WO2014008257A2 - Processes for the preparation of 3,5-disubstituted-1,2,4-oxadiazoles - Google Patents
Processes for the preparation of 3,5-disubstituted-1,2,4-oxadiazoles Download PDFInfo
- Publication number
- WO2014008257A2 WO2014008257A2 PCT/US2013/049060 US2013049060W WO2014008257A2 WO 2014008257 A2 WO2014008257 A2 WO 2014008257A2 US 2013049060 W US2013049060 W US 2013049060W WO 2014008257 A2 WO2014008257 A2 WO 2014008257A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- oxadiazole
- water
- disubstituted
- reaction mixture
- Prior art date
Links
- 0 *c1n[o]c([Al])n1 Chemical compound *c1n[o]c([Al])n1 0.000 description 1
- JZVVIHHKORLLIR-UHFFFAOYSA-N Cc(cc1)cc(F)c1-c1n[o]c(-c2c[s]cc2)n1 Chemical compound Cc(cc1)cc(F)c1-c1n[o]c(-c2c[s]cc2)n1 JZVVIHHKORLLIR-UHFFFAOYSA-N 0.000 description 1
- NXXURVXTYOJCSP-UHFFFAOYSA-N Cc(cc1)ccc1-c1n[o]c(-c2ccc[o]2)n1 Chemical compound Cc(cc1)ccc1-c1n[o]c(-c2ccc[o]2)n1 NXXURVXTYOJCSP-UHFFFAOYSA-N 0.000 description 1
- AINKNWCODGPLKV-UHFFFAOYSA-N c1c[o]c(-c2nc(-c3ccccc3)n[o]2)c1 Chemical compound c1c[o]c(-c2nc(-c3ccccc3)n[o]2)c1 AINKNWCODGPLKV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/06—1,2,4-Oxadiazoles; Hydrogenated 1,2,4-oxadiazoles
Definitions
- 1,2,4-Oxadiazoles and in particular, 3,5-disubstituted-l,2,4-oxadiazoles, have been shown to have biological activity in pharmaceutical and agricultural fields.
- 3,5-disubstituted-l,2,4-oxadiazoles are disclosed as disease suppression and treatment agents (U.S. Pat. No. 6,992,096), as therapeutic agents for hepatitis C (Pub. No. US 2005/0075375 Al), and for nematode control in agriculture (Pub. No. US 2009/004831 1 Al).
- 3,5-Disubstituted-l,2,4-oxadiazoles can be prepared in a number of ways.
- Ar 1 is phenyl, pyridyl, pyrazyl, oxazolyl or isoxazolyl, each of which can be optionally independently substituted with one or more substituents e.g. alkyl, haloalkyl, alkoxy, haloalkoxy, halogen, acyl, ester, and nitrile; and Ar 2 is thienyl, furanyl, oxazolyl, isoxazolyl, or phenyl, each of which can be optionally independently substituted with one or more substituents e.g. fluorine, chlorine, CH 3 , and OCF 3 .
- the process comprises reacting a N- hydroxyamidine of Formula (Ila) or (lib), respectively, or a tautomeric form thereof, wherein
- Ar 1 and Ar 2 are defined as above,
- the present disclosure is directed to improved processes for the preparation of 3, 5 -disubstituted- 1,2,4-oxadiazoles and salts thereof.
- Various embodiments of the process enable greater ease of production, milder reaction conditions, reduced reaction time cycles, fewer reaction intermediates, and/or significantly reduced capital equipment requirements.
- Ar 1 is phenyl, pyridyl, pyrazyl, oxazolyl or isoxazolyl, each of which can be optionally independently substituted with one or more substituents.
- Ar 1 is optionally independently substituted with one or more substituents selected from the group consisting of alkyl, haloalkyl, alkoxy, haloalkoxy, halogen, acyl, ester, and nitrile each of which can be optionally independently substituted.
- Ar 1 is optionally independently substituted with one or more substituents selected from the group consisting of halogen, CF 3 , CH 3 , OCF 3 , OCH 3 , CN and C(H)0.
- Ar 2 is thienyl, furanyl, oxazolyl, isoxazolyl, or phenyl, each of which can be optionally independently substituted. In one embodiment, Ar 2 is optionally independently substituted with one or more substituents selected from the group consisting of fluorine, chlorine, CH 3 , and OCF 3 .
- Ar 1 is unsubstituted phenyl.
- Ar 1 is monosubstituted phenyl wherein the substituent is a halogen.
- Ar 1 is a disubstituted chloroalkylphenyl.
- Ar 2 is substituted thienyl or substituted furanyl.
- Ar 2 is unsubstituted thienyl or unsubstituted furanyl.
- the 3, 5 -disubstituted- 1,2,4-oxadiazole is a compound of Formula (Ia-i) or a salt thereof,
- R 1 and R 5 are independently selected from hydrogen, CH 3 , F, CI, Br, CF 3 and OCF 3 ;
- R 2 and R 4 are independently selected from hydrogen, F, CI, Br, and CF 3 ;
- R 3 is selected from hydrogen, CH 3 , CF 3 , F, CI, Br, OCF 3 , OCH 3 , CN, and C(H)0;
- R 7 and R 8 are independently selected from hydrogen and fluorine;
- R 9 is selected from hydrogen, F, CI, CH 3 , and OCF 3 ; and E is O, N or S.
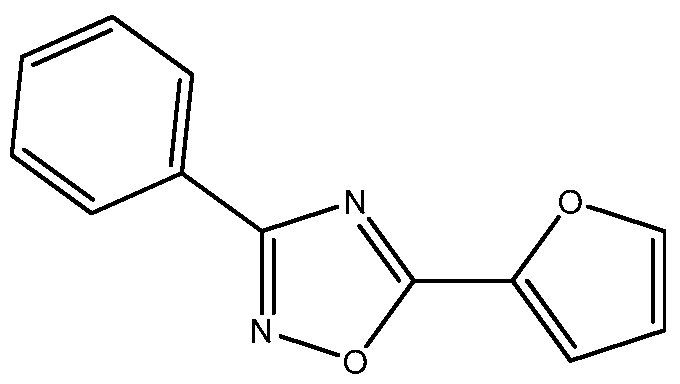
- Non-limiting examples of 3,5-disubstituted-l,2,4-oxadiazoles that can be prepared in accordance with the present disclosure include 3-phenyl-5-(2-thienyl)-l,2,4- oxadiazole of Formula (Ia-ii),
- the 3,5-disubstituted-l,2,4-oxadiazole is a compound of Formula (Ib-i) or a salt thereof,
- R 1 through R 5 , R 7 through R 9 and E are defined as above.
- 3,5-disubstituted-l,2,4-oxadiazoles that can be prepared in accordance with the present disclosure include 3-(thiophen-2-yl)-5-(p-tolyl)- 1,2,4-oxadiazole of Formul -ii),
- the process comprises reacting a N-hydroxyamidine of
- the condensation reaction of the N-hydroxyamidine with the acyl chloride is conducted in a reaction mixture comprising an organic solvent and an aqueous base.
- a reaction mixture comprising an organic solvent and an aqueous base.
- the reaction of the N-hydroxyamidine and the acyl chloride is believed to produce an oxime ester intermediate of Formula (IVa) or (IVb), a salt thereof, or a tautomeric form thereof,
- the presence of an aqueous base facilitates cyclization of the oxime ester at relatively low temperatures to produce the 3,5-disubstituted-l,2,4-oxadiazole product.
- the condensation and/or cyclization reaction is allowed to proceed directly to the final 3,5-disubstituted-l,2,4- oxadiazole product, without isolation or purification of the oxime ester intermediate.
- the oxime ester intermediate, or a portion thereof may be isolated and/or purified prior to formation of the 3,5-disubstituted-l,2,4-oxadiazole product.
- solvents used to form the reaction mixture are selected on the basis of one or more criteria, to facilitate simplification and overall economics of the process.
- the organic solvent employed is capable of solubilizing the N-hydroxyamidine of Formula (Ila) or (lib) and the 3,5-disubstituted-l,2,4- oxadiazole product of Formula (la) or (lb).
- the organic solvent is capable of solubilizing the intermediate oxime ester of Formula (IVa) or (IVb) as well.
- an organic solvent to solubilize the N-hydroxyamidine starting material as well as the oxime ester intermediate and the 3,5-disubstituted-l,2,4-oxadiazole product reduces or eliminates the need for isolation of the N-hydroxyamidine and/or the oxime ester, and reduces or eliminates the need for a solvent switch prior to cyclization to form the 3,5-disubstituted-l,2,4-oxadiazole product.
- the organic solvent may advantageously be used to transfer the N- hydroxyamidine reactant from one reaction vessel to another.
- a water-immiscible organic solvent is used for isolation of the N-hydroxyamidine of Formula (Ila) or (lib) and production of the 3,5-disubstituted-l,2,4-oxadiazole product.
- the use of a water-immiscible organic solvent allows for separation of the aqueous and organic phases of the reaction mixture, which enables a more expedient transfer of the N-hydroxyamidine reactant from one vessel to another and facilitates the recovery of the final 3,5-disubstituted-l,2,4-oxadiazole product, as set forth in greater detail below.
- the water-immiscible organic solvent forms an azeotrope with water.
- an azeotrope facilitates removal, via e.g. evaporation or distillation, of the solvent for purposes of isolation of the intermediate oxime ester or the 3,5-disubstituted-l,2,4-oxadiazole product.
- more than one organic solvent can be used, for example, a water-immiscible solvent for production of the oxime ester intermediate of Formula (IVa) or (IVb) and a water-miscible solvent for the production of the 3,5-disubstituted-l,2,4-oxadiazole product.
- Non-limiting examples of suitable organic solvents include acetone, 2-butanone, ethyl acetate, isopropyl acetate, butyl acetate, chloroform, dichloromethane, diethylether, methyl-tert-butyl ether, dibutylether, anisole, tetrahydrofuran, 2- methyltetrahydrofuran, xylene, and toluene.
- the organic solvent includes acetone, 2-butanone, tetrahydrofuran, 2-methyltetrahydrofuran, or butyl acetate.
- the organic solvent is butyl acetate or 2-methyltetrahydrofuran.
- the aqueous base comprises a chemical reagent which reacts with the acid byproduct of the condensation reaction between the N-hydroxyamidine and acyl chloride.
- aqueous base catalyzes the cyclization of the oxime ester of Formula (IVa) or (IVb) to produce the
- Aqueous base is defined herein as a strong base or a base that completely or almost completely dissociates in water. The use of a strong base may advantageously result in faster reaction times.
- the aqueous base is an alkali base comprising an alkali metal or alkaline earth metal, and which dissociates to form hydroxide ions in aqueous solution.
- suitable aqueous bases include aqueous solutions of inorganic bases such as sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide, and mixtures thereof.
- the aqueous base solution added to the reaction mixture comprises the base reagent in a concentration of from about 5% to about 60% by weight.
- the aqueous base is an alkaline hydroxide which reacts with the acid by-product of the condensation reaction between N-hydroxyamidine and acyl chloride to form water (water forms only from the hydroxide base) and a salt.
- the reaction mixture optionally comprises a phase transfer catalyst.
- a phase transfer catalyst is a chemical reagent that facilitates the migration of a reactant from the aqueous phase into the organic phase, or vice versa.
- phase transfer catalyst may advantageously result in faster reaction times and higher conversions or yields.
- the phase transfer catalyst is a quaternary ammonium salt, a phosphonium salt, or a crown ether.
- suitable phase transfer catalysts include tetrabutylammonium hydroxide, benzyltrimethylammonium hydroxide, tetrabutylammonium bromide, tetrabutylammonium chloride, tetrabutylammonium iodide, tetrabutylphosphonium bromide, and tetrabutylphosphonium chloride.
- the phase transfer catalyst is tetrabutylammonium hydroxide.
- the phase transfer catalyst is added to the reaction mixture in the form of an aqueous solution (e.g., a 40% by weight aqueous solution of the phase transfer catalyst) such that the final concentration is about 0.5-3 mol% based on acyl chloride, more preferably in a range of about 0.5-1.5 mol% or of about 1.5-2 mol% based on acyl chloride.
- an aqueous solution e.g., a 40% by weight aqueous solution of the phase transfer catalyst
- the N-hydroxyamidine of Formula (Ila) or (lib) is dissolved in a water-immiscible organic solvent and the solution is introduced into a reaction vessel.
- Water, at least a portion of the aqueous base, and the optional phase transfer catalyst are added to the reaction vessel. This may be simultaneous and/or sequentially. This results in the formation of a two-phase reaction mixture, in which the N-hydroxyamidine remains dissolved in the organic phase comprising a water-immiscible organic solvent.
- the acyl chloride reactant of Formula (Ilia) or (Illb) and any remaining portion of the aqueous base are added to the reaction vessel.
- the acyl chloride and the remaining portion of the aqueous base may be added simultaneously or sequentially.
- the N-hydroxyamidine is added to the reaction mixture in an amount equal to or in excess of the acyl chloride on a molar basis. Additional water may be added to the reaction mixture at any time during the course of the reaction.
- the mixture may be agitated.
- the reaction could be conducted continuously, for example, in a continuous stirred-tank reactor.
- the concentration of base in the aqueous phase of the reaction mixture is preferably sufficient to achieve a pH of at least about 8, at least about 9, or at least about 10 with higher pH resulting in increased reaction rate.
- a pH of from about 12 to about 13 is a pH range resulting in an increased reaction rate.
- the pH of the aqueous phase may be maintained or increased during the process by addition of additional aqueous base to the reaction mixture.
- the temperature of the reaction mixture is from about 25°C to about 85°C.
- the temperature of the reaction mixture is in some embodiments allowed to rise as a result of the reaction exotherm, and is preferably from about 55°C to about 75°C.
- the reaction may be allowed to proceed in the absence of external heat applied to the reaction mixture, although heat from an extraneous source may be supplied as needed to maintain the desired reaction temperature and rate.
- the rate of addition of the acyl chloride and the remaining aqueous base is dependent on reaction scale and ability to control the temperature.
- the rate of addition of acyl chloride may be between 30 min and 4 hours.
- the reaction mixture is advantageously allowed to heat as a result of the reaction exotherm, as described above, and may be maintained at from about 55°C to about 75°C for about 30 minutes to about 2 hours to allow the reaction to proceed to completion.
- the aqueous and organic phases are typically allowed to separate while hot.
- the aqueous phase may be optionally neutralized to a lower pH (e.g., pH lowered to less than about 9) to assist in subsequent formulation of the 3,5-disubstituted-l,2,4-oxadiazole product.
- a portion of the aqueous phase is removed from the reaction mixture and the organic solvent is removed by evaporation or distillation.
- the azeotrope, or pairing of the water and solvent assists in driving out traces of solvent.
- the distillation may be conducted at atmospheric pressure, or under vacuum. Additional water may be added to the reaction mixture in a volume sufficient to replace the distilled solvent and to facilitate the isolation of the final product.
- the 3,5-disubstituted- 1,2,4-oxadiazole product is precipitated in the aqueous layer, resulting in a slurry of solids in the aqueous layer.
- the solid 3,5- disubstituted- 1,2,4-oxadiazole product may be recovered by filtration, centrifugation, and/or decanting.
- the 3, 5 -disubstituted- 1,2,4-oxadiazole product may not consistently precipitate as a fine crystalline solid and may form beads (e.g., several mm in diameter), large irregular hard lumps, or encrusted solids on the reactor walls and agitator, which is a problem for discharging from the reactor and subsequent processing.
- beads e.g., several mm in diameter
- large irregular hard lumps e.g., large irregular hard lumps
- encrusted solids e.g., encrusted solids on the reactor walls and agitator
- One solution is to not remove the byproduct salt aqueous phase.
- high residual salt (e.g., NaCl) levels in the 3, 5-disubstituted- 1,2,4- oxadiazole product may cause problems in formulating the product.
- An alternative embodiment is to use a surfactant to stabilize droplets of product (possibly a metastable melt or a very concentrated solution) and allow the droplets to solidify as a precipitate in the aqueous layer without coalescing into larger droplets which otherwise ultimately may "crash out” as the large lumps and/or encrusted solids.
- Suitable surfactants may be selected from the group consisting of anionic or nonionic dispersants, anionic or nonionic detergents, anionic or nonionic surfactants, and combinations thereof.
- Non-limiting examples of suitable surfactants include Morwet D-425 (an anionic dispersant, an alkyl naphthalene sulfonate condensate sodium salt, from AkzoNobel), Morwet D-425 and Greenworks (sodium lauryl sulfate (anionic detergent), alkyl polyglucoside (nonionic surfactant), lauramine oxide (nonionic detergent), and glycerine), Morwet D-425 and Pluronic L-35 (a surfactant, a PEG- PPG-PEG block copolymer of polyethylene glycol-polypropylene glycol-polyethylene glycol, Mn ⁇ 1900), Pluronic L-35, and Triton X-100 (a nonionic detergent, an octylphenol ethoxylate (n ⁇ 10)).
- the surfactant is added to the reaction mixture after the reaction is substantially complete and prior to removal of at least a portion of the water- immiscible organic solvent
- the solvent may be recovered and recycled for re-use in the process (e.g., for use in dissolving the N-hydroxyamidine in a subsequent cycle of the process).
- the N-hydroxyamidine of Formula (Ila) or (lib) used in the preparation of 3,5- disubstituted-l,2,4-oxadiazole as described above can be obtained from commercial sources, or may be prepared using methods known to one skilled in the art.
- an arylamide oxime e.g.
- benzamide oxime or optionally independently substituted arylamide oxime may be prepared by reacting the corresponding aryl nitrile with hydroxylamine hydrochloride and an aqueous base.
- aqueous base for this purpose is sodium hydroxide.
- hydroxylamine hydrochloride is slurried in an alcoholic solvent, such as methanol or ethanol and is combined with an equivalent molar amount of the aqueous base.
- the aryl nitrile is added in an amount such that the hydroxylamine is in excess, for example, from about 1.01 to about 1.25 molar equivalents.
- the resulting reaction is mildly exothermic, and the resulting reaction mixture is heated to a temperature of from about 20°C to about 75°C, from about 20°C to about 65°C, from about 50°C to about 75°C, or from about 50°C to about 60°C.
- the aryl nitrile is added.
- the reaction mixture is then in some embodiments allowed to continue heating until the reaction is complete.
- the arylamide oxime product may be isolated for use in the process of the present disclosure.
- the alcoholic solvent may be removed by distillation, leaving a reaction mixture comprising a product melt phase and an aqueous phase. While the reaction mixture is still hot, an organic solvent as described above is added to dissolve the arylamide oxime product in an organic phase. The aqueous phase may then be separated and removed.
- the organic solvent satisfies the criteria for the organic solvent used in the production of the 3,5-disubstituted-l,2,4-oxadiazole, as set forth in detail above.
- the organic solvent phase comprising the dissolved arylamide oxime product may be forwarded directly to the condensation/cyclization reaction for production of the 3,5-disubstituted-l,2,4-oxadiazole without further processing.
- the methods described herein may be readily adapted to produce other N-hydroxyamidine compounds of Formula (Ila) or (lib).
- an arylamide oxime starting material e.g., benzamide oxime
- aqueous hydroxylamine includes but is not limited to aqueous hydroxylamine free base or aqueous hydroxylamine generated from the hydrochloride.
- a solvent can be used including an alcoholic solvent, such as methanol or ethanol.
- the solvent used may be a water-immiscible organic solvent or combination of solvents selected on the basis of one or more of the criteria as set forth in detail above.
- the organic solvent may be butyl acetate or 2-methyltetrahydrofuran.
- the arylamide oxime starting material is prepared by combining and reacting the corresponding aryl nitrile with aqueous hydroxylamine in the absence of a solvent, after the reaction is complete, the resulting product may be dissolved in an organic solvent as described above for further processing.
- the acyl chloride of Formula (Ilia) or (Illb) (e.g., 2-thiophenecarbonyl chloride or 2-furancarbonyl chloride), used in the processes for production of 3,5-disubstituted-l,2,4- oxadiazole as described above, can be obtained from commercial sources, or may be prepared using methods known to one skilled in the art.
- thiophene may be used to produce either 2-acetylthiophene or 2-thiophenecarboxaldehyde. Each of these may be used to produce a 2-thiophene carboxylic acid and, subsequently, 2-thiophenecarbonyl chloride.
- Other methods may be suitably employed to produce other acyl chloride compounds within the scope of Formula (III) or (Illb).
- Benzamide oxime (5.00 g, 36.7 mmol) was dissolved in acetone (50 mL) at 0°C. The solution was stirred and 50% aqueous sodium hydroxide (4.5 mL) added. An exotherm was noted and a precipitate formed in the reaction mixture. The exotherm lasted for about 15 minutes and the slurry became thick. The solution was warmed to 20°C and external cooling was added. The 2-thiophenecarbonyl chloride (5.36, 36.7 mmol) was added over 15 minutes maintaining the reaction mixture below 30°C. The reaction was maintained at 30°C for 15 minutes and the slurry became thinner. The reaction mixture was cooled to room temperature and stirred for 20 additional minutes.
- Benzonitrile (103.1 g, 1.00 moles) was added and the reaction was heated to 65-71°C for 4 hours. HPLC showed that the reaction was complete by the disappearance of benzonitrile. A portion of the methanol (366 mL) was removed by distillation, DI water was added (200 mL), and the remaining methanol was distilled at 200 torr to give two liquid phases (benzamide oxime melt and salt water). Butyl acetate (650 mL) and DI water (100 mL) were added, and the aqueous salt phase was drained off.
- Benzonitrile (50.0 g, 0.485 mol) was added to a 250 mL flask and heated to 70°C with stirring.
- Deionized water (1.0 g) was added and the temperature was lined out at 70°C.
- Aqueous hydroxylamine (50%, 37.50 g, 34.8 ml, assay was 53.3%, 0.605 moles) was added via a syringe pump over 1.5 hours while maintaining the temperature at 70° ⁇ 2°C.
- HPLC analysis indicated that the conversion of benzonitrile was >99% after about 2.5 hours of reaction time.
- the batch was heated for a total of 3.5 hours at 70°C, then was cooled to 60°C.
- the batch was transferred to a 500 mL flask with 2-methyltetrahydrofuran (208.98 g), followed by 20 wt % aqueous NaCl (40.0 g).
- the mixture was heated to 60°C, and the phases were separated at 60°C to give a solution of benzamide oxime in 2-methyltetrahydrofuran (281.80 g) and aqueous waste (44.61 g).
- the 2-methyltetrahydrofuran solution assayed for 21.73 wt% benzamide oxime.
- Hot deionized water (10 mL) was added to dissolve salts, followed by separation, washing with 10 mL of hot water and separation.
- Deionized water (50 mL) was added and the butyl acetate was removed by azeotropic distillation at 66-75°C and 300 torr. The 3-phenyl-5-(thiophen-2-yl)-l,2,4-oxadiazole precipitated during the distillation.
- benzyltrimethylammonium hydroxide 100 uL was added and the solution heated to 50°C.
- Benzamide oxime (2.00 g, 14.7 mmol) and a 40% aqueous solution of benzyltrimethylammonium hydroxide (100 uL) were combined in 2-methyltetrahydrofuran (20 mL) and water (2 mL).
- the solution was warmed to 50°C and 50% aqueous solution of sodium hydroxide (1.52 g, 19.1 mmol) and 2-furoyl chloride (2.00 g, 15.3 mmol) were added dropwise simultaneously over 10 min.
- the mixture was stirred at 60°C for 48 hours and the solids dissolved.
- the mixture was cooled to room temperature and poured into water (25 mL).
- embodiment 1 is a process for preparing a 3,5-disubstituted 1,2,4- oxadiazole of Formula (la), or a salt thereof,
- reaction mixture comprising a water-immiscible organic solvent and an aqueous base, wherein the temperature of the reaction mixture is no greater than about 85°C;
- Ar 1 is phenyl, pyridyl, pyrazyl, oxazolyl or isoxazolyl, each of which can be optionally independently substituted with one or more substituents selected from the group consisting of halogen, CF 3 , CH 3 , OCF 3 , OCH 3 , CN and C(H)0; and
- Ar 2 is thienyl, furanyl, oxazolyl or isoxazolyl, each of which can be optionally independently substituted with one or more substituents selected from the group consisting of fluorine, chlorine, CH 3 , and OCF 3 .
- Embodiment 2 is the process of embodiment 1 wherein the reaction mixture further comprises a phase transfer catalyst.
- Embodiment 3 is the process of embodiment 2 wherein the phase transfer catalyst is selected from the group consisting of quaternary ammonium salts, phosphonium salts, and crown ethers.
- Embodiment 4 is the process of embodiment 2 or 3 wherein the phase transfer catalyst is selected from the group consisting of tetrabutylammonium hydroxide,
- Embodiment 5 is the process of embodiment 4 wherein the phase transfer catalyst is tetrabutylammonium hydroxide.
- Embodiment 6 is the process of any one of embodiments 1 to 5 wherein the water-immiscible organic solvent solubilizes the N-hydroxyamidine of Formula (Ila) and the 3,5-disubstituted-l,2,4-oxadiazole of Formula (la).
- Embodiment 7 is the process of any one of embodiments 1 to 6 wherein the water-immiscible organic solvent forms an azeotrope with water.
- Embodiment 8 is the process of any one of embodiments 1 to 7 wherein the water-immiscible organic solvent is selected from the group consisting of 2- methyltetrahydrofuran and butyl acetate.
- Embodiment 9 is the process of embodiment 8 wherein the water- immiscible organic solvent is 2-methyltetrahydrofuran.
- Embodiment 10 is the process of any one of embodiments 1 to 9 wherein the aqueous base is selected from the group consisting of sodium hydroxide, potassium hydroxide, lithium hydroxide, and calcium hydroxide.
- Embodiment 1 1 is the process of embodiment 10 wherein the aqueous base is selected from the group consisting of sodium hydroxide and potassium hydroxide.
- Embodiment 12 is the process of any one of embodiments 1 to 11 wherein the reaction mixture comprises an organic phase and an aqueous phase.
- Embodiment 13 is the process of embodiment 12 wherein the pH of the aqueous phase is greater than about 8.
- Embodiment 14 is the process of embodiment 12 wherein the pH of the aqueous phase is greater than about 10.
- Embodiment 15 is the process of any one of embodiments 12 to 14 wherein the pH of the aqueous phase is increased or maintained by adding additional aqueous base to the reaction mixture.
- Embodiment 16 is the process of any one of embodiments 1 to 15 wherein the temperature of the reaction mixture is maintained at from about 55°C to about 75°C.
- Embodiment 17 is the process of any one of embodiments 1 to 15 wherein the N- hydroxyamidine of Formula (Ila) is dissolved in the water-immiscible organic solvent prior to adding the acyl chloride of Formula (Ilia) to form the reaction mixture.
- Embodiment 18 is the process of any one of embodiments 1 to 17 further comprising recovering the 3,5-disubstituted-l,2,4-oxadiazole of Formula (la) from the reaction mixture as a precipitate from an aqueous layer after removal of at least a portion of the water- immiscible organic solvent from the reaction mixture.
- Embodiment 19 is the process of embodiment 18 further comprising adding a surfactant to the reaction mixture.
- Embodiment 20 is the process of embodiment 19 wherein the surfactant added to the reaction mixture stabilizes droplets of the 3,5-disubstituted-l,2,4-oxadiazole of Formula (la) in an aqueous layer and allows the droplets to solidify without coalescing into larger droplets.
- Embodiment 21 is the process of embodiment 19 or 20 wherein the surfactant is added to the reaction mixture prior to removal of at least a portion of the water- immiscible organic solvent from the reaction mixture.
- Embodiment 22 is the process of any one of embodiments 19 to 21 wherein the surfactant is added to the reaction mixture after the reaction is substantially complete.
- Embodiment 23 is the process of any one of embodiments 19 to 22 wherein the surfactant is selected from the group consisting of anionic or nonionic dispersants, anionic or nonionic detergents, anionic or nonionic surfactants, and combinations thereof.
- the surfactant is selected from the group consisting of anionic or nonionic dispersants, anionic or nonionic detergents, anionic or nonionic surfactants, and combinations thereof.
- Embodiment 24 is the process of any one of embodiments 1 to 23 wherein the reaction of the N-hydroxyamidine of Formula (Ila) and the acyl chloride of Formula (Ilia) produces an oxime ester intermediate of Formula (IVa), a salt thereof, or a tautomeric form thereof,
- Ar 1 and Ar 2 are defined as in embodiment 1.
- Embodiment 25 is the process of embodiment 24 wherein the water-immiscible organic solvent solubilizes the N-hydroxyamidine of Formula (Ila), the oxime ester intermediate of Formula (IVa), and the 3,5-disubstituted 1,2,4-oxadiazole of Formula (la).
- Embodiment 26 is the process of embodiment 24 or 25 wherein the oxime ester intermediate of Formula (IVa) is isolated prior to formation of the 3,5-disubstituted 1,2,4- oxadiazole of Formula (la).
- Embodiment 27 is the process of any one of embodiments 1 to 26 wherein the N- hydroxyamidine of Formula (Ila) is a benzamide oxime formed by reacting a substituted or unsubstituted benzonitrile and hydroxylamine hydrochloride in an alcoholic solvent with sodium hydroxide and the benzamide oxime is separated from the alcohol solvent and subsequently dissolved in a solvent selected from the group consisting of 2-methyltetrahydrofuran and butyl acetate.
- a solvent selected from the group consisting of 2-methyltetrahydrofuran and butyl acetate selected from the group consisting of 2-methyltetrahydrofuran and butyl acetate.
- Embodiment 28 is the process of any one of embodiments 1 to 26 wherein the N- hydroxyamidine of Formula (Ila) is a benzamide oxime formed by reacting a substituted or unsubstituted benzonitrile and hydroxylamine and the formed benzamide oxime is dissolved in a solvent selected from the group consisting of 2-methyltetrahydrofuran and butyl acetate.
- Embodiment 29 is the process of embodiment 27 or 28 wherein the reaction to form the benzamide oxime of Formula (Ila) is carried out at a temperature of from about 20°C to about 75°C.
- Embodiment 30 is the process of any one of embodiments 1 to 26 wherein the N- hydroxyamidine of Formula (Ila) is a benzamide oxime formed by combining and reacting a substituted or unsubstituted benzonitrile and aqueous hydroxylamine.
- Embodiment 31 is the process of embodiment 30 wherein the substituted or unsubstituted benzonitrile and aqueous hydroxylamine are combined and reacted in the absence of a water- immiscible organic solvent.
- Embodiment 32 is the process of embodiment 30 wherein the substituted or unsubstituted benzonitrile and aqueous hydroxylamine are combined and reacted in a water- immiscible organic solvent and the benzamide oxime is dissolved in the solvent.
- Embodiment 33 is the process of embodiment 32 wherein the water-immiscible organic solvent is selected from the group consisting of 2-methyltetrahydrofuran and butyl acetate.
- Embodiment 34 is the process of any one of embodiments 1 to 33 wherein the process comprises:
- Embodiment 35 is the process of embodiment 34 wherein the water-immiscible organic solvent is selected from the group consisting of 2-methyltetrahydrofuran and butyl acetate.
- Embodiment 36 is the process of embodiment 34 or 35 wherein the acyl chloride and the additional aqueous base are added to the reaction vessel simultaneously.
- Embodiment 37 is the process of embodiment 34 or 35 wherein the acyl chloride and the additional aqueous base are added to the reaction vessel sequentially.
- Embodiment 38 is the process of any one of embodiments 1 to 37 wherein Ar 1 is unsubstituted phenyl.
- Embodiment 39 is the process of any one of embodiments 1 to 37 wherein Ar 1 is monosubstituted phenyl wherein the substituent is a halogen.
- Embodiment 40 is the process of any one of embodiments 1 to 37 wherein Ar 1 is a disubstituted chloroalkylphenyl.
- Embodiment 41 is the process of any one of embodiments 1 to 40 wherein Ar 2 is substituted or unsubstituted thiophene or furan.
- Embodiment 42 is the process of embodiment 41 wherein Ar 2 is unsubstituted thiophene.
- Embodiment 43 is the process of any one of embodiments 1 to 37 wherein the 3,5-disubstituted-l,2,4-oxadiazole of Formula (la) is 3-phenyl-5-(2-thienyl)-l,2,4-oxadiazole.
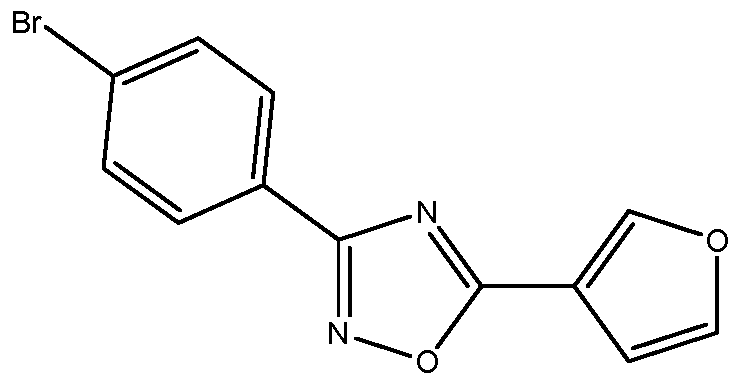
- Embodiment 44 is the process of any one of embodiments 1 to 37 wherein the 3,5-disubstituted-l,2,4-oxadiazole of Formula (la) is 3-(4-chlorophenyl)-5-(2-furanyl)-l,2,4- oxadiazole.
- Embodiment 45 is the process of any one of embodiments 1 to 37 wherein the 3,5-disubstituted-l,2,4-oxadiazole of Formula (la) is 3-(4-chloro-2-methylphenyl)-5-(2- furanyl)-l,2,4-oxadiazole.
- Embodiment 46 is the process of any one of embodiments 1 to 37 wherein the 3,5-disubstituted-l,2,4-oxadiazole of Formula (la) is 3-phenyl-5-(2-furanyl)-l,2,4-oxadiazole.
- Embodiment 47 is a process for preparing a 3,5-disubstituted 1,2,4-oxadiazole of Formula (lb), or a salt thereof
- reaction mixture comprising a water-immiscible organic solvent and an aqueous base, wherein the temperature of the reaction mixture is no greater than about 85°C;
- Ar 1 is phenyl, pyridyl, pyrazyl, oxazolyl or isoxazolyl, each of which can be optionally independently substituted with one or more substituents selected from the group consisting of halogen, CF 3 , CH 3 , OCF 3 , OCH 3 , CN and C(H)0; and
- Ar 2 is thienyl, furanyl, oxazolyl or isoxazolyl, each of which can be optionally independently substituted with one or more substituents selected from the group consisting of fluorine, chlorine, CH 3 , and OCF 3 .
- Embodiment 48 is the process of embodiment 47 wherein the reaction mixture further comprises a phase transfer catalyst.
- Embodiment 49 is the process of embodiment 48 wherein the phase transfer catalyst is selected from the group consisting of quaternary ammonium salts, phosphonium salts, and crown ethers.
- Embodiment 50 is the process of embodiment 48 or 49 wherein the phase transfer catalyst is selected from the group consisting of tetrabutylammonium hydroxide,
- Embodiment 51 is the process of embodiment 50 wherein the phase transfer catalyst is tetrabutylammonium hydroxide.
- Embodiment 52 is the process of any one of embodiments 47 to 51 wherein the water- immiscible organic solvent solubilizes the N-hydroxyamidine of Formula (lib) and the 3,5-disubstituted-l,2,4-oxadiazole of Formula (lb).
- Embodiment 53 is the process of any one of embodiments 47 to 52 wherein the water-immiscible organic solvent forms an azeotrope with water.
- Embodiment 54 is the process of any one of embodiments 47 to 53 wherein the water- immiscible organic solvent is selected from the group consisting of 2- methyltetrahydrofuran and butyl acetate.
- Embodiment 55 is the process of embodiment 54 wherein the water-immiscible organic solvent is 2-methyltetrahydrofuran.
- Embodiment 56 is the process of any one of embodiments 47 to 55 wherein the aqueous base is selected from the group consisting of sodium hydroxide, potassium hydroxide, lithium hydroxide, and calcium hydroxide.
- Embodiment 57 is the process of embodiment 56 wherein the aqueous base is selected from the group consisting of sodium hydroxide and potassium hydroxide.
- Embodiment 58 is the process of any one of embodiments 47 to 57 wherein the reaction mixture comprises an organic phase and an aqueous phase.
- Embodiment 59 is the process of embodiment 58 wherein the pH of the aqueous phase is greater than about 8.
- Embodiment 60 is the process of embodiment 58 wherein the pH of the aqueous phase is greater than about 10.
- Embodiment 61 is the process of any one of embodiments 58 to 60 wherein the pH of the aqueous phase is increased or maintained by adding additional aqueous base to the reaction mixture.
- Embodiment 62 is the process of any one of embodiments 47 to 61 wherein the temperature of the reaction mixture is maintained at from about 55°C to about 75°C.
- Embodiment 63 is the process of any one of embodiments 47 to 62 wherein the N-hydroxyamidine of Formula (lib) is dissolved in the water- immiscible organic solvent prior to adding the acyl chloride of Formula (Illb) to form the reaction mixture.
- Embodiment 64 is the process of any one of embodiments 47 to 63 further comprising recovering the 3,5-disubstituted-l,2,4-oxadiazole of Formula (lb) from the reaction mixture as a precipitate from an aqueous layer after removal of at least a portion of the water- immiscible organic solvent from the reaction mixture.
- Embodiment 65 is the process of embodiment 64 further comprising adding a surfactant to the reaction mixture.
- Embodiment 66 is the process of embodiment 65 wherein the surfactant added to the reaction mixture stabilizes droplets of the 3,5-disubstituted-l,2,4-oxadiazole of Formula (lb) in an aqueous layer and allows the droplets to solidify without coalescing into larger droplets.
- Embodiment 67 is the process of embodiment 65 or 66 wherein the surfactant is added to the reaction mixture prior to removal of at least a portion of the water- immiscible organic solvent from the reaction mixture.
- Embodiment 68 is the process of any one of embodiments 65 to 67 wherein the surfactant is added to the reaction mixture after the reaction is substantially complete.
- Embodiment 69 is the process of any one of embodiments 65 to 68 wherein the surfactant is selected from the group consisting of anionic or nonionic dispersants, anionic or nonionic detergents, anionic or nonionic surfactants, and combinations thereof.
- the surfactant is selected from the group consisting of anionic or nonionic dispersants, anionic or nonionic detergents, anionic or nonionic surfactants, and combinations thereof.
- Embodiment 70 is the process of any one of embodiments 47 to 69 wherein the reaction of the N-hydroxyamidine of Formula (lib) and the acyl chloride of Formula (Illb) produces an oxime ester intermediate of Formula (IVb), a salt thereof, or a tautomeric form thereof,
- Embodiment 71 is the process of embodiment 70 wherein the water-immiscible organic solvent solubilizes the N-hydroxyamidine of Formula (lib), the oxime ester intermediate of Formula (IVb), and the 3,5-disubstituted 1,2,4-oxadiazole of Formula (lb).
- Embodiment 72 is the process of embodiment 70 or 71 wherein the oxime ester intermediate of Formula (IVb) is isolated prior to formation of the 3,5-disubstituted 1,2,4- oxadiazole of Formula (lb).
- Embodiment 73 is the process of any one of embodiments 47 to 72 wherein the N-hydroxyamidine of Formula (lib) is a benzamide oxime formed by reacting a substituted or unsubstituted benzonitrile and hydroxylamine hydrochloride in an alcoholic solvent with sodium hydroxide and the benzamide oxime is separated from the alcohol solvent and subsequently dissolved in a solvent selected from the group consisting of 2-methyltetrahydrofuran and butyl acetate.
- a solvent selected from the group consisting of 2-methyltetrahydrofuran and butyl acetate selected from the group consisting of 2-methyltetrahydrofuran and butyl acetate.
- Embodiment 74 is the process of any one of embodiments 47 to 72 wherein the N-hydroxyamidine of Formula (lib) is a benzamide oxime formed by reacting a substituted or unsubstituted benzonitrile and hydroxylamine and the formed benzamide oxime is dissolved in a solvent selected from the group consisting of 2-methyltetrahydrofuran and butyl acetate.
- Embodiment 75 is the process of embodiment 73 or 74 wherein the reaction to form the benzamide oxime of Formula (lib) is carried out at a temperature of from about 20°C to about 75 °C.
- Embodiment 76 is the process of any one of embodiments 47 to 72 wherein the N-hydroxyamidine of Formula (lib) is a benzamide oxime formed by combining and reacting a substituted or unsubstituted benzonitrile and aqueous hydroxylamine.
- the N-hydroxyamidine of Formula (lib) is a benzamide oxime formed by combining and reacting a substituted or unsubstituted benzonitrile and aqueous hydroxylamine.
- Embodiment 77 is the process of embodiment 76 wherein the substituted or unsubstituted benzonitrile and aqueous hydroxylamine are combined and reacted in the absence of a water- immiscible organic solvent.
- Embodiment 78 is the process of embodiment 76 wherein the substituted or unsubstituted benzonitrile and aqueous hydroxylamine are combined and reacted in a water- immiscible organic solvent and the benzamide oxime is dissolved in the solvent.
- Embodiment 79 is the process of embodiment 78 wherein the water-immiscible organic solvent is selected from the group consisting of 2-methyltetrahydrofuran and butyl acetate.
- Embodiment 80 is the process of any one of embodiments 47 to 79 wherein the process comprises:
- Embodiment 81 is the process of embodiment 80 wherein the water-immiscible organic solvent is selected from the group consisting of 2-methyltetrahydrofuran and butyl acetate.
- Embodiment 82 is the process of embodiment 80 or 81 wherein the acyl chloride and the additional aqueous base are added to the reaction vessel simultaneously.
- Embodiment 83 is the process of embodiment 80 or 81 wherein the acyl chloride and the additional aqueous base are added to the reaction vessel sequentially.
- Embodiment 84 is the process of any one of embodiments 47 to 83 wherein Ar 1 is unsubstituted phenyl.
- Embodiment 85 is the process of any one of embodiments 47 to 83 wherein Ar 1 is monosubstituted phenyl wherein the substituent is a halogen.
- Embodiment 86 is the process of any one of embodiments 47 to 83 wherein Ar 1 is a disubstituted chloroalkylphenyl.
- Embodiment 87 is the process of any one of embodiments 47 to 86 wherein Ar 2 is substituted or unsubstituted thiophene or furan.
- Embodiment 88 is the process of embodiment 87 wherein Ar 2 is unsubstituted thiophene.
- Embodiment 89 is the process of any one of embodiments 47 to 83 wherein the 3,5-disubstituted-l,2,4-oxadiazole of Formula (lb) is 3-(thiophen-2-yl)-5-(p-tolyl)-l,2,4- oxadiazole.
- Embodiment 90 is the process of any one of embodiments 47 to 83 wherein the 3,5-disubstituted-l,2,4-oxadiazole of Formula (lb) is 5-(3-chlorophenyl)-3-(thiophen-2-yl)- 1,2,4-oxadiazole.
- Embodiment 91 is the process of any one of embodiments 47 to 83 wherein the 3,5-disubstituted-l,2,4-oxadiazole of Formula (lb) is 5-(4-chloro-2-methylphenyl)-3-(furan-2- yl)- 1 ,2,4-oxadiazole.
- Embodiment 92 is the process of embodiment 27 or 73 wherein the alcohol solvent is methanol.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description























Claims



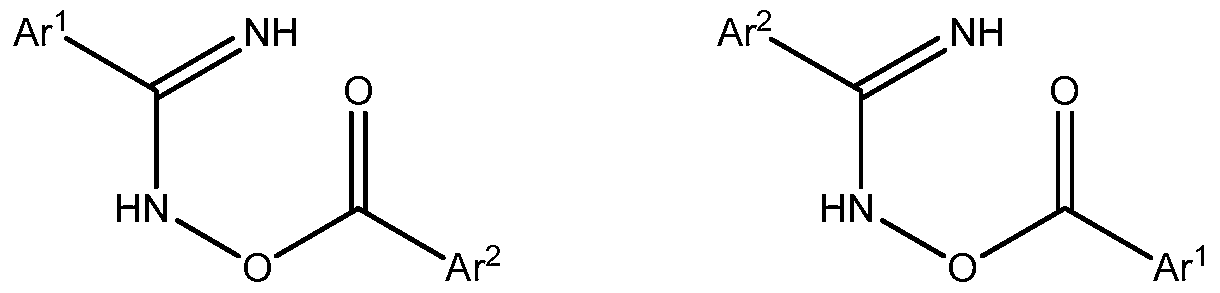
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201380033222.7A CN104428296B (en) | 2012-07-02 | 2013-07-02 | The method for preparing 3,5 two 1,2,4 oxadiazoles of substitution |
| UAA201500768A UA114915C2 (en) | 2012-07-02 | 2013-07-02 | Processes for the preparation of 3,5-disubstituted-1,2,4-oxadiazoles |
| MX2014015794A MX352351B (en) | 2012-07-02 | 2013-07-02 | Processes for the preparation of 3,5-disubstituted-1,2,4-oxadiazo les. |
| BR112014032956-7A BR112014032956B1 (en) | 2012-07-02 | 2013-07-02 | PROCESSES FOR THE PREPARATION OF 1,2,4-OXADIAZOILS 3,5-DISSUBSTITUED |
| DK13813965.4T DK2867230T3 (en) | 2012-07-02 | 2013-07-02 | METHOD OF PREPARING 3,5-DISUBSTITUTED 1,2,4-OXADIAZOLES |
| AU2013286860A AU2013286860B2 (en) | 2012-07-02 | 2013-07-02 | Processes for the preparation of 3,5-disubstituted-1,2,4-oxadiazoles |
| CA2876215A CA2876215A1 (en) | 2012-07-02 | 2013-07-02 | Processes for the preparation of 3,5-disubstituted-1,2,4-oxadiazoles |
| EP13813965.4A EP2867230B1 (en) | 2012-07-02 | 2013-07-02 | Process for the preparation of 3,5-disubstituted-1,2,4-oxadiazoles |
| ES13813965T ES2782509T3 (en) | 2012-07-02 | 2013-07-02 | Procedure for the preparation of 3,5-disubstituted 1,2,4-oxadiazoles |
| ZA2014/09000A ZA201409000B (en) | 2012-07-02 | 2014-12-08 | Processes for the preparation of 3,5-disubstituted-1,2,4-oxadiazoles |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261667361P | 2012-07-02 | 2012-07-02 | |
| US61/667,361 | 2012-07-02 | ||
| US201361777210P | 2013-03-12 | 2013-03-12 | |
| US61/777,210 | 2013-03-12 | ||
| US201361783466P | 2013-03-14 | 2013-03-14 | |
| US61/783,466 | 2013-03-14 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2014008257A2 true WO2014008257A2 (en) | 2014-01-09 |
| WO2014008257A3 WO2014008257A3 (en) | 2014-02-27 |
Family
ID=49882590
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2013/049060 WO2014008257A2 (en) | 2012-07-02 | 2013-07-02 | Processes for the preparation of 3,5-disubstituted-1,2,4-oxadiazoles |
Country Status (13)
| Country | Link |
|---|---|
| US (2) | US9040711B2 (en) |
| EP (1) | EP2867230B1 (en) |
| CN (1) | CN104428296B (en) |
| AR (1) | AR091655A1 (en) |
| AU (1) | AU2013286860B2 (en) |
| BR (1) | BR112014032956B1 (en) |
| CA (1) | CA2876215A1 (en) |
| DK (1) | DK2867230T3 (en) |
| ES (1) | ES2782509T3 (en) |
| MX (1) | MX352351B (en) |
| UA (1) | UA114915C2 (en) |
| WO (1) | WO2014008257A2 (en) |
| ZA (1) | ZA201409000B (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103980265A (en) * | 2014-05-14 | 2014-08-13 | 武汉工程大学 | Synthetic technology for 3-phenyl-5-(thiophene-2-yl)-1,2,4-oxadiazole |
| US9051309B2 (en) | 2013-03-15 | 2015-06-09 | Monsanto Technology Llc | 3,5-disubstituted-1,3,4-oxadiazol-2(3H)-ones and compositions and methods for controlling nematode pests |
| US9173401B2 (en) | 2013-03-15 | 2015-11-03 | Monsanto Technology Llc | N-,C-disubstituted azoles and compositions and methods for controlling nematode pests |
| WO2016143654A1 (en) * | 2015-03-06 | 2016-09-15 | 旭硝子株式会社 | Production method for 1,2,4-oxadiazole derivative |
| WO2016150937A1 (en) | 2015-03-25 | 2016-09-29 | Lonza Ltd | Method for preparation of thiophenecarbonyl chlorides |
| WO2017076844A1 (en) * | 2015-11-04 | 2017-05-11 | Lonza Ltd | Method for preparation of thiophene-2-carbonyl chlorides with oxalyl chloride |
| KR20170122795A (en) | 2015-03-06 | 2017-11-06 | 아사히 가라스 가부시키가이샤 | Method for producing 1,2,4-oxadiazole derivative |
| EP3433242A4 (en) * | 2016-03-24 | 2020-02-26 | Monsanto Technology LLC | Processes for the preparation of heteroaryl carboxylic acids |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HUE037661T2 (en) | 2007-08-13 | 2018-09-28 | Monsanto Technology Llc | Compositions and methods for controlling nematodes |
| UA118254C2 (en) | 2012-12-04 | 2018-12-26 | Монсанто Текнолоджи Ллс | NEMATOCIDAL WATER COMPOSITIONS OF SUSPENSION CONCENTRATE |
| WO2015179378A2 (en) | 2014-05-20 | 2015-11-26 | Monsanto Technology Llc | Methods for improving plant resistance to soybean cyst nematode and compositions thereof |
| UY36456A (en) * | 2014-12-19 | 2016-07-29 | Monsanto Technology Llc | COMPOSITIONS AND METHODS TO IMPROVE AGRICULTURAL CHARACTERISTICS OF PLANTS |
| AR104159A1 (en) * | 2015-03-31 | 2017-06-28 | Monsanto Technology Llc | PROCESSES TO PREPARE 2-THIOPHENOCARBONYL CHLORIDE |
| CN111675672A (en) * | 2020-05-12 | 2020-09-18 | 石家庄市度智医药科技有限公司 | Method for preparing astallurens |
Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3211742A (en) | 1962-04-17 | 1965-10-12 | Union Carbide Corp | Process for preparing oxadiazoles |
| US20030055085A1 (en) | 1999-08-19 | 2003-03-20 | Wagenen Bradford Van | Heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists |
| US20050075375A1 (en) | 2003-05-14 | 2005-04-07 | Anadys Pharmaceuticals, Inc. | Heterocyclic compounds for treating hepatitis C virus |
| US6992096B2 (en) | 2003-04-11 | 2006-01-31 | Ptc Therapeutics, Inc. | 1,2,4-oxadiazole benzoic acid compounds and their use for nonsense suppression and the treatment of disease |
| US7041685B2 (en) | 2001-06-08 | 2006-05-09 | Cytovia, Inc. | Substituted 3-aryl-5-aryl-[1,2,4]-oxadiazoles and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| US20080113961A1 (en) | 2005-04-22 | 2008-05-15 | Daiichi Sankyo Company, Limited | Heterocyclic compound |
| JP2008120794A (en) | 2006-10-16 | 2008-05-29 | Daiichi Sankyo Co Ltd | Pharmaceutical composition containing heterocyclic compound |
| US20080269236A1 (en) | 2006-12-12 | 2008-10-30 | Abbott Laboratories | Novel 1,2,4 Oxadiazole Compounds and Methods of Use Thereof |
| US20090048311A1 (en) | 2007-08-13 | 2009-02-19 | Williams Deryck J | Compostions and Methods for Controlling Nematodes |
| WO2009148452A1 (en) | 2008-06-06 | 2009-12-10 | Abbott Laboratories | Novel 1,2,4 oxadiazole compounds and methods of use thereof |
| US20100048648A1 (en) | 2006-09-07 | 2010-02-25 | Actelion Pharmaceuticals Ltd. | Thiophene derivatives as S1P1/EDGE1 receptor agonists |
| US7678922B2 (en) | 2006-09-08 | 2010-03-16 | Ptc Therapeutics, Inc. | Processes for the preparation of 1,2,4-oxadiazole benzoic acids |
| WO2011085406A1 (en) | 2010-01-11 | 2011-07-14 | Mithridion, Inc. | Compounds and compositions for cognition-enhancement, methods of making, and methods of treating |
| WO2011141326A1 (en) | 2010-05-08 | 2011-11-17 | Bayer Pharma Aktiengesellschaft | Substituted heterocyclyl benzyl pyrazoles, and use thereof |
| WO2012012477A1 (en) | 2010-07-20 | 2012-01-26 | Bristol-Myers Squibb Company | Substituted 3-phenyl-1,2,4-oxadiazole compounds |
| US20120022109A1 (en) | 2009-04-03 | 2012-01-26 | Merck Serono Sa | Oxadiazole derivatives |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL256329A (en) | 1959-09-29 | |||
| US3227725A (en) | 1962-04-17 | 1966-01-04 | Union Carbide Corp | Certain 3,5-disubstituted 1,2,4-oxadiazole compounds |
| NL291628A (en) | 1962-04-17 | |||
| US3218331A (en) | 1962-04-17 | 1965-11-16 | Union Carbide Corp | Preparation of substituted oxadiazoles |
| US3264318A (en) | 1964-04-29 | 1966-08-02 | Union Carbide Corp | Process for the production of substituted oxadiazoles |
| JPS62241946A (en) | 1986-04-14 | 1987-10-22 | Tonen Sekiyukagaku Kk | Thermoplastic resin composition |
| JPH03258779A (en) | 1990-03-06 | 1991-11-19 | Mitsui Toatsu Chem Inc | Imidazole derivative and anticonvulsant containing the same imidazole derivative as active ingredient |
| DE19725450A1 (en) | 1997-06-16 | 1998-12-17 | Hoechst Schering Agrevo Gmbh | 4-Haloalkyl-3-heterocyclylpyridines and 4-haloalkyl-5-heterocyclylpyrimidines, processes for their preparation, compositions containing them and their use as pesticides |
| CN100364531C (en) | 2002-12-18 | 2008-01-30 | 西托维亚公司 | 3,5-disubstituted-[1,2,4]-oxadiazoles and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| AU2005271669A1 (en) * | 2004-08-03 | 2006-02-16 | Merck & Co., Inc. | 1,3-disubstituted heteroaryl NMDA/NR2B antagonists |
| GB0601744D0 (en) | 2006-01-27 | 2006-03-08 | Novartis Ag | Organic compounds |
| WO2009041972A1 (en) | 2007-09-27 | 2009-04-02 | University Of Notre Dame Du Lac | Antibacterial compounds and methods of using same |
| GB0725102D0 (en) | 2007-12-21 | 2008-01-30 | Glaxo Group Ltd | Compounds |
| KR20110082569A (en) | 2008-11-14 | 2011-07-19 | 바이엘 쉐링 파마 악티엔게젤샤프트 | Heterocyclically substituted aryl compounds as hif inhibitors |
| EP2202232A1 (en) | 2008-12-26 | 2010-06-30 | Laboratorios Almirall, S.A. | 1,2,4-oxadiazole derivatives and their therapeutic use |
| WO2010093650A2 (en) | 2009-02-10 | 2010-08-19 | Divergence, Inc. | Compositions and methods for controlling nematodes |
| WO2010138600A2 (en) | 2009-05-29 | 2010-12-02 | Abbott Laboratories | Pharmaceutical compositions for the treatment of pain |
| AR076984A1 (en) | 2009-06-08 | 2011-07-20 | Merck Serono Sa | DERIVATIVES OF PIRAZOL OXADIAZOL |
| EP2585458B1 (en) | 2010-06-22 | 2015-01-07 | Chiesi Farmaceutici S.p.A. | Alkaloid aminoester derivatives and medicinal compositions thereof |
| ES2740965T3 (en) | 2010-09-02 | 2020-02-07 | Monsanto Technology Llc | New compositions and procedures against nematode pests |
-
2013
- 2013-07-02 CA CA2876215A patent/CA2876215A1/en not_active Abandoned
- 2013-07-02 DK DK13813965.4T patent/DK2867230T3/en active
- 2013-07-02 CN CN201380033222.7A patent/CN104428296B/en not_active Expired - Fee Related
- 2013-07-02 US US13/933,616 patent/US9040711B2/en not_active Expired - Fee Related
- 2013-07-02 MX MX2014015794A patent/MX352351B/en active IP Right Grant
- 2013-07-02 UA UAA201500768A patent/UA114915C2/en unknown
- 2013-07-02 EP EP13813965.4A patent/EP2867230B1/en active Active
- 2013-07-02 WO PCT/US2013/049060 patent/WO2014008257A2/en active Application Filing
- 2013-07-02 BR BR112014032956-7A patent/BR112014032956B1/en not_active IP Right Cessation
- 2013-07-02 AU AU2013286860A patent/AU2013286860B2/en not_active Ceased
- 2013-07-02 ES ES13813965T patent/ES2782509T3/en active Active
- 2013-07-02 AR ARP130102359 patent/AR091655A1/en active IP Right Grant
-
2014
- 2014-12-08 ZA ZA2014/09000A patent/ZA201409000B/en unknown
-
2015
- 2015-04-20 US US14/690,537 patent/US9273037B2/en not_active Expired - Fee Related
Patent Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3211742A (en) | 1962-04-17 | 1965-10-12 | Union Carbide Corp | Process for preparing oxadiazoles |
| US20030055085A1 (en) | 1999-08-19 | 2003-03-20 | Wagenen Bradford Van | Heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists |
| US7041685B2 (en) | 2001-06-08 | 2006-05-09 | Cytovia, Inc. | Substituted 3-aryl-5-aryl-[1,2,4]-oxadiazoles and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| US6992096B2 (en) | 2003-04-11 | 2006-01-31 | Ptc Therapeutics, Inc. | 1,2,4-oxadiazole benzoic acid compounds and their use for nonsense suppression and the treatment of disease |
| US20050075375A1 (en) | 2003-05-14 | 2005-04-07 | Anadys Pharmaceuticals, Inc. | Heterocyclic compounds for treating hepatitis C virus |
| US20080113961A1 (en) | 2005-04-22 | 2008-05-15 | Daiichi Sankyo Company, Limited | Heterocyclic compound |
| US20100048648A1 (en) | 2006-09-07 | 2010-02-25 | Actelion Pharmaceuticals Ltd. | Thiophene derivatives as S1P1/EDGE1 receptor agonists |
| US7678922B2 (en) | 2006-09-08 | 2010-03-16 | Ptc Therapeutics, Inc. | Processes for the preparation of 1,2,4-oxadiazole benzoic acids |
| JP2008120794A (en) | 2006-10-16 | 2008-05-29 | Daiichi Sankyo Co Ltd | Pharmaceutical composition containing heterocyclic compound |
| US20080269236A1 (en) | 2006-12-12 | 2008-10-30 | Abbott Laboratories | Novel 1,2,4 Oxadiazole Compounds and Methods of Use Thereof |
| US20090048311A1 (en) | 2007-08-13 | 2009-02-19 | Williams Deryck J | Compostions and Methods for Controlling Nematodes |
| WO2009148452A1 (en) | 2008-06-06 | 2009-12-10 | Abbott Laboratories | Novel 1,2,4 oxadiazole compounds and methods of use thereof |
| US20120022109A1 (en) | 2009-04-03 | 2012-01-26 | Merck Serono Sa | Oxadiazole derivatives |
| WO2011085406A1 (en) | 2010-01-11 | 2011-07-14 | Mithridion, Inc. | Compounds and compositions for cognition-enhancement, methods of making, and methods of treating |
| WO2011141326A1 (en) | 2010-05-08 | 2011-11-17 | Bayer Pharma Aktiengesellschaft | Substituted heterocyclyl benzyl pyrazoles, and use thereof |
| WO2012012477A1 (en) | 2010-07-20 | 2012-01-26 | Bristol-Myers Squibb Company | Substituted 3-phenyl-1,2,4-oxadiazole compounds |
Non-Patent Citations (5)
| Title |
|---|
| BIOORG. MED CHEM LETT., vol. 19, pages 4410 |
| KEMNITZER, W. ET AL., BIOORG. & MED. CHEM. LETTERS, vol. 19, no. 15, 2009 |
| LUKYANOV, S.M. ET AL., ARKIVOC, vol. IV, 2009 |
| See also references of EP2867230A4 |
| ZHANG, H.-Z. ET AL., J. MED. CHEM., vol. 48, no. 16, 2005 |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9763449B2 (en) | 2013-03-15 | 2017-09-19 | Monsanto Technology Llc | N-,C-disubstituted azoles and compositions and methods for controlling nematode pests |
| US9051309B2 (en) | 2013-03-15 | 2015-06-09 | Monsanto Technology Llc | 3,5-disubstituted-1,3,4-oxadiazol-2(3H)-ones and compositions and methods for controlling nematode pests |
| US9173401B2 (en) | 2013-03-15 | 2015-11-03 | Monsanto Technology Llc | N-,C-disubstituted azoles and compositions and methods for controlling nematode pests |
| US9402397B2 (en) | 2013-03-15 | 2016-08-02 | Monsanto Technology Llc | N-,C-disubstituted azoles and compositions and methods for controlling nematode pests |
| US10398144B2 (en) | 2013-03-15 | 2019-09-03 | Monsanto Technology Llc | N-,C-disubstituted azoles and compositions and methods for controlling nematode pests |
| CN103980265A (en) * | 2014-05-14 | 2014-08-13 | 武汉工程大学 | Synthetic technology for 3-phenyl-5-(thiophene-2-yl)-1,2,4-oxadiazole |
| KR20170122795A (en) | 2015-03-06 | 2017-11-06 | 아사히 가라스 가부시키가이샤 | Method for producing 1,2,4-oxadiazole derivative |
| US10035793B2 (en) | 2015-03-06 | 2018-07-31 | Asahi Glass Company, Limited | Method for producing 1,2,4-oxadiazole derivative |
| WO2016143654A1 (en) * | 2015-03-06 | 2016-09-15 | 旭硝子株式会社 | Production method for 1,2,4-oxadiazole derivative |
| WO2016150937A1 (en) | 2015-03-25 | 2016-09-29 | Lonza Ltd | Method for preparation of thiophenecarbonyl chlorides |
| WO2017076844A1 (en) * | 2015-11-04 | 2017-05-11 | Lonza Ltd | Method for preparation of thiophene-2-carbonyl chlorides with oxalyl chloride |
| KR20180037301A (en) * | 2015-11-04 | 2018-04-11 | 론자 리미티드 | Preparation of thiophene-2-carbonyl chloride using oxalyl chloride |
| KR101880881B1 (en) * | 2015-11-04 | 2018-07-20 | 론자 리미티드 | Preparation of thiophene-2-carbonyl chloride using oxalyl chloride |
| JP2018530519A (en) * | 2015-11-04 | 2018-10-18 | ロンザ・リミテッド | Method for preparing thiophene-2-carbonyl chlorides using oxalyl chloride |
| US10112921B2 (en) | 2015-11-04 | 2018-10-30 | Lonza Ltd | Method for preparation of thiophene-2-carbonyl chlorides with oxalyl chloride |
| EP3433242A4 (en) * | 2016-03-24 | 2020-02-26 | Monsanto Technology LLC | Processes for the preparation of heteroaryl carboxylic acids |
| US10745376B2 (en) | 2016-03-24 | 2020-08-18 | Monsanto Technology Llc | Processes for the preparation of heteroaryl carboxylic acids |
Also Published As
| Publication number | Publication date |
|---|---|
| CN104428296B (en) | 2017-11-14 |
| US9273037B2 (en) | 2016-03-01 |
| DK2867230T3 (en) | 2020-04-14 |
| BR112014032956B1 (en) | 2020-02-27 |
| MX2014015794A (en) | 2015-03-10 |
| WO2014008257A3 (en) | 2014-02-27 |
| EP2867230B1 (en) | 2020-02-12 |
| US9040711B2 (en) | 2015-05-26 |
| CA2876215A1 (en) | 2014-01-09 |
| MX352351B (en) | 2017-11-21 |
| CN104428296A (en) | 2015-03-18 |
| EP2867230A4 (en) | 2016-04-13 |
| UA114915C2 (en) | 2017-08-28 |
| ZA201409000B (en) | 2017-04-26 |
| BR112014032956A2 (en) | 2017-06-27 |
| AU2013286860B2 (en) | 2017-10-26 |
| US20140039197A1 (en) | 2014-02-06 |
| AR091655A1 (en) | 2015-02-18 |
| EP2867230A2 (en) | 2015-05-06 |
| US20150225382A1 (en) | 2015-08-13 |
| AU2013286860A1 (en) | 2015-01-22 |
| ES2782509T3 (en) | 2020-09-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9273037B2 (en) | Processes for the preparation of 3,5-disubstituted-1,2,4-oxadiazoles | |
| KR20130004257A (en) | Production method | |
| AU2006300422A1 (en) | Triarylcarboxylic acid derivative | |
| IL167315A (en) | Synthesis of thiophenecarboxylic acid esters for the production of ranelic acid salts | |
| CN104487436B (en) | Improved process for preparing rivaroxaban using intermediates | |
| TW200540160A (en) | Process for the production of 5-difluoromethoxy -4-thiomethylpyrazoles | |
| JP6559798B2 (en) | Process for preparing 2-thiophenecarbonyl chloride | |
| CN107936006B (en) | Synthetic method of rivaroxaban | |
| KR20010092799A (en) | Method for the preparation of citalopram | |
| KR100837582B1 (en) | Process for the manufacture of thiazole derivatives with pesticidal activity | |
| KR100519132B1 (en) | Processes for the preparation of thiazolidinedione derivatives and intermediates | |
| KR850000096B1 (en) | Process for preparing 5-substituted dialuric acid | |
| HU197003B (en) | Process for producing new isoxazole derivatives and pharmaceuticals comprising same | |
| JPH08501307A (en) | Synthesis of useful intermediates of substituted imidazole compounds | |
| EP2699560B1 (en) | 5-carbamoyl-adamantan-2-yl amide derivatives, pharmaceutically acceptable salts thereof and preparation process thereof | |
| CA2306910C (en) | Synthesis of 3-carbomethoxy-4,5-dimethylthiophene | |
| US3391154A (en) | Process for producing 5-methylisoxazole | |
| CN108250193B (en) | Method for preparing rivaroxaban by one-pot method | |
| Kumar et al. | Synthesis of novel 1, 3, 4-oxadiazole analogues with expected antibacterial activity | |
| US9950997B2 (en) | Method for preparing high-purity sulfonamide compound, and intermediate of sulfonamide compound | |
| JP2008525507A (en) | Process for producing substituted 2-alkoxycarbonyl-3-aminothiophenes | |
| JP2007506671A (en) | Method for producing insulin sensitizer and intermediate compound thereof | |
| WO2014121439A1 (en) | Synthesis method of thiadiazolylamide derivative | |
| US20090221834A1 (en) | Synthesis of 5-Beta-Keto-1,2,4-Oxadiazoles and Conversion of 5-Beta-Keto-1,2,4 Oxadiazoles to N-Pyrazolyl Amidoximes | |
| JP2004018480A (en) | Method for producing 5-substituted-1,2,4-triazole-3-carboxylic acid ester derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13813965 Country of ref document: EP Kind code of ref document: A2 |
|
| ENP | Entry into the national phase |
Ref document number: 2876215 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013813965 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2014/015794 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2013286860 Country of ref document: AU Date of ref document: 20130702 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201500768 Country of ref document: UA |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014032956 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112014032956 Country of ref document: BR Kind code of ref document: A2 Effective date: 20141230 |