WO2014060908A1 - Improved process for preparation of revaprazan hydrochloride - Google Patents
Improved process for preparation of revaprazan hydrochloride Download PDFInfo
- Publication number
- WO2014060908A1 WO2014060908A1 PCT/IB2013/059225 IB2013059225W WO2014060908A1 WO 2014060908 A1 WO2014060908 A1 WO 2014060908A1 IB 2013059225 W IB2013059225 W IB 2013059225W WO 2014060908 A1 WO2014060908 A1 WO 2014060908A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- process according
- tetra
- butyl
- revaprazan
- Prior art date
Links
- AXGDZZVIIPMCEJ-UHFFFAOYSA-N CC(c1ccccc1CC1)N1c1c(C)c(C)nc(N(CC2)C(C)c3c2cccc3)n1 Chemical compound CC(c1ccccc1CC1)N1c1c(C)c(C)nc(N(CC2)C(C)c3c2cccc3)n1 AXGDZZVIIPMCEJ-UHFFFAOYSA-N 0.000 description 1
- LECZXZOBEZITCL-UHFFFAOYSA-N CC(c1ccccc1CC1)N1c1c(C)c(C)nc(Nc(cc2)ccc2F)n1 Chemical compound CC(c1ccccc1CC1)N1c1c(C)c(C)nc(Nc(cc2)ccc2F)n1 LECZXZOBEZITCL-UHFFFAOYSA-N 0.000 description 1
- STZSYZKNKUYOEB-UHFFFAOYSA-N CC(c1ccccc1CC1)N1c1nc(Cl)c(C)c(C)n1 Chemical compound CC(c1ccccc1CC1)N1c1nc(Cl)c(C)c(C)n1 STZSYZKNKUYOEB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to improved process for preparation of revaprazan hydrochloride in a high yield and purity.
- Revaprazan hydrochloride is chemically known as 4-(3,4-dihydro-l-mefhyl-2(lH)- isoquinolinyl)-N-(4-fluorophenyl)-5,6-dimethyl-2-pyrimidinamine hydrochloride and represented by formula I.
- Revaprazan hydrochloride (I) inhibits gastric acid secretion by means of a reversible proton-pump inhibiting effect and, therefore, can be used as an anti-ulcer agent.
- Revaprazan hydrochloride (I) was first disclosed in patent US 5,750,531 , along with its process for preparation as depicted in scheme 1.
- the synthesis involve reaction of 5, 6- dimethyl-2, 4-dichloro pyrimidine (II) and 1 -methyl- 1, 2, 3,4-tetrahydroisoquinoline (III) to produce 5, 6-dimethyl-4-(l -methyl- l ,2,3,4-tetrahydroisoquinolin-2-yl)-2- chloropyrimidine (IV) which is purified by column chromatography. Pure compound (IV) is reacted with 4-fluorophenyl amine (V) to obtain revaprazan (VI).
- Scheme 1 Process for preparation ofrevaprazan hydrochloride (I) as given in US 5,750,531.
- Scheme 2 Process for preparation ofrevaprazan hydrochloride as given in US 6,252,076.
- Figure 1 X-ray powder diffraction pattern of revaprazan hydrochloride.
- the present invention provides process for preparation of revaprazan hydrochloride (I) comprising reaction of l-methyl-l ,2,3,4-tetrahydroisoquinoline (III) compound (VII)
- R is a good leaving group selected from in presence of phase transfer catalyst and solvent to give revaprazan (VI) and followed by treatment with hydrochloride acid.
- Revaprazan hydrochloride (I) can be optionally purified.
- the present invention provides process for preparation of revaprazan hydrochloride (I) comprising reaction of 1 -methyl- 1,2,3,4- tetrahydroisoquinoline (III) with compound (VII) wherein R is a good leaving group selected from is F, CI, Br, I,
- the phase transfer catalyst is selected from tetrabutylarmnomurn bromide ⁇ ⁇ tetra butyl ammonium chloride, benzyl tri butyl ammonium chloride, benzyl tri ethyl ammonium chloride, tetra ethyl ammonium bromide, tetra ethyl ammonium chloride, tetra propyl ammonium bromide, cetyltrimethyl ammonium bromide, ethyl tri phenyl phosphonium bromide, tetra butyl phosphonium chloride, tetra butyl phosphonium bromide, tetra phenyl phosphonium bromide and methyl tri phenyl phosphonium bromide, polyethylene glycol; preferably TBAB.
- the reaction can be carried out in solvent selected form esters such as ethyl acetate, butyl acetate etc.; chlorinated hydrocarbons such as dichlorome thane, chloroform, ethylene dichloride etc.; ketones such as acetone, methyl ethyl ketone, methyl isobutyl ketone etc., nitriles such as acetonitrile, propionitrile etc.; ether such as diethyl ether, diisopropyl ether, t-butyl methyl ether, tetrahydrofuran, dioxan etc.; alcohols such as tert-butyl alcohol etc.; amides such as dimethylformamide, dimethylacetamide, N-methyl pyrrolidone etc.; dimethyl sulfoxide, sulfolane; aromatic hydrocarbons such as benzene, toluene, xylene etc.; or mixtures thereof; preferably methyl isobutyl
- the phase transfer catalyst can be employed in the range of 5 to 20%, preferably 10-20%, weight/weight of4-halide-2-(4-fluorophenylamino)-5, 6- dimethylpyrimidine (VII).
- the compound 1 -methyl- 1, 2, 3,4-tetrahydroisoquinoline (III) can be used in slight molar excess, preferably 1.1 to 1.5 moles, of 4-halide-2-(4- fluorophenylamino)-5,6-dimethylpyrimidine (VII).
- Revaprazan (VI) is converted to revaprazan hydrochloride (I) by treating with hydrochloride acid.
- Revaprazan hydrochloride (I) can be isolated from the reaction mixture by techniques known in art like filtration, concentration, evaporation of the solvent etc. Yield of revaprazan hydrochloride (I) was 97-100%.
- Revaprazan hydrochloride (I) can be optionally purified by crystallization froman alcoholic solvent like methanol, isopropanol, tert-butanol etc., preferably methanol.
- the present invention provides revaprazan hydrochloride (I) with purity of >99.9%, by HPLC.
- the X-ray powder diffraction pattern of revaprazan hydrochloride (I) is as depicted in figure- 1. It shows characteristic peaks at 7.41, 10.33, 12.26, 13.34, 16.34, 16.97, 17.66, 19.66, 20.84, 22.22, 23.35, 24.26, 25.51, 25.73, 26.05, 27.12 ⁇ °2Theta.
- the X-ray diffraction pattern was measured using PANalytical, X'pert PRO machine with parameters as: scan axis: gonio; step size: 0.0083°; scan mode: continuous; anode material: Cu; radiation type: K-alpha 1 and scan angle (°): 3.5 to 39.98.
- Revaprazan hydrochloride (I) obtained by the present invention exhibit a particle size of d(0.5) of about 20-30 micron and d(0.9) of about 120-130 micron.
- the micronized revaprazan hydrochloride exhibit a particle size of d(0.5) of about 2-5 micron and d(0.9) of about 6-8 micron.
- the manufacture of revaprazan hydrochloride (I) as per the process of present invention is advantageous in that the process iseconomical due to high yield, provides revaprazan hydrochloride with high purity and is commercially viable.
- the present invention is further illustrated by the following representative examples and does not limit the scope of the invention.
- Example 1 process for preparation of revaprazan hydrochloride (I):
- revaprazan hydrochloride (I) (4.58 Kg) and methanol (27.5 L) was heated to 60-65°C to get clear solution.
- the solution was filtered through hyflo bed.
- the solution was heated to 60-65°C and methanol (about 22 L) was distilled out under atmospheric pressure.
- the mixture was cooled to 0-5°C.
- the solid was filtered, washed with methanol and dried under vacuum. Yield: 3.72 Kg (81%); HPLC purity: 99.95%.
Abstract
The present invention provides improved process for preparation of revaprazan hydrochloride (I) comprising reaction of l-methyl-l,2,3,4-tetrahydroisoquinoline (III) withcompound (VII) wherein R is a good leaving group selected from F, Cl, Br, I, (1), (2), (3) in presence of phase transfer catalyst and solvent to give revaprazan (VI) and followed by treatment with hydrochloride acid.
Description
IMPROVED PROCESS FOR PREPARATION OF REVAPRAZAN
HYDROCHLORIDE
FIELD OF INVENTION
The present invention relates to improved process for preparation of revaprazan hydrochloride in a high yield and purity. BACKGROUND OF THE INVENTION
Revaprazan hydrochloride is chemically known as 4-(3,4-dihydro-l-mefhyl-2(lH)- isoquinolinyl)-N-(4-fluorophenyl)-5,6-dimethyl-2-pyrimidinamine hydrochloride and represented by formula I.

Revaprazan hydrochloride (I) inhibits gastric acid secretion by means of a reversible proton-pump inhibiting effect and, therefore, can be used as an anti-ulcer agent.
Revaprazan hydrochloride (I) was first disclosed in patent US 5,750,531 , along with its process for preparation as depicted in scheme 1. The synthesis involve reaction of 5, 6- dimethyl-2, 4-dichloro pyrimidine (II) and 1 -methyl- 1, 2, 3,4-tetrahydroisoquinoline (III) to produce 5, 6-dimethyl-4-(l -methyl- l ,2,3,4-tetrahydroisoquinolin-2-yl)-2- chloropyrimidine (IV) which is purified by column chromatography. Pure compound (IV) is reacted with 4-fluorophenyl amine (V) to obtain revaprazan (VI).

(VI) (IV)
Scheme 1: Process for preparation ofrevaprazan hydrochloride (I) as given in US 5,750,531.
Another patent US 6,252,076 provides process for preparation of revaprazan hydrochloride (I) as depicted in scheme 2. Reaction of 1 -methyl- 1,2,3,4- tetrahydroisoquinoline (III) and 4-chloro-2-(4-fluorophenylamino)-5,6- dimethylpyrimidine (VII, R=C1) is carried out in presence of base and solvent to give revaprazan (VI) followed by treatmentwith hydrochloride acid to provide revaprazan hydrochloride (I) in a yield upto91%.

Scheme 2: Process for preparation ofrevaprazan hydrochloride as given in US 6,252,076.
The synthesis of US 5,750,531, suffers a severe drawback of side reactions which leads to formation of contaminants like 2-(4-chloro-5,6-dimethylpyrimidin-2-yl)-l-methyl-
1 ,2,3,4-tetrahydroisoquinoline (VIII); 2,2'-(5,6-dimethylpyrimidine-2,4-diyl)bis(l- methyl-l ,2,3,4-tetrahydroisoquinoline) (IX) and the patent US 6,252,076 does not disclose purit of revaprazan hydrochloride (I) obtained.

DESCRIPTION OF DRAWING
Figure 1 :X-ray powder diffraction pattern of revaprazan hydrochloride.
SUMMARY OF THE INVENTION
The present invention provides process for preparation of revaprazan hydrochloride (I) comprising reaction of l-methyl-l ,2,3,4-tetrahydroisoquinoline (III) compound (VII)

(vii) wherein R is a good leaving group selected from
 in presence of phase transfer catalyst and solvent to give revaprazan (VI) and followed by treatment with hydrochloride acid. Revaprazan hydrochloride (I) can be optionally purified. DETAILED DES CRIPTION OF THE INVENTION
in presence of phase transfer catalyst and solvent to give revaprazan (VI) and followed by treatment with hydrochloride acid. Revaprazan hydrochloride (I) can be optionally purified. DETAILED DES CRIPTION OF THE INVENTION
In a preferred embodiment, the present invention provides process for preparation of revaprazan hydrochloride (I) comprising reaction of 1 -methyl- 1,2,3,4- tetrahydroisoquinoline (III) with compound (VII) wherein R is a good leaving group selected from is F, CI, Br, I,

in presence of phase transfer catalyst and solvent to give revaprazan (VI) andfollowed by treatment with hydrochloride acid, wherein R is a good
The phase transfer catalyst is selected from tetrabutylarmnomurn bromide Π ΗΛΗ tetra butyl ammonium chloride, benzyl tri butyl ammonium chloride, benzyl tri ethyl ammonium chloride, tetra ethyl ammonium bromide, tetra ethyl ammonium chloride, tetra propyl ammonium bromide, cetyltrimethyl ammonium bromide, ethyl tri phenyl phosphonium bromide, tetra butyl phosphonium chloride, tetra butyl phosphonium bromide, tetra phenyl phosphonium bromide and methyl tri phenyl phosphonium bromide, polyethylene glycol; preferably TBAB.
The reaction can be carried out in solvent selected form esters such as ethyl acetate, butyl acetate etc.; chlorinated hydrocarbons such as dichlorome thane, chloroform, ethylene dichloride etc.; ketones such as acetone, methyl ethyl ketone, methyl isobutyl ketone etc., nitriles such as acetonitrile, propionitrile etc.; ether such as diethyl ether, diisopropyl ether, t-butyl methyl ether, tetrahydrofuran, dioxan etc.; alcohols such as tert-butyl alcohol etc.; amides such as dimethylformamide, dimethylacetamide, N-methyl pyrrolidone etc.; dimethyl sulfoxide, sulfolane; aromatic hydrocarbons such as benzene, toluene, xylene etc.; or mixtures thereof; preferably methyl isobutyl ketone and xylene.
Thereaction is carried out at a temperature of 75 to 200°C, preferably 100 to 150°C.
In the reaction, the phase transfer catalyst can be employed in the range of 5 to 20%, preferably 10-20%, weight/weight of4-halide-2-(4-fluorophenylamino)-5, 6- dimethylpyrimidine (VII). The compound 1 -methyl- 1, 2, 3,4-tetrahydroisoquinoline (III) can be used in slight molar excess, preferably 1.1 to 1.5 moles, of 4-halide-2-(4- fluorophenylamino)-5,6-dimethylpyrimidine (VII).
Revaprazan (VI) is converted to revaprazan hydrochloride (I) by treating with hydrochloride acid. Revaprazan hydrochloride (I) can be isolated from the reaction mixture by techniques known in art like filtration, concentration, evaporation of the solvent etc. Yield of revaprazan hydrochloride (I) was 97-100%.
Revaprazan hydrochloride (I) can be optionally purified by crystallization froman alcoholic solvent like methanol, isopropanol, tert-butanol etc., preferably methanol.
The present invention provides revaprazan hydrochloride (I) with purity of >99.9%, by HPLC. The X-ray powder diffraction pattern of revaprazan hydrochloride (I) is as depicted in figure- 1. It shows characteristic peaks at 7.41, 10.33, 12.26, 13.34, 16.34, 16.97, 17.66, 19.66, 20.84, 22.22, 23.35, 24.26, 25.51, 25.73, 26.05, 27.12 ±°2Theta.The X-ray diffraction pattern was measured using PANalytical, X'pert PRO machine with parameters as: scan axis: gonio; step size: 0.0083°; scan mode: continuous; anode material: Cu; radiation type: K-alpha 1 and scan angle (°): 3.5 to 39.98.
Revaprazan hydrochloride (I) obtained by the present invention exhibit a particle size of d(0.5) of about 20-30 micron and d(0.9) of about 120-130 micron. The micronized revaprazan hydrochloride exhibit a particle size of d(0.5) of about 2-5 micron and d(0.9) of about 6-8 micron.
The manufacture of revaprazan hydrochloride (I) as per the process of present invention is advantageous in that the process iseconomical due to high yield, provides revaprazan hydrochloride with high purity and is commercially viable. The present invention is further illustrated by the following representative examples and does not limit the scope of the invention.
EXAMPLES:
Example 1: process for preparation of revaprazan hydrochloride (I):
A mixture of4-chloro-2-(4-fluorophenylamino)-5,6-dimethylpyrimidine (VII) (1.5 Kg), 1- methyl-l,2,3,4-tetrahydroisoquinoline (III) (1.32 Kg),and TBAB (0.15 Kg) in methyl isobutyl ketone (12 L) was heated to 115-119°C for 48 hours. The reaction mixture was cooled to 25-30°C and water (3.2 L) was added to it. The pH of the reaction mixture was adjusted to about 0.6 with cone. HC1. The reaction mixture was cooled to 0-5°C and stirred for 3 hours. The solid was filtered, washed with water and dried under vacuum. Yield: 2.31 Kg (97.46%); HPLC purity: 98.34%.
Example 2: process for preparation of revaprazan hydrochloride (I):
A mixture of4-chloro-2-(4-fluorophenylamino)-5,6-dimethylpyrimidine (VII) (50 g), 1- methyl-l,2,3,4-tetrahydroisoquinoline (III) (43.86 g),and TBAB (5 g) in xylene (400 ml) was heated to 135-140°C for 33 hours. The reaction mixture was cooled to 25-30°C and water (100 ml) was added to it. The pH of the reaction mixture was adjusted to about 0.6 with cone. HC1. The reaction mixture was cooled to 0-5°C and stirred for 2 hours. The solid was filtered, washed with water and dried under vacuum. Yield: quantitative.
Example 3: purification of revaprazan hydrochloride (I):
A mixture of revaprazan hydrochloride (I) (4.58 Kg) and methanol (27.5 L) was heated to 60-65°C to get clear solution. The solution was filtered through hyflo bed. The solution was heated to 60-65°C and methanol (about 22 L) was distilled out under atmospheric pressure. The mixture was cooled to 0-5°C. The solid was filtered, washed with methanol and dried under vacuum. Yield: 3.72 Kg (81%); HPLC purity: 99.95%.
Claims
1. A process for preparation of revaprazan hydrochloride (I)

comprising reaction of l-methyl-l,2,3,4-tetrahydroisoquinoline (III)

(HI)
with 4-halide-2-(4-fluorophenylamino)-5,6-dimethylpyrimidine (VII)

(vi i)
from is F, CI, Br, I,

in presence of phase transfer catalyst and solvent to give revaprazan (VI)
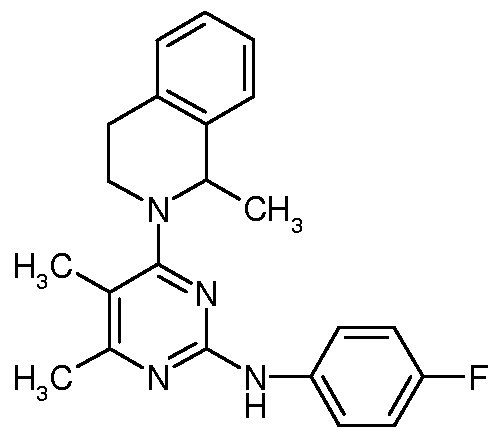
(VI) and followed by treatment with hydrochloride acid.
A process according to claim 1 wherein, the phase transfer catalyst is selected from tetra butyl ammonium bromide, tetra butyl ammonium chloride, benzyl tri butyl ammonium chloride, benzyl tri ethyl ammonium chloride, tetra ethyl ammonium bromide, tetra ethyl ammonium chloride, tetra propyl ammonium bromide, cetyl tri methyl ammonium bromide, ethyl tri phenyl phosphonium bromide, tetra butyl phosphonium chloride, tetra butyl phosphonium bromide, tetra phenyl phosphonium bromide and methyl tri phenyl phosphonium bromide, polyethylene glycol.
A process according to claim 2 wherein, the phase transfer catalyst istetra butyl ammonium bromide.
A process according to claim 1 wherein, solvent is selected form esters such as ethyl acetate, butyl acetate; chlorinated hydrocarbons such as dichloromethane, chloroform, ethylene dichloride; ketones such as acetone, methyl ethyl ketone, methyl isobutyl ketone; nitriles such as acetonitrile, propionitrile; ether such as diethyl ether, diisopropyl ether, t-butyl methyl ether, tetrahydrofuran, dioxan; alcohols such as tert-butyl alcohol; amides such as dimethylformamide, dimethylacetamide, N-methyl pyrrolidone; dimethyl sulfoxide; sulfolane; aromatic hydrocarbons such as benzene, toluene, xylene; or mixtures thereof.
A process according to claim 4 wherein, solvent is methyl isobutyl ketone or xylene.
A process according to claim 1 wherein,reaction is carried out at a temperature of 75 to 200°C.
A process according to claim 1 wherein, the phase transfer catalyst is employed as 5 to 20% weight/weight of 4-halide-2-(4-fluorophenylamino)-5, 6- dimethylpyrimidine (VII).
A process according to claim 1 wherein, 1 -methyl- 1, 2, 3, 4- tetrahydroisoquinoline (III) is employed in 1.1 to 1.5 moles of 4-halide-2-(4- fluorophenylamino)-5,6-dimethylpyrimidine (VII).
A process according to claim 1 wherein, revaprazan hydrochloride can be optionally purified by crystallization from an alcoholic solvent like methanol, isopropanol, tert-butanol.
A process according to claim 9 wherein, revaprazan hydrochloride is purified by crystallizationfrom methanol.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1171KO2012 | 2012-10-17 | ||
| IN1171/KOL/2012 | 2012-10-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014060908A1 true WO2014060908A1 (en) | 2014-04-24 |
Family
ID=49515440
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2013/059225 WO2014060908A1 (en) | 2012-10-17 | 2013-10-09 | Improved process for preparation of revaprazan hydrochloride |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2014060908A1 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105399727A (en) * | 2015-12-02 | 2016-03-16 | 芷威(上海)化学科技有限公司 | Novel revaprazan hydrochloride crystalline form |
| CN105601611A (en) * | 2014-11-19 | 2016-05-25 | 江苏天士力帝益药业有限公司 | Revaprzan hydrochloride polymorphic substance and preparation method thereof |
| WO2016078542A1 (en) * | 2014-11-19 | 2016-05-26 | 江苏天士力帝益药业有限公司 | Preparation method for revaprazan hydrochloride |
| CN106995431A (en) * | 2016-01-25 | 2017-08-01 | 广东华南药业集团有限公司 | A kind of auspicious pula life hydrate crystal forms and its production and use |
| CN106995432A (en) * | 2016-01-25 | 2017-08-01 | 广东华南药业集团有限公司 | A kind of auspicious pula life is without hydrate crystal forms and its production and use |
| CN107759562A (en) * | 2016-08-21 | 2018-03-06 | 常州四药制药有限公司 | A kind of preparation method of hydrochloric acid revaprzan |
| WO2019221522A1 (en) * | 2018-05-18 | 2019-11-21 | Yuhan Corporation | Novel processes for preparing a diaminopyrimidine derivative or acid addition salt thereof |
| JP7474754B2 (en) | 2018-05-18 | 2024-04-25 | ユーハン・コーポレイション | Novel process for producing diaminopyrimidine derivatives or acid addition salts thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996005177A1 (en) * | 1994-08-13 | 1996-02-22 | Yuhan Corporation | Novel pyrimidine derivatives and processes for the preparation thereof |
| US6252076B1 (en) | 1996-05-04 | 2001-06-26 | Yuhan Corporation | Process for preparation of pyrimidine derivatives |
-
2013
- 2013-10-09 WO PCT/IB2013/059225 patent/WO2014060908A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996005177A1 (en) * | 1994-08-13 | 1996-02-22 | Yuhan Corporation | Novel pyrimidine derivatives and processes for the preparation thereof |
| US5750531A (en) | 1994-08-13 | 1998-05-12 | Yuhan Corporation | Pyrimidine derivatives and processes for the preparation thereof |
| US6252076B1 (en) | 1996-05-04 | 2001-06-26 | Yuhan Corporation | Process for preparation of pyrimidine derivatives |
Non-Patent Citations (2)
| Title |
|---|
| H.H. FREEDMAN: "Industrial applications of phase transfer catalysis (PTC): past, present and future", PURE & APPL. CHEM., vol. 58, no. 6, 1 January 1986 (1986-01-01), pages 857 - 868, XP055071123 * |
| MAGGIOLO A ET AL: "The reaction of alkylamines with chloroheterocyclic compounds. II. 2-Amino-4-chloro-6-methylpyrimidine", THE JOURNAL OF ORGANIC CHEMISTRY, AMERICAN CHEMICAL SOCIETY [NOT]ETC. , US, vol. 16, no. 3, 1 January 1951 (1951-01-01), pages 3763 - 82, XP001180523, ISSN: 0022-3263, DOI: 10.1021/JO01143A003 * |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9981917B2 (en) | 2014-11-19 | 2018-05-29 | Jiangsu Tasly Diyi Pharmaceutical Co., Ltd. | Preparation method for revaprazan hydrochloride |
| CN105601611A (en) * | 2014-11-19 | 2016-05-25 | 江苏天士力帝益药业有限公司 | Revaprzan hydrochloride polymorphic substance and preparation method thereof |
| WO2016078543A1 (en) * | 2014-11-19 | 2016-05-26 | 江苏天士力帝益药业有限公司 | Revaprazan hydrochloride polymorphs and preparation method therefor |
| WO2016078542A1 (en) * | 2014-11-19 | 2016-05-26 | 江苏天士力帝益药业有限公司 | Preparation method for revaprazan hydrochloride |
| RU2702625C2 (en) * | 2014-11-19 | 2019-10-09 | Цзянсу Тасли Дии Фармасьютикал Ко., Лтд. | Preparation method for revaprazan hydrochloride |
| JP2017534635A (en) * | 2014-11-19 | 2017-11-24 | 江蘇天士力帝益薬業有限会社Jiangsu Tasly Diyi Pharmaceutical Co., Ltd. | Crystalline polymorph of revaprazan hydrochloride and its preparation method |
| JP2017534637A (en) * | 2014-11-19 | 2017-11-24 | 江蘇天士力帝益薬業有限会社Jiangsu Tasly Diyi Pharmaceutical Co., Ltd. | Preparation method of revaprazan hydrochloride |
| US10005757B2 (en) | 2014-11-19 | 2018-06-26 | Jiangsu Tasly Diyi Pharmaceutical Co., Ltd. | Revaprazan hydrochloride polymorphs and preparation method therefor |
| CN105399727A (en) * | 2015-12-02 | 2016-03-16 | 芷威(上海)化学科技有限公司 | Novel revaprazan hydrochloride crystalline form |
| CN106995432A (en) * | 2016-01-25 | 2017-08-01 | 广东华南药业集团有限公司 | A kind of auspicious pula life is without hydrate crystal forms and its production and use |
| CN106995431A (en) * | 2016-01-25 | 2017-08-01 | 广东华南药业集团有限公司 | A kind of auspicious pula life hydrate crystal forms and its production and use |
| CN107759562A (en) * | 2016-08-21 | 2018-03-06 | 常州四药制药有限公司 | A kind of preparation method of hydrochloric acid revaprzan |
| WO2019221522A1 (en) * | 2018-05-18 | 2019-11-21 | Yuhan Corporation | Novel processes for preparing a diaminopyrimidine derivative or acid addition salt thereof |
| KR20190131983A (en) * | 2018-05-18 | 2019-11-27 | 주식회사유한양행 | Novel processes for preparing a diaminopyrimidine derivative or acid addition salt thereof |
| CN112135820A (en) * | 2018-05-18 | 2020-12-25 | 柳韩洋行 | Novel process for preparing diaminopyrimidine derivatives or their acid addition salts |
| JP2021524500A (en) * | 2018-05-18 | 2021-09-13 | ユーハン・コーポレイションYUHAN Corporation | A novel method for producing a diaminopyrimidine derivative or an acid addition salt thereof |
| US11434224B2 (en) | 2018-05-18 | 2022-09-06 | Yuhan Corporation | Processes for preparing a diaminopyrimidine derivative or acid addition salt thereof |
| KR102441327B1 (en) * | 2018-05-18 | 2022-09-07 | 주식회사유한양행 | Novel processes for preparing a diaminopyrimidine derivative or acid addition salt thereof |
| US11623925B2 (en) | 2018-05-18 | 2023-04-11 | Yuhan Corporation | Processes for preparing a diaminopyrimidine derivative or acid addition salt thereof |
| CN112135820B (en) * | 2018-05-18 | 2024-01-02 | 柳韩洋行 | Novel process for preparing diaminopyrimidine derivatives or acid addition salts thereof |
| JP7474754B2 (en) | 2018-05-18 | 2024-04-25 | ユーハン・コーポレイション | Novel process for producing diaminopyrimidine derivatives or acid addition salts thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2014060908A1 (en) | Improved process for preparation of revaprazan hydrochloride | |
| EP2334645B1 (en) | Process for the synthesis of fluorinated cyclic compounds | |
| ES2905973T3 (en) | Process for the preparation of 5-chloro-N2-(2-isopropoxy-5-methyl-4-piperidin-4-yl-phenyl)-N4-[2-(propane-2-sulfonyl)-phenyl]-pyrimidine dihydrochloride -2,4-diamine | |
| US8399668B2 (en) | Process for the preparation of 5-(2-amino-pyrimidin-4-yl)-2-aryl-1H-pyrrole-3-carboxamides | |
| JP5594743B2 (en) | Method for producing bosentan potassium salt | |
| US8309726B2 (en) | Substituted piperazine compounds of formula 8 | |
| EP2763981B1 (en) | Method for preparing 1-(4-(4-(3,4-dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6-yloxy)piperidin-1-yl)-prop-2-en-1-one hydrochloride and intermediates used therein | |
| JP5863789B2 (en) | Method for producing pyrazole derivative | |
| WO2013111163A2 (en) | Process for the preparation of dabigatran etexilate mesylate and polymorphs of intermediates thereof | |
| JP4953822B2 (en) | Method for producing muscarinic receptor antagonist and intermediate thereof | |
| US20230018429A1 (en) | Process for synthesis of (3-chloro-2-pyridyl)hydrazine | |
| KR20220124679A (en) | Synthesis of 6-methyl-N1-(4-(pyridin-3-yl)pyrimidin-2-yl)benzene-1,3-diamine | |
| EP2608791A1 (en) | A process for the preparation of imatinib base | |
| HU226752B1 (en) | Process for preparation of pyrimidine derivatives | |
| CA2682384C (en) | Benzophenone hybrids as potential anticancer agents and a process for the preparation thereof | |
| US20110306763A1 (en) | Process for the preparation of imatinib and salts thereof | |
| WO2012090221A1 (en) | Novel salts of imatinib | |
| KR101457453B1 (en) | Process for preparing gefitinib and an intermediate used for preparing thereof | |
| JP5640283B2 (en) | Process for the preparation of pyrimidine derivatives | |
| US10562889B2 (en) | Process for the preparation of 1-(arylmethyl)quinazoline-2,4(1H,3H)-diones | |
| JP4887454B2 (en) | Method for producing pyrimidinylpyrazole compound | |
| WO2007069265A1 (en) | A novel process for the synthesis of lamotrigine and its intermediate | |
| TWI478914B (en) | Process for the preparation of 5-(2-amino-pyrimidin-4-yl)-2-aryl-1h-pyrrole-3-carboxamides | |
| US20180230116A1 (en) | Process for the preparation of vortioxetine and salts thereof | |
| KR101635724B1 (en) | An improved process for the preparation of gefitinib |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13785658 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13785658 Country of ref document: EP Kind code of ref document: A1 |