WO2014193937A1 - The kras variant and response to cancer therapy - Google Patents
The kras variant and response to cancer therapy Download PDFInfo
- Publication number
- WO2014193937A1 WO2014193937A1 PCT/US2014/039763 US2014039763W WO2014193937A1 WO 2014193937 A1 WO2014193937 A1 WO 2014193937A1 US 2014039763 W US2014039763 W US 2014039763W WO 2014193937 A1 WO2014193937 A1 WO 2014193937A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- kras
- variant
- cancer
- cell
- mutation
- Prior art date
Links
- 0 CCC(C1)C(C*)C*11C[C@](C)C(CC)C1 Chemical compound CCC(C1)C(C*)C*11C[C@](C)C(CC)C1 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/106—Pharmacogenomics, i.e. genetic variability in individual responses to drugs and drug metabolism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/156—Polymorphic or mutational markers
Definitions
- This disclosure relates generally to the fields of cancer and molecular biology.
- the disclosure provides methods for predicting a subject's sensitivity or resistance to therapeutic agents used to treat cancer by determining the presence or absence of a genetic marker.
- the disclosure provides methods for prognosing a subject based at least in part upon the subject's predicted response to treatment.
- the KRAS Variant is a SNP located in the let-7 complementary site 6 (LCS6) of the 3' UTR of the KRAS gene in which the nucleotide at position 4 of the LCS6 undergoes a uracil (U) or thymine (T) to guanine (G) transition.
- the KRAS Variant provides superior predictive power over known germ-line mutations used as cancer biomarkers in the art because the presence of the KRAS Variant is predictive of the response of a cancer cell of any type or subtype to a particular cancer treatment or therapeutic agent.
- Variant within a cancer cell indicates resistance to known agents including, but not limited to, cisplatin ((5P-4-2)-diamminedichloridoplatinum, also known as cisplatinum, or cis- diamminedichloroplatinum(II) (CDDP)), carboplatin (czs-diammine(cyclobutane-l,l- dicarboxylate-0,O platinum(II) also known as cis-Diammine(l,l- cyclobutanedicarboxylato)platinum(II)), gefitinib (N-(3-chloro-4-fluoro-phenyl)-7-methoxy- 6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine), and a combination therapy of cetuximab (monoclonal antibody) and irinotecan ((5)-4,l l-diethyl-3,4,12,14-tetra
- Cancer and cancer cells of the disclosure include, but are not limited to those cells derived from a(n) AIDS-related cancer, breast cancer, cancer of the digestive/gastrointestinal tract, anal cancer, appendix cancer, bile duct cancer, colon cancer, colorectal cancer, esophageal cancer, gallbladder cancer, islet cell tumors, pancreatic neuroendocrine tumors, liver cancer, pancreatic cancer, rectal cancer, small intestine cancer, stomach (gastric) cancer, endocrine system cancer, adrenocortical carcinoma, parathyroid cancer, pheochromocytoma, pituitary tumor, thyroid cancer, eye cancer, intraocular melanoma, retinoblastoma, bladder cancer, kidney (renal cell) cancer, penile cancer, prostate cancer, transitional cell renal pelvis and ureter cancer, testicular cancer, urethral cancer, Wilms' tumor, other childhood kidney tumors, germ cell cancer, central nervous system cancer, extracranial germ cell tumor,
- the tumor or cancer is metastatic.
- a non-cancer and/or a cancer cells may be evaluated in vivo, in vitro, or ex vivo to determine the presence or absence of thr KRAS variant. Because the KRAS variant is a germline mutation, the KRAS variant mutation is present and detectable in any cell of the subject, including non-cancer as well as cancer cells. Alternatively, or in addition, DNA isolated from any cell in the body of the subject may be evaluated for the presence or absence of the KRAS variant.
- non-cancer and/or cancer cells may be evaluated in vivo, in vitro, or ex vivo.
- the cell may be obtained from a subject.
- the subject has cancer.
- the subject may be of any age. In certain embodiments of this method the subject is at least 30, 35, 40, 45, 50, 55, 60, 65, 70, 75 years of age, or any age in between.
- the cell When a non-cancer and/or cancer cell is evaluated in vitro, the cell may be isolated, reproduced, cloned or derived from an established cell lines, including, but not limited to, a cancer cell line included in the NCI-60 panel.
- cancer cells may be isolated, reproduced, cloned or derived from any cancer cell line, including, but not limited to, those cell lines that carry the KRAS- variant, either alone, or in combination with a second or additional mutation in KRAS or another gene.
- the methods described herein may be applied to subjects of all ages, in certain embodiments of this method, the subject is a newborn, child, adult, or senior (aged 65 or above). Although the methods described herein may be applied to subjects of both sexes, male and female, in certain embodiments of this method, the subject is female. Alternatively, the subject may be male. Female subjects may be either pre-menopausal or post-menopausal. Post-menopausal subjects may be aged 52 years or more. In this disclosure the terms subject and patient are used interchangeably.
- survival rate may be an overall survival rate (for instance, some examples, include, but are not limited to, survival rates calculated from the time of cancer development or diagnosis until the subject succumbs to the cancer
- the terms "resistant” or “sensitive” may be used as relative terms when compared to a known or control value or compared to a measurement from a control subject.
- Control subjects may include healthy individuals and those women who have EOC, but who do not carry the KRAS-variant.
- the control subject can be a national average based upon the expected survival of women born in the same year as the test subject, or who belong to the same generation as the test subject. In a preferred embodiment, this control value does not include those individuals who carry the KRAS-variant.
- the survival rate is an overall survival rate (for instance, some examples, include, but are not limited to, survival rates calculated from the time of cancer development or diagnosis until the subject succumbs to the cancer (death), enters remission, or a doctor declares the subject cured or clean of all cancer cells), five-year survival rate or one-year survival. Shorter survival periods are calculated, for instance, from either the development or diagnosis of the cancer until a determined time, such as one or five years.
- the KRAS variant confers resistance to platinum-based chemotherapy
- the disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to platinum-based chemotherapy.
- treatment of cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to platinum-based chemotherapy.
- treatment of epithelial ovarian cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to platinum-based chemotherapy.
- the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR ofKRAS.
- the methods of the disclosure can be applied to all subjects, in certain embodiments of these methods, the subject is a woman. Although the methods of the disclosure can be applied to subjects of all ages, in certain embodiments of these methods, the subject is post-menopausal or 52 years of age or older.
- the methods described herein may be used to optimize the treatment of epithelial ovarian cancer (EOC) by assaying for the presence of the KRAS Variant and, if present, predicting resistance to platinum-based chemotherapy.
- EOC epithelial ovarian cancer
- the methods described herein may be used to optimize the treatment of epithelial ovarian cancer (EOC) in a female, post-menopausal subject by assaying for the presence of the KRAS Variant and, if present, predicting resistance to platinum-based chemotherapy.
- EOC epithelial ovarian cancer
- the disclosure provides a method of predicting the response of an epithelial ovarian cancer (EOC) cell to a platinum-based chemotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and wherein the presence of the mutation indicates a resistance to platinum-based chemotherapy.
- the EOC cell may be evaluated in vitro or ex vivo. When the EOC cell is evaluated ex vivo, the cell is obtained from a subject.
- the subject may be of any age, however, in a preferred embodiment, the subject is either post-menopausal or at least 52 years old. Alternatively, in the same embodiment, the subject is at least 30, 35, 40, 45, 50, 55, 60, 65, 70, 75 years of age, or any age in between. In other aspects of this method, the subject is not postmenopausal, but presents a similar hormonal profile due to a second medical condition or medical treatment.
- An exemplary, but non-limiting menopausal hormonal profile includes decreased levels of estrogen and progesterone hormone, as determined by, for instance, assessment of a sample of the subject's blood or urine.
- Exemplary, but non-limiting, secondary medical conditions that induce a menopausal hormonal profile are surgical removal of at least one ovary (ovariectomy, also known as surgical menopause), cervical, uterine or ovarian cancer that necessitates a hysterectomy (especially if removal of the uterus is combined with removal of the Fallopian tubes and one or both ovaries).
- Exemplary, but non-limiting, secondary medical conditions that induce a menopausal hormonal profile are chemotherapy and anti-estrogen treatments.
- the cell When the EOC cell is evaluated in vitro, the cell is isolated, reproduced, or derived from the BGl, CAOV3, or IGR-OVl cell lines. These cell lines are non-limiting examples of ovarian cancer cell lines.
- An EOC cell may be isolated, reproduced, or derived from any ovarian cancer cell line, including, but not limited to, those cell lines that carry the KRAS- variant, a deleterious BRCAI mutation, a deleterious BRCA2 mutation, or any combination thereof.
- a deleterious BRCAI or BRCA2 mutation is a mutation that increases the risk or likelihood that it's carrier will develop cancer, and, in preferred embodiments, breast or ovarian cancer.
- a deleterious BRCAI or BRCA2 mutation is a mutation that also increases the risk or likelihood that it's carrier will develop cancer at a younger age (i.e. experience an earlier onset of cancer), and, in preferred embodiments, the cancer is breast or ovarian cancer.
- treatment of recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to platinum-based chemotherapy.
- the platinum-based chemotherapy is cisplatin, however, the methods may be applied to any platinum-based chemotherapy.
- the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR of KRAS.
- R/M recurrent and/or metastatic head and neck squamous cell carcinoma
- FSSCC head and neck squamous cell carcinoma
- the disclosure provides a method of predicting the response of head and neck squamous cell carcinoma (HNSCC ) cell and/or a recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) cell to a platinum-based chemotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and wherein the presence of the mutation indicates a resistance to platinum-based chemotherapy.
- HNSCC head and neck squamous cell carcinoma
- R/M metastatic head and neck squamous cell carcinoma
- the disclosure provides a method of predicting the response of head and neck squamous cell carcinoma (HNSCC ) cell and/or a recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) cell to a monoclonal anti-EGFR antibody therapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and wherein the presence of the mutation indicates sensitivity or an enhanced response to monoclonal anti-EGFR antibody therapy, either as a monotherapy or as a combined therapy with another chemotherapeutic agent or medical procedure (e.g. radiation or surgery).
- HNSCC head and neck squamous cell carcinoma
- R/M metastatic head and neck squamous cell carcinoma
- the HNSCC cell may be evaluated in vivo, in vitro or ex vivo. When the HNSCC cell is evaluated ex vivo, the cell is obtained from a subject.
- the preferred platinum-based chemotherapy is cisplatin, carboplatin or paclitaxel, however, the term "platinum-based chemotherapy” encompasses all chemotherapy agent that incorporate platinum or a platinum salt to treat or prevent cancer. In certain aspects of these methods, the platinum-based chemotherapy is an adjuvant therapy. Therefore, the methods described herein predict a patient's response to the use of a platinum-based chemotherapy as either a monotherapy or a combination therapy with other anti-cancer agents or techniques (e.g. radiation and surgery, for example).
- the KRAS variant confers resistance to small molecule EGFR inhibitors
- the disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors).
- treatment of cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors).
- treatment of non-small cell lung cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors and small molecule EGFR inhibitors).
- the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR ofKRAS.
- chemotherapeutic agents that inhibit an activity of EGFR include, but are not limited to, gefinitib and erlotinib.
- chemotherapeutic agents that inhibit an activity of EGFR include, but are not limited to, gefinitib.
- the KRAS variant confers sensitivity to MAP kinase pathway inhibitors
- the disclosure provides a method of treating a cancer patient, the method comprising administering a MAP kinase pathway inhibitor to a patient after a determination that the patient has a mutation in let-7 complementary site LCS6 of human KRAS, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine transition at position 4 of LCS6.
- the disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors).
- treatment of cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors).
- treatment of non-small cell lung cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors).
- the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR ofKRAS.
- chemotherapeutic agents that inhibit an activity of the MAP kinase pathway include, but are not limited to, inhibitors of tyrosine protein kinases and serine/threonine kinases.
- tyrosine protein kinases include the VEGFR (vascular endothelial growth factor receptor) and PDGFR (platelet-derived growth factor receptor).
- VEGFR vascular endothelial growth factor receptor
- PDGFR platelet-derived growth factor receptor
- serine/threonine protein kinases include C-raf (encoded by the RAFl gene) and B-raf (encoded by the BRAF gene).
- Chemotherapeutic agents that inhibit an activity of the MAP kinase pathway include, but are not limited to, sorafenib.
- the KRAS variant confers sensitivity to monoclonal anti-EGFR antibody monotherapy
- the disclosure provides a method of treating a cancer patient, the method comprising administering a MAP kinase pathway inhibitor to a patient after a determination that the patient has a mutation in let-7 complementary site LCS6 of human KRAS, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine transition at position 4 of LCS6.
- the MAP kinase pathway inhibitor is a
- the disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to monoclonal anti- EGFR antibody therapy.
- treatment of cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting sensitivity to monoclonal anti-EGFR antibody therapy.
- treatment of colorectal cancer (CRC) or metastatic CRC (mCRC) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting sensitivity to monoclonal anti-EGFR antibody therapy.
- the KRAS variant is defined as a SNP comprising a uracil (U) or thymine
- the disclosure provides a method of predicting the response of a cancer cell to a monoclonal anti-EGFR antibody monotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, wherein the presence of the mutation indicates a sensitivity to monoclonal anti-EGFR antibody monotherapy.
- the cancer cell is a colorectal cancer (CRC) cell.
- the cancer cell may be evaluated in vitro or ex vivo.
- a non- limiting example of the monoclonal antibody monotherapy is Cetuximab.
- the disclosure provides a method of predicting the response of a cancer cell to a monoclonal anti-EGFR antibody monotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 (the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 (the
- KRAS Variant KRAS Variant
- RASSFIA hypermethylation of RASSFIA
- the cancer cell is a colorectal cancer (CRC) cell.
- CRC colorectal cancer
- the cancer cell may be evaluated in vitro or ex vivo.
- a non- limiting example of the monoclonal anti-EGFR antibody monotherapy is Cetuximab.
- KRAS Variant in combination with (epi)genetic alterations in genes from the Ras pathway, induce senescence cancer cells, including, but not limited to, early-stage colorectal cancer cells (CRC cells), thereby enhancing or promoting survival of the subject.
- CRC cells early-stage colorectal cancer cells
- treatment of head and neck squamous cell carcinoma (FINSCC ) or a recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting sensitivity to monoclonal anti-EGFR antibody therapy.
- the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR of KRAS.
- the disclosure provides a method of predicting the response of a head and neck squamous cell carcinoma (FiNSCC ) cell to a monoclonal anti-EGFR antibody monotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, wherein the presence of the mutation indicates a sensitivity to monoclonal anti-EGFR antibody monotherapy.
- the head and neck squamous cell carcinoma (FiNSCC ) cell may be evaluated in vivo, in vitro or ex vivo.
- a non-limiting example of the monoclonal anti-EGFR antibody monotherapy is Cetuximab.
- EGF a potent ligand of EGFR
- cetuximab is beneficial for these patients because the anti- EGFR antibody blocks the pro-growth signal provided by an upregulation of EGF in these tumors.
- MACF1 is upregulated in our current analysis. MACF1 interacts with ErbB2 to control microtubule capture during cell migration.
- EGFR-specific growth stimulatory ligand EGF
- SYNJ2 pro-migratory phosphatase
- DST, MACF1, EML6 various components of the microtubule/cytoskeletal architecture
- methods of the disclosure include detection of the KRAS variant in combination with detection of upregulation of EGF, wherein the combined presence of the KRAS variant and upregulated EGF mRNA or protein indicates sensitivity of the cancer cell to a monoclonal anti-EGFR antibody therapy.
- the KRAS variant confers resistance to a combinatorial therapy comprised of a monoclonal anti-EGFR antibody and a chemotherapeutic agent.
- the disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to a combinatorial therapy comprised of a monoclonal anti-EGFR antibody and a chemotherapeutic agent.
- treatment of cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to a combinatorial therapy comprised of a monoclonal anti-EGFR antibody and a chemotherapeutic agent.
- treatment of colorectal cancer (CRC) or metastatic CRC (mCRC) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to a combinatorial therapy comprised of a monoclonal anti-EGFR antibody and a chemotherapeutic agent.
- the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR of KRAS.
- the disclosure provides a method of predicting the response of a cancer cell to the combination of a chemotherapy and monoclonal anti-EGFR antibody therapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and wherein the presence of the mutation indicates a resistance to the combination.
- the cancer cell is a colorectal cancer (CRC) cell.
- the cancer cell may be evaluated in vitro or ex vivo.
- a non- limiting example of the monoclonal anti-EGFR antibody monotherapy is Cetuximab.
- the chemotherapy may be a cytotoxic agent.
- a non-limiting example of the cytotoxic agent is irinotecan.
- a chemotherapeutic agent e.g. irinotecan
- reporter expression is compared following irinitecan exposure in KRAS-variant versus non-variant cancer cells, no change was found in expression of the wild-type 3 'UTR reporter.
- a statistically-significant increase in expression in the KRAS- variant 3'UTR reporter was discovered ( Figures 14A and 14B). The data indicates that irinotecan exposure changes the cellular context in a manner that activates the KRAS- variant allele.
- Methods of the disclosure encompass any monoclonal anti-EGFR antibody used to treat or prevent cancer.
- the monoclonal anti-EGFR antibody is in part or entirely human (e.g. chimeric) or humanized.
- Methods of the disclosure encompass any
- the chemotherapy or cytotoxic chemotherapy is an adjuvant therapy.
- Methods of the disclosure predict the response of a cancer cell or a subject having a cancer cell to treatment, including monotherapies or combination therapies.
- the methods of the disclosure predict the response of a cancer cell or a subject having a cancer cell to treatment with a monoclonal anti-EGFR antibody and/or a chemotherapy used in conjunction with an additional medical procedure. Additional medical procedures include, but are not limited to, radiation and surgery.
- Figure 2 is a graph showing that the KRAS variant is associated with suboptimal debulking after neoadjuvant chemotherapy. Surgical debulking after neoadjuvant
- Figure 3A is a signature of 50 differentially expression gene candidates in KRAS variant (KV) triple-negative breast tumors (TNBC KRAS Signature) that shows higher scores in KV EOC samples than in non- variant samples.
- KV KRAS variant
- TNBC KRAS Signature triple-negative breast tumors
- Figure 3B is a signature of genes associated with KRAS-addicted tumors (KRAS Addiction Signature), which are upregulated in KV EOC tumors.
- Figure 3C a signature of differential expression of the top 20 genes in KV EOC tumors, reflecting a re-analysis of differential gene expression in carboplatin-sensitive and carboplatin-resistant EOC cells.
- Figure 3D is a heat map of the top differentially expressed genes between KV (dark gray) and non- variant (light gray) tumor samples.
- the color key depicts a spectrum from blue
- FIG. 4 is a graph showing that the KRAS variant confers resistance to carboplatin and carboplatin/taxol chemotherapy in cell lines.
- Cell lines with the KRAS variant (BGl) and without the KRAS variant (CAOV3) were treated with chemotherapy and half-maximal inhibitory concentration (IC50) is shown on the Y axis, and chemotherapeutic agent on the X axis. Higher IC50 represents resistance to the tested chemotherapeutic agent.
- BGl KRAS variant/ ⁇ CA wild-type cell line
- CAOV3 non-variant/ ⁇ CA wild-type cell line
- IGR-OV1 KRAS-vanant/BRCAl mutant cell line. Error bars are RSE.
- Figure 5 A is a graph showing decreased cell survival in the KRAS- variant line, BGl (*P ⁇ 0.001), with no effect on the non-variant line, CAOV3.
- Figure 5B is a graph showing decreased KRAS protein expression in BGl (right) concordant with the decrease in cell survival, with no effect on CAOV3 (left).
- siRNAs are denoted by numbers.
- Figure 6 is a graph depicting Cell lines with the KRAS variant (BG-1 and IGROV1) have significantly lower levels of let-7b compared to a non- variant cell line (CaOV3).
- Figure 7A-B is a schematic depicting an alignment of the KRAS- variant sequence with non-variant sequences.
- Panel A depicts a non-variant sequence of KRAS.
- Panel B depicts exemplary variant siRNA oligos targeted to the KRAS-yaxiant sequence.
- the underlined sequence depicts the let- 7 binding site.
- the boxed nucleotide represents either the wild type (non- variant) nucleotide (A) or the KRAS variant single nucleotide polymorphism (B). siRNAs are shown starting with their 3' end.
- Figure 8A is a graph depicting the median progression free survival according to the KRAS LCS6 genotype status in patients treated with anti-EGFR mAbs monotherapy or in combination with chemotherapy as salvage treatment.
- Figure 8B is a graph depicting the median overall survival according to the KRAS LCS6 genotype status in patients treated with anti-EGFR mAbs monotherapy or in combination with chemotherapy as salvage treatment.
- Figure 9A is a graph depicting the median progression-free survival according to the KRAS LCS6 genotype status in all patients treated with anti-EGFR mAbs monotherapy as salvage treatment.
- Figure 9B is a graph depicting the median progression-free survival according to the KRAS LCS6 genotype status in all patients treated with anti-EGFR mAbs based combination chemotherapy as salvage treatment.
- Figure 9C is a graph depicting the median progression-free survival according to type of therapy in all KRAS variant carriers.
- Figure 9D is a graph depicting the median progression-free survival according to type of therapy in all non-KRAS variant carriers.
- Figure 1 OA is a graph depicting the median progression-free survival according to the KRAS LCS6 genotype status in the double (KRAS and BRAF) wt patients' population treated with anti-EGFR mAbs monotherapy as salvage treatment.
- Figure 1 OB is a graph depicting the median progression-free survival according to the
- KRAS LCS6 genotype status in the double (KRAS and BRAF) wt patients' population treated with anti-EGFR mAbs based combination chemotherapy as salvage treatment.
- Figure IOC is a graph depicting the Median progression- free survival according to type of therapy in the double (KRAS and BRAF) wt KRAS variant carriers
- Figure 10D is a graph depicting the Median progression- free survival according to type of therapy in the double (KRAS and BRAF) wt non-KRAS variant carriers.
- Figure 11 A is a graph depicting the median overall survival according to the KRAS
- LCS6 genotype status in all patients treated with anti-EGFR mAbs monotherapy as salvage treatment.
- Figure 1 IB is a graph depicting the median overall survival according to the KRAS LCS6 genotype status in all patients treated with anti-EGFR mAbs based combination chemotherapy as salvage treatment.
- Figure 11C is a graph depicting the median overall survival according to type of therapy in all KRAS variant carriers.
- Figure 1 ID is a graph depicting the median overall survival according to type of therapy in all non-KRAS variant carriers.
- Figure 12A is a graph depicting the median overall survival according to the KRAS LCS6 genotype status in the double (KRAS and BRAF) wt patients' population treated with anti-EGFR mAbs monotherapy as salvage treatment.
- Figure 12B is a graph depicting the median overall survival according to the KRAS
- LCS6 genotype status in the double (KRAS and BRAF) wt patients' population treated with anti-EGFR mAbs based combination chemotherapy as salvage treatment.
- Figure 12C is a graph depicting the median overall survival according to type of therapy in the double (KRAS and BRAF) wt KRAS variant carriers.
- Figure 12D is a graph depicting the median overall survival according to type of therapy in the double (KRAS and BRAF) wt non- KRAS variant carriers.
- Figure 13A is a graph depicting the median progression-free survival according to type of therapy in the KRAS and BRAF mutated KRAS variant carriers.
- Figure 13B is a graph depicting the median progression-free survival according to type of therapy in the KRAS and BRAF mutated non-KRAS variant carriers.
- Figure 13C is a graph depicting the median overall survival according to type of therapy in the KRAS and BRAF mutated KRAS variant carriers.
- Figure 13D is a graph depicting the median overall survival according to type of therapy in the KRAS and BRAF mutated non- KRAS variant carriers.
- Figure 14A is a graph depicting the normalized luciferase expression in wild type
- KRAS and KRAS- variant cancer cells following treatment with the chemotherapeutic agent irinotecan.
- Figure 14B is a graph depicting the fold repression (expressed as KRAS
- variant/KRAS wild type as a function of irinotecan concentration, when cancer cells are treated with irinotecan.
- Figure 15A-B is a pair of graphs depicting the overall survival (OS) of non-small cell lung cancer (NSCLC) patients following treatment with either Vandetanib (A, left panel) or Sorafenib (B, right panel).
- OS overall survival
- NSCLC non-small cell lung cancer
- Figure 16 is a graph depicting progression free survival (PFS) of non-small cell lung cancer (NSCLC) patients treated with sorafenib, including both carriers and non-carriers of the KRAS variant.
- PFS progression free survival
- NSCLC non-small cell lung cancer
- Figure 17A-F is a series of graphs depicting Kaplan-Meier survival plots by KRAS- variant status (variant TG/GG vs. non-variant TT).
- HN0501 phase II trial of docetaxel+bortezomib in R/M HNSCC patients
- B overall survival from HN0501 (phase II trial of docetaxel+bortezomib in R/M HNSCC patients)
- C Progression-free survival from E5397 (randomized phase III trial of cisplatin+placebo vs. cisplatin+cetuximab in R/M HNSCC patients)
- D overall survival from E5397 (randomized phase III trial of cisplatin+placebo vs. cisplatin+cetuximab in R/M HNSCC patients)
- E Progression-free survival by KRAS- variant status (variant TG/GG vs.
- non-variant TT non-variant TT
- E5397 treatment arms cisplatin+placebo vs. cisplatin+cetuximab
- F overall survival by KRAS- variant status (variant TG/GG vs. non- variant TT) and E5397 treatment arms
- Figure 18 A-C is a series of graphs depicting MTS growth assays of KRAS variant and non-variant HNSCC cell lines treated with cisplatin.
- Figure 19 is a heatmap of a gene expression profile that is differentially expressed between AT ⁇ S-variant (TG/GG) and non-variant (TT) in 22 HNSCC tumors with available microarray data.
- Figure 20 is a graph depicting MEK inhibition in colorectal cancer cell lines.
- Figure 21 is a schematic and a graph showing that MEK inhibitor specificially target
- Figure 22 is a schematic and a graph showing Biomarker integrated Approaches of Targeted Therapy of Lung Cancer Elimination.
- microRNAs are an abundant class of highly conserved, endogenous, non- coding, small RNA molecules, 18-25 nucleotides in length, which negatively regulate gene expression by binding to partially complementary sites in the 3 '-untranslated region (UTR) of their target mRNAs.
- miRNAs Upon processing by Dicer and Drosha RNase III endonucleases, mature miRNAs can suppress mRNA translation by directing an RNA-induced silencing complex to the target mRNA.
- MiRNAs regulate of a number of genes involved in basic biological processes such as proliferation, cellular differentiation and apoptosis, and act as important players in cancer development and progression by behaving either as oncogenes or as tumor suppressors. Although more than 700 miRNA sequences have been recognized in the human genome to date, this number is expected to double. Furthermore, each miRNA can control hundreds of genes by regulating many mRNAs simultaneously.
- MiRNA binding to mRNAs is critical for the regulation process of mRNA levels and subsequent protein expression, and this regulation can be affected by single-nucleotide polymorphisms (SNPs) occurring in the miRNA target sites.
- SNPs single-nucleotide polymorphisms
- These SNPs can either create erroneous binding sites or abolish (eliminate) the correct ones, leading to resistance to miRNA regulation and reflecting another kind of genetic variability capable of playing a role in human diseases like cancer (or conferring an increased risk for certain diseases like cancer).
- Emerging research focuses on the systematic genomic evaluation of these sites and the functional and biological relevance of the detected SNPs, which are significant molecular markers in the rapidly growing area of personalized medicine. Such SNPs appear to affect not only gene expression, but also tumor biology and drug response and drug resistance.
- a single miRNA can regulate many mRNAs simultaneously. Moreover, miRNAs can act as both tumor suppressors and oncogenes.
- the lethal-7 (let-7) family of miRNAs is one of the first miRNA families to be discovered.
- the expression of let-7 family miRNAs is altered in many cancers. For example, in lung cancer, let-7 is poorly expressed and overexpression of let- 7 inhibits cell growth in vitro and in vivo, suggesting that let- 7 miRNAs may act as tumor suppressors.
- Disruption of miRNAs' regulation of oncogenes or tumor suppressor genes impact cancer risk, tumor development, and response to treatment.
- MiRNAs may regulate oncogenes or tumor suppressor genes directly or indirectly.
- KRAS mRNA The KRAS variant affects let- 7 mediated regulation of KRAS expression.
- the occurrence of the variant G-allele ⁇ i.e., the KRAS variant) leads to higher KRAS levels and lower let-7 levels as compared to the wild type.
- G-allele carriers have an increased lung cancer risk in moderate smokers, an increased ovarian cancer risk (particularly for postmenopausal women), an increased risk of developing breast cancer (and, in particular, the triple negative breast cancer subtype), and a reduced survival in oral cancers but not in lung cancer.
- Acquired KRAS mutations are not the same as the KRAS variant, which is a congenital mutation, and, therefore, has a different effect on tumor development, biology, and thus prognosis.
- the KRAS variant disrupts regulation of KRAS by the let- 7 family of miRNAs.
- /et-7-mediated regulation of KRAS is disrupted; however, there are secondary effects of the KRAS variant.
- Disruption of the let-7 I KRAS interaction upstream perpetuates aberrant signaling to downstream factors.
- components of signaling pathways other than the canonical RAS pathway are affected.
- the presence of the KRAS variant increases angiogenesis, survival (even under hypoxic conditions), metastasis, and predicts resistance or sensitivity to frequently used chemotherapy agents.
- epigenetic changes in the cancer cell influence the development, survival, and response to treatment of a cancer cell positive for the KRAS variant.
- the cellular consequences of the KRAS variant are independent of other mutations in KRAS, including, for example, acquired mutations in a coding region of KRAS.
- the occurrence of the KRAS variant is mutually exclusive with the occurrence of other KRAS mutations.
- the KRAS variant is a germ-line mutation.
- the KRAS variant is a heritable biomarker of tumor cell biology.
- the presence of the KRAS Variant indicates poor clinical outcomes in various cancers, including, but not limited to, breast, colon, ovarian, head and neck cancer, and lung cancer. This indication is due, at least in part, to the ability of the KRAS Variant to confer resistance or sensitivity to certain cancer therapies to either a cancer cell or subject containing the cancer cell. Thus, when the KRAS variant confers resistance to a standard
- the occurrence of the KRAS variant predicts a worse outcome for a carrier of the mutation than for a non-carrier.
- the KRAS variant confers sensitivity to a standard chemotherapeutic agent
- the occurrence of the KRAS variant predicts a better outcome for a carrier of the mutation than for a non-carrier.
- the presence of the KRAS Variant predicts either resistance or sensitivity to traditional chemotherapeutic agents (including monoclonal anti-EGFR antibody therapy and combinations thereof).
- the presence of the KRAS Variant may predict sensitivity for a cancer therapy when use as a monotherapy, and the opposite response, resistance, to the combination of the same therapy when used in combination with another treatment (e.g. chemotherapy or monoclonal anti-EGFR antibody) or medical procedure (e.g. radiation or surgery). This apparently contradiction may be resolved by understanding the specific gene or gene product targeted by each of these therapies.
- the occurrence of the KRAS variant may indicate that the use of cancer treatments that specifically target genes or gene products located upstream of KRAS in a cell signaling pathway will be successful; however, that the use of cancer treatments that specifically target genes or gene products located downstream of KRAS in a cell signaling pathway may be ineffective (e.g. conventional chemotherapeutic agents that target cell cycle checkpoints, which are downstream of KRAS).
- the KRAS variant increases a subject's sensitivity to the monoclonal anti-EGFR antibody therapy, Cetuximab, when delivered as the only treatment (a monotherapy), which targets an upstream regulator of the KRAS pathway (EGFR).
- the KRAS Variant confers resistance to platinum-based chemotherapy.
- Platinum-based chemotherapeutic agents crosslink DNA molecules to prevent DNA replication, ultimately triggering apoptosis.
- DNA replication is a process that occurs downstream of KRAS activation, and, therefore, may be ineffective to treat cancer, particularly in light of data showing the recruitment of signaling pathways other than RAS.
- chemotherapeutic agent that kills cancer cells may also damage or weaken the patient's heart.
- the doctor will be able to choose an optimal treatment, or at least avoid losing time and causing patient discomfort by using an ineffective treatment.
- the KRAS variant confers resistance to platinum-based chemotherapy
- Methods of the disclosure encompass assaying for the KRAS Variant to predict a subject's resistance to platinum-based chemotherapy when used as a treatment for any cancer.
- the cancer is epithelial ovarian cancer (EOC).
- EOC Epithelial ovarian cancer
- the standard chemotherapy regimen to treat EOC currently used is carboplatin and paclitaxel, based on prospective randomized trials. Although some patients are initially resistant to platinum-based chemotherapy (referred to as 'platinum resistant'), developing recurrence within 6 months of treatment, it is the first line treatment given to all EOC patients.
- rs61764370 An example of such a functional variant is rs61764370, referred to herein as the KRAS variant, which is located in the KRAS 3' UTR in a let- 7 miRNA complementary site.
- rs61764370 is associated with an increased risk of developing epithelial ovarian cancer
- Relative survival varies by age, with older women twice as likely to die within 5 years of diagnosis of EOC, further supporting the hypothesis that postmenopausal women may have biologically different tumors than younger women.
- the role of the KRAS variant in cancer risk and biology may further depend on miRNA expression alterations in response to physiologic conditions, such as menopause. For instance, women with the KRAS variant may be first at risk for breast cancer and then, subsequently, be at risk for developing postmenopausal ovarian cancer.
- KRAS variant does not predict poor outcome in a cohort of EOC patients with known deleterious BRCA mutations.
- BRCA mutations are associated with sensitivity to platinum-based chemotherapy. Without wishing to be bound by theory, downstream cell signaling consequences of BRCA mutations that confer sensitivity to platinum-based chemotherapeutic agents may affect targets upstream of KRAS, thereby bypassing KRAS-Variant mediated resistance to platinum-based
- treatment of recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to platinum-based chemotherapy.
- the platinum-based chemotherapy is cisplatin, however, the methods may be applied to any platinum-based chemotherapy.
- the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR of KRAS.
- treatment of recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting sensitivity or an enhanced response to monoclonal anti-EGFR antibody therapy, either as a monotherapy or as a combined therapy with another chemotherapeutic agent or medical procedure (e.g. radiation or surgery).
- R/M recurrent and/or metastatic head and neck squamous cell carcinoma
- the disclosure provides a method of predicting the response of head and neck squamous cell carcinoma (HNSCC ) cell and/or a recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) cell to a platinum-based chemotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and wherein the presence of the mutation indicates a resistance to platinum-based chemotherapy.
- HNSCC head and neck squamous cell carcinoma
- R/M metastatic head and neck squamous cell carcinoma
- the disclosure provides a method of predicting the response of head and neck squamous cell carcinoma (HNSCC ) cell and/or a recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) cell to a monoclonal anti-EGFR antibody therapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and wherein the presence of the mutation indicates sensitivity or an enhanced response to monoclonal anti-EGFR antibody therapy, either as a monotherapy or as a combined therapy with another chemotherapeutic agent or medical procedure (e.g. radiation or surgery).
- HNSCC head and neck squamous cell carcinoma
- R/M metastatic head and neck squamous cell carcinoma
- the FiNSCC cell may be evaluated in vivo, in vitro or ex vivo. When the FiNSCC cell is evaluated ex vivo, the cell is obtained from a subject.
- the KRAS variant confers resistance to small molecule EGFR inhibitors
- the disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors).
- treatment of cancer is optimized by assaying for the presence of the KRAS
- EGFR inhibitors Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors).
- treatment of non-small cell lung cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors and small molecule EGFR inhibitors).
- the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR ofKRAS.
- chemotherapeutic agents that inhibit an activity of EGFR include, but are not limited to, gefinitib and erlotinib.
- chemotherapeutic agents that inhibit an activity of EGFR include, but are not limited to, gefinitib.
- the KRAS variant confers sensitivity to MAP kinase pathway inhibitors
- the disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors).
- treatment of cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors).
- treatment of non-small cell lung cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors).
- the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR ofKRAS.
- chemotherapeutic agents that inhibit an activity of the MAP kinase pathway include, but are not limited to, inhibitors of tyrosine protein kinases and serine/threonine kinases.
- tyrosine protein kinases include the VEGFR (vascular endothelial growth factor receptor) and PDGFR (platelet-derived growth factor receptor).
- VEGFR vascular endothelial growth factor receptor
- PDGFR platelet-derived growth factor receptor
- serine/threonine protein kinases include C-raf (encoded by the RAFl gene) and B-raf (encoded by the BRAF gene).
- Chemotherapeutic agents that inhibit an activity of the MAP kinase pathway include, but are not limited to, sorafenib.
- the KRAS variant confers sensitivity to monoclonal anti-EGFR antibody monotherapy
- BRAF V600E mutation may confer resistance to anti-EGFR mAbs.
- KRAS variant predicts a positive response to monoclonal anti-EGFR antibody (mAb) monotherapy, without any additional benefit of cytotoxic chemotherapy.
- the studies presented herein demonstrate a statistically significant improvement in median progression- free survival (PFS) for all KRAS variant carriers with metastatic colon cancer (and a trend towards improved overall survival (OS) in the double wildtype patients) who received anti-EGFR mAb monotherapy.
- KRAS variant (G allele) carriers appeared to have no benefit from chemotherapy in addition to anti-EGFR mAb therapy. This was in contrast to non-KRAS variant patients, who derived a significant benefit from the addition of
- the KRAS genotypes were equally distributed among the KRAS wt and mutated groups, but, in the BRAF mutated group, the frequency of the KRAS variant was statistically significantly increased, i.e., twice as high compared to wild type.
- the KRAS variant allele may mediate the selection of less differentiated and more aggressive clones that carry BRAF mutations. Additionally, a selective pressure may favor the development of KRAS or BRAF mutations in the presence of the KRAS variant, depending on exposure to specific therapies.
- Cetuximab regardless of patients also having a KRAS or a BRAF mutation, suggesting that these groups need re-evaluation for the potential of Cetuximab treatment.
- chemotherapeutic agents in combination with anti-EGFR mAb-based therapy is detrimental to patients who carry the KRAS Variant.
- KRAS variant-positive tumors derive no benefit from the addition of cytotoxic therapy to monoclonal anti-EGFR antibody monotherapy.
- Chemotherapeutic agents may further decrease let-7 levels (which are already low due to the presence of the KRAS Variant) and, consequently, allow greater KRAS expression (especially in the presence of the KRAS variant).
- treatment of KRAS variant-positive tumors with chemotherapeutic agents may increase activation of this mutant allele, thereby removing the ability of upstream anti-EGFR mAb therapy to overcome KRAS pathway activation.
- treatment of head and neck squamous cell carcinoma (FINSCC ) or a recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (FINSCC ) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting sensitivity to monoclonal anti-EGFR antibody therapy.
- the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR of KRAS.
- the disclosure provides a method of predicting the response of a head and neck squamous cell carcinoma (FiNSCC ) cell to a monoclonal anti-EGFR antibody monotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, wherein the presence of the mutation indicates a sensitivity to monoclonal anti-EGFR antibody monotherapy.
- the head and neck squamous cell carcinoma (FiNSCC ) cell may be evaluated in vivo, in vitro or ex vivo.
- a non-limiting example of the monoclonal anti-EGFR antibody monotherapy is Cetuximab.
- EGF a potent ligand of EGFR
- cetuximab is beneficial for these patients because the anti- EGFR antibody blocks the pro-growth signal provided by an upregulation of EGF in these tumors.
- MACF1 is upregulated in our current analysis. MACF1 interacts with ErbB2 to control microtubule capture during cell migration.
- EGFR-specific growth stimulatory ligand EGF
- SYNJ2 pro-migratory phosphatase
- DST, MACF1, EML6 various components of the microtubule/cytoskeletal architecture
- methods of the disclosure include detection of the KRAS variant in combination with detection of upregulation of EGF, wherein the combined presence of the KRAS variant and upregulated EGF mRNA or protein indicates sensitivity of the cancer cell to a monoclonal anti-EGFR antibody therapy.
- the disclosure is based, in part, upon the unexpected discovery that the presence of a SNP in the 3' untranslated region (UTR) oiKRAS, referred to herein as the "LCS6 SNP” or the “KRAS variant,” which is predictive of an individual's risk of developing cancer and an individual's response to treatment for cancer.
- the KRAS variant is located in LCS6, the wild type and variant sequence of which is provided below.
- the KRAS variant may be represented by one or more of the following sequences.
- the KRAS variant may be defined by the GenBank accession number rs61764370 and the sequence GTCTCGAACTCCTGACCTCAAGTGATGCACCCACCTTGGCCTCATAAACCTG (SEQ ID NO: 22, in which the SNP is bolded and underlined).
- each human RAS gene comprises multiple miRNA complementary sites in the 3'UTR of their mRNA transcripts.
- each human RAS gene comprises multiple let-7 complementary sites (LCSs).
- the let-7 family-of-microRNAs includes global genetic regulators important in controlling lung cancer oncogene expression by binding to the 3'UTRs (untranslated regions) of their target messenger RNAs (mRNAs).
- let-7 complementary site is meant to describe any region of a gene or gene transcript that binds a member of the let-7 family of miRNAs. Moreover, this term encompasses those sequences within a gene or gene transcript that are complementary to the sequence of a let-7 family miRNA.
- complementary describes a threshold of binding between two sequences wherein a majority of nucleotides in each sequence are capable of binding to a majority of nucleotides within the other sequence in trans.
- LCS1-LCS8 The Human KRAS 3' UTR comprises 8 LCSs named LCS1-LCS8, respectively.
- T thymine
- U uracil
- LCS1 comprises the sequence GACAGUGGAAGUUUUUUUUCCUCG (SEQ ID NO: 1).
- LCS2 comprises the sequence AUUAGUGUCAUCUUGCCUC (SEQ ID NO: 2).
- LCS3 comprises the sequence AAUGCCCUACAUCUUAUUUUCCUCA (SEQ ID NO: 3).
- LCS4 comprises the sequence GGUUCAAGCGAUUCUCGUGCCUCG (SEQ ID NO: 4).
- LCS5 comprises the sequence GGCUGGUCCGAACUCCUGACCUCA (SEQ ID NO: 5).
- LCS6 comprises the sequence GAUUCACCCACCUUGGCCUCA (SEQ ID NO: 6).
- LCS7 comprises the sequence GGGUGUUAAG ACUUG AC AC AGUAC CUC G (SEQ ID NO: 7).
- LCS8 comprises the sequence AGUGCUUAUGAGGGGAUAUUUAGGCCUC (SEQ ID NO: 8).
- Human KRAS has two wild type forms, encoded by transcripts a and b, which are provided below as SEQ ID NOs: 9 and 10, respectively.
- the sequences of each human KRAS transcript, containing the LCS6 SNP, are provided below as SEQ ID NOs: 11 and 12.
- Human KRAS, transcript variant a is encoded by the following mRNA sequence (NCBI Accession No. NM 033360 and SEQ ID NO: 9) (untranslated regions are bolded, LCS6 is underlined):
- Human KRAS, transcript variant b is encoded by the following mRNA sequence (NCBI Accession No. NM 004985 and SEQ ID NO: 10)(untranslated regions are bolded, LCS6 is underlined):
- Human KRAS, transcript variant a, comprising the LCS6 SNP is encoded by the following mRNA sequence (SEQ ID NO: 11) (untranslated regions are bolded, LCS6 is underlined, SNP is capitalized):
- Human KRAS, transcript variant b, comprising the LCS6 SNP is encoded by the following mRNA sequence (SEQ ID NO: 12)(untranslated regions are bolded, LCS6 is underlined, SNP is capitalized):
- the KRAS variant is the result of a substitution of a G for a U at position 4 of SEQ ID NO: 6 of LCS6.
- This KRAS variant comprises the sequence
- the KRAS variant leads to altered KRAS expression by disrupting the miRNA regulation of a KRAS.
- the identification and characterization of the KRAS variant is further described in International Application No. PCT/US08/65302 (and corresponding publication number WO 2008/151004), the contents of which are incorporated by reference in their entirety.
- let-7 family miRNAs Expression of let-7 family miRNAs is decreased in cells that carry the KRAS variant. Interestingly, the let-7 family of miRNAs bind to the let-7 complementary site in which the KRAS variant in located. The presence of the KRAS variant interferes with let-7 binding to KRAS. By interfering, the KRAS variant either induces let-7 to bind more or less tightly to LCS6 of KRAS. It was discovered that cells containing the KRAS variant have lower levels of KRAS mRNA compared to wild type cells, and increased levels of the KRAS protein. Thus, while not wishing to be bound by theory, the presence of the KRAS variant within cells may interfere with the ability of let-7 to bind to KRAS and inhibit protein translation, allowing higher KRAS protein levels.
- let-7 miRNAs include, but are not limited to, let-7 a (let-7 a-l, let-7 a-2, let-7 a-3), let-7 b, let-7 c, let-7 d, let-7 e, let-7 f (let-7 f- 1 and let-7 f-2), let-7 g, and let-7 i.
- thymine (T) may be substituted for uracil (U).
- let-7 a comprises the sequence UUGAUAUGUUGGAUGAUGGAGU (SEQ ID NO: 14).
- let-7b comprises the sequence UUGGUGUGUUGGAUGAUGGAGU (SEQ ID NO: 15).
- let-7 c comprises the sequence UUGGUAUGUUGGAUGAUGGAGU (SEQ ID NO: 16).
- let-7d comprises the sequence UGAUACGUUGGAUGAUGGAGA (SEQ ID NO: 17).
- let-7e comprises the sequence UAUAUGUUGGAGGAUGGAGU (SEQ ID NO: 18).
- let- 7f comprises the sequence UUGAUAUGUUAGAUGAUGGAGU (SEQ ID NO: 19).
- let-7g comprises the sequence GACAUGUUUGAUGAUGGAGU (SEQ ID NO: 20).
- let-7 i comprises the sequence UGUCGUGUUUGUUGAUGGAGU (SEQ ID NO: 21).
- poor prognosis is meant that the probability of the individual surviving the development of a particularly aggressive, high-risk, severe, or inherited form of cancer (e.g., triple negative breast cancer), or that the probability of surviving the development or progression of an aggressive, high-risk, severe, or inherited form is less than the probability of surviving the development or progression of a more benign form.
- a particularly aggressive, high-risk, severe, or inherited form of cancer e.g., triple negative breast cancer
- Poor prognosis is also meant to describe a less satisfactory recovery, longer recovery period, more invasive or high-risk therapeutic regimen, or an increased probability of reoccurrence of cancer or a metastasis thereof.
- triple negative breast cancer or a metastasis thereof is correlated with the worst prognosis of breast cancer subtypes, resulting in a poor prognosis for the subject.
- a subject is preferably a mammal.
- the mammal can be a human, non-human primate, mouse, rat, dog, cat, horse, or cow, but are not limited to these examples.
- a subject is male or female.
- a subject may not have been previously diagnosed as having cancer, a particular type of cancer (e.g., breast cancer), or a subtype of cancer (e.g., triple negative breast cancer as a subtype of breast cancer).
- the subject may exhibit one or more risk factors for cancer, a particular type of cancer or a subtype of cancer. Alternatively, the subject does not exhibit a risk factor for cancer, a particular type of cancer or a subtype of cancer.
- the methods described herein may require obtaining a sample from a subject.
- the sample can be any tissue or fluid that contains nucleic acids.
- Various embodiments include, but are not limited to, paraffin imbedded tissue, frozen tissue, surgical fine needle aspirations, and cells of the breast (including cells harvested from a duct, a lobule, or connective tissue), a lymph node (including a sentinel or axillary node), a thoracic or abdominal muscle or connective tissue, an organ (including any potential deposit site for a potential metastatic cell, such as the brain, liver, kidney, stomach, intestines, bone marrow, pancreas, colon, or lung).
- Other embodiments include fluid samples such as blood, plasma, serum, lymph fluid, ascites, serous fluid, and urine.
- the KRAS variant is a single nucleotide polymorphism that occurs within the 3 ' UTR of the human KRAS gene.
- Linkage disequilibrium refers to the co-inheritance of alleles (e.g., alternative nucleotides) at two or more different SNP sites at frequencies greater than would be expected from the separate frequencies of occurrence of each allele in a given population.
- the expected frequency of co-occurrence of two alleles that are inherited independently is the frequency of the first allele multiplied by the frequency of the second allele. Alleles that co-occur at expected frequencies are said to be in "linkage equilibrium".
- LD refers to any non-random genetic association between allele(s) at two or more different SNP sites, which is generally due to the physical proximity of the two loci along a chromosome. LD can occur when two or more SNPs sites are in close physical proximity to each other on a given chromosome and therefore alleles at these SNP sites will tend to remain unseparated for multiple generations with the consequence that a particular nucleotide (allele) at one SNP site will show a non-random association with a particular nucleotide (allele) at a different SNP site located nearby. Hence, genotyping one of the SNP sites will give almost the same information as genotyping the other SNP site that is in LD.
- polymorphisms e.g., SNPs and/or haplotypes
- the genotype of the polymorphism(s) that is/are in LD with the causative polymorphism is predictive of the genotype of the causative polymorphism and, consequently, predictive of the phenotype (e.g., disease) that is influenced by the causative SNP(s).
- polymorphic markers that are in LD with causative polymorphisms are useful as markers, and are particularly useful when the actual causative polymorphism(s) is/are unknown.
- the screening techniques of the present disclosure may employ a variety of methodologies to determine whether a test subject has a SNP or a SNP pattern associated with an increased or decreased risk of developing a detectable trait or whether the individual suffers from a detectable trait as a result of a particular polymorphism/mutation, including, for example, methods which enable the analysis of individual chromosomes for haplotyping, family studies, single sperm DNA analysis, or somatic hybrids.
- the trait analyzed using the diagnostics of the disclosure may be any detectable trait that is commonly observed in pathologies and disorders.
- SNP genotyping is referred to as SNP genotyping.
- the present disclosure provides methods of SNP genotyping, such as for use in screening for a variety of disorders, or determining predisposition thereto, or determining responsiveness to a form of treatment, or prognosis, or in genome mapping or SNP association analysis, etc.
- Nucleic acid samples can be genotyped to determine which allele(s) is/are present at any given genetic region (e.g., SNP position) of interest by methods well known in the art.
- the neighboring sequence can be used to design SNP detection reagents such as
- oligonucleotide probes which may optionally be implemented in a kit format.
- SNP genotyping methods are described in Chen et al, “Single nucleotide polymorphism genotyping: biochemistry, protocol, cost and throughput", Pharmaco genomics J. 2003;
- Common SNP genotyping methods include, but are not limited to, TaqMan assays, molecular beacon assays, nucleic acid arrays, allele-specific primer extension, allele-specific PCR, arrayed primer extension, homogeneous primer extension assays, primer extension with detection by mass spectrometry, pyrosequencing, multiplex primer extension sorted on genetic arrays, ligation with rolling circle amplification, homogeneous ligation, OLA (U.S. Pat. No. 4,988,167), multiplex ligation reaction sorted on genetic arrays, restriction-fragment length
- polymorphism single base extension-tag assays, and the Invader assay.
- detection mechanisms such as, for example, luminescence or chemiluminescence detection, fluorescence detection, time-resolved fluorescence detection, fluorescence resonance energy transfer, fluorescence polarization, mass spectrometry, and electrical detection.
- RNA/RNA RNA/RNA
- methods for detecting polymorphisms include, but are not limited to, methods in which protection from cleavage agents is used to detect mismatched bases in RNA/RNA or
- SNP genotyping is performed using the TaqMan assay, which is also known as the 5' nuclease assay (U.S. Pat. Nos. 5,210,015 and 5,538,848).
- the TaqMan assay detects the accumulation of a specific amplified product during PCR.
- the TaqMan assay utilizes an oligonucleotide probe labeled with a fluorescent reporter dye and a quencher dye.
- the reporter dye is excited by irradiation at an appropriate wavelength, it transfers energy to the quencher dye in the same probe via a process called fluorescence resonance energy transfer (FRET). When attached to the probe, the excited reporter dye does not emit a signal.
- FRET fluorescence resonance energy transfer
- the proximity of the quencher dye to the reporter dye in the intact probe maintains a reduced fluorescence for the reporter.
- the reporter dye and quencher dye may be at the 5' most and the 3' most ends, respectively, or vice versa.
- the reporter dye may be at the 5 ' or 3' most end while the quencher dye is attached to an internal nucleotide, or vice versa.
- both the reporter and the quencher may be attached to internal nucleotides at a distance from each other such that fluorescence of the reporter is reduced.
- DNA polymerase cleaves the probe, thereby separating the reporter dye and the quencher dye and resulting in increased fluorescence of the reporter. Accumulation of PCR product is detected directly by monitoring the increase in fluorescence of the reporter dye.
- the DNA polymerase cleaves the probe between the reporter dye and the quencher dye only if the probe hybridizes to the target SNP-containing template which is amplified during PCR, and the probe is designed to hybridize to the target SNP site only if a particular SNP allele is present.
- SNP and associated nucleic acid sequence information provided herein.
- a number of computer programs such as Primer Express (Applied Biosystems, Foster City, Calif), can be used to rapidly obtain optimal primer/probe sets. It will be apparent to one of skill in the art that such primers and probes for detecting the SNPs of the present disclosure are useful in prognostic assays for a variety of disorders including cancer, and can be readily incorporated into a kit format.
- the present disclosure also includes modifications of the Taqman assay well known in the art such as the use of Molecular Beacon probes (U.S. Pat. Nos. 5,118,801 and 5,312,728) and other variant formats (U.S. Pat. Nos. 5,866,336 and 6,1 17,635).
- polymorphisms may also be determined using a mismatch detection technique, including but not limited to the RNase protection method using riboprobes (Winter et al, Proc. Natl. Acad Sci. USA 82:7575, 1985; Meyers et al, Science 230: 1242, 1985) and proteins which recognize nucleotide mismatches, such as the E. coli mutS protein (Modrich, P. Ann. Rev. Genet. 25:229-253, 1991).
- riboprobes Winter et al, Proc. Natl. Acad Sci. USA 82:7575, 1985; Meyers et al, Science 230: 1242, 1985
- proteins which recognize nucleotide mismatches such as the E. coli mutS protein (Modrich, P. Ann. Rev. Genet. 25:229-253, 1991).
- variant alleles can be identified by single strand conformation polymorphism (SSCP) analysis (Orita et al., Genomics 5:874-879, 1989; Humphries et al, in Molecular Diagnosis of Genetic Diseases, R. Elles, ed., pp. 321- 340, 1996) or denaturing gradient gel electrophoresis (DGGE) (Wartell et al, Nuci. Acids Res. 18:2699-2706, 1990; Sheffield et al, Proc. Natl. Acad. Sci. USA 86:232-236, 1989).
- SSCP single strand conformation polymorphism
- DGGE denaturing gradient gel electrophoresis
- a polymerase-mediated primer extension method may also be used to identify the polymorphism(s).
- Several such methods have been described in the patent and scientific literature and include the "Genetic Bit Analysis” method (W092/15712) and the
- ligase/polymerase mediated genetic bit analysis U.S. Pat. No. 5,679,524. Related methods are disclosed in WO91/02087, WO90/09455, W095/17676, U.S. Pat. Nos. 5,302,509, and 5,945,283. Extended primers containing a polymorphism may be detected by mass spectrometry as described in U.S. Pat. No. 5,605,798. Another primer extension method is allele-specific PCR (Ruano et al, Nucl. Acids Res. 17:8392, 1989; Ruano et al, Nucl. Acids Res.
- multiple polymorphic sites may be investigated by simultaneously amplifying multiple regions of the nucleic acid using sets of allele-specific primers as described in Wallace et al. (WO89/10414).
- Another preferred method for genotyping the KRAS variant is the use of two oligonucleotide probes in an OLA (see, e.g., U.S. Pat. No. 4,988,617). In this method, one probe hybridizes to a segment of a target nucleic acid with its 3' most end aligned with the
- a second probe hybridizes to an adjacent segment of the target nucleic acid molecule directly 3' to the first probe.
- the two juxtaposed probes hybridize to the target nucleic acid molecule, and are ligated in the presence of a linking agent such as a ligase if there is perfect complementarity between the 3' most nucleotide of the first probe with the
- the ligated probes are separated from the target nucleic acid molecule, and detected as indicators of the presence of a SNP.
- spectrometry takes advantage of the unique mass of each of the four nucleotides of DNA. SNPs can be unambiguously genotyped by mass spectrometry by measuring the differences in the mass of nucleic acids having alternative SNP alleles.
- MALDI-TOF Microx Assisted Laser Desorption Ionization— Time of Flight
- mass spectrometry technology is preferred for extremely precise determinations of molecular mass, such as SNPs. Numerous approaches to SNP analysis have been developed based on mass spectrometry. Preferred mass
- spectrometry-based methods of SNP genotyping include primer extension assays, which can also be utilized in combination with other approaches, such as traditional gel-based formats and microarrays.
- the primer extension assay involves designing and annealing a primer to a template PCR amplicon upstream (5') from a target SNP position.
- ddNTPs dideoxynucleotide triphosphates
- dNTPs deoxynucleotide triphosphates
- Extension of the primer terminates at the first position in the template where a nucleotide complementary to one of the ddNTPs in the mix occurs.
- the primer can be either immediately adjacent (i.e., the nucleotide at the 3' end of the primer hybridizes to the nucleotide next to the target SNP site) or two or more nucleotides removed from the SNP position. If the primer is several nucleotides removed from the target SNP position, the only limitation is that the template sequence between the 3' end of the primer and the SNP position cannot contain a nucleotide of the same type as the one to be detected, or this will cause premature termination of the extension primer.
- primers are designed to bind one nucleotide upstream from the SNP position (i.e., the nucleotide at the 3' end of the primer hybridizes to the nucleotide that is immediately adjacent to the target SNP site on the
- ddNTPs can be employed in the primer extension reactions in place of unmodified ddNTPs. This increases the mass difference between primers extended with these ddNTPs, thereby providing increased sensitivity and accuracy, and is particularly useful for typing heterozygous base positions.
- Mass-tagging also alleviates the need for intensive sample-preparation procedures and decreases the necessary resolving power of the mass spectrometer.
- the extended primers can then be purified and analyzed by MALDI-TOF mass spectrometry to determine the identity of the nucleotide present at the target SNP position.
- the products from the primer extension reaction are combined with light absorbing crystals that form a matrix.
- the matrix is then hit with an energy source such as a laser to ionize and desorb the nucleic acid molecules into the gas-phase.
- the ionized molecules are then ejected into a flight tube and accelerated down the tube towards a detector.
- the time between the ionization event, such as a laser pulse, and collision of the molecule with the detector is the time of flight of that molecule.
- the time of flight is precisely correlated with the mass-to-charge ratio (m/z) of the ionized molecule. Ions with smaller m/z travel down the tube faster than ions with larger m/z and therefore the lighter ions reach the detector before the heavier ions. The time-of- flight is then converted into a corresponding, and highly precise, m/z. In this manner, SNPs can be identified based on the slight differences in mass, and the corresponding time of flight differences, inherent in nucleic acid molecules having different nucleotides at a single base position.
- SNPs can also be scored by direct DNA sequencing.
- a variety of automated sequencing procedures can be utilized ((1995) Biotechniques 19:448), including sequencing by mass spectrometry (see, e.g., PCT International Publication No. WO94/16101; Cohen et al., Adv. Chromatogr. 36: 127-162 (1996); and Griffin et al., Appl. Biochem. Biotechnol. 38: 147-159 (1993)).
- the nucleic acid sequences of the present disclosure enable one of ordinary skill in the art to readily design sequencing primers for such automated sequencing procedures.
- Commercial instrumentation such as the Applied Biosystems 377, 3100, 3700, 3730, and 3730.times. l DNA Analyzers (Foster City, Calif), is commonly used in the art for automated sequencing.
- KRAS variants include single-strand conformational polymorphism (SSCP), and denaturing gradient gel electrophoresis (DGGE)
- SSCP identifies base differences by alteration in electrophoretic migration of single stranded PCR products, as described in Orita et al, Proc.
- Single-stranded PCR products can be generated by heating or otherwise denaturing double stranded PCR products.
- Single-stranded nucleic acids may refold or form secondary structures that are partially dependent on the base sequence.
- the different electrophoretic mobilities of single-stranded amplification products are related to base- sequence differences at SNP positions.
- DGGE differentiates SNP alleles based on the different sequence-dependent stabilities and melting properties inherent in polymorphic DNA and the corresponding differences in electrophoretic migration patterns in a denaturing gradient gel (Erlich, ed., PCR Technology, Principles and Applications for DNA
- Sequence-specific ribozymes (U.S. Pat. No. 5,498,531) can also be used to score SNPs based on the development or loss of a ribozyme cleavage site. Perfectly matched sequences can be distinguished from mismatched sequences by nuclease cleavage digestion assays or by differences in melting temperature. If the SNP affects a restriction enzyme cleavage site, the SNP can be identified by alterations in restriction enzyme digestion patterns, and the corresponding changes in nucleic acid fragment lengths determined by gel electrophoresis
- SNP genotyping can include the steps of, for example, collecting a biological sample from a human subject (e.g., sample of tissues, cells, fluids, secretions, etc.), isolating nucleic acids (e.g., genomic DNA, mRNA or both) from the cells of the sample, contacting the nucleic acids with one or more primers which specifically hybridize to a region of the isolated nucleic acid containing a target SNP under conditions such that hybridization and amplification of the target nucleic acid region occurs, and determining the nucleotide present at the SNP position of interest, or, in some assays, detecting the presence or absence of an amplification product (assays can be designed so that hybridization and/or amplification will only occur if a particular SNP allele is present or absent).
- the size of the amplification product is detected and compared to the length of a control sample; for example, deletions and insertions can be detected by a change in size of the ampl
- Example 1 Prevalence of the KRAS variant in various cancer cell lines
- Taqman genotyping was performed to determine the presence of the KRAS variant allele as described previously (Bussey KJ, et al. Mol Cancer Ther 2006; 5:853-67). Cells were cultured under standard conditions (see,
- the presence of the KRAS variant is a genetic marker for prediction of risk and tumor biology as well as response to treatment in multiple cancers.
- the presence of the KRAS variant results in altered regulation by the KRAS 3' UTR.
- This study elucidates the biological significance of the KRAS variant in cancer cells.
- the data provided herein elucidate exemplary molecular pathways that are affected by the presence of the KRAS variant.
- the NCI-60 panel of cell lines were categorized based on the presence of either an acquired dominant mutation in the KRAS coding region (KRAS mutation) or the presence of the KRAS variant, it was determined that all seven cell lines that contained the KRAS variant were negative for the presence of KRAS-activating mutations. Similarly, the cell lines that carried a KRAS coding sequence mutation lacked the KRAS variant allele. Thus, the presence or occurrence of either a KRAS coding mutation or the KRAS variant allele is mutually exclusive in these cell lines. Furthermore, because this mutual exclusivity occurs in cell lines derived from a variety of cancer types, this mutual exclusivity is not specific to a particular tissue type.
- Cancer Res 2008; 68:8535-40 and in ovarian cancer patients (Ratner E, Cancer Res 2010; 70:6509-15), but not in colon cancer patients (Zhang W, et al. Ann Oncol 2011; 22:484-5; Zhang W, et al. Ann Oncol 2011; 22: 104-9).
- MiR-23 and miR-27 are expressed from the same cluster and advance progression of angiogenesis and metastasis (Zhou Q, et al. Proc Natl Acad Sci USA 2011; 108:8287-92).
- miR-23 and miR-27 are enriched in endothelial cells and highly vascularized tissue.
- miR-23 and miR-27 elevate signaling pathways that are essential for angiogenesis by reducing the expression of Sprouty2 and Sema6A, which have anti- angiogenic functions.
- miR-210 is statistically significantly correlated with the presence of the KRAS variant allele in cells.
- MiR-210 is a marker of chronic hypoxia.
- miR-210 is associated with proliferation and metastasis of breast and melanoma tumors as well as poor prognosis.
- MiR-210 is a direct transcriptional target of HIF proteins. Elevated levels of miR- 210 are required for tumor cell survival under conditions of hypoxia.
- MiR-210 directly regulates the expression of MNT, a MYC antagonist that is required for cell cycle arrest under hypoxia. Consequently, increased levels of miR-210 contribute to an override of cell cycle arrest under conditions of hypoxic stress in tumor cells. Because increased miR-210 expression is associated with the presence of the KRAS variant, tumor cells containing the KRAS variant survive and proliferate under hypoxic conditions.
- the data provided herein demonstrate that the KRAS variant contributes to or initiates aberrant signaling pathways that control the expression of several miRNAs (including, for example, miR-23, miR-27 and miR-210). Perturbation of signaling pathways that regulate expression of miRNAs, such as miR-23, miR-27 and miR-210, results in the initiation, development, maintenance or augmentation of tumor proliferation and metastatic
- Promoter methylation is one mechanism through which gene expression is silenced in many cancers because changes in the methylation status of gene promoters lead to reduction in gene expression. Specifically, DNA methylation is an epigenetic effect caused when CpG dinucleotides are methylated, often in the promoter region of genes. Because methylation blocks access to the promoter by molecules that mediate gene transcription, methylation of the promoter results in gene silencing. Different cancers show distinct methylation patterns, the result of which is alterations in gene expression signatures.
- the methylation status of these cell lines was compared with the non-KRAS variant lines in the NCI-60 panel (Ehrich M, et al. Proc Natl Acad Sci USA 2008; 105:4844- 9).
- the presence of the KRAS variant allele shows a statistically significant positive correlation with increased methylation of the promoter of many genes, including, for example, Notchl, cyclin D3 and CNBP (also known as ZNF9) (Table 3).
- Notchl activation results in an increase in invasive and migratory characteristics of breast cancer cells.
- Notchl overexpression in a MYC background induces adenomas in the mouse lung, leading to the formation of lung adenocarcinoma.
- the evidence indicates that Notchl may function as an oncogene.
- Notchl may also function as a tumor suppressor.
- inhibitory mutations in Notchl have been identified in squamous cell carcinomas of the head and neck. Depletion of Notchl in mouse skin keratinocytes results in enhanced tumorigenesis by chemical carcinogens or by oncogenic Ras.
- Notchl expression is decreased when compared with normal adjacent tissue.
- Overexpression of activated Notchl in HPV-positive cervical cancers and neuroblastoma cells leads to growth inhibition.
- Notchl is dysregulated in many cancers and, in some instances, may function as a putative tumor suppressor. Because methylation of the Notchl promoter is increased in KRAS variant-positive cancer cells,
- Notchl expression may be reduced in cells carrying the KRAS variant allele, and, therefore, KRAS- variant cell lines may induce or maintain their tumorigenic potential by inhibiting the tumor suppressing effects of Notchl.
- Cyclin D3 is the member of the cyclin family of cell cycle proteins that is required for the Gi/S transition of the cell cycle.
- promoter methylation of cyclin D3 is increased, which indicates repression of cyclin D3 transcription. Consequently, the evidence suggests two exemplary mechanisms in which either cyclin D3 is not required for the transformed phenotype of these cell lines or methylation of the cyclin D3 promoter blocks a transcriptional repressor of cyclin D3.
- CNBP cellular nucleic acid binding protein
- ZNF9 cellular nucleic acid binding protein
- the thetal isoform has been implicated in several cancers. For example, increased expression of GSTTl is statistically significantly correlated with aggressive bladder cancers. In other different tumors types, GSTTl is nonfunctional or absent due to genetic polymorphism, thus leading to increased risk of carcinogenesis and poor prognosis as a result of an accumulation or increased accumulation of toxic metabolites.
- Mitogen-activated protein kinase 3 is a member of the MAP kinase family. Moreover, increased expression of mitogen-activated protein kinase 3 (MAPK3) is associated with the KRAS variant in cancer cells. MAPK3 transduces signals from extracellular cues to regulate intracellular processes, such as cell proliferation and differentiation. For example, increased expression of phosphorylated MAPK3 has been associated with aggressive colorectal tumors and metastatic meduloblastoma. Increased levels of KRAS in KRAS variant positive cancer cells are associated with an increase in MAPK3 mRNA. At least in part, increased MAPK3 expression induces an increase cellular proliferation and neoplastic progression in these cells.
- MAPK4 MAPK 4
- MAPK3 and/or MAP2K4 KRAS and MAPK 4
- KRAS and MAPK may contribute to a synergistic interaction between KRAS and MAPK signaling in KRAS-vaxiant cancer cells that induces or enhances cell proliferation and/or neoplastic progression.
- synaptotagmin-12 and increased expression of inter-a globulin inhibitor-Hl are positively correlated with the presence of the KRAS variant in cancer cell lines.
- synaptotagmins regulate calcium-dependent membrane trafficking during synaptic transmission.
- overexpression of synaptotagmin-13, a family member of synaptotagmin-12 suppresses a transformed phenotype of cells derived from a rat liver tumor cell line.
- Overexpression of synaptotagmin-12 in KRAS variant-positive cancer cell lines indicates a deregulation of novel pathways involving syntaptotagmins in cancer cells.
- the inter-a (globulin) inhibitor HI is the heavy chain of the plasma serine protease inhibitor. Functionally, the inter-a (globulin) inhibitor HI is required for extracellular matrix stability. Though the role of the inter-a (globulin) inhibitor HI in cancer remains unexplored, recent evidence indicates that the expression of inter-a (globulin) inhibitor HI is either lost or repressed in various solid tumors, including, for example, tumors of the lung, colon and breast.
- stage 1 ovarian cancer patients receive adjuvant chemotherapy, when substage information was not available for patients with stage 1 tumors, these patients were excluded from the analysis. Otherwise, stage IB and 1C tumors were included with stages 2- 4. To minimize inadvertent inclusion of borderline tumors, tumors with an unknown grade were excluded from this analysis.
- the date of pathological diagnosis was considered the start date of treatment.
- the date of surgery was considered the start date of treatment.
- a total of 386 patients with wild-type BRCA or not tested for BRCA mutations and 79 patients with documented BRCA mutations fit the above-described parameters and were included in the two survival analyses.
- Platinum resistance was defined as progression-free survival of ⁇ 6 months from the completion of platinum-containing adjuvant chemotherapy to the date of recurrence.
- Table 8 describes the clinicopatho logical parameters of these patients.
- KRAS variant DNA was isolated using standard methods from tumor, blood or saliva.
- the KRAS variant does not appear to be somatically acquired nor does it require a loss of heterozygosity (Chin LJ, et al. (2008). Cancer Res 68: 8535-8540); hence, blood and saliva, for example, are appropriate to test and the results are identical regardless of the tissue tested.
- the KRAS variant allele was detected using a primer specific to the KRAS variant and a TaqMan (Applied Biosystems, Foster City, CA, USA) PCR assay on all samples. Genotyping was performed at the YNHH, except for on samples from COH, for which the genotyping was performed in their facility.
- KRAS- variant carriers Less than 3% of populations carry 2 copies of the KRAS variant (Chin LJ, et al. (2008). Cancer Res 68: 8535-8540). As such, patients who carried at least one copy of the KRAS variant allele were classified as KRAS- variant carriers.
- signature scores were computed as Pearson's correlation between the respective signature vector of gene contributions and each sample's expression profile for these genes. Differences between signature scores in and non-variant
- IC50s were determined using a sigmoidal equilibrium model regression using XLfit version 5.2 (ID Business Solutions Ltd). The IC50 was defined as the concentration of drug required for a 50% reduction in growth/viability. All experiments were carried out a minimum of three times.
- Targeting the KRAS variant Small-interfering RNA sequences were designed to target the KRAS- variant sequence by placing the single-nucleotide polymorphism at varying positions of the 6 nucleotides at the 5 ' end of the siRNA guide strand corresponding to the so-called 'seed sequence'. Blast searches were performed to minimize cross-reactivity. In some of the siRNA sequences, DNA nucleotides were introduced to optimize
- thermoenergetic features for preferred incorporation of the guide strand into the argonaute effector complex or to increase specificity for the variant.
- the negative control used was purchased from Qiagen (Valencia, CA, USA) (AllStars Negative-Control siRNA). Knockdown efficiency and specificity to the KRAS variant of these sequences were confirmed using a dual luciferase assay (see WO/2009/155100, the contents of which are incorporated herein by reference). Oligonucleotide combinations were annealed using standard conditions and then transfected into cells using standard protocols. Cell survival was assayed using MTT assays and experiments were conducted in quadruplicate, and repeated in four independent experiments for all lines. Cell lysates were collected 72 hours after transfection and KRAS protein levels measured by western analysis using a probe specific to KRAS as described previously (Chin LJ, et al. (2008). Cancer Res 68: 8535-8540).
- J R Stat Soc 34: 187-220 was used to assess the impact of the KRAS variant and demographic and prognostic variables (such as age, stage, grade and histology) on overall survival.
- Multivariate logistic regression analyses (Cox D. (1970). The Analysis of Binary Data. Methuen, London) were used to determine the impact of the KRAS variant and other demographic and prognostic factors on the probability of suboptimal cytoreduction.
- Multivariate logistic regression analyses (Cox D. (1970). The Analysis of Binary Data. Methuen, London) were used to assess the association of the KRAS variant and other prognostic factors on the probability of platinum resistance. All statistical analyses were performed using SAS 9.1.3 (SAS Institute Inc., Cary, NC, USA) and in R 2.12.1 (R Foundation for Statistical Computing)
- let-7b miRNA expression data were not available on tumor samples, the expression of let-7b miRNA in two cell lines with the KRAS variant (BG-1 and IGROVl) was compared with the expression of let- 7b in a non-KRAS variant line (CAOV3).
- the expression of let-7b miRNA is altered in KRAS variant-positive lung tumors (Chin LJ, et al. (2008). Cancer Res 68: 8535-8540) and triple-negative breast tumors (Paranjape T, et al. (2011). Lancet Oncol 12: 377-386).
- EOC cell lines with and without the KRAS variant were used to test their sensitivity to different chemotherapeutic agents.
- a cell line that is KRAS variant positive/5i?C4 wild- type (BG1), a non-variant/5i? CA wild-type cell line (CAOV3) and a cell line KRAS- variant positive/ BRCA1 mutant (IGR-OV1) were tested. It was determined that the KRAS- variant line, BG1, was statistically significantly resistant to carboplatin (P ⁇ 0.04) and
- carboplatin/paclitaxel combination chemotherapy (P ⁇ 0.0001) compared with CAOV3, the cell line without the KRAS variant.
- IGROVl the cell line with the KRAS variant and a deleterious BRCA1 mutation, was not resistant to these agents when compared with
- agents frequently used as second line therapy for patients who have failed carboplatin/paclitaxel chemotherapy were evaluated. These second line therapeutic agents included doxorubicin, topotecan and gemcitabine.
- the KRAS-variant line, BG1 was significantly resistant to each of these agents compared with CAOV3, the nonvariant cell line (Table 13).
- Example 3 The KRAS variant and Response to Monoclonal Anti-EGFR Antibody
- VIC reporter probe 5 '-CTCAAGTGATTCACCCA C-3' (SEQ ID NO: 29), VIC reporter probe: 5 '-CTCAAGTGATTCACCCA C-3' (SEQ ID NO:
- FAM reporter probe 5 '-CAAGTGATTCACCCAC- 3' (SEQ ID NO: 31).
- Cell lines were treated with Cetuximab ( ⁇ ) or none and dilutions of Irinotecan (lmg/ml- lOOmg/ml). Cells were plated, treated with agents 24 hours after plating, media was changed after a 24 hour exposure, and then survival was scored 48 hours later using the MTT assay.
- GG for the KRAS variant allele were considered positive for the LCS6 (KRAS-variant or G allele) and entered the analyses as one group of at least one KRAS variant (G allele) genotypes.
- PFS and OS were measured as previously described (De RW, et al. Lancet Oncol 2010; l l(8):753-762; Zhang W, al. Ann Oncol 2011; 22(1): 104-109)
- KRAS LCS6 in the entire patient cohort.
- KRAS mutations in codons 12, 13 and 61 were found in 184 patients (33%) and the BRAF V600E was found in 29 patients (5.3%). All patients had received anti-EGFR mAbs-based salvage treatment, 169 as monotherapy and 377 in combination with chemotherapy. No statistically significant differences were found between KRAS wt and KRAS variant carriers for sex and age at diagnosis. The characteristics of the 559 patients have been previously published (De RW, et al. Lancet Oncol 2010;
- 3 '-UTR LCS6, 3 ' untranslated region of the Let-7 complementary site WT, wild type; PFS, progression-free survival; OS, overall survival; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease.
- Progression free survival analysis correlated with treatment. Patients who received mAbs monotherapy and mAbs combination therapy were analyzed separately. From the 501 patients evaluable for LCS6 SNP genotyping and treatment administration, 160 (32%) received anti-EGFR mAbs as monotherapy. Of the monotherapy patients, 128 (80%) were carriers of the LCS6 wt TT genotype and 32 (20%) were carriers of the LCS6 G variant genotype. There were 341 (68%) patients who received multiple chemotherapy combinations. Of the combination treatment patients, 266 (78%) were carriers of the LCS6 wt TT genotype and 75 (22%) were carriers of the LCS6 at least one G variant genotype.
- the LCS6 variant is prognostic in KRAS and BRAF mutated patients.
- KRAS and BRAF mutated patients' population no statistical significant differences regarding PFS and OS were observed in patients treated with both anti-EGFR mAbs monotherapy and in combination with chemotherapy.
- individualized targeted therapy for NSCLC patients for whom standard therapy had failed and this trial prospectively examined patient tumor biomarker profiles, which were obtained from a mandated fresh core tumor biopsy, and assigned them to corresponding targeted therapies (erlotinib, vandetanib, bexarotene+erlotinib, and sorafenib) in the hopes of yielding better clinical outcomes.
- the primary end-point was 8-week disease control (DC) rate, with a secondary endpoint of overall survival (OS).
- the data support a conclusion that the KRAS- variant meaningfully subgroups patients as responders or non-responders regardless of tumor acquired mutations in NSCLC, similar as to colon cancer.
- the KRAS- variant significantly separated the group, with double mutant patients (tumor acquired KRAS and KRAS- variant) having significantly longer PFS than patients with only tumor acquired KRAS mutations ( Figure 16).
- Example 5 The KRAS Variant and Resistance to Cisplatin [237] Methods of the disclosure predict the resistance of any cancer cell, tumor cell, or subject having any cancer, to treatment with cisplatin following detection of the KRAS
- the power of the KRAS Variant to predict resistance to cisplatin treatment is demonstrated below for an exemplary cancer.
- the data provided herein demonstrate that, in patients with recurrent and/or metastatic head neck squamous cell carcinoma, the KRAS Variant predicts resistance to cisplatin treatment.
- HNSCC Head and neck squamous cell carcinoma
- Risk factors for HNSCC include tobacco and alcohol consumption, as well as human papillomavirus (HPV) infection.
- HPV human papillomavirus
- Recent studies have concluded that HPV-positive and -negative HNSCC represent distinct diseases.
- -20% of HPV-positive and -50% of HPV-negative HNSCC patients experience treatment failure and subsequent disease-related death.
- the current standard of care for first- line recurrent and/or metastatic (R/M) disease is a platinum-based combination regimen (e.g. cisplatin), 5-fluorouracil (5-FU) and cetuximab.
- R/M metastatic
- treatment efficacy is still limited in these patients, with overall survival ranging from 7-10 months.
- the KRAS- variant is specifically associated with therapeutic resistance.
- ovarian cancer patients demonstrate enhanced platinum resistance and poor outcome (see Example 2). Additionally,
- colon cancer patients are more resistant to cetuximab when given in combination with irinotecan chemotherapy, but more sensitive to cetuximab is given as a monotherapy (see Example 3).
- the KRAS Variant predicts resistance to a platinum-based
- Tumor samples were collected from three previously published clinical trials and one exempted tissue collection study; 1) 24 samples from FTN0582
- NCT00003809 a randomized, double-blind, placebo controlled phase III evaluation of cisplatin+placebo versus cisplatin+C225 in patients with R/M FINSCC ( Burtness B, et al. J Clin Oncol 2005;23 : 8646-54), and 4) 83 samples from an exempted retrospective tissue collection study.
- Genomic DNA from the cell lines and formalin-fixed paraffin-embedded tumors was isolated as previously described and 100 ng of DNA from blinded samples was run on a CLIA-certified AT ⁇ S-variant assay (Weidhaas JB, et al. Cancer Res 2007;67: 1 1 1 1 1-6).
- pl6 status determination In this study, pl6 status was used as a control to ensure the validity of experimental results. pl6 status was determined by immunohistochemistry using a pl6 mouse monoclonal antibody (prediluted, MTM-CINtech, 9518). pl6 positivity was determined by diffuse staining with greater than 70% of the tumor staining positive as previously described (Ang K , et al. N Engl J Med 2010;363 : 24-35).
- HNSCC cell lines Eight HNSCC cell lines (Cal27,
- Cisplatin was purchased from Cell Signaling Technology (Danvers, MA). [247] MTS assay: To assess in vitro proliferation in the presence of cisplatin, MTS assay was used to estimate relative cell growth. Cells were seeded at a density of 1 ,000 cells/well in 96-well plates. Drug-containing media was added the following day to these cultures. Cells were then allowed to proliferate 72-96 hours before adding PMS (phenazine methosulfate) and MTS according to the manufacturer's protocol (Promega, Madison, WI).
- PMS phenazine methosulfate
- KRAS- variant incidence was reported by frequency and percentage of TG/GG samples with respect to total samples with known status. Descriptive statistics were used to characterize patient demographics and disease characteristics. Associations between KRAS- variant status and patient characteristics/response were evaluated using Fisher's exact tests. Progression- free survival (PFS) was defined as the time between date of study entry and date of progression or death from any cause, censored at date of last disease assessment. Overall survival (OS) was defined as the time between date of study entry and date of death, censored at last contact. Event-time distributions were estimated by the Kaplan-Meier method and compared using log-rank tests.
- Microarrays were normalized using frozen robust multiarray analysis (fRMA) (McCall MN, et al. Biostatistics 2010;11 : 242-53).
- fRMA frozen robust multiarray analysis
- Empirical Bayes moderated t-statistics were computed to compare gene expression from KRAS variant (TG/GG) samples to wild type (TT) samples using LIMMA (Smyth GK. Statistical applications in genetics and molecular biology 2004; 3: Article 3). Benjamini- Hotchberg multiple testing correction was applied to the corresponding p-values (Benjamini Y and Hochberg Y. J Roy Statist Soc Ser B 1995;57: 289-300).
- KRAS- variant status is associated with cisplatin resistance in R/M HNSCC patients: To determine whether status is associated with cisplatin resistance in R M HNSCC patients, survival analyses were performed for patients from HN0501 and E5397. Both of these studies were powered to determine survival benefit and also enrolled similar first-line R/M patients (Burtness B, et al. J Clin Oncol 2005; 23 : 8646-54 and Chung CH, et al. Ann Oncol 2009). HN0582 was not included in these analyses because the primary end point of this study was a clinical response rate of the induction chemotherapy rather than survival outcomes. It was determined that no association was evident between KRAS- variant status and disease control response (DCR) or objective response (ORR) (Table 17).
- DCR disease control response
- ORR objective response
- Table 17 Response by the KRAS- variant genotypes and the treatment regimens in HN0501 and E5397.
- KRAS-vanant status was compared with progression-free survival (PFS) and overall survival (OS) in these studies separately, as patients were differently treated.
- PFS progression-free survival
- OS overall survival
- HRs hazard ratios
- KRAS-variant status was further examined in E5397, between the cisplatin + placebo or cisplatin + cetuximab treatment arms (Table 18 and Figure 17E).
- PFS was significantly improved in KRAS- variant (TG/GG) patients who received cetuximab in univariate analysis (median: 1.9 months in cisplatin+placebo vs.
- Table 18 Event-time distribution by the KRAS- variant genotypes and the treatment regimens in HN0501 and E5397.
- N 21 in the cisplatin+placebo arm and in the cisplatin+cetuximab arm.
- KRAS-variant HNSCC cell lines are more resistant to cisplatin in vitro: To determine if KRAS-variant status affects in vitro cisplatin sensitivity, eight FiNSCC cell lines were evaluated by MTS assay with increasing doses of cisplatin ( ⁇ - ⁇ ). Six of these cell lines were HPV-negative (CAL27, UNC7, FaDu, SK 3, SCC6 and SCC61), while two were HPV-positive (UPCLSCC90 and UMSCC47). KRAS 3'UTR analysis determined three of these cell lines had the A ⁇ S-variant: CAL27, UNC7, and UMSCC47.
- epidermal growth factor (EGF) is upregulated (logFC: 0.77; p-value: 6.00xl0 "5 ) in KRAS-vanant compared to non-variant tumors, suggesting that increased KRAS activity through the KRAS-variant is dependent on upstream growth signal through EGFR.
- the dependence of the KRAS variant upon upstream growth signal through EGFR may explain the enhanced disease control observed in these patients with cetuximab.
- Another interesting trend observed in KRAS- variant tumor gene expression is the upregulation of various cytoskeleton and microtubule associated proteins.
- DST (dystonin or bullous pemphigoid antigen 1, logFC: 1.88; p-value: 3.98xl0 "6 ) and MACFl (logFC: 0.90; p-value: 4.22xl0 "5 ) are both members of the plakin family, responsible for junctional complex and cytoskeleton function reviewed in (Sonnenberg A and Liem RK. Exp Cell Res
- EML6 echinoderm microtubule-associated protein-like 6, logFC: 0.93; p-value: 2.07xl0 ⁇ 4
- EML6 echinoderm microtubule-associated protein-like 6, logFC: 0.93; p-value: 2.07xl0 ⁇ 4
- Houtman SH et al. Neuroscience 2007; 144: 1373-82
- SYNJ2 (synaptojanin 2, logFC: 1.08; p- value: 2.58xl0 ⁇ 4 ) is a regulatory lipid phosphatase which dephosphorylates PIP 3 to PIP 2 at the D-5 position in a manner similar to SHIP1 or SHIP2 (Nemoto Y, et al. J Biol Chem 1997; 272: 30817-21). While its function is most important for vesicle uncoating in neurons (Verstreken P, et al. Neuron 2003;40: 733-48), SYNJ2 upregulation has previously been associated with metastatic spread (Roesli C, et al.
- Table 19 Top 25 most differentially expressed probes between the KRAS-variant (TG/GG) and non- variant (TT)
- the presence of the KRAS- variant was significantly associated with poor survival, and these effects were mostly observed in oral cancer, but not pharyngeal or laryngeal tumors.
- the higher incidence rate (24%) of this mutation indicates two possibilities: 1) the presence of a KRAS- variant is associated with particularly aggressive de novo disease or 2) this variant confers increased resistance to first-line therapy, enriching this variant within this sample population. Current evidence suggests that these mechanisms may not be mutually exclusive.
- Identifying biomarkers of cisplatin resistance in HNSCC is critically important as cisplatin is the most commonly used chemotherapy in HNSCC and often administered in the
- the upregulation of an EGFR-specific growth stimulatory ligand (EGF), a pro-migratory phosphatase (SYNJ2), and various components of the microtubule/cytoskeletal architecture (DST, MACF1, EML6) indicates the presence of an enhanced migratory or metastatic gene expression profile associated with KRAS-vanant FiNSCCs as well as explains an enhanced sensitivity of KRAS -variant cancer cells (e.g. FiNSCCs) to cetuximab treatment.
- EGF EGFR-specific growth stimulatory ligand
- SYNJ2 pro-migratory phosphatase
- DST, MACF1, EML6 various components of the microtubule/cytoskeletal architecture
- TG/GG KRAS- variant is a biomarker of altered response to cisplatin and cetuximab treatment. Consequently, platinum- based regimens provide suboptimal disease control in these patients, and treatment with cetuximab should be considered.
- the KRAS variant confers a unique biology to tumors of various subtypes.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Pathology (AREA)
- Analytical Chemistry (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Physics & Mathematics (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Hospice & Palliative Care (AREA)
- Biophysics (AREA)
- Oncology (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The disclosure provides methods for predicting a subject's response to cancer treatment by determining the presence or absence of a SNP in the KRAS oncogene, known as the KRAS variant.
Description
THE KRAS VARIANT AND RESPONSE TO CANCER THERAPY
FIELD OF THE DISCLOSURE
[01] This disclosure relates generally to the fields of cancer and molecular biology. The disclosure provides methods for predicting a subject's sensitivity or resistance to therapeutic agents used to treat cancer by determining the presence or absence of a genetic marker.
Moreover, the disclosure provides methods for prognosing a subject based at least in part upon the subject's predicted response to treatment.
BACKGROUND
[02] The heterogeneity of cancer is reflected by the variable risk factors, treatment response and outcome in patients. While prognostic gene expression markers are highly divergent, several modules such as DNA repair deficiency, signatures of immune response or epithelial-to-mesenchymal transition are commonly found to be relevant for a subset of tumors. Thus, there is a need in the art for the identification of the drivers of these
transcriptional modules as a promising approach for the discovery of specific and
personalized therapies.
SUMMARY
[03] This disclosure identifies a gene marker, the KRAS Variant, which is predictive of a subject's response to cancer therapy. The KRAS Variant is a SNP located in the let-7 complementary site 6 (LCS6) of the 3' UTR of the KRAS gene in which the nucleotide at position 4 of the LCS6 undergoes a uracil (U) or thymine (T) to guanine (G) transition. The KRAS Variant provides superior predictive power over known germ-line mutations used as cancer biomarkers in the art because the presence of the KRAS Variant is predictive of the response of a cancer cell of any type or subtype to a particular cancer treatment or therapeutic agent.
[04] The data provided in this disclosure demonstrate that the presence of the KRAS
Variant within a cancer cell indicates resistance to known agents including, but not limited to, cisplatin ((5P-4-2)-diamminedichloridoplatinum, also known as cisplatinum, or cis- diamminedichloroplatinum(II) (CDDP)), carboplatin (czs-diammine(cyclobutane-l,l- dicarboxylate-0,O platinum(II) also known as cis-Diammine(l,l-
cyclobutanedicarboxylato)platinum(II)), gefitinib (N-(3-chloro-4-fluoro-phenyl)-7-methoxy- 6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine), and a combination therapy of cetuximab (monoclonal antibody) and irinotecan ((5)-4,l l-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14- dioxolH-pyrano[3',4' :6,7]-indolizino[l,2-b]q^^
chemotherapy in all cancer types. Moreover, the data provided in this disclosure demonstrate that the presence of the KRAS Variant within a cancer cell indicates sensitivity to sorafenib
(4-[4- [ [4-chloro-3 -(trifluoromethyl)phenyl] carbamoylamino]phenoxy] -N-methyl-pyridine-2- carboxamide) and cetuximab (as a monotherapy) in all cancer types.
[05] Cancer and cancer cells of the disclosure include, but are not limited to those cells derived from a(n) AIDS-related cancer, breast cancer, cancer of the digestive/gastrointestinal tract, anal cancer, appendix cancer, bile duct cancer, colon cancer, colorectal cancer, esophageal cancer, gallbladder cancer, islet cell tumors, pancreatic neuroendocrine tumors, liver cancer, pancreatic cancer, rectal cancer, small intestine cancer, stomach (gastric) cancer, endocrine system cancer, adrenocortical carcinoma, parathyroid cancer, pheochromocytoma, pituitary tumor, thyroid cancer, eye cancer, intraocular melanoma, retinoblastoma, bladder cancer, kidney (renal cell) cancer, penile cancer, prostate cancer, transitional cell renal pelvis and ureter cancer, testicular cancer, urethral cancer, Wilms' tumor, other childhood kidney tumors, germ cell cancer, central nervous system cancer, extracranial germ cell tumor, extragonadal germ cell tumor, ovarian germ cell tumor, gynecologic cancer, cervical cancer, endometrial cancer, gestational trophoblastic tumor, ovarian epithelial cancer, uterine sarcoma, vaginal cancer, vulvar cancer, head and neck cancer, hypopharyngeal cancer, laryngeal cancer, lip and oral cavity cancer, metastatic squamous neck cancer with occult primary, mouth cancer, nasopharyngeal cancer, oropharyngeal cancer, paranasal sinus and nasal cavity cancer, pharyngeal cancer, salivary gland cancer, throat cancer, musculoskeletal cancer, bone cancer, Ewing's sarcoma, gastrointestinal stromal tumors (GIST), osteosarcoma, malignant fibrous histiocytoma of bone, rhabdomyosarcoma, soft tissue sarcoma, uterine sarcoma, neurologic cancer, brain tumor, astrocytoma, brain stem glioma, central nervous system atypical teratoid/rhabdoid tumor, central nervous system embryonal tumors, central nervous system germ cell tumor, craniopharyngioma, ependymoma, meduUoblastoma, spinal cord tumor, supratentorial primitive neuroectodermal tumors and pineoblastoma,
neuroblastoma, respiratory cancer, thoracic cancer, non-small cell lung cancer, small cell lung cancer, malignant mesothelioma, thymoma, thymic carcinoma, skin cancer, Kaposi's
sarcoma, melanoma, or Merkel cell carcinoma. Alternatively, or in addition, the tumor or cancer is metastatic.
[06] When performing the methods of the disclosure, a non-cancer and/or a cancer cells may be evaluated in vivo, in vitro, or ex vivo to determine the presence or absence of thr KRAS variant. Because the KRAS variant is a germline mutation, the KRAS variant mutation is present and detectable in any cell of the subject, including non-cancer as well as cancer cells. Alternatively, or in addition, DNA isolated from any cell in the body of the subject may be evaluated for the presence or absence of the KRAS variant.
[07] When performing the methods of the disclosure, non-cancer and/or cancer cells may be evaluated in vivo, in vitro, or ex vivo. When a non-cancer and/or cancer cell is evaluated ex vivo, the cell may be obtained from a subject. Preferably, the subject has cancer. The subject may be of any age. In certain embodiments of this method the subject is at least 30, 35, 40, 45, 50, 55, 60, 65, 70, 75 years of age, or any age in between.
[08] When a non-cancer and/or cancer cell is evaluated in vitro, the cell may be isolated, reproduced, cloned or derived from an established cell lines, including, but not limited to, a cancer cell line included in the NCI-60 panel.
[09] When performing the methods of the disclosure, cancer cells may be isolated, reproduced, cloned or derived from any cancer cell line, including, but not limited to, those cell lines that carry the KRAS- variant, either alone, or in combination with a second or additional mutation in KRAS or another gene.
[10] Although the methods described herein may be applied to subjects of all ages, in certain embodiments of this method, the subject is a newborn, child, adult, or senior (aged 65 or above). Although the methods described herein may be applied to subjects of both sexes, male and female, in certain embodiments of this method, the subject is female. Alternatively, the subject may be male. Female subjects may be either pre-menopausal or post-menopausal. Post-menopausal subjects may be aged 52 years or more. In this disclosure the terms subject and patient are used interchangeably.
[11] As described in this disclosure, the term "survival rate" may be an overall survival rate (for instance, some examples, include, but are not limited to, survival rates calculated from the time of cancer development or diagnosis until the subject succumbs to the cancer
(death), enters remission, or a doctor declares the subject cured or clean of all cancer cells), five-year survival rate or one-year survival. Shorter survival periods are calculated, for
instance, from either the development or diagnosis of the cancer until a determined time, such as one or five years.
[12] As used herein, the terms "resistant" or "sensitive" may be used as relative terms when compared to a known or control value or compared to a measurement from a control subject. Control subjects may include healthy individuals and those women who have EOC, but who do not carry the KRAS-variant. Moreover, the control subject can be a national average based upon the expected survival of women born in the same year as the test subject, or who belong to the same generation as the test subject. In a preferred embodiment, this control value does not include those individuals who carry the KRAS-variant. In certain aspects of this method, the survival rate is an overall survival rate (for instance, some examples, include, but are not limited to, survival rates calculated from the time of cancer development or diagnosis until the subject succumbs to the cancer (death), enters remission, or a doctor declares the subject cured or clean of all cancer cells), five-year survival rate or one-year survival. Shorter survival periods are calculated, for instance, from either the development or diagnosis of the cancer until a determined time, such as one or five years. The KRAS variant confers resistance to platinum-based chemotherapy
[13] The disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to platinum-based chemotherapy. In certain embodiments of these methods, treatment of cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to platinum-based chemotherapy.
[14] In certain aspects of this embodiment, treatment of epithelial ovarian cancer (EOC) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to platinum-based chemotherapy. For all aspects of these methods, the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR ofKRAS.
[15] Although the methods of the disclosure can be applied to all subjects, in certain embodiments of these methods, the subject is a woman. Although the methods of the disclosure can be applied to subjects of all ages, in certain embodiments of these methods, the subject is post-menopausal or 52 years of age or older. For example, the methods described herein may be used to optimize the treatment of epithelial ovarian cancer (EOC) by assaying for the presence of the KRAS Variant and, if present, predicting resistance to platinum-based
chemotherapy. Moreover, the methods described herein may be used to optimize the treatment of epithelial ovarian cancer (EOC) in a female, post-menopausal subject by assaying for the presence of the KRAS Variant and, if present, predicting resistance to platinum-based chemotherapy.
[16] The disclosure provides a method of predicting the response of an epithelial ovarian cancer (EOC) cell to a platinum-based chemotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and wherein the presence of the mutation indicates a resistance to platinum-based chemotherapy. The EOC cell may be evaluated in vitro or ex vivo. When the EOC cell is evaluated ex vivo, the cell is obtained from a subject. The subject may be of any age, however, in a preferred embodiment, the subject is either post-menopausal or at least 52 years old. Alternatively, in the same embodiment, the subject is at least 30, 35, 40, 45, 50, 55, 60, 65, 70, 75 years of age, or any age in between. In other aspects of this method, the subject is not postmenopausal, but presents a similar hormonal profile due to a second medical condition or medical treatment. An exemplary, but non-limiting menopausal hormonal profile includes decreased levels of estrogen and progesterone hormone, as determined by, for instance, assessment of a sample of the subject's blood or urine. Exemplary, but non-limiting, secondary medical conditions that induce a menopausal hormonal profile are surgical removal of at least one ovary (ovariectomy, also known as surgical menopause), cervical, uterine or ovarian cancer that necessitates a hysterectomy (especially if removal of the uterus is combined with removal of the Fallopian tubes and one or both ovaries). Exemplary, but non-limiting, secondary medical conditions that induce a menopausal hormonal profile are chemotherapy and anti-estrogen treatments.
[17] When the EOC cell is evaluated in vitro, the cell is isolated, reproduced, or derived from the BGl, CAOV3, or IGR-OVl cell lines. These cell lines are non-limiting examples of ovarian cancer cell lines. An EOC cell may be isolated, reproduced, or derived from any ovarian cancer cell line, including, but not limited to, those cell lines that carry the KRAS- variant, a deleterious BRCAI mutation, a deleterious BRCA2 mutation, or any combination thereof. A deleterious BRCAI or BRCA2 mutation is a mutation that increases the risk or likelihood that it's carrier will develop cancer, and, in preferred embodiments, breast or ovarian cancer. A deleterious BRCAI or BRCA2 mutation is a mutation that also increases the
risk or likelihood that it's carrier will develop cancer at a younger age (i.e. experience an earlier onset of cancer), and, in preferred embodiments, the cancer is breast or ovarian cancer.
[18] In certain aspects of this embodiment, treatment of recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to platinum-based chemotherapy. In certain aspects of this embodiment, the platinum-based chemotherapy is cisplatin, however, the methods may be applied to any platinum-based chemotherapy. For all aspects of these methods, the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR of KRAS. Alternatively, or in addition, treatment of recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (FINSCC ) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting sensitivity or an enhanced response to monoclonal anti-EGFR antibody therapy, either as a monotherapy or as a combined therapy with another chemotherapeutic agent or medical procedure (e.g. radiation or surgery).
[19] The disclosure provides a method of predicting the response of head and neck squamous cell carcinoma (HNSCC ) cell and/or a recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) cell to a platinum-based chemotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and wherein the presence of the mutation indicates a resistance to platinum-based chemotherapy. In certain aspects of this embodiment, the disclosure provides a method of predicting the response of head and neck squamous cell carcinoma (HNSCC ) cell and/or a recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) cell to a monoclonal anti-EGFR antibody therapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and wherein the presence of the mutation indicates sensitivity or an enhanced response to monoclonal anti-EGFR antibody therapy, either as a monotherapy or as a combined therapy with another chemotherapeutic agent or medical procedure (e.g. radiation or surgery).
[20] The HNSCC cell may be evaluated in vivo, in vitro or ex vivo. When the HNSCC cell is evaluated ex vivo, the cell is obtained from a subject.
[21] For the methods described herein, the preferred platinum-based chemotherapy is cisplatin, carboplatin or paclitaxel, however, the term "platinum-based chemotherapy" encompasses all chemotherapy agent that incorporate platinum or a platinum salt to treat or prevent cancer. In certain aspects of these methods, the platinum-based chemotherapy is an adjuvant therapy. Therefore, the methods described herein predict a patient's response to the use of a platinum-based chemotherapy as either a monotherapy or a combination therapy with other anti-cancer agents or techniques (e.g. radiation and surgery, for example).
The KRAS variant confers resistance to small molecule EGFR inhibitors
[22] The disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors). In certain embodiments of these methods, treatment of cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors).
[23] In certain aspects of this embodiment, treatment of non-small cell lung cancer (NSCLC) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors and small molecule EGFR inhibitors). For all aspects of these methods, the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR ofKRAS.
[24] In certain aspects of this embodiment, chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors or small molecule EGFR inhibitors), include, but are not limited to, gefinitib and erlotinib. In certain aspects of this embodiment, chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors or small molecule EGFR inhibitors), include, but are not limited to, gefinitib.
The KRAS variant confers sensitivity to MAP kinase pathway inhibitors
[25] The disclosure provides a method of treating a cancer patient, the method comprising administering a MAP kinase pathway inhibitor to a patient after a determination that the patient has a mutation in let-7 complementary site LCS6 of human KRAS, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine transition at position 4 of LCS6.
[26] The disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors). In certain embodiments of these methods, treatment of cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors).
[27] In certain aspects of this embodiment, treatment of non-small cell lung cancer (NSCLC) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors). For all aspects of these methods, the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR ofKRAS.
[28] In certain aspects of this embodiment, chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors), include, but are not limited to, inhibitors of tyrosine protein kinases and serine/threonine kinases. Nonlimiting examples of tyrosine protein kinases include the VEGFR (vascular endothelial growth factor receptor) and PDGFR (platelet-derived growth factor receptor). Nonlimiting examples of
serine/threonine protein kinases include C-raf (encoded by the RAFl gene) and B-raf (encoded by the BRAF gene). Chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors), include, but are not limited to, sorafenib.
The KRAS variant confers sensitivity to monoclonal anti-EGFR antibody monotherapy
[29] The disclosure provides a method of treating a cancer patient, the method comprising administering a MAP kinase pathway inhibitor to a patient after a determination that the patient has a mutation in let-7 complementary site LCS6 of human KRAS, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine transition at position 4 of LCS6. In certain aspects of this method, the MAP kinase pathway inhibitor is a
monoclonal anti-EGFR antibody.
[30] The disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to monoclonal anti- EGFR antibody therapy. In certain embodiments of these methods, treatment of cancer is
optimized by assaying for the presence of the KRAS Variant and, if present, predicting sensitivity to monoclonal anti-EGFR antibody therapy.
[31] In certain aspects of this embodiment, treatment of colorectal cancer (CRC) or metastatic CRC (mCRC) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting sensitivity to monoclonal anti-EGFR antibody therapy. For all aspects of these methods, the KRAS variant is defined as a SNP comprising a uracil (U) or thymine
(T) to guanine (G) transition at position 4 of LCS6 in the 3 ' UTR of KRAS.
[32] The disclosure provides a method of predicting the response of a cancer cell to a monoclonal anti-EGFR antibody monotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, wherein the presence of the mutation indicates a sensitivity to monoclonal anti-EGFR antibody monotherapy. In certain embodiments of this method, the cancer cell is a colorectal cancer (CRC) cell. The cancer cell may be evaluated in vitro or ex vivo. A non- limiting example of the monoclonal antibody monotherapy is Cetuximab.
[33] The disclosure provides a method of predicting the response of a cancer cell to a monoclonal anti-EGFR antibody monotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 (the
KRAS Variant), and either a mutation in BRAF or hypermethylation of RASSFIA, wherein the presence of the KRAS Variant and either a mutation in BRAF or hypermethylation of
RASSFIA indicates a sensitivity to monoclonal anti-EGFR antibody monotherapy. In certain embodiments of this method, the cancer cell is a colorectal cancer (CRC) cell. The cancer cell may be evaluated in vitro or ex vivo. A non- limiting example of the monoclonal anti-EGFR antibody monotherapy is Cetuximab. When combining (epi)genetic events, the better outcome of patients with a combination of the KRAS variant (G-allele) and an alternation of
KRAS, BRAF, or RASSFIA was even more enhanced. Thus, Ras overexpression due to the
KRAS Variant (G-allele), in combination with (epi)genetic alterations in genes from the Ras pathway, induce senescence cancer cells, including, but not limited to, early-stage colorectal cancer cells (CRC cells), thereby enhancing or promoting survival of the subject.
[34] In certain aspects of this embodiment, treatment of head and neck squamous cell carcinoma (FINSCC ) or a recurrent and/or metastatic (R/M) head and neck squamous cell
carcinoma (HNSCC ) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting sensitivity to monoclonal anti-EGFR antibody therapy. For all aspects of these methods, the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR of KRAS.
[35] The disclosure provides a method of predicting the response of a head and neck squamous cell carcinoma (FiNSCC ) cell to a monoclonal anti-EGFR antibody monotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, wherein the presence of the mutation indicates a sensitivity to monoclonal anti-EGFR antibody monotherapy. The head and neck squamous cell carcinoma (FiNSCC ) cell may be evaluated in vivo, in vitro or ex vivo. A non-limiting example of the monoclonal anti-EGFR antibody monotherapy is Cetuximab.
[36] EGF, a potent ligand of EGFR, is upregulated in KRAS- variant positive FiNSCC tumors. It is hypothesized that cetuximab is beneficial for these patients because the anti- EGFR antibody blocks the pro-growth signal provided by an upregulation of EGF in these tumors. Furthermore, a number of genes associated with microtubule and cytoskeleton function are also upregulated. More specifically, MACF1 is upregulated in our current analysis. MACF1 interacts with ErbB2 to control microtubule capture during cell migration. Taken together, the upregulation of an EGFR-specific growth stimulatory ligand (EGF), a pro-migratory phosphatase (SYNJ2), and various components of the microtubule/cytoskeletal architecture (DST, MACF1, EML6) indicates the presence of an enhanced migratory or metastatic gene expression profile associated with
 FTNSCCs as well as explains an enhanced sensitivity of KRAS -variant cancer cells (e.g. FTNSCCs) to cetuximab treatment. Furthermore, to optimize cancer treatment, methods of the disclosure include detection of the KRAS variant in combination with detection of upregulation of EGF, wherein the combined presence of the KRAS variant and upregulated EGF mRNA or protein indicates sensitivity of the cancer cell to a monoclonal anti-EGFR antibody therapy.
FTNSCCs as well as explains an enhanced sensitivity of KRAS -variant cancer cells (e.g. FTNSCCs) to cetuximab treatment. Furthermore, to optimize cancer treatment, methods of the disclosure include detection of the KRAS variant in combination with detection of upregulation of EGF, wherein the combined presence of the KRAS variant and upregulated EGF mRNA or protein indicates sensitivity of the cancer cell to a monoclonal anti-EGFR antibody therapy.
The KRAS variant confers resistance to a combinatorial therapy comprised of a monoclonal anti-EGFR antibody and a chemotherapeutic agent.
[37] The disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to a combinatorial therapy comprised of a monoclonal anti-EGFR antibody and a chemotherapeutic agent. In
certain embodiments of these methods, treatment of cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to a combinatorial therapy comprised of a monoclonal anti-EGFR antibody and a chemotherapeutic agent. In certain aspects of this embodiment, treatment of colorectal cancer (CRC) or metastatic CRC (mCRC) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to a combinatorial therapy comprised of a monoclonal anti-EGFR antibody and a chemotherapeutic agent. For all aspects of these methods, the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR of KRAS.
[38] The disclosure provides a method of predicting the response of a cancer cell to the combination of a chemotherapy and monoclonal anti-EGFR antibody therapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and wherein the presence of the mutation indicates a resistance to the combination. In certain embodiments of this method, the cancer cell is a colorectal cancer (CRC) cell. The cancer cell may be evaluated in vitro or ex vivo. A non- limiting example of the monoclonal anti-EGFR antibody monotherapy is Cetuximab. The chemotherapy may be a cytotoxic agent. A non-limiting example of the cytotoxic agent is irinotecan.
[39] In certain embodiments of the methods described herein, treatment of a subject carrying the KRAS-vanant with a chemotherapeutic agent (e.g. irinotecan) results in increased expression of the KRAS-variant. When reporter expression is compared following irinitecan exposure in KRAS-variant versus non-variant cancer cells, no change was found in expression of the wild-type 3 'UTR reporter. However, a statistically-significant increase in expression in the KRAS- variant 3'UTR reporter was discovered (Figures 14A and 14B). The data indicates that irinotecan exposure changes the cellular context in a manner that activates the KRAS- variant allele.
[40] Methods of the disclosure encompass any monoclonal anti-EGFR antibody used to treat or prevent cancer. Preferably, the monoclonal anti-EGFR antibody is in part or entirely human (e.g. chimeric) or humanized. Methods of the disclosure encompass any
chemotherapy agent that is used to treat or prevent cancer. In certain aspects of this method, the chemotherapy or cytotoxic chemotherapy is an adjuvant therapy. Methods of the
disclosure predict the response of a cancer cell or a subject having a cancer cell to treatment, including monotherapies or combination therapies. For example, the methods of the disclosure predict the response of a cancer cell or a subject having a cancer cell to treatment with a monoclonal anti-EGFR antibody and/or a chemotherapy used in conjunction with an additional medical procedure. Additional medical procedures include, but are not limited to, radiation and surgery.
BRIEF DESCRIPTION OF THE DRAWINGS
[41] Figure 1 is a graph showing that the KRAS variant predicts significantly worse overall survival for postmenopausal ovarian cancer patients over 52 years of age. Overall survivals for ovarian cancer patients with (n= 59) and without (n= 220) the KRAS variant are compared using the Kaplan -Meier analysis. Outcome is significantly worse for KRAS variant positive EOC patients over 52 years of age by log-rank test (P = 0.0399).
[42] Figure 2 is a graph showing that the KRAS variant is associated with suboptimal debulking after neoadjuvant chemotherapy. Surgical debulking after neoadjuvant
chemotherapy is compared in ovarian cancer patients (n = 116) with the KRAS variant (n = 26) or without (n = 90). By χ analysis, KRAS-vaxiant patients are significantly more likely to be suboptimally debulked with greater residual disease (RD) than are non- variant patients (P = 0.044).
[43] Figure 3A is a signature of 50 differentially expression gene candidates in KRAS variant (KV) triple-negative breast tumors (TNBC KRAS Signature) that shows higher scores in KV EOC samples than in non- variant samples.
[44] Figure 3B is a signature of genes associated with KRAS-addicted tumors (KRAS Addiction Signature), which are upregulated in KV EOC tumors.
[45] Figure 3C a signature of differential expression of the top 20 genes in KV EOC tumors, reflecting a re-analysis of differential gene expression in carboplatin-sensitive and carboplatin-resistant EOC cells.
[46] Figure 3D is a heat map of the top differentially expressed genes between KV (dark gray) and non- variant (light gray) tumor samples. The color key depicts a spectrum from blue
(values 0 to 5) to white (approximately 5), and from white to red (5 to 10). For a color version of this heat map, see Ratner ES, et al. Oncogene, (5 December 2011), 1-8; the contents of which are incorporated herein by reference).
[47] Figure 4 is a graph showing that the KRAS variant confers resistance to carboplatin and carboplatin/taxol chemotherapy in cell lines. Cell lines with the KRAS variant (BGl) and without the KRAS variant (CAOV3) were treated with chemotherapy and half-maximal inhibitory concentration (IC50) is shown on the Y axis, and chemotherapeutic agent on the X axis. Higher IC50 represents resistance to the tested chemotherapeutic agent. BGl = KRAS variant/^ CA wild-type cell line; CAOV3 = non-variant/^ CA wild-type cell line; IGR-OV1 = KRAS-vanant/BRCAl mutant cell line. Error bars are RSE.
[48] Figure 5 A is a graph showing decreased cell survival in the KRAS- variant line, BGl (*P < 0.001), with no effect on the non-variant line, CAOV3. Cell lines, with (BGl) and without (CAOV3) the KRAS variant, were treated with siRNA/miRNA combinations that bind selectively to the variant allele.
[49] Figure 5B is a graph showing decreased KRAS protein expression in BGl (right) concordant with the decrease in cell survival, with no effect on CAOV3 (left). Cell lines, with (BGl) and without (CAOV3) the KRAS variant, were treated with siRNA/miRNA
combinations that bind selectively to the variant allele. Different siRNAs are denoted by numbers.
[50] Figure 6 is a graph depicting Cell lines with the KRAS variant (BG-1 and IGROV1) have significantly lower levels of let-7b compared to a non- variant cell line (CaOV3).
Statistical analysis was done with a one way Anovea and Tukey's Multiple comparison test.
[51] Figure 7A-B is a schematic depicting an alignment of the KRAS- variant sequence with non-variant sequences. Panel A depicts a non-variant sequence of KRAS. Panel B depicts exemplary variant siRNA oligos targeted to the KRAS-yaxiant sequence. In both panels, the underlined sequence depicts the let- 7 binding site. In both panels, the boxed nucleotide represents either the wild type (non- variant) nucleotide (A) or the KRAS variant single nucleotide polymorphism (B). siRNAs are shown starting with their 3' end.
[52] Figure 8A is a graph depicting the median progression free survival according to the KRAS LCS6 genotype status in patients treated with anti-EGFR mAbs monotherapy or in combination with chemotherapy as salvage treatment.
[53] Figure 8B is a graph depicting the median overall survival according to the KRAS LCS6 genotype status in patients treated with anti-EGFR mAbs monotherapy or in combination with chemotherapy as salvage treatment.
[54] Figure 9A is a graph depicting the median progression-free survival according to the KRAS LCS6 genotype status in all patients treated with anti-EGFR mAbs monotherapy as salvage treatment.
[55] Figure 9B is a graph depicting the median progression-free survival according to the KRAS LCS6 genotype status in all patients treated with anti-EGFR mAbs based combination chemotherapy as salvage treatment.
[56] Figure 9C is a graph depicting the median progression-free survival according to type of therapy in all KRAS variant carriers.
[57] Figure 9D is a graph depicting the median progression-free survival according to type of therapy in all non-KRAS variant carriers.
[58] Figure 1 OA is a graph depicting the median progression-free survival according to the KRAS LCS6 genotype status in the double (KRAS and BRAF) wt patients' population treated with anti-EGFR mAbs monotherapy as salvage treatment.
[59] Figure 1 OB is a graph depicting the median progression-free survival according to the
KRAS LCS6 genotype status in the double (KRAS and BRAF) wt patients' population treated with anti-EGFR mAbs based combination chemotherapy as salvage treatment.
[60] Figure IOC is a graph depicting the Median progression- free survival according to type of therapy in the double (KRAS and BRAF) wt KRAS variant carriers
[61] Figure 10D is a graph depicting the Median progression- free survival according to type of therapy in the double (KRAS and BRAF) wt non-KRAS variant carriers.
[62] Figure 11 A is a graph depicting the median overall survival according to the KRAS
LCS6 genotype status in all patients treated with anti-EGFR mAbs monotherapy as salvage treatment.
[63] Figure 1 IB is a graph depicting the median overall survival according to the KRAS LCS6 genotype status in all patients treated with anti-EGFR mAbs based combination chemotherapy as salvage treatment.
[64] Figure 11C is a graph depicting the median overall survival according to type of therapy in all KRAS variant carriers.
[65] Figure 1 ID is a graph depicting the median overall survival according to type of therapy in all non-KRAS variant carriers.
[66] Figure 12A is a graph depicting the median overall survival according to the KRAS LCS6 genotype status in the double (KRAS and BRAF) wt patients' population treated with anti-EGFR mAbs monotherapy as salvage treatment.
[67] Figure 12B is a graph depicting the median overall survival according to the KRAS
LCS6 genotype status in the double (KRAS and BRAF) wt patients' population treated with anti-EGFR mAbs based combination chemotherapy as salvage treatment.
[68] Figure 12C is a graph depicting the median overall survival according to type of therapy in the double (KRAS and BRAF) wt KRAS variant carriers.
[69] Figure 12D is a graph depicting the median overall survival according to type of therapy in the double (KRAS and BRAF) wt non- KRAS variant carriers.
[70] Figure 13A is a graph depicting the median progression-free survival according to type of therapy in the KRAS and BRAF mutated KRAS variant carriers.
[71] Figure 13B is a graph depicting the median progression-free survival according to type of therapy in the KRAS and BRAF mutated non-KRAS variant carriers.
[72] Figure 13C is a graph depicting the median overall survival according to type of therapy in the KRAS and BRAF mutated KRAS variant carriers.
[73] Figure 13D is a graph depicting the median overall survival according to type of therapy in the KRAS and BRAF mutated non- KRAS variant carriers.
[74] Figure 14A is a graph depicting the normalized luciferase expression in wild type
KRAS and KRAS- variant cancer cells following treatment with the chemotherapeutic agent irinotecan.
[75] Figure 14B is a graph depicting the fold repression (expressed as KRAS
variant/KRAS wild type) as a function of irinotecan concentration, when cancer cells are treated with irinotecan.
[76] Figure 15A-B is a pair of graphs depicting the overall survival (OS) of non-small cell lung cancer (NSCLC) patients following treatment with either Vandetanib (A, left panel) or Sorafenib (B, right panel).
[77] Figure 16 is a graph depicting progression free survival (PFS) of non-small cell lung cancer (NSCLC) patients treated with sorafenib, including both carriers and non-carriers of the KRAS variant. The KRAS Variant predicts significantly better PFS in tumors having tumor-acquired KRAS mutations.
[78] Figure 17A-F is a series of graphs depicting Kaplan-Meier survival plots by KRAS- variant status (variant TG/GG vs. non-variant TT). A) Progression-free survival from
HN0501 (phase II trial of docetaxel+bortezomib in R/M HNSCC patients), B) overall survival from HN0501 (phase II trial of docetaxel+bortezomib in R/M HNSCC patients), C) Progression-free survival from E5397 (randomized phase III trial of cisplatin+placebo vs. cisplatin+cetuximab in R/M HNSCC patients), D) overall survival from E5397 (randomized phase III trial of cisplatin+placebo vs. cisplatin+cetuximab in R/M HNSCC patients), E) Progression-free survival by KRAS- variant status (variant TG/GG vs. non-variant TT) and E5397 treatment arms (cisplatin+placebo vs. cisplatin+cetuximab), F) overall survival by KRAS- variant status (variant TG/GG vs. non- variant TT) and E5397 treatment arms
(cisplatin+placebo vs. cisplatin+cetuximab).
[79] Figure 18 A-C is a series of graphs depicting MTS growth assays of KRAS variant and non-variant HNSCC cell lines treated with cisplatin. A) Dose-response
 (red line) and non-variant (black line) HPV-negative HNSCC cell lines, B) growth inhibition with respect to control at the highest cisplatin dose (10 DM). C) Dose-response of KRAS- variant (red line) and non-variant (black line) HPV-positive HNSCC cell lines.
(red line) and non-variant (black line) HPV-negative HNSCC cell lines, B) growth inhibition with respect to control at the highest cisplatin dose (10 DM). C) Dose-response of KRAS- variant (red line) and non-variant (black line) HPV-positive HNSCC cell lines.
[80] Figure 19 is a heatmap of a gene expression profile that is differentially expressed between AT^S-variant (TG/GG) and non-variant (TT) in 22 HNSCC tumors with available microarray data.
[81] Figure 20 is a graph depicting MEK inhibition in colorectal cancer cell lines.
[82] Figure 21 is a schematic and a graph showing that MEK inhibitor specificially target
HCC1937 rs61774270 positive cells.
[83] Figure 22 is a schematic and a graph showing Biomarker integrated Approaches of Targeted Therapy of Lung Cancer Elimination.
DETAILED DESCRIPTION
[84] A functional variant in a let- 7 microRNA complementary site in the 3 'UTR of the KRAS oncogene (rs61764370) associated with cancer was previously identified (International Patent Application No. PCT/US2008/065302, the contents of which are incorporated herein by reference in their entirety). This functional variant is also referred to herein as the KRAS Variant. An investigation of the association of this variant with cancer tumor biology is described herein.
[85] MicroRNAs (miRNAs) are an abundant class of highly conserved, endogenous, non- coding, small RNA molecules, 18-25 nucleotides in length, which negatively regulate gene expression by binding to partially complementary sites in the 3 '-untranslated region (UTR) of their target mRNAs. Upon processing by Dicer and Drosha RNase III endonucleases, mature miRNAs can suppress mRNA translation by directing an RNA-induced silencing complex to the target mRNA. MiRNAs regulate of a number of genes involved in basic biological processes such as proliferation, cellular differentiation and apoptosis, and act as important players in cancer development and progression by behaving either as oncogenes or as tumor suppressors. Although more than 700 miRNA sequences have been recognized in the human genome to date, this number is expected to double. Furthermore, each miRNA can control hundreds of genes by regulating many mRNAs simultaneously.
[86] MiRNA binding to mRNAs is critical for the regulation process of mRNA levels and subsequent protein expression, and this regulation can be affected by single-nucleotide polymorphisms (SNPs) occurring in the miRNA target sites. These SNPs can either create erroneous binding sites or abolish (eliminate) the correct ones, leading to resistance to miRNA regulation and reflecting another kind of genetic variability capable of playing a role in human diseases like cancer (or conferring an increased risk for certain diseases like cancer). Emerging research focuses on the systematic genomic evaluation of these sites and the functional and biological relevance of the detected SNPs, which are significant molecular markers in the rapidly growing area of personalized medicine. Such SNPs appear to affect not only gene expression, but also tumor biology and drug response and drug resistance.
[87] A single miRNA can regulate many mRNAs simultaneously. Moreover, miRNAs can act as both tumor suppressors and oncogenes. The lethal-7 (let-7) family of miRNAs is one of the first miRNA families to be discovered. The expression of let-7 family miRNAs is altered in many cancers. For example, in lung cancer, let-7 is poorly expressed and overexpression of let- 7 inhibits cell growth in vitro and in vivo, suggesting that let- 7 miRNAs may act as tumor suppressors. Disruption of miRNAs' regulation of oncogenes or tumor suppressor genes impact cancer risk, tumor development, and response to treatment. MiRNAs may regulate oncogenes or tumor suppressor genes directly or indirectly.
[88] Let-7 induces RAS downregulation after binding to specific sites in the 3'-UTR of
KRAS mRNA. The KRAS variant affects let- 7 mediated regulation of KRAS expression. The occurrence of the variant G-allele {i.e., the KRAS variant) leads to higher KRAS levels and
lower let-7 levels as compared to the wild type. G-allele carriers have an increased lung cancer risk in moderate smokers, an increased ovarian cancer risk (particularly for postmenopausal women), an increased risk of developing breast cancer (and, in particular, the triple negative breast cancer subtype), and a reduced survival in oral cancers but not in lung cancer. Acquired KRAS mutations are not the same as the KRAS variant, which is a congenital mutation, and, therefore, has a different effect on tumor development, biology, and thus prognosis.
[89] The KRAS variant disrupts regulation of KRAS by the let- 7 family of miRNAs. In this case, /et-7-mediated regulation of KRAS is disrupted; however, there are secondary effects of the KRAS variant. Disruption of the let-7 I KRAS interaction upstream perpetuates aberrant signaling to downstream factors. Furthermore, components of signaling pathways other than the canonical RAS pathway are affected. The presence of the KRAS variant increases angiogenesis, survival (even under hypoxic conditions), metastasis, and predicts resistance or sensitivity to frequently used chemotherapy agents. Moreover, epigenetic changes in the cancer cell, such as changes to promoter methylation of tumor suppressor and cell cycle genes, influence the development, survival, and response to treatment of a cancer cell positive for the KRAS variant. The cellular consequences of the KRAS variant are independent of other mutations in KRAS, including, for example, acquired mutations in a coding region of KRAS. For many cancer cells, the occurrence of the KRAS variant is mutually exclusive with the occurrence of other KRAS mutations. Unlike acquired mutations in KRAS, the KRAS variant is a germ-line mutation. Thus, the KRAS variant is a heritable biomarker of tumor cell biology.
[90] The presence of the KRAS Variant indicates poor clinical outcomes in various cancers, including, but not limited to, breast, colon, ovarian, head and neck cancer, and lung cancer. This indication is due, at least in part, to the ability of the KRAS Variant to confer resistance or sensitivity to certain cancer therapies to either a cancer cell or subject containing the cancer cell. Thus, when the KRAS variant confers resistance to a standard
chemotherapeutic agent, the occurrence of the KRAS variant predicts a worse outcome for a carrier of the mutation than for a non-carrier. Conversely, when the KRAS variant confers sensitivity to a standard chemotherapeutic agent, the occurrence of the KRAS variant predicts a better outcome for a carrier of the mutation than for a non-carrier.
[91] The presence of the KRAS Variant predicts either resistance or sensitivity to traditional chemotherapeutic agents (including monoclonal anti-EGFR antibody therapy and combinations thereof). Interestingly, the presence of the KRAS Variant may predict sensitivity for a cancer therapy when use as a monotherapy, and the opposite response, resistance, to the combination of the same therapy when used in combination with another treatment (e.g. chemotherapy or monoclonal anti-EGFR antibody) or medical procedure (e.g. radiation or surgery). This apparently contradiction may be resolved by understanding the specific gene or gene product targeted by each of these therapies.
[92] The occurrence of the KRAS variant may indicate that the use of cancer treatments that specifically target genes or gene products located upstream of KRAS in a cell signaling pathway will be successful; however, that the use of cancer treatments that specifically target genes or gene products located downstream of KRAS in a cell signaling pathway may be ineffective (e.g. conventional chemotherapeutic agents that target cell cycle checkpoints, which are downstream of KRAS). For example, the KRAS variant increases a subject's sensitivity to the monoclonal anti-EGFR antibody therapy, Cetuximab, when delivered as the only treatment (a monotherapy), which targets an upstream regulator of the KRAS pathway (EGFR). Conversely, the KRAS Variant confers resistance to platinum-based chemotherapy. Platinum-based chemotherapeutic agents crosslink DNA molecules to prevent DNA replication, ultimately triggering apoptosis. However, DNA replication is a process that occurs downstream of KRAS activation, and, therefore, may be ineffective to treat cancer, particularly in light of data showing the recruitment of signaling pathways other than RAS.
[93] These discoveries about KRAS tumor biology, and, in particular, the effect of the KRAS variant on KRAS signaling, have significant clinical value because many known cancer therapies (e.g. chemotherapy) are very hard on the patient. Chemotherapeutic agents present side effects that not only add to the patient's discomfort, but also introduce complications with otherwise properly functioning bodily systems. For instance, a
chemotherapeutic agent that kills cancer cells may also damage or weaken the patient's heart. Thus, if it is determined that a patient carries the KRAS variant, then the doctor will be able to choose an optimal treatment, or at least avoid losing time and causing patient discomfort by using an ineffective treatment.
The KRAS variant confers resistance to platinum-based chemotherapy
[94] Methods of the disclosure encompass assaying for the KRAS Variant to predict a subject's resistance to platinum-based chemotherapy when used as a treatment for any cancer. In preferred embodiments, the cancer is epithelial ovarian cancer (EOC).
[95] Epithelial ovarian cancer (EOC) is the second most common female pelvic
reproductive organ cancer in the United States, and carries the highest mortality in this category in the Western world. It is the fifth overall leading cause of cancer death in females in the United States, with 13,850 women dying from this disease yearly. Despite multiple new approaches to treatment, the high rates of death from EOC have remained largely unchanged for many years, with a 5-year overall survival of only 30-39%.
[96] The standard chemotherapy regimen to treat EOC currently used is carboplatin and paclitaxel, based on prospective randomized trials. Although some patients are initially resistant to platinum-based chemotherapy (referred to as 'platinum resistant'), developing recurrence within 6 months of treatment, it is the first line treatment given to all EOC patients.
[97] Additional insight into the importance of miRNAs in cancer has come from the discovery of inherited single-nucleotide polymorphisms that disrupt miRNA coding sequences and miRNA-binding sites in the 3' untranslated regions (3 ' UTRs) of oncogenes.
An example of such a functional variant is rs61764370, referred to herein as the KRAS variant, which is located in the KRAS 3' UTR in a let- 7 miRNA complementary site.
rs61764370 is associated with an increased risk of developing epithelial ovarian cancer
(EOC) (see, International Patent Publication No. WO 2008/151004 and International Patent
Publication No. WO 2010/101696; the contents of which are each herein incorporated in their entireties). This disclosure demonstrates that the KRAS Variant is a biomarker chemotherapy resistance, and therefore, clinical outcome, in epithelial ovarian cancer (EOC).
[98] The biological differences between
 EOC and nonvariant EOC tumors are supported by gene expression data, demonstrating KRAS addiction and AKT -mediated platinum resistance in KRAS- variant-associated EOC. Moreover, the KRAS variant is more significantly associated with post-menopausal, as opposed to pre-menopausal, ovarian cancer.
EOC and nonvariant EOC tumors are supported by gene expression data, demonstrating KRAS addiction and AKT -mediated platinum resistance in KRAS- variant-associated EOC. Moreover, the KRAS variant is more significantly associated with post-menopausal, as opposed to pre-menopausal, ovarian cancer.
Relative survival varies by age, with older women twice as likely to die within 5 years of diagnosis of EOC, further supporting the hypothesis that postmenopausal women may have biologically different tumors than younger women. Thus, the role of the KRAS variant in cancer risk and biology may further depend on miRNA expression alterations in response to
physiologic conditions, such as menopause. For instance, women with the KRAS variant may be first at risk for breast cancer and then, subsequently, be at risk for developing postmenopausal ovarian cancer.
[99] The presence of the KRAS variant does not predict poor outcome in a cohort of EOC patients with known deleterious BRCA mutations. In contrast to the KRAS Variant, BRCA mutations are associated with sensitivity to platinum-based chemotherapy. Without wishing to be bound by theory, downstream cell signaling consequences of BRCA mutations that confer sensitivity to platinum-based chemotherapeutic agents may affect targets upstream of KRAS, thereby bypassing KRAS-Variant mediated resistance to platinum-based
chemotherapeutic agents.
[100] In certain aspects of this embodiment, treatment of recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to platinum-based chemotherapy. In certain aspects of this embodiment, the platinum-based chemotherapy is cisplatin, however, the methods may be applied to any platinum-based chemotherapy. For all aspects of these methods, the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR of KRAS. Alternatively, or in addition, treatment of recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting sensitivity or an enhanced response to monoclonal anti-EGFR antibody therapy, either as a monotherapy or as a combined therapy with another chemotherapeutic agent or medical procedure (e.g. radiation or surgery).
[101] The disclosure provides a method of predicting the response of head and neck squamous cell carcinoma (HNSCC ) cell and/or a recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) cell to a platinum-based chemotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and wherein the presence of the mutation indicates a resistance to platinum-based chemotherapy. In certain aspects of this embodiment, the disclosure provides a method of predicting the response of head and neck squamous cell carcinoma (HNSCC ) cell and/or a recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC ) cell to a monoclonal anti-EGFR antibody therapy,
including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and wherein the presence of the mutation indicates sensitivity or an enhanced response to monoclonal anti-EGFR antibody therapy, either as a monotherapy or as a combined therapy with another chemotherapeutic agent or medical procedure (e.g. radiation or surgery).
[102] The FiNSCC cell may be evaluated in vivo, in vitro or ex vivo. When the FiNSCC cell is evaluated ex vivo, the cell is obtained from a subject.
The KRAS variant confers resistance to small molecule EGFR inhibitors
[103] The disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors). In certain embodiments of these methods, treatment of cancer is optimized by assaying for the presence of the KRAS
Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors).
[104] In certain aspects of this embodiment, treatment of non-small cell lung cancer (NSCLC) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors and small molecule EGFR inhibitors). For all aspects of these methods, the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR ofKRAS.
[105] In certain aspects of this embodiment, chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors or small molecule EGFR inhibitors), include, but are not limited to, gefinitib and erlotinib. In certain aspects of this embodiment, chemotherapeutic agents that inhibit an activity of EGFR (i.e. EGFR inhibitors or small molecule EGFR inhibitors), include, but are not limited to, gefinitib.
The KRAS variant confers sensitivity to MAP kinase pathway inhibitors
[106] The disclosure provides methods of prognosing subjects with cancer and, furthermore, methods of optimizing treatment by predicting the subject's response to chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors). In certain embodiments of these methods, treatment of cancer is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to
chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors).
[107] In certain aspects of this embodiment, treatment of non-small cell lung cancer (NSCLC) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting resistance to chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors). For all aspects of these methods, the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR ofKRAS.
[108] In certain aspects of this embodiment, chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors), include, but are not limited to, inhibitors of tyrosine protein kinases and serine/threonine kinases. Nonlimiting examples of tyrosine protein kinases include the VEGFR (vascular endothelial growth factor receptor) and PDGFR (platelet-derived growth factor receptor). Nonlimiting examples of
serine/threonine protein kinases include C-raf (encoded by the RAFl gene) and B-raf (encoded by the BRAF gene). Chemotherapeutic agents that inhibit an activity of the MAP kinase pathway (i.e. MAP kinase pathway inhibitors), include, but are not limited to, sorafenib.
The KRAS variant confers sensitivity to monoclonal anti-EGFR antibody monotherapy
[109] The presence of tumor-acquired KRAS mutations predicts resistance to anti-EGFR mAbs therapy and, therefore, is associated with a worse prognosis and a shorter survival.
However, approximately 50-65% of the metastatic colorectal cancer (mCRC) patients with tumors having wild type KRAS sequences derive no benefit from these treatments, implying the existence of additional genetic determinants of resistance or sensitivity. For example, the
BRAF V600E mutation may confer resistance to anti-EGFR mAbs.
[110] In addition to the tumoral genetic characteristics, the germ-line genome of patients plays a role in granting resistance or sensitivity to anti-EGFR mAb therapy. In support of this notion, polymorphisms in the genes encoding for FcyRIIa and FcyRIIIa, EGFR, EGF, cyclinDl and COX-2 have been associated with outcome in mCRC patients treated with cetuximab, administered both as monotherapy and in combination with chemotherapy.
[Ill] The KRAS variant (G allele) predicts a positive response to monoclonal anti-EGFR antibody (mAb) monotherapy, without any additional benefit of cytotoxic chemotherapy. The studies presented herein demonstrate a statistically significant improvement in median
progression- free survival (PFS) for all KRAS variant carriers with metastatic colon cancer (and a trend towards improved overall survival (OS) in the double wildtype patients) who received anti-EGFR mAb monotherapy. KRAS variant (G allele) carriers appeared to have no benefit from chemotherapy in addition to anti-EGFR mAb therapy. This was in contrast to non-KRAS variant patients, who derived a significant benefit from the addition of
chemotherapy to anti-EGFR mAb therapy across all cohorts. For non-carries of the KRAS Variant, the addition of chemotherapy brought their prognosis to the same level of KRAS variant allele carriers who received anti-EGFR mAb monotherapy. Cell lines studies showed the same lack of benefit of combination therapy in KRAS variant cell lines compared to non- variant cell lines. These findings demonstrate that KRAS variant allele patients with metastatic colon cancer should avoid the toxic (and potentially deadly) side effects of chemotherapy treatment. In other words, the data provided herein demonstrate that carriers of the KRAS- Variant can be treated with anti-EGFR mAb monotherapy alone. Moreover, the KRAS Variant predicts a good prognosis in both early stage colon cancer as well as metastatic colon cancer patients when treated with Cetuximab monotherapy.
[112] A different distribution of the KRAS variant genotypes according to the KRAS and
BRAF mutational status was observed in this study with respect to the mCRC patient population compared to prior reports. In this study, the KRAS genotypes were equally distributed among the KRAS wt and mutated groups, but, in the BRAF mutated group, the frequency of the KRAS variant was statistically significantly increased, i.e., twice as high compared to wild type. In the later stages of CRC carcinogenesis, the KRAS variant allele may mediate the selection of less differentiated and more aggressive clones that carry BRAF mutations. Additionally, a selective pressure may favor the development of KRAS or BRAF mutations in the presence of the KRAS variant, depending on exposure to specific therapies.
Patients with the KRAS variant (G allele) have a different prognosis when treated with
Cetuximab regardless of patients also having a KRAS or a BRAF mutation, suggesting that these groups need re-evaluation for the potential of Cetuximab treatment.
[113] When the survival outcomes were analyzed following anti-EGFR mAbs monotherapy, the KRAS variant genotype carriers had a statistically significantly longer PFS (p = 0.019 and p = 0.039, respectively) compared to the total subject pool and compared to the double wildtype (wildtype KRAS and BRAF) subpopulation. Compared to the total monotherapy patient population, the KRAS variant genotype carriers had a longer OS of 45 weeks
compared to 28.85 weeks of the wild type carriers (nevertheless this difference did not reach statistical significance). In the double wild type (KRAS and BRAF) patient population, a trend towards statistical significance (p = 0.087) was observed with a longer OS in favor of the KRAS variant carriers (55.43 vs. 35.71 weeks).
[114] When treated with a combination of cetuximab (monoclonal anti-EGFR mAb) and irinotecan (a chemotherapeutic agent) carriers of the KRAS variant (G-allele) genotype showed a significantly worse PFS of 6.4 weeks compared to 12 weeks in those patients with the wild type genotype (non-carriers of the KRAS Variant) (p = 0.037, log-rank test). In the anti-EGFR mAb-based combination chemotherapy group, where patients were treated with a variety of chemotherapeutic agents in addition to an anti-EGFR mAb, no statistically significant differences were found in PFS or OS in any population between the KRAS variant and wt genotype carriers. There was a trend for worse survival (23 versus 28 weeks) in KRAS variant carriers with additional KRAS or BRAF mutations when they received a combination therapy including an anti-EGFR mAb and a chemotherapeutic agent versus anti-EGFR mAb monotherapy, respectively. These results demonstrate that the use of certain
chemotherapeutic agents in combination with anti-EGFR mAb-based therapy is detrimental to patients who carry the KRAS Variant.
[115] KRAS variant-positive tumors derive no benefit from the addition of cytotoxic therapy to monoclonal anti-EGFR antibody monotherapy. Chemotherapeutic agents may further decrease let-7 levels (which are already low due to the presence of the KRAS Variant) and, consequently, allow greater KRAS expression (especially in the presence of the KRAS variant). Thus, based upon the data presented herein, treatment of KRAS variant-positive tumors with chemotherapeutic agents may increase activation of this mutant allele, thereby removing the ability of upstream anti-EGFR mAb therapy to overcome KRAS pathway activation.
[116] In certain aspects of this embodiment, treatment of head and neck squamous cell carcinoma (FINSCC ) or a recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (FINSCC ) is optimized by assaying for the presence of the KRAS Variant and, if present, predicting sensitivity to monoclonal anti-EGFR antibody therapy. For all aspects of these methods, the KRAS variant is defined as a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6 in the 3' UTR of KRAS.
[117] The disclosure provides a method of predicting the response of a head and neck squamous cell carcinoma (FiNSCC ) cell to a monoclonal anti-EGFR antibody monotherapy, including detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, wherein the presence of the mutation indicates a sensitivity to monoclonal anti-EGFR antibody monotherapy. The head and neck squamous cell carcinoma (FiNSCC ) cell may be evaluated in vivo, in vitro or ex vivo. A non-limiting example of the monoclonal anti-EGFR antibody monotherapy is Cetuximab.
[118] EGF, a potent ligand of EGFR, is upregulated in AT^S-variant positive FiNSCC tumors. It is hypothesized that cetuximab is beneficial for these patients because the anti- EGFR antibody blocks the pro-growth signal provided by an upregulation of EGF in these tumors. Furthermore, a number of genes associated with microtubule and cytoskeleton function are also upregulated. More specifically, MACF1 is upregulated in our current analysis. MACF1 interacts with ErbB2 to control microtubule capture during cell migration. Taken together, the upregulation of an EGFR-specific growth stimulatory ligand (EGF), a pro-migratory phosphatase (SYNJ2), and various components of the microtubule/cytoskeletal architecture (DST, MACF1, EML6) indicates the presence of an enhanced migratory or metastatic gene expression profile associated with
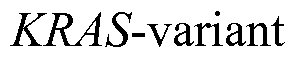 FTNSCCS as well as explains an enhanced sensitivity of KRAS -variant cancer cells (e.g. FTNSCCs) to cetuximab treatment. Furthermore, to optimize cancer treatment, methods of the disclosure include detection of the KRAS variant in combination with detection of upregulation of EGF, wherein the combined presence of the KRAS variant and upregulated EGF mRNA or protein indicates sensitivity of the cancer cell to a monoclonal anti-EGFR antibody therapy.
FTNSCCS as well as explains an enhanced sensitivity of KRAS -variant cancer cells (e.g. FTNSCCs) to cetuximab treatment. Furthermore, to optimize cancer treatment, methods of the disclosure include detection of the KRAS variant in combination with detection of upregulation of EGF, wherein the combined presence of the KRAS variant and upregulated EGF mRNA or protein indicates sensitivity of the cancer cell to a monoclonal anti-EGFR antibody therapy.
KRAS Variant
[119] The disclosure is based, in part, upon the unexpected discovery that the presence of a SNP in the 3' untranslated region (UTR) oiKRAS, referred to herein as the "LCS6 SNP" or the "KRAS variant," which is predictive of an individual's risk of developing cancer and an individual's response to treatment for cancer. The KRAS variant is located in LCS6, the wild type and variant sequence of which is provided below.
[120] The KRAS variant may be represented by one or more of the following sequences. For example, the KRAS variant may be defined by the GenBank accession number rs61764370 and the sequence
GTCTCGAACTCCTGACCTCAAGTGATGCACCCACCTTGGCCTCATAAACCTG (SEQ ID NO: 22, in which the SNP is bolded and underlined).
[121] There are three human RAS genes comprising HRAS, KRAS, and NRAS. Each gene comprises multiple miRNA complementary sites in the 3'UTR of their mRNA transcripts. Specifically, each human RAS gene comprises multiple let-7 complementary sites (LCSs). The let-7 family-of-microRNAs (miRNAs) includes global genetic regulators important in controlling lung cancer oncogene expression by binding to the 3'UTRs (untranslated regions) of their target messenger RNAs (mRNAs).
[122] Specifically, the term "let-7 complementary site" is meant to describe any region of a gene or gene transcript that binds a member of the let-7 family of miRNAs. Moreover, this term encompasses those sequences within a gene or gene transcript that are complementary to the sequence of a let-7 family miRNA. The term "complementary" describes a threshold of binding between two sequences wherein a majority of nucleotides in each sequence are capable of binding to a majority of nucleotides within the other sequence in trans.
[123] The Human KRAS 3' UTR comprises 8 LCSs named LCS1-LCS8, respectively. For the following sequences, thymine (T) may be substituted for uracil (U). LCS1 comprises the sequence GACAGUGGAAGUUUUUUUUUCCUCG (SEQ ID NO: 1). LCS2 comprises the sequence AUUAGUGUCAUCUUGCCUC (SEQ ID NO: 2). LCS3 comprises the sequence AAUGCCCUACAUCUUAUUUUCCUCA (SEQ ID NO: 3). LCS4 comprises the sequence GGUUCAAGCGAUUCUCGUGCCUCG (SEQ ID NO: 4). LCS5 comprises the sequence GGCUGGUCCGAACUCCUGACCUCA (SEQ ID NO: 5). LCS6 comprises the sequence GAUUCACCCACCUUGGCCUCA (SEQ ID NO: 6). LCS7 comprises the sequence GGGUGUUAAG ACUUG AC AC AGUAC CUC G (SEQ ID NO: 7). LCS8 comprises the sequence AGUGCUUAUGAGGGGAUAUUUAGGCCUC (SEQ ID NO: 8).
[124] Human KRAS has two wild type forms, encoded by transcripts a and b, which are provided below as SEQ ID NOs: 9 and 10, respectively. The sequences of each human KRAS transcript, containing the LCS6 SNP, are provided below as SEQ ID NOs: 11 and 12.
[125] Human KRAS, transcript variant a, is encoded by the following mRNA sequence (NCBI Accession No. NM 033360 and SEQ ID NO: 9) (untranslated regions are bolded, LCS6 is underlined):
1 ggccgcggcg gcggaggcag cagcggcggc ggcagtggcg gcggcgaagg tggcggcggc 61 tcggccagta ctcccggccc ccgccatttc ggactgggag cgagcgcggc gcaggcactg 121 aaggcggcgg cggggccaga ggctcagcgg ctcccaggtg cgggagagag gcctgctgaa
181 aatgactgaa tataaacttg tggtagttgg agctggtggc gtaggcaaga gtgccttgac
241 gatacagcta attcagaatc attttgtgga cgaatatgat ccaacaatag aggattccta
301 caggaagcaa gtagtaattg atggagaaac ctgtctcttg gatattctcg acacagcagg
361 tcaagaggag tacagtgcaa tgagggacca gtacatgagg actggggagg gctttctttg
421 tgtatttgcc ataaataata ctaaatcatt tgaagatatt caccattata gagaacaaat
481 taaaagagtt aaggactctg aagatgtacc tatggtccta gtaggaaata aatgtgattt
541 gccttctaga acagtagaca caaaacaggc tcaggactta gcaagaagtt atggaattcc
601 ttttattgaa acatcagcaa agacaagaca gagagtggag gatgcttttt atacattggt
661 gagggagatc cgacaataca gattgaaaaa aatcagcaaa gaagaaaaga ctcctggctg
721 tgtgaaaatt aaaaaatgca ttataatgta atctgggtgt tgatgatgcc ttctatacat
781 tagttcgaga aattcgaaaa cataaagaaa agatgagcaa agatggtaaa aagaagaaaa
841 agaagtcaaa gacaaagtgt gtaattatgt aaatacaatt tgtacttttt tcttaaggca
901 tactagtaca agtggtaatt tttgtacatt acactaaatt attagcattt gttttagcat
961 tacctaattt ttttcctgct ccatgcagac tgttagcttt taccttaaat gcttatttta
1021 aaatgacagt ggaagttttt ttttcctcta agtgccagta ttcccagagt tttggttttt
1081 gaactagcaa tgcctgtgaa aaagaaactg aatacctaag atttctgtct tggggttttt
1141 ggtgcatgca gttgattact tcttattttt cttaccaatt gtgaatgttg gtgtgaaaca
1201 aattaatgaa gcttttgaat catccctatt ctgtgtttta tctagtcaca taaatggatt
1261 aattactaat ttcagttgag accttctaat tggtttttac tgaaacattg agggaacaca
1321 aatttatggg cttcctgatg atgattcttc taggcatcat gtcctatagt ttgtcatccc
1381 tgatgaatgt aaagttacac tgttcacaaa ggttttgtct cctttccact gctattagtc
1441 atggtcactc tccccaaaat attatatttt ttctataaaa agaaaaaaat ggaaaaaaat
1501 tacaaggcaa tggaaactat tataaggcca tttccttttc acattagata aattactata
1561 aagactccta atagcttttc ctgttaaggc agacccagta tgaaatgggg attattatag
1621 caaccatttt ggggctatat ttacatgcta ctaaattttt ataataattg aaaagatttt
1681 aacaagtata aaaaattctc ataggaatta aatgtagtct ccctgtgtca gactgctctt
1741 tcatagtata actttaaatc ttttcttcaa cttgagtctt tgaagatagt tttaattctg
1801 cttgtgacat taaaagatta tttgggccag ttatagctta ttaggtgttg aagagaccaa
1861 ggttgcaagg ccaggccctg tgtgaacctt tgagctttca tagagagttt cacagcatgg
1921 actgtgtccc cacggtcatc cagtgttgtc atgcattggt tagtcaaaat ggggagggac
1981 tagggcagtt tggatagctc aacaagatac aatctcactc tgtggtggtc ctgctgacaa
2041 atcaagagca ttgcttttgt ttcttaagaa aacaaactct tttttaaaaa ttacttttaa
2101 atattaactc aaaagttgag attttggggt ggtggtgtgc caagacatta attttttttt
2161 taaacaatga agtgaaaaag ttttacaatc tctaggtttg gctagttctc ttaacactgg
2221 ttaaattaac attgcataaa cacttttcaa gtctgatcca tatttaataa tgctttaaaa
2281 taaaaataaa aacaatcctt ttgataaatt taaaatgtta cttattttaa aataaatgaa
2341 gtgagatggc atggtgaggt gaaagtatca ctggactagg aagaaggtga cttaggttct
2401 agataggtgt cttttaggac tctgattttg aggacatcac ttactatcca tttcttcatg
2461 ttaaaagaag tcatctcaaa ctcttagttt ttttttttta caactatgta atttatattc
2521 catttacata aggatacact tatttgtcaa gctcagcaca atctgtaaat ttttaaccta
2581 tgttacacca tcttcagtgc cagtcttggg caaaattgtg caagaggtga agtttatatt
2641 tgaatatcca ttctcgtttt aggactcttc ttccatatta gtgtcatctt gcctccctac
2701 cttccacatg ccccatgact tgatgcagtt ttaatacttg taattcccct aaccataaga
2761 tttactgctg ctgtggatat ctccatgaag ttttcccact gagtcacatc agaaatgccc
2821 tacatcttat ttcctcaggg ctcaagagaa tctgacagat accataaagg gatttgacct
2881 aatcactaat tttcaggtgg tggctgatgc tttgaacatc tctttgctgc ccaatccatt
2941 agcgacagta ggatttttca aacctggtat gaatagacag aaccctatcc agtggaagga
3001 gaatttaata aagatagtgc tgaaagaatt ccttaggtaa tctataacta ggactactcc
3061 tggtaacagt aatacattcc attgttttag taaccagaaa tcttcatgca atgaaaaata
3121 ctttaattca tgaagcttac tttttttttt tggtgtcaga gtctcgctct tgtcacccag
3181 gctggaatgc agtggcgcca tctcagctca ctgcaacctc catctcccag gttcaagcga
3241 ttctcgtgcc tcggcctcct gagtagctgg gattacaggc gtgtgccact acactcaact
3301 aatttttgta tttttaggag agacggggtt tcaccctgtt ggccaggctg gtctcgaact
3361 cctgacctca agtgattcac ccaccttggc ctcataaacc tgttttgcag aactcattta
3421 ttcagcaaat atttattgag tgcctaccag atgccagtca ccgcacaagg cactgggtat
3481 atggtatccc caaacaagag acataatccc ggtccttagg tagtgctagt gtggtctgta
3541 atatcttact aaggcctttg gtatacgacc cagagataac acgatgcgta ttttagtttt
3601 gcaaagaagg ggtttggtct ctgtgccagc tctataattg ttttgctacg attccactga
3661 aactcttcga tcaagctact ttatgtaaat cacttcattg ttttaaagga ataaacttga 3721 ttatattgtt tttttatttg gcataactgt gattctttta ggacaattac tgtacacatt 3781 aaggtgtatg tcagatattc atattgaccc aaatgtgtaa tattccagtt ttctctgcat 3841 aagtaattaa aatatactta aaaattaata gttttatctg ggtacaaata aacaggtgcc 3901 tgaactagtt cacagacaag gaaacttcta tgtaaaaatc actatgattt ctgaattgct 3961 atgtgaaact acagatcttt ggaacactgt ttaggtaggg tgttaagact tacacagtac 4021 ctcgtttcta cacagagaaa gaaatggcca tacttcagga actgcagtgc ttatgagggg 4081 atatttaggc ctcttgaatt tttgatgtag atgggcattt ttttaaggta gtggttaatt 4141 acctttatgt gaactttgaa tggtttaaca aaagatttgt ttttgtagag attttaaagg 4201 gggagaattc tagaaataaa tgttacctaa ttattacagc cttaaagaca aaaatccttg 4261 ttgaagtttt tttaaaaaaa gctaaattac atagacttag gcattaacat gtttgtggaa 4321 gaatatagca gacgtatatt gtatcatttg agtgaatgtt cccaagtagg cattctaggc 4381 tctatttaac tgagtcacac tgcataggaa tttagaacct aacttttata ggttatcaaa 4441 actgttgtca ccattgcaca attttgtcct aatatataca tagaaacttt gtggggcatg 4501 ttaagttaca gtttgcacaa gttcatctca tttgtattcc attgattttt tttttcttct 4561 aaacattttt tcttcaaaca gtatataact ttttttaggg gatttttttt tagacagcaa 4621 aaactatctg aagatttcca tttgtcaaaa agtaatgatt tcttgataat tgtgtagtaa 4681 tgttttttag aacccagcag ttaccttaaa gctgaattta tatttagtaa cttctgtgtt 4741 aatactggat agcatgaatt ctgcattgag aaactgaata gctgtcataa aatgaaactt 4801 tctttctaaa gaaagatact cacatgagtt cttgaagaat agtcataact agattaagat 4861 ctgtgtttta gtttaatagt ttgaagtgcc tgtttgggat aatgataggt aatttagatg 4921 aatttagggg aaaaaaaagt tatctgcaga tatgttgagg gcccatctct ccccccacac 4981 ccccacagag ctaactgggt tacagtgttt tatccgaaag tttccaattc cactgtcttg 5041 tgttttcatg ttgaaaatac ttttgcattt ttcctttgag tgccaatttc ttactagtac 5101 tatttcttaa tgtaacatgt ttacctggaa tgtattttaa ctatttttgt atagtgtaaa 5161 ctgaaacatg cacattttgt acattgtgct ttcttttgtg ggacatatgc agtgtgatcc 5221 agttgttttc catcatttgg ttgcgctgac ctaggaatgt tggtcatatc aaacattaaa 5281 aatgaccact cttttaattg aaattaactt ttaaatgttt ataggagtat gtgctgtgaa 5341 gtgatctaaa atttgtaata tttttgtcat gaactgtact actcctaatt attgtaatgt 5401 aataaaaata gttacagtga caaaaaaaaa aaaaaa
[126] Human KRAS, transcript variant b, is encoded by the following mRNA sequence (NCBI Accession No. NM 004985 and SEQ ID NO: 10)(untranslated regions are bolded, LCS6 is underlined):
1 ggccgcggcg gcggaggcag cagcggcggc ggcagtggcg gcggcgaagg tggcggcggc 61 tcggccagta ctcccggccc ccgccatttc ggactgggag cgagcgcggc gcaggcactg 121 aaggcggcgg cggggccaga ggctcagcgg ctcccaggtg cgggagagag gcctgctgaa
181 aatgactgaa tataaacttg tggtagttgg agctggtggc gtaggcaaga gtgccttgac 241 gatacagcta attcagaatc attttgtgga cgaatatgat ccaacaatag aggattccta 301 caggaagcaa gtagtaattg atggagaaac ctgtctcttg gatattctcg acacagcagg 361 tcaagaggag tacagtgcaa tgagggacca gtacatgagg actggggagg gctttctttg 421 tgtatttgcc ataaataata ctaaatcatt tgaagatatt caccattata gagaacaaat 481 taaaagagtt aaggactctg aagatgtacc tatggtccta gtaggaaata aatgtgattt 541 gccttctaga acagtagaca caaaacaggc tcaggactta gcaagaagtt atggaattcc 601 ttttattgaa acatcagcaa agacaagaca gggtgttgat gatgccttct atacattagt 661 tcgagaaatt cgaaaacata aagaaaagat gagcaaagat ggtaaaaaga agaaaaagaa 721 gtcaaagaca aagtgtgtaa ttatgtaaat acaatttgta cttttttctt aaggcatact 781 agtacaagtg gtaatttttg tacattacac taaattatta gcatttgttt tagcattacc 841 taattttttt cctgctccat gcagactgtt agcttttacc ttaaatgctt attttaaaat 901 gacagtggaa gttttttttt cctctaagtg ccagtattcc cagagttttg gtttttgaac 961 tagcaatgcc tgtgaaaaag aaactgaata cctaagattt ctgtcttggg gtttttggtg 1021 catgcagttg attacttctt atttttctta ccaattgtga atgttggtgt gaaacaaatt 1081 aatgaagctt ttgaatcatc cctattctgt gttttatcta gtcacataaa tggattaatt 1141 actaatttca gttgagacct tctaattggt ttttactgaa acattgaggg aacacaaatt 1201 tatgggcttc ctgatgatga ttcttctagg catcatgtcc tatagtttgt catccctgat
1261 gaatgtaaag ttacactgtt cacaaaggtt ttgtctcctt tccactgcta ttagtcatgg
1321 tcactctccc caaaatatta tattttttct ataaaaagaa aaaaatggaa aaaaattaca
1381 aggcaatgga aactattata aggccatttc cttttcacat tagataaatt actataaaga
1441 ctcctaatag cttttcctgt taaggcagac ccagtatgaa atggggatta ttatagcaac
1501 cattttgggg ctatatttac atgctactaa atttttataa taattgaaaa gattttaaca
1561 agtataaaaa attctcatag gaattaaatg tagtctccct gtgtcagact gctctttcat
1621 agtataactt taaatctttt cttcaacttg agtctttgaa gatagtttta attctgcttg
1681 tgacattaaa agattatttg ggccagttat agcttattag gtgttgaaga gaccaaggtt
1741 gcaaggccag gccctgtgtg aacctttgag ctttcataga gagtttcaca gcatggactg
1801 tgtccccacg gtcatccagt gttgtcatgc attggttagt caaaatgggg agggactagg
1861 gcagtttgga tagctcaaca agatacaatc tcactctgtg gtggtcctgc tgacaaatca
1921 agagcattgc ttttgtttct taagaaaaca aactcttttt taaaaattac ttttaaatat
1981 taactcaaaa gttgagattt tggggtggtg gtgtgccaag acattaattt tttttttaaa
2041 caatgaagtg aaaaagtttt acaatctcta ggtttggcta gttctcttaa cactggttaa
2101 attaacattg cataaacact tttcaagtct gatccatatt taataatgct ttaaaataaa
2161 aataaaaaca atccttttga taaatttaaa atgttactta ttttaaaata aatgaagtga
2221 gatggcatgg tgaggtgaaa gtatcactgg actaggaaga aggtgactta ggttctagat
2281 aggtgtcttt taggactctg attttgagga catcacttac tatccatttc ttcatgttaa
2341 aagaagtcat ctcaaactct tagttttttt tttttacaac tatgtaattt atattccatt
2401 tacataagga tacacttatt tgtcaagctc agcacaatct gtaaattttt aacctatgtt
2461 acaccatctt cagtgccagt cttgggcaaa attgtgcaag aggtgaagtt tatatttgaa
2521 tatccattct cgttttagga ctcttcttcc atattagtgt catcttgcct ccctaccttc
2581 cacatgcccc atgacttgat gcagttttaa tacttgtaat tcccctaacc ataagattta
2641 ctgctgctgt ggatatctcc atgaagtttt cccactgagt cacatcagaa atgccctaca
2701 tcttatttcc tcagggctca agagaatctg acagatacca taaagggatt tgacctaatc
2761 actaattttc aggtggtggc tgatgctttg aacatctctt tgctgcccaa tccattagcg
2821 acagtaggat ttttcaaacc tggtatgaat agacagaacc ctatccagtg gaaggagaat
2881 ttaataaaga tagtgctgaa agaattcctt aggtaatcta taactaggac tactcctggt
2941 aacagtaata cattccattg ttttagtaac cagaaatctt catgcaatga aaaatacttt
3001 aattcatgaa gcttactttt tttttttggt gtcagagtct cgctcttgtc acccaggctg
3061 gaatgcagtg gcgccatctc agctcactgc aacctccatc tcccaggttc aagcgattct
3121 cgtgcctcgg cctcctgagt agctgggatt acaggcgtgt gccactacac tcaactaatt
3181 tttgtatttt taggagagac ggggtttcac cctgttggcc aggctggtct cgaactcctg
3241 acctcaagtg attcacccac cttggcctca taaacctgtt ttgcagaact catttattca
3301 gcaaatattt attgagtgcc taccagatgc cagtcaccgc acaaggcact gggtatatgg
3361 tatccccaaa caagagacat aatcccggtc cttaggtagt gctagtgtgg tctgtaatat
3421 cttactaagg cctttggtat acgacccaga gataacacga tgcgtatttt agttttgcaa
3481 agaaggggtt tggtctctgt gccagctcta taattgtttt gctacgattc cactgaaact
3541 cttcgatcaa gctactttat gtaaatcact tcattgtttt aaaggaataa acttgattat
3601 attgtttttt tatttggcat aactgtgatt cttttaggac aattactgta cacattaagg
3661 tgtatgtcag atattcatat tgacccaaat gtgtaatatt ccagttttct ctgcataagt
3721 aattaaaata tacttaaaaa ttaatagttt tatctgggta caaataaaca ggtgcctgaa
3781 ctagttcaca gacaaggaaa cttctatgta aaaatcacta tgatttctga attgctatgt
3841 gaaactacag atctttggaa cactgtttag gtagggtgtt aagacttaca cagtacctcg
3901 tttctacaca gagaaagaaa tggccatact tcaggaactg cagtgcttat gaggggatat
3961 ttaggcctct tgaatttttg atgtagatgg gcattttttt aaggtagtgg ttaattacct
4021 ttatgtgaac tttgaatggt ttaacaaaag atttgttttt gtagagattt taaaggggga
4081 gaattctaga aataaatgtt acctaattat tacagcctta aagacaaaaa tccttgttga
4141 agttttttta aaaaaagcta aattacatag acttaggcat taacatgttt gtggaagaat
4201 atagcagacg tatattgtat catttgagtg aatgttccca agtaggcatt ctaggctcta
4261 tttaactgag tcacactgca taggaattta gaacctaact tttataggtt atcaaaactg
4321 ttgtcaccat tgcacaattt tgtcctaata tatacataga aactttgtgg ggcatgttaa
4381 gttacagttt gcacaagttc atctcatttg tattccattg attttttttt tcttctaaac
4441 attttttctt caaacagtat ataacttttt ttaggggatt tttttttaga cagcaaaaac
4501 tatctgaaga tttccatttg tcaaaaagta atgatttctt gataattgtg tagtaatgtt
4561 ttttagaacc cagcagttac cttaaagctg aatttatatt tagtaacttc tgtgttaata
4621 ctggatagca tgaattctgc attgagaaac tgaatagctg tcataaaatg aaactttctt
4681 tctaaagaaa gatactcaca tgagttcttg aagaatagtc ataactagat taagatctgt
4741 gttttagttt aatagtttga agtgcctgtt tgggataatg ataggtaatt tagatgaatt
4801 taggggaaaa aaaagttatc tgcagatatg ttgagggccc atctctcccc ccacaccccc
4861 acagagctaa ctgggttaca gtgttttatc cgaaagtttc caattccact gtcttgtgtt
4921 ttcatgttga aaatactttt gcatttttcc tttgagtgcc aatttcttac tagtactatt
4981 tcttaatgta acatgtttac ctggaatgta ttttaactat ttttgtatag tgtaaactga
5041 aacatgcaca ttttgtacat tgtgctttct tttgtgggac atatgcagtg tgatccagtt
5101 gttttccatc atttggttgc gctgacctag gaatgttggt catatcaaac attaaaaatg
5161 accactcttt taattgaaat taacttttaa atgtttatag gagtatgtgc tgtgaagtga
5221 tctaaaattt gtaatatttt tgtcatgaac tgtactactc ctaattattg taatgtaata
5281 aaaatagtta cagtgacaaa aaaaaaaaaa aa
[127] Human KRAS, transcript variant a, comprising the LCS6 SNP, is encoded by the following mRNA sequence (SEQ ID NO: 11) (untranslated regions are bolded, LCS6 is underlined, SNP is capitalized):
1 ggccgcggcg gcggaggcag cagcggcggc ggcagtggcg gcggcgaagg tggcggcggc 61 tcggccagta ctcccggccc ccgccatttc ggactgggag cgagcgcggc gcaggcactg 121 aaggcggcgg cggggccaga ggctcagcgg ctcccaggtg cgggagagag gcctgctgaa
181 aatgactgaa tataaacttg tggtagttgg agctggtggc gtaggcaaga gtgccttgac 241 gatacagcta attcagaatc attttgtgga cgaatatgat ccaacaatag aggattccta 301 caggaagcaa gtagtaattg atggagaaac ctgtctcttg gatattctcg acacagcagg 361 tcaagaggag tacagtgcaa tgagggacca gtacatgagg actggggagg gctttctttg 421 tgtatttgcc ataaataata ctaaatcatt tgaagatatt caccattata gagaacaaat 481 taaaagagtt aaggactctg aagatgtacc tatggtccta gtaggaaata aatgtgattt 541 gccttctaga acagtagaca caaaacaggc tcaggactta gcaagaagtt atggaattcc 601 ttttattgaa acatcagcaa agacaagaca gagagtggag gatgcttttt atacattggt 661 gagggagatc cgacaataca gattgaaaaa aatcagcaaa gaagaaaaga ctcctggctg 721 tgtgaaaatt aaaaaatgca ttataatgta atctgggtgt tgatgatgcc ttctatacat 781 tagttcgaga aattcgaaaa cataaagaaa agatgagcaa agatggtaaa aagaagaaaa 841 agaagtcaaa gacaaagtgt gtaattatgt aaatacaatt tgtacttttt tcttaaggca 901 tactagtaca agtggtaatt tttgtacatt acactaaatt attagcattt gttttagcat 961 tacctaattt ttttcctgct ccatgcagac tgttagcttt taccttaaat gcttatttta 1021 aaatgacagt ggaagttttt ttttcctcta agtgccagta ttcccagagt tttggttttt 1081 gaactagcaa tgcctgtgaa aaagaaactg aatacctaag atttctgtct tggggttttt 1141 ggtgcatgca gttgattact tcttattttt cttaccaatt gtgaatgttg gtgtgaaaca 1201 aattaatgaa gcttttgaat catccctatt ctgtgtttta tctagtcaca taaatggatt 1261 aattactaat ttcagttgag accttctaat tggtttttac tgaaacattg agggaacaca 1321 aatttatggg cttcctgatg atgattcttc taggcatcat gtcctatagt ttgtcatccc 1381 tgatgaatgt aaagttacac tgttcacaaa ggttttgtct cctttccact gctattagtc 1441 atggtcactc tccccaaaat attatatttt ttctataaaa agaaaaaaat ggaaaaaaat 1501 tacaaggcaa tggaaactat tataaggcca tttccttttc acattagata aattactata 1561 aagactccta atagcttttc ctgttaaggc agacccagta tgaaatgggg attattatag 1621 caaccatttt ggggctatat ttacatgcta ctaaattttt ataataattg aaaagatttt 1681 aacaagtata aaaaattctc ataggaatta aatgtagtct ccctgtgtca gactgctctt 1741 tcatagtata actttaaatc ttttcttcaa cttgagtctt tgaagatagt tttaattctg 1801 cttgtgacat taaaagatta tttgggccag ttatagctta ttaggtgttg aagagaccaa 1861 ggttgcaagg ccaggccctg tgtgaacctt tgagctttca tagagagttt cacagcatgg 1921 actgtgtccc cacggtcatc cagtgttgtc atgcattggt tagtcaaaat ggggagggac 1981 tagggcagtt tggatagctc aacaagatac aatctcactc tgtggtggtc ctgctgacaa 2041 atcaagagca ttgcttttgt ttcttaagaa aacaaactct tttttaaaaa ttacttttaa 2101 atattaactc aaaagttgag attttggggt ggtggtgtgc caagacatta attttttttt 2161 taaacaatga agtgaaaaag ttttacaatc tctaggtttg gctagttctc ttaacactgg 2221 ttaaattaac attgcataaa cacttttcaa gtctgatcca tatttaataa tgctttaaaa 2281 taaaaataaa aacaatcctt ttgataaatt taaaatgtta cttattttaa aataaatgaa 2341 gtgagatggc atggtgaggt gaaagtatca ctggactagg aagaaggtga cttaggttct 2401 agataggtgt cttttaggac tctgattttg aggacatcac ttactatcca tttcttcatg
2461 ttaaaagaag tcatctcaaa ctcttagttt ttttttttta caactatgta atttatattc 2521 catttacata aggatacact tatttgtcaa gctcagcaca atctgtaaat ttttaaccta 2581 tgttacacca tcttcagtgc cagtcttggg caaaattgtg caagaggtga agtttatatt 2641 tgaatatcca ttctcgtttt aggactcttc ttccatatta gtgtcatctt gcctccctac 2701 cttccacatg ccccatgact tgatgcagtt ttaatacttg taattcccct aaccataaga 2761 tttactgctg ctgtggatat ctccatgaag ttttcccact gagtcacatc agaaatgccc 2821 tacatcttat ttcctcaggg ctcaagagaa tctgacagat accataaagg gatttgacct 2881 aatcactaat tttcaggtgg tggctgatgc tttgaacatc tctttgctgc ccaatccatt 2941 agcgacagta ggatttttca aacctggtat gaatagacag aaccctatcc agtggaagga 3001 gaatttaata aagatagtgc tgaaagaatt ccttaggtaa tctataacta ggactactcc 3061 tggtaacagt aatacattcc attgttttag taaccagaaa tcttcatgca atgaaaaata 3121 ctttaattca tgaagcttac tttttttttt tggtgtcaga gtctcgctct tgtcacccag 3181 gctggaatgc agtggcgcca tctcagctca ctgcaacctc catctcccag gttcaagcga 3241 ttctcgtgcc tcggcctcct gagtagctgg gattacaggc gtgtgccact acactcaact 3301 aatttttgta tttttaggag agacggggtt tcaccctgtt ggccaggctg gtctcgaact 3361 cctgacctca agtgatGcac ccaccttggc ctcataaacc tgttttgcag aactcattta 3421 ttcagcaaat atttattgag tgcctaccag atgccagtca ccgcacaagg cactgggtat 3481 atggtatccc caaacaagag acataatccc ggtccttagg tagtgctagt gtggtctgta 3541 atatcttact aaggcctttg gtatacgacc cagagataac acgatgcgta ttttagtttt 3601 gcaaagaagg ggtttggtct ctgtgccagc tctataattg ttttgctacg attccactga 3661 aactcttcga tcaagctact ttatgtaaat cacttcattg ttttaaagga ataaacttga 3721 ttatattgtt tttttatttg gcataactgt gattctttta ggacaattac tgtacacatt 3781 aaggtgtatg tcagatattc atattgaccc aaatgtgtaa tattccagtt ttctctgcat 3841 aagtaattaa aatatactta aaaattaata gttttatctg ggtacaaata aacaggtgcc 3901 tgaactagtt cacagacaag gaaacttcta tgtaaaaatc actatgattt ctgaattgct 3961 atgtgaaact acagatcttt ggaacactgt ttaggtaggg tgttaagact tacacagtac 4021 ctcgtttcta cacagagaaa gaaatggcca tacttcagga actgcagtgc ttatgagggg 4081 atatttaggc ctcttgaatt tttgatgtag atgggcattt ttttaaggta gtggttaatt 4141 acctttatgt gaactttgaa tggtttaaca aaagatttgt ttttgtagag attttaaagg 4201 gggagaattc tagaaataaa tgttacctaa ttattacagc cttaaagaca aaaatccttg 4261 ttgaagtttt tttaaaaaaa gctaaattac atagacttag gcattaacat gtttgtggaa 4321 gaatatagca gacgtatatt gtatcatttg agtgaatgtt cccaagtagg cattctaggc 4381 tctatttaac tgagtcacac tgcataggaa tttagaacct aacttttata ggttatcaaa 4441 actgttgtca ccattgcaca attttgtcct aatatataca tagaaacttt gtggggcatg 4501 ttaagttaca gtttgcacaa gttcatctca tttgtattcc attgattttt tttttcttct 4561 aaacattttt tcttcaaaca gtatataact ttttttaggg gatttttttt tagacagcaa 4621 aaactatctg aagatttcca tttgtcaaaa agtaatgatt tcttgataat tgtgtagtaa 4681 tgttttttag aacccagcag ttaccttaaa gctgaattta tatttagtaa cttctgtgtt 4741 aatactggat agcatgaatt ctgcattgag aaactgaata gctgtcataa aatgaaactt 4801 tctttctaaa gaaagatact cacatgagtt cttgaagaat agtcataact agattaagat 4861 ctgtgtttta gtttaatagt ttgaagtgcc tgtttgggat aatgataggt aatttagatg 4921 aatttagggg aaaaaaaagt tatctgcaga tatgttgagg gcccatctct ccccccacac 4981 ccccacagag ctaactgggt tacagtgttt tatccgaaag tttccaattc cactgtcttg 5041 tgttttcatg ttgaaaatac ttttgcattt ttcctttgag tgccaatttc ttactagtac 5101 tatttcttaa tgtaacatgt ttacctggaa tgtattttaa ctatttttgt atagtgtaaa 5161 ctgaaacatg cacattttgt acattgtgct ttcttttgtg ggacatatgc agtgtgatcc 5221 agttgttttc catcatttgg ttgcgctgac ctaggaatgt tggtcatatc aaacattaaa 5281 aatgaccact cttttaattg aaattaactt ttaaatgttt ataggagtat gtgctgtgaa 5341 gtgatctaaa atttgtaata tttttgtcat gaactgtact actcctaatt attgtaatgt 5401 aataaaaata gttacagtga caaaaaaaaa aaaaaa
[128] Human KRAS, transcript variant b, comprising the LCS6 SNP, is encoded by the following mRNA sequence (SEQ ID NO: 12)(untranslated regions are bolded, LCS6 is underlined, SNP is capitalized):
1 ggccgcggcg gcggaggcag cagcggcggc ggcagtggcg gcggcgaagg tggcggcggc
61 tcggccagta ctcccggccc ccgccatttc ggactgggag cgagcgcggc gcaggcactg
121 aaggcggcgg cggggccaga ggctcagcgg ctcccaggtg cgggagagag gcctgctgaa
181 aatgactgaa tataaacttg tggtagttgg agctggtggc gtaggcaaga gtgccttgac
241 gatacagcta attcagaatc attttgtgga cgaatatgat ccaacaatag aggattccta
301 caggaagcaa gtagtaattg atggagaaac ctgtctcttg gatattctcg acacagcagg
361 tcaagaggag tacagtgcaa tgagggacca gtacatgagg actggggagg gctttctttg
421 tgtatttgcc ataaataata ctaaatcatt tgaagatatt caccattata gagaacaaat
481 taaaagagtt aaggactctg aagatgtacc tatggtccta gtaggaaata aatgtgattt
541 gccttctaga acagtagaca caaaacaggc tcaggactta gcaagaagtt atggaattcc
601 ttttattgaa acatcagcaa agacaagaca gggtgttgat gatgccttct atacattagt
661 tcgagaaatt cgaaaacata aagaaaagat gagcaaagat ggtaaaaaga agaaaaagaa
721 gtcaaagaca aagtgtgtaa ttatgtaaat acaatttgta cttttttctt aaggcatact
781 agtacaagtg gtaatttttg tacattacac taaattatta gcatttgttt tagcattacc
841 taattttttt cctgctccat gcagactgtt agcttttacc ttaaatgctt attttaaaat
901 gacagtggaa gttttttttt cctctaagtg ccagtattcc cagagttttg gtttttgaac
961 tagcaatgcc tgtgaaaaag aaactgaata cctaagattt ctgtcttggg gtttttggtg
1021 catgcagttg attacttctt atttttctta ccaattgtga atgttggtgt gaaacaaatt
1081 aatgaagctt ttgaatcatc cctattctgt gttttatcta gtcacataaa tggattaatt
1141 actaatttca gttgagacct tctaattggt ttttactgaa acattgaggg aacacaaatt
1201 tatgggcttc ctgatgatga ttcttctagg catcatgtcc tatagtttgt catccctgat
1261 gaatgtaaag ttacactgtt cacaaaggtt ttgtctcctt tccactgcta ttagtcatgg
1321 tcactctccc caaaatatta tattttttct ataaaaagaa aaaaatggaa aaaaattaca
1381 aggcaatgga aactattata aggccatttc cttttcacat tagataaatt actataaaga
1441 ctcctaatag cttttcctgt taaggcagac ccagtatgaa atggggatta ttatagcaac
1501 cattttgggg ctatatttac atgctactaa atttttataa taattgaaaa gattttaaca
1561 agtataaaaa attctcatag gaattaaatg tagtctccct gtgtcagact gctctttcat
1621 agtataactt taaatctttt cttcaacttg agtctttgaa gatagtttta attctgcttg
1681 tgacattaaa agattatttg ggccagttat agcttattag gtgttgaaga gaccaaggtt
1741 gcaaggccag gccctgtgtg aacctttgag ctttcataga gagtttcaca gcatggactg
1801 tgtccccacg gtcatccagt gttgtcatgc attggttagt caaaatgggg agggactagg
1861 gcagtttgga tagctcaaca agatacaatc tcactctgtg gtggtcctgc tgacaaatca
1921 agagcattgc ttttgtttct taagaaaaca aactcttttt taaaaattac ttttaaatat
1981 taactcaaaa gttgagattt tggggtggtg gtgtgccaag acattaattt tttttttaaa
2041 caatgaagtg aaaaagtttt acaatctcta ggtttggcta gttctcttaa cactggttaa
2101 attaacattg cataaacact tttcaagtct gatccatatt taataatgct ttaaaataaa
2161 aataaaaaca atccttttga taaatttaaa atgttactta ttttaaaata aatgaagtga
2221 gatggcatgg tgaggtgaaa gtatcactgg actaggaaga aggtgactta ggttctagat
2281 aggtgtcttt taggactctg attttgagga catcacttac tatccatttc ttcatgttaa
2341 aagaagtcat ctcaaactct tagttttttt tttttacaac tatgtaattt atattccatt
2401 tacataagga tacacttatt tgtcaagctc agcacaatct gtaaattttt aacctatgtt
2461 acaccatctt cagtgccagt cttgggcaaa attgtgcaag aggtgaagtt tatatttgaa
2521 tatccattct cgttttagga ctcttcttcc atattagtgt catcttgcct ccctaccttc
2581 cacatgcccc atgacttgat gcagttttaa tacttgtaat tcccctaacc ataagattta
2641 ctgctgctgt ggatatctcc atgaagtttt cccactgagt cacatcagaa atgccctaca
2701 tcttatttcc tcagggctca agagaatctg acagatacca taaagggatt tgacctaatc
2761 actaattttc aggtggtggc tgatgctttg aacatctctt tgctgcccaa tccattagcg
2821 acagtaggat ttttcaaacc tggtatgaat agacagaacc ctatccagtg gaaggagaat
2881 ttaataaaga tagtgctgaa agaattcctt aggtaatcta taactaggac tactcctggt
2941 aacagtaata cattccattg ttttagtaac cagaaatctt catgcaatga aaaatacttt
3001 aattcatgaa gcttactttt tttttttggt gtcagagtct cgctcttgtc acccaggctg
3061 gaatgcagtg gcgccatctc agctcactgc aacctccatc tcccaggttc aagcgattct
3121 cgtgcctcgg cctcctgagt agctgggatt acaggcgtgt gccactacac tcaactaatt
3181 tttgtatttt taggagagac ggggtttcac cctgttggcc aggctggtct cgaactcctg
3241 acctcaagtg atGcacccac cttggcctca taaacctgtt ttgcagaact catttattca
3301 gcaaatattt attgagtgcc taccagatgc cagtcaccgc acaaggcact gggtatatgg
3361 tatccccaaa caagagacat aatcccggtc cttaggtagt gctagtgtgg tctgtaatat
3421 cttactaagg cctttggtat acgacccaga gataacacga tgcgtatttt agttttgcaa
3481 agaaggggtt tggtctctgt gccagctcta taattgtttt gctacgattc cactgaaact
3541 cttcgatcaa gctactttat gtaaatcact tcattgtttt aaaggaataa acttgattat 3601 attgtttttt tatttggcat aactgtgatt cttttaggac aattactgta cacattaagg 3661 tgtatgtcag atattcatat tgacccaaat gtgtaatatt ccagttttct ctgcataagt 3721 aattaaaata tacttaaaaa ttaatagttt tatctgggta caaataaaca ggtgcctgaa 3781 ctagttcaca gacaaggaaa cttctatgta aaaatcacta tgatttctga attgctatgt 3841 gaaactacag atctttggaa cactgtttag gtagggtgtt aagacttaca cagtacctcg 3901 tttctacaca gagaaagaaa tggccatact tcaggaactg cagtgcttat gaggggatat 3961 ttaggcctct tgaatttttg atgtagatgg gcattttttt aaggtagtgg ttaattacct 4021 ttatgtgaac tttgaatggt ttaacaaaag atttgttttt gtagagattt taaaggggga 4081 gaattctaga aataaatgtt acctaattat tacagcctta aagacaaaaa tccttgttga 4141 agttttttta aaaaaagcta aattacatag acttaggcat taacatgttt gtggaagaat 4201 atagcagacg tatattgtat catttgagtg aatgttccca agtaggcatt ctaggctcta 4261 tttaactgag tcacactgca taggaattta gaacctaact tttataggtt atcaaaactg 4321 ttgtcaccat tgcacaattt tgtcctaata tatacataga aactttgtgg ggcatgttaa 4381 gttacagttt gcacaagttc atctcatttg tattccattg attttttttt tcttctaaac 4441 attttttctt caaacagtat ataacttttt ttaggggatt tttttttaga cagcaaaaac 4501 tatctgaaga tttccatttg tcaaaaagta atgatttctt gataattgtg tagtaatgtt 4561 ttttagaacc cagcagttac cttaaagctg aatttatatt tagtaacttc tgtgttaata 4621 ctggatagca tgaattctgc attgagaaac tgaatagctg tcataaaatg aaactttctt 4681 tctaaagaaa gatactcaca tgagttcttg aagaatagtc ataactagat taagatctgt 4741 gttttagttt aatagtttga agtgcctgtt tgggataatg ataggtaatt tagatgaatt 4801 taggggaaaa aaaagttatc tgcagatatg ttgagggccc atctctcccc ccacaccccc 4861 acagagctaa ctgggttaca gtgttttatc cgaaagtttc caattccact gtcttgtgtt 4921 ttcatgttga aaatactttt gcatttttcc tttgagtgcc aatttcttac tagtactatt 4981 tcttaatgta acatgtttac ctggaatgta ttttaactat ttttgtatag tgtaaactga 5041 aacatgcaca ttttgtacat tgtgctttct tttgtgggac atatgcagtg tgatccagtt 5101 gttttccatc atttggttgc gctgacctag gaatgttggt catatcaaac attaaaaatg 5161 accactcttt taattgaaat taacttttaa atgtttatag gagtatgtgc tgtgaagtga 5221 tctaaaattt gtaatatttt tgtcatgaac tgtactactc ctaattattg taatgtaata 5281 aaaatagtta cagtgacaaa aaaaaaaaaa aa
[129] The KRAS variant is the result of a substitution of a G for a U at position 4 of SEQ ID NO: 6 of LCS6. This KRAS variant comprises the sequence
GAUGCACCCACCUUGGCCUCA (SNP bolded for emphasis) (SEQ ID NO: 13).
[130] The KRAS variant leads to altered KRAS expression by disrupting the miRNA regulation of a KRAS. The identification and characterization of the KRAS variant is further described in International Application No. PCT/US08/65302 (and corresponding publication number WO 2008/151004), the contents of which are incorporated by reference in their entirety.
Let-7 family miRNAs
[131] Expression of let-7 family miRNAs is decreased in cells that carry the KRAS variant. Interestingly, the let-7 family of miRNAs bind to the let-7 complementary site in which the KRAS variant in located. The presence of the KRAS variant interferes with let-7 binding to KRAS. By interfering, the KRAS variant either induces let-7 to bind more or less tightly to LCS6 of KRAS. It was discovered that cells containing the KRAS variant have lower levels of KRAS mRNA compared to wild type cells, and increased levels of the
KRAS protein. Thus, while not wishing to be bound by theory, the presence of the KRAS variant within cells may interfere with the ability of let-7 to bind to KRAS and inhibit protein translation, allowing higher KRAS protein levels.
[132] Exemplary let-7 miRNAs include, but are not limited to, let-7 a (let-7 a-l, let-7 a-2, let-7 a-3), let-7 b, let-7 c, let-7 d, let-7 e, let-7 f (let-7 f- 1 and let-7 f-2), let-7 g, and let-7 i. For the following sequences, thymine (T) may be substituted for uracil (U). let-7 a comprises the sequence UUGAUAUGUUGGAUGAUGGAGU (SEQ ID NO: 14). let-7b comprises the sequence UUGGUGUGUUGGAUGAUGGAGU (SEQ ID NO: 15). let-7 c comprises the sequence UUGGUAUGUUGGAUGAUGGAGU (SEQ ID NO: 16). let-7d comprises the sequence UGAUACGUUGGAUGAUGGAGA (SEQ ID NO: 17). let-7e comprises the sequence UAUAUGUUGGAGGAUGGAGU (SEQ ID NO: 18). let- 7f comprises the sequence UUGAUAUGUUAGAUGAUGGAGU (SEQ ID NO: 19). let-7g comprises the sequence GACAUGUUUGAUGAUGGAGU (SEQ ID NO: 20). let-7 i comprises the sequence UGUCGUGUUUGUUGAUGGAGU (SEQ ID NO: 21).
[133] Sequences of additional let-7 family members are publicly available from miRBase at (www.mirbase.org).
Therapeutic Methods
[134] By poor prognosis is meant that the probability of the individual surviving the development of a particularly aggressive, high-risk, severe, or inherited form of cancer (e.g., triple negative breast cancer), or that the probability of surviving the development or progression of an aggressive, high-risk, severe, or inherited form is less than the probability of surviving the development or progression of a more benign form.
[135] Poor prognosis is also meant to describe a less satisfactory recovery, longer recovery period, more invasive or high-risk therapeutic regimen, or an increased probability of reoccurrence of cancer or a metastasis thereof. For example, triple negative breast cancer or a metastasis thereof is correlated with the worst prognosis of breast cancer subtypes, resulting in a poor prognosis for the subject.
[136] The terms subject, patient, and individual are used interchangeably throughout the description. A subject is preferably a mammal. The mammal can be a human, non-human primate, mouse, rat, dog, cat, horse, or cow, but are not limited to these examples. A subject is male or female. A subject may not have been previously diagnosed as having cancer, a particular type of cancer (e.g., breast cancer), or a subtype of cancer (e.g., triple negative
breast cancer as a subtype of breast cancer). The subject may exhibit one or more risk factors for cancer, a particular type of cancer or a subtype of cancer. Alternatively, the subject does not exhibit a risk factor for cancer, a particular type of cancer or a subtype of cancer.
[137] The methods described herein may require obtaining a sample from a subject. The sample can be any tissue or fluid that contains nucleic acids. Various embodiments include, but are not limited to, paraffin imbedded tissue, frozen tissue, surgical fine needle aspirations, and cells of the breast (including cells harvested from a duct, a lobule, or connective tissue), a lymph node (including a sentinel or axillary node), a thoracic or abdominal muscle or connective tissue, an organ (including any potential deposit site for a potential metastatic cell, such as the brain, liver, kidney, stomach, intestines, bone marrow, pancreas, colon, or lung). Other embodiments include fluid samples such as blood, plasma, serum, lymph fluid, ascites, serous fluid, and urine.
SNP Genotyping Methods
[138] The KRAS variant is a single nucleotide polymorphism that occurs within the 3 ' UTR of the human KRAS gene. Linkage disequilibrium (LD) refers to the co-inheritance of alleles (e.g., alternative nucleotides) at two or more different SNP sites at frequencies greater than would be expected from the separate frequencies of occurrence of each allele in a given population. The expected frequency of co-occurrence of two alleles that are inherited independently is the frequency of the first allele multiplied by the frequency of the second allele. Alleles that co-occur at expected frequencies are said to be in "linkage equilibrium". In contrast, LD refers to any non-random genetic association between allele(s) at two or more different SNP sites, which is generally due to the physical proximity of the two loci along a chromosome. LD can occur when two or more SNPs sites are in close physical proximity to each other on a given chromosome and therefore alleles at these SNP sites will tend to remain unseparated for multiple generations with the consequence that a particular nucleotide (allele) at one SNP site will show a non-random association with a particular nucleotide (allele) at a different SNP site located nearby. Hence, genotyping one of the SNP sites will give almost the same information as genotyping the other SNP site that is in LD.
[139] For the purposes of screening individuals for genetic disorders, if a particular SNP site is found to be useful for screening a disorder, other SNP sites which are in LD with this SNP site are useful for screening the condition. Various degrees of LD can be encountered between two or more SNPs with the result being that some SNPs are more closely associated
(i.e., in stronger LD) than others. Furthermore, the physical distance over which LD extends along a chromosome differs between different regions of the genome, and therefore the degree of physical separation between two or more SNP sites necessary for LD to occur can differ between different regions of the genome.
[140] For screening applications, polymorphisms (e.g., SNPs and/or haplotypes) that are not the actual disease-causing (causative) polymorphisms, but are in LD with such causative polymorphisms, are also useful. In such instances, the genotype of the polymorphism(s) that is/are in LD with the causative polymorphism is predictive of the genotype of the causative polymorphism and, consequently, predictive of the phenotype (e.g., disease) that is influenced by the causative SNP(s). Thus, polymorphic markers that are in LD with causative polymorphisms are useful as markers, and are particularly useful when the actual causative polymorphism(s) is/are unknown.
[141] Linkage disequilibrium in the human genome is reviewed in: Wall et al., "Haplotype blocks and linkage disequilibrium in the human genome", Nat Rev Genet. 2003 August; 4(8):587-97; Gamer et al, "On selecting markers for association studies: patterns of linkage disequilibrium between two and three diallelic loci", Genet Epidemiol. 2003 January;
24(l):57-67; Ardlie et al., "Patterns of linkage disequilibrium in the human genome", Nat Rev Genet. 2002 April; 3(4):299-309 (erratum in Nat Rev Genet 2002 July; 3(7):566); and Remm et al., "High-density genotyping and linkage disequilibrium in the human genome using chromosome 22 as a model"; Curr Opin Chem Biol. 2002 February; 6(l):24-30.
[142] The screening techniques of the present disclosure may employ a variety of methodologies to determine whether a test subject has a SNP or a SNP pattern associated with an increased or decreased risk of developing a detectable trait or whether the individual suffers from a detectable trait as a result of a particular polymorphism/mutation, including, for example, methods which enable the analysis of individual chromosomes for haplotyping, family studies, single sperm DNA analysis, or somatic hybrids. The trait analyzed using the diagnostics of the disclosure may be any detectable trait that is commonly observed in pathologies and disorders.
[143] The process of determining which specific nucleotide (i.e., allele) is present at each of one or more SNP positions, such as a SNP position in a nucleic acid molecule disclosed in
SEQ ID NO: 11, 12, 13 or 22, is referred to as SNP genotyping. The present disclosure provides methods of SNP genotyping, such as for use in screening for a variety of disorders,
or determining predisposition thereto, or determining responsiveness to a form of treatment, or prognosis, or in genome mapping or SNP association analysis, etc.
[144] Nucleic acid samples can be genotyped to determine which allele(s) is/are present at any given genetic region (e.g., SNP position) of interest by methods well known in the art. The neighboring sequence can be used to design SNP detection reagents such as
oligonucleotide probes, which may optionally be implemented in a kit format. Exemplary SNP genotyping methods are described in Chen et al, "Single nucleotide polymorphism genotyping: biochemistry, protocol, cost and throughput", Pharmaco genomics J. 2003;
3(2):77-96; Kwok et al, "Detection of single nucleotide polymorphisms", Curr Issues Mol. Biol. 2003 April; 5(2):43-60; Shi, "Technologies for individual genotyping: detection of genetic polymorphisms in drug targets and disease genes", Am J Pharmacogenomics. 2002; 2(3): 197-205; and Kwok, "Methods for genotyping single nucleotide polymorphisms", Annu Rev Genomics Hum Genet 2001; 2: 235-58. Exemplary techniques for high-throughput SNP genotyping are described in Marnellos, "High-throughput SNP analysis for genetic association studies", Curr Opin Drug Discov Devel. 2003 May; 6(3):317-21. Common SNP genotyping methods include, but are not limited to, TaqMan assays, molecular beacon assays, nucleic acid arrays, allele-specific primer extension, allele-specific PCR, arrayed primer extension, homogeneous primer extension assays, primer extension with detection by mass spectrometry, pyrosequencing, multiplex primer extension sorted on genetic arrays, ligation with rolling circle amplification, homogeneous ligation, OLA (U.S. Pat. No. 4,988,167), multiplex ligation reaction sorted on genetic arrays, restriction-fragment length
polymorphism, single base extension-tag assays, and the Invader assay. Such methods may be used in combination with detection mechanisms such as, for example, luminescence or chemiluminescence detection, fluorescence detection, time-resolved fluorescence detection, fluorescence resonance energy transfer, fluorescence polarization, mass spectrometry, and electrical detection.
[145] Various methods for detecting polymorphisms include, but are not limited to, methods in which protection from cleavage agents is used to detect mismatched bases in RNA/RNA or
RNA/DNA duplexes (Myers et al, Science 230: 1242 (1985); Cotton et al, PNAS 85:4397
(1988); and Saleeba et al, Meth. Enzymol. 217:286-295 (1992)), comparison of the electrophoretic mobility of variant and wild type nucleic acid molecules (Orita et al., PNAS
86:2766 (1989); Cotton et al, Mutat. Res. 285: 125-144 (1993); and Hayashi et al, Genet.
Anal. Tech. Appl. 9:73-79 (1992)), and assaying the movement of polymorphic or wild-type fragments in polyacrylamide gels containing a gradient of denaturant using denaturing gradient gel electrophoresis (DGGE) (Myers et al, Nature 313:495 (1985)). Sequence variations at specific locations can also be assessed by nuclease protection assays such as RNase and SI protection or chemical cleavage methods.
[146] In a preferred embodiment, SNP genotyping is performed using the TaqMan assay, which is also known as the 5' nuclease assay (U.S. Pat. Nos. 5,210,015 and 5,538,848). The TaqMan assay detects the accumulation of a specific amplified product during PCR. The TaqMan assay utilizes an oligonucleotide probe labeled with a fluorescent reporter dye and a quencher dye. The reporter dye is excited by irradiation at an appropriate wavelength, it transfers energy to the quencher dye in the same probe via a process called fluorescence resonance energy transfer (FRET). When attached to the probe, the excited reporter dye does not emit a signal. The proximity of the quencher dye to the reporter dye in the intact probe maintains a reduced fluorescence for the reporter. The reporter dye and quencher dye may be at the 5' most and the 3' most ends, respectively, or vice versa. Alternatively, the reporter dye may be at the 5 ' or 3' most end while the quencher dye is attached to an internal nucleotide, or vice versa. In yet another embodiment, both the reporter and the quencher may be attached to internal nucleotides at a distance from each other such that fluorescence of the reporter is reduced.
[147] During PCR, the 5' nuclease activity of DNA polymerase cleaves the probe, thereby separating the reporter dye and the quencher dye and resulting in increased fluorescence of the reporter. Accumulation of PCR product is detected directly by monitoring the increase in fluorescence of the reporter dye. The DNA polymerase cleaves the probe between the reporter dye and the quencher dye only if the probe hybridizes to the target SNP-containing template which is amplified during PCR, and the probe is designed to hybridize to the target SNP site only if a particular SNP allele is present.
[148] Preferred TaqMan primer and probe sequences can readily be determined using the
SNP and associated nucleic acid sequence information provided herein. A number of computer programs, such as Primer Express (Applied Biosystems, Foster City, Calif), can be used to rapidly obtain optimal primer/probe sets. It will be apparent to one of skill in the art that such primers and probes for detecting the SNPs of the present disclosure are useful in prognostic assays for a variety of disorders including cancer, and can be readily incorporated
into a kit format. The present disclosure also includes modifications of the Taqman assay well known in the art such as the use of Molecular Beacon probes (U.S. Pat. Nos. 5,118,801 and 5,312,728) and other variant formats (U.S. Pat. Nos. 5,866,336 and 6,1 17,635).
[149] The identity of polymorphisms may also be determined using a mismatch detection technique, including but not limited to the RNase protection method using riboprobes (Winter et al, Proc. Natl. Acad Sci. USA 82:7575, 1985; Meyers et al, Science 230: 1242, 1985) and proteins which recognize nucleotide mismatches, such as the E. coli mutS protein (Modrich, P. Ann. Rev. Genet. 25:229-253, 1991). Alternatively, variant alleles can be identified by single strand conformation polymorphism (SSCP) analysis (Orita et al., Genomics 5:874-879, 1989; Humphries et al, in Molecular Diagnosis of Genetic Diseases, R. Elles, ed., pp. 321- 340, 1996) or denaturing gradient gel electrophoresis (DGGE) (Wartell et al, Nuci. Acids Res. 18:2699-2706, 1990; Sheffield et al, Proc. Natl. Acad. Sci. USA 86:232-236, 1989).
[150] A polymerase-mediated primer extension method may also be used to identify the polymorphism(s). Several such methods have been described in the patent and scientific literature and include the "Genetic Bit Analysis" method (W092/15712) and the
ligase/polymerase mediated genetic bit analysis (U.S. Pat. No. 5,679,524). Related methods are disclosed in WO91/02087, WO90/09455, W095/17676, U.S. Pat. Nos. 5,302,509, and 5,945,283. Extended primers containing a polymorphism may be detected by mass spectrometry as described in U.S. Pat. No. 5,605,798. Another primer extension method is allele-specific PCR (Ruano et al, Nucl. Acids Res. 17:8392, 1989; Ruano et al, Nucl. Acids Res. 19, 6877-6882, 1991; WO 93/22456; Turki et al, J Clin. Invest. 95: 1635-1641, 1995). In addition, multiple polymorphic sites may be investigated by simultaneously amplifying multiple regions of the nucleic acid using sets of allele-specific primers as described in Wallace et al. (WO89/10414).
[151] Another preferred method for genotyping the KRAS variant is the use of two oligonucleotide probes in an OLA (see, e.g., U.S. Pat. No. 4,988,617). In this method, one probe hybridizes to a segment of a target nucleic acid with its 3' most end aligned with the
SNP site. A second probe hybridizes to an adjacent segment of the target nucleic acid molecule directly 3' to the first probe. The two juxtaposed probes hybridize to the target nucleic acid molecule, and are ligated in the presence of a linking agent such as a ligase if there is perfect complementarity between the 3' most nucleotide of the first probe with the
SNP site. If there is a mismatch, ligation would not occur. After the reaction, the ligated
probes are separated from the target nucleic acid molecule, and detected as indicators of the presence of a SNP.
[152] The following patents, patent applications, and published international patent applications, which are all hereby incorporated by reference, provide additional information pertaining to techniques for carrying out various types of OLA: U.S. Pat. Nos. 6,027,889, 6,268,148, 5494810, 5830711, and 6054564 describe OLA strategies for performing SNP detection; WO 97/31256 and WO 00/56927 describe OLA strategies for performing SNP detection using universal arrays, wherein a zipcode sequence can be introduced into one of the hybridization probes, and the resulting product, or amplified product, hybridized to a universal zip code array; U.S. application US01/17329 (and Ser. No. 09/584,905) describes OLA (or LDR) followed by PCR, wherein zipcodes are incorporated into OLA probes, and amplified PCR products are determined by electrophoretic or universal zipcode array readout; U.S. application 60/427,818, 60/445,636, and 60/445,494 describe SNPlex methods and software for multiplexed SNP detection using OLA followed by PCR, wherein zipcodes are incorporated into OLA probes, and amplified PCR products are hybridized with a zipchute reagent, and the identity of the SNP determined from electrophoretic readout of the zipchute. In some embodiments, OLA is carried out prior to PCR (or another method of nucleic acid amplification). In other embodiments, PCR (or another method of nucleic acid amplification) is carried out prior to OLA.
[153] Another method for SNP genotyping is based on mass spectrometry. Mass
spectrometry takes advantage of the unique mass of each of the four nucleotides of DNA. SNPs can be unambiguously genotyped by mass spectrometry by measuring the differences in the mass of nucleic acids having alternative SNP alleles. MALDI-TOF (Matrix Assisted Laser Desorption Ionization— Time of Flight) mass spectrometry technology is preferred for extremely precise determinations of molecular mass, such as SNPs. Numerous approaches to SNP analysis have been developed based on mass spectrometry. Preferred mass
spectrometry-based methods of SNP genotyping include primer extension assays, which can also be utilized in combination with other approaches, such as traditional gel-based formats and microarrays.
[154] Typically, the primer extension assay involves designing and annealing a primer to a template PCR amplicon upstream (5') from a target SNP position. A mix of
dideoxynucleotide triphosphates (ddNTPs) and/or deoxynucleotide triphosphates (dNTPs) are
added to a reaction mixture containing template (e.g., a SNP-containing nucleic acid molecule which has typically been amplified, such as by PCR), primer, and DNA
polymerase. Extension of the primer terminates at the first position in the template where a nucleotide complementary to one of the ddNTPs in the mix occurs. The primer can be either immediately adjacent (i.e., the nucleotide at the 3' end of the primer hybridizes to the nucleotide next to the target SNP site) or two or more nucleotides removed from the SNP position. If the primer is several nucleotides removed from the target SNP position, the only limitation is that the template sequence between the 3' end of the primer and the SNP position cannot contain a nucleotide of the same type as the one to be detected, or this will cause premature termination of the extension primer. Alternatively, if all four ddNTPs alone, with no dNTPs, are added to the reaction mixture, the primer will always be extended by only one nucleotide, corresponding to the target SNP position. In this instance, primers are designed to bind one nucleotide upstream from the SNP position (i.e., the nucleotide at the 3' end of the primer hybridizes to the nucleotide that is immediately adjacent to the target SNP site on the
5' side of the target SNP site). Extension by only one nucleotide is preferable, as it minimizes the overall mass of the extended primer, thereby increasing the resolution of mass differences between alternative SNP nucleotides. Furthermore, mass-tagged ddNTPs can be employed in the primer extension reactions in place of unmodified ddNTPs. This increases the mass difference between primers extended with these ddNTPs, thereby providing increased sensitivity and accuracy, and is particularly useful for typing heterozygous base positions.
Mass-tagging also alleviates the need for intensive sample-preparation procedures and decreases the necessary resolving power of the mass spectrometer.
[155] The extended primers can then be purified and analyzed by MALDI-TOF mass spectrometry to determine the identity of the nucleotide present at the target SNP position. In one method of analysis, the products from the primer extension reaction are combined with light absorbing crystals that form a matrix. The matrix is then hit with an energy source such as a laser to ionize and desorb the nucleic acid molecules into the gas-phase. The ionized molecules are then ejected into a flight tube and accelerated down the tube towards a detector. The time between the ionization event, such as a laser pulse, and collision of the molecule with the detector is the time of flight of that molecule. The time of flight is precisely correlated with the mass-to-charge ratio (m/z) of the ionized molecule. Ions with smaller m/z travel down the tube faster than ions with larger m/z and therefore the lighter
ions reach the detector before the heavier ions. The time-of- flight is then converted into a corresponding, and highly precise, m/z. In this manner, SNPs can be identified based on the slight differences in mass, and the corresponding time of flight differences, inherent in nucleic acid molecules having different nucleotides at a single base position. For further information regarding the use of primer extension assays in conjunction with MALDI-TOF mass spectrometry for SNP genotyping, see, e.g., Wise et al, "A standard protocol for single nucleotide primer extension in the human genome using matrix-assisted laser
desorption/ionization time-of-flight mass spectrometry", Rapid Commun Mass Spectrom. 2003; 17(11): 1195-202.
[156] The following references provide further information describing mass spectrometry- based methods for SNP genotyping: Bocker, "SNP and mutation discovery using base- specific cleavage and MALDI-TOF mass spectrometry", Bioinformatics. 2003 July; 19 Suppl 1 : 144-153; Storm et al, "MALDI-TOF mass spectrometry-based SNP genotyping", Methods Mol. Biol. 2003;212:241-62; Jurinke et al, "The use of MassARRAY technology for high throughput genotyping", Adv Biochem Eng Biotechnol. 2002; 77:57-74; and Jurinke et al, "Automated genotyping using the DNA MassArray technology", Methods Mol. Biol. 2002; 187: 179-92.
[157] SNPs can also be scored by direct DNA sequencing. A variety of automated sequencing procedures can be utilized ((1995) Biotechniques 19:448), including sequencing by mass spectrometry (see, e.g., PCT International Publication No. WO94/16101; Cohen et al., Adv. Chromatogr. 36: 127-162 (1996); and Griffin et al., Appl. Biochem. Biotechnol. 38: 147-159 (1993)). The nucleic acid sequences of the present disclosure enable one of ordinary skill in the art to readily design sequencing primers for such automated sequencing procedures. Commercial instrumentation, such as the Applied Biosystems 377, 3100, 3700, 3730, and 3730.times. l DNA Analyzers (Foster City, Calif), is commonly used in the art for automated sequencing.
[158] Other methods that can be used to genotype the KRAS variant include single-strand conformational polymorphism (SSCP), and denaturing gradient gel electrophoresis (DGGE)
(Myers et al, Nature 313:495 (1985)). SSCP identifies base differences by alteration in electrophoretic migration of single stranded PCR products, as described in Orita et al, Proc.
Nat. Acad. Single-stranded PCR products can be generated by heating or otherwise denaturing double stranded PCR products. Single-stranded nucleic acids may refold or form
secondary structures that are partially dependent on the base sequence. The different electrophoretic mobilities of single-stranded amplification products are related to base- sequence differences at SNP positions. DGGE differentiates SNP alleles based on the different sequence-dependent stabilities and melting properties inherent in polymorphic DNA and the corresponding differences in electrophoretic migration patterns in a denaturing gradient gel (Erlich, ed., PCR Technology, Principles and Applications for DNA
Amplification, W. H. Freeman and Co, New York, 1992, Chapter 7).
[159] Sequence-specific ribozymes (U.S. Pat. No. 5,498,531) can also be used to score SNPs based on the development or loss of a ribozyme cleavage site. Perfectly matched sequences can be distinguished from mismatched sequences by nuclease cleavage digestion assays or by differences in melting temperature. If the SNP affects a restriction enzyme cleavage site, the SNP can be identified by alterations in restriction enzyme digestion patterns, and the corresponding changes in nucleic acid fragment lengths determined by gel electrophoresis
[160] SNP genotyping can include the steps of, for example, collecting a biological sample from a human subject (e.g., sample of tissues, cells, fluids, secretions, etc.), isolating nucleic acids (e.g., genomic DNA, mRNA or both) from the cells of the sample, contacting the nucleic acids with one or more primers which specifically hybridize to a region of the isolated nucleic acid containing a target SNP under conditions such that hybridization and amplification of the target nucleic acid region occurs, and determining the nucleotide present at the SNP position of interest, or, in some assays, detecting the presence or absence of an amplification product (assays can be designed so that hybridization and/or amplification will only occur if a particular SNP allele is present or absent). In some assays, the size of the amplification product is detected and compared to the length of a control sample; for example, deletions and insertions can be detected by a change in size of the amplified product compared to a normal genotype.
EXAMPLES
Example 1 : Prevalence of the KRAS variant in various cancer cell lines
Materials and Methods
[161] Genotyping. DNA from the NCI-60 cell line panel was obtained from the NCI's
Developmental Therapeutics Program. Taqman genotyping was performed to determine the
presence of the KRAS variant allele as described previously (Bussey KJ, et al. Mol Cancer Ther 2006; 5:853-67). Cells were cultured under standard conditions (see,
dtp.cancer.gov/branches/btb/ivclsp.html; Monks A, et al. J Natl Cancer Inst 1991; 83:757- 66), for a maximum of 20 passages from frozen stock. DNA was isolated using the Qiagen QIAamp DNA blood maxi kit procedure (cat. 51192).
[162] Statistical analyses. The KRAS variant allele data were coded numerically, with 1 representing the presence of KRAS variant allele and 0 representing the absence of the KRAS variant allele. This pattern was used as a "seed" in COMPARE analyses (Paull KD, et al. J Natl Cancer Inst 1989; 81 : 1088-9248) to probe the existing NCI-60 data sets in the NCI-DTP databases. Correlations included, for example, miRNA measurements and DNA methylation measurements. A positive correlation indicates, for example, that cell lines with the variant allele tend to have higher expression of the miRNA/mRNA or greater percentage DNA methylation. Conversely, negative correlations indicate that cell lines with the variant allele tend to have lower expression of a given miRNA/mRNA or lower percentage DNA methylation at the indicated gene. These data sets can be queried or downloaded at dtp.cancer.gov.
[163] The presence of the KRAS variant is a genetic marker for prediction of risk and tumor biology as well as response to treatment in multiple cancers. The presence of the KRAS variant results in altered regulation by the KRAS 3' UTR. This study elucidates the biological significance of the KRAS variant in cancer cells. The data provided herein elucidate exemplary molecular pathways that are affected by the presence of the KRAS variant. To simultaneously analyze a broad range of cancer types, the comprehensive NCI-60 panel of cancer cell lines (Blower PE, et al. Mol Cancer Ther 2007; 6: 1483-91; Liu H, et al. Mol
Cancer Ther 2010; 9: 1080-91) was used. Various molecular parameters were studied to determine which molecular events correlate with the presence of the KRAS variant in these cancer cell lines (Kundu, S.T. et al. 2012 Jan 15. Cell Cycle 11 :2, 361-366).
[164] Seven of 60 cell lines in the NCI-60 panel harbor the KRAS variant allele (Table 1).
When the NCI-60 panel of cell lines were categorized based on the presence of either an acquired dominant mutation in the KRAS coding region (KRAS mutation) or the presence of the KRAS variant, it was determined that all seven cell lines that contained the KRAS variant were negative for the presence of KRAS-activating mutations. Similarly, the cell lines that carried a KRAS coding sequence mutation lacked the KRAS variant allele. Thus, the presence
or occurrence of either a KRAS coding mutation or the KRAS variant allele is mutually exclusive in these cell lines. Furthermore, because this mutual exclusivity occurs in cell lines derived from a variety of cancer types, this mutual exclusivity is not specific to a particular tissue type. Rather, this mutual exclusivity is a common feature of these cancer cell lines regardless of origin. These results indicate that the occurrence of either of these two events alone (i.e., the occurrence of the KRAS variant or the occurrence of a KRAS coding mutation), is sufficient to affect tumorigenesis in these cancer types. These results also indicate that the level of KRAS activation caused by a canonical coding sequence mutation is functionally comparable to the elevated KRAS expression induced by the presence of the KRAS variant in the 3' UTR. This mutual exclusivity of acquired KRAS coding mutations and the KRAS variant was also found in non-small cell lung cancer patients (Chin LJ, et al. Cancer Res 2008; 68:8535-40) and in ovarian cancer patients (Ratner E, Cancer Res 2010; 70:6509-15), but not in colon cancer patients (Zhang W, et al. Ann Oncol 2011; 22:484-5; Zhang W, et al. Ann Oncol 2011; 22: 104-9).
[165] Table 1. Cell lines in the NCI-60 panel that harbor the KRAS variant allele or a functional mutation in the coding sequence of KRAS.

•0
. . . .
1:
Λ
w ^ m
ii I
w ^ m
, >·i;
W ii iiiiiiiiiiiiiii
Ϊ
■ ■ . ■ ■■ ¾i w m i
■0 1:
.i ·:·:· ·: ·:
€f?
■ >\<- ...
w i m
is
!!!!!!llll!!!
ί ύ
! - , : 11111111111111111
[166] To determine whether the cell lines having the KRAS variant allele show a conserved alteration in the expression of miRNAs, a statistical analysis was performed on the miRNA expression profiles that were generated from seven cell lines that contain the KRAS variant allele compared with the miRNA expression profiles of the remaining cell lines of the NCI- 60 panel (Blower PE, et al. Mol Cancer Ther 2007; 6: 1483-91; Gaur A, et al. Cancer Res 2007; 67:2456-68). The presence of the KRAS variant allele shows a statistically significant positive correlation with increased expression of miR-23, miR-27 and miR-210 (Table 2). MiR-23 and miR-27 are expressed from the same cluster and advance progression of angiogenesis and metastasis (Zhou Q, et al. Proc Natl Acad Sci USA 2011; 108:8287-92). For example, miR-23 and miR-27 are enriched in endothelial cells and highly vascularized tissue. Moreover, miR-23 and miR-27 elevate signaling pathways that are essential for angiogenesis by reducing the expression of Sprouty2 and Sema6A, which have anti- angiogenic functions. Blocking the function of either miR-23 or miR-27 leads to a decrease in capillary tube formation and migration in response to VEGF in vitro and reduced vascularization of postnatal retinas in vivo (Zhou Q, et al. Proc Natl Acad Sci USA 2011; 108:8287-92). The statistically significant positive correlation of the KRAS variant with increased expression of miR-23, miR-27 suggests that tumor cells having the KRAS variant allele are prone to growth and metastatic progression as a result of elevated levels of miR-23 and miR-27.
[167] Table 2. MicroRNAs with statistically significant increased expression in cell lines having the KRAS- variant allele.

[168] The expression of miR-210 is statistically significantly correlated with the presence of the KRAS variant allele in cells. MiR-210 is a marker of chronic hypoxia. Moreover, miR-210 is associated with proliferation and metastasis of breast and melanoma tumors as well as poor prognosis. MiR-210 is a direct transcriptional target of HIF proteins. Elevated levels of miR- 210 are required for tumor cell survival under conditions of hypoxia. MiR-210 directly
regulates the expression of MNT, a MYC antagonist that is required for cell cycle arrest under hypoxia. Consequently, increased levels of miR-210 contribute to an override of cell cycle arrest under conditions of hypoxic stress in tumor cells. Because increased miR-210 expression is associated with the presence of the KRAS variant, tumor cells containing the KRAS variant survive and proliferate under hypoxic conditions.
[169] The data provided herein demonstrate that the KRAS variant contributes to or initiates aberrant signaling pathways that control the expression of several miRNAs (including, for example, miR-23, miR-27 and miR-210). Perturbation of signaling pathways that regulate expression of miRNAs, such as miR-23, miR-27 and miR-210, results in the initiation, development, maintenance or augmentation of tumor proliferation and metastatic
transformation.
[170] Promoter methylation is one mechanism through which gene expression is silenced in many cancers because changes in the methylation status of gene promoters lead to reduction in gene expression. Specifically, DNA methylation is an epigenetic effect caused when CpG dinucleotides are methylated, often in the promoter region of genes. Because methylation blocks access to the promoter by molecules that mediate gene transcription, methylation of the promoter results in gene silencing. Different cancers show distinct methylation patterns, the result of which is alterations in gene expression signatures. Therefore, to determine whether there is an alteration in DNA methylation patterns in the tumor cell lines having the KRAS variant, the methylation status of these cell lines was compared with the non-KRAS variant lines in the NCI-60 panel (Ehrich M, et al. Proc Natl Acad Sci USA 2008; 105:4844- 9). The presence of the KRAS variant allele shows a statistically significant positive correlation with increased methylation of the promoter of many genes, including, for example, Notchl, cyclin D3 and CNBP (also known as ZNF9) (Table 3).
[171] Table 3. Genes with statistically significant promoter hyper-methylation in KRAS variant positive cell lines.
!iiiii l sii!!
ξ: ίί:>: :>;·> ¾ν i^i¾ v .:&
. ■
:
·¾ 5S.¾iXi? is;s¾ : «· : :>: - *
.·»
m mii
.¾"·:: ¾·*.? ®,
S5

¾s? .·· ¾>··*<>· 5 sjiss
: : : : : : : : : : : : : : : : : : : : : : : : : : : : : : : : : : : : : : : : : : : :::::: : : : ¾ ::::: :¾$ :::::: i : i i^i ^i^J : i : i ^ ^i^i^ ^iL^ i : i : i : :
« 5 Ϊ* ;ι: :· ·¾·ί
5* ;;;;;;;: 1I§«
[172] The role of Notchl expression in cancers is diverse. In many tumors, Notchl
overexpression or activation drives cancer progression and metastasis. For example, Notchl activation results in an increase in invasive and migratory characteristics of breast cancer cells. Alternatively, Notchl overexpression in a MYC background induces adenomas in the mouse lung, leading to the formation of lung adenocarcinoma. Thus, the evidence indicates that Notchl may function as an oncogene. In contrast, Notchl may also function as a tumor suppressor. For example, inhibitory mutations in Notchl have been identified in squamous cell carcinomas of the head and neck. Depletion of Notchl in mouse skin keratinocytes results in enhanced tumorigenesis by chemical carcinogens or by oncogenic Ras. In cervical cancers positive for the human papillomavirus (HPV), Notchl expression is decreased when compared with normal adjacent tissue. Overexpression of activated Notchl in HPV-positive cervical cancers and neuroblastoma cells (Zage PE, et al. Pediatr Blood Cancer 2011) leads to growth inhibition. Considered together, the evidence show that Notchl is dysregulated in many cancers and, in some instances, may function as a putative tumor suppressor. Because methylation of the Notchl promoter is increased in KRAS variant-positive cancer cells,
Notchl expression may be reduced in cells carrying the KRAS variant allele, and, therefore, KRAS- variant cell lines may induce or maintain their tumorigenic potential by inhibiting the tumor suppressing effects of Notchl.
[173] Cyclin D3 is the member of the cyclin family of cell cycle proteins that is required for the Gi/S transition of the cell cycle. In KRAS variant cell lines, promoter methylation of cyclin D3 is increased, which indicates repression of cyclin D3 transcription. Consequently,
the evidence suggests two exemplary mechanisms in which either cyclin D3 is not required for the transformed phenotype of these cell lines or methylation of the cyclin D3 promoter blocks a transcriptional repressor of cyclin D3.
[174] In contrast to Notchl and cyclin D3, CNBP (cellular nucleic acid binding protein), also called ZNF9, is not associated with the development or progression of cancer. However, CNBP/ ZNF9 is part of a complex that binds to the MYC promoter. When expression of MYC is dysregulated, MYC contributes to the development and progression of cancer. The mechanism by which the association of the KRAS variant with the methylation status of ZNF9 contributes to cancer progression in AT^S-variant cells is unclear.
[175] Gene expression in the seven cell lines harboring the KRAS variant allele was compared with the profiles of the remaining cell lines in the NCI-60 panel to determine specific alterations in gene expression in these cell lines. As shown in Table 4, a gene whose elevated expression is statistically significantly correlated with the presence of the KRAS variant in the cell lines is glutathione S-transferase thetal (GSTTl). The GSTTl gene encodes a member of the glutathione S transferase family of human phase II detoxifying enzymes, which detoxifies complex metabolic byproducts, xenobiotics and drugs by conjugating a glutathione group to these compounds, thus making them more soluble and easily excreted out of the cell. The thetal isoform has been implicated in several cancers. For example, increased expression of GSTTl is statistically significantly correlated with aggressive bladder cancers. In other different tumors types, GSTTl is nonfunctional or absent due to genetic polymorphism, thus leading to increased risk of carcinogenesis and poor prognosis as a result of an accumulation or increased accumulation of toxic metabolites.
[176] Table 4. Genes with statistically significantly higher mR A expression in KRAS positive cell lines.

mm ί ί ί¾¾ί¾ ί ί ί
!ii¾s «ίίί^ϊϊϊϊί H:

¾*ss¾^s 1111111111 lllilllll ^¾^ :Μ
;·/>?« .:>;:.
:li¾ll 11111:1:1 ϊίχ-ί
^^^^^^
Μ$
iCS! »
ί :::::::^^:^:::::::::::::::::::
ss:
¾:·
'■•■'■i
*S¾

[177] Mitogen-activated protein kinase 3 (MAPK3) is a member of the MAP kinase family. Moreover, increased expression of mitogen-activated protein kinase 3 (MAPK3) is associated with the KRAS variant in cancer cells. MAPK3 transduces signals from extracellular cues to regulate intracellular processes, such as cell proliferation and differentiation. For example, increased expression of phosphorylated MAPK3 has been associated with aggressive colorectal tumors and metastatic meduloblastoma. Increased levels of KRAS in KRAS variant positive cancer cells are associated with an increase in MAPK3 mRNA. At least in part, increased MAPK3 expression induces an increase cellular proliferation and neoplastic progression in these cells. Similarly, the expression of another MAPK (MAP2K4) was increased in the AT^S-variant positive expression profile. Furthermore, KRAS and MAPK (MAPK3 and/or MAP2K4) may contribute to a synergistic interaction between KRAS and
MAPK signaling in KRAS-vaxiant cancer cells that induces or enhances cell proliferation and/or neoplastic progression.
[178] Increased expression of Synaptotagmin-12 and increased expression of inter-a globulin inhibitor-Hl are positively correlated with the presence of the KRAS variant in cancer cell lines. Under normal conditions, synaptotagmins regulate calcium-dependent membrane trafficking during synaptic transmission. Although there is no evidence of an involvement of synaptotagmin-12 with cancer, overexpression of synaptotagmin-13, a family member of synaptotagmin-12, suppresses a transformed phenotype of cells derived from a rat liver tumor cell line. Overexpression of synaptotagmin-12 in KRAS variant-positive cancer cell lines indicates a deregulation of novel pathways involving syntaptotagmins in cancer cells. The inter-a (globulin) inhibitor HI is the heavy chain of the plasma serine protease inhibitor. Functionally, the inter-a (globulin) inhibitor HI is required for extracellular matrix stability. Though the role of the inter-a (globulin) inhibitor HI in cancer remains unexplored, recent evidence indicates that the expression of inter-a (globulin) inhibitor HI is either lost or repressed in various solid tumors, including, for example, tumors of the lung, colon and breast.
Example 2: The KRAS variant and Resistance to Platinum-Based Chemotherapy
Materials and Methods
[179] Overall survival analysis cohorts. Complete clinical data and DNA from women diagnosed with EOC without known BRCA mutations were included from the following three institutions under individual International Review Board approvals. All protocols accrued patients prospectively at the time of their diagnosis to avoid selection bias.
References indicate previous detailed descriptions of these patients: (1) Turin, Italy #1 (n = 197) (Lu L, et al. (2007). Cancer Res 67: 10117-10122), (2) Brescia, Italy #2 (n = 59) (Ratner E, et al. (2010). Cancer Res 15: 6509-6515), and (3) the Yale New Haven Hospital (YNHH) (n = 198). Yale patients were collected prospectively on two clinical trials at the Yale Medical School of newly diagnosed EOC patients diagnosed between 2000 and 2009 (Table 5).
[180] Table 5. Clinicopathologic parameters for overall survival analysis.

[181] Documented BRCA mutant EOC cases with known outcome were collected from the following two institutions: (1) the YNHH (n = 17) and (2) the City of Hope Comprehensive Cancer Center (n = 62) (Table 6).
[182] Table 6. Clinicopathologic parameters for BRCA mutant EOC patients.
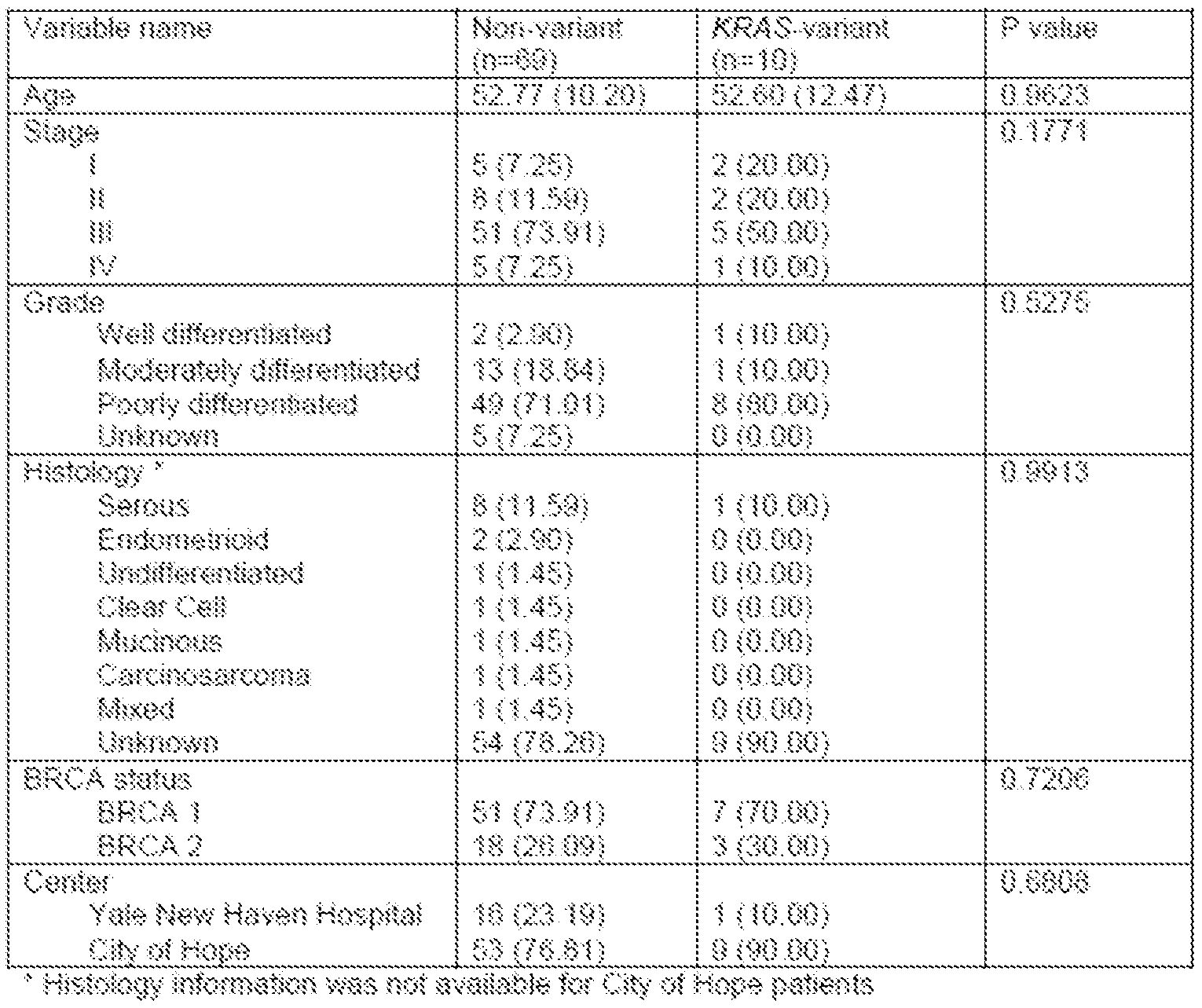
[183] As not all stage 1 ovarian cancer patients receive adjuvant chemotherapy, when substage information was not available for patients with stage 1 tumors, these patients were excluded from the analysis. Otherwise, stage IB and 1C tumors were included with stages 2- 4. To minimize inadvertent inclusion of borderline tumors, tumors with an unknown grade were excluded from this analysis. For women treated with neoadjuvant chemotherapy, the date of pathological diagnosis was considered the start date of treatment. For women treated with adjuvant chemotherapy, the date of surgery was considered the start date of treatment. A total of 386 patients with wild-type BRCA or not tested for BRCA mutations and 79 patients with documented BRCA mutations fit the above-described parameters and were included in the two survival analyses.
[184] Neoadjuvant chemotherapy cohort. Women with EOC who received neoadjuvant platinum-based chemotherapy followed by cytoreductive surgery at the YNHH between 1996 and 2010 were identified on an International Review Board-approved protocol (n = 125) (Table 7). This cohort of patients received chemotherapy as a primary treatment due to tumor burden that was too extensive for optimal surgical debulking at presentation. After
chemotherapy, patients underwent cytoreductive surgery and additional adjuvant treatment. Only patients treated with four or more cycles of neoadjuvant platinum-containing combinations were included in this analysis (n = 116). Optimal cytoreduction was defined as residual disease measuring < 1 cm remaining after surgery, whereas suboptimal cytoreduction was defined as residual disease measuring > 1 cm at the completion of surgery. Only women operated on at Yale by the same group of surgeons were included to avoid bias in surgical skill as a factor impacting residual disease.
[185] Table 7. Clinicopathologic parameters of patients receiving neoadjuvant
chemotherapy.
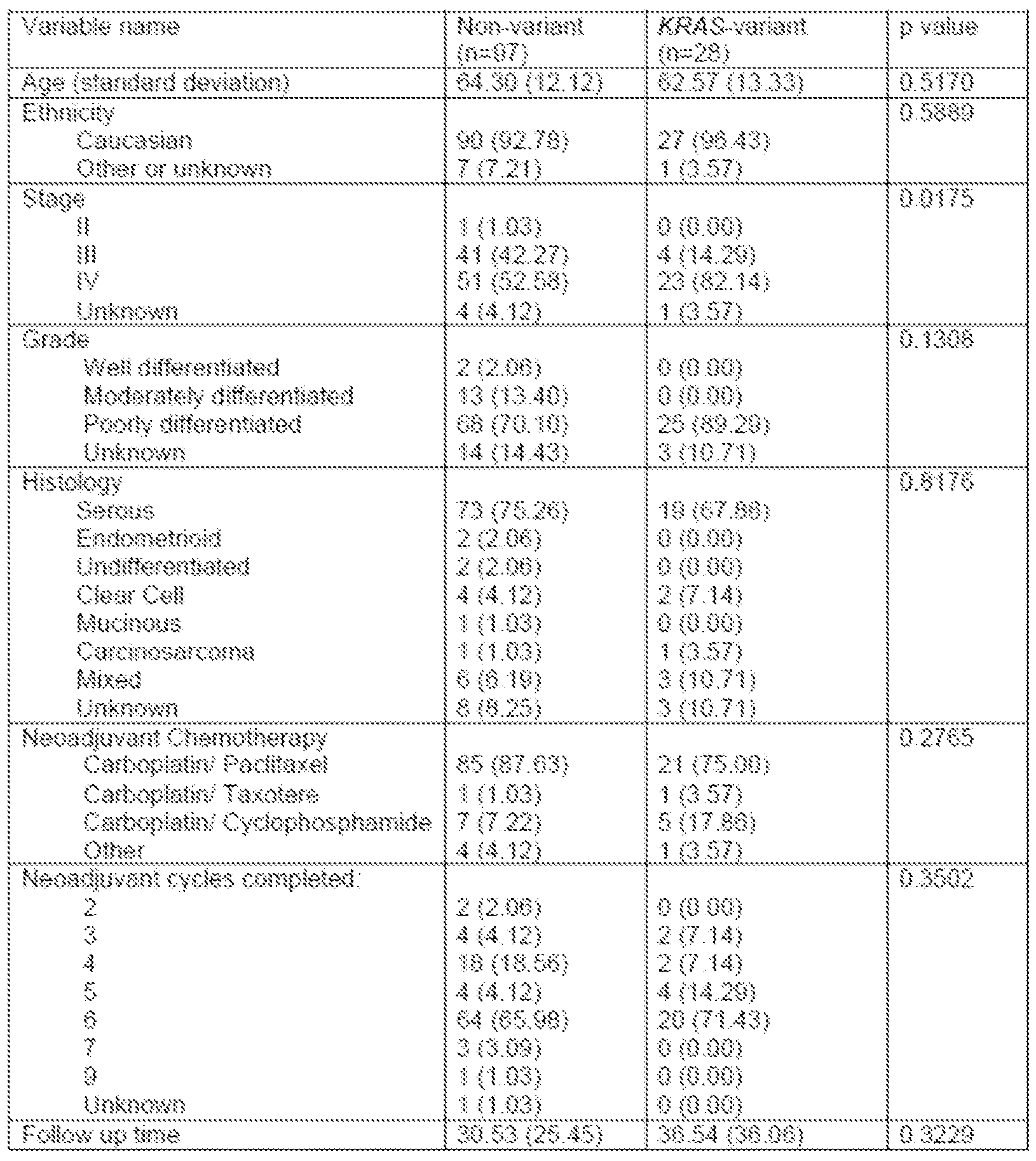
[187] Table 8. Clinicopathologic parameters for platinum resistance analysis.

[188] Detection of the KRAS variant. DNA was isolated using standard methods from tumor, blood or saliva. The KRAS variant does not appear to be somatically acquired nor does it require a loss of heterozygosity (Chin LJ, et al. (2008). Cancer Res 68: 8535-8540); hence, blood and saliva, for example, are appropriate to test and the results are identical regardless of the tissue tested. The KRAS variant allele was detected using a primer specific to the KRAS variant and a TaqMan (Applied Biosystems, Foster City, CA, USA) PCR assay on all samples. Genotyping was performed at the YNHH, except for on samples from COH, for
which the genotyping was performed in their facility. Less than 3% of populations carry 2 copies of the KRAS variant (Chin LJ, et al. (2008). Cancer Res 68: 8535-8540). As such, patients who carried at least one copy of the KRAS variant allele were classified as KRAS- variant carriers.
[189] Gene expression analysis of EOC with and without the KRAS variant. Gene expression in fresh-frozen tumor samples obtained from 16 patients (9 non-variant and 7 KRAS variant) was profiled on the Affymetrix GeneChip Human Genome U133 Plus 2.0 platform (Affymetrix, Santa Clara, CA, USA). All samples were from high-grade serous epithelial ovarian tumors that were stage IIIC or IV. Images were processed with the MAS5 algorithm and probes that were judged absent in at least 75% of the samples were removed. Intensity values were log transformed and quantile normalized. Differential gene expression was assessed in samples obtained from patients over 52 years of age (n = 6 non-variant and 4 KRAS variant) using a linear model and empirical Bayesian error moderation as implemented in the LIMMA package for R statistical software (R Foundation for Statistical Computing, publicly available at www.r-project.org) (Smyth G. (2005). Limma. in Gentleman R, et al. (eds) Bioinformatics and Computational Biology Solutions using R and Bioconductor.
Springer: New York, pp. 397-420).
[190] Association of published results with the KRAS variant in this data set was assessed using a signature approach to reduce cross-platform effects (Paranjape T, et al. (2011). Lancet
Oncol 12: 377-386). In brief, signature scores were computed as Pearson's correlation between the respective signature vector of gene contributions and each sample's expression profile for these genes. Differences between signature scores in
 and non-variant
and non-variant
EOC samples were assessed using the paired Kolmogorov-Smirnov test. Unless otherwise indicated, gene lists from the respective publications were used as signature vectors. Data from the study by Peters et al. (Mol Cancer Ther 4: 1605-1616) were obtained from the Gene
Expression Omnibus (GSE1926) and re-analyzed to generate a signature from the 50 most significantly differentially expressed genes between platinum sensitive and resistant samples.
[191] Chemosensitivity and cell viability assays. The activity of drugs alone or in combination was determined by a high-through-put CellTiter-Blue cell viability assay. For these assays, 1.2 x 10 cells were plated in each well of 384-well plates using a Precision XS liquid handling station (Bio-Tek Instruments Inc., Winooski, VT, USA) and allowed to attach overnight with incubation at 37°C, 5% C02 . Using the liquid handling station, all drugs were
serially diluted 2:3 or 1 :2 in media, and 5 μΐ of these dilutions were added to appropriate wells at indicated times. Four replicate wells were used for each drug concentration and an additional four control wells received a diluent control without drug. At the end of the incubation period with drugs, 5 μΐ CellTiter-Blue reagent (Promega Corp., Madison, WI, USA) was added to each well. Cell viability was assessed by the ability of the remaining viable cells to bioreduce resazurin to resorufin. The fluorescence of resorufin (579 nm Ex/584 nm Em) was measured using a Synergy 4 microplate reader (Bio-Tek Instruments Inc.). The fluorescence data were transferred to Microsoft Excel (Microsoft) to calculate the percentage viability relative to the four replicate cell wells that did not receive the drug. IC50s were determined using a sigmoidal equilibrium model regression using XLfit version 5.2 (ID Business Solutions Ltd). The IC50 was defined as the concentration of drug required for a 50% reduction in growth/viability. All experiments were carried out a minimum of three times.
[192] Targeting the KRAS variant. Small-interfering RNA sequences were designed to target the KRAS- variant sequence by placing the single-nucleotide polymorphism at varying positions of the 6 nucleotides at the 5 ' end of the siRNA guide strand corresponding to the so-called 'seed sequence'. Blast searches were performed to minimize cross-reactivity. In some of the siRNA sequences, DNA nucleotides were introduced to optimize
thermoenergetic features for preferred incorporation of the guide strand into the argonaute effector complex or to increase specificity for the variant.
[193] Small-interfering RNA guide strand sequences used in the experiments are as follows (lower case = RNA, upper case = DNA; GS = guide strand, PS = passenger strand):
2-1 GS ugcaucacuugaggucaggag (SEQ ID NO: 23)
2-1 PS ccugaccucaagugaugcacc(SEQ ID NO: 24)
2- 3 GS TGCATCACuugaggucaggag(SEQ ID NO: 25) (passenger strand same as 2-1)
3- 2 GS ucaucacuugaggucaggagu(SEQ ID NO: 26)
3-2 PS uccugaccucaagugaugcac(SEQ ID NO: 27)
[194] The negative control used was purchased from Qiagen (Valencia, CA, USA) (AllStars Negative-Control siRNA). Knockdown efficiency and specificity to the KRAS variant of these sequences were confirmed using a dual luciferase assay (see WO/2009/155100, the contents of which are incorporated herein by reference). Oligonucleotide combinations were annealed
using standard conditions and then transfected into cells using standard protocols. Cell survival was assayed using MTT assays and experiments were conducted in quadruplicate, and repeated in four independent experiments for all lines. Cell lysates were collected 72 hours after transfection and KRAS protein levels measured by western analysis using a probe specific to KRAS as described previously (Chin LJ, et al. (2008). Cancer Res 68: 8535-8540).
[195] Statistics. To assess the significance of demographic variables, a χ test or a two-sided Fisher's exact test was used for categorical variables. A t-test was used for continuous variables, such as age. The overall survival time
 and wild-type patients was compared using the Kaplan-Meier method (Kaplan E and Meier P. (1958). J Am Stat Assoc 53: 457-481), and the statistical significance of the survival curves was determined by the log-rank test (Mantel N. (1966). Cancer Chemother Rep 50: 163-170). A Cox proportional hazards regression model (Cox D. (1972). J R Stat Soc 34: 187-220) was used to assess the impact of the KRAS variant and demographic and prognostic variables (such as age, stage, grade and histology) on overall survival. Multivariate logistic regression analyses (Cox D. (1970). The Analysis of Binary Data. Methuen, London) were used to determine the impact of the KRAS variant and other demographic and prognostic factors on the probability of suboptimal cytoreduction. Multivariate logistic regression analyses (Cox D. (1970). The Analysis of Binary Data. Methuen, London) were used to assess the association of the KRAS variant and other prognostic factors on the probability of platinum resistance. All statistical analyses were performed using SAS 9.1.3 (SAS Institute Inc., Cary, NC, USA) and in R 2.12.1 (R Foundation for Statistical Computing)
and wild-type patients was compared using the Kaplan-Meier method (Kaplan E and Meier P. (1958). J Am Stat Assoc 53: 457-481), and the statistical significance of the survival curves was determined by the log-rank test (Mantel N. (1966). Cancer Chemother Rep 50: 163-170). A Cox proportional hazards regression model (Cox D. (1972). J R Stat Soc 34: 187-220) was used to assess the impact of the KRAS variant and demographic and prognostic variables (such as age, stage, grade and histology) on overall survival. Multivariate logistic regression analyses (Cox D. (1970). The Analysis of Binary Data. Methuen, London) were used to determine the impact of the KRAS variant and other demographic and prognostic factors on the probability of suboptimal cytoreduction. Multivariate logistic regression analyses (Cox D. (1970). The Analysis of Binary Data. Methuen, London) were used to assess the association of the KRAS variant and other prognostic factors on the probability of platinum resistance. All statistical analyses were performed using SAS 9.1.3 (SAS Institute Inc., Cary, NC, USA) and in R 2.12.1 (R Foundation for Statistical Computing)
Data and Results
[196] The association of the KRAS variant with overall survival in 454 EOC patients either tested and negative or untested for deleterious BRCA mutations was evaluated. When the entire cohort was considered, the KRAS variant did not predict worse survival by Kaplan- Meier analysis. Because the KRAS variant is most strongly associated with postmenopausal ovarian cancer (Chin LJ, et al. (2008). Cancer Res 68: 8535-8540), survival in women over 52 years of age (n = 279) were evaluated. Over and including 52 years of age is considered to be an appropriate surrogate for menopausal status. By Kaplan-Meier analysis, survival was significantly reduced in postmenopausal KRAS- variant EOC patients (n = 59) compared with non- variant EOC patients (n = 220, Figure 1, logrank P = 0.0399, non-KRAS- variant survival median 60 months,
 survival median 34 months). When other variables
including age, stage, grade, histology and treatment center were included with KRAS- variant status in a multivariate Cox proportional hazards regression model, the KRAS variant was a statistically significant predictor of reduced overall survival for postmenopausal women with EOC (Table 9); the hazard ratio for the KRAS variant was 1.67 (95% confidence interval: 1.09-2.57, = 0.019).
survival median 34 months). When other variables
including age, stage, grade, histology and treatment center were included with KRAS- variant status in a multivariate Cox proportional hazards regression model, the KRAS variant was a statistically significant predictor of reduced overall survival for postmenopausal women with EOC (Table 9); the hazard ratio for the KRAS variant was 1.67 (95% confidence interval: 1.09-2.57, = 0.019).
[197] Table 9. The KRAS variant is associated with reduced survival in postmenopausal (> 52 years of age) ovarian cancer patients (n = 279).

[198] The association of the KRAS variant with survival in a separate cohort of EOC patients carrying deleterious BRCA1 or BRCA2 mutations (n = 79) was evaluated. EOC patients carrying BRCA mutations were statistically significantly younger than EOC patients without BRCA mutations (52.7 vs 60.8 years of age, P < 0.0001). In addition, EOC patients with BRCA mutations had a significantly longer median survival by multivariate analysis controlling for age, stage, grade and histology than did EOC patients without BRCA mutations (120 vs 52 months, P = 0.0036). There was no significant difference in survival between EOC patients with BRCA mutations with or without the KRAS variant in a multivariate analysis using a multivariate Cox proportional hazards regression model (Table 10, KRAS-variant hazard ratio = 0.75, 95% confidence interval: 0.21-2.72, P = 0.66). In this study, there were too few patients to evaluate the impact of the KRAS variant on survival in postmenopausal EOC patients with deleterious BRCA mutations.
[199] Table 10. The KRAS-vaxiant and overall survival in EOC patients with deleterious
BRCA mutations (n = 79).

MR: a ds ratio obt n d from Cox proportional Hazards multivariate anal sis CI: confidence Interval
Studies included: Yale New Haven Hospital, Cit of Hope
[200] To explain the reduced survival in postmenopausal KRAS variant-positive EOC patients, the association of AT^S-variant positivity with response to platinum-based chemotherapy was evaluated. Platinum-based chemotherapy is the standard first-line chemotherapy in the treatment of EOC. First, all women with EOC who were treated at the Yale-New Haven Hospital (YNHH) with neoadjuvant platinum-containing chemotherapy followed by surgical cytoreduction (n = 116) were evaluated. Residual disease after surgery (cytoreduction) was used as a surrogate marker of patient response to chemotherapy. It was determined that 15.4% of KRAS- variant patients (n = 26) were suboptimally cytoreduced (41 cm of residual disease after surgery), compared with only 3.33% of non- variant patients (n = 90) (Figure 2, P = 0.044). The KRAS variant was also significantly associated with suboptimal cytoreduction after neoadjuvant chemotherapy and surgery in a multivariate logistic regression model controlling for age, stage, grade and histology (Table 11 , odds ratio = 9.36, 95% confidence interval: 1.34-65.22, P = 0.024).
[201] Table 11. The KRAS- variant predicts suboptimal debulking after neoadjuvant chemotherapy (n = 116).

2. Oi'. confidence !fs sf ¾i
3. fc?u¾f¥sriaie sd«* ed for ag¾, stage, grade, histo ogy, type of c emo erap
sgimsii, and numbers ©f cycles »eei sd rior to su gery..
[202] To determine whether the cause of poor response to neoadjuvant platinum-based chemotherapy seen in AT^S-variant EOC patients was due to resistance to platinum chemotherapy, platinum resistance in all EOC patients treated adjuvantly with platinum chemotherapy without documented BRCA mutations with available response data (n = 291) were evaluated. It was determined that platinum resistance (defined in this example as disease recurrence within 6 months of receiving platinum-based chemotherapy) was significantly more likely in KRAS variant-positive EOC patients than in non-KRAS variant EOC patients (16.67 vs 7.56%, P = 0.034). The KRAS variant was a statistically significant predictor for platinum resistance for EOC patients of all ages in a multivariate logistic regression analysis controlling for residual disease remaining after cytoreductive surgery, stage, histology, age and grade (Table 12, odds ratio = 3.18, 95% confidence interval: 1.31—7.72, P = 0.0106).
[203] Table 12. The KRAS variant is associated with platinum resistance.
¾JSSf,*SS?$ S8f »»»»»»»»»»»»»»»»»»»»»»»»»™.. in ^ im

[204] Gene expression studies were performed on a small cohort of ovarian cancer patients who had fresh-frozen tissue available (Brescia cohort), and compared between seven serous EOC samples with the KRAS variant and nine without the KRAS variant (n = 16). Within this
cohort, in postmenopausal EOC patients over 52 years of age with EOC (n = 10), a gene signature previously found to be associated with KRAS variant-associated TNBC (Paranjape T, et al. (2011). Lancet Oncol 12: 377-86) was also upregulated in KRAS variant-associated EOC (Figure 3 A). Similar to the previous analysis in TNBC, overexpression of KRAS- associated downstream pathways in EOC KRAS-variant tumors was discovered, which is consistent with 'KRAS addiction' (Singh A, et al. (2009). Cancer Cell 15: 489-500) (Figure 3B).
[205] Using previous analyses of gene expression data identifying platinum-resistant vs sensitive signatures (Peters D, et al. (2005). Mol Cancer Ther 4: 1605-1616), it was determined that KRAS- variant EOC samples had a lower carboplatin sensitivity signature compared with non-variant EOC samples (Figure 3C). In agreement with findings showing that the activation of the AKT pathway was frequently involved in platinum resistance, it was determined that AKT3 was one of the most significantly upregulated transcripts in KRAS- variant EOC tumors (Figure 3D).
[206] Although miRNA expression data were not available on tumor samples, the expression of let-7b miRNA in two cell lines with the KRAS variant (BG-1 and IGROVl) was compared with the expression of let- 7b in a non-KRAS variant line (CAOV3). The expression of let-7b miRNA is altered in KRAS variant-positive lung tumors (Chin LJ, et al. (2008). Cancer Res 68: 8535-8540) and triple-negative breast tumors (Paranjape T, et al. (2011). Lancet Oncol 12: 377-386).
[207] It was determined that let-7b was statistically significantly lower in cells with the KRAS variant (Figure 6).
[208] To confirm altered chemosensitivity in the presence of the KRAS variant, EOC cell lines with and without the KRAS variant were used to test their sensitivity to different chemotherapeutic agents. For example, a cell line that is KRAS variant positive/5i?C4 wild- type (BG1), a non-variant/5i? CA wild-type cell line (CAOV3) and a cell line KRAS- variant positive/ BRCA1 mutant (IGR-OV1) were tested. It was determined that the KRAS- variant line, BG1, was statistically significantly resistant to carboplatin (P < 0.04) and
carboplatin/paclitaxel combination chemotherapy (P < 0.0001) compared with CAOV3, the cell line without the KRAS variant. In contrast, IGROVl, the cell line with the KRAS variant and a deleterious BRCA1 mutation, was not resistant to these agents when compared with
CAOV3 (Figure 4). These results agree with corresponding clinical results demonstrating that
the KRAS variant is associated with platinum resistance, but not in the presence of deleterious BRCA mutations.
[209] Additionally, agents frequently used as second line therapy for patients who have failed carboplatin/paclitaxel chemotherapy were evaluated. These second line therapeutic agents included doxorubicin, topotecan and gemcitabine. The KRAS-variant line, BG1, was significantly resistant to each of these agents compared with CAOV3, the nonvariant cell line (Table 13).
[210] Table 13. Chemosensitivity in a KRAS-variant cell line (BG1) vs a non-variant line (CAOV3).

[211] Because the data presented herein demonstrate a continued use of KRAS signaling in KRAS variant-associated tumors, the impact of directly targeting the AT^S-variant was evaluated. Small-interfering RNA (siRNA)/miRNA-like complexes were designed to directly bind the altered allele in KRAS variant transcripts, but not bind to non-KRAS- variant transcripts (Figure 7). It was determined that transfecting these oligonucleotide duplexes that target the KRAS variant caused a statistically significant decrease in cell survival in the KRAS variant carrying BG1 cell line (P < 0.001), but had no effect in CAOV3 (Figure 5 A) or SKOV3, two non- variant EOC cell lines. This result is concordant with a moderate decrease in KRAS protein levels by western blot in BG1, but not in CAOV3 (Figure 5B) or
SKOV3 after treatment.
Example 3 : The KRAS variant and Response to Monoclonal Anti-EGFR Antibody
Treatment.
Materials and methods
[212] Patient characteristics. A total of 559 mCRC patients, 300 treated in the University Hospital of Leuven with anti-EGFR mAb monotherapy and MAb in combination with chemotherapy, as well as 148 patients from the Universite Paris Descartes treated with cetuximab-based salvage combination chemotherapy ( De RW, et al. Lancet Oncol 2010;
11(8):753-762), and 111 previously published (Zhang W, al. Ann Oncol 2011; 22(1): 104- 109) mCRC patients treated with cetuximab monotherapy after failing fluoropyrimidine, irinotecan and oxaliplatin containing regimens ( Zhang W, al. Ann Oncol 2011; 22(1): 104- 109; Lenz HJ, et al. J Clin Oncol 2006; 24(30):4914-4921) had tissue available and amenable for analysis of the KRAS variant polymorphism. The mutational status of the KRAS and BRAF genes in the above mentioned patient populations is publicly available (De RW, et al. Lancet Oncol 2010; l l(8):753-762; Zhang W, al. Ann Oncol 2011; 22(1): 104-109). The above mentioned molecular characteristics were correlated with ORR, PFS and OS. From the 559 mCRC patients entered in the study the KRAS 3'-UTR LCS6 variant was determined in 512 due to exhaustion of available DNA from other molecular testing.
[213] Genetic analyses. Formalin-fixed, paraffin-embedded normal tissue from the patients' specimens was macroscopically dissected using a scalpel blade and DNA was isolated as previously described (De RW, et al. Lancet Oncol 2010; 11(8):753-762; Zhang W, al. Ann
Oncol 2011; 22(1): 104-109). DNA was amplified using, as previously described (Hollestelle
A, et al. Breast Cancer Res Treat 2010), a custom-made Taqman genotyping assay (Applied
Biosystems, Foster City, CA) designed specifically to identify the T or variant G allele of the ^S-variant (rs61764370) with the forward primer: 5 '-GCCAGGCTGGTCTCGAA-3 '
(SEQ ID NO: 28), reverse primer: 5 ' -CTGAAT AAATGAGTTCTGC AAAAC AGGT T-
3'(SEQ ID NO: 29), VIC reporter probe: 5 '-CTCAAGTGATTCACCCA C-3' (SEQ ID NO:
30), and FAM reporter probe: 5 '-CAAGTGATTCACCCAC- 3' (SEQ ID NO: 31). The
KRAS and BRAF mutational status was determined as previously described (De RW, et al.
Lancet Oncol 2010; l l(8):753-762; Zhang W, al. Ann Oncol 2011; 22(1): 104-109).
[214] Cell line studies. A cell line with the KRAS variant (G-allele) (HCC2998) and a cell line without the allele and without a KRAS tumor acquired mutation (HT-29) were studied to evaluate the impact of treatment with chemotherapy alone or in combination with Cetuximab.
Cell lines were treated with Cetuximab (ΙΟΟηΜ) or none and dilutions of Irinotecan (lmg/ml- lOOmg/ml). Cells were plated, treated with agents 24 hours after plating, media was changed after a 24 hour exposure, and then survival was scored 48 hours later using the MTT assay.
[215] Statistical analyses. The distribution of genotypes was tested for Hardy-Weinberg
Equilibrium and the χ test was p = 0.8. Because of the low frequency of homozygotes for the
KRAS variant allele, patient samples that were either heterozygous (TG) or homozygous
(GG) for the KRAS variant allele were considered positive for the LCS6 (KRAS-variant or G
allele) and entered the analyses as one group of at least one KRAS variant (G allele) genotypes. PFS and OS were measured as previously described (De RW, et al. Lancet Oncol 2010; l l(8):753-762; Zhang W, al. Ann Oncol 2011; 22(1): 104-109)
[216] The two-tailed Fisher's exact test was used to compare proportions between carriers of the wild-type (wt) TT genotype and carriers of at least one G allele genotypes (TG and GG). PFS and OS were estimated with the use of the Kaplan-Meier method and their association with genotypes was tested with the use of the log-rank test. The association of genotypes with objective response was determined by contingency table and the Fisher's exact test. To fully explore the possible influence of the KRAS variant, analyses were performed in the whole mCRC population, in the patients harboring no mutations in the KRAS and BRAF genes (double wt population) and in the KRAS variant population. The level of significance was set at a two-sided p value of <0.05. All statistical tests were performed using the statistical package SPSS version 13.
Results
[217] KRAS LCS6 in the entire patient cohort. In these 512 mCRC patients there were 403 carriers of the wt LCS6 TT genotype (72%), 102 (18%>) carriers of the heterozygous KRAS variant TG allele and 7 (1.3%) of the homozygous KRAS variant GG allele, thus 109 (19.5%) carriers of at least one G allele genotype. KRAS mutations in codons 12, 13 and 61 were found in 184 patients (33%) and the BRAF V600E was found in 29 patients (5.3%). All patients had received anti-EGFR mAbs-based salvage treatment, 169 as monotherapy and 377 in combination with chemotherapy. No statistically significant differences were found between KRAS wt and KRAS variant carriers for sex and age at diagnosis. The characteristics of the 559 patients have been previously published (De RW, et al. Lancet Oncol 2010;
l l(8):753-762; Zhang W, al. Ann Oncol 2011; 22(1): 104-109).
[218] As shown in Table 14 the distribution of the KRAS genotypes was different among patients harboring KRAS and BRAF mutations. In particular, whereas the percentage of at least one G variant allele genotype was equally distributed among the KRAS wt and mutant groups (20% in each), the KRAS variant (G allele) was twice as frequent in the BRAF V600E mutated group (40%>) compared to the wt one (20%>), resulting in a statistically significant difference (Fisher's exact test p = 0.030).
[219] Table 14. Distribution of the KRAS 3 '-UTR LCS6 genotypes according to KRAS and BRAF mutational status in the mCRC patients' cohort.

Abbreviations: 3'-UTR LCS6, 3' untranslated region of the Let-7 complementary site, WT, wild type
[220] Outcome and Survival analysis in the entire patient cohort. In the cohort as a whole, for patients with PFS and OS information and LCS6 genotyping («=510 and 503, respectively) no significant differences were detected regarding median PFS and OS between the LCS6 wt TT genotype carriers and the LCS6 G variant (KRAS variant) genotype carriers (Figure 8A and 8B). Similarly, no differences in PFS and OS were observed in the double (KRAS and BRAF) wt or in the KRAS variant patient cohort. Furthermore, no significant correlations regarding response (n=483) and skin rash (n=359) were observed between the KRAS variant and wt carriers in the whole and in the double wt patients' cohorts (Table 15).
[221] Table 15. Outcome and survival analysis according to KRAS genotypes and other clinical variables for the entire population.

Abbreviations: 3 '-UTR LCS6, 3 ' untranslated region of the Let-7 complementary site; WT, wild type; PFS, progression-free survival; OS, overall survival; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease.
[222] Progression free survival analysis correlated with treatment. Patients who received mAbs monotherapy and mAbs combination therapy were analyzed separately. From the 501 patients evaluable for LCS6 SNP genotyping and treatment administration, 160 (32%) received anti-EGFR mAbs as monotherapy. Of the monotherapy patients, 128 (80%) were carriers of the LCS6 wt TT genotype and 32 (20%) were carriers of the LCS6 G variant
genotype. There were 341 (68%) patients who received multiple chemotherapy combinations. Of the combination treatment patients, 266 (78%) were carriers of the LCS6 wt TT genotype and 75 (22%) were carriers of the LCS6 at least one G variant genotype.
[223] The median PFS of the whole monotherapy patients' population was 10.43 weeks (95%o CI: 7.73-13.12 weeks) and a statistically significant difference (p = 0.019, log-rank test) was observed between the LCS6 wt TT genotype carriers, 7.85 weeks (95%> CI: 3.897-11.817 weeks), and the LCS6 G variant (KRAS variant) genotype carriers, 16.86 weeks (95%> CI: 10.2-23.51 weeks) (Figure 9A). The median PFS of the whole combination therapy patients' population was 18 weeks (95%> CI: 15.87-20.12 weeks) and no statistically significant difference (p = 0.760, log-rank test) was observed between the LCS6 wt TT genotype carriers, 18.43 weeks (95%> CI: 16.16-20.69 weeks), and the LCS6 G variant genotype carriers, 18 weeks (95%> CI: 9.97-26.02 weeks) (Figure 9B). There was also no significant difference (p = 0.291, log-rank test) between PFS for KRAS variant patients that received mAbs therapy [16.86 weeks, (95%> CI: 8.55-25.18 weeks)] versus combination therapy [18 weeks, (95%> CI: 13.37-22.64 weeks)] (Figure 9C), while there was a significant benefit with the addition of chemotherapy for non-KRAS variant patients [p < 0.0001, log-rank test, PFS for mAbs monotherapy 7.86 weeks, (95%> CI: 3.9-11.82 weeks) versus combination therapy 19.29 weeks, (95%> CI: 17-21.58 weeks) (Figure 9D). Of note, there was no significant difference in PFS between KRAS variant patients treated with monotherapy therapy versus non-KRAS variant patients treated with combination therapy.
[224] In the double (KRAS and BRAF) wt patients' population the median PFS of the monotherapy patients was 12 weeks (95%> CI: 8.38-15.61 weeks) and a statistically significant difference (p = 0.039, log-rank test) was again observed between the LCS6 wt TT genotype carriers, 10.43 weeks (95%> CI: 6.74-14.11 weeks), and the LCS6 G variant genotype carriers, 18 weeks (95%> CI: 5.16-30.83 weeks) (Figure 10A). In the double wt patients' population the median PFS of the combination therapy patients was 28.71 weeks
(95%) CI: 24.98-32.43 weeks) and no statistically significant difference (p = 0.39, log-rank test) was observed between the LCS6 wt TT genotype carriers, 28.3 weeks (95%> CI: 24.15-
32.45 weeks), and the LCS6 G variant genotype carriers, 28.85 weeks (95% CI: 14.82-42.87 weeks) (Figure 10B). There was no significant improvement (p = 0.096, log-rank test) between PFS for LCS6 variant patients that received mAbs monotherapy [23 weeks, (95%
CI: 9.5-36.5 weeks)] versus combination therapy [28 weeks, (95%> CI: 14.83-42.87 weeks)]
(Figure IOC), while there was for non-LCS6 patients {p < 0.0001, log-rank test, PFS for mAbs monotherapy 10.43 weeks, (95% CI: 6.75-14.15 weeks) versus combination therapy 28.71 weeks, (95% CI: 24.8-32.6 weeks) (Figure 10D). There was no difference in PFS between KRAS variant (G allele) patients receiving mAbs monotherapy and non-KRAS variant patients receiving combination therapy.
[225] Overall survival analysis correlated with treatment. The median OS of the whole monotherapy patients' population was 33.14 weeks (95%> CI: 26.70-39.57 weeks) and no statistically significant difference (p = 0.139, log-rank test) was observed between the LCS6 wt TT genotype carriers, 28.85 weeks (95%> CI: 22.53-35.18 weeks), and the LCS6 G variant genotype carriers, 45 weeks (95%> CI: 35.02-54.97 weeks) (Figure 11A). The median OS of the whole combination therapy patients' population was 44 weeks (95% CI: 40.11-47.88 weeks) and no statistically significant difference (p = 0.759, log-rank test) was observed between the LCS6 wt TT genotype carriers, 44 weeks (95%> CI: 40.06-47.93 weeks), and the LCS6 at least one G variant genotype carriers, 43 weeks (95%> CI: 29.8-56.2 weeks) (Figure 1 IB). Again, there was no significant improvement (p = 0.574, log-rank test) between OS for KRAS variant patients that received mAbs monotherapy [45 weeks, (95%> CI: 35-55 weeks)] versus combination therapy [43 weeks, (95%> CI: 29.8-56.2 weeks)] (Figure 11C), while there was a benefit of chemotherapy addition for non-KRAS variant patients [p < 0.0001, log-rank test, OS for mAbs monotherapy 28.86 weeks, (95%> CI: 22.53-35.18 weeks) versus combination therapy 44 weeks, (95%> CI: 40-47.93 weeks) (Figure 1 ID). Again, there was no significant difference in OS between LCS6 G variant carriers treated with monotherapy, and non-KRAS variant carriers treated with combination therapy.
[226] In the double (KRAS and BRAF) wt patients' population the median OS of the monotherapy patients was 37 weeks (95 %> CI: 30.82-43.17 weeks) and a trend towards a statistically significant difference (p = 0.087, log-rank test) was observed between the LCS6 wt TT genotype carriers, 35.71 weeks (95%> CI: 32.03-39.4 weeks), and the LCS6 at least one G variant genotype carriers, 55.43 weeks (95%> CI: 36.98-73.87 weeks) (Figure 12A). In the double wt patients' population, the median OS of the combination therapy patients was 55 weeks (95%> CI: 48.3-61.7 weeks) and no statistically significant difference (p = 0.649, log- rank test) was observed between the LCS6 wt TT genotype carriers, 57 weeks (95%> CI: 49.4- 64.6 weeks), and the LCS6 at least one G variant genotype carriers, 54 weeks (95%> CI:
45.46-62.53 weeks) (Figure 12B). There was no significant improvement (p = 0.705, log-rank
test) between OS for KRAS variant (G allele) patients that received mAbs monotherapy
[55.43 weeks, (95% CI: 37-73.87 weeks)] versus combination therapy [54 weeks, (95%> CI: 45.47-62.54 weeks)] (Figure 12C), while there was for non- KRAS variant patients [p < 0.0001, log-rank test, OS for mAbs monotherapy 35.71 weeks, (95% CI: 32-39.4 weeks) versus combination therapy 57 weeks, (95%> CI: 49.4-64.6 weeks) (Figure 12D). There was no significant difference between double wild-type patients KRAS variant carriers treated with monotherapy versus non-LCS6 carriers treated with combination therapy.
[227] The LCS6 variant is prognostic in KRAS and BRAF mutated patients. In the KRAS and BRAF mutated patients' population no statistical significant differences regarding PFS and OS were observed in patients treated with both anti-EGFR mAbs monotherapy and in combination with chemotherapy. Median PFS times were identical between KRAS variant and non- KRAS variant patients, with no significant improvement (p = 0.641, log-rank test) between PFS for KRAS variant patients that received mAbs monotherapy [6 weeks, (95% CI: 0-13.25 weeks)] versus combination therapy [12 weeks, (95%> CI: 6.45-17.56 weeks)] (Figure 13 A). There was a significant improvement in PFS for non- KRAS variant patients [p < 0.0001, log-rank test, PFS for mAbs monotherapy 6 weeks, (95%> CI: 4.46-7.53 weeks) versus combination therapy 12 weeks, (95%> CI: 9.72-14.28 weeks) (Figure 13B). For OS, there was no significant difference (p = 0.303, log-rank test) between OS for KRAS variant (G allele) patients that received mAbs monotherapy [28.43 weeks, (95%> CI: 9.47-47.39 weeks)] versus combination therapy [23 weeks, (95%> CI: 10.8-35.19 weeks)] (Figure 13C), while there was for non- KRAS variant patients [p = 0.002, log-rank test, OS for mAbs monotherapy 21.29 weeks, (95%> CI: 15-27.55 weeks) versus combination therapy 31 weeks, (95%> CI: 25.65-36.34 weeks) (Figure 13D).
[228] The KRAS variant and response. From the whole population of 483 patients that were evaluable for both response and KRAS variant genotyping, 147 (30.4%) had received anti- EGFR mAbs as monotherapy and 336 (69.6%) with multiple chemotherapy combinations. In the monotherapy group 123 (83.6%>) patients were non-responders (SD and PD), 104 LCS6 wt and 19 LCS6 variant (KRAS variant) carriers, and 24 (16.4%) were responders (PR and CR), 13 LCS6 wt and 11 LCS6 variant (KRAS variant) carriers. A statistically significant difference was observed between the wt and KRAS variant genotype carriers distribution in the responders and non-responders groups (Fisher's exact test /?=0.002). In the combination with chemotherapy group 252 (75%>) patients were non-responders (SD and PD) and 84
(25%) were responders (PR and CR). No statistically significant difference was observed between the wt and KRAS variant genotype carriers, 197 vs. 55 non-responders and 66 vs. 18 responders, respectively (Fisher's exact test p= ).
[229] In the 270 double (KRAS and BRAF) wt population 90 (33.3%) had received anti- EGFR mAbs as monotherapy and 180 (66.6%) with multiple chemotherapy combinations. In the monotherapy group 71 (78.8%) patients were non-responders (SD and PD), 60 LCS6 wt and 11 LCS6 variant (KRAS variant) carriers and 19 (21.2%) were responders (PR and CR), 10 LCS6 wt and 9 LCS6 variant (KRAS variant) carriers. A statistically significant difference was observed between the wt and KRAS variant genotype carriers distribution in the responders and non-responders groups (Fisher's exact test /?=0.010). In the combination with chemotherapy group 102 (56.6%) patients were non-responders (SD and PD) and 78 (43.4%) were responders (PR and CR). No statistically significant difference was observed between the wt and KRAS variant genotype carriers, 81 vs. 21 non-responders and 62 vs. 16 responders, respectively (Fisher's exact test p= ).
[230] Cell line studies of the effect of mAbs monotherapy and combination therapy and the LCS6 variant. To confirm that the presence of the KRAS variant (G allele) predicts response to mAbs monotherapy, without any benefit of additional cytotoxic therapy, the impact of monotherapy versus combination therapy in colon cancer cell lines with and without the LCS6 G variant was evaluated. It was discovered that in non-KRAS variant cell lines, the addition of Cetuximab to cytotoxic therapy, both radiation as well as irinotecan
chemotherapy, increased cell death as compared to cytotoxic therapy alone. In contrast, in a cell line with the KRAS variant (G allele), there was no additional cell kill with the addition of Cetuximab to cytotoxic therapy, and in the case of radiation in fact higher cell survival when Cetuximab was added. These findings are consistent with our in vivo findings, that there is no benefit of the combination of Cetuximab with cytotoxic therapy in KRAS variant (G allele) carriers.
Example 4: The KRAS Variant and Sensitivity to Sorafenib
[231] A group at MDACC proposed to implement a translational lung cancer research program entitled, "BATTLE: Biomarker-integrated Approaches of Targeted Therapy of
Lung Cancer Elimination". The BATTLE program was developed to establish
individualized targeted therapy for NSCLC patients for whom standard therapy had failed, and this trial prospectively examined patient tumor biomarker profiles, which were obtained
from a mandated fresh core tumor biopsy, and assigned them to corresponding targeted therapies (erlotinib, vandetanib, bexarotene+erlotinib, and sorafenib) in the hopes of yielding better clinical outcomes. The primary end-point was 8-week disease control (DC) rate, with a secondary endpoint of overall survival (OS).
[232] BATTLE- 1 clinical study samples were tested for the KRAS- variant, and 161 patients had available DNA. Overall, for this exploratory analysis, there were 16 (heterozygous=15, homozygous=l) patients with the KRAS- variant identified. For the primary objective of the study of 8-week DC, 48.5% non-variant and 28.6%
 patients were observed to reach this primary endpoint. KRAS- variant patients rarely developed tumors that went on to develop additional EGFR or KRAS mutations (4% and 9%, respectively).
patients were observed to reach this primary endpoint. KRAS- variant patients rarely developed tumors that went on to develop additional EGFR or KRAS mutations (4% and 9%, respectively).
[233] For the erlotinib arm, 42% non-variant patients (13/31) achieved 8-week DC while 0% (0/4) of the AT^S-variant patients reached this early response endpoint. A similar poor response by KRAS- variant patients was observed in the vandetanib arm, with 31% of the non- variant patients (9/32) reaching the 8-week DC endpoint versus 0% KRAS- variant patients (0/3) reaching this endpoint. There were no AT^S-variant patients in the arm combining erlotinib and bexarotene.
[234] In contrast to the poor response of KRAS-variant patients to erlotinib and/or bexarotene, in the Sorafenib arm the KRAS-variant patients reached the 8-week DC endpoint as well as the non-variant patients (56.9% non-variant [33/58] versus 57.1% KRAS-variant patients [4/7]).
[235] In the Overall Survival (OS) analysis, the vandetanib treatment group demonstrated statistically significant worse survival for the KRAS- variant patients receiving this treatment (p=0.029).
[236] Conversely, in the Sorafenib arm, the KRAS-variant patients achieved significantly longer OS than non-variant patients (p=0.056, Figure 15). The data support a conclusion that the KRAS- variant meaningfully subgroups patients as responders or non-responders regardless of tumor acquired mutations in NSCLC, similar as to colon cancer. In patients with tumor acquired KRAS mutations, the KRAS- variant significantly separated the group, with double mutant patients (tumor acquired KRAS and KRAS- variant) having significantly longer PFS than patients with only tumor acquired KRAS mutations (Figure 16).
Example 5 : The KRAS Variant and Resistance to Cisplatin
[237] Methods of the disclosure predict the resistance of any cancer cell, tumor cell, or subject having any cancer, to treatment with cisplatin following detection of the KRAS
Variant within the cancer cell. The power of the KRAS Variant to predict resistance to cisplatin treatment is demonstrated below for an exemplary cancer. The data provided herein demonstrate that, in patients with recurrent and/or metastatic head neck squamous cell carcinoma, the KRAS Variant predicts resistance to cisplatin treatment.
[238] Head and neck squamous cell carcinoma (HNSCC) is the sixth most frequent cancer worldwide. Risk factors for HNSCC include tobacco and alcohol consumption, as well as human papillomavirus (HPV) infection. Recent studies have concluded that HPV-positive and -negative HNSCC represent distinct diseases. Despite recent advances in multimodality treatment, -20% of HPV-positive and -50% of HPV-negative HNSCC patients experience treatment failure and subsequent disease-related death. The current standard of care for first- line recurrent and/or metastatic (R/M) disease is a platinum-based combination regimen (e.g. cisplatin), 5-fluorouracil (5-FU) and cetuximab. However, treatment efficacy is still limited in these patients, with overall survival ranging from 7-10 months.
[239] Aside from general trends in survival, the KRAS- variant is specifically associated with therapeutic resistance. For example,
 ovarian cancer patients demonstrate enhanced platinum resistance and poor outcome (see Example 2). Additionally,
ovarian cancer patients demonstrate enhanced platinum resistance and poor outcome (see Example 2). Additionally,

colon cancer patients are more resistant to cetuximab when given in combination with irinotecan chemotherapy, but more sensitive to cetuximab is given as a monotherapy (see Example 3). Similarly, the KRAS Variant predicts resistance to a platinum-based
chemotherapy (cisplatin) and an enhanced response (i.e. increased sensitivity) to cetuximab in R/M HNSCC patients.
[240] A retrospective study was conducted of 186 HNSCCs from three previously reported clinical trials and an exempted tissue collection study. In this study, the association of the
 with clinical outcome in R/M HNSCC patients treated with cisplatin + cetuximab or docetaxel + bortezomib as well as cisplatin sensitivity and altered gene expression in HNSCC cell lines was evaluated. The data from this study showed that patients with the AT^S-variant had worse outcomes when given cisplatin, yet improved responses to cetuximab.
[241] It is hypothesized that the oncogenic and therapeutic significance of KRAS- variant is dependent on upstream EGFR signaling. In this study, data from experiments with cell lines supported clinical findings of cisplatin resistance and dependence on EGFR signaling.
with clinical outcome in R/M HNSCC patients treated with cisplatin + cetuximab or docetaxel + bortezomib as well as cisplatin sensitivity and altered gene expression in HNSCC cell lines was evaluated. The data from this study showed that patients with the AT^S-variant had worse outcomes when given cisplatin, yet improved responses to cetuximab.
[241] It is hypothesized that the oncogenic and therapeutic significance of KRAS- variant is dependent on upstream EGFR signaling. In this study, data from experiments with cell lines supported clinical findings of cisplatin resistance and dependence on EGFR signaling.
[242]
 genotyping was performed for these tumors and 8 FINSCC cell lines using a PCR-based assay. pl6 expression was determined in 129 of these tumors by immunohistochemistry. Microarray analysis was also utilized to elucidate differentially expressed genes between AT^S-variant and non-variant tumors.
genotyping was performed for these tumors and 8 FINSCC cell lines using a PCR-based assay. pl6 expression was determined in 129 of these tumors by immunohistochemistry. Microarray analysis was also utilized to elucidate differentially expressed genes between AT^S-variant and non-variant tumors.
Methods
[243] Patient characteristics: Tumor samples were collected from three previously published clinical trials and one exempted tissue collection study; 1) 24 samples from FTN0582
(NCT00503997): a phase II trial for efficacy and toxicity of induction pemetrexed
(ALIMTA) and oxaliplatin (ELOXATIN) in patients with locally advanced HNSCC ( Gilbert J, et al. Cancer 2012), 2) 22 samples from HN0501 (NCT00425750): phase II trial of combination weekly bortezomib (VELCADE®) and docetaxel (TAXOTERE) in patients with R/M HNSCC ( Chung CH, et al. Ann Oncol 2009), 3) 57 samples from E5397
(NCT00003809): a randomized, double-blind, placebo controlled phase III evaluation of cisplatin+placebo versus cisplatin+C225 in patients with R/M FINSCC ( Burtness B, et al. J Clin Oncol 2005;23 : 8646-54), and 4) 83 samples from an exempted retrospective tissue collection study.
[244]
 determination: Genomic DNA from the cell lines and formalin-fixed paraffin-embedded tumors was isolated as previously described and 100 ng of DNA from blinded samples was run on a CLIA-certified AT^S-variant assay (Weidhaas JB, et al. Cancer Res 2007;67: 1 1 1 1 1-6).
determination: Genomic DNA from the cell lines and formalin-fixed paraffin-embedded tumors was isolated as previously described and 100 ng of DNA from blinded samples was run on a CLIA-certified AT^S-variant assay (Weidhaas JB, et al. Cancer Res 2007;67: 1 1 1 1 1-6).
[245] pl6 status determination: In this study, pl6 status was used as a control to ensure the validity of experimental results. pl6 status was determined by immunohistochemistry using a pl6 mouse monoclonal antibody (prediluted, MTM-CINtech, 9518). pl6 positivity was determined by diffuse staining with greater than 70% of the tumor staining positive as previously described (Ang K , et al. N Engl J Med 2010;363 : 24-35).
[246] Head and neck squamous cell carcinoma cell lines: Eight HNSCC cell lines (Cal27,
UNC7, FaDu, SKN3, SCC6, SCC61 , SCC90 and SCC47) were obtained and maintained in cell culture as previously published ( Hatakeyama H, et al. PLoS One 2010;5 : el 2702).
Cisplatin was purchased from Cell Signaling Technology (Danvers, MA).
[247] MTS assay: To assess in vitro proliferation in the presence of cisplatin, MTS assay was used to estimate relative cell growth. Cells were seeded at a density of 1 ,000 cells/well in 96-well plates. Drug-containing media was added the following day to these cultures. Cells were then allowed to proliferate 72-96 hours before adding PMS (phenazine methosulfate) and MTS according to the manufacturer's protocol (Promega, Madison, WI). After 2-4 hours of incubation at 37°C, 100 μΐ of reactant was removed from the wells and absorbance was analyzed at 450 nm using the GloMax Multi Detection System plate reader (Promega, Madison, WI). Dose-response curves were analyzed as the percent change in absorbance at each concentration with respect to control growth (0.1% DMSO).
[248] R A isolation and DNA microarray analysis: Each tumor was examined by H&E staining to ensure the presence of tumor and subsequently enriched by macrodissection. Total RNA was purified from frozen tumors (-10-20 milligrams of wet tissue per sample) using Qiagen RNeasy Mini Kit according to the manufacturer's recommendations (Qiagen, Valencia, CA). Gene expression data was generated using Affymetrix Human Genome U133 plus 2.0 GeneChip (Slebos RJ, et al. Clin Cancer Res 2006; 12: 701-9) and is available from GEO (GSE361 10).
[249] Statistical analysis of KRAS- variant status and clinical correlations: KRAS- variant incidence was reported by frequency and percentage of TG/GG samples with respect to total samples with known
 status. Descriptive statistics were used to characterize patient demographics and disease characteristics. Associations between KRAS- variant status and patient characteristics/response were evaluated using Fisher's exact tests. Progression- free survival (PFS) was defined as the time between date of study entry and date of progression or death from any cause, censored at date of last disease assessment. Overall survival (OS) was defined as the time between date of study entry and date of death, censored at last contact. Event-time distributions were estimated by the Kaplan-Meier method and compared using log-rank tests. Association of KRAS- variant genotype with OS and PFS was evaluated using univariate Cox proportional hazards regression modeling. If any association achieved statistical significance, multivariable Cox regression models were further fitted by controlling important patient/clinical variables [PS (0 vs. 1), disease status (previously untreated vs. recurrent), cell differentiation (well/moderately vs. poorly differentiation), primary site (oropharynx vs. non- oropharynx), smoking history (<40 packs-years vs. >40 packs-years), alcohol consumption (< 10 oz whiskey/week vs. > 10 oz whiskey/week), and
treatment (when appropriate)]. All p-values are two-sided and a level of p <0.05 was considered statistically significant.
status. Descriptive statistics were used to characterize patient demographics and disease characteristics. Associations between KRAS- variant status and patient characteristics/response were evaluated using Fisher's exact tests. Progression- free survival (PFS) was defined as the time between date of study entry and date of progression or death from any cause, censored at date of last disease assessment. Overall survival (OS) was defined as the time between date of study entry and date of death, censored at last contact. Event-time distributions were estimated by the Kaplan-Meier method and compared using log-rank tests. Association of KRAS- variant genotype with OS and PFS was evaluated using univariate Cox proportional hazards regression modeling. If any association achieved statistical significance, multivariable Cox regression models were further fitted by controlling important patient/clinical variables [PS (0 vs. 1), disease status (previously untreated vs. recurrent), cell differentiation (well/moderately vs. poorly differentiation), primary site (oropharynx vs. non- oropharynx), smoking history (<40 packs-years vs. >40 packs-years), alcohol consumption (< 10 oz whiskey/week vs. > 10 oz whiskey/week), and
treatment (when appropriate)]. All p-values are two-sided and a level of p <0.05 was considered statistically significant.
[250] Statistical analyses of microarray data: Microarrays were normalized using frozen robust multiarray analysis (fRMA) (McCall MN, et al. Biostatistics 2010;11 : 242-53).
Empirical Bayes moderated t-statistics were computed to compare gene expression from KRAS variant (TG/GG) samples to wild type (TT) samples using LIMMA (Smyth GK. Statistical applications in genetics and molecular biology 2004; 3: Article 3). Benjamini- Hotchberg multiple testing correction was applied to the corresponding p-values (Benjamini Y and Hochberg Y. J Roy Statist Soc Ser B 1995;57: 289-300).
[251] KRAS-variant status was determined in 173/186 (93%) of the HNSCC tumor samples and the allelic frequency of TG/GG was 24.3%. Three of the HNSCC cell lines (3/8) also had the KRAS- variant. No association between KRAS- variant status and pl6 expression was observed in this study cohort (Fisher's exact test, p=0.37). With respect to patient outcome, KRAS- variant tumors were associated with poor progression-free survival when patients were treated with cisplatin (log-rank p=0.002). Conversely, KRAS-vanant patients experienced improved disease control when cetuximab was added to their platinum-based regimen (log- rank p=0.04).
Results
[252] Tumor Samples and KRAS Genotypes: Detailed patient characteristics of the clinical trial study populations are published (Burtness B, et al. J Clin Oncol 2005;23: 8646-54;
Gilbert J, et al. Cancer 2012; and Chung CH, et al. Ann Oncol 2009). Of the combined 186 samples evaluated, KRAS variant status was determined in 173/186 (93%>). More specifically, 19/22 samples (86%) from HN0501 and 54/57 (95%) from E5397 yielded interpretable KRAS status information (Table 16). The overall allelic frequency of KRAS-variant (TG/GG) tumors was 24.3%) (42/173). pl6 status was available in 129/186 samples (34 positive and 95 negative). However, consistent with the role of pl6 as a negative control, there was no association between pl6 expression and KRAS variant status (Fisher exact test, p=0.37).
[253] Table 16: Patient characteristics of two clinical trials conducted in
recurrent/metastatic head and neck squamous cell carcinoma; HN0501 (phase II docetaxel and bortezomib) and E5397 (phase III cisplatin with/without cetuximab)

[254] KRAS- variant status is associated with cisplatin resistance in R/M HNSCC patients: To determine whether
 status is associated with cisplatin resistance in R M HNSCC patients, survival analyses were performed for patients from HN0501 and E5397. Both of these studies were powered to determine survival benefit and also enrolled similar first-line R/M patients (Burtness B, et al. J Clin Oncol 2005; 23 : 8646-54 and Chung CH, et al. Ann Oncol 2009). HN0582 was not included in these analyses because the primary end point of this study was a clinical response rate of the induction chemotherapy rather than survival outcomes. It was determined that no association was evident between KRAS- variant status and disease control response (DCR) or objective response (ORR) (Table 17).
status is associated with cisplatin resistance in R M HNSCC patients, survival analyses were performed for patients from HN0501 and E5397. Both of these studies were powered to determine survival benefit and also enrolled similar first-line R/M patients (Burtness B, et al. J Clin Oncol 2005; 23 : 8646-54 and Chung CH, et al. Ann Oncol 2009). HN0582 was not included in these analyses because the primary end point of this study was a clinical response rate of the induction chemotherapy rather than survival outcomes. It was determined that no association was evident between KRAS- variant status and disease control response (DCR) or objective response (ORR) (Table 17).
[255] Table 17: Response by the KRAS- variant genotypes and the treatment regimens in HN0501 and E5397.

a DCR: Disease control response
b ORR: Objective response
[256] Because of the predictive power of the KRAS- variant, KRAS-vanant status was compared with progression-free survival (PFS) and overall survival (OS) in these studies separately, as patients were differently treated. The median survival data and hazard ratios (HRs) with respect to KRAS- variant status are summarized in Table 18. In HN0501 (VELCADE and TAXOTERE), KRAS-Y&xi&nt status was not associated with PFS (median:
1.6 months in TG/GG vs. 1.7 months in TT; log-rank p=0.89; Figure 17A) or OS (median:
6.7 months in TG/GG vs. 5.1 months in TT; log-rank p=0.60; Figure 17B). In contrast, in E5397, when the two treatment arms were combined (cisplatin+placebo and
cisplatin+cetuximab), KRAS- variant status was a statistically significant predictor for poor PFS (median: 2.2 months in TG/GG vs. 4.7 months in TT; log-rank p=0.002; Figure 17C).
Furthermore, in multivariable analyses, the HR for TG/GG vs. TT with respect to PFS was 3.85 (95% CI: 1.59-9.35; Wald p=0.003). While this association was significant for PFS, KRAS-variant status did not significantly associate with OS (median: 7.3 months in TG/GG vs. 8.2 months in TT, log-rank p=0.11; Figure 17D).
[257] Addition of cetuximab to cisplatin may benefit R/M FiNSCC patients with the KRAS- variant: KRAS-variant status was further examined in E5397, between the cisplatin + placebo or cisplatin + cetuximab treatment arms (Table 18 and Figure 17E). PFS was significantly improved in KRAS- variant (TG/GG) patients who received cetuximab in univariate analysis (median: 1.9 months in cisplatin+placebo vs. 3.9 months in cisplatin+cetuximab; log-rank p=0.03), while this effect was not seen in the non- variant (TT) group (median: 3.9 months in cisplatin+placebo vs. 5.8 months in cisplatin+cetuximab; log-rank p=0.57). While KRAS- variant patients experienced additional disease control from the addition of cetuximab, this effect was not statistically significant for OS (TG/GG median: 5.4 months in
cisplatin+placebo vs. 8.0 months in cisplatin+cetuximab, log-rank p=0.37; TT median: 8.1 months in cisplatin+placebo vs. 8.2 months in cisplatin+cetuximab, log-rank p=0.96; Figure 17F). The data support a conclusion that the KRAS- variant predicts cisplatin resistance yet cetuximab sensitivity in R/M FiNSCC patients.
Table 18: Event-time distribution by the KRAS- variant genotypes and the treatment regimens in HN0501 and E5397.

a Covariates included PS (0 vs. 1 ), disease status (previously untreated vs. recurrent), cell differentiation (well/moderately vs. poorly differentiation), primary site (oropharynx vs. non- oropharynx), smoking history (<40 packs-years vs. >40 packs- years), alcohol consumption (< 10 oz whiskey/week vs.≥ 10 oz whiskey/week) and treatment (when appropriate). N=21 in the cisplatin+placebo arm and in the cisplatin+cetuximab arm.
[259] KRAS-variant HNSCC cell lines are more resistant to cisplatin in vitro: To determine if KRAS-variant status affects in vitro cisplatin sensitivity, eight FiNSCC cell lines were evaluated by MTS assay with increasing doses of cisplatin (ΙηΜ-ΙΟμΜ). Six of these cell lines were HPV-negative (CAL27, UNC7, FaDu, SK 3, SCC6 and SCC61), while two were HPV-positive (UPCLSCC90 and UMSCC47). KRAS 3'UTR analysis determined three of these cell lines had the A^S-variant: CAL27, UNC7, and UMSCC47. Following dose- dependent in vitro exposure to cisplatin, it was determined that CAL27 and UNC7 were two of the most cisplatin-resistant cell lines evaluated (Figure 18 A). In fact, UNC7 demonstrated complete resistance to cisplatin at the highest dosage utilized (10 μΜ) (Figure 18B).
Additionally, two of the three non-variant cell lines (SCC61 and SCC6) began demonstrating cisplatin sensitivity at the lowest doses in the study (1 and ΙΟηΜ). FaDu, a highly metastatic FiNSCC cell line developed from a hypopharygeal tumor (Giard DJ, et al. J Natl Cancer Inst 1973;51 : 1417-23), was the only non- variant cell line that demonstrated significant cisplatin resistance (Figure 18A). While the KRAS- variant appears to confer resistance to cisplatin in HPV-negative cells, this does not occur in HPV-positive cell lines (Figure 18C).
[260] Differentially expressed genes based on the KRAS-variant status in HNSCC: To determine a possible genetic basis for the observed differences in clinical response, the gene expression profiles of 17 non- variant and 5 KRAS- variant tumors were compared with available microarray data. After normalizing the datasets and employing empirical Bayes moderated t-statistics, 25 probes with the smallest FDR adjusted p-values were examined (Table 19 and Figure 19). Among the genes with known molecular functions, a few interesting trends emerge. Most notably, epidermal growth factor (EGF) is upregulated (logFC: 0.77; p-value: 6.00xl0"5) in KRAS-vanant compared to non-variant tumors, suggesting that increased KRAS activity through the KRAS-variant is dependent on upstream growth signal through EGFR. The dependence of the KRAS variant upon upstream growth signal through EGFR may explain the enhanced disease control observed in these patients with cetuximab. Another interesting trend observed in KRAS- variant tumor gene expression is the upregulation of various cytoskeleton and microtubule associated proteins. DST (dystonin or bullous pemphigoid antigen 1, logFC: 1.88; p-value: 3.98xl0"6) and MACFl (logFC: 0.90; p-value: 4.22xl0"5) are both members of the plakin family, responsible for junctional complex and cytoskeleton function reviewed in (Sonnenberg A and Liem RK. Exp Cell Res
2007;313: 2189-203). EML6 (echinoderm microtubule-associated protein-like 6, logFC:
0.93; p-value: 2.07xl0~4) belongs to a family of microtubule-associating proteins (Houtman SH, et al. Neuroscience 2007; 144: 1373-82), although its function has been speculated by homology and has not been experimentally verified. SYNJ2 (synaptojanin 2, logFC: 1.08; p- value: 2.58xl0~4) is a regulatory lipid phosphatase which dephosphorylates PIP3 to PIP2 at the D-5 position in a manner similar to SHIP1 or SHIP2 (Nemoto Y, et al. J Biol Chem 1997; 272: 30817-21). While its function is most important for vesicle uncoating in neurons (Verstreken P, et al. Neuron 2003;40: 733-48), SYNJ2 upregulation has previously been associated with metastatic spread (Roesli C, et al. Cancer Res 2009;69: 5406-14; and Chuang YY, et al. Cancer Res 2004;64: 8271-5). Taken together, this gene expression profile suggests KRAS-variant HNSCC cells have a pro-metastatic gene signature due to enhanced expression of migratory machinery. These signaling mechanisms occur in all cancer cells, and, therefore, this gene expression profile suggests, more generally, that KRAS-variant cancer cells have a pro-metastatic gene signature due to enhanced expression of migratory machinery.
[261] Table 19: Top 25 most differentially expressed probes between the KRAS-variant (TG/GG) and non- variant (TT)

220154_at DST dystonin microtubule plus- 1.04 0.000226 0.584 Up end binding
241073_at AA883820 AA883820 Unknown 1.01 5.10E-05 0.41 Up echinoderm
microtubule
242785_at EML6 microtubule 0.93 0.000207 0.584 Up associated protein
like 6
microtubule-actin cytoskeletal linker
208634_s_at MACF1 0.90 4.22E-05 0.41 Up crosslinking factor 1 protein
CDNA
clone
238824_at FLJ30581 Unknown 0.90 0.000259 0.584 Up
BRAWH2007069
fis
golgi reassembly
243677_at G0RASP1 stacking protein 1 , membrane protein 0.86 3.31 E-05 0.41 Up
65kDa
microtubule-actin cytoskeletal linker
214894_x_at MACF1 0.86 0.000137 0.554 Up crosslinking factor 1 protein
Interact with
enkurin, TRPC
calmodulin and
237314_at ENKUR channel interacting 0.82 0.000259 0.584 Up cation channel
protein
proteins
regulatory lipid
212828_at SYNJ2 synaptojanin 2 0.82 5.39E-05 0.41 Up phosphatase
Epidermal growth EGFR binding,
206254_at EGF 0.77 6.00E-05 0.41 Up factor growth factor activity
NADH
dehydrogenase
Mitochondrial inner
21 1407_at NDUFB7 (ubiquinone) 1 beta 0.76 0.000143 0.554 Up membrane protein
subcomplex, 7,
18kDa
Transcription factor
40284_at F0XA2 forkhead box A2 0.56 0.00021 0.584 Up complex
transmembrane and
1560292_a_at TMC04 membrane protein -0.52 8.12E-05 0.447 Down coiled-coil domains 4
solute carrier family Mitochondrial inner
205716_at SLC25A40 -0.81 9.87E-05 0.456 Down
25, member 40 membrane protein
Nuclear protein,
ash2 (absent, small,
histone
209517_s_at ASH2L or homeotic)-like -0.82 0.000159 0.554 Down methyl transferase
(Drosophila)
complex
metal response
209704_at MTF2 element binding DNA binding protein -0.96 0.000165 0.554 Down transcription factor 2
cation
227560_at SFXN2 Sideroflexin-2 transmembrane -1 .07 0.000277 0.584 Down transporter activity
[263] The results of this study support a conclusion that HNSCC patients with the KRAS- variant exhibit increased cisplatin resistance and poor survival. Additionally, these patients may benefit from the addition of cetuximab to their treatment regimen. Consequently, the KRAS- variant is a predictive biomarker in HNSCC to specific therapy, and not necessarily only prognostic. Within this analysis, the results of cell lines studies agreed with the clinical findings analyzed, providing a potential mechanism for cetuximab sensitivity in these patients.
[264] This study is the first to examine the allelic frequency in a patient population comprised of mostly R/M disease. The worldwide allelic frequency of the TG/GG KRAS- variant is approximately 6%, with incidence rates as high as 23% in newly diagnosed cancers where it associated with increased cancer risk, including non-small cell lung cancer, ovarian cancer, and triple negative breast cancer. It has also been reported the frequency of TG/GG is approximately 15% in newly diagnosed patients. An association between incidence of HNSCC and specific subsites (oral, pharyngeal or laryngeal cancer) was not observed after stratification by potential confounders. However, as in this study, the presence of the KRAS- variant was significantly associated with poor survival, and these effects were mostly observed in oral cancer, but not pharyngeal or laryngeal tumors. Given the patient population in this study, the higher incidence rate (24%) of this mutation indicates two possibilities: 1) the presence of a KRAS- variant is associated with particularly aggressive de novo disease or 2) this variant confers increased resistance to first-line therapy, enriching this variant within this sample population. Current evidence suggests that these mechanisms may not be mutually exclusive.
[265] Identifying biomarkers of cisplatin resistance in HNSCC is critically important as cisplatin is the most commonly used chemotherapy in HNSCC and often administered in the
R/M setting. The association between the KRAS- variant and cisplatin resistance has been well-established in ovarian cancer (see Example 2). Experiments genotyping 536 epithelial ovarian cancers for the KRAS- variant and subset analysis determined that variant patients exhibited significant resistance to platinum-based chemotherapies. In vivo clinical data and in vitro cell line data support the conclusion that cisplatin therapy should be reconsidered in

[266] AT^S-variant patients experience improved disease control when cetuximab is added to their treatment. After determining the gene expression profile associated with KRAS-
variant tumors, it was noted that EGF, a potent ligand of EGFR, was upregulated in these tumors. It is hypothesized that cetuximab is beneficial for these patients because the anti- EGFR antibody blocks the pro-growth signal provided by an upregulation of EGF in these tumors. Furthermore, a number of genes associated with microtubule and cytoskeleton function are also upregulated. More specifically, MACF1 is upregulated in our current analysis. MACF1 interacts with ErbB2 to control microtubule capture during cell migration. Taken together, the upregulation of an EGFR-specific growth stimulatory ligand (EGF), a pro-migratory phosphatase (SYNJ2), and various components of the microtubule/cytoskeletal architecture (DST, MACF1, EML6) indicates the presence of an enhanced migratory or metastatic gene expression profile associated with KRAS-vanant FiNSCCs as well as explains an enhanced sensitivity of KRAS -variant cancer cells (e.g. FiNSCCs) to cetuximab treatment.
[267] The fundamental question regarding cetuximab sensitivity in KRAS- variant tumors centers on whether the KRAS variant behaves more like an activating KRAS mutant allele or simply results in higher expression of wild-type KRAS protein, which remains dependent upon activation by intact upstream regulatory mechanisms. The former mechanism would likely result in KRAS- variant cetuximab resistant tumors, while the latter may allow for cetuximab sensitivity through a continued reliance on upstream receptor activity. This continued reliance on EGFR signaling, a feature not required in tumors with KRAS activating mutations, may allow cetuximab to mitigate the effects of KRAS- variant cisplatin resistance, and provide enhanced disease control in these patients.
[268] The results of this study support a conclusion that the TG/GG KRAS- variant is a biomarker of altered response to cisplatin and cetuximab treatment. Consequently, platinum- based regimens provide suboptimal disease control in these patients, and treatment with cetuximab should be considered. The KRAS variant confers a unique biology to tumors of various subtypes.
OTHER EMBODIMENTS
[269] While the disclosure has been described in conjunction with the detailed description thereof, the foregoing description is intended to illustrate and not limit the scope of the disclosure, which is defined by the scope of the appended claims. Other aspects, advantages, and modifications are within the scope of the following claims.
[270] The patent and scientific literature referred to herein establishes the knowledge that is available to those with skill in the art. All United States patents and published or unpublished United States patent applications cited herein are incorporated by reference. All published foreign patents and patent applications cited herein are hereby incorporated by reference. Genbank and NCBI submissions indicated by accession number cited herein are hereby incorporated by reference. All other published references, documents, manuscripts and scientific literature cited herein are hereby incorporated by reference.
[271] While this disclosure has been particularly shown and described with references to preferred embodiments thereof, it will be understood by those skilled in the art that various changes in form and details may be made therein without departing from the scope of the disclosure encompassed by the appended claims.
Claims
1. A method of predicting the response of a head and neck squamous cell carcinoma (HNSCC) cell to a platinum-based chemotherapy, comprising detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, wherein the presence of the mutation indicates resistance to platinum-based chemotherapy.
2. The method of claim 1, wherein the head and neck squamous cell carcinoma
(HNSCC) cell is a recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC) cell.
3. The method of claim 1 or 2, wherein the HNSCC cell is evaluated in vivo, in vitro or ex vivo.
4. The method of any one of claims 1-3, wherein the platinum-based chemotherapy is cisplatin, carboplatin or paclitaxel.
5. The method of claim 4, wherein the platinum-based chemotherapy is cisplatin.
6. The method of any one of claims 1-5, wherein the platinum-based chemotherapy is an adjuvant therapy.
7. A method of predicting the response of a cancer cell to a monoclonal anti-EGFR antibody monotherapy, comprising detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and wherein the presence of the mutation indicates sensitivity to monoclonal anti-EGFR antibody monotherapy.
8. The method of claim 7, wherein the cancer cell is a colorectal cancer (CRC) cell.
9. The method of claim 7, wherein the cancer cell is a head and neck squamous cell carcinoma (HNSCC) cell.
10. The method of claim 9, wherein the head and neck squamous cell carcinoma
(HNSCC) cell is a recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC) cell.
11. The method of any one of claims 7-10, wherein the cancer cell is evaluated in vivo, in vitro or ex vivo.
12. The method of any one of claims 7-11, wherein the monoclonal anti-EGFR antibody monotherapy is Cetuximab.
13. A method of predicting the response of a cancer cell to an EGFR-inhibitor, comprising detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, wherein the presence of the mutation indicates resistance to an EGFR-inhibitor.
14. The method of claim 13, wherein the cancer cell is a non- small cell lung cancer (NSCLC) cell.
15. The method of claim 13 or 14, wherein the cancer cell is evaluated in vivo, in vitro or ex vivo.
16. The method of any one of claims 13-15, wherein the EGFR-inhibitor is gefitinib.
17. A method of predicting the response of a cancer cell to a MAP kinase pathway inhibitor, comprising detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, wherein the presence of the mutation indicates sensitivity to a MAP kinase pathway inhibitor.
18. The method of claim 17, wherein the cancer cell is a non-small cell lung cancer (NSCLC) cell.
19. The method of claim 17 or 18, wherein the cancer cell is evaluated in vivo, in vitro or ex vivo.
20. The method of any one of claims 17-19, wherein the EGFR-inhibitor is MAP kinase pathway inhibitor is sorafenib.
21. A method of treating as subject having head and neck squamous cell carcinoma (FiNSCC), the method comprising detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and treating the subject as follows:
(i) if the mutation is present, administering a monoclonal anti-EGFR antibody monotherapy to the subject and avoiding platinum-based chemotherapy; and
(ii) if the mutation is absent, administering platinum-based chemotherapy to the patient and avoiding monoclonal anti-EGFR antibody monotherapy.
22. A method of treating a subject having non-small cell lung cancer (NSCLC), the method comprising detecting a mutation in let-7 complementary site LCS6 of human KRAS in a patient sample, wherein the mutation is a SNP comprising a uracil (U) or thymine (T) to guanine (G) transition at position 4 of LCS6, and treating the subject as follows:
(i) if the mutation is present, administering a MAP kinase pathway inhibitor to the subject and avoiding EGFR inhibitors.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361829766P | 2013-05-31 | 2013-05-31 | |
| US61/829,766 | 2013-05-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014193937A1 true WO2014193937A1 (en) | 2014-12-04 |
Family
ID=51989359
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2014/039763 WO2014193937A1 (en) | 2013-05-31 | 2014-05-28 | The kras variant and response to cancer therapy |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2014193937A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017189906A1 (en) * | 2016-04-27 | 2017-11-02 | Mira Dx, Inc. | Immune-based treatment of kras-variant cancer patients |
| US10278976B2 (en) | 2014-12-12 | 2019-05-07 | Mira Dx, Inc. | Methods for treating or preventing cancer in a KRAS-variant patient and for diagnosing risk of developing multiple primary breast tumors |
| EP3638308A4 (en) * | 2017-06-15 | 2021-06-16 | Miradx | Biomarkers for predicting tumor response to and toxicity of immunotherapy |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012129352A1 (en) * | 2011-03-21 | 2012-09-27 | Yale University | The kras variant and tumor biology |
-
2014
- 2014-05-28 WO PCT/US2014/039763 patent/WO2014193937A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012129352A1 (en) * | 2011-03-21 | 2012-09-27 | Yale University | The kras variant and tumor biology |
Non-Patent Citations (4)
| Title |
|---|
| CHRISTENSEN ET AL.: "A let-7 microRNA-binding site polymorphism in the KRAS 3' UTR is associated with reduced survival in oral cancers.", CARCINOGENESIS, vol. 30, no. 6, 2009, pages 1003 - 1007, XP002597740, DOI: doi:10.1093/CARCIN/BGP099 * |
| KUNDU ET AL.: "KRAS alleles: The LCS6 3' UTR variant and KRAS coding sequence mutations in the NCI-60 panel.", CELL CYCLE, vol. 11, no. 2, 2012, pages 361 - 366, XP008152916, DOI: doi:10.4161/cc.11.2.18794 * |
| RYAN ET AL.: "KRAS-LCS6 genotype as a prognostic marker in early-stage CRC-letter.", CLINICAL CANCER RESEARCH, vol. 18, no. 12, 2012, pages 3487 - 3488 * |
| SLABY ET AL.: "Genetic polymorphisms and microRNAs: new direction in molecular epidemiology of solid cancer.", JOURNAL OF CELLULAR AND MOLECULAR MEDICINE, vol. 16, no. 1, 2012, pages 8 - 21 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10278976B2 (en) | 2014-12-12 | 2019-05-07 | Mira Dx, Inc. | Methods for treating or preventing cancer in a KRAS-variant patient and for diagnosing risk of developing multiple primary breast tumors |
| WO2017189906A1 (en) * | 2016-04-27 | 2017-11-02 | Mira Dx, Inc. | Immune-based treatment of kras-variant cancer patients |
| CN109790581A (en) * | 2016-04-27 | 2019-05-21 | 米拉迪克斯有限公司 | KRAS- variant cancer patient based on immune treatment |
| JP2019515035A (en) * | 2016-04-27 | 2019-06-06 | ミラ ディーエックス, インコーポレイテッド | Immunity-based treatment of KRAS variant cancer patients |
| EP3638308A4 (en) * | 2017-06-15 | 2021-06-16 | Miradx | Biomarkers for predicting tumor response to and toxicity of immunotherapy |
| US11578371B2 (en) | 2017-06-15 | 2023-02-14 | Mira Dx, Inc. | Biomarkers for predicting tumor response to and toxicity of immunotherapy |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20140065615A1 (en) | The KRAS Variant and Tumor Biology | |
| Xie et al. | miR-7 inhibits the invasion and metastasis of gastric cancer cells by suppressing epidermal growth factor receptor expression | |
| Tu et al. | The association between genetic polymorphism and the processing efficiency of miR-149 affects the prognosis of patients with head and neck squamous cell carcinoma | |
| EP2714927B1 (en) | Methods and devices for prognosis of cancer relapse | |
| AU2008260029B2 (en) | A genetic lesion associated with cancer | |
| US20140024590A1 (en) | KRAS-Variant And Endometriosis | |
| Zhang et al. | A genetic variant in pre-miR-27a is associated with a reduced breast cancer risk in younger Chinese population | |
| CN103547683A (en) | The KRAS variant and tumor biology | |
| Zhao et al. | MicroRNA-411 inhibits malignant biological behaviours of colorectal cancer cells by directly targeting PIK3R3 Retraction in/10.3892/or. 2022.8283 | |
| Fung et al. | Identification of epidermal growth factor receptor (EGFR) genetic variants that modify risk for head and neck squamous cell carcinoma | |
| JP2013212052A (en) | Kras variant and tumor biology | |
| WO2014193937A1 (en) | The kras variant and response to cancer therapy | |
| US20130252832A1 (en) | KRAS Variant and Tumor Biology | |
| US20120028254A1 (en) | SNP Marker of Breast and Ovarian Cancer Risk | |
| US20200325234A1 (en) | Immune-based treatment of kras-variant cancer patients | |
| CA2772338A1 (en) | The kras variant and tumor biology | |
| US20220088033A1 (en) | Methods for treating or preventing cancer in a kras-variant patient and for diagnosing risk of developing multiple primary breast tumors | |
| Soldevilla et al. | MicroRNA signature and integrative omics analyses define prognostic clusters and key pathways driving prognosis in patients with neuroendocrine neoplasms | |
| Zang et al. | Association between microRNA-125a rs12976445 C> T polymorphism and 18F-Fluorodeoxyglucose (18FDG) uptake: clinical and metabolic response in patients with non-small cell lung cancer | |
| WO2017201291A1 (en) | Microrna expression signatures for doublecortin-like kinase 1 (dclk1) activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14803470 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14803470 Country of ref document: EP Kind code of ref document: A1 |