WO2013154799A1 - Tricyclic nucleosides and oligomeric compounds prepared therefrom - Google Patents
Tricyclic nucleosides and oligomeric compounds prepared therefrom Download PDFInfo
- Publication number
- WO2013154799A1 WO2013154799A1 PCT/US2013/033151 US2013033151W WO2013154799A1 WO 2013154799 A1 WO2013154799 A1 WO 2013154799A1 US 2013033151 W US2013033151 W US 2013033151W WO 2013154799 A1 WO2013154799 A1 WO 2013154799A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- alkyl
- oligomeric compound
- group
- compound
- Prior art date
Links
- 0 CC(C(OC)(P)P)(C1(*)O*)C(P=C)=C(*)C(*)(*)C1(*)P* Chemical compound CC(C(OC)(P)P)(C1(*)O*)C(P=C)=C(*)C(*)(*)C1(*)P* 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
Definitions
- the present disclosure provides tricyclic nucleosides and oligomeric compounds prepared therefrom. More particularly, the tricyclic nucleosides provided herein comprise a tricyclic ribosyl sugar moiety having a bridge between the 4' and 2' ring carbon atoms and a further fused carbocyclic or heterocyclic ring at the 3' and 4' carbon atoms.
- the tricyclic nucleosides are expected to be useful for enhancing one or more properties of the oligomeric compounds they are
- the oligomeric compounds provided herein are expected to hybridize to a portion of a target RNA resulting in loss of normal function of the target RNA.
- Antisense technology is an effective means for reducing the expression of one or more specific gene products and can therefore prove to be uniquely useful in a number of therapeutic, diagnostic, and research applications.
- Chemically modified nucleosides are routinely used for incorporation into antisense sequences to enhance one or more properties such as for example nuclease resistance.
- One such group of chemical modifications includes bicyclic nucleosides wherein the furanose portion of the nucleoside includes a bridge connecting two atoms on the furanose ring thereby forming a bicyclic ring system.
- Such bicyclic nucleosides have various names including BNA's and LNA's for bicyclic nucleic acids or locked nucleic acids respectively.
- Tricyclo-DNA has also been prepared as monomers, has been incorporated into oligomeric compounds and has been evaluated in certain biological assays (see: Steffens et al, Helvetica Chimica Acta., 1997, 80, 2426-2439; Steffens et al, J. Am. Chem. Soc, 1997, 119, 11548-11549; Steffens et al, J. Org. Chem., 1999, 121, 3249-3255; Renneberg et al, J. Am. Chem. Soc, 2002, 124, 5993-6002; Ittig et al, Nucl. Acids Res., 2004, 32(1), 346-353; Ittig et al, Collection Symposium Series, 2005, 12, 8014-8023; and Ivanova et al, Oligonucleotides, 2007, 17, 54-65).
- novel tricyclic nucleosides and antisense oligomeric compounds prepared therefrom. More particularly, novel tricyclic nucleosides are provided wherein each furanose ring comprises a 4'-CH 2 -0-2' bridge and a further carbocyclic or heterocyclic ring fused to the 3' and 4' atoms. Oligomeric compounds comprising one or more of the tricyclic nucleosides are expected to be useful for modulating gene expression pathways, including those relying on mechanisms of action such as RNaseH, RNAi and dsRNA enzymes, as well as other antisense mechanisms based on target degradation or target occupancy.
- RNaseH RNAi and dsRNA enzymes
- tricyclic nucleosides are provided having Formula I:
- Bx is a heterocyclic base moiety
- one of Ti and T 2 is hydroxyl or a protected hydroxyl and the other of Ti and T 2 is H;
- T 3 is hydroxyl, a protected hydroxyl, an H-phosphonate or a phosphoramidite group
- Ri, R 2 , R 3 and R 4 are each, independently, H, hydroxyl, halogen, Ci-C 6 alkyl, substituted Ci- C 6 alkyl, Ci-C 6 alkoxy, substituted Ci-C 6 alkoxy, amino or substituted amino;
- R 5 is H, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, C 2 -C 6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted C 2 -C 6 alkynyl, substituted acyl, aryl, substituted aryl, a heterocyclic radical, a substituted heterocyclic radical or a protecting group;
- each Ji and J 2 is, independently, H, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, Ci-C 6 aminoalkyl or a protecting group;
- Bx is an optionally protected pyrimidine, substituted pyrimidine, purine or substituted purine. In certain embodiments, Bx is an optionally protected uracil, thymine, cytosine, 5-methylcytosine, adenine or guanine.
- one of Ti and T 2 is a 4,4'-dimethoxytrityl protected hydroxyl group.
- T 3 is diisopropylcyanoethoxy phosphoramidite.
- n is 0. In certain embodiments, m is 1.
- Q is O
- Q is S.
- Q is R 5 .
- R5 is H, Ci-C 6 alkyl or substituted Ci-C 6 alkyl. In certain embodiments, R5 is methyl.
- n is 1. In certain embodiments, n is 2.
- tricyclic nucleosides having the configuration of Formula la:
- tricyclic nucleosides are provided having the configuration of Formula la, Ti is 4,4'-dimethoxytrityl protected hydroxyl and T 3 is diisopropylcyanoethoxy phosphoramidite. In certain embodiments, tricyclic nucleosides are provided having the
- T 2 is 4,4'-dimethoxytrityl protected hydroxyl and T 3 is
- oligomeric compounds comprising at least one tricyclic nucleoside having Formula II:
- Bx is a heterocyclic base moiety
- T 4 and T5 is hydroxyl, a protected hydroxyl, a 5' terminal group or an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound and the other of T 4 and T 5 is H;
- T 6 is hydroxyl, a protected hydroxyl, a 3' terminal group or an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound;
- Ri, R 2 , R 3 and R 4 are each, independently, H, hydroxyl, halogen, Ci-C 6 alkyl, substituted Ci- C 6 alkyl, Ci-C 6 alkoxy, substituted Ci-C 6 alkoxy, amino or substituted amino;
- R5 is H, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, C 2 -C6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted C 2 -C 6 alkynyl, substituted acyl, aryl, substituted aryl, a heterocyclic radical, a substituted heterocyclic radical or a protecting group;
- each Ji and J 2 is, independently, H, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, Ci-C 6 aminoalkyl or a protecting group;
- T 4 , T5 and T 6 is an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound.
- Q is CRiR 2 CR 3 R 4 .
- one of Ri, R 2 , R 3 and R 4 is Ci-C 6 alkyl for each tricyclic nucleoside having Formula II.
- one of Ri, R 2 , R 3 and R 4 IS CH 3 for each tricyclic nucleoside having Formula II.
- one of Ri, R 2 , R 3 and R 4 IS substituted Ci-C 6 alkyl for each tricyclic nucleoside having Formula II.
- one of Ri and R 2 is Ci-C 6 alkyl or substituted Ci-C 6 alkyl and one of R 3 and R 4 IS Ci-C 6 alkyl or substituted Ci-C 6 alkyl for each tricyclic nucleoside having Formula II.
- each Q is CH 2 CH 2 .
- each m is 0. In certain embodiments, each m is 1.
- each Q is O. In certain embodiments, each Q is S.
- each Q is R 5 .
- each R 5 is H, Ci-C 6 alkyl or substituted Ci-C 6 alkyl. In certain embodiments, each R 5 is methyl.
- each n is 1. In certain embodiments, n is 2.
- oligomeric compounds comprising at least one tricyclic nucleoside wherein each tricyclic nucleoside has the configuration of Formula Ila:
- oligomeric compounds comprising at least one tricyclic nucleoside of Formula II or Ila wherein each tricyclic nucleoside is uniformly modified having the same stereochemistry wherein the heterocyclic base is variable.
- tricyclic nucleosides are identical except that they can have different heterocyclic bases and that they can be at terminal or internal positions within the oligomeric compound such that T 4 , T5 and T 6 may be the same or different between each tricyclic nucleoside.
- oligomeric compounds are provided wherein each internucleoside linking group between adjacent monomeric subunits is, independently, a phosphodiester internucleoside linking group or a phosphorothioate internucleoside linking group. In certain embodiments, oligomeric compounds are provided wherein each internucleoside linking group between adjacent monomeric subunits is a phosphorothioate internucleoside linking group
- oligomeric compounds comprising a first region having at least two contiguous tricyclic nucleosides having Formula II or Ila.
- oligomeric compounds comprising a first region having at least two contiguous tricyclic nucleosides having Formula II or Ila and further comprising a second region having at least two contiguous monomeric subunits wherein each monomeric subunit in the second region is a modified nucleoside different from the tricyclic nucleosides of said first region.
- oligomeric compounds comprising a first region having at least two contiguous tricyclic nucleosides having Formula II or Ila, a second region having at least two contiguous monomeric subunits wherein each monomeric subunit in the second region is a modified nucleoside different from the tricyclic nucleosides of said first region and further comprising a third region located between said first and second regions wherein each monomer subunit in the third region is independently, a nucleoside or a modified nucleoside that is different from each tricyclic nucleoside of said first region and each monomer subunit the second region.
- gapped oligomeric compounds comprising, an internal region of from 6 to 14 contiguous monomer subunits flanked on each side by an external region of from 1 to 5 contiguous monomer subunits wherein each monomer subunit in each external region is tricyclic nucleoside of Formula II or Ila, and each monomer subunit in the internal region is, independently, a nucleoside or modified nucleoside.
- the internal region comprises from about 8 to about 14 contiguous P-D-2'-deoxyribonucleosides.
- the internal region comprises from about 9 to about 12 contiguous ⁇ - ⁇ -2'- deoxyribonucleosides.
- methods of inhibiting gene expression comprising contacting a cell with an oligomeric compound as provided herein wherein said oligomeric compound comprises from about 8 to about 40 monomeric subunits and is complementary to a target RNA.
- the cell is in an animal.
- the cell is in a human.
- the target RNA is selected from mRNA, pre-mRNA and micro RNA.
- the target RNA is mRNA.
- the target RNA is human mRNA.
- the target RNA is cleaved thereby inhibiting its function.
- the method further comprises detecting the levels of target RNA.
- an in vitro method of inhibiting gene expression comprising contacting one or more cells or a tissue with an oligomeric compound as provided herein.
- oligomeric compounds are provided for use in an in vivo method of inhibiting gene expression said method comprising contacting one or more cells, a tissue or an animal with an oligomeric compound as provided herein.
- methods of preparing tricyclic nucleosides comprising: reacting a first reactant and a second reactant in the presence of a solvent; wherein the first reactant comprises a compound represented by formula V:
- each R13 and Ri 4 are indepdendently selected from the group consisting of H, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, C 2 -C6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted C 2 -C 6 alkynyl and a protecting group.
- composition wherein the
- composition comprises the compound of any of formulas I-V and a
- a method of modulating target mRNA in a cell comprising contacting the cell with an oligomeric compound according to any of formulas I- V or a pharmaceutical composition comprising an oligomeric compound according to any of formulas I-V.
- the cell is in vitro.
- the cell is in an animal. In certain embodiments, the animal is a human.
- novel tricyclic nucleosides and oligomeric compounds prepared therefrom are expected to be useful for enhancing one or more properties of the oligomeric compounds they are incorporated into such as for example nuclease resistance.
- the oligomeric compounds provided herein are expected to hybridize to a portion of a target RNA resulting in loss of normal function of the target RNA.
- the tricyclic nucleosides provided herein can be incorporated into antisense oligomeric compounds to reduce target RNA, such as messenger RNA, in vitro and in vivo.
- target RNA such as messenger RNA
- the reduction of target RNA is useful for inhibition of gene expression via numerous pathways. Such pathways include for example the steric blocking of transcription or translation and cleavage of mRNA via single or double stranded oligomeric compounds.
- the oligomeric compounds provided herein are also expected to be useful as primers and probes in diagnostic applications.
- oligomeric compounds comprising at least one of the tricyclic nucleosides provided herein are expected to be useful as aptamers which are oligomeric compounds capable of binding to aberrant proteins in an in vivo setting.
- tricyclic nucleosides are provided having Formula I:
- Bx is a heterocyclic base moiety
- one of Ti and T 2 is hydroxyl or a protected hydroxyl and the other of Ti and T 2 is H;
- T 3 is hydroxyl, a protected hydroxyl, an H-phosphonate or a phosphoramidite group
- Ri, R 2 , R 3 and R4 are each, independently, H, hydroxyl, halogen, Ci-C 6 alkyl, substituted Ci- C 6 alkyl, Ci-C 6 alkoxy, substituted Ci-C 6 alkoxy, amino or substituted amino;
- R5 is H, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, C 2 -C 6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted C 2 -C 6 alkynyl, substituted acyl, aryl, substituted aryl, a heterocyclic radical, a substituted heterocyclic radical or a protecting group;
- each Ji and J 2 is, independently, H, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, Ci-C 6 aminoalkyl or a protecting group;
- Bx is an optionally protected pyrimidine, substituted pyrimidine, purine or substituted purine.
- Bx is an optionally protected uracil, thymine, cytosine, 5- methylcytosine, adenine or guanine.
- one of Ti and T 2 is a 4,4'- dimethoxytrityl protected hydroxyl group.
- T 3 is diisopropylcyanoethoxy phosphoramidite.
- Ri, R 2 , R 3 and R4 are Ci-C 6 alkyl. In certain embodiments, at least one of Ri, R 2 , R 3 and R4 IS CH 3 . In certain embodiments, at least one of Ri, R 2 , R 3 and R4 IS substituted Ci-C 6 alkyl. In certain embodiments, one of Ri and R 2 is Ci-C 6 alkyl or substituted Ci-C 6 alkyl and one of R 3 and R4 IS Ci-C 6 alkyl or substituted Ci-C 6 alkyl. In certain embodiments, Ri, R 2 , R 3 and R4 are each H. In certain embodiments, m is 0. In certain
- m is 1. In certain embodiments, Q is O. In certain embodiments, Q is S. In certain embodiments, Q is R 5 . In certain embodiments, R5 is H, Ci-C 6 alkyl or substituted Ci-C 6 alkyl. In certain embodiments, R5 is methyl. In certain embodiments, n is 1. In certain embodiments, n is 2.
- tricyclic nucleosides of Formula I having the configuration of Formula la:
- Ti is 4,4'-dimethoxytrityl protected hydroxyl and T3 is
- T 2 is 4,4'-dimethoxytrityl protected hydroxyl and T3 is diisopropylcyanoethoxy phosphoramidite.
- oligomeric compounds having at least one tricyclic nucleoside having formula II:
- Bx is a heterocyclic base moiety
- T 4 and T5 is hydroxyl, a protected hydroxyl, a 5' terminal group or an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound and the other of T 4 and
- T 5 is H
- T 6 is hydroxyl, a protected hydroxyl, a 3' terminal group or an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound;
- Ri, R 2 , R 3 and R 4 are each, independently, H, hydroxyl, halogen, Ci-C 6 alkyl, substituted Ci- C 6 alkyl, Ci-C 6 alkoxy, substituted Ci-C 6 alkoxy, amino or substituted amino;
- R5 is H, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, C 2 -C 6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted C 2 -C 6 alkynyl, substituted acyl, aryl, substituted aryl, a heterocyclic radical, a substituted heterocyclic radical or a protecting group; wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, Ci-C 6 alkyl, C 2 -C6 alkenyl, C 2 -C 6 alkynyl, OJi, SJi,
- each Ji and J 2 is, independently, H, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, Ci-C 6 aminoalkyl or a protecting group;
- T 4 , T5 and T 6 is an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound.
- one of Ri, R 2 , R 3 and R4 IS Ci-C 6 alkyl for each tricyclic nucleoside having Formula II.
- one of Ri, R 2 , R 3 and R4 is CH 3 for each tricyclic nucleoside having Formula II.
- one of Ri, R 2 , R 3 and R4 is substituted Ci-C 6 alkyl for each tricyclic nucleoside having Formula II.
- one of Ri and R 2 is Ci-C 6 alkyl or substituted Ci-C 6 alkyl and one of R 3 and R4 IS Ci-C 6 alkyl or substituted Ci-C 6 alkyl for each tricyclic nucleoside having Formula II.
- each Q is CH 2 CH 2 .
- each m is 0.
- each m is 1.
- each Q is O.
- each Q is S.
- each Q is R 5 .
- each R 5 is H, Ci-C 6 alkyl or substituted Ci-C 6 alkyl.
- each R 5 is methyl.
- each n is i .
- each n is 2.
- oligomeric compounds comprising at least one tricyclic nucleoside of Formula II wherein each tricyclic nucleoside of Formula II has the configuration of Formula Ila:
- oligomeric compounds comprising at least one tricyclic nucleoside of Formula II or Ila wherein each tricyclic nucleoside is uniformly modified having the same stereochemistry wherein the heterocyclic base is variable.
- tricyclic nucleosides are identical except that they can have different heterocyclic bases and they can be at different positions such as internally or terminally within the oligomeric compound.
- T 4 , T5 and T 6 may be the same or different between one tricyclic nucleoside and the next.
- oligomeric compounds are provided wherein each internucleoside linking group between adjacent monomeric subunits is, independently, a phosphodiester
- internucleoside linking group or a phosphorothioate internucleoside linking group.
- essentially each internucleoside linking group between adjacent monomeric subunits is a phosphorothioate internucleoside linking group.
- oligomeric compounds comprising a first region having at least two contiguous tricyclic nucleosides having Formula II or Ila.
- oligomeric compounds comprising a second region having at least two contiguous monomeric subunits wherein each monomeric subunit in the second region is a modified nucleoside different from the tricyclic nucleosides of said first region.
- oligomeric compounds are provided comprising a third region located between said first and second regions wherein each monomer subunit in the third region is independently, a nucleoside or a modified nucleoside that is different from each tricyclic nucleoside of said first region and each monomer subunit the second region.
- oligomeric compounds comprising a gapped oligomeric compound having an internal region of from 6 to 14 contiguous monomer subunits flanked on each side by an external region of from 1 to 5 contiguous monomer subunits wherein each monomer subunit in each external region is tricyclic nucleoside of Formula II or Ila and each monomer subunit in the internal region is, independently, a nucleoside or modified nucleoside.
- said internal region comprises from about 8 to about 14 contiguous ⁇ - ⁇ -2'- deoxyribonucleosides.
- said internal region comprises from about 9 to about 12 contiguous P-D-2'-deoxyribonucleosides.
- tricyclic nucleosides having Formula III are provided:
- Bx is a heterocyclic base moiety
- each Zi, Z 2 , and Z 3 is independently selected from the group consisting of CH 2 -CH 2 ,
- CH CH, CR 7 R 8 , O, S, or R 9 ;
- each R 7 and R 8 are indepdendently selected from the group consisting of H, hydroxyl, protected hydroxyl, halogen, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, C 2 -C 6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted C 2 -C 6 alkynyl, Ci-C 6 alkoxy, Ci-C 6 substituted alkoxy, an alkyl ester, and a protecting group;
- R9 is H, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, C 2 -C 6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted C 2 -C 6 alkynyl or a protecting group;
- each Ji and J 2 is, independently, H, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, Ci-C 6 aminoalkyl or a protecting group;
- T 7 and T 8 is hydroxyl or a protected hydroxyl and the other of T 7 and T 8 is H; and T 9 is hydroxyl, a protected hydroxyl, an H-phosphonate or a phosphoramidite group.
- oligomeric compounds comprising at least one tricyclic nucleoside having Formula IV:
- Bx is a heterocyclic base moiety
- Ri 2 is H, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, C 2 -C 6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted C 2 -C 6 alkynyl or a protecting group;
- each Ji and J 2 is, independently, H, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, Ci-C 6 aminoalkyl or a protecting group;
- Tio and Tn is hydroxyl, a protected hydroxyl, a 5' terminal group or an
- internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound and the other of Tio and Tn is H;
- T 12 is hydroxyl, a protected hydroxyl, a 3' terminal group or an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound;
- Tio, Tn and Ti 2 is an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound.
- methods of preparing a tricyclic nucleoside comprising: reacting a first reactant and a second reactant in the presence of a solvent; wherein the first reactant comprises a compound represented by formula V:
- each Ri 3 and Ri 4 are indepdendently selected from the group consisting of H, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, C 2 -C 6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted C 2 -C 6 alkynyl and a protecting group.
- oligomeric compounds are provided comprising the protecting group is selected from the group consisting of benzyl, phenyl, naphthyl, triflate, trimethylsilyl, tert- butyldiphenylsilyl, tert-butyldimethylsilyl, and benzyl chloromethyl ether.
- the second reactant is an amino acid. In certain embodiments, the second reactant is L-proline.
- the solvent is selected from among a polar solvent, a polar aprotic solvent, a polar protic solvent, an apolar solvent, an apolar aprotic solvent, and an apolar protic solvent. In certain embodiments the solvent is selected from the group consisting of DMF and DMSO.
- compositions comprising the compound of any of Formulas I-IV and a pharmaceutically acceptable salt, carrier, or diluent.
- methods of modulating target mRNA in a cell comprising contacting the cell with an oligomeric compound according to any of Formulas I-IV or a pharmaceutical composition thereof.
- the cell is in vitro.
- the cell is in an animal.
- the animal is a human.
- incorporation of one or more of the tricyclic nucleosides, as provided herein, into an oligomeric compound is expected to enhance one or more desired properties of the resulting oligomeric compound.
- properties include without limitation stability, nuclease resistance, binding affinity, specificity, absorption, cellular distribution, cellular uptake, charge, pharmacodynamics and pharmacokinetics.
- the tricyclic nucleosides provided herein are incorporated into oligomeric compounds such that a motif results.
- the placement of tricyclic nucleosides into oligomeric compounds to provide particular motifs can enhance the desired properties of the resulting oligomeric compounds for activity using a particular mechanism such as RNaseH or RNAi.
- Such motifs include without limitation, gapmer motifs, hemimer motifs, blockmer motifs, uniformly fully modified motifs, positionally modified motifs and alternating motifs.
- a wide variety of internucleoside linkages can also be used including but not limited to phosphodiester and phosphorothioate internucleoside linkages which can be incorporated uniformly or in various combinations.
- the oligomeric compounds can further include a 5' and or 3' terminal group such as a conjugate or reporter group.
- a 5' and or 3' terminal group such as a conjugate or reporter group.
- the term "motif refers to the pattern created by the relative positioning of monomer subunits within an oligomeric compound wherein the pattern is determined by comparing the sugar moieties of the linked monomer subunits.
- the only determinant for the motif of an oligomeric compound is the differences or lack of differences between the sugar moieties.
- the internucleoside linkages, heterocyclic bases and further groups such as terminal groups are not considered when determining the motif of an oligomeric compound.
- alternating motif refers to an oligomeric compound comprising a contiguous sequence of linked monomer subunits wherein the monomer subunits have two different types of sugar moieties that alternate for essentially the entire sequence of the oligomeric compound.
- Oligomeric compounds having an alternating motif can be described by the formula: 5'-A(-L-B-L- A) n (-L-B) nn -3' where A and B are monomer subunits that have different sugar moieties, each L is, independently, an internucleoside linking group, n is from about 4 to about 12 and nn is 0 or 1.
- the heterocyclic base and internucleoside linkage is independently variable at each position.
- the motif further optionally includes the use of one or more other groups including but not limited to capping groups, conjugate groups and other 5' and or 3'-terminal groups. This permits alternating oligomeric compounds from about 9 to about 26 monomer subunits in length. This length range is not meant to be limiting as longer and shorter oligomeric compounds are also amenable to oligomeric compounds provided herein.
- each A or each B comprise tricyclic nucleosides as provided herein.
- the term "uniformly fully modified motif refers to an oligomeric compound comprising a contiguous sequence of linked monomer subunits that each have the same type of sugar moiety. The heterocyclic base and internucleoside linkage is independently variable at each position.
- the motif further optionally includes the use of one or more other groups including but not limited to capping groups, conjugate groups and other 5' and or 3'-terminal groups.
- the uniformly fully modified motif includes a contiguous sequence of tricyclic nucleosides.
- one or both of the 5' and 3 '-ends of the contiguous sequence of tricyclic nucleosides comprise 5' and or 3'-terminal groups such as one or more unmodified nucleosides.
- hemimer motif refers to an oligomeric compound comprising a contiguous sequence of monomer subunits that each have the same type of sugar moiety with a further short contiguous sequence of monomer subunits located at the 5' or the 3' end that have a different type of sugar moiety.
- the heterocyclic base and internucleoside linkage is independently variable at each position.
- the motif further optionally includes the use of one or more other groups including but not limited to capping groups, conjugate groups and other 5' and or 3'-terminal groups.
- a hemimer is an oligomeric compound of uniform sugar moieties further comprising a short region (1, 2, 3, 4 or about 5 monomer subunits) having uniform but different sugar moieties located on either the 3' or the 5' end of the oligomeric compound.
- the hemimer motif comprises a contiguous sequence of from about 10 to about 28 monomer subunits having one type of sugar moiety with from 1 to 5 or from 2 to about 5 monomer subunits having a second type of sugar moiety located at one of the termini.
- the hemimer is a contiguous sequence of from about 8 to about 20 ⁇ -D-2'- deoxyribonucleosides having from 1-12 contiguous tricyclic nucleosides located at one of the termini.
- the hemimer is a contiguous sequence of from about 8 to about 20 ⁇ -D-2'-deoxyribonucleosides having from 1-5 contiguous tricyclic nucleosides located at one of the termini. In certain embodiments, the hemimer is a contiguous sequence of from about 12 to about 18 ⁇ -D-2'-deoxyribonucleosides having from 1-3 contiguous tricyclic nucleosides located at one of the termini. In certain embodiments, the hemimer is a contiguous sequence of from about 10 to about 14 ⁇ -D-2'-deoxyribonucleosides having from 1-3 contiguous tricyclic nucleosides located at one of the termini.
- blockmer motif and “blockmer” refer to an oligomeric compound comprising an otherwise contiguous sequence of monomer subunits wherein the sugar moieties of each monomer subunit is the same except for an interrupting internal block of contiguous monomer subunits having a different type of sugar moiety.
- the heterocyclic base and internucleoside linkage is independently variable at each position of a blockmer.
- the motif further optionally includes the use of one or more other groups including but not limited to capping groups, conjugate groups and other 5' or 3'-terminal groups.
- a blockmer overlaps somewhat with a gapmer in the definition but typically only the monomer subunits in the block have non-naturally occurring sugar moieties in a blockmer and only the monomer subunits in the external regions have non-naturally occurring sugar moieties in a gapmer with the remainder of monomer subunits in the blockmer or gapmer being ⁇ - D-2'-deoxyribonucleosides or ⁇ -D-ribonucleosides.
- blockmers are provided herein wherein all of the monomer subunits comprise non-naturally occurring sugar moieties.
- positionally modified motif is meant to include an otherwise contiguous sequence of monomer subunits having one type of sugar moiety that is interrupted with two or more regions of from 1 to about 5 contiguous monomer subunits having another type of sugar moiety.
- Each of the two or more regions of from 1 to about 5 contiguous monomer subunits are independently uniformly modified with respect to the type of sugar moiety.
- each of the two or more regions have the same type of sugar moiety.
- each of the two or more regions have a different type of sugar moiety.
- each of the two or more regions independently, have the same or a different type of sugar moiety.
- the heterocyclic base and internucleoside linkage is independently variable at each position of a positionally modified oligomeric compound.
- the motif further optionally includes the use of one or more other groups including but not limited to capping groups, conjugate groups and other 5' or 3'- terminal groups.
- positionally modified oligomeric compounds are provided comprising a sequence of from 8 to 20 P-D-2'-deoxyribonucleosides that further includes two or three regions of from 2 to about 5 contiguous tricyclic nucleosides each.
- Positionally modified oligomeric compounds are distinguished from gapped motifs, hemimer motifs, blockmer motifs and alternating motifs because the pattern of regional substitution defined by any positional motif does not fit into the definition provided herein for one of these other motifs.
- the term positionally modified oligomeric compound includes many different specific substitution patterns.
- gapmer or “gapped oligomeric compound” refers to an oligomeric compound having two external regions or wings and an internal region or gap. The three regions form a contiguous sequence of monomer subunits with the sugar moieties of the external regions being different than the sugar moieties of the internal region and wherein the sugar moiety of each monomer subunit within a particular region is essentially the same. In certain embodiments, each monomer subunit within a particular region has the same sugar moiety.
- the gapmer is a symmetric gapmer and when the sugar moiety used in the 5 '-external region is different from the sugar moiety used in the 3 '-external region, the gapmer is an asymmetric gapmer.
- the external regions are small (each
- the monomer subunits independently 1, 2, 3, 4 or about 5 monomer subunits) and the monomer subunits comprise non- naturally occurring sugar moieties with the internal region comprising P-D-2'-deoxyribonucleosides.
- the external regions each, independently, comprise from 1 to about 5 monomer subunits having non-naturally occurring sugar moieties and the internal region comprises from 6 to 18 unmodified nucleosides.
- the internal region or the gap generally comprises ⁇ - ⁇ -2'- deoxyribonucleosides but can comprise non-naturally occurring sugar moieties.
- the heterocyclic base and internucleoside linkage is independently variable at each position of a gapped oligomeric compound.
- the motif further optionally includes the use of one or more other groups including but not limited to capping groups, conjugate groups and other 5' or 3'-terminal groups.
- the gapped oligomeric compounds comprise an internal region of P-D-2'-deoxyribonucleosides with one of the external regions comprising tricyclic nucleosides as disclosed herein. In certain embodiments, the gapped oligomeric compounds comprise an internal region of P-D-2'-deoxyribonucleosides with both of the external regions comprising tricyclic nucleosides as provided herein. In certain embodiments, gapped oligomeric compounds are provided herein wherein all of the monomer subunits comprise non-naturally occurring sugar moieties.
- gapped oligomeric compounds comprising one or two tricyclic nucleosides at the 5'-end, two or three tricyclic nucleosides at the 3'-end and an internal region of from 10 to 16 P-D-2'-deoxyribonucleosides. In certain embodiments, gapped oligomeric compounds are provided comprising one tricyclic nucleoside at the 5'-end, two tricyclic nucleosides at the 3 '-end and an internal region of from 10 to 16 P-D-2'-deoxyribonucleosides.
- gapped oligomeric compounds comprising one tricyclic nucleosides at the 5'-end, two tricyclic nucleosides at the 3'-end and an internal region of from 10 to 14 ⁇ - ⁇ -2'- deoxyribonucleosides.
- gapped oligomeric compounds are provided that are from about 10 to about 21 monomer subunits in length. In certain embodiments, gapped oligomeric compounds are provided that are from about 12 to about 16 monomer subunits in length. In certain embodiments, gapped oligomeric compounds are provided that are from about 12 to about 14 monomer subunits in length. In certain embodiments, gapped oligomeric compounds are provided that are from about 14 to about 16 monomer subunits in length.
- alkyl refers to a saturated straight or branched hydrocarbon radical containing up to twenty four carbon atoms.
- alkyl groups include without limitation, methyl, ethyl, propyl, butyl, isopropyl, n-hexyl, octyl, decyl, dodecyl and the like.
- Alkyl groups typically include from 1 to about 24 carbon atoms, more typically from 1 to about 12 carbon atoms (C1-C12 alkyl) with from 1 to about 6 carbon atoms being more preferred.
- the term "lower alkyl” as used herein includes from 1 to about 6 carbon atoms.
- Alkyl groups as used herein may optionally include one or more further substituent groups.
- alkenyl refers to a straight or branched hydrocarbon chain radical containing up to twenty four carbon atoms and having at least one carbon-carbon double bond.
- alkenyl groups include without limitation, ethenyl, propenyl, butenyl, l-methyl-2- buten-l-yl, dienes such as 1,3-butadiene and the like.
- Alkenyl groups typically include from 2 to about 24 carbon atoms, more typically from 2 to about 12 carbon atoms with from 2 to about 6 carbon atoms being more preferred.
- Alkenyl groups as used herein may optionally include one or more further substituent groups.
- alkynyl refers to a straight or branched hydrocarbon radical containing up to twenty four carbon atoms and having at least one carbon-carbon triple bond.
- alkynyl groups include, without limitation, ethynyl, 1-propynyl, 1-butynyl, and the like.
- Alkynyl groups typically include from 2 to about 24 carbon atoms, more typically from 2 to about 12 carbon atoms with from 2 to about 6 carbon atoms being more preferred.
- Alkynyl groups as used herein may optionally include one or more further substituent groups.
- acyl refers to a radical formed by removal of a hydroxyl group from an organic acid and has the general Formula -C(0)-X where X is typically aliphatic, alicyclic or aromatic. Examples include aliphatic carbonyls, aromatic carbonyls, aliphatic sulfonyls, aromatic sulfinyls, aliphatic sulfinyls, aromatic phosphates, aliphatic phosphates and the like. Acyl groups as used herein may optionally include further substituent groups.
- alicyclic refers to a cyclic ring system wherein the ring is aliphatic.
- the ring system can comprise one or more rings wherein at least one ring is aliphatic.
- Preferred alicyclics include rings having from about 5 to about 9 carbon atoms in the ring.
- Alicyclic as used herein may optionally include further substituent groups.
- aliphatic refers to a straight or branched hydrocarbon radical containing up to twenty four carbon atoms wherein the saturation between any two carbon atoms is a single, double or triple bond.
- An aliphatic group preferably contains from 1 to about 24 carbon atoms, more typically from 1 to about 12 carbon atoms with from 1 to about 6 carbon atoms being more preferred.
- the straight or branched chain of an aliphatic group may be interrupted with one or more heteroatoms that include nitrogen, oxygen, sulfur and phosphorus.
- Such aliphatic groups interrupted by heteroatoms include without limitation, polyalkoxys, such as polyalkylene glycols, polyamines, and polyimines. Aliphatic groups as used herein may optionally include further substituent groups.
- alkoxy refers to a radical formed between an alkyl group and an oxygen atom wherein the oxygen atom is used to attach the alkoxy group to a parent molecule.
- alkoxy groups include without limitation, methoxy, ethoxy, propoxy, isopropoxy, n- butoxy, sec-butoxy, tert-butoxy, n-pentoxy, neopentoxy, n-hexoxy and the like.
- Alkoxy groups as used herein may optionally include further substituent groups.
- aminoalkyl refers to an amino substituted C 1 -C 12 alkyl radical.
- the alkyl portion of the radical forms a covalent bond with a parent molecule.
- the amino group can be located at any position and the aminoalkyl group can be substituted with a further substituent group at the alkyl and/or amino portions.
- aralkyl and “arylalkyl,” refer to an aromatic group that is covalently linked to a C 1 -C 12 alkyl radical.
- the alkyl radical portion of the resulting aralkyl (or arylalkyl) group forms a covalent bond with a parent molecule. Examples include without limitation, benzyl, phenethyl and the like.
- Aralkyl groups as used herein may optionally include further substituent groups attached to the alkyl, the aryl or both groups that form the radical group.
- aryl and “aromatic,” refer to a mono- or poly cyclic carbocyclic ring system radicals having one or more aromatic rings.
- aryl groups include without limitation, phenyl, naphthyl, tetrahydronaphthyl, indanyl, idenyl and the like.
- Preferred aryl ring systems have from about 5 to about 20 carbon atoms in one or more rings.
- Aryl groups as used herein may optionally include further substituent groups.
- halo and “halogen,” refer to an atom selected from fluorine, chlorine, bromine and iodine.
- heteroaryl and “heteroaromatic,” refer to a radical comprising a mono- or poly-cyclic aromatic ring, ring system or fused ring system wherein at least one of the rings is aromatic and includes one or more heteroatoms. Heteroaryl is also meant to include fused ring systems including systems where one or more of the fused rings contain no heteroatoms.
- Heteroaryl groups typically include one ring atom selected from sulfur, nitrogen or oxygen.
- heteroaryl groups include without limitation, pyridinyl, pyrazinyl, pyrimidinyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isooxazolyl, thiadiazolyl, oxadiazolyl, thiophenyl, furanyl, quinolinyl, isoquinolinyl, benzimidazolyl, benzooxazolyl, quinoxalinyl and the like.
- Heteroaryl radicals can be attached to a parent molecule directly or through a linking moiety such as an aliphatic group or hetero atom.
- Heteroaryl groups as used herein may optionally include further substituent groups.
- heteroarylalkyl refers to a heteroaryl group as previously defined that further includes a covalently attached C1-C12 alkyl radical.
- the alkyl radical portion of the resulting heteroarylalkyl group is capable of forming a covalent bond with a parent molecule.
- heteroarylalkyl groups as used herein may optionally include further substituent groups on one or both of the heteroaryl or alkyl portions.
- heterocyclic radical refers to a radical mono-, or poly-cyclic ring system that includes at least one heteroatom and is unsaturated, partially saturated or fully saturated, thereby including heteroaryl groups. Heterocyclic is also meant to include fused ring systems wherein one or more of the fused rings contain at least one heteroatom and the other rings can contain one or more heteroatoms or optionally contain no heteroatoms.
- a heterocyclic radical typically includes at least one atom selected from sulfur, nitrogen or oxygen.
- heterocyclic radicals include, [l,3]dioxolanyl, pyrrolidinyl, pyrazolinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, piperidinyl, piperazinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl, isothiazolidinyl, quinoxalinyl, pyridazinonyl, tetrahydrofuryl and the like.
- Heterocyclic groups as used herein may optionally include further substituent groups.
- protecting group refers to a labile chemical moiety which is known in the art to protect reactive groups including without limitation, hydroxyl, amino and thiol groups, against undesired reactions during synthetic procedures.
- Protecting groups are typically used selectively and/or orthogonally to protect sites during reactions at other reactive sites and can then be removed to leave the unprotected group as is or available for further reactions.
- Protecting groups as known in the art are described generally in Greene's Protective Groups in Organic
- Groups can be selectively incorporated into oligomeric compounds as provided herein as precursors.
- an amino group can be placed into a compound as provided herein as an azido group that can be chemically converted to the amino group at a desired point in the synthesis.
- groups are protected or present as precursors that will be inert to reactions that modify other areas of the parent molecule for conversion into their final groups at an appropriate time. Further representative protecting or precursor groups are discussed in Agrawal et al, Protocols for Oligonucleotide Conjugates, Humana Press; New Jersey, 1994, 26, 1-72.
- orthogonal protected refers to functional groups which are protected with different classes of protecting groups, wherein each class of protecting group can be removed in any order and in the presence of all other classes (see, Barany et al, J. Am. Chem. Soc, 1977, 99, 7363- 7365; Barany et al, J. Am. Chem. Soc, 1980, 102, 3084-3095).
- Orthogonal protection is widely used in for example automated oligonucleotide synthesis.
- a functional group is deblocked in the presence of one or more other protected functional groups which is not affected by the deblocking procedure. This deblocked functional group is reacted in some manner and at some point a further orthogonal protecting group is removed under a different set of reaction conditions. This allows for selective chemistry to arrive at a desired compound or oligomeric compound.
- hydroxyl protecting groups include without limitation, acetyl, t-butyl, t- butoxymethyl, methoxymethyl, tetrahydropyranyl, 1-ethoxyethyl, l-(2-chloroethoxy)ethyl, p- chlorophenyl, 2,4-dinitrophenyl, benzyl, 2,6-dichlorobenzyl, diphenylmethyl, p-nitrobenzyl, bis(2- acetoxyethoxy)methyl (ACE), 2-trimethylsilylethyl, trimethylsilyl, triethylsilyl, t-butyldimethylsilyl, t-butyldiphenylsilyl, triphenylsilyl, [(triisopropylsilyl)oxy]methyl (TOM), benzoylformate, chloroacetyl, trichloroacetyl, trifluoroacetyl, pivaloyl, benzo
- hydroxyl protecting groups include without limitation, benzyl, 2,6-dichlorobenzyl, t-butyldimethylsilyl, t-butyl- diphenylsilyl, benzoyl, mesylate, tosylate, dimethoxytrityl (DMT), 9-phenylxanthine-9-yl (Pixyl) and 9-(p-methoxyphenyl)xanthine-9-yl (MOX).
- amino protecting groups include without limitation, carbamate-protecting groups, such as 2-trimethylsilylethoxycarbonyl (Teoc), 1 -methyl- l-(4-biphenylyl)ethoxycarbonyl (Bpoc), t-butoxycarbonyl (BOC), allyloxycarbonyl (Alloc), 9-fluorenylmethyloxycarbonyl (Fmoc), and benzyloxycarbonyl (Cbz); amide-protecting groups, such as formyl, acetyl, trihaloacetyl, benzoyl, and nitrophenylacetyl; sulfonamide-protecting groups, such as 2-nitrobenzenesulfonyl; and imine- and cyclic imide-protecting groups, such as phthalimido and dithiasuccinoyl.
- carbamate-protecting groups such as 2-trimethylsilylethoxycarbonyl (Teoc), 1 -methyl- l-
- thiol protecting groups include without limitation, triphenylmethyl (trityl), benzyl (Bn), and the like.
- the compounds described herein contain one or more asymmetric centers and thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)- or (S)-, a or ⁇ , or as (D)- or (L)- such as for amino acids. Included herein are all such possible isomers, as well as their racemic and optically pure forms. Optical isomers may be prepared from their respective optically active precursors by the procedures described above, or by resolving the racemic mixtures. The resolution can be carried out in the presence of a resolving agent, by chromatography or by repeated crystallization or by some combination of these techniques which are known to those skilled in the art.
- substituted and substituteduent group are meant to include groups that are typically added to other groups or parent compounds to enhance desired properties or provide other desired effects. Substituent groups can be protected or unprotected and can be added to one available site or to many available sites in a parent compound. Substituent groups may also be further substituted with other substituent groups and may be attached directly or via a linking group such as an alkyl or hydrocarbyl group to a parent compound.
- each R aa , R bb and R cc is, independently, H, an optionally linked chemical functional group or a further substituent group with a preferred list including without limitation, H, alkyl, alkenyl, alkynyl, aliphatic, alkoxy, acyl, aryl, aralkyl, heteroaryl, alicyclic, heterocyclic and heteroarylalkyl. Selected substituents within the compounds described herein are present to a recursive degree.
- recursive substituent means that a substituent may recite another instance of itself. Because of the recursive nature of such substituents, theoretically, a large number may be present in any given claim.
- One of ordinary skill in the art of medicinal chemistry and organic chemistry understands that the total number of such substituents is reasonably limited by the desired properties of the compound intended. Such properties include, by way of example and not limitation, physical properties such as molecular weight, solubility or logP, application properties such as activity against the intended target and practical properties such as ease of synthesis.
- Recursive substituents are an intended aspect of the invention.
- One of ordinary skill in the art of medicinal and organic chemistry understands the versatility of such substituents.
- stable compound and “stable structure” as used herein are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent. Only stable compounds are contemplated herein.
- nucleobase refers to unmodified or naturally occurring nucleobases which include, but are not limited to, the purine bases adenine (A) and guanine (G), and the pyrimidine bases thymine (T), cytosine (C) and uracil (U).
- heterocyclic base moiety refers to unmodified or naturally occurring nucleobases as well as modified or non-naturally occurring nucleobases and synthetic mimetics thereof (such as for example phenoxazines).
- a heterocyclic base moiety is any heterocyclic system that contains one or more atoms or groups of atoms capable of hydrogen bonding to a heterocyclic base of a nucleic acid.
- heterocyclic base moieties include without limitation modified nucleobases such as 5-methylcytosine (5-me-C), 5-hydroxymethyl cytosine, xanthine, hypoxanthine, 2-aminoadenine, 6-methyl and other alkyl derivatives of adenine and guanine, 2-propyl and other alkyl derivatives of adenine and guanine, 2-thiouracil, 2-thiothymine and 2-thiocytosine, 5- halouracil and cytosine, 5-propynyl (-C ⁇ C-CH 3 ) uracil and cytosine and other alkynyl derivatives of pyrimidine bases, 6-azo uracil, cytosine and thymine, 5-uracil (pseudouracil), 4-thiouracil, 8-halo, 8- amino, 8-thiol, 8-thioalkyl, 8-hydroxyl and other 8-substituted adenines and
- heterocyclic base moieties include without limitation tricyclic pyrimidines such as l,3-diazaphenoxazine-2-one, l,3-diazaphenothiazine-2-one and 9-(2- aminoethoxy)-l,3-diazaphenoxazine-2-one (G-clamp).
- Heterocyclic base moieties also include those in which the purine or pyrimidine base is replaced with other heterocycles, for example 7- deaza-adenine, 7-deazaguanosine, 2-aminopyridine and 2-pyridone.
- Further heterocyclic base moieties include without limitation those known to the art skilled (see for example: United States Patent No.
- sugar moiety refers to naturally occurring sugars having a furanose ring, synthetic or non-naturally occurring sugars having a modified furanose ring and sugar surrogates wherein the furanose ring has been replaced with a cyclic ring system such as for example a morpholino or hexitol ring system or a non-cyclic sugar surrogate such as that used in peptide nucleic acids.
- sugar moieties useful in the preparation of oligomeric compounds include without limitation, ⁇ -D-ribose, P-D-2'-deoxyribose, substituted sugars (such as 2', 5' and bis substituted sugars), 4'-S-sugars (such as 4'-S-ribose, 4'-S-2'-deoxyribose and 4 -S-2'- substituted ribose), bicyclic modified sugars (such as the 2'-0-CH 2 -4' or 2'-0-(CH 2 ) 2 -4' bridged ribose derived bicyclic sugars) and sugar surrogates (such as for example when the ribose ring has been replaced with a morpholino, a hexitol ring system or an open non-cyclic system).
- substituted sugars such as 2', 5' and bis substituted sugars
- 4'-S-sugars such as 4'-S-ribose, 4'-
- sugar substituent group refers to groups that are covalently attached to sugar moieties.
- examples of sugar substituent groups include without limitation 2'-F, 2'-allyl, 2'-amino, 2'-azido, 2'-thio, 2'-0-allyl, 2'-OCF 3 , 2'-O-Ci-Ci 0 alkyl, 2'- OCH 3 , 2'-0(CH 2 ) n CH 3 , 2'-OCH 2 CH 3 , 2'-0-(CH 2 ) 2 CH 3 , 2'-0-(CH 2 ) 2 -0-CH 3 (MOE), 2'- 0[(CH 2 ) n O] m CH 3 , 2'-0(CH 2 ) 2 SCH 3 , 2'-0-(CH 2 ) 3 -N(R p )(R q ), 2'-0(CH 2 ) n H 2 , 2'-0-(CH
- examples of sugar substituent groups include without limitation substituted silyl, an RNA cleaving group, a reporter group, an intercalator, a group for improving pharmacokinetic properties, or a group for improving the pharmacodynamic properties of an oligomeric compound, and other substituents having similar properties.
- oligomeric compounds include modifed nucleosides comprising 2'-MOE substituent groups (Baker et al, J. Biol. Chem., 1997, 272, 11944-12000).
- Sugar moieties can be substituted with combinations of sugar substituent groups including without limitation 2'-F-5'-methyl substituted nucleosides (see PCT International Application WO 2008/101157, published on 8/21/08 for other disclosed 5', 2'-bis substituted nucleosides). Other combinations are also possible, including without limitation, replacement of the ribosyl ring oxygen atom with S and further substitution at the 2'-position (see published U.S.
- Patent Application US2005-0130923, published on June 16, 2005) and 5'-substitution of a bicyclic nucleoside see PCT International Application WO 2007/134181, published on 11/22/07 wherein a 4'-CH 2 -0-2' bicyclic nucleoside is further substituted at the 5' position with a 5'-methyl or a 5'-vinyl group).
- nucleoside refers to a nucleobase-sugar combination.
- the two most common classes of such nucleobases are purines and pyrimidines.
- nucleotide refers to a nucleoside further comprising a modified or unmodified phosphate internucleoside linking group or a non-phosphate internucleoside linking group.
- the internucleoside linking group can be linked to either the 2', 3' or 5' hydroxyl moiety of the sugar.
- the phosphate and or a non-phosphate internucleoside linking groups are routinely used to covalently link adjacent nucleosides to one another to form a linear polymeric compound.
- PNA peptide nucleic acids
- the heterocyclic base at each position is maintained for hybridization to a nucleic acid target but the sugar and linkage is replaced with surrogate groups that are expected to function similar to native groups but have one or more enhanced properties.
- nucleoside mimetic is intended to include those structures used to replace the sugar and the base at one or more positions of an oligomeric compound.
- nucleoside mimetics include without limitation nucleosides wherein the heterocyclic base moiety is replaced with a phenoxazine moiety (for example the 9-(2-aminoethoxy)-l,3-diazaphenoxazine-2- one group, also referred to as a G-clamp which forms four hydrogen bonds when hybridized with a guanosine base) and further replacement of the sugar moiety with a group such as for example a morpholino, a cyclohexenyl or a bicyclo[3.1.0]hexyl.
- a group such as for example a morpholino, a cyclohexenyl or a bicyclo[3.1.0]hexyl.
- modified nucleoside is meant to include all manner of modified nucleosides that can be incorporated into an oligomeric compound using oligomer synthesis.
- the term is intended to include modifications made to a nucleoside such as modified stereochemical configurations, one or more substitutions, and deletion of groups as opposed to the use of surrogate groups which are described elsewhere herein.
- the term includes nucleosides having a furanose sugar (or 4'-S analog) portion and can include a heterocyclic base or can be an abasic nucleoside.
- modified nucleosides includes without limitation, substituted nucleosides (such as 2', 5', and/or 4' substituted nucleosides) 4'-S-modified nucleosides, (such as 4'- S-ribonucleosides, 4'-S-2'-deoxyribonucleosides and 4'-S-2'-substituted ribonucleosides), bicyclic modified nucleosides (such as for example, bicyclic nucleosides wherein the sugar moiety has a 2'- 0-CHR a -4' bridging group, wherein R a is H, alkyl or substituted alkyl) and base modified nucleosides.
- substituted nucleosides such as 2', 5', and/or 4' substituted nucleosides
- 4'-S-modified nucleosides such as 4'- S-ribonucleosides, 4'-S-2'-deoxyribonucleo
- the sugar can be modified with more than one of these modifications listed such as for example a bicyclic modified nucleoside further including a 5'-substitution or a 5' or 4' substituted nucleoside further including a 2' substituent.
- modified nucleoside also includes combinations of these modifications such as base and sugar modified nucleosides. These modifications are meant to be illustrative and not exhaustive as other modifications are known in the art and are also envisioned as possible modifications for the modified nucleosides described herein.
- the term "monomer subunit” is meant to include all manner of monomer units that are amenable to oligomer synthesis with one preferred list including monomer subunits such as ⁇ -D-ribonucleosides, P-D-2'-deoxyribnucleosides, modified nucleosides, including substituted nucleosides (such as 2', 5' and bis substituted nucleosides), 4'-S-modified nucleosides, (such as 4'-S- ribonucleosides, 4'-S-2'-deoxyribonucleosides and 4'-S-2'-substituted ribonucleosides), bicyclic modified nucleosides (such as bicyclic nucleosides wherein the sugar moiety has a 2'-0-CHR a -4' bridging group, wherein R a is H, alkyl or substituted alkyl), other modified nucleosides, nucleoside mimetics
- bicyclic nucleoside refers to a nucleoside comprising at least a bicyclic sugar moiety.
- bicyclic nucleosides include without limitation nucleosides having a furanosyl sugar that comprises a bridge between two of the non-geminal carbons, preferably the 4' and the 2' carbon atoms.
- oligomeric compounds provided herein include one or more 4' to 2' bridged bicyclic nucleosides.
- 4' to 2' bridged bicyclic nucleosides include but are not limited to one of formulae: 4'-(CH 2 )-0-2' (LNA); 4'-(CH 2 )- S-2'; 4'-(CH 2 ) 2 -0-2' (ENA); 4'-CH(CH 3 )-0-2' and 4'-CH(CH 2 OCH 3 )-0-2' (and analogs thereof see U.S.
- Each of the foregoing bicyclic nucleosides can be prepared having one or more stereochemical sugar configurations including for example a-L-ribofuranose and ⁇ -D-ribofuranose (see PCT international application PCT/DK98/00393, published on March 25, 1999 as WO 99/14226).
- the bridge of a bicyclic sugar moiety is , -[C(R a )(Rb)] n -,
- the bridge is 4'-CH 2 -2', 4'-(CH 2 )2-2', 4'-(CH 2 )3-2', 4'-CH 2 -0-2', 4'-(CH 2 )2-0-2', 4'-CH 2 -0-N(R)-2' and 4'-CH 2 - N(R)-0-2'- wherein each R is, independently, H, a protecting group or C1-C12 alkyl.
- bicyclic nucleosides are further defined by isomeric configuration.
- a nucleoside comprising a 4'-(CH 2 )-0-2' bridge may be in the a-L configuration or in the ⁇ -D configuration.
- a-L-methyleneoxy (4'-CH 2 -0-2') BNA's have been incorporated into antisense oligonucleotides that showed antisense activity (Frieden et al, Nucleic Acids Research, 2003, 21, 6365-6372).
- bicyclic nucleosides include those having a 4' to 2' bridge wherein such bridges include without limitation, a-L-4'-(CH 2 )-0-2', P-D-4'-CH 2 -0-2', 4'-(CH 2 )2-0-2', 4'- CH 2 -0-N(R)-2', 4'-CH 2 -N(R)-0-2', 4'-CH(CH 3 )-0-2', 4'-CH 2 -S-2', 4'-CH 2 -N(R)-2', 4'-CH 2 - CH(CH 3 )-2', and 4'-(CH 2 ) 3 -2', wherein R is H, a protecting group or C1-C12 alkyl.
- bicyclic nucleosides have the formula:
- Bx is a heterocyclic base moiety
- Rc is C1-C12 alkyl or an amino protecting group
- T a and Tb are each, independently H, a hydroxyl protecting group, a conjugate group, a reactive phosphorus group, a phosphorus moiety or a covalent attachment to a support medium.
- bicyclic nucleosides have the formula:
- Bx is a heterocyclic base moiety
- T a and Tb are each, independently H, a hydroxyl protecting group, a conjugate group, a reactive phosphorus group, a phosphorus moiety or a covalent attachment to a support medium;
- Z a is Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted Ci-C 6 alkyl, substituted C 2 -C 6 alkenyl, substituted C 2 -C 6 alkynyl, acyl, substituted acyl, substituted amide, thiol or substituted thiol.
- bicyclic nucleosides have the formula:
- Bx is a heterocyclic base moiety
- T a and Tb are each, independently H, a hydroxyl protecting group, a conjugate group, a reactive phosphorus group, a phosphorus moiety or a covalent attachment to a support medium;
- bicyclic nucleosides have the formula:
- T a and Tb are each, independently H, a hydroxyl protecting group, a conjugate group, a reactive phosphorus group, a phosphorus moiety or a covalent attachment to a support medium;
- Rd is Ci-C 6 alkyl, substituted Ci-C 6 alkyl, C 2 -C 6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl or substituted C 2 -C 6 alkynyl;
- each q a , qb, q c and qd is, independently, H, halogen, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, C 2 -
- Ci-C 6 alkenyl substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl or substituted C 2 -C 6 alkynyl, Ci-C 6 alkoxyl, substituted Ci-C 6 alkoxyl, acyl, substituted acyl, Ci-C 6 aminoalkyl or substituted Ci-C 6 aminoalkyl;
- bicyclic nucleosides have the formula:
- Bx is a heterocyclic base moiety
- T a and Tb are each, independently H, a hydroxyl protecting group, a conjugate group, a reactive phosphorus group, a phosphorus moiety or a covalent attachment to a support medium;
- q g and q h are each, independently, H, halogen, Ci-Ci 2 alkyl or substituted Ci-Ci 2 alkyl.
- bicyclic nucleosides have the formula:
- Bx is a heterocyclic base moiety
- bicyclic nucleosides include, but are not limited to, (A) a-L- methyleneoxy (4'-CH 2 -0-2') BNA , (B) ⁇ -D-methyleneoxy (4'-CH 2 -0-2') BNA , (C) ethyleneoxy (4'-(CH 2 )2-0-2') BNA , (D) aminooxy (4'-CH 2 -0-N(R)-2') BNA, (E) oxyamino (4'-CH 2 -N(R)-0- 2') BNA, (F) methyl(methyleneoxy) (4'-CH(CH 3 )-0-2') BNA (also referred to as constrained ethyl or cEt), (G) methylene-thio (4'-CH 2 -S-2') BNA, (H) methylene-amino (4'-CH 2 -N(R)-2') BNA, (I) methyl carbocyclic (4'-CH 2
- Bx is the base moiety and R is, independently, H, a protecting group, Ci-C 6 alkyl or Ci-C 6 alkoxy.
- sugar surrogate refers to replacement of the nucleoside furanose ring with a non-furanose (or 4'-substituted furanose) group with another structure such as another ring system or open system.
- Such structures can be as simple as a six membered ring as opposed to the five membered furanose ring or can be more complicated such as a bicyclic or tricyclic ring system or a non-ring system used in peptide nucleic acid.
- sugar surrogates include without limitation sugar surrogate groups such as morpholinos, cyclohexenyls and cyclohexitols.
- nucleosides having sugar surrogate groups include without limitation, replacement of the ribosyl ring with a sugar surrogate such as a tetrahydropyranyl ring system (also referred to as hexitol) as illustrated below:
- sugar surrogates are selected having the formula:
- Bx is a heterocyclic base moiety
- T 3 and T 4 are each, independently, an internucleoside linking group linking the
- internucleoside linking group linking the tetrahydropyran nucleoside analog to an oligomeric compound or oligonucleotide and the other of T3 and T 4 is H, a hydroxyl protecting group, a linked conjugate group or a 5' or 3'-terminal group;
- qi, q 2 , q3, q 4 , qs, q 6 and q 7 are each independently, H, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, C 2 -C6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl or substituted C 2 -C 6 alkynyl; and
- qi, q 2 , q3, q 4 , qs, q 6 and q 7 are each H. In certain embodiments, at least one of qi, q 2 , q3, q 4 , qs, q 6 and q 7 is other than H. In certain embodiments, at least one of qi, q 2 , q3, q 4 , qs, q 6 and q 7 is methyl.
- THP nucleosides are provided wherein one of Ri and R 2 is F. In certain embodiments, Ri is fluoro and R 2 is H; Ri is methoxy and R 2 is H, and Ri is methoxyethoxy and R 2 is H.
- Modified THP nucleosides include, but are not limited to, what is referred to in the art as hexitol nucleic acid (HNA), altritol nucleic acid (ANA), and mannitol nucleic acid (MNA) (see Leumann, C. J., Bioorg. & Med. Chem., 2002, 10, 841-854).
- HNA hexitol nucleic acid
- ANA altritol nucleic acid
- MNA mannitol nucleic acid
- oligomeric compounds comprise one or more modified
- cyclohexenyl nucleosides which is a nucleoside having a six-membered cyclohexenyl in place of the pentofuranosyl residue in naturally occurring nucleosides.
- Modified cyclohexenyl nucleosides include, but are not limited to those described in the art (see for example commonly owned, published PCT Application WO 2010/036696, published on April 10, 2010, Robeyns et al, J. Am. Chem. Soc, 2008, 130(6), 1979-1984; Horvath et al, Tetrahedron Letters, 2007, 48, 3621-3623; Nauwelaerts et al, J. Am. Chem. Soc, 2007, 129(30), 9340-9348; Gu et al.,, Nucleosides,
- Bx is a heterocyclic base moiety
- tricyclic nucleosides provided herein can be prepared by any of the applicable techniques of organic synthesis, as, for example, illustrated in the examples below. Many such techniques are well known in the art. However, many of the known techniques are elaborated in
- reactive phosphorus is meant to include groups that are covalently linked to a monomer subunit that can be further attached to an oligomeric compound that are useful for forming internucleoside linkages including for example phosphodiester and phosphorothioate internucleoside linkages.
- Such reactive phosphorus groups are known in the art and contain phosphorus atoms in P m or P v valence state including, but not limited to, phosphoramidite, H- phosphonate, phosphate triesters and phosphorus containing chiral auxiliaries.
- reactive phosphorus groups are selected from diisopropylcyanoethoxy
- a preferred synthetic solid phase synthesis utilizes phosphoramidites (P in chemistry) as reactive phosphites.
- the intermediate phosphite compounds are subsequently oxidized to the phosphate or thiophosphate (P v chemistry) using known methods to yield, phosphodiester or phosphorothioate internucleoside linkages.
- oligonucleotide refers to a compound comprising a plurality of linked nucleosides. In certain embodiments, one or more of the plurality of nucleosides is modified. In certain embodiments, an oligonucleotide comprises one or more ribonucleosides (RNA) and/or deoxyribonucleosides (DNA).
- RNA ribonucleosides
- DNA deoxyribonucleosides
- oligonucleoside refers to a sequence of nucleosides that are joined by
- Internucleoside linkages that do not have phosphorus atoms.
- Internucleoside linkages of this type include short chain alkyl, cycloalkyl, mixed heteroatom alkyl, mixed heteroatom cycloalkyl, one or more short chain heteroatomic and one or more short chain heterocyclic.
- internucleoside linkages include without limitation, siloxane, sulfide, sulfoxide, sulfone, acetyl, formacetyl, thioformacetyl, methylene formacetyl, thioformacetyl, alkeneyl, sulfamate, methyleneimino, methylenehydrazino, sulfonate, sulfonamide, amide and others having mixed N, O, S and CH 2 component parts.
- oligomeric compound refers to a contiguous sequence of linked monomer subunits. Each linked monomer subunit normally includes a heterocyclic base moiety but monomer subunits also includes those without a heterocyclic base moiety such as abasic monomer subunits. At least some and generally most if not essentially all of the heterocyclic bases in an oligomeric compound are capable of hybridizing to a nucleic acid molecule, normally a preselected RNA target.
- the term “oligomeric compound” therefore includes oligonucleotides, oligonucleotide analogs and oligonucleosides. It also includes polymers having one or a plurality of nucleoside mimetics and or nucleosides having sugar surrogate groups.
- oligomeric compounds comprise a plurality of monomer subunits independently selected from naturally occurring nucleosides, non-naturally occurring nucleosides, modified nucleosides, nucleoside mimetics, and nucleosides having sugar surrogate groups.
- oligomeric compounds are single stranded.
- oligomeric compounds are double stranded comprising a double-stranded duplex.
- oligomeric compounds comprise one or more conjugate groups and/or terminal groups.
- oligomeric compounds having specific motifs as disclosed herein it can be advantageous to mix non-naturally occurring monomer subunits such as the tricyclic nucleosides as provided herein with other non-naturally occurring monomer subunits, naturally occurring monomer subunits (nucleosides) or mixtures thereof.
- oligomeric compounds are provided herein comprising a contiguous sequence of linked monomer subunits wherein at least one monomer subunit is a tricyclic nucleoside as provided herein.
- oligomeric compounds are provided comprising a plurality of tricyclic nucleosides as provided herein.
- Oligomeric compounds are routinely prepared linearly but can also be joined or otherwise prepared to be circular and/or can be prepared to include branching. Oligomeric compounds can form double stranded constructs such as for example two strands hybridized to form a double stranded composition. Double stranded compositions can be linked or separate and can include various other groups such as conjugates and/or overhangs on the ends.
- antisense compound refers to an oligomeric compound, at least a portion of which is at least partially complementary to a target nucleic acid to which it hybridizes. In certain embodiments, an antisense compound modulates (increases or decreases) expression or amount of a target nucleic acid. In certain embodiments, an antisense compound alters splicing of a target pre- mRNA resulting in a different splice variant. In certain embodiments, an antisense compound modulates expression of one or more different target proteins. Antisense mechanisms contemplated herein include, but are not limited to an RNase H mechanism, RNAi mechanisms, splicing modulation, translational arrest, altering RNA processing, inhibiting microRNA function, or mimicking microRNA function.
- antisense activity refers to any detectable and/or measurable activity attributable to the hybridization of an antisense compound to its target nucleic acid.
- such activity may be an increase or decrease in an amount of a nucleic acid or protein.
- such activity may be a change in the ratio of splice variants of a nucleic acid or protein.
- Detection and/or measuring of antisense activity may be direct or indirect.
- antisense activity is assessed by detecting and/or measuring the amount of target protein or the relative amounts of splice variants of a target protein.
- antisense activity is assessed by detecting and/or measuring the amount of target nucleic acids and/or cleaved target nucleic acids and/or alternatively spliced target nucleic acids. In certain embodiments, antisense activity is assessed by observing a phenotypic change in a cell or animal.
- oligomeric compounds as provided herein can be prepared having one or more internucleoside linkages containing modified e.g. non-naturally occurring
- internucleoside linkages The two main classes of internucleoside linkages are defined by the presence or absence of a phosphorus atom.
- Modified internucleoside linkages having a phosphorus atom include without limitation, phosphorothioates, chiral phosphorothioates, phosphorodithioates, phosphotriesters, aminoalkylphosphotriesters, methyl and other alkyl phosphonates including 3'- alkylene phosphonates, 5'-alkylene phosphonates and chiral phosphonates, phosphinates, phos- phoramidates including 3 '-amino phosphoramidate and aminoalkylphosphoramidates,
- Oligonucleotides having inverted polarity can comprise a single 3' to 3' linkage at the 3 '-most internucleotide linkage i.e. a single inverted nucleoside residue which may be abasic (the nucleobase is missing or has a hydroxyl group in place thereof).
- Various salts, mixed salts and free acid forms are also included.
- oligomeric compounds as provided herein can be prepared having one or more non-phosphorus containing internucleoside linkages.
- Such oligomeric compounds include without limitation, those that are formed by short chain alkyl or cycloalkyl internucleoside linkages, mixed heteroatom and alkyl or cycloalkyl internucleoside linkages, or one or more short chain heteroatomic or heterocyclic internucleoside linkages.
- siloxane backbones include those having siloxane backbones; sulfide, sulfoxide and sulfone backbones; formacetyl and thioformacetyl backbones; methylene formacetyl and thioformacetyl backbones; riboacetyl backbones; alkene containing backbones; sulfamate backbones; methyleneimino and methylenehydrazino backbones; sulfonate and sulfonamide backbones; amide backbones; and others having mixed N, O, S and CH 2 component parts.
- neutral internucleoside linkage is intended to include internucleoside linkages that are non-ionic.
- Further neutral internucleoside linkages include nonionic linkages comprising siloxane
- oligomeric compounds as provided herein can be prepared having one or more optionally protected phosphorus containing internucleoside linkages.
- Representative protecting groups for phosphorus containing internucleoside linkages such as phosphodiester and phosphorothioate linkages include ⁇ -cyanoethyl, diphenylsilylethyl, ⁇ -cyanobutenyl, cyano p-xylyl (CPX), N-methyl-N-trifluoroacetyl ethyl (MET A), acetoxy phenoxy ethyl (APE) and butene-4-yl groups. See for example U.S. Patents Nos. 4,725,677 and Re.
- linking groups and “bifunctional linking moieties” are meant to include groups known in the art that are useful for attachment of chemical functional groups, conjugate groups, reporter groups and other groups to selective sites in a parent compound such as for example an oligomeric compound.
- a bifunctional linking moiety comprises a hydrocarbyl moiety having two functional groups. One of the functional groups is selected to bind to a parent molecule or compound of interest and the other is selected to bind to essentially any selected group such as a chemical functional group or a conjugate group.
- the linker comprises a chain structure or a polymer of repeating units such as ethylene glycols or amino acid units.
- bifunctional linking moieties examples include without limitation, electrophiles for reacting with nucleophilic groups and nucleophiles for reacting with electrophilic groups.
- bifunctional linking moieties include amino, hydroxyl, carboxylic acid, thiol, unsaturations (e.g., double or triple bonds), and the like.
- bifunctional linking moieties include 8-amino-3,6-dioxaoctanoic acid (ADO), succinimidyl 4-(N-maleimidomethyl) cyclohexane-l-carboxylate (SMCC) and 6- aminohexanoic acid (AHEX or AHA).
- ADO 8-amino-3,6-dioxaoctanoic acid
- SMCC succinimidyl 4-(N-maleimidomethyl) cyclohexane-l-carboxylate
- AHEX or AHA 6- aminohexanoic acid
- linking groups include without limitation, substituted Ci-Cio alkyl, substituted or unsubstituted C 2 -Cio alkenyl or substituted or unsubstituted C 2 -Cio alkynyl, wherein a nonlimiting list of preferred substituent groups includes hydroxyl, amino, alkoxy, carboxy, benzyl, phenyl, nitro, thiol, thioalkoxy, halogen, alkyl, aryl, alkenyl and alkynyl.
- the oligomeric compounds as provided herein can be modified by covalent attachment of one or more conjugate groups.
- conjugate groups modify one or more properties of the oligomeric compounds they are attached to.
- Such oligonucleotide properties include without limitation, pharmacodynamics, pharmacokinetics, binding, absorption, cellular distribution, cellular uptake, charge and clearance.
- Conjugate groups are routinely used in the chemical arts and are linked directly or via an optional linking moiety or linking group to a parent compound such as an oligomeric compound.
- conjugate groups includes without limitation, intercalators, reporter molecules, polyamines, polyamides, polyethylene glycols, thioethers, poly ethers, cholesterols, thiocholesterols, cholic acid moieties, folate, lipids,
- the oligomeric compounds as provided herein can be modified by covalent attachment of one or more terminal groups to the 5' or 3'-terminal groups.
- a terminal group can also be attached at any other position at one of the terminal ends of the oligomeric compound.
- the terms "5'-terminal group”, “3'-terminal group”, “terminal group” and combinations thereof are meant to include useful groups known to the art skilled that can be placed on one or both of the terminal ends, including but not limited to the 5' and 3 '-ends of an oligomeric compound respectively, for various purposes such as enabling the tracking of the oligomeric compound (a fluorescent label or other reporter group), improving the pharmacokinetics or pharmacodynamics of the oligomeric compound (such as for example: uptake and/or delivery) or enhancing one or more other desirable properties of the oligomeric compound (a group for improving nuclease stability or binding affinity).
- 5' and 3 '-terminal groups include without limitation, modified or unmodified nucleosides; two or more linked nucleosides that are independently, modified or unmodified; conjugate groups; capping groups; phosphate moieties; and protecting groups.
- phosphate moiety refers to a terminal phosphate group that includes phosphates as well as modified phosphates.
- the phosphate moiety can be located at either terminus.
- the phosphate moiety is located at the 5'-terminal nucleoside.
- the terminal phosphate is modified such that one or more of the O and OH groups are replaced with H, O, S, N(R) or alkyl where R is H, an amino protecting group or unsubstituted or substituted alkyl.
- the 5' and or 3' terminal group can comprise from 1 to 3 phosphate moieties that are each, independently, unmodified (di or tri-phosphates) or modified.
- phosphorus moiety refers to a group having the formula:
- R x and R y are each, independently, hydroxyl, protected hydroxyl group, thiol, protected thiol group, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, Ci-C 6 alkoxy, substituted Ci-C 6 alkoxy, a protected amino or substituted amino; and
- R z is O or S.
- a monomer such as a phosphoramidite or H-phosphonate the protected phosphorus moiety is preferred to maintain stability during oligomer synthesis.
- the phosphorus moiety can include deprotected groups.
- Phosphorus moieties included herein can be attached to a monomer, which can be used in the preparation of oligomeric compounds, wherein the monomer may be attached using O, S, NRa or CR e f , wherein Ra includes without limitation H, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, Ci-C 6 alkoxy, substituted Ci-C 6 alkoxy, C 2 -C6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted C 2 -C 6 alkynyl or substituted acyl, and R e and R f each, independently, include without limitation H, halogen, Ci-C 6 alkyl, substituted Ci-C 6 alkyl, Ci-C 6 alkoxy or substituted Ci-C 6 alkoxy.
- Such linked phosphorus moieties include without limitation, phosphates, modified phosphates, thiophosphates, modified thiophosphates, phosphonates, modified phosphonates, phosphoramidates and modified phosphoramidates.
- RNA duplexes exist in what has been termed “A Form” geometry while DNA duplexes exist in “B Form” geometry.
- RNA:RNA duplexes are more stable, or have higher melting temperatures (T m ) than DNA:DNA duplexes (Sanger et al, Principles of Nucleic Acid Structure,
- RNA has been attributed to several structural features, most notably the improved base stacking interactions that result from an A- form geometry (Searle et al, Nucleic Acids Res., 1993, 21, 2051-2056).
- the presence of the 2' hydroxyl in RNA biases the sugar toward a C3' endo pucker, i.e., also designated as Northern pucker, which causes the duplex to favor the A-form geometry.
- RNA duplex the 2' hydroxyl groups of RNA can form a network of water mediated hydrogen bonds that help stabilize the RNA duplex (Egli et al, Biochemistry, 1996, 35, 8489-8494).
- deoxy nucleic acids prefer a C2' endo sugar pucker, i.e., also known as Southern pucker, which is thought to impart a less stable B-form geometry (Sanger, W. (1984) Principles of Nucleic Acid Structure, Springer- Verlag, New York, NY).
- the relative ability of a chemically-modified oligomeric compound to bind to complementary nucleic acid strands, as compared to natural oligonucleotides, is measured by obtaining the melting temperature of a hybridization complex of said chemically-modified oligomeric compound with its complementary unmodified target nucleic acid.
- the melting temperature (T m ) a characteristic physical property of double helixes, denotes the temperature in degrees centigrade at which 50% helical versus coiled (unhybridized) forms are present.
- T m also commonly referred to as binding affinity
- Base stacking which occurs during hybridization, is accompanied by a reduction in UV absorption (hypochromicity). Consequently a reduction in UV absorption indicates a higher T m .
- RNA target duplex can be modulated through incorporation of chemically-modified nucleosides into the antisense compound.
- Sugar-modified nucleosides have provided the most efficient means of modulating the T m of an antisense compound with its target RNA.
- Sugar-modified nucleosides that increase the population of or lock the sugar in the C3' '-endo (Northern, RNA-like sugar pucker) configuration have predominantly provided a per modification T m increase for antisense compounds toward a complementary RNA target.
- Sugar-modified nucleosides that increase the population of or lock the sugar in the C2'-endo (Southern, DNA-like sugar pucker) configuration predominantly provide a per modification Tm decrease for antisense compounds toward a complementary RNA target.
- the sugar pucker of a given sugar-modified nucleoside is not the only factor that dictates the ability of the nucleoside to increase or decrease an antisense compound's T m toward complementary RNA.
- the sugar-modified nucleoside tricycloDNA is predominantly in the C2'-endo conformation, however it imparts a 1.9 to 3° C per modification increase in T m toward a
- RNA complementary RNA.
- a sugar-modified high-affinity nucleoside that does not adopt the C3'-endo conformation is a-L-LNA (described in more detail herein).
- T m means melting temperature which is the temperature at which the two strands of a duplex nucleic acid separate. T m is often used as a measure of duplex stability or the binding affinity of an antisense compound toward a complementary RNA molecule.
- complementarity in reference to nucleobases refers to a nucleobase that is capable of base pairing with another nucleobase.
- adenine A
- complementary nucleobase refers to a nucleobase of an antisense compound that is capable of base pairing with a nucleobase of its target nucleic acid. For example, if a nucleobase at a certain position of an antisense compound is capable of hydrogen bonding with a nucleobase at a certain position of a target nucleic acid, then the position of hydrogen bonding between the oligonucleotide and the target nucleic acid is considered to be complementary at that nucleobase pair. Nucleobases or more broadly, heterocyclic base moieties, comprising certain modifications may maintain the ability to pair with a counterpart nucleobase and thus, are still capable of complementarity.
- non-complementary in reference to nucleobases refers to a pair of nucleobases that do not form hydrogen bonds with one another or otherwise support hybridization.
- complementary in reference to linked nucleosides, oligonucleotides, oligomeric compounds, or nucleic acids, refers to the capacity of an oligomeric compound to hybridize to another oligomeric compound or nucleic acid through nucleobase or more broadly, heterocyclic base, complementarity.
- an antisense compound and its target are complementary to each other when a sufficient number of corresponding positions in each molecule are occupied by nucleobases that can bond with each other to allow stable association between the antisense compound and the target.
- nucleobases that can bond with each other to allow stable association between the antisense compound and the target.
- antisense compounds may comprise up to about 20% nucleotides that are mismatched (i.e., are not nucleobase complementary to the corresponding nucleotides of the target).
- the antisense compounds contain no more than about 15%, more preferably not more than about 10%, most preferably not more than 5% or no mismatches.
- nucleobase complementary or otherwise do not disrupt hybridization e.g., universal bases.
- compounds provided herein are at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%), at least 99% or 100% complementary to a target nucleic acid.
- oligomeric compounds need not be 100% complementary to that of its target nucleic acid to be specifically hybridizable. Moreover, an oligomeric compound may hybridize over one or more segments such that intervening or adjacent segments are not involved in the hybridization event (e.g., a loop structure or hairpin structure). In certain embodiments, oligomeric compounds can comprise at least about 70%, at least about 80%, at least about 90%, at least about 95%, or at least about 99% sequence complementarity to a target region within the target nucleic acid sequence to which they are targeted.
- an oligomeric compound in which 18 of 20 nucleobases of the oligomeric compound are complementary to a target region, and would therefore specifically hybridize would represent 90 percent complementarity.
- the remaining noncomplementary nucleobases may be clustered or interspersed with complementary nucleobases and need not be contiguous to each other or to complementary nucleobases.
- an oligomeric compound which is 18 nucleobases in length having 4 (four) noncomplementary nucleobases which are flanked by two regions of complete complementarity with the target nucleic acid would have 77.8% overall complementarity with the target nucleic acid and would thus fall within this scope.
- Percent complementarity of an oligomeric compound with a region of a target nucleic acid can be determined routinely using BLAST programs (basic local alignment search tools) and PowerBLAST programs known in the art (Altschul et al, J. Mol. Biol, 1990, 215, 403-410; Zhang and Madden, Genome Res., 1997, 7, 649- 656).
- hybridization refers to the pairing of complementary oligomeric compounds (e.g., an antisense compound and its target nucleic acid). While not limited to a particular mechanism, the most common mechanism of pairing involves hydrogen bonding, which may be Watson-Crick, Hoogsteen or reversed Hoogsteen hydrogen bonding, between
- nucleobases complementary nucleoside or nucleotide bases
- the natural base adenine is nucleobase complementary to the natural nucleobases thymidine and uracil which pair through the formation of hydrogen bonds.
- the natural base guanine is nucleobase complementary to the natural bases cytosine and 5-methyl cytosine. Hybridization can occur under varying circumstances.
- target nucleic acid refers to any nucleic acid molecule the expression, amount, or activity of which is capable of being modulated by an antisense compound.
- the target nucleic acid is DNA or RNA.
- the target RNA is mRNA, pre-mRNA, non-coding RNA, pri-microRNA, pre-microRNA, mature microRNA, promoter-directed RNA, or natural antisense transcripts.
- the target nucleic acid can be a cellular gene (or mRNA transcribed from the gene) whose expression is associated with a particular disorder or disease state, or a nucleic acid molecule from an infectious agent.
- target nucleic acid is a viral or bacterial nucleic acid.
- oligomeric compounds such as antisense oligomeric compounds, antisense oligonucleotides, ribozymes, external guide sequence (EGS) oligonucleotides, alternate splicers, primers, probes, and other oligomeric compounds which hybridize to at least a portion of the target nucleic acid.
- these oligomeric compounds may be introduced in the form of single- stranded, double- stranded, circular or hairpin oligomeric compounds and may contain structural elements such as internal or terminal bulges or loops.
- the oligomeric compounds provided herein may elicit the action of one or more enzymes or structural proteins to effect modification of the target nucleic acid.
- the oligomeric compound may inhibit the activity the target nucleic acid through an occupancy-based method, thus interfering with the activity of the target nucleic acid.
- RNAse H a cellular endonuclease which cleaves the RNA strand of an RNA:DNA duplex. It is known in the art that single- stranded oligomeric compounds which are "DNA-like" elicit RNAse H. Activation of RNase H, therefore, results in cleavage of the RNA target, thereby greatly enhancing the efficiency of oligonucleotide- mediated inhibition of gene expression. Similar roles have been postulated for other ribonucleases such as those in the RNase III and ribonuclease L family of enzymes.
- oligomeric compound is a single- stranded antisense oligonucleotide
- double-stranded RNA (dsRNA) molecules has been shown to induce potent and specific antisense-mediated reduction of the function of a gene or its associated gene products. This phenomenon occurs in both plants and animals and is believed to have an evolutionary connection to viral defense and transposon silencing.
- modulation refers to a perturbation of amount or quality of a function or activity when compared to the function or activity prior to modulation.
- modulation includes the change, either an increase (stimulation or induction) or a decrease (inhibition or reduction) in gene expression.
- modulation of expression can include perturbing splice site selection of pre-mRNA processing, resulting in a change in the amount of a particular splice-variant present compared to conditions that were not perturbed.
- modulation includes perturbing translation of a protein.
- pharmaceutically acceptable salts refers to salts that retain the desired activity of the compound and do not impart undesired toxicological effects thereto.
- pharmaceutically acceptable salt includes a salt prepared from pharmaceutically acceptable non-toxic acids or bases, including inorganic or organic acids and bases.
- oligomeric compounds described herein may be prepared by methods well-known in the art. For a review of pharmaceutically acceptable salts, see Stahl and Wermuth, Handbook of Pharmaceutical Salts: Properties, Selection and Use (Wiley- VCH, Weinheim, Germany, 2002). Sodium salts of antisense oligonucleotides are useful and are well accepted for therapeutic administration to humans. Accordingly, in one embodiment the oligomeric compounds described herein are in the form of a sodium salt. In certain embodiments, oligomeric compounds provided herein comprise from about 8 to about 80 monomer subunits in length.
- oligomeric compounds of 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, or 80 monomer subunits in length, or any range therewithin.
- oligomeric compounds provided herein comprise from about 8 to 40 monomer subunits in length.
- oligomeric compounds of 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 or 40 monomer subunits in length, or any range therewithin.
- oligomeric compounds provided herein comprise from about 8 to 20 monomer subunits in length.
- oligomeric compounds of 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 monomer subunits in length, or any range therewithin.
- oligomeric compounds provided herein comprise from about 8 to 16 monomer subunits in length.
- oligomeric compounds of 8, 9, 10, 11, 12, 13, 14, 15 or 16 monomer subunits in length, or any range therewithin.
- oligomeric compounds provided herein comprise from about 10 to
- oligomeric compounds provided herein comprise from about 10 to 18 monomer subunits in length.
- oligomeric compounds of 10, 11, 12, 13, 14, 15, 16, 17 or 18 monomer subunits in length, or any range therewithin.
- oligomeric compounds provided herein comprise from about 10 to 21 monomer subunits in length.
- oligomeric compounds provided herein comprise from about 12 to 14 monomer subunits in length.
- this embodies oligomeric compounds of 12, 13 or 14 monomer subunits in length, or any range therewithin.
- oligomeric compounds provided herein comprise from about 12 to
- oligomeric compounds provided herein comprise from about 12 to 21 monomer subunits in length.
- oligomeric compounds of 12, 13, 14, 15, 16, 17, 18, 19, 20 or 21 monomer subunits in length, or any range therewithin.
- oligomeric compounds provided herein comprise from about 14 to 18 monomer subunits in length.
- oligomeric compounds of 14, 15, 16, 17 or 18 monomer subunits in length, or any range therewithin.
- oligomeric compounds of any of a variety of ranges of lengths of linked monomer subunits are provided.
- oligomeric compounds are provided consisting of X-Y linked monomer subunits, where X and Y are each independently selected from 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, and 50; provided that X ⁇ Y.
- this provides oligomeric compounds comprising: 8-9, 8-10, 8-11, 8-12, 8-13, 8-14, 8-15, 8-16, 8-17, 8-18, 8-19, 8-20, 8-21, 8-22, 8-23, 8-24, 8-25, 8-26, 8-27, 8-28, 8-29, 8-30, 9-10, 9-11, 9-12, 9-13, 9-14, 9-15, 9-16, 9-17, 9-18, 9-19, 9-20, 9-21, 9-22, 9-23, 9-24, 9-25, 9-26, 9-27, 9-28, 9-29, 9-30, 10-11, 10-12, 10-13, 10-14, 10-15, 10-16, 10-17, 10-18, 10- 19, 10-20, 10-21, 10-22, 10-23, 10-24, 10-25, 10-26, 10-27, 10-28, 10-29, 10-30, 11-12, 11-13, 11- 14, 11-15, 11-16, 11-17, 11-18, 11-19, 11-20, 11-21, 11-22, 11-23, 11-24, 10-30, 11-12, 11
- the ranges for the oligomeric compounds listed herein are meant to limit the number of monomer subunits in the oligomeric compounds, however such oligomeric compounds may further include 5' and/or 3'-terminal groups including but not limited to protecting groups such as hydroxyl protecting groups, optionally linked conjugate groups and/or other substituent groups.
- the preparation of oligomeric compounds as disclosed herein is performed according to literature procedures for DNA: Protocols for Oligonucleotides and Analogs, Agrawal, Ed., Humana Press, 1993, and/or RNA: Scaringe, Methods, 2001, 23, 206-217; Gait et al, Applications of Chemically synthesized RNA in RNA:Protein Interactions, Smith, Ed., 1998, 1-36; Gallo et al, Tetrahedron , 2001, 57, 5707-5713. Additional methods for solid-phase synthesis may be found in Caruthers U.S. Patents Nos. 4,415,732; 4,458,066; 4,500,707; 4,668,777; 4,973,679; and 5, 132,418; and Koster U.S. Patents Nos. 4,725,677 and Re. 34,069.
- Oligomeric compounds are routinely prepared using solid support methods as opposed to solution phase methods.
- Commercially available equipment commonly used for the preparation of oligomeric compounds that utilize the solid support method is sold by several vendors including, for example, Applied Biosystems (Foster City, CA). Any other means for such synthesis known in the art may additionally or alternatively be employed.
- Suitable solid phase techniques, including automated synthesis techniques, are described in Oligonucleotides and Analogues, a Practical Approach, F. Eckstein, Ed., Oxford University Press, New York, 1991.
- RNA and related analogs have been increasing as efforts in RNA interference and micro RNA increase.
- the primary RNA synthesis strategies that are presently being used commercially include 5'-0-DMT-2'-0-t- butyldimethylsilyl (TBDMS), 5'-0-DMT-2'-0-[l(2-fluorophenyl)-4-methoxypiperidin-4-yl] (FPMP), 2'-0-[(triisopropylsilyl)oxy]methyl (2'-0-CH 2 -0-Si(iPr) 3 (TOM) and the 5'-0-silyl ether- 2'-ACE (5'-0-bis(trimethylsiloxy)cyclododecyloxysilyl ether (DOD)-2'-0-bis(2- acetoxyethoxy)methyl (ACE).
- TDMS 5'-0-DMT-2'-0-t- butyldimethylsilyl
- FPMP l(2-fluorophenyl)-4-meth
- RNA synthesis activator advertised to reduce coupling times especially with TOM and TBDMS chemistries.
- the primary groups being used for commercial RNA synthesis are:
- TBDMS 5'-0-DMT-2'-0-t-butyldimethylsilyl
- TOM 2'-0-[(triisopropylsilyl)oxy]methyl
- DOD/ACE (5'-0-bis(trimethylsiloxy)cyclododecyloxysilyl ether-2'-0-bis(2-acetoxyethoxy)methyl
- FPMP 5'-0-DMT-2'-0-[l(2-fluorophenyl)-4-ethoxypiperidin-4-yl].
- each of the aforementioned RNA synthesis strategies can be used herein.
- the aforementioned RNA synthesis strategies can be performed together in a hybrid fashion e.g. using a 5 '-protecting group from one strategy with a 2'-0-protecting from another strategy.
- suitable target segments may be employed in a screen for additional oligomeric compounds that modulate the expression of a selected protein.
- Modules are those oligomeric compounds that decrease or increase the expression of a nucleic acid molecule encoding a protein and which comprise at least an 8-nucleobase portion which is complementary to a suitable target segment.
- the screening method comprises the steps of contacting a suitable target segment of a nucleic acid molecule encoding a protein with one or more candidate modulators, and selecting for one or more candidate modulators which decrease or increase the expression of a nucleic acid molecule encoding a protein. Once it is shown that the candidate modulator or modulators are capable of modulating (e.g.
- the modulator may then be employed herein in further investigative studies of the function of the peptide, or for use as a research, diagnostic, or therapeutic agent.
- candidate modulators may be evaluated by the extent to which they increase the expression of a microRNA target RNA or protein (as interference with the activity of a microRNA will result in the increased expression of one or more targets of the microRNA).
- expression refers to the process by which a gene ultimately results in a protein. Expression includes, but is not limited to, transcription, splicing, post-transcriptional modification, and translation.
- Suitable target segments may also be combined with their respective complementary oligomeric compounds provided herein to form stabilized double-stranded (duplexed) oligonucleotides.
- double stranded oligonucleotide moieties have been shown in the art to modulate target expression and regulate translation as well as RNA processing via an antisense mechanism.
- the double-stranded moieties may be subject to chemical modifications (Fire et al, Nature, 1998, 391, 806-811; Timmons and Fire, Nature, 1998, 395, 854; Timmons et al, Gene, 2001, 263, 103-112; Tabara et al, Science, 1998, 282, 430-431; Montgomery et al, Proc. Natl Acad. Sci. USA, 1998, 95, 15502-15507; Tuschl et al, Genes Dev., 1999, 13, 3191-3197;
- oligomeric compounds provided herein can also be applied in the areas of drug discovery and target validation.
- provided herein is the use of the oligomeric compounds and targets identified herein in drug discovery efforts to elucidate
- oligomeric compounds are provided for use in therapy.
- oligomeric compounds are provided for use in therapy.
- the therapy is reducing target messenger RNA.
- a dose refers to a specified quantity of a pharmaceutical agent provided in a single administration.
- a dose may be administered in two or more boluses, tablets, or injections.
- the desired dose requires a volume not easily accommodated by a single injection.
- two or more injections may be used to achieve the desired dose.
- a dose may be administered in two or more injections to minimize injection site reaction in an individual.
- Oligomeric compounds may be admixed with pharmaceutically acceptable active and/or inert substances for the preparation of pharmaceutical compositions or formulations.
- Compositions and methods for the formulation of pharmaceutical compositions are dependent upon a number of criteria, including, but not limited to, route of administration, extent of disease, or dose to be administered.
- Oligomeric compounds can be utilized in pharmaceutical compositions by combining such oligomeric compounds with a suitable pharmaceutically acceptable diluent or carrier.
- a pharmaceutically acceptable diluent includes phosphate-buffered saline (PBS).
- PBS is a diluent suitable for use in compositions to be delivered parenterally.
- employed in the methods described herein is a pharmaceutical composition comprising an antisense compound and a pharmaceutically acceptable diluent.
- the pharmaceutically acceptable diluent is PBS.
- compositions comprising oligomeric compounds encompass any pharmaceutically acceptable salts, esters, or salts of such esters.
- pharmaceutical compositions comprising oligomeric compounds comprise one or more oligonucleotide which, upon administration to an animal, including a human, is capable of providing (directly or indirectly) the biologically active metabolite or residue thereof.
- the disclosure is also drawn to pharmaceutically acceptable salts of antisense compounds, prodrugs, pharmaceutically acceptable salts of such prodrugs, and other bioequivalents.
- Suitable pharmaceutically acceptable salts include, but are not limited to, sodium and potassium salts.
- a prodrug can include the incorporation of additional nucleosides at one or both ends of an oligomeric compound which are cleaved by endogenous nucleases within the body, to form the active oligomeric compound.
- Lipid-based vectors have been used in nucleic acid therapies in a variety of methods.
- the nucleic acid is introduced into preformed liposomes or lipoplexes made of mixtures of cationic lipids and neutral lipids.
- DNA complexes with mono- or poly-cationic lipids are formed without the presence of a neutral lipid.
- preparations are made that include a polyamine compound or a lipid moiety complexed with a nucleic acid. Such preparations are described in PCT publication
- chemically-modified oligomeric compounds are provided herein that may have a higher affinity for target RNAs than does non-modified DNA.
- higher affinity in turn provides increased potency allowing for the administration of lower doses of such compounds, reduced potential for toxicity, improvement in therapeutic index and decreased overall cost of therapy.
- RNAi activity Effect of nucleoside modifications on RNAi activity is evaluated according to existing literature (Elbashir et al, Nature, 2001, 411, 494-498; Nishikura et al, Cell, 2001, 707, 415-416; and Bass et al, Cell, 2000, 101, 235-238.)
- oligomeric compounds provided herein can be utilized for diagnostics, therapeutics, prophylaxis and as research reagents and kits. Furthermore, antisense oligonucleotides, which are able to inhibit gene expression with 17, specificity, are often used by those of ordinary skill to elucidate the function of particular genes or to distinguish between functions of various members of a biological pathway. In certain embodiments, oligomeric compounds provided herein can be utilized either alone or in combination with other oligomeric compounds or other therapeutics as tools in differential and/or combinatorial analyses to elucidate expression patterns of a portion or the entire complement of genes expressed within cells and tissues. Oligomeric compounds can also be effectively used as primers and probes under conditions favoring gene amplification or detection, respectively.
- primers and probes are useful in methods requiring the specific detection of nucleic acid molecules encoding proteins and in the amplification of the nucleic acid molecules for detection or for use in further studies.
- Hybridization of oligomeric compounds as provided herein, particularly the primers and probes, with a nucleic acid can be detected by means known in the art. Such means may include conjugation of an enzyme to the oligonucleotide, radiolabelling of the oligonucleotide or any other suitable detection means. Kits using such detection means for detecting the level of selected proteins in a sample may also be prepared.
- the tricyclic oligomeric compounds disclosed herein may be used treat a disease or condition. In certain embodiments, the oligomeric compounds disclosed herein may be used to ameliorate one or more symptoms of a disease or condition. In certain
- a tricyclic nucleoside monomer may be used as an anti- viral agent. In certain embodiments, a tricyclic nucleoside monomer may be used ameliorate one or more symptoms caused by a virus. In certain embodiments, a tricyclic nucleoside monomer, a salt thereof, or a mono-, di-, or tri-phosphate analog thereof, may be used as an anti-viral agent. In certain embodiments, a tricyclic nucleoside monomer, a salt thereof, or a mono-, di-, or tri-phosphate analog thereof, may be used ameliorate one or more symptoms caused by a virus.
- expression patterns within cells or tissues treated with one or more of the oligomeric compounds provided herein are compared to control cells or tissues not treated with oligomeric compounds and the patterns produced are analyzed for differential levels of gene expression as they pertain, for example, to disease association, signaling pathway, cellular localization, expression level, size, structure or function of the genes examined. These analyses can be performed on stimulated or unstimulated cells and in the presence or absence of other compounds and or oligomeric compounds which affect expression patterns.
- Examples of methods of gene expression analysis known in the art include DNA arrays or microarrays (Brazma and Vilo, FEB S Lett., 2000, 480, 17-24; Celis, et al, FEB S Lett, 2000, 480, 2- 16), SAGE (serial analysis of gene expression)(Madden, et al, Drug Discov. Today, 2000, 5, 415- 425), READS (restriction enzyme amplification of digested cDNAs) (Prashar and Weissman, Methods EnzymoL, 1999, 303, 258-72), TOGA (total gene expression analysis) (Sutcliffe, et al., Proc. Natl. Acad. Sci.
- oligomeric compounds comprising a contiguous sequence of linked monomer subunits, of essentially any viable length to practice the methods disclosed herein.
- Such oligomeric compounds will include at least one and preferably a plurality of the tricyclic nucleosides provided herein and may also include other monomer subunits including but not limited to nucleosides, modified nucleosides, nucleosides comprising sugar surrogate groups and nucleoside mimetics.
- an oligonucleotide comprising a nucleoside comprising a 2'-OH sugar moiety and a thymine base
- a DNA having a modified sugar (2'-OH for the natural 2'-H of DNA) or as an RNA having a modified base (thymine (methylated uracil) for natural uracil of RNA).
- oligomeric compounds provided herein can be utilized as described, the following examples serve only to illustrate and are not intended to be limiting.
- nucleoside phosphoramidites The preparation of nucleoside phosphoramidites is performed following procedures that are illustrated herein and in the art such as but not limited to US Patent 6,426,220 and published PCT WO 02/36743.
- oligomeric compounds used in accordance with this invention may be conveniently and routinely made through the well-known technique of solid phase synthesis.
- Equipment for such synthesis is sold by several vendors including, for example, Applied Biosystems (Foster City, CA).
- the oligomeric compounds are recovered by precipitating with greater than 3 volumes of ethanol from a 1 M H 4 OAc solution.
- Phosphinate internucleoside linkages can be prepared as described in U.S. Patent 5,508,270.
- Alkyl phosphonate internucleoside linkages can be prepared as described in U.S. Patent 4,469,863.
- 3 '-Deoxy-3 ' -methylene phosphonate internucleoside linkages can be prepared as described in U.S. Patents 5,610,289 or 5,625,050.
- Phosphoramidite internucleoside linkages can be prepared as described in U.S. Patent,
- Alkylphosphonothioate internucleoside linkages can be prepared as described in published PCT applications PCT/US94/00902 and PCT/US93/06976 (published as WO 94/17093 and WO 94/02499, respectively).
- Phosphotriester internucleoside linkages can be prepared as described in U.S. Patent 5,023,243.
- Borano phosphate internucleoside linkages can be prepared as described in U.S. Patents 5, 130,302 and 5, 177, 198.
- Formacetal and thioformacetal internucleoside linkages can be prepared as described in U.S. Patents 5,264,562 and 5,264,564.
- Ethylene oxide internucleoside linkages can be prepared as described in U.S. Patent
- the oligomeric compounds including without limitation oligonucleotides and oligonucleosides, are recovered by precipitation out of 1 M NH 4 OAc with >3 volumes of ethanol. Synthesized oligomeric compounds are analyzed by electrospray mass spectroscopy (molecular weight determination) and by capillary gel electrophoresis. The relative amounts of phosphorothioate and phosphodiester linkages obtained in the synthesis is determined by the ratio of correct molecular weight relative to the -16 amu product (+/-32 +/-48).
- oligomeric compounds are purified by FIPLC, as described by Chiang et al, J. Biol. Chem. 1991, 266, 18162-18171. Results obtained with HPLC-purified material are generally similar to those obtained with non-HPLC purified material.
- Oligomeric compounds can be synthesized via solid phase P(III) phosphoramidite chemistry on an automated synthesizer capable of assembling 96 sequences simultaneously in a 96-well format.
- Phosphodiester internucleoside linkages are afforded by oxidation with aqueous iodine.
- Phosphorothioate internucleoside linkages are generated by sulfurization utilizing 3,H-1,2 benzodithiole-3-one 1, 1 dioxide (Beaucage Reagent) in anhydrous acetonitrile.
- Standard base-protected beta-cyanoethyl-diiso-propyl phosphoramidites can be purchased from commercial vendors (e.g.
- Non-standard nucleosides are synthesized as per standard or patented methods and can be functionalized as base protected beta-cyanoethyldiisopropyl phosphoramidites.
- Oligomeric compounds can be cleaved from support and deprotected with concentrated NH 4 OH at elevated temperature (55-60 °C) for 12-16 hours and the released product then dried in vacuo. The dried product is then re-suspended in sterile water to afford a master plate from which all analytical and test plate samples are then diluted utilizing robotic pipettors.
- the concentration of oligomeric compounds in each well can be assessed by dilution of samples and UV absorption spectroscopy.
- the full-length integrity of the individual products can be evaluated by capillary electrophoresis (CE) in either the 96-well format (Beckman P/ACETM MDQ) or, for individually prepared samples, on a commercial CE apparatus (e.g., Beckman P/ACETM 5000, ABI 270). Base and backbone composition is confirmed by mass analysis of the oligomeric compounds utilizing electrospray-mass spectroscopy. All assay test plates are diluted from the master plate using single and multi-channel robotic pipettors. Plates are judged to be acceptable if at least 85% of the oligomeric compounds on the plate are at least 85% full length.
- oligomeric compounds on target nucleic acid expression is tested in any of a variety of cell types provided that the target nucleic acid is present at measurable levels. This can be routinely determined using, for example, PCR or Northern blot analysis. Cell lines derived from multiple tissues and species can be obtained from American Type Culture Collection (ATCC, Manassas, VA).
- the following cell type is provided for illustrative purposes, but other cell types can be routinely used, provided that the target is expressed in the cell type chosen. This can be readily determined by methods routine in the art, for example Northern blot analysis, ribonuclease protection assays or RT-PCR.
- b.END cells The mouse brain endothelial cell line b.END was obtained from Dr. Werner Risau at the Max Plank Institute (Bad Nauheim, Germany). b.END cells are routinely cultured in DMEM, high glucose (Invitrogen Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen Life Technologies, Carlsbad, CA). Cells are routinely passaged by trypsinization and dilution when they reached approximately 90% confluence. Cells are seeded into 96-well plates (Falcon-Primaria #353872, BD Biosciences, Bedford, MA) at a density of approximately 3000 cells/well for uses including but not limited to oligomeric compound transfection experiments.
- oligomeric compounds When cells reached 65-75% confluency, they are treated with one or more oligomeric compounds.
- the oligomeric compound is mixed with LIPOFECTINTM Invitrogen Life Tech- nologies, Carlsbad, CA) in Opti-MEMTM-l reduced serum medium (Invitrogen Life Technologies, Carlsbad, CA) to achieve the desired concentration of the oligomeric compound(s) and a
- transfection reagents include, but are not limited to,
- CYTOFECTINTM LIPOFECTAMINETM, OLIGOFECTAMINETM, and FUGENETM.
- Other suitable transfection methods known in the art include, but are not limited to, electroporation.
- Quantitation of target mRNA levels is accomplished by real-time quantitative PCR using the ABI PRISMTM 7600, 7700, or 7900 Sequence Detection System (PE-Applied Biosystems, Foster City, CA) according to manufacturer's instructions.
- ABI PRISMTM 7600, 7700, or 7900 Sequence Detection System PE-Applied Biosystems, Foster City, CA
- This is a closed-tube, non-gel-based, fluorescence detection system which allows high-throughput quantitation of polymerase chain reaction (PCR) products in real-time.
- PCR polymerase chain reaction
- products in real-time quantitative PCR are quantitated as they accumulate. This is accomplished by including in the PCR reaction an oligonucleotide probe that anneals specifically between the forward and reverse PCR primers, and contains two fluorescent dyes.
- a reporter dye e.g., FAM or JOE, obtained from either PE-Applied Biosystems, Foster City, CA, Operon Technologies Inc., Alameda, CA or Integrated DNA Technologies Inc., Coralville, IA
- a quencher dye e.g., TAMRA, obtained from either PE- Applied Biosystems, Foster City, CA, Operon Technologies Inc., Alameda, CA or Integrated DNA Technologies Inc., Coralville, IA
- TAMRA obtained from either PE- Applied Biosystems, Foster City, CA, Operon Technologies Inc., Alameda, CA or Integrated DNA Technologies Inc., Coralville, IA
- annealing of the probe to the target sequence creates a substrate that can be cleaved by the 5'-exonuclease activity of Taq polymerase.
- cleavage of the probe by Taq polymerase releases the reporter dye from the remainder of the probe (and hence from the quencher moiety) and a sequence-specific fluorescent signal is generated.
- additional reporter dye molecules are cleaved from their respective probes, and the fluorescence intensity is monitored at regular intervals by laser optics built into the ABI PRISMTM Sequence Detection System.
- a series of parallel reactions containing serial dilutions of mRNA from untreated control samples generates a standard curve that is used to quantitate the percent inhibition after antisense oligonucleotide treatment of test samples.
- primer-probe sets specific to the target gene being measured are evaluated for their ability to be "multiplexed" with a GAPDH amplification reaction.
- multiplexing both the target gene and the internal standard gene GAPDH are amplified concurrently in a single sample.
- mRNA isolated from untreated cells is serially diluted. Each dilution is amplified in the presence of primer-probe sets specific for GAPDH only, target gene only ("single-plexing"), or both (multiplexing).
- standard curves of GAPDH and target mRNA signal as a function of dilution are generated from both the single-plexed and multiplexed samples.
- the primer-probe set specific for that target is deemed multiplexable.
- Other methods of PCR are also known in the art.
- RT and PCR reagents are obtained from Invitrogen Life Technologies (Carlsbad, CA).
- RT real-time PCR is carried out by adding 20 ⁇ .
- PCR cocktail 2.5x PCR buffer minus MgCl 2 , 6.6 mM MgCl 2 , 375 ⁇ each of dATP, dCTP, dCTP and dGTP, 375 nM each of forward primer and reverse primer, 125 nM of probe, 4 Units RNAse inhibitor, 1.25 Units PLATINUM® Taq, 5 Units MuLV reverse transcriptase, and 2.5x ROX dye) to 96-well plates containing 30 ⁇ L ⁇ total RNA solution (20- 200 ng).
- the RT reaction is carried out by incubation for 30 minutes at 48°C. Following a 10 minute incubation at 95°C to activate the PLATINUM® Taq, 40 cycles of a two-step PCR protocol are carried out: 95°C for 15 seconds (denaturation) followed by 60°C for 1.5 minutes (annealing/- extension).
- Gene target quantities obtained by RT, real-time PCR are normalized using either the expression level of GAPDH, a gene whose expression is constant, or by quantifying total RNA using RIBOGREENTM (Molecular Probes, Inc. Eugene, OR).
- GAPDH expression is quantified by real time RT-PCR, by being run simultaneously with the target, multiplexing, or separately.
- Total RNA is quantified using RiboGreenTM RNA quantification reagent (Molecular Probes, Inc. Eugene, OR). Methods of RNA quantification by RIBOGREENTM are taught in Jones, L.J., et al, (Analytical Biochemistry, 1998, 265, 368-374).
- RIBOGREENTM working reagent 170 ⁇ _ of RIBOGREENTM working reagent (RIBOGREENTM reagent diluted 1 :350 in lOmM Tris-HCl, 1 mM EDTA, pH 7.5) is pipetted into a 96-well plate containing 30 ⁇ _ purified, cellular RNA.
- the plate is read in a CytoFluor 4000 (PE Applied Biosystems) with excitation at 485nm and emission at 530nm.
- Antisense modulation of a target expression can be assayed in a variety of ways known in the art.
- a target mRNA levels can be quantitated by, e.g., Northern blot analysis, competitive polymerase chain reaction (PCR), or real-time PCR.
- Real-time quantitative PCR is presently desired.
- RNA analysis can be performed on total cellular RNA or poly(A)+ mRNA.
- One method of RNA analysis of the present disclosure is the use of total cellular RNA as described in other examples herein. Methods of RNA isolation are well known in the art.
- Northern blot analysis is also routine in the art.
- Real-time quantitative (PCR) can be conveniently accomplished using the commercially available ABI PRISMTM 7600, 7700, or 7900 Sequence Detection System, available from PE- Applied Biosystems, Foster City, CA and used according to manufacturer's instructions.
- Protein levels of a target can be quantitated in a variety of ways well known in the art, such as immunoprecipitation, Western blot analysis (immunoblotting), enzyme-linked immunosorbent assay (ELISA) or fluorescence-activated cell sorting (FACS).
- Antibodies directed to a target can be identified and obtained from a variety of sources, such as the MSRS catalog of antibodies (Aerie Corporation, Birmingham, MI), or can be prepared via conventional monoclonal or polyclonal antibody generation methods well known in the art. Methods for preparation of polyclonal antisera are taught in, for example, Ausubel, F.M. et al, Current Protocols in Molecular Biology, Volume 2, pp. 11.12.1-11.12.9, John Wiley & Sons, Inc., 1997. Preparation of monoclonal antibodies is taught in, for example, Ausubel, F.M. et al., Current Protocols in Molecular Biology, Volume 2, pp.
- Immunoprecipitation methods are standard in the art and can be found at, for example, Ausubel, F.M. et al., Current Protocols in Molecular Biology, Volume 2, pp. 10.16.1-10.16.11, John Wiley & Sons, Inc., 1998.
- Western blot (immunoblot) analysis is standard in the art and can be found at, for example, Ausubel, F.M. et al, Current Protocols in Molecular Biology, Volume 2, pp. 10.8.1-10.8.21, John Wiley & Sons, Inc., 1997.
- Enzyme-linked immunosorbent assays ELISA are standard in the art and can be found at, for example, Ausubel, F.M. et al, Current Protocols in Molecular Biology, Volume 2, pp. 11.2.1-11.2.22, John Wiley & Sons, Inc., 1991.
- the oligomeric compounds are further investigated in one or more phenotypic assays, each having measurable endpoints predictive of efficacy in the treatment of a particular disease state or condition.
- Phenotypic assays, kits and reagents for their use are well known to those skilled in the art and are herein used to investigate the role and/or association of a target in health and disease.
- phenotypic assays which can be purchased from any one of several commercial vendors, include those for determining cell viability, cytotoxicity, proliferation or cell survival (Molecular Probes, Eugene, OR; PerkinElmer, Boston, MA), protein-based assays including enzymatic assays (Panvera, LLC, Madison, WI; BD Biosciences, Franklin Lakes, NJ; Oncogene Research Products, San Diego, CA), cell regulation, signal transduction, inflammation, oxidative processes and apoptosis (Assay Designs Inc., Ann Arbor, MI), triglyceride accumulation (Sigma- Aldrich, St.
- angiogenesis assays i.e., MCF-7 cells selected for breast cancer studies; adipocytes for obesity studies
- a target inhibitors identified from the in vitro studies as well as control compounds at optimal concentrations which are determined by the methods described above.
- treated and untreated cells are analyzed by one or more methods specific for the assay to determine phenotypic outcomes and endpoints.
- Phenotypic endpoints include changes in cell morphology over time or treatment dose as well as changes in levels of cellular components such as proteins, lipids, nucleic acids, hormones, saccharides or metals. Measurements of cellular status which include pH, stage of the cell cycle, intake or excretion of biological indicators by the cell, are also endpoints of interest.
- Measurement of the expression of one or more of the genes of the cell after treatment is also used as an indicator of the efficacy or potency of the target inhibitors.
- Hallmark genes or those genes suspected to be associated with a specific disease state, condition, or phenotype, are measured in both treated and untreated cells.
- the individual subjects of the in vivo studies described herein are warm-blooded vertebrate animals, which includes humans.
- Poly(A)+ mRNA is isolated according to Miura et al, (Clin. Chem., 1996, 42, 1758-1764). Other methods for poly(A)+ mRNA isolation are routine in the art. Briefly, for cells grown on 96- well plates, growth medium is removed from the cells and each well is washed with 200 xL cold PBS. 60 ⁇ -L lysis buffer (10 mM Tris-HCl, pH 7.6, 1 mM EDTA, 0.5 M NaCl, 0.5% NP-40, 20 mM vanadyl-ribonucleoside complex) is added to each well, the plate is gently agitated and then incubated at room temperature for five minutes.
- 60 ⁇ -L lysis buffer (10 mM Tris-HCl, pH 7.6, 1 mM EDTA, 0.5 M NaCl, 0.5% NP-40, 20 mM vanadyl-ribonucleoside complex
- lysate is transferred to Oligo d(T) coated 96-well plates (AGCT Inc., Irvine CA). Plates are incubated for 60 minutes at room temperature, washed 3 times with 200 ⁇ -L of wash buffer (10 mM Tris-HCl pH 7.6, 1 mM EDTA, 0.3 M NaCl). After the final wash, the plate is blotted on paper towels to remove excess wash buffer and then air- dried for 5 minutes. 60 xL of elution buffer (5 mM Tris-HCl pH 7.6), preheated to 70°C, is added to each well, the plate is incubated on a 90°C hot plate for 5 minutes, and the eluate is then transferred to a fresh 96-well plate.
- wash buffer 10 mM Tris-HCl pH 7.6, 1 mM EDTA, 0.3 M NaCl
- 60 xL of elution buffer (5 mM Tris-HCl pH 7.6), preheated to 70°C, is added to each well,
- Cells grown on 100 mm or other standard plates may be treated similarly, using appropriate volumes of all solutions.
- Total RNA is isolated using an RNEASY 96TM kit and buffers purchased from Qiagen Inc. (Valencia, CA) following the manufacturer's recommended procedures. Briefly, for cells grown on 96-well plates, growth medium is removed from the cells and each well is washed with 200 xL cold PBS. 150 xL Buffer RLT is added to each well and the plate vigorously agitated for 20 seconds. 150 xL of 70% ethanol is then added to each well and the contents mixed by pipetting three times up and down. The samples are then transferred to the RNEASY 96TM well plate attached to a
- QIAVACTM manifold fitted with a waste collection tray and attached to a vacuum source. Vacuum is applied for 1 minute. 500 of Buffer RW1 is added to each well of the RNEASY 96TM plate and incubated for 15 minutes and the vacuum is again applied for 1 minute. An additional 500 xL of Buffer RW1 is added to each well of the RNEASY 96TM plate and the vacuum is applied for 2 minutes. 1 mL of Buffer RPE is then added to each well of the RNEASY 96TM plate and the vacuum applied for a period of 90 seconds. The Buffer RPE wash is then repeated and the vacuum is applied for an additional 3 minutes. The plate is then removed from the QIAVACTM manifold and blotted dry on paper towels.
- RNA is then eluted by pipetting 140 xL of RNAse free water into each well, incubating 1 minute, and then applying the vacuum for 3 minutes.
- the repetitive pipetting and elution steps may be automated using a QIAGEN Bio-Robot 9604 (Qiagen, Inc., Valencia CA). Essentially, after lysing of the cells on the culture plate, the plate is transferred to the robot deck where the pipetting, DNase treatment and elution steps are carried out.
- Probes and primers may be designed to hybridize to a target sequence, using published sequence information.
- the following primer-probe set was designed using published sequence information (GENBANKTM accession number U92436.1, SEQ ID NO: 1).
- Reverse primer TGCACATATCATTACACCAGTTCGT (SEQ ID NO: 3)
- FAM-TTGCAGCAATTCACTGTAAAGCTGGAAAGG-TAMRA (SEQ ID NO: 4), where FAM is the fluorescent dye and TAMRA is the quencher dye.
- Diacetone glucose, Compound 1 is commercially available.
- Compound 5 is prepared as per the procedures illustrated in Example 13. In the tritylation step, other hydroxyl protecting groups can be used in place of DMTC1 (e.g. levulinic acid).
- tert-Butyldiphenylchlorosilane (305.0 mmol, 84.0 mL) was added to a cold (0 °C) stirring solution of Compound 47 (278.0 mmol, 100.0 g) and triethylamine (305 mmol, 43.0 mL) in dichloromethane (600 mL). After the addition was complete, the reaction was warmed to room temperature and the stirring was continued for 16 hours. MeOH (50 mL) was added (to quench the excess TBDPSC1) to the reaction and the stirring was continued for another 2 hours at room temperature. The reaction was then diluted with chloroform and the organic layer was washed with 10% HC1, saturated NaHC0 3 , brine, dried (Na 2 S0 4 ) and concentrated to provide a thick oil.
- Tetrabutylammonium fluoride 70 mL of a 1M solution in THF was added to a cold (0 °C) stirring solution of Compound 49 (62.7 mmol, 37.5 g) in THF (250 mL) after which, the reaction was allowed to warm to room temperature gradually. After stirring for an additional 72 hours, the reaction was concentrated under vacuum and the residue was poured onto crushed ice. The flask was rinsed with some additional THF (3 times) and added to the above suspension. The supernatant was removed by decantation and the solid at the bottom was added to a stirring mixture of hexanes (200 mL) and water (200 mL). After stirring for 2 hours, the flocculent solid was collected by filtration, washed with additional water and hexanes and dried under high vacuum to provide Compound 47 (20 g, 89%) as a white solid.
- Compound 48 was prepared as per the procedures illustrated in Example 18. To a solution of alcohol, Compound 48 (5.0 g, 8.35 mmol) in DCM (60 mL) was added pyridinium
- reaction mixture was poured into a cooled, aqueous solution of 1 M HC1 (20 mL) and then partitioned with dichloro methane (15 mL). The organic phase was collected and the aqueous portion was back-extracted with DCM (3 ⁇ 10 mL). The organic phases were combined, washed with a saturated aqueous solution of sodium bicarbonate (15 mL), dried through a phase separator cartridge and concentrated under reduced pressure.
- Compound 58 was prepared as per the procedures illustrated in Example 19. Analytical data of Compound 76 was consistent with the structure. The transformation of the anhydronucleoside, Compound 76 to tricylic nucleoside,
- Compound 69 is then carried out by desilylation, mesylation followed by basic hydrolysis and ring closing in the presence of NaOH.
- R d H, Ci-C 6 alkyl, substituted C 1 -C5 alkyl, C 2 -C6 alkenyl, substituted C 2 -C6 alkenyl, C 2 -C 6 alkynyl, substituted C 2 -C 6 alkynyl, substituted acyl, aryl, substituted aryl, a heterocyclic radical, a substituted heterocyclic radical or a protecting group.
- ⁇ Rd H, Ci-C 6 alkyl, substituted C 1 -C5 alkyl, C 2 -C6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted C 2 -C 6 alkynyl, substituted acyl, aryl, substituted aryl, a heterocyclic radical, a substituted heterocyclic radical or a protecting group.
- LNA locked nucleic acid
- oc-L-TriNA a-L-tricyclic nucleic acid
- oligomeric compounds presented in Table 1 were synthesized on a 1 ⁇ scale using the UnyLinkerTM polystyrene support on an ABI 394 DNA/RNA synthesizer. Standard conditions were used to incorporate phosphoramidite building blocks of the unmodified nucleosides which include for example T, G, A and C residues. Solvents and reagents used during synthesis were prepared according to the indications of the manufacturer. A 0.
- the coupling was carried out manually using two syringes by dissolving the tricyclic phosphoramidite (33 ⁇ ) in dichloromethane (400 ⁇ ), mixing with 0. 5 M solution of ETT in acetonitrile (600 ⁇ ) and contacting with the solid support for 30 min. The coupling time was extended to 30 min with the coupling efficiency of approximately 90%.
- compositions of the oligonucleotides are described in Table 1.
- Nucleosides without a subscript are -D-2'-deoxyribonucleosides.
- Nucleosides followed by a subscript "1", "a” or "b” are defined below.
- the oligomeric compounds in Table 1 were evaluated for thermal stability (T m ) using the method described herein.
- T m thermal stability
- the T m of the modified 12mer oligomeric compounds were compared to an unmodified 12mer DNA oligonucleotide when duplexed to DNA or RNA complement.
- Tm's were determined using a Cary 100 Bio spectrophotometer with the Cary Win UV thermal program was used to measure absorbance vs. temperature.
- the oligomeric compounds were prepared at a concentration of 8 ⁇ in 10 mM sodium phosphate buffer (pH 7) containing 100 mM NaCl and 0.1 mM EDTA. The concentrations of each oligomeric compound was determined at 85 °C in T m buffer.
- the oligomeric compounds were hybridized with a complimentaryDNA or RNA by heating the duplex to 90 °C for 5 minutes and then cooling to room temperature. T m measurements were taken using a spectrophotometer while the duplex solution was heated in a cuvette at a rate of 0.5 °C/min starting at 15 °C until the temperature was 85 °C. T m values were determined using Vant Hoff calculations (A 260 vs temperature curve) using non self-complementary sequences where the minimum absorbance related to the duplex and the maximum absorbance related to the non-duplex single strand are manually integrated into the program. Thermal stability of the oligomeric compounds versus the mismatched RNA complement was also measured and the results are presented in Table 2.
- the oligomeric compound, ISIS 583517 was prepared in same manner as described previously and was evaluated for thermal stability in a different T m sequence than the one described above.
- the T m measurement was performed using similar protocol as illustrated in Example 24.
- the T m of the modified 12mer oligomeric compounds were compared to an unmodified 12mer DNA oligonucleotide when duplexed to DNA or RNA complement. The results are presented in Table 3.
- the sequence for DNA complement, ISIS A01 is designated herein as SEQ ID No. : 10, 5'- d(GC AT ATC ACTGG)-3 ' .
- the sequence for RNA complement, ISIS A02 is designated herein as SEQ ID No. : 11, 5'-r(GCAUAUCACUGG)-3'.
- compositions of the oligonucleotides are described in Table 3.
- Nucleosides without a subscript are -D-2'-deoxyribonucleosides.
- Nucleosides followed by a subscript "b" or "1" is defined in the same manner as illustrated in Example 23.
Abstract
The present disclosure provides tricyclic nucleosides and oligomeric compounds prepared therefrom. More particularly, the tricyclic nucleosides provided herein comprise a tricyclic ribosyl sugar moiety having a bridge between the 4' and 2' ring carbon atoms and a further fused carbocyclic or heterocyclic ring at the 3' and 4' carbon atoms. The tricyclic nucleosides are expected to be useful for enhancing properties of oligomeric compounds including for example binding affinity and nuclease resistance.
Description
TRICYCLIC NUCLEOSIDES AND OLIGOMERIC
COMPOUNDS PREPARED THEREFROM
SEQUENCE LISTING
The present application is being filed along with a Sequence Listing in electronic format. The Sequence Listing is provided as a file entitled CHEM0082WOSEQ.TXT created March 20, 2013, which is 8 Kb in size. The information in the electronic format of the sequence listing is incorporated herein by reference in its entirety.
FIELD OF THE INVENTION
The present disclosure provides tricyclic nucleosides and oligomeric compounds prepared therefrom. More particularly, the tricyclic nucleosides provided herein comprise a tricyclic ribosyl sugar moiety having a bridge between the 4' and 2' ring carbon atoms and a further fused carbocyclic or heterocyclic ring at the 3' and 4' carbon atoms. The tricyclic nucleosides are expected to be useful for enhancing one or more properties of the oligomeric compounds they are
incorporated into such as for example nuclease resistance. In certain embodiments, the oligomeric compounds provided herein are expected to hybridize to a portion of a target RNA resulting in loss of normal function of the target RNA.
BACKGROUND OF THE INVENTION
Antisense technology is an effective means for reducing the expression of one or more specific gene products and can therefore prove to be uniquely useful in a number of therapeutic, diagnostic, and research applications. Chemically modified nucleosides are routinely used for incorporation into antisense sequences to enhance one or more properties such as for example nuclease resistance. One such group of chemical modifications includes bicyclic nucleosides wherein the furanose portion of the nucleoside includes a bridge connecting two atoms on the furanose ring thereby forming a bicyclic ring system. Such bicyclic nucleosides have various names including BNA's and LNA's for bicyclic nucleic acids or locked nucleic acids respectively.
Various BNA's have been prepared and reported in the patent literature as well as in scientific literature, see for example: Singh et al, Chem. Commun., 1998, 4, 455-456; Koshkin et al, Tetrahedron, 1998, 54, 3607-3630; Wahlestedt et al, Proc. Natl. Acad. Sci. U. S. A, 2000, 97,
5633-5638; Kumar et al, Bioorg. Med. Chem. Lett., 1998, 8, 2219-2222; Wengel et al, PCT
International Application WO 98-DK393 19980914; Singh et al, J. Org. Chem., 1998, 63, 10035- 10039, the text of each is incorporated by reference herein, in their entirety. Examples of issued US patents and published applications include for example: U.S. Patents 7,053,207, 6,770,748,
6,268,490 and 6,794,499 and published U.S. applications 20040219565, 20040014959,
20030207841, 20040192918, 20030224377, 20040143114 and 20030082807; the text of each is incorporated by reference herein, in their entirety.
In a recent in vivo study with LNA in mice, hepatotoxicity was reported. See, e.g., Swayze et al, Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals, Nucl. Acids Res., 2007, 35(2), 687-700.
Although less studied, tricyclic nucleosides "Tricyclo-DNA" has also been prepared as monomers, has been incorporated into oligomeric compounds and has been evaluated in certain biological assays (see: Steffens et al, Helvetica Chimica Acta., 1997, 80, 2426-2439; Steffens et al, J. Am. Chem. Soc, 1997, 119, 11548-11549; Steffens et al, J. Org. Chem., 1999, 121, 3249-3255; Renneberg et al, J. Am. Chem. Soc, 2002, 124, 5993-6002; Ittig et al, Nucl. Acids Res., 2004, 32(1), 346-353; Ittig et al, Collection Symposium Series, 2005, 12, 8014-8023; and Ivanova et al, Oligonucleotides, 2007, 17, 54-65).
BRIEF SUMMARY OF THE INVENTION
Disclosed herein are novel tricyclic nucleosides and antisense oligomeric compounds prepared therefrom. More particularly, novel tricyclic nucleosides are provided wherein each furanose ring comprises a 4'-CH2-0-2' bridge and a further carbocyclic or heterocyclic ring fused to the 3' and 4' atoms. Oligomeric compounds comprising one or more of the tricyclic nucleosides are expected to be useful for modulating gene expression pathways, including those relying on mechanisms of action such as RNaseH, RNAi and dsRNA enzymes, as well as other antisense mechanisms based on target degradation or target occupancy. One having skill in the art, once armed with this disclosure will be able, without undue experimentation, to identify, prepare and exploit antisense compounds for these uses.
The variables are defined individually in further detail herein. It is to be understood that the tricyclic nucleosides and oligomeric compounds prepared therefrom provided herein include all combinations of the embodiments disclosed and variables defined herein.
In certain embodiments, tricyclic nucleosides are provided having Formula I:

wherein:
Bx is a heterocyclic base moiety;
one of Ti and T2 is hydroxyl or a protected hydroxyl and the other of Ti and T2 is H;
T3 is hydroxyl, a protected hydroxyl, an H-phosphonate or a phosphoramidite group;
Q is CRiR2CR3R4, CH=CH, O, S or R5;
Ri, R2, R3 and R4 are each, independently, H, hydroxyl, halogen, Ci-C6 alkyl, substituted Ci- C6 alkyl, Ci-C6 alkoxy, substituted Ci-C6 alkoxy, amino or substituted amino;
R5 is H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl, substituted acyl, aryl, substituted aryl, a heterocyclic radical, a substituted heterocyclic radical or a protecting group;
wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, OJi, SJi, NJiJ2, N3, CN, C(=0)OJi, C(=0)NJiJ2, C(=0)Ji, 0-C(=0)NJiJ2, N(H)C(=0)NJiJ2, N(H)C(=S)NJiJ2 and a protecting group;
each Ji and J2 is, independently, H, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C6 aminoalkyl or a protecting group;
when Q is CRiR2CR3R4 or CH=CH then n is 1 and m is 0 or 1; and
when Q is O, S or R5 then m is 1 and n is 1 or 2.
In certain embodiments, Bx is an optionally protected pyrimidine, substituted pyrimidine, purine or substituted purine. In certain embodiments, Bx is an optionally protected uracil, thymine, cytosine, 5-methylcytosine, adenine or guanine.
In certain embodiments, one of Ti and T2 is a 4,4'-dimethoxytrityl protected hydroxyl group. In certain embodiments, T3 is diisopropylcyanoethoxy phosphoramidite.
In certain embodiments, Q is CH=CH. In certain embodiments, Q is CRiR2CR3R4. In certain embodiments, at least one of Ri, R2, R3 and R4 IS Ci-C6 alkyl. In certain embodiments, at least one of Ri, R2, R3 and R4 is CH3. In certain embodiments, at least one of Ri, R2, R3 and R4 is substituted Ci-C6 alkyl. In certain embodiments, one of Ri and R2 is Ci-C6 alkyl or substituted Ci-
C6 alkyl and one of R3 and R^s Ci-C6 alkyl or substituted Ci-C6 alkyl. In certain embodiments, Ri, R2, R3 and R4 are each H.
In certain embodiments, m is 0. In certain embodiments, m is 1.
In certain embodiments, Q is O.
In certain embodiments, Q is S.
In certain embodiments, Q is R5. In certain embodiments, R5 is H, Ci-C6 alkyl or substituted Ci-C6 alkyl. In certain embodiments, R5 is methyl.
In certain embodiments, n is 1. In certain embodiments, n is 2.
In certain embodiments, tricyclic nucleosides are provided having the configuration of Formula la:

In certain embodiments, tricyclic nucleosides are provided having the configuration of Formula la, Ti is 4,4'-dimethoxytrityl protected hydroxyl and T3 is diisopropylcyanoethoxy phosphoramidite. In certain embodiments, tricyclic nucleosides are provided having the
configuration of Formula la, T2 is 4,4'-dimethoxytrityl protected hydroxyl and T3 is
diisopropylcyanoethoxy phosphoramidite.
In certain embodiments, oligomeric compounds are provided comprising at least one tricyclic nucleoside having Formula II:

wherein independently for each tricyclic nucleoside of Formula II:
Bx is a heterocyclic base moiety;
one of T4 and T5 is hydroxyl, a protected hydroxyl, a 5' terminal group or an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound and the other of T4 and T5 is H;
T6 is hydroxyl, a protected hydroxyl, a 3' terminal group or an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound;
Q is CR1R2CR3R4, CH=CH, O, S or R5;
Ri, R2, R3 and R4 are each, independently, H, hydroxyl, halogen, Ci-C6 alkyl, substituted Ci- C6 alkyl, Ci-C6 alkoxy, substituted Ci-C6 alkoxy, amino or substituted amino;
R5 is H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl, substituted acyl, aryl, substituted aryl, a heterocyclic radical, a substituted heterocyclic radical or a protecting group;
wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, OJi, SJi, NJiJ2, N3, CN, C(=0)OJi, C(=0)NJiJ2, C(=0)Ji, 0-C(=0)NJiJ2, N(H)C(=0)NJiJ2, N(H)C(=S)NJiJ2 and a protecting group;
each Ji and J2 is, independently, H, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C6 aminoalkyl or a protecting group;
when Q is CRiR2CR3R4 or CH=CH then n is 1 and m is 0 or 1;
when Q is O, S or R5 then m is 1 and n is 1 or 2; and
wherein at least one of T4, T5 and T6 is an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound.
In certain embodiments, each Bx is, independently, an optionally protected pyrimidine, substituted pyrimidine, purine or substituted purine. In certain embodiments, each Bx is, independently, an optionally protected uracil, thymine, cytosine, 5-methylcytosine, adenine or guanine.
In certain embodiments, each Q is CH=CH.
In certain embodiments, Q is CRiR2CR3R4. In certain embodiments, one of Ri, R2, R3 and R4 is Ci-C6 alkyl for each tricyclic nucleoside having Formula II. In certain embodiments, one of Ri, R2, R3 and R4 IS CH3 for each tricyclic nucleoside having Formula II. In certain embodiments, one of Ri, R2, R3 and R4 IS substituted Ci-C6 alkyl for each tricyclic nucleoside having Formula II. In certain embodiments, one of Ri and R2 is Ci-C6 alkyl or substituted Ci-C6 alkyl and one of R3 and R4 IS Ci-C6 alkyl or substituted Ci-C6 alkyl for each tricyclic nucleoside having Formula II.
In certain embodiments, each Q is CH2CH2.
In certain embodiments, each m is 0. In certain embodiments, each m is 1.
In certain embodiments, each Q is O.
In certain embodiments, each Q is S.
In certain embodiments, each Q is R5. In certain embodiments, each R5 is H, Ci-C6 alkyl or substituted Ci-C6 alkyl. In certain embodiments, each R5 is methyl.
In certain embodiments, each n is 1. In certain embodiments, n is 2.
In certain embodiments, oligomeric compounds are provided comprising at least one tricyclic nucleoside wherein each tricyclic nucleoside has the configuration of Formula Ila:

In certain embodiments, oligomeric compounds are provided comprising at least one tricyclic nucleoside of Formula II or Ila wherein each tricyclic nucleoside is uniformly modified having the same stereochemistry wherein the heterocyclic base is variable. Such tricyclic nucleosides are identical except that they can have different heterocyclic bases and that they can be at terminal or internal positions within the oligomeric compound such that T4, T5 and T6 may be the same or different between each tricyclic nucleoside.
In certain embodiments, oligomeric compounds are provided wherein each internucleoside linking group between adjacent monomeric subunits is, independently, a phosphodiester internucleoside linking group or a phosphorothioate internucleoside linking group. In certain embodiments, oligomeric compounds are provided wherein each internucleoside linking group between adjacent monomeric subunits is a phosphorothioate internucleoside linking group
In certain embodiments, oligomeric compounds are provided comprising a first region having at least two contiguous tricyclic nucleosides having Formula II or Ila. In certain
embodiments, oligomeric compounds are provided comprising a first region having at least two contiguous tricyclic nucleosides having Formula II or Ila and further comprising a second region having at least two contiguous monomeric subunits wherein each monomeric subunit in the second region is a modified nucleoside different from the tricyclic nucleosides of said first region. In certain embodiments, oligomeric compounds are provided comprising a first region having at least two contiguous tricyclic nucleosides having Formula II or Ila, a second region having at least two contiguous monomeric subunits wherein each monomeric subunit in the second region is a modified nucleoside different from the tricyclic nucleosides of said first region and further comprising a third region located between said first and second regions wherein each monomer subunit in the third
region is independently, a nucleoside or a modified nucleoside that is different from each tricyclic nucleoside of said first region and each monomer subunit the second region.
In certain embodiments, gapped oligomeric compounds are provided comprising, an internal region of from 6 to 14 contiguous monomer subunits flanked on each side by an external region of from 1 to 5 contiguous monomer subunits wherein each monomer subunit in each external region is tricyclic nucleoside of Formula II or Ila, and each monomer subunit in the internal region is, independently, a nucleoside or modified nucleoside. In certain embodiments, the internal region comprises from about 8 to about 14 contiguous P-D-2'-deoxyribonucleosides. In certain
embodiments, the internal region comprises from about 9 to about 12 contiguous β-ϋ-2'- deoxyribonucleosides.
In certain embodiments, methods of inhibiting gene expression are provided comprising contacting a cell with an oligomeric compound as provided herein wherein said oligomeric compound comprises from about 8 to about 40 monomeric subunits and is complementary to a target RNA. In certain embodiments, the cell is in an animal. In certain embodiments, the cell is in a human. In certain embodiments, the target RNA is selected from mRNA, pre-mRNA and micro RNA. In certain embodiments, the target RNA is mRNA. In certain embodiments, the target RNA is human mRNA. In certain embodiments, the target RNA is cleaved thereby inhibiting its function. In certain embodiments, the method further comprises detecting the levels of target RNA.
In certain embodiments, an in vitro method of inhibiting gene expression is provided comprising contacting one or more cells or a tissue with an oligomeric compound as provided herein.
In certain embodiments, oligomeric compounds are provided for use in an in vivo method of inhibiting gene expression said method comprising contacting one or more cells, a tissue or an animal with an oligomeric compound as provided herein.
In certain embodiments, methods of preparing tricyclic nucleosides are provided, said methods comprising: reacting a first reactant and a second reactant in the presence of a solvent; wherein the first reactant comprises a compound represented by formula V:

and wherein each R13 and Ri4 are indepdendently selected from the group consisting of H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl and a protecting group.
In certain embodiments a pharmaceutical composition is provided, wherein the
pharmaceutical composition comprises the compound of any of formulas I-V and a
pharmaceutically acceptable salt, carrier, or diluent.
In certain embodiments a method of modulating target mRNA in a cell is provided, said method comprising contacting the cell with an oligomeric compound according to any of formulas I- V or a pharmaceutical composition comprising an oligomeric compound according to any of formulas I-V. In certain embodiments the cell is in vitro. In certain embodiments, the cell is in an animal. In certain embodiments, the animal is a human.
DETAILED DESCRIPTION OF THE INVENTION
Provided herein are novel tricyclic nucleosides and oligomeric compounds prepared therefrom. The tricyclic nucleosides are expected to be useful for enhancing one or more properties of the oligomeric compounds they are incorporated into such as for example nuclease resistance. In certain embodiments, the oligomeric compounds provided herein are expected to hybridize to a portion of a target RNA resulting in loss of normal function of the target RNA.
In certain embodiments, the tricyclic nucleosides provided herein can be incorporated into antisense oligomeric compounds to reduce target RNA, such as messenger RNA, in vitro and in vivo. In one aspect the reduction of target RNA is useful for inhibition of gene expression via numerous pathways. Such pathways include for example the steric blocking of transcription or translation and cleavage of mRNA via single or double stranded oligomeric compounds. The oligomeric compounds provided herein are also expected to be useful as primers and probes in diagnostic applications. In certain embodiments, oligomeric compounds comprising at least one of the tricyclic nucleosides provided herein are expected to be useful as aptamers which are oligomeric compounds capable of binding to aberrant proteins in an in vivo setting.
In certain embodiments, tricyclic nucleosides are provided having Formula I:


wherein:
Bx is a heterocyclic base moiety;
one of Ti and T2 is hydroxyl or a protected hydroxyl and the other of Ti and T2 is H;
T3 is hydroxyl, a protected hydroxyl, an H-phosphonate or a phosphoramidite group;
Q is CRiR2CR3R4, CH=CH, O, S or R5;
Ri, R2, R3 and R4 are each, independently, H, hydroxyl, halogen, Ci-C6 alkyl, substituted Ci- C6 alkyl, Ci-C6 alkoxy, substituted Ci-C6 alkoxy, amino or substituted amino;
R5 is H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl, substituted acyl, aryl, substituted aryl, a heterocyclic radical, a substituted heterocyclic radical or a protecting group;
wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, OJi, SJi, NJiJ2, N3, CN, C(=0)OJi, C(=0)NJiJ2, C(=0)Ji, 0-C(=0)NJiJ2, N(H)C(=0)NJiJ2, N(H)C(=S)NJiJ2 and a protecting group;
each Ji and J2 is, independently, H, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C6 aminoalkyl or a protecting group;
when Q is CRiR2CR3R4 or CH=CH then n is 1 and m is 0 or 1; and
when Q is O, S or R5 then m is 1 and n is 1 or 2.
The tricyclic nucleoside of claim 1 wherein Bx is an optionally protected pyrimidine, substituted pyrimidine, purine or substituted purine.
In certain embodiments, Bx is an optionally protected uracil, thymine, cytosine, 5- methylcytosine, adenine or guanine. In certain embodiments, one of Ti and T2 is a 4,4'- dimethoxytrityl protected hydroxyl group. In certain embodiments, T3 is diisopropylcyanoethoxy phosphoramidite. In certain embodiments, Q is CH=CH. In certain embodiments, Q is
CRiR2CR3R4. In certain embodiments, at least one of Ri, R2, R3 and R4 is Ci-C6 alkyl. In certain embodiments, at least one of Ri, R2, R3 and R4 IS CH3. In certain embodiments, at least one of Ri, R2, R3 and R4 IS substituted Ci-C6 alkyl. In certain embodiments, one of Ri and R2 is Ci-C6 alkyl or substituted Ci-C6 alkyl and one of R3 and R4 IS Ci-C6 alkyl or substituted Ci-C6 alkyl. In certain embodiments, Ri, R2, R3 and R4 are each H. In certain embodiments, m is 0. In certain
embodiments, m is 1. In certain embodiments, Q is O. In certain embodiments, Q is S. In certain
embodiments, Q is R5. In certain embodiments, R5 is H, Ci-C6 alkyl or substituted Ci-C6 alkyl. In certain embodiments, R5 is methyl. In certain embodiments, n is 1. In certain embodiments, n is 2.
In certain embodiments, tricyclic nucleosides of Formula I are provided having the configuration of Formula la:

In certain embodiments, Ti is 4,4'-dimethoxytrityl protected hydroxyl and T3 is
diisopropylcyanoethoxy phosphoramidite. In certain embodiments, T2 is 4,4'-dimethoxytrityl protected hydroxyl and T3 is diisopropylcyanoethoxy phosphoramidite.
In certain embodiments, oligomeric compounds are provided having at least one tricyclic nucleoside having formula II:

wherein independently for each tricyclic nucleoside of Formula II:
Bx is a heterocyclic base moiety;
one of T4 and T5 is hydroxyl, a protected hydroxyl, a 5' terminal group or an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound and the other of T4 and
T5 is H;
T6 is hydroxyl, a protected hydroxyl, a 3' terminal group or an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound;
Q is CRiR2CR3R4, CH=CH, O, S or R5;
Ri, R2, R3 and R4 are each, independently, H, hydroxyl, halogen, Ci-C6 alkyl, substituted Ci- C6 alkyl, Ci-C6 alkoxy, substituted Ci-C6 alkoxy, amino or substituted amino;
R5 is H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl, substituted acyl, aryl, substituted aryl, a heterocyclic radical, a substituted heterocyclic radical or a protecting group;
wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, OJi, SJi,
NJiJ2, N3, CN, C(=0)OJi, C(=0)NJiJ2, C(=0)Ji, 0-C(=0)NJiJ2, N(H)C(=0)NJiJ2, N(H)C(=S)NJiJ2 and a protecting group;
each Ji and J2 is, independently, H, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C6 aminoalkyl or a protecting group;
when Q is CRiR2CR3R4 or CH=CH then n is 1 and m is 0 or 1;
when Q is O, S or R5 then m is 1 and n is 1 or 2; and
wherein at least one of T4, T5 and T6 is an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound.
In certain embodiments, each Bx is, independently, an optionally protected pyrimidine, substituted pyrimidine, purine or substituted purine. In certain embodiments, each Bx is, independently, an optionally protected uracil, thymine, cytosine, 5-methylcytosine, adenine or guanine. In certain embodiments, each Q is CH=CH. In certain embodiments, each Q is
CRiR2CR3R . In certain embodiments, one of Ri, R2, R3 and R4 IS Ci-C6 alkyl for each tricyclic nucleoside having Formula II. In certain embodiments, one of Ri, R2, R3 and R4 is CH3 for each tricyclic nucleoside having Formula II. In certain embodiments, one of Ri, R2, R3 and R4 is substituted Ci-C6 alkyl for each tricyclic nucleoside having Formula II. In certain embodiments, one of Ri and R2 is Ci-C6 alkyl or substituted Ci-C6 alkyl and one of R3 and R4 IS Ci-C6 alkyl or substituted Ci-C6 alkyl for each tricyclic nucleoside having Formula II. In certain embodiments, each Q is CH2CH2. In certain embodiments, each m is 0. In certain embodiments, each m is 1. In certain embodiments, each Q is O. In certain embodiments, each Q is S. In certain embodiments, each Q is R5. In certain embodiments, each R5 is H, Ci-C6 alkyl or substituted Ci-C6 alkyl. In certain embodiments, each R5 is methyl. In certain embodiments, each n is i . In certain
embodiments, each n is 2.
In certain embodiments, oligomeric compounds are provided comprising at least one tricyclic nucleoside of Formula II wherein each tricyclic nucleoside of Formula II has the configuration of Formula Ila:
In certain embodiments, oligomeric compounds are provided comprising at least one tricyclic nucleoside of Formula II or Ila wherein each tricyclic nucleoside is uniformly modified having the same stereochemistry wherein the heterocyclic base is variable. Such tricyclic nucleosides are identical except that they can have different heterocyclic bases and they can be at different positions such as internally or terminally within the oligomeric compound. Thus T4, T5 and T6 may be the same or different between one tricyclic nucleoside and the next.
In certain embodiments oligomeric compounds are provided wherein each internucleoside linking group between adjacent monomeric subunits is, independently, a phosphodiester
internucleoside linking group or a phosphorothioate internucleoside linking group. In certain embodiments, essentially each internucleoside linking group between adjacent monomeric subunits is a phosphorothioate internucleoside linking group.
In certain embodiments, oligomeric compounds are provided comprising a first region having at least two contiguous tricyclic nucleosides having Formula II or Ila. In certain
embodiments, oligomeric compounds are provided comprising a second region having at least two contiguous monomeric subunits wherein each monomeric subunit in the second region is a modified nucleoside different from the tricyclic nucleosides of said first region. In certain embodiments, oligomeric compounds are provided comprising a third region located between said first and second regions wherein each monomer subunit in the third region is independently, a nucleoside or a modified nucleoside that is different from each tricyclic nucleoside of said first region and each monomer subunit the second region.
In certain embodiments, oligomeric compounds are provided comprising a gapped oligomeric compound having an internal region of from 6 to 14 contiguous monomer subunits flanked on each side by an external region of from 1 to 5 contiguous monomer subunits wherein each monomer subunit in each external region is tricyclic nucleoside of Formula II or Ila and each monomer subunit in the internal region is, independently, a nucleoside or modified nucleoside. In certain embodiments, said internal region comprises from about 8 to about 14 contiguous β-ϋ-2'- deoxyribonucleosides. In certain embodiments, said internal region comprises from about 9 to about 12 contiguous P-D-2'-deoxyribonucleosides.
In certain embodiments, tricyclic nucleosides having Formula III are provided:

Bx is a heterocyclic base moiety;
each Zi, Z2, and Z3 is independently selected from the group consisting of CH2-CH2,
CH=CH, CR7R8, O, S, or R9;
each R7 and R8 are indepdendently selected from the group consisting of H, hydroxyl, protected hydroxyl, halogen, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl, Ci-C6 alkoxy, Ci-C6 substituted alkoxy, an alkyl ester, and a protecting group;
R9 is H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl or a protecting group;
wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, OJi, SJi, NJiJ2, N3, CN, C(=0)OJi, C(=0)NJiJ2, C(=0)Ji, 0-C(=0)NJiJ2, N(H)C(=0)NJiJ2, N(H)C(=S)NJiJ2 and a protecting group;
each Ji and J2 is, independently, H, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C6 aminoalkyl or a protecting group;
one of T7 and T8 is hydroxyl or a protected hydroxyl and the other of T7 and T8 is H; and T9 is hydroxyl, a protected hydroxyl, an H-phosphonate or a phosphoramidite group.
In certain embodiments, oligomeric compounds are provided comprising at least one tricyclic nucleoside having Formula IV:

wherein independently for each tricyclic nucleoside having Formula II:
Bx is a heterocyclic base moiety;
each Z4, Z5, and Z6 is independently selected from the group consisting of CH2-CH2, CH=CH, CR10R11, O, S, or Ri2;
each Rio and Rn are indepdendently selected from the group consisting of H, hydroxyl, protected hydroxyl, halogen, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl, Ci-C6 alkoxy, Ci-C6 substituted alkoxy, an alkyl ester, and a protecting group;
Ri2 is H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl or a protecting group;
wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, OJi, SJi, NJiJ2, N3, CN, C(=0)OJi, C(=0)NJiJ2, C(=0)Ji, 0-C(=0)NJiJ2, N(H)C(=0)NJiJ2, N(H)C(=S)NJiJ2 and a protecting group;
each Ji and J2 is, independently, H, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C6 aminoalkyl or a protecting group;
one of Tio and Tn is hydroxyl, a protected hydroxyl, a 5' terminal group or an
internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound and the other of Tio and Tn is H;
T12 is hydroxyl, a protected hydroxyl, a 3' terminal group or an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound; and
wherein at least one of Tio, Tn and Ti2 is an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound.
In certain embodiments, methods of preparing a tricyclic nucleoside are provided, said methods comprising: reacting a first reactant and a second reactant in the presence of a solvent; wherein the first reactant comprises a compound represented by formula V:

wherein each Ri3 and Ri4 are indepdendently selected from the group consisting of H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl and a protecting group.
In certain embodiments, oligomeric compounds are provided comprising the protecting group is selected from the group consisting of benzyl, phenyl, naphthyl, triflate, trimethylsilyl, tert- butyldiphenylsilyl, tert-butyldimethylsilyl, and benzyl chloromethyl ether. In certain embodiments, the second reactant is an amino acid. In certain embodiments, the second reactant is L-proline. In certain embodiments the solvent is selected from among a polar solvent, a polar aprotic solvent, a polar protic solvent, an apolar solvent, an apolar aprotic solvent, and an apolar protic solvent. In certain embodiments the solvent is selected from the group consisting of DMF and DMSO.
In certain embodiments the tricyclic nucleoside comprises a compound having Formula VI:

In certain embodiments pharmaceutical compositions are provided comprising the compound of any of Formulas I-IV and a pharmaceutically acceptable salt, carrier, or diluent.
In certain embodiments methods of modulating target mRNA in a cell are provided, said methods comprising contacting the cell with an oligomeric compound according to any of Formulas I-IV or a pharmaceutical composition thereof. In certain embodiments the cell is in vitro. In certain embodiments the cell is in an animal. In certain embodiments the animal is a human.
Incorporation of one or more of the tricyclic nucleosides, as provided herein, into an oligomeric compound is expected to enhance one or more desired properties of the resulting oligomeric compound. Such properties include without limitation stability, nuclease resistance, binding affinity, specificity, absorption, cellular distribution, cellular uptake, charge, pharmacodynamics and pharmacokinetics.
In certain embodiments, the tricyclic nucleosides provided herein are incorporated into oligomeric compounds such that a motif results. The placement of tricyclic nucleosides into oligomeric compounds to provide particular motifs can enhance the desired properties of the resulting oligomeric compounds for activity using a particular mechanism such as RNaseH or RNAi. Such motifs include without limitation, gapmer motifs, hemimer motifs, blockmer motifs, uniformly fully modified motifs, positionally modified motifs and alternating motifs. In conjunction with these motifs a wide variety of internucleoside linkages can also be used including but not limited to phosphodiester and phosphorothioate internucleoside linkages which can be incorporated
uniformly or in various combinations. The oligomeric compounds can further include a 5' and or 3' terminal group such as a conjugate or reporter group. The positioning of the tricyclic nucleosides provided herein, the use of linkage strategies and 5' and or 3' terminal groups can be easily optimized to enhance a desired activity for a selected target.
As used herein the term "motif refers to the pattern created by the relative positioning of monomer subunits within an oligomeric compound wherein the pattern is determined by comparing the sugar moieties of the linked monomer subunits. The only determinant for the motif of an oligomeric compound is the differences or lack of differences between the sugar moieties. The internucleoside linkages, heterocyclic bases and further groups such as terminal groups are not considered when determining the motif of an oligomeric compound.
Representative U.S. patents that teach the preparation of motifs include without limitation, 5,013,830; 5, 149,797; 5,220,007; 5,256,775; 5,366,878; 5,403,711; 5,491, 133; 5,565,350;
5,623,065; 5,652,355; 5,652,356; and 5,700,922, certain of which are commonly owned with the instant application, and each of which is herein incorporated by reference in its entirety. Motifs are also disclosed in International Applications PCT/US2005/019219, filed June 2, 2005 and published as WO 2005/121371 on December 22, 2005 and PCT/US2005/019220, filed June 2, 2005 and published as WO 2005/121372 on December 22, 2005; each of which is incorporated by reference herein in its entirety.
As used herein the term "alternating motif refers to an oligomeric compound comprising a contiguous sequence of linked monomer subunits wherein the monomer subunits have two different types of sugar moieties that alternate for essentially the entire sequence of the oligomeric compound. Oligomeric compounds having an alternating motif can be described by the formula: 5'-A(-L-B-L- A)n(-L-B)nn-3' where A and B are monomer subunits that have different sugar moieties, each L is, independently, an internucleoside linking group, n is from about 4 to about 12 and nn is 0 or 1. The heterocyclic base and internucleoside linkage is independently variable at each position. The motif further optionally includes the use of one or more other groups including but not limited to capping groups, conjugate groups and other 5' and or 3'-terminal groups. This permits alternating oligomeric compounds from about 9 to about 26 monomer subunits in length. This length range is not meant to be limiting as longer and shorter oligomeric compounds are also amenable to oligomeric compounds provided herein. In certain embodiments, each A or each B comprise tricyclic nucleosides as provided herein.
As used herein the term "uniformly fully modified motif refers to an oligomeric compound comprising a contiguous sequence of linked monomer subunits that each have the same type of sugar moiety. The heterocyclic base and internucleoside linkage is independently variable at each position. The motif further optionally includes the use of one or more other groups including but not limited to capping groups, conjugate groups and other 5' and or 3'-terminal groups. In certain embodiments, the uniformly fully modified motif includes a contiguous sequence of tricyclic nucleosides. In certain embodiments, one or both of the 5' and 3 '-ends of the contiguous sequence of tricyclic nucleosides, comprise 5' and or 3'-terminal groups such as one or more unmodified nucleosides.
As used herein the term "hemimer motif refers to an oligomeric compound comprising a contiguous sequence of monomer subunits that each have the same type of sugar moiety with a further short contiguous sequence of monomer subunits located at the 5' or the 3' end that have a different type of sugar moiety. The heterocyclic base and internucleoside linkage is independently variable at each position. The motif further optionally includes the use of one or more other groups including but not limited to capping groups, conjugate groups and other 5' and or 3'-terminal groups. In general, a hemimer is an oligomeric compound of uniform sugar moieties further comprising a short region (1, 2, 3, 4 or about 5 monomer subunits) having uniform but different sugar moieties located on either the 3' or the 5' end of the oligomeric compound.
In certain embodiments, the hemimer motif comprises a contiguous sequence of from about 10 to about 28 monomer subunits having one type of sugar moiety with from 1 to 5 or from 2 to about 5 monomer subunits having a second type of sugar moiety located at one of the termini. In certain embodiments, the hemimer is a contiguous sequence of from about 8 to about 20 β-D-2'- deoxyribonucleosides having from 1-12 contiguous tricyclic nucleosides located at one of the termini. In certain embodiments, the hemimer is a contiguous sequence of from about 8 to about 20 β-D-2'-deoxyribonucleosides having from 1-5 contiguous tricyclic nucleosides located at one of the termini. In certain embodiments, the hemimer is a contiguous sequence of from about 12 to about 18 β-D-2'-deoxyribonucleosides having from 1-3 contiguous tricyclic nucleosides located at one of the termini. In certain embodiments, the hemimer is a contiguous sequence of from about 10 to about 14 β-D-2'-deoxyribonucleosides having from 1-3 contiguous tricyclic nucleosides located at one of the termini.
As used herein the terms "blockmer motif and "blockmer" refer to an oligomeric compound comprising an otherwise contiguous sequence of monomer subunits wherein the sugar moieties of
each monomer subunit is the same except for an interrupting internal block of contiguous monomer subunits having a different type of sugar moiety. The heterocyclic base and internucleoside linkage is independently variable at each position of a blockmer. The motif further optionally includes the use of one or more other groups including but not limited to capping groups, conjugate groups and other 5' or 3'-terminal groups. A blockmer overlaps somewhat with a gapmer in the definition but typically only the monomer subunits in the block have non-naturally occurring sugar moieties in a blockmer and only the monomer subunits in the external regions have non-naturally occurring sugar moieties in a gapmer with the remainder of monomer subunits in the blockmer or gapmer being β- D-2'-deoxyribonucleosides or β-D-ribonucleosides. In certain embodiments, blockmers are provided herein wherein all of the monomer subunits comprise non-naturally occurring sugar moieties.
As used herein the term "positionally modified motif is meant to include an otherwise contiguous sequence of monomer subunits having one type of sugar moiety that is interrupted with two or more regions of from 1 to about 5 contiguous monomer subunits having another type of sugar moiety. Each of the two or more regions of from 1 to about 5 contiguous monomer subunits are independently uniformly modified with respect to the type of sugar moiety. In certain embodiments, each of the two or more regions have the same type of sugar moiety. In certain embodiments, each of the two or more regions have a different type of sugar moiety. In certain embodiments, each of the two or more regions, independently, have the same or a different type of sugar moiety. The heterocyclic base and internucleoside linkage is independently variable at each position of a positionally modified oligomeric compound. The motif further optionally includes the use of one or more other groups including but not limited to capping groups, conjugate groups and other 5' or 3'- terminal groups. In certain embodiments, positionally modified oligomeric compounds are provided comprising a sequence of from 8 to 20 P-D-2'-deoxyribonucleosides that further includes two or three regions of from 2 to about 5 contiguous tricyclic nucleosides each. Positionally modified oligomeric compounds are distinguished from gapped motifs, hemimer motifs, blockmer motifs and alternating motifs because the pattern of regional substitution defined by any positional motif does not fit into the definition provided herein for one of these other motifs. The term positionally modified oligomeric compound includes many different specific substitution patterns.
As used herein the term "gapmer" or "gapped oligomeric compound" refers to an oligomeric compound having two external regions or wings and an internal region or gap. The three regions form a contiguous sequence of monomer subunits with the sugar moieties of the external regions
being different than the sugar moieties of the internal region and wherein the sugar moiety of each monomer subunit within a particular region is essentially the same. In certain embodiments, each monomer subunit within a particular region has the same sugar moiety. When the sugar moieties of the external regions are the same the gapmer is a symmetric gapmer and when the sugar moiety used in the 5 '-external region is different from the sugar moiety used in the 3 '-external region, the gapmer is an asymmetric gapmer. In certain embodiments, the external regions are small (each
independently 1, 2, 3, 4 or about 5 monomer subunits) and the monomer subunits comprise non- naturally occurring sugar moieties with the internal region comprising P-D-2'-deoxyribonucleosides. In certain embodiments, the external regions each, independently, comprise from 1 to about 5 monomer subunits having non-naturally occurring sugar moieties and the internal region comprises from 6 to 18 unmodified nucleosides. The internal region or the gap generally comprises β-ϋ-2'- deoxyribonucleosides but can comprise non-naturally occurring sugar moieties. The heterocyclic base and internucleoside linkage is independently variable at each position of a gapped oligomeric compound. The motif further optionally includes the use of one or more other groups including but not limited to capping groups, conjugate groups and other 5' or 3'-terminal groups.
In certain embodiments, the gapped oligomeric compounds comprise an internal region of P-D-2'-deoxyribonucleosides with one of the external regions comprising tricyclic nucleosides as disclosed herein. In certain embodiments, the gapped oligomeric compounds comprise an internal region of P-D-2'-deoxyribonucleosides with both of the external regions comprising tricyclic nucleosides as provided herein. In certain embodiments, gapped oligomeric compounds are provided herein wherein all of the monomer subunits comprise non-naturally occurring sugar moieties.
In certain embodiments, gapped oligomeric compounds are provided comprising one or two tricyclic nucleosides at the 5'-end, two or three tricyclic nucleosides at the 3'-end and an internal region of from 10 to 16 P-D-2'-deoxyribonucleosides. In certain embodiments, gapped oligomeric compounds are provided comprising one tricyclic nucleoside at the 5'-end, two tricyclic nucleosides at the 3 '-end and an internal region of from 10 to 16 P-D-2'-deoxyribonucleosides. In certain embodiments, gapped oligomeric compounds are provided comprising one tricyclic nucleosides at the 5'-end, two tricyclic nucleosides at the 3'-end and an internal region of from 10 to 14 β-ϋ-2'- deoxyribonucleosides.
In certain embodiments, gapped oligomeric compounds are provided that are from about 10 to about 21 monomer subunits in length. In certain embodiments, gapped oligomeric compounds
are provided that are from about 12 to about 16 monomer subunits in length. In certain embodiments, gapped oligomeric compounds are provided that are from about 12 to about 14 monomer subunits in length. In certain embodiments, gapped oligomeric compounds are provided that are from about 14 to about 16 monomer subunits in length.
As used herein the term "alkyl," refers to a saturated straight or branched hydrocarbon radical containing up to twenty four carbon atoms. Examples of alkyl groups include without limitation, methyl, ethyl, propyl, butyl, isopropyl, n-hexyl, octyl, decyl, dodecyl and the like. Alkyl groups typically include from 1 to about 24 carbon atoms, more typically from 1 to about 12 carbon atoms (C1-C12 alkyl) with from 1 to about 6 carbon atoms being more preferred. The term "lower alkyl" as used herein includes from 1 to about 6 carbon atoms. Alkyl groups as used herein may optionally include one or more further substituent groups.
As used herein the term "alkenyl," refers to a straight or branched hydrocarbon chain radical containing up to twenty four carbon atoms and having at least one carbon-carbon double bond. Examples of alkenyl groups include without limitation, ethenyl, propenyl, butenyl, l-methyl-2- buten-l-yl, dienes such as 1,3-butadiene and the like. Alkenyl groups typically include from 2 to about 24 carbon atoms, more typically from 2 to about 12 carbon atoms with from 2 to about 6 carbon atoms being more preferred. Alkenyl groups as used herein may optionally include one or more further substituent groups.
As used herein the term "alkynyl," refers to a straight or branched hydrocarbon radical containing up to twenty four carbon atoms and having at least one carbon-carbon triple bond.
Examples of alkynyl groups include, without limitation, ethynyl, 1-propynyl, 1-butynyl, and the like. Alkynyl groups typically include from 2 to about 24 carbon atoms, more typically from 2 to about 12 carbon atoms with from 2 to about 6 carbon atoms being more preferred. Alkynyl groups as used herein may optionally include one or more further substituent groups.
As used herein the term "acyl," refers to a radical formed by removal of a hydroxyl group from an organic acid and has the general Formula -C(0)-X where X is typically aliphatic, alicyclic or aromatic. Examples include aliphatic carbonyls, aromatic carbonyls, aliphatic sulfonyls, aromatic sulfinyls, aliphatic sulfinyls, aromatic phosphates, aliphatic phosphates and the like. Acyl groups as used herein may optionally include further substituent groups.
As used herein the term "alicyclic" refers to a cyclic ring system wherein the ring is aliphatic. The ring system can comprise one or more rings wherein at least one ring is aliphatic.
Preferred alicyclics include rings having from about 5 to about 9 carbon atoms in the ring. Alicyclic as used herein may optionally include further substituent groups.
As used herein the term "aliphatic," refers to a straight or branched hydrocarbon radical containing up to twenty four carbon atoms wherein the saturation between any two carbon atoms is a single, double or triple bond. An aliphatic group preferably contains from 1 to about 24 carbon atoms, more typically from 1 to about 12 carbon atoms with from 1 to about 6 carbon atoms being more preferred. The straight or branched chain of an aliphatic group may be interrupted with one or more heteroatoms that include nitrogen, oxygen, sulfur and phosphorus. Such aliphatic groups interrupted by heteroatoms include without limitation, polyalkoxys, such as polyalkylene glycols, polyamines, and polyimines. Aliphatic groups as used herein may optionally include further substituent groups.
As used herein the term "alkoxy," refers to a radical formed between an alkyl group and an oxygen atom wherein the oxygen atom is used to attach the alkoxy group to a parent molecule. Examples of alkoxy groups include without limitation, methoxy, ethoxy, propoxy, isopropoxy, n- butoxy, sec-butoxy, tert-butoxy, n-pentoxy, neopentoxy, n-hexoxy and the like. Alkoxy groups as used herein may optionally include further substituent groups.
As used herein the term "aminoalkyl" refers to an amino substituted C1-C12 alkyl radical. The alkyl portion of the radical forms a covalent bond with a parent molecule. The amino group can be located at any position and the aminoalkyl group can be substituted with a further substituent group at the alkyl and/or amino portions.
As used herein the terms "aralkyl" and "arylalkyl," refer to an aromatic group that is covalently linked to a C1-C12 alkyl radical. The alkyl radical portion of the resulting aralkyl (or arylalkyl) group forms a covalent bond with a parent molecule. Examples include without limitation, benzyl, phenethyl and the like. Aralkyl groups as used herein may optionally include further substituent groups attached to the alkyl, the aryl or both groups that form the radical group.
As used herein the terms "aryl" and "aromatic," refer to a mono- or poly cyclic carbocyclic ring system radicals having one or more aromatic rings. Examples of aryl groups include without limitation, phenyl, naphthyl, tetrahydronaphthyl, indanyl, idenyl and the like. Preferred aryl ring systems have from about 5 to about 20 carbon atoms in one or more rings. Aryl groups as used herein may optionally include further substituent groups.
As used herein the terms "halo" and "halogen," refer to an atom selected from fluorine, chlorine, bromine and iodine.
As used herein the terms "heteroaryl," and "heteroaromatic," refer to a radical comprising a mono- or poly-cyclic aromatic ring, ring system or fused ring system wherein at least one of the rings is aromatic and includes one or more heteroatoms. Heteroaryl is also meant to include fused ring systems including systems where one or more of the fused rings contain no heteroatoms.
Heteroaryl groups typically include one ring atom selected from sulfur, nitrogen or oxygen.
Examples of heteroaryl groups include without limitation, pyridinyl, pyrazinyl, pyrimidinyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isooxazolyl, thiadiazolyl, oxadiazolyl, thiophenyl, furanyl, quinolinyl, isoquinolinyl, benzimidazolyl, benzooxazolyl, quinoxalinyl and the like. Heteroaryl radicals can be attached to a parent molecule directly or through a linking moiety such as an aliphatic group or hetero atom. Heteroaryl groups as used herein may optionally include further substituent groups.
As used herein the term "heteroarylalkyl," refers to a heteroaryl group as previously defined that further includes a covalently attached C1-C12 alkyl radical. The alkyl radical portion of the resulting heteroarylalkyl group is capable of forming a covalent bond with a parent molecule.
Examples include without limitation, pyridinylmethyl, pyrimidinylethyl, napthyridinylpropyl and the like. Heteroarylalkyl groups as used herein may optionally include further substituent groups on one or both of the heteroaryl or alkyl portions.
As used herein the term "heterocyclic radical" refers to a radical mono-, or poly-cyclic ring system that includes at least one heteroatom and is unsaturated, partially saturated or fully saturated, thereby including heteroaryl groups. Heterocyclic is also meant to include fused ring systems wherein one or more of the fused rings contain at least one heteroatom and the other rings can contain one or more heteroatoms or optionally contain no heteroatoms. A heterocyclic radical typically includes at least one atom selected from sulfur, nitrogen or oxygen. Examples of heterocyclic radicals include, [l,3]dioxolanyl, pyrrolidinyl, pyrazolinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, piperidinyl, piperazinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl, isothiazolidinyl, quinoxalinyl, pyridazinonyl, tetrahydrofuryl and the like. Heterocyclic groups as used herein may optionally include further substituent groups.
As used herein the term "oxo" refers to the group (=0).
As used herein the term "protecting group," refers to a labile chemical moiety which is known in the art to protect reactive groups including without limitation, hydroxyl, amino and thiol groups, against undesired reactions during synthetic procedures. Protecting groups are typically used selectively and/or orthogonally to protect sites during reactions at other reactive sites and can
then be removed to leave the unprotected group as is or available for further reactions. Protecting groups as known in the art are described generally in Greene's Protective Groups in Organic
Synthesis, 4th edition, John Wiley & Sons, New York, 2007.
Groups can be selectively incorporated into oligomeric compounds as provided herein as precursors. For example an amino group can be placed into a compound as provided herein as an azido group that can be chemically converted to the amino group at a desired point in the synthesis. Generally, groups are protected or present as precursors that will be inert to reactions that modify other areas of the parent molecule for conversion into their final groups at an appropriate time. Further representative protecting or precursor groups are discussed in Agrawal et al, Protocols for Oligonucleotide Conjugates, Humana Press; New Jersey, 1994, 26, 1-72.
The term "orthogonally protected" refers to functional groups which are protected with different classes of protecting groups, wherein each class of protecting group can be removed in any order and in the presence of all other classes (see, Barany et al, J. Am. Chem. Soc, 1977, 99, 7363- 7365; Barany et al, J. Am. Chem. Soc, 1980, 102, 3084-3095). Orthogonal protection is widely used in for example automated oligonucleotide synthesis. A functional group is deblocked in the presence of one or more other protected functional groups which is not affected by the deblocking procedure. This deblocked functional group is reacted in some manner and at some point a further orthogonal protecting group is removed under a different set of reaction conditions. This allows for selective chemistry to arrive at a desired compound or oligomeric compound.
Examples of hydroxyl protecting groups include without limitation, acetyl, t-butyl, t- butoxymethyl, methoxymethyl, tetrahydropyranyl, 1-ethoxyethyl, l-(2-chloroethoxy)ethyl, p- chlorophenyl, 2,4-dinitrophenyl, benzyl, 2,6-dichlorobenzyl, diphenylmethyl, p-nitrobenzyl, bis(2- acetoxyethoxy)methyl (ACE), 2-trimethylsilylethyl, trimethylsilyl, triethylsilyl, t-butyldimethylsilyl, t-butyldiphenylsilyl, triphenylsilyl, [(triisopropylsilyl)oxy]methyl (TOM), benzoylformate, chloroacetyl, trichloroacetyl, trifluoroacetyl, pivaloyl, benzoyl, p-phenylbenzoyl, 9-fluorenylmethyl carbonate, mesylate, tosylate, triphenylmethyl (trityl), monomethoxytrityl, dimethoxytrityl (DMT), trimethoxytrityl, l(2-fluorophenyl)-4-methoxypiperidin-4-yl (FPMP), 9-phenylxanthine-9-yl (Pixyl) and 9-(p-methoxyphenyl)xanthine-9-yl (MOX). Wherein more commonly used hydroxyl protecting groups include without limitation, benzyl, 2,6-dichlorobenzyl, t-butyldimethylsilyl, t-butyl- diphenylsilyl, benzoyl, mesylate, tosylate, dimethoxytrityl (DMT), 9-phenylxanthine-9-yl (Pixyl) and 9-(p-methoxyphenyl)xanthine-9-yl (MOX).
Examples of amino protecting groups include without limitation, carbamate-protecting groups, such as 2-trimethylsilylethoxycarbonyl (Teoc), 1 -methyl- l-(4-biphenylyl)ethoxycarbonyl (Bpoc), t-butoxycarbonyl (BOC), allyloxycarbonyl (Alloc), 9-fluorenylmethyloxycarbonyl (Fmoc), and benzyloxycarbonyl (Cbz); amide-protecting groups, such as formyl, acetyl, trihaloacetyl, benzoyl, and nitrophenylacetyl; sulfonamide-protecting groups, such as 2-nitrobenzenesulfonyl; and imine- and cyclic imide-protecting groups, such as phthalimido and dithiasuccinoyl.
Examples of thiol protecting groups include without limitation, triphenylmethyl (trityl), benzyl (Bn), and the like.
The compounds described herein contain one or more asymmetric centers and thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)- or (S)-, a or β, or as (D)- or (L)- such as for amino acids. Included herein are all such possible isomers, as well as their racemic and optically pure forms. Optical isomers may be prepared from their respective optically active precursors by the procedures described above, or by resolving the racemic mixtures. The resolution can be carried out in the presence of a resolving agent, by chromatography or by repeated crystallization or by some combination of these techniques which are known to those skilled in the art. Further details regarding resolutions can be found in Jacques, et ah, Enantiomers, Racemates, and Resolutions, John Wiley & Sons, 1981. When the compounds described herein contain olefinic double bonds, other unsaturation, or other centers of geometric asymmetry, and unless specified otherwise, it is intended that the compounds include both E and Z geometric isomers or cis- and trans-isomers. Likewise, all tautomeric forms are also intended to be included. The configuration of any carbon- carbon double bond appearing herein is selected for convenience only and is not intended to limit a particular configuration unless the text so states.
The terms "substituent" and "substituent group," as used herein, are meant to include groups that are typically added to other groups or parent compounds to enhance desired properties or provide other desired effects. Substituent groups can be protected or unprotected and can be added to one available site or to many available sites in a parent compound. Substituent groups may also be further substituted with other substituent groups and may be attached directly or via a linking group such as an alkyl or hydrocarbyl group to a parent compound.
Substituent groups amenable herein include without limitation, halogen, hydroxyl, alkyl, alkenyl, alkynyl, acyl (-C(0)Raa), carboxyl (-C(0)0-Raa), aliphatic groups, alicyclic groups, alkoxy, substituted oxy (-0-Raa), aryl, aralkyl, heterocyclic radical, heteroaryl, heteroarylalkyl, amino
(-N(Rbb)(Rcc)), imino(= Rbb), amido (-C(0)N(Rbb)(Rcc) or -N(Rbb)C(0)Raa), azido (-N3), nitro (-NO2), cyano (-CN), carbamido (-OC(0)N(Rbb)(Rcc) or -N(Rbb)C(0)ORaa), ureido (-N(Rbb)C(0)- N(Rbb)(Rcc)), thioureido (-N(Rbb)C(S)N(Rbb)(RcC)), guanidinyl (-N(Rbb)C(= Rbb)N(Rbb)(Rcc)), amidinyl (-C(= Rbb)N(Rbb)(RcC) or -N(Rbb)C(= Rbb)(Raa)), thiol (-SRbb), sulfinyl (-S(0)Rbb), sulfonyl (-S(0)2Rbb) and sulfonamidyl (-S(0)2N(Rbb)(Rcc) or -N(Rbb)S(0)2Rbb). Wherein each Raa, Rbb and Rcc is, independently, H, an optionally linked chemical functional group or a further substituent group with a preferred list including without limitation, H, alkyl, alkenyl, alkynyl, aliphatic, alkoxy, acyl, aryl, aralkyl, heteroaryl, alicyclic, heterocyclic and heteroarylalkyl. Selected substituents within the compounds described herein are present to a recursive degree.
In this context, "recursive substituent" means that a substituent may recite another instance of itself. Because of the recursive nature of such substituents, theoretically, a large number may be present in any given claim. One of ordinary skill in the art of medicinal chemistry and organic chemistry understands that the total number of such substituents is reasonably limited by the desired properties of the compound intended. Such properties include, by way of example and not limitation, physical properties such as molecular weight, solubility or logP, application properties such as activity against the intended target and practical properties such as ease of synthesis.
Recursive substituents are an intended aspect of the invention. One of ordinary skill in the art of medicinal and organic chemistry understands the versatility of such substituents. To the degree that recursive substituents are present in a claim of the invention, the total number will be determined as set forth above.
The terms "stable compound" and "stable structure" as used herein are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent. Only stable compounds are contemplated herein.
As used herein, the term "nucleobase" refers to unmodified or naturally occurring nucleobases which include, but are not limited to, the purine bases adenine (A) and guanine (G), and the pyrimidine bases thymine (T), cytosine (C) and uracil (U).
As used herein the term "heterocyclic base moiety" refers to unmodified or naturally occurring nucleobases as well as modified or non-naturally occurring nucleobases and synthetic mimetics thereof (such as for example phenoxazines). In one embodiment, a heterocyclic base moiety is any heterocyclic system that contains one or more atoms or groups of atoms capable of hydrogen bonding to a heterocyclic base of a nucleic acid.
In certain embodiments, heterocyclic base moieties include without limitation modified nucleobases such as 5-methylcytosine (5-me-C), 5-hydroxymethyl cytosine, xanthine, hypoxanthine, 2-aminoadenine, 6-methyl and other alkyl derivatives of adenine and guanine, 2-propyl and other alkyl derivatives of adenine and guanine, 2-thiouracil, 2-thiothymine and 2-thiocytosine, 5- halouracil and cytosine, 5-propynyl (-C≡C-CH3) uracil and cytosine and other alkynyl derivatives of pyrimidine bases, 6-azo uracil, cytosine and thymine, 5-uracil (pseudouracil), 4-thiouracil, 8-halo, 8- amino, 8-thiol, 8-thioalkyl, 8-hydroxyl and other 8-substituted adenines and guanines, 5-halo particularly 5-bromo, 5-trifluoromethyl and other 5-substituted uracils and cytosines, 7- methylguanine and 7-methyladenine, 2-F-adenine, 2-amino-adenine, 8-azaguanine and 8- azaadenine, 7-deazaguanine and 7-deazaadenine, 3-deazaguanine and 3-deazaadenine, universal bases, hydrophobic bases, promiscuous bases, size-expanded bases, and fluorinated bases as defined herein.
In certain embodiments, heterocyclic base moieties include without limitation tricyclic pyrimidines such as l,3-diazaphenoxazine-2-one, l,3-diazaphenothiazine-2-one and 9-(2- aminoethoxy)-l,3-diazaphenoxazine-2-one (G-clamp). Heterocyclic base moieties also include those in which the purine or pyrimidine base is replaced with other heterocycles, for example 7- deaza-adenine, 7-deazaguanosine, 2-aminopyridine and 2-pyridone. Further heterocyclic base moieties include without limitation those known to the art skilled (see for example: United States Patent No. 3,687,808; Swayze et al, The Medicinal Chemistry of Oligonucleotides in Antisense a Drug Technology, Chapter 6, pages 143-182, Crooke, S.T., ed., 2008); The Concise Encyclopedia Of Polymer Science And Engineering, Kroschwitz, J.I., Ed., John Wiley & Sons, 1990, 858-859; Englisch et al., Angewandte Chemie, International Edition, 1991, 30, 613; Sanghvi, Y.S., Chapter 15, Antisense Research and Applications, Crooke, S.T. and Lebleu, B., Eds., CRC Press, 1993, 273- 302). Modified polycyclic heterocyclic compounds useful as heterocyclic base moieties are disclosed in the above noted U.S. 3,687,808, as well as U.S. : 4,845,205; 5, 130,302; 5, 134,066; 5, 175,273; 5,367,066; 5,432,272; 5,434,257; 5,457, 187; 5,459,255; 5,484,908; 5,502, 177;
5,525,711; 5,552,540; 5,587,469; 5,594, 121, 5,596,091; 5,614,617; 5,645,985; 5,646,269;
5,681,941; 5,750,692; 5,763,588; 5,830,653; 6,005,096; and U.S. Patent Application Publication 20030158403, each of which is incorporated herein by reference in its entirety.
As used herein the term "sugar moiety" refers to naturally occurring sugars having a furanose ring, synthetic or non-naturally occurring sugars having a modified furanose ring and sugar surrogates wherein the furanose ring has been replaced with a cyclic ring system such as for example a
morpholino or hexitol ring system or a non-cyclic sugar surrogate such as that used in peptide nucleic acids. Illustrative examples of sugar moieties useful in the preparation of oligomeric compounds include without limitation, β-D-ribose, P-D-2'-deoxyribose, substituted sugars (such as 2', 5' and bis substituted sugars), 4'-S-sugars (such as 4'-S-ribose, 4'-S-2'-deoxyribose and 4 -S-2'- substituted ribose), bicyclic modified sugars (such as the 2'-0-CH2-4' or 2'-0-(CH2)2-4' bridged ribose derived bicyclic sugars) and sugar surrogates (such as for example when the ribose ring has been replaced with a morpholino, a hexitol ring system or an open non-cyclic system).
As used herein the term "sugar substituent group" refers to groups that are covalently attached to sugar moieties. In certain embodiments, examples of sugar substituent groups include without limitation 2'-F, 2'-allyl, 2'-amino, 2'-azido, 2'-thio, 2'-0-allyl, 2'-OCF3, 2'-O-Ci-Ci0 alkyl, 2'- OCH3, 2'-0(CH2)nCH3, 2'-OCH2CH3, 2'-0-(CH2)2CH3, 2'-0-(CH2)2-0-CH3 (MOE), 2'- 0[(CH2)nO]mCH3, 2'-0(CH2)2SCH3, 2'-0-(CH2)3-N(Rp)(Rq), 2'-0(CH2)n H2, 2'-0-(CH2)2-0- N(Rp)(Rq), 0(CH2)nON[(CH2)nCH3]2, 2'-0(CH2)nO H2, 2'-0-(CH2)2-0-(CH2)2-N(Rp)(Rq), 2'-0- CH2C(=0)-N(Rp)(Rq), 2'-OCH2C(=0)N(H)CH3, 2'-0-CH2C(=0)-N(H)-(CH2)2-N(Rp)(Rq) and 2'-0- CH2-N(H)-C(= Rr)[N(Rp)(Rq)], 5'-vinyl, 5'-methyl (R or S) and 4*-S wherein each Rp, Rq and Rr is, independently, H, substituted or unsubstituted Ci-Cio alkyl or a protecting group and where n and m are from 1 to about 10. Further examples of modified sugar moieties include without limitation bicyclic sugars used in bicyclic nucleosides.
In certain embodiments, examples of sugar substituent groups include without limitation substituted silyl, an RNA cleaving group, a reporter group, an intercalator, a group for improving pharmacokinetic properties, or a group for improving the pharmacodynamic properties of an oligomeric compound, and other substituents having similar properties. In certain embodiments, oligomeric compounds include modifed nucleosides comprising 2'-MOE substituent groups (Baker et al, J. Biol. Chem., 1997, 272, 11944-12000). Such 2'-MOE substitution has been described as having improved binding affinity compared to unmodified nucleosides and to other modified nucleosides, such as 2'-0-methyl, 2'-0-propyl, and 2'-0-aminopropyl. Oligonucleotides having the 2'-MOE substituent also have been shown to be antisense inhibitors of gene expression with promising features for in vivo use (Martin, V ., Helv. Chim. Acta, 1995, 78, 486-504; Altmann et al, Chimia, 1996, 50, 168-176; Altmann et al, Biochem. Soc. Trans., 1996, 24, 630-637; and Altmann et al, Nucleosides Nucleotides, 1997, 16, 917-926).
Sugar moieties can be substituted with combinations of sugar substituent groups including without limitation 2'-F-5'-methyl substituted nucleosides (see PCT International Application WO
2008/101157, published on 8/21/08 for other disclosed 5', 2'-bis substituted nucleosides). Other combinations are also possible, including without limitation, replacement of the ribosyl ring oxygen atom with S and further substitution at the 2'-position (see published U.S. Patent Application US2005-0130923, published on June 16, 2005) and 5'-substitution of a bicyclic nucleoside (see PCT International Application WO 2007/134181, published on 11/22/07 wherein a 4'-CH2-0-2' bicyclic nucleoside is further substituted at the 5' position with a 5'-methyl or a 5'-vinyl group).
As used herein, the term "nucleoside" refers to a nucleobase-sugar combination. The two most common classes of such nucleobases are purines and pyrimidines.
As used herein, the term nucleotide refers to a nucleoside further comprising a modified or unmodified phosphate internucleoside linking group or a non-phosphate internucleoside linking group. For nucleotides that include a pentofuranosyl sugar, the internucleoside linking group can be linked to either the 2', 3' or 5' hydroxyl moiety of the sugar. The phosphate and or a non-phosphate internucleoside linking groups are routinely used to covalently link adjacent nucleosides to one another to form a linear polymeric compound.
The term "nucleotide mimetic" as used herein is meant to include monomers that incorporate into oligomeric compounds with sugar and linkage surrogate groups, such as for example peptide nucleic acids (PNA) or morpholinos (linked by -N(H)-C(=0)-0-). In general, the heterocyclic base at each position is maintained for hybridization to a nucleic acid target but the sugar and linkage is replaced with surrogate groups that are expected to function similar to native groups but have one or more enhanced properties.
As used herein the term "nucleoside mimetic" is intended to include those structures used to replace the sugar and the base at one or more positions of an oligomeric compound. Examples of nucleoside mimetics include without limitation nucleosides wherein the heterocyclic base moiety is replaced with a phenoxazine moiety (for example the 9-(2-aminoethoxy)-l,3-diazaphenoxazine-2- one group, also referred to as a G-clamp which forms four hydrogen bonds when hybridized with a guanosine base) and further replacement of the sugar moiety with a group such as for example a morpholino, a cyclohexenyl or a bicyclo[3.1.0]hexyl.
As used herein the term "modified nucleoside" is meant to include all manner of modified nucleosides that can be incorporated into an oligomeric compound using oligomer synthesis. The term is intended to include modifications made to a nucleoside such as modified stereochemical configurations, one or more substitutions, and deletion of groups as opposed to the use of surrogate groups which are described elsewhere herein. The term includes nucleosides having a furanose
sugar (or 4'-S analog) portion and can include a heterocyclic base or can be an abasic nucleoside. One group of representative modified nucleosides includes without limitation, substituted nucleosides (such as 2', 5', and/or 4' substituted nucleosides) 4'-S-modified nucleosides, (such as 4'- S-ribonucleosides, 4'-S-2'-deoxyribonucleosides and 4'-S-2'-substituted ribonucleosides), bicyclic modified nucleosides (such as for example, bicyclic nucleosides wherein the sugar moiety has a 2'- 0-CHRa-4' bridging group, wherein Ra is H, alkyl or substituted alkyl) and base modified nucleosides. The sugar can be modified with more than one of these modifications listed such as for example a bicyclic modified nucleoside further including a 5'-substitution or a 5' or 4' substituted nucleoside further including a 2' substituent. The term modified nucleoside also includes combinations of these modifications such as base and sugar modified nucleosides. These modifications are meant to be illustrative and not exhaustive as other modifications are known in the art and are also envisioned as possible modifications for the modified nucleosides described herein.
As used herein the term "monomer subunit" is meant to include all manner of monomer units that are amenable to oligomer synthesis with one preferred list including monomer subunits such as β-D-ribonucleosides, P-D-2'-deoxyribnucleosides, modified nucleosides, including substituted nucleosides (such as 2', 5' and bis substituted nucleosides), 4'-S-modified nucleosides, (such as 4'-S- ribonucleosides, 4'-S-2'-deoxyribonucleosides and 4'-S-2'-substituted ribonucleosides), bicyclic modified nucleosides (such as bicyclic nucleosides wherein the sugar moiety has a 2'-0-CHRa-4' bridging group, wherein Ra is H, alkyl or substituted alkyl), other modified nucleosides, nucleoside mimetics, nucleosides having sugar surrogates and the tricyclic nucleosides as provided herein.
As used herein the term "bicyclic nucleoside" refers to a nucleoside comprising at least a bicyclic sugar moiety. Examples of bicyclic nucleosides include without limitation nucleosides having a furanosyl sugar that comprises a bridge between two of the non-geminal carbons, preferably the 4' and the 2' carbon atoms. In certain embodiments, oligomeric compounds provided herein include one or more 4' to 2' bridged bicyclic nucleosides. Examples of such 4' to 2' bridged bicyclic nucleosides, include but are not limited to one of formulae: 4'-(CH2)-0-2' (LNA); 4'-(CH2)- S-2'; 4'-(CH2)2-0-2' (ENA); 4'-CH(CH3)-0-2' and 4'-CH(CH2OCH3)-0-2' (and analogs thereof see U.S. Patent 7,399,845, issued on July 15, 2008); 4'-C(CH3)(CH3)-0-2' (and analogs thereof see published International Application WO/2009/006478, published January 8, 2009); 4'-CH2- N(OCH3)-2' (and analogs thereof see published International Application WO/2008/150729, published December 11, 2008); 4'-CH2-0-N(CH3)-2' (see published U.S. Patent Application
US2004-0171570, published September 2, 2004 ); 4'-CH2-N(R)-0-2', wherein R is H, Ci-Ci2 alkyl,
or a protecting group (see U.S. Patent 7,427,672, issued on September 23, 2008); 4'-CH2-C- (H)(CH3)-2* (see Chattopadhyaya, et al, J. Org. Chem., 2009, 74, 118-134); and 4'-CH2-C(=CH2)-2' (and analogs thereof see published International Application WO 2008/154401, published on December 8, 2008). Further bicyclic nucleosides have been reported in published literature (see for example: Srivastava et al, J. Am. Chem. Soc, 2007, 129(26) 8362-8379; Frieden et al., Nucleic Acids Research, 2003, 21, 6365-6372; Elayadi et al, Curr. Opinion Inverts. Drugs, 2001, 2, 558- 561; Braasch et al., Chem. Biol, 2001, 8, 1-7; Orum et al., Curr. Opinion Mol. Ther., 2001, 3, 239- 243; Wahlestedt et al, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5633-5638; Singh et al, Chem. Commun., 1998, 4, 455-456; Koshkin et al, Tetrahedron, 1998, 54, 3607-3630; Kumar et al, Bioorg. Med. Chem. Lett., 1998, 8, 2219-2222; Singh et al, J. Org. Chem., 1998, 63, 10035-10039; U.S. Patents Nos. : 7,399,845; 7,053,207; 7,034, 133; 6,794,499; 6,770,748; 6,670,461; 6,525, 191; 6,268,490; U.S. Patent Publication Nos. : US2008-0039618; US2007-0287831; US2004-0171570; U.S. Patent Applications, Serial Nos. : 12/129, 154; 61/099,844; 61/097,787; 61/086,231;
61/056,564; 61/026,998; 61/026,995; 60/989,574; International applications WO 2007/134181; WO 2005/021570; WO 2004/106356; WO 94/14226; and PCT International Applications Nos. :
PCT/US2008/068922; PCT/US2008/066154; and PCT/US2008/064591). Each of the foregoing bicyclic nucleosides can be prepared having one or more stereochemical sugar configurations including for example a-L-ribofuranose and β-D-ribofuranose (see PCT international application PCT/DK98/00393, published on March 25, 1999 as WO 99/14226).
In certain embodiments, bicyclic nucleosides comprise a bridge between the 4' and the 2' carbon atoms of the pentofuranosyl sugar moiety including without limitation, bridges comprising 1 or from 1 to 4 linked groups independently selected from -[C(Ra)(Rb)]n-, -C(Ra)=C(Rb)-, -C(Ra)=N-, -C(=NRa)-, -C(=0)-, -C(=S)-, -0-, -Si(Ra)2-, -S(=0)x-, and -N(Ra)-; wherein: x is 0, 1, or 2; n is 1, 2, 3, or 4; each Ra and Rb is, independently, H, a protecting group, hydroxyl, Ci-Ci2 alkyl, substituted Ci-Ci2 alkyl, C2-Ci2 alkenyl, substituted C2-Ci2 alkenyl, C2-Ci2 alkynyl, substituted C2-Ci2 alkynyl, C5-C2o aryl, substituted Cs-C2o aryl, heterocycle radical, substituted heterocycle radical, heteroaryl, substituted heteroaryl, C5-C7 alicyclic radical, substituted C5-C7 alicyclic radical, halogen, OJi, NJiJ2, SJi, N3, COOJi, acyl (C(=0)-H), substituted acyl, CN, sulfonyl (S(=0)2-Ji), or sulfoxyl (S(=0)-Ji); and
each Ji and J2 is, independently, H, Ci-Ci2 alkyl, substituted Ci-Ci2 alkyl, C2-Ci2 alkenyl, substituted C2-Ci2 alkenyl, C2-Ci2 alkynyl, substituted C2-Ci2 alkynyl, Cs-C2o aryl, substituted C5- C2o aryl, acyl (C(=0)-H), substituted acyl, a heterocycle radical, a substituted heterocycle radical,
C1-C12 aminoalkyl, substituted C1-C12 aminoalkyl or a protecting group.
In certain embodiments, the bridge of a bicyclic sugar moiety is , -[C(Ra)(Rb)]n-,
-[C(Ra)(Rb)]n-0-, -C(RaRb)-N(R)-0- or -C(RaRb)-0-N(R)-. In certain embodiments, the bridge is 4'-CH2-2', 4'-(CH2)2-2', 4'-(CH2)3-2', 4'-CH2-0-2', 4'-(CH2)2-0-2', 4'-CH2-0-N(R)-2' and 4'-CH2- N(R)-0-2'- wherein each R is, independently, H, a protecting group or C1-C12 alkyl.
In certain embodiments, bicyclic nucleosides are further defined by isomeric configuration. For example, a nucleoside comprising a 4'-(CH2)-0-2' bridge, may be in the a-L configuration or in the β-D configuration. Previously, a-L-methyleneoxy (4'-CH2-0-2') BNA's have been incorporated into antisense oligonucleotides that showed antisense activity (Frieden et al, Nucleic Acids Research, 2003, 21, 6365-6372).
In certain embodiments, bicyclic nucleosides include those having a 4' to 2' bridge wherein such bridges include without limitation, a-L-4'-(CH2)-0-2', P-D-4'-CH2-0-2', 4'-(CH2)2-0-2', 4'- CH2-0-N(R)-2', 4'-CH2-N(R)-0-2', 4'-CH(CH3)-0-2', 4'-CH2-S-2', 4'-CH2-N(R)-2', 4'-CH2- CH(CH3)-2', and 4'-(CH2)3-2', wherein R is H, a protecting group or C1-C12 alkyl.
In certain embodiments, bicyclic nucleosides have the formula:

wherein:
Bx is a heterocyclic base moiety;
-Qa-Qb-Qc- is -CH2-N(Rc)-CH2-, -C(=0)-N(Rc)-CH2-, -CH2-0-N(Rc)-, -CH2-N(Rc)-0- or -
N(Rc)-0-CH2;
Rc is C1-C12 alkyl or an amino protecting group; and
Ta and Tb are each, independently H, a hydroxyl protecting group, a conjugate group, a reactive phosphorus group, a phosphorus moiety or a covalent attachment to a support medium.
In certain embodiments, bicyclic nucleosides have the formula:

wherein:
Bx is a heterocyclic base moiety;
Ta and Tb are each, independently H, a hydroxyl protecting group, a conjugate group, a reactive phosphorus group, a phosphorus moiety or a covalent attachment to a support medium;
Za is Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, substituted Ci-C6 alkyl, substituted C2-C6 alkenyl, substituted C2-C6 alkynyl, acyl, substituted acyl, substituted amide, thiol or substituted thiol.
In one embodiment, each of the substituted groups, is, independently, mono or poly substituted with substituent groups independently selected from halogen, oxo, hydroxyl, OJc, NJ d, SJc, N3, OC(=X)Jc, and NJeC(=X)NJcJd, wherein each Jc, Jd and Je is, independently, H, Ci-C6 alkyl, or substituted Ci-C6 alkyl and X is O or NJC.
In certain embodiments, bicyclic nucleosides have the formula:

wherein:
Bx is a heterocyclic base moiety;
Ta and Tb are each, independently H, a hydroxyl protecting group, a conjugate group, a reactive phosphorus group, a phosphorus moiety or a covalent attachment to a support medium;
Zb is Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, substituted Ci-C6 alkyl, substituted C2-C6 alkenyl, substituted C2-C6 alkynyl or substituted acyl (C(=0)-).
In certain embodiments, bicyclic nucleosides have the formula:

wherein:
Bx is a heterocyclic base moiety;
Ta and Tb are each, independently H, a hydroxyl protecting group, a conjugate group, a reactive phosphorus group, a phosphorus moiety or a covalent attachment to a support medium;
Rd is Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl or substituted C2-C6 alkynyl;
each qa, qb, qc and qd is, independently, H, halogen, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-
C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl or substituted C2-C6 alkynyl, Ci-C6 alkoxyl, substituted Ci-C6 alkoxyl, acyl, substituted acyl, Ci-C6 aminoalkyl or substituted Ci-C6 aminoalkyl;
In certain embodiments, bicyclic nucleosides have the formula:

Bx is a heterocyclic base moiety;
Ta and Tb are each, independently H, a hydroxyl protecting group, a conjugate group, a reactive phosphorus group, a phosphorus moiety or a covalent attachment to a support medium; qa, qb, qe and qf are each, independently, hydrogen, halogen, Ci-Ci2 alkyl, substituted Ci-Ci2 alkyl, C2-Ci2 alkenyl, substituted C2-Ci2 alkenyl, C2-Ci2 alkynyl, substituted C2-Ci2 alkynyl, Ci-Ci2 alkoxy, substituted Ci-Ci2 alkoxy, OJj, SJj, SOJj, S02Jj, NJjJk, N3, CN, C(=0)OJj, C(=0)NJjJk, C(=0)Jj, 0-C(=0)NJjJk, N(H)C(= H)NJjJk, N(H)C(=0)NJjJk orN(H)C(=S)NJjJk;
or qe and qf together are =C(qg)(qh);
qg and qh are each, independently, H, halogen, Ci-Ci2 alkyl or substituted Ci-Ci2 alkyl.
The synthesis and preparation of adenine, cytosine, guanine, 5-methyl-cytosine, thymine and uracil bicyclic nucleosides having a 4'-CH2-0-2' bridge, along with their oligomerization, and nucleic acid recognition properties have been described (Koshkin et al, Tetrahedron, 1998, 54, 3607-3630). The synthesis of bicyclic nucleosides has also been described in WO 98/39352 and WO 99/14226.
Analogs of various bicyclic nucleosides that have 4' to 2' bridging groups such as 4'-CH2-0- 2' and 4'-CH2-S-2', have also been prepared (Kumar et al, Bioorg. Med. Chem. Lett., 1998, 8, 2219- 2222). Preparation of oligodeoxyribonucleotide duplexes comprising bicyclic nucleosides for use as
substrates for nucleic acid polymerases has also been described (Wengel et al, WO 99/14226 ). Furthermore, synthesis of 2'-amino-BNA, a novel conformationally restricted high-affinity oligonucleotide analog has been described in the art (Singh et al, J. Org. Chem., 1998, 63, 10035- 10039). In addition, 2'-amino- and 2'-methylamino-BNA's have been prepared and the thermal stability of their duplexes with complementary RNA and DNA strands has been previously reported.
In certain embodiments, bicyclic nucleosides have the formula:

Bx is a heterocyclic base moiety;
Ta and Tb are each, independently H, a hydroxyl protecting group, a conjugate group, a reactive phosphorus group, a phosphorus moiety or a covalent attachment to a support medium; each φ, c , qk and qi is, independently, H, halogen, C1-C12 alkyl, substituted C1-C12 alkyl, C2- C12 alkenyl, substituted C2-C12 alkenyl, C2-C12 alkynyl, substituted C2-C12 alkynyl, C1-C12 alkoxyl, substituted C1-C12 alkoxyl, OJj, SJj, SOJj, S02Jj, NJjJk, N3, CN, C(=0)OJj, C(=0)NJjJk, C(=0)Jj, O- C(=0)NJjJk, N(H)C(=NH)NJjJk, N(H)C(=0)NJjJk orN(H)C(=S)NJjJk; and
qi and qj or qi and qk together are =C(qg)(q ), wherein qg and q are each, independently, H, halogen, C1-C12 alkyl or substituted C1-C12 alkyl.
One carbocyclic bicyclic nucleoside having a 4'-(CH2)3-2' bridge and the alkenyl analog bridge 4'-CH=CH-CH2-2' have been described (Frier et al, Nucleic Acids Research, 1997, 25(22), 4429-4443 and Albaek et al, J. Org. Chem., 2006, 71, 7731-7740). The synthesis and preparation of carbocyclic bicyclic nucleosides along with their oligomerization and biochemical studies have also been described (Srivastava et al, J. Am. Chem. Soc. 2007, 129(26), 8362-8379).
In certain embodiments, bicyclic nucleosides include, but are not limited to, (A) a-L- methyleneoxy (4'-CH2-0-2') BNA , (B) β-D-methyleneoxy (4'-CH2-0-2') BNA , (C) ethyleneoxy (4'-(CH2)2-0-2') BNA , (D) aminooxy (4'-CH2-0-N(R)-2') BNA, (E) oxyamino (4'-CH2-N(R)-0- 2') BNA, (F) methyl(methyleneoxy) (4'-CH(CH3)-0-2') BNA (also referred to as constrained ethyl or cEt), (G) methylene-thio (4'-CH2-S-2') BNA, (H) methylene-amino (4'-CH2-N(R)-2') BNA, (I)
methyl carbocyclic (4'-CH2-CH(CH3)-2') BNA, (J) propylene carbocyclic (4'-(CH2)3-2') BNA, and (K) vinyl BNA as depicted below.

As used herein the term "sugar surrogate" refers to replacement of the nucleoside furanose ring with a non-furanose (or 4'-substituted furanose) group with another structure such as another ring system or open system. Such structures can be as simple as a six membered ring as opposed to the five membered furanose ring or can be more complicated such as a bicyclic or tricyclic ring system or a non-ring system used in peptide nucleic acid. In certain embodiments, sugar surrogates include without limitation sugar surrogate groups such as morpholinos, cyclohexenyls and cyclohexitols. In general the heterocyclic base is maintained even when the sugar moiety is a sugar surrogate so that the resulting monomer subunit will be able to hybridize.
In certain embodiments, nucleosides having sugar surrogate groups include without limitation, replacement of the ribosyl ring with a sugar surrogate such as a tetrahydropyranyl ring system (also referred to as hexitol) as illustrated below:

In certain embodiments, sugar surrogates are selected having the formula:

wherein:
Bx is a heterocyclic base moiety;
T3 and T4 are each, independently, an internucleoside linking group linking the
tetrahydropyran nucleoside analog to the oligomeric compound or one of T3 and T4 is an
internucleoside linking group linking the tetrahydropyran nucleoside analog to an oligomeric compound or oligonucleotide and the other of T3 and T4 is H, a hydroxyl protecting group, a linked conjugate group or a 5' or 3'-terminal group;
qi, q2, q3, q4, qs, q6 and q7 are each independently, H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl or substituted C2-C6 alkynyl; and
one of Ri and R2 is hydrogen and the other is selected from halogen, substituted or unsubstituted alkoxy, NJiJ2, SJi, N3, OC(=X)Ji, OC(=X)NJiJ2, NJ3C(=X)NJiJ2 and CN, wherein X is O, S or NJi and each Ji, J2 and J3 is, independently, H or Ci-C6 alkyl.
In certain embodiments, qi, q2, q3, q4, qs, q6 and q7 are each H. In certain embodiments, at least one of qi, q2, q3, q4, qs, q6 and q7 is other than H. In certain embodiments, at least one of qi, q2, q3, q4, qs, q6 and q7 is methyl. In certain embodiments, THP nucleosides are provided wherein one of Ri and R2 is F. In certain embodiments, Ri is fluoro and R2 is H; Ri is methoxy and R2 is H, and Ri is methoxyethoxy and R2 is H.
Such sugar surrogates can be referred to as a "modified tetrahydropyran nucleoside" or "modified THP nucleoside". Modified THP nucleosides include, but are not limited to, what is
referred to in the art as hexitol nucleic acid (HNA), altritol nucleic acid (ANA), and mannitol nucleic acid (MNA) (see Leumann, C. J., Bioorg. & Med. Chem., 2002, 10, 841-854).
In certain embodiments, oligomeric compounds comprise one or more modified
cyclohexenyl nucleosides, which is a nucleoside having a six-membered cyclohexenyl in place of the pentofuranosyl residue in naturally occurring nucleosides. Modified cyclohexenyl nucleosides include, but are not limited to those described in the art (see for example commonly owned, published PCT Application WO 2010/036696, published on April 10, 2010, Robeyns et al, J. Am. Chem. Soc, 2008, 130(6), 1979-1984; Horvath et al, Tetrahedron Letters, 2007, 48, 3621-3623; Nauwelaerts et al, J. Am. Chem. Soc, 2007, 129(30), 9340-9348; Gu et al.,, Nucleosides,
Nucleotides & Nucleic Acids, 2005, 24(5-7), 993-998; Nauwelaerts et al, Nucleic Acids Research, 2005, 33(8), 2452-2463; Robeyns et al, Acta Crystallographica, Section F: Structural Biology and Crystallization Communications, 2005, F61(6), 585-586; Gu et al, Tetrahedron, 2004, 60(9), 2111- 2123; Gu et al, Oligonucleotides, 2003, 13(6), 479-489; Wang et al, J. Org. Chem., 2003, 68, 4499-4505; Verbeure et al, Nucleic Acids Research, 2001, 29(24), 4941-4947; Wang et al, J. Org. Chem., 2001, 66, 8478-82; Wang et al, Nucleosides, Nucleotides & Nucleic Acids, 2001, 20(4-7), 785-788; Wang et al, J. Am. Chem., 2000, 122, 8595-8602; Published PCT application, WO 06/047842; and Published PCT Application WO 01/049687; the text of each is incorporated by reference herein, in their entirety). Certain modified cyclohexenyl nucleosides have Formula X.

wherein independently for each of said at least one cyclohexenyl nucleoside analog of Formula X:
Bx is a heterocyclic base moiety;
T3 and T4 are each, independently, an internucleoside linking group linking the cyclohexenyl nucleoside analog to an antisense compound or one of T3 and T is an internucleoside linking group linking the tetrahydropyran nucleoside analog to an antisense compound and the other of T3 and T is H, a hydroxyl protecting group, a linked conjugate group, or a 5'-or 3'-terminal group; and qi, q2, q3, q4, qs, q6, q7, qs and qg are each, independently, H, C1-C5 alkyl, substituted C1-C5 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl or other sugar substituent group.
Many other monocyclic, bicyclic and tricyclic ring systems are known in the art and are suitable as sugar surrogates that can be used to modify nucleosides for incorporation into oligomeric compounds as provided herein (see for example review article: Leumann, Christian J. Bioorg. &
Med. Chem., 2002, 10, 841-854). Such ring systems can undergo various additional substitutions to further enhance their activity.
Some representative U.S. patents that teach the preparation of such modified sugars include without limitation, U.S.: 4,981,957; 5, 118,800; 5,319,080; 5,359,044; 5,393,878; 5,446, 137;
5,466,786; 5,514,785; 5,519, 134; 5,567,811; 5,576,427; 5,591,722; 5,597,909; 5,610,300;
5,627,053; 5,639,873; 5,646,265; 5,670,633; 5,700,920; 5,792,847 and 6,600,032 and International Application PCT/US2005/019219, filed June 2, 2005 and published as WO 2005/121371 on
December 22, 2005 certain of which are commonly owned with the instant application, and each of which is herein incorporated by reference in its entirety.
The tricyclic nucleosides provided herein can be prepared by any of the applicable techniques of organic synthesis, as, for example, illustrated in the examples below. Many such techniques are well known in the art. However, many of the known techniques are elaborated in
Compendium of Organic Synthetic Methods, John Wiley & Sons, New York: Vol. 1, Ian T. Harrison and Shuyen Harrison, 1971; Vol. 2, Ian T. Harrison and Shuyen Harrison, 1974; Vol. 3, Louis S.
Hegedus and Leroy Wade, 1977; Vol. 4, Leroy G. Wade Jr., 1980; Vol. 5, Leroy G. Wade Jr., 1984; and Vol. 6, Michael B. Smith; as well as March, J., Advanced Organic Chemistry, 3rd Edition, John Wiley & Sons, New York, 1985; Comprehensive Organic Synthesis. Selectivity, Strategy &
Efficiency in Modern Organic Chemistry, in 9 Volumes, Barry M. Trost, Editor-in-Chief, Pergamon
Press, New York, 1993; Advanced Organic Chemistry, Part B: Reactions and Synthesis, 4th Edition;
Carey and Sundberg, Kluwer Academic/Plenum Publishers, New York, 2001; Advanced Organic
Chemistry, Reactions, Mechanisms, and Structure, 2nd Edition, March, McGraw Hill, 1977; Greene, T.W., and Wutz, P.G.M., Protecting Groups in Organic Synthesis, 4th Edition, John Wiley & Sons,
New York, 1991; and Larock, R.C., Comprehensive Organic Transformations, 2nd Edition, John
Wiley & Sons, New York, 1999.
As used herein the term "reactive phosphorus" is meant to include groups that are covalently linked to a monomer subunit that can be further attached to an oligomeric compound that are useful for forming internucleoside linkages including for example phosphodiester and phosphorothioate internucleoside linkages. Such reactive phosphorus groups are known in the art and contain phosphorus atoms in Pm or Pv valence state including, but not limited to, phosphoramidite, H-
phosphonate, phosphate triesters and phosphorus containing chiral auxiliaries. In certain embodiments, reactive phosphorus groups are selected from diisopropylcyanoethoxy
phosphoramidite (-0*-P[N[(CH(CH3)2]2]0(CH2)2CN) and H-phosphonate (-0*-P(=0)(H)OH), wherein the O* is provided from the Markush group for the monomer. A preferred synthetic solid phase synthesis utilizes phosphoramidites (Pin chemistry) as reactive phosphites. The intermediate phosphite compounds are subsequently oxidized to the phosphate or thiophosphate (Pv chemistry) using known methods to yield, phosphodiester or phosphorothioate internucleoside linkages.
Additional reactive phosphates and phosphites are disclosed in Tetrahedron Report Number 309 (Beaucage and Iyer, Tetrahedron, 1992, 48, 2223-2311).
As used herein, "oligonucleotide" refers to a compound comprising a plurality of linked nucleosides. In certain embodiments, one or more of the plurality of nucleosides is modified. In certain embodiments, an oligonucleotide comprises one or more ribonucleosides (RNA) and/or deoxyribonucleosides (DNA).
The term "oligonucleoside" refers to a sequence of nucleosides that are joined by
internucleoside linkages that do not have phosphorus atoms. Internucleoside linkages of this type include short chain alkyl, cycloalkyl, mixed heteroatom alkyl, mixed heteroatom cycloalkyl, one or more short chain heteroatomic and one or more short chain heterocyclic. These internucleoside linkages include without limitation, siloxane, sulfide, sulfoxide, sulfone, acetyl, formacetyl, thioformacetyl, methylene formacetyl, thioformacetyl, alkeneyl, sulfamate, methyleneimino, methylenehydrazino, sulfonate, sulfonamide, amide and others having mixed N, O, S and CH2 component parts.
As used herein, the term "oligomeric compound" refers to a contiguous sequence of linked monomer subunits. Each linked monomer subunit normally includes a heterocyclic base moiety but monomer subunits also includes those without a heterocyclic base moiety such as abasic monomer subunits. At least some and generally most if not essentially all of the heterocyclic bases in an oligomeric compound are capable of hybridizing to a nucleic acid molecule, normally a preselected RNA target. The term "oligomeric compound" therefore includes oligonucleotides, oligonucleotide analogs and oligonucleosides. It also includes polymers having one or a plurality of nucleoside mimetics and or nucleosides having sugar surrogate groups.
In certain embodiments, oligomeric compounds comprise a plurality of monomer subunits independently selected from naturally occurring nucleosides, non-naturally occurring nucleosides, modified nucleosides, nucleoside mimetics, and nucleosides having sugar surrogate groups. In
certain embodiments, oligomeric compounds are single stranded. In certain embodiments, oligomeric compounds are double stranded comprising a double-stranded duplex. In certain embodiments, oligomeric compounds comprise one or more conjugate groups and/or terminal groups.
When preparing oligomeric compounds having specific motifs as disclosed herein it can be advantageous to mix non-naturally occurring monomer subunits such as the tricyclic nucleosides as provided herein with other non-naturally occurring monomer subunits, naturally occurring monomer subunits (nucleosides) or mixtures thereof. In certain embodiments, oligomeric compounds are provided herein comprising a contiguous sequence of linked monomer subunits wherein at least one monomer subunit is a tricyclic nucleoside as provided herein. In certain embodiments, oligomeric compounds are provided comprising a plurality of tricyclic nucleosides as provided herein.
Oligomeric compounds are routinely prepared linearly but can also be joined or otherwise prepared to be circular and/or can be prepared to include branching. Oligomeric compounds can form double stranded constructs such as for example two strands hybridized to form a double stranded composition. Double stranded compositions can be linked or separate and can include various other groups such as conjugates and/or overhangs on the ends.
As used herein, "antisense compound" refers to an oligomeric compound, at least a portion of which is at least partially complementary to a target nucleic acid to which it hybridizes. In certain embodiments, an antisense compound modulates (increases or decreases) expression or amount of a target nucleic acid. In certain embodiments, an antisense compound alters splicing of a target pre- mRNA resulting in a different splice variant. In certain embodiments, an antisense compound modulates expression of one or more different target proteins. Antisense mechanisms contemplated herein include, but are not limited to an RNase H mechanism, RNAi mechanisms, splicing modulation, translational arrest, altering RNA processing, inhibiting microRNA function, or mimicking microRNA function.
As used herein, "antisense activity" refers to any detectable and/or measurable activity attributable to the hybridization of an antisense compound to its target nucleic acid. In certain embodiments, such activity may be an increase or decrease in an amount of a nucleic acid or protein. In certain embodiments, such activity may be a change in the ratio of splice variants of a nucleic acid or protein. Detection and/or measuring of antisense activity may be direct or indirect. For example, in certain embodiments, antisense activity is assessed by detecting and/or measuring the amount of target protein or the relative amounts of splice variants of a target protein. In certain
embodiments, antisense activity is assessed by detecting and/or measuring the amount of target nucleic acids and/or cleaved target nucleic acids and/or alternatively spliced target nucleic acids. In certain embodiments, antisense activity is assessed by observing a phenotypic change in a cell or animal.
As used herein the term "internucleoside linkage" or "internucleoside linking group" is meant to include all manner of internucleoside linking groups known in the art including but not limited to, phosphorus containing internucleoside linking groups such as phosphodiester and phosphorothioate, and non-phosphorus containing internucleoside linking groups such as formacetyl and methyleneimino. Internucleoside linkages also includes neutral non-ionic internucleoside linkages such as amide-3 (3'-CH2-C(=O)-N(H)-5'), amide-4 (3'-CH2-N(H)-C(=O)-5') and
methylphosphonate wherein a phosphorus atom is not always present.
In certain embodiments, oligomeric compounds as provided herein can be prepared having one or more internucleoside linkages containing modified e.g. non-naturally occurring
internucleoside linkages. The two main classes of internucleoside linkages are defined by the presence or absence of a phosphorus atom. Modified internucleoside linkages having a phosphorus atom include without limitation, phosphorothioates, chiral phosphorothioates, phosphorodithioates, phosphotriesters, aminoalkylphosphotriesters, methyl and other alkyl phosphonates including 3'- alkylene phosphonates, 5'-alkylene phosphonates and chiral phosphonates, phosphinates, phos- phoramidates including 3 '-amino phosphoramidate and aminoalkylphosphoramidates,
thionophosphoramidates, thionoalkylphosphonates, thionoalkylphosphotriesters, selenophosphates and boranophosphates having normal 3 '-5' linkages, 2 -5' linked analogs of these, and those having inverted polarity wherein one or more internucleotide linkages is a 3' to 3', 5' to 5' or 2' to 2' linkage. Oligonucleotides having inverted polarity can comprise a single 3' to 3' linkage at the 3 '-most internucleotide linkage i.e. a single inverted nucleoside residue which may be abasic (the nucleobase is missing or has a hydroxyl group in place thereof). Various salts, mixed salts and free acid forms are also included.
Representative U.S. patents that teach the preparation of the above phosphorus containing linkages include without limitation, U.S. : 3,687,808; 4,469,863; 4,476,301; 5,023,243; 5, 177, 196; 5, 188,897; 5, 194,599; 5,264,423; 5,276,019; 5,278,302; 5,286,717; 5,321, 131; 5,399,676;
5,405,939; 5,453,496; 5,455,233; 5,466,677; 5,476,925; 5,519, 126; 5,527,899; 5,536,821;
5,541,306; 5,550, 111; 5,563,253; 5,565,555; 5,571,799; 5,587,361; 5,625,050; 5,672,697 and
5,721,218, certain of which are commonly owned with this application, and each of which is herein incorporated by reference.
In certain embodiments, oligomeric compounds as provided herein can be prepared having one or more non-phosphorus containing internucleoside linkages. Such oligomeric compounds include without limitation, those that are formed by short chain alkyl or cycloalkyl internucleoside linkages, mixed heteroatom and alkyl or cycloalkyl internucleoside linkages, or one or more short chain heteroatomic or heterocyclic internucleoside linkages. These include those having siloxane backbones; sulfide, sulfoxide and sulfone backbones; formacetyl and thioformacetyl backbones; methylene formacetyl and thioformacetyl backbones; riboacetyl backbones; alkene containing backbones; sulfamate backbones; methyleneimino and methylenehydrazino backbones; sulfonate and sulfonamide backbones; amide backbones; and others having mixed N, O, S and CH2 component parts.
Representative U.S. patents that teach the preparation of the above oligonucleosides include without limitation, U.S.: 5,034,506; 5, 166,315; 5, 185,444; 5,214, 134; 5,216, 141; 5,235,033;
5,264,562; 5,264,564; 5,405,938; 5,434,257; 5,466,677; 5,470,967; 5,489,677; 5,541,307;
5,561,225; 5,596,086; 5,602,240; 5,608,046; 5,610,289; 5,618,704; 5,623,070; 5,663,312;
5,633,360; 5,677,437; 5,677,439; 5,646,269 and 5,792,608, certain of which are commonly owned with this application, and each of which is herein incorporated by reference.
As used herein "neutral internucleoside linkage" is intended to include internucleoside linkages that are non-ionic. Neutral internucleoside linkages include without limitation, phosphotriesters, methylphosphonates, MMI (3'-CH2-N(CH3)-0-5'), amide-3 (3'-CH2-C(=0)-N(H)- 5'), amide-4 (3'-CH2-N(H)-C(=0)-5'), formacetal (3'-0-CH2-0-5'), and thioformacetal (3'-S-CH2-0- 5'). Further neutral internucleoside linkages include nonionic linkages comprising siloxane
(dialkylsiloxane), carboxylate ester, carboxamide, sulfide, sulfonate ester and amides (See for example: Carbohydrate Modifications in Antisense Research; Y.S. Sanghvi and P.D. Cook, Eds., ACS Symposium Series 580; Chapters 3 and 4, 40-65). Further neutral internucleoside linkages include nonionic linkages comprising mixed N, O, S and CH2 component parts.
In certain embodiments, oligomeric compounds as provided herein can be prepared having one or more optionally protected phosphorus containing internucleoside linkages. Representative protecting groups for phosphorus containing internucleoside linkages such as phosphodiester and phosphorothioate linkages include β-cyanoethyl, diphenylsilylethyl, δ-cyanobutenyl, cyano p-xylyl (CPX), N-methyl-N-trifluoroacetyl ethyl (MET A), acetoxy phenoxy ethyl (APE) and butene-4-yl
groups. See for example U.S. Patents Nos. 4,725,677 and Re. 34,069 (β-cyanoethyl); Beaucage et al, Tetrahedron, 1993, 49(10), 1925-1963; Beaucage et al, Tetrahedron, 1993, 49(46), 10441- 10488; Beaucage et al, Tetrahedron, 1992, 48(12), 2223-2311.
As used herein the terms "linking groups" and "bifunctional linking moieties" are meant to include groups known in the art that are useful for attachment of chemical functional groups, conjugate groups, reporter groups and other groups to selective sites in a parent compound such as for example an oligomeric compound. In general, a bifunctional linking moiety comprises a hydrocarbyl moiety having two functional groups. One of the functional groups is selected to bind to a parent molecule or compound of interest and the other is selected to bind to essentially any selected group such as a chemical functional group or a conjugate group. In some embodiments, the linker comprises a chain structure or a polymer of repeating units such as ethylene glycols or amino acid units. Examples of functional groups that are routinely used in bifunctional linking moieties include without limitation, electrophiles for reacting with nucleophilic groups and nucleophiles for reacting with electrophilic groups. In some embodiments, bifunctional linking moieties include amino, hydroxyl, carboxylic acid, thiol, unsaturations (e.g., double or triple bonds), and the like.
Some nonlimiting examples of bifunctional linking moieties include 8-amino-3,6-dioxaoctanoic acid (ADO), succinimidyl 4-(N-maleimidomethyl) cyclohexane-l-carboxylate (SMCC) and 6- aminohexanoic acid (AHEX or AHA). Other linking groups include without limitation, substituted Ci-Cio alkyl, substituted or unsubstituted C2-Cio alkenyl or substituted or unsubstituted C2-Cio alkynyl, wherein a nonlimiting list of preferred substituent groups includes hydroxyl, amino, alkoxy, carboxy, benzyl, phenyl, nitro, thiol, thioalkoxy, halogen, alkyl, aryl, alkenyl and alkynyl.
In certain embodiments, the oligomeric compounds as provided herein can be modified by covalent attachment of one or more conjugate groups. In general, conjugate groups modify one or more properties of the oligomeric compounds they are attached to. Such oligonucleotide properties include without limitation, pharmacodynamics, pharmacokinetics, binding, absorption, cellular distribution, cellular uptake, charge and clearance. Conjugate groups are routinely used in the chemical arts and are linked directly or via an optional linking moiety or linking group to a parent compound such as an oligomeric compound. A preferred list of conjugate groups includes without limitation, intercalators, reporter molecules, polyamines, polyamides, polyethylene glycols, thioethers, poly ethers, cholesterols, thiocholesterols, cholic acid moieties, folate, lipids,
phospholipids, biotin, phenazine, phenanthridine, anthraquinone, adamantane, acridine, fluoresceins, rhodamines, coumarins and dyes.
In certain embodiments, the oligomeric compounds as provided herein can be modified by covalent attachment of one or more terminal groups to the 5' or 3'-terminal groups. A terminal group can also be attached at any other position at one of the terminal ends of the oligomeric compound. As used herein the terms "5'-terminal group", "3'-terminal group", "terminal group" and combinations thereof are meant to include useful groups known to the art skilled that can be placed on one or both of the terminal ends, including but not limited to the 5' and 3 '-ends of an oligomeric compound respectively, for various purposes such as enabling the tracking of the oligomeric compound (a fluorescent label or other reporter group), improving the pharmacokinetics or pharmacodynamics of the oligomeric compound (such as for example: uptake and/or delivery) or enhancing one or more other desirable properties of the oligomeric compound (a group for improving nuclease stability or binding affinity). In certain embodiments, 5' and 3 '-terminal groups include without limitation, modified or unmodified nucleosides; two or more linked nucleosides that are independently, modified or unmodified; conjugate groups; capping groups; phosphate moieties; and protecting groups.
As used herein the term "phosphate moiety" refers to a terminal phosphate group that includes phosphates as well as modified phosphates. The phosphate moiety can be located at either terminus. In certain embodiments, the phosphate moiety is located at the 5'-terminal nucleoside. In certain embodiments, the terminal phosphate is unmodified having the formula -0-P(=0)(OH)OH. In another aspect, the terminal phosphate is modified such that one or more of the O and OH groups are replaced with H, O, S, N(R) or alkyl where R is H, an amino protecting group or unsubstituted or substituted alkyl. In certain embodiments, the 5' and or 3' terminal group can comprise from 1 to 3 phosphate moieties that are each, independently, unmodified (di or tri-phosphates) or modified.
As used herein, the term "phosphorus moiety" refers to a group having the formula:

wherein:
Rx and Ry are each, independently, hydroxyl, protected hydroxyl group, thiol, protected thiol group, Ci-C6 alkyl, substituted Ci-C6 alkyl, Ci-C6 alkoxy, substituted Ci-C6 alkoxy, a protected amino or substituted amino; and
Rz is O or S.
As a monomer such as a phosphoramidite or H-phosphonate the protected phosphorus moiety is preferred to maintain stability during oligomer synthesis. After incorporation into an oligomeric compound the phosphorus moiety can include deprotected groups.
Phosphorus moieties included herein can be attached to a monomer, which can be used in the preparation of oligomeric compounds, wherein the monomer may be attached using O, S, NRa or CRe f, wherein Ra includes without limitation H, Ci-C6 alkyl, substituted Ci-C6 alkyl, Ci-C6 alkoxy, substituted Ci-C6 alkoxy, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl or substituted acyl, and Re and Rf each, independently, include without limitation H, halogen, Ci-C6 alkyl, substituted Ci-C6 alkyl, Ci-C6 alkoxy or substituted Ci-C6 alkoxy. Such linked phosphorus moieties include without limitation, phosphates, modified phosphates, thiophosphates, modified thiophosphates, phosphonates, modified phosphonates, phosphoramidates and modified phosphoramidates.
RNA duplexes exist in what has been termed "A Form" geometry while DNA duplexes exist in "B Form" geometry. In general, RNA:RNA duplexes are more stable, or have higher melting temperatures (Tm) than DNA:DNA duplexes (Sanger et al, Principles of Nucleic Acid Structure,
1984, Springer- Verlag; New York, NY.; Lesnik et al, Biochemistry, 1995, 34, 10807-10815; Conte et al, Nucleic Acids Res., 1997, 25, 2627-2634). The increased stability of RNA has been attributed to several structural features, most notably the improved base stacking interactions that result from an A- form geometry (Searle et al, Nucleic Acids Res., 1993, 21, 2051-2056). The presence of the 2' hydroxyl in RNA biases the sugar toward a C3' endo pucker, i.e., also designated as Northern pucker, which causes the duplex to favor the A-form geometry. In addition, the 2' hydroxyl groups of RNA can form a network of water mediated hydrogen bonds that help stabilize the RNA duplex (Egli et al, Biochemistry, 1996, 35, 8489-8494). On the other hand, deoxy nucleic acids prefer a C2' endo sugar pucker, i.e., also known as Southern pucker, which is thought to impart a less stable B-form geometry (Sanger, W. (1984) Principles of Nucleic Acid Structure, Springer- Verlag, New York, NY).
The relative ability of a chemically-modified oligomeric compound to bind to complementary nucleic acid strands, as compared to natural oligonucleotides, is measured by obtaining the melting temperature of a hybridization complex of said chemically-modified oligomeric compound with its complementary unmodified target nucleic acid. The melting temperature (Tm), a characteristic physical property of double helixes, denotes the temperature in degrees centigrade at which 50% helical versus coiled (unhybridized) forms are present. Tm (also commonly referred to as
binding affinity) is measured by using the UV spectrum to determine the formation and breakdown (melting) of hybridization. Base stacking, which occurs during hybridization, is accompanied by a reduction in UV absorption (hypochromicity). Consequently a reduction in UV absorption indicates a higher Tm.
It is known in the art that the relative duplex stability of an antisense compound:RNA target duplex can be modulated through incorporation of chemically-modified nucleosides into the antisense compound. Sugar-modified nucleosides have provided the most efficient means of modulating the Tm of an antisense compound with its target RNA. Sugar-modified nucleosides that increase the population of or lock the sugar in the C3' '-endo (Northern, RNA-like sugar pucker) configuration have predominantly provided a per modification Tm increase for antisense compounds toward a complementary RNA target. Sugar-modified nucleosides that increase the population of or lock the sugar in the C2'-endo (Southern, DNA-like sugar pucker) configuration predominantly provide a per modification Tm decrease for antisense compounds toward a complementary RNA target. The sugar pucker of a given sugar-modified nucleoside is not the only factor that dictates the ability of the nucleoside to increase or decrease an antisense compound's Tm toward complementary RNA. For example, the sugar-modified nucleoside tricycloDNA is predominantly in the C2'-endo conformation, however it imparts a 1.9 to 3° C per modification increase in Tm toward a
complementary RNA. Another example of a sugar-modified high-affinity nucleoside that does not adopt the C3'-endo conformation is a-L-LNA (described in more detail herein).
As used herein, "Tm" means melting temperature which is the temperature at which the two strands of a duplex nucleic acid separate. Tm is often used as a measure of duplex stability or the binding affinity of an antisense compound toward a complementary RNA molecule.
As used herein, "complementarity" in reference to nucleobases refers to a nucleobase that is capable of base pairing with another nucleobase. For example, in DNA, adenine (A) is
complementary to thymine (T). For example, in RNA, adenine (A) is complementary to uracil (U). In certain embodiments, complementary nucleobase refers to a nucleobase of an antisense compound that is capable of base pairing with a nucleobase of its target nucleic acid. For example, if a nucleobase at a certain position of an antisense compound is capable of hydrogen bonding with a nucleobase at a certain position of a target nucleic acid, then the position of hydrogen bonding between the oligonucleotide and the target nucleic acid is considered to be complementary at that nucleobase pair. Nucleobases or more broadly, heterocyclic base moieties, comprising certain modifications may maintain the ability to pair with a counterpart nucleobase and thus, are still
capable of complementarity.
As used herein, "non-complementary" " in reference to nucleobases refers to a pair of nucleobases that do not form hydrogen bonds with one another or otherwise support hybridization.
As used herein, "complementary" in reference to linked nucleosides, oligonucleotides, oligomeric compounds, or nucleic acids, refers to the capacity of an oligomeric compound to hybridize to another oligomeric compound or nucleic acid through nucleobase or more broadly, heterocyclic base, complementarity. In certain embodiments, an antisense compound and its target are complementary to each other when a sufficient number of corresponding positions in each molecule are occupied by nucleobases that can bond with each other to allow stable association between the antisense compound and the target. One skilled in the art recognizes that the inclusion of mismatches is possible without eliminating the ability of the oligomeric compounds to remain in association. Therefore, described herein are antisense compounds that may comprise up to about 20% nucleotides that are mismatched (i.e., are not nucleobase complementary to the corresponding nucleotides of the target). Preferably the antisense compounds contain no more than about 15%, more preferably not more than about 10%, most preferably not more than 5% or no mismatches.
The remaining nucleotides are nucleobase complementary or otherwise do not disrupt hybridization (e.g., universal bases). One of ordinary skill in the art would recognize the compounds provided herein are at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%), at least 99% or 100% complementary to a target nucleic acid.
It is understood in the art that the sequence of an oligomeric compound need not be 100% complementary to that of its target nucleic acid to be specifically hybridizable. Moreover, an oligomeric compound may hybridize over one or more segments such that intervening or adjacent segments are not involved in the hybridization event (e.g., a loop structure or hairpin structure). In certain embodiments, oligomeric compounds can comprise at least about 70%, at least about 80%, at least about 90%, at least about 95%, or at least about 99% sequence complementarity to a target region within the target nucleic acid sequence to which they are targeted. For example, an oligomeric compound in which 18 of 20 nucleobases of the oligomeric compound are complementary to a target region, and would therefore specifically hybridize, would represent 90 percent complementarity. In this example, the remaining noncomplementary nucleobases may be clustered or interspersed with complementary nucleobases and need not be contiguous to each other or to complementary nucleobases. As such, an oligomeric compound which is 18 nucleobases in length having 4 (four) noncomplementary nucleobases which are flanked by two regions of complete
complementarity with the target nucleic acid would have 77.8% overall complementarity with the target nucleic acid and would thus fall within this scope. Percent complementarity of an oligomeric compound with a region of a target nucleic acid can be determined routinely using BLAST programs (basic local alignment search tools) and PowerBLAST programs known in the art (Altschul et al, J. Mol. Biol, 1990, 215, 403-410; Zhang and Madden, Genome Res., 1997, 7, 649- 656).
As used herein, "hybridization" refers to the pairing of complementary oligomeric compounds (e.g., an antisense compound and its target nucleic acid). While not limited to a particular mechanism, the most common mechanism of pairing involves hydrogen bonding, which may be Watson-Crick, Hoogsteen or reversed Hoogsteen hydrogen bonding, between
complementary nucleoside or nucleotide bases (nucleobases). For example, the natural base adenine is nucleobase complementary to the natural nucleobases thymidine and uracil which pair through the formation of hydrogen bonds. The natural base guanine is nucleobase complementary to the natural bases cytosine and 5-methyl cytosine. Hybridization can occur under varying circumstances.
As used herein, "target nucleic acid" refers to any nucleic acid molecule the expression, amount, or activity of which is capable of being modulated by an antisense compound. In certain embodiments, the target nucleic acid is DNA or RNA. In certain embodiments, the target RNA is mRNA, pre-mRNA, non-coding RNA, pri-microRNA, pre-microRNA, mature microRNA, promoter-directed RNA, or natural antisense transcripts. For example, the target nucleic acid can be a cellular gene (or mRNA transcribed from the gene) whose expression is associated with a particular disorder or disease state, or a nucleic acid molecule from an infectious agent. In certain embodiments, target nucleic acid is a viral or bacterial nucleic acid.
Further included herein are oligomeric compounds such as antisense oligomeric compounds, antisense oligonucleotides, ribozymes, external guide sequence (EGS) oligonucleotides, alternate splicers, primers, probes, and other oligomeric compounds which hybridize to at least a portion of the target nucleic acid. As such, these oligomeric compounds may be introduced in the form of single- stranded, double- stranded, circular or hairpin oligomeric compounds and may contain structural elements such as internal or terminal bulges or loops. Once introduced to a system, the oligomeric compounds provided herein may elicit the action of one or more enzymes or structural proteins to effect modification of the target nucleic acid. Alternatively, the oligomeric compound
may inhibit the activity the target nucleic acid through an occupancy-based method, thus interfering with the activity of the target nucleic acid.
One non-limiting example of such an enzyme is RNAse H, a cellular endonuclease which cleaves the RNA strand of an RNA:DNA duplex. It is known in the art that single- stranded oligomeric compounds which are "DNA-like" elicit RNAse H. Activation of RNase H, therefore, results in cleavage of the RNA target, thereby greatly enhancing the efficiency of oligonucleotide- mediated inhibition of gene expression. Similar roles have been postulated for other ribonucleases such as those in the RNase III and ribonuclease L family of enzymes.
While one form of oligomeric compound is a single- stranded antisense oligonucleotide, in many species the introduction of double-stranded structures, such as double-stranded RNA (dsRNA) molecules, has been shown to induce potent and specific antisense-mediated reduction of the function of a gene or its associated gene products. This phenomenon occurs in both plants and animals and is believed to have an evolutionary connection to viral defense and transposon silencing.
As used herein, "modulation" refers to a perturbation of amount or quality of a function or activity when compared to the function or activity prior to modulation. For example, modulation includes the change, either an increase (stimulation or induction) or a decrease (inhibition or reduction) in gene expression. As a further example, modulation of expression can include perturbing splice site selection of pre-mRNA processing, resulting in a change in the amount of a particular splice-variant present compared to conditions that were not perturbed. As a further example, modulation includes perturbing translation of a protein.
As used herein, the term "pharmaceutically acceptable salts" refers to salts that retain the desired activity of the compound and do not impart undesired toxicological effects thereto. The term "pharmaceutically acceptable salt" includes a salt prepared from pharmaceutically acceptable non-toxic acids or bases, including inorganic or organic acids and bases.
Pharmaceutically acceptable salts of the oligomeric compounds described herein may be prepared by methods well-known in the art. For a review of pharmaceutically acceptable salts, see Stahl and Wermuth, Handbook of Pharmaceutical Salts: Properties, Selection and Use (Wiley- VCH, Weinheim, Germany, 2002). Sodium salts of antisense oligonucleotides are useful and are well accepted for therapeutic administration to humans. Accordingly, in one embodiment the oligomeric compounds described herein are in the form of a sodium salt.
In certain embodiments, oligomeric compounds provided herein comprise from about 8 to about 80 monomer subunits in length. One having ordinary skill in the art will appreciate that this embodies oligomeric compounds of 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, or 80 monomer subunits in length, or any range therewithin.
In certain embodiments, oligomeric compounds provided herein comprise from about 8 to 40 monomer subunits in length. One having ordinary skill in the art will appreciate that this embodies oligomeric compounds of 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 or 40 monomer subunits in length, or any range therewithin.
In certain embodiments, oligomeric compounds provided herein comprise from about 8 to 20 monomer subunits in length. One having ordinary skill in the art will appreciate that this embodies oligomeric compounds of 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 monomer subunits in length, or any range therewithin.
In certain embodiments, oligomeric compounds provided herein comprise from about 8 to 16 monomer subunits in length. One having ordinary skill in the art will appreciate that this embodies oligomeric compounds of 8, 9, 10, 11, 12, 13, 14, 15 or 16 monomer subunits in length, or any range therewithin.
In certain embodiments, oligomeric compounds provided herein comprise from about 10 to
14 monomer subunits in length. One having ordinary skill in the art will appreciate that this embodies oligomeric compounds of 10, 11, 12, 13 or 14 monomer subunits in length, or any range therewithin.
In certain embodiments, oligomeric compounds provided herein comprise from about 10 to 18 monomer subunits in length. One having ordinary skill in the art will appreciate that this embodies oligomeric compounds of 10, 11, 12, 13, 14, 15, 16, 17 or 18 monomer subunits in length, or any range therewithin.
In certain embodiments, oligomeric compounds provided herein comprise from about 10 to 21 monomer subunits in length. One having ordinary skill in the art will appreciate that this embodies oligomeric compounds of 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or 21 monomer subunits in length, or any range therewithin.
In certain embodiments, oligomeric compounds provided herein comprise from about 12 to 14 monomer subunits in length. One having ordinary skill in the art will appreciate that this embodies oligomeric compounds of 12, 13 or 14 monomer subunits in length, or any range therewithin.
In certain embodiments, oligomeric compounds provided herein comprise from about 12 to
18 monomer subunits in length. One having ordinary skill in the art will appreciate that this embodies oligomeric compounds of 12, 13, 14, 15, 16, 17 or 18 monomer subunits in length, or any range therewithin.
In certain embodiments, oligomeric compounds provided herein comprise from about 12 to 21 monomer subunits in length. One having ordinary skill in the art will appreciate that this embodies oligomeric compounds of 12, 13, 14, 15, 16, 17, 18, 19, 20 or 21 monomer subunits in length, or any range therewithin.
In certain embodiments, oligomeric compounds provided herein comprise from about 14 to 18 monomer subunits in length. One having ordinary skill in the art will appreciate that this embodies oligomeric compounds of 14, 15, 16, 17 or 18 monomer subunits in length, or any range therewithin.
In certain embodiments, oligomeric compounds of any of a variety of ranges of lengths of linked monomer subunits are provided. In certain embodiments, oligomeric compounds are provided consisting of X-Y linked monomer subunits, where X and Y are each independently selected from 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, and 50; provided that X < Y. For example, in certain embodiments, this provides oligomeric compounds comprising: 8-9, 8-10, 8-11, 8-12, 8-13, 8-14, 8-15, 8-16, 8-17, 8-18, 8-19, 8-20, 8-21, 8-22, 8-23, 8-24, 8-25, 8-26, 8-27, 8-28, 8-29, 8-30, 9-10, 9-11, 9-12, 9-13, 9-14, 9-15, 9-16, 9-17, 9-18, 9-19, 9-20, 9-21, 9-22, 9-23, 9-24, 9-25, 9-26, 9-27, 9-28, 9-29, 9-30, 10-11, 10-12, 10-13, 10-14, 10-15, 10-16, 10-17, 10-18, 10- 19, 10-20, 10-21, 10-22, 10-23, 10-24, 10-25, 10-26, 10-27, 10-28, 10-29, 10-30, 11-12, 11-13, 11- 14, 11-15, 11-16, 11-17, 11-18, 11-19, 11-20, 11-21, 11-22, 11-23, 11-24, 11-25, 11-26, 11-27, 11- 28, 11-29, 11-30, 12-13, 12-14, 12-15, 12-16, 12-17, 12-18, 12-19, 12-20, 12-21, 12-22, 12-23, 12- 24, 12-25, 12-26, 12-27, 12-28, 12-29, 12-30, 13-14, 13-15, 13-16, 13-17, 13-18, 13-19, 13-20, 13- 21, 13-22, 13-23, 13-24, 13-25, 13-26, 13-27, 13-28, 13-29, 13-30, 14-15, 14-16, 14-17, 14-18, 14- 19, 14-20, 14-21, 14-22, 14-23, 14-24, 14-25, 14-26, 14-27, 14-28, 14-29, 14-30, 15-16, 15-17, 15- 18, 15-19, 15-20, 15-21, 15-22, 15-23, 15-24, 15-25, 15-26, 15-27, 15-28, 15-29, 15-30, 16-17, 16-
18, 16-19, 16-20, 16-21, 16-22, 16-23, 16-24, 16-25, 16-26, 16-27, 16-28, 16-29, 16-30, 17-18, 17-
19, 17-20, 17-21, 17-22, 17-23, 17-24, 17-25, 17-26, 17-27, 17-28, 17-29, 17-30, 18-19, 18-20, 18- 21, 18-22, 18-23, 18-24, 18-25, 18-26, 18-27, 18-28, 18-29, 18-30, 19-20, 19-21, 19-22, 19-23, 19-
24, 19-25, 19-26, 19-27, 19-28, 19-29, 19-30, 20-21, 20-22, 20-23, 20-24, 20-25, 20-26, 20-27, 20- 28, 20-29, 20-30, 21-22, 21-23, 21-24, 21-25, 21-26, 21-27, 21-28, 21-29, 21-30, 22-23, 22-24, 22-
25, 22-26, 22-27, 22-28, 22-29, 22-30, 23-24, 23-25, 23-26, 23-27, 23-28, 23-29, 23-30, 24-25, 24-
26, 24-27, 24-28, 24-29, 24-30, 25-26, 25-27, 25-28, 25-29, 25-30, 26-27, 26-28, 26-29, 26-30, 27- 28, 27-29, 27-30, 28-29, 28-30, or 29-30 linked monomer subunits.
In certain embodiments, the ranges for the oligomeric compounds listed herein are meant to limit the number of monomer subunits in the oligomeric compounds, however such oligomeric compounds may further include 5' and/or 3'-terminal groups including but not limited to protecting groups such as hydroxyl protecting groups, optionally linked conjugate groups and/or other substituent groups.
In certain embodiments, the preparation of oligomeric compounds as disclosed herein is performed according to literature procedures for DNA: Protocols for Oligonucleotides and Analogs, Agrawal, Ed., Humana Press, 1993, and/or RNA: Scaringe, Methods, 2001, 23, 206-217; Gait et al, Applications of Chemically synthesized RNA in RNA:Protein Interactions, Smith, Ed., 1998, 1-36; Gallo et al, Tetrahedron , 2001, 57, 5707-5713. Additional methods for solid-phase synthesis may be found in Caruthers U.S. Patents Nos. 4,415,732; 4,458,066; 4,500,707; 4,668,777; 4,973,679; and 5, 132,418; and Koster U.S. Patents Nos. 4,725,677 and Re. 34,069.
Oligomeric compounds are routinely prepared using solid support methods as opposed to solution phase methods. Commercially available equipment commonly used for the preparation of oligomeric compounds that utilize the solid support method is sold by several vendors including, for example, Applied Biosystems (Foster City, CA). Any other means for such synthesis known in the art may additionally or alternatively be employed. Suitable solid phase techniques, including automated synthesis techniques, are described in Oligonucleotides and Analogues, a Practical Approach, F. Eckstein, Ed., Oxford University Press, New York, 1991.
The synthesis of RNA and related analogs relative to the synthesis of DNA and related analogs has been increasing as efforts in RNA interference and micro RNA increase. The primary RNA synthesis strategies that are presently being used commercially include 5'-0-DMT-2'-0-t- butyldimethylsilyl (TBDMS), 5'-0-DMT-2'-0-[l(2-fluorophenyl)-4-methoxypiperidin-4-yl] (FPMP), 2'-0-[(triisopropylsilyl)oxy]methyl (2'-0-CH2-0-Si(iPr)3 (TOM) and the 5'-0-silyl ether-
2'-ACE (5'-0-bis(trimethylsiloxy)cyclododecyloxysilyl ether (DOD)-2'-0-bis(2- acetoxyethoxy)methyl (ACE). A current list of some of the major companies currently offering RNA products include Pierce Nucleic Acid Technologies, Dharmacon Research Inc., Ameri Biotechnologies Inc., and Integrated DNA Technologies, Inc. One company, Princeton Separations, is marketing an RNA synthesis activator advertised to reduce coupling times especially with TOM and TBDMS chemistries. The primary groups being used for commercial RNA synthesis are:
TBDMS: 5'-0-DMT-2'-0-t-butyldimethylsilyl; TOM: 2'-0-[(triisopropylsilyl)oxy]methyl;
DOD/ACE: (5'-0-bis(trimethylsiloxy)cyclododecyloxysilyl ether-2'-0-bis(2-acetoxyethoxy)methyl; and FPMP: 5'-0-DMT-2'-0-[l(2-fluorophenyl)-4-ethoxypiperidin-4-yl]. In certain embodiments, each of the aforementioned RNA synthesis strategies can be used herein. In certain embodiments, the aforementioned RNA synthesis strategies can be performed together in a hybrid fashion e.g. using a 5 '-protecting group from one strategy with a 2'-0-protecting from another strategy.
In some embodiments, "suitable target segments" may be employed in a screen for additional oligomeric compounds that modulate the expression of a selected protein. "Modulators" are those oligomeric compounds that decrease or increase the expression of a nucleic acid molecule encoding a protein and which comprise at least an 8-nucleobase portion which is complementary to a suitable target segment. The screening method comprises the steps of contacting a suitable target segment of a nucleic acid molecule encoding a protein with one or more candidate modulators, and selecting for one or more candidate modulators which decrease or increase the expression of a nucleic acid molecule encoding a protein. Once it is shown that the candidate modulator or modulators are capable of modulating (e.g. either decreasing or increasing) the expression of a nucleic acid molecule encoding a peptide, the modulator may then be employed herein in further investigative studies of the function of the peptide, or for use as a research, diagnostic, or therapeutic agent. In the case of oligomeric compounds targeted to microRNA, candidate modulators may be evaluated by the extent to which they increase the expression of a microRNA target RNA or protein (as interference with the activity of a microRNA will result in the increased expression of one or more targets of the microRNA).
As used herein, "expression" refers to the process by which a gene ultimately results in a protein. Expression includes, but is not limited to, transcription, splicing, post-transcriptional modification, and translation.
Suitable target segments may also be combined with their respective complementary oligomeric compounds provided herein to form stabilized double-stranded (duplexed)
oligonucleotides. Such double stranded oligonucleotide moieties have been shown in the art to modulate target expression and regulate translation as well as RNA processing via an antisense mechanism. Moreover, the double-stranded moieties may be subject to chemical modifications (Fire et al, Nature, 1998, 391, 806-811; Timmons and Fire, Nature, 1998, 395, 854; Timmons et al, Gene, 2001, 263, 103-112; Tabara et al, Science, 1998, 282, 430-431; Montgomery et al, Proc. Natl Acad. Sci. USA, 1998, 95, 15502-15507; Tuschl et al, Genes Dev., 1999, 13, 3191-3197;
Elbashir et al, Nature, 2001, 411, 494-498; Elbashir et al, Genes Dev., 2001, 15, 188-200). For example, such double-stranded moieties have been shown to inhibit the target by the classical hybridization of antisense strand of the duplex to the target, thereby triggering enzymatic
degradation of the target (Tijsterman et al, Science, 2002, 295, 694-697).
The oligomeric compounds provided herein can also be applied in the areas of drug discovery and target validation. In certain embodiments, provided herein is the use of the oligomeric compounds and targets identified herein in drug discovery efforts to elucidate
relationships that exist between proteins and a disease state, phenotype, or condition. These methods include detecting or modulating a target peptide comprising contacting a sample, tissue, cell, or organism with one or more oligomeric compounds provided herein, measuring the nucleic acid or protein level of the target and/or a related phenotypic or chemical endpoint at some time after treatment, and optionally comparing the measured value to a non-treated sample or sample treated with a further oligomeric compound as provided herein. These methods can also be performed in parallel or in combination with other experiments to determine the function of unknown genes for the process of target validation or to determine the validity of a particular gene product as a target for treatment or prevention of a particular disease, condition, or phenotype. In certain embodiments, oligomeric compounds are provided for use in therapy. In certain
embodiments, the therapy is reducing target messenger RNA.
As used herein, the term "dose" refers to a specified quantity of a pharmaceutical agent provided in a single administration. In certain embodiments, a dose may be administered in two or more boluses, tablets, or injections. For example, in certain embodiments, where subcutaneous administration is desired, the desired dose requires a volume not easily accommodated by a single injection. In such embodiments, two or more injections may be used to achieve the desired dose. In certain embodiments, a dose may be administered in two or more injections to minimize injection site reaction in an individual.
Oligomeric compounds may be admixed with pharmaceutically acceptable active and/or
inert substances for the preparation of pharmaceutical compositions or formulations. Compositions and methods for the formulation of pharmaceutical compositions are dependent upon a number of criteria, including, but not limited to, route of administration, extent of disease, or dose to be administered.
Oligomeric compounds, including antisense compounds, can be utilized in pharmaceutical compositions by combining such oligomeric compounds with a suitable pharmaceutically acceptable diluent or carrier. A pharmaceutically acceptable diluent includes phosphate-buffered saline (PBS). PBS is a diluent suitable for use in compositions to be delivered parenterally. Accordingly, in certain embodiments, employed in the methods described herein is a pharmaceutical composition comprising an antisense compound and a pharmaceutically acceptable diluent. In certain embodiments, the pharmaceutically acceptable diluent is PBS.
Pharmaceutical compositions comprising oligomeric compounds encompass any pharmaceutically acceptable salts, esters, or salts of such esters. In certain embodiments, pharmaceutical compositions comprising oligomeric compounds comprise one or more oligonucleotide which, upon administration to an animal, including a human, is capable of providing (directly or indirectly) the biologically active metabolite or residue thereof. Accordingly, for example, the disclosure is also drawn to pharmaceutically acceptable salts of antisense compounds, prodrugs, pharmaceutically acceptable salts of such prodrugs, and other bioequivalents. Suitable pharmaceutically acceptable salts include, but are not limited to, sodium and potassium salts.
A prodrug can include the incorporation of additional nucleosides at one or both ends of an oligomeric compound which are cleaved by endogenous nucleases within the body, to form the active oligomeric compound.
Lipid-based vectors have been used in nucleic acid therapies in a variety of methods. In one method, the nucleic acid is introduced into preformed liposomes or lipoplexes made of mixtures of cationic lipids and neutral lipids. In another method, DNA complexes with mono- or poly-cationic lipids are formed without the presence of a neutral lipid.
In certain methods, preparations are made that include a polyamine compound or a lipid moiety complexed with a nucleic acid. Such preparations are described in PCT publication
WO/2008/042973; and in Akinc et al, Nature Biotechnology 26, 561 - 569 (01 May 2008), which are herein incorporated by reference in their entirety.
In certain embodiments, chemically-modified oligomeric compounds are provided herein that may have a higher affinity for target RNAs than does non-modified DNA. In certain such
embodiments, higher affinity in turn provides increased potency allowing for the administration of lower doses of such compounds, reduced potential for toxicity, improvement in therapeutic index and decreased overall cost of therapy.
Effect of nucleoside modifications on RNAi activity is evaluated according to existing literature (Elbashir et al, Nature, 2001, 411, 494-498; Nishikura et al, Cell, 2001, 707, 415-416; and Bass et al, Cell, 2000, 101, 235-238.)
In certain embodiments, oligomeric compounds provided herein can be utilized for diagnostics, therapeutics, prophylaxis and as research reagents and kits. Furthermore, antisense oligonucleotides, which are able to inhibit gene expression with exquisite specificity, are often used by those of ordinary skill to elucidate the function of particular genes or to distinguish between functions of various members of a biological pathway. In certain embodiments, oligomeric compounds provided herein can be utilized either alone or in combination with other oligomeric compounds or other therapeutics as tools in differential and/or combinatorial analyses to elucidate expression patterns of a portion or the entire complement of genes expressed within cells and tissues. Oligomeric compounds can also be effectively used as primers and probes under conditions favoring gene amplification or detection, respectively. These primers and probes are useful in methods requiring the specific detection of nucleic acid molecules encoding proteins and in the amplification of the nucleic acid molecules for detection or for use in further studies. Hybridization of oligomeric compounds as provided herein, particularly the primers and probes, with a nucleic acid can be detected by means known in the art. Such means may include conjugation of an enzyme to the oligonucleotide, radiolabelling of the oligonucleotide or any other suitable detection means. Kits using such detection means for detecting the level of selected proteins in a sample may also be prepared.
In certain embodiments, the tricyclic oligomeric compounds disclosed herein may be used treat a disease or condition. In certain embodiments, the oligomeric compounds disclosed herein may be used to ameliorate one or more symptoms of a disease or condition. In certain
embodiments, a tricyclic nucleoside monomer may be used as an anti- viral agent. In certain embodiments, a tricyclic nucleoside monomer may be used ameliorate one or more symptoms caused by a virus. In certain embodiments, a tricyclic nucleoside monomer, a salt thereof, or a mono-, di-, or tri-phosphate analog thereof, may be used as an anti-viral agent. In certain embodiments, a tricyclic nucleoside monomer, a salt thereof, or a mono-, di-, or tri-phosphate analog thereof, may be used ameliorate one or more symptoms caused by a virus.
As one nonlimiting example, expression patterns within cells or tissues treated with one or more of the oligomeric compounds provided herein are compared to control cells or tissues not treated with oligomeric compounds and the patterns produced are analyzed for differential levels of gene expression as they pertain, for example, to disease association, signaling pathway, cellular localization, expression level, size, structure or function of the genes examined. These analyses can be performed on stimulated or unstimulated cells and in the presence or absence of other compounds and or oligomeric compounds which affect expression patterns.
Examples of methods of gene expression analysis known in the art include DNA arrays or microarrays (Brazma and Vilo, FEB S Lett., 2000, 480, 17-24; Celis, et al, FEB S Lett, 2000, 480, 2- 16), SAGE (serial analysis of gene expression)(Madden, et al, Drug Discov. Today, 2000, 5, 415- 425), READS (restriction enzyme amplification of digested cDNAs) (Prashar and Weissman, Methods EnzymoL, 1999, 303, 258-72), TOGA (total gene expression analysis) (Sutcliffe, et al., Proc. Natl. Acad. Sci. USA, 2000, 97, 1976-81), protein arrays and proteomics (Celis, et al., FEBS Lett., 2000, 480, 2-16; Jungblut, et al, Electrophoresis, 1999, 20, 2100-10), expressed sequence tag (EST) sequencing (Celis, et al, FEBS Lett., 2000, 480, 2-16; Larsson, et al, J. Biotechnol, 2000, 80, 143-57), subtractive RNA fingerprinting (SuRF) (Fuchs, et al, Anal. Biochem., 2000, 286, 91- 98; Larson, et al., Cytometry, 2000, 41, 203-208), subtractive cloning, differential display (DD) (Jurecic and Belmont, Curr. Opin. Microbiol., 2000, 3, 316-21), comparative genomic hybridization (Carulli, et al., J. Cell Biochem. Suppl., 1998, 31, 286-96), FISH (fluorescent in situ hybridization) techniques (Going and Gusterson, Eur. J. Cancer, 1999, 35, 1895-904) and mass spectrometry methods (To, Comb. Chem. High Throughput Screen, 2000, 3, 235-41).
Those skilled in the art, having possession of the present disclosure will be able to prepare oligomeric compounds, comprising a contiguous sequence of linked monomer subunits, of essentially any viable length to practice the methods disclosed herein. Such oligomeric compounds will include at least one and preferably a plurality of the tricyclic nucleosides provided herein and may also include other monomer subunits including but not limited to nucleosides, modified nucleosides, nucleosides comprising sugar surrogate groups and nucleoside mimetics.
While certain compounds, compositions and methods described herein have been described with specificity in accordance with certain embodiments, the following examples serve only to illustrate the compounds described herein and are not intended to limit the same. Each of the references, GenBank accession numbers, and the like recited in the present application is incorporated herein by reference in its entirety.
Although the sequence listing accompanying this filing identifies each sequence as either "RNA" or "DNA" as required, in reality, those sequences may be modified with any combination of chemical modifications. One of skill in the art will readily appreciate that such designation as "RNA" or "DNA" to describe modified oligonucleotides is, in certain instances, arbitrary. For example, an oligonucleotide comprising a nucleoside comprising a 2'-OH sugar moiety and a thymine base could be described as a DNA having a modified sugar (2'-OH for the natural 2'-H of DNA) or as an RNA having a modified base (thymine (methylated uracil) for natural uracil of RNA).
While in certain embodiments, oligomeric compounds provided herein can be utilized as described, the following examples serve only to illustrate and are not intended to be limiting.
Examples (General)
1H and 13C NMR spectra were recorded on a 300 MHz and 75 MHz Bruker spectrometer, respectively.
Example 1
Synthesis of Nucleoside Phosphoramidites
The preparation of nucleoside phosphoramidites is performed following procedures that are illustrated herein and in the art such as but not limited to US Patent 6,426,220 and published PCT WO 02/36743.
Example 2
Synthesis of Oligomeric Compounds
The oligomeric compounds used in accordance with this invention may be conveniently and routinely made through the well-known technique of solid phase synthesis. Equipment for such synthesis is sold by several vendors including, for example, Applied Biosystems (Foster City, CA).
Any other means for such synthesis known in the art may additionally or alternatively be employed.
It is well known to use similar techniques to prepare oligonucleotides such as alkylated derivatives and those having phosphorothioate linkages.
Oligomeric compounds: Unsubstituted and substituted phosphodiester (P=0) oligomeric compounds, including without limitation, oligonucleotides can be synthesized on an automated
DNA synthesizer (Applied Biosystems model 394) using standard phosphoramidite chemistry with oxidation by iodine.
In certain embodiments, phosphorothioate internucleoside linkages (P=S) are synthesized similar to phosphodiester internucleoside linkages with the following exceptions: thiation is effected by utilizing a 10% w/v solution of 3,H-l,2-benzodithiole-3-one 1, 1-dioxide in acetonitrile for the oxidation of the phosphite linkages. The thiation reaction step time is increased to 180 sec and preceded by the normal capping step. After cleavage from the CPG column and deblocking in concentrated ammonium hydroxide at 55°C (12-16 hr), the oligomeric compounds are recovered by precipitating with greater than 3 volumes of ethanol from a 1 M H4OAc solution. Phosphinate internucleoside linkages can be prepared as described in U.S. Patent 5,508,270.
Alkyl phosphonate internucleoside linkages can be prepared as described in U.S. Patent 4,469,863.
3 '-Deoxy-3 ' -methylene phosphonate internucleoside linkages can be prepared as described in U.S. Patents 5,610,289 or 5,625,050.
Phosphoramidite internucleoside linkages can be prepared as described in U.S. Patent,
5,256,775 or U.S. Patent 5,366,878.
Alkylphosphonothioate internucleoside linkages can be prepared as described in published PCT applications PCT/US94/00902 and PCT/US93/06976 (published as WO 94/17093 and WO 94/02499, respectively).
3 '-Deoxy-3 '-amino phosphoramidate internucleoside linkages can be prepared as described in
U.S. Patent 5,476,925.
Phosphotriester internucleoside linkages can be prepared as described in U.S. Patent 5,023,243.
Borano phosphate internucleoside linkages can be prepared as described in U.S. Patents 5, 130,302 and 5, 177, 198.
Oligomeric compounds having one or more non-phosphorus containing internucleoside linkages including without limitation methylenemethylimino linked oligonucleosides, also identified as MMI linked oligonucleosides, methylenedimethylhydrazo linked oligonucleosides, also identified as MDH linked oligonucleosides, methylenecarbonylamino linked oligonucleosides, also identified as amide-3 linked oligonucleosides, and methyleneaminocarbonyl linked oligonucleosides, also identified as amide-4 linked oligonucleosides, as well as mixed backbone oligomeric compounds
having, for instance, alternating MMI and P=0 or P=S linkages can be prepared as described in U.S. Patents 5,378,825, 5,386,023, 5,489,677, 5,602,240 and 5,610,289.
Formacetal and thioformacetal internucleoside linkages can be prepared as described in U.S. Patents 5,264,562 and 5,264,564.
Ethylene oxide internucleoside linkages can be prepared as described in U.S. Patent
5,223,618.
Example 3
Isolation and Purification of Oligomeric Compounds
After cleavage from the controlled pore glass solid support or other support medium and deblocking in concentrated ammonium hydroxide at 55°C for 12-16 hours, the oligomeric compounds, including without limitation oligonucleotides and oligonucleosides, are recovered by precipitation out of 1 M NH4OAc with >3 volumes of ethanol. Synthesized oligomeric compounds are analyzed by electrospray mass spectroscopy (molecular weight determination) and by capillary gel electrophoresis. The relative amounts of phosphorothioate and phosphodiester linkages obtained in the synthesis is determined by the ratio of correct molecular weight relative to the -16 amu product (+/-32 +/-48). For some studies oligomeric compounds are purified by FIPLC, as described by Chiang et al, J. Biol. Chem. 1991, 266, 18162-18171. Results obtained with HPLC-purified material are generally similar to those obtained with non-HPLC purified material.
Example 4
Synthesis of Oligomeric Compounds using the 96 Well Plate Format
Oligomeric compounds, including without limitation oligonucleotides, can be synthesized via solid phase P(III) phosphoramidite chemistry on an automated synthesizer capable of assembling 96 sequences simultaneously in a 96-well format. Phosphodiester internucleoside linkages are afforded by oxidation with aqueous iodine. Phosphorothioate internucleoside linkages are generated by sulfurization utilizing 3,H-1,2 benzodithiole-3-one 1, 1 dioxide (Beaucage Reagent) in anhydrous acetonitrile. Standard base-protected beta-cyanoethyl-diiso-propyl phosphoramidites can be purchased from commercial vendors (e.g. PE-Applied Biosystems, Foster City, CA, or Pharmacia, Piscataway, NJ). Non-standard nucleosides are synthesized as per standard or patented methods and can be functionalized as base protected beta-cyanoethyldiisopropyl phosphoramidites.
Oligomeric compounds can be cleaved from support and deprotected with concentrated NH4OH at elevated temperature (55-60 °C) for 12-16 hours and the released product then dried in vacuo. The dried product is then re-suspended in sterile water to afford a master plate from which all analytical and test plate samples are then diluted utilizing robotic pipettors.
Example 5
Analysis of Oligomeric Compounds using the 96-Well Plate Format
The concentration of oligomeric compounds in each well can be assessed by dilution of samples and UV absorption spectroscopy. The full-length integrity of the individual products can be evaluated by capillary electrophoresis (CE) in either the 96-well format (Beckman P/ACE™ MDQ) or, for individually prepared samples, on a commercial CE apparatus (e.g., Beckman P/ACE™ 5000, ABI 270). Base and backbone composition is confirmed by mass analysis of the oligomeric compounds utilizing electrospray-mass spectroscopy. All assay test plates are diluted from the master plate using single and multi-channel robotic pipettors. Plates are judged to be acceptable if at least 85% of the oligomeric compounds on the plate are at least 85% full length.
Example 6
In Vitro Treatment of Cells with Oligomeric Compounds
The effect of oligomeric compounds on target nucleic acid expression is tested in any of a variety of cell types provided that the target nucleic acid is present at measurable levels. This can be routinely determined using, for example, PCR or Northern blot analysis. Cell lines derived from multiple tissues and species can be obtained from American Type Culture Collection (ATCC, Manassas, VA).
The following cell type is provided for illustrative purposes, but other cell types can be routinely used, provided that the target is expressed in the cell type chosen. This can be readily determined by methods routine in the art, for example Northern blot analysis, ribonuclease protection assays or RT-PCR.
b.END cells: The mouse brain endothelial cell line b.END was obtained from Dr. Werner Risau at the Max Plank Institute (Bad Nauheim, Germany). b.END cells are routinely cultured in DMEM, high glucose (Invitrogen Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen Life Technologies, Carlsbad, CA). Cells are routinely passaged by trypsinization and dilution when they reached approximately 90% confluence. Cells are seeded into
96-well plates (Falcon-Primaria #353872, BD Biosciences, Bedford, MA) at a density of approximately 3000 cells/well for uses including but not limited to oligomeric compound transfection experiments.
Experiments involving treatment of cells with oligomeric compounds:
When cells reach appropriate confluency, they are treated with oligomeric compounds using a transfection method as described.
LIPOFECTIN™
When cells reached 65-75% confluency, they are treated with one or more oligomeric compounds. The oligomeric compound is mixed with LIPOFECTIN™ Invitrogen Life Tech- nologies, Carlsbad, CA) in Opti-MEM™-l reduced serum medium (Invitrogen Life Technologies, Carlsbad, CA) to achieve the desired concentration of the oligomeric compound(s) and a
LIPOFECTIN™ concentration of 2.5 or 3 μg/mL per 100 nM oligomeric compound(s). This transfection mixture is incubated at room temperature for approximately 0.5 hours. For cells grown in 96-well plates, wells are washed once with 100 OPTI-MEM™-l and then treated with 130 of the transfection mixture. Cells grown in 24-well plates or other standard tissue culture plates are treated similarly, using appropriate volumes of medium and oligomeric compound(s). Cells are treated and data are obtained in duplicate or triplicate. After approximately 4-7 hours of treatment at 37°C, the medium containing the transfection mixture is replaced with fresh culture medium. Cells are harvested 16-24 hours after treatment with oligomeric compound(s).
Other suitable transfection reagents known in the art include, but are not limited to,
CYTOFECTIN™, LIPOFECTAMINE™, OLIGOFECTAMINE™, and FUGENE™. Other suitable transfection methods known in the art include, but are not limited to, electroporation.
Example 7
Real-time Quantitative PCR Analysis of target mRNA Levels
Quantitation of target mRNA levels is accomplished by real-time quantitative PCR using the ABI PRISM™ 7600, 7700, or 7900 Sequence Detection System (PE-Applied Biosystems, Foster City, CA) according to manufacturer's instructions. This is a closed-tube, non-gel-based, fluorescence detection system which allows high-throughput quantitation of polymerase chain reaction (PCR) products in real-time. As opposed to standard PCR in which amplification products are quantitated after the PCR is completed, products in real-time quantitative PCR are quantitated as they accumulate. This is accomplished by including in the PCR reaction an oligonucleotide probe
that anneals specifically between the forward and reverse PCR primers, and contains two fluorescent dyes. A reporter dye (e.g., FAM or JOE, obtained from either PE-Applied Biosystems, Foster City, CA, Operon Technologies Inc., Alameda, CA or Integrated DNA Technologies Inc., Coralville, IA) is attached to the 5' end of the probe and a quencher dye (e.g., TAMRA, obtained from either PE- Applied Biosystems, Foster City, CA, Operon Technologies Inc., Alameda, CA or Integrated DNA Technologies Inc., Coralville, IA) is attached to the 3' end of the probe. When the probe and dyes are intact, reporter dye emission is quenched by the proximity of the 3' quencher dye. During amplification, annealing of the probe to the target sequence creates a substrate that can be cleaved by the 5'-exonuclease activity of Taq polymerase. During the extension phase of the PCR amplification cycle, cleavage of the probe by Taq polymerase releases the reporter dye from the remainder of the probe (and hence from the quencher moiety) and a sequence-specific fluorescent signal is generated. With each cycle, additional reporter dye molecules are cleaved from their respective probes, and the fluorescence intensity is monitored at regular intervals by laser optics built into the ABI PRISM™ Sequence Detection System. In each assay, a series of parallel reactions containing serial dilutions of mRNA from untreated control samples generates a standard curve that is used to quantitate the percent inhibition after antisense oligonucleotide treatment of test samples.
Prior to quantitative PCR analysis, primer-probe sets specific to the target gene being measured are evaluated for their ability to be "multiplexed" with a GAPDH amplification reaction. In multiplexing, both the target gene and the internal standard gene GAPDH are amplified concurrently in a single sample. In this analysis, mRNA isolated from untreated cells is serially diluted. Each dilution is amplified in the presence of primer-probe sets specific for GAPDH only, target gene only ("single-plexing"), or both (multiplexing). Following PCR amplification, standard curves of GAPDH and target mRNA signal as a function of dilution are generated from both the single-plexed and multiplexed samples. If both the slope and correlation coefficient of the GAPDH and target signals generated from the multiplexed samples fall within 10% of their corresponding values generated from the single-plexed samples, the primer-probe set specific for that target is deemed multiplexable. Other methods of PCR are also known in the art.
RT and PCR reagents are obtained from Invitrogen Life Technologies (Carlsbad, CA). RT, real-time PCR is carried out by adding 20 μΐ. PCR cocktail (2.5x PCR buffer minus MgCl2, 6.6 mM MgCl2, 375 μΜ each of dATP, dCTP, dCTP and dGTP, 375 nM each of forward primer and reverse primer, 125 nM of probe, 4 Units RNAse inhibitor, 1.25 Units PLATINUM® Taq, 5 Units MuLV
reverse transcriptase, and 2.5x ROX dye) to 96-well plates containing 30 μL· total RNA solution (20- 200 ng). The RT reaction is carried out by incubation for 30 minutes at 48°C. Following a 10 minute incubation at 95°C to activate the PLATINUM® Taq, 40 cycles of a two-step PCR protocol are carried out: 95°C for 15 seconds (denaturation) followed by 60°C for 1.5 minutes (annealing/- extension).
Gene target quantities obtained by RT, real-time PCR are normalized using either the expression level of GAPDH, a gene whose expression is constant, or by quantifying total RNA using RIBOGREEN™ (Molecular Probes, Inc. Eugene, OR). GAPDH expression is quantified by real time RT-PCR, by being run simultaneously with the target, multiplexing, or separately. Total RNA is quantified using RiboGreen™ RNA quantification reagent (Molecular Probes, Inc. Eugene, OR). Methods of RNA quantification by RIBOGREEN™ are taught in Jones, L.J., et al, (Analytical Biochemistry, 1998, 265, 368-374).
In this assay, 170 μΙ_ of RIBOGREEN™ working reagent (RIBOGREEN™ reagent diluted 1 :350 in lOmM Tris-HCl, 1 mM EDTA, pH 7.5) is pipetted into a 96-well plate containing 30 μΙ_ purified, cellular RNA. The plate is read in a CytoFluor 4000 (PE Applied Biosystems) with excitation at 485nm and emission at 530nm.
Example 8
Analysis of inhibition of target expression
Antisense modulation of a target expression can be assayed in a variety of ways known in the art. For example, a target mRNA levels can be quantitated by, e.g., Northern blot analysis, competitive polymerase chain reaction (PCR), or real-time PCR. Real-time quantitative PCR is presently desired. RNA analysis can be performed on total cellular RNA or poly(A)+ mRNA. One method of RNA analysis of the present disclosure is the use of total cellular RNA as described in other examples herein. Methods of RNA isolation are well known in the art. Northern blot analysis is also routine in the art. Real-time quantitative (PCR) can be conveniently accomplished using the commercially available ABI PRISM™ 7600, 7700, or 7900 Sequence Detection System, available from PE- Applied Biosystems, Foster City, CA and used according to manufacturer's instructions.
Protein levels of a target can be quantitated in a variety of ways well known in the art, such as immunoprecipitation, Western blot analysis (immunoblotting), enzyme-linked immunosorbent assay (ELISA) or fluorescence-activated cell sorting (FACS). Antibodies directed to a target can be identified and obtained from a variety of sources, such as the MSRS catalog of antibodies (Aerie
Corporation, Birmingham, MI), or can be prepared via conventional monoclonal or polyclonal antibody generation methods well known in the art. Methods for preparation of polyclonal antisera are taught in, for example, Ausubel, F.M. et al, Current Protocols in Molecular Biology, Volume 2, pp. 11.12.1-11.12.9, John Wiley & Sons, Inc., 1997. Preparation of monoclonal antibodies is taught in, for example, Ausubel, F.M. et al., Current Protocols in Molecular Biology, Volume 2, pp.
11.4.1-11.11.5, John Wiley & Sons, Inc., 1997.
Immunoprecipitation methods are standard in the art and can be found at, for example, Ausubel, F.M. et al., Current Protocols in Molecular Biology, Volume 2, pp. 10.16.1-10.16.11, John Wiley & Sons, Inc., 1998. Western blot (immunoblot) analysis is standard in the art and can be found at, for example, Ausubel, F.M. et al, Current Protocols in Molecular Biology, Volume 2, pp. 10.8.1-10.8.21, John Wiley & Sons, Inc., 1997. Enzyme-linked immunosorbent assays (ELISA) are standard in the art and can be found at, for example, Ausubel, F.M. et al, Current Protocols in Molecular Biology, Volume 2, pp. 11.2.1-11.2.22, John Wiley & Sons, Inc., 1991. Example 9
Design of phenotypic assays and in vivo studies for the use of target inhibitors
Phenotypic assays
Once target inhibitors have been identified by the methods disclosed herein, the oligomeric compounds are further investigated in one or more phenotypic assays, each having measurable endpoints predictive of efficacy in the treatment of a particular disease state or condition.
Phenotypic assays, kits and reagents for their use are well known to those skilled in the art and are herein used to investigate the role and/or association of a target in health and disease.
Representative phenotypic assays, which can be purchased from any one of several commercial vendors, include those for determining cell viability, cytotoxicity, proliferation or cell survival (Molecular Probes, Eugene, OR; PerkinElmer, Boston, MA), protein-based assays including enzymatic assays (Panvera, LLC, Madison, WI; BD Biosciences, Franklin Lakes, NJ; Oncogene Research Products, San Diego, CA), cell regulation, signal transduction, inflammation, oxidative processes and apoptosis (Assay Designs Inc., Ann Arbor, MI), triglyceride accumulation (Sigma- Aldrich, St. Louis, MO), angiogenesis assays, tube formation assays, cytokine and hormone assays and metabolic assays (Chemicon International Inc., Temecula, CA; Amersham Biosciences, Piscataway, NJ).
In one non-limiting example, cells determined to be appropriate for a particular phenotypic assay (i.e., MCF-7 cells selected for breast cancer studies; adipocytes for obesity studies) are treated with a target inhibitors identified from the in vitro studies as well as control compounds at optimal concentrations which are determined by the methods described above. At the end of the treatment period, treated and untreated cells are analyzed by one or more methods specific for the assay to determine phenotypic outcomes and endpoints.
Phenotypic endpoints include changes in cell morphology over time or treatment dose as well as changes in levels of cellular components such as proteins, lipids, nucleic acids, hormones, saccharides or metals. Measurements of cellular status which include pH, stage of the cell cycle, intake or excretion of biological indicators by the cell, are also endpoints of interest.
Measurement of the expression of one or more of the genes of the cell after treatment is also used as an indicator of the efficacy or potency of the target inhibitors. Hallmark genes, or those genes suspected to be associated with a specific disease state, condition, or phenotype, are measured in both treated and untreated cells.
In vivo studies
The individual subjects of the in vivo studies described herein are warm-blooded vertebrate animals, which includes humans.
Example 10
RNA Isolation
Poly (A) + mRNA isolation
Poly(A)+ mRNA is isolated according to Miura et al, (Clin. Chem., 1996, 42, 1758-1764). Other methods for poly(A)+ mRNA isolation are routine in the art. Briefly, for cells grown on 96- well plates, growth medium is removed from the cells and each well is washed with 200 xL cold PBS. 60 μ-L lysis buffer (10 mM Tris-HCl, pH 7.6, 1 mM EDTA, 0.5 M NaCl, 0.5% NP-40, 20 mM vanadyl-ribonucleoside complex) is added to each well, the plate is gently agitated and then incubated at room temperature for five minutes. 55 xL of lysate is transferred to Oligo d(T) coated 96-well plates (AGCT Inc., Irvine CA). Plates are incubated for 60 minutes at room temperature, washed 3 times with 200 μ-L of wash buffer (10 mM Tris-HCl pH 7.6, 1 mM EDTA, 0.3 M NaCl). After the final wash, the plate is blotted on paper towels to remove excess wash buffer and then air- dried for 5 minutes. 60 xL of elution buffer (5 mM Tris-HCl pH 7.6), preheated to 70°C, is added to
each well, the plate is incubated on a 90°C hot plate for 5 minutes, and the eluate is then transferred to a fresh 96-well plate.
Cells grown on 100 mm or other standard plates may be treated similarly, using appropriate volumes of all solutions.
Total RNA Isolation
Total RNA is isolated using an RNEASY 96™ kit and buffers purchased from Qiagen Inc. (Valencia, CA) following the manufacturer's recommended procedures. Briefly, for cells grown on 96-well plates, growth medium is removed from the cells and each well is washed with 200 xL cold PBS. 150 xL Buffer RLT is added to each well and the plate vigorously agitated for 20 seconds. 150 xL of 70% ethanol is then added to each well and the contents mixed by pipetting three times up and down. The samples are then transferred to the RNEASY 96™ well plate attached to a
QIAVAC™ manifold fitted with a waste collection tray and attached to a vacuum source. Vacuum is applied for 1 minute. 500 of Buffer RW1 is added to each well of the RNEASY 96™ plate and incubated for 15 minutes and the vacuum is again applied for 1 minute. An additional 500 xL of Buffer RW1 is added to each well of the RNEASY 96™ plate and the vacuum is applied for 2 minutes. 1 mL of Buffer RPE is then added to each well of the RNEASY 96™ plate and the vacuum applied for a period of 90 seconds. The Buffer RPE wash is then repeated and the vacuum is applied for an additional 3 minutes. The plate is then removed from the QIAVAC™ manifold and blotted dry on paper towels. The plate is then re-attached to the QIAVAC™ manifold fitted with a collection tube rack containing 1.2 mL collection tubes. RNA is then eluted by pipetting 140 xL of RNAse free water into each well, incubating 1 minute, and then applying the vacuum for 3 minutes.
The repetitive pipetting and elution steps may be automated using a QIAGEN Bio-Robot 9604 (Qiagen, Inc., Valencia CA). Essentially, after lysing of the cells on the culture plate, the plate is transferred to the robot deck where the pipetting, DNase treatment and elution steps are carried out.
Example 11
Target-specific primers and probes
Probes and primers may be designed to hybridize to a target sequence, using published sequence information.
For example, for human PTEN, the following primer-probe set was designed using published sequence information (GENBANK™ accession number U92436.1, SEQ ID NO: 1).
Forward primer: AATGGCTAAGTGAAGATGACAATCAT (SEQ ID NO: 2)
Reverse primer: TGCACATATCATTACACCAGTTCGT (SEQ ID NO: 3)
And the PCR probe:
FAM-TTGCAGCAATTCACTGTAAAGCTGGAAAGG-TAMRA (SEQ ID NO: 4), where FAM is the fluorescent dye and TAMRA is the quencher dye.
Example 12
Western blot analysis of target protein levels
Western blot analysis (immunoblot analysis) is carried out using standard methods. Cells are harvested 16-20 h after oligonucleotide treatment, washed once with PBS, suspended in Laemmli buffer (100 μΐ/well), boiled for 5 minutes and loaded on a 16% SDS-PAGE gel. Gels are run for 1.5 hours at 150 V, and transferred to membrane for western blotting. Appropriate primary antibody directed to a target is used, with a radiolabeled or fluorescently labeled secondary antibody directed against the primary antibody species. Bands are visualized using a PHOSPHORIMAGER™
(Molecular Dynamics, Sunnyvale CA).
Example 13
Preparation of Compounds 7 and 8

Diacetone glucose, Compound 1 is commercially available.
Example 14
Preparation of Compounds 17 and 18


Compounds 7 and 8 are prepared as per the procedures illustrated in Example 13.
Example 15
Preparation of Compounds 21 and 22

Compounds 15 and 16 are prepared as per the procedures illustrated in Example
Example 16
Preparation of Compounds 33 and 34

Compound 5 is prepared as per the procedures illustrated in Example 13. In the tritylation step, other hydroxyl protecting groups can be used in place of DMTC1 (e.g. levulinic acid).
Example 17
Preparation of Compounds 41 and 42


Compounds 35 and 36 are prepared as per the procedures illustrated in Example 16. In the tritylation step, other hydroxyl protecting groups can be used in place of DMTC1 (e.g. levulinic acid).
Example 18
Preparation of Compound 48

a) Preparation of Compound 44
Commercially available l,2;5,6-di-0-isopropylidene-a-D-allofuranose, Compound 43, (135 g, 519.0 mmol) and 2-(bromomethyl)-naphthalene (126 g, 570.0 mmol) were dissolved in DMF (500 mL) in a three-necked flask (500 mL) and the reaction was cooled in an ice bath. Sodium hydride (60% w/w, 29 g, 727.0 mmol) was carefully added (6 g portions every 10 minutes) to the reaction and the stirring was continued for another 60 minutes after the addition was complete. At this time TLC analysis showed no more starting Compound 43. The reaction was carefully poured onto crushed ice (ca. 500 g) and the resulting slurry was stirred vigorously until all the ice melted. The resulting off-white solid was collected by filtration and suspended in water. The suspension was stirred vigorously using a mechanical stirrer for 30 minutes after which the solid was collected
by filtration and suspended in hexanes. The suspension was stirred vigorously for 30 minutes after which the solid was collected by filtration and air dried for 4-6 hours and then dried under high vacuum over P2O3 for 16 hours to provide Compound 44 (206.0 g, 99%) as an off-white solid.
Compound 44: 1H MR (300 MHz, CDC13): δ: 7.85 (m, 4H), 7.48 (m, 3H), 5.74 (s, 1H), 4.92 (d, 1H, 7= 11.7), 4.75 (d, 1H, 7= 11.6), 4.58 (m, 1H), 4.36 (m, 1H), 4.15 (m, 1H), 4.03-3.86 (m, 3H), 1.61 (s, 3H), 1.36 (s, 9H). b) Preparation of Compound 45
Compound 44 (200.0 g, 0.5 moles) was added in small portions to a solution of acetic acid (2.2 L) and water (740 mL). The reaction was stirred at room temperature for 16 h after which, TLC analysis (30% EtOAc/hexanes) indicated complete consumption of Compound 44. The reaction was then concentrated under reduced pressure until most of the acetic acid was removed. The remaining solution was poured into a stirred mixture of EtOAc (IL) and water (IL). Solid KOH was then added to the above mixture until the aqueous layer was strongly basic (pH>12). The organic layer was then separated, washed with saturated sodium bicarbonate solution, brine, dried (Na2SO4), filtered and concentrated under reduced pressure to provide Compound 45 as a yellow foam, which was used without any further purification. c) Preparation of Compound 46
A solution of NaI04 (107.0 g) in water (3 L) was added over 40 minutes to a stirred
(mechanical stirrer) solution of Compound 45 (crude from above) in dioxane (1.5 L) After 60 minutes the reaction mixture was poured into EtOAc (1.5 L) and the organic layer was separated, washed with water (IL), brine (IL), dried (Na2SO4) and concentrated to provide Compound 46 as a yellow oil, which was used without any further purification. d) Preparation of Compound 47
Compound 46 (crude from above) was dissolved in a mixture of THF (500) and water (500 mL) and the reaction was cooled in an ice bath. 2N NaOH (600 mL) and formaldehyde (250 mL of a 37%) aqueous solution) were added to the reaction and the stirring was continued at room temperature for 3 days. The reaction was then poured into EtOAc (1 L) and washed with water (1 L), brine (1 L) and evaporated under reduced pressure until approximately 200 mL of EtOAc was left (a white precipitate was formed in the process). Hexanes (300 mL) was added to the precipitate
and the mixture was allowed to stand for 16 hours after which the white solid was collected by filtration, washed with hexanes and dried under high vacuum over P2O5 to provide Compound 47 as a white solid (124 g, 66% from Compound 48).
Compound 47: 1H MR (300 MHz, CDC13): δ: 7.85 (m, 4H), 7.48 (m, 3H), 5.75 (d, 1H, J = 3.9), 4.96 (d, 1H. J= 11.8), 4.75 (d, 1H, J= 11.8), 4.66 (m, 1H), 4.26 (d, 1H, J= 5.2), 3.95 (m, 2H),
3.79 (m, 1H), 3.63 (m, 1H), 2.39 (m, 1H, OH), 1.66 (s, 3H), 1.34 (s, 3H). e) Preparation of Compounds 48 and 49
tert-Butyldiphenylchlorosilane (305.0 mmol, 84.0 mL) was added to a cold (0 °C) stirring solution of Compound 47 (278.0 mmol, 100.0 g) and triethylamine (305 mmol, 43.0 mL) in dichloromethane (600 mL). After the addition was complete, the reaction was warmed to room temperature and the stirring was continued for 16 hours. MeOH (50 mL) was added (to quench the excess TBDPSC1) to the reaction and the stirring was continued for another 2 hours at room temperature. The reaction was then diluted with chloroform and the organic layer was washed with 10% HC1, saturated NaHC03, brine, dried (Na2S04) and concentrated to provide a thick oil.
Hexanes (150 mL) was added to the oil and the mixture was sonicated until a solution resulted. The solution was now seeded with a small amount of Compound 48 (previously isolated by column chromatography). After standing for 16 hours additional hexanes was added to the thick slurry and the solid was collected by filtration. The solid was then resuspended in hexanes and stirred vigorously for 30 minutes. The solid was collected by filtration to provide Compound 48 (80.5,
48%) g) after drying under high vacuum for 16 hours. The filtrates were combined and concentrated under reduced pressure. The resulting oil was redissolved in minimum amount of hexanes and passed through a plug of silica gel (eluting with 20%> EtOAc in hexanes). Fractions containing Compound 48 were combined, concentrated and crystallized as described above to provide a second crop of Compound 48 (20 g, 12%>) as a white solid. Further elution of the silica gel plug with 50%> EtOAc in hexanes provided pure Compound 48 (40.0 g, 24%>) as a thick oil. In addition, a mixture of Compounds 48 and 49 (ca 15 g, 9%>) was also isolated as a thick oil.
Compound 48: 1H NMR (300 MHz, CDC13): δ: 7.83 (m, 4H), 7.56 (m, 7H), 7.30 (m, 6H),
5.80 (s, 1H), 4.97 (d, 1H, J= 11.4), 4.70 (m, 2H), 4.46 (m, 1H), 3.92-3.66 (m, 4H), 2.39 (m, 1H, OH), 1.67 (s, 3H), 1.37 (s, 3H), 0.92 (s, 9H).
Compound 49: 1H NMR (300 MHz, CDC13): δ: 7.9-7.3 (m, 17H), 5.71 (d, 1H, J= 3.9), 4.86 (d, 1H, J= 12.2), 4.74 (d, 1H, J= 12.2), 4.56 (m, 1H), 4.22 (d, 1H, 7= 11.1), 4.18 (m, 1H), 4.07 (d,
1H, J= 11.1), 4.02 (dd, 1H, 7= 4.2, 12.0), 3.64 (dd, 1H, 7= 9.4, 11.9), 1.89 (m, 1H), 1.25 (s, 6H), 1.05 (s, 9H). f) Recover Compound 47 from Compound 49
Tetrabutylammonium fluoride (70 mL of a 1M solution in THF) was added to a cold (0 °C) stirring solution of Compound 49 (62.7 mmol, 37.5 g) in THF (250 mL) after which, the reaction was allowed to warm to room temperature gradually. After stirring for an additional 72 hours, the reaction was concentrated under vacuum and the residue was poured onto crushed ice. The flask was rinsed with some additional THF (3 times) and added to the above suspension. The supernatant was removed by decantation and the solid at the bottom was added to a stirring mixture of hexanes (200 mL) and water (200 mL). After stirring for 2 hours, the flocculent solid was collected by filtration, washed with additional water and hexanes and dried under high vacuum to provide Compound 47 (20 g, 89%) as a white solid. Example 19
Pre aration of Compound 70



Compound 48 was prepared as per the procedures illustrated in Example 18. To a solution of alcohol, Compound 48 (5.0 g, 8.35 mmol) in DCM (60 mL) was added pyridinium
chlorochromate (PCC, 5.4 g, 23.20 mmol). After that stirring for 13 h at 40 °C, the reaction mixture was cooled to room temperature and filtered through a plug of silica gel (4x7 cm). The plug was washed thoroughly with DCM and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 10:90% ethyl acetate-hexanes to afford Compound 50 (4.73 g, 95%) as a white solid.
Compound 50: Rf : 0.32 (ethyl acetate :hexanes = 10: 90%). [a]D +5.4 (c 0.25, CHC13). IR (NaCl) u 3051, 2931, 2857, 1730, 1427, 1383, 1215, 1112, 1021, 822, 752, 703 cm"1. 1H NMR (CDC13, 400 MHz): δ 0.91 (s, 9H), 1.37 (s, 3H), 1.64 (s, 3H), 3.89 (d, 7= 11.4 Hz, 1H), 3.89 (d, 7 = 11.4 Hz, 1H), 4.57 (d, 7= 4.4 Hz, 1H), 4.65-4.67 (m, 1H), 4.78 (d, 7= 12.3 Hz, 1H), 4.90 (d, 7 = 12.3 Hz, 1H), 5.85 (d, 7= 3.3 Hz, 1H), 7.28-7.35 (m, 4H), 7.35-7.43 (m, 2H), 7.45-7.51 (m, 3H), 7.53-7.60 (m, 4H), 7.74-7.84 (m, 4H), 9.93 (s, 1H). 13C NMR (CDC13, 100 MHz): δ 26.4; 26.9; 63.3; 73.2; 78.8, 79.2; 90.9; 105.1; 114.4; 125.9; 126.4, 126.5, 127.2, 127.9, 128.0, 128.1, 128.7, 130.0, 130.1, 132.7, 133.0, 133.4, 133.5, 134.6, 135.7, 135.8, 200.5. HRMS (ESIMS): calcd for C36H4oNa06Si: [M+Na]+' 619.24864; found: 619.25006.
b) Preparation of Compound 51
To a solution of Compound 50 (3.0 g, 4.7 mmol) in DCM (45 mL) at -40 °C was added BF3.OEt2 (1.26 mL, 9.8 mmol) dropwise followed by allyltrimethylsilane (1.20 ml, 7.6 mmol). After stirring at -41 °C for 70 min, a solution of saturated aqueous NaHC03 (15 mL) was added and the reaction mixture was allowed to warm to room temperature. Additional water (40 mL) was added and the organic phase was collected. The aqueous portion was back-extracted with with DCM (40 mL) and the combined organic phase was dried over MgS04, filtered and evaporated. The residue was purified by flash column chromatography on silica gel with a solvent system of 8:92% ethyl acetate-hexanes to afford Compound 51 (2.83 g, 88%) as a white solid.
Compound 51 : Rf : 0.31 (EtOAc:hexanes = 10:90%). [a]D +9.9 (c 0.15, CHC13). IR (NaCl) υ 3543, 3071, 2933, 2857, 1641, 1471, 1428, 1382, 1373, 1214, 1166, 1112, 1026, 820, 753, 703 cm"1. 1H MR (CDC13, 400 MHz): δ 0.90 (s, 9H), 1.38 (s, 3H), 1.63 (s, 3H), 1.82-1.95 (m, 1H), 2.51 (dd, Jl = 14.4 Hz, J2 = 6.7 Hz, 1H), 3.33 (t, J= 2.2 Hz, 1H), 3.79 (d, J= 11.2 Hz, 1H), 3.95 (d, J= 11.2 Hz, 1H), 4.46 (td, Jl = 2.1 Hz, J2 = 11.0 Hz, 1H), 4.67 (d, J= 5.2 Hz, 1H), 4.68 (d, J = 11.8 Hz, 1H), 4.75 (dd, Jl = 5.2 Hz, J2 = 3.7 Hz, 1H), 4.95-5.00 (m, 1H), 5.94 (d, J= 11.9 Hz, 1H), 5.00-5,07 (m, 1H), 5.77-5.89 (m, 1H), 5.83 (d, J= 3.8, 1H), 7,25-7.35 (m, 4H), 7.35-7.43 (m, 2H), 7.45-7.55 (m, 5H), 7.55-7.60 (m, 2H), 7.75-7.85 (m, 4H). 13C MR (CDC13, 100 MHz): δ 19.3, 26.9, 34.8, 62.7, 72.8, 73.3, 78.3, 79.4, 88.4, 104.9, 114.0, 116.4, 125.9, 126.5, 126.6, 127.5, 127.88, 127.93, 127.6, 128.2, 128.9, 129.85, 129.94, 133.2, 133.3, 133.4, 133.5, 134.5, 135.7, 135.8, 136.6. HRMS (ESIMS): calcd for C^iLgNaOeSi: [M+Na]+, 661.29559; found: 661.29705. c) Preparation of Compound 52
To a solution of Compound 51 (2.6 g, 4.1 mmol) in anhydrous DCM (100 mL) at 0 °C was sequentially added pyridine (1 mL, 12.3 mmol) and TBSOTf (1.4 mL, 6.1 mmol). The reaction mixture was allowed to warm to room temperature and the stirring was continued for 3 h. The mixture was partitioned between a saturated aqueous solution of NaHC03 (70 mL) and DCM. The organic phase was collected and the aqueous portion was back-extracted with DCM (2x50 mL). The organic phases were combined and dried over MgS04, filtered and evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 5:95% ethyl acetate-hexanes to afford Compound 52 (3.01 g, 98%) as a colorless oil.
Compound 52: Rf . 0.56 (EtOAc:hexanes = 10: 90%). [a]D +7.7 (c 0.11, CHC13). IR (NaCl) υ 3071, 2954, 2930, 2891, 2856, 1471, 1428, 1381, 1360, 1253, 1166, 1104, 1027, 903, 834, 775,
741, 702 cm"1. 1H MR (CDC13, 400 MHz): (5 -0.16 (s, 3H), -0.07 (s, 3H), 0.70 (s, 9H), 0.96 (s, 9H), 1.41 (s, 3H), 1.62 (s, 3H), 2.38-2.49 (m, 1H), 2.60-2.68 (m, 1H), 3.79 (d, J= 11.1 Hz, 1H), 3.92 (d, J= 11.1 Hz, 1H), 4.38 (d, J= 5.6 Hz, 1H), 4.56 (dd, Jl = 5.6 Hz, J2 = 4.1 Hz, 1H), 4.67 (d, J= 12.0 Hz, 1H), 4.85 (dd, Jl = 5.2 Hz, J2 = 4.4 Hz, 1H), 4.90 (brd, J= 10.1 Hz, 1H), 5.04 (brd, J = 16.8 Hz, 1H), 5.06 (d, J= 12.0 Hz, 1H), 5.78-5.91 (m, 1H), 6.02 (d, J= 4.1 Hz, 1H), 7.32-7.38 (m, 4H), 7.39-7.45 (m, 2H), 7.46-7.52 (m, 2H), 7.53-7.57 (m, 1H), 7.58-7.66 (m, 4H), 7.79-7.87 (m, 4H). 13C MR (CDC13, 100 MHz): (5 -4.6, -3.5, 18.3, 19.3, 25.9, 26.1, 27.0, 27.3, 27.4, 38.6, 66.6, 72.0, 72.6, 77.4, 78.1, 81.0, 91.2, 105.6, 113.9, 116.7, 126.0, 126.3, 126.8, 127.90, 127.94, 128.0, 128.2, 129.8, 129.9, 133.1, 133.2, 133.3, 133.4, 135.73, 135.79, 136.8. HRMS (ESIMS): calcd for C45H6oNa06Si2: [M+Na]+, 775.38206; found: 775.38208. d) Preparation of Compound 53
To a solution of Compound 52 (3.0 g, 4.0 mmol) in DCM (90 mL) and H20 (10 mL) was added DDQ (1.81 g, 8.0 mmol). After stirring at rt for 5.5 h, the mixture was poured into a saturated aqueous solution of NaHC03 (180 mL). The organic phase was collected and the aqueous portion was back-extracted with DCM (3x80 mL). The organic phases were combined and dried over MgS04, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on silica gel with a solvent system of 2:98% ethyl acetate-hexanes to afford Compound 53 (2.14 g, 88%) as a colorless oil.
Compound 53 : Rf : 0.62 (EtOAc:hexanes = 5:95%). [a]D +38.5 (c 0.07, CHC13). IR (NaCl) υ 3498, 3051, 2955, 2931, 2890, 2857, 1471, 1428, 1383, 1252, 1160, 1093, 1026, 913, 836, 778, 702 cm"1. 1H MR (CDC13, 400 MHz): δ 0.01 (s, 3H), 0.06 (s, 3H), 0.74 (s, 9H), 1.05 (s, 9H), 1.44 (s, 3H), 1.60 (s, 3H), 1.99-2.09 (m, 1H), 2.45- 2.53 (m, 1H), 2.92 (d, J= 4.0 Hz, 1H), 3.82 (d, J = 10.9 Hz, 1H), 3.91 (d, J= 10.9 Hz, 1H), 4.23 (dd, Jl = 6.4 Hz, J2 = 4.1 Hz, 1H), 4.30 (dd, Jl = 7.6 Hz, J2 = 3.4 Hz, 1H), 4.91 (brd, J= 10.1 Hz, 1H), 4.87-5.00 (m, 2H), 5.70-5.85 (m, 1H), 6.14 (d, J = 4.5 Hz, 1H), 7.34-7.46 (m, 6H), 7.62-7.68 (m, 4H). 13C NMR (CDC13, 100 MHz): δ -4.7, -3.9, 18.4, 19.3, 26.2, 27.1, 27.4, 27.8, 37.8, 67.3, 70.6, 72.0, 82.4, 94.1, 106.4, 114.8, 116.7, 128.1, 130.0, 130.1, 132.8, 135.5, 136.8. HRMS (ESIMS): calcd for C34H52Na06Si2: [M+Na]+, 635.31946; found: 635.32037.
e) Preparation of Compound 54
To a solution of Compound 53 (2.2 g, 3.6 mmol) in DCM (55 mL) was added solid NaHC03 (0.5 g, 5.9 mmol) followed by DMP (2.34 g, 5.5 mmol). After stirring for 17.5 h, the reaction mixture was filtered through a plug of silica gel (8 cm x 2.5 cm) and all volatiles of the filtrate was evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 5:95% ethyl acetate-hexanes to afford Compound 54 (2.18 g, 97%) as a colorless oil.
Compound 54: Rf : 0.61 (EtOAc:hexanes = 5:95%). [a]D +24.6 (c 0.05, CHC13). IR (NaCl) υ 3073, 2956, 2931, 2892, 1773, 1472, 1428, 1383, 1257, 1214, 1159, 1113, 1055, 1005, 913, 836, 776, 702 cm"1. 1H NMR (CDC13, 400 MHz): δ -0.25 (s, 3H), -0.09 (s, 3H), 0.69 (s, 9H), 0.99 (s, 9H), 1.38 (s, 3H), 1.55 (s, 3H), 2.22-2.39 (m, 2H), 3.82 (d, J= 10.4 Hz, 1H), 3.85 (d, J= 10.4 Hz, 1H), 3.92 (dd, Jl = 8.0 Hz, J2 = 4.2 Hz, 1H), 4.42 (d, J= 4.2 Hz, 1H), 4.92-4.97 (m, 1H), 4.97-5.03 (m, 1H), 6.29 (d, J= 4.2 Hz, 1H), 7.32-7.45 (m, 6H), 7.53-7.58 (m, 2H), 7.61-7.66 (m, 2H). 13C NMR (CDC13, 100 MHz): δ -4.6, -3.3, 18.2, 19.3, 26.0, 26.8, 27.0, 27.1, 38.5, 68.1, 73.9, 78.1, 93.3, 103.1, 115.3, 117.6, 128.1, 130.09, 130.14, 132.3, 132.5, 135.7, 135.8, 209.5. HRMS (ESIMS): calcd for C34H5oNa06Si2: [M+Na]+, 633.30381; found: 633.30586. f) Preparation of Compound 55
To a solution of Compound 54 (2.23 g, 3.66 mmol) in DMA (39 mL) and H20 (6 mL) was added Cu(AcO)2 (0.134 g, 0.74 mmol) and PdCl2 (0.065 g, 3.67 mmol). After stirring for 23.5 h at 40 °C, the reaction mixture was partitioned between Et20 (4x200 mL) and saturated aqueous NaHC03 (400 mL). The organic phase was collected, dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 5:95% ethyl acetate-hexanes to afford Compound 55 (2.18 g, 95%) as a yellowish oil.
Compound 55: Rf : 0.52 (EtOAc:hexanes = 10:90%). [a]D +26.7 (c 0.09, CHC13); IR (NaCl) u 3072, 2955, 2931, 2892, 1775, 1721, 1472, 1428, 1375, 1256, 1215, 1159, 1113, 1029, 1005, 869, 838, 779, 703 cm"1. 1H MR (CDC13, 400 MHz): δ -0.17 (s, 3H), ), -0.09 (s, 3H), 0.68 (s, 9H), 0.99 (s, 9H), 1.38 (s, 3H), 1.59 (s, 3H), 2.08 (s, 3H), 2.56 (dd, Jl = 18.0 Hz, J2 = 7.5 Hz, 1H), 2.73 (dd, Jl = 18.0 Hz, J2 = 2.8 Hz, 1H), 3.74 (d, J= 10.5 Hz, 1H), 3.98 (d, J= 10.5 Hz, 1H), 4.41 (d, J= 4.2 Hz, 1H), 4.56 (dd, Jl = 7.2 Hz, J2 = 2.8 Hz, 1H), 6.21 (d, J= 4.2 Hz, 1H), 7.33-7.46 (m, 6H), 7.56-7.60 (m, 2H), 7.63-7.67 (m, 2H). 13C NMR (CDC13, 100 MHz): δ -4.6, -3.3, 18.2,
19.3, 26.0, 26.8, 27.0, 27.1, 38.5, 68.1, 73.9, 78.1, 93.3, 103.1, 115.3, 117.6, 128.1, 130.09, 130.14, 132.3, 132.5, 135.7, 135.8, 209.6. HRMS (ESIMS): calcd for C34H50NaO7Si2: [M+Na]+,
649.29873; found: 649.30014. g) Preparation of Compound 56
To a solution of Compound 55 (2.1 g, 3.35 mmol) in DMSO (90 mL) was added L-proline (0.116 g, 1.0 mmol). After stirring at 55 °C for 53.5 h, the reaction mixture was cooled to rt and partitioned between ethyl acetate and a solution of saturated aqueous NaHC03 (200 mL). The aqueous portion was back-extracted with EtOAc (4x200 mL) and the combined
oganic phase was washed with brine (200 mL) followed by water (2x200 mL). The organic phase was collected, dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 10:90% ethyl acetate-hexanes to afford the desired Compound 56 (1.72 g, 82%) as a white solid and the starting material Compound 55 (0.155 g, 7%).
Compound 56: Rf : 0.25 (EtOAc:hexanes = 10:90%). Melting point = 39.4-43.0 °C. [<x]D
+52.3 (c 0.12, CHC13); IR (NaCl) υ 3467, 3072, 2954, 2931, 2889, 2858, 1724, 1472, 1428, 1383, 1254, 1215, 1166, 1113, 1031, 940, 836, 778, 739, 703 cm"1. 1H NMR (CDC13, 400 MHz): (5 -0.18 (s, 3H), -0.09 (s, 3H), 0.69 (s, 9H), 1.09 (s, 9H), 1.29 (s, 3H), 1.49 (s, 3H), 2.34 (dd, Jl = 18.2 Hz, J2 = 3.8 Hz, 1H), 2.38 (d, J= 16.3 Hz, 1H), 2.70 (dd, Jl = 18.2 Hz, J2 = 9.9 Hz, 1H), 3.76 (d, J = 10.8 Hz, 1H), 3.89 (dd, Jl = 9.9 Hz, J2 = 3.9 Hz, 1H), 4.07 (d, J= 10.8 Hz, 1H), 4.61 (d, J= 3.9 Hz, 1H), 4.62 (s, 1H), 6.19 (d, J= 3.9 Hz, 1H), 7.36-7.48 (m, 6H), 7.60-7.64 (m, 2H), 7.66-7.70 (m, 2H). 13C NMR (CDC13, 100 MHz): δ -4.84, -4.40, 18.2, 19.2, 25.8, 26.69, 26.71, 27.2, 43.0, 46.5, 68.0, 69.1, 80.7, 90.9, 91.2, 103.9, 114.6, 128.3, 128.4, 130.4, 130.6, 131.3, 131.6, 135.6, 135.7, 206.3. HRMS (ESIMS): calcd for C34H5oNa07Si2: [M+Na]+, 649.29873; found: 649.30023.
The preparation of Compound 56 was also prepared using similar reaction conditions as described above, except DMF was used in place of DMSO. h) Preparation of Compound 57
To a suspension of tetramethylammonium triacetoxyborohydride (Me4NBH(OAc)3, 2.55 g, 9.69 mmol) in acetonitrile (45 mL) at 0 °C was added anhydrous AcOH (1.11 mL, 19.36 mmol). After stirring for 15 min, a solution of Compound 56 (1.21 g, 1.93 mmol) in acetonitrile (80 mL) was added. The reaction mixture was allowed to warm to rt and left stirring for another 7 h. The
mixture was then partitioned with DCM and H20 (200 mL). The aqueous portion was back- extracted with DCM (3x200 mL) and the combined organic phase was washed with saturated aqueous solution of NaHC03 (2x200 mL). The organic phase was collected, dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 30:70% ethyl acetate-hexanes to afford the desired Compound 57 (1.13 g, 93%) as a white solid.
Compound 57: Rf : 0.20 (EtOAc:hexanes = 30:70%). Melting point = 38.6-41.9 °C. [a]D +32.9 (c 0.14, CHC13). IR (NaCl) υ 3428, 2956, 2932, 2857, 1471, 1462, 1428, 1374, 1256, 1208, 1112, 1059, 1005, 910, 837, 775, 702 cm"1. 1H NMR (CDC13, 400 MHz): (5 -0.17 (s, 3H); -0.06 (s, 3H), 0.72 (s, 9H), 1.08 (s, 9H), 1.31 (s, 3H), 1.60 (s, 3H), 1.80-2.00 (m, 3H), 2.11 (dd, Jl = 14.8 Hz, J2 = 6.0 Hz, 1H), 2.28-2.34 (m, 1H), 3.44 (s, 1H), 3.57 (dd, Jl = 9.7 Hz, J2 = 3.6 Hz, 1H), 3.68 (d, J = 10.8 Hz, 1H), 3.93 (d, J= 10.8 Hz, 1H), 3.88-3.97 (m, 1H), 4.85 (d, J= 4.8 Hz, 1H), 6.14 (d, J = 4.8 Hz, 1H), 7.32-7.45 (m, 6H), 7.61-7.71 (m, 4H). 13C NMR (CDC13, 100 MHz): δ -4.6, -4.3, 18.2, 19.2, 26.0, 26.6, 26.9, 27.1, 37.4, 39.7, 65.1, 68.1, 69.4, 81.0, 90.1, 90.8, 104.1, 114.2, 128.2, 130.2, 130.4, 131.9, 132.1, 135.7, 135.8. HRMS (ESIMS): calcd for C34H52Na07Si2: [M+Na]+, 651.31438; found: 651.31722.
The preparation of Compound 57 was also prepared using similar reaction conditions as described above, except sodium triacetoxyborohydride (NaBH(OAc)3) was used in place of tetramethylammonium triacetoxyborohydride (Me4NB(OAc)3. i) Preparation of Compound 58
To a solution of Compound 57 (2.41, 3.83 mmol) in DCM (125 mL) was added DMAP (0.24 mg, 1.96 mmol) followed by thiocarbodiimidazole (0.89 g, 4.5 mmol). After stirring for 19 h at 40 °C, the reaction mixture was allowed cooled to rt and treated with 0.5% aqueous HC1 (350 mL). The organic phase was collected and the aqueous portion was back-extracted with DCM (3x100 mL). The organic phases were combined, washed with saturated aqueous NaHC03 (200 mL), dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 30:70%) ethyl acetate-hexanes to afford the carbothioate (2.35 g, 83%>) as a white solid.
Carbothioate: f : 0.23 (EtO Ac : Hexanes = 33 :66%). Melting point = 88.1-90.3 °C. [a]D
+23.2 (c 0.07, CHC13). IR (NaCl) υ 3136, 2953, 2932, 2888, 1471, 1428, 1384, 1335, 1284, 1255, 1208, 1111, 1052, 1024, 984, 776, 735, 707 cm"1. 1H NMR (CDC13, 400 MHz): δ -0.17 (s, 3H), -
0.07 (s, 3H), 0.73 (s, 9H), 1.09 (s, 9H), 1.32 (s, 3H), 1.57 (s, 3H), 2.01-2.11 (m, 1H), 2.12-2.20 (m, 1H), 2.30 (s, 1H), 2.33 (s, 1H), 3.56 (s, 1H), 3.65 (dd, Jl = 8.6 Hz, J2 = 3.6 Hz, 1H), 3.73 (d, J= 11 Hz, 1H), 3.92 (d, J= 11 Hz, 1H), 4.99 (d, J= 11 Hz, 1H), 4.99 (d, J= 5.1 Hz, 1H), 5.64-5.74 (m, 1H), 6.19 (d, J= 5.1 Hz, 1H), 7.01 (s, 1H), 7.35-7.47 (m, 6H), 7.59 (s, 1H), 7.64-7.72 (m, 4H), 8.34 (s, 1H). 13C MR (CDC13, 100 MHz): (5 -4.7, -4.4, 18.2, 19.2, 25.9, 26.6, 27.1, 34.5, 34.9, 68.1, 68.6, 77.4, 78.5, 80.4, 89.8, 90.8, 104.3, 114.2, 118.0, 128.2, 130.2, 130.4, 130.8, 131.9, 132.0, 135.7, 135.8, 137.1, 183.5. HRMS (ESIMS): calcd for C38H54N2O7SS12: [M+H]+, 739.32630;
found: 739.32742.
To a solution of carbothioate from above (2.26 g, 3.1 mmol) in anhydrous toluene (175 mL) under reflux, was added dropwise a suspension of BU3S11H (6.49 mL, 24.5 mmol) followed AIBN (50 mg, 0.31 mmol) in toluene (10 mL) via syringe. The reaction mixture was left stirring for an additional 15 min and allowed to warm to rt. Toluene was then removed under reduced pressure with a water bath heated at approximately 70 °C. The resulting residue was purified by flash column chromatography on silica gel with a solvent system of 5:95% , then gradually increase to 10:90% ethyl acetate-hexanes to afford Compound 58 (1.65 g, 87%) as a colorless oil which in turn slowly solidified at rt.
Compound 58: Rf : 0.66 (EtOAc:hexanes = 15:85%). [a]D +36.8.2 (c 0.1, CHC13). IR (NaCl) u 3436, 2932, 2857, 1471, 1428, 1382, 1253, 1208, 1162, 1111, 1080, 1055, 915, 836, 775, 702 cm"1. 1H MR (CDCI3, 400 MHz): δ -0.26 (s, 3H), -0.11 (s, 3H), 0.66 (s, 9H), 1.09 (s, 9H), 1.29 (s, 3H), 1.44-1.58 (m, 5H), 1.55 (s, 3H), 1.66-1.77 (m, 2H), 1.93-2.03 (m, 1H), 3.00 (s, 1H), 3.60 (dd, Jl =10 Hz, J2 = 5.2 Hz, 1H), 3.72 (d, J= 10.9 Hz, 1H), 4.06 (d, J= 10.9 Hz, 1H), 4.90 (d, J= 5.0 Hz, 1H), 6.13 (d, J= 5.0 Hz, 1H), 7.35-7.45 (m, 6H), 7.62-7.70 (m, 4H). 13C NMR (CDC13, 100 MHz): δ -4.9, -3.9, 13.8, 18.0, 18.1, 19.2, 25.9, 26.2, 26.8, 27.2, 28.4, 29.9, 68.6, 70.7, 80.6, 90.1, 92.0, 104.0, 113.7, 128.1, 130.1, 130.2, 132.2, 132.3, 135.7, 135.8. HRMS (ESIMS): calcd for C34H52Na06Si2: [M+Na]+, 635.31946; found: 635.32141. j) Preparation of Compound 59
To a cool (0 °C) solution of Compound 58 (1.72 g, 2.81 mmol) in anhydrous THF (125 mL) was added TBAF (1.0 M in THF 3.23 mL, 3.23 mmol) dropwise. After stirring for 70 min, the reaction mixture was partitioned between DCM (30 mL) and H20 (40 mL). The organic phase was collected and the aqueous portion was back-extracted with DCM (2x30 mL). The combined organic phase was washed with brine (50 mL), dried over MgS04, filtered and concentrated under reduced
pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 35:65% ethyl acetate-hexanes to afford Compound 59 (0.97 g, 92%) as a colorless oil.
Compound 59: Rf : 0.33 (EtOAc:hexanes = 40:60%). [a]D +23.3 (c 0.11, CHC13). IR (NaCl) umax 3456, 2929, 2856, 1462, 1383, 1252, 1209, 1165, 1080, 836, 776 cm"1. 1H MR (CDC13, 400 MHz): δ 0.01 (s, 3H), 0.02 (s, 3H), 0.83 (s, 9H), 1.27 (s, 3H), 1.42-1.52 (m, 2H), 1.50 (s, 3H), 1.60-1.72 (m, 3H), 1.80-1.90 (m, 1H), 3.25-3.35 (m, 1H), 3.73-3.83 (m, 2H), 3.85-3.95 (m, 1H), 4.77 (d, J= 4.7 Hz, 1H), 5.89 (d, J= 4.7 Hz, 1H). 13C MR (CDC13, 100 MHz): (5 -4.7, -4.1, 18.2, 18.5, 26.0, 26.1, 26.8, 28.8, 29.8, 30.7, 65.0, 70.3, 81.1, 89.3, 89.6, 103.4, 114.3. HRMS (ESIMS): calcd for Ci8H34Na06Si: [M+Na]+, 397.20169; found: 397.20102. k) Preparation of Compound 60
To a cool (0 °C ) solution of Compound 59 (0.875 g, 2.34 mmol) in anhydrous THF (16 mL) and DMF (16 mL) was sequentially added NaH (60% in mineral oil, 0.1 g, 2.50 mmol), NapBr (0.568 g, 2.57 mmol) and TBAI (0.432 g, 1.16 mmol). After stirring for 2 h, the reaction was quenched with MeOH (7 mL) and partitioned between DCM (30 mL) and H20 (30 mL). The aqueous portion was back-extracted with DCM (3x30 mL) and the combined organic phase was washed with saturated aqueous solution of NaHC03 (4x40 mL) and brine (40 mL). The organic phase was collected, dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 10:90%) ethyl acetate-hexanes to afford Compound 60 (1.154 g, 96%>) as a colorless oil.
Compound 60: Rf : 0.17 (EtOAc:hexanes = 10:90%). [a]D +15.8 (c 0.05, CHC13). IR (NaCl) umax 3479, 2933, 2856, 1471, 1461, 1382, 1372, 1252, 1208, 1166, 1107, 1068, 1028, 890, 835, 815, 775 cm"1. 1H MR (CDC13, 400 MHz): δ 0.02 (s, 3H), 0.03 (s, 3H), 0.85 (s, 9H), 1.25- 1.30 (m, 2H), 1.30 (s, 3H), 1.55 (s, 3H), 1.65-1.77 (m, 3H), 1.85-1.95 (m, 1H), 3.47 (s, 1H), 3.81 (s, 2H), 3.85 (dd, Jl = 10.0Hz, J2 = 5.0 Hz, 1H), 4.70 (s, 3H), 5.96 (d, J= 4.9 Hz, 1H), 7.39-7.45 (brd, J= 8.3 Hz, 1H), 7.45-7.52 (m, 2H), 7.74 (s, 1H), 7.79-7.87 (m, 3H). 13C NMR (CDC13, 100 MHz): δ -4.6, -3.8, 18.2, 18.3, 26.1, 26.3, 26.8, 28.3, 29.7, 29.9, 70.7, 74.0, 74.4, 80.8, 89.9, 90.1, 113.8, 114.1, 125.7, 126.4, 126.5, 127.2, 127.9, 128.1, 128.8, 133.3, 133.4, 134.5. HRMS (ESIMS): calcd for C29H42Na06Si: [M+Na]+, 538.26717; found: 538.26709.
1) Preparation of Compound 61
To a solution of Compound 60 (0.948 g, 1.84 mmol) in anhydrous THF (80 mL) was added TBAF (1.0 M in THF, 5.53 mL, 5.53 mmol). After stirring at rt for 24 h, the reaction mixture was partitioned between DCM (80 mL) and H20 (100 mL). The organic phase was collected and the aqueous portion was back-extracted with DCM (2x50 mL). The combined organic phase was washed with brine (80 mL), dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 40:60% ethyl acetate-hexanes to afford Compound 61 (0.700 g, 95%) as a light yellow solid.
Compound 61 : Rf : 0.27 (EtOAc:hexanes = 40:60%). Melting point = 101.3-102.0 °C. [<x]D +32.8 (c 0.16, CHC13). IR (NaCl) umax 3460, 3055, 2977, 2936, 2969, 1509, 1455, 1375, 1264, 1209, 1166, 1069, 1026, 903, 858, 818, 733 cm"1. 1H MR (CDC13, 400 MHz): δ 1.29 (s, 3H ), 1.44-1.62 (m, 2H), 1.57 (s, 3H), 1.64-1.84 (m, 3H), 2.06-2.16 (m, 1H), 2.63 (d, J= 5.1 Hz, 1H), 3.52 (s, 1H), 3.70-3.77 (m, 1H), 3.75 (s, 2H), 4.62 (d, J= 4.5 Hz, 1H), 4.70 (s, 2H), 5.96 (d, J= 4.5 Hz, 1H), 7.41 (dd, Jl = 8.4 Hz, J2 = 1.5 Hz, 1H), 7.45-7.50 (m, 2H), 7.73 (s, 1H), 7.80-7.85 (m, 3H). 13C MR (CDC13, 100 MHz): δ 16.9, 26.1, 26.3, 28.0, 31.3, 70.0, 73.9, 74.3, 80.5, 89.9, 90.5, 104.2, 114.0, 125.6, 126.4, 126.5, 127.0, 127.9, 128.1, 128.7, 133.26, 133.33, 134.5. HRMS (ESIMS): calcd for C23H28Na06: [M+Na]+, 423.17781; found: 423.17703 m) Preparation of Compound 62
To a cool (0 °C) solution of Compound 61 (0.652 g, 1.63 mmol) in anhydrous THF (25 mL) and DMF (25 mL) was sequentially added NaH (60% in mineral oil, 0.25 g, 6.25 mmol), BnBr (0.775 g, 4.53 mmol) and TBAI (0.602g, 1.63 mmol). The reaction mixture was allowed to warm up to rt and quenched with MeOH (10 mL) after 2.5 h. The solution was partitioned between DCM (40 mL) and H20 (40 mL). The organic phase was collected and the aqueous portion was back- extracted with DCM (3x50 mL). The combined organic phase was washed with saturated aqueous solution of NaHC03 (4x45 mL), brine (50 mL), dried over MgS04 and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 10:90%> ethyl acetate-hexanes to afford Compound 62 (0.861 g, 91%>) as a colorless oil.
Compound 62: Rf . 0.70 (EtOAc:hexanes = 1 : 1). [a]D +120.0 (c 0.035, CHC13). IR (NaCl) umax 3029, 2936, 2865, 1603, 1508, 1454, 1382, 1374, 1310, 1253, 1208, 1163, 1068, 902, 818, 736, 697 cm"1. 1H MR (CDC13, 400 MHz): δ 1.29 (s, 3H), 1.32-1.42 (m, 1H), 1.56 (s, 3H), 1.63-1.78 (m, 3H), 1.89-1.97 (m, 1H), 2.02-2.10 (m, 1H), 3.35 (dd, Jl = 11.3 Hz, J2 = 5.0 Hz, 1H), 3.75 (d, J
= 9.9 Hz, 1H), 4.02 (d, J= 9.9 Hz, 1H), 4.42 (d, J= 12.0 Hz, 1H), 4.48 (d, J= 11.7 Hz, 1H), 4.53 (d, J= 11.7 Hz, 1H), 4.61 (d, J= 7.8 Hz, 1H), 4.63 (d, J= 7.8 Hz, 1H), 4.70 (d, J= 12.0 Hz, 1H), 5.17 (d, J= 5.4 Hz, 1H), 6.14 (d, J= 5.4 Hz, 1H), 7.17-7.32 (m, 10H), 7.40 (dd, Jl = 8.5 Hz, J2 = 1.2 Hz, 1H), 7.43-7.48 (m, 2H), 7.70 (s, 1H), 7.72-7.70 (m, 2H), 7.79-7.83 (m, 1H). 13C NMR (CDC13, 100 MHz): δ 18.9, 25.8, 26.1, 27.0, 66.8, 71.3, 73.2, 74.1, 85.4, 85.5, 91.7, 105.1, 114.0, 125.7, 126.0, 126.2, 126.3, 127.2, 127.5, 127.7, 127.9, 128.1, 128.2, 128.3, 128.4, 133.1, 133.5, 136.2, 138.6, 139.2. HRMS (ESIMS): calcd for C37H4oNa06: [M+Na]+, 603.27171; found: 603.27142. n) Preparation of Compound 63
To a cool (0 °C) solution of Compound 62 (0.901 g, 1.55 mmol) in anhydrous DCM (80 mL) was added FeCl3-6H20 (0.419 g, 1.55 mmol). The reaction mixture was allowed to warm to rt and after 1 hr, a saturated aqueous solution of NaHC03 (50 mL) was added. The organic phase was collected and the aqueous portion was back-extracted with DCM (3x50 mL). The combined organic phase was dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 35:75% ethyl acetate-hexanes to afford a diol intermediate which was used for acetylation after being dried under high vacuum for 1 h.
To a cool (0 °C) solution of the diol from above in anhydrous DCM (75 mL) was added pyridine (0.025 mL, 0.3 mmol) and DMAP (19 mg, 0.15 mmol) followed by a dropwise addition of Ac20 (0.585 mL, 6.2 mmol). After 1 h when the reaction mixture warmed up to rt, a solution of aqueous HCl (1.0 M, 8 mL) and water (60 mL) were added. The organic phase was collected and the aqueous portion was back-extracted with DCM (3x60 mL). The combined organic phase was washed with a solution of saturated aqueous NaHC03 (50 mL), brine (50 mL), dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 20:80% ethyl acetate-hexanes to afford Compound 63 (0.698 g, 72%) as an oil.
Compound 63 : Rf : 0.27 (EtOAc:hexanes = 35:65%). [a]D +25.3 (c 0.075, CHC13). IR (NaCl) umax 3088, 3061, 3029, 2939, 2867, 1748, 1604, 1497, 1454, 1371, 1242, 1220, 1091, 1064, 1010, 962, 888, 818, 752, 698 cm"1. 1H NMR (CDC13, 400 MHz): δ 1.41-1.53 (m, 1H), 1.68-1.78 (m, 2H), 1.92-2.02 (m, 2H), 2.10 (s, 3H), 2.11 (s, 3H), 2.26-2.34 (m. 1H), 3.37 (dd, Jl = 10.8 Hz, J2 = 4.4 Hz, 1H), 3.79 (d, J= 9.8 Hz, 1H), 3.99 (d, J= 9.8 Hz, 1H), 4.42 (d, J= 11.9 Hz, 1H), 4.49 (d, J= 11.1 Hz, 1H), 4.59 (d, J= 11.1 Hz, 1H), 4.67 (d, J= 11.9 Hz, 1H), 4.72 (d, J= 12.6 Hz, 1H),
4.85 (d, J= 12.6 Hz, 1H), 6.29 (d, J= 6.0 Hz, 1H), 6.79 (d, J= 6.0 Hz, 1H), 7.20-7.32 (m, 10H), 7.45-7.52 (m, 3H), 7.77-7.92 (m, 4H). 13C MR (CDC13, 100 MHz): (5 18.7, 20.7, 21.1, 24.7, 25.5, 66.1, 71.7, 73.8, 76.6, 76.9, 82.5, 86.3, 93.3, 125.5, 125.7, 126.02, 126.04, 127.4, 127.57, 127.63, 127.7, 128.0, 128.2, 128.3, 128.4, 133.0, 133.5, 135.8, 138.2, 138.5, 169.0, 169.4. HRMS (ESIMS): calcd for C38H4oNa08: [M+Na]+, 647.26154; found: 647.26096. o) Preparation of Compound 64
To a suspension of thymine (0.311 g, 2.5 mmol) in 1,2 dichloroethane (DCE, 65 mL) was added BSA (6.05 mL, 25 mmol). The suspension was allowed to go under reflux for 1 h until a clear solution formed. The solution was cooled to 0 °C, then Compound 63 (0.687 g, 0.995 mmol) and TMSOTf (0.447 mL, 2.5 mmol) were added sequentially. After stirring for 13 h at 55 °C, the reaction mixture was allowed to cool to rt and a solution of saturated aqueous NaHC03 (70 mL) was added. The organic phase was collected and the aqueous portion was back-extracted with DCM (2x40 mL). The organic phases were combined, washed with brine (100 mL), dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified flash column
chromatography on silica gel with a solvent system of 50:50% ethyl acetate-hexanes to afford Compound 64 (0.615 g, 81%) as a white solid.
Compound 64: Rf : 0.15 (EtOAc:hexanes = 40:60%). Melting point = 71.3-76.4 °C. [a]D +27.9 (c 0.14, CHC13). IR (NaCl) umax 3186, 3061, 2929, 1747, 1693, 1454, 1372, 1261, 1229, 1128, 1067, 910, 818, 752, 698 cm"1. 1H NMR (CDC13, 400 MHz): δ 1.29 (s, 3H), 1.40-1.50 (m,
1H), 1.71-1.91 (m, 3H), 1.95-2.05 (m, 1H), 2.04 (s, 3H), 2.12-2.21 (m, 1H), 3.64-3.70 (m, 1H), 3.77 (d, J= 10.6 Hz, 1H), 4.04 (d, J= 10.6 Hz, 1H), 4.44 (d, J= 10.8 Hz, 1H), 4.52-4.58 (m, 2H), 4.60- 4.70 (m, 2H), 4.78 (d, J= 12.2 Hz, 1H), 6.13 (d, J= 5.9 Hz, 1H), 6.39 (d, J= 5.9 Hz, 1H), 7.17-7.37 (m, 10H), 7.40-7.50 (m, 3H), 7.75-7.87 (m, 5H). 8.96 (s, 1H). 13C NMR (CDC13, 100 MHz): δ 11.9, 17.9, 20.9, 25.5, 26.8, 65.5, 72.2, 72.5, 73.8, 77.3, 78.8, 83.1, 86.7, 88.4, 111.0, 125.3, 126.1, 126.3, 126.4, 127.6, 127.8, 127.90, 127.93, 128.0, 128.3, 128.4, 128.5, 128.6, 133.1, 133.5, 135.0, 136.8, 137.8, 138.1, 151.2, 163.9, 170.3. HRMS (ESIMS): calcd for C-uHdNfeNaOg: [M+Na]+, 713.28334; found: 713.28465. p) Preparation of Compound 65
To a solution of Compound 64 (0.561 g, 0.812 mmol) in MeOH (80 mL) was added K2C03 After stirring at rt for for 18 h, a solution of saturated NH C1 (25 mL) was added. The reaction
mixture was partitioned between DCM (80 mL) and H20 (35 mL). The organic phase was collected and the aqueous portion was back-extracted with DCM (2x40 mL). The organic phases were combined, dried over MgS04, filtered and concentrated under reduced pressure to provide the crude alcohol (0.542 g, 93%) as a white solid, which was used in the next step without further purification.
Crude alcohol: Rf : 0.26 (EtOAc:hexanes = 40:60%). Melting point = 79.2-83.8 °C. [<x]D
+25.8 (c 0.06, CHC13). IR (NaCl) umax 3407, 3188, 3061, 2928, 2867, 1690, 1466, 1454, 1405, 1368, 1273, 1214, 1073, 922, 857, 819, 753 699 cm"1. 1H MR (CDC13, 400 MHz): δ 1.33 (s, 3H),
I .50-1.60 (m, 2H), 1.75-1.85 (m, 3H), 2.22-2.32 (m, 1H), 3.86 (d, J= 11.4 Hz, 1H), 3.94 (d, J =
I I .4 Hz, 1H), 4.15-4.19 (brd, J= 3.8 Hz, 1H), 4.31 (d, J= 12.0 Hz, 1H), 4.37-4.42 (m, 1H), 4.53 (d, J= 12.0 Hz, 1H), 4.60 (d, J= 12.1 Hz, H), 4.67-4.77 (m, 4H), 6.07 (d, J= 2.0 Hz, 1H), 7.10-7.14
(m, 2H), 7.18-7.25 (m, 3H), 7.29-7.40 (m, 7H), 7.44-7.49 (m, 2H), 7.66 (s, 1H), 7.73-7.78 (m, 2H), 7.79-7.83 (m, 1H), 8.67 (s, 1H). 13C NMR (CDC13, 100 MHz): δ 12.2, 18.1, 25.7, 27.4, 29.9, 71.0, 73.2, 73.9, 81.1, 84.9, 90.7, 93.8, 125.8, 126.3, 126.5, 126.8, 127.5, 127.9, 128.0, 128.4, 128.5, 128.6, 128.9, 133.2, 133.3, 135.2, 136.8, 137.0, 137.8, 151.0, 163.9. HRMS (ESIMS): calcd for C39H4oN2Na07: [M+Na]+, 671.27277; found: 671.27383.
To a solution of crude alcohol from above (0.542 g, 0.835 mmol) in anhydrous pyridine (75 mL) was added DMAP (10 mg, 0.082 mmol). The reaction mixture was cooled to 0 °C, then MsCl (0.193 mL, 2.51 mmol) was added dropwise. The reaction mixture was then allowed warm up to rt and the stirring was continued for 16 h. After that time, the mixture was partitioned between DCM (80 mL) and a solution of saturated aqueous NaHC03 (40 mL). The organic phase was collected and the aqueous portion was back-extracted with DCM (3x60 mL). The organic phases were combined, washed with aqueous HC1 (1.0 M, 2x40 mL), brine (80 mL), dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified flash column chromatography on silica gel with a solvent system of 50:50% ethyl acetate-hexanes to afford the mesylate
Compound 65 (0.569 g, 94%) as a white solid.
Compound 65: Rf . 0.33 (EtOAc:hexanes = 50:50%). Melting point = 83.3-86.1 °C. [a]D +48.1 (c 0.07, CHC13). IR (NaCl) umax 3182, 3030, 2933, 2868, 1692, 1497, 1455, 1352, 1279, 1259, 1176, 1086, 1026, 964, 854, 819, 754, 699 cm"1. 1H NMR (CDC13, 400 MHz): δ 1.23 (d, J = 1.1 Hz, 3H), 1.45-1.55 (m, 1H), 1.59-1.68 (m, 1H), 1.80-1.90 (m, 1H), 1.91-2.06 (m, 2H), 2.13-2.23 (m, 1H), 2.84 (s, 3H), 3.82 (d, J= 11.0 Hz, 1H), 3.92-3.96 (m, 1H), 3.94 (d, J= 11.0 Hz, 1H), 4.51 (d, J= 10.9 Hz, 1H), 4.52 (d, J= 11.2 Hz, 1H), 4.59 (d, J= 11.2 Hz, 1H), 4.61 (d, J= 11.2 Hz, 1H), 4.65 (d, J= 12.1 Hz, 1H), 4.77 (d, J= 12.0 Hz, 1H), 5.53 (d, J= 4.4 Hz, 1H); 6.45 (d, J= 4.5 Hz,
1H), 7.15-7.23 (m, 5H), 7.25-7.28 (m, 1H), 7.29-7.34 (m, 2H), 7.35-7.39 (m, 2H), 7.40-7.42 (m, 1H), 7.45-7.49 (m, 2H), 7.55 (s, 1H), 7.75-7.82 (m, 4H), 8.78 (s, 1H). 13C NMR (CDC13, 100 MHz): δ 12.0, 17.5, 25.3, 27.4, 39.1, 65.1, 71.6, 72.3, 73.9, 77.2, 83.5, 85.2, 87.5, 89.4, 111.6, 125.6, 126.3, 126.5, 126.7, 127.7, 127.9, 128.0, 128.08, 128, 12, 128.56, 128.58, 128.6, 128.7, 133.2, 133.5, 135.0, 136.2, 137,4, 138.2, 151.0, 163.6. HRMS (ESIMS): calcd for C4oH42N2Na09S: [M+Na]+, 749.25032; found: 749.25213. q) Preparation of Compound 66
To a solution of Compound 65 (0.569 g, 0.783 mmol) in MeCN (45 mL) was added DBU (0.435 mL, 3.13 mmol). The reaction mixture was heated at 80 °C for 75 h and then allowed to cool to 0 °C. The mixture was partitioned between DCM (45 mL) and aqueous HC1 (0.6 M, 40 mL).
The organic phase was collected and the aqueous portion was back-extracted with DCM (2x25 mL).
The organic phases were combined, washed with brine, dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified flash column chromatography on silica gel with a solvent system of 3 :97% ethanol-ethyl acetate to afford the desired Compound 66 (0.35 g, 71%) as a white solid.
Compound 66: Rf : 0.35 (EtOH:EtOAc = 7:93%). Melting point = 79.3-83.6 °C. [<x]D -20.7 (c 0.06, CHC13). IR (NaCl) umax 3029, 2926, 2867, 1667, 1647, 1559, 1479, 1380, 1290, 1259, 1124, 1086, 1039, 883, 820, 796, 783, 752 cm"1. 1H NMR (CDC13, 400 MHz): δ 1.30-1.40 (m, 1H), 1.55-1.65 (m, 1H), 1.65-1.77 (m, 2H), 1.84-1.93 (m, 1H), 1.91 (s, 3H), 2.26-2.35 (m, 1H), 3.64 (d, J = 11.0 Hz, 1H), 3.82 (d, 7= 11.0 Hz, 1H), 4.15 (d, 7= 3.0 Hz, 1H), 4.49 (d, 7= 11.2 Hz, 1H), 4.53 (d, 7= 12.1 Hz, 1H), 4.58 (d, 7= 11.2 Hz, 1H), 4.59 (d, 7= 11.8 Hz, 1H), 4.68 (d, 7= 11.7 Hz, 1H), 4.75 (d, 7= 12.1 Hz, 1H), 5.33 (d, 7= 6.2 Hz, 1H), 5.93 (d, 7= 6.2 Hz, 1H), 7.05-7.12 (m, 2H), 7.13-7.20 (m, 4H), 7.25-7.37 (m, 5H), 7.43-7.52 (m, 2H), 7.67 (s, 1H), 7.75-7.85 (m, 3H). 13C NMR (CDC13, 100 MHz): δ 14.3, 16.2, 25.5, 28.1, 29.9, 67.1, 70.4, 72.6, 74.1, 83.6, 85.0, 89.5, 91.0, 118.8, 126.0, 126.2, 126.4, 127.0, 127.59, 127.63, 127.8, 127.9, 128.1, 128.2, 128.4, 128.8, 130.5, 133.2, 133.4, 135.2, 138.0, 138.6, 159.8, 172.5. HRMS (ESIMS): calcd for C39H38N2Na06: [M+Na]+, 653.26221; found: 653.26267. r) Preparation of Compound 67
To a solution of Compound 66 (0.286 g, 0.461 mmol) in DCM (25 mL) and H20 (2.75
mL) was added DDQ (0.315 g, 1.39 mmol). After stirring at rt for 2.5 h, a solution of saturated aqueous NaHC03 (100 mL) was added. The organic phase was collected and the aqueous portion was back-extracted with DCM (2x100 mL). The organic phases were combined, dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 7:93% ethanol-ethyl acetate to afford the alcohol (0.220 g, 96%) as a white solid.
Alcohol: Rf : 0.43 (EtOH:EtOAc = 7:93%). Melting point. = 105.3-106.9 oC. [<x]D -14.3 (c 0.06, CHC13). IR (NaCl) umax 3536, 3335, 3065, 3031, 2947, 2870, 1666, 1646, 1558, 1482, 1381, 1259, 1218, 1130, 1070, 1040, 910, 797, 784, 734, 698 cm"1. 1H MR (CDC13, 400 MHz): δ 1.37- 1.65 (m, 4H), 1.68-1.78 (m. 1H), 1.77 (s, 3H), 1.94-2.04 (m, 1H), 2.65 (dd, Jl = 8.6 Hz, J2 = 2.8 Hz, 1H), 3.58 (dd, Jl = 11.8 Hz, J2 = 2.9 Hz, 1H), 3.65-3.75 (m, 1H), 3.91 (dd, Jl = 8.4 Hz, J2 = 2.8 Hz, 1H), 4.40 (d, J= 11.4 Hz, 1H), 4.44 (d, J= 11.3 Hz, 1H), 4.49 (d, J= 10.4 Hz, 1H), 4.52 (d, J = 11.1 Hz, 1H), 5.08 (d, J= 5.7 Hz, 1H), 5.65 (d, J= 5.7 Hz, 1H), 7.10-7.24 (m, 10H). 13C MR (CDC13, 100 MHz): δ 14.0, 17.7, 25.4, 28.6, 63.1, 67.4, 72.6, 77.0, 84.3, 85.9, 88.2, 89.3, 118.5, 127.8, 128.0, 128.1, 128.5, 128.6, 131.0, 137.1, 137.7, 159.5, 172.4. HRMS (ESIMS): calcd for C28H3oN2Na06: [M+Na]+, 513.19961; found: 513.19968.
To a solution of alcohol from above (0.221 g, 0.451 mmol) in pyridine (35 mL), DMAP (5.5 mg, 0.045 mmol) was added. The solution was cooled to 0 °C, then MsCl (0.088 ml, 1.14 mmol) was added dropwise. After 12 h of stirring, the reaction mixture warmed up to rt and was partitioned between DCM (150 mL) and saturated aqueous solution of NaHC03 (150 mL). The organic phase was collected and the aqueous portion was back-extracted with DCM (2x100 mL). The organic phases were combined, washed with 1.0 M aqueous HC1 (100 mL), brine (150 mL), dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 7:93% ethanol-ethyl acetate to afford the desire mesylate Compound 67 (0.226 g, 93%) as a white solid.
Compound 67: Rf : 0.28 (EtOH:EtOAc = 10:90%). Melting point = 98.7.3-101.5 °C. [a]D +29.5 (c 0.064, CHC13). IR (NaCl) umax 2947, 1667, 1646, 1561, 1480, 1357, 1291, 1259, 1217, 1176, 1130, 1072, 1039, 964, 878, 829, 783, 753, 699 cm"1. 1H MR (CDC13, 400 MHz): δ 1.45- 1.60 (m, 2H), 1.63-1.70 (m, 1H), 1.73-1.85 (m, 2H), 1.83 (s, 3H), 2.25-2.10 (m, 1H), 2.68 (s, 3H), 3.84-3.88 (m, 1H), 4.18 (d, 7= 11.1 Hz, 1H), 4.37 (d, J= 11.1 Hz, 1H), 4.42 (d, J= 11.1 Hz, 1H), 4.51 (d, J= 11.2 Hz, 1H), 4.54 (s, 2H), 5.23 (d, J= 5.2 Hz, 1H), 5.89 (d, J= 5.9 Hz, 1H), 7.08-7.28 (m, 11H). 13C NMR (CDC13, 100 MHz): δ 14.1, 16.3, 25.1, 28.0, 37.0, 67.3, 69.1, 72.6, 75.9, 84.2,
84.4, 88.3, 89.6, 118.8, 127.8, 127.9, 128.0, 128.1, 128.5, 128.6, 130.5, 137.4, 137.5, 159.6, 172.4. HRMS (ESIMS): calcd for C29H32N2Na08S: [M+Na]+, 591.17716; found: 591.17676. s) Preparation of Compound 68
To a suspension of Compound 67 (0.220 g, 0.387 mmol) in anhydrous ethanol (25 mL) and
H20 (25 mL) was added an aqueous solution of 2 M NaOH (1.9 mL). The reaction mixture was heated to 100 °C and allowed to cool to rt after 35 min. DCM (100 mL) was added and the organic phase was collected. The aqueous portion was back-extracted with DCM (2x100 mL). The organic phases were combined, washed with saturated aqueous NaHC03 (100 mL), dried over MgS04, filtered and concentrated in vacuo. The residue was purified by flash column
chromatography on silica gel with a solvent system of 5:95% ethanol-ethyl acetate to afford
Compound 68 (0.181 g, 96%) as a white solid.
Compound 68: Rf : 0.92 (EtOH:EtO Ac = 5:95%). Melting point = 85.2.3-99.7 °C. [a]D +121.4 (c 0.1, CHC13). IR (NaCl) umax 3194, 3031, 2951, 2867, 1689, 1454, 1353, 1283, 1271, 1176, 1057, 949, 891, 851, 750, 698 cm"1. 1H MR (CDC13, 400 MHz): δ 1.50-1.67 (m, 3H), 1.72- 1.79 (m, 1H), 1.87 (s, 3H), 1.91-1.97 (m, 1H), 2.07 (brd, J= 11.6 Hz, 1H), 3.60-3.70 (m, 1H), 4.04 (d, J= 8.1 Hz, 1H), 4.09 (d, J= 8.0 Hz, 1H), 4.35-4.45 (m, 3H), 4.59 (d, J= 12.1 Hz, 1H), 4.65 (s, 1H), 5.94 (s, 1H), 7.15-7.30 (m, 10H), 7.47 (s, 1H), 9.50 (s, 1H). 13C NMR (CDC13, 100 MHz): δ 12.8, 20.4, 23.3, 26.1, 67.5, 71.5, 74.4, 74.8, 77.3, 86.3, 89.1, 89.7, 109.7, 127.68, 127.72, 127.9, 128.0, 128.5, 128.6, 135.8, 137.8, 138.2, 150.8, 164.2. HRMS (ESIMS): calcd for C28H3oN2Na06: [M+Na]+, 513.19961; found: 513.19931. t) Preparation of Compound 69
To a solution of Compound 68 (0.184 g, 0.375 mmol) in a 1 : 1 mixture of EtOH (15 mL) and MeOH (15 mL) was added Pd(OH)2 on carbon. A hydrogen filled ballon was placed over the reaction and allowed to stir for 15 h at rt. After that, the reaction mixture was filtered and the filtrate was evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel with a solvent system of 5:95% ethanol-ethyl acetate to afford Compound 69 ((0.114 g, 98%)) as a white solid.
Compound 69: Rf : 0.22 (EtOH:EtO Ac = 5:95%). Melting point = 209.2-.210 °C. [a]D
+158.3 (c 0.06, CHC13). IR (NaCl) umax 3391, 2948, 1691, 1660, 1472, 1356, 1275, 1199, 1105, 1041, 1004, 952, 941, 894, 841, 785 cm"1. 1H NMR (CD3OD, 400 MHz): δ 1.73-1.87 (m, 6H), 1.92
(, d, J= 1.1 Hz, 3H), 3.89 (dd, J= 10.7 Hz, J2 = 4.8 Hz, 1H), 4.09 (s, 2H), 4.28 (d, 7= 1.1 Hz, 1H), 5.91 (d, 7= 1.0 Hz, 1H), 7.98 (d, 7= 1.0 Hz, 1H). ). 13C NMR (CD3OD, 100 MHz): δ 12.6, 21.7,
28.8, 30.7, 78.4, 75.0, 81.9, 82.1, 90.1, 90.8, 110.0, 138.4, 152.3, 166.8. HRMS (ESIMS): calcd for Ci4Hi8N2Na06: [M+Na]+, 333.10571; found: 333.10613. u) Preparation of Compound 70
To a solution of Compound 69 (0.070 g, 0.226 mmol) and levulinic acid (28 μΐ., 0.27 mmol) in acetonitrile at rt (11.3 mL) was added Mukaiyama's reagent (0.075 g, 0.29 mmol). The solution was stirred under an argon atmosphere for 15 min before triethylamine (94 μΐ., 0.68 mmol) and DMAP(0.008 g, 0.07 mmol) were added. After stirring for approximate 16 h upon which the starting material was completely consumed as indicated by TLC, the reaction mixture was poured into a cooled, aqueous solution of 1 M HC1 (20 mL) and then partitioned with dichloro methane (15 mL). The organic phase was collected and the aqueous portion was back-extracted with DCM (3 χ 10 mL). The organic phases were combined, washed with a saturated aqueous solution of sodium bicarbonate (15 mL), dried through a phase separator cartridge and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel with a solvent system of 1 :20 methanol-dichloromethane to afford the desired Compound 70 (0.070 g, 76% yield) as a colorless solid. The stereochemistry was confirmed by X-ray crystallography.
Compound 70: Rf : 0.24 (methanol :dichloromethane = 1 : 15). Melting point = 238-239 °C (dichloromethane). [a]D 2° +34.0 (c = 0.25, chloroform). IR (KBr disc) v 3382, 2949, 1738, 1704, 1653, 1469, 1384, 1279, 1183, 1103, 1036, 1025, 941, 889 cm"1. 1H NMR (400 MHz, CD2C12): δ 8.32 (s, 1H), 7.80 (s, 1H), 5.99 (s, 1H), 5.33 (s, 1H), 5.06 (dd, 7= 8.0, 7.1, 1H), 4.29 (s, 1H), 4.01 (q, 7= 9.8, 2H), 2.86 (ddd, 7= 18.7, 9.1, 4.4, 1H), 2.75 (ddd, 7= 18.6, 6.4, 4.1, 1H), 2.57 (ddd, 7 =
16.9, 9.1, 4.1, 1H), 2.45 (ddd, 7= 17.0, 6.6, 4.4, 1H), 2.16 (s, 3H), 1.98 (s, 3H), 1.96-1.78 (m, 6H). 13C NMR (75 MHz, CD2C12): 5 207.6, 172.9, 164.1, 150.7, 136.6, 110.1, 89.3, 88.3, 82.4, 80.2,
74.3, 70.0, 38.8, 29.9, 28.7, 27.9, 26.6, 20.9, 12.5. HRMS (ESI): calc'd for Ci9H25N208 [M+H]+ m/z 409.1605, found 409.1596. v) Preparation of phosphoramidite, Compound 70a
To a solution of tricyclic nucleoside, Compound 70 (64 mg, 0.15 mmol), DIPEA (0.1 mL,
0.4 mmol) and N-methylimidazole (0.07 mL, 0.4 mmol) in dichloromethane (1.5 mL), was added 2- cyanoethoxy-(N,N-diisopropylamino)-chlorophosphine (0.1 mL, 0.4 mmol). After stirring for 2 h at
rt, additional phosphine reagent (0.03 mL) and DIEPA (0.04 mL) were added. The reaction was allowed to stir for another 10 min and then quenched with DIPEA (0.1 mL) and methanol (0.8 mL). The volatile solvents were removed under reduced pressure and the residue was purified by flash column chromatography on silica gel with a solvent system of 20 to 30% acetone in
dichloromethane to afford the phosphoramidite, Compound 70a. The analytical data was consistent with the desired structure.
Example 20
Alternative method for the preparation of Compound 69

Compound 58 was prepared as per the procedures illustrated in Example 19. Analytical data of Compound 76 was consistent with the structure.
The transformation of the anhydronucleoside, Compound 76 to tricylic nucleoside,
Compound 69 is then carried out by desilylation, mesylation followed by basic hydrolysis and ring closing in the presence of NaOH.
Example 21
Preparation of Compounds 79-84
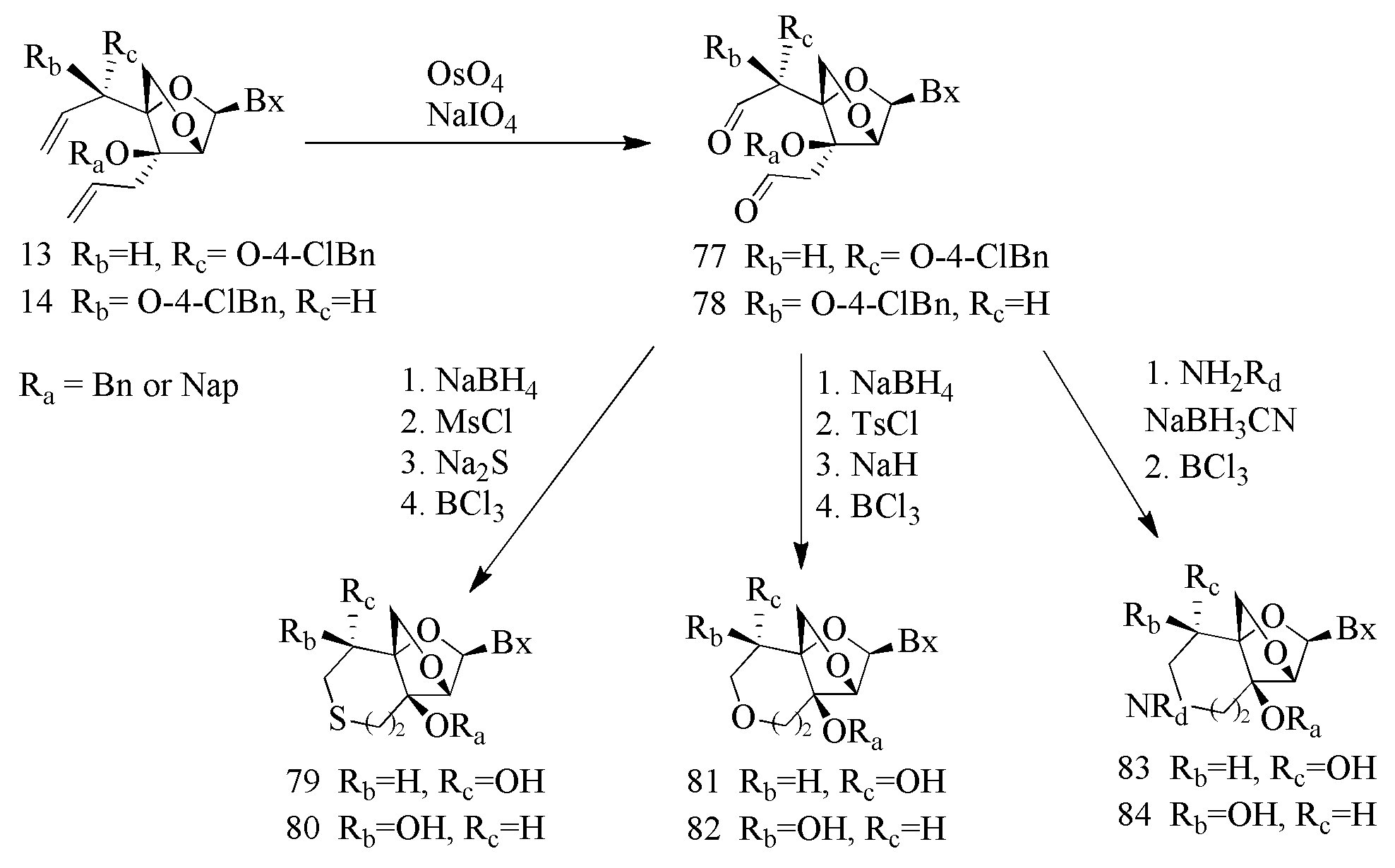
Rd = H, Ci-C6 alkyl, substituted C1-C5 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl, substituted acyl, aryl, substituted aryl, a heterocyclic radical, a substituted heterocyclic radical or a protecting group.
Compounds 13 and 14 are prepared as per the procedures illustrated in Example 14.
Example 22
Preparation of Compounds 121-126

Compounds 79-84 are prepared as per the procedures illustrated in Example 21. In the tritylation step, other hydroxyl protecting groups can be used in place of DMTC1 (e.g. levulinic acid).
Example 23
General method for the preparation of oligomeric compounds comprising locked nucleic acid (LNA), a-L-LNA or a-L-tricyclic nucleic acid (oc-L-TriNA) modification
The synthesis of oligomeric compounds presented in Table 1 was performed on a 1 μιηοΐ scale using the UnyLinker™ polystyrene support on an ABI 394 DNA/RNA synthesizer. Standard conditions were used to incorporate phosphoramidite building blocks of the unmodified nucleosides which include for example T, G, A and C residues. Solvents and reagents used during synthesis were prepared according to the indications of the manufacturer. A 0. 5 M solution of 5-ethylthio-l- H-tetrazole (ETT) in acetonitrile was used as an activator and a 0.5 M solution of hydrazine in pyridine: acetic acid (5 mL, 3 :2 v/v) was used as a deblocking reagent. For DNA building blocks, a 0.1 M solution of unmodified phosphoramidite in acetonitrile was used with the coupling time of 2 x 3 min. A I M solution of DCI and 0.1 M NMI in acetonitrile was used as a coupling activator. For deblocking, a solution of dichloroacetic acid (3%) in dichloromethane was used. For capping, a solution of acetic acid in THF (Cap A) and 1-methylimidazole (10%) in tetrahydrofuran/pyridine (Cap B) were used. For oxidation, a 0.02 M solution of iodine in tetrahydrofuran/pyridine/water was used.
For the incorporation of LNA or a-L-LNA phosphoramidite building block, similar protocol as described above was carried out except the coupling time was extended to 2 x 6 min.
For the the incorporation of a-L-TriNA phosphoramidite building block, the coupling was carried out manually using two syringes by dissolving the tricyclic phosphoramidite (33 μπιοΐ) in dichloromethane (400 μί), mixing with 0. 5 M solution of ETT in acetonitrile (600 μί) and contacting with the solid support for 30 min. The coupling time was extended to 30 min with the coupling efficiency of approximately 90%. Following capping and oxidation on the machine, deblocking was carried out manually over 10 min using two syringes containing a freshly prepared 0.5 M solution of hydrazine in pyridine: acetic acid (5 mL, 3 :2 v/v) (see Romieu et al, J. Chem. Soc.
Perkin Trans. 1, 1999, 1257). After construction of full length product was completed, the final DNA was deblocked and the cyanoethyl protecting groups were removed using
triethylamine:acetonitrile (1 : 1, v/v). The remaining protecting groups were removed using concentrated aqueous ammonia at 55 °C for 8 h. The crude samples were purified by IE-HPLC using a linear gradient of buffer A (50 mM NaHC03 in acetonitrile:water 3 :7) and B (1.5 M NaBr, 50 mM NaHC03 in acetonitrile:water 3 :7). The purified oligomeric compounds were desalted using C18 reverse phase cartridges. The integrity of the oligonucleotides was confirmed and analyzed by ESI-mass spectrometry.
The compositions of the oligonucleotides are described in Table 1. The internucleoside linkages throughout each oligomeric compound are phosphodiester linkages (P=0). Nucleosides without a subscript are -D-2'-deoxyribonucleosides. Nucleosides followed by a subscript "1", "a" or "b" are defined below.


Example 24
Thermal stability (Tm) measurements
The oligomeric compounds in Table 1 were evaluated for thermal stability (Tm) using the method described herein. The Tm of the modified 12mer oligomeric compounds were compared to an unmodified 12mer DNA oligonucleotide when duplexed to DNA or RNA complement.
Tm's were determined using a Cary 100 Bio spectrophotometer with the Cary Win UV thermal program was used to measure absorbance vs. temperature. For the Tm experiments, the oligomeric compounds were prepared at a concentration of 8 μΜ in 10 mM sodium phosphate buffer (pH 7) containing 100 mM NaCl and 0.1 mM EDTA. The concentrations of each oligomeric compound was determined at 85 °C in Tm buffer. The oligomeric compounds were hybridized with a complimentaryDNA or RNA by heating the duplex to 90 °C for 5 minutes and then cooling to room temperature. Tm measurements were taken using a spectrophotometer while the duplex solution was heated in a cuvette at a rate of 0.5 °C/min starting at 15 °C until the temperature was 85 °C. Tm values were determined using Vant Hoff calculations (A260 vs temperature curve) using non self-complementary sequences where the minimum absorbance related to the duplex and the maximum absorbance related to the non-duplex single strand are manually integrated into the program. Thermal stability of the oligomeric compounds versus the mismatched RNA complement was also measured and the results are presented in Table 2.
The sequence for matched DNA complement, ISIS 483795 is designated herein as SEQ ID No.: 6, 5'-d(AGCAAAXAACGC)-3', wherein X = A. The sequence for matched RNA
complement, ISIS 438704 is designated herein as SEQ ID No. : 6, 5'-r(AGCAAAYAACGC)-3', wherein Y = A. The sequence for mismatched RNA complements is designated herein as the following: ISIS 447578, SEQ ID No. : 7, 5'-r(AGCAAAZAACGC)-3', wherein Z= G; ISIS 447580, SEQ ID No. : 8, wherein Z = U; and ISIS 447579, SEQ ID No. : 9, wherein Z = C.

Additional Tm measurements
The oligomeric compound, ISIS 583517 was prepared in same manner as described previously and was evaluated for thermal stability in a different Tm sequence than the one described above. The Tm measurement was performed using similar protocol as illustrated in Example 24. The Tm of the modified 12mer oligomeric compounds were compared to an unmodified 12mer DNA oligonucleotide when duplexed to DNA or RNA complement. The results are presented in Table 3.
The sequence for DNA complement, ISIS A01 is designated herein as SEQ ID No. : 10, 5'- d(GC AT ATC ACTGG)-3 ' . The sequence for RNA complement, ISIS A02 is designated herein as SEQ ID No. : 11, 5'-r(GCAUAUCACUGG)-3'.
The compositions of the oligonucleotides are described in Table 3. The internucleoside linkages throughout each oligomeric compound are phosphodiester linkages (P=0). Nucleosides without a subscript are -D-2'-deoxyribonucleosides. Nucleosides followed by a subscript "b" or "1" is defined in the same manner as illustrated in Example 23.

Claims
1. A tricyclic nucleoside having Formula I:

wherein:
Bx is a heterocyclic base moiety;
one of Ti and T2 is hydroxyl or a protected hydroxyl and the other of Ti and T2 is H;
T3 is hydroxyl, a protected hydroxyl, an H-phosphonate or a phosphoramidite group;
Q is CRiR2CR3R4, CH=CH, O, S or R5;
Ri, R2, R3 and R4 are each, independently, H, hydroxyl, halogen, Ci-C6 alkyl, substituted Ci- C6 alkyl, Ci-C6 alkoxy, substituted Ci-C6 alkoxy, amino or substituted amino;
R5 is H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl, substituted acyl, aryl, substituted aryl, a heterocyclic radical, a substituted heterocyclic radical or a protecting group;
wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, OJi, SJi, NJiJ2, N3, CN, C(=0)OJi, C(=0)NJiJ2, C(=0)Ji, 0-C(=0)NJiJ2, N(H)C(=0)NJiJ2, N(H)C(=S)NJiJ2 and a protecting group;
each Ji and J2 is, independently, H, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C6 aminoalkyl or a protecting group;
when Q is CRiR2CR3R4 or CH=CH then n is 1 and m is 0 or 1; and
when Q is O, S or R5 then m is 1 and n is 1 or 2.
2. The tricyclic nucleoside of claim 1 wherein Bx is an optionally protected pyrimidine, substituted pyrimidine, purine or substituted purine.
3. The tricyclic nucleoside of any one of claims 1 or 2 wherein Bx is an optionally protected uracil, thymine, cytosine, 5-methylcytosine, adenine or guanine.
4. The tricyclic nucleoside of any one of claims 1 to 3 wherein one of Ti and T2 is a 4,4'- dimethoxytrityl protected hydroxyl group.
5. The tricyclic nucleoside of any one of claims 1 to 4 wherein T3 is diisopropylcyanoethoxy phosphoramidite.
6. The tricyclic nucleoside of any one of claims 1 to 5 wherein Q is CH=CH.
7. The tricyclic nucleoside of any one of claims 1 to 5 wherein Q is CRiR2CR3R4.
8. The tricyclic nucleoside of claim 7 wherein at least one of Ri, R2, R3 and R4 is Ci-C6 alkyl.
9. The tricyclic nucleoside of any one of claims 7 or 8 wherein at least one of Ri, R2, R3 and R4 is CH3.
10. The tricyclic nucleoside of any one of claims 7 to 9 wherein at least one of Ri, R2, R3 and R4 is substituted Ci-C6 alkyl.
11. The tricyclic nucleoside of any one of claims 7 to 10 wherein one of Ri and R2 is Ci-C6 alkyl or substituted Ci-C6 alkyl and one of R3 and R4 IS Ci-C6 alkyl or substituted Ci-C6 alkyl.
12. The tricyclic nucleoside of claim 7 wherein Ri, R2, R3 and R4 are each H.
13. The tricyclic nucleoside of any one of claims 1 to 12 wherein m is 0.
14. The tricyclic nucleoside of any one of claims 1 to 12 wherein m is 1.
15. The tricyclic nucleoside of any one of claims 1 to 5 wherein Q is O.
16. The tricyclic nucleoside of any one of claims 1 to 5 wherein Q is S.
17. The tricyclic nucleoside of any one of claims 1 to 5 wherein Q is R5.
18. The tricyclic nucleoside of claim 17 wherein R5 is H, Ci-C6 alkyl or substituted Ci-C6 alkyl.
19. The tricyclic nucleoside of any one of claims 17 or 18 wherein R5 is methyl.
20. The tricyclic nucleoside of any one of claims 15 to 19 wherein n is 1.
21. The tricyclic nucleoside of any one of claims 15 to 19 wherein n is 2.
22. The tricyclic nucleoside of any one of claims 1 to 21 having the configuration of Formula la:

23. The tricyclic nucleoside of claim 22 wherein Ti is 4,4'-dimethoxytrityl protected hydroxyl and T3 is diisopropylcyanoethoxy phosphoramidite.
24. The tricyclic nucleoside of claim 22 wherein T2 is 4,4'-dimethoxytrityl protected hydroxyl and T3 is diisopropylcyanoethoxy phosphoramidite.
25. gomeric compound comprising at least one tricyclic nucleoside having Formula II:

wherein independently for each tricyclic nucleoside of Formula II:
Bx is a heterocyclic base moiety;
one of T4 and T5 is hydroxyl, a protected hydroxyl, a 5' terminal group or an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound and the other of T4 and
T5 is H; T6 is hydroxyl, a protected hydroxyl, a 3' terminal group or an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound;
Q is CR1R2CR3R4, CH=CH, O, S or R5;
Ri, R2, R3 and R4 are each, independently, H, hydroxyl, halogen, Ci-C6 alkyl, substituted Ci- C6 alkyl, Ci-C6 alkoxy, substituted Ci-C6 alkoxy, amino or substituted amino;
R5 is H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl, substituted acyl, aryl, substituted aryl, a heterocyclic radical, a substituted heterocyclic radical or a protecting group;
wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, OJi, SJi, NJiJ2, N3, CN, C(=0)OJi, C(=0)NJiJ2, C(=0)Ji, 0-C(=0)NJiJ2, N(H)C(=0)NJiJ2, N(H)C(=S)NJiJ2 and a protecting group;
each Ji and J2 is, independently, H, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C6 aminoalkyl or a protecting group;
when Q is CRiR2CR3R4 or CH=CH then n is 1 and m is 0 or 1;
when Q is O, S or R5 then m is 1 and n is 1 or 2; and
wherein at least one of T4, T5 and T6 is an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound.
26. The oligomeric compound of claim 25 wherein each Bx is, independently, an optionally protected pyrimidine, substituted pyrimidine, purine or substituted purine.
27. The oligomeric compound of any one of claims 25 or 26 wherein each Bx is, independently, an optionally protected uracil, thymine, cytosine, 5-methylcytosine, adenine or guanine.
28. The oligomeric compound of any one of claims 25 to 27 wherein each Q is CH=CH.
29. The oligomeric compound of any one of claims 25 to 27 wherein each Q is CRiR2CR3R4.
30. The oligomeric compound of claim 29 wherein one of Ri, R2, R3 and R4 is Ci-C6 alkyl for each tricyclic nucleoside having Formula II.
31. The oligomeric compound of any one of claims 29 or 30 wherein one of Ri, R2, R3 and R4 is CH3 for each tricyclic nucleoside having Formula II.
32. The oligomeric compound of any one of claims 29 to 31 wherein one of Ri, R2, R3 and R4 is substituted Ci-C6 alkyl for each tricyclic nucleoside having Formula II.
33. The oligomeric compound of any one of claims 31 to 32 wherein one of Ri and R2 is Ci-C6 alkyl or substituted Ci-C6 alkyl and one of R3 and R4 IS Ci-C6 alkyl or substituted Ci-C6 alkyl for each tricyclic nucleoside having Formula II.
34. The oligomeric compound of claim 29 wherein each Q is CH2CH2.
35. The oligomeric compound of any one of claims 25 to 34 wherein each m is 0.
36. The oligomeric compound of any one of claims 25 to 34 wherein each m is 1.
37. The oligomeric compound of any one of claims 25 to 27 wherein each Q is O.
38. The oligomeric compound of any one of claims 25 to 27 wherein each Q is S.
39. The oligomeric compound of any one of claims 25 to 27 wherein each Q is R5.
40. The oligomeric compound of claim 39 wherein each R5 is H, Ci-C6 alkyl or substituted Ci-C6 alkyl.
41. The oligomeric compound of any one of claims 39 or 40 wherein each R5 is methyl.
42. The oligomeric compound of any one of claims 37 to 41 wherein each n is 1.
43. The oligomeric compound of any one of claims 37 to 41 wherein each n is 2.
44. The oligomeric compound of any one of claims 25 to 43 wherein each tricyclic nucleoside has Formula Il

45. The oligomeric compound of any one of claims 25 to 44 wherein each tricyclic nucleoside is uniformly modified having the same stereochemistry wherein the heterocyclic base is variable.
46. The oligomeric compound of any one of claims 25 to 45 wherein each internucleoside linking group between adjacent monomeric subunits is, independently, a phosphodiester
internucleoside linking group or a phosphorothioate internucleoside linking group.
47. The oligomeric compound of any one of claims 25 to 45 wherein essentially each internucleoside linking group between adjacent monomeric subunits is a phosphorothioate internucleoside linking group.
48. The oligomeric compound of any one of claims 25 to 47 comprising a first region having at least two contiguous tricyclic nucleosides having Formula II or Ila.
49. The oligomeric compound of claim 48 comprising a second region having at least two contiguous monomeric subunits wherein each monomeric subunit in the second region is a modified nucleoside different from the tricyclic nucleosides of said first region.
50. The oligomeric compound of claim 49 further comprising a third region located between said first and second regions wherein each monomer subunit in the third region is independently, a nucleoside or a modified nucleoside that is different from each tricyclic nucleoside of said first region and each monomer subunit the second region.
51. The oligomeric compound of any one of claims 25 to 47 comprising a gapped oligomeric compound having an internal region of from 6 to 14 contiguous monomer subunits flanked on each side by an external region of from 1 to 5 contiguous monomer subunits wherein each monomer subunit in each external region is tricyclic nucleoside of Formula II or Ila and each monomer subunit in the internal region is, independently, a nucleoside or modified nucleoside.
52. The oligomeric compound of claim 51 wherein said internal region comprises from about 8 to about 14 contiguous P-D-2'-deoxyribonucleosides.
53. The oligomeric compound of claim 51 wherein said internal region comprises from about 9 to about 12 contiguous P-D-2'-deoxyribonucleosides.
54. A tricyclic nucleoside having Formula III:

wherein:
Bx is a heterocyclic base moiety;
each Zi, Z2, and Z3 is independently selected from the group consisting of CH2-CH2, CH=CH, CR7R8, O, S, or R9;
each R7 and R8 are indepdendently selected from the group consisting of H, hydroxyl, protected hydroxyl, halogen, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl, Ci-C6 alkoxy, Ci-C6 substituted alkoxy, an alkyl ester, and a protecting group;
R9 is H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl or a protecting group;
wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, OJi, SJi, NJiJ2, N3, CN, C(=0)OJi, C(=0)NJiJ2, C(=0)Ji, 0-C(=0)NJiJ2, N(H)C(=0)NJiJ2, N(H)C(=S)NJiJ2 and a protecting group;
each Ji and J2 is, independently, H, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C6 aminoalkyl or a protecting group; one of T7 and T8 is hydroxyl or a protected hydroxyl and the other of T7 and T8 is H; and T9 is hydroxyl, a protected hydroxyl, an H-phosphonate or a phosphoramidite group.
55. An oligomeric compound comprising at least one tricyclic nucleoside having Formula IV:

wherein independently for each tricyclic nucleoside having Formula II:
Bx is a heterocyclic base moiety;
each Z4, Z5, and Z6 is independently selected from the group consisting of CH2-CH2, CH=CH, CR10R11, O, S, or Ri2;
each Rio and Rn are indepdendently selected from the group consisting of H, hydroxyl, protected hydroxyl, halogen, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl, Ci-C6 alkoxy, Ci-C6 substituted alkoxy, an alkyl ester, and a protecting group;
Ri2 is H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl or a protecting group;
wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, OJi, SJi, NJiJ2, N3, CN, C(=0)OJi, C(=0)NJiJ2, C(=0)Ji, 0-C(=0)NJiJ2, N(H)C(=0)NJiJ2, N(H)C(=S)NJiJ2 and a protecting group;
each Ji and J2 is, independently, H, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C6 aminoalkyl or a protecting group;
one of T10 and Tn is hydroxyl, a protected hydroxyl, a 5' terminal group or an
internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound and the other of T10 and Tn is H;
T12 is hydroxyl, a protected hydroxyl, a 3' terminal group or an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound; and
wherein at least one of T10, Tn and Ti2 is an internucleoside linking group attaching the tricyclic nucleoside to the oligomeric compound.
56. A method of preparing a tricyclic nucleoside, comprising: reacting a first reactant and a second reactant in the presence of a solvent; wherein the first reactant comprises a compound represented by formula V:

and wherein each R13 and Ri4 are indepdendently selected from the group consisting of H, Ci-C6 alkyl, substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl, substituted C2-C6 alkynyl and a protecting group.
57. The method of claim 56, wherein the protecting group is selected from the group consisting of benzyl, phenyl, naphthyl, triflate, trimethylsilyl, tert-butyldiphenylsilyl, tert-butyldimethylsilyl, and benzyl chloromethyl ether.
58. The method of any of claims 56 to 57 wherein the second reactant is an amino acid.
59. The method of any of claims 56 to 58 wherein the second reactant is L-proline.
60. The method of any of claims 56 to 59, wherein the solvent is selected from among a polar solvent, a polar aprotic solvent, a polar protic solvent, an apolar solvent, an apolar aprotic solvent, and an apolar protic solvent.
61. The method of claim 60, wherein the solvent is selected from the group consisting of DMF and DMSO.
62. The method of any of claims 56 to 59, wherein the tricyclic nucleoside comprises a compound having Formula VI: 
63. A pharmaceutical composition comprising the compound of any of claims 1 to 55 and a pharmaceutically acceptable salt, carrier, or diluent.
64. A method of modulating target mRNA in a cell, comprising contacting the cell with an oligomeric compound according to any of claims 1 to 55 or 63.
65. The method of claim 64, wherein the cell is in vitro.
66. The method of claim 65, wherein the cell is in an animal.
67. The method of claim 66, wherein the animal is a human.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261621855P | 2012-04-09 | 2012-04-09 | |
| US61/621,855 | 2012-04-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013154799A1 true WO2013154799A1 (en) | 2013-10-17 |
Family
ID=48045790
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2013/033151 WO2013154799A1 (en) | 2012-04-09 | 2013-03-20 | Tricyclic nucleosides and oligomeric compounds prepared therefrom |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2013154799A1 (en) |
Citations (149)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2699508A (en) | 1951-12-21 | 1955-01-11 | Selectronics Inc | Method of mounting and construction of mounting for low frequency piezoelectric crystals |
| US2699808A (en) | 1944-10-06 | 1955-01-18 | Mark W Lowe | Apparatus for peeling tomatoes |
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4415732A (en) | 1981-03-27 | 1983-11-15 | University Patents, Inc. | Phosphoramidite compounds and processes |
| US4458066A (en) | 1980-02-29 | 1984-07-03 | University Patents, Inc. | Process for preparing polynucleotides |
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US4476301A (en) | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| US4500707A (en) | 1980-02-29 | 1985-02-19 | University Patents, Inc. | Nucleosides useful in the preparation of polynucleotides |
| US4668777A (en) | 1981-03-27 | 1987-05-26 | University Patents, Inc. | Phosphoramidite nucleoside compounds |
| US4725677A (en) | 1983-08-18 | 1988-02-16 | Biosyntech Gmbh | Process for the preparation of oligonucleotides |
| US4845205A (en) | 1985-01-08 | 1989-07-04 | Institut Pasteur | 2,N6 -disubstituted and 2,N6 -trisubstituted adenosine-3'-phosphoramidites |
| US4973679A (en) | 1981-03-27 | 1990-11-27 | University Patents, Inc. | Process for oligonucleo tide synthesis using phosphormidite intermediates |
| US4981957A (en) | 1984-07-19 | 1991-01-01 | Centre National De La Recherche Scientifique | Oligonucleotides with modified phosphate and modified carbohydrate moieties at the respective chain termini |
| US5013830A (en) | 1986-09-08 | 1991-05-07 | Ajinomoto Co., Inc. | Compounds for the cleavage at a specific position of RNA, oligomers employed for the formation of said compounds, and starting materials for the synthesis of said oligomers |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| US5118800A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | Oligonucleotides possessing a primary amino group in the terminal nucleotide |
| US5130302A (en) | 1989-12-20 | 1992-07-14 | Boron Bilogicals, Inc. | Boronated nucleoside, nucleotide and oligonucleotide compounds, compositions and methods for using same |
| US5132418A (en) | 1980-02-29 | 1992-07-21 | University Patents, Inc. | Process for preparing polynucleotides |
| US5134066A (en) | 1989-08-29 | 1992-07-28 | Monsanto Company | Improved probes using nucleosides containing 3-dezauracil analogs |
| USRE34069E (en) | 1983-08-18 | 1992-09-15 | Biosyntech Gmbh | Process for the preparation of oligonucleotides |
| US5149797A (en) | 1990-02-15 | 1992-09-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of rna and production of encoded polypeptides |
| US5166315A (en) | 1989-12-20 | 1992-11-24 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5175273A (en) | 1988-07-01 | 1992-12-29 | Genentech, Inc. | Nucleic acid intercalating agents |
| US5177196A (en) | 1990-08-16 | 1993-01-05 | Microprobe Corporation | Oligo (α-arabinofuranosyl nucleotides) and α-arabinofuranosyl precursors thereof |
| US5177198A (en) | 1989-11-30 | 1993-01-05 | University Of N.C. At Chapel Hill | Process for preparing oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5185444A (en) | 1985-03-15 | 1993-02-09 | Anti-Gene Deveopment Group | Uncharged morpolino-based polymers having phosphorous containing chiral intersubunit linkages |
| US5188897A (en) | 1987-10-22 | 1993-02-23 | Temple University Of The Commonwealth System Of Higher Education | Encapsulated 2',5'-phosphorothioate oligoadenylates |
| US5194599A (en) | 1988-09-23 | 1993-03-16 | Gilead Sciences, Inc. | Hydrogen phosphonodithioate compositions |
| US5214134A (en) | 1990-09-12 | 1993-05-25 | Sterling Winthrop Inc. | Process of linking nucleosides with a siloxane bridge |
| US5216141A (en) | 1988-06-06 | 1993-06-01 | Benner Steven A | Oligonucleotide analogs containing sulfur linkages |
| US5220007A (en) | 1990-02-15 | 1993-06-15 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of RNA and production of encoded polypeptides |
| US5223618A (en) | 1990-08-13 | 1993-06-29 | Isis Pharmaceuticals, Inc. | 4'-desmethyl nucleoside analog compounds |
| US5235033A (en) | 1985-03-15 | 1993-08-10 | Anti-Gene Development Group | Alpha-morpholino ribonucleoside derivatives and polymers thereof |
| US5256775A (en) | 1989-06-05 | 1993-10-26 | Gilead Sciences, Inc. | Exonuclease-resistant oligonucleotides |
| US5264564A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences | Oligonucleotide analogs with novel linkages |
| US5264423A (en) | 1987-03-25 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5264562A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| US5276019A (en) | 1987-03-25 | 1994-01-04 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5278302A (en) | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| WO1994002499A1 (en) | 1992-07-27 | 1994-02-03 | Hybridon, Inc. | Oligonucleotide alkylphosphonothioates |
| US5319080A (en) | 1991-10-17 | 1994-06-07 | Ciba-Geigy Corporation | Bicyclic nucleosides, oligonucleotides, process for their preparation and intermediates |
| US5321131A (en) | 1990-03-08 | 1994-06-14 | Hybridon, Inc. | Site-specific functionalization of oligodeoxynucleotides for non-radioactive labelling |
| WO1994014226A1 (en) | 1992-12-14 | 1994-06-23 | Honeywell Inc. | Motor system with individually controlled redundant windings |
| WO1994017093A1 (en) | 1993-01-25 | 1994-08-04 | Hybridon, Inc. | Oligonucleotide alkylphosphonates and alkylphosphonothioates |
| US5359044A (en) | 1991-12-13 | 1994-10-25 | Isis Pharmaceuticals | Cyclobutyl oligonucleotide surrogates |
| US5367066A (en) | 1984-10-16 | 1994-11-22 | Chiron Corporation | Oligonucleotides with selectably cleavable and/or abasic sites |
| US5378825A (en) | 1990-07-27 | 1995-01-03 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs |
| US5386023A (en) | 1990-07-27 | 1995-01-31 | Isis Pharmaceuticals | Backbone modified oligonucleotide analogs and preparation thereof through reductive coupling |
| US5399676A (en) | 1989-10-23 | 1995-03-21 | Gilead Sciences | Oligonucleotides with inverted polarity |
| US5403711A (en) | 1987-11-30 | 1995-04-04 | University Of Iowa Research Foundation | Nucleic acid hybridization and amplification method for detection of specific sequences in which a complementary labeled nucleic acid probe is cleaved |
| US5405939A (en) | 1987-10-22 | 1995-04-11 | Temple University Of The Commonwealth System Of Higher Education | 2',5'-phosphorothioate oligoadenylates and their covalent conjugates with polylysine |
| US5405938A (en) | 1989-12-20 | 1995-04-11 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5432272A (en) | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| US5434257A (en) | 1992-06-01 | 1995-07-18 | Gilead Sciences, Inc. | Binding compentent oligomers containing unsaturated 3',5' and 2',5' linkages |
| US5446137A (en) | 1993-12-09 | 1995-08-29 | Syntex (U.S.A.) Inc. | Oligonucleotides containing 4'-substituted nucleotides |
| US5457187A (en) | 1993-12-08 | 1995-10-10 | Board Of Regents University Of Nebraska | Oligonucleotides containing 5-fluorouracil |
| US5459255A (en) | 1990-01-11 | 1995-10-17 | Isis Pharmaceuticals, Inc. | N-2 substituted purines |
| US5466677A (en) | 1993-03-06 | 1995-11-14 | Ciba-Geigy Corporation | Dinucleoside phosphinates and their pharmaceutical compositions |
| US5466786A (en) | 1989-10-24 | 1995-11-14 | Gilead Sciences | 2'modified nucleoside and nucleotide compounds |
| US5470967A (en) | 1990-04-10 | 1995-11-28 | The Dupont Merck Pharmaceutical Company | Oligonucleotide analogs with sulfamate linkages |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| US5484908A (en) | 1991-11-26 | 1996-01-16 | Gilead Sciences, Inc. | Oligonucleotides containing 5-propynyl pyrimidines |
| US5489677A (en) | 1990-07-27 | 1996-02-06 | Isis Pharmaceuticals, Inc. | Oligonucleoside linkages containing adjacent oxygen and nitrogen atoms |
| US5491133A (en) | 1987-11-30 | 1996-02-13 | University Of Iowa Research Foundation | Methods for blocking the expression of specifically targeted genes |
| US5502177A (en) | 1993-09-17 | 1996-03-26 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| US5508270A (en) | 1993-03-06 | 1996-04-16 | Ciba-Geigy Corporation | Nucleoside phosphinate compounds and compositions |
| US5514785A (en) | 1990-05-11 | 1996-05-07 | Becton Dickinson And Company | Solid supports for nucleic acid hybridization assays |
| US5519126A (en) | 1988-03-25 | 1996-05-21 | University Of Virginia Alumni Patents Foundation | Oligonucleotide N-alkylphosphoramidates |
| US5519134A (en) | 1994-01-11 | 1996-05-21 | Isis Pharmaceuticals, Inc. | Pyrrolidine-containing monomers and oligomers |
| US5525711A (en) | 1994-05-18 | 1996-06-11 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Pteridine nucleotide analogs as fluorescent DNA probes |
| US5541307A (en) | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| US5550111A (en) | 1984-07-11 | 1996-08-27 | Temple University-Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| US5552540A (en) | 1987-06-24 | 1996-09-03 | Howard Florey Institute Of Experimental Physiology And Medicine | Nucleoside derivatives |
| US5561225A (en) | 1990-09-19 | 1996-10-01 | Southern Research Institute | Polynucleotide analogs containing sulfonate and sulfonamide internucleoside linkages |
| US5565350A (en) | 1993-12-09 | 1996-10-15 | Thomas Jefferson University | Compounds and methods for site directed mutations in eukaryotic cells |
| US5567811A (en) | 1990-05-03 | 1996-10-22 | Amersham International Plc | Phosphoramidite derivatives, their preparation and the use thereof in the incorporation of reporter groups on synthetic oligonucleotides |
| US5571799A (en) | 1991-08-12 | 1996-11-05 | Basco, Ltd. | (2'-5') oligoadenylate analogues useful as inhibitors of host-v5.-graft response |
| US5576427A (en) | 1993-03-30 | 1996-11-19 | Sterling Winthrop, Inc. | Acyclic nucleoside analogs and oligonucleotide sequences containing them |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5591722A (en) | 1989-09-15 | 1997-01-07 | Southern Research Institute | 2'-deoxy-4'-thioribonucleosides and their antiviral activity |
| US5594121A (en) | 1991-11-07 | 1997-01-14 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified purines |
| US5596086A (en) | 1990-09-20 | 1997-01-21 | Gilead Sciences, Inc. | Modified internucleoside linkages having one nitrogen and two carbon atoms |
| US5596091A (en) | 1994-03-18 | 1997-01-21 | The Regents Of The University Of California | Antisense oligonucleotides comprising 5-aminoalkyl pyrimidine nucleotides |
| US5597909A (en) | 1994-08-25 | 1997-01-28 | Chiron Corporation | Polynucleotide reagents containing modified deoxyribose moieties, and associated methods of synthesis and use |
| US5602240A (en) | 1990-07-27 | 1997-02-11 | Ciba Geigy Ag. | Backbone modified oligonucleotide analogs |
| US5608046A (en) | 1990-07-27 | 1997-03-04 | Isis Pharmaceuticals, Inc. | Conjugated 4'-desmethyl nucleoside analog compounds |
| US5610300A (en) | 1992-07-01 | 1997-03-11 | Ciba-Geigy Corporation | Carbocyclic nucleosides containing bicyclic rings, oligonucleotides therefrom, process for their preparation, their use and intermediates |
| US5610289A (en) | 1990-07-27 | 1997-03-11 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| US5614617A (en) | 1990-07-27 | 1997-03-25 | Isis Pharmaceuticals, Inc. | Nuclease resistant, pyrimidine modified oligonucleotides that detect and modulate gene expression |
| US5618704A (en) | 1990-07-27 | 1997-04-08 | Isis Pharmacueticals, Inc. | Backbone-modified oligonucleotide analogs and preparation thereof through radical coupling |
| US5623065A (en) | 1990-08-13 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Gapped 2' modified oligonucleotides |
| US5623070A (en) | 1990-07-27 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5625050A (en) | 1994-03-31 | 1997-04-29 | Amgen Inc. | Modified oligonucleotides and intermediates useful in nucleic acid therapeutics |
| US5627053A (en) | 1994-03-29 | 1997-05-06 | Ribozyme Pharmaceuticals, Inc. | 2'deoxy-2'-alkylnucleotide containing nucleic acid |
| US5633360A (en) | 1992-04-14 | 1997-05-27 | Gilead Sciences, Inc. | Oligonucleotide analogs capable of passive cell membrane permeation |
| US5639873A (en) | 1992-02-05 | 1997-06-17 | Centre National De La Recherche Scientifique (Cnrs) | Oligothionucleotides |
| US5646265A (en) | 1990-01-11 | 1997-07-08 | Isis Pharmceuticals, Inc. | Process for the preparation of 2'-O-alkyl purine phosphoramidites |
| US5646269A (en) | 1994-04-28 | 1997-07-08 | Gilead Sciences, Inc. | Method for oligonucleotide analog synthesis |
| US5645985A (en) | 1991-11-26 | 1997-07-08 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified pyrimidines |
| US5652355A (en) | 1992-07-23 | 1997-07-29 | Worcester Foundation For Experimental Biology | Hybrid oligonucleotide phosphorothioates |
| US5652356A (en) | 1995-08-17 | 1997-07-29 | Hybridon, Inc. | Inverted chimeric and hybrid oligonucleotides |
| US5656408A (en) | 1996-04-29 | 1997-08-12 | Xerox Corporation | Coated carrier particles |
| US5663312A (en) | 1993-03-31 | 1997-09-02 | Sanofi | Oligonucleotide dimers with amide linkages replacing phosphodiester linkages |
| US5670633A (en) | 1990-01-11 | 1997-09-23 | Isis Pharmaceuticals, Inc. | Sugar modified oligonucleotides that detect and modulate gene expression |
| US5672697A (en) | 1991-02-08 | 1997-09-30 | Gilead Sciences, Inc. | Nucleoside 5'-methylene phosphonates |
| US5677439A (en) | 1990-08-03 | 1997-10-14 | Sanofi | Oligonucleotide analogues containing phosphate diester linkage substitutes, compositions thereof, and precursor dinucleotide analogues |
| US5677437A (en) | 1990-07-27 | 1997-10-14 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5681941A (en) | 1990-01-11 | 1997-10-28 | Isis Pharmaceuticals, Inc. | Substituted purines and oligonucleotide cross-linking |
| US5700922A (en) | 1991-12-24 | 1997-12-23 | Isis Pharmaceuticals, Inc. | PNA-DNA-PNA chimeric macromolecules |
| US5721218A (en) | 1989-10-23 | 1998-02-24 | Gilead Sciences, Inc. | Oligonucleotides with inverted polarity |
| US5750692A (en) | 1990-01-11 | 1998-05-12 | Isis Pharmaceuticals, Inc. | Synthesis of 3-deazapurines |
| US5792608A (en) | 1991-12-12 | 1998-08-11 | Gilead Sciences, Inc. | Nuclease stable and binding competent oligomers and methods for their use |
| WO1998039352A1 (en) | 1997-03-07 | 1998-09-11 | Takeshi Imanishi | Novel bicyclonucleoside and oligonucleotide analogues |
| US5830653A (en) | 1991-11-26 | 1998-11-03 | Gilead Sciences, Inc. | Methods of using oligomers containing modified pyrimidines |
| WO1999014226A2 (en) | 1997-09-12 | 1999-03-25 | Exiqon A/S | Bi- and tri-cyclic nucleoside, nucleotide and oligonucleotide analogues |
| WO2000066604A2 (en) * | 1999-05-04 | 2000-11-09 | Exiqon A/S | L-ribo-lna analogues |
| WO2001049687A2 (en) | 1999-12-30 | 2001-07-12 | K. U. Leuven Research & Development | Cyclohexene nucleic acids |
| WO2002036743A2 (en) | 2000-10-30 | 2002-05-10 | Isis Pharmaceuticals, Inc. | Antisense modulation of calreticulin expression |
| US6525191B1 (en) | 1999-05-11 | 2003-02-25 | Kanda S. Ramasamy | Conformationally constrained L-nucleosides |
| US20030082807A1 (en) | 1999-03-18 | 2003-05-01 | Jesper Wengel | Xylo-LNA analogues |
| US6600032B1 (en) | 1998-08-07 | 2003-07-29 | Isis Pharmaceuticals, Inc. | 2′-O-aminoethyloxyethyl-modified oligonucleotides |
| US20030158403A1 (en) | 2001-07-03 | 2003-08-21 | Isis Pharmaceuticals, Inc. | Nuclease resistant chimeric oligonucleotides |
| US20030207841A1 (en) | 1999-02-12 | 2003-11-06 | Sankyo Company Limited | Novel nucleoside and oligonucleotide analogues |
| US20030224377A1 (en) | 2001-09-04 | 2003-12-04 | Jesper Wengel | Novel LNA compositions and uses thereof |
| US6670461B1 (en) | 1997-09-12 | 2003-12-30 | Exiqon A/S | Oligonucleotide analogues |
| US20040014959A1 (en) | 2002-05-08 | 2004-01-22 | Sorensen Mads Detlef | Synthesis of locked nucleic acid derivatives |
| US20040143114A1 (en) | 1999-07-22 | 2004-07-22 | Sankyo Company, Limited | Novel bicyclonucleoside analogues |
| US6770748B2 (en) | 1997-03-07 | 2004-08-03 | Takeshi Imanishi | Bicyclonucleoside and oligonucleotide analogue |
| US20040171570A1 (en) | 2002-11-05 | 2004-09-02 | Charles Allerson | Polycyclic sugar surrogate-containing oligomeric compounds and compositions for use in gene modulation |
| US20040192918A1 (en) | 2000-08-29 | 2004-09-30 | Takeshi Imanishi | Novel nucleoside analogs and oligonucleotide derivatives containing these analogs |
| US20040219565A1 (en) | 2002-10-21 | 2004-11-04 | Sakari Kauppinen | Oligonucleotides useful for detecting and analyzing nucleic acids of interest |
| WO2004106356A1 (en) | 2003-05-27 | 2004-12-09 | Syddansk Universitet | Functionalized nucleotide derivatives |
| WO2005021570A1 (en) | 2003-08-28 | 2005-03-10 | Gene Design, Inc. | Novel artificial nucleic acids of n-o bond crosslinkage type |
| US20050130923A1 (en) | 2003-09-18 | 2005-06-16 | Balkrishen Bhat | 4'-thionucleosides and oligomeric compounds |
| WO2005121372A2 (en) | 2004-06-03 | 2005-12-22 | Isis Pharmaceuticals, Inc. | Double strand compositions comprising differentially modified strands for use in gene modulation |
| WO2006047842A2 (en) | 2004-11-08 | 2006-05-11 | K.U. Leuven Research And Development | Modified nucleosides for rna interference |
| WO2007134181A2 (en) | 2006-05-11 | 2007-11-22 | Isis Pharmaceuticals, Inc. | 5'-modified bicyclic nucleic acid analogs |
| US20080039618A1 (en) | 2002-11-05 | 2008-02-14 | Charles Allerson | Polycyclic sugar surrogate-containing oligomeric compounds and compositions for use in gene modulation |
| WO2008042973A2 (en) | 2006-10-03 | 2008-04-10 | Alnylam Pharmaceuticals, Inc. | Lipid containing formulations |
| US7399845B2 (en) | 2006-01-27 | 2008-07-15 | Isis Pharmaceuticals, Inc. | 6-modified bicyclic nucleic acid analogs |
| WO2008101157A1 (en) | 2007-02-15 | 2008-08-21 | Isis Pharmaceuticals, Inc. | 5'-substituted-2'-f modified nucleosides and oligomeric compounds prepared therefrom |
| WO2008150729A2 (en) | 2007-05-30 | 2008-12-11 | Isis Pharmaceuticals, Inc. | N-substituted-aminomethylene bridged bicyclic nucleic acid analogs |
| WO2008154401A2 (en) | 2007-06-08 | 2008-12-18 | Isis Pharmaceuticals, Inc. | Carbocyclic bicyclic nucleic acid analogs |
| WO2009006478A2 (en) | 2007-07-05 | 2009-01-08 | Isis Pharmaceuticals, Inc. | 6-disubstituted bicyclic nucleic acid analogs |
| WO2010036696A1 (en) | 2008-09-24 | 2010-04-01 | Isis Pharmaceuticals, Inc. | Cyclohexenyl nucleic acid analogs |
| US8623108B2 (en) | 2009-01-14 | 2014-01-07 | Boehler Edelstahl Gmbh & Co Kg | Wear-resistant material |
| US9778708B1 (en) | 2016-07-18 | 2017-10-03 | Lenovo Enterprise Solutions (Singapore) Pte. Ltd. | Dual sided latching retainer for computer modules |
| US9984408B1 (en) | 2012-05-30 | 2018-05-29 | Amazon Technologies, Inc. | Method, medium, and system for live video cooperative shopping |
-
2013
- 2013-03-20 WO PCT/US2013/033151 patent/WO2013154799A1/en active Application Filing
Patent Citations (174)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2699808A (en) | 1944-10-06 | 1955-01-18 | Mark W Lowe | Apparatus for peeling tomatoes |
| US2699508A (en) | 1951-12-21 | 1955-01-11 | Selectronics Inc | Method of mounting and construction of mounting for low frequency piezoelectric crystals |
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4458066A (en) | 1980-02-29 | 1984-07-03 | University Patents, Inc. | Process for preparing polynucleotides |
| US4500707A (en) | 1980-02-29 | 1985-02-19 | University Patents, Inc. | Nucleosides useful in the preparation of polynucleotides |
| US5132418A (en) | 1980-02-29 | 1992-07-21 | University Patents, Inc. | Process for preparing polynucleotides |
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US4973679A (en) | 1981-03-27 | 1990-11-27 | University Patents, Inc. | Process for oligonucleo tide synthesis using phosphormidite intermediates |
| US4415732A (en) | 1981-03-27 | 1983-11-15 | University Patents, Inc. | Phosphoramidite compounds and processes |
| US4668777A (en) | 1981-03-27 | 1987-05-26 | University Patents, Inc. | Phosphoramidite nucleoside compounds |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US4476301A (en) | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| USRE34069E (en) | 1983-08-18 | 1992-09-15 | Biosyntech Gmbh | Process for the preparation of oligonucleotides |
| US4725677A (en) | 1983-08-18 | 1988-02-16 | Biosyntech Gmbh | Process for the preparation of oligonucleotides |
| US5118800A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | Oligonucleotides possessing a primary amino group in the terminal nucleotide |
| US5550111A (en) | 1984-07-11 | 1996-08-27 | Temple University-Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| US4981957A (en) | 1984-07-19 | 1991-01-01 | Centre National De La Recherche Scientifique | Oligonucleotides with modified phosphate and modified carbohydrate moieties at the respective chain termini |
| US5367066A (en) | 1984-10-16 | 1994-11-22 | Chiron Corporation | Oligonucleotides with selectably cleavable and/or abasic sites |
| US4845205A (en) | 1985-01-08 | 1989-07-04 | Institut Pasteur | 2,N6 -disubstituted and 2,N6 -trisubstituted adenosine-3'-phosphoramidites |
| US5235033A (en) | 1985-03-15 | 1993-08-10 | Anti-Gene Development Group | Alpha-morpholino ribonucleoside derivatives and polymers thereof |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| US5185444A (en) | 1985-03-15 | 1993-02-09 | Anti-Gene Deveopment Group | Uncharged morpolino-based polymers having phosphorous containing chiral intersubunit linkages |
| US5013830A (en) | 1986-09-08 | 1991-05-07 | Ajinomoto Co., Inc. | Compounds for the cleavage at a specific position of RNA, oligomers employed for the formation of said compounds, and starting materials for the synthesis of said oligomers |
| US5276019A (en) | 1987-03-25 | 1994-01-04 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5264423A (en) | 1987-03-25 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5286717A (en) | 1987-03-25 | 1994-02-15 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5552540A (en) | 1987-06-24 | 1996-09-03 | Howard Florey Institute Of Experimental Physiology And Medicine | Nucleoside derivatives |
| US5405939A (en) | 1987-10-22 | 1995-04-11 | Temple University Of The Commonwealth System Of Higher Education | 2',5'-phosphorothioate oligoadenylates and their covalent conjugates with polylysine |
| US5188897A (en) | 1987-10-22 | 1993-02-23 | Temple University Of The Commonwealth System Of Higher Education | Encapsulated 2',5'-phosphorothioate oligoadenylates |
| US5403711A (en) | 1987-11-30 | 1995-04-04 | University Of Iowa Research Foundation | Nucleic acid hybridization and amplification method for detection of specific sequences in which a complementary labeled nucleic acid probe is cleaved |
| US5491133A (en) | 1987-11-30 | 1996-02-13 | University Of Iowa Research Foundation | Methods for blocking the expression of specifically targeted genes |
| US5519126A (en) | 1988-03-25 | 1996-05-21 | University Of Virginia Alumni Patents Foundation | Oligonucleotide N-alkylphosphoramidates |
| US5278302A (en) | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| US5453496A (en) | 1988-05-26 | 1995-09-26 | University Patents, Inc. | Polynucleotide phosphorodithioate |
| US5216141A (en) | 1988-06-06 | 1993-06-01 | Benner Steven A | Oligonucleotide analogs containing sulfur linkages |
| US5175273A (en) | 1988-07-01 | 1992-12-29 | Genentech, Inc. | Nucleic acid intercalating agents |
| US5194599A (en) | 1988-09-23 | 1993-03-16 | Gilead Sciences, Inc. | Hydrogen phosphonodithioate compositions |
| US5565555A (en) | 1988-09-23 | 1996-10-15 | Gilead Sciences, Inc. | Nucleoside hydrogen phosphonodithioate diesters and activated phosphonodithioate analogues |
| US5256775A (en) | 1989-06-05 | 1993-10-26 | Gilead Sciences, Inc. | Exonuclease-resistant oligonucleotides |
| US5134066A (en) | 1989-08-29 | 1992-07-28 | Monsanto Company | Improved probes using nucleosides containing 3-dezauracil analogs |
| US5591722A (en) | 1989-09-15 | 1997-01-07 | Southern Research Institute | 2'-deoxy-4'-thioribonucleosides and their antiviral activity |
| US5721218A (en) | 1989-10-23 | 1998-02-24 | Gilead Sciences, Inc. | Oligonucleotides with inverted polarity |
| US5527899A (en) | 1989-10-23 | 1996-06-18 | Gilead Sciences, Inc. | Oligonucleotides with inverted polarity |
| US5399676A (en) | 1989-10-23 | 1995-03-21 | Gilead Sciences | Oligonucleotides with inverted polarity |
| US5466786B1 (en) | 1989-10-24 | 1998-04-07 | Gilead Sciences | 2' Modified nucleoside and nucleotide compounds |
| US5264564A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences | Oligonucleotide analogs with novel linkages |
| US5466786A (en) | 1989-10-24 | 1995-11-14 | Gilead Sciences | 2'modified nucleoside and nucleotide compounds |
| US5792847A (en) | 1989-10-24 | 1998-08-11 | Gilead Sciences, Inc. | 2' Modified Oligonucleotides |
| US5264562A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| US5455233A (en) | 1989-11-30 | 1995-10-03 | University Of North Carolina | Oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5177198A (en) | 1989-11-30 | 1993-01-05 | University Of N.C. At Chapel Hill | Process for preparing oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5166315A (en) | 1989-12-20 | 1992-11-24 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5130302A (en) | 1989-12-20 | 1992-07-14 | Boron Bilogicals, Inc. | Boronated nucleoside, nucleotide and oligonucleotide compounds, compositions and methods for using same |
| US5405938A (en) | 1989-12-20 | 1995-04-11 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5670633A (en) | 1990-01-11 | 1997-09-23 | Isis Pharmaceuticals, Inc. | Sugar modified oligonucleotides that detect and modulate gene expression |
| US5587469A (en) | 1990-01-11 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides containing N-2 substituted purines |
| US5646265A (en) | 1990-01-11 | 1997-07-08 | Isis Pharmceuticals, Inc. | Process for the preparation of 2'-O-alkyl purine phosphoramidites |
| US5750692A (en) | 1990-01-11 | 1998-05-12 | Isis Pharmaceuticals, Inc. | Synthesis of 3-deazapurines |
| US5681941A (en) | 1990-01-11 | 1997-10-28 | Isis Pharmaceuticals, Inc. | Substituted purines and oligonucleotide cross-linking |
| US5459255A (en) | 1990-01-11 | 1995-10-17 | Isis Pharmaceuticals, Inc. | N-2 substituted purines |
| US5220007A (en) | 1990-02-15 | 1993-06-15 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of RNA and production of encoded polypeptides |
| US5149797A (en) | 1990-02-15 | 1992-09-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of rna and production of encoded polypeptides |
| US5366878A (en) | 1990-02-15 | 1994-11-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of RNA and production of encoded polypeptides |
| US5321131A (en) | 1990-03-08 | 1994-06-14 | Hybridon, Inc. | Site-specific functionalization of oligodeoxynucleotides for non-radioactive labelling |
| US5541306A (en) | 1990-03-08 | 1996-07-30 | Worcester Foundation For Biomedical Research | Aminoalkylphosphotriester oligonucleotide derivatives |
| US5563253A (en) | 1990-03-08 | 1996-10-08 | Worcester Foundation For Biomedical Research | Linear aminoalkylphosphoramidate oligonucleotide derivatives |
| US5536821A (en) | 1990-03-08 | 1996-07-16 | Worcester Foundation For Biomedical Research | Aminoalkylphosphorothioamidate oligonucleotide deratives |
| US5470967A (en) | 1990-04-10 | 1995-11-28 | The Dupont Merck Pharmaceutical Company | Oligonucleotide analogs with sulfamate linkages |
| US5567811A (en) | 1990-05-03 | 1996-10-22 | Amersham International Plc | Phosphoramidite derivatives, their preparation and the use thereof in the incorporation of reporter groups on synthetic oligonucleotides |
| US5514785A (en) | 1990-05-11 | 1996-05-07 | Becton Dickinson And Company | Solid supports for nucleic acid hybridization assays |
| US5677437A (en) | 1990-07-27 | 1997-10-14 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5623070A (en) | 1990-07-27 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5608046A (en) | 1990-07-27 | 1997-03-04 | Isis Pharmaceuticals, Inc. | Conjugated 4'-desmethyl nucleoside analog compounds |
| US5489677A (en) | 1990-07-27 | 1996-02-06 | Isis Pharmaceuticals, Inc. | Oligonucleoside linkages containing adjacent oxygen and nitrogen atoms |
| US5614617A (en) | 1990-07-27 | 1997-03-25 | Isis Pharmaceuticals, Inc. | Nuclease resistant, pyrimidine modified oligonucleotides that detect and modulate gene expression |
| US5610289A (en) | 1990-07-27 | 1997-03-11 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| US5618704A (en) | 1990-07-27 | 1997-04-08 | Isis Pharmacueticals, Inc. | Backbone-modified oligonucleotide analogs and preparation thereof through radical coupling |
| US5541307A (en) | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| US5378825A (en) | 1990-07-27 | 1995-01-03 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs |
| US5602240A (en) | 1990-07-27 | 1997-02-11 | Ciba Geigy Ag. | Backbone modified oligonucleotide analogs |
| US5386023A (en) | 1990-07-27 | 1995-01-31 | Isis Pharmaceuticals | Backbone modified oligonucleotide analogs and preparation thereof through reductive coupling |
| US5677439A (en) | 1990-08-03 | 1997-10-14 | Sanofi | Oligonucleotide analogues containing phosphate diester linkage substitutes, compositions thereof, and precursor dinucleotide analogues |
| US5223618A (en) | 1990-08-13 | 1993-06-29 | Isis Pharmaceuticals, Inc. | 4'-desmethyl nucleoside analog compounds |
| US5623065A (en) | 1990-08-13 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Gapped 2' modified oligonucleotides |
| US5177196A (en) | 1990-08-16 | 1993-01-05 | Microprobe Corporation | Oligo (α-arabinofuranosyl nucleotides) and α-arabinofuranosyl precursors thereof |
| US5214134A (en) | 1990-09-12 | 1993-05-25 | Sterling Winthrop Inc. | Process of linking nucleosides with a siloxane bridge |
| US5561225A (en) | 1990-09-19 | 1996-10-01 | Southern Research Institute | Polynucleotide analogs containing sulfonate and sulfonamide internucleoside linkages |
| US5596086A (en) | 1990-09-20 | 1997-01-21 | Gilead Sciences, Inc. | Modified internucleoside linkages having one nitrogen and two carbon atoms |
| US5432272A (en) | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| US5672697A (en) | 1991-02-08 | 1997-09-30 | Gilead Sciences, Inc. | Nucleoside 5'-methylene phosphonates |
| US5571799A (en) | 1991-08-12 | 1996-11-05 | Basco, Ltd. | (2'-5') oligoadenylate analogues useful as inhibitors of host-v5.-graft response |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5319080A (en) | 1991-10-17 | 1994-06-07 | Ciba-Geigy Corporation | Bicyclic nucleosides, oligonucleotides, process for their preparation and intermediates |
| US5393878A (en) | 1991-10-17 | 1995-02-28 | Ciba-Geigy Corporation | Bicyclic nucleosides, oligonucleotides, process for their preparation and intermediates |
| US5594121A (en) | 1991-11-07 | 1997-01-14 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified purines |
| US5645985A (en) | 1991-11-26 | 1997-07-08 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified pyrimidines |
| US5484908A (en) | 1991-11-26 | 1996-01-16 | Gilead Sciences, Inc. | Oligonucleotides containing 5-propynyl pyrimidines |
| US5830653A (en) | 1991-11-26 | 1998-11-03 | Gilead Sciences, Inc. | Methods of using oligomers containing modified pyrimidines |
| US5792608A (en) | 1991-12-12 | 1998-08-11 | Gilead Sciences, Inc. | Nuclease stable and binding competent oligomers and methods for their use |
| US5359044A (en) | 1991-12-13 | 1994-10-25 | Isis Pharmaceuticals | Cyclobutyl oligonucleotide surrogates |
| US5700922A (en) | 1991-12-24 | 1997-12-23 | Isis Pharmaceuticals, Inc. | PNA-DNA-PNA chimeric macromolecules |
| US5639873A (en) | 1992-02-05 | 1997-06-17 | Centre National De La Recherche Scientifique (Cnrs) | Oligothionucleotides |
| US5633360A (en) | 1992-04-14 | 1997-05-27 | Gilead Sciences, Inc. | Oligonucleotide analogs capable of passive cell membrane permeation |
| US5434257A (en) | 1992-06-01 | 1995-07-18 | Gilead Sciences, Inc. | Binding compentent oligomers containing unsaturated 3',5' and 2',5' linkages |
| US5610300A (en) | 1992-07-01 | 1997-03-11 | Ciba-Geigy Corporation | Carbocyclic nucleosides containing bicyclic rings, oligonucleotides therefrom, process for their preparation, their use and intermediates |
| US5700920A (en) | 1992-07-01 | 1997-12-23 | Novartis Corporation | Carbocyclic nucleosides containing bicyclic rings, oligonucleotides therefrom, process for their preparation, their use and intermediates |
| US5652355A (en) | 1992-07-23 | 1997-07-29 | Worcester Foundation For Experimental Biology | Hybrid oligonucleotide phosphorothioates |
| WO1994002499A1 (en) | 1992-07-27 | 1994-02-03 | Hybridon, Inc. | Oligonucleotide alkylphosphonothioates |
| WO1994014226A1 (en) | 1992-12-14 | 1994-06-23 | Honeywell Inc. | Motor system with individually controlled redundant windings |
| WO1994017093A1 (en) | 1993-01-25 | 1994-08-04 | Hybridon, Inc. | Oligonucleotide alkylphosphonates and alkylphosphonothioates |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| US5508270A (en) | 1993-03-06 | 1996-04-16 | Ciba-Geigy Corporation | Nucleoside phosphinate compounds and compositions |
| US5466677A (en) | 1993-03-06 | 1995-11-14 | Ciba-Geigy Corporation | Dinucleoside phosphinates and their pharmaceutical compositions |
| US5576427A (en) | 1993-03-30 | 1996-11-19 | Sterling Winthrop, Inc. | Acyclic nucleoside analogs and oligonucleotide sequences containing them |
| US5663312A (en) | 1993-03-31 | 1997-09-02 | Sanofi | Oligonucleotide dimers with amide linkages replacing phosphodiester linkages |
| US5763588A (en) | 1993-09-17 | 1998-06-09 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| US5502177A (en) | 1993-09-17 | 1996-03-26 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| US6005096A (en) | 1993-09-17 | 1999-12-21 | Gilead Sciences, Inc. | Pyrimidine derivatives |
| US5457187A (en) | 1993-12-08 | 1995-10-10 | Board Of Regents University Of Nebraska | Oligonucleotides containing 5-fluorouracil |
| US5446137B1 (en) | 1993-12-09 | 1998-10-06 | Behringwerke Ag | Oligonucleotides containing 4'-substituted nucleotides |
| US5446137A (en) | 1993-12-09 | 1995-08-29 | Syntex (U.S.A.) Inc. | Oligonucleotides containing 4'-substituted nucleotides |
| US5565350A (en) | 1993-12-09 | 1996-10-15 | Thomas Jefferson University | Compounds and methods for site directed mutations in eukaryotic cells |
| US5519134A (en) | 1994-01-11 | 1996-05-21 | Isis Pharmaceuticals, Inc. | Pyrrolidine-containing monomers and oligomers |
| US5596091A (en) | 1994-03-18 | 1997-01-21 | The Regents Of The University Of California | Antisense oligonucleotides comprising 5-aminoalkyl pyrimidine nucleotides |
| US5627053A (en) | 1994-03-29 | 1997-05-06 | Ribozyme Pharmaceuticals, Inc. | 2'deoxy-2'-alkylnucleotide containing nucleic acid |
| US5625050A (en) | 1994-03-31 | 1997-04-29 | Amgen Inc. | Modified oligonucleotides and intermediates useful in nucleic acid therapeutics |
| US5646269A (en) | 1994-04-28 | 1997-07-08 | Gilead Sciences, Inc. | Method for oligonucleotide analog synthesis |
| US5525711A (en) | 1994-05-18 | 1996-06-11 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Pteridine nucleotide analogs as fluorescent DNA probes |
| US5597909A (en) | 1994-08-25 | 1997-01-28 | Chiron Corporation | Polynucleotide reagents containing modified deoxyribose moieties, and associated methods of synthesis and use |
| US5652356A (en) | 1995-08-17 | 1997-07-29 | Hybridon, Inc. | Inverted chimeric and hybrid oligonucleotides |
| US5656408A (en) | 1996-04-29 | 1997-08-12 | Xerox Corporation | Coated carrier particles |
| US6268490B1 (en) | 1997-03-07 | 2001-07-31 | Takeshi Imanishi | Bicyclonucleoside and oligonucleotide analogues |
| WO1998039352A1 (en) | 1997-03-07 | 1998-09-11 | Takeshi Imanishi | Novel bicyclonucleoside and oligonucleotide analogues |
| US6770748B2 (en) | 1997-03-07 | 2004-08-03 | Takeshi Imanishi | Bicyclonucleoside and oligonucleotide analogue |
| WO1999014226A2 (en) | 1997-09-12 | 1999-03-25 | Exiqon A/S | Bi- and tri-cyclic nucleoside, nucleotide and oligonucleotide analogues |
| US7034133B2 (en) | 1997-09-12 | 2006-04-25 | Exiqon A/S | Oligonucleotide analogues |
| US6794499B2 (en) | 1997-09-12 | 2004-09-21 | Exiqon A/S | Oligonucleotide analogues |
| US6670461B1 (en) | 1997-09-12 | 2003-12-30 | Exiqon A/S | Oligonucleotide analogues |
| US6600032B1 (en) | 1998-08-07 | 2003-07-29 | Isis Pharmaceuticals, Inc. | 2′-O-aminoethyloxyethyl-modified oligonucleotides |
| US20030207841A1 (en) | 1999-02-12 | 2003-11-06 | Sankyo Company Limited | Novel nucleoside and oligonucleotide analogues |
| US20030082807A1 (en) | 1999-03-18 | 2003-05-01 | Jesper Wengel | Xylo-LNA analogues |
| WO2000066604A2 (en) * | 1999-05-04 | 2000-11-09 | Exiqon A/S | L-ribo-lna analogues |
| US7053207B2 (en) | 1999-05-04 | 2006-05-30 | Exiqon A/S | L-ribo-LNA analogues |
| US6525191B1 (en) | 1999-05-11 | 2003-02-25 | Kanda S. Ramasamy | Conformationally constrained L-nucleosides |
| US20040143114A1 (en) | 1999-07-22 | 2004-07-22 | Sankyo Company, Limited | Novel bicyclonucleoside analogues |
| WO2001049687A2 (en) | 1999-12-30 | 2001-07-12 | K. U. Leuven Research & Development | Cyclohexene nucleic acids |
| US20040192918A1 (en) | 2000-08-29 | 2004-09-30 | Takeshi Imanishi | Novel nucleoside analogs and oligonucleotide derivatives containing these analogs |
| US6426220B1 (en) | 2000-10-30 | 2002-07-30 | Isis Pharmaceuticals, Inc. | Antisense modulation of calreticulin expression |
| WO2002036743A2 (en) | 2000-10-30 | 2002-05-10 | Isis Pharmaceuticals, Inc. | Antisense modulation of calreticulin expression |
| US20030158403A1 (en) | 2001-07-03 | 2003-08-21 | Isis Pharmaceuticals, Inc. | Nuclease resistant chimeric oligonucleotides |
| US20030224377A1 (en) | 2001-09-04 | 2003-12-04 | Jesper Wengel | Novel LNA compositions and uses thereof |
| US20040014959A1 (en) | 2002-05-08 | 2004-01-22 | Sorensen Mads Detlef | Synthesis of locked nucleic acid derivatives |
| US20040219565A1 (en) | 2002-10-21 | 2004-11-04 | Sakari Kauppinen | Oligonucleotides useful for detecting and analyzing nucleic acids of interest |
| US20080039618A1 (en) | 2002-11-05 | 2008-02-14 | Charles Allerson | Polycyclic sugar surrogate-containing oligomeric compounds and compositions for use in gene modulation |
| US20040171570A1 (en) | 2002-11-05 | 2004-09-02 | Charles Allerson | Polycyclic sugar surrogate-containing oligomeric compounds and compositions for use in gene modulation |
| WO2004106356A1 (en) | 2003-05-27 | 2004-12-09 | Syddansk Universitet | Functionalized nucleotide derivatives |
| US7427672B2 (en) | 2003-08-28 | 2008-09-23 | Takeshi Imanishi | Artificial nucleic acids of n-o bond crosslinkage type |
| WO2005021570A1 (en) | 2003-08-28 | 2005-03-10 | Gene Design, Inc. | Novel artificial nucleic acids of n-o bond crosslinkage type |
| US20050130923A1 (en) | 2003-09-18 | 2005-06-16 | Balkrishen Bhat | 4'-thionucleosides and oligomeric compounds |
| WO2005121372A2 (en) | 2004-06-03 | 2005-12-22 | Isis Pharmaceuticals, Inc. | Double strand compositions comprising differentially modified strands for use in gene modulation |
| WO2005121371A2 (en) | 2004-06-03 | 2005-12-22 | Isis Pharmaceuticals, Inc. | Double strand compositions comprising differentially modified strands for use in gene modulation |
| WO2006047842A2 (en) | 2004-11-08 | 2006-05-11 | K.U. Leuven Research And Development | Modified nucleosides for rna interference |
| US7399845B2 (en) | 2006-01-27 | 2008-07-15 | Isis Pharmaceuticals, Inc. | 6-modified bicyclic nucleic acid analogs |
| WO2007134181A2 (en) | 2006-05-11 | 2007-11-22 | Isis Pharmaceuticals, Inc. | 5'-modified bicyclic nucleic acid analogs |
| US20070287831A1 (en) | 2006-05-11 | 2007-12-13 | Isis Pharmaceuticals, Inc | 5'-modified bicyclic nucleic acid analogs |
| WO2008042973A2 (en) | 2006-10-03 | 2008-04-10 | Alnylam Pharmaceuticals, Inc. | Lipid containing formulations |
| WO2008101157A1 (en) | 2007-02-15 | 2008-08-21 | Isis Pharmaceuticals, Inc. | 5'-substituted-2'-f modified nucleosides and oligomeric compounds prepared therefrom |
| WO2008150729A2 (en) | 2007-05-30 | 2008-12-11 | Isis Pharmaceuticals, Inc. | N-substituted-aminomethylene bridged bicyclic nucleic acid analogs |
| WO2008154401A2 (en) | 2007-06-08 | 2008-12-18 | Isis Pharmaceuticals, Inc. | Carbocyclic bicyclic nucleic acid analogs |
| WO2009006478A2 (en) | 2007-07-05 | 2009-01-08 | Isis Pharmaceuticals, Inc. | 6-disubstituted bicyclic nucleic acid analogs |
| WO2010036696A1 (en) | 2008-09-24 | 2010-04-01 | Isis Pharmaceuticals, Inc. | Cyclohexenyl nucleic acid analogs |
| US8623108B2 (en) | 2009-01-14 | 2014-01-07 | Boehler Edelstahl Gmbh & Co Kg | Wear-resistant material |
| US9984408B1 (en) | 2012-05-30 | 2018-05-29 | Amazon Technologies, Inc. | Method, medium, and system for live video cooperative shopping |
| US9778708B1 (en) | 2016-07-18 | 2017-10-03 | Lenovo Enterprise Solutions (Singapore) Pte. Ltd. | Dual sided latching retainer for computer modules |
Non-Patent Citations (117)
| Title |
|---|
| AGRAWAL ET AL.: "Protocols for Oligonucleotide Conjugates", vol. 26, 1994, HUMANA PRESS, pages: 1 - 72 |
| AGRAWAL,: "DNA: Protocols for Oligonucleotides and Analogs", 1993, HUMANA PRESS |
| AKINC ET AL., NATURE BIOTECHNOLOGY, vol. 26, 1 May 2008 (2008-05-01), pages 561 - 569 |
| ALBAEK ET AL., J. ORG. CHEM., vol. 71, 2006, pages 7731 - 7740 |
| ALTMANN ET AL., BIOCHEM. SOC. TRANS., vol. 24, 1996, pages 630 - 637 |
| ALTMANN ET AL., CHIMIA, vol. 50, 1996, pages 168 - 176 |
| ALTMANN ET AL., NUCLEOSIDES NUCLEOTIDES, vol. 16, 1997, pages 917 - 926 |
| ALTSCHUL ET AL., J. MOL. BIOL., vol. 215, 1990, pages 403 - 410 |
| AUSUBEL, F.M. ET AL.: "Current Protocols in Molecular Biology", vol. 2, 1991, JOHN WILEY & SONS, INC., pages: 11.2.1 - 11.2.22 |
| AUSUBEL, F.M. ET AL.: "Current Protocols in Molecular Biology", vol. 2, 1997, JOHN WILEY & SONS, INC., pages: 10.8.1 - 10.8.21 |
| AUSUBEL, F.M. ET AL.: "Current Protocols in Molecular Biology", vol. 2, 1997, JOHN WILEY & SONS, INC., pages: 11.12.1 - 11.12.9 |
| AUSUBEL, F.M. ET AL.: "Current Protocols in Molecular Biology", vol. 2, 1997, JOHN WILEY & SONS, INC., pages: 11.4.1 - 11.11.5 |
| AUSUBEL, F.M. ET AL.: "Current Protocols in Molecular Biology", vol. 2, 1998, JOHN WILEY & SONS, INC., pages: 10.16.1 - 10.16.11 |
| BAKER ET AL., J. BIOL. CHEM., vol. 272, 1997, pages 11944 - 12000 |
| BARANY ET AL., J. AM. CHEM. SOC., vol. 102, 1980, pages 3084 - 3095 |
| BARANY ET AL., J. AM. CHEM. SOC., vol. 99, 1977, pages 7363 - 7365 |
| BARRY M. TROST,: "Comprehensive Organic Synthesis. Selectivity, Strategy & Efficiency in Modern Organic Chemistry", vol. 9, 1993, PERGAMON |
| BASS ET AL., CELL, vol. 101, 2000, pages 235 - 238 |
| BEAUCAGE ET AL., TETRAHEDRON, vol. 48, no. 12, 1992, pages 2223 - 2311 |
| BEAUCAGE ET AL., TETRAHEDRON, vol. 49, no. 10, 1993, pages 1925 - 1963 |
| BEAUCAGE ET AL., TETRAHEDRON, vol. 49, no. 46, 1993, pages 10441 - 10488 |
| BEAUCAGE; IYER, TETRAHEDRON, vol. 48, 1992, pages 2223 - 2311 |
| BRAASCH ET AL., CHEM. BIOL., vol. 8, 2001, pages 1 - 7 |
| BRAZMA; VILO, FEBS LETT., vol. 480, 2000, pages 17 - 24 |
| CAREY; SUNDBERG: "Advanced Organic Chemistry, Part B: Reactions and Synthesis, 4th Edition;", 2001 |
| CARULLI ET AL., J. CELL BIOCHEM. SUPPL., vol. 31, 1998, pages 286 - 96 |
| CELIS ET AL., FEBS LETT., vol. 480, 2000, pages 2 - 16 |
| CELIS ET AL., FEBSLETT., vol. 480, 2000, pages 2 - 16 |
| CHATTOPADHYAYA ET AL., J ORG. CHEM., vol. 74, 2009, pages 118 - 134 |
| CHIANG ET AL., J. BIOL. CHEM., vol. 266, 1991, pages 18162 - 18171 |
| CONTE ET AL., NUCLEIC ACIDS RES., vol. 25, 1997, pages 2627 - 2634 |
| EGLI ET AL., BIOCHEMISTRY, vol. 35, 1996, pages 8489 - 8494 |
| ELAYADI ET AL., CURR. OPINION INVENS. DRUGS, vol. 2, 2001, pages 558 - 561 |
| ELBASHIR ET AL., GENES DEV., vol. 15, 2001, pages 188 - 200 |
| ELBASHIR ET AL., NATURE, vol. 411, 2001, pages 494 - 498 |
| ENGLISCH ET AL.: "Angewandte Chemie", vol. 30, 1991, pages: 613 |
| F. ECKSTEIN,: "Oligonucleotides and Analogues, a Practical Approach", 1991, OXFORD UNIVERSITY PRESS |
| FIRE ET AL., NATURE, vol. 391, 1998, pages 806 - 811 |
| FRIEDEN ET AL., NUCLEIC ACIDS RESEARCH, vol. 21, 2003, pages 6365 - 6372 |
| FRIER ET AL., NUCLEIC ACIDS RESEARCH, vol. 25, no. 22, 1997, pages 4429 - 4443 |
| FUCHS ET AL., ANAL. BIOCHEM., vol. 286, 2000, pages 91 - 98 |
| GAIT ET AL.: "Applications of Chemically synthesized RNA in RNA:Protein Interactions", 1998, pages: 1 - 36 |
| GALLO ET AL., TETRAHEDRON, vol. 57, 2001, pages 5707 - 5713 |
| GOING; GUSTERSON, EUR. J CANCER, vol. 35, 1999, pages 1895 - 904 |
| GREENE, T.W.; WUTZ, P.G.M.: "Protecting Groups in Organic Synthesis, 4th Edition", 1991, JOHN WILEY & SONS |
| GU ET AL., NUCLEOSIDES, NUCLEOTIDES & NUCLEIC ACIDS, vol. 24, no. 5-7, 2005, pages 993 - 998 |
| GU ET AL., OLIGONUCLEOTIDES, vol. 13, no. 6, 2003, pages 479 - 489 |
| GU ET AL., TETRAHEDRON, vol. 60, no. 9, 2004, pages 2111 - 2123 |
| HORVÁTH ET AL., TETRAHEDRON LETTERS, vol. 48, 2007, pages 3621 - 3623 |
| IAN T. HARRISON; SHUYEN HARRISON: "COMPENDING OF ORGANIC SYNTHETIC METHODS", vol. 2, 1974 |
| IAN T. HARRISON; SHUYEN HARRISON: "Compendium of Organic Synthetic Methods", vol. 1, 1971, JOHN WILEY & SONS |
| ITTIG ET AL., COLLECTION SYMPOSIUM SERIES, vol. 12, 2005, pages 8014 - 8023 |
| ITTIG ET AL., NUCL. ACIDS RES., vol. 32, no. 1, 2004, pages 346 - 353 |
| IVANOVA ET AL., OLIGONUCLEOTIDES, vol. 17, 2007, pages 54 - 65 |
| JONES, L.J. ET AL., ANALYTICAL BIOCHEMISTRY, vol. 265, 1998, pages 368 - 374 |
| JUNGBLUT ET AL., ELECTROPHORESIS, vol. 20, 1999, pages 2100 - 10 |
| JURECIC; BELMONT, CURR. OPIN. MICROBIOL., vol. 3, 2000, pages 316 - 21 |
| KOSHKIN ET AL., TETRAHEDRON, vol. 54, 1998, pages 3607 - 3630 |
| KROSCHWITZ, J.L,: "The Concise Encyclopedia Of Polymer Science And Engineering", 1990, JOHN WILEY & SONS, pages: 858 - 859 |
| KUMAR ET AL., BIOORG. MED CHEM. LETT., vol. 8, 1998, pages 2219 - 2222 |
| KUMAR ET AL., BIOORG. MED. CHEM. LETT., vol. 8, 1998, pages 2219 - 2222 |
| LAROCK, R.C.: "Comprehensive Organic Transformations, 2nd Edition,", 1999, JOHN WILEY & SONS |
| LARSON ET AL., CYTOMETRY, vol. 41, 2000, pages 203 - 208 |
| LARSSON ET AL., J BIOTECHNOL., vol. 80, 2000, pages 143 - 57 |
| LEROY G. WADE JR.: "COMPENDING OF ORGANIC SYNTHETIC METHODS", vol. 4, 1980 |
| LEROY G. WADE JR.: "COMPENDING OF ORGANIC SYNTHETIC METHODS", vol. 5, 1984 |
| LESNIK ET AL., BIOCHEMISTRY, vol. 34, 1995, pages 10807 - 10815 |
| LEUMANN, C. J., BIOORG. & MED. CHEM., vol. 10, 2002, pages 841 - 854 |
| LEUMANN, CHRISTIAN J., BIOORG. & MED CHEM., vol. 10, 2002, pages 841 - 854 |
| LOUIS S. HEGEDUS; LEROY WADE: "COMPENDING OF ORGANIC SYNTHETIC METHODS", vol. 3, 1977 |
| MADDEN ET AL., DRUG DISCOV. TODAY, vol. 5, 2000, pages 415 - 425 |
| MARCH, J.: "Advanced Organic Chemistry 3rd Edition,", 1985, JOHN WILEY & SONS |
| MARCH: "Advanced Organic Chemistry, Reactions, Mechanisms, and Structure, 2nd Edition,", MCGRAW HILL |
| MARTIN, P., HELV. CHIM. ACTA, vol. 78, 1995, pages 486 - 504 |
| MICHAEL B. SMITH: "COMPENDING OF ORGANIC SYNTHETIC METHODS", vol. 6 |
| MIURA ET AL., CLIN. CHEM., vol. 42, 1996, pages 1758 - 1764 |
| MONTGOMERY ET AL., PROC. NATL. ACAD SCI. USA, vol. 95, 1998, pages 15502 - 15507 |
| NAUWELAERTS ET AL., J. AM. CHEM. SOC., vol. 129, no. 30, 2007, pages 9340 - 9348 |
| NAUWELAERTS ET AL., NUCLEIC ACIDS RESEARCH, vol. 33, no. 8, 2005, pages 2452 - 2463 |
| NISHIKURA ET AL., CELL, vol. 107, 2001, pages 415 - 416 |
| ORUM ET AL., CURR. OPINION MOL. THER., vol. 3, 2001, pages 239 - 243 |
| PRASHAR; WEISSMAN, METHODS ENZYMOL., vol. 303, 1999, pages 258 - 72 |
| RAVN J ET AL: "A conformationally locked tricyclic nucleoside. Synthesis, crystal structure and incorporation into oligonucleotides", JOURNAL OF THE CHEMICAL SOCIETY, PERKIN TRANSACTIONS 1, CHEMICAL SOCIETY, LETCHWORTH; GB, no. 16, 1 January 2001 (2001-01-01), pages 1855 - 1861, XP002962136, ISSN: 0300-922X, DOI: 10.1039/B104438A * |
| RENNEBERG ET AL., J. AM. CHEM. SOC., vol. 124, 2002, pages 5993 - 6002 |
| ROBEYNS ET AL., ACTA CRYSTALLOGRAPHICA, SECTION F: STRUCTURAL BIOLOGY AND CRYSTALLIZATION COMMUNICATIONS, vol. F61, no. 6, 2005, pages 585 - 586 |
| ROBEYNS ET AL., J AM. CHEM. SOC., vol. 130, no. 6, 2008, pages 1979 - 1984 |
| ROMIEU ET AL., J. CHEM. SOC. PERKIN TRANS. 1, 1999, pages 1257 |
| SANGER ET AL.: "Principles of Nucleic Acid Structure", 1984, SPRINGER-VERLAG |
| SANGER, W.: "Principles of Nucleic Acid Structure", 1984, SPRINGER-VERLAG |
| SANGHVI, Y.S.: "Antisense Research and Applications", vol. 15, 1993, CRC PRESS, pages: 273 - 302 |
| SCARINGE: "Methods", vol. 23, 2001, article "RNA", pages: 206 - 217 |
| SEARLE ET AL., NUCLEIC ACIDS RES., vol. 21, 1993, pages 2051 - 2056 |
| SINGH ET AL., CHEM. COMMUN., vol. 4, 1998, pages 455 - 456 |
| SINGH ET AL., J. ORG. CHEM., vol. 63, 1998, pages 10035 - 10039 |
| SRIVASTAVA ET AL., J. AM. CHEM. SOC., vol. 129, no. 26, 2007, pages 8362 - 8379 |
| STAHL; WERMUTH: "Handbook of Pharmaceutical Salts: Properties, Selection and Use", 2002, WILEY-VCH |
| STEFFENS ET AL., HELVETICA CHIMICA ACTA., vol. 80, 1997, pages 2426 - 2439 |
| STEFFENS ET AL., J. ORG. CHEM., vol. 121, 1999, pages 3249 - 3255 |
| STEFFENS, J AM. CHEM. SOC., vol. 119, 1997, pages 11548 - 11549 |
| SUTCLIFFE ET AL., PROC. NATL. ACAD. SCI. USA, vol. 97, 2000, pages 1976 - 81 |
| SWAYZE ET AL.: "Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals", NUCL. ACIDS RES., vol. 35, no. 2, 2007, pages 687 - 700, XP002539513, DOI: doi:10.1093/nar/gkl1071 |
| SWAYZE ET AL.: "The Medicinal Chemistry of Oligonucleotides in Antisense a Drug Technology", pages: 143 - 182 |
| TABARA ET AL., SCIENCE, vol. 282, 1998, pages 430 - 431 |
| TIJ STERMAN ET AL., SCIENCE, vol. 295, 2002, pages 694 - 697 |
| TIMMONS ET AL., GENE, vol. 263, 2001, pages 103 - 112 |
| TIMMONS; FIRE, NATURE, vol. 395, 1998, pages 854 |
| TO, COMB. CHEM. HIGH THROUGHPUT SCREEN, vol. 3, 2000, pages 235 - 41 |
| TUSCHL ET AL., GENES DEV., vol. 13, 1999, pages 3191 - 3197 |
| VERBEURE ET AL., NUCLEIC ACIDS RESEARCH, vol. 29, no. 24, 2001, pages 4941 - 4947 |
| WAHLESTEDT ET AL., PROC. NATL. ACAD SCI. U.S.A., vol. 97, 2000, pages 5633 - 5638 |
| WAHLESTEDT ET AL., PROC. NATL. ACAD. SCI. U. S. A., vol. 97, 2000, pages 5633 - 5638 |
| WANG ET AL., J ORG. CHEM., vol. 66, 2001, pages 8478 - 82 |
| WANG ET AL., J ORG. CHEM., vol. 68, 2003, pages 4499 - 4505 |
| WANG ET AL., J. AM. CHEM., vol. 122, 2000, pages 8595 - 8602 |
| WANG ET AL., NUCLEOSIDES, NUCLEOTIDES & NUCLEIC ACIDS, vol. 20, no. 4-7, 2001, pages 785 - 788 |
| Y. S. SANGHVI AND P.D. COOK,: "Carbohydrate Modifications in Antisense Research", pages: 40 - 65 |
| ZHANG; MADDEN, GENOME RES., vol. 7, 1997, pages 649 - 656 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2850092B1 (en) | Tricyclic nucleic acid analogs | |
| EP2580228B1 (en) | Substituted 2'-amino and 2'-thio-bicyclic nucleosides and oligomeric compounds prepared therefrom | |
| EP2361256B1 (en) | Cyclohexenyl nucleic acid analogs | |
| EP2356129B1 (en) | Substituted alpha-l-bicyclic nucleosides | |
| EP2285819B1 (en) | Oligomeric compounds comprising neutrally linked terminal bicyclic nucleosides | |
| EP2188298B1 (en) | Tetrahydropyran nucleic acid analogs | |
| EP2462153B1 (en) | Bicyclic cyclohexose nucleic acid analogs | |
| US9688707B2 (en) | Bicyclic morpholino compounds and oligomeric compounds prepared therefrom | |
| WO2011115818A1 (en) | 5'-substituted bicyclic nucleosides and oligomeric compounds prepared therefrom | |
| WO2011085102A1 (en) | Base modified bicyclic nucleosides and oligomeric compounds prepared therefrom | |
| EP3119789B1 (en) | Bicyclic carbocyclic nucleosides and oligomeric compounds prepared therefrom | |
| EP2358397A2 (en) | 5' and 2' bis-substituted nucleosides and oligomeric compounds prepared therefrom | |
| US9029335B2 (en) | Substituted 2′-thio-bicyclic nucleosides and oligomeric compounds prepared therefrom | |
| US8957200B2 (en) | Bicyclic nucleosides and oligomeric compounds prepared therefrom | |
| US10865413B2 (en) | Oligomeric compounds comprising α-β-constrained nucleic acid | |
| WO2012170347A1 (en) | Bicyclic nucleosides and oligomeric compounds prepared therefrom | |
| WO2013154799A1 (en) | Tricyclic nucleosides and oligomeric compounds prepared therefrom |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13714165 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13714165 Country of ref document: EP Kind code of ref document: A1 |