WO2014109258A1 - Method for producing living polymer on base, block copolymer, and film having microphase-separated structure produced using same and method for production thereof - Google Patents
Method for producing living polymer on base, block copolymer, and film having microphase-separated structure produced using same and method for production thereof Download PDFInfo
- Publication number
- WO2014109258A1 WO2014109258A1 PCT/JP2013/085051 JP2013085051W WO2014109258A1 WO 2014109258 A1 WO2014109258 A1 WO 2014109258A1 JP 2013085051 W JP2013085051 W JP 2013085051W WO 2014109258 A1 WO2014109258 A1 WO 2014109258A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- monomer
- support
- living
- producing
- polymer
- Prior art date
Links
- 0 C*C1=CCC(C)(C)C=C1 Chemical compound C*C1=CCC(C)(C)C=C1 0.000 description 2
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/38—Polymerisation using regulators, e.g. chain terminating agents, e.g. telomerisation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F293/00—Macromolecular compounds obtained by polymerisation on to a macromolecule having groups capable of inducing the formation of new polymer chains bound exclusively at one or both ends of the starting macromolecule
- C08F293/005—Macromolecular compounds obtained by polymerisation on to a macromolecule having groups capable of inducing the formation of new polymer chains bound exclusively at one or both ends of the starting macromolecule using free radical "living" or "controlled" polymerisation, e.g. using a complexing agent
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2438/00—Living radical polymerisation
- C08F2438/01—Atom Transfer Radical Polymerization [ATRP] or reverse ATRP
Definitions
- the present invention relates to a method for producing a living polymer on a support, a block copolymer, a microphase-separated structure membrane using the same, and a method for producing the same. More specifically, a method for producing a living polymer on a support (particularly, a production of a living polymer on a support capable of producing a block copolymer for forming a microphase-separated structure film on the support) Method), a block copolymer produced by a method for producing a living polymer on a support, a production method for producing a microphase separation structure film on a support, and a microphase separation structure film obtained by the production method About.
- Patent Documents 1 and 2 a block copolymer in which a hydrophilic polymer component (A) and a hydrophobic polymer component (B) are linked by a covalent bond, and the molecular weight distribution of A and B is ⁇ 1.3. Certain block copolymers have been described.
- Patent Document 2 a self-supporting polymer thin film made of a block copolymer formed by covalently bonding a hydrophilic polymer component and a hydrophobic polymer component having a crosslinkable structure, wherein the self-supporting polymer thin film is A self-supporting polymer thin film having a cylinder made of the hydrophilic polymer component oriented in a certain direction in the film and having the hydrophobic polymer component crosslinked is described.
- the methods described in these documents were epoch-making in that a microphase-separated structure can be produced in about several minutes.
- a hydrophilic compound is synthesized by atom transfer radical polymerization (hereinafter abbreviated as ATRP) with a liquid crystalline monomer after synthesizing a hydrophilic macroinitiator in Patent Document 1, for example.
- ATRP atom transfer radical polymerization
- a block copolymer in which hydrophobic portions are linked is obtained.
- patent document 2 after obtaining the living polymerization part only by a hydrophobic part by ATRP, the terminal is modified and finally the hydrophilic part is produced by polymer reaction.
- Non-Patent Document 2 describes a method and material for synthesizing a block copolymer by living radical polymerization by reversible addition-fragmentation chain transfer (hereinafter referred to as RAFT) polymerization.
- RAFT reversible addition-fragmentation chain transfer
- an appropriate chain transfer agent also called RAFT agent
- RAFT agent a reaction related to RAFT equilibrium is added to general free radical polymerization of substituted monomers, and the polymerization reaction is controlled by a reversible chain transfer reaction.
- Patent Document 2 describes a method of introducing a group capable of photodimerization into the hydrophobic polymer chain of the block copolymer and irradiating with ionizing radiation.
- Patent Documents 3 and 4 describe a method of introducing an oxetanyl group for immobilization of the obtained phase separation structure, and further related compounds are described in Patent Document 5.
- the problem to be solved by the present invention is to provide a method for producing a living polymer having excellent economic rationality.
- a living polymer can be provided on the support by providing a living polymer having a high economic rationality. .
- a method for producing a living polymer on a support comprising a step of subjecting the monomer-containing composition to living polymerization on the support.
- the living polymerization is preferably living radical polymerization.
- the step of living polymerizing the monomer-containing composition on the support may be 1000 seconds or less. preferable.
- the method for producing a living polymer on a support according to any one of [1] to [3], wherein the monomer consumption rate in the step of living polymerizing the monomer-containing composition on the support Is preferably controlled to 10 to 100%.
- the living polymer preferably has a number average molecular weight Mn of 1,000 to 100,000.
- the support is preferably a metal support.
- the metal is preferably copper.
- the living polymerization is preferably reversible addition-fragmentation chain transfer polymerization.
- the monomer-containing composition contains a polymerizable rod-like liquid crystal compound as a monomer component. preferable.
- the living polymer is preferably a block copolymer.
- the following steps (1) and (2) are preferably performed on the support.
- a monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms, and a monomer component containing an alkylene oxide chain and a carbon number of 2 (2) applying the monomer-containing composition onto the support using at least one of the monomer-containing compositions containing a polymer component containing 20 to 20 alkylene chains (2) the monomer-containing composition on the support [15]
- the manufacturing method of the living polymer on the support of the following steps (1 ′) and (2) In is preferably performed.
- a monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms is used as the monomer-containing composition.
- the monomer-containing composition is living-polymerized on the support so that the polymer component containing an alkylene oxide chain and the polymer component containing an alkylene chain having 2 to 20 carbon atoms are covalently bonded.
- Step [16] for obtaining a linked block copolymer [16] The method for producing a living polymer on a support according to [14], wherein the monomer component containing an alkylene chain having 2 to 20 carbon atoms is used.
- a polymerizable rod-like liquid crystal compound is preferable.
- [17] The method for producing a living polymer on a support according to any one of [14] to [16], wherein the block copolymer is a block copolymer for forming a microphase-separated structure film. It is preferably a coalescence.
- a method for producing a microphase separation structure membrane A method for producing a microphase separation structure membrane.
- the step of forming the microphase separation structure is preferably heating at 40 to 250 ° C.
- the method for producing a microphase-separated structure film according to [19] or [20] is further characterized in that (4) a block copolymer having the microphase-separated structure is crosslinked or polymerized on the support to form a micro It is preferable to include a step of immobilizing the phase separation structure.
- the method for producing a microphase separation structure film according to any one of [19] to [21] preferably includes a step of peeling the microphase separation structure film from the support.
- the thickness of the microphase separation structure film is preferably 1 to 2000 nm.
- FIG. 1 is a schematic view showing a GPC measurement area of a block copolymer in a GPC chart when a residual monomer and a residual initiator (when the initiator is a polymer) are mixed into the block copolymer.
- FIG. 2 is an AFM photograph of the microphase-separated structure film produced in Example 46.
- FIG. 3 is an AFM photograph of the microphase-separated structure film produced in Example 47.
- FIG. 4 is an AFM photograph of the microphase-separated structure film produced in Example 48.
- FIG. 5 is a schematic view of an example of an apparatus for forming a microphase-separated structure film by an example of a method for producing a living polymer on a support of the present invention.
- a numerical range expressed using “to” means a range including numerical values described before and after “to” as a lower limit value and an upper limit value.
- (meth) acrylate is used in the meaning containing both acrylate and methacrylate.
- the method for producing a living polymer on the support of the present invention includes a step of living polymerizing the monomer-containing composition on the support. With such a configuration, a method for producing a living polymer having excellent economic rationality can be provided. Generally, living polymerization in solution requires several hours (depending on the substrate), and the polymerization on the support in the production method of the present invention is economically reasonable from the viewpoint of time.
- the support used in the method for producing a living polymer on the support of the present invention is not particularly limited, and may be a glass substrate, a metallic support, or a resin film. .
- the living polymerization method is ATRP, it is preferable to use a metallic support as the support, but details will be described later.
- the living polymerization method is RAFT polymerization, it is preferable to use a glass substrate and a resin film.
- the monomer-containing composition contains a monomer component used for living polymerization.
- the said monomer containing composition may also contain another component as needed.
- a monomer component including an alkylene oxide chain may be used, or a monomer component including an alkylene chain having 2 to 20 carbon atoms may be used. It is preferable to use a monomer component containing an alkylene chain having 2 to 20 carbon atoms.
- the monomer component containing an alkylene oxide chain is a hydrophilic monomer, and the monomer component containing an alkylene chain having 2 to 20 carbon atoms is a hydrophobic monomer.
- the monomer component containing an alkylene oxide chain there is no restriction
- the monomer component containing a well-known alkylene oxide chain can be used.
- Examples of the monomer component containing an alkylene oxide chain include a compound in which methacrylic acid and acrylic acid and a compound obtained by alkyl etherifying one end of a polyol compound are linked by an ester bond.
- “A compound obtained by alkyl etherifying one end of a polyol compound” is obtained by etherifying a polyol compound described below with an alkyl compound having 1 to 10 carbon atoms, preferably 1 to 5 carbon atoms, more preferably methyl. Etherified (carbon number 1).
- polyol compound examples include polyether polyol, polyester polyol, polycarbonate polyol, polycaprolactone polyol, aliphatic hydrocarbon having two or more hydroxyl groups in the molecule, and fat having two or more hydroxyl groups in the molecule. Cyclic hydrocarbons, unsaturated hydrocarbons having two or more hydroxyl groups in the molecule, and the like are used. These polyols can be used alone or in combination of two or more.
- polyether polyol examples include aliphatic polyether polyols, alicyclic polyether polyols, and aromatic polyether polyols.
- aliphatic polyether polyol for example, polyethylene glycol, polypropylene glycol, polytetramethylene glycol, polyhexamethylene glycol, polyheptamethylene glycol, polydecamethylene glycol, pentaerythritol, dipentaerythritol, trimethylolpropane, and Ethylene oxide addition triol of trimethylolpropane, propylene oxide addition triol of trimethylolpropane, ethylene oxide and propylene oxide addition triol of trimethylolpropane, ethylene oxide addition tetraol of pentaerythritol, ethylene oxide addition hexaol of dipentaerythritol, etc.
- Polyhydric alcohol such as alkylene oxide addition polyol , Or two or more types of ionic-polymerizable cyclic compounds by ring-opening polymerization polyether polyols obtained can be mentioned.
- Examples of the ion-polymerizable cyclic compound include ethylene oxide, propylene oxide, butene-1-oxide, isobutene oxide, 3,3-bis (chloromethyl) oxetane, tetrahydrofuran, 2-methyltetrahydrofuran, dioxane, trioxane, tetraoxane, and cyclohexene.
- Examples include cyclic ethers.
- Specific combinations of the two or more types of ion-polymerizable cyclic compounds include tetrahydrofuran and ethylene oxide, tetrahydrofuran and propylene oxide, tetrahydrofuran and 2-methyltetrahydrofuran, tetrahydrofuran and 3-methyltetrahydrofuran, ethylene oxide and propylene oxide, butene-1- Examples thereof include oxide and ethylene oxide, tetrahydrofuran, butene-1-oxide, and ethylene oxide.
- a polyether polyol obtained by ring-opening copolymerization of the above ion-polymerizable cyclic compound with a cyclic imine such as ethyleneimine, a cyclic lactone acid such as ⁇ -propiolactone or glycolic acid lactide, or dimethylcyclopolysiloxane can also be used.
- Examples of the alicyclic polyether polyol include hydrogenated bisphenol A alkylene oxide addition diol, hydrogenated bisphenol F alkylene oxide addition diol, 1,4-cyclohexanediol alkylene oxide addition diol, and the like.
- aromatic polyether polyol examples include alkylene oxide addition diol of bisphenol A, alkylene oxide addition diol of bisphenol F, alkylene oxide addition diol of hydroquinone, alkylene oxide addition diol of naphthohydroquinone, alkylene oxide addition diol of anthrahydroquinone, etc. It is done.
- polyether polyol for example, as an aliphatic polyether polyol, PTMG650, PTMG1000, PTMG2000 (manufactured by Mitsubishi Chemical Corporation), PPG1000, EXCENOL1020, EXCENOL2020, EXCENOL3020, EXCENOL4020 (above, Asahi Glass Co., Ltd.) )), PEG1000, Unisafe DC1100, Unisafe DC1800, Unisafe DCB1100, Unisafe DCB1800 (above, manufactured by NOF Corporation), PPTG1000, PPTG2000, PPTG4000, PTG400, PTG650, PTG2000, PTG3000, PTGL1000, PTGL2000 (above, Hodogaya Chemical Industry Co., Ltd.), PPG400, PBG400, Z-300 -4, Z-3001-5, PBG2000, PBG2000B (Daiichi Kogyo Seiyaku Co., Ltd.), TMP
- the polyester polyol is obtained by reacting a polyhydric alcohol and a polybasic acid.
- a polyhydric alcohol ethylene glycol, polyethylene glycol, propylene glycol, polypropylene glycol, tetramethylene glycol, polytetramethylene glycol, 1,4-butanediol, 1,5-pentanediol, 1,6-hexanediol 1,7-heptanediol, 1,8-octanediol, neopentyl glycol, 1,4-cyclohexanediol, 1,4-cyclohexanedimethanol, 1,2-bis (hydroxyethyl) cyclohexane, 2,2-diethyl 1,3-propanediol, 3-methyl-1,5-pentanediol, 1,9-nonanediol, 2-methyl-1,8-octanediol, glycerin, trimethyl
- polyester polyols examples include phthalic acid, isophthalic acid, terephthalic acid, maleic acid, fumaric acid, adipic acid, sebacic acid and the like.
- examples of commercially available products of these polyester polyols include Kurapol P1010, Kurapol P2010, PMIPA, PKA-A, PKA-A2, PNA-2000 (manufactured by Kuraray Co., Ltd.) and the like.
- polycarbonate polyol examples include polycarbonate diols represented by the following general formula (i).
- R 1 represents an alkylene group having 2 to 20 carbon atoms, a (poly) ethylene glycol residue, a (poly) propylene glycol residue or a (poly) tetramethylene glycol residue, and m is 1 An integer in the range of ⁇ 30.
- R 1 include residues obtained by removing both terminal hydroxyl groups from the following compounds, that is, 1,4-butanediol, 1,5-pentanediol, neopentyl glycol, 1,6-hexanediol, 1,4 -Cyclohexanedimethanol, 1,7-heptanediol, 1,8-octanediol, 1,9-nonanediol, ethylene glycol, diethylene glycol, triethylene glycol, tetraethylene glycol, propylene glycol, dipropylene glycol, tripropylene glycol, Examples thereof include a residue obtained by removing a hydroxyl group from tetrapropylene glycol or the like.
- polycarbonate polyols Commercial products of these polycarbonate polyols include DN-980, DN-981, DN-982, DN-983 (manufactured by Nippon Polyurethane Industry Co., Ltd.), PC-8000 (manufactured by PPG), PNOC1000, PNOC2000, PMC100, PMC2000 (above, manufactured by Kuraray Co., Ltd.), Plaxel CD-205, CD-208, CD-210, CD-220, CD-205PL, CD-208PL, CD-210PL, CD-220PL, CD-205HL, CD-208HL, CD-210HL, CD-220HL, CD-210T, CD-221T (above, manufactured by Daicel Chemical Industries, Ltd.) and the like can be used.
- polycaprolactone polyol examples include ⁇ -caprolactone such as ethylene glycol, polyethylene glycol, propylene glycol, polypropylene glycol, tetramethylene glycol, polytetramethylene glycol, 1,2-polybutylene glycol, 1,6-hexanediol, neo
- polycaprolactone diols obtained by addition reaction with diols such as pentyl glycol, 1,4-cyclohexanedimethanol and 1,4-butanediol.
- Plaxel 205, 205AL, 212, 212AL, 220, 220AL above, manufactured by Daicel Chemical Industries, Ltd.
- Examples of the aliphatic hydrocarbon having two or more hydroxyl groups in the molecule include ethylene glycol, propylene glycol, 1,4-butanediol, 1,5-pentanediol, 1,6-hexanediol, and 1,7-heptanediol.
- Examples of the alicyclic hydrocarbon having two or more hydroxyl groups in the molecule include 1,4-cyclohexanediol, 1,4-cyclohexanedimethanol, 1,2-bis (hydroxyethyl) cyclohexane, dimethylol such as dicyclopentadiene. Compound, tricyclodecane dimethanol and the like.
- Examples of the unsaturated hydrocarbon having two or more hydroxyl groups in the molecule include hydroxy-terminated polybutadiene and hydroxy-terminated polyisoprene.
- examples of polyols other than the above include ⁇ -methyl- ⁇ -valerolactone diol, castor oil-modified diol, terminal diol compound of polydimethylsiloxane, polydimethylsiloxane carbitol-modified diol, and the like.
- These polyol compounds preferably have a weight average molecular weight of 1,000 to 10,000, particularly preferably 1,000 to 9000.
- the mass average molecular weight is a value measured by gel permeation chromatography (GPC) by dissolving a part of the polymer in tetrahydrofuran (THF).
- GPC gel permeation chromatography
- THF tetrahydrofuran
- the mass average molecular weight in the present invention is a value using polystyrene as a standard substance.
- polyethylene glycol monomethyl ether is polyethylene glycol monomethyl ether, and its number average molecular weight is from 100 to 10,000, preferably from 200 to 5,000, most preferably from 300 to 1,000.
- the monomer component including an alkylene chain having 2 to 20 carbon atoms is not particularly limited, and a known monomer component including an alkylene chain having 2 to 20 carbon atoms can be used.
- the monomer component containing an alkylene chain having 2 to 20 carbon atoms is preferably a liquid crystalline monomer from the viewpoint of low volatility and easy production of a microphase separation structure film described later.
- the reaction rate of living polymerization can be increased by performing polymerization in a liquid crystal field.
- the block copolymer of the present invention preferably has a mesogenic side chain having 6 to 50 carbon atoms in the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms. More preferably, the chain has at least one polymerizable group in one of the mesogenic side chains.
- the preferred range of the polymerizable group is the same as the preferred range of the polymerizable group represented by Q 1 and Q 2 in the general formula (X) described later.
- the mesogen side chain having 6 to 50 carbon atoms is more preferably a mesogen side chain having 6 to 40 carbon atoms, and particularly preferably a mesogen side chain having 6 to 30 carbon atoms.
- “Mesogenic group” includes a group capable of forming a core part of a liquid crystal compound, and examples of the compound having a mesogenic group include a liquid crystalline compound and also has a mesogenic group but does not form a liquid crystal. Sexual compounds are also included. The mesogenic group will be further described.
- the mesogenic group is a group showing a main skeleton of liquid crystal molecules that contribute to liquid crystal formation.
- the liquid crystal molecules exhibit liquid crystallinity that is an intermediate state (mesophase) between a crystalline state and an isotropic liquid state.
- the mesogenic group is not particularly limited.
- “Flushage Kristall in Tablen II” VEB Deutsche Verlagfur Grundoff Industrie, Leipzig, published in 1984
- pages 7 to 16 You can refer to the editions of the association, Liquid Crystal Handbook (Maruzen, 2000), especially the description in Chapter 3.
- a residue of a thermotropic liquid crystal is preferable, and a residue of a rod-like liquid crystal and a discotic liquid crystal is more preferable.
- a liquid crystal residue showing a nematic phase and a smectic A phase is more preferable, and in a discotic liquid crystal, a liquid crystal residue showing a discotic nematic phase is more preferable.
- Preferred examples of the residue of the discotic liquid crystal include benzene, triphenylene, truxene, trioxatruxene, anthraquinone, phthalocyanine or porphyrin, macrocyclene, bis (1,3-diketone) copper complex, tetraarylbipyranylidene, Tetrathiafulvalene and inositol are included.
- the main skeleton of the liquid crystal molecules contributing to the formation of a rigid liquid crystal is -Cy 1 -L 2- ( cy 2 -L 3) n -Cy 3 -L 4 - is preferably a group represented by.
- the mesogenic group is preferably a group represented by -Cy 1 -L 2- (Cy 2 -L 3 ) n -Cy 3 -L 4- in the general formula (X) described later.
- the more preferable range of the group represented by the above -Cy 1 -L 2- (Cy 2 -L 3 ) n -Cy 3 -L 4- is the same as the preferable range of each group in the general formula (X) described later. is there.
- a rod-like liquid crystal compound is more preferable.
- the remaining monomer component or polymer such as an initiator-derived polymer
- the liquid crystal monomer remains and the fluidity is increased, so that a beautiful cylindrical microphase separation structure free from defects can be formed.
- rod-like liquid crystal compounds examples include azomethines, azoxys, cyanobiphenyls, cyanophenyl esters, benzoic acid esters, cyclohexanecarboxylic acid phenyl esters, cyanophenylcyclohexanes, cyano-substituted phenylpyrimidines, alkoxy-substituted phenylpyrimidines, Phenyldioxanes, tolanes and alkenylcyclohexylbenzonitriles are preferably used.
- high-molecular liquid crystalline molecules can also be used.
- a polymerizable rod-like liquid crystal compound that can fix the orientation of the rod-like liquid crystal compound by polymerization is more preferable.
- the polymerizable rod-like liquid crystal compound is preferably a compound represented by the following general formula (X) and a compound represented by the following general formula (V), and a compound represented by the above general formula (X) Is more preferable.
- Q 1 and Q 2 are each independently a polymerizable group
- L 1 and L 4 are each independently a divalent linking group
- at least one of L 1 and L 4 is Including at least an alkylene group having 2 to 20 carbon atoms
- L 2 and L 3 are each independently a single bond or a divalent linking group
- Cy 1 , Cy 2 and Cy 3 are divalent cyclic groups
- n is 0, 1, 2, or 3.
- the polymerizable rod-like liquid crystal compound represented by the general formula (X) will be described below.
- Q 1 and Q 2 are each independently a polymerizable group.
- the polymerization reaction of the polymerizable group is preferably addition polymerization (including ring-opening polymerization) or condensation polymerization.
- the polymerizable group is preferably a functional group capable of addition polymerization reaction or condensation polymerization reaction. Examples of polymerizable groups are shown below.
- at least one of Q 1 and Q 2 is And the other of Q 1 and Q 2 is preferably a group having an oxetane ring (hereinafter also referred to as oxetanyl group).
- L 1 and L 4 are each independently a divalent linking group, and at least one of L 1 and L 4 includes at least an alkylene group having 2 to 20 carbon atoms.
- L 1 and L 4 each independently comprise —O—, —S—, —CO—, —NR—, —C ⁇ N—, a divalent chain group, a divalent cyclic group, and combinations thereof.
- a divalent linking group selected from the group is preferred.
- R is an alkyl group having 1 to 7 carbon atoms or a hydrogen atom.
- the example of the bivalent coupling group which consists of a combination is shown below. Here, the left side is coupled to Q (Q 1 or Q 2 ), and the right side is coupled to Cy (Cy 1 or Cy 3 ).
- L-1 —CO—O—divalent chain group —O— L-2: —CO—O—divalent chain group —O—CO— L-3: —CO—O—divalent chain group —O—CO—O— L-4: —CO—O—divalent chain group—O—divalent cyclic group— L-5: —CO—O—divalent chain group —O—divalent cyclic group —CO—O— L-6: —CO—O—divalent chain group —O—divalent cyclic group —O—CO— L-7: —CO—O—Divalent chain group—O—Divalent cyclic group—Divalent chain group— L-8: —CO—O—divalent chain group—O—divalent cyclic group—divalent chain group —CO—O— L-9: —CO—O—Divalent chain group—O—Divalent cyclic group—Divalent chain group—O—CO— L-10: —CO
- alkylene group, a substituted alkylene group, an alkenylene group and a substituted alkenylene group are preferred, and an alkylene group and an alkenylene group are more preferred.
- the alkylene group may have a branch.
- the alkylene group preferably has 2 to 20 carbon atoms, more preferably 3 to 18 carbon atoms, still more preferably 4 to 16 carbon atoms, and most preferably 5 to 15 carbon atoms.
- the total number of carbon atoms of all alkylene groups contained in the divalent linking group represented by L 1 and L 4 is preferably in the above range.
- a divalent chain group in L-1 to L-22 When a plurality of are included, the total carbon number of the divalent chain group which is an alkylene group is preferably within the above range.
- the alkylene part of the substituted alkylene group is the same as the above alkylene group.
- Examples of the substituent include a halogen atom.
- the alkenylene group may have a branch.
- the alkenylene group preferably has 2 to 12 carbon atoms, more preferably 2 to 10 carbon atoms, and most preferably 2 to 8 carbon atoms.
- the alkylene part of the substituted alkylene group is the same as the above alkylene group. Examples of the substituent include a halogen atom.
- the alkynylene group may have a branch.
- the alkynylene group preferably has 2 to 12 carbon atoms, more preferably 2 to 10 carbon atoms, and most preferably 2 to 8 carbon atoms.
- the alkynylene part of the substituted alkynylene group is the same as the above alkynylene group.
- Examples of the substituent include a halogen atom.
- Specific examples of the divalent chain group include ethylene, trimethylene, propylene, tetramethylene, 2-methyl-tetramethylene, pentamethylene, hexamethylene, octamethylene, 2-butenylene, 2-butynylene and the like.
- the definition and examples of the divalent cyclic group are the same as those of Cy 1 , Cy 2 and Cy 3 described later.
- One of the linking groups bonded to the oxetane group out of L 1 and L 4 is preferably L-21, and the other one of L 1 and L 4 is preferably L-1.
- R 2 is preferably an alkyl group having 1 to 4 carbon atoms or a hydrogen atom, more preferably a methyl group, an ethyl group or a hydrogen atom, and a hydrogen atom. Is most preferred.
- L 2 and L 3 are each independently a single bond or a divalent linking group.
- L 2 and L 3 each independently comprises —O—, —S—, —CO—, —NR—, —C ⁇ N—, a divalent chain group, a divalent cyclic group, and combinations thereof. It is preferably a divalent linking group or a single bond selected from the group.
- R is an alkyl group having 1 to 7 carbon atoms or a hydrogen atom, preferably an alkyl group having 1 to 4 carbon atoms or a hydrogen atom, and more preferably a methyl group, an ethyl group or a hydrogen atom. Preferably, it is a hydrogen atom.
- the divalent chain group and the divalent cyclic group are synonymous with the definitions of L 1 and L 4 .
- Preferred divalent linking groups for L 2 or L 3 include —COO—, —OCO—, —OCOO—, —OCONR—, —COS—, —SCO—, —CONR—, —NRCO—, —CH 2. CH 2 —, —C ⁇ C—COO—, —C ⁇ N—, —C ⁇ N—N ⁇ C—, and the like.
- L 2 and L 3 are preferably each independently —COO— or —OCO—.
- n is 0, 1, 2, or 3.
- two L 3 may be the same or different, and two Cy 2 may be the same or different.
- n is preferably 1 or 2, and more preferably 1.
- Cy 1 , Cy 2 and Cy 3 are each independently a divalent cyclic group.
- the ring contained in the cyclic group is preferably a 5-membered ring, 6-membered ring, or 7-membered ring, more preferably a 5-membered ring or 6-membered ring, and most preferably a 6-membered ring.
- the ring contained in the cyclic group may be a condensed ring. However, it is more preferably a monocycle than a condensed ring.
- the ring contained in the cyclic group may be any of an aromatic ring, an aliphatic ring, and a heterocyclic ring.
- Examples of the aromatic ring include a benzene ring and a naphthalene ring.
- Examples of the aliphatic ring include a cyclohexane ring.
- Examples of the heterocyclic ring include a pyridine ring and a pyrimidine ring.
- As the cyclic group having a benzene ring 1,4-phenylene is preferable.
- As the cyclic group having a naphthalene ring naphthalene-1,5-diyl and naphthalene-2,6-diyl are preferable.
- the cyclic group having a cyclohexane ring is preferably 1,4-cyclohexylene.
- the cyclic group having a pyridine ring is preferably pyridine-2,5-diyl.
- the cyclic group having a pyrimidine ring is preferably pyrimidine-2,5-diyl.
- the cyclic group may have a substituent. Examples of the substituent include a halogen atom, a cyano group, a nitro group, an alkyl group having 1 to 5 carbon atoms, a halogen-substituted alkyl group having 1 to 5 carbon atoms, and an alkoxy group having 1 to 5 carbon atoms.
- Examples of the polymerizable liquid crystal compound represented by the general formula (X) are shown below. The present invention is not limited to these.
- M 1 and M 2 are each independently a hydrogen atom, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aryl group, a heterocyclic group, a cyano group, a halogen, —SCN, —CF 3 , a nitro group, or Q 1 is represented, but at least one of M 1 and M 2 represents a group other than Q 1 .
- Q 1 , L 1 , L 2 , L 3 , L 4 , Cy 1 , Cy 2 , Cy 3 and n have the same meaning as the group represented by the general formula (X).
- P and q are 0 or 1.
- M 1 and M 2 do not represent Q 1 , it is preferably a hydrogen atom, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aryl group, or a cyano group, more preferably a carbon number It is preferably an alkyl group of 1 to 4 or a phenyl group, and p and q are preferably 0.
- one of the compounds having two or more acrylic groups and methacrylic groups may potentially contain a polymerizable group. That is, since a polymerizable group can be expressed from a compound as described below by a chemical reaction described in EP2130817, after one of the compounds having two or more acrylic groups and methacrylic groups is used for living polymerization, it is potentially polymerized. A portion containing a functional group may be treated by a chemical reaction to generate a polymerizable group.
- the polymerizable liquid crystal compounds 1 and 2 are described without any distinction, and are regarded as synonymous in the present invention.
- any solvent that does not inhibit the reaction can be used as the solvent that can be used in the monomer-containing composition.
- aromatic hydrocarbon solvents such as benzene, toluene and xylene
- aliphatic hydrocarbon solvents such as pentane, hexane, heptane, octane, nonane and decane
- cyclohexane methylcyclohexane and decahydronaphthalene.
- Alicyclic hydrocarbon solvents such as chlorobenzene, dichlorobenzene, trichlorobenzene, methylene chloride, chloroform, carbon tetrachloride and tetrachloroethylene, methanol, ethanol, n-propanol, iso-propanol, n Alcohol solvents such as butanol, sec-butanol and tert-butanol, ketone solvents such as acetone, methyl ethyl ketone and methyl isobutyl ketone, and esters such as ethyl acetate, butyl acetate and dimethyl phthalate System solvent, dimethyl ether, diethyl ether, and di -n- amyl ether, diphenyl ether, ether solvents such as tetrahydrofuran and anisole, dimethyl sulfoxide, N- dimethylformamide, dioxane or the like.
- chlorinated hydrocarbon solvents
- suspension polymerization and emulsion polymerization can be performed using water as a solvent.
- solvents may be used alone or in admixture of two or more.
- the reaction liquid becomes a homogeneous phase by using these solvents, but a plurality of non-uniform phases may be used.
- One of the preferable embodiments of the method for producing a living polymer on the support of the present invention is an embodiment in which the living polymerization is atom transfer radical polymerization, and the support is a metal support.
- ATRP Atom Transfer Radical Polymerization
- ATRP Atom Transfer Radical Polymerization
- This is a method of radical polymerization of a radically polymerizable monomer using a simple substance as a catalyst.
- ATRP As a preferable aspect of ATRP, specifically, for example, Matyjazewski et al., Chem. Rev. 101, 2921 (2001), WO 96/30421, WO 97/18247, WO 98/01480, WO 98/40415, WO 00/15695, or Sawamoto et al., Chem. Rev. 101, 3689 (2001), JP-A-8-411117, JP-A-9-208616, JP-A 2000-264914, JP-A 2001-316410, JP-A 2002-80523, JP No. 2004-307872 and the like.
- the initiator used for ATRP is not particularly limited, and examples thereof include organic halides and sulfonyl halide compounds.
- carbon-carbon double bonds or carbon atoms present at the ⁇ -position of carbon-oxygen double bonds A structure in which a halogen bond or a plurality of halogen atoms are added on one carbon atom is suitable as the initiator structure.
- a carbon-halogen bond present at the ⁇ -position of a carbon-carbon double bond, or a structure in which a plurality of halogens are added on one carbon atom can be used as an initiator structure.
- the initiator used for ATRP is preferably a macroinitiator containing a hydrophilic unit, more preferably a macroinitiator containing a polyalkyleneoxy structure, from the viewpoint of facilitating production of a microphase separation structure membrane.
- the polyalkyleneoxy structure is not particularly limited, but the alkyleneoxy group is preferably an alkyleneoxy group having 1 to 1000 carbon atoms, more preferably an alkyleneoxy group having 10 to 400 carbon atoms, Particularly preferred is a group.
- the number of repeating units of the alkyleneoxy structure is not particularly limited, but is preferably 1 to 500, more preferably 3 to 250, and particularly preferably 5 to 200.
- a commercially available initiator for ATRP may be used.
- examples of the initiator for low molecular weight ATRP include ethyl- ⁇ -bromoisobutyrate (manufactured by Aldrich).
- the ATRP initiator that is a macroinitiator may be produced by synthesis.
- the initiator for ATRP which is a macroinitiator, for example, the following can be used, but the present invention is not limited by the following specific examples.
- the amount of the ATRP initiator added is preferably 1 to 60% by mass, more preferably 5 to 55% by mass, and more preferably 10 to 45% by mass with respect to the monomer component. % Is particularly preferred.
- ligand Macromolecules, 2006, 39, 4953.
- the ligands described in (1) can be preferably used. 2,2′-bipyridine, 1,1,4,7,10,10-hexamethyltriethylenetetramine, N, N, N ′, N′-tetrakis (2-pyridylmethyl) ethylenediamine, tris [2- (dimethyl It is particularly preferred to use amino) ethyl] amine.
- the amount of the ligand added is preferably 0.1 to 40% by mass, more preferably 0.5 to 20% by mass, with respect to the monomer component. 10% by mass is particularly preferable.
- the method for producing a living polymer on the support of the present invention is not particularly limited as a polymerization catalyst for ATRP.
- transition metal complex used as a polymerization catalyst of ATRP is a metal complex which uses a periodic table group 7, 8, 9, 10, or 11 element as a central metal. More preferable examples include a complex of zero-valent copper, monovalent copper, divalent ruthenium, divalent iron, or divalent nickel. Of these, a copper complex is preferable. Specific examples of monovalent copper compounds include cuprous chloride, cuprous bromide, cuprous iodide, cuprous cyanide, cuprous oxide, cuprous perchlorate, etc. is there.
- a copper compound 2,2′-bipyridyl or a derivative thereof, 1,10-phenanthroline or a derivative thereof, or tetramethylethylenediamine, pentamethyldiethylenetriamine or hexamethyltris (2-aminoethyl) amine is used to increase the catalytic activity.
- Polyamines such as are added as ligands.
- a tristriphenylphosphine complex of divalent ruthenium chloride (RuCl 2 (PPh 3 ) 3 ) is also suitable as a catalyst.
- RuCl 2 (PPh 3 ) 3 is also suitable as a catalyst.
- an aluminum alkoxide is added as an activator.
- a divalent iron bistriphenylphosphine complex FeCl 2 (PPh 3 ) 2
- a divalent nickel bistriphenylphosphine complex NiCl 2 (PPh 3 ) 2
- a divalent nickel bistributylphosphine A complex NiBr 2 (PBu 3 ) 2
- the simple substance of the transition metal used as the polymerization catalyst for ATRP is not particularly limited, but is preferably a metal belonging to Group 7, 8, 9, 10, or 11 of the periodic table. Further preferred are copper, ruthenium, iron or nickel. Of these, copper is preferable.
- a metal support is used as the support, and the metal support is also used as an ATRP polymerization catalyst.
- Living polymerization on a metal support uses a metal support as a catalyst to save the trouble of removing the metal from the composition after ordinary ATRP in which the metal catalyst is added to the monomer-containing composition.
- the metal support is preferably a metallic film.
- a metallic film having a thickness of 5 to 50 ⁇ m is preferably used from the viewpoint of operation and economic rationality.
- a metal film having a width of 100 to 500 mm is preferably used from the viewpoint of continuous production.
- the metal in the metal support is preferably copper.
- a copper film can be preferably used.
- the metallic support a commercially available support may be used.
- examples of the copper film include Nippon Foil Co., Ltd., rolled copper foil TCU, and the like.
- the surface of the monomer-containing composition that is not in contact with the metal support is covered with another support, and the monomer-containing composition is sandwiched. It is preferable to heat from the viewpoint of making it difficult to volatilize the materials used (the monomer component and the initiator). From the viewpoint of further providing the ATRP polymerization catalytic activity, it is preferable to use another sheet of the above-mentioned metal support.
- the metal in the metal film is preferably copper.
- the preferable range of the metal film used in this case is the same as the preferable range of the metallic support.
- the pressure applied when the monomer-containing composition is sandwiched is not particularly limited, but is preferably 0.1 to 500 KPa, for example, from the viewpoint of production.
- the copper ion concentration in the said living polymer is 5 ppm or less with respect to the said living polymer.
- the copper ion concentration in the living polymer is 100 ppm with respect to the living polymer. It was a degree exceeding.
- RAFT Reversible Addition Fragmentation Chain Transfer
- One of the preferable embodiments of the method for producing a living polymer on the support of the present invention is an embodiment in which the living polymerization is a reversible addition-fragmentation chain transfer polymerization.
- the monomer-containing composition preferably contains a polymerizable rod-like liquid crystal compound as a monomer component.
- RAFT agent used by RAFT polymerization
- a well-known RAFT agent can be used.
- Macromolecules, 2006, 39, 4953. Can be mentioned.
- RAFT agent a commercially available RAFT agent may be used.
- 4-cyano-4-[(dodecylsulfanyl-thiocarbonyl) sulfanyl] pentanoic acid or 2-cyano-2-propylbenzodithioate is used as a low molecular weight RAFT agent. (Both manufactured by Aldrich).
- the addition amount of the RAFT agent is preferably 1 to 60% by mass, more preferably 5 to 55% by mass, and more preferably 10 to 45% by mass with respect to the monomer component. It is particularly preferred.
- radical polymerization initiator A commercially available radical polymerization initiator can be used as the radical polymerization initiator. V-70, V-60 (AIBN), V-40, V-65, V-601, V-59, V-30, V-501 and the like manufactured by Wako Pure Chemical Industries are preferred, and AIBN is particularly preferred.
- the addition amount of the radical polymerization initiator is preferably 0.01 to 10% by mass, more preferably 0.1 to 5% by mass, based on the monomer component. It is particularly preferably 2 to 1% by mass.
- RAFT polymerization examples include the same monomers as those described above.
- a photopolymerization initiator when forming a microphase-separated structure film using the method for producing a living polymer on the support of the present invention, a polymerizable rod-like liquid crystal compound is used as the monomer component, and a photopolymerization initiator is further used. Is preferably added.
- the phase separation structure can be easily fixed by using the polymerizable rod-like liquid crystal compound and the photopolymerization initiator in combination.
- a photopolymerization initiator a compound known as a photoacid generator is preferable.
- a photoacid generator described in JP2012-150428A can be used.
- the photoacid generator for example, the following can be used, but the present invention is not limited by the following specific examples.
- the addition amount of the photopolymerization initiator is preferably 0.01 to 10% by mass, more preferably 0.1 to 5% by mass with respect to the monomer component.
- the content is particularly preferably 0.2 to 1% by mass.
- additives may be added to the monomer-containing composition.
- additives include additives that can be used in combination with a polymerizable rod-like liquid crystal compound as described in Liquid Crystal Handbook, Liquid Crystal Handbook Editorial Committee, Maruzen Co., Ltd., and the like.
- the polymerization conditions and the polymerization method are not particularly limited as long as the living polymerization can be performed on the support. Bulk polymerization, suspension polymerization, emulsion polymerization, block Turbid polymerization or the like can be applied.
- film formation by a solution process refers to a method in which an organic compound is dissolved in a solvent capable of dissolving, and the solution is applied onto a substrate and dried to form a film.
- the present invention it is more preferable to use a casting method, a spin coating method, and an ink jet method.
- a thin film having a smooth surface and a large area can be produced at a low cost.
- the spin coating is preferably performed, for example, at 100 to 5000 revolutions / minute for 5 to 60 seconds.
- the preferable range of the thickness after application of the monomer-containing composition in the step of applying the monomer-containing composition on the support is the same as the preferable range of the thickness of the microphase-separated structure film of the present invention described later.
- surface modification or guides may or may not be performed on the support. It is preferable not to perform from a viewpoint of chemical conversion.
- a micro phase separation structure described later is formed without surface modification or guide to the support. Can do.
- the amphiphilic liquid crystal block copolymer described later is formed by the production method of the present invention, it is possible to form a clean cylindrical microphase separation structure without defects even without surface modification or guide to the support. it can.
- the step of living polymerizing the monomer-containing composition on the support is preferably 1000 seconds or less, more preferably 800 seconds or less. Preferably, it is particularly preferably 600 seconds or less.
- the lower limit of the time for the step of living polymerizing the monomer-containing composition on the support is not particularly limited, but is preferably 5 seconds or more.
- reaction temperature may be any temperature as long as the polymerization reaction proceeds, and is not uniform depending on the desired degree of polymerization of the polymer and the type and amount of the polymerization initiator and solvent used. ° C.
- the step of living polymerizing the monomer-containing composition on the support is preferably heating at 50 to 200 ° C., more preferably 60 ° C to 180 ° C, more preferably 80 ° C to 160 ° C.
- a heating method A well-known method can be used, For example, the method of mounting the said support body on a hotplate and heating the said monomer containing composition with the said support body etc. can be mentioned.
- after heating it is preferable to cool to room temperature finally and to obtain a living polymer.
- the living polymerization reaction in the step of living polymerizing the monomer-containing composition on the support can be carried out under reduced pressure, normal pressure, or increased pressure depending on the case.
- the polymerization reaction is preferably carried out under a flow of an inert gas such as nitrogen or argon, more preferably under a flow of nitrogen gas, particularly when the initiator is a low molecular weight initiator.
- an inert gas such as nitrogen or argon
- nitrogen gas preferably under a flow of nitrogen gas, particularly when the initiator is a low molecular weight initiator.
- the flow conditions of the inert gas it may be, for example, 0.001 to 50 L / min.
- the monomer consumption rate in the step of living polymerizing the monomer-containing composition on the support is controlled to 10 to 100%. From the viewpoint of rapid formation, it is preferable.
- the monomer consumption rate is more preferably 20 to 100%, and particularly preferably 30 to 100%.
- the monomer consumption rate described above can be determined by performing 1 H-NMR (BRUKER-300 MHZ) measurement on an extract obtained by extracting the obtained living polymer with deuterated THF.
- a living polymer is manufactured by the manufacturing method of the living polymer on the support body of this invention.
- the living polymer is preferably formed into a film on the support.
- the number average molecular weight Mn of the resulting living polymer is preferably 1,000 to 100,000, more preferably 2,000 to 50,000, and more preferably 3,000 to 30,000. It is particularly preferred.
- the method for producing the living polymer on the support of the present invention is such that the ratio Mw / Mn of the weight average molecular weight Mw and the number average molecular weight Mn of the resulting living polymer extract exceeds 1.3 is a residual monomer. Is preferable from the viewpoint of repairing defects in the phase separation structure, and the residual macroinitiator and the residual macro RAFT agent from controlling the size of the phase separation structure. On the other hand, Mw / Mn is preferably 1.3 or less from the viewpoint of stably forming the phase separation structure.
- the number average molecular weight Mn of a block copolymer, a polymer component containing an alkylene oxide chain, a polymer component containing an alkylene chain having 2 to 20 carbon atoms, and the like is a value measured by the following method.
- the polymer was dissolved in THF, and a high-speed GPC (HLC-8220GPC) manufactured by Tosoh was used.
- the number average molecular weight Mn was calculated in terms of polystyrene.
- the weight average molecular weight Mw of the block copolymer, the polymer component containing an alkylene oxide chain, the polymer component containing an alkylene chain having 2 to 20 carbon atoms, and the like is a value measured by the following method. .
- the polymer was dissolved in THF, and a high-speed GPC (HLC-8220GPC) manufactured by Tosoh was used.
- the weight average molecular weight Mw was calculated in terms of polystyrene.
- the living polymer is preferably a block copolymer.
- the manufacturing method of the living polymer on the support body of this invention it is more preferable to implement the following process (1) and (2) on a support body.
- a monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms, and a monomer component containing an alkylene oxide chain and a carbon number of 2 (2) applying the monomer-containing composition onto the support using at least one of the monomer-containing compositions containing a polymer component containing 20 to 20 alkylene chains (2) the monomer-containing composition on the support Is a step of living polymerizing to obtain a block copolymer in which a polymer component containing an alkylene oxide chain and a polymer component containing an alkylene chain having 2 to 20 carbon atoms are linked by a covalent bond
- the following steps (1 ′) and (2) are particularly preferably carried out on the support.
- (1 ′) A monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms is used as the monomer-containing composition.
- the monomer-containing composition is living-polymerized on the support so that the polymer component containing an alkylene oxide chain and the polymer component containing an alkylene chain having 2 to 20 carbon atoms are covalently bonded.
- Preferred production conditions for the steps (1) and (1 ′) are the same as the preferred production conditions for the step (i).
- Preferred production conditions for the step (2) are the same as the preferred production conditions for the step (ii).
- the block copolymer is preferably a block copolymer for forming a microphase-separated structure film.
- the block copolymer reaction mixture is once taken out and purified, and then the block copolymer is applied to a substrate (or support) to form a phase separation structure. It is common to make it.
- the phase separation structure can be formed on the support as it is after the living polymerization is performed on the support, which is further economically rational. .
- the block copolymer of the present invention is characterized in that, in the method for producing a living polymer on the support of the present invention, the block copolymer is obtained under the condition that the living polymer becomes a block copolymer.
- the conditions for the living polymer to be a block copolymer include the method of performing the steps (1) and (2) described above, and the method of performing the steps (1 ′) and (2) described above. Is preferred.
- the preferred molecular weight range of the obtained block copolymer is the same as the preferred molecular weight range of the living polymer obtained by the method for producing a living polymer on the support of the present invention.
- block copolymer of the present invention is not particularly limited, but the block copolymer of the present invention is preferably a block copolymer for forming a microphase-separated structure film.
- the block copolymer of the present invention is not particularly limited in structure, but the polymer component (A) containing an alkylene oxide chain and the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms are incompatible with each other. It is a block copolymer in which a polymer is bonded by a covalent bond, and the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms preferably has a mesogenic side chain having 6 to 50 carbon atoms. Such a block copolymer is sometimes called an amphiphilic liquid crystal block copolymer.
- the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms is preferably a poly ((meth) acrylate) having a mesogenic side chain having 6 to 50 carbon atoms, More preferably, it contains a polymer component derived from the polymerizable rod-like liquid crystal compound.
- the polymer component (A) containing an alkylene oxide chain preferably contains a polyalkyleneoxy structure, and more preferably contains a polymer component derived from the macroinitiator.
- the block copolymer of the present invention can be bonded via a linking group. It may be bonded. Because of less coloring, in the light-related application, the following divalent linking group is bonded to the linking part of the polymer component (A) containing an alkylene oxide chain and the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms. It may be a block copolymer containing any of them.
- the method for introducing at least one of the divalent linking groups represented by the following general formulas (11) and (12) into the linking part of the polymer component (A) and the polymer component (B) is not particularly limited. Absent. For example, a method using a polymer component (A) containing an alkylene oxide chain, that is, a hydrophilic macro RAFT agent having at least one of divalent linking groups represented by the following general formulas (11) and (12) Can be mentioned.
- Examples of the block copolymer of the present invention include those represented by the following general formulas (I-1) and (I-2).
- X represents the structure described in the following general formula (3) and a halogen atom (iodine, bromine, chlorine atom) or a hydrogen atom.
- n and m each independently represent an integer of 1 to 500
- L 11 and L 12 each independently represents a single bond or an alkylene group, an arylene group, an alkenylene.
- L 12 comprises an alkylene group having at least carbon atoms 2 ⁇ 20.
- R 1 each independently represent a hydrogen atom or a methyl group
- R 2 is independently an oxetanyl group
- an epoxy group an acrylate group Represents a methacrylate group, an optionally substituted alkyl group or an optionally substituted alkoxy group
- M represents a mesogenic group having 6 to 50 carbon atoms
- Z 3 represents substituted or unsubstituted.
- An aryl group and a substituted or unsubstituted alkyl group, provided that m repeating units may be the same or different.
- Z 3 is preferably a substituted or unsubstituted aryl group, a substituted or unsubstituted alkyl group, and preferably a substituted or unsubstituted alkyl group. More preferably, it is an alkyl group having 4 to 20 carbon atoms.
- n is preferably 1 to 500, more preferably 3 to 250, and particularly preferably 5 to 200.
- m is preferably 3 to 200, more preferably 5 to 150, and particularly preferably 10 to 100.
- L 11 and L 12 are each independently a single bond, a functional group described below, or two hydrogen atoms removed from a functional group described below.
- the divalent linking group represented by For example, phenylene, naphthylene), alkenylene group (carbon number 2 to 20, for example, ethenylene, propenylene), alkynylene group (carbon number 2 to 20, for example, ethynylene, propynylene), metallocenylene group (for example, ferrocene), —CO—N (R 101 ) -, - CO-O - , - SO 2 -N (R 102) -, - SO 2 -O -, - N (R 103) -CO-N (R 104) -, SO 2 -, - SO -, - S -, - O -, - CO -, - N (R 105) -, heterylene group (carbon number 1 to 26, for example 6-chloro-1,3,5-triazyl - 2,4-diyl group, pyrimidine-2,4-diyl group) or a combination of one
- L 12 contains at least an alkylene group having 2 to 20 carbon atoms.
- the alkylene group contained in L 12 preferably has 3 to 18 carbon atoms, more preferably 4 to 16 carbon atoms, and particularly preferably 5 to 15 carbon atoms.
- R 101 , R 102 , R 103 , R 104 , and R 105 each independently represent a hydrogen atom, a substituted or unsubstituted alkyl group, or a substituted or unsubstituted aryl group.
- One or more linking groups represented by L 11 and L 12 may be present, and a plurality (preferably two) of the linking groups may be bonded to form a ring.
- R 1 is preferably independently a hydrogen atom or a methyl group from the viewpoint of versatility.
- M is preferably a mesogenic group having 6 to 50 carbon atoms, more preferably a mesogenic group having 6 to 40 carbon atoms, Particular preference is given to 30 mesogenic groups.
- a preferable range of the mesogenic group having 6 to 50 carbon atoms represented by M is a preferable range of the above -Cy 1 -L 2- (Cy 2 -L 3 ) n -Cy 3 -L 4-in the general formula (X). It is the same.
- R 2 is preferably a polymerizable group, such as an oxetanyl group, an epoxy group, an acrylate group, or a methacrylate group, and preferably an oxetanyl group or an epoxy group. More preferred.
- the block copolymer of the present invention has a copolymerization ratio (number average molecular weight ratio) of the polymer component (A) containing an alkylene oxide chain and the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms is 65. : 35 to 1:99 is preferable, 55:45 to 5:95 is more preferable, and 45:55 to 10:90 is particularly preferable.
- the method for producing a microphase-separated structure film of the present invention includes (3) forming the microphase-separated structure by promoting phase separation of the block copolymer of the present invention on the support by heat, light, solvent vapor or electric field. Including the step of: As a method of promoting phase separation by heat, light, solvent vapor or electric field to the block copolymer of the present invention for forming a microphase separation structure, a method of promoting phase separation by heating is preferable.
- FIG. 5 shows a schematic diagram of an example of an apparatus for producing a microphase separation structure membrane.
- the step of feeding the support 21 from the feed roll 11, applying the monomer-containing composition 22 onto the support by the coating device 12, and applying the monomer-containing composition onto the support is performed.
- the monomer-containing composition provided on the support is subjected to living polymerization of the monomer-containing composition on the support in the first aging zone 14 after passing through the drying zone 13.
- a step of forming a microphase separation structure on the support is performed, and the monomer-containing composition is subjected to high-speed phase separation.
- the micro phase separation structure is fixed in the UV irradiation zone 16.
- the micro phase separation structure film is peeled off from the support 21 by the peeling roll 17.
- the support 21 may be wound up by the winding roll 18.
- the method for producing a microphase-separated structure film of the present invention is such that (3) the step of heating the block copolymer of the present invention on the support to form a microphase-separated structure is 5 to 1000 seconds. It is preferably 10 to 600 seconds, more preferably 30 to 200 seconds.
- the method for producing a microphase-separated structure film of the present invention is such that (3) the step of heating the block copolymer of the present invention on the support to form a microphase-separated structure has a heating start temperature of 40 to 250. It is preferably 0 ° C., more preferably 50 ° C. to 200 ° C., and still more preferably 60 ° C. to 190 ° C.
- a heating start temperature 40 to 250. It is preferably 0 ° C., more preferably 50 ° C. to 200 ° C., and still more preferably 60 ° C. to 190 ° C.
- a heating method in the said (3) process A well-known method can be used, for example, the said support body is mounted on a hotplate and the said monomer containing composition is heated with the said support body etc. Can be mentioned.
- the temperature is finally lowered to the start temperature (irradiation temperature of active radiation) of the step (4) described later.
- the rate of temperature decrease from the start of heating to the UV irradiation temperature is preferably 1 to 100 ° C./min, more preferably 5 to 80 ° C./min, and particularly preferably 10 to 50 ° C./min. .
- the method for producing a microphase separation structure membrane of the present invention further includes (4) a step of crosslinking or polymerizing the block copolymer having the microphase separation structure on the support to fix the microphase separation structure. It is preferable. There is no particular limitation on the method of fixing the microphase separation structure by crosslinking or polymerizing the block copolymer having the microphase separation structure on the support, but the block copolymer having the microphase separation structure is not limited. It is preferable that the polymer is irradiated with actinic radiation, and UV irradiation is more preferable.
- the method for producing a microphase-separated structure film of the present invention comprises (4) actinic radiation in the step of immobilizing the microphase-separated structure by crosslinking or polymerizing the block copolymer having the microphase-separated structure on the support.
- the irradiation temperature is preferably 40 to 200 ° C., more preferably 50 to 200 ° C., and still more preferably 60 to 200 ° C.
- the irradiation of the active radiation is preferably from 50 ⁇ 2000mJ / cm 2, is preferably 100 ⁇ 1500mJ / cm 2, 200 ⁇ 1000mJ / cm 2 It is particularly preferred that There is no restriction
- the method for producing a microphase separation structure membrane of the present invention preferably includes a step of peeling the microphase separation structure membrane from the support.
- the microphase-separated structure film of the present invention is manufactured by the method for manufacturing a microphase-separated structure film of the present invention.
- AFM atomic force microscope
- the microphase separation structure film obtained by the method for producing a microphase separation structure film of the present invention is formed on a substrate, but may be used after peeling from the substrate.
- the thickness of the microphase separation structure film is preferably 1 to 2000 nm.
- the microphase separation structure film is preferably a cylinder type microphase separation structure film.
- the cylinder type micro phase separation structure membrane means a membrane in which a large number of cylinders to which the polymer component containing the alkylene oxide chain is fixed are arranged in a plane, and each cylinder is aligned at equal intervals. preferable.
- the height of each cylinder is preferably 1 to 2000 nm, and more preferably 1 to 500 nm.
- the diameter of each cylinder is preferably 1 to 1000 nm, and more preferably 1 to 200 nm.
- the microphase-separated structure film obtained by the method for producing a microphase-separated structure film of the present invention is a high-density recording such as an optical / electronic functional material (for example, a brightness enhancement film), an energy-related material, a surface modifying material, and a patterned medium
- Microphase separation consisting of controlled-oriented block copolymers useful as materials, various nanofilters (permeation membranes, ultrafiltration membranes, nanoreactors), anisotropic ion conductive membranes, anisotropic conductive films, etc. It is a structural film.
- Such a microphase separation structure membrane can also be used as a porous structure by removing the cylinder structure portion.
- Porous structures obtained from microphase separation structure membranes are anisotropic, such as polymer electrolytes for fuel cells, ion exchange resins, thin films for microreactors, protein separation membranes, organic zeolites, and templates for various pillars. It can be used as a conductive ion conductive material. Such a microphase-separated structure film can also introduce another substance by removing the cylinder structure portion.
- Ethyl- ⁇ -bromoisobutyrate (manufactured by Aldrich) is used as a commercially available low-molecular ATRP initiator (3).
- Hydrophilic macro RAFT agent A-2 A commercially available hydrophilic macro RAFT agent A-2 (poly (ethylene glycol) methyl ether (4-cyano-4- (dodecylsulfanylthiocarbonyl) sulfanyl) pentanoate) (Mn10000, manufactured by Aldrich)) is used.
- Hydrophilic macroinitiator A-3 30 g of commercially available poly (ethylene glycol) methyl ether (Mn5000, manufactured by Aldrich) and 0.81 g of N, N′-dimethylaminopyridine were dissolved in 180 ml of methylene chloride, and 2-bromoisolacsan bromide (manufactured by Tokyo Chemical Industry Co., Ltd.) 1 .52 g was dissolved in 20 ml of methylene chloride and then added dropwise at room temperature, followed by heating at 40 ° C. and stirring for 48 hours.
- Mn5000 poly (ethylene glycol) methyl ether
- 2-bromoisolacsan bromide manufactured by Tokyo Chemical Industry Co., Ltd.
- Liquid crystalline monomer B-1 was synthesized by the synthesis method described in Transactions of Materials Research Society of Japan, 28 [3], 553-556, (2003).
- Liquid crystalline monomer B-2 was synthesized by the synthesis method described in Example 1 of JP-A-2008-127336.
- Liquid crystalline monomer B-4 was synthesized based on the synthesis method described in JP 2010-116463 A.
- Liquid crystalline monomer B-5 was synthesized according to the synthesis method described below.
- Hydroquinone monomethyl ether (37 mg) was added to a THF solution (17 mL) of methanesulfonyl chloride (33.0 mmol, 2.6 mL), and the internal temperature was cooled to ⁇ 5 ° C.
- a THF solution (16 mL) of a-1 (31.5 mmol, 8.33 g) and diisopropylethylamine (33.0 mmol, 5.75 mL) was added dropwise so that the internal temperature did not rise above 0 ° C. After stirring at ⁇ 5 ° C.
- Hydroquinone monomethyl ether (7 mg) was added to a THF solution (3 mL) of methanesulfonyl chloride (6.0 mmol, 0.46 mL), and the internal temperature was cooled to ⁇ 5 ° C.
- a THF solution (6 mL) of carboxylic acid d-1 (5.5 mmol, 2.1 g) and diisopropylethylamine (6.0 mmol, 1.1 mL) was added dropwise so that the internal temperature did not rise above 0 ° C. After stirring at ⁇ 5 ° C.
- Example 1 Living polymer synthesis example>
- Low molecular weight RAFT agent (1) (0.0081 g), liquid crystalline monomer B-1 (0.493 g), and azobisisobutyronitrile (hereinafter abbreviated as AIBN, 0.0028 g) were dissolved in 5 ml of toluene.
- AIBN azobisisobutyronitrile
- 50 ⁇ l of the obtained toluene solution was applied onto a glass substrate (2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds using a spin coater (SPINCATOR 1H-D7 manufactured by MIKASA).
- Example 1 Under a nitrogen flow (0.5 L / min), heating was performed according to the table below on a hot plate (DATAPLATE manufactured by PMC). Thereafter, the mixture was allowed to cool to room temperature to obtain a living polymer 1 (Example 1).
- the film-like living polymer of Example 1 was extracted with deuterated THF, and monomer consumption and GPC (HLC-8220GPC manufactured by TOSOH) were analyzed by 1 H-NMR (BRUKER-300 MHZ) measurement.
- Mw represents the weight average molecular weight
- Mn represents the number average molecular weight
- PDI represents Mw / Mn, which means the molecular weight distribution.
- the results obtained are listed in the table below.
- Examples 2 to 5 Living polymer synthesis examples> Living polymers were obtained in the same manner as in Synthesis Example 1 except that equimolar amounts of the liquid crystalline monomer used in Example 1 were used and B-1 was changed to B-2 to 5 (Examples 2 to 5). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
- Examples 6 to 10 Living polymer synthesis examples> Living polymers were obtained in the same manner as in Example 1 except that equimolar amounts of the low molecular weight RAFT agent (1) used in Example 1 were used to change to the low molecular weight RAFT agent (2) (Examples 6 to 10). ). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
- Example 11 to 22 Examples of block copolymer synthesis> Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g) and AIBN (0.0028 g) were dissolved in 5 ml of toluene. 50 ⁇ l of the obtained toluene solution was applied onto a glass substrate (2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds using a spin coater (SPINCATOR 1H-D7 manufactured by MIKASA). Under a nitrogen flow (0.5 L / min), heating was performed according to the table below on a hot plate (DATAPLATE manufactured by PMC).
- SPINCATOR 1H-D7 manufactured by MIKASA
- Example 11 to 22 block copolymers 11 to 22 (Examples 11 to 22).
- the film-like block copolymers 11 to 22 of Examples 11 to 22 were extracted with deuterated THF, and monomer consumption and GPC (HLC-8220GPC manufactured by TOSOH) were measured by 1 H-NMR (BRUKER-300 MHZ) measurement. The analysis was conducted. The results obtained are listed in the table below.
- Examples 23 to 31 Examples of block copolymer synthesis Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g) and AIBN (0.0028 g) were dissolved in 5 ml of toluene. 50 ⁇ l of the obtained toluene solution was applied on a glass substrate (2 cm square) and spin-coated by rotating at 1500 rpm for 30 seconds. In the atmosphere, the glass substrate was heated on a hot plate according to the table below. Thereafter, the mixture was allowed to cool to room temperature to obtain block copolymers 23 to 31 (Examples 23 to 31). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
- Examples 32 to 35 Examples of block copolymer synthesis Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystal monomer B-2 (0.677 g), 1,1′-azobis (cyclohexane-1-carbonitrile) (hereinafter abbreviated as V-40) 0043 g) was dissolved in 5 ml of toluene. 50 ⁇ l of the obtained toluene solution was applied on a glass substrate (2 cm square). As a protective cover for the glass substrate, a slide glass was placed and heated according to the following table on a hot plate under a nitrogen flow (0.5 L / min). Thereafter, the mixture was allowed to cool to room temperature to obtain block copolymers 32-35 (Examples 32-35). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
- Examples 36 to 42 Synthesis example of block copolymer> Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g), and V-40 (0.0043 g) were dissolved in 5 ml of toluene. 50 ⁇ l of the obtained toluene solution was applied on a glass substrate (2 cm square). It heated according to the following table
- Examples 43 to 45 Block copolymer synthesis examples> Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g), and V-40 (0.0043 g) were dissolved in 5 ml of toluene. 50 ⁇ l of the obtained toluene solution was applied on a glass substrate (2 cm square). Heating was performed according to the following table on a hot plate in the atmosphere. Thereafter, the mixture was allowed to cool to room temperature to obtain block copolymers 43 to 45 (Examples 43 to 45). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
- a glass substrate is heated on a hot plate under a nitrogen flow (0.5 L / min) (ripening 1), and the temperature is lowered from a predetermined starting temperature to a UV irradiation temperature in accordance with the following table in the atmosphere using another hot plate. (Maturation 2), and cured at 500 mJ / cm 2 with a UV irradiation apparatus (EXECURE 3000 manufactured by HOYA).
- AFM measurement The block copolymer on the glass substrate produced by the method for producing a living polymer on the glass substrate (Examples 46 to 48) was measured with AFM (SPI3800N manufactured by SII). Representative examples of the measured surface shape are shown in FIGS. Lamella and cylindrical microphase separation structures were observed.
- Examples 46 to 48 it was possible to peel the microphase separation structure film from the glass substrate.
- the thicknesses of the microphase-separated structure films peeled in Examples 46 to 48 were 150 nm, 200 nm, and 210 nm, respectively.
- a living polymer (particularly a block copolymer) is used in the case of RAFT polymerization. It was found that it can be economically and reasonably manufactured in a short time. It was also found that a microphase separation structure membrane can be produced economically rationally by using the method for producing a living polymer on the support of the present invention.
- Example 101 Living polymer synthesis example> ATRP initiator A-3 (0.0039 g), liquid crystalline monomer B-1 (0.492 g), N, N, N ′, N ′′, N ′′ -pentamethyldiethylenetriamine (hereinafter abbreviated as PMDETA) 021 g was dissolved in 5 ml of toluene. 50 ⁇ l of the obtained toluene solution was applied onto a copper film (Nihon Foil Co., Ltd., rolled copper foil TCU, thickness: 18 ⁇ m, 2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds.
- PMDETA N, N, N ′, N ′′, N ′′ -pentamethyldiethylenetriamine
- Example 101 As a protective cover for the monomer-containing composition on the copper film, the same copper film as that used as the support was placed on the monomer-containing composition, and N 2 flow (0.5 L / min) was applied while pressing at 5.3 KPa. ) The copper film was heated on a hot plate according to the table below. Then, it stood to cool to room temperature, and obtained the block copolymer 101 (Example 101). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
- Examples 102 to 105 Living polymer synthesis examples> Living in the same manner as the living polymer synthesis example of Example 101 except that equimolar amounts of the liquid crystalline monomer used in the living polymer synthesis example of Example 101 were used and changed from B-1 to B-2 to 5 respectively. Polymers were obtained (Examples 102 to 105). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
- Examples 106 to 117 Block copolymer synthesis example> Hydrophilic macroinitiator A-3 (0.103 g), liquid crystal monomer B-2 (0.677 g), and PMDETA 0.021 g were dissolved in 5 ml of toluene. 50 ⁇ l of the obtained toluene solution was applied onto a copper film (Nihon Foil Co., Ltd., rolled copper foil TCU, thickness: 18 ⁇ m, 2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds.
- a copper film Nihon Foil Co., Ltd., rolled copper foil TCU, thickness: 18 ⁇ m, 2 cm square
- Example 119 Block copolymer synthesis example> Hydrophilic macroinitiator A-3 (0.103 g), liquid crystal monomer B-2 (0.677 g), 1,1,4,7,10,10-hexamethyltriethylenetetramine 0.028 g in 5 ml of toluene Dissolved. 50 ⁇ l of the obtained toluene solution was applied on a copper film (2 cm square) and spin-coated by rotating at 1500 rpm for 30 seconds.
- Example 120 Block copolymer synthesis example> 5 ml of toluene with hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-2 (0.677 g), 0.051 g of N, N, N ′, N′-tetrakis (2-pyridylmethyl) ethylenediamine It was dissolved in. 50 ⁇ l of the obtained toluene solution was applied on a copper film (2 cm square) and spin-coated by rotating at 1500 rpm for 30 seconds.
- Example 121 Block copolymer synthesis example> Hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-2 (0.677 g) and 0.028 g of tris [2- (dimethylamino) ethyl] amine were dissolved in 5 ml of toluene. 50 ⁇ l of the obtained toluene solution was applied on a copper film, and spin-coated by rotating at 1500 rpm for 30 seconds.
- a living polymer (particularly a block copolymer) is used in the case of ATRP. It was found that it can be economically and reasonably manufactured in a short time. It was also found that a microphase separation structure membrane can be produced economically rationally by using the method for producing a living polymer on the support of the present invention. Furthermore, in the living polymers (block copolymers) obtained in Examples 101 to 121, the remaining amount of copper ions was 5 ppm or less with respect to the block copolymer. In the present specification, the remaining amount of copper ions was determined by the method described in Gunma Prefectural Industrial Technology Center Research Report 2010.
Abstract
A method for producing a living polymer on a base, characterized by comprising a step of carrying out the living polymerization of a monomer-containing composition on the base. The method has excellent economic performance.
Description
本発明は、支持体上でのリビング重合体の製造方法、ブロック共重合体、それを用いたミクロ相分離構造膜およびその製造方法に関する。より詳しくは、支持体上でのリビング重合体の製造方法(特にミクロ相分離構造膜の形成用のブロック共重合体を支持体上で製造することができる支持体上でのリビング重合体の製造方法)、支持体上でのリビング重合体の製造方法で製造されたブロック共重合体、ミクロ相分離構造膜を支持体上で製造する製造方法および該製造方法で得られたミクロ相分離構造膜に関する。
The present invention relates to a method for producing a living polymer on a support, a block copolymer, a microphase-separated structure membrane using the same, and a method for producing the same. More specifically, a method for producing a living polymer on a support (particularly, a production of a living polymer on a support capable of producing a block copolymer for forming a microphase-separated structure film on the support) Method), a block copolymer produced by a method for producing a living polymer on a support, a production method for producing a microphase separation structure film on a support, and a microphase separation structure film obtained by the production method About.
ミクロ相分離構造を作製する方法としてリビング重合により製造したブロック共重合体を用い、得られた相分離構造の固定化をする方法が知られている。
As a method for producing a microphase-separated structure, a method of immobilizing the obtained phase-separated structure using a block copolymer produced by living polymerization is known.
従来、ミクロ相分離構造を作製するためには高温・長時間(200℃、5時間程度)必要としていた。これに対し、ブロック共重合体が相分離で形成するナノスケールのヘキサゴナルシリンダー構造の配向を、「両親媒性」と「液晶」を導入することで制御した例が知られている(非特許文献1、特許文献1、2)。具体的には、特許文献1では、親水性ポリマー成分(A)と疎水性ポリマー成分(B)が共有結合によって連結されたブロック共重合体で、A及びBの分子量分布が≦1.3であるブロック共重合体が記載されている。特許文献2では、親水性ポリマー成分と、架橋可能な構造を有する疎水性ポリマー成分とが共有結合してなるブロック共重合体からなる自立性高分子薄膜であって、上記自立性高分子薄膜がその膜中に一定方向に配向した上記親水性ポリマー成分からなるシリンダーを有しており、上記疎水性ポリマー成分が架橋している自立性高分子薄膜が記載されている。これらの文献に記載の方法はミクロ相分離構造を、数分程度で作製することができるようになった点で画期的であった。必要な化合物の合成は、例えば特許文献1では、親水的なマクロ開始剤を合成した後に、液晶性モノマーとの原子移動ラジカル重合(Atom Transfer Radical Polymerization、以下、ATRPと略する)により親水部-疎水部が連結されたブロック共重合体を得ている。また、特許文献2では、疎水部のみをATRPによりリビング重合部分を得たのちに、末端を修飾し、最後に高分子反応により親水部を作製している。
Conventionally, high temperature and long time (200 ° C., about 5 hours) have been required to produce a microphase separation structure. On the other hand, there is known an example in which the orientation of the nanoscale hexagonal cylinder structure formed by phase separation of the block copolymer is controlled by introducing “amphiphilicity” and “liquid crystal” (non-patent literature). 1, Patent Documents 1 and 2). Specifically, in Patent Document 1, a block copolymer in which a hydrophilic polymer component (A) and a hydrophobic polymer component (B) are linked by a covalent bond, and the molecular weight distribution of A and B is ≦ 1.3. Certain block copolymers have been described. In Patent Document 2, a self-supporting polymer thin film made of a block copolymer formed by covalently bonding a hydrophilic polymer component and a hydrophobic polymer component having a crosslinkable structure, wherein the self-supporting polymer thin film is A self-supporting polymer thin film having a cylinder made of the hydrophilic polymer component oriented in a certain direction in the film and having the hydrophobic polymer component crosslinked is described. The methods described in these documents were epoch-making in that a microphase-separated structure can be produced in about several minutes. For example, in Patent Document 1, a hydrophilic compound is synthesized by atom transfer radical polymerization (hereinafter abbreviated as ATRP) with a liquid crystalline monomer after synthesizing a hydrophilic macroinitiator in Patent Document 1, for example. A block copolymer in which hydrophobic portions are linked is obtained. Moreover, in patent document 2, after obtaining the living polymerization part only by a hydrophobic part by ATRP, the terminal is modified and finally the hydrophilic part is produced by polymer reaction.
一方、非特許文献2には、可逆的付加開裂連鎖移動(Reversible Addition-Fragmentation Chain Transfer、以下、RAFTと称する)重合によるリビングラジカル重合により、ブロック共重合体を合成する方法や材料が記載されている。RAFT重合では、適切な連鎖移動剤(RAFT剤とも言う)の存在下で、置換モノマーの一般的なフリーラジカル重合にRAFT平衡に関する反応が加わり、可逆的な連鎖移動反応によって重合反応を制御している。
On the other hand, Non-Patent Document 2 describes a method and material for synthesizing a block copolymer by living radical polymerization by reversible addition-fragmentation chain transfer (hereinafter referred to as RAFT) polymerization. Yes. In RAFT polymerization, in the presence of an appropriate chain transfer agent (also called RAFT agent), a reaction related to RAFT equilibrium is added to general free radical polymerization of substituted monomers, and the polymerization reaction is controlled by a reversible chain transfer reaction. Yes.
また、得られた相分離構造の固定化については、特許文献2にブロック共重合体の疎水性ポリマー鎖に光二量化反応できる基を導入し、電離放射線照射を行う方法が記載されている。また、特許文献3、4には得られた相分離構造の固定化のためにオキセタニル基を導入する方法が記載されており、さらに関連する化合物が特許文献5に記載されている。
Also, regarding the immobilization of the obtained phase separation structure, Patent Document 2 describes a method of introducing a group capable of photodimerization into the hydrophobic polymer chain of the block copolymer and irradiating with ionizing radiation. Patent Documents 3 and 4 describe a method of introducing an oxetanyl group for immobilization of the obtained phase separation structure, and further related compounds are described in Patent Document 5.
これらの文献に記載の方法では、予めリビング重合を反応容器中で実施した後にブロック共重合体を得ることが必須である。
ここで、一般に反応容器中でのリビング重合は反応時間が長時間必要であり、触媒除去やモノマー除去工程が必要のため高価であり、実使用上の制約となっていた。 In the methods described in these documents, it is essential to obtain a block copolymer after conducting living polymerization in a reaction vessel in advance.
Here, in general, living polymerization in a reaction vessel requires a long reaction time, and is expensive because a catalyst removal step and a monomer removal step are necessary, which is a limitation in practical use.
ここで、一般に反応容器中でのリビング重合は反応時間が長時間必要であり、触媒除去やモノマー除去工程が必要のため高価であり、実使用上の制約となっていた。 In the methods described in these documents, it is essential to obtain a block copolymer after conducting living polymerization in a reaction vessel in advance.
Here, in general, living polymerization in a reaction vessel requires a long reaction time, and is expensive because a catalyst removal step and a monomer removal step are necessary, which is a limitation in practical use.
本発明の解決しようとする課題は、経済合理性の優れたリビング重合体の製造方法を提供することである。
The problem to be solved by the present invention is to provide a method for producing a living polymer having excellent economic rationality.
上記の課題を解決するために鋭意検討を行った結果、本発明者らは、支持体上でリビング重合を行うことにより、経済合理性の優れたリビング重合体の製造方法を提供できることを見出した。
As a result of intensive studies to solve the above problems, the present inventors have found that a living polymer can be provided on the support by providing a living polymer having a high economic rationality. .
[1] 支持体上でモノマー含有組成物をリビング重合させる工程を含むことを特徴とする支持体上でのリビング重合体の製造方法。
[2] [1]に記載の支持体上でのリビング重合体の製造方法は、前記リビング重合が、リビングラジカル重合であることが好ましい。
[3] [1]または[2]に記載の支持体上でのリビング重合体の製造方法は、前記支持体上で前記モノマー含有組成物をリビング重合させる工程が、1000秒以下であることが好ましい。
[4] [1]~[3]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記支持体上で前記モノマー含有組成物をリビング重合させる工程のモノマー消費率を、10~100%に制御することが好ましい。
[5] [1]~[4]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記支持体上で前記モノマー含有組成物をリビング重合させる工程が、50~200℃での加熱であることが好ましい。
[6] [1]~[5]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記リビング重合体の数平均分子量Mnが1000~100000であることが好ましい。
[7] [1]~[6]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記リビング重合が原子移動ラジカル重合であり、かつ、
前記支持体が金属製の支持体であることが好ましい。
[8] [7]に記載の支持体上でのリビング重合体の製造方法は、前記モノマー含有組成物の前記金属製の支持体と接していない面を別の支持体で覆い、前記モノマー含有組成物を挟圧しながら加熱することが好ましい。
[9] [7]または[8]に記載の支持体上でのリビング重合体の製造方法は、前記金属が銅であることが好ましい。
[10] [7]~[9]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記リビング重合体中の銅イオン濃度が、前記リビング重合体に対して5ppm以下であることが好ましい。
[11] [1]~[6]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記リビング重合が可逆的付加開裂連鎖移動重合であることが好ましい。
[12] [1]~[11]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記モノマー含有組成物が、モノマー成分として重合性棒状液晶化合物を含むことが好ましい。
[13] [1]~[12]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記リビング重合体が、ブロック共重合体であることが好ましい。
[14] [13]に記載の支持体上でのリビング重合体の製造方法は、下記(1)および(2)の工程を、支持体上で実施することが好ましい。
(1)前記モノマー含有組成物として、アルキレンオキサイド鎖を含むポリマー成分および炭素数2~20のアルキレン鎖を含むモノマー成分を含むモノマー含有組成物、ならびに、アルキレンオキサイド鎖を含むモノマー成分および炭素数2~20のアルキレン鎖を含むポリマー成分を含むモノマー含有組成物のうち少なくとも一方を用いて、該モノマー含有組成物を前記支持体上に適用する工程
(2)前記支持体上で前記モノマー含有組成物をリビング重合させて、アルキレンオキサイド鎖を含むポリマー成分と炭素数2~20のアルキレン鎖を含むポリマー成分とが共有結合にて連結されたブロック共重合体を得る工程
[15] [13]に記載の支持体上でのリビング重合体の製造方法は、下記(1’)および(2)の工程を、支持体上で実施することが好ましい。
(1’)前記モノマー含有組成物として、アルキレンオキサイド鎖を含むポリマー成分および炭素数2~20のアルキレン鎖を含むモノマー成分を含むモノマー含有組成物を用いて、該モノマー含有組成物を前記支持体上に適用する工程
(2)前記支持体上で前記モノマー含有組成物をリビング重合させて、アルキレンオキサイド鎖を含むポリマー成分と炭素数2~20のアルキレン鎖を含むポリマー成分とが共有結合にて連結されたブロック共重合体を得る工程
[16] [14]または[15]に記載の支持体上でのリビング重合体の製造方法は、前記炭素数2~20のアルキレン鎖を含むモノマー成分が重合性棒状液晶化合物であることが好ましい。
[17] [14]~[16]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記ブロック共重合体が、ミクロ相分離構造膜の形成用のブロック共重合体であることが好ましい。
[18] [14]~[17]のいずれか一項に記載の支持体上でのリビング重合体の製造方法で得られたことを特徴とするブロック共重合体。
[19] (3)[18]に記載のブロック共重合体を前記支持体上で熱、光、溶媒蒸気または電場により相分離を促進させ、ミクロ相分離構造を形成する工程を含むことを特徴とするミクロ相分離構造膜の製造方法。
[20] [19]に記載のミクロ相分離構造膜の製造方法は、前記ミクロ相分離構造を形成する工程が、40~250℃での加熱であることが好ましい。
[21] [19]または[20]に記載のミクロ相分離構造膜の製造方法は、さらに(4)前記ミクロ相分離構造を有するブロック共重合体を前記支持体上で架橋または重合させてミクロ相分離構造を固定化させる工程を含むことが好ましい。
[22] [19]~[21]のいずれか一項に記載のミクロ相分離構造膜の製造方法は、前記ミクロ相分離構造膜を、前記支持体から剥ぎ取る工程を含むことが好ましい。
[23] [19]~[22]のいずれか一項に記載のミクロ相分離構造膜の製造方法は、前記ミクロ相分離構造膜の厚みが、1~2000nmであることが好ましい。
[24] [19]~[23]のいずれか一項に記載のミクロ相分離構造膜の製造方法で製造されたことを特徴とするミクロ相分離構造膜。 [1] A method for producing a living polymer on a support, comprising a step of subjecting the monomer-containing composition to living polymerization on the support.
[2] In the method for producing a living polymer on a support according to [1], the living polymerization is preferably living radical polymerization.
[3] In the method for producing a living polymer on a support according to [1] or [2], the step of living polymerizing the monomer-containing composition on the support may be 1000 seconds or less. preferable.
[4] The method for producing a living polymer on a support according to any one of [1] to [3], wherein the monomer consumption rate in the step of living polymerizing the monomer-containing composition on the support Is preferably controlled to 10 to 100%.
[5] The method for producing a living polymer on a support according to any one of [1] to [4], wherein the step of living polymerizing the monomer-containing composition on the support comprises 50 to 50 Heating at 200 ° C. is preferable.
[6] In the method for producing a living polymer on a support according to any one of [1] to [5], the living polymer preferably has a number average molecular weight Mn of 1,000 to 100,000.
[7] The method for producing a living polymer on a support according to any one of [1] to [6], wherein the living polymerization is atom transfer radical polymerization, and
The support is preferably a metal support.
[8] In the method for producing a living polymer on a support according to [7], a surface of the monomer-containing composition that is not in contact with the metal support is covered with another support, and the monomer-containing composition is provided. It is preferable to heat while pressing the composition.
[9] In the method for producing a living polymer on a support according to [7] or [8], the metal is preferably copper.
[10] The method for producing a living polymer on a support according to any one of [7] to [9], wherein the copper ion concentration in the living polymer is 5 ppm relative to the living polymer. The following is preferable.
[11] In the method for producing a living polymer on a support according to any one of [1] to [6], the living polymerization is preferably reversible addition-fragmentation chain transfer polymerization.
[12] In the method for producing a living polymer on a support according to any one of [1] to [11], the monomer-containing composition contains a polymerizable rod-like liquid crystal compound as a monomer component. preferable.
[13] In the method for producing a living polymer on a support according to any one of [1] to [12], the living polymer is preferably a block copolymer.
[14] In the method for producing a living polymer on a support according to [13], the following steps (1) and (2) are preferably performed on the support.
(1) As the monomer-containing composition, a monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms, and a monomer component containing an alkylene oxide chain and a carbon number of 2 (2) applying the monomer-containing composition onto the support using at least one of the monomer-containing compositions containing a polymer component containing 20 to 20 alkylene chains (2) the monomer-containing composition on the support [15] A process for obtaining a block copolymer in which a polymer component containing an alkylene oxide chain and a polymer component containing an alkylene chain having 2 to 20 carbon atoms are covalently linked to each other by living polymerizing [15] [13] The manufacturing method of the living polymer on the support of the following steps (1 ′) and (2) In is preferably performed.
(1 ′) A monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms is used as the monomer-containing composition. (2) The monomer-containing composition is living-polymerized on the support so that the polymer component containing an alkylene oxide chain and the polymer component containing an alkylene chain having 2 to 20 carbon atoms are covalently bonded. Step [16] for obtaining a linked block copolymer [16] The method for producing a living polymer on a support according to [14], wherein the monomer component containing an alkylene chain having 2 to 20 carbon atoms is used. A polymerizable rod-like liquid crystal compound is preferable.
[17] The method for producing a living polymer on a support according to any one of [14] to [16], wherein the block copolymer is a block copolymer for forming a microphase-separated structure film. It is preferably a coalescence.
[18] A block copolymer obtained by the method for producing a living polymer on a support according to any one of [14] to [17].
[19] (3) A step comprising accelerating phase separation of the block copolymer according to [18] by heat, light, solvent vapor or electric field on the support to form a microphase separation structure. A method for producing a microphase separation structure membrane.
[20] In the method for producing a microphase separation structure film according to [19], the step of forming the microphase separation structure is preferably heating at 40 to 250 ° C.
[21] The method for producing a microphase-separated structure film according to [19] or [20] is further characterized in that (4) a block copolymer having the microphase-separated structure is crosslinked or polymerized on the support to form a micro It is preferable to include a step of immobilizing the phase separation structure.
[22] The method for producing a microphase separation structure film according to any one of [19] to [21] preferably includes a step of peeling the microphase separation structure film from the support.
[23] In the method for producing a microphase separation structure film according to any one of [19] to [22], the thickness of the microphase separation structure film is preferably 1 to 2000 nm.
[24] A microphase-separated structure film produced by the method for producing a microphase-separated structure film according to any one of [19] to [23].
[2] [1]に記載の支持体上でのリビング重合体の製造方法は、前記リビング重合が、リビングラジカル重合であることが好ましい。
[3] [1]または[2]に記載の支持体上でのリビング重合体の製造方法は、前記支持体上で前記モノマー含有組成物をリビング重合させる工程が、1000秒以下であることが好ましい。
[4] [1]~[3]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記支持体上で前記モノマー含有組成物をリビング重合させる工程のモノマー消費率を、10~100%に制御することが好ましい。
[5] [1]~[4]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記支持体上で前記モノマー含有組成物をリビング重合させる工程が、50~200℃での加熱であることが好ましい。
[6] [1]~[5]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記リビング重合体の数平均分子量Mnが1000~100000であることが好ましい。
[7] [1]~[6]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記リビング重合が原子移動ラジカル重合であり、かつ、
前記支持体が金属製の支持体であることが好ましい。
[8] [7]に記載の支持体上でのリビング重合体の製造方法は、前記モノマー含有組成物の前記金属製の支持体と接していない面を別の支持体で覆い、前記モノマー含有組成物を挟圧しながら加熱することが好ましい。
[9] [7]または[8]に記載の支持体上でのリビング重合体の製造方法は、前記金属が銅であることが好ましい。
[10] [7]~[9]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記リビング重合体中の銅イオン濃度が、前記リビング重合体に対して5ppm以下であることが好ましい。
[11] [1]~[6]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記リビング重合が可逆的付加開裂連鎖移動重合であることが好ましい。
[12] [1]~[11]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記モノマー含有組成物が、モノマー成分として重合性棒状液晶化合物を含むことが好ましい。
[13] [1]~[12]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記リビング重合体が、ブロック共重合体であることが好ましい。
[14] [13]に記載の支持体上でのリビング重合体の製造方法は、下記(1)および(2)の工程を、支持体上で実施することが好ましい。
(1)前記モノマー含有組成物として、アルキレンオキサイド鎖を含むポリマー成分および炭素数2~20のアルキレン鎖を含むモノマー成分を含むモノマー含有組成物、ならびに、アルキレンオキサイド鎖を含むモノマー成分および炭素数2~20のアルキレン鎖を含むポリマー成分を含むモノマー含有組成物のうち少なくとも一方を用いて、該モノマー含有組成物を前記支持体上に適用する工程
(2)前記支持体上で前記モノマー含有組成物をリビング重合させて、アルキレンオキサイド鎖を含むポリマー成分と炭素数2~20のアルキレン鎖を含むポリマー成分とが共有結合にて連結されたブロック共重合体を得る工程
[15] [13]に記載の支持体上でのリビング重合体の製造方法は、下記(1’)および(2)の工程を、支持体上で実施することが好ましい。
(1’)前記モノマー含有組成物として、アルキレンオキサイド鎖を含むポリマー成分および炭素数2~20のアルキレン鎖を含むモノマー成分を含むモノマー含有組成物を用いて、該モノマー含有組成物を前記支持体上に適用する工程
(2)前記支持体上で前記モノマー含有組成物をリビング重合させて、アルキレンオキサイド鎖を含むポリマー成分と炭素数2~20のアルキレン鎖を含むポリマー成分とが共有結合にて連結されたブロック共重合体を得る工程
[16] [14]または[15]に記載の支持体上でのリビング重合体の製造方法は、前記炭素数2~20のアルキレン鎖を含むモノマー成分が重合性棒状液晶化合物であることが好ましい。
[17] [14]~[16]のいずれか一項に記載の支持体上でのリビング重合体の製造方法は、前記ブロック共重合体が、ミクロ相分離構造膜の形成用のブロック共重合体であることが好ましい。
[18] [14]~[17]のいずれか一項に記載の支持体上でのリビング重合体の製造方法で得られたことを特徴とするブロック共重合体。
[19] (3)[18]に記載のブロック共重合体を前記支持体上で熱、光、溶媒蒸気または電場により相分離を促進させ、ミクロ相分離構造を形成する工程を含むことを特徴とするミクロ相分離構造膜の製造方法。
[20] [19]に記載のミクロ相分離構造膜の製造方法は、前記ミクロ相分離構造を形成する工程が、40~250℃での加熱であることが好ましい。
[21] [19]または[20]に記載のミクロ相分離構造膜の製造方法は、さらに(4)前記ミクロ相分離構造を有するブロック共重合体を前記支持体上で架橋または重合させてミクロ相分離構造を固定化させる工程を含むことが好ましい。
[22] [19]~[21]のいずれか一項に記載のミクロ相分離構造膜の製造方法は、前記ミクロ相分離構造膜を、前記支持体から剥ぎ取る工程を含むことが好ましい。
[23] [19]~[22]のいずれか一項に記載のミクロ相分離構造膜の製造方法は、前記ミクロ相分離構造膜の厚みが、1~2000nmであることが好ましい。
[24] [19]~[23]のいずれか一項に記載のミクロ相分離構造膜の製造方法で製造されたことを特徴とするミクロ相分離構造膜。 [1] A method for producing a living polymer on a support, comprising a step of subjecting the monomer-containing composition to living polymerization on the support.
[2] In the method for producing a living polymer on a support according to [1], the living polymerization is preferably living radical polymerization.
[3] In the method for producing a living polymer on a support according to [1] or [2], the step of living polymerizing the monomer-containing composition on the support may be 1000 seconds or less. preferable.
[4] The method for producing a living polymer on a support according to any one of [1] to [3], wherein the monomer consumption rate in the step of living polymerizing the monomer-containing composition on the support Is preferably controlled to 10 to 100%.
[5] The method for producing a living polymer on a support according to any one of [1] to [4], wherein the step of living polymerizing the monomer-containing composition on the support comprises 50 to 50 Heating at 200 ° C. is preferable.
[6] In the method for producing a living polymer on a support according to any one of [1] to [5], the living polymer preferably has a number average molecular weight Mn of 1,000 to 100,000.
[7] The method for producing a living polymer on a support according to any one of [1] to [6], wherein the living polymerization is atom transfer radical polymerization, and
The support is preferably a metal support.
[8] In the method for producing a living polymer on a support according to [7], a surface of the monomer-containing composition that is not in contact with the metal support is covered with another support, and the monomer-containing composition is provided. It is preferable to heat while pressing the composition.
[9] In the method for producing a living polymer on a support according to [7] or [8], the metal is preferably copper.
[10] The method for producing a living polymer on a support according to any one of [7] to [9], wherein the copper ion concentration in the living polymer is 5 ppm relative to the living polymer. The following is preferable.
[11] In the method for producing a living polymer on a support according to any one of [1] to [6], the living polymerization is preferably reversible addition-fragmentation chain transfer polymerization.
[12] In the method for producing a living polymer on a support according to any one of [1] to [11], the monomer-containing composition contains a polymerizable rod-like liquid crystal compound as a monomer component. preferable.
[13] In the method for producing a living polymer on a support according to any one of [1] to [12], the living polymer is preferably a block copolymer.
[14] In the method for producing a living polymer on a support according to [13], the following steps (1) and (2) are preferably performed on the support.
(1) As the monomer-containing composition, a monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms, and a monomer component containing an alkylene oxide chain and a carbon number of 2 (2) applying the monomer-containing composition onto the support using at least one of the monomer-containing compositions containing a polymer component containing 20 to 20 alkylene chains (2) the monomer-containing composition on the support [15] A process for obtaining a block copolymer in which a polymer component containing an alkylene oxide chain and a polymer component containing an alkylene chain having 2 to 20 carbon atoms are covalently linked to each other by living polymerizing [15] [13] The manufacturing method of the living polymer on the support of the following steps (1 ′) and (2) In is preferably performed.
(1 ′) A monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms is used as the monomer-containing composition. (2) The monomer-containing composition is living-polymerized on the support so that the polymer component containing an alkylene oxide chain and the polymer component containing an alkylene chain having 2 to 20 carbon atoms are covalently bonded. Step [16] for obtaining a linked block copolymer [16] The method for producing a living polymer on a support according to [14], wherein the monomer component containing an alkylene chain having 2 to 20 carbon atoms is used. A polymerizable rod-like liquid crystal compound is preferable.
[17] The method for producing a living polymer on a support according to any one of [14] to [16], wherein the block copolymer is a block copolymer for forming a microphase-separated structure film. It is preferably a coalescence.
[18] A block copolymer obtained by the method for producing a living polymer on a support according to any one of [14] to [17].
[19] (3) A step comprising accelerating phase separation of the block copolymer according to [18] by heat, light, solvent vapor or electric field on the support to form a microphase separation structure. A method for producing a microphase separation structure membrane.
[20] In the method for producing a microphase separation structure film according to [19], the step of forming the microphase separation structure is preferably heating at 40 to 250 ° C.
[21] The method for producing a microphase-separated structure film according to [19] or [20] is further characterized in that (4) a block copolymer having the microphase-separated structure is crosslinked or polymerized on the support to form a micro It is preferable to include a step of immobilizing the phase separation structure.
[22] The method for producing a microphase separation structure film according to any one of [19] to [21] preferably includes a step of peeling the microphase separation structure film from the support.
[23] In the method for producing a microphase separation structure film according to any one of [19] to [22], the thickness of the microphase separation structure film is preferably 1 to 2000 nm.
[24] A microphase-separated structure film produced by the method for producing a microphase-separated structure film according to any one of [19] to [23].
本発明によれば、経済性に優れるリビング重合体の製造方法を提供することができる。
According to the present invention, it is possible to provide a method for producing a living polymer that is excellent in economic efficiency.
以下において、本発明について詳細に説明する。以下に記載する構成要件の説明は、代表的な実施形態や具体例に基づいてなされることがあるが、本発明はそのような実施形態に限定されるものではない。なお、本明細書において「~」を用いて表される数値範囲は「~」前後に記載される数値を下限値および上限値として含む範囲を意味する。
また、(メタ)アクリレートは、アクリレートとメタクリレートの両方を含む意味で使われる。 Hereinafter, the present invention will be described in detail. The description of the constituent elements described below may be made based on representative embodiments and specific examples, but the present invention is not limited to such embodiments. In the present specification, a numerical range expressed using “to” means a range including numerical values described before and after “to” as a lower limit value and an upper limit value.
Moreover, (meth) acrylate is used in the meaning containing both acrylate and methacrylate.
また、(メタ)アクリレートは、アクリレートとメタクリレートの両方を含む意味で使われる。 Hereinafter, the present invention will be described in detail. The description of the constituent elements described below may be made based on representative embodiments and specific examples, but the present invention is not limited to such embodiments. In the present specification, a numerical range expressed using “to” means a range including numerical values described before and after “to” as a lower limit value and an upper limit value.
Moreover, (meth) acrylate is used in the meaning containing both acrylate and methacrylate.
[支持体上でのリビング重合体の製造方法]
本発明の支持体上でのリビング重合体の製造方法(以下、本発明の製造方法とも言う)は、支持体上でモノマー含有組成物をリビング重合させる工程を含むことを特徴とする。
このような構成により、経済合理性の優れたリビング重合体の製造方法を提供できる。一般的に溶液中でのリビング重合は(基質にも依存するが)数時間は必要であり、本発明の製造方法における支持体上での重合は、時間の観点からも経済合理性がある。モノマー含有組成物を支持体上でリビング重合を行うことは従来知られておらず、いかなる理論に拘泥するものでもないが、モノマー含有組成物を薄膜の状態としてリビング重合を行うことにより、溶液中でリビング重合を行うよりも、経済合理性の点が優れる。 [Method for producing living polymer on support]
The method for producing a living polymer on the support of the present invention (hereinafter also referred to as the production method of the present invention) includes a step of living polymerizing the monomer-containing composition on the support.
With such a configuration, a method for producing a living polymer having excellent economic rationality can be provided. Generally, living polymerization in solution requires several hours (depending on the substrate), and the polymerization on the support in the production method of the present invention is economically reasonable from the viewpoint of time. It is not known in the art to perform living polymerization on a support on a monomer-containing composition, and it is not bound by any theory, but by performing living polymerization with the monomer-containing composition in a thin film state, Compared with living polymerization, the economic rationality is superior.
本発明の支持体上でのリビング重合体の製造方法(以下、本発明の製造方法とも言う)は、支持体上でモノマー含有組成物をリビング重合させる工程を含むことを特徴とする。
このような構成により、経済合理性の優れたリビング重合体の製造方法を提供できる。一般的に溶液中でのリビング重合は(基質にも依存するが)数時間は必要であり、本発明の製造方法における支持体上での重合は、時間の観点からも経済合理性がある。モノマー含有組成物を支持体上でリビング重合を行うことは従来知られておらず、いかなる理論に拘泥するものでもないが、モノマー含有組成物を薄膜の状態としてリビング重合を行うことにより、溶液中でリビング重合を行うよりも、経済合理性の点が優れる。 [Method for producing living polymer on support]
The method for producing a living polymer on the support of the present invention (hereinafter also referred to as the production method of the present invention) includes a step of living polymerizing the monomer-containing composition on the support.
With such a configuration, a method for producing a living polymer having excellent economic rationality can be provided. Generally, living polymerization in solution requires several hours (depending on the substrate), and the polymerization on the support in the production method of the present invention is economically reasonable from the viewpoint of time. It is not known in the art to perform living polymerization on a support on a monomer-containing composition, and it is not bound by any theory, but by performing living polymerization with the monomer-containing composition in a thin film state, Compared with living polymerization, the economic rationality is superior.
<支持体>
本発明の支持体上でのリビング重合体の製造方法に用いられる支持体としては特に制限はなく、ガラス基板であっても、金属性の支持体であっても、樹脂フィルムであってもよい。リビング重合の方式がATRPの場合は、前記支持体として金属性の支持体を用いることが好ましいが、詳細は後述する。リビング重合の方式がRAFT重合の場合は、ガラス基板及び樹脂フィルムを用いることが好ましい。 <Support>
The support used in the method for producing a living polymer on the support of the present invention is not particularly limited, and may be a glass substrate, a metallic support, or a resin film. . When the living polymerization method is ATRP, it is preferable to use a metallic support as the support, but details will be described later. When the living polymerization method is RAFT polymerization, it is preferable to use a glass substrate and a resin film.
本発明の支持体上でのリビング重合体の製造方法に用いられる支持体としては特に制限はなく、ガラス基板であっても、金属性の支持体であっても、樹脂フィルムであってもよい。リビング重合の方式がATRPの場合は、前記支持体として金属性の支持体を用いることが好ましいが、詳細は後述する。リビング重合の方式がRAFT重合の場合は、ガラス基板及び樹脂フィルムを用いることが好ましい。 <Support>
The support used in the method for producing a living polymer on the support of the present invention is not particularly limited, and may be a glass substrate, a metallic support, or a resin film. . When the living polymerization method is ATRP, it is preferable to use a metallic support as the support, but details will be described later. When the living polymerization method is RAFT polymerization, it is preferable to use a glass substrate and a resin film.
<モノマー成分>
本発明の支持体上でのリビング重合体の製造方法では、前記モノマー含有組成物中にリビング重合に供するモノマー成分を含む。なお、前記モノマー含有組成物は必要に応じて他の成分を含んでもよい。
前記モノマー成分としては、アルキレンオキサイド鎖を含むモノマー成分を用いても、炭素数2~20のアルキレン鎖を含むモノマー成分を用いてもよいが、後述するミクロ相分離構造膜を製造しやすい観点から炭素数2~20のアルキレン鎖を含むモノマー成分を用いることが好ましい。
なお、アルキレンオキサイド鎖を含むモノマー成分は親水性モノマーであり、炭素数2~20のアルキレン鎖を含むモノマー成分は疎水性モノマーである。 <Monomer component>
In the method for producing a living polymer on the support of the present invention, the monomer-containing composition contains a monomer component used for living polymerization. In addition, the said monomer containing composition may also contain another component as needed.
As the monomer component, a monomer component including an alkylene oxide chain may be used, or a monomer component including an alkylene chain having 2 to 20 carbon atoms may be used. It is preferable to use a monomer component containing an alkylene chain having 2 to 20 carbon atoms.
The monomer component containing an alkylene oxide chain is a hydrophilic monomer, and the monomer component containing an alkylene chain having 2 to 20 carbon atoms is a hydrophobic monomer.
本発明の支持体上でのリビング重合体の製造方法では、前記モノマー含有組成物中にリビング重合に供するモノマー成分を含む。なお、前記モノマー含有組成物は必要に応じて他の成分を含んでもよい。
前記モノマー成分としては、アルキレンオキサイド鎖を含むモノマー成分を用いても、炭素数2~20のアルキレン鎖を含むモノマー成分を用いてもよいが、後述するミクロ相分離構造膜を製造しやすい観点から炭素数2~20のアルキレン鎖を含むモノマー成分を用いることが好ましい。
なお、アルキレンオキサイド鎖を含むモノマー成分は親水性モノマーであり、炭素数2~20のアルキレン鎖を含むモノマー成分は疎水性モノマーである。 <Monomer component>
In the method for producing a living polymer on the support of the present invention, the monomer-containing composition contains a monomer component used for living polymerization. In addition, the said monomer containing composition may also contain another component as needed.
As the monomer component, a monomer component including an alkylene oxide chain may be used, or a monomer component including an alkylene chain having 2 to 20 carbon atoms may be used. It is preferable to use a monomer component containing an alkylene chain having 2 to 20 carbon atoms.
The monomer component containing an alkylene oxide chain is a hydrophilic monomer, and the monomer component containing an alkylene chain having 2 to 20 carbon atoms is a hydrophobic monomer.
(アルキレンオキサイド鎖を含むモノマー成分)
前記アルキレンオキサイド鎖を含むモノマー成分としては特に制限はなく、公知のアルキレンオキサイド鎖を含むモノマー成分を用いることができる。
前記アルキレンオキサイド鎖を含むモノマー成分としては、メタクリル酸及びアクリル酸と、ポリオール化合物の片末端をアルキルエーテル化した化合物が、エステル結合で連結された化合物を挙げることができる。
「ポリオール化合物の片末端をアルキルエーテル化した化合物」とは下記に記載するポリオール化合物を炭素数1~10のアルキル化合物でエーテル化したものであり、好ましくは炭素数1~5、より好ましくはメチルエーテル化(炭素数1)したものである。
(i)ポリオール化合物
ポリオール化合物としては、ポリエーテルポリオール、ポリエステルポリオール、ポリカーボネートポリオール、ポリカプロラクトンポリオール、分子中に2個以上の水酸基を有する脂肪族炭化水素、分子中に2個以上の水酸基を有する脂環式炭化水素、分子中に2個以上の水酸基を有する不飽和炭化水素等が用いられる。これらのポリオールは単独で用いることも、2種類以上併用することもできる。 (Monomer component containing an alkylene oxide chain)
There is no restriction | limiting in particular as a monomer component containing the said alkylene oxide chain, The monomer component containing a well-known alkylene oxide chain can be used.
Examples of the monomer component containing an alkylene oxide chain include a compound in which methacrylic acid and acrylic acid and a compound obtained by alkyl etherifying one end of a polyol compound are linked by an ester bond.
“A compound obtained by alkyl etherifying one end of a polyol compound” is obtained by etherifying a polyol compound described below with an alkyl compound having 1 to 10 carbon atoms, preferably 1 to 5 carbon atoms, more preferably methyl. Etherified (carbon number 1).
(I) Polyol compound Examples of the polyol compound include polyether polyol, polyester polyol, polycarbonate polyol, polycaprolactone polyol, aliphatic hydrocarbon having two or more hydroxyl groups in the molecule, and fat having two or more hydroxyl groups in the molecule. Cyclic hydrocarbons, unsaturated hydrocarbons having two or more hydroxyl groups in the molecule, and the like are used. These polyols can be used alone or in combination of two or more.
前記アルキレンオキサイド鎖を含むモノマー成分としては特に制限はなく、公知のアルキレンオキサイド鎖を含むモノマー成分を用いることができる。
前記アルキレンオキサイド鎖を含むモノマー成分としては、メタクリル酸及びアクリル酸と、ポリオール化合物の片末端をアルキルエーテル化した化合物が、エステル結合で連結された化合物を挙げることができる。
「ポリオール化合物の片末端をアルキルエーテル化した化合物」とは下記に記載するポリオール化合物を炭素数1~10のアルキル化合物でエーテル化したものであり、好ましくは炭素数1~5、より好ましくはメチルエーテル化(炭素数1)したものである。
(i)ポリオール化合物
ポリオール化合物としては、ポリエーテルポリオール、ポリエステルポリオール、ポリカーボネートポリオール、ポリカプロラクトンポリオール、分子中に2個以上の水酸基を有する脂肪族炭化水素、分子中に2個以上の水酸基を有する脂環式炭化水素、分子中に2個以上の水酸基を有する不飽和炭化水素等が用いられる。これらのポリオールは単独で用いることも、2種類以上併用することもできる。 (Monomer component containing an alkylene oxide chain)
There is no restriction | limiting in particular as a monomer component containing the said alkylene oxide chain, The monomer component containing a well-known alkylene oxide chain can be used.
Examples of the monomer component containing an alkylene oxide chain include a compound in which methacrylic acid and acrylic acid and a compound obtained by alkyl etherifying one end of a polyol compound are linked by an ester bond.
“A compound obtained by alkyl etherifying one end of a polyol compound” is obtained by etherifying a polyol compound described below with an alkyl compound having 1 to 10 carbon atoms, preferably 1 to 5 carbon atoms, more preferably methyl. Etherified (carbon number 1).
(I) Polyol compound Examples of the polyol compound include polyether polyol, polyester polyol, polycarbonate polyol, polycaprolactone polyol, aliphatic hydrocarbon having two or more hydroxyl groups in the molecule, and fat having two or more hydroxyl groups in the molecule. Cyclic hydrocarbons, unsaturated hydrocarbons having two or more hydroxyl groups in the molecule, and the like are used. These polyols can be used alone or in combination of two or more.
上記ポリエーテルポリオールとしては、脂肪族ポリエーテルポリオール、脂環式ポリエーテルポリオール、芳香族ポリエーテルポリオールを挙げることができる。
Examples of the polyether polyol include aliphatic polyether polyols, alicyclic polyether polyols, and aromatic polyether polyols.
ここで、脂肪族ポリエーテルポリオールとしては、例えばポリエチレングリコール、ポリプロピレングリコール、ポリテトラメチレングリコール、ポリヘキサメチレングリコール、ポリヘプタメチレングリコール、ポリデカメチレングリコール、ペンタエリスリトール、ジペンタエリスリトール、トリメチロールプロパン、およびトリメチロールプロパンのエチレンオキサイド付加トリオール、トリメチロールプロパンのプロピレンオキサイド付加トリオール、トリメチロールプロパンのエチレンオキサイドとプロピレンオキサイド付加トリオール、ペンタエリスリトールのエチレンオキサイド付加テトラオール、ジペンタエリスリトールのエチレンオキサイド付加ヘキサオール等のアルキレンオキサイド付加ポリオール等の多価アルコール、あるいは2種類以上のイオン重合性環状化合物を開環重合させて得られるポリエーテルポリオール等が挙げられる。
Here, as the aliphatic polyether polyol, for example, polyethylene glycol, polypropylene glycol, polytetramethylene glycol, polyhexamethylene glycol, polyheptamethylene glycol, polydecamethylene glycol, pentaerythritol, dipentaerythritol, trimethylolpropane, and Ethylene oxide addition triol of trimethylolpropane, propylene oxide addition triol of trimethylolpropane, ethylene oxide and propylene oxide addition triol of trimethylolpropane, ethylene oxide addition tetraol of pentaerythritol, ethylene oxide addition hexaol of dipentaerythritol, etc. Polyhydric alcohol such as alkylene oxide addition polyol , Or two or more types of ionic-polymerizable cyclic compounds by ring-opening polymerization polyether polyols obtained can be mentioned.
なお、イオン重合性環状化合物としては、例えばエチレンオキシド、プロピレンオキシド、ブテン-1-オキシド、イソブテンオキシド、3,3-ビス(クロロメチル)オキセタン、テトラヒドロフラン、2-メチルテトラヒドロフラン、ジオキサン、トリオキサン、テトラオキサン、シクロヘキセンオキシド、スチレンオキシド、エピクロルヒドリン、グリシジルエーテル、アリルグリシジルエーテル、アリルグリシジルカーボネート、ブタジエンモノオキシド、イソプレンモノオキシド、ビニルオキセタン、ビニルテトラヒドロフラン、ビニルシクロヘキセンオキシド、フェニルグリシジルエーテル、ブチルグリシジルエーテル、安息香酸グリシジルエステル等の環状エーテル類が挙げられる。上記二種類以上のイオン重合性環状化合物の具体的な組み合わせとしては、テトラヒドロフランとエチレンオキシド、テトラヒドロフランとプロピレンオキシド、テトラヒドロフランと2-メチルテトラヒドロフラン、テトラヒドロフランと3-メチルテトラヒドロフラン、エチレンオキシドとプロピレンオキシド、ブテン-1-オキシドとエチレンオキシド、テトラヒドロフランとブテン-1-オキシドとエチレンオキシド等を挙げることができる。
Examples of the ion-polymerizable cyclic compound include ethylene oxide, propylene oxide, butene-1-oxide, isobutene oxide, 3,3-bis (chloromethyl) oxetane, tetrahydrofuran, 2-methyltetrahydrofuran, dioxane, trioxane, tetraoxane, and cyclohexene. Such as oxide, styrene oxide, epichlorohydrin, glycidyl ether, allyl glycidyl ether, allyl glycidyl carbonate, butadiene monooxide, isoprene monooxide, vinyl oxetane, vinyl tetrahydrofuran, vinyl cyclohexene oxide, phenyl glycidyl ether, butyl glycidyl ether, benzoic acid glycidyl ester, etc. Examples include cyclic ethers. Specific combinations of the two or more types of ion-polymerizable cyclic compounds include tetrahydrofuran and ethylene oxide, tetrahydrofuran and propylene oxide, tetrahydrofuran and 2-methyltetrahydrofuran, tetrahydrofuran and 3-methyltetrahydrofuran, ethylene oxide and propylene oxide, butene-1- Examples thereof include oxide and ethylene oxide, tetrahydrofuran, butene-1-oxide, and ethylene oxide.
また、上記イオン重合性環状化合物と、エチレンイミン等の環状イミン類、β-プロピオラクトン、グリコール酸ラクチド等の環状ラクトン酸、あるいはジメチルシクロポリシロキサン類とを開環共重合させたポリエーテルポリオールを使用することもできる。
Further, a polyether polyol obtained by ring-opening copolymerization of the above ion-polymerizable cyclic compound with a cyclic imine such as ethyleneimine, a cyclic lactone acid such as β-propiolactone or glycolic acid lactide, or dimethylcyclopolysiloxane. Can also be used.
脂環式ポリエーテルポリオールとしては、例えば水添ビスフェノールAのアルキレンオキシド付加ジオール、水添ビスフェノールFのアルキレンオキシド付加ジオール、1,4-シクロヘキサンジオールのアルキレンオキシド付加ジオール等が挙げられる。
Examples of the alicyclic polyether polyol include hydrogenated bisphenol A alkylene oxide addition diol, hydrogenated bisphenol F alkylene oxide addition diol, 1,4-cyclohexanediol alkylene oxide addition diol, and the like.
芳香族ポリエーテルポリオールとしては、例えばビスフェノールAのアルキレンオキシド付加ジオール、ビスフェノールFのアルキレンオキシド付加ジオール、ハイドロキノンのアルキレンオキシド付加ジオール、ナフトハイドロキノンのアルキレンオキシド付加ジオール、アントラハイドロキノンのアルキレンオキシド付加ジオール等が挙げられる。
Examples of the aromatic polyether polyol include alkylene oxide addition diol of bisphenol A, alkylene oxide addition diol of bisphenol F, alkylene oxide addition diol of hydroquinone, alkylene oxide addition diol of naphthohydroquinone, alkylene oxide addition diol of anthrahydroquinone, etc. It is done.
上記ポリエーテルポリオールの市販品としては、例えば脂肪族ポリエーテルポリオールとしては、PTMG650、PTMG1000、PTMG2000(以上、三菱化学(株)製)、PPG1000、EXCENOL1020、EXCENOL2020、EXCENOL3020、EXCENOL4020(以上、旭硝子(株)製)、PEG1000、ユニセーフDC1100、ユニセーフDC1800、ユニセーフDCB1100、ユニセーフDCB1800(以上、日本油脂(株)製)、PPTG1000、PPTG2000、PPTG4000、PTG400、PTG650、PTG2000、PTG3000、PTGL1000、PTGL2000(以上、保土谷化学工業(株)製)、PPG400、PBG400、Z-3001-4、Z-3001-5、PBG2000、PBG2000B(以上、第一工業製薬(株)製)、TMP30、PNT4グリコール、EDA P4、EDA P8(以上、日本乳化剤(株)製)、クオドロール(旭電化(株)製)が挙げられる。芳香族ポリエーテルポリオールとしてはユニオールDA400、DA700、DA1000、DB400(以上、日本油脂(株)製)等を挙げることができる。
As a commercially available product of the above polyether polyol, for example, as an aliphatic polyether polyol, PTMG650, PTMG1000, PTMG2000 (manufactured by Mitsubishi Chemical Corporation), PPG1000, EXCENOL1020, EXCENOL2020, EXCENOL3020, EXCENOL4020 (above, Asahi Glass Co., Ltd.) )), PEG1000, Unisafe DC1100, Unisafe DC1800, Unisafe DCB1100, Unisafe DCB1800 (above, manufactured by NOF Corporation), PPTG1000, PPTG2000, PPTG4000, PTG400, PTG650, PTG2000, PTG3000, PTGL1000, PTGL2000 (above, Hodogaya Chemical Industry Co., Ltd.), PPG400, PBG400, Z-300 -4, Z-3001-5, PBG2000, PBG2000B (Daiichi Kogyo Seiyaku Co., Ltd.), TMP30, PNT4 glycol, EDA P4, EDA P8 (Nippon Emulsifier Co., Ltd.), Quadrol (Asahi Denka) Product). Examples of the aromatic polyether polyol include Uniol DA400, DA700, DA1000, DB400 (manufactured by NOF Corporation) and the like.
また、上記ポリエステルポリオールは、多価アルコールと多塩基酸とを反応させて得られる。ここで、多価アルコールとしては、エチレングリコール、ポリエチレングリコール、プロピレングリコール、ポリプロピレングリコール、テトラメチレングリコール、ポリテトラメチレングリコール、1,4-ブタンジオール、1,5-ペンタンジオール、1,6-ヘキサンジオール、1,7-ヘプタンジオール、1,8-オクタンジオール、ネオペンチルグリコール、1,4-シクロヘキサンジオール、1,4-シクロヘキサンジメタノール、1,2-ビス(ヒドロキシエチル)シクロヘキサン、2,2-ジエチル-1,3-プロパンジオール、3-メチル-1,5-ペンタンジオール、1,9-ノナンジオール、2-メチル-1,8-オクタンジオール、グリセリン、トリメチロールプロパン、トリメチロールプロパンのエチレンオキシド付加体、トリメチロールプロパンのプロピレンオキシド付加体、トリメチロールプロパンのエチレンオキシドとプロピレンオキシドの付加体、ソルビトール、ペンタエリスリトール、ジペンタエリスリトール、アルキレンオキシド付加ポリオール等が挙げられる。また、多塩基酸としては、例えばフタル酸、イソフタル酸、テレフタル酸、マレイン酸、フマル酸、アジピン酸、セバシン酸等を挙げることができる。これらのポリエステルポリオールの市販品としては、クラポールP1010、クラポールP2010、PMIPA、PKA-A、PKA-A2、PNA-2000(以上、(株)クラレ製)等を使用することができる。
The polyester polyol is obtained by reacting a polyhydric alcohol and a polybasic acid. Here, as the polyhydric alcohol, ethylene glycol, polyethylene glycol, propylene glycol, polypropylene glycol, tetramethylene glycol, polytetramethylene glycol, 1,4-butanediol, 1,5-pentanediol, 1,6-hexanediol 1,7-heptanediol, 1,8-octanediol, neopentyl glycol, 1,4-cyclohexanediol, 1,4-cyclohexanedimethanol, 1,2-bis (hydroxyethyl) cyclohexane, 2,2-diethyl 1,3-propanediol, 3-methyl-1,5-pentanediol, 1,9-nonanediol, 2-methyl-1,8-octanediol, glycerin, trimethylolpropane, ethyleneoxy of trimethylolpropane Adducts, propylene oxide adducts of trimethylol propane, the adduct of ethylene oxide and propylene oxide trimethylolpropane, sorbitol, pentaerythritol, dipentaerythritol, alkylene oxide addition polyol, and the like. Examples of the polybasic acid include phthalic acid, isophthalic acid, terephthalic acid, maleic acid, fumaric acid, adipic acid, sebacic acid and the like. Examples of commercially available products of these polyester polyols include Kurapol P1010, Kurapol P2010, PMIPA, PKA-A, PKA-A2, PNA-2000 (manufactured by Kuraray Co., Ltd.) and the like.
また、上記ポリカーボネートポリオールとしては、例えば下記一般式(i)で示されるポリカーボネートジオールが挙げられる。
Further, examples of the polycarbonate polyol include polycarbonate diols represented by the following general formula (i).

一般式(i)中、R1は、炭素数2~20のアルキレン基、(ポリ)エチレングリコール残基、(ポリ)プロピレングリコール残基または(ポリ)テトラメチレングリコール残基を示し、mは1~30の範囲の整数である。
In general formula (i), R 1 represents an alkylene group having 2 to 20 carbon atoms, a (poly) ethylene glycol residue, a (poly) propylene glycol residue or a (poly) tetramethylene glycol residue, and m is 1 An integer in the range of ~ 30.
R1の具体例としては、次の化合物から両末端水酸基を除いた残基、すなわち1,4-ブタンジオール、1,5-ペンタンジオール、ネオペンチルグリコール、1,6-ヘキサンジオール、1,4-シクロヘキサンジメタノール、1、7-ヘプタンジオール、1,8-オクタンジオール、1,9-ノナンジオール、エチレングリコール、ジエチレングリコール、トリエチレングリコール、テトラエチレングリコール、プロピレングリコール、ジプロピレングリコール、トリプロピレングリコール、テトラプロピレングリコール等から水酸基を除いた残基が挙げられる。これらのポリカーボネートポリオールの市販品としては、DN-980、DN-981、DN-982、DN-983(以上、日本ポリウレタン工業(株)製)、PC-8000(PPG社製)、PNOC1000、PNOC2000、PMC100、PMC2000(以上、(株)クラレ製)、プラクセルCD-205、CD-208、CD-210、CD-220、CD-205PL、CD-208PL、CD-210PL、CD-220PL、CD-205HL、CD-208HL、CD-210HL、CD-220HL、CD-210T、CD-221T(以上、ダイセル化学工業(株)製)等を使用することができる。
Specific examples of R 1 include residues obtained by removing both terminal hydroxyl groups from the following compounds, that is, 1,4-butanediol, 1,5-pentanediol, neopentyl glycol, 1,6-hexanediol, 1,4 -Cyclohexanedimethanol, 1,7-heptanediol, 1,8-octanediol, 1,9-nonanediol, ethylene glycol, diethylene glycol, triethylene glycol, tetraethylene glycol, propylene glycol, dipropylene glycol, tripropylene glycol, Examples thereof include a residue obtained by removing a hydroxyl group from tetrapropylene glycol or the like. Commercial products of these polycarbonate polyols include DN-980, DN-981, DN-982, DN-983 (manufactured by Nippon Polyurethane Industry Co., Ltd.), PC-8000 (manufactured by PPG), PNOC1000, PNOC2000, PMC100, PMC2000 (above, manufactured by Kuraray Co., Ltd.), Plaxel CD-205, CD-208, CD-210, CD-220, CD-205PL, CD-208PL, CD-210PL, CD-220PL, CD-205HL, CD-208HL, CD-210HL, CD-220HL, CD-210T, CD-221T (above, manufactured by Daicel Chemical Industries, Ltd.) and the like can be used.
上記ポリカプロラクトンポリオールとしては、εーカプロラクトンを例えば、エチレングリコール、ポリエチレングリコール、プロピレングリコール、ポリプロピレングリコール、テトラメチレングリコール、ポリテトラメチレングリコール、1,2-ポリブチレングリコール、1,6-ヘキサンジオール、ネオペンチルグリコール、1,4-シクロヘキサンジメタノール、1,4-ブタンジオール等のジオールに付加反応させて得られるポリカプロラクトンジオールが挙げられる。これらの市販品としては、プラクセル205、205AL、212、212AL、220、220AL(以上、ダイセル化学工業(株)製)等を使用することができる。
Examples of the polycaprolactone polyol include ε-caprolactone such as ethylene glycol, polyethylene glycol, propylene glycol, polypropylene glycol, tetramethylene glycol, polytetramethylene glycol, 1,2-polybutylene glycol, 1,6-hexanediol, neo Examples thereof include polycaprolactone diols obtained by addition reaction with diols such as pentyl glycol, 1,4-cyclohexanedimethanol and 1,4-butanediol. As these commercial products, Plaxel 205, 205AL, 212, 212AL, 220, 220AL (above, manufactured by Daicel Chemical Industries, Ltd.) or the like can be used.
分子中に2個以上の水酸基を有する脂肪族炭化水素としては、エチレングリコール、プロピレングリコール、1,4-ブタンジオール、1,5-ペンタンジオール、1,6-ヘキサンジオール、1、7-ヘプタンジオール、1,8-オクタンジオール、1,9-ノナンジオール、ネオペンチルグリコール、2,2-ジエチル-1,3-プロパンジオール、3-メチル-1,5-ペンタンジオール、2-メチル-1,8-オクタンジオール、ヒドロキシ末端水添ポリブタジエン、グリセリン、トリメチロールプロパン、ペンタエリスリトール、ソルビトール等が挙げられる。
Examples of the aliphatic hydrocarbon having two or more hydroxyl groups in the molecule include ethylene glycol, propylene glycol, 1,4-butanediol, 1,5-pentanediol, 1,6-hexanediol, and 1,7-heptanediol. 1,8-octanediol, 1,9-nonanediol, neopentyl glycol, 2,2-diethyl-1,3-propanediol, 3-methyl-1,5-pentanediol, 2-methyl-1,8 -Octanediol, hydroxy-terminated hydrogenated polybutadiene, glycerin, trimethylolpropane, pentaerythritol, sorbitol and the like.
分子中に2個以上の水酸基を有する脂環式炭化水素としては、例えば1,4-シクロヘキサンジオール、1,4-シクロヘキサンジメタノール、1,2-ビス(ヒドロキシエチル)シクロヘキサン、ジシクロペンタジエンのジメチロール化合物、トリシクロデカンジメタノール等が挙げられる。
Examples of the alicyclic hydrocarbon having two or more hydroxyl groups in the molecule include 1,4-cyclohexanediol, 1,4-cyclohexanedimethanol, 1,2-bis (hydroxyethyl) cyclohexane, dimethylol such as dicyclopentadiene. Compound, tricyclodecane dimethanol and the like.
分子中に2個以上の水酸基を有する不飽和炭化水素としては、例えばヒドロキシ末端ポリブタジエン、ヒドロキシ末端ポリイソプレン等が挙げられる。
Examples of the unsaturated hydrocarbon having two or more hydroxyl groups in the molecule include hydroxy-terminated polybutadiene and hydroxy-terminated polyisoprene.
さらにまた、上記以外のポリオールとしては、例えばβ-メチル-δ-バレロラクトンジオール、ひまし油変性ジオール、ポリジメチルシロキサンの末端ジオール化合物、ポリジメチルシロキサンカルビトール変性ジオール等が挙げられる。
Furthermore, examples of polyols other than the above include β-methyl-δ-valerolactone diol, castor oil-modified diol, terminal diol compound of polydimethylsiloxane, polydimethylsiloxane carbitol-modified diol, and the like.
これらのポリオール化合物の好ましい質量平均分子量は1000~10000、特に好ましくは1000~9000である。質量平均分子量は、ポリマーの一部をテトラヒドロフラン(THF)に溶解し、ゲルパーミエイションクロマトグラフィー(GPC)によって測定される値とする。本発明における質量平均分子量は、ポリスチレンを標準物質とした値である。
These polyol compounds preferably have a weight average molecular weight of 1,000 to 10,000, particularly preferably 1,000 to 9000. The mass average molecular weight is a value measured by gel permeation chromatography (GPC) by dissolving a part of the polymer in tetrahydrofuran (THF). The mass average molecular weight in the present invention is a value using polystyrene as a standard substance.
最も好ましくは、ポリエチレングリコールモノメチルエーテルであり、その数平均分子量は100~10000であり、好ましくは200~5000であり、最も好ましくは300~1000である。
Most preferably, it is polyethylene glycol monomethyl ether, and its number average molecular weight is from 100 to 10,000, preferably from 200 to 5,000, most preferably from 300 to 1,000.
(炭素数2~20のアルキレン鎖を含むモノマー成分)
前記炭素数2~20のアルキレン鎖を含むモノマー成分としては特に制限はなく、公知の炭素数2~20のアルキレン鎖を含むモノマー成分を用いることができる。前記炭素数2~20のアルキレン鎖を含むモノマー成分は液晶性モノマーであることが、揮散性が低い観点や、後述するミクロ相分離構造膜を製造しやすい観点から好ましい。いかなる理論に拘泥するものでもないが、液晶性モノマーを用いる場合、リビング重合後の残存モノマー成分が相分離配向を促進できることができる。
また、液晶場での重合とすることで、リビング重合の反応速度を速くすることもできる。 (Monomer component containing an alkylene chain having 2 to 20 carbon atoms)
The monomer component including an alkylene chain having 2 to 20 carbon atoms is not particularly limited, and a known monomer component including an alkylene chain having 2 to 20 carbon atoms can be used. The monomer component containing an alkylene chain having 2 to 20 carbon atoms is preferably a liquid crystalline monomer from the viewpoint of low volatility and easy production of a microphase separation structure film described later. Without being bound by any theory, when a liquid crystalline monomer is used, the residual monomer component after living polymerization can promote phase separation alignment.
Moreover, the reaction rate of living polymerization can be increased by performing polymerization in a liquid crystal field.
前記炭素数2~20のアルキレン鎖を含むモノマー成分としては特に制限はなく、公知の炭素数2~20のアルキレン鎖を含むモノマー成分を用いることができる。前記炭素数2~20のアルキレン鎖を含むモノマー成分は液晶性モノマーであることが、揮散性が低い観点や、後述するミクロ相分離構造膜を製造しやすい観点から好ましい。いかなる理論に拘泥するものでもないが、液晶性モノマーを用いる場合、リビング重合後の残存モノマー成分が相分離配向を促進できることができる。
また、液晶場での重合とすることで、リビング重合の反応速度を速くすることもできる。 (Monomer component containing an alkylene chain having 2 to 20 carbon atoms)
The monomer component including an alkylene chain having 2 to 20 carbon atoms is not particularly limited, and a known monomer component including an alkylene chain having 2 to 20 carbon atoms can be used. The monomer component containing an alkylene chain having 2 to 20 carbon atoms is preferably a liquid crystalline monomer from the viewpoint of low volatility and easy production of a microphase separation structure film described later. Without being bound by any theory, when a liquid crystalline monomer is used, the residual monomer component after living polymerization can promote phase separation alignment.
Moreover, the reaction rate of living polymerization can be increased by performing polymerization in a liquid crystal field.
前記液晶性モノマーの中でも重合性液晶化合物を用いることが好ましい。いかなる理論に拘泥するものでもないが、重合性液晶化合物を用いる場合、リビング重合後の残存モノマー成分を、後述する相分離構造を固定化する工程で架橋または重合させることによって、相分離構造を固定化しやすくすることができる。すなわち、本発明のブロック共重合体は、前記炭素数2~20のアルキレン鎖を含むポリマー成分(B)中、炭素数6~50のメソゲン側鎖を有していることが好ましく、前記メソゲン側鎖が、前記メソゲン側鎖1本の中に少なくとも1個の重合性基を有することがより好ましい。前記重合性基の好ましい範囲は、後述の一般式(X)中の前記Q1およびQ2が表す重合性基の好ましい範囲と同様である。
炭素数6~50のメソゲン側鎖は、炭素数6~40のメソゲン側鎖であることがより好ましく、炭素数6~30のメソゲン側鎖であることが特に好ましい。「メソゲン基」は、液晶化合物のコア部を形成し得る基を含み、メソゲン基を有する化合物の例には、液晶性化合物が含まれるとともに、メソゲン基を有するが液晶形成をしない、即ち非液晶性化合物、も含まれる。メソゲン基についてさらに説明する。本発明において、メソゲン基とは、液晶形成に寄与する液晶分子の主要骨格を示す基である。液晶分子は、結晶状態と等方性液体状態の中間の状態(メソフェーズ)である液晶性を示す。前記メソゲン基については特に制限はなく、例えば、「Flussige Kristalle in Tabellen II」(VEB Deutsche Verlag fur Grundstoff Industrie,Leipzig、1984年刊)、特に第7頁~第16頁の記載、及び、液晶便覧編集委員会編、液晶便覧(丸善、2000年刊)、特に第3章の記載、を参照することができる。好ましくは、サーモトロピック液晶の残基であり、さらに好ましくは、棒状液晶及びディスコティック液晶の残基である。棒状液晶ではネマティック相及びスメクティックA相を示す液晶の残基がより好ましく、ディスコティック液晶ではディスコティックネマティック相を示す液晶の残基がより好ましい。
ディスコティック液晶の残基の好ましい例には、ベンゼン、トリフェニレン、トルキセン、トリオキサトルキセン、アントラキノン、フタロシアニン又はポリフィリン、マクロサイクレン、ビス(1,3-ジケトン)銅錯体、テトラアリールビピラニリデン、テトラチアフルバレン、及びイノシトールが含まれる。
棒状液晶の残基、即ち、棒状液晶のメソゲン基あるいはコア部と呼ばれる剛直な液晶形成に寄与する液晶分子の主要骨格としては、後述する一般式(X)中の-Cy1-L2-(Cy2-L3)n-Cy3-L4-で表される基であることが好ましい。
これらの中でも前記メソゲン基としては、後述する一般式(X)中の-Cy1-L2-(Cy2-L3)n-Cy3-L4-で表される基であることが好ましく、前記-Cy1-L2-(Cy2-L3)n-Cy3-L4-で表される基のより好ましい範囲は後述の一般式(X)における各基の好ましい範囲と同様である。 Among the liquid crystal monomers, it is preferable to use a polymerizable liquid crystal compound. Without being bound by any theory, when a polymerizable liquid crystal compound is used, the phase separation structure is fixed by crosslinking or polymerizing the residual monomer component after living polymerization in the step of fixing the phase separation structure described later. Can be made easier. That is, the block copolymer of the present invention preferably has a mesogenic side chain having 6 to 50 carbon atoms in the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms. More preferably, the chain has at least one polymerizable group in one of the mesogenic side chains. The preferred range of the polymerizable group is the same as the preferred range of the polymerizable group represented by Q 1 and Q 2 in the general formula (X) described later.
The mesogen side chain having 6 to 50 carbon atoms is more preferably a mesogen side chain having 6 to 40 carbon atoms, and particularly preferably a mesogen side chain having 6 to 30 carbon atoms. “Mesogenic group” includes a group capable of forming a core part of a liquid crystal compound, and examples of the compound having a mesogenic group include a liquid crystalline compound and also has a mesogenic group but does not form a liquid crystal. Sexual compounds are also included. The mesogenic group will be further described. In the present invention, the mesogenic group is a group showing a main skeleton of liquid crystal molecules that contribute to liquid crystal formation. The liquid crystal molecules exhibit liquid crystallinity that is an intermediate state (mesophase) between a crystalline state and an isotropic liquid state. The mesogenic group is not particularly limited. For example, “Flushage Kristall in Tablen II” (VEB Deutsche Verlagfur Grundoff Industrie, Leipzig, published in 1984), pages 7 to 16 You can refer to the editions of the association, Liquid Crystal Handbook (Maruzen, 2000), especially the description inChapter 3. A residue of a thermotropic liquid crystal is preferable, and a residue of a rod-like liquid crystal and a discotic liquid crystal is more preferable. In the rod-like liquid crystal, a liquid crystal residue showing a nematic phase and a smectic A phase is more preferable, and in a discotic liquid crystal, a liquid crystal residue showing a discotic nematic phase is more preferable.
Preferred examples of the residue of the discotic liquid crystal include benzene, triphenylene, truxene, trioxatruxene, anthraquinone, phthalocyanine or porphyrin, macrocyclene, bis (1,3-diketone) copper complex, tetraarylbipyranylidene, Tetrathiafulvalene and inositol are included.
The main skeleton of the liquid crystal molecules contributing to the formation of a rigid liquid crystal called a mesogenic group or core portion of the rod-like liquid crystal, that is, the residue of the rod-like liquid crystal, is -Cy 1 -L 2- ( cy 2 -L 3) n -Cy 3 -L 4 - is preferably a group represented by.
Among these, the mesogenic group is preferably a group represented by -Cy 1 -L 2- (Cy 2 -L 3 ) n -Cy 3 -L 4- in the general formula (X) described later. The more preferable range of the group represented by the above -Cy 1 -L 2- (Cy 2 -L 3 ) n -Cy 3 -L 4- is the same as the preferable range of each group in the general formula (X) described later. is there.
炭素数6~50のメソゲン側鎖は、炭素数6~40のメソゲン側鎖であることがより好ましく、炭素数6~30のメソゲン側鎖であることが特に好ましい。「メソゲン基」は、液晶化合物のコア部を形成し得る基を含み、メソゲン基を有する化合物の例には、液晶性化合物が含まれるとともに、メソゲン基を有するが液晶形成をしない、即ち非液晶性化合物、も含まれる。メソゲン基についてさらに説明する。本発明において、メソゲン基とは、液晶形成に寄与する液晶分子の主要骨格を示す基である。液晶分子は、結晶状態と等方性液体状態の中間の状態(メソフェーズ)である液晶性を示す。前記メソゲン基については特に制限はなく、例えば、「Flussige Kristalle in Tabellen II」(VEB Deutsche Verlag fur Grundstoff Industrie,Leipzig、1984年刊)、特に第7頁~第16頁の記載、及び、液晶便覧編集委員会編、液晶便覧(丸善、2000年刊)、特に第3章の記載、を参照することができる。好ましくは、サーモトロピック液晶の残基であり、さらに好ましくは、棒状液晶及びディスコティック液晶の残基である。棒状液晶ではネマティック相及びスメクティックA相を示す液晶の残基がより好ましく、ディスコティック液晶ではディスコティックネマティック相を示す液晶の残基がより好ましい。
ディスコティック液晶の残基の好ましい例には、ベンゼン、トリフェニレン、トルキセン、トリオキサトルキセン、アントラキノン、フタロシアニン又はポリフィリン、マクロサイクレン、ビス(1,3-ジケトン)銅錯体、テトラアリールビピラニリデン、テトラチアフルバレン、及びイノシトールが含まれる。
棒状液晶の残基、即ち、棒状液晶のメソゲン基あるいはコア部と呼ばれる剛直な液晶形成に寄与する液晶分子の主要骨格としては、後述する一般式(X)中の-Cy1-L2-(Cy2-L3)n-Cy3-L4-で表される基であることが好ましい。
これらの中でも前記メソゲン基としては、後述する一般式(X)中の-Cy1-L2-(Cy2-L3)n-Cy3-L4-で表される基であることが好ましく、前記-Cy1-L2-(Cy2-L3)n-Cy3-L4-で表される基のより好ましい範囲は後述の一般式(X)における各基の好ましい範囲と同様である。 Among the liquid crystal monomers, it is preferable to use a polymerizable liquid crystal compound. Without being bound by any theory, when a polymerizable liquid crystal compound is used, the phase separation structure is fixed by crosslinking or polymerizing the residual monomer component after living polymerization in the step of fixing the phase separation structure described later. Can be made easier. That is, the block copolymer of the present invention preferably has a mesogenic side chain having 6 to 50 carbon atoms in the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms. More preferably, the chain has at least one polymerizable group in one of the mesogenic side chains. The preferred range of the polymerizable group is the same as the preferred range of the polymerizable group represented by Q 1 and Q 2 in the general formula (X) described later.
The mesogen side chain having 6 to 50 carbon atoms is more preferably a mesogen side chain having 6 to 40 carbon atoms, and particularly preferably a mesogen side chain having 6 to 30 carbon atoms. “Mesogenic group” includes a group capable of forming a core part of a liquid crystal compound, and examples of the compound having a mesogenic group include a liquid crystalline compound and also has a mesogenic group but does not form a liquid crystal. Sexual compounds are also included. The mesogenic group will be further described. In the present invention, the mesogenic group is a group showing a main skeleton of liquid crystal molecules that contribute to liquid crystal formation. The liquid crystal molecules exhibit liquid crystallinity that is an intermediate state (mesophase) between a crystalline state and an isotropic liquid state. The mesogenic group is not particularly limited. For example, “Flushage Kristall in Tablen II” (VEB Deutsche Verlagfur Grundoff Industrie, Leipzig, published in 1984), pages 7 to 16 You can refer to the editions of the association, Liquid Crystal Handbook (Maruzen, 2000), especially the description in
Preferred examples of the residue of the discotic liquid crystal include benzene, triphenylene, truxene, trioxatruxene, anthraquinone, phthalocyanine or porphyrin, macrocyclene, bis (1,3-diketone) copper complex, tetraarylbipyranylidene, Tetrathiafulvalene and inositol are included.
The main skeleton of the liquid crystal molecules contributing to the formation of a rigid liquid crystal called a mesogenic group or core portion of the rod-like liquid crystal, that is, the residue of the rod-like liquid crystal, is -Cy 1 -L 2- ( cy 2 -L 3) n -Cy 3 -L 4 - is preferably a group represented by.
Among these, the mesogenic group is preferably a group represented by -Cy 1 -L 2- (Cy 2 -L 3 ) n -Cy 3 -L 4- in the general formula (X) described later. The more preferable range of the group represented by the above -Cy 1 -L 2- (Cy 2 -L 3 ) n -Cy 3 -L 4- is the same as the preferable range of each group in the general formula (X) described later. is there.
前記液晶性モノマーの中でも棒状液晶化合物であることがより好ましい。支持体上でリビング重合する際に、残存するモノマー成分もしくはポリマー(開始剤由来のポリマーなど)がミクロ相分離構造の形成に不利に作用することが予想された。しかし特定の棒状液晶化合物を液晶性モノマーとして使用した場合には、液晶モノマーが残存し、流動性が高まるため欠陥のない綺麗なシリンダー型ミクロ相分離構造を形成することができる。
Among the liquid crystalline monomers, a rod-like liquid crystal compound is more preferable. When living polymerizing on a support, it was expected that the remaining monomer component or polymer (such as an initiator-derived polymer) adversely affects the formation of a microphase separation structure. However, when a specific rod-like liquid crystal compound is used as the liquid crystal monomer, the liquid crystal monomer remains and the fluidity is increased, so that a beautiful cylindrical microphase separation structure free from defects can be formed.
-棒状液晶化合物-
棒状液晶化合物としては、アゾメチン類、アゾキシ類、シアノビフェニル類、シアノフェニルエステル類、安息香酸エステル類、シクロヘキサンカルボン酸フェニルエステル類、シアノフェニルシクロヘキサン類、シアノ置換フェニルピリミジン類、アルコキシ置換フェニルピリミジン類、フェニルジオキサン類、トラン類およびアルケニルシクロヘキシルベンゾニトリル類が好ましく用いられる。以上のような低分子液晶性分子だけではなく、高分子液晶性分子も用いることができる。
前記棒状液晶化合物の中でも、棒状液晶化合物を重合によって配向を固定することができる重合性棒状液晶化合物であることがより好ましい。 -Rod-shaped liquid crystal compounds-
Examples of the rod-like liquid crystal compound include azomethines, azoxys, cyanobiphenyls, cyanophenyl esters, benzoic acid esters, cyclohexanecarboxylic acid phenyl esters, cyanophenylcyclohexanes, cyano-substituted phenylpyrimidines, alkoxy-substituted phenylpyrimidines, Phenyldioxanes, tolanes and alkenylcyclohexylbenzonitriles are preferably used. In addition to the above low-molecular liquid crystalline molecules, high-molecular liquid crystalline molecules can also be used.
Among the rod-like liquid crystal compounds, a polymerizable rod-like liquid crystal compound that can fix the orientation of the rod-like liquid crystal compound by polymerization is more preferable.
棒状液晶化合物としては、アゾメチン類、アゾキシ類、シアノビフェニル類、シアノフェニルエステル類、安息香酸エステル類、シクロヘキサンカルボン酸フェニルエステル類、シアノフェニルシクロヘキサン類、シアノ置換フェニルピリミジン類、アルコキシ置換フェニルピリミジン類、フェニルジオキサン類、トラン類およびアルケニルシクロヘキシルベンゾニトリル類が好ましく用いられる。以上のような低分子液晶性分子だけではなく、高分子液晶性分子も用いることができる。
前記棒状液晶化合物の中でも、棒状液晶化合物を重合によって配向を固定することができる重合性棒状液晶化合物であることがより好ましい。 -Rod-shaped liquid crystal compounds-
Examples of the rod-like liquid crystal compound include azomethines, azoxys, cyanobiphenyls, cyanophenyl esters, benzoic acid esters, cyclohexanecarboxylic acid phenyl esters, cyanophenylcyclohexanes, cyano-substituted phenylpyrimidines, alkoxy-substituted phenylpyrimidines, Phenyldioxanes, tolanes and alkenylcyclohexylbenzonitriles are preferably used. In addition to the above low-molecular liquid crystalline molecules, high-molecular liquid crystalline molecules can also be used.
Among the rod-like liquid crystal compounds, a polymerizable rod-like liquid crystal compound that can fix the orientation of the rod-like liquid crystal compound by polymerization is more preferable.
重合性棒状液晶化合物としては、Makromol. Chem., 190巻、2255頁(1989年)、Advanced Materials 5巻、107頁(1993年)、米国特許4683327号、同5622648号、同5770107号、WO95/22586号、同95/24455号、同97/00600号、同98/23580号、同98/52905号、特開平1-272551号、同6-16616号、同7-110469号、同11-80081号、および特願2001-64627号(特開2001-328973号公報)、特開2004-1235979号公報、Transactions of Materials Reseach Society of Japan, 28[3], 553-556(2003)、特開2008-127336号公報、特開2010-116463号公報などに記載の化合物、ならびに、後述の一般式(X)にて表される化合物および後述の一般式(V)で表される化合物などを用いることができる。
As the polymerizable rod-like liquid crystal compound, Makromol. Chem. 190, 2255 (1989), Advanced Materials, 5, 107 (1993), U.S. Pat. Nos. 4,683,327, 5,622,648 and 5,770,107, WO 95/22586, 95/24455, 97/97. No. 0600, No. 98/23580, No. 98/52905, JP-A-1-272551, JP-A-6-16616, JP-A-7-110469, JP-A-11-80081, and Japanese Patent Application No. 2001-64627 2001-328893), JP 2004-1235979 A, Transactions of Materials Research Society of Japan, 28 [3], 553-556 (2003), JP 2008-127336 A, JP Compounds described like 010-116463 JP, and the like can be used compounds represented by the general formula below compounds and later the general formula represented by (X) (V).
また、重合性棒状液晶化合物として好ましくは、下記一般式(X)にて表される化合物および後述の一般式(V)で表される化合物であり、前記一般式(X)で表される化合物がより好ましい。
The polymerizable rod-like liquid crystal compound is preferably a compound represented by the following general formula (X) and a compound represented by the following general formula (V), and a compound represented by the above general formula (X) Is more preferable.
一般式(X) Q1-L1-Cy1-L2-(Cy2-L3)n-Cy3-L4-Q2
一般式(X)中、Q1およびQ2はそれぞれ独立に重合性基であり、L1、およびL4はそれぞれ独立に二価の連結基であり、L1およびL4のうち少なくとも一方は、少なくとも炭素数2~20のアルキレン基を含み、L2およびL3はそれぞれ独立に単結合または二価の連結基であり、Cy1、Cy2およびCy3は二価の環状基であり、nは0、1、2、または3である。
以下にさらに一般式(X)で表される重合性棒状液晶化合物について説明する。
前記一般式(X)中、Q1およびQ2はそれぞれ独立に重合性基である。重合性基の重合反応は、付加重合(開環重合を含む)または縮合重合であることが好ましい。言い換えると、重合性基は、付加重合反応または縮合重合反応が可能な官能基であることが好ましい。以下に重合性基の例を示す。

これらの重合性基の中でも、Q1およびQ2のうち少なくとも一方が

のいずれかであることが好ましく、Q1およびQ2のうち他の一方がオキセタン環を有する基(以下、オキセタニル基とも言う)であることが好ましい。
Formula (X) Q 1 -L 1 -Cy 1 -L 2- (Cy 2 -L 3 ) n -Cy 3 -L 4 -Q 2
In general formula (X), Q 1 and Q 2 are each independently a polymerizable group, L 1 and L 4 are each independently a divalent linking group, and at least one of L 1 and L 4 is Including at least an alkylene group having 2 to 20 carbon atoms, L 2 and L 3 are each independently a single bond or a divalent linking group, Cy 1 , Cy 2 and Cy 3 are divalent cyclic groups, n is 0, 1, 2, or 3.
The polymerizable rod-like liquid crystal compound represented by the general formula (X) will be described below.
In the general formula (X), Q 1 and Q 2 are each independently a polymerizable group. The polymerization reaction of the polymerizable group is preferably addition polymerization (including ring-opening polymerization) or condensation polymerization. In other words, the polymerizable group is preferably a functional group capable of addition polymerization reaction or condensation polymerization reaction. Examples of polymerizable groups are shown below.
Among these polymerizable groups, at least one of Q 1 and Q 2 is
And the other of Q 1 and Q 2 is preferably a group having an oxetane ring (hereinafter also referred to as oxetanyl group).
一般式(X)中、Q1およびQ2はそれぞれ独立に重合性基であり、L1、およびL4はそれぞれ独立に二価の連結基であり、L1およびL4のうち少なくとも一方は、少なくとも炭素数2~20のアルキレン基を含み、L2およびL3はそれぞれ独立に単結合または二価の連結基であり、Cy1、Cy2およびCy3は二価の環状基であり、nは0、1、2、または3である。
以下にさらに一般式(X)で表される重合性棒状液晶化合物について説明する。
前記一般式(X)中、Q1およびQ2はそれぞれ独立に重合性基である。重合性基の重合反応は、付加重合(開環重合を含む)または縮合重合であることが好ましい。言い換えると、重合性基は、付加重合反応または縮合重合反応が可能な官能基であることが好ましい。以下に重合性基の例を示す。
In general formula (X), Q 1 and Q 2 are each independently a polymerizable group, L 1 and L 4 are each independently a divalent linking group, and at least one of L 1 and L 4 is Including at least an alkylene group having 2 to 20 carbon atoms, L 2 and L 3 are each independently a single bond or a divalent linking group, Cy 1 , Cy 2 and Cy 3 are divalent cyclic groups, n is 0, 1, 2, or 3.
The polymerizable rod-like liquid crystal compound represented by the general formula (X) will be described below.
In the general formula (X), Q 1 and Q 2 are each independently a polymerizable group. The polymerization reaction of the polymerizable group is preferably addition polymerization (including ring-opening polymerization) or condensation polymerization. In other words, the polymerizable group is preferably a functional group capable of addition polymerization reaction or condensation polymerization reaction. Examples of polymerizable groups are shown below.
前記一般式(X)中、L1およびL4はそれぞれ独立に二価の連結基であり、L1およびL4のうち少なくとも一方は、少なくとも炭素数2~20のアルキレン基を含む。L1およびL4はそれぞれ独立に、-O-、-S-、-CO-、-NR-、-C=N-、二価の鎖状基、二価の環状基およびそれらの組み合わせからなる群より選ばれる二価の連結基であることが好ましい。上記Rは炭素原子数が1から7のアルキル基または水素原子である。
組み合わせからなる二価の連結基の例を以下に示す。ここで、左側がQ(Q1またはQ2)に、右側がCy(Cy1またはCy3)に結合する。
L-1:-CO-O-二価の鎖状基-O-
L-2:-CO-O-二価の鎖状基-O-CO-
L-3:-CO-O-二価の鎖状基-O-CO-O-
L-4:-CO-O-二価の鎖状基-O-二価の環状基-
L-5:-CO-O-二価の鎖状基-O-二価の環状基-CO-O-
L-6:-CO-O-二価の鎖状基-O-二価の環状基-O-CO-
L-7:-CO-O-二価の鎖状基-O-二価の環状基-二価の鎖状基-
L-8:-CO-O-二価の鎖状基-O-二価の環状基-二価の鎖状基-CO-O-
L-9:-CO-O-二価の鎖状基-O-二価の環状基-二価の鎖状基-O-CO-
L-10:-CO-O-二価の鎖状基-O-CO-二価の環状基-
L-11:-CO-O-二価の鎖状基-O-CO-二価の環状基-CO-O-
L-12:-CO-O-二価の鎖状基-O-CO-二価の環状基-O-CO-
L-13:-CO-O-二価の鎖状基-O-CO-二価の環状基-二価の鎖状基-
L-14:-CO-O-二価の鎖状基-O-CO-二価の環状基-二価の鎖状基-CO-O-
L-15:-CO-O-二価の鎖状基-O-CO-二価の環状基-二価の鎖状基-O-CO-
L-16:-CO-O-二価の鎖状基-O-CO-O-二価の環状基-
L-17:-CO-O-二価の鎖状基-O-CO-O-二価の環状基-CO-O-
L-18:-CO-O-二価の鎖状基-O-CO-O-二価の環状基-O-CO-
L-19:-CO-O-二価の鎖状基-O-CO-O-二価の環状基-二価の鎖状基-
L-20:-CO-O-二価の鎖状基-O-CO-O-二価の環状基-二価の鎖状基-CO-O-
L-21:-CO-O-二価の鎖状基-O-CO-O-二価の環状基-二価の鎖状基-O-CO-
L-22:-二価の鎖状基-O-二価の鎖状基-O-
二価の鎖状基は、アルキレン基、置換アルキレン基、アルケニレン基、置換アルケニレン基、アルキニレン基、置換アルキニレン基を意味する。アルキレン基、置換アルキレン基、アルケニレン基、置換アルケニレン基が好ましく、アルキレン基およびアルケニレン基がさらに好ましい。
アルキレン基は、分岐を有していてもよい。アルキレン基の炭素数は2~20であることが好ましく、3乃至18であることがより好ましく、4乃至16であることがさらに好ましく、5乃至15であることが最も好ましい。L1およびL4が表す二価の連結基中に含まれるすべてのアルキレン基の炭素数の合計が上記範囲であることが好ましく、例えばL-1~L-22中に二価の鎖状基が複数含まれる場合はアルキレン基である二価の鎖状基の合計の炭素数が上記範囲であることが好ましい。
置換アルキレン基のアルキレン部分は、上記アルキレン基と同様である。置換基の例としてはハロゲン原子が含まれる。
アルケニレン基は、分岐を有していてもよい。アルケニレン基の炭素数は2乃至12であることが好ましく、2乃至10であることがさらに好ましく、2乃至8であることがもっとも好ましい。
置換アルキレン基のアルキレン部分は、上記アルキレン基と同様である。置換基の例としてはハロゲン原子が含まれる。
アルキニレン基は、分岐を有していてもよい。アルキニレン基の炭素数は2乃至12であることが好ましく、2乃至10であることがさらに好ましく、2乃至8であることがもっとも好ましい。
置換アルキニレン基のアルキニレン部分は、上記アルキニレン基と同様である。置換基の例としてはハロゲン原子が含まれる。
二価の鎖状基の具体例としては、エチレン、トリメチレン、プロピレン、テトラメチレン、2-メチル-テトラメチレン、ペンタメチレン、ヘキサメチレン、オクタメチレン、2-ブテニレン、2-ブチニレンなどが挙げられる。
二価の環状基の定義および例は、後述するCy1、Cy2およびCy3の定義および例と同様である。
L1およびL4のうちオキセタン基に結合する一方の連結基がL-21であることが好ましく、L1およびL4のうち他の一方はL-1であることが好ましい。 In the general formula (X), L 1 and L 4 are each independently a divalent linking group, and at least one of L 1 and L 4 includes at least an alkylene group having 2 to 20 carbon atoms. L 1 and L 4 each independently comprise —O—, —S—, —CO—, —NR—, —C═N—, a divalent chain group, a divalent cyclic group, and combinations thereof. A divalent linking group selected from the group is preferred. R is an alkyl group having 1 to 7 carbon atoms or a hydrogen atom.
The example of the bivalent coupling group which consists of a combination is shown below. Here, the left side is coupled to Q (Q 1 or Q 2 ), and the right side is coupled to Cy (Cy 1 or Cy 3 ).
L-1: —CO—O—divalent chain group —O—
L-2: —CO—O—divalent chain group —O—CO—
L-3: —CO—O—divalent chain group —O—CO—O—
L-4: —CO—O—divalent chain group—O—divalent cyclic group—
L-5: —CO—O—divalent chain group —O—divalent cyclic group —CO—O—
L-6: —CO—O—divalent chain group —O—divalent cyclic group —O—CO—
L-7: —CO—O—Divalent chain group—O—Divalent cyclic group—Divalent chain group—
L-8: —CO—O—divalent chain group—O—divalent cyclic group—divalent chain group —CO—O—
L-9: —CO—O—Divalent chain group—O—Divalent cyclic group—Divalent chain group —O—CO—
L-10: —CO—O—divalent chain group—O—CO—divalent cyclic group—
L-11: —CO—O—divalent chain group —O—CO—divalent cyclic group —CO—O—
L-12: —CO—O—divalent chain group —O—CO—divalent cyclic group —O—CO—
L-13: —CO—O—Divalent chain group—O—CO—Divalent cyclic group—Divalent chain group—
L-14: —CO—O—divalent chain group—O—CO—divalent cyclic group—divalent chain group—CO—O—
L-15: —CO—O—Divalent chain group—O—CO—Divalent cyclic group—Divalent chain group—O—CO—
L-16: —CO—O—divalent chain group—O—CO—O—divalent cyclic group—
L-17: —CO—O—divalent chain group —O—CO—O—divalent cyclic group —CO—O—
L-18: —CO—O—divalent chain group —O—CO—O—divalent cyclic group —O—CO—
L-19: —CO—O—divalent chain group—O—CO—O—divalent cyclic group—divalent chain group—
L-20: —CO—O—divalent chain group—O—CO—O—divalent cyclic group—divalent chain group—CO—O—
L-21: —CO—O—divalent chain group—O—CO—O—divalent cyclic group—divalent chain group—O—CO—
L-22: -Divalent chain group -O-Divalent chain group -O-
The divalent chain group means an alkylene group, a substituted alkylene group, an alkenylene group, a substituted alkenylene group, an alkynylene group, or a substituted alkynylene group. An alkylene group, a substituted alkylene group, an alkenylene group and a substituted alkenylene group are preferred, and an alkylene group and an alkenylene group are more preferred.
The alkylene group may have a branch. The alkylene group preferably has 2 to 20 carbon atoms, more preferably 3 to 18 carbon atoms, still more preferably 4 to 16 carbon atoms, and most preferably 5 to 15 carbon atoms. The total number of carbon atoms of all alkylene groups contained in the divalent linking group represented by L 1 and L 4 is preferably in the above range. For example, a divalent chain group in L-1 to L-22 When a plurality of are included, the total carbon number of the divalent chain group which is an alkylene group is preferably within the above range.
The alkylene part of the substituted alkylene group is the same as the above alkylene group. Examples of the substituent include a halogen atom.
The alkenylene group may have a branch. The alkenylene group preferably has 2 to 12 carbon atoms, more preferably 2 to 10 carbon atoms, and most preferably 2 to 8 carbon atoms.
The alkylene part of the substituted alkylene group is the same as the above alkylene group. Examples of the substituent include a halogen atom.
The alkynylene group may have a branch. The alkynylene group preferably has 2 to 12 carbon atoms, more preferably 2 to 10 carbon atoms, and most preferably 2 to 8 carbon atoms.
The alkynylene part of the substituted alkynylene group is the same as the above alkynylene group. Examples of the substituent include a halogen atom.
Specific examples of the divalent chain group include ethylene, trimethylene, propylene, tetramethylene, 2-methyl-tetramethylene, pentamethylene, hexamethylene, octamethylene, 2-butenylene, 2-butynylene and the like.
The definition and examples of the divalent cyclic group are the same as those of Cy 1 , Cy 2 and Cy 3 described later.
One of the linking groups bonded to the oxetane group out of L 1 and L 4 is preferably L-21, and the other one of L 1 and L 4 is preferably L-1.
組み合わせからなる二価の連結基の例を以下に示す。ここで、左側がQ(Q1またはQ2)に、右側がCy(Cy1またはCy3)に結合する。
L-1:-CO-O-二価の鎖状基-O-
L-2:-CO-O-二価の鎖状基-O-CO-
L-3:-CO-O-二価の鎖状基-O-CO-O-
L-4:-CO-O-二価の鎖状基-O-二価の環状基-
L-5:-CO-O-二価の鎖状基-O-二価の環状基-CO-O-
L-6:-CO-O-二価の鎖状基-O-二価の環状基-O-CO-
L-7:-CO-O-二価の鎖状基-O-二価の環状基-二価の鎖状基-
L-8:-CO-O-二価の鎖状基-O-二価の環状基-二価の鎖状基-CO-O-
L-9:-CO-O-二価の鎖状基-O-二価の環状基-二価の鎖状基-O-CO-
L-10:-CO-O-二価の鎖状基-O-CO-二価の環状基-
L-11:-CO-O-二価の鎖状基-O-CO-二価の環状基-CO-O-
L-12:-CO-O-二価の鎖状基-O-CO-二価の環状基-O-CO-
L-13:-CO-O-二価の鎖状基-O-CO-二価の環状基-二価の鎖状基-
L-14:-CO-O-二価の鎖状基-O-CO-二価の環状基-二価の鎖状基-CO-O-
L-15:-CO-O-二価の鎖状基-O-CO-二価の環状基-二価の鎖状基-O-CO-
L-16:-CO-O-二価の鎖状基-O-CO-O-二価の環状基-
L-17:-CO-O-二価の鎖状基-O-CO-O-二価の環状基-CO-O-
L-18:-CO-O-二価の鎖状基-O-CO-O-二価の環状基-O-CO-
L-19:-CO-O-二価の鎖状基-O-CO-O-二価の環状基-二価の鎖状基-
L-20:-CO-O-二価の鎖状基-O-CO-O-二価の環状基-二価の鎖状基-CO-O-
L-21:-CO-O-二価の鎖状基-O-CO-O-二価の環状基-二価の鎖状基-O-CO-
L-22:-二価の鎖状基-O-二価の鎖状基-O-
二価の鎖状基は、アルキレン基、置換アルキレン基、アルケニレン基、置換アルケニレン基、アルキニレン基、置換アルキニレン基を意味する。アルキレン基、置換アルキレン基、アルケニレン基、置換アルケニレン基が好ましく、アルキレン基およびアルケニレン基がさらに好ましい。
アルキレン基は、分岐を有していてもよい。アルキレン基の炭素数は2~20であることが好ましく、3乃至18であることがより好ましく、4乃至16であることがさらに好ましく、5乃至15であることが最も好ましい。L1およびL4が表す二価の連結基中に含まれるすべてのアルキレン基の炭素数の合計が上記範囲であることが好ましく、例えばL-1~L-22中に二価の鎖状基が複数含まれる場合はアルキレン基である二価の鎖状基の合計の炭素数が上記範囲であることが好ましい。
置換アルキレン基のアルキレン部分は、上記アルキレン基と同様である。置換基の例としてはハロゲン原子が含まれる。
アルケニレン基は、分岐を有していてもよい。アルケニレン基の炭素数は2乃至12であることが好ましく、2乃至10であることがさらに好ましく、2乃至8であることがもっとも好ましい。
置換アルキレン基のアルキレン部分は、上記アルキレン基と同様である。置換基の例としてはハロゲン原子が含まれる。
アルキニレン基は、分岐を有していてもよい。アルキニレン基の炭素数は2乃至12であることが好ましく、2乃至10であることがさらに好ましく、2乃至8であることがもっとも好ましい。
置換アルキニレン基のアルキニレン部分は、上記アルキニレン基と同様である。置換基の例としてはハロゲン原子が含まれる。
二価の鎖状基の具体例としては、エチレン、トリメチレン、プロピレン、テトラメチレン、2-メチル-テトラメチレン、ペンタメチレン、ヘキサメチレン、オクタメチレン、2-ブテニレン、2-ブチニレンなどが挙げられる。
二価の環状基の定義および例は、後述するCy1、Cy2およびCy3の定義および例と同様である。
L1およびL4のうちオキセタン基に結合する一方の連結基がL-21であることが好ましく、L1およびL4のうち他の一方はL-1であることが好ましい。 In the general formula (X), L 1 and L 4 are each independently a divalent linking group, and at least one of L 1 and L 4 includes at least an alkylene group having 2 to 20 carbon atoms. L 1 and L 4 each independently comprise —O—, —S—, —CO—, —NR—, —C═N—, a divalent chain group, a divalent cyclic group, and combinations thereof. A divalent linking group selected from the group is preferred. R is an alkyl group having 1 to 7 carbon atoms or a hydrogen atom.
The example of the bivalent coupling group which consists of a combination is shown below. Here, the left side is coupled to Q (Q 1 or Q 2 ), and the right side is coupled to Cy (Cy 1 or Cy 3 ).
L-1: —CO—O—divalent chain group —O—
L-2: —CO—O—divalent chain group —O—CO—
L-3: —CO—O—divalent chain group —O—CO—O—
L-4: —CO—O—divalent chain group—O—divalent cyclic group—
L-5: —CO—O—divalent chain group —O—divalent cyclic group —CO—O—
L-6: —CO—O—divalent chain group —O—divalent cyclic group —O—CO—
L-7: —CO—O—Divalent chain group—O—Divalent cyclic group—Divalent chain group—
L-8: —CO—O—divalent chain group—O—divalent cyclic group—divalent chain group —CO—O—
L-9: —CO—O—Divalent chain group—O—Divalent cyclic group—Divalent chain group —O—CO—
L-10: —CO—O—divalent chain group—O—CO—divalent cyclic group—
L-11: —CO—O—divalent chain group —O—CO—divalent cyclic group —CO—O—
L-12: —CO—O—divalent chain group —O—CO—divalent cyclic group —O—CO—
L-13: —CO—O—Divalent chain group—O—CO—Divalent cyclic group—Divalent chain group—
L-14: —CO—O—divalent chain group—O—CO—divalent cyclic group—divalent chain group—CO—O—
L-15: —CO—O—Divalent chain group—O—CO—Divalent cyclic group—Divalent chain group—O—CO—
L-16: —CO—O—divalent chain group—O—CO—O—divalent cyclic group—
L-17: —CO—O—divalent chain group —O—CO—O—divalent cyclic group —CO—O—
L-18: —CO—O—divalent chain group —O—CO—O—divalent cyclic group —O—CO—
L-19: —CO—O—divalent chain group—O—CO—O—divalent cyclic group—divalent chain group—
L-20: —CO—O—divalent chain group—O—CO—O—divalent cyclic group—divalent chain group—CO—O—
L-21: —CO—O—divalent chain group—O—CO—O—divalent cyclic group—divalent chain group—O—CO—
L-22: -Divalent chain group -O-Divalent chain group -O-
The divalent chain group means an alkylene group, a substituted alkylene group, an alkenylene group, a substituted alkenylene group, an alkynylene group, or a substituted alkynylene group. An alkylene group, a substituted alkylene group, an alkenylene group and a substituted alkenylene group are preferred, and an alkylene group and an alkenylene group are more preferred.
The alkylene group may have a branch. The alkylene group preferably has 2 to 20 carbon atoms, more preferably 3 to 18 carbon atoms, still more preferably 4 to 16 carbon atoms, and most preferably 5 to 15 carbon atoms. The total number of carbon atoms of all alkylene groups contained in the divalent linking group represented by L 1 and L 4 is preferably in the above range. For example, a divalent chain group in L-1 to L-22 When a plurality of are included, the total carbon number of the divalent chain group which is an alkylene group is preferably within the above range.
The alkylene part of the substituted alkylene group is the same as the above alkylene group. Examples of the substituent include a halogen atom.
The alkenylene group may have a branch. The alkenylene group preferably has 2 to 12 carbon atoms, more preferably 2 to 10 carbon atoms, and most preferably 2 to 8 carbon atoms.
The alkylene part of the substituted alkylene group is the same as the above alkylene group. Examples of the substituent include a halogen atom.
The alkynylene group may have a branch. The alkynylene group preferably has 2 to 12 carbon atoms, more preferably 2 to 10 carbon atoms, and most preferably 2 to 8 carbon atoms.
The alkynylene part of the substituted alkynylene group is the same as the above alkynylene group. Examples of the substituent include a halogen atom.
Specific examples of the divalent chain group include ethylene, trimethylene, propylene, tetramethylene, 2-methyl-tetramethylene, pentamethylene, hexamethylene, octamethylene, 2-butenylene, 2-butynylene and the like.
The definition and examples of the divalent cyclic group are the same as those of Cy 1 , Cy 2 and Cy 3 described later.
One of the linking groups bonded to the oxetane group out of L 1 and L 4 is preferably L-21, and the other one of L 1 and L 4 is preferably L-1.
前記一般式(X)中、R2は、炭素原子数1から4のアルキル基または水素原子であることが好ましく、メチル基、エチル基または水素原子であることがさらに好ましく、水素原子であることがもっとも好ましい。
In the general formula (X), R 2 is preferably an alkyl group having 1 to 4 carbon atoms or a hydrogen atom, more preferably a methyl group, an ethyl group or a hydrogen atom, and a hydrogen atom. Is most preferred.
前記一般式(X)中、L2またはL3はそれぞれ独立に単結合または二価の連結基である。L2およびL3はそれぞれ独立に、-O-、-S-、-CO-、-NR-、-C=N-、二価の鎖状基、二価の環状基およびそれらの組み合わせからなる群より選ばれる二価の連結基または単結合であることが好ましい。上記Rは炭素原子数が1から7のアルキル基または水素原子であり、炭素原子数1から4のアルキル基または水素原子であることが好ましく、メチル基、エチル基または水素原子であることがさらに好ましく、水素原子であることがもっとも好ましい。二価の鎖状基、および二価の環状基についてはL1およびL4の定義と同義である。
L2またはL3として好ましい二価の連結基としては、-COO-、-OCO-、-OCOO-、-OCONR-、-COS-、-SCO-、-CONR-、-NRCO-、-CH2CH2-、-C=C-COO-、-C=N-、-C=N-N=C-、等が挙げられる。
L2およびL3はそれぞれ独立に-COO-または-OCO-であることが好ましい。 In the general formula (X), L 2 and L 3 are each independently a single bond or a divalent linking group. L 2 and L 3 each independently comprises —O—, —S—, —CO—, —NR—, —C═N—, a divalent chain group, a divalent cyclic group, and combinations thereof. It is preferably a divalent linking group or a single bond selected from the group. R is an alkyl group having 1 to 7 carbon atoms or a hydrogen atom, preferably an alkyl group having 1 to 4 carbon atoms or a hydrogen atom, and more preferably a methyl group, an ethyl group or a hydrogen atom. Preferably, it is a hydrogen atom. The divalent chain group and the divalent cyclic group are synonymous with the definitions of L 1 and L 4 .
Preferred divalent linking groups for L 2 or L 3 include —COO—, —OCO—, —OCOO—, —OCONR—, —COS—, —SCO—, —CONR—, —NRCO—, —CH 2. CH 2 —, —C═C—COO—, —C═N—, —C═N—N═C—, and the like.
L 2 and L 3 are preferably each independently —COO— or —OCO—.
L2またはL3として好ましい二価の連結基としては、-COO-、-OCO-、-OCOO-、-OCONR-、-COS-、-SCO-、-CONR-、-NRCO-、-CH2CH2-、-C=C-COO-、-C=N-、-C=N-N=C-、等が挙げられる。
L2およびL3はそれぞれ独立に-COO-または-OCO-であることが好ましい。 In the general formula (X), L 2 and L 3 are each independently a single bond or a divalent linking group. L 2 and L 3 each independently comprises —O—, —S—, —CO—, —NR—, —C═N—, a divalent chain group, a divalent cyclic group, and combinations thereof. It is preferably a divalent linking group or a single bond selected from the group. R is an alkyl group having 1 to 7 carbon atoms or a hydrogen atom, preferably an alkyl group having 1 to 4 carbon atoms or a hydrogen atom, and more preferably a methyl group, an ethyl group or a hydrogen atom. Preferably, it is a hydrogen atom. The divalent chain group and the divalent cyclic group are synonymous with the definitions of L 1 and L 4 .
Preferred divalent linking groups for L 2 or L 3 include —COO—, —OCO—, —OCOO—, —OCONR—, —COS—, —SCO—, —CONR—, —NRCO—, —CH 2. CH 2 —, —C═C—COO—, —C═N—, —C═N—N═C—, and the like.
L 2 and L 3 are preferably each independently —COO— or —OCO—.
前記一般式(X)において、nは0、1、2、または3である。nが2または3の場合、二つのL3は同じであっても異なっていてもよく、二つのCy2も同じであっても異なっていてもよい。nは1または2であることが好ましく、1であることがさらに好ましい。
In the general formula (X), n is 0, 1, 2, or 3. When n is 2 or 3, two L 3 may be the same or different, and two Cy 2 may be the same or different. n is preferably 1 or 2, and more preferably 1.
前記一般式(X)において、Cy1、Cy2およびCy3は、それぞれ独立に、二価の環状基である。
環状基に含まれる環は、5員環、6員環、または7員環であることが好ましく、5員環または6員環であることがさらに好ましく、6員環であることが最も好ましい。
環状基に含まれる環は、縮合環であっても良い。ただし、縮合環よりも単環であることがより好ましい。
環状基に含まれる環は、芳香族環、脂肪族環、および複素環のいずれでもよい。芳香族環の例には、ベンゼン環およびナフタレン環が含まれる。脂肪族環の例には、シクロヘキサン環が含まれる。複素環の例には、ピリジン環およびピリミジン環が含まれる。
ベンゼン環を有する環状基としては、1,4-フェニレンが好ましい。ナフタレン環を有する環状基としては、ナフタレン-1,5-ジイルおよびナフタレン-2,6-ジイルが好ましい。シクロヘキサン環を有する環状基としては1,4-シクロへキシレンであることが好ましい。ピリジン環を有する環状基としてはピリジン-2,5-ジイルが好ましい。ピリミジン環を有する環状基としては、ピリミジン-2,5-ジイルが好ましい。
環状基は、置換基を有していてもよい。置換基の例には、ハロゲン原子、シアノ基、ニトロ基、炭素原子数が1乃至5のアルキル基、炭素原子数が1乃至5のハロゲン置換アルキル基、炭素原子数が1乃至5のアルコキシ基、炭素原子数が1乃至5のアルキルチオ基、炭素原子数が2乃至6のアシルオキシ基、炭素原子数が2乃至6のアルコキシカルボニル基、カルバモイル基、炭素原子数が2乃至6のアルキル置換カルバモイル基および炭素原子数が2乃至6のアシルアミノ基が含まれる。 In the general formula (X), Cy 1 , Cy 2 and Cy 3 are each independently a divalent cyclic group.
The ring contained in the cyclic group is preferably a 5-membered ring, 6-membered ring, or 7-membered ring, more preferably a 5-membered ring or 6-membered ring, and most preferably a 6-membered ring.
The ring contained in the cyclic group may be a condensed ring. However, it is more preferably a monocycle than a condensed ring.
The ring contained in the cyclic group may be any of an aromatic ring, an aliphatic ring, and a heterocyclic ring. Examples of the aromatic ring include a benzene ring and a naphthalene ring. Examples of the aliphatic ring include a cyclohexane ring. Examples of the heterocyclic ring include a pyridine ring and a pyrimidine ring.
As the cyclic group having a benzene ring, 1,4-phenylene is preferable. As the cyclic group having a naphthalene ring, naphthalene-1,5-diyl and naphthalene-2,6-diyl are preferable. The cyclic group having a cyclohexane ring is preferably 1,4-cyclohexylene. The cyclic group having a pyridine ring is preferably pyridine-2,5-diyl. The cyclic group having a pyrimidine ring is preferably pyrimidine-2,5-diyl.
The cyclic group may have a substituent. Examples of the substituent include a halogen atom, a cyano group, a nitro group, an alkyl group having 1 to 5 carbon atoms, a halogen-substituted alkyl group having 1 to 5 carbon atoms, and an alkoxy group having 1 to 5 carbon atoms. An alkylthio group having 1 to 5 carbon atoms, an acyloxy group having 2 to 6 carbon atoms, an alkoxycarbonyl group having 2 to 6 carbon atoms, a carbamoyl group, and an alkyl-substituted carbamoyl group having 2 to 6 carbon atoms And an acylamino group having 2 to 6 carbon atoms.
環状基に含まれる環は、5員環、6員環、または7員環であることが好ましく、5員環または6員環であることがさらに好ましく、6員環であることが最も好ましい。
環状基に含まれる環は、縮合環であっても良い。ただし、縮合環よりも単環であることがより好ましい。
環状基に含まれる環は、芳香族環、脂肪族環、および複素環のいずれでもよい。芳香族環の例には、ベンゼン環およびナフタレン環が含まれる。脂肪族環の例には、シクロヘキサン環が含まれる。複素環の例には、ピリジン環およびピリミジン環が含まれる。
ベンゼン環を有する環状基としては、1,4-フェニレンが好ましい。ナフタレン環を有する環状基としては、ナフタレン-1,5-ジイルおよびナフタレン-2,6-ジイルが好ましい。シクロヘキサン環を有する環状基としては1,4-シクロへキシレンであることが好ましい。ピリジン環を有する環状基としてはピリジン-2,5-ジイルが好ましい。ピリミジン環を有する環状基としては、ピリミジン-2,5-ジイルが好ましい。
環状基は、置換基を有していてもよい。置換基の例には、ハロゲン原子、シアノ基、ニトロ基、炭素原子数が1乃至5のアルキル基、炭素原子数が1乃至5のハロゲン置換アルキル基、炭素原子数が1乃至5のアルコキシ基、炭素原子数が1乃至5のアルキルチオ基、炭素原子数が2乃至6のアシルオキシ基、炭素原子数が2乃至6のアルコキシカルボニル基、カルバモイル基、炭素原子数が2乃至6のアルキル置換カルバモイル基および炭素原子数が2乃至6のアシルアミノ基が含まれる。 In the general formula (X), Cy 1 , Cy 2 and Cy 3 are each independently a divalent cyclic group.
The ring contained in the cyclic group is preferably a 5-membered ring, 6-membered ring, or 7-membered ring, more preferably a 5-membered ring or 6-membered ring, and most preferably a 6-membered ring.
The ring contained in the cyclic group may be a condensed ring. However, it is more preferably a monocycle than a condensed ring.
The ring contained in the cyclic group may be any of an aromatic ring, an aliphatic ring, and a heterocyclic ring. Examples of the aromatic ring include a benzene ring and a naphthalene ring. Examples of the aliphatic ring include a cyclohexane ring. Examples of the heterocyclic ring include a pyridine ring and a pyrimidine ring.
As the cyclic group having a benzene ring, 1,4-phenylene is preferable. As the cyclic group having a naphthalene ring, naphthalene-1,5-diyl and naphthalene-2,6-diyl are preferable. The cyclic group having a cyclohexane ring is preferably 1,4-cyclohexylene. The cyclic group having a pyridine ring is preferably pyridine-2,5-diyl. The cyclic group having a pyrimidine ring is preferably pyrimidine-2,5-diyl.
The cyclic group may have a substituent. Examples of the substituent include a halogen atom, a cyano group, a nitro group, an alkyl group having 1 to 5 carbon atoms, a halogen-substituted alkyl group having 1 to 5 carbon atoms, and an alkoxy group having 1 to 5 carbon atoms. An alkylthio group having 1 to 5 carbon atoms, an acyloxy group having 2 to 6 carbon atoms, an alkoxycarbonyl group having 2 to 6 carbon atoms, a carbamoyl group, and an alkyl-substituted carbamoyl group having 2 to 6 carbon atoms And an acylamino group having 2 to 6 carbon atoms.
以下に、前記一般式(X)で表される重合性液晶化合物の例を示す。本発明はこれらに限定されるものではない。
Examples of the polymerizable liquid crystal compound represented by the general formula (X) are shown below. The present invention is not limited to these.




また、棒状液晶化合物としては、下記一般式(V)で表される化合物も好ましい。
一般式(V) M1-(L1)p-Cy1-L2-(Cy2-L3)n-Cy3-(L4)q-M2
一般式(V)中、M1、および、M2はそれぞれ独立に、水素原子、置換もしくは無置換のアルキル基、置換もしくは無置換のアリール基、ヘテロ環基、シアノ基、ハロゲン、-SCN、-CF3、ニトロ基、または、Q1を表すが、M1、および、M2の少なくとも一つは、Q1以外の基を表す。
ただし、Q1、L1、L2、L3、L4、Cy1、Cy2、Cy3およびnは前記一般式(X)で表される基と同義である。また、pおよびqは0、または1である。
M1、および、M2が、Q1を表さない場合、水素原子、置換もしくは無置換のアルキル基、置換もしくは無置換のアリール基、シアノ基であることが好ましく、より好ましくは、炭素数1~4のアルキル基、もしくは、フェニル基であり、pおよびqは0であることが好ましい。 Moreover, as a rod-shaped liquid crystal compound, the compound represented by the following general formula (V) is also preferable.
Formula (V) M 1- (L 1 ) p-Cy 1 -L 2- (Cy 2 -L 3 ) n -Cy 3- (L 4 ) q-M 2
In general formula (V), M 1 and M 2 are each independently a hydrogen atom, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aryl group, a heterocyclic group, a cyano group, a halogen, —SCN, —CF 3 , a nitro group, or Q 1 is represented, but at least one of M 1 and M 2 represents a group other than Q 1 .
However, Q 1 , L 1 , L 2 , L 3 , L 4 , Cy 1 , Cy 2 , Cy 3 and n have the same meaning as the group represented by the general formula (X). P and q are 0 or 1.
When M 1 and M 2 do not represent Q 1 , it is preferably a hydrogen atom, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aryl group, or a cyano group, more preferably a carbon number It is preferably an alkyl group of 1 to 4 or a phenyl group, and p and q are preferably 0.
一般式(V) M1-(L1)p-Cy1-L2-(Cy2-L3)n-Cy3-(L4)q-M2
一般式(V)中、M1、および、M2はそれぞれ独立に、水素原子、置換もしくは無置換のアルキル基、置換もしくは無置換のアリール基、ヘテロ環基、シアノ基、ハロゲン、-SCN、-CF3、ニトロ基、または、Q1を表すが、M1、および、M2の少なくとも一つは、Q1以外の基を表す。
ただし、Q1、L1、L2、L3、L4、Cy1、Cy2、Cy3およびnは前記一般式(X)で表される基と同義である。また、pおよびqは0、または1である。
M1、および、M2が、Q1を表さない場合、水素原子、置換もしくは無置換のアルキル基、置換もしくは無置換のアリール基、シアノ基であることが好ましく、より好ましくは、炭素数1~4のアルキル基、もしくは、フェニル基であり、pおよびqは0であることが好ましい。 Moreover, as a rod-shaped liquid crystal compound, the compound represented by the following general formula (V) is also preferable.
Formula (V) M 1- (L 1 ) p-Cy 1 -L 2- (Cy 2 -L 3 ) n -Cy 3- (L 4 ) q-M 2
In general formula (V), M 1 and M 2 are each independently a hydrogen atom, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aryl group, a heterocyclic group, a cyano group, a halogen, —SCN, —CF 3 , a nitro group, or Q 1 is represented, but at least one of M 1 and M 2 represents a group other than Q 1 .
However, Q 1 , L 1 , L 2 , L 3 , L 4 , Cy 1 , Cy 2 , Cy 3 and n have the same meaning as the group represented by the general formula (X). P and q are 0 or 1.
When M 1 and M 2 do not represent Q 1 , it is preferably a hydrogen atom, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aryl group, or a cyano group, more preferably a carbon number It is preferably an alkyl group of 1 to 4 or a phenyl group, and p and q are preferably 0.
なお、前記一般式(X)で表される重合性棒状液晶化合物と前記一般式(V)で表される化合物を混合して用いてもよい。また、前記一般式(X)で表される重合性液晶化合物と、一般式(V)で表される化合物の混合物中における、前記一般式(V)で表される化合物の好ましい混合比率としては、0.1%~40%であり、より好ましくは、1%~30%であり、更に好ましくは、5%~20%である。
In addition, you may mix and use the polymeric rod-shaped liquid crystal compound represented by the said general formula (X), and the compound represented by the said general formula (V). Moreover, as a preferable mixing ratio of the compound represented by the general formula (V) in the mixture of the polymerizable liquid crystal compound represented by the general formula (X) and the compound represented by the general formula (V), 0.1% to 40%, more preferably 1% to 30%, and still more preferably 5% to 20%.
以下に、前記一般式(V)で表される化合物の好ましい例を示すが、本発明はこれらに限定されるものではない。
Preferred examples of the compound represented by the general formula (V) are shown below, but the present invention is not limited to these.


また本発明に用いることができる重合性液晶化合物のうち二つ以上のアクリル基及びメタクリル基を有する化合物は、そのうち一つが潜在的に重合性基を内在していても構わない。すなわち以下のような化合物からEP2130817記載の化学反応により重合性基を発現させることができるため、二つ以上のアクリル基及びメタクリル基を有する化合物の一方をリビング重合に用いた後に、潜在的に重合性基を内在している部分を化学反応により処理して重合性基を発生させても良い。
Of the polymerizable liquid crystal compounds that can be used in the present invention, one of the compounds having two or more acrylic groups and methacrylic groups may potentially contain a polymerizable group. That is, since a polymerizable group can be expressed from a compound as described below by a chemical reaction described in EP2130817, after one of the compounds having two or more acrylic groups and methacrylic groups is used for living polymerization, it is potentially polymerized. A portion containing a functional group may be treated by a chemical reaction to generate a polymerizable group.

本発明中の各一般式では上記重合性液晶化合物1及び2をなんら区別なく記載しており、本発明中では同義とみなしている。
In the general formulas in the present invention, the polymerizable liquid crystal compounds 1 and 2 are described without any distinction, and are regarded as synonymous in the present invention.
<溶媒>
本発明の支持体上でのリビング重合体の製造方法において、前記モノマー含有組成物に使用できる溶媒としては、反応を阻害しないものであればいずれでも使用することができる。
具体的には、ベンゼン、トルエンおよびキシレン等の芳香族炭化水素系溶媒、ペンタン、ヘキサン、ヘプタン、オクタン、ノナンおよびデカン等の脂肪族炭化水素系溶媒、シクロヘキサン、メチルシクロヘキサンおよびデカヒドロナフタレンのような脂環族炭化水素系溶媒、クロロベンゼン、ジクロロベンゼン、トリクロロベンゼン、塩化メチレン、クロロホルム、四塩化炭素およびテトラクロルエチレン等の塩素化炭化水素系溶媒、メタノール、エタノール、n-プロパノール、iso-プロパノール、n-ブタノール、sec-ブタノールおよびtert-ブタノール等のアルコール系溶媒、アセトン、メチルエチルケトンおよびメチルイソブチルケトン等のケトン系溶媒、酢酸エチル、酢酸ブチルおよびジメチルフタレート等のエステル系溶媒、ジメチルエーテル、ジエチルエーテル、ジ-n-アミルエーテル、ジフェニルエーテル、テトラヒドロフランおよびアニソールのようなエーテル系溶媒、ジメチルスルホキシド、N-ジメチルホルムアミド、ジオキサン等を挙げることができる。 <Solvent>
In the method for producing a living polymer on the support of the present invention, any solvent that does not inhibit the reaction can be used as the solvent that can be used in the monomer-containing composition.
Specifically, aromatic hydrocarbon solvents such as benzene, toluene and xylene, aliphatic hydrocarbon solvents such as pentane, hexane, heptane, octane, nonane and decane, cyclohexane, methylcyclohexane and decahydronaphthalene. Alicyclic hydrocarbon solvents, chlorinated hydrocarbon solvents such as chlorobenzene, dichlorobenzene, trichlorobenzene, methylene chloride, chloroform, carbon tetrachloride and tetrachloroethylene, methanol, ethanol, n-propanol, iso-propanol, n Alcohol solvents such as butanol, sec-butanol and tert-butanol, ketone solvents such as acetone, methyl ethyl ketone and methyl isobutyl ketone, and esters such as ethyl acetate, butyl acetate and dimethyl phthalate System solvent, dimethyl ether, diethyl ether, and di -n- amyl ether, diphenyl ether, ether solvents such as tetrahydrofuran and anisole, dimethyl sulfoxide, N- dimethylformamide, dioxane or the like.
本発明の支持体上でのリビング重合体の製造方法において、前記モノマー含有組成物に使用できる溶媒としては、反応を阻害しないものであればいずれでも使用することができる。
具体的には、ベンゼン、トルエンおよびキシレン等の芳香族炭化水素系溶媒、ペンタン、ヘキサン、ヘプタン、オクタン、ノナンおよびデカン等の脂肪族炭化水素系溶媒、シクロヘキサン、メチルシクロヘキサンおよびデカヒドロナフタレンのような脂環族炭化水素系溶媒、クロロベンゼン、ジクロロベンゼン、トリクロロベンゼン、塩化メチレン、クロロホルム、四塩化炭素およびテトラクロルエチレン等の塩素化炭化水素系溶媒、メタノール、エタノール、n-プロパノール、iso-プロパノール、n-ブタノール、sec-ブタノールおよびtert-ブタノール等のアルコール系溶媒、アセトン、メチルエチルケトンおよびメチルイソブチルケトン等のケトン系溶媒、酢酸エチル、酢酸ブチルおよびジメチルフタレート等のエステル系溶媒、ジメチルエーテル、ジエチルエーテル、ジ-n-アミルエーテル、ジフェニルエーテル、テトラヒドロフランおよびアニソールのようなエーテル系溶媒、ジメチルスルホキシド、N-ジメチルホルムアミド、ジオキサン等を挙げることができる。 <Solvent>
In the method for producing a living polymer on the support of the present invention, any solvent that does not inhibit the reaction can be used as the solvent that can be used in the monomer-containing composition.
Specifically, aromatic hydrocarbon solvents such as benzene, toluene and xylene, aliphatic hydrocarbon solvents such as pentane, hexane, heptane, octane, nonane and decane, cyclohexane, methylcyclohexane and decahydronaphthalene. Alicyclic hydrocarbon solvents, chlorinated hydrocarbon solvents such as chlorobenzene, dichlorobenzene, trichlorobenzene, methylene chloride, chloroform, carbon tetrachloride and tetrachloroethylene, methanol, ethanol, n-propanol, iso-propanol, n Alcohol solvents such as butanol, sec-butanol and tert-butanol, ketone solvents such as acetone, methyl ethyl ketone and methyl isobutyl ketone, and esters such as ethyl acetate, butyl acetate and dimethyl phthalate System solvent, dimethyl ether, diethyl ether, and di -n- amyl ether, diphenyl ether, ether solvents such as tetrahydrofuran and anisole, dimethyl sulfoxide, N- dimethylformamide, dioxane or the like.
また、水を溶媒として、懸濁重合、乳化重合することもできる。これらの溶媒は、単独でもまたは2種以上を混合して使用してもよい。また、これらの溶媒の使用によって、反応液が均一相となることが好ましいが、不均一な複数の相となっても構わない。
Also, suspension polymerization and emulsion polymerization can be performed using water as a solvent. These solvents may be used alone or in admixture of two or more. Moreover, it is preferable that the reaction liquid becomes a homogeneous phase by using these solvents, but a plurality of non-uniform phases may be used.
(ATRP(原子移動ラジカル重合))
本発明の支持体上でのリビング重合体の製造方法の好ましい態様の一つは、前記リビング重合が原子移動ラジカル重合であり、かつ、前記支持体が金属製の支持体である態様である。 (ATRP (Atom Transfer Radical Polymerization))
One of the preferable embodiments of the method for producing a living polymer on the support of the present invention is an embodiment in which the living polymerization is atom transfer radical polymerization, and the support is a metal support.
本発明の支持体上でのリビング重合体の製造方法の好ましい態様の一つは、前記リビング重合が原子移動ラジカル重合であり、かつ、前記支持体が金属製の支持体である態様である。 (ATRP (Atom Transfer Radical Polymerization))
One of the preferable embodiments of the method for producing a living polymer on the support of the present invention is an embodiment in which the living polymerization is atom transfer radical polymerization, and the support is a metal support.
本明細書中におけるATRP(原子移動ラジカル重合)とは、リビングラジカル重合の一つであり、有機ハロゲン化物又はハロゲン化スルホニル化合物などを開始剤、遷移金属を中心金属とする金属錯体又は遷移金属の単体を触媒としてラジカル重合性単量体をラジカル重合する方法である。
ATRP (Atom Transfer Radical Polymerization) in the present specification is one of living radical polymerizations, and is a metal complex or transition metal having an organic halide or a sulfonyl halide compound as an initiator and a transition metal as a central metal. This is a method of radical polymerization of a radically polymerizable monomer using a simple substance as a catalyst.
ATRPの好ましい態様としては、具体的には、例えば、Matyjaszewskiら、Chem. Rev., 101, 2921 (2001)、WO96/30421号公報、WO97/18247号公報、WO98/01480号公報、WO98/40415号公報、WO00/156795号公報、あるいは澤本ら、Chem. Rev., 101, 3689 (2001)、特開平8-41117号公報、特開平9-208616号公報、特開2000-264914号公報、特開2001-316410号公報、特開2002-80523号公報、特開2004-307872号公報などが挙げられる。
As a preferable aspect of ATRP, specifically, for example, Matyjazewski et al., Chem. Rev. 101, 2921 (2001), WO 96/30421, WO 97/18247, WO 98/01480, WO 98/40415, WO 00/15695, or Sawamoto et al., Chem. Rev. 101, 3689 (2001), JP-A-8-411117, JP-A-9-208616, JP-A 2000-264914, JP-A 2001-316410, JP-A 2002-80523, JP No. 2004-307872 and the like.
-開始剤-
ATRPに用いられる開始剤としては、特に制限はなく、例えば有機ハロゲン化物やハロゲン化スルホニル化合物が挙げられるが、特に炭素-炭素二重結合または炭素-酸素二重結合のα位に存在する炭素-ハロゲン結合、あるいは一つの炭素原子上に複数のハロゲンが付加した構造が開始剤構造として好適である。本発明においては、炭素-炭素二重結合のα位に存在する炭素-ハロゲン結合、あるいは一つの炭素原子上に複数のハロゲンが付加した構造を開始剤構造として利用することができる。 -Initiator-
The initiator used for ATRP is not particularly limited, and examples thereof include organic halides and sulfonyl halide compounds. In particular, carbon-carbon double bonds or carbon atoms present at the α-position of carbon-oxygen double bonds A structure in which a halogen bond or a plurality of halogen atoms are added on one carbon atom is suitable as the initiator structure. In the present invention, a carbon-halogen bond present at the α-position of a carbon-carbon double bond, or a structure in which a plurality of halogens are added on one carbon atom can be used as an initiator structure.
ATRPに用いられる開始剤としては、特に制限はなく、例えば有機ハロゲン化物やハロゲン化スルホニル化合物が挙げられるが、特に炭素-炭素二重結合または炭素-酸素二重結合のα位に存在する炭素-ハロゲン結合、あるいは一つの炭素原子上に複数のハロゲンが付加した構造が開始剤構造として好適である。本発明においては、炭素-炭素二重結合のα位に存在する炭素-ハロゲン結合、あるいは一つの炭素原子上に複数のハロゲンが付加した構造を開始剤構造として利用することができる。 -Initiator-
The initiator used for ATRP is not particularly limited, and examples thereof include organic halides and sulfonyl halide compounds. In particular, carbon-carbon double bonds or carbon atoms present at the α-position of carbon-oxygen double bonds A structure in which a halogen bond or a plurality of halogen atoms are added on one carbon atom is suitable as the initiator structure. In the present invention, a carbon-halogen bond present at the α-position of a carbon-carbon double bond, or a structure in which a plurality of halogens are added on one carbon atom can be used as an initiator structure.
ATRPに用いられる開始剤は、ミクロ相分離構造膜を製造しやすくする観点からは、親水性ユニットを含むマクロ開始剤であることが好ましく、ポリアルキレンオキシ構造を含むマクロ開始剤であることがより好ましい。
前記ポリアルキレンオキシ構造としては特に制限はないが、アルキレンオキシ基が炭素数1~1000のアルキレンオキシ基であることが好ましく、炭素数10~400のアルキレンオキシ基であることがより好ましく、エチレンオキシ基であることが特に好ましい。
前記ポリアルキレンオキシ構造中、アルキレンオキシ構造の繰り返し単位数は特に制限はないが、1~500であることが好ましく、3~250であることがより好ましく、5~200であることが特に好ましい。 The initiator used for ATRP is preferably a macroinitiator containing a hydrophilic unit, more preferably a macroinitiator containing a polyalkyleneoxy structure, from the viewpoint of facilitating production of a microphase separation structure membrane. preferable.
The polyalkyleneoxy structure is not particularly limited, but the alkyleneoxy group is preferably an alkyleneoxy group having 1 to 1000 carbon atoms, more preferably an alkyleneoxy group having 10 to 400 carbon atoms, Particularly preferred is a group.
In the polyalkyleneoxy structure, the number of repeating units of the alkyleneoxy structure is not particularly limited, but is preferably 1 to 500, more preferably 3 to 250, and particularly preferably 5 to 200.
前記ポリアルキレンオキシ構造としては特に制限はないが、アルキレンオキシ基が炭素数1~1000のアルキレンオキシ基であることが好ましく、炭素数10~400のアルキレンオキシ基であることがより好ましく、エチレンオキシ基であることが特に好ましい。
前記ポリアルキレンオキシ構造中、アルキレンオキシ構造の繰り返し単位数は特に制限はないが、1~500であることが好ましく、3~250であることがより好ましく、5~200であることが特に好ましい。 The initiator used for ATRP is preferably a macroinitiator containing a hydrophilic unit, more preferably a macroinitiator containing a polyalkyleneoxy structure, from the viewpoint of facilitating production of a microphase separation structure membrane. preferable.
The polyalkyleneoxy structure is not particularly limited, but the alkyleneoxy group is preferably an alkyleneoxy group having 1 to 1000 carbon atoms, more preferably an alkyleneoxy group having 10 to 400 carbon atoms, Particularly preferred is a group.
In the polyalkyleneoxy structure, the number of repeating units of the alkyleneoxy structure is not particularly limited, but is preferably 1 to 500, more preferably 3 to 250, and particularly preferably 5 to 200.
本発明では市販のATRP用開始剤を用いてもよく、例えば、低分子ATRP用開始剤としてエチル-α-ブロモイソブチレート(アルドリッチ社製)などを挙げることができる。
In the present invention, a commercially available initiator for ATRP may be used. Examples of the initiator for low molecular weight ATRP include ethyl-α-bromoisobutyrate (manufactured by Aldrich).
一方、マクロ開始剤であるATRP用開始剤は、合成により製造してもよい。マクロ開始剤であるATRP用開始剤としては、例えば、以下のものを用いることができるが、本発明は以下の具体例によって制限されるものではない。

On the other hand, the ATRP initiator that is a macroinitiator may be produced by synthesis. As the initiator for ATRP, which is a macroinitiator, for example, the following can be used, but the present invention is not limited by the following specific examples.
前記モノマー組成物中、前記ATRP用開始剤の添加量は、前記モノマー成分に対して、1~60質量%であることが好ましく、5~55質量%であることがより好ましく、10~45質量%であることが特に好ましい。
In the monomer composition, the amount of the ATRP initiator added is preferably 1 to 60% by mass, more preferably 5 to 55% by mass, and more preferably 10 to 45% by mass with respect to the monomer component. % Is particularly preferred.
配位子としては、Macromolecules、2006、39、4953.に記載の配位子を好適に使用することができる。
2,2’-ビピリジン、1,1,4,7,10,10-ヘキサメチルトリエチレンテトラミン、N,N,N’,N’-テトラキス(2-ピリジルメチル)エチレンジアミン、トリス[2-(ジメチルアミノ)エチル]アミンを用いることが特に好ましい。
前記モノマー組成物中、配位子の添加量は、前記モノマー成分に対して、0.1~40質量%であることが好ましく、0.5~20質量%であることがより好ましく、1~10質量%であることが特に好ましい。 As a ligand, Macromolecules, 2006, 39, 4953. The ligands described in (1) can be preferably used.
2,2′-bipyridine, 1,1,4,7,10,10-hexamethyltriethylenetetramine, N, N, N ′, N′-tetrakis (2-pyridylmethyl) ethylenediamine, tris [2- (dimethyl It is particularly preferred to use amino) ethyl] amine.
In the monomer composition, the amount of the ligand added is preferably 0.1 to 40% by mass, more preferably 0.5 to 20% by mass, with respect to the monomer component. 10% by mass is particularly preferable.
2,2’-ビピリジン、1,1,4,7,10,10-ヘキサメチルトリエチレンテトラミン、N,N,N’,N’-テトラキス(2-ピリジルメチル)エチレンジアミン、トリス[2-(ジメチルアミノ)エチル]アミンを用いることが特に好ましい。
前記モノマー組成物中、配位子の添加量は、前記モノマー成分に対して、0.1~40質量%であることが好ましく、0.5~20質量%であることがより好ましく、1~10質量%であることが特に好ましい。 As a ligand, Macromolecules, 2006, 39, 4953. The ligands described in (1) can be preferably used.
2,2′-bipyridine, 1,1,4,7,10,10-hexamethyltriethylenetetramine, N, N, N ′, N′-tetrakis (2-pyridylmethyl) ethylenediamine, tris [2- (dimethyl It is particularly preferred to use amino) ethyl] amine.
In the monomer composition, the amount of the ligand added is preferably 0.1 to 40% by mass, more preferably 0.5 to 20% by mass, with respect to the monomer component. 10% by mass is particularly preferable.
-重合触媒-
本発明の支持体上でのリビング重合体の製造方法は、ATRPの重合触媒としては特に制限はない。 -Polymerization catalyst-
The method for producing a living polymer on the support of the present invention is not particularly limited as a polymerization catalyst for ATRP.
本発明の支持体上でのリビング重合体の製造方法は、ATRPの重合触媒としては特に制限はない。 -Polymerization catalyst-
The method for producing a living polymer on the support of the present invention is not particularly limited as a polymerization catalyst for ATRP.
ATRPの重合触媒として用いられる遷移金属錯体としては特に限定されないが、好ましくは周期律表第7族、8族、9族、10族、または11族元素を中心金属とする金属錯体である。更に好ましいものとして、0価の銅、1価の銅、2価のルテニウム、2価の鉄又は2価のニッケルの錯体が挙げられる。なかでも、銅の錯体が好ましい。1価の銅化合物を具体的に例示するならば、塩化第一銅、臭化第一銅、ヨウ化第一銅、シアン化第一銅、酸化第一銅、過塩素酸第一銅等である。銅化合物を用いる場合、触媒活性を高めるために2,2′-ビピリジル若しくはその誘導体、1,10-フェナントロリン若しくはその誘導体、又はテトラメチルエチレンジアミン、ペンタメチルジエチレントリアミン若しくはヘキサメチルトリス(2-アミノエチル)アミン等のポリアミン等が配位子として添加される。また、2価の塩化ルテニウムのトリストリフェニルホスフィン錯体(RuCl2(PPh3)3)も触媒として好適である。ルテニウム化合物を触媒として用いる場合は、活性化剤としてアルミニウムアルコキシド類が添加される。更に、2価の鉄のビストリフェニルホスフィン錯体(FeCl2(PPh3)2)、2価のニッケルのビストリフェニルホスフィン錯体(NiCl2(PPh3)2)、及び、2価のニッケルのビストリブチルホスフィン錯体(NiBr2(PBu3)2)も、触媒として好適である。
Although it does not specifically limit as a transition metal complex used as a polymerization catalyst of ATRP, Preferably it is a metal complex which uses a periodic table group 7, 8, 9, 10, or 11 element as a central metal. More preferable examples include a complex of zero-valent copper, monovalent copper, divalent ruthenium, divalent iron, or divalent nickel. Of these, a copper complex is preferable. Specific examples of monovalent copper compounds include cuprous chloride, cuprous bromide, cuprous iodide, cuprous cyanide, cuprous oxide, cuprous perchlorate, etc. is there. When a copper compound is used, 2,2′-bipyridyl or a derivative thereof, 1,10-phenanthroline or a derivative thereof, or tetramethylethylenediamine, pentamethyldiethylenetriamine or hexamethyltris (2-aminoethyl) amine is used to increase the catalytic activity. Polyamines such as are added as ligands. A tristriphenylphosphine complex of divalent ruthenium chloride (RuCl 2 (PPh 3 ) 3 ) is also suitable as a catalyst. When a ruthenium compound is used as a catalyst, an aluminum alkoxide is added as an activator. Furthermore, a divalent iron bistriphenylphosphine complex (FeCl 2 (PPh 3 ) 2 ), a divalent nickel bistriphenylphosphine complex (NiCl 2 (PPh 3 ) 2 ), and a divalent nickel bistributylphosphine A complex (NiBr 2 (PBu 3 ) 2 ) is also suitable as a catalyst.
ATRPの重合触媒として用いられる遷移金属の単体としては特に限定されないが、好ましくは周期律表第7族、8族、9族、10族、または11族元素の金属である。更に好ましいものとして、銅、ルテニウム、鉄又はニッケルが挙げられる。なかでも、銅が好ましい。
The simple substance of the transition metal used as the polymerization catalyst for ATRP is not particularly limited, but is preferably a metal belonging to Group 7, 8, 9, 10, or 11 of the periodic table. Further preferred are copper, ruthenium, iron or nickel. Of these, copper is preferable.
本発明の製造方法では、前記支持体として金属製の支持体を用い、この金属製の支持体をATRPの重合触媒としても利用することが好ましい。
金属製の支持体上でのリビング重合は金属製の支持体を触媒として用いることにより、モノマー含有組成物に金属触媒を添加する通常のATRP後の組成物から金属を除去する手間を省けるために経済合理性がある。
本発明の支持体上でのリビング重合体の製造方法は、金属製の支持体が、金属性のフィルムであることが好ましい。金属性のフィルムは、厚み5~50μmのものを用いることが操作上と経済合理性の観点から好ましい。金属性のフィルムは、幅が100~500mmのものを用いることが連続生産の観点から好ましい。
本発明の支持体上でのリビング重合体の製造方法は、金属製の支持体における金属が銅であることが好ましい。この場合に用いられる支持体としては、銅フィルムを好ましく用いることができる。
前記金属性の支持体としては市販の支持体を用いてもよく、例えば、銅フィルムとして日本製箔株式会社製、圧延銅箔TCUなどを挙げることができる。 In the production method of the present invention, it is preferable that a metal support is used as the support, and the metal support is also used as an ATRP polymerization catalyst.
Living polymerization on a metal support uses a metal support as a catalyst to save the trouble of removing the metal from the composition after ordinary ATRP in which the metal catalyst is added to the monomer-containing composition. There is economic rationality.
In the method for producing a living polymer on the support of the present invention, the metal support is preferably a metallic film. A metallic film having a thickness of 5 to 50 μm is preferably used from the viewpoint of operation and economic rationality. A metal film having a width of 100 to 500 mm is preferably used from the viewpoint of continuous production.
In the method for producing a living polymer on the support of the present invention, the metal in the metal support is preferably copper. As the support used in this case, a copper film can be preferably used.
As the metallic support, a commercially available support may be used. For example, examples of the copper film include Nippon Foil Co., Ltd., rolled copper foil TCU, and the like.
金属製の支持体上でのリビング重合は金属製の支持体を触媒として用いることにより、モノマー含有組成物に金属触媒を添加する通常のATRP後の組成物から金属を除去する手間を省けるために経済合理性がある。
本発明の支持体上でのリビング重合体の製造方法は、金属製の支持体が、金属性のフィルムであることが好ましい。金属性のフィルムは、厚み5~50μmのものを用いることが操作上と経済合理性の観点から好ましい。金属性のフィルムは、幅が100~500mmのものを用いることが連続生産の観点から好ましい。
本発明の支持体上でのリビング重合体の製造方法は、金属製の支持体における金属が銅であることが好ましい。この場合に用いられる支持体としては、銅フィルムを好ましく用いることができる。
前記金属性の支持体としては市販の支持体を用いてもよく、例えば、銅フィルムとして日本製箔株式会社製、圧延銅箔TCUなどを挙げることができる。 In the production method of the present invention, it is preferable that a metal support is used as the support, and the metal support is also used as an ATRP polymerization catalyst.
Living polymerization on a metal support uses a metal support as a catalyst to save the trouble of removing the metal from the composition after ordinary ATRP in which the metal catalyst is added to the monomer-containing composition. There is economic rationality.
In the method for producing a living polymer on the support of the present invention, the metal support is preferably a metallic film. A metallic film having a thickness of 5 to 50 μm is preferably used from the viewpoint of operation and economic rationality. A metal film having a width of 100 to 500 mm is preferably used from the viewpoint of continuous production.
In the method for producing a living polymer on the support of the present invention, the metal in the metal support is preferably copper. As the support used in this case, a copper film can be preferably used.
As the metallic support, a commercially available support may be used. For example, examples of the copper film include Nippon Foil Co., Ltd., rolled copper foil TCU, and the like.
本発明の支持体上でのリビング重合体の製造方法は、前記モノマー含有組成物の前記金属製の支持体と接していない面を別の支持体で覆い、前記モノマー含有組成物を挟圧しながら加熱することが、用いる材料(前記モノマー成分や前記開始剤)を揮散させにくくする観点から好ましい。前記別の支持体は、さらにATRPの重合触媒活性を付与する観点から、前述の金属製の支持体をもう1枚用いることが好ましい。
本発明の支持体上でのリビング重合体の製造方法は、前記金属製の膜における金属が銅であることが好ましい。この場合に用いられる金属製の膜の好ましい範囲は、前記金属性の支持体の好ましい範囲と同様である。
前記モノマー含有組成物を挟圧するときにかける圧力としては特に制限はないが、例えば0.1~500KPaとすることが製造上の観点から好ましい。 In the method for producing a living polymer on the support of the present invention, the surface of the monomer-containing composition that is not in contact with the metal support is covered with another support, and the monomer-containing composition is sandwiched. It is preferable to heat from the viewpoint of making it difficult to volatilize the materials used (the monomer component and the initiator). From the viewpoint of further providing the ATRP polymerization catalytic activity, it is preferable to use another sheet of the above-mentioned metal support.
In the method for producing a living polymer on the support of the present invention, the metal in the metal film is preferably copper. The preferable range of the metal film used in this case is the same as the preferable range of the metallic support.
The pressure applied when the monomer-containing composition is sandwiched is not particularly limited, but is preferably 0.1 to 500 KPa, for example, from the viewpoint of production.
本発明の支持体上でのリビング重合体の製造方法は、前記金属製の膜における金属が銅であることが好ましい。この場合に用いられる金属製の膜の好ましい範囲は、前記金属性の支持体の好ましい範囲と同様である。
前記モノマー含有組成物を挟圧するときにかける圧力としては特に制限はないが、例えば0.1~500KPaとすることが製造上の観点から好ましい。 In the method for producing a living polymer on the support of the present invention, the surface of the monomer-containing composition that is not in contact with the metal support is covered with another support, and the monomer-containing composition is sandwiched. It is preferable to heat from the viewpoint of making it difficult to volatilize the materials used (the monomer component and the initiator). From the viewpoint of further providing the ATRP polymerization catalytic activity, it is preferable to use another sheet of the above-mentioned metal support.
In the method for producing a living polymer on the support of the present invention, the metal in the metal film is preferably copper. The preferable range of the metal film used in this case is the same as the preferable range of the metallic support.
The pressure applied when the monomer-containing composition is sandwiched is not particularly limited, but is preferably 0.1 to 500 KPa, for example, from the viewpoint of production.
本発明の支持体上でのリビング重合体の製造方法は、前記リビング重合体中の銅イオン濃度が、前記リビング重合体に対して5ppm以下であることが好ましい。
なお、モノマー含有組成物に重合触媒として金属触媒を添加していた通常のATRPでは、例えば金属触媒として銅を用いる場合は、前記リビング重合体中の銅イオン濃度が前記リビング重合体に対して100ppmを超える程度であった。 As for the manufacturing method of the living polymer on the support body of this invention, it is preferable that the copper ion concentration in the said living polymer is 5 ppm or less with respect to the said living polymer.
In addition, in normal ATRP in which a metal catalyst is added as a polymerization catalyst to the monomer-containing composition, for example, when copper is used as the metal catalyst, the copper ion concentration in the living polymer is 100 ppm with respect to the living polymer. It was a degree exceeding.
なお、モノマー含有組成物に重合触媒として金属触媒を添加していた通常のATRPでは、例えば金属触媒として銅を用いる場合は、前記リビング重合体中の銅イオン濃度が前記リビング重合体に対して100ppmを超える程度であった。 As for the manufacturing method of the living polymer on the support body of this invention, it is preferable that the copper ion concentration in the said living polymer is 5 ppm or less with respect to the said living polymer.
In addition, in normal ATRP in which a metal catalyst is added as a polymerization catalyst to the monomer-containing composition, for example, when copper is used as the metal catalyst, the copper ion concentration in the living polymer is 100 ppm with respect to the living polymer. It was a degree exceeding.
-モノマー-
ATRPで用いられるモノマー(ラジカル重合性単量体)としては、前述のモノマー成分と同様のものが挙げられる。 -monomer-
Examples of the monomer (radically polymerizable monomer) used in ATRP include the same monomers as those described above.
ATRPで用いられるモノマー(ラジカル重合性単量体)としては、前述のモノマー成分と同様のものが挙げられる。 -monomer-
Examples of the monomer (radically polymerizable monomer) used in ATRP include the same monomers as those described above.
(RAFT(可逆的付加開裂連鎖移動)重合)
本発明の支持体上でのリビング重合体の製造方法の好ましい態様の一つは、前記リビング重合が可逆的付加開裂連鎖移動重合である態様である。 (RAFT (Reversible Addition Fragmentation Chain Transfer) Polymerization)
One of the preferable embodiments of the method for producing a living polymer on the support of the present invention is an embodiment in which the living polymerization is a reversible addition-fragmentation chain transfer polymerization.
本発明の支持体上でのリビング重合体の製造方法の好ましい態様の一つは、前記リビング重合が可逆的付加開裂連鎖移動重合である態様である。 (RAFT (Reversible Addition Fragmentation Chain Transfer) Polymerization)
One of the preferable embodiments of the method for producing a living polymer on the support of the present invention is an embodiment in which the living polymerization is a reversible addition-fragmentation chain transfer polymerization.
本発明の支持体上でのリビング重合体の製造方法は、前記モノマー含有組成物が、モノマー成分として重合性棒状液晶化合物を含むことが好ましい。
In the method for producing a living polymer on the support of the present invention, the monomer-containing composition preferably contains a polymerizable rod-like liquid crystal compound as a monomer component.
-RAFT剤-
RAFT重合で用いられるRAFT剤としては、特に制限はなく、公知のRAFT剤を用いることができる。例えば、Macromolecules、2006、39、4953.に記載のものなどを挙げることができる。 -RAFT agent-
There is no restriction | limiting in particular as RAFT agent used by RAFT polymerization, A well-known RAFT agent can be used. For example, Macromolecules, 2006, 39, 4953. Can be mentioned.
RAFT重合で用いられるRAFT剤としては、特に制限はなく、公知のRAFT剤を用いることができる。例えば、Macromolecules、2006、39、4953.に記載のものなどを挙げることができる。 -RAFT agent-
There is no restriction | limiting in particular as RAFT agent used by RAFT polymerization, A well-known RAFT agent can be used. For example, Macromolecules, 2006, 39, 4953. Can be mentioned.
本発明では市販のRAFT剤を用いてもよく、例えば、低分子RAFT剤として4-シアノ-4-[(ドデシルスルファニル-チオカルボニル)スルファニル]ペンタノイック酸や、2-シアノ-2-プロピルベンゾジチオエート(いずれもアルドリッチ社製)などを挙げることができる。また、マクロRAFT剤として、ポリ(エチレングリコール)メチルエーテル(4-シアノ-4-ペンタノエート ドデシル トリチオカーボネートや、ポリ(エチレングリコール)メチルエーテル(4-シアノ-4-(ドデシルスルファニルチオカルボニル)スルファニル)ペンタノエート(いずれもアルドリッチ社製)などを挙げることができる。
In the present invention, a commercially available RAFT agent may be used. For example, 4-cyano-4-[(dodecylsulfanyl-thiocarbonyl) sulfanyl] pentanoic acid or 2-cyano-2-propylbenzodithioate is used as a low molecular weight RAFT agent. (Both manufactured by Aldrich). As macro RAFT agents, poly (ethylene glycol) methyl ether (4-cyano-4-pentanoate dodecyl trithiocarbonate and poly (ethylene glycol) methyl ether (4-cyano-4- (dodecylsulfanylthiocarbonyl) sulfanyl) Examples include pentanoate (all manufactured by Aldrich).
前記モノマー組成物中、前記RAFT剤の添加量は、前記モノマー成分に対して、1~60質量%であることが好ましく、5~55質量%であることがより好ましく、10~45質量%であることが特に好ましい。
In the monomer composition, the addition amount of the RAFT agent is preferably 1 to 60% by mass, more preferably 5 to 55% by mass, and more preferably 10 to 45% by mass with respect to the monomer component. It is particularly preferred.
-ラジカル重合開始剤-
ラジカル重合開始剤としては市販のラジカル重合開始剤を用いることが可能である。和光純薬製V-70、V-60(AIBN)、V-40、V-65、V-601、V-59、V-30、V-501などが好ましく、AIBNを用いることが特に好ましい。
前記モノマー組成物中、ラジカル重合開始剤の添加量は、前記モノマー成分に対して、0.01~10質量%であることが好ましく、0.1~5質量%であることがより好ましく、0.2~1質量%であることが特に好ましい。 -Radical polymerization initiator-
A commercially available radical polymerization initiator can be used as the radical polymerization initiator. V-70, V-60 (AIBN), V-40, V-65, V-601, V-59, V-30, V-501 and the like manufactured by Wako Pure Chemical Industries are preferred, and AIBN is particularly preferred.
In the monomer composition, the addition amount of the radical polymerization initiator is preferably 0.01 to 10% by mass, more preferably 0.1 to 5% by mass, based on the monomer component. It is particularly preferably 2 to 1% by mass.
ラジカル重合開始剤としては市販のラジカル重合開始剤を用いることが可能である。和光純薬製V-70、V-60(AIBN)、V-40、V-65、V-601、V-59、V-30、V-501などが好ましく、AIBNを用いることが特に好ましい。
前記モノマー組成物中、ラジカル重合開始剤の添加量は、前記モノマー成分に対して、0.01~10質量%であることが好ましく、0.1~5質量%であることがより好ましく、0.2~1質量%であることが特に好ましい。 -Radical polymerization initiator-
A commercially available radical polymerization initiator can be used as the radical polymerization initiator. V-70, V-60 (AIBN), V-40, V-65, V-601, V-59, V-30, V-501 and the like manufactured by Wako Pure Chemical Industries are preferred, and AIBN is particularly preferred.
In the monomer composition, the addition amount of the radical polymerization initiator is preferably 0.01 to 10% by mass, more preferably 0.1 to 5% by mass, based on the monomer component. It is particularly preferably 2 to 1% by mass.
-モノマー-
RAFT重合で用いられるモノマー(ラジカル重合性単量体)としては、前述のモノマー成分と同様のものが挙げられる。 -monomer-
Examples of the monomer (radically polymerizable monomer) used in RAFT polymerization include the same monomers as those described above.
RAFT重合で用いられるモノマー(ラジカル重合性単量体)としては、前述のモノマー成分と同様のものが挙げられる。 -monomer-
Examples of the monomer (radically polymerizable monomer) used in RAFT polymerization include the same monomers as those described above.
<光重合開始剤>
本発明では、本発明の支持体上でのリビング重合体の製造方法を用いてミクロ相分離構造膜を形成する場合は、前記モノマー成分として重合性棒状液晶化合物を用いて、さらに光重合開始剤を添加することが好ましい。ミクロ相分離構造膜を形成するときに、重合性棒状液晶化合物と光重合開始剤を併用することで、相分離構造を容易に固定することができる。前記光重合開始剤としては、光酸発生剤として公知の化合物が好ましく、例えば、特開2012-150428号公報に記載されている光酸発生剤などを用いることができる。
前記光酸発生剤としては、例えば、以下のものを用いることができるが、本発明は以下の具体例によって制限されるものではない。

<Photopolymerization initiator>
In the present invention, when forming a microphase-separated structure film using the method for producing a living polymer on the support of the present invention, a polymerizable rod-like liquid crystal compound is used as the monomer component, and a photopolymerization initiator is further used. Is preferably added. When forming the micro phase separation structure film, the phase separation structure can be easily fixed by using the polymerizable rod-like liquid crystal compound and the photopolymerization initiator in combination. As the photopolymerization initiator, a compound known as a photoacid generator is preferable. For example, a photoacid generator described in JP2012-150428A can be used.
As the photoacid generator, for example, the following can be used, but the present invention is not limited by the following specific examples.
本発明では、本発明の支持体上でのリビング重合体の製造方法を用いてミクロ相分離構造膜を形成する場合は、前記モノマー成分として重合性棒状液晶化合物を用いて、さらに光重合開始剤を添加することが好ましい。ミクロ相分離構造膜を形成するときに、重合性棒状液晶化合物と光重合開始剤を併用することで、相分離構造を容易に固定することができる。前記光重合開始剤としては、光酸発生剤として公知の化合物が好ましく、例えば、特開2012-150428号公報に記載されている光酸発生剤などを用いることができる。
前記光酸発生剤としては、例えば、以下のものを用いることができるが、本発明は以下の具体例によって制限されるものではない。
In the present invention, when forming a microphase-separated structure film using the method for producing a living polymer on the support of the present invention, a polymerizable rod-like liquid crystal compound is used as the monomer component, and a photopolymerization initiator is further used. Is preferably added. When forming the micro phase separation structure film, the phase separation structure can be easily fixed by using the polymerizable rod-like liquid crystal compound and the photopolymerization initiator in combination. As the photopolymerization initiator, a compound known as a photoacid generator is preferable. For example, a photoacid generator described in JP2012-150428A can be used.
As the photoacid generator, for example, the following can be used, but the present invention is not limited by the following specific examples.
前記モノマー組成物中、前記光重合開始剤の添加量は、前記モノマー成分に対して、0.01~10質量%であることが好ましく、0.1~5質量%であることがより好ましく、0.2~1質量%であることが特に好ましい。
In the monomer composition, the addition amount of the photopolymerization initiator is preferably 0.01 to 10% by mass, more preferably 0.1 to 5% by mass with respect to the monomer component. The content is particularly preferably 0.2 to 1% by mass.
<その他の添加剤>
前記モノマー含有組成物には、その他の添加剤を添加してもよい。その他の添加剤としては、例えば、液晶便覧、液晶便覧編集委員会編、丸善株式会社に記載されているような重合性棒状液晶化合物に併用することができる添加剤などを挙げることができる。 <Other additives>
Other additives may be added to the monomer-containing composition. Examples of other additives include additives that can be used in combination with a polymerizable rod-like liquid crystal compound as described in Liquid Crystal Handbook, Liquid Crystal Handbook Editorial Committee, Maruzen Co., Ltd., and the like.
前記モノマー含有組成物には、その他の添加剤を添加してもよい。その他の添加剤としては、例えば、液晶便覧、液晶便覧編集委員会編、丸善株式会社に記載されているような重合性棒状液晶化合物に併用することができる添加剤などを挙げることができる。 <Other additives>
Other additives may be added to the monomer-containing composition. Examples of other additives include additives that can be used in combination with a polymerizable rod-like liquid crystal compound as described in Liquid Crystal Handbook, Liquid Crystal Handbook Editorial Committee, Maruzen Co., Ltd., and the like.
<重合条件>
本発明の支持体上でのリビング重合体の製造方法において、支持体上でリビング重合ができる限りは重合条件や重合方法は特に限定されず、塊状重合、懸濁重合、乳化重合、塊状・懸濁重合などを適用することができる。
本発明の支持体上でのリビング重合体の製造方法では、
(i)前記モノマー含有組成物を前記支持体上に適用する工程と、
(ii)前記支持体上で前記モノマー含有組成物をリビング重合させる工程
を含むことが好ましい。 <Polymerization conditions>
In the method for producing a living polymer on the support of the present invention, the polymerization conditions and the polymerization method are not particularly limited as long as the living polymerization can be performed on the support. Bulk polymerization, suspension polymerization, emulsion polymerization, block Turbid polymerization or the like can be applied.
In the method for producing a living polymer on the support of the present invention,
(I) applying the monomer-containing composition onto the support;
(Ii) It is preferable to include a step of living polymerizing the monomer-containing composition on the support.
本発明の支持体上でのリビング重合体の製造方法において、支持体上でリビング重合ができる限りは重合条件や重合方法は特に限定されず、塊状重合、懸濁重合、乳化重合、塊状・懸濁重合などを適用することができる。
本発明の支持体上でのリビング重合体の製造方法では、
(i)前記モノマー含有組成物を前記支持体上に適用する工程と、
(ii)前記支持体上で前記モノマー含有組成物をリビング重合させる工程
を含むことが好ましい。 <Polymerization conditions>
In the method for producing a living polymer on the support of the present invention, the polymerization conditions and the polymerization method are not particularly limited as long as the living polymerization can be performed on the support. Bulk polymerization, suspension polymerization, emulsion polymerization, block Turbid polymerization or the like can be applied.
In the method for producing a living polymer on the support of the present invention,
(I) applying the monomer-containing composition onto the support;
(Ii) It is preferable to include a step of living polymerizing the monomer-containing composition on the support.
(適用方法)
前記モノマー含有組成物を前記支持体上に適用する工程における適用方法としては特に制限はないが、溶液プロセスにより成膜することが特に好ましい。
溶液プロセスによる成膜とは、ここでは有機化合物を溶解させることができる溶媒中に溶解させ、その溶液を基板上に塗布し乾燥させて成膜する方法を指す。具体的には、キャスト法、ブレードコーティング法、ワイヤーバーコーティング法、スプレーコーティング法、ディッピング(浸漬)コーティング法、ビードコーティング法、エアーナイフコーティング法、カーテンコーティング法、インクジェット法、スピンコート法、ラングミュア-ブロジェット(Langmuir-Blodgett)(LB)法などの通常の方法を用いることができる。本発明においては、キャスト法、スピンコート法、およびインクジェット法を用いることがさらに好ましい。このような溶液プロセスにより、表面が平滑で大面積の薄膜を低コストで生産することが可能となる。
スピンコートは、例えば、100~5000回転/分で、5~60秒間行うことが好ましい。
前記モノマー含有組成物を前記支持体上に適用する工程における前記モノマー含有組成物の適用後の厚みの好ましい範囲は、後述する本発明のミクロ相分離構造膜の厚みの好ましい範囲と同様である。 (Method of applying)
Although there is no restriction | limiting in particular as an application method in the process of applying the said monomer containing composition on the said support body, It is especially preferable to form into a film by a solution process.
Here, film formation by a solution process refers to a method in which an organic compound is dissolved in a solvent capable of dissolving, and the solution is applied onto a substrate and dried to form a film. Specifically, casting method, blade coating method, wire bar coating method, spray coating method, dipping (dipping) coating method, bead coating method, air knife coating method, curtain coating method, inkjet method, spin coating method, Langmuir- A usual method such as a Langmuir-Blodgett (LB) method can be used. In the present invention, it is more preferable to use a casting method, a spin coating method, and an ink jet method. By such a solution process, a thin film having a smooth surface and a large area can be produced at a low cost.
The spin coating is preferably performed, for example, at 100 to 5000 revolutions / minute for 5 to 60 seconds.
The preferable range of the thickness after application of the monomer-containing composition in the step of applying the monomer-containing composition on the support is the same as the preferable range of the thickness of the microphase-separated structure film of the present invention described later.
前記モノマー含有組成物を前記支持体上に適用する工程における適用方法としては特に制限はないが、溶液プロセスにより成膜することが特に好ましい。
溶液プロセスによる成膜とは、ここでは有機化合物を溶解させることができる溶媒中に溶解させ、その溶液を基板上に塗布し乾燥させて成膜する方法を指す。具体的には、キャスト法、ブレードコーティング法、ワイヤーバーコーティング法、スプレーコーティング法、ディッピング(浸漬)コーティング法、ビードコーティング法、エアーナイフコーティング法、カーテンコーティング法、インクジェット法、スピンコート法、ラングミュア-ブロジェット(Langmuir-Blodgett)(LB)法などの通常の方法を用いることができる。本発明においては、キャスト法、スピンコート法、およびインクジェット法を用いることがさらに好ましい。このような溶液プロセスにより、表面が平滑で大面積の薄膜を低コストで生産することが可能となる。
スピンコートは、例えば、100~5000回転/分で、5~60秒間行うことが好ましい。
前記モノマー含有組成物を前記支持体上に適用する工程における前記モノマー含有組成物の適用後の厚みの好ましい範囲は、後述する本発明のミクロ相分離構造膜の厚みの好ましい範囲と同様である。 (Method of applying)
Although there is no restriction | limiting in particular as an application method in the process of applying the said monomer containing composition on the said support body, It is especially preferable to form into a film by a solution process.
Here, film formation by a solution process refers to a method in which an organic compound is dissolved in a solvent capable of dissolving, and the solution is applied onto a substrate and dried to form a film. Specifically, casting method, blade coating method, wire bar coating method, spray coating method, dipping (dipping) coating method, bead coating method, air knife coating method, curtain coating method, inkjet method, spin coating method, Langmuir- A usual method such as a Langmuir-Blodgett (LB) method can be used. In the present invention, it is more preferable to use a casting method, a spin coating method, and an ink jet method. By such a solution process, a thin film having a smooth surface and a large area can be produced at a low cost.
The spin coating is preferably performed, for example, at 100 to 5000 revolutions / minute for 5 to 60 seconds.
The preferable range of the thickness after application of the monomer-containing composition in the step of applying the monomer-containing composition on the support is the same as the preferable range of the thickness of the microphase-separated structure film of the present invention described later.
前記モノマー含有組成物を前記支持体上に適用する工程の前に、前記支持体への表面修飾やガイドなど(ラビングなど)を行っても、行わなくてもよいが、製造コストおよび工程の短縮化の観点から、行わないことが好ましい。特に、前記モノマー成分および前記開始剤として、互いに非相溶性の疎水性および親水性のものを組み合わせて用いる場合、支持体への表面修飾やガイドなしで、後述のミクロ相分離構造を形成することができる。特に、本発明の製造方法により、後述する両親媒性・液晶ブロックコポリマーを形成する場合は、支持体への表面修飾やガイドなしでも欠陥のない綺麗なシリンダー型ミクロ相分離構造を形成することができる。
Prior to the step of applying the monomer-containing composition on the support, surface modification or guides (such as rubbing) may or may not be performed on the support. It is preferable not to perform from a viewpoint of chemical conversion. In particular, when the monomer component and the initiator are used in combination with hydrophobic and hydrophilic substances that are incompatible with each other, a micro phase separation structure described later is formed without surface modification or guide to the support. Can do. In particular, when the amphiphilic liquid crystal block copolymer described later is formed by the production method of the present invention, it is possible to form a clean cylindrical microphase separation structure without defects even without surface modification or guide to the support. it can.
(重合時間)
本発明の支持体上でのリビング重合体の製造方法は、前記支持体上で前記モノマー含有組成物をリビング重合させる工程が、1000秒以下であることが好ましく、800秒以下であることがより好ましく、600秒以下であることが特に好ましい。
前記支持体上で前記モノマー含有組成物をリビング重合させる工程の時間の下限値は特に制限されるものではないが、5秒以上とすることが好ましい。 (Polymerization time)
In the method for producing a living polymer on the support of the present invention, the step of living polymerizing the monomer-containing composition on the support is preferably 1000 seconds or less, more preferably 800 seconds or less. Preferably, it is particularly preferably 600 seconds or less.
The lower limit of the time for the step of living polymerizing the monomer-containing composition on the support is not particularly limited, but is preferably 5 seconds or more.
本発明の支持体上でのリビング重合体の製造方法は、前記支持体上で前記モノマー含有組成物をリビング重合させる工程が、1000秒以下であることが好ましく、800秒以下であることがより好ましく、600秒以下であることが特に好ましい。
前記支持体上で前記モノマー含有組成物をリビング重合させる工程の時間の下限値は特に制限されるものではないが、5秒以上とすることが好ましい。 (Polymerization time)
In the method for producing a living polymer on the support of the present invention, the step of living polymerizing the monomer-containing composition on the support is preferably 1000 seconds or less, more preferably 800 seconds or less. Preferably, it is particularly preferably 600 seconds or less.
The lower limit of the time for the step of living polymerizing the monomer-containing composition on the support is not particularly limited, but is preferably 5 seconds or more.
(反応温度)
反応温度は重合反応が進行する温度であればいずれでも構わず、所望する重合体の重合度、使用する重合開始剤および溶媒の種類や量によって一様ではないが、通常、-100℃~250℃である。
本発明の支持体上でのリビング重合体の製造方法は、前記支持体上で前記モノマー含有組成物をリビング重合させる工程が、50~200℃での加熱であることが好ましく、より好ましくは60℃~180℃であり、更に好ましくは80℃~160℃である。
加熱方法としては特に制限はなく、公知の方法を用いることができ、例えば前記支持体をホットプレート上に載せて、前記モノマー含有組成物を前記支持体ごと加熱する方法などを挙げることができる。
なお、加熱後は、最終的に室温まで放冷してリビング重合体を得ることが好ましい。 (Reaction temperature)
The reaction temperature may be any temperature as long as the polymerization reaction proceeds, and is not uniform depending on the desired degree of polymerization of the polymer and the type and amount of the polymerization initiator and solvent used. ° C.
In the method for producing a living polymer on the support of the present invention, the step of living polymerizing the monomer-containing composition on the support is preferably heating at 50 to 200 ° C., more preferably 60 ° C to 180 ° C, more preferably 80 ° C to 160 ° C.
There is no restriction | limiting in particular as a heating method, A well-known method can be used, For example, the method of mounting the said support body on a hotplate and heating the said monomer containing composition with the said support body etc. can be mentioned.
In addition, after heating, it is preferable to cool to room temperature finally and to obtain a living polymer.
反応温度は重合反応が進行する温度であればいずれでも構わず、所望する重合体の重合度、使用する重合開始剤および溶媒の種類や量によって一様ではないが、通常、-100℃~250℃である。
本発明の支持体上でのリビング重合体の製造方法は、前記支持体上で前記モノマー含有組成物をリビング重合させる工程が、50~200℃での加熱であることが好ましく、より好ましくは60℃~180℃であり、更に好ましくは80℃~160℃である。
加熱方法としては特に制限はなく、公知の方法を用いることができ、例えば前記支持体をホットプレート上に載せて、前記モノマー含有組成物を前記支持体ごと加熱する方法などを挙げることができる。
なお、加熱後は、最終的に室温まで放冷してリビング重合体を得ることが好ましい。 (Reaction temperature)
The reaction temperature may be any temperature as long as the polymerization reaction proceeds, and is not uniform depending on the desired degree of polymerization of the polymer and the type and amount of the polymerization initiator and solvent used. ° C.
In the method for producing a living polymer on the support of the present invention, the step of living polymerizing the monomer-containing composition on the support is preferably heating at 50 to 200 ° C., more preferably 60 ° C to 180 ° C, more preferably 80 ° C to 160 ° C.
There is no restriction | limiting in particular as a heating method, A well-known method can be used, For example, the method of mounting the said support body on a hotplate and heating the said monomer containing composition with the said support body etc. can be mentioned.
In addition, after heating, it is preferable to cool to room temperature finally and to obtain a living polymer.
前記支持体上で前記モノマー含有組成物をリビング重合させる工程でのリビング重合反応は場合によって減圧、常圧または加圧のいずれでも実施できる。
また、上記重合反応は、特に開始剤が低分子開始剤であるときは窒素やアルゴン等の不活性ガスのフロー下で行うことが好ましく、窒素ガスのフロー下で行うことがより好ましい。不活性ガスのフロー条件としては特に制限はないが、例えば、0.001~50L/minとすることができる。 The living polymerization reaction in the step of living polymerizing the monomer-containing composition on the support can be carried out under reduced pressure, normal pressure, or increased pressure depending on the case.
The polymerization reaction is preferably carried out under a flow of an inert gas such as nitrogen or argon, more preferably under a flow of nitrogen gas, particularly when the initiator is a low molecular weight initiator. There are no particular restrictions on the flow conditions of the inert gas, but it may be, for example, 0.001 to 50 L / min.
また、上記重合反応は、特に開始剤が低分子開始剤であるときは窒素やアルゴン等の不活性ガスのフロー下で行うことが好ましく、窒素ガスのフロー下で行うことがより好ましい。不活性ガスのフロー条件としては特に制限はないが、例えば、0.001~50L/minとすることができる。 The living polymerization reaction in the step of living polymerizing the monomer-containing composition on the support can be carried out under reduced pressure, normal pressure, or increased pressure depending on the case.
The polymerization reaction is preferably carried out under a flow of an inert gas such as nitrogen or argon, more preferably under a flow of nitrogen gas, particularly when the initiator is a low molecular weight initiator. There are no particular restrictions on the flow conditions of the inert gas, but it may be, for example, 0.001 to 50 L / min.
(モノマー消費率)
本発明の支持体上でのリビング重合体の製造方法は、前記支持体上で前記モノマー含有組成物をリビング重合させる工程のモノマー消費率を、10~100%に制御することが、相分離を迅速に形成する観点から、好ましい。前記モノマー消費率は、20~100%であることがより好ましく、30~100%であることが特に好ましい。
上述のモノマー消費率は、得られたリビング重合体を重THFにて抽出した抽出液について、1H-NMR(BRUKER-300MHZ)測定を行うことにより求めることができる。 (Monomer consumption rate)
In the method for producing a living polymer on a support according to the present invention, the monomer consumption rate in the step of living polymerizing the monomer-containing composition on the support is controlled to 10 to 100%. From the viewpoint of rapid formation, it is preferable. The monomer consumption rate is more preferably 20 to 100%, and particularly preferably 30 to 100%.
The monomer consumption rate described above can be determined by performing 1 H-NMR (BRUKER-300 MHZ) measurement on an extract obtained by extracting the obtained living polymer with deuterated THF.
本発明の支持体上でのリビング重合体の製造方法は、前記支持体上で前記モノマー含有組成物をリビング重合させる工程のモノマー消費率を、10~100%に制御することが、相分離を迅速に形成する観点から、好ましい。前記モノマー消費率は、20~100%であることがより好ましく、30~100%であることが特に好ましい。
上述のモノマー消費率は、得られたリビング重合体を重THFにて抽出した抽出液について、1H-NMR(BRUKER-300MHZ)測定を行うことにより求めることができる。 (Monomer consumption rate)
In the method for producing a living polymer on a support according to the present invention, the monomer consumption rate in the step of living polymerizing the monomer-containing composition on the support is controlled to 10 to 100%. From the viewpoint of rapid formation, it is preferable. The monomer consumption rate is more preferably 20 to 100%, and particularly preferably 30 to 100%.
The monomer consumption rate described above can be determined by performing 1 H-NMR (BRUKER-300 MHZ) measurement on an extract obtained by extracting the obtained living polymer with deuterated THF.
<リビング重合体>
本発明の支持体上でのリビング重合体の製造方法により、リビング重合体が製造される。リビング重合体は、前記支持体上で、フィルム状に形成されることが好ましい。 <Living polymer>
A living polymer is manufactured by the manufacturing method of the living polymer on the support body of this invention. The living polymer is preferably formed into a film on the support.
本発明の支持体上でのリビング重合体の製造方法により、リビング重合体が製造される。リビング重合体は、前記支持体上で、フィルム状に形成されることが好ましい。 <Living polymer>
A living polymer is manufactured by the manufacturing method of the living polymer on the support body of this invention. The living polymer is preferably formed into a film on the support.
本発明の支持体上でのリビング重合体の製造方法は、得られるリビング重合体の数平均分子量Mnが1000~100000であることが好ましく、2000~50000であることがより好ましく、3000~30000であることが特に好ましい。
In the method for producing a living polymer on the support of the present invention, the number average molecular weight Mn of the resulting living polymer is preferably 1,000 to 100,000, more preferably 2,000 to 50,000, and more preferably 3,000 to 30,000. It is particularly preferred.
本発明の支持体上でのリビング重合体の製造方法は、得られるリビング重合体の抽出液の重量平均分子量Mwと数平均分子量Mnの比Mw/Mnが1.3を超えることが、残存モノマーが相分離構造の欠陥を補修する点、残存マクロ開始剤及び残存マクロRAFT剤が相分離構造のサイズを制御する点からは好ましい。
一方、Mw/Mnが1.3以下であることが、相分離構造を安定的に形成させる観点からは好ましい。
本明細書中、ブロック共重合体、アルキレンオキサイド鎖を含むポリマー成分、炭素数2~20のアルキレン鎖を含むポリマー成分などの数平均分子量Mnは、以下の方法で測定した値を採用する。
ポリマーをTHFに溶解させ、東ソー製高速GPC(HLC-8220GPC)を用いて行った。数平均分子量Mnはポリスチレン換算で計算した。
また、本明細書中、ブロック共重合体、アルキレンオキサイド鎖を含むポリマー成分、炭素数2~20のアルキレン鎖を含むポリマー成分などの重量平均分子量Mwは、以下の方法で測定した値を採用する。
ポリマーをTHFに溶解させ、東ソー製高速GPC(HLC-8220GPC)を用いて行った。重量平均分子量Mwはポリスチレン換算で計算した。 The method for producing the living polymer on the support of the present invention is such that the ratio Mw / Mn of the weight average molecular weight Mw and the number average molecular weight Mn of the resulting living polymer extract exceeds 1.3 is a residual monomer. Is preferable from the viewpoint of repairing defects in the phase separation structure, and the residual macroinitiator and the residual macro RAFT agent from controlling the size of the phase separation structure.
On the other hand, Mw / Mn is preferably 1.3 or less from the viewpoint of stably forming the phase separation structure.
In the present specification, the number average molecular weight Mn of a block copolymer, a polymer component containing an alkylene oxide chain, a polymer component containing an alkylene chain having 2 to 20 carbon atoms, and the like is a value measured by the following method.
The polymer was dissolved in THF, and a high-speed GPC (HLC-8220GPC) manufactured by Tosoh was used. The number average molecular weight Mn was calculated in terms of polystyrene.
In this specification, the weight average molecular weight Mw of the block copolymer, the polymer component containing an alkylene oxide chain, the polymer component containing an alkylene chain having 2 to 20 carbon atoms, and the like is a value measured by the following method. .
The polymer was dissolved in THF, and a high-speed GPC (HLC-8220GPC) manufactured by Tosoh was used. The weight average molecular weight Mw was calculated in terms of polystyrene.
一方、Mw/Mnが1.3以下であることが、相分離構造を安定的に形成させる観点からは好ましい。
本明細書中、ブロック共重合体、アルキレンオキサイド鎖を含むポリマー成分、炭素数2~20のアルキレン鎖を含むポリマー成分などの数平均分子量Mnは、以下の方法で測定した値を採用する。
ポリマーをTHFに溶解させ、東ソー製高速GPC(HLC-8220GPC)を用いて行った。数平均分子量Mnはポリスチレン換算で計算した。
また、本明細書中、ブロック共重合体、アルキレンオキサイド鎖を含むポリマー成分、炭素数2~20のアルキレン鎖を含むポリマー成分などの重量平均分子量Mwは、以下の方法で測定した値を採用する。
ポリマーをTHFに溶解させ、東ソー製高速GPC(HLC-8220GPC)を用いて行った。重量平均分子量Mwはポリスチレン換算で計算した。 The method for producing the living polymer on the support of the present invention is such that the ratio Mw / Mn of the weight average molecular weight Mw and the number average molecular weight Mn of the resulting living polymer extract exceeds 1.3 is a residual monomer. Is preferable from the viewpoint of repairing defects in the phase separation structure, and the residual macroinitiator and the residual macro RAFT agent from controlling the size of the phase separation structure.
On the other hand, Mw / Mn is preferably 1.3 or less from the viewpoint of stably forming the phase separation structure.
In the present specification, the number average molecular weight Mn of a block copolymer, a polymer component containing an alkylene oxide chain, a polymer component containing an alkylene chain having 2 to 20 carbon atoms, and the like is a value measured by the following method.
The polymer was dissolved in THF, and a high-speed GPC (HLC-8220GPC) manufactured by Tosoh was used. The number average molecular weight Mn was calculated in terms of polystyrene.
In this specification, the weight average molecular weight Mw of the block copolymer, the polymer component containing an alkylene oxide chain, the polymer component containing an alkylene chain having 2 to 20 carbon atoms, and the like is a value measured by the following method. .
The polymer was dissolved in THF, and a high-speed GPC (HLC-8220GPC) manufactured by Tosoh was used. The weight average molecular weight Mw was calculated in terms of polystyrene.
<ブロック共重合体の形成>
本発明の支持体上でのリビング重合体の製造方法は、前記リビング重合体が、ブロック共重合体であることが好ましい。この場合、本発明の支持体上でのリビング重合体の製造方法は、下記(1)および(2)の工程を、支持体上で実施することがより好ましい。
(1)前記モノマー含有組成物として、アルキレンオキサイド鎖を含むポリマー成分および炭素数2~20のアルキレン鎖を含むモノマー成分を含むモノマー含有組成物、ならびに、アルキレンオキサイド鎖を含むモノマー成分および炭素数2~20のアルキレン鎖を含むポリマー成分を含むモノマー含有組成物のうち少なくとも一方を用いて、該モノマー含有組成物を前記支持体上に適用する工程
(2)前記支持体上で前記モノマー含有組成物をリビング重合させて、アルキレンオキサイド鎖を含むポリマー成分と炭素数2~20のアルキレン鎖を含むポリマー成分とが共有結合にて連結されたブロック共重合体を得る工程 <Formation of block copolymer>
In the method for producing a living polymer on the support of the present invention, the living polymer is preferably a block copolymer. In this case, as for the manufacturing method of the living polymer on the support body of this invention, it is more preferable to implement the following process (1) and (2) on a support body.
(1) As the monomer-containing composition, a monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms, and a monomer component containing an alkylene oxide chain and a carbon number of 2 (2) applying the monomer-containing composition onto the support using at least one of the monomer-containing compositions containing a polymer component containing 20 to 20 alkylene chains (2) the monomer-containing composition on the support Is a step of living polymerizing to obtain a block copolymer in which a polymer component containing an alkylene oxide chain and a polymer component containing an alkylene chain having 2 to 20 carbon atoms are linked by a covalent bond
本発明の支持体上でのリビング重合体の製造方法は、前記リビング重合体が、ブロック共重合体であることが好ましい。この場合、本発明の支持体上でのリビング重合体の製造方法は、下記(1)および(2)の工程を、支持体上で実施することがより好ましい。
(1)前記モノマー含有組成物として、アルキレンオキサイド鎖を含むポリマー成分および炭素数2~20のアルキレン鎖を含むモノマー成分を含むモノマー含有組成物、ならびに、アルキレンオキサイド鎖を含むモノマー成分および炭素数2~20のアルキレン鎖を含むポリマー成分を含むモノマー含有組成物のうち少なくとも一方を用いて、該モノマー含有組成物を前記支持体上に適用する工程
(2)前記支持体上で前記モノマー含有組成物をリビング重合させて、アルキレンオキサイド鎖を含むポリマー成分と炭素数2~20のアルキレン鎖を含むポリマー成分とが共有結合にて連結されたブロック共重合体を得る工程 <Formation of block copolymer>
In the method for producing a living polymer on the support of the present invention, the living polymer is preferably a block copolymer. In this case, as for the manufacturing method of the living polymer on the support body of this invention, it is more preferable to implement the following process (1) and (2) on a support body.
(1) As the monomer-containing composition, a monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms, and a monomer component containing an alkylene oxide chain and a carbon number of 2 (2) applying the monomer-containing composition onto the support using at least one of the monomer-containing compositions containing a polymer component containing 20 to 20 alkylene chains (2) the monomer-containing composition on the support Is a step of living polymerizing to obtain a block copolymer in which a polymer component containing an alkylene oxide chain and a polymer component containing an alkylene chain having 2 to 20 carbon atoms are linked by a covalent bond
本発明の支持体上でのリビング重合体の製造方法は、下記(1’)および(2)の工程を、支持体上で実施することが特に好ましい。
(1’)前記モノマー含有組成物として、アルキレンオキサイド鎖を含むポリマー成分および炭素数2~20のアルキレン鎖を含むモノマー成分を含むモノマー含有組成物を用いて、該モノマー含有組成物を前記支持体上に適用する工程
(2)前記支持体上で前記モノマー含有組成物をリビング重合させて、アルキレンオキサイド鎖を含むポリマー成分と炭素数2~20のアルキレン鎖を含むポリマー成分とが共有結合にて連結されたブロック共重合体を得る工程 In the method for producing a living polymer on the support of the present invention, the following steps (1 ′) and (2) are particularly preferably carried out on the support.
(1 ′) A monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms is used as the monomer-containing composition. (2) The monomer-containing composition is living-polymerized on the support so that the polymer component containing an alkylene oxide chain and the polymer component containing an alkylene chain having 2 to 20 carbon atoms are covalently bonded. Process for obtaining linked block copolymer
(1’)前記モノマー含有組成物として、アルキレンオキサイド鎖を含むポリマー成分および炭素数2~20のアルキレン鎖を含むモノマー成分を含むモノマー含有組成物を用いて、該モノマー含有組成物を前記支持体上に適用する工程
(2)前記支持体上で前記モノマー含有組成物をリビング重合させて、アルキレンオキサイド鎖を含むポリマー成分と炭素数2~20のアルキレン鎖を含むポリマー成分とが共有結合にて連結されたブロック共重合体を得る工程 In the method for producing a living polymer on the support of the present invention, the following steps (1 ′) and (2) are particularly preferably carried out on the support.
(1 ′) A monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms is used as the monomer-containing composition. (2) The monomer-containing composition is living-polymerized on the support so that the polymer component containing an alkylene oxide chain and the polymer component containing an alkylene chain having 2 to 20 carbon atoms are covalently bonded. Process for obtaining linked block copolymer
前記(1)および(1’)工程の好ましい製造条件は、前記(i)工程の好ましい製造条件と同様である。前記(2)工程の好ましい製造条件は、前記(ii)工程の好ましい製造条件と同様である。
Preferred production conditions for the steps (1) and (1 ′) are the same as the preferred production conditions for the step (i). Preferred production conditions for the step (2) are the same as the preferred production conditions for the step (ii).
本発明の支持体上でのリビング重合体の製造方法は、前記ブロック共重合体が、ミクロ相分離構造膜の形成用のブロック共重合体であることが好ましい。
得られたブロック共重合体を機能材料として用いるためには、ブロック共重合体反応混合物を一旦取り出し精製した後に、ブロック共重合体を基材(または支持体)に塗布して相分離構造を形成させるのが一般的である。一方、本発明のリビング重合体の製造方法によりブロック共重合体を得る場合は支持体上でリビング重合した後に、そのまま支持体上に相分離構造を形成させることができるため更に経済合理性に富む。 In the method for producing a living polymer on the support of the present invention, the block copolymer is preferably a block copolymer for forming a microphase-separated structure film.
In order to use the obtained block copolymer as a functional material, the block copolymer reaction mixture is once taken out and purified, and then the block copolymer is applied to a substrate (or support) to form a phase separation structure. It is common to make it. On the other hand, when a block copolymer is obtained by the method for producing a living polymer according to the present invention, the phase separation structure can be formed on the support as it is after the living polymerization is performed on the support, which is further economically rational. .
得られたブロック共重合体を機能材料として用いるためには、ブロック共重合体反応混合物を一旦取り出し精製した後に、ブロック共重合体を基材(または支持体)に塗布して相分離構造を形成させるのが一般的である。一方、本発明のリビング重合体の製造方法によりブロック共重合体を得る場合は支持体上でリビング重合した後に、そのまま支持体上に相分離構造を形成させることができるため更に経済合理性に富む。 In the method for producing a living polymer on the support of the present invention, the block copolymer is preferably a block copolymer for forming a microphase-separated structure film.
In order to use the obtained block copolymer as a functional material, the block copolymer reaction mixture is once taken out and purified, and then the block copolymer is applied to a substrate (or support) to form a phase separation structure. It is common to make it. On the other hand, when a block copolymer is obtained by the method for producing a living polymer according to the present invention, the phase separation structure can be formed on the support as it is after the living polymerization is performed on the support, which is further economically rational. .
[ブロック共重合体]
本発明のブロック共重合体は、本発明の支持体上でのリビング重合体の製造方法において、前記リビング重合体がブロック共重合体となる条件で得られたことを特徴とする。前記リビング重合体がブロック共重合体となる条件としては、上述の(1)および(2)の工程を行う方法を挙げることができ、上述の(1’)および(2)の工程を行う方法が好ましい。
得られたブロック共重合体の好ましい分子量の範囲は、本発明の支持体上でのリビング重合体の製造方法で得られるリビング重合体の好ましい分子量の範囲と同様である。 [Block copolymer]
The block copolymer of the present invention is characterized in that, in the method for producing a living polymer on the support of the present invention, the block copolymer is obtained under the condition that the living polymer becomes a block copolymer. Examples of the conditions for the living polymer to be a block copolymer include the method of performing the steps (1) and (2) described above, and the method of performing the steps (1 ′) and (2) described above. Is preferred.
The preferred molecular weight range of the obtained block copolymer is the same as the preferred molecular weight range of the living polymer obtained by the method for producing a living polymer on the support of the present invention.
本発明のブロック共重合体は、本発明の支持体上でのリビング重合体の製造方法において、前記リビング重合体がブロック共重合体となる条件で得られたことを特徴とする。前記リビング重合体がブロック共重合体となる条件としては、上述の(1)および(2)の工程を行う方法を挙げることができ、上述の(1’)および(2)の工程を行う方法が好ましい。
得られたブロック共重合体の好ましい分子量の範囲は、本発明の支持体上でのリビング重合体の製造方法で得られるリビング重合体の好ましい分子量の範囲と同様である。 [Block copolymer]
The block copolymer of the present invention is characterized in that, in the method for producing a living polymer on the support of the present invention, the block copolymer is obtained under the condition that the living polymer becomes a block copolymer. Examples of the conditions for the living polymer to be a block copolymer include the method of performing the steps (1) and (2) described above, and the method of performing the steps (1 ′) and (2) described above. Is preferred.
The preferred molecular weight range of the obtained block copolymer is the same as the preferred molecular weight range of the living polymer obtained by the method for producing a living polymer on the support of the present invention.
本発明のブロック共重合体の用途としては特に制限はないが、本発明のブロック共重合体はミクロ相分離構造膜の形成用のブロック共重合体であることが好ましい。
The use of the block copolymer of the present invention is not particularly limited, but the block copolymer of the present invention is preferably a block copolymer for forming a microphase-separated structure film.
本発明のブロック共重合体は、構造上の制限は特にないが、アルキレンオキサイド鎖を含むポリマー成分(A)および炭素数2~20のアルキレン鎖を含むポリマー成分(B)の互いに非相溶性のポリマーが共有結合によって結合したブロック共重合体であって、前記炭素数2~20のアルキレン鎖を含むポリマー成分(B)が炭素数6~50のメソゲン側鎖を有していることが好ましい。このようなブロック共重合体は、両親媒性・液晶ブロックコポリマーと呼ばれることもある。
本発明のブロック共重合体は、前記炭素数2~20のアルキレン鎖を含むポリマー成分(B)が炭素数6~50のメソゲン側鎖を有するポリ((メタ)アクリレート)であることが好ましく、前記重合性棒状液晶化合物由来のポリマー成分を含むことがより好ましい。
本発明のブロック共重合体は、前記アルキレンオキサイド鎖を含むポリマー成分(A)が、ポリアルキレンオキシ構造を含むことが好ましく、前記マクロ開始剤由来のポリマー成分を含むことがより好ましい。 The block copolymer of the present invention is not particularly limited in structure, but the polymer component (A) containing an alkylene oxide chain and the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms are incompatible with each other. It is a block copolymer in which a polymer is bonded by a covalent bond, and the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms preferably has a mesogenic side chain having 6 to 50 carbon atoms. Such a block copolymer is sometimes called an amphiphilic liquid crystal block copolymer.
In the block copolymer of the present invention, the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms is preferably a poly ((meth) acrylate) having a mesogenic side chain having 6 to 50 carbon atoms, More preferably, it contains a polymer component derived from the polymerizable rod-like liquid crystal compound.
In the block copolymer of the present invention, the polymer component (A) containing an alkylene oxide chain preferably contains a polyalkyleneoxy structure, and more preferably contains a polymer component derived from the macroinitiator.
本発明のブロック共重合体は、前記炭素数2~20のアルキレン鎖を含むポリマー成分(B)が炭素数6~50のメソゲン側鎖を有するポリ((メタ)アクリレート)であることが好ましく、前記重合性棒状液晶化合物由来のポリマー成分を含むことがより好ましい。
本発明のブロック共重合体は、前記アルキレンオキサイド鎖を含むポリマー成分(A)が、ポリアルキレンオキシ構造を含むことが好ましく、前記マクロ開始剤由来のポリマー成分を含むことがより好ましい。 The block copolymer of the present invention is not particularly limited in structure, but the polymer component (A) containing an alkylene oxide chain and the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms are incompatible with each other. It is a block copolymer in which a polymer is bonded by a covalent bond, and the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms preferably has a mesogenic side chain having 6 to 50 carbon atoms. Such a block copolymer is sometimes called an amphiphilic liquid crystal block copolymer.
In the block copolymer of the present invention, the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms is preferably a poly ((meth) acrylate) having a mesogenic side chain having 6 to 50 carbon atoms, More preferably, it contains a polymer component derived from the polymerizable rod-like liquid crystal compound.
In the block copolymer of the present invention, the polymer component (A) containing an alkylene oxide chain preferably contains a polyalkyleneoxy structure, and more preferably contains a polymer component derived from the macroinitiator.
本発明のブロック共重合体は、前記アルキレンオキサイド鎖を含むポリマー成分(A)および前記炭素数2~20のアルキレン鎖を含むポリマー成分(B)が直接結合していても、連結基を介して結合していてもよい。着色が少ないため、光関連用途では、前記アルキレンオキサイド鎖を含むポリマー成分(A)および前記炭素数2~20のアルキレン鎖を含むポリマー成分(B)の連結部に、下記2価の連結基のいずれかを含むブロック共重合体であってもよい。前記ポリマー成分(A)および前記ポリマー成分(B)の連結部に、下記一般式(11)および(12)で表される2価の連結基のうち少なくとも一方を導入する方法としては特に制限はない。例えば、アルキレンオキサイド鎖を含むポリマー成分(A)、すなわち親水性マクロRAFT剤として下記一般式(11)および(12)で表される2価の連結基のうち少なくとも一方を有するものを用いる方法を挙げることができる。


Even if the polymer component (A) containing an alkylene oxide chain and the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms are directly bonded to each other, the block copolymer of the present invention can be bonded via a linking group. It may be bonded. Because of less coloring, in the light-related application, the following divalent linking group is bonded to the linking part of the polymer component (A) containing an alkylene oxide chain and the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms. It may be a block copolymer containing any of them. The method for introducing at least one of the divalent linking groups represented by the following general formulas (11) and (12) into the linking part of the polymer component (A) and the polymer component (B) is not particularly limited. Absent. For example, a method using a polymer component (A) containing an alkylene oxide chain, that is, a hydrophilic macro RAFT agent having at least one of divalent linking groups represented by the following general formulas (11) and (12) Can be mentioned.
本発明のブロック共重合体としては、下記一般式(I-1)、(I-2)などで表されるものなどを挙げることができる。


ここでXは下記一般式(3)に記載の構造およびハロゲン原子(ヨウ素、臭素、塩素原子)もしくは水素原子を表す。

(一般式(I-1)、(I-2)中、nおよびmはそれぞれ独立に1~500の整数を表し、L11およびL12はそれぞれ独立に単結合またはアルキレン基、アリーレン基、アルケニレン基、アルキニレン基、メタロセニレン基、-CO-N(R101)-、-CO-O-、-SO2-N(R102)-、-SO2-O-、-N(R103)-CO-N(R104)-、-SO2-、-SO-、-S-、-O-、-CO-、-N(R105)-、ヘテリレン基を1つまたはそれ以上組み合わせて構成される炭素数0~100以下、好ましくは1以上20以下の連結基を表し(R101、R102、R103、R104、R105は、各々独立に、水素原子、置換または無置換のアルキル基、置換または無置換のアリール基を表す。ただし、L12は少なくとも炭素数2~20のアルキレン基を含む。)、R1はそれぞれ独立に水素原子またはメチル基を表し、R2はそれぞれ独立にオキセタニル基、エポキシ基、アクリレート基、メタクリレート基、置換基を有していてもよいアルキル基または置換基を有していてもよいアルコキシ基を表し、Mは炭素数6~50のメソゲン基を表し、Z3は置換もしくは無置換のアリール基、置換もしくは無置換のアルキル基を表す。但し、m個の繰り返し単位は互いに同一であっても異なっていてもよい。)
ここで一般式(I-1)および(I-2)、中、Z3は置換もしくは無置換のアリール基、置換もしくは無置換のアルキル基であることが好ましく、置換もしくは無置換のアルキル基であることがより好ましく、炭素数4~20のアルキル基であることが特に好ましい。 Examples of the block copolymer of the present invention include those represented by the following general formulas (I-1) and (I-2).
Here, X represents the structure described in the following general formula (3) and a halogen atom (iodine, bromine, chlorine atom) or a hydrogen atom.
(In the general formulas (I-1) and (I-2), n and m each independently represent an integer of 1 to 500, and L 11 and L 12 each independently represents a single bond or an alkylene group, an arylene group, an alkenylene. Group, alkynylene group, metallocenylene group, —CO—N (R 101 ) —, —CO—O—, —SO 2 —N (R 102 ) —, —SO 2 —O—, —N (R 103 ) —CO -N (R 104 )-, -SO 2- , -SO-, -S-, -O-, -CO-, -N (R 105 )-, or a combination of one or more heterylene groups Represents a linking group having 0 to 100 carbon atoms, preferably 1 to 20 carbon atoms (R 101 , R 102 , R 103 , R 104 , and R 105 each independently represents a hydrogen atom, a substituted or unsubstituted alkyl group, Substituted or unsubstituted aryl The represented. However, L 12 comprises an alkylene group having atleast carbon atoms 2 ~ 20.), R 1 each independently represent a hydrogen atom or a methyl group, R 2 is independently an oxetanyl group, an epoxy group, an acrylate group Represents a methacrylate group, an optionally substituted alkyl group or an optionally substituted alkoxy group, M represents a mesogenic group having 6 to 50 carbon atoms, and Z 3 represents substituted or unsubstituted. An aryl group and a substituted or unsubstituted alkyl group, provided that m repeating units may be the same or different.
In the general formulas (I-1) and (I-2), Z 3 is preferably a substituted or unsubstituted aryl group, a substituted or unsubstituted alkyl group, and preferably a substituted or unsubstituted alkyl group. More preferably, it is an alkyl group having 4 to 20 carbon atoms.
(一般式(I-1)、(I-2)中、nおよびmはそれぞれ独立に1~500の整数を表し、L11およびL12はそれぞれ独立に単結合またはアルキレン基、アリーレン基、アルケニレン基、アルキニレン基、メタロセニレン基、-CO-N(R101)-、-CO-O-、-SO2-N(R102)-、-SO2-O-、-N(R103)-CO-N(R104)-、-SO2-、-SO-、-S-、-O-、-CO-、-N(R105)-、ヘテリレン基を1つまたはそれ以上組み合わせて構成される炭素数0~100以下、好ましくは1以上20以下の連結基を表し(R101、R102、R103、R104、R105は、各々独立に、水素原子、置換または無置換のアルキル基、置換または無置換のアリール基を表す。ただし、L12は少なくとも炭素数2~20のアルキレン基を含む。)、R1はそれぞれ独立に水素原子またはメチル基を表し、R2はそれぞれ独立にオキセタニル基、エポキシ基、アクリレート基、メタクリレート基、置換基を有していてもよいアルキル基または置換基を有していてもよいアルコキシ基を表し、Mは炭素数6~50のメソゲン基を表し、Z3は置換もしくは無置換のアリール基、置換もしくは無置換のアルキル基を表す。但し、m個の繰り返し単位は互いに同一であっても異なっていてもよい。)
ここで一般式(I-1)および(I-2)、中、Z3は置換もしくは無置換のアリール基、置換もしくは無置換のアルキル基であることが好ましく、置換もしくは無置換のアルキル基であることがより好ましく、炭素数4~20のアルキル基であることが特に好ましい。 Examples of the block copolymer of the present invention include those represented by the following general formulas (I-1) and (I-2).
(In the general formulas (I-1) and (I-2), n and m each independently represent an integer of 1 to 500, and L 11 and L 12 each independently represents a single bond or an alkylene group, an arylene group, an alkenylene. Group, alkynylene group, metallocenylene group, —CO—N (R 101 ) —, —CO—O—, —SO 2 —N (R 102 ) —, —SO 2 —O—, —N (R 103 ) —CO -N (R 104 )-, -SO 2- , -SO-, -S-, -O-, -CO-, -N (R 105 )-, or a combination of one or more heterylene groups Represents a linking group having 0 to 100 carbon atoms, preferably 1 to 20 carbon atoms (R 101 , R 102 , R 103 , R 104 , and R 105 each independently represents a hydrogen atom, a substituted or unsubstituted alkyl group, Substituted or unsubstituted aryl The represented. However, L 12 comprises an alkylene group having at
In the general formulas (I-1) and (I-2), Z 3 is preferably a substituted or unsubstituted aryl group, a substituted or unsubstituted alkyl group, and preferably a substituted or unsubstituted alkyl group. More preferably, it is an alkyl group having 4 to 20 carbon atoms.
一般式(I-1)、(I-2)中、nは1~500であることが好ましく、3~250であることがより好ましく、5~200であることが特に好ましい。
一般式(I-1)、(I-2)中、mは3~200であることが好ましく、5~150であることがより好ましく、10~100であることが特に好ましい。
一般式(I-1)、(I-2)中、L11、L12は各々独立に、単結合、下記に記載する官能基または下記に記載する官能基から水素原子を2つ除去してなる2価の連結基を表すことが好ましく、特に限定は無いが、好ましくはアルキレン基(炭素数2~20、例えばメチレン、エチレン、プロピレン、ブチレン、ペンチレン)、アリーレン基(炭素数6~26、例えばフェニレン、ナフチレン)、アルケニレン基(炭素数2~20、例えばエテニレン、プロペニレン)、アルキニレン基(炭素数2~20、例えばエチニレン、プロピニレン)、メタロセニレン基(例えばフェロセン)、-CO-N(R101)-、-CO-O-、-SO2-N(R102)-、-SO2-O-、-N(R103)-CO-N(R104)-、-SO2-、-SO-、-S-、-O-、-CO-、-N(R105)-、ヘテリレン基(炭素数1~26、例えば6-クロロ-1,3,5-トリアジル-2,4-ジイル基、ピリミジン-2,4-ジイル基)を1つまたはそれ以上組み合わせて構成される炭素数0~100以下、好ましくは1以上20以下の連結基を表す。
ただし、L12は少なくとも炭素数2~20のアルキレン基を含む。L12が含むアルキレン基の炭素数は3~18であることが好ましく、4~16であることがより好ましく、5~15であることが特に好ましい。
上記、R101、R102、R103、R104、R105は、各々独立に、水素原子、置換または無置換のアルキル基、置換または無置換のアリール基を表す。また、L11、L12で表される連結基は、1つ以上複数個存在していてもよく、複数個(好ましくは2つ)が結合して環を形成してもよい。
一般式(I-1)、(I-2)中、R1はそれぞれ独立に水素原子、メチル基であることが汎用性の観点から好ましい。
一般式(I-1)、(I-2)中、Mは炭素数6~50のメソゲン基であることが好ましく、炭素数6~40のメソゲン基であることがより好ましく、炭素数6~30のメソゲン基であることが特に好ましい。Mが表す炭素数6~50のメソゲン基の好ましい範囲は、前記一般式(X)中の前記-Cy1-L2-(Cy2-L3)n-Cy3-L4-の好ましい範囲と同様である。
一般式(I-1)、(I-2)中、R2は重合性基である、オキセタニル基、エポキシ基、アクリレート基、メタクリレート基であることが好ましく、オキセタニル基、エポキシ基であることがより好ましい。 In general formulas (I-1) and (I-2), n is preferably 1 to 500, more preferably 3 to 250, and particularly preferably 5 to 200.
In general formulas (I-1) and (I-2), m is preferably 3 to 200, more preferably 5 to 150, and particularly preferably 10 to 100.
In general formulas (I-1) and (I-2), L 11 and L 12 are each independently a single bond, a functional group described below, or two hydrogen atoms removed from a functional group described below. The divalent linking group represented by For example, phenylene, naphthylene), alkenylene group (carbon number 2 to 20, for example, ethenylene, propenylene), alkynylene group (carbon number 2 to 20, for example, ethynylene, propynylene), metallocenylene group (for example, ferrocene), —CO—N (R 101 ) -, - CO-O - , - SO 2 -N (R 102) -, - SO 2 -O -, - N (R 103) -CO-N (R 104) -, SO 2 -, - SO -, - S -, - O -, - CO -, - N (R 105) -, heterylene group (carbon number 1 to 26, for example 6-chloro-1,3,5-triazyl - 2,4-diyl group, pyrimidine-2,4-diyl group) or a combination of one or more thereof represents a linking group having 0 to 100 carbon atoms, preferably 1 to 20 carbon atoms.
However, L 12 contains at least an alkylene group having 2 to 20 carbon atoms. The alkylene group contained in L 12 preferably has 3 to 18 carbon atoms, more preferably 4 to 16 carbon atoms, and particularly preferably 5 to 15 carbon atoms.
R 101 , R 102 , R 103 , R 104 , and R 105 each independently represent a hydrogen atom, a substituted or unsubstituted alkyl group, or a substituted or unsubstituted aryl group. One or more linking groups represented by L 11 and L 12 may be present, and a plurality (preferably two) of the linking groups may be bonded to form a ring.
In general formulas (I-1) and (I-2), R 1 is preferably independently a hydrogen atom or a methyl group from the viewpoint of versatility.
In general formulas (I-1) and (I-2), M is preferably a mesogenic group having 6 to 50 carbon atoms, more preferably a mesogenic group having 6 to 40 carbon atoms, Particular preference is given to 30 mesogenic groups. A preferable range of the mesogenic group having 6 to 50 carbon atoms represented by M is a preferable range of the above -Cy 1 -L 2- (Cy 2 -L 3 ) n -Cy 3 -L 4-in the general formula (X). It is the same.
In general formulas (I-1) and (I-2), R 2 is preferably a polymerizable group, such as an oxetanyl group, an epoxy group, an acrylate group, or a methacrylate group, and preferably an oxetanyl group or an epoxy group. More preferred.
一般式(I-1)、(I-2)中、mは3~200であることが好ましく、5~150であることがより好ましく、10~100であることが特に好ましい。
一般式(I-1)、(I-2)中、L11、L12は各々独立に、単結合、下記に記載する官能基または下記に記載する官能基から水素原子を2つ除去してなる2価の連結基を表すことが好ましく、特に限定は無いが、好ましくはアルキレン基(炭素数2~20、例えばメチレン、エチレン、プロピレン、ブチレン、ペンチレン)、アリーレン基(炭素数6~26、例えばフェニレン、ナフチレン)、アルケニレン基(炭素数2~20、例えばエテニレン、プロペニレン)、アルキニレン基(炭素数2~20、例えばエチニレン、プロピニレン)、メタロセニレン基(例えばフェロセン)、-CO-N(R101)-、-CO-O-、-SO2-N(R102)-、-SO2-O-、-N(R103)-CO-N(R104)-、-SO2-、-SO-、-S-、-O-、-CO-、-N(R105)-、ヘテリレン基(炭素数1~26、例えば6-クロロ-1,3,5-トリアジル-2,4-ジイル基、ピリミジン-2,4-ジイル基)を1つまたはそれ以上組み合わせて構成される炭素数0~100以下、好ましくは1以上20以下の連結基を表す。
ただし、L12は少なくとも炭素数2~20のアルキレン基を含む。L12が含むアルキレン基の炭素数は3~18であることが好ましく、4~16であることがより好ましく、5~15であることが特に好ましい。
上記、R101、R102、R103、R104、R105は、各々独立に、水素原子、置換または無置換のアルキル基、置換または無置換のアリール基を表す。また、L11、L12で表される連結基は、1つ以上複数個存在していてもよく、複数個(好ましくは2つ)が結合して環を形成してもよい。
一般式(I-1)、(I-2)中、R1はそれぞれ独立に水素原子、メチル基であることが汎用性の観点から好ましい。
一般式(I-1)、(I-2)中、Mは炭素数6~50のメソゲン基であることが好ましく、炭素数6~40のメソゲン基であることがより好ましく、炭素数6~30のメソゲン基であることが特に好ましい。Mが表す炭素数6~50のメソゲン基の好ましい範囲は、前記一般式(X)中の前記-Cy1-L2-(Cy2-L3)n-Cy3-L4-の好ましい範囲と同様である。
一般式(I-1)、(I-2)中、R2は重合性基である、オキセタニル基、エポキシ基、アクリレート基、メタクリレート基であることが好ましく、オキセタニル基、エポキシ基であることがより好ましい。 In general formulas (I-1) and (I-2), n is preferably 1 to 500, more preferably 3 to 250, and particularly preferably 5 to 200.
In general formulas (I-1) and (I-2), m is preferably 3 to 200, more preferably 5 to 150, and particularly preferably 10 to 100.
In general formulas (I-1) and (I-2), L 11 and L 12 are each independently a single bond, a functional group described below, or two hydrogen atoms removed from a functional group described below. The divalent linking group represented by For example, phenylene, naphthylene), alkenylene group (
However, L 12 contains at least an alkylene group having 2 to 20 carbon atoms. The alkylene group contained in L 12 preferably has 3 to 18 carbon atoms, more preferably 4 to 16 carbon atoms, and particularly preferably 5 to 15 carbon atoms.
R 101 , R 102 , R 103 , R 104 , and R 105 each independently represent a hydrogen atom, a substituted or unsubstituted alkyl group, or a substituted or unsubstituted aryl group. One or more linking groups represented by L 11 and L 12 may be present, and a plurality (preferably two) of the linking groups may be bonded to form a ring.
In general formulas (I-1) and (I-2), R 1 is preferably independently a hydrogen atom or a methyl group from the viewpoint of versatility.
In general formulas (I-1) and (I-2), M is preferably a mesogenic group having 6 to 50 carbon atoms, more preferably a mesogenic group having 6 to 40 carbon atoms, Particular preference is given to 30 mesogenic groups. A preferable range of the mesogenic group having 6 to 50 carbon atoms represented by M is a preferable range of the above -Cy 1 -L 2- (Cy 2 -L 3 ) n -Cy 3 -L 4-in the general formula (X). It is the same.
In general formulas (I-1) and (I-2), R 2 is preferably a polymerizable group, such as an oxetanyl group, an epoxy group, an acrylate group, or a methacrylate group, and preferably an oxetanyl group or an epoxy group. More preferred.
本発明のブロック共重合体は、前記アルキレンオキサイド鎖を含むポリマー成分(A)および前記炭素数2~20のアルキレン鎖を含むポリマー成分(B)の共重合割合(数平均分子量比)は、65:35~1:99であることが好ましく、55:45~5:95であることがより好ましく、45:55~10:90であることが特に好ましい。
The block copolymer of the present invention has a copolymerization ratio (number average molecular weight ratio) of the polymer component (A) containing an alkylene oxide chain and the polymer component (B) containing an alkylene chain having 2 to 20 carbon atoms is 65. : 35 to 1:99 is preferable, 55:45 to 5:95 is more preferable, and 45:55 to 10:90 is particularly preferable.
[ミクロ相分離構造膜の製造方法]
本発明のミクロ相分離構造膜の製造方法は、(3)本発明のブロック共重合体を前記支持体上で熱、光、溶媒蒸気または電場により相分離を促進させ、ミクロ相分離構造を形成する工程を含むことを特徴とする。ミクロ相分離構造を形成するための、本発明のブロック共重合体への熱、光、溶媒蒸気または電場により相分離を促進させる方法としては、加熱により相分離を促進する方法が好ましい。 [Production method of micro phase separation structure membrane]
The method for producing a microphase-separated structure film of the present invention includes (3) forming the microphase-separated structure by promoting phase separation of the block copolymer of the present invention on the support by heat, light, solvent vapor or electric field. Including the step of: As a method of promoting phase separation by heat, light, solvent vapor or electric field to the block copolymer of the present invention for forming a microphase separation structure, a method of promoting phase separation by heating is preferable.
本発明のミクロ相分離構造膜の製造方法は、(3)本発明のブロック共重合体を前記支持体上で熱、光、溶媒蒸気または電場により相分離を促進させ、ミクロ相分離構造を形成する工程を含むことを特徴とする。ミクロ相分離構造を形成するための、本発明のブロック共重合体への熱、光、溶媒蒸気または電場により相分離を促進させる方法としては、加熱により相分離を促進する方法が好ましい。 [Production method of micro phase separation structure membrane]
The method for producing a microphase-separated structure film of the present invention includes (3) forming the microphase-separated structure by promoting phase separation of the block copolymer of the present invention on the support by heat, light, solvent vapor or electric field. Including the step of: As a method of promoting phase separation by heat, light, solvent vapor or electric field to the block copolymer of the present invention for forming a microphase separation structure, a method of promoting phase separation by heating is preferable.
図5に、ミクロ相分離構造膜の製造装置の一例の概略図を示す。図5では、支持体21を送り出しロール11から送り出し、塗布装置12により支持体上にモノマー含有組成物22が塗布され、前記モノマー含有組成物を前記支持体上に適用する工程が行われる。
支持体上に設けられたモノマー含有組成物は、乾燥ゾーン13を経て、第一の熟成ゾーン14で、前記支持体上で前記モノマー含有組成物をリビング重合させる工程が行われる。
その後、第二の熟成ゾーン15にて、前記支持体上でミクロ相分離構造を形成する工程が行われ、前記モノマー含有組成物は高速相分離される。
その後、UV照射ゾーン16にて、ミクロ相分離構造が固定化される。
その後、剥ぎ取りロール17により、支持体21からミクロ相分離構造膜が剥ぎ取られる。
なお、支持体21は、巻取りロール18で巻取られてもよい。
以下、各工程の好ましい態様を説明する。 FIG. 5 shows a schematic diagram of an example of an apparatus for producing a microphase separation structure membrane. In FIG. 5, the step of feeding thesupport 21 from the feed roll 11, applying the monomer-containing composition 22 onto the support by the coating device 12, and applying the monomer-containing composition onto the support is performed.
The monomer-containing composition provided on the support is subjected to living polymerization of the monomer-containing composition on the support in the first agingzone 14 after passing through the drying zone 13.
Thereafter, in the second agingzone 15, a step of forming a microphase separation structure on the support is performed, and the monomer-containing composition is subjected to high-speed phase separation.
Thereafter, the micro phase separation structure is fixed in theUV irradiation zone 16.
Thereafter, the micro phase separation structure film is peeled off from thesupport 21 by the peeling roll 17.
Thesupport 21 may be wound up by the winding roll 18.
Hereinafter, preferred embodiments of each step will be described.
支持体上に設けられたモノマー含有組成物は、乾燥ゾーン13を経て、第一の熟成ゾーン14で、前記支持体上で前記モノマー含有組成物をリビング重合させる工程が行われる。
その後、第二の熟成ゾーン15にて、前記支持体上でミクロ相分離構造を形成する工程が行われ、前記モノマー含有組成物は高速相分離される。
その後、UV照射ゾーン16にて、ミクロ相分離構造が固定化される。
その後、剥ぎ取りロール17により、支持体21からミクロ相分離構造膜が剥ぎ取られる。
なお、支持体21は、巻取りロール18で巻取られてもよい。
以下、各工程の好ましい態様を説明する。 FIG. 5 shows a schematic diagram of an example of an apparatus for producing a microphase separation structure membrane. In FIG. 5, the step of feeding the
The monomer-containing composition provided on the support is subjected to living polymerization of the monomer-containing composition on the support in the first aging
Thereafter, in the second aging
Thereafter, the micro phase separation structure is fixed in the
Thereafter, the micro phase separation structure film is peeled off from the
The
Hereinafter, preferred embodiments of each step will be described.
<ミクロ相分離構造を形成する工程>
(反応時間)
本発明のミクロ相分離構造膜の製造方法は、前記(3)本発明のブロック共重合体を前記支持体上で加熱して、ミクロ相分離構造を形成する工程の時間は、5~1000秒であることが好ましく、10~600秒であることがより好ましく、30~200秒であることが特に好ましい。 <Step of forming a microphase separation structure>
(Reaction time)
The method for producing a microphase-separated structure film of the present invention is such that (3) the step of heating the block copolymer of the present invention on the support to form a microphase-separated structure is 5 to 1000 seconds. It is preferably 10 to 600 seconds, more preferably 30 to 200 seconds.
(反応時間)
本発明のミクロ相分離構造膜の製造方法は、前記(3)本発明のブロック共重合体を前記支持体上で加熱して、ミクロ相分離構造を形成する工程の時間は、5~1000秒であることが好ましく、10~600秒であることがより好ましく、30~200秒であることが特に好ましい。 <Step of forming a microphase separation structure>
(Reaction time)
The method for producing a microphase-separated structure film of the present invention is such that (3) the step of heating the block copolymer of the present invention on the support to form a microphase-separated structure is 5 to 1000 seconds. It is preferably 10 to 600 seconds, more preferably 30 to 200 seconds.
(反応温度)
本発明のミクロ相分離構造膜の製造方法は、前記(3)本発明のブロック共重合体を前記支持体上で加熱して、ミクロ相分離構造を形成する工程の加熱開始温度が40~250℃であることが好ましく、より好ましくは50℃~200℃であり、更に好ましくは60℃~190℃である。
前記(3)工程における加熱方法としては特に制限はなく、公知の方法を用いることができ、例えば前記支持体をホットプレート上に載せて、前記モノマー含有組成物を前記支持体ごと加熱する方法などを挙げることができる。
なお、加熱開始後は、最終的に後述する(4)工程の開始温度(活性放射線の照射温度)まで降温することが好ましい。加熱開始後からUV照射温度までの降温速度は、1~100℃/分であることが好ましく、5~80℃/分であることがより好ましく、10~50℃/分であることが特に好ましい。 (Reaction temperature)
The method for producing a microphase-separated structure film of the present invention is such that (3) the step of heating the block copolymer of the present invention on the support to form a microphase-separated structure has a heating start temperature of 40 to 250. It is preferably 0 ° C., more preferably 50 ° C. to 200 ° C., and still more preferably 60 ° C. to 190 ° C.
There is no restriction | limiting in particular as a heating method in the said (3) process, A well-known method can be used, for example, the said support body is mounted on a hotplate and the said monomer containing composition is heated with the said support body etc. Can be mentioned.
After the start of heating, it is preferable that the temperature is finally lowered to the start temperature (irradiation temperature of active radiation) of the step (4) described later. The rate of temperature decrease from the start of heating to the UV irradiation temperature is preferably 1 to 100 ° C./min, more preferably 5 to 80 ° C./min, and particularly preferably 10 to 50 ° C./min. .
本発明のミクロ相分離構造膜の製造方法は、前記(3)本発明のブロック共重合体を前記支持体上で加熱して、ミクロ相分離構造を形成する工程の加熱開始温度が40~250℃であることが好ましく、より好ましくは50℃~200℃であり、更に好ましくは60℃~190℃である。
前記(3)工程における加熱方法としては特に制限はなく、公知の方法を用いることができ、例えば前記支持体をホットプレート上に載せて、前記モノマー含有組成物を前記支持体ごと加熱する方法などを挙げることができる。
なお、加熱開始後は、最終的に後述する(4)工程の開始温度(活性放射線の照射温度)まで降温することが好ましい。加熱開始後からUV照射温度までの降温速度は、1~100℃/分であることが好ましく、5~80℃/分であることがより好ましく、10~50℃/分であることが特に好ましい。 (Reaction temperature)
The method for producing a microphase-separated structure film of the present invention is such that (3) the step of heating the block copolymer of the present invention on the support to form a microphase-separated structure has a heating start temperature of 40 to 250. It is preferably 0 ° C., more preferably 50 ° C. to 200 ° C., and still more preferably 60 ° C. to 190 ° C.
There is no restriction | limiting in particular as a heating method in the said (3) process, A well-known method can be used, for example, the said support body is mounted on a hotplate and the said monomer containing composition is heated with the said support body etc. Can be mentioned.
After the start of heating, it is preferable that the temperature is finally lowered to the start temperature (irradiation temperature of active radiation) of the step (4) described later. The rate of temperature decrease from the start of heating to the UV irradiation temperature is preferably 1 to 100 ° C./min, more preferably 5 to 80 ° C./min, and particularly preferably 10 to 50 ° C./min. .
<ミクロ相分離構造を固定化させる工程>
本発明のミクロ相分離構造膜の製造方法は、さらに(4)前記ミクロ相分離構造を有するブロック共重合体を前記支持体上で架橋または重合させてミクロ相分離構造を固定化させる工程を含むことが好ましい。
前記ミクロ相分離構造を有するブロック共重合体を前記支持体上で架橋または重合させてミクロ相分離構造を固定化させる工程の方法としては特に制限はないが、前記ミクロ相分離構造を有するブロック共重合体への活性放射線の照射であることが好ましく、UV照射であることがより好ましい。 <Step of immobilizing the microphase separation structure>
The method for producing a microphase separation structure membrane of the present invention further includes (4) a step of crosslinking or polymerizing the block copolymer having the microphase separation structure on the support to fix the microphase separation structure. It is preferable.
There is no particular limitation on the method of fixing the microphase separation structure by crosslinking or polymerizing the block copolymer having the microphase separation structure on the support, but the block copolymer having the microphase separation structure is not limited. It is preferable that the polymer is irradiated with actinic radiation, and UV irradiation is more preferable.
本発明のミクロ相分離構造膜の製造方法は、さらに(4)前記ミクロ相分離構造を有するブロック共重合体を前記支持体上で架橋または重合させてミクロ相分離構造を固定化させる工程を含むことが好ましい。
前記ミクロ相分離構造を有するブロック共重合体を前記支持体上で架橋または重合させてミクロ相分離構造を固定化させる工程の方法としては特に制限はないが、前記ミクロ相分離構造を有するブロック共重合体への活性放射線の照射であることが好ましく、UV照射であることがより好ましい。 <Step of immobilizing the microphase separation structure>
The method for producing a microphase separation structure membrane of the present invention further includes (4) a step of crosslinking or polymerizing the block copolymer having the microphase separation structure on the support to fix the microphase separation structure. It is preferable.
There is no particular limitation on the method of fixing the microphase separation structure by crosslinking or polymerizing the block copolymer having the microphase separation structure on the support, but the block copolymer having the microphase separation structure is not limited. It is preferable that the polymer is irradiated with actinic radiation, and UV irradiation is more preferable.
(活性放射線の照射)
本発明のミクロ相分離構造膜の製造方法は、(4)前記ミクロ相分離構造を有するブロック共重合体を前記支持体上で架橋または重合させてミクロ相分離構造を固定化させる工程の活性放射線の照射温度は、40~200℃であることが好ましく、より好ましくは50℃~200℃であり、更に好ましくは60℃~200℃である。
本発明のミクロ相分離構造膜の製造方法は、前記活性放射線の照射が、50~2000mJ/cm2であることが好ましく、100~1500mJ/cm2であることが好ましく、200~1000mJ/cm2であることが特に好ましい。
前記(4)工程における活性放射線の照射装置としては特に制限はなく、公知の装置を用いることができ、例えばHOYA社製 EXECURE3000などを挙げることができる。
なお、活性放射線の照射後は、最終的に室温まで放冷してミクロ相分離構造膜を得ることが好ましい。 (Irradiation with actinic radiation)
The method for producing a microphase-separated structure film of the present invention comprises (4) actinic radiation in the step of immobilizing the microphase-separated structure by crosslinking or polymerizing the block copolymer having the microphase-separated structure on the support. The irradiation temperature is preferably 40 to 200 ° C., more preferably 50 to 200 ° C., and still more preferably 60 to 200 ° C.
Method for producing a microphase-separated structure membrane of the present invention, the irradiation of the active radiation is preferably from 50 ~ 2000mJ / cm 2, is preferably 100 ~ 1500mJ / cm 2, 200 ~ 1000mJ /cm 2 It is particularly preferred that
There is no restriction | limiting in particular as an irradiation apparatus of the actinic radiation in the said (4) process, A well-known apparatus can be used, for example, EXERE3000 by HOYA etc. can be mentioned.
In addition, after irradiation of actinic radiation, it is preferable to cool to room temperature finally and to obtain a micro phase-separation structure film.
本発明のミクロ相分離構造膜の製造方法は、(4)前記ミクロ相分離構造を有するブロック共重合体を前記支持体上で架橋または重合させてミクロ相分離構造を固定化させる工程の活性放射線の照射温度は、40~200℃であることが好ましく、より好ましくは50℃~200℃であり、更に好ましくは60℃~200℃である。
本発明のミクロ相分離構造膜の製造方法は、前記活性放射線の照射が、50~2000mJ/cm2であることが好ましく、100~1500mJ/cm2であることが好ましく、200~1000mJ/cm2であることが特に好ましい。
前記(4)工程における活性放射線の照射装置としては特に制限はなく、公知の装置を用いることができ、例えばHOYA社製 EXECURE3000などを挙げることができる。
なお、活性放射線の照射後は、最終的に室温まで放冷してミクロ相分離構造膜を得ることが好ましい。 (Irradiation with actinic radiation)
The method for producing a microphase-separated structure film of the present invention comprises (4) actinic radiation in the step of immobilizing the microphase-separated structure by crosslinking or polymerizing the block copolymer having the microphase-separated structure on the support. The irradiation temperature is preferably 40 to 200 ° C., more preferably 50 to 200 ° C., and still more preferably 60 to 200 ° C.
Method for producing a microphase-separated structure membrane of the present invention, the irradiation of the active radiation is preferably from 50 ~ 2000mJ / cm 2, is preferably 100 ~ 1500mJ / cm 2, 200 ~ 1000mJ /
There is no restriction | limiting in particular as an irradiation apparatus of the actinic radiation in the said (4) process, A well-known apparatus can be used, for example, EXERE3000 by HOYA etc. can be mentioned.
In addition, after irradiation of actinic radiation, it is preferable to cool to room temperature finally and to obtain a micro phase-separation structure film.
<ミクロ相分離構造膜を剥ぎ取る工程>
本発明のミクロ相分離構造膜の製造方法は、前記ミクロ相分離構造膜を、前記支持体から剥ぎ取る工程を含むことが好ましい。
前記ミクロ相分離構造膜を、前記支持体から剥ぎ取る方法としては特に制限はない。 <Step of peeling the micro phase separation structure membrane>
The method for producing a microphase separation structure membrane of the present invention preferably includes a step of peeling the microphase separation structure membrane from the support.
There is no restriction | limiting in particular as a method of peeling the said micro phase-separation structure film | membrane from the said support body.
本発明のミクロ相分離構造膜の製造方法は、前記ミクロ相分離構造膜を、前記支持体から剥ぎ取る工程を含むことが好ましい。
前記ミクロ相分離構造膜を、前記支持体から剥ぎ取る方法としては特に制限はない。 <Step of peeling the micro phase separation structure membrane>
The method for producing a microphase separation structure membrane of the present invention preferably includes a step of peeling the microphase separation structure membrane from the support.
There is no restriction | limiting in particular as a method of peeling the said micro phase-separation structure film | membrane from the said support body.
[ミクロ相分離構造膜]
本発明のミクロ相分離構造膜は、本発明のミクロ相分離構造膜の製造方法で製造されたことを特徴とする。ここで、本明細書中、原子間力顕微鏡(AFMとも言う)で観察した場合に、ラメラ構造やシリンダー構造が確認できたときは「ミクロ相分離構造」を有していることとみなす。
本発明のミクロ相分離構造膜の製造方法で得られるミクロ相分離構造膜は、基板上に形成されるが、基板から剥離して用いてもよい。
前記ミクロ相分離構造膜の厚みが、1~2000nmであることが好ましい。用途によって前記ミクロ相分離構造膜のより好ましい厚みは異なるが、例えば1~500nmであることがより好ましい。
前記ミクロ相分離構造膜は、シリンダー型ミクロ相分離構造膜であることが好ましい。シリンダー型ミクロ相分離構造膜とは、前記アルキレンオキサイド鎖を含むポリマー成分が固定されたシリンダーが、面内に多数並んでいる膜のことを言い、各シリンダーは等間隔で整列していることが好ましい。各シリンダーの高さは、1~2000nmであることが好ましく、1~500nmであることがより好ましい。各シリンダーの直径は、1~1000nmであることが好ましく、1~200nmであることがより好ましい。 [Micro phase separation structure membrane]
The microphase-separated structure film of the present invention is manufactured by the method for manufacturing a microphase-separated structure film of the present invention. Here, in this specification, when a lamellar structure or a cylinder structure can be confirmed when observed with an atomic force microscope (also referred to as AFM), it is regarded as having a “microphase separation structure”.
The microphase separation structure film obtained by the method for producing a microphase separation structure film of the present invention is formed on a substrate, but may be used after peeling from the substrate.
The thickness of the microphase separation structure film is preferably 1 to 2000 nm. Although the more preferable thickness of the microphase-separated structure membrane varies depending on the application, it is more preferably 1 to 500 nm, for example.
The microphase separation structure film is preferably a cylinder type microphase separation structure film. The cylinder type micro phase separation structure membrane means a membrane in which a large number of cylinders to which the polymer component containing the alkylene oxide chain is fixed are arranged in a plane, and each cylinder is aligned at equal intervals. preferable. The height of each cylinder is preferably 1 to 2000 nm, and more preferably 1 to 500 nm. The diameter of each cylinder is preferably 1 to 1000 nm, and more preferably 1 to 200 nm.
本発明のミクロ相分離構造膜は、本発明のミクロ相分離構造膜の製造方法で製造されたことを特徴とする。ここで、本明細書中、原子間力顕微鏡(AFMとも言う)で観察した場合に、ラメラ構造やシリンダー構造が確認できたときは「ミクロ相分離構造」を有していることとみなす。
本発明のミクロ相分離構造膜の製造方法で得られるミクロ相分離構造膜は、基板上に形成されるが、基板から剥離して用いてもよい。
前記ミクロ相分離構造膜の厚みが、1~2000nmであることが好ましい。用途によって前記ミクロ相分離構造膜のより好ましい厚みは異なるが、例えば1~500nmであることがより好ましい。
前記ミクロ相分離構造膜は、シリンダー型ミクロ相分離構造膜であることが好ましい。シリンダー型ミクロ相分離構造膜とは、前記アルキレンオキサイド鎖を含むポリマー成分が固定されたシリンダーが、面内に多数並んでいる膜のことを言い、各シリンダーは等間隔で整列していることが好ましい。各シリンダーの高さは、1~2000nmであることが好ましく、1~500nmであることがより好ましい。各シリンダーの直径は、1~1000nmであることが好ましく、1~200nmであることがより好ましい。 [Micro phase separation structure membrane]
The microphase-separated structure film of the present invention is manufactured by the method for manufacturing a microphase-separated structure film of the present invention. Here, in this specification, when a lamellar structure or a cylinder structure can be confirmed when observed with an atomic force microscope (also referred to as AFM), it is regarded as having a “microphase separation structure”.
The microphase separation structure film obtained by the method for producing a microphase separation structure film of the present invention is formed on a substrate, but may be used after peeling from the substrate.
The thickness of the microphase separation structure film is preferably 1 to 2000 nm. Although the more preferable thickness of the microphase-separated structure membrane varies depending on the application, it is more preferably 1 to 500 nm, for example.
The microphase separation structure film is preferably a cylinder type microphase separation structure film. The cylinder type micro phase separation structure membrane means a membrane in which a large number of cylinders to which the polymer component containing the alkylene oxide chain is fixed are arranged in a plane, and each cylinder is aligned at equal intervals. preferable. The height of each cylinder is preferably 1 to 2000 nm, and more preferably 1 to 500 nm. The diameter of each cylinder is preferably 1 to 1000 nm, and more preferably 1 to 200 nm.
(用途)
本発明のミクロ相分離構造膜の製造方法で得られるミクロ相分離構造膜は、光・電子機能材料(例えば輝度向上膜)、エネルギー関連材料、表面修飾材料、パターンドメディアのような高密度記録材料、種々のナノフィルター(透過膜、限外ろ過膜、ナノリアクター)、異方性イオン伝導膜、異方性導電膜、等として有用な配向の制御されたブロック共重合体からなるミクロ相分離構造膜である。
このようなミクロ相分離構造膜は、シリンダー構造部分を除去することにより、多孔質構造体として用いることもできる。ミクロ相分離構造膜から得られる多孔質構造体は、燃料電池用高分子電解質、イオン交換樹脂、マイクロリアクター用薄膜、蛋白質の分離膜、有機ゼオライトや種々のピラー用高配向用テンプレートなどの異方性イオン伝導材料として利用できる。
このようなミクロ相分離構造膜は、シリンダー構造部分を除去して、別の物質を導入することもできる。 (Use)
The microphase-separated structure film obtained by the method for producing a microphase-separated structure film of the present invention is a high-density recording such as an optical / electronic functional material (for example, a brightness enhancement film), an energy-related material, a surface modifying material, and a patterned medium Microphase separation consisting of controlled-oriented block copolymers useful as materials, various nanofilters (permeation membranes, ultrafiltration membranes, nanoreactors), anisotropic ion conductive membranes, anisotropic conductive films, etc. It is a structural film.
Such a microphase separation structure membrane can also be used as a porous structure by removing the cylinder structure portion. Porous structures obtained from microphase separation structure membranes are anisotropic, such as polymer electrolytes for fuel cells, ion exchange resins, thin films for microreactors, protein separation membranes, organic zeolites, and templates for various pillars. It can be used as a conductive ion conductive material.
Such a microphase-separated structure film can also introduce another substance by removing the cylinder structure portion.
本発明のミクロ相分離構造膜の製造方法で得られるミクロ相分離構造膜は、光・電子機能材料(例えば輝度向上膜)、エネルギー関連材料、表面修飾材料、パターンドメディアのような高密度記録材料、種々のナノフィルター(透過膜、限外ろ過膜、ナノリアクター)、異方性イオン伝導膜、異方性導電膜、等として有用な配向の制御されたブロック共重合体からなるミクロ相分離構造膜である。
このようなミクロ相分離構造膜は、シリンダー構造部分を除去することにより、多孔質構造体として用いることもできる。ミクロ相分離構造膜から得られる多孔質構造体は、燃料電池用高分子電解質、イオン交換樹脂、マイクロリアクター用薄膜、蛋白質の分離膜、有機ゼオライトや種々のピラー用高配向用テンプレートなどの異方性イオン伝導材料として利用できる。
このようなミクロ相分離構造膜は、シリンダー構造部分を除去して、別の物質を導入することもできる。 (Use)
The microphase-separated structure film obtained by the method for producing a microphase-separated structure film of the present invention is a high-density recording such as an optical / electronic functional material (for example, a brightness enhancement film), an energy-related material, a surface modifying material, and a patterned medium Microphase separation consisting of controlled-oriented block copolymers useful as materials, various nanofilters (permeation membranes, ultrafiltration membranes, nanoreactors), anisotropic ion conductive membranes, anisotropic conductive films, etc. It is a structural film.
Such a microphase separation structure membrane can also be used as a porous structure by removing the cylinder structure portion. Porous structures obtained from microphase separation structure membranes are anisotropic, such as polymer electrolytes for fuel cells, ion exchange resins, thin films for microreactors, protein separation membranes, organic zeolites, and templates for various pillars. It can be used as a conductive ion conductive material.
Such a microphase-separated structure film can also introduce another substance by removing the cylinder structure portion.
以下に実施例と比較例を挙げて本発明の特徴をさらに具体的に説明する。以下の実施例に示す材料、使用量、割合、処理内容、処理手順等は、本発明の趣旨を逸脱しない限り適宜変更することができる。したがって、本発明の範囲は以下に示す具体例により限定的に解釈されるべきものではない。
Hereinafter, the features of the present invention will be described more specifically with reference to examples and comparative examples. The materials, amounts used, ratios, processing details, processing procedures, and the like shown in the following examples can be changed as appropriate without departing from the spirit of the present invention. Therefore, the scope of the present invention should not be construed as being limited by the specific examples shown below.
以下の実施例では、得られるリビング重合体(ブロック共重合体)に残存モノマーや残存開始剤(開始剤が高分子の場合)が混入するために図1のようなGPCチャートが得られるが、その際にリビング重合体(ブロック共重合体)のMw(重量平均分子量)、Mn(数平均分子量)、分子量分布(Mw/Mn)は矢印のGPC測定エリアの値を記載している。
また、モノマー消費率は、モノマー添加量に対して、1H-NMRの面積比から求められる残存モノマー量の割合(モル比)から計算した。 In the following examples, since the residual monomer and residual initiator (when the initiator is a polymer) are mixed into the resulting living polymer (block copolymer), a GPC chart as shown in FIG. 1 is obtained. At that time, Mw (weight average molecular weight), Mn (number average molecular weight), and molecular weight distribution (Mw / Mn) of the living polymer (block copolymer) describe the values in the GPC measurement area indicated by arrows.
The monomer consumption rate was calculated from the ratio (molar ratio) of the residual monomer amount determined from the area ratio of 1 H-NMR with respect to the monomer addition amount.
また、モノマー消費率は、モノマー添加量に対して、1H-NMRの面積比から求められる残存モノマー量の割合(モル比)から計算した。 In the following examples, since the residual monomer and residual initiator (when the initiator is a polymer) are mixed into the resulting living polymer (block copolymer), a GPC chart as shown in FIG. 1 is obtained. At that time, Mw (weight average molecular weight), Mn (number average molecular weight), and molecular weight distribution (Mw / Mn) of the living polymer (block copolymer) describe the values in the GPC measurement area indicated by arrows.
The monomer consumption rate was calculated from the ratio (molar ratio) of the residual monomer amount determined from the area ratio of 1 H-NMR with respect to the monomer addition amount.
まず、各実施例および比較例で使用した材料およびその調製方法を以下に示す。
First, the materials used in each Example and Comparative Example and their preparation methods are shown below.
<低分子RAFT剤の調製>
(低分子RAFT剤(1))
市販の低分子RAFT剤(1)として、4-シアノ-4-[(ドデシルスルファニル-チオカルボニル)スルファニル]ペンタノイック酸(アルドリッチ社製)を利用する。

<Preparation of low molecular weight RAFT agent>
(Low molecular weight RAFT agent (1))
As a commercially available low molecular weight RAFT agent (1), 4-cyano-4-[(dodecylsulfanyl-thiocarbonyl) sulfanyl] pentanoic acid (manufactured by Aldrich) is used.
(低分子RAFT剤(1))
市販の低分子RAFT剤(1)として、4-シアノ-4-[(ドデシルスルファニル-チオカルボニル)スルファニル]ペンタノイック酸(アルドリッチ社製)を利用する。
(Low molecular weight RAFT agent (1))
As a commercially available low molecular weight RAFT agent (1), 4-cyano-4-[(dodecylsulfanyl-thiocarbonyl) sulfanyl] pentanoic acid (manufactured by Aldrich) is used.
(低分子RAFT剤(2))
市販の低分子RAFT剤(2)として、2-シアノ-2-プロピルベンゾジチオエート(アルドリッチ社製)を利用する。

(Low molecular weight RAFT agent (2))
As a commercially available low molecular weight RAFT agent (2), 2-cyano-2-propylbenzodithioate (Aldrich) is used.
市販の低分子RAFT剤(2)として、2-シアノ-2-プロピルベンゾジチオエート(アルドリッチ社製)を利用する。
As a commercially available low molecular weight RAFT agent (2), 2-cyano-2-propylbenzodithioate (Aldrich) is used.
(低分子ATRP開始剤(3))
市販の低分子ATRP開始剤(3)として、エチル-α-ブロモイソブチレート(アルドリッチ社製)を利用する。

(Low molecular ATRP initiator (3))
Ethyl-α-bromoisobutyrate (manufactured by Aldrich) is used as a commercially available low-molecular ATRP initiator (3).
市販の低分子ATRP開始剤(3)として、エチル-α-ブロモイソブチレート(アルドリッチ社製)を利用する。
Ethyl-α-bromoisobutyrate (manufactured by Aldrich) is used as a commercially available low-molecular ATRP initiator (3).
<親水性部分の調製>
(親水性マクロRAFT剤A-1)
市販の親水性マクロRAFT剤A-1(ポリ(エチレングリコール)メチルエーテル(4-シアノ-4-ペンタノエート ドデシル トリチオカーボネート)(Mn5400、アルドリッチ社製))を利用する(分子量Mw8000、Mn7500、Mw/Mn=1.07:GPCポリスチレン換算)。

<Preparation of hydrophilic part>
(Hydrophilic macro RAFT agent A-1)
A commercially available hydrophilic macro RAFT agent A-1 (poly (ethylene glycol) methyl ether (4-cyano-4-pentanoate dodecyl trithiocarbonate) (Mn5400, manufactured by Aldrich)) is used (molecular weight Mw 8000, Mn 7500, Mw / Mn = 1.07: GPC polystyrene conversion).
(親水性マクロRAFT剤A-1)
市販の親水性マクロRAFT剤A-1(ポリ(エチレングリコール)メチルエーテル(4-シアノ-4-ペンタノエート ドデシル トリチオカーボネート)(Mn5400、アルドリッチ社製))を利用する(分子量Mw8000、Mn7500、Mw/Mn=1.07:GPCポリスチレン換算)。
<Preparation of hydrophilic part>
(Hydrophilic macro RAFT agent A-1)
A commercially available hydrophilic macro RAFT agent A-1 (poly (ethylene glycol) methyl ether (4-cyano-4-pentanoate dodecyl trithiocarbonate) (Mn5400, manufactured by Aldrich)) is used (molecular weight Mw 8000, Mn 7500, Mw / Mn = 1.07: GPC polystyrene conversion).
(親水性マクロRAFT剤A-2)
市販の親水性マクロRAFT剤A-2(ポリ(エチレングリコール)メチルエーテル(4-シアノ-4-(ドデシルスルファニルチオカルボニル)スルファニル)ペンタノエート)(Mn10000、アルドリッチ社製))を利用する。

(Hydrophilic macro RAFT agent A-2)
A commercially available hydrophilic macro RAFT agent A-2 (poly (ethylene glycol) methyl ether (4-cyano-4- (dodecylsulfanylthiocarbonyl) sulfanyl) pentanoate) (Mn10000, manufactured by Aldrich)) is used.
市販の親水性マクロRAFT剤A-2(ポリ(エチレングリコール)メチルエーテル(4-シアノ-4-(ドデシルスルファニルチオカルボニル)スルファニル)ペンタノエート)(Mn10000、アルドリッチ社製))を利用する。
A commercially available hydrophilic macro RAFT agent A-2 (poly (ethylene glycol) methyl ether (4-cyano-4- (dodecylsulfanylthiocarbonyl) sulfanyl) pentanoate) (Mn10000, manufactured by Aldrich)) is used.
(親水性マクロ開始剤A-3)
市販のポリ(エチレングリコール)メチルエーテル(Mn5000、アルドリッチ社製)30g、N,N’-ジメチルアミノピリジン0.81gを塩化メチレン180mlに溶解させ、2-ブロモイソラクサンブロミド(東京化成社製)1.52gを塩化メチレン20mlに溶解させた後に室温で滴下し、40℃で加熱環流して48時間攪拌した。
1規定の塩酸水溶液、0.5規定の炭酸水素ナトリウム水溶液、1規定の塩酸水溶液の順番に分液操作を施し、硫酸マグネシウムで乾燥後、ジエチルエーテルで再結晶した。
目的とするATRP親水性マクロ開始剤A-3を22g(分子量Mw8000、Mn7700、Mw/Mn=1.04:GPCポリスチレン換算)を得た。

(Hydrophilic macroinitiator A-3)
30 g of commercially available poly (ethylene glycol) methyl ether (Mn5000, manufactured by Aldrich) and 0.81 g of N, N′-dimethylaminopyridine were dissolved in 180 ml of methylene chloride, and 2-bromoisolacsan bromide (manufactured by Tokyo Chemical Industry Co., Ltd.) 1 .52 g was dissolved in 20 ml of methylene chloride and then added dropwise at room temperature, followed by heating at 40 ° C. and stirring for 48 hours.
Separation operations were carried out in the order of 1 N aqueous hydrochloric acid solution, 0.5 N aqueous sodium hydrogen carbonate solution and 1 N aqueous hydrochloric acid solution, dried over magnesium sulfate, and recrystallized from diethyl ether.
22 g (molecular weight Mw 8000, Mn 7700, Mw / Mn = 1.04: GPC polystyrene conversion) of the target ATRP hydrophilic macroinitiator A-3 were obtained.
市販のポリ(エチレングリコール)メチルエーテル(Mn5000、アルドリッチ社製)30g、N,N’-ジメチルアミノピリジン0.81gを塩化メチレン180mlに溶解させ、2-ブロモイソラクサンブロミド(東京化成社製)1.52gを塩化メチレン20mlに溶解させた後に室温で滴下し、40℃で加熱環流して48時間攪拌した。
1規定の塩酸水溶液、0.5規定の炭酸水素ナトリウム水溶液、1規定の塩酸水溶液の順番に分液操作を施し、硫酸マグネシウムで乾燥後、ジエチルエーテルで再結晶した。
目的とするATRP親水性マクロ開始剤A-3を22g(分子量Mw8000、Mn7700、Mw/Mn=1.04:GPCポリスチレン換算)を得た。
(Hydrophilic macroinitiator A-3)
30 g of commercially available poly (ethylene glycol) methyl ether (Mn5000, manufactured by Aldrich) and 0.81 g of N, N′-dimethylaminopyridine were dissolved in 180 ml of methylene chloride, and 2-bromoisolacsan bromide (manufactured by Tokyo Chemical Industry Co., Ltd.) 1 .52 g was dissolved in 20 ml of methylene chloride and then added dropwise at room temperature, followed by heating at 40 ° C. and stirring for 48 hours.
Separation operations were carried out in the order of 1 N aqueous hydrochloric acid solution, 0.5 N aqueous sodium hydrogen carbonate solution and 1 N aqueous hydrochloric acid solution, dried over magnesium sulfate, and recrystallized from diethyl ether.
22 g (molecular weight Mw 8000, Mn 7700, Mw / Mn = 1.04: GPC polystyrene conversion) of the target ATRP hydrophilic macroinitiator A-3 were obtained.
<疎水性モノマーの調製>
(疎水性モノマー合成例1)
Transactions of Materials Reseach Society of Japan, 28[3], 553-556, (2003)に記載の合成方法により液晶性モノマーB-1を合成した。

<Preparation of hydrophobic monomer>
(Hydrophobic monomer synthesis example 1)
Liquid crystalline monomer B-1 was synthesized by the synthesis method described in Transactions of Materials Research Society of Japan, 28 [3], 553-556, (2003).
(疎水性モノマー合成例1)
Transactions of Materials Reseach Society of Japan, 28[3], 553-556, (2003)に記載の合成方法により液晶性モノマーB-1を合成した。
(Hydrophobic monomer synthesis example 1)
Liquid crystalline monomer B-1 was synthesized by the synthesis method described in Transactions of Materials Research Society of Japan, 28 [3], 553-556, (2003).
(疎水性モノマー合成例2)
特開2008-127336号公報の実施例1に記載の合成法により液晶性モノマーB-2を合成した。

(Hydrophobic monomer synthesis example 2)
Liquid crystalline monomer B-2 was synthesized by the synthesis method described in Example 1 of JP-A-2008-127336.
特開2008-127336号公報の実施例1に記載の合成法により液晶性モノマーB-2を合成した。
Liquid crystalline monomer B-2 was synthesized by the synthesis method described in Example 1 of JP-A-2008-127336.
(疎水性モノマー合成例3)
特開2008-127336号公報の実施例4に記載の合成法において、用いるカルボン酸化合物10の置換基の鎖長を変更した以外は同様の合成法により液晶性モノマーB-3を合成した。

(Hydrophobic monomer synthesis example 3)
In the synthesis method described in Example 4 of JP-A-2008-127336, a liquid crystalline monomer B-3 was synthesized by the same synthesis method except that the chain length of the substituent of the carboxylic acid compound 10 used was changed.
特開2008-127336号公報の実施例4に記載の合成法において、用いるカルボン酸化合物10の置換基の鎖長を変更した以外は同様の合成法により液晶性モノマーB-3を合成した。
In the synthesis method described in Example 4 of JP-A-2008-127336, a liquid crystalline monomer B-3 was synthesized by the same synthesis method except that the chain length of the substituent of the carboxylic acid compound 10 used was changed.
(疎水性モノマー合成例4)
特開2010-116463号公報に記載の合成法に基づいて、液晶性モノマーB-4を合成した。

(Hydrophobic monomer synthesis example 4)
Liquid crystalline monomer B-4 was synthesized based on the synthesis method described in JP 2010-116463 A.
特開2010-116463号公報に記載の合成法に基づいて、液晶性モノマーB-4を合成した。
Liquid crystalline monomer B-4 was synthesized based on the synthesis method described in JP 2010-116463 A.
(疎水性モノマー合成例5)
以下に記載の合成法に従い、液晶性モノマーB-5を合成した。

(Hydrophobic monomer synthesis example 5)
Liquid crystalline monomer B-5 was synthesized according to the synthesis method described below.
以下に記載の合成法に従い、液晶性モノマーB-5を合成した。
Liquid crystalline monomer B-5 was synthesized according to the synthesis method described below.

アルデヒドc-1のアセトニトリル溶液(67mL)に対し、亜塩素酸ナトリウム(42.0mmol,3.80g)の水溶液(32mL)、リン酸二水素ナトリウム二水和物(6.0mmol,0.94g)の水溶液(8.2mL)、過酸化水素水(4.0mL)を加え、室温で12時間撹拌した。1N 塩酸水溶液を100mL加えた後に、ろ過した。残渣をメタノールで少量のアセトニトリルで洗浄することにより、カルボン酸d-1を定量的に得た。
メタンスルホニルクロリド(6.0mmol,0.46mL)のTHF溶液(3mL)にヒドロキノンモノメチルエーテル(7mg)を加え、内温を-5℃まで冷却した。そこに、カルボン酸d-1(5.5mmol,2.1g)とジイソプロピルエチルアミン(6.0mmol,1.1mL)のTHF溶液(6mL)を内温が0℃以上に上昇しないように滴下した。-5℃で30分撹拌した後、ジイソプロピルエチルアミン(6.0mmol,1.1mL)、4-エチルフェノールであるe-1(5.0mmol,0.82g)のTHF溶液(4mL)、DMAP(スパチュラ一杯)を加えた。その後、室温で2時間撹拌した。メタノール(5mL)を加えて反応を停止した後に、水と酢酸エチルを加えた。酢酸エチルで抽出した有機層を、ロータリーエバポレーターで溶媒を除去し液晶性モノマーB-5の粗生成物を得た。酢酸エチルとメタノールで再結晶を行い、液晶性モノマーB-5を78%の収率で得た。
1H-NMR(溶媒:CDCl3)δ(ppm):1.2(t,3H)、1.8-2.0(m,4H), 2.6(d,2H), 4.1-4.3(m,4H), 5.8(d,1H), 6.1(dd,1H), 6.4(d,1H), 6.9-7.0(m,2H), 7.1-7.2(m,2H), 7.2-7.3(m,2H), 7.3-7.4(m,2H), 8.1-8.2(m,2H), 8.2-8.3(m,2H)
An aqueous solution (32 mL) of sodium chlorite (42.0 mmol, 3.80 g), sodium dihydrogen phosphate dihydrate (6.0 mmol, 0.94 g) against an acetonitrile solution (67 mL) of aldehyde c-1 Aqueous solution (8.2 mL) and hydrogen peroxide (4.0 mL) were added, and the mixture was stirred at room temperature for 12 hours. After adding 100 mL of 1N hydrochloric acid aqueous solution, it filtered. The residue was washed with methanol with a small amount of acetonitrile to quantitatively obtain carboxylic acid d-1.
Hydroquinone monomethyl ether (7 mg) was added to a THF solution (3 mL) of methanesulfonyl chloride (6.0 mmol, 0.46 mL), and the internal temperature was cooled to −5 ° C. A THF solution (6 mL) of carboxylic acid d-1 (5.5 mmol, 2.1 g) and diisopropylethylamine (6.0 mmol, 1.1 mL) was added dropwise so that the internal temperature did not rise above 0 ° C. After stirring at −5 ° C. for 30 minutes, diisopropylethylamine (6.0 mmol, 1.1 mL), 4-ethylphenol e-1 (5.0 mmol, 0.82 g) in THF (4 mL), DMAP (spatula) I added a cup). Then, it stirred at room temperature for 2 hours. Methanol (5 mL) was added to stop the reaction, and water and ethyl acetate were added. From the organic layer extracted with ethyl acetate, the solvent was removed by a rotary evaporator to obtain a crude product of liquid crystalline monomer B-5. Recrystallization was performed with ethyl acetate and methanol to obtain liquid crystal monomer B-5 at a yield of 78%.
1 H-NMR (solvent: CDCl 3 ) δ (ppm): 1.2 (t, 3H), 1.8-2.0 (m, 4H), 2.6 (d, 2H), 4.1 4.3 (m, 4H), 5.8 (d, 1H), 6.1 (dd, 1H), 6.4 (d, 1H), 6.9-7.0 (m, 2H), 7 1-7.2 (m, 2H), 7.2-7.3 (m, 2H), 7.3-7.4 (m, 2H), 8.1-8.2 (m, 2H) , 8.2-8.3 (m, 2H)
[実施例1~48:支持体上でのRAFT重合]
<実施例1:リビング重合体合成例>
低分子RAFT剤(1)(0.0081g)、液晶性モノマーB-1(0.493g)、アゾビスイソブチロニトリル(以下AIBNと略、0.0028g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板上(2cm角)に塗布して、スピンコーター(MIKASA社製 SPINCOATER1H-D7)を用いて1500回転/分、30秒間回転させてスピンコートした。窒素フロー(0.5L/min)下、ホットプレート(PMC社製 DATAPLATE)上で下表に従い加熱した。その後、室温まで放冷し、リビング重合体1を得た(実施例1)。該フィルム状の実施例1のリビング重合体を重THFにて抽出して、1H-NMR(BRUKER-300MHZ)測定によりモノマー消費量とGPC(TOSOH社製 HLC-8220GPC)の分析を実施した。ここでMwとは重量平均分子量を、Mnとは数平均分子量、PDIはMw/Mnを表し、分子量分布の意味である。得られた結果を下記表に記載した。 [Examples 1 to 48: RAFT polymerization on a support]
<Example 1: Living polymer synthesis example>
Low molecular weight RAFT agent (1) (0.0081 g), liquid crystalline monomer B-1 (0.493 g), and azobisisobutyronitrile (hereinafter abbreviated as AIBN, 0.0028 g) were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied onto a glass substrate (2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds using a spin coater (SPINCATOR 1H-D7 manufactured by MIKASA). Under a nitrogen flow (0.5 L / min), heating was performed according to the table below on a hot plate (DATAPLATE manufactured by PMC). Thereafter, the mixture was allowed to cool to room temperature to obtain a living polymer 1 (Example 1). The film-like living polymer of Example 1 was extracted with deuterated THF, and monomer consumption and GPC (HLC-8220GPC manufactured by TOSOH) were analyzed by 1 H-NMR (BRUKER-300 MHZ) measurement. Here, Mw represents the weight average molecular weight, Mn represents the number average molecular weight, and PDI represents Mw / Mn, which means the molecular weight distribution. The results obtained are listed in the table below.
<実施例1:リビング重合体合成例>
低分子RAFT剤(1)(0.0081g)、液晶性モノマーB-1(0.493g)、アゾビスイソブチロニトリル(以下AIBNと略、0.0028g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板上(2cm角)に塗布して、スピンコーター(MIKASA社製 SPINCOATER1H-D7)を用いて1500回転/分、30秒間回転させてスピンコートした。窒素フロー(0.5L/min)下、ホットプレート(PMC社製 DATAPLATE)上で下表に従い加熱した。その後、室温まで放冷し、リビング重合体1を得た(実施例1)。該フィルム状の実施例1のリビング重合体を重THFにて抽出して、1H-NMR(BRUKER-300MHZ)測定によりモノマー消費量とGPC(TOSOH社製 HLC-8220GPC)の分析を実施した。ここでMwとは重量平均分子量を、Mnとは数平均分子量、PDIはMw/Mnを表し、分子量分布の意味である。得られた結果を下記表に記載した。 [Examples 1 to 48: RAFT polymerization on a support]
<Example 1: Living polymer synthesis example>
Low molecular weight RAFT agent (1) (0.0081 g), liquid crystalline monomer B-1 (0.493 g), and azobisisobutyronitrile (hereinafter abbreviated as AIBN, 0.0028 g) were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied onto a glass substrate (2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds using a spin coater (SPINCATOR 1H-D7 manufactured by MIKASA). Under a nitrogen flow (0.5 L / min), heating was performed according to the table below on a hot plate (DATAPLATE manufactured by PMC). Thereafter, the mixture was allowed to cool to room temperature to obtain a living polymer 1 (Example 1). The film-like living polymer of Example 1 was extracted with deuterated THF, and monomer consumption and GPC (HLC-8220GPC manufactured by TOSOH) were analyzed by 1 H-NMR (BRUKER-300 MHZ) measurement. Here, Mw represents the weight average molecular weight, Mn represents the number average molecular weight, and PDI represents Mw / Mn, which means the molecular weight distribution. The results obtained are listed in the table below.
<実施例2~5:リビング重合体合成例>
実施例1で使用した液晶性モノマーを等モル使用してB-1からB-2~5に変更する以外は合成例1と同様にしてリビング重合体を得た(実施例2~5)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 <Examples 2 to 5: Living polymer synthesis examples>
Living polymers were obtained in the same manner as in Synthesis Example 1 except that equimolar amounts of the liquid crystalline monomer used in Example 1 were used and B-1 was changed to B-2 to 5 (Examples 2 to 5). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
実施例1で使用した液晶性モノマーを等モル使用してB-1からB-2~5に変更する以外は合成例1と同様にしてリビング重合体を得た(実施例2~5)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 <Examples 2 to 5: Living polymer synthesis examples>
Living polymers were obtained in the same manner as in Synthesis Example 1 except that equimolar amounts of the liquid crystalline monomer used in Example 1 were used and B-1 was changed to B-2 to 5 (Examples 2 to 5). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.

<実施例6~10:リビング重合体合成例>
実施例1で使用した低分子RAFT剤(1)を等モル使用して低分子RAFT剤(2)に変更する以外は実施例1と同様にしてリビング重合体を得た(実施例6~10)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 <Examples 6 to 10: Living polymer synthesis examples>
Living polymers were obtained in the same manner as in Example 1 except that equimolar amounts of the low molecular weight RAFT agent (1) used in Example 1 were used to change to the low molecular weight RAFT agent (2) (Examples 6 to 10). ). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
実施例1で使用した低分子RAFT剤(1)を等モル使用して低分子RAFT剤(2)に変更する以外は実施例1と同様にしてリビング重合体を得た(実施例6~10)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 <Examples 6 to 10: Living polymer synthesis examples>
Living polymers were obtained in the same manner as in Example 1 except that equimolar amounts of the low molecular weight RAFT agent (1) used in Example 1 were used to change to the low molecular weight RAFT agent (2) (Examples 6 to 10). ). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.

<実施例11~22:ブロック共重合体合成例>
親水性マクロRAFT剤A-1(0.216g)、液晶性モノマーB-2(0.677g)、AIBN(0.0028g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板上(2cm角)に塗布して、スピンコーター(MIKASA社製 SPINCOATER1H-D7)を用いて1500回転/分、30秒間回転させてスピンコートした。窒素フロー(0.5L/min)下、ホットプレート(PMC社製 DATAPLATE)上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体11~22を得た(実施例11~22)。該フィルム状の実施例11~22のブロック共重合体11~22を重THFにて抽出して、1H-NMR(BRUKER-300MHZ)測定によりモノマー消費量とGPC(TOSOH社製 HLC-8220GPC)の分析を実施した。得られた結果を下記表に記載した。 <Examples 11 to 22: Examples of block copolymer synthesis>
Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g) and AIBN (0.0028 g) were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied onto a glass substrate (2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds using a spin coater (SPINCATOR 1H-D7 manufactured by MIKASA). Under a nitrogen flow (0.5 L / min), heating was performed according to the table below on a hot plate (DATAPLATE manufactured by PMC). Thereafter, the mixture was allowed to cool to room temperature to obtainblock copolymers 11 to 22 (Examples 11 to 22). The film-like block copolymers 11 to 22 of Examples 11 to 22 were extracted with deuterated THF, and monomer consumption and GPC (HLC-8220GPC manufactured by TOSOH) were measured by 1 H-NMR (BRUKER-300 MHZ) measurement. The analysis was conducted. The results obtained are listed in the table below.
親水性マクロRAFT剤A-1(0.216g)、液晶性モノマーB-2(0.677g)、AIBN(0.0028g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板上(2cm角)に塗布して、スピンコーター(MIKASA社製 SPINCOATER1H-D7)を用いて1500回転/分、30秒間回転させてスピンコートした。窒素フロー(0.5L/min)下、ホットプレート(PMC社製 DATAPLATE)上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体11~22を得た(実施例11~22)。該フィルム状の実施例11~22のブロック共重合体11~22を重THFにて抽出して、1H-NMR(BRUKER-300MHZ)測定によりモノマー消費量とGPC(TOSOH社製 HLC-8220GPC)の分析を実施した。得られた結果を下記表に記載した。 <Examples 11 to 22: Examples of block copolymer synthesis>
Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g) and AIBN (0.0028 g) were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied onto a glass substrate (2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds using a spin coater (SPINCATOR 1H-D7 manufactured by MIKASA). Under a nitrogen flow (0.5 L / min), heating was performed according to the table below on a hot plate (DATAPLATE manufactured by PMC). Thereafter, the mixture was allowed to cool to room temperature to obtain

<実施例23~31:ブロック共重合体合成例>
親水性マクロRAFT剤A-1(0.216g)、液晶性モノマーB-2(0.677g)、AIBN(0.0028g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板上(2cm角)に塗布して、1500回転/分、30秒間回転させてスピンコートした。大気下、上記ガラス基板をホットプレート上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体23~31を得た(実施例23~31)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 Examples 23 to 31: Examples of block copolymer synthesis
Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g) and AIBN (0.0028 g) were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied on a glass substrate (2 cm square) and spin-coated by rotating at 1500 rpm for 30 seconds. In the atmosphere, the glass substrate was heated on a hot plate according to the table below. Thereafter, the mixture was allowed to cool to room temperature to obtainblock copolymers 23 to 31 (Examples 23 to 31). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
親水性マクロRAFT剤A-1(0.216g)、液晶性モノマーB-2(0.677g)、AIBN(0.0028g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板上(2cm角)に塗布して、1500回転/分、30秒間回転させてスピンコートした。大気下、上記ガラス基板をホットプレート上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体23~31を得た(実施例23~31)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 Examples 23 to 31: Examples of block copolymer synthesis
Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g) and AIBN (0.0028 g) were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied on a glass substrate (2 cm square) and spin-coated by rotating at 1500 rpm for 30 seconds. In the atmosphere, the glass substrate was heated on a hot plate according to the table below. Thereafter, the mixture was allowed to cool to room temperature to obtain

<実施例32~35:ブロック共重合体合成例>
親水性マクロRAFT剤A-1(0.216g)、液晶性モノマーB-2(0.677g)、1,1’-アゾビス(シクロヘキサン-1-カルボニトリル)(以下V-40と略す。0.0043g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板上(2cm角)に塗布した。上記ガラス基板の保護カバーとしてスライドガラスをのせ、窒素フロー(0.5L/min)下、ホットプレート上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体32~35を得た(実施例32~35)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 Examples 32 to 35: Examples of block copolymer synthesis
Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystal monomer B-2 (0.677 g), 1,1′-azobis (cyclohexane-1-carbonitrile) (hereinafter abbreviated as V-40) 0043 g) was dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied on a glass substrate (2 cm square). As a protective cover for the glass substrate, a slide glass was placed and heated according to the following table on a hot plate under a nitrogen flow (0.5 L / min). Thereafter, the mixture was allowed to cool to room temperature to obtain block copolymers 32-35 (Examples 32-35). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
親水性マクロRAFT剤A-1(0.216g)、液晶性モノマーB-2(0.677g)、1,1’-アゾビス(シクロヘキサン-1-カルボニトリル)(以下V-40と略す。0.0043g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板上(2cm角)に塗布した。上記ガラス基板の保護カバーとしてスライドガラスをのせ、窒素フロー(0.5L/min)下、ホットプレート上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体32~35を得た(実施例32~35)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 Examples 32 to 35: Examples of block copolymer synthesis
Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystal monomer B-2 (0.677 g), 1,1′-azobis (cyclohexane-1-carbonitrile) (hereinafter abbreviated as V-40) 0043 g) was dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied on a glass substrate (2 cm square). As a protective cover for the glass substrate, a slide glass was placed and heated according to the following table on a hot plate under a nitrogen flow (0.5 L / min). Thereafter, the mixture was allowed to cool to room temperature to obtain block copolymers 32-35 (Examples 32-35). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.

<実施例36~42:ブロック共重合体合成例>
親水性マクロRAFT剤A-1(0.216g)、液晶性モノマーB-2(0.677g)、V-40(0.0043g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板上(2cm角)に塗布した。窒素フロー(0.5L/min)下、ホットプレート上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体36~42を得た(実施例36~42)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 <Examples 36 to 42: Synthesis example of block copolymer>
Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g), and V-40 (0.0043 g) were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied on a glass substrate (2 cm square). It heated according to the following table | surface on the hotplate under nitrogen flow (0.5 L / min). Thereafter, the mixture was allowed to cool to room temperature to obtain block copolymers 36 to 42 (Examples 36 to 42). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
親水性マクロRAFT剤A-1(0.216g)、液晶性モノマーB-2(0.677g)、V-40(0.0043g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板上(2cm角)に塗布した。窒素フロー(0.5L/min)下、ホットプレート上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体36~42を得た(実施例36~42)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 <Examples 36 to 42: Synthesis example of block copolymer>
Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g), and V-40 (0.0043 g) were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied on a glass substrate (2 cm square). It heated according to the following table | surface on the hotplate under nitrogen flow (0.5 L / min). Thereafter, the mixture was allowed to cool to room temperature to obtain block copolymers 36 to 42 (Examples 36 to 42). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.

<実施例43~45:ブロック共重合体合成例>
親水性マクロRAFT剤A-1(0.216g)、液晶性モノマーB-2(0.677g)、V-40(0.0043g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板上(2cm角)に塗布した。大気下、ホットプレート上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体43~45を得た(実施例43~45)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 <Examples 43 to 45: Block copolymer synthesis examples>
Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g), and V-40 (0.0043 g) were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied on a glass substrate (2 cm square). Heating was performed according to the following table on a hot plate in the atmosphere. Thereafter, the mixture was allowed to cool to room temperature to obtain block copolymers 43 to 45 (Examples 43 to 45). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
親水性マクロRAFT剤A-1(0.216g)、液晶性モノマーB-2(0.677g)、V-40(0.0043g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板上(2cm角)に塗布した。大気下、ホットプレート上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体43~45を得た(実施例43~45)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 <Examples 43 to 45: Block copolymer synthesis examples>
Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g), and V-40 (0.0043 g) were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied on a glass substrate (2 cm square). Heating was performed according to the following table on a hot plate in the atmosphere. Thereafter, the mixture was allowed to cool to room temperature to obtain block copolymers 43 to 45 (Examples 43 to 45). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.

<実施例46~48:ブロック共重合体合成例・ミクロ相分離構造の確認・硬化>
親水性マクロRAFT剤A-1(0.216g)、液晶性モノマーB-2(0.677g)、AIBN(0.0028g)、下記構造の光酸発生剤E-1(0.0040g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板(支持体)上(2cm角)に塗布して、スピンコーターを用いて1500回転/分、30秒間回転させてスピンコートした。窒素フロー(0.5L/min)下、ホットプレート上でガラス基板を加熱し(熟成1)、さらに別のホットプレートを用いて大気下にて下表に従い既定の開始温度からUV照射温度まで降温させて(熟成2)、UV照射装置(HOYA社製 EXECURE3000)にて500mJ/cm2にて硬化させた。

<Examples 46 to 48: Example of block copolymer synthesis / confirmation and curing of microphase separation structure>
Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g), AIBN (0.0028 g), photoacid generator E-1 (0.0040 g) having the following structure was added to toluene. Dissolved in 5 ml. 50 μl of the obtained toluene solution was applied on a glass substrate (support) (2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds using a spin coater. A glass substrate is heated on a hot plate under a nitrogen flow (0.5 L / min) (ripening 1), and the temperature is lowered from a predetermined starting temperature to a UV irradiation temperature in accordance with the following table in the atmosphere using another hot plate. (Maturation 2), and cured at 500 mJ / cm 2 with a UV irradiation apparatus (EXECURE 3000 manufactured by HOYA).
親水性マクロRAFT剤A-1(0.216g)、液晶性モノマーB-2(0.677g)、AIBN(0.0028g)、下記構造の光酸発生剤E-1(0.0040g)をトルエン5mlに溶解させた。得られたトルエン溶液50μlをガラス基板(支持体)上(2cm角)に塗布して、スピンコーターを用いて1500回転/分、30秒間回転させてスピンコートした。窒素フロー(0.5L/min)下、ホットプレート上でガラス基板を加熱し(熟成1)、さらに別のホットプレートを用いて大気下にて下表に従い既定の開始温度からUV照射温度まで降温させて(熟成2)、UV照射装置(HOYA社製 EXECURE3000)にて500mJ/cm2にて硬化させた。
Hydrophilic macro RAFT agent A-1 (0.216 g), liquid crystalline monomer B-2 (0.677 g), AIBN (0.0028 g), photoacid generator E-1 (0.0040 g) having the following structure was added to toluene. Dissolved in 5 ml. 50 μl of the obtained toluene solution was applied on a glass substrate (support) (2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds using a spin coater. A glass substrate is heated on a hot plate under a nitrogen flow (0.5 L / min) (ripening 1), and the temperature is lowered from a predetermined starting temperature to a UV irradiation temperature in accordance with the following table in the atmosphere using another hot plate. (Maturation 2), and cured at 500 mJ / cm 2 with a UV irradiation apparatus (EXECURE 3000 manufactured by HOYA).

(AFM測定)
上記ガラス基板上でのリビング重合体の製造方法(実施例46~48)で製造された、ガラス基板上のブロック共重合体を、AFM(SII社製 SPI3800N)にて測定した。測定した表面形状の代表例を図2~図4に記す。ラメラやシリンダー状のミクロ相分離構造が観察された。 (AFM measurement)
The block copolymer on the glass substrate produced by the method for producing a living polymer on the glass substrate (Examples 46 to 48) was measured with AFM (SPI3800N manufactured by SII). Representative examples of the measured surface shape are shown in FIGS. Lamella and cylindrical microphase separation structures were observed.
上記ガラス基板上でのリビング重合体の製造方法(実施例46~48)で製造された、ガラス基板上のブロック共重合体を、AFM(SII社製 SPI3800N)にて測定した。測定した表面形状の代表例を図2~図4に記す。ラメラやシリンダー状のミクロ相分離構造が観察された。 (AFM measurement)
The block copolymer on the glass substrate produced by the method for producing a living polymer on the glass substrate (Examples 46 to 48) was measured with AFM (SPI3800N manufactured by SII). Representative examples of the measured surface shape are shown in FIGS. Lamella and cylindrical microphase separation structures were observed.
(支持体からのミクロ相分離構造膜の剥離)
実施例46~48において、ガラス基板からミクロ相分離構造膜を剥離することも可能であった。
実施例46~48で剥離したミクロ相分離構造膜の厚みは、それぞれ150nm、200nm、210nmであった。 (Peeling of micro phase separation structure film from support)
In Examples 46 to 48, it was possible to peel the microphase separation structure film from the glass substrate.
The thicknesses of the microphase-separated structure films peeled in Examples 46 to 48 were 150 nm, 200 nm, and 210 nm, respectively.
実施例46~48において、ガラス基板からミクロ相分離構造膜を剥離することも可能であった。
実施例46~48で剥離したミクロ相分離構造膜の厚みは、それぞれ150nm、200nm、210nmであった。 (Peeling of micro phase separation structure film from support)
In Examples 46 to 48, it was possible to peel the microphase separation structure film from the glass substrate.
The thicknesses of the microphase-separated structure films peeled in Examples 46 to 48 were 150 nm, 200 nm, and 210 nm, respectively.
[比較例1~14:溶液中でのRAFT重合]
(ブロック共重合体比較合成例1)
液晶性モノマーB-1(0.493g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、ヘキサン200mlに反応溶液を添加して、濾過した。さらに得られたポリマーをエタノール40mlに懸濁して、1時間後にろ過した。ブロック共重合体D-1を0.33g得た。GPCの分析結果からMw22200、Mn17500、Mw/Mn=1.27であった。

[Comparative Examples 1 to 14: RAFT polymerization in solution]
(Block copolymer comparative synthesis example 1)
Liquid crystalline monomer B-1 (0.493 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g) and nitrogen flow was started. The mixture was stirred for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 200 ml of hexane and filtered. Further, the obtained polymer was suspended in 40 ml of ethanol and filtered after 1 hour. 0.33 g of block copolymer D-1 was obtained. It was Mw22200, Mn17500, Mw / Mn = 1.27 from the analysis result of GPC.
(ブロック共重合体比較合成例1)
液晶性モノマーB-1(0.493g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、ヘキサン200mlに反応溶液を添加して、濾過した。さらに得られたポリマーをエタノール40mlに懸濁して、1時間後にろ過した。ブロック共重合体D-1を0.33g得た。GPCの分析結果からMw22200、Mn17500、Mw/Mn=1.27であった。
(Block copolymer comparative synthesis example 1)
Liquid crystalline monomer B-1 (0.493 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g) and nitrogen flow was started. The mixture was stirred for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 200 ml of hexane and filtered. Further, the obtained polymer was suspended in 40 ml of ethanol and filtered after 1 hour. 0.33 g of block copolymer D-1 was obtained. It was Mw22200, Mn17500, Mw / Mn = 1.27 from the analysis result of GPC.
(ブロック共重合体比較合成例2)
液晶性モノマーB-1(0.739g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-2を0.6g得た。GPCの分析結果からMw31100、Mn25600、Mw/Mn=1.22であった。 (Block copolymer comparative synthesis example 2)
Liquid crystalline monomer B-1 (0.739 g), AIBN (0.0028 g), xylene 5 ml, hydrophilic macro RAFT agent A-1 (0.108 g) was added and nitrogen flow was started. The mixture was stirred for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.6 g of block copolymer D-2 was obtained. From the analysis results of GPC, it was Mw31100, Mn25600, Mw / Mn = 1.22.
液晶性モノマーB-1(0.739g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-2を0.6g得た。GPCの分析結果からMw31100、Mn25600、Mw/Mn=1.22であった。 (Block copolymer comparative synthesis example 2)
Liquid crystalline monomer B-1 (0.739 g), AIBN (0.0028 g), xylene 5 ml, hydrophilic macro RAFT agent A-1 (0.108 g) was added and nitrogen flow was started. The mixture was stirred for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.6 g of block copolymer D-2 was obtained. From the analysis results of GPC, it was Mw31100, Mn25600, Mw / Mn = 1.22.
(ブロック共重合体比較合成例3)
液晶性モノマーB-1(0.985g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-3を0.8g得た。GPCの分析結果からMw37100、Mn29100、Mw/Mn=1.28であった。 (Block copolymer comparative synthesis example 3)
Liquid crystalline monomer B-1 (0.985 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g) and nitrogen flow was started. The mixture was stirred for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.8 g of block copolymer D-3 was obtained. It was Mw37100, Mn29100, Mw / Mn = 1.28 from the analysis result of GPC.
液晶性モノマーB-1(0.985g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-3を0.8g得た。GPCの分析結果からMw37100、Mn29100、Mw/Mn=1.28であった。 (Block copolymer comparative synthesis example 3)
Liquid crystalline monomer B-1 (0.985 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g) and nitrogen flow was started. The mixture was stirred for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.8 g of block copolymer D-3 was obtained. It was Mw37100, Mn29100, Mw / Mn = 1.28 from the analysis result of GPC.
(ブロック共重合体比較合成例4)
液晶性モノマーB-1(0.493g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-2(0.200g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン200mlに反応溶液を添加して、濾過した。さらに得られたポリマーをエタノール40mlに懸濁して、1時間後にろ過した。ブロック共重合体D-4を0.4g得た。GPCの分析結果からMw22600、Mn17200、Mw/Mn=1.31であった。

(Block copolymer comparative synthesis example 4)
Liquid crystalline monomer B-1 (0.493 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-2 (0.200 g) and nitrogen flow was started. The mixture was stirred for 11 hours. Thereafter, the reaction solution was allowed to cool to room temperature, and the reaction solution was added to 200 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in 40 ml of ethanol and filtered after 1 hour. 0.4 g of block copolymer D-4 was obtained. It was Mw22600, Mn17200, Mw / Mn = 1.31 from the analysis result of GPC.
液晶性モノマーB-1(0.493g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-2(0.200g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン200mlに反応溶液を添加して、濾過した。さらに得られたポリマーをエタノール40mlに懸濁して、1時間後にろ過した。ブロック共重合体D-4を0.4g得た。GPCの分析結果からMw22600、Mn17200、Mw/Mn=1.31であった。
Liquid crystalline monomer B-1 (0.493 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-2 (0.200 g) and nitrogen flow was started. The mixture was stirred for 11 hours. Thereafter, the reaction solution was allowed to cool to room temperature, and the reaction solution was added to 200 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in 40 ml of ethanol and filtered after 1 hour. 0.4 g of block copolymer D-4 was obtained. It was Mw22600, Mn17200, Mw / Mn = 1.31 from the analysis result of GPC.
(ブロック共重合体比較合成例5)
液晶性モノマーB-2(0.677g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・9時間加熱攪拌した。その後、室温まで放冷し、THF2mlを添加後冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-5を0.4g得た。GPCの分析結果からMw24200、Mn17800、Mw/Mn=1.35であった。

(Block copolymer comparative synthesis example 5)
Liquid crystalline monomer B-2 (0.677 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g), and nitrogen flow was started. The mixture was heated and stirred for 9 hours. Thereafter, the mixture was allowed to cool to room temperature, and 2 ml of THF was added and then the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.4 g of block copolymer D-5 was obtained. From the analysis results of GPC, it was Mw24200, Mn17800, Mw / Mn = 1.35.
液晶性モノマーB-2(0.677g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・9時間加熱攪拌した。その後、室温まで放冷し、THF2mlを添加後冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-5を0.4g得た。GPCの分析結果からMw24200、Mn17800、Mw/Mn=1.35であった。
Liquid crystalline monomer B-2 (0.677 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g), and nitrogen flow was started. The mixture was heated and stirred for 9 hours. Thereafter, the mixture was allowed to cool to room temperature, and 2 ml of THF was added and then the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.4 g of block copolymer D-5 was obtained. From the analysis results of GPC, it was Mw24200, Mn17800, Mw / Mn = 1.35.
(ブロック共重合体比較合成例6)
液晶性モノマーB-2(1.015g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・9時間加熱攪拌した。その後、室温まで放冷し、THF5mlを添加後、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-6を0.7g得た。GPCの分析結果からMw32700、Mn24700、Mw/Mn=1.32であった。 (Block copolymer comparative synthesis example 6)
Liquid crystalline monomer B-2 (1.015 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g) and nitrogen flow was started. The mixture was heated and stirred for 9 hours. Thereafter, the mixture was allowed to cool to room temperature, and after adding 5 ml of THF, the reaction solution was added to 300 ml of cooled hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.7 g of block copolymer D-6 was obtained. From the analysis results of GPC, it was Mw32700, Mn24700, Mw / Mn = 1.32.
液晶性モノマーB-2(1.015g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・9時間加熱攪拌した。その後、室温まで放冷し、THF5mlを添加後、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-6を0.7g得た。GPCの分析結果からMw32700、Mn24700、Mw/Mn=1.32であった。 (Block copolymer comparative synthesis example 6)
Liquid crystalline monomer B-2 (1.015 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g) and nitrogen flow was started. The mixture was heated and stirred for 9 hours. Thereafter, the mixture was allowed to cool to room temperature, and after adding 5 ml of THF, the reaction solution was added to 300 ml of cooled hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.7 g of block copolymer D-6 was obtained. From the analysis results of GPC, it was Mw32700, Mn24700, Mw / Mn = 1.32.
(ブロック共重合体比較合成例7)
液晶性モノマーB-2(1.354g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・9時間加熱攪拌した。その後、室温まで放冷し、THF5mlを添加後、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-7を1.06g得た。GPCの分析結果からMw38600、Mn27200、Mw/Mn=1.42であった。 (Block copolymer comparative synthesis example 7)
Liquid crystalline monomer B-2 (1.354 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g), and nitrogen flow was started. The mixture was heated and stirred for 9 hours. Thereafter, the mixture was allowed to cool to room temperature, and after adding 5 ml of THF, the reaction solution was added to 300 ml of cooled hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 1.06 g of block copolymer D-7 was obtained. From the analysis results of GPC, it was Mw38600, Mn27200, Mw / Mn = 1.42.
液晶性モノマーB-2(1.354g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・9時間加熱攪拌した。その後、室温まで放冷し、THF5mlを添加後、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-7を1.06g得た。GPCの分析結果からMw38600、Mn27200、Mw/Mn=1.42であった。 (Block copolymer comparative synthesis example 7)
Liquid crystalline monomer B-2 (1.354 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g), and nitrogen flow was started. The mixture was heated and stirred for 9 hours. Thereafter, the mixture was allowed to cool to room temperature, and after adding 5 ml of THF, the reaction solution was added to 300 ml of cooled hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 1.06 g of block copolymer D-7 was obtained. From the analysis results of GPC, it was Mw38600, Mn27200, Mw / Mn = 1.42.
(ブロック共重合体比較合成例8)
液晶性モノマーB-2(0.846g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・9時間加熱攪拌した。その後、室温まで放冷し、THF5mlを添加後、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-8を0.6g得た。GPCの分析結果からMw29200、Mn23100、Mw/Mn=1.27であった。 (Block copolymer comparative synthesis example 8)
Liquid crystalline monomer B-2 (0.846 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g), and nitrogen flow was started. The mixture was heated and stirred for 9 hours. Thereafter, the mixture was allowed to cool to room temperature, and after adding 5 ml of THF, the reaction solution was added to 300 ml of cooled hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.6 g of block copolymer D-8 was obtained. It was Mw29200, Mn23100, Mw / Mn = 1.27 from the analysis result of GPC.
液晶性モノマーB-2(0.846g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・9時間加熱攪拌した。その後、室温まで放冷し、THF5mlを添加後、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-8を0.6g得た。GPCの分析結果からMw29200、Mn23100、Mw/Mn=1.27であった。 (Block copolymer comparative synthesis example 8)
Liquid crystalline monomer B-2 (0.846 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g), and nitrogen flow was started. The mixture was heated and stirred for 9 hours. Thereafter, the mixture was allowed to cool to room temperature, and after adding 5 ml of THF, the reaction solution was added to 300 ml of cooled hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.6 g of block copolymer D-8 was obtained. It was Mw29200, Mn23100, Mw / Mn = 1.27 from the analysis result of GPC.
(ブロック共重合体比較合成例9)
液晶性モノマーB-2(1.015g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・7時間加熱攪拌した。その後、室温まで放冷し、THF5mlを添加後、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-9を0.4g得た。GPCの分析結果からMw26300、Mn21800、Mw/Mn=1.21であった。 (Block copolymer comparative synthesis example 9)
Liquid crystalline monomer B-2 (1.015 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g) and nitrogen flow was started. The mixture was heated and stirred for 7 hours. Thereafter, the mixture was allowed to cool to room temperature, and after adding 5 ml of THF, the reaction solution was added to 300 ml of cooled hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.4 g of block copolymer D-9 was obtained. From the analysis results of GPC, it was Mw26300, Mn21800, Mw / Mn = 1.21.
液晶性モノマーB-2(1.015g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・7時間加熱攪拌した。その後、室温まで放冷し、THF5mlを添加後、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-9を0.4g得た。GPCの分析結果からMw26300、Mn21800、Mw/Mn=1.21であった。 (Block copolymer comparative synthesis example 9)
Liquid crystalline monomer B-2 (1.015 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g) and nitrogen flow was started. The mixture was heated and stirred for 7 hours. Thereafter, the mixture was allowed to cool to room temperature, and after adding 5 ml of THF, the reaction solution was added to 300 ml of cooled hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.4 g of block copolymer D-9 was obtained. From the analysis results of GPC, it was Mw26300, Mn21800, Mw / Mn = 1.21.
(ブロック共重合体比較合成例10)
液晶性モノマーB-2(0.677g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-2(0.200g)を加えて窒素フローを開始し、オイルバスで60℃・9時間加熱攪拌した。その後、室温まで放冷し、THF2mlを添加後冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-10を0.6g得た。GPCの分析結果からMw24500、Mn18100、Mw/Mn=1.35であった。
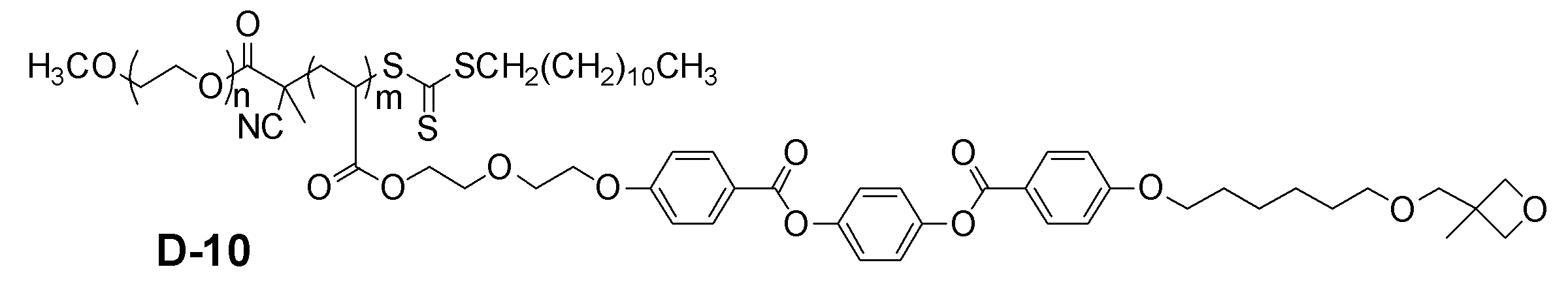
(Block copolymer comparative synthesis example 10)
Liquid crystalline monomer B-2 (0.677 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-2 (0.200 g), and nitrogen flow was started. The mixture was heated and stirred for 9 hours. Thereafter, the mixture was allowed to cool to room temperature, and 2 ml of THF was added and then the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.6 g of block copolymer D-10 was obtained. It was Mw24500, Mn18100, Mw / Mn = 1.35 from the analysis result of GPC.
液晶性モノマーB-2(0.677g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-2(0.200g)を加えて窒素フローを開始し、オイルバスで60℃・9時間加熱攪拌した。その後、室温まで放冷し、THF2mlを添加後冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-10を0.6g得た。GPCの分析結果からMw24500、Mn18100、Mw/Mn=1.35であった。
Liquid crystalline monomer B-2 (0.677 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-2 (0.200 g), and nitrogen flow was started. The mixture was heated and stirred for 9 hours. Thereafter, the mixture was allowed to cool to room temperature, and 2 ml of THF was added and then the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.6 g of block copolymer D-10 was obtained. It was Mw24500, Mn18100, Mw / Mn = 1.35 from the analysis result of GPC.
(ブロック共重合体比較合成例11)
液晶性モノマーB-3(0.329g)、液晶性モノマーB-1(0.246g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-11を0.4g得た。GPCの分析結果からMw27400、Mn21500、Mw/Mn=1.28であった。

(Block copolymer comparative synthesis example 11)
Liquid crystalline monomer B-3 (0.329 g), liquid crystalline monomer B-1 (0.246 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g). Nitrogen flow was started, and the mixture was heated and stirred in an oil bath at 60 ° C. for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.4g of block copolymer D-11 was obtained. From the analysis results of GPC, it was Mw27400, Mn21500, Mw / Mn = 1.28.
液晶性モノマーB-3(0.329g)、液晶性モノマーB-1(0.246g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-11を0.4g得た。GPCの分析結果からMw27400、Mn21500、Mw/Mn=1.28であった。
Liquid crystalline monomer B-3 (0.329 g), liquid crystalline monomer B-1 (0.246 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g). Nitrogen flow was started, and the mixture was heated and stirred in an oil bath at 60 ° C. for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.4g of block copolymer D-11 was obtained. From the analysis results of GPC, it was Mw27400, Mn21500, Mw / Mn = 1.28.
(ブロック共重合体比較合成例12)
液晶性モノマーB-3(0.066g)、液晶性モノマーB-1(0.443g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-12を0.3g得た。GPCの分析結果からMw25300、Mn19500、Mw/Mn=1.30であった。 (Block copolymer comparative synthesis example 12)
Liquid crystalline monomer B-3 (0.066 g), liquid crystalline monomer B-1 (0.443 g), AIBN (0.0028 g), and 5 ml of xylene were added hydrophilic macro RAFT agent A-1 (0.108 g). Nitrogen flow was started, and the mixture was heated and stirred in an oil bath at 60 ° C. for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.3 g of block copolymer D-12 was obtained. From the analysis results of GPC, it was Mw25300, Mn19500, Mw / Mn = 1.30.
液晶性モノマーB-3(0.066g)、液晶性モノマーB-1(0.443g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-12を0.3g得た。GPCの分析結果からMw25300、Mn19500、Mw/Mn=1.30であった。 (Block copolymer comparative synthesis example 12)
Liquid crystalline monomer B-3 (0.066 g), liquid crystalline monomer B-1 (0.443 g), AIBN (0.0028 g), and 5 ml of xylene were added hydrophilic macro RAFT agent A-1 (0.108 g). Nitrogen flow was started, and the mixture was heated and stirred in an oil bath at 60 ° C. for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.3 g of block copolymer D-12 was obtained. From the analysis results of GPC, it was Mw25300, Mn19500, Mw / Mn = 1.30.
(ブロック共重合体比較合成例13)
液晶性モノマーB-4(0.329g)、液晶性モノマーB-1(0.246g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-13を0.3g得た。GPCの分析結果からMw23000、Mn19000、Mw/Mn=1.21であった。
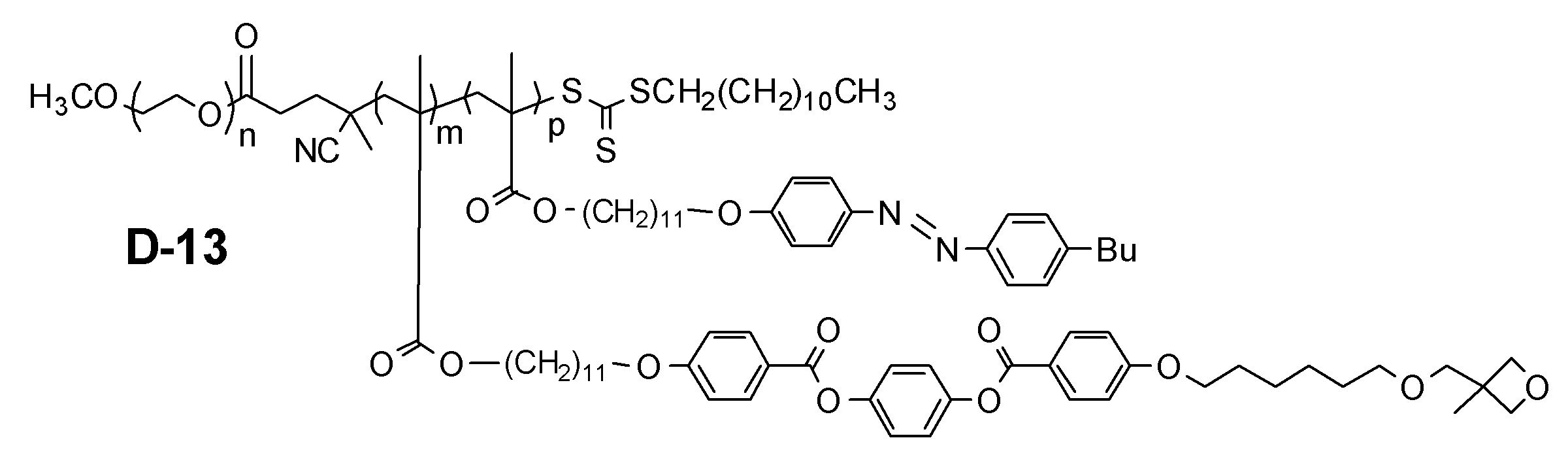
(Block copolymer comparative synthesis example 13)
Liquid crystalline monomer B-4 (0.329 g), liquid crystalline monomer B-1 (0.246 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g). Nitrogen flow was started, and the mixture was heated and stirred in an oil bath at 60 ° C. for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.3 g of block copolymer D-13 was obtained. It was Mw23000, Mn19000, Mw / Mn = 1.21 from the analysis result of GPC.
液晶性モノマーB-4(0.329g)、液晶性モノマーB-1(0.246g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-13を0.3g得た。GPCの分析結果からMw23000、Mn19000、Mw/Mn=1.21であった。
Liquid crystalline monomer B-4 (0.329 g), liquid crystalline monomer B-1 (0.246 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g). Nitrogen flow was started, and the mixture was heated and stirred in an oil bath at 60 ° C. for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.3 g of block copolymer D-13 was obtained. It was Mw23000, Mn19000, Mw / Mn = 1.21 from the analysis result of GPC.
(ブロック共重合体比較合成例14)
液晶性モノマーB-5(0.329g)、液晶性モノマーB-3(0.246g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-14を0.4g得た。GPCの分析結果からMw25000、Mn19000、Mw/Mn=1.32であった。

(Block copolymer comparative synthesis example 14)
Liquid crystalline monomer B-5 (0.329 g), liquid crystalline monomer B-3 (0.246 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g). Nitrogen flow was started, and the mixture was heated and stirred in an oil bath at 60 ° C. for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.4 g of block copolymer D-14 was obtained. It was Mw25000, Mn19000, Mw / Mn = 1.32 from the analysis result of GPC.
液晶性モノマーB-5(0.329g)、液晶性モノマーB-3(0.246g)、AIBN(0.0028g)、キシレン5mlに親水性マクロRAFT剤A-1(0.108g)を加えて窒素フローを開始し、オイルバスで60℃・11時間加熱攪拌した。その後、室温まで放冷し、冷却したヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後にろ過した。ブロック共重合体D-14を0.4g得た。GPCの分析結果からMw25000、Mn19000、Mw/Mn=1.32であった。
Liquid crystalline monomer B-5 (0.329 g), liquid crystalline monomer B-3 (0.246 g), AIBN (0.0028 g) and xylene 5 ml were added with hydrophilic macro RAFT agent A-1 (0.108 g). Nitrogen flow was started, and the mixture was heated and stirred in an oil bath at 60 ° C. for 11 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of cooled hexane, followed by filtration. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.4 g of block copolymer D-14 was obtained. It was Mw25000, Mn19000, Mw / Mn = 1.32 from the analysis result of GPC.
以上の実施例1~48と比較例1~14の比較から、本発明の支持体上でのリビング重合体の製造方法によれば、RAFT重合の場合にリビング重合体(特にブロック共重合体)を短時間で経済合理的に製造できることがわかった。
また、本発明の支持体上でのリビング重合体の製造方法を用いることにより、ミクロ相分離構造膜を経済合理的に製造できることがわかった。 From the comparison of Examples 1 to 48 and Comparative Examples 1 to 14, according to the method for producing a living polymer on a support of the present invention, a living polymer (particularly a block copolymer) is used in the case of RAFT polymerization. It was found that it can be economically and reasonably manufactured in a short time.
It was also found that a microphase separation structure membrane can be produced economically rationally by using the method for producing a living polymer on the support of the present invention.
また、本発明の支持体上でのリビング重合体の製造方法を用いることにより、ミクロ相分離構造膜を経済合理的に製造できることがわかった。 From the comparison of Examples 1 to 48 and Comparative Examples 1 to 14, according to the method for producing a living polymer on a support of the present invention, a living polymer (particularly a block copolymer) is used in the case of RAFT polymerization. It was found that it can be economically and reasonably manufactured in a short time.
It was also found that a microphase separation structure membrane can be produced economically rationally by using the method for producing a living polymer on the support of the present invention.
[実施例101~121:支持体上でのATRP検討]
<実施例101:リビング重合体合成例>
ATRP開始剤A-3(0.0039g)、液晶性モノマーB-1(0.492g)、N,N,N’,N’’,N’’-ペンタメチルジエチレントリアミン(以下PMDETAと略)0.021gをトルエン5mlに溶解させた。得られたトルエン溶液50μlを銅フィルム(日本製箔株式会社、圧延銅箔TCU、厚み;18μm、2cm角)上に塗布して、1500回転/分、30秒間回転させてスピンコートした。上記銅フィルム上のモノマー含有組成物の保護カバーとしてさらにモノマー含有組成物上に支持体として用いたものを同じ銅フィルムをのせ、5.3KPaで加圧しながら、N2フロー(0.5L/min)下、上記銅フィルムをホットプレート上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体101を得た(実施例101)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 [Examples 101 to 121: ATRP examination on support]
<Example 101: Living polymer synthesis example>
ATRP initiator A-3 (0.0039 g), liquid crystalline monomer B-1 (0.492 g), N, N, N ′, N ″, N ″ -pentamethyldiethylenetriamine (hereinafter abbreviated as PMDETA) 021 g was dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied onto a copper film (Nihon Foil Co., Ltd., rolled copper foil TCU, thickness: 18 μm, 2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds. As a protective cover for the monomer-containing composition on the copper film, the same copper film as that used as the support was placed on the monomer-containing composition, and N 2 flow (0.5 L / min) was applied while pressing at 5.3 KPa. ) The copper film was heated on a hot plate according to the table below. Then, it stood to cool to room temperature, and obtained the block copolymer 101 (Example 101). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
<実施例101:リビング重合体合成例>
ATRP開始剤A-3(0.0039g)、液晶性モノマーB-1(0.492g)、N,N,N’,N’’,N’’-ペンタメチルジエチレントリアミン(以下PMDETAと略)0.021gをトルエン5mlに溶解させた。得られたトルエン溶液50μlを銅フィルム(日本製箔株式会社、圧延銅箔TCU、厚み;18μm、2cm角)上に塗布して、1500回転/分、30秒間回転させてスピンコートした。上記銅フィルム上のモノマー含有組成物の保護カバーとしてさらにモノマー含有組成物上に支持体として用いたものを同じ銅フィルムをのせ、5.3KPaで加圧しながら、N2フロー(0.5L/min)下、上記銅フィルムをホットプレート上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体101を得た(実施例101)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 [Examples 101 to 121: ATRP examination on support]
<Example 101: Living polymer synthesis example>
ATRP initiator A-3 (0.0039 g), liquid crystalline monomer B-1 (0.492 g), N, N, N ′, N ″, N ″ -pentamethyldiethylenetriamine (hereinafter abbreviated as PMDETA) 021 g was dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied onto a copper film (Nihon Foil Co., Ltd., rolled copper foil TCU, thickness: 18 μm, 2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds. As a protective cover for the monomer-containing composition on the copper film, the same copper film as that used as the support was placed on the monomer-containing composition, and N 2 flow (0.5 L / min) was applied while pressing at 5.3 KPa. ) The copper film was heated on a hot plate according to the table below. Then, it stood to cool to room temperature, and obtained the block copolymer 101 (Example 101). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
<実施例102~105:リビング重合体合成例>
実施例101のリビング重合体合成例で使用した液晶性モノマーを等モル使用してB-1からB-2~5にそれぞれ変更する以外は実施例101のリビング重合体合成例と同様にしてリビング重合体を得た(実施例102~105)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 <Examples 102 to 105: Living polymer synthesis examples>
Living in the same manner as the living polymer synthesis example of Example 101 except that equimolar amounts of the liquid crystalline monomer used in the living polymer synthesis example of Example 101 were used and changed from B-1 to B-2 to 5 respectively. Polymers were obtained (Examples 102 to 105). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
実施例101のリビング重合体合成例で使用した液晶性モノマーを等モル使用してB-1からB-2~5にそれぞれ変更する以外は実施例101のリビング重合体合成例と同様にしてリビング重合体を得た(実施例102~105)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 <Examples 102 to 105: Living polymer synthesis examples>
Living in the same manner as the living polymer synthesis example of Example 101 except that equimolar amounts of the liquid crystalline monomer used in the living polymer synthesis example of Example 101 were used and changed from B-1 to B-2 to 5 respectively. Polymers were obtained (Examples 102 to 105). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.

<実施例106~117:ブロック共重合体合成例>
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-2(0.677g)、PMDETA0.021gをトルエン5mlに溶解させた。得られたトルエン溶液50μlを銅フィルム(日本製箔株式会社、圧延銅箔TCU、厚み;18μm、2cm角)上に塗布して、1500回転/分、30秒間回転させてスピンコートした。上記銅フィルム上のモノマー含有組成物の保護カバーとしてさらにモノマー含有組成物上に支持体として用いたものを同じ銅フィルムをのせ、5.3KPaで加圧しながら、N2フロー(0.5L/min)下、上記銅フィルムをホットプレート上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体106~117を得た(実施例106~117)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 <Examples 106 to 117: Block copolymer synthesis example>
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystal monomer B-2 (0.677 g), and PMDETA 0.021 g were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied onto a copper film (Nihon Foil Co., Ltd., rolled copper foil TCU, thickness: 18 μm, 2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds. As a protective cover for the monomer-containing composition on the copper film, the same copper film as that used as the support was placed on the monomer-containing composition, and N 2 flow (0.5 L / min) was applied while pressing at 5.3 KPa. ) The copper film was heated on a hot plate according to the table below. Thereafter, the mixture was allowed to cool to room temperature to obtain block copolymers 106 to 117 (Examples 106 to 117). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-2(0.677g)、PMDETA0.021gをトルエン5mlに溶解させた。得られたトルエン溶液50μlを銅フィルム(日本製箔株式会社、圧延銅箔TCU、厚み;18μm、2cm角)上に塗布して、1500回転/分、30秒間回転させてスピンコートした。上記銅フィルム上のモノマー含有組成物の保護カバーとしてさらにモノマー含有組成物上に支持体として用いたものを同じ銅フィルムをのせ、5.3KPaで加圧しながら、N2フロー(0.5L/min)下、上記銅フィルムをホットプレート上で下表に従い加熱した。その後、室温まで放冷し、ブロック共重合体106~117を得た(実施例106~117)。実施例1と同様に1H-NMR測定によりモノマー消費量とGPCの分析を行い、得られた結果を下記表に記載した。 <Examples 106 to 117: Block copolymer synthesis example>
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystal monomer B-2 (0.677 g), and PMDETA 0.021 g were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied onto a copper film (Nihon Foil Co., Ltd., rolled copper foil TCU, thickness: 18 μm, 2 cm square), and spin-coated by rotating at 1500 rpm for 30 seconds. As a protective cover for the monomer-containing composition on the copper film, the same copper film as that used as the support was placed on the monomer-containing composition, and N 2 flow (0.5 L / min) was applied while pressing at 5.3 KPa. ) The copper film was heated on a hot plate according to the table below. Thereafter, the mixture was allowed to cool to room temperature to obtain block copolymers 106 to 117 (Examples 106 to 117). The monomer consumption and GPC were analyzed by 1 H-NMR measurement in the same manner as in Example 1, and the results obtained are listed in the following table.

<実施例118:ブロック共重合体合成例>
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-2(0.677g)、2,2’-ビピリジン0.038gをトルエン5mlに溶解させた。得られたトルエン溶液50μlを銅フィルム上(2cm角)に塗布して、1500回転/分、30秒間回転させてスピンコートした。上記銅フィルム上のモノマー含有組成物の保護カバーとしてさらにモノマー含有組成物上に支持体として用いたものを同じ銅フィルム(2cm角)をのせ、5.3KPaで加圧しながら、N2フロー(0.5L/min)下、上記銅フィルムをホットプレート上で120℃・30秒加熱した。その後、室温まで放冷し、ブロック共重合体118を得た(実施例118)。GPCの分析結果からMw55500、Mn30000、Mw/Mn=1.85であった。 <Example 118: Synthesis example of block copolymer>
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-2 (0.677 g), and 0.038 g of 2,2′-bipyridine were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied on a copper film (2 cm square) and spin-coated by rotating at 1500 rpm for 30 seconds. As a protective cover for the monomer-containing composition on the copper film, the same copper film (2 cm square) that was used as a support was placed on the monomer-containing composition and N 2 flow (0 .5 L / min), the copper film was heated on a hot plate at 120 ° C. for 30 seconds. Then, it stood to cool to room temperature, and obtained the block copolymer 118 (Example 118). It was Mw55500, Mn30000, Mw / Mn = 1.85 from the analysis result of GPC.
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-2(0.677g)、2,2’-ビピリジン0.038gをトルエン5mlに溶解させた。得られたトルエン溶液50μlを銅フィルム上(2cm角)に塗布して、1500回転/分、30秒間回転させてスピンコートした。上記銅フィルム上のモノマー含有組成物の保護カバーとしてさらにモノマー含有組成物上に支持体として用いたものを同じ銅フィルム(2cm角)をのせ、5.3KPaで加圧しながら、N2フロー(0.5L/min)下、上記銅フィルムをホットプレート上で120℃・30秒加熱した。その後、室温まで放冷し、ブロック共重合体118を得た(実施例118)。GPCの分析結果からMw55500、Mn30000、Mw/Mn=1.85であった。 <Example 118: Synthesis example of block copolymer>
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-2 (0.677 g), and 0.038 g of 2,2′-bipyridine were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied on a copper film (2 cm square) and spin-coated by rotating at 1500 rpm for 30 seconds. As a protective cover for the monomer-containing composition on the copper film, the same copper film (2 cm square) that was used as a support was placed on the monomer-containing composition and N 2 flow (0 .5 L / min), the copper film was heated on a hot plate at 120 ° C. for 30 seconds. Then, it stood to cool to room temperature, and obtained the block copolymer 118 (Example 118). It was Mw55500, Mn30000, Mw / Mn = 1.85 from the analysis result of GPC.
<実施例119:ブロック共重合体合成例>
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-2(0.677g)、1,1,4,7,10,10-ヘキサメチルトリエチレンテトラミン0.028gをトルエン5mlに溶解させた。得られたトルエン溶液50μlを銅フィルム上(2cm角)に塗布して、1500回転/分、30秒間回転させてスピンコートした。上記銅フィルム上のモノマー含有組成物の保護カバーとしてさらにモノマー含有組成物上に支持体として用いたものを同じ銅フィルム(2cm角)をのせ、5.3KPaで加圧しながら、N2フロー(0.5L/min)下、上記銅フィルムをホットプレート上で120℃・30秒加熱した。その後、室温まで放冷し、ブロック共重合体119を得た(実施例119)。GPCの分析結果からMw36000、Mn23500、Mw/Mn=1.53であった。 <Example 119: Block copolymer synthesis example>
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystal monomer B-2 (0.677 g), 1,1,4,7,10,10-hexamethyltriethylenetetramine 0.028 g in 5 ml of toluene Dissolved. 50 μl of the obtained toluene solution was applied on a copper film (2 cm square) and spin-coated by rotating at 1500 rpm for 30 seconds. As a protective cover for the monomer-containing composition on the copper film, the same copper film (2 cm square) that was used as a support was placed on the monomer-containing composition and N 2 flow (0 .5 L / min), the copper film was heated on a hot plate at 120 ° C. for 30 seconds. Then, it stood to cool to room temperature, and obtained the block copolymer 119 (Example 119). It was Mw36000, Mn23500, Mw / Mn = 1.53 from the analysis result of GPC.
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-2(0.677g)、1,1,4,7,10,10-ヘキサメチルトリエチレンテトラミン0.028gをトルエン5mlに溶解させた。得られたトルエン溶液50μlを銅フィルム上(2cm角)に塗布して、1500回転/分、30秒間回転させてスピンコートした。上記銅フィルム上のモノマー含有組成物の保護カバーとしてさらにモノマー含有組成物上に支持体として用いたものを同じ銅フィルム(2cm角)をのせ、5.3KPaで加圧しながら、N2フロー(0.5L/min)下、上記銅フィルムをホットプレート上で120℃・30秒加熱した。その後、室温まで放冷し、ブロック共重合体119を得た(実施例119)。GPCの分析結果からMw36000、Mn23500、Mw/Mn=1.53であった。 <Example 119: Block copolymer synthesis example>
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystal monomer B-2 (0.677 g), 1,1,4,7,10,10-hexamethyltriethylenetetramine 0.028 g in 5 ml of toluene Dissolved. 50 μl of the obtained toluene solution was applied on a copper film (2 cm square) and spin-coated by rotating at 1500 rpm for 30 seconds. As a protective cover for the monomer-containing composition on the copper film, the same copper film (2 cm square) that was used as a support was placed on the monomer-containing composition and N 2 flow (0 .5 L / min), the copper film was heated on a hot plate at 120 ° C. for 30 seconds. Then, it stood to cool to room temperature, and obtained the block copolymer 119 (Example 119). It was Mw36000, Mn23500, Mw / Mn = 1.53 from the analysis result of GPC.
<実施例120:ブロック共重合体合成例>
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-2(0.677g)、N,N,N’,N’-テトラキス(2-ピリジルメチル)エチレンジアミン0.051gをトルエン5mlに溶解させた。得られたトルエン溶液50μlを銅フィルム上(2cm角)に塗布して、1500回転/分、30秒間回転させてスピンコートした。上記銅フィルム上のモノマー含有組成物の保護カバーとしてさらにモノマー含有組成物上に支持体として用いたものを同じ銅フィルム(2cm角)をのせ、5.3KPaで加圧しながら、N2フロー(0.5L/min)下、上記銅フィルムをホットプレート上で120℃・30秒加熱した。その後、室温まで放冷し、ブロック共重合体120を得た(実施例120)。GPCの分析結果からMw60000、Mn28000、Mw/Mn=2.14であった。 <Example 120: Block copolymer synthesis example>
5 ml of toluene with hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-2 (0.677 g), 0.051 g of N, N, N ′, N′-tetrakis (2-pyridylmethyl) ethylenediamine It was dissolved in. 50 μl of the obtained toluene solution was applied on a copper film (2 cm square) and spin-coated by rotating at 1500 rpm for 30 seconds. As a protective cover for the monomer-containing composition on the copper film, the same copper film (2 cm square) that was used as a support was placed on the monomer-containing composition and N 2 flow (0 .5 L / min), the copper film was heated on a hot plate at 120 ° C. for 30 seconds. Then, it stood to cool to room temperature, and obtained the block copolymer 120 (Example 120). It was Mw60000, Mn28000, Mw / Mn = 2.14 from the analysis result of GPC.
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-2(0.677g)、N,N,N’,N’-テトラキス(2-ピリジルメチル)エチレンジアミン0.051gをトルエン5mlに溶解させた。得られたトルエン溶液50μlを銅フィルム上(2cm角)に塗布して、1500回転/分、30秒間回転させてスピンコートした。上記銅フィルム上のモノマー含有組成物の保護カバーとしてさらにモノマー含有組成物上に支持体として用いたものを同じ銅フィルム(2cm角)をのせ、5.3KPaで加圧しながら、N2フロー(0.5L/min)下、上記銅フィルムをホットプレート上で120℃・30秒加熱した。その後、室温まで放冷し、ブロック共重合体120を得た(実施例120)。GPCの分析結果からMw60000、Mn28000、Mw/Mn=2.14であった。 <Example 120: Block copolymer synthesis example>
5 ml of toluene with hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-2 (0.677 g), 0.051 g of N, N, N ′, N′-tetrakis (2-pyridylmethyl) ethylenediamine It was dissolved in. 50 μl of the obtained toluene solution was applied on a copper film (2 cm square) and spin-coated by rotating at 1500 rpm for 30 seconds. As a protective cover for the monomer-containing composition on the copper film, the same copper film (2 cm square) that was used as a support was placed on the monomer-containing composition and N 2 flow (0 .5 L / min), the copper film was heated on a hot plate at 120 ° C. for 30 seconds. Then, it stood to cool to room temperature, and obtained the block copolymer 120 (Example 120). It was Mw60000, Mn28000, Mw / Mn = 2.14 from the analysis result of GPC.
<実施例121:ブロック共重合体合成例>
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-2(0.677g)、トリス[2-(ジメチルアミノ)エチル]アミン0.028gをトルエン5mlに溶解させた。得られたトルエン溶液50μlを銅フィルム上に塗布して、1500回転/分、30秒間回転させてスピンコートした。上記銅フィルム上のモノマー含有組成物の保護カバーとしてさらにモノマー含有組成物上に支持体として用いたものを同じ銅フィルム(2cm角)をのせ、5.3KPaで加圧しながら、N2フロー(0.5L/min)下、上記銅フィルムをホットプレート上で120℃・30秒加熱した。その後、室温まで放冷し、ブロック共重合体121を得た(実施例121)。GPCの分析結果からMw29500、Mn21000、Mw/Mn=1.40であった。 <Example 121: Block copolymer synthesis example>
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-2 (0.677 g) and 0.028 g of tris [2- (dimethylamino) ethyl] amine were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied on a copper film, and spin-coated by rotating at 1500 rpm for 30 seconds. As a protective cover for the monomer-containing composition on the copper film, the same copper film (2 cm square) that was used as a support was placed on the monomer-containing composition and N 2 flow (0 .5 L / min), the copper film was heated on a hot plate at 120 ° C. for 30 seconds. Then, it stood to cool to room temperature, and obtained the block copolymer 121 (Example 121). From the analysis results of GPC, it was Mw29500, Mn21000, Mw / Mn = 1.40.
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-2(0.677g)、トリス[2-(ジメチルアミノ)エチル]アミン0.028gをトルエン5mlに溶解させた。得られたトルエン溶液50μlを銅フィルム上に塗布して、1500回転/分、30秒間回転させてスピンコートした。上記銅フィルム上のモノマー含有組成物の保護カバーとしてさらにモノマー含有組成物上に支持体として用いたものを同じ銅フィルム(2cm角)をのせ、5.3KPaで加圧しながら、N2フロー(0.5L/min)下、上記銅フィルムをホットプレート上で120℃・30秒加熱した。その後、室温まで放冷し、ブロック共重合体121を得た(実施例121)。GPCの分析結果からMw29500、Mn21000、Mw/Mn=1.40であった。 <Example 121: Block copolymer synthesis example>
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-2 (0.677 g) and 0.028 g of tris [2- (dimethylamino) ethyl] amine were dissolved in 5 ml of toluene. 50 μl of the obtained toluene solution was applied on a copper film, and spin-coated by rotating at 1500 rpm for 30 seconds. As a protective cover for the monomer-containing composition on the copper film, the same copper film (2 cm square) that was used as a support was placed on the monomer-containing composition and N 2 flow (0 .5 L / min), the copper film was heated on a hot plate at 120 ° C. for 30 seconds. Then, it stood to cool to room temperature, and obtained the block copolymer 121 (Example 121). From the analysis results of GPC, it was Mw29500, Mn21000, Mw / Mn = 1.40.
[比較例101~105:溶液中でのATRP]
(ブロック共重合体比較合成例101)
親水性マクロ開始剤A-3(0.4122g)、液晶性モノマーB-1(1.97g)、N,N,N’,N’’,N’’-ペンタメチルジエチレントリアミン(PMDETA)0.0832g、臭化銅(I)(0.069g)をアニソール20mlに溶解させた。窒素フローを開始し、オイルバス80℃・10時間加熱攪拌した。その後、室温まで放冷し、アルミナ処理した後にTHF200mlにて洗浄し、濾過・濃縮した。さらに得られたポリマーをヘキサン250mlに懸濁して、1時間後に濾過した。ブロック共重合体D-15を0.50g得た。GPCの分析結果からMw26900、Mn22000、Mw/Mn=1.22であった。アルミナ処理後の銅イオンの残存は130ppmであった。

[Comparative Examples 101 to 105: ATRP in solution]
(Block copolymer comparative synthesis example 101)
Hydrophilic macroinitiator A-3 (0.4122 g), liquid crystalline monomer B-1 (1.97 g), N, N, N ′, N ″, N ″ -pentamethyldiethylenetriamine (PMDETA) 0.0832 g Copper (I) bromide (0.069 g) was dissolved in 20 ml of anisole. Nitrogen flow was started and the oil bath was heated and stirred at 80 ° C. for 10 hours. Thereafter, the mixture was allowed to cool to room temperature, treated with alumina, washed with 200 ml of THF, filtered and concentrated. Further, the obtained polymer was suspended in 250 ml of hexane and filtered after 1 hour. 0.50 g of block copolymer D-15 was obtained. From the analysis results of GPC, it was Mw26900, Mn22000, Mw / Mn = 1.22. The residual copper ion after the alumina treatment was 130 ppm.
(ブロック共重合体比較合成例101)
親水性マクロ開始剤A-3(0.4122g)、液晶性モノマーB-1(1.97g)、N,N,N’,N’’,N’’-ペンタメチルジエチレントリアミン(PMDETA)0.0832g、臭化銅(I)(0.069g)をアニソール20mlに溶解させた。窒素フローを開始し、オイルバス80℃・10時間加熱攪拌した。その後、室温まで放冷し、アルミナ処理した後にTHF200mlにて洗浄し、濾過・濃縮した。さらに得られたポリマーをヘキサン250mlに懸濁して、1時間後に濾過した。ブロック共重合体D-15を0.50g得た。GPCの分析結果からMw26900、Mn22000、Mw/Mn=1.22であった。アルミナ処理後の銅イオンの残存は130ppmであった。
(Block copolymer comparative synthesis example 101)
Hydrophilic macroinitiator A-3 (0.4122 g), liquid crystalline monomer B-1 (1.97 g), N, N, N ′, N ″, N ″ -pentamethyldiethylenetriamine (PMDETA) 0.0832 g Copper (I) bromide (0.069 g) was dissolved in 20 ml of anisole. Nitrogen flow was started and the oil bath was heated and stirred at 80 ° C. for 10 hours. Thereafter, the mixture was allowed to cool to room temperature, treated with alumina, washed with 200 ml of THF, filtered and concentrated. Further, the obtained polymer was suspended in 250 ml of hexane and filtered after 1 hour. 0.50 g of block copolymer D-15 was obtained. From the analysis results of GPC, it was Mw26900, Mn22000, Mw / Mn = 1.22. The residual copper ion after the alumina treatment was 130 ppm.
(ブロック共重合体比較合成例102)
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-2(0.677g)、PMDETA0.021g、臭化銅(I)(0.017g)をキシレン5mlに溶解させた。窒素フローを開始し、オイルバス80℃・18時間加熱攪拌した。その後、室温まで放冷し、ヘキサン200mlに反応溶液を添加して、濾過した。さらに得られたブロック共重合体D-16をヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後に濾過した。ブロック共重合体D-16を0.56g得た。GPCの分析結果からMw26800、Mn20500、Mw/Mn=1.31であった。

得られたブロック共重合体D-16を0.1g、THF5mlに完溶させた後、吸着剤としてアルミナ、KW700SN、アンバーリスト15 DRYを0.1g加え、室温で1h撹拌。セライトろ過の後にTHF10mlでの洗浄後、溶媒留去した。その後ブロック共重合体D-16の1H-NMRを測定したところいずれもオキセタニル部位が分解していることを確認した。これは吸着剤がルイス酸として働き、オキセタニル基の開環を促進したと考えている。アルミナ処理後の銅イオンの残存はブロック共重合体D-16に対して50ppmであった。 (Block copolymer comparative synthesis example 102)
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-2 (0.677 g), PMDETA 0.021 g, and copper (I) bromide (0.017 g) were dissolved in 5 ml of xylene. Nitrogen flow was started and the oil bath was heated and stirred at 80 ° C. for 18 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 200 ml of hexane and filtered. Further, the obtained block copolymer D-16 was suspended in hexane / ethyl acetate = 20 ml / 30 ml, and filtered after 1 hour. 0.56 g of block copolymer D-16 was obtained. It was Mw26800, Mn20500, Mw / Mn = 1.31 from the analysis result of GPC.
After 0.1 g of the obtained block copolymer D-16 was completely dissolved in 5 ml of THF, 0.1 g of alumina, KW700SN andAmberlyst 15 DRY as adsorbents was added, and the mixture was stirred at room temperature for 1 h. After celite filtration, the solvent was distilled off after washing with 10 ml of THF. Thereafter, 1 H-NMR of the block copolymer D-16 was measured, and it was confirmed that all of the oxetanyl sites were decomposed. This is because the adsorbent worked as a Lewis acid and promoted the ring opening of the oxetanyl group. The residual copper ion after the alumina treatment was 50 ppm with respect to the block copolymer D-16.
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-2(0.677g)、PMDETA0.021g、臭化銅(I)(0.017g)をキシレン5mlに溶解させた。窒素フローを開始し、オイルバス80℃・18時間加熱攪拌した。その後、室温まで放冷し、ヘキサン200mlに反応溶液を添加して、濾過した。さらに得られたブロック共重合体D-16をヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後に濾過した。ブロック共重合体D-16を0.56g得た。GPCの分析結果からMw26800、Mn20500、Mw/Mn=1.31であった。
得られたブロック共重合体D-16を0.1g、THF5mlに完溶させた後、吸着剤としてアルミナ、KW700SN、アンバーリスト15 DRYを0.1g加え、室温で1h撹拌。セライトろ過の後にTHF10mlでの洗浄後、溶媒留去した。その後ブロック共重合体D-16の1H-NMRを測定したところいずれもオキセタニル部位が分解していることを確認した。これは吸着剤がルイス酸として働き、オキセタニル基の開環を促進したと考えている。アルミナ処理後の銅イオンの残存はブロック共重合体D-16に対して50ppmであった。 (Block copolymer comparative synthesis example 102)
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-2 (0.677 g), PMDETA 0.021 g, and copper (I) bromide (0.017 g) were dissolved in 5 ml of xylene. Nitrogen flow was started and the oil bath was heated and stirred at 80 ° C. for 18 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 200 ml of hexane and filtered. Further, the obtained block copolymer D-16 was suspended in hexane / ethyl acetate = 20 ml / 30 ml, and filtered after 1 hour. 0.56 g of block copolymer D-16 was obtained. It was Mw26800, Mn20500, Mw / Mn = 1.31 from the analysis result of GPC.
After 0.1 g of the obtained block copolymer D-16 was completely dissolved in 5 ml of THF, 0.1 g of alumina, KW700SN and
(比較例103)
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-3(0.658g)、PMDETA0.021g、臭化銅(I)(0.017g)をアニソール5mlに溶解させた。窒素フローを開始し、オイルバス80℃・12時間加熱攪拌した。その後、室温まで放冷し、ヘキサン200mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後に濾過した。ブロック共重合体D-17を0.45g得た。GPCの分析結果からMw26800、Mn20500、Mw/Mn=1.31であった。

得られたブロック共重合体D-17を0.1g、THF5mlに完溶させた後、吸着剤としてアルミナを0.1g加え、室温で1h撹拌。セライトろ過の後にTHF10mlでの洗浄後、溶媒留去した。その後ブロック共重合体D-17の1H-NMRを測定したところいずれもオキセタニル部位が分解していることを確認した。これは吸着剤がルイス酸として働き、オキセタニル基の開環を促進したと考えている。アルミナ処理後の銅イオンの残存はブロック共重合体D-17に対して120ppmであった。 (Comparative Example 103)
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-3 (0.658 g), PMDETA 0.021 g, and copper (I) bromide (0.017 g) were dissolved in 5 ml of anisole. Nitrogen flow was started, and the oil bath was heated and stirred at 80 ° C. for 12 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 200 ml of hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.45 g of block copolymer D-17 was obtained. It was Mw26800, Mn20500, Mw / Mn = 1.31 from the analysis result of GPC.
After 0.1 g of the obtained block copolymer D-17 was completely dissolved in 5 ml of THF, 0.1 g of alumina was added as an adsorbent and stirred at room temperature for 1 h. After celite filtration, the solvent was distilled off after washing with 10 ml of THF. Thereafter, 1 H-NMR of the block copolymer D-17 was measured, and it was confirmed that all of the oxetanyl sites were decomposed. This is because the adsorbent worked as a Lewis acid and promoted the ring opening of the oxetanyl group. The residual copper ion after the alumina treatment was 120 ppm with respect to the block copolymer D-17.
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-3(0.658g)、PMDETA0.021g、臭化銅(I)(0.017g)をアニソール5mlに溶解させた。窒素フローを開始し、オイルバス80℃・12時間加熱攪拌した。その後、室温まで放冷し、ヘキサン200mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後に濾過した。ブロック共重合体D-17を0.45g得た。GPCの分析結果からMw26800、Mn20500、Mw/Mn=1.31であった。
得られたブロック共重合体D-17を0.1g、THF5mlに完溶させた後、吸着剤としてアルミナを0.1g加え、室温で1h撹拌。セライトろ過の後にTHF10mlでの洗浄後、溶媒留去した。その後ブロック共重合体D-17の1H-NMRを測定したところいずれもオキセタニル部位が分解していることを確認した。これは吸着剤がルイス酸として働き、オキセタニル基の開環を促進したと考えている。アルミナ処理後の銅イオンの残存はブロック共重合体D-17に対して120ppmであった。 (Comparative Example 103)
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-3 (0.658 g), PMDETA 0.021 g, and copper (I) bromide (0.017 g) were dissolved in 5 ml of anisole. Nitrogen flow was started, and the oil bath was heated and stirred at 80 ° C. for 12 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 200 ml of hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.45 g of block copolymer D-17 was obtained. It was Mw26800, Mn20500, Mw / Mn = 1.31 from the analysis result of GPC.
After 0.1 g of the obtained block copolymer D-17 was completely dissolved in 5 ml of THF, 0.1 g of alumina was added as an adsorbent and stirred at room temperature for 1 h. After celite filtration, the solvent was distilled off after washing with 10 ml of THF. Thereafter, 1 H-NMR of the block copolymer D-17 was measured, and it was confirmed that all of the oxetanyl sites were decomposed. This is because the adsorbent worked as a Lewis acid and promoted the ring opening of the oxetanyl group. The residual copper ion after the alumina treatment was 120 ppm with respect to the block copolymer D-17.
(比較例104)
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-4(0.773g)、PMDETA0.021g、臭化銅(I)(0.017g)をアニソール5mlに溶解させた。窒素フローを開始し、オイルバス80℃・17時間加熱攪拌した。その後、室温まで放冷し、ヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後に濾過した。ブロック共重合体D-18を0.48g得た。GPCの分析結果からMw30500、Mn23800、Mw/Mn=1.28であった。

得られたブロック共重合体D-18を0.1g、THF5mlに完溶させた後、吸着剤としてアルミナを0.1g加え、室温で1h撹拌。セライトろ過の後にTHF10mlでの洗浄後、溶媒留去した。その後ブロック共重合体D-18の1H-NMRを測定したところいずれもオキセタニル部位が分解していることを確認した。これは吸着剤がルイス酸として働き、オキセタニル基の開環を促進したと考えている。アルミナ処理後の銅イオンの残存はブロック共重合体D-18に対して220ppmであった。 (Comparative Example 104)
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-4 (0.773 g), PMDETA 0.021 g, and copper (I) bromide (0.017 g) were dissolved in 5 ml of anisole. Nitrogen flow was started and the oil bath was heated and stirred at 80 ° C. for 17 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.48 g of block copolymer D-18 was obtained. It was Mw30500, Mn23800, Mw / Mn = 1.28 from the analysis result of GPC.
After 0.1 g of the obtained block copolymer D-18 and 5 ml of THF were completely dissolved, 0.1 g of alumina was added as an adsorbent and stirred at room temperature for 1 h. After celite filtration, the solvent was distilled off after washing with 10 ml of THF. Thereafter, 1 H-NMR of the block copolymer D-18 was measured, and it was confirmed that all of the oxetanyl sites were decomposed. This is because the adsorbent worked as a Lewis acid and promoted the ring opening of the oxetanyl group. The residual copper ion after the alumina treatment was 220 ppm with respect to the block copolymer D-18.
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-4(0.773g)、PMDETA0.021g、臭化銅(I)(0.017g)をアニソール5mlに溶解させた。窒素フローを開始し、オイルバス80℃・17時間加熱攪拌した。その後、室温まで放冷し、ヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/30mlに懸濁して、1時間後に濾過した。ブロック共重合体D-18を0.48g得た。GPCの分析結果からMw30500、Mn23800、Mw/Mn=1.28であった。
得られたブロック共重合体D-18を0.1g、THF5mlに完溶させた後、吸着剤としてアルミナを0.1g加え、室温で1h撹拌。セライトろ過の後にTHF10mlでの洗浄後、溶媒留去した。その後ブロック共重合体D-18の1H-NMRを測定したところいずれもオキセタニル部位が分解していることを確認した。これは吸着剤がルイス酸として働き、オキセタニル基の開環を促進したと考えている。アルミナ処理後の銅イオンの残存はブロック共重合体D-18に対して220ppmであった。 (Comparative Example 104)
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystalline monomer B-4 (0.773 g), PMDETA 0.021 g, and copper (I) bromide (0.017 g) were dissolved in 5 ml of anisole. Nitrogen flow was started and the oil bath was heated and stirred at 80 ° C. for 17 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 30 ml and filtered after 1 hour. 0.48 g of block copolymer D-18 was obtained. It was Mw30500, Mn23800, Mw / Mn = 1.28 from the analysis result of GPC.
After 0.1 g of the obtained block copolymer D-18 and 5 ml of THF were completely dissolved, 0.1 g of alumina was added as an adsorbent and stirred at room temperature for 1 h. After celite filtration, the solvent was distilled off after washing with 10 ml of THF. Thereafter, 1 H-NMR of the block copolymer D-18 was measured, and it was confirmed that all of the oxetanyl sites were decomposed. This is because the adsorbent worked as a Lewis acid and promoted the ring opening of the oxetanyl group. The residual copper ion after the alumina treatment was 220 ppm with respect to the block copolymer D-18.
(比較例105)
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-5(0.4885g)、PMDETA0.021g、臭化銅(I)(0.017g)をトルエン5mlに溶解させた。窒素フローを開始し、オイルバス80℃・24時間加熱攪拌した。その後、室温まで放冷し、ヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/50mlに懸濁して、1時間後に濾過した。ブロック共重合体D-19を0.06g得た。GPCの分析結果からMw12300、Mn9800、Mw/Mn=1.25であった。

得られたブロック共重合体D-19を0.1g、THF5mlに完溶させた後、吸着剤としてアルミナを0.1g加え、室温で1h撹拌。セライトろ過の後にTHF10mlでの洗浄後、溶媒留去した。アルミナ処理後の銅イオンの残存はブロック共重合体D-19に対して150ppmであった。 (Comparative Example 105)
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystal monomer B-5 (0.4885 g), PMDETA 0.021 g, and copper (I) bromide (0.017 g) were dissolved in 5 ml of toluene. Nitrogen flow was started, and the oil bath was heated and stirred at 80 ° C. for 24 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 50 ml and filtered after 1 hour. 0.06 g of block copolymer D-19 was obtained. It was Mw12300, Mn9800, Mw / Mn = 1.25 from the analysis result of GPC.
After 0.1 g of the obtained block copolymer D-19 was completely dissolved in 5 ml of THF, 0.1 g of alumina was added as an adsorbent and stirred at room temperature for 1 h. After celite filtration, the solvent was distilled off after washing with 10 ml of THF. The residual copper ion after the alumina treatment was 150 ppm with respect to the block copolymer D-19.
親水性マクロ開始剤A-3(0.103g)、液晶性モノマーB-5(0.4885g)、PMDETA0.021g、臭化銅(I)(0.017g)をトルエン5mlに溶解させた。窒素フローを開始し、オイルバス80℃・24時間加熱攪拌した。その後、室温まで放冷し、ヘキサン300mlに反応溶液を添加して、濾過した。さらに得られたポリマーをヘキサン/酢酸エチル=20ml/50mlに懸濁して、1時間後に濾過した。ブロック共重合体D-19を0.06g得た。GPCの分析結果からMw12300、Mn9800、Mw/Mn=1.25であった。
得られたブロック共重合体D-19を0.1g、THF5mlに完溶させた後、吸着剤としてアルミナを0.1g加え、室温で1h撹拌。セライトろ過の後にTHF10mlでの洗浄後、溶媒留去した。アルミナ処理後の銅イオンの残存はブロック共重合体D-19に対して150ppmであった。 (Comparative Example 105)
Hydrophilic macroinitiator A-3 (0.103 g), liquid crystal monomer B-5 (0.4885 g), PMDETA 0.021 g, and copper (I) bromide (0.017 g) were dissolved in 5 ml of toluene. Nitrogen flow was started, and the oil bath was heated and stirred at 80 ° C. for 24 hours. Thereafter, the mixture was allowed to cool to room temperature, and the reaction solution was added to 300 ml of hexane and filtered. Further, the obtained polymer was suspended in hexane / ethyl acetate = 20 ml / 50 ml and filtered after 1 hour. 0.06 g of block copolymer D-19 was obtained. It was Mw12300, Mn9800, Mw / Mn = 1.25 from the analysis result of GPC.
After 0.1 g of the obtained block copolymer D-19 was completely dissolved in 5 ml of THF, 0.1 g of alumina was added as an adsorbent and stirred at room temperature for 1 h. After celite filtration, the solvent was distilled off after washing with 10 ml of THF. The residual copper ion after the alumina treatment was 150 ppm with respect to the block copolymer D-19.
以上の実施例101~121と比較例101~105の比較から、本発明の支持体上でのリビング重合体の製造方法によれば、ATRPの場合にリビング重合体(特にブロック共重合体)を短時間で経済合理的に製造できることがわかった。
また、本発明の支持体上でのリビング重合体の製造方法を用いることにより、ミクロ相分離構造膜を経済合理的に製造できることがわかった。
さらに、実施例101~121で得られたリビング重合体(ブロック共重合体)では、銅イオンの残存量はブロック共重合体に対して5ppm以下であった。なお、本明細書中では、銅イオンの残存量は群馬県立産業技術センター研究報告2010に記載の方法で求めた。 From the comparison of Examples 101 to 121 and Comparative Examples 101 to 105, according to the method for producing a living polymer on a support of the present invention, a living polymer (particularly a block copolymer) is used in the case of ATRP. It was found that it can be economically and reasonably manufactured in a short time.
It was also found that a microphase separation structure membrane can be produced economically rationally by using the method for producing a living polymer on the support of the present invention.
Furthermore, in the living polymers (block copolymers) obtained in Examples 101 to 121, the remaining amount of copper ions was 5 ppm or less with respect to the block copolymer. In the present specification, the remaining amount of copper ions was determined by the method described in Gunma Prefectural Industrial Technology Center Research Report 2010.
また、本発明の支持体上でのリビング重合体の製造方法を用いることにより、ミクロ相分離構造膜を経済合理的に製造できることがわかった。
さらに、実施例101~121で得られたリビング重合体(ブロック共重合体)では、銅イオンの残存量はブロック共重合体に対して5ppm以下であった。なお、本明細書中では、銅イオンの残存量は群馬県立産業技術センター研究報告2010に記載の方法で求めた。 From the comparison of Examples 101 to 121 and Comparative Examples 101 to 105, according to the method for producing a living polymer on a support of the present invention, a living polymer (particularly a block copolymer) is used in the case of ATRP. It was found that it can be economically and reasonably manufactured in a short time.
It was also found that a microphase separation structure membrane can be produced economically rationally by using the method for producing a living polymer on the support of the present invention.
Furthermore, in the living polymers (block copolymers) obtained in Examples 101 to 121, the remaining amount of copper ions was 5 ppm or less with respect to the block copolymer. In the present specification, the remaining amount of copper ions was determined by the method described in Gunma Prefectural Industrial Technology Center Research Report 2010.
1 リビング重合体(ブロック共重合体)のピーク
2 残存開始剤のピーク
3 残存モノマーのピーク
4 GPC測定エリア
11 送り出しロール
12 塗布装置
13 乾燥ゾーン
14 第一の熟成ゾーン
15 第二の熟成ゾーン
16 UV照射ゾーン
17 剥ぎ取りロール
18 巻取りロール
21 支持体
22 モノマー含有組成物
23 ミクロ相分離構造膜 1 Peak of Living Polymer (Block Copolymer) 2 Peak ofResidual Initiator 3 Peak of Residual Monomer 4 GPC Measurement Area 11 Feed Roll 12 Coating Device 13 Drying Zone 14 First Aging Zone 15 Second Aging Zone 16 UV Irradiation zone 17 Stripping roll 18 Winding roll 21 Support 22 Monomer-containing composition 23 Microphase separation structure membrane
2 残存開始剤のピーク
3 残存モノマーのピーク
4 GPC測定エリア
11 送り出しロール
12 塗布装置
13 乾燥ゾーン
14 第一の熟成ゾーン
15 第二の熟成ゾーン
16 UV照射ゾーン
17 剥ぎ取りロール
18 巻取りロール
21 支持体
22 モノマー含有組成物
23 ミクロ相分離構造膜 1 Peak of Living Polymer (Block Copolymer) 2 Peak of
Claims (24)
- 支持体上でモノマー含有組成物をリビング重合させる工程を含むことを特徴とする支持体上でのリビング重合体の製造方法。 A method for producing a living polymer on a support, comprising a step of living polymerizing a monomer-containing composition on the support.
- 前記リビング重合が、リビングラジカル重合であることを特徴とする請求項1に記載の支持体上でのリビング重合体の製造方法。 The method for producing a living polymer on a support according to claim 1, wherein the living polymerization is living radical polymerization.
- 前記支持体上で前記モノマー含有組成物をリビング重合させる工程が、1000秒以下であることを特徴とする請求項1または2に記載の支持体上でのリビング重合体の製造方法。 The method for producing a living polymer on a support according to claim 1 or 2, wherein the step of living polymerizing the monomer-containing composition on the support is 1000 seconds or less.
- 前記支持体上で前記モノマー含有組成物をリビング重合させる工程のモノマー消費率を、10~100%に制御することを特徴とする請求項1~3のいずれか一項に記載の支持体上でのリビング重合体の製造方法。 The support according to any one of claims 1 to 3, wherein the monomer consumption rate in the step of living polymerizing the monomer-containing composition on the support is controlled to 10 to 100%. A method for producing a living polymer.
- 前記支持体上で前記モノマー含有組成物をリビング重合させる工程が、50~200℃での加熱であることを特徴とする請求項1~4のいずれか一項に記載の支持体上でのリビング重合体の製造方法。 The living on the support according to any one of claims 1 to 4, wherein the step of living polymerizing the monomer-containing composition on the support is heating at 50 to 200 ° C. A method for producing a polymer.
- 前記リビング重合体の数平均分子量Mnが1000~100000であることを特徴とする請求項1~5のいずれか一項に記載の支持体上でのリビング重合体の製造方法。 6. The method for producing a living polymer on a support according to claim 1, wherein the living polymer has a number average molecular weight Mn of 1,000 to 100,000.
- 前記リビング重合が原子移動ラジカル重合であり、かつ、
前記支持体が金属製の支持体であることを特徴とする請求項1~6のいずれか一項に記載の支持体上でのリビング重合体の製造方法。 The living polymerization is atom transfer radical polymerization, and
The method for producing a living polymer on a support according to any one of claims 1 to 6, wherein the support is a metal support. - 前記モノマー含有組成物の前記金属製の支持体と接していない面を別の支持体で覆い、前記モノマー含有組成物を挟圧しながら加熱することを特徴とする請求項7に記載の支持体上でのリビング重合体の製造方法。 The surface of the monomer-containing composition that is not in contact with the metal support is covered with another support and heated while sandwiching the monomer-containing composition. For producing living polymer in Japan.
- 前記金属が銅であることを特徴とする請求項7または8に記載の支持体上でのリビング重合体の製造方法。 The method for producing a living polymer on a support according to claim 7 or 8, wherein the metal is copper.
- 前記リビング重合体中の銅イオン濃度が、前記リビング重合体に対して5ppm以下であることを特徴とする請求項7~9のいずれか一項に記載の支持体上でのリビング重合体の製造方法。 The production of the living polymer on the support according to any one of claims 7 to 9, wherein a concentration of copper ions in the living polymer is 5 ppm or less with respect to the living polymer. Method.
- 前記リビング重合が可逆的付加開裂連鎖移動重合であることを特徴とする請求項1~6のいずれか一項に記載の支持体上でのリビング重合体の製造方法。 The method for producing a living polymer on a support according to any one of claims 1 to 6, wherein the living polymerization is a reversible addition-fragmentation chain transfer polymerization.
- 前記モノマー含有組成物が、モノマー成分として重合性棒状液晶化合物を含むことを特徴とする請求項1~11のいずれか一項に記載の支持体上でのリビング重合体の製造方法。 The method for producing a living polymer on a support according to any one of claims 1 to 11, wherein the monomer-containing composition contains a polymerizable rod-like liquid crystal compound as a monomer component.
- 前記リビング重合体が、ブロック共重合体であることを特徴とする請求項1~12のいずれか一項に記載の支持体上でのリビング重合体の製造方法。 The method for producing a living polymer on a support according to any one of claims 1 to 12, wherein the living polymer is a block copolymer.
- 下記(1)および(2)の工程を、支持体上で実施することを特徴とする請求項13に記載の支持体上でのリビング重合体の製造方法。
(1)前記モノマー含有組成物として、アルキレンオキサイド鎖を含むポリマー成分および炭素数2~20のアルキレン鎖を含むモノマー成分を含むモノマー含有組成物、ならびに、アルキレンオキサイド鎖を含むモノマー成分および炭素数2~20のアルキレン鎖を含むポリマー成分を含むモノマー含有組成物のうち少なくとも一方を用いて、該モノマー含有組成物を前記支持体上に適用する工程
(2)前記支持体上で前記モノマー含有組成物をリビング重合させて、アルキレンオキサイド鎖を含むポリマー成分と炭素数2~20のアルキレン鎖を含むポリマー成分とが共有結合にて連結されたブロック共重合体を得る工程 14. The method for producing a living polymer on a support according to claim 13, wherein the following steps (1) and (2) are carried out on the support.
(1) As the monomer-containing composition, a monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms, and a monomer component containing an alkylene oxide chain and a carbon number of 2 (2) applying the monomer-containing composition onto the support using at least one of the monomer-containing compositions containing a polymer component containing 20 to 20 alkylene chains (2) the monomer-containing composition on the support Is a step of living polymerizing to obtain a block copolymer in which a polymer component containing an alkylene oxide chain and a polymer component containing an alkylene chain having 2 to 20 carbon atoms are linked by a covalent bond - 下記(1’)および(2)の工程を、支持体上で実施することを特徴とする請求項13に記載の支持体上でのリビング重合体の製造方法。
(1’)前記モノマー含有組成物として、アルキレンオキサイド鎖を含むポリマー成分および炭素数2~20のアルキレン鎖を含むモノマー成分を含むモノマー含有組成物を用いて、該モノマー含有組成物を前記支持体上に適用する工程
(2)前記支持体上で前記モノマー含有組成物をリビング重合させて、アルキレンオキサイド鎖を含むポリマー成分と炭素数2~20のアルキレン鎖を含むポリマー成分とが共有結合にて連結されたブロック共重合体を得る工程 14. The method for producing a living polymer on a support according to claim 13, wherein the following steps (1 ′) and (2) are carried out on the support.
(1 ′) A monomer-containing composition containing a polymer component containing an alkylene oxide chain and a monomer component containing an alkylene chain having 2 to 20 carbon atoms is used as the monomer-containing composition. (2) The monomer-containing composition is living-polymerized on the support so that the polymer component containing an alkylene oxide chain and the polymer component containing an alkylene chain having 2 to 20 carbon atoms are covalently bonded. Process for obtaining linked block copolymer - 前記炭素数2~20のアルキレン鎖を含むモノマー成分が重合性棒状液晶化合物であることを特徴とする請求項14または15に記載の支持体上でのリビング重合体の製造方法。 16. The method for producing a living polymer on a support according to claim 14, wherein the monomer component containing an alkylene chain having 2 to 20 carbon atoms is a polymerizable rod-like liquid crystal compound.
- 前記ブロック共重合体が、ミクロ相分離構造膜の形成用のブロック共重合体であることを特徴とする請求項14~16のいずれか一項に記載の支持体上でのリビング重合体の製造方法。 The production of a living polymer on a support according to any one of claims 14 to 16, wherein the block copolymer is a block copolymer for forming a microphase-separated structure film. Method.
- 請求項14~17のいずれか一項に記載の支持体上でのリビング重合体の製造方法で得られたことを特徴とするブロック共重合体。 A block copolymer obtained by the method for producing a living polymer on a support according to any one of claims 14 to 17.
- (3)請求項18に記載のブロック共重合体を前記支持体上で熱、光、溶媒蒸気または電場により相分離を促進させ、ミクロ相分離構造を形成する工程を含むことを特徴とするミクロ相分離構造膜の製造方法。 (3) A step comprising accelerating phase separation of the block copolymer according to claim 18 on the support by heat, light, solvent vapor or electric field to form a microphase separation structure. A method for producing a phase separation structure membrane.
- 前記ミクロ相分離構造を形成する工程が、40~250℃での加熱であることを特徴とする請求項19に記載のミクロ相分離構造膜の製造方法。 The method for producing a microphase-separated structure film according to claim 19, wherein the step of forming the microphase-separated structure is heating at 40 to 250 ° C.
- さらに(4)前記ミクロ相分離構造を有するブロック共重合体を前記支持体上で架橋または重合させてミクロ相分離構造を固定化させる工程を含むことを特徴とする請求項19または20に記載のミクロ相分離構造膜の製造方法。 21. The method according to claim 19 or 20, further comprising the step of (4) immobilizing the microphase separation structure by crosslinking or polymerizing the block copolymer having the microphase separation structure on the support. Manufacturing method of micro phase separation structure membrane.
- 前記ミクロ相分離構造膜を、前記支持体から剥ぎ取る工程を含むことを特徴とする請求項19~21のいずれか一項に記載のミクロ相分離構造膜の製造方法。 The method for producing a microphase-separated structure film according to any one of claims 19 to 21, further comprising a step of peeling the microphase-separated structure film from the support.
- 前記ミクロ相分離構造膜の厚みが、1~2000nmであることを特徴とする請求項19~22のいずれか一項に記載のミクロ相分離構造膜の製造方法。 The method for producing a microphase-separated structure film according to any one of claims 19 to 22, wherein the thickness of the microphase-separated structure film is 1 to 2000 nm.
- 請求項19~23のいずれか一項に記載のミクロ相分離構造膜の製造方法で製造されたことを特徴とするミクロ相分離構造膜。 A microphase separation structure membrane produced by the method for producing a microphase separation structure membrane according to any one of claims 19 to 23.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014556388A JP6196635B2 (en) | 2013-01-08 | 2013-12-27 | Method for producing living polymer on support, block copolymer, microphase separation structure membrane using the same, and method for producing the same |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013000876 | 2013-01-08 | ||
| JP2013-000876 | 2013-01-08 | ||
| JP2013092188 | 2013-04-25 | ||
| JP2013-092188 | 2013-04-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014109258A1 true WO2014109258A1 (en) | 2014-07-17 |
Family
ID=51166914
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/085051 WO2014109258A1 (en) | 2013-01-08 | 2013-12-27 | Method for producing living polymer on base, block copolymer, and film having microphase-separated structure produced using same and method for production thereof |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP6196635B2 (en) |
| WO (1) | WO2014109258A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014148659A (en) * | 2013-01-08 | 2014-08-21 | Fujifilm Corp | Block copolymer, microphase separation structure film using the same, and their production method |
| WO2019154740A1 (en) * | 2018-02-06 | 2019-08-15 | Merck Patent Gmbh | Liquid-crystal medium |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002053630A (en) * | 2000-08-08 | 2002-02-19 | Sekisui Chem Co Ltd | Semiconductor substrate and method for producing the same |
| JP2008285617A (en) * | 2007-05-21 | 2008-11-27 | Institute Of Physical & Chemical Research | Preparation of polymer thin film and polymer thin film |
| JP2009035640A (en) * | 2007-08-02 | 2009-02-19 | Mitsui Chemicals Inc | Antibacterial film |
| JP2010275349A (en) * | 2009-05-26 | 2010-12-09 | Tokyo Institute Of Technology | Free-standing polymer thin film |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4321599B2 (en) * | 2007-02-06 | 2009-08-26 | セイコーエプソン株式会社 | Detection element |
| JP2010116466A (en) * | 2008-11-12 | 2010-05-27 | Nippon Oil Corp | Micro phase separation structure membrane, nano porous membrane, and their production method |
-
2013
- 2013-12-27 WO PCT/JP2013/085051 patent/WO2014109258A1/en active Application Filing
- 2013-12-27 JP JP2014556388A patent/JP6196635B2/en not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002053630A (en) * | 2000-08-08 | 2002-02-19 | Sekisui Chem Co Ltd | Semiconductor substrate and method for producing the same |
| JP2008285617A (en) * | 2007-05-21 | 2008-11-27 | Institute Of Physical & Chemical Research | Preparation of polymer thin film and polymer thin film |
| JP2009035640A (en) * | 2007-08-02 | 2009-02-19 | Mitsui Chemicals Inc | Antibacterial film |
| JP2010275349A (en) * | 2009-05-26 | 2010-12-09 | Tokyo Institute Of Technology | Free-standing polymer thin film |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014148659A (en) * | 2013-01-08 | 2014-08-21 | Fujifilm Corp | Block copolymer, microphase separation structure film using the same, and their production method |
| WO2019154740A1 (en) * | 2018-02-06 | 2019-08-15 | Merck Patent Gmbh | Liquid-crystal medium |
| CN111712556A (en) * | 2018-02-06 | 2020-09-25 | 默克专利股份有限公司 | Liquid-crystalline medium |
| CN111712556B (en) * | 2018-02-06 | 2023-10-24 | 默克专利股份有限公司 | Liquid-crystalline medium |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6196635B2 (en) | 2017-09-13 |
| JPWO2014109258A1 (en) | 2017-01-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2014199932A1 (en) | Composition and microphase-separated structure membrane produced using same, and method for producing microphase-separated structure membrane | |
| JP5168976B2 (en) | Biphenyl and terphenyl compounds and polymerizable liquid crystal compositions containing the compounds | |
| EP2965803A1 (en) | Functional polymer membrane, manufacturing method therefor, ion exchange membrane and proton conducting membrane equipped with functional polymer membrane, and ion exchange device | |
| JP2772116B2 (en) | Fluorescent curing degree monitor | |
| WO2014050992A1 (en) | Functional polymer membrane, and method for producing same | |
| JP2010116466A (en) | Micro phase separation structure membrane, nano porous membrane, and their production method | |
| JP2003119179A (en) | Polymerizable iridium complex, its polymer and method for producing the same | |
| WO2012144331A1 (en) | Polymerizable compound having lateral substituent group at terminal ring structure | |
| Pannell et al. | Ferrocenyl containing polysilanes | |
| JP6769131B2 (en) | Polymerizable compounds and optical anisotropics | |
| JP6196635B2 (en) | Method for producing living polymer on support, block copolymer, microphase separation structure membrane using the same, and method for producing the same | |
| US6049000A (en) | Compounds and their application as well as a method of producing liquid crystalline polymers therefrom | |
| CN116322951A (en) | Compounds, compositions and polymeric films | |
| JPH10310612A (en) | Compounds and method for producing liquid crystal polymer from these compounds | |
| CN116234630A (en) | Composition and polymer film | |
| JP6100597B2 (en) | Block copolymer, microphase-separated structure film using the same, and production method thereof | |
| WO2018128084A1 (en) | Polymerizable compound and optically anisotropic body | |
| Wang et al. | Topochemical polymerization of unsymmetrical aryldiacetylene supramolecules with nitrophenyl substituents utilizing C–H⋯ π interactions | |
| JP2008239958A (en) | Polysilane compound and its synthetic method, ultraviolet-ray curable ink composition using the polysilane compound, inkjet recording method using the ultraviolet-ray curable ink composition, and ink container and inkjet recording apparatus, both containing the ultraviolet ray-curable ink composition | |
| Chikada et al. | Intercalation and polymerization in chenodeoxycholic acid channels with retention of a crystalline state | |
| JP2018035126A (en) | Polymerizable compound and optically anisotropic body | |
| JP6958877B2 (en) | Block copolymer | |
| JP2023547043A (en) | film | |
| JP4314351B2 (en) | Polymer-encapsulated Lewis acid metal catalyst | |
| JP7027668B2 (en) | Neutral layer composition |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13870931 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014556388 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13870931 Country of ref document: EP Kind code of ref document: A1 |