WO2014083432A2 - Female intranasal lower dosage strength testosterone gel formulations and use thereof for treating anorgasmia or hypoactive sexual desire disorder - Google Patents
Female intranasal lower dosage strength testosterone gel formulations and use thereof for treating anorgasmia or hypoactive sexual desire disorder Download PDFInfo
- Publication number
- WO2014083432A2 WO2014083432A2 PCT/IB2013/003121 IB2013003121W WO2014083432A2 WO 2014083432 A2 WO2014083432 A2 WO 2014083432A2 IB 2013003121 W IB2013003121 W IB 2013003121W WO 2014083432 A2 WO2014083432 A2 WO 2014083432A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- testosterone
- gel formulation
- formulation
- subject
- nasal administration
- Prior art date
Links
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 title claims abstract description 1626
- 229960003604 testosterone Drugs 0.000 title claims abstract description 784
- 239000000203 mixture Substances 0.000 title claims abstract description 520
- 238000009472 formulation Methods 0.000 title claims abstract description 500
- 208000006262 Psychological Sexual Dysfunctions Diseases 0.000 title claims abstract description 174
- 206010024419 Libido decreased Diseases 0.000 title claims abstract description 165
- 208000017020 hypoactive sexual desire disease Diseases 0.000 title claims abstract description 165
- 206010002652 Anorgasmia Diseases 0.000 title claims abstract description 153
- 238000000034 method Methods 0.000 claims abstract description 120
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical group O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 36
- 239000004359 castor oil Substances 0.000 claims description 34
- 235000019438 castor oil Nutrition 0.000 claims description 34
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 claims description 34
- 230000001965 increasing effect Effects 0.000 claims description 33
- 229940075614 colloidal silicon dioxide Drugs 0.000 claims description 32
- 150000003839 salts Chemical class 0.000 claims description 22
- 239000003795 chemical substances by application Substances 0.000 claims description 19
- 239000003981 vehicle Substances 0.000 claims description 19
- 125000002811 oleoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])/C([H])=C([H])\C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 17
- 239000002904 solvent Substances 0.000 claims description 17
- 239000000080 wetting agent Substances 0.000 claims description 16
- 239000008194 pharmaceutical composition Substances 0.000 claims description 8
- 239000000651 prodrug Substances 0.000 claims 21
- 229940002612 prodrug Drugs 0.000 claims 21
- 239000003937 drug carrier Substances 0.000 claims 7
- 238000011282 treatment Methods 0.000 abstract description 162
- 239000000499 gel Substances 0.000 description 238
- 230000001568 sexual effect Effects 0.000 description 213
- 238000012360 testing method Methods 0.000 description 90
- 239000000902 placebo Substances 0.000 description 82
- 229940068196 placebo Drugs 0.000 description 82
- 230000035946 sexual desire Effects 0.000 description 79
- 239000003814 drug Substances 0.000 description 72
- 229940079593 drug Drugs 0.000 description 68
- 230000000694 effects Effects 0.000 description 67
- 230000000638 stimulation Effects 0.000 description 63
- 230000004044 response Effects 0.000 description 60
- 230000007935 neutral effect Effects 0.000 description 55
- 238000004458 analytical method Methods 0.000 description 39
- 230000037007 arousal Effects 0.000 description 38
- 238000012216 screening Methods 0.000 description 34
- 239000000523 sample Substances 0.000 description 33
- 210000004369 blood Anatomy 0.000 description 32
- 239000008280 blood Substances 0.000 description 32
- 210000004392 genitalia Anatomy 0.000 description 30
- 230000003304 psychophysiological effect Effects 0.000 description 29
- 230000036299 sexual function Effects 0.000 description 29
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 28
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 28
- 230000009429 distress Effects 0.000 description 28
- 238000002560 therapeutic procedure Methods 0.000 description 28
- 239000003098 androgen Substances 0.000 description 26
- 229940011871 estrogen Drugs 0.000 description 26
- 239000000262 estrogen Substances 0.000 description 26
- 239000000825 pharmaceutical preparation Substances 0.000 description 26
- 230000003285 pharmacodynamic effect Effects 0.000 description 25
- 201000001880 Sexual dysfunction Diseases 0.000 description 24
- 229940126534 drug product Drugs 0.000 description 22
- 231100000872 sexual dysfunction Toxicity 0.000 description 21
- NVKAWKQGWWIWPM-ABEVXSGRSA-N 17-β-hydroxy-5-α-Androstan-3-one Chemical compound C1C(=O)CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@H]21 NVKAWKQGWWIWPM-ABEVXSGRSA-N 0.000 description 20
- 229960003473 androstanolone Drugs 0.000 description 20
- 230000007423 decrease Effects 0.000 description 20
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 18
- 108010089417 Sex Hormone-Binding Globulin Proteins 0.000 description 18
- 102100030758 Sex hormone-binding globulin Human genes 0.000 description 18
- 229940088597 hormone Drugs 0.000 description 18
- 239000005556 hormone Substances 0.000 description 18
- 206010040483 Sexual inhibition Diseases 0.000 description 17
- 230000005284 excitation Effects 0.000 description 17
- 230000036470 plasma concentration Effects 0.000 description 16
- 230000036259 sexual stimuli Effects 0.000 description 16
- 208000024891 symptom Diseases 0.000 description 16
- 230000002411 adverse Effects 0.000 description 15
- 210000004727 amygdala Anatomy 0.000 description 15
- 201000010099 disease Diseases 0.000 description 15
- 235000021251 pulses Nutrition 0.000 description 15
- 210000002966 serum Anatomy 0.000 description 15
- 241000282414 Homo sapiens Species 0.000 description 14
- 238000000540 analysis of variance Methods 0.000 description 14
- 230000009245 menopause Effects 0.000 description 14
- 239000000047 product Substances 0.000 description 14
- 210000002700 urine Anatomy 0.000 description 14
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 13
- 229940030486 androgens Drugs 0.000 description 13
- 229960005309 estradiol Drugs 0.000 description 13
- 229930182833 estradiol Natural products 0.000 description 13
- 210000003928 nasal cavity Anatomy 0.000 description 13
- 238000007619 statistical method Methods 0.000 description 13
- 201000009032 substance abuse Diseases 0.000 description 13
- 230000000007 visual effect Effects 0.000 description 13
- BPYKTIZUTYGOLE-IFADSCNNSA-N Bilirubin Chemical compound N1C(=O)C(C)=C(C=C)\C1=C\C1=C(C)C(CCC(O)=O)=C(CC2=C(C(C)=C(\C=C/3C(=C(C=C)C(=O)N\3)C)N2)CCC(O)=O)N1 BPYKTIZUTYGOLE-IFADSCNNSA-N 0.000 description 12
- 102000003946 Prolactin Human genes 0.000 description 12
- 108010057464 Prolactin Proteins 0.000 description 12
- 210000004556 brain Anatomy 0.000 description 12
- 230000003247 decreasing effect Effects 0.000 description 12
- 208000035475 disorder Diseases 0.000 description 12
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 12
- 230000001976 improved effect Effects 0.000 description 12
- 201000004197 inhibited female orgasm Diseases 0.000 description 12
- 229940097325 prolactin Drugs 0.000 description 12
- 238000001356 surgical procedure Methods 0.000 description 12
- OBHRVMZSZIDDEK-UHFFFAOYSA-N urobilinogen Chemical compound CCC1=C(C)C(=O)NC1CC1=C(C)C(CCC(O)=O)=C(CC2=C(C(C)=C(CC3C(=C(CC)C(=O)N3)C)N2)CCC(O)=O)N1 OBHRVMZSZIDDEK-UHFFFAOYSA-N 0.000 description 12
- 102000009027 Albumins Human genes 0.000 description 11
- 108010088751 Albumins Proteins 0.000 description 11
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 11
- 208000005176 Hepatitis C Diseases 0.000 description 11
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 11
- 238000010241 blood sampling Methods 0.000 description 11
- 230000008859 change Effects 0.000 description 11
- 208000014840 female orgasmic disease Diseases 0.000 description 11
- 239000008103 glucose Substances 0.000 description 11
- 208000002672 hepatitis B Diseases 0.000 description 11
- 230000003054 hormonal effect Effects 0.000 description 11
- 210000000265 leukocyte Anatomy 0.000 description 11
- 230000035807 sensation Effects 0.000 description 11
- 235000019615 sensations Nutrition 0.000 description 11
- 229940078749 testosterone nasal gel Drugs 0.000 description 11
- 230000001225 therapeutic effect Effects 0.000 description 11
- 102000001554 Hemoglobins Human genes 0.000 description 10
- 108010054147 Hemoglobins Proteins 0.000 description 10
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 10
- 239000008186 active pharmaceutical agent Substances 0.000 description 10
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 10
- 238000013461 design Methods 0.000 description 10
- 238000003745 diagnosis Methods 0.000 description 10
- -1 illness Substances 0.000 description 10
- 238000002483 medication Methods 0.000 description 10
- 239000000546 pharmaceutical excipient Substances 0.000 description 10
- 239000004480 active ingredient Substances 0.000 description 9
- 230000008901 benefit Effects 0.000 description 9
- 239000000227 bioadhesive Substances 0.000 description 9
- 229940088679 drug related substance Drugs 0.000 description 9
- 230000007717 exclusion Effects 0.000 description 9
- 230000006870 function Effects 0.000 description 9
- 238000005259 measurement Methods 0.000 description 9
- 239000000186 progesterone Substances 0.000 description 9
- 229960003387 progesterone Drugs 0.000 description 9
- 239000012896 selective serotonin reuptake inhibitor Substances 0.000 description 9
- 230000036332 sexual response Effects 0.000 description 9
- 241000725303 Human immunodeficiency virus Species 0.000 description 8
- 238000000585 Mann–Whitney U test Methods 0.000 description 8
- 238000010521 absorption reaction Methods 0.000 description 8
- 230000001154 acute effect Effects 0.000 description 8
- 239000011575 calcium Substances 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 210000003029 clitoris Anatomy 0.000 description 8
- 239000007924 injection Substances 0.000 description 8
- 238000002347 injection Methods 0.000 description 8
- 238000005461 lubrication Methods 0.000 description 8
- 239000003921 oil Substances 0.000 description 8
- 235000019198 oils Nutrition 0.000 description 8
- 239000002245 particle Substances 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 208000020016 psychiatric disease Diseases 0.000 description 8
- 230000035484 reaction time Effects 0.000 description 8
- 230000009257 reactivity Effects 0.000 description 8
- 230000002829 reductive effect Effects 0.000 description 8
- 238000011160 research Methods 0.000 description 8
- 229940124834 selective serotonin reuptake inhibitor Drugs 0.000 description 8
- 230000000699 topical effect Effects 0.000 description 8
- 238000012384 transportation and delivery Methods 0.000 description 8
- 238000002562 urinalysis Methods 0.000 description 8
- 230000036642 wellbeing Effects 0.000 description 8
- 208000019901 Anxiety disease Diseases 0.000 description 7
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 7
- 206010054089 Depressive symptom Diseases 0.000 description 7
- 230000036506 anxiety Effects 0.000 description 7
- 229910052791 calcium Inorganic materials 0.000 description 7
- 235000001465 calcium Nutrition 0.000 description 7
- 230000006378 damage Effects 0.000 description 7
- 230000004064 dysfunction Effects 0.000 description 7
- 230000005484 gravity Effects 0.000 description 7
- 230000006872 improvement Effects 0.000 description 7
- 208000014674 injury Diseases 0.000 description 7
- 230000033001 locomotion Effects 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 230000036651 mood Effects 0.000 description 7
- 208000012201 sexual and gender identity disease Diseases 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 208000004483 Dyspareunia Diseases 0.000 description 6
- 102000012673 Follicle Stimulating Hormone Human genes 0.000 description 6
- 108010079345 Follicle Stimulating Hormone Proteins 0.000 description 6
- 102000009151 Luteinizing Hormone Human genes 0.000 description 6
- 108010073521 Luteinizing Hormone Proteins 0.000 description 6
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 6
- 208000029899 Sexual aversion disease Diseases 0.000 description 6
- 206010046914 Vaginal infection Diseases 0.000 description 6
- 230000009471 action Effects 0.000 description 6
- 230000036772 blood pressure Effects 0.000 description 6
- ZPUCINDJVBIVPJ-LJISPDSOSA-N cocaine Chemical compound O([C@H]1C[C@@H]2CC[C@@H](N2C)[C@H]1C(=O)OC)C(=O)C1=CC=CC=C1 ZPUCINDJVBIVPJ-LJISPDSOSA-N 0.000 description 6
- 230000009850 completed effect Effects 0.000 description 6
- CZWCKYRVOZZJNM-USOAJAOKSA-N dehydroepiandrosterone sulfate Chemical compound C1[C@@H](OS(O)(=O)=O)CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC=C21 CZWCKYRVOZZJNM-USOAJAOKSA-N 0.000 description 6
- 229960003638 dopamine Drugs 0.000 description 6
- 239000002552 dosage form Substances 0.000 description 6
- 238000011156 evaluation Methods 0.000 description 6
- 229940028334 follicle stimulating hormone Drugs 0.000 description 6
- 230000036541 health Effects 0.000 description 6
- 150000002576 ketones Chemical class 0.000 description 6
- 229940040129 luteinizing hormone Drugs 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 238000002156 mixing Methods 0.000 description 6
- 201000006417 multiple sclerosis Diseases 0.000 description 6
- 230000003287 optical effect Effects 0.000 description 6
- 230000002093 peripheral effect Effects 0.000 description 6
- 229950009829 prasterone sulfate Drugs 0.000 description 6
- 239000000583 progesterone congener Substances 0.000 description 6
- 210000002307 prostate Anatomy 0.000 description 6
- 238000013102 re-test Methods 0.000 description 6
- 230000000306 recurrent effect Effects 0.000 description 6
- 208000015891 sexual disease Diseases 0.000 description 6
- 210000001685 thyroid gland Anatomy 0.000 description 6
- DNXHEGUUPJUMQT-UHFFFAOYSA-N (+)-estrone Natural products OC1=CC=C2C3CCC(C)(C(CC4)=O)C4C3CCC2=C1 DNXHEGUUPJUMQT-UHFFFAOYSA-N 0.000 description 5
- 241000283073 Equus caballus Species 0.000 description 5
- 208000021663 Female sexual arousal disease Diseases 0.000 description 5
- 206010057671 Female sexual dysfunction Diseases 0.000 description 5
- 208000002193 Pain Diseases 0.000 description 5
- CZWCKYRVOZZJNM-UHFFFAOYSA-N Prasterone sodium sulfate Natural products C1C(OS(O)(=O)=O)CCC2(C)C3CCC(C)(C(CC4)=O)C4C3CC=C21 CZWCKYRVOZZJNM-UHFFFAOYSA-N 0.000 description 5
- AUYYCJSJGJYCDS-LBPRGKRZSA-N Thyrolar Chemical compound IC1=CC(C[C@H](N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-LBPRGKRZSA-N 0.000 description 5
- 238000008050 Total Bilirubin Reagent Methods 0.000 description 5
- 208000027418 Wounds and injury Diseases 0.000 description 5
- 230000002159 abnormal effect Effects 0.000 description 5
- 239000000427 antigen Substances 0.000 description 5
- 102000036639 antigens Human genes 0.000 description 5
- 108091007433 antigens Proteins 0.000 description 5
- 238000004364 calculation method Methods 0.000 description 5
- 239000004202 carbamide Substances 0.000 description 5
- 238000009535 clinical urine test Methods 0.000 description 5
- 229940109239 creatinine Drugs 0.000 description 5
- 230000002354 daily effect Effects 0.000 description 5
- 238000013498 data listing Methods 0.000 description 5
- 238000009826 distribution Methods 0.000 description 5
- 210000003743 erythrocyte Anatomy 0.000 description 5
- 239000011888 foil Substances 0.000 description 5
- 238000005534 hematocrit Methods 0.000 description 5
- 230000003993 interaction Effects 0.000 description 5
- 229940100652 nasal gel Drugs 0.000 description 5
- 210000005036 nerve Anatomy 0.000 description 5
- 239000002858 neurotransmitter agent Substances 0.000 description 5
- 150000002826 nitrites Chemical class 0.000 description 5
- 230000036407 pain Effects 0.000 description 5
- 230000035935 pregnancy Effects 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 208000020431 spinal cord injury Diseases 0.000 description 5
- 206010046947 vaginismus Diseases 0.000 description 5
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 4
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 4
- 102100036475 Alanine aminotransferase 1 Human genes 0.000 description 4
- 108010082126 Alanine transaminase Proteins 0.000 description 4
- 101710189683 Alkaline protease 1 Proteins 0.000 description 4
- 101710154562 Alkaline proteinase Proteins 0.000 description 4
- 208000035285 Allergic Seasonal Rhinitis Diseases 0.000 description 4
- 102100021253 Antileukoproteinase Human genes 0.000 description 4
- 101710170876 Antileukoproteinase Proteins 0.000 description 4
- 108010003415 Aspartate Aminotransferases Proteins 0.000 description 4
- 102000004625 Aspartate Aminotransferases Human genes 0.000 description 4
- 206010003694 Atrophy Diseases 0.000 description 4
- 101710112538 C-C motif chemokine 27 Proteins 0.000 description 4
- 102000004420 Creatine Kinase Human genes 0.000 description 4
- 108010042126 Creatine kinase Proteins 0.000 description 4
- 108020004206 Gamma-glutamyltransferase Proteins 0.000 description 4
- 108010010234 HDL Lipoproteins Proteins 0.000 description 4
- 102000015779 HDL Lipoproteins Human genes 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- 108010007622 LDL Lipoproteins Proteins 0.000 description 4
- 102000007330 LDL Lipoproteins Human genes 0.000 description 4
- 208000026344 Nasal disease Diseases 0.000 description 4
- 206010028980 Neoplasm Diseases 0.000 description 4
- 208000030880 Nose disease Diseases 0.000 description 4
- 229910019142 PO4 Inorganic materials 0.000 description 4
- 239000004698 Polyethylene Chemical class 0.000 description 4
- 102000011923 Thyrotropin Human genes 0.000 description 4
- 108010061174 Thyrotropin Proteins 0.000 description 4
- LEHOTFFKMJEONL-UHFFFAOYSA-N Uric Acid Chemical compound N1C(=O)NC(=O)C2=C1NC(=O)N2 LEHOTFFKMJEONL-UHFFFAOYSA-N 0.000 description 4
- TVWHNULVHGKJHS-UHFFFAOYSA-N Uric acid Natural products N1C(=O)NC(=O)C2NC(=O)NC21 TVWHNULVHGKJHS-UHFFFAOYSA-N 0.000 description 4
- 208000003728 Vulvodynia Diseases 0.000 description 4
- 206010069055 Vulvovaginal pain Diseases 0.000 description 4
- 230000001919 adrenal effect Effects 0.000 description 4
- 210000004100 adrenal gland Anatomy 0.000 description 4
- 239000003263 anabolic agent Substances 0.000 description 4
- 229940070021 anabolic steroids Drugs 0.000 description 4
- 238000013103 analytical ultracentrifugation Methods 0.000 description 4
- 230000037444 atrophy Effects 0.000 description 4
- 229940049706 benzodiazepine Drugs 0.000 description 4
- 150000001557 benzodiazepines Chemical class 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 210000000481 breast Anatomy 0.000 description 4
- 201000011510 cancer Diseases 0.000 description 4
- 230000001684 chronic effect Effects 0.000 description 4
- 238000004590 computer program Methods 0.000 description 4
- 230000008602 contraction Effects 0.000 description 4
- 239000006071 cream Substances 0.000 description 4
- FMGSKLZLMKYGDP-USOAJAOKSA-N dehydroepiandrosterone Chemical compound C1[C@@H](O)CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC=C21 FMGSKLZLMKYGDP-USOAJAOKSA-N 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 238000000151 deposition Methods 0.000 description 4
- 206010012601 diabetes mellitus Diseases 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- JKKFKPJIXZFSSB-CBZIJGRNSA-N estrone 3-sulfate Chemical compound OS(=O)(=O)OC1=CC=C2[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1 JKKFKPJIXZFSSB-CBZIJGRNSA-N 0.000 description 4
- 238000002599 functional magnetic resonance imaging Methods 0.000 description 4
- 102000006640 gamma-Glutamyltransferase Human genes 0.000 description 4
- 239000011521 glass Substances 0.000 description 4
- 210000002216 heart Anatomy 0.000 description 4
- 201000001881 impotence Diseases 0.000 description 4
- 239000005414 inactive ingredient Substances 0.000 description 4
- 238000009533 lab test Methods 0.000 description 4
- 230000008450 motivation Effects 0.000 description 4
- 210000002850 nasal mucosa Anatomy 0.000 description 4
- 239000000820 nonprescription drug Substances 0.000 description 4
- 210000001672 ovary Anatomy 0.000 description 4
- 230000027758 ovulation cycle Effects 0.000 description 4
- 229960005489 paracetamol Drugs 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 4
- 239000010452 phosphate Substances 0.000 description 4
- 229920000573 polyethylene Chemical class 0.000 description 4
- 238000009597 pregnancy test Methods 0.000 description 4
- 229940063238 premarin Drugs 0.000 description 4
- 230000000284 resting effect Effects 0.000 description 4
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 238000010561 standard procedure Methods 0.000 description 4
- 150000003431 steroids Chemical class 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 230000001839 systemic circulation Effects 0.000 description 4
- 229940037128 systemic glucocorticoids Drugs 0.000 description 4
- 239000005495 thyroid hormone Substances 0.000 description 4
- 229940036555 thyroid hormone Drugs 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 230000017756 tolerance induction in nasopharyngeal-associated lymphoid tissue Effects 0.000 description 4
- 150000003626 triacylglycerols Chemical class 0.000 description 4
- 229940035722 triiodothyronine Drugs 0.000 description 4
- 229940116269 uric acid Drugs 0.000 description 4
- SHXWCVYOXRDMCX-UHFFFAOYSA-N 3,4-methylenedioxymethamphetamine Chemical compound CNC(C)CC1=CC=C2OCOC2=C1 SHXWCVYOXRDMCX-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 206010061623 Adverse drug reaction Diseases 0.000 description 3
- 208000007848 Alcoholism Diseases 0.000 description 3
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 3
- 201000008283 Atrophic Rhinitis Diseases 0.000 description 3
- 208000020925 Bipolar disease Diseases 0.000 description 3
- 206010005133 Bleeding tendencies Diseases 0.000 description 3
- 208000014644 Brain disease Diseases 0.000 description 3
- XUIIKFGFIJCVMT-GFCCVEGCSA-N D-thyroxine Chemical compound IC1=CC(C[C@@H](N)C(O)=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-GFCCVEGCSA-N 0.000 description 3
- 206010011953 Decreased activity Diseases 0.000 description 3
- 206010051055 Deep vein thrombosis Diseases 0.000 description 3
- 206010012289 Dementia Diseases 0.000 description 3
- 208000030453 Drug-Related Side Effects and Adverse reaction Diseases 0.000 description 3
- 208000030814 Eating disease Diseases 0.000 description 3
- 208000002091 Febrile Seizures Diseases 0.000 description 3
- 208000019454 Feeding and Eating disease Diseases 0.000 description 3
- 208000004230 Gender Dysphoria Diseases 0.000 description 3
- 208000029810 Gender identity disease Diseases 0.000 description 3
- 208000031886 HIV Infections Diseases 0.000 description 3
- 208000037357 HIV infectious disease Diseases 0.000 description 3
- 208000031226 Hyperlipidaemia Diseases 0.000 description 3
- 206010058359 Hypogonadism Diseases 0.000 description 3
- 208000005615 Interstitial Cystitis Diseases 0.000 description 3
- 206010022998 Irritability Diseases 0.000 description 3
- 241001313288 Labia Species 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 206010028164 Multiple allergies Diseases 0.000 description 3
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 3
- 206010028762 Nasal septum deviation Diseases 0.000 description 3
- 208000001132 Osteoporosis Diseases 0.000 description 3
- 206010033888 Paraphilia Diseases 0.000 description 3
- 208000029082 Pelvic Inflammatory Disease Diseases 0.000 description 3
- 229920005439 Perspex® Polymers 0.000 description 3
- 239000004743 Polypropylene Substances 0.000 description 3
- 208000028017 Psychotic disease Diseases 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- 206010039088 Rhinitis atrophic Diseases 0.000 description 3
- 206010039094 Rhinitis perennial Diseases 0.000 description 3
- 208000036284 Rhinitis seasonal Diseases 0.000 description 3
- 208000030047 Sexual desire disease Diseases 0.000 description 3
- 208000006011 Stroke Diseases 0.000 description 3
- 208000032109 Transient ischaemic attack Diseases 0.000 description 3
- 208000030886 Traumatic Brain injury Diseases 0.000 description 3
- 208000006374 Uterine Cervicitis Diseases 0.000 description 3
- 201000008100 Vaginitis Diseases 0.000 description 3
- 229960001138 acetylsalicylic acid Drugs 0.000 description 3
- 230000006978 adaptation Effects 0.000 description 3
- 201000007930 alcohol dependence Diseases 0.000 description 3
- 125000003158 alcohol group Chemical group 0.000 description 3
- 201000010105 allergic rhinitis Diseases 0.000 description 3
- 239000005030 aluminium foil Substances 0.000 description 3
- 229940069428 antacid Drugs 0.000 description 3
- 239000003159 antacid agent Substances 0.000 description 3
- 208000024823 antisocial personality disease Diseases 0.000 description 3
- 239000002199 base oil Substances 0.000 description 3
- 235000013405 beer Nutrition 0.000 description 3
- 230000017531 blood circulation Effects 0.000 description 3
- 230000036765 blood level Effects 0.000 description 3
- 210000000988 bone and bone Anatomy 0.000 description 3
- 229930003827 cannabinoid Natural products 0.000 description 3
- 239000003557 cannabinoid Substances 0.000 description 3
- 229940065144 cannabinoids Drugs 0.000 description 3
- 206010008323 cervicitis Diseases 0.000 description 3
- 230000004087 circulation Effects 0.000 description 3
- 229960003920 cocaine Drugs 0.000 description 3
- 238000004891 communication Methods 0.000 description 3
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 3
- 230000008021 deposition Effects 0.000 description 3
- 235000014632 disordered eating Nutrition 0.000 description 3
- 238000012377 drug delivery Methods 0.000 description 3
- 230000002996 emotional effect Effects 0.000 description 3
- 230000002708 enhancing effect Effects 0.000 description 3
- 206010015037 epilepsy Diseases 0.000 description 3
- 208000001780 epistaxis Diseases 0.000 description 3
- 230000002349 favourable effect Effects 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 230000009975 flexible effect Effects 0.000 description 3
- 239000012530 fluid Substances 0.000 description 3
- 239000003163 gonadal steroid hormone Substances 0.000 description 3
- 208000019622 heart disease Diseases 0.000 description 3
- 230000000423 heterosexual effect Effects 0.000 description 3
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 3
- 230000002989 hypothyroidism Effects 0.000 description 3
- 208000003532 hypothyroidism Diseases 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 229910052500 inorganic mineral Inorganic materials 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 208000017169 kidney disease Diseases 0.000 description 3
- 238000002372 labelling Methods 0.000 description 3
- 201000002364 leukopenia Diseases 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 208000019423 liver disease Diseases 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 208000024714 major depressive disease Diseases 0.000 description 3
- 239000011707 mineral Substances 0.000 description 3
- 235000010755 mineral Nutrition 0.000 description 3
- 210000004400 mucous membrane Anatomy 0.000 description 3
- 210000003205 muscle Anatomy 0.000 description 3
- 239000000133 nasal decongestant Substances 0.000 description 3
- 230000004770 neurodegeneration Effects 0.000 description 3
- 208000015122 neurodegenerative disease Diseases 0.000 description 3
- 210000001331 nose Anatomy 0.000 description 3
- 229940127240 opiate Drugs 0.000 description 3
- 208000022719 perennial allergic rhinitis Diseases 0.000 description 3
- 208000033808 peripheral neuropathy Diseases 0.000 description 3
- 230000002085 persistent effect Effects 0.000 description 3
- 239000004926 polymethyl methacrylate Substances 0.000 description 3
- 208000015768 polyposis Diseases 0.000 description 3
- 229920001155 polypropylene Polymers 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000009256 replacement therapy Methods 0.000 description 3
- 230000004043 responsiveness Effects 0.000 description 3
- 238000012552 review Methods 0.000 description 3
- 208000017022 seasonal allergic rhinitis Diseases 0.000 description 3
- 230000001932 seasonal effect Effects 0.000 description 3
- 201000002859 sleep apnea Diseases 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- 230000009974 thixotropic effect Effects 0.000 description 3
- 201000005060 thrombophlebitis Diseases 0.000 description 3
- 229940034208 thyroxine Drugs 0.000 description 3
- XUIIKFGFIJCVMT-UHFFFAOYSA-N thyroxine-binding globulin Natural products IC1=CC(CC([NH3+])C([O-])=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-UHFFFAOYSA-N 0.000 description 3
- 201000010875 transient cerebral ischemia Diseases 0.000 description 3
- 210000001635 urinary tract Anatomy 0.000 description 3
- 210000001215 vagina Anatomy 0.000 description 3
- 238000010200 validation analysis Methods 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- AHOUBRCZNHFOSL-YOEHRIQHSA-N (+)-Casbol Chemical compound C1=CC(F)=CC=C1[C@H]1[C@H](COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-YOEHRIQHSA-N 0.000 description 2
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 2
- 206010067484 Adverse reaction Diseases 0.000 description 2
- 206010002261 Androgen deficiency Diseases 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 206010063659 Aversion Diseases 0.000 description 2
- 206010004146 Basal cell carcinoma Diseases 0.000 description 2
- 208000017667 Chronic Disease Diseases 0.000 description 2
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 2
- DNXHEGUUPJUMQT-CBZIJGRNSA-N Estrone Chemical compound OC1=CC=C2[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1 DNXHEGUUPJUMQT-CBZIJGRNSA-N 0.000 description 2
- 208000036993 Frustration Diseases 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 208000029549 Muscle injury Diseases 0.000 description 2
- 206010033296 Overdoses Diseases 0.000 description 2
- 240000005373 Panax quinquefolius Species 0.000 description 2
- 235000003140 Panax quinquefolius Nutrition 0.000 description 2
- 208000010340 Sleep Deprivation Diseases 0.000 description 2
- PDMMFKSKQVNJMI-BLQWBTBKSA-N Testosterone propionate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](OC(=O)CC)[C@@]1(C)CC2 PDMMFKSKQVNJMI-BLQWBTBKSA-N 0.000 description 2
- 240000000143 Turnera diffusa Species 0.000 description 2
- 230000005856 abnormality Effects 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- 230000006838 adverse reaction Effects 0.000 description 2
- 230000032683 aging Effects 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229940094957 androgens and estrogen Drugs 0.000 description 2
- 210000001971 anterior hypothalamus Anatomy 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 238000004820 blood count Methods 0.000 description 2
- 230000004094 calcium homeostasis Effects 0.000 description 2
- 238000004422 calculation algorithm Methods 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 150000005829 chemical entities Chemical class 0.000 description 2
- 235000019504 cigarettes Nutrition 0.000 description 2
- 230000001113 coital effect Effects 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 239000002131 composite material Substances 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 230000002596 correlated effect Effects 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- 230000008451 emotion Effects 0.000 description 2
- 230000002124 endocrine Effects 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 230000002964 excitative effect Effects 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 210000001508 eye Anatomy 0.000 description 2
- 208000037869 female anorgasmia Diseases 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 230000037406 food intake Effects 0.000 description 2
- 235000012631 food intake Nutrition 0.000 description 2
- 210000000245 forearm Anatomy 0.000 description 2
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 2
- 229920001903 high density polyethylene Polymers 0.000 description 2
- 239000004700 high-density polyethylene Substances 0.000 description 2
- 238000009802 hysterectomy Methods 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- 230000007794 irritation Effects 0.000 description 2
- 230000003907 kidney function Effects 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 230000003908 liver function Effects 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 238000013507 mapping Methods 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 229940126601 medicinal product Drugs 0.000 description 2
- 210000004165 myocardium Anatomy 0.000 description 2
- 210000000492 nasalseptum Anatomy 0.000 description 2
- 231100001079 no serious adverse effect Toxicity 0.000 description 2
- 229960002748 norepinephrine Drugs 0.000 description 2
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 230000002611 ovarian Effects 0.000 description 2
- 238000004806 packaging method and process Methods 0.000 description 2
- 229960002296 paroxetine Drugs 0.000 description 2
- 210000003903 pelvic floor Anatomy 0.000 description 2
- 229940127557 pharmaceutical product Drugs 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 210000003814 preoptic area Anatomy 0.000 description 2
- 238000003825 pressing Methods 0.000 description 2
- 230000037452 priming Effects 0.000 description 2
- 238000000275 quality assurance Methods 0.000 description 2
- 238000011472 radical prostatectomy Methods 0.000 description 2
- 230000001850 reproductive effect Effects 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 230000033764 rhythmic process Effects 0.000 description 2
- 231100000279 safety data Toxicity 0.000 description 2
- 238000009517 secondary packaging Methods 0.000 description 2
- 229940076279 serotonin Drugs 0.000 description 2
- VGKDLMBJGBXTGI-SJCJKPOMSA-N sertraline Chemical compound C1([C@@H]2CC[C@@H](C3=CC=CC=C32)NC)=CC=C(Cl)C(Cl)=C1 VGKDLMBJGBXTGI-SJCJKPOMSA-N 0.000 description 2
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 235000012239 silicon dioxide Nutrition 0.000 description 2
- 210000002027 skeletal muscle Anatomy 0.000 description 2
- 230000007958 sleep Effects 0.000 description 2
- 238000012066 statistical methodology Methods 0.000 description 2
- 239000003270 steroid hormone Substances 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 230000035882 stress Effects 0.000 description 2
- 230000000153 supplemental effect Effects 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- 229960001712 testosterone propionate Drugs 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 238000009804 total hysterectomy Methods 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000008733 trauma Effects 0.000 description 2
- 238000011269 treatment regimen Methods 0.000 description 2
- 230000001960 triggered effect Effects 0.000 description 2
- 235000004952 turnera diffusa Nutrition 0.000 description 2
- 208000019206 urinary tract infection Diseases 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- 235000019154 vitamin C Nutrition 0.000 description 2
- 239000011718 vitamin C Substances 0.000 description 2
- 235000019165 vitamin E Nutrition 0.000 description 2
- 239000011709 vitamin E Substances 0.000 description 2
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 1
- DIWRORZWFLOCLC-HNNXBMFYSA-N (3s)-7-chloro-5-(2-chlorophenyl)-3-hydroxy-1,3-dihydro-1,4-benzodiazepin-2-one Chemical compound N([C@H](C(NC1=CC=C(Cl)C=C11)=O)O)=C1C1=CC=CC=C1Cl DIWRORZWFLOCLC-HNNXBMFYSA-N 0.000 description 1
- UCTWMZQNUQWSLP-VIFPVBQESA-N (R)-adrenaline Chemical compound CNC[C@H](O)C1=CC=C(O)C(O)=C1 UCTWMZQNUQWSLP-VIFPVBQESA-N 0.000 description 1
- 229930182837 (R)-adrenaline Natural products 0.000 description 1
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 description 1
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 1
- OIQOAYVCKAHSEJ-UHFFFAOYSA-N 2-[2,3-bis(2-hydroxyethoxy)propoxy]ethanol;hexadecanoic acid;octadecanoic acid Chemical compound OCCOCC(OCCO)COCCO.CCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O OIQOAYVCKAHSEJ-UHFFFAOYSA-N 0.000 description 1
- ZBMRKNMTMPPMMK-UHFFFAOYSA-N 2-amino-4-[hydroxy(methyl)phosphoryl]butanoic acid;azane Chemical compound [NH4+].CP(O)(=O)CCC(N)C([O-])=O ZBMRKNMTMPPMMK-UHFFFAOYSA-N 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- ZKHQWZAMYRWXGA-KQYNXXCUSA-N Adenosine triphosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-N 0.000 description 1
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 1
- 206010001854 Altered state of consciousness Diseases 0.000 description 1
- 206010003055 Application site reaction Diseases 0.000 description 1
- 241000972773 Aulopiformes Species 0.000 description 1
- 206010048909 Boredom Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 241000252095 Congridae Species 0.000 description 1
- ZZZCUOFIHGPKAK-UHFFFAOYSA-N D-erythro-ascorbic acid Natural products OCC1OC(=O)C(O)=C1O ZZZCUOFIHGPKAK-UHFFFAOYSA-N 0.000 description 1
- 206010012335 Dependence Diseases 0.000 description 1
- 208000020401 Depressive disease Diseases 0.000 description 1
- 206010058929 Disturbance in sexual arousal Diseases 0.000 description 1
- 206010013654 Drug abuse Diseases 0.000 description 1
- 108010049140 Endorphins Proteins 0.000 description 1
- 102000009025 Endorphins Human genes 0.000 description 1
- 208000010228 Erectile Dysfunction Diseases 0.000 description 1
- 206010016275 Fear Diseases 0.000 description 1
- 206010067035 Fear of pregnancy Diseases 0.000 description 1
- 241000282324 Felis Species 0.000 description 1
- 235000011201 Ginkgo Nutrition 0.000 description 1
- 235000008100 Ginkgo biloba Nutrition 0.000 description 1
- 244000194101 Ginkgo biloba Species 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- XKMLYUALXHKNFT-UUOKFMHZSA-N Guanosine-5'-triphosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XKMLYUALXHKNFT-UUOKFMHZSA-N 0.000 description 1
- 240000002045 Guettarda speciosa Species 0.000 description 1
- 235000001287 Guettarda speciosa Nutrition 0.000 description 1
- GVGLGOZIDCSQPN-PVHGPHFFSA-N Heroin Chemical compound O([C@H]1[C@H](C=C[C@H]23)OC(C)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4OC(C)=O GVGLGOZIDCSQPN-PVHGPHFFSA-N 0.000 description 1
- 101000711466 Homo sapiens SAM pointed domain-containing Ets transcription factor Proteins 0.000 description 1
- 208000033830 Hot Flashes Diseases 0.000 description 1
- 206010060800 Hot flush Diseases 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-N L-arginine Chemical compound OC(=O)[C@@H](N)CCCN=C(N)N ODKSFYDXXFIFQN-BYPYZUCNSA-N 0.000 description 1
- 235000014852 L-arginine Nutrition 0.000 description 1
- 229930064664 L-arginine Natural products 0.000 description 1
- 239000011786 L-ascorbyl-6-palmitate Substances 0.000 description 1
- QAQJMLQRFWZOBN-LAUBAEHRSA-N L-ascorbyl-6-palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](O)[C@H]1OC(=O)C(O)=C1O QAQJMLQRFWZOBN-LAUBAEHRSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 241000283953 Lagomorpha Species 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- YJPIGAIKUZMOQA-UHFFFAOYSA-N Melatonin Natural products COC1=CC=C2N(C(C)=O)C=C(CCN)C2=C1 YJPIGAIKUZMOQA-UHFFFAOYSA-N 0.000 description 1
- 208000019022 Mood disease Diseases 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 208000010428 Muscle Weakness Diseases 0.000 description 1
- 206010028372 Muscular weakness Diseases 0.000 description 1
- 206010061533 Myotonia Diseases 0.000 description 1
- RTHCYVBBDHJXIQ-UHFFFAOYSA-N N-methyl-3-phenyl-3-[4-(trifluoromethyl)phenoxy]propan-1-amine Chemical compound C=1C=CC=CC=1C(CCNC)OC1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-UHFFFAOYSA-N 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- IOVCWXUNBOPUCH-UHFFFAOYSA-M Nitrite anion Chemical compound [O-]N=O IOVCWXUNBOPUCH-UHFFFAOYSA-M 0.000 description 1
- 208000026251 Opioid-Related disease Diseases 0.000 description 1
- XNOPRXBHLZRZKH-UHFFFAOYSA-N Oxytocin Natural products N1C(=O)C(N)CSSCC(C(=O)N2C(CCC2)C(=O)NC(CC(C)C)C(=O)NCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(CCC(N)=O)NC(=O)C(C(C)CC)NC(=O)C1CC1=CC=C(O)C=C1 XNOPRXBHLZRZKH-UHFFFAOYSA-N 0.000 description 1
- 102400000050 Oxytocin Human genes 0.000 description 1
- 101800000989 Oxytocin Proteins 0.000 description 1
- 235000005035 Panax pseudoginseng ssp. pseudoginseng Nutrition 0.000 description 1
- AHOUBRCZNHFOSL-UHFFFAOYSA-N Paroxetine hydrochloride Natural products C1=CC(F)=CC=C1C1C(COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-UHFFFAOYSA-N 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 208000000450 Pelvic Pain Diseases 0.000 description 1
- ORNBQBCIOKFOEO-YQUGOWONSA-N Pregnenolone Natural products O=C(C)[C@@H]1[C@@]2(C)[C@H]([C@H]3[C@@H]([C@]4(C)C(=CC3)C[C@@H](O)CC4)CC2)CC1 ORNBQBCIOKFOEO-YQUGOWONSA-N 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 208000001431 Psychomotor Agitation Diseases 0.000 description 1
- 206010038743 Restlessness Diseases 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 102100034018 SAM pointed domain-containing Ets transcription factor Human genes 0.000 description 1
- 208000019802 Sexually transmitted disease Diseases 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 1
- 206010041243 Social avoidant behaviour Diseases 0.000 description 1
- 229920002334 Spandex Polymers 0.000 description 1
- SECKRCOLJRRGGV-UHFFFAOYSA-N Vardenafil Chemical compound CCCC1=NC(C)=C(C(N=2)=O)N1NC=2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(CC)CC1 SECKRCOLJRRGGV-UHFFFAOYSA-N 0.000 description 1
- GXBMIBRIOWHPDT-UHFFFAOYSA-N Vasopressin Natural products N1C(=O)C(CC=2C=C(O)C=CC=2)NC(=O)C(N)CSSCC(C(=O)N2C(CCC2)C(=O)NC(CCCN=C(N)N)C(=O)NCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(CCC(N)=O)NC(=O)C1CC1=CC=CC=C1 GXBMIBRIOWHPDT-UHFFFAOYSA-N 0.000 description 1
- 108010004977 Vasopressins Proteins 0.000 description 1
- 102000002852 Vasopressins Human genes 0.000 description 1
- 229930003268 Vitamin C Natural products 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- WAHQVRCNDCHDIB-QZYSPNBYSA-N [(3s,8r,9s,10r,13s,14s,17r)-17-acetyl-17-acetyloxy-6,10,13-trimethyl-1,2,3,8,9,11,12,14,15,16-decahydrocyclopenta[a]phenanthren-3-yl] 3-cyclopentylpropanoate Chemical group O([C@@H]1C=C2C(C)=C[C@H]3[C@@H]4CC[C@]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)(OC(=O)C)C(C)=O)C(=O)CCC1CCCC1 WAHQVRCNDCHDIB-QZYSPNBYSA-N 0.000 description 1
- 210000001015 abdomen Anatomy 0.000 description 1
- 230000037374 absorbed through the skin Effects 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- OIPILFWXSMYKGL-UHFFFAOYSA-N acetylcholine Chemical compound CC(=O)OCC[N+](C)(C)C OIPILFWXSMYKGL-UHFFFAOYSA-N 0.000 description 1
- 229960004373 acetylcholine Drugs 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 238000012863 analytical testing Methods 0.000 description 1
- 210000003484 anatomy Anatomy 0.000 description 1
- 238000009165 androgen replacement therapy Methods 0.000 description 1
- 230000001548 androgenic effect Effects 0.000 description 1
- 230000001430 anti-depressive effect Effects 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 229940005513 antidepressants Drugs 0.000 description 1
- 229940125715 antihistaminic agent Drugs 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- KBZOIRJILGZLEJ-LGYYRGKSSA-N argipressin Chemical compound C([C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CSSC[C@@H](C(N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N1)=O)N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)NCC(N)=O)C1=CC=CC=C1 KBZOIRJILGZLEJ-LGYYRGKSSA-N 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 235000010385 ascorbyl palmitate Nutrition 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229940125717 barbiturate Drugs 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000002146 bilateral effect Effects 0.000 description 1
- 238000009809 bilateral salpingo-oophorectomy Methods 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 230000037182 bone density Effects 0.000 description 1
- 235000021324 borage oil Nutrition 0.000 description 1
- 239000010474 borage seed oil Substances 0.000 description 1
- 230000008309 brain mechanism Effects 0.000 description 1
- 239000007936 buccal or sublingual tablet Substances 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 206010007821 cauda equina syndrome Diseases 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 230000035606 childbirth Effects 0.000 description 1
- 229960004407 chorionic gonadotrophin Drugs 0.000 description 1
- 230000009194 climbing Effects 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- 239000012568 clinical material Substances 0.000 description 1
- 239000007957 coemulsifier Substances 0.000 description 1
- 230000019771 cognition Effects 0.000 description 1
- 238000009225 cognitive behavioral therapy Methods 0.000 description 1
- 235000020242 coleus extract Nutrition 0.000 description 1
- 238000011970 concomitant therapy Methods 0.000 description 1
- 229940124301 concurrent medication Drugs 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 239000003433 contraceptive agent Substances 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 238000013523 data management Methods 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 229960002069 diamorphine Drugs 0.000 description 1
- 229960003529 diazepam Drugs 0.000 description 1
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 1
- 238000007599 discharging Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 230000000857 drug effect Effects 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 229920001971 elastomer Polymers 0.000 description 1
- 229960005139 epinephrine Drugs 0.000 description 1
- 229960003399 estrone Drugs 0.000 description 1
- 235000008524 evening primrose extract Nutrition 0.000 description 1
- 239000010475 evening primrose oil Substances 0.000 description 1
- 229940089020 evening primrose oil Drugs 0.000 description 1
- 239000004744 fabric Substances 0.000 description 1
- 238000005429 filling process Methods 0.000 description 1
- 238000010579 first pass effect Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229960002464 fluoxetine Drugs 0.000 description 1
- 210000005153 frontal cortex Anatomy 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 239000003349 gelling agent Substances 0.000 description 1
- 235000008434 ginseng Nutrition 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 229940049906 glutamate Drugs 0.000 description 1
- 125000005456 glyceride group Chemical class 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 210000004013 groin Anatomy 0.000 description 1
- 210000004209 hair Anatomy 0.000 description 1
- 230000005802 health problem Effects 0.000 description 1
- 230000003862 health status Effects 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- IIRDTKBZINWQAW-UHFFFAOYSA-N hexaethylene glycol Chemical class OCCOCCOCCOCCOCCOCCO IIRDTKBZINWQAW-UHFFFAOYSA-N 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 238000002657 hormone replacement therapy Methods 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 230000000642 iatrogenic effect Effects 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 206010022437 insomnia Diseases 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 229940074928 isopropyl myristate Drugs 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 238000001325 log-rank test Methods 0.000 description 1
- 229960004391 lorazepam Drugs 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 1
- 238000013160 medical therapy Methods 0.000 description 1
- 229960003987 melatonin Drugs 0.000 description 1
- DRLFMBDRBRZALE-UHFFFAOYSA-N melatonin Chemical compound COC1=CC=C2NC=C(CCNC(C)=O)C2=C1 DRLFMBDRBRZALE-UHFFFAOYSA-N 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000004630 mental health Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 238000001690 micro-dialysis Methods 0.000 description 1
- DDLIGBOFAVUZHB-UHFFFAOYSA-N midazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NC=C2CN=C1C1=CC=CC=C1F DDLIGBOFAVUZHB-UHFFFAOYSA-N 0.000 description 1
- 229960003793 midazolam Drugs 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 210000004877 mucosa Anatomy 0.000 description 1
- 230000004118 muscle contraction Effects 0.000 description 1
- 229940037525 nasal preparations Drugs 0.000 description 1
- 230000001722 neurochemical effect Effects 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 206010029410 night sweats Diseases 0.000 description 1
- 230000036565 night sweats Effects 0.000 description 1
- 210000002445 nipple Anatomy 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000000346 nonvolatile oil Substances 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- 238000009806 oophorectomy Methods 0.000 description 1
- 201000005040 opiate dependence Diseases 0.000 description 1
- 230000002450 orbitofrontal effect Effects 0.000 description 1
- 208000024309 orgasm disease Diseases 0.000 description 1
- 230000011599 ovarian follicle development Effects 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- XNOPRXBHLZRZKH-DSZYJQQASA-N oxytocin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CSSC[C@H](N)C(=O)N1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)NCC(N)=O)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 XNOPRXBHLZRZKH-DSZYJQQASA-N 0.000 description 1
- 229960001723 oxytocin Drugs 0.000 description 1
- 238000012858 packaging process Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 238000005192 partition Methods 0.000 description 1
- 231100000915 pathological change Toxicity 0.000 description 1
- 230000036285 pathological change Effects 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 210000004197 pelvis Anatomy 0.000 description 1
- 210000003899 penis Anatomy 0.000 description 1
- 230000008447 perception Effects 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 229960000249 pregnenolone Drugs 0.000 description 1
- ORNBQBCIOKFOEO-QGVNFLHTSA-N pregnenolone Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 ORNBQBCIOKFOEO-QGVNFLHTSA-N 0.000 description 1
- 206010036596 premature ejaculation Diseases 0.000 description 1
- 239000000955 prescription drug Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000009516 primary packaging Methods 0.000 description 1
- 238000004886 process control Methods 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 150000003180 prostaglandins Chemical class 0.000 description 1
- 238000011471 prostatectomy Methods 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 229940035613 prozac Drugs 0.000 description 1
- 239000001944 prunus armeniaca kernel oil Substances 0.000 description 1
- 230000010490 psychological well-being Effects 0.000 description 1
- 210000004061 pubic symphysis Anatomy 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- XKMLYUALXHKNFT-UHFFFAOYSA-N rGTP Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O XKMLYUALXHKNFT-UHFFFAOYSA-N 0.000 description 1
- 230000011514 reflex Effects 0.000 description 1
- 230000029058 respiratory gaseous exchange Effects 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 230000001020 rhythmical effect Effects 0.000 description 1
- 235000019515 salmon Nutrition 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 229960002073 sertraline Drugs 0.000 description 1
- 229960003310 sildenafil Drugs 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 208000020352 skin basal cell carcinoma Diseases 0.000 description 1
- 201000010106 skin squamous cell carcinoma Diseases 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 239000004759 spandex Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 230000007480 spreading Effects 0.000 description 1
- 238000003892 spreading Methods 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 238000000528 statistical test Methods 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 239000002951 street drug Substances 0.000 description 1
- 239000006190 sub-lingual tablet Substances 0.000 description 1
- 208000011117 substance-related disease Diseases 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 229960000835 tadalafil Drugs 0.000 description 1
- IEHKWSGCTWLXFU-IIBYNOLFSA-N tadalafil Chemical compound C1=C2OCOC2=CC([C@@H]2C3=C([C]4C=CC=CC4=N3)C[C@H]3N2C(=O)CN(C3=O)C)=C1 IEHKWSGCTWLXFU-IIBYNOLFSA-N 0.000 description 1
- 238000004885 tandem mass spectrometry Methods 0.000 description 1
- 229940088956 testosterone transdermal system Drugs 0.000 description 1
- 229960000746 testosterone undecanoate Drugs 0.000 description 1
- UDSFVOAUHKGBEK-CNQKSJKFSA-N testosterone undecanoate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](OC(=O)CCCCCCCCCC)[C@@]1(C)CC2 UDSFVOAUHKGBEK-CNQKSJKFSA-N 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 239000003017 thermal stabilizer Substances 0.000 description 1
- WZDGZWOAQTVYBX-XOINTXKNSA-N tibolone Chemical compound C([C@@H]12)C[C@]3(C)[C@@](C#C)(O)CC[C@H]3[C@@H]1[C@H](C)CC1=C2CCC(=O)C1 WZDGZWOAQTVYBX-XOINTXKNSA-N 0.000 description 1
- 229960001023 tibolone Drugs 0.000 description 1
- 230000005919 time-dependent effect Effects 0.000 description 1
- 229930003799 tocopherol Natural products 0.000 description 1
- 235000010384 tocopherol Nutrition 0.000 description 1
- 229960001295 tocopherol Drugs 0.000 description 1
- 239000011732 tocopherol Substances 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 238000012549 training Methods 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 229960002381 vardenafil Drugs 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 229960003726 vasopressin Drugs 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 235000019155 vitamin A Nutrition 0.000 description 1
- 239000011719 vitamin A Substances 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 230000001755 vocal effect Effects 0.000 description 1
- 239000000341 volatile oil Substances 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 230000003245 working effect Effects 0.000 description 1
- 229940020965 zoloft Drugs 0.000 description 1
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/565—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids not substituted in position 17 beta by a carbon atom, e.g. estrane, estradiol
- A61K31/568—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids not substituted in position 17 beta by a carbon atom, e.g. estrane, estradiol substituted in positions 10 and 13 by a chain having at least one carbon atom, e.g. androstanes, e.g. testosterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/14—Esters of carboxylic acids, e.g. fatty acid monoglycerides, medium-chain triglycerides, parabens or PEG fatty acid esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/44—Oils, fats or waxes according to two or more groups of A61K47/02-A61K47/42; Natural or modified natural oils, fats or waxes, e.g. castor oil, polyethoxylated castor oil, montan wax, lignite, shellac, rosin, beeswax or lanolin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0043—Nose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/06—Ointments; Bases therefor; Other semi-solid forms, e.g. creams, sticks, gels
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
Definitions
- the present invention relates to lower dosage strength intranasal testosterone gels for providing intranasal delivery of testosterone to a female and intranasal treatment methods for treating females with anorgasmia and/or hypoactive sexual desire disorder (HSDD).
- the present invention relates to improved methods and lower dosage strength intranasal testosterone gel formulations for treating female anorgasmia and/or HSDD.
- the present invention also relates to a system for
- testosterone and/or estrogen may result in female sexual disorder, which include clinical symptoms such as lack of sex drive, lack of arousal or pleasure, decreased energy levels or fatigue with blunted motivation, flat mood or depression, reduced sense of well-being, insomnia, irritability, partial decreases in vaginal lubrication, and osteoporosis.
- clinical symptoms such as lack of sex drive, lack of arousal or pleasure, decreased energy levels or fatigue with blunted motivation, flat mood or depression, reduced sense of well-being, insomnia, irritability, partial decreases in vaginal lubrication, and osteoporosis.
- reduced levels of estrogen and/or progesterone in women often result in clinical symptoms including hot flashes, night sweats, vaginal atrophy, decreased libido, and osteoporosis.
- Testosterone has historically been thought of as a male hormone, but it is also synthesized in women in small amounts, primarily by the ovaries and adrenal glands.
- the physiological functions of testosterone in women include, among others, development of pubic and axillary hair, sexual libido; effects on bone density and muscle tone, sexual libido, and overall vitality and sense of psychological well-being.
- Testosterone plasma concentrations in pre-menopausal women normally fluctuate during the menstrual cycle, with the total testosterone plasma concentrations generally ranging between about 15 ng/dL and about 65 ng/dL.
- Hypoactive sexual desire disorder the most common women's sexual problem, is a condition characterized by the lack or absence of sexual fantasies and desire for sexual activity which causes marked distress or interpersonal difficulties.
- the sexual dysfunction is not accounted for by another psychiatric disorder nor is it a result of direct physiological effects of a substance (i.e., drug abuse) or a general medical condition.
- Anorgasmia the second most frequently reported women's sexual problem, is considered to be the persistent or recurrent delay in, or absence of, orgasm following a normal sexual excitement phase, causing marked distress or interpersonal difficulty.
- sexual activity may become a chore or a duty rather than a mutually satisfying, intimate experience. This may also lead to secondary loss of sexual interest and/or interpersonal difficulties.
- hypoactive sexual disorder disease and anorgasmia affect millions of women in the United States.
- Sexual response is a complex and finely tuned process that can be disrupted at various time points in the reproductive life cycle (pre and postpartum, peri and postmenopausal) which likely accounts for the high prevalence of reported sexual dysfunction in the general population of healthy women. See, e.g., Laumann et al., Supra.
- testosterone has central and peripheral effects on sexual function.
- the decline of androgen levels following surgically induced menopause has supported the hypothesis that a decrease in testosterone levels is related to a decrease in sexual desire.
- Testosterone the primary circulation androgen in women, is a naturally occurring steroid.
- androgens are derived from three sources: the adrenal glands, the ovaries and peripheral conversion. Androgens are secreted by the ovaries and the adrenal glands. Contrary to the sudden drop in estrogen during menopause, serum levels of androgens fall gradually as women age, primarily due to a decrease in the production of adrenal androgen precursors. See, e.g. Goldstat et al.
- Transdermal testosterone therapy improves well-being, mood, and sexual function in pre-menopausal women; Menopause, 10(5): 390-398 (2003). As indicated above, this is likely due to a decline in ovarian and adrenal function with age.
- testosterone plays a role in mood, body composition, and bone mineral density and has central and peripheral effects on sexual function. See, e.g., Davis et al.: Androgen replacement in women: a commentary; Menopause, J Clin Endocrinol Metab, 84(6): 1886-1891 (1999); and Goldstat et al., Supra.
- testosterone is required for nitric oxide to stimulate vasocongestion for the engorgement of clitoral tissue and vaginal lubrication during sexual arousal.
- Testosterone stimulates dopamine release in various brain structures implicated in motivation and reward systems, including sexual desire. Testosterone was found to stimulate dopamine release in the medial preoptic area of the anterior hypothalamus under basal conditions and with sexual stimulation in rats. See, e.g., Halaris A. : Neurochemical aspects of the sexual response cycle; CNS Spectrums, 9: 21 1 -216 (2003).
- An fMRI study in healthy women of different ages showed a testosterone level dependent modulation of amygdala activity, suggesting that an age-related decline in androgen levels contributes to the decrease in amygdala reactivity.
- Testosterone increases amygdala reactivity in middle-aged women to a young adulthood level; Neuropsychopharmacology, Feb: 34(3) 539-547 (2009).
- Intrinsa ® is a testosterone slow-release transdermal patch. Intrinsa ® is indicated for the treatment of hypoactive sexual desire disorder (HSDD) in bilaterally
- Testosterone Patch for low sexual desire in surgically menopausal women a
- Testosterone patch increases sexual activity and desire in surgically menopausal women with hyposactive sexual desire disorder; J Clin Endocrinol Metab, 90(9) 5226- 5233 (2005); Davis et al. : Efficacy and safety of a testosterone patch for the treatment of hypoactive sexual desire disorder in surgically menopausal women: a randomized, placebo-controlled trial; Menopause, 13(3): 387-396 (2006); and Shifren et al. :
- Testosterone patch for the treatment of hypoactive sexual desire disorder in naturally menopausal women results from the INTIMATE1 study; Menopause, 143:770-779 (2006).
- LibiGel is a gel formulation of testosterone that is applied on the upper arm of a female. It is reported that treatment with LibiGel increases the number of satisfying sexual events versus baseline and placebo treated individuals It is further reported that the effective dose of LibiGel produces testosterone blood levels within the normal range for pre-menopausal women. See, e.g., www.libigel.org.
- Zestra® is a blend of botanical oils and extracts, including: Borage Seed Oil, Evening Primrose Oil, Angelica Extract, Coleus Forskohlii Extract, Theobromine, Antioxidants ⁇ Ascorbyl Palmitate (Vitamin C), Tocopherol (Vitamin E) ⁇ ; and Flavor (U.S. 6,737,084) that may benefit some women with anorgasmia. Zestra® has
- ArginMaxTM is a mixture of L-arginine, ginseng, ginkgo, damiana, calcium, and iron. ArginMaxTM for Women was formulated specifically for women. It contains calcium and iron to help relieve fatigue issues specific to women. The American ginseng in the men's product has been replaced with Damiana, an aromatic herb which helps calm anxiety and induce a relaxed state of mind. ArginMaxTM for Women provides 100% of the RDA of vitamins A, C, E and the B-complex vitamins.
- ArginMaxTM safely enhances the female sexual experience by improving circulation. Sufficient blood flow is critical to female arousal, engorgement and lubrication. See, e.g., www.arginimax.com.
- the present invention overcomes the limitations and disadvantages associated with the treatment of anorgasmia and/or HDDD using available therapies through the discovery of novel lower dosage strength pernasal testosterone gels and methods of use to treat HSDD and/or anorgasmia.
- the present invention overcomes the limitations and disadvantages of currently available options for administration of testosterone through the discovery of novel and improved lower dosage strength testosterone gel formulations specifically designed for intranasal administration to deliver therapeutically effective amounts of testosterone to treat females who suffer from and/or have been diagnosed with HSDD and/or anorgasmia.
- a therapeutically effective amount means an amount of testosterone sufficient to induce a therapeutic or prophylactic effect for use in testosterone
- FSD female sexual dysfunction
- HSDD hypoactive sexual desire disorder
- anorgasmia female orgasmic disorder
- the present invention provides for new and improved, substantially less-irritating, lower dosage strength testosterone gel formulations formulated with testosterone in amounts ranging from between about 0.10% to about 1 .5% by weight, for nasal administration to deliver a therapeutically effective amount of testosterone to effectively treat anorgasmia and/or HSDD.
- the present invention is also directed to novel methods for pernasal administration of the nasal testosterone gels.
- the novel methods involve depositing the intranasal testosterone gels topically into the nasal cavity of each nostril to deliver a therapeutically effective amount of testosterone, e.g., from about 150 mcg/nostril to about 600 mcg/nostril per application, over dose life for providing constant effective testosterone brain and/or blood levels for use in testosterone replacement or supplemental therapy, especially for effectively treating females in need of testosterone to treat anorgasmia and/or HSDD.
- a therapeutically effective amount of testosterone e.g., from about 150 mcg/nostril to about 600 mcg/nostril per application
- the intranasal testosterone gels are topically deposited on the outer external walls (opposite the nasal septum) inside the naval cavity of each nostril, preferably at about the middle to about the upper section of the outer external wall (opposite the nasal septum) just under the cartilage section of the outer external wall inside the naval cavity of each nostril.
- the outer nose is then gently and carefully squeezed and/or rubbed by the subject, so that the deposited gel remains in contact with the mucosal membranes within the nasal cavity for sustained release of the testosterone over dose life.
- Typical testosterone gel dosage amounts deposited pernasal application is between about 50 to about 150 microliters per nostril, and preferably about 100 microliters per nostril.
- a lower dosage strength testosterone gel of the present invention is applied to each nostril of a subject once or twice daily, e.g., for one, two, three, four or more consecutive weeks, or for two, three, four, five or six
- testosterone treatment limiting reactions or related adverse events is contemplated by the present invention.
- the present invention therefore provides for a new and improved treatment for anorgasmia and/or HSDD, wherein nasal administration of a lower dosage strength testosterone gel formulation of the present invention provides for: (1 ) rapid delivery of testosterone due to the highly permeable nasal tissue both systemically and across the blood-brain barrier into the brain; (2) fast onset of action; (3) avoidance of hepatic first- pass metabolism; (4) ease of administration to improve sexual experience; (5) avoidance of irritation from transdermal administration, particularly, no exposition to contacts, no transference from topical gels, and no local irritability from topical patch products; and (6) a more pleasant mode of administration, as compared to injections and buccal or sublingual tablets.
- the present invention provides for a new and improved
- anorgasmia and/or HSDD treatment that (a) is easy and convenient to use either according to a prescribed treatment regimen or on-demand, (b) rapidly delivers therapeutically effective amounts of testosterone, thereby improving female sexual function in a timely manner, (c) provides for simple use, (d) has reduced side effects associated with prior exogenous systemic testosterone therapies, (e) avoids local irritability associated with prior topical gels and topical patches, and (f) eliminates the need for invasive and painful testosterone injections.
- the present invention provides numerous surprising advantages over currently available therapies for anorgasmia and HSDD.
- the present invention provides for (1 ) a rapid increase in the plasma testosterone plasma level (e.g., an increase in the plasma testosterone to a level of at least about 0.4 ng/ml within about 15 minutes immediately after nasal administration of the testosterone gel formulation of the invention); (2) a sustained increase in the plasma testosterone plasma level (e.g., an increase in the plasma testosterone level that is maintained in a subject for at least about 6 hours following nasal administration of the testosterone gel formulation of the invention); and (3) a higher maximum level of plasma testosterone as compared to the maximum level of plasma testosterone following administration of Intrinsa ® within about 100 minutes immediately following administration (e.g., an increase in the plasma testosterone level to at least about 0.7 ng/ml as compared to about 0.1 ng/ml for Intrinsa®.).
- an improved testosterone gel formulation of the invention provide advantages over therapies for treating anorgasmia and HSDD that are currently available.
- a testosterone gel formulation of the invention comprising about 0.6% testosterone by weight of the gel formulation is administered intranasally to subjects.
- control subjects who are treated with an Intrinsa® patch, receive a testosterone dose of about 2100-2800 meg/day, up to approximately 3.5-4.5 times the testosterone received by women treated with the lower dosage strength testosterone gels of the present invention (i.e., about 600, 1800, or 2400 meg/day, for the 0.15%, 0.45%, and 0.6% testosterone gels of the invention, respectively, or about 600, 1200 or 1800 meg/day, for the 0.24%, 0.48% or 0.72% testosterone gels of the invention, respectively).
- testosterone levels return to baseline after about 12 hours after treatment with the lower dosage strength testosterone gels of the present invention (at least for the 0.15% and 0.45% gel formulations of the invention).
- the improved 0.6% testosterone gel formulation for nasal administration of the present invention provides a plasma testosterone concentration, following nasal administration wherein (a) a plasma testosterone level of at least about 0.4 ng/ml is achieved; (b) a plasma testosterone level of at least about 0.7 ng/ml is achieved; (c) an increase in plasma testosterone level is achieved within at least about 10 minutes following nasal administration to a subject; (d) a plasma testosterone level of at least about 0.4 ng/ml is achieved and maintained for at least about 6 hours immediately following nasal administration to a subject; (e) a plasma testosterone level of at least about 0.3 ng/ml is achieved and maintained for at least about 13 hours immediately following nasal administration to a subject; and (f) a plasma testosterone level of at least about 0.7 ng/ml is achieved within about 100 minutes immediately following nasal administration to the subjects (see Figure 1 ).
- the plasma testosterone level does not increase until at least about 3 hours following administration of Intrinsa ® . Even then, a plasma testosterone concentration of at least about 0.4 ng/ml is not observed in the Intrinsa ® treated subjects until at least about 6.5 hours following administration. The maximum plasma testosterone level of a subject treated with Intrinsa ® (only about 0.68 ng/ml) is not observed until about 12 hours following administration of Intrinsa ® .
- one improved testosterone (0.6%) gel formulation for nasal administration of the present invention may provide one or more of the following plasma testosterone concentrations, following nasal administration:
- a plasma testosterone level of at least about 0.3 ng/ml is achieved and maintained for at least about 13 hours immediately following nasal administration to a subject;
- a plasma testosterone concentration of at least about 0.7 ng/ml is achieved within about 100 minutes immediately following nasal administration to a subject (see Figure 1 ).
- the present invention overcomes certain of the limitations associated with the treatment of anorgasmia and/or HSDD using currently available therapies, for example, Intrinsa ® , and addresses current medical needs for (1 ) a pharmaceutical formulation that is conveniently, easily and unobtrusively administered; (2) a rapidly acting formulation that improves female sexual dysfunction in a timely manner; (3) a decrease in the incidence of application site reactions; (4) a formulation that has reduced side effects; and (5) a formulation that can be used either according to a prescribed treatment regimen or on demand; to treat anorgasmia and/or HSDD.
- therapies for example, Intrinsa ®
- Example 1 1 A safety study was also conducted in accordance with the present invention, as reported in Example 1 1 , wherein women were treated intranasally with a 0.72% testosterone gel (about 1200 meg/dose administration) of the present invention t.i.d. for two consecutive days and qd on the third consecutive day resulting in a total daily dose of testosterone of about 3600 meg/day for the first two days and about 1200 meg on the third consecutive day. As shown in Example 1 1 , the intranasal testosterone gels of the present invention are believed to be safe and well tolerated.
- the salient elements of the novel intranasal testosterone gels according to the present invention comprise (a) testosterone in a therapeutically effective amount, (b) a solvent, (c) a wetting agent, and (d) a viscosity increasing agent.
- the improved lower dosage strength testosterone gel formulations of the present invention may be formulated with testosterone in amounts by weight of between about 0.10% to about 1 .5%, e.g., about 0.15%, 0.24%, 0.45%, 0.48%, 0.6% and 0.72%, and more preferably between about 0.24% and 0.72%.
- Exemplary nasally administered testosterone gel formulations contemplated by the present invention include:
- the improved testosterone gel formulations for nasal administration of the invention may further comprise any pharmaceutically acceptable vehicle, excipient and/or other active ingredient.
- the present invention contemplates testosterone gel formulations for nasal administration that are pharmaceutically equivalent, therapeutically equivalent, bioequivalent and/or interchangeable, regardless of the method selected to
- the present invention contemplates testosterone gel formulations for nasal administration that are bioequivalent, pharmaceutically equivalent and/or therapeutically equivalent, especially testosterone gel formulations for nasal administration that are 0.15% testosterone by weight of the gel formulation, 0.24% testosterone by weight of the gel formulation, 0.45% testosterone by weight of the gel formulation, 0.48% testosterone by weight of the gel formulation, 0.6% testosterone by weight of the gel formulation and 0.72% testosterone by weight of the gel formulation, when used in accordance with the therapy of the present invention to treat anorgasmia and/or HSDD by intranasal administration.
- the present invention contemplates: (a) pharmaceutically equivalent testosterone gel formulations for nasal administration which contain the same amount of testosterone in the same dosage form; (b) bioequivalent testosterone gel formulations for nasal administration which are chemically equivalent and which, when administered to the same individuals in the same dosage regimens, result in comparable bioavailabilities; (c) therapeutic equivalent testosterone gel formulations for nasal administration which, when administered to the same individuals in the same dosage regimens, provide essentially the same efficacy and/or toxicity; and (d)
- interchangeable testosterone gel formulations for nasal administration of the present invention which are pharmaceutically equivalent, bioequivalent and therapeutically equivalent.
- intranasal testosterone gels of the present invention are preferred pharmaceutical preparations when practicing the novel methods of the present invention, it should be understood that the novel topical intranasal gel formulations and methods of the present invention also contemplate the pernasal administration of any suitable active ingredient, either alone or in combination with testosterone or other active ingredients, such as neurosteroids or sexual hormones (e.g., androgens and progestins, like).
- active ingredient such as neurosteroids or sexual hormones (e.g., androgens and progestins, like
- neurotransmitters e.g., acetylcholine, epinephrine, norepinephrine, dopamine, serotonin, melatonin, histamine, glutamate, gamma aminobutyric acid, aspartate, glycine, adenosine, ATP, GTP, oxytocin, vasopressin, endorphin, nitric oxide, pregnenolone, etc.), prostaglandin, benzodiazepines like diazepam, midazolam, lorazepam, etc., and PDEF inhibitors like sildenafil, tadalafil, vardenafil, etc., in any suitable pharmaceutical preparation, such as a liquid, cream, ointment, salve or gel. Examples of additional topical formulations for practice in
- the present invention is also directed to packaged pharmaceuticals comprising the novel and improved testosterone gel formulations for nasal administration of the invention.
- the present invention contemplates pre-filled, single or multi-dose applicator systems for pernasal administration to strategically and uniquely deposit the nasal testosterone gels at the preferred locations within the nasal cavity for practicing the novel methods and teachings of the present invention.
- the applicator systems of the present invention are, e.g., airless fluid, dip-tube fluid dispensing systems, pumps, syringes or any other system suitable for practicing the methods of the present invention.
- the applicator systems or pumps include, for example, a chamber, pre-filled with a single dose or multiple doses of an intranasal testosterone gel of the present invention, that is closed by an actuator nozzle or cap.
- the actuator nozzle may comprise an outlet channel and tip, wherein the actuator nozzle is shaped to conform to the interior surface of a user's nostril for (a) consistent delivery of uniform dose amounts of an intranasal testosterone gel of the present invention during pernasal application within the nasal cavity, and (b) deposition at the instructed location within each nostril of a patient as contemplated by the novel methods and teachings of the present invention.
- the pump design when inserted into a nasal cavity, is configured to help ensure that the nasal tip is properly positioned within the nasal cavity so that, when the gel is dispensed, the gel is dispensed within the appropriate location within the nasal cavity.
- the pump design when inserted into a nasal cavity, is configured to help ensure that the nasal tip is properly positioned within the nasal cavity so that, when the gel is dispensed, the gel is dispensed within the appropriate location within the nasal cavity. See Steps 3 and 8 in Fig. 39.
- the nozzles of te pumps are preferably designed to dispense the gels from from the side in a swirl direction, i.e., the tips of the nozzles are designed to dispense in a side distribution direction, as opposed to a direct distribution direction, onto the nasal mucosa, as shown in steps 4 and 9 of Fig. 39. It is believed that the swirl action allows for better gel adhesion and side distribution from the nozzle tip avoids the dispensed gel from splashing back onto the tip. Finally, it is preferrred to design the nozzle and tip to allow for any residual gel on the nozzle/tip to be wiped off as the tip is removed from the nasal cavity. See, e.g., Fig. 39.
- pre-filled, multi-dose applicator systems examples include, e.g., (a) the COMOD system available from Ursatec,maschine-GmbH, Schillerstr. 4, 66606 St. Wendel, Germany, (b) the Albion or Digital airless applicator systems available from
- the amount of testosterone in a lower dosage strength intranasal testosterone gel of the present invention that will be therapeutically effective in a specific situation will depend upon such things as the dosing regimen, the application site, the particular gel formulation, dose longevity and the condition being treated. As such, it is generally not practical to identify specific administration amounts herein; however, it is believed that those skilled in the art will be able to determine appropriate therapeutically effective amounts based on the guidance provided herein, information available in the art pertaining to testosterone replacement therapy, and routine testing.
- Fig. 1 shows the effects of a single dose testosterone nasal gel formulation of the invention (0.6% testosterone by weight of the gel formulation) as compared to Intrinsa® on the plasma testosterone levels in subjects diagnosed with HSDD (triangles-100 microliters 0.6% testosterone nasal gel formulation of the invention in each nostril;
- Fig. 2 shows the effects of a testosterone nasal gel formulation of the invention after the fifth 12 hourly dose (0.6% testosterone by weight of the gel formulation) as compared to Intrinsa® on the plasma testosterone levels in subjects diagnosed with anorgasmia (TBS-2 line-0.6% testosterone nasal gel formulation of the invention( 100 mcl * 2); solid line- Intrinsa®);
- Fig. 3 shows the effects of a testosterone nasal gel formulation of the invention (0.15, 0.45 and 0.6% testosterone by weight of the gel formulation) after the first and the 5th 12 hourly dose as compared to IntrinsaR single dose on the plasma testosterone levels in subjects diagnosed with HSDD;
- Fig. 4 shows the effects of a testosterone nasal gel formulation of the invention at day zero (open triangles) and day 3 (dose 5 - closed square -of 0.6% testosterone by weight of the gel formulation) as compared to Intrinsa® on the plasma testosterone levels in subjects diagnosed with HSDD (open squares day 0- lines day 3 of 0.6% squares- Intrinsa®;
- Fig 5 is a copy of Fig 1 , but with comparative data for the 0.6% testosterone gel and Intinsa® from days 1 and 3;
- Fig 6 is comparative data from treatment study for HSDD for the 0.6%
- Fig 7 shows the effects of a testosterone nasal gel formulation of the invention (0.15, 0.45 and 0.6% testosterone by weight of the gel formulation) after the first and the 5th 12 hourly dose as compared to Intrinsa® single dose on the plasma testosterone levels in subjects diagnosed with anorgasmia;
- Fig 8 shows the effects of a testosterone nasal gel formulation of the invention (0.45% day 3 (Med Day 1 ) and 0.6% day zero and Day 3 (ANOR) testosterone by weight of the gel formulation) after the first and the 5 th 12 hourly dose;
- Fig 9 shows the accumulation effect effects of a testosterone nasal gel formulation of the invention (0.15, 0.45 and 0.6% testosterone by weight of the gel formulation) before each bi-daily dose of testosterone gel on the plasma testosterone levels at time (0) in subjects diagnosed with HSDD or anorgasmia;
- Fig. 1 1 presents trough data for subjects diagnosed with anorgasmia or HSDD and treated with testosterone nasal gel formulations of the invention (0.15%, 0.45% or 0.6% testosterone by weight of the gel formulation)or placebo (anorgasmia) or Intrinsa® (HSDD);
- Fig. 12 presents distribution of testosterone mean concentration for TBS-2 treatments for the first (AUC_0-12) and the last dose (AUC_48-60).
- the boxplots show the median (thick solid line), the inter-quartile range (box) and the extreme values (whiskers).
- the horizontal solid grey line indicates the median C_mean during treatment with Intrinsa from 48 to 60 hrs, and the horizontal dotted lines indicate the minimum and maximum C_mean during treatment with Intrinsa from 48-60 hrs;
- Fig. 13 presents scores on the AFSDQ 30 minutes (left) and 4.5 hours (right) after dosing.
- Fig. 14 concerns mean testisterone levels following TBS-2 high, TBS-2 medium and TBS-2 low dose administration or placebo adminsitration (hours 0-12) in women with anorgasmia.
- TBS-2 high, TBS-2 medium and TBS-2 low dose administration or placebo adminsitration hours 0-12
- placebo adminsitration hours 0-12
- three different dosage strengths of TBS-2 testosterone bio-adhesive gel formulations of the invention are adminstered intranasally to a hybrid group of 12 healthy and anorgasmic women.
- the testosterone serum levels are compared for the three different testosterone bio-adhesive gel formulations of the invention (0.15% - , 0.45% and 0.6% testosterone by weight of the gel formulation, as reported in Examples 1 -5), during 2 hours following a single application of each TBS-2 formualtion or a placebo to each of the 12 women.
- the total testosterone dosage strength that is adminstered is either 1 .2 mg (0.6% - 0.6 mg/100 ⁇ /nostril), 0.9 mg (0.45% - 0.45 mg/100 ⁇ /nostril) or 0.3 mg (0.15% - 0.15 mg/100 ⁇ /nostril).
- the testosterone serum level is measured and compared.
- the C max and C aV g for testosterone following single dose administration for each of the three dosage strengths do not exceed the normal testosterone serum level in women (3 - 80 ng/dL);
- Fig. 15 depicts a study design (Example 10), in which 56 anorgasmic women are enrolled for a Vibrotactile Stimulation Study (VTS) that concerns the three different testosterone bio-adhesive gel formulations of the invention, i.e., the 0.15% - , 0.45% and 0.6% testosterone by weight of the gel formulation, as reported in Examples 1 -5;
- VTS Vibrotactile Stimulation Study
- Fig. 16 depicts orgasm result of the VTS study of Example 10, wherein the number of orgasms achieved during the treatment phase and the post-treatment phase are compared;
- Fig. 17 depicts sexual response results of the VTS study of Example 10 for the three different testosterone bio-adhesive gel formulations of the invention, i.e., the 0.15% - , 0.45% and 0.6% testosterone by weight of the gel formulation, as reported in Examples 1 -5, as compared to placebo;
- Fig. 18 depicts the VTS appreciation score of the VTS study of Example 10 for the three different testosterone bio-adhesive gel formulations of the invention, i.e., the 0.15%, 0.45% and 0.6% testosterone by weight of the gel formulation, as reported in Examples 1 -5, as compared to placebo;
- Fig. 19 depicts Mean Corrected Free Testosterone Concentrations (Single-Dose Population).
- Fig. 20 depicts Mean Corrected Total Testosterone Concentrations (Single-Dose Population);
- Fig. 21 depicts Mean Corrected Dihydrotestosterone Concentrations (Single- Dose Population);
- Fig. 22 depicts Mean Corrected Estradiol Concentrations (Single-Dose
- Fig. 23 depicts Mean Corrected SHBG Concentrations (Single-Dose Population);
- Fig. 24 depicts Mean Observed Free Testosterone Concentrations (Single-Dose Population);
- Fig. 25 depicts Mean Observed Total Testosterone Concentrations (Single-Dose Population);
- Fig. 26 depicts Mean Observed Dihydrotestosterone Concentrations (Single- Dose Population);
- Fig. 27 depicts Mean Observed Estradiol Concentrations (Single-Dose
- Fig. 28 depicts Mean Observed SHBG Concentrations (Single-Dose Population);
- Fig. 29 depicts Mean Free Testosterone Plasma Concentrations (Multi-Dose Population);
- Fig. 30 depicts Mean Total Testosterone Plasma Concentrations (Multi-Dose Population).
- Fig. 31 depicts Mean Dihydrotestosterone Plasma Concentrations (Multi-Dose Population).
- Fig. 32 depicts Mean Estradiol Plasma Concentrations (Multi-Dose Population);
- Fig. 33 depicts Mean SHBG Plasma Concentrations (Multi-Dose Population);
- Fig. 34 depicts Spaghetti Concentration Plots with Mean for Free Testosterone Plasma Concentrations (Multi-Dose Population);
- Fig. 35 depicts Spaghetti Concentration Plots with Mean for Total Testosterone Plasma Concentrations (Multi-Dose Population);
- Fig. 36 depicts Spaghetti Concentration Plots with Mean for Dihydrotestosterone Plasma Concentrations (Multi-Dose Population);
- Fig. 37 depicts Spaghetti Concentration Plots with Mean for Estradiol Plasma Concentrations (Multi-Dose Population);
- Fig. 38 depicts Spaghetti Concentration Plots with Mean for SHBG Plasma Concentrations (Multi-Dose Population).
- Fig. 39 depicts an intranasal applicator contemplated by and used in accordance with the present invention.
- bioequivalence or “bioequivalent” refers to nasally
- testosterone gel formulations or drug products which are pharmaceutically equivalent and their bioavailabilities (rate and extent of absorption) after administration in the same molar dosage or amount are similar to such a degree that their therapeutic effects, as to safety and efficacy, are essentially the same.
- bioequivalence or bioequivalent means the absence of a significant difference in the rate and extent to which testosterone becomes available from such formulations at the site of testosterone action when administered at the same molar dose under similar conditions, e.g., the rate at which testosterone can leave such a formulation and the rate at which testosterone can be absorbed and/or become available at the site of action to affect anorgasmia and/or HSDD.
- the rate at which testosterone can leave such a formulation and the rate at which testosterone can be absorbed and/or become available at the site of action to affect anorgasmia and/or HSDD.
- there is a high degree of similarity in the bioavailabilities of two testosterone gel formulation pharmaceutical products for nasal administration (of the same galenic form) from the same molar dose that are unlikely to produce clinically relevant differences in therapeutic effects, or adverse reactions, or both.
- bioequivalence as well as “pharmaceutical equivalence” and “therapeutic equivalence” are also used herein as defined and/or used by (a) the FDA, (b) the Code of Federal Regulations ("C.F.R.”), Title 21 , (c) Health Canada, (d) European Medicines Agency (EMEA), and/or (e) the Japanese Ministry of Health and Welfare.
- C.F.R. Code of Federal Regulations
- EMEA European Medicines Agency
- testosterone gel formulations for nasal administration or drug products that may be bioequivalent to other testosterone gel formulations for nasal administration or drug products of the present invention.
- a first testosterone gel formulation for nasal administration or drug product is bioequivalent to a second testosterone gel formulation for nasal administration or drug product, in accordance with the present invention, when the measurement of at least one pharmacokinetic parameter(s), such as a Cmax, Tmax, AUC, etc., of the first testosterone gel formulation for nasal administration or drug product varies by no more than about ⁇ 25%, when compared to the measurement of the same pharmacokinetic parameter for the second testosterone gel formulation for nasal administration or drug product of the present invention.
- pharmacokinetic parameter(s) such as a Cmax, Tmax, AUC, etc.
- bioavailability means generally the rate and extent of absorption of testosterone into the systemic circulation and, more specifically, the rate or measurements intended to reflect the rate and extent to which testosterone becomes available at the site of action or is absorbed from a drug product and becomes available at the site of action.
- the extent and rate of testosterone absorption from a lower dosage strength gel formulation for nasal administration of the present invention as reflected by a time-concentration curve of testosterone in systemic circulation.
- the terms “pharmaceutical equivalence” or “pharmaceutically equivalent”, refer to testosterone gel formulations for nasal administration or drug products of the present invention that contain the same amount of testosterone, in the same dosage forms, but not necessarily containing the same inactive ingredients, for the same route of administration and meeting the same or comparable compendial or other applicable standards of identity, strength, quality, and purity, including potency and, where applicable, content uniformity and /or stability.
- the present invention contemplates testosterone gel formulations for nasal administration or drug products that may be pharmaceutically equivalent to other testosterone gel formulations for nasal administration or drug products used in accordance with the present invention.
- therapeutic equivalence or “therapeutically equivalent” means those testosterone gel formulations for nasal administration or drug products which (a) will produce the same clinical effect and safety profile when utilizing testosterone drug product to treat anorgasmia or HSDD in accordance with the present invention and (b) are pharmaceutical equivalents, e.g., they contain testosterone in the same dosage form, they have the same route of administration; and they have the same testosterone strength.
- therapeutic equivalence means that a chemical equivalent of a lower dosage strength testosterone formulation of the present invention (i.e., containing the same amount of testosterone in the same dosage form when administered to the same individuals in the same dosage regimen) will provide essentially the same efficacy and toxicity.
- testosterone gel formulation for nasal administration means a formulation comprising testosterone in combination with a solvent, a wetting agent, and a viscosity increasing agent.
- the plasma testosterone level is 1 , 2, 3, 4, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 100, 500, 1000 or 10,000-fold or more greater in a subject that has been treated with a testosterone gel formulation for nasal administration of the invention as compared to the plasma testosterone level in the subject prior to treatment.
- plasma testosterone level means the level of testosterone in the plasma of a subject.
- the plasma testosterone level is determined by methods known in the art.
- Diagnosis refers to the use of information (e.g., biological or chemical information from biological samples, signs and symptoms, physical exam findings, psychological exam findings, etc.) to anticipate the most likely outcomes, timeframes, and/or responses to a particular treatment for a given disease, disorder, or condition, based on comparisons with a plurality of individuals sharing symptoms, signs, family histories, or other data relevant to consideration of a patient's health status, or the confirmation of a subject's affliction, e.g., with anorgasmia and/or HSDD.
- information e.g., biological or chemical information from biological samples, signs and symptoms, physical exam findings, psychological exam findings, etc.
- a "subject” is an individual whose signs and symptoms, physical exams findings and/or psychological exam findings are to be determined and recorded in conjunction with the individual's condition (i.e., disease or disorder status) and/or response to a candidate drug or treatment.
- Subject is preferably, but not necessarily limited to, a human subject.
- the subject may be male or female, and is preferably female, and may be of any race or ethnicity, including, but not limited to, Caucasian, African-American, African, Asian, Hispanic, Indian, etc.
- Subject as used herein may also include an animal, particularly a mammal such as a canine, feline, bovine, caprine, equine, ovine, porcine, rodent (e.g., a rat and mouse), a lagomorph, a primate (including non-human primate), etc., that may be treated in accordance with the methods of the present invention or screened for veterinary medicine or pharmaceutical drug development purposes.
- a subject according to some embodiments of the present invention include a patient, human or otherwise, in need of therapeutic treatment for anorgasmia and/or HSDD.
- Treatment includes any drug, drug product, method, procedure, lifestyle change, or other adjustment introduced in attempt to effect a change in a particular aspect of a subject's health (i.e. , directed to a particular disease, disorder, or condition).
- drug or “drug substance,” as used herein, refers to an active ingredient, such as a chemical entity or biological entity, or combinations of chemical entities and/or biological entities, suitable to be administered to a subject to (a) treat anorgasmia and/or (b) treat HSDD.
- the drug or drug substance is testosterone or a pharmaceutically acceptable salt or ester thereof.
- drug product is synonymous with the terms
- a drug product is approved by a government agency for use in accordance with the methods of the present invention.
- a drug product, in accordance with the present invention is an intranasal gel formulated with a drug substance, i.e., testosterone.
- Diseases or conditions are commonly recognized in the art and designate the presence of signs and/or symptoms in an individual or patient that are generally recognized as abnormal and/or undesirable. Diseases or conditions may be diagnosed and categorized based on pathological changes. The disease or condition may be selected from the types of diseases listed in standard texts, such as Harrison's Principles of Internal Medicine, 1 997, or Robbins Pathologic Basis of Disease, 1 998.
- anorgasmia or HSDD refers to a process of determining if an individual is afflicted with anorgasmia or HSDD.
- control subject means a subject that has not been diagnosed with anorgasmia and/or HSDD and/or does not exhibit any detectable symptoms associated with these diseases.
- a “control subject” also means a subject that is not at risk of developing anorgasmia and/or HSDD, as defined herein.
- Anorgasmia is a type of sexual dysfunction in which a person cannot regularly achieve orgasm, even with adequate stimulation. In males the condition is often related to delayed ejaculation. Anorgasmia can often cause sexual frustration. Anorgasmia is far more common in females than in males and is especially rare in younger men.
- Anorgasmia is a very common occurrence in women, affecting 1 in 5 women worldwide.
- the condition is sometimes classified as a psychiatric disorder.
- it can also be caused by medical problems such as diabetic neuropathy, multiple sclerosis, genital mutilation, complications from genital surgery, pelvic trauma (such as from a straddle injury caused by falling on the bars of a climbing frame, bicycle or gymnastics beam), hormonal imbalances, total hysterectomy, spinal cord injury, cauda equina syndrome, uterine embolisation, childbirth trauma (vaginal tearing through the use of forceps or suction or a large or unclosed episiotomy), vulvodynia and cardiovascular disease.
- medical problems such as diabetic neuropathy, multiple sclerosis, genital mutilation, complications from genital surgery, pelvic trauma (such as from a straddle injury caused by falling on the bars of a climbing frame, bicycle or gymnastics beam), hormonal imbalances, total hysterectomy, spinal cord injury, cauda equina syndrome, uterine emb
- DSM-IV-TR The Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Text Revision (DSM-IV-TR) defines female orgasmic disorder (FOD, formerly inhibited female orgasm) as a persistent or recurrent delay in, or absence of, orgasm following a normal sexual excitement phase.
- FOD formerly inhibited female orgasm
- the type or intensity of stimulation that triggers female orgasm varies widely among women. Therefore, the diagnosis of female orgasmic disorder, according to the DSM-IV-TR, is based on these 3 criteria:
- Criterion A A clinician must judge that a woman's orgasmic capacity is less than what is reasonable for her age, sexual experience, and the adequacy of sexual stimulation she receives.
- Criterion B The disturbance must cause marked distress or interpersonal difficulty.
- Criterion C Another axis I disorder (except another sexual dysfunction) does not account for the orgasmic dysfunction better than female orgasmic disorder does, and the orgasmic dysfunction is not exclusively due to the direct physiologic effects of a substance (e.g., drug of abuse, medication) or a general medical condition.
- a substance e.g., drug of abuse, medication
- female orgasmic disorder specifiers include the following:
- the presence of a normal sexual excitement phase is a prerequisite for female orgasmic disorder.
- diagnoses such as hypoactive sexual desire disorder, sexual aversion disorder, or female sexual arousal disorder, respectively, might be more appropriate, even if anorgasmia is the common final outcome.
- Certain prescription drugs including fluoxetine (Prozac), paroxetine (Paxil), and sertraline (Zoloft)
- Hormonal disorders hormonal changes due to menopause, and chronic illnesses that affect general health and sexual interest Medical conditions that affect the nerve supply to the pelvis (such as multiple sclerosis, diabetic neuropathy, and spinal cord injury)
- Orgasm Medical diseases. Any illness can affect this part of an individual's sexuality, including diabetes and neurological diseases, such as multiple sclerosis. Orgasm may also be affected by gynecologic surgeries, such as hysterectomy or cancer surgeries. In addition, lack of orgasm often goes hand in hand with other sexual problems, such as painful intercourse.
- SSRIs selective serotonin reuptake inhibitors
- Alcohol and drugs A glass of wine may make you feel amorous, but too much alcohol can cramp your ability to climax; the same is true of street drugs.
- SSRIs serotonin reuptake inhibitors
- PSSD serotonin reuptake inhibitors
- anorgasmia As a side effect of SSRIs is not precise, it is estimated that 15-50% of users of such medications are affected by this condition.
- the chemical amantadine has been shown to relieve SSRI-induced anorgasmia in some, but not all, people.
- Another cause of anorgasmia is opiate addiction, particularly to heroin.
- anorgasmia The major symptoms of anorgasmia are inability to experience orgasm or long delays in reaching orgasm. Different types of anorgasmia have been identified.
- Primary anorgasmia is a condition where one has never experienced an orgasm. This is significantly more common in women, although it can occur in men who lack the gladipudendal (bulbocavernosus) reflex. Women with this condition can sometimes achieve a relatively low level of sexual excitement. Frustration, restlessness, and pelvic pain or a heavy pelvic sensation may occur because of vascular engorgement.
- an idea such as this may be a component of treatment as one consideration among many, but responsible clinical practice should not be guided, based on, or informed by it.
- Secondary anorgasmia is the loss of the ability to have orgasms. Particularly, you used to have orgasms, but now experience difficulty reaching climax. The cause may be alcoholism, depression, grief, pelvic surgery (such as total hysterectomy) or injuries, certain medications, illness, estrogen deprivation associated with menopause or an event that has violated the patient's sexual value system.
- a person may have an orgasm from one type of stimulation but not from another, a person may achieve orgasm with one partner but not another, or have an orgasm only under certain conditions or only with a certain type or amount of foreplay. These common variations are within the range of normal sexual expression and should not be considered problematic.
- Factors that may affect whether or not an individual is orgasmic include fatigue, emotional concerns, feeling pressured to have sex when he or she is not interested, or a partner's sexual dysfunction.
- some sex therapists recommend that couples incorporate manual or vibrator stimulation during intercourse, or using the female-above position as it may allow for greater stimulation of the clitoris by the penis or pubic symphysis or both, and it allows the woman better control of movement.
- orgasmic dysfunction The symptom of orgasmic dysfunction is being unable to reach orgasm, taking longer than you want to reach orgasm, or having only unsatisfying orgasms.
- Treatment can involve education, cognitive behavioral therapy, teaching orgasm by focusing on pleasurable stimulation, and directed masturbation.
- treatment may include communication training and relationship enhancement work.
- hypoactive sexual desire disorder is considered as a sexual
- HSDD is characterized as a lack or absence of sexual fantasies and desire for sexual activity for some period of time. For this to be regarded as a disorder, it must cause marked distress or interpersonal difficulties and not be better accounted for by another mental disorder (i.e. depression), a drug (legal or illegal), or some other medical condition.
- HSDD can be general (general lack of sexual desire) or situational (still has sexual desire, but lacks sexual desire for current partner), and it can be acquired (HSDD started after a period of normal sexual functioning) or lifelong (the person has always had no/low sexual desire.)
- Low sexual desire is not equivalent to HSDD because of the requirement that the low sexual desire causes marked distress and interpersonal difficulty and because of the requirement that the low desire is not better accounted for by another disorder in the DSM or by a general medical problem, so it is difficult to say exactly what causes HSDD. It is easier to describe, instead, what causes low sexual desire.
- men there are theoretically more types of HSDD/low sexual desire, typically men are only diagnosed with one of three subtypes.
- the man has little or no desire for sexual stimulation (with a partner or alone) and never has.
- the man was previously sexually interested in his present partner but now lacks sexual interest in them but has desire for sexual stimulation (i.e. alone or with someone other than his present partner.)
- sexual desire is controlled by a balance between inhibitory and excitatory factors. This is thought to be expressed via neurotransmitters in selective brain areas. A decrease in sexual desire may therefore be due to an imbalance between neurotransmitters with excitatory activity like dopamine and norepinephrine and neurotransmitters with inhibitory activity, like serotonin.
- Low sexual desire can also be a side effect of various medications.
- possible causes include intimacy difficulty, relationship problems, sexual addiction, and chronic illness of the man's partner. The evidence for these is somewhat in question.
- Some claimed causes of low sexual desire are based on empirical evidence. However, some are based merely on clinical observation. In many cases, the cause of HSDD is simply unknown.
- the steroid hormone testosterone is the active ingredient in the testosterone gel formulations of the invention.
- the manufacture of the drug substance presents no potential risk for humans; the synthesis route is well-characterized.
- testosterone gel formulations of the invention performance of the drug product, testosterone gel formulations of the invention.
- the solubility of the drug substance in the matrix is especially favorable.
- the testosterone drug can be in, for instance, crystalline, amorphous, micronized, non-micronized, powder, small particle or large particle form when formulating to intranasal testosterone gels of the present invention.
- An Exemplary range of testosterone particle sizes include from about 0.5 microns to about 200 microns.
- the testosterone particle size is in a range of from about 5 microns to about 100 microns, and the testosterone is in crystalline or amorphous and non-micronized or micronized form.
- the testosterone is in crystalline or amorphous micronized form.
- testosterone contains no functional groups that can be protonated or deprotonated in the physiological pH-range. Therefore testosterone is to be considered as a neutral molecule with no pKa value in the range 1 -14. Because it is neutral, testosterone is compatible with excipients.
- the testosterone gel formulations of the invention are viscous and thixotropic, oil- based formulations containing a solution of testosterone intended for intranasal application.
- the non-irritating formulation is designed to adhere to the inner nose. In addition, it acts as a controlling matrix, thus allowing sustained drug delivery through the nasal mucosa.
- polyoxylglycerides are used as hydrophilic oil for topicals, injectables and nasals. In FDA-approved medicinal products it is used as co-emulsifier in topical
- oleoyi polyoxylglycerides is suitable for an application route where safety and tolerability are of highest importance (e.g. injectables and nasal or vaginal preparations).
- Oleoyi macrogolglycerides are also referred to as Labrafil M 1944 CS, apricot kernel oil PEG-6 esters, Peglicol-5-oleate, mixture of glycerides and polyethylene esters.
- the castor oil which is used as a solvent for testosterone gel formulations of the invention is a fixed oil.
- Such oils have the advantage of being non-volatile or spreading (in contrast to essential oils or liquid paraffin), but have the disadvantage of being hydrophobic.
- the nasal mucosa contains 95-97% water. Without the oleoyl macrogol-glycerides, the castor oil containing the active ingredient would form a non- interactive layer on the mucous membrane. In order to achieve adequate contact between the castor oil layer and the mucous membrane, the hydrophilic oleoyl macrogol-glycerides oil is added to the formulation to form an emulsion between the castor oil and the mucosa fluid.
- Oleoyl macrogol-glycerides are used in semi-solids at concentrations ranging from about 3 to 20%, depending on the application.
- the amount of oleoyl macrogol- glycerides in testosterone gel formulations of the invention is high enough to allow for a better contact of the carrier oil with the mucous membrane and low enough to have minimal impact on the amount of testosterone that can be incorporated into the carrier oil.
- a favourable concentration of oleoyl microgol-glycerides in testosterone gel formulations of the invention is found to be 4% of the formulation.
- colloidal silicon dioxide is used as an oil adsorbent, thermal stabilizer and gellant.
- FDA-approved medicinal products it is used in dental gels, sublingual tablets, endocervical gel, suppositories, vaginal emulsions/creams/tablets/tampons and capsules for inhalation.
- it is used as an excipient in "Testoderm with adhesives” (Alza Corporation, approved in 1996) a testosterone transdermal patch.
- colloidal silicon dioxide is suitable for an application route where safety and tolerability are of highest importance (e.g. inhalations, endocervical, vaginal or rectal preparations).
- testosterone intranasal gel is supplied in unit-dose syringes consisting of a syringe body made from polypropylene, a plunger molded from polyethylene and a syringe cap made from high density polyethylene.
- the syringes are wrapped in aluminum foil as secondary packaging.
- the content of a syringe (125 mg) amounts to 0.10 to 1 .5 mg of testosterone.
- the oil in testosterone gel formulations of the invention is thickened with colloidal silicon dioxide, which acts as a gel-forming agent. This compound is used commonly for stiffening oleogels.
- the intended dosage form for testosterone gel formulations of the invention is a semi-solid, not a liquid.
- the formulation is thickened with colloidal silicon dioxide. It is believed that colloidal silicon dioxide contributes to the thixotropic properties of the gel, simplifying drug delivery to the nostril.
- Colloidal silicon dioxide is generally an inert material which is well tolerated as an excipient in mucosal applications such as suppositories. Colloidal silicon dioxide is typically used in these preparations at concentrations ranging from about 0.5 to 10%. The concentration of colloidal silicon dioxide in testosterone gel formulations of the invention is high enough to achieve gel formation but at a level that has minimal impact on testosterone incorporation into the carrier oil.
- the intranasal testosterone gels of the present invention have in general, a viscosity in the range of between about 3,000 cps and about 27,000 cps. It should nevertheless be understood by those versed in this art that, while the above- mentioned viscosity range is believed to be a preferred viscosity range, any suitable viscosities or viscosity ranges that do not defeat the objectives of the present invention are contemplated.
- Table 3 Composition of a testosterone gel formulation of the invention
- the testosterone gel formulations of the invention are stored at room
- the stability data available to date are conclusive to support a 24- month shelf life.
- Unit dose syringes are chosen for the primary packaging of the clinical materials for this clinical trial to allow for ease of dosing, ability to generate multiple doses by varying the fill volume and consistency of dose delivered.
- the syringe consists of a syringe body, a plunger and a syringe cap.
- the syringes body is molded from polypropylene
- the plunger is molded from polyethylene
- the cap is HDPE.
- the syringes are packed in a foil-laminate overwrap pouch.
- the syringes and caps are designed for use in a clinical setting and meet the requirements of the EU Medical Devices Directive 93/42/EEC of June 14, 1993 and as amended. As this container closure is only intended for use in this portion of the clinical program, no additional studies are performed on the syringe and syringe components. See also Example 1 1 and Fig. 39.
- two syringes are contained in secondary packaging consisting of an aluminium foil pouch. Two syringes are packaged in the aluminium foil pouch and each pouch is sealed.
- the pouch consists of a flexible, 3-layered-foil-laminate of a) polyester 12 micron, b) aluminum 12 micron and c) a polyethylene 75 micron. It is manufactured by Floeter Flexibles GmbH, and supplied under the name "CLIMAPAC II 12-12-75".
- Procter & Gamble developed a transdermal therapeutic system containing testosterone as active substance for the treatment of HSDD (SD Intrinsa ® ).
- SD Intrinsa ® a transdermal therapeutic system containing testosterone as active substance for the treatment of HSDD.
- Four controlled clinical studies were performed (2 in Phase II b, 2 in Phase III).
- the 300 ⁇ g testosterone transdermal system is effective in the treatment of HSDD in surgically menopausal women on concomitant estrogen therapy.
- the women who received testosterone experienced increased frequency of satisfying sexual activity, increased sexual desire, and decreased distress compared with women who received placebo. Improvements were also seen in all other efficacy endpoints (i.e., arousal, pleasure, orgasm, responsiveness, self-image, concerns).
- the testosterone serum levels were increased to the physiological range of premenopausal women, but did not exceed this range.
- Intrinsa significantly increased the frequency of total satisfying episodes compared to placebo (p ⁇ 0.05) and also experienced a significantly greater increase in sexual desire domain of PSFS (profile of female sexual function) and a significantly greater decrease in personal distress (p ⁇ 0.05) in patients on placebo.
- Intrinsa ® is approved for the treatment of HSDD in bilaterally oophorectomised and hysterectomised women receiving concomitant estrogen therapy in the European Union.
- Biosante developed a testosterone gel designed to be quickly absorbed through the skin after a once-daily application on the upper arm, delivering testosterone to the bloodstream evenly over time for the treatment of HSDD.
- One Phase II study has been performed and 2 Phase III studies are ongoing.
- the invention provides for gel formulations of testosterone to be administered intranasally, wherein the dosage of the formulation is from about 0.15% testosterone by weight of said gel formulation to about 0.6% testosterone by weight of said gel formulation, for example, 0.15% testosterone by weight of the gel formulation, 0.45% testosterone by weight of said gel formulation and 0.6% by weight of the gel formulation.
- the methods of the invention are used to treat anorgasmia and/or HSDD in a diagnosed with one or both of these conditions.
- the invention also provides for intranasal testosterone gel formulations that can be used to treat anorgasmia or HSDD in a patient diagnosed with one or both of these conditions.
- compositions of three different concentrations of the drug product to be administered in this clinical trial are provided in the tables below.
- the testosterone gel formulations of the invention are viscous and thixotropic, oil- based formulations containing solubilized testosterone intended for intranasal application.
- the drug product is formulated with the compendial inactive ingredients: castor oil, oleoyl polyoxyl-glycerides and colloidal silicon dioxide.
- testosterone gel formulations of the invention Three different doses of the testosterone gel formulations of the invention are intranasally administered: 0.15% w/w, 0.45% w/w and 0.6% w/w.
- An overage is added to each syringe to account for the gel that is retained in the syringe after dosing. This overage remains consistent at 23 ⁇ , regardless of volume of gel in the syringe.
- Table 4 Components, Quantity, Quality Standards and Function - testosterone gel formulation of the invention
- Testosterone gel formulations of the invention are supplied in unit-dose polypropylene syringes. Two syringes of each dosage are packaged in a protective aluminium foil pouch.
- the testosterone gel formulations of the invention are formulations of testosterone in an intranasal gel proposed for assessing the pharmacokinetic and pharmacodynamics of three different doses of testosterone gel formulations of the invention, compared to Intrinsa ® and placebo for testosterone gel formulations of the invention in women with hypoactive sexual desire disorder (HSDD) and secondary anorgasmia (SA).
- HSDD hypoactive sexual desire disorder
- SA secondary anorgasmia
- the active ingredient, testosterone is sourced from Bayer Schering.
- Testosterone is indicated for the treatment of HSDD in bilaterally
- oophorectomised and hysterectomised women receiving concomitant estrogen therapy. It is also indicated for the treatment of hormone replacement therapy in the treatment of hypogonadism in men.
- the currently available options for administration of testosterone are oral, buccal, injectable, implantable and transdermal.
- TBS-1 gel An intranasal testosterone (3.2%) gel, TBS-1 gel, is developed for the treatment of hypogonadism in men and has been administered to hypogonadal men in several clinical trials (Mattern, C. et al., 2008 The Aging Male 1 1 (4):171 -178 (Dec 2008), which is incorporated herein by reference in its entirety).
- the intranasal testosterone gel for women, testosterone gel formulations of the present invention are developed at concentrations ranging from about 0.15% to about 0.6% testosterone.
- the testosterone gel formulations of the invention has a viscosity in the range of 3,000 to 10,000 mPa x sec.
- the viscosity is important because it facilitates
- the viscosity is less than approximately 3,000 mPa x sec (i.e., 3,000 centipoise), the gel tends to be drawn by gravity out of the nasal cavity.
- Table 5 Batch Formulae for 0.15%, 0.45% and 0.6% testosterone gel formulations of the invention at the 8 kg Batch Size
- Oleoyl polyoxylglycerides Ph.
- Material is manufactured according to the following process.
- the Pre-Mix is prepared by mixing, with a propeller mixer, the full amount of Testosterone with portion 1 of the castor oil for 10 minutes.
- Mixture I is prepared by adding the Pre-Mix to the remaining castor oil and mixing for 60 minutes. The product temperature is maintained below 50 °C for the entire mixing process.
- the oleoyl polyxoylglycerides are pre-heated to 40 - 50 °C and mixed for 10 minutes before being added to Mixture I. This is identified as Mixture II. It is mixed for 45 minutes while maintaining product temperature below 50 °C. Mixture II is then screened through a sieve to remove any un-dissolved Testosterone aggregates.
- Mixture III is prepared by adding the colloidal silicon dioxide to Mixture II and mixing for 15 minutes while maintaining product temperature below 50 °C. A visual check is conducted after this step, to ensure that the gel is clear.
- the gel is stirred and cooled to a product temperature below 30 °C.
- the product is then discharged into stainless steel drums and the bulk gel sample is taken for analytical testing.
- the filling and packaging process is carried out by filling a pre-determined volume into the syringe followed by the application of the syringe cap. Two syringes are packaged into a foil pouch.
- the syringes are filled using a pipette with the gel taken from a holding tank. The tip of the pipette is discarded after the syringe is filled and the syringe cap is applied. Each syringe is individually labelled.
- testosterone gel formulations of the invention are evaluated in studies of women with anorgasmia.
- the effect of testosterone gel formulations of the invention on sexual stimuli in women with anorgasmia is also determined.
- testosterone gel formulations of the invention Three doses of testosterone gel formulations of the invention are investigated: 150 ⁇ g, 450 ⁇ g and 600 ⁇ g per nostril. A total of 5 doses of a testosterone gel formulation of the invention is administered BID intranasally to women. Placebo testosterone gel formulation of the invention is administered as a control.
- Test subjects receive 5 doses of a testosterone gel formulation of the invention over a three day period.
- the plasma concentrations of total testosterone and dihydrotestosterone are measured using validated LC/MS/MS.
- the following pharmacokinetic parameters are determined for all subjects:
- Subjects in the ANOR cohort are randomized to receive either a testosterone gel formulation of the invention (3 dose levels) or placebo. Randomization is according to the design allocation below.
- the analysis is both a double-blind and open label study, depending on the treatment cohort.
- the study is placebo-controlled and double-blinded.
- testosterone gel formulation of the invention 0.15%, 0.45% or 0.6%) or testosterone gel formulation of the invention placebo on five (5) occasions during the study: Day 1 at 2000 hours, Day 2 at 800 and 2000 hours, and Day 3 at 800 and 2000 hours.
- the intranasal gel is administered to both nostrils (1 syringe (100 ⁇ volume) per nostril).
- Study medication consists of a testosterone gel formulation of the invention and placebo gel and is packed in single use syringes designed to expel 100 ⁇ of gel. Two syringes are packaged in a foil pouch.
- Subjects are randomized into the dosing regimen that is administered during a four day (three night) in-patient treatment period and receive either an intranasal
- testosterone gel formulation of the invention (3 dose levels) or placebo (ANOR) according to the design allocation:
- the randomization scheme is created for each study center and will consist of blocks of four treatments per cohort.
- the study is a four day study.
- the study starts with Study Drug dosing between 2000 and 2100 hours on Day 1 (Baseline).
- Blood samples for plasma testosterone and dihydrotestosterone profiles are drawn at -60, 0, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360 and 480 minutes following the evening dose on Days 1 and 3 and at 0 and 60 minutes post administration following the morning/evening doses on Day 2 and morning dose on Day 3.
- PD testing takes place on Day 2, 30 minutes and 4.5 hours after the morning dose (psychophysiological testing) and Day 3, 30 minutes after the morning dose (computer testing). Subjects undergo a practice psychophysiological session before the first dosing.
- the subjects in this study are women with ANOR. Subjects are recruited from the medical practice or the general population through advertisements in local newspapers with additional information available on a website. Before scheduling the screening visit, subjects are asked a series of standardized questions by telephone to assess whether they are likely to be suitable for the study.
- DSM-IV criteria for Sexual Aversion Disorder, Substance-Induced Sexual Dysfunction, Dyspareunia (not caused by inadequate foreplay stimulation or alleviated by lubricants), Vaginismus, Gender Identity Disorder, Paraphilia, or for sexual Dysfunction Due to a General Medical Condition,
- sex steroid hormones such as androgens, estrogens other than in low dose combined ET/P, or gestagens (e.g. anabolic steroids, DHEA, Premarin® (conjugated equine estrogens)).
- any form of diabetes mellitus (subjects using antacids or with treated hyperlipidaemia or treated hypothyroidism will not be excluded provided they have been stable on their drug dose for at least six (6) months).
- History of nasal disorders e.g., seasonal or perennial allergic rhinitis, atrophic rhinitis, polyposis, abuse of nasal decongestants, clinically relevant nasal septum deviation, recurrent epistaxis) or sleep apnea.
- Hepatitis B a positive test for Hepatitis B surface antigen
- a history of Hepatitis C a positive test for Hepatitis C antibody
- a history of HIV infection or demonstration of HIV antibodies a positive test for Hepatitis B surface antigen
- Visit 1 (Day -15) - Screening Subjects for Inclusion and Exclusion Criteria:
- Pre-study screening is carried out within two (2) weeks prior to the start of the treatment. Subjects, after having voluntarily signed the Informed Consent Form, and before enrolment, are interviewed by the Clinical Investigator or his/her designee physician who will take the medical, sexual and physical history, record demographic data, and perform a routine physical examination including vital signs (blood pressure, resting heart rate, body weight, and height).
- FSFI and FSDS-R are administered, as well as MMQ, BDI-II, ISS, SIDI-II, SESII-W.
- CBC hemoglobin, hemoglobin A1 c, hematocrit, MCV, MCHC, RBC, WBC & differential
- Clinical Chemistry profile Na/K, glucose, urea, creatinine, total bilirubin, albumin, calcium, phosphate, uric acid, LDL, HDL, triglycerides, AST, ALT, ALP, GGT and CK).
- follicle stimulating hormone (percent), follicle stimulating hormone, luteinizing hormone, prolactin, progesterone, sex hormone binding globulin, total testosterone, and dehydroepiandrosterone sulfate are collected.
- Urine is collected for measuring specific gravity, glucose, ketones, bilirubin, pH, urobilinogen, leukocytes, nitrites.
- a urine drug screen is performed for amphetamines, benzodiazepines,
- cannabinoids cannabinoids, cocaine, opiates, MDMA. Subjects with positive test are not enrolled. • Ethanol will be screened for by breathalyzer.
- Visit 2 (Day 1) - Start of Baseline, Randomization, PK Blood Sampling and PD Testing:
- OTC and prescription drugs, alcohol or cigarettes.
- Subjects are requested to abstain from alcohol for 48 hours prior to admission to the clinic. Alcohol consumption is strictly forbidden at any time during the overnight stay in the clinic. There are no restrictions with respect to food intake during the blood collections for the PK profile.
- Urine for urinalysis and urine drug screen is collected along with performing an alcohol breath test.
- a venous cannula will be placed in a forearm vein, and blood sampling starts one hour before the evening administration of the Study Drug.
- Blood samples are drawn for plasma testosterone and dihydrotestosterone levels at -60, 0, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360, and 480 minutes post administration.
- Subjects are dosed with Study Drug between 800 and 900 and between 2000 and 2100 hours (unless on Intrinsa ® ). Blood samples are drawn for plasma testosterone and dihydrotestosterone levels at times 0 and 60 minutes post administration.
- Subjects are dosed with Study Drug at 800 and 900 and between 2000 and 2100 hours. Blood samples are drawn for plasma testosterone and
- dihydrotestosterone levels at times 0 and 60 minutes following administration of the morning dose and at 0, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360, and 480 minutes following administration of the evening dose.
- Visit 5 (Day 4) - Discharge-Close Out:
- Venous blood is collected, after an overnight fast, for a CBC (hemoglobin, hemoglobin A1 c, hematocrit, MCV, MCHC, RBC, WBC & differential), clinical chemistry profile (Na/K, glucose, urea, creatinine, total bilirubin, albumin, calcium, phosphate, uric acid, LDL, HDL, triglycerides, AST, ALT, ALP, GGT and CK).
- CBC hemoglobin, hemoglobin A1 c, hematocrit, MCV, MCHC, RBC, WBC & differential
- clinical chemistry profile Na/K, glucose, urea, creatinine, total bilirubin, albumin, calcium, phosphate, uric acid, LDL, HDL, triglycerides, AST, ALT, ALP, GGT and CK).
- progesterone progesterone, sex hormone binding globulin, total testosterone, and
- dehydroepiandrosterone sulfate are collected.
- Urine is collected for measuring specific gravity, glucose, ketones, bilirubin, pH, urobilinogen, leukocytes, nitrites.
- the 21 -item BDI-II is administered (Beck, Steer, & Brown. 1996), Dutch adaptation (Van der Does, 2002).
- the range for the BDI total score is 0-63, with higher scores indicating more depressive symptoms.
- the Maudsley Marital Questionnaire (MMQ; Crowe, 1978) is a 20-item self-report instrument measuring dissatisfaction with the general relationship, with the sexual relationship, and with life in general.
- the MMQ has shown good reliability and validity.
- the psychometric qualities of the Dutch version of the MMQ were also found to be satisfactory (Arrindell, Boelens, & Lambert. 1983). Higher scores represent larger dissatisfaction.
- the level of the woman's sexual functioning is assessed by the Female Sexual Function Index (FSFI ; Rosen, Brown, Heiman, et al. 2000).
- the FSFI ® is a self- administered questionnaire that consists of 19 questions.
- the scale contains six domains: desire, arousal, lubrication, orgasm, satisfaction, and pain.
- the range for the total score is 2-36, with lower scores representing worse sexual function.
- the psychometric quality of the FSFI is satisfactory (Wiegel, Meston, & Rosen. 2005).
- the woman's level of personal distress due to sexual dysfunction is assessed by the Female Sexual Distress Scale-Revised (FSDS-R ® ; Derogatis, Clayton, Lewis- D'Agostino, et al. 2008).
- FSDS-R ® Female Sexual Distress Scale-Revised
- the items inquire about negative feelings and problems that are bothersome or cause distress during the past 30 days. Reliability and validity of the FSDS ® (12-item version) has been evaluated in different samples of sexually functional and
- the FSDS ® showed a high degree of discrimination between sexually dysfunctional and functional women in each of its three validation studies. Results in a Dutch sample supported the unidimensional structure of the FSDS and its reliability and psychometric validity (ter Kuile, Brauer, & Laan, 2006).
- An additional question (question 13) has been added to the validated FSDS ® . This question is about distress specifically related to sexual desire. The maximum total score of the FSDS-R ® indicating the maximum level of sexual distress is '52'. Both the FSDS-R ® total score and the Question 13 score alone will be analyzed.
- the level of the woman's sexual desire is assessed by the Sexual Desire
- SDI-II Inventory-ll
- Spector, Carey, & Steinberg. 1 996 The SDI-II consists of two seven-item self-report scales: the Dyadic Sexual Desire scale, which measures an individual's desire for sexual activity with a partner, and the Solitary Sexual Desire scale, which measures an individual's desire for autoerotic sexual activity.
- the SESII-W has eight lower-order factors, which in turn load on two higher-order factors, Sexual Excitation and Sexual Inhibition.
- the questionnaire shows good test-retest reliability and convergent and discriminant validity and Sexual
- Conform Wigboldus et al. (2005), the stIAT used in this study is designed to assess subjects' affective associations with sexual stimuli (Brauer, van Leeuwen, Janssen, et al. submitted). Subjects are instructed to classify pictures portraying sexual acts (i.e., target stimuli) and words representing "positive” or “negative” meanings (i.e., attribute stimuli) to the appropriate superordinate category (i.e., "sex", "positive”, “negative”) as quickly as possible by pressing only a left or right response key on a keyboard. These labels used for these categories (sex, positive, negative) are continuously visible on the computer screen.
- the stIAT consists of a combination of practice and experimental blocks (see Greenwald, McGhee & Schwartz. 1998 for detailed methodology).
- the experimental blocks consist of one 'incongruent' and one 'congruent' block of trials. In the incongruent block, "sex" and “negative” are mapped on a single key and “positive” on the other, while in the congruent block, "sex" and
- the target category consists of 5 exemplar stimuli of sexual images from the International Affective Picture System (IAPS; Center for the Study of Emotion and Attention, 1995), with the following numbers: 4800, 4652, 4658, 4659, and 4672.
- the attribute categories consists of 20 generally positive and 20 generally negative words (Dotsch & Wigboldus, 2008; Dotsch, Wigboldus, Langner et al. 2008), thus reflecting more global affective associations with sex. These words were controlled for length and frequency. With respect to the validity, the stIAT's strength lies in high effect sizes due to double opposing categories often leading to slower reaction times (the categorization decision requires effort as there are several possibilities to consider).
- target words are preceded by another word or image that influences the categorisation speed of the target word.
- PAT Hermans, De Houwer, & Eelen, 1994
- Subjects are further instructed to focus on the words that appeared on the screen and not to attend to the background images as these are of no importance for the task and the categories to which the pictorial stimuli belong (sex, neutral) are not explained.
- the PAT captures the unintentional influence of the affective value of the pictorial background stimuli on task performance.
- the time to select the correct response to the words is influenced by the match between the valence of the word and the valence of the background image (sex or neutral), thereby revealing indirectly the valence of the picture for the subjects.
- the word categories consist of 10 positive words and 10 negative words.
- the PAT consists of positive and negative words that are applicable to a sexual situation, but that do not exclusively refer to sexual experiences (e.g., enjoyable, wonderful, dirty, disgusting) in order to create a conceptual overlap between the content of the words and the content triggered by the sexual pictures.
- the words appear at one of four randomized locations on the picture to avoid expectation-related responses and to make sure subjects would move their eyes over the image.
- the sexual pictures were taken from another study on implicit associations with sexual stimuli in women with dyspareunia (Brauer, de Jong, Huijding et al., 2009). These pictures display a variety of sexual acts (e.g., kissing, cunnilingus, fellatio, coitus). Based on each sexual picture, a control picture was created by scrambling the sexual image, leaving a neutral stimulus. All pictures are standardized to 600 x 480 pixels and matched for brightness and contrast. Each stimulus remains on the screen until subjects make a decision or until 3,000 ms has elapsed. After 10 practice trials, 80 experimental trials are presented.
- Each word is paired randomly with a sexual picture and a neutral picture, resulting in four different combinations each presented 20 times: positive words and sexual images, negative words and sexual images, positive words and neutral images, negative words and neutral images.
- the order of presentation of the trials is counterbalanced within, and response key mappings (i.e., positive/negative or negative/positive) are
- the computer records the accuracy and latency of each response.
- the strength of the PAT is that it is not sensitive to a possible interpretation bias due to the need to attend to the different stimulus categories at the same time, as is the case in the stIAT.
- the dot-probe task assesses attentional preference for sexual and neutral visual stimuli.
- subjects are shown two images side by side on a computer screen for 500 ms. When the two images disappear, a target stimulus represented by a small dot appears in the place of one of the images. Subjects are asked to indicate the location (side) of the dot.
- Mean RTs are calculated for three categories: 1 ) neutral neutral 2) neutral sex with the dot under neutral 3) neutral sex with the dot under sex. If reaction times are faster when the dot appears in the place of a certain class of stimuli this indicates an attentional bias towards this class of stimuli.
- Psychophysiological testing consists of assessment of genital response (vaginal pulse amplitude) and subjective sexual arousal during sexual to self-induced erotic fantasy (3 min), a low-intensity erotic film clip (5 min), and a high-intensity erotic film clip (5 min) (Laan et al., in preparation).
- the erotic conditions are separated by variable interstimulus intervals during which subjects complete a concentration task (simple arithmetic problems) to allow for return-to-baseline.
- the erotic stimulus testing is preceded by a 8 min neutral film to establish baseline levels.
- VPA is measured using a vaginal photoplethysmograph developed by Bert Molenkamp (Technical Support, Department of Psychology, University of Amsterdam) based on instruments initially developed by Sintchak and Geer (1975).
- a signal-conditioning amplifier separates the VPA from the direct current component using a 12 dB/octave, 0.7-Hz filter. Additional filtering for VPA is 24 dB/octave, 0.4 Hz high-pass.
- the VPA signal is digitalized at 100 Hz with a Keithley KPCI3107 A/D converter, running on a Windows 2000 PC system. Depth of the probe and orientation of the light source is controlled by a device (a 9-x2-cm FDA-approved perspex plate) attached to the cable within 5cm of the optical sensor. Subjects are instructed to insert the probe until the plate touched their labia. The probe and plate are sterilized according to standard department protocol. .
- AUC Area under the concentration curve
- C aV g are calculated for the 12 hour period as well as ⁇ when appropriate. For subjects on Intrinsa ® , a 24 hour calculation is performed.
- Peak Trough Fluctuation (PTF) and Peak Trough Swing (PTS) are calculated as follows:
- the stIAT effect is analyzed with an analysis of variance with fixed factor treatment, group (HSDD and SA) and the interaction treatment by group.
- the contrasts are calculated within the model.
- the two PAT variables (RT positive and RT negative) are analyzed with an analysis of variance with fixed factors treatment, group (ANOR) and group by treatment. The contrasts will be calculated within the model.
- DOT effect mean neutral sex with dot under neutral - mean neutral sex with dot under sex.
- the DOT effect is analyzed with an analysis of variance with fixed factor treatment, group (ANOR) and the interaction treatment by group.
- the contrasts are calculated within the model.
- VPA is averaged every 30 seconds during several conditions: neutral film (8 min), self induced erotic fantasy (3 min), low intensity erotic film clip (5 min) and high intensity erotic film clip (5 min). All conditions are offered twice: once 0.5 hours after application of the nasal gel and once 4.5 hours after application of the nasal gel.
- VPA during the erotic fantasy the low intensity film and the high intensity film are analyzed separately and the different moments (0.5 hours after and 4.5 hours after dosing) are also be analyzed separately, resulting in 6 analyses.
- VPA during a condition and a moment is analyzed with a mixed model analysis of variance with fixed factors treatment, group (ANOR), time, group by treatment, treatment by time and random factor subject and the average VPA score during the neutral film as covariate. Contrasts are calculated within the model.
- ASFDQ score after erotic stimulation during a moment is analyzed with an analysis of variance with factors treatment, group (ANOR), and group by treatment; and the score prior to sexual stimuli as covariate. Contrasts are calculated within the model.
- analyses are not feasible according to the described models with the given data, analyses are adjusted. If considered useful extra exploratory analyses are conducted.
- Nasal Tolerance Nasal tolerance data is presented in summary tables. No statistical analysis will be performed.
- testosterone gel formulations of the invention are evaluated in studies of women with HSDD.
- the effect of testosterone gel formulations of the invention on sexual stimuli in women with HSDD is also determined.
- Three doses of a testosterone gel formulation of the invention are investigated: 150 ⁇ g, 450 ⁇ g and 600 ⁇ g per nostril.
- a total of 5 doses of a testosterone gel formulation of the invention is administered BID intranasally to women.
- the Intrinsa ® patch 300 ⁇ g testosterone is administered as a control in the HSDD cohort.
- Test subjects receive 5 doses of a testosterone gel formulation of the invention over a three day period.
- Subjects in the HSDD cohort are randomized to receive either a testosterone gel formulation of the invention (3 dose levels) or the Intrinsa ® patch. Randomization is according to the design allocation below.
- Three-quarters (75%) of the subjects in the HSDD cohort are administered a testosterone gel formulation of the invention (0.15%, 0.45% or 0.6%) on five (5) occasions during the study: Day 1 at 2000 hours, Day 2 at 800 and 2000 hours, and Day 3 at 800 and 2000 hours.
- the intranasal gel is administered to both nostrils (1 syringe (100 ⁇ volume) per nostril).
- To the remaining one-quarter (25%) of the subjects is administered the Intrinsa ® patch at 2000 hours on Day 1 which will remain on the subject's lower abdomen for the duration of the study. The patch is removed on Day 4 prior to discharging the subject from the clinic.
- Study medication consists of testosterone gel formulations of the invention and testosterone gel formulations of the invention placebo gel and is packed in single use syringes designed to expel 100 ⁇ of gel. Two syringes are packaged in a foil pouch.
- the active control for the HSDD cohort, Intrinsa ® remains in its original packaging from the manufacturer.
- Subjects are randomized into the dosing regimen that is administered during a four day (three night) in-patient treatment period and receive either intranasal testosterone gel formulation of the invention (3 dose levels) or Intrinsa ® patch (HSDD) according to the design allocation:
- the randomization scheme is created for each study center and consists of blocks of four treatments per cohort.
- the study is a four day study.
- the study start with Study Drug dosing between 2000 and 2100 hours on Day 1 (Baseline).
- Blood samples for plasma testosterone and dihydrotestosterone profiles are drawn at -60, 0, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360 and 480 minutes following the evening dose on Days 1 and 3 and at 0 and 60 minutes (except Intrinsa arm - no 60 minute on Day 2 and Day 3 morning draws) post administration following the morning/evening doses on Day 2 and morning dose on Day 3.
- PD testing will take place on Day 2, 30 minutes and 4.5 hours after the morning dose (psychophysiological testing) and Day 3, 30 minutes after the morning dose (computer testing). Subjects will have undergone a practice
- the subjects in this study are women with HSDD. Subjects are recruited from the medical practice or the general population through advertisements in local newspapers with additional information available on a website. Before scheduling the screening visit, subjects are asked a series of standardized questions by telephone to assess whether they are likely to be suitable for the study.
- sex steroid hormones such as androgens, estrogens other than in low dose combined ET/P, or gestagens (e.g. anabolic steroids, DHEA, Premarin® (conjugated equine estrogens)).
- thyroid hormones Treatment with thyroid hormones (only for stable replacement therapy).
- Significant intercurrent disease of any type, in particular liver, kidney, or heart disease, or any form of diabetes mellitus subjects using antacids or with treated hyperlipidaemia or treated hypothyroidism will not be excluded provided they have been stable on their drug dose for at least six months).
- nasal disorders e.g., seasonal or perennial allergic rhinitis, atrophic rhinitis, polyposis, abuse of nasal decongestants, clinically relevant nasal septum deviation, recurrent epistaxis) or sleep apnea.
- Hepatitis B a positive test for Hepatitis B surface antigen
- a history of Hepatitis C a positive test for Hepatitis C antibody
- a history of HIV infection or demonstration of HIV antibodies a positive test for Hepatitis B surface antigen
- Visit 1 (Day -15) - Screening Subjects for Inclusion and Exclusion Criteria:
- Pre-study screening is carried out within two (2) weeks prior to the start of the treatment. Subjects, after having voluntarily signed the Informed Consent Form, and before enrolment, are interviewed by the Clinical Investigator or his/her designee physician who takes the medical, sexual and physical history, record demographic data, and performs a routine physical examination including vital signs (blood pressure, resting heart rate, body weight, and height).
- FSFI and FSDS-R are administered, as well as MMQ, BDI-II, ISS, SIDI-II, SESII-W.
- CBC hemoglobin, hemoglobin A1 c, hematocrit, MCV, MCHC, RBC, WBC & differential
- Clinical Chemistry profile Na/K, glucose, urea, creatinine, total bilirubin, albumin, calcium, phosphate, uric acid, LDL, HDL, triglycerides, AST, ALT, ALP, GGT and CK).
- follicle stimulating hormone (percent), follicle stimulating hormone, luteinizing hormone, prolactin, progesterone, sex hormone binding globulin, total testosterone, and dehydroepiandrosterone sulfate are collected.
- Urine is collected for measuring specific gravity, glucose, ketones, bilirubin, pH, urobilinogen, leukocytes, nitrites. • Subjects also undergo Hepatitis B, C and HIV testing (Hepatitis B surface antigen, Hepatitis C antibody, HIV antibodies in plasma).
- a urine drug screen is performed for amphetamines, benzodiazepines,
- cannabinoids cannabinoids, cocaine, opiates, MDMA. Subjects with positive test are not enrolled.
- Visit 2 (Day 1) - Start of Baseline, Randomization, PK Blood Sampling and PD Testing:
- OTC and prescription drugs, alcohol or cigarettes.
- Subjects are requested to abstain from alcohol for 48 hours prior to admission to the clinic. Alcohol consumption is strictly forbidden at any time during the overnight stay in the clinic. There are no restrictions with respect to food intake during the blood collections for the PK profile.
- Urine for urinalysis and urine drug screen is collected along with performing an alcohol breath test.
- a venous cannula is placed in a forearm vein, and blood sampling starts one hour before the evening administration of the Study Drug.
- Subjects are dosed between 2000 and 2100 hours. • Blood samples are drawn for plasma testosterone and dihydrotestosterone levels at -60, 0, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360, and 480 minutes post administration.
- Subjects are dosed with Study Drug between 800 and 900 and between 2000 and 2100 hours (unless on Intrinsa ® ). Blood samples are drawn for plasma testosterone and dihydrotestosterone levels at times 0 and 60 minutes post administration. For subjects on Intrinsa ® , a time 0 draw between 800-900 and 2000-2100 hours is obtained.
- invention -arms or the placebo gel, psychophysiological testing is performed 30 minutes and 4.5 hours post-dosing in the morning
- Subjects are dosed with Study Drug at 800 and 900 and between 2000 and 2100 hours (unless on Intrinsa ® ). Blood samples are drawn for plasma testosterone and dihydrotestosterone levels at times 0 and 60 minutes following administration of the morning dose and at 0, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360, and 480 minutes following administration of the evening dose. Subjects receiving Intrinsa ® have the time 0 draw for the morning dose and all times in the evening as if they received a new dose. • For subjects allocated to the three testosterone gel formulations of the invention -arms or the placebo gel, computer testing is performed 30 minutes post-dosing in the morning. For the women randomized to the Intrinsa ® patch, psychophysiological testing takes place on Day 3 between 800 and 900 hours. Computer testing follows in the afternoon of Day 3 between 1600 and 1700 hours.
- Visit 5 (Day 4) - Discharge-Close Out:
- Venous blood is collected, after an overnight fast, for a CBC (hemoglobin, hemoglobin A1 c, hematocrit, MCV, MCHC, RBC, WBC & differential), clinical chemistry profile (Na/K, glucose, urea, creatinine, total bilirubin, albumin, calcium, phosphate, uric acid, LDL, HDL, triglycerides, AST, ALT, ALP, GGT and CK).
- CBC hemoglobin, hemoglobin A1 c, hematocrit, MCV, MCHC, RBC, WBC & differential
- clinical chemistry profile Na/K, glucose, urea, creatinine, total bilirubin, albumin, calcium, phosphate, uric acid, LDL, HDL, triglycerides, AST, ALT, ALP, GGT and CK).
- estradiol free testosterone
- free testosterone percent
- follicle stimulating hormone luteinizing hormone
- prolactin prolactin
- dehydroepiandrosterone sulfate dehydroepiandrosterone sulfate
- Urine is collected for measuring specific gravity, glucose, ketones, bilirubin, pH, urobilinogen, leukocytes, nitrites.
- the 21 -item BDI-II is administered (Beck, Steer, & Brown. 1996), Dutch adaptation (Van der Does, 2002).
- the range for the BDI total score is 0-63, with higher scores indicating more depressive symptoms.
- the Maudsley Marital Questionnaire (MMQ; Crowe, 1978) is a 20-item self-report instrument measuring dissatisfaction with the general relationship, with the sexual relationship, and with life in general.
- the MMQ has shown good reliability and validity.
- the psychometric qualities of the Dutch version of the MMQ were also found to be satisfactory (Arrindell, Boelens, & Lambert. 1983). Higher scores represent larger dissatisfaction.
- the level of the woman's sexual functioning is assessed by the Female Sexual Function Index (FSFI ; Rosen, Brown, Heiman, et al. 2000).
- the FSFI ® is a self- administered questionnaire that consists of 19 questions.
- the scale contains six domains: desire, arousal, lubrication, orgasm, satisfaction, and pain.
- the range for the total score is 2-36, with lower scores representing worse sexual function.
- the woman's level of personal distress due to sexual dysfunction is assessed by the Female Sexual Distress Scale-Revised (FSDS-R ® ; Derogatis, Clayton, Lewis- D'Agostino, et al. 2008).
- FSDS-R ® Female Sexual Distress Scale-Revised
- the items inquire about negative feelings and problems that are bothersome or cause distress during the past 30 days. Reliability and validity of the FSDS ® (12-item version) has been evaluated in different samples of sexually functional and
- the FSDS ® showed a high degree of discrimination between sexually dysfunctional and functional women in each of its three validation studies. Results in a Dutch sample supported the unidimensional structure of the FSDS and its reliability and psychometric validity (ter Kuile, Brauer, & Laan, 2006).
- An additional question (question 13) has been added to the validated FSDS ® . This question is about distress specifically related to sexual desire. The maximum total score of the FSDS-R ® indicating the maximum level of sexual distress is '52'. Both the FSDS-R ® total score and the Question 13 score alone will be analyzed.
- the level of the woman's sexual desire is assessed by the Sexual Desire
- SDI-II Inventory-ll
- Spector, Carey, & Steinberg. 1 996 The SDI-II consists of two seven-item self-report scales: the Dyadic Sexual Desire scale, which measures an individual's desire for sexual activity with a partner, and the Solitary Sexual Desire scale, which measures an individual's desire for autoerotic sexual activity.
- the SESII-W has eight lower-order factors, which in turn load on two higher-order factors, Sexual Excitation and Sexual Inhibition.
- the questionnaire shows good test-retest reliability and convergent and discriminant validity and Sexual
- Conform Wigboldus et al. (2005), the stIAT used in this study is designed to assess subjects' affective associations with sexual stimuli (Brauer, van Leeuwen, Janssen, et al. submitted). Subjects are instructed to classify pictures portraying sexual acts (i.e., target stimuli) and words representing "positive” or “negative” meanings (i.e., attribute stimuli) to the appropriate superordinate category (i.e., "sex", "positive”, “negative”) as quickly as possible by pressing only a left or right response key on a keyboard. These labels used for these categories (sex, positive, negative) are continuously visible on the computer screen.
- the stIAT consists of a combination of practice and experimental blocks (see Greenwald, McGhee & Schwartz. 1998 for detailed methodology).
- the experimental blocks consist of one 'incongruent' and one 'congruent' block of trials. In the incongruent block, "sex" and “negative” are mapped on a single key and “positive” on the other, while in the congruent block, "sex" and
- the target category consists of 5 exemplar stimuli of sexual images from the International Affective Picture System (IAPS; Center for the Study of Emotion and Attention, 1995), with the following numbers: 4800, 4652, 4658, 4659, and 4672.
- the attribute categories consists of 20 generally positive and 20 generally negative words (Dotsch & Wigboldus, 2008; Dotsch, Wigboldus, Langner et al. 2008), thus reflecting more global affective associations with sex. These words were controlled for length and frequency. With respect to the validity, the stIAT's strength lies in high effect sizes due to double opposing categories often leading to slower reaction times (the categorization decision requires effort as there are several possibilities to consider).
- target words are preceded by another word or image that influences the categorisation speed of the target word.
- PAT Hermans, De Houwer, & Eelen, 1994
- Subjects are further instructed to focus on the words that appeared on the screen and not to attend to the background images as these are of no importance for the task and the categories to which the pictorial stimuli belong (sex, neutral) are not explained.
- the PAT captures the unintentional influence of the affective value of the pictorial background stimuli on task performance.
- the time to select the correct response to the words is influenced by the match between the valence of the word and the valence of the background image (sex or neutral), thereby revealing indirectly the valence of the picture for the subjects.
- the word categories consist of 10 positive words and 10 negative words.
- the PAT consists of positive and negative words that are applicable to a sexual situation, but that do not exclusively refer to sexual experiences (e.g., enjoyable, wonderful, dirty, disgusting) in order to create a conceptual overlap between the content of the words and the content triggered by the sexual pictures.
- the words appear at one of four randomized locations on the picture to avoid expectation-related responses and to make sure subjects would move their eyes over the image.
- the sexual pictures were taken from another study on implicit associations with sexual stimuli in women with dyspareunia (Brauer, de Jong, Huijding et al., 2009). These pictures display a variety of sexual acts (e.g., kissing, cunnilingus, fellatio, coitus). Based on each sexual picture, a control picture was created by scrambling the sexual image, leaving a neutral stimulus. All pictures are standardized to 600 x 480 pixels and matched for brightness and contrast. Each stimulus remains on the screen until subjects make a decision or until 3,000 ms has elapsed. After 10 practice trials, 80 experimental trials are presented.
- Each word is paired randomly with a sexual picture and a neutral picture, resulting in four different combinations each presented 20 times: positive words and sexual images, negative words and sexual images, positive words and neutral images, negative words and neutral images.
- the order of presentation of the trials is counterbalanced within, and response key mappings (i.e., positive/negative or negative/positive) are
- the computer records the accuracy and latency of each response.
- the strength of the PAT is that it is not sensitive to a possible interpretation bias due to the need to attend to the different stimulus categories at the same time, as is the case in the stIAT.
- the dot-probe task assesses attentional preference for sexual and neutral visual stimuli.
- subjects are shown two images side by side on a computer screen for 500 ms. When the two images disappear, a target stimulus represented by a small dot appears in the place of one of the images. Subjects are asked to indicate the location (side) of the dot.
- Mean RTs are calculated for three categories: 1 ) neutral neutral 2) neutral sex with the dot under neutral 3) neutral sex with the dot under sex. If reaction times are faster when the dot appears in the place of a certain class of stimuli this indicates an attentional bias towards this class of stimuli.
- Psychophysiological testing consists of assessment of genital response (vaginal pulse amplitude) and subjective sexual arousal during sexual to self-induced erotic fantasy (3 min), a low-intensity erotic film clip (5 min), and a high-intensity erotic film clip (5 min) (Laan et al., in preparation).
- the erotic conditions are separated by variable interstimulus intervals during which subjects complete a concentration task (simple arithmetic problems) to allow for return-to-baseline.
- the erotic stimulus testing is preceded by a 8 min neutral film to establish baseline levels.
- VPA is measured using a vaginal photoplethysmograph developed by Bert Molenkamp (Technical Support, Department of Psychology, University of Amsterdam) based on instruments initially developed by Sintchak and Geer (1975).
- a signal-conditioning amplifier separates the VPA from the direct current component using a 12 dB/octave, 0.7-Hz filter. Additional filtering for VPA is 24 dB/octave, 0.4 Hz high-pass.
- the VPA signal is digitalized at 100 Hz with a Keithley KPCI3107 A/D converter, running on a Windows 2000 PC system. Depth of the probe and orientation of the light source is controlled by a device (a 9-x2-cm FDA-approved perspex plate) attached to the cable within 5cm of the optical sensor. Subjects are instructed to insert the probe until the plate touched their labia. The probe and plate are sterilized according to standard department protocol. .
- AUC Area under the concentration curve
- Peak Trough Fluctuation (PTF) and Peak Trough Swing (PTS) are calculated as follows:
- the stIAT effect is analyzed with an analysis of variance with fixed factor treatment, group (HSDD and SA) and the interaction treatment by group.
- the contrasts are calculated within the model.
- the two PAT variables (RT positive and RT negative) are analyzed with an analysis of variance with fixed factors treatment, group (ANOR) and group by treatment. The contrasts will be calculated within the model.
- DOT effect mean neutral sex with dot under neutral - mean neutral sex with dot under sex.
- ANOR fixed factor treatment
- VPA VPA artefact deletion
- Bert Molenkamp Plant Support, Department of Psychology, University of Amsterdam
- peak-to-trough amplitude is calculated for each remaining pulse.
- VPA is averaged every 30 seconds during several conditions: neutral film (8 min), self induced erotic fantasy (3 min), low intensity erotic film clip (5 min) and high intensity erotic film clip (5 min). All conditions are offered twice: once 0.5 hours after application of the nasal gel and once 4.5 hours after application of the nasal gel.
- VPA during the erotic fantasy the low intensity film and the high intensity film are analyzed separately and the different moments (0.5 hours after and 4.5 hours after dosing) are also be analyzed separately, resulting in 6 analyses.
- VPA during a condition and a moment is analyzed with a mixed model analysis of variance with fixed factors treatment, group (ANOR), time, group by treatment, treatment by time and random factor subject and the average VPA score during the neutral film as covariate. Contrasts are calculated within the model.
- ASFDQ score after erotic stimulation during a moment is analyzed with an analysis of variance with factors treatment, group (ANOR), and group by treatment; and the score prior to sexual stimuli as covariate. Contrasts are calculated within the model.
- results are back-transformed and reported as % change.
- Graphs of least square means estimates over time by treatment are presented with error bars indicating the upper and lower 95% confidence interval for the highest and lowest profile respectively. Least square means of the contrasts are tabulated.
- analyses are not feasible according to the described models with the given data, analyses are adjusted. If considered useful extra exploratory analyses are conducted.
- Nasal Tolerance Nasal tolerance data is presented in summary tables. No statistical analysis will be performed.
- Figs. 2, 3 and 7-8 show or compare the results between the effects of the testosterone gel nasal formulations of the invention on subjects diagnosed with anorgasmia or HSDD.
- PK Pharmacokinetic
- PD Pharmacodynamic
- this PK and PD study is to assess the serum testosterone pharmacokinetic profile and the pharmacodynamic response measuring Vaginal Pulse Amplitude ("VPA") following a single dose administration of each of the testosterone bio- adhesive gel formulations of the invention (0.15% - , 0.45% and 0.6% testosterone by weight of the gel formulation, as reported in Examples 1 -5), as compared to placebo, in women with anorgasmia.
- VPA Vaginal Pulse Amplitude
- Each women receives four different single intranasal doses of 100 ⁇ per nostril adminsitered via a unit-dose syringe on four different days (that is, each of the 12 women in this study receives a TBS-2 high dose (1 .2 mg - 0.6% by weight testosterone - 0.6mg/100 ⁇ /nostril), a TBS-2 medium dose (0.9 mg - 0.45% by weight testosterone - 0.45mg/100 ⁇ /nostril) or a TBS-2 low dose (0.3 mg - 0.15% by weight testosterone - 0.15mg/100 ⁇ /nostril), and placebo TBS-2 (anorgasmia cohort).
- VPA vaginal pulse amplitude
- the PK results show that there is an increase in plasma testosterone levels with increasing dose levels. See Fig. 14.
- Mean concentrations of plasma testosterone 0-12 hours after dosing are: (a) TBS-2 high dose (1 .2 mg - 0.6% by weight testosterone - 0.6 mg/100 ⁇ /nostril) - about 70 ng/dL after about the first 100 minutes of adminsitration, about 50ng/dl_ after about the first 250 minutes of adminsitration, and about 40 ng/dL after about 350 minutes following adminsitration and thereafter; (b) TBS-2 medium dose (0.9 mg - 0.45% by weight testosterone - 0.45 mg/100 ⁇ /nostril) - about 55 ng/dL after about the first 25 minutes of adminsitration, about 35ng/dL after about the first 250 minutes of adminsitration, and about 35-30 ng/dL after about 350 minutes following adminsitration and thereafter; and (c) TBS-2 low dose (0.3 mg - 0.15% by weight testosterone - 0.15 mg/
- the testosterone level returns to baseline following adminsitration of the single dose.
- the PD aspect of the study there are favorable statistically significant differences in VPA response in the women for each of the TBS-2 dosage strengths vs. placebo at 0.5 hours and at 4.5 hours post-dose.
- the TBS-2 nasally applied testosterone bio-adhesive gels of the preset invention may be uniquely taken on a prn basis, i.e., on demand, (ii) have an ideal safety profile, i.e., there appears to be no androgen-related side effects, there ia a low testosterone drug load, and (iii) present no risk of testosterone transference.
- VTS Vibrotactile Stimulation Study
- each women is administered an intranasal single high-dose of TBS-2 (1 .2 mg - 0.6% by weight testosterone - 0.6 mg/100 ⁇ /nostril).
- Fig. 16 more women report, treated with high dose TBS-2, report orgasms, as compared to placebo.
- women who are treated with high dose TBS-2 8 report a total of 8 orgasms, as compared to no orgasms for those women who receive placebo.
- women who are treated with high dose TBS-2 8 report a total of 4 orgasms, as compared to 2 orgasms for those women who receive placebo. See Fig. 16.
- Mean free testosterone levels are about 6.35 pg/mL (chronic treatments achieve about 3.1 -4.0 pg/mL);
- TBS-2 when administered prn, is believed to elicit and enhance sexual response in women • TBS-2 is believed to maintain total and free testosterone levels in the normal range
- TBS-2 is believed to be safe and none of the adverse events commonly observed with chronic testosterone treatments are observed
- Comfortable environment (home setting) and partner interaction may play a role in acheivement of orgasm
- Anorgasmia is the second most common sexual disorder among women, with prevalence figures ranging from 16% to 28% in the United States, Europe, and Central/South America, and 30-46% in Asia.
- the androgen testosterone is known to play a role in mood, body composition, bone mineral density and has central and peripheral effects on sexual function.
- the only registered testosterone product for use in women is Intrinsa, a testosterone slow- release transdermal patch.
- TBS-2 The product under investigation in this study, is a testosterone intranasal gel that may allow for direct uptake of testosterone into the brain and rapid absorption into systemic circulation.
- the delivery of testosterone to the brain and the rapid systemic absorption is hypothesized to be effective in enhancing sexual desire and orgasm in an "as needed" way, thus avoiding chronic exposure to testosterone.
- the primary objective of this study was to evaluate the effect of a single dose of TBS-2 on the occurrence of orgasm in primary and secondary anorgasmic women during sexual stimulation at one of four different time points after administration of the study drug compared to a placebo group.
- Sexual stimulation consisted of vibrotactile stimulation to the glans clitoris (VTS) in combination with visual sexual stimulation (VSS).
- This randomized, single-blind, placebo-controlled five arms parallel group study was conducted at a single study center to evaluate the effect of a single dose of TBS-2, 1200 meg (600 meg per nostril), on the occurrence of orgasm at 0.5, 2.0, 4.0 and 8.0 hours post-dose administration in female subjects with primary or secondary anorgasmia using VTS and VSS.
- the study consisted of two screening visits and one treatment visit. TBS-2 or placebo was administered at Visit 3.
- VTS/VSS In the treatment visit, subjects received either TBS-2 or placebo and underwent VTS/VSS at 0.5, 2.0, 4.0 and 8.0 hours after dosing. VTS/VSS lasted 20 minutes or until orgasm was reached, after which stimulation was stopped. Safety was monitored throughout the study and PK data were collected. The study was conducted with approval of the Medical Ethics Committee of the Academic Medical Center.
- TBS-2 was administered to 45 subjects, while 1 1 subjects received placebo. At baseline, none of the subjects experienced serious relationship problems, and none of the subjects had a depressive disorder. Placebo and TBS-2 groups were comparable with respect to sexual function, sexual distress, their current level of sexual desire, and sexual satisfaction in the relationship.
- Two (2) subjects in the placebo group self-reported an orgasm.
- an additional eight (8) subjects reported sensations indicative of an actual orgasm at exit interview but had not recorded the orgasm during the treatment phase. No additional patients in the placebo arm provided such reports at exit interview.
- VPA Vaginal Pulse Amplitude
- TBS-2 was well tolerated. All AEs were mild and completely resolved at the end of the study. Physical examination, vital signs and clinical laboratory evaluations results did not reveal any additional clinically significant findings related to study treatment.
- IEC Independent Ethics Committee
- IRB Institutional Review Board
- FOD Female Orgasm Disorder
- Testosterone the primary circulation androgen in women, is a naturally occurring steroid secreted by the ovaries and the adrenal glands. Contrary to the sudden drop in estrogen during menopause, serum levels of androgens fall gradually as women age primarily due to a decrease in the production of adrenal androgen precursors (Goldstat, Briganti, Tran, Wolfe, & Davis, 2003). Testosterone plays a role in mood, body composition, bone mineral density and has central and peripheral effects on sexual function (Davis 1999, Goldstat et al., 2003).
- Testosterone stimulates dopamine release in various brain structures implicated in motivation and reward systems, including sexual desire. Testosterone was found to stimulate dopamine release in the medial preoptic area of the anterior hypothalamus under basal conditions and with sexual stimulation in rats (Halaris 2003).
- Testosterone therapy resulted in statistically significant improvements in the composite scores of the Psychological General Weil-Being Index, the Sabbatsberg Sexual Self-Rating Scale and the Beck Depression Inventory when compared with placebo. These effects were found while the mean total testosterone levels were in the low end of the normal range before treatment, and at the high end of the normal range during treatment. On the different sub-scales of the Sabbatsberg Sexuality Scale there was a significant effect of testosterone treatment on orgasm. This study suggests that although most previous studies with testosterone have addressed decreased sexual desire and especially included postmenopausal women, there are also measurable effects in premenopausal women both on general sexual wellbeing and on orgasm specifically.
- testosterone influences processes further "down the line” that lead to these responses.
- Fewer studies are available that directly demonstrate an enhancing effect of testosterone on orgasm.
- 300 women who received bilateral salpingo-oophorectomy and hysterectomy 300 mg of testosterone patch showed improvements in FOD symptoms (Braunstein, Sundwall, Katz, Shifren, Buster, Simon, et al., 2005).
- 10 mg of testosterone gel had positive effects on orgasm (Davis, Moreau, Kroll, Bouchard, Panay, Gass, et al., 2008).
- tibolone a synthetic steroid available in Europe, has shown improvements in orgasmic functioning (Kamenov, Todorova, & Christov, 2007).
- Intrinsa a testosterone slow-release transdermal patch. Intrinsa is indicated for the treatment of hypoactive sexual desire disorder (HSDD) in bilaterally oophorectomised and hysterectomised (surgically induced menopause) women receiving concomitant estrogen therapy.
- HSDD hypoactive sexual desire disorder
- TBS-2 The product under investigation in this study, is a testosterone intranasal gel. Contrary to the transdermal administration, administration of the bioadhesive TBS-2 through the nasal route may allow for direct uptake of testosterone into the brain and rapid absorption into systemic circulation (Banks, Morley, Niehoff, & Mattern, 2009). The delivery of testosterone to the brain and systemic absorption is hypothesized to be effective in enhancing sexual desire and orgasm. In addition, TBS-2 may prove effective in alleviating anorgasmia in an "as needed" way, thus avoiding chronic exposure to testosterone.
- Van Wingen et al. investigated whether the age-related decline in androgen levels is associated with reduced amygdala activity, and whether exogenous testosterone can restore amygdala activity.
- Healthy young and middle-aged women participated during the early follicular phase of the menstrual cycle, and amygdala responses to biologically salient stimuli were measured with functional magnetic resonance imaging (fMRI).
- Androgen levels were lower in middle-aged than young women, which was associated with decreased amygdala reactivity.
- Endogenous testosterone levels correlated positively with amygdala reactivity across the young and middle-aged women.
- TBS-2 pharmacokinetic profile of three doses of TBS-2 (300mcg, 900mcg and 1200mcg) in 32 female subjects diagnosed with hypoactive sexual desire disorder (HSDD) and anorgasmia (ANOR) (van Gorsel, Laan, Both, van Lunsen, van Gerven, Tkachenko, Dickstein, & Kreppner, 201 1 ).
- This study also assessed pharmacodynamic efficacy based on computer tasks with psychophysiological testing involving sexual imagery and visual sexual stimuli presentation along with genital arousal (vaginal pulse amplitude).
- TBS-2 was well absorbed following nasal administration of the three different doses. The maximum serum concentration was reached approximately 30 minutes to 2 hours post administration.
- TBS-2 Pharmacodynamic efficacy was evaluated using psychophysiological testing involving sexual imagery, visual sexual stimuli presentations and genital arousal (measured by vaginal pulse amplitude) as a dependent measure 30 minutes and 4.5 hours post administration of TBS-2.
- TBS-2 significantly increased genital arousal in ANOR women post TBS-2 administration.
- Trends in the subjective measures were observed in both the ANOR and HSDD cohort post TBS-2 administration.
- the primary objective of this study was to evaluate the effect of a single dose of TBS-2 on the occurrence of orgasm at 0.5, 2.0, 4.0 or 8.0 hours post-dose in female subjects with primary or secondary anorgasmia. Placebo was used as a control at 0.5 hour post-dose.
- TBS-2 time to orgasm
- This randomized, single-blind placebo-controlled five arms parallel group study was conducted at a single study center to evaluate the effect of a single dose of TBS-2, 1200 meg (600 meg per nostril), on the occurrence of orgasm at 0.5, 2.0, 4.0 and 8.0 hours post- dose in female subjects with primary or secondary anorgasmia using vibrotactile stimulation (VTS) to the glans clitoris and visual sexual stimulation (VSS).
- VTS vibrotactile stimulation
- VTS visual sexual stimulation
- the study was conducted with approval of the Medical Ethics Committee of the Academic Medical Center.
- Subjects were recruited from the general population through advertisement in local newspapers. Subjects were pre-screened by telephone using a structured interview with questions regarding sexual complaints and general health. Subjects who seemed eligible were invited for a screening visit. Written informed consent was obtained at the start of the first screening visit.
- BDI-II depressive symptoms
- MMQ relationship quality
- BDI-II Beck Depression Inventory-ll
- Protocol Protocol, appendix 2
- 21 -item BDI-II was administered (Beck, Steer, & Brown, 1996), Dutch adaptation (Van der Does, 2002).
- the range for the BDI total score is 0-63, with higher scores indicating more depressive symptoms. Subjects with a score of >14 on the BDI total score were excluded.
- the Maudslev Marital Questionnaire (MMQ, Protocol, appendix 3): the MMQ (Crowe, 1978) is a 20-item self-report instrument measuring dissatisfaction with the general relationship, with the sexual relationship, and with life in general.
- the MMQ has shown good reliability and validity.
- the psychometric qualities of the Dutch version of the MMQ were also found to be satisfactory (Arrindell, Boelens, & Lambert, 1983). Higher scores represent larger dissatisfaction. Subjects with a score on the MMQ-M subscale of > 20 were excluded.
- FSFI Female Sexual Function Index
- Protocol The level of the woman's sexual functioning was assessed by the FSFI (Rosen, Brown, Heiman, Leiblum, Meston, Shabsigh, et al. 2000).
- the FSFI is a self-administered questionnaire that consists of 19 questions.
- the scale contains six domains: desire, arousal, lubrication, orgasm, satisfaction, and pain.
- the range for the total score is 2-36, with lower scores representing worse sexual function.
- the psychometric quality of the FSFI is satisfactory (Wiegel, Meston, & Rosen, 2005).
- FSDS-R Female Sexual Distress Scale- Revised (FSDS-R, Protocol, appendix 5): The woman's level of personal distress due to sexual dysfunction was assessed by the FSDS-R (Derogatis, Clayton, Lewis-D'Agostino, Wunderlich, & Fu, 2008). The items inquire about negative feelings and problems that were bothersome or caused distress during the past 30 days. Reliability and validity of the FSDS (12-item version) was evaluated in different samples of sexually functional and dysfunctional women (Derogatis, Rosen, Leiblum, Burnett & Heiman, 2002). For the FSDS, results indicated a unidimensional factor structure, a high degree of internal consistency, and test-retest reliability.
- the FSDS showed a high degree of discrimination between sexually dysfunctional and functional women in each of its three validation studies. Results in a Dutch sample supported the unidimensional structure of the FSDS and its reliability and psychometric validity (ter Kuile, Brauer, & Laan, 2006). An additional question (Question 13) has been added to the validated FSDS. This question is about distress specifically related to sexual desire. The maximum total score of the FSDS-R indicating the maximum level of sexual distress is 52.
- ISS Index of Sexual Satisfaction
- ISS Protocol appendix 6
- This 25-item questionnaire asks subjects to evaluate various aspects of their sexual relationship, leading to a sum score that can range between 0 and 100. Higher scores correspond to greater sexual satisfaction.
- This measure has been shown to have good face, convergent, and discriminant validity with various samples. Example items are "I feel that my partner enjoys our sex life,” “I think that sex is wonderful,” and "My partner is sexually very exciting.”
- the ISS was translated into Dutch for an earlier study initiated by this Sponsor, psychometric data are not yet available.
- SDI-II Sexual Desire Inventory-ll
- Protocol Protocol, appendix 7
- the SDI-II consists of two seven-item self-report scales: the Dyadic Sexual Desire scale, which measures an individual's desire for sexual activity with a partner, and the Solitary Sexual Desire scale, which measures an individual's desire for autoerotic sexual activity.
- Psychometric data for the Dutch translation are not yet available.
- SESII-W Sexual Excitation/Sexual Inhibition
- Protocol, appendix 8 The SESII-W (Graham, Sanders, & Milhausen, 2006) was used to assess individuals' propensity for sexual excitation and sexual inhibition. It consists of 36 items, referring to stimulus situations that could affect sexual inhibition and sexual excitation or to general statements about arousability and inhibition. The instructions ask women to report what would be the most typical reaction now or how they think they would respond if the item does not apply to them. Items are rated on a 4-point Likert-rating scale, from "strongly disagree" to "strongly agree.” The SESII-W has eight lower-order factors, which in turn load on two higher-order factors, Sexual Excitation and Sexual Inhibition.
- the questionnaire shows good test-retest reliability and convergent and discriminant validity and Sexual Excitation and Sexual Inhibition appear to be relatively independent factors.
- the translated list is already in use in the Netherlands, and psychometric properties are currently being investigated in a large sample of women with and without sexual complaints (Bloemendaal & Laan, 2012).
- Intrauterine device in place for at least 3 months prior to study initiation Barrier method (condom use by partner)
- vaginal infection/vaginitis, cervicitis, interstitial cystitis, vulvodynia, or significant vaginal atrophy is a concern for vaginal infection/vaginitis, cervicitis, interstitial cystitis, vulvodynia, or significant vaginal atrophy.
- sex steroid hormones such as androgens, estrogens other than in low dose combined ET/P, or gestagens (e.g. anabolic steroids, DHEA, Premarin® (conjugated equine estrogens)).
- any form of diabetes mellitus (subjects with antacids or treated hyperlipidaemia or treated hypothyroidism will not be excluded provided they have been stable on their drug dose for at least six (6) months).
- nasal disorders e.g., seasonal or perennial allergic rhinitis, atrophic rhinitis, polyposis, abuse of nasal decongestants, clinically relevant nasal septum deviation, recurrent epistaxis) or sleep apnea.
- Subjects could leave the study at any time for any reason if they wished to do so without any consequences.
- the responsible investigator could also withdraw a subject if continuing participation was, in her opinion, deleterious for the subject's wellbeing. Subjects could also be withdrawn in case of protocol violation and non-compliance.
- TBS-2 an intranasal testosterone gel.
- a description of its physical, chemical, pharmaceutical properties and formulation can be found in the Investigator's Brochure. (1 ) Active drug;
- TBS-2 Syringes are pre-fied to contain 800 meg
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Epidemiology (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Diabetes (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Otolaryngology (AREA)
- Endocrinology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
The present invention relates to lower dosage strength pernasal testosterone gel formulations for intranasal administration and treatment methods for using the lower dosage strength pernasal testosterone gel formulations for treating a female subject with anorgasmia and/or hypoactive sexual desire disorder.
Description
FEMALE INTRANASAL LOWER DOSAGE STRENGTH TESTOSTERONE GEL FORMULATIONS AND USE THEREOF FOR TREATING ANORGASMIA OR HYPOACTIVE SEXUAL DESIRE DISORDER
Related Applications
This application claims the benefit of and priority to U.S. Provisional Application Serial No. 61 /726,570, filed November 14, 2012, U.S. Provisional Application Serial No. 61 /727,1 12, filed November 15, 2012, and U.S. Provisional Application Serial No.
61 /729,324, filed November 21 , 2012. The contents of each of the foregoing
applications are incorporated by reference herein in their entirety.
Field of the Invention
The present invention relates to lower dosage strength intranasal testosterone gels for providing intranasal delivery of testosterone to a female and intranasal treatment methods for treating females with anorgasmia and/or hypoactive sexual desire disorder (HSDD). In particular, the present invention relates to improved methods and lower dosage strength intranasal testosterone gel formulations for treating female anorgasmia and/or HSDD. The present invention also relates to a system for
dispensing intranasally a precise dosage amount of such gels at an optimal anatomical location within each nostril of the female, so that an effective amount of testosterone is deposited within each nostril at the optimal anatomical location to effectively treat female anorgasmia and/or HSDD.
Background
Reduced levels of endogenous steroid hormones in humans often lead to a variety of undesirable clinical symptoms. For example, low testosterone levels in men (hypogonadism) may result in clinical symptoms including impotence, lack of sex drive, muscle weakness, and osteoporosis. Similarly, in women, reduced levels of
testosterone and/or estrogen may result in female sexual disorder, which include clinical symptoms such as lack of sex drive, lack of arousal or pleasure, decreased energy levels or fatigue with blunted motivation, flat mood or depression, reduced sense of well-being, insomnia, irritability, partial decreases in vaginal lubrication, and
osteoporosis. Moreover, reduced levels of estrogen and/or progesterone in women, as observed during menopause, often result in clinical symptoms including hot flashes, night sweats, vaginal atrophy, decreased libido, and osteoporosis.
Testosterone has historically been thought of as a male hormone, but it is also synthesized in women in small amounts, primarily by the ovaries and adrenal glands. The physiological functions of testosterone in women include, among others, development of pubic and axillary hair, sexual libido; effects on bone density and muscle tone, sexual libido, and overall vitality and sense of psychological well-being. Testosterone plasma concentrations in pre-menopausal women normally fluctuate during the menstrual cycle, with the total testosterone plasma concentrations generally ranging between about 15 ng/dL and about 65 ng/dL. However, in the years leading to post-menopause, levels of circulating testosterone begin to decline, generally thought to be due to age-related reductions in ovarian and adrenal secretion. Generally, women with testosterone deficiency have total testosterone levels of less than about 20-25 ng/dL, while oophorectomized women can have testosterone levels of less than about 10 ng/dL.
In the National health and Social Life Survey of over 1 ,700 women aged 18-59, 43% acknowledged a form of female sexual dysfunction (FSD). See, e.g., Laumann et al. : Sexual dysfunction in the United States: prevalence and predictors; JAMA, 281 : 537-544 (1999).
Hypoactive sexual desire disorder (HSDD), the most common women's sexual problem, is a condition characterized by the lack or absence of sexual fantasies and desire for sexual activity which causes marked distress or interpersonal difficulties. The sexual dysfunction is not accounted for by another psychiatric disorder nor is it a result of direct physiological effects of a substance (i.e., drug abuse) or a general medical condition.
Anorgasmia, the second most frequently reported women's sexual problem, is considered to be the persistent or recurrent delay in, or absence of, orgasm following a normal sexual excitement phase, causing marked distress or interpersonal difficulty. When a woman has sexual activity that is not accompanied by good quality orgasmic release, sexual activity may become a chore or a duty rather than a mutually satisfying,
intimate experience. This may also lead to secondary loss of sexual interest and/or interpersonal difficulties.
Hypoactive sexual disorder disease and anorgasmia affect millions of women in the United States.
Sexual response is a complex and finely tuned process that can be disrupted at various time points in the reproductive life cycle (pre and postpartum, peri and postmenopausal) which likely accounts for the high prevalence of reported sexual dysfunction in the general population of healthy women. See, e.g., Laumann et al., Supra.
It is hypothesized that testosterone has central and peripheral effects on sexual function. The decline of androgen levels following surgically induced menopause has supported the hypothesis that a decrease in testosterone levels is related to a decrease in sexual desire. Testosterone, the primary circulation androgen in women, is a naturally occurring steroid. In women, androgens are derived from three sources: the adrenal glands, the ovaries and peripheral conversion. Androgens are secreted by the ovaries and the adrenal glands. Contrary to the sudden drop in estrogen during menopause, serum levels of androgens fall gradually as women age, primarily due to a decrease in the production of adrenal androgen precursors. See, e.g. Goldstat et al. : Transdermal testosterone therapy improves well-being, mood, and sexual function in pre-menopausal women; Menopause, 10(5): 390-398 (2003). As indicated above, this is likely due to a decline in ovarian and adrenal function with age.
A recent cross sectional study in woman ages 18-75 shows that total and free testosterone levels significantly decrease with age starting in the early reproductive years. In contrast to naturally occurring menopause, women who have undergone bilateral oopherectomy experience a dramatic decline in testosterone production with levels decreasing as much as 50%.
It has been reported that testosterone plays a role in mood, body composition, and bone mineral density and has central and peripheral effects on sexual function. See, e.g., Davis et al.: Androgen replacement in women: a commentary; Menopause, J Clin Endocrinol Metab, 84(6): 1886-1891 (1999); and Goldstat et al., Supra. In the
periphery, testosterone is required for nitric oxide to stimulate vasocongestion for the engorgement of clitoral tissue and vaginal lubrication during sexual arousal.
Recent studies have shown that testosterone is effective in increasing the number of sexually satisfying events, increasing sexual desire and decreasing personal distress in bilaterally oophorectomized and hysterectomized women suffering from HSDD.
Central effects of testosterone are less well characterized. Testosterone stimulates dopamine release in various brain structures implicated in motivation and reward systems, including sexual desire. Testosterone was found to stimulate dopamine release in the medial preoptic area of the anterior hypothalamus under basal conditions and with sexual stimulation in rats. See, e.g., Halaris A. : Neurochemical aspects of the sexual response cycle; CNS Spectrums, 9: 21 1 -216 (2003). An fMRI study in healthy women of different ages showed a testosterone level dependent modulation of amygdala activity, suggesting that an age-related decline in androgen levels contributes to the decrease in amygdala reactivity. In addition, it has been reported that a decreased amygdala reactivity in older women may be restored to levels of young women with intranasal exogenous testosterone. See e.g., van Wingen et al. :
Testosterone increases amygdala reactivity in middle-aged women to a young adulthood level; Neuropsychopharmacology, Feb: 34(3) 539-547 (2009).
The use of androgens to increase women's sexual libido was reported in 1940 by Loeser. Salmon (1942) observed that a number of young, married women who formerly considered themselves "frigid" were able to experience "a marked increase in coital gratification, culminating in an orgasm" after testosterone propionate injections. The effects wore off within several weeks after the discontinuation of the injections. See, e.g., Traish et al. : Testosterone therapy in women with gynecological and sexual disorders: a triumph of clinical endocrinology from 1938 to 2008; J Sex Med 4:609-619 (2009). In the 1980s, the role of androgens in maintaining sexual function was studied in oophrectomized women. See, e.g., Sherwin et al. : The role of androgen in the maintenance of sexual functioning in oophorectomized women; Psychosomatic
Medicine, 49:397-409 (1987). In the Sherwin et al. study, a three (3) month prospective open-label study of 44 women, it was reported that monthly injections of estrogen and
testosterone increased rates of sexual desire, sexual arousal, and number of fantasies. It was further reported in the Sherwin et al. study that rates of intercourse and orgasm were higher in women treated with androgens and estrogen compared to the controls.
Over the past two decades, over 80 studies have been conducted in postmenopausal women with HSDD using exogenous testosterone through the oral, transdermal, sublingual or parental route of administration with or without concomitant estrogen therapy, in which some degree of an increase in sexual desire, arousal, frequency of satisfactory sexual activity, pleasure and responsiveness was allegedly observed. See, Traish et al. Supra, 2009.
Fewer studies have been performed in pre-menopausal women with low libido. Goldstat et al., Supra, apparently studied the effects of transdermal testosterone therapy on well-being, mood and sexual function in eugonadal, pre-menopausal women presenting with low libido. Testosterone therapy resulted in statistically significant improvements in the composite scores of the Psychological General Weil-Being Index, the Sabbatsberg Sexual Self-Rating Scale and the Beck Depression Inventory when compared with placebo. These effects were found while the mean total testosterone levels were in the low end of the normal range before treatment, and at the high end of the normal range during treatment. On the different sub-scales of the Sabbatsberg Sexuality Scale, however, there was a significant effect of testosterone treatment on orgasm. This study suggests that, although most previous studies with testosterone have addressed decreased sexual desire in post-menopausal women, there are also measurable effects in pre-menopausal women both on general sexual well-being and on orgasm specifically.
Intrinsa® is a testosterone slow-release transdermal patch. Intrinsa® is indicated for the treatment of hypoactive sexual desire disorder (HSDD) in bilaterally
oophorectomized and hysterectomized (surgically induced menopause) women receiving concomitant estrogen therapy. Clinical studies using Intrinsa® have shown enhancement of sexual desire and number of satisfactory sexual events with mild androgenic skin effects as the primary safety concern in post-menopausal women with HSDD. See, e.g., Shifren et al. : Transdermal testosterone treatment in women with impaired sexual function after oophorectomy; N Eng N Med, 343(10): 682-688 (2000);
Braunstein et al.: Safety and efficacy of a testosterone patch for the treatment of hypoacytive sexual desire disorder in surgically menopausal women: a randomized placebo-controlled trial; Arch Intern Med, 165(14): 1582-1589 (2005); Buster et al. :
Testosterone Patch for low sexual desire in surgically menopausal women: a
randomized trial; Obstet Gynecol, 105(5 Pt 1 ): 944-952 (2005); Simon et al. :
Testosterone patch increases sexual activity and desire in surgically menopausal women with hyposactive sexual desire disorder; J Clin Endocrinol Metab, 90(9) 5226- 5233 (2005); Davis et al. : Efficacy and safety of a testosterone patch for the treatment of hypoactive sexual desire disorder in surgically menopausal women: a randomized, placebo-controlled trial; Menopause, 13(3): 387-396 (2006); and Shifren et al. :
Testosterone patch for the treatment of hypoactive sexual desire disorder in naturally menopausal women: results from the INTIMATE1 study; Menopause, 143:770-779 (2006).
LibiGel is a gel formulation of testosterone that is applied on the upper arm of a female. It is reported that treatment with LibiGel increases the number of satisfying sexual events versus baseline and placebo treated individuals It is further reported that the effective dose of LibiGel produces testosterone blood levels within the normal range for pre-menopausal women. See, e.g., www.libigel.org.
Zestra® is a blend of botanical oils and extracts, including: Borage Seed Oil, Evening Primrose Oil, Angelica Extract, Coleus Forskohlii Extract, Theobromine, Antioxidants {Ascorbyl Palmitate (Vitamin C), Tocopherol (Vitamin E)}; and Flavor (U.S. 6,737,084) that may benefit some women with anorgasmia. Zestra® has
demonstrated significant improvements in the measures of desire, arousal and sexual satisfaction in women. See, e.g., www.zestra.com.
ArginMax™ is a mixture of L-arginine, ginseng, ginkgo, damiana, calcium, and iron. ArginMax™ for Women was formulated specifically for women. It contains calcium and iron to help relieve fatigue issues specific to women. The American ginseng in the men's product has been replaced with Damiana, an aromatic herb which helps calm anxiety and induce a relaxed state of mind. ArginMax™ for Women provides 100% of the RDA of vitamins A, C, E and the B-complex vitamins.
ArginMax™ safely enhances the female sexual experience by improving circulation.
Sufficient blood flow is critical to female arousal, engorgement and lubrication. See, e.g., www.arginimax.com.
In view of the fact that millions of women in the United States, as well as through out the world, suffer from HSDD and anorgasmia, there is a real and immediate need for an effective medical therapy that can treat these diseases, so that the quality of life of these individuals can be improved. One therapeutic goal of such therapy to solve this immediate need might be to restore testosterone levels in women to young adulthood levels or to at least the natural pre-menopausal state in hopes to alleviate the symptoms generally associated with HSDD and/or anaorgasmia due possibly to testosterone deficiency.
Brief Summary Of The Invention
The present invention overcomes the limitations and disadvantages associated with the treatment of anorgasmia and/or HDDD using available therapies through the discovery of novel lower dosage strength pernasal testosterone gels and methods of use to treat HSDD and/or anorgasmia. Particularly, the present invention overcomes the limitations and disadvantages of currently available options for administration of testosterone through the discovery of novel and improved lower dosage strength testosterone gel formulations specifically designed for intranasal administration to deliver therapeutically effective amounts of testosterone to treat females who suffer from and/or have been diagnosed with HSDD and/or anorgasmia.
The term "a therapeutically effective amount" means an amount of testosterone sufficient to induce a therapeutic or prophylactic effect for use in testosterone
replacement or supplemental therapy to treat female sexual dysfunction ("FSD"), namely, hypoactive sexual desire disorder ("HSDD") and/or female orgasmic disorder ("anorgasmia") in females.
Thus, generally speaking, the present invention provides for new and improved, substantially less-irritating, lower dosage strength testosterone gel formulations formulated with testosterone in amounts ranging from between about 0.10% to about 1 .5% by weight, for nasal administration to deliver a therapeutically effective amount of testosterone to effectively treat anorgasmia and/or HSDD.
The present invention is also directed to novel methods for pernasal administration of the nasal testosterone gels. Generally speaking, the novel methods involve depositing the intranasal testosterone gels topically into the nasal cavity of each nostril to deliver a therapeutically effective amount of testosterone, e.g., from about 150 mcg/nostril to about 600 mcg/nostril per application, over dose life for providing constant effective testosterone brain and/or blood levels for use in testosterone replacement or supplemental therapy, especially for effectively treating females in need of testosterone to treat anorgasmia and/or HSDD.
In accordance with the novel methods of the present invention, the intranasal testosterone gels are topically deposited on the outer external walls (opposite the nasal septum) inside the naval cavity of each nostril, preferably at about the middle to about the upper section of the outer external wall (opposite the nasal septum) just under the cartilage section of the outer external wall inside the naval cavity of each nostril. Once gel deposition is complete within each nostril of the nose, the outer nose is then gently and carefully squeezed and/or rubbed by the subject, so that the deposited gel remains in contact with the mucosal membranes within the nasal cavity for sustained release of the testosterone over dose life. Typical testosterone gel dosage amounts deposited pernasal application is between about 50 to about 150 microliters per nostril, and preferably about 100 microliters per nostril.
In carrying out the methods of the present invention, approximately between 50 microliters and about 150 microliters of a lower dosage strength testosterone gel of the present invention is applied to each nostril of a subject once or twice daily, e.g., for one, two, three, four or more consecutive weeks, or for two, three, four, five or six
consecutive days or more, or intermittently such as every other day or once, twice or three times weekly, or on demand once or twice during the same day, to treat HSDD and/or anorgasmia.
While the present invention has identified what it believes to be preferred concentrations of intranasal testosterone gel formulations, numbers of applications per day, durations of therapy, pernasal methods and pre-filled, multi-dose applicator systems, it should be understood by those versed in this art that any effective low dosage concentration of testosterone, i.e., between about 0.10% and about 1 .5% by
weight, in an intranasal gel formulation that delivers an effective amount of testosterone and any numbers of applications per day, week, month or year, as described herein, that can effectively treat anorgasmia and/or HSDD without causing unwanted
testosterone treatment limiting reactions or related adverse events is contemplated by the present invention.
The present invention therefore provides for a new and improved treatment for anorgasmia and/or HSDD, wherein nasal administration of a lower dosage strength testosterone gel formulation of the present invention provides for: (1 ) rapid delivery of testosterone due to the highly permeable nasal tissue both systemically and across the blood-brain barrier into the brain; (2) fast onset of action; (3) avoidance of hepatic first- pass metabolism; (4) ease of administration to improve sexual experience; (5) avoidance of irritation from transdermal administration, particularly, no exposition to contacts, no transference from topical gels, and no local irritability from topical patch products; and (6) a more pleasant mode of administration, as compared to injections and buccal or sublingual tablets.
In other words, the present invention provides for a new and improved
anorgasmia and/or HSDD treatment that (a) is easy and convenient to use either according to a prescribed treatment regimen or on-demand, (b) rapidly delivers therapeutically effective amounts of testosterone, thereby improving female sexual function in a timely manner, (c) provides for simple use, (d) has reduced side effects associated with prior exogenous systemic testosterone therapies, (e) avoids local irritability associated with prior topical gels and topical patches, and (f) eliminates the need for invasive and painful testosterone injections.
The present invention, in one embodiment, provides numerous surprising advantages over currently available therapies for anorgasmia and HSDD. For example, the present invention provides for (1 ) a rapid increase in the plasma testosterone plasma level (e.g., an increase in the plasma testosterone to a level of at least about 0.4 ng/ml within about 15 minutes immediately after nasal administration of the testosterone gel formulation of the invention); (2) a sustained increase in the plasma testosterone plasma level (e.g., an increase in the plasma testosterone level that is maintained in a subject for at least about 6 hours following nasal administration of the testosterone gel
formulation of the invention); and (3) a higher maximum level of plasma testosterone as compared to the maximum level of plasma testosterone following administration of Intrinsa® within about 100 minutes immediately following administration (e.g., an increase in the plasma testosterone level to at least about 0.7 ng/ml as compared to about 0.1 ng/ml for Intrinsa®.).
As demonstrated in Figure 1 , an improved testosterone gel formulation of the invention provide advantages over therapies for treating anorgasmia and HSDD that are currently available. For example, a testosterone gel formulation of the invention comprising about 0.6% testosterone by weight of the gel formulation is administered intranasally to subjects. In comparison, control subjects, who are treated with an Intrinsa® patch, receive a testosterone dose of about 2100-2800 meg/day, up to approximately 3.5-4.5 times the testosterone received by women treated with the lower dosage strength testosterone gels of the present invention (i.e., about 600, 1800, or 2400 meg/day, for the 0.15%, 0.45%, and 0.6% testosterone gels of the invention, respectively, or about 600, 1200 or 1800 meg/day, for the 0.24%, 0.48% or 0.72% testosterone gels of the invention, respectively). Importantly, unlike the Intrinsa® patch, testosterone levels return to baseline after about 12 hours after treatment with the lower dosage strength testosterone gels of the present invention (at least for the 0.15% and 0.45% gel formulations of the invention).
One unique pharmacokinetic profile, as compared to Intrinsa®, is presented in Figure 1 . The improved 0.6% testosterone gel formulation for nasal administration of the present invention provides a plasma testosterone concentration, following nasal administration wherein (a) a plasma testosterone level of at least about 0.4 ng/ml is achieved; (b) a plasma testosterone level of at least about 0.7 ng/ml is achieved; (c) an increase in plasma testosterone level is achieved within at least about 10 minutes following nasal administration to a subject; (d) a plasma testosterone level of at least about 0.4 ng/ml is achieved and maintained for at least about 6 hours immediately following nasal administration to a subject; (e) a plasma testosterone level of at least about 0.3 ng/ml is achieved and maintained for at least about 13 hours immediately following nasal administration to a subject; and (f) a plasma testosterone level of at least
about 0.7 ng/ml is achieved within about 100 minutes immediately following nasal administration to the subjects (see Figure 1 ).
In contrast, following treatment with Intrinsa®, as illustrated in Figure 1 , the plasma testosterone level does not increase until at least about 3 hours following administration of Intrinsa®. Even then, a plasma testosterone concentration of at least about 0.4 ng/ml is not observed in the Intrinsa® treated subjects until at least about 6.5 hours following administration. The maximum plasma testosterone level of a subject treated with Intrinsa® (only about 0.68 ng/ml) is not observed until about 12 hours following administration of Intrinsa®.
Thus, one improved testosterone (0.6%) gel formulation for nasal administration of the present invention may provide one or more of the following plasma testosterone concentrations, following nasal administration:
(a) a plasma testosterone concentration of at least about 0.4 ng/ml within less than hour following administration;
(b) a plasma testosterone concentration of at least about 0.7 ng/ml within less than about 100 minutes following administration;
(c) a plasma testosterone concentration increase within at least about 10 minutes following nasal administration to a subject;
(d) a plasma testosterone concentration of at least about 0.4 ng/ml, wherein the increase is achieved and maintained for at least about 6 hours immediately following nasal administration to a subject;
(e) a plasma testosterone level of at least about 0.3 ng/ml is achieved and maintained for at least about 13 hours immediately following nasal administration to a subject; and/or
(f) a plasma testosterone concentration of at least about 0.7 ng/ml is achieved within about 100 minutes immediately following nasal administration to a subject (see Figure 1 ).
Thus, as exemplified in Figure 1 , the present invention overcomes certain of the limitations associated with the treatment of anorgasmia and/or HSDD using currently available therapies, for example, Intrinsa®, and addresses current medical needs for (1 ) a pharmaceutical formulation that is conveniently, easily and unobtrusively
administered; (2) a rapidly acting formulation that improves female sexual dysfunction in a timely manner; (3) a decrease in the incidence of application site reactions; (4) a formulation that has reduced side effects; and (5) a formulation that can be used either according to a prescribed treatment regimen or on demand; to treat anorgasmia and/or HSDD.
A safety study was also conducted in accordance with the present invention, as reported in Example 1 1 , wherein women were treated intranasally with a 0.72% testosterone gel (about 1200 meg/dose administration) of the present invention t.i.d. for two consecutive days and qd on the third consecutive day resulting in a total daily dose of testosterone of about 3600 meg/day for the first two days and about 1200 meg on the third consecutive day. As shown in Example 1 1 , the intranasal testosterone gels of the present invention are believed to be safe and well tolerated.
The salient elements of the novel intranasal testosterone gels according to the present invention comprise (a) testosterone in a therapeutically effective amount, (b) a solvent, (c) a wetting agent, and (d) a viscosity increasing agent. The improved lower dosage strength testosterone gel formulations of the present invention may be formulated with testosterone in amounts by weight of between about 0.10% to about 1 .5%, e.g., about 0.15%, 0.24%, 0.45%, 0.48%, 0.6% and 0.72%, and more preferably between about 0.24% and 0.72%. Exemplary nasally administered testosterone gel formulations contemplated by the present invention include:
(a) 0.15% testosterone, 91 .85% castor oil, 4.0% oleoyl polyoxylglycerides, and 4% colloidal silicon dioxide;
(b) 0.24% testosterone, 91 .76% castor oil, 4.0% oleoyl polyoxylglycerides, and 4% colloidal silicon dioxide
(c) 0.45% testosterone, 91 .55% castor oil, 4.0% oleoyl polyoxylglycerides, and 4% colloidal silicon dioxide;
(d) 0.48% testosterone, 91 .52% castor oil, 4.0% oleoyl polyoxylglycerides, and 4% colloidal silicon dioxide
(e) 0.6% testosterone, 91 .4% castor oil, 4.0% oleoyl polyoxylglycerides, and 4% colloidal silicon dioxide; and
(f) 0.72% testosterone, 91 .28% castor oil, 4.0% oleoyl polyoxylglycerides, and 4% colloidal silicon dioxide.
Thus, the improved testosterone gel formulations for nasal administration of the invention may further comprise any pharmaceutically acceptable vehicle, excipient and/or other active ingredient.
In addition, the present invention contemplates testosterone gel formulations for nasal administration that are pharmaceutically equivalent, therapeutically equivalent, bioequivalent and/or interchangeable, regardless of the method selected to
demonstrate equivalents or bioequivalence, such as pharmacokinetic methodologies, microdialysis, in vitro and in vivo methods and/or clinical endpoints described herein.
Thus, the present invention contemplates testosterone gel formulations for nasal administration that are bioequivalent, pharmaceutically equivalent and/or therapeutically equivalent, especially testosterone gel formulations for nasal administration that are 0.15% testosterone by weight of the gel formulation, 0.24% testosterone by weight of the gel formulation, 0.45% testosterone by weight of the gel formulation, 0.48% testosterone by weight of the gel formulation, 0.6% testosterone by weight of the gel formulation and 0.72% testosterone by weight of the gel formulation, when used in accordance with the therapy of the present invention to treat anorgasmia and/or HSDD by intranasal administration. Thus, the present invention contemplates: (a) pharmaceutically equivalent testosterone gel formulations for nasal administration which contain the same amount of testosterone in the same dosage form; (b) bioequivalent testosterone gel formulations for nasal administration which are chemically equivalent and which, when administered to the same individuals in the same dosage regimens, result in comparable bioavailabilities; (c) therapeutic equivalent testosterone gel formulations for nasal administration which, when administered to the same individuals in the same dosage regimens, provide essentially the same efficacy and/or toxicity; and (d)
interchangeable testosterone gel formulations for nasal administration of the present invention which are pharmaceutically equivalent, bioequivalent and therapeutically equivalent.
While the intranasal testosterone gels of the present invention are preferred pharmaceutical preparations when practicing the novel methods of the present invention,
it should be understood that the novel topical intranasal gel formulations and methods of the present invention also contemplate the pernasal administration of any suitable active ingredient, either alone or in combination with testosterone or other active ingredients, such as neurosteroids or sexual hormones (e.g., androgens and progestins, like
testosterone, estradiol, estrogen, oestrone, progesterone, etc.), neurotransmitters, (e.g., acetylcholine, epinephrine, norepinephrine, dopamine, serotonin, melatonin, histamine, glutamate, gamma aminobutyric acid, aspartate, glycine, adenosine, ATP, GTP, oxytocin, vasopressin, endorphin, nitric oxide, pregnenolone, etc.), prostaglandin, benzodiazepines like diazepam, midazolam, lorazepam, etc., and PDEF inhibitors like sildenafil, tadalafil, vardenafil, etc., in any suitable pharmaceutical preparation, such as a liquid, cream, ointment, salve or gel. Examples of additional topical formulations for practice in
accordance with the novel methods of the present invention include the topical pernasal formulations disclosed in, for example, U.S. Patent Nos. 5,578,588, 5,756,071 and
5,756,071 and U.S. Patent Publication Nos. 2005/0100564, 2007/0149454 and
2009/0227550, all of which are incorporated herein by reference in their entireties.
The present invention is also directed to packaged pharmaceuticals comprising the novel and improved testosterone gel formulations for nasal administration of the invention. For example, the present invention contemplates pre-filled, single or multi-dose applicator systems for pernasal administration to strategically and uniquely deposit the nasal testosterone gels at the preferred locations within the nasal cavity for practicing the novel methods and teachings of the present invention. Generally, speaking the applicator systems of the present invention are, e.g., airless fluid, dip-tube fluid dispensing systems, pumps, syringes or any other system suitable for practicing the methods of the present invention. The applicator systems or pumps include, for example, a chamber, pre-filled with a single dose or multiple doses of an intranasal testosterone gel of the present invention, that is closed by an actuator nozzle or cap. The actuator nozzle may comprise an outlet channel and tip, wherein the actuator nozzle is shaped to conform to the interior surface of a user's nostril for (a) consistent delivery of uniform dose amounts of an intranasal testosterone gel of the present invention during pernasal application within the nasal cavity, and (b) deposition at the instructed location within each nostril of a patient as contemplated by the novel methods and teachings of the present invention. Preferably, when inserted
into a nasal cavity, the pump design is configured to help ensure that the nasal tip is properly positioned within the nasal cavity so that, when the gel is dispensed, the gel is dispensed within the appropriate location within the nasal cavity. Preferably, when inserted into a nasal cavity, the pump design is configured to help ensure that the nasal tip is properly positioned within the nasal cavity so that, when the gel is dispensed, the gel is dispensed within the appropriate location within the nasal cavity. See Steps 3 and 8 in Fig. 39. Additionally, the nozzles of te pumps are preferably designed to dispense the gels from from the side in a swirl direction, i.e., the tips of the nozzles are designed to dispense in a side distribution direction, as opposed to a direct distribution direction, onto the nasal mucosa, as shown in steps 4 and 9 of Fig. 39. It is believed that the swirl action allows for better gel adhesion and side distribution from the nozzle tip avoids the dispensed gel from splashing back onto the tip. Finally, it is preferrred to design the nozzle and tip to allow for any residual gel on the nozzle/tip to be wiped off as the tip is removed from the nasal cavity. See, e.g., Fig. 39. Examples of pre-filled, multi-dose applicator systems include, e.g., (a) the COMOD system available from Ursatec, Verpackung-GmbH, Schillerstr. 4, 66606 St. Wendel, Germany, (b) the Albion or Digital airless applicator systems available from
Airlessystems, RD 149 27380 Charleval, France or 250 North Route 303 Congers, NY 10950 (See, for example, Fig. 39), (c) the nasal applicators from Neopac, The Tube, Hoffmann Neopac AG, Burgdorfstrasse 22, Postfach, 3672 Oberdiessbach, Switzerland, or (d) the syringes described in the Examples herein below.
It should be understood by those versed in this art that the amount of testosterone in a lower dosage strength intranasal testosterone gel of the present invention that will be therapeutically effective in a specific situation will depend upon such things as the dosing regimen, the application site, the particular gel formulation, dose longevity and the condition being treated. As such, it is generally not practical to identify specific administration amounts herein; however, it is believed that those skilled in the art will be able to determine appropriate therapeutically effective amounts based on the guidance provided herein, information available in the art pertaining to testosterone replacement therapy, and routine testing.
It should be further understood that the above summary of the present invention is not intended to describe each disclosed embodiment or every implementation of the
present invention. The description further exemplifies illustrative embodiments. In several places throughout the specification, guidance is provided through examples, which examples can be used in various combinations. In each instance, the examples serve only as representative groups and should not be interpreted as exclusive examples.
Brief Description of the Drawings
The patent or application file contains at least one drawing executed in color. Copies of this patent or patent application publication with color drawing(s) will be provided by the Office upon request and payment of the necessary fee.
The foregoing and other objects, advantages and features of the present invention, and the manner in which the same are accomplished, will become more readily apparent upon consideration of the following detailed description of the invention taken in conjunction with the accompanying figures and examples, which illustrate embodiments, wherein:
Fig. 1 shows the effects of a single dose testosterone nasal gel formulation of the invention (0.6% testosterone by weight of the gel formulation) as compared to Intrinsa® on the plasma testosterone levels in subjects diagnosed with HSDD (triangles-100 microliters 0.6% testosterone nasal gel formulation of the invention in each nostril;
squares- Intrinsa®);
Fig. 2 shows the effects of a testosterone nasal gel formulation of the invention after the fifth 12 hourly dose (0.6% testosterone by weight of the gel formulation) as compared to Intrinsa® on the plasma testosterone levels in subjects diagnosed with anorgasmia (TBS-2 line-0.6% testosterone nasal gel formulation of the invention( 100 mcl*2); solid line- Intrinsa®);
Fig. 3 shows the effects of a testosterone nasal gel formulation of the invention (0.15, 0.45 and 0.6% testosterone by weight of the gel formulation) after the first and the 5th 12 hourly dose as compared to IntrinsaR single dose on the plasma testosterone levels in subjects diagnosed with HSDD;
Fig. 4 shows the effects of a testosterone nasal gel formulation of the invention at day zero (open triangles) and day 3 (dose 5 - closed square -of 0.6% testosterone by weight of the gel formulation) as compared to Intrinsa® on the plasma testosterone
levels in subjects diagnosed with HSDD (open squares day 0- lines day 3 of 0.6% squares- Intrinsa®;
Fig 5 is a copy of Fig 1 , but with comparative data for the 0.6% testosterone gel and Intinsa® from days 1 and 3;
Fig 6 is comparative data from treatment study for HSDD for the 0.6%
testosterone gel and Intinsa® from days 1 and 3;
Fig 7 shows the effects of a testosterone nasal gel formulation of the invention (0.15, 0.45 and 0.6% testosterone by weight of the gel formulation) after the first and the 5th 12 hourly dose as compared to Intrinsa® single dose on the plasma testosterone levels in subjects diagnosed with anorgasmia;
Fig 8 shows the effects of a testosterone nasal gel formulation of the invention (0.45% day 3 (Med Day 1 ) and 0.6% day zero and Day 3 (ANOR) testosterone by weight of the gel formulation) after the first and the 5th 12 hourly dose;
Fig 9 shows the accumulation effect effects of a testosterone nasal gel formulation of the invention (0.15, 0.45 and 0.6% testosterone by weight of the gel formulation) before each bi-daily dose of testosterone gel on the plasma testosterone levels at time (0) in subjects diagnosed with HSDD or anorgasmia;
Fig 10 (same as 12 but table on AUC Cavg and Cmax) vs Intrinsa® (note - because Intrinsa® is not approved for Anorgasmia, it was not assessed in the study);
Fig. 1 1 presents trough data for subjects diagnosed with anorgasmia or HSDD and treated with testosterone nasal gel formulations of the invention (0.15%, 0.45% or 0.6% testosterone by weight of the gel formulation)or placebo (anorgasmia) or Intrinsa® (HSDD);
Fig. 12 presents distribution of testosterone mean concentration for TBS-2 treatments for the first (AUC_0-12) and the last dose (AUC_48-60). The boxplots show the median (thick solid line), the inter-quartile range (box) and the extreme values (whiskers). The horizontal solid grey line indicates the median C_mean during treatment with Intrinsa from 48 to 60 hrs, and the horizontal dotted lines indicate the minimum and maximum C_mean during treatment with Intrinsa from 48-60 hrs;
Fig. 13 presents scores on the AFSDQ 30 minutes (left) and 4.5 hours (right) after dosing. White bars = start of session; solid bars = end of session. Groups left to
right: ANOR placebo, ANOR low, ANOR medium, ANOR high, HSDD Intrinsa patch (at 30 min. only), HSDD low, HSDD medium, HSDD high;
Fig. 14 concerns mean testisterone levels following TBS-2 high, TBS-2 medium and TBS-2 low dose administration or placebo adminsitration (hours 0-12) in women with anorgasmia. In this pharmacokinetic study, as described in Example 9, three different dosage strengths of TBS-2 testosterone bio-adhesive gel formulations of the invention are adminstered intranasally to a hybrid group of 12 healthy and anorgasmic women. As illustrated in Fig.14, the testosterone serum levels are compared for the three different testosterone bio-adhesive gel formulations of the invention (0.15% - , 0.45% and 0.6% testosterone by weight of the gel formulation, as reported in Examples 1 -5), during 2 hours following a single application of each TBS-2 formualtion or a placebo to each of the 12 women. The total testosterone dosage strength that is adminstered is either 1 .2 mg (0.6% - 0.6 mg/100 μΙ/nostril), 0.9 mg (0.45% - 0.45 mg/100 μΙ/nostril) or 0.3 mg (0.15% - 0.15 mg/100 μΙ/nostril). Following adminsitration, the testosterone serum level is measured and compared. As shown in Fig. 14, the Cmax and CaVg for testosterone following single dose administration for each of the three dosage strengths do not exceed the normal testosterone serum level in women (3 - 80 ng/dL);
Fig. 15 depicts a study design (Example 10), in which 56 anorgasmic women are enrolled for a Vibrotactile Stimulation Study (VTS) that concerns the three different testosterone bio-adhesive gel formulations of the invention, i.e., the 0.15% - , 0.45% and 0.6% testosterone by weight of the gel formulation, as reported in Examples 1 -5;
Fig. 16 depicts orgasm result of the VTS study of Example 10, wherein the number of orgasms achieved during the treatment phase and the post-treatment phase are compared;
Fig. 17 depicts sexual response results of the VTS study of Example 10 for the three different testosterone bio-adhesive gel formulations of the invention, i.e., the 0.15% - , 0.45% and 0.6% testosterone by weight of the gel formulation, as reported in Examples 1 -5, as compared to placebo;
Fig. 18 depicts the VTS appreciation score of the VTS study of Example 10 for the three different testosterone bio-adhesive gel formulations of the invention, i.e., the
0.15%, 0.45% and 0.6% testosterone by weight of the gel formulation, as reported in Examples 1 -5, as compared to placebo;
Fig. 19 depicts Mean Corrected Free Testosterone Concentrations (Single-Dose Population);
Fig. 20 depicts Mean Corrected Total Testosterone Concentrations (Single-Dose Population);
Fig. 21 depicts Mean Corrected Dihydrotestosterone Concentrations (Single- Dose Population);
Fig. 22 depicts Mean Corrected Estradiol Concentrations (Single-Dose
Population);
Fig. 23 depicts Mean Corrected SHBG Concentrations (Single-Dose Population); Fig. 24 depicts Mean Observed Free Testosterone Concentrations (Single-Dose Population);
Fig. 25 depicts Mean Observed Total Testosterone Concentrations (Single-Dose Population);
Fig. 26 depicts Mean Observed Dihydrotestosterone Concentrations (Single- Dose Population);
Fig. 27 depicts Mean Observed Estradiol Concentrations (Single-Dose
Population);
Fig. 28 depicts Mean Observed SHBG Concentrations (Single-Dose Population); Fig. 29 depicts Mean Free Testosterone Plasma Concentrations (Multi-Dose Population);
Fig. 30 depicts Mean Total Testosterone Plasma Concentrations (Multi-Dose Population);
Fig. 31 depicts Mean Dihydrotestosterone Plasma Concentrations (Multi-Dose Population);
Fig. 32 depicts Mean Estradiol Plasma Concentrations (Multi-Dose Population); Fig. 33 depicts Mean SHBG Plasma Concentrations (Multi-Dose Population); Fig. 34 depicts Spaghetti Concentration Plots with Mean for Free Testosterone Plasma Concentrations (Multi-Dose Population);
Fig. 35 depicts Spaghetti Concentration Plots with Mean for Total Testosterone Plasma Concentrations (Multi-Dose Population);
Fig. 36 depicts Spaghetti Concentration Plots with Mean for Dihydrotestosterone Plasma Concentrations (Multi-Dose Population);
Fig. 37 depicts Spaghetti Concentration Plots with Mean for Estradiol Plasma Concentrations (Multi-Dose Population);
Fig. 38 depicts Spaghetti Concentration Plots with Mean for SHBG Plasma Concentrations (Multi-Dose Population); and
Fig. 39 depicts an intranasal applicator contemplated by and used in accordance with the present invention.
Detailed Description
By way of illustrating and providing a more complete appreciation of the present invention and many of the attendant advantages thereof, the following detailed description and examples are given concerning the novel lower dosage strength intranasal testosterone gels, application devices and methods of the present invention.
/. Definitions
As used in the description of the invention and the appended claims, the singular forms "a", "an" and "the" are used interchangeably and intended to include the plural forms as well and fall within each meaning, unless the context clearly indicates otherwise. Also, as used herein, "and/or" refers to and encompasses any and all possible combinations of one or more of the listed items, as well as the lack of combinations when interpreted in the alternative ("or").
As used herein, "at least one" is intended to mean "one or more" of the listed elements.
Singular word forms are intended to include plural word forms and are likewise used herein interchangeably where appropriate and fall within each meaning, unless expressly stated otherwise.
Except where noted otherwise, capitalized and non-capitalized forms of all terms fall within each meaning.
Unless otherwise indicated, it is to be understood that all numbers expressing quantities, ratios, and numerical properties of ingredients, reaction conditions, and so forth used in the specification and claims are contemplated to be able to be modified in all instances by the term "about".
All parts, percentages, ratios, etc. herein are by weight unless indicated otherwise.
As used herein, "bioequivalence" or "bioequivalent", refers to nasally
administered testosterone gel formulations or drug products which are pharmaceutically equivalent and their bioavailabilities (rate and extent of absorption) after administration in the same molar dosage or amount are similar to such a degree that their therapeutic effects, as to safety and efficacy, are essentially the same. In other words,
bioequivalence or bioequivalent means the absence of a significant difference in the rate and extent to which testosterone becomes available from such formulations at the site of testosterone action when administered at the same molar dose under similar conditions, e.g., the rate at which testosterone can leave such a formulation and the rate at which testosterone can be absorbed and/or become available at the site of action to affect anorgasmia and/or HSDD. In other words, there is a high degree of similarity in the bioavailabilities of two testosterone gel formulation pharmaceutical products for nasal administration (of the same galenic form) from the same molar dose, that are unlikely to produce clinically relevant differences in therapeutic effects, or adverse reactions, or both. The terms "bioequivalence", as well as "pharmaceutical equivalence" and "therapeutic equivalence" are also used herein as defined and/or used by (a) the FDA, (b) the Code of Federal Regulations ("C.F.R."), Title 21 , (c) Health Canada, (d) European Medicines Agency (EMEA), and/or (e) the Japanese Ministry of Health and Welfare. Thus, it should be understood that the present invention
contemplates testosterone gel formulations for nasal administration or drug products that may be bioequivalent to other testosterone gel formulations for nasal administration or drug products of the present invention. By way of example, a first testosterone gel formulation for nasal administration or drug product is bioequivalent to a second testosterone gel formulation for nasal administration or drug product, in accordance with the present invention, when the measurement of at least one pharmacokinetic
parameter(s), such as a Cmax, Tmax, AUC, etc., of the first testosterone gel formulation for nasal administration or drug product varies by no more than about ±25%, when compared to the measurement of the same pharmacokinetic parameter for the second testosterone gel formulation for nasal administration or drug product of the present invention.
As used herein, "bioavailability" or "bioavailable", means generally the rate and extent of absorption of testosterone into the systemic circulation and, more specifically, the rate or measurements intended to reflect the rate and extent to which testosterone becomes available at the site of action or is absorbed from a drug product and becomes available at the site of action. In other words, and by way of example, the extent and rate of testosterone absorption from a lower dosage strength gel formulation for nasal administration of the present invention as reflected by a time-concentration curve of testosterone in systemic circulation.
As used herein, the terms "pharmaceutical equivalence" or "pharmaceutically equivalent", refer to testosterone gel formulations for nasal administration or drug products of the present invention that contain the same amount of testosterone, in the same dosage forms, but not necessarily containing the same inactive ingredients, for the same route of administration and meeting the same or comparable compendial or other applicable standards of identity, strength, quality, and purity, including potency and, where applicable, content uniformity and /or stability. Thus, it should be
understood that the present invention contemplates testosterone gel formulations for nasal administration or drug products that may be pharmaceutically equivalent to other testosterone gel formulations for nasal administration or drug products used in accordance with the present invention.
As used herein, "therapeutic equivalence" or "therapeutically equivalent", means those testosterone gel formulations for nasal administration or drug products which (a) will produce the same clinical effect and safety profile when utilizing testosterone drug product to treat anorgasmia or HSDD in accordance with the present invention and (b) are pharmaceutical equivalents, e.g., they contain testosterone in the same dosage form, they have the same route of administration; and they have the same testosterone strength. In other words, therapeutic equivalence means that a chemical equivalent of a
lower dosage strength testosterone formulation of the present invention (i.e., containing the same amount of testosterone in the same dosage form when administered to the same individuals in the same dosage regimen) will provide essentially the same efficacy and toxicity.
As used herein a "testosterone gel formulation for nasal administration" means a formulation comprising testosterone in combination with a solvent, a wetting agent, and a viscosity increasing agent.
As used herein, "increases" as it refers to the plasma testosterone level, also means that the plasma testosterone level is 1 , 2, 3, 4, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 100, 500, 1000 or 10,000-fold or more greater in a subject that has been treated with a testosterone gel formulation for nasal administration of the invention as compared to the plasma testosterone level in the subject prior to treatment.
As used herein, "plasma testosterone level" means the level of testosterone in the plasma of a subject. The plasma testosterone level is determined by methods known in the art.
"Diagnosis" or "prognosis," as used herein, refers to the use of information (e.g., biological or chemical information from biological samples, signs and symptoms, physical exam findings, psychological exam findings, etc.) to anticipate the most likely outcomes, timeframes, and/or responses to a particular treatment for a given disease, disorder, or condition, based on comparisons with a plurality of individuals sharing symptoms, signs, family histories, or other data relevant to consideration of a patient's health status, or the confirmation of a subject's affliction, e.g., with anorgasmia and/or HSDD.
A "subject" according to some embodiments is an individual whose signs and symptoms, physical exams findings and/or psychological exam findings are to be determined and recorded in conjunction with the individual's condition (i.e., disease or disorder status) and/or response to a candidate drug or treatment.
"Subject," as used herein, is preferably, but not necessarily limited to, a human subject. The subject may be male or female, and is preferably female, and may be of any race or ethnicity, including, but not limited to, Caucasian, African-American, African, Asian, Hispanic, Indian, etc. Subject as used herein may also include an animal,
particularly a mammal such as a canine, feline, bovine, caprine, equine, ovine, porcine, rodent (e.g., a rat and mouse), a lagomorph, a primate (including non-human primate), etc., that may be treated in accordance with the methods of the present invention or screened for veterinary medicine or pharmaceutical drug development purposes. A subject according to some embodiments of the present invention include a patient, human or otherwise, in need of therapeutic treatment for anorgasmia and/or HSDD.
"Treatment," as used herein, includes any drug, drug product, method, procedure, lifestyle change, or other adjustment introduced in attempt to effect a change in a particular aspect of a subject's health (i.e. , directed to a particular disease, disorder, or condition).
"Drug" or "drug substance," as used herein, refers to an active ingredient, such as a chemical entity or biological entity, or combinations of chemical entities and/or biological entities, suitable to be administered to a subject to (a) treat anorgasmia and/or (b) treat HSDD. In accordance with the present invention, the drug or drug substance is testosterone or a pharmaceutically acceptable salt or ester thereof.
The term "drug product," as used herein, is synonymous with the terms
"medicine," "medicament," "therapeutic intervention," or "pharmaceutical product." Most preferably, a drug product is approved by a government agency for use in accordance with the methods of the present invention. A drug product, in accordance with the present invention, is an intranasal gel formulated with a drug substance, i.e., testosterone.
"Disease," "disorder," and "condition" are commonly recognized in the art and designate the presence of signs and/or symptoms in an individual or patient that are generally recognized as abnormal and/or undesirable. Diseases or conditions may be diagnosed and categorized based on pathological changes. The disease or condition may be selected from the types of diseases listed in standard texts, such as Harrison's Principles of Internal Medicine, 1 997, or Robbins Pathologic Basis of Disease, 1 998.
As used herein, "diagnosing" or "identifying a patient or subject having
anorgasmia or HSDD" refers to a process of determining if an individual is afflicted with anorgasmia or HSDD.
As used herein, "control subject" means a subject that has not been diagnosed with anorgasmia and/or HSDD and/or does not exhibit any detectable symptoms associated with these diseases. A "control subject" also means a subject that is not at risk of developing anorgasmia and/or HSDD, as defined herein.
//. Diseases
Anorgasmia
Anorgasmia is a type of sexual dysfunction in which a person cannot regularly achieve orgasm, even with adequate stimulation. In males the condition is often related to delayed ejaculation. Anorgasmia can often cause sexual frustration. Anorgasmia is far more common in females than in males and is especially rare in younger men.
Anorgasmia is a very common occurrence in women, affecting 1 in 5 women worldwide.
The condition is sometimes classified as a psychiatric disorder. However, it can also be caused by medical problems such as diabetic neuropathy, multiple sclerosis, genital mutilation, complications from genital surgery, pelvic trauma (such as from a straddle injury caused by falling on the bars of a climbing frame, bicycle or gymnastics beam), hormonal imbalances, total hysterectomy, spinal cord injury, cauda equina syndrome, uterine embolisation, childbirth trauma (vaginal tearing through the use of forceps or suction or a large or unclosed episiotomy), vulvodynia and cardiovascular disease.
A comprehensive definition of female orgasm, as Meston et al. proposed, is as follows:
"[A] variable, transient peak sensation of intense pleasure creating an altered state of consciousness, usually accompanied by involuntary, rhythmic contractions of the pelvic striated circumvaginal musculature, often with concomitant uterine and anal contractions and myotonia that resolves the sexually-induced vasocongestion
(sometimes only partially), usually with an induction of well-being and contentment."
The Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Text Revision (DSM-IV-TR) defines female orgasmic disorder (FOD, formerly inhibited female orgasm) as a persistent or recurrent delay in, or absence of, orgasm following a normal sexual excitement phase.
The type or intensity of stimulation that triggers female orgasm varies widely among women. Therefore, the diagnosis of female orgasmic disorder, according to the DSM-IV-TR, is based on these 3 criteria:
Criterion A: A clinician must judge that a woman's orgasmic capacity is less than what is reasonable for her age, sexual experience, and the adequacy of sexual stimulation she receives.
Criterion B: The disturbance must cause marked distress or interpersonal difficulty.
Criterion C: Another axis I disorder (except another sexual dysfunction) does not account for the orgasmic dysfunction better than female orgasmic disorder does, and the orgasmic dysfunction is not exclusively due to the direct physiologic effects of a substance (e.g., drug of abuse, medication) or a general medical condition.
In the DSM-IV-TR, female orgasmic disorder specifiers include the following:
Lifelong or acquired
Generalized or situational
Due to psychological or combined factors
The presence of a normal sexual excitement phase is a prerequisite for female orgasmic disorder. In other words, if the absence of orgasm follows a time of decreased desire for sexual activity, an aversion to genital sexual contact, or a decreased lubrication-swelling response, diagnoses such as hypoactive sexual desire disorder, sexual aversion disorder, or female sexual arousal disorder, respectively, might be more appropriate, even if anorgasmia is the common final outcome.
A wide range of physical and psychological causes of anorgasmia have been identified.
They include the following:
A history of sexual abuse or rape
Boredom and monotony in sexual activity
Certain prescription drugs, including fluoxetine (Prozac), paroxetine (Paxil), and sertraline (Zoloft)
Hormonal disorders, hormonal changes due to menopause, and chronic illnesses that affect general health and sexual interest
Medical conditions that affect the nerve supply to the pelvis (such as multiple sclerosis, diabetic neuropathy, and spinal cord injury)
Negative attitudes toward sex (usually learned in childhood or adolescence)
Shyness or embarrassment about asking for whatever type of stimulation works best
Strife or lack of emotional closeness within the relationship Physical causes
A wide range of illnesses, physical changes and medications can interfere with orgasm:
Medical diseases. Any illness can affect this part of an individual's sexuality, including diabetes and neurological diseases, such as multiple sclerosis. Orgasm may also be affected by gynecologic surgeries, such as hysterectomy or cancer surgeries. In addition, lack of orgasm often goes hand in hand with other sexual problems, such as painful intercourse.
Medications. Many prescription and over-the-counter medications can interfere with orgasm. This includes blood pressure medications, antihistamines and
antidepressants— particularly selective serotonin reuptake inhibitors (SSRIs). In men, SSRIs can actually result in both anorgasmia and inability to obtain an adequate erection for satisfactory sexual activity (erectile dysfunction).
Alcohol and drugs. A glass of wine may make you feel amorous, but too much alcohol can cramp your ability to climax; the same is true of street drugs.
The aging process. As you age, normal changes in your anatomy, hormones, neurological system and circulatory system can affect your sexuality. The drop in estrogen that occurs during the transition to menopause can be a particularly notable foe of orgasm. Lower levels of this female hormone can decrease sensations in the clitoris, nipples and skin and impede blood flow to the vagina and clitoris, which can delay or stop orgasm entirely. Still, anorgasmia isn't limited to older women. And many women say sex becomes more satisfying with age.
Psychological causes
Many psychological factors play a role in your ability to orgasm, including:
Mental health problems, such as anxiety or depression
Performance anxiety
Stress and financial pressures
Cultural and religious beliefs
Fear of pregnancy or sexually transmitted diseases
Embarrassment
Guilt about enjoying sexual experiences
Relationship issues
Many couples who are experiencing problems outside of the bedroom will also experience problems in the bedroom. These overarching issues may include:
Lack of connection with your partner
Unresolved conflicts or fights
Poor communication of sexual needs and preferences
Infidelity or breach of trust
A common cause of situational anorgasmia, in both men and women, is the use of anti-depressants, particularly selective serotonin reuptake inhibitors (SSRIs). Post- SSRI sexual dysfunction (PSSD) is a name given to a reported iatrogenic sexual dysfunction caused by the previous use of SSRI antidepressants. Though reporting of anorgasmia as a side effect of SSRIs is not precise, it is estimated that 15-50% of users of such medications are affected by this condition. The chemical amantadine has been shown to relieve SSRI-induced anorgasmia in some, but not all, people.
Another cause of anorgasmia is opiate addiction, particularly to heroin.
About 15% of women report difficulties with orgasm, and as many as 10% of women in the United States have never climaxed. Many women who orgasm regularly only climax about 50-70% of the time.
The major symptoms of anorgasmia are inability to experience orgasm or long delays in reaching orgasm. Different types of anorgasmia have been identified.
Primary anorgasmia
Primary anorgasmia is a condition where one has never experienced an orgasm. This is significantly more common in women, although it can occur in men who lack the gladipudendal (bulbocavernosus) reflex.
Women with this condition can sometimes achieve a relatively low level of sexual excitement. Frustration, restlessness, and pelvic pain or a heavy pelvic sensation may occur because of vascular engorgement.
On occasion, there may be no obvious reason why orgasm is unobtainable. In such cases, women report that they are unable to orgasm even if they have a caring, skilled partner, adequate time and privacy, and an absence of medical issues which would affect sexual satisfaction. It should be noted that the attention and skill of one's partner are not inextricably linked to woman's internal, implicit comfort level. Thus, anorgasmia in a woman whose partner is adequately attentive and skilled should not be regarded as a clinical mystery.
Some social theorists believe that inability to orgasm may be related to residual psychosocial perceptions that female sexual desire is somehow 'wrong,' and that this stems from the age of Victorian repression. It is thought that this view may impede some women - perhaps those raised in a more repressed environment - from being able to experience natural and healthy sexual feeling. While such proposals may have a place in academic social theory, they have not been established scientifically.
Therefore, an idea such as this may be a component of treatment as one consideration among many, but responsible clinical practice should not be guided, based on, or informed by it.
Primary male anorgasmia is more common among circumsized men than intact men. As many men age, they lose the ability to orgasm even if they still ejaculate. The pleasure of orgasm may diminish in older men.
Secondary anorgasmia
Secondary anorgasmia is the loss of the ability to have orgasms. Particularly, you used to have orgasms, but now experience difficulty reaching climax. The cause may be alcoholism, depression, grief, pelvic surgery (such as total hysterectomy) or injuries, certain medications, illness, estrogen deprivation associated with menopause or an event that has violated the patient's sexual value system.
Secondary anorgasmia is close to 50% among males undergoing prostatectomy; 80% among radical prostatectomies. This is a serious adverse result because radical prostatectomies are usually given to younger males who are expected to live more than
10 years. At more advanced ages, the prostate is more unlikely to grow during that person's remaining lifetime. This is generally caused by damage to the primary nerves serving the penile area, which pass near the prostate gland. Removal of the prostate frequently damages or even completely removes these nerves, making sexual response unreasonably difficult.
Due to the existence of these nerves in the prostate, surgeons performing sex reassignment surgery on transsexual male to female patients avoid removing the prostate. This leaves the nerves that will then lead to the newly-formed clitoris, and decreases the chances that the patient will not respond to clitoral stimulation after surgery. Additionally, by leaving the prostate in the patient, the surgeon allows it to be situated close to the wall of the newly-formed vagina, which may potentially increase stimulation during vaginal intercourse after the procedure.
Situational anorgasmia
People who are orgasmic in some situations may not be in others. This means that you are able to orgasm only during certain circumstances, such as during oral sex or masturbation. This is very common in women. In fact, about 80% of women experience orgasm from stimulation of the clitoris.
A person may have an orgasm from one type of stimulation but not from another, a person may achieve orgasm with one partner but not another, or have an orgasm only under certain conditions or only with a certain type or amount of foreplay. These common variations are within the range of normal sexual expression and should not be considered problematic.
Factors that may affect whether or not an individual is orgasmic, include fatigue, emotional concerns, feeling pressured to have sex when he or she is not interested, or a partner's sexual dysfunction. In the relatively common case of female situational anorgasmia during penile-vaginal intercourse, some sex therapists recommend that couples incorporate manual or vibrator stimulation during intercourse, or using the female-above position as it may allow for greater stimulation of the clitoris by the penis or pubic symphysis or both, and it allows the woman better control of movement.
General Anorgasmia
This means an individual cannot orgasm in any situation with any partner.
Random anorgasmia
Some people are orgasmic but not in enough instances to satisfy their sense of what is appropriate or desirable.
Prevention
A healthy attitude toward sex, and education about sexual stimulation and response will minimize problems.
Couples who clearly communicate their sexual needs and desires, verbally or nonverbally, will experience orgasmic dysfunction less frequently.
It is also important to realize that sexual response is a complex coordination of the mind and the body, and both need to be functioning well for orgasms to happen. Symptoms
The symptom of orgasmic dysfunction is being unable to reach orgasm, taking longer than you want to reach orgasm, or having only unsatisfying orgasms.
Signs and Tests
A complete medical history and physical examination needs to be done, but results are almost always normal. If the problem began after starting a medication, this should be discussed with the doctor who prescribed the drug. A qualified specialist in sex therapy may be helpful.
Treatment
Treatment can involve education, cognitive behavioral therapy, teaching orgasm by focusing on pleasurable stimulation, and directed masturbation.
Most women require clitoral stimulation to reach an orgasm. Incorporating this into sexual activity may be all that is necessary. If this doesn't solve the problem, then teaching the woman to masturbate may help her understand what she needs to become sexually excited.
A series of couple exercises to practice communication, more effective stimulation, and playfulness can help. If relationship difficulties play a role, treatment may include communication training and relationship enhancement work.
Medical problems, new medications, or untreated depression may need evaluation and treatment in order for orgasmic dysfunction to improve.
If other sexual dysfunctions (such as lack of interest and pain during intercourse) are happening at the same time, these need to be addressed as part of the treatment plan.
HSDD
Hypoactive sexual desire disorder (HSDD), is considered as a sexual
dysfunction and is listed under the Sexual and Gender Identity Disorders of the DSM-IV. It was first included in the DSM-III under the name Inhibited Sexual Desire Disorder, but the name was changed in the DSM-III-R.
HSDD is characterized as a lack or absence of sexual fantasies and desire for sexual activity for some period of time. For this to be regarded as a disorder, it must cause marked distress or interpersonal difficulties and not be better accounted for by another mental disorder (i.e. depression), a drug (legal or illegal), or some other medical condition.
There are various subtypes. HSDD can be general (general lack of sexual desire) or situational (still has sexual desire, but lacks sexual desire for current partner), and it can be acquired (HSDD started after a period of normal sexual functioning) or lifelong (the person has always had no/low sexual desire.)
In the early versions of the DSM, there were only two sexual dysfunctions listed: frigidity (for women) and impotence (for men).
In 1970, Masters and Johnson published their book Human Sexual Inadequacy describing sexual dysfunctions, though these included only dysfunctions dealing with the function of genitals such as premature ejaculation and impotence for men, and anorgasmia and vaginismus for women. Prior to Masters and Johnson's research, female orgasm was assumed to originate primarily from vaginal, rather than clitoral, stimulation. Consequently, feminists have argued that "frigidity" was "defined by men as the failure of women to have vaginal orgasms".
Following this book, sex therapy increased throughout the 1970s. Reports from sex-therapists about people with low sexual desire are reported from at least 1972, but labeling this as a specific disorder did not occur until 1977. In that year, sex therapists Helen Singer Kaplan and Harold Lief independently of each other proposed creating a specific category for people with low or no sexual desire. Lief named it "Inhibited Sexual
Desire," and Kaplan named it "Hypoactive Sexual Desire." The primary motivation for this was that previous models for sex therapy assumed certain levels of sexual interest in one's partner and that problems were only caused by abnormal functioning/non- functioning of the genitals or performance anxiety but that therapies based on those problems were ineffective for people who did not sexually desire their partner. The following year, 1978, Lief and Kaplan together made a proposal to the APA's taskforce for sexual disorders for the DSM III, of which Kaplan and Lief were both members. The diagnosis of Inhibited Sexual Desire (ISD) was added to the DSM when the 3rd edition was published in 1980.
For understanding this diagnosis, it is important to recognize the social context in which it was created. In some cultures, low sexual desire may be considered normal and high sexual desire is problematic. In others, this may be reversed. Some cultures try hard to restrain sexual desire. Others try to excite it. Concepts of "normal" levels of sexual desire are culturally dependent and rarely value-neutral. In the 1970s, there were strong cultural messages that sex is good for you and "the more the better." Within this context, people who were habitually uninterested in sex, who in previous times may not have seen this as a problem, were more likely to feel that this was a situation that needed to be fixed. They may have felt alienated by dominant messages about sexuality and increasingly people went to sex-therapists complaining of low sexual desire. It was within this context that the diagnosis of ISD was created.
In the revision of the DSM-III, published in 1987 (DSM-III-R), ISD was subdivided into two categories: Hypoactive Sexual Desire Disorder and Sexual Aversion Disorder (SAD). The former is a lack of interest in sex and the latter is a phobic aversion to sex. In addition to this subdivision, one reason for the change is that the committee involved in revising the psychosexual disorders for the DMS-III-R thought that term "inhibited" suggests psychodynamic etiology (i.e. that the conditions for sexual desire are present, but the person is, for some reason, inhibiting their own sexual interest.) The term
"hypoactive sexual desire" is more awkward, but more neutral with respect to the cause. The DSM-III-R estimated that about 20% of the population had HSDD. In the DSM-IV (1994), the criterion that the diagnosis requires "marked distress or interpersonal difficulty" was added.
Causes
Low sexual desire is not equivalent to HSDD because of the requirement that the low sexual desire causes marked distress and interpersonal difficulty and because of the requirement that the low desire is not better accounted for by another disorder in the DSM or by a general medical problem, so it is difficult to say exactly what causes HSDD. It is easier to describe, instead, what causes low sexual desire.
In men, there are theoretically more types of HSDD/low sexual desire, typically men are only diagnosed with one of three subtypes.
Lifelong/generalized:
The man has little or no desire for sexual stimulation (with a partner or alone) and never has.
Acquired/situational:
The man was previously sexually interested in his present partner but now lacks sexual interest in them but has desire for sexual stimulation (i.e. alone or with someone other than his present partner.)
Acquired/generalized:
The man previously had sexual interest in his present partner, but lacks interest in sexual activity, partnered or solitary.
Though it can sometimes be difficult to distinguish between these types, they do not necessarily have the same etiology. The cause of lifelong/generalized HSDD is unknown. In the case of acquired/generalized low sexual desire, possible causes include various medical/health problems, psychiatric problems, low levels of
testosterone or high levels of prolactin. One theory suggests that sexual desire is controlled by a balance between inhibitory and excitatory factors. This is thought to be expressed via neurotransmitters in selective brain areas. A decrease in sexual desire may therefore be due to an imbalance between neurotransmitters with excitatory activity like dopamine and norepinephrine and neurotransmitters with inhibitory activity, like serotonin. Low sexual desire can also be a side effect of various medications. In the case of acquired/situational HSDD, possible causes include intimacy difficulty, relationship problems, sexual addiction, and chronic illness of the man's partner. The evidence for these is somewhat in question. Some claimed causes of low sexual desire
are based on empirical evidence. However, some are based merely on clinical observation. In many cases, the cause of HSDD is simply unknown.
There are some factors that are believed to be possible causes of HSDD in women. As with men, various medical problems, psychiatric problems (such as mood disorders), or increased amounts of prolactin can cause HSDD. Other hormones are believed to be involved as well. Additionally, factors such as relationship problems or stress are believed to be possible causes of reduced sexual desire in women.
///. Testosterone
The steroid hormone testosterone is the active ingredient in the testosterone gel formulations of the invention. The manufacture of the drug substance presents no potential risk for humans; the synthesis route is well-characterized.
Table 1 : Nomenclature Testosterone

Structural Formula

Molecular Formula
Relative Molecular Mass
288.4
The physical chemical properties of testosterone are listed in Table 2. Table 2: General Properties of Testosterone

Testosterone, for testosterone gel formulations of the invention, appears as white or slightly creamy white crystals or crystalline powder. It is freely soluble in methanol and ethanol, soluble in acetone and isopropanol and insoluble in n-heptane. It can also be considered as insoluble in water (S2o°c=2.41 x 10"2 g/L ± 0.04 x 10"2 g/L); its n- Octanol/Water partition coefficient (log Pow determined by HPLC) is 2.84. The solubility of testosterone in oils was determined to be 0.8% in isopropylmyristate, 0.5% in peanut oil, 0.6% in soybean oil, 0.5% in corn oil, 0.7% in cottonseed oil and up to 4% in castor oil.
Because testosterone is fully dissolved within the formulations of the present invention, physical characteristics of the drug substance do not influence the
performance of the drug product, testosterone gel formulations of the invention. The manufacturability of testosterone gel formulations of the invention, however is
influenced by the particle size of testosterone. When using a particle size of 50% < 25 microns, 90% < 50 microns the solubility of the drug substance in the matrix is especially favorable.
In accordance with the present invention, the testosterone drug can be in, for instance, crystalline, amorphous, micronized, non-micronized, powder, small particle or large particle form when formulating to intranasal testosterone gels of the present invention. An Exemplary range of testosterone particle sizes include from about 0.5 microns to about 200 microns. Preferably, the testosterone particle size is in a range of from about 5 microns to about 100 microns, and the testosterone is in crystalline or
amorphous and non-micronized or micronized form. Preferably, the testosterone is in crystalline or amorphous micronized form.
The molecular structure of testosterone contains no functional groups that can be protonated or deprotonated in the physiological pH-range. Therefore testosterone is to be considered as a neutral molecule with no pKa value in the range 1 -14. Because it is neutral, testosterone is compatible with excipients.
Drug Product
The testosterone gel formulations of the invention are viscous and thixotropic, oil- based formulations containing a solution of testosterone intended for intranasal application. The non-irritating formulation is designed to adhere to the inner nose. In addition, it acts as a controlling matrix, thus allowing sustained drug delivery through the nasal mucosa.
Other pharmacologically inactive ingredients in the testosterone intranasal gel are castor oil USP, oleoyi macrogolglycerides EP and colloidal silicon dioxide NF. None of these excipients are of human or animal origin. All excipients are well-known and listed in the "Inactive Ingredient" list for Approved Drug Products issued by the FDA.
According to the "Handbook of Pharmaceutical Additives" oleoyi
polyoxylglycerides are used as hydrophilic oil for topicals, injectables and nasals. In FDA-approved medicinal products it is used as co-emulsifier in topical
emulsions/lotions/creams and in vaginal emulsions/creams. In France this excipient is approved for nasal preparations such as "Rhino-Sulforgan" (Laboratoire Jolly-Jatel, France; containing 10% oleoyi polyoxylglycerides) and "Huile Gomenolee 2%
("Laboratoire Gomenol, France; containing 10% oleoyi polyoxylglycerides). Hence, like for castor oil it can be deduced that oleoyi polyoxylglycerides is suitable for an application route where safety and tolerability are of highest importance (e.g. injectables and nasal or vaginal preparations).
Oleoyi macrogolglycerides are also referred to as Labrafil M 1944 CS, apricot kernel oil PEG-6 esters, Peglicol-5-oleate, mixture of glycerides and polyethylene esters. The castor oil, which is used as a solvent for testosterone gel formulations of the invention is a fixed oil. Such oils have the advantage of being non-volatile or spreading (in contrast to essential oils or liquid paraffin), but have the disadvantage of
being hydrophobic. The nasal mucosa contains 95-97% water. Without the oleoyl macrogol-glycerides, the castor oil containing the active ingredient would form a non- interactive layer on the mucous membrane. In order to achieve adequate contact between the castor oil layer and the mucous membrane, the hydrophilic oleoyl macrogol-glycerides oil is added to the formulation to form an emulsion between the castor oil and the mucosa fluid.
Oleoyl macrogol-glycerides are used in semi-solids at concentrations ranging from about 3 to 20%, depending on the application. The amount of oleoyl macrogol- glycerides in testosterone gel formulations of the invention is high enough to allow for a better contact of the carrier oil with the mucous membrane and low enough to have minimal impact on the amount of testosterone that can be incorporated into the carrier oil. A favourable concentration of oleoyl microgol-glycerides in testosterone gel formulations of the invention is found to be 4% of the formulation.
According to the "Handbook of Pharmaceutical Additives" colloidal silicon dioxide is used as an oil adsorbent, thermal stabilizer and gellant. In FDA-approved medicinal products it is used in dental gels, sublingual tablets, endocervical gel, suppositories, vaginal emulsions/creams/tablets/tampons and capsules for inhalation. Furthermore, it is used as an excipient in "Testoderm with adhesives" (Alza Corporation, approved in 1996) a testosterone transdermal patch. Hence, it can be deduced that colloidal silicon dioxide is suitable for an application route where safety and tolerability are of highest importance (e.g. inhalations, endocervical, vaginal or rectal preparations).
For clinical trial supplies, testosterone intranasal gel is supplied in unit-dose syringes consisting of a syringe body made from polypropylene, a plunger molded from polyethylene and a syringe cap made from high density polyethylene. The syringes are wrapped in aluminum foil as secondary packaging. The content of a syringe (125 mg) amounts to 0.10 to 1 .5 mg of testosterone.
The oil in testosterone gel formulations of the invention is thickened with colloidal silicon dioxide, which acts as a gel-forming agent. This compound is used commonly for stiffening oleogels.
The intended dosage form for testosterone gel formulations of the invention is a semi-solid, not a liquid. The formulation is thickened with colloidal silicon dioxide. It is
believed that colloidal silicon dioxide contributes to the thixotropic properties of the gel, simplifying drug delivery to the nostril.
Colloidal silicon dioxide is generally an inert material which is well tolerated as an excipient in mucosal applications such as suppositories. Colloidal silicon dioxide is typically used in these preparations at concentrations ranging from about 0.5 to 10%. The concentration of colloidal silicon dioxide in testosterone gel formulations of the invention is high enough to achieve gel formation but at a level that has minimal impact on testosterone incorporation into the carrier oil.
Preferably, the intranasal testosterone gels of the present invention have in general, a viscosity in the range of between about 3,000 cps and about 27,000 cps. It should nevertheless be understood by those versed in this art that, while the above- mentioned viscosity range is believed to be a preferred viscosity range, any suitable viscosities or viscosity ranges that do not defeat the objectives of the present invention are contemplated.
A detailed description of batches of a testosterone gel formulation of the invention is shown in Table 3.
Table 3: Composition of a testosterone gel formulation of the invention

The testosterone gel formulations of the invention are stored at room
temperature (20-25 °C or 68 to 77 °F). See also Example 1 1 . Temperature excursions from 15 to 30 °C or 59 to 86 °F are permissible for the testosterone gel formulations of the inventions. The stability data available to date are conclusive to support a 24- month shelf life. Unit dose syringes are chosen for the primary packaging of the clinical materials for this clinical trial to allow for ease of dosing, ability to generate multiple doses by varying the fill volume and consistency of dose delivered. The syringe consists of a syringe body, a plunger and a syringe cap. The syringes body is molded
from polypropylene, the plunger is molded from polyethylene and the cap is HDPE. These syringes are designed and manufactured to deliver sterile and non-sterile solutions, liquids and gels at low volumes. For additional protection from the
environment (i.e., exposure to dirt, light, humidity and oxygen), the syringes are packed in a foil-laminate overwrap pouch.
The syringes and caps are designed for use in a clinical setting and meet the requirements of the EU Medical Devices Directive 93/42/EEC of June 14, 1993 and as amended. As this container closure is only intended for use in this portion of the clinical program, no additional studies are performed on the syringe and syringe components. See also Example 1 1 and Fig. 39.
For a further element of protection, two syringes are contained in secondary packaging consisting of an aluminium foil pouch. Two syringes are packaged in the aluminium foil pouch and each pouch is sealed.
The pouch consists of a flexible, 3-layered-foil-laminate of a) polyester 12 micron, b) aluminum 12 micron and c) a polyethylene 75 micron. It is manufactured by Floeter Flexibles GmbH, and supplied under the name "CLIMAPAC II 12-12-75".
INTRINSA®
Procter & Gamble developed a transdermal therapeutic system containing testosterone as active substance for the treatment of HSDD (SD Intrinsa®). Four controlled clinical studies were performed (2 in Phase II b, 2 in Phase III).
The 300 μg testosterone transdermal system is effective in the treatment of HSDD in surgically menopausal women on concomitant estrogen therapy. The women who received testosterone experienced increased frequency of satisfying sexual activity, increased sexual desire, and decreased distress compared with women who received placebo. Improvements were also seen in all other efficacy endpoints (i.e., arousal, pleasure, orgasm, responsiveness, self-image, concerns). The testosterone serum levels were increased to the physiological range of premenopausal women, but did not exceed this range.
Overall, treatment benefits were seen as early as 4 weeks with maximal effects in total satisfying episodes and sexual desire seen by approximately 12 weeks.
Beneficial effects were maintained for the remainder of the 24-week efficacy period. At
week 24, trial results showed that Intrinsa significantly increased the frequency of total satisfying episodes compared to placebo (p<0.05) and also experienced a significantly greater increase in sexual desire domain of PSFS (profile of female sexual function) and a significantly greater decrease in personal distress (p<0.05) in patients on placebo. Intrinsa® is approved for the treatment of HSDD in bilaterally oophorectomised and hysterectomised women receiving concomitant estrogen therapy in the European Union.
LIBIGEL®
Biosante developed a testosterone gel designed to be quickly absorbed through the skin after a once-daily application on the upper arm, delivering testosterone to the bloodstream evenly over time for the treatment of HSDD. One Phase II study has been performed and 2 Phase III studies are ongoing.
The Phase II trial results showed LibiGel significantly increased the number of satisfying sexual events by 238% versus baseline (p<0.0001 ); this increase also was significant versus placebo (p<0.05). In this study, the effective dose of LibiGel produced testosterone blood levels within the normal range for pre-menopausal women and had a safety profile similar to that observed in the placebo group. In addition, no serious adverse events and no discontinuations due to adverse events occurred in any subject receiving LibiGel.
IV. Dosages and Modes of Administration
The invention provides for gel formulations of testosterone to be administered intranasally, wherein the dosage of the formulation is from about 0.15% testosterone by weight of said gel formulation to about 0.6% testosterone by weight of said gel formulation, for example, 0.15% testosterone by weight of the gel formulation, 0.45% testosterone by weight of said gel formulation and 0.6% by weight of the gel formulation.
V. Uses
The methods of the invention are used to treat anorgasmia and/or HSDD in a diagnosed with one or both of these conditions. The invention also provides for intranasal testosterone gel formulations that can be used to treat anorgasmia or HSDD in a patient diagnosed with one or both of these conditions.
Unless otherwise defined, all technical and scientific terms used herein have the
same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. Although methods and materials similar or equivalent to those described herein can be used in the practice or testing of the present invention, suitable methods and materials are described below. All publications, patent applications, patents, and other references mentioned herein are incorporated by reference in their entirety. In case of conflict, the present specification, including definitions, will control. In addition, the materials, methods, and examples are illustrative only and not intended to be limiting.
EXAMPLES
Having now generally described the invention, the same will be more readily understood through reference to the following Examples which are provided by way of illustration, and are not intended to be limiting of the present invention, unless specified.
The following examples are put forth for illustrative purposes only and are not intended to limit the scope of what the inventors regard as their invention.
EXAMPLE 1
Description and Composition of Testosterone
Gel Formulations of the Invention
The compositions of three different concentrations of the drug product to be administered in this clinical trial are provided in the tables below.
Description of Dosage Form
The testosterone gel formulations of the invention are viscous and thixotropic, oil- based formulations containing solubilized testosterone intended for intranasal application. The drug product is formulated with the compendial inactive ingredients: castor oil, oleoyl polyoxyl-glycerides and colloidal silicon dioxide.
Three different doses of the testosterone gel formulations of the invention are intranasally administered: 0.15% w/w, 0.45% w/w and 0.6% w/w. An overage is added to each syringe to account for the gel that is retained in the syringe after dosing. This overage remains consistent at 23 μΙ, regardless of volume of gel in the syringe.
Lower Dosage Strength Intranasal Testosterone Compositions
Table 4: Components, Quantity, Quality Standards and Function - testosterone gel formulation of the invention

Viscosity NF
Colloidal silicon
4.0% 4.92 4.0 increasing
dioxide
agent
Total 100% 123 mg 100 mg
Table 5 Components, Quantity, Quality Standards and Function - 0.45% testosterone gel formulation of the invention

Total 100% 123 mg 100 mg
Table 6 Components, Quantity, Quality Standards and Function - 0.6% testosterone gel formulation of the invention

Testosterone gel formulations of the invention are supplied in unit-dose polypropylene syringes. Two syringes of each dosage are packaged in a protective aluminium foil pouch.
EXAMPLE 2
Intranasal Testosterone Gel Formulations
The testosterone gel formulations of the invention, are formulations of testosterone in an intranasal gel proposed for assessing the pharmacokinetic and pharmacodynamics of three different doses of testosterone gel formulations of the invention, compared to Intrinsa® and placebo for testosterone gel formulations of the invention in women with hypoactive sexual desire disorder (HSDD) and secondary anorgasmia (SA).
The active ingredient, testosterone, is sourced from Bayer Schering.
Challenges for nasal delivery include:
• requirements for larger particles than pulmonary administration (i.e., only
particles > 10 μιτι are sufficiently heavy to avoid entering the respiratory tract);
• concentrations must be higher due to the smaller volumes that can be
administered;
• rapid clearance of the therapeutic agent from the site of deposition results in a shorter time available for absorption;
• potential for local tissue irritation; and
• limited formulation manipulation possibilities to alter drug delivery profiles.
Testosterone is indicated for the treatment of HSDD in bilaterally
oophorectomised and hysterectomised (surgically induced menopausal) women receiving concomitant estrogen therapy. It is also indicated for the treatment of hormone replacement therapy in the treatment of hypogonadism in men. The currently
available options for administration of testosterone are oral, buccal, injectable, implantable and transdermal.
An intranasal testosterone (3.2%) gel, TBS-1 gel, is developed for the treatment of hypogonadism in men and has been administered to hypogonadal men in several clinical trials (Mattern, C. et al., 2008 The Aging Male 1 1 (4):171 -178 (Dec 2008), which is incorporated herein by reference in its entirety). The intranasal testosterone gel for women, testosterone gel formulations of the present invention, are developed at concentrations ranging from about 0.15% to about 0.6% testosterone.
EXAMPLE 3
Overages
[Testosterone Gel Formulations of the Invention!
No overage is added to the formulation. An overage is added to each syringe to account for the gel that is retained in the syringe after dosing. This overage remains consistent at 23 μΙ, regardless of volume of gel in the syringe. The theoretical fill and dispensed amounts for testosterone gel formulations of the invention are provided below.
Syringe Dosage Theoretical Fill Theoretical Dispensed
Volume (μΐ) Volume (μΐ)
0.15% Testosterone 123 100
Gel formulation of
the Invention
0.45% Testosterone 123 100
Gel formulation of
the Invention
0.6% Testosterone 123 100
Gel formulation of
the Invention
EXAMPLE 4
Physicochemical and Biological Properties
[Testosterone Gel Formulations of the Invention!
The testosterone gel formulations of the invention has a viscosity in the range of 3,000 to 10,000 mPa x sec. The viscosity is important because it facilitates
maintenance of the gel in the nasal cavity in contact with the nasal mucosa. When the viscosity is less than approximately 3,000 mPa x sec (i.e., 3,000 centipoise), the gel tends to be drawn by gravity out of the nasal cavity.
EXAMPLE 5
Batch Formula
[testosterone gel formulations of the invention]
Three different concentrations of testosterone gel formulations of the invention, 0.15%, 0.45% and 0.6%, are manufactured for the proposed clinical trial. The batch formulae for these batches are presented in Table 5 below.
Table 5: Batch Formulae for 0.15%, 0.45% and 0.6% testosterone gel formulations of the invention at the 8 kg Batch Size
Quantity per Batch (g)
Components
0.15% 0.45% 0.6%
Testosterone, USP 12 36 48
Castor oil, USP 7348 7324 7312
Oleoyl polyoxylglycerides, Ph.
320 320 320 Eur./NF
Colloidal silicon dioxide, NF 320 320 320
EXAMPLE 6
Manufacturing Process and Process Controls
[Testosterone Gel Formulations of the Invention!
Material is manufactured according to the following process.
Flow Diagram of the Manufacturing Process
Compound Activity Control

Mixing of the Ingredients - Bulk Gel
The Pre-Mix is prepared by mixing, with a propeller mixer, the full amount of Testosterone with portion 1 of the castor oil for 10 minutes.
Mixture I is prepared by adding the Pre-Mix to the remaining castor oil and mixing for 60 minutes. The product temperature is maintained below 50 °C for the entire mixing process.
The oleoyl polyxoylglycerides are pre-heated to 40 - 50 °C and mixed for 10 minutes before being added to Mixture I. This is identified as Mixture II. It is mixed for 45 minutes while maintaining product temperature below 50 °C. Mixture II is then screened through a sieve to remove any un-dissolved Testosterone aggregates.
Mixture III is prepared by adding the colloidal silicon dioxide to Mixture II and mixing for 15 minutes while maintaining product temperature below 50 °C. A visual check is conducted after this step, to ensure that the gel is clear.
At the completion of mixing the gel is stirred and cooled to a product temperature below 30 °C. The product is then discharged into stainless steel drums and the bulk gel sample is taken for analytical testing.
Filling and Packaging - Clinical Supplies
After release of the final gel mixture by the quality control laboratory, the filling and packaging process is carried out by filling a pre-determined volume into the syringe followed by the application of the syringe cap. Two syringes are packaged into a foil pouch.
The syringes are filled using a pipette with the gel taken from a holding tank. The tip of the pipette is discarded after the syringe is filled and the syringe cap is applied. Each syringe is individually labelled.
Following the application of the label, two syringes are packaged in a pre-formed foil pouch and the pouch is sealed. Each pouch is labeled.
EXAMPLE 7
Evaluation of Testosterone Gel Formulations
of the Invention In Women with Anorgasmia
The pharmacokinetics and pharmacodynamic efficacy of testosterone gel formulations of the invention are evaluated in studies of women with anorgasmia. The effect of testosterone gel formulations of the invention on sexual stimuli in women with anorgasmia is also determined.
PATIENT POPULATION
Otherwise healthy females, aged 18 to 65 years, presenting with anorgasmia are evaluated. Sixteen (16) subjects are recruited.
DOSING
Three doses of testosterone gel formulations of the invention are investigated: 150 μg, 450 μg and 600 μg per nostril. A total of 5 doses of a testosterone gel formulation of the invention is administered BID intranasally to women. Placebo testosterone gel formulation of the invention is administered as a control.
TREATMENT DURATION
Test subjects receive 5 doses of a testosterone gel formulation of the invention over a three day period.
ENDPOINTS
Primary end-point:
The plasma concentrations of total testosterone and dihydrotestosterone are measured using validated LC/MS/MS. The following pharmacokinetic parameters are determined for all subjects:
• Cmin, Cmax, tmax, PTF and PTS are determined, for each dosing interval
• AUCO-τ, and Cavg, are calculated for each dosing interval.
• The percentage of time within, below, and above the physiological reference range for plasma testosterone and dihydrotestosterone.
Secondary end-points:
• Efficacy is determined by a battery of computer and psychophysiological tests.
• Safety is monitored according to the following parameters:
o Complete blood counts at Baseline and the Close-Out Visit.
o Clinical chemistry and urinalysis testing at Baseline and Close-Out assesses selected endocrine parameters, renal function, liver function, skeletal/heart muscle damage, lipid abnormalities, and changes in calcium homeostasis,
o Measurement of plasma testosterone, dihydrotestosterone and various hormones at Baseline, study days and Close-Out.
o Adverse Events.
RANDOMIZATION
Subjects in the ANOR cohort are randomized to receive either a testosterone gel formulation of the invention (3 dose levels) or placebo. Randomization is according to the design allocation below.

LD (0.15%) - low dose; MD (0.45%) - mid dose; HD (0.6%) - high dose BLINDING
The analysis is both a double-blind and open label study, depending on the treatment cohort. For subjects in the ANOR group, the study is placebo-controlled and double-blinded.
DOSAGE AND DOSAGE REGIMEN
All subjects are administered a testosterone gel formulation of the invention (0.15%, 0.45% or 0.6%) or testosterone gel formulation of the invention placebo on five (5) occasions during the study: Day 1 at 2000 hours, Day 2 at 800 and 2000 hours, and Day 3 at 800 and 2000 hours. The intranasal gel is administered to both nostrils (1 syringe (100 μΙ volume) per nostril).
PACKAGE AND LABELLING
Study medication consists of a testosterone gel formulation of the invention and placebo gel and is packed in single use syringes designed to expel 100 μΙ of gel. Two syringes are packaged in a foil pouch.
TREATMENT SCHEDULE
Subjects are randomized into the dosing regimen that is administered during a four day (three night) in-patient treatment period and receive either an intranasal
testosterone gel formulation of the invention (3 dose levels) or placebo (ANOR) according to the design allocation:

The randomization scheme is created for each study center and will consist of blocks of four treatments per cohort.
The study is a four day study. The study starts with Study Drug dosing between 2000 and 2100 hours on Day 1 (Baseline). Blood samples for plasma testosterone and dihydrotestosterone profiles are drawn at -60, 0, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360 and 480 minutes following the evening dose on Days 1 and 3 and at 0 and 60 minutes post administration following the morning/evening doses on Day 2 and morning dose on Day 3.
For subjects allocated to the three testosterone gel formulation of the invention arms or the placebo gel, PD testing takes place on Day 2, 30 minutes and 4.5 hours after the morning dose (psychophysiological testing) and Day 3, 30 minutes after the morning dose (computer testing). Subjects undergo a practice psychophysiological session before the first dosing.
Because subjects are expected to not sleep well during their first overnight stay with repeated nightly blood draws, the order of psychophysiological testing and computer testing is not counterbalanced. Computer testing is expected to be more
negatively affected by sleep deprivation than psychophysiological testing. Therefore, on Day 2 psychophysiological testing takes place, and computer testing occurs on Day 3.
Adverse events are assessed and reported.
PATIENT SELECTION AND WITHDRAWAL
The subjects in this study are women with ANOR. Subjects are recruited from the medical practice or the general population through advertisements in local newspapers with additional information available on a website. Before scheduling the screening visit, subjects are asked a series of standardized questions by telephone to assess whether they are likely to be suitable for the study.
INCLUSION CRITERIA
o Women aged 18-65 years.
o Diagnosis of Female Orgasmic Disorder (Anorgasmia) according to the DSM-IV criteria. The current episode must be at least 24 weeks in duration by the Screening Visit. Subtype should be generalized and not due to etiological factors that would prohibit treatment response (e.g., depression, alcoholism, surgery, injury). HSDD as a co-morbid disorder is allowed only if it began after the anorgasmia.
o BMI < 35.
o Women must have a score of >1 1 on the FSDS-R at the Screen Visit together with a score of < 26.55 on the FSFI.
o Women in a steady relationship of at least 12 months or single women who use masturbation as the primary way to attempt to reach orgasm.
o Pre-and post-menopausal women - for physiological and surgical postmenopausal women -estrogen/progestagen substitution (low dose combined ET/P) for at least three (3) months before study entry or post-menopausal women na'ive to ET/P substitution.
o Premenopausal heterosexual women will need to be on a reliable birth control method (i.e., OCPs or partner must use condoms).
o Normal thyroid function, physiological prolactin concentration.
o Normal otorhinolaryngologic examination.
o Provide written informed consent.
EXCLUSION CRITERIA
History of any other clinically relevant psychiatric disorders that could impact sexual function, risks patient's safety, or may impact compliance are as assessed. This includes bipolar disorders, psychotic disorders, severe anxiety, eating disorders, antisocial personality disorders, etc.
o History of Major Depressive Disorder within six (6) months prior the Screening
Visit or a score of >14 on the Beck Depression Inventory II.
o Subjects who meet DSM-IV criteria (APA) for Sexual Aversion Disorder, Substance-Induced Sexual Dysfunction, Dyspareunia (not caused by inadequate foreplay stimulation or alleviated by lubricants), Vaginismus, Gender Identity Disorder, Paraphilia, or for Sexual Dysfunction Due to a General Medical Condition,
o Subjects experiencing relational discord as indicated by a score of > 20 on the MMQ.
o Subjects with known active pelvic inflammatory disease, urinary tract or vaginal infection/vaginitis, cervicitis, interstitial cystitis, vulvodynia, or significant vaginal atrophy,
o Subjects who are breast feeding or have breast fed within the last six (6) months prior to the Baseline Visit,
o Subjects who are pregnant (by serum pregnancy test at the Screen Visit) or have been pregnant within the last 12 months prior to the Baseline Visit, o Treatment with systemic glucocorticoids.
o Treatment with sex steroid hormones such as androgens, estrogens other than in low dose combined ET/P, or gestagens (e.g. anabolic steroids, DHEA, Premarin® (conjugated equine estrogens)).
o Treatment with thyroid hormones (only for stable replacement therapy).
o Significant intercurrent disease of any type, in particular liver, kidney, or heart disease, any form of diabetes mellitus (subjects using antacids or with treated hyperlipidaemia or treated hypothyroidism will not be excluded provided they have been stable on their drug dose for at least six (6) months).
History of nasal disorders (e.g., seasonal or perennial allergic rhinitis, atrophic rhinitis, polyposis, abuse of nasal decongestants, clinically relevant nasal septum deviation, recurrent epistaxis) or sleep apnea.
Subjects with a history of dementia or other neurodegenerative diseases, organic brain disease, stroke, transient ischemic attacks, brain surgery, significant brain trauma, multiple sclerosis, spinal cord injury, peripheral neuropathy, and epilepsy (febrile seizures limited to childhood do not exclude subjects).
History of cancer, excluding basal cell carcinoma.
History of severe or multiple allergies, severe adverse drug reaction or leucopenia.
History of abnormal bleeding tendencies or thrombophlebitis unrelated to venepuncture or intravenous cannulation.
History of DVT.
History of Hepatitis B, a positive test for Hepatitis B surface antigen, a history of Hepatitis C, a positive test for Hepatitis C antibody, a history of HIV infection or demonstration of HIV antibodies.
Recent history of significant sleeping problems. Shift-worker need to have adequate day-night rhythms for three weeks before study entry.
Regular drinkers of more than three (3) units of alcohol daily (1 unit = 300 ml beer, 1 glass wine, 1 measure spirit).
History of, or current evidence of, abuse of alcohol or any drug substance, licit or illicit; or positive urine drug and alcohol screen for drugs of abuse and alcohol.
Difficulty in abstaining from OTC medication (except occasional paracetamol/aspirin) for the duration of the study.
Poor compilers or subjects unlikely to attend study visits.
Receipt of any drug as part of a research study within 30 days of initial dose administration in this study.
Blood donation (usually 550 ml) within the 12 week period before the initial study dose.
TREATMENT OF SUBJECTS
Study Visits
Visit 1 (Day -15) - Screening Subjects for Inclusion and Exclusion Criteria:
• Pre-study screening is carried out within two (2) weeks prior to the start of the treatment. Subjects, after having voluntarily signed the Informed Consent Form, and before enrolment, are interviewed by the Clinical Investigator or his/her designee physician who will take the medical, sexual and physical history, record demographic data, and perform a routine physical examination including vital signs (blood pressure, resting heart rate, body weight, and height).
• FSFI and FSDS-R are administered, as well as MMQ, BDI-II, ISS, SIDI-II, SESII-W.
• The otolaryngologic examination is done by an ENT specialist.
• Venous blood is collected, after an overnight fast, for a CBC (hemoglobin, hemoglobin A1 c, hematocrit, MCV, MCHC, RBC, WBC & differential), Clinical Chemistry profile (Na/K, glucose, urea, creatinine, total bilirubin, albumin, calcium, phosphate, uric acid, LDL, HDL, triglycerides, AST, ALT, ALP, GGT and CK).
• Venous blood samples for estradiol, free testosterone, free testosterone
(percent), follicle stimulating hormone, luteinizing hormone, prolactin, progesterone, sex hormone binding globulin, total testosterone, and dehydroepiandrosterone sulfate are collected.
• A blood sample for TSH, total and free tri-iodothyronine, total and free
thyroxine is obtained.
• Urine is collected for measuring specific gravity, glucose, ketones, bilirubin, pH, urobilinogen, leukocytes, nitrites.
• Subjects undergo Hepatitis B, C and HIV testing (Hepatitis B surface antigen, Hepatitis C antibody, HIV antibodies in plasma).
• A urine drug screen is performed for amphetamines, benzodiazepines,
cannabinoids, cocaine, opiates, MDMA. Subjects with positive test are not enrolled.
• Ethanol will be screened for by breathalyzer.
Visit 2 (Day 1) - Start of Baseline, Randomization, PK Blood Sampling and PD Testing:
• Subjects are admitted to the clinic in the afternoon for three overnights stays.
• A check-in examination is conducted to check for disallowed medications
(OTC and prescription), drugs, alcohol or cigarettes. Subjects are requested to abstain from alcohol for 48 hours prior to admission to the clinic. Alcohol consumption is strictly forbidden at any time during the overnight stay in the clinic. There are no restrictions with respect to food intake during the blood collections for the PK profile.
• Urine tests for the same drugs of abuse as at Screening are repeated.
• Pregnancy is excluded using a urine test (if applicable).
• Vital signs (blood pressure, resting heart rate, and body weight) are checked.
• Blood is drawn for a CBC, chemistry profile, hormone profile, and pregnancy testing.
• Urine for urinalysis and urine drug screen is collected along with performing an alcohol breath test.
• Before dosing and blood sampling, subjects undergo a psychophysiological familiarization test in which neutral and erotic films are shown and VPA is recorded to get acquainted with the experimental procedures and being exposed to explicit erotic stimuli. The data obtained is not be used for analysis.
• A venous cannula will be placed in a forearm vein, and blood sampling starts one hour before the evening administration of the Study Drug.
• Subjects are dosed between 2000 and 2100 hours.
• Blood samples are drawn for plasma testosterone and dihydrotestosterone levels at -60, 0, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360, and 480 minutes post administration.
• Randomization to the treatment schemes is done at this visit.
• Safety assessment is recorded.
• Subjects remain in the clinic overnight.
Visit 3 (Day 2) - PK Blood Sampling:
• Vital signs are obtained.
• A hormone profile is collected.
• Subjects are dosed with Study Drug between 800 and 900 and between 2000 and 2100 hours (unless on Intrinsa®). Blood samples are drawn for plasma testosterone and dihydrotestosterone levels at times 0 and 60 minutes post administration.
• For subjects allocated to the three testosterone gel formulation of the invention -arms or the placebo gel, psychophysiological testing is performed 30 minutes and 4.5 hours post-dosing in the morning
• Safety assessment is recorded.
• Subjects remain in the clinic overnight.
Visit 4 (Day 3) - PK Blood Sampling and PD Testing:
• Vital signs are obtained.
• A hormone profile is collected.
• Subjects are dosed with Study Drug at 800 and 900 and between 2000 and 2100 hours. Blood samples are drawn for plasma testosterone and
dihydrotestosterone levels at times 0 and 60 minutes following administration of the morning dose and at 0, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360, and 480 minutes following administration of the evening dose.
• For subjects allocated to the three testosterone gel formulation of the invention - arms or the placebo gel, computer testing is performed 30 minutes post-dosing in the morning.
• Safety assessment is recorded.
Visit 5 (Day 4) - Discharge-Close Out:
• Physical examination including vital signs.
• Venous blood is collected, after an overnight fast, for a CBC (hemoglobin, hemoglobin A1 c, hematocrit, MCV, MCHC, RBC, WBC & differential), clinical chemistry profile (Na/K, glucose, urea, creatinine, total bilirubin, albumin, calcium, phosphate, uric acid, LDL, HDL, triglycerides, AST, ALT, ALP, GGT and CK).
• Appropriate blood samples for estradiol, free testosterone, free testosterone
(percent), follicle stimulating hormone, luteinizing hormone, prolactin,
progesterone, sex hormone binding globulin, total testosterone, and
dehydroepiandrosterone sulfate are collected.
• Urine is collected for measuring specific gravity, glucose, ketones, bilirubin, pH, urobilinogen, leukocytes, nitrites.
• Safety assessment is recorded.
CLINICAL ASSESSMENTS
Screening and Covariate Questionnaires are used for clinical assessments.
BDI
To index the current level of depressive symptoms, the 21 -item BDI-II is administered (Beck, Steer, & Brown. 1996), Dutch adaptation (Van der Does, 2002). The range for the BDI total score is 0-63, with higher scores indicating more depressive symptoms.
MMQ
The Maudsley Marital Questionnaire (MMQ; Crowe, 1978) is a 20-item self-report instrument measuring dissatisfaction with the general relationship, with the sexual relationship, and with life in general. The MMQ has shown good reliability and validity. The psychometric qualities of the Dutch version of the MMQ were also found to be satisfactory (Arrindell, Boelens, & Lambert. 1983). Higher scores represent larger dissatisfaction.
FSFI
The level of the woman's sexual functioning is assessed by the Female Sexual Function Index (FSFI ; Rosen, Brown, Heiman, et al. 2000). The FSFI® is a self- administered questionnaire that consists of 19 questions. The scale contains six domains: desire, arousal, lubrication, orgasm, satisfaction, and pain. The range for the
total score is 2-36, with lower scores representing worse sexual function. The psychometric quality of the FSFI is satisfactory (Wiegel, Meston, & Rosen. 2005).
Based on a Dutch sample consisting of approximately 350 women with and without sexual complaints, the internal consistency and stability of the FSFI were found to be satisfactory-to-good. The FSFI's ability to discriminate between sexually functional and dysfunctional women was excellent as was the ability to predict the presence or absence of sexual complaints (ter Kuile, Brauer, & Laan, 2006).
FSDS-R
The woman's level of personal distress due to sexual dysfunction is assessed by the Female Sexual Distress Scale-Revised (FSDS-R®; Derogatis, Clayton, Lewis- D'Agostino, et al. 2008).
The items inquire about negative feelings and problems that are bothersome or cause distress during the past 30 days. Reliability and validity of the FSDS® (12-item version) has been evaluated in different samples of sexually functional and
dysfunctional women. For the FSDS®, results indicated a unidimensional factor structure, a high degree of internal consistency, and test-retest reliability. The FSDS® showed a high degree of discrimination between sexually dysfunctional and functional women in each of its three validation studies. Results in a Dutch sample supported the unidimensional structure of the FSDS and its reliability and psychometric validity (ter Kuile, Brauer, & Laan, 2006). An additional question (question 13) has been added to the validated FSDS®. This question is about distress specifically related to sexual desire. The maximum total score of the FSDS-R® indicating the maximum level of sexual distress is '52'. Both the FSDS-R® total score and the Question 13 score alone will be analyzed.
Covariate Questionnaires
Index of Sexual Satisfaction (ISS)
The woman's level of sexual satisfaction as assessed by the Index of Sexual Satisfaction (ISS; Hudson, Harrison, & Crosscup. 1981 ). This 25-item questionnaire asks subjects to evaluate various aspects of their sexual relationship, leading to a sum score that can range between 0 and 100. Higher scores correspond to greater sexual satisfaction. This measure has been shown to have good face, convergent, and
discriminant validity with various samples. Example items are "I feel that my partner enjoys our sex life," "I think that sex is wonderful," and "My partner is sexually very exciting." For the purpose of the present study, the ISS is translated into Dutch.
SDI-II
The level of the woman's sexual desire is assessed by the Sexual Desire
Inventory-ll (SDI-II ; Spector, Carey, & Steinberg. 1 996). The SDI-II consists of two seven-item self-report scales: the Dyadic Sexual Desire scale, which measures an individual's desire for sexual activity with a partner, and the Solitary Sexual Desire scale, which measures an individual's desire for autoerotic sexual activity. The two subscales were internally consistent (Cronbach's a: Dyadic scale = 0.86; Solitary scale = 0.96).
Sexual Excitation/Sexual Inhibition (SESII-W)
The Sexual Inhibition/Excitation Inventory for women (SESII-W; Graham,
Sanders, & Milhausen. 2006) is used to assess individuals' propensity for sexual excitation and sexual inhibition. It consists of 36 items, referring to stimulus situations that could affect sexual inhibition and sexual excitation or to general statements about arousability and inhibition. The instructions ask women to report what would be the most typical reaction now or how they think they would respond if the item does not apply to them. Items are rated on a 4-point Likert-rating scale, from "strongly disagree" to
"strongly agree." The SESII-W has eight lower-order factors, which in turn load on two higher-order factors, Sexual Excitation and Sexual Inhibition. The questionnaire shows good test-retest reliability and convergent and discriminant validity and Sexual
Excitation and Sexual Inhibition appear to be relatively independent factors. The list is already in use in the Netherlands, but psychometric properties have not been
investigated yet.
Efficacy Pharmacodynamic Testing
Computer testing
Single target Implicit Association Task (StIAT):
Conform Wigboldus et al. (2005), the stIAT used in this study is designed to assess subjects' affective associations with sexual stimuli (Brauer, van Leeuwen, Janssen, et al. submitted). Subjects are instructed to classify pictures portraying sexual
acts (i.e., target stimuli) and words representing "positive" or "negative" meanings (i.e., attribute stimuli) to the appropriate superordinate category (i.e., "sex", "positive", "negative") as quickly as possible by pressing only a left or right response key on a keyboard. These labels used for these categories (sex, positive, negative) are continuously visible on the computer screen. The stIAT consists of a combination of practice and experimental blocks (see Greenwald, McGhee & Schwartz. 1998 for detailed methodology). The experimental blocks consist of one 'incongruent' and one 'congruent' block of trials. In the incongruent block, "sex" and "negative" are mapped on a single key and "positive" on the other, while in the congruent block, "sex" and
"positive" are mapped on the same key and "negative" on the other. The difference in reaction times between the two experimental blocks is assumed to reflect whether sex is associated more strongly with either positive or negative. Faster responses in the congruent block (compared to the other block) reflect stronger associations between positive and sex, and faster responses in the incongruent block reflect stronger associations between negative and sex. The target-attribute combinations that share response keys (i.e., block order), and left or right key response requirements are counterbalanced. Each critical block consists of 40 trials of which responses were divided equally over the two response keys. The target category consists of 5 exemplar stimuli of sexual images from the International Affective Picture System (IAPS; Center for the Study of Emotion and Attention, 1995), with the following numbers: 4800, 4652, 4658, 4659, and 4672. The attribute categories consists of 20 generally positive and 20 generally negative words (Dotsch & Wigboldus, 2008; Dotsch, Wigboldus, Langner et al. 2008), thus reflecting more global affective associations with sex. These words were controlled for length and frequency. With respect to the validity, the stIAT's strength lies in high effect sizes due to double opposing categories often leading to slower reaction times (the categorization decision requires effort as there are several possibilities to consider).
Picture Association Task (PAT
This task, developed by van Leeuwen and Macrae (2004), is based on the Affective Priming Task (e.g., Bargh, Chaiken, Govender, et al. 1992; Fazio,
Sanbonmatsu, Powell, et al. 1986; Hermans, De Houwer, & Eelen, 1994) where target
words are preceded by another word or image that influences the categorisation speed of the target word. In the PAT, however, target words and images appear
simultaneously. In the PAT used in this study, subjects are presented with positive or negative words superimposed on either sexual or neutral pictures (Brauer, van
Leeuwen, Janssen, et al. submitted). They are instructed to categorize the words as fast as possible as either positive or negative by pressing one of two computer keys.
Subjects are further instructed to focus on the words that appeared on the screen and not to attend to the background images as these are of no importance for the task and the categories to which the pictorial stimuli belong (sex, neutral) are not explained.
Thus, the PAT captures the unintentional influence of the affective value of the pictorial background stimuli on task performance. The time to select the correct response to the words (positive or negative) is influenced by the match between the valence of the word and the valence of the background image (sex or neutral), thereby revealing indirectly the valence of the picture for the subjects. The word categories consist of 10 positive words and 10 negative words. Whereas for the stIAT general positive and negative words are selected (e.g., peace, respect, war, hate), the PAT consists of positive and negative words that are applicable to a sexual situation, but that do not exclusively refer to sexual experiences (e.g., enjoyable, wonderful, dirty, disgusting) in order to create a conceptual overlap between the content of the words and the content triggered by the sexual pictures. These words are taken from a pilot study in the Netherlands in which female subjects {N = 20) were asked to indicate on a 7-point Likert scale for each positive and negative word how well it described a positive or a negative sexual situation, respectively (Brauer & Laan, 2008). The words appear at one of four randomized locations on the picture to avoid expectation-related responses and to make sure subjects would move their eyes over the image. The sexual pictures were taken from another study on implicit associations with sexual stimuli in women with dyspareunia (Brauer, de Jong, Huijding et al., 2009). These pictures display a variety of sexual acts (e.g., kissing, cunnilingus, fellatio, coitus). Based on each sexual picture, a control picture was created by scrambling the sexual image, leaving a neutral stimulus. All pictures are standardized to 600 x 480 pixels and matched for brightness and contrast. Each stimulus remains on the screen until subjects make a decision or until
3,000 ms has elapsed. After 10 practice trials, 80 experimental trials are presented. Each word is paired randomly with a sexual picture and a neutral picture, resulting in four different combinations each presented 20 times: positive words and sexual images, negative words and sexual images, positive words and neutral images, negative words and neutral images. The order of presentation of the trials is counterbalanced within, and response key mappings (i.e., positive/negative or negative/positive) are
counterbalanced across subjects. The computer records the accuracy and latency of each response. With respect to validity, the strength of the PAT is that it is not sensitive to a possible interpretation bias due to the need to attend to the different stimulus categories at the same time, as is the case in the stIAT.
Dot probe task (DOT)
The dot-probe task (DOT) assesses attentional preference for sexual and neutral visual stimuli. In this task, subjects are shown two images side by side on a computer screen for 500 ms. When the two images disappear, a target stimulus represented by a small dot appears in the place of one of the images. Subjects are asked to indicate the location (side) of the dot. Mean RTs are calculated for three categories: 1 ) neutral neutral 2) neutral sex with the dot under neutral 3) neutral sex with the dot under sex. If reaction times are faster when the dot appears in the place of a certain class of stimuli this indicates an attentional bias towards this class of stimuli.
Psychophysiological testing
Genital response (VPA)
Psychophysiological testing consists of assessment of genital response (vaginal pulse amplitude) and subjective sexual arousal during sexual to self-induced erotic fantasy (3 min), a low-intensity erotic film clip (5 min), and a high-intensity erotic film clip (5 min) (Laan et al., in preparation). The erotic conditions are separated by variable interstimulus intervals during which subjects complete a concentration task (simple arithmetic problems) to allow for return-to-baseline. The erotic stimulus testing is preceded by a 8 min neutral film to establish baseline levels. VPA is measured using a vaginal photoplethysmograph developed by Bert Molenkamp (Technical Support, Department of Psychology, University of Amsterdam) based on instruments initially developed by Sintchak and Geer (1975). The light source (3mm LED, λ=620ηιη) and
optical sensor (Texas Instruments TSL250) are produced in batches of 100, resulting in all photoplethysmographs used in this study having nearly equal electronic
characteristics. A signal-conditioning amplifier separates the VPA from the direct current component using a 12 dB/octave, 0.7-Hz filter. Additional filtering for VPA is 24 dB/octave, 0.4 Hz high-pass. The VPA signal is digitalized at 100 Hz with a Keithley KPCI3107 A/D converter, running on a Windows 2000 PC system. Depth of the probe and orientation of the light source is controlled by a device (a 9-x2-cm FDA-approved perspex plate) attached to the cable within 5cm of the optical sensor. Subjects are instructed to insert the probe until the plate touched their labia. The probe and plate are sterilized according to standard department protocol. .
Sexual feelings and affect (SAQ).
Prior to and immediately after erotic stimulus subjects fill out a questionnaire measuring sexual feelings and affect during sexual stimulation, consisting of 5 scales: sexual arousal (Cronbach's cc=0.87); genital sensations (Cronbach's a =0.96);
sensuality (Cronbach's a =0.73); positive affect (Cronbach's a =0.93); and negative affect (Cronbach's a =0.65). Each question is preceded by the sentence: "During the film, I felt:" after which a positive, negative, physical or sexual experience is described, for instance, pleasant; worried; genital pulsing or throbbing; sexually aroused. The items are measured on a 1 (not at all) to 7 (intensely) scale.
Acute Female Sexual Desire (AFSDQ)
Prior to and following psychophysiological testing subjects fill out the Acute Female Sexual Desire Questionnaire (Laan, Heiman, unpublished). This questionnaire assesses sexual interest in erotic stimuli and has shown to discriminate between women with acquired HSDD and sexually functional controls (Laan et al., in preparation)
STATISTICAL METHODOLOGY
Calculation of Pharmacokinetic Parameters
• Cmin, Cmax, and tmax are taken from the actual measured values. Values are determined relative to the testosterone administration time in treated subjects.
• Area under the concentration curve (AUC) are estimated for the 0 to 24 hour time interval, as well as the BID dosing intervals, using the trapezoidal rule.
• PK evaluations after Day 1 evening dose for testosterone gel formulation of the invention and placebo and Day 3 evening dose for testosterone gel formulation of the invention, placebo and Intrinsa® patch (which was applied on Day 1 ) - AUC, concentrations of total and free testosterone, DHT, estradiol, SHBG. Analyses of Cavg,
 Cmax, tmax, AUC0-t, PTF, and PTS. CaVg are calculated for the 12 hour period as well as τ when appropriate. For subjects on Intrinsa®, a 24 hour calculation is performed.
Cmax, tmax, AUC0-t, PTF, and PTS. CaVg are calculated for the 12 hour period as well as τ when appropriate. For subjects on Intrinsa®, a 24 hour calculation is performed.
• The average concentration in the dosing interval (Cavg) is calculated from the AUC using the following formula: Cavg = AUC0- T / τ, with τ = dosing interval time.
• Peak Trough Fluctuation (PTF) and Peak Trough Swing (PTS) are calculated as follows:
O PTF = (Cmax " Cmin ) / Cavg
O PTS = (Cmax - Cmin ) / Cmin
• Percent time that the plasma testosterone concentration is above, within, and below the reference range of 10 to 70 ng/dl, is calculated.
Statistical Analysis of Pharmacodynamic Data
stIAT:
Incorrect responses are excluded from analyses. In addition, RTs shorter than 300 ms or longer than 3000 ms are excluded from analyses. With respect to the stIAT data, Wigboldus, Holland & van Knippenberg (2005) is followed in that, for each subject, the median response latency of the correct responses to the attribute items in congruent and incongruent blocks is used. Following this, median reaction times of the two experimental blocks are subtracted from one another to obtain a stIAT effect (i.e., stIAT effect = median (Sex/Negative) - median (Sex/Positive)). Negative stIAT effects indicate relatively stronger negative associations with sexual stimuli.
The stIAT effect is analyzed with an analysis of variance with fixed factor treatment, group (HSDD and SA) and the interaction treatment by group. The contrasts are calculated within the model.
PAT:
Median response latencies of the correct responses are calculated, following van Leeuwen and Macrae (2004). To correct for baseline reactions to positive and negative words, difference scores are calculated by subtracting RTs for neutral words
superimposed on sexual pictures from positive words superimposed on sexual pictures. The same is done for negative words superimposed on sexual and neutral pictures (i.e., Sex/+ = RT (sex/positive words) - RT (neutral/positive words) and Sex/- = RT
(sex/negative words) - RT (neutral/negative words. Sex/+ < Sex/- = automatic positive associations with sex).
The two PAT variables (RT positive and RT negative) are analyzed with an analysis of variance with fixed factors treatment, group (ANOR) and group by treatment. The contrasts will be calculated within the model.
DOT:
For each subject the difference between mean RT for the category neutral sex with the dot under sex and the mean RT for the category neutral sex with the dot under neutral is calculated to obtain a DOT effect (i.e., DOT effect = mean neutral sex with dot under neutral - mean neutral sex with dot under sex). Higher DOT scores indicate relatively stronger attention for sexual stimuli.
The DOT effect is analyzed with an analysis of variance with fixed factor treatment, group (ANOR) and the interaction treatment by group. The contrasts are calculated within the model.
VPA:
After VPA artefact deletion, done by a computer program developed Bert
Molenkamp (Technical Support, Department of Psychology, University of Amsterdam), peak-to-trough amplitude is calculated for each remaining pulse. VPA is averaged every 30 seconds during several conditions: neutral film (8 min), self induced erotic fantasy (3 min), low intensity erotic film clip (5 min) and high intensity erotic film clip (5 min). All conditions are offered twice: once 0.5 hours after application of the nasal gel and once 4.5 hours after application of the nasal gel.
VPA during the erotic fantasy, the low intensity film and the high intensity film are analyzed separately and the different moments (0.5 hours after and 4.5 hours after dosing) are also be analyzed separately, resulting in 6 analyses. VPA during a condition
and a moment is analyzed with a mixed model analysis of variance with fixed factors treatment, group (ANOR), time, group by treatment, treatment by time and random factor subject and the average VPA score during the neutral film as covariate. Contrasts are calculated within the model.
SAQ:
For each of the five SAQ scale mean response during a condition and a moment are analyzed with an analysis of variance with factors treatment, group (ANOR) and group by treatment, with the score prior to sexual stimuli as covariate. Contrasts are calculated within the model.
AFSDQ:
ASFDQ score after erotic stimulation during a moment is analyzed with an analysis of variance with factors treatment, group (ANOR), and group by treatment; and the score prior to sexual stimuli as covariate. Contrasts are calculated within the model.
If necessary to meet requirements for analysis of variance data is log- transformed. Results are back-transformed and reported as % change.
Graphs of least square means estimates over time by treatment are presented with error bars indicating the upper and lower 95% confidence interval for the highest and lowest profile respectively. Least square means of the contrasts are tabulated.
If analyses are not feasible according to the described models with the given data, analyses are adjusted. If considered useful extra exploratory analyses are conducted.
Statistical Analysis of Safety Data
Nasal Tolerance: Nasal tolerance data is presented in summary tables. No statistical analysis will be performed.
Vital signs and clinical laboratory parameters:
A table summarizing all laboratory test values and changes from Baseline is presented for each treatment group. In case parameters are ± 20% of their reference range, the clinical significance of these findings are evaluated.
The results of this analysis are presented in Figs. 1 , and 4-6. The following Figs. 3 and 9-1 1 compare the results between the effects of the lower dosage strength testosterone gel nasal formulations of the present invention in subjects diagnosed with anorgasmia or HSDD.
EXAMPLE 8
Evaluation of a Testosterone Nasal Gel
Formulation of the Invention In Women with HSDD
The pharmacokinetics and pharmacodynamic efficacy of testosterone gel formulations of the invention are evaluated in studies of women with HSDD. The effect of testosterone gel formulations of the invention on sexual stimuli in women with HSDD is also determined.
DOSING
Three doses of a testosterone gel formulation of the invention are investigated: 150 μg, 450 μg and 600 μg per nostril. A total of 5 doses of a testosterone gel formulation of the invention is administered BID intranasally to women. The Intrinsa® patch (300μg testosterone) is administered as a control in the HSDD cohort.
PATIENT POPULATION
Otherwise healthy females, aged 18 to 65 years, presenting with HSDD are evaluated. Sixteen (16) subjects are be recruited to each indication.
TREATMENT DURATION
Test subjects receive 5 doses of a testosterone gel formulation of the invention over a three day period.
ENDPOINTS
Primary end-point:
The plasma concentrations of total testosterone and dihydrotestosterone are measured
using validated LC/MS/MS. The following pharmacokinetic parameters are determined for all subjects:
• Cmin, Cmax, tmax, PTF and PTS are determined, for each dosing interval
• AUCO-τ, and Cavg, are calculated for each dosing interval.
• The percentage of time within, below, and above the physiological reference range for plasma testosterone and dihydrotestosterone.
Secondary end-points:
• Efficacy is determined by a battery of computer and psychophysiological tests.
• Safety is monitored according to the following parameters:
o Complete blood counts at Baseline and the Close-Out Visit. o Clinical chemistry and urinalysis testing at Baseline and Close-Out assesses selected endocrine parameters, renal function, liver function, skeletal/heart muscle damage, lipid abnormalities, and changes in calcium homeostasis,
o Measurement of plasma testosterone, dihydrotestosterone and various hormones at Baseline, study days and Close-Out.
o Adverse Events.
RANDOMIZATION
Subjects in the HSDD cohort are randomized to receive either a testosterone gel formulation of the invention (3 dose levels) or the Intrinsa® patch. Randomization is according to the design allocation below.

(0.15%) - low dose; MD (0.45%)- mid dose; HD (0.6%) - high dose
BLINDING
This is both a double-blind and open label study, depending on the treatment cohort. For the HSDD cohort, this is a partly open-label study, as blinding of intra-nasal dosing versus patch dosing is not feasible. The testosterone gel formulation of the invention dose in the HSDD cohort is blinded.
DOSAGE AND DOSAGE REGIMEN
Three-quarters (75%) of the subjects in the HSDD cohort are administered a testosterone gel formulation of the invention (0.15%, 0.45% or 0.6%) on five (5) occasions during the study: Day 1 at 2000 hours, Day 2 at 800 and 2000 hours, and Day 3 at 800 and 2000 hours. The intranasal gel is administered to both nostrils (1
syringe (100 μΙ volume) per nostril). To the remaining one-quarter (25%) of the subjects is administered the Intrinsa® patch at 2000 hours on Day 1 which will remain on the subject's lower abdomen for the duration of the study. The patch is removed on Day 4 prior to discharging the subject from the clinic.
PACKAGE AND LABELLING
Study medication consists of testosterone gel formulations of the invention and testosterone gel formulations of the invention placebo gel and is packed in single use syringes designed to expel 100 μΙ of gel. Two syringes are packaged in a foil pouch. The active control for the HSDD cohort, Intrinsa®, remains in its original packaging from the manufacturer.
TREATMENT SCHEDULE
Subjects are randomized into the dosing regimen that is administered during a four day (three night) in-patient treatment period and receive either intranasal testosterone gel formulation of the invention (3 dose levels) or Intrinsa® patch (HSDD) according to the design allocation:

The randomization scheme is created for each study center and consists of blocks of four treatments per cohort.
The study is a four day study. The study start with Study Drug dosing between 2000 and 2100 hours on Day 1 (Baseline). Blood samples for plasma testosterone and dihydrotestosterone profiles are drawn at -60, 0, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360 and 480 minutes following the evening dose on Days 1 and 3 and at 0 and 60 minutes (except Intrinsa arm - no 60 minute on Day 2 and Day 3 morning draws) post administration following the morning/evening doses on Day 2 and morning dose on Day 3.
For subjects allocated to the three testosterone gel formulations of the invention arms or the placebo gel, PD testing will take place on Day 2, 30 minutes and 4.5 hours after the morning dose (psychophysiological testing) and Day 3, 30 minutes after the morning dose (computer testing). Subjects will have undergone a practice
psychophysiological session before the first dosing.
Because subjects are expected to not sleep well during their first overnight stay with repeated nightly blood draws, the order of psychophysiological testing and computer testing is not counterbalanced. Computer testing is expected to be more negatively affected by sleep deprivation than psychophysiological testing. Therefore, on Day 2 psychophysiological testing takes place, and computer testing occurs on Day 3. For the subjects randomized to the Intrinsa® patch, psychophysiological testing take place on Day 3 between 800 and 900 hours and computer testing follows in the afternoon of Day 3 between 1600 and 1700 hours.
Adverse events are assessed and reported.
PATIENT SELECTION AND WITHDRAWAL
The subjects in this study are women with HSDD. Subjects are recruited from the medical practice or the general population through advertisements in local newspapers with additional information available on a website. Before scheduling the screening visit, subjects are asked a series of standardized questions by telephone to assess whether they are likely to be suitable for the study.
INCLUSION CRITERIA
• Women up to 65 years.
• Postmenopausal women with the primary diagnosis of HSDD, generalized acquired type, according to DSM-IV criteria at the Screening Visit. The current episode must be at least 24 weeks in duration by the Screening Visit. Secondary Female Sexual Arousal Disorder and/or Female Orgasmic Disorder are allowed if co-morbid. This inclusion criterion is met only if the HSDD began prior to Female Sexual Arousal Disorder and/or Female Orgasmic Disorder and the HSDD is of more importance to the subject, in the investigators' judgment.
• BMI < 35.
• Women must have a score of >1 1 on the FSDS-R at the Screen Visit together with a score of < 26.55 on the FSFI.
• Women in a steady relationship of at least 12 months
• Physiological and surgical post-menopausal women - estrogen/progesterone substitution (low dose combined ET/P) for at least three (3) months before study entry or post-menopausal women na'ive to ET/P substitution.
• Normal thyroid function, physiological prolactin concentration.
• Normal otorhinolaryngologic examination.
• Provide written informed consent.
EXCLUSION CRITERIA - HSDD
• History of any other clinically relevant psychiatric disorders that could impact sexual function, risk patient's safety, or may impact compliance, as assessed by the MINI. This includes bipolar disorders, psychotic disorders, severe anxiety, eating disorders, antisocial personality disorders, etc.
• History of Major Depressive Disorder within six (6) months prior to Screening Visit or a score of >14 on the Beck Depression Inventory II.
• Subjects who meet DSM-IV criteria (APA) for Sexual Aversion Disorder,
Substance-Induced Sexual Dysfunction, Dyspareunia (not caused by
inadequate foreplay stimulation or alleviated by lubricants), Vaginismus, Gender Identity Disorder, Paraphilia, or for Sexual Dysfunction Due to a
General Medical Condition.
• Subjects with known active pelvic inflammatory disease, urinary tract or vaginal infection/vaginitis, cervicitis, interstitial cystitis, vulvodynia, or significant vaginal atrophy.
• Women with relationship discord as indicated by a score of > 20 on the MMQ.
• Treatment with systemic glucocorticoids.
• Treatment with sex steroid hormones such as androgens, estrogens other than in low dose combined ET/P, or gestagens (e.g. anabolic steroids, DHEA, Premarin® (conjugated equine estrogens)).
• Treatment with thyroid hormones (only for stable replacement therapy).
Significant intercurrent disease of any type, in particular liver, kidney, or heart disease, or any form of diabetes mellitus (subjects using antacids or with treated hyperlipidaemia or treated hypothyroidism will not be excluded provided they have been stable on their drug dose for at least six months).
History of nasal disorders (e.g., seasonal or perennial allergic rhinitis, atrophic rhinitis, polyposis, abuse of nasal decongestants, clinically relevant nasal septum deviation, recurrent epistaxis) or sleep apnea.
Subjects with a history of dementia or other neurodegenerative diseases, organic brain disease, stroke, transient ischemic attacks, brain surgery, significant brain trauma, multiple sclerosis, spinal cord injury, peripheral neuropathy, and epilepsy (febrile seizures limited to childhood do not exclude subjects)
History of cancer, excluding basal cell carcinoma
History of severe or multiple allergies, severe adverse drug reaction or leucopenia.
History of abnormal bleeding tendencies or thrombophlebitis unrelated to venepuncture or intravenous cannulation.
History of DVT.
History of Hepatitis B, a positive test for Hepatitis B surface antigen, a history of Hepatitis C, a positive test for Hepatitis C antibody, a history of HIV infection or demonstration of HIV antibodies.
Recent history of significant sleeping problems, shift-workers need to have adequate day-night rhythms for three weeks before study entry.
Regular drinkers of more than three (3) units of alcohol daily (1 unit = 300 ml beer, 1 glass wine, 1 measure spirit).
History of, or current evidence of, abuse of alcohol or any drug substance, licit or illicit; or positive urine drug and alcohol screen for drugs of abuse and alcohol.
Difficulty in abstaining from OTC medication (except occasional
paracetamol/aspirin) for the duration of the study.
Poor compilers or subjects unlikely to attend study visits.
• Receipt of any drug as part of a research study within 30 days of initial dose administration in this study.
• Blood donation (usually 550 ml) within the 12 week period before the initial study dose.
TREATMENT OF SUBJECTS
Study Visits
Visit 1 (Day -15) - Screening Subjects for Inclusion and Exclusion Criteria:
• Pre-study screening is carried out within two (2) weeks prior to the start of the treatment. Subjects, after having voluntarily signed the Informed Consent Form, and before enrolment, are interviewed by the Clinical Investigator or his/her designee physician who takes the medical, sexual and physical history, record demographic data, and performs a routine physical examination including vital signs (blood pressure, resting heart rate, body weight, and height).
• FSFI and FSDS-R are administered, as well as MMQ, BDI-II, ISS, SIDI-II, SESII-W.
• The otolaryngologic examination is done by an ENT specialist.
• Venous blood is collected, after an overnight fast, for a CBC (hemoglobin, hemoglobin A1 c, hematocrit, MCV, MCHC, RBC, WBC & differential), Clinical Chemistry profile (Na/K, glucose, urea, creatinine, total bilirubin, albumin, calcium, phosphate, uric acid, LDL, HDL, triglycerides, AST, ALT, ALP, GGT and CK).
• Venous blood samples for estradiol, free testosterone, free testosterone
(percent), follicle stimulating hormone, luteinizing hormone, prolactin, progesterone, sex hormone binding globulin, total testosterone, and dehydroepiandrosterone sulfate are collected.
• A blood sample for TSH, total and free tri-iodothyronine, total and free
thyroxine is obtained.
• Urine is collected for measuring specific gravity, glucose, ketones, bilirubin, pH, urobilinogen, leukocytes, nitrites.
• Subjects also undergo Hepatitis B, C and HIV testing (Hepatitis B surface antigen, Hepatitis C antibody, HIV antibodies in plasma).
• A urine drug screen is performed for amphetamines, benzodiazepines,
cannabinoids, cocaine, opiates, MDMA. Subjects with positive test are not enrolled.
• Ethanol is screened for by breathalyzer.
Visit 2 (Day 1) - Start of Baseline, Randomization, PK Blood Sampling and PD Testing:
• Subjects are admitted to the clinic in the afternoon for three overnights stays.
• A check-in examination is conducted to check for disallowed medications
(OTC and prescription), drugs, alcohol or cigarettes. Subjects are requested to abstain from alcohol for 48 hours prior to admission to the clinic. Alcohol consumption is strictly forbidden at any time during the overnight stay in the clinic. There are no restrictions with respect to food intake during the blood collections for the PK profile.
• Urine tests for the same drugs of abuse as at Screening are repeated.
• Pregnancy is excluded using a urine test (if applicable).
• Vital signs (blood pressure, resting heart rate, and body weight) are checked.
• Blood is drawn for a CBC, chemistry profile, hormone profile, and pregnancy testing.
• Urine for urinalysis and urine drug screen is collected along with performing an alcohol breath test.
• Before dosing and blood sampling, subjects undergo a psychophysiological familiarization test in which neutral and erotic films are shown and VPA is recorded to get acquainted with the experimental procedures and being exposed to explicit erotic stimuli. The data obtained is not used for analysis.
• A venous cannula is placed in a forearm vein, and blood sampling starts one hour before the evening administration of the Study Drug.
• Subjects are dosed between 2000 and 2100 hours.
• Blood samples are drawn for plasma testosterone and dihydrotestosterone levels at -60, 0, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360, and 480 minutes post administration.
• Randomization to the treatment schemes is done at this visit.
• Safety assessment is recorded.
• Subjects remain in the clinic overnight.
Visit 3 (Day 2) - PK Blood Sampling:
• Vital signs are obtained.
• A hormone profile is collected.
• Subjects are dosed with Study Drug between 800 and 900 and between 2000 and 2100 hours (unless on Intrinsa®). Blood samples are drawn for plasma testosterone and dihydrotestosterone levels at times 0 and 60 minutes post administration. For subjects on Intrinsa®, a time 0 draw between 800-900 and 2000-2100 hours is obtained.
• For subjects allocated to the three testosterone gel formulations of the
invention -arms or the placebo gel, psychophysiological testing is performed 30 minutes and 4.5 hours post-dosing in the morning
• Safety assessment is recorded.
• Subjects remain in the clinic overnight.
Visit 4 (Day 3) - PK Blood Sampling and PD Testing:
• Vital signs are obtained.
• A hormone profile is collected.
• Subjects are dosed with Study Drug at 800 and 900 and between 2000 and 2100 hours (unless on Intrinsa®). Blood samples are drawn for plasma testosterone and dihydrotestosterone levels at times 0 and 60 minutes following administration of the morning dose and at 0, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360, and 480 minutes following administration of the evening dose. Subjects receiving Intrinsa® have the time 0 draw for the morning dose and all times in the evening as if they received a new dose.
• For subjects allocated to the three testosterone gel formulations of the invention -arms or the placebo gel, computer testing is performed 30 minutes post-dosing in the morning. For the women randomized to the Intrinsa® patch, psychophysiological testing takes place on Day 3 between 800 and 900 hours. Computer testing follows in the afternoon of Day 3 between 1600 and 1700 hours.
• Safety assessment is recorded.
Visit 5 (Day 4) - Discharge-Close Out:
• Physical examination including vital signs.
• Venous blood is collected, after an overnight fast, for a CBC (hemoglobin, hemoglobin A1 c, hematocrit, MCV, MCHC, RBC, WBC & differential), clinical chemistry profile (Na/K, glucose, urea, creatinine, total bilirubin, albumin, calcium, phosphate, uric acid, LDL, HDL, triglycerides, AST, ALT, ALP, GGT and CK).
• Appropriate blood samples for estradiol, free testosterone, free testosterone (percent), follicle stimulating hormone, luteinizing hormone, prolactin, progesterone, sex hormone binding globulin, total testosterone, and dehydroepiandrosterone sulfate are collected.
• Urine is collected for measuring specific gravity, glucose, ketones, bilirubin, pH, urobilinogen, leukocytes, nitrites.
• The Intrinsa® patch is removed (as appropriate).
• Safety assessment is recorded.
CLINICAL ASSESSMENTS
Screening and Covariate Questionnaires are used for clinical assessments.
BDI
To index the current level of depressive symptoms, the 21 -item BDI-II is administered (Beck, Steer, & Brown. 1996), Dutch adaptation (Van der Does, 2002). The range for the BDI total score is 0-63, with higher scores indicating more depressive symptoms.
MMQ
The Maudsley Marital Questionnaire (MMQ; Crowe, 1978) is a 20-item self-report instrument measuring dissatisfaction with the general relationship, with the sexual relationship, and with life in general. The MMQ has shown good reliability and validity. The psychometric qualities of the Dutch version of the MMQ were also found to be satisfactory (Arrindell, Boelens, & Lambert. 1983). Higher scores represent larger dissatisfaction.
FSFI
The level of the woman's sexual functioning is assessed by the Female Sexual Function Index (FSFI ; Rosen, Brown, Heiman, et al. 2000). The FSFI® is a self- administered questionnaire that consists of 19 questions. The scale contains six domains: desire, arousal, lubrication, orgasm, satisfaction, and pain. The range for the total score is 2-36, with lower scores representing worse sexual function. The
psychometric quality of the FSFI is satisfactory (Wiegel, Meston, & Rosen. 2005).
Based on a Dutch sample consisting of approximately 350 women with and without sexual complaints, the internal consistency and stability of the FSFI were found to be satisfactory-to-good. The FSFI's ability to discriminate between sexually functional and dysfunctional women was excellent as was the ability to predict the presence or absence of sexual complaints (ter Kuile, Brauer, & Laan, 2006).
FSDS-R
The woman's level of personal distress due to sexual dysfunction is assessed by the Female Sexual Distress Scale-Revised (FSDS-R®; Derogatis, Clayton, Lewis- D'Agostino, et al. 2008).
The items inquire about negative feelings and problems that are bothersome or cause distress during the past 30 days. Reliability and validity of the FSDS® (12-item version) has been evaluated in different samples of sexually functional and
dysfunctional women. For the FSDS®, results indicated a unidimensional factor structure, a high degree of internal consistency, and test-retest reliability. The FSDS® showed a high degree of discrimination between sexually dysfunctional and functional women in each of its three validation studies. Results in a Dutch sample supported the unidimensional structure of the FSDS and its reliability and psychometric validity (ter
Kuile, Brauer, & Laan, 2006). An additional question (question 13) has been added to the validated FSDS®. This question is about distress specifically related to sexual desire. The maximum total score of the FSDS-R® indicating the maximum level of sexual distress is '52'. Both the FSDS-R® total score and the Question 13 score alone will be analyzed.
Index of Sexual Satisfaction (ISS)
The woman's level of sexual satisfaction as assessed by the Index of Sexual Satisfaction (ISS; Hudson, Harrison, & Crosscup. 1981 ). This 25-item questionnaire asks subjects to evaluate various aspects of their sexual relationship, leading to a sum score that can range between 0 and 100. Higher scores correspond to greater sexual satisfaction. This measure has been shown to have good face, convergent, and discriminant validity with various samples. Example items are "I feel that my partner enjoys our sex life," "I think that sex is wonderful," and "My partner is sexually very exciting." For the purpose of the present study, the ISS is translated into Dutch.
SDI-II
The level of the woman's sexual desire is assessed by the Sexual Desire
Inventory-ll (SDI-II ; Spector, Carey, & Steinberg. 1 996). The SDI-II consists of two seven-item self-report scales: the Dyadic Sexual Desire scale, which measures an individual's desire for sexual activity with a partner, and the Solitary Sexual Desire scale, which measures an individual's desire for autoerotic sexual activity. The two subscales were internally consistent (Cronbach's a: Dyadic scale = 0.86; Solitary scale = 0.96).
Sexual Excitation/Sexual Inhibition (SESII-W)
The Sexual Inhibition/Excitation Inventory for women (SESII-W; Graham,
Sanders, & Milhausen. 2006) is used to assess individuals' propensity for sexual excitation and sexual inhibition. It consists of 36 items, referring to stimulus situations that could affect sexual inhibition and sexual excitation or to general statements about arousability and inhibition. The instructions ask women to report what would be the most typical reaction now or how they think they would respond if the item does not apply to them. Items are rated on a 4-point Likert-rating scale, from "strongly disagree" to
"strongly agree." The SESII-W has eight lower-order factors, which in turn load on two
higher-order factors, Sexual Excitation and Sexual Inhibition. The questionnaire shows good test-retest reliability and convergent and discriminant validity and Sexual
Excitation and Sexual Inhibition appear to be relatively independent factors. The list is already in use in the Netherlands, but psychometric properties have not been investigated yet.
Efficacy Pharmacodynamic Testing
Computer testing
Single target Implicit Association Task (StIAT):
Conform Wigboldus et al. (2005), the stIAT used in this study is designed to assess subjects' affective associations with sexual stimuli (Brauer, van Leeuwen, Janssen, et al. submitted). Subjects are instructed to classify pictures portraying sexual acts (i.e., target stimuli) and words representing "positive" or "negative" meanings (i.e., attribute stimuli) to the appropriate superordinate category (i.e., "sex", "positive", "negative") as quickly as possible by pressing only a left or right response key on a keyboard. These labels used for these categories (sex, positive, negative) are continuously visible on the computer screen. The stIAT consists of a combination of practice and experimental blocks (see Greenwald, McGhee & Schwartz. 1998 for detailed methodology). The experimental blocks consist of one 'incongruent' and one 'congruent' block of trials. In the incongruent block, "sex" and "negative" are mapped on a single key and "positive" on the other, while in the congruent block, "sex" and
"positive" are mapped on the same key and "negative" on the other. The difference in reaction times between the two experimental blocks is assumed to reflect whether sex is associated more strongly with either positive or negative. Faster responses in the congruent block (compared to the other block) reflect stronger associations between positive and sex, and faster responses in the incongruent block reflect stronger associations between negative and sex. The target-attribute combinations that share response keys (i.e., block order), and left or right key response requirements are counterbalanced. Each critical block consists of 40 trials of which responses were divided equally over the two response keys. The target category consists of 5 exemplar stimuli of sexual images from the International Affective Picture System (IAPS; Center for the Study of Emotion and Attention, 1995), with the following numbers: 4800, 4652,
4658, 4659, and 4672. The attribute categories consists of 20 generally positive and 20 generally negative words (Dotsch & Wigboldus, 2008; Dotsch, Wigboldus, Langner et al. 2008), thus reflecting more global affective associations with sex. These words were controlled for length and frequency. With respect to the validity, the stIAT's strength lies in high effect sizes due to double opposing categories often leading to slower reaction times (the categorization decision requires effort as there are several possibilities to consider).
Picture Association Task (PAT
This task, developed by van Leeuwen and Macrae (2004), is based on the Affective Priming Task (e.g., Bargh, Chaiken, Govender, et al. 1992; Fazio,
Sanbonmatsu, Powell, et al. 1986; Hermans, De Houwer, & Eelen, 1994) where target words are preceded by another word or image that influences the categorisation speed of the target word. In the PAT, however, target words and images appear
simultaneously. In the PAT used in this study, subjects are presented with positive or negative words superimposed on either sexual or neutral pictures (Brauer, van
Leeuwen, Janssen, et al. submitted). They are instructed to categorize the words as fast as possible as either positive or negative by pressing one of two computer keys.
Subjects are further instructed to focus on the words that appeared on the screen and not to attend to the background images as these are of no importance for the task and the categories to which the pictorial stimuli belong (sex, neutral) are not explained.
Thus, the PAT captures the unintentional influence of the affective value of the pictorial background stimuli on task performance. The time to select the correct response to the words (positive or negative) is influenced by the match between the valence of the word and the valence of the background image (sex or neutral), thereby revealing indirectly the valence of the picture for the subjects. The word categories consist of 10 positive words and 10 negative words. Whereas for the stIAT general positive and negative words are selected (e.g., peace, respect, war, hate), the PAT consists of positive and negative words that are applicable to a sexual situation, but that do not exclusively refer to sexual experiences (e.g., enjoyable, wonderful, dirty, disgusting) in order to create a conceptual overlap between the content of the words and the content triggered by the sexual pictures. These words are taken from a pilot study in the Netherlands in which
female subjects (Λ/ = 20) were asked to indicate on a 7-point Likert scale for each positive and negative word how well it described a positive or a negative sexual situation, respectively (Brauer & Laan, 2008). The words appear at one of four randomized locations on the picture to avoid expectation-related responses and to make sure subjects would move their eyes over the image. The sexual pictures were taken from another study on implicit associations with sexual stimuli in women with dyspareunia (Brauer, de Jong, Huijding et al., 2009). These pictures display a variety of sexual acts (e.g., kissing, cunnilingus, fellatio, coitus). Based on each sexual picture, a control picture was created by scrambling the sexual image, leaving a neutral stimulus. All pictures are standardized to 600 x 480 pixels and matched for brightness and contrast. Each stimulus remains on the screen until subjects make a decision or until 3,000 ms has elapsed. After 10 practice trials, 80 experimental trials are presented. Each word is paired randomly with a sexual picture and a neutral picture, resulting in four different combinations each presented 20 times: positive words and sexual images, negative words and sexual images, positive words and neutral images, negative words and neutral images. The order of presentation of the trials is counterbalanced within, and response key mappings (i.e., positive/negative or negative/positive) are
counterbalanced across subjects. The computer records the accuracy and latency of each response. With respect to validity, the strength of the PAT is that it is not sensitive to a possible interpretation bias due to the need to attend to the different stimulus categories at the same time, as is the case in the stIAT.
Dot probe task (DOT)
The dot-probe task (DOT) assesses attentional preference for sexual and neutral visual stimuli. In this task, subjects are shown two images side by side on a computer screen for 500 ms. When the two images disappear, a target stimulus represented by a small dot appears in the place of one of the images. Subjects are asked to indicate the location (side) of the dot. Mean RTs are calculated for three categories: 1 ) neutral neutral 2) neutral sex with the dot under neutral 3) neutral sex with the dot under sex. If reaction times are faster when the dot appears in the place of a certain class of stimuli this indicates an attentional bias towards this class of stimuli.
Psychophysiological testing
Genital response (VPA)
Psychophysiological testing consists of assessment of genital response (vaginal pulse amplitude) and subjective sexual arousal during sexual to self-induced erotic fantasy (3 min), a low-intensity erotic film clip (5 min), and a high-intensity erotic film clip (5 min) (Laan et al., in preparation). The erotic conditions are separated by variable interstimulus intervals during which subjects complete a concentration task (simple arithmetic problems) to allow for return-to-baseline. The erotic stimulus testing is preceded by a 8 min neutral film to establish baseline levels. VPA is measured using a vaginal photoplethysmograph developed by Bert Molenkamp (Technical Support, Department of Psychology, University of Amsterdam) based on instruments initially developed by Sintchak and Geer (1975). The light source (3mm LED, λ=620ηιη) and optical sensor (Texas Instruments TSL250) are produced in batches of 100, resulting in all photoplethysmographs used in this study having nearly equal electronic
characteristics. A signal-conditioning amplifier separates the VPA from the direct current component using a 12 dB/octave, 0.7-Hz filter. Additional filtering for VPA is 24 dB/octave, 0.4 Hz high-pass. The VPA signal is digitalized at 100 Hz with a Keithley KPCI3107 A/D converter, running on a Windows 2000 PC system. Depth of the probe and orientation of the light source is controlled by a device (a 9-x2-cm FDA-approved perspex plate) attached to the cable within 5cm of the optical sensor. Subjects are instructed to insert the probe until the plate touched their labia. The probe and plate are sterilized according to standard department protocol. .
Sexual feelings and affect (SAQ).
Prior to and immediately after erotic stimulus subjects fill out a questionnaire measuring sexual feelings and affect during sexual stimulation, consisting of 5 scales: sexual arousal (Cronbach's cc=0.87); genital sensations (Cronbach's a =0.96);
sensuality (Cronbach's a =0.73); positive affect (Cronbach's a =0.93); and negative affect (Cronbach's a =0.65). Each question is preceded by the sentence: "During the film, I felt:" after which a positive, negative, physical or sexual experience is described, for instance, pleasant; worried; genital pulsing or throbbing; sexually aroused. The items are measured on a 1 (not at all) to 7 (intensely) scale.
Acute Female Sexual Desire (AFSDQ)
Prior to and following psychophysiological testing subjects fill out the Acute Female Sexual Desire Questionnaire (Laan, Heiman, unpublished). This questionnaire assesses sexual interest in erotic stimuli and has shown to discriminate between women with acquired HSDD and sexually functional controls (Laan et al., in
preparation).
STATISTICAL METHODOLOGY
Calculation of Pharmacokinetic Parameters
• Cmin, Cmax, and tmax are taken from the actual measured values. Values are determined relative to the testosterone administration time in treated subjects.
• Area under the concentration curve (AUC) are estimated for the 0 to 24 hour time interval, as well as the BID dosing intervals, using the trapezoidal rule.
• PK evaluations after Day 1 evening dose for testosterone gel formulations of the invention and placebo and Day 3 evening dose for testosterone gel formulations of the invention, placebo and Intrinsa® patch (which was applied on Day 1 ) - AUC, concentrations of total and free testosterone, DHT, estradiol, SHBG. Analyses of Cavg,
 Cmax, tmax, AUC0-t, PTF, and PTS. CaVg are calculated for the 12 hour period as well as τ when appropriate. For subjects on Intrinsa®, a 24 hour calculation is performed.
Cmax, tmax, AUC0-t, PTF, and PTS. CaVg are calculated for the 12 hour period as well as τ when appropriate. For subjects on Intrinsa®, a 24 hour calculation is performed.
• The average concentration in the dosing interval (Cavg) is calculated from the AUC using the following formula: Cavg = AUC0- T / τ, with τ = dosing interval time.
• Peak Trough Fluctuation (PTF) and Peak Trough Swing (PTS) are calculated as follows:
O PTF = (Cmax - Cmin ) / CaVg
O PTS = (Cmax - Cmin ) / Cmin
• Percent time that the plasma testosterone concentration is above, within, and below the reference range of 10 to 70 ng/dl, is calculated.
Statistical Analysis of Pharmacodynamic Data
stIAT:
Incorrect responses are excluded from analyses. In addition, RTs shorter than 300 ms or longer than 3000 ms are excluded from analyses. With respect to the stIAT data, Wigboldus, Holland & van Knippenberg (2005) is followed in that, for each subject, the median response latency of the correct responses to the attribute items in congruent and incongruent blocks is used. Following this, median reaction times of the two experimental blocks are subtracted from one another to obtain a stIAT effect (i.e., stIAT effect = median (Sex/Negative) - median (Sex/Positive)). Negative stIAT effects indicate relatively stronger negative associations with sexual stimuli.
The stIAT effect is analyzed with an analysis of variance with fixed factor treatment, group (HSDD and SA) and the interaction treatment by group. The contrasts are calculated within the model.
PAT:
Median response latencies of the correct responses are calculated, following van Leeuwen and Macrae (2004). To correct for baseline reactions to positive and negative words, difference scores are calculated by subtracting RTs for neutral words
superimposed on sexual pictures from positive words superimposed on sexual pictures. The same is done for negative words superimposed on sexual and neutral pictures (i.e., Sex/+ = RT (sex/positive words) - RT (neutral/positive words) and Sex/- = RT
(sex/negative words) - RT (neutral/negative words. Sex/+ < Sex/- = automatic positive associations with sex).
The two PAT variables (RT positive and RT negative) are analyzed with an analysis of variance with fixed factors treatment, group (ANOR) and group by treatment. The contrasts will be calculated within the model.
DOT:
For each subject the difference between mean RT for the category neutral sex with the dot under sex and the mean RT for the category neutral sex with the dot under neutral is calculated to obtain a DOT effect (i.e., DOT effect = mean neutral sex with dot under neutral - mean neutral sex with dot under sex). Higher DOT scores indicate relatively stronger attention for sexual stimuli.
The DOT effect is analyzed with an analysis of variance with fixed factor treatment, group (ANOR) and the interaction treatment by group. The contrasts are calculated within the model.
VPA:
After VPA artefact deletion, done by a computer program developed Bert Molenkamp (Technical Support, Department of Psychology, University of Amsterdam), peak-to-trough amplitude is calculated for each remaining pulse. VPA is averaged every 30 seconds during several conditions: neutral film (8 min), self induced erotic fantasy (3 min), low intensity erotic film clip (5 min) and high intensity erotic film clip (5 min). All conditions are offered twice: once 0.5 hours after application of the nasal gel and once 4.5 hours after application of the nasal gel.
VPA during the erotic fantasy, the low intensity film and the high intensity film are analyzed separately and the different moments (0.5 hours after and 4.5 hours after dosing) are also be analyzed separately, resulting in 6 analyses. VPA during a condition and a moment is analyzed with a mixed model analysis of variance with fixed factors treatment, group (ANOR), time, group by treatment, treatment by time and random factor subject and the average VPA score during the neutral film as covariate. Contrasts are calculated within the model.
SAQ:
For each of the five SAQ scale mean response during a condition and a moment are analyzed with an analysis of variance with factors treatment, group (ANOR) and group by treatment, with the score prior to sexual stimuli as covariate. Contrasts are calculated within the model.
AFSDQ:
ASFDQ score after erotic stimulation during a moment is analyzed with an analysis of variance with factors treatment, group (ANOR), and group by treatment; and the score prior to sexual stimuli as covariate. Contrasts are calculated within the model.
If necessary to meet requirements for analysis of variance data is log- transformed. Results are back-transformed and reported as % change.
Graphs of least square means estimates over time by treatment are presented with error bars indicating the upper and lower 95% confidence interval for the highest and lowest profile respectively. Least square means of the contrasts are tabulated.
If analyses are not feasible according to the described models with the given data, analyses are adjusted. If considered useful extra exploratory analyses are conducted.
Statistical Analysis of Safety Data
Nasal Tolerance: Nasal tolerance data is presented in summary tables. No statistical analysis will be performed.
Vital signs and clinical laboratory parameters:
A table summarizing all laboratory test values and changes from Baseline is presented for each treatment group. In case parameters are ± 20% of their reference range, the clinical significance of these findings are evaluated.
The results of this analysis is presented in Figs. 2, 3 and 7-8. Figs. 3, 9-1 1 show or compare the results between the effects of the testosterone gel nasal formulations of the invention on subjects diagnosed with anorgasmia or HSDD.
EXAMPLE 9
Pharmacokinetic ("PK") and Pharmacodynamic ("PD") Study concerning 53 Anorgasmia Women and the three different testosterone bio-adhesive gel formulations of the invention (0.15% - , 0.45% and 0.6% testosterone
by weight of the gel formulation, as reported in Examples 1-5)
Objective:
As shown in Fig. 14, this PK and PD study is to assess the serum testosterone pharmacokinetic profile and the pharmacodynamic response measuring Vaginal Pulse Amplitude ("VPA") following a single dose administration of each of the testosterone bio- adhesive gel formulations of the invention (0.15% - , 0.45% and 0.6% testosterone by weight of the gel formulation, as reported in Examples 1 -5), as compared to placebo, in women with anorgasmia.
Method:
A total of 12 women with anorgasmia (n=12) are included in this placebo and active comparator PK and PD study. Each women receives four different single intranasal doses of 100 μΙ per nostril adminsitered via a unit-dose syringe on four different days (that is, each of the 12 women in this study receives a TBS-2 high dose (1 .2 mg - 0.6% by weight testosterone - 0.6mg/100 μΙ/nostril), a TBS-2 medium dose (0.9 mg - 0.45% by weight testosterone - 0.45mg/100 μΙ/nostril) or a TBS-2 low dose (0.3 mg - 0.15% by weight testosterone - 0.15mg/100 μΙ/nostril), and placebo TBS-2 (anorgasmia cohort). Frequent PK serum amples are collected from each woman during the first 12 hours after intranasal dose adminsitration. Initial pharmacodynamic efficacy is explored using vaginal pulse amplitude (VPA). VPA is a measure of blood flow to the vagina (engorgement). Safety is monitored throughout the PK and PD study.
Results:
The PK results show that there is an increase in plasma testosterone levels with increasing dose levels. See Fig. 14. Mean concentrations of plasma testosterone 0-12 hours after dosing are: (a) TBS-2 high dose (1 .2 mg - 0.6% by weight testosterone - 0.6 mg/100 μΙ/nostril) - about 70 ng/dL after about the first 100 minutes of adminsitration, about 50ng/dl_ after about the first 250 minutes of adminsitration, and about 40 ng/dL after about 350 minutes following adminsitration and thereafter; (b) TBS-2 medium dose (0.9 mg - 0.45% by weight testosterone - 0.45 mg/100 μΙ/nostril) - about 55 ng/dL after about the first 25 minutes of adminsitration, about 35ng/dL after about the first 250 minutes of adminsitration, and about 35-30 ng/dL after about 350 minutes following adminsitration and thereafter; and (c) TBS-2 low dose (0.3 mg - 0.15% by weight testosterone - 0.15 mg/100 μΙ/nostril) - about 28 ng/dL after about the first 100 minutes of adminsitration, about 23ng/dL after about the first 250 minutes of adminsitration, and about 20 ng/dL after about 350 minutes following adminsitration and thereafter.
The testosterone Cmax and Cavg for testosterone following single dose
administration for each of the three TBS-2 dosage strengths do not exceed the normal testosterone serum level in women (3 - 80 ng/dL).
For all TBS-2 dosage strengths, the testosterone level returns to baseline following adminsitration of the single dose.
As to the PD aspect of the study, there are favorable statistically significant differences in VPA response in the women for each of the TBS-2 dosage strengths vs. placebo at 0.5 hours and at 4.5 hours post-dose.
Conclusion:
It is presently believed that the TBS-2 nasally applied testosterone bio-adhesive gels of the preset invention (i) may be uniquely taken on a prn basis, i.e., on demand, (ii) have an ideal safety profile, i.e., there appears to be no androgen-related side effects, there ia a low testosterone drug load, and (iii) present no risk of testosterone transference.
EXAMPLE 10A
Vibrotactile Stimulation Study ("VTS") of 56 Women with Anorqasmia
Objective:
To evaluate the effect of a single dose of high dose TBS-2 (1 .2 mg - 0.6% by weight testosterone - 0.6 mg/100 μΙ/nostril, as reported in Examples 1 -5) on orgasm at 0.5, 2.0 4.0 and 8.0 hours post-dose administration in 56 women with anorgasmia. Another objective of this study is to evaluate the time to orgasm and the quality of the orgasm following TBS-2 intranasal administration. Further, an objective of this VTS study is to determine the effect of high dose TBS-2 on arousal, sensuality and genital stimulation and to assess safety. See Fig. 15.
Method:
This VTS study is a single-center, randomized, single-blind, placebo-controlled, five-arm parallel group study using vibrotactile stimulation that is combined with visual sexual stimulation (n=56). See Fig. 15.
In accordance with this VTS study protocol, each women is administered an intranasal single high-dose of TBS-2 (1 .2 mg - 0.6% by weight testosterone - 0.6 mg/100 μΙ/nostril).
The demographics for this VTS study are, see Fig. 15:
• 59 women randomized, 56 completed the study
• All 3 drop-outs due to protocol violations
• Average age 27.8 years
• 87.5% Primary Anorgasmia, 12.5% Secondary Anorgasmia
• All randomized subjects met entrance criteria for Martial Quality and
Depression
• 3% of patients (3 out of 97) excluded due to orgasm at Visit 2
Results:
As shown in Fig. 16, more women report, treated with high dose TBS-2, report orgasms, as compared to placebo. In fact, during the 4 weeks post-treatment phase, women who are treated with high dose TBS-2 8 report a total of 8 orgasms, as compared to no orgasms for those women who receive placebo. With respect to the treatment phase, women who are treated with high dose TBS-2 8 report a total of 4 orgasms, as compared to 2 orgasms for those women who receive placebo. See Fig. 16.
Additional VTS study results and findings include:
• The time to orgasm ranges from 12.17 minutes to 18.22 minutes following high dose TBS-2 administration;
• The orgasms by women, who are administered TBS-2, are reported as more pleasant and more intense as compared to the orgasms by those women who are administered placebo;
• More TBS-2 women report high arousal compared to Placebo (83.3% vs.
16.7%). See Figs. 17 and 18;
• TBS-2 women report more sexual desire (AFSDQ Scores). See also Figs.
17 and 18;
• Positive response to stimulation more pronounced for TBS-2. See Fig. 18;
• As reported in Example 9, there is statistically significant differences in VPA between TBS-2 high, medium and low dose and Placebo (mean change from baseline);
• Total testosterone levels are increased to upper end of normal (mean
about 66.7 ng/dL);
Mean free testosterone levels are about 6.35 pg/mL (chronic treatments achieve about 3.1 -4.0 pg/mL);
Other Findings - Patient Feedback
(a) Women prefer home setting
(i) thoughts about hospital setting, experimenter, are distracting
(ii) still a little tense, becasue of the setting
(iii) home is a better setting
(iv) at home, more relaxed
(v) a little limited in movement due to position
(b) Partner Involvement Important to some women
(i) with real life partner, it would be better
(ii) felt guilty he is (partner) is not here
(iii) nicer with a man
Safety:
No Serious Adverse Events
Total of 18 Adverse Events reported
(i) mild and resolved by end of study
(ii) 5 Not Related to study medication
(iii) 3 of unknown etiology (Possibly Related)
(iv) no association with active treatment and adverse events
(v) placebo had 50% of women report adverse events vs. 19.1 % for TBS- 2
No difference in endorcinology results (SHBG, Albumin, Hemogolobin)
Conclusion
Adequate clitoral stimulation alone is not sufficient to treat anorgasmia The data from Examples 10 and 1 1 point to the success of TBS-2 pharmaceutical intervention in addition to effective stimulation TBS-2 data indicates a positive response between 2 and 8 hours post- dose
TBS-2, when administered prn, is believed to elicit and enhance sexual response in women
• TBS-2 is believed to maintain total and free testosterone levels in the normal range
• TBS-2 is believed to be safe and none of the adverse events commonly observed with chronic testosterone treatments are observed
• Comfortable environment (home setting) and partner interaction may play a role in acheivement of orgasm
EXAMPLE 10B
Single-Center, Randomized, Single-Blind, Placebo Controlled, Five-Arm Parallel Group Study to Assess Efficacy of a Single Dose of TBS-2 Intranasal Gel at Four
Time Points Post-Dose using Vibrotactile Stimulation combined with Visual Sexual Stimulation in Female Subjects with Primary or Secondary Anorgasmia
2. SYNOPSIS
Background
Anorgasmia is the second most common sexual disorder among women, with prevalence figures ranging from 16% to 28% in the United States, Europe, and Central/South America, and 30-46% in Asia. The androgen testosterone is known to play a role in mood, body composition, bone mineral density and has central and peripheral effects on sexual function. The only registered testosterone product for use in women is Intrinsa, a testosterone slow- release transdermal patch.
The product under investigation in this study, TBS-2, is a testosterone intranasal gel that may allow for direct uptake of testosterone into the brain and rapid absorption into systemic circulation. The delivery of testosterone to the brain and the rapid systemic absorption is hypothesized to be effective in enhancing sexual desire and orgasm in an "as needed" way, thus avoiding chronic exposure to testosterone. The primary objective of this study was to evaluate the effect of a single dose of TBS-2 on the occurrence of orgasm in primary and secondary anorgasmic women during sexual stimulation at one of four different time points after administration of the study drug compared to a placebo group. Sexual stimulation consisted of vibrotactile stimulation to the glans clitoris (VTS) in combination with visual sexual stimulation (VSS).
Methods
This randomized, single-blind, placebo-controlled five arms parallel group study was conducted at a single study center to evaluate the effect of a single dose of TBS-2, 1200 meg (600 meg per nostril), on the occurrence of orgasm at 0.5, 2.0, 4.0 and 8.0 hours post-dose administration in female subjects with primary or secondary anorgasmia using VTS and VSS. The study consisted of two screening visits and one treatment visit. TBS-2 or placebo was administered at Visit 3.
At Visit 1 , all screening assessments were done and subjects were familiarized with the VTS technique and VSS. At Visit 2, women underwent VTS/VSS stimulation for 20 minutes to determine if they were able to achieve orgasm via stimulation alone. Women who did not achieve orgasm during the VTS/VSS procedure and met all other eligibility criteria were randomized to one of the five treatment arms. Fifty-nine (59) women on hormonal or non- hormonal contraception methods were enrolled in a 2 :1 ratio and stratified over treatment/time post dose combinations.
In the treatment visit, subjects received either TBS-2 or placebo and underwent VTS/VSS at 0.5, 2.0, 4.0 and 8.0 hours after dosing. VTS/VSS lasted 20 minutes or until orgasm was reached, after which stimulation was stopped. Safety was monitored throughout the study and PK data were collected. The study was conducted with approval of the Medical Ethics Committee of the Academic Medical Center.
Results
TBS-2 was administered to 45 subjects, while 1 1 subjects received placebo. At baseline, none of the subjects experienced serious relationship problems, and none of the subjects had a depressive disorder. Placebo and TBS-2 groups were comparable with respect to sexual function, sexual distress, their current level of sexual desire, and sexual satisfaction in the relationship. Four (4) subjects receiving TBS-2 self-reported an orgasm during the study of which two (2) of the subjects were in the 4.0 hr arm and the other 2 subjects in the 8.0 hr arm. Two (2) subjects in the placebo group self-reported an orgasm. In the TBS-2 treated group, an additional eight (8) subjects reported sensations indicative of an actual orgasm at exit interview but had not recorded the orgasm during the treatment phase. No additional patients in the placebo arm provided such reports at exit interview. Therefore, a total of 12 patients (27%) in the TBS-2 treated group either self reported an orgasm at Visit 3 (4 patients)
or were suspected of having an orgasm following their exit interview (8 patients) versus only 2 patients (18%) in the placebo arm. This result did not achieve statistical significance.
Sexual Arousal, Genital Sensations, and Sensuality were measured at Visit 2 and Visit 3 using The Sexual Feelings and Affect Questionnaire. For the placebo group, no effect was observed with the difference between scores at Visit 2 and Visit 3 being small, and mostly negative. This indicates that feelings became less intense over visits. In the TBS-2 treated subjects a positive difference was observed for all items. Relative to placebo subjects, TBS- 2 treated subjects reported more intense sexual feelings (sexual arousal, genital sensations, and sensuality) at Visit 3, in which active treatment was received. The difference between the two treatment groups was statistically significant for sexual arousal (p=0.02, Mann- Whitney U test), but not for genital sensations (p=0.25, Mann-Whitney U test). Differences were marginally significant for feelings of sensuality (p=0.06, Mann-Whitney U test).
Sexual Desire was measured using the Acute Female Sexual Desire Questionnaire. Placebo treated subjects showed a small negative average difference between Visit 3 and Visit 2, indicative of a reduction in reported feelings of sexual desire at Visit 3. For the TBS2 treated subjects, an increase in sexual desire was observed at Visit 3. In addition, the TBS-2 treated subjects reported more feelings of sexual desire than the placebo group at Visit 3 and this difference between the two treatment groups was marginally significant (p=0.09, Mann-Whitney U test).
Genital Response was assessed using Vaginal Pulse Amplitude (VPA) which was measured using a vaginal photoplethysmograph. Median VPA mean was comparable between the placebo group and the TBS-2 group at Visit 2, whereas at Visit 3, median VPA mean was higher in the TBS-2 group. The difference in change in VPA mean between the placebo and TBS-2 groups was statistically significant (Mann-Whitney U test p=0.0493).
Both pre- and post-dose SHBG and albumin levels were comparable for the placebo and the TBS-2 treated subjects. Free and total testosterone were elevated in subjects receiving TBS-2, whereas no change in testosterone levels was observed in the placebo group. Testosterone levels post-dose were most pronounced in the 0.5 hr treatment arm. This pattern of findings suggests that TBS-2 produces a rapid onset elevation of free- and total testosterone levels without affecting SHBG and albumin levels. Nevertheless, largest effects
of TBS-2 were seen in the 2 and 4 hr groups, providing further evidence of a delay-effect of testosterone on sexual arousal.
TBS-2 was well tolerated. All AEs were mild and completely resolved at the end of the study. Physical examination, vital signs and clinical laboratory evaluations results did not reveal any additional clinically significant findings related to study treatment.
Conclusions
Although more patients treated with TBS-2 reported an orgasm or were suspected of having an orgasm following their exit interview when compared to placebo, this result was not statistically significant. However, subjects treated with TBS-2 did demonstrate a statistically significant improvement in Sexual Arousal and Genital response as well as a marginally significant result in terms of Sensuality and Sexual Desire. These findings are considered encouraging as they may be indicative of a true drug effect. More large scale research is certainly warranted.
5. ETHICS
5.1 Independent Ethics Committee (IEC) or Institutional Review Board (IRB)
The protocol of this study was submitted to the Medical Ethics Committee of the Academic Medical Center (AMC) and the CCMO (Competent Authority). The protocol was approved by the Medical Ethics Committee on March 3, 201 1 .
5.2 Ethical Conduct of the Study
The study was conducted according to the principles of the "Declaration of Helsinki" (as amended in Tokyo, Venice and Hong Kong, Somerset West, Edinburgh and Washington), the ΈΜΕΑ/CPMP position statement on the use of placebo in clinical trials with regard to the revised Declaration of Helsinki" and in accordance with the Guideline for Good Clinical Practice (CPMP/ICH/135/95 - 17th July 1996), and the pertaining Dutch law.
5.2.1 Patient Information and Consent
The study did not start until formal CCMO and AMC ethical approval was granted. Subjects who showed interest in study participation were given verbal and written explanations about the study by the investigators at the information visit. After they had provided written acknowledgement of informed consent to participate, a medical screening took place. Although the subjects were told they were free to leave the study at any time, an attempt
was made to recruit subjects who were likely to continue the study to completion. After approval of the subjects, their general practitioners were notified. All subjects were sufficiently insured to any injury arising from the study treatment, as required by the Dutch Directive of the Central Committee on Medical research involving Human Subjects (see Appendix 16.1 ).
7. INTRODUCTION
In the 4th edition of the Diagnostic and Statistical Manual of Mental Disorders, Text Revision (DSM-IV-TR) Female Orgasm Disorder (FOD) is defined as a persistent or recurrent delay in, or absence of, orgasm following a normal sexual excitement phase, causing marked distress or interpersonal difficulty (American Psychiatric Association, 2000). A review of research on the prevalence and incidence of FOD shows that this disorder is the second most common among women, closely following Hypoactive Sexual Desire Disorder (Lewis, Fugl-Meyer, Corona, Hayes, Laumann, Moreira, et al., 2010). Reported prevalence figures for FOD range from 16% to 28% in the United States, Europe, and Central/South America, but are as high as 30-46% in Asia. As with all other sexual disorders, the proportion of women with problems reaching an orgasm is double that for women with this difficulty who report associated distress (Graham, 2010). A factor that impacts the assessment and treatment of this condition is the relatively common occurrence of other, concurrent sexual dysfunctions (Segraves & Segraves, 1991 ). It has been estimated that among women with FOD, 31 % also report difficulties with sexual arousal, 18% with lubrication, 14% with desire, 12% with pain, and 0.9% with vaginismus (Nobre, Pinto-Gouveia, & Gomes, 2006). Because of this high level of co-morbidity, it is often hard to determine risk factors and treatment efficacy specific to FOD. It also means that most clients will present with a complex combination of problems, requiring a comprehensive assessment that takes into consideration the known correlates of FOD, as well as other relevant biopsychosocial factors, in an appropriate cultural context.
It is hypothesized that testosterone has central and peripheral effects on sexual function. Testosterone, the primary circulation androgen in women, is a naturally occurring steroid secreted by the ovaries and the adrenal glands. Contrary to the sudden drop in estrogen during menopause, serum levels of androgens fall gradually as women age primarily due to a decrease in the production of adrenal androgen precursors (Goldstat, Briganti, Tran,
Wolfe, & Davis, 2003). Testosterone plays a role in mood, body composition, bone mineral density and has central and peripheral effects on sexual function (Davis 1999, Goldstat et al., 2003). In the periphery, testosterone is required for nitric oxide to stimulate vasocongestion of clitoral tissue and vaginal lubrication during sexual arousal. Central effects are less well characterized. Testosterone stimulates dopamine release in various brain structures implicated in motivation and reward systems, including sexual desire. Testosterone was found to stimulate dopamine release in the medial preoptic area of the anterior hypothalamus under basal conditions and with sexual stimulation in rats (Halaris 2003). An fMRI study in healthy women of different ages showed a testosterone level dependent modulation of amygdala activity, suggesting that an age-related decline in androgen levels contribute to the decrease in amygdala reactivity (van Wingen, Pieters, Mattern, Verkes, Buitelaar, & Fernandez, 2009). Decreased amygdala reactivity in older women could be restored to levels of young women with intranasal exogenous testosterone. Exogenous intranasal testosterone thus may enhance healthy sexual functioning through complex brain mechanisms.
The use of androgens to increase women's sexual desire was reported in 1940. It was observed that a number of young, married women who formerly considered themselves "frigid" were able to experience "a marked increase in coital gratification, culminating in an orgasm" after testosterone propionate injections, and the effects wore off within several weeks after the discontinuation of the injections. In the 1980s, the role of androgens in maintaining sexual functioning was studied in oophrectomized women (Sherwin & Gelfand, 1987). In this three month prospective open-label study of 44 women, monthly injections of estrogen and testosterone increased rates of sexual desire, sexual arousal, and number of sexual fantasies. Furthermore, rates of intercourse and orgasm were higher in women treated with androgens and estrogen compared to the controls.
Over the two past decades, over 80 studies have been conducted in postmenopausal women with hypoactive sexual desire disorder using exogenous testosterone through the oral, transdermal, sublingual or parental route of administration with or without concomitant estrogen therapy, resulting in an increase in sexual desire, arousal, frequency of satisfactory sexual activity, pleasure and responsiveness (see Traish et al., 2009, for a review).
Fewer studies have been performed in premenopausal women with low sexual desire. Goldstat et al. (2003) studied the effects of transdermal testosterone therapy on well-being, mood and sexual function in eugonadal, premenopausal women presenting with complaints of low sexual desire. Testosterone therapy resulted in statistically significant improvements in the composite scores of the Psychological General Weil-Being Index, the Sabbatsberg Sexual Self-Rating Scale and the Beck Depression Inventory when compared with placebo. These effects were found while the mean total testosterone levels were in the low end of the normal range before treatment, and at the high end of the normal range during treatment. On the different sub-scales of the Sabbatsberg Sexuality Scale there was a significant effect of testosterone treatment on orgasm. This study suggests that although most previous studies with testosterone have addressed decreased sexual desire and especially included postmenopausal women, there are also measurable effects in premenopausal women both on general sexual wellbeing and on orgasm specifically.
Only a limited number of studies have investigated potential beneficial effects of testosterone on sexual response. In a placebo-controlled study in hypogonadotropic hypogonadal women, treatment with testosterone undecanoate, 40 mg per day orally during an 8-week period, enhanced genital arousal as measured by vaginal pulse amplitude (VPA) (Tuiten, Laan, Everaerd, Panhuysen, de Haan, Koppeschaar, & Vroon, 1996). Because women in this study swallowed the capsules each morning and the measurements were performed in the afternoon, it was assumed that this effect on genital sexual response would be caused by a time-dependent effect of testosterone. To test this hypothesis, eugonadal and sexually functional women were administered a single dose of testosterone sublingually (0.5 mg). Such pulsed testosterone delivery produced supraphysiological testosterone levels 15 minutes after treatment, with levels returning to normal within 1 .5 hours (Tuiten, van Honk, Koppeschaar, Bernaards, Thijssen, & Verbaten, 2000). Despite the return to baseline level of testosterone in the blood, the investigators found a delay- effect of testosterone on VPA 4.5 hours after testosterone administration (Tuiten et al., 2000, Tuiten, van Honk, Verbaten, Laan, & Everaerd, 2002). This finding was replicated in another laboratory (Heard-Davison, Heiman, & Kuffel, 2007). To date there is no clear explanation what causes this effect, but clearly testosterone influences processes further "down the line" that lead to these responses.
Fewer studies are available that directly demonstrate an enhancing effect of testosterone on orgasm. Among 300 women who received bilateral salpingo-oophorectomy and hysterectomy, 300 mg of testosterone patch showed improvements in FOD symptoms (Braunstein, Sundwall, Katz, Shifren, Buster, Simon, et al., 2005). Similarly, 10 mg of testosterone gel had positive effects on orgasm (Davis, Moreau, Kroll, Bouchard, Panay, Gass, et al., 2008). Additionally, tibolone, a synthetic steroid available in Europe, has shown improvements in orgasmic functioning (Kamenov, Todorova, & Christov, 2007).
At present, the only registered testosterone product for use in women is Intrinsa, a testosterone slow-release transdermal patch. Intrinsa is indicated for the treatment of hypoactive sexual desire disorder (HSDD) in bilaterally oophorectomised and hysterectomised (surgically induced menopause) women receiving concomitant estrogen therapy. Clinical studies using Intrinsa have shown enhancement of sexual desire and concern in post-menopausal women with HSDD (Braunstein et al., 2005; Buster, Kingsberg, Aguirre, Brown, Breaux, Buch, et al., 2005; Davis, van der Mooren, van Lunsen, Lopes, Ribot, Rees et al., 2006; Simon, Braunstein, Nachtigall, Utian, Katz, Miller, et al., 2005; Shifren, Braunstein, Simon, Casson, Buster, Redmond, et al., 2000; Shifren, Davis, Moreau, Waldbaum, Bouchard, DeRogatis, et al., 2006).
The product under investigation in this study, TBS-2, is a testosterone intranasal gel. Contrary to the transdermal administration, administration of the bioadhesive TBS-2 through the nasal route may allow for direct uptake of testosterone into the brain and rapid absorption into systemic circulation (Banks, Morley, Niehoff, & Mattern, 2009). The delivery of testosterone to the brain and systemic absorption is hypothesized to be effective in enhancing sexual desire and orgasm. In addition, TBS-2 may prove effective in alleviating anorgasmia in an "as needed" way, thus avoiding chronic exposure to testosterone.
Two clinical trials have been completed to date using TBS-2 in healthy females. Van Wingen et al. (2009) investigated whether the age-related decline in androgen levels is associated with reduced amygdala activity, and whether exogenous testosterone can restore amygdala activity. Healthy young and middle-aged women participated during the early follicular phase of the menstrual cycle, and amygdala responses to biologically salient stimuli were measured with functional magnetic resonance imaging (fMRI). Androgen levels were lower in middle-aged than young women, which was associated with
decreased amygdala reactivity. Endogenous testosterone levels correlated positively with amygdala reactivity across the young and middle-aged women. The middle-aged women received a single nasal dose of testosterone in a double-blind, placebo controlled, crossover manner, which rapidly increased amygdala reactivity to a level comparable to the young women. The enhanced testosterone levels correlated positively with superior frontal cortex responses and negatively with orbitofrontal cortex responses across individuals, which may reflect testosterone-induced changes in amygdala regulation. These results lend further neurobiological support for the important modulatory role of testosterone in attributing significance to emotional cues, although the cues in this study were not sexual in nature. The study also shows that intranasal exogenous testosterone is able to influence the amygdala in a consistent way.
A second study was recently completed which evaluated the pharmacokinetic profile of three doses of TBS-2 (300mcg, 900mcg and 1200mcg) in 32 female subjects diagnosed with hypoactive sexual desire disorder (HSDD) and anorgasmia (ANOR) (van Gorsel, Laan, Both, van Lunsen, van Gerven, Tkachenko, Dickstein, & Kreppner, 201 1 ). This study also assessed pharmacodynamic efficacy based on computer tasks with psychophysiological testing involving sexual imagery and visual sexual stimuli presentation along with genital arousal (vaginal pulse amplitude). TBS-2 was well absorbed following nasal administration of the three different doses. The maximum serum concentration was reached approximately 30 minutes to 2 hours post administration. Pharmacodynamic efficacy was evaluated using psychophysiological testing involving sexual imagery, visual sexual stimuli presentations and genital arousal (measured by vaginal pulse amplitude) as a dependent measure 30 minutes and 4.5 hours post administration of TBS-2. TBS-2 significantly increased genital arousal in ANOR women post TBS-2 administration. Trends in the subjective measures were observed in both the ANOR and HSDD cohort post TBS-2 administration.
This study explored the effect of TBS-2 on orgasm occurrence in primary and secondary anorgasmic women during sexual stimulation at four different time points post-dose. Sexual stimulation consisted of vibrotactile stimulation (VTS) in combination with visual sexual stimulation (VSS). Female VTS was tested in a previous study (Laan & van Lunsen, 2002) and showed that VTS in combination with visual sexual stimulation (VSS) reliably could trigger orgasm in healthy female volunteers with no self-reported difficulties in
achieving orgasm. Time to orgasm (TTO) showed an acceptable intra-subject variability. In the present study, a similar clitoral vibrator was used.
8. STUDY OBJECTIVES
8.1 . Primary Objective
The primary objective of this study was to evaluate the effect of a single dose of TBS-2 on the occurrence of orgasm at 0.5, 2.0, 4.0 or 8.0 hours post-dose in female subjects with primary or secondary anorgasmia. Placebo was used as a control at 0.5 hour post-dose.
8.2. Secondary Objectives
The secondary objectives of this study were to evaluate the safety of TBS-2, and the effect of a single dose of TBS-2 on time to orgasm (TTO) and quality of orgasm.
9. INVESTIGATIONAL PLAN
9.1 Overall Study Design and Plan
9.1.1 Description of Study Design
This randomized, single-blind placebo-controlled five arms parallel group study was conducted at a single study center to evaluate the effect of a single dose of TBS-2, 1200 meg (600 meg per nostril), on the occurrence of orgasm at 0.5, 2.0, 4.0 and 8.0 hours post- dose in female subjects with primary or secondary anorgasmia using vibrotactile stimulation (VTS) to the glans clitoris and visual sexual stimulation (VSS). The study was conducted with approval of the Medical Ethics Committee of the Academic Medical Center.
The study consisted of two screening visits and one treatment visit. At Visit 1 , all screening assessments were done and subjects were familiarized with the VTS technique and VSS. At Visit 2, women underwent VTS/VSS stimulation for 20 minutes to determine if they were anorgasmic during the VTS/VSS procedure. Women who did not achieve orgasm during the VTS/VSS procedure and met all other eligibility criteria were randomized to one of the five treatment arms. Women on hormonal or non-hormonal contraception methods were enrolled in a 2:1 ratio and stratified over treatment/time post dose combinations. In the treatment visit, subjects received either TBS-2 or placebo and underwent VTS/VSS at 0.5, 2.0, 4.0 and 8.0 hours after dosing. VTS/VSS lasted 20 minutes or until orgasm was reached, after which stimulation was stopped.
9.1.2 Selection of Study Population
Potential subjects aged 18-50 years, with regular menstrual cycle and with diagnosis of primary or secondary Female Orgasmic Disorder (Anorgasmia) according to DSM-IV criteria and with the current episode at least 24 weeks in duration by the Screening Visit, were eligible for study entry, provided all other inclusion/exclusion criteria were satisfied.
Subjects were recruited from the general population through advertisement in local newspapers. Subjects were pre-screened by telephone using a structured interview with questions regarding sexual complaints and general health. Subjects who seemed eligible were invited for a screening visit. Written informed consent was obtained at the start of the first screening visit.
9.1 .2.1 Screening Questionnaires
Two questionnaires were used to assess any depressive symptoms (BDI-II) and relationship quality (MMQ).
Beck Depression Inventory-ll (BDI-II, Protocol, appendix 2): To index the current level of depressive symptoms, the 21 -item BDI-II was administered (Beck, Steer, & Brown, 1996), Dutch adaptation (Van der Does, 2002). The range for the BDI total score is 0-63, with higher scores indicating more depressive symptoms. Subjects with a score of >14 on the BDI total score were excluded.
The Maudslev Marital Questionnaire (MMQ, Protocol, appendix 3): the MMQ (Crowe, 1978) is a 20-item self-report instrument measuring dissatisfaction with the general relationship, with the sexual relationship, and with life in general. The MMQ has shown good reliability and validity. The psychometric qualities of the Dutch version of the MMQ were also found to be satisfactory (Arrindell, Boelens, & Lambert, 1983). Higher scores represent larger dissatisfaction. Subjects with a score on the MMQ-M subscale of > 20 were excluded.
9.1 .2.2. Covariate Questionnaires
Five questionnaires were administered to characterize the sample in terms of symptom severity and sexual distress, as a check on randomization of subjects into one of the five treatment arms, and to be able to study the relationship with the constructs described below with response to treatment.
Female Sexual Function Index (FSFI, Protocol, appendix 4): The level of the woman's sexual functioning was assessed by the FSFI (Rosen, Brown, Heiman, Leiblum, Meston,
Shabsigh, et al. 2000). The FSFI is a self-administered questionnaire that consists of 19 questions. The scale contains six domains: desire, arousal, lubrication, orgasm, satisfaction, and pain. The range for the total score is 2-36, with lower scores representing worse sexual function. The psychometric quality of the FSFI is satisfactory (Wiegel, Meston, & Rosen, 2005). Based on a Dutch sample consisting of approximately 350 women with and without sexual complaints, the internal consistency and stability of the FSFI were found to be satisfactory-to-good. The FSFI's ability to discriminate between sexually functional and dysfunctional women was excellent as was the ability to predict the presence or absence of sexual complaints (ter Kuile, Brauer, & Laan, 2006).
Female Sexual Distress Scale- Revised (FSDS-R, Protocol, appendix 5): The woman's level of personal distress due to sexual dysfunction was assessed by the FSDS-R (Derogatis, Clayton, Lewis-D'Agostino, Wunderlich, & Fu, 2008). The items inquire about negative feelings and problems that were bothersome or caused distress during the past 30 days. Reliability and validity of the FSDS (12-item version) was evaluated in different samples of sexually functional and dysfunctional women (Derogatis, Rosen, Leiblum, Burnett & Heiman, 2002). For the FSDS, results indicated a unidimensional factor structure, a high degree of internal consistency, and test-retest reliability. The FSDS showed a high degree of discrimination between sexually dysfunctional and functional women in each of its three validation studies. Results in a Dutch sample supported the unidimensional structure of the FSDS and its reliability and psychometric validity (ter Kuile, Brauer, & Laan, 2006). An additional question (Question 13) has been added to the validated FSDS. This question is about distress specifically related to sexual desire. The maximum total score of the FSDS-R indicating the maximum level of sexual distress is 52.
index of Sexual Satisfaction (ISS, Protocol appendix 6): The woman's level of sexual satisfaction was assessed by the ISS (Hudson, Harrison, & Crosscup, 1981 ). This 25-item questionnaire asks subjects to evaluate various aspects of their sexual relationship, leading to a sum score that can range between 0 and 100. Higher scores correspond to greater sexual satisfaction. This measure has been shown to have good face, convergent, and discriminant validity with various samples. Example items are "I feel that my partner enjoys our sex life," "I think that sex is wonderful," and "My partner is sexually very exciting." The
ISS was translated into Dutch for an earlier study initiated by this Sponsor, psychometric data are not yet available.
Sexual Desire Inventory-ll (SDI-II, Protocol, appendix 7): The level of the woman's sexual desire was assessed by the SDI-II (Spector, Carey, & Steinberg, 1996). The SDI-II consists of two seven-item self-report scales: the Dyadic Sexual Desire scale, which measures an individual's desire for sexual activity with a partner, and the Solitary Sexual Desire scale, which measures an individual's desire for autoerotic sexual activity. The two subscales were internally consistent (Cronbach's a: Dyadic scale = 0.86; Solitary scale = 0.96). Psychometric data for the Dutch translation are not yet available.
Sexual Excitation/Sexual Inhibition {SESII-W, Protocol, appendix 8): The SESII-W (Graham, Sanders, & Milhausen, 2006) was used to assess individuals' propensity for sexual excitation and sexual inhibition. It consists of 36 items, referring to stimulus situations that could affect sexual inhibition and sexual excitation or to general statements about arousability and inhibition. The instructions ask women to report what would be the most typical reaction now or how they think they would respond if the item does not apply to them. Items are rated on a 4-point Likert-rating scale, from "strongly disagree" to "strongly agree." The SESII-W has eight lower-order factors, which in turn load on two higher-order factors, Sexual Excitation and Sexual Inhibition. The questionnaire shows good test-retest reliability and convergent and discriminant validity and Sexual Excitation and Sexual Inhibition appear to be relatively independent factors. The translated list is already in use in the Netherlands, and psychometric properties are currently being investigated in a large sample of women with and without sexual complaints (Bloemendaal & Laan, 2012).
9.1.3 Inclusion Criteria
• Heterosexual women with a regular menstrual cycle aged 18-50 years.
• Diagnosis of primary or secondary Female Orgasmic Disorder (Anorgasmia) according to the DSM-IV criteria. The current episode must be at least 24 weeks in duration by the Screening Visit. Subtype should be generalized and not due to etiological factors that would prohibit treatment response (e.g., depression, alcoholism, surgery, injury). HSDD as a co-morbid disorder is allowed only if it began after the anorgasmia.
• BMI < 35.
• Women in a steady relationship of at least 12 months.
• Heterosexual women of childbearing potential must agree to use one of the following reliable birth control methods:
Surgically sterile
Intrauterine device (IUD) in place for at least 3 months prior to study initiation Barrier method (condom use by partner)
• Stable hormonal contraceptive for at least 3 months prior to study and through study completion
• Normal thyroid function. Physiological prolactin concentration.
• Normal otorhinolaryngologic examination.
• Able to understand and provide written informed consent.
• Inability to achieve orgasm on the VTS/VSS at screening.
• A score of > 1 on the VTS/VSS Appreciation Questionnaire at Visit 2.
Exclusion Criteria
• History of any other clinically relevant psychiatric disorders that could impact sexual function, risks patient's safety, or may impact compliance as assessed by the MINI. This includes bipolar disorders, psychotic disorders, severe anxiety, eating disorders, antisocial personality disorders, etc.
• History of Major Depressive Disorder within six (6) months prior the Screening Visit or a score of >14 on the BDI-II.
• Subjects who meet DSM-IV criteria (APA) for Sexual Aversion Disorder, Substance- Induced Sexual Dysfunction, Dyspareunia (not caused by inadequate foreplay stimulation or alleviated by lubricants), Vaginismus, Gender Identity Disorder, Paraphilia, or for Sexual Dysfunction Due to a General Medical Condition.
• □ Subjects experiencing relational discord as indicated by a score of > 20 on the MMQ-M, a subscale of the MMQ.
• Subjects with known active pelvic inflammatory disease, urinary tract or
vaginal infection/vaginitis, cervicitis, interstitial cystitis, vulvodynia, or significant vaginal atrophy.
• Subjects who are breast feeding or have breast fed within the last six (6)
months prior to the screening Visit.
• Subjects who are pregnant (by serum pregnancy test at the Screen Visit).
Women who have been pregnant within the last 12 months prior to the
Baseline Visit, unless the anorgasmia was already present before pregnancy and delivery.
Treatment with systemic glucocorticoids.
Treatment with sex steroid hormones such as androgens, estrogens other than in low dose combined ET/P, or gestagens (e.g. anabolic steroids, DHEA, Premarin® (conjugated equine estrogens)).
Treatment with thyroid hormones (only for stable replacement therapy).
Significant intercurrent disease of any type, in particular liver, kidney, or heart disease, any form of diabetes mellitus (subjects with antacids or treated hyperlipidaemia or treated hypothyroidism will not be excluded provided they have been stable on their drug dose for at least six (6) months).
History of nasal disorders (e.g., seasonal or perennial allergic rhinitis, atrophic rhinitis, polyposis, abuse of nasal decongestants, clinically relevant nasal septum deviation, recurrent epistaxis) or sleep apnea.
Subjects with a history of dementia or other neurodegenerative diseases, organic brain disease, stroke, transient ischemic attacks, brain surgery, significant brain trauma, multiple sclerosis, spinal cord injury, peripheral neuropathy, and epilepsy (febrile seizures limited to childhood do not exclude subjects).
History of cancer, excluding basal and squamous cell carcinoma of the skin curatively treated by surgery.
History of severe or multiple allergies, severe adverse drug reaction or leucopenia.
History of abnormal bleeding tendencies or thrombophlebitis unrelated to venepuncture or intravenous cannulation.
History of DVT.
History of Hepatitis B, a positive test for Hepatitis B surface antigen, a history of Hepatitis C, a positive test for Hepatitis C antibody, a history of HIV infection or demonstration of HIV antibodies.
• Regular drinkers of more than three (3) units of alcohol daily (1 unit = 300 ml beer, 1 glass wine, 1 measure spirit).
• History of, or current evidence of, abuse of alcohol or any drug substance, licit or illicit; or positive urine drug and alcohol screen for drugs of abuse and alcohol.
• Difficulty in abstaining from OTC medication (except occasional
paracetamol/aspirin) for the duration of the study.
• Poor compilers or subjects unlikely to attend study visits.
• Receipt of any drug as part of a research study within 30 days of initial dose administration in this study.
• Blood donation (usually 550 ml) within the 12 week period before the initial study dose.
9.1.5 Removal of Patients from Therapy or Assessment
Subjects could leave the study at any time for any reason if they wished to do so without any consequences. The responsible investigator could also withdraw a subject if continuing participation was, in her opinion, deleterious for the subject's wellbeing. Subjects could also be withdrawn in case of protocol violation and non-compliance.
Two subjects withdrew consent, one was lost to follow up, one was ineligible to continue, and three were non-compliant.
9.2 Treatments
9.2.1 Treatments Administered
The dose, 1200 meg, was based on the results of TBS-2-PK-2010-01 study demonstrating a suitable PK profile for a PRN product to treat anorgasmia and an acceptable safety profile (van Gorsel et al., 201 1 ).
In the current study, women received a total of 1200 meg of TBS-2 (600 meg per nostril). The placebo was identical to the active gel with the exception that an increased quantity of castor oil replaces the active pharmaceutical ingredient testosterone.
9.2.2 Identity of Investigational Products
The investigational product in this trial was TBS-2, an intranasal testosterone gel. A description of its physical, chemical, pharmaceutical properties and formulation can be found in the Investigator's Brochure.
(1 ) Active drug;
ame of the drug; TBS-2 (Syringes are pre-fied to contain 800 meg,
of testosterone/syringe)
Pharmaceutical form; Gel for nasal administration
Content Active ingredient: testosterone
Excipients silicon dioxide, castor oil, Labrafir*
Mode of administration: intranasal as a single dose to each nostril
Manufacturer: Haupt P arrna Amareg
Batch number: 2264
Storage conditions: Between 20 ~ 25 SC.
{2} Placebo:
Name of the drug; TBS-2 Placebo Gel (Syringes are pre-fied to
contain only the gel without testosterone)
Pharmaceutical form: Gel for nasal administration
Content: Active ingredient: None
Exctpients: silicon dioxide, castor oil, LabrafB8
Mode of administration: intranasal as a single dose to each nostril
Manufacturer: Haupt Ptiarma Amareg
Batch number: 2265
Storage conditions: Between 20 25 «C.
9.2.3 Randomization
Women on hormonal and non-hormonal contraception were enrolled in a 2:1 ratio and stratified over treatment/time post dose combinations. Women who did not achieve orgasm during the VTS/VSS, and who did not report the VTS/VSS procedure to be very unpleasant as evidenced by a score >1 on the VTS/VSS Appreciation Scale (VAQ) and met all other eligibility criteria, were randomized to one of the five treatment arms. Randomization was done using a minimization technique to prevent imbalance in the distributions of women on hormonal and non-hormonal contraception methods. Subjects received either a single dose of TBS-2 or a single dose of placebo and were subjected to VTS/VSS at 0.5, 2.0, 4.0 or 8.0 hours post-dose as specified below:
TBS~2/0,5 hour post-dose VTS VSS
ieS-2 2.0 hours posf-ctosi VTS/VSS
TBS»:2 4.0 hou s post-dose- VTS V
TBS-2$L0 hours os!-etase VTS VSS
Placebo/ O hour post-dose VTS VSS
9.2.4 Blinding
For all subjects, the study was blinded.
9.2.5 Prior and Concomitant Therapy
Subjects were instructed not to use concomitant medication (with the exception of occasional paracetamol) during the study period. If a subject used other medication, it was assessed if this could influence the outcome measures or kinetics of the study drugs. If this was the case, the subject was either excluded or the study day was postponed.
9.2.6 Treatment Compliance
All study medication was administered under direct supervision, according to standard operating procedures. Any study procedure that did not take place adequately or within 10% of the planned time interval was recorded with a note in the CRF explaining why this was the case.
9.3 Pharmacokinetics, Pharmacodynamics and Safety
During the study, several measures for safety, pharmacokinetics and pharmacodynamics were performed. An overview of the study is presented in the flow chart below. A further description of the different outcome measure and their rationale can be found in the following sections.
9.3.1 Flowchart
Flowchart of time points and measurements
 ) Informed consent was signed prior to Screening Visit.
) Informed consent was signed prior to Screening Visit.
) Urinalysis Specific gravity, PH, Protein, Glucose, Ketones, Nitrite, bilirubin, urobilinogen Leukocytes,
) Hematology: hemoglobin, hemoglobin A1 c, hematocrit, MCV, MCHC, RBC, WBC & differential
) Clinical Chemistry profile (Na/K, glucose, urea, creatinine, total bilirubin, albumin, calcium,
5) Thyroid Panel: TSH, total and free triiodothyronine, total and free thyroxine
6) Urine Drug Screen: Amphetamines, Barbiturates, Benzodiazepines, Cannabinoids, Cocaine, Opiates, MDMA.
7) Pregnancy Test: Serum Beta-Subunit Human Chorionic Gonadotrophin - Qualitative at Screening. Urine test before treatment and serum test before discharge at Visit 3.
8) Hormone Assays: Estradiol, Free Testosterone, Free Testosterone (percent), Follicle Stimulating Hormone, Luteinizing Hormone, Prolactin, Progesterone, Sex Hormone Binding Globulin, Total Testosterone, Dehydroepiandrosterone Sulfate
9) Dosing : TBS-2/Placebo, Day 1 .
10) Psychophysiological Testing - performed in a psychophysiological laboratory - involved the VSS/VTS procedure that is described on page 23.
9.3.2 Pharmacokinetics
Blood samples for concentration measurements of plasma total testosterone, plasma free testosterone and SHBG were taken at the following time points (at Visit 3): prior to administration of the nasal gel and immediately after VTS/VSS-testing.
9.3.3 VTS/VSS Efficacy Testing
VTS/VSS testing consisted of assessment of genital response (vaginal pulse amplitude, VPA) and subjective sexual arousal (as measured by the Sexual feelings and Affect Questionnaire, SAQ, and Acute Female Sexual Desire Questionnaire, AFSDQ) during VSS/VTS-testing.
Genital response (VPA): VPA was measured using a vaginal photoplethysmograph developed by Bert Molenkamp (Technical Support, Department of Psychology, University of Amsterdam) based on instruments initially developed by Sintchak and Geer (1975). The light source (3mm LED, D=620nm) and optical sensor (Texas Instruments TSL250) were produced in batches of 100, resulting in all photoplethysmographs used in this study having nearly equal electronic characteristics. A signal-conditioning amplifier separated the VPA from the direct current component using a 12 dB/octave, 0.7-Hz filter. Additional filtering for VPA was 24 dB/octave, 0.4 Hz high-pass. The VPA signal was digitalized at 100 Hz with a Keithley KPCI3107 A/D converter, running on a Windows 2000 PC system. Depth of the probe and orientation of the light source were controlled by a device (a 9 x 2cm FDA-
approved perspex plate) attached to the cable within 5cm of the optical sensor. Subjects were instructed to insert the probe until the plate touched their labia. The probe and plate were sterilized according to standard department protocol.
VTS/VSS: The vibrotactile stimulation (VTS) consisted of a clitoral vibrator with a rubber stopper of 2 cm in diameter mounted on a flexible metal strap lined with washable lycra cloth. It was designed such that minimal interference on physiological measurements of genital response occurs. Before the start of VTS/VSS the frequency of the vibration was set at 6 on a 1 -10 frequency scale which could be altered by the subjects themselves at the familiarization session at Visit 1 ; they were encouraged to increase frequency of the vibrator so as to provide optimal stimulation. Visual sexual stimulation (VSS) consisted of 20 minutes of woman-friendly erotic film with sexually explicit material (Laan, Everaerd, van Bellen, & Hanewald, 1994).
The non-erotic and erotic films were presented digitally. Two separate film sequences with different non-erotic and erotic film excerpts were used in a random order such that either the first film sequence was used in Visit 2 and the second film sequence in Visit 3, or vice versa. Vaginal pulse amplitude (VPA) was recorded starting from 8 minutes before VTS/VSS until orgasm was reached, or until 20 minutes after start VTS/VSS. VPA was used to support subjects' report of orgasm. Orgasm was characterised, among other things, by involuntary contractions of the pelvic floor muscles. These contractions are visible as movement artefacts in the VPA signal. Subjects were carefully instructed of what should be considered an orgasm using the following instruction: The clitoral vibration and watching the film may lead to very strong sexual arousal, which can bring about an orgasm. You may notice that the muscles in your groin contract spontaneously. This is usually accompanied by a very pleasurable feeling. If you have not had an orgasm before, this may be an overwhelming experience. Don't try to block the feeling, it will fade away by itself.'
All VTS/VSS testing was done individually.
Quality of Orgasm Questionnaire (QOQ, Protocol, appendix 9): This questionnaire was designed specifically for this study and consists of 4 items, with items 2-4 being measured on a 1 -5 scale. Item 2, orgasm pleasure, is the secondary endpoint, items 3 and 4 were designed to tap perceived orgasm intensity and perceived ease of orgasm.
Acute Female Sexual Desire Questionnaire (AFSDQ, Protocol, appendix 10): prior to and following psychophysiological testing subjects filled out the Acute Female Sexual Desire Questionnaire (Laan, Brauer, Janssen, Hahn, & Heiman, in preparation). This questionnaire assesses sexual interest in erotic stimuli and has shown to discriminate between women with acquired HSDD and sexually functional controls.
Sexual feelings and Affect Questionnaire (SAQ, Protocol, appendix 1 1 ): prior to and immediately after an erotic stimulus subjects filled out a questionnaire measuring sexual feelings and affect during sexual stimulation, consisting of 5 scales: sexual arousal (Cronbach's a=0.87); genital sensations (Cronbach's a=0.96); sensuality (Cronbach's a=0.73); positive affect (Cronbach's a=0.93); and negative affect (Cronbach's a=0.65) (Heiman, 1977). Each question was preceded by the sentence: "During the film, I felt:" after which a positive, negative, physical or sexual experience was described, for instance: "pleasant", "worried", "genital pulsing or throbbing", "sexually aroused". The items were measured on a 1 (not at all) to 7 (intensely) scale.
VTS/VSS Appreciation Questionnaire fVAQ, Protocol, appendix 12): After the AFSDQ, subjects filled out the VTS/VSS Appreciation Questionnaire (VAQ). The VAQ was designed specifically for this study to tap any unpleasant feelings that may result from the VTS/VSS procedure. Subjects who scored 1 (very unpleasant) during the VTS/VSS screening session were excluded after Visit 2, under the assumption that a sexual stimulation method that was experienced as very unpleasant will not generate an orgasm. The questionnaire was also administered during Visit 3 (TBS 2/Placebo treatment) to explore whether appreciation of the procedure changed with repeated exposure.
Exit interview: After each VTS/VSS visit, a member of the study personnel performed a structured exit interview with the subjects. This interview entailed questions about the subject's'experience with the testing set up, including the vibrator, the VPA-probe and the erotic movies; whether any of these aspects (negatively) influenced them; whether there was a difference between the feelings of sexual arousal they experienced in this setting compared to what they'd experienced thus far; whether (after Visit 3) their feelings at this visit differed from those at Visit 2. In case, at Visit 2, orgasm was not reached, they were asked two additional questions: 1. what they think they would need to be able to experience orgasm in this setting, and 2. whether they felt that they would have had a greater chance at
orgasm with longer stimulus duration. These two additional questions were asked to help understand, in case orgasm rate would turn out to be low, what, from the perspective of the subjects themselves, could have explained this low orgasm rate.
VTS/VSS Procedure: In order for the subjects to be confident and at ease with the workings and use of the vibratory equipment and timing device, at Screening Visit 1 they were shown the equipment at a familiarization test. After explaining how the equipment worked, they were asked to perform the VTS/VSS test as described here. They were familiarized with the content and nature of the material used for visual sexual stimulation and shown the vibrotactile device. They were asked to apply the vibrator and to alter the frequency that was set at a predetermined level of 6 to a level they found sexually arousing. They were then presented with 5 minutes of VTS/VSS stimulation. The optimum settings of the vibrator frequency for that subject were noted and recorded; these settings were used in subsequent visits involving VTS/VSS.
After placing the vibrator against the clitoris and inserting the VPP probe the subjects completed the AFSDQ and SAQ presented on the television monitor. This was followed by an 8-min neutral film, the last 2 minutes of which represented the baseline recording. Then the erotic film (VSS) and vibrator (at the frequency that was determined at the familiarization session at Visit 1 ) were started simultaneously. In case of an orgasm, the subjects was required to press a button allowing for an automatic recording of orgasm duration. Immediately following orgasm, the subject again pressed this button allowing for an automatic recording of orgasm duration. In case orgasm was reached, subjects completed the QOQ, All subjects were asked to again complete the SAQ and AFSDQ after VTS/VSStesting.
9.3.4 Safety
Qualified medical staff were always present during the subjects' stay at the study center. Safety analysis were performed on all subjects by monitoring adverse events, clinical laboratory test results, hormone profiles, physical and ENT examinations, and vital signs. All symptoms noted by subjects were recorded in the CRF. All adverse events were recorded by the investigator from the screening visit until discharge from the study center. If adverse events were ongoing at time of discharge, subjects were followed-up until the event had resolved.
9.3.4.1 Blood Sampling and Sample Handling
Blood samples for safety analysis (chemistry, hematology) and hormones other than PK analyses were performed by the Central Laboratory for Clinical Chemistry of the Academic Medical Center, Amsterdam, the Netherlands. Samples were analyzed within 48 hours of draw time and results were reported back to investigators on an ongoing basis.
A detailed description of sample handling and processing can be found in the Central Laboratory Services Manual.
Venous blood samples for PK analysis were centrifuged at 2500g for 15 minutes at room temperature within 30 minutes of blood draw. The plasma was transferred into the 2 aliquots (0.75mL aliquot tube red/green). The aliquots were stored in the freezer at <-18°C and shipped frozen in batches at the end of the study. Frozen shipments on dry ice were delivered at the Analytical Biochemical Laboratory (ABL, Assen, the Netherlands) for analysis. The two aliquots per time point were shipped in separate batches. At ABL samples were analyzed using a validated, highly sensitive and reliable liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) method for the determination of T in human plasma. With a calibration range of 0.0500 - 20.0 ng/mL this assay proved to be most adequate for the determination of T in human male as well as female plasma.
9.4 Quality Assurance
The study was conducted in compliance with the pertaining CRO Clinical Research Unit's (CRU) Standard Operating Procedures (SOP) and standards of Good Clinical Practice. Data management and statistical analysis were performed in compliance with the CRU's SOPs and Quality Assurance procedures.
9.5 Statistical Methods Planned in the Protocol
9.5.1 Statistical and Analytical Plans
Before the analyses were executed, a detailed statistical analysis plan was written. The following items were described in this plan.
It was planned to summarize demographic variables using descriptive statistics; to summarize categorical variables by means of frequency counts, whereas for continuous variables descriptive statistics (i.e. mean, standard deviation, minimum, maximum, median, number of subjects and number of subject with missing values) were planned.
All variables were to be presented per treatment arm and per treatment group.
For the analysis of this study, 2 populations were planned:
) Intent to Treat population
) Safety population
Intent-to- Treat population
This population was defined as all subjects who were randomized and received study medication. In addition, at least one efficacy assessment had to be available.
Safety population
The safety population was defined as all subjects who received at least one dose of study medication.
The analysis of efficacy was to be based on the intent-to-treat population, whereas the safety analysis was to be based on the safety population.
Prior to the analysis of the study, the following were defined as protocol violations:
Major protocol violations:
• Violation of at least one of the in- and/or exclusion criteria
• VTS/VSS test not performed
• Use of any of the following exclusionary medications: Systemic glucocorticoids, Sex steroids hormones such as androgens, estrogens and gestagens (e.g. anabolic steroids, DHEA, Premarin® (conjugated equine estrogens))
• Positive serum pregnancy test at visit 3 (after dosing)
Minor protocol deviations:
• Minor changes in thyroid hormone levels during the study.
A number of variables were defined before the analysis started:
• Age:
Age is defined as the (date of screening visit MINUS date of birth) divided by 365.25
• Time to orgasm
Time to orgasm (TTO) using VTS/VSS. TTO is defined as the time from onset of VTS/VSS stimulation until the start of orgasm. Subjects were instructed to press a button at the start of an orgasm. The TTO was automatically determined by the computer program which also recorded Vaginal Pulse Amplitude (VPA). The TTO is expressed in minutes. In the event that a subject did not press the button at the point of orgasm but indicated at exit- interview that she did have an orgasm, the occurrence of orgasm was, whenever possible,
corroborated by movement-artifacts in the VPA signal indicative of the involuntary pelvic floor muscle contractions that typically accompany orgasm. In these cases, TTO was calculated as time of onset of VTS/VSS stimulation until the start of movement artifacts in the VPA signal that are indicative of orgasm.
• SAQ-scores / AFSDS-score
For the SAQ, the score on each individual item was recorded by the computer program mentioned above. The SAQ consists of 5 scales, items belonging to each scale were averaged using a computer-based algorithm to produce 5 SAQ scores (Laan, van Driel & van Lunsen, 2008). The AFSDS-score consists of the average of all 1 1 AFSDS items which were obtained using a computer-based algorithm.
• VPAmax and VPAmean scores
VPA was sampled continuously during the entire VTS/VSS assessment until, and including, orgasm or until the end of the 20 minute VTS/VSS procedure. Data were entered into a data reduction program that deletes movement artefacts, computes peak-to-through amplitude of each VPA pulse, and produces 30-second means. Baseline was calculated as the mean of the last 3 minutes of neutral film presentation, immediately prior to VTS/VSS start. The maximum 30-second mean from VTS/VSS start to orgasm or of the entire 20 minutes VTS/VSS presentation (minus mean preceding baseline) was recorded (VPAmax). In addition, the mean of the 30-second means from VTS/VSS start to orgasm or of the entire 20 minutes VTS/VSS presentation (minus mean preceding baseline) was calculated ( VPAmean). Both variables were used for analysis.
• Exit interview
The following questions were asked at the exit interview after Visit 2 and Visit 3.
1 . What did you think of the session?
2. Did the probe or vibrator-device bother you at any point?
3. What was it like for you to use the clitoral vibrator (pleasurable, arousing?) What do you think about how the clitoral vibrator was placed; would you have preferred to have it done another way?
4. Is there a difference between the feelings of sexual arousal you experienced here and what you usually experience at home?
5. (after V3): How were your experiences during this visit compared to visit two, in terms of experienced feelings?
6. Do you have any questions/remarks.
Other questions if no orgasm was obtained (after Visit 2)
7. What do you think you would need to be able to experience orgasm in this stimulus situation?
8. (If duration of stimulation was not mentioned): Do you think that it would have helped if the sexual stimulation had been of longer duration? If so, how much longer?
9.5.1.1 . Analysis of Efficacy
Occurrence of orgasm
For the analysis of the primary objective, a frequency table with the occurrence of orgasm per treatment group at Visit 3 was planned. A X2 test was planned to analyze the data.
Time to orgasm
A survival analysis was planned to compare time to orgasm (TTO) between the treatment and placebo group, using the Kaplan Meier method. A graphical presentation was planned as well as a presentation of descriptive statistics on TTO per treatment group and treatment arm.
A non-parametric Log-Rank test was planned to compare TBS-2 treated and placebo- treated subjects. Before start of this analysis, the assumption of proportional hazards had to be verified.
SAS procedure LIFETEST was planned to obtain the Kaplan-Meier estimate of the survivor function. For subjects not reaching an orgasm, data are considered censored, with censored survival time set to zero.
Presentation of estimated median survival time (including the 95% confidence limits) was planned per treatment group, as well as the range (excluding censored observations).
Quality of Orgasm
Tabulation of frequency counts on item 2 (orgasm pleasure) of the Quality of Orgasm Questionnaire was planned per treatment arm and treatment group. It was planned to test
the difference between the TBS-2 treated and the placebo-treated group using a Mann- Whitney U test.
SAQ /AFSDQ
For each of the five SAQ scales mean response was calculated for each treatment group at Visit 2 and Visit 3. For the AFSDQ, an AFSDQ total score was calculated consisting of the mean of all items. The change in SAQ and AFSDQ scores per subject at Visit 2 and Visit 3 was also determined. Descriptive statistics are presented per treatment group and treatment arm. Statistical significance between groups was determined using the Mann- Whitney U test.
VTS/VSS Appreciation Scale (VAQ)
Results from the VAQ were tabulated for each treatment group at Visit 2 and Visit 3. Descriptive statistics are presented per treatment group. Statistical analysis of the VAQ was not planned.
VPAmax and VPAmean
The mean response for VPAmax and VPAmean was calculated for each treatment group at Visit 2 and Visit 3. The change in VPAmax and VPAmean scores per subject at Visit 2 and Visit 3 was also determined. Descriptive statistics are presented per treatment group and treatment arm. For both variables, statistical significance between groups was determined using the Mann-Whitney U test.
Testosterone, SHBG and Albumin
Testosterone, SHBG an albumin levels were analyze at Visit 3 before and after dosing. The mean values for free testosterone, total testosterone, and albumin were calculated for each treatment group and treatment arm.
Exit Interview
Data collected at the exit interview were translated and are presented by data listings.
9.5.1 .2. Safety Analysis
In order to assess the safety of TBS-2, the following parameters were assessed:
Adverse events
Tables summarizing all adverse reactions based on MedDRA coding including severity and relationship to study drug are presented.
Laboratory Results
Hematology, Clinical Chemistry & Urinalysis
Tables summarizing all laboratory test values are presented for each treatment group. In cases where laboratory values were outside of the reference range, separate listings are provided and the clinical significance of these findings was evaluated.
Urinalysis
Specific gravity, glucose, ketones, bilirubin, pH, urobilinogen, leukocytes, nitrites were assessed at Visit 1 and Visit 3. Tables summarizing the urinalysis test values are presented for each treatment group.
Thyroid panel
TSH, total and free triiodothyronine, total and free thyroxin were assessed at Visit 1 and 3. Tables summarizing the thyroid panel are presented for each treatment group at each time point. In cases where parameters were ± 20% of their reference range, separate listing are provided and the clinical significance of these findings was evaluated.
Hormone profiles
Estradiol, free testosterone, free testosterone (percent), follicle stimulating hormone, luteinizing hormone, prolactin, progesterone, sex hormone binding globulin, total testosterone, and dehydroepiandrosterone sulphate were assessed at Visit 1 and 3. In cases where parameters were ± 20% of their reference range, separate listing are provided and the clinical significance of these findings was evaluated
Physical and otorhinolaryngologic examinations
Results of physical and ENT examinations are provided in the data listings.
Hepatitis B, C and HIV testing
Data on viral tests are presented in data listings.
Vital signs
Data on pulse rate, temperature, respiration rate and blood pressure were recorded at Visit 2 and 3. At Visit 3, vital signs were assessed prior to dosing and after completion of VTS/VSS. were collected. Descriptive statistics are presented for each time point and per treatment group, as well as individual data listings.
9.5.1.3 Data Analysis and Presentation of Findings
A 5% level of significance with two-sided testing is used. In case of missing data no imputation techniques are used.
Individual patient data listings are included in section 16.4. These listings include data of all screened subjects.
Descriptive statistics for continuous variables are presented (i.e. n, mean, standard deviation, minimum, maximum, median and number of subjects with missing data). For categorical variables frequency counts are presented. In case of (laboratory) values with a '>' sign or a '<' sign, the actual value will be used in computations, i.e. in case of a laboratory result '<2' the value 2 is used for computations.
9.5.2 Determination of Sample Size
The sample size calculation for this study was based on the assumption that a subject in the placebo group will have a 0% chance of obtaining an orgasm, whereas in the TBS-2 group, the likelihood of obtaining an orgasm was estimated at 50%.
A two group X2 test with an alpha 0.05 two-sided significance level will have 80% power to detect the difference between a patient in the placebo group and a patient in any of the four active treatment groups when the sample size in each group is 12. Consequently, it was planned to randomize a total number of 60 patients.
9.5.3 Changes in Conduct of the Study or Planned Analyses
Changes were made to the VTS/VSS instructions and the statistical analysis of the study. In the sex laboratory, detailed instructions on the use of the device were provided. These instructions, provided to the first 5 subjects (#1001 , #1002, #1003, #1008, #1009), are described in section 9.3.3.
Changes in the VTS/VSS instructions
After the 5th subject, more specific instructions were provided to the subjects because orgasm rates lagged behind of what was expected at Visit 2. To find optimally pleasurable frequency settings for the clitoral vibrator, the vibrator was now set at maximum (level 10). If the subject experienced this frequency as unpleasurable, she was allowed to reduce the frequency. This procedure produced overall higher vibrator frequency settings (see Listing 14). In addition to the sexual history, the subjects with primary anorgasmia were asked to describe their expectation of an orgasm; the subjects with secondary anorgasmia were asked to describe a previous orgasm. Both groups were asked whether they had associated negative cognitions or fears. Subsequently, the Masters & Johnson (1966) sexual response cycle was explained to them to diminish the likelihood that primary anorgasmic women's
uncertainty about what to expect in terms of sexual feelings would prevent them from experiencing an orgasm. Subjects were encouraged to focus on their own sensations and to take this opportunity for their own benefit. These instructions were given to subject numbers 1006, 1007, 1010, 1012 and 1018.
The remainder of the subjects were given even more specific instructions. Subjects were encouraged to apply more pressure to the vibrator and to make pelvic movements so as to increase the likelihood of orgasm. While this might compromise the VPA signal as this produces movement artefacts that may obscure actual genital response, it could encourage the likelihood of orgasm, the primary endpoint of this study.
Changes in the statistical analysis of the study
An interim analysis was performed after 30 subjects had completed the study. This comprised of descriptive statistics on efficacy variables. No statistical tests were performed. Therefore no p-value adjustments are needed in the final analysis.
During the study, it became clear that even with the specific instructions given prior to VTS/VSS, subjects who self-reported orgasm at exit interview had not used the orgasm button to record their orgasm. They either expressed initial uncertainty about orgasm occurrence, or expressed curiosity about what would follow after orgasm, and did not press the orgasm button for that reason. Based on this finding, concerns were raised that there may be another subset of the primary anorgasmic subjects in the study who may have confused orgasm with high arousal. To test this possibility, 4 subgroups were identified, based on their responses at exit interview:
1) Subjects who self-reported orgasm (the originally planned efficacy measure)
2) Subjects who were suspected to have had an orgasm based on expressing
uncertainty about having had an orgasm at exit interview
3) Subjects who reported increased sexual arousal at Visit 3 relative to Visit 2
4) Remaining subjects
Because the assumption is that only primary anorgasmic women may be at risk of confusing orgasm with high arousal, group 2 consists of primary anorgasmic women only. For these 4 subgroups, descriptive statistics on mean and maximum VPA scores, prolactin levels and questionnaire data are presented. If primary anorgasmic women indeed confuse orgasm
with high arousal, subjects in group 2 and 3 should exhibit higher levels of VPA and subjective sexual arousal than subjects in group 4.
Furthermore, it was decided to change the population for the statistical analysis into the Per Protocol Population, as the number of subjects with a major protocol violation was higher than expected. Due to the limited sample size, the inclusion of subjects with major protocol violation(s) would have a larger impact on the accuracy of the result than desirable.
10. STUDY PATIENTS
10.1 Disposition of Subjects
In total 97 subjects were screened. Of the 97 subjects screened, 59 were randomized. Thirty-three (33) subjects did not fulfill the in- and exclusion criteria. Exclusions included relationship discord (N=10), nasal disorders and/or hay fever (N=6), and other medical conditions (N=8). Five (5) subjects had co-morbid sexual dysfunctions. Two (2) subjects (#1016, #1025) experienced an orgasm at Visit 2. Finally, 2 subjects had a positive drug test at screening.
An additional 5 subjects were excluded for other reasons. One subject had her first orgasm at home after Visit 2 but before Visit 3, which made her ineligible to continue, 2 subjects withdrew consent, 1 subject had a positive drug test at Visit 3, and 1 subject was lost-to- follow-up. Details are presented in Listing 30 (section 16).
Of the 59 subjects randomized, 1 (#1085) could not perform the VTS/VSS procedure as the vibrator was not working, this subject was replaced (#1098). A randomization error was made in 2 subjects: Subject #1063 was randomized to placebo but received TBS-2, whereas #1069 was randomized to TBS-2 (2hr) but received placebo. Fifty-six (56) subjects completed the study according to the protocol. A graphical illustration of subject disposition is given in Figure 1.
10.2 Protocol Deviations
All 59 subjects randomized were screened for protocol violations. All subjects had a VTS/VSS test performed. However, 3 major protocol violations occurred during the conduct of the study.
Subject #1085: Subject #1085 did undergo the VTS/VSS test but at the end of the test it appeared that the vibrator had malfunctioned.
Subject # 1063: Randomization error. Subject #1063 was randomized to receive placebo but received TBS-2 (0.5 hr group).
Subject #1069: Randomization error. Subject #1069 was randomized to receive
TBS-2 (2hrs) but received placebo.
The following minor protocol violations occurred during the conduct of the study.
• Subjects # 1006, #1024, #1034, #1056, #1071 had visits which were outside the predefined time window.
• Subject #1005, #1044 had elevated prolactin levels at Visit 1 .
• Subject # 1086 had elevated leucocytes at Visit 1 .
• Subject # 1077 presented with mild hay fever, which was inactive at the time of the study.
• Subject #1088 tested positive for gardenerella and had a visit outside the predefined time window. This subjected was excluded at Visit 3 because she reported at this visit that she had had an orgasm in the days following Visit 2.
All of the above described deviations were considered minor and not to have any impact on the outcome of the study.
Figure 1 , Flow Diagram of S&Kly Population

11 EFFICACY EVALUATION
11 .1 Data Sets Analysed
Of the 59 subjects who were randomized and received study drug, 3 had a major protocol violation. As such, the Per Protocol Population consists of 56 subjects. All 59 subjects who received the study drug are included in the intent-to-treat population and are evaluated for safety.
11.2 Demographic and Other Baseline Characteristics
Subject demographics are summarized in Tables 1 and 2 (see also Section 16, Listing 1 ). The mean (sd) age of the subjects in the placebo arm was 27.2 (7.9) years was comparable with the subjects treated with TBS-2: 28.2 (8.1 ) years. There were no differences in height, weight and BMI between the five treatment arms.
Ta te i. Ag©
T
TBS- T8S- I S*
mo haw Sows
0 tew p ~ pest-
TBS-2
VTSiVSS (to a!) 1 VTSiVSS
Taste 2, - Weight i snd SMI
TBS- TBS- TBS- TSS- as 2-7.0 2t/48 z si
6.S how
iX-'Si-
TBS-2 dose
VTS/VSS VTSi'VSS
H it ¾ is 12 it
Tit .8 ?$.& 15¾.4 ?as 1 it.
$.§ ■S.2 ¾.? S.4 5.3
;y ?s? 88
1 ?S 1S3 in
t im
a .;
N 12 12
ss.s ss.1 S4.S SS.2 ss.s
IS.55 3 ΐΐ f ¾ ^ :¾ V-
Mini m Ϊ® 4S S4 8? ;;::>
S S3 S S3 1">
:·-, SS is; 82 l&SS
1
22.3 •v> >'
3.4 2.3 2,8
58 IS t& is
28
8 s
The majority of the subjects were Dutch (51/56, 91.1%). Five subjects were Surinamese, or had another ethnicity. In the placebo arm, 1 subject was Surinamese (1/11, 9.1%) and in the TBS-2 arm, 3 women were Surinamese (3/45, 6.7%) and one (1/45, 2.2%) had another ethnicity. Data are presented in Table 3.
Ta&te 3, Ethnicity
OTHER
Teiaf

All of the subjects in the placebo arm were primary anorgasmic, whereas 84.4% (38/45) of the subjects receiving TBS-2 had primary anorgasmia.
Because subjects on hormonal and non-hormonal contraception were enrolled in a 2:1 ratio and stratified over treatment/time post dose combinations, the number of subjects using hormonal contraception was comparable between the placebo (81.8%) and TBS-2 treated subjects (75.6%). Data are presented in Table 4. Details on the contraception method used are presented in Listing 9.
TaDte 4, Frat atiey of Primary a wd Second r Armrgasmia ami t sa erf
% %
Ew m Orgasm?
11
At screening, subjects completed several questionnaires: the MMQ, BDI-II, FSDS-R, FSFI (for descriptive statistics see Table 5) and the ISS, SDI-2 and the SESII-W (see Table 6). Women reporting serious relational discord as indicated by a score of ~ 20 on the MMQ-M were excluded from participating. In addition, women with a score of ~ 14 on the BDI-II, indicative of depressive disorder, were excluded. Per Table 5, the MMQ-M and the BDI-II mean and maximum scores of all subjects in all groups are below the respective cut-off scores, indicating that none of the subjects experienced serious relationship problems, and none of the subjects had a depressive disorder.
Total scores of > 1 1 on the FSDS-R and of < 26.55 on the FSFI are indicative of sexual dysfunction (Derogatis et al., 2008). The cut-off score for the FSDS-R was not enforced as formal exclusion criterion in this study, because many subjects in an earlier study with this compound and this indication (van Gorsel et al., 201 1 ) indicated that they underscored their actual level of sexual distress as a cognitive strategy to relieve the psychological burden associated with the complaints. This resulted in many eligible women being excluded from that study. As can be seen from Table 5, in all groups but the placebo group, the minimum score on the FSDS-R was above the cut-off score. In the placebo group, one subject scored below the cut-off (this subject scored 9). This means that all but one subject in this study scored above the formal cut-off on the FSDS-R, indicating sexual distress.
As no formal cut-off score is available for the Orgasm domain of the FSFI, it was decided not to enforce the formal cut-off score on the FSFI total score because the indication for this study was anorgasmia, which may or may not be related to sexual problems on other domains. On the FSFI, 27 of 56 women (48%) scored below the formal cut-off score of < 26.55, indicative of sexual dysfunction. Mean scores centered around the formal cut-off score and were comparable between groups.
There was no apparent difference between the placebo treated subjects and TBS-2 treated subjects, for these questionnaire items.
Table & Qyesiif >ίίΓϊ8ϊΓ0 SCOE¾S 81.1 Serening - ama- / BDi ii i f FSDS J¾
& FSFi
Trealmem
TSS- TBS- TBS- . m%
hows pesl- dose T S-2
¥?$.'¥SS ■VTSiVSS VT&'VSS
N ii 12 18 12 sees*
1S. ■' ·; ii.'i 47 8.7 as
Standard
4.S s.e 4.S 3.1 S.S S.2
¾.?St5!:f?KiO 2 s 2 1 :5 1
1S.S S.. 4.3 4.8 11.3 1ft 3
18 is 15 11 18 IS s 3 8 0 S
SDi « n 1 43 12 13 2 ii
2.3 2.1 33 23 2.13 1.8
3.4 3.3 4.3 4.¾ .2.3
Wmkwm s 0 3 0
2.0 1.S S S,5 ■53 s.s s 13 2 13 13 >3 s 8 0 !>
FSBS..R 11 43 12 13 12 1 ·
24.2 2S.8 24.8 2¾: 2S.3 s. S3 73 S.S 11.S s 4 h 1? 4
24.S SS.S ss.3 22.¾ 24 i: *0
Maximum 3 47 3? ss 4?
S 0 3 3 3 n 15 S 12 S 12 11
23.3 2S.S .2S.S 27 24,? 24.S
3.3 4.3 £.5 2M •S 5 3.8
£1 S 24 13 6
27.2 as as 27.1 27.?
Maxim m 33 32 28 32 i
Ta&te 6. Questionnaire Scams at Scr© ISS / SOf-2 fc SESJL
TBS- TBS- TBS- TBS-
0· ©J 2®M 2/2.0 m,Q 2 8.0

¥TS;V$ (*B¾ VT&VS V SiVS
S ai) s s s s
5SS scor : ·: S 12 1: i 12 11
"?
12<: il
21 1&S i;?.s ≤2.S
sa
Stefan
1: 1-S.¾ ¾ "si"- ^ •2 a
Mil 18S ΰ ΰ s s 8
• -Z ® H 1 ·: 1 ¾ -^
5.Λ 7.4 7.4
8,3 5.7 s.s 5
¾ a i.'.
SfeKan 4.Ϊ5 S.S as
22 IS 1S i:S S a a 5
SE§Jf . H 12 11
27 <* i .3 2S.J S i .s s,≤ 3.1
23 155 is £1 21
≤?.£> is 25.5 28.5 '^; :"i 2S¾
38 -32 36
¾ S a
The ISS evaluates subjects' satisfaction with various aspects of the sexual relationship. Scores on this questionnaire range from 0 (never satisfied) to 150 (always satisfied). Mean scores for the placebo group (1 12.9) and the TBS-2 groups (1 17.3) are comparable, representing sexual satisfaction 'a good deal of the time'. The TBS-2 4.0 hours group
appeared somewhat more satisfied, with the average score representing sexual satisfaction 'most of the time'.
The SDI-2 scores represent solitary sexual desire, with scores ranging from 0 (no desire) to 23 (strong desire). Mean scores for the placebo group (5.4) and the TBS-2 groups (7.4) are comparable, and scores are indicative of low levels of sexual desire. Scores in the TBS-2 2.0 hour group seem somewhat higher, but variability of the scores in this group is higher as well.
Of the SESII-W the arousability factor is used for analysis, with scores for this factor ranging from 9 (low arousability) to 36 (high arousability). Scores appear comparable, with scores in the placebo group (27.4) seeming to be somewhat higher than in the TBS-2 groups (25.3). The results of the medical history for all subjects that were screened are presented in Listing 2. Any abnormalities are summarized in Listing 3. Subjects with abnormalities were excluded from participation (see section 10.1 for an overview of subjects that were excluded based on medical history).
Details on current relationship duration, menstrual cycle phase, hormonal contraception use and type of anorgasmia problem (primary or secondary) are provided in Listing 4. Listing 5 represents a detailed description of the nature of the anorgasmia problem (situational or generalized) and other related sexual problems.
At the screening visit, a physical examination was performed on each subject. Results are presented in Listing 6. Clinical findings found during the physical examination are provided in Listing 7. Use of concomitant medication during the study is listed in Listing 8.
Specifications on type of contraception are included in Listing 9. Individual data from the screening questionnaires for all subjects that were screened are provided in Listing 10. A summary of in- and exclusion criteria is provided in Listings 11 and 12, showing that no non- eligible subjects were entered into the study.
11.3 Measurements of Treatment Compliance
The study drug was administered to the subject at the hospital by the investigator or the co- investigator. A record was kept for the disposition of the study drug, identifying, by subject number, the person to whom the drug has been dispensed and the quantity and date of dispensing. In addition, a confirmation of returned used syringes and unused investigational drug was recorded. No further measures on compliance were taken.
11.4 Efficacy Results and Tabulations of Individual Subject Data
For the analysis of efficacy, the Per Protocol population was used. In section 16, all subject data are listed.
11.4.1 Analysis of Efficacy
11.4.1.1 Primary Efficacy
The primary objective of this study was to evaluate the effect of a single dose of TBS-2 on the occurrence of orgasm at one of four time-points after the administration of the study drug compared to a placebo group. TBS-2 or placebo was administered at Visit 3 (for details see Listing 13).
The frequency of subjects experiencing an orgasm is summarized in Table 7. Details on the VTS/VSS procedure and the extent to which the subject experienced sexual pleasure are provided in Listing 14. equency of Or sm
NO IS
f¾ % % %
2 is.s
T8S-2 1 SI. t 8.S
·(¾ s IS tiSLS l s z %m IS 05.S T S M 2 tS,?
im um > i ΐδδ,ο
Four (4) subjects receiving TBS-2 (8.9%) reported an orgasm. Two (2) of the subjects were in the 4.0 hr arm, whereas the other 2 subjects were in the 8.0 hr arm. Two (2) subjects in the placebo group (18.2%) reported an orgasm.
1 1 .4.1 .2 Secondary Efficacy
Time to Orgasm
Since the number of orgasms observed in this study was too limited, the analysis on time to orgasm as planned, was not feasible. Instead, the time to orgasm is listed per subject in Table 8 below.
For subject #1013 the time to orgasm is missing, since in the VPA signal the indication of orgasm could not be detected. Subject #1013 reported this orgasm at exit interview.
Frgqu&ncy of Orgasm
Syfcfget Q $a$m Htm te Qtgwrn
KVi3 b 8,5 tot $K*SN3 £« VT¾¥S5
183? T -m i posW^ VTS/VSS
T@S-¾'¾© teMt's pesMoee V7 /VSS i m 53 sec
Quality of Orgasm
The Quality of Orgasm questionnaire (item 2) was used to evaluate the quality of orgasm Subject #1010 indicated she had an orgasm at exit interview, but qualified it as a 'mini- orgasm; the questionnaire was not completed. Most of the subjects judged their orgasm as 'pleasant' or 'very pleasant'. The results of the Quality of Orgasm Questionnaire, for the 6 patients who experienced an orgasm, is given below (Table 9).
Tafete 9. Quality of Orgasm
H»w≠emmt#- Mm Mmm
wm ψΜ® ¥**8 How *«*¥ ¥tm m orgasm? sjrgssm? ergasm? mm*
mw WfcB. iSSifii-sii / tm. assy i j 5¾t¾ ix¾' s.sS / !5¾! assy m
W7Z
Easy
1 1 .4.1 .3 Additional Analysis
Sexual feelings and Affect Questionnaire
The Sexual feelings and Affect Questionnaire was completed by the subjects at Visit 2 and Visit 3. This questionnaire measures sexual feelings and affect during sexual stimulation. Five scales are distinguished: sexual arousal, genital sensations, sensuality, positive affect and negative affect. The items are measured on a 1 (not at all) to 7 (intensely) scale.
Descriptive statistics on all items of this questionnaire at Visit 2 are presented in Tables 10 and 1 1 .
Taisfe i& SAQ Results at Visit 2: exual Arousal i Genital Sensations & Sensuality
TBS- TBS- TBS- TBS- roseate;'
0J hew um hours fx>§l- pmi-
VT-Si'VSS VTSVSS
1 12 ¾ 12
4.. Ϊ S.S i.rs • 22 Ι.ΰίϊ ΐ.2-3 153 ΐ.14
,¾S 3.33 23Z z:
■am S.DS S.SO S.I? 4.83 SJS7
Mm mm 8JS7 ¾ ¾ ?M ΰ 5.S3
8 0 8 8 s
1 12 ¾ 12
4.53 ·:·· ί; :·2 4,?S 4.S4
(ViS« If
Mittimus* ί :·:· ¾; 2.¾S 3.1? t.iSS 2.S?
£.08 a.33
Hm>&mm S-Sfi: 7.SS ¾ ·¾ ?.i¾ ?.3iS S.S7 s s 0 8 8 8

1.34 ?J6 a.s≥ 114
1 *¾ :·: ;;;; 2.SS
4.m *.§8 4.S3
Mmitmm sss S.8S S-.SS e.es S.SS s 8 S 8 δ S
SAO fitawi*¾t¾& at Vmt :i¥> S B * TBS- TBS- TBS-
2-2,0 me
0,5 ew tor hears
post-
TBS-2
V S/VSS itetaf VTSiVSS
n tt 44 12 w 51: 11
4- 1 •Ϊ ¾_ ά Ti ass 4.
■■ - 1 1.32 .S2 .Ω
1 S: 2-4S i -¾ 2 »¾ 2..«5
.*D ass 5 Ϊ3 s.&¾ ?,03 ■S.SS
issing a 1 S 0 i 3

SSartiiaiit
S.2S S.2¾ a..l i 0.S3 8.2.3 s.f?
1.05 S53 1M i.e¾ i.SS δΰ 1 JSS 1.00 ii? t. 1.08
MMiftmm 1.83 2S? IS 2.®? i .s; -S.5S
S a a a 5
At Visit 2, the average scores on all items are comparable between the placebo and the TBS-2 treated subjects.
Descriptive statistics on all items of this questionnaire at Visit 3 are presented in Tables 12 and 13. Individual data are presented in Listing 15.
Table 1 SAQ Results at Visit 3: Sexual Arousal Qenital Sensations &

QS hour hour ha s post
J B-Z doss
VT¾:VSS VTS fSS VT&'VSS VTS/VSS VTSVSS
H 11 45 12: is
Visii 3| ■4. IS $m 81 S.SS
Stewart
1.85 1.23 1.S2 1.12' 1.84 1.41
Wis tarn £SS 2.8§ 3.S? .S3 3*5
4. S3 5.33 S 77 7S5 S.SS 58?
70S 7.50 855 7.35 7feS 7SS s 0 3 r; 45 IS rs
4.2? &. SS S-.4S. 7.73 i.i4 7 ¾ s 5·
S.OS s.sa- .3.6? 2.00
3.S3 ass ass ass ass S.S0 a S3 7 IS 5.83 757 7.50 a S a fi is
m 11 4S 1 13 12 51
(Visit 3)
425 5.SS S.5S 5.12 SIS
Stands^
1.TSS S.S2 572 &S7 ii.SS ΐι·
7.4.5 7 5 425 3.83 3.48
445 5.25 S.SS ?ff 5.33 &§S
S.S 5 «7 ass &S0 S.60
0 0 o a 0 5
Tsisal
TBS- res- TBS- TBS-
2.¾.S 2:·2.0 28.0
8.S how ho hours hours hews p«si- priSsosS- des©
¥T¾¥SS Cists! VTSiVSS VTSiVSS
ass
i"S vfi
■i f
■.%0
ass s.sa s os
D
iss lis as? . SS5
u¾≥ loo
·:?
Q
At Visit 3, the average scores in the placebo group are comparable to the scores at Visit 2, whereas subjects who received TBS-2 showed a higher average score on all items as compared with the scores obtained at Visit 2.
Differences between Visit 2 and Visit 3 on each item of the SAQ are presented in Tables 14 and 15. On average, the difference between scores at Visit 2 and Visit 3 in placebo treated subjects were small, and mostly negative. This indicates that feelings became less intense over visits. In the TBS-2 treated subjects a positive difference was observed for all items, with the exception of negative affect. Concerning negative affect, the score at Visit 3 was similar to the score at Visit 2.
Change in SAG F Sexual Arousal Geni al ierisations &
Sensuality
Treatment
TBS* IBS- S-

{total} ss ss $s VTSiVS® S IS 1.2 11
s.n a 48
1.1? S.31 iM 113 SS4
-≤.33 •ii.S? · ·; 33
S.33 s.s? 0 33 as?
2 i 2.ss s m 1.23
6 s a 8
Cftangs in n 48 IS: 12 1
0 ;.:> S.S3 ¾ 33
1.81 %.m US 133 S?S
.•ass ■3.i>3 ••1.S? ¾ ■- S?
0.3iS s.s¾ S.33 S.S?
3J? 2.1? 33 2.1? 1.SS ΰ s S ύ 8
C ange i n a 45- 12 8 V£ u
•S c ? S :<? ¾.ss 8.23 & S3 d SS sis? 1.S4

.if¾ ΰ f ΐ.¾.\ s aiii*.
¾ .38
Mi ing
Relative to placebo subjects, TBS-2 treated subjects reported more intense sexual feelings (sexual arousal, genital sensations, and sensuality) at Visit 3, in which active treatment was received (see Table 14). The difference between the two treatment groups was statistically significant for sexual arousal (p=0.02, Mann-Whitney U test), but not for genital sensations
(p=0.25, Mann-Whitney U test). Differences were marginally significant for feelings of sensuality (p=0.06, Mann-Whitney U test).
The average difference in reported intensity of sexual arousal, genital sensations and positive affect was most obvious in the 4 hours treatment arm. Feelings of sensuality showed a greater difference from placebo at 8 hrs and at 2 hrs.
Ta&fe 1& Change in SAQ Results: Positive m4 ^ i Affect
TBS- TBS- TBS- TBS- a 2/4.0
hoar
TBS* peat* post-
2
vrssv ¥TSi¥
VTSfVSS !
12 1*3 11
Affeei {¥3»V¾
S.4S 'Mi &SS- t.13 S;?5
MMmm -2.23 • ? 5 •■ξ.δδ •ass
&M 8.4S &2S &4S &?¾ & s.m £ ?S ISS ≤4i3 ■ 20
■5 2 0 0 2 0
Vtom®* in S¾¾ !i¾s H 11 4S 12 S 12 11
-s.st -SM •ass
&13 CM
•CM ;* -¾.S? •s.r? •as?
S.8» a. as a. as a. as
S i ? at?
5 5 Q
TBS-2 treated subjects reported more positive but less negative affect in Visit 3 relative to Visit 2 (see Table 15). There was no statistically significant difference between the two groups with regard to positive affect or negative affect (p=0.10 and p=0.47 respectively).
Acute Female Sexual Desire Questionnaire
The results of the AFSDQ showed a similar pattern as was described for the SAQ. Descriptive statistics on the average AFSDQ score at Visit 2 and Visit 3, as well as the difference in average score between Visit 2 and Visit 3 are presented in Table 16 below.
Tstife 16·, Descriptive Statistics on FSDG
TBS- TBS- T8S-
2·: ,δ
post- post- post- vi ® VT&¥SS VTSAJSS
H 1:1 44 12 18 11
M rt S4 3,?¾ :·. ¾ 4.Ϊ3Ϊ ess
2.8S :2.DS ZSS :¾ i'?
.® &8≥
,£7 &¾s * ?3 5. S3
S 1 ii s ϋ
Ή 44 12
3.34 « ί:; ASS .1S 3.» ft. Si s.s? δ®?'
1. ?S 2J& ass
.S9 4,α-5 &8≥ 4.ΰδ
* ¾·< &¾S 4,33 4,31 5. S3
S: 1 3 s i S
U ί1 43 !.≤ S 11 iV3-V2} 15 22 a.*s ass; s.s?
3 5* S.?4 ¾,sa 1 i:¾ as? ft.-ί?
i's ¾ 8.1-S a. £s 8.1S
2JS S.GiS 1.33 0.S4 a ≤ a S 2 S
Placebo treated subjects showed a small negative average difference between Visit 3 and Visit 2, indicative of a reduction in reported feelings of sexual desire at Visit 3, whereas in TBS-2 treated subjects there was an increase in sexual desire at Visit 3, relative to Visit 2. The individual results are presented in Listing 16.
At Visit 3, in which active treatment was received, TBS-2 treated subjects reported more feelings of sexual desire than the placebo group (see Table 16). This difference between
the two treatment groups was marginally significant (p=0.09, Mann-Whitney U test).
Increase in desire was most pronounced in the TBS2 2hrs group.
The difference in change in AFSDQ total score between the placebo and TBS-2 groups was not statistically significant (Mann-Withney U test, p=0.51 ).
VTS / VSS Appreciation Scale
At the end of Visit 2 and at Visit 3, VTS/VSS-procedure pleasure ratings were collected. The majority of the subjects (placebo 7/1 1 , 63.6%; TBS-2 35/45,77.8%) rated the procedure as 'pleasant' or Very pleasant' at Visit 2. At Visit 3, 8 out of 1 1 placebo subjects (72.7%) and 38/45 of the TBS-2 subjects (84,4%) rated the procedure as 'pleasant' or 'very pleasant' (Listing 14).The 'very pleasant' ratings doubled in Visit 3 relative to Visit 2, with most notable increases in TBS-2 2 and 8 hrs groups. In the placebo group 'very pleasant' ratings decreased in Visit 3 relative to Visit 2. The results are summarized in Table 17.
TBS ¾Jsf TSS 2,8¾s> TBS 4te TBS n % H . H % H n ¾
VSs½ VAQ
ΐίί 22 16.2
Ptes*iStf 4 3S. - 28 §44 83, S ΐ 8-3 1 tS.SS 2 16.7 2 1&≥ Τδΐδΐ 11 1 S3.fi <*» io&s
3 VAQ
l * S«ss,¾fst ¾ ¾ 2.2 ύ (! i 8.3 0
S 13,3 2 23 S 2 IS.? 1 ¾ ΐ
2S sr.s IS 4 4i; ··; ¾■
V ry pteasss!S 2 ¾12 1 8 3 4 4S.S 3 2S.S 4 T«¾3i 11 5¾5.δ S 1S¾X¾
VPA
For subject #1022, VPA data were missing at Visit 2 and at Visit 3. The VPA signal showed an immediate start of movement artifacts at vibrator onset, which went on during the entire 20 minutes of stimulation, and disappeared after the vibrator was turned off. It was not possible to distinguish response from artifact. In this particular subject, the VPA
cord must have made contact with the vibrator, which made it impossible to use her VPA signal. The same may be true for subject #1012, whose data showed a similar pattern at Visit 3. Another explanation for the (more random) missing values in VPA present in the data of other subjects is related to the instructions that were given to the women to increase the likelihood of orgasm during VTS/VSS, which we started using after 10 subjects had already completed the study (see 9.3.3. for a description of the changes in instructions that were given to subjects after study start). These instructions included moving the clitoral vibrator and moving the pelvis to increase clitoral stimulation. In support of this reasoning, Listing 16 shows that the majority of missings were in subjects who received these instructions.
VPAmax
Descriptive statistics for VPAmax at Visit 2 and Visit 3 are presented in Table 18. This comprises maximum VPA up to, but not including orgasm. With regard to the median VPAmax, a large variation is observed. Furthermore, the data were not normally distributed. The average difference in VPAmax between Visit 3 and Visit 2 was largest in the TBS-2 4.0 hr treatment arm, indicating a larger VPAmax increase in the latter group at Visit 3 relative to Visit 2. Details on the difference in VPAmax between Visit 3 and Visit 2 for all treatment arms are presented in Table 19.
Statistical testing to compare differences in VPAmax between the TBS-2 and the placebo groups revealed no significant difference (Mann-Whitney U test, p=0.13). Individual data are presented in Listing 16.
Tabte 18. Descriptive Statistics on YPA-tnax
T M &ni
PSacebo 0.5 TBS-2/2.8 TBS-2/4.0 TBS-2/S.0 hour past- TBS-2/G.S fsour hours
dose post-dose pos!-ifese pest-dose pest -dose VTS/VSS TBS-2 ftotai) TS/VSS VTS/VSS VTS/VSS VTS/VSS
WA-m&x (Visit 2) N 11 41 9 10 2 10
Mean isne.ss 12048.29 121S 5 3 Π 151 ,83 1 J440.4S 13546.80
Standard D®¾? iaiisft S714.S3 3738,55 3137.30 ««,22 8538.84 3892.17
Wrsimum 178.® 92-1.67 somots 395. ! 7 921.6 1780.SS3
Media 10290.17 10863.1? 9763.33 Sat? 75 1 12 .42 1284S.S8
Rfexirrjufn 1S1S3.S7 322?3,S? 2788433 32279,6?
0 4 3 0 0 1
VPA-m (Visit 3) N 0 4C< 0 9 2 g
17 13984.1 1280.40 1210A.S3 15736 84
Standard DeviaBori SS08.4S $38? M SS354.8S 5725.28 t0252.¾8 1 54 1.01
Minimum 3738.00 78 Y 78.S? 4067.69 3338,83 476.17
8357.67 12724.2S 8018.50 11482.00 ΚΪ708 78 191 OS.67
Maximum 17738. S3 40849.17 31189.33 20185.33 40840.17 34320.67 issing 1 5 2 1 D 2
Table 19. Descriptive Statistics on Change in VPA-msx
T eatment
Piaeebs TBS-2/0.S TSS-2 2.0 TBS-2i4.0 TBS-2/S.0 0.5 hour hour posi- hours pest- hours posi- hours post-
TBS-2 dosa dose
VTS/¥SS VTSiVSS VTSfVSS VT&VSS VT8 VSS
Change in VPA- U ts 9 3 12 s max
(V3-V2 fcfears - 163.98 1952.63 - 184.93 386.72 «645.00 2SS7.28
Standard
Dmlsi n SmSS IW&S.QS 3217,21 1:3231,82
Minimum -13S7S.5Q -28318.83 -18δ0?.δδ 53S?¾.cO -28319.83 -16968.00
Median 3845.83 mm 4SS1.00 SS3S.87 SSSS.83
Maximum - . ¾6?3,8'¾ 24823.80 i3S¾8. 7
Missing 6 3 1 0 2:
VPAmean
Descriptive statistics for VPAmean at Visit 2 and Visit 3 are presented in Table 20. This again comprises maximum VPA up to, but not including orgasm. With regard to the median VPAmean, a large variation is observed. Again, the data were not normally distributed. Median VPAmean was comparable between the placebo group and the TBS-2 group at Visit 2, whereas at Visit 3, median VPAmean was higher in the TBS-2 group.
The descriptive statistics on change in VPAmean from Visit 2 to Visit 3 are presented in Table 21. Similar to what was observed for VPAmax, the average difference in VPAmean between Visit 3 and Visit 2 was largest in the TBS-2 4.0 hr treatment arm, indicating a larger VPAmean increase in the latter group at Visit 3 relative to Visit 2. The difference in change in VPAmean between the placebo and TBS-2 groups was statistically significant (Mann-Whitney U test p=0.0493).
Table 2D, Descriptive Statistics on VPA-meare

•i¾5S :Sl 11* ,SS>
4. 3 ft 1 $ 2
ί>4·: ::: <*··

<ΐ*ί¾ 1iisS6,S
2 1 if a
Descriptive Statistics on change in VPA-mean
TreatmsRf
TBS- TBS- TBS-
Placebo 2/2.0 2/4.0 .o
0.S hour hours hours post- TBS-2iO.S post- post- post- dose hour post- do s« dose doss
VTS/VS TBS-2 dose VTS V VTS/V VTS/V
S VTS/VSS SS SS SS
Changs N 10 39 S 52 g
VPA-mean
(¥3-V¾ f,teao -S3S.60 -36.63
Standard
Dsviaiian 3≤36.96 ¾ ·ί · ¾·, /■>« · ; 6¾8£.92 -S267.S9 S-KM.S3 S /"SS.;37 kfe iian -2SS S -;¾ss 3≤4?..3 3S1 f .4S 1638.23 fcSaximum
233*.»3 % j · ? 54SS.S?
Mssing 3 6 3 1
1 1 .5 PK Data
Testosterone levels, free- and total testosterone were measured before and after dosing at Visit 3. Pre-dose free- and total testosterone levels were comparable for the placebo and the TBS-2 groups. After dosing, both free- and total testosterone increased in the TBS-2 treated subjects, whereas no increase was observed in the placebo treated subjects. Results are presented in Table 22.
Descriptive Statistics on Free and Tola! Testosterone - Placebo ¥S TBS-2 iming
Pre-Qose $¾st-Dos<s
Free Totati Fre© Tots!
Testosterone Testosterone Testosterone Test»steron«
(rtg dL) (rtg/dL) ng L)
Treatment
Piaeeira N i ίΐ
0.2481 se.osse 0.23SS 24,8327
Standard Deviation 0.58 2 i3,S0l 0. 796 52 i-:..'
Minimum QMM f J .§080 aoists
8A$W 20.0088 0.1 stO 23.9000
Mmimum 0.6520 56.3080 0.S370 4S.fi(X30
0 & 0 &
TBS-2 44 44
(at!)
Mem 27.234 · 3 S34S :? m
Standard Deviation 12.:52>¾!i 0.4S20 33.SS0S
Minimum Q. rn f S .2030 a si« 20,0000
Median 0.228S 24.SSD0 0.4S30 55.2000
Maximum 0.7770 7S.0OS0 24800 2ϊ8,0Ο Ο fussing t 1 0 S arsges for Free Testosteroi !w: 0.2 - 3.1 ^di.; :: ormsi fsfigi »s tor ToSa! T«¾ sferone: 3· SO rtg dt
Sex Hormone Binding Globulin (SHBG) and albumin data are presented in Table 23. Both pre- and post-dose SHBG and albumin levels were comparable for the placebo and the TBS2 treated subjects.
Table 23. Descriptive Statistics on SHBG nd Albumin - Placebo vs TBS-2
Treatment
acefoo TBS-2 {af!}
Timing Timing
Pre-Dgse Pos -Bsse Pre-Oose Post-Doss
SHBG {nmoi/L} H n 11 ^ 45
Standard Dswatisn S4,7 65.5 S3.≤ 85,5
Minimum 38.7 21 ,9 tSS
Median 03.5 H-.A 82.9
Maximum 2SS.0 2SS.0 358.0
Missing 0 0 0 0
1 1 ¾ - S
33.3 33.1 38-3 38 8
Standard Deviation t .§ 2.2 2,3
Minimum 36.1 3?.0 34.5 33.3
edian 38.1 38.0 3?.a 3@,4
Maximum 44.1 42.8 44.8 44.7
Missing « 0 a 0
The results for (free) testosterone, SHBG and albumin in the TBS-2 group are presented for the 4 different treatment arms in Table 24 and 25. Testosterone levels post-dose were most pronounced in the 0.5 hr treatment arm, which is indicative of a rapid onset.
Descriptive Statistics on Free anci Total Testosterone - TBS 2 treated ubjects
Timing
Pre-Oose Post-Dose
Free Yotai Free Total
Testosterone Testosterone Testosterone estosierQ
| g/dL)
Treatment
TBS-2 2 12 12 S
0.5hr
Q. 33,741? 1.0053 H 0OS
Standard 3.1330 0.8868 &'5.ί642
Minimum 0.0888 S.OOOO 0.360 55.4000
34.1500 o.ssss 107.95OO
Ma imum 0.7130 75.0003 2.4303 25O.O¾ 0
Missing 0 0 0 ø
TBS-2 9 8 13 10
3,1 Ss2 20.8S07 0.5408 57.6100
Standard Deviaiion 0.0735 7.67SS 0.207B 13.3823 Minimum 0 0SSS ■SI .2003 44.2000
0.58» 28.KW 0.6 S5 53.SS0G 3.2SS0 36.6000 0.S7S& 7S.6 O0 issing ί 1 0 0
TBS-2 2 12 12 1
4.0hrs
.21 23.891? 0.4S83 5 J6S7
Standard Deviation 0.3975 ?.7251 0..1SS2 15.0813 Mi ni mo ra 3.1 mo 14.4000 0.2320 34.3000
0,2255 20.0000 β.4685 43.9000
MaM niufii 3,3820 34.7000 0.937S SS.iOOQ issing ø β a 0
Timing
Prs-Dose Post- Dose
Free Total Free Totai
Testosterone Testosterone Testosterone Testosterone
TBS-2 N i t 1 11 11
Sfean 2&. s 0.439$ 43.2SO0
Standard Deviaiion 0.1 S¾8 8.2791
O. J06S 55.0800 0JSS8 £0.8000
Median 2?.£S0QO S,44/0 -8«B
61.1000 i .tDOO 78.0800 iss ng Q o 0 8
Mais: norma ranges for Free Testosierone: 0.2 ~ 3.1 ray'dl; norma! ra ges for Total Testosterone: 3-80 ng dL
"Table ts. Descriptive Statistics on SHBG a id Albumirt - TBS-2 treated subjects
Treatment
TBS-2 G.5hr T85-2 2,0hrs TBS-24.0 rs TBS-2 S.8hi¾
Timing Timing Timing Timing
Pre- PGSt- Prs- Post- Prs- Post- Pre- Post- Dose Dose o&8 Dose Dsse Doss Dose Dose
SHBG :N s≥ 2 10 ■¾ ^ 12 IS i i
{nmel L}
Mean 13S : -136.S 3 ,7 1 .3 90.2
Standard
Deviation 1:tf ,4 113.4 34.3 3 73,8 634 91.0 101-4 inimum 4£.S 43.2 40.2 Ϊ .3 53,0 61 .3 21.3 13.S
Media 38-3 34.S 503,0 S3.3 37,0 3S 4 99.3 S9.S
Maximum 303 \· 385.0 S 3.0 3δ.θ 30S.3 202.0 :¾:> '· 368.3 Missing 0 0 3 3
2 10 12: 1 ! 11
Mean 38.S 39.2 38.1 38,1 SS.-S 38.3 38.7
Stan ard
DssiaJion 2.1 2: 0 1 .S 1.7 3 inimum 36,0 36.5 34.3 35.0 36.7 36.2 34.S 33,3
Median 33,5 33.S 33.4 37.8 33.4 37.7 36.3
Maximum 43.0 43, > 41,7 41.S 41.S 41 .0 44,8 44,7 issing 0 0 0 ;"5 ft 0 0 0
This pattern of findings suggests that TBS-2 produces a rapid onset elevation of free- and total testosterone levels without affecting SHBG and albumin levels. Individual data on (free) testosterone, SHBG and albumin at Visit 3 prior to, and after dosing, are provided in Listing 29.
11.6 Exit Interview
The (translated) results of the exit interview are presented in section 14.12.
As pointed out in section 9.5.3, the study population was stratified in four groups, based on their exit interview responses at Visit 3, reflecting the likelihood of the occurrence of an orgasm.
The following definition of groups was used:
1 ) Subjects who self-reported orgasm during the study (the originally planned efficacy measure): n=6 (subjects 1010; 1013; 1037; 1040; 1072; 1093)
2) Subjects who were suspected to have had an orgasm based on them expressing, at exit interview, uncertainty about having had an orgasm: n=8 (subjects 1006; 1018; 1034; 1053; 1075; 1077; 1080; 1084)
3) Subjects who reported increased sexual arousal at Visit 3 relative to Visit 2: n=18 (subjects 1002; 1005; 1007; 1008; 1011 ; 1033; 1042; 1043; 1046; 1049; 1051 ; 1052; 1056; 1057; 1071 ; 1082; 1097; 1098)
4) Remaining subjects: n=21 (subjects 1009; 1012; 1014; 1019; 1020; 1022; 1024; 1028;
1030; 1048; 1050; 1058; 1066; 1076; 1078; 1079; 1081 ; 1083; 1086; 1089; 1095)
Three subjects (#1001 , #1003, #1044) were not included in any of the groups, because their responses at exit interview did not allow for inclusion in any of the groups.
This stratification was the basis for the orgasm analysis and additional exploratory analysis on each group designed to better understand the benefits of application of the study drug in women with anorgasmia. This allocation of subjects to subgroups was conducted prior to unblinding.
11.7 Exploratory Analysis
11.7.1 Statistical/Analytical Issues
1 1.7.1.1 Adjustments for Covariates
No adjustments for covariates were made.
1 1.7.1.2 Handling of Dropouts or Missing Data
During the study dropouts were replaced. One subject (1085) was replaced because the clitoral vibrator failed during VTS-VSS in Visit 3. No imputation techniques for missing data were used.
1 1.7.1.3 Interim Analyses and Data Monitoring
The study protocol did not include an interim analysis. An interim analysis, which included only descriptive information, was performed following the inclusion of 30 subjects. As no statistical tests were performed in the interim analysis, a p-value adjustment is not required for the final analysis of this study.
1 1.7.1.4 Multicenter Studies
As this is a single center study, this section is not applicable.
1 1.7.1.5 Multiple Comparisons / Multiplicity
This section is not applicable.
1 1.7.1.6 Use of an Efficacy Subset of Patients
This section is not applicable.
1 1.7.1.7 Active-Control Studies Intended to Show Equivalence
This section is not applicable.
1 1.7.1.8 Examination of Subgroups
11.7.1.8.1 Subgroup based on exit interview responses
To better understand the effect of the administration of the study drug, an additional analysis was performed by creating subgroups based on the category of response. Based on the responses at exit interview, patients were assigned to one of the 4 subgroups listed in 11.6. Subjects in group 1 were those that self-reported an orgasm during the study. Because the assumption is that only primary anorgasmic women may be at risk of confusing orgasm with high arousal, group 2 consists of primary anorgasmic women only. For these 4 subgroups, descriptive statistics on mean and maximum VPA scores, prolactin levels and questionnaire data are presented. If primary anorgasmic women indeed confuse orgasm with high arousal, subjects in group 2 and 3 should exhibit higher levels of VPA and subjective sexual arousal than subjects in group 4.
The rationale for this subgroup analysis and a description of each subgroup is provided in sections 9.5.3 and 1 1.6. The number of subjects in each subgroup and their randomization group is presented in Table 26.
In the placebo group, 5 out of the 10 subjects included in the analysis (50.0%) self-reported, at exit interview, an increase in sexual arousal during Visit 3, relative to Visit 2, which resulted in an orgasm during Visit 3 in 2 cases. In TBS-2 subjects, 27 women out of 43 (62.8%) presented an increase in arousal, 12 women (27.9%) reported an orgasm or were suspected to have had an orgasm.
Table 26. Frequency Count of Assignment to Subgroups
Treatment
TBS-
2ϊ»Λ
Ptecebof TBS-2.¾.S TSS-2/2,0 TBS-2 .0 hours 0.S hour hour post- hours hours post-
TBS-2 das® past-dose posi-dose dose
VT&'VSS (Total) VT¾'VSS VTSiVSS VTS VSS VTS/VSS fi % N % H % N % N % N %
Subgroup
Orgasm reported
by patient 2 33.3 ·'· 6β.? 0 8 0 0 2 33.3 2 33.3
Orgasm indicative
based! o« Exit
interview 0 0 8 ίΟ .ίϊ 2 25.0 o s 3 VI
V2-V3 Increased
Arejagai !:= 83.3 22.2 4 ≤2.2 2 11.1
Other |V2-V3 no
increased^ arousal 5 ≤3.0 $ 72.6 5 28.$ 3 14,3 $ 23.3 2 3.S
TotaS 10 1¾.S 43 Si , 3 12 22.S tO 58.3 1 ¾ § 1/.0
The findings summarized in this table suggest that if the subjective reports at exit interview of the 8 subjects in subgroup 2 are taken to be indicative of an actual orgasm, then there appears to be some involvement of TBS-2, because all of those 8 women were TBS-2 treated subjects.
An overview of subject numbers assigned to each subgroup is presented in Listing 34.
For each subgroup, descriptive statistics on VPA, SAQ, AFSDQ and prolactin are included in section 14.
11.7.1.8.2 Subgroups based on baseline Total Testosterone
To investigate whether the primary efficacy variable, orgasm occurrence, is related to baseline total testosterone values, this section describes whether total testosterone at study start is related to treatment efficacy. Subjects who have total testosterone values near the low end or below the lower limit of normal at study start, may benefit more from TBS-2 treatment than subjects with total testosterone values within normal ranges.
Deficient total testosterone values are noted as missing values in Table 42.
Section 14.2.2.3, Table 42, lists descriptive data for total testosterone as part of the safety analysis. Safety total testosterone was assessed at screening and post-dose at Visit 3, in the same post-VTS/VSS draw as the PK data. PK total testosterone was assayed by ABL, with
total testosterone being expressed as ng/dL. Safety total testosterone was assayed at the Central Laboratory of the Academic Medical Center, with total testosterone expressed as nmol/L. At the Sexology clinic of the Academic Medical Center, a total testosterone value of < 0.03 nmol/L is considered evidence of a testosterone deficiency, which may result in reduced sexual arousability. For the ABL assay, it is less clear how values in the lower range correlate with clinically significant findings.
Taking the Central Laboratory testosterone values as a criterion for testosterone deficiency, eight (8) subjects in the TBS-2 groups had deficient total testosterone values at screening: 2 in the TBS-2 0.5hr group (subjects 1012 and 1095); 2 in the TBS-2 2hr group (subjects 1005 and 1078); 2 in the TBS-2 4hr group (subjects 1014 and 1024); and 2 in the TBS-2 8hr group (subjects 1053 and 1057). All but one subject had post-dose total testosterone values within the normal range, suggesting that TBS-2 was effective in elevating testosterone levels. None of these subjects had an orgasm at Visit 3. Therefore, in terms of efficacy, testosterone deficient subjects who had normal total testosterone levels as a result of TBS-2 treatment were not more likely to orgasm during VTS/VSS.
The one subject that also had a deficient total testosterone value at close out (1053) was in the TBS-2 8hr group. This subjects' post-dose total testosterone as assayed by ABL was lowest of all TBS-2 treated subjects, so there appears to be consistency between measures. This suggests that, as expected, testosterone levels may no longer be elevated 8 hours after dosing.
Of all the TBS-2 treated subjects with deficient testosterone values, only subject 1095 was not on hormonal contraception.
Three (3) placebo subjects (1048; 1069; 1071 ) had deficient total testosterone values at screening and at close out. Of those placebo subjects, subject 1069, was excluded due to a randomization error. Three other placebo subjects had deficient total testosterone values at close out only (1019; 1042; 1093). Of those, subject 1093 had an orgasm at Visit 3. Her total testosterone as assayed by ABL was also in the low range.
Of all the placebo subjects with deficient testosterone values, only subject 1071 was not on hormonal contraception.
Future analyses should exploratorily investigate the relationship between total testosterone and the other endpoints in this study.
11.7.2 Tabulation of Individual Response Data
All patient data have been listed on an individual basis in section 16.
This section is not applicable.
11.7.3 Drug-Drug and Drug-Disease Interactions
No drug-drug or drug-disease interactions has been observed.
11.7.4 By-Patient Displays
This section is not applicable.
11.7.5 Efficacy Conclusions
Four (4) subjects receiving TBS-2 self-reported an orgasm during the study of which two (2) of the subjects were in the 4.0 hr arm and the other 2 subjects in the 8.0 hr arm. Two (2) subjects in the placebo group self-reported an orgasm. In the TBS-2 treated group, an additional eight (8) subjects reported sensations indicative of an actual orgasm at exit interview but had not recorded the orgasm during the treatment phase. No additional patients in the placebo arm provided such reports at exit interview. Therefore, a total of 12 patients (27%) in the TBS-2 treated group either self reported an orgasm at Visit 3 (4 patients) or were suspected of having an orgasm following their exit interview (8 patients) versus only 2 patients (18%) in the placebo arm. Although more patients treated with TBS-2 reported an orgasm or were suspected of having an orgasm following their exit interview when compared to placebo, this result was not statistically significant.
TBS-2 showed positive effects on subjective and genital indices of sexual arousal. Several subjective and genital indices of sexual response in the TBS-2 groups were greater in Visit 3 relative to Visit 2. In contrast, these responses remained the same or decreased in Visit 3 relative to Visit 2 in the placebo group. The finding that in Visit 3, relative to Visit 2, subjective and genital arousal responses decreased in the group that did not receive active treatment and that subjective and genital indices of sexual arousal increased in the TBS-2 treated subjects may be indicative of a true drug-effect.
Both pre- and post-dose SHBG and albumin levels were comparable for the placebo and the TBS-2 treated subjects. Free and total testosterone were elevated in subjects receiving TBS- 2, whereas no change in testosterone levels was observed in the placebo group. Testosterone levels post-dose were most pronounced in the 0.5 hr treatment arm. This
pattern of findings suggests that TBS-2 produces a rapid onset elevation of free- and total testosterone levels without affecting SHBG and albumin levels.
12. SAFETY EVALUATION
12.1. Extent of Exposure
This was a single-dose study. All 59 subjects who were randomized received a single dose of
TBS-2 or placebo and were included in the safety evaluation. Twelve (12) subjects received placebo, while 47 subjects were treated with TBS-2. After the administration of the study drug, patients fulfilled the remainder of the study procedures (at the same visit). No additional follow-up data were collected.
122. Adverse Events (AES)
12.2.1 Brief Summary of Adverse Events
The safety analysis showed that TBS-2 was well tolerated by the subjects. There were no serious AEs or discontinuations due to AEs in the study. A total of 18 AEs were reported, 15 of which were classified as not related to study medication and 3 were of unknown etiology. In the TBS-2 treated group, 9/47 (19.1 %) of the subjects experienced at least one adverse event, whereas in the placebo group 6/12 subjects (50.0%) reported at least one adverse event.
The frequency of adverse events observed per treatment arm is presented in Table 27.
A detailed adverse event listing with subject identification is included in section 14 (Listings 31 and 32).
Yafote .27, Frequenc of Adverse Events
Any adverse ©¥ents?
NO YES Total n % fi % %
Treateetii
PSacsboi 0.5 tour post-doss VTS VSS · 6 50.3 12 190.0
TBS-mS hour post-tfose VTS/VSS 8 61 -S 5 3S.S IS 00.S
TBS-2 2.6 hours post-dose VTS/VSS 80.0 ί 10.0 io 100.0
TBS-2/4.0 hours post-dose VTS/VSS ¾··> ··'· ·!-: ·: 13 s os.e
TBS-2 ae hours o t-dose VTS/VSS 18 80.0 ■s 11 ί 00..
Total 74.S 25. 100.0
12.2.2 Display of Adverse Events
In total 18 adverse events were observed in 15 subjects. All adverse events were considered mild and the majority of the adverse events were considered not related to study drug. The relationship of 3 adverse events to the study drug was considered unknown: patient number 1042 (placebo group) experienced a drop in hemoglobin and a drop in platelets, for which the investigator referred her to the general practitioner. As the subject refused to return to the clinic, the outcome was unknown. Subject number 1018 (TBS-2, 2hr) experienced headache, for which the relation to the study medication was also unknown. In one case (subject number 1080 / TBS-2 0.5 hr, urinary tract infection) the relationship to the study drug was judged 'unlikely' by the investigator. Details are presented in Listing 31 . In Listing 32 all coding related to adverse events is provided.
The listing of all reported adverse events per system organ class and preferred term per treatment arm is presented in Table 28.
Table 28. Adverse Events
Treatment
Place T8S- TBS- THiS- bo/ 0.S m,5 2/2.0 2/4.0
hour hour hours TBS-S/S.O post- post- post- past- hours dose dose dose dose post-
VTS. V VTSiVS VTSiVS VTS/VS dose
ss s S s VTS VSS Tota! fj N N H
System Organ Preferred Term
Ciass
Gas!reintsBt n Abdominal pain 0 ί 0 0
mi disorders
Gastritis 0 ί 0 0
Orai pain 0 0 0 0
infections and Cystitis o 1 0 0
(fifeststioos
Herpes simplex 0 1 0 0 0 influenza ■j 0 0 0
Nasopharyngitis 0 2 0 0 0
Urinary tract
infection 0 1 0 1 β
Virai pharyngitis 1 0 0 0
investigations Haemoglobin
decreased 0 0 0
Platelet count
decreased 0 0 s 0
Nervous Headache o 15 1 1 3 system
disorders Psraesthesfa (5 0 0 0
Reproductive Pruritus gersits!
system and
brsas!
disorders ■j <3 0 ΰ
Vascuiar HaemaioRis
disorders ■5 0 0 Q 0
Toiai
12.2.3 Analysis of Adverse Events
No formal statistical analysis of adverse event data was performed.
12.2.4 Listings of Adverse Events by Patient
A detailed listing of all adverse events on a per subject basis is provided in section 16.2.7. 12.3. Deaths, Other Serious Adverse Events and Other Significant Adverse
Events
No serious adverse events or deaths occurred during the study.
12.3.1 Listing of Deaths, Other Serious Adverse Events and Other Significant
Adverse Events
This section is not applicable.
12.3.2 Narratives of Deaths Other Serious Adverse Events and Certain
Other Significant Adverse Events
This section is not applicable.
12.4. Clinical Laboratory Evaluation
12.4.1 Listing of Individual Laboratory Measurements by Patient and each
Abnormal Laboratory Value
A chemistry profile, hematology profile and urinalysis were performed at screening and at Visit 3. At similar time points, thyroid panel and hormone assays were performed. Furthermore, alcohol breath tests and a urine drug screen were performed at all visits.
Hematology
All hematology results are presented per subject in Listing 17. In Listing 18, all hematology results which were outside the reference range are listed as well as their clinical relevance. In two subjects (#1086, #1098; both TBS-2, 4hr) clinically relevant abnormal values were reported, but in both cases this was at the screening visit.
One (1 ) subject (#1042, placebo group) showed clinically relevant abnormalities in hematology at Visit 3. This has been reported as an adverse event (see section 12.2).
Descriptive statistics on all hematology parameters are given in section 14.
Biochemistry
Biochemistry results are presented on a per patient basis in Listing 19.
Results outside the laboratory reference range are presented in Listing 20. In this listing, the clinical relevance of the abnormality as judged by the investigator is also included.
None of the values outside of the laboratory reference range were considered clinically relevant. Summary statistics on all biochemistry parameters at screening and at the close-out visit are presented in section 14.
Urinalysis
Listing 25 shows the individual urinalysis results. Out of range results are presented in Listing 26. In the safety population, no clinically relevant abnormalities were observed.
Thyroid Panel and Hormone Profile
The results on thyroid panel and hormone profile are included in Listing 22. Listing 23 presents the values which were outside the reference range and their clinical relevance. In several cases the clinical relevance was unknown (see Listing 23).
Three cases had clinically relevant abnormal values: #1085 (TBS-2, 4hr group) had clinically relevant abnormal values for SHBG, at Visit 1 as well as at Visit 3. Subject number 1093 (placebo) had a clinically relevant elevated prolactin value at Visit 3.
Descriptive statistics on hormone and thyroid parameters are included in section 14.
12.4.2 Evaluation of each Laboratory Parameter
Descriptive statistics on all laboratory parameters at Visit 1 and at Visit 3 are included in section 14.
At Visit 1 and Visit 3 a physical examination was carried out. The results are presented in Listing 6. All abnormalities are specified in Listing 7. No serious abnormalities were found. The results on vital signs are presented in Listing 28. Pulse, temperature and blood pressure were measured at all visits as well as before and after dosing. Descriptive statistics on these parameters per time point are included in section 14.
No deviations were observed.
125. Vital Signs, Physical Findings and Other Observations
A physical examination was performed at Visit 1 and 3. None of the measured vital signs or findings on physical examination changed in a consistent manner. No serious abnormalities were observed. The results are presented in Listing 6. All abnormalities are specified in Listing 7.
The results on vital signs are presented in Listing 28. Pulse, temperature and blood pressure were measured at all visits as well as before and after dosing. No deviations were observed. Descriptive statistics on these parameters per time point are included in Section 14.
126. Safety Conclusions
The safety analysis showed that TBS-2 was well tolerated by subjects. There were no serious AEs or discontinuations due to AEs in this study. All AEs were mild and completely resolved at the end of the study. In total 18 adverse events were observed, of which 1 1 occurred in 9 TBS-2 treated subjects. All adverse events in the TBS-2 treated subjects were considered not related to the study drug or of unknown etiology. Furthermore, all adverse
events in TBS-2 treated subjects were mild and completely resolved at the end of the study. Physical examination, vital signs and clinical laboratory evaluations results did not reveal any additional clinically significant findings related to study treatment.
Overall, no safety concerns were identified in this study.
13 DISCUSSION AND OVERALL CONCLUSION
This 5-arm study assessed the efficacy and safety of TBS-2 nasal gel in women with primary or secondary anorgasmia. Fifty-six (56) subjects were included in the per protocol patient analysis. TBS-2 was administered to 45 subjects, while 1 1 subjects received placebo. At baseline, none of the subjects experienced serious relationship problems, and none of the subjects had a depressive disorder. Placebo and TBS-2 groups were comparable with respect to sexual function, sexual distress, their current level of sexual desire, and sexual satisfaction in the relationship. Overall, the VTS/VSS procedure was evaluated as 'pleasant' and 'very pleasant', with the 'very pleasant' ratings decreasing at Visit 3 relative to Visit 2 in the placebo group. In contrast, for the TBS-2 treated subjects the 'very pleasant' ratings doubled in Visit 3 relative to Visit 2, with most notable increases in TBS-2 2 and 8 hr groups.
Four (4) subjects receiving TBS-2 self-reported an orgasm during the study of which two (2) of the subjects were in the 4.0 hr arm and the other 2 subjects in the 8.0 hr arm. Two (2) subjects in the placebo group self-reported an orgasm. In the TBS-2 treated group, an additional eight (8) subjects reported sensations indicative of an actual orgasm at exit interview but had not recorded the orgasm during the treatment phase. No additional patients in the placebo arm provided such reports at exit interview. Therefore, a total of 12 patients (27%) in the TBS-2 treated group either self reported an orgasm at Visit 3 (4 patients) or were suspected of having an orgasm following their exit interview (8 patients) versus only 2 patients (18%) in the placebo arm. This result did not achieve statistical significance.
Sexual Arousal, Genital Sensations, and Sensuality were measured at Visit 2 and Visit 3 using The Sexual Feelings and Affect Questionnaire. For the placebo group, no effect was observed with the difference between scores at Visit 2 and Visit 3 being small, and mostly negative. This indicates that feelings became less intense over visits. In the TBS-2 treated subjects a positive difference was observed for all items. Relative to placebo subjects, TBS-2 treated subjects reported more intense sexual feelings (sexual arousal, genital sensations, and sensuality) at Visit 3, in which active treatment was received. The difference between the
two treatment groups was statistically significant for sexual arousal (p=0.02, Mann-Whitney U test), but not for genital sensations (p=0.25, Mann-Whitney U test). Differences were marginally significant for feelings of sensuality (p=0.06, Mann-Whitney U test).
Sexual Desire was measured using the Acute Female Sexual Desire Questionnaire. Placebo treated subjects showed a small negative average difference between Visit 3 and Visit 2, indicative of a reduction in reported feelings of sexual desire at Visit 3. For the TBS-2 treated subjects, an increase in sexual desire was observed at Visit 3. In addition, the TBS-2 treated subjects reported more feelings of sexual desire than the placebo group at Visit 3 and this difference between the two treatment groups was marginally significant (p=0.09, Mann- Whitney U test).
Genital Response was assessed using Vaginal Pulse Amplitude (VPA) which was measured using a vaginal photoplethysmograph. Genital response in the placebo group decreased in Visit 3 relative to Visit 2. This reduction in genital response with repeated testing can be interpreted as a habituation effect, and is seen in all drug-related studies in our laboratory that involve more than one session (e.g. Laan, van Lunsen, & Everaerd, 2001 ; Laan, van Lunsen, Everaerd, Boolell, & Riley, 2002). This was in contrast to the increase in genital response that was observed in the TBS-2 groups. For VPAmean the largest average increase in Visit 3 relative to Visit 2 was also found in the TBS-2 4 hr group, and for this index of genital response, the difference between placebo and TBS-2 groups reached significance (Mann- Whitney U test p=0.0493). VPAmax was greater in Visit 3 relative to Visit 2, and was largest in the TBS-2 4 hr treatment arm. These differences between TBS-2 groups and placebo were not statistically significant.
The finding that subjective and genital arousal responses decreased in Visit 3 in the group that did not receive active treatment and that subjective and genital indices of sexual arousal increased in the TBS-2 treated subjects in Visit 3 may be indicative of a true drug-effect.
Both pre- and post-dose SHBG and albumin levels were comparable for the placebo and the TBS-2 treated subjects. In contrast, free and total testosterone were elevated in all subjects receiving TBS-2. Testosterone levels post-dose were most pronounced in the 0.5 hr treatment arm. No change in testosterone levels was observed in the placebo group. This pattern of findings suggests that TBS-2 produces a rapid onset elevation of free- and total testosterone levels without affecting SHBG and albumin levels. Nevertheless, largest effects of TBS-2 were
seen in the 2 and 4 hr groups, providing further evidence of a delay-effect of testosterone on sexual arousal (cf. Heard-Davison et al., 2007; Tuiten et al., 2000; 2002).
The analysis of sub-groups based on the exit interview results identified eight women who may have had an orgasm but did not report it during the study. At exit interview, these women reported uncertainty about orgasm occurrence and reported having sexual feelings they had never experienced before. Because these women were primary anorgasmic, which means that they had never had an orgasm, they may have confused orgasm with high arousal and therefore, as a group, underreported orgasm occurrence. All of these eight women, who were identified prior to unblinding, were TBS-2 treated subjects (2 in the 0.5 hr group, 3 in the 2 hr group and 3 in the 8 hr group). If these subjective reports at the exit interview are taken as indicative of an actual orgasm, then there does appear to be some involvement of TBS-2 in orgasm occurrence. This line of reasoning deserves further study.
In contrast, another set of exploratory analyses found no direct relationship between low or deficient testosterone values at screening and response to active treatment in terms of orgasm occurrence. In fact, the only placebo subject that had an orgasm at Visit 3 was a subject with low to deficient testosterone values (based on the ABL and the Central Laboratory assays, respectively). None of the 8 testosterone deficient subjects who had normal total testosterone levels as a result of TBS-2 treatment experienced an orgasm during VTS/VSS.
The fact that a positive effect of TBS-2 compared to placebo on subjective and genital indices of sexual arousal was found, but not on orgasm occurrence, may suggest that anorgasmia is not simply related to lack of sexual arousal. This suggestion is supported by the fact that contrary to expectation, only 2 of 59 subjects (3%) had an orgasm at Visit 2. This is in sharp contrast with the 50% that was expected to experience an orgasm in the VTS/VSS procedure. This projected 50% was not based on data of anorgasmic women, because this is the very first study of its kind, but was extrapolated from the 100% orgasm occurrence rate that was found with this sexual stimulation procedure in women without orgasm difficulties (Laan & van Lunsen, 2002).
Lack of adequate sexual stimulation is a recognized cause of primary and secondary anorgasmia in otherwise healthy women. That the VTS/VSS procedure provides adequate sexual stimulation is evidenced by the 100% orgasm occurrence rate in orgasmic women
mentioned above. The low orgasm rate in both visits suggests that this procedure was not sufficiently sexually arousing to overcome the orgasm problems in the anorgasmic women that participated in this study. TBS-2 was not more successful than placebo in increasing orgasm occurrence given sexual stimulation that is, in principle, adequate.
As a recent review indicates (Laan, Rellini and Barnes, 2012), very little is known about the prevalence of another hypothesized cause of anorgasmia, which is fear of losing control. The VTS/VSS procedure was, apparently, not able to help women lose control, despite several efforts to help them overcome their fear of losing control (such as specific instructions to increase sexual stimulation, inquiring about fears and negative cognitions regarding orgasms, and education about what to expect when orgasm is reached). Further study of the exit interview responses of this study may help to gain insight into factors that impede loss of control in these anorgasmic women.
Overall, no safety concerns were identified in this study. TBS-2 was well tolerated. All AEs were mild and completely resolved at the end of the study. Physical examination, vital signs and clinical laboratory evaluations results did not reveal any additional clinically significant findings related to study treatment.
In summary, although more patients treated with TBS-2 reported an orgasm or were suspected of having an orgasm following their exit interview when compared to placebo, this result was not statistically significant. However, subjects treated with TBS-2 did demonstrate a statistically significant improvement in Sexual Arousal and Genital response as well as a marginally significant result in terms of Sensuality and Sexual Desire. These findings are considered encouraging as they may be indicative of a true drug effect. More large scale research is certainly warranted.
15. REFERENCES
American Psychiatric Association (2000). Diagnostic and Statistical Manual of Mental Disorders - Text Revision (4th ed). Washington, DC, American Psychiatric Association.
Arrindell, W.A., Boelens, W., & Lambert, H. (1983). On the psychometric properties of the
Maudsley Marital Questionnaire (MMQ): Evaluation of self-ratings in distressed and
"normal" volunteer couples based on the Dutch version. Personality and Individual
Differences, 4, 293-306.
Banks, W.A., Morley, J.E., Niehoff, M.L., & Mattern, C. (2009). Delivery of testosterone to the brain by intranasal administration: comparison to intravenous testosterone. Journal of
Drug Targeting, 17, 91 -97.
Beck, AT., Steer, R.A., & Brown, G.K. (1996). Beck Depression Index-ll. San Antonio, TX:
Harcourt, Brace and Company.
Bloemendaal, L, & Laan, E. (2012). The psychometric properties of the Sexual
Excitation/Sexual Inhibition Inventory for Women (SESII-W) within a Dutch population.
Manuscript submitted for publication.
Braunstein, G. D., Sundwall, D. A., Katz, M., Shifren, J. L, Buster, J. E., Simon, J. A. et al.,
(2005). Safety and efficacy of a testosterone patch for the treatment of hypoactive sexual desire disorder in surgically menopausal women: A randomized, placebo-controlled trial.
Archives of Internal Medicine, 165, 1582-1589.
Buster, J.E., Kingsberg, S.A., Aguirre, O., Brown, C, Breaux, J.G., Buch, A., Rodenberg, C.A.,
Wekselman, K., & Casson, P. (2005). Testosterone patch for low sexual desire in surgically menopausal women: a randomized trial. Obstetrics and Gynecology, 105, 944-
952.
Crowe, M. J. (1983). Conjoint marital-therapy-controlled outcome study. Psychological Medicine, 8, 623-636.
Davis, S. (1999). Androgen replacement in women: a commentary. Journal of Clinical Endocrinology and Metabolism, 84, 1886-1891.
Davis, S.R., van der Mooren, M.J., van Lunsen, R.H., Lopes, P., Ribot, C, Rees, M., Moufarege, A., Rodenberg, C, Buch, A., & Purdie, D.W. (2006). Efficacy and safety of a testosterone patch for the treatment of hypoactive sexual desire disorder in surgically menopausal women: a randomized, placebo-controlled trial. Menopause, 13, 387-396.
Davis, S. R., Moreau, M., Kroll, R., Bouchard, C, Panay, N., Gass, M. et al., (2008).
Testosterone for low libido in postmenopausal women not taking estrogen. New England
Journal of Medicine, 359, 2005-2017.
Derogatis, L, Clayton, A., Lewis-D'Agostino, D., Wunderlich, G., & Fu, Y. (2008). Validation of the female sexual distress scale-revised for assessing distress in women with hypoactive sexual desire disorder. Journal of Sexual Medicine, 5, 357-364.
Derogatis, L, Rosen, R., Leiblum, S., Burnett, A., & Heiman, J. (2002). The Female Sexual
Distress Scale (FSDS): initial validation of a standardized scale for assessment of sexually related personal distress in women. Journal of Sex and Marital therapy, 28, 317-
330.
Does, A. W. J. van der (2002). Handleiding bij de Nederlandse bewerking van de BDI - II [Manual of the Dutch version of the BDI-II]. San Antonio, TX/Lisse, The Netherlands: The Psychological Corporation/Swets Test Publishers.
Goldstat, R., Briganti, E., Tran, J., Wolfe, R., & Davis, S.R. (2003). Transdermal testosterone therapy improves well-being, mood, and sexual function in premenopausal women. Menopause, 10, 390-398.
Gorsel, H. van, Laan, E., Both, S., van Lunsen, H.W., van Gerven, J.M.A., Tkachenko, N., Dickstein, J., & Kreppner, W. (201 1 ). Two-Center, In-Patient, Randomized, Placebo and Active Comparator Evaluation of the Pharmacokinetics (PK) and Safety, Along with Initial Pharmacodynamic (PD) Efficacy of Three Dose Levels of TBS-2 Intranasal Gel Applied Twice-Daily (BID) for up to Three Days in Female Patients with Hypoactive Sexual Desire Disorder (HSDD) or Anorgasmia (ANOR). Internal Report CHDR 1009. Leiden, the Netherlands.
Graham, C. A. (2010). The DSM Diagnostic Criteria for Female Orgasmic Disorder. Archives of
Sexual Behavior, 39, 256-270.
Graham, C.A., Sanders, S.A., & Milhausen, R. (2006). The Sexual Excitation and Sexual
Inhibition Inventory for Women: Psychometric properties. Archives of Sexual Behavior,
35, 397-410.
Halaris, A. (2003). Neurochemical aspects of the sexual response cycle. CNS Spectrums, 9, 21 1 -216.
Heard-Davison, A., Heiman, J.R., Kuffel, S. (2007). Genital and subjective measurement of the time course effects of an acute dose of testosterone vs. placebo in postmenopausal women. Journal of Sexual Medicine, 4, 209-217.
Heiman, J.R. (1977). A psychophysiological exploration of sexual arousal patterns in females and males. Psychophysiology, 14, 266-274.
Hudson, W.W., Harrison, D.F., & Crosscup, P.C. (1981 ). The Index of Sexual Satisfaction: a short-form scale to measure sexual discord in dyadic relationships. Journal of Sex
Research, 17, 157-174.
Kamenov, Todorova, & Christov, (2007). Effect of tibolone on sexual function in late postmenopausal women. Folia Medicine, 49, 41 -48.
Kuile, M.M., ter, Brauer, M., & Laan, E. (2006). The Female Sexual Function Index (FSFI) and the Female Sexual Distress Scale (FSDS): Psychometric properties within a Dutch population. Journal of Sex and Marital Therapy, 32, 289-304.
Laan, E., Brauer, M., Janssen, E., Hahn, S., & Heiman, J.R. (in preparation). Genital and subjective sexual responsivity in US and Dutch women with Hypoactive Sexual Desire
Disorder.
Laan, E., van Driel, E.M., & van Lunsen, R.H.W. (2008). Genital responsiveness in healthy women with and without sexual arousal disorder. Journal of Sexual Medicine, 5, 1424- 1435.
Laan, E., Everaerd, W., Bellen, G. van, & Hanewald, G. (1994). Women's sexual and emotional responses to male- and female-produced erotica. Archives of Sexual Behavior, 23, 153-170.
Laan, E., & van Lunsen, R.H.W. (2002, June). Orgasm latency, duration and quality in women:
Validation of a laboratory sexual stimulation technique. Poster presented at 28th Conference of the International Academy of Sex Research, Hamburg, Germany.
Laan, E., van Lunsen, R.H.W., & Everaerd, W. (2001 ). The effects of tibolone on vaginal blood flow, sexual desire and arousability in postmenopausal women. Climacteric, 4, 28- 41 .
Laan, E., van Lunsen, R.H.W., Everaerd, W., Boolell, M., & Riley, A. (2002). The enhancement of vaginal vasocongestion by sildenafil in healthy premenopausal women. Journal of Women's Health and Gender-based Medicine, 1 1 , 357-365.
Laan, E., Rellini, A.H., & Barnes, T. (2012). Standard Operating Procedures for Female
Orgasmic Disorder: Consensus of the International Society for Sexual Medicine. Journal of Sexual Medicine, doi: 10.1 1 1 1/j.1743-6109.2012.02880.x
Lewis, R.W., Fugl-Meyer, K.S., Corona, G., Hayes, R.D., Laumann, E.O., Moreira, E.D., Jr.,
Rellini, A.H., & Segraves, T. (2010). Definitions/epidemiology/risk factors for sexual dysfunction. Journal of Sexual Medicine, 7, 1598-1607.
Masters, W. H, & Johnson, V. E. (1966). Human Sexual Response. Boston: Little, Brown. Nobre, P.J., Pinto-Gouveia, J., & Gomes, F.A. (2006). Prevalence and comorbidity of sexual dysfunctions in a Portuguese clinical sample. Journal of Sex and Marital Therapy, 32,
173- 182.
Rosen, R., Brown, C, Heiman, J., Leiblum, S., Meston, C, Shabsigh, R., Ferguson, D., &
D'Agostino, R. Jr (2000). The Female Sexual Function Index (FSFI): A multidimensional self-report instrument for the assessment of female sexual function. Journal of Sex and
Marital Therapy, 26,191-208.
Segraves, K.B., & Segraves, R.T. (1991 ). Hypoactive sexual desire disorder: Prevalence and comorbidity in 906 subjects. Journal of Sex and Marital Therapy, 17, 55-58.
Sherwin, B.B., & Gelfand, M.M. (1987). The role of androgen in the maintenance of sexual functioning in oophorectomized women. Psychosomatic Medicine, 49, 397-409.
Shifren, J.L, Braunstein, G.D., Simon, J.A., Casson, P.R., Buster, J.E., Redmond, G.P., Burki,
R.E., Ginsburg, E.S., Rosen, R.C., Leiblum, S.R., Caramelli, K.E., & Mazer, N.A. (2000).
Transdermal testosterone treatment in women with impaired sexual function after oophorectomy. New England Journal of Medicine, 343, 682-688.
Shifren, J.L, Davis, S.R., Moreau, M., Waldbaum, A., Bouchard, C, DeRogatis, L, Derzko, C,
Bearnson, P., Kakos, N., O'Neill, S., Levine, S., Wekselman, K., Buch, A., Rodenberg, C,
& Kroll, R. (2006). Testosterone patch for the treatment of hypoactive sexual desire disorder in naturally menopausal women: results from the INTIMATE NM1 study.
Menopause, 143, 770-779.
Simon, J., Braunstein, G., Nachtigall, L, Utian, W, Katz, M., Miller, S., Waldbaum, A.,
Bouchard, C, Derzko, C, Buch, A., Rodenberg, C, Lucas, J., & Davis, S. (2005).
Testosterone Patch Increases Sexual Activity and Desire in Surgically Menopausal
Women with Hypoactive Sexual Desire Disorder. Journal of Clinical Endocrinology and Metabolism, 90, 5226-5233.
Sintchak, G, Geer, J.H. (1975). A vaginal photoplethysmograph system. Psychophysiology 12, 1 13-1 15.
Spector, I. P., Carey, M.P., & Steinberg, L. (1996). The Sexual Desire Inventory: Development, factor structure, and evidence for reliability. Journal of Sex & Marital Therapy, 22, 175- 190.
Traish, A.M., Feeley, R.J., & Guay, AT. (2009). Testosterone therapy in women with gynecological and sexual disorders: A triumph of clinical endocrinology from 1938 to 2008. Journal of Sexual Medicine, 6, 334-351 .
Tuiten, A., Laan, E., Everaerd, W., Panhuysen, G., de Haan, E., Koppeschaar, H., & Vroon, P.
(1996). Discrepancies between genital responses and subjective sexual function during testosterone substitution in women with hypothalamic amenorrhea. Psychosomatic Medicine, 58, 234-241.
Tuiten, A,, van Honk, J., Koppeschaar, H, Bernaards, C, Thijssen, J., & Verbaten, R. (2000).
Time course of effects of testosterone administration on sexual arousal in women. Archives of General Psychiatry, 57, 149-153.
Tuiten, A., van Honk, J., Verbaten, R., Laan, E., & Everaerd, W. (2002). Can sublingual testosterone increase subjective and physiological measures of laboratory-induced sexual arousal? Archives of General Psychiatry, 59, 465-473.
Wiegel, M., Meston, C, & Rosen, R. (2005). The Female Sexual Function Index (FSFI): Cross- validation and development of clinical cutoff scores. Journal of Sex and Marital Therapy, 31 , 1-20.
Wingen, G.A. van, Zylicz, S.A., Pieters, S., Mattern, C, Verkes, R.J., Buitelaar, J.K., & Fernandez, G. (2009). Testosterone increases amygdala reactivity in middle-aged women to a young adulthood level. Neuropsychopharmacology, 34, 539-547.
16. APPENDICES
16.1 Study Information
The English version is for information only. The Dutch text will be given to the subjects. This is NOT an official translation and in any disputes regarding the interpretation of this document the Dutch text will take precedence.
Title
A study on the effectiveness of testosterone intranasal gel on the ability to reach orgasm in women with anorgasmia.
Introduction
Studies indicate that the hormone testosterone plays an important role in regulating, among others, mood and sexual function in women. In women with long-lasting and distressing sexual complaints, studies indicate that the administration of the hormone testosterone resulted in increased desire, arousal, frequency of satisfactory sexual activity, pleasure and orgasm. Currently, the only testosterone replacement product that is approved for use in women with sexual problems is Intrinsa, which is a slow-release testosterone skin patch.
This study evaluates the effect of a single dose of a testosterone intranasal gel, TBS-2, on the ability to reach orgasm during sexual stimulation. During this test you will be alone in a private room in our sexology laboratory, with the (female) researcher present in an adjacent room, such that your privacy will be secured. The sexual stimulation involves watching female friendly erotic film excerpts in combination with clitoral vibration. This vibration occurs by means of a small rubber stopper that is mounted on a flexible metal strap lined with washable lycra cloth. You will be asked to attach the metal strap such that the rubber stopper touches your clitoris. Subsequently you will insert a vaginal probe into your vagina through an opening in the cloth, that allows for measurement of vaginal blood flow, a reliable index for genital sexual arousal. Once inserted, the probe hardly causes discomfort and the metal strap and the probe can be worn under loose-fitting clothing. After 20 minutes of sexual stimulation, or after you've reached orgasm, you are asked to fill out a questionnaire regarding your erotic feelings during stimulation. Study setup
You will visit the AMC three times. The first two visits a screening will take place to assess whether you are eligible and whether you are healthy. During the first visit a medical and sexological screening will be done. During the second visit you will undergo the sexual stimulation test without any medication. During the third visit, the study visit, you will undergo the sexual stimulation test in combination with a dosing of intranasal gel.
A total of 60 women will be enrolled in this study, all women with difficulty or inability to achieve orgasm (anorgasmia). Participation in this study might provide health benefits for participants. This is uncertain, but the study may contribute to development of an improved treatment for anorgasmia that may benefit women in the future. The nasal gel will not be available after the study is terminated.
This study is approved by the Medical Ethics Committee of the Academic Medical
Center (AMC).
Drugs
Participants receive either (1 ) TBS-2 testosterone intranasal gel or (2) placebo (no active ingredient) intranasal gel. Subjects are randomly assigned to one of the treatments. You will remain unaware of which of two treatments is given to you during the study visit (you remain 'blind' with respect to treatment received); unless there are medical reasons to undo the blinding earlier.
Side-effects
Some of the side effects that are known to be associated with testosterone given in a skin patch are: irritation of the skin at the patch application site, acne, excessive facial hair growth, migraine, voice deepening, breast pain, weight gain, hair loss, difficulty sleeping (insomnia), increased sweating, anxiety, nasal congestion, dry mouth, increased appetite, double vision, vaginal burning or itching, enlargement of the clitoris or palpitations. These side effects are also possible with testosterone intranasal gel. Generally these side effects are mild and emerge after longer treatment duration than this study.
During the medical screening and at the beginning and end of the third visit an intravenous venipuncture will be done. The insertion of the needle can be unpleasant and sometimes lead to a bruise.
What happens during the study
General
The study consists of three visits: a medical and sexological screening, a second screening visit with the sexual stimulation test, and the study visit with the sexual stimulation test in combination with the dosing of nasal gel (approximately a week apart). At the beginning of each visit you will be tested on the use of alcohol and drugs.
Blood will be drawn three times during the study, once at the first screening visit and twice at the study visit. Collected blood samples will not be preserved.
First Screening Visit: medical and sexological examination
You will be screened to determine if you are suitable for the study. The duration of the screening visit is approximately two hours.
The screening includes but is not limited to:
- questions regarding your sexual problems, general health and use of
medication, alcohol, tobacco and/or illicit drugs;
- general physical examination, including weight, height and blood
pressure measurements;
- ear nose throat examination;
- blood draw (5 tubes, 23 ml) and urine screening for general health (like kidney and liver function), Hepatitis B and C, HIV (AI DS virus), hormone levels and a pregnancy test.
It is possible that you are healthy but are not found suitable for the study. Sometimes the screening process uncovers something that was unknown and needs further medical attention. If so, you and you general practitioner will be informed. Charges for further medical assistance are covered by your own health insurance.
During this first visit you will become acquainted with the measurement procedures (the erotic film and the vibrator) and the vibratory frequency that is most pleasurable for you will be determined. To this end, you will be watching 5 minutes of erotic film while clitoral vibration is performed. You will also insert the vaginal probe for the purpose of becoming acquainted with it.
Second Screening Visit: sexual stimulation test
During this visit you will undergo sexual stimulation for a maximum of 20 minutes. If you experience an orgasm during the sexual stimulation, the study ends for you at this point. You will receive a brief medical examination after which you will be discharged. Study visit
In case you did not reach orgasm during the second screening visit, you will be invited to a third AMC visit, about a week later. You will again undergo the sexual stimulation test, prior to which you will receive a dose of intranasal gel. This can be a placebo (an
inactive agent) or the intranasal gel with testosterone; because your expectations that a drug may help you can influence the findings you will not be told what kind of treatment you receive. At this visit, the time between dosing and sexual stimulation may vary. Random choice will determine whether you will receive the sexual stimulation half an hour, 2 hours, 4 hours or 8 hours after dosing. This waiting period will be spent in a room in the AMC and we will supply you with some food and drink. After the sexual stimulation test a medical and ear nose throat examination will be done. Both before dosing and after the medical examination a blood will be drawn (4 tubes, 21 ml in total) to test for general health (like kidney and liver function), hormone levels and blood testosterone levels before and after dosing. Prior to this study visit a pregnancy test will be performed. After the final blood draw you will be discharged.
Lifestyle regulations
If you are using any medication, it is important that you discuss this with one of the study physicians beforehand as not all medication is allowed in the study. It is important that you refrain from alcohol within 48 hours of the start of the study. You may not drink alcohol or use drugs during the study. Smoking is not allowed inside the hospital. There are no food or drink restrictions during the study.
(Please see Attachment 1 for an overview of lifestyle regulations)
Safety
During your stay at the research center, a physician or nurse is always available. It is important for both yourself and the study that you inform them about all potential side effects or other problems.
The study center will consult your general practitioner about your participation. You cannot participate if you do not have a general practitioner or if you do not allow the study center to inform your general practitioner. To participate in this study you must have valid health insurance.
Because blood will be drawn, recent blood donation is not allowed. We follow the blood bank (Sanquin) regulations.
Termination of the study
You may withdraw from this study at any time without providing a reason and without consequences. The physician may also end the study if your health requires this.
We ask you to adhere to the lifestyle regulations for your safety and for the success of the study. If you do not adhere to the lifestyle regulations or house regulations of the research center, you may be excluded from further participation.
Confidentiality
All research and medical information is kept confidential. The data will be used for the purpose of the study in which you are participating and possibly for supplementary research. The research data are stored with a code, only the researchers have access to the code-key. The sponsors of the study only have access to the anonymous study data. Your personal details are also kept confidential in the publication of study results. After study termination all study files are stored in the AMC for a period of 20 years.
I case of an official enquiry, access to the research and medical data will be provided to supervisory personnel belonging to the Dutch or a foreign government, the Sponsor or an international organization authorized by the Sponsor. By signing the informed consent, you permit this.
Your personal data is processed in accordance with the Personal Records Act { Wet Bescherming Persoonsgegevens) as registered under number M1 020567 with the College Bescherming Persoonsgegevens.
Attachment 1 Lifestyle regulations
11 Use of medication:
12 The use of medication taken for chronic diseases should be stable for at least three months before the start of the study.
13 If you need to take other medication, you are requested to consult the investigator by telephone beforehand.
14 Use of illicit drugs and alcohol :
Recreational drugs, including cannabis, may affect the measurements and are therefore not allowed. Your urine will be tested during the screening visit and on the first study day. A positive result will cancel further study participation. Alcohol is not allowed from 48 hours before the study until study completion.
• Use of tobacco:
Inside the AMC smoking is prohibited. You are allowed to smoke an occasional cigarette outside the building, if it does not interfere with the study procedures.
• Contraception:
Women who are pregnant or breastfeeding cannot participate in the study. During the screening visit and the study visit pregnancy tests will be performed. Women of child- bearing potential agree to use reliable contraception for the duration of the study.
• Dress code:
During all visits to the AMC we recommend to wear comfortable, wide clothing to accommodate the execution of the tests.
EXAMPLE 11 A
1. Title
An Open-Label, Single and Multiple-Application of Intranasal Testosterone Gel (TBS-2) in Healthy Pre-menopausal Female Subjects at Three Dose Levels
2. Synopsis
The primary objective is to assess the bioavailability of total testosterone through pharmacokinetic (PK) profiles obtained following single administration of an intranasal
application of TBS-2 at doses of 600 1200 μ9, and 1800 μ9, and multiple administration of 1200 μ9 of TBS-2 given three times a day (t.i.d.) for 3 days. TBS-2 is a bioadhesive intranasal testosterone gel.
The secondary objectives are to assess the bioavailability of free testosterone, dihydrotestosterone, sex hormone-binding globulin (SHBG), and estradiol through PK profiles obtained following single administration of an intranasal application of TBS-2 at doses of 600 μg (0.24%), 1200 μg (0.48%), and 1800 μg (0.72%), and multiple administration of 1200 μg (0.72%) TBS-2 given for 3 days (t.i.d. for the first 2 days, and once in the morning of the third day) and to assess the safety of TBS-2
A. Methodology
This was a phase 1 , single-center, randomized, open-label parallel-group study designed to evaluate the safety, tolerability, and PK of TBS-2 in healthy, normal-cycling adult women. Subjects were randomly assigned on a 1 :1 :1 basis to 1 of 3 treatment groups (Cohort 1 , Cohort 2, or Cohort 3) during Period 1 and were administered a single dose of an intranasal application of TBS-2 at doses of 600 μg, 1200 μg, or 1800 μg (single doses of 300 μg, 600 μg, and 900 μg per nostril). At the end of Period 1 , a total of 8 subjects sampled from these 3 cohorts, who were willing and able to continue with the multiple-dose portion of the study were selected to participate in Period 2. During Period 2, subjects were administered 1200 μg TBS-2 (600 μg per nostril) t.i.d. for 2 days and once on the morning of the third day. Subjects were screened (Visit 1 ) for eligibility up to 3 weeks prior to dosing in Period 1 , and were admitted to the Clinical Research Unit (CRU) at 0700 hours on the day prior to dosing (Visit 2, Day 1 ). On Day 2, subjects were administered a single dose of TBS-2 and remained in the CRU for 72 hours post- dose (Day 4) for safety monitoring and PK assessments. During Period 2, subjects were admitted to the CRU at 0700 hours on the day of dosing (Visit 3, Day 1 ). On Days 1 and 2, subjects were administered TBS-2 at 0800 hours (±30 minutes), 1600 hours
(±30 minutes), and 2400 hours (±30 minutes). On Day 3, subjects were administered TBS-2 at 0800 hours (±30 minutes).
During Period 1 , blood samples for determination of baseline testosterone (free and total), SHBG, dihydrotestosterone (DHT), and estradiol concentration were collected on Day 1 at 0745 hours and then at 15, 30, and 45 minutes and at 1 , 1 .5, 2, 4, 6, 8, 12, 16,
20, and 23.5 hours relative to an 0800 hour clock time. Blood samples for determination of testosterone (free and total), SHBG, dihydrotestosterone, and estradiol plasma concentration were collected on Day 2 (15, 30, and 45 minutes and 1 , 1 .5, 2, 4, 6, 8, 12, 16, and 20 hours post-dose) and Day 3 (24, 32, 40, and 48 hours post-dose) during the confinement period.
During Period 2, a blood sample for baseline testosterone (free and total), SHBG, DHT, and estradiol concentration was collected at 0745 hours (i.e., 15 minutes prior to study drug administration). Blood samples for determination of testosterone (free and total), SHBG, dihydrotestosterone, and estradiol concentration were collected on Day 1 (pre-dose [15 minutes prior to dosing] and at 1545 and 2345 hours), Day 2 (1545 and 2345 hours), Day 3 (15, 30, and 45 minutes and 1 , 1 .5, 2, 4, 6, 8, 12, 16, and 20 hours), and Day 4 (24, 32, 40, and 48 hours) during the confinement period.
Other assessments performed during the study included the monitoring of adverse events (AEs), clinical laboratory evaluations (chemistry [including hormone profile], hematology, and urinalysis), vital signs assessments (systolic and diastolic blood pressure [BP], heart rate [HR], respiratory rate [RR], and body temperature), and physical examinations. In addition, otorhinolaryngological examination findings, 12-lead electrocardiogram (ECG) readings, medical history, and concomitant medication use were recorded.
B. Number of Subjects (planned and analyzed)
Planned: A total of 24 subjects were planned to be enrolled.
Enrolled: A total of 24 subjects were enrolled and randomly assigned to treatment in Period 1 : 8 subjects in Cohort 1 , 8 subjects in Cohort 2, 8 subjects in Cohort 3. A total of 8 subjects sampled from these 3 cohorts, who were willing and able to continue with the multiple-dose portion of the study, were selected to participate in Period 2.
Analyzed: All 24 subjects were included in the safety analyses and 24 subjects were included in the PK analyses.
C. Diagnosis and Main Criteria for Inclusion
Healthy, normal-cycling women between the ages of 18 and 40 years (inclusive) who were premenopausal, had a body mass index (BMI) of 18.5 to 35 kg/m2 (inclusive),
met all of the inclusion and none of the exclusion criteria, and provided informed consent were included in the study.
D. Test Product, Dose and Mode of Administration, Batch Number
The TBS-2 used in this study was an intranasal testosterone gel supplied in prefilled dispensers with 0.24% testosterone gel to deliver a single intranasal dose of 300 μg of testosterone per nostril (Cohort 1 ), 0.48% testosterone gel to deliver a single intranasal dose of 600 μg of testosterone per nostril (Cohort 2 [single-dose] and
Multidose group), and 0.72% testosterone gel to deliver a single intranasal dose of 900 μg of testosterone per nostril (Cohort 3). The lot numbers of TBS-2 drug substance used in this study were IMP1 1008, IMP1 1009 and IMP1 1010.
The compositions of the three different concentrations of the drug product to be used in this clinical trial are provided in the CMC Section below and in Tables 3.2. P.1 -1 - 3.
E. Duration of Treatment
The study involved 1 period for Cohorts 1 , 2, and 3 and the duration of individual subject participation from the start of screening until the post-study visit, was
approximately 25 days. This study involved 2 periods totaling 30 to 36 days for the Multi-dose group.
F. Criteria for Evaluation
Safety: Safety was assessed throughout the study and included the monitoring of AEs, clinical laboratory evaluations (chemistry [including hormone profile],
hematology, and urinalysis), vital signs, and 12-lead ECGs. Physical and
otorhinolaryngological examinations were performed and medical history and
concomitant medication use were recorded.
Pharmacokinetics: Whole blood samples for determination of plasma
concentrations of testosterone (free and total), SHBG, DHT, and estradiol were collected at specified time points. Actual sampling time points were recorded and used for PK calculations. Pharmacokinetic parameters for testosterone (free and total), SHBG, DHT, and estradiol were calculated by standard non-compartmental methods for all subjects as data permitted. The PK parameters evaluated for plasma concentrations of testosterone (free and total), SHBG, dihydrotestosterone, and estradiol following the
single-dose cohorts (Cohorts 1 , 2, and 3) included area under the plasma concentration time curve from time zero to the last measurable concentration time point (AUCo-t), area under the plasma concentration time curve from time zero to infinity (AUC maximum concentration observed after dosing (Cmax), time of observed Cmax relative to the time of dosing (tmax), terminal elimination rate constant (λζ), and elimination half-life (t½). The PK parameters evaluated for plasma testosterone (free and total), SHBG,
dihydrotestosterone, and estradiol concentrations following the multiple-dose cohort included area under the concentration-time curve from time zero to the dosing interval (AUCo— τ, where τ=8 hours), Cmax, tmax, minimum concentration over a dosing interval during multiple dosing (Cmir,), pre-dose concentration determined immediately before a dose at steady state (Cpd), average steady-state concentration (Cavg), % peak to trough fluctuation (PTF), and % peak to trough swing (PTS).
G. Statistical Methods
This study evaluated the PK properties as well as the safety and tolerability of TBS-2. Power calculations were not performed. The sample size for this study was not determined on the basis of statistical hypothesis testing. Based on typical, early-stage PK studies, groups of 8 subjects per cohort provided adequate clinical information to satisfy the objectives of the study. Data were summarized by using descriptive statistics (sample size, mean, median, standard deviation [SD], minimum, and maximum) for each of the safety variables by treatment group and overall. Data from all visits during the study were displayed in the data listings.
Concentration-time data for 5 analytes (testosterone [total and free], SHBG, dihydrotestosterone, and estradiol) were determined by a validated assay method and PK parameters were calculated. Actual sampling time points were recorded and used in calculation of the actual elapsed time from dose to sample for PK calculations. As each administration was to each of the nostrils, the time of dosing was the time of the first nostril administration. Baseline analyte concentrations from the 24-hour pre-dose profile were subtracted from the time-matched analyte concentrations following dose administration before calculation of the PK parameters.
Individual PK parameters were estimated for each subject's profile in the PK population by using WinNonlin (Pharsight Corporation) and were displayed in the data
listings. Data were summarized by using descriptive statistics (mean, SD, % coefficient of variation [CV], confidence interval (CI), median, minimum, and maximum) and are presented by treatment group. Geometric means were included for AUC and Cmax estimations and were included for some other PK parameters. By using Generalized Linear Model (GLM) procedure in SAS®, an analysis of variance (ANOVA) was performed on natural logarithmic (In) transformed parameters AUC0-t, AUC AUCo-τ, CaVg, and Cmax and on untransformed parameters t½, and λζ at the significance level of 0.05. The intrasubject CV was calculated for AUC0-t, AUC AUCo-τ, and Cmax by using the ANOVA residual error.
Dose linearity following single-dose administration (Period 1 ) was assessed after natural-log transformation for the AUC0-t, AUC and Cmax.
The following Period 1 comparisons for PK parameters were made:
• Comparison 1 : 600 μg (0.24%) TBS-2 versus 1200 μg 0.48% TBS-2;
• Comparison 2: 600 μg (0.24%) TBS-2 versus 1800 μg 0.72% TBS-2;
• Comparison 3: 1200 μg (0.48%) TBS-2 versus 1800 μg 0.72% TBS-2.
3. List of Tables and Figures
Table 9 1 : Study Schedule
Table 9 2: Schedule of Pharmacokinetic Sample Collection
Table 1 1 1 : Medical History (Single-Dose Population)
Figure 19: Mean Corrected Free Testosterone Concentrations (Single-Dose
Population)
Figure 20: Mean Corrected Total Testosterone Concentrations (Single-Dose
Population)
Figure 21 : Mean Corrected Dihydrotestosterone Concentrations (Single-Dose
Population)
Figure 22: Mean Corrected Estradiol Concentrations (Single-Dose Population) Figure 23: Mean Corrected SHBG Concentrations (Single-Dose Population)
Figure 24: Mean Observed Free Testosterone Concentrations (Single-Dose
Population)
Figure 25: Mean Observed Total Testosterone Concentrations (Single-Dose
Population)
Figure 26: Mean Observed Dihydrotestosterone Concentrations (Single-Dose Population)
Figure 27 Mean Observed Estradiol Concentrations (Single-Dose Population) Figure 28 Mean Observed SHBG Concentrations (Single-Dose Population)
Figure 29 Mean Free Testosterone Plasma Concentrations (Multi-Dose Population) Figure 30 Mean Total Testosterone Plasma Concentrations (Multi-Dose Population) Figure 31 Mean Dihydrotestosterone Plasma Concentrations (Multi-Dose
Population)
Figure 32 Mean Estradiol Plasma Concentrations (Multi-Dose Population)
Figure 33 Mean SHBG Plasma Concentrations (Multi-Dose Population)
Figure 34 Spaghetti Concentration Plots with Mean for Free Testosterone Plasma
Concentrations (Multi-Dose Population)
Figure 35: Spaghetti Concentration Plots with Mean for Total Testosterone Plasma
Concentrations (Multi-Dose Population)
Figure 36: Spaghetti Concentration Plots with Mean for Dihydrotestosterone Plasma
Concentrations (Multi-Dose Population)
Figure 37: Spaghetti Concentration Plots with Mean for Estradiol Plasma
Concentrations (Multi-Dose Population)
Figure 38: Spaghetti Concentration Plots with Mean for SHBG Plasma
Concentrations (Multi-Dose Population)
Table 1 1 22 Free Testosterone Summary (Single-Dose Population)
Table 1 1 23 Total Testosterone Summary (Single-Dose Population)
Table 1 1 24 Dihydrotestosterone Summary (Single-Dose Population)
Table 1 1 25 Estradiol Summary (Single-Dose Population)
Table 1 1 26 SHBG Summary (Single-Dose Population)
Table 1 1 27 Free Testosterone Summary for Multiple Dose Profile (Multi-Dose
Population)
Table 1 1 28 Free Testosterone Concentration at Baseline (Single-Dose Population)
Table 1 1 29: Total Testosterone Summary for Multiple Dose Profile (Multi-Dose
Population)
Table 1 1 30: Total Testosterone Concentration at Baseline (Single-Dose Population) Table 1 1 31 : Dihydrotestosterone Summary for Multiple Dose Profile (Multi-Dose
Population)
Table 1 1 32 !: Dihydrotestosterone Concentration at Baseline (Single-Dose Population)
Table 1 1 33 i: Estradiol Summary for Multiple Dose Profile (Multi-Dose Population)
Table 1 1 34 k Estradiol Concentration at Baseline (Single-Dose Population)
Table 1 1 35 i: SHBG Summary for Multiple Dose Profile (Multi-Dose Population)
Table 1 1 36 i: SHBG Concentration at Baseline (Single-Dose Population)
Table 1 1 37 ': Dose Proportionality Analysis (Single-Dose Population)
Table 1 1 38 i: Analysis of Variance for Some Pharmacokinetic Parameters (Single-Dose
Population)
Table 1 1 39: Paired t-Test Results for Pharmacokinetic Parameters AUCO-8 and AUC0-
24 for Subjects Who Had 1200 μg TBS-2 in Period 1 and Period 2
Table 12 1 : Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term (Single-Dose Population)
Table 12 2: Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term (Multi-Dose Population)
Table 12 3: Subjects with Adverse Reactions (Single- and Multi-Dose Populations) Table 12 4:Subjects with New Abnormal Hematology Laboratory Evaluation Results
Post Dose (Single- and Multi-Dose Populations)
Table 12 5: Subjects with New Abnormal Chemistry Laboratory Evaluation Results
Post Dose (Single- and Multi-Dose Populations)
Table 12 6: Subjects with New Abnormal Urinalysis Laboratory Evaluation Results
Post Dose (Single- and Multi-Dose Populations)
Table 12 7: Subjects with Abnormal Basic Ear, Nose, and Throat Examination Results
(Single- and Multi-Dose Populations)
4. Glossary of Abbreviations and Terms
AE adverse event
ALT alanine transaminase
ANOVA analysis of variance
AST aspartate transaminase
AUC area under the plasma concentration time curve
AUC0-∞ AUC from time zero to infinity
AUCO-t AUC from time zero to the last measurable concentration time point
AUCo-τ AUC from time zero to the dosing interval (where τ=8 hours) during multi-dose period
AUCo-8 AUC from time zero to 8 hours during single-dose period
BMI body mass index
BP blood pressure
BUN blood urea nitrogen
Cavg average steady-state concentration
CFR Code of Federal Regulations
CI confidence interval
CK creatine kinase
CL Clearance
Cmax maximum concentration observed after dosing
Cmin minimum concentration over a dosing interval during multiple dosing
ConcBase baseline concentration
ConcBC baseline corrected concentration
ConcBLQ Active dose concentrations corrected for BLQ
Cpd pre-dose concentration determined immediately before a dose at steady state
CRF case report form
CRU clinical research unit
CS clinically significant
CV coefficient of variation
DHT Dihydrotestosterone
ECG Electrocardiogram
eCRF electronic case report form
FDA Food and Drug Administration
FSD female sexual dysfunction
GCP good clinical practice
GGT gamma-glutamyl transferase
GLM Generalized Linear Model
HbsAg hepatitis B surface antigen
HCV hepatitis C virus
HIV human immunodeficiency virus
HR heart rate
ICF informed consent form
IRB institutional review board
In Logarithmic
MedDRA Medical Dictionary for Regulatory Activities
NCS not clinically significant
OTC over-the-counter
PD Pharmacodynamic
PI Principal Investigator
PK Pharmacokinetic
PTF % peak to trough fluctuation
PTS % peak to trough swing
RR respiratory rate
SAP statistical analysis plan
SD standard deviation
SHBG sex hormone-binding globulin
t½ elimination half-life
tmax time of observed Cmax relative to the time of dosing
VPA vaginal pulse amplitude
λζ terminal elimination rate constant Ethics
A. Institutional Review Board (IRB)
The study and any amendments were reviewed by the Institutional Review Board (IRB) for each center. Any subsequent protocol amendments or informed consent revisions were approved by the IRB before any changes were initiated.
B. Ethical Conduct of the Study
This study was conducted in accordance with the ethical principles originating from the Declaration of Helsinki and current good clinical practice (GCP) and in compliance with local regulatory requirements and 21 CFR 312.
C. Subject Information and Consent
The format and content of the subject information sheet and informed consent form (ICF) were agreed upon by the Principal Investigator (PI), the IRB, and Trimel Pharmaceuticals Corp. (hereafter referred to as Trimel). Each subject's written informed consent to participate in the study was obtained before any study specific procedures were performed. The PI or a medically qualified member of the study team provided the subject with a full explanation of the study drugs and manner of treatment allocation, the possible risks and benefits, and the compensation or treatment available in the event of study-related injury.
Subjects who agreed to participate in the study signed and dated an ICF. The ICF was also signed and dated by designated site personnel. The original and all amended signed and dated ICFs were retained at the study site, and copies of these forms were provided to the subject.
6. Investigators and Study Administrative Structure
This study was conducted at 1 investigative site in the United States. The PI at the investigative site was responsible for the validity and accuracy of the data supplied on the case report form (CRF). Delegation of authority for completion of the CRF was permitted, but the PI was responsible for its accurate completion and was required to sign the completed CRF that it was a true and accurate reflection of the subject's participation in the study.
The PI at the investigative site signed the protocol signature page. By signing this page, the PI agreed to conduct the study in accordance with the study protocol and also
to comply with the requirements regarding the obligations of clinical investigators and all other pertinent requirements of applicable regulatory agencies.
7. Introduction
TBS-2 is developed for the treatment of anorgasmia.
A. Background
Anorgasmia is the second most frequently reported women's sexual problem after hypoactive sexual desire disorder. The Global Survey of Sexual Attitudes and Behaviors assessed sexual problems in 9000 women aged 40 to 80 years (inclusive) in 29 countries. The prevalence of inability to reach orgasm ranged from 17.7% (in
Northern Europe) to 41 .2% (in Southeast Asia). In the PRESIDE survey of over 31 ,000 women, approximately 10% reported low desire with distress and almost 5% report difficulty reaching orgasm with distress. Anorgasmia is considered to be the persistent or recurrent delay in, or absence of, orgasm following a normal sexual excitement phase, causing marked distress or interpersonal difficulty. When a woman has sexual activity that is not accompanied by good quality orgasmic release, sexual activity may become a chore or a duty rather than a mutually satisfying, intimate experience. This may also lead to secondary loss of sexual interest and/or interpersonal difficulties.
Testosterone, the primary circulating androgen in women, is a naturally occurring steroid secreted by the ovaries and the adrenal glands. Contrary to the sudden drop in estrogen during menopause, serum levels of androgens fall gradually as women age primarily due to a decrease in the production of adrenal androgen precursors.
Testosterone plays a role in regulation of mood, body composition, and bone mineral density, and has central and peripheral effects on sexual function. In the periphery, testosterone is required for nitric oxide to stimulate vasocongestion for the engorgement of clitoral tissue and vaginal lubrication during sexual arousal. Testosterone stimulates dopamine release in various brain structures implicated in motivation and reward systems, including sexual desire. Testosterone was found to stimulate dopamine release in the medial preoptic area of the anterior hypothalamus under basal conditions and with sexual stimulation in rats.
The use of androgens to increase women's sexual desire was first reported in 1940 by Loeser. Salmon observed that a number of young married women who formerly considered themselves "frigid" were able to experience "a marked increase in coital gratification, culminating in an orgasm" after testosterone propionate injections, with the effects wearing off within several weeks after the discontinuation of the injections. In the 1980s, the role of androgens in maintaining sexual functioning was studied in
oophorectomized women. In this 3-month, prospective open-label study of 44 women, monthly injections of estrogen and testosterone increased rates of sexual desire, sexual arousal, and number of fantasies. Furthermore, rates of intercourse and orgasm were higher in women treated with androgens and estrogen compared to the controls. Over the two past decades, over 80 studies have been conducted in women with hypoactive sexual desire disorder by using exogenous testosterone through the oral, transdermal, sublingual or parenteral route of administration with or without concomitant estrogen therapy, resulting in an increase in sexual desire, orgasm, arousal, frequency of satisfactory sexual activity, pleasure and responsiveness.
Trimel has developed an intranasal testosterone gel containing 0.24% to 0.72% testosterone with castor oil, oleoyl polyoxylglycerides, and colloidal silicon dioxide as excipients. TBS-2 is administered as a dose applied equally into each nostril. The formulation has many advantageous features including rapid absorption into systemic circulation and rapid clearance, the lack of first-pass metabolism, the avoidance of transference from one person to another, and ease of use. It is a logical next step in a research program dedicated to investigating the role of testosterone, to investigate whether TBS-2, in the absence of other FSDs, has a direct effect on sexual function in general and anorgasmia in particular.
Two pharmacokinetic (PK)/pharmacodynamic (PD) studies have been performed to evaluate the effect of testosterone on the amygdala reactivity and PD endpoints associated with sexual stimulation. The first PD study (CMO-nr: 2004/144) investigated whether the age-related decline in androgen levels was associated with reduced amygdala activity, and whether exogenous testosterone could restore amygdala activity. The enhanced testosterone levels correlated positively with superior frontal cortex responses and negatively with orbitofrontal cortex responses across individuals, which
may reflect testosterone-induced changes in amygdala regulation. These results support the modulatory role of testosterone on emotional cues suggesting testosterone helps to enhance sensation that is necessary to trigger orgasm.
The second study (TBS-2-2010-01 ) evaluated the PK of 3 dose levels of TBS-2 and sexual function PD compared to a testosterone patch and placebo. The PK results demonstrated a linear increase in plasma testosterone levels with increasing dose levels. During the first PK series, the mean plasma testosterone concentration and area under the plasma concentration-time curve (AUC) in the TBS-2 high-dose group reached the same level as the mean concentration and AUC of the testosterone patch at steady state. During the second PK series, the mean plasma testosterone
concentration and AUC in the TBS-2 high-dose group reached levels higher than the mean plasma testosterone concentration and AUC in the testosterone patch group, but still within the upper limit of the normal physiological range. Sexual function PD efficacy was explored by assessing the role of testosterone on vaginal pulse amplitude (VPA), subjective arousal questionnaires, and validated computer tasks. Significant differences were observed in VPA response following testosterone administration in women in the anorgasmia cohort. Women who received TBS-2 (see Examples 9 and 10 above) had greater responsiveness in genital response and subjective sexual measurement compared to women receiving the testosterone patch.
The product under investigation in this study, TBS-2 at 0.24%, 0.48% and 0.72% strengths, is a bioadhesive intranasal testosterone gel. Contrary to the transdermal administration (Intrinsa, a testosterone transdermal patch that has been approved in the European Union for the treatment of hypoactive sexual desire disorder (HSDD) in bilaterally oophorectomized and hysterectomized [surgically induced menopausal] women receiving concomitant estrogen therapy), administration of the bioadhesive TBS-2 through the nasal mucosa allows for rapid absorption into the systemic circulation. The rapid onset and higher peak concentration are hypothesized to be more effective in enhancing sexual desire and orgasm with lower total concentrations of testosterone needed, thus increasing efficacy and decreasing side effects. In addition, TBS-2 may prove effective in alleviating anorgasmia in an "as needed" way, thus avoiding chronic exposure to testosterone.
8. Study Objectives
A. Primary Objective
The primary objective of this study was to assess the bioavailability of total testosterone through PK profiles obtained following single administration of an intranasal application of TBS-2 at doses of 600 μg, 1200 μg, and 1800 μg, and multiple administration of 1200 μg TBS-2 given 3 times daily (t.i.d) for the first 2 days and once on the morning of the third day.
B. Secondary Objectives
The secondary objectives of this study were:
• To assess the bioavailability of free testosterone, sex hormone-binding globulin (SHBG), dihydrotestosterone (DHT), and estradiol through PK profiles obtained following single administration of an intranasal application of TBS-2 at doses of 600 1200 μ9, and 1800 μ9, and multiple administration of 1200 μg TBS-2 given for 3 days (t.i.d. for the first 2 days and once on the morning of the third day).
• To evaluate the safety of TBS-2.
9. Investigational Plan
A. Overall Design and Plan Description
This was a phase 1 , single-center, randomized, open-label parallel-group study in healthy, normal-cycling adult women. Approximately 24 healthy adult women were to be enrolled. Subjects were randomly assigned on a 1 :1 :1 basis to 1 of 3 treatment groups (Cohort 1 , Cohort 2, or Cohort 3) during Period 1 and were administered a single dose of an intranasal application of TBS-2 at doses of 600 μ9, 1200 μ9, or 1800 μg (single doses of 300 μ9, 600 μ9, and 900 μg per nostril). At the end of Period 1 , a total of 8 subjects sampled from these 3 cohorts, who were willing and able to continue with the multiple-dose portion of the study were selected to participate in Period 2. During Period 2, subjects were administered 1200 μg TBS-2 (600 μg per nostril) t.i.d. for 2 days and once on the morning of the third day.
Subjects were screened (Visit 1 ) for eligibility up to 3 weeks prior to dosing in Period 1 , and were admitted to the Clinical Research Unit (CRU) at 0700 hours on the day prior to dosing (Visit 2, Day 1 ) for baseline testosterone measurement. On Day 2, subjects were administered a single dose of TBS-2 and remained in the CRU for 72 hours post-dose (Day 4) for safety monitoring and PK assessments. Subjects were discharged from the clinic on Day 4, and subjects who did not continue into Period 2 also underwent close-out assessments. Depending on their cycle, subjects selected to participate in Period 2 returned to the CRU approximately 26 to 32 days following the conclusion of Period 1 for Visit 3 (Period 2). During Period 2, subjects were admitted to the CRU at 0700 hours on the day of dosing (Visit 3, Day 1 ). On Days 1 and 2, subjects were administered TBS-2 at 0800 hours (±30 minutes), 1600 hours (±30 minutes), and 2400 hours (±30 minutes). On Day 3, subjects were administered TBS-2 at 0800 hours. Subjects remained in the CRU for 48 hours following dosing on Day 3 for safety monitoring and PK assessments. Subjects were discharged from the clinic on Day 5.
During Period 1 , blood samples for determination of baseline testosterone (free and total), SHBG, dihydrotestosterone (DHT), and estradiol concentration were collected on Day 1 at 0745 hours and then at 15, 30, and 45 minutes and at 1 , 1 .5, 2, 4, 6, 8, 12, 16, 20, and 23.5 hours relative to an 0800 hour clock time. Blood samples for determination of testosterone (free and total), SHBG, dihydrotestosterone, and estradiol concentration were collected on Day 2 (15, 30, and 45 minutes and at 1 , 1 .5, 2, 4, 6, 8, 12, 16, and 20 hours post-dose) and Day 3 (24, 32, 40, and 48 hours post-dose) during the
confinement period. During Period 2, a blood sample for a baseline serum testosterone concentration was collected at 0745 hours (ie, 15 minutes prior to study drug
administration). Blood samples for determination of testosterone (free and total), SHBG, dihydrotestosterone, and estradiol plasma concentration were collected on Day 1 (pre- dosepre-dose [15 minutes prior to dosing] and at 1545 and 2345 hours), Day 2 (1545 and 2345 hours), Day 3 (15, 30, and 45 minutes and at, 1 , 1 .5, 2, 4, 6, 8, 12, 16, and 20 hours), and Day 4 (24, 32, 40, and 48 hours) during the confinement period.
Other assessments performed during the study included the monitoring of adverse events (AEs), clinical laboratory evaluations (chemistry [including hormone profile], hematology, and urinalysis), vital signs assessments (systolic and diastolic
blood pressure [BP], heart rate [HR], respiratory rate [RR], and body temperature), and physical examinations. In addition, otorhinolaryngological examination findings, 12-lead electrocardiogram (ECG) readings, medical history, and concomitant medication use were recorded.
B. Discussion of Study Design and Choice of Control Groups
This was a phase 1 , single-center, randomized, open-label parallel-group study of TBS-2 in 3 cohorts of subjects (Cohorts 1 , 2, and 3) in Period 1 (single-dose) and a multiple-dose cohort in Period 2. Approximately 24 healthy women were to receive a single intranasal dose of 600 μg, 1200 μg, or 1800 μg TBS-2 to evaluate the safety, tolerability, and PK of TBS-2. Subjects were confined in a CRU for 4 days during
Period 1 and 5 days during Period 2.
C. Selection of Study Population
Inclusion Criteria
Subjects were eligible for study inclusion if they met all of the following inclusion criteria:
1 . Female subjects between 18 and 40 years of age.
2. Subjects with regular menstrual cycles between 26 and 32 days.
3. Women of childbearing potential must have agreed to use 1 of the following reliable birth control methods prior to the study, during the study, and up until 1 month after the end of the study:
a. Surgically sterile (tubal ligation).
b. Intrauterine device in place for at least 3 months prior to study initiation.
c. Barrier method (condom with spermicidal agent use by partner).
d. Abstinence.
4. Subjects negative for drugs of abuse, hepatitis B-surface antigen (HbsAg), hepatitis C, human immunodeficiency virus (HIV), and pregnancy (serum β-HCG).
5. Subjects with a Body Mass Index (BMI) between 18.5 kg/m2 and 35 kg/m2 (inclusive).
6. Subjects with a normal ear, nose, throat (ENT) examination.
7. Subjects with normal thyroid-stimulating hormone (TSH) values.
8. Subjects with no clinically significant findings in the physical examination, 12-lead ECG, and vital signs.
9. Subjects with normal thyroid function. Physiological prolactin concentration.
10. Subjects with no clinical laboratory values outside of the acceptable range, unless the PI decided that they were not clinically significant.
1 1 . Subjects who were able to understand and provide written informed consent.
12. Subjects who were available for the entire study period and were willing to adhere to protocol requirements, as evidenced by a signed ICF.
Exclusion Criteria
Subjects were excluded from study participation if they met any of the following exclusion criteria:
1 . Known history of hypersensitivity to testosterone (eg, Intrinsa patch) and/or related drugs.
2. Known history of polycystic ovarian syndrome.
3. Known history or presence of cardiac, pulmonary, gastrointestinal, endocrine,
musculoskeletal, neurological, psychiatric, hematological, reproductive, liver, or kidney disease, unless judged not clinically significant by the PI or medical designate.
4. Presence of or known history of estrogen-responsive tumors such as breast cancer and/or history of any cancer, excluding basal cell carcinoma.
5. Known history of frequent clinically significant acne.
6. Known history of hirsutism.
7. History of nasal surgery, specifically turbinoplasty, septoplasty, rhinoplasty, "nose job," or sinus surgery.
8. Prior nasal fractures.
9. Active allergies, such as rhinitis, rhinorrhea, and nasal congestion.
10. Mucosal inflammatory disorders, specifically pemphigus and Sjogren's syndrome.
1 1 . Sinus disease, specifically acute sinusitis, chronic sinusitis, or allergic fungal
sinusitis.
12. History of nasal disorders (eg, polyposis, recurrent epistaxis [>1 nose bleed per month], abuse of nasal decongestants) or sleep apnea.
Use of any form of intranasal medication delivery, specifically nasal corticosteroids and oxymetazoline containing nasal sprays (eg, Dristan 12-Hour Nasal Spray).
History of hepatitis B, a positive test for HbsAg, a history of hepatitis C, a positive test for hepatitis C antibody, a history of HIV infection, or demonstration of HIV antibodies.
Any history of severe allergic reaction including drugs, food, insect bites,
environmental allergens, or any condition known to interfere with the absorption, distribution, metabolism, or excretion of drugs.
Any history of drug abuse or alcohol abuse as per the Diagnostic and Statistical Manual of Mental Disorders (fourth edition) criteria within 6 months of study drug administration.
Current treatment with any hormonal therapy within previous 12 months, treatment with drugs that interfere with metabolism of testosterone within 30 days of study drug administration and/or any other prescription medications. Difficulty in abstaining from over-the-counter (OTC) medication use for the duration of study.
Use of oral, transdermal, and implant contraceptives within 30 days prior to study drug administration or a depot contraceptives injection within 1 year prior to study drug administration.
Evidence of pregnancy or lactation.
Subjects who were breast feeding or had breast fed within 6 months prior to the screening visit.
Administration of another investigational drug within 30 days prior to study drug administration.
Blood donation within 56 days prior to study drug administration.
Any participation as a plasma donor in a plasmapheresis program within 7 days preceding this study.
Intolerance to venipuncture.
History of abnormal bleeding tendencies or thrombophlebitis unrelated to
venipuncture or intravenous cannulation.
History of deep venous thrombosis or coagulation disorders.
Removal of Subjects from Therapy or Assessment
Subjects withdrawn from the study after receiving study drug were not replaced, regardless of the reason for withdrawal.
A subject may have been prematurely discontinued from the study for any of the following reasons:
• Significant noncompliance with the study protocol and procedures.
• Intercurrent illness that interfered with the progress of the study.
• Intolerable AE, including clinically significant abnormal laboratory findings, which in the opinion of the PI could have interfered with the subject's safety.
• Principal Investigator's decision that the withdrawal from the study was in the best interest of the subject.
D. Treatments
Treatments Administered
Subjects were randomly assigned on a 1 :1 :1 basis to 1 of 3 treatment groups (Cohort 1 , Cohort 2, or Cohort 3) during Period 1 and were administered a single dose of an intranasal application of TBS-2 at doses of 600 μg, 1200 μg, or 1800 μg (single doses of 300 μg, 600 μg, and 900 μg per nostril). At the end of Period 1 , a total of 8 subjects sampled from these 3 cohorts, who were willing and able to continue with the multiple-dose portion of the study were selected to participate in Period 2. During Period 2, subjects were administered 1200 μg TBS-2 (600 μg per nostril) t.i.d. for 2 days and once on the morning of the third day.
Treatment 1 : TBS-2 dispensers prefilled with 0.24% testosterone gel to deliver a single dose of 300 μg of testosterone per nostril, for a total dose of 600 μg given at 0800 hours (±30 minutes) on Day 2 of Period 1 for Cohort 1 .
Treatment 2: TBS-2 dispensers prefilled with 0.48% testosterone gel to deliver a single dose of 600 μg of testosterone per nostril, for a total dose of 1200 μg given at 0800 hours (±30 minutes) on Day 2 of Period 1 for Cohort 2.
Treatment 3: TBS-2 dispensers prefilled with 0.72% testosterone gel to deliver a single dose of 900 μg of testosterone per nostril, for a total dose of 1800 μg given at 0800 hours (±30 minutes) on Day 2 of Period 1 for Cohort 3.
Treatment 4: TBS-2 dispensers prefilled with 0.48% testosterone gel to deliver a single dose of 600 μg of testosterone per nostril, for a total dose of 1200 μg given t.i.d. daily at 0800 hours (± 30 minutes), 1600 hours (± 30 minutes), and 2400 hours (± 30 minutes) on Days 1 and 2 of Period 2, and once in the morning at 0800 hours (±30 minutes) on Day 3 of Period 2 (Multidose group).
Subjects were instructed on the proper procedure for applying the TBS-2 gel intranasally by using the prefilled dispensers. Study drug was self-administered at 0800 hours on Day 2 of Period 1 and on Days 1 , 2, and 3 of Period 2. Self-administration of TBS-2 was monitored by study personnel. Subjects were instructed to not blow their nose or sniff immediately, and for the first hour following study drug administration.
Identity of Investigational Products
Active study drug was supplied in prefilled dispensers containing 0.24%, 0.48%, or 0.72% TBS-2.
The TBS-2 gels were packaged in prefilled dispensers. A multidose dispenser was used for gel deposition into the nasal cavity. The dispenser was a finger actuated dispensing system designed to dispense 125 μΙ_ of TBS-2 gel per actuation from a non- pressurized container into the nasal cavity. The key components of the multiple-dose dispenser included a barrel, base, pump, and actuator, which were composed of polypropylene and a piston, which was composed of polyethylene. See Fig.39 and CMC Section below.
All study drug boxes and dispensers were labeled and supplied according to applicable regulatory requirements. Qualified licensed study personnel prepared the unit doses for the study as per the randomization schedule. Study drug was provided to the subjects in appropriate unit-dose foil pouches, clearly labeled as to the amount of drug being given.
Study drug was stored in a secure location at a controlled room temperature of 15°C to 25 °C (59°F to 77°F). The storage location was a locked room with limited access, available to appropriate study personnel only.
The PI, or an approved representative, (eg, co-investigator), ensured that all study drug was stored in a secured area, under recommended storage conditions, and in accordance with applicable regulatory requirements.
Upon the completion or termination of the study, and upon written authorization from Trimel, all unused and/or partially used study drug was returned or destroyed at the investigational site, as specified by Trimel.
Method of Assigning Subjects to Treatment Groups
A randomization schedule was prepared by Premier Research and was provided to the PI at the CRU prior to the start of the study.
Subjects who met the enrollment criteria were randomly assigned on a 1 :1 :1 basis to 1 of 3 treatment groups.
Selection of Doses in the Study
The doses of 600 μg, 1200 μg, and 1800 μg were selected for this study based on clinical pharmacology data and are appropriate for a phase 1 study. The animal toxicology studies completed to date do not suggest any unusual or unexpected toxicities related to TBS-2 exposure. Subjects were not expected to experience side effects with TBS-2.
Selection and Timing of Dose for Each Subject
Study drug was administered on Day 2 of Period 1 (single-dose). During Period 2 (multi-dose), study drug was administered t.i.d. on Days 1 and 2 and on the morning of Day 3. Subjects were confined in a CRU for 4 days during Period 1 and 5 days during Period 2.
Blinding
This was an open-label study; subjects were not blinded to treatment
assignments.
Prior and Concomitant Therapy
The use of any prescription or nonprescription medication or treatment with drugs that interfere with the metabolism of testosterone was prohibited within 30 days of study drug administration and until the final study visit. The use of any form of intranasal medication delivery, specifically nasal corticosteroids and oxymetazoline-containing nasal sprays (eg, Dristan 12-Hour Nasal Spray) was prohibited until the final study visit. Current treatment with any hormonal therapy was prohibited 12 months prior to study drug administration and until the final study visit. Also prohibited was the use of oral, transdermal, or implant contraceptives within 30 days prior to drug administration or a
depot contraceptives injection within 1 year prior to study drug administration. In addition, the administration of another investigational drug was prohibited within 30 days prior to study drug administration. The use of illegal drugs was not permitted while the subjects were enrolled in this study.
Throughout the study, the PI could prescribe any concomitant therapy deemed necessary to provide adequate supportive care. The PI was to notify the sponsor of any subject using medications within 30 days prior to Day 1 or if concomitant medications were required during the study. The decision to allow the subject to be enrolled into the study or take medications during the study was made jointly by the sponsor and PI and was based on their opinion that the use of the medication was unlikely to compromise the safety of the subject or the interpretation of the study data. All medications taken within 30 days prior to dosing and all concomitant therapy were recorded on the appropriate section of the CRF.
Treatment Compliance
Subjects who received doses of study drug were monitored by study personnel for 1 hour post-dose to assure conformity to the TBS-2 instructions. Subjects were confined in a CRU for 4 days during Period 1 and 5 days during Period 2 and were closely monitored. A drug dispensing log was maintained. Pharmacokinetic results were available to confirm compliance.
Study drug was administered on Day 2 of Period 1 (single-dose). During Period 2 (multi-dose), study drug was administered t.i.d. on Days 1 and 2 and on the morning of Day 3. Subjects were confined in a CRU for 4 days during Period 1 and 5 days during Period 2.
E. Pharmacokinetic and Safety Variables
Pharmacokinetic and Safety Measurements Assessed and Flow Chart
The Study Schedule is presented in Table 9-1 .
Table 9-1: Study Schedule

Abbreviations: AE = adverse event; CBC = complete blood count; DHT = dihydrotestosterone; ECG = electrocardiogram; ENT = ear, nose, throat; FSH = follicle-stimulating hormone; HbsAg = hepatitis B surface antigen; HIV = human immunodeficiency virus; LH = luteinizing hormone; PK = pharmacokinetic; SHBG = sex hormone-binding globulin; TSH = thyroid-stimulating hormone.
a. A total of 8 subjects who were willing and able to continue on to the multiple dose portion of the study were selected from Cohorts 1, 2, and 3 and comprised the Multi-Dose group.
b. Vital signs (heart rate, blood pressure, temperature, and respiratory rate).
c. The site physician examined subjects and identified any clinically significant changes to the nasal mucosa at follow-up.
d. Chemistry, hematology, urinalysis.
e. Hormone profile: TSH, total and free tri-iodothyronine, total and free thyroxine, FSH, prolactin, and progesterone.
f. Study drug administered at 0800 hours (+30 minutes).
g. Study drug administered at 0800 hours (+30 minutes), 1600 hours (+30 minutes), and 2400 hours (+30 minutes).
h. Study drug a administered at 0800 hours (+30 minutes).
i. Blood samples for the 24-hour baseline profile (total and free testosterone, SHBG, DHT, and estradiol) were collected at 0745 hours and then at 15, 30, and 45 minutes and at 1, 1.5, 2, 4, 6, 8, 12, 16, 20, and 23.5 hours relative to an 0800 hour clock time.
j. Blood samples for the 48-hour PK profile (total and free testosterone, SHBG, DHT, and estradiol) were collected at 15, 30, and 45 minutes and at 1, 1.5, 2,
4, 6, 8, 12, 16, 20, 24, 32, 40, and 48 hours,
k. A baseline serum testosterone blood sample was collected at 0745 hour (ie, 15 minutes prior to dosing).
1. PK sampling for trough level measurement was done prior to dosing at 0800, 1600, and 2400 hours.
m. The 48-hour PK profile blood samples were collected pre-dose at 0745 hours and at 15, 30, and 45 minutes and at 1, 1.5, 2, 4, 6, 8, 12, 16, 20, 24, 32, 40, and 48 hours.
Appropriateness of Measurements
The measurements included in this study were typical for a phase I single- and multiple-dose study.
Pharmacokinetic Drug Concentration Measurements
Whole blood samples for determination of testosterone (free and total), SHBG, dihydrotestosterone, and estradiol concentration were collected at each time point indicated in Table 9-2Table 9-2. The date and time of collection were recorded on the CRF to the nearest minute. Whole blood samples were obtained through direct venipuncture; the use of a heparin lock or IV indwelling catheter was allowed for early time point PK sample collections. The volume of plasma samples collected for PK analysis for each subject was approximately 120 mL during Period 1 and 92 mL during Period 2.
Table 9-2: Schedule of Pharmacokinetic Sample Collection

Abbreviations: DHT = dihydrotestosterone; PK = pharmacokinetic; SHBG - sex hormone-binding globulin; t.i.d. = three times daily
Note: During Period 1 dosing occurred on the morning of Day 2. During Period 2, dosing occurred t.i.d. on Days 1 and 2, and once on the morning of Day 3.
Pharmacokinetic Parameter Estimates
Concentration-time data for 5 analytes [testosterone (Total and Free), SHBG, dihydrotestosterone and estradiol] were determined and PK parameters were calculated from them. Actual sampling time points were recorded and used in calculation of the actual elapsed time from dose to sample for PK calculations. As each administration was made to each of the nostrils, the time of dosing was the time of the first nostril administration. The units of concentration for each of the 5 different analytes may have differed so the final units of concentration reflected the bioanalytical laboratory data reported on the concentrations. Units of PK parameters were derived from the analyte concentration units. Baseline analyte concentrations from the 24-hour pre-dose profile were subtracted from the reported analyte concentrations before calculation of the PK parameters.
The following PK parameters were calculated following single-dose (Cohorts 1 , 2, and 3) for PK characterization. Profile intervals were from the start of the first dosing and continued to the last sample collected on the dosing day. PK parameters were estimated by standard methods used by the WinNonlin program and short descriptions are given below. The determination of the terminal elimination phase was more fully described in the WinNonlin documentation. Note that Cmin, Cmax, and tmax were taken from the actual measured values but after baseline correction for Cmin and Cmax.
Parameter Description and Calculation
AUCo-t Area under the plasma concentration time curve from time zero to the last
measurable concentration time point, calculated by using the combination of the linear and log trapezoid rules. The linear trapezoidal rule would be used from the time of dose to tmax and the log trapezoidal rule would be used following tmax.
AUCo-8 Area under the plasma concentration time curve from time zero to 8 hours.
AUCo-24 Area under the plasma concentration time curve from time zero to 24 hours.
Area under the plasma concentration time curve from time zero to infinity, calculated as where iast is the last measurable concentration, and is the terminal elimination rate constant calculated from the log-linear terminal phase.
Cmax Maximum concentration observed after dosing.
Co Concentration at pre-dose.
C24 Concentration at 24 hours.
tmax Time of observed maximum concentration (Cmax) relative to the time of dosing. λζ Terminal elimination rate constant.
j-j.j ) .
tl/2
Elimination half-life calculated as Ξ
The following PK parameters were calculated following multiple-dose (Multi-
Dose group):
Parameter Description and Calculation
AUCo-τ Area under the concentration-time curve from time zero to the dosing interval (τ = 8 hours).
AUCo-8 Area under the plasma concentration time curve from time zero to 8 hours.
AUCo-24 Area under the plasma concentration time curve from time zero to 24 hours.
c ^max Maximum concentration observed after dosing.
tmax Time of observed maximum concentration (Cmax) relative to the time of dosing. c ^rmn Minimum concentration over a dosing interval during multiple dosing.
Cpd Pre-dose concentration determined immediately before a dose at steady state
Co Concentration at pre-dose.
C24 Concentration at 24 hours.
Parameter Description and Calculation
c ^avg
Average steady- state concentration, calculated as t ,
where τ = 8 hours.
PTF (€ΏΜΧ - ca,ja 0)*ia©
% Peak to Trough Fluctuation: ι-·'^
PTS % Peak to Trough Swing is the degree of concentration swing at a steady state:
C ■ "
Additional exploratory analyses of PK parameters may have been performed as necessary.
Safety Measurements
Safety measurements included the monitoring of AEs, clinical laboratory evaluations (chemistry [including hormone profile], hematology, and urinalysis), vital signs, and 12-lead ECGs. In addition, physical and otorhinolaryngological examinations were performed and medical history and concomitant medication use were recorded. Safety measurements were performed at the times specified in Table 9-1 . The PI followed all clinically-significant abnormal findings after study drug treatment until resolution or return to baseline.
Adverse Events
An AE is any untoward, undesired, unplanned clinical event in the form of signs, symptoms, disease, or laboratory or physiological observations occurring in a human participating in a clinical study with a Trimel product, regardless of causal relationship. A pre-existing condition is one that is present prior to study drug administration and is reported as part of the subject's medical history. Pre-existing conditions were reported as an AE only if the frequency, intensity, or character of the pre-existing condition worsened during the course of the study.
Laboratory abnormalities were not considered AEs unless they were associated with clinical signs or symptoms, or required medical intervention. However, a laboratory abnormality (eg, a clinically significant change detected in clinical chemistry [including hormone profile], hematology, urinalysis) that was independent from the underlying
medical condition and that required medical or surgical intervention, or led to study drug interruption or discontinuation, was considered an AE.
All AEs judged to be clinically significant, including clinically-significant laboratory abnormalities, were followed until resolution or return to baseline.
F. Severity Rating
All AEs or SAEs were assessed for severity, by using the following grading scale:
Mild An event easily tolerated by the subject; transient or mild discomfort
(usually <48 hours); no medical intervention/therapy required.
Moderate An event that may interfere with normal, everyday activities; some
assistance may be needed; no or minimal medical intervention/therapy required.
Severe An event that prevented the subject from performing their normal,
everyday activities; some assistance usually required; medical
intervention/therapy required, hospitalizations possible.
When changes in the severity of an AE occurred more frequently than once a day, the maximum severity for the experience that day was noted. If the severity category changed over a number of days, then those changes were recorded separately (with distinct onset dates). The severity of the AE was recorded in the appropriate section of the AE page of the CRF. The evaluation of severity was distinguished from the evaluation of "seriousness." A severe event might not have met the criteria for seriousness and a serious event might have been evaluated as mild.
G. Causality Rating
For each reported adverse reaction, the PI made an assessment of the relationship of the event to the study drug by using the following scale:
Definite There is a plausible temporal relationship with drug administration and withdrawal, and reappears after drug restart.
Probable There is a plausible temporal relationship with drug administration.
Possible There is a plausible temporal relationship with drug administration but can reasonably be associated to other factors.
Unlikely There is no plausible temporal relationship with drug administration.
Unknown There are no sufficient elements to establish a correlation with drug intake.
Not Related Cannot be correlated to study drug administration.
H. Serious Adverse Events
The PI classified each AE as either serious or not serious. A serious adverse event (SAE) was defined as any AE occurring at any dose that resulted in any of the following outcomes:
• Death3
• Was life-threatening13
• Required inpatient hospitalization or prolongation of existing hospitalization
• Resulted in a persistent or significant disability/incapacity0
• Resulted in a congenital anomaly/ birth defect
• Additionally, important medical events that may not have resulted in death, been life-threatening, or required hospitalization may be considered an SAE when, based upon appropriate medical judgment, they may jeopardize the subject and may require medical or surgical intervention to prevent one of the outcomes listed above. Example: allergic bronchospasm requiring intensive treatment in an emergency room or at home.
NOTES:
a. Death was an outcome and was NOT the AE. In the event of death, the cause of death was recorded as the AE. The only exception was "sudden death" when the cause was unknown.
b. Life-threatening AEs included any adverse drug experience, which, in the view of the PI, placed the subject at immediate risk of death from the reaction as it occurred. It did not include a reaction that, had it occurred in a more serious form, might have caused death,
c Disability was defined as a substantial disruption in a person's ability to conduct normal life functions.
All SAEs that resulted in death or were life-threatening, regardless of causal relationship, were reported to Trimel (or designee) within 24 hours of the site's knowledge of the event. A copy of the initial SAE report was to have been received within 1 business day. All other SAEs or other events reportable to Trimel were forwarded to Trimel (or designee) within 1 business day. If there was any doubt whether the information constituted an SAE, the information was treated as an SAE for the purposes of this study.
Clinical Laboratory Tests
Clinical laboratory tests were performed according to the schedule provided in Table 9-1 .
Serum chemistry evaluations included sodium, potassium, chloride, glucose, urea, creatinine, calcium, phosphate, uric acid, total bilirubin, albumin, aspartate transaminase (AST), alanine transaminase (ALT), alkaline phosphatase,
gamma-glutamyl transferase (GGT), creatine kinase (CK), and cholesterol, and hormone profiles.
Hormone profiles included TSH, total and free tri-iodothyronine, total and free thyroxine, FSH, prolactin, and progesterone.
Hematology evaluations included white blood count, hemoglobin, and
hematocrit.
Urinalysis included glucose, bilirubin, ketones, specific gravity, blood, pH, protein, urobilinogen, nitrites, leukocytes and, if necessary, microscopic examination.
At Screening and on admission to the CRU, a urine specimen was obtained to test for drugs of abuse (marijuana, cocaine, opiates, amphetamines, phencyclidine, benzodiazepines, and barbiturates) and alcohol was tested for by use of an alcohol breath test.
Testing for HbsAg, antiHCV, and HIV were performed at Screening (Visit 1 ) only. Samples were analyzed by MEDTOX Laboratories, Inc.
Vital Signs
Vital sign measurements included systolic and diastolic BP and HR, body temperature, and RR. Vital signs assessments were performed at the times specified in Table 9-1 .
12-Lead Electrocardiogram
A standard 12-lead ECG was assessed at Screening (Visit 1 ) only.
Other Safety Measurements
Physical examinations were performed at the times specified in Table 9-1
An ENT (otorhinolaryngological) nasal endoscopic examination was performed at Screening. Basic ENT (otorhinolaryngological) examinations were performed at the times specified in Table 9-1 ; the site physician examined subjects and identified any clinically significant changes to the nasal mucosa at follow-up.
Concomitant medications were monitored throughout the study.
I. Data Quality Assurance
This study was monitored by trained experienced personnel in accordance with GCPs. The clinical study monitor reviewed study records to verify adherence to the protocol, accuracy, completeness, and consistency of the data; and adherence to local regulations on the conduct of clinical research. The clinical monitor maintained regular contact with the site and had access to subject medical records and other study-related records needed to verify the entries on the CRFs.
Electronic CRFs (eCRFs) were used for this study and included only the subject's initials, date of birth, and an assigned subject number on the eCRFs as identifiers. The PI ensured the availability and reliability of source documents from which the information on the eCRF was derived, and was required to comply with document retention procedures as outlined in the protocol. Case report forms were reviewed for accuracy and signed and dated by the PI.
J. Statistical Methods and Determination of Sample Size
Statistical and Analytical Plans
The statistical methods presented in the study protocol were superseded by those described in the statistical analysis plan (SAP). This study evaluated the PK properties as well as the safety and tolerability of TBS-2. Power calculations were not performed. Data were summarized by using descriptive statistics (sample size, mean, median, standard deviation [SD], minimum, and maximum) for each of the safety variables by treatment group and overall. Data from all visits during the study were displayed in the data listings.
Concentration-time data for 5 analytes (testosterone [total and free], SHBG, dihydrotestosterone, and estradiol) were determined by a validated assay method and PK parameters were calculated. Actual sampling time points were recorded and used in calculation of the actual elapsed time from dose to sample for PK calculations. As each administration was to each of the nostrils, the time of dosing was the time of the first nostril administration. Baseline analyte concentrations from the 24-hour pre-dose profile were subtracted from the time-matched analyte concentrations following dose administration before calculation of the PK parameters.
The plasma TBS-2 concentration-time data were analyzed by using Phoenix WinNonlin (Pharsight Corporation). The PK parameters (refer to Pharmacokinetic Parameter Estimates) for testosterone (free and total), SHBG, DHT, and estradiol were calculated by standard noncompartmental methods for all subjects as data permitted. Individual PK parameters estimated for each subject's profile in the PK population were displayed in the data listings. Data were summarized by using descriptive statistics (mean, SD, % coefficient of variation [CV], confidence interval (CI), median, minimum, and maximum) and are presented by treatment group.
Geometric means were included for AUC and Cmax estimations and were included for some other PK parameters. By using Generalized Linear Model (GLM) procedure in SAS®, an analysis of variance (ANOVA) was performed on natural logarithmic (In) transformed parameters AUCo-t, AUC AUCo-τ, Cavg, and Cmax and on untransformed parameters t½, and λζ at the significance level of 0.05. The intrasubject CV was calculated for AUCo-t, AUC AUCo-τ, and Cmax by using the ANOVA residual error.
Dose linearity following single-dose administration (Period 1 ) was assessed after normalizing the AUC0-t, AUC and Cmax to the dose administered.
The following Period 1 comparisons for PK parameters were made:
• Comparison 1 : 600 μg 0.24% TBS-2 versus 1200 μg 0.48% TBS-2;
• Comparison 2: 600 μg 0.24% TBS-2 versus 1800 μg 0.72% TBS-2;
• Comparison 3: 1200 μg 0.48% TBS-2 versus 1800 μg 0.72% TBS-2.
Determination of Sample Size
The sample size for this study was not determined on the basis of statistical hypothesis testing. Based on typical, early-stage PK studies, groups of 8 subjects per cohort were sufficient to provide adequate clinical information to satisfy the objectives of the study.
Interim Analysis
No formal interim analysis was performed in this study. However, when the concentration data were received from the bioanalytical laboratory following Period 1 and again following Period 2, all PK TLFs were provided to the limit of data available by using scheduled sampling times rather than the actual scheduled sampling times. Multiple-dose data were made available after Period 2. No quality control testing of the preliminary PK analysis or the preliminary tables, listings, and figures were completed for the interim analysis and no testing of the interim PK parameters were completed for the interim analysis.
K. Changes in the Conduct of the Study or Planned Analyses
There were changes to the SAP detailing the planned PK analysis of the data after the first draft analysis was conducted. These fell under the category of additional exploratory analyses of PK parameters as provided for in the SAP.
The first analysis followed the SAP (corrected tau = 8 hours) but found that the elimination half-life could not be adequately estimated during the entire 48 hours of the collection period.
Pharmacokinetic consultants directed the PK analysis and made changes to the PK analysis plans to best present the PK results. In the final analysis a number of additional PK parameters were added to the analysis and additional tests were performed.
The first round of consultant comments requested these additions to the PK analysis:
1 . Individual figures of the 3 subjects who received the 1200 μg dose in both periods.
Data include plots of the BLQ-corrected raw data from the Bioanalyst for free, total and dihydro- testosterone. Total of 9 figures with 3 for each of the 3 analytes.
2. Recalculate the AUCo-s and AUCo on the BLQ-corrected data from the Bioanalyst (ConcBLQ). These parameters were compared by ANOVA between Period 1 and Period 2 in a new table. There were only 3 subjects who received the 1200 μ9 dose in Period 1 (single-dose) and Period 2 (multi-dose). The ANOVA requested was on the 3 subjects who received the 1200 μ9 dose and also on all 8 subjects' data.
The second round of consultant comments requested the following. Both consultants reviewed the PK section of the SAP and had the following comments.
1 . The AUC was requested for either AUC0-s or AUC using baseline corrected for single dose and not baseline corrected for multiple dose. Both were supplied.
2. AUCo was requested to be limited to the 24 hours that was baseline corrected for single dose.
3. Concentration at C24 was to be reported as a PK parameter for single-dose.
4. The error in the SAP was noted where tau was incorrectly given as 12 hours when it was 8 hours.
5. Requested C0 and C24 from the multiple-dose to be added to the PK parameters
6. Noted that the Cavg was to be based on an 8 hour dosing interval (tau).
10. Study Subjects
A. Disposition of Subjects
A total of 24 subjects were enrolled in Period 1 ; 8 subjects each were
randomized into Cohort 1 , Cohort 2, and Cohort 3. All 24 subjects completed Period 1 .
Following Period 1 , a total of 8 subjects continued to Period 2. All 8 subjects completed Period 2.
B. Protocol Deviations
The protocol deviations reported during the study were minor and were not expected to affect the outcome of the study. A total of 6 of 24 subjects (25.0%) had protocol deviations of vital signs assessments missed or PK blood draws out of window by 2 or 3 minutes.
11. Pharmacokinetics Evaluation
A. Data Sets Analyzed
• Single-Dose Population: All subjects who were randomized and received at least 1 dose of TBS-2 during the study; 24 subjects (100%) were included in this population. (Section 14.1 , Table 14.1 .1 a)
• Multi-Dose Population: All subjects in the Single-Dose Population who were selected to continue in study Period 2; 8 subjects (100%) were included in this population. (Section 14.1 , Table 14.1 .1 b)
B. Demographic and Other Baseline Characteristics
Demographic Data
Most subjects in Period 1 were white (19 of 24 subjects [79.2%]), followed in percentage by subjects who were black (4 of 24 subjects [16.7%]) and those who were American Indian or Alaskan Native/white (1 of 24 subjects [4.2%]). A similar number of subjects were not Hispanic or Latino (13 of 24 subjects [54.2%]) and Hispanic or Latino (1 1 of 24 subjects [45.8%]). Mean (SD) age was 29.8 (5.86) years. Mean (SD) height was 161 .02 (6.667) cm, weight was 66.36 (1 1 .378) kg, and BMI was
25.61 (4.079) kg/m2. Most subjects had a normal BMI (13 of 24 subjects [54.2%]), followed in percentage by subjects who were overweight (7 of 24 subjects [29.2%]) and subjects who were obese (4 of 24 subjects [16.7%]).
Most subjects in Period 2 were white (5 of 8 subjects [62.5%]), followed in percentage by those who were black (2 of 8 subjects [25.0%]) and those who were American Indian or Alaskan Native/white (1 of 8 subjects [12.5%]). A similar number of subjects were not Hispanic or Latino (5 of 8 subjects [62.5%]) and Hispanic or Latino (3 of 8 subjects [37.5%]). Mean (SD) age was 30.3 (6.48) years. Mean (SD) height was 160.8 (3.99) cm, weight was 61 .13 (9.815) kg, and BMI was 23.65 (3.454) kg/m2. Most subjects had a normal BMI (6 of 8 subjects [75.0%]), followed in percentage by those who were overweight (2 of 8 subjects [25.0%]).
Baseline Characteristics
Medical History
Subject medical history reported at Screening is summarized by body system in Table 1 1 .1 .
Currently active non-exclusionary medical history conditions were reported by at least 10% of subjects overall in the following body systems: nervous system (15 of 24 subjects [62.5%]), genitourinary system (7 of 24 subjects [29.2%]), integumentary system (4 of 24 subjects [16.7%]), and allergic conditions (4 of 24 subjects [16.7%]).
Table 11-1: Medical History (Single-Dose Population)

HEENT = head, eyes, ears, nose, and throat
D. Prior and Concomitant Medications
No concomitant medications were reported during the study.
Overall, 10 subjects reported prior medications. Most reported prior medications were over-the-counter analgesic or anti-inflammatory medications and multivitamins.
E. Measurement of Treatment Compliance
All subjects were observed during dosing at the study site and were compliant with the study drug administration.
F. Tabulations of Individual Subject Data
Analysis of Pharmacokinetics
There were 3 doses profiled pharmacokinetically in this study: 3 doses were profiled for the single-dose period and 1 dose was profiled for the multi-dose period. The single-dose profile included doses of 600, 1200, and 1800 g testosterone in separate cohorts while the multi-dose period included only the 1200 g testosterone dose. Each dose was profiled for free and total testosterone, dihydrotestosterone, estradiol, and SHBG. There were 8 subjects in each dose group.
The results are presented first for the concentrations and then for the PK parameters.
G. Concentration Results
Period 1: Single Dose Profile
The mean concentration profiles are presented in the following figures for each of the 5 analytes after baseline correction. The free testosterone and total testosterone concentration figures present the clearest relationship of concentration to increasing testosterone dose (Figure 19 and Figure 20, respectively). The dihydrotestosterone analyte provides a clear distinction between the 1800 g dose compared with the lower doses which are not clearly differentiated from each other (Figure 21 ). The obverse is observed in the estradiol concentrations where it is the lowest dose of 600 g, which is clearly distinct from the two higher doses, which are not clearly differentiated from each other (Figure 22). The SHBG concentrations are not clearly distinguished between administered testosterone doses (Figure 23).
The free and total testosterone and to a lesser extent the dihydrotestosterone concentrations are more clearly differentiated between the baseline and the active dose (Figure 24, Figure 25, and Figure 26, respectively). The estradiol and SHBG
concentrations have post-dose curves that overlap with the baseline concentrations in the same doses (Figure 27 and Figure 28, respectively).
Period 2: Multiple Dose Profiles
The free testosterone and total testosterone mean figures present the clearest dose effect with the highest concentrations at the time of dose and decreasing to a plateau at about 12 - 16 hours. The concentrations seem to get back to baseline levels
after 10 hours post dose. These two plots track so closely that they seem to be superimposable once the scale is adjusted (Figure 19 and Figure 20, respectively). The dihydrotestosterone concentrations also indicate a peak at the time of dose but it is not as clear as with the free and total testosterone (Figure 21 ). Little difference in estradiol and SHBG concentrations can been seen over the observation period of the multi-dose profile that can be attributed to the absence of a spike in concentration when the dose was administered. (Figure 32 and Figure 33, respectively).
Subject 522-03 had free testosterone concentrations that didn't show a similar decrease as compared to the other 7 subjects 12 hours after the final dose (Figure 34). The free testosterone concentrations for Subject 522-03 were also the highest concentrations after 12 hours but not as different from the other 7 subjects as were the free testosterone concentrations (Figure 35). Relationship to dose is increasingly harder to find for the dihydrotestosterone, estradiol, and SHBG in that order with SHBG concentrations essentially constant for each subject but at different concentrations (Figure 36, Figure 37, and Figure 38, respectively).
H. Pharmacokinetic Parameter Estimates
Pharmacokinetic parameters were estimated by using Phoenix WinNonlin Version 6.2.
Period 1: Single Dose Pharmacokinetic Parameter Estimates
All PK parameter estimates were performed on the first 24 hours with the baseline-corrected concentrations. The PK parameters estimated from the single-dose profiles for each individual are presented in Appendix 16.2, Listing 16.2.5.1 .2a and summarized in Section 14.2, Table 14.4.2a. The following table (Table 1 1 -22) presents the data for the single-dose PK parameters but the SD and median have been omitted.
All 24 subjects had profiles that estimated the λζ (terminal elimination rate) for free and total testosterone. Only 12 subjects had profiles that estimated the λζ for dihydrotestosterone, 1 1 for estradiol, and 9 for SHBG. Without λζ, the elimination half- life, t½, and AUCo could not be calculated.
Free Testosterone
For free testosterone, the mean Cmax closely tracked the administered dose suggesting proportionality to dose. The mean AUC0-s was proportional from the 600 μg
dose to the 1200 μ9 dose (1 .94 ratio compared to 2.00 dose ratio) but was somewhat lower for the 1800 μ9 dose (3.55 ratio compared to 3.00 dose ratio). Other measures of AUC were less clearly related to dose and did not suggest a proportional relationship to dose as strongly as the AUCo-s- This was probably due to the increase in concentration from the dose being most prominent in the first 8 hours as seen in the figures. The λζ values were similar across doses.
Table 11-22: Free Testosterone Summary (Single-Dose Population)
Dose c AUC„.8 AUC AUCo., AUC λζ ConcBase ConcBLQ ConcBC
(ng/dL) (hr) (hr*ng/dL) (hr*ng/dL) (hr*ng/dL) (hr*ng/dL) (1/hr) (hr) (ng/dL) (ng/dL) (ng/dL)
600 N 8 8 8 8 8 8 8 8 8 8 8
Mean 0.56 0.438 1.85 3.104 3.006 4.489 0.101 10.291 0.448 0.481 0.052
CV 74.2 59.1 61.5 58.1 55.2 63.8 90.5 48.9 91.9 75 107.2
Min 0.16 0.25 0.94 1.45 1.41 1.47 0.04 2.21 0.18 0.2 0
Max 1.45 1 4.32 6.18 5.51 8.76 0.31 15.7 1.44 1.3 0.16
GeoMean 0.442 1.622 2.711 2.652 3.758 0.079 8.784 0.357 0.401
1200 N 8 8 8 8 8 8 8 8 8 8 8
Mean 1.274 0.656 3.598 5.265 5.265 6.099 0.104 7.289 0.346 0.423 0.077
CV 63.8 73.2 60.8 54.8 54.8 49.3 36.8 27.8 39.7 35.9 42.1
Min 0.32 0.25 0.87 1.28 1.28 1.64 0.07 3.79 0.2 0.24 0.04
Max 2.51 1.5 8.16 10.61 10.61 11.42 0.18 9.59 0.53 0.63 0.12
GeoMean 1.017 3.021 4.488 4.488 5.354 0.099 6.995 0.322 0.4 0.071
1800 N 8 8 8 8 8 8 8 8 8 8 8
Mean 1.708 1.406 6.562 10.958 10.926 12.314 0.123 6.411 0.314 0.436 0.125
CV 44.2 137.4 36.8 50.1 50.5 51.1 43.3 34.4 24 25.5 79.5
Min 0.41 0.25 2.36 3.68 3.68 4.04 0.07 2.9 0.19 0.28 0
Max 2.53 6 9.46 21.22 21.22 23.23 0.24 10.32 0.41 0.62 0.28
GeoMean 1.5 6.053 9.615 9.561 10.689 0.115 6.049 0.306 0.423
AUCo-s = Area under the plasma concentration time curve from time zero to 8 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to 24 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to the last measurable concentration time point; AUCo^, = Area under the plasma concentration time curve from time zero to infinity;
C ConcBase = baseline concentration; C ConcBLQ = concentration of active dose; C ConcBC = baseline-corrected concentration; Cmax = maximum concentration observed after dosing; CV = % coefficient of variation; GeoMean = geometric mean; = terminal elimination rate constant; ty2 = eimination half-life; tmax = time of observed maximum concentration relative to the time of dosing
Note: The geometric mean is not normally reported for tmax.
Note: The geometric mean cannot be calculated if Min = 0.
Total Testosterone
Total testosterone Cmax parameters increased with increasing dose as did all of the AUC measures. The λζ measures were similar across doses. (Table 1 1 -23).
Table 11-23: Total Testosterone Summary (Single-Dose Population)
Dose c tmax AUC„.8 AUC AUCo-, AUC-co λζ t½ ConcBase ConcBLQ ConcBC
(ng/dL) (hr) (hr*ng/dL) (hr*ng/dL) (hr*ng/dL) (hr*ng/dL) (1/hr) (hr) (ng/dL) (ng/dL) (ng/dL)
600 N 8 8 8 8 8 8 8 8 8 8 8
Mean 40.625 0.438 135.313 229.095 223.981 327.448 0.108 9.434 30.138 33.138 4.175
CV 61.1 59.1 31.6 38.3 39 45.1 91.2 48.6 44.7 36.6 98.9
Min 15.8 0.25 84.58 127.73 127.79 157.01 0.04 2.03 17.1 16 0
Max 86.7 1 195.66 396.56 396.56 553.97 0.34 16.5 58 50.7 10.4
GeoMean 34.058 129.311 215.781 210.961 296.981 0.086 8.106 27.873 31.141
1200 N 8 8 8 8 8 8 8 8 8 8 8
Mean 71.063 0.688 225.758 328.002 328.002 372.176 0.118 6.472 23.538 28.013 4.475
CV 48.6 66.6 41.9 35.8 35.8 27.5 39.5 28.5 43.3 35.2 67.3
Min 21.9 0.25 65.78 102.59 102.59 161.89 0.07 3.09 13.3 17.2 1.4
Max 133 1.5 377.85 448.61 448.61 487.25 0.22 9.34 41.1 44.6 11.4
GeoMean 62.88 203.648 301.821 301.821 355.747 0.112 6.199 21.791 26.582 3.809
1800 N 8 8 8 8 8 8 8 8 8 8 8
Mean 130.863 1.031 515.848 837.083 834.391 947.786 0.133 6.143 24.35 34.55 10.625
CV 44.3 118.4 44.6 49.3 49.7 54.5 52.1 34.7 28.8 32.1 100.1
Min 27.2 0.25 169.05 273.14 273.14 315.07 0.09 2.57 17 25 0
Max 194 4 899.74 1593.93 1593.93 1989.73 0.27 8.07 38.8 59.8 34
GeoMean 113.912 464.728 742.547 739.181 830.493 0.121 5.716 23.552 33.314
AUCo- = Area under the plasma concentration time curve from time zero to 8 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to 24 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to the last measurable concentration time point; AUCo^, = Area under the plasma concentration time curve from time zero to infinity;
C ConcBase = baseline concentration; C ConcBLQ = concentration of active dose; C ConcBC = : baseline-corrected concentration; maximum concentration observed after dosing ; CV = % coefficient of variation; GeoMean = Geometric mean; = terminal elimination rate constant; ty2 = eimination half-life; t max = time of observed maximum concentration relative to the time of dosing
Note: The geometric mean is not normally reported for tmax.
Note: The geometric mean cannot be calculated if Min = 0.
Pi hydro-testosterone
Dihydrotestosterone PK parameters were missing a number of parameter values and some entire profiles were missing after baseline correction; only 18 of the 24 profiles were present and λζ could be estimated in only 12 of the 24 profiles. Mean Cmax, AUCo-8, and AUC values decreased from the 600 μg dose to the 1200 μg dose but then increased for the 1800 μg dose. The other AUC measures demonstrated increasing exposure with increasing dose. The λζ values were roughly similar for each dose. (Table 1 1 -24)
Table 11-24: Dihydrotestosterone Summary (Single-Dose Population)
Dose c AUC„.8 AUC AUCo., AUC λζ ConcBase ConcBLQ ConcBC
(μ$) (ng/dL) (hr) (hr*ng/dL) (hr*ng/dL) (hr*ng/dL) (hr*ng/dL) (1/hr) (hr) (ng/dL) (ng/dL) (ng/dL)
600 N 6 6 5 5 6 4 4 4 5 5 5
Mean 7.078 1.75 26.902 39.038 31.844 83.96 0.153 16.229 14.274 14.302 0.828
CV 70.1 122.6 74.8 61.7 74.9 98.6 127.7 105.6 44.2 31.4 151.3
Min 2.66 0.25 9.44 11.51 5.61 13.85 0.02 1.58 8.23 8.61 0
Max 16.27 6 59.82 71.37 70.18 202.98 0.44 39.89 22.1 19 2.97
GeoMean 5.888 21.859 32.493 23.515 55.582 0.077 9.009 13.144 13.697
1200 N 6 6 6 6 6 3 3 3 6 6 6
Mean 6.23 5.083 23.909 45.617 43.261 72.971 0.057 12.43 12.048 12.203 0.867
CV 43.6 182.4 48.4 52.4 49.1 22.2 19.6 20.6 54.9 39.7 116
Min 2.27 1 8.32 16.9 16.9 54.27 0.05 10.22 8.03 8.32 0
Max 9.62 24 37.33 79.02 69.98 82.43 0.07 15.24 25.3 21.8 2.27
GeoMean 5.646 20.858 39.952 38.457 71.647 0.057 12.259 11.004 11.589
1800 N 6 6 6 6 6 5 5 5 6 6 6
Mean 11.392 2.792 52.921 98.838 98.078 158.68 0.1 11.855 11.865 13.615 1.918
CV 59.3 101.1 68.5 63.4 64.2 73.4 71.6 78 40 37.6 124.6
Min 3.93 0.75 19.3 41.71 41.71 70.03 0.03 3.7 5.55 8.06 0
Max 23.4 8 117.76 217.97 217.97 327.95 0.19 23.31 20 21.4 6.4
GeoMean 9.82 43.767 86.043 85.18 128.512 0.077 8.971 11.068 12.834
AUCo = Area under the plasma concentration time curve from time zero to 8 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to 24 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to the last measurable concentration time point; AUCo^, = Area under the plasma concentration time curve from time zero to infinity;
C ConcBase = baseline concentration; C ConcBLQ = concentration of active dose; C ConcBC = baseline-corrected concentration; Cmax = maximum concentration observed after dosing; CV = % coefficient of variation; GeoMean = geometric mean; = terminal elimination rate constant; ty2 = eimination half-life; tmax = time of observed maximum concentration relative to the time of dosing
Note: The geometric mean is not normally reported for tmax.
Note: The geometric mean cannot be calculated if Min = 0.
Estradiol
Estradiol PK parameters for Cmax and AUC measures were similar for the 1200 μ9 and 1800 μ9 doses, but consistently less than the values from the 600 μ9 dose. For example the mean C ratios were 0.61 and 0.64 for 600 μ9 dose compared to the 1200 μ9 and 1800 μ9 doses, respectively. Similar ratios for AUC0-s were 0.31 and 0.47, respectively, where ratios of 2.00 and 3.00 would indicate proportionality. Also note that the 600 μ9 dose had an N=8 while both the 1200 μ9 and 1800 μ9 doses had an N=7. The λζ values were similar. (Table 1 1 -25)
Table 11-25: Estradiol Summary (Single-Dose Population)

600 N 5 5 5
Mean 23.275 10.816 112.508 236.334 230.951 718.407 0.063 30.458 50.1 47.438 6.863 CV 128.4 90.3 159 120.4 118.7 69 106.8 119.5 65.4 41.4 81.1 Min 3.4 0.25 0 10 10 194.98 0.01 3.88 22.9 27.9 0 Max 92.8 24 528.41 723 679.81 1351.44 0.18 93.63 122 90.6 18
GeoMean 13.468 106.562 105.763 553.278 0.039 17.818 43.319 44.477
1200 N 7 7 7 7 2 2 2
Mean 14.186 9.854 34.928 134.419 133.848 383.708 0.067 21.96 40.288 42.6 7.1 CV 33.9 100.7 110.2 81.3 82.2 51.7 102.7 102.7 40.9 33.1 106.4 Min 7.8 1 0 9.3 7.3 243.46 0.02 6.02 22.5 26.6 0 Max 22.2 24 101.1 322.73 322.73 523.95 0.12 37.9 75 70.2 19.9
GeoMean 13.474 80.078 75.97 357.16 0.046 15.104 37.815 40.76
1800 N 7 7 7 7 7 4 4 4
Mean 14.843 7.176 52.593 134.952 132.894 323.544 0.166 7.886 42.55 34.713 4.038 CV 92.3 143.1 177.8 192.4 197.9 166.1 109.4 63.3 38.3 65 213 Min 5.4 0.25 1.88 9.74 8.11 22.4 0.06 1.59 23.3 16.5 0 Max 44.5 24 262.18 722.27 727.84 1128.44 0.44 12.24 65.4 88.2 24.1
GeoMean 11.47 18.811 48.85 43.97 105.227 0.114 6.103 39.849 30.552
AUCo = Area under the plasma concentration time curve from time zero to 8 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to 24 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to the last measurable concentration time point; AUCo^, = Area under the plasma concentration time curve from time zero to infinity;
C ConcBase = baseline concentration; C ConcBLQ = concentration of active dose; C ConcBC = baseline-corrected concentration; Cmax = maximum concentration observed after dosing; CV = % coefficient of variation; GeoMean = geometric mean; = terminal elimination rate constant; ty2 = eimination half-life; tmax = time of observed maximum concentration relative to the time of dosing
Note: The geometric mean is not normally reported for tmax.
Note: The geometric mean cannot be calculated if Min = 0.
SHBG
The SHBG C and AUCo-s, AUCo-24, and AUCo-t values were similar for the 600 μ9 and the 1200 μ9 doses. The λζ was not characterized for the majority of these subjects. (Table 1 1 -26)
Table 11-26: SHBG Summary (Single-Dose Population)
C
Dose c AUCo-, ConcBase ConcBLQ ConcBC
(nmol L) (hr) (hr*nmol/L) (hr*nmol/L) (hr* nmol L) (hr* nmol L) (1/hr) (hr) (nmol/L) (nmol/L) (nmol/L)
600 N 8 8 8 8 8 4 4 4 8 8 8
Mean 7.138 4.25 22.987 44.1 40.805 195.593 0.126 20.766 61.413 61.25 1.275
CV 53.9 84.3 66.7 63.1 72.5 121.3 129.1 93 36.3 37.9 258.4
Min 2.6 0.25 8.74 13.63 8.02 35.66 0.02 1.92 18.7 17.6 0
Max 14.3 8 48.46 92.06 92.06 548.32 0.36 40.79 97.2 93.7 9.4
GeoMean 6.284 19.12 37.116 31.916 117.278 0.06 11.53 56.534 55.893
1200 N 8 8 8 8 8 2 2 2 8 8 8
Mean 7.525 5.406 23.798 45.416 40.309 99.885 0.323 4.869 51.738 49.663 0.713
CV 52.2 91.5 109 81.4 97.7 16.6 105.7 105.7 45.9 43.5 282.8
Min 1.2 0.25 0.2 7.1 5.29 88.18 0.08 1.23 17.4 17.2 0
Max 11.8 16 80.45 111.62 111.42 111.59 0.56 8.51 86.7 77 5.7
GeoMean 6.155 9.296 32.213 24.806 99.197 0.214 3.235 46.256 44.723
1800 N 8 7 7 6 8 3 3 3 8 8 8
Mean 10.888 9.013 13.161 34.62 32.988 205.537 0.053 29.135 63.388 64.513 2.25
CV 101 114.8 76.4 82.5 89.6 65.7 117.5 71.6 52.8 47.1 112.9
Min 0 0.25 0.54 7.33 0 63.1 0.02 5.54 27.9 31.1 0
Max 29.3 24.07 26.82 71.26 71.26 331.45 0.13 45.09 116 109 7.3
GeoMean 8.128 23.258 166.844 0.033 20.941 56.059 58.42
AUCo- = Area under the plasma concentration time curve from time zero to 8 hours after dosing; AUCo Area under the plasma concentration time curve from time zero to 24 hours after dosing ; AUCo = Area under the plasma concentration time curve from time zero to the last measurable concentration time point; AUCo^, = Area under the plasma concentration time curve from time zero to infinity;
C ConcBase = baseline concentration; C ConcBLQ = concentration of active dose; C ConcBC = : baseline-corrected concentration;
maximum concentration observed after dosing; CV = % coefficient of variation; GeoMean = geometric mean; = terminal elimination rate constant; : eimination half-life; tmax = time of observed maximum concentration relative to the time of dosing
Note: The geometric mean is not normally reported for tmax.
Note: The geometric mean cannot be calculated if Min = 0.
/. Period 2: Multiple Dose Pharmacokinetic Parameter Estimates
The PK parameters estimated from the multi-dose profiles for each individual are presented in Appendix 16.2, Listing 16.2.5.1 .2b and summarized in Section 14.2, Table 14.4.2b. The following tables present the data summarized in Section 14.2, Table 14.4.2b for the multi-dose PK parameters but the SD and median have been omitted. All subjects received 7 doses of 1200 μg testosterone.
Free Testosterone
Since the doses of 1200 μg testosterone were used in both the single- and multi- dose periods of this study, it is would seem useful to compare their PK parameters. However, this would not be useful as the single-dose data is based on baseline- corrected data while the multi-dose data is not. However, the baseline concentration data from the single-dose period may be reviewed in comparison to the multi-dose data. Since the data in TableTable 1 1 -28 are from baseline, the dose is not relevant as all concentrations are prior to dose.
The minimum and maximum values observed in the multi-dose period are greater than the minimum and maximum values observed in the baseline period for all
3 dose cohorts. This demonstrates that the testosterone multiple dosing are increasing both the minimum and the maximum free testosterone concentrations over the baseline values and provides the expected increase in free testosterone concentrations after multiple dosing. (Table 1 1 -27) and (Table 1 1 -28Table).
The AUCo-8 and the AUCo-τ are identical as the dosing interval is 8 hours. The AUCo-24 hours spans the last dose period of 8 hours but also includes the preceding 16 hours. The %PTF and %PTS indicate the percent difference between the Cmax and Cmin when divided by the Cavg or
 respectively.
respectively.
Table 11-27: Free Testosterone Summary for Multiple Dose Profile (Multi-Dose Population) c C„ C■ c AUC„.8 AUC AUCo-, %PTF %PTS
(ng/dL) (hr) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (hr*ng/dL) (hr*ng/dL) (hr*ng/dL)
N 8 8 8 8 8 8 8 8 8 8 8
Mean 1.886 0.378 1.155 0.704 1.006 0.578 8.046 17.259 8.046 140.636 214.058 CV 12.4 49.6 34.2 55.1 40.6 79.8 40.6 55.0 40.6 55.2 55.0 Min 1.65 0.25 0.77 0.40 0.59 0.28 4.69 9.74 4.69 30.60 35.00 Max 2.18 0.75 1.82 1.60 1.83 1.71 14.64 39.66 14.64 295.75 437.88
GeoMean 1.874 0.344 1.098 0.638 0.944 0.492 7.554 15.722 7.554 119.579 176.905
AUCo-8 = Area under the plasma concentration time curve from time zero to 8 hours after dosing; AUCo-24 = Area under the plasma concentration time curve from time zero to 24 hours after dosing; AUCO-T = Area under the concentration-time curve from time zero to the dosing interval;
= maximum concentration observed after dosing; C0 = maximum concentration at baseline; C = Minimum concentration over a dosing interval during multiple dosing; C„e = Average steady-state concentration; C24 = maximum concentration at 24 hours after dosing;
CV = % coefficient of variation; GeoMean = geometric mean; %PTF = percent peak to trough fluctuation; %PTS = percent peak to trough swing;
= time of observed maximum concentration relative to the time of dosing.
Table 11-28: Free Testosterone Concentration at Baseline (Single-Dose Population)

ConcBase = baseline concentration
Total Testosterone
The total testosterone PK Cmax range of 85.80 to 242.00 ng/dL following multiple dosing compares to the maximum testosterone concentration range of 41 .10 to 58.00 ng/dL observed at baseline. The Cmin range of 25.20 to 79.50 ng/dL total testosterone following multiple dosing compares to the 8.00 to 12.70 ng/dL range of minimum value of total testosterone observed at baseline. Both the Cmax and the Cmin indicate an effect of the testosterone dose administration; 2- to 4-fold effect on Cmax and a 3-fold effect on
Cmin- (Table 1 1 -29 and Table 1 1 -30), however, this accumulation disappears within 24 hours after the last administration.
Table 11-29: Total Testosterone Summary for Multiple Dose Profile (Multi-Dose
Population)
c C■ c
(ng/dL) (hr) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (hr*ng/dL) (hr*ng/dL) (hr*ng/dL)
N 8 8 8 8 8 8 8 8 8 8 8
Mean 148.038 0.440 85.450 49.088 73.802 38.513 590.419 1224.571 590.419 141.760 216.089 CV 41.5 49.8 32.1 36.4 41.4 47.2 41.4 38.5 41.4 53.8 53.5 Min 85.80 0.25 40.50 25.20 41.12 18.70 329.00 679.18 329.00 48.83 64.47 Max 242.00 0.75 131.00 79.50 141.00 77.30 1127.99 2026.22 1127.99 301.43 447.51
GeoMean 137.555 0.395 81.313 46.136 69.166 35.360 553.325 1147.566 553.325 125.100 187.544
AUCo = Area under the plasma concentration time curve from time zero to 8 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to 24 hours after dosing; AUCo = Area under the concentration-time curve from time zero to the dosing interval;
= maximum concentration observed after dosing; Co = maximum concentration at baseline; C = Minimum concentration over a dosing interval during multiple dosing; C„e = Average steady-state concentration; C = maximum concentration at 24 hours after dosing;
CV = % coefficient of variation; GeoMean = geometric mean; %PTF = percent peak to trough fluctuation; %PTS = percent peak to trough swing;
= time of observed maximum concentration relative to the time of dosing.
Table 11-30: Total Testosterone Concentration at Baseline (Single-Dose Population)

Dihydrotestosterone
The dihydrotestosterone observations do not indicate much difference from the baseline observations. The maximum dihydrotestosterone Cmax from the multi-dose period of 30.50 ng/dL is comparable to with the 32.60 C observed at baseline. The minimum Cmin from the multi-dose period of 6.32 ng/dL is comparable to the 5.02 ng/dL observed at baseline. No effect of the testosterone administration is seen in this comparison of dihydrotestosterone concentrations to baseline. (Table 1 1 -31 and Table 1 1 -32)
Table 11-31: Dihydrotestosterone Summary for Multiple Dose Profile (Multi-Dose Population)
c C„ c . c AUC„.8 AUC AUC-, %PTF %PTS
(ng/dL) (hr) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (hr*ng/dL) (hr*ng/dL) (hr*ng/dL)
N 8 8 8 8 8 8 8 8 8 8
Mean 22.300 0.500 20.724 13.465 16.628 14.774 133.024 338.888 133.024 57.240 73.956
CV 35.9 53.5 39.3 44.9 40.0 40.1 40.0 38.6 40.0 44.1 48.7
Min 10.20 0.25 7.39 6.32 7.21 6.33 57.66 145.85 57.66 13.66 14.90
Max 30.50 1.00 31.70 20.80 24.30 24.70 194.43 494.97 194.43 96.25 125.53
GeoMean 20.792 0.442 18.927 12.192 15.274 13.637 122.194 313.198 122.194 50.641 63.444
AUCo = Area under the plasma concentration time curve from time zero to 8 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to 24 hours after dosing; AUCo = Area under the concentration-time curve from time zero to the dosing interval;
= maximum concentration observed after dosing; Co = maximum concentration at baseline; C = Minimum concentration over a dosing interval during multiple dosing; C„e = Average steady-state concentration; C = maximum concentration at 24 hours after dosing;
CV = % coefficient of variation; GeoMean = geometric mean; %PTF = percent peak to trough fluctuation; %PTS = percent peak to trough swing;
= time of observed maximum concentration relative to the time of dosing.
Table 11-32: Dihydrotestosterone Concentration at Baseline (Single-Dose Population)

Estradiol
The estradiol Cmin concentration observed over 24 hours during the baseline observation period was 17.00 pg/mL (17.00 to 20.90 pg/mL range of 24 subjects in the single-dose period) which compares to the 12.70 pg/mL (12.70 to 95.20 pg/mL range of 8 subjects) in the multi-dose period. The estradiol Cmax concentration was 122.00 during the baseline period and 145.00 during the multi-dose period. While there is some difference in these concentrations, they are similar. (Table 1 1 -33 and Tbale 1 1 - 34).
Table 11-33: Estradiol Summary for Multiple Dose Profile (Multi-Dose Population) c C■ c
(pg/mL) (hr) (pg/mL) (pg/mL) (pg/mL) (pg/mL) (hr*pg/mL) (hr*pg/mL) (hr*pg/mL)
N 8 8 8 8 8 8 8 8 8 8 8 Mean 70.800 3.231 54.113 48.875 58.019 64.113 464.148 1759.342 464.148 35.465 42.164 CV 54.9 79.7 44.3 50.6 54.0 44.2 54.0 41.3 54.0 44.8 46.9 Min 16.60 0.27 16.30 12.70 14.96 18.60 119.67 503.91 119.67 20.16 22.22 Max 145.00 8.00 84.20 95.20 120.64 104.00 965.13 2895.80 965.13 66.76 79.82 GeoMean 60.034 2.024 48.062 42.569 50.033 56.820 400.264 1587.754 400.264 32.768 38.514
AUCo = Area under the plasma concentration time curve from time zero to 8 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to 24 hours after dosing; AUCo = Area under the concentration-time curve from time zero to the dosing interval;
= maximum concentration observed after dosing; Co = maximum concentration at baseline; Cmin = Minimum concentration over a dosing interval during multiple dosing; C„e = Average steady-state concentration; C = maximum concentration at 24 hours after dosing;
CV = % coefficient of variation; GeoMean = geometric mean; %PTF = percent peak to trough fluctuation; %PTS = percent peak to trough swing;
= time of observed maximum concentration relative to the time of dosing.
Table 11-34: Estradiol Concentration at Baseline (Single-Dose Population)

SHBG
The SHBG Cmin from the baseline observation period was 15.30 nmol/L while the Cmin from the multi-dose period was 19.90 nmol/L. The SHBG Cmax was 137.00 nmol/L while the Cmax from the multi-dose period was 100.00 nmol/L. No effect from the testosterone dosing on SHBG concentrations is obvious. (Table 1 1 -35 and Table 1 1 - 36).
Table 11-35: SHBG Summary for Multiple Dose Profile (Multi-Dose Population)

= time of observed maximum concentration relative to the time of dosing.
Table 11-36: SHBG Concentratio at Baseline (Single-Dose Population)

ConcBase = baseline concentration
J. Pharmacokinetic Parameter Statistical Testing
Dose Proportionality Analysis on Single Dose
Section 14.2, Table 14.4.3 presents the analysis of dose proportionality. Table
1 1 -37 is abstracted from this table to present the linear regression analysis of the log- transformed PK parameters.
Dose proportionality occurs when increases in the administered dose are accompanied by proportional increases in a measure of exposure (PK parameter). The linear regression model is fitted to each of the interested PK parameters by using dose (600, 1200, and 1800 as the predictor variable:
PK = Slope * Dose + Intercept.
A dose proportionality hypothesis cannot be rejected if the intercept's 95% confidence limit includes 0.
Dose proportionality was not rejected for only AUC0-24, AUCo-s, and AUC0-t for free testosterone.
Table 11-37: Dose Proportionality Analysis (Single-Dose Population)
95% Confidence Limit for
Estimate
Log Transformed Standard N /
Analyte PK Parameter Regression Estimate Error Lower Limit Upper Limit R-Square
Free Testosterone AUCo Intercept 0.3211 0.3154 -0.3331 0.9753 24
Slope 0.0011 0.0002 0.0006 0.0016 0.4607
AUCo Intercept -0.1868 0.2951 -0.7988 0.4252 24
Slope 0.0011 0.0002 0.0006 0.0016 0.5136
AUCo Intercept 0.7450 0.3266 0.0676 1.4224 24
Slope 0.0009 0.0003 0.0003 0.0014 0.352
AUCo Intercept 0.2956 0.3134 -0.3544 0.9456 24
Slope 0.0011 0.0002 0.0006 0.0016 0.4703 cm» Intercept -1.3533 0.3811 -2.1436 -0.5630 24
Slope 0.0010 0.0003 0.0004 0.0016 0.3529
Total Testosterone AUCo Intercept 4.6622 0.2607 4.1216 5.2029 24
Slope 0.0010 0.0002 0.0006 0.0014 0.5437
AUCo Intercept 4.1608 0.2525 3.6371 4.6845 24
Slope 0.0011 0.0002 0.0007 0.0015 0.5765
AUCo Intercept 5.0683 0.2653 4.5181 5.6185 24
Slope 0.0009 0.0002 0.0004 0.0013 0.4435
AUCo-, Intercept 4.6351 0.2597 4.0965 5.1738 24
Slope 0.0010 0.0002 0.0006 0.0015 0.5528 cmax Intercept 2.9275 0.3285 2.2463 3.6088 24
Slope 0.0010 0.0003 0.0005 0.0015 0.4174
Dihydrotestosterone AUCo Intercept 2.8766 0.4092 2.0043 3.7488 17
Slope 0.0008 0.0003 0.0002 0.0015 0.3234
AUCo Intercept 2.5722 0.4515 1.6098 3.5347 17
Slope 0.0006 0.0003 -0.0001 0.0013 0.1713
AUCo Intercept 3.5478 0.5806 2.2542 4.8415 12
Slope 0.0007 0.0004 -0.0003 0.0017 0.2131
AUCo-, Intercept 2.4635 0.4276 1.5571 3.3699 18
Slope 0.0011 0.0003 0.0004 0.0018 0.3979 cmax Intercept 1.4180 0.3725 0.6283 2.2076 18
Slope 0.0004 0.0003 -0.0002 0.0010 0.1209
AUCo = Area under the plasma concentration time curve from time zero to 8 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to 24 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to the last measurable concentration time point; AUCo-„ = Area under the plasma concentration time curve from time zero to infinity; Cmax = maximum concentration observed after dosing.
Dose Pair-wise Test Using Analysis of Variance
Table 1 1 -38 presents pair-wise comparisons of single-dose PK parameters using ANOVA after dose normalizing. Results found that there were differences in the dose normalized values for all analytes for at least 1 of the 3 pair-wise comparisons except for free testosterone AUC measures for AUCo-24, AUCo-s, and AUCo-t.
Table 11-38: Analysis of Variance for Some Pharmacokinetic Parameters (Single-Dose Population)
P-Value
Intra- subject 600 μg 600 μg 1200 μg
Coefficient vs. vs. vs.
Analyte PK Parameter of Variation1 1200 μg 1800 μg 1800 μg
Free Testosterone AUC0.24 45.8505 0.2114 0.2182 0.9845
AUC0.8 67.4794 0.7110 0.5338 0.7996
AUC0.∞ 43.0039 0.0324 0.0197 0.8185
AUC0.t 45.3344 0.2439 0.2477 0.9921
c -223.3208 0.0034 0.0010 0.6182 λζ 84.7568 0.0908 0.0406 0.6863 t½ 55.9189 0.0002 <.0001 0.3257
Total Testosterone AUC0.24 7.8951 <.0001 <.0001 0.0001
AUC0.8 8.2332 <.0001 <.0001 0.0001
AUC0.∞ 8.7190 <.0001 <.0001 0.0002
AUC0.t 7.8792 <.0001 <.0001 0.0001
c 17.9163 <.0001 <.0001 0.0320 λζ 85.1223 0.1153 0.0454 0.6333 t½ 55.6641 0.0002 <.0001 0.3940
Dihydrotestosterone AUC0.24 19.8167 <.0001 <.0001 0.1756
AUC0.8 23.5230 <.0001 <.0001 0.3291
AUC0.∞ 26.0151 0.0048 0.0004 0.3131
AUC0.t 26.5016 0.0011 0.0001 0.3144
c 37.3379 0.0021 0.0009 0.6763 λζ 158.0250 0.1869 0.1523 0.9553 t½ 117.5657 0.2274 0.1041 0.7665
AUCo = Area under the plasma concentration time curve from time zero to 8 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to 24 hours after dosing; AUCo = Area under the plasma concentration time curve from time zero to the last measurable concentration time point; AUCo^, = Area under the plasma concentration time curve from time zero to infinity; Cmax = maximum concentration observed after dosing; = terminal elimination rate constant; ty2 = eimination half-life a Coefficient of Variation = 100 x ANOVA residual error (Root Mean Square Error) / PK parameter mean. Pair-wise comparisons are from the ANOVA model with Dose as a class (categorical) variable and PK parameter estimate values as response.
Note: AUCo AUCo-∞, AUCo and Cmax parameter values are natural logarithmic transformed.
Paired A-Test Comparison of AUC0.s and AUCo-24 from Single to Multiple Dose
There were 3 subjects who participated in both the single- and multi-dose periods at the 1200 μg dose. Uncorrected parameters for AUC0-s and AUCo-24 were calculated from the free testosterone, total testosterone, and dihydrotestosterone analyte concentrations for comparison to uncorrected parameters from the multi-dose profile. Table Table 1 1 -39 compares the AUC0-s and AUCo-24 from these 3 subjects by using a paired f-test for free testosterone, total testosterone, and dihydrotestosterone. The results of the log transformed comparisons are provided.
Table 11-39: Paired t-Test Results for Pharmacokinetic Parameters AUCo-8 and AUCo-24 for Subjects Who Had 1200 g TBS-2 in Period 1 and Period 2
Natural-logarithmic 95% Confidence Limit for
transformed PK Parameter Mean Difference
Mean of Standard Error
Analyte Name N Difference of Difference Lower Limit Upper Limit P-Value
Free Testosterone AUCo 3 0.6041 0.4255 -1.2267 2.4349 0.2915
AUCo 3 0.9312 0.3853 -0.7266 2.5890 0.1369
Total Testosterone AUCo 3 0.5782 0.1780 -0.1876 1.3440 0.0831
AUCo 3 0.9058 0.1683 0.1815 1.6301 0.0328
Dihydrotestosterone AUCo 3 2.1025 0.2060 1.2160 2.9889 0.0095
AUCo 3 2.1709 0.4044 0.4311 3.9108 0.0330
AUCo = Area under the plasma concentration time curve from time zero to 8 hours; AUCo = Area under the plasma concentration time curve from time zero to 24 hours; PK = pharmacokinetic.
The free testosterone comparisons of 95% CI included zero in the difference for both AUC measures so equivalence between the single- and multi-dose AUC
measures could not be rejected. Total testosterone 95% CIs included zero for the AUCo-8 but not for the AUCo-24 so equivalence could not be rejected for AUC0-s but was for AUCo-24- In the analyte dihydrotestosterone, neither 95% CI included zero, so equivalence was rejected for both AUCs.
Statistical and Analytical Issues
K. Adjustments for Covariate
Not applicable.
L. Handling of Dropouts or Missing Data
To handle missing or partial AE and concomitant medication dates, the following rules were applied:
1 . For partial AE and concomitant medication start dates, if the year was unknown, then a missing value was assigned. If the month was unknown and the year matches the year of the first dose date, then the month and day of the first dose date was imputed; otherwise January was assigned. If the day was unknown and the month and year matched the month and year of the first dose date, then the day of the first dose date was imputed; otherwise "01 " was assigned.
2. For partial AE and concomitant medication end dates, if the year was unknown, then a missing value was assigned. If the month was unknown, then 'December' was assigned. If the day was unknown, then the last day of the month was assigned.
After implementing the rules above, to determine whether AEs (or medications) with missing start or stop dates are pretreatment or on/after treatment, the following strategy was used:
1 . If the start date and stop date were both missing, then the most conservative approach was taken and the AE (or medication) was considered to be treatment-emergent (or concomitant).
2. If the start date was missing but the stop date was not missing and was after the day of study dose administration, then the most conservative approach was taken and the AE (or medication) was considered to be treatment-emergent (or concomitant).
3. If the start date was missing but the stop date was not missing and was on or before the day of study dose and after the date of signed informed consent, then the AE (or medication) was considered to be pretreatment (or prior).
4. If the start date was not missing but the stop date was missing, then the most conservative approach was taken and medication was considered to be concomitant while the AE was defined by start date.
The missing severity of an AE was imputed to the greatest severity; the missing study drug causality to an AE was imputed to "related."
M. Interim Analysis and Data Monitoring
Interim analyses are discussed in the Interim Analysis section above.
N. Multi-center Studies
Not applicable
O. Multipe Comparison/ Multiplicity
A few pair-wise comparisons were performed, but no Type 1 error rate was adjusted since they were exploratory analyses.
P. Use of a "Pharmacokinetics Subset" of Subjects
All subjects were included in the PK analyses.
Q. Active-Control Studies Intended to Show Equivalence
Not applicable.
R. Examination of Subgroups
Not applicable.
Tabulation of Individual Response Data
Not applicable.
Drug Dose, Drug Concentration, and Relationship to Response
Not applicable.
Drug-Drug and Drug-Disease Interactions
Not applicable.
By-Subject Displays
Not applicable.
Pharmacokinetics Conclusions
1 . The single-dose baseline-corrected free and total testosterone concentrations have the clearest relationship of concentration to increasing testosterone dose. The dihydrotestosterone analyte concentrations provide a clear distinction between the 1800 iQ dose compared with the lower doses, which are not clearly differentiated from each other. A slight change is observed in the estradiol concentrations where it is the lowest dose of 600 g, which is clearly distinct from the two higher doses, which are not clearly differentiated from each other. The SHBG concentrations are not clearly distinguished between administered testosterone doses.
2. The multi-dose concentrations were not baseline corrected but have similar
graphical evaluations. The free and total testosterone concentrations have the clearest demonstration of the concentration increase due to dose. A more modest increase in dihydrotestosterone concentration is observed at the time of dose, but little increase in concentration of estradiol and SHBG is seen at the time of dose. When given BID at these dose, Testosterone accumulates 2-4 fold, but returned to baseline after 12-15 hours
3. Pharmacokinetic parameters were listed individually and summarized for single- dose and multi-dose profiles. Formal statistical testing of dose proportionality was performed on the single-dose profiles.
4. Testing of the single-dose profiles using linear regression across all 3 doses
indicated that the free testosterone PK parameters AUCo-s, AUCo-24, and AUCo-t were the only analyte parameters for which dose proportionality was not rejected.
5. Pair-wise testing of doses from the single-dose profile using ANOVA agreed with the linear regression and also indicated that the free testosterone AUCo-s, AUCo-24, and AUCo-t parameters were the only analyte parameters that indicated for which dose proportionality could not be rejected for at least one dose comparison.
6. The 3 subjects who participated in both the single- and multi-dose profiles at the same 1200 μg dose level were compared using a paired f-test for AUC0-s and AUCo- 24. Equivalence between the uncorrected single- and multi-dose profiles was rejected for both parameters for dihydrotestosterone, and for AUC0-24 with total testosterone, but equivalence between the profiles was not rejected for either of the free testosterone parameters.
12. Safety Evaluation
A. Extent of Exposure
Study drug administration was performed at the clinical site under study personnel supervision
B. Adverse Events
Summary of Treatment Emergent Adverse Events
There were no deaths, SAEs, or AEs leading to discontinuation during the study. Most TEAEs were in the system organ classes of general disorders (nasal related events) and administration site conditions and respiratory, thoracic, and mediastinal disorders. Most TEAEs were mild in severity and were unlikely or not related to study medication.
Display of Treatment Emergent Adverse Events
The incidence of TEAEs is shown for Period 1 in Table 12-1 and Table 12-2.
Table 12-1: Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term (Single-Dose Population)

MedDRA = Medical Dictionary for Regulatory Activities; TEAE = treatment-emergent adverse event
Note: Subjects reporting more than 1 TEAE in each level (system organ class or preferred term) were only counted once.
Table 12-2: Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term (Multi-Dose Population)

MedDRA = Medical Dictionary for Regulatory Activities; TEAE = treatment-emergent adverse event
Note: Subjects reporting more than 1 TEAE in each level (system organ class or preferred term) were only counted once.
Analysis of Adverse Events
Adverse Events in Period 1
Overall, 1 1 of 24 subjects (45.8%) experienced at least one TEAE in Period 1 . Most subjects experienced TEAEs of general disorders and administration site conditions (experienced by 7 of 24 subjects [29.2%]) and respiratory, thoracic, and mediastinal disorders (experienced by 5 of 24 subjects [20.8%]). TEAEs experienced by more than 1 subject were catheter site erythema (experienced by 3 of 24 subjects [12.5%]) and catheter site hemorrhage, catheter site inflammation, dizziness, and nasal congestion (each experienced by 2 of 24 subjects [8.3%]). (Table 12-1 Table ).
The majority of TEAEs in Period 1 were of mild severity. A total of 10 of 24 subjects (41 .7%) experienced mild TEAEs. Only 1 of 24 subjects (4.2%) experienced moderate TEAEs. Subject 522-53 (Cohort 2) experienced headache and dizziness of moderate severity on Day 1 . Both TEAEs resolved without treatment the next day and were considered as unlikely to be related to the study drug.
Overall, 4 of 24 subjects (16.7%) experienced TEAEs that were considered possibly related to the study medication. A total of 3 of 24 subjects (12.5%) had TEAEs
that were considered to be unlikely related to the study medication and 4 of 24 subjects (16.7%) had TEAEs that were considered not related to the study medication.
Adverse Events in Period 2
Overall, 4 of 8 subjects (50.0%) experienced at least 1 TEAE in Period 2.
General disorders and administration site conditions occurred in 2 of 8 subjects
(25.0%). The TEAEs that were experienced by 1 of 8 subjects (12.5%) each were catheter site pain, catheter site phlebitis, dyspepsia, headache, and rhinalgia. (Table 12-2).
The majority of TEAEs in Period 2 were of mild severity. Overall, 3 of 8 subjects (37.5%) experienced mild TEAEs. Only 1 of 8 subjects (1 2.5%) experienced a moderate TEAE. Subject 522-29 experienced an increase in headache intensity of moderate severity on Day 2. The TEAE resolved without treatment 2 days later and was considered as possibly related to the study drug.
Overall, 2 of 8 subjects (25.0%) experienced TEAEs that were considered possibly related to the study medication and 2 of 8 subjects (25.0%) experienced TEAEs that were considered not related to the study medication.
Adverse Reactions in Period 1 and Period 2
Adverse reactions (defined as TEAEs considered possibly, probably, or definitely related to study medication) in Period 1 and Period 2 are shown in Table 12-3.
A total of 6 subjects experienced adverse reactions during the study: 4 of 24 subjects (16.7%) in Period 1 and 2 of 8 subjects (25.0%) in Period 2. All adverse reactions were considered possibly related to study medication. Subject 522-29 (Period 2) had 2 adverse reactions of headache that were each recorded as a TEAE: a mild headache that increased in severity to moderate.
Table 12-3: Subjects with Adverse Reactions (Single- and Multi-Dose Populations)

MedDRA = Medical Dictionary for Regulatory Activities; TEAE = treatment-emergent adverse event a. The onset was calculated, respectively, as [Onset Date - Treatment Date of Period 1 + 1], if the onset was during Period 1, or [Onset Date - Treatment Date of Period 2 + 1], if the onset was during Period 2.
b. Severity was rated by the Principal Investigator as mild, moderate, or severe.
Listing of Adverse Events by Subject
Adverse events are listed by subject in Appendix 16.2, Listing 16.2.7.1 . Adverse reactions (defined as TEAEs considered possibly, probably, or definitely related to study medication) in Period 1 and Period 2 are listed by subject in Appendix 16.2, Listing 16.2.7.4.
C. Deaths, Other Serious Adverse Events, and Other Significant Adverse Events
There were no deaths, SAEs, or AEs leading to discontinuation during the study.
D. Clinical Laboratory Evaluations
valuation of Each Laboratory Parameter
Laboratory Values Over Time
Changes over time in laboratory values were not calculated.
Individual Subject Changes
Subjects with abnormal laboratory values that were not present at screening are shown in Table 12-4 (hematology), Table 12-5 (chemistry), and Table 12-26
(urinalysis).
Table 12-24: Subjects with New Abnormal Hematology Laboratory Evaluation Results Post Dose (Single- and Multi-Dose Populations)
Study Study Low/
Subject Period Day Test (Normal Range) Result High
Cohort 1, 600 g
522-15 1 4 Hematocrit (32.5%-46.9%) 32.0% Low
1 4 Red Blood Cell Count (3.70-5.46 3.55 Low mil/mm ) mil/mm3
522-40 1 4 Lymphocytes (15.0%-50.0%) 50.6% High
522-49 1 4 Absolute Basophil Count (0-125 140 High cells/mm3) cells/mm3
Cohort 2, 1200 g
522-28 2 5 Absolute Basophil Count (0-125 160 High cells/mm3) cells/mm3
2 5 Basophil Percentage (0.0-2.0%) 2.6% High
522-38 2 5 Absolute Basophil Count (0-125 160 High cells/mm3) cells/mm3
Cohort 3, 1800 g
522-24 1 4 Eosinophils Percentage (0.0%-6.0%) 7.0% High
Table 12-25: Subjects with New Abnormal Chemistry Laboratory Evaluation Results Post Dose (Single- and Multi-Dose Populations)

CK = creatine kinase; TSH = thyroid-stimulating hormone
Table 12-26: Subjects with New Abnormal Urinalysis Laboratory Evaluation Results Post Dose (Single- and Multi-Dose Populations)

Individual Clinically Significant Abnormalities
No abnormal clinical laboratory results were recorded as TEAEs.
E. Vital Signs, Physical Examinations, and Other Observations Related to Safety
Vital Signs
No abnormal vital signs were reported by the PI or were recorded as a TEAE. Physical Examination
Three subjects had abnormal physical examination results related to the PK blood sample venipuncture site. None of these results were considered clinically significant.
• Subject 522-35 (1200 - Period 2, Day 5: Slight phlebitis to left antecubital site
• Subject 522-38 (1200 μg) - Period 2, Day 5: Tenderness/soreness on left antecubital area
• Subject 522-51 (1200 - Period 1 , Day 4: Slight tenderness left AC IV site
Except for those related to HEENT or the PK blood sample venipuncture site, no other abnormal physical examination results were reported.
Nasal Endoscopic Examination
No ENT nasal endoscopic examination findings were reported at Screening (Appendix 16.2, Listing 16.2.4.3.2).
Two subjects had ENT examination findings on Day 4 in Period 1 that were interpreted as clinically significant by the PI : Subject 522-37 (600 μg) had slight rhinorrhea and Subject 522-53 (1200 μg) had mild erythema to the left nostril mucosa.
All abnormal findings in basic ENT examinations are presented in Table 12-27.
Table 11-27: Subjects with Abnormal Basic Ear, Nose, and Throat Examination Results (Single- and Multi-Dose Populations)

= clinically significant; ENT = ears, nose, and throat; NCS = not clinically significant; TEAE = treatment emergent adverse event
a. A TEAE of rhinorrhea (mild, possibly related to study medication) was reported on Day 2 and resolved on Day 5.
b. A TEAE of nasal mucosal disorder (mild, possibly related to study medication) was reported on Day 3 and was ongoing at study completion.
F. Safety Conclusions
There were no deaths, SAEs, or AEs leading to discontinuation during the study. Most TEAEs were in the system organ classes of general disorders and administration site conditions and respiratory, thoracic, and mediastinal disorders (nasal related
events). Most TEAEs were mild in severity and were unlikely or not related to study medication.
In Period 1 , a total of 1 1 of 24 subjects (45.8%) experienced at least 1 TEAE. Those experienced by more than 1 subject were catheter site erythema (experienced by 3 of 24 subjects [12.5%]) and catheter site hemorrhage, catheter site inflammation, dizziness, and nasal congestion (each experienced by 2 of 24 subjects [8.3%]).
The majority of TEAEs in Period 1 were of mild severity. A total of 10 of 24 subjects (41 .7%) experienced mild TEAEs. Only 1 of 24 subjects (4.2%) experienced moderate TEAEs. Subject 522-53 (1200 μg) experienced headache and dizziness of moderate severity on Day 1 . Both TEAEs resolved without treatment the next day and were considered as unlikely to be related to the study drug.
In Period 2, a total of 4 of 8 subjects (50.0%) experienced at least 1 TEAE. General disorders and administration site conditions occurred in 2 of 8 subjects (25.0%). The TEAEs that were experienced by 1 of 8 subjects (12.5%) each were catheter site pain, catheter site phlebitis, dyspepsia, headache, and rhinalgia.
The majority of TEAEs in Period 2 were of mild severity. Overall, 3 of 8 subjects (37.5%) experienced mild TEAEs. Only 1 of 8 subjects (1 2.5%) experienced a moderate TEAE. Subject 522-29 experienced an increase in headache intensity of moderate severity on Day 2. The TEAE resolved without treatment 2 days later and was considered as possibly related to the study drug.
A total of 6 subjects experienced adverse reactions (defined as TEAEs considered possibly, probably, or definitely related to study medication) during the study; 4 subjects in Period 1 and 2 subjects in Period 2. All adverse reactions were considered possibly related to study medication. Subject 522-29 (Period 2, 600 μg) had 2 adverse reactions of headache that were each recorded as a TEAE: a mild headache that increased in severity to moderate.
A total of 6 subjects experienced adverse reactions during the study: 4 of 24 subjects (16.7%) in Period 1 and 2 of 8 subjects (25.0%) in Period 2. All adverse reactions were considered possibly related to study medication. Subject 522-29 (Period 2) had 2 adverse reactions of headache that were each recorded as a TEAE: a mild headache that increased in severity to moderate.
There were no TEAEs associated with clinical laboratory tests or vital signs. Two subjects had ENT examination findings that were interpreted as clinically significant by the PI on Day 4 in Period 1 : Subject 522-37 (600 had slight rhinorrhea and
Subject 522-53 (1200 μg) had mild erythema to the left nostril mucosa.
13. CMC Section
Testosterone batch, 80402960, used in the clinical batches was fully tested by the applicant in accordance with USP specifications.
The drug product, TBS-2 is a viscous bioadhesive oil-based formulation containing solubilized testosterone intended for intranasal application for the treatment of anorgasmia in women.
The drug product is formulated with the following compendial inactive ingredients: castor oil, oleoyl polyoxylglycerides, and colloidal silicon dioxide.
TBS-2 is supplied in a white, non-aerosol, multi-dose, metered pump container. The containers are supplied separately as barrels (Albion 15ml) and pumps
(VP39/140H) with cap attached. The container closure is manufactured using materials acceptable for pharmaceutical use. See Fig 39.
The container closure relies on atmospheric pressure and device design to deliver the required dose. When the actuator of the VP39/140 pump is pressed, a valve is opened in the pump mechanism. This allows atmospheric pressure to act on the piston, through the base of the gel-containing Albion 15ml barrel, forcing it upwards. Consequently, the gel is forced upwards, through the tip of the device. The accurate and concise quantity of gel delivered is a function of the design of VP39/140 pump. When the actuation is stopped, a spring in the pump mechanism closes the valve, stops the effect of atmospheric pressure on the piston and returns the actuator to its starting position.
The patient is instructed to place his finger on the pump of the actuator and to advance the tip of the actuator until the finger on the pump reaches the base of the nose. The opening on the tip of the actuator must face the nasal mucosa. The patient depresses the pump and the gel is expelled onto the nasal mucosa. Each dose consists of two actuations, one actuation per nostril.
The compositions of the three different concentrations of the drug product to be used in this clinical trial are provided in Tables 3.2. P.1 -1 - 3.
Table 3.2.P.1-1: Components, Quality, Quality Standards and Function - 0.24% TBS-2 Gel

Table 3.2.P.1-2: Components, Quality, Quality Standards and Function
TBS-2 Gel
Component Amount Amount per Amount Function Quality
(%w/w) multiple dose delivered per Standard dispenser (g) actuation
Testosterone 0.48 0.048 0.60 Active USP
Ingredient
Castor oil 91.52 9.152 114.40 Solvent USP
Oleoyl 4.00 0.400 5.00 Wetting Ph. Eur./NF polyoxylglycerides agent
(hydrophilic
oil)
Colloidal silicon 4.00 0.400 5.00 Viscosity NF dioxide increasing
agent
Total 100.00 10.0 g 125.00
Table 3.2.P.1-3: Components, Quality, Quality Standards and Function - 0.72% TBS-2 Gel

A study is completed in accordance with USP<51 > Antimicrobial
Effectiveness Testing to establish that microbial growth is absent in the container closure system after filling and during use. This study is performed with a 4.5% testosterone gel filled in the multiple dose dispenser.
Sixteen (1 6) multiple dose dispensers are tested in this study. Each multiple dose dispensers is inoculated with 0.05 ml_ of one of the standardized microbial suspensions {Candida albicans, Aspergillus brasiliensis,
Sraphylococcus aureus, Pseudomonas aeruginosa) through the tip of the piston. These microorganisms are chosen as they are the microorganisms recommended in USP <51 > Antimicrobial Effectiveness Testing for nasal products. The theoretical concentration of the organisms used for the inoculation is between 1 00,000 and 1 ,000,000 microorganisms per ml_. For the following 21 days, two actuations are
performed on each multiple dose dispenser daily to reflect real use (2 actuations is equivalent to 1 dose).
The inoculated dispensers are incubated upright at 20 - 25 °C. Each of the samples is examined at 1 , 7, 14 and 21 days, subsequent to inoculation. The study is conducted over twenty-one (21 ) days. The Day 1 timepoint is chosen to assess the worst case scenario. Testing of the samples does occur following the daily actuations. For all the timepoints, the gel remaining in the barrel is tested. The piston is removed and the remaining gel in the barrel was subjected to further testing to determine the number of test organisms present, using 10 grams of the sample. The sample is examined at these intervals for any observed changes in appearance (i.e. colour, viscosity). Each dispenser is discarded after sampling. The results are presented in Table 3.2. P.2.5-1 .
Table 3.2.P.2.5-1: Number of Microorganisms Surviving During Testing Period

ND - none detected; No changes were observed in the appearance of the product at each timepoint during the testing interval
Table 3.2.P.2.5-2: Criteria for Tested Microorganisms (Catergory 2)
Criteria for Tested Microorganisms (Category 2)
Bacteria Not less than 2.0 log reduction form the initial calculated count at 14 days, and no increase from the 14 day count at 28 days.
Yeast and Molds No increase from the initial calculated count at 14 and 28 days
In summary, the bacteria S. Aureus and P. Aeruginosa showed a reduction of not less than 2.0 from the initial count at 14 days and no increase from the 14 days count at 21 days. The yeast C. Albicans and A. Brasiliensis showed no increase from the initial calculated count at 14 and 21 days. Testosterone nasal gel packaged in a multiple dose dispenser meets the criteria for the Anitmicrobial Effectiveness Test.
Three different concentrations of TBS-2 clinical material, 0.24%, 0.48% and 0.72%, will be manufactured for the proposed clinical trial. The batch formulae for these batches are presented in Table 3.2. P.3.2-1 .
Table 3.2.P.3.2-1: Batch Formulae for 0.24%, 0.48% and 0.72% TBS-2 at the 100 kg Batch Size

The TBS-2 bulk gel is tested to the following specifications for batch
release.
Table 3.2.P.5.1-1: Specification for TBS-2 Bulk Gel
Test Parameter Method/Reference Acceptance Criteria
Appearance STM.GEN.001 Slightly yellow gel
Identification A STM.TBS2.001 Retention time of the peak in the sample corresponds to that of the Testosterone standard.
Identification B STM.TBS2.001 UV spectrum of the sample corresponds to that of the Testosterone standard
Assay STM.TBS2.001 95.0-105.0%
Related STM.TBS2.002 TBS1 RC4 , 0.2% Compounds TBS1 RC5 , 0.5%
Single unknown impurity , 0.2% Total impurities , 1.0%
Viscosity USP<91 1> Report results
Water USP<921> Method I Report results
Residual Solvents USP<467> The product complies with USP<467>
requirements
TBS1 RC4 - 17.-hydroxyandrosta-4,6-dien-3-one (Delta-6-testosterone);
EP Impurity I TBS1 RC5- 17,-hydroxyandrost-4-en-3one
(Epitestosterone); EP Impurity C
The TBS-2 gel packaged in the multiple dose dispenser is tested to the following specifications for batch release.
Table 3.2.P.5.1-2: Specification for TBS-2 Gel Packaged in Multiple Dose Dispenser
Test Parameter Method/Reference Acceptance Criteria
Appearance STM.GEN.001 White barrel and cap, filled with slightly
yellow gel
Identification A STM.TBS2.001 Retention time and spectrum corresponds to standard
Identification B STM.TBS2.001 UV spectrum matches reference spectrum
Delivered Dose STM.TBS2.003 Meets USP <601>
Uniformity Mean (beginning doses & end doses)
Min:
Max:
%RSD:
Related STM.TBS2.002 TBS1 RC4 , 0.2% Compounds TBS1 RC5 , 0.5%
Single unknown impurity , 0.2% Total impurities , 1.0%
Residual USP<467> Complies with USP<467>
Solvents
Microbial USP<61>& <62> Total aerobic microbial count < 10 cfu/g Limits Total combined yeasts/mould count < 10 cfu/g
P. aeruginosa 0/g
TBS1 RC4 - 17.-hydroxyandrosta-4,6-dien-3-one (Delta-6-testosterone);
EP Impurity I TBS1 RC5- 17,-hydroxyandrost-4-en-3one
(Epitestosterone); EP Impurity C
Delivered Dose Uniformity as per USP has been added as a release test to verify the performance of the dispsenser. In addition, the analytical procedure to test for related compounds has been modified. The viscosity and water content of the gel are tested upon release for information purposes only. These analytical procedures are described in this section.
Ten individual barrels are taken and tested for Delivered Dose Uniformity according to the method summarized in Table 3.2. P.5.2.1 .1 -1 . Each dispenser is primed by actuating the pump 10 times. The drug content of the first dose, i.e. actuations #1 1 and #12 and the final dose (based on label claim of 1 0 doses, i.e. 20 actuations), i.e. actuation #29 and #30 are tested. The mean of the 10 doses (20 actuations) are reported.
Table 3.2.P.5.2.1.1-1: Summary of Chromatographic Conditions for Delivered Dose Uniformity

The related compounds TBS1 RC4 (Impurity Ι/Δ-6-testosterone) and TBS1 RC5 (Impurity C/epitestosterone) in the finished product are analysed by HPLC, as well as the unknown impurities according to the method summarized in Table 3.2. P.5.2.1 .2- 1 .
Table 3.2.P.5.2.1.2-1: Summary of Chromatographic Conditions for Related Compounds Method

The measurement of the viscosity of TBS-2 is performed using a rotational viscosimeter. The results are reported for information purposes.
The content of water in the gel is determined using a direct titration measurement as per USP Method 1 a. The results of the water content are reported for information purposes.
Assay and Delivered Dose Uniformity as per USP has been added as a release test to verify the performance of the dispsenser. In addition, the analytical procedure to test for related compounds has been modified. The validation of these analytical procedures are described in this section.
The Assay and Delivered Dose method has been validated according to the parameters listed in Table 3.2. P.5.3.1 -1 .
Table 3.2.P.5.3.1-1: Assay and Delivered Dose Validation
Test Acceptance Criteria Results
Linearity r2 > 0.998 r2 = 0.999
0.24% and 0.48% Gel
Mean = 100.6% (for all levels
50%, 100% and 150%)
Mean Recovery 98 - %RD = 0.6%
Accuracy 102% Individual 98 -
0.72 % Gel
102% Mean = 100.0% (for all levels
50%, 100% and 150%)
%RD = 0.2%
RSD < 2.0% At ^g/mL RSD 0.2%
System Precision At 30μg/mL RSD 0.4%
0.24% TBS-2
Chemist 1 Chemist2
Mean 98.2% 98.2%
RSD 0.1% 0.3%
Absolute
Difference Q Q
Repeatability and 0.48% TBS-2
Intermediate Precision
Chemist 1 Chemist2
Mean 96.6%
Assay for 6 samples RSD < 97.0%
Method Precision 2.0%
RSD 0.1% 0.2%
Absolute
Absolute Difference
Difference 0.4
between
Chemist 1 and Chemist 2 is
NMT 2%
0.72% TBS-2
Chemist 1 Chemist2
Mean 99.0% 98.3%
RSD 0.2% 0.1%
Absolute
Difference 0.7
Standard Solution for 5 Days
0.24% and 0.48%
Standard Solution for
5 Days
0.72%
Stability of
Sample Solution for 5 Days
0.24% and 0.48%
Sample Solution for 0.72% 5 Days
No interference of
Selectivity excipient mixture with Complies
testosterone peak
Related Compounds/Degradation Products by HPLC. The method has been validated to the performance characteristics presented in Table 3.2. P.5.3.2-1 and 3.2.P.5.3.2-2.
Table 3.2.P.5.3.2-1: Analytical Validation for the Determination of Related Compounds by

Chemist 1 Chemist 1
% RSD of Mean = 0.20% Mean = 0.24% mean result %RSD = 1.3 %RSD = 1.4 values of
known related Chemist 2 Chemist 2 compound Mean = 0.19% Mean = 0.24% obtained from %RSD = 1.6 %RSD = 1.3 six samples:
< 10%
Table 3.2.P.5.3.2-2: Analytical Validation for the Determination of Related Compounds

Selectivity are free of interference. Conforms
It can be concluded that the analytical method meets the acceptance criteria described in the validation plan.
For the Phase I trial, three bulk gel batches of TBS-2 have been
manufactured and are summarized in Table 3.2.P.5.4-1 . Batch analysis data from the bulk gel batches IMP1 1005, IMP1 1006 and IMP1 1007 are presented in Tables 3.2.P.5.4-2 and batch analysis data from finished drug product batches IMP1 1008, IMP1 1009 and IMP1 1010 are presented in Table 3.2.P.5.4-3.
Table 3.2.P.5.4-1: Description of TBS-2 Batches

Net fill weight lO.g + l.Og

TBS1 RC4 - 17.-hydroxyandrosta-4,6-dien-3-one (Delta-6-testosterone); EP Impurity I TBS1 RC5 - 17,-hydroxyandrost-4-en-3one
(Epitestosterone); EP Impurity C
BRT - below reporting threshold
Table 3.2.P.5.4-3: Batch Analysis - TBS-2 Finished Product Filled in Multiple Dose Dispensers Batches EVIP1 1008, EVIP1 1009 and IMPl 1010
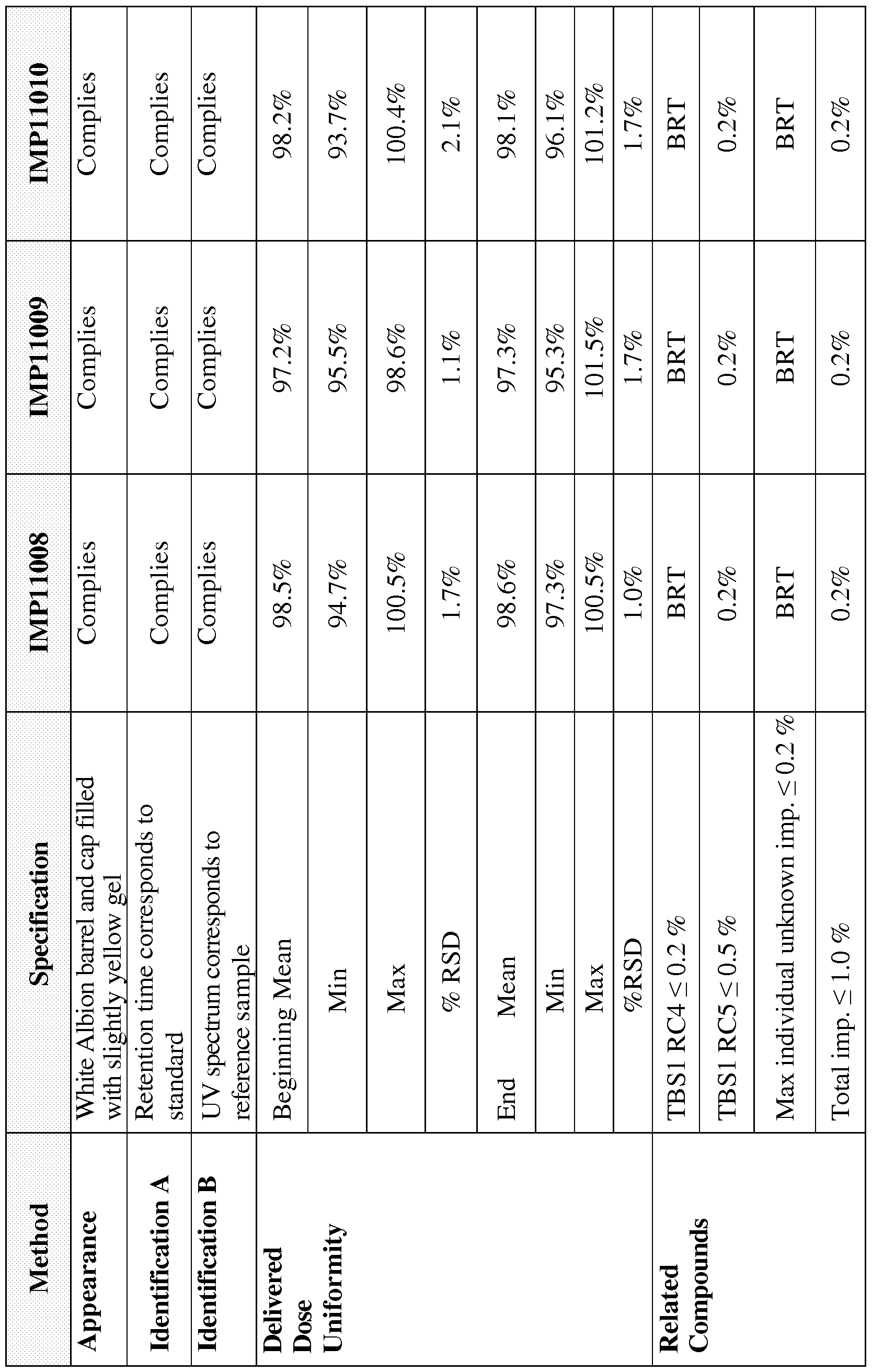

TBS1 RC4 - 17.-hydroxyandrosta-4,6-dien-3-one (Delta-6-testosterone); EP Impurity I TBS 1 RC5- 17 -hydroxyandrost-4-en-3one
(Epi testosterone); EP Impurity C
TAMC - total aerobic microbial count; TYMC - total combined yeasts/mould count BRT - below reporting threshold
The only change made to the previous specification is the additional test for Delivered Dose Uniformity as per USP, to verify the performance of the dispenser. The following is a description of this test in the specification with a discussion
concerning their suitability for intended use and a justification for the acceptable criterion.
The delivered dose of the gel discharged from the nasal actuator is analysed for the active content from a sample at the beginning and at the end of an individual multiple dose dispenser. Each dispenser is primed by actuating the pump 1 0 times. The drug content of the first dose, i.e. actuations #1 1 and #12 and the final dose (based on label claim of 1 0 doses, i.e. 20 actuations), i.e. actuation #29 and #30 are tested using an in house HPLC method to determine the amount of active delivered from the nasal actuator, which is expressed as a percentage of the label claim.
The delivered dose uniformity test demonstrates the uniformity of the dose per actuation, and per total dose (2 actuations) and is consistent with the label claim, discharged from the nasal actuator.
The acceptance criteria proposed are based on USP <601 > for Delivered Dose Uniformity and Guidance for Nasal Spray and Inhalation Solution, Suspension, and Spray Drug Products - Chemistry, Manufacturing and Controls Documentation:
The amount of active per determination is not outside of 80 to 120% of the LC for more than 2 of 20 determinations from 10 containers; none of the determinations is outside of 75 to 125% of the LC and the mean for each of the beginning and end determinations are not outside of 85 to 1 15% of LC.
TBS-2 is packaged in an Albion 15 ml barrel composed of polypropylene with a VP39/140H pump and cap. The barrel is filled with gel under vacuum using an Airlesssystems Laboratory filling unit (Airlesssystems, RD 149 Charleval 27380, France) to effectively expel 125 mg of gel.
The target fill weight for the Albion 15 ml barrel is 10.0 ± 1 .0g.
The quality control measures are proposed for the packaging components. Material information on the Albion 1 5 ml Barrel with VP 39/140H Pump and Cap is provided in Table 3.2. P.7-1 . In addition to the supplier release CoA, the container closure system is retested upon receipt to conform to the specifications summarized
in Table 3.2. P.7-2 The CoA for the Albion 15 ml Barrel Lot 1 0318UV121 01 is provided in Appendix 5. A Figure of the 15 ml Albion Bottle and the PMP VP 39/140H 15 Albion + Digital Actuator + Cap 15 ml Albion Rounded is provided at Figure 39.
Table 3.2.P.7-1: Albion 15 ml Barrel with VP 39/140H Pump and Cap - Material Information
Component Material
Barrel Polypropylene: PPR 7220
White colorant: PP 00121522
Piston Polytethylene PEHD PURELL GC 7260
Base Polypropylene: PPR 7220
White colourant: PP 00121522
Insert Polypropylene : PPH 7060
Pen actuator Polypropylene : PPC 11712
White colourant : PP0012 1522
Snap on Polypropylene : PPH 5060
White colourant : PP0012 1522
Neck gasket Thermoplastic : F217-5 906
Stem gasket Elastomer : 522C
Pump
Body PBT VALOX HX312C-1H1001
Ball support HD Polyethylene : Purell GB7250
Ball STAINLESS STEEL AISI 304L
Spring cap POM HOSTAFORM C27021
Return spring STAINLESS STEEL 1.43 10
Piston HD Polyethylene : HOSTALEN GF4750
SILBIONE DM300 GMP
Stem POM DELRIN 900PNC 10
Pre compression spring STAINLESS STEEL 1,43 10
Turret Polypropylene: PPH5060
White colourant: 85276
Table 3.2.P.7-2: Albion 15 ml Barrel with VP 39/140 Pump and Cap - Specifications

Three clinical batches bulk batches of TBS-2 (IMP1 1005, IMP1 1006 and IMP1 1007) were manufactured in a GMP facility. Each batch was then filled in the multiple dose dispenser as summarized in Table 3.2. P.8.1 .
Table 3.2.P.8. 1: Bulk Gel and Finished Product Batches
Concentration 0.24% 0.48% 0.72%
Bulk Batch No. IMP11005 IMP11006 IMP11007
Finished IMP11008 IMP11009 IMP11010
Product Batch No.
The bulk gel will stored under real time conditions 25 ± 2 °C, 60 ± 5 % RH and will be tested at the time intervals presented in Table 3.2. P.8.3. -2.
Table 3.2.P.8.3-2: Stability Schedule for TBS-2 Batches IMPl 1005, IMPl 1006 and IMPl 1007

Table 3.2.P.8.1-3: Stability Study Test Parameters of TBS-2 Bulk Gel and
Corresponding Acceptance Criteria

TBS1 RC4 - 17.-hydroxyandrosta-4,6-dien-3-one (Delta-6- testosterone); EP Impurity I TBS1 RC5- 17,-hydroxyandrost-4-en- 3one (Epitestosterone); EP Impurity C
The multiple dose dispensers are stored under real time storage (25 ± 2°C, 60 ± 5 % RH), intermediate time storage (30± 2°C, 65 ± 5 % RH) and accelerated
storage conditions (40 ± 2°C, 75 ± 5 % RH) and tested to the schedule presented in Table 3.2. P.8.1 -4. Ten (10) dispensers will be analyzed at each time point.
Table 3.2.P.8.1-4: Stability Schedule COMPLEO Finished Product Filled in Multiple
Dose Dispenser
Completed Test
Storage Conditions (°C, Batch
Strength Product Type Intervals % RH) Number (Outstanding Test
Intervals")
Finished Om, (3m, 6m, 9m,
IMPl product in
25 ± 2°C, 60 ± 5 % RH 0.24% 12m, 18m, 24m,
1008 multiple dose 36m)
dispenser
Finished
IMPl product in
30± 2°C, 65 ± 5 % RH 0.24% 0m, (3m, 6m, 9m, 12m,)
1008 multiple dose
dispenser
Finished
IMPl product in
40 ± 2°C, 75 ± 5 % RH 0.24% iple dose 0m, (lm, 2m, 3m, 6m)
1008 mult
dispenser
Finished 0m, (3m, 6m, 9m,
IMPl product in 12m, 18m, 24m,
25 ± 2°C, 60 ± 5 % RH 048%
1009 multiple dose 36m)
dispenser
Finished
IMPl product in
30± 2°C, 65 ± 5 % RH 0.48%
1009 multiple dose 0m, (3m, 6m, 9m, 12m) dispenser
Finished
IMPl product in
40 ± 2°C, 75 ± 5 % RH 0.48% , (lm, 2m, 3m, 6m)
1009 multiple dose 0m
dispenser
Finished
0m, (3m, 6m, 9m,
25 ± 2°C, 60 ± 5 % IMPl product in
0.72%
RH 1010 multiple dose 12m, 18m, 24m,
dispenser 36m)
Finished
30± 2°C, 65 ± 5 % IMPl product in
0.72%
RH 1010 multiple dose 0m, (3m, 6m, 9m, 12m) dispenser
Finished
40 ± 2°C, 75 ± 5 % IMPl product in
0.72% (lm, 2m, 3m, 6m) RH 1010 multiple dose 0m,
dispenser
The attributes used to confirm the quality of TBS-2 finished product and
corresponding acceptance criteria are listed in Table 3.2.P.8. 1 -5.
Table 3.2.P.8.1-5: Stability Study Test Parameters of TBS-2 Finished Product and Corresponding Acceptance Criteria

The results from the stability studies will be evaluated for any trends and a shelf life proposed after data has been generated. Room temperature,
intermediate and accelerated stability studies are ongoing and updated data are available upon request.
TBS-2 bulk and finished product will be placed on a stability program as described in section 3.2. P.8.1 . Stability updates will be provided as available.
A 4.5% testosterone gel inaccordance with this invention is packaged in the identical container closure system, the Albion 15 ml barrel composed of polypropylene with a VP39/140H pump and cap. The qualitative formulation is the same for the 4.5% gel as for the 0.24% 0.48% and 0.72% gels, only the proportion of castor oil and testosterone are different. Tables 3.2. P.8.3-1 and 3.2. P.8.3-2 provides supportive data for the stability of testosterone gel packaged in the Albion 15 ml barrel composed of polypropylene with a VP39/140H pump and cap. This supportive data indicates that the gel packaged in the dispenser provides adequate protection for potency and purity and are within acceptance criteria.
Table 3.2.P.8.3 1: Stability Data 4.5% Testosterone Gel Batch 1969 in Multiple Dose


BRT - below reporting threshold
Table 3.2.P.8.3-2: Stability Data 4.5% Testosterone Gel Batch 1969 in Multiple Dose

BRT - below reporting threshold
14. Discussion and Overall Conclusions
A. Summary of Subjects
This was a phase 1 , single-center, randomized, open-label parallel-group study of TBS-2 in 3 cohorts of subjects (Cohorts 1 , 2, and 3) in Period 1 (single-dose) and a multiple-dose cohort in Period 2. A total of 24 healthy women received intranasal 600 1200 or 1800 μg TBS-2 to evaluate the safety, tolerability, and PK of TBS-2.
B. Pharmacokinetic Conclusions
Results from the study indicate that the free testosterone concentrations and AUCo-8, AUCo-24, and AUCo-t parameters are dose proportional. While the total testosterone concentrations and parameters are clearly reflective of the dose, the strongest dose proportionality relationship is with the free testosterone. Dose
proportionality is tested both by linear regression and by pair-wise dose comparison of parameters and both tests are equivocal in that free testosterone concentrations were dose proportional. Other analyte concentrations are less changed with the dose in the order of dihydrotestosterone with a minor change to estradiol with less and finally SHBG with the lest change.
Additionally equivalence between the single- and multi-dose profiles is not rejected for either of the free testosterone AUCo-s and AUCo-24 parameters suggesting little if any accumulation of free testosterone following multiple dosing. However, this result is limited to the 3 subjects who received 1200 μg on each profile. Single-dose parameter calculations are performed on uncorrected concentrations of free
testosterone, total testosterone and dihydrotestosterone for comparison to multiple-dose parameters. The pharmacokinetic analysis is based on 5 endogenous analytes that have reported normal ranges as well as baseline ranges. The following table presents the normal ranges, the baseline ranges observed and a break-down of the baseline ranges by dose cohort.
Normal Pooled Dose Cohort
Analyte Range Baseline 600 (μ8) 1200 (μ8) 1800 (μ8)
Free Testosterone (ng/dL) Min 0.166 0.10 0.10 0.13 0.15
N=112 Max 1.33 1.44 1.44 0.53 0.45
Total Testosterone (ng/dL) Min 6.00 7.41 8.00 7.41 12.70
N=112 Max 86.00 58.00 58.00 41.10 43.90
Dihydrotestosterone (ng/dL) Min 4.00 5.02 5.29 5.04 5.02
N=75 Max 22.00 32.60 22.10 25.30 32.60
Estradiol (pg/mL) Min 20.00 17.00 20.90 17.00 17.70
N=112 Max 241.00 122.00 122.00 119.00 97.40
SHBG (nmol/L) Min 18.00 15.30 15.30 16.00 25.20
N=112 Max 114.00 137.00 102.00 86.70 137.00
These data comparisons of the normal ranges with the pooled baseline ranges indicate that the pooled baseline ranges fall outside the normal ranges for free testosterone (min and max), dihydrotestosterone (max), estradiol (min) and SHBG (min and max).
C. Safety Conclusions
There have been no deaths, SAEs, or AEs leading to discontinuation during the study. Most TEAEs are in the system organ classes of general disorders (nasal related events) and administration site conditions and respiratory, thoracic, and mediastinal disorders. This is not unexpected given the venipuncture for PK blood sample draws and the intranasal study drug administration. Most TEAEs are mild in severity and are unlikely or not related to study medication.
TBS-2 is believed to be safe and well tolerated with respect to AEs and vital signs.
EXAMPLE 11 B
An Open-Label, Single and Multiple- Application of IntranasalTestosterone Gel (TBS-2) in Healthy Pre-menopausal
Female Subjects at Three Dose Levels
Methodology:
This was a phase 1 , single-center, randomized, open-label parallel-group study designed to evaluate the safety, tolerability, and PK of TBS-2 in healthy, normal-cycling adult pre-menopausal women. Subjects were randomly assigned on a 1 :1 :1 basis to 1 of 3 treatment groups (Cohort 1 , Cohort 2, or Cohort 3) during Period 1 and were
administered a single dose of an intranasal application of TBS-2 at doses of 600 pg, 1200 μg! or 1800 μg (single doses of 300 ]ig, 600 μg! and 900 ^g per nostril). At the end of Period 1 , a total of 8 subjects sampled from these 3 cohorts, who were willing and able to continue with the multiple-dose portion of the study were selected to participate in Period 2. During Period 2, subjects were administered 1200 lig TBS-2 (600 μg per nostril) t.i.d. for 2 days and once on the morning of the third day. Subjects were screened (Visit 1 ) for eligibility up to 3 weeks prior to dosing in Period 1 , and were admitted to the Clinical Research Unit (CRU) at 0700 hours on the day prior to dosing (Visit 2, Day 1 ) for baseline testosterone measurement.. Subjects started Period 1 on day 1 -2 of their menstrual cycle. On study Day 2, subjects were administered a single dose of TBS-2 at 0800 and remained in the CRU for 48 hours post-dose for safety monitoring and PK assessments. Subjects were discharged from the clinic on Day 4, and subjects who did not continue into Period 2 also underwent close-out assessments. Depending on their menstrual cycle, subjects selected to participate in Period 2 returned to the CRU approximately 26 to 32 days following the conclusion of Period 1 for Visit 3 (Period 2). Subjects started Period 2 on day 1 -2 of their menstrual cycle. During Period 2, subjects were admitted to the CRU at 0700 hours on the day of dosing (Visit 3, Day 1 ) On study Days 1 and 2, subjects were administered TBS-2 at 0800 hours (+30 minutes), 1600 hours (+30 minutes), and 2400 hours (+30 minutes). On Day 3, subjects were administered TBS-2 at 0800 hours (+30 minutes). Subjects remained in the CRU for 48 hours following dosing on Day 3 for safety monitoring and PK assessments. Subjects were discharged from the clinic on Day 5.
During Period 1 , blood samples for determination of baseline testosterone (free and total), SHBG, DHT, and estradiol concentration were collected on Day 1 at 0745 hours and then at 15, 30, and 45 minutes and at 1 , 1 .5, 2, 4, 6, 8, 12, 16, 20, and 23.5 hours relative to an 0800 hour clock time. Blood samples for determination of testosterone (free and total), SHBG, DHT, and estradiol plasma concentration were collected on Day 2 (15, 30, and 45 minutes and 1 , 1 .5, 2, 4, 6, 8, 12, 16, and 20 hours post-dose) and Day 3 (24, 32, 40, and 48 hours post-dose) during the confinement period.
During Period 2, a blood sample for baseline testosterone (free and total), SHBG, DHT, and estradiol concentration was collected at 0745 hours (i.e., 15 minutes prior to
study drug administration). Blood samples for determination of testosterone (free and total), SHBG, DHT, and estradiol concentration were collected on Day 1 (pre-dose [15 minutes prior to dosing] and at 1545 and 2345 hours), Day 2 (1545 and 2345 hours), Day 3 (15, 30, and 45 minutes and 1 , 1 .5, 2, 4, 6, 8, 12, 16, and 20 hours), and Day 4 (24, 32, 40, and 48 hours) during the confinement period.
Other assessments performed during the study included the monitoring of adverse events (AEs), clinical laboratory evaluations (chemistry [including hormone profile], hematology, and urinalysis), vital signs assessments (systolic and diastolic blood pressure [BP], heart rate [HR], respiratory rate [RR], and body temperature), and physical examinations. In addition, otorhinolaryngological examination findings, 12-lead electrocardiogram (ECG) readings, medical history, and concomitant medication use were recorded.
Number of Subjects (planned and analyzed)
Planned: A total of 24 subjects were planned to be enrolled.
Enrolled: A total of 24 subjects were enrolled and randomly assigned to treatment in Period 1 : 8 subjects in Cohort 1 , 8 subjects in Cohort 2, 8 subjects in Cohort 3. A total of 8 subjects sampled from these 3 cohorts, who were willing and able to continue with the multiple-dose portion of the study, were selected to participate in
Period 2.
Analyzed: All 24 subjects were included in the safety analyses and 24 subjects were included in the PK analyses.
Diagnosis and Main Criteria for Inclusion
Healthy, normal-cycling women between the ages of 1 8 and 40 years (inclusive) who were premenopausal, had a body mass index (BMI) of 18.5 to 35 kg/m2 (inclusive), met all of the inclusion and none of the exclusion criteria, and provided informed consent were included in the study.
Test Product, Dose and Mode of Administration, Batch Number
The TBS-2 used in this study was an intranasal testosterone gel supplied in prefilled dispensers with 0.24% testosterone gel to deliver a single intranasal dose of 300 pig of testosterone per nostril (Cohort 1 ), 0.48% testosterone gel to deliver a single intranasal dose of 600 pig of testosterone per nostril (Cohort 2 [single-dose] and Multi- dose group), and 0.72% testosterone gel to deliver a single intranasal dose of 900 i.tg of testosterone per nostril (Cohort 3). The lot numbers of TBS-2 drug substance used in this study were IMP1 1008, IMP1 1009 and IMP1 1010.
Duration of Treatment
The study involved I period for Cohorts 1 , 2, and 3 and the duration of individual subject participation from the start of screening until the post-study visit, was
approximately 25 days. This study involved 2 periods totaling 30 to 36 days for the Multi- dose group.
CRITERIA FOR EVALUATION
Safety: Safety was assessed throughout the study and included the monitoring of AEs, clinical laboratory evaluations (chemistry [including hormone profile], hematology, and urinalysis), vital signs, and 12-lead ECGs. Physical and otorhinolaryngological examinations were performed and medical history and concomitant medication use were recorded.
Pharmacokinetics: Whole blood samples for determination of plasma concentrations of testosterone (free and total), SHBG, DHT, and estradiol were collected at specified time points. Actual sampling time points were recorded and used for PK calculations. Pharmacokinetic parameters for testosterone (free and total), SHBG, DHT, and estradiol were calculated by standard noncompartmental methods for all subjects as data permitted. The PK parameters evaluated for plasma concentrations (uncorrected and baseline-corrected) of testosterone (free and total), SHBG, DHT, and estradiol following the single-dose cohorts (Cohorts 1 , 2, and 3) included area under the plasma concentration time curve from time zero to the last measurable concentration time point (AUC0_t), area under the plasma concentration time curve from time zero to infinity
(AUCo-∞,), maximum concentration observed after dosing (Cmax), time of observed Cmax relative to the time of dosing (tmax), terminal elimination rate constant (λζ), and elimination half- life (t½). The PK parameters evaluated for plasma testosterone (free and total), SHBG, DHT, and estradiol concentrations (uncorrected) following the multiple-dose cohort included area under the concentration-time curve from time zero to the dosing interval (AUCo-τ, where τ=8 hours), Cmax, , minimum concentration over a dosing interval during multiple dosing (Cmir,), pre-dose concentration determined immediately before a dose at steady state (Cpd), average steady-state concentration (Cavg), % peak to trough fluctuation (PTF), and % peak to trough swing (PTS).
STATISTICAL METHODS
This study evaluated the PK properties as well as the safety and tolerability of TBS-2. Power calculations were not performed. The sample size for this study was not determined on the basis of statistical hypothesis testing. Based on typical, early-stage PK studies, groups of 8 subjects per cohort provided adequate clinical information to satisfy the objectives of the study. Data were summarized by using descriptive statistics (sample size, mean, median, standard deviation [SD], minimum, and maximum) for each of the safety variables by treatment group and overall. Data from all visits during the study were displayed in the data listings. Concentration-time data for 5 analytes (testosterone [total and free], SHBG, DHT, and estradiol) were determined by a validated assay method and PK parameters were calculated. Actual sampling time points were recorded and used in calculation of the actual elapsed time from dose to sample for PK calculations. As each administration was to each of the nostrils, the time of dosing was the time of the first nostril administration. Baseline analyte concentrations from the 24-hour pre- dose profile were subtracted from the time-matched analyte concentrations following dose administration before calculation of the PK parameters.
Individual PK parameters were estimated for each subject's profile in the PK population by using Phoenix WinNonlin (Pharsight Corporation) version 6.2 and were displayed in the data listings. Data were summarized by using descriptive statistics (mean, SD, % coefficient of variation [CV], confidence interval (CI), median, minimum,
and maximum) and are presented by treatment group. Geometric means were included for AUC and Cmax, estimations and were included for some other PK parameters. By using Generalized Linear Model (GLM) procedure in SAS®, an analysis of variance (ANOVA) was performed on natural logarithmic (In) transformed parameters AUC0_t, AUCo- AUCo-t„ Cavg, and Cmax, and on untransformed parameters t-i/2, and λζ, at the significance level of 0.05. The intrasubject CV was calculated for AUC0-t, AUC0-∞, AUCo-τ, and Cmax by using the ANOVA residual error.
RESULTS
Pharmacokinetics:
1 . Period 1 -Single Dose
a. The mean baseline pre-dose concentrations from Period 1 were consistently within the normal range for all analytes.
b. The uncorrected 600, 1200, and 1800 jig dose concentrations of free and total testosterone and to a lesser extent of DHT were more clearly differentiated from the baseline pre-dose concentrations. The estradiol and SHBG concentrations had post-dose curves that overlapped with the baseline pre-dose concentrations for the same dose.
c. TBS-2 was rapidly absorbed after dose administration. The median T. values of free and total testosterone for 600, 1200, and 1800 ug doses were consistent between the two analytes and reported as 0.38, 0.5 and 0.63 hrs, and 0.38, 0.63 and 0.63 hrs respectively. The geometric mean uncorrected Cmax values of free and total testosterone were 0.761 , 1 .302, and 1 .822 ng/dL, and 58.438, 81 .287 and 139.742 ng/dL respectively.
d. For the most part, the observed post-dose uncorrected mean concentrations of all 5 analytes were within their normal ranges. The only exceptions were during the first 2 hours for the 1800 jig testosterone dose in free (0.5 hrs— 2.0 hrs) and total testosterone (0.25 hrs — 2.0 hrs) in a 24 hour period. Testosterone concentrations exceeding the upper limit of the physiological range (upper limit of normal, ULN) for a limited amount of time following dosing can be expected due
to the peak and trough PK profile of TBS2, and the endogenous nature of testosterone.
e. Mean uncorrected free and total testosterone concentrations at 24 hours after dosing were comparable to pre-dose mean concentrations, with little difference across the three doses.
f. Dose proportionality was not rejected for baseline-corrected AUco-24, AUco-s, and AUco-t of free testosterone. Cmax values of baseline-corrected free and total testosterone also increased with the dose increase (0.442, 1 .017, and 1 .500 ng/dL and 34.058, 62.88 and 1 13.912 ng/dL respectively), but did not demonstrate statistical significance. The DHT PK parameters as well as those of estradiol and SHBG did not pass the statistical test for dose proportionality. eriod 2 -Multiple Dose
a. The free and total testosterone concentrations had the clearest demonstration of administration-related concentration increase. A more modest administration- related increase in DHT concentration, but little administration-related increase in estradiol and SHBG concentrations, was observed. b. Peak concentrations of free and total testosterone were achieved within 15 to 45 minutes following the last (7th) dose administration. The ranges of Tmax, were 0.25-0.75 hrs for both and geometric mean Cmax values were 1 .874 and 137.555 ng/dL for free and total testosterone, respectively. Mean Cavg values of 1 .006 ng/dL, and geometric mean Cavg of 0.944 ng/dL for free testosterone and mean Cavg value of 73.802 ng/dL, and geometric mean Cavg of 69.166 ng/dL for total testosterone were well within the normal range for women.
c. The mean concentration values for free and total testosterone after the multiple (1200 pg every 8 hours) TBS-2 administration exceeded ULN approximately for 1 .0 and 1 .5 hours respectively in the 24-hour period following the last (7th) dose administration, which can be expected due to the peak and trough PK profile of TBS-2.
d. Accumulation analysis did not detect substantial evidence of accumulation for any analytes.
e. Equivalence between the single- and multiple-dose profiles in the 3 subjects who participated in both profiles was not rejected for AUC0-s and AUC0-24 of free testosterone and for AUCo-s of total testosterone, also suggesting minimal accumulation. Equivalence was rejected for AUCo-24 of total testosterone, which can be explained by possible perturbation of the data by the diurnal variation in testosterone levels around 24 and 48 hours post-dose. Since there was no accumulation at 8 and at 16 hours, it would appear logical that the apparent significant difference between available total testosterone at 24 hours was possibly due to the diurnal variation in testosterone levels.
Safety
There were no deaths, SAEs, or AEs leading to discontinuation during the study. Most TEAEs were in the system organ classes of general disorders and administration site conditions and respiratory, thoracic, and mediastinal disorders (nasal related events). Most TEAEs were mild in severity and were unlikely or not related to study medication. In Period 1 , a total of 1 1 of 24 subjects (45.8%) experienced at least 1 TEAE. Those experienced by more than 1 subject were catheter site erythema (experienced by 3 of 24 subjects [12.5%]) and catheter site hemorrhage, catheter site inflammation, dizziness, and nasal congestion (each experienced by 2 of 24 subjects [8.3%]).
The majority of TEAEs in Period 1 were of mild severity. A total of 10 of 24 subjects (41 .7%) experienced mild TEAEs. Only 1 of 24 subjects (4.2%) experienced moderate TEAEs. Subject 522-53 (1200 pg) experienced headache and dizziness of moderate severity on Day 1 . Both TEAEs resolved without treatment the next day and were considered as unlikely to be related to the study drug.
In Period 2, a total of 4 of 8 subjects (50.0%) experienced at least 1 TEAE. General disorders and administration site conditions occurred in 2 of 8 subjects (25.0%). The
TEAEs that were experienced by 1 of 8 subjects (12.5%) each were catheter site pain, catheter site phlebitis, dyspepsia, headache, and rhinalgia.
The majority of TEAEs in Period 2 were of mild severity. Overall, 3 of 8 subjects (37.5%) experienced mild TEAEs. Only 1 of 8 subjects (12.5%) experienced a moderate TEAE. Subject 522-29 experienced an increase in headache intensity of moderate severity on Day 2. The TEAE resolved without treatment 2 days later and was considered as possibly related to the study drug.
A total of 6 subjects experienced adverse reactions (defined as TEAEs considered possibly, probably, or definitely related to study medication) during the study; 4 subjects in Period 1 and 2 subjects in Period 2. All adverse reactions were considered possibly related to study medication. Subject 522-29 (Period 2, 600 pg) had 2 adverse reactions of headache that were each recorded as a TEAE: a mild headache that increased in severity to moderate. A total of 6 subjects experienced adverse reactions during the study: 4 of 24 subjects (16.7%) in Period 1 and 2 of 8 subjects (25.0%) in Period 2. All adverse reactions were considered possibly related to study medication. In Period 2, Subject 522-29 (1200 pg) had 2 adverse reactions of headache that were each recorded as a TEAE: a mild headache that increased in severity to moderate.
There were no TEAEs associated with clinical laboratory tests or vital signs. Two subjects had ENT examination findings that were interpreted as clinically significant by the PI on Day 4 in Period 1 : Subject 522-37 (600 lig) had slight rhinorrhea and Subject 522-53 (1200 ug) had mild erythema to the left nostril mucosa.
CONCLUSIONS
Summary of Subjects
This was a phase 1 , single-center, randomized, open-label parallel-group study of TBS- 2 in 3 cohorts of subjects (Cohorts 1 , 2, and 3) in Period 1 (single-dose) and a multiple-
dose group in Period 2. A total of 24 healthy women received intranasal 600 μg, 1200 ng, or 1800 μg TBS-2 to evaluate the safety, tolerability, and PK of TBS-2.
Pharmacokinetic Conclusions
Results from the study indicate that the free testosterone concentrations and AUC0.8, AUCo-24, and AUCat parameters were dose proportional and the total testosterone concentrations and parameters were clearly reflective of the dose strength. Other analyte concentrations were less changed with the dose strength in the order of DHT with a minor change to estradiol with less and finally SHBG with the least change. No substantial evidence of accumulation was detected for any analyte following multiple dose administration. Additionally equivalence between the single- and multi-dose profiles was not rejected for free and total testosterone, also suggesting little
accumulation.
Although peak levels of serum free and total testosterone exceeded the upper level of normal for a limited amount of time following the single administration of the highest dose strength and after the multiple dosing, the observed mean concentrations of these two and 3 other analytes were well within their normal ranges for all dose strengths.
Safety Conclusions
There were no deaths, SAEs, or AEs leading to discontinuation during the study. Most TEAEs were in the system organ classes of general disorders and administration site conditions and respiratory, thoracic, and mediastinal disorders (nasal related events). This was not unexpected given the venipuncture for PK blood sample draws and the intranasal study drug administration. Most TEAEs were mild in severity and were unlikely or not related to study medication.
TBS-2 was safe and well tolerated with respect to AEs and vital signs.
List of Figures
Figure 11-1: Mean Observed Free Testosterone Concentrations (Single-Dose Population)
Figure 11-2: Mean Observed Total Testosterone Concentrations (Single-Dose Population)
Figure 11-3: Mean Observed Dihydrotestosterone Concentrations (Single-Dose Population)
Figure 11-4: Mean Observed Estradiol Concentrations (Single-Dose Population)
Figure 11-5: Mean Observed SHBG Concentrations (Single-Dose Population)
Figure 11-6: Mean Corrected Free Testosterone Concentrations (Single-Dose Population)
Figure 11-7: Mean Corrected Total Testosterone Concentrations (Single-Dose Population)
Figure 11-8: Mean Corrected Dihydrotestosterone Concentrations (Single-Dose Population)
Figure 11-9: Mean Corrected Estradiol Concentrations (Single-Dose Population)
Figure 11-10: Mean Corrected SHBG Concentrations (Single-Dose Population)
Figure 11-11: Baseline-Corrected Free Testosterone Spaghetti Figure, 600 μg (Single-Dose Population)
Figure 11-12: Baseline-Corrected Free Testosterone Spaghetti Figure, 1200 μg (Single-Dose Population)
Figure 11-13: Baseline-Corrected Free Testosterone Spaghetti Figure, 1800 μg (Single-Dose Population)
Figure 11-14: Trough Total Testosterone Linear Regression Figure (Multi-Dose Population) Figure 11-15: Mean Free Testosterone Plasma Concentrations (Multi-Dose Population) Figure 11-16: Mean Total Testosterone Plasma Concentrations (Multi-Dose Population) Figure 11-17: Mean Dihydrotestosterone Plasma Concentrations (Multi-Dose Population) Figure 11-18: Mean Estradiol Plasma Concentrations (Multi-Dose Population)
Figure 11-19: Mean SHBG Plasma Concentrations (Multi-Dose Population)
Figure 11-20: Spaghetti Concentration Plots with Mean for Free Testosterone Plasma Concentrations (Multi-Dose Population)
Figure 11-21: Spaghetti Concentration Plots with Mean for Total Testosterone Plasma Concentrations (Multi-Dose Population)
Figure 11-22: Spaghetti Concentration Plots with Mean for Dihydrotestosterone Plasma Concentrations (Multi-Dose Population)
Figure 11-23: Spaghetti Concentration Plots with Mean for Estradiol Plasma Concentrations
(Multi-Dose Population)
Figure 11-24: Spaghetti Concentration Plots with Mean for SHBG Plasma Concentrations
(Multi-Dose Population)
Figure 11-25: Trough Free Testosterone Linear Regression Figure (Multi-Dose Population)
3.2 List of Tables
Table 9-1: Study Schedule
Table 9-2: Schedule of Pharmacokinetic Sample Collection
Table 11-1: Medical History (Single-Dose Population)
Table 11-2: Baseline Analyte Concentrations (Single-Dose Population)..
Table 11-3: Trough Concentration Summary (Multi-Dose Population)
Table 11-4: Free Testosterone Summary (Single-Dose Population)
Table 11-5: Total Testosterone Summary (Single-Dose Population)
Table 11-6: Dihydrotestosterone Summary (Single-Dose Population) Table 11-7: Estradiol Summary (Single- Table 11-8: SHBG Summary (Single-Dose Population)
Table 11-9: Free Testosterone Summary for Multiple-Dose Profile (Multi-Dose Population)
Table 11-10: Total Testosterone Summary for Multiple-Dose Profile (Multi-Dose Population)
Table 11-11: Dihydrotestosterone Summary for Multiple Dose Profile (Multi-Dose Population)
Table 11-12: Estradiol Summary for Multiple-Dose Profile (Multi-Dose Population)
Table 11-13: SHBG Summary for Multiple Dose Profile (Multi-Dose Population)
Table 11-14: Dose Proportionality Analysis (Single-Dose Population)
Table 11-15: Analysis of Variance for Some Pharmacokinetic Parameters (Single-Dose
Population)
Table 11-16: Paired t-Test Results for Pharmacokinetic Parameters AUCO-8 and AUCO-24 for
Subjects Who Had 1200 μg
Table 12-1: Incidence of Treatment- Emergent Adverse Events by System Organ Class and
Preferred Term (Single-Dose Population)
Table 12-2: Incidence of Treatment- Emergent Adverse Events by System Organ Class and Preferred Term (Multi-Dose Population)
Table 12-3: Subjects with Adverse Reactions (Single- and Multi-Dose Populations)
Table 12-4: Subjects with New Abnormal Hematology Laboratory Evaluation Results Post-Dose (Single- and Multi-Dose Populations)
Table 12-5: Subjects with New Abnormal Chemistry Laboratory Evaluation Results Post-Dose (Single- and Multi-Dose Populations)
Table 12-6: Subjects with New Abnormal Urinalysis Laboratory Evaluation Results Post-Dose (Single- and Multi-Dose Populations)
Table 12-7: Subjects with Abnormal Basic Ear, Nose, and Throat Examination Results (Single- and Multi-Dose Populations)
3.3 List of Appendices
16.1 Study Information
16.1.1 Protocol and Protocol Amendments
16.1.2 Sample Case Report Form
16.1.3 Institutional Review Boards (IRBs) and Subject Consents and Information
16.1.4 Description of Investigators
16.1.5 Signature Page
16.1.6 Listing of Batch Numbers by Subject
16.1.7 Randomization Scheme and Codes
16.1.8 Audit Certificates (if available)
16.1.9 Statistical Analysis Plan
16.1.10 Documentation of Inter-laboratory Standardization Methods and Quality Assurance Procedures, if Used
16.1.11 Publications Based on the Study
16.1.12 Important Publications Referenced in the Report
16.2 Subject Data Listings
16.2.1.1 Analysis Population and Subject Disposition
16.2.1.2a Study Completion: Period 1
16.2.1.2b Study Completion: Period 2
16.2.4.1 Demographics
16.2.4.2 Medical History
16.2.4.3.1 Physical Examination
16.2.4.3.2 ENT Nasal Endoscopic Examination
16.2.4.3.3 Basic ENT Examination
16.2.5.1a Single-Dose - Study Drug Administration
16.2.5.1b Multi-Dose - Study Drug Administration
16.2.5.1.1a Single-Dose - Pharmacokinetic Sampling Times and Plasma Baseline, Observed, and
Baseline-Corrected Concentrations
16.2.5.1.1b Multi-Dose - Pharmacokinetic Sampling Times and Plasma Baseline, Observed, and
Baseline-Corrected Concentrationsl6.2.5.1.2a Single-Dose - Pharmacokinetic Plasma Parameter Estimates
16.2.5.1.2b Multi-Dose - Pharmacokinetic Plasma Parameter Estimates
16.2.6 Hormone Profile Laboratory Evaluations
16.2.7.1 Adverse Events
16.2.7.4 Adverse Reactions
16.2.8.1 Hematology Laboratory Evaluations
16.2.8.2 Chemistry Laboratory Evaluations
16.2.8.3 Urinalysis Laboratory Evaluations
16.2.8.4 Vital Signs
16.2.9.1 Prior and Concomitant Medications
16.3 Case Report Forms
16.4 Additional Information
4 GLOSSARY OF ABBREVIATIONS AND TERMS
AE adverse event
ALT alanine transaminase
ANOVA analysis of variance
AST aspartate transaminase
AUC area under the plasma concentration time curve
AUCo-∞ AUC from time zero to infinity
AUCO-t AUC from time zero to the last measurable concentration time point
AUCO-τ AUC from time zero to the dosing interval (where τ=8 hours) during the multi- dose period
AUCo-8 AUC from time zero to 8 hours after dosing during either the single- or multi-dose periods
AUCo-24 AUC from time zero to 24 hours after dosing during either the single- or multi-dose periods
BMI body mass index
BP blood pressure
BUN blood urea nitrogen
CaVg average steady- state concentration
CFR Code of Federal Regulations
CI confidence interval
CK creatine kinase
CL clearance
Cmax maximum concentration observed after dosing
Cmin minimum concentration over a dosing interval during multiple dosing
ConcBase baseline concentration
ConcBC baseline corrected concentration
ConcBLQ active dose concentrations corrected for BLQ
Cpd pre-dose concentration determined immediately before a dose at steady state
CRF case report form
CRU clinical research unit
CS clinically significant
CV coefficient of variation
DHT dihydrotestosterone
ECG electrocardiogram
eCrf electronic case report form
FDA Food and Drug Administration
FSD female sexual dysfunction
GCP good clinical practice
GGT gamma-glutamyl transferase
GLM Generalized Linear Model
HbsAg hepatitis B surface antigen
HCV hepatitis C virus
HIV human immunodeficiency virus
HR heart rate
ICF informed consent form
IRB institutional review board
In logarithmic
MedDRA Medical Dictionary for Regulatory Activities
NCS not clinically significant
OTC over-the-counter
PD pharmacodynamic
PI Principal Investigator
PK pharmacokinetic
PTF % peak to trough fluctuation
PTS % peak to trough swing
RR respiratory rate
Rsq linear correlation coefficient
SAP statistical analysis plan
SD standard deviation
SHBG sex hormone-binding globulin
t½ elimination half-life
tmax time of observed Cmax relative to the time of dosing
VPA vaginal pulse amplitude
λζ terminal elimination rate constant
6 INVESTIGATORS AND STUDY ADMINISTRATIVE STRUCTURE
This study was conducted at 1 study center in the United States. The PI at the study center was responsible for the validity and accuracy of the data supplied on the case report form (CRF). Delegation of authority for completion of the CRF was permitted, but the PI was responsible for its accurate completion and was required to sign the completed CRF that it was a true and accurate reflection of the subject's participation in the study.
The PI at the study center signed the protocol signature page. By signing this page, the PI agreed to conduct the study in accordance with the study protocol and also to comply with the requirements regarding the obligations of clinical investigators and all other pertinent requirements of applicable regulatory agencies.
7 INTRODUCTION
TBS-2 is being developed for the treatment of Female Orgasmic Disorder. 7.1 Background
Anorgasmia is the second most frequently reported women's sexual problem after hypoactive sexual desire disorder. The Global Survey of Sexual Attitudes and Behaviors 1 assessed sexual problems in 9000 women aged 40 to 80 years (inclusive) in 29 countries. The prevalence of inability to reach orgasm ranged from 17.7% (in Northern Europe) to 41.2% (in Southeast Asia).
In the PRESIDE survey of over 31,000 women, approximately 10% reported low desire with distress and almost 5% report difficulty reaching orgasm with distress.2 Anorgasmia is considered to be the persistent or recurrent delay in, or absence of, orgasm following a normal sexual excitement phase, causing marked distress or interpersonal difficulty. When a woman has sexual activity that is not accompanied by good quality orgasmic release, sexual activity may become a chore or a duty rather than a mutually satisfying, intimate experience. This may also lead to secondary loss of sexual interest and/or interpersonal difficulties.
Testosterone, the primary circulating androgen in women, is a naturally occurring steroid secreted by the ovaries and the adrenal glands. Contrary to the sudden drop in estrogen during menopause, serum levels of androgens fall gradually as women age primarily due to a decrease in the production of adrenal androgen precursors. Testosterone plays a role in regulation of mood, body composition, and bone mineral density, and has central and peripheral effects on sexual function.3'4 In the periphery, testosterone is required for nitric oxide to stimulate vasocongestion for the engorgement of clitoral tissue and vaginal lubrication during sexual arousal. Testosterone stimulates dopamine release in various brain structures implicated in motivation and reward systems, including sexual desire. Testosterone was found to stimulate dopamine release in the medial preoptic area of the anterior hypothalamus under basal conditions and with sexual stimulation in rats.6
The use of androgens to increase women's sexual desire was first reported in 1940 by Loeser. Salmon8 observed that a number of young married women who formerly considered themselves "frigid" were able to experience "a marked increase in coital gratification, culminating in an orgasm" after testosterone propionate injections, with the effects wearing off within several weeks after the discontinuation of the injections. In the 1980s, the role of androgens in maintaining sexual functioning was studied in oophorectomized women.9 In this 3-month, prospective open-label study of 44 women, monthly injections of estrogen and testosterone increased rates of sexual desire, sexual arousal, and number of fantasies. Furthermore, rates of intercourse and orgasm were higher in women treated with androgens and estrogen compared to the controls. Over the two past decades, over 80 studies have been conducted in women with hypoactive sexual desire disorder by using exogenous testosterone through the oral, transdermal, sublingual or parenteral route of administration with or without concomitant estrogen therapy, resulting in an increase in sexual desire, orgasm, arousal, frequency of satisfactory sexual activity, pleasure and responsiveness.10
An intranasal testosterone gel containing 0.24% to 0.72% testosterone with castor oil, oleoyl polyoxylglycerides, and colloidal silicon dioxide as excipients has been developed. 0.24 % and 0.72% TBS-2 is administered as a dose applied equally into each nostril. The formulation has many advantageous features including rapid absorption into systemic circulation and rapid clearance, the lack of firstpass metabolism, the avoidance of transference from one person to another, and ease of use. It is a logical next step in a research program dedicated to investigating the role of testosterone, to investigate whether TBS-2, in the absence of other FSDs, has a direct effect on sexual function in general and anorgasmia in particular.
Two pharmacokinetic (PK)/pharmacodynamic (PD) studies have been performed by evaluating effect of testosterone on the amygdala reactivity and PD endpoints associated with sexual stimulation. The first PD study (CMO-nr: 2004/144) investigated whether the age-related decline
in androgen levels was associated with reduced amygdala activity, and whether exogenous testosterone could restore amygdala activity. The enhanced testosterone levels correlated positively with superior frontal cortex responses and negatively with orbitofrontal cortex responses across individuals, which may reflect testosterone-induced changes in amygdale regulation. These results support the modulatory role of testosterone on emotional cues suggesting testosterone helps to enhance sensation that is necessary to trigger orgasm.
The second study (TBS-2-2010-01) evaluated the PK of 3 dose levels of TBS-2 and sexual function PD compared to a testosterone patch and placebo. The PK results demonstrated a linear increase in plasma testosterone levels with increasing dose levels. During the first PK series, the mean plasma testosterone concentration and area under the plasma concentration-time curve (AUC) in the TBS-2 high-dose group reached the same level as the mean concentration and AUC of the testosterone patch at steady state. During the second PK series, the mean plasma testosterone concentration and AUC in the TBS-2 high-dose group reached levels higher than the mean plasma testosterone concentration and AUC in the testosterone patch group, but still within the upper limit of the normal physiological range. Sexual function PD efficacy was explored by assessing the role of testosterone on vaginal pulse amplitude (VPA), subjective arousal questionnaires, and validated computer tasks. Significant differences were observed in VPA response following testosterone administration in women in the anorgasmia cohort. Women who received TBS-2 had greater responsiveness in genital response and subjective sexual
measurement compared to women receiving the testosterone patch.
The product under investigation in this study, TBS-2, is a testosterone intranasal gel. Contrary to the transdermal administration (Intrinsa, a testosterone transdermal patch that has been approved in the European Union for the treatment of hypoactive sexual desire disorder (HSDD) in bilaterally oophorectomized and hysterectomized [surgically induced menopausal] women receiving concomitant estrogen therapy), administration of the bioadhesive TBS-2 through the nasal mucosa allows for rapid absorption into the systemic circulation. The rapid onset and higher peak concentration are hypothesized to be more effective in enhancing sexual desire and orgasm with lower total concentrations of testosterone needed, thus increasing efficacy and decreasing side effects. In addition, TBS-2 may prove effective in alleviating anorgasmia in an "as needed" way, thus avoiding chronic exposure to testosterone.
8 STUDY OBJECTIVES
8.1 Primary Objective
The primary objective of this study was to assess the bioavailability of total testosterone through PK profiles obtained following single administration of an intranasal application of TBS-2 at doses of 600 μg, 1200 μg, and 1800 μg, and multiple administration of 1200 μg TBS-2 given 3 times daily (t.i.d) for the first 2 days and once on the morning of the third day.
8.2 Secondary Objectives
The secondary objectives of this study were:
• To assess the bioavailability of free testosterone, sex hormone-binding globulin (SHBG), dihydrotestosterone (DHT), and estradiol through PK profiles obtained
following single administration of an intranasal application of TBS-2 at doses of 600 μg, 1200 μg, and 1800 μg, and multiple administration of 1200 μg TBS-2 given for 3 days (t.i.d. for the first 2 days and once on the morning of the third day).
• To evaluate the safety of TBS-2.
9 INVESTIGATIONAL PLAN
9.1 Overall Design and Plan Description
This was a phase 1, single-center, randomized, open-label parallel-group study in healthy, normal-cycling adult women. Approximately 24 healthy adult pre-menopausal women were to be enrolled. Subjects were randomly assigned on a 1: 1: 1 basis to 1 of 3 treatment groups (Cohort 1, Cohort 2, or Cohort 3) during Period 1 and were administered a single dose of an intranasal application of TBS-2 at doses of 600 μg, 1200 μg, or 1800 μg (single doses of 300 μg, 600 μg, and 900 μg per nostril). At the end of Period 1, a total of 8 subjects sampled from these 3 cohorts, who were willing and able to continue with the multiple-dose portion of the study were selected to participate in Period 2. During Period 2, subjects were administered 1200 μg TBS-2 (600 μg per nostril) t.i.d. for 2 days and once on the morning of the third day.
Subjects were screened (Visit 1) for eligibility up to 3 weeks prior to dosing in Period 1, and were admitted to the Clinical Research Unit (CRU) at 0700 hours on the day prior to dosing (Visit 2, Day 1) for baseline testosterone measurement. Subjects started Period 1 on day 1-2 of their menstrual cycle. On study Day 2, subjects were administered a single dose of TBS-2 at 0800 hours and remained in the CRU for 48 hours post-dose for safety monitoring and PK assessments. Subjects were discharged from the clinic on Day 4, and subjects who did not continue into Period 2 also underwent close-out assessments. Depending on their menstrual cycle, subjects who selected to participate in Period 2 returned to the CRU approximately 26 to 32 days following the conclusion of Period 1 for Visit 3 (Period 2). Subjects started Period 2 on day 1-2 of their menstrual cycle. During Period 2, subjects were admitted to the CRU at 0700 hours on the day of dosing (Visit 3, Day 1). On study Days 1 and 2, subjects were administered TBS-2 at 0800 hours (+30 minutes), 1600 hours (+30 minutes), and 2400 hours (+30 minutes). On Day 3, subjects were administered TBS-2 at 0800 hours. Subjects remained in the CRU for 48 hours following dosing on Day 3 for safety monitoring and PK assessments.
Subjects were discharged from the clinic on Day 5.
During Period 1, blood samples for determination of baseline testosterone (free and total), SHBG, DHT, and estradiol concentration were collected on Day 1 at 0745 hours and then at 15, 30, and 45 minutes and at 1, 1.5, 2, 4, 6, 8, 12, 16, 20, and 23.5 hours relative to an 0800 hour clock time. Blood samples for determination of testosterone (free and total), SHBG, DHT, and estradiol concentration were collected on Day 2 (15, 30, and 45 minutes and at 1, 1.5, 2, 4, 6, 8, 12, 16, and 20 hours post-dose) and Day 3 (24, 32, 40, and 48 hours post-dose) during the confinement period. During Period 2, a blood sample for a baseline serum testosterone concentration was collected at 0745 hours (i.e., 15 minutes prior to study drug administration). Blood samples for determination of testosterone (free and total), SHBG, DHT, and estradiol
plasma concentration were collected on Day 1 (pre-dose [15 minutes prior to dosing] and at 1545 and 2345 hours), Day 2 (1545 and 2345 hours), Day 3 (15, 30, and 45 minutes and at, 1, 1.5, 2, 4, 6, 8, 12, 16, and 20 hours), and Day 4 (24, 32, 40, and 48 hours) during the confinement period.
Other assessments performed during the study included the monitoring of adverse events (AEs), clinical laboratory evaluations (chemistry [including hormone profile], hematology, and urinalysis), vital signs assessments (systolic and diastolic blood pressure [BP], heart rate [HR], respiratory rate [RR], and body temperature), and physical examinations. In addition, otorhinolaryngological examination findings, 12-lead electrocardiogram (ECG) readings, medical history, and concomitant medication use were recorded.
9.2 Discussion of Study Design and Choice of Control Groups
This was a phase 1, single-center, randomized, open-label parallel-group study of TBS-2 in 3 cohorts of subjects (Cohorts 1, 2, and 3) in Period 1 (single-dose) and one multiple-dose group of subjects in Period 2. Approximately 24 healthy pre-menopausal women were to receive a single intranasal administration of 600 μg, 1200 μg, or 1800 μg of TBS-2 in Period 1 and 8 women were to receive multiple administrations of 1200 μg of TBS-2 in Period 2 to evaluate the bioavailability through pharmacokinetic profile and safety of TBS-2. Subjects were confined in a CRU for 4 days during Period 1 and 5 days during Period 2.
The rationale for dose selection is discussed in Section 9.4.4 and Section 9.4.5.
9.3 Selection of Study Population
9.3.1 Inclusion Criteria
Subjects were eligible for study inclusion if they met all of the following inclusion criteria:
1. Female subjects between 18 and 40 years of age.
2. Subjects with regular menstrual cycles between 26 and 32 days.
3. Women of childbearing potential must have agreed to use 1 of the following reliable birth control methods prior to the study, during the study, and up until 1 month after the end of the study:
a. Surgically sterile (tubal ligation).
b. Intrauterine device in place for at least 3 months prior to study initiation.
c. Barrier method (condom with spermicidal agent use by partner).
d. Abstinence.
4. Subjects negative for drugs of abuse, hepatitis B-surface antigen (HbsAg), hepatitis C, human immunodeficiency virus (HIV), and pregnancy (serum β-HCG).
5. Subjects with a Body Mass Index (BMI) between 18.5 kg/m2 and 35 kg/m2 (inclusive).
6. Subjects with a normal ear, nose, throat (ENT) examination.
7. Subjects with normal thyroid- stimulating hormone (TSH) values.
8. Subjects with no clinically significant findings in the physical examination, 12-lead ECG, and vital signs.
9. Subjects with normal thyroid function. Physiological prolactin concentration.
10. Subjects with no clinical laboratory values outside of the acceptable range, unless the PI decided that they were not clinically significant.
11. Subjects who were able to understand and provide written informed consent.
12. Subjects who were available for the entire study period and were willing to adhere to protocol requirements, as evidenced by a signed ICF.
9.3.2 Exclusion Criteria
Subjects were excluded from study participation if they met any of the following exclusion criteria:
1. Known history of hypersensitivity to testosterone (e.g., Intrinsa patch) and/or related drugs.
2. Known history of polycystic ovarian syndrome.
3. Known history or presence of cardiac, pulmonary, gastrointestinal, endocrine,
musculoskeletal, neurological, psychiatric, hematological, reproductive, liver, or kidney disease, unless judged not clinically significant by the PI or medical designate.
4. Presence of or known history of estrogen-responsive tumors such as breast cancer and/or history of any cancer, excluding basal cell carcinoma.
5. Known history of frequent clinically significant acne.
6. Known history of hirsutism.
7. History of nasal surgery, specifically turbinoplasty, septoplasty, rhinoplasty, "nose job," or sinus surgery.
8. Prior nasal fractures.
9. Active allergies, such as rhinitis, rhinorrhea, and nasal congestion.
10. Mucosal inflammatory disorders, specifically pemphigus and Sjogren's syndrome.
Sinus disease, specifically acute sinusitis, chronic sinusitis, or allergic fungal sinusitis. History of nasal disorders (e.g., polyposis, recurrent epistaxis [>1 nose bleed per month], abuse of nasal decongestants) or sleep apnea.
Use of any form of intranasal medication delivery, specifically nasal corticosteroids and oxymetazoline containing nasal sprays (e.g., Dristan 12-Hour Nasal Spray).
History of hepatitis B, a positive test for HbsAg, a history of hepatitis C, a positive test for hepatitis C antibody, a history of HIV infection, or demonstration of HIV antibodies. Any history of severe allergic reaction including drugs, food, insect bites, environmental allergens, or any condition known to interfere with the absorption, distribution, metabolism, or excretion of drugs.
Any history of drug abuse or alcohol abuse as per the Diagnostic and Statistical Manual of Mental Disorders (fourth edition) criteria within 6 months of study drug
administration.
Current treatment with any hormonal therapy within previous 12 months, treatment with drugs that interfere with metabolism of testosterone within 30 days of study drug administration and/or any other prescription medications. Difficulty in abstaining from over the-counter (OTC) medication use for the duration of study.
Use of oral, transdermal, and implant contraceptives within 30 days prior to study drug administration or a depot contraceptives injection within 1 year prior to study drug administration.
Evidence of pregnancy or lactation.
Subjects who were breast feeding or had breast fed within 6 months prior to the screening visit.
Administration of another investigational drug within 30 days prior to study drug administration.
Blood donation within 56 days prior to study drug administration.
Any participation as a plasma donor in a plasmapheresis program within 7 days preceding this study.
Intolerance to venipuncture.
History of abnormal bleeding tendencies or thrombophlebitis unrelated to venipuncture or intravenous cannulation.
History of deep venous thrombosis or coagulation disorders.
Removal of Subjects from Therapy or Assessment
Subjects withdrawn from the study after receiving study drug were not to be replaced, regardless of the reason for withdrawal.
A subject may have been prematurely discontinued from the study for any of the following reasons:
• Significant noncompliance with the study protocol and procedures.
• Intercurrent illness that interfered with the progress of the study.
• Intolerable AE, including clinically significant abnormal laboratory findings, which in the opinion of the PI could have interfered with the subject's safety.
• Principal Investigator' s decision that the withdrawal from the study was in the best interest of the subject.
9.4 Treatments
9.4.1 Treatments Administered
Subjects were randomly assigned on a 1: 1: 1 basis to 1 of 3 treatment groups (Cohort 1, Cohort 2, or Cohort 3) during Period 1 and were administered a single dose of an intranasal application of TBS-2 at doses of 600 μg, 1200 μg, or 1800 μg (300 μg, 600 μg, and 900 μg per nostril). At the end of Period 1, a total of 8 subjects sampled from these 3 cohorts, who were willing and able to continue with the multiple-dose portion of the study were selected to participate in Period 2. During Period 2, subjects were administered 1200 μg TBS-2 (600 μg per nostril) t.i.d. for 2 days and once on the morning of the third day.
Treatment 1: TBS-2 dispensers prefilled with 0.24% testosterone gel to deliver a single dose of 300 μg of testosterone per nostril, for a total dose of 600 μg given at 0800 hours (+30 minutes) on Day 2 of Period 1 for Cohort 1.
Treatment 2: TBS-2 dispensers prefilled with 0.48% testosterone gel to deliver a single dose of 600 μg of testosterone per nostril, for a total dose of 1200 μg given at 0800 hours (+30 minutes) on Day 2 of Period 1 for Cohort 2.
Treatment 3: TBS-2 dispensers prefilled with 0.72% testosterone gel to deliver a single dose of 900 μg of testosterone per nostril, for a total dose of 1800 μg given at 0800 hours (+30 minutes) on Day 2 of Period 1 for Cohort 3.
Treatment 4: TBS-2 dispensers prefilled with 0.48% testosterone gel to deliver a single dose of 600 μg of testosterone per nostril, for a total dose of 1200 μg given t.i.d. daily at 0800 hours (+ 30 minutes), 1600 hours (+ 30 minutes), and 2400 hours (+ 30 minutes) on Days 1 and 2 of Period 2, and once in the morning at 0800 hours (+ 30 minutes) on Day 3 of Period 2 (Multi-dose group).
Subjects were instructed on the proper procedure for applying the TBS-2 gel intranasally by using the prefilled dispensers. Study drug was self-administered at 0800 hours on Day 2 of
Period 1 and on Days 1, 2, and 3 of Period 2. Self-administration of TBS-2 was monitored by study personnel. Subjects were instructed to not blow their nose or sniff immediately, and for the first hour following study drug administration.
9.4.2 Identity of Investigational Products
Active study drug was supplied in prefilled dispensers containing 0.24% TBS-2, 0.48% TBS-2, or 0.72% TBS-2.
The TBS-2 prefilled dispensers were packaged. A multi-dose dispenser was used for gel deposition into the nasal cavity. The dispenser was a finger actuated dispensing system designed to dispense 125
 of TBS-2 gel per actuation from a nonpressurized container into the nasal cavity. The key components of the multiple-dose dispenser included a barrel, base, pump, and actuator, which were composed of polypropylene and a piston, which was composed of polyethylene.
of TBS-2 gel per actuation from a nonpressurized container into the nasal cavity. The key components of the multiple-dose dispenser included a barrel, base, pump, and actuator, which were composed of polypropylene and a piston, which was composed of polyethylene.
All study drug boxes and dispensers were labeled and supplied according to applicable regulatory requirements. Qualified licensed study personnel prepared the unit doses for the study as per the randomization schedule. Study drug was provided to the subjects in appropriate unitdose foil pouches, clearly labeled as to the amount of drug being given.
Study drug was stored in a secure location at a controlled room temperature of 15°C to 25 °C (59°F to 77°F). The storage location was a locked room with limited access, available to appropriate study personnel only.
The PI, or an approved representative, (e.g., co-investigator), ensured that all study drug was stored in a secured area, under recommended storage conditions, and in accordance with applicable regulatory requirements.
9.4.3 Method of Assigning Subjects to Treatment Groups
A randomization schedule was prepared by Premier Research and was provided to the PI at the CRU prior to the start of the study.
Subjects who met the enrollment criteria were randomly assigned on a 1: 1: 1 basis to 1 of 3 treatment groups.
9.4.4 Selection of Doses in the Study
The doses of 600 μg, 1200 μg, and 1800 μg were selected for this study based on the current pharmacokinetics of the gel from the study TBS-2-2010-01 where women received three doses (300 meg, 900 meg and 1200 meg) of TBS-2. The AUC of testosterone following intranasal administration was within normal limits for women. TBS-2 had a favorable safety profile at these doses. (Data is on file with the Sponsor.)
The animal toxicology studies completed to date do not suggest any unusual or unexpected toxicities related to TBS-2 exposure. Subjects were not expected to experience side effects with TBS-2.
9.4.5 Selection and Timing of Dose for Each Subject
Study drug was administered on Day 2 of Period 1 (single-dose). During Period 2 (multi-dose), study drug was administered t.i.d. on Days 1 and 2 and once on the morning of Day 3. Subjects were confined in a CRU for 4 days during Period 1 and 5 days during Period 2.
9.4.6 Blinding
This was an open-label study; subjects were not blinded to treatment assignments.
9.4.7 Prior and Concomitant Therapy
The use of any prescription or nonprescription medication or treatment with drugs that interfere with the metabolism of testosterone was prohibited within 30 days of study drug administration and until the final study visit. The use of any form of intranasal medication delivery, specifically nasal corticosteroids and oxymetazoline-containing nasal sprays (e.g., Dristan 12-Hour Nasal Spray) was prohibited until the final study visit. Current treatment with any hormonal therapy was prohibited 12 months prior to study drug administration and until the final study visit. Also prohibited was the use of oral, transdermal, or implant contraceptives within 30 days prior to drug administration or a depot contraceptives injection within 1 year prior to study drug administration. In addition, the administration of another investigational drug was prohibited within 30 days prior to study drug administration. The use of illegal drugs was not permitted while the subjects were enrolled in this study.
Throughout the study, the PI could prescribe any concomitant therapy deemed necessary to provide adequate supportive care. The PI was to notify the sponsor if concomitant medications were required during the study. The decision to allow the subject to be enrolled into the study or take medications during the study was to be made jointly by the sponsor and PI and was based on their opinion that the use of the medication was unlikely to compromise the safety of the subject or the interpretation of the study data. All medications taken within 30 days prior to dosing and all concomitant therapy were recorded on the appropriate section of the CRF.
9.4.8 Treatment Compliance
Subjects who received doses of study drug were monitored by study personnel for 1 hour postdose to assure conformity to the TBS-2 instructions. Subjects were confined in a CRU for 4 days during Period 1 and 5 days during Period 2 and were closely monitored. A drug dispensing log was maintained. Pharmacokinetic results were available to confirm compliance.
Study drug was administered on Day 2 of Period 1 (single-dose). During Period 2 (multi-dose), study drug was administered t.i.d. on Days 1 and 2 and on the morning of Day 3. Subjects were confined in a CRU for 4 days during Period 1 and 5 days during Period 2.
9.5 Pharmacokinetic and Safety Variables
9.5.1 Pharmacokinetic and Safety Measurements Assessed and Flow Chart
The Study Schedule is presented in Table 9- 1.
Table 9-1: Study Schedule


Abbreviations: AE = adverse event; CBC = complete blood count; DHT = dihydrotestosterone; ECG = electrocardiogram; ENT = ear, nose, throat; FSH = follicle-stimulating hormone; HbsAg = hepatitis B surface antigen; HIV = human immunodeficiency virus; LH = luteinizing hormone; PK = pharmacokinetic; SHBG = sex hormone-binding globulin; TSH = thyroid- stimulating hormone.
a. A total of 8 subjects who were willing and able to continue on to the multiple dose portion of the study were selected from Cohorts 1, 2, and 3 and comprised the Multi-Dose group.
b. Vital signs (heart rate, blood pressure, temperature, and respiratory rate).
c. The study center physician examined subjects and identified any clinically significant changes to the nasal mucosa at follow- up.
d. Chemistry, hematology, urinalysis.
e. Hormone profile: TSH, total and free tri-iodothyronine, total and free thyroxine, FSH, prolactin, and progesterone.
f. Study drug administered at 0800 hours (+30 minutes).
g. Study drug administered at 0800 hours (+30 minutes), 1600 hours (+30 minutes), and 2400 hours (+30 minutes).
h. Study drug a administered at 0800 hours (+30 minutes).
i. Blood samples for the 24-hour baseline profile (total and free testosterone, SHBG, DHT, and estradiol) were collected at 0745
hours and then at 15, 30, and 45 minutes and at 1, 1.5, 2, 4, 6, 8, 12, 16, 20, and 23.5 hours relative to an 0800 hour clock time.
j. Blood samples for the 48-hour PK profile (total and free testosterone, SHBG, DHT, and estradiol) were collected at 15, 30, and
45 minutes and at 1, 1.5, 2, 4, 6, 8, 12, 16, 20, 24, 32, 40, and 48 hours,
k. A baseline serum testosterone blood sample was collected at 0745 hour (i.e., 15 minutes prior to dosing).
1. PK sampling for trough level measurement was done prior to dosing at 0800, 1600, and 2400 hours.
m. The 48-hour PK profile blood samples were collected pre-dose at 0745 hours and at 15, 30, and 45 minutes and at 1, 1.5, 2, 4,
6, 8, 12, 16, 20, 24, 32, 40, and 48 hours.
9.5.2 Appropriateness of Measurements
The measurements included in this study were typical for a phase I single- and multiple-dose study.
9.5.3 Pharmacokinetic Drug Concentration Measurements
Whole blood samples for determination of testosterone (free and total), SHBG, DHT, and estradiol concentration were collected at each time point indicated in Table 9-2. The date and time of collection were recorded on the CRF to the nearest minute. Whole blood samples were obtained through direct venipuncture; the use of a heparin lock or IV indwelling catheter was allowed for early time point PK sample collections. The volume of plasma samples collected for PK analysis for each subject was approximately 120 mL during Period 1 and 92 mL during Period 2.
Table 9-2: Schedule of Pharmacokinetic Sample Collection
Visit/Day Treatment PK Sample Collection
Visit 1/Day 1 None; Baseline measurement Blood samples for 24-hour baseline profile (total and free testosterone, SHBG, DHT, and estradiol) were collected at 0745 hours and then at 15, 30, and 45 minutes and at 1, 1.5, 2, 4, 6, 8, 12, 16, 20, and 23.5 hours relative to an 0800 hour clock time.
Visit 2/Days 2-4 Single-dose (Cohorts 1, 2, and Blood samples for the 48-hour PK
3) profile (total and free testosterone,
SHBG, DHT, and estradiol) were collected at 15, 30, and 45 minutes and at l, 1.5, 2, 4, 6, 8, 12, 16, 20, 24, 32, 40, and 48 hours post-drug
administration.
Visit 3/Day 1 Multiple-dose A baseline serum blood sample was collected at 0745 hour (i.e., 15 minutes prior to dosing).
Visit 3/Days 1-2 Multiple-dose PK sampling for each trough level measurement was drawn 15 minutes prior to dosing at 0745 hours, 1545 hours and 2345 hours (total 5
samples).
Visit 3/Days 3-5 Multiple-dose The 48-hour PK profile blood samples were collected pre-dose at 0745 hours (pre-dose) and at 15, 30, and 45 minutes and at 1, 1.5, 2, 4, 6, 8, 12, 16, 20, 24, 32, 40, and 48 hours post-drug administration.
Abbreviations: DHT = dihydrotestosterone; PK = pharmacokinetic; SHBG = sex hormone- binding globulin; t.i.d. = three times daily
Note: During Period 1 dosing occurred on the morning of Day 2. During Period 2, dosing occurred t.i.d. on Days 1 and 2, and once on the morning of Day 3.
9.5.4 Pharmacokinetic Parameter Estimates
Concentration-time data for 5 analytes [testosterone (Total and Free), SHBG, DHT and estradiol] were determined and PK parameters were calculated from them. Actual sampling time points were recorded and used in calculation of the actual elapsed time from dose to sample for PK calculations. As each administration was made to each of the nostrils, the time of dosing was the time of the first nostril administration. Baseline analyte concentrations from the 24-hour pre-dose profile were subtracted from the reported analyte concentrations for the single dose treatment before calculation of the PK parameters. Additionally PK parameters were calculated on uncorrected concentrations for free testosterone, total testosterone, and DHT for the single dose treatment.
The following PK parameters (uncorrected and baseline-corrected) were calculated following single-dose treatment (Cohorts 1, 2, and 3) for PK characterization. Profile intervals were from the start of the first dosing and continued to the last sample collected over 24 hours. PK parameters were estimated by standard methods used by the Phoenix WinNonlin program (Pharsight, Cary, NC) version 6.2 and short descriptions are given below. The determination of the terminal elimination phase was more fully described in the WinNonlin documentation. Note that Cmin, Cmax, and tmax were taken from the actual measured values but after baseline correction for Cmin and Cmax.
Parameter Description and Calculation
AUCo-t Area under the plasma concentration time curve from time zero to the last
measurable concentration time point, calculated by using the combination of the linear and log trapezoid rules. The linear trapezoidal rule would be used from the time of dose to tmax and the log trapezoidal rule would be used following tmax-
AUCo-8 Area under the plasma concentration time curve from time zero to 8 hours.
AUCo-24 Area under the plasma concentration time curve from time zero to 24 hours.
AUCo-oo Area under the plasma concentration time curve from time zero to infinity,
calculated as AUCa^ = AUCo-t + where Ciast is
the last
measurable
concentration, terminal
elimination
rate constant
calculated
from the log- linear
terminal
phase. c ^max Maximum concentration observed after dosing.
Co Concentration at pre-dose.
C24 Concentration at 24 hours. ax Time of observed maximum concentration (Cmax) relative to the time of dosing. λζ Terminal elimination rate constant.
ln(2)
tl/2 Elimination half-life calculated as
The following PK parameters (uncorrected) were calculated following multiple-dose treatment:
Parameter Description and Calculation
AUCo-x Area under the concentration-time curve from
time zero to the dosing interval (τ = 8 hours).
AUCo-8 Area under the plasma concentration time
curve from time zero to 8 hours.
AUCo-24 Area under the plasma concentration time
curve from time zero to 24 hours. c ^max Maximum concentration observed after dosing. tmax Time of observed maximum concentration
(Cmax) relative to the time of dosing. c ^min Minimum concentration over a dosing interval
during multiple dosing.
Cpd Pre-dose concentration determined
immediately before a dose at steady state
Co Concentration at pre-dose.
C24 Concentration at 24 hours. c ^avg Avprn cje stead -state concentration, calculated
as AUCO- , where τ = 8 hours.
τ PTF % Peak to Trough Fluctuation:
(Cmax-C Cmin)* 100
c PTS % Peak to Trough Swing is the degree of
concentration swing at a steady state:
(Cmax-C Cmin) * 100
c ^min
Additional exploratory analyses of PK parameters have been performed as necessary.
9.5.5 Safety Measurements
Safety measurements included the monitoring of AEs, clinical laboratory evaluations (chemistry [including hormone profile], hematology, and urinalysis), and vital signs. In addition, physical and otorhinolaryngological examinations were performed and medical history and concomitant medication use were recorded. Safety measurements were performed at the times specified in
Table 9-1. The PI followed all clinically- significant abnormal findings after study drug treatment until resolution or return to baseline.
9.5.6 Adverse Events
An AE is any untoward, undesired, unplanned clinical event in the form of signs, symptoms, disease, or laboratory or physiological observations occurring in a human participating in a clinical study, regardless of causal relationship. A pre-existing condition is one that is present prior to study drug administration and is reported as part of the subject's medical history. Preexisting conditions were reported as an AE only if the frequency, intensity, or character of the pre-existing condition worsened during the course of the study. Laboratory abnormalities were not considered AEs unless they were associated with clinical signs or symptoms, or required medical intervention. However, a laboratory abnormality (e.g., a clinically significant change detected in clinical chemistry [including hormone profile], hematology, urinalysis) that was independent from the underlying medical condition and that required medical or surgical intervention, or led to study drug interruption or discontinuation, was considered an AE.
All AEs judged to be clinically significant, including clinically- significant laboratory
abnormalities, were followed until resolution or return to baseline.
9.5.6.1 Severity Rating
All AEs or SAEs were assessed for severity, by using the following grading scale:
Mild An event easily tolerated by the subject; transient or mild discomfort (usually <48 hours); no medical intervention/therapy required.
Moderate An event that may interfere with normal, everyday activities; some assistance may be needed; no or minimal medical intervention/therapy required.
Severe An event that prevented the subject from performing their normal, everyday
activities; some assistance usually required; medical intervention/therapy required, hospitalizations possible.
When changes in the severity of an AE occurred more frequently than once a day, the maximum severity for the experience that day was noted. If the severity category changed over a number of days, then those changes were recorded separately (with distinct onset dates). The severity of the AE was recorded in the appropriate section of the AE page of the CRF. The evaluation of severity was distinguished from the evaluation of "seriousness." A severe event might not have met the criteria for seriousness and a serious event might have been evaluated as mild.
9.5.6.2 Causality Rating
For each reported adverse reaction, the PI made an assessment of the relationship of the event to the study drug by using the following scale:
Definite There is a plausible temporal relationship with drug administration and
withdrawal, and reappears after drug restart.
Probable There is a plausible temporal relationship with drug administration.
Possible There is a plausible temporal relationship with drug administration but can
reasonably be associated to other factors.
Unlikely There is no plausible temporal relationship with drug administration.
Unknown There are no sufficient elements to establish a correlation with drug intake.
Not Related Cannot be correlated to study drug administration.
9.5.6.3 Serious Adverse Events
The PI classified each AE as either serious or not serious. A serious adverse event (SAE) was defined as any AE occurring at any dose that resulted in any of the following outcomes:
Death1
Was life-threateningb
Required inpatient hospitalization or prolongation of existing hospitalization Resulted in a persistent or significant disability/incapacity0
Resulted in a congenital anomaly/ birth defect
Additionally, important medical events that may not have resulted in death, been life-threatening, or required hospitalization may be considered an SAE when, based upon appropriate medical judgment, they may jeopardize the subject and may require medical or surgical intervention to prevent one of the outcomes listed above. Example: allergic bronchospasm requiring intensive treatment in an emergency room or at home.
Death was an outcome and was NOT the AE. In the event of death, the cause of death was recorded as the AE. The only exception was "sudden death" when the cause was unknown.
Life-threatening AEs included any adverse drug experience, which, in the view of the PI, placed the subject at immediate risk of death from the reaction as it occurred. It did not include a reaction that, had it occurred in a more serious form, might have caused death.
Disability was defined as a substantial disruption in a person's ability to conduct normal life functions.
All SAEs that resulted in death or were life-threatening, regardless of causal relationship, were reported (or designee) within 24 hours of the study center's knowledge of the event. A copy of
the initial SAE report was to have been received within 1 business day. All other SAEs were forwarded (or designee) within 1 business day. If there was any doubt whether the information constituted an SAE, the information was treated as an SAE for the purposes of this study.
9.5.7 Clinical Laboratory Tests
Clinical laboratory tests were performed according to the schedule provided in Table 9-1.
Serum chemistry evaluations included sodium, potassium, chloride, glucose, urea, creatinine, calcium, phosphate, uric acid, total bilirubin, albumin, aspartate transaminase (AST), alanine transaminase (ALT), alkaline phosphatase, gamma-glutamyl transferase (GGT), creatine kinase (CK), and cholesterol, and hormone profiles.
Hormone profiles included TSH, total and free tri-iodothyronine, total and free thyroxine, FSH, prolactin, and progesterone.
Hematology evaluations included white blood count, hemoglobin, and hematocrit.
Urinalysis included glucose, bilirubin, ketones, specific gravity, blood, pH, protein, urobilinogen, nitrites, leukocytes and, if necessary, microscopic examination.
At Screening and on admission to the CRU, a urine specimen was obtained to test for drugs of abuse (marijuana, cocaine, opiates, amphetamines, phencyclidine, benzodiazepines, and barbiturates) and alcohol was tested for by use of an alcohol breath test.
Testing for HbsAg, antiHCV, and HIV were performed at Screening (Visit 1) only.
Samples were analyzed by MEDTOX Laboratories, Inc.
9.5.8 Vital Signs
Vital sign measurements included systolic and diastolic BP and HR, body temperature, and RR. Vital signs assessments were performed at the times specified in Table 9-1.
9.5.9 12-Lead Electrocardiogram
A standard 12-lead ECG was assessed at Screening (Visit 1) only.
9.5.10 Other Safety Measurements
Physical examinations were performed at the times specified in Table 9-1.
An ENT (otorhinolaryngological) nasal endoscopic examination was performed by ENT specialist at Screening. Basic ENT (otorhinolaryngological) examinations were performed at the times specified in Table 9-1; the study center physician examined subjects and identified any clinically significant changes to the nasal mucosa at follow-up.
Concomitant medications were monitored throughout the study.
9.6 Data Quality Assurance
This study was monitored by trained experienced personnel in accordance with GCPs. The clinical study monitor reviewed study records to verify adherence to the protocol, accuracy, completeness, and consistency of the data; and adherence to local regulations on the conduct of clinical research. The clinical monitor maintained regular contact with the study center and had access to subject medical records and other study-related records needed to verify the entries on the CRFs.
Electronic CRFs (eCRFs) were used for this study and included only the subject's initials, date of birth, and an assigned subject number on the eCRFs as identifiers. (A sample eCRF can be found in Appendix 16.1.2.) The PI ensured the availability and reliability of source documents from which the information on the eCRF was derived, and was required to comply with document retention procedures as outlined in the protocol. Case report forms were reviewed for accuracy and signed and dated by the PI.
The database lock for this study was achieved on 12 January 2012.
9.7 Statistical Methods and Determination of Sample Size
9.7.1 Statistical and Analytical Plans
The statistical methods presented in the study protocol were superseded by those described in the statistical analysis plan (SAP). This study evaluated the PK properties as well as the safety of TBS-2. Power calculations were not performed. Data were summarized by using descriptive statistics (sample size, mean, median, standard deviation [SD], minimum, and maximum) for each of the safety variables by treatment group and overall. Data from all visits during the study were displayed in the data listings.
Concentration-time data for 5 analytes (testosterone [total and free], SHBG, DHT, and estradiol) were determined by a validated assay method and PK parameters were calculated. Actual sampling time points were recorded and used in calculation of the actual elapsed time from dose to sample for PK calculations. As each administration was to each of the nostrils, the time of dosing was the time of the first nostril administration. Baseline analyte concentrations from the 24-hour pre-dose profile were subtracted from the time-matched analyte concentrations following dose administration before calculation of the PK parameters.
The plasma TBS-2 concentration-time data were analyzed by using Phoenix WinNonlin
(Pharsight Corporation) version 6.2. The PK parameters (refer to Section 9.5.4) for testosterone (free and total), SHBG, DHT, and estradiol were calculated by standard noncompartmental methods for all subjects as data permitted. Individual PK parameters estimated for each subject's profile in the PK population were displayed in the data listings. Data were summarized by using descriptive statistics (mean, SD, % coefficient of variation [CV], confidence interval (CI), median, minimum, and maximum) and are presented by treatment group.
Geometric means were included for AUC and Cmax estimations and were included for some other PK parameters. By using Generalized Linear Model (GLM) procedure in SAS®, an analysis of variance (ANOVA) was performed on natural logarithmic (In) transformed
parameters AUCo-t, AUCo-∞, AUCo-τ, Cavg, and Cmax and on untransformed parameters t½, and λζ at the significance level of 0.05. The intrasubject CV was calculated for AUCo-t, AUCo-∞, AUCo-τ, and Cmax by using the ANOVA residual error.
Dose linearity following single-dose administration (Period 1) was assessed after normalizing the AUCo-t, AUCo-oo, and Cmax to the dose administered.
The following Period 1 comparisons for PK parameters were made:
Comparison 1: 600 μg 0.24% TBS-2 versus 1200 μg 0.48% TBS-2;
Comparison 2: 600 μg 0.24% TBS-2 versus 1800 μg 0.72% TBS-2;
Comparison 3: 1200 μg 0.48% TBS-2 versus 1800 μg 0.72% TBS-2.
9.7.2 Determination of Sample Size
The sample size for this study was not determined on the basis of statistical hypothesis testing. Based on typical, early-stage PK studies, groups of 8 subjects per cohort were sufficient to provide adequate clinical information to satisfy the objectives of the study.
9.7.3 Interim Analysis
No formal interim analysis was performed in this study. However, when the concentration data were received from the bioanalytical laboratory following Period 1 and again following Period 2, all PK TLFs were provided to the limit of data available by using scheduled sampling times rather than the actual scheduled sampling times. Multiple-dose data were made available after Period 2. No quality control testing of the preliminary PK analysis or the preliminary tables, listings, and figures were completed for the interim analysis and no testing of the interim PK parameters were completed for the interim analysis.
9.8 Changes in the Conduct of the Study or Planned Analyses
No changes were made to the protocol procedures during this study (Appendix 16.1.1).
There were changes to the PK analysis planned in the SAP after the first draft analysis was conducted. These changes fell under the categories of additional exploratory analyses of PK parameters and additional PK parameters to be calculated as requested.
In order to more fully explore any accumulation of free testosterone, total testosterone and DHT, PK parameters were calculated using uncorrected data from the single-dose treatment (supported by a listing and a table); additionally, an ANOVA table contrasting selected single-dose and multiple-dose parameters was created and two new figures were created. Two different methods of inquiry were used as described in the following.
There were 3 subjects who participated in the single-dose and the multiple-dose treatments at the 1200 μg dose. Since the multiple-dose treatment was an uncorrected profile while the singledose treatment was a corrected profile, the PK parameter based on single-dose uncorrected values
were estimated and supported by a new listing and table. Selected PK parameters from the 3 subjects were then analyzed in an ANOVA table for estimation of accumulation. A set of new figures was created for these 3 subjects to present the baseline, single-dose profile, and multipledose profile by subject for free testosterone, total testosterone, and DHT.
The second method of exploring accumulation involved the trough concentrations from the 8 subjects in the multiple-dose treatment; all 8 subjects received the 1200 μg dose of testosterone. A set of scatter-plot figures of the trough samples was created using the 8-hour dosing interval between doses as the x-axis and the analyte concentration as the y-axis. Linear regression included, for all of the 5 analytes reported, the linear correlation coefficient (Rsq) as well as the slope and intercept of the regression line.
After review of preliminary results, additional PK parameters were requested and calculated; AUCo-8 and AUCo-24 parameters were calculated for the single-dose and multiple-dose profiles, single-dose PK calculation was limited to 24 hours, and C24 was reported as a PK parameter.
10 STUDY SUBJECTS
10.1 Disposition of Subjects
A total of 24 subjects were enrolled in Period 1; 8 subjects each were randomized into Cohort 1, Cohort 2, and Cohort 3. All 24 subjects completed Period 1 (Section 14.1, Table 14.1.1a).
Following Period 1, a total of 8 subjects continued to Period 2. All 8 subjects completed Period 2 (Section 14.1, Table 14.1.1b).
Analysis population and disposition by subject are provided in Appendix 16.2, Listing 16.2.1.1. Study completion by subject is provided in Appendix 16.2, Listing 16.2.1.2a (Period 1) and Appendix 16.2, Listing 16.2.1.2b (Period 2).
10.2 Protocol Deviations
The protocol deviations reported during the study were minor and were not expected to affect the outcome of the study. A total of 6 of 24 subjects (25.0%) had protocol deviations of vital signs assessments missed or PK blood draws out of window by 2 or 3 minutes. A list of protocol deviations is provided in Appendix 16.4.
11 PHARMACOKINETICS EVALUATIONS
11.1 Data Sets Analyzed
• Single-Dose Population: All subjects who were randomized and received at least 1 dose of TBS-2 during the study; 24 subjects (100%) were included in this population (Section 14.1, Table 14.1.1a).
• Multi-Dose Population: All subjects in the Single-Dose Population who were selected to continue in study Period 2; 8 subjects (100%) were included in this population (Section 14.1, Table 14.1.1b).
11.2 Demographic and Other Baseline Characteristics
11.2.1 Demographic Data
Most subjects in Period 1 were white (19 of 24 subjects [79.2%]), followed in percentage by subjects who were black (4 of 24 subjects [16.7%]) and those who were American Indian or Alaskan Native/white (1 of 24 subjects [4.2%]). A similar number of subjects were not Hispanic or Latino (13 of 24 subjects [54.2%]) and Hispanic or Latino (11 of 24 subjects [45.8%]). Mean (SD) age was 29.8 (5.86) years. Mean (SD) height was 161.02 (6.667) cm, weight was 66.36 11.378) kg, and BMI was 25.61 (4.079) kg/m2. Most subjects had a normal BMI (> 18.5 kg/m2) (13 of 24 subjects [54.2%]), followed in percentage by subjects who were overweight (BMI 25.0-29.9 kg/m2) (7 of 24 subjects [29.2%]) and subjects who were obese (> 30 kg/m2) (4 of 24 subjects [16.7%] ; Section 14.1, Table 14.1.2a).
Most subjects in Period 2 were white (5 of 8 subjects [62.5%]), followed in percentage by those who were black (2 of 8 subjects [25.0%]) and those who were American Indian or Alaskan Native/white (1 of 8 subjects [12.5%]). A similar number of subjects were not Hispanic or Latino (5 of 8 subjects [62.5%]) and Hispanic or Latino (3 of 8 subjects [37.5%]). Mean (SD) age was 30.3 (6.48) years. Mean (SD) height was 160.8 (3.99) cm, weight was 61.13 (9.815) kg, and BMI was 23.65 (3.454) kg/m2. Most subjects had a normal BMI ( > 18.5 kg/m2) (6 of 8 subjects
[75.0%]), followed in percentage by those who were overweight (BMI 25.0-29.9 kg/m2) (2 of 8 subjects [25.0%] ; Section 14.1, Table 14.1.2b).
Subject demographics are provided in Appendix 16.2, Listing 16.2.4.1. 11.2.2 Baseline Characteristics
11.2.2.1 Medical History
Subject medical history reported at Screening is summarized by body system in Table 11- 1 and in Section 14.1, Table 14.1.3.1 and Appendix 16.2, Listing 16.2.4.2.
Currently active non-exclusionary medical history conditions were reported by at least 10% of subjects overall in the following body systems: nervous system (15 of 24 subjects [62.5%]), genitourinary system (7 of 24 subjects [29.2%]), integumentary system (4 of 24 subjects
[16.7%]), and allergic conditions (4 of 24 subjects [16.7%]).
Subject medical history is provided in Section 14.1, Table 14.1.3.1 and in Appendix 16.2, Listing 16.2.4.2.
Table 11-1: Medical History (Single-Dose Population)
Cohort 1 Cohort 2 Cohort 2 Total
(600 μΒ) (1200 μΒ) (1200 μΒ)
(n = 8) (n = 8) (n = 8) (n = 24)
Body System Any Currently Any Currently Any Currently Any Currently History Active History Active History Active History Active
HEENT 1 0 1 0 2 0 4 0
(12.5%) (12.5%) (25.0%) (16.7%)
Respiratory 0 0 1 0 3 0 4 0 System (12.5%) (37.5%) (16.7%)
Musculoskeletal 3 0 0 2 0 2 1 7 1 System (37.5%) (25.0%) (25.0%) (12.5%) (29.2%) (4.2%)
Integumentary 0 0 2 2 2 2 4 4 System (skin, (25.0%) (25.0%) (25.0%) (25.0%) (16.7%) (16.7%) hair, nails)
Gastrointestinal 2 1 1 0 1 1 4 2 (8.3%) System (25.0%) (12.5%) (12.5%) (12.5%) (12.5%) (16.7%)
Genitourinary 2 0 4 4 3 3 9 7 System (25.0%) (50.0%) (50.0%) (37.5%) (37.5%) (37.5%) (29.2%)
Nervous 5 5 6 6 4 4 15 15 System (62.5%) (62.5%) (75.0%) (75.0%) (50.0%) (50.0%) (62.5%) (62.5%)
Endocrine 1 0 0 0 0 0 1 0 System (12.5%) (4.2%)
Psychiatric/ 1 0 0 0 1 0 2 0
Neurological (12.5%) (12.5%) (8.3%)
System
Allergic 1 1 0 0 4 3 5 4 Conditions (12.5%) (12.5%) (50.0%) (37.5%) (20.8%) (16.7%)
HEENT = head, eyes, ears, nose, and throat
Source: Section 14.1, Table 14.1.3.1
11.2.2.2 Prior and Concomitant Medications
A listing of medications used by each subject before and during the study is provided in
Appendix 16.2, Listing 16.2.9.1.
No concomitant medications were reported during the study.
Overall, 10 subjects reported prior medications. Most reported prior medications were over-the- counter analgesic or anti-inflammatory medications and multivitamins.
11.2.2.3 Period 1 (Single dose) Baseline Hormone Profile
During Period 1, blood samples for 24-hour baseline profile (total and free testosterone, SHBG, DHT, and estradiol) were collected at 0745 hours and then at 15, 30, and 45 minutes and at 1, 1.5, 2, 4, 6, 8, 12, 16, 20, and 23.5 hours relative to an 0800 hour clock time.
Table 11-2 presents the normal ranges, the pooled baseline ranges observed from Period 1 (single dose) and a break-down of the these baseline ranges by dose cohort. These data comparisons of the normal ranges with the pooled baseline ranges indicate that the pooled baseline values fall outside the normal ranges for free testosterone (min and max), DHT (max), estradiol (min) and SHBG (min and max).
Table 11-2: Baseline Analyte Concentrations (Single-Dose Population)
Dose Cohort
Normal Pooled
Analyte Range Baseline 600 (μ§) 1200 (μ§) 1800 (μ§)
Free Testosterone Min 0.166 0.10 0.10 0.13 0.15 (ng/dL); n = 336
Max 1.33 1.44 1.44 0.53 0.45
Total Testosterone Min 6.00 7.41 8.00 7.41 12.70 (ng/dL); n = 336
Max 86.00 58.00 58.00 41.10 43.90
Dihydrotestosterone Min 4.00 5.02 5.29 5.04 5.02 (ng/dL); n = 231
Max 22.00 32.60 22.10 25.30 32.60
Estradiol (pg/mL); Min 20.00 17.00 20.90 17.00 17.70 n = 336
Max 241.00 122.00 122.00 119.00 97.40
SHBG (nmol/L); n Min 18.00 15.30 15.30 16.00 25.20 = 336
Max 114.00 137.00 102.00 86.70 137.00
SHBG = sex hormone-binding globulin
Source: Appendix 16.1.10; Appendix 16.2, Listing 16.2.5.1.1a
11.3 Measurement of Treatment Compliance
All subjects were observed during dosing at the study center and were compliant with the study drug administration.
11.4 Tabulations of Individual Subject Data
11.4.1 Analysis of Pharmacokinetics
Pharmacokinetic (PK) profiles were obtained from 2 study periods in this study: single dose PK during Period 1, and multiple-dose PK during Period 2. The single-dose profile included administration of 600, 1200, or 1800 μg TBS-2 in separate cohorts while the multiple-dose profile included multiple administration of only 1200 μg TBS-2. For each dose, PK profiles for free and total testosterone, DHT, estradiol, and SHBG were estimated. There were 8 subjects in each dose strength cohort with 3 single-dose cohorts and 1 multiple-dose group.
The results are presented first for the concentrations and then for the PK parameters.
11.4.1.1 Concentration Results
11.4.1.1.1 Period 1: Single-Dose Treatment
Appendix 16.2, Listing 16.2.5.1.1a presents the dosing and concentration data from the singledose treatment for each of the 5 analytes as actual sampling clock time, scheduled elapsed sampling time, actual elapsed sampling time, time difference between scheduled and actual elapsed sampling times, baseline concentration, reported active dose concentrations, difference between baseline and active concentrations, and baseline-corrected concentrations with negative values replaced with zero. Section 14.4, Table 14.4.1a summarizes these data for baseline, active-dose, and baseline-corrected concentrations by administered dose for each scheduled sampling time.
11.4.1.1.1.1 Period 1: Baseline and Uncorrected Single-Dose Profile
The baseline and active-dose mean concentrations according to the normal y-axis scale are presented in the following graphs (source: Section 14.4, Figure 14.4.3.1a). The mean baseline concentrations (ConcBase) from Period 1 were consistently within the normal range for all analytes. They were flat with little change during the 24-hour observation period particularly for free testosterone, total testosterone, and SHBG but to a more limited extent with estradiol and DHT. Estradiol variation may have been unduly exaggerated since the full normal range was not
included for the figure. The observed baseline total and free testosterone as well as DHT concentrations had a consistent peak at 24 hours across all 3 dose cohorts (See ConcBase mean data in Figure 11-1, Figure 11-2, Figure 11-3, Figure 11-4, and Figure 11-5.)
The uncorrected active-dose concentrations for free and total testosterone were clearly differentiated by increasing dose, while this was not true for the other 3 analytes. The
uncorrected active-dose concentrations of free and total testosterone and to a lesser extent DHT were more clearly differentiated from the baseline concentrations (Figure 11-1, Figure 11-2, and Figure 11-3, respectively). The estradiol and SHBG concentrations had post-dose curves that overlapped with the baseline concentrations for the same dose (Figure 11-4 and Figure 11-5, respectively).
TBS-2 was rapidly absorbed after dose administration. The median Tmax values of free and total testosterone for 600, 1200, and 1800 μg doses were consistent between the two analytes and reported as 0.38, 0.5 and 0.63 hrs, and 0.38, 0.63 and 0.63 hrs respectively. The geometric mean uncorrected Cmax values of free and total testosterone were 0.761, 1.302, and 1.822 ng/dL, and 58.438, 81.287 and 139.742 ng/dL respectively (Table 14.4.2c).
For the most part, the observed mean concentrations of all 5 analytes were within their normal ranges. The only exceptions were during the first 2 hours for the 1800 μg TBS-2 dose in free (0.5 hrs - 2.0 hrs; Figure 11-1) and total testosterone (0.25 hrs - 2.0 hrs; Figure 11-2).
Figure 11-1: Mean Observed Free Testosterone Concentrations (Single-Dose Population)

ConcBase = baseline concentration; ConcBLQ = concentration of active dose
Source: Section 14.4, Figure 14.4.3.1a
Figure 11-2: Mean Observed Total Testosterone Concentrations (Single-Dose Population)

ConcBase = baseline concentration; ConcBLQ = concentration of active dose
Source: Section 14.4, Figure 14.4.3.1a
Figure 11-3: Mean Observed Dihydrotestosterone Concentrations (Single-Dose Population)

ConcBase = baseline concentration; ConcBLQ = concentration of active dose
Source: Section 14.4, Figure 14.4.3.1a
Figure 11-4: Mean Observed Estradiol Concentrations (Single-Dose Population)

ConcBase = baseline concentration; ConcBLQ = concentration of active dose Source: Section 14.4, Figure 14.4.3.1a
Figure 11-5: Mean Observed SHBG Concentrations (Single-Dose Population)

ConcBase = baseline concentration; ConcBLQ = concentration of active dose; SHBG = sex hormone-binding globulin
Source: Section 14.4, Figure 14.4.3.1a
11.4.1.1.1.2 Period 1: Single-Dose Baseline-Corrected Profiles
The baseline-corrected profiles from the single-dose treatment are presented as mean data and by spaghetti figures in this section.
11.4.1.1.1.2.1 Mean Figures
The mean concentration profiles are presented in the following figures for each of the 5 analytes after baseline correction (source: Section 14.4, Figure 14.4.3.2a). Individual concentration figures are presented in Section 14.4, Figure 14.4.1a. The free testosterone and total testosterone concentration figures presented the clearest relationship of concentration to increasing testosterone dose (Figure 11-6 and Figure 11-7, respectively). The DHT analyte provided a clear distinction between the 1800 μg dose compared with the lower doses, which were not clearly differentiated from each other (Figure 11-8). The obverse was observed in the estradiol concentrations where it was the lowest dose of 600 μg that was clearly distinct from the 2 higher doses, which were not clearly differentiated from each other (Figure 11-9). The SHBG concentrations were not clearly distinguished between administered testosterone doses (Figure 11-10).
Review of the terminal phase of the profiles suggest that it would be difficult to determine a terminal elimination rate constant for estradiol and SHBG and to some extent for DHT. Without estimation of a terminal elimination rate constant (λζ), the elimination half-life (t½), and area under the plasma concentration-time curve to infinity (AUC0-∞) could not be estimated.
Figure 11-6: Mean Corrected Free Testosterone Concentrations (Single-Dose Population)

ConcBC = baseline-corrected concentration
Source: Section 14.4, Figure 14.4.3,2a
Figure 11-7: Mean Corrected Total Testosterone Concentrations (Single-Dose Population)

ConcBC = baseline-corrected concentration
Source: Section 14.4, Figure 14.4.3.2a
Figure 11-8: Mean Corrected Dihydrotestosterone Concentrations (Single-Dose Population)

ConcBC = baseline-corrected concentration
Source: Section 14.4, Figure 14.4.3.2a
Figure 11-9: Mean Corrected Estradiol Concentrations (Single-Dose Population)

ConcBC = baseline-corrected concentration
Source: Section 14.4, Figure 14.4.3.2a
Figure 11-10: Mean Corrected SHBG Concentrations (Single-Dose Population)

ConcBC = baseline-corrected concentration; SHBG = sex hormone-binding globulin
Source: Section 14.4, Figure 14.4.3.2a
These mean concentration profiles are presented, using a log y-axis scale in Section 14.4, Figure 14.4.3.2b. Additionally the mean concentration profiles of the baseline without a dose and the concentrations following the dose are presented in both normal scale and log y-axis scale as Section 14.4, Figure 14.4.3.1a and Section 14.4, Figure 14.4.3.1b, respectively. It should be noted that the baseline data were collected only for the 24 hours before the active dose so the baseline data ends at 24 hours while the active dose continues.
11.4.1.1.1.2.2 Spaghetti Figures
Spaghetti figures (source: Section 14.4, Figure 14.4.5a) of the single-dose concentration profiles present the individual plots on the same analyte figure along with the mean data by dose in order to put each individual's profile in perspective. The free testosterone spaghetti figures are included for each of the 3 dose cohorts (Figure 11-11, Figure 11-12, and Figure 11-13);
comparisons between dose cohorts have been facilitated by using the same y-axis scale.
Figure 11-11: Baseline- Corrected Free Testosterone Spaghetti Figure, 600 μg (Single-Dose Population)

ConcBC = baseline-corrected concentration
Source: Section 14.4, Figure 14.4.5a
Figure 11-12: Baseline- Corrected Free Testosterone Spaghetti Figure, 1200 μg (Single-Dose Population)

ConcBC = baseline-corrected concentration
Source: Section 14.4, Figure 14.4.5a
Figure 11-13: Baseline- Corrected Free Testosterone Spaghetti Figure, 1800 μg (Single-Dose Population)

ConcBC = baseline-corrected concentration
Source: Section 14.4, Figure 14.4.5a
11.4.1.1.2 Period 2: Multiple-Dose Treatment
The multiple-dose treatment period included 7 TBS-2 1200 μg doses administered 8 hours apart. Pre-dose samples were collected for each of these doses and are presented in the following table (Table 11 -3). Appendix 16.2, Listing 16.2.5.1.1b presents dosing and concentration data from the multiple-dose treatment for each of the 5 analytes as actual sampling clock time, actual elapsed sampling time, time difference between scheduled and actual elapsed sampling times, and reported active dose concentrations. Section 14.4, Table 14.4.1b summarizes the active-dose analyte concentrations for each scheduled sampling time. This listing and this table also include the trough samples collected before each of the multiple doses.
11.4.1.1.2.1 Trough Concentrations Summary and Linear Regression
The predose or trough samples collected before each dose administration are summarized in Table 11-3.
Table 11-3: Trough Concentration Summary (Multi-Dose Population)

= coe c ent of variance; SD = standard deviation; SHBG ^ sex hormone-binding globulin
Note: All subjects received TBS-2 1200 μg
Source: Section 14.4, Table 14.4.1 b
Section 14.4, Figure 14.4.7 presents the individual concentrations for each analyte with linear regression analysis on the 7 predose or trough samples collected prior to the profile dose. It was noted that the trough values for total testosterone at 24 and 48 hours were atypically higher than those at other time points. Figure 11-14 below presents the total testosterone plot. The slope of the linear regression line for total testosterone was 0.8364. The slopes were very low for free testosterone (0.0110) and DHT (0.0851). Both estradiol (-0.3679) and SHBG (-0.0214) had slightly negative slopes, but there was large variability in the data as suggested by the R2 values (0.0134 for estradiol and 0.0002 for SHBG).
Figure 11-14: Trough Total Testosterone Linear Regression Figure (Multi-Dose Populatio
Trough Total lestosterone Linear Regression figure (Muiti-Dose Population)
Total Testosterone

R3 = linear correlation coefficient
Note: All subjects received TBS-2 1200 μ
Source: Section 14.4, Figure 14.4.7
11.4.1.1.2.2 Multiple-Dose Profile Concentrations
The mean concentration profiles are presented in the following figures for each of the 5 analytes after multiple dosing (source: Section 14.4, Figure 14.4.4a for normal y-axis). Individual figures are presented in Section 14.4, Figure 14.4.2a.
11.4.1.1.2.3 Mean Multiple-Dose Profile Figures
The free testosterone and total testosterone mean figures present the clearest relationship of dose administration effect with the highest concentrations occurring shortly after the dose
administration and decreasing to a plateau at about 12 to 16 hours. These 2 plots tracked so closely that they seemed to be superimposable once the scale was adjusted (Figure 11-15 and Figure 11-16, respectively). The DHT concentration also indicated a peak shortly after the dose administration but it was not as clear as with the free and total testosterone (Figure 11-17). Little difference in estradiol and SHBG concentrations were seen over the observation period of the multi-dose profile, which could have been attributed to the absence of a spike in concentration when the dose was administered. (Figure 11-18 and Figure 11-19, respectively). These mean figures are presented in Section 14.4, Figure 14.4.4.b fo the log y-axis scale and in Section 14.4 Figure 14.4.2b for the individual figures with the log y-axis scale.
Figure 11-15: Mean Free Testosterone Plasma Concentrations (Multi-Dose Population)

Source: Section 14.4, Figure 14.4.4a
Figure 11-16: Mean Total Testosterone Plasma Concentrations (Multi-Qosc Population)

Source: Section 14.4, Figure 14.4.4a
Figure 11-17: Mean Dihydrotesiosterone Plasma Concentrations (Multi-Dose Population)

Source: Section 14.4, Figure 14.4.4a
Figure 11-18: Mean Estradiol Plasma Concentrations (Multi-Dose Population)

Elapsed Time (hr)
Source: Section 14.4, Figure 14.4.4a
Figure 11-19: Mean SHBG Plasma Concentrations (Multi-Dose Population)

Elapsed Time (hr)
SHBG = sex hormone-binding globulin
Source: Section 14.4, Figure 14.4.4a
11.4.1.1.2.4 Multiple-Dose Profile Spaghetti Figures
The following spaghetti figures (source: Section 14.4, Figure 14.4.5a) of the multi-dose concentration profiles present the individual plots on the same analyte figure along with the mean data in order to put each individual concentration in perspective. Both free and total testosterone concentrations followed the mean values in having a positive relationship with a concentration peak due to the dose. Subject 522-03 had free testosterone concentrations that didn't show a similar decrease as compared to the other 7 subjects for 48 hours after the final dose (Figure 11-20). The total testosterone concentrations for Subject 522-03 were also the highest concentrations after 12 hours but not as different from the other 7 subjects as were the free testosterone concentrations (Figure 11-21). Relationship to dose was increasingly more difficult to detect for DHT, estradiol, and SHBG, with SHBG concentrations essentially constant for each subject but at different concentrations (Figure 11-22, Figure 11-23, and Figure 11-24, respectively).
Figure 11-20: Spaghetti Concentration Plots with Mean for Free Testosterone Plasma Concentrations (Multi-Dose Population)
I I I I I I I I I I ] I I I I I I I [ I I I I I I I I I I I I I" I I I I I I I I I I Γ I I I I I I I
(hr)
Source: Section 14.4, Figure 14.4.5a
Figure 11-21: Spaghetti Concentration Plots with Mean for Total Testosterone Plasma Concentrations (Multi-Dose Population)

(hr)
Source: Section 14.4, Figure 14.4.5a
Figure 11-22: Spaghetti Concentration Plots with Mean for Dihydrotestosterone Plasma Concentrations (Multi-Dose Population)
"Ί IT" I I I" I' I' I I I I I n [ I I I I I r I I ! I T I I I I I I Ι ' Γ 'Μ I I I M I I I I I
Source: Section 14.4, Figure 14.4.5a
Figure 11-23: Spaghetti Concentration Plots with Mean for Estradiol Plasma
Concentrations (Multi-Dose Population)

Source: Section 14.4, Figure 14.4.5a
Figure 11-24: Spaghetti Concentration Plots with Mean for SHBG Plasma Concentrations (Multi-Dose Population)

SHBG = sex hormone-binding globulin
Source: Section 14.4, Figure 14.4.5a
11.4.1.2 Pharmacokinetic Parameter Estimates
Pharmacokinetic parameters were estimated by using Phoenix WinNonlin version 6.2.
11.4.1.2.1 Period 1: Single Dose Pharmacokinetic Parameter Estimates
All PK parameter estimates were performed on the first 24 hours with the baseline-corrected concentrations. The PK parameters estimated from the single-dose profiles for each individual are presented in Appendix 16.2, Listing 16.2.5.1.2a and summarized in Section 14.4, Table 14.4.2a. The following table (Table 11-4) presents the data summarized in Section 14.4, Table 14.4.2a for the single-dose PK parameters but the SD and median have been omitted.
The terminal elimination rate constant (λζ) could be estimated for all 24 subjects for free and total testosterone. The terminal elimination rate constant could only be estimated for 12 subjects for DHT, for 11 subjects for estradiol, and for 9 subjects for SHBG. Without λζ estimate, the elimination half-life, t½, and AUC0-∞ could not be calculated.
11.4.1.2.1.1 Free Testosterone
For free testosterone, the geometric mean Cmax values (0.442, 1.017, and 1.500 ng/dL) closely tracked the administered doses (600, 1200, and 1800 μg, respectively), (Section 14.4, Table 14.4.2a).
Geometric mean AUCO-8 was proportional from the 600 μg dose to the 1200 μg dose (ratio of 1.86 compared to the 2.00 dose ratio) but was somewhat higher for the 1800 μg dose (ratio of 3.73 compared to the 3.00 dose ratio). Other measures of AUC were less clearly related to dose and did not suggest a proportional relationship to dose as strongly as AUC0-8. The λζ values were similar across doses.
Mean uncorrected concentrations returned to pre-dose values after 12 hours for the 600 μg dose and after 16 hours for the 1200 μg dose (Table 14.4.1a and Figure 14.4.3.1a), but showed an increase towards the 24 hour time point. For the 1800 μg dose, mean uncorrected concentrations were consistently decreasing up to 20 hours post-dose, but demonstrated an increase at 24 hours similarly to the two other doses.
Arithmetic mean uncorrected concentrations at 24 hours after dosing (C24ConcBLQ of 0.481, 0.423, and 0.436 ng/dL) were comparable to pre-dose mean concentrations (C24ConcBase of 0.448, 0.346, and 0.314 ng/dL), with little difference across the three doses.
Table 11-4: Free Testosterone Summary (Single-Dose Population)

11.4.1.2.1.2 Total Testosterone
Total testosterone geometric mean Cmax parameters increased with increasing dose (34.058, 62.88, and 113.912 ng/dL for the 600, 1200, and 1800 μg doses, respectively). AUC values increased with dose as well.
Similarly to free testosterone, mean uncorrected concentrations returned to pre-dose values after 12 hours for the 600 μg dose and after 16 hours for the 1200 μg dose (Table 14.4.1a and Figure 14.4.3.1a), but showed an increase towards the 24 hour time point. For the 1800 μg dose, mean uncorrected concentrations were consistently decreasing up to 20 hours post-dose, but demonstrated an increase at 24 hours similarly to the two other doses.
Arithmetic mean uncorrected concentrations at 24 hours after dosing (C24ConcBLQ of 33.138, 28.013, and 34.55 ng/dL) were comparable to baseline mean concentrations (C24ConcBase of
30.138, 23.538, and 24.35 ng/dL), also with no demonstration of either proportional or significant difference across the three doses (Table 11-5). The CV was > 28.8% for all 6 arithmetic mean values.
Table 11-5: Total Testosterone Summary (Single-Dose Population)

11.4.1.2.1.3 Dihydrotestosterone
The PK parameters for DHT were missing a number of parameter values and some entire profiles were missing after baseline correction; only 18 of the 24 profiles were evaluable and λζ could be estimated in only 12 of the 24 profiles. Geometric mean Cmax (5.888, 5.646, and 9.82 ng/dL), AUCO-8 (21.859, 20.858, and 43.767 hr*ng/dL), decreased from the 600 μg dose to the 1200 μg dose but then increased for the 1800 μg dose. For AUC0-∞, the values were 55.582, 71.647, and 128.512 hr*ng/dL, respectively, for the 600, 1200 and 1800 μg doses. The other AUC measures demonstrated increasing exposure with increasing dose strength (Table 11-6).
Table 11-6: Dihydrotestosterone Summary (Single-Dose Population)


11.4.1.2.1.4 Estradiol
Estradiol geometric mean PK parameters for Cmax (13.468, 13.474, and 11.47 ng/niL, respectively, for the 600, 1200 and 1800 μg doses) were similar across doses. The geometric mean AUCO-24 measures (106.562, 80.078 and 48.85, respectively, for the 600, 1200 and 1800 μg doses) followed an inverse relationship with dose. Arithmetic mean uncorrected concentrations at 24 hours after dosing (C24ConcBLQ of 47.438, 42.6, and 34.713 ng/dL) did not have a clear relationship to baseline mean concentrations (C24ConcBase of 50.1, 40.288, and 42.55 ng/dL, respectively, for the 600, 1200 and 1800 μg doses) (Table 11-7).
Table 11-7: Estradiol Summary (Single-Dose Population)

11.4.1.2.1.5 SHBG
The geometric mean Cmax values for SHBG (6.284, 6.155, and 8.046 nmol/L) were similar for the 600 μg and the 1200 μg doses and slightly higher for the 1800 μg dose. No clear relationship between dose and geometric mean values for AUC0-8, AUC0-24, and AUCO-t were observed.. The elimination rate constant, λζ, could not be estimated for the majority of these subjects (Table 11-8).
Table 11-8: SHBG Summary (Single-Dose Population)

11.4.1.2.2 Period 2: Multiple-Dose Pharmacokinetic Parameter Estimates
The PK parameters estimated from the multiple-dose profiles for each individual are presented in Appendix 16.2, Listing 16.2.5.1.2b and summarized in Section 14.4, Table 14.4.2b. The tables in the following sections present the data summarized in Section 14.4, Table 14.4.2b for the multiple-dose PK parameters but the SD and median have been omitted. All subjects received 7 doses of 1200 μg testosterone in the multiple-dose treatment.
Because the multiple-dose treatment parameter estimates were uncorrected for baseline, an additional estimate of the single-dose treatment uncorrected parameters was conducted for comparison with the uncorrected multiple-dose treatment estimates. These uncorrected PK parameter estimates from the single-dose treatment were limited to free testosterone, total testosterone and DHT. Pharmacokinetic analyte parameter estimations on uncorrected data for the single-dose treatment are listed in Appendix 16.2, Listing 16.2.5.1.1a and summarized in Section 14.4, Table 14.4.2c. Comparisons of the single-dose to multiple doses of uncorrected parameter estimates were most appropriate at the only common dose strength, 1200 μg.
11.4.1.2.2.1 Free Testosterone
Free testosterone summary for the multiple-dose population is shown in Table 11-9. Peak concentrations were achieved within 15 to 45 minutes (range of Tmax 0.25-0.75 hrs) following the last (7th) dose administration. The multiple-dose profile of free testosterone PK had a mean
Cmax (arithmetic mean 1.886 ng/dl; geometric mean 1.874 ng/dL) that was above the upper normal limit of 1.33 ng/dL. The mean Cmin (arithmetic mean 0.704 ng/dl; geometric mean 0.638 ng/dL) was within the range of 0.166 - 1.33 ng/dL. The mean Cavg of 1.006 ng/dL, and geometric mean Cavg of 0.944 ng/dL were well within the normal range for women (0.166 to 1.33 ng/dL).
The mean concentration values for free testosterone concentration exceeded ULN approximately for 1 hour post-dose in the 24-hour period following the 48 hours of 1200 μg doses every
8 hours.
The AUCO-8 and the AUCO-τ were identical, because the dosing interval was 8 hours (Table 11- 9). The AUCO-24 profile spanned the first 8 hours of the dosing interval following the last dose but also included the following 16 hours, which totaled 24 hours. The percentage peak to trough fluctuation (%PTF) signifies the percentage fluctuation from the average concentration to the peak or trough, and had a geometric mean of 119.58%. The percentage peak to trough swing (%PTS) signifies the percentage concentration swing between the peak and trough concentration, and had a geometric mean of 176.91 .
Table 11-9: Free Testosterone Summary for Multiple-Dose Profile (Multi-Dose Population)
Statistic <:,,, v C %rrw
iagSU w (»g,¾L) (¾r½ ¾
8 s 8 S s 8 8 H s H
M¾sss ! S86 0. 78 \. 5 0.704 i 006 0.57S S.046 17 259 S 046 ; 40 676 .2S4.05S
CV •2 4 49.6 .: 55. ; 40 6 7- S 40.6 55.0 40 6 55.2 55.0 a • 65 0.25 0.77 0.40 0.59 0.2S 4.69 9 74 4 69 30.60 35.00
Mas 2.58 0.75 i .S2. 5 .60 I .S3 1.71 1 14 39.66 54.64 295.75 437.88
Geo ean • S74 0.344 um 0.638 .944 0.492 7.554 • 5 722 7 554 • · 9 579 576 905
AUC " A:Bs ii!isie!' i¾t p!tisssia eencemratioii time cur e ftom time -sro to 8 hours afi« dosing AUC^ ~ Area under i!w pimta coaeeatraaen lims e«3\¾ ftom iifise scat© » 24 hams isSva de-ssag, AUCW - ,%sa mdet is* aiusi-Urrse carve (am. ii∞e m<3 » tbe dosing aitervafc
CS5i : tna iffsuii? e«iCBfi!;aitftr! <it ser ed at½r dosi;sg; 0 " ;?5is«im«i« cc ceafrsiioss at bsssSifis, C^^-Mm a eoaeemratioa ovsr a essag miervai dafia¾«adiipie dosiag; C9 ~ Average aaa y-staw ccraentution, Ci5 - m_xtm<it» eotseemr&tiari si 24 hours after &s»tt&
CV ---- % toeffidisU of ratisuoa. Geo easi ----- geometric mean; "aPTF "· percent peak io ίτοίΐ Πιιίίιϋίίίαι; %PTS ---p&rcsfii peas ΐο trough swing
W" iii of observed maximum concentration reistrve t» die thm of dosiag
Source-. Session \d 4, Ta !e 1442b
Accumulation was estimated by a linear regression analysis of pre-dose/trough concentrations. This comparison is presented in Figure 11-25 and indicated that only a small fraction of predose/ trough concentration variability (Rsq = 0.1601) could be explained by the linear regression line, which had a slope (Slope = 0.011) very close to zero. This accumulation analysis did not detect substantial evidence of accumulation from the pre-dose sample drawn before any doses were given through the 6 trough concentrations drawn before each of those doses. In line with total testosterone values, the trough concentrations for the 4th and 7th doses were atypically higher than the other 5 dose pre-dose/trough concentrations.
Figure 11-25: Trough Free Testosterone Linear Regression Figure (Multi-Dose Population)
Trough Free Testosterone Linear Regression Figure (Mufti-Dose Population)
Free Testosterone

Source: Section Ϊ .4, Figure 14.4.7 11.4.1.2.2.2 Total Testosterone
A total testosterone summary for the multiple-dose population is shown in Table 11-10.
Peak concentrations of total testosterone were achieved within 15 to 45 minutes (range of Tmax 0.25-0.75 hrs) following the last (7th) dose administration suggesting a rapid absorption of the nasal gel. The multiple-dose profile of total testosterone PK had a mean Cmax (arithmetic mean 148.038 ng/dl; geometric mean 137.555 ng/dL) that was above the upper normal limit of 86 ng/mL. The mean Cmin (arithmetic mean 49.088 ng/dl; geometric mean 46.136 ng/dL) was within the range of 6.00 - 86.00 ng/dL. The mean value of Cavg (arithmetic mean 73.802 ng/dL; geometric mean 69.166) was well within the normal range. The mean concentration values for total testosterone after the multiple (1200 μg every 8 hours) TBS-2 administration exceeded ULN approximately for 1.5 hours in the 24-hour period following the last (7th ) dose
administration.
A linear regression analysis of the 7 pre-dose/trough concentrations of the multiple-dose treatment were previously discussed in Section 11.4.1.2.2.1.
Table 11-10: Total Testosterone Summary for Multiple-Dose Profile (Multi-Dose Population)

- tiaw of observed rtmisaaa eotieeturatiesi relati e to the time of dosing
Souse*; S«s»e» (4.4. Table 14.4.2b
11.4.1.2.2.3 Dihydrotestosterone
A DHT summary for the multiple-dose population is shown in Table 11-11.
Peak DHT concentrations were achieved within 15 minutes to one hour following the last (7th) dose administration. The mean value of Cavg (arithmetic mean 16.628 ng/dL; geometric mean 15.274 ng/mL) was within the normal range. The arithmetic mean Cmax (22.3 ng/dL) slightly exceeded ULN (22.0 ng/dL), however, the geometric mean Cmax (20.792 ng/dL) was below the ULN.
Table 11-11: Dihydrotestosterone Summary for Multiple Dose Profile (Multi-Dose Population)

Soiei Sic ou 14 4, liibte 14 4 2b
11.4.1.2.2.4 Estradiol
An estradiol summary for the multiple-dose population is shown in Table 11-12.
No effect from TBS-2 dosing on estradiol concentrations was obvious. Mean Cmax (arithmetic mean 70.8 pg/mL; geometric mean 60.034 pg/mL) and Cavg (arithmetic mean 58.019 pg/mL; geometric mean 50.033 pg/mL) were within the normal range (20.00 to 241.00 pg/mL).
Table 11-12: Estradiol Summary for Multiple-Dose Profile (Multi-Dose Population)
Sttftrfte *»» c„ AL¾;;s At ( , ; > At < >, ¾!'fi P S
! r) ip&¼U (pg/mL} <hr*pgfciL)
s 8 s 8 s 8 S s 8 8 s
Mean 70.800 3.23! 54413 48.875 58 019 64. S B 464.148 Ϊ 759.3 2 464448 35465 42. 164
CV 54.9 79.7 44.3 S . 54.0 442 54 0 J .J 54.0 44,8 46 9
Mm •6 60 87.7 16.30 12 70 14 % I8 60 i ;9.67 503.91 1 19.67 20.16 22.22
Max i 45.00 S.99 84.20 95.20 1 20.64 104.00 5>65.13 2895 80 965. ί 3 66.76 79 82
GeoMeast 60.054 2.024 >. 42.50? 5 .033 S6.S20 400 :■(.■ 1 ί 587.754 400.2S4 32.76S 5S.514
Area uader th plasms fi.aniratiijr time i om t me sro to 8 dosing, AUC " Area lindi.".' she plasms canoe* iff as on tw&e curve from tw&e »¾«> to 24 hours after dosing, AUC^ - Area under the conce»i>»tS<>K-tfine carve from time awe » the dosing ffitejyai;
C,„>:"- cis enirjs o observed aftsr dosing. C¾∞ maximu «*κ»«Κ3όο« at baseline; C!K<.-∞Mj«jmuffs concentration over a desang iiucjvsi c!utmg m ddle dosissg; C»¾ - Average co«c«att*t«on. C¾t -· tnaximutti eoneefiisalkm at 24 hours after
CV ~ ¾ eoefficsetnef venation,, GeoMean - »«ean; %PTF - percent peas trough ilt ¾PTS -· pi-r an to trough swing; t.,ss - urns of maximum relative the of ifcjang
&>»««· Setiioa 5.4.4. Jahk- i4.4.2¾
11.4.1.2.2.5 SHBG
A summary of SHBG PK parameters for the multiple-dose population is shown in Table 11-13.
No effect from the testosterone dosing on SHBG concentrations was obvious. Mean Cmax (arithmetic mean 62.725 nmol/L; geometric mean 56.270 nmol/L), Cmin (arithmetic mean 55.300 nmol/L; geometric mean 49.024 nmol/L), and Cavg (arithmetic mean 58.548 nmol/L; geometric mean 52.530 nmol/L), were within the normal range (18.00 to 114.00 nmol/L).
Table 11-13: SHBG Summary for Multiple Dose Profile (Multi-Dose Population)

11.4.1.3 Pharmacokinetic Parameter Statistical Testing
11.4.1.3.1 Dose Proportionality Analysis on Single Dose
Section 14.4, Table 14.4.3 presents the analysis of dose proportionality. Table 11-14 is abstracted from this table to present the linear regression analysis of the log-transformed PK parameters.
Dose proportionality occurs when increases in the administered dose are accompanied by proportional increases in a measure of exposure (PK parameter). The linear regression model is fitted to each of the interested PK parameters by using dose (600, 1200, and 1800 μg) as the predictor variable:
PK = Slope * Dose + Intercept.
A dose proportionality hypothesis cannot be rejected if the intercept's 95% confidence limit includes 0.
Dose proportionality was not rejected for AUC0-8, AUC0-24, and AUCO-t for free testosterone. Cmax values of free and total testosterone also increased with each dose increase, but did not demonstrate statistical significance. None of the PK parameters of any of the other analytes (DHT, estradiol, and SHBG) passed the statistical test for dose proportionality.
Table 11-14: Dose Proportionality Analysis (Single-Dose Population)

11.4.1.3.2 Dose Pair- wise Test Using Analysis of Variance
Table 11-15 presents pair-wise comparisons of single-dose PK parameters by using ANOVA after dose normalizing. Results showed that there were differences in the dose normalized values for all analytes for at least 1 of the 3 pair- wise comparisons except for free testosterone AUC measures for AUC0-24, AUC0-8, and AUCO-t. It can only be concluded from this analysis that most of the PK outcomes from the three dose levels were not statistically identical.
Table 11-15: Analysis of Variance for Some Pharmacokinetic Parameters (Single-Dose
P- Value
Intrs-sabject 600 pg 600 pg 120» fig
Coefficient vs. vs. vs.
Anslyte PK Pai¾«et< M- of Variation* n m 1800„g 1800
Free Teslosterwe AUC^* 45.8505 0.2114 0.2182 0.9845
AUCC.S 67.4794 e».7uo 0.5338 0.799S
AUC^. 41M39 0 0197 0.8185
Αΐ.¾, 45.3344 0.2439 0.2477 0.9921
-223,3208 9, 0034 0,0010 0.6182
Total Testosterone 7.895 A <.ooei < .00 1 0.0001
8.23?2 0001 • .000 i 0.0001
AUCV 8.7190 _0001 <.000 0.0002
7.S792 .000 ! .000 l 0.000 ! c,,..t . 17.9163 < 00 1 0001 0.0320
Diii droteittssiei ne AUCi ¾. 1 9.8167 .00 1 .00 1 0.1756
AUG,S 23.5230 .000 i 00 1 0.3291
AUG*.., 26.015 1 0.004S 0.0004 0. 131
AUCW 26.5016 0.001 ! 0.0001 0.3144
37.3379 0.002 1 0.0009 0.6763
AUC.s, - Ares ihe$¼$m& eor.eenttsiioo time cBt * rk«;s tittw . S to S !·«¾!> after dosing. AUG** Area ur
emcmm m ims cuive .from tim?. «ss» to 24 mfs after dosiag AUC** -- At«a u»der ths !ssma eofica-arat k»s &ts≤. carvs from time «¾ to the !stst mea&atM concentrsiian time pwm; AUi^ . Atea ussiier the plasma concentration time curve fi»m tx i zsto to Lsisiiiiy; , ascsB»8« etsneetttratien observed after dosi«¾:
a Coefficient of Vansttoe - 100 s A NOVA sesiduai atm (Soot Mean Square Eftet)/ PK parameter s!!ean. Pait-wise oowipansorrs are roas ίΐϊβ A NOVA model wth Dose its a class (caiegorksii aria l ∞d PK parawsiafif estimate values as response. Nate; AUC** AUCS,.„ AUCM isisd Cw, ptsaameser alues ate ssaiura! logarithm: ti¾¾si rmeo.
Source: S«ttion S4.4. Tairfe 14.4.3
11.4.1.3.3 Paired t-Test Comparison of AUCO-8 and AUCO-24 from Single to Multiple Dose
There were 3 subjects who participated in both the single- and multi-dose periods at the 1200 μg dose. Uncorrected parameters for AUCO-8 and AUCO-24 were calculated from the free testosterone, total testosterone, and DHT analyte concentrations for comparison to uncorrected parameters from the multi-dose profile. Table 11-16 compares the AUCO-8 and AUCO-24 from these 3 subjects by using a paired t-test for free testosterone, total testosterone, and DHT. The results of the log transformed comparisons are provided.
Table 11-16: Paired t-Test Results for Pharmacokinetic Parameters AUCO-8 and AUCO-24 for Subjects Who Had 1200 μg TBS-2 in Period 1 and Period 2
: ain rat 4 ogtiriifcrnk 95% CenSutence Limit for
transfermeti PK Parameter Mea« PiiT retice
of Standard E ror
AiSiiiyie Name ?i iMfteretu* iff DtSerence lower Limit Vpper Limit P-Vaiue
Free lesies&te 8.684! 0.42ίί -i..22>¾7 2.4349
Λ: ·: .. . . Ϊ 0.93 ! 2 '"J.3S53 0.1369
Tettd Testosie«sBe AUC« ? S.57S2 (U7S& -0 38?& i . 448 S OS !
AU( 3 8.9858 0.16S3 .1¾Ϊ 5 1.S301 8.8328
Oih siioiestostertifii AIJCS.S 3 2 ! 825 1.2160 2.9889 8.8095
ΑΙ¾.:¾ 3 0.43 H 3.9388
Alj ;..;, ----- Area aajfet ¾fi piastiJ3 toncsijwafion tms curve item nrne - o la 8 hours. AUCs,^ Afea uader Ai- plasma eoiiteatfishfjii time curve fiam time xm> to 24 SJ&LKS: JPK j.*»rnaco)«»eue
Sousee: Seciioa 34 4. 34.4.5
The free testosterone comparisons of 95% CI included zero in the difference for both AUC measures so equivalence between the single- and multi-dose AUC measures could not be rejected. Total testosterone 95% CIs included zero for the AUCO-8 but not for the AUCO-24 so equivalence could not be rejected for AUCO-8 but was for AUCO-24. In the analyte DHT, neither 95% CI included zero, so equivalence was rejected for both AUCs.
11.4.2 Statistical and Analytical Issues
11.4.2.1 Adjustments for Covariate
Not applicable.
11.4.2.2 Handling of Dropouts or Missing Data
Handling of missing or partial AE and concomitant medication dates is described the SAP (Appendix 16.1.9).
11.4.2.3 Interim Analysis and Data Monitoring
Interim analyses are discussed in Section 9.7.3.
11.4.2.4 Multi-center Studies
Not applicable.
11.4.2.5 Multiple Comparison/ Multiplicity
A few pair- wise comparisons were performed, but no Type 1 error rate was adjusted since they were exploratory analyses.
11.4.2.6 Use of a "Pharmacokinetics Subset" of Subjects
All subjects were included in the PK analyses.
Active-Control Studies Intended to Show Equivalence
licable.
Examination of Subgroups
licable.
abulation of Individual Response Data
licable.
rug Dose, Drug Concentration, and Relationship to Response
licable.
rug-Drug and Drug-Disease Interactions
licable.
y-Subject Displays
licable.
harmacokinetics Conclusions
Period 1 -Single Dose
a. The mean baseline pre-dose concentrations from Period 1 were consistently
within the normal range for all analytes.
b. The uncorrected 600, 1200, and 1800 μg dose concentrations of free and total testosterone and to a lesser extent of DHT were more clearly differentiated from the baseline pre-dose concentrations. The estradiol and SHBG concentrations had postdose curves that overlapped with the baseline pre-dose concentrations for the same dose.
c. TBS-2 was rapidly absorbed after dose administration. The median Tmax values of free and total testosterone for 600, 1200, and 1800 μg doses were consistent between the two analytes and reported as 0.38, 0.5 and 0.63 hrs, and 0.38, 0.63 and 0.63 hrs respectively. The geometric mean uncorrected Cmax values of free and total testosterone were 0.761, 1.302, and 1.822 ng/dL, and 58.438, 81.287 and 139.742 ng/dL respectively.
d. For the most part, the observed post-dose uncorrected mean concentrations of all 5 analytes were within their normal ranges. The only exceptions were during the first 2 hours for the 1800 μg testosterone dose in free (0.5 hrs - 2.0 hrs) and total testosterone (0.25 hrs - 2.0 hrs). Testosterone concentrations exceeding the upper limit of the physiological range (upper limit of normal, ULN) for a limited
amount of time following dosing can be expected due to the peak and trough PK profile of TBS-2, and the endogenous nature of testosterone.
e. Mean uncorrected free and total testosterone concentrations at 24 hours after dosing were comparable to pre-dose mean concentrations, with little difference across the three doses.
f. Dose proportionality was not rejected for baseline-corrected AUCO-24, AUCO-8, and AUCO-t of free testosterone. Cmax values of baseline-corrected free and total testosterone also increased with the dose increase (0.442, 1.017, and 1.500 ng/dL and 34.058, 62.88 and 113.912 ng/dL respectively), but did not demonstrate statistical significance. The DHT PK parameters as well those of estradiol and SHBG did not pass the statistical test for dose proportionality.
Period 2 - Multiple Dose
a. The free and total testosterone concentrations had the clearest demonstration of administration-related concentration increase. A more modest administration- related increase in DHT concentration, but little administration-related increase in estradiol and SHBG concentrations was observed.
b. Peak concentrations of free and total testosterone were achieved within 15 to 45 minutes following the last (7th) dose administration. The ranges of Tmax were 0.25-0.75 hrs for both and geometric mean Cmax values were 1.874 and 137.555 ng/dL for free and total testosterone, respectively. Mean Cavg values of 1.006 ng/dL, and geometric mean Cavg of 0.944 ng/dL for free testosterone and mean Cavg value of 73.802 ng/dL, and geometric mean Cavg of 69.166 ng/dL for total testosterone were well within the normal range for women.
c. The mean concentration values for free and total testosterone after the multiple (1200 μg every 8 hours) TBS-2 administration exceeded ULN approximately for 1.0 and 1.5 hours respectively in the 24-hour period following the last (7th ) dose administration, which can be expected due to the peak and trough PK profile of TBS-2.
d. Accumulation analysis did not detect substantial evidence of accumulation for any analytes.
e. Equivalence between the single- and multiple-dose profiles in the 3 subjects who participated in both profiles was not rejected for AUC0-8 and AUCO-24 of free testosterone and for AUC0-8 of total testosterone, also suggesting minimal accumulation. Equivalence was rejected for AUCO-24 of total testosterone, which can be explained by possible perturbation of the data by the diurnal variation in testosterone levels around 24 and 48 hours post-dose. Since there was no accumulation at 8 and at 16 hours, it would appear logical that the apparent significant difference between available total testosterone at 24 hours was possibly due to the diurnal variation in testosterone levels.
12 SAFETY EVALUATION
12.1 Extent of Exposure
Study drug administration was performed at the clinical site under study personnel supervision and is listed by subject in Appendix 16.2, Listing 16.2.5.1a (Period 1) and Appendix 16.2, Listing 16.2.5.1b (Period 2).
12.2 Adverse Events
12.2.1 Summary of Treatment Emergent Adverse Events
There were no deaths, SAEs, or AEs leading to discontinuation during the study. Most TEAEs were in the system organ classes of general disorders and administration site conditions and respiratory, thoracic, and mediastinal disorders (nasal related events). Most TEAEs were mild in severity and were unlikely or not related to study medication.
12.2.2 Display of Treatment Emergent Adverse Events
The incidence of TEAEs is shown for Period 1 in Table 12-1 and Period 2 in Table 12-2.
Table 12-1: Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term (Single-Dose Population)

ecDSA ······ Medical Dictionary for Regulatory Activities; TEAE - treatment-emergent adverse event
Nose: Subjects reporting mote than 1 TEAE in each level (system otgan class or preferred term) were only counted once.
Source: Section 14.3.1, Table 14.3.1.2a
Table 12-2: Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term (Multi-Dose Population)

MedDRA = Medical Dictionary for Regulatory Activities; TEAE = treatment-emergent adverse event
Note: Subjects reporttog sno e than 1 TEAE in each level (system organ class or preferred term) were only counted once
Source: Section i4.3, 1 , Table H.3. i ,2b
12.2.3 Analysis of Adverse Events
12.2.3.1 Adverse Events in Period 1
Overall, 11 of 24 subjects (45.8%) experienced at least one TEAE in Period 1. Most subjects experienced TEAEs of general disorders and administration site conditions (experienced by 7 of 24 subjects [29.2%]) and respiratory, thoracic, and mediastinal disorders (experienced by 5 of 24 subjects [20.8%]). TEAEs experienced by more than 1 subject were catheter site erythema (experienced by 3 of 24 subjects [12.5%]) and catheter site hemorrhage, catheter site
inflammation, dizziness, and nasal congestion (each experienced by 2 of 24 subjects [8.3%]) (Table 12-1 and Section 14.3.1, Table 14.3.1.2a).
The majority of TEAEs in Period 1 were of mild severity. A total of 10 of 24 subjects (41.7%) experienced mild TEAEs. Only 1 of 24 subjects (4.2%) experienced moderate TEAEs.
(Section 14.3.1, Table 14.3.1.4a) Subject 522-53 (Cohort 2) experienced headache and dizziness of moderate severity on Day 1. Both TEAEs resolved without treatment the next day and were considered as unlikely to be related to the study drug (Appendix 16.2, Listing 16.2.7.1).
Overall, 4 of 24 subjects (16.7%) experienced TEAEs that were considered possibly related to the study medication. A total of 3 of 24 subjects (12.5%) had TEAEs that were considered to be unlikely related to the study medication and 4 of 24 subjects (16.7%) had TEAEs that were considered not related to the study medication (Section 14.3.1, Table 14.3.1.5a).
12.2.3.2 Adverse Events in Period 2
Overall, 4 of 8 subjects (50.0%) experienced at least 1 TEAE in Period 2. General disorders and administration site conditions occurred in 2 of 8 subjects (25.0%). The TEAEs that were experienced by 1 of 8 subjects (12.5%) each were catheter site pain, catheter site phlebitis, dyspepsia, headache, and rhinalgia (Table 12-2 and Section 14.3.1, Table 14.3.1.2b).
The majority of TEAEs in Period 2 were of mild severity. Overall, 3 of 8 subjects (37.5%) experienced mild TEAEs. Only 1 of 8 subjects (12.5%) experienced a moderate TEAE. (Section 14.3.1, Table 14.3.1.4b) Subject 522-29 experienced an increase in headache intensity of moderate severity on Day 2. The TEAE resolved without treatment 2 days later and was considered as possibly related to the study drug (Appendix 16.2, Listing 16.2.7.1).
Overall, 2 of 8 subjects (25.0%) experienced TEAEs that were considered possibly related to the study medication and 2 of 8 subjects (25.0%) experienced TEAEs that were considered not related to the study medication (Section 14.3.1, Table 14.3.1.5b).
12.2.3.3 Adverse Reactions in Period 1 and Period 2
Adverse reactions (defined as TEAEs considered possibly, probably, or definitely related to study medication) in Period 1 and Period 2 are shown in Table 12-3.
A total of 6 subjects experienced adverse reactions during the study: 4 of 24 subjects (16.7%) in Period 1 and 2 of 8 subjects (25.0%) in Period 2. All adverse reactions were considered possibly related to study medication. (Appendix 16.2, Listing 16.2.7.4) During Period 2, Subject 522-29 (1200 μg) had 2 adverse reactions of headache that were each recorded as a TEAE: a mild headache that increased in severity to moderate (Appendix 16.2, Listing 16.2.7.1 and Appendix 16.2, Listing 16.2.7.4).
The incidence of adverse reactions is shown in Section 14.3.1, Table 14.3.2.4a (Period 1) and Section 14.3.1, Table 14.3.2.4b (Period 2).
Table 12-3: Subjects with Adverse Reactions (Single- and Multi-Dose Populations)

MedDRA = Medical Dictionary for Regulatory Activities; TEAE = treatment-emergent adverse event
a. Values indicate the study period and dose the subject was receiving at the time of the event.
b. The onset was calculated, respectively, as (Onset Date - Treatment Date of Period I t ij, if the onset was
during Period L or [Onset Date - Treatment Dale of Period 2 ■ i j, if the onset was during Period 2.
c. Severity was rated by the Principal i nvestigator as mild, moderate, or severe.
Source: Appendix 16.2, Listing ! 6 2.7.4
12.2.4 Listing of Adverse Events by Subject
Adverse events are listed by subject in Appendix 16.2, Listing 16.2.7.1. Adverse reactions (defined as TEAEs considered possibly, probably, or definitely related to study medication) in Period 1 and Period 2 are listed by subject in Appendix 16.2, Listing 16.2.7.4.
12.3 Deaths, Other Serious Adverse Events, and Other Significant Adverse Events
There were no deaths, SAEs, or AEs leading to discontinuation during the study (Section 14.3.1, Table 14.3.2.2a; Section 14.3.1, Table 14.3.2.2b; and Appendix 16.2, Listing 16.2.7.1).
12.4 Clinical Laboratory Evaluations
12.4.1 Listing of Individual Laboratory Measurements by Subjects and Each Abnormal
Laboratory Value
Clinical laboratory evaluation measurements and abnormal values are listed in Appendix 16.2, Listing 16.2.8.1 (hematology), Appendix 16.2, Listing 16.2.8.2 (chemistry), and Appendix 16.2, Listing 16.2.8.3 (urinalysis).
12.4.2 Evaluation of Each Laboratory Parameter
12.4.2.1 Laboratory Values Over Time
Changes over time in laboratory values were not calculated.
12.4.2.2 Individual Subject Changes
Subjects with abnormal laboratory values that were not present at screening are shown in Table 12-4 (hematology), Table 12-5 (chemistry), and Table 12-6 (urinalysis).
Table 12-4: Subjects with New Abnormal Hematology Laboratory Evaluation Results Post- Dose (Single- and Multi-Dose Populations)

Note: Values were only included if they were normal before study drug dose and became abnormal after study
drug dose. Abnormal values at baseline were not included,
a. 'Values indicate the study period and dose the subject was receiving at the time of the event.
Source: Appendix 16.2, Listing 16.2.8. i
Table 12-5: Subjects with New Abnormal Chemistry Laboratory Evaluation Results Post- Dose (Single- and Multi-Dose Populations)

CK creatine kinase; TSF! = thjroid-stiwulating hormone
Note: Vaiues were only included if they were normal before study drug dose md became abnormal after study drug dose. Abnormal vaiues at baseline were not included,
a. I ndicates the study period and dose the subject was receiving a! the time of the event
Source: Appendix 16.2, listing 16.2.8.2
Table 12-6: Subjects with New Abnormal Urinalysis Laboratory Evaluation Results Post- Dose (Single- and Multi-Dose Populations)

Note: Values were only included if they were normal before study drug dose and became abnormal after study- drug dose. Abnormal values at baseline were not included,
a. indicates the study period and dose the subject was receiving at the time of the event.
Source: Appendix ! 6,2, Listing 16.2.8.3
12.4.2.3 Individual Clinically Significant Abnormalities
No abnormal clinical laboratory results were recorded as TEAEs.
12.5 Vital Signs, Physical Examinations, and Other Observations Related to Safety
12.5.1 Vital Signs
No abnormal vital signs were reported by the PI (Appendix 16.2, Listing 16.2.8.4) or were recorded as a TEAE (Appendix 16.2, Listing 16.2.7.1).
12.5.2 Physical Examination
Abnormal HEENT results are discussed in Section 12.5.3.
Three subjects had abnormal physical examination results related to the PK blood sample venipuncture site. None of these results were considered clinically significant.
• Subject 522-35 (1200 μg) - Period 2, Day 5: Slight phlebitis to left antecubital site
• Subject 522-38 (1200 μg) - Period 2, Day 5: Tenderness/soreness on left antecubital area
• Subject 522-51 (1200 μg) - Period 1, Day 4: Slight tenderness left AC IV site
Except for those related to HEENT or the PK blood sample venipuncture site, no other abnormal physical examination results were reported (Appendix 16.2, Listing 16.2.4.3.1).
12.5.3 Nasal Endoscopic Examination
No ENT nasal endoscopic examination findings were reported at Screening (Appendix 16.2, Listing 16.2.4.3.2).
Two subjects had ENT examination findings on Day 4 in Period 1 that were interpreted as clinically significant by the PI: Subject 522-37 (600 μg) had slight rhinorrhea and Subject 522-53 (1200 μg) had mild erythema to the left nostril mucosa.
All abnormal findings in basic ENT examinations are presented in Table 12-7.
Table 12-7: Subjects with Abnormal Basic Ear, Nose, and Throat Examination Results (Single- and Multi-Dose Populations)

CS - clinically significant; ENT ·- ears, nose, and throat; NCS -~ not clinically significant; TEAE - treatment
emergent adverse event
a. Indicates the study period and dose the subject was receiving at the time of the event,
h. A TEAE of rhinorrhea (.mild, possiblv related to studv medication} was reported on Day 2 and resolved on
Day 5.
c. A TEAK of nasal mucosal disorder (mild, possibly related to study medication) was reported on Day 3 and
was ongoing at study completion.
Source: Appendix 16.2. Listing 16.2 4.3.2. Appendix 16.2, Listing 16.2 7 1
12.6 Safety Conclusions
There were no deaths, SAEs, or AEs leading to discontinuation during the study. Most TEAEs were in the system organ classes of general disorders and administration site conditions and
respiratory, thoracic, and mediastinal disorders (nasal related events). Most TEAEs were mild in severity and were unlikely or not related to study medication.
In Period 1, a total of 11 of 24 subjects (45.8%) experienced at least 1 TEAE. Those experienced by more than 1 subject were catheter site erythema (experienced by 3 of 24 subjects [12.5%]) and catheter site hemorrhage, catheter site inflammation, dizziness, and nasal congestion (each experienced by 2 of 24 subjects [8.3%]).
The majority of TEAEs in Period 1 were of mild severity. A total of 10 of 24 subjects (41.7%) experienced mild TEAEs. Only 1 of 24 subjects (4.2%) experienced moderate TEAEs. Subject 522-53 (1200 μg) experienced headache and dizziness of moderate severity on Day 1. Both TEAEs resolved without treatment the next day and were considered as unlikely to be related to the study drug.
In Period 2, a total of 4 of 8 subjects (50.0%) experienced at least 1 TEAE. General disorders and administration site conditions occurred in 2 of 8 subjects (25.0%). The TEAEs that were experienced by 1 of 8 subjects (12.5%) each were catheter site pain, catheter site phlebitis, dyspepsia, headache, and rhinalgia.
The majority of TEAEs in Period 2 were of mild severity. Overall, 3 of 8 subjects (37.5%) experienced mild TEAEs. Only 1 of 8 subjects (12.5%) experienced a moderate TEAE. Subject 522-29 experienced an increase in headache intensity of moderate severity on Day 2. The TEAE resolved without treatment 2 days later and was considered as possibly related to the study drug.
A total of 6 subjects experienced adverse reactions (defined as TEAEs considered possibly, probably, or definitely related to study medication) during the study; 4 subjects in Period 1 and 2 subjects in Period 2. All adverse reactions were considered possibly related to study medication. Subject 522-29 (Period 2, 600 μg) had 2 adverse reactions of headache that were each recorded as a TEAE: a mild headache that increased in severity to moderate.
A total of 6 subjects experienced adverse reactions during the study: 4 of 24 subjects (16.7%) in Period 1 and 2 of 8 subjects (25.0%) in Period 2. All adverse reactions were considered possibly related to study medication. In Period 2, Subject 522-29 (1200 μg) had 2 adverse reactions of
headache that were each recorded as a TEAE: a mild headache that increased in severity to moderate.
There were no TEAEs associated with clinical laboratory tests or vital signs. Two subjects had ENT examination findings that were interpreted as clinically significant by the PI on Day 4 in Period 1: Subject 522-37 (600 μg) had slight rhinorrhea and Subject 522-53 (1200 μg) had mild erythema to the left nostril mucosa.
13 DISCUSSION AND OVERALL CONCLUSIONS
13.1 Discussion
In this phase 1, 2-period, randomized study of TBS-2 in 3 cohorts of subjects: Cohorts 1, 2, and 3 in Period 1 (single-dose) and a multiple-dose group in Period 2, a total of 24 healthy women received intranasal 600 μg, 1200 μg, or 1800 μg TBS-2 to evaluate bioavailability through pharmacokinetic profile and the safety of TBS-2.
Study results demonstrated that the mean baseline concentrations were within the normal range for all analytes.
In both treatment periods, the free and total testosterone had the clearest demonstration of TBS-2 administration-related concentration increase. A more modest administration-related increase in DHT concentration was observed. The estradiol and SHBG concentrations had post-dose curves that overlapped with the baseline pre-dose concentrations for the same dose.
TBS-2 was rapidly absorbed after dose administration. Peak concentrations of free and total testosterone were achieved within 15 to 45 minutes.
For the most part, the observed mean concentrations of all 5 analytes were within their normal ranges. The only exceptions were free and total testosterone peak concentrations that exceeded the ULN (upper limit of normal) during the first 2 hours, for 1.5 and 1.75 hrs respectively, post 1800 μg dose in Period 1 and for 1.0 h and 1.5 hrs respectively after multiple dosing in Period 2. Testosterone concentrations exceeding the upper limit of the physiological range for a limited amount of time following dosing can be expected due to the peak and trough PK profile of TBS- 2, and the endogenous nature of testosterone.
In Period 1, mean uncorrected concentrations returned to pre-dose values after 12 hours for the 600 μg dose and after 16 hours for the 1200 μg dose. For the 1800 μg dose, mean uncorrected concentrations for both free and total testosterone were consistently decreasing up to 20 hours post-dose, but demonstrated an increase at 24 hours similarly to the two other doses. The rise in concentrations could be due to an overlap with the diurnal increase in testosterone levels at about the 24 hour time point.
Mean uncorrected free and total testosterone concentrations at 24 hours after dosing were comparable to pre-dose mean concentrations, with little difference across the three doses.
During Period 2, it was also noted that the trough values for free and total testosterone at 24 and 48 hours were atypically higher than those at other time points, in line with the reported diurnal variations of testosterone levels. 11 ' 12 ' 13
A diurnal variation in mean testosterone levels can also be seen at 24 hours but may not be apparent at 0 hours in baseline profile likely due to the pattern in testosterone levels during the menstrual cycle that demonstrates a consistent increase towards midcycle from the lowest concenrations at the start of the period (at Day 1 or 2, when the baseline levels were obtained).14
Mean Cavg values for free and total testosterone were well within the normal range for women.
Dose proportionality was not rejected for AUC0-8, AUC0-24, and AUCO-t for free testosterone. Cmax values of free and total testosterone also increased with each dose increase, but did not demonstrate statistical significance. None of the PK parameters of any of the other analytes (DHT, estradiol, and SHBG) passed the statistical test for dose proportionality.
Accumulation analysis did not detect substantial evidence of accumulation for any analytes.
Additionally, equivalence between the single- and multiple-dose profiles in the 3 subjects who participated in both profiles was not rejected for AUC0-8 and AUC0-24 of free testosterone and for AUC0-8 of total testosterone, also suggestive of minimal accumulation. Equivalence was rejected for AUC0-24 of total testosterone, which can be explained by possible perturbation of the data by the diurnal variation in testosterone levels around 24 and 48 hours post-dose. Since there was no accumulation at 8 and at 16 hours, it would appear logical that the apparent
significant difference between available total testosterone at 24 hours was possibly due to the diurnal variation in testosterone levels.
13.2 Overall Conclusions
13.2.1 Pharmacokinetic Conclusions
Results from the study indicate that the free testosterone concentrations and AUCO-8, AUCO-24, and AUCO-t parameters were dose proportional and the total testosterone concentrations and parameters were clearly reflective of the dose strength. Other analyte concentrations were less changed with the dose strength in the order of DHT with a minor change to estradiol with less and finally SHBG with the least change.
No substantial evidence of accumulation was detected for all analytes following multiple dose administration.
Additionally equivalence between the single- and multi-dose profiles was not rejected for free and total testosterone, also suggesting little accumulation.
Although peak levels of serum free and total testosterone exceeded the upper level of normal for a limited amount of time following the single administration of the highest dose strength and after the multiple dosing, the observed mean concentrations of these two and 3 other analytes were well within their normal ranges for all dose strengths.
13.2.2 Safety Conclusions
There were no deaths, SAEs, or AEs leading to discontinuation during the study. Most TEAEs were in the system organ classes of general disorders and administration site conditions and respiratory, thoracic, and mediastinal disorders (nasal related events). Most TEAEs were mild in severity and were unlikely or not related to study medication. TBS-2 was safe and well tolerated.
14 TABLES, FIGURES, AND GRAPHS REFERRED TO BUT NOT INCLUDED IN THE TEXT
14.1 Demographic and Baseline Characteristics Summary Tables
Table 14.1.1a Analysis Population and Subject Disposition (Population: All Subjects)
Table 14.1.1b Analysis Population and Subject Disposition (Population: Multi-Dose) Table 14.1.2a Demographics (Population: Single-Dose)
Table 14.1.2b Demographics (Population: Multi-Dose)
Table 14.1.3.1 Medical History (Population: Single-Dose)
14.2 Efficacy
Not applicable
14.3 Safety Data
14.3.1 Safety Tables
Table 14.3.1.2a Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term (Population: Single-Dose)
Table 14.3.1.2b Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term (Population: Multi-Dose)
Table 14.3.1.4a Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Severity (Population: Single-Dose)
Table 14.3.1.4b Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Severity (Population: Multi-Dose)
Table 14.3.1.5a Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Study Drug Causality (Population: Single-Dose)
Table 14.3.1.5b Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Study Drug Causality (Population: Multi-Dose)
Table 14.3.2.2a Incidence of Serious Adverse Events by System Organ Class and
Preferred Term (Population: Single-Dose)
Table 14.3.2.2b Incidence of Serious Adverse Events by System Organ Class and
Preferred Term (Population: Multi-Dose)
Table 14.3.2.4a Incidence of Adverse Reactions by System Organ Class and Preferred
Term (Population: Single-Dose)
Table 14.3.2.4b Incidence of Adverse Reactions by System Organ Class and Preferred
Term (Population: Multi-Dose)
14.3.2 Narratives of Deaths, Other Serious, and Other Significant Adverse Events
Not applicable.
14.4 Pharmacokinetics Data
Table 14.4.1a Pharmacokinetics: Analyte Plasma Concentrations (PK Population: Sin
Dose)
Table 14.4.1b Pharmacokinetics: Analyte Plasma Concentration Summary (PK
Population: Multiple Dose)
Table 14.4.2a Pharmacokinetics: Analyte Parameter on Baseline Corrected Data
(ConcBC) (PK Population: Single Dose)
Table 14.4.2b Analyte Pharmacokinetic Parameter Summary for Multiple Dose Profile
(Dose of 1200 μg) (Population: Pharmacokinetic)
Table 14.4.2c Pharmacokinetics: Analyte Parameter on Uncorrected Data (ConcBLQ)
(PK Population: Single Dose)
Table 14.4.3 Dose Proportionality Analysis (Population: Single-Dose)
Table 14.4.4 Analysis of Variance for Some PK Parameters (Population: Single-Dose) Table 14.4.5 Paired T-Test Results for PK Parameters AUCO-8 and AUCO-24
(Population: Subjects of 1200 μg in Both Periods 1 and 2)
Figure 14.4.1a Individual Analyte Plasma Concentrations (PK Population: Single Dose) Figure 14.4.2a Individual Analyte Plasma Concentrations (PK Population: Multiple
Dose)
Figure 14.4.2b Individual Analyte Plasma Concentrations (Log y-axis Scale) (PK
Population: Multiple Dose)
Figure 14.4.3.1a Pharmacokinetics: Mean Observed Analyte Concentrations (Uncorrected)
(PK Population: Single Dose)
Figure 14.4.3.1b Pharmacokinetics: Mean Observed Analyte Concentrations (Log y-axis
Scale) (PK Population: Single Dose)
Figure 14.4.3.2a Pharmacokinetics: Mean Corrected Analyte Concentrations (PK
Population: Single Dose)
Figure 14.4.3.2b Pharmacokinetics: Mean Corrected Analyte Concentrations (Log y-axis
Scale) (PK Population: Single Dose)
Mean Analyte Plasma Concentrations (PK Population: Multiple Dose) Protocol TBS-2-2011-01 Page 91 of 92
Individual Analyte Plasma Concentrations (Log y-axis scale) (PK Population: Multiple Dose)
Pharmacokinetics: Analyte Parameter Summary (PK Population: Single Dose)
Spaghetti Concentration Plots with Mean for Analyte Plasma
Concentrations (PK Population: Multiple Dose)
Free, Total and Dihydro- Testosterone Concentrations in Subjects Receiving 1200 mg Dose in Single and Multiple Dose Cohorts (PK Population: Subjects 522-28, 522-35, and 522-38)
Pharmacokinetics: Accumulation Estimate by Linear Regression on Trough Concentrations (PK Population: Multiple Dose)
Table 14.1.1a
Analysis Population and Subject Disposition
Population: All Subjects
Cohort i Cohort 2 Cohort 3
600 moo 1S00 m¾q 18·¾ meg Tola! !,u = a? { = 8} :'N = si
Signed intotmed Consens form S6 Sens-ert Failure 32 Single-Dose Poptiiaiion 3 (100%} 8 (100%} 8 (100%) 24 (100%) Compiled S5udy Period 1 a (ioo%) e (100%) 8 (100%) 24 (100%) Wising to Participate in Study mctj 2 8 (100%) 7 (87.5%) S3 (91. ? ) Early Ter ination of Study Period 0 0 0 0
Note: All the percentages are out of the number of subjects in Single-Dose population within each Cohort.
Reference Listing(s): 16.2.1.1, 16.2.1.2a
Table 14.1.1b
Analysis Population and Subject Disposition
Population: Multi-Dose
TcsaS { = 8)
Subjects Continued on Study Period Ξ S
Completed the Study Period 2 8 {10D¾¾)
Earfy Termination or Study Period 2 0
Note: All the percentages are out of the number of subjects who continued on study period 2.
Reference Listing(s): 16.2.1.2b
Table 14.1.2a
Demographics
Population: Single-Dose
Cohori 1 Cohort 2 Cohort 3
690 meg 1200 it: eg ■J80O meg fotaf
Category or Statistics ( = 8} (N = 8! = 3) i = 24)
Ethnicity
Hispanic or Latino 2 (250%} 5 (82.5%) 4 (50.0%} 1 1 (45.8%) Noi Hispanic of Latino 6 (75.0%) 3 (37.5%} 4 {50.0%) 13 (54 2%)
Race h]
Anwtcar fKJian or Aiaa a Native/White 0 0 1 (12.5%} 1 ( 4.2%) Asian 0 0 0 0
8facK or African American 1 (12.5%} 2 (25.0%) 1 02.5%} 4 ί.16.7%} or Pacific i Q 0 0 Q
7 (87 5%} 6 (7S.0¾) 6 (75.0%} 19 (79.2%)
Other 0 0 0 0
Gender
Femaie 8 (100%) 8 (100%} 8 (100%) 24 (100%)
Age (Yrs)
n 8 24
Mean (SO) 32.5 (5.73} 29.8 (4 43) 27 (S 57) 29.8 (6.88} Median 34 30 27 30 Mi«, Max 22, 33 24, 36 IS, 39 8. 39
Height (cm)
n 8 24
Mean (SD) 165.5 (6.74) 160,94 (6.71 1) 16-6.63 (3,249) 161.02 (6,667)
163 161 15S 160 n, Max 158.0, -530.0 150 0, 173.0 162.0. 160.0 -150.0, 180.0
[1] Subjects could select more than one race category.
Note: The percentages are out of the total number of subjects in each Cohort.
Reference Listing(s): 16.2.4.1
Table 14.1.2a
Demographics
Population: Single-Dose
Cohort 1 Cohort 2 Cohort 3
Parameter/ 600 meg 1200 meg 1800 meg Totai Category or Statistics (N = 3) (N = 8} (N = 8} (H = 24} e¾hi (kg)
n e 8 8 24
Mean (SO) 63 43 (12.71 1} 88.8 (7.23?) 80.85 (12 823} 6836 (11.378) Median 66-6 68.6 57.05 66.6
Min. !te 54.4, 95.5 60 9. 844 46.2. 77.3 48. ¾ 95.5
(Kg/m25
8 s 24
(SDi 25.3 (3.689} 26.8 (4.047) 24.73 (4.697} 25.61 (4,079}
24.6 25.5 24.2 24.7 20.3, 313 23,0, 34,1 3,2, 30. 19.2, 34.1
SMI (Category)
Underweight ( 18.S) 0 0 o
orma! (18.6-24.9;. 5 (62,5%) A (50.0%} (50.0%) 13 (54.2%} Overweight (250-29.9) 2 (25.0%) 2 (25.0%; 3 (37,5%) 7 (28.2%) Obese i °¾) 1 (12.5%) 2 (25.0%; 1 (12 5%) 4 (18.7%)
[1] Subjects could select more than one race category.
Note: The percentages are out of the total number of subjects in each Cohort.
Reference Listing(s): 16.2.4.1
Table 14.1.2b
Demographics
Population: Multi-Dose
Parameter/ Tola! Category or Statistics M = 8)
Etftnfcly
Hispanic or latino 3 (37.5%) Not Hispanic or latino 5 (62.5%)
Race m
American^ ndiart or Alaska NatheSWhHe 1 (125%)
0
Biack or Aftfcar* American 2 (250%) ative Hawaiian or Pacific isiarnJer 0
5 (62.5%)
Other 0 Gender
6 (100%)
Age <Yrs}
n s
: (SD) 30.3 (648) an 30
Mifi. Max 21 , 39
Height (cm)
n s i (S0> 160.8 (3.99)
161 5 152, 165
[1] Subjects could select more than one race category.
Note: The percentages are out of the total number of subjects in each Cohort.
Reference Listing(s): 16.2.4.1
Table 14.1.2b
Demographics
Population: Multi-Dose
Category or Sialics
Weight ; kg)
a 8
Mean (SD) 6113 (0.81$) Median 62,85 Mm, Max 4S 2, 75 5
BMS QtgtmZ)
3
Mean ;SD} 23.65 (3.454) Median 24.25 m, Max 192.. 29 5
BM! (Cafegoty)
Umferweight («13.5} 0 Uorma (1&5-24.9) 6 {75.0%}
Overs ght (26.0-29,$) 2 (2S.0%; Obese =305 0
[1] Subjects could select more than one race category.
Note: The percentages are out of the total number of subjects in each Cohort.
Reference Listing(s): 16.2.4.1
Table 14.1.3.1
Medical History
Population: Single-Dose
Cohort 2 Co ort s
( 200 meg ; { 1800 mca> Total
5> (* : = 24)
Any Currently Any Cu nc Any Currently Any Currently
Body System History Active History Active History Active History Active
Head, eyes, ears, nose, throat 1 (12.5%) 0 1 {12 5%) 0 2 (25.0%) 0 (16 7%) 0
Respiratory System 0 0 1 (12.5%) 0 3 (37 5%) 0 4 (16.7%) 0
Muscutostotetet System 3 ί375%> 0 2 (25.0%) 0 2 (25 0%} 1 (1.2.5%) 7 (2S.2%) 1 ( 4.2%) integumentary Sysiem(skin. hair, nails) 0 ■3 2 {25.0%} 2 (25.9%; 2 (25.0%) 2 {25.0%; 4 (16.7%.) 4 (16.7%;
Gastrointestinal System 2 (25.0%) 1 (12.5%; 1 (12.5%) 0 1 (12.5%) 1 (12.5%; 4 (16.7%) 2 ( 8 3%)
Genitourinary System 2 (25.0%) 0 4 ίδθ 0%> 4 {50.0%; 3 (37.5%) 3 (37 5% l 9 (37 5%) 7 (29.2%; ervous System 5 (625%} 5 (62-5%) 6 (75.0%) 6 (75.0%} 4 (50 0%} 4 (50.0%) 15 (62 5%) 15 (62.5%;
Endocrine System 1 (1Z5%) 0 0 0 0 0 1 ( 4.2%; 0
Lymphatic System 0 0 0 0 0 0 0 0 immundogscal Sy stern 0 0 0 0 0 0 0 0
Circuiatory System 0 0 0 0 0 0 0
Psyc iatrtcWeuroiogi ai System 1 ( 12-5%) 0 0 0 1 (12.5%) 0 2 ( 8.3%) 0
Allergic Coodsons 1 {125%) 1 02.5%; 0 0 4 (50 0%) 3 {37.5%; 5 (20.8%) (16 7%;
Note: The percentages are out of the total number of subjects in each Cohort.
Reference Listing(s): 16.2.4.2
Table 14.3.1.2a
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term
Population: Single-Dose
Cohort 1 Cohort 2 Cohort 3
S stem Organ Class/ 60*3 srsco ■J 200 moq 1300 mca Totel
Preferred Term (MeflDRA) (N = 8) (N « 3) (N = 24;
Totel Num er of Sutsecis wish at !easS or¾e TEAE 4 (50.0%) 4 (500%) 3 (37.5%) 11 (45.8%)
GasiroiriestinaS disorders 1 (12.5%) 0 0 1 ( 4.2%)
Abdomiret peta upper 1 ( 12.5%) 0 0 1 ( 4,2%)
Genera! disorders ancJ administ ation ite conditions 3 (37,5%) 2 (26.0%) 2 (25,9%) 7 (29.2%)
Catheter site erythema 1 Π25%) 1 (12.5%) 1 (12 5%} 3 (12.5%)
CaSheier sise haemormage 0 0 2 (25,0%) 2 ( 3%)
Catheter site inflammation 2 (25,0%) 0 0 2 ( 8 3%)
Catheter site pain 0 1 (12.5%) 0 1 ( 42%) infections and infesiattons 1 (12,5%) 0 0 1 ( 4.2%)
Vulvitis 1 ( 2,5%) 0 0 1 ( 4,2%)
Nervous system disorders 0 1 (12.5%) 1 (12,5%ί 2 ( 8,3%)
Dizziness 0 ΐ (12.5%) 1 (12 5%) 2 ( 83%)
Headache 0 5 (12 5%) 0 1 ( 2%)
Respiratory, thoracic and me_fes6nat disor ers 1 (12 5%) 3 (37 5%) 1 (12.5%) 5 (20.8%)
Hasal congestion 0 2 (25.0%) 0 2 ( 8 3%)
Hasa! mixsosal disorder 0 1 (12.5%) 0 1 ( 42%)
Rh!naigla 0 0 t (12,5%) 1 ( 4,2%) hinorrhoea 1 (12.5%) 0 0 1 ( 4.2%)
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once.
Reference Listing(s): 16.2.7.1
Table 14.3.1.2b
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term
Population: Multi-Dose
Svs&m Otoan Ctassf Totsi
P;«f6E!«d Term ( SKSORA m = B)
Total umber of Subj cts with at ieast one T£A£ 4 (500¾!
Gaarointestinai aiswders {12,5%;
Dyspepsia 1 (12.5%;
General disorders and administration site conditions 2 (250%)
Catheter site 1 {12,5%;
Gstftet 8Γ Si e :Bfc>: :S 1 (15 5%!
Nervous s stem disorders 1 (12 5%i
Headache 1 (12.5%;
Respiratory, thoracic and metiasitnal diso ders 1 (12.5%;
Rhinaigia 1 {12,5%;
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once.
Reference Listing(s): 16.2.7.1
Table 14.3.1.4a
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Severity
Population: Single-Dose
Cohort 1 Cohort 2 Cohort 3
Body System/ 600 meg 200 met! 1800 meg Tosai
Prefe red Term (MedDRA) Severity -·· ¾ (N = 8) (N = S) (N = 24;
Num er of Subjects with at least one TEAE Overall 4 (50.0%) 4 (50.0%) 3 (37 5%) 11 (45.8%)
!V!iid 4 (55.0%) 3 (37.5%) 3 (37.5%) 10 (417%)
Moderate 0 1 (12.5%) 0 1 ( 4.2%}
Severe 0 0 0 0
Gastrointestinal disorders Overall 1 (12.5%) Q 0 1 ( 4.2%}
! Ssia 1 ( 2 5%) a 0 1 { 4.2%}
Moderate 0 Q 0 0
Severe 0 0 0 0
Abdomtnai pain upper Overall 1 (12.5%) a 0 1 ί 4.2%}
1 (12.5%) 0 0 1 ί 4.2%}
Moderate 0 0 O 0
Severe 0 0 0 0
G«neraf olsorders and adrninsiraSon sfte conditions Overall 3 (37,5%) 2 (25.0%) 2 (25.0%) 7 (2θ,2%)
Mitt 3 (37.5% ) 2 (25.0%) 2 (25.0%) 7 (2S.2%5
Moderate 0 0 0 0
0 Q 0 0
Catheter Overall 1 (12 5%) 1 (12 6%) 1 (12.5%) 3 (12.5%}
<s 1 (1 .5%) 1 (12.5%) 1 ( 2 5%) 3 (12.5%}
Moderate 0 0 0 0
Severe 0 0 0 0
Cashetw site haemorrhage Ovetafi 0 0 2 (25.0%) 2 ( 3.3%} ild 0 0 2 (250%) 2 ί s.3%; oderate 0 0 0 0
0 0 0 0
Catheter site inflarrsmaiion OveiaR 2 (250%) 0 0 2 { 3.3%}
Mild 2 (25 Q¾) 0 0 2 ( 8 3 %.'i
Moderate 0 0 0 O
Severe 0 0 0 0
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once with the greatest severity.
Reference Listing(s): 16.2.7.1
Table 14.3.1.4a
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Severity
Population: Single-Dose
Cohort 1 Cor>of> 2 Cohort 3
Body System/ 600 meg 1200 rr.cq 1809 meg Tola!
Preferred Term (MetiD A} Severity i = 3) in = 8} (N = $} ' (N = 24)
Catheter site ain Overs!! D 1 (12.5%) 0 '· ( 4.2%) mn 0 1 (1.2.5%) 0 1 ( 4.2%)
Moderate 0 0 0 0
0 0 0 0
Infections and irtfesiasorts OveraH 1 (12.5%} 0 0 1 ( 4.2%) ms 1 (12.5%; 0 0 1 ( 4.2%)
Moderate C' 0 0 0
Severe 0 0 •3 Q
VuiVitiS Overs!! 1 {12.5%; 0 0 1 ( 4.2%) iid 1 (12 5%} 0 0 1 f 4.2%)
Moderate 0 0 0 0
Severe 0 0 0 0 ervous sy em disorders OveraSf 0 1 (1.2,5%.! 1 {12.$%} 2 ( 8,3%) ms 0 0 1 (12,5%) 1 ( 4.2%)
Moderate 0 1 (12.5%) 0 1 ( .2%)
Severe Q 0 •3 0
Dizziness Overs!! 0 1 (12 5%) 1 (12.5%) 2 ( 8.3%)
Miid D 0 1 (12.5%) 1 ( 4.2%)
Moderate 0 1 (12,5%) 0 1 f 4.2%)
Severe 0 0 0 0
Headache OveraSf 0 1 (1.2,5%! •3 1 f 4.2%) ma Q 0 3 0
Moderate 0 1 (12.5%) 0 1 ( 4.2%)
Severe 0 0 0 0
Respiratory, thoracic and mediastinal disorders O ers!! 1 (12.5%) 3 (37.5%) 1 (12.5%) 5 (20,3%)
MiSd i12 5%5 3 (37.5%) 1 (12.5%} 5 (20.8%)
Moderate 0 0 0 0
Severe 0 0 0 0
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once with the greatest severity.
Reference Listing(s): 16.2.7.1
Table 14.3.1.4a
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Severity
Population: Single-Dose
Cohort 1 Cohort 2 Cohort 3
Body System/ 600 meg 1800 meg Total
Preferred Term (MedDRA) Severity m = &) (N = Si (N = Si i = 24)
Nasal congestion Overall Q 2 (25 0%) 0 2 ( 8 3%) rn 0 2 (25.0%) 0 2 ( 8.3%) feder te 0 0 0 O
Severe 0 0 0 0
Nasal mucosal disorder Overall 0 1 ( 12.5%) 0 1 { 4.2%)
WM 0 1 (12.5%) 0 1 ( 4 2%)
Moderate 0 0 0 0
Severe 0 0 0 O hinaSgia Overall 0 0 1 (12.5%) 1 i 4 2%)
MM 0 0 1 (12.5%) 1 ί 4.2%)
Moderate 0 0 0 0
Severe 0 0 0 0 inorrhoea Overall 1 (12 5%) 0 0 1 ( 2%)
MiW 1 (12.5%) 0 0 1 ( ,2%) f lod rai 0 0 0 0
Severe 0 0 0 0
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once with the greatest severity.
Reference Listing(s): 16.2.7.1
Table 14.3.1.4b
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Severity
Population: Multi-Dose
Booy System/ Total
Preferred Term (MedDRA) Severity ;M = ¾
Number of Subjects with at least one TEAE Overall 4 (50.0%;
MM 3 (37.5%}
Moderate 1 (12.5%;
Severe 0
Gasiroinsesiinai disorders Overaft 1 ( 2.$% J
MM 1 (12.5%}
Moderate 0
Severs 0
D spepsia Overall 1 f12.5¾%>
MM 1 (12.5%}
Moderate 0
Severs 0
Genera* disorders and administration site conditions O er^ii 2 (25.0%}
Mm 2 (25.0%5
Moderate 0
Severe 0
Catheter site pain Overall 1 (12.5%}
MM 1 (12.5%; oderate 0
Severe 0
Catheter s¾e pftsebii Is Overall 1 (12.5%5 urn 1 (12.5%; ode aie 0
Severe 0
Nervous system tiiswtefs Overall 1 (12.5%;
Mild 0
Moderate 1 (12.5%5
Severe 0
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once with the greatest severity. Reference Listing(s): 16.2.7.1
Table 14.3.1.4b
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Severity
Population: Multi-Dose
Body SysSsm/ ToSai
Preferred Term (MedDRA) Severity {N = S!
Heartache OveraiS 1 m.m ms 0
Moderate 1 {12.5%} f
Respiratory, thoracic aod mediasit sis ( disor e s Gyeraii t (12.5%)

Severe 0
OveraiS 1 {12.5%}
Miid 1 {12,5%)
Moderate 0
Severe 0
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once with the greatest severity. Reference Listing(s): 16.2.7.1
Table 14.3.1.5a
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Study Drug Causality
Population: Single-Dose
Cohort 1 Cohort 2 Cohort 3
8o y System/ 600 mm 1200 meg 1800 meg Total
Preferred Term (Med OR A) Causaiiiy {H = 8} (N = 8) (N = 8) i ··· 24)
Number of Subjects with at !easi one TEAE Overall 4 (50.0%) 4 (S>.0% 3 (37.5% } 1 1 (45.3%)
Definite 0 0 0 0
Probable Q Q 0 0
Possibly 1 (12.5%) 3 (37.5%) 0 4 ( 6 7%) unBkeiy 2 5250%} 0 1 (12.5%) 3 (12.5%)
Unknow 0 0 0 O ot Related 1 (12.5%) 1 (12.5%) 2 (25.0%) 4 (16.7%)
Gasirointestinai disorders Overall 1 (12.5%) Q 0 1 £ 4.2%)
Definite 0 0 0 O
Probable 0 0 0 0
Possibly 0 0 0 0
Unlikely 1 (12.5%) 0 0 1 ( 4.2%)
Unknown Q Q 0 0 ot Related 0 0 0 0
Abdominal pain upper Overall 1 (12 5%} 0 0 1 ί 4 2%)
Definite 0 0 0 O
Probable 0 0 0 0
Possibly Q Q 0 0
Unlikely 1 (12.5%) 0 0 1 ( 4.2%)
Unknown 0 0 0 0
Net Related 0 0 0 O
Genera! disorders and administration sits conditions Overall 3 (37.5%) 2 (25,0%) 2 (25.0%) 7 £29.2%)
Definite Q Q 0 0
Pre sble 0 0 0 O
Possibly 0 0 0 0
Unlikely 0 0 0 0
Unknown 0 0 0 0
Hot Related 3 (37.5%) 2 ' 25 ø%) 2 (25.0%) 7 (20.2%)
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once with the most related term.
Reference Listing(s): 16.2.7.1
Table 14.3.1.5a
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Study Drug Causality
Population: Single-Dose
Coiwrl 1 Cohort 2 Cohort 3
Body System/ 600 mco 1200 mca 1800 mca Toisi
Preferred Term (MeflDRA) Causality (N = 8) ( = 8) ( = 24!
Caiheier site erythema Oman 1 (12.5%) t (12.5%) 1 (12.5%) 3 (12.5%)
Definite 0 0 0 0
Probable Q 0 0 0
Possibly 0 0 0 0
Unlikely 0 0 0 0
Unknown 0 0 0 0
Ncs Related 5 (12 5%} 5 (12.5%} 1 (12.5%} 3 (12.5%!
Catheser site aemorrhage Overall 0 0 2 (25.0%) 2 ( 8.3%!
Definite 0 0 0 0
Probable Q 0 0 0
Possibly 0 0 0 0
Unilkeiy 0 0 0 0
Unknown 0 0 0 0
Not Related Q 0 2 (25.0%) 2 ( 8.3%)
Catheser ate inflammation 2 (25.0%) 0 0 2 ( 8.3%!
Defin 0 0 0 0
Probable 0 0 0 0
Possibly 0 0 0 0
Unlikely Q 0 0 0
Unknown 0 0 0 0
Not Related 2 25.0%) 0 0 2 ( 8.3%)
Catheter sfts pain Overall 0 1 (12.5%) 0 1 ( 4.2%)
OeffnS* 0 0 0 0
Probable 0 0 0 0
Possi&V 0 0 0 0
Unlikely 0 0 0 0
Unknown 0 0 0 0
0 t (12.5%) 0 1 ( 4.2%!
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once with the most related term.
Reference Listing(s): 16.2.7. Γ
Table 14.3.1.5a
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Study Drug Causality
Population: Single-Dose
Cohort 1 Cohort 2 Cohort 3
Body Svefem/ 600 rrscq 1200 mco 1800 roco Total
Preferred Term (MedD A) Causafiiy (N = 8) (M = 81 (¾ = 3) (N = 24) infecsiorts and infestations Overall 1 (12.5%} 0 0 1 ( 4.2%)
Definit 0 0 0 0
Probable 0 0 0 0
Possib!y 0 0 0 0
UnftKef 1 (12.5%! 0 0 1 ( 4.2%)
U know 0 0 0 0
Not Related 0 0 0 0
Vul itis Overaif 1 (12.6%} 0 0 1 ( 4.2%)
Define 0 0 0 0
Probable 0 0 0 0 0¾¾i i>' 0 0 0 0
1 (12.5%} Q Q 1 ( 4.2%)
Unknown 0 0 0 0 ot Related 0 0 0 0
Nervous s stem dieor sets Overaif 0 1 (12.5%) 1 (12.5%} 2 ( 8.3%)
Definite 0 0 0 0
Probable 0 0 0 0
Possib!y 0 0 0 0
Unlikely 0 (12.5%) Q 1 ( 4.2%)
Unknown 0 0 0 0 f ot Related 0 Q 1 (12,5%) 1 ( 4.2%)
Oaziness Overall 0 1 (12.5%t 1 (12.5%} 2 ( 8.3%)
Oefs l e 0 0 0 0
Probable 0 Q Q 0
Possib!y 0 O O 0
UnfiKsfy 0 1 (12.5%) 0 1 ( 4.2%)
Unknown 0 0 0 0
Not Related 0 0 (I2.s%; 1 ( 4.2%)
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once with the most related term.
Reference Listing(s): 16.2.7. Γ
Table 14.3.1.5a
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Study Drug Causality
Population: Single-Dose
Cohort Cohort 2 Cohort 3
Sod-/ Svsisrn/ SCO rrico 1200 meg ■ 800 moo Total
Preferred Term (MedDRA) Causality (N = 8} ( = 8) ( = 8)~ {N = 24}
Headache Overall 0 1 (12.5%) Q 1 ( 4.2%}
Definite 0 0 0 0
Probable 0 0 0 0
Possfcfr 0 0 Q 0
Unlikely 0 1 (12.5%) 0 1 ( 4.2%)
Unknown 0 0 Q 0
Not Re!aieo" 0 0 0 0 espiraiory, thoracic and mediastinal disorders Overall 1 (12.5%} 3 (37.5%} 1 (12 5%} 5 (20.8%)
Definite 0 0 Q 0
Probable 0 0 0 0
Possibly 1 (12.5%) 3 (37,5%) Q 4 (16.7%)
Unlikely 0 0 1 (12.5%) 1 ( 4.2%)
Unkn wn 0 0 0 0
Not Re!aieo" 0 0 0 0
Nasal congesta Overall 0 2 (25.9%) 0 2 ( 8.3%)
Definite 0 0 Q 0
Probable 0 0 0 0
Possibly 0 2 (25.0%} 0 2 ( 8.3%}
Unlikely 0 0 Q 0
Unkno n 0 0 0 0
Not elated 0 0 Q 0
Naaa! irtijco&a! disorder Overall 0 1 (12.5%) 0 1 ( 4.2%)
Definite 0 0 0 0
Probable 0 0 Q 0
Possibly 0 1 (12.5%) 0 1 ( 4.2%)
Unlikely 0 0 0 0
Unknown 0 0 0 0
Not Related 0 0 0 0
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once with the most related term.
Reference Listing(s): 16.2.7.1
Table 14.3.1.5a
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Study Drug Causality
Population: Single-Dose
Cohort 1 Cohort 2 Cohort 3
Body System/ 600 meg 1200 mo 1800 i!tco Toiai
Preferred Term (HiiedDRA) CausaSt* ( i = 8} (N = 8) (N = B) W = ?4; umania Overall 0 0 1 (52.5%) I { 4.2%}
Definite 0 0 0 0
Probable Q 0 0 0
Posstt 0 0 0 0
UnSkeSy 0 0 1 ) 1 ( .2%)
Unknown 0 0 0 0
Hot Re!aisd 0 0 0 0
Rhsnormoea Overali •i (12.5%) 0 0 5 { 4.2¾)
Definite 0 0 0 0
Probable Q 0 0 0
Possibly 1 (12.5%) 0 0 1 ( 4.2%)
UnSkeSy 0 0 0 0
Unknown 0 0 0 0
Not Reiaisd 0 0 0 0
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once with the most related term.
Reference Listing(s): 16.2.7.1
Table 14.3.1.5b
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Study Drug Causality
Population: Multi-Dose
Body System/ Total
Preferred Term { edORA) Causal: S
Number of Streets wish at feast one TEAE Overall 4 50.0%)
Definite 0
P obata 0
Possibly 2 {25.0%}
Unlikely 0
Unk own 0
Not Related 2 {25,0%;
Gastrointestinal ds«K fers Overall 1 {12.5%}
Definite 0
Probable 0
PojSlbiy 0
Unlikely 0
Unknown 0
Not elated 1 {12.5%}
Dyspepsia Overall 1 i12.8%>
Definite 0
Probable 0
Possibly 0
Unlikely 0
Unknown 0
Not Related 1 (12.6%)
General disorders and admfr&tratton ssta conciiiier? Overall 2 {25.0%}
Defini e 0
Probable 0
Possibly 0
Unlikely 0
U k o n 0
Not Related
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once with the most related term. Reference Listing(s): 16.2.7.1
Table 14.3.1.5b
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Study Drug Causality
Population: Multi-Dose
SD<¾' System Total
Preferred Term {SSedORA} Causality (N = S)
Catheter site pain QveraS 1 (12.5%)
Define 0
Probable 0
Possi&i 0 ni-keiy 0
Unknown 0
5s!Qi Related 1 {12.5%)
Cai eter sife pntebftis Ove s* 1 (52.5%)
Definite 0
Probable 0
Possibly 0
Unlikel 0
Not Related t (12.5%)
Nervous system disorders Overall 1 (12.$%}
Definite 0
Probable 0
Possibly 1 (12 5% )
Unfikei 0
Π
Hot eSaie i 0
Headache Overs*! 1 (12.5%)
Definite 0
Probable 0
Possibly 1 (12.5%)
Unlikeiy 0
Unknown 0
Hot Related Q
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once with the most related term. Reference Listing(s): 16.2.7.1
Table 14.3.1.5b
Incidence of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term by Study Drug Causality
Population: Multi-Dose
Body Svsfem/
Preferred "
espiratory, ih ractc and mediastinal dfeorde Overall
Defsnise
Probable
Possi ly
Unknown
Not Related
inasgia Overall
Definite
Probable
Possibly
Unlikely
Note: TEAE = Treatment-emergent adverse event.
Subjects reporting more than one TEAE in each level (System Organ Class or Preferred Term) are only counted once with the most related term. Reference Listing(s): 16.2.7.1
Table 14.3.2.2a
Incidence of Serious Adverse Events by System Organ Class and Preferred Term
Population: Single-Dose
Ho SAE observed during siixiy Period 1.
Note: Subjects reporting more than one SAE in each level (System Organ Class or Preferred Term) are only counted once. Reference Listing(s): 16.2.7.1
Table 14.3.2.2b
Incidence of Serious Adverse Events by System Organ Class and Preferred Term
Population: Multi-Dose
No SAE o ser ed forir¾ stud Period 2.
Note: Subjects reporting more than one SAE in each level (System Organ Class or Preferred Term) are only counted once. Reference Listing(s): 16.2.7.1
Table 14.3.2.4a
Incidence of Adverse Reactions by System Organ Class and Preferred Term
Population: Single-Dose
Cohort 1 Cohort s Cohort 3
System Organ Ciassr 600 meg 1200 moo ■5800 meg Totai
Preferred Term (MedP A) S = 8)
Toia! Number of Subjects with at feast one Adverse Reaction 1 (125%) 3 {37.5%} 0 fie ?¾) Respiratory, thoracic and mediastinal disorders 1 ( 2,5%) 3 S3?.6%) 0 A (16.7%)
0 2 (25.0%) 0 2 ί β.3. } 0 1 (12.5%) 0
hinorrhoea 1 (1.2.5%) 0 1 { 4,2%;
Note: Adverse Reactions include AEs whose study drug causality fall into Definite, Probable or Possible categories reported on AE CRF page.
Subjects reporting more than one Adverse Reactions in each level (System Organ Class or Preferred Term) are only counted once with the most related term.
Reference Listing(s): 16.2.7.4
Table 14.3.2.4b
Incidence of Adverse Reactions by System Organ Class and Preferred Term
Population: Multi-Dose
System Organ Class; Total
Preferred Term ;svSedD Aj
Toiai Mumiserof Subjects with at ieast one Adverse Reaction 2 (25.0%)
fster ous system disof¾Jsrs 1 in )
Headache 1 {12.5%)
Respiratory, thoracis and mediastinal disorders 1 i« 5%i
Rhsnaigla 1 (12.5%)
Note: Adverse Reactions include AEs whose study drug causality fall into Definite, Probable or Possible categories reported in AE CRF page.
Subjects reporting more than one Adverse Reactions in each level (System Organ Class or Preferred Term) are only counted once with the most related term.
Reference Listing(s): 16.2.7.4
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (jug) Dose 1200 {μ ) Dose 1800 (f*g) Dose
Analyte Time ConcBase ConcBLQ ConcBC ConcBase ConcBLQ ConcBC ConcBase ConcBLQ ConcBC
(fir) [ng/dL) (ng/dL) (ng'dL) (ng/dL) (ng/dL) (ng/dL) (ne/ciL) (ng/dL) (ng/dL)
Free Testosterone -0.25 H 8 a 8 8 8 8 8 8 8
Mean 0.387 0.448 0.06S 0.281 0,346 0.OS6 0 , 280 0,314 0.043
SD 0.344 0.412 0.072 0.092 0.137 0.052 0.060 0,076 0.028 cv% 8S.0 91.8 110.7 32.7 3S.7 79.3 2 .3 24,0 66.2
Median 0.31 0.31 0.05 0.28 0.31 0.06 0.25 0.31 0.04 tein 0,15 0,18 0.00 0.16 0.20 0.00 0,22 0.19 0.00
Max 1.21 1.44 0.23 0.40 0.63 0.14 0.39 0.41 0.08
Seometric He an 0.309 0.35? 0.268 0.322 0.275 0.306
Free Testosterone 0.25 N 8 8 8 8 8 8 8 8 8
Mean 0.367 0.845 0.478 0.271 1.231 0,980 0.298 1 > 339 ,041
SO 0,340 0 , 754 0.431 0.088 0.674 0.644 0 , 086 0.80S 0.85B cv% 92.8 8:9.2 90.2 33.0 54.7 67.1 28.8 60,2 82.2
Median 0,27 0,56 0.29 0.26 ! .04 0.71 0.27 1.12 0.84 iJin 0, 14 0.33 0.14 0.15 0.47 0.32 0.20 0.65 0.16
1,19 2,84 1.45 0.38 2.43 2.07 0.45 2.81 2.53
Geometric Ntean 0.292 0.674 0,363 0-258 1,077 0,785 0,288 1,143 0,691
Free Testosterone 0,50 8 8 a 8 8 a 8 8 8
Mean 0,329 0.822 0.403 0.281 1.254 0.973 0.286 1.627 1.342 so 0.263 0.661 0.415 0.092 0,744 0,691 0.O81 0 , 694 0,720 cv% 80.0 80.4 84,2 32,7 59,3 71 ,1 28,2 42,7 53,6
Median 0,25 0.65 0.47 0.29 1.14 0.79 0.26 1.69 1.37
Kin 0,15 0,34 0.09 0.15 0.46 0.31 0.20 0.73 0.34
Wax 0,96 2.38 1.42 0.40 2.73 2.34 0.39 2.49 2,30
Geometric Wean 0.276 0.676 0.364 0.26S 1.077 0.782 0.276 1.487 1,135
Free Testosterone 0.75 « 8 8 8 8 8 8 8 8 8
Msan 0.346 0.731 0.385 0.258 1 , 193 0,935 0.270 1 , 48 1,479
SO 0,302 0,582 0.291 0.085 0.768 0.720 0.067 0.720 0.736 cv% 87.5 76.9 76,6 33,1 64.4 77.0 24.8 41 ,2 49.8
Median o,as 0,54 0.39 0.26 1.02 0.75 0.26 1.83 1.57
(Sin 0,13 0.32 0.05 0.14 0.43 0.29 0.18 0.78 0,40
BSax 1,06 2,03 G.S7 0.37 2.88 2.51 0.38 2.73 2.46
Geometric Mean 0.276 0.607 0.278 0,245 1,013 0.745 0,263 1,602 1,276
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
S00 [^g) Dose 120Q (fjg) Dose 1800 (pg) Dose
Analyte Tiae ConcBaae ConcBLQ ConcBC ConcBaae ConcBLQ ConcBC ConcBaae ConcBLQ ConcBC
(hr) (ng/dL) {ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL)
Free Testosterone .00 N 8 8 8 8 8 8 8 8 8
Mean 0.344 0,889 0.344 0.273 1.307 1,034 0.268 1.577 1.309
SO 0.31 ? 0.524 0.246 0.087 0.845 0.787 0.073 0.601 0.641 cv 92, £ 78.1 71.5 31.8 S4.6 78.2 27.1 38,1 43.0
Median 0,25 0.42 0.26 0.29 ,07 0,82 0.25 1 ,88 ,39
Min 0.11 0.30 0.10 0,17 0.36 0.19 0,20 0.79 0.37
Max 1.10 1 ,8S 0.75 0.41 2.68 2,27 0.41 £.48 2,27
Geometric Mean 0.270 o.se? 0.271 0.261 1,084 0.786 0.281 1.468 1.142
Free Testosterone 1.50 H 8 8 8 8 8 S 8 8 S
Sfeari 0.353 0.634 O.2S0 0.287 1.033 0,766 0,257 1,270 1.013
SO 0.318 o.sao 0.231 0.078 0,553 G,fi05 0.054 0.437 0.465 cv% 89.9 82,1 82,4 29.4 53,5 65.9 21.0 34,4 45.3
Median 0.27 0,38 0,17 0.27 0.9S 0,70 0.24 1.23 1.01
Bin 0.13 0.29 0,08 0.17 0.33 0,14 0.21 0,73 0,36
Max 1.11 1.82 0.71 0,38 2.12 1.75 0,37 1.98 1.72
Geometric Sean 0,280 0,512 0,213 0.256 0,906 0,61 0.253 1,204 0.907
Free Testosterone £.00 N 8 8 8 8 8 8 8 6 8
Mean 0 , 364 0.611 0.247 0.271 0,863 0.592 0.2S6 1,334 1,078
SO 0.321 0.51S 0,204 0.080 0.417 0.381 0,062 0.426 0.447
CV% 88.3 84.3 82.4 29,3 48.3 64.3 24.4 32,0 41.4
Median 0.28 0.42 0,19 0,29 0.81 0,50 0,25 1.25 1.00 win 0.16 0,31 0,07 0.17 0.35 0,17 0.18 0.78 0.41
Max 1.13 1.85 0.72 0.36 .77 1 ,42 0.37 .99 1 ,75
Geometric sssan 0 , 292 0.504 0.198 0.2S0 0,788 0.501 0.249 1,274 0.988
Free T&stasterone 4,00 N 8 8 8 8 8 8 8 8 8
Mean 0.338 0,878 0.240 0.275 0.826 0,351 0.252 0.977 0.726
SO 0,273 0.462 0.192 0.102 0,318 0.238 0.087 0,286 0,290 cv% 80,6 79,8 79, S 37.1 50,8 67,7 26.5 £9,3 40,0 aedisn 0.25 0,44 0.18 0.25 0,51 0,28 0.25 0,88 0,67
Kin 0.16 0.26 0,09 0,17 0.25 0,08 0,15 0.86 0,37
Max 0.98 1 ,67 0,69 0.46 1.17 0,82 0.35 1.35 1,06
Geometric Mean 0.2S1 0.480 0.196 0.259 0.558 0.285 0.243 0.942 0.673
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (jug) Dose 1200 (f/g) DOS8 1800 (i.<g) Dose
Analyte Jim ConcBase COOCBLQ ConcBC ConcBase ConcBLQ CDnoB0 CodeBase ConcBLQ ConcBC
(ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL)
Free Testosterone 6.00 u 8 8 8 B B 8 8 8 8
Stean 0.33S 0,434 0,158 0.263 0,48? 0.22S 0.260 0,889 0,629
SD 0.307 0.340 0,103 0.096 0,234 0.156 0.076 0,364 0.354
GV¾ 91.6 69.0 65.1 36,4 48,1 68.9 29.1 3S.8 56.3
Median 0.23 0.38 0.13 0,24 0.46 0.23 0.25 0.80 0.55
(Jin 0,13 0.19 0,06 0.15 0.19 0.04 0.16 0,44 0.18
Max 1 ,07 1.23 0,40 0.40 0.80 0,47 0.37 1.43 1.11
Geometric Mean Q.26S 0.418 0.138 0-247 0.435 0.167 0.2S1 0.827 0 , S30
Free Testosterone 8.00 N 8 8 8 8 8 8 8 8 8
Mean 0,346 0 , 82 0.136 0.256 0,475 0.219 0,268 0,812 0.544
SO 0.339 0.345 0,049 0.087 0,215 0.161 0.074 0,494 0 , 487
GV% 98.0 71.6 36.2 33.9 45.3 73.5 27.6 60,8 89.5
Median 0.26 0,36 0.13 0,27 0,50 0.19 0,25 0,65 0.42
SSin 0,10 0.24 0,06 0.16 0.19 0.03 0.20 0,32 0.13
Max 1,16 .89 Q.22 0.35 0.83 0,50 0,42 1.96 1.6S
Geometric Mean 0.264 0,413 0.127 0,242 0,429 0.160 0,260 0,718 0,417
Free Testosterone 12.00 N 8 8 8 8 8 8 8 8 8
Sean 0.376 0,448 0.090 0.259 0,378 0.119 0.243 0,823 0 , 387
SO 0.393 0.356 0,137 0,107 0.159 0,062 0.064 0,275 0.276
GV 104,6 79.5 151 , 8 41.3 42,0 52.2 26. 43,7 71.3
Median 0.25 0.27 0.04 0,24 0,40 0.14 0.22 0,63 0.36
Win 0,13 0.19 0,00 0,13 0.15 0.02 0.18 0.28 0.08
Ma 1,33 1,19 0,41 0,42 0.55 0,18 0,37 1.20 0.92
Geometric Mean 0.284 0,359 0,239 0.343 0.097 0,236 0.580 0,294
Fres Testosterone 16.00 N 8 8 8 8 8 8 8 8 8
»ean 0,370 0.406 0,047 0.273 0,350 0,077 0.25S 0,458 0,206
SO 0.349 0.3S2 0,040 0,102 0,145 0,046 0.G7 0,196 0. 77 cv% 94.4 89.2 86.2 37.5 41.5 60.1 28.9 42,9 8S.0
Median 0,24 0.30 0,05 0,30 0,40 0.08 0.25 0.46 0.17
(Sin 0,16 0.17 0,00 0,13 0.15 0.01 0,15 0.23 0.00
Max 1.21 1 ,27 0.12 0,39 0.52 0.13 0,39 0.89 0.57
Geometric Mean 0.291 0,324 0.254 0,318 0.058 0,246 0.426
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (PQ) Dose 1200 (fjg) Dose 1800 (fi ) Dose
Analyte Time CodeBase ConcBLQ ConcBC ConcBase ConcBLQ ConcBC ConcBass ConcBLQ ConcBC
(hr) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (rsg/dL) (ng/dL) {ng/dL} (ng/dL) (ng/dL)
Free Testosterone 48.00 H 0 8 0 0 8 0 0 8 0
Sfean 0.467 0.377 0.410
SD 0.336 0.1S1 Q.099
CV% 71 ,9 40.1 24.3
Median 0,36 0,33 0.41
Kin 0.16 0.19 0.27 fciax 1.21 0,63 0,5?
Geometric Mstn 0.389 0.3S1 0.399
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (jjQ) Dose 1200 {pg} Dose 18 30 (μ@) Dose
Analyte Time Coocsase ConcBLQ ConcBC eoncBass COfiOSLQ ConcBC ConcBase ConcBLQ ConcBC
(hr) (ng/dL) {ng/dL) {ng/dL) £ng/dL) (ng/dL) {ng/dL} {ng/dL) (ng/dL) (ng/dL)
Total Testosterone -0.25 8 8 8 8 8 8 8 8 8
Mean 25,513 30.138 5.213 19.113 23.538 4.425 23.138 24.350 2.S75
SO 10.001 13.486 3.375 6.641 10.193 3.959 S.7D6 7.019 2.193
CV% 39,2 44.7 74.3 34.7 43,3 89,5 37.6 28.8 73.7
Median 24. §0 27. ao 5.40 17.85 20.20 3.85 20.15 24.90 2: , 70 toin 12.50 17.10 0.00 1 ,30 13.30 0.10 15,90 17,00 0,00
Hax 45.10 58.00 12,80 29.00 41.10 12,10 39. SO 38.80 6.30
Seometric Mean 23.854 27,873 18.148 21,791 2,364 2 .920 23.552
Total Testosterone 0,25 N 8 8 8 8 8 8 8 8 8
Mean 23.538 59,213 35,575 13.300 79, 13 60.813 23,463 106,275 82.813 sa 9.792 31.795 25.397 6.989 34,655 33,857 9.554 6S.785 6 , 321 cv¾ 41.4 53.7 71.4 38,1 43.8 55.7 40,7 62.8 80.1
Median 21 ,60 43,70 28 , 15 16.50 74.55 48,15 20.90 80,90 54.45
Hin 11.oo 34.20 15.80 10.80 32.50 21.70 13.70 43,10 19.00
Hax 43.90 111 ,00 B6.70 28,50 153,00 133,00 41 ,60 218. GO 194.00
Geometric Mean 22.023 52,861 29-423 17.219 72.97S 53.751 21.968 89.101 60.284
Total Testosterone 0,50 Pi 8 8 8 8 8 8 8 8 8
Wear) 22.325 56.350 34.025 18.825 79,138 60.313 23.575 1 7,563 103.988
SO 7.153 22,970 18,464 6,483 34, 103 32.513 9,674 57.911 56 , 802 cv 32.0 40.8 54.3 34,4 43.1 53.9 41 ,0 45.4 54.6
Median 21.00 57,70 40.80 17,90 78.45 55.35 20,05 118. GO 102.50
Kin 12,40 27.10 8,00 11.00 32.90 21 ,90 14.20 47,50 24.30
Ka 36,20 92.20 56.00 29,80 144.00 123,60 39.00 210,00 193.10
Geometric Mean 21.379 52,023 26.212 17.883 72,712 53.097 21 ,934 1 4,524 87. 63
Total Testosterone 0.75 N 8 8 8 8 8 6 8 8 8
Mean 23.080 50,400 27.320 17.156 73,300 56.144 22.225 13 ,388 109.163
SO 9.925 19.973 14.408 6,385 31.883 3 .678 7,315 49.913 51.359 cv% 43,0 39.6 52.7 37.2 43,5 56.4 32.9 38.0 47.0
Median 20,50 51.85 34.28 17,20 71.SO 49,70 20.65 127,50 102.50
Hin §.84 26,60 5,30 9.65 29.30 19,65 14.00 49.10 24.30
(Sax 41.60 77.50 41.30 30.50 132.00 114.90 33.50 198 , 00 182,00
Geometric Mean 21.298 46.750 22.453 15.223 87,157 48.794 21.224 121,577 95 , 184
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (fjg) Otsse 1200 (jjg) Dose 1800 {μ ) Dose
Analyte Time ConcBase ConcBLQ COftcBO ConcBase ConcBLQ ConcBC ConcBase ConcBLQ ConcBC
(hr) (ng dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL} (ng/dL) (ng/dL) ( ng/dL) (ng/dL)
Total Testosterone 1 ,00 N 8 8 8 8 8 8 8 8 8
Mean 22.631 47.400 24.769 18,013 75.738 57.725 21 ,988 117,625 95.638
SD S.287 20.178 13.813 5.186 32. §95 31 ,575 7,403 43.964 45.152 cv% 41.0 42.6 55,8 28,8 43.6 54.7 33,7 37.4 47.2
Median 21 ,65 41 ,90 24,68 18.10 68.75 47,70 19.10 124.50 96.45
8.8S a? . 0 8.70 12.30 24.80 12.50 15.10 53.30 2? .20
Max 36.70 70.50 47.30 28.30 122.00 103.80 34.50 180.00 163.48
Seoisetric Mean 20.868 43,634 21.102 7.40 S8.389 48 , 684 20.879 109.473 84.326
Total Testosterone 1.50 ti 8 8 S 8 8 8 8 8 8
Mean 23.538 43,388 19,850 17.588 64,825 47.238 21.775 104,488 82,713
SD 0,078 19,158 12.217 5.228 25,885 25 , 625 7,713 38.959 35.640 cv¾ 42.8 44.2 61.5 29,7 39,9 54.2 35.4 37,3 43,1
Median 20,30 37.10 18,75 16,60 62.05 36,70 20,25 104.15 80.80
Min 10,10 24.20 6,00 12.60 24,00 1 ,40 13.60 48,90 24.90
Max 41.90 70.00 43.60 27.30 106.00 39.40 34,00 154.00 122.60
Geometric Mean 21.709 39,870 16,553 16.978 59.681 40.438 20,643 97.234 73.955
Total Testosterone 2.00 N 8 8 8 S 8 8 8 S 8
Bean 23,938 41 ,513 17.575 18,125 56.225 38,100 20.550 103,175 8Z.625
SO 9.595 1 ,775 5.858 6.036 19.893 19.669 6,902 39,675 38.400 cv% 40.1 35.8 30,0 33.3 35,4 8 ,6 28,7 38.4 46,6
Median 20.25 36.80 18.10 16,35 57.30 36.15 19,00 97,85 74.60
Min 12,90 25.90 6,40 12.30 24.70 12,40 14.80 50,20 26.80
Max 42,30 70. SO 28.60 29.60 93,50 77,40 30.80 152,00 130,80
Geometric (fean 22.421 39.480 16. 89 17,356 52.880 33.746 19.856 96 , 121 73,717
Total Testosterone 4.00 N 8 8 8 8 8 8 8 8 8 ean 23,413 39,275 5.863 9,000 41.700 22 , 00 20.350 79,025 58,675
SO 8,424 12,901 4.689 8.463 17,945 10,849 4.660 37,821 36,341
GV¾ 36,0 32.8 29.6 44.5 43,0 47,8 22.9 47.9 61 ,9
Median 20,95 33.70 13.80 15,90 41 ,90 24,10 19,80 6© < 55 SO , 75
Min 13,90 26,20 11,60 11 ,60 18.20 6.60 15,40 39,50 24.10
Ma 37,90 63.40 25.50 32.70 72,90 40,20 27.40 157.00 136,10
Geometric Mean 22,177 37.829 15.342 17,589 38,202 19,921 19.890 72,346 50,598
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 Dose 1200 (^g) Dose 1800 (fig} Dose
Analyte Time CodeBase ConcBLQ Cof!ODC ConcBase ConcBLQ ConcBC CodeBase ConcBLQ ConcBC
(hr) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) {ng/dL} (ng/dL) (ng/dL)
Total Testosterone 6.00 N 8 a 8 8 8 8 8 8 8
Wean 21.471 35.063 13.691 17.870 32,463 14.693 20.576 72.813 52.238
SD 9.21? 13.417 7.691 8.433 13.917 7.298 5.339 38.893 38.373 cy% 42,9 38.3 56.6 47.2 42.9 50.0 25.9 53.4 73.6
Median 18,06 30.95 11.50 14,85 29.40 15.70 19.76 59.55 42.70
Min 9.7? 18.00 6.23 9,46 14.40 4.94 15.20 38.90 15,60
Max 38.90 57.90 31.60 34.10 64.40 26.30 30.90 158.00 135.80 geometric Mean 13.841 32.793 12.252 6.360 29.9S6 12.635 20,018 65.80S 42.547
Total Testosterone 8.00 8 S Θ 6 a 8 8 8 8 fcears 22.150 33.125 10.975 .289 31 , 125 13 , 838 22.325 62.4SS 39,663
SD 11.003 5,980 4.O70 6.980 12.323 8,115 10,201 30,794 27.309 ov% 49.7 30,1 37,1 40.4 39.6 58, S 44.7 49.3 68,9
Median 19.60 29,75 12.30 1 S .65 28, 65 14.50 20.00 50,60 33.00
Min 8,00 20,70 4,50 7.41 13,20 1.00 13,50 33,80 13.50
Max 45.20 49.70 17.80 28,10 52.40 28.20 43. SO 120.00 85.00
Geometric Sean 19,944 31 , 860 10.221 5.971 28.952 10.250 21.139 56,952 32.129
Total Testosterone 12.00 N 8 e 8 8 8 a 8 8 a
Sfean 23,800 30.850 7.600 .379 25.363 7,965 19,863 47.438 27.575
3D 12.231 16,060 10.933 7.587 10,607 3,694 5,741 20.003 9,388
CV¾ 51 ,4 52.1 145,8 43.7 41.8 46.3 28,9 42.2 70,3
Median 19.70 23.50 3,55 15, SO 22,40 7.75 20.05 40,60 21.65
Kin 11.10 16,10 0,00 9.62 1 , 80 2.18 13.10 29,90 6.30
Max 51.20 60.80 33.20 29.30 44.40 15.10 26.90 86.10 59.90
Geometric Mean 21,663 27.716 6,080 23.556 7,118 19,118 44.210 21.117
Total Testosterone 16.00 N β 8 8 8 8 8 8 8 8
Mean 24,213 26,463 3.S63 17.279 22,088 4,809 19,875 34,938 15.600
SD 10,979 11. 40 2.978 5.883 8,124 3,009 6,167 12.443 13.204 cv% 45,3 44,4 83,6 34.0 36.8 62.6 31,0 35.6 84,6
Median 20.2E 22,80 3>90 16.10 19,85 5.05 16,46 29,30 12.70 fc!in 12.30 13,30 0,00 9.23 10,70 .20 12.70 24.00 o.oo
Wax 44.10 49,70 7,80 26.60 34, SO 9,40 31.30 59,50 39.30
Seorastric Mean 22.214 2 .430 18.406 20,809 3,838 19.092 33.346
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (vg) Dose 1200 {jig) Dose 1600 (fig) Dose
Analyte Time ConcBasa ConcBLQ ConcSC ConeBase ConcBLQ Cot!CBC CoocSase ConcBLQ ConcBC
(hr) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL)
Total Testosterone 20.00 tit 8 8 a 8 8 8 8 8 8
Mean 23.325 28.038 4.825 19.756 23.388 3.631 21.625 33.213 12.100
SO 9.5S0 13.757 5,114 8.240 9.802 2.56? 7.144 6.441 8.S91
GV% 40,9 49, ί 106,0 41 ,7 41 ,9 70,7 33.0 19,4 74.3
Median 20.7S 23.00 3.OS 17.SS 20,25 2 - 88 22.10 31.60 10.00
Min 10.30 13.40 0.00 9.75 12.60 0.80 12,80 23,70 0.00
Max 40.80 66.30 15.50 36.90 42.30 8.90 35.70 42.00 29.20
Geometric fearc 21.626 25.62? 18.435 21.849 2.894 20.655 32.664
Total Testosterone 24.00 N a a a a 8 8 8 8 8
Mean 30.138 33.138 4.175 23.538 28.013 4.475 24.350 34.650 10,625
SO 13.466 12.127 4,127 10.193 9.872 3.011 7.019 , 07 10.831
GV% 44.7 36,6 98.3 43,3 35.2 67,3 28.S 32.1 100.1
Median 27,80 30,10 4.10 20,20 25 , 20 3,80 24.30 30.50 9.25
Min 17.10 16.00 0>00 13.30 17.20 1.40 7,00 25,00 o.oo
Max S8.00 SO.70 10.40 41.10 4 . SO 11.40 38.80 59.80 34.00
Geometric ¾eart 27.873 31.141 21.791 26.582 3.809 23 , 552 33,314
Total Testosterone 32.00 H 0 8 0 0 8 0 0 8 0
Hean 29.088 21.150 29.350
SO 11.259 9,331 7,290
ov% 38,7 44,1 24,S
Median 29,80 18,50 28.25
Min 12.30 12.50 22.20
Max 44.60 40 , 30 40.80
Geometric ¾ear> 26.928 10,681 28,608
Total Testosterone 40 , 00 N 0 a 0 0 8 0 0 8 0 yean 27.188 19,650 25,525
SO 15.106 9,075 5,932
cv 55,6 45.7 23.4
Median 20.15 16.30 25.70
Min 12.80 11 ,50 16,70
Max 58.00 36 , 0 36.10
Geomet ic Stearc 24.171 18.339 24.905
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 {ug) Dose 1200 {jug} Dose 1800 (fig) Dose
Analyte Time ConcBasa ConcBLQ ConcBC Con cease ConcBLQ ConcBC CodeBase ConcBLQ ConcBC
<hr) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL)
Total Testosterone 48.00 0 8 0 0 8 0 0 S 0 fcteari 34.113 25.700 32.350
SO 13.460 11,313 12.182
cv% 39.5 44.0 37.8
Median 36.00 23.25 28.95
Bin 14.00 12,90 1S.80
Max 50,90 44. SO 54.10
Geometric Mean 31.421 23.639 30,535
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (jug) Dose 1200 {jjg) Dose 1800 f^g) Dose
Analyte Time ConcBase COfWBLQ ConcBC ConcBase ConcBLQ ConcBC ConcBase ConcBLQ ConcBC
(he) (ng/dL) (οβ/'dL) (ng/dL) (ng/dL) ins/cfL} {ng/dL) (ng/dL) (ng/dL) (ng/dL)
Dihydrotestostersns •0.2S ti 6 6 6 6 6 6 6 6 6
Mean 9.625 13.16S 3.590 10.118 12.048 1.967 10.642 1 ,865 2,040
SO 3.30S 6.260 4.218 4.431 6.614 2.389 4.1 OS 4,745 1.S84
GV¾ 34.3 47.5 117.5 43. S 54.9 120.2 38.6 40.0 97.2
Median 9.21 10.87 2.13 9.04 9.59 1,22 9.39 11 ,60 1.87 fiin S.79 7.62 0.00 6.10 8,03 0.00 5,31 5.55 0.00
Max 14.70 22.10 11.60 18.70 25.30 6,60 16.10 20,00 S,00
Geometric Mean 9.181 2.002 9.481 11.004 S .964 11 ,068
Ditiydrotestosterone 0,25 H 6 6 6 6 6 6 6 6 β
Mean 9,33s 13.853 4.518 9,375 12,130 2.813 10,473 13,870 3,613
SO 4.259 6.848 4.651 4.741 4.997 .597 4.054 6,408 3,507 cv% 45,6 49,4 102.S 50.6 41 ,2 56.7 38.7 46.2 97,0
Median 7.9S 11.80 2.95 7,95 11.50 3,11 9,22 13,10 2,60
Min 5,29 7.65 .44 5,04 7.15 0,00 6.08 7.28 0,00
Max 16.20 23,10 13,69 18.60 21 ,70 4, as 16,10 25,50 9.40
Geometric Mean 8,597 12,491 3.S32 8,607 11,426 9.851 12,788
Dtiiyirotestosterone 0,50 ti 6 6 6 6 6 6 6 6 6
Mean 9,658 14,925 5,267 8,912 13,550 4.638 10.620 16,370 5,933
$0 4,099 7,678 5,932 3.369 4.805 1.864 4.76S 0.S95 7,846 cv% 42,4 51.4 106,9 37,8 35.5 40.2 44.9 66,6 132.2
Median 8,45 13,01 3.75 7,87 12.56 4,84 10,05 13,46 3.04
Kirs 5.69 7,05 0.28 5.87 8,60 1 ,46 5,20 6.02 0.00
Max 16.60 26.00 16.27 15,30 22.50 7,20 16.30 37.10 20,80 geometric Mean 9,000 13,314 3,047 8.476 12,941 4.198 9.693 13,971
Oltiydrotestosterone 0.75 H 6 6 $ 6 8 6 6 6 6
Mean 9,595 14,263 4,668 8.830 13,747 4.S17 10,612 16.238 7,665 as 4,393 6.132 2.631 3.721 5.6Q3 2.818 3,843 1 .564 8,621 cv% 45,8 43.0 56,4 42.1 40,8 57.3 36.2 83.4 114.8
Median 9.12 13,10 4,32 8,15 11.90 5,12 10.18 15,20 5,43
Min 5,56 8.19 2.06 5,18 7,96 1 ,01 6.24 6.93 0,00
SSax 16.20 21 ,50 9,40 16.00 23,80 8,00 16,50 39,90 23.40
Geometric Mean 6,778 13,166 4,125 6.301 12-919 3.987 10,042 15,731
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (jug) Dose 1200 (pg) D0S8 1800 (f/g) Dose
Analyte Time ConcBass ConoBLQ ConcBO ConcBase ConcBLQ ConcBC ConcBase ConoBLQ ConcBC
(hr) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL)
Di ydrotestosterane 1.00 N β 6 6 6 6 6 s 5 S
Mean 9.813 14.457 4.643 8.665 13.103 4.436 10,924 20.820 9.896
SD 4.064 6.772 3.350 4.291 4.867 1,711 3.453 7.577 6.201
CV¾ 41.6 46,8 72,2 43.5 37.1 38,6 31.6 38.4 62.7
Median 9.05 12, 88 4.46 7,48 12,05 4,97 1.00 18.10 10,58
Bin S,S9 7,79 0.90 £.23 8,72 1,19 S.72 13,70 2,70
Max 56. SO 22.70 10.10 17,10 22.00 £.96 14.80 32.90 18.10
Seometr c tears 9.1 S3 13.13S 3.514 8.00S 12.458 3.S67 10.406 9.837 8.053
Dihydrotsstosterorie 1.50 N 6 6 6 6 6 6 5 5 S sjearc 9.403 13.938 4.535 8.295 3,587 5.272 10,950 19.680 8,730
SO .432 S.357 3.793 4.263 6.461 3.779 5.S20 9,261 5.175
C¥% 47.1 45.6 83.6 51.4 47.6 71.7 51.3 47,1 §9.3
Median 8,25 13.49 2.58 7,19 10,75 5,49 10,10 17,40 8,84
W n 5.30 7.96 1,81 5,20 8.34 0.07 5,56 10.90 3,00
Wax 16, SO 20.60 10.88 16,80 26,60 9,62 18.70 34,90 16.20
Seometrio Mear» 8 , 600 12.881 3.487 7.6 1 12.650 2.661 9.804 18.174 7.425
Oihydrotestosterone 2,00 N 6 6 6 4 6 4 5 5 5
Mean 3.355 12,413 3,068 9.498 12.632 2,868 1.366 19,652 8,286
SD 4,062 8,043 2.376 5.674 4.653 ,330 4.234 10.22? 6,305
CV% 43.4 40,6 77,7 59.7 36.8 SI ,8 37.3 52.0 76.1
Median 8.72 11 ,59 2,15 7,32 9.9S 2,06 9.87 17,70 6,S0
Kin 5.47 S.94 1.47 5,46 9.21 1.63 7,6S 9.86 2.17
Max 15.70 18,60 7,70 17.90 18,80 4.53 18.60 38.50 17,90
Geometric Mean 8.655 11 ,SSI 2.536 8.501 12,024 2,360 10.841 17,813 8,413
Dinydrotestostgrong 4.00 N e 6 6 5 5 S 8 6 6
M an 9.783 11.887 a.103 10,014 12,900 2,886 9.833 17.427 7,593 so 4.760 5,334 1,763 5.234 5.505 1,026 3.153 S,806 8.245 cv 48.7 45,4 83,8 52,3 42.7 35,6 32.1 50,5 82.2
Median 8.75 11 ,oo 1,61 7,74 10,50 3.40 9,37 18.40 7.3S w n 5.44 6,14 0,70 6,75 9.90 1,45 S.44 8.76 0,94
Kax 1 .00 18.60 5.60 19,30 22.70 3.75 15,60 32.20 16.60
Geometric Mean 8.856 10,845 1.688 9.218 12.191 2.711 9,460 15.716 4.954
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 Cfjg} Case 1200 {fig} Dosa 1800 (p ) Dose
Analyte lime CoiicSase CoiieBLQ CQftcBC ConcBass ConcBLQ ConcBC CodeBase ConcBLQ CffiCBC
(hr) (ng/dL) (ng/cfL) (ng/dL) (ng/dL) (ne/dL) (ng/dL) (ng/dL) (ng/dL) (ng/cfL)
Oitiydrotestosterone 6,00 fit 5 S S 5 5 5 5 5 5
Msan 8.834 12.386 3.554 9.2S2 10.578 .818 10.570 16.776 6,206
SD 3.264 6.239 3.803 4.176 4.737 1.687 5.626 8.B11 3.466
CV% 36,9 50.4 107.0 45,2 44.8 92,8 53.2 50,7 55.8
Median 8.14 13.10 1.60 8.67 8.30 2.60 8.84 13,80 5,25 kSirc 5.45 6.13 0.13 5.27 7.2S 0,00 5.45 9.88 2.09
Max 12.90 20. SO 7.70 IS.20 18.80 3.46 20.20 31.30 11.10
Seometric Mean .353 11.052 1.528 8.604 9,815 9.602 15.403 5,366
Diiiydrotestostsrotis 8,00 N 4 4 4 e 6 e 6 6 6
Mean 9.188 11 ,868 2.680 8,522 9.772 1,472 13.263 15.133 4,470
SD 8.568 3.930 1.713 3.618 4,273 1,391 10,030 6,090 4,151
CV% 28.0 33.1 63.9 42,4 43.7 94, B 75,6 40,2 92.9
Median 9.32 12.65 2.73 7,54 8. as 1,52 9.78 13.00 3.27
Mirc 5.92 6,57 0,65 5.16 §,94 0.00 6,22 10.40 0,00
Max 12.20 15.60 4.62 15,40 8.00 3,20 32.60 26, SO 11.70
Geometric Mean 8,89? 11.289 2.139 8,003 9.157 11.011 14.308
OifiycSrotestosteron* 12.00 H 4 4 4 6 6 6 5 5 5 fie an 9.615 10,808 1.240 7,613 9,117 1 , 553 10.122 3.392 3,270
SO 2.996 4,754 2.058 3,515 4.430 1,227 4.301 5.96 1,813
GV 31.2 44,0 165.9 46.2 48.6 79.0 42, S 44.2 55,4 iflediari 10.04 10.25 0.33 6,34 7,93 1.59 8,96 10.70 2,12 fiin 5,58 5,64 0.00 S.20 5.92 0.00 6.37 8.36 1,74
(Sax 12.80 17.10 4.30 14.50 7,90 3.40 17,00 22.30 5,30
Geometric fSsan 9.210 10.020 7,107 8,460 9,479 12.453 2,892
Ditiydrote stasis rone IS, 00 N 4 4 4 5 6 5 6 6 6
Mean 10.975 9,985 0.000 7.892 8.195 0,922 8.625 .537 2,977
SD 3,160 2,921 O.0O0 3,683 4.774 1,523 3.567 5,834 2,821
CV% 28,8 29.3 46,7 58.3 165.2 41 ,4 50,3 34.8
Median 11 ,50 1.30 0.00 6.54 S.41 0.33 8.06 S.75 2,38 liiri 6.70 5.64 0,00 5.18 5.72 0.00 5.02 6.49 0,00
Ma 14.20 1.70 O.OQ 14.30 17.90 3.60 15.20 22.70 7,50
Geometric Mean 10.584 9,578 7,358 7.437 8.097 10.637
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
606 Dose 1200 (yg) Dose 1800 (μ3) Dose
Artaiyte Tine© CodeBase ConcBLQ COftCBC CodeBase ConcBLQ ConcBC ConoBase COfloBLQ ConcBC
(hr) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL)
Dihydrotestoeterons SO.00 N S S 6 S 6 5 5 6 5
Ms&n 11.448 10.160 0.176 3.734 9,743 1,640 10.112 1,852 1.938
SO 3,952 3.S18 G.3S4 4.933 6. OSS 1.789 3,435 5,668 2,302 cv% 34. S 34,6 223, e 56,6 62.1 10S.1 34.0 47,8 117.0
Median 1 ,90 11 ,80 0,00 6.44 7.73 1.42 8.69 10.10 1.41 fctin 5.Θ2 6.10 0.00 6.00 S.7S 0.00 7.22 7.50 0.00
Max 1S.30 14,10 0.88 17. £0 22.00 4.50 16.00 21 ,70 5,70
Geoisetrio Mean 10,81? 9,623 7.938 8.722 9,722 11.008
Dxhydrotestosterene 24.00 ti 5 5 5 6 6 6 S 6 8
Mean 14.274 14.302 0.828 12.048 12,203 0,867 1.865 13.615 1.318
SD 8.305 4.4S7 1 ,253 6.614 4.848 1,005 4,745 5,125 2,391 cv% 44.2 31.4 15 ,3 54,9 33.7 116.0 40.0 37,6 124,6
Median 13,40 14.30 0.27 S.5S 10.50 0.62 11 ,80 12.80 1.30 ssin 8,23 8,61 0,00 8.03 8,32 o.oo 5,65 8.06 0.00
Ma 22.10 19,00 2.37 25,30 21.80 2.27 20.00 21 ,40 6,40
Geometric Mean 13,14 13.697 1 .004 1 .589 1 ,068 2.834
Oinydrotestoaterone 32,00 Pi 0 5 0 0 8 0 0 6 0
SJearc 11.098 8,757 11.200
80 4.574 4,237 5,310
cv% 41.2 48,4 47.4
Medisn 12,90 7,72 3.37
Kin 6,09 5.31 7.47
Bflax 16.60 17,10 21.60
Geodietric Mean 10.274 8,118 10.415
Olfiydi-otestosterone 40,00 N 0 5 0 0 6 0 0 6 0 iSsan 10.348 8.15S 10.147
SO 3,204 3,796 6,082
cv 31.0 46,5 50.1
Median 10.90 6,82 S.75
M n 6,70 5,18 6,22
.80 15,40 20.20
Geometric Heart 9,948 7,581 9.370
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (fjg) Dose 1200 (fjg) Dose 1800 (PQ) Dose
Analyte Time ConcBase ConcBLQ ConcBC CodeBase ConcBLQ ConcBC ConcBase ConcBLQ ConcBC
(ng/dL) (ng/dLj (ng/dL) (ng/dL) (ng/dL) (ng/dL) (ng/dL)
Di ydrotestosterone 48.00 N 5 6 o
Mean 15.834 12.012 13.557
ss 7.SG7 6.263 6.482
GV% 47.4 §2.1 47,8
Medi n 17.20 ΙΟ.βδ 12,55
Bi 7,77 7,03 7.24
Max 24.20 24.40 24.80
Geometric Sean 14.246 1 .018 12.3SS
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 { g) Dase 1200 {μ ) Dose 1800 {fjg} Dose
Analyte Time GoncBase ConcBLQ ConcBC ConcBase ConcBLQ ConcBC GoncBass ConcBLQ ConcBC
(hr> (Ρβ/aiL) (pg/mL) (pg/iJiL) {pg/mL; (pg/siL) {pg/ntL) {pg niL) (pg/mL) (pgW
Estradiol -0.25 M 8 S 8 s 8 8 8 8 8
Bean 34.925 SO. 00 17.750 38.863 40.288 7.238 40.950 42.550 9.760
SO 4,902 32.762 3 .8S4 32,649 16.468 6.765 25.999 16.284 10.418
CV¾ 14.0 65.4 179.5 84.0 40.9 93.5 63.5 38.3 108.8
Median 35.75 39.20 4,90 29,35 34.40 7. SO 31 ,35 38,70 8.55 fcfin 29.00 22.80 0.00 19.70 22.80 0,00 18.10 23.30 0.00
Max 42.30 122,00 90.90 119,00 76.00 18.70 97.40 6S.40 26.30
Geometric Mean 34,625 43,310 32,574 37,815 35,414 39,849
Estradiol 0,25 tt 8 8 8 8 8 8 8 S 8
He an 33.000 47.68fl 16.825 38.213 40.100 5.825 40.888 41.213 7,638
SO 4,325 32,261 32,688 31 ,635 21 ,987 8.111 22,347 15,414 S.08S
0V% 13,1 67.7 34, 3 82,9 54.8 104.9 84,7 37.4 519,0
Median 33.10 37,40 0,30 28,35 33,70 5,70 34,50 35,20 e.oo
Kin 27.20 20.40 0.00 18.80 20.20 0.00 18.60 22.30 o.oo
Max 35,20 120,00 92,80 16,00 90,20 17,20 84,50 66,80 26,00
Geometric Wears 32.751 40,913 32. 2 36,283 36.217 38,783
Estradiol 0,50 tt 8 8 S a 8 a a a 8
¾iearc 34,325 45,213 14,850 39,063 39.2 3 5.775 39,750 40,538 8,100
SO 4,784 28,422 27,846 32,600 20 , 528 6.542 21 ,081 6,085 8,050 cv% 13,9 62,9 188,2 83 , 5 52.4 113,3 53,0 39.7 99.4
Median 35.65 34,15 0,00 29,55 33,60 3.9S 35.50 35,10 7.25
Min 25.70 21.50 0,00 19.20 19,00 0.00 17.70 20,90 0.00
Max 39.70 103.00 77 , 30 119.00 83,50 16,90 84,80 82,80 21 ,00
Geometric Hie an 34,010 39.201 32,770 35.456 35.673 37,720
Estradiol 0,75 H 8 8 8 3 8 8 8 8 a
Mean 34,780 43,388 13,213 36,663 36,613 6,925 39,886 38,700 6,513
SD 4,820 28. 43 25,908 29,158 16.577 6,013 19.852 5,884 7,044 cv 13.3 65.1 138.1 79.5 46,5 101.5 49.8 41.0 108,2
Median 36.20 33.20 0,00 28,25 33,25 4,50 36.70 33, SO 5,40
Min 28.10 21.50 0.00 20.00 17,00 0.00 19,90 20.80 0.00
Max 4 .10 103.00 73 , 70 108.00 89,60 13,60 81 ,50 64,20 19,00
Geometric Mean 34.473 37.307 31,152 32.828 36.229 35.943
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (fig) Boss 1200 (fig) Dose 1800 ( g) Dose
Analyte Time ConcBase ConcBLQ ConcBC ConcBase ConcBLQ ConcBC Co no Base ConcBLQ ConcBC
(hr) (pg/aL) (pg/isL) (pg/niL) {pg/aiL) ipg/ntL) (pg/aLJ (pg/BtL)
Estradiol 1.00 H 8 8 a e 8 8 8 8 8
Mean 35.660 43.463 13.000 35.275 35.450 5.938 40.688 38.700 6.750
SD 4.775 27.447 25.322 24.944 14.832 5.367 2O.0S0 16.837 7.729
CV% 13.4 63.2 194.8 70.7 41.8 100. s 49.4 43,0 114.5
Median 37.40 3S.40 0.S5 27,70 33.10 £.35 37.30 35.35 5.30 fciin 28. SO 21 ,00 0.00 19.80 17.20 0.00 18.00 20.30 0.00
Wax 40. ΐθ 99.60 71.10 S6.10 65.70 14.60 81.90 64.00 19.60
Seouietric *tean 3S , 3S6 37.483 30.847 32. SSS 36,904 3S.622
Estradiol 1 ,so N B 8 8 8 8 8 8 8 8
Mean 38.175 43.363 13.988 35,400 33.263 3.288 39.775 37.850 6,388
3D 4,283 30,364 26,710 20,937 15.074 5,399 18,258 1 .383 10,094 cv% 11.8 70,0 591.0 69.1 45,3 164.2 46,8 46,9 158.0
Median 36.30 33, 55 0,00 30,75 28.45 0,95 38,65 33.15 2,05
63if 30.90 16, 60 0,00 18,80 18.00 0,00 20.80 20.70 0,00 ma 42.10 106,00 75.10 es.70 64.80 5,20 76,10 72.30 2S.10
Geometric Mean 35 , 953 36.198 31,904 30.796 36.485 34.798
estradiol 2.00 N 8 8 8 e 8 8 8 8 8
Mean 43 > 000 43,300 13,126 34. 88 30.113 2.9S0 36,588 37.125 7.000
SO 17.325 31.204 27.658 22,048 12.603 4.534 14.077 18.468 13,278
CV% 41.2 72.1 210,7 63,9 41.9 153.7 38.5 49,7 189.7
Med an 35.20 32.75 0.30 26,85 25,70 2.00 33.00 30.85 2,25
Kin 31.60 18,70 0.00 20,40 18.10 0,00 21 ,80 19,70 o.oo
SSax 63.30 in ,οο 79.40 87,70 66,50 13,70 65.10 75.60 39.10
Sewwrtric Sisan 39,810 36.127 30,737 28.254 34,552 33,804
Estradiol 4.00 N 8 8 8 8 8 8 3 8 8
Mean 35 , 950 46.513 14.963 35,350 35.3S3 4.000 36.975 36.663 7,063
S3 7.598 26,640 2 .962 17.125 15.840 6,208 12.676 20.406 15.290
CV 21.1 57,3 1 6.8 48.4 44. B 1S5.2 34,3 55.7 216.5
Median 31.75 40,20 6.80 32.10 30.40 0,00 34.10 28.35 1.05
8Jin 29.50 16.20 0.00 17,50 19,30 0.00 22.40 19.90 0,00
Ka 49.80 S7,00 64.70 74.10 66.30 16,80 55,60 82.00 44.50
Geometric Mean 35,308 40.291 32,511 32.801 35. 03 33.006
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (^ ) Dose 1200 (jug) Doss 1800 ()ig} Dose
Arsaiyte Jim eoncBase C fiOBLQ ConcBC ConcBase ConcBLQ ConcBC CodeBase ConcBLQ COficBC
(hr) (P8/!HL) (pg/mL) (pQ/nL) (pg/siL) (pg/mL) (pg/mL) (pg/t!!L) (pg/mL) (pg/mL)
Estradiol 6.00 N 8 S s S 8 8 8 8 8
Mean 31,125 42.768 14.500 3 .700 30,775 4,450 41.525 34.275 3.338
SD 4.51? 24.6S9 20.2BQ 16,784 9,401 6,731 20.164 ,842 7. §82
CV% 14.5 57.6 139.7 52.9 30,5 181 ,3 48.8 43.3 239.1
Median 31.60 35.50 6.10 27,75 27.25 0,00 35.45 28.05 0.00
Uin 24,10 18,80 0.00 17.00 19.10 0.00 19,70 22,00 0,00
Max 37.40 94.60 60,30 71 ,80 46.00 17.10 74.30 63.30 22.80
Geometric Mean 30,830 37.629 29,086 29.566 37.402 31,898
Estradiol 8,00 N 8 S 8 S 8 8 8 8 8 filsan 33,975 45.300 14.125 35 , 025 33 , 300 1.725 34 , 500 35.075 5,163
SD 4.784 25.471 18,082 17,4S2 10,401 3,481 11.810 18.698 14,202
CV% 14.1 56.2 528,0 49.9 31.2 201.8 34.2 53.3 275.1
Median 33.90 40.65 6.75 31 ,15 29,05 0,00 31.00 29.95 0.00
Kin 28.30 16.70 0.00 17,30 22.70 0,00 21 ,50 14,30 0,00
Max 40.90 94. SO 54,00 75,00 5 , 60 3,70 53.1 75,40 40.30
Geometric Mean 33,683 3S.277 32,103 32,072 32.794 31.439
Estradiol 12,00 N 8 8 8 Θ 8 8 8 8 8
35.550 43.088 8.688 3S .238 3 , 588 3.863 38 , 25 33.686 2,363
SD 1 ,304 22.621 12,795 22 , 541 1 ,182 3,521 13.626 14.176 5.088 cv% 31.8 52.5 147,3 62.2 32.3 91 ,2 35,2 42.1 215.4
Median 34.15 38.15 3.25 30,35 32.55 4,45 36.30 32.30 0.00
631 n 20.90 22,20 0,00 21 ,00 24.20 0,00 23,80 13.70 0,00
Max 59.30 81.50 35.10 90,70 58,70 8.50 60.00 64,10 14.40
Geometric Mean 34,150 38.669 32 , 40 33,257 36.663 31.517
Estradiol 16,00 Pi 8 8 8 8 8 8 8 8 8
Mean 44.450 48.000 5,400 39.963 40, 1S8 8.538 41 ,S75 40,538 6,288
SD 18,674 21.117 9,318 2 ,293 10,943 5,910 14.648 22,827 13 , 598 cv% 42.0 44.0 172.6 53.3 27,3 90,4 35,1 56.3 216,3
Median 42.05 45.20 1.35 33,00 36.50 7,60 37.95 35 , 55 1.05
24.70 21 ,90 0,00 22,40 26.00 0,00 26,20 21.40 0,00
Max 84.30 81.00 26.90 89.80 57.70 14,70 66,20 93,80 39.60
Geometric Mean 41,555 44.033 36,516 38,915 39.517 36,632
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
800 ifjg) Dose 1200 dig) Doss 1800 [μ§) Dose
Analyte Time CodeBase ConcBLQ ConcBC ConeSase ConcBLQ ConcBC Cone Base ConcBLQ ConcBC
(he) (pg/!sL) (P9./mL) (PQ/»LJ (pg/mi-) {pg/fflL} (pg mL) (pg/tsL)
Estradiol 20.00 ill Θ 8 8 8 8 8 8 B 8
Mean 48.213 49.988 6.375 37.338 42,538 8.938 42.050 38.475 4,938
SO 23.249 21.182 10.920 8.2S7 12.602 7.594 18.732 26.012 9.907
GV¾ 48,2 42,4 171.3 22.1 29. S 109. S 44. S 67.6 200.6
Median 42,36 48,00 1.75 35,45 39.70 6.80 33.65 30.7S 0.00
2S.7D 2S.10 0.00 24.70 31.20 0.00 23.90 18.00 0.00 k(ax 89.90 88,40 31.10 60.80 68.40 22.20 72.30 101.00 28.70
Geometric Mean 4 , 325 46.167 36,533 41.109 38.680 33.703
Estradiol 24.00 H 8 8 8 8 8 8 8 8 8
Mean 60.100 47,438 8.863 40,288 42 , 600 7,100 42.650 34,713 4 , 038
SO 32.762 19.652 5.563 16,466 14.117 7.553 16.284 £2,553 8.599
GV¾ 65,4 41 ,4 81 ,1 40.9 33.1 106.4 36.3 65.0 213,0
Median 39,20 45,60 5,50 34.40 39.45 5,75 38,70 28,60 0.00
Kin 22.90 27.90 0.00 22.50 26.60 0.00 23.30 16,50 0.00
Max 122,00 SO, 80 18,00 75.00 70.20 19. SO 65.40 88.20 24.10
Geometric Mean 43.319 44.477 37,815 40 , 760 39.649 30.552
Estradiol 32.00 N 0 8 0 0 8 0 0 e 0
Mean 46.400 35,313 34.188
SO 17.204 10, loo 22.449
GV% 37,1 28.6 65.7
Median 41 ,20 33.25 2S, OS
Min 31.50 25.20 17,10
Ma 85,50 50.80 87.30
Gwwttrie Mean 4 .240 3 , 063 30.008
Estradiol 40.00 N 0 8 0 0 8 0 0 e 0
Mean 48,988 37 , 563 39.275
SO 14.785 12,631 22.281
GV% 30,2 33.6 56.7
Median 45,25 32.70 35,35
liiti 32.20 25.20 16,80
Max 81 ,20 58.90 89.40
Georcstric Mean 47.320 35 , 843 34.957
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (jjjj) Dosa 12GQ (fjg ) Dose 1800 (fjg) Dose
Analyte Tiiste Concsase ConcBLQ ConcBC Con DBase ConcBLQ CONCBC Cone Base ConcBLQ ConcBC
( hr) (pg/rsL) { g 'oiL> ( pg/sL) (pg/mL) (pg mL) (pg/mL)
Estradiol 48.00 8 8
48.325 36.225 41 .750
SD 18.32? 9.788 24.37S cv% 37 .9 27.0 58.4
Median 45.85 37.46 3S .25 hfin 27 ,80 SO . 10 14. SO fcfax S4.70 48.40 97.10
Geometric Mean 45.630 34.936 36.7S2
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 {jjg) Dose 1200 (ug) Dose 1800 (μ@) Dose
Analyte CoocBase ConcBLQ CcmcBC CodeBase ConcBLQ ConcBC ConcBase ConoBLQ ConoBC
(hr) (rnnol/L) (nmoi/L) (nmol/L) !nOOl/L; (nmol/L) (nmoi/L) {nmoi/L} (nmoi/L) (niSOi/L)
SHBG -0.25 8 8 8 8 8 8 8 8 8
Mean 60.663 61.413 2.438 51.325 51.738 1.963 67.400 63.388 1.013
SO 22.919 22.272 3.026 22.728 23.757 2.984 38.763 33-477 1.942
GV% 37,8 36.3 124.1 44.3 45,9 152.1 57.6 52.8 191.8
Median 61.20 SS.30 i.as 51 ,00 48.10 0.00 56.50 53.15 0,00
Win 16.40 18.70 0.00 18.80 7.40 0.00 29,20 27.90 0.00
Wax 101.00 ST.20 8,80 78.80 86.70 7.90 137.00 116.00 5.00
Se metric Mean 55.265 56,534 46.037 46,256 58.918 58.059
SHBG. 0,25 ti 8 8 β 8 8 a 8 8 8
Mean 58.988 62.783 4.888 49.725 52,763 3.475 6 .550 65.450 5.575
3D 21.961 25,102 4,316 2 .040 23.563 4,172 29.701 32.420 10.980
CV% 37.2 40.0 88,3 42.3 44,7 120.1 48,3 49.5 197,0
Median 57,30 50,15 4.35 52.00 SO.25 2.25 59.70 57.50 0,00 ssin 16.10 18.70 0.00 18,20 18.20 0,00 30.20 29,00 0,00
Max 95.70 105,00 12,00 77,00 83.30 11.50 116,00 116.00 29.30
Geometric Mean 63.808 67,139 44.993 47.211 55.696 58.803
SHBG 0.50 8 8 a 8 8 8 8 8 8 fciearc 60.875 60,500 1,228 5 , 438 52-238 2,213 66.938 64,500 2,583
SO 21.467 £2,059 1,923 21.533 23,474 2.909 39,402 30.515 3.143 cv¾ 35.3 38.5 57.0 42,7 44,9 131.5 58.9 47,3 122,7
Med an 61 ,00 59.85 0.25 51 ,40 50.95 0.90 56.50 66,40 ,70
Win 17,00 18,00 0.00 18.30 17.40 0.00 25.20 33,80 0,00 iSax 93,30 98.40 5.20 77,00 8 . SO 7, SO 128,00 110,00 8,60
Geometric (tear* 55.831 55,587 45,551 46,579 57.778 58,646
SHBG 0.75 N 8 8 8 8 8 8 8 8 8
Mea 60.175 60,863 i,763 49.663 50.890 2.150 67.813 64,383 1.150
SO 22.510 22,700 2,387 22. 57 23,310 4,050 37.934 32.302 2.195
CV% 37,4 37,3 135.5 44.6 46,1 188.4 55.9 50.2 190,9
Median 58,25 57.80 0.65 50.70 48.50 0.2S 58.45 57.50 0,00
Kin 16,30 17.60 0.00 17,70 16.80 0.00 27.00 30,60 0,00
Wax 95,80 97,70 5.60 76.40 87.90 11 ,50 130.00 116.00 5,60
Geometric Me n 5 .735 55.714 4 .383 45,199 59.317 57.840
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
S0G {j/g) Dose 1 00 ( 8) Dose 1300 (μβ) Dose
Analyte Time CoficSass ConcBLQ ConcBC ConcBase COficBLQ ConcBC CodeBase ConcBLQ ConcBC
(hr) (nmol/L) (nmol/L) (nmoi/L) (nmol/L) (mttoi/L) (nmol/L) (rtmol/L) {nutol/L)
SHBG 1.00 w Θ 8 8 8 8 8 8 8 8
Mean 61.300 61.025 2,013 50.550 47.926 1.025 66,563 83.900 1.163 so 23 , 392 23.768 3.879 23.237 23.9S9 1.262 35.960 36.509 2.153
CV% 38,2 38,9 132.7 46.0 50.1 123,2 54.0 55.6 185.2
Median 59, 3S 58,00 0.00 51.05 44.80 0,60 57.60 51.75 0.00
Hiin 15.30 16.80 0.00 10,90 18.10 0.00 30.30 26,00 0,00
Max 98.70 38.10 11.10 85.80 67.80 3.40 1 8.00 124.00 4.70
Geometric Mean 56.396 55.431 44.966 42.4S1 69.089 55,927
SHB5 1 ,so N a 8 8 6 8 a 8 8 8
Mean 60. 25 61.850 3,275 49.663 49,863 2.088 68,700 67.275 3.888
SO 21.738 23. 77 5,010 2 .989 22.622 3, 58 39.715 29.512 8.363 cv% 36.0 38.4 153,0 44,3 45.4 151.3 57.8 43,9 215.1
Median 59.45 SO, 75 0,75 48,65 48.25 0.05 58.85 54,70 0,75
Hln 18.10 17.60 0.00 17,70 17.80 0.00 27.40 34.90 0,00
Mm 92.90 98,70 14.30 78,30 86.70 8.40 137.00 118,00 24.40
Geometric «ear> 55.139 56,353 44.719 44.737 59,730 62.277
SHBG 2,00 H 8 8 8 8 8 8 S 8 8
Mean 60.313 62.275 3.70O 50,250 51 , 700 2,488 56.113 63.163 1 ,013
SO 2S.066 23.861 4 , 279 22.044 23,719 3,979 38,074 32,065 1.530 cv¾ 36.6 38.3 115,7 43,9 45. S 160,0 54,6 50,8 161.0
Median 58.20 53,60 1,70 48,30 49.50 0.70 56,40 55,55 ο,οο
Bin 16.90 17,70 0,00 17,30 17.00 0.00 32.20 29,30 0,00 ssax 91.SO 103.00 11.10 77,30 S8.90 11.60 125,00 114,00 4,40 aeoraetric Sear! 55.133 56,826 45,142 46,265 58,576 56.512
SHBG 4.00 N e 8 8 S S 8 8 8 s
Mean 62 , 075 62.250 2,075 50.975 52,575 3,438 67.463 64.450 0.125
SO 23.457 25.058 3.606 22,710 24,035 4,497 34.540 33.692 0,354
CV¾ 37,8 40,3 173.8 44.6 46. S 130.8 51.2 52,3 282,6
Median 62.85 63.20 0.20 49,50 49,65 1.20 58.85 57,30 0,00
Ntin 16.90 17,30 0,00 17,40 17.00 0.00 31 ,60 28,60 0,00 flax 102,00 104.00 10.30 77.30 89,10 1 ,80 122.00 117,00 1.00
Geometr c ssead 56. §35 56.330 45.584 46,856 60,448 57.416
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (fjg) Dose 1200 {<¾} Dose 1800 ( Q) Dose
Analyte Time ConcBase ConcBLQ ConcBC ConcBase ConcBLQ ConcBC ConeBasss GOFICBLQ CODCBC
(hr> (nmol/L) (nmol/L) (nmol/L) (nmol/L) (nmol/L) (nmol/L) (nmol/L) (nmol/L)
SHBG 6,00 H 8 8 8 8 8 8 8 8 8
Msan 58.463 61.888 3.575 49.800 52.926 3.438 66.675 65.675 2.113
SD 22.341 23.294 2.738 22.171 24.320 4.273 37.65S 31.434 2.728
CV% 38.2 37.6 76.6 44. S 46.0 124.3 56.5 47.8 129.1
Siedian SS.8S 59.75 2.95 4 .3D 50.85 1.85 56.25 58.80 1.20
SSin 15.90 18.10 0.00 18.30 17. SO 0.00 27. SO 31.80 0.00
Max 92.30 99.90 7.60 81.00 92.00 11.00 128.00 113.00 7. SO
Geometric Mean S3.136 56.631 44.847 47.371 58 , 348 59.4S6
SHBQ 8.00 ti 8 8 8 8 8 8 8 8 8
Mean 59.163 60.613 2. 75 49.663 50.638 3.488 67,425 65,538 1.075
SD 22.806 22.631 2.988 £3.835 23.754 4.324 3 , 931 31.733 2.332 cv% 38.5 37.2 111,7 48.0 46.7 124,0 51,8 48.4 216.9
Median 55,65 59,60 2.05 55,15 46.25 1.65 61 ,55 57.80 0,00
Min 17.40 17.40 0.00 17.00 18.00 0.00 30.30 31.80 o.oo
Max 95,00 99,00 7.50 79,80 88,80 10,90 124.00 113.00 6,70 fieowBtric tiean 54,000 55,659 43,639 45.579 60.102 59.1 2
SHBS 12.00 N a S a 3 a a S 8 8
Mean 59.638 60.338 1.288 50.726 51.525 .600 67 , 288 61.963 0.637
SO 21.833 21.588 1.186 22.918 22.733 2.132 36.465 29.86S 1.205 cv¾ 36,7 35,8 92,1 45,2 44,1 133.3 54.2 48.2 189.0
Median 59.85 60.25 1.45 49.80 52.10 0.25 59.40 57,05 0,00
Mill 16.80 17.90 0.00 17.40 17.30 0.00 32,00 27,70 o.oo
Max 94,00 S3, 90 2.90 83,80 86 , 80 5,00 130.00 109.00 3,00
Geometric Mean 54.546 55.511 45.270 46.348 59,612 55,731
SHBG 16.00 N 8 8 8 8 8 8 8 8 8
Mean 60.338 59.038 1.113 48.363 48.788 1.325 63.363 63.625 ,700
SO 23.332 22.326 2.351 21.573 21.116 1.279 32.249 30.147 2.043 cv% 36.7 37.8 211.3 44.6 43.3 96. S 50.9 47.4 120.2
Median 60,70 58,75 0.00 48 , 35 51 ,35 1.30 56.60 58,60 1,00
SJirt 15,70 17,60 0.00 16,00 16,90 0,00 29.90 31 ,90 0,00
Max 94,00 93,80 6.70 78.10 78.20 3.30 118,00 114.00 5,30
Geometric Mean 54.538 54.045 43.314 44.001 56 , 796 57,816
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose
600 (fjg) Dose 1200 (fig) Dose 1800 (jiQ) Dose
Analyte Time CodeBase CoflcBLG ConcBC ConcSase ConcBLQ ConcBC CodeBase ConcBLQ ConcBC
( r) (nntoi L) (πίϋθΐί' L) (nnwl/L) (nmol/L) (rimol L) {fU!10l/L) ¾nittoi/L) (nmol LJ
SHSO 20.00 ft 8 8 8 8 8 8 8 8 8 kiean 58.400 S7.913 0.913 48.900 46.960 0.388 62.225 61,675 1.713
SO 2 .S20 21 ,403 1 ,378 22.371 21,076 1,096 33.751 29.1S5 2,073 cv% 3?.5 3T.0 S: .0 45.7 44.9 282,8 54,2 47.3 121.1
Median 58.70 57.75 0,06 46.40 45.90 0.00 54.20 57.05 0.9S t£iii 16.20 17.70 0.00 16.80 16.50 0.00 29.50 30. SO 0.00
Max 82,60 90.20 3.70 81.10 76.50 3.10 116,00 111.00 5.00
Geometric Mean 53.226 S3. 02 43.772 42.123 55.003 55.947
SHBfi 24,00 ti 8 8 8 8 8 8 8 8 8
Mean 61,413 61 ,250 1.275 S1.738 49,653 0.713 63.388 64,513 2.250
SO 22.273 23. 96 3.295 23.757 21,626 2.018 33.477 30,402 2.540 cv% 36.3 37,0 258.4 45.9 43,6 282.8 52.8 47,1 112.9
Median 59,30 59.00 0,00 48,10 49.10 0.00 S3 , 1 S 57.70 1,75
18,70 17.60 0,00 17,40 17.20 0.00 27.90 31.10 0,00
Max 97.20 93,70 9,40 86.70 77,00 5,70 116,00 109,00 7,30
Geometric Mean S6.SS 55.393 46.266 44,723 56.068 58.420
SHBG 32.00 N 0 a 0 0 a 0 0 8 0
Mean 62.063 50,050 64, 38
SO 23,582 21,998 32.090
CV% 38.0 44.0 50.0
Median 62.26 £2.10 54.95
Min 17.70 17.20 30.70
Sax 94. SO 75.30 1 3.00
Geometric Mean 56.565 44,909 57.509
SHBS 40.00 N 0 8 0 0 8 0 0 8 0
Mean 58.125 49-83 62.288
SB 20.263 23,616 28,296
CV% 34.9 47,4 45,4
Median 59.70 50.85 57.05
in 16.40 16.40 30.00
Max 84.40 8$, SO 105,00
Geometric Mean 58.359 44,224 56.948
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1a
Pharmacokinetics: Analyte Plasma Concentrations
PK Population: Single Dose

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.1b
Pharmacokinetics: Analyte Plasma Concentration Summary
PK Population: Multiple Dose
Testosterone Testosterone Testosterone
DOSE Sample Sampling Time Free Total Di ydro- Estradiol SHB8
(US) umber (hr) (ng dL) (ng/di,) ingidl) (pg/itsL) ;n(IK)l/L}
1200 31 {trough.* -0,25 N a 8 7 8 8
Arithawtic m 0.404 28.025 2.826 77.325 52,413
SP 0.362 1 .556 3.88S SO.182 30.949 cv% 8S.4 51,9 30.1 77,2 4S.6
Median G.25 28.10 14,20 5? , SO 81.40
Mio 0,18 12,40 6.28 20.40 25.20 mx 1.27 58.30 17,70 175.00 03,00
Gee ■aetric Sean 0.326 25.082 12.313 58, 6S 55,268
1200 32 (trough) ■0.25 N S 8 S 8 S
Ari hssetie ifcan 0.536 38.S3S 11.890 64,650 61,538
SO 0.331 5.530 4,303 49.822 28.745 cv% 31.6 40.0 56.2 77.1 48.3
Sedian 0.4S 32.60 12.65 4 >·..:¾ SO.10
Sin 0.27 20.10 7.00 20.50 24.70 mx I .32 65.30 18.40 176,00 106.00
Geometric Sean 0.478 36.369 .173 52.496 54.955
1200 33 (troueft) -0.25 n 8 8 7 8 8
Arit saetic ¾3san 0.542 37.813 2.033 79,088 58,613
SD 0.318 13.888 3.541 81.547 27,438 ov% 68.7 S7.0 29.4 103.1 4S,8
M dian 0,48 34,20 12.60 57.10 56.30
0.24 8.70 6.95 20,50 24.50
Si ax 1 ,28 56,20 16. SC- 273,00 102.00
Geo met ic Sean 0.491 35.453 ll .554 57,589 52.887
1200 34 (trough) -0.25 u 8 S 8 s 8
Ar t hfnetic ^ean 1.148 SO .075 • 9.689 72.913 ■32,388
SD 0.432 43.644 8,911 63,918 30,047
GV 40.2 55.1 45.2 87.7 48.2
Median 1 , 18 72.70 19.80 52.15 60.35
Sin 0.4S 37.50 5.29 14.80 26.30
S5a:< 1.73 195,00 32. SO 210.00 102.00
Geo netric Dean 1. OSS SO . 20 17.423 53.996 55.736
Note: There were 7 dihydrotestosterone concentrations that were <LLOQ and were not included in this table (3 from the trough values).
Table 14.4.1b
Pharmacokinetics: Analyte Plasma Concentration Summary
PK Population: Multiple Dose
Testosterone Testosterone Testosterone
DOSE Sarople Sampling Ti»8 free Total DiHydro- Estradiol SHBG
(«8) number (ttr) (rsg/dL) (ng/dL) {ng/dL} (PS/ Bit.) (nrool/L)
• 200 35 ftrougn) •0.25 Pi 8 s 8 8 8
Arithmetic faean 0.61 44.863 14. 26 52.413 61. 25
SO 0.323 16.035 6.1 OS 32.504 30.2S0
CV% 52.8 35.7 43.2 62.0 48.9
Med an 0.54 4? . · 5 •4.40 4?. S 83.00
Sin 0.24 25,20 5.57 21.40 26,80
Max 1 ,35 56.30 21.10 26,00 05,00
Geometric Mea 0.552 42.225 12,804 45.532 54,782
1200 36 (trough) -0,25 H 8 8 ? 8 8:
Arithmetic 0.61 42.850 12.747 S5.238 56. SCO
SO 0.325 15.156 4,873 37 , 783 25,056 cv% 52. S 35.4 38.2 S?.S 44.3
Median 0.51 42,45 13.00 60,40 57.55
Min 0,36 24.00 5,76 21,80 25,40
S x 1.36 62,60 19,10 143,00 92,00
Geometric 0.550 40.408 11 , 824 56.334 51,458
• 200 37 ftrougn) •0.25 Pi 8 S 8 8 8
Arithmetic man 1.155 85.450 20,724 54. 13 53,700
SD 0.394 27.406 8,150 23,936 27.801
CV¾ 34.2 32, 1 39.3 44.3 46.2 edian 1 ,08 82.25 21.45 57,45 53.70 lain 0.7? 40,50 7,39 6,30 25.40
Sax 1.82 31.00 31.70 84.20 S3. SO
Geometric Mean 1.098 81.313 18.927 48.062 53.S30
3200 38 0.26 u 8 8 8 8 S
Arithmetic ?iSea« ! .835 344.788 21,146 50.738 60,650
SD 62.094 7.682 30.700 28,702 cv% 15.5 42.9 3S.3 50.5 47.3 fiSedian 1.82 133,00 23. OS 65.70 S2.50
Sin 1.44 77,7 8.27 14.20 25,50
¾¾ax; 2.16 242.00 23.80 107.00 10 .00
Geometric Mean 1.815 133,736 9,586 51.778 54, 6
Note: There were 7 dihydrotestosterone concentrations that were <LLOQ and were not included in this table (3 from the trough values).
Table 14.4.1b
Pharmacokinetics: Analyte Plasma Concentration Summary
PK Population: Multiple Dose
Testosterone Testosterone Testosterone
DOSE Sample Sampling Time Free Total Dihydro- Estradiol SHsa
( ) Number (t»r) (ng/dt) Wat) {og/cJL} (fimo L)
1200 39 0.so Pi s 8 g 8 8
Arit 1 , 658 21.419 63.738 60.275
SO 0.265 39.139 8,038 36,985 28,084 cv¾ 16.0 31.2 37,5 58,0 45. S
1.66 123. SO 23.65 67. S5 61.00 .28 85,20 9.15 12,90 27,40
Sax 2.11 205.00 30. SO 133.00 37.80
G83fliat"ic aean • . S39 120.446 19.8 3 52.643 54. 61
1 £00 40 0,75 κι 8 8 8 8 8
Ac it i!setic Mean • .545 115.425 20.338 63.275 59.413
SD 0.442 43.102 7.430 36,489 28.036 ev% 28,6 42. S 36,5 67 , 47.2 ssedian 1.54 1 2.50 22.60 67.10 61.00
«ir> 0.92 74.00 10.20 14.40 25.70 fex 2.18 231.00 30,00 131 , 00 33, 50 tetric Seats ! .487 108.600 18.985 52.725 53.047
1200 41 1.00 N 8 S S 8 8
Ar t i!setic Mean • .482 11 .425 18.93S 61.838 59.713
SD 0,456 50.764 ?. 0 51.772 27.747
CV 31.5 45. S 37.7 s; .4 46.5
Median 1.39 1QS.85 20,60 73.90 61,15
K n 0.70 S5.SG 9.01 13.40 27.20
«ax 2.12 224,00 30,50 110,00 32, 30
Gear is trie Sfear, 1. 1 · 103.395 17.SS0 £2, 153 53.568
1200 42 1.60 H 8 8 8 8 8
Arithmetic Mean • .200 89,300 7.918 55,813 53.563
SD 0.384 38.21S 7.245 25.242 28.378
CV% 32.0 42.3 40.4 47.6 47.6 ae Sian 1. 6 79.65 21.10 5S.40 SO.8S te 0.63 53.40 8. 6 12.70 24.50
1.73 176.00 27.40 95.20 94.90
Ceor letrie mean 1.142 S3. 80 16.4QS 43.293 52.879
Note: There were 7 dihydrotestosterone concentrations that were <LLOQ and were not included in this table (3 from the trough values).
Table 14.4.1b
Pharmacokinetics: Analyte Plasma Concentration Summary
PK Population: Multiple Dose
Testosterone Testosterone Testosterone
DOSE Sample San tpling Time Free Total Dihydro- Estradiol SHSG
Number (hr) (ng/dL) (ng/dt) {ng/dL) (nraol/L) 200 43 2.00 8 8 8 8 8
Arithmetic ue&n 1.103 80. ?88 7.191 64.513 67.5S0 so 0.40· 33. S 12 6.733 28.20S 28.41S cv% 36,2 41 ,6 33.2 48.1 45, S
Median 1.05 75,50 19.95 57.95 60.45 f&in 0.58 45.80 7.24 13.40 23.20 :\: 1.82 165.00 24,00 103.00 94. !O
Seosaetric Mean 1 , 04S 75 , 785 15 , 794 47,679 51 ,683
44 4.00 8 8 8 8 8
Arithmetic ean 0. S87 72.613 17.359 62.438 68.838
SD 0. S3S 38.375 6.867 38.7 1 28.206
CV'% S4.? 62.8 30.7 S2.0 4 . S
Median 0 , 73 61.10 18,50 57.30 60,75 fci 0.53 37.10 7.17 IS.50 26.10
2,08 156.00 25, 10 1 5.00 9:2,80
Seosaetric Sean 0.88S SS . S6S • 6. 87 52. ?9S S3.2S8 200 45 6.00 8 8
Arithmetic Mean 0.742 S2.450 4.790 62.988 68. ISO so 0.334 22.410 7.08S 23.827 2:7. 1 S cv¾ 42.7 47.9 56.3 48.0
Media 0.61 46.15 15,25 51,70 59,50 fci n 0.40 25.20 5.32 15.40 19,90
Mas* • .SO 07.30 23.80 116.00 92.20
Seosaetric Sean 0.670 48. SS2 • 3.134 46.033 SI .394 200 4S 8.00 8 8 8 8 8
Arithmetic Mean 0.700 50.275 3.770 81.088 68.288 so 0.389 19.335 S.739 31.024 2:6.343 cv¾ 55.6 38, 5 41.7 5 .3 43.5
Median 0 , 56 44.10 13, 15 SO, 40 64,50 fciin 0.42 20.10 8.43 15.70 25.70
Max • .SO ?S .60 22: . SO 121.00 87.90
Seosaetric ¾5e a ?t 0.634 47.132 12.60S 52.845 52.775
Note: There were 7 dihydrotestosterone concentrations that were <LLOQ and were not included in this table (3 from the trough values).
Table 14.4.1b
Pharmacokinetics: Analyte Plasma Concentration Summary
PK Population: Multiple Dose
Testosterone Testosterone Testosterone
DOSE Sample Sampling Tiffle Free Total Di ydro- Estradiol SHBG
Number (hr) (ng/dt) {ng/dt} {nc./d {pg/mL) (nmol/L)
1200 47 12.00 8 8 7 8 8
Arithmetic itean 0.663 39.950 13,00} 60,875 57,925
SD 0.326 14.516 5.S06 25,06? 26,711 ev% £7,9 36,3 42.3 41 ,2 46,1
Median Q.4S 37.75 13.90 63.26 60.60
Mir. 0.33 21.00 5.00 16.60 24.60
Max 1.35 60.90 20.30 97.90 88.80 seosaetric ean 0.511 37,610 11.807 54,750 51.850
1200 48 16,00 s 3 8 8 3
Ar thmet c mm 0.521 35,538 11.640 69,600 56.300 so 0.376 1S.975 4.701 26.232 2S.002
CV'% 72.3 44,9 40,4 37,7 44,4
0.4· 32 , SO 2. as 73.85 S8.3S
Mi: 0.26 16.60 5.00 19.50 25.10 m&y. 1.43 55.20 17. SO 104,00 36.40
Geometric SSean 0.4S1 32.S87 0.675 63,247 50,920
1 £00 9 20. Off 8 8 7 3 8
Ari thee tic an 0.580 38.613 13,994 68.625 56,275
SD 0.50? 21 ,298 4 , S48 26,607 24.189
CV'% 87, 3 55,2 32.5 38,8 43,8 iSedian 0.39 34,60 13.50 73.96 53.70
Uin 0.32 17.20 7, 6S 23.00 25.00
Max 1.82 82.00 22.40 98. 0 85.00
QfcOBtstric ean 0.479 34.137 13,375 62.635 50,1 7
• 200 SO 24.00 ¾i 3 3 8 8 3
Arithmetic utean 0.578 38,513 1 .774 54,113 56.275
SD 0.461 18.167 S.921 28.365 25.630 cv% 79,3 47,2 40,1 44.2 45,5
SSediarc 0,44 34.45 16.00 65.55 59,65 i¾¾if: 0.23 13,70 6,33 18. SO 25.10 fsiax 1.71 77.30 24.70 104,00 37.30
Geometric fcSeafi 0.492 35.360 13.637 56,820 SO, 636
Note: There were 7 dihydrotestosterone concentrations that were <LLOQ and were not included in this table (3 from the trough values).
Table 14.4.1b
Pharmacokinetics: Analyte Plasma Concentration Summary
PK Population: Multiple Dose
Testosterone Testosterone Testosterone
DOSE Sample Sampling Time Free Total Dihycfro- Estradiol SHBQ number (hr) (ng/dL) (OQ/dL) {ng/dL} (PS/ml) (nrool/L)
·, 200 51 32,00 8 8 8 8 8
Arithmetic fcsean 0,462 31 ,488 11.396 55,925 57 , 350
3D 0.303 11.461 4,370 29.161 25.978
CV% 6S, 7 36,4 38,4 52,2 45,3
0.38 28,40 11 ,25 52,88 60, 80 fciin 0.21 18.20 6.25 28,40 23.40
1.18 54,10 18,00 116,00 33.00
Geometric Mean 0. 05 29.864 10.6 49.824 51.469
1200 52 40.00 M 8 8 7 8 S
Arithmetic Sean 0. 23 28.038 0.563 58.588 56.525
3D 0.38S 16.216 3.8S5 28.36S 24.388
CV% 91.3 5?.8 36.6 48.4 43.9
Sedian 0.28 23,60 11. io 53, SO 58.60
¾in 0.23 12.70 5.34 27.20 25.40 ssax 1.3?" 83. SO 16.10 1 7.00 85.80
Osoeietric Mean 0.345 24.914 9,917 53.273 50.289
1200 53 48,00 ίΐ 8 S 7 8 8
Arithmetic Sean 0.481 33.188 15.866 61.550 58.088
3D 0.3S4 13,432 5.615 34,850 25,643 cv% 73.6 40. S 35.4 56.6 44.2
0.38 30.40 15.80 53. S5 63.75
Mifi 0.28 17,30 8.20 25.40 24,00
M x 1.35 82. 0 22.30 36.00 91.70
<3e owe trie &ean 0.418 31. 48 14. SOS 54.513 52.37S
Note: There were 7 dihydrotestosterone concentrations that were <LLOQ and were not included in this table (3 from the trough values).
Table 14.4.2a
Pharmacokinetics: Analyte Parameter on Baseline Corrected Data (ConcBC)
PK Population: Single Dose
Pifiydrotestostercme
C2 C24 C24
DOSE Cm ax Tmasc AUCO-S AtffiO-24 AUCO-t AUCinf Lasibda_z t¾ CodeBase ConoBLO ConcSC in) (ng dL) < r) (r<r*fi9/cJL) (iir*ng/dL) (hr*ng/dL5 (hr>*ng/dL) {1/ftr} (hr) (ng/dL) (ng iL) (og/dL)
300 8 8 5 5 6 4 4 4 5 5 5
Mean 7.073 1 , 75 26,902 39.033 31.344 33,96 0,153 16.229 14,274 14.302 0.828
SD 4,364 2, 145 20. -2 24.104 23,866 32,803 0.195 17.135 6.305 4,437 ; ,253 cv¾ 70.1 122.6 74.8 61.7 74.9 98,6 127,7 105.6 44,2 31,4 151.3
Heef an 6.22 1.13 18.91 32. 5 28,43 53.51 0.08 11.72 13.4 14.3 0.27
Min 2.66 0.25 9,44 11.51 5.61 13.85 0.02 1.58 8.23 8.61 0 faax 16.27 6 63.32 71 , 37 70,18 202.98 0.44 39,33 22.1 19 2.97
GeoMean 5.8 SB 2 . SS9 32,483 23.515 55.582 0.077 9.009 13, 4 13.697
1200 8 8 S 6 8 3 3 3 6 6 6
Mean 6.23 5,033 23.903 45.5 7 43,261 72,971 0.05? 12,43 12.048 12,203 0,36?
SD 2,717 9,27 11 , 582 23.888 21.23 16.194 0,011 2,563 6,614 4.848 1.005 cv¾ 43,6 182,4 48,4 52,4 43.1 22.2: 13,8 20,6 54,9 39.7 116
8eJian 5.73 • ,S 28.32 44.88 42.33 82.22 0.06 11.83 3.53 10.5 0.62
Mrs 2.27 1 8.32 16,9 16.9 54.27 0,05 10,22 8.03 8,32 0
Max 9.62 24 37,33 79 , 02 69.98 82.43 0,07 15.24 25,3 21.3 2.27
Gso¾esr) a, 846 20.858 39.952 38.45? 1.647 0.05? 12.259 11.004 1,589
1800 f 6 6 6 6 6 5 5 5 6 6 6
Mean 11 , 392 2.792 52,9 98.838 98.078 158,68 0.1 11.855 1.865 13.615 1.318
SD 6, 751 2,021 36,24 62,638 62 , 97 118.436 0.072 9.247 4.745 5,125 2,331 cv¾ 59.3 101. J 88.5 63.4 64.2 73.4 71.6 7S 40 37.6 12 .6
Median 10.72 1.5 46.34 75,88 73,88 85.15 0,09 7,7 1,8 12.8 1.3 fen 3.93 0.75 18.3 41.71 .71 70.03 0,03 3.7 5,55 8.06 0 faax 23.4 8 11 .76 217,97 2· 7.9? 327.95 0.19 23.31 20 21.4 6.4
9.82 43,767 S6.043 85.18 128.512 0,077 8,971 11.068 12.834
Note: GeoMean = Geometric Mean
Note: Geometric Mean is not normally reported for Tmax.
Note: Geometric Mean cannot be calculated with Min = 0.
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2a
Pharmacokinetics: Analyte Parameter on Baseline Corrected Data (ConcBC)
PK Population: Single Dose
Free Testosterone
C24 C24 C24
DOSE Cisax Tisax AUCO-S Ayeo-2 Ayco - t AUCinf larttbda_z t¾ ConoBase ConcBLQ ConcBG if) {ng/dL} (Hr) {hr* ng/dL) ( r*rig/ciL} (ftr*fig/cJL) (1/Hr) (ng/dL) {rtg dti (ng/tJL)
800 N a a 8 a 8 8 8 8 8 8 8
SSesn 0.56 0.438 1.35 3.104 3.006 4,439 0,101 10,291 0,448 0,481 0,052
SO 0.415 0.258 • .138 1.804 } .659 2.363 0.081 5,036 0,412 0.361 0,055 cv% 74.2 59.1 S .5 ss.i 55,2 63.8 90.5 48. 91,9 75 107.2
Median 0.54 0.38 1.36 2.07 2.03 3.27 0.05 • 1,18 0,31 0.37 0,06 nm 0.16 0.25 0.94 1.45 1.41 1.47 0.04 2.21 0. 8 0,2 0
1.45 1 4.32 6.18 5,51 8.76 0.31 15,7 1.44 1.3 0. 6
Geafctean 0.442 1 ,622 2,711 2,652 3.758 0.079 8.784 0,357 0.401
1200 H 8 8 8 8 8 8 8 3 8 8 8 m 1.£7 0.656 3.588 5.255 5.265 6.098 0.104 7.289 0.346 0, 23 0.077 so 0.S13 0.481 s. jse 2.886 2.886 3,008 0.038 2.026 0.137 0.1 0.033 cv% 63.8 73.2 60.8 54.8 54.8 49.3 36.8 27.8 39.7 35,9 42.1
1.24 0.5 3.45 5.16 5. IS 5.94 0.09 7.81 0.31 0.39 0.03 i 0.32 0.2:5 0.57 1.23 1.28 • .64 0.07 3,79 0,2 0.24 0,04 mx 2.51 5,5 8. 6 10.81 10.61 11 , 42 0,18 9.59 0.53 0.S3 0.12
1.017 3.021 4.43S 4.4S8 5.354 0,099 5,995 0,322 0,4 0.071
-} 800 a 8 8 8 8 3 8 8 8 8 8 8
Hears 1 ,703 1 ,405 6.552 10.853 10.S26 •2.314 0.123 8.4 0.314 0.436 0.12:5
SD 0.755 1.932 2.416 5.457 5.516 6.292 0.053 2.205 0.076 0,1 1 0.089
CV% 4.2 137.4 36.3 50.1 50.5 51.1 43,3 34.4 24 25,6 79.5 fiSediart 1.86 0.83 6.96 11 ,02 1 ,02 ,3 0,11 6,37 0,31 0.48 0,13 am 0,41 0,25 2,36 3,68 3,68 4,04 0,07 2.3 0.19 0,28 0
2.53 8 9.48 21.22 21 , 22 23.23 0,24 0.32 0,41 0,82 0,28
Geosaean 1,5 6,053 9,615 9,561 0,689 0.115 6.049 0.306 0,423
Note: GeoMean = Geometric Mean
Note: Geometric Mean is not normally reported for Tmax.
Note: Geometric Mean cannot be calculated with Min = 0.
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2a
Pharmacokinetics: Analyte Parameter on Baseline Corrected Data (ConcBC)
PK Population: Single Dose
Total Testosterone
C24 C24 C24
DOSE Cesax Ttsax AUGO-S AUCQ-24 AUCO-t AUCinf larttbda_z t¾ ConoBLQ ConcBC
00 (ne/dL) (f»r) {Hr*ng,'dL} ( r*tig dL) (Hr*fig/dL) {1/tir) { r} (fig/dL) (ng/til) (ftg/dL)
600 8 8 s 8 8 8 8 8 8 8 s
¾ean 40,625 0,438 535,313 229.095 223.981 327.448 0.103 9.434 30.138 33.138 .175
SO 24.818 0,255 42,698 87,74 37.805 147.544 0,099 4.584 13,486 12.127
GV¾ 61.1 59.1 3 i , 6 38,3 39 45. SI .2 48,6 44,7 36.6 98.8 ssdian 40,85 0.33 134,49 197,29 195,32 343.35 0,07 9,62 27,8 30.1 4,1
SSin 1S.S 0.25 84.58 127.73 127.79 157.01 0.04 2.03 1 ,1 16 0 ax ee.7 1 85,66 386,86 386,86 553,97 0,34 16.5 58 50,7 10.4
Qeofaean 34,058 129,31 J 215.731 210.361 298,981 0.038 8,106 27,873 31.141
1200 3 s 8 8 8 8 8 8 8 8 8
Sean 7 ,063 0,688 225,758 328,002 328,002 372.178 0.113 6.472 23.533 23.013 4.475
SO 34,5 3 0.458 94,878 117.301 117.301 102 , 36 0.047 ,343 10.1 3 9.372 3.011 cv% 48. S 66. S 1 , 35 , 8 35.8 27,5 39.5 23.5 43.3 3S.2 67.3
¾eciian 73,1 0.S3 227.74 351.81 351.81 393.15 0.1 6.87 20,2 25.2 3.3
Mifl 21 ,9 0.25 65,78 102,53 102,53 161.39 0,07 3,09 13,3 17.2 1,4 &y, 133 1.5 377.86 448.61 448.61 4S7.25 0.22 9.34 41.1 44.6 11.4
Qeotsaan 62, SS 203,648 301.82 301.82 355.747 0.112 6.193 21.791 26.582 3.809
J 800 8 S 8 8 8 S 8 3 3 S 8
¾¾san 130,863 1 ,031 515.848 337.033 834.391 347,786 0.133 8,143 24.35 34 , 55 i 0.825
SD 57,929 1 ,221 230,105 12.846 41 .555 516.318 0.089 2.131 7.019 11.107 10.631
CV% 44.3 118.4 44.6 49.3 49.7 54.5 52.1 34.7 28.8 82.1 100.1
137.55 0.63 476,35 840,22 835.54 SOS.91 0,1 6.92: ?A,B 30.5 9.25 ton 27.2 0.25 169,05 273,14 273,14 316.07 0,09 2,57 17 5 0
94 4 898.74 1538,93 1533,93 889.73 0.27 3.07 38.8 53.8 34
Geo&san •13.912 464,728 742,847 788,181 830.483 0, 21 5.716 23,552 38.31
Note: GeoMean = Geometric Mean
Note: Geometric Mean is not normally reported for Tmax.
Note: Geometric Mean cannot be calculated with Min = 0.
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2a
Pharmacokinetics: Analyte Parameter on Baseline Corrected Data (ConcBC)
PK Population: Single Dose
Estradiol
C24 C24 C24
DOSE Cmax Tmax AUCO-8 AUCO-24 AUC0-t AUCinf lambda_z t½ ConcBase ConcBL ConcBC
{¾} (pg/»L) (hr) (hr*f>g/it!L} (hr*pg/8L) ( r*pg/mL) (ftr) {ρβ/rnL) ί PQ/SSL} (P3/aL)
6 GO !ii 8 8 8 8 8 5 5 8 8 8 m 23.275 10.816 112.503 235.334 230.951 718.407 0.063 30.458 50.1 47.438 6.863
3D 29.874 9.763 178.889 284,577 274.24 495.771 0,067 36.383 32,762 19.652 5.583
128.4 80.3 59 120.4 113.7 69 106.8 1 9,5 65,4 41 ,4 31 ,1
10.6 S 38.8 83.54 83.6 80 J , 34 0.05 13.92 39.2 45.6 5.5
¾in 3,4 0.25 0 10 JO 134.88 0.01 3,88 22.9 27.9 0
¾ , 8 24 528.41 723 879.8! 1351.44 0.18 93 , 63 122 90.6 IS
13.466 106.562 105.763 553,278 0.038 17.818 43.313 44.477
1200 7 7 7 7 7 2 2 2 8 S 8
Mean •4.188 8.884 34,928 J34. J9 3.848 383.708 0,067 21.98 40,288 42.6 7.1
SD 4.811 9,921 38.503 108.251 } 03, 84 198.335 0.068 22.543 18.466 14.517 7.553
CV% 33,9 100,7 1 ίθ.2 8i,3 82.2 51,7 102.7 102.7 40.9 33.1 106.4
Median •4.6 5 16.83 120.97 120,87 383.71 0.07 21.36 34.4 39.45 5. 5
7.8 0 9.3 7.3 243.46 0.02 6.02 22.5 26,6 0
Sax 22.2 24 101.1 322,73 322.73 523,95 0.12 37.9 75 70.2 19.9
<3eo¾isan 13,474 SO.078 75, S? 357.16 0.048 15.104 37.315 40.78
1300 7 7 7 7 7 4 4 4 3 8 3
Se n 4.343 7. 6 52.593 134,852 132.894 323,544 0.166 7.386 42.55 34.713 4.038
SD •3.699 10.266 93.515 259.625 262.975 537.276 0.182 4.339 16.284 22.553 8.599 cv 93 , 3 1 3.1 177.3 192.4 197,9 186.1 • OS.4 63.3 38.3 65 213 ssdian 3.2 14.63 52.95 49,63 71 ,87 0.0S 3.86 38,7 28.6 0 a 5.4 0.25 J.S8 9.74 8. J1 22,4 0.06 1.59 23.3 16.5 0
44.5 24 262,18 722.27 727.84 1128.44 0.44 12.24 65,4 88.2 24,1
Geoiaean 11. 7 18,811 48,85 43,97 105.227 0,114 6.103 39,849 30.552
Note: GeoMean = Geometric Mean
Note: Geometric Mean is not normally reported for Tmax.
Note: Geometric Mean cannot be calculated with Min = 0.
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2a
Pharmacokinetics: Analyte Parameter on Baseline Corrected Data (ConcBC)
PK Population: Single Dose
SHBG
C24 G24 C24
Cssax AUCG-8 AUCO-24 AUC0-t AUCinf t% CodeBase ConcBLQ ConcBC
( ) (nmol/L) (hr) {tir*rwol/l} (hr*n*ol/t> { r*rwol/l) (hr*nnioi/L5 iu {hr} (nnol/L) (nmol/L)
600 s s 8 8 8 4 4 4 8 6 8
7.138 4.25 £2.337 44.1 40.805 195.593 0.126 £0.765 Si.4 3 61. £5 1.£75
SD 3.849 3.563 15.325 27.308 23.53? 237.281 0.162 •3,318 22.272 23.136 3.295 cv% S3.S 84.3 66,7 63,1 72,5 121,3 129.1 33 35.3 37,3 258.
Mediae 6.05 4 16.01 32.3 29.83 39.2 0.08 20. IS 53 , 3 59 0 tain 2.6 0.25 8.74 53.63 3.02 35.66 0,02 1.92 18.7 17.5 0 isiax 14.3 8 48.46 92.06 92.06 548,32 0.3S 40,79 97.2 93,7 9.4
S . £84 19,12 37.116 31.316 117.S7S 0.06 1 · .53 56,534 55.833
1200 ti 8 3 S S 3 2 2 2 8 6 S
7.535 5.408 23. 98 45.416 40.309 83.885 0.323 .869 5 ί , 738 48.683 0.7 3
SD 3.927 4.946 25.337 33.363 33.331 13.559 0,341 5.147 23.757 si ,6se 2.0
CVS. 52.2 S ,5 109 31.4 37.7 15.6 105.7 105.7 45.9 43.5 262.3 f>¾ cii an 8 4 2· ,62 32,48 19,93 99,89 0.32 4,87 48. · 49.1 0
1 ,2 0,25 O.S 7,1 5.29 es.is 0.08 1.S3 17.4 17.2 0
M&x 11.8 16 80.45 111.62 111.42 111.59 0,55 8.51 85.7 77 5.7
Qeoaean 6.155 9.296 38.213 24,306 99.197 0.214 3.235 46,256 4 .723
1800 M 8 ? 7 6 8 3 3 3 8 e S
Sean 10. see 9.013 13.161 3 .es 32,388 205,537 0.053 2S, 135 63.383 34.518 £.25
SO 10,997 50.35 ,08 28.545 23.55· 34.938 0.062 20.854 33,477 30.402 2,54 ev% 101 11 .3 75.4 32.5 39.6 35.7 117.5 71.6 52,8 47 , 1 1 2.9
Mesial 7 6 11.4 31.73 31.73 222.05 0,02 36.78 53.15 57.7 1.75 . 0 0,25 0.54 7.33 0 63,1 0.02 5,54 27.9 31.1 0 y 2S.3 24 ,07 26.82 71.£6 71.£6 331.45 0.13 45.09 e 03 7.3
8,128 23.253 166.644 0.033 20.9 1 56.059 58,42
Note: GeoMean = Geometric Mean
Note: Geometric Mean is not normally reported for Tmax.
Note: Geometric Mean cannot be calculated with Min = 0.
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Analyte Pharmacokinetic Parameter Summary for Multiple Dose Profile (Dose of 1200 μg)
Population: Pharmacokinetic
Free Testosterone
Csiax Tmax CO Drain Cavg C24 AUCG-S AUCO-24 AUCO-tau PTF %PTS
(ng/dL) (hr) {ng/dt} (ng/dL) (ng/dl) (ilQ/CfL) {hr*ng/dL) (hr*rtg/dl} ( r*ne/dL>
8 8 8 8 8 s a 8 8 8
¾¾ean 1 ,886 0.3?8 1, 155 0,704 , oos 0.578 a.0 S 17.259 8.04S 140,635 214,058
SD 0,233 0.18? 0.394 0,388 0.408 0.461 3.266 9,498 3,266 77.680 1 ,818 cv¾ 12,4 49. S 34.2 5S.1 40, S 79.8 40.5 -.5.0 4 , 6 5S,2 5S,0 itedian 1 ,32 0.26 1.OB 0. s? 0.86 0.44 6.88 13,48 S.89 144. S? 21?.91 sain • ,SS 0.25 0.77 0.40 0.69 0.28 4.68 S,74 4.69 30.60 36.00
2.18 0.75 1.82 1.60 1.83 1 .71 4.64 39.66 14,54 29S.7S 43?. S3
1.874 0.344 1,098 0.638 0.944 0.492 ? .554 15.722 7,554 31S.579 1 6.905
Total Testosterone
Csiax Ttiia CO Grain Cavg C24 AUCO-8 AUCO-24 AUCO-tau %PTF %PTS
(ng/dL) (hr) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (hr*ng dL) (hr*fig/dL) (hr*ng/dL)
8 8 8 8 8 8 8 8 8 8 8
548.038 0.440 85.450 49.088 73.802 38.513 590.419 1224.571 590,4 9 141 ,760 256,089
SD 6 ,395 0.219 27.406 7.870 30.57? 18. 67 244.612 471.964 244,6 2 76.222 115,709 cv% 41.5 49.8 32.1 38.4 41,4 47,2 41.4 38.5 41.4 53,8 53,5
Stedian •38,00 0,39 32,25 46.15 59,21 34.45 S53.68 1176.03 553.58 133,23 220,4?
85,30 0,25 40 , 50 25.20 41 ,12 18.70 329.00 679.18 323,00 48,83 84,4?
Max 242,00 0,75 131.00 79. SO 141,00 77.30 1127,99 2026.22 1 27,99 301,43 447,51
137.565 0.395 81.313 46.135 69. 68 35.380 5S3.325 1 47.586 S53.325 125 , 00 187.544
Dihydrotestosterone
CiliaX Tma CO Grain CavQ C24 AUCO-8 AUCO-24 AUCO-tau %PTF %PTS
(ng/dL) (hr) (ng/dL) (ng/dL) (ng/dL) (ng/dL) (nr*ng/dL) (hr*ng/dL) {hr*ng/dL)
H 8 8 8 8 s 8 8 8 8 8 8
22.300 0 , SOO 20,724 3.465 ■6.628 14 , 774 133,024 338.888 33.024 57.240 73,955
SD 8.005 0,267 8. ISO 5.041 6.658 5.S21 53.262 130.886 53,262 2S.2 7 36.035
GV% 35.9 53.5 39.3 44.9 40.0 40.1 40.0 38,5 40.0 44.1 48.7
23.90 0.50 21.45 13.15 18.14 16.00 145.09 361.3? 45.09 £4.12 68,72 airt 10.20 0.25 7.39 5.32 7.21 6.33 57.66 145.8S 5? .66 13.66 14.90
30.50 1.00 31.70 20,80 24.30 24.70 194.43 494.9? 94.43 96.25 125.53 o - m 20,792 0.442 18.92? 12.192 15.274 13.637 122.194 313. 98 122,194 50.641 63.444
Geo-Mean = Geometric Mean; Tau = 8 hours; %PTF =Percent Peak to Trough Fluctuation; %PTS = Percent Peak to Trough Swing
Table 14.4.2b
Analyte Pharmacokinetic Parameter Summary for Multiple Dose Profile (Dose of 1200 μg)
Population: Pharmacokinetic
Estradiol
Cfflax Tni&x CO Cmin Cavg 024 AUCO-8 AUCO-24 AUCJFAU %PTF %PTS
(pg aiL) <hr> (pg/iBL) (pg/!Bt) fpg/mt) {hr*pg/mL) {hr*pg/iSL} (hr*pg/ml) n s 8 s 8 8 8 8 8 8 8 8 &an 70.800 3.231 54. 13 48.875 58.019 64. 13 464. 43 1759.342 464. 48 36.465 42. 64
SD 38,885 2.576 23 , 936 2 .749 31.328 28.365 250,621 727.142 260,521 16.304 19.763
CV% 54.9 79.7 44.3 SO.6 54.0 44,2 54.0 41.3 54.0 44.8 46.9
76.75 4.00 57.45 49. SO S8.94 S6.5S 47 .53 1768.32 471.53 29.77 3S.S4 ton 16.60 0.27 16.30 12.70 14.36 18.60 119.67 503.91 119.67 20.16 22.22
Sax ■545,00 8,00 84.20 95,20 120.84 104.00 956,13 2895 , 80 985.13 68.76 79,82
Geo- (Sean 60.034 2.024 48.062 42.5S9 60,033 5S.82Q 400.264 1587.754 400.264 32,768 38.514
SHBG
Cfflax Tmax CO Grain Cavg C24 AUCO-B AUCO-24 AUC_TA0 %PTF %PTS
(nmol/L) ( r) (r!Riol/L) (nisol/L) (MOl L) (nraol/L) (hr*nmol/L) (hr*nstol/L} {hr*nmol L
H 8 8 8 8 8 8 8 & 8 8 8
62.725 1.998 59.700 55.300 58.548 58.275 468.387 1375.463 488.38? 3.778 15.114
SD 28,971 2.716 27,501 26.279 26,885 25.630 25,07? 69.735 215,077 7.805 9,781 cy% 46.2 136.0 46.2 47.5 45. S 45.5 45.9 45.1 45.9 56.6 84.6
S4.5S 0.75 63.70 53,36 60.92 59.65 487.37 1446.26 48? .37 11.24 11.86
27.40 0.25 26.40 19.90 23.91 25.10 19 .29 591.68 81.29 S.10 S.4S
Sa 100,00 7,88 93.90 87,90 82.85 87.30 741 ,16 225 , 93 741. 6 31.37 37.89
Geo · fiiean 56.270 0.964 53.630 49.024 52.530 50.536 420.235 1239.069 420.236 12.392 13.279
Geo-Mean = Geometric Mean; Tau = 8 hours; %PTF =Percent Peak to Trough Fluctuation; %PTS = Percent Peak to Trough Swing
Table 14.4.2c
Pharmacokinetics: Analyte Parameter on Uncorrected Data (ConcBLQ)
PK Population: Single Dose
Frse Testosterone
DOSE Caax Tsax AUCO-8 AUC0-24 AUCQ-t AUCinf Lambda^ ΐ½ in) (ns/dL) (hr) (ftr*ng/<a,} (hr*ng/dl) (hr*ng dL5 {(ir*fi8/dL) (1/hr) { r?
600 8 8 8 8 8 8 8 8
0.921 0,44 4.563 1 . est 22.2S2 53.491 0,022 46,153
SD 0,729 0,26 3.4S2 S.398 3.37 42.421 0,01 41.022 cv¾ 79, 50.1 76..5 80,5 82.4 79,3 46,0 S8.9
Median 0.75 0,38 3.16 a 15.62 37 , 52 0 , 02 31.53
0.35 0.25 2.43 5.51 9.56 15.16 0 19.09
Max 2.64 12.7? 33,6? 66.31 1 8.64 0.04 141.79
<3ββΜ63Π 0.761 0,33 3.S44 9.635 8.15? 40,555 0,01 36.668
J 200 8 8 8 8 8 8 8 8
Sean 1.537 0.66 5.70? J .920 20. J 44 3 . 7 0.044 20.304
SD 0,865 0.48 2.683 5,014 3.366 5.835 0.024 11.456
CV% 56.3 73.2 46,8 42 41.5 49.1 65,2 54.8
Median 1.45 0.5 5,57 12.07 •9,4 30.18 0,04 18.17
Si in 0,47 0,25 2.16 4.9 9,35 15.5 0.02 8.33
¾ax 2.88 1.5 10.83 9.79 31.59 57 0 , 08 38.1
1.302 0,51 5. 62 JO.923 18.57? 23.39 0,038 18.126 iaoo 8 8 8 8 δ 3 8 8
Sean • , 372 1.28 8.6 6 17.261 26.389 35.582 0.061 14.057
SD 0,746 5.92 2.349 5.506 5,548 8, 85 0.025 7.939 cv% 37,8 150 27.3 31.9 20.6 23 41 56.5
Median 2.17 0.63 8,65 16.96 25.01 34.43 0,08 10.79
Kin 0,79 0.26 5.32 8.85 22 26.33 0.03 7,7?
2.81 6 11 , 86 28.48 33.6 46,2 0 , 09 27.46
Qeefsfean 1.822 0,76 8.315 16.643 26.55 34,755 0,056 12.475
Note: GeoMean = Geometric Mean
Note: Geometric Mean is not normally reported for Tmax.
Note: Geometric Mean cannot be calculated with Min = 0.
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2c
Pharmacokinetics: Analyte Parameter on Uncorrected Data (ConcBLQ)
PK Population: Single Dose
Total Testosterone
DOSS Cmax Tuax AUCO-8 AUCQ-24 AUCG-ΐ AUCirvf LaB eia_2 t½
W (ng W ifir) ihr*ng/dL) ihr*ng/dL) {fir*ftg/cSL) (iir'ne/dL) {1 /hr) (hr)
600 H 8 8 8 8 8 8 8 8
Sean 64.413 0.433 31 ,68 788.804 1 SOS.036 3088,726 0.Q2S 32.08·
SD 30,338 0.258 107.529 302.684 598.271 1371.43 0,011 15,524 cv¾ 47.1 59.1 34.3 38. 39.7 44.4 42.4 48.4
Titian 61.2 0,38 275.76 655.53 1325.21 2979.19 0,02 30.35
34.3 0.25 200.03 440.57 761.42 131.25 0.01 1 .61
111 1 492,48 1307.75 2520.83 5:26.34 0.04 es. ?
Geo¾ean 58.438 0.385 293,591 74 J .257 1407.828 2731.041 0.024 23,124
1200 H 8 8 8 8 8 8 8 8 an 88,413 0.688 388.542 770.154 1313,3 22 9.324 0,033 24, 53
SD 35.988 0.453 23.433 261.457 476.097 1029,231 0.031 10.197 cv% 40.7 68.6 33.5 33.3 36.3 46,4 79.1 42,2
Media 87,2 0.63 369, 1 727,4 1192,92 1858,84 0.03 26.27 fain 32.9 0,25 154.25 356.66 595.88 1257.6 0,02 6,36
M x 153 1.5 507,38 143,41 2040,36 3867,97 0.1 35,4 eso&san 81.287 0.553 346,535 727.8 1238,515 2038.713 0.032 21.336 800 8 8 8 8 8 8 8 8
52.038 1.281 582.658 1339,176 2137.518 2722.518 0.063 13.626
SD 57,796 5 , 92 243.898 446.561 652.74 543.78 0,025 7.554 cv¾ 38 149,9 36,6 33.3 30 , 5 23.6 39 , S 55. sseefian J 64 0,53 657.36 1322.32 1972.93 2553.01 0,07 8.3 iiin 53,3 0.25 359,38 851,3 1370,95 20 8,49 0.03 7.86
SSax 218 5 1121.18 £230.33 3289.28 3902.68 0,08 25.4?
Gao¾¾as 39.742 0.755 642.877 1279,5 1 2055.138 2660,045 0.057 12.149
Note: GeoMean = Geometric Mean
Note: Geometric Mean is not normally reported for Tmax.
Note: Geometric Mean cannot be calculated with Min = 0.
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2c
Pharmacokinetics: Analyte Parameter on Uncorrected Data (ConcBLQ)
PK Population: Single Dose
Difiyd rotestoste ram
DOSE Gmax Tmax AUCG-8 AUCO-2 AUCO-t AUCinf t½
M {ng/cti.) (hr) ( r*ng dt) {Hr*ng/<Jt} {hr*rtg/dL) (1/hp) (fir)
600 ti 8 6 6 6 6 4 4 4
16.55 IS.583 04.453 24-0.171 474.385 2374.844 0,033 82.239
3D 7.86 8,866 44.625 1 3.081 2SS.397 3048.362 0.04 1 .2:41
GV% 47.6 156.3 47.2 48.6 60. S 123.3 119,3 136,5
Median 15.65 1.25 85.81 234,4 432,09 1300.46 0.02 36,02 sain 8.74 0.5 48.34 33,95 27.88 94.36 0 7.56
Sax 26 43 149,81 389.38 811,06 6803,59 0.09 243,36
QeoMean 14.311 2,749 85.535 2 1,826 324.386 962,729 0.018 39.495
1200 !4 8 6 8 6 6 3 3 3
Me 14.75 4.75 91,513 243,734 476,044 926.355 0,022 33.132 so 5.708 9,445 35.341 5 4.276 22:0.361 2:02.7 1 0.006 10.923 cv% 38.7 98.8 39.? 46.9 46,4 2:1 ,9 28 32.9 sc!ian 12.6 1, 13 79.48 206.39 393.65 849.53 0.02: 27.91
Sin 10.3 0.25 67,31 68.29 317.4? 773,25 0,02 25.89
Max 25.6 24 1 S3.3 474.68 919.73 1156,28 0.03 45.75
QeoMean 14,009 1 , 31 86.973 227,956 445.288 912,406 0.022 32.094
1800 H 8 6 6 6 6 6 8 6 ean 21.05 2.792 133.293 332.708 652.145 1 20.714 0,024 67,51
SD •0.047 2,821 64.6:56 49.72 274.719 1304,629 0.017 90.393
CV¾ 47.7 101.1 48.5 45 42, 1 94 S3 , 1 34.8
Ma iian 18.4 1.5 118,61 280.57 609.37 1088,26 0,02: 30.2:4 n S.7 0.75 77.61 218,2 397,76 731.22 0 13,99
Max 39.9 8 254.64 6 7.43 1136.66 5465,44 0.05 250.28
19.44 .908 122.744 310.621 60S.328 1471.226 0,018 39.485
Note: GeoMean = Geometric Mean
Note: Geometric Mean is not normally reported for Tmax.
Note: Geometric Mean cannot be calculated with Min = 0.
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.3
Dose Proportionality Analysis
Population: Single-Dose
95% Confidence Lima for
PK Parameter
Standard H I
Astaryte PK Parameter Form Estimate Error Lower Limit Upper Umit :■■ ·■■■¾:■-<¾ F-Square
Toisi Testosterone AUG 0-24 Orsginai) Estimate 415.4053 198..6384 3.471 1 827.3394 0.0483 24
Stop* 0.4588 Q.I 532 0.7766 0.0087 0.2395
NaturaMog transformed intercept 6.2383 0.2033 5.8167 6.6539 < Q0D1 24
Stop* Q.O0O5 Q.00Q2 0.0O01 O OOOS 0.0083 0.2766
AUG o-sn Oilginai) Estimate 8? 3153 97 3538 -114.6048 289.2354 0.37S5 24
Stop* 0.3068 0.0751 0.1509 0 4624 0.0005 0.431
NaturaMag fransformecS intercept 5.2419 0 2051 4 8166 5.6672 •i.0001 24
Stope 0.OQ0S 0.0002 0.0003 0 0010 O.0Q06 0.4237
AUG C-!nf {Original Estimate 3043.0640 587.0430 1825.6113 4260.5167 <.0001 24
Stope -0.3052 0.4529 -1.2445 0 63 1 O.S075 0.0202 MatUFsMag transformed interc< 7.8S1S 0.2269 7.3909 3.3322 O05 24
Stope -0.0000 0.0002 -0.0004 0,0003 0.82 1 0.0024
A U G 04 {Original) Estimate intercept 1023.6488 334.3854 329.0392 1718.0667 0.0058 24
Stope 0.5245 0.2534 -0.0113 1 0603 0.0546 0. 5578 MatUFsMag transformed 8.9551 0.2OS8 8.5282 7.3820 O05 24
Stope 0.0003 0.0002 -O.OQOO ø 0306 0.0597 0.152
Cmax (Original) Estimate 13.9958 23.3101 -34.3464 62 3380 0.5544 24
Stope 0.0730 0.0130 0.0357 0 1103 9.0005 0.4234 NaturaMog transformed inte s 3 5958 0.2497 3.0789 4.1147 <.0O01 24
Stope 0.O007 O.0OO2 0.8003 0.0011 0.801 1 0.3926
Linear regression model is fitted to a PK parameter by using Dose (= 600, 1200, 1800 ) as a predictor variable.
Reference Listing(s): 16.2.5.2a
Table 14.4.3
Dose Proportionality Analysis
Population: Single-Dose
85% Confidence limit for
PK Pa;¾rfi¾ier Estimate
Standard H t
Anaiyte PK Parameter Form Parameter Estimate Error Lower Limit Upper Limit P-Vaiue R-SqLiare
Free Testosterone: AUC 0-24f> {Ort ral} Estimate intercept 8 0443 3.7148 03403 15 7482 0.0415 24
Slops 0.0048 0.0023 -0.00-13 0.0106 0 1 90 0 1069
NaiursMog transformed intercept 1.9470 0.2539 1 204 2 4736 f. OO 24
Slops 0.0005 Q.O0Q2 O.OOOO 0.0009 0.0313 0 193S
AUG 0-8h (Orig rial) Estimate intercept 2.2426 1.5363 -0.9436 5.428? 0.1565 24
Slope 0.0034 0.0012 0.0009 0.0053 00093 2695 aturaMoo transformed intercept 093OS 0.2469 04185 1 4425 0.00 24
Slope 0.O0 6 0.0002 0.0002 0.CO10 0.002? 0.3412
AUC O-tnf (Original) Estimate intercept 53.3563 14.3921 23.5090 83.2037 0.0005 24
Stops -0.0149 0.01 1 1 -0.O360 0.0061 0.1926 0.0759
M atur a! -log tra reform ed intercept 3.6925 0 304? 3.0616 4,3234 « 0001 24
Slope -0.0001 0.0002 -0.0006 0.0004 0.5892 0.0135
AUC 0 (Origa-iaf) Estimate intercept 8.4454 6.4871 4.3919 31.8969 0.0035 24
Slope 00035 O.0050 -00065 0 0143 0.4423 0.027
Natural-log transformed intercept 2.6558 0.2430 2.1493 3.1614 O0O1 24
Slope 000O3 0.0002 -0.0001 O.OOO? 0.1 ceo 0.1132
Crrsax (Original) Estimate intercept 0.4260 0. 136 -0431S 1 2337 0.31 2 24
Slope oxxm 0,0003 0.0002 0.0015 0.01 8 0.255
Naturai-fog fransformecl intercept -0.6772 0.3067 -1.3132 -0.0412 0.0380 24
Slope O.OOO? 0.0002 0.0002 0.0012 00055 Q.X1
Linear regression model is fitted to a PK parameter by using Dose (= 600, 1200, 1800 ) as a predictor variable.
Reference Listing(s): 16.2.5.2a
Table 14.4.3
Dose Proportionality Analysis
Population: Single-Dose
95% Confidence limit for
PK Parameter Estimate
Standard
AnaSjfte PK Parameter Parameter Estimate Error Lower Limit Upper Limit P-Va!ue R-Sqrtare
Oi ydraiesiost e-ore (OHT) AUG 0-24 (Original) Estimate Sntsreepi 73.5673 73.8150 2.5375 346 7463 0.0367 18
Stops 0.0771 0.0608 -0.0518 0 2060 0.2229 00913
NalursMog transform^ 5 1250 0 2776 4.5366 5.7134 <.0O01 13
Stops 0.O003 0.0002 -0.0001 0.00O6 0.1557 0 1218
AUG o-an (Original) Estimate Sntsreepi 67.5802 30.9702 1.S263 133 2340 0.0444 18
Stops 0.0324 0.0239 -0.0183 0.0830 0.1944 0.1029
NatursMcg trai»form«i intercept 4 2137 0.2596 3.6630 4.7643 <.0O01 13
Stops 0,0003 0,0002 -0,0001 o.ooo? 0,1526 0.1236
AUG O-inf (Original) Estimate intercept 2194.7671 1350.7672 -1213.4488 5607 9827 0. 847 13
Stops -0,2615 1 ,1 138 7330 2.1700 0,8051 0.0058
NaturaMog fransformecs intercept 6.5682 0.3010 4.806.2 8,3321 <.0O01 13
Stops 0.0004 0.0006 -0.0009 0 0016 0.5284 0.0371
AUG 04 (Original) Estimate intercept 358.4352 161.0962 14.9132 697 9441 0.0418 18
Stops 0,1461 0,1243 -0,1 153 0.4116 0,2507 0.0315
NaturaMog fransformecs intercept 5.4672 0.4766 4.4566 6,4779 <.0O01 13
Stops 0,0005 0,0004 -0,0003 0.0013 0,1724 0.1131
CIT<8>: (Original} Estimate intercept 12.9100 5.0315 2.2436 23 5764 O.0207 18
Stops 0.0038 0.0039 -0.0045 0.0 20 0.3453 0.0553 aiurai4og transrormseci intercept 2.5045 0.2636 1.S453 3 0637 --,0001 IS
Stops 0.0002 0.0002 -0.C0D2 0.00O7 0.2936 Q.OSSS
Linear regression model is fitted to a PK parameter by using Dose (= 600, 1200, 1800 ) as a predictor variable.
Reference Listing(s): 16.2.5.2a
Table 14.4.4
Analysis of Variance for Some PK Parameters
Population: Single-Dose
Cohort i {600 meg} Cohort i ;6O0 megs Cohort 2 {1200 meg}
Coffteienl vs. vs.
Analyse PK of arsaston Cohort 2 (1200 meg) Cohort 3 {5300 meg) Cohan 3 (1800 rre¾)
DSH Y DROTESTOSTERONE AUG o-z-m 19.818? < 00Q1 < OQ01 0.5756
AUC 0-6 235230 « 0001 «.G0O1 0.3291 AUG 0-inf 26.0 51 0.0048 0.0004 0.3131 AUC 0 26.5016 0.00 0,0001 0.3144
37.3379 0.0021 0.0009 0.6763 568.0250 0.5869 0.1523 0.9553
: 1/2 117.565? 0.2274 0.1041 0.7665
FREE TESTOSTERONE AUC 0-24h 458505 0 2114 0 2182 09845
AUG 0-Sh 87.4794 0.7510 0.5338 0.7996 AUC 0*f 43.0039 0.0324 0,0137 0.813a AUG 04 45.334 0.2439 0.2477 0.992 Cmax -223.3208 0.0034 0.0010 0.6132 Lambda„2 84.7668 0.0908 0.0406 0.6S63 1 /2 65.9189 O.0002 «,ΟΟΟΙ 0.3267
TOTAL. TESTOSTERONE AUG 0-24h 7.S951 0001 .00O1 O.0O01
AUC 0-8h 8.2332 «.0001 « 0001 0.0001 AUG 0-inf 8.7190 00Q1 O0O1 0.00Q2 AUC 04 78792 « 0001 «.0001 0.0001 Cmax 17.9163 « 0001 < 0001 0.0320 lam da .z 35.1223 0.1163 G.04S4 0.6333 i 1/2 56.6641 0.0002 «.0001 0.3940
Coefficient of Variation = 100 x ANOVA residual error (Root Mean Square Error) / PK parameter mean.
Pair-wise comparisons are from the ANOVA model with Dose as a class (categorical) variable and PK parameter estimate as response, where AUC 0-8, AUC 0- 24, AUC 0-inf, AUC 0-t and Cmax parameter values are natural logarithmic (In) transformed, and all param
Reference Listing(s): 16.2.5.2a
Table 14.4.5
Paired T-Test Results for PK Parameters AUCO-8 and AUCO-24
Population: Subjects of 1200 meg in Both Periods 1 and 2
95% Confidence limit for an
PK Parameter Difference
Mea of Standard Error
Ansiyie Name Fonts N Di ference of Difference Lower Lints Upper Limit
D! H Y D OT ESTOST E RQ NE AUG 03h iOfiginsi) Estimate 3 21.713? 20.3497 -658439 109.2714 0.3977
Maiurai-!og, transformed 3 Q.T731 0.2183 -0.7662 1.1124 0,5 1
AUC 0-24 (Original) Estimate 3 33.3448 50.7850 -186.1657 251.8548 0,5739
NatureMtH) transformed 3 0.0815 0.2217 -0.8725 1.0355 0.7435
F EE TESTUb ! EHOHE AUC 0-Srt (Osigina!) Estimate 3 -Q.3705 1.S416 -3,7246 7.S83S 0.8683 s\iaiurai-k¾ transformed 3 0.0564 0.31 OS -1 2322 1.3930 03750
AUG 0-24H iOriginai) Estimate 3 -0,61 S3 3.3448 -15,9076 13.7759 0.8709
atuiaMog transformed 3 0.0331 0.2732 -1. 24 12086 0.0147
TOTAL TESTOSTERONE AUG iMfti iOfiginsi} Estimate 3 5.9324 2 .3340 ■>■ mm 101.7253 08S73
NatuiaMog transform d 3 0.019? 0.0499 -0.1049 0,2343 0.7312
AUC 0-24h (Original) Estimate 3 6.3295 25,6895 -104.2036 6,8626 0.8284
Naturai-!cg transformed 3 -0.0003 0.0274 -0.1183 0.1171 0.9736
The difference is Period 2 - Period 1.
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose— 600 μ§)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone 'Individual Concentrations (Dose = 600 g)

ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concen trations (Dose = 600 μ%)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone individual Concentrations (Dose = 600 |ig)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone individual Concentrations (Dose - 600 jig)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose - 600 jig) ¾¾ss y?:™¾?ms¾*:. M&§8§s su s&s

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose - 600 ug)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone 1 m lividual Concentrations (Dose = 600 (tig)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose - 1200 jtg)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose— 1200 u¾)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose = 1200 μ§)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose—— 1200 pg)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose = 120(1 μg)

5¾
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose = 1200

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose— 1200 μ¾)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone ftidividitai Concentrations (Dose = 1200 jttg)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosteron e Individual Concentrations (Dose = 1 00 jtg)

"S- *x>:;S.xSf v? ξ;Α?¾:; i n'£ ¾·*· x ^'¾>>
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose = 1800 fig)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concent rations (Dose = 1800 pg)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose = 18ΘΘ g)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose = 1800 ¾)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose = 1800 μ¾)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual C oncenf rations (Dose = 1800 μ¾)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Free Testosterone Individual Concentrations (Dose = 1 00 ug)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose = 6ΘΘ μ }

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose Ξ 600 μ§)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (I ose = 600

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone individual Concentrations (Dose = 600 μ-g)

¾«w£f¾t ¾.A:? .?J> ~«S™ orsic *? C«:¾ >£ lyysfggs
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose - 600 |ig)
*· >5.Ϊ5ί »: ίί-ί ! £5 ί \>isx i '¾ί

Χ ί
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concent rations (Dose

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosteron individual Concentrations (Dose = 6(10 μ-g)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose = 6(H) μ§)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosteron Individual Concentrations (Dose = 1200

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose - 1200

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone individual Concentrations (Dose = 1200 ρ

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose = 120(1 μ«)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone individual Concentrations (Dose = ! 200 jig)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose - 1200 μ¾)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose = 1200 μ }

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concent rations (Dose— 1200

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose = 1 00 u¾)

C¾X¾,Q v § "*>~ ? ¾# fe& X? »¾«X »;s X>¾¾P
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose = 1800 μ§)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose = 1 00 ¾)

··«*··· iiX Si YS S :¾ ¾: ··'*··· C S S ':· ? $L¾?¾!C
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose = 1800 pg)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose = 1.800 j*g)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentrations (Dose = !SOO μ§)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Total Testosterone Individual Concentratrotis (Dose—— 1800 p.g)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Difaydrotestosterone Individual Concentrations (Dose

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Dihydrotestosterone individual Concentrations (Dose = 6(1(1 fig)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Dih drotestosterone individuai Concentrations (Dose = 600 u«)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Dthydrotestosterone Individual Concentrations (Dose - 600 μ§)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Dihydrotestosterone Individual Concentrations (Dose = 600 pg)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Dihydrotestosterone Individual Conceotrations (Dose = 600 ug)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Dihydrotestosterone Individual Conceotrations (Dose = 1200 μ§)

i"S?i ¾S. SlA S ···»··· O SC s-S
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Dihydrofestosterone individual Concentrations (Dose = 1200 j& )

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Dili yd rotes toster tie Individual Concentrations (Dose = 1200

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
l>ihyd rotes toste rone individual Concentrations (Dose = 1200 j& )

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Dihydrotestosterone Individual Concentrations (Dose = 1200

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Difcydrotesfosterone 'Individual Concentrations (Dose = 1800 j&g)

···*-·· ¾¾&y v;; ¾»Si> ··:·»··· £«««§«8« v? a,;¾i¾:>
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Dih drotestosterone individual Concentratioti s (Dose = 1200 M )

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Dih drotestosterone Individual Coocentratiotis (I>ose = 1200 pg)

www
··*··· C "¾i«k $ wu ··**··· c¾»*J* «$ ¾*>¾S
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Dihydrotestosterone Individual Coiicentratioiis (Dose = 1200 ug)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Dihydrotestosterone Individual Concentrations (Dose = 1200 ,ug)
22: M
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
D lsyilr tesiosterene Ittdividoal Concentrations (Base

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
(Table not in pdf - was blank)
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Individual Coaceisfr loas (Dose— 1)®

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Individual Conc ntrations ( ose = C*t i p¾)

3
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol individua Concent ations (Dose ~ i© μ

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Individual Concentrations Dose™ 6(Mi jqg)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Individual C««ee«irati«¾s Dose™ MW jig)

··<*·· :'. f:s:<?A. : ssSA^SS
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Individual ConceirtratioHS (Dose = 60© fig

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Est adiol ½di¥td«ai Cosieeairatiees ( ose = <*0O fig)
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Individual Concentrations (Bme 6§§ fig)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol individual C¾« ceo trat less ( ese— 1200 jsg)

■··«··· Csssli w H¾?¾S .··*·*. ;'.'··> i ί f.'i..¾¾ ::0
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol ¾dlvld«al Coitceatr&iieiis Dose ~ il® μ

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol is.di¾islita! Co»c€»trat.ioas (Dose ~ ίίϋ ρι)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol la dividual Conceit traties3$ (l «se - I2§ p.g)
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Individual Con ent ations. (Dose = 12OT jig)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Individual orrceHiratloas {Doi« ~ !2§§ pg)

···*··· ·:'.':> ;:¾·:.' ν§ Π .¾«··''·
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Iiidtvislitai Co»c«iilralio»s {.Dose - 1200 g)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Individual CoBcentratioss (i o&e J M) jig

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Individual Coseentraii ttis Do&e— ISM ^g)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Est radio! IM dividual Cosi eeu trafloas (Dose '18$® ρ

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
stradiol Individual Coneeiitratioss e !$§§ fig)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Individual Concentrations (Dose - 1 SIM! ig|

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Individual Concentrations (Dose 855 !&©& fig)

ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
stra iol Individual Coaeeiiiratlotis (Bu . ~ 1S 0 g

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Indivi ti&E Coneeiitratiotis (Dose■» 18ίβ© μφ

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Estradiol Ixidivldsial Co8C€»fratio»f (D«se ~ f NS jig)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SHBG individual Gra eeut rations (Dose

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Sil.BG In ivi ual Caiiceittratioiis l>ese = dO© g)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SH G IndMdnai Coiiee«tratio«s (Dose— 6(i§ jttg)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
§M G Ifldivi ita! Concentrations (Dose ~ 6-0D ftfg)

··*··· i g ^ss ··«··■ C««s :ss* ! iSO ··«·· »s
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SH.8G ledmdual Concentrations (Dose - 6$ i g)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SIIBG i»d tvi toa! Coacentrafleas (Dose 6§θ g)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SMBG fediv tiiiJ Concentrations { ose - i MJ fig)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SHIM* iadtvidoal Co cesa trailers |.l ose - 6§ pg)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SH G liitllvklwal€e»eei!irat &ii3 <B»ie ! 2 §

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SH G liitllvklwal€e»eei!irat &ii3 <B»ie ! 2 §

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
Sf!BG Individual Concentrations (Dose

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SM G liidividttiil Concentrations Bos* ~ I2## μ

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SliBG ladiv'tciti-ti Coaeeiitr ioiis (Dose™ OCM) tg)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SM G Individual Csncentrati&as (Do.se = 124ϊ# g)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SIIBG individual Coiiceatmtieiis (Dose - 12§© jtg)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SI1 G Indivtdti&l. CeacealraticHii (fkise ~ !2© g)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
S IBG Individual Coiiceiiir ions (Dose - Ι2Φ0 μ

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
§11 BG Individ «al oncentrations ( ose ** g)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SfiBG ladi'vidtia! Coiicenlraiieas —■ !SIMf p§

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SltBG indtvid«al oncentrations (Base ~ 18¾0 .g)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
S.HBG Individual ConcestrafMiiis (Dose » 1SI jtg)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SHBG individual Concentrations (Dose = ϊ� «)

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
S1IBG isdivtdtm! Ce«€eatraiie»$ (Dose— IS§# g)

ote: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Table 14.4.2b
Pharmacokinetics: Analyte Parameter Summary
PK Population: Multiple Dose
SliBG Individual Cottceiitr ioiis (Dose 35 ISCNf g
Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Figure 14.4.2a
Individual Analyte Plasma Concentrations
PK Population: Multiple Dose
rree lestosterone
PCTEST=f SE TESTOSTERONE, USUB3_D=522-¾1

Note: ConcBase = Baseline Concentration; ConcBLQ = Active Dose Concentration with BLQ zero or missing; ConcBC = Baseline Corrected Concentration
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Free Testosterone

Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Free Testosterone
:PC7EST=FREE TESTOSTERO E, USU85IO= 522-17

Elapsed ! ree [Hours}
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Free Testosterone

Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
-tee ieslosieraae
PC7EST= FREE ESTO STE OL E, U SU Β3Ε>=522-28

dapsed li 8 *cu?
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Free Testosterone

Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Free Testosterone
P€TEST=;F*EE TESTOSTERONE. USUSJID=522-¾5

dapsed Thse (hours
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
.Free estoster ne
PCTcST=FREE TESTOSTERONE, USU:E iD=522-S8

Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Total Testosterone

Figure 14.4.2a
Individual Analyte Plasma Concentrations
PK Population: Multiple Dose
'ϊύί Testosterone

36 ¾! -)·$ 52
Elapsed TsrrffiChcisfs}
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Total Testosterone

Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Total Tesoterone
PCTEET=T07¾L TESTS STE O E, USU BHD =522-34

la sed sirseClTCtiiis)
Figure 14.4.2a
Individual Analyte Plasma Concentrations
PK Population: Multiple Dose
otai Ί eslosterone
-C EST=TQmL TESTOSTERONE., U5U¾ID = 522-28

4 S 12 IS- 2Q Z-% 2S ; 2 3 & ¾F -ag. Si
Elapsed Tirfse ( airs)
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Ί ota! Testosteron
PCTEST=TOTAL TESTS ETTEROME, USU83ID = 522-29

El apsed Ti e{hc-ii sf
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Total Testosterone
«ΠΕ5Τ=Τ0!41 TESTOSTERONE, USUSJiD =522-35

Eiapssci 7¾>ε (hours)
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
PCTEST=TGTAL TESTOSTERONE, USUS3∑D=522-38

Siscsed fmeiSours)
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
PCTEST-DI i YDROTE£TOS^RSNE. U S EJIS- 522-31

Figure 14.4.2a
Individual Analyte Plasma Concentrations
PK Population: Multiple Dose
PCT:E5T~-DiH ' R0TEST0ST£R0 :E, USUSj;IC-522~23-

4 8 12 i€ IS- 24 28 32 36 4fi +4 S 52
Elspsi r (rouri)
Figure 14.4.2a
Individual Analyte Plasma Concentrations
PK Population: Multiple Dose
Oitsydrotestosfer-cMig
PCTEST-D IH YDROTESTOS ERS E.. U SU83 Η5-522·· 17

C: 4 S 12 1.6 :0 24 25 32 36 40 44 4S
Figure 14.4.2a
Individual Analyte Plasma Concentrations
PK Population: Multiple Dose
:pCrEST~DiH¥DROTE£TOST5i5.0¾ E S!J B3IC— 522 -24

S 4 3 12 IS 25 24 21 32 36 4S 44 -S 52
E!ipsid T) :"!:=: (hours)
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
P TEE —OXH ¥Di¾OTE£TOS75 ON£. :U SU S3∑D- 522-38

Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Dsbydrotestosterone

id ¾p s ±■:■ Ti!rs irons }
Figure 14.4.2a
Individual Analyte Plasma Concentrations
PK Population: Multiple Dose
PCTEST-D iHYD QTE£TOST£ Cff-'d g. J 522-35

4 8 12 16 .23 2:4 2:3 32 35 40 4 4§ 52
Sapsed : ir«s{hsi:iE>
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose

Elapsed : »":¾{teis"s)
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Estrad ol

Figure 14.4.2a
Individual Analyte Plasma Concentrations
PK Population: Multiple Dose
Estradiol
PCT 5T=E5?R 3I3L, USUB332=522-83

48 E2
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose

Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
ioi.

Figure 14.4.2a
Individual Analyte Plasma Concentrations
PK Population: Multiple Dose
Estradiol
FCTEST= EJTR.--. 0 L U5UE-.0=.522-2S

8
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose

Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Estradiol.

Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
Estradiol
PCIEST=£S RAOIO L, U SUBS Ώ =522-38

Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
FCTEET-SHB-G... USLB iD-S22- 0i
is a ses T^ai ours}
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
SI1BG

Baps sd I i τί≤ f c'ii! .}
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
SHBG

EspS ·.:! I '2 i> !fs)
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
SHBG

S aps ed Ti m* (hours)
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
SliBG

Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
SHBG
FCnEST-SHBG, USUB1SD - S22--2?-

Eapss<5 T¾RS (hours)
Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
S1I.BG
PCTS£T-5HBG, USUBT>D~S2Z-3i

Figure 14.4.2a
Individual Analyte Plasma Concentrations PK Population: Multiple Dose
SffBG

S a ps ssd Ti srs Hours}
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Free Tes ost rone
PCTEST— FREE 7Ξ·Π05 ΈΚΟ*<£, USJ 3.13 ~ S22 -C'l

lapsed Vfxn ^ftsttn)
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Free Testosterone
FCTEST-SREE TEiTDSHT O ¾. USJ3J Ιΐί - 522-53

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
¾se Testosterone
■REE TSSTOSr_KO¾ ξ, :.
<9*
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Free Testosterone

Sailed Tsfss (kails
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Free Testosterone

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Free Testosterone
FCTEET- FREE TESTOSTERON E, S 3.3D-S-22-3S

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Free Testosterone
PCTEST ¾EE 1SSTQSTB¾3!^^SJ3332-522-33

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Total TestosteijRe
PCT T=TO ¾.} . TE^l^BRiJ F, ϋ 5U 3.1 ¾D =575-!} 5

WK sedTsffs'f ( s 's
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Total Testosterone


Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Total Testoserone
PCTEST=T07Sl TE5TO TgR i¾: . U5U53?D=5?5-}7

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Total Testos erone

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Total Teslosfsfoae

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Total Testosterone

73
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Total Testosterone
 TESTOSTE ONE, USUSJiD = 5¾2-55
TESTOSTE ONE, USUSJiD = 5¾2-55

4 g 34 2:S 32 36 4ft 5
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Total Testosterone


Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
D¾ydr0t€st stefoti€
:PCT£ST= DIH YD O 1Έ5Τ0 STER0?3¾ :USUaJ ID = 522-01

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
=OIHYDR0 TESTOSTERONE, USU3JID=522-I7

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
PCTEST=0 H¥D ROTESTO 5TER0N£, 050 S33D = 522-2:4

Elao sedTirae 3¾si;:rs }
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
O ilivdrotestos erone
PCTt5T=DMYD:RQTEST0 STSRSfs E; USU 3 JID =522-28

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
PCTEST =OIH YD R 0 TESTS S1E <3 f¾ £>■ USUSJiD =522-25

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
FC1KT=DiH¥DR0TEST0 STE O E, U5U3JID =522-35

Saps &d Tins s ( o^rs
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Di ydroiestosfcerone
PC1TST=D_HYDRQ1∑STOSTER0ME, USj3J-D = S'22-38

Eiapsed Tms (;~z-■■.■■'£}
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Estradiol

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Estradiol
two

4 8 ;3 16 2Q 24 Ti · ζ $ 4 45
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Estradiol
PC EST= ES ADIOL, !!SJE! 3D =53 ?

4 8 U. IS- 20: 34 2-¾ 3 3fi 4ff 4 4¾ 52
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Estradiol

4 8 12 IS- 2Q 54 2.1 32 .36 S «4 K fc! ii='-S .ma ['"■:: J.TS 5
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Estradiol
:ΡΓΠ?5Τ= EST DIQ i , U SiiBJiD =527-29

g! <-p seij H n {hours}'
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Estradiol

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
»ττ γ..[.Ύ..,... |..ττ..|.τ..Γ
23
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
SHBG
C!'EST=SHeG, US!J:-uiD=.-22-a i

4 S 12 lis .28 24 ZS 52 3t ¾ 48 52
Ei 3p s ad "ΠΪΤΚ (he ufs }
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
>CTEST=SH B G , U:-U8: iD= 522-0 i
I
24 25
Elapsed Tons, hows)
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
SMBG
Pi:TE"=SHEG, U≤US3ID=5.22-i7

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
SB BO
P-:TES7=5HEG, USUE3'£D=52--24

8 12 IS 30 24 2s .32 3S C 44 ¾ 52
Elapsed Ti rne i
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
S!!BO
FCTEST=SH8G, USUEOID =52.-2!

Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
SHBG
PCTEST=5HBG , USUSJ 10=522-2*

9 12 IS. 2fi _∑-¾ 23 22 3-S 4C 44 4$ iE!anss Time, (fcisiafs
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
SHBG
PCTEST=SKBG , USU53 ID =522-35
Figure 14.4.2b
Individual Analyte Plasma Concentrations (Log y7-axis Scale)
PK Population: Multiple Dose
Si BG

Figure 14.4.3.1a
Pharmacokinetics: Mean Observed Analyte concentrations (Uncorrected)
PK Population: Single Dose
Free Testestereiie Mesa. Omce-iirai.ons

Figure 14.4.3.1a
Pharmacokinetics: Mean Observed Analyte concentrations (Uncorrected)
PK Population: Single Dose
637

Pharmacokinetics: Mean Observed Analyte concentrations (Uncorrected)
PK Population: Single Dose
i l kyd imimtt ae Mea n Co aceatraiioiis

Figure 14.4.3.1a
Pharmacokinetics: Mean Observed Analyte concentrations (Uncorrected)
PK Population: Single Dose
Estradiol Mean C«»eeirtraii«»s

Figure 14.4.3.1a
Pharmacokinetics: Mean Observed Analyte concentrations (Uncorrected)
PK Population: Single Dose

55
Figure 14.4.3.1a
Pharmacokinetics: Mean Observed Analyte concentrations (Uncorrected)
PK Population: Single Dose

55
Figure 14.4.3.1b
Pharmacokinetics: Mean Observed Analyte Concentrations (Log y-axis Scale)
PK Population: Single Dose
e Testosterone Mean€©i-«entratis»s
? ^ST= FREE" TCS$JS75tSRE

Figure 14.4.3.1b
Pharaiacokinetics: Mean Observed Analyte Concentrations (Log y-axis Scale)
PK Population: Single Dose
Total e tosterttne Mean « a centra turns
PS EST-iSrAL 7E57G STE OLS

Figure 14.4.3.1b
Pharmacokinetics: Mean Observed Analyte Concentrations (Log y-axis Scale)
PK Population: Single Dose

Figure 14.4.3.1b
Pharmacokinetics: Mean Observed Analyte Concentrations (Log y-axis Scale)
PK Population: Single Dose
Estradiol Mean Ceneenftrations


Pharmacokinetics: Mean Correctred Analyte Concentrations
PK Population: Single Dose

Figure 14.4.3.2a
Pharmacokinetics: Mean Correctred Analyte Concentrations
PK Population: Single Dose
Total Testosieroae esa oMC€Htrsti<iiis

Figure 14.4.3.2a
Pharmacokinetics: Mean Correctred Analyte Concentrations
PK Population: Single Dose
.Hibvdretestosieraite ean Corjeetiirstieiis

Figure 14.4.3.2a
Pharmacokinetics: Mean Correctred Analyte Concentrations
PK Population: Single Dose
Estrswliisi M m o-icentratiws-
Figure 14.4.3.2a
Pharmacokinetics: Mean Corrected Analyte Concentrations
PK Population: Single Dose
SMBG Mean Conceit tratkms
Ρ£ΤΕ5Τ=5ίβ-3

Figure 14.4.3.2b
Pharmacokinetics: Mean Correctred Analyte Concentrations (Log y-axis Scale)
PK Population: Single Dose
Free Tesfesst rane Meaa C»iiceitf ratitms

Figure 14.4.3.2b
Pharmacokinetics: Mean Correctred Analyte Concentrations (Log y-axis Scale)
PK Population: Single Dose

Figure 14.4.3.2b
Pharmacokinetics: Mean Correctred Analyte Concentrations (Log y-axis Scale)
PK Population: Single Dose
Bihydrotestosierone Mean o iceiitrat¾»s
Figure 14.4.3.2b
Pharmacokinetics: Mean Correctred Analyte Concentrations (Log y-axis Scale)
PK Population: Single Dose
Estradiol Mesit Concen rations
,
Figure 14.4.3.2b
Pharmacokinetics: Mean Correctred Analyte Concentrations (Log y-axis Scale)
PK Population: Single Dose

Figure 14.4.4a
Mean Analyte Plasma Concentrations PK Population: Multiple Dose
Free Test ster ne

Figure 14.4.4a
Mean Analyte Plasma Concentrations PK Population: Multiple Dose

Figure 14.4.4a
Mean Analyte Plasma Concentrations PK Population: Multiple Dose
DOwdro testosterone

Figure 14.4.4a
Mean Analyte Plasma Concentrations PK Population: Multiple Dose

Elapsed l¾r¾e (jhr)
Figure 14.4.4a
Mean Analyte Plasma Concentrations PK Population: Multiple Dose s

Figure 14.4.4a
Mean Analyte Plasma Concentrations PK Population: Multiple Dose
Free Testosterone

Figure 14.4.4a
Mean Analyte Plasma Concentrations PK Population: Multiple Dose
Total Te^osteroae

Figure 14.4.4a
Mean Analyte Plasma Concentrations PK Population: Multiple Dose

Figure 14.4.4a
Mean Analyte Plasma Concentrations PK Population: Multiple Dose

Figure 14.4.4a
Mean Analyte Plasma Concentrations PK Population: Multiple Dose
SHBG

Figure 14.4.4b
Individual Analyte Plasma Concentrations (Log y-axis scale)
PK Population: Multiple Dose

Figure 14.4.4b
Individual Analyte Plasma Concentrations (Log y-axis scale)
PK Population: Multiple Dose
Total Testosterone

Figure 14.4.4b
Individual Analyte Plasma Concentrations (Log y-axis scale)
PK Population: Multiple Dose
f CTEST=D IHYDROTESTDSTE G ME, MSJ B31D =M ΜΆ

Figure 14.4.4b
Individual Analyte Plasma Concentrations (Log y-axis scale)
PK Population: Multiple Dose
.Estradiol
I I I I ' 1 I I I I I I I I I I 1 I I I I I I 1 I I 1 I I I M I I | 1 I I | 1 I I I
28 32: 36
Figure 14.4.4b
Individual Analyte Plasma Concentrations (Log y-axis scale)
PK Population: Multiple Dose
SHBG
PCTE5T=SHBS USyB3ID=S¾ gets

Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose
Free Tesi s!eroee Meaa€¾aee»trstiims
Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose
Free Testosterone Mean Concentrations

Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose
F ree Teste si erese Mean C» ceiif r tiims

Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose
Total Testestenjiie Mean Coii€S«trafi«i*s

Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose

Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose
Total Tesiester ae Meaa C siceiiirailflijs

Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose
Pilijlre stost r ae Mean Ce-iceatraiioiis
Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose

Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose
!i ky dm i imierm Mm » cnicot i ra tm a s

Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose

Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose

Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose

Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose

Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose

Figure 14.4.5a
Pharmacokinetics: Analyte Parameter Summary
PK Population: Single Dose

Figure 14.4.5b
Spaghetti Concentration Plots with Mean for Analyte Plasma Concentrations
PK Population: Multiple Dose
Free Testosterone

Figure 14.4.5b
Spaghetti Concentration Plots with Mean for Analyte Plasma Concentrations
PK Population: Multiple Dose
Total Testosterone

Figure 14.4.5b
Spaghetti Concentration Plots with Mean for Analyte Plasma Concentrations
PK Population: Multiple Dose
F :TEST=DiH¥ 01i5TO:STEE;i3l¾

Figure 14.4.5b
Spaghetti Concentration Plots with Mean for Analyte Plasma Concentrations
PK Population: Multiple Dose

Figure 14.4.5b
Spaghetti Concentration Plots with Mean for Analyte Plasma Concentrations
PK Population: Multiple Dose
SHBG

Figure 14.4.6
Free, Total and Dihydro-Testosterone Concentrations in Subjects Receiving 1200 mg Dose iin Single and Multiple Dose Cohorts
PK Population: Subjects 522-28, 522-35, and 522-38
Free Tes osteFose
Subject.522-28

!¾ '«ΐΐκ »«>¾sS 5oss »¾e colisfed for 24 bums sS¾i½g as th* da¾ ^rj« so t s sjsak ifcsea ssidsas j isss V« sksgkdese:.
Figure 14.4.6
Free, Total and Dihydro-Testosterone Concentrations in Subjects Receiving 1200 mg Dose iin Single and Multiple Dose Cohorts
PK Population: Subjects 522-28, 522-35, and 522-38
Free Tesiosierc e
Subject 522-35

Figure 14.4.6
Free, Total and Dihydro-Testosterone Concentrations in Subjects Receiving 1200 mg Dose iin Single and Multiple Dose Cohorts
PK Population: Subjects 522-28, 522-35, and 522-38
F ee Testosterone
Subject 522-38
WS« lH~$5m«i& «0WBMI2*fc 0095>;&»

···*··· M¾;;:; ¾ 0« Si
 Mgfc sfesessd <sk$kg|« fc&xe ¾he *½k
Mgfc sfesessd <sk$kg|« fc&xe ¾he *½k
Figure 14.4.6
Free, Total and Dihydro-Testosterone Concentrations in Subjects Receiving 1200 mg Dose iin Single and Multiple Dose Cohorts
PK Population: Subjects 522-28, 522-35, and 522-38
Total Testoster ie
Subject 522 28

Figure 14.4.6
Free, Total and Dihydro-Testosterone Concentrations in Subjects Receiving 1200 mg Dose iin Single and Multiple Dose Cohorts
PK Population: Subjects 522-28, 522-35, and 522-38

Figure 14.4.6
Free, Total and Dihydro-Testosterone Concentrations in Subjects Receiving 1200 mg Dose iin Single and Multiple Dose Cohorts
PK Population: Subjects 522-28, 522-35, and 522-38
Total Tesiosiero«
Sofeject 522-38

Figure 14.4.6
Free, Total and Dihydro-Testosterone Concentrations in Subjects Receiving 1200 mg Dose iin Single and Multiple Dose Cohorts
PK Population: Subjects 522-28, 522-35, and 522-38
Subject 522-28

BSiid UK «i sx¾si:i;¾k!S3S ν· ··ν a ; ssl ΐ-i ? .24 h-i-xf t s o -h day ¾>;·¾<:«· s« ssfc fe &? ssssJ «sM¾s & j ¾¾ db κ; s.b sssssk dose..
Figure 14.4.6
Free, Total and Dihydro-Testosterone Concentrations in Subjects Receiving 1200 mg Dose iin Single and Multiple Dose Cohorts
PK Population: Subjects 522-28, 522-35, and 522-38
S¾¾eci 522-35

Basd e «SKXCTMS!!HS. «se tolsctoi ikr 24 teats «¾as feg ®: : ¾! mrts -the ¾lj>;.;k do-*- ¾·>·5 sssxfs&g jest l;d¾ c she s^k- ose.
Figure 14.4.6
Free, Total and Dihydro-Testosterone Concentrations in Subjects Receiving 1200 mg Dose iin Single and Multiple Dose Cohorts
PK Population: Subjects 522-28, 522-35, and 522-38
Subject 522-38

Figure 14.4.6
Pharmacokinetics: Accumulation Estimate by Linear Regression on Trough Concentrations
PK Population: Multiple Dose
704

Pharmacokinetics: Accumulation Estimate by Linear Regression on Trough Concentrations
PK Population: Multiple Dose
Total T stosterone Troagli Concentrations
Figure 14.4.6
Pharmacokinetics: Accumulation Estimate by Linear Regression on Trough Concentrations
PK Population: Multiple Dose
Bih y<f ro testosterone Trettgb Co seen i tie its
Figure 14.4.6
Pharmacokinetics: Accumulation Estimate by Linear Regression on Trough Concentrations
PK Population: Multiple Dose
Estradiol Trough Concentrations

Figure 14.4.6
Pharmacokinetics: Accumulation Estimate by Linear Regression on Trough Concentrations
PK Population: Multiple Dose
< * :> :>; ; <:.S.
a

REFERENCE LIST
Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA 1999;281:537-544.
Shifren JL, Monz BU, Russo PA, et al. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol. 2008;112:970-978.
Davis S. Androgen replacement in women: a commentary. J Clin Endocrinol Metab. 1999;84(6):1886-1891.
Goldstat R, Briganti E, Tran J, Wolfe R, Davis SR. Transdermal testosterone therapy improves wellbeing, mood, and sexual function in premenopausal women; Menopause 2003;10(5):390-398.
Traish AM, Kim SW, Stankovic M, Goldstein I, Kim NN. Testosterone increase blood flow and expression of androgen and estrogen receptors in the rat vagina. J Sex Med. 2007;4:609-619.
Halaris A Neurochemical aspects of the sexual response cycle. CNS spectrums.
2003;9:211-216.
Loeser AA. Subcutaneous implantation of female and male hormones in tablet form in women. Br med J. 1940;1:479-482.
Salmon UJ, Geist SH. Effect of androgens upon libido in women. J Clinical
Endocrinol. 1943;3:235-238.
Sherwin BB, Gelfand MM. The role of androgen in the maintenance of sexual functioning in oophorectomized women. Psychosomatic Medicine. 1987;49:397-409. Traish AM, Feeley RJ, GuayAT, Testosterone therapy in women with gynecological and sexual disorders: a triumph of clinical endocrinology from 1938 to 2008. J Sex Med 2009. 6(2):334-351.
Rickenlund A, Thoren M, Carlstrom K, et al. Diurnal Profiles of Testosterone and Pituitary Hormones Suggest Different Mechanisms for Menstrual Disturbances in Endurance Athletes. J. Clin. Endocrinol. Metab. 2004, 89: 702-707.
Javanbakht M, Singh AB, Mazer NA,et al. Pharmacokinetics of a Novel Testosterone Matrix Transdermal System in Healthy, Premenopausal Women and Women Infected with the Human Immunodeficiency Virus. J Clin Endocrinol Metab 2000, 85:2395- 2401.
Vierhapper H, Nowotny P and Waldhausl W. Determination of Testosterone
Production Rates in Men and Women Using Stable Isotope/Dilution and Mass Spectrometry. J. Clin. Endocrinol. Metab. 1997, 82: 1492-1496.
Abraham G. Ovarian and Adrenal Contribution to Peripheral Androgens During the Menstrual Cycle. J. Clin. Endocrinol. Metab. 1974, 39 (2): 340-346.
Example 12
Two-Center, In-Patient, Randomized, Placebo and Active Comparator
Evaluation of the Pharmacokinetics (PK) and Safety, Along with Initial
Pharmacodynamic (PD) Efficacy of Three Dose Levels of TBS-2 Intranasal Gel Applied Twice-Daily (BID) for up to Three Days in Female Patients with
Hypoactive Sexual Desire Disorder (HSDD) or Anorgasmia (ANOR).
Background
Approximately 43 percent of women acknowledge a form of female sexual dysfunction. Hypoactive sexual desire disorder (HSDD) is the most common women's sexual problem; anorgasmia is the second most common. The androgen testosterone is know to play an important role in sexual function. The only registered testosterone product for use in women is Intrinsa, a testosterone slow-release transdermal patch. This Phase I study was designed to assess the pharmacokinetic profile of testosterone and dihydrotestosterone (DHT) following administration of a new product, TBS-2 testosterone intranasal gel, in women with HSDD and anorgasmia. This study also explored initial pharmacodynamic efficacy and assessed the safety of TBS-2.
Methods
This was a two-center, in-patient, parallel groups, randomized, placebo and active comparator controlled evaluation of the pharmacokinetics (PK) and exploratory pharmacodynamics of three dose levels of TBS-2 testosterone intranasal gel. Thirty two women participated in two cohorts (HSDD and anorgasmia), depending on primary complaints. Subjects received five doses of TBS-2, placebo TBS-2 (anorgasmia cohort) or a single Intrinsa patch (HSDD cohort), in three consecutive study days. Frequent sampling PK series were collected after the first dose (0-12 hours) and after the fifth dose (48-60 hours after first dose). Initial pharmacodynamic efficacy was explored using vaginal pulse amplitude (VPA), subjective arousal questionnaires (SAQ, AFSDQ) and validated computer tasks. Safety was monitored throughout the study.
Results
The pharmacokinetic results show that there is a linear increase in plasma testosterone levels with increasing dose levels. Mean concentrations of plasma
testosterone 0-12 hours after dosing were: 53.38ng/dl_ after TBS-2 high dose, 34.55ng/dl_ after TBS-2 medium dose and 21 .35ng/dl_ after TBS-2 low dose. The mean concentration of plasma testosterone is consistently higher after five administrations (48-60 hours post first dose): 65.52ng/dl_ after TBS-2 high dose, 44.49ng/dl_ after TBS-2 medium dose and 21 .81 ng/dl_ after TBS-2 low dose. Accumulation seems to occur with dosing intervals of 12 hours in the higher dosage groups.
During the second PK series, the mean plasma testosterone concentration and area under the curve (AUC) in the TBS-2 high dose group reached levels higher than the mean plasma testosterone concentration and AUC in the Intrinsa group, but still within the upper limit of the normal physiological range. Although peak plasma testosterone concentrations reached levels higher than the upper limit of the reference range in the TBS-2 high dose and medium dose treatment groups, the mean concentration was within the normal reference range for all treatment groups.
The plasma concentrations for DHT were often too low to detect; pharmacokinetic parameters that were calculated for testosterone could therefore not be calculated for DHT.
Baseline VPA and genital response to sexual stimuli were consistently lower in the HSDD cohort, when compared to the anorgasmia cohort. No dose-related trend in efficacy was observed in either cohort. In the anorgasmia cohort there was a statistically significant difference between TBS-2 high dose and placebo TBS-2 after 30 minutes, which was not maintained after 4.5 hours. After 4.5 hours there was only a statistically significant difference between TBS-2 low dose and placebo in the anorgasmia cohort. In the HSDD cohort there were no significant differences. None of the other pharmacodynamic measurements (SAQ, AFSDQ, computer tasks) showed consistent statistically significant effects, even at high doses of TBS-2.
Treatment was generally well-tolerated, with headache as the most common side effect.
Conclusions
Plasma testosterone levels showed a reliable linear increase after administration of increasing dose levels of TBS-2. Accumulation seemed to occur at the higher doses and did not seem to have reached a steady-state plateau after the five doses administered. Since the higher doses are likely to be the effective doses, the extent to which accumulation occurs needs to be taken into consideration in future studies.
This study was not powered to show any significant pharmacodynamic effects, although the graphs suggest that there may be an effect of testosterone on VPA in the anorgasmia cohort without changes in subjective response; while in the HSDD cohort VPA response is very low in all treatment groups, but with some increase in subjective response .
The mean plasma testosterone concentration and AUC in the TBS-2 high dose group reached levels higher than the mean plasma testosterone concentration and AUC in the Intrinsa group during the second PK series. Although peak levels of plasma testosterone reached levels higher than the upper level of normal, the mean plasma testosterone concentration was within the normal reference range for all treatment groups, limiting safety concerns. Overall, no safety concerns were identified in this study.
EXAMPLE 13
Introduction
Female orgasmic disorder (FOD) is the second most prevalent sexual disorder in women after hypoactive sexual desire disorder. A conservative estimate of prevalence is 5% with many epidemiologic studies reporting higher rates (Laumann et al., 1999, Rosen et al., 1993, Read et at., 1997, Shifren et al., 2008) Two of the most cited epidemiologic surveys of female sexual problems are the National Health and Social Life Survey (NHSLS) (Laumann et al., 1999) and The Prevalence of Female Sexual Problems Associated with Distress and Determinants of Treatment Seeking (PRESIDE) (Shifren et al., 2008). The NHSLS surveyed a random sample of 1 ,749 U.S. women between the ages of 18-59 and found that 24% reported a lack of orgasm in the past year for at least 12 months or more. The PRESIDE survey included interviews of 31 ,581 women aged 18 years and older from 50,002
households sampled from a national research panel representative of U.S. women. Twenty-one percent of the women surveyed reported having problems with orgasm and 4.6% reported sexually related personal distress as measured by the Female Sexual Distress Scale (FSDS) (Shifren et al., 2008, Derogatis et al., 2002). However, a precise estimate is difficult due to a paucity of well-controlled studies and a lack of consistency in definitions and diagnostic criteria (Meston et al., 2004). Despite the high prevalence of FOD, women suffering from this disorder have not been well characterized. One reason for so few well-controlled studies is the lack of available treatment options. Currently cognitive behavior therapy (CBT) is the most effective treatment for FOD but there is little published data on success rates (Meston et al., 2004). These CBT methods include directed masturbation, sensate focus exercises and systematic desensitization. The goal of most of these strategies is to help women become more comfortable with their own genitals and sexuality by altering negative attitudes, decreasing anxiety and learning self-stimulation (Kingsberg and Knudson 201 1 ). There are currently no pharmacologic treatment options for women with FOD.
The Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM IV) and the most recent revision (DSM-IV-TR) defines FOD as "the persistent or recurrent delay in, or absence of, orgasm following normal sexual excitement phase that causes marked distress or interpersonal difficulties and is not better accounted for by another Axis I disorder and is not due exclusively to the direct physiological effects of a substance or a general medical condition". It is further specified as lifelong or acquired.
Marked distress is central to the diagnosis of all sexual disorders including FOD. In practice, the term "distress" for use as a criterion for a clinical diagnosis is a medical construct and may not correlate with the language used by women with FOD to describe what they are experiencing (Goldstein et al., 2009). Nevertheless, the diagnostic criteria includes a measurement of distress and, in clinical trials of treatments for sexual disorders a measurement tool is required to evaluate a therapeutic change in marked distress. The Third International Consultation on Sexual Medicine's committee on standards for clinical trials in sexual dysfunction in women recommends that clinical trials include a validated measure of distress
(Clayton et al., 2010). Currently the gold standard measure of distress is the FSDS-R (Derogatis et al., 2008). This 13 item distress scale has been consistently used as the measure of distress in most clinical trials for treatments of female sexual disorders, particularly HSDD. This scale has been further revised to include items specific to arousal and orgasm as clinical endpoints and is called the FSDS-DAO (Desire, Arousal, Orgasm). This most recent revision is currently being validated. In addition, the Sexual Satisfaction Scale for Women (SSS-W), designed to measure women's sexual satisfaction and distress, has been validated in women with sexual dysfunction (Meston et al., 2005). Most other validated measures of distress are primarily directed toward measuring distress related to HSDD (DeRogatis et al., 2008).
In anticipation of conducting trials to assess the safety and efficacy of a new pharmacologic treatment for FOD, a better understanding was needed of how women with FOD experience the disorder and the language they use to describe the impact that FOD has on their lives. This article describes the results of 12 focus groups (6 in Cincinnati and 6 in New York City) evaluating women with self-reported FOD conducted at two geographic locations in the United States. The specific objective of each focus group was to explore the terminology used by women to describe their feelings associated with difficulties in achieving orgasm. To achieve this, the focus groups were designed to understand women's experience with FOD and explore the impact of a decrease in satisfying orgasmic attainment on women's emotional well- being, sense of self, relationship and overall life. The groups were structured to include exercises that captured how women verbally characterized their experience of having orgasmic difficulty, how they visually expressed their feelings and self- perception (picture sort) and an exercise to evaluate women's concurrence with the concepts of the Female Sexual Distress Scale-Desire, Arousal, Orgasm (FSDS- DAO) (Derogatis unpublished).
Methods
Women aged 20 - 60 were recruited in Cincinnati and New York City (NYC) using a consumer database by a market research group. Inclusion into the focus group was based on a screening questionnaire administered over the phone. The questionnaire was designed to capture age, ethnicity, income, relationship status, satisfaction of
current relationship, how often women engage in sexual activity that would typically be expected to result in orgasm, ability/difficulty in achieving orgasm, most recent orgasm experienced, duration of difficulties having an orgasm, bothersomeness of their inability to achieve orgasm and the word they would choose to describe their experience associated with having orgasm difficulties.
Women were invited to participate in the Cincinnati focus group, if they were currently in a sexual relationship, experiencing difficulties in achieving orgasm, their inability/difficulty to achieve orgasm negatively affected them, were somewhat, very or extremely bothered by lack of orgasm and their condition was not caused by concomitant medication, medical condition or partner medical condition and they had no major psychiatric or medical problems, and had no drug or alcohol dependency. The inclusion criteria for the focus group in New York City were further refined to exclude women with lifelong orgasmic disorder and who had an orgasm over the past 6 months.
Each focus group was 90 minutes in duration, included 4-6 women and was facilitated by a single facilitator. Written consent was obtained from all participants. Women were compensated for their participation in the focus group.
Characterization Exercise
Participants were asked to recall a specific scenario that epitomized their sexual peak.
They were then asked to write down words, phrases, thoughts, visual images and to think of one celebrity or character that captured their self-perception at the time of their peak sexuality.
Participants were then asked to recall the point in time when they identified that they were dissatisfied with their ability to achieve a satisfying orgasm and were then asked to write down words, phrases, thoughts, and to create visual images to describe their emotion about this and their self-perception. They were further asked to identify an individual or character that best matched their self-perception at the time of their
realization of dissatisfaction/distress with a decrease/lack of orgasm.
Participants were then requested to identify an individual or character that personified how they would like their sexuality to be now.
Picture Sort Exercises - Impact of decrease in orgasm
Participants were provided photographs of images pulled from various sources and asked to select pictures that reflected: (1 ) themselves sexually and their relationship at present; and (2) what best represented how they want their sex life to be if they could experience orgasm to a degree that it would no longer be an issue. Women were asked to show selected images and discuss why the pictures described their feelings and experiences.
The images from the second task (which depicted their sex life if they could achieve an orgasm with ease) were also discussed in depth with the group. The discussion was guided to determine if the return of orgasm would be important for themselves, for the relationship or for both and to describe how the return of orgasm was important to them.
Language Exploration Exercise - Emotional associations with decreased orgasm
Participants were asked to discuss the feelings that they experience as a result of a decline/lack of orgasm. The words to describe these feelings were recorded on a chart pad. Women were then asked to choose which of the following three words best described their feelings: bothered, frustrated or distressed and rate the intensity of this feeling on a scale of 1 - 5 where 1 was least and 5 was greatest on the scale.
Sexual Distress Scale - FSDS-DAO - Question 15
Participants were asked to complete the FSDS-DAO questionnaire. The FSDS-DAO is a self administered questionnaire consisting of 15 items that related to different aspects of sexual distress. The FSDS-DAO is a revision of the validated FSDS-R with the addition of two questions. Question 15 specifically addresses distress related to orgasm, "How often do you feel frustrated by problems with orgasm?". Every question is answered based on a 0-4 scale (never [0], rarely [1 ], occasionally [2], frequently [3] or always [4]. They were then asked to provide their interpretation of
the items and the extent to which the FSDS-DAO accurately reflected their personal experience and feelings.
Results
Screening
Screening occurred in two different geographical areas of the United States: Cincinnati, OH (mid-west), and New York City, NY (east coast).
A) Cincinnati site
657 women were contacted to participate in the research focus groups. Women were initially included based on age and then screened using a questionnaire. Of the 657 women, 208 (32%) were currently experiencing difficulties in achieving orgasm. Selected demographics of the 208 women experiencing orgasm difficulties are presented in Table 1 .
Table 1 : Selected demographic characteristics of women contacted experiencing difficulties in achieving orgasm in Cincinnati


No comment 57 (27.5%)
Of the 208 women, 192 (92%) were somewhat bothered, very bothered or extremely bothered by their difficulties/inability to achieve an orgasm. Of the 1 92 women, 167 women responded to the question "What one word would you use to describe your orgasm difficulties?" and of these 1 67 women, 77 women (46%) chose the term "frustrating" to best describe their feeling associated with difficulties in achieving orgasm. Other common terms (n>4 and n<7) used by women who responded to the questionnaire included "Stressful", "Unfulfilled" and "Sad". There were 55 other words used describing their feeling associated with difficulties in achieving orgasm. Of the 167 women who provided a term describing their orgasm difficulties, 1 14 (68%) claimed they were "very or extremely bothered" by their inability to achieve orgasm.
Figure A: Word used by women who have orgasm difficulties (Cincinnati) to best describe their feeling associated with difficulties in achieving orgasm

One hundred forty nine (149) women were contacted to participate in the research focus groups. Women were chosen based on age, an annual income of at least 50K and then screened using a questionnaire. Of the 149 women contacted, 121 (81 %) were currently experiencing difficulties in achieving orgasm. Selected demographics of the 121 women experiencing orgasm difficulties are presented in Table 2.
Table 2: Selected demographic characteristics of women contacted experiencing difficulties in achieving orgasm in New York City

Of the 1 21 women, 1 19 (98%) were somewhat bothered, very bothered or extremely bothered by their difficulties in achieving orgasm. Of the 1 18 women who responded to the question "what one word would you use to describe your orgasm difficulties" 68 (58%) used the term "frustrating" to best describe their feeling associated with difficulties in achieving orgasm. The next most common term was "annoying". There were 32 other words used describing their feeling associated with difficulties in achieving orgasm.
Figure B: Word used by women who have orgasm difficulties (NYC) to best describe their feeling associated with difficulties in achieving orgasm

Focus Groups
A total of 12 focus groups were conducted: 6 in Cincinnati and 6 in New York City. The focus group participants are described in Table 3.
Table 3: Characteristics of focus rou artici ants

Of the 72 women from both Cincinnati and NYC who qualified and agreed to participate in the focus group, 66 women with what appears to be acquired FOD responded to the question "What single word would you use to describe your experience with orgasm difficulties/inability?" The six women with lifelong anorgasmia did not participate in this exercise. Sixty-seven percent of the participants who responded to the question chose the term "frustrating" to best describe their feeling associated with difficulties in achieving orgasm. Other common terms (n=2) used by women who responded to the questionnaire included
"Bothersome", "Embarrassed" and "Sad". There were 16 other words used describing their feeling associated with difficulties in achieving orgasm.
Figure C: Word used by women who have orgasm difficulties (who participated in the focus group in Cincinnati and NYC) to best describe their feeling associated with difficulties in achieving orgasm

Characterization Exercise
The characterisation exercise was conducted with the Cincinnati focus groups only. This was omitted from the NYC focus groups so more time could be spent on the picture sort exercises. When asked to describe themselves when they had been in their sexual peak, subjects often described being "carefree" and unencumbered with life's responsibilities. They also reported living a more adventurous life, having more self-confidence, and being more fulfilled in life, relationships, and sexual experiences. Subjects were asked to draw celebrities and characters that best represented the person they felt like at the time of their sexual peak. These celebrities are all known for being considered sexy, confident, free, bouncy, beautiful, young, fun and flirty at the peak of their success and included Marilyn Monroe, Farrah Fawcett, Jennifer Garner, Raquel Welch, Halle Berre, Beyonce and Christie Brinkley. Subjects stated that these images also reflected their own confidence during their sexual peak, at a time when they were comfortable with themselves and felt vibrant, skinny, attractive and successful.
In contrast, women often associated lack of orgasm or declining orgasmic attainment with feeling overwhelmed by life's responsibilities, and consequently stressed and fatigued. These women describe themselves now as frustrated, depressed/sad, overwhelmed, and lacking confidence. Individuals/characters selected by subjects to represent the person they feel like because of their dissatisfaction/distress with a decrease/lack of orgasm were associated with being cranky, bitchy, unhappy, stressed, fat, broken, unattractive or otherwise not sexy. These celebrities/characters included Peg Bundy, Roseanne Barr, Whoopi Goldberg, Eeyore, the Wicked Witch of the West, and Humpty Dumpty.
Subjects were also asked to describe their sexual ideal. Descriptions included sexual ideal "feeling sexy and enjoying sex", "having fun in their sexual relationship", "being beautiful, attractive to the opposite sex", "energetic", "vibrant", and often a little "naughty". Celebrities/characters selected to represent this included Kim Cattrall, Sophia Vergara, Madonna, Jennifer Lopez, Jennifer Aniston, Angelina Jolie, Rihanna and Wonder Woman. Several subjects chose couples to represent their ideal, based on the perception that these couples were exciting and having fun and spontaneous sex (e.g. Kyra Sedgwick and Kevin Bacon). Younger women often described a sexy man to represent their ideal, with these images bringing to mind arousal and pleasure. These celebrities included Brad Pitt, Blair Underwood and Denzel Washington.
Picture sort exercise
The picture sort exercise was conducted at both the Cincinnati and NYC focus groups. In selecting images to represent their current sex life, most women focus on their personal frustrations around FOD, rather than their relational or situational frustrations. The most frequent selected images of a windy road, a woman chained on top of a summit, a woman sitting in a dark room with streams of light penetrating the blinds, a pensive picture of a woman, a man and woman standing with a barrier between them look out (not at each other) a cartoon image of a woman crying with no thoughts (in the thought bubble), a toilet seat, and a man catering to a cardboard woman lying in bed. Subjects describe these images as feeling "trapped" in the problem / unable to escape the nightmare, a never ending lack of fulfillment, feeling
broken / something is wrong with me, feeling depressed / sad and isolated, stressed, aging, guilt, embarrassment and insecurity.
Images chosen to depict the relationship with their partner included a man and a woman standing back to back with their arms crossed, a man catering to a cardboard woman lying in bed, a group of friends partying at the beach, a man and woman about to kiss in bed, a jovial couple gazing into each other's eyes. In general, the women in the NYC focus group appeared to be more satisfied with their relationship with their partner than the women in the Cincinnati group based on the images chosen in the picture sort exercise. Images chosen by the women in Cincinnati were associated with feelings of frustration often accompanied by guilt, dissatisfaction, stress, avoidance, lack of interest in sex and sometimes anger.
These women described their partners as being easily satisfied, while they themselves remain unfulfilled. They also viewed their anorgasmia as not only a personal loss but a loss for their partner and for the two of them together as a couple. They see this loss as evidence that something is "broken" in their relationship. Women in the NYC focus group chose similar images to that of the Cincinnati groups in addition to images representing their relationship as loving, companionable, close, and very important. They derive great happiness from their relationships and enjoy sharing their lives with their partners. However, decrease/lack in orgasm, causes tension and fighting. Many women described feeling frustration or sadness around the sexual experience, which decreases fulfillment and ultimately interest in engaging in sex.
When asked to describe their orgasmic ideal, women chose images of fireworks, a lightning bolt, a sensual picture of a man and woman about to kiss in bed, a group of friends partying at the beach and a slot machine. Subjects associated these images with a feeling of euphoria, a sense of accomplishment, confidence, happiness, stress-free, celebration, wholeness, restoring balance to a relationship, shared pleasure, intimacy and excitement about engaging in sex more often. Women described an orgasm as an event with a physical release, which lifts the mood and infuses confidence and satisfaction, as well as a sense of completeness. Women also described an orgasm as an important part of an intimate bond with their partner.
Language exploration exercise
Women were asked to provide a comprehensive list of their feelings that they experience as a result of a decline/lack of orgasm. Emotional associations with decreased orgasm are presented in Table 4. Other words used to describe lack of orgasm included stressed, cranky, lonely, empty, guilty, unhappy, devastated, incomplete, confused, angry, embarrassed, unwanted and stuck.
Table 4: Emotional associates with decreased orgasm
Emotion Word that modify Subject Quotes
associations the emotion
with
decreased
orgasm
Frustration Loss, worry, guilt, "You don't know why and you want to fix it and it's dissatisfied, not necessarily easy to be fixed. " disappointed "You have anticipation, hope, then disappointment and both you and your partner feel frustrated. " "He's relaxed and enjoying himself and I'm frustrated because 1 haven't. "
Worried / An ongoing problem "I'm scared it might be an ongoing problem and Scared Husband could cheat not just stress. "
Stressed • Distracted Childcare, errands, work, they all create stress.
• Overly responsible And the lack of orgasm too because you're
thinking about it. You feel like you can do better. "
Depressed Loss (of youth, "I'm getting older but 1 don't want this to be one of closeness, the things that 1 have to live without. " spontaneity)
Insecure Failure, uncertain, low "It makes me even more insecure about myself self esteem, poor because I'm not delivering sexually. " body image "Not only do 1 feel like a failure but at times he feels like a failure. It has nothing to do with him at all but he thinks that. "
"1 just feel insecure with how my body looks after having the baby. "
Confused What is wrong with "Thinking something is wrong with you health- me? wise. "
Phony Faking orgasm "You realize it's not going to happen for you but it's hopefully going to happen for him, so you take yourself out of it and concentrate on him and you wonder if he knows. "
Broken Something is wrong "My sex life is shattered. 1 feel broken. "
Cheated Missing out, "I paid the price of admission but didn't like the disappointed ride. He's getting what he needs and is happy but
I'm frustrated. "
Unfulfilled Dissatisfied, "You feel less of a woman. You're not getting what
incomplete you need. You are empty. "
Isolated / • Alone just feel alone. When people make it seem so lonely • Different easy, you can't say the same. You feel like you're
• Distanced the only person who hasn't [had an orgasm]. "
Failure • Failing your partner "You feel like you're letting the other person down.
• Failing yourself They feel like they've failed too and they aren't
• Disappointed pleasing or satisfying you. "
• What's wrong with "I resent myself. Is this normal? What is wrong me? with me?"
Time • A project "Sometimes you don't want to keep going and consuming • A chore keep trying and you just want to pretend, so you
• Pressured can move on and do something else. "
Women were then asked to choose which of the following three words best described their feelings: bothered, frustrated or distressed. They then rated the intensity of this feeling on a scale of 1 - 5 where 1 is the low end of the scale and 5 is the high end of the scale as presented in Table 5. s a


Sexual Distress Scale - FSDS-DAO - Question 15
Women in the NYC focus group only were asked to complete the FSDS-DAO questionnaire. Responses to question 15 ranged from 1 - 4 with a mean rating of 3.0 indicating that women were frequently frustrated by problems with orgasm. Women scored highest in the orgasm frustration domain of the FSDS-DAO compared to any of the other domains of the questionnaire.
How often did you feel: Avera
ge score
Distressed about your sex life 19
2 Unhappy about your sexual relationship 2.4
3 Guilty about sexual difficulties 5
4 Frustrated by your sexual problems 2J_
5 Stressed about sex 19
6 Inferior because of sexual problems 19
7 Worried about sex L2
8 Sexually inadequate Λ
9 Regrets about your sexual functioning 21
Embarrassed about sexual problems 2
Dissatisfied with your sex life 23
Angry about your sex life 13
Bothered by low sexual desire 2Λ
Concerned by difficulties with sexual arousal 2A
Frustrated by problems with orgasm 3.0
Discussion
The results of these focus groups have enhanced the understanding of how FOD is experienced by women as well as identifying the language used to describe their experience and the impact that FOD has on their lives and relationships. The term "frustrated' emerges as the most common and relevant emotion women feel when they have difficulties achieving orgasm. This was observed during initial telephone screening in women stating they have orgasm issues, and specifically in the women who met the inclusion criteria for the focus group. The term "frustrated" was also the most common word used by the participants during the language exploration exercise. Additionally, the women consistently supported the content validity of question 15 of the FSDS-DAO. The vast majority reported that the FSDS-DAO and question 15 adequately reflected their characterization of distress related to having problems with reaching orgasm.
In the language exploration exercise, when asked to choose which word of the three words "Bothered", "Frustrated" and "Distressed" best describes their feeling of difficulty/inability to have an orgasm, "Frustration" was the most common word used by women to describe their feelings associated with difficulty achieving orgasm. We observed that these women seemed to think of these three words along a spectrum of intensity, with "bothered" being the least intense and "distressed" being the most intense and being saved for extremely serious medical or personal experiences, i.e. a sick or injured child.
"Bothered" represented an annoyance, and for most of the women, was not a strong enough expression for how the significance of the loss they feel and how the lack of sexual satisfaction impacts their lives. The women who chose the term "bothered" to describe the impact of their decrease in orgasm reported that other things are more important in their relationship or that their orgasmic difficulties are affecting their relationship or even the quality of their sex life. These women were likely to view the problem as one which could be resolved, something they can deal with or a compartmentalized problem that they could choose to face only when engaging in sex. More of the women in the 45- 60 age group described themselves as bothered than did the younger women ages 20-44. "Frustrated" and "distressed" appear to be too extreme for roughly half of respondents ages 45-60. Bothered better represented the limited impact that decrease in orgasm was having on their lives, their feelings about their own sexuality, and the health of their relationships. The other half of women ages 45-60 who chose frustrated as best capturing their emotional response to decrease in orgasm were more verbally expressive of the loss they had experienced and had more intense emotions described than those who chose the word bothered.
The women in the focus groups who resonated with the term "Frustrated" felt that the term encompassed feeling dissatisfied or disappointed, angry and stressed. For several women, frustrated also implies action and therefore represents their intention to "fight" to fix the problem. This was in contrast to distressed, which for many implies a lack of hope or solution.
Overall, "Frustrated" emerged as the best descriptor of the feelings associated with declining / lack of orgasm for the majority of the participants.
Most of the participants described FOD as having a substantial negative impact on their overall quality of life. They found this to be frustrating and/or bothersome. Many participants reported that they experience pleasure in the other aspects of the sexual act, yet feel frustrated and unhappy when they cannot reach their sexual climax. Some acknowledged resentment, sadness or anger that their partners are always satisfied, while they are not. Despite the use of the term "distress" in the DSM-IV-TR criteria for FOD, the term reflects the medical construct required to become a sexual dysfunction and does not appear to be an accurate representation of most women's description of their feelings surrounding the experience of a decrease in orgasm. "Distress as a lay term had an urgent quality and a sense of fatalism that does not fit with how women think and feel about their own difficulty achieving orgasm.
In contrast to the focus on how difficulty achieving orgasm negatively impacted women's lives, the participants clearly stated that the ability to achieve orgasm was associated with euphoria, happiness, relaxation, and sexual excitement. The ability to reliably achieve orgasm enhances women's sense of well-being and lifts their mood, while also restoring balance in their relationship and relieving tension (both physically and psychologically). Sexual fulfillment is perceived as strengthening the bond between partners and increasing women's interest in engaging in sex.
Orgasmic attainment strongly impacts a woman's quality of life, her self-perception and her sexual relationships. Women are clearly able to articulate the impact that orgasmic difficulties have on important life domains.
References
Clayton AH, Goldmeier D, Nappi RE, Wunderlich G, Lewis-D'Agostino DJ, Pyke R. Validation of the sexual interest and desire inventory-female in hypoactive sexual desire disorder. J Sex Med. (2010) 7(12):3918-28
Derogatis L, Clayton A, Lewis-D'Agostino D, Wunderlich G, Fu Y. Validation of the female sexual distress scale-revised for assessing distress in women with hypoactive sexual desire disorder. J Sex Med. (2008) 5(2):357-64
Derogatis L, Rosen R, Leiblum S, Burnett A, Heiman J. The Female Sexual Distress Scale (FSDS): initial validation of a standardized scale for assessment of sexually related personal distress in women. J Sex Marital Ther. (2002) 28(4):317-30.
Goldstein I, Lines C, Pyke R, Scheld JS. National differences in patient-clinician communication regarding hypoactive sexual desire disorder. (2009) J Sex Med 6(5) 1349-57.
Kingsberg SA, Knudson G. Female Sexual Disorders: Assessment, Diagnosis, and Treatment. CNS Spectr. 201 1
Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA. (1999) 1 0;281 (6):537-44.
Meston C, Trapnell P. Development and validation of a five-factor sexual satisfaction and distress scale for women: the Sexual Satisfaction Scale for Women (SSS-W). J Sex Med. (2005) 2(1 ):66-81 .
Meston CM, Hull E, Levin RJ, Sipski M. Disorders of orgasm in women. J Sex Med. (2004) 1 (1 ):66-8.
Read S, King M, Watson J. Sexual dysfunction in primary medical care: prevalence, characteristics and detection by the general practitioner. J Public Health. 19 (4): 387- 391
Rosen RC, Taylor JF, Leiblum SR, Bachmann GA. Prevalence of sexual dysfunction in women: results of a survey study of 329 women in an outpatient gynaecological clinic. J Sex Marital Ther. (1 993) 19(3):171 -88.
Shifren JL, Monz BU, Russo PA, Segreti A, Johannes CB. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol. (2008) 1 12(5):970-8.
Appendix 1 - Screener from Cincinnati
FEMALE PATIENTS
RESPONDENT FIRST NAME & LAST INITIAL:
INTRODUCTION
Hello, my name is and I am calling from We are contacting women between the ages of 20 to 60 years old in your area to request their participation in a research study. Is there anyone in the household that fits this description that I may speak with?
Many of the questions we're going to ask pertain to women' s health and sexual function and are very personal, but it is specific to the research question and will be anonymous and strictly confidential.
May I please have your age?
Under 20 Years Old (TERMINATE)
20 to 35 (RECORD EXACTAGE)
35 to 44 (RECORD EXACTAGE)
45 to 49 (RECORD EXACT AGE)
50 to 60 (RECORD EXACT AGE) I EXACT AGE:
Over 60 Years Old (TERMINATE)
What is your ethnicity or race? Are you (READ LIST)
African American / Black
Asian
Caucasian / White
Hispanic
Some Other Ethnicity / Race
NOTE: RECRUIT 1 -2 NON-CAUCASIANS PER GROUP
3. Which of the following best describes your relationship status:
A. Married
B. Co-habitating
C. Single in a monogamous relationship
D. Single, not in a monogamous relationship
4. Are you currently in a sexual relationship?
Yes No
5. How long have you been in this current relationship?
Less than 6 months
6 months or more
5a. On a scale of 1-10 (1 being not happy and 10 being extremely happy) how happy are you in your current relationship?
1 2 3 4 5 6 7 8 9 10
RESPONDENT MUST ANSWER AT LEAST 6 TO CONTINUE
6. Are you currently experiencing difficulties in achieving orgasms?
Yes (CONTINUE)
No (THANK AND TERMINATE)
6a. Have you ever experienced an orgasm?
Yes
No (skip to question 7)
6b. How often do you experience difficulties achieving an orgasm?
NEVER (TERMINATE)
RARELY SOMETIMES OFTEN ALWAYS
Have you always had difficulty reaching an orgasm?
Yes
No
6d. How bothered are you by your inability to experience an orgasm?
Extremely bothered
Very bothered
Somewhat bothered
A little bothered (TERMINATE)
Not at all bothered (TERMINATE)
6e. When was the last time you experienced an orgasm?
Within the past week
Over a week but less than a month ago
Over a month but less than 6 months ago
Over 6 months but less than a year ago
A year ago or longer
6f. How long have you been experiencing difficulties in having an orgasm?
(write duration)
What one word would you use to describe your orgasm difficulties?
(write down word)
7. Do you or does your spouse or significant other suffer from or receive medical treatment for any of the following medical conditions, which interfere with their normal sexual function or sexual activity, or not? (READ LIST)
A psychiatric / psychological disorder (such as clinical depression, multiple personality disorder, or a neurological disorder) (THANK AND TERMINATE)
A medical/physical problem (THANK AND TERMINATE)
Drug or alcohol dependency (THANK AND TERMINATE)
No (CONTINUE)
8. 1 would like to ask you some questions about your menstrual status? Which one of these statements best describes you?
I am still having regular or irregular menstrual periods and am not menopausal (go to Q 9)
I became menopausal after I had surgery to remove my uterus and ovaries, i.e., I am surgically menopausal (go to Q 10)
I am currently going through menopause naturally (go to Q 10)
I no longer experience the symptoms of menopause (go to Q 10)
9. Are you currently on any type of birth control?
Yes
No
10. Are you currently on any type of hormone therapy?
Yes
No (go to Q.12)
11. What hormone therapy are you currently using?
12. What is your annual household income? (READ LIST, ACCEPT ONE ANSWER)
Under $20,000
$20,000-839,999
$40,000-859,999
$60,000 or above
Prefer not to answer (DO NOT READ)
13. Would you be willing to participate in a research study exploring women's health issues? Some of the questions will be very sensitive in nature. All responses would be kept confidential. Your input is necessary to help understand women's health issues.
Yes
No (THANK & TERMINATE)
INVITATION
We are inviting individuals like you to participate in a group discussion with an experienced female moderator. The interview is for research purposes only. The discussion will remain confidential and no sales of any kind will take place. The discussion will last approximately MINUTES, and for your participation you will be paid $ the discussion is being held at [LOCATION]. The interview will be conducted on
[DATE]. Will you be able to join us?
Yes No (THANK AND TERMINATE)
PLEASE PRINT:
NAME:
ADDRESS:
STATE: ZIP CODE:
SCHEDULED FOR DATE: TIME:
I would like to read you a brief statement for your permission regarding this research. Although I will always remain anonymous, I hereby give permission to use the information provided during my interview, with the FDA in order to establish medical treatment for women and women' s health issues. I am aware that I may be audio and videotaped for the purposes of documenting my interview. A pharmaceutical company may use these tapes for the above purpose and/or for development purposes for future medications but will not be released to the public in anyway. Again, I am aware that my name will not be associated in any way with these tapes.
Yes, I give permission
No, I do not give permission (TERMINATE)
Screener from NYC
FEMALE PATIENTS
RESPONDENT FIRST NAME & LAST INITIAL:
INTRODUCTION
Hello, my name is and I am calling from We are contacting women between the ages of 20 to 60 years old in your area to request their participation in a research study. Is there anyone in the household that fits this description that I may speak with?
Many of the questions we're going to ask pertain to women' s health and sexual function and are very personal, but it is specific to the research question and will be anonymous and strictly confidential.
1. May I please have your age?
Under 20 Years Old (TERMINATE)
20 to 35 (RECORD EXACTAGE)
35 to 44 (RECORD EXACTAGE)
45 to 49 (RECORD EXACT AGE)
50 to 60 (RECORD EXACT AGE) EXACT AGE:
Over 60 Years Old (TERMINATE)
2. What is your annual household income? (READ LIST, ACCEPT ONE ANSWER)
Under $50,000 (TERMINATE)
$50,000-$75,000
S75,000 - 100,000
8100,000 or above
Prefer not to answer (TERMINATE)
3. What is your ethnicity or race? Are you (READ LIST)
African American / Black
Asian
Caucasian / White
Hispanic
Some Other Ethnicity / Race
NOTE: RECRUIT 1-2 NON-CAUCASIANS PER GROUP
4. Which of the following best describes your relationship status:
A. Married
B. Co-habitating
C. Single in a monogamous relationship
D. Single, not in a monogamous relationship
5. Are you currently in a sexual relationship?
Yes No
6. How long have you been in this current relationship?
Less than 6 months
6 months or more
6a. On a scale of 1-10 (1 being not happy and 10 being extremely happy) how happy are you in your current relationship?
1 2 3 4 5 6 7 8 9 10
RESPONDENT MUST ANSWER AT LEAST 6 TO CONTINUE
7. Have you ever experienced an orgasm?
Yes
No (THANK AND TERMINATE)
7a. Are you currently experiencing difficulties in achieving orgasms or an absence of orgasm when you engage in sexual stimulation including sexual intercourse, oral sex or masturbation?
Yes (CONTINUE)
No (THANK AND TERMINATE)
7b. How many times a month to you engage in sexual activity that would typically be expected to result in an orgasm?
#/month
7c. Have you always had difficulty reaching an orgasm?
Yes
No
7d. How bothered are you by your difficulties or inability to experience an orgasm?
Extremely bothered
Very bothered
Somewhat bothered
A little bothered (TERMINATE)
Not at all bothered (TERMINATE)
7e. How long have you been experiencing difficulties or inability in having an orgasm?
(write duration)
7f. What one word would you use to describe your orgasm difficulties/inability?
(write down word)
7g. When was the last time you experienced an orgasm by any type of sexual stimulation?
Within the past week (THANK AND TERMINATE)
Over a week but less than 6 months ago (THANK AND TERMINATE)
Over 6 months (CONTINUE)
Do you or does your spouse or significant other suffer from or receive medical treatment for any of the following medical conditions, which interfere with their normal sexual function or sexual activity, or not? (READ LIST)
A psychiatric / psychological disorder (such as clinical depression, multiple personality disorder, or neurological disorder) (THANK AND TERMINATE)
A medical/physical problem (THANK AND TERMINATE)
Drug or alcohol dependency (THANK AND TERMINATE)
No (CONTINUE)
9. 1 would like to ask you some questions about your menstrual status? Which one of these statements best describes you?
I am still having regular or irregular menstrual periods and am not menopausal (go to Q 10) I became menopausal after I had surgery to remove my uterus and ovaries, i.e., I am surgically menopausal (go to Q 11)
I am currently going through menopause naturally (go to Q 11)
I no longer experience the symptoms of menopause (go to Q 11)
10. Are you currently on any type of birth control?
Yes
No
11. Are you currently on any type of hormone therapy?
Yes
No (go to Q.12)
12. What hormone therapy are you currently using?
13. Would you be willing to participate in a research study exploring women's health issues? Some of the questions will be very sensitive in nature. All responses would be kept confidential. Your input is necessary to help understand women's health issues.
Yes
No (THANK & TERMINATE)
INVITATION
We are inviting individuals like you to participate in a group discussion with an experienced female moderator. The interview is for research purposes only. The discussion will remain confidential and no sales of any kind will take place. The discussion will last approximately MINUTES, and for your participation you will be paid $ the discussion is being held at [LOCATION]. The interview will be conducted on
[DATE]. Will you be able to join us?
Yes No (THANK AND TERMINATE)
PLEASE PRINT:
NAME:
ADDRESS:
CITY: STATE: ZIP CODE:
HOME PHONE:
WORK PHONE:
CELL PHONE:
E-MAIL ADDRESS:
SCHEDULED FOR DATE: TIME:
I would like to read you a brief statement for your permission regarding this research. Although I will always remain anonymous, I hereby give permission to use the information provided during my interview, with the FDA in order to establish medical treatment for women and women's health issues. I am aware that I may be audio and videotaped for the purposes of documenting my interview. A pharmaceutical company may use these tapes for the above purpose and/or for development purposes for future medications but will not be released to the public in anyway. Again, I am aware that my name will not be associated in any way with these tapes.
Yes, I give permission
No, I do not give permission (TERMINATE)
EXAMPLE 14
TBS-2 Testosterone Intranasal Gel
For the Treatment of Female Orgasmic Disorder)
SUMMARY
TBS-2 is a bioadhesive testosterone intranasal gel for the treatment of female orgasmic disorder (FOD). The testosterone intranasal gel has been referred to as TBS-2 or Noseafix.
TBS-2 is an innovative galenic formulation of testosterone for nasal administration. The formulation has many advantageous features including rapid absorption into
systemic circulation and rapid clearance, the lack of first pass metabolism, the avoidance of transference from one person to another, and the ease of use.
Testosterone is indicated for the treatment of hypogonadism in men and hypoactive sexual desire disorder (HSDD) in bilaterally oophorectomised and hysterectomised (surgically induced menopausal) women receiving concomitant estrogen therapy in the European Union. An overwhelming amount of pharmacology and toxicology studies have been conducted to support the safety of testosterone.
Pharmacological studies in monkeys showed that intranasal administration of TBS-2 induced sexual behaviour consisting of eyebrow raising, chest rubbing, courtship behaviour, and masturbation. TBS-2 is well tolerated in animals. No evidence of systemic drug related toxicity was observed in rabbits after repeated daily administration over a 90 day period at a dose level up to 10-fold the maximum clinical daily dose. Local tolerance studies in rat and rabbit following single dose and repeated dose administrations showed no irritation. Testosterone intranasal gel was classified as a non-irritant in the HET-CAM test. Per the literature, a NOEL of 0.1 mg was reported in a reproductive toxicology study following s.c. administration of testosterone propionate to rats. Assuming a rat weight of 300 g, this is a dose of 333 μg/kg/day. For comparison, the maximum daily dose of testosterone to be administered in the clinical trial from the nasal gel is 1200 μg. Thus, a 75 kg woman would be exposed to 24 μg/kg/day, over 10 fold less than the NOEL in rats.
The use of androgens to increase women's sexual libido was initially reported in 1 940 by Loeser. Several well designed, randomized, placebo controlled clinical trials have been performed over the last decade illustrating the efficacy and safety of androgen therapy in post menopausal woman who have low sexual desire.
Four clinical trials have been performed using TBS-2 in healthy pre-menopausal women and in post-menopausal women with HSDD and pre- and postmenopausal women with FOD . TBS-2 is rapidly absorbed following intranasal administration restoring the serum testosterone levels to normal serum testosterone levels of young women. The pharmacokinetics of 3 doses of TBS-2, 0.6 mg, 1 .2 mg and 1 .8 mg, have been determined. The 24 hour AUC of total and free testosterone were within
the normal ranges for all 3 doses. Multiple dose administration over 3 days showed minimal accumulation and TBS-2 was completely cleared within 24 hours. Pharmacodynamic studies in healthy middle aged women revealed that intranasal testosterone increased amygdala reactivity (a centre in the brain associated with emotional and sexual responses) to a level comparable to that of young women. Exploratory pharmacodynamic studies revealed increases in vaginal pulse amplitude (VPA) in women with FOD and increases in subjective measures of sexual arousal and positive affect in women with HSDD following TBS-2 administration. In a controlled environment study using vibrotactile stimulation to the glans clitoris (VTS) and visual sexual stimulation (VSS), TBS-2 was effective in inducing orgasm with VTS/VSS in women with FOD and patients treated with TBS-2 also showed a statistically significant improvement in VPA versus placebo, and elevation of sexual arousal, as well as positive trends in terms of elevating sensuality and pleasurable genital sensation. The greatest effects of TBS-2 were seen in the 2 and 4 hr groups, suggesting a delay-effect of testosterone on sexual arousal.
The clinical development program is intended to investigate the efficacy and safety of TBS-2 for the treatment of female orgasmic disorder.
TBS-2 is currently not approved for marketing in any country.
INTRODUCTION
Female orgasmic disorder (FOD) is the second most frequently reported women's sexual problem after hypoactive sexual desire disorder (HSDD). The Global Survey of Sexual Attitudes and Behaviours (Laumann et al., 1999) assessed sexual problems in 9,000 women aged 40-80 years in 29 countries. In the PRESIDE survey of over 31 ,000 women, approximately 10% reported low desire with distress and almost 5% report difficulty reaching orgasm with distress (Shifren et al., 2008). The prevalence of 'inability to reach orgasm' ranged from 17.7% (in Northern Europe) to 41 .2% (in Southeast Asia). FOD is considered to be the persistent or recurrent delay in, or absence of, orgasm following a normal sexual excitement phase, causing marked distress or interpersonal difficulty (DSM IV). When a woman has sexual activity that is not accompanied by good quality orgasmic release, sexual activity may
become a chore or a duty rather than a mutually satisfying, intimate experience. This may also lead to secondary loss of sexual interest and/or interpersonal difficulties.
Testosterone and Sexual Function
Testosterone, the primary circulating androgen in women, is a secreted by the ovaries and the adrenal glands. Contrary to the sudden drop in oestrogen during menopause, serum levels of androgens fall gradually as women age primarily due to a decrease in the production of adrenal androgen precursors. Testosterone plays a role in mood, body composition, bone mineral density and has central and peripheral effects on sexual function (Davis 1999, Goldstat 2003). In the periphery, testosterone is required for nitric oxide to stimulate vasocongestion for the engorgement of clitoral tissue and vaginal lubrication during sexual arousal (Traish 2007). Testosterone stimulates dopamine release in various brain structures implicated in motivation and reward systems, including sexual desire. Testosterone was found to stimulate dopamine release in the medial preoptic area of the anterior hypothalamus under basal conditions and with sexual stimulation in rats (Halaris 2003).
The use of androgens to increase women's sexual desire was first reported in 1940 by Loeser. Salmon (1943) observed that a number of young, married women who formerly considered themselves "frigid" were able to experience "a marked increase in coital gratification, culminating in an orgasm" after testosterone propionate injections and the effects wore off within several weeks after the discontinuation of the injections. In the 1980s, the role of androgens in maintaining sexual functioning was studied in oophrectomized women (Sherwin and Gelfand, 1987). In this three (3) month prospective open-label study of 44 women, monthly injections of estrogen and testosterone increased rates of sexual desire, sexual arousal, and number of fantasies. Furthermore, rates of intercourse and orgasm were higher in women treated with androgens and estrogen compared to the controls.
Several large randomized clinical trials have shown the efficacy of daily transdermal testosterone (Intrinsa) on the primary endpoint of desire for postmenopausal women with HSDD. These women also report greater arousal, orgasm, pleasure and responsiveness and less personal distress.
It is unlikely that orgasm improved simply as a result of improved desire. Most women with primary or secondary anorgasmia present clearly stating that desire was there, at least initially (for some women the anorgasmia is so distressing that it results in the secondary development of HSDD) and that HSDD was not the reason that they could not orgasm. Therefore, testosterone has an impact specifically on orgasmic attainment.
It is a logical next step in a research program dedicated to investigating the role of testosterone, to investigate whether testosterone, in the absence of other FSDs, has a direct effect on sexual function in general and FOD in particular. TBS-2 is a testosterone nasal gel for the treatment of female orgasmic disorder. The product, TBS-2 is a bioadhesive intranasal gel containing 0.24% to 0.72% testosterone with castor oil, oleoyl polyoxylglycerides, and colloidal silicon dioxide as excipients. TBS- 2 is administered as a dose administered equally into each nostril. The formulation has many advantageous features including the lack of first pass metabolism, the avoidance of transference from one person to another, in addition to the ease of use and the small quantity of active ingredient required.
TBS-2 is intended to be administered as required (p.r.n.) but not more than once- daily in women with female orgasmic disorder.
References to the Introduction
Davis, S. Androgen replacement in women: a commentary. J Clin Endocrinol. Metab. 1999; 84(6):1 886-1891 .
Diagnostic and Statistical Manual of Mental Disorders (fourth edition). American Psychiatric Association, 2000, Section 302.73.
Goldstat R, Briganti E, Tran J, Wolfe R, Davis SR. Transdermal testosterone therapy improves wellbeing, mood, and sexual function in premenopausal women; Menopause 2003; 1 0(5):390-8
Halaris A Neurochemical aspects of the sexual response cycle. CNS spectrums. 2003;9:21 1 -6.
Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA 1999;281 :537-544.
Loeser AA. Subcutaneous implantation of female and male hormones in tablet form in women. Br med J. 1940;1 :479-82.
Salmon UJ, Geist SH. Effect of androgens upon libido in women. J Clinical Endocrinol 1943;3:235-8.
Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA 1999;281 :537-544.
Loeser AA. Subcutaneous implantation of female and male hormones in tablet form in women. Br med J. 1940;1 :479-82.
Salmon UJ, Geist SH. Effect of androgens upon libido in women. J Clinical Endocrinol 1943;3:235-8.
Traish AM, Kim SW, Stankovic M, Goldstein I, Kim NN. Testosterone increase blood flow and expression of androgen and estrogen receptors in the rat vagina. J Sex Med., 2007;4:609 - 61 9.
PHYSICAL, CHEMICAL, AND PHARMACEUTICAL PROPERTIES AND FORMULATION
TBS-2 is a bioadhesive intranasal gel containing 0.24% to 0.72% testosterone. Drug Substance
Testosterone is the active ingredient in TBS-2 gel. The manufacture of the drug substance presents no potential risk for humans; the synthesis route is well- characterized.
Table 3.1 -1 : Nomenclature of Testosterone
INN name Testosterone
Compendial name Testosterone
Chemical name 1 7 -Hydroxyandrost-4-en-3-one
Other non-proprietary names Androst-4-en-3-one, 1 7-hydroxy-, (17β)-
Trans-testosterone
A4-androsten-1 7 -ol-3-one
CAS registry number 58-22-0
Proquina code 8139
Figure 3.1 -1 : Structural Formula

Molecular Formula
Relative Molecular Mass
288.4
Physical Chemical Properties
The physical chemical properties of testosterone are listed in Table 3.1 .1 -1 .
Table 3.1 .1 -1 : General Pro erties of Testosterone

Drug Product
Composition
The drug product TBS-2, testosterone nasal gel, is a viscous and thixotropic oil- based formulation containing solubilised testosterone intended for intranasal application. Three different formulations of TBS-2 will be used in this clinical trial, 0.24%, 0.48% or 0.72% w/w testosterone gel.
Other pharmacologically inactive ingredients in the testosterone intranasal gel are castor oil USP, oleoyl polyoxylglycerides Ph.Eur./NF and colloidal silicon dioxide NF. None of these excipients are of human or animal origin. All excipients are well- known and listed in the "Inactive Ingredient" list for Approved Drug Products issued by the FDA.
Castor oil
Castor oil is used as an excipient in several approved medicinal products applied by different administration routes, e.g. i.m. injection, ophthalmic emulsion, sublingual tablet or topical emulsions/creams/ointments/solutions. Hence, it can be deduced that castor oil is suitable for an application route where safety and tolerability are of highest importance (e.g. injectables and ophthalmic preparations) although it has not been approved in nasally applied preparations yet.
Oleoyl polvoxylqlvcerides
According to the "Handbook of Pharmaceutical Additives" oleoyl polyoxylglycerides are used as hydrophilic oil for topicals, injectables and nasals. In FDA-approved medicinal products it is used as co-emulsifier in topical emulsions/lotions/creams and in vaginal emulsions/creams. In France this excipient is approved for nasal preparations such as "Rhino-Sulforgan" (Laboratoire Jolly-Jatel, France; containing 10% oleoyl polyoxylglycerides) and "Huile Gomenolee 2%" ("Laboratoire Gomenol, France; containing 10% oleoyl polyoxylglycerides). Hence, it can be deduced that oleoyl polyoxylglycerides is suitable for an application route where safety and tolerability are of highest importance (e.g. injectables and nasal or vaginal preparations).
Colloidal silicon dioxide
According to the "Handbook of Pharmaceutical Additives" colloidal silicon dioxide is used as an oil adsorbent, thermal stabiliser and gellant. In FDA-approved medicinal
products it is used in dental gels, sublingual tablets, endocervical gel, suppositories, vaginal emulsions/creams/tablets/tampons and capsules for inhalation. Furthermore, it is used as an excipient in "Testoderm with adhesives" (Alza Corporation, approved in 1996) a testosterone transdermal patch. Hence, it can be deduced that colloidal silicon dioxide is suitable for an application route where safety and tolerability are of highest importance (e.g. inhalations, endocervical, vaginal or rectal preparations).
A detailed description of the batches is shown in Table 3.2.1 -1 .
Table 3.2.1 -1 : Composition of TBS-2

For clinical trial supplies, testosterone intranasal gel is supplied in a multiple dose dispenser. The dispenser is a finger actuated dispensing system designed to dispense 122.25 mg of TBS-2 gel per actuation from a non-pressurized container into the nasal cavity. One dose consists of 2 actuations. The dispenser is designed to deliver 45 doses (90 actuations) of TBS-2. The key components of the multiple dose dispenser include a barrel, base, pump, and actuator, which are composed of polypropylene and a piston, which is composed of polyethylene.
3.2.2 Administration of TBS-2
No preparation of TBS-2 is required before administration.
TBS-2 is administered by the patient. Specific administration instructions are appended to each protocol. In general, the patient is instructed to:
• Blow your nose to clear your nostrils.
• Place your finger on the pump of the actuator and, in front of a mirror, advance the tip of the actuator into your left nostril until the finger on the pump reaches the base of the nose.
• Aim the tip of the actuator to the inner corner of your left eye. The opening on the tip of the actuator must face the skin that lines the inner nose.
• Depress the pump until it stops.
• Slowly remove the actuator from your nose.
• Wipe away any gel that remains on the tip of the actuator using a clean dry swab.
• Repeat process for the right nostril.
• Press the nostrils together lightly and massage for 1 second.
• Do not blow your nose or sniff for 1 hour after administration of the medication.
3.2.3 Recommended Storage Conditions
TBS-2 is stored at room temperature (20 - 25 °C or 68 - 77 °F). Temperature excursions from 15 to 30°C or 59 to 86 °F are permissible for TBS-2.
NON-CLINICAL STUDIES
A large number of non-clinical pharmacology and toxicology studies have been conducted to support the safety of testosterone in humans. While the majority of these studies have been performed using male animals, substantial information is available from female animal studies. In addition to the studies referenced in the literature, non-clinical studies are conducted to assess the primary pharmacodynamics, repeat dose toxicity and single and repeat dose local tolerance of TBS-2.
Pharmacology
Primary Pharmacodynamic Studies Performed by the Sponsor
Non-clinical pharmacodynamic studies have been performed to determine the effect of nasally administered testosterone on sexual behaviour in female capuchin monkeys.
4.1.1.1 Effects of intranasally administered testosterone in female capuchin monkeys
The study was performed in female capuchin monkeys living with male cohorts to determine the effect of TBS-2 on sexual behaviour (Tavares, 2007).
During eight days of baseline, 10 females were observed for sexual and non-sexual behaviour. Monkeys then received daily administrations of TBS-2 (0.24 mg per nostril, n = 5 per group) or placebo gel for 5 days, followed by five days of wash-out, followed by 5 days of daily intranasal administration, whereby the animals that had received testosterone before, now received placebo and vice versa. Diverse sexual and non-sexual behaviours were scored and blood samples were collected and analysed for testosterone, estradiol, dihydrotestosterone and progesterone.
TBS-2 induced sexual behaviour (eyebrow raising, chest rubbing, courtship behaviour, masturbation) in the females during testosterone treatment, which seemed to be prolonged even when testosterone treatment was discontinued. These behavioural results were accompanied by an increase in plasma testosterone levels. This study demonstrates the efficacy of TBS-2 in enhancing sexual behaviour in female capuchin monkeys following intranasal administration.
4.1.1.2 Prolonged effects of intranasally administered testosterone in female capuchin monkeys
A second study was performed to determine prolonged effects of TBS-2 in female capuchin monkeys (Topic, 2007).
During five days of baseline, females were observed for their sexual and non-sexual behaviour. Monkeys received daily administrations of TBS-2 (0.24 mg per nostril) for 7 days, followed by 15 days of TBS-2 every third day or were left undisturbed (n=5 per group). Diverse sexual and non-sexual behaviours were scored and blood samples were collected and analysed for testosterone, estradiol, dihydrotestosterone and progesterone.
Similar to the initial study, an increase in sexual proceptive behaviour was revealed during TBS-2 treatment, which persisted about two weeks beyond treatment in the
group receiving TBS-2 every third day as well as in the group that no longer received any treatment. These behavioural results were accompanied by an increase in plasma testosterone levels. This study demonstrates the efficacy of TBS-2 in enhancing sexual behaviour in female capuchin monkeys following intranasal administration, and provided evidence that daily substance application is not necessary to maintain beneficial effects on sexual behaviour in monkeys.
Primary Pharmacodynamic Studies on Testosterone in the Literature
Little is known of the role that androgens play on sexual behaviour in animals because the classic laboratory models for sexual behaviour are rodents in which sexual receptivity is regulated by oestrogen and progesterone rather than androgens. However, androgens have been implicated in the expression of sexual behaviour in females of several non-rodent species (musk shrews, horses, monkeys and humans). Studies have shown that testosterone and its metabolites regulate sexual behaviour in the musk shrew. Sharma and Rissman (1994) demonstrated that testosterone implants in the medial preoptic area and in the dorsalmedial hypothalamus induced sexual receptivity in the female musk shrew including tail wagging and receptivity to receive mounts and ejaculation from male musk shrews.
Female sexual arousal is characterized by increased genital blood flow, genital sensation and vaginal lubrication. A number of studies have recently been performed in animals elucidating the biochemical and physiological role of androgens in female genital tissue and sexual function. Testosterone has been shown to play a critical role in the development and maintenance of vaginal tissue structure. Kim et al. (2004) demonstrated that testosterone prevents the atrophy of the muscularis layer of the vaginal wall in addition to maintaining the relaxatory response of the vaginal wall to endogenous neurotransmitters.
Testosterone has been implicated in increasing genital blood flow. Clitoral and vaginal blood flow is primarily mediated by the nitric oxide/cyclic guanosine monophate pathway (Giuliano 2002). Traish et al. (2003) demonstrated that testosterone administration to ovarietomized animals increased protein expression and enzyme activity of nitric oxide synthase in the proximal vaginal tissue. In addition, testosterone treatment enhanced the vaginal blood flow response and
resulted in upregulation of androgen and estrogen receptors in ovarectomized rats relative to vehicle-infused ovariectomized animals (Traish 2007). These observations are consistent with the hypothesis that androgens directly modulate vaginal function.
Secondary Pharmacodynamics
In men, testosterone significantly influences the body composition. Androgen deficiency is associated with reduced bone mineral density, increase in body fat and decrease in lean body weight. Studies have been performed in animals to determine the role of testosterone on body composition. Supraphysiological testosterone administration successfully prevents orchiectomy-induced skeletal catabolism (Turner 1990) and enhances bone strength and mass in gonadectomised male and female rats (Yarrow et al., 2008). Gentile et al. (2010) showed that aged castrated rats develop increased fat mass, reduced muscle mass and strength and lower bone mass. Treatment with testosterone or DHT reverses the effects on muscle and adipose tissue while only aromatizable testosterone increased bone mass.
4.3 Pharmacokinetics and Metabolism in Animals
4.3.1 Nasal Administration
Toxicokinetic study are conducted as supportive data to the 90-day repeat-dose intranasal toxicity studies in female rabbits (#227422). In the pivotal repeat-dose toxicology study, blood samples were collected on Day 1 and Day 90 for determination of serum levels of testosterone and its metabolites, DHT and estradiol. Testosterone was rapidly absorbed and exhibited dose-proportional kinetics. After 90 days of dosing and in comparison to Day 1 , there were no significant differences between the serum exposure and TK parameters for testosterone at the low TBS-2 gel dose, however there was an apparent 30% reduction of Cmax and 31 % reduction of the AUCo-8 hr- At the high dose TBS-2 gel and in comparison to Day 1 , the serum exposure to testosterone was lower as indicated by a significant reduction of AUCo-shr (59%) and Cmax (45%) accompanied by a significant increase (2.6-fold) in the Vz/F (apparent fluid volume in the body necessary to maintain the total dose of drug at the concentration found in the serum) and an apparent increase (2.3-fold) in CL/F (apparent volume of serum per unit time from which drug is completely removed). Thus, 90-day dosing with low and high dose TBS-2 gel did not result in accumulating
levels of testosterone in the serum. Rather, 90-day dosing with TBS-2 gel resulted in dose-dependent decreases of the serum levels of testosterone compared to Day 1 likely due to the increased clearance and volume of distribution of testosterone.
DHT levels were above the limit of detection in most but not all serum samples on Day 1 post treatment with high dose TBS-2 gel, while at the lower dose, DHT levels were below either the limit of quantification or limit of detection. At Day 90 for both the high and low doses of TBS-2 gel, the majority of serum samples were below the limit of detection (BLD) for DHT. Estradiol could not be detected in any of the serum samples analyzed.
Figure 4.3.1 -1 : Mean Serum Concentration-Time Profiles of Testosterone on
Day 1 and Day 90: Testosterone concentration-time profiles are shown following low (0.6%) and high (0.9%) dose TBS-2 gel treatment to rabbits on (A) Day 1 and (B) Day 90. Data are presented as the mean ± SE.

Time (hr)
B

0 1 2 3 4 5 6 7 8
Time (hr)
4.3.2 Metabolism
Testosterone is extensively metabolized by cytochrome P450 3A4 (CYP3A4) aromatase, and 5-alpha-reductase. CYP3A4 metabolizes testosterone primarily in the liver by conversion to various hydroxyl steroids. The active metabolites of testosterone are DHT and estradiol. Testosterone is metabolized to DHT by steroid 5-alpha reductase in many tissues. Testosterone is metabolized to estradiol by aromatase.
4.3.2.1 Metabolism in Nasal Mucosa
The nasal mucosa is a metabolically active tissue and is able to metabolize xenobiotics.
Experiments in animals showed that testosterone is minimally metabolized in the nose. The main metabolites of testosterone after nasal administration were 4- androstene-3, 17-dione, 5cc-dihydrotestosterone, 3a-hydroxy-5cc-androstane-1 7-one, and 5cc-androstane-3alpha, 17beta-diol (Brittebo 1984). These metabolites were identified in in vitro investigations using the nasal septa of F-344 rats and indicated the presence of 5cc-reductase, 3- and 17-hydroxysteroid dehydrogenase in the nasal mucosa.
The main metabolites in men and minipigs were dihydrotestosterone and estradiol, indicating that the minipig is a very suitable model for testosterone kinetics in men,
including absorption behavior. In rhesus monkeys, dihydrotestosterone is also an important metabolite. In contrast to humans, minipigs and rhesus monkeys, DHT is not a main metabolite in dogs.
4.3.3 Distribution
Circulating testosterone in plasma exists either tightly bound to SHBG (sex hormone binding globulin; a plasma protein synthesized in the liver that specifically binds steroid sex hormones - approximately 65-80%), weakly and reversibly bound to albumin (approximately 20-30%) or as free testosterone (0.5 - 2%). The albumin bound fraction and the unbound fractions are collectively termed "bioavailable" testosterone.
4.3.4 Elimination
About 90% of a dose of testosterone given intramuscularly is excreted in the urine as glucoronide and sulfate conjugates of testosterone and its metabolites; about 6% of a dose is excreted in the faeces, mostly in unconjugated form.
4.4 Safety Pharmacology
4.4.1 Safety Pharmacology Studies Performed by the Sponsor
Separate safety studies have not been performed; however, safety pharmacology evaluations of TBS-2, including effects on function of the nervous system, cardiovascular and respiratory system were incorporated into a 90 day repeat dose toxicity study in rabbits (Study #227422).
A neurological examination for motor activity and sensory activity to visual, audio and proprioceptive stimuli was conducted in this GLP compliant 90-day repeat-dose toxicity study. In this study, rabbits received TBS-2 daily for 90-days at a dose of 0 (vehicle control), 0.12 mg/kg, 0.24 mg/kg or no treatment. Neurological evaluation (FOB - Functional Observation Battery testing) which included approach response, touch response, auditory response, pain perception, eye blink response, righting reflex, pupil response, pinna reflex and proprioception were performed on all animals during the pre-treatment period, once on study Day 45 and again during the last week of dosing, and on all recovery animals during the last week of the study.
Cardiovascular and respiratory safety assessments were also performed evaluating heart rate, blood pressure and respiratory rate.
There were no significant differences in the neurological, cardiovascular and respiratory evaluations between the control groups and the two TBS-2 groups.
4.4.2 Safety Pharmacology Studies Reported in the Literature
4.4.2.1 Influence on the cardiovascular and renal system
Androgen treatment in men has been linked to increases in blood pressure. Experiments were conducted by Seachrist (2000) in spontaneously hypertensive rats (SHR) to determine the mechanism by which testosterone elevates blood pressure. In the SHR rats, testosterone significantly increased blood pressure while castration significantly decreased blood pressure. Testosterone replacement in the castrated rats increased blood pressure but not completely to that of the control levels. The increase in blood pressure was partially caused by a structural change of increased collagen deposition to the coronary arteries in addition to a testosterone induced increase of angiotensin II. Further evidence of the influence of androgen with the rennin-angiotensin system has been demonstrated in Wistar Kyoto rats. Castration lowered angiotensinogen mRNA levels in male rats while female rats with T implants showed an increase in renal angiotensinogen mRNA levels. Testosterone has also been shown to increase plasma levels of endothelin-1 which can act as a vasoconstrictor (Polderman, 1993).
4.4.2.2 Influence on lipid and liver function
In men, androgen therapy is associated with a shift in lipoprotein fractions. This shift is a known risk factor for coronary artery disease. Furthermore, androgen use is known to increase liver transaminases in certain ethnic populations. Therefore, there is a need to monitor the effects of repeated and long-term use of androgen on lipid and liver function parameters.
A study was performed by Tyagi et al. (1999a, 1999b) in rhesus monkeys to further understand the influence of long term testosterone intramuscular administration on lipid metabolism and liver function. Monkeys received bimonthly administration of 50
mg testosterone enanthate (TE) for 32 months. TE increased serum testosterone levels into the supraphysiological range one day after injection and peak levels were seen on Day 3, followed by a decrease to above baseline levels by Day 14.
Long-term treatment with intramuscular testosterone enanthate (TE) altered the pattern of testosterone metabolism by the liver. In control animals, the liver converted testosterone to androstenedione as the major metabolite whereas androsterone was the major metabolite in chronically TE treated animals. The percentage of DHT formed was significantly higher in animals chronically treated with TE. The percentage of testosterone which remained un-metabolized was similar in TE treated and control animals.
Long-term treatment with TE also altered lipid parameters. In TE treated animals, high density lipoprotein cholesterol (HDL-C) levels decreased gradually compared to baseline values and the decrease was significant from the 19th month of injections until the first month of recovery. This decrease in HDL-C was significant when compared to control animals from the 12th month of injection until the first month of recovery. Low density lipoprotein cholesterol (LDL-C) levels increased significantly in TE treated animals compared to control levels from the 12th month of injections until the end of the study period. The LDL/HDL-C ratio was also elevated following TE injections.
TE injections did not change the levels of triglycerides, alkaline phosphatise or bilirubin compared to baseline or control animals. Transaminase (SGOT and SGPT) levels increased following TE injections and remained elevated until the end of injections followed by a return to baseline values or below during the recovery period.
4.5 Toxicology
The toxicology profile of testosterone has been characterized in numerous studies referenced in literature. To supplement the toxicology studies referenced in the literature, a series of studies are performed to assess the toxicology and local tolerance of the TBS-1 and TBS-2 testosterone intranasal gels. The only difference between TBS-1 and TBS-2 is the concentration of testosterone. Therefore, the
applicant considers the local tolerance studies conducted with TBS-1 applicable for TBS-2.
4.5.1 Toxicity after single dose administration
Data on the toxicity of testosterone after single-dose administration have been compiled in Table 4.5.1 -1 .
Table 4.5.1 -1 : Toxicity of Testosterone after Single Dose Administration
Species Parameter Route Dose Symptoms/effect Reference
(mg/kg)
Arcatia LCio water 2.5 mg/l Not reported. [Andersen, H. tonsa R. 2001 ]
(copepod)
Arcatia LC50 water 5.6 mg/l Not reported. [Andersen, H. tonsa R. 2001 ]
(copepod)
Mammal, LD50 p.o > 5,000 Not reported. [RTECS not 2002] specified
Rat LDLo i.p. 326 Lungs, thorax, [RTECS
respiration (other 2002] effects)
4.5.2 Repeat Dose Toxicity Studies
4.5.2.1 Repeat dose toxicity study of the sponsor
Repeat-dose toxicity study of high dose testosterone intranasal gel in male rabbits (Study number 227120417)
Testosterone intranasal gel was administered intranasally to male rabbits twice a day for three month (90 days) at concentrations of 0.093 mg/kg, 0.550 mg/kg and 1 .867 mg/kg (1 x, 5x and 1 0x the proposed clinical concentration for the treatment of hypogonadism in males; approximately 10x, 50x and 100x the proposed clinical concentration for the treatment of female orgasmic disorder). Study controls included a negative control (no treatment) and a placebo control (gel base minus testosterone). Testosterone and DHT levels were assessed on Day 0, 45 and 90 of the study. Animals were observed daily for signs of systemic toxicity and sacrificed at the end of the study to determine any signs of tissue pathology. This study was performed in accordance to GLP requirements.
The rabbits did not show any evidence of systemic toxicity during the in-life observations in the treatment and control animals. There were no abnormal histopathological findings in the nasal turbinates, brain, testes, heart, kidneys, and lungs. Incidence and severity of centrilobular/midzonal glycogenic vacuolation in the liver were marginally greater in the high-dose testosterone group in comparison with the untreated control. All other changes were considered to be part of the normal background, including minimal inflammatory foci seen in the nasal turbinates, heart, lungs, and liver.
In summary, administration of testosterone intranasal gel for three months at doses up to the 100-fold of the proposed female dose was not associated with any local or systemic toxicity in the organs examined, including the nasal turbinates, brain, testes, heart, kidneys, and lungs.
Repeat dose toxicity study of low dose testosterone intranasal gel in female rabbits (Study number 227422)
The toxicity of intranasal TBS-2 was investigated in a 90-day repeat dose study in female rabbits. The rabbits were dosed intranasally at the following dose levels 0.1 2 mg/kg and 0.24 mg/kg daily for 90 days. A third group of animals received a placebo gel (TBS-2 gel minus testosterone) and a fourth group (control) were not dosed.
Each group of rabbits (n=10) consisted of a Main Study group (n=5) and a Recovery period group (n=5).
At the end of the treatment period, all Main Study animals were sacrificed and submitted for gross necropsy. The Recovery animals were sacrificed at the end of the 30-day recovery period. Clinical reactions to treatment were monitored as well as body weight assessment, food consumption, ophthalmology, clinical pathology, gross pathology and organ weights. Blood samples were collected on Day 1 and Day 90 for determination of serum levels of testosterone and its metabolites. Histopathology evaluations were performed on a comprehensive range of organs from the placebo and high-dose groups (Main Study and Recovery). The study was performed in accordance to GLP requirements.
The rabbits did not show any evidence of systemic toxicity during the in-life
observations in the treatment and control animals. There were no abnormal histopathological findings in any organ/tissue examined.
Testosterone was rapidly absorbed and exhibited dose-proportional toxicokinetics. DHT levels in the serum were approximately 15% of serum testosterone levels, which is consistent with the literature. Estradiol could not be detected in the serum.
In summary, under the conditions of this experiment, the no observed effect level (NOEL) of TBS-2 testosterone gel, when administered intranasally to female rabbits for 90 days, was considered to be equal to 0.24 mg/kg/day, the highest dose tested, based on lack of toxicity during the 90-day study period. The dose levels of 0.12 mg/kg and 0.24 mg/kg represent approximately 7.5x and 15x anticipated human therapeutic dose of 1 .2 mg (based on 75 kg woman).
4.5.2.2 Repeat dose toxicity studies from the literature
Repeat-dose toxicity studies were performed on rodents. The results have been compiled in Table 4.5.2.2-1 . Depending on the mode of administration, the lowest toxic dose ranged from 6.25 mg/kg to 400 mg/kg. The main results were the development of tumours in the experimental animals. The role of testosterone in tumourigenicity is discussed in detail under "Carcinogenicity" (including supportive toxicokinetics evaluations).
The toxicity of repeated doses of testosterone depends on the dose and duration of administration. Testosterone stimulated the tumourigenesis in rodents. No special problems concerning the gastrointestinal tract, blood and blood formation or any other frequent adverse events of drug use were observed.
Table 4.5.2.2-1 : Repeat Dose Toxicity in Animals
Species Parameter Route Dose Symptoms/effect Reference
(mg/kg)
Mouse TDLo p.o. 6,25ο1 Tumourigenic - ovarian [RTECS
tumours 2002]
Mouse TDLo s.c. 302 Tumourigenic - adrenal [RTECS
cortex tumours, other 2002] reproductive system
1 52 d continuous
2 5 d intermittent
tumors
Mouse TDLo Implant 4003 Tumourigenic - ovarian [RTECS
tumours 2002] Mouse4 Implant 1 -2 mg5 Cervical/uterine [HSDB 2002] tumours (61 .9%),
metastases/ infiltration
into the lungs (38.5% of
animals with tumours)
Rat6 Implant 1 -3x10 prostatic carcinoma [HSDB 2002] mg7 (0%, 16.7%, 20%),
metastasis (about
40%), incidence in
untreated controls
0.48%
4.5.3 Genotoxicity
The genotoxicity profile of testosterone has been evaluated in a wide range of well designed (including all ICH endpoints) in vivo and in vitro studies reported in the literature and have been compiled in Table 4.5.3-1 .
Table 4.5.3-1 : Mutagenic and Genotoxic Activity of Testosterone
Type of test Route of exposure Test system Dose duration Reference
Cell in vitro Syrian hamster 1 -30 μ9/ιτιΙ [Tsutusi, T. transformation embryo cells 1995]
Cytogenetic in vitro Human kidney 100 μιηοΙ/Ι [RTECS 2002] analysis
DNA damage in vitro Mammal 1 μιηοΙ/Ι [RTECS 2002]
(unspecified)
liver
DNA damage in vitro Mammal 10 μιηοΙ/Ι [RTECS 2002]
(unspecified)
lymphocytes
DNA damage in vitro Mouse liver 100 μιηοΙ/Ι [RTECS 2002]
DNA inhibition in vitro Human kidney 100 μηηοΙ/Ι [RTECS 2002]
DNA inhibition in vitro Human 50 μηηοΙ/Ι [RTECS 2002] lymphocyte
DNA inhibition in vitro Rat liver 100 μηηοΙ/Ι [RTECS 2002]
Mitotic Acute/chronic Drosophila 0-4 mmol/l [Vogel, E. W. recombination feeding, melanogaster 1993]
surface (eye mosaic
treatment, assay)
inhalation
3 50 d continuous
4 Strain: C57BLXDBA
5 twice weekly for life span
6 Strain: NB
7 up to 91 weeks, change of implants every 6-8 weeks
Type of test Route of exposure Test system Dose duration Reference
Morphological in vitro Hamster 5 mg/l [RTECS 2002] transformation embryo
Recombination in vitro Saccharomyces 0-10 mg/ml [Fahrig, R.
(without cerevisiae MPI 1996] metabolic
activation)
Recombination in vitro Saccharomyces 0-10 mg/ml [Fahrig, R.
(with metabolic cerevisiae MPI 1996] activation)
Sperm Parenteral Non- 12 mg/8 wk, [RTECS 2002] morphology mammalian intermittent
species
Unscheduled Parenteral Rat 10 mg/kg [RTECS 2002]
DNA synthesis
Testosterone was reported as being non-mutagenic in a range of assays evaluating effects on chromosomes: in the Ames test in bacteria, in a mouse lymphoma assay and in chromosomal aberration tests (HeLa cells, human synovial cells, Chinese hamster Don cells) except in the Chinese hamster lung cells where a clastogenic response was observed at the highest dose tested corresponding to cytotoxic dose levels. Three mouse micronucleus tests were reported from different laboratories, and all were negative. An absence of DNA adducts was further reported.
In in vivo tests, testosterone was non-mutagenic in two micronucleus tests in mice and in one chromosomal aberration test (sperm head morphology and chromosomal aberrations bone marrow) in rats.
The results of these studies consistently confirm the lack of a genotoxic effect with testosterone (Intrinsa SBA).
4.5.4 Carcinogenicity
A substantial body of literature demonstrates that, as expected for a hormonally active compound, tumour incidence can be increased in hormonally responsive organs and tissues including endometrium, mammary glands, liver and prostate. A summary of these animal studies is listed in Table 4.5.4-1 .
In 1987, the International Agency for Research on Cancer (IARC Supplement 7) concluded in the overall evaluation that androgenic (anabolic) steroids are probably carcinogenic in humans.
Table 4.5.4-1 : Carcinogenicity of Testosterone
Species Organ Route Dose Symptoms/effect Reference system (mg/kg)
Rat (Noble) Mammary TP 120 mg Mammary gland [Xie 1999] implant carcinogenesis
Rat (Wistar) Mammary TP 50 mg Acinotubular [Chambo- implant differentiation, gland Filho
hyperplasia 2005]
Guinea pig Ovarian TC 232 - benign cysts, small [Silva
500ng/m adenomas in the 1997] ovary parenchyma
and papillomas on
the ovarian surface
Mice Cervical- TP 1 -2mg Cervical uterine [HSDB
(C757BLXDBA) uterine implant 2x/wk tumours 2002] Rat (Lobund Prostate TP Depot 50mg Prostate [Pollard Wistar) + N-nitrioso- every adenocarcinoma 1986] N-methylurea 2m
Rat (Wistar) Prostate BOP + 100 Prostate [Pour
TP mg/kg adenocarcinoma, 1987] injection every 21 urinary bladder
or Pellet d papillomas and
35-45 carcinomas
mg
every 6
w
Rat (F344) Prostate TP Every Reduced incidence [Cui 1998] implant 40d of atypical
hyperplasia of the
ventral prostate
Rat (Noble) Prostate TP 30 Increase in [Noble implant mg/6-8 carcinoma of the 1977] w prostate
Rat (F344) Prostate TP 40 mg/6 Decrease in [Shirai
DMAB weeks incidence of typical 1994] hyperplasia of the
ventral prostate and
seminal vesicles;
increased the
incidence of atypical
hyperplasia and
carcinomas of the
dorsolateral and
anterior prostate
Rat (NBL/Cr Prostate TP Every Prostatic [Bosland and Sprague estradiol 6m adenocarcima 1995] Dawley) 17β (lower incidence in
Sprague Dawley
rats)
Rat Testicular T implant Addition of T [Waalkes
(F344/NCr) Cadmium abolished cadmium 1997] induction of leydig
cell tumours.
Increased incidence
of histiocytic
sarcoma; reduced
incidence of
fibrosarcoma
TP - testosterone propionate
BOP - N-Nitrosobis(2-oxopropyl)amine
MNU - N-nitrioso-N-methylurea
DMAB - 3.2'-dimethyl-4-aminobiphenyl
4.5.5 Reproductive and Developmental Toxicity
4.5.5.1 Prenatal development
Mammalians -Fertility and early embryonic development
Both female and male animals showed changes in the structure and function of the genital organs if the animals were treated with testosterone before mating (RTECS 2002). These disturbances probably contributed to the effects observed with regard to the fertility of the animals.
The administration of testosterone before mating or after conception disturbed different parameters of fertility (e.g. female fertility index, increased pre-implantation mortality). An overview is given in Table 4.5.5-1 . These effects were observed in rodents (e.g. rats, rabbits, hamsters) and other domestic animals (RTECS 2002).
Mammalians - Embryo-fetal development
Besides a higher incidence of fetal death in testosterone-treated animals, specific developmental abnormalities were observed in animals and human beings (RTECS 2002).
The specific developmental abnormalities included the urogenital tract, endocrine organs and skin and skin appendages (Table 4.5.5-1 ) Female offspring was masculinised dependent on the dose and time of testosterone administration.
In a study in rats, high levels of testosterone treatment on the 14 day of gestation caused the mammary glands in the offspring of both sexes to be smaller or absent, and the nipples were absent in female offspring. Testosterone had no effect on female masculinisation at 0.1 mg (HSDB 2002).
4.5.5.2 Postnatal development
In mice, it was shown that the live birth index and the viability index were lower; in other animals behavioural disturbances were observed (Table 4.5.5-1 ).
Table 4.5.5-1 Reproductive Toxicity of Testosterone (Andersen, H. R. 2001 , HSDB 2002, RTECS 2002)
Species/sex Parameter Dose/ Route Effects
duration
Human/male TDLo 17 mg/kg/26 wk Implant Spermatogenesis (incl. genetic material, morphology, motility, count), other effects
Human/female TDLo 34.6 mg/kg No information specific developmental abnormalities (urogenital system)
7-13 wk p. concept.
Rat/female TDLo 100 mg/kg p.o. specific developmental abnormalities (urogenital system)
17-20d p. concept.
Rat/male TDLo 64 mg/kg p.o. Paternal effects (prostate, seminal vesicle, Cowper's
10 d premating gland, accessory glands)
Rat/female TDLo 25 mg/kg s.c. Effects on newborn (physical, delayed effects)
17 d p. concept.
Rat/female TDLo 7 mg/kg s.c. Fertility - abortion
10-16 d p. concept.
Rat/female TDLo 4 mg/kg s.c. Maternal effects (parturition), post-implantation mortality
9 d p. concept. (e.g. dead and/or resorbed implants per total number of implants)
Rat/female TDLo 20 mg/kg s.c. Preimplantation-mortality (e.g. recution in number of
5 d p. concept. implants per female, total number of implants per corpora lutea)
Rat/male TDLo 8.4 mg/kg s.c. Spermatogenesis (incl. genetic material, morphology,
21 d premating motility, count), other effects
Rat/female TDLo 1 .4 mg/kg s.c. Fertility (other measures of fertility)
14 d premating
Rat/female TDLo 0.7 mg/kg s.c. Maternal effects (ovaries, fallopian tubes, uterus, cervix,
14 days premating vagina)
Rat/female TDLo 60 mg/kg i.m. Preimplantation-mortality (e.g. recution in number of
3-7 d after implants per female, total number of implants per corpora
conception lutea)
Rat/female TDLo 8 mg/kg i.m. Specific developmental abnormalities (skin/skin
13-20 d after appendages, urogenital system)
conception
Rat/male TDLo 0.28 mg/kg i.m. Paternal effects (testes, epididymis, sperm duct, prostate,
14 d premating seminal vesicle, Cowper's gland, accessory glands)
Rat/female TDLo 2.5 mg/kg parenteral Maternal effects (ovaries, fallopian tubes)
10 d premating
(continued)
Species/sex Parameter Dose/ Route Effects
duration
Rat/male TDLo 4 mg/kg parenteral Paternal effects (spermatogenesis, incl. genetic material,
3 wk premating sperm morphology, motility and count)
Rat/male TDLo 8 mg/kg parenteral Paternal effects (spermatogenesis, incl. genetic material,
3 wk premating sperm morphology, motility and count)
Rat/male TDLo 10.4 mg/kg Implant Paternal effects (testes, epididymis, sperm duct)
30 d premating
Rat/male TDLo 27 mg/kg Implant Paternal effects (spermatogenesis, incl. genetic material,
90 d premating sperm morphology, motility and count, testes, epididymis, sperm duct), fertility (male fertility index8)
Rat/male TDLo 10.9 mg/kg Implant Paternal effects (prostate, seminal vesicle, Cowper's
91 d premating gland, accessory gland)
Rat/male TDLo 33.3 mg/kg Implant Paternal effects (other effects on male), fertility (pre-
15 wk premating implantation mortality9, litter size10)
Rat/male TDLo 24 mg/kg Intratesticular Paternal effects (testes, epididymis, sperm duct, prostate,
30 d premating seminal vesicle, Cowper's gland, accessory glands)
Mouse/female TDLo 15 g/kg p.o. Effects on new-born( live birth index, viability index)
8-12 d after
conception
Mouse/female TDLo 40 mg/kg s.c. Maternal effects (uterus, cervix, vagina, other effects)
10 d premating
Mouse/female TDLo 168 mg/kg s.c. Maternal effects (uterus, cervix, vagina)
3 d premating
Mouse/female TDLo 10 mg/kg s.c. Maternal effects (uterus, cervix, vagina), Fertility (female
5 d premating fertility index11) males impregnating females per males exposed to fertile non-pregnant females
9 e.g. reduction in number of implants per female, total number of implants per corpora lutea
10 e.g. fetuses per litter, measured before birth
(continued)
Species/sex Parameter Dose/ Route Effects
duration
Mouse/male TDLo 4.5 mg/kg Parenteral Paternal effects (testes, epididymis, sperm duct)
19 d premating
Mouse/female TDLo 9.6 Parenteral Maternal effects (uterus, cervix, vagina)
1 d premating
Monkey/male TDLo 1 .4 mg/kg Implant Paternal effects (spermatogenesis, incl. genetic material,
70 d premating sperm morphology, motility and count)
Rabbit/female TDLo 30 mg/kg s.c. Fertility (preimplantation mortality77)
1 -3 d after
conception
Rabbit/female TDLo 6 mg/kg s.c. Fertility (preimplantation mortality77)
1 -3 d after
conception
Guinea pig/female TDLo 86 mg/kg s.c. Specific developmental abnormalities (endocrine system,
18-60 d after urogenital system)
conception
Hamster/fe- male TDLo 180 mg/kg s.c. Fertility (female fertility index79)
3-8 d after
conception
Mammal domestic/ TDLo 13.3 mg/kg s.c. Effects on newborn (behavioral)
female 50 d after conception
Mammal domestic/ TDLo 6.4 mg/kg Implant Specific developmental abnormalities (urogenital system) female 30-80 d after
conception
Mammal domestic/ TDLo 6.5 mg/kg Implant Effects on embryo/fetus (fetal death)
female 13-20 wk after
11 females pregnant per sperm positive females or females pregnant per females m!ita
conception
Mammal domestic/ TDLo 18 l-ig/kg Implant Fertility (mating performance12)
female 7-14 wk after
conception
Rats/female13 n.i. 1 -55 mg s.c. Resorptions, necrosis, lethality, post-partum mortality,
12th-19th d of masculinization of female offspring14
gestation
12 Sperm positive females per females mated or copulations per estrus cycles
13 [HSDB 2002]
14 Effect directly correlated with dose and period of administration
(continued)
Species/sex Parameter Dose/ Route Effects
duration
Rat/female13 n.i. 100 mg Parenteral Small or absent mammary glands in offspring of both
14th day of gestation sexed, abbsence of nipples in female offspring (100%) Arcatia tonsa15 EC10 EC50 water Inhibition of naupliar development (EC10 0.74 mg/l, EC50
1 .5 mg/l; LC50/EC50 3.6)
15 [Andersen, H. R. 2001 ]
4.5.6 Local tolerance
Experimental studies in an ex vivo model and animal experiments in rats and rabbits were performed to determine the local tolerance of a high dose of testosterone intranasal gel (Confarma Study Report 208040401 , 208040402, 208040403). All studies were performed according to European Union medicinal product testing guidelines. The high dose testosterone intranasal 3.5% gel, TBS-1 , was used in the local tolerance studies. The only difference between TBS-1 and TBS-2 is a greater concentration of testosterone. Therefore, the results of local tolerance studies using the high dose testosterone intranasal gel also apply for the low dose testosterone intranasal gel.
HET-CAM (#208040403)
This ex vivo test system16 is widely used for assessing the dermal and mucosal tolerance. The test was carried out according to GLP both for the testosterone intranasal gel test product and for the neutral oil (Mygliol DAC) used as a negative control. The overall result showed that TBS-1 is not an irritant.
Local tolerance after single-dose administration (#208040401)
Single dose local tolerance studies have been conducted in rats and rabbits, in which 0.1 ml of 3.5% testosterone intranasal gel was introduced in one nostril and the same amount of control (neutral oil Myogliol DAC) was applied to the other nostril. Local reactions were observed for a period of 4 hours whereupon the animals were sacrificed and the nasal mucosa was subjected to histopathology examination. No reactions were observed during the 4 hours observation period and no abnormal histopathological findings were observed following administration of testosterone intranasal gel or the control. Hence, the testosterone intranasal gel is well tolerated intranasally following single-dose administration.
Local tolerance after repeat-dose administration (#208040402)
16 The chorion allantois membrane of chicken eggs incubated for 10 days is exposed. Although this already contains blood vessels, it does not yet contain any neuronal pathways. Solutions or suspensions applied on this thin membrane induce hyperalgia, thrombosis or bleeding in the blood vessels within seconds to minutes.
Repeat dose local tolerance studies have been conducted in rats and rabbits, in which 0.1 ml of 3.5% testosterone intranasal gel was introduced in one nostril and the same amount of control (neutral oil Myogliol DAC) was applied to the other nostril for a duration of 14 days. Local reactions were observed throughout the 2-week study following which the animals were sacrificed and the nasal mucosa was subjected to histopathology examination. No reactions were observed during the 2 week study period and no abnormal histopathological findings were observed following administration of the testosterone intranasal gel (3.5%) or the control. Hence, testosterone intranasal gel (3.5%) is well tolerated intranasally following repeat-dose administration.
4.6 References to the Nonclinical Chapter
Andersen, H. R., Wollenberger, L, Halling-Sorensen, B. et al. Development of copepod nauplii to copepodites--a parameter for chronic toxicity including endocrine disruption. Environ. Toxicol Chem 2001 : 20, 2821 -2829.
Bosland, MC, Ford H, Horton L. Induction at high incidence of ductal prostate adenocarcinomas in NBL/Cr and Sprague-Dawley Hsd:SD rats treated with a combination of testosterone and estradiol-17 b or diethylstilbestrol Carcinogenesis 1995:16, 131 1 -17.
Brittebo EB, Rafter JJ. Steroid metabolism by rat nasal mucosa: studies on progesterone and testosterone. J Steroid Biochem. 1984:20(5), 1 147-51 .
Chambo-Filho A, Camargos AF, Pereira FEL. Morphological changes induced by testosterone in the mammary gladns of female Wistar rats. Br J Med Biol Res. 2005: 38, 553-58.
Confarma Study Report 208040401 : Study of local tolerance single application. 2004
Confarma Study Report 208040402: Study of local tolerance repeated application during 2 weeks. 2004
Confarma Study Report 208040403: Study of HETCAM according to the official method for the assessment of the irritation potential through application of the chorio-allantois membrane of the Hen's Egg. 2004
Confarma Study Report 227120417: Three month toxicity study. 2005
Cui L, Mori T, Takahashi S, et al., Slight promotion effects of intermittent administration of testosterone propionate and/or diethylstilbestrol on 3,2'-dimethyl-4-aminobiphenyl- initiated rat prostate carcinogenesis. Cancer Letters 1998: 122, 195-199
Fahrig, R. Anti-mutagenic agents are also co-recombinogenic and can be converted into co-mutagens. Mutat.Res 1996: 350 (1 ), 59-67.
Gentile MA, Nantermet PV, Vogel RL Androgen-mediated improvement of body composition and muscle function involves a novel early transcriptional program including IGF1 , mechano growth factor and induction of D-catenin. J Mol Endo. 2010: 44, 55-73
Giuliano F, Rampin O, Allard J. Neurophysiology and pharmacology of female genital sexual response. J Sex Marital Ther. 2002;28 Suppl 1 :101 -21 .
HSDB. Testosterone. Hazardous Substances Data Bank 2002. 1 -34.
IARC Monographs on the evaluation of carcinogenic risks to humans overall evaluations of carcinogenicity: An updating of ISRC monographs Volume 1 to 42. Supplement 7. 1987.
Kim, N. N., Min, K., Pessina, M. A. et al. Effects of ovariectomy and steroid hormones on vaginal smooth muscle contractility. Int. J Impot. Res 2004: 16 (1 ), 43-50.
Kim, N.N., Min, K., Pessina, M.A. et al. Effects of ovariectomy and steroid hormones on vaginal smooth muscle contractility. Int. J Impot. Res 2004: 16(1 ), 43-50.
Noble RL. The development of prostatic adenocarcinoma in Nb rats following prolonged Sex hormone administration. Cancer Research. 1977: 37, 1929-33.
Nucro-technics Study Report 227422: A 90-day repeated intranasal dose toxicity study with TBS-2 gel in rabbits followed by a 30-day recovery period. 201 1 .
Polderman KH, Stehouwer CD, van Kamp gJ et al. Influence of sex hormones on plasma endothelin levels. Ann Intern Med. 1993: 1 18(6), 429-32.
Pollard M, Luckert PH. Production of autochthonous prostate cancer in Lobund-Wistar rats by treatments with N-Nitroso-N-methylurea and testosterone. JNCI 1986:77,583- 587
Pour PM, Stepan K. Induction of prostatic carcinomas and lower urinary tract neoplasms by combined treatment of intact and castrated rats with testosterone propionate and N-Nitrosobis(2-oxopropyl)amine. Cancer Research 1987:47, 5699- 5706.
RTECS. Testosterone. Registry of Toxic Effects of Chemical Substances 2002. 1 -1 1 .
Seachrist, D., Dunphy, G., Daneshvar, H. et al. Testosterone increases blood pressure and cardiovascular and renal pathology in spontaneously hypertensive rats. Blood Press 2000: 9 (4), 227-238.
Sharma UR, Rissman EF. Testosterone implants in specific neural sites activate female sexual behaviour. J Neuroendocrinology 1994: 6, 423-432.
Shirai T, Sano M, Imaida K et al. Duration dependent induction of invasive prostatic carcinomas with pharmacological dose of testosterone propionate in rats pretreated with 3,2'-dimethyl-4-aminobiphenyl and development of androgen-independent carcinomas after castration. Cancer Letters 1994: 83,1 1 1 -16.
Silva EG, Tornos C, Fritsche HA et al. The induction of benign epithelial neoplasms of the ovaries of guinea pigs by testosterone stimulation: a potent animal model. Mol Pathol. 1997:10(9)879-83.
Tavares, M. C, Topic, B., Abreu, C. et al. Effects of intra-nasally administered testosterone on sexual proceptive behavior in female capuchin monkeys (Cebus apella). Behav.Brain Res 2007: 179 (1 ), 33-42.
Topic, B., Tavares, M. C, Tomaz, C. et al. Prolonged effects of intra-nasally administered testosterone on proceptive behavior in female capuchin monkeys (Cebus apella). Behav.Brain Res 2007: 179 (1 ), 60-68.
Turner RT, Wakley GK, Hannon KS. Differential effects of androgens on cortical bone histomorphometry in gonadectomized male and female rats. J Ortho Res. 1990:8(4), 612-7.
Traish AM, Kim NN, Huang YH. Sex steroids hormones differentially regulate nitric oxide synthase and arginase activity in the proximal and distal rabbit vagina. Int J Impot Res. 2003:15(6), 397-404.
Traish AM, Kim SW, Stankovic M et al. Testosterone increases blood flow and expression of androgen and estrogen receptors in the rat vagina. J Sex Med. 2007: 4, 609-619.
Tsutusi, T., Komine, A., Huff, J. et al. Effects of testosterone, testosterone propionate, 17beta-trenbolone and progesterone on cell transformation and mutagenesis in Syrian hamster embryo cells. Carcinogenesis (Oxford) 1995: 16 (6), 1329-1333.
Tyagi A, Rajalakshmi M, Bajaj JS, Mohan Kumar V. Effects of long term treatment with testosterone enanthate in rhesus monkeys: I. Pharmacokinetics of testosterone, testicular volume and liver metabolism of testosterone. Int J Andrology. 1999: 22, 139- 147.
Tyagi A, Rajalakshmi M, Jeyaraj DA, Sharma RS, Bajaj JS. Effects of long-term use of testosterone enanthate. II. Effects on lipids, high and low density lipoprotein cholesterol and liver function parameters. Int J Andrology. 1999: 22, 347-355.
Vogel, E. W. and Nivard, M. J. Performance of 181 chemicals in a Drosophila assay predominantly monitoring interchromosomal mitotic recombination. Mutagenesis 1993: 8 (1 ), 57-81 .
Waalkes MP, Rehm S, Devor DE. The effects of continuous testosterone exposure on spontaneous and cadmium induced tumors in the male fischer (F344/NCr) rat: Loss of testicular response. Tox and Appl Pharm 1997:142, 40-46.
Xie B, Tsao SW, Wong YC. Sex hormones induced mammary carcinogenesis in female Noble rats: the role of androgens. Carcinogenesis 1999: 20, 1597 - 1606.
Yarrow JD, Conover CF, Purandare AV et al. Supraphysiological testosterone enanthate administration prevents bone loss and augments bone strength in
gonadectomised male and female rats. Am J Physiol Endocrinol Metab. 2008: 295, E1213 - E1222.
EFFECTS IN HUMANS
Over the past 70 years, testosterone has been studied as a therapy to improve sexual functioning in women. A number of different testosterone formulations have been evaluated for the treatment of HSDD including a testosterone patch, testosterone cream, testosterone enanthate injections, oral methyltestosterone and subcutaneous pellets. The goal of these therapies has been to restore testosterone levels to those compatible with a woman's physiology thereby improving sexual desire, arousal and orgasm. The testosterone production in premenopausal women ranges from 100 - 400 □ g/day and normal testosterone levels range between 10 -70 ng/dL.
The role of androgens in female sexuality has been explored over the past seven decades. In the late 1930's, clinicians started treating women with testosterone for the treatment of gynecological and sexual disorders. In 1940, Loeser showed that testosterone implantations (300 mg) resulted in enlargement of the clitoris, an increase in libido and a heightened sense of well being. In the mid 1980's the role of androgens in female sexual health was further explored. In 1987, Sherwin and Gelfand investigated the role of testosterone on female sexual health. Surgically menopausal women received either an estrogen-androgen preparation intramuscularly, estrogen alone or were untreated. Women receiving the estrogen-androgen preparation reported higher rates of sexual desire, sexual arousal and number of fantasies than those receiving estrogen alone or untreated. In addition, rates of intercourse and orgasm were higher in women treated with estrogen and androgens.
Several well designed, randomized, placebo controlled clinical trials have been performed over the last decade illustrating the efficacy and safety of androgen therapy in post menopausal woman. Four clinical studies are performed for evaluating the
pharmacokinetics, pharmacodynamics and safety of TBS-2, intranasal testosterone gel, in pre- and post-menopausal women.
Testosterone Clinical Pharmacology
Testosterone, the primary circulation androgen in women, is secreted by the ovaries and the adrenal glands. In premenopausal women, the rate of production of testosterone is 100 to 400 micrograms/day, of which, half is contributed by the ovary as either testosterone or a precursor. Serum levels of androgens fall as women age.
In women, circulating testosterone is primarily bound in the serum to SHBG (65 - 80 %) and to albumin (20 - 30 %) leaving only about 0.5 - 2 % as the free fraction. The affinity of binding to serum SHBG is relatively high and the SHBG bound fraction is regarded as not contributing to biological activity. Binding to albumin is of relatively low affinity and is reversible.
Testosterone is metabolised primarily in the liver by cytochrome P450 3A4 (CYP3A4) aromatase, and 5-alpha-reductase. CYP3A4 metabolizes testosterone primarily in the liver by conversion to various hydroxyl steroids. The active metabolites of testosterone are DHT and estradiol. Testosterone is metabolized to DHT by steroid 5-alpha reductase in many tissues. Testosterone is metabolized to estradiol by aromatase.
Testosterone is mainly excreted in the urine as glucuronide and sulfate conjugates of testosterone and its metabolites. About 6% of a dose is excreted in the faeces, mostly in unconjugated form.
Androgens are not recommended to breast-feeding women. Testosterone Intranasal Gel Clinical Efficacy and Safety
Clinical experience with TBS-2 include a Phase I single dose and multiple dose steady state pharmacokinetic study in healthy pre-menopausal women, a Phase l/l I assessing
amygdala reactivity in healthy middle aged women to TBS-2 and two Phase II studies assessing the pharmacokinetics and efficacy of TBS-2 in women with FOD and HSDD.
Table 5.2-1 : Summary of Completed Clinical Studies with TBS-2

5.2.1 Phase I: An open label single and multiple application of intranasal testosterone gel (TBS-2) in healthy pre-menopausal female subjects at three dose levels (TBS-2-2011-01)
This was a Phase I, single-centre, randomized, open label, parallel group study in healthy, normal-cycling women to evaluate the pharmacokinetic profile of testosterone following a single administration of TBS-2 at doses of 0.6 mg, 1 .2 mg and 1 .8 mg and multiple administration of 1 .2 mg TBS-2 given three times a day for 3 days. Women were randomly assigned on a 1 :1 :1 basis to one of the three single dose administration treatment groups (Period 1 ). A total of 8 subjects from Period 1 continued with the multiple dose portion of the study (Period 2).
Blood samples to determine the PK profile for free and total testosterone, DHT, SHBG and estradiol were collected after the first dose in Period 1 (0-48 hours). For Period 2,
blood samples were collected twice daily on Day 1 and Day 2 and 16 times over the course of 48 hours beginning post administration on Day 3.
Twenty-four (24) women (mean age 29.8 ± 5.86 yrs) participated in the Period 1 and 8 women (mean age 30.3 ± 6.48 yrs) participated in Period 2.
.2.1.1 Efficacy results
The pharmacokinetics data showed a linear increase in free and total testosterone levels with increasing concentrations of TBS-2. The 24 hour AUC of total and free testosterone were within the normal ranges for all 3 doses. Multiple dose administration over 3 days showed minimal accumulation and TBS-2 was completely cleared within 24 hours.
Table 5.2.1 .1 -1 : Total Testosterone Pharmacokinetic Summary (Single Dose
Administration)

Table 5.2.1 .1 -2: Free Testosterone Pharmacokinetic Summary (Single Dose
Administration)

5.2.1.2 Safety results
Twenty-four (24) women received a single dose of TBS-2 and 8 women received 7 doses in 3 days of TBS-2. Safety analysis showed that TBS-2 was well tolerated by subjects. There were no serious AEs or discontinuations due to AEs in this study. In Period 1 , a total of 18 AEs were reported of which 4 were classified as possibly related to study medication and 14 were classified as not related to study. In Period 2, a total of 5 AEs were reported of which 2 were classified as possibly related to study medication and 3 were classified as not related to study treatment. Drug related adverse event
included nasal congestion, rhinorrhea and rhinalgia, which were all mild in intensity and a mild headache that increased in severity to moderate.
5.2.2 Phase II: Single centre single blind placebo run in and randomized single blind placebo controlled five arm parallel group study to assess efficacy of a single dose of TBS-2 intranasal gel at four time points post dose using vibrotactile stimulation combined with visual sexual stimulation in female subjects with primary or secondary anorgasmia (TBS-2- VTS-2010-01)
This was a randomized, single-blind, single site, placebo-controlled five arms parallel group study designed to evaluate the effect of a single dose of TBS-2, 1 .2 mg (0.6 mg per nostril), on the occurrence of orgasm at 0.5, 2.0, 4.0 and 8.0 hours post-dose in premenopausal female subjects with primary or secondary FOD using vibrotactile stimulation to the glans clitoris (VTS) and visual sexual stimulation (VSS) in a laboratory setting. The study consisted of two screening visits and one treatment visit. At Visit 1 , all screening assessments were done and subjects were familiarized with the VTS technique and VSS. At Visit 2, women underwent VTS/VSS stimulation for 20 minutes to determine if they were anorgasmic during the VTS/VSS procedure. Women who did not achieve orgasm during the VTS/VSS procedure and met all other eligibility criteria were randomized to one of the five treatment arms. In the treatment visit, subjects received either TBS-2 or placebo and underwent VTS/VSS at 0.5, 2.0, 4.0 and 8.0 hours after dosing. VTS/VSS lasted 20 minutes or until orgasm was reached, after which stimulation was stopped. Patient reports of orgasms, Vaginal Pulse Amplitude (VPA) a physiological measurement of blood flow in the vagina corresponding with engorgement of female genitalia, as well as clinically accepted patient questionnaires were used to measure the response.
Fifty six (56) women participated in this study, 45 women (mean age 27.2 yrs; range 20-46) received TBS-2 and 1 1 women (mean age 28.2 yrs; range 18-47) received placebo.
5.2.2.1 Efficacy results
Study analysis concluded that during the VTS treatment phase four women who were administered TBS-2 experienced orgasms, while an additional eight patients treated with TBS-2 were also determined to have experienced orgasms based on a post treatment assessment. Of the patients in the placebo arm, two patients reported experiencing orgasms during the VTS treatment, however one patient seemed to have experienced an orgasm during the screening portion of the study, and should have been excluded from proceeding into the treatment phase.
Patients treated with TBS-2 also showed a statistically significant improvement in VPA versus placebo, and elevation of sexual arousal, as well as positive trends in terms of elevating sensuality and pleasurable genital sensation. The greatest effects of TBS-2 were seen in the 2 and 4 hr groups, suggesting a delay-effect of testosterone on sexual arousal.
5.2.2.2 Safety results
Forty-seven (47) women received a single dose to TBS-2. Safety analysis showed that TBS-2 was well tolerated by subjects. There were no serious AEs or discontinuations due to AEs in this study. A total of 18 AEs were reported, 15 of which were classified as not related to study medication and 3 were of unknown etiology. In the TBS-2 treated group, 9/47 (19.1 %) of the subjects experienced at least one adverse event, whereas in the placebo group 6/12 subjects (50.0%) reported at least one adverse event.
5.2.3 Phase II: Two-center, randomized, placebo and active comparator evaluation of the pharmacokinetics and safety along with initial pharmacodynamic efficacy of three dose levels of TBS-2 applied twice daily for up to three days in female subjects with HSDD or FOP (TBS-2-PK-2010-01)
This was a two-center, in-patient, parallel groups, randomized, placebo and active comparator controlled evaluation of the pharmacokinetics (PK) exploratory pharmacodynamics (PD) and safety of three dose levels of TBS-2. Women in the FOD cohort were randomized to receive either TBS-2 (3 dose levels; 0.3 mg, 0.9 mg or 1 .2 mg) or placebo TBS-2 administered as half the dose to each nostril. Women in the HSDD cohort were randomized to receive either TBS-2 (same 3 dose levels) or Intrinsa. Subjects randomized to the nasal gel received a total of five doses of study medication. Subjects randomized to the Intrinsa patch received one patch for the duration of the study.
The pharmacokinetic profile of testosterone and DHT was determined following TBS-2 administration. The primary objective for the PK analysis was to determine the AUC, CaVg, Cmin, Cmax, Tmax, PTF and PTS of testosterone and DHT following single dose and multiple doses. Blood samples were collected after the first dose (0-12 hours) and after the fifth dose (48-60 hours after first dose). PK draw times were at 0, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360, and 480 minutes post dose. The analysis of testosterone and DHT was performed using a highly sensitive and validated LC-MS/MS method for the determination of T and DHT in human plasma.
Thirty two (32) women participated in the two study cohorts: 15 post menopausal women with HSDD (mean age 56.1 yr; range 46 - 63) and 17 pre- and post menopausal women with ANOR (mean age 77.8 yr; range 21 -64).
5.2.3.1 Efficacy results
5.2.3.1.1 Pharmacokinetics
The baseline levels of testosterone in both the HSDD and FOD women were in the lower third of the normal reference range of testosterone (normal testosterone range 10 - 70 ng/dL). The pharmacokinetic data showed a linear increase in plasma testosterone levels with increasing concentrations of TBS-2. During the first PK sampling series the mean concentration of the TBS-2 high dose group reached
approximately the same level as the overall mean concentration of the testosterone patch (Cmean 53.38 and 52.1 ng/dL respectively). During the second PK series, the mean plasma testosterone concentration and AUC of TBS-2 high dose reached levels slightly higher than the mean concentration of the testosterone patch during the same time period (Cmean 65.52 and 57.88 ng/dL respectively). These values are within the upper limit of the normal physiological testosterone range thus limiting safety concerns. The mean concentration of testosterone in the women treated with placebo remained in the lower third of the normal reference range. The mean concentration of plasma testosterone is consistently higher after five administrations of the high and medium dose of TBS-2 (48-60 hours post first dose): 65.52 ng/dL after TBS-2 high dose and 44.49 ng/dL after TBS-2 medium dose. The plasma concentrations for DHT were often too low to detect; pharmacokinetic parameters that were calculated for testosterone were not calculated for DHT.
Table 5.2.3.1 -1 : Mean Pharmacokinetic Parameters Following the First Dose (0 - 1 2 hours)

Table 5.2.3.1 -2: Mean Pharmacokinetic Parameters Following the Fifth Dose (48 - 60 hours)

5.2.3.1.2 Pharmacodynamics
A statistically significant contrast in VPA after 30 minutes is observed in the TBS-2 high dose group compared to placebo in the FOD cohort (61 % difference, p = 0.04). After 4.5 hours a statistically significant contrast in VPA is observed in the TBS-2 low dose compared to placebo in the FOD cohort (82% difference, p = 0.039).
A statistically significant increase in sexual arousal was observed in women who received TBS-2 high dose compared to women who received Intrinsa (p=0.039). Women receiving high dose TBS-2 showed a significant increase in sensuality
compared to women receiving the Intrinsa patch (P=0.032). Similarly, a statistically significant increase in positive affect was observed between HSDD women receiving TBS-2 high dose and women receiving Intrinsa after 30 minutes and 4.5 hours post dose (p=0.034 and p=0.031 respectively).
5.2.3.2 Safety results
24 women were exposed to TBS-2; 8 patients received the high dose TBS-2 (1 .2 mg/dose), 8 patients received the medium dose TBS-2 (0.9 mg/dose) and 8 patients received the low dose TBS-2 (0.3 mg/dose). All patients received the full course of study drug with the exception of one patient in the low dose group who discontinued the study after 4 doses due to difficulties in drawing blood for PK analysis.
The safety analysis showed that all treatments were well tolerated by subjects. There were no serious AEs or discontinuations due to AEs in this study. A total of 42 AEs were reported, of which 2 was classified as probably related to study drug treatment, 23 were classified as possibly related to study treatment, 8 were classified as unlikely to be related, 8 were classified as unrelated (6 before treatment administration, 2 following treatment administration) and 1 event (mild Headache) was of unknown etiology. Two (2) AEs were moderate in intensity (migraine and headache in ANOR cohort, patients on Low and Medium TBS-2 dose respectively), the remaining 40 were considered mild.
The most common study drug related adverse event was headache; 9 subjects (excluding migraine headache) reported having headaches, either single episode or intermittent. Headaches occurred at variable intervals after dosing. No local nasal reactions were reported for subjects receiving TBS-2. A summary of TBS-2 related events is presented in Table 5.2.3.2-1 .
Table 5.2.3.2-1 : Summary of Adverse Events TBS-2 Related Events (Probable or Possible) by Patient
HSDD ANOR
LD MD HD Intrinsa LD MD HD Placebo

Dry skin 1 (3.2%)
5.2.4 Phase l/ll Acute effects of TBS-2 on amygdala reactivity and memory in healthy middle-aged women- a placebo controlled cross over study using fMRI. (CMO-nr: 2004/144)
This Phase l/l I study was designed to investigate whether nasally applied TBS-2 could rapidly increase amygdala reactivity in healthy, naturally cycling middle aged women, using a double blinded, placebo controlled, cross-over design. To investigate whether the diminished endogenous levels of androgens during middle adulthood influenced amygdala activity, the placebo arm was compared to a placebo arm in young healthy, naturally cycling women who were expected to have higher androgen levels (van Wingen, 2008).
The women participated during the early follicular phase of the menstrual cycle, and amygdala responses to biologically salient stimuli were measured with functional magnetic resonance imaging. Testosterone levels were measured at baseline, 30 minutes after drug intake (prior to scanning) and 150 minutes after drug intake (at the time of the final completion of the sexual arousal questionnaire).
5.2.4.1 Efficacy results
Androgen levels were lower in middle-aged compared to young women, which was associated with decreased amygdala reactivity. Endogenous testosterone levels correlated positively with amygdala reactivity across the young and middle aged women. The middle aged women received a single dose of TBS-2 in a double blind, placebo controlled, cross-over manner, which rapidly increased amygdala reactivity to a level comparable to the young women. The enhanced testosterone levels correlated positively with superior frontal cortex responses and negatively with orbitofrontal cortex responses across individuals, which may reflect testosterone-induced changes in amygdala regulation.
5.2.4.2 Safety results
Twenty five women were exposed to a single dose of 0.9 mg TBS-2 gel. There were no deaths in the study and none of the subjects experienced any SAEs. Four subjects receiving TBS-2 and one subject receiving placebo gel experienced one or more AEs.
None of the AEs were considered study drug-related and none of the subjects were discontinued from treatment because of an AE.
Information on Published Testosterone Studies
INTRINSA®
Intrinsa, a testosterone transdermal patch, has been approved in the European Union for the treatment of hypoactive sexual desire disorder (HSDD) in bilaterally oophorectomised and hysterectomised (surgically induced menopausal) women receiving concomitant estrogen therapy.
Table 5.3-1 lists the published studies demonstrating efficacy of the Intrinsa patch in postmenopausal women. A 24-week placebo controlled, parallel group clinical trial was performed to investigate the effect of 3 dosage levels of testosterone administered via patch to women with hypoactive sexual desire disorder after surgically induced menopause (Braunstein et al. 2005). Women receiving the 300 Dg/day testosterone patch had significantly greater increases from baseline in sexual desire and in the frequency of satisfying sexual activity, total number of sexual events and number of orgasms. Simon et al. (2005) conducted a large placebo controlled clinical trial comparing the 300 D g/day testosterone patch to placebo and found that surgically post-menopausal women with hypoactive sexual desire disorder receiving a testosterone patch treatment 300 D g/day with concomitant estrogen therapy for 24 weeks experienced greater total satisfying sexual activity, sexual desire and less personal distress. These women also reported greater arousal, orgasm, pleasure and responsiveness in comparison to women in the placebo group.
The efficacy and safety of the 300 Dg/day patch was further evaluated in a 52 week placebo controlled study for low sexual desire in naturally menopausal women not
taking estrogen supplements (Davis et al. 2008). Women treated with testosterone had significant improvement in the mean frequency of satisfying sexual episodes and were associated with increases in sexual desire, arousal, orgasm and pleasure and a reduction in personal distress. Panay et al. (2010) also reported similar findings in a study using the testosterone patch for low sexual desire in naturally menopausal women with and without estrogen hormone therapy.
Table 5.3-1 : Published Studies Demonstrating Efficacy of 300 Dg/day Testosterone atch in Postmeno ausal Women on or off estro en

In the Intrinsa pivotal studies, 882 women were exposed to a 300 μg dose for more than 20 weeks and 348 women for more than 48 weeks. The 300 μg/day testosterone patch demonstrated a favourable safety profile in surgically menopausal women with HDSS with up to 78 weeks of exposure. The adverse reaction most often reported (30.4%) was application site reaction. The majority of these adverse reactions consisted of mild erythema and itching and did not result in patient withdrawal. The overall incidence of adverse events, serious adverse events and withdrawals was similar in the Intrinsa and placebo groups (Intrinsa SPA).
Hirsutism was also very commonly reported to the chin and upper lip (> 90% were mild) and was reversible in the majority of patients. Other androgenic effects commonly reported were acne, voice deepening and alopecia. The overall incident of androgenic adverse events (acne, alopecia, hirsutism and voice deepening) was higher in the Intrinsa group than in the placebo group in the combined studies (Intrinsa 123 patients, 17.7%; placebo 101 patients, 14.4%). The specific events that accounted for most of this difference were acne and hirsutism. There was a statistically significant association between the probability of having hirsutism and maximal free testosterone levels (p=0.014). More than 90% of these reported were considered mild and were reversible in the majority of patients. Other adverse events occurring in less than 10% of the population treated with Intrinsa included insomnia, migraines, abdominal pain, breast pain and increased weight.
Intrinsa was granted marketing authorization by the CHMP on condition that the potential long term risks be carefully monitored according to a Risk Management Plan. The Intrinsa safety data base included only a very small number of patients treated beyond one year and did not adequately address the potential long term risks associated with estrogen and testosterone on cardiovascular disease or breast cancer. A long-term safety study evaluating the long term effects of the testosterone patch (2 yr exposure) on cardiovascular, breast cancer and metabolic syndrome has been performed. One (1 ) woman in the 150 μg/day and three women in the 300 μg/day cohort developed clitoral enlargement. All cases were considered mild by the site investigator and none of these women withdrew from the study. No cases of endometrial hyperplasia or carcinoma were diagnosed.
Breast cancer was diagnosed in four women who received testostereone as compared with none who received placebo. One of the four women received the diagnosis in the first 4 months of the study period and one, in retrospect had symptoms before undergoing randomization. Based on the 1 -year rate of breast cancer in post
menopausal women, the number of events in the Phase I II trials was not higher than expected (Davis 2008).
LIBIGEL®
Biosante is developing a testosterone gel designed to be quickly absorbed through the skin after a once-daily application on the upper arm, delivering 300 Dg/day testosterone to the bloodstream evenly over time for the treatment of HSDD.
The Phase II trial results showed LibiGel significantly increased the number of satisfying sexual events by 238% versus baseline (p<0.0001 ); this increase also was significant versus placebo (p<0.05). In this study, the effective dose of LibiGel produced testosterone blood levels within the normal range for pre-menopausal women and had a safety profile similar to that observed in the placebo group. In addition, no serious adverse events and no discontinuations due to adverse events occurred in any subject receiving LibiGel.
The Phase II trial results showed that LibiGel showed a difference in sexual satisfying events compared to baseline but did not differentiate from a robust placebo effect in postmenopausal women with HSDD. LibiGel was well tolerated with a safety profile comparable to placebo. In the safety extension, to date, there have been 17 adjudicated cardiovascular (CV) events, which is lower than anticipated rate of approximately 0.57 percent. There have been eight breast cancers reported, a rate of approximately 0.27 percent.
Marketing Experience
Testosterone intranasal gel, TBS-2, is not approved for marketing authorisation and currently not marketed.
References: Effects in Humans
Braunstein GD, Sundwall DA, Katz M, et al. Safety and efficacy of a testosterone patch for treatment of hypoactive sexual desire disorder in surgically menopausal women: a randomized placebo controlled trial. Arch Intern Med 2005;165:1582-9
Buster J, Kingsberg SA, Aguirre O, et al. Testosterone patch for low sexual desire in surgically menopausal women: a randomized trial. Obstet Gynecol 2005;105:944-52 CMO-nr: 2004/144, Phase l/ll, Acute effects of TBS-2 on amygdala reactivity and memory in healthy middle-aged women (2009)
Davis SR, Moreau M, Kroll R, et al. Testosterone for low libido in postmenopausal women not taking estrogen. N Engl J Med 2008;359: 2005-17
Davis SR, van der Mooren MJ, van Lunsen RH, et al. Efficacy and safety of a testosterone patch for the treatment of hypoactive sexual desire disorder in surgically menopausal women: a randomized placebo controlled trial. Menopause 2006;13:387- 96
Liu JH, Kingsberg K, Aguirre O, et al. Treatment of naturally menopausal women with hypoactive sexual desire disorder: effect of transdermal testosterone patch in the NM2 trial. Abstract presented at the ISSWSH Meeting, 2008
Loeser AA. Subcutaneous implantation of female and male hormones in tablet form in women. Br med J. 1940;1 :479-82
Panay N, Al-Azzawi F, Bouchard C, et al. Testosterone treatment of HSDD in naturally menopausal women: the ADORE study. Climacteric. 2010;1 3(2):121 -31
Salmon UJ, Geist SH. Effect of androgens upon libido in women. J Clinical Endocrinol 1943;3:235-8
Scientific Discussion, Intrinsa
http://www.ema.europa.eu/humandocs/PDFs/EPAR/intrinsa/063406en6.pdf
Sherwin BB, and Gelfand MM. The role of androgen in the maintenance of sexual functioning in oophorectomized women. Psychosomatic Medicine. 1987;49:397-409
Shifren JL, Monz BU, Russo PA, et al. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol. 2008;1 12:970-978
Shifren JL, Davis SR, Moreau M, et al. Testosterone patch for the treatment of hypoactive sexual desire disorder in naturally menopausal women: results from the INTIMATE NMI study. Menopause 2006;13:1 -10
Shifren JL, Braunstein GD, Simon JA, et al. Transdermal testosterone treatment in women with impaired sexual function after oophorectomy. N Engl J Med 2000;343:682- 8
Simon J, Braunstein G, Nachtigall L, et al. Testosterone patch increases sexual activity and desire in surgically menopausal women with hypoactive sexual desire disorder. J Clin Endocrinol Metab 2005;90:5226-33
TBS-2-201 1 -01 : An open-label, single and multiple application of intranasal testosterone gel (TBS-2) in healthy pre-menopausal female subjects at three dose levels, 2012
TBS-2-PK-2010-01 : Two-center, in-patient, randomized, placebo and active comparator evaluation of the pharmacokinetics and safety along with initial pharmacodynamics
efficacy of three dose levels of TBS-2 intranasal gel applied twice daily for up to three days in female patients with hypoactive sexual desire disorder or anorgasmia, 201 1
TBS-2- VTS-2010-01 : Single center, randomized, single-blind, placebo controlled, five arm parallel group study to assess efficacy of a single dose of TBS-2 intranasal gel at four time points post dose using vibrotactile stimulation combined with visual sexual stimulation in female subjects with primary or secondary anorgasmia, 2012 van Wingen GA, Zylicz SA, Pieters S, Mattern C, Verkes RJ, Buitelaar JK, Fernandez G; Testosterone increases amygdala reactivity in middle-aged women to a young adulthood level; Neuropsychopharmacology. 2009;34(3):539-47
USP. Androgens (Systemic). In: USP Dl - Drug Information for the Health Care
Professional; Edition: 23, Publisher: Micromedex. USP Dl Editorial Group, Englewood, 1 -35, 2003 www.biosantepharma.com
SUMMARY OF DATA AND GUIDANCE FOR THE INVESTIGATOR
6.1 Discussion and Conclusion on Non-clinical and Clinical Data
The animal and human data for TBS-2 have not demonstrated toxicity related concerns which would preclude the use of TBS-2 in the clinical trial program. A large number of non-clinical pharmacology and toxicology studies have been conducted to support the efficacy and safety of testosterone cited in the literature and in previously submitted applications.
No evidence of systemic drug related toxicity was observed in rabbits after repeated daily administration of TBS-2 over a 90 day period up to a dose level of 100-fold the maximum clinical daily dose. Local tolerance studies in rat and rabbit following single dose and repeated dose administrations showed that testosterone intranasal gel was
well tolerated. Testosterone intranasal gel was classified as a non-irritant in the HET- CAM test.
Four clinical studies have been completed in healthy pre-menopausal women, pre and pos-menopausalwomen with FOD and post-menopausal women with HSDD with TBS- 2. In each of these studies, TBS-2 was rapidly absorbed (maximum testosterone serum concentration is reached between one and two hours after administration) and eliminated from the body within 24 hours. Total and free testosterone did not exceed the normal physiological range of testosterone in women. Initial pharmacodynamic data indicate that TBS-2 increases sexual arousal, sensuality and genital sensation in addition to increasing the incidence of orgasm in women with FOD in a controlled environment. In the vibrotactile stimulation (VTS) study, the greatest effects of TBS-2 were seen in the 2 and 4 hr groups, suggesting a delay-effect of testosterone on sexual arousal.
6.2 Guidance for the Investigator
TBS-2 is an innovative pharmaceutical formulation of testosterone for nasal administration which is being developed for the treatment of FOD. The formulation has many advantageous features including rapid absorption into systemic circulation and rapid clearance, the lack of first pass metabolism, the avoidance of transference from one person to another, and the ease of use.
6.2.1 Potential Side Effects
Based on prior clinical experiences and the human studies conducted to date, the risks associated with the use of TBS-2 are minimal. The most common side effects reported in clinical studies using the testosterone patch were application site reactions and hirsutism. Other common side effects include hair loss, acne, migraine, insomnia, voice deepening, breast pain, weight gain and abdominal pain. These events were generally mild and did not result in discontinuation of treatment. No clinically important changes in laboratory parameters were observed with testosterone treatment.
TBS-2 was well tolerated in healthy women and women with HSDD and female orgasmic disorder. The most common adverse event reported with TBS-2 was headache with the majority of adverse events being transient and mild in intensity.
6.2.2 Precautions
No specific drug-drug interaction studies have been conducted for testosterone in women. Drug-drug interaction studies with testosterone replacement therapies have been performed in men. The following information on drug-drug interactions is included in the approved labeling of testosterone hormone replacement therapies for men marketed in the US (for example, see Testim).
Oxyphenbutazone: Concurrent administration of oxyphenbutazone and androgens may result in elevated serum levels of oxyphenbutazone.
Insulin: In diabetic patients, the metabolic effects of androgens may decrease blood glucose and, therefore, insulin requirements.
Propranolol: In a published pharmacokinetic study of an injectable testosterone product, administration of testosterone cypionate led to an increased clearance of propranolol in the majority of men tested.
Corticosteroids: The concurrent administration of testosterone with ACTH or corticosteroids may enhance edema formation; thus these drugs should be administered cautiously, particularly in patients with cardiac or hepatic disease.
6.2.3 Medical Considerations/Contraindications
Testosterone should not be used in women who:
1 . Are pregnant or may become pregnant. Testosterone may cause fetal harm when administered to a pregnant woman. If this drug is used during
pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
2. Are breast feeding.
3. Have known or suspected cancer of the breast or estrogen-dependent neoplasia.
4. Have known history of hypersensitivity to testosterone and/or related drugs.
5. Have known history of polycystic ovarian syndrome, hirsutism, frequent clinically significant acne.
The disclosures of the patents, patent documents, articles, abstracts and other publications cited herein are incorporated herein by reference in their entireties as if each were individually incorporated. In case of conflict, the present specification, including definitions, shall control. Various modifications and alterations to this invention will become apparent to those skilled in the art without departing from the scope and spirit of this invention. Illustrative embodiments and examples are provided as examples only and are not intended to limit the scope of the present invention. The scope of the invention is limited only by the claims set forth as follows.
Claims
1 . A lower dosage strength testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. between about 0.1 % to about 1 .5% testosterone by weight of said gel formulation; and
b. a pharmaceutically acceptable vehicle.
2. The testosterone gel formulation of any one of claim 2, wherein said gel formula further comprises a solvent, a wetting agent, and a viscosity increasing agent.
3. The testosterone gel formulation of claim 2, wherein said solvent is castor oil.
4. The testosterone gel formulation of claim 2, wherein said wetting agent is an oleoyi polyoxylglyceride.
5. The testosterone gel formulation of claim 2, wherein said viscosity increasing agent is colloidal silicon dioxide.
6. The testosterone gel formulation of any one of claims 1 or 2, wherein said gel formulation further comprises castor oil, oleoyi polyoxylglycerides and colloidal silicon dioxide.
7. The testosterone gel formulation of any one of claims 1 -6, wherein said gel formulation is a bioequivalent formulation.
8. The testosterone gel formulation of any one of claims 1 -6, wherein said gel formulation is a pharmaceutically equivalent formulation.
9. The testosterone gel formulation of any one of claims 1 -6, wherein said gel formulation is a therapeutically equivalent formulation.
10. A packaged pharmaceutical comprising:
(a) a lower dosage strength testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises between about 0.1 % and 1 .5% testosterone by weight; and
(b) associated instructions for using said testosterone gel formulation to treat anorgasmia.
1 1 . A packaged pharmaceutical comprising:
(a) a lower dosage strength testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises between about 0.1 % and 1 .5% testosterone by weight; and
(b) associated instructions for using said testosterone gel formulation to treat hypoactive sexual desire disorder (HSDD).
12. The packaged pharmaceutical of claim 10 or 1 1 , wherein said testosterone is present as a pharmaceutical composition comprising a therapeutically effective amount of testosterone or a pharmaceutically acceptable salt or prodrug thereof and a pharmaceutically acceptable carrier.
13. The packaged pharmaceutical of claim 10 or 1 1 further comprising a step of identifying a subject in need of said pharmaceutical.
14. A method of treating anorgasmia comprising administering intranasally to a subject said gel formulation of any one of claims 1 -9 to deliver a therapeutically effective amount of testosterone to effectively treat HSDD.
15. A method of treating hypoactive sexual desire disorder (HSDD), comprising administering intranasally to each nostril of a subject said gel formulation of any one of claims 1 -9 to deliver a therapeutically effective amount of testosterone to effectively treat HSDD.
16. An intranasal method of treating a subject diagnosed with anorgasmia with a lower dosage strength testosterone gel formulation for nasal administration comprising between about 0.1 % and 1 .5% testosterone by weight of said gel formulation, comprising: applying the testosterone gel formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the anorgasmia.
17. The method of claim 16, wherein said subject receives said testosterone gel intranasally twice daily.
18. An intranasal method of treating a subject diagnosed with HSDD with a lower dosage strength testosterone gel formulation for nasal administration comprising between about 0.1 % and 1 .5% testosterone by weight of said gel formulation, comprising: applying the testosterone gel formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the HSDD.
19. The method of claim 18, wherein said subject receives said testosterone gel intranasally twice daily.
20. The method of claim 18, wherein the lower dosage strength testosterone gel formulation comprises between about 0.24% and 0.72% testosterone by weight of said gel formulation.
21 . The method of claim 18, wherein the lower dosage strength testosterone gel formulation comprises about 0.24% testosterone by weight of said gel formulation.
22. The method of claim 18, wherein the lower dosage strength testosterone gel formulation comprises about 0.48% testosterone by weight of said gel formulation.
23. The method of claim 18, wherein the lower dosage strength testosterone gel formulation comprises about 0.72% testosterone by weight of said gel formulation.
24. A lower dosage strength testosterone gel formulation of claim 1 , wherein the lower dosage strength testosterone gel formulation comprises between about 0.24% and 0.72% testosterone by weight of said gel formulation.
25. The method of claim 1 , wherein the lower dosage strength testosterone gel formulation comprises about 0.24% testosterone by weight of said gel formulation.
26. The method of claim 1 , wherein the lower dosage strength testosterone gel formulation comprises about 0.48% testosterone by weight of said gel formulation.
27. The method of claim 1 , wherein the lower dosage strength testosterone gel formulation comprises about 0.72% testosterone by weight of said gel formulation.
28. A packaged pharmaceutical of claim 10, wherein the lower dosage strength testosterone gel formulation comprises between about 0.24% and 0.72% testosterone by weight of said gel formulation.
29. The method of claim 10, wherein the lower dosage strength testosterone gel formulation comprises about 0.24% testosterone by weight of said gel formulation.
30. The method of claim 10, wherein the lower dosage strength testosterone gel formulation comprises about 0.48% testosterone by weight of said gel formulation.
31 . The method of claim 10, wherein the lower dosage strength testosterone gel formulation comprises about 0.72% testosterone by weight of said gel formulation.
32. A packaged pharmaceutical of claim 1 1 , wherein the lower dosage strength testosterone gel formulation comprises between about 0.24% and 0.72% testosterone by weight of said gel formulation.
33. The method of claim 1 1 , wherein the lower dosage strength testosterone gel formulation comprises about 0.24% testosterone by weight of said gel formulation.
34. The method of claim 1 1 , wherein the lower dosage strength testosterone gel formulation comprises about 0.48% testosterone by weight of said gel formulation.
35. The method of claim 1 1 , wherein the lower dosage strength testosterone gel formulation comprises about 0.72% testosterone by weight of said gel formulation.
36. A testosterone gel formulation for nasal administration, wherein said gel formulation increases the plasma testosterone level to at least about 0.4 ng/ml within about 10 minutes following the immediate nasal administration of said testosterone gel formulation to a subject.
37. A testosterone gel formulation for nasal administration, wherein said gel formulation increases the plasma testosterone level to at least about 0.7 ng/ml within about 100 minutes following the immediate nasal administration of said testosterone gel formulation to a subject.
38 A testosterone gel formulation for nasal administration, wherein administration of said gel formulation increases the plasma testosterone level within at least about 10 minutes following the immediate nasal administration of said testosterone gel formulation to a subject.
39 A testosterone gel formulation for nasal administration, wherein nasal administration of said formulation increases the plasma testosterone level to a level of at least about 0.3 ng/ml, and wherein said increase is maintained in a subject for about 6 hours immediately following the nasal administration of said testosterone gel formulation to said subject.
40. A testosterone gel formulation for nasal administration, wherein nasal administration of said formulation to a subject increases the plasma testosterone level to at least about 0.4 ng/ml, and wherein said increase is maintained for at least 6 hours immediately following nasal administration to a subject.
41 . A testosterone gel formulation for nasal administration wherein nasal
administration of said formulation to a subject increases the plasma testosterone level to at least 0.7 ng/ml about 60 minutes immediately following the nasal administration of said testosterone gel formulation to a subject.
42. A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. about 0.6% testosterone by weight of said gel formulation; and
b. a pharmaceutically acceptable vehicle.
43. The testosterone gel formulation of any one of claims 36-42 wherein said gel formulation further comprises a pharmaceutically acceptable vehicle.
44. A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. testosterone; and
b. a pharmaceutically acceptable vehicle
wherein a plasma testosterone level of at least about 0.4 ng/ml is maintained in a subject for about 6 hours immediately following the nasal administration of said gel formulation to a subject.
45. A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. testosterone; and
b. a pharmaceutically acceptable vehicle;
wherein, a plasma testosterone level of at least about 0.7 ng/ml is achieved in a subject within about 60 minutes immediately following the nasal administration of said gel formulation to a subject.
46. The testosterone gel formulation of any one of claims 36-45, wherein said gel formula further comprises a solvent, a wetting agent, and a viscosity increasing agent.
47. The testosterone gel formulation of claim 46, wherein said solvent is castor oil.
48. The testosterone gel formulation of claim 46, wherein said wetting agent is an oleoyi polyoxylglyceride.
49. The testosterone gel formulation of claim 46, wherein said viscosity increasing agent is colloidal silicon dioxide.
50. The testosterone gel formulation of any one of claims 36-45, wherein said gel formulation further comprises castor oil, oleoyi polyoxylglycerides and colloidal silicon dioxide.
51 . The testosterone gel formulation of any one of claims 36-50, wherein said gel formulation is a bioequivalent formulation.
52. The testosterone gel formulation of any one of claims 36-50, wherein said gel formulation is a pharmaceutically equivalent formulation.
53. The testosterone gel formulation of any one of claims 36-50, wherein said gel formulation is a therapeutically equivalent formulation.
54. A packaged pharmaceutical comprising:
(a) a testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises about 0.6% by weight; and
(b) associated instructions for using said testosterone gel formulation to treat anorgasmia.
55. A packaged pharmaceutical comprising:
(a) a testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises about 0.6% by weight; and
(b) associated instructions for using said testosterone gel formulation to treat hypoactive sexual desire disorder (HSDD).
56. The packaged pharmaceutical of claim 54 or 55, wherein said testosterone is present as a pharmaceutical composition comprising a therapeutically effective amount of testosterone or a pharmaceutically acceptable salt or prodrug thereof and a pharmaceutically acceptable carrier.
57. The packaged pharmaceutical of claim 54 or 55 further comprising a step of identifying a subject in need of said pharmaceutical.
58. A method of treating anorgasmia comprising administering intranasally to each nostril of a subject said gel formulation of any one of claims 36-57 to deliver a therapeutically effective amount of testosterone to effectively treat anorgasmia.
59. A method of treating hypoactive sexual desire disorder (HSDD), comprising administering intranasally to each nostril of a subject said gel formulation of any one of claims 36-57 to deliver a therapeutically effective amount of testosterone to effectively treat HSDD.
60. An intranasal method of treating a subject suffering from or diagnosed with anorgasmia with a testosterone gel formulation for nasal administration comprising about 0.6% testosterone by weight of said gel formulation, comprising: applying the testosterone gel formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the anorgasmia.
61 . The method of claim 60, wherein said subject receives said testosterone gel intranasally twice daily.
62. An intranasal method of treating a subject suffering from or diagnosed with HSDD with a testosterone gel formulation for nasal administration comprising about 0.6% testosterone by weight of said gel formulation, comprising: applying the testosterone gel formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the HSDD.
63. The method of claim 62, wherein said subject receives said testosterone gel intranasally twice daily.
64. A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. about 0.72% testosterone by weight of said gel formulation; and
b. a pharmaceutically acceptable vehicle,
wherein said gel formulation increases the plasma testosterone level to at least about 0.4 ng/ml within about 10 minutes following the immediate nasal administration of said testosterone gel formulation to a subject.
65. A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. about 0.72% testosterone by weight of said gel formulation; and
b. a pharmaceutically acceptable vehicle,
wherein said gel formulation increases the plasma testosterone level to at least about 0.7 ng/ml within about 100 minutes following the immediate nasal administration of said testosterone gel formulation to a subject.
66. A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. about 0.72% testosterone by weight of said gel formulation; and
b. a pharmaceutically acceptable vehicle,
wherein administration of said gel formulation increases the plasma testosterone level within at least about 10 minutes following the immediate nasal administration of said testosterone gel formulation to a subject.
67. A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. about 0.72% testosterone by weight of said gel formulation; and
b. a pharmaceutically acceptable vehicle,
wherein nasal administration of said formulation increases the plasma testosterone level to a level of at least about 0.3 ng/ml, and wherein said increase is maintained in a subject for about 6 hours immediately following the nasal administration of said testosterone gel formulation to said subject.
68. A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. about 0.72% testosterone by weight of said gel formulation; and
b. a pharmaceutically acceptable vehicle,
wherein nasal administration of said formulation to a subject increases the plasma testosterone level to at least about 0.4 ng/ml, and wherein said increase is maintained for at least 6 hours immediately following nasal administration to a subject.
69. A testosterone gel formulation for nasal administration said testosterone gel formulation comprising:
a. about 0.72% testosterone by weight of said gel formulation; and
b. a pharmaceutically acceptable vehicle,
wherein nasal administration of said formulation to a subject increases the plasma testosterone level to at least 0.7 ng/ml about 60 minutes immediately following the nasal administration of said testosterone gel formulation to a subject.
70. The testosterone gel formulation of any one of claims 64-69, wherein said gel formulation further comprises a pharmaceutically acceptable vehicle.
71 . A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. about 0.72% testosterone; and
b. a pharmaceutically acceptable vehicle
wherein a plasma testosterone level of at least about 0.4 ng/ml is maintained in a subject for about 6 hours immediately following the nasal administration of said gel formulation to a subject.
72. A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. About 0.72% testosterone; and
b. a pharmaceutically acceptable vehicle;
wherein, a plasma testosterone level of at least about 0.7 ng/ml is achieved in a subject within about 60 minutes immediately following the nasal administration of said gel formulation to a subject.
73. The testosterone gel formulation of any one of claims 63-72, wherein said gel formula further comprises a solvent, a wetting agent, and a viscosity increasing agent.
74. The testosterone gel formulation of claim 73, wherein said solvent is castor oil.
75. The testosterone gel formulation of claim 73, wherein said wetting agent is an oleoyi polyoxylglyceride.
76. The testosterone gel formulation of claim 73, wherein said viscosity increasing agent is colloidal silicon dioxide.
77. The testosterone gel formulation of any one of claims 64-72, wherein said gel formulation further comprises castor oil, oleoyi polyoxylglycerides and colloidal silicon dioxide.
78. The testosterone gel formulation of any one of claims 64-77, wherein said gel formulation is a bioequivalent formulation.
79. The testosterone gel formulation of any one of claims 64-77, wherein said gel formulation is a pharmaceutically equivalent formulation.
80. The testosterone gel formulation of any one of claims 64-77, wherein said gel formulation is a therapeutically equivalent formulation.
81 . A packaged pharmaceutical comprising:
(a) a testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises about 0.72% by weight; and
(b) associated instructions for using said testosterone gel formulation to treat anorgasmia.
82. A packaged pharmaceutical comprising:
(a) a testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises about 0.72% by weight; and
(b) associated instructions for using said testosterone gel formulation to treat hypoactive sexual desire disorder (HSDD).
83. The packaged pharmaceutical of claim 81 or 82, wherein said testosterone is present as a pharmaceutical composition comprising a therapeutically effective amount of testosterone or a pharmaceutically acceptable salt or prodrug thereof and a pharmaceutically acceptable carrier.
84. The packaged pharmaceutical of claim 82 or 83 further comprising a step of identifying a subject in need of said pharmaceutical.
85. A method of treating anorgasmia comprising administering intranasally to each nostril of a subject said gel formulation of any one of claims 64-84 to deliver a therapeutically effective amount of testosterone to effectively treat anorgasmia.
86. A method of treating hypoactive sexual desire disorder (HSDD), comprising administering intranasally to each nostril of a subject said gel formulation of any one of claims 29-49 to deliver a therapeutically effective amount of testosterone to effectively treat HSDD.
87. An intranasal method of treating a subject suffering from or diagnosed with anorgasmia with a testosterone gel formulation for nasal administration comprising about 0.72% testosterone by weight of said gel formulation, comprising: applying the testosterone gel formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the anorgasmia.
88. The method of claim 87, wherein said subject receives said testosterone gel intranasally twice daily.
89. An intranasal method of treating a subject suffering from or diagnosed with HSDD with a testosterone gel formulation for nasal administration comprising about 0.72% testosterone by weight of said gel formulation, comprising: applying the testosterone gel formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the HSDD.
90. The method of claim 89, wherein said subject receives said testosterone gel intranasally twice daily.
91 . A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. about 0.45% testosterone by weight of said gel formulation; and b. a pharmaceutically acceptable vehicle.
92. The testosterone gel formulation of claim 91 , wherein said gel formula further comprises a solvent, a wetting agent, and a viscosity increasing agent.
93. The testosterone gel formulation of claim 92, wherein said solvent is castor oil.
94. The testosterone gel formulation of claim 93, wherein said wetting agent is an oleoyl polyoxylglyceride.
95. The testosterone gel formulation of claim 93, wherein said viscosity increasing agent is colloidal silicon dioxide.
96. The testosterone gel formulation of any one of claims 91 or 92, wherein said gel formulation further comprises castor oil, oleoyi polyoxylglycerides and colloidal silicon dioxide.
97. The testosterone gel formulation of any one of claims 91 -96, wherein said gel formulation is a bioequivalent formulation.
98. The testosterone gel formulation of any one of claims 91 -96, wherein said gel formulation is a pharmaceutically equivalent formulation.
99. The testosterone gel formulation of any one of claims 91 -96, wherein said gel formulation is a therapeutically equivalent formulation.
100. A packaged pharmaceutical comprising:
(a) a testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises about 0.45% by weight; and
(b) associated instructions for using said testosterone gel formulation to treat anorgasmia.
101 . A packaged pharmaceutical comprising:
(a) a testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises about 0.45% by weight; and
(b) associated instructions for using said testosterone gel formulation to treat hypoactive sexual desire disorder (HSDD).
102. The packaged pharmaceutical of claim 100 or 101 , wherein said testosterone is present as a pharmaceutical composition comprising a therapeutically effective amount of testosterone or a pharmaceutically acceptable salt or prodrug thereof and a pharmaceutically acceptable carrier.
103. The packaged pharmaceutical of claim 100 or 101 , further comprising a step of identifying a subject in need of said pharmaceutical.
104. A method of treating anorgasmia comprising administering intranasally to a subject said gel formulation of any one of claims 91 -99 to deliver a therapeutically effective amount of testosterone to effectively treat HSDD.
105. A method of treating hypoactive sexual desire disorder (HSDD), comprising administering intranasally to each nostril of a subject said gel formulation of any one of claims 1 -9 to deliver a therapeutically effective amount of testosterone to effectively treat HSDD.
106. An intranasal method of treating a subject diagnosed with anorgasmia with a testosterone gel formulation for nasal administration comprising about 0.45%
testosterone by weight of said gel formulation, comprising: applying the testosterone gel formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the anorgasmia.
107. The method of claim 106, wherein said subject receives said testosterone gel intranasally twice daily.
108. An intranasal method of treating a subject diagnosed with HSDD with a testosterone gel formulation for nasal administration comprising about 0.45%
testosterone by weight of said gel formulation, comprising: applying the testosterone gel
formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the HSDD.
109. The method of claim 108, wherein said subject receives said testosterone gel intranasally twice daily.
1 10. A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. about 0.48% testosterone by weight of said gel formulation; and b. a pharmaceutically acceptable vehicle.
1 1 1 . The testosterone gel formulation of claim 1 10, wherein said gel formula further comprises a solvent, a wetting agent, and a viscosity increasing agent.
1 12. The testosterone gel formulation of claim 1 1 1 , wherein said solvent is castor oil.
1 13. The testosterone gel formulation of claim 1 12, wherein said wetting agent is an oleoyi polyoxylglyceride.
1 14. The testosterone gel formulation of claim 1 12, wherein said viscosity increasing agent is colloidal silicon dioxide.
1 15. The testosterone gel formulation of any one of claims 100 or 1 1 1 , wherein said gel formulation further comprises castor oil, oleoyi polyoxylglycerides and colloidal silicon dioxide.
1 16. The testosterone gel formulation of any one of claims 1 10-1 15, wherein said gel formulation is a bioequivalent formulation.
1 17. The testosterone gel formulation of any one of claims 1 10-1 15, wherein said gel formulation is a pharmaceutically equivalent formulation.
1 18. The testosterone gel formulation of any one of claims 1 10-1 15, wherein said gel formulation is a therapeutically equivalent formulation.
1 19. A packaged pharmaceutical comprising:
(a) a testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises about 0.48% by weight; and
(b) associated instructions for using said testosterone gel formulation to treat anorgasmia.
120. A packaged pharmaceutical comprising:
(a) a testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises about 0.48% by weight; and
(b) associated instructions for using said testosterone gel formulation to treat hypoactive sexual desire disorder (HSDD).
121 . The packaged pharmaceutical of claim 1 19 or 120, wherein said testosterone is present as a pharmaceutical composition comprising a therapeutically effective amount of testosterone or a pharmaceutically acceptable salt or prodrug thereof and a pharmaceutically acceptable carrier.
122. The packaged pharmaceutical of claim 1 19 or 1200, further comprising a step of identifying a subject in need of said pharmaceutical.
123 A method of treating anorgasmia comprising administering intranasally to a subject said gel formulation of any one of claims 1 10-1 18 to deliver a therapeutically effective amount of testosterone to effectively treat HSDD.
124 A method of treating hypoactive sexual desire disorder (HSDD), comprising administering intranasally to each nostril of a subject said gel formulation of any one of claims 1 10-1 18 to deliver a therapeutically effective amount of testosterone to
effectively treat HSDD.
125. An intranasal method of treating a subject diagnosed with anorgasmia with a testosterone gel formulation for nasal administration comprising about 0.48%
testosterone by weight of said gel formulation, comprising: applying the testosterone gel formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the anorgasmia.
126 The method of claim 125, wherein said subject receives said testosterone gel intranasally twice daily.
127 An intranasal method of treating a subject diagnosed with HSDD with a testosterone gel formulation for nasal administration comprising about 0.48%
testosterone by weight of said gel formulation, comprising: applying the testosterone gel formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the HSDD.
128 The method of claim 127, wherein said subject receives said testosterone gel intranasally twice daily.
129 A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. about 0.15% testosterone by weight of said gel formulation; and
b. a pharmaceutically acceptable vehicle.
130. The testosterone gel formulation of claim 129, wherein said gel formula further comprises a solvent, a wetting agent, and a viscosity increasing agent.
131 . The testosterone gel formulation of claim 130, wherein said solvent is castor oil.
132. The testosterone gel formulation of claim 130, wherein said wetting agent is an oleoyi polyoxylglyceride.
133. The testosterone gel formulation of claim 130, wherein said viscosity increasing agent is colloidal silicon dioxide.
134. The testosterone gel formulation of any one of claims 129 or 130, wherein said gel formulation further comprises castor oil, oleoyi polyoxylglycerides and colloidal silicon dioxide.
135. The testosterone gel formulation of any one of claims 129 or 134, wherein said gel formulation is a bioequivalent formulation.
136. The testosterone gel formulation of any one of claims 129 or 134, wherein said gel formulation is a pharmaceutically equivalent formulation.
137. The testosterone gel formulation of any one of claims 129 or 134, wherein said gel formulation is a therapeutically equivalent formulation.
138. A packaged pharmaceutical comprising:
(a) a testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises about 0.15% by weight; and
(b) associated instructions for using said testosterone gel formulation to treat anorgasmia.
139. A packaged pharmaceutical comprising:
(a) a testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises about 0.15% by weight; and
(b) associated instructions for using said testosterone gel formulation to treat hypoactive sexual desire disorder (HSDD).
140. The packaged pharmaceutical of claim 138 or 139, wherein said testosterone is present as a pharmaceutical composition comprising a therapeutically effective amount of testosterone or a pharmaceutically acceptable salt or prodrug thereof and a pharmaceutically acceptable carrier.
141 . The packaged pharmaceutical of claim 138 or 139, further comprising a step of identifying a subject in need of said pharmaceutical.
142. A method of treating anorgasmia comprising administering intranasally to each nostril of a subject said gel formulation of any one of claims 129-137 to deliver a therapeutically effective amount of testosterone to effectively treat anorgasmia.
143. A method of treating hypoactive sexual desire disorder (HSDD), comprising administering intranasally to each nostril of a subject said gel formulation of any one of claims 1 -9 to deliver a therapeutically effective amount of testosterone to effectively treat HSDD.
144. An intranasal method of treating a subject diagnosed with anorgasmia with a testosterone gel formulation for nasal administration comprising about 0.15%
testosterone by weight of said gel formulation, comprising: applying the testosterone gel
formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the anorgasmia.
145. The method of claim 144 wherein said subject receives said testosterone gel intranasally twice daily.
146. An intranasal method of treating a subject diagnosed with HSDD with a testosterone gel formulation for nasal administration comprising about 0.15%
testosterone by weight of said gel formulation, comprising: applying the testosterone gel formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the HSDD.
147. The method of claim 146, wherein said subject receives said testosterone gel intranasally twice daily.
148. A testosterone gel formulation for nasal administration, said testosterone gel formulation comprising:
a. about 0.24% testosterone by weight of said gel formulation; and
b. a pharmaceutically acceptable vehicle.
149. The testosterone gel formulation of claim 148, wherein said gel formula further comprises a solvent, a wetting agent, and a viscosity increasing agent.
150. The testosterone gel formulation of claim 149, wherein said solvent is castor oil.
151 . The testosterone gel formulation of claim 150, wherein said wetting agent is an oleoyl polyoxylglyceride.
152. The testosterone gel formulation of claim 150, wherein said viscosity increasing agent is colloidal silicon dioxide.
153. The testosterone gel formulation of any one of claims 148 or 149, wherein said gel formulation further comprises castor oil, oleoyl polyoxylglycerides and colloidal silicon dioxide.
154. The testosterone gel formulation of any one of claims 148-153, wherein said gel formulation is a bioequivalent formulation.
155. The testosterone gel formulation of any one of claims 148-153, wherein said gel formulation is a pharmaceutically equivalent formulation.
156. The testosterone gel formulation of any one of claims 148-153, wherein said gel formulation is a therapeutically equivalent formulation.
157. A packaged pharmaceutical comprising:
(a) a testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises about 0.24% by weight; and
(b) associated instructions for using said testosterone gel formulation to treat anorgasmia.
158. A packaged pharmaceutical comprising:
(a) a testosterone gel formulation for nasal administration or a pharmaceutically acceptable salt or prodrug thereof, wherein said gel formulation comprises about 0.24% by weight; and
(b) associated instructions for using said testosterone gel formulation to treat hypoactive sexual desire disorder (HSDD).
159. The packaged pharmaceutical of claim 157 or 158, wherein said testosterone is present as a pharmaceutical composition comprising a therapeutically effective amount
of testosterone or a pharmaceutically acceptable salt or prodrug thereof and a pharmaceutically acceptable carrier.
160. The packaged pharmaceutical of claim 157 or 158, further comprising a step of identifying a subject in need of said pharmaceutical.
161 . A method of treating anorgasmia comprising administering intranasally to a subject said gel formulation of any one of claims 148-156 to deliver a therapeutically effective amount of testosterone to effectively treat HSDD.
162. A method of treating hypoactive sexual desire disorder (HSDD), comprising administering intranasally to each nostril of a subject said gel formulation of any one of claims 148-156 to deliver a therapeutically effective amount of testosterone to
effectively treat HSDD.
163. An intranasal method of treating a subject diagnosed with anorgasmia with a testosterone gel formulation for nasal administration comprising about 0.24%
testosterone by weight of said gel formulation, comprising: applying the testosterone gel formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the anorgasmia.
164. The method of claim 163, wherein said subject receives said testosterone gel intranasally twice daily.
165. An intranasal method of treating a subject diagnosed with HSDD with a testosterone gel formulation for nasal administration comprising about 0.24%
testosterone by weight of said gel formulation, comprising: applying the testosterone gel formulation into each nostril of said subject at least once a day to deliver an effective amount of testosterone to treat the HSDD.
166. The method of claim 165, wherein said subject receives said testosterone gel intranasally twice daily.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261726570P | 2012-11-14 | 2012-11-14 | |
| US61/726,570 | 2012-11-14 | ||
| US201261727112P | 2012-11-15 | 2012-11-15 | |
| US61/727,112 | 2012-11-15 | ||
| US201261729324P | 2012-11-21 | 2012-11-21 | |
| US61/729,324 | 2012-11-21 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2014083432A2 true WO2014083432A2 (en) | 2014-06-05 |
| WO2014083432A3 WO2014083432A3 (en) | 2014-10-30 |
Family
ID=50828553
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2013/003121 WO2014083432A2 (en) | 2012-11-14 | 2013-11-14 | Female intranasal lower dosage strength testosterone gel formulations and use thereof for treating anorgasmia or hypoactive sexual desire disorder |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2014083432A2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017035619A1 (en) * | 2015-08-28 | 2017-03-09 | F.B.M. Indústria Farmacêutica Ltda | Nasal pharmaceutical testosterone formulation and kit designed as a card of unit nasal devices for the application of testosterone monodoses |
| WO2019175290A1 (en) | 2018-03-13 | 2019-09-19 | Beckley Canopy Therapeutics Limited | Cannabis or cannabis derived compositions for promoting cessation of chemical dependence |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7404965B2 (en) * | 2000-08-03 | 2008-07-29 | Antares Pharma Ipl Ag | Composition for transdermal and/or transmucosal administration of active compounds that ensures adequate therapeutic levels |
| US20100311707A1 (en) * | 2003-11-11 | 2010-12-09 | Claudia Mattern | Controlled release delivery system for nasal applications and methods of treatment |
| US8067399B2 (en) * | 2005-05-27 | 2011-11-29 | Antares Pharma Ipl Ag | Method and apparatus for transdermal or transmucosal application of testosterone |
| WO2012022446A1 (en) * | 2010-08-16 | 2012-02-23 | Innotesto Bvba | Testosterone solutions for the treatment of testosterone deficiency |
-
2013
- 2013-11-14 WO PCT/IB2013/003121 patent/WO2014083432A2/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7404965B2 (en) * | 2000-08-03 | 2008-07-29 | Antares Pharma Ipl Ag | Composition for transdermal and/or transmucosal administration of active compounds that ensures adequate therapeutic levels |
| US20100311707A1 (en) * | 2003-11-11 | 2010-12-09 | Claudia Mattern | Controlled release delivery system for nasal applications and methods of treatment |
| US8067399B2 (en) * | 2005-05-27 | 2011-11-29 | Antares Pharma Ipl Ag | Method and apparatus for transdermal or transmucosal application of testosterone |
| WO2012022446A1 (en) * | 2010-08-16 | 2012-02-23 | Innotesto Bvba | Testosterone solutions for the treatment of testosterone deficiency |
Non-Patent Citations (1)
| Title |
|---|
| MILLER, HB ET AL.: 'Female Sexual Dysfunction: Review of the Disorder and Evidence for Available Treatment Alternatives.' JOUMAL OF PHARMACY PRACTICE. vol. 16, no. 3, pages 200 - 208 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017035619A1 (en) * | 2015-08-28 | 2017-03-09 | F.B.M. Indústria Farmacêutica Ltda | Nasal pharmaceutical testosterone formulation and kit designed as a card of unit nasal devices for the application of testosterone monodoses |
| WO2019175290A1 (en) | 2018-03-13 | 2019-09-19 | Beckley Canopy Therapeutics Limited | Cannabis or cannabis derived compositions for promoting cessation of chemical dependence |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2014083432A3 (en) | 2014-10-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2020264363B2 (en) | Intranasal lower dosage strength estosterone gel formulations and use thereof for treating anorgasmia or hypoactive sexual desire disorder | |
| CN103796636B (en) | Intranasal testosterone bioadhesiveness gel preparation and its purposes for treating male hypogonadism | |
| Anawalt et al. | Combined nestorone–testosterone gel suppresses serum gonadotropins to concentrations associated with effective hormonal contraception in men | |
| US20160250228A1 (en) | Intranasal 0.45% and 0.48% testosterone gel formulations and use thereof for treating anorgasmia or hypoactive sexual desire disorder | |
| US10668084B2 (en) | Intranasal lower dosage strength testosterone gel formulations and use thereof for treating anorgasmia or hypoactive sexual desire disorder | |
| US10111888B2 (en) | Intranasal 0.15% and 0.24% testosterone gel formulations and use thereof for treating anorgasmia or hypoactive sexual desire disorder | |
| US9757388B2 (en) | Intranasal methods of treating women for anorgasmia with 0.6% and 0.72% testosterone gels | |
| Fooladi et al. | An update on the pharmacological management of female sexual dysfunction | |
| WO2014080283A2 (en) | Male testosterone titration methods, male intranasal testosterone bio-adhesive gel formulations and use thereof for treating hypogonadism and trt | |
| US20240316063A1 (en) | Methods of treating hypogonadism with transnasal testosterone bio-adhesive gel formulations in male with allergic rhinitis, and methods for preventing an allergic rhinitis event | |
| WO2014080282A2 (en) | One-and two-point titration methods to determine daily treatment regimens to treat hypogonadism or male testosterone deficiency with an intranasal testosterone bio-adhesive gel, and primary and secondary efficacy and safety endpoints | |
| WO2014083432A2 (en) | Female intranasal lower dosage strength testosterone gel formulations and use thereof for treating anorgasmia or hypoactive sexual desire disorder | |
| Krapf et al. | Management of hypoactive sexual desire disorder (HSDD) | |
| MX2013013236A (en) | Intranasal lower dosage strength testosterone gel formulations and use thereof for treating anorgasmia or hypoactive sexual desire disorder. | |
| BR112013029335A2 (en) | testosterone gel formulations of lower intranasal dosage intensity and use thereof for the treatment of anorgasmia or hypoactive sexual desire disorder |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13857940 Country of ref document: EP Kind code of ref document: A2 |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13857940 Country of ref document: EP Kind code of ref document: A2 |