WO2014128588A1 - Solid forms of a selective cdk4/6 inhibitor - Google Patents
Solid forms of a selective cdk4/6 inhibitor Download PDFInfo
- Publication number
- WO2014128588A1 WO2014128588A1 PCT/IB2014/058865 IB2014058865W WO2014128588A1 WO 2014128588 A1 WO2014128588 A1 WO 2014128588A1 IB 2014058865 W IB2014058865 W IB 2014058865W WO 2014128588 A1 WO2014128588 A1 WO 2014128588A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- free base
- μηι
- methyl
- pyridin
- cyclopentyl
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- This invention relates to the free base of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin- 1-yl-pyridin-2-ylamino)-8/-/-pyrido[2,3-c]pyrimidin-7-one having improved physicochemical properties.
- the invention also relates to pharmaceutical compositions and dosage forms comprising the free base, and to methods for making and using such compounds, compositions and dosage forms in the treatment of cell proliferative diseases, such as cancer.
- Compound 1_ is a potent and selective inhibitor of CDK4 and CDK6.
- compound 1_ is a potent and selective CDK4/CDK6 inhibitor
- its use as a free base presented challenges for pharmaceutical development.
- the free base provided by traditional salt break procedures, e.g., as in Example 4 of WO 2005/005426, was highly static prone and formed small primary particles, which agglomerated into large, hard agglomerates that were difficult to disperse by sieving and were unsuitable for further development.
- the present invention provides compound 1. free base having larger primary particle size that demonstrates improved physicochemical and manufacturability properties.
- the free base of compound 1_ 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl- pyridin-2-ylamino)-8/-/-pyrido[2,3-c]pyrimidin-7-one, can exist in one or more polymorphic forms, including Form A and Form B, wherein Form A is the more stable crystalline form.
- the free base may be anhydrous, or may contain varying amounts of water or one or more solvents.
- the present invention provides the crystalline free base of compound ⁇ having larger primary particle size, greatly reduced specific surface area, and lower surface energy measurements than the free base provided by traditional salt break methods described in the art.
- the large particle size compound 1. free base disclosed herein is distinguishable by a variety of methods.
- the polymorphic and solid forms of the invention can be distinguished by powder X-ray diffractometry (PXRD), solid state NMR (ssNMR), differential scanning calorimetry (DSC), vibrational spectroscopy (e.g., IR and Raman spectroscopy), polarized light microscopy (PLM), scanning electron microscopy (SEM), hot stage optical microscopy, electron crystallography, single crystal X-ray diffractometry, quantitative analysis, particle size analysis (PSA) (e.g., particle size, particle size distribution (PSD), and particle shape), specific surface area (SSA) analysis, surface energy analysis (e.g., inverse gas chromatography or IGC), by solubility studies and dissolution studies, or a combination of these techniques.
- PXRD powder X-ray diffractometry
- ssNMR solid state NMR
- DSC differential scanning calorimetry
- vibrational spectroscopy e.g., IR and Raman spectroscopy
- PLM polarized light
- the invention provides a crystalline free base of 6-acetyl-8-cyclopentyl-5- methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8/-/-pyrido[2,3-c]pyrimidin-7-one having a specific surface area of ⁇ 2 m 2 /g.
- the free base has a specific surface area of ⁇ 1 m 2 /g.
- the crystalline free base of compound 1. is a polymorph Form A of the free base.
- the crystalline free base has a PXRD pattern comprising a peak at diffraction angle (2 ⁇ ) of 10.1 ⁇ 0.2.
- the crystalline free base has a PXRD pattern comprising peaks at diffraction angles (2 ⁇ ) of 8.0 ⁇ 0.2 and 10.1 ⁇ 0.2.
- the crystalline free base has a PXRD pattern comprising peaks at diffraction angles (2 ⁇ ) of 8.0 ⁇ 0.2, 10.1 ⁇ 0.2, and 1 1.5 ⁇ 0.2.
- the crystalline free base has a PXRD pattern comprising peaks at diffraction angles (2 ⁇ ) of 8.0 ⁇ 0.2, 10.1 ⁇ 0.2, 10.3 ⁇ 0.2, and 11.5 ⁇ 0.2. In further embodiments, the crystalline free base has a PXRD pattern comprising peaks at diffraction angles (2 ⁇ ) essentially the same as shown in Figure 1.
- the crystalline free base of compound ⁇ has a 13 C solid state NMR (ssNMR) spectrum comprising the following resonance (ppm) values: 12.5 ppm ⁇ 0.2 ppm.
- the crystalline free base has a 13 C solid state NMR spectrum comprising the following resonance (ppm) values: 12.5 ppm and 1 12.4 ppm ⁇ 0.2 ppm.
- the crystalline free base has a 13 C solid state NMR spectrum comprising the following resonance (ppm) values: or 12.5 ppm, 1 12.4 ppm and 143.2 ppm ⁇ 0.2 ppm.
- the compound 1. free base of the invention is distinguished by particle size analysis.
- the crystalline free base has a primary particle size of from about 5 ⁇ to about 150 ⁇ , preferably from about 10 ⁇ to about 100 ⁇ , or more preferably from about 15 ⁇ to about 80 ⁇ .
- the crystalline free base has a primary particle size distribution characterized by: (i) a D10 value of from about 5 ⁇ to about 10 ⁇ ; (ii) a D50 value of from about 10 ⁇ to about 45 ⁇ ; or (iii) a D90 value of from about 30 ⁇ to about 125 ⁇ ; or a combination of (i), (ii) and (iii).
- the crystalline free base has a primary particle size distribution ratio of (D90-D10)/D50 of from about 2 to about 3. In further embodiments, the crystalline free base has a volume mean diameter (D[4,3]) of from about 15 ⁇ to about 125 ⁇ .
- the crystalline free base of compound 1. is anhydrous. In other embodiments, the crystalline free base of compound 1. is a solvate, in particular a hydrate.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a crystalline free base of compound ⁇ , having the large primary particle size according to the invention, and a pharmaceutically acceptable carrier, diluent or excipient.
- the pharmaceutical composition comprises polymorph Form A of the free base.
- the invention further provides a capsule comprising such a pharmaceutical composition of the invention.
- the capsule comprises from 0.1 to 200 mg, and preferably from 25 to 150 mg, of compound 1. free base (preferably as polymorph Form A), having the large primary particle size as described herein.
- the invention provides a method of treating cancer in a mammal, preferably a human, comprising administering to the mammal a therapeutically effective amount of a pharmaceutical composition of the invention.
- the method of treatment may further comprise administration of compound 1_ in combination with one or more additional therapeutic agents.
- the invention provides methods of making the free base of compound 1_ having a large primary particle size, as described herein.
- One method involves dissolving the small particle size free base of compound 1_ in mixture of a first solvent and a second solvent and heating to achieve dissolution, cooling to appropriate temperature, providing seed crystals of compound 1_ free base (Form A), followed by crystallization to provide the large particle size free base of compound
- the small particle size free base used in this process may be isolated from a traditional salt break procedure, e.g., by acidic hydrolysis of the intermediate vinyl ether to provide an acid addition salt, followed by basification, as described in Example 5.
- Another method involves acidic hydrolysis of the intermediate vinyl ether in a mixture of water and a first solvent, which may require heating to obtain dissolution, addition of a second solvent and basification to provide a second mixture comprising the free base generated in situ, heating if required to obtain dissolution and to distill off water, and providing seed crystals of compound 1_ free base (Form A) at an appropriate temperature, followed by crystallization to provide the free base of compound 1_ having a large primary particle size.
- the invention further provides the free base of compound 1_ prepared by these methods, having the properties described herein.
- the first solvent is an alcohol and the second solvent is an aromatic solvent.
- Suitable alcohols include, but are not limited to, relatively high boiling alcohols such as n-butanol, t-butanol, n-propanol, pentanol, 1 ,4-butanediol or propylene glycol, and the like.
- Suitable aromatic solvents include, but are not limited to, anisole, mesitylene, m-xylene, chlorobenzene, pyridine, and the like.
- the methods may include heating or cooling to temperatures above or below room temperature.
- the reaction mixtures may be heated to temperatures ranging from about 30°C to about 150°C, and more frequently from about 50°C to about 120°C to achieve dissolution.
- it may be desirable to cool the reaction mixture to a temperature that is at or below room temperature, for example between about 0°C and about 30°C, preferably to about 5°C, about 10°C, about 15°C, or about 20°C.
- Figure 1 shows a PXRD pattern of compound 1. free base, polymorph Form A.
- Figure 2 shows the Carbon CPMAS spectrum of compound 1. free base, polymorph Form A. Peaks marked by asterisks are spinning sidebands.
- Figure 3 shows a PXRD pattern of compound 1_ free base, polymorph Form B.
- Figure 4 shows the Carbon CPMAS spectrum of compound 1_ free base, polymorph Form B. Peaks marked by asterisks are spinning sidebands.
- Figure 5 shows a scanning electron microscopy (200x magnification) image of compound 1_ free base API, polymorph Form A, recrystallized from 40% n-BuOH/anisole.
- Figure 6 shows a scanning electron microscopy (1500x magnification) image of compound 1_ free base API, polymorph Form A, isolated from a standard free basing process.
- Figure 7 shows the particle size distribution of compound 1_ free base API, polymorph Form A, recrystallized from 40% n-BuOH/anisole.
- Figure 8 shows the particle size distribution of compound 1_ free base API, polymorph Form A, isolated from a standard free basing process.
- Figure 9 shows a polarized light microscopy (PLM) image (200x) of compound ⁇ free base API, polymorph Form A, recrystallized from 40% n-BuOH/anisole.
- PLM polarized light microscopy
- the term "about” means within a statistically meaningful range of a value, such as a stated concentration range, time frame, molecular weight, particle size, temperature or pH. Such a range can be within an order of magnitude, typically within 20%, more typically within 10%, and even more typically within 5% of the indicated value or range. Sometimes, such a range can be within the experimental error typical of standard methods used for the measurement and/or determination of a given value or range. The allowable variation encompassed by the term "about” will depend upon the particular system under study, and can be readily appreciated by one of ordinary skill in the art. Whenever a range is recited within this application, every whole number integer within the range is also contemplated as an embodiment of the invention.
- abnormal cell growth refers to cell growth that is independent of normal regulatory mechanisms (e.g., loss of contact inhibition).
- Abnormal cell proliferative diseases are diseases characterized by abnormal cell growth, such as cancer.
- cancer includes both solid tumors and hematological malignancies.
- Cancers include, but are not limited to, breast cancer, ovarian cancer, cervical cancer, endometrial cancer, prostate cancer, testicular cancer, pancreatic cancer, esophageal cancer, head and neck cancer, gastric cancer, bladder cancer, lung cancer (e.g., adenocarcinoma, NSCLC and SCLC), bone cancer (e.g., osteosarcoma), colon cancer, rectal cancer, thyroid cancer, brain and central nervous system cancers, glioblastoma, neuroblastoma, neuroendocrine cancer, rhabdoid cancer, keratoacanthoma, epidermoid carcinoma, seminoma, melanoma, sarcoma (e.g., liposarcoma), bladder cancer, liver cancer (e.g., hepatocellular carcinoma), kidney cancer (e.g., renal cell carcinoma), myeloid disorders (e.g., AML, CML, mye
- phrases "pharmaceutically acceptable” refers to substances, which are within the scope of sound medical judgment, suitable for use in contact with the tissues of patients without undue toxicity, irritation, allergic response, and the like, commensurate with a reasonable benefit/risk ratio, and effective for their intended use.
- mammal may be a human or non-human mammal (e.g., dog, cat, rabbit, rat, mouse, horse, monkey, other lower-order primate, etc.).
- mammal is a human.
- treating means reversing, alleviating, inhibiting the progress of, or preventing the disorder or condition to which such term applies, or one or more symptoms of such disorder or condition.
- treatment refers to the act of treating as “treating” as defined immediately above.
- an "effective" amount refers to an amount of a compound, agent, substance, formulation or composition that is of sufficient quantity to result in a decrease in severity of disease symptoms, an increase in frequency and duration of disease symptom-free periods, or a prevention of impairment or disability due to the disease affliction.
- the amount may be as a single dose or according to a multiple dose regimen, alone or in combination with other compounds, agents or substances.
- One of ordinary skill in the art would be able to determine such amounts based on such factors as a subject's size, the severity of a subject's symptoms, and the particular composition or route of administration selected.
- Unit dosage form refers to a physically discrete unit of inventive formulation appropriate for the subject to be treated. It will be understood, however, that the total daily usage of the compositions of the present invention will be decided by the attending physician within the scope of sound medical judgment.
- the specific effective dose level for any particular subject will depend upon a variety of factors including the disorder being treated and the severity of the disorder; specific composition employed; age, body weight, general health, sex and diet of the subject; time of administration, duration of the treatment; drugs and/or additional therapies used in combination or coincidental with the inventive compositions, and like factors well known in the medical arts.
- the term "essentially the same" with reference to X-ray diffraction peak positions means that typical peak position and intensity variability are taken into account.
- the peak positions (2 ⁇ ) will show some inter- apparatus variability, typically as much as 0.2° or 0.1 °.
- relative peak intensities will show inter-apparatus variability as well as variability due to degree of crystallinity, preferred orientation, prepared sample surface, and other factors known to those skilled in the art, and should be taken as qualitative measures only.
- solvate refers to a crystal form of a substance which contains solvent.
- hydrate refers to a solvate wherein the solvent is water.
- seeding means the addition of crystals to a crystallization system, for the purpose of initiating or enhancing nucleation or acting as substrate for further crystallization.
- API active pharmaceutical ingredient
- active pharmaceutical ingredient refers to the free base of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8/-/-pyrido[2,3- d]pyrimidin-7-one.
- primary particles refers to individual API crystals.
- agglomerates refers to tightly bound API crystals that are difficult to disperse into primary particles during processing and particle size analysis.
- the present invention provides compound 1. free base having larger primary particle size, greatly reduced specific surface area, and lower surface energy measurements than the free base provided by traditional salt break methods.
- the compound 1. free base provided by the invention may sometimes be referred to herein as the "large (primary) particle size” free base. This is in contrast to the free base of compound 1_ prepared through traditional salt break methods, which is sometimes referred to as the "small (primary) particle size” free base.
- the reference to "small particle size” in this case refers to the particle size of individual API crystals, and does not take into account the propensity of the "small” particles to form large agglomerates.
- the crystalline free base of compound 1. is distinguished by specific surface area (SSA).
- SSA specific surface area
- the invention provides a crystalline free base of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2- ylamino)-8/-/-pyrido[2,3-c]pyrimidin-7-one having a specific surface area (SSA) of ⁇ 2 m 2 /g.
- the free base has a specific surface area (SSA) of ⁇ 1 m 2 /g.
- the free base of compound 1_ has an SSA of ⁇ 0.9 m 2 /g, ⁇ 0.8 m 2 /g or ⁇ 0.7 m 2 /g.
- the free base of compound 1_ has an SSA of between 0.2 m 2 /g and 2 m 2 /g, between 0.5 m 2 /g and 1.5 m 2 /g, or between 0.5 m 2 /g and 1 m 2 /g.
- the crystalline free base of compound 1_ is distinguished by dispersive surface energy.
- the invention provides a crystalline free base of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)- 8/-/-pyrido[2,3-c]pyrimidin-7-one having a dispersive surface energy of ⁇ 60 mJ/m 2 .
- the free base has a dispersive surface energy of ⁇ 55 mJ/m 2 , ⁇ 50 mJ/m 2 , ⁇ 45 mJ/m 2 or ⁇ 40 mJ/m 2 .
- the free base of compound 1_ has a dispersive surface energy of between 20 mJ/m 2 and 60 mJ/m 2 , between 25 mJ/m 2 and 50 mJ/m 2 , or between 30 mJ/m 2 and 50 mJ/m 2 .
- the crystalline free base of compound 1_ is a polymorph Form A of the free base.
- the crystalline form has a PXRD pattern comprising a peak at diffraction angle (2 ⁇ ) of 10.1 ⁇ 0.2.
- the crystalline form has a PXRD pattern comprising peaks at diffraction angles (2 ⁇ ) of 8.0 ⁇ 0.2 and 10.1 ⁇ 0.2.
- the crystalline form has a PXRD pattern comprising peaks at diffraction angles (2 ⁇ ) of 8.0 ⁇ 0.2, 10.1 ⁇ 0.2, and 1 1.5 ⁇ 0.2.
- the crystalline form has a PXRD pattern comprising peaks at diffraction angles (2 ⁇ ) of 8.0 ⁇ 0.2, 10.1 ⁇ 0.2, 10.3 ⁇ 0.2, and 11.5 ⁇ 0.2. In other embodiments, the crystalline form has a PXRD pattern comprising peaks at diffraction angles (2 ⁇ ) of 5.1 ⁇ 0.2, 8.0 ⁇ 0.2, 10.1 ⁇ 0.2, and 1 1.5 ⁇ 0.2. In further embodiments, the crystalline form has a PXRD pattern comprising peaks at diffraction angles (2 ⁇ ) of 8.0 ⁇ 0.2, 10.1 ⁇ 0.2, 11.5 ⁇ 0.2, and 19.7 ⁇ 0.2.
- the crystalline form has a PXRD pattern comprising peaks at diffraction angles (2 ⁇ ) of 8.0 ⁇ 0.2, 10.1 ⁇ 0.2, 11.5 ⁇ 0.2, and 22.5 ⁇ 0.2. In further embodiments, the crystalline form has a PXRD pattern comprising peaks at diffraction angles (2 ⁇ ) essentially the same as shown in Figure 1.
- the crystalline free base of compound 1 has a 13 C solid state NMR spectrum comprising the following resonance (ppm) values: 12.5 ppm ⁇ 0.2 ppm.
- the crystalline form has a 13 C solid state NMR spectrum comprising the following resonance (ppm) values: 12.5 ppm and 112.4 ppm ⁇ 0.2 ppm.
- the crystalline form has a 13 C solid state NMR spectrum comprising the following resonance (ppm) values: or 12.5 ppm, 1 12.4 ppm and 143.2 ppm ⁇ 0.2 ppm.
- the crystalline free base of compound 1. is distinguished by particle size analysis.
- the free base has a primary particle size of from about 5 ⁇ to about 150 ⁇ , preferably from about 10 ⁇ to about 100 ⁇ , and more preferably from about 15 ⁇ to about 80 ⁇ .
- the free base has a primary particle size distribution characterized by: (i) a D10 value of from about 5 ⁇ to about 10 ⁇ ; (ii) a D50 value of from about 10 ⁇ to about 45 ⁇ ; or (iii) a D90 value of from about 30 ⁇ to about 125 ⁇ ; or a combination of (i), (ii) and (iii).
- the free base has a primary particle size distribution ratio of (D90-D10)/D50 of from about 2 to about 3.
- the free base has a volume mean diameter (D[4,3]) of from about 15 ⁇ to about 125 ⁇ .
- the invention provides a crystalline free base of 6-acetyl-8-cyclopentyl-5- methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8/-/-pyrido[2,3-c]pyrimidin-7-one, having a primary particle size of greater than about 5 ⁇ .
- the free base has a primary particle size of greater than about 7.5 ⁇ .
- the free base has a primary particle size of greater than about 10 ⁇ .
- the free base has a primary particle size of greater than about 12.5 ⁇ .
- the free base has a primary particle size of greater than about 15 ⁇ .
- the invention provides a crystalline free base of 6-acetyl-8-cyclopentyl-
- the free base has a primary particle size of: from about 5 ⁇ to about 175 ⁇ ; from about 5 ⁇ to about 150 ⁇ ; from about 5 ⁇ to about 125 ⁇ ; from about 5 ⁇ to about 100 ⁇ ; from about 5 ⁇ to about 75 ⁇ ; from about 10 ⁇ to about 200 ⁇ ; from about 10 ⁇ to about 175 ⁇ ; from about 10 ⁇ to about 150 ⁇ ; from about 10 ⁇ to about 125 ⁇ ; from about 10 ⁇ to about 100 ⁇ ; from about 10 ⁇ to about 75 ⁇ ; from about 15 ⁇ to about 200 ⁇ ; from about 15 ⁇ to about 175 ⁇ ; from about 15 ⁇ to about 150 ⁇ ; from about 15 ⁇ to about 125 ⁇ ; from about 15 ⁇ to about 100 ⁇ ; or from about 15 ⁇ to about 75 ⁇ .
- the invention provides a crystalline free base of 6-acetyl-8-cyclopentyl-
- the free base has a D10 value of from about 5 ⁇ to about 10 ⁇ . In other such embodiments, the free base has a D90 value of from about 30 ⁇ to about 125 ⁇ . In other such embodiments, the free base has a D50 value of from about 10 ⁇ to about 45 ⁇ . In some such embodiments, the free base has a D10 value of from about 5 ⁇ to about 10 ⁇ and a D90 value of from about 30 ⁇ to about 125 ⁇ . In further embodiments, the free base has a D10 value of from about 5 ⁇ to about 10 ⁇ , a D90 value of from about 30 ⁇ to about 125 ⁇ , and a D50 value of from about 10 ⁇ to about 45 ⁇ .
- the invention provides a crystalline free base of 6-acetyl-8-cyclopentyl- 5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8/-/-pyrido[2,3-c]pyrimidin-7-one, having a primary particle size distribution having at least one of: (d) a D10 value of from about 5 ⁇ to about 10 ⁇ ;
- the free base has a D10 value of from about 5 ⁇ to about 10 ⁇ . In other such embodiments, the free base has a D90 value of from about 30 ⁇ to about 75 ⁇ . In other such embodiments, the free base has a D50 value of from about 10 ⁇ to about 25 ⁇ . In some such embodiments, the free base has a D10 value of from about 5 ⁇ to about 10 ⁇ and a D90 value of from about 30 ⁇ to about 75 ⁇ . In further embodiments, the free base has a D10 value of from about 5 ⁇ to about 10 ⁇ , a D90 value of from about 30 ⁇ to about 755 ⁇ , and a D50 value of from about 10 ⁇ to about 25 ⁇ .
- the free base has a primary particle size distribution having a D10 value of: from about 5 ⁇ to about 7.5 ⁇ ; from about 5 ⁇ to about 10 ⁇ ; from about 5 ⁇ to about 12.5 ⁇ ; or from about 5 ⁇ to about 15 ⁇ .
- the free base has a primary particle size distribution having a D50 value of: from about 10 ⁇ to about 50 ⁇ ; from about 10 ⁇ to about 45 ⁇ ; from about 10 ⁇ to about 40 ⁇ ; from about 10 ⁇ to about 35 ⁇ ; from about 10 ⁇ to about 30 ⁇ ; from about 10 ⁇ to about 25 ⁇ ; or from about 10 ⁇ to about 20 ⁇ ..
- the free base has a primary particle size distribution having a D90 value of: from about 30 ⁇ to about 175 ⁇ ; from about 30 ⁇ to about 160 ⁇ ; from about 30 ⁇ to about 150 ⁇ ; from about 30 ⁇ to about 140 ⁇ ; from about 30 ⁇ to about 130 ⁇ ; from about 30 ⁇ to about 125 ⁇ ; from about 30 ⁇ to about 120 ⁇ ; from about 30 ⁇ to about 115 ⁇ ; from about 30 ⁇ to about 110 ⁇ ; from about 30 ⁇ to about 100 ⁇ ; from about 30 ⁇ to about 75 ⁇ ; from about 30 ⁇ to about 70 ⁇ ; from about 30 ⁇ to about 65 ⁇ ; from about 30 ⁇ to about 60 ⁇ ; from about 30 ⁇ to about 55 ⁇ ; from about 30 ⁇ to about 50 ⁇ ; or from about 30 ⁇ to about 45 ⁇ .
- Each of the foregoing values of embodiments for D10 can be combined with any value for D50 and/or D90 value not inconsistent with it.
- Each of the foregoing values of embodiments for D50 can be combined with any value for D10 and/or D90 value not inconsistent with it.
- Each of the foregoing values of embodiments for D90 can be combined with any value for D10 and/or D50 value not inconsistent with it.
- the invention provides a crystalline free base of 6-acetyl-8-cyclopentyl- 5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8/-/-pyrido[2,3-c]pyrimidin-7-one, having a primary particle size distribution ratio of (D90-D10)/D50 of from about 2 to about 3.
- the free base has a primary particle size of from about 5 ⁇ to about 150 ⁇ .
- the free base has a primary particle size distribution ratio of (D90-D10)/D50 of: from about 2 to about 2.75; from about 2 to about 2.5; from about 2 to about 2.25. In other embodiments, the ratio is about 2.0, about 2.1 , about 2.2, about 2.3, about 2.4, about 2.5, about 2.6, about 2.7, about 2.8, about 2.9, or about 3.0.
- the invention provides a crystalline free base of 6-acetyl-8- cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-c]pyrimidin-7-one, having a volume mean diameter (D[4,3]) of from about 15 ⁇ to about 125 ⁇ .
- the free base has a D[4,3] of from about 50 ⁇ to about 100 ⁇ .
- the free base has a D[4,3] of from about 15 ⁇ to about 30 ⁇ .
- the free base has a D[4,3] of: from about 15 ⁇ to about 100 ⁇ ; from about 15 ⁇ to about 90 ⁇ ; from about 15 ⁇ to about 80 ⁇ ; from about 15 ⁇ to about 70 ⁇ ; from about 15 ⁇ to about 60 ⁇ ; from about 15 ⁇ to about 50 ⁇ ; from about 15 ⁇ to about 40 ⁇ ; from about 25 ⁇ to about 120 ⁇ ; from about 25 ⁇ to about 100 ⁇ ; from about 25 ⁇ to about 90 ⁇ ; from about 25 ⁇ to about 80 ⁇ ; from about 25 ⁇ to about 70 ⁇ ; from about 25 ⁇ to about 60 ⁇ ; from about 25 ⁇ to about 50 ⁇ ; from about 25 ⁇ to about 40 ⁇ ; about 25 ⁇ ; about 30 ⁇ ; about 35 ⁇ ; about 40 ⁇ ; about 45 ⁇ ; about 50 ⁇ ; about 55 ⁇ ; about 60 ⁇ ; about 65 ⁇ ; about 70 ⁇ ; about 75 ⁇ ; to about 80 ⁇ ; about 90 ⁇ ; about 100 ⁇
- the invention provides a pharmaceutical composition comprising the free base of the invention, and a pharmaceutically acceptable carrier, diluent or excipient.
- the invention further provides capsule comprising such a pharmaceutical composition of the invention.
- the capsule comprises from 0.1 to 200 mg of polymorph Form A of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8/-/-pyrido[2,3- c]pyrimidin-7-one. In other embodiments, the capsule comprises from 25 to 150 mg of the polymorph Form A of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8/-/- pyrido[2,3-c]pyrimidin-7-one.
- the capsule comprises from 50 to 150 mg of the polymorph Form A of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2- ylamino)-8/-/-pyrido[2,3-c]pyrimidin-7-one. In other embodiments, the capsule comprises from 50 to 100 mg of the polymorph Form A of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl- pyridin-2-ylamino)-8/-/-pyrido[2,3-c]pyrimidin-7-one.
- the capsule comprises from 75 to 150 mg of the polymorph Form A of 6-acetyl-8-cyclopentyl-5-methyl-2-(5- piperazin-1-yl-pyridin-2-ylamino)-8/-/-pyrido[2,3-c]pyrimidin-7-one
- the invention provides a method of treating cancer in a mammal, including a human, comprising administering to the mammal a therapeutically effective amount of a pharmaceutical composition of the invention.
- the pharmaceutical composition is administered in a capsule.
- the capsule may comprise from 0.1 to 200 mg of the polymorph Form A of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin- 2-ylarTiino)-8/-/-pyrido[2,3-c ]pyrimidin-7-one free base.
- the capsule may comprise from 25 to 150 mg of the polymorph Form A of 6-acetyl-8-cyclopentyl-5-methyl-2-(5- piperazin-1-yl-pyridin-2-ylamino)-8/-/-pyrido[2,3-c ]pyrimidin-7-one free base.
- the capsule may comprise from 50 to 150 mg of the polymorph Form A of 6- acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1 -yl-pyridin-2-ylamino)-8H-pyrido[2,3-c ]pyrimidin-7- one free base.
- Techniques for characterizing the crystalline free base of compound 1_ according to the invention include, but are not limited to, powder X-ray diffractometry (PXRD), solid state NMR (ssNMR), differential scanning calorimetry (DSC), vibrational spectroscopy (e.g. , I R and Raman spectroscopy), polarized light microscopy (PLM), scanning electron microscopy (SEM), hot stage optical microscopy, electron crystallography, single crystal X-ray diffractometry, quantitative analysis, particle size analysis (PSA) (e.g. , particle size, particle size distribution (PSD), and particle shape), specific surface area (SSA) analysis, surface energy analysis (e.g., inverse gas chromatography or IGC), by solubility studies and dissolution studies, or a combination of these techniques.
- PXRD powder X-ray diffractometry
- ssNMR solid state NMR
- DSC differential scanning calorimetry
- vibrational spectroscopy e.g. , I R
- the invention provides methods of making the free base of compound 1_ having a large primary particle size, as described herein.
- One method involves dissolving the small particle size free base of compound 1. in mixture of a first solvent and a second solvent and heating to achieve dissolution, cooling to appropriate temperature, providing seed crystals of compound 1. free base (Form A), followed by crystallization to provide the large particle size free base of compound
- the small particle size free base used in this process may be isolated from a traditional salt break procedure, e.g., by acidic hydrolysis of the intermediate vinyl ether to provide an acid addition salt, followed by basification, as described in Example 5.
- the invention provides a method of making the large particle size free base of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8/-/- pyrido[2,3-c ]pyrimidin-7-one (Form A), comprising: (a) suspending 6-acetyl-8-cyclopentyl-5- methyl-2-(5-piperazin-1 -yl-pyridin-2-ylamino)-8/-/-pyrido[2,3-c ]pyrimidin-7-one free base in mixture of a first solvent and a second solvent and heating to achieve dissolution; (b) cooling to an appropriate temperature and providing seed crystals of 6-acetyl-8-cyclopentyl-5-methyl-2-(5- piperazin-1-yl-pyridin-2-ylamino)-8/-/-pyrido[2,3-c ]pyrimidin-7-one free base (Form A), compris
- the invention provides a method of making the large particle size free base of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8/-/- pyrido[2,3-c ]pyrimidin-7-one (Form A), comprising: (a) suspending 6-acetyl-8-cyclopentyl-5- methyl-2-(5-piperazin-1 -yl-pyridin-2-ylamino)-8/-/-pyrido[2,3-c ]pyrimidin-7-one free base in mixture of n-butanol and anisole and heating to about 95-100°C to achieve dissolution; (b) cooling to about 80 °C and providing seed crystals of 6-acetyl-8-cyclopentyl-5-methyl-2-(5- piperazin-1-yl-pyridin-2-ylamino)-8/-/-pyrido[2,3-c]pyrimidin-7-
- Another method involves acidic hydrolysis of the intermediate vinyl ether in a mixture of water and a first solvent, which may require heating to obtain dissolution, addition of a second solvent and basification to provide a second mixture comprising the free base generated in situ, heating if required to obtain dissolution and to distill off water, cooling to appropriate temperature, providing seed crystals of compound 1_ free base (Form A), followed by crystallization to provide the free base of compound 1_ having a large primary particle size
- the invention provides a method of making the large particle size free base of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8/-/- pyrido[2,3-c]pyrimidin-7-one (Form A), comprising: (a) suspending 4- ⁇ 6-[6-(1-butoxyl-vinyl)-8- cyclopentyl-5-methyl-7-oxo-7,8-dihydropyrido[2,3-c]pyrimidin-2-ylamino]-pyridin-3-yl ⁇ -piperazine- 1-carboxylic acid te/f-butyl ester in a mixture of water and a first solvent and heating to achieve dissolution; (b) addition of acid and reaction to produce the acid addition salt of 6-acetyl-8- cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamin
- the invention provides a method of making the large particle size free base of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8/-/- pyrido[2,3-c]pyrimidin-7-one (Form A), comprising: (a) suspending 4- ⁇ 6-[6-(1-butoxyl-vinyl)-8- cyclopentyl-5-methyl-7-oxo-7,8-dihydropyrido[2,3-c]pyrimidin-2-ylamino]-pyridin-3-yl ⁇ -piperazine- 1-carboxylic acid terf-butyl ester in a mixture of water and n-butanol and heating to about 70°C to achieve dissolution; (b) addition of concentrated HCI and heating at about 70°C for 4-6 hrs; (c) addition of anisole and aqueous NaOH to achieve a biphasic mixture having
- the method provides the free base of compound 1_ having a specific surface area of ⁇ 2 m 2 /g. In other embodiments of each of the foregoing methods, the method provides the free base of compound 1_ having a specific surface area of ⁇ 1 m 2 /g. In other embodiments of each of the foregoing methods, the method provides the free base of compound 1_ having a primary particle size of from about 5 ⁇ to about 150 ⁇ , preferably from about 10 ⁇ to about 100 ⁇ , and more preferably from about 15 ⁇ to about 80 ⁇ . In other embodiments of each of the foregoing methods, the method provides the free base of compound 1.
- the method provides the free base of compound 1_ having a primary particle size distribution ratio of (D90-D10)/D50 of from about 2 to about 3.
- the method provides the free base of compound 1. having a volume mean diameter (D[4,3]) of from about 15 ⁇ to about 125 ⁇ .
- the invention provides the free base of compound 1., as described herein, prepared according to one of these methods.
- the invention provides the crystalline free base of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin- 2-ylamino)-8/-/-pyrido[2,3-c]pyrimidin-7-one (Form A), prepared according to any of the methods described herein.
- the free base prepared by the methods described herein may be characterized by its SSA, PSA, or surface energy, or a combination of these methods, alone or in further combination with PXRD or ssNMR.
- the crystalline free base has a residual solvent content of between 0.05-0.25 wt% anisole and/or between 0.05-0.25 wt% n-butanol. In other such embodiments, the crystalline free base has a residual solvent content of ⁇ 0.5 wt% anisole and ⁇ 0.5 wt% n-butanol, and preferably ⁇ 0.25 wt% anisole and ⁇ 0.25 wt% n-butanol.
- the first solvent is an alcohol and the second solvent is an aromatic solvent.
- Suitable alcohols include, but are not limited to, relatively high boiling alcohols such as n-butanol, t-butanol, n-propanol, pentanol, 1 ,4-butanediol or propylene glycol, and the like.
- Suitable aromatic solvents include, but are not limited to, anisole, mesitylene, m-xylene, chlorobenzene, pyridine, and the like.
- the solvent mixture comprises 10% alcohol, 15% alcohol,
- the solvent mixture comprises 90% aromatic, 85% aromatic, 80% aromatic, 75% aromatic, 70% aromatic, 65% aromatic, 60% aromatic, 55% aromatic, 50% aromatic, 40% aromatic, 30% aromatic, or ⁇ 30% aromatic, with the balance being the alcohol solvent.
- the first solvent is n-butanol.
- the second solvent is anisole.
- the first solvent is n-butanol and the second solvent is anisole.
- the solvent mixture comprises 10% n-butanol/anisole, 15% n-butanol/anisole, 20% n-butanol/anisole, 25% n-butanol/anisole, 30% n-butanol/anisole, 35% n-butanol/anisole, 40% n-butanol/anisole, 45% n-butanol/anisole, 50% n-butanol/anisole, 60% n-butanol/anisole, 70% n-butanol/anisole, or >70% n-butanol/anisole.
- the solvent mixture comprises from about 20 to about 50% n-butanol/anisole. In a particularly preferred embodiment, the solvent mixture comprises about 40%
- the methods may include heating or cooling to temperatures above or below room temperature.
- the reaction mixtures may be heated to temperatures ranging from about 30°C to about 150°C, and more frequently from about 50°C to about 120°C to achieve dissolution.
- it may be desirable to cool the reaction mixture to a temperature that is at or below room temperature, for example between about 0°C and about 30°C, preferably to about 5°C, about 10°C, about 15°C, or about 20°C.
- the free base of compound 1. is polymorph Form A having a powder X-ray diffraction pattern comprising a peak at diffraction angle (2 ⁇ ) of 10.1 ⁇ 0.2.
- the crystalline form has a powder X-ray diffraction pattern comprising peaks at diffraction angles (2 ⁇ ) of 10.1 ⁇ 0.2 and 22.5 ⁇ 0.2.
- the crystalline form has a powder X-ray diffraction pattern comprising peaks at diffraction angles (2 ⁇ ) of 5.1 ⁇ 0.2, 10.1 ⁇ 0.2, and 22.5 ⁇ 0.2.
- the crystalline form has a powder X-ray diffraction pattern comprising peaks at diffraction angles (2 ⁇ ) of 5.1 ⁇ 0.2, 10.1 ⁇ 0.2, 19.7 ⁇ 0.2, and 22.5 ⁇ 0.2.
- the crystalline form has a powder X-ray diffraction pattern comprising peaks at diffraction angles (2 ⁇ ) of 5.1 ⁇ 0.2, 10.1 ⁇ 0.2, 17.1 ⁇ 0.2, 19.7 ⁇ 0.2, and 22.5 ⁇ 0.2.
- the crystalline form has a powder X-ray diffraction pattern comprising peaks at diffraction angles (2 ⁇ ) of 5.1 ⁇ 0.2, 10.1 ⁇ 0.2, 1 1.5 ⁇ 0.2, 17.1 ⁇ 0.2, 19.7 ⁇ 0.2, and 22.5 ⁇ 0.2.
- the crystalline form has a powder X-ray diffraction pattern comprising peaks at diffraction angles (2 ⁇ ) of 5.1 ⁇ 0.2, 10.1 ⁇ 0.2, 1 1.5 ⁇ 0.2, 17.1 ⁇ 0.2, 18.7 ⁇ 0.2, 19.7 ⁇ 0.2, and 22.5 ⁇ 0.2.
- the crystalline form has a powder X-ray diffraction (PXRD) pattern comprising peaks at diffraction angles (2 ⁇ ) essentially the same as shown in Figure 1.
- the invention provides a crystalline free base of compound 1., wherein the crystalline free base is a polymorph Form B of the free base of compound 1_.
- the crystalline form has a powder X-ray diffraction pattern comprising a peak at diffraction angle (2 ⁇ ) of 6.0 ⁇ 0.2.
- the crystalline form has a powder X-ray diffraction pattern comprising peaks at diffraction angles (2 ⁇ ) of 6.0 ⁇ 0.2 and 19.8 ⁇ 0.2.
- the crystalline form has a powder X-ray diffraction pattern comprising peaks at diffraction angles (2 ⁇ ) of 6.0 ⁇ 0.2, 19.8 ⁇ 0.2, and 26.7 ⁇ 0.2. In further embodiments, the crystalline form has a powder X-ray diffraction pattern comprising peaks at diffraction angles (2 ⁇ ) of 6.0 ⁇ 0.2, 16.4 ⁇ 0.2, 19.8 ⁇ 0.2, and 26.7 ⁇ 0.2.
- the crystalline form has a powder X-ray diffraction pattern comprising peaks at diffraction angles (2 ⁇ ) of 6.0 ⁇ 0.2, 12.8 ⁇ 0.2, 16.4 ⁇ 0.2, 19.8 ⁇ 0.2, and 26.7 ⁇ 0.2.
- the crystalline form has a powder X-ray diffraction pattern comprising peaks at diffraction angles (2 ⁇ ) of 6.0 ⁇ 0.2, 12.8 ⁇ 0.2, 16.4 ⁇ 0.2, 19.8 ⁇ 0.2, 22.6 ⁇ 0.2, and 26.7 ⁇ 0.2.
- the crystalline form has a powder X-ray diffraction pattern comprising peaks at diffraction angles (2 ⁇ ) of 6.0 ⁇ 0.2, 10.9 ⁇ 0.2, 12.8 ⁇ 0.2, 16.4 ⁇ 0.2, 19.8 ⁇ 0.2, 22.6 ⁇ 0.2, and 26.7 ⁇ 0.2.
- the crystalline form has a PXRD pattern comprising peaks at diffraction angles (2 ⁇ ) essentially the same as shown in Figure 3.
- the powder X-ray diffraction (PXRD) pattern of free base polymorph Form B is shown in Figure 3 and the corresponding data is tabulated in Table 3.
- a sample of a free base was placed into a cavity located on a planar surface of the holder, and a glass slide was used to level the surface of the sample.
- the holder which contains the sample, was placed in the diffractometer, and the source of the X-ray beam irradiated the sample, initially at a small angle relative to the planar surface of the holder.
- the X-ray beam was subsequently moved through an arc in a stepwise manner, which successively increased the angle between the incident beam and the planar surface of the holder.
- the scintillation counter detected the amount of diffracted radiation, which was recorded as a function of 2 ⁇ (°).
- the instrument software displays the diffracted radiation results of the scan as intensity versus 2 ⁇ (°).
- Tables 1 and 3 list significant PXRD peaks (i.e., those exhibiting peak height to noise ratio greater than 3.5) for the free base of compound 1. having polymorph Form A or Form B, respectively.
- the list of characteristic peaks provided is not the only possible list of characteristic peaks. Persons of ordinary skill in the art of polymorph identification may choose other sets of characteristic peaks that will also distinguish one polymorph from another.
- Differences in PXRD patterns among separate measurements of the same polymorph may arise for many reasons.
- Sources of error include variations in sample preparation (e.g. sample height), instrument errors, calibration errors, and operator errors (including errors in determining peak locations).
- Preferential orientation i.e., a lack of random orientation of crystals in the PXRD sample, can result in significant differences in relative peak heights.
- Calibration errors and sample height errors often result in a shift of all of the peaks of the diffractogram in the same direction and by the same amount. Small differences in sample height on a flat holder may lead to large displacements in PXRD peak positions.
- sample height differences of 1 mm may lead to peak shifts as high as 1° 2 ⁇ , see Chen et al., J. Pharmaceutical and Biomedical Analysis (2001) 26:63.
- peak shifts among diffraction patterns resulting from systematic error can be eliminated by compensating for the shift (e.g., applying a correction factor to all peak position values) or by recalibrating the diffractometer.
- the same techniques can be used to compensate for differences among diffractometers so that PXRD peak positions obtained from two different instruments can be brought into agreement.
- the peak positions for a particular polymorph will usually agree to within about ⁇ 0.2° 2 ⁇ .
- the disclosed compounds embrace all pharmaceutically acceptable isotopic variations.
- An isotopic variation is a compound in which at least one atom is replaced by an atom having the same atomic number, but an atomic mass different from the atomic mass usually found in nature.
- Useful isotopes include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, and chlorine. Exemplary isotopes thus include, without limitation, 2 H, 3 H, 13 C, 14 C, 15 N, 17 0, 18 0, 32 P, 35 S, 18 F, and 36 CI.
- isotopes such as deuterium, i.e. 2 H
- isotopes such as deuterium, i.e. 2 H
- isotopic variations for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies.
- Isotopic variations of the disclosed compounds can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples using appropriate isotopic variations of suitable reagents.
- Pharmaceutically acceptable solvates of the disclosed compounds include those in which the solvent of crystallization may be isotopically substituted, e.g. D 2 0, d 6 -acetone, d 6 - DMSO.
- U.S. Patent No. 7,345, 171 reported that the free base of compound ⁇ , prepared by a traditional salt break procedure, had poor water solubility (9 ⁇ g/mL) at pH 7.9 and exhibited low bioavailability in animal studies. The free base was reported to be in its most stable crystal phase according to slurry experiments (i.e.., Form A).
- Figure 17 of U.S. Patent No. 7,345, 171 provided the water adsorption/desorption isotherms for the free base of Form A. As noted previously, this material corresponds to the small particle size free base of compound 1. described herein.
- the free base of compound 1. has a high propensity for punch sticking in the drug particle manufacturing process. As punch sticking is related to API surface area, API particle size control is critical for minimizing sticking during drug product manufacturing. In addition to issues with punch sticking, compound ⁇ free base isolated directly from a standard salt break process was found to be highly static prone and found to form large (approximately 500 microns) hard agglomerates that were not dispersed by sieving. Free base API with similarly poor physical properties was produced by free basing of the existing isethionate salt API or by neutralization of the in situ salt formed in the final step of the API synthesis. In either process, small API primary particles were produced due to the rapid crystallization caused by the dramatic change in solubility with adjustment of the pH. In all cases the free base was isolated as the more stable polymorph of Form A.
- Figure 6 shows a scanning electron microscopy (SEM) image of typical small primary particles formed by the free basing and neutralization experiments described above.
- SEM scanning electron microscopy
- anisole became the focus of additional crystallization and solubility studies as the particles produced were large and as anisole is an ICH Class III solvent.
- This screening study also identified pyridine, m-xylene, and mesitylene as potential solvent systems based on the particles produced, although none of these solvents also have the ICH class III listing similar to anisole.
- Particle sizes for the recrystallized materials were assessed using laser diffraction methods.
- Laser diffraction is recognized by standards and guidance agencies including ISO and ASTM and is widely used to determine particle size distributions.
- the sample is passed through a laser beam which results in laser light scattered at a range of angles. Detectors placed at fixed angles measure the intensity of light scattered at that position.
- a mathematical model (Mie or Fraunhoffer Theory) is then applied to generate a particle size distribution.
- the particle size was analyzed using the laser diffraction (or small angle light scattering) technique by dispersing the dry sample powder with compressed air. Specifically, the particle size distribution was analyzed using the Sympatec HELOS RODOS system equipped with a
- an Aspiros micro-dosing device was used, and the powder sample was dispersed with a dispersion pressure of 0.2bar.
- a suitable lens was selected to cover the particle size range of each sample.
- the median value is defined as the value where half of the population resides above this point, and half resides below this point.
- the median is called the D50.
- the D50 is the size in microns that splits the distribution with half above and half below this diameter.
- the expression Dv50 or D[v,0.5] is sometimes used for the median of a volume distribution.
- the mode is the peak of a frequency distribution.
- a particle distribution may include more than one mode, e.g., where the particles exist as primary particles and agglomerations.
- the span is sometimes used as a measurement of distribution width, and is defined as the ratio of (D[v,0.9]- D[v,0.1]) / D[v,0.5] or (D90-D10)/D50.
- the distribution width may also be characterized by citing one, two or preferably three values on the x-axis, typically some combination of the D10, D50, and D90.
- the D50 the median, has been defined above as the diameter where half of the population lies below this value.
- 90 percent of the distribution lies below the D90, and 10 percent of the population lies below the D10.
- D[4,3] refers to the volume mean or mass moment mean. Laser diffraction results are reported on a volume basis and the volume mean can be used to define the central point of the distribution. The D[4,3] value is sensitive to the presence of large particles in the distribution.
- the present invention also relates to pharmaceutical compositions comprising the free base polymorph Form A of compound ⁇ described herein.
- Pharmaceutical compositions of the present invention may, for example, be in a form suitable for oral administration as a tablet, capsule, pill, powder, sustained release formulations, solution, suspension, for parenteral injection as a sterile solution, suspension or emulsion, for topical administration as an ointment or cream or for rectal administration as a suppository.
- the pharmaceutical composition may be in unit dosage forms suitable for single administration of precise dosages.
- the pharmaceutical composition will include a conventional pharmaceutical carrier or excipient and a compound according to the invention as an active ingredient. In addition, it may include other medicinal or pharmaceutical agents, carriers, adjuvants, etc.
- Suitable pharmaceutical carriers include inert diluents or fillers, water and various organic solvents.
- the pharmaceutical compositions may, if desired, contain additional ingredients such as flavorings, binders, excipients and the like.
- excipients such as citric acid
- disintegrants such as starch, alginic acid and certain complex silicates
- binding agents such as sucrose, gelatin and acacia.
- lubricating agents such as magnesium stearate, sodium lauryl sulfate and talc are often useful for tableting purposes.
- Solid compositions of a similar type may also be employed in soft and hard filled gelatin capsules.
- Preferred materials include lactose or milk sugar and high molecular weight polyethylene glycols.
- the active compound therein may be combined with various sweetening or flavoring agents, coloring matters or dyes and, if desired, emulsifying agents or suspending agents, together with diluents such as water, ethanol, propylene glycol, glycerin, or combinations thereof.
- the disclosed compound may be administered alone or in combination with other drugs and will generally be administered as a formulation in association with one or more pharmaceutically acceptable excipients.
- excipient describes any ingredient other than compound 1. and its salts. The choice of excipient will to a large extent depend on the particular mode of administration.
- the disclosed compounds may be administered orally.
- Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, or buccal or sublingual administration may be employed by which the compound enters the blood stream directly from the mouth.
- Formulations suitable for oral administration include solid formulations such as tablets, capsules containing particulates, liquids, or powders, lozenges (including liquid-filled), chews, multi- and nano-particulates, gels, solid solution, liposome, films (including muco-adhesive), ovules, sprays and liquid formulations.
- Liquid formulations include suspensions, solutions, syrups and elixirs.
- Such formulations may be employed as fillers in soft or hard capsules and typically comprise a carrier, for example, water, EtOH, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents.
- Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet.
- the disclosed compounds may also be used in fast-dissolving, fast-disintegrating dosage forms such as those described in Liang and Chen, Expert Opinion in Therapeutic Patents (2001) 11 (6):981-986.
- the drug may make up from 1 wt % to 80 wt % of the dosage form, more typically from 5 wt % to 60 wt % of the dosage form.
- tablets generally contain a disintegrant.
- disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methylcellulose, microcrystalline cellulose, lower alkyl-substituted hydroxypropyl cellulose, starch, pregelatinized starch, and sodium alginate.
- the disintegrant will comprise from 1 wt % to 25 wt %, preferably from 5 wt % to 20 wt % of the dosage form.
- Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinized starch, hydroxypropyl cellulose, and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch, and dibasic calcium phosphate dihydrate.
- lactose monohydrate, spray-dried monohydrate, anhydrous and the like
- mannitol xylitol
- dextrose sucrose
- sorbitol microcrystalline cellulose
- starch and dibasic calcium phosphate dihydrate.
- Tablets may also optionally include surface-active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc.
- surface-active agents such as sodium lauryl sulfate and polysorbate 80
- glidants such as silicon dioxide and talc.
- surface-active agents may comprise from 0.2 wt % to 5 wt % of the tablet, and glidants may comprise from 0.2 wt % to 1 wt % of the tablet.
- Tablets also generally contain lubricants such as magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulfate.
- Lubricants generally comprise from 0.25 wt % to 10 wt %, preferably from 0.5 wt % to 3 wt % of the tablet.
- Other ingredients may include preservatives, anti-oxidants, flavors, and colorants.
- Tablet blends may be directly compressed to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting.
- the final formulation may comprise one or more layers and may be coated or uncoated. Exemplary tablets contain up to about 80 % drug, from about 10 wt % to about 90 wt % binder, from about 0 wt % to about 85 wt % diluent, from about 2 wt % to about 10 wt % disintegrant, and from about 0.25 wt % to about 10 wt % lubricant.
- Pharmaceutical Dosage Forms Tablets, Vol. 1 (1980).
- Solid formulations for oral administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted-, and programmed-release.
- suitable modified release formulations see US Patent No. 6, 106,864.
- Other useful release technologies such as high energy dispersions and osmotic and coated particles, see Verma et al, Pharmaceutical Technology On-line (2001) 25(2): 1-14.
- WO 00/35298 For a discussion of the use of chewing gum to achieve controlled release.
- the disclosed compounds may also be administered directly into the blood stream, into muscle, or into an internal organ.
- Suitable means for parenteral administration include intravenous, intra-arterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular, and subcutaneous.
- Suitable devices for parenteral administration include needle (including micro-needle) injectors, needle-free injectors and infusion techniques.
- Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates, and buffering agents (preferably to a pH of from 3 to 9), but for some applications, they may be more suitably formulated as a sterile non-aqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
- a suitable vehicle such as sterile, pyrogen-free water.
- the preparation of parenteral formulations under sterile conditions for example, by lyophilization, may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
- Exemplary parenteral administration forms include solutions or suspensions of active compounds in sterile aqueous solutions, for example, aqueous propylene glycol or dextrose solutions. Such dosage forms can be suitably buffered, if desired.
- solubility of the disclosed compounds used in the preparation of parenteral solutions may be increased by the use of appropriate formulation techniques, such as the incorporation of solubility-enhancing agents.
- Formulations for parenteral administration may be formulated to be immediate and/or modified release as described above.
- the disclosed compounds may be formulated in a more solid form for administration as an implanted depot providing long-term release of the active compound.
- the compounds of the invention may also be administered topically to the skin or mucosa, either dermally or transdermally.
- Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibers, bandages, and microemulsions.
- Liposomes may also be used.
- Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol.
- Topical formulations may also include penetration enhancers. See, for example, Finnin and Morgan, J Pharm Sci (1999) 88(10):955-958.
- Topical administration examples include delivery by iontophoresis, electroporation, phonophoresis, sonophoresis and needle-free (e.g. POWDERJECT) or micro-needle injection.
- Formulations for topical administration may be formulated to be immediate and/or modified release as described above.
- the disclosed compounds can also be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids) from a dry powder inhaler or as an aerosol spray from a pressurized container, pump, spray, atomizer (preferably an atomizer using electrohydrodynamics to produce a fine mist), or nebulizer, with or without the use of a suitable propellant, such as dichlorofluoromethane.
- a dry powder either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids
- atomizer preferably an atomizer using electrohydrodynamics to produce a fine mist
- nebulizer with or without the use of a suitable propellant, such as dichlorofluoromethane.
- the pressurized container, pump, spray, atomizer, or nebulizer contains a solution or suspension, which comprises the active compound, an agent for dispersing, solubilizing, or extending release of the active compound (e.g., EtOH or aqueous EtOH), one or more solvents, which serve as a propellant, and an optional surfactant, such as sorbitan trioleate or an oligolactic acid.
- a solution or suspension which comprises the active compound, an agent for dispersing, solubilizing, or extending release of the active compound (e.g., EtOH or aqueous EtOH), one or more solvents, which serve as a propellant, and an optional surfactant, such as sorbitan trioleate or an oligolactic acid.
- the drug product Prior to use in a dry powder or suspension formulation, the drug product is micronized to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenization, or spray drying.
- comminuting method such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenization, or spray drying.
- Capsules, blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the active compound, a suitable powder base such as lactose or starch, and a performance modifier such as L-leucine, mannitol, or magnesium stearate.
- the lactose may be anhydrous or, preferably, monohydrated.
- Other suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
- a suitable solution formulation for use in an atomizer using electrohydrodynamics to produce a fine mist may contain from 1 ⁇ g to 20 mg of the compound of the invention per actuation and the actuation volume may vary from 1 ⁇ to 100 ⁇ .
- a typical formulation may comprise compound 1_, propylene glycol, sterile water, EtOH, and NaCI.
- Alternative solvents, which may be used instead of propylene glycol, include glycerol and polyethylene glycol.
- Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, poly(DL-lactic-coglycolic acid (PGLA).
- PGLA poly(DL-lactic-coglycolic acid
- Suitable flavors, such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium may be added to formulations intended for inhaled/intranasal administration.
- the dosage unit is determined by means of a valve that delivers a metered amount.
- Units in accordance with the invention are typically arranged to administer a metered dose or "puff" containing from 100 to 1000 ⁇ g of the active pharmaceutical ingredient.
- the overall daily dose will typically be in the range 100 ⁇ g to 10 mg which may be administered in a single dose or, more usually, as divided doses throughout the day.
- the active compounds may be administered rectally or vaginally, for example, in the form of a suppository, pessary, or enema. Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate.
- Formulations for rectal/vaginal administration may be formulated to be immediate and/or modified release as described above.
- the disclosed compounds may also be administered directly to the eye or ear, typically in the form of drops of a micronized suspension or solution in isotonic, pH-adjusted, sterile saline.
- Other formulations suitable for ocular and aural administration include ointments, biodegradable (e.g. absorbable gel sponges, collagen) and non-biodegradable (e.g. silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes.
- a polymer such as crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer (e.g., hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose), or a heteropolysaccharide polymer (e.g., gelan gum), may be incorporated together with a preservative, such as benzalkonium chloride.
- a preservative such as benzalkonium chloride.
- Such formulations may also be delivered by iontophoresis.
- Formulations for ocular/andial administration may be formulated to be immediate and/or modified release as described above.
- the disclosed compounds may be combined with soluble macromolecular entities such as cyclodextrin or polyethylene glycol-containing polymers to improve their solubility, dissolution rate, taste masking, bioavailability and/or stability.
- Drug-cyclodextrin complexes for example, are found to be generally useful for most dosage forms and administration routes. Both inclusion and non-inclusion-complexes may be used.
- the cyclodextrin may be used as an auxiliary additive, i.e. as a carrier, diluent, or solubilizer.
- Alpha-, beta- and gamma-cyclodextrins are commonly used for these purposes. See, for example, International Patent Applications WO 91/11 172, WO 94/02518, and WO 98/55148.
- the therapeutically effective dose of compound 1_ will vary from approximately
- Typical adult doses will be approximately 0.1 mg to approximately 3000 mg per day.
- the quantity of active component in a unit dose preparation may be varied or adjusted from approximately 0.1 mg to approximately 500 mg, preferably from about 0.6 mg to 100 mg according to the particular application and the potency of the active component.
- the composition can, if desired, also contain other compatible therapeutic agents.
- a subject in need of treatment is administered a dosage of about 0.6 to about 500 mg per day, either singly or in multiple doses over a 24-hour period. Such treatment may be repeated at successive intervals for as long as necessary.
- disorders or conditions caused by abnormal cell proliferation include cancer and vascular smooth muscle proliferation associated with atherosclerosis, post-surgical vascular stenosis and restenosis, and endometriosis.
- Autoimmune diseases include psoriasis, inflammation-like rheumatoid arthritis, lupus, type 1 diabetes, diabetic nephropathy, multiple sclerosis, glomerulonephritis, and organ transplant rejection, including host versus graft disease.
- the present invention provides a method of treating abnormal cell growth in a mammal, including a human, in need of such treatment comprising, administering to said mammal a therapeutically effective amount of a crystalline free base of compound 1_ according to the invention described herein.
- the free base is a polymorph of Form A.
- the abnormal cell growth is cancer, including both solid tumors and hematological malignancies.
- the cancer is selected from breast cancer, ovarian cancer, cervical cancer, endometrial cancer, prostate cancer, testicular cancer, pancreatic cancer, esophageal cancer, head and neck cancer, gastric cancer, bladder cancer, lung cancer (e.g., adenocarcinoma, NSCLC and SCLC), bone cancer (e.g., osteosarcoma), colon cancer, rectal cancer, thyroid cancer, brain and central nervous system cancers, glioblastoma, neuroblastoma, neuroendocrine cancer, rhabdoid cancer, keratoacanthoma, epidermoid carcinoma, seminoma, melanoma, sarcoma (e.g., liposarcoma), bladder cancer, liver cancer (e.g., hepatocellular carcinoma), kidney cancer (e.g., renal cell carcinoma), myeloid disorders (e.g., AML
- PXRD data were collected according to the following protocol. A sample (2 mg) was placed on a microscopic slide with zero background. The sample was then placed in a Discover D8 (Bruker AXS Instruments) equipped with a GADDS detector. The system used a copper X- ray source maintained at 40kV and 40 mA to provide CUa1 emission at 1.5406 angstroms. Data were collected from 4 to 40 °2 ⁇ using a step scan of 0.02° with a step time of 60.1 seconds. Diffraction peaks are typically measured with an error of ⁇ 0.2 degrees (2 ⁇ ).
- SSNMR data were collected according to the following protocol. Spectra were collected on Bruker-Biospin 4 mm and 7 mm BL CPMAS probe positioned into a wide-bore Bruker- Biospin Avance III 500 MHz NMR spectrometer. The 4 mm rotors were oriented at the magic angle and spun at 15.0 kHz. The 7 mm rotors were oriented at the magic angle and spun at 7.0 kHz. All spectra were acquired at ambient conditions (temperature uncontrolled).
- DSC measurements are carried out using a Q1000, Thermal Analysis Instruments.
- a sample is placed in a hermetically sealed aluminum pan with a pinhole.
- a typical sample weight is 1.6 mg.
- the sample is equilibrated to 25°C and then ramped to 250°C at a scan rate of 10°C/min. Dry nitrogen is used as the purge gas.
- BET Brunauer, Emmet and Teller
- SSA Specific Surface area
- SSA measurements were collected according to the following protocol. Monolayer formation of gas molecules on the crystal surface was used to determine the specific surface area of a dry powder of active pharmaceutical ingredient.
- the sample was made free of moisture and atmospheric vapours by applying heat and purging with nitrogen gas. The sample temperature was then reduced to that of liquid nitrogen for the adsorbate gas (nitrogen) to be adsorbed. The quantity of adsorbed gas and pressure data were used to generate an adsorption isotherm plot.
- the data were then converted into specific surface area value using a mathematical algorithm based on the so-called Brunauer, Emmett, and Teller (BET) theory (see, e.g., J. Am. Chem. Soc, 1938, 60:309). Specific surface area was measured using a static multi-point or single-point gas adsorption method, as fully described in ISO 9277:2010 and in the experimental below.
- BET Brunauer, Emmett, and Teller
- a plot of the retention time (corrected for the 'dead volume' of interstitial space within the packed column) versus a function of the cross sectional area and surface tension of the alkane vapour probe molecules used yielded a line with a slope indicative of the surface energy of the solid powder under examination.
- Step A Preparation of 4-(6-nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester
- Step B Preparation of 4-(6-amino-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester
- the product mixture was filtered and washed with ethyl acetate (2 x 1.5 mL). The combined filtrate was concentrated under reduced pressure to a volume of 6 mL (2 vol.). To the solution was added n-heptane (54 mL, 4.5 vol.) and the mixture was distilled under reduced pressure to a volume of 6 mL (2 vol.). To the solution was added n-heptane (54 mL, 4.5 vol.). The resulting thick slurry was cooled to 20-25°C and allowed to stir for 2 hours. The slurry was filtered and the filter cake washed with n-heptane (36 mL, 3 vol.). The solids were allowed to dry overnight in a vacuum oven at 50-55°C.
- Acetic anhydride (6.8 mL, 2.0 equiv) was added to the reaction mixture. The reaction was allowed to react at 65 °C until starting material was consumed (usually 1-2 hours). The reaction mixture was cooled to 20°C and H 2 0 (100 mL, 10 vol) was added to dissolve triethylamine-HBr salts and precipitate out 2-chloro-8-cyclopentyl-5-methyl-8/-/- pyrido[2,3-c]pyrimidin-7-one. The material was granulated at 20 °C for 1 hour.
- Step C Preparation of 6-bromo-2-chloro-8-cvclopentyl-5-methyl-8/-/-pyridor2,3-dlpyrimidin-7-one
- 2-chloro-8-cyclopentyl-5-methyl-8/-/-pyrido[2,3-c]pyrimidin-7- one (9.35 g, 1.0 equiv.) along with acetonitrile (65 mL, 7.0 vol).
- N-Bromosuccinimide (9.67 g, 1.5 equiv.
- oxalic acid (0.65 g, 0.2 equiv.
- the product was further purified by recrystallization from toluene and n-heptanes.
- Toluene (60 mL, 6 vol) and 6-bromo-2-chloro-8-cyclopentyl-5-methyl-8/-/-pyrido[2,3-c]pyrimidin- 7-one (10.00 g, 1 equiv) were added to a reaction vessel and heated to 80°C.
- the warm reaction mixture was filtered through an appropriate cartridge to ensure the removal of insoluble Pd and other insoluble contaminants.
- the filter cartridge was washed with 80°C toluene (5 mL, 0.5 vol). The slurry was cooled to 25°C at 1 °C/min.
- n-Heptane 70 mL, 7 vol was added to the reaction slurry at 1 mL/min. The slurry was further cooled to 0°C at 1 °C/min. The slurry was granulated at 0°C for at least 1 hour.
- a dry, nitrogen purged reactor was charged with tetrahydrofuran (900 ml_, 15 ml_/g). The batch temperature was set at 20°C and agitation at 250 RPM was started. The reactor was charged with 4-(6-amino-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester (63.4g, 0.2278 moles, 1.3 equiv.) and the mixture held at 20°C for 30 min to dissolve the starting material. The reactor was charged with isopropylmagnesium chloride (93.9 g, 0.193 moles, 1 st charge 1.1 eq) (2.0M in THF, 1.1 equiv.) by pump over 30 min.
- the batch was maintained at 20°C for 40 min.
- the reactor was charged with 6-bromo-2-chloro-8-cyclopentyl-5-methyl-8/-/-pyrido[2,3- c]pyrimidin-7-one (60.1g, 0.1755 moles, 1 eq.) all at once and rinsed with THF (50 ml_ rinse).
- An additional charge of isopropylmagnesium chloride (93.9g, 0.193 moles, 1.1 eq - 2nd charge (2.0M in THF, 1.1 equiv.) was added by pump over 30 min.
- the batch was held at 20°C for 90 min. and then heated from 20°C to 60°C.
- Diisopropylethylamine (5.3g, 0.041 moles, 2.4 eq) was added and the mixture was sparged with nitrogen through a sparge tube for 30 minutes.
- Palladium acetate (0.16g, 0.00068 moles, 0.0400 eq) and bis(2-diphenyphosphinophenyl)ether (0.45g, 0.00082 moles, 0.04800 eq) were added.
- the mixture was heated to 95°C over 30 minutes and the batch was stirred at 95°C for 2 hours.
- the mixture was cooled to 80°C and sampled to monitor reaction completion. Following completion, water (15 mL, 1.5 ml_/g) and 1-butanol (30 mL, 3 ml_/g) were added.
- the solution was filtered through a 0.45 micron filter to remove precipitated palladium.
- the mixture was cooled to 50°C over one hour and held at 50°C for three hours.
- the mixture was cooled to 30°C over three hours and held at 30°C for two hours, then cooled to 20°C over four hours and held at 20°C for four hours.
- the slurry was filtered and washed with 1-butanol (10 mL, 1 ml_/g).
- the filter cake was blown down and the mixture was charged with 1-butanol (10 mL, 1 mL/g) and the slurry was stirred at 20°C for 1 hour.
- the filter cake was blown down.
- the intermediate butoxyl-vinyl ether may be isolated in one of several polymorphic forms.
- Form A was isolated as the kinetic product in the absence of seeding, while Form B was isolated in a few cases but is rarely observed.
- Form C The most stable crystalline form of the butoxyl- vinyl ether, Form C, was obtained by seeding the reaction mixture with Form C crystals. Any of these polymorphic forms may be utilized in the preparation of Compound 1. free base, but polymorph Form C of the butoxyl-vinyl ether is preferred for ease of filterability.
- the reaction mixture was cooled to 35°C, and a mixture of 5 wt% sodium hydroxide solution was added in portions to the reactor to raise the reaction mixture to a pH > 9.
- the reactor was cooled to between 20°C and 25°C, granulated, and filtered. The cake was washed with water followed by acetone and dried under vacuum.
- This method generated the small primary particle size free base of compound 1_, which was equivalent to the material prepared from treatment of the compound 1_ hydrochloride salt with aqueous NaOH in Example 4 of WO 2005/005426.
- compound 1_ free base (20g, 44.69 mmol, 1.0 eq.), prepared according to Example 5, followed by 1-butanol (320 ml, 16 ml/g) and anisole (480 ml, 24 ml/g).
- 1-butanol 320 ml, 16 ml/g
- anisole 480 ml, 24 ml/g
- the yellow slurry was warmed to between 95°C and 100°C to achieve dissolution.
- the reactor was cooled to 80°C.
- a seed slurry containing compound 1_ free base (Form A) seed crystals (0.1 g, 0.2 mmol, 0.005 eq.) suspended in 1-butanol (5 mL, 0.25 mL/g of starting material) was charged to induce crystallization.
- the resulting slurry was stirred at 80°C for 3 hours.
- the slurry was cooled to 10°C at 0.2 °C/min over 350 minutes, granulated, and filtered.
- the cake was washed with anisole followed by heptane, and dried under vacuum.
- the phases were separated and the organic phase was washed with water twice.
- the batch was then heated to 80°C and speck-free filtered into the crystallizing vessel, rinsing the filter with butanol.
- the batch was then distilled to remove water and achieve a temperature of 120°C.
- the batch was then cooled to 80 °C and seeded with a seed slurry of compound 1_free base (Form A) seed crystals (0.015 g, 0.033 mmol, 0.1 wt.% wrt compound 1j and 1-BuOH (10 mL, 0.5 mL/g).
- the batch was then cooled to 30°C at 0.2°C /min and then ripened with three cycles where the temperature was stepped down by 10°C each time. On the final cycle, the batch was cooled to 10°C, granulated and filtered.
- the cake was washed with twice with heptane and dried under vacuum. After drying, the sample was confirmed to be
- Carbon spectra on Form A were acquired on a 4 mm rotor for 2048 scans with recycle delay of 25 seconds and 2 ms of cross polarization. 100 kHz of proton decoupling was applied during acquisition. Carbon spectra on Form B were acquired on a 4 mm rotor for 2048 scans for 128 scans were collected with recycle delay of 4.5 seconds with 2 ms of cross polarization. 70 kHz of proton decoupling and total suppress of spinning sideband (TOSS) was applied during acquisition.
- TOSS total suppress of spinning sideband
- CPMAS proton phase modulated decoupled cross-polarization magic angle spinning
- the carbon spectrum of compound 1_ Form A was acquired for 512 scans with a 25 second recycle delay.
- the spectrum is shown Figure 2 and the data is tabulated in Table 2.
- the carbon spectrum of compound ⁇ Form B was acquired for 2048 scans with a 4.5 second recycle delay.
- the carbon spectra were referenced using an external standard of crystalline adamantane, setting its upfield resonance to 29.5 ppm.
- the spectrum is shown Figure 4 and the data is tabulated in Table 4.
- the particle size was analyzed using the laser diffraction (or small angle light scattering) technique by dispersing the dry sample powder with compressed air. Specifically, the particle size distribution was analyzed using the Sympatec HELOS RODOS system equipped with a Vibri dry powder feeder. The powder sample was dispersed with a dispersion pressure of 0.5bar. In some instances, an Aspiros micro-dosing device was used, and the powder sample was dispersed with a dispersion pressure of 0.2bar. A suitable lens was selected to cover the particle size range of each sample.
- Comparative data for four batches of API are provided in Table 1 1 below, using either the Vibri or Aspiros devices to disperse the sample.
- Batch No. 4 had a D90 of around 75 ⁇ , whereas Batch Nos. 1 and 2 both had a D90 of approximately 45 ⁇ .
- the laser diffraction particle size data confirms the SEM observations for these batches.
- Figure 5 provides a SEM (200x magnification) image of compound 1_ free base Form A recrystallized from 40% BuOH/anisole.
- Figure 6 provides a SEM (1500x magnification) image of compound 1_ free base Form A isolated from a standard free basing process
- the MASS Magnetic Adhesion Screen for Sticking
- Punch was developed in-house to quantitatively assess the sticking propensity of tablet formulations by weighing the amount of adhered powder on removable punch tip after a series of compressions. This test enables formulators to objectively and quickly evaluate the risk of punch sticking during drug product development and troubleshoot sticking observed during clinical tablet manufacturing.
- the MASS Punch profile for compound 1_ free base mixed in the standard blend showed a positive response. Photos of the punch tips at the end of the compression runs confirmed that powder adhered to the tips (data not shown). For reference, a control sample of the standard blend is not sticky and would have less than 10 ⁇ g powdered adhered. The test method was found to rank the sticking propensity of new API lots relative to those of known materials. Specific Surface Area (SSA) Measurement (BET Nitrogen)
- SSA Specific Surface Area
- Sample tube 3/8" mm flat bottom cell with glass filler rods
- Isothermal collection points 1 1 point BET in the range 0.05-0.30 P/Po
- Isothermal data analysis range 7 point BET in the range 0.05-0.20 P/Po
- the mass of sample varies according to the particle size of the test sample. For samples where the particle size is relatively small, approximately 0.50g of material was required to 3 ⁇ 4 fill the cell bulb, and where the particle size of the sample is relatively large, 0.75g of material was required to 3 ⁇ 4 fill the cell bulb.
- the specific surface area was reported in the range 0.05-0.20 P/Po using 7 point BET from a triplicate determination.
- the sample mass, specific surface area, BET constant ('C value) and correlation coefficient for each replicate were determined.
- Table 12 provides BET-N2 SSA for four batches of compound 1_ free base API, one comprising the small primary particle size API prepared by the traditional salt break method (batch 5), and three batches comprising the large particle size API prepared according to the present invention.
- Batch 5 contained compound 1. free base having small primary particles and large agglomerates, which was very static-prone and sticky.
- Batch 6 was prepared using temperature cycling and had a typical particle size distribution (PSD) for the large particle size free base of compound 1_, with a VMD of approximately 17 ⁇ .
- PSD particle size distribution
- Batch 7 demonstrated a similar PSD to batch 6.
- Batch 8 is a representative ICH batch of the large particle size free base of compound 1., also prepared by temperature cycling. The same batches were used in the surface energy determinations below.
- a sufficient quantity of sample was packed into a silanised glass column with the powder mass secured within the column by glass wool plugs inserted at both ends.
- the column was conditioned by flowing a stream of dry nitrogen through the powder mass for sufficient time for any surface adsorbates to be removed. Measurements were made by injecting a series of alkane vapour probes (Nonane, Octane, Heptane and Hexane) into the carrier gas stream at concentrations low enough to assume infinite dilution of the alkane vapour in the nitrogen stream and recording the time taken for each vapour to elute through the column.
- alkane vapour probes Nonane, Octane, Heptane and Hexane
- a plot of the retention time (corrected for the 'dead volume' of interstitial space within the packed column) versus a function of the cross sectional area and surface tension of the alkane vapour probe molecules used yields a line with a slope indicative of the surface energy of the solid powder under examination.
- Table 13 provides Dispersive Surface Energy (mJ/m 2 ) data generated for the four batches of compound 1_ free base, i.e., batches 5-8, described above with respect to the SSA data.
- Batch 5 is the small particle size free base
- batches 6-8 include the large particle size of the free base API.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description




















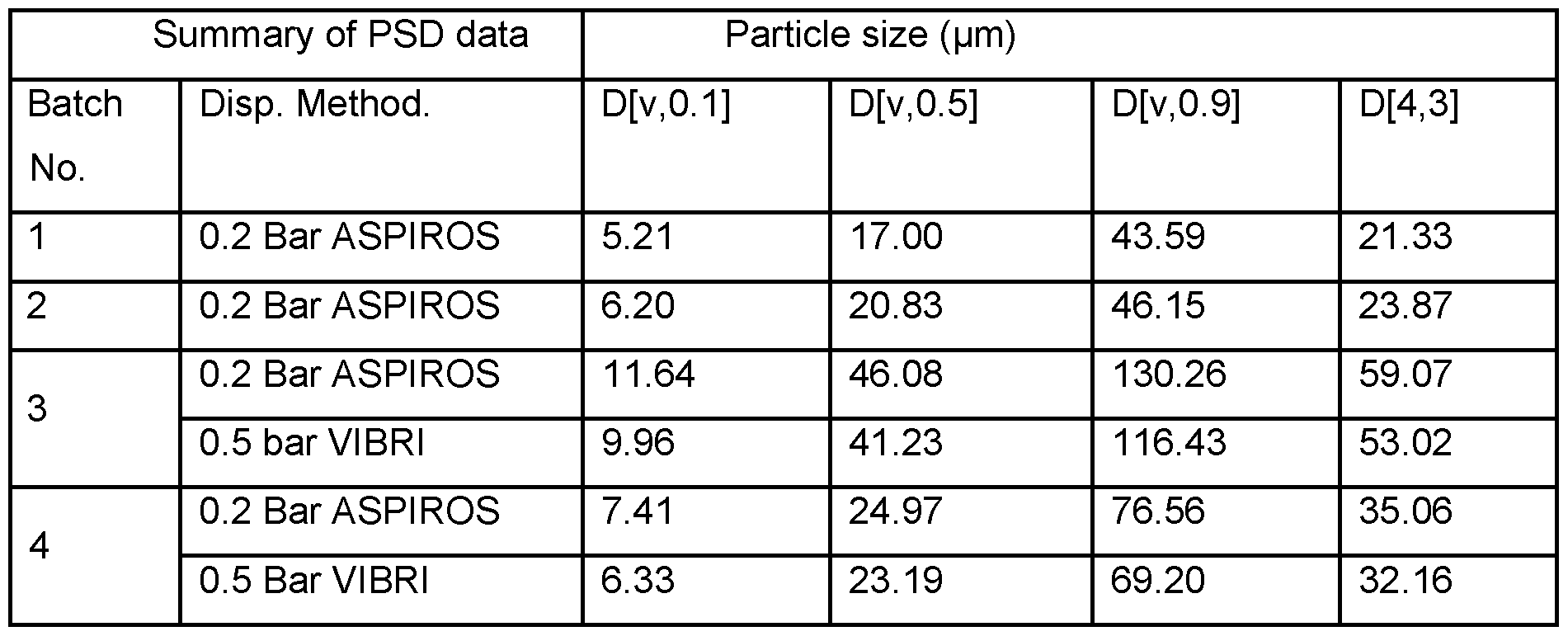


Claims
Priority Applications (23)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14705884.6A EP2958916B1 (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective cdk4/6 inhibitor |
| PL14705884T PL2958916T3 (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective cdk4/6 inhibitor |
| DK14705884.6T DK2958916T3 (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective CDK4 / 6 inhibitor |
| US14/769,038 US20160002223A1 (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective cdk4/6 inhibitor |
| KR1020157022487A KR20150107872A (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective cdk4/6 inhibitor |
| BR112015019508A BR112015019508A8 (en) | 2013-02-21 | 2014-02-08 | solid forms of a selective cdk4 / 6 inhibitor |
| EP18186675.7A EP3431475B1 (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective cdk4/6 inhibitor |
| NZ710138A NZ710138B2 (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective cdk4/6 inhibitor |
| CN201480009556.5A CN105008357A (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective CDK4/6 inhibitor |
| PL18186675T PL3431475T3 (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective cdk4/6 inhibitor |
| AU2014220354A AU2014220354B2 (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective CDK4/6 inhibitor |
| KR1020177022021A KR101858913B1 (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective cdk4/6 inhibitor |
| MX2015010858A MX363715B (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective cdk4/6 inhibitor. |
| RU2015132371A RU2619944C2 (en) | 2013-02-21 | 2014-02-08 | Solid forms of selective cdk4/6 inhibitor |
| ES14705884.6T ES2694787T3 (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective CDK4 / 6 inhibitor |
| SG11201505680RA SG11201505680RA (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective cdk4/6 inhibitor |
| SI201430929T SI2958916T1 (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective cdk4/6 inhibitor |
| CA2900322A CA2900322C (en) | 2013-02-21 | 2014-02-08 | Solid forms of the selective cdk4/6 inhibitor compound acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8h-pyrido[2,3-d]pyrimidin-7-one |
| IL24027715A IL240277B (en) | 2013-02-21 | 2015-07-30 | Solid forms of a selective cdk4/6 inhibitor |
| HK15111953.5A HK1211032A1 (en) | 2013-02-21 | 2015-12-03 | Solid forms of a selective cdk4/6 inhibitor cdk4/6 |
| US15/808,577 US10723730B2 (en) | 2013-02-21 | 2017-11-09 | Solid forms of a selective CDK4/6 inhibitor |
| CY181101034T CY1120734T1 (en) | 2013-02-21 | 2018-10-05 | SOLID FORMS OF AN ECLECTIVE SUSPENDER CDK4 / 6 |
| CY20211100336T CY1124068T1 (en) | 2013-02-21 | 2021-04-16 | SOLID FORMS OF A SELECTIVE CDK4/6 INHIBITOR |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361767761P | 2013-02-21 | 2013-02-21 | |
| US61/767,761 | 2013-02-21 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/769,038 A-371-Of-International US20160002223A1 (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective cdk4/6 inhibitor |
| US15/808,577 Continuation US10723730B2 (en) | 2013-02-21 | 2017-11-09 | Solid forms of a selective CDK4/6 inhibitor |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014128588A1 true WO2014128588A1 (en) | 2014-08-28 |
Family
ID=50151343
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2014/058865 WO2014128588A1 (en) | 2013-02-21 | 2014-02-08 | Solid forms of a selective cdk4/6 inhibitor |
Country Status (24)
| Country | Link |
|---|---|
| US (2) | US20160002223A1 (en) |
| EP (2) | EP3431475B1 (en) |
| JP (3) | JP6381016B2 (en) |
| KR (2) | KR101858913B1 (en) |
| CN (3) | CN107759594A (en) |
| AR (1) | AR094842A1 (en) |
| AU (1) | AU2014220354B2 (en) |
| BR (1) | BR112015019508A8 (en) |
| CA (1) | CA2900322C (en) |
| CY (2) | CY1120734T1 (en) |
| DK (2) | DK3431475T3 (en) |
| ES (2) | ES2869277T3 (en) |
| HK (2) | HK1211032A1 (en) |
| HU (2) | HUE040434T2 (en) |
| IL (1) | IL240277B (en) |
| MX (2) | MX363715B (en) |
| PL (2) | PL2958916T3 (en) |
| PT (2) | PT3431475T (en) |
| RU (1) | RU2619944C2 (en) |
| SG (1) | SG11201505680RA (en) |
| SI (2) | SI2958916T1 (en) |
| TR (1) | TR201816077T4 (en) |
| TW (2) | TWI670269B (en) |
| WO (1) | WO2014128588A1 (en) |
Cited By (71)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104610254A (en) * | 2015-01-26 | 2015-05-13 | 新发药业有限公司 | Low-cost preparation method for palbociclib |
| CN105085517A (en) * | 2015-08-06 | 2015-11-25 | 天津华洛康生物科技有限公司 | Crystal-type palbociclib free-alkali hydrate and preparation method thereof |
| CN105153149A (en) * | 2015-07-29 | 2015-12-16 | 江苏中邦制药有限公司 | Preparation method for selective kinases inhibitor Palbociclib |
| WO2016024249A1 (en) * | 2014-08-14 | 2016-02-18 | Sun Pharmaceutical Industries Limited | Crystalline forms of palbociclib |
| WO2016030439A1 (en) * | 2014-08-28 | 2016-03-03 | Ratiopharm Gmbh | Method of producing palbociclib and pharmaceutical compositions comprising the same |
| CN105524059A (en) * | 2016-01-06 | 2016-04-27 | 北京修正创新药物研究院有限公司 | Palbociclib impurity preparation method |
| WO2016090257A1 (en) * | 2014-12-05 | 2016-06-09 | Crystal Pharmatech Inc. | Salts and crystalline forms of 6-acetyl-8-cyclopentyl-5-methyl-2((5-(piperazin-1-yl)pyridin-2-yl)amino)pyrido[2,3-d] pyrimidin-7(8h)-one (palbociclib) |
| WO2016092442A1 (en) * | 2014-12-08 | 2016-06-16 | Sun Pharmaceutical Industries Limited | Processes for the preparation of crystalline forms of palbociclib acetate |
| WO2016127963A1 (en) | 2015-02-11 | 2016-08-18 | Zentiva, K.S. | Solid forms of palbociclib salts |
| CN105924439A (en) * | 2016-06-24 | 2016-09-07 | 石家庄海瑞药物科技有限公司 | Preparation method for Palbociclib |
| WO2016156070A1 (en) | 2015-04-02 | 2016-10-06 | Sandoz Ag | Modified particles of palbociclib |
| EP3078663A1 (en) | 2015-04-09 | 2016-10-12 | Sandoz Ag | Modified particles of palbociclib |
| CN106117199A (en) * | 2015-05-04 | 2016-11-16 | 江苏恒瑞医药股份有限公司 | The dihydroxy ethyl sulfonate of a kind of cell cycle protein dependent kinase inhibitor, its crystal form and preparation method thereof |
| WO2016193860A1 (en) | 2015-06-04 | 2016-12-08 | Pfizer Inc. | Solid dosage forms of palbociclib |
| CN106317053A (en) * | 2015-06-29 | 2017-01-11 | 北大方正集团有限公司 | Preparation method of palbociclib crystal form A |
| CN106336411A (en) * | 2016-04-27 | 2017-01-18 | 上海医药集团股份有限公司 | Preparation method and use of CDK4/6 inhibitor palbociclib highly-pure raw drug |
| WO2017021111A1 (en) * | 2015-08-05 | 2017-02-09 | Ratiopharm Gmbh | New crystalline form and acetic acid adducts of palbociclib |
| CN106608876A (en) * | 2015-10-21 | 2017-05-03 | 新发药业有限公司 | Preparation method of high-purity palbociclib |
| WO2017072543A1 (en) | 2015-10-28 | 2017-05-04 | Egis Gyógyszergyár Zrt. | Palbociclib salts |
| WO2017115315A1 (en) * | 2015-12-30 | 2017-07-06 | Dr. Reddy's Laboratories Limited | Solid forms of palbociclib |
| WO2017130219A1 (en) | 2016-01-25 | 2017-08-03 | Mylan Laboratories Limited | Amorphous solid dispersion of palbociclib |
| WO2017145054A1 (en) * | 2016-02-24 | 2017-08-31 | Lupin Limited | Modified particles of crystalline palbociclib free base and process for the preparation thereof |
| WO2018009735A1 (en) | 2016-07-07 | 2018-01-11 | Plantex Ltd. | Solid state forms of palbociclib dimesylate |
| WO2018049233A1 (en) | 2016-09-08 | 2018-03-15 | Nicolas Stransky | Inhibitors of the fibroblast growth factor receptor in combination with cyclin-dependent kinase inhibitors |
| WO2018065999A1 (en) | 2016-10-07 | 2018-04-12 | Mylan Laboratories Limited | Novel polymorph of an intermediate for palbociclib synthesis |
| WO2018073687A1 (en) | 2016-10-20 | 2018-04-26 | Pfizer Inc. | Anti-proliferative agents for treating pah |
| WO2018073574A1 (en) | 2016-10-20 | 2018-04-26 | Cipla Limited | Polymorphic forms of palbociclib |
| WO2018091999A1 (en) | 2016-11-16 | 2018-05-24 | Pfizer Inc. | Combination of an egfr t790m inhibitor and a cdk inhibitor for the treatment of non-small cell lung cancer |
| EP3216450A4 (en) * | 2014-11-07 | 2018-06-13 | Jiangsu Hansoh Pharmaceutical Group Co., Ltd. | Pharmaceutical preparation comprising cyclin inhibitor and preparation method thereof |
| US10000490B2 (en) | 2014-01-15 | 2018-06-19 | Blueprint Medicines Corporation | Inhibitors of the fibroblast growth factor receptor |
| WO2018108167A1 (en) | 2016-12-16 | 2018-06-21 | 基石药业 | Cdk4/6 inhibitor |
| CN108299422A (en) * | 2018-02-28 | 2018-07-20 | 杭州福斯特药业有限公司 | A kind of preparation method of pa pool former times profit cloth intermediate |
| WO2018191950A1 (en) * | 2017-04-21 | 2018-10-25 | Alnova Pharmaceuticals, Ltd. | Palbociclib compositions and methods thereof |
| WO2019020715A1 (en) | 2017-07-28 | 2019-01-31 | Synthon B.V. | Pharmaceutical composition comprising palbociclib |
| US10196436B2 (en) | 2012-07-11 | 2019-02-05 | Blueprint Medicines Corporation | Inhibitors of the fibroblast growth factor receptor |
| US10221154B2 (en) | 2013-10-25 | 2019-03-05 | Blueprint Medicines Corporation | Inhibitors of the fibroblast growth factor receptor |
| EP3372603A4 (en) * | 2015-11-02 | 2019-04-24 | Zhejiang Huahai Pharmaceutical Co., Ltd | Preparation methods for palbociclib free base crystal form a and crystal form b |
| WO2019082143A1 (en) | 2017-10-27 | 2019-05-02 | Fresenius Kabi Oncology Ltd. | An improved process for the preparation of ribociclib and its salts |
| WO2019166928A1 (en) | 2018-02-27 | 2019-09-06 | Pfizer Inc. | Combination of a cyclin dependent kinase inhibitor and a bet-bromodomain inhibitor |
| US10449195B2 (en) | 2016-03-29 | 2019-10-22 | Shenzhen Pharmacin Co., Ltd. | Pharmaceutical formulation of palbociclib and a preparation method thereof |
| WO2019220253A1 (en) | 2018-05-14 | 2019-11-21 | Pfizer Inc. | Oral solution formulation |
| WO2019224194A1 (en) | 2018-05-24 | 2019-11-28 | Synthon B.V. | A process for making palbociclib |
| WO2019238088A1 (en) * | 2018-06-13 | 2019-12-19 | 基石药业 | Salt form and crystal form of pyridopyridone derivative |
| WO2020058820A1 (en) | 2018-09-18 | 2020-03-26 | Pfizer Inc. | Combinations of tgfb inhibitors and cdk inhibitors for the treatment of breast cancer |
| WO2020157709A1 (en) | 2019-02-01 | 2020-08-06 | Pfizer Inc. | Combination of a cdk inhibitor and a pim inhibitor |
| US10800783B2 (en) | 2016-08-15 | 2020-10-13 | Pfizer Inc. | CDK2/4/6 inhibitors |
| WO2020239558A1 (en) | 2019-05-24 | 2020-12-03 | Pfizer Inc. | Combination therapies using cdk inhibitors |
| WO2020240360A1 (en) | 2019-05-24 | 2020-12-03 | Pfizer Inc. | Combination therapies using cdk inhibitors |
| CN112274493A (en) * | 2020-11-18 | 2021-01-29 | 石药集团中奇制药技术(石家庄)有限公司 | Preparation method of pipera cypress xili capsule |
| WO2021176349A1 (en) | 2020-03-05 | 2021-09-10 | Pfizer Inc. | Combination of an anaplastic lymphoma kinase inhibitor and a cyclin dependent kinase inhibitor |
| WO2022013369A1 (en) | 2020-07-15 | 2022-01-20 | Pfizer Inc. | Kat6 inhibitor methods and combinations for cancer treatment |
| WO2022018596A1 (en) | 2020-07-20 | 2022-01-27 | Pfizer Inc. | Combination therapy |
| CN114195784A (en) * | 2021-12-29 | 2022-03-18 | 斯坦德标准技术研究(湖北)有限公司 | Pabociclib related substance, preparation method and application thereof |
| WO2022091001A1 (en) | 2020-10-29 | 2022-05-05 | Pfizer Ireland Pharmaceuticals | Process for preparation of palbociclib |
| WO2022123419A1 (en) | 2020-12-08 | 2022-06-16 | Pfizer Inc. | Treatment of luminal subtypes of hr-positive, her2-negative early breast cancer with palbociclib |
| US11467142B2 (en) | 2016-01-25 | 2022-10-11 | Sweden Development Research Pharma (sdr) Ab | Inverse gas chromatography standard solutions, device and method |
| US11471418B2 (en) | 2020-09-29 | 2022-10-18 | Shenzhen Pharmacin Co., Ltd. | Pharmaceutical compositions of amorphous solid dispersions and methods of preparation thereof |
| WO2023111810A1 (en) | 2021-12-14 | 2023-06-22 | Pfizer Inc. | Combination therapies and uses for treating cancer |
| EP4302755A1 (en) | 2022-07-07 | 2024-01-10 | Lotus Pharmaceutical Co., Ltd. | Palbociclib formulation containing an amino acid |
| EP4302832A1 (en) | 2022-07-07 | 2024-01-10 | Lotus Pharmaceutical Co., Ltd. | Palbociclib formulation containing glucono delta lactone |
| WO2024023703A1 (en) | 2022-07-29 | 2024-02-01 | Pfizer Inc. | Dosing regimens comprising a kat6 inhibitor for the treatment of cancer |
| WO2024049926A1 (en) | 2022-08-31 | 2024-03-07 | Arvinas Operations, Inc. | Dosage regimens of estrogen receptor degraders |
| US11976077B2 (en) | 2015-10-16 | 2024-05-07 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-α]pyrrolo[2,3-e]-pyrazin-8-yl)-n-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms therof |
| US11993606B2 (en) | 2015-10-16 | 2024-05-28 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-n-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| US11993605B2 (en) | 2015-10-16 | 2024-05-28 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-n-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| WO2024133726A1 (en) | 2022-12-22 | 2024-06-27 | Synthon B.V. | Pharmaceutical composition comprising palbociclib |
| WO2024132652A1 (en) | 2022-12-22 | 2024-06-27 | Synthon B.V. | Pharmaceutical composition comprising palbociclib |
| US12043630B2 (en) | 2017-08-18 | 2024-07-23 | Cothera Bioscience, Inc. | Polymorphic form of TG02 for treating cancer |
| US12077545B2 (en) | 2015-10-16 | 2024-09-03 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| WO2024201334A1 (en) | 2023-03-30 | 2024-10-03 | Pfizer Inc. | Kat6a as a predictive biomarker for treatment with a kat6a inhibitor and methods of treatment thereof |
| WO2024201340A1 (en) | 2023-03-30 | 2024-10-03 | Pfizer Inc. | Kat6a as a predictive biomarker for treatment of breast cancer with a cdk4 inhibitor and an antiestrogen and methods of treatment thereof |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG11201505680RA (en) | 2013-02-21 | 2015-09-29 | Pfizer | Solid forms of a selective cdk4/6 inhibitor |
| CN106397431A (en) * | 2015-07-28 | 2017-02-15 | 苏州国匡医药科技有限公司 | Now crystal form of antitumor drug, preparation method and uses thereof |
| CN106831759A (en) * | 2015-12-03 | 2017-06-13 | 上海星泰医药科技有限公司 | The preparation method of Pabuk former times profit cloth and its intermediate |
| CN105541832A (en) * | 2015-12-15 | 2016-05-04 | 南京艾德凯腾生物医药有限责任公司 | Preparation method of Palbociclib isethionate |
| WO2017197904A2 (en) * | 2016-05-08 | 2017-11-23 | 上海诚妙医药科技有限公司 | New crystal forms of palbociclib, and preparation method and use therefor |
| CN106146494B (en) * | 2016-06-29 | 2018-02-06 | 重庆华邦制药有限公司 | For preparing the brilliant solvent of Pabuk former times profit cloth A types and preparation method |
| CN106220627A (en) * | 2016-07-31 | 2016-12-14 | 合肥远志医药科技开发有限公司 | A kind of industrialized process for preparing of high-purity Pabuk former times profit cloth |
| CN106220626A (en) * | 2016-07-31 | 2016-12-14 | 合肥远志医药科技开发有限公司 | A kind of polymorphic of Pabuk former times profit cloth and preparation method thereof |
| CN108017630B (en) * | 2016-10-31 | 2022-10-11 | 上海创诺制药有限公司 | Preparation method of small-specific-surface-area palbociclib free base |
| CN108117550B (en) * | 2016-11-29 | 2020-08-14 | 上海医药工业研究院 | Preparation method of pyrido [2,3-d ] pyrimidine compound |
| CN108864078B (en) * | 2017-05-10 | 2021-10-15 | 江苏豪森药业集团有限公司 | Preparation method of palbociclib crystal form B |
| CA3069523A1 (en) | 2017-07-11 | 2019-01-17 | Actym Therapeutics, Inc. | Engineered immunostimulatory bacterial strains and uses thereof |
| CN109897034A (en) * | 2017-12-07 | 2019-06-18 | 南京卡文迪许生物工程技术有限公司 | A kind of high-purity crystal form A Pabuk former times benefit cloth and preparation method thereof and pharmaceutical composition |
| CN108586452A (en) * | 2018-01-12 | 2018-09-28 | 重庆市碚圣医药科技股份有限公司 | A kind of synthetic method of Pa Boxini intermediates |
| CN108558745A (en) * | 2018-05-17 | 2018-09-21 | 苏州莱克施德药业有限公司 | A kind of pa wins the synthetic method of XiLin intermediate |
| WO2020047161A2 (en) | 2018-08-28 | 2020-03-05 | Actym Therapeutics, Inc. | Engineered immunostimulatory bacterial strains and uses thereof |
| EP3870178A4 (en) | 2018-10-24 | 2022-08-03 | Effector Therapeutics Inc. | Crystalline forms of mnk inhibitors |
| CN109336886A (en) * | 2018-12-07 | 2019-02-15 | 重庆三圣实业股份有限公司 | A kind of preparation method of Pa Boxini and products thereof |
| WO2020148635A1 (en) * | 2019-01-17 | 2020-07-23 | Pfizer Inc. | Crystalline form of a cdk inhibitor |
| CN110256424A (en) * | 2019-07-03 | 2019-09-20 | 武汉工程大学 | A kind of synthetic method of Pa Boxini key intermediate V |
| CN110551063A (en) * | 2019-10-17 | 2019-12-10 | 山东邹平大展新材料有限公司 | Method for synthesizing 5- (N-BOC-piperazine-1-yl) pyridine-2-amine |
| CN112457311B (en) * | 2020-12-04 | 2022-07-12 | 江苏豪森药业集团有限公司 | Preparation method of compound containing chloro-bromo-pyrrole-pyrimidone structure |
| CN112661753B (en) * | 2020-12-29 | 2022-06-03 | 山东铂源药业股份有限公司 | Preparation method of palbociclib intermediate |
| CN112778303A (en) * | 2020-12-31 | 2021-05-11 | 武汉九州钰民医药科技有限公司 | Preparation method of CDK4/6 kinase inhibitor SHR6390 |
| CN112898299B (en) * | 2021-01-26 | 2021-11-26 | 山东铂源药业有限公司 | Preparation method of palbociclib intermediate |
| EP4356398A1 (en) | 2021-06-14 | 2024-04-24 | Preh Holding, LLC | Connected body surface care module |
| CN113845520A (en) * | 2021-09-09 | 2021-12-28 | 安徽皓元药业有限公司 | Palbociclib orotate and preparation method thereof |
| CN113999227A (en) * | 2021-11-26 | 2022-02-01 | 常州大学 | Preparation method of palbociclib intermediate |
| IL313422A (en) | 2021-12-10 | 2024-08-01 | Lilly Co Eli | Cdk4 and 6 inhibitor in combination with fulvestrant for the treatment of hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced or metastatic breast cancer in patients previously treated with a cdk4 and 6 inhibitor |
| WO2023114264A1 (en) | 2021-12-15 | 2023-06-22 | Eli Lilly And Company | Combination for treatment of high-risk metastatic hormone-sensitive prostate cancer |
| WO2024097206A1 (en) | 2022-11-02 | 2024-05-10 | Petra Pharma Corporation | Allosteric chromenone inhibitors of phosphoinositide 3-kinase (pi3k) for the treatment of disease |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991011172A1 (en) | 1990-01-23 | 1991-08-08 | The University Of Kansas | Derivatives of cyclodextrins exhibiting enhanced aqueous solubility and the use thereof |
| WO1994002518A1 (en) | 1992-07-27 | 1994-02-03 | The University Of Kansas | Derivatives of cyclodextrins exhibiting enhanced aqueous solubility and the use thereof |
| WO1998055148A1 (en) | 1997-06-05 | 1998-12-10 | Janssen Pharmaceutica N.V. | Pharmaceutical compositions comprising cyclodextrins |
| WO2000035298A1 (en) | 1996-11-27 | 2000-06-22 | Wm. Wrigley Jr. Company | Chewing gum containing medicament active agents |
| US6106864A (en) | 1995-09-15 | 2000-08-22 | Pfizer Inc. | Pharmaceutical formulations containing darifenacin |
| WO2003062236A1 (en) | 2002-01-22 | 2003-07-31 | Warner-Lambert Company Llc | 2-(PYRIDIN-2-YLAMINO)-PYRIDO[2,3d]PYRIMIDIN-7-ONES |
| WO2005005426A1 (en) | 2003-07-11 | 2005-01-20 | Warner-Lambert Company Llc | Isethionate salt of a selective cdk4 inhibitor |
| WO2008032157A2 (en) | 2006-09-08 | 2008-03-20 | Pfizer Products Inc. | Synthesis of 2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11138004A (en) * | 1997-11-06 | 1999-05-25 | Mitsui Chem Inc | Copper catalyst for hydration of nitrile and production of copper catalyst |
| HUP0300136A2 (en) * | 2000-03-06 | 2003-05-28 | Warner Lambert Co | 5-alkylpyrido [2,3-d]pyrimidines tyrosine kinase inhibitors, pharmaceutical compositions containing them and their use |
| JP2004074041A (en) * | 2002-08-20 | 2004-03-11 | Cabot Supermetal Kk | Recovering method of fluorine |
| JP2004149472A (en) * | 2002-10-31 | 2004-05-27 | Sumitomo Chem Co Ltd | Crystal-form phosphorous ester, and method for producing the same |
| CN1976921B (en) * | 2004-06-29 | 2013-08-21 | 蓝宝石治疗公司 | Crystal forms of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-(phenylmethyl)-3-piperidinecarboxylic acid 1,2,2-trimethylhydrazide |
| GT200600315A (en) * | 2005-07-20 | 2007-03-19 | CRYSTAL FORMS OF 4-METHYL-N- [3- (4-METHYL-IMIDAZOL-1-ILO) -5-TRIFLUOROMETILO-PHENYL] -3- (4-PYRIDINA-3-ILO-PIRIMIDINA-2-ILOAMINO) -BENZAMIDA | |
| JP5261487B2 (en) * | 2007-08-03 | 2013-08-14 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Crystal forms of dihydropteridinone derivatives |
| TW201014830A (en) * | 2008-09-30 | 2010-04-16 | Theravance Inc | Crystalline form of an alkoxyimidazol-1-ylmethyl biphenyl carboxylic acid |
| PT2453894E (en) * | 2009-07-15 | 2016-02-02 | Theravance Biopharma R&D Ip Llc | Crystalline freebase form of a biphenyl compound |
| AU2010295717B2 (en) * | 2009-09-20 | 2014-09-11 | Abbvie Inc. | ABT-263 crystalline forms and solvates for use in treating Bcl-2 protein related diseases |
| CZ201039A3 (en) | 2010-01-19 | 2011-07-27 | Zentiva, K. S | Method of industrial production of amorphous form of (3R,5R) 7-[3-phenyl-4-phenylcarbamoyl-2-(4-fluorophenyl)-5-isopropylpyrrol-1-yl]-3,5-dihydroxyheptanoic acid hemicalcium salt (atorvastatin) with low specific surface and use thereof in medicamento |
| WO2012064838A1 (en) * | 2010-11-09 | 2012-05-18 | Zafgen Corporation | Crystalline solids of a metap-2 inhibitor and methods of making and using same |
| SG11201505680RA (en) | 2013-02-21 | 2015-09-29 | Pfizer | Solid forms of a selective cdk4/6 inhibitor |
| EP3180007A1 (en) | 2014-08-14 | 2017-06-21 | Sun Pharmaceutical Industries Ltd | Crystalline forms of palbociclib |
-
2014
- 2014-02-08 SG SG11201505680RA patent/SG11201505680RA/en unknown
- 2014-02-08 MX MX2015010858A patent/MX363715B/en unknown
- 2014-02-08 DK DK18186675.7T patent/DK3431475T3/en active
- 2014-02-08 SI SI201430929T patent/SI2958916T1/en unknown
- 2014-02-08 TR TR2018/16077T patent/TR201816077T4/en unknown
- 2014-02-08 CN CN201711141072.8A patent/CN107759594A/en active Pending
- 2014-02-08 EP EP18186675.7A patent/EP3431475B1/en active Active
- 2014-02-08 KR KR1020177022021A patent/KR101858913B1/en active IP Right Grant
- 2014-02-08 CN CN202010094827.9A patent/CN111253394A/en active Pending
- 2014-02-08 WO PCT/IB2014/058865 patent/WO2014128588A1/en active Application Filing
- 2014-02-08 DK DK14705884.6T patent/DK2958916T3/en active
- 2014-02-08 PL PL14705884T patent/PL2958916T3/en unknown
- 2014-02-08 ES ES18186675T patent/ES2869277T3/en active Active
- 2014-02-08 KR KR1020157022487A patent/KR20150107872A/en active Application Filing
- 2014-02-08 CA CA2900322A patent/CA2900322C/en active Active
- 2014-02-08 HU HUE14705884A patent/HUE040434T2/en unknown
- 2014-02-08 HU HUE18186675A patent/HUE054212T2/en unknown
- 2014-02-08 EP EP14705884.6A patent/EP2958916B1/en active Active
- 2014-02-08 RU RU2015132371A patent/RU2619944C2/en active
- 2014-02-08 PL PL18186675T patent/PL3431475T3/en unknown
- 2014-02-08 PT PT181866757T patent/PT3431475T/en unknown
- 2014-02-08 ES ES14705884.6T patent/ES2694787T3/en active Active
- 2014-02-08 US US14/769,038 patent/US20160002223A1/en not_active Abandoned
- 2014-02-08 PT PT14705884T patent/PT2958916T/en unknown
- 2014-02-08 CN CN201480009556.5A patent/CN105008357A/en active Pending
- 2014-02-08 BR BR112015019508A patent/BR112015019508A8/en not_active Application Discontinuation
- 2014-02-08 AU AU2014220354A patent/AU2014220354B2/en active Active
- 2014-02-08 SI SI201431819T patent/SI3431475T1/en unknown
- 2014-02-18 JP JP2014028092A patent/JP6381016B2/en active Active
- 2014-02-18 TW TW106131041A patent/TWI670269B/en active
- 2014-02-18 TW TW103105287A patent/TWI633103B/en active
- 2014-02-20 AR ARP140100540A patent/AR094842A1/en unknown
-
2015
- 2015-07-30 IL IL24027715A patent/IL240277B/en active IP Right Review Request
- 2015-08-20 MX MX2019003605A patent/MX2019003605A/en unknown
- 2015-12-03 HK HK15111953.5A patent/HK1211032A1/en unknown
- 2015-12-03 HK HK18107562.3A patent/HK1248217A1/en unknown
-
2017
- 2017-07-18 JP JP2017139197A patent/JP6524152B2/en active Active
- 2017-11-09 US US15/808,577 patent/US10723730B2/en active Active
-
2018
- 2018-10-05 CY CY181101034T patent/CY1120734T1/en unknown
-
2019
- 2019-04-26 JP JP2019085273A patent/JP2019116512A/en active Pending
-
2021
- 2021-04-16 CY CY20211100336T patent/CY1124068T1/en unknown
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991011172A1 (en) | 1990-01-23 | 1991-08-08 | The University Of Kansas | Derivatives of cyclodextrins exhibiting enhanced aqueous solubility and the use thereof |
| WO1994002518A1 (en) | 1992-07-27 | 1994-02-03 | The University Of Kansas | Derivatives of cyclodextrins exhibiting enhanced aqueous solubility and the use thereof |
| US6106864A (en) | 1995-09-15 | 2000-08-22 | Pfizer Inc. | Pharmaceutical formulations containing darifenacin |
| WO2000035298A1 (en) | 1996-11-27 | 2000-06-22 | Wm. Wrigley Jr. Company | Chewing gum containing medicament active agents |
| WO1998055148A1 (en) | 1997-06-05 | 1998-12-10 | Janssen Pharmaceutica N.V. | Pharmaceutical compositions comprising cyclodextrins |
| US7456168B2 (en) | 2002-01-22 | 2008-11-25 | Warner-Lambert Company | 2-(pyridin-2-ylamino)-pyrido[2,3, d]pyrimidin-7-ones |
| WO2003062236A1 (en) | 2002-01-22 | 2003-07-31 | Warner-Lambert Company Llc | 2-(PYRIDIN-2-YLAMINO)-PYRIDO[2,3d]PYRIMIDIN-7-ONES |
| US6936612B2 (en) | 2002-01-22 | 2005-08-30 | Warner-Lambert Company | 2-(Pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones |
| US7208489B2 (en) | 2002-01-22 | 2007-04-24 | Warner-Lambert Company | 2-(pyridin-2-ylamino)-pyrido [2,3-d]pyrimidin-7-ones |
| WO2005005426A1 (en) | 2003-07-11 | 2005-01-20 | Warner-Lambert Company Llc | Isethionate salt of a selective cdk4 inhibitor |
| US7345171B2 (en) | 2003-07-11 | 2008-03-18 | Warner-Lambert Company Llc | Isethionate salt of a selective CKD4 inhibitor |
| US7863278B2 (en) | 2003-07-11 | 2011-01-04 | Warner-Lambert CompanyLLC | Isethionate salt of a selective CDK4 inhibitor |
| WO2008032157A2 (en) | 2006-09-08 | 2008-03-20 | Pfizer Products Inc. | Synthesis of 2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones |
| US7781583B2 (en) | 2006-09-08 | 2010-08-24 | Pfizer Inc | Synthesis of 2-(pyridin-2-ylamino)-pyrido[2,3-d] pryimidin-7-ones |
Non-Patent Citations (7)
| Title |
|---|
| "Remington's Pharmaceutical Sciences", 1975, MACK PUBLISHING COMPANY |
| CHEN ET AL., J. PHARMACEUTICAL AND BIOMEDICAL ANALYSIS, vol. 26, 2001, pages 63 |
| FINNIN; MORGAN, J PHARM SCI, vol. 88, no. 10, 1999, pages 955 - 958 |
| H. LIEBERMAN; L. LACHMAN, PHARMACEUTICAL DOSAGE FORMS: TABLETS, vol. 1, 1980 |
| J. AM. CHEM. SOC., vol. 60, 1938, pages 309 |
| LIANG; CHEN, EXPERT OPINION IN THERAPEUTIC PATENTS, vol. 11, no. 6, 2001, pages 981 - 986 |
| VERMA ET AL., PHARMACEUTICAL TECHNOLOGY ON-LINE, vol. 25, no. 2, 2001, pages 1 - 14 |
Cited By (112)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10196436B2 (en) | 2012-07-11 | 2019-02-05 | Blueprint Medicines Corporation | Inhibitors of the fibroblast growth factor receptor |
| US10875837B2 (en) | 2013-10-25 | 2020-12-29 | Blueprint Medicines Corporation | Inhibitors of the fibroblast growth factor receptor |
| US10221154B2 (en) | 2013-10-25 | 2019-03-05 | Blueprint Medicines Corporation | Inhibitors of the fibroblast growth factor receptor |
| US10000490B2 (en) | 2014-01-15 | 2018-06-19 | Blueprint Medicines Corporation | Inhibitors of the fibroblast growth factor receptor |
| WO2016024249A1 (en) * | 2014-08-14 | 2016-02-18 | Sun Pharmaceutical Industries Limited | Crystalline forms of palbociclib |
| WO2017025784A1 (en) * | 2014-08-14 | 2017-02-16 | Sun Pharmaceutical Industries Limited | A carbon dioxide adduct of palbociclib |
| WO2016030439A1 (en) * | 2014-08-28 | 2016-03-03 | Ratiopharm Gmbh | Method of producing palbociclib and pharmaceutical compositions comprising the same |
| EP3216450A4 (en) * | 2014-11-07 | 2018-06-13 | Jiangsu Hansoh Pharmaceutical Group Co., Ltd. | Pharmaceutical preparation comprising cyclin inhibitor and preparation method thereof |
| WO2016090257A1 (en) * | 2014-12-05 | 2016-06-09 | Crystal Pharmatech Inc. | Salts and crystalline forms of 6-acetyl-8-cyclopentyl-5-methyl-2((5-(piperazin-1-yl)pyridin-2-yl)amino)pyrido[2,3-d] pyrimidin-7(8h)-one (palbociclib) |
| WO2016092442A1 (en) * | 2014-12-08 | 2016-06-16 | Sun Pharmaceutical Industries Limited | Processes for the preparation of crystalline forms of palbociclib acetate |
| CN104610254A (en) * | 2015-01-26 | 2015-05-13 | 新发药业有限公司 | Low-cost preparation method for palbociclib |
| WO2016127963A1 (en) | 2015-02-11 | 2016-08-18 | Zentiva, K.S. | Solid forms of palbociclib salts |
| WO2016156070A1 (en) | 2015-04-02 | 2016-10-06 | Sandoz Ag | Modified particles of palbociclib |
| EP3078663A1 (en) | 2015-04-09 | 2016-10-12 | Sandoz Ag | Modified particles of palbociclib |
| CN106117199A (en) * | 2015-05-04 | 2016-11-16 | 江苏恒瑞医药股份有限公司 | The dihydroxy ethyl sulfonate of a kind of cell cycle protein dependent kinase inhibitor, its crystal form and preparation method thereof |
| EP3302565B1 (en) | 2015-06-04 | 2019-11-06 | Pfizer Inc | Solid dosage forms of palbociclib |
| US11065250B2 (en) | 2015-06-04 | 2021-07-20 | Pfizer Inc. | Solid dosage forms of palbociclib |
| KR20200006633A (en) * | 2015-06-04 | 2020-01-20 | 화이자 인코포레이티드 | Solid dosage forms of palbociclib |
| KR102369405B1 (en) * | 2015-06-04 | 2022-03-02 | 화이자 인코포레이티드 | Solid dosage forms of palbociclib |
| WO2016193860A1 (en) | 2015-06-04 | 2016-12-08 | Pfizer Inc. | Solid dosage forms of palbociclib |
| EP3636283A1 (en) | 2015-06-04 | 2020-04-15 | Pfizer Inc | Solid dosage forms of palbociclib |
| AU2019204689B2 (en) * | 2015-06-04 | 2020-12-03 | Pfizer Inc. | Solid dosage forms of palbociclib |
| CN106317053A (en) * | 2015-06-29 | 2017-01-11 | 北大方正集团有限公司 | Preparation method of palbociclib crystal form A |
| CN105153149A (en) * | 2015-07-29 | 2015-12-16 | 江苏中邦制药有限公司 | Preparation method for selective kinases inhibitor Palbociclib |
| WO2017021111A1 (en) * | 2015-08-05 | 2017-02-09 | Ratiopharm Gmbh | New crystalline form and acetic acid adducts of palbociclib |
| US10526326B2 (en) | 2015-08-05 | 2020-01-07 | Ratiopharm Gmbh | Crystalline form and acetic acid adducts of palbociclib |
| EP3543235A1 (en) | 2015-08-05 | 2019-09-25 | ratiopharm GmbH | Crystalline form and acetic acid adduct of palbociclib |
| CN105085517A (en) * | 2015-08-06 | 2015-11-25 | 天津华洛康生物科技有限公司 | Crystal-type palbociclib free-alkali hydrate and preparation method thereof |
| US12110297B2 (en) | 2015-10-16 | 2024-10-08 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-n-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| US11976077B2 (en) | 2015-10-16 | 2024-05-07 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-α]pyrrolo[2,3-e]-pyrazin-8-yl)-n-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms therof |
| US12116373B2 (en) | 2015-10-16 | 2024-10-15 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| US12103933B2 (en) | 2015-10-16 | 2024-10-01 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| US11993606B2 (en) | 2015-10-16 | 2024-05-28 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-n-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| US11993605B2 (en) | 2015-10-16 | 2024-05-28 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-n-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| US12077545B2 (en) | 2015-10-16 | 2024-09-03 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| US12091415B2 (en) | 2015-10-16 | 2024-09-17 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-n-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| CN106608876A (en) * | 2015-10-21 | 2017-05-03 | 新发药业有限公司 | Preparation method of high-purity palbociclib |
| CN106608876B (en) * | 2015-10-21 | 2018-06-19 | 新发药业有限公司 | A kind of preparation method of high-purity Pa Boxini |
| WO2017072543A1 (en) | 2015-10-28 | 2017-05-04 | Egis Gyógyszergyár Zrt. | Palbociclib salts |
| US10766895B2 (en) | 2015-11-02 | 2020-09-08 | Zhejiang Huahai Pharmaceutical Co., Ltd. | Preparation methods for palbociclib free base crystal form A and crystal form B |
| EP3372603A4 (en) * | 2015-11-02 | 2019-04-24 | Zhejiang Huahai Pharmaceutical Co., Ltd | Preparation methods for palbociclib free base crystal form a and crystal form b |
| WO2017115315A1 (en) * | 2015-12-30 | 2017-07-06 | Dr. Reddy's Laboratories Limited | Solid forms of palbociclib |
| CN105524059A (en) * | 2016-01-06 | 2016-04-27 | 北京修正创新药物研究院有限公司 | Palbociclib impurity preparation method |
| WO2017130219A1 (en) | 2016-01-25 | 2017-08-03 | Mylan Laboratories Limited | Amorphous solid dispersion of palbociclib |
| US11467142B2 (en) | 2016-01-25 | 2022-10-11 | Sweden Development Research Pharma (sdr) Ab | Inverse gas chromatography standard solutions, device and method |
| WO2017145054A1 (en) * | 2016-02-24 | 2017-08-31 | Lupin Limited | Modified particles of crystalline palbociclib free base and process for the preparation thereof |
| US11464779B2 (en) | 2016-03-29 | 2022-10-11 | Shenzhen Pharmacin Co., Ltd. | Pharmaceutical formulation of palbociclib and a preparation method thereof |
| US10813937B2 (en) | 2016-03-29 | 2020-10-27 | Shenzhen Pharmacin Co., Ltd. | Pharmaceutical formulation of palbociclib and a preparation method thereof |
| US10894049B2 (en) | 2016-03-29 | 2021-01-19 | Shenzhen Pharmacin Co., Ltd. | Pharmaceutical formulation of palbociclib and a preparation method thereof |
| US10449195B2 (en) | 2016-03-29 | 2019-10-22 | Shenzhen Pharmacin Co., Ltd. | Pharmaceutical formulation of palbociclib and a preparation method thereof |
| CN106336411B (en) * | 2016-04-27 | 2018-03-06 | 上海医药集团股份有限公司 | The preparation technology and purposes of CDK4/6 inhibitor Pa Boxini high-purity raw medicines |
| CN106336411A (en) * | 2016-04-27 | 2017-01-18 | 上海医药集团股份有限公司 | Preparation method and use of CDK4/6 inhibitor palbociclib highly-pure raw drug |
| CN105924439B (en) * | 2016-06-24 | 2017-11-24 | 石家庄海瑞药物科技有限公司 | A kind of preparation method of Pabuk former times profit cloth |
| CN105924439A (en) * | 2016-06-24 | 2016-09-07 | 石家庄海瑞药物科技有限公司 | Preparation method for Palbociclib |
| US10597393B2 (en) | 2016-07-07 | 2020-03-24 | Plantex Ltd. | Solid state forms of Palbociclib dimesylate |
| WO2018009735A1 (en) | 2016-07-07 | 2018-01-11 | Plantex Ltd. | Solid state forms of palbociclib dimesylate |
| US11332467B2 (en) | 2016-07-07 | 2022-05-17 | Plantex Ltd. | Solid state forms of palbociclib dimesylate |
| US10800783B2 (en) | 2016-08-15 | 2020-10-13 | Pfizer Inc. | CDK2/4/6 inhibitors |
| US11396512B2 (en) | 2016-08-15 | 2022-07-26 | Pfizer Inc. | CDK2/4/6 inhibitors |
| WO2018049233A1 (en) | 2016-09-08 | 2018-03-15 | Nicolas Stransky | Inhibitors of the fibroblast growth factor receptor in combination with cyclin-dependent kinase inhibitors |
| US10710999B2 (en) | 2016-10-07 | 2020-07-14 | Mylan Laboratories Limited | Polymorph of an intermediate for palbociclib synthesis |
| WO2018065999A1 (en) | 2016-10-07 | 2018-04-12 | Mylan Laboratories Limited | Novel polymorph of an intermediate for palbociclib synthesis |
| WO2018073687A1 (en) | 2016-10-20 | 2018-04-26 | Pfizer Inc. | Anti-proliferative agents for treating pah |
| JP2021038265A (en) * | 2016-10-20 | 2021-03-11 | ファイザー・インク | Anti-proliferative agents for treating pah |
| TWI656876B (en) * | 2016-10-20 | 2019-04-21 | 美商輝瑞股份有限公司 | Anti-proliferative agents for treating pah |
| US10849903B2 (en) | 2016-10-20 | 2020-12-01 | Pfizer Inc. | Anti-proliferative agents for treating PAH |
| US11439646B2 (en) | 2016-10-20 | 2022-09-13 | Pfizer Inc. | Anti-proliferative agents for treating PAH |
| JP2018076290A (en) * | 2016-10-20 | 2018-05-17 | ファイザー・インク | Anti-proliferative agents for treating pah |
| EP3804724A1 (en) | 2016-10-20 | 2021-04-14 | Pfizer Inc. | Cdk inhibitors for treating pah |
| WO2018073574A1 (en) | 2016-10-20 | 2018-04-26 | Cipla Limited | Polymorphic forms of palbociclib |
| WO2018091999A1 (en) | 2016-11-16 | 2018-05-24 | Pfizer Inc. | Combination of an egfr t790m inhibitor and a cdk inhibitor for the treatment of non-small cell lung cancer |
| US10676474B2 (en) | 2016-12-16 | 2020-06-09 | Cstone Pharmaceuticals | 1,6-naphthyridine derivatives as CDK4/6 inhibitor |
| WO2018108167A1 (en) | 2016-12-16 | 2018-06-21 | 基石药业 | Cdk4/6 inhibitor |
| WO2018191950A1 (en) * | 2017-04-21 | 2018-10-25 | Alnova Pharmaceuticals, Ltd. | Palbociclib compositions and methods thereof |
| US11529353B2 (en) | 2017-07-28 | 2022-12-20 | Synthon B.V. | Pharmaceutical composition comprising Palbociclib |
| WO2019020715A1 (en) | 2017-07-28 | 2019-01-31 | Synthon B.V. | Pharmaceutical composition comprising palbociclib |
| US12043630B2 (en) | 2017-08-18 | 2024-07-23 | Cothera Bioscience, Inc. | Polymorphic form of TG02 for treating cancer |
| US11440912B2 (en) | 2017-10-27 | 2022-09-13 | Fresenius Kabi Oncology Ltd | Process for the preparation of ribociclib and its salts |
| WO2019082143A1 (en) | 2017-10-27 | 2019-05-02 | Fresenius Kabi Oncology Ltd. | An improved process for the preparation of ribociclib and its salts |
| WO2019166928A1 (en) | 2018-02-27 | 2019-09-06 | Pfizer Inc. | Combination of a cyclin dependent kinase inhibitor and a bet-bromodomain inhibitor |
| CN108299422A (en) * | 2018-02-28 | 2018-07-20 | 杭州福斯特药业有限公司 | A kind of preparation method of pa pool former times profit cloth intermediate |
| EP4309656A2 (en) | 2018-05-14 | 2024-01-24 | Pfizer Inc. | Oral solution formulation |
| WO2019220253A1 (en) | 2018-05-14 | 2019-11-21 | Pfizer Inc. | Oral solution formulation |
| US11911383B2 (en) | 2018-05-14 | 2024-02-27 | Pfizer Inc. | Oral solution formulation |
| EP4289844A2 (en) | 2018-05-24 | 2023-12-13 | Synthon B.V. | A process for making palbociclib |
| US11858928B2 (en) | 2018-05-24 | 2024-01-02 | Synthon B.V. | Process for making palbociclib |
| EP4289844A3 (en) * | 2018-05-24 | 2024-04-10 | Synthon B.V. | A process for making palbociclib |
| WO2019224194A1 (en) | 2018-05-24 | 2019-11-28 | Synthon B.V. | A process for making palbociclib |
| CN112292379B (en) * | 2018-06-13 | 2022-03-04 | 基石药业(苏州)有限公司 | Salt form and crystal form of pyridopyridone derivative |
| CN112292379A (en) * | 2018-06-13 | 2021-01-29 | 基石药业(苏州)有限公司 | Salt form and crystal form of pyridopyridone derivative |
| WO2019238088A1 (en) * | 2018-06-13 | 2019-12-19 | 基石药业 | Salt form and crystal form of pyridopyridone derivative |
| WO2020058820A1 (en) | 2018-09-18 | 2020-03-26 | Pfizer Inc. | Combinations of tgfb inhibitors and cdk inhibitors for the treatment of breast cancer |
| WO2020157709A1 (en) | 2019-02-01 | 2020-08-06 | Pfizer Inc. | Combination of a cdk inhibitor and a pim inhibitor |
| WO2020239558A1 (en) | 2019-05-24 | 2020-12-03 | Pfizer Inc. | Combination therapies using cdk inhibitors |
| WO2020240360A1 (en) | 2019-05-24 | 2020-12-03 | Pfizer Inc. | Combination therapies using cdk inhibitors |
| WO2021176349A1 (en) | 2020-03-05 | 2021-09-10 | Pfizer Inc. | Combination of an anaplastic lymphoma kinase inhibitor and a cyclin dependent kinase inhibitor |
| WO2022013369A1 (en) | 2020-07-15 | 2022-01-20 | Pfizer Inc. | Kat6 inhibitor methods and combinations for cancer treatment |
| WO2022018596A1 (en) | 2020-07-20 | 2022-01-27 | Pfizer Inc. | Combination therapy |
| US11471418B2 (en) | 2020-09-29 | 2022-10-18 | Shenzhen Pharmacin Co., Ltd. | Pharmaceutical compositions of amorphous solid dispersions and methods of preparation thereof |
| WO2022091001A1 (en) | 2020-10-29 | 2022-05-05 | Pfizer Ireland Pharmaceuticals | Process for preparation of palbociclib |
| CN112274493A (en) * | 2020-11-18 | 2021-01-29 | 石药集团中奇制药技术(石家庄)有限公司 | Preparation method of pipera cypress xili capsule |
| WO2022123419A1 (en) | 2020-12-08 | 2022-06-16 | Pfizer Inc. | Treatment of luminal subtypes of hr-positive, her2-negative early breast cancer with palbociclib |
| WO2023111810A1 (en) | 2021-12-14 | 2023-06-22 | Pfizer Inc. | Combination therapies and uses for treating cancer |
| CN114195784A (en) * | 2021-12-29 | 2022-03-18 | 斯坦德标准技术研究(湖北)有限公司 | Pabociclib related substance, preparation method and application thereof |
| EP4302755A1 (en) | 2022-07-07 | 2024-01-10 | Lotus Pharmaceutical Co., Ltd. | Palbociclib formulation containing an amino acid |
| EP4302832A1 (en) | 2022-07-07 | 2024-01-10 | Lotus Pharmaceutical Co., Ltd. | Palbociclib formulation containing glucono delta lactone |
| WO2024023703A1 (en) | 2022-07-29 | 2024-02-01 | Pfizer Inc. | Dosing regimens comprising a kat6 inhibitor for the treatment of cancer |
| WO2024049926A1 (en) | 2022-08-31 | 2024-03-07 | Arvinas Operations, Inc. | Dosage regimens of estrogen receptor degraders |
| WO2024132652A1 (en) | 2022-12-22 | 2024-06-27 | Synthon B.V. | Pharmaceutical composition comprising palbociclib |
| WO2024133726A1 (en) | 2022-12-22 | 2024-06-27 | Synthon B.V. | Pharmaceutical composition comprising palbociclib |
| WO2024201334A1 (en) | 2023-03-30 | 2024-10-03 | Pfizer Inc. | Kat6a as a predictive biomarker for treatment with a kat6a inhibitor and methods of treatment thereof |
| WO2024201340A1 (en) | 2023-03-30 | 2024-10-03 | Pfizer Inc. | Kat6a as a predictive biomarker for treatment of breast cancer with a cdk4 inhibitor and an antiestrogen and methods of treatment thereof |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10723730B2 (en) | Solid forms of a selective CDK4/6 inhibitor | |
| US20230167128A1 (en) | Polymorphs and solid forms of (s)-2-((2-((s)-4-(difluoromethyl)-2-oxooxazolidin-3-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)amino)propanamide, and methods of production | |
| NZ710138B2 (en) | Solid forms of a selective cdk4/6 inhibitor | |
| KR20160099101A (en) | Crystals (1) of pyrazino[2, 1-c][ 1, 2, 4]triazine compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14705884 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2900322 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 240277 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2014220354 Country of ref document: AU Date of ref document: 20140208 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014705884 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14769038 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 20157022487 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2015/010858 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112015019508 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2015132371 Country of ref document: RU Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 112015019508 Country of ref document: BR Kind code of ref document: A2 Effective date: 20150813 |