例化合物47(0.020g、0.050mmol)を用いて実施例2〔ステップ3〕と同様の反応を行うことにより、(R)-N-(2,4-ジクロロベンジル)-1-((R)-3-ヒドロキシ-3-メチル-2-(メチルスルホンアミド)ブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物41)を0.020g(85%)得た。 Example 41 (R) -N- (2,4-dichlorobenzyl) -1-((R) -3-hydroxy-3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide Synthesis:
The same reaction as in Example 2 [Step 3] was carried out using Reference Compound 47 (0.020 g, 0.050 mmol) to give (R) -N- (2,4-dichlorobenzyl) -1- ( 0.020 g (85%) of (R) -3-hydroxy-3-methyl-2- (methylsulfonamido) butanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 41) was obtained.
(実施例42) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-ヒドロキシプロパノイル)ピペリジン-3-カルボキサミドの合成:
(R)-2-ヒドロキシプロパン酸ナトリウム(0.019g、0.17mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-ヒドロキシプロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物42)を0.045g(74%)得た。 Example 42 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2-hydroxypropanoyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2) was obtained by carrying out the same reaction as in Example 1 [Step 9] using (R) -2-hydroxypropanoic acid sodium salt (0.019 g, 0.17 mmol). 0.045 g (74%) of-(trifluoromethoxy) benzyl) -1-((R) -2-hydroxypropanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 42) was obtained.
(R)-2-ヒドロキシプロパン酸ナトリウム(0.019g、0.17mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-ヒドロキシプロパノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物42)を0.045g(74%)得た。 Example 42 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2-hydroxypropanoyl) piperidine-3-carboxamide:
(R) -N- (4-cyano-2) was obtained by carrying out the same reaction as in Example 1 [Step 9] using (R) -2-hydroxypropanoic acid sodium salt (0.019 g, 0.17 mmol). 0.045 g (74%) of-(trifluoromethoxy) benzyl) -1-((R) -2-hydroxypropanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 42) was obtained.
(実施例43) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-ヒドロキシブタノイル)ピペリジン-3-カルボキサミドの合成:
(R)-2-ヒドロキシブタン酸(0.017g、0.17mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-ヒドロキシブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物43)を0.056g(89%)得た。 Example 43 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2-hydroxybutanoyl) piperidine-3-carboxamide:
(R) -2-N- (4-cyano-2-) is prepared by carrying out the same reaction as in Example 1 [Step 9] using (R) -2-hydroxybutanoic acid (0.017 g, 0.17 mmol). 0.056 g (89%) of (trifluoromethoxy) benzyl) -1-((R) -2-hydroxybutanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 43) was obtained.
(R)-2-ヒドロキシブタン酸(0.017g、0.17mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-ヒドロキシブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物43)を0.056g(89%)得た。 Example 43 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2-hydroxybutanoyl) piperidine-3-carboxamide:
(R) -2-N- (4-cyano-2-) is prepared by carrying out the same reaction as in Example 1 [Step 9] using (R) -2-hydroxybutanoic acid (0.017 g, 0.17 mmol). 0.056 g (89%) of (trifluoromethoxy) benzyl) -1-((R) -2-hydroxybutanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 43) was obtained.
(実施例44) (R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-ヒドロキシ-3-メチルブタノイル)ピペリジン-3-カルボキサミドの合成:
(R)-2-ヒドロキシ-3-メチルブタン酸(0.018g、0.15mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-ヒドロキシ-3-メチルブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物44)を0.042g(64%)得た。 Example 44 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2-hydroxy-3-methylbutanoyl) piperidine-3-carboxamide :
(R) -2-Hydroxy-3-methylbutanoic acid (0.018 g, 0.15 mmol) was used for the same reaction as in Example 1 [Step 9] to give (R) -N- (4-cyano 0.042 g (64%) of 2- (trifluoromethoxy) benzyl) -1-((R) -2-hydroxy-3-methylbutanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 44) was obtained. It was.
(R)-2-ヒドロキシ-3-メチルブタン酸(0.018g、0.15mmol)を用いて実施例1〔ステップ9〕と同様の反応を行うことにより、(R)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)-1-((R)-2-ヒドロキシ-3-メチルブタノイル)ピペリジン-3-カルボキサミド(以下、実施例化合物44)を0.042g(64%)得た。 Example 44 Synthesis of (R) -N- (4-cyano-2- (trifluoromethoxy) benzyl) -1-((R) -2-hydroxy-3-methylbutanoyl) piperidine-3-carboxamide :
(R) -2-Hydroxy-3-methylbutanoic acid (0.018 g, 0.15 mmol) was used for the same reaction as in Example 1 [Step 9] to give (R) -N- (4-cyano 0.042 g (64%) of 2- (trifluoromethoxy) benzyl) -1-((R) -2-hydroxy-3-methylbutanoyl) piperidine-3-carboxamide (hereinafter, Example Compound 44) was obtained. It was.
(比較例1) (R)-N-(5-(tert-ブチル)イソキサゾール-3-イル)-1-(1-ヒドロキシシクロヘキサンカルボニル)ピペリジン-3-カルボキサミドの合成:
〔ステップ1〕
(R)-tert-ブチル 3-((5-(tert-ブチル)イソキサゾール-3-イル)カルバモイル)ピペリジン-1-カルボキシラートの合成:
氷冷下、(R)-1-(tert-ブトキシカルボニル)ピペリジン-3-カルボン酸(0.80g、3.5mmol)のTHF(12mL)溶液に、DMF(2.0mL、26mmol)、オキサリルクロリド(0.33mL、3.7mmol)を内温が5℃を超えないように加えた。氷冷下で30分間撹拌した後、-25℃下、反応溶液に、5-(tert-ブチル)イソキサゾール-3-アミン(0.49g、3.5mmol)、ピリジン(0.71mL、8.7mmol)のTHF(5mL)溶液を内温が-20℃を超えないように加えた。氷冷下で3時間撹拌した後、反応溶液に、飽和塩化ナトリウム水溶液を内温が5℃を超えないように加え、ジクロロメタンで抽出した。有機層を飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=20:1→1:1)で精製し、(R)-tert-ブチル 3-((5-(tert-ブチル)イソキサゾール-3-イル)カルバモイル)ピペリジン-1-カルボキシラート(以下、参考例化合物48)を0.93g(76%)得た。 Comparative Example 1 Synthesis of (R) —N- (5- (tert-butyl) isoxazol-3-yl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of (R) -tert-butyl 3-((5- (tert-butyl) isoxazol-3-yl) carbamoyl) piperidine-1-carboxylate:
Under ice cooling, a solution of (R) -1- (tert-butoxycarbonyl) piperidine-3-carboxylic acid (0.80 g, 3.5 mmol) in THF (12 mL) was added to DMF (2.0 mL, 26 mmol), oxalyl chloride. (0.33 mL, 3.7 mmol) was added so that the internal temperature did not exceed 5 ° C. After stirring for 30 minutes under ice cooling, the reaction solution was added to 5- (tert-butyl) isoxazol-3-amine (0.49 g, 3.5 mmol), pyridine (0.71 mL, 8.7 mmol) at −25 ° C. ) In THF (5 mL) was added so that the internal temperature did not exceed -20 ° C. After stirring for 3 hours under ice cooling, a saturated aqueous sodium chloride solution was added to the reaction solution so that the internal temperature did not exceed 5 ° C., and the mixture was extracted with dichloromethane. The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 20: 1 → 1: 1), and (R) -tert-butyl 3-((5- (tert-butyl) 0.93 g (76%) of isoxazol-3-yl) carbamoyl) piperidine-1-carboxylate (hereinafter referred to as Reference Example Compound 48) was obtained.
〔ステップ1〕
(R)-tert-ブチル 3-((5-(tert-ブチル)イソキサゾール-3-イル)カルバモイル)ピペリジン-1-カルボキシラートの合成:
氷冷下、(R)-1-(tert-ブトキシカルボニル)ピペリジン-3-カルボン酸(0.80g、3.5mmol)のTHF(12mL)溶液に、DMF(2.0mL、26mmol)、オキサリルクロリド(0.33mL、3.7mmol)を内温が5℃を超えないように加えた。氷冷下で30分間撹拌した後、-25℃下、反応溶液に、5-(tert-ブチル)イソキサゾール-3-アミン(0.49g、3.5mmol)、ピリジン(0.71mL、8.7mmol)のTHF(5mL)溶液を内温が-20℃を超えないように加えた。氷冷下で3時間撹拌した後、反応溶液に、飽和塩化ナトリウム水溶液を内温が5℃を超えないように加え、ジクロロメタンで抽出した。有機層を飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=20:1→1:1)で精製し、(R)-tert-ブチル 3-((5-(tert-ブチル)イソキサゾール-3-イル)カルバモイル)ピペリジン-1-カルボキシラート(以下、参考例化合物48)を0.93g(76%)得た。 Comparative Example 1 Synthesis of (R) —N- (5- (tert-butyl) isoxazol-3-yl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide:
[Step 1]
Synthesis of (R) -tert-butyl 3-((5- (tert-butyl) isoxazol-3-yl) carbamoyl) piperidine-1-carboxylate:
Under ice cooling, a solution of (R) -1- (tert-butoxycarbonyl) piperidine-3-carboxylic acid (0.80 g, 3.5 mmol) in THF (12 mL) was added to DMF (2.0 mL, 26 mmol), oxalyl chloride. (0.33 mL, 3.7 mmol) was added so that the internal temperature did not exceed 5 ° C. After stirring for 30 minutes under ice cooling, the reaction solution was added to 5- (tert-butyl) isoxazol-3-amine (0.49 g, 3.5 mmol), pyridine (0.71 mL, 8.7 mmol) at −25 ° C. ) In THF (5 mL) was added so that the internal temperature did not exceed -20 ° C. After stirring for 3 hours under ice cooling, a saturated aqueous sodium chloride solution was added to the reaction solution so that the internal temperature did not exceed 5 ° C., and the mixture was extracted with dichloromethane. The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 20: 1 → 1: 1), and (R) -tert-butyl 3-((5- (tert-butyl) 0.93 g (76%) of isoxazol-3-yl) carbamoyl) piperidine-1-carboxylate (hereinafter referred to as Reference Example Compound 48) was obtained.
〔ステップ2〕
(R)-N-(5-(tert-ブチル)イソキサゾール-3-イル)ピペリジン-3-カルボキサミド塩酸塩の合成:
氷冷下、参考例化合物48(0.85g、2.4mmol)に、4規定塩化水素酢酸エチル溶液(5mL、20mmol)を加えた。室温下で2時間撹拌した後、反応溶液を減圧濃縮し、(R)-N-(5-(tert-ブチル)イソキサゾール-3-イル)ピペリジン-3-カルボキサミド塩酸塩(以下、参考例化合物49)を0.69g(100%)得た。 [Step 2]
Synthesis of (R) -N- (5- (tert-butyl) isoxazol-3-yl) piperidine-3-carboxamide hydrochloride:
Under ice-cooling, 4N hydrogen chloride ethyl acetate solution (5 mL, 20 mmol) was added to Reference Example compound 48 (0.85 g, 2.4 mmol). After stirring at room temperature for 2 hours, the reaction solution was concentrated under reduced pressure to give (R) -N- (5- (tert-butyl) isoxazol-3-yl) piperidine-3-carboxamide hydrochloride (hereinafter referred to as Reference Example Compound 49). ) Was obtained 0.69 g (100%).
(R)-N-(5-(tert-ブチル)イソキサゾール-3-イル)ピペリジン-3-カルボキサミド塩酸塩の合成:
氷冷下、参考例化合物48(0.85g、2.4mmol)に、4規定塩化水素酢酸エチル溶液(5mL、20mmol)を加えた。室温下で2時間撹拌した後、反応溶液を減圧濃縮し、(R)-N-(5-(tert-ブチル)イソキサゾール-3-イル)ピペリジン-3-カルボキサミド塩酸塩(以下、参考例化合物49)を0.69g(100%)得た。 [Step 2]
Synthesis of (R) -N- (5- (tert-butyl) isoxazol-3-yl) piperidine-3-carboxamide hydrochloride:
Under ice-cooling, 4N hydrogen chloride ethyl acetate solution (5 mL, 20 mmol) was added to Reference Example compound 48 (0.85 g, 2.4 mmol). After stirring at room temperature for 2 hours, the reaction solution was concentrated under reduced pressure to give (R) -N- (5- (tert-butyl) isoxazol-3-yl) piperidine-3-carboxamide hydrochloride (hereinafter referred to as Reference Example Compound 49). ) Was obtained 0.69 g (100%).
〔ステップ3〕
(R)-N-(5-(tert-ブチル)イソキサゾール-3-イル)-1-(1-ヒドロキシシクロヘキサンカルボニル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物49(0.30g、1.0mmol)、1-ヒドロキシシクロヘキサンカルボン酸(0.14g、0.95mmol)、DIPEA(0.50mL、2.8mmol)のDMF(7.0mL)溶液に、HATU(0.43g、1.1mmol)を加えた。室温下で14時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=5:1→1:2)で精製し、(R)-N-(5-(tert-ブチル)イソキサゾール-3-イル)-1-(1-ヒドロキシシクロヘキサンカルボニル)ピペリジン-3-カルボキサミド(以下、比較例化合物1)を0.29g(80%)得た。 [Step 3]
Synthesis of (R) -N- (5- (tert-butyl) isoxazol-3-yl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide:
DMF (7.0 mL) of Reference Example Compound 49 (0.30 g, 1.0 mmol), 1-hydroxycyclohexanecarboxylic acid (0.14 g, 0.95 mmol), DIPEA (0.50 mL, 2.8 mmol) under ice-cooling. ) To the solution was added HATU (0.43 g, 1.1 mmol). After stirring at room temperature for 14 hours, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 5: 1 → 1: 2), and (R) -N- (5- (tert-butyl) isoxazole-3- Yl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide (hereinafter, Comparative Compound 1) was obtained in an amount of 0.29 g (80%).
(R)-N-(5-(tert-ブチル)イソキサゾール-3-イル)-1-(1-ヒドロキシシクロヘキサンカルボニル)ピペリジン-3-カルボキサミドの合成:
氷冷下、参考例化合物49(0.30g、1.0mmol)、1-ヒドロキシシクロヘキサンカルボン酸(0.14g、0.95mmol)、DIPEA(0.50mL、2.8mmol)のDMF(7.0mL)溶液に、HATU(0.43g、1.1mmol)を加えた。室温下で14時間撹拌した後、反応溶液に、水、1規定塩酸を加え、ジエチルエーテルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(溶離液;ヘキサン:酢酸エチル=5:1→1:2)で精製し、(R)-N-(5-(tert-ブチル)イソキサゾール-3-イル)-1-(1-ヒドロキシシクロヘキサンカルボニル)ピペリジン-3-カルボキサミド(以下、比較例化合物1)を0.29g(80%)得た。 [Step 3]
Synthesis of (R) -N- (5- (tert-butyl) isoxazol-3-yl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide:
DMF (7.0 mL) of Reference Example Compound 49 (0.30 g, 1.0 mmol), 1-hydroxycyclohexanecarboxylic acid (0.14 g, 0.95 mmol), DIPEA (0.50 mL, 2.8 mmol) under ice-cooling. ) To the solution was added HATU (0.43 g, 1.1 mmol). After stirring at room temperature for 14 hours, water and 1N hydrochloric acid were added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (eluent: hexane: ethyl acetate = 5: 1 → 1: 2), and (R) -N- (5- (tert-butyl) isoxazole-3- Yl) -1- (1-hydroxycyclohexanecarbonyl) piperidine-3-carboxamide (hereinafter, Comparative Compound 1) was obtained in an amount of 0.29 g (80%).
(比較例2) (R)-1-(2-アセトアミドアセチル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミドの合成:
室温下、参考例化合物8(0.10g、0.31mmol)、N-アセチルグリシン(0.036g、0.31mmol)、DIPEA(0.16mL、0.92mmol)のDMF(5mL)溶液にHATU(0.14g、0.37mmol)を加えた。室温下で14時間撹拌した後、氷冷下、反応溶液に、1規定希塩酸を加え、ヘキサン:酢酸エチル(ヘキサン:酢酸エチル=1:2)混合溶媒で抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をジエチルエーテル(1.0mL)に溶解させ、ヘキサン(4.0mL)を加えた。析出したゲル状物質をろ取し、(R)-1-(2-アセトアミドアセチル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、比較例化合物2)を0.040g(31%)得た。 Comparative Example 2 Synthesis of (R) -1- (2-acetamidoacetyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
At room temperature, HATU (5 mL) was added to a solution of Reference Example Compound 8 (0.10 g, 0.31 mmol), N-acetylglycine (0.036 g, 0.31 mmol), DIPEA (0.16 mL, 0.92 mmol). 0.14 g, 0.37 mmol) was added. After stirring at room temperature for 14 hours, 1N dilute hydrochloric acid was added to the reaction solution under ice cooling, and the mixture was extracted with a mixed solvent of hexane: ethyl acetate (hexane: ethyl acetate = 1: 2). The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was dissolved in diethyl ether (1.0 mL), and hexane (4.0 mL) was added. The precipitated gel substance was collected by filtration, and (R) -1- (2-acetamidoacetyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Comparative Example Compound) 0.040 g (31%) of 2) was obtained.
室温下、参考例化合物8(0.10g、0.31mmol)、N-アセチルグリシン(0.036g、0.31mmol)、DIPEA(0.16mL、0.92mmol)のDMF(5mL)溶液にHATU(0.14g、0.37mmol)を加えた。室温下で14時間撹拌した後、氷冷下、反応溶液に、1規定希塩酸を加え、ヘキサン:酢酸エチル(ヘキサン:酢酸エチル=1:2)混合溶媒で抽出した。有機層を水、飽和塩化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた粗生成物をジエチルエーテル(1.0mL)に溶解させ、ヘキサン(4.0mL)を加えた。析出したゲル状物質をろ取し、(R)-1-(2-アセトアミドアセチル)-N-(4-シアノ-2-(トリフルオロメトキシ)ベンジル)ピペリジン-3-カルボキサミド(以下、比較例化合物2)を0.040g(31%)得た。 Comparative Example 2 Synthesis of (R) -1- (2-acetamidoacetyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide:
At room temperature, HATU (5 mL) was added to a solution of Reference Example Compound 8 (0.10 g, 0.31 mmol), N-acetylglycine (0.036 g, 0.31 mmol), DIPEA (0.16 mL, 0.92 mmol). 0.14 g, 0.37 mmol) was added. After stirring at room temperature for 14 hours, 1N dilute hydrochloric acid was added to the reaction solution under ice cooling, and the mixture was extracted with a mixed solvent of hexane: ethyl acetate (hexane: ethyl acetate = 1: 2). The organic layer was washed with water and a saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained crude product was dissolved in diethyl ether (1.0 mL), and hexane (4.0 mL) was added. The precipitated gel substance was collected by filtration, and (R) -1- (2-acetamidoacetyl) -N- (4-cyano-2- (trifluoromethoxy) benzyl) piperidine-3-carboxamide (hereinafter referred to as Comparative Example Compound) 0.040 g (31%) of 2) was obtained.
表1-1~表1-6には、実施例化合物1~44の物性データを示し、表2には、比較例化合物1及び2の物性データを示し、表3-1~表3-5には、参考例化合物1~49の物性データを示した。なお、表中のN.D.は「データなし」を表す。
Tables 1-1 to 1-6 show physical property data of Example compounds 1 to 44, Table 2 shows physical property data of Comparative compounds 1 and 2, and Tables 3-1 to 3-5 The physical property data of Reference Example compounds 1 to 49 are shown in FIG. In the table, N.I. D. Represents “no data”.
1H-NMRデータにおいて、プロトン積分値が整数でないものは、回転異性体の存在などによるものである。
In the 1H-NMR data, the proton integral value that is not an integer is due to the presence of a rotational isomer.
1H-NMRデータ中の溶媒名は、測定に使用した溶媒を示している。また、400 MHzNMRスペクトルは、JNM-AL400型核磁気共鳴装置(日本電子社)を用いて測定した。ケミカルシフトは、テトラメチルシランを基準としてδ(単位:ppm)で表し、シグナルはそれぞれs(一重線)、d(二重線)、t(三重線)、q(四重線)、m(多重線)、brs(幅広)、dd(二重二重線)、dt(二重三重線)、ddd(二重二重二重線)、dq(二重四重線)、td(三重二重線)又はtt(三重三重線)で表した。溶媒は全て市販のものを用いた。ESI-MSスペクトルは、Agilent Technologies 1200 Series、G6130A(AgilentTechnology社)を用いて測定した。
The solvent name in the 1H-NMR data indicates the solvent used for the measurement. The 400 MHz NMR spectrum was measured using a JNM-AL400 type nuclear magnetic resonance apparatus (JEOL Ltd.). The chemical shift is represented by δ (unit: ppm) based on tetramethylsilane, and the signals are s (single line), d (double line), t (triple line), q (quadruple line), m ( Multiple line), brs (wide), dd (double double line), dt (double triple line), ddd (double double line), dq (double quadruple line), td (triple double line) (Multiple line) or tt (triple triple line). All solvents were commercially available. The ESI-MS spectrum was measured using Agilent Technologies 1200 Series, G6130A (Agilent Technology).









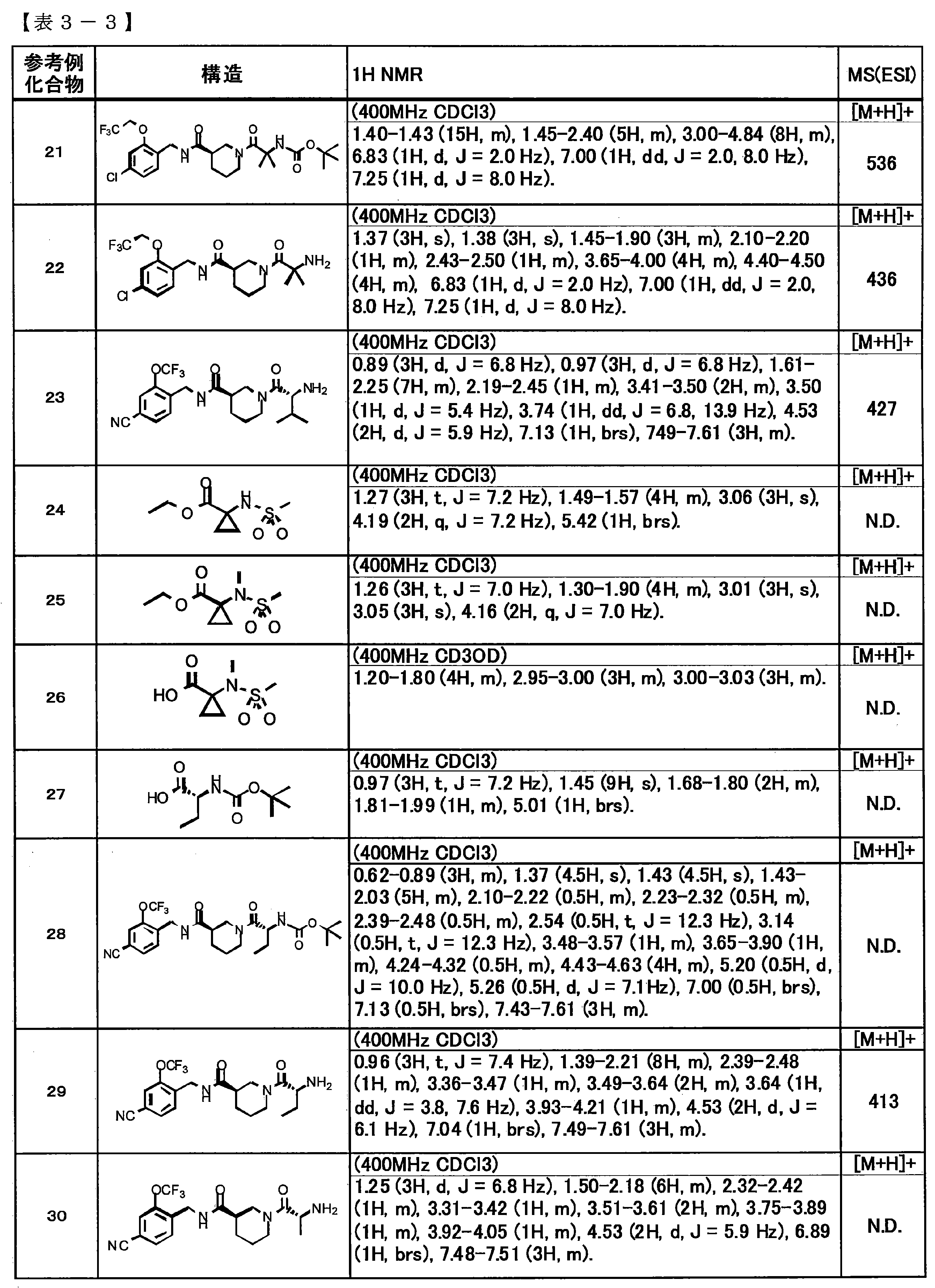


(実施例45) In vitroにおけるsEH阻害活性の評価試験:
公知文献(Analytical Biochemistry、2005年、第343巻、p.66-75)記載の方法に基づき、ヒトsEHを用いて、ニペコチン酸誘導体(I)又はその薬理学的に許容される塩のsEH阻害活性を評価した。 (Example 45) Evaluation test of sEH inhibitory activity in vitro:
Inhibition of sEH of nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof using human sEH based on the method described in known literature (Analytical Biochemistry, 2005, 343, p. 66-75) Activity was evaluated.
公知文献(Analytical Biochemistry、2005年、第343巻、p.66-75)記載の方法に基づき、ヒトsEHを用いて、ニペコチン酸誘導体(I)又はその薬理学的に許容される塩のsEH阻害活性を評価した。 (Example 45) Evaluation test of sEH inhibitory activity in vitro:
Inhibition of sEH of nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof using human sEH based on the method described in known literature (Analytical Biochemistry, 2005, 343, p. 66-75) Activity was evaluated.
リコンビナント・ヒトsEH(最終濃度0.026μg/mL;Cayman社)を、被験化合物とともに、0.1mg/mL BSAを含む25mM Bis-Tris-HCl緩衝液(pH7.0)中にて室温で30分間インキュベートした。その後、蛍光基質としてシアノ(6-メトキシナフタレン-2-イル)メチル 2-(3-フェニルオキシラン-2-イル)アセタート(最終濃度6.25μmol/L;Cayman社)を加え、さらに室温で20分間インキュベートし、ZnSO4(最終濃度0.2mol/L)を加え反応を停止させ、蛍光強度(Fusionα(パッカード社);Excitation:330nm、Emission:485nm)を測定した。
Recombinant human sEH (final concentration 0.026 μg / mL; Cayman) together with the test compound in 25 mM Bis-Tris-HCl buffer (pH 7.0) containing 0.1 mg / mL BSA for 30 minutes at room temperature Incubated. Thereafter, cyano (6-methoxynaphthalen-2-yl) methyl 2- (3-phenyloxiran-2-yl) acetate (final concentration 6.25 μmol / L; Cayman) was added as a fluorescent substrate, and further at room temperature for 20 minutes. After incubation, ZnSO 4 (final concentration 0.2 mol / L) was added to stop the reaction, and the fluorescence intensity (Fusion α (Packard); Excitation: 330 nm, Emission: 485 nm) was measured.
sEH非添加かつ被験化合物非添加の蛍光強度を0%sEH酵素反応率とし、sEH添加かつ被験化合物非添加の蛍光強度を100%sEH酵素反応率として、得られた蛍光強度から、各被験化合物の各sEH酵素反応率を算出し、IC50を求めた。その結果を表4に示す。
The fluorescence intensity with no sEH added and no test compound added was defined as 0% sEH enzyme reaction rate, and the fluorescence intensity with sEH added and no test compound added was defined as 100% sEH enzyme reaction rate. Each sEH enzyme reaction rate was calculated and IC 50 was determined. The results are shown in Table 4.

その結果、実施例化合物1~44は、比較例化合物1及び2と比較して、ヒトsEHの酵素反応に対して非常に強い阻害活性を示した。
As a result, Example compounds 1 to 44 showed a very strong inhibitory activity against the enzymatic reaction of human sEH, as compared with Comparative compounds 1 and 2.
したがって、ニペコチン酸誘導体(I)又はその薬理学的に許容される塩が、ヒトsEHの酵素反応に対して非常に強い阻害活性を示すことが明らかとなった。
Therefore, it has been clarified that the nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof exhibits a very strong inhibitory activity on the enzyme reaction of human sEH.
(実施例46) マウス酢酸ライジングモデルでの薬効評価試験:
侵害受容性疼痛を評価できるマウス酢酸ライジング法を用い、ニペコチン酸誘導体(I)又はその薬理学的に許容される塩の鎮痛作用を検討した。 (Example 46) Drug efficacy evaluation test in mouse acetate rising model:
The analgesic action of nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof was examined using a mouse acetate rising method capable of evaluating nociceptive pain.
侵害受容性疼痛を評価できるマウス酢酸ライジング法を用い、ニペコチン酸誘導体(I)又はその薬理学的に許容される塩の鎮痛作用を検討した。 (Example 46) Drug efficacy evaluation test in mouse acetate rising model:
The analgesic action of nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof was examined using a mouse acetate rising method capable of evaluating nociceptive pain.
マウス酢酸ライジング法は、既存の方法(Endohら、Life Science、1999年、第65巻、p.1685-1694)に準じて行った。ddY系雄性マウス、5~6週齢(日本エスエルシー)を自由飲水摂食下で飼育し、被験化合物溶液又はその溶媒(Vehicle)を経口投与(10mL/kg)した。被験化合物溶液の溶媒としてTween80:メチルセルロース(以下、MC):蒸留水(0.5:0.5:99)を使用した。被験化合物溶液又はその溶媒の投与1時間45分後に0.6%酢酸溶液(10mL/kg)を腹腔内に投与し、ライジング反応(体を伸ばしたり、反らしたりする行動)を誘発させた。酢酸溶液投与10分後から10分間に生じたライジング反応を計数し、その回数を痛みの指標とした。その結果を図1に示す。なお、溶媒を投与した群を「溶媒投与群」、実施例化合物1を30mg/kgの投与量で投与した群を「実施例化合物1投与群」、比較例化合物1を30mg/kgの投与量で投与した群を「比較例化合物1投与群」、及び、比較例化合物2を30mg/kgの投与量で投与した群を「比較例化合物2投与群」とした。
The mouse acetate rising method was performed according to an existing method (Endoh et al., Life Science, 1999, Vol. 65, p. 1685-1694). ddY male mice, 5-6 weeks old (Japan SLC) were bred under free drinking, and the test compound solution or its solvent (Vehicle) was orally administered (10 mL / kg). Tween 80: methyl cellulose (hereinafter referred to as MC): distilled water (0.5: 0.5: 99) was used as a solvent for the test compound solution. One hour and 45 minutes after administration of the test compound solution or its solvent, a 0.6% acetic acid solution (10 mL / kg) was administered intraperitoneally to induce a rising reaction (behavior to stretch or warp the body). Rising reactions that occurred 10 minutes after administration of the acetic acid solution were counted, and the number of times was used as an index of pain. The result is shown in FIG. It should be noted that the group administered with the solvent was the “solvent administered group”, the group administered with Example Compound 1 at a dose of 30 mg / kg was the “Example Compound 1 administered group”, and the comparative compound 1 was administered at a dose of 30 mg / kg. The group administered in 1) was designated as “Comparative Example Compound 1 Administration Group”, and the group administered with Comparative Compound 2 at a dose of 30 mg / kg was designated as “Comparative Example Compound 2 Administration Group”.
図の横軸は各群を示し、縦軸はライジング反応回数(平均値±標準誤差、n=8)を示し、数値が高いほど痛みがあることを表す。図中の*印は、溶媒投与群との比較で統計学的に有意であることを示す(Student’s t検定、p<0.05)。
In the figure, the horizontal axis represents each group, and the vertical axis represents the number of rising reactions (average value ± standard error, n = 8). The higher the value, the more painful. The * mark in the figure indicates that it is statistically significant in comparison with the solvent administration group (Student's t test, p <0.05).
実施例化合物1の30mg/kgの経口投与により、溶媒投与群と比較して有意なライジング反応回数の減少が観察された。一方、比較例化合物1又は比較例化合物2の30mg/kgの経口投与では、溶媒投与群と比較して有意なライジング反応回数の減少作用は認められなかった。以上の結果から、実施例化合物1が、侵害受容性疼痛に対して鎮痛効果を有することが示され、ニペコチン酸誘導体(I)又はその薬理学的に許容される塩が、侵害受容性疼痛に対する鎮痛作用を有することが示された。
When the Example Compound 1 was orally administered at 30 mg / kg, a significant decrease in the number of rising reactions was observed as compared with the solvent administration group. On the other hand, when 30 mg / kg of Comparative Example Compound 1 or Comparative Example Compound 2 was orally administered, no significant reduction in the number of rising reactions was observed compared to the solvent administration group. From the above results, Example Compound 1 was shown to have an analgesic effect on nociceptive pain, and nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof was effective against nociceptive pain. It was shown to have analgesic action.
(実施例47) マウス坐骨神経部分結紮モデルでの薬効評価試験:
神経障害性疼痛を評価できるマウス坐骨神経部分結紮モデル(Seltzerモデル)を用い、ニペコチン酸誘導体(I)又はその薬理学的に許容される塩の鎮痛作用を検討した。 (Example 47) Drug efficacy evaluation test in mouse sciatic nerve partial ligation model:
Using a mouse partial sciatic nerve ligation model (Seltzer model) that can evaluate neuropathic pain, the analgesic action of nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof was examined.
神経障害性疼痛を評価できるマウス坐骨神経部分結紮モデル(Seltzerモデル)を用い、ニペコチン酸誘導体(I)又はその薬理学的に許容される塩の鎮痛作用を検討した。 (Example 47) Drug efficacy evaluation test in mouse sciatic nerve partial ligation model:
Using a mouse partial sciatic nerve ligation model (Seltzer model) that can evaluate neuropathic pain, the analgesic action of nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof was examined.
マウス坐骨神経部分結紮モデルは、Seltzerらの方法(Malmbergら、Pain、1998年、第76巻、p.215-222)に従って作製した。ICR系雄性マウス、5週齢(日本エスエルシー)をイソフルランにて吸入麻酔し、右側後肢大腿部の坐骨神経を露出させ、顕微鏡下で8-0の絹糸(夏目製作所)を用いて坐骨神経を半周だけ強度に三回結紮した群を「結紮群」とし、坐骨神経を露出しただけで、結紮しなかった群を「偽手術群」とした。
The mouse partial sciatic nerve ligation model was prepared according to the method of Seltzer et al. (Malberg et al., Pain, 1998, Vol. 76, p. 215-222). An ICR male mouse, 5 weeks old (Japan SLC), is inhaled by anesthesia with isoflurane to expose the sciatic nerve of the right hind leg thigh, and sciatic nerve using 8-0 silk thread (Natsume Seisakusho) under the microscope A group that was ligated three times in half-strength was called a “ligation group”, and a group that only exposed the sciatic nerve but did not ligate was called a “sham surgery group”.
神経障害性疼痛の評価(以下、von Frey試験)は、金網上に設置した測定用アクリル製ケージ(夏目製作所)内でマウスを最低1時間馴化させた後、0.16gの圧がかかるフィラメント(North Coast Medical, Inc. CA, USA)を用い、両側後肢の足底にフィラメントを3秒間押し当てる機械的触刺激を3秒間隔で3回繰り返し行い、機械的触刺激を加えたときの逃避行動の強度をスコア化(0:無反応、1:刺激に対して緩徐でわずかな逃避行動、2:flinching(足をすばやく連続的に振る行動)やlicking(足舐め行動)を伴わない刺激に対する素早い逃避行動、3:flinching又はlickingを伴う素早い逃避行動)し、その3回のスコアの合計値(以下、von Frey試験総スコア)を痛みの指標とした。von Frey試験は、坐骨神経結紮手術7日後の被験化合物経口投与前にpre値を測定し、von Frey試験総スコアが均一となるように結紮群のマウスを溶媒投与群と被験化合物投与群に群分けした。
Evaluation of neuropathic pain (hereinafter referred to as von Frey test) was performed by acclimating a mouse for at least 1 hour in a measurement acrylic cage (Natsume Seisakusho) installed on a wire mesh and then applying a filament (0.16 g pressure) Using North Coast Medical, Inc. CA, USA), mechanical tactile stimulation that repeatedly presses the filament against the sole of both hind limbs for 3 seconds is repeated 3 times at intervals of 3 seconds, and escape behavior when mechanical tactile stimulation is applied (0: no response, 1: slow and slight escape behavior to the stimulus, 2: quick response to the stimulus without flinching (behavior to shake the foot quickly) or licking (foot licking behavior) Escape behavior, 3: Fast escape behavior with flinching or licking), and the total of the three scores (below) The von Frey test total score) was used as an indicator of pain. In the von Frey test, the pre value was measured before oral administration of the test compound 7 days after the sciatic nerve ligation operation, and the mice in the ligation group were divided into the solvent administration group and the test compound administration group so that the total score of the von Frey test was uniform. Divided.
坐骨神経結紮手術7日後に結紮群のマウスに、被験化合物溶液又はその溶媒(Vehicle)を経口投与(10mL/kg)し、経口投与1時間後及び2時間後にそれぞれvon Frey試験を実施し、その値を鎮痛作用の指標とした。被験化合物溶液の溶媒としてTween80:MC:蒸留水(0.5:0.5:99)を使用した。その結果を図2及び3に示す。なお、溶媒を投与した群を「溶媒投与群」、実施例化合物1を30mg/kgの投与量で投与した群を「実施例化合物1投与群」、比較例化合物1を30mg/kgの投与量で投与した群を「比較例化合物1投与群」、及び、比較例化合物2を30mg/kgの投与量で投与した群を「比較例化合物2投与群」とした。
Seven days after the sciatic nerve ligation surgery, the test compound solution or its solvent (Vehicle) was orally administered (10 mL / kg) to mice in the ligation group, and von Frey tests were performed 1 hour and 2 hours after oral administration, respectively. The value was used as an index of analgesic action. Tween 80: MC: distilled water (0.5: 0.5: 99) was used as a solvent for the test compound solution. The results are shown in FIGS. It should be noted that the group administered with the solvent was the “solvent administered group”, the group administered with Example Compound 1 at a dose of 30 mg / kg was the “Example Compound 1 administered group”, and the comparative compound 1 was administered at a dose of 30 mg / kg. The group administered in 1) was designated as “Comparative Example Compound 1 Administration Group”, and the group administered with Comparative Compound 2 at a dose of 30 mg / kg was designated as “Comparative Example Compound 2 Administration Group”.
図の横軸は各群を示し、縦軸はvon Frey試験総スコア(平均値±標準誤差、n=4~6)を示し、数値が高いほど痛みが強いことを示す。図中の*印は、溶媒投与群との比較で統計学的に有意であることを示す(Student’s t検定、p<0.05)。
In the figure, the horizontal axis shows each group, and the vertical axis shows the von Frey test total score (mean ± standard error, n = 4 to 6). The higher the value, the stronger the pain. The * mark in the figure indicates that it is statistically significant in comparison with the solvent administration group (Student's t test, p <0.05).
実施例化合物1の30mg/kgの経口投与により、経口投与1時間後及び2時間後において溶媒投与群と比較して統計学的に有意なvon Frey試験総スコアの減少作用を示した。一方、比較例化合物1又は比較例化合物2の30mg/kgの経口投与では、溶媒投与群と比較して有意なvon Frey試験総スコアの減少作用は認められなかった。以上の結果から、実施例化合物1が、神経障害性疼痛に対して鎮痛効果を有することが示され、ニペコチン酸誘導体(I)又はその薬理学的に許容される塩が、神経障害性疼痛に対する鎮痛作用を有することが示された。
The oral administration of Example Compound 1 at 30 mg / kg showed a statistically significant von Frey test total score reduction effect 1 hour and 2 hours after oral administration compared to the solvent administration group. On the other hand, when 30 mg / kg of Comparative Example Compound 1 or Comparative Example Compound 2 was orally administered, no significant von Frey test total score reduction effect was observed compared to the solvent administration group. From the above results, Example Compound 1 was shown to have an analgesic effect on neuropathic pain, and nipecotic acid derivative (I) or a pharmacologically acceptable salt thereof was effective against neuropathic pain. It was shown to have analgesic action.
本発明の鎮痛剤は、ニペコチン酸誘導体又はその薬理学的に許容される塩を有効成分とし、sEH阻害活性に基づいて薬効を発揮する、疼痛、特に、侵害受容性疼痛及び/又は神経障害性疼痛の鎮痛剤として用いることができる。
The analgesic of the present invention comprises a nipecotic acid derivative or a pharmacologically acceptable salt thereof as an active ingredient, and exhibits a medicinal effect based on sEH inhibitory activity, in particular nociceptive pain and / or neuropathy. It can be used as an analgesic for pain.
Claims (5)
- 以下の一般式(I)で示されるニペコチン酸誘導体又はその薬理学的に許容される塩を有効成分として含有する、鎮痛剤。
R2及びR3は、それぞれ独立して、水素原子、炭素数1~6のアルキル基又は炭素数2~7のアルキルオキシアルキル基(該アルキル基及びアルキルオキシアルキル基は、1~3個の水素原子が、それぞれ独立して、ハロゲン原子、水酸基又はシアノ基で置換されていてもよい)を表すか、又は、一緒になって-(CH2)l-若しくは-(CH2)m-O-(CH2)n-を表すが、同時に水素原子を表すことはなく、
R4及びR5は、それぞれ独立して、水素原子、ハロゲン原子、シアノ基、炭素数1~6のアルキル基若しくはアルキルオキシ基、炭素数3~6のシクロアルキル基若しくはシクロアルキルオキシ基(該アルキル基、アルキルオキシ基、シクロアルキル基及びシクロアルキルオキシ基は、1~3個の水素原子が、それぞれ独立して、ハロゲン原子で置換されていてもよい)又は-C(=O)NH2を表すが、同時にアルキルオキシ基を表すことはなく、
R6は、水素原子又は炭素数1~6のアルキル基を表し、
R7は、炭素数1~6のアルキル基、炭素数3~6のシクロアルキル基、炭素数2~7のアルキルオキシアルキル基又は炭素数4~7のシクロアルキルアルキル基(該アルキル基、シクロアルキル基、アルキルオキシアルキル基及びシクロアルキルアルキル基は、1~3個の水素原子が、それぞれ独立して、ハロゲン原子、水酸基又はシアノ基で置換されていてもよい)を表し、
lは、2~5の整数を表し、
m及びnは、それぞれ独立して、1又は2を表す。] An analgesic comprising, as an active ingredient, a nipecotic acid derivative represented by the following general formula (I) or a pharmacologically acceptable salt thereof.
R 2 and R 3 are each independently a hydrogen atom, an alkyl group having 1 to 6 carbon atoms or an alkyloxyalkyl group having 2 to 7 carbon atoms (the alkyl group and the alkyloxyalkyl group have 1 to 3 carbon atoms) Each independently represents a halogen atom, a hydroxyl group or a cyano group, or together, — (CH 2 ) 1 — or — (CH 2 ) m —O — (CH 2 ) n —, but not simultaneously representing a hydrogen atom,
R 4 and R 5 each independently represents a hydrogen atom, a halogen atom, a cyano group, an alkyl group or alkyloxy group having 1 to 6 carbon atoms, a cycloalkyl group or cycloalkyloxy group having 3 to 6 carbon atoms In the alkyl group, alkyloxy group, cycloalkyl group and cycloalkyloxy group, 1 to 3 hydrogen atoms may be each independently substituted with a halogen atom) or —C (═O) NH 2 Represents an alkyloxy group at the same time,
R 6 represents a hydrogen atom or an alkyl group having 1 to 6 carbon atoms,
R 7 represents an alkyl group having 1 to 6 carbon atoms, a cycloalkyl group having 3 to 6 carbon atoms, an alkyloxyalkyl group having 2 to 7 carbon atoms, or a cycloalkylalkyl group having 4 to 7 carbon atoms (the alkyl group, An alkyl group, an alkyloxyalkyl group and a cycloalkylalkyl group each represents 1 to 3 hydrogen atoms, each of which may be independently substituted with a halogen atom, a hydroxyl group or a cyano group;
l represents an integer of 2 to 5;
m and n each independently represent 1 or 2. ] - R2及びR3は、それぞれ独立して、水素原子若しくは炭素数1~6のアルキル基を表すか、又は、一緒になって-(CH2)l-を表すが、同時に水素原子を表すことはなく、
R4は、ベンゼン環上の2位の置換基を表し、
R5は、ベンゼン環上の4位の置換基を表す、請求項1記載の鎮痛剤。 R 2 and R 3 each independently represent a hydrogen atom or an alkyl group having 1 to 6 carbon atoms, or together represent — (CH 2 ) 1 —, but simultaneously represent a hydrogen atom. Not,
R 4 represents a substituent at the 2-position on the benzene ring;
The analgesic according to claim 1, wherein R 5 represents a substituent at the 4-position on the benzene ring. - R1は、-N(R6)C(=O)R7又は-N(R6)S(=O)2R7を表し、
R4は、ハロゲン原子又は炭素数1~6のアルキル基若しくはアルキルオキシ基を表し、
R5は、ハロゲン原子、シアノ基又は炭素数1~6のアルキル基若しくはアルキルオキシ基を表し、
R6は、水素原子を表す、請求項1又は2記載の鎮痛剤。 R 1 represents —N (R 6 ) C (═O) R 7 or —N (R 6 ) S (═O) 2 R 7 ,
R 4 represents a halogen atom, an alkyl group having 1 to 6 carbon atoms or an alkyloxy group,
R 5 represents a halogen atom, a cyano group, an alkyl group having 1 to 6 carbon atoms or an alkyloxy group,
The analgesic according to claim 1 or 2, wherein R 6 represents a hydrogen atom. - 侵害受容性疼痛の鎮痛剤である、請求項1~3のいずれか一項記載の鎮痛剤。 The analgesic according to any one of claims 1 to 3, which is an analgesic for nociceptive pain.
- 神経障害性疼痛の鎮痛剤である、請求項1~3のいずれか一項記載の鎮痛剤。
The analgesic according to any one of claims 1 to 3, which is an analgesic for neuropathic pain.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014547193A JPWO2015046405A1 (en) | 2013-09-26 | 2014-09-26 | Painkiller |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-199852 | 2013-09-26 | ||
| JP2013199852 | 2013-09-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015046405A1 true WO2015046405A1 (en) | 2015-04-02 |
Family
ID=52743531
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/075571 WO2015046405A1 (en) | 2013-09-26 | 2014-09-26 | Analgesic |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JPWO2015046405A1 (en) |
| WO (1) | WO2015046405A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111909067A (en) * | 2020-08-28 | 2020-11-10 | 浙江凯普化工有限公司 | Organic total synthesis method of D-penicillamine |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009070497A1 (en) * | 2007-11-28 | 2009-06-04 | Smithkline Beecham Corporation | SEH AND 11 β-HSD1 INHIBITORS AND THEIR USE |
| JP2010521456A (en) * | 2007-03-13 | 2010-06-24 | アレテ セラピューティクス, インコーポレイテッド | Soluble epoxide hydrolase inhibitor |
| JP2011016742A (en) * | 2009-07-07 | 2011-01-27 | Astellas Pharma Inc | Cyclic amino compound, or salt thereof |
| WO2013147161A1 (en) * | 2012-03-29 | 2013-10-03 | 東レ株式会社 | Nipecotic acid derivative and use thereof for medical purposes |
-
2014
- 2014-09-26 WO PCT/JP2014/075571 patent/WO2015046405A1/en active Application Filing
- 2014-09-26 JP JP2014547193A patent/JPWO2015046405A1/en active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010521456A (en) * | 2007-03-13 | 2010-06-24 | アレテ セラピューティクス, インコーポレイテッド | Soluble epoxide hydrolase inhibitor |
| WO2009070497A1 (en) * | 2007-11-28 | 2009-06-04 | Smithkline Beecham Corporation | SEH AND 11 β-HSD1 INHIBITORS AND THEIR USE |
| JP2011016742A (en) * | 2009-07-07 | 2011-01-27 | Astellas Pharma Inc | Cyclic amino compound, or salt thereof |
| WO2013147161A1 (en) * | 2012-03-29 | 2013-10-03 | 東レ株式会社 | Nipecotic acid derivative and use thereof for medical purposes |
Non-Patent Citations (2)
| Title |
|---|
| INCEOGLU BORA ET AL.: "Analgesia mediated by soluble epoxide hydrolase inhibitors is dependent on cAMP", PNAS, vol. 108, no. 12, 2011, pages 5093 - 5097 * |
| XING LI ET AL.: "al.Discovery of Potent Inhibitors of Soluble Epoxide Hydrolase by Combinatorial Library Design and Structure-Based Virtual Screening", J.MED.CHEM., vol. 54, 2011, pages 1211 - 1222 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111909067A (en) * | 2020-08-28 | 2020-11-10 | 浙江凯普化工有限公司 | Organic total synthesis method of D-penicillamine |
| CN111909067B (en) * | 2020-08-28 | 2022-03-15 | 浙江凯普化工有限公司 | Organic total synthesis method of D-penicillamine |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2015046405A1 (en) | 2017-03-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8765783B2 (en) | Pharmaceutical composition for treatment of disease due to vascular constriction or vasodilation | |
| JP3022951B2 (en) | Aroyl-piperidine derivatives | |
| RU2351588C2 (en) | N-phenyl(piperidine-2-yl)methyl-benzamide derivatives, and their application in therapy | |
| JP3099072B2 (en) | (1S, 2S) -1- (4-hydroxyphenyl) -2- (4-hydroxy-4-phenylpiperidin-1-yl) -1-propanol methanesulfonate trihydrate | |
| CA2822453C (en) | Novel morphinans useful as analgesics | |
| US20070015828A1 (en) | Highly selective serotonin and norepinephrine dual reuptake inhibitor and use thereof | |
| JP2012509352A (en) | BACE1 Quinazoline Inhibitors and Methods of Use | |
| JP2013028538A (en) | Novel amide derivative | |
| US20160317479A1 (en) | Method of treating or preventing pain | |
| AU2008323026B2 (en) | Drug active in neuropathic pain | |
| US20030236253A1 (en) | Beta-thioamino acids | |
| WO2013147161A1 (en) | Nipecotic acid derivative and use thereof for medical purposes | |
| US9463187B2 (en) | Methylphenidate derivatives and uses of them | |
| KR101108222B1 (en) | Indazolamides with analgesic activity | |
| TW200948805A (en) | Enol carbamate derivatives as modulators of fatty acid amide hydrolase | |
| EP2475361B1 (en) | N-substituted benzenepropanamides for use in the treatment of pain and inflammation | |
| WO2015046405A1 (en) | Analgesic | |
| JP2012211086A (en) | Curative medicine or preventive medicine of articular rheumatism | |
| RU2347775C2 (en) | Piperidine derivative or its pharmaceutical salt | |
| TWI769493B (en) | Deuterated peptidamide compound and its preparation method and use in medicine | |
| WO2015046404A1 (en) | Therapeutic agent or prophylactic agent for pulmonary hypertension | |
| TW201302736A (en) | Cathepsin C inhibitors | |
| JP2008001596A (en) | Sodium channel inhibitor | |
| AU2011348043B9 (en) | Novel morphinans useful as analgesics | |
| EP2185525B1 (en) | Pyrazole 3,5 carboxylate derivatives preparation and therapeutic application thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2014547193 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14849165 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14849165 Country of ref document: EP Kind code of ref document: A1 |