WO2013112741A1 - Apoptosis signal-regulating kinase inhibitor - Google Patents
Apoptosis signal-regulating kinase inhibitor Download PDFInfo
- Publication number
- WO2013112741A1 WO2013112741A1 PCT/US2013/022997 US2013022997W WO2013112741A1 WO 2013112741 A1 WO2013112741 A1 WO 2013112741A1 US 2013022997 W US2013022997 W US 2013022997W WO 2013112741 A1 WO2013112741 A1 WO 2013112741A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- pharmaceutically acceptable
- salt
- effective amount
- Prior art date
Links
- 230000006907 apoptotic process Effects 0.000 title abstract description 8
- 229940043355 kinase inhibitor Drugs 0.000 title description 4
- 239000003757 phosphotransferase inhibitor Substances 0.000 title description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 179
- 238000011282 treatment Methods 0.000 claims abstract description 16
- 208000007342 Diabetic Nephropathies Diseases 0.000 claims abstract description 15
- 208000033679 diabetic kidney disease Diseases 0.000 claims abstract description 15
- 206010023421 Kidney fibrosis Diseases 0.000 claims abstract description 10
- 150000003839 salts Chemical class 0.000 claims description 41
- 238000000034 method Methods 0.000 claims description 26
- 239000003814 drug Substances 0.000 claims description 19
- 208000020832 chronic kidney disease Diseases 0.000 claims description 18
- 239000008194 pharmaceutical composition Substances 0.000 claims description 10
- 208000005069 pulmonary fibrosis Diseases 0.000 claims description 7
- 239000003937 drug carrier Substances 0.000 claims description 5
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 4
- 238000004519 manufacturing process Methods 0.000 claims description 3
- 208000019425 cirrhosis of liver Diseases 0.000 claims description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 2
- 238000002560 therapeutic procedure Methods 0.000 claims description 2
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 claims 1
- ANMJVFKRCBKCFN-UHFFFAOYSA-N 5-(4-cyclopropylimidazol-1-yl)-2-fluoro-4-methylbenzoic acid Chemical compound CC1=CC(F)=C(C(O)=O)C=C1N1C=C(C2CC2)N=C1 ANMJVFKRCBKCFN-UHFFFAOYSA-N 0.000 claims 1
- 201000010099 disease Diseases 0.000 abstract description 15
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 15
- 208000017169 kidney disease Diseases 0.000 abstract description 12
- 108091000080 Phosphotransferase Proteins 0.000 abstract description 10
- 102000020233 phosphotransferase Human genes 0.000 abstract description 10
- 230000002401 inhibitory effect Effects 0.000 abstract description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 72
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 42
- 238000012360 testing method Methods 0.000 description 40
- 210000004027 cell Anatomy 0.000 description 34
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 31
- 102100033127 Mitogen-activated protein kinase kinase kinase 5 Human genes 0.000 description 28
- 108010075639 MAP Kinase Kinase Kinase 5 Proteins 0.000 description 26
- 230000005764 inhibitory process Effects 0.000 description 23
- 239000000203 mixture Substances 0.000 description 22
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 21
- 241000700159 Rattus Species 0.000 description 20
- 238000006243 chemical reaction Methods 0.000 description 20
- 239000000758 substrate Substances 0.000 description 20
- 238000002360 preparation method Methods 0.000 description 17
- 239000002904 solvent Substances 0.000 description 17
- 239000003112 inhibitor Substances 0.000 description 16
- 238000003556 assay Methods 0.000 description 15
- 102000004169 proteins and genes Human genes 0.000 description 15
- 108090000623 proteins and genes Proteins 0.000 description 15
- 239000007787 solid Substances 0.000 description 15
- 239000000872 buffer Substances 0.000 description 14
- 125000000217 alkyl group Chemical group 0.000 description 13
- 230000000694 effects Effects 0.000 description 13
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 13
- 230000002829 reductive effect Effects 0.000 description 13
- 239000000523 sample Substances 0.000 description 13
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 13
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 12
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 12
- 125000003118 aryl group Chemical group 0.000 description 12
- 125000000623 heterocyclic group Chemical group 0.000 description 12
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 12
- 239000000243 solution Substances 0.000 description 12
- 230000015572 biosynthetic process Effects 0.000 description 11
- 230000002440 hepatic effect Effects 0.000 description 11
- 125000001072 heteroaryl group Chemical group 0.000 description 11
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 11
- 239000013641 positive control Substances 0.000 description 11
- 229940124597 therapeutic agent Drugs 0.000 description 11
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 10
- 125000000753 cycloalkyl group Chemical group 0.000 description 10
- -1 heteroaryl amide Chemical class 0.000 description 10
- 210000003734 kidney Anatomy 0.000 description 10
- 102000004190 Enzymes Human genes 0.000 description 9
- 108090000790 Enzymes Proteins 0.000 description 9
- 125000005843 halogen group Chemical group 0.000 description 9
- 210000001853 liver microsome Anatomy 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- 229940126062 Compound A Drugs 0.000 description 8
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 8
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 8
- 208000004608 Ureteral Obstruction Diseases 0.000 description 8
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- 238000002877 time resolved fluorescence resonance energy transfer Methods 0.000 description 8
- 241001465754 Metazoa Species 0.000 description 7
- 125000004093 cyano group Chemical group *C#N 0.000 description 7
- 238000002474 experimental method Methods 0.000 description 7
- 238000001914 filtration Methods 0.000 description 7
- 230000035699 permeability Effects 0.000 description 7
- 108090000765 processed proteins & peptides Proteins 0.000 description 7
- 108010081668 Cytochrome P-450 CYP3A Proteins 0.000 description 6
- 102100039205 Cytochrome P450 3A4 Human genes 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- 125000003545 alkoxy group Chemical group 0.000 description 6
- 238000009739 binding Methods 0.000 description 6
- 230000037396 body weight Effects 0.000 description 6
- 206010012601 diabetes mellitus Diseases 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 238000011534 incubation Methods 0.000 description 6
- 239000000543 intermediate Substances 0.000 description 6
- 230000003228 microsomal effect Effects 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- LVTJOONKWUXEFR-FZRMHRINSA-N protoneodioscin Natural products O(C[C@@H](CC[C@]1(O)[C@H](C)[C@@H]2[C@]3(C)[C@H]([C@H]4[C@@H]([C@]5(C)C(=CC4)C[C@@H](O[C@@H]4[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@@H](O)[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@H](CO)O4)CC5)CC3)C[C@@H]2O1)C)[C@H]1[C@H](O)[C@H](O)[C@H](O)[C@@H](CO)O1 LVTJOONKWUXEFR-FZRMHRINSA-N 0.000 description 6
- 125000001424 substituent group Chemical group 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 102000008186 Collagen Human genes 0.000 description 5
- 108010035532 Collagen Proteins 0.000 description 5
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 5
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 239000011324 bead Substances 0.000 description 5
- 230000027455 binding Effects 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 229920001436 collagen Polymers 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 239000002207 metabolite Substances 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- 239000003981 vehicle Substances 0.000 description 5
- 208000024172 Cardiovascular disease Diseases 0.000 description 4
- 108010020070 Cytochrome P-450 CYP2B6 Proteins 0.000 description 4
- 102000009666 Cytochrome P-450 CYP2B6 Human genes 0.000 description 4
- 108010026925 Cytochrome P-450 CYP2C19 Proteins 0.000 description 4
- 108010000543 Cytochrome P-450 CYP2C9 Proteins 0.000 description 4
- 108010001237 Cytochrome P-450 CYP2D6 Proteins 0.000 description 4
- 102100029363 Cytochrome P450 2C19 Human genes 0.000 description 4
- 102100029358 Cytochrome P450 2C9 Human genes 0.000 description 4
- 102100021704 Cytochrome P450 2D6 Human genes 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 4
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 description 4
- 229920001213 Polysorbate 20 Polymers 0.000 description 4
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- HDYANYHVCAPMJV-LXQIFKJMSA-N UDP-alpha-D-glucuronic acid Chemical compound C([C@@H]1[C@H]([C@H]([C@@H](O1)N1C(NC(=O)C=C1)=O)O)O)OP(O)(=O)OP(O)(=O)O[C@H]1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O HDYANYHVCAPMJV-LXQIFKJMSA-N 0.000 description 4
- HDYANYHVCAPMJV-UHFFFAOYSA-N Uridine diphospho-D-glucuronic acid Natural products O1C(N2C(NC(=O)C=C2)=O)C(O)C(O)C1COP(O)(=O)OP(O)(=O)OC1OC(C(O)=O)C(O)C(O)C1O HDYANYHVCAPMJV-UHFFFAOYSA-N 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 238000004364 calculation method Methods 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 238000001802 infusion Methods 0.000 description 4
- 208000036971 interstitial lung disease 2 Diseases 0.000 description 4
- 238000000021 kinase assay Methods 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 235000019341 magnesium sulphate Nutrition 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 230000002503 metabolic effect Effects 0.000 description 4
- 210000000651 myofibroblast Anatomy 0.000 description 4
- 125000004043 oxo group Chemical group O=* 0.000 description 4
- 230000026731 phosphorylation Effects 0.000 description 4
- 238000006366 phosphorylation reaction Methods 0.000 description 4
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 4
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 239000003642 reactive oxygen metabolite Substances 0.000 description 4
- 238000011084 recovery Methods 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- VWSFOEHQKFAAGN-UHFFFAOYSA-N 5-(4-cyclopropylimidazol-1-yl)-2-fluoro-4-methylbenzonitrile Chemical compound CC1=CC(F)=C(C#N)C=C1N1C=C(C2CC2)N=C1 VWSFOEHQKFAAGN-UHFFFAOYSA-N 0.000 description 3
- GPWZGVIWTGDEMY-UHFFFAOYSA-N 5-amino-2-fluoro-4-methylbenzonitrile Chemical compound CC1=CC(F)=C(C#N)C=C1N GPWZGVIWTGDEMY-UHFFFAOYSA-N 0.000 description 3
- 102000007469 Actins Human genes 0.000 description 3
- 108010085238 Actins Proteins 0.000 description 3
- 208000023275 Autoimmune disease Diseases 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 108010000561 Cytochrome P-450 CYP2C8 Proteins 0.000 description 3
- 208000009011 Cytochrome P-450 CYP3A Inhibitors Diseases 0.000 description 3
- 102100029359 Cytochrome P450 2C8 Human genes 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 206010020772 Hypertension Diseases 0.000 description 3
- 108060001084 Luciferase Proteins 0.000 description 3
- 239000005089 Luciferase Substances 0.000 description 3
- 102000001708 Protein Isoforms Human genes 0.000 description 3
- 108010029485 Protein Isoforms Proteins 0.000 description 3
- 102000001253 Protein Kinase Human genes 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 108010090804 Streptavidin Proteins 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 238000009825 accumulation Methods 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 108010004469 allophycocyanin Proteins 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 238000000502 dialysis Methods 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 125000004438 haloalkoxy group Chemical group 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- 210000003494 hepatocyte Anatomy 0.000 description 3
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 3
- 230000002757 inflammatory effect Effects 0.000 description 3
- 210000000231 kidney cortex Anatomy 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 3
- 210000004072 lung Anatomy 0.000 description 3
- UEGPKNKPLBYCNK-UHFFFAOYSA-L magnesium acetate Chemical compound [Mg+2].CC([O-])=O.CC([O-])=O UEGPKNKPLBYCNK-UHFFFAOYSA-L 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 210000003205 muscle Anatomy 0.000 description 3
- 230000009871 nonspecific binding Effects 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 230000036542 oxidative stress Effects 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 238000010791 quenching Methods 0.000 description 3
- 230000000171 quenching effect Effects 0.000 description 3
- 201000002793 renal fibrosis Diseases 0.000 description 3
- 238000001356 surgical procedure Methods 0.000 description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 3
- 241000701161 unidentified adenovirus Species 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- XMAYWYJOQHXEEK-OZXSUGGESA-N (2R,4S)-ketoconazole Chemical compound C1CN(C(=O)C)CCN1C(C=C1)=CC=C1OC[C@@H]1O[C@@](CN2C=NC=C2)(C=2C(=CC(Cl)=CC=2)Cl)OC1 XMAYWYJOQHXEEK-OZXSUGGESA-N 0.000 description 2
- UUUHXMGGBIUAPW-UHFFFAOYSA-N 1-[1-[2-[[5-amino-2-[[1-[5-(diaminomethylideneamino)-2-[[1-[3-(1h-indol-3-yl)-2-[(5-oxopyrrolidine-2-carbonyl)amino]propanoyl]pyrrolidine-2-carbonyl]amino]pentanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-methylpentanoyl]pyrrolidine-2-carbon Chemical compound C1CCC(C(=O)N2C(CCC2)C(O)=O)N1C(=O)C(C(C)CC)NC(=O)C(CCC(N)=O)NC(=O)C1CCCN1C(=O)C(CCCN=C(N)N)NC(=O)C1CCCN1C(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C1CCC(=O)N1 UUUHXMGGBIUAPW-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- AXTUTVOXCLUJMK-UHFFFAOYSA-N 5-(4-cyclopropylimidazol-1-yl)-2-fluoro-4-methylbenzoic acid;hydrochloride Chemical compound Cl.CC1=CC(F)=C(C(O)=O)C=C1N1C=C(C2CC2)N=C1 AXTUTVOXCLUJMK-UHFFFAOYSA-N 0.000 description 2
- YSWOWAIEZYDOGB-UHFFFAOYSA-N 5-(5-cyclopropyl-2-sulfanylidene-1h-imidazol-3-yl)-2-fluoro-4-methylbenzonitrile Chemical compound CC1=CC(F)=C(C#N)C=C1N1C(S)=NC(C2CC2)=C1 YSWOWAIEZYDOGB-UHFFFAOYSA-N 0.000 description 2
- KWHGHFBULZPCCX-UHFFFAOYSA-N 5-[(2-cyclopropyl-2-oxoethyl)amino]-2-fluoro-4-methylbenzonitrile Chemical compound CC1=CC(F)=C(C#N)C=C1NCC(=O)C1CC1 KWHGHFBULZPCCX-UHFFFAOYSA-N 0.000 description 2
- DNCLVDGUXUSPTL-UHFFFAOYSA-N 5-bromo-4-fluoro-2-methylaniline Chemical compound CC1=CC(F)=C(Br)C=C1N DNCLVDGUXUSPTL-UHFFFAOYSA-N 0.000 description 2
- VFMMPHCGEFXGIP-UHFFFAOYSA-N 7,8-Benzoflavone Chemical compound O1C2=C3C=CC=CC3=CC=C2C(=O)C=C1C1=CC=CC=C1 VFMMPHCGEFXGIP-UHFFFAOYSA-N 0.000 description 2
- 102000015936 AP-1 transcription factor Human genes 0.000 description 2
- 108050004195 AP-1 transcription factor Proteins 0.000 description 2
- 108010005094 Advanced Glycation End Products Proteins 0.000 description 2
- 108010009551 Alamethicin Proteins 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 101100497948 Caenorhabditis elegans cyn-1 gene Proteins 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 2
- 208000002249 Diabetes Complications Diseases 0.000 description 2
- 206010012655 Diabetic complications Diseases 0.000 description 2
- 241000588724 Escherichia coli Species 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 229940126656 GS-4224 Drugs 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 2
- 239000007993 MOPS buffer Substances 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 101710164337 Mitogen-activated protein kinase kinase kinase 5 Proteins 0.000 description 2
- 235000009421 Myristica fragrans Nutrition 0.000 description 2
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 2
- 102000004270 Peptidyl-Dipeptidase A Human genes 0.000 description 2
- 108090000882 Peptidyl-Dipeptidase A Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 239000004793 Polystyrene Substances 0.000 description 2
- 238000011529 RT qPCR Methods 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 2
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 2
- 229960000583 acetic acid Drugs 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- LGHSQOCGTJHDIL-UTXLBGCNSA-N alamethicin Chemical compound N([C@@H](C)C(=O)NC(C)(C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)NC(C)(C)C(=O)N[C@H](C(=O)NC(C)(C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)NC(C)(C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NC(C)(C)C(=O)NC(C)(C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](CO)CC=1C=CC=CC=1)C(C)C)C(=O)C(C)(C)NC(=O)[C@@H]1CCCN1C(=O)C(C)(C)NC(C)=O LGHSQOCGTJHDIL-UTXLBGCNSA-N 0.000 description 2
- 239000000908 ammonium hydroxide Substances 0.000 description 2
- 239000012491 analyte Substances 0.000 description 2
- 239000012911 assay medium Substances 0.000 description 2
- 230000001363 autoimmune Effects 0.000 description 2
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 2
- 238000012742 biochemical analysis Methods 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 230000017531 blood circulation Effects 0.000 description 2
- 239000006143 cell culture medium Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 238000011260 co-administration Methods 0.000 description 2
- 238000011284 combination treatment Methods 0.000 description 2
- 230000002860 competitive effect Effects 0.000 description 2
- 230000002596 correlated effect Effects 0.000 description 2
- 230000000875 corresponding effect Effects 0.000 description 2
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 2
- 238000009509 drug development Methods 0.000 description 2
- 230000002526 effect on cardiovascular system Effects 0.000 description 2
- HKSZLNNOFSGOKW-UHFFFAOYSA-N ent-staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 HKSZLNNOFSGOKW-UHFFFAOYSA-N 0.000 description 2
- 238000011067 equilibration Methods 0.000 description 2
- 230000001747 exhibiting effect Effects 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 238000003306 harvesting Methods 0.000 description 2
- 208000027866 inflammatory disease Diseases 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 229960002725 isoflurane Drugs 0.000 description 2
- 229960004125 ketoconazole Drugs 0.000 description 2
- 239000001115 mace Substances 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 208000030159 metabolic disease Diseases 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 210000001589 microsome Anatomy 0.000 description 2
- DDLIGBOFAVUZHB-UHFFFAOYSA-N midazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NC=C2CN=C1C1=CC=CC=C1F DDLIGBOFAVUZHB-UHFFFAOYSA-N 0.000 description 2
- 229960003793 midazolam Drugs 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 208000015122 neurodegenerative disease Diseases 0.000 description 2
- 230000000626 neurodegenerative effect Effects 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 238000003305 oral gavage Methods 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 102000002574 p38 Mitogen-Activated Protein Kinases Human genes 0.000 description 2
- 108010068338 p38 Mitogen-Activated Protein Kinases Proteins 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 230000003285 pharmacodynamic effect Effects 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 229920000139 polyethylene terephthalate Polymers 0.000 description 2
- 239000005020 polyethylene terephthalate Substances 0.000 description 2
- 229920002223 polystyrene Polymers 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 239000008057 potassium phosphate buffer Substances 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 108060006633 protein kinase Proteins 0.000 description 2
- LOUPRKONTZGTKE-LHHVKLHASA-N quinidine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@H]2[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-LHHVKLHASA-N 0.000 description 2
- 208000023504 respiratory system disease Diseases 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 229940054269 sodium pyruvate Drugs 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 239000012265 solid product Substances 0.000 description 2
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical compound C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 2
- CGPUWJWCVCFERF-UHFFFAOYSA-N staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(OC)O1 CGPUWJWCVCFERF-UHFFFAOYSA-N 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 229960003604 testosterone Drugs 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 208000037999 tubulointerstitial fibrosis Diseases 0.000 description 2
- IGLYMJRIWWIQQE-QUOODJBBSA-N (1S,2R)-2-phenylcyclopropan-1-amine (1R,2S)-2-phenylcyclopropan-1-amine Chemical compound N[C@H]1C[C@@H]1C1=CC=CC=C1.N[C@@H]1C[C@H]1C1=CC=CC=C1 IGLYMJRIWWIQQE-QUOODJBBSA-N 0.000 description 1
- QHSMEGADRFZVNE-UHFFFAOYSA-N 1-hydroxymidazolam Chemical compound C12=CC(Cl)=CC=C2N2C(CO)=NC=C2CN=C1C1=CC=CC=C1F QHSMEGADRFZVNE-UHFFFAOYSA-N 0.000 description 1
- ZMPAPJBFYQSNFM-UHFFFAOYSA-N 1-sulfanylimidazole Chemical compound SN1C=CN=C1 ZMPAPJBFYQSNFM-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- KISWVXRQTGLFGD-UHFFFAOYSA-N 2-[[2-[[6-amino-2-[[2-[[2-[[5-amino-2-[[2-[[1-[2-[[6-amino-2-[(2,5-diamino-5-oxopentanoyl)amino]hexanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-(diaminomethylideneamino)p Chemical compound C1CCN(C(=O)C(CCCN=C(N)N)NC(=O)C(CCCCN)NC(=O)C(N)CCC(N)=O)C1C(=O)NC(CO)C(=O)NC(CCC(N)=O)C(=O)NC(CCCN=C(N)N)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(C(=O)NC(CC(C)C)C(O)=O)CC1=CC=C(O)C=C1 KISWVXRQTGLFGD-UHFFFAOYSA-N 0.000 description 1
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 1
- WCCCDMWRBVVYCQ-UHFFFAOYSA-N 2-bromo-1-cyclopropylethanone Chemical compound BrCC(=O)C1CC1 WCCCDMWRBVVYCQ-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- SJRHYONYKZIRPM-UHFFFAOYSA-N 4-hydroxytolbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(CO)C=C1 SJRHYONYKZIRPM-UHFFFAOYSA-N 0.000 description 1
- RZTAMFZIAATZDJ-HNNXBMFYSA-N 5-o-ethyl 3-o-methyl (4s)-4-(2,3-dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound CCOC(=O)C1=C(C)NC(C)=C(C(=O)OC)[C@@H]1C1=CC=CC(Cl)=C1Cl RZTAMFZIAATZDJ-HNNXBMFYSA-N 0.000 description 1
- 102100031126 6-phosphogluconolactonase Human genes 0.000 description 1
- 108010029731 6-phosphogluconolactonase Proteins 0.000 description 1
- NDCWHEDPSFRTDA-VEHVCKHKSA-N 6a-hydrox-ypaclitaxel Chemical compound C12C3(OC(C)=O)COC3C(O)C(O)C2(C)C(=O)C(OC(=O)C)C(C2(C)C)=C(C)C(OC(=O)C(O)C(NC(=O)C=3C=CC=CC=3)C=3C=CC=CC=3)CC2(O)C1OC(=O)[13C]1=[13CH][13CH]=[13CH][13CH]=[13CH]1 NDCWHEDPSFRTDA-VEHVCKHKSA-N 0.000 description 1
- CRCWUBLTFGOMDD-UHFFFAOYSA-N 7-ethoxyresorufin Chemical compound C1=CC(=O)C=C2OC3=CC(OCC)=CC=C3N=C21 CRCWUBLTFGOMDD-UHFFFAOYSA-N 0.000 description 1
- 229940127254 ASK1 inhibitor Drugs 0.000 description 1
- 208000030090 Acute Disease Diseases 0.000 description 1
- 206010001580 Albuminuria Diseases 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 239000005528 B01AC05 - Ticlopidine Substances 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000002947 C09CA04 - Irbesartan Substances 0.000 description 1
- 101150041968 CDC13 gene Proteins 0.000 description 1
- 101150022946 CYP3 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 229940126639 Compound 33 Drugs 0.000 description 1
- 108010074922 Cytochrome P-450 CYP1A2 Proteins 0.000 description 1
- 208000006075 Cytochrome P-450 CYP2B6 Inhibitors Diseases 0.000 description 1
- 208000007667 Cytochrome P-450 CYP2C19 Inhibitors Diseases 0.000 description 1
- 208000006619 Cytochrome P-450 CYP2C8 Inhibitors Diseases 0.000 description 1
- 208000005487 Cytochrome P-450 CYP2C9 Inhibitors Diseases 0.000 description 1
- 208000007220 Cytochrome P-450 CYP2D6 Inhibitors Diseases 0.000 description 1
- 102100026533 Cytochrome P450 1A2 Human genes 0.000 description 1
- NBSCHQHZLSJFNQ-GASJEMHNSA-N D-Glucose 6-phosphate Chemical compound OC1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H](O)[C@H]1O NBSCHQHZLSJFNQ-GASJEMHNSA-N 0.000 description 1
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine Chemical compound NCCCC[C@@H](N)C(O)=O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 239000012848 Dextrorphan Substances 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 101100137368 Dictyostelium discoideum cypD gene Proteins 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- 108010039471 Fas Ligand Protein Proteins 0.000 description 1
- 102000015212 Fas Ligand Protein Human genes 0.000 description 1
- 102000016359 Fibronectins Human genes 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- VFRROHXSMXFLSN-UHFFFAOYSA-N Glc6P Natural products OP(=O)(O)OCC(O)C(O)C(O)C(O)C=O VFRROHXSMXFLSN-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 108010018962 Glucosephosphate Dehydrogenase Proteins 0.000 description 1
- 108060003393 Granulin Proteins 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 101000628647 Homo sapiens Serine/threonine-protein kinase 24 Proteins 0.000 description 1
- 101000880439 Homo sapiens Serine/threonine-protein kinase 3 Proteins 0.000 description 1
- 101001050288 Homo sapiens Transcription factor Jun Proteins 0.000 description 1
- AKOAEVOSDHIVFX-UHFFFAOYSA-N Hydroxybupropion Chemical compound OCC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 AKOAEVOSDHIVFX-UHFFFAOYSA-N 0.000 description 1
- OQPLORUDZLXXPD-UHFFFAOYSA-N Hydroxymephenytoin Chemical compound C=1C=C(O)C=CC=1C1(CC)NC(=O)N(C)C1=O OQPLORUDZLXXPD-UHFFFAOYSA-N 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- 238000012404 In vitro experiment Methods 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 1
- MKXZASYAUGDDCJ-SZMVWBNQSA-N LSM-2525 Chemical compound C1CCC[C@H]2[C@@]3([H])N(C)CC[C@]21C1=CC(OC)=CC=C1C3 MKXZASYAUGDDCJ-SZMVWBNQSA-N 0.000 description 1
- 241000254158 Lampyridae Species 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 102000001291 MAP Kinase Kinase Kinase Human genes 0.000 description 1
- 108030005453 Mitogen-activated protein kinase kinase kinases Proteins 0.000 description 1
- UCHDWCPVSPXUMX-TZIWLTJVSA-N Montelukast Chemical compound CC(C)(O)C1=CC=CC=C1CC[C@H](C=1C=C(\C=C\C=2N=C3C=C(Cl)C=CC3=CC=2)C=CC=1)SCC1(CC(O)=O)CC1 UCHDWCPVSPXUMX-TZIWLTJVSA-N 0.000 description 1
- 102000047918 Myelin Basic Human genes 0.000 description 1
- 101710107068 Myelin basic protein Proteins 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- 101800000135 N-terminal protein Proteins 0.000 description 1
- 229910017833 NH2NH2.H2O Inorganic materials 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 239000005480 Olmesartan Substances 0.000 description 1
- 101800001452 P1 proteinase Proteins 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 235000006040 Prunus persica var persica Nutrition 0.000 description 1
- 240000006413 Prunus persica var. persica Species 0.000 description 1
- 208000001647 Renal Insufficiency Diseases 0.000 description 1
- 206010062237 Renal impairment Diseases 0.000 description 1
- 206010061481 Renal injury Diseases 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 238000003120 Steady-Glo Luciferase Assay System Methods 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- JLRGJRBPOGGCBT-UHFFFAOYSA-N Tolbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(C)C=C1 JLRGJRBPOGGCBT-UHFFFAOYSA-N 0.000 description 1
- 102100023132 Transcription factor Jun Human genes 0.000 description 1
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102100040247 Tumor necrosis factor Human genes 0.000 description 1
- 206010046406 Ureteric obstruction Diseases 0.000 description 1
- HSCJRCZFDFQWRP-UHFFFAOYSA-N Uridindiphosphoglukose Natural products OC1C(O)C(O)C(CO)OC1OP(O)(=O)OP(O)(=O)OCC1C(O)C(O)C(N2C(NC(=O)C=C2)=O)O1 HSCJRCZFDFQWRP-UHFFFAOYSA-N 0.000 description 1
- 239000003070 absorption delaying agent Substances 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 229960004308 acetylcysteine Drugs 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 206010069351 acute lung injury Diseases 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229960000528 amlodipine Drugs 0.000 description 1
- HTIQEAQVCYTUBX-UHFFFAOYSA-N amlodipine Chemical compound CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl HTIQEAQVCYTUBX-UHFFFAOYSA-N 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000001430 anti-depressive effect Effects 0.000 description 1
- 230000003064 anti-oxidating effect Effects 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 229940005513 antidepressants Drugs 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940030600 antihypertensive agent Drugs 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 239000002249 anxiolytic agent Substances 0.000 description 1
- 239000006286 aqueous extract Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 230000003190 augmentative effect Effects 0.000 description 1
- 239000012148 binding buffer Substances 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 229960001058 bupropion Drugs 0.000 description 1
- SNPPWIUOZRMYNY-UHFFFAOYSA-N bupropion Chemical compound CC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 SNPPWIUOZRMYNY-UHFFFAOYSA-N 0.000 description 1
- QWCRAEMEVRGPNT-UHFFFAOYSA-N buspirone Chemical compound C1C(=O)N(CCCCN2CCN(CC2)C=2N=CC=CN=2)C(=O)CC21CCCC2 QWCRAEMEVRGPNT-UHFFFAOYSA-N 0.000 description 1
- 229960002495 buspirone Drugs 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000007894 caplet Substances 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 230000005800 cardiovascular problem Effects 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 239000013553 cell monolayer Substances 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940000425 combination drug Drugs 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 230000001054 cortical effect Effects 0.000 description 1
- 230000001186 cumulative effect Effects 0.000 description 1
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 description 1
- 101150055214 cyp1a1 gene Proteins 0.000 description 1
- 230000016396 cytokine production Effects 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 229960001985 dextromethorphan Drugs 0.000 description 1
- JAQUASYNZVUNQP-PVAVHDDUSA-N dextrorphan Chemical compound C1C2=CC=C(O)C=C2[C@@]23CCN(C)[C@@H]1[C@H]2CCCC3 JAQUASYNZVUNQP-PVAVHDDUSA-N 0.000 description 1
- 229950006878 dextrorphan Drugs 0.000 description 1
- 125000004663 dialkyl amino group Chemical group 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000012149 elution buffer Substances 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 208000028208 end stage renal disease Diseases 0.000 description 1
- 201000000523 end stage renal failure Diseases 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 238000013401 experimental design Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 229960003580 felodipine Drugs 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 230000003176 fibrotic effect Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 230000024924 glomerular filtration Effects 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 1
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 201000001421 hyperglycemia Diseases 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 238000003364 immunohistochemistry Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 229960002198 irbesartan Drugs 0.000 description 1
- YCPOHTHPUREGFM-UHFFFAOYSA-N irbesartan Chemical compound O=C1N(CC=2C=CC(=CC=2)C=2C(=CC=CC=2)C=2[N]N=NN=2)C(CCCC)=NC21CCCC2 YCPOHTHPUREGFM-UHFFFAOYSA-N 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 201000006370 kidney failure Diseases 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 238000002350 laparotomy Methods 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- RLAWWYSOJDYHDC-BZSNNMDCSA-N lisinopril Chemical compound C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 RLAWWYSOJDYHDC-BZSNNMDCSA-N 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 229960000906 mephenytoin Drugs 0.000 description 1
- GMHKMTDVRCWUDX-UHFFFAOYSA-N mephenytoin Chemical compound C=1C=CC=CC=1C1(CC)NC(=O)N(C)C1=O GMHKMTDVRCWUDX-UHFFFAOYSA-N 0.000 description 1
- 210000003584 mesangial cell Anatomy 0.000 description 1
- 238000006241 metabolic reaction Methods 0.000 description 1
- OHIHEJTUXNQOPM-UHFFFAOYSA-N methyl 6-aminopyridine-2-carboxylate Chemical compound COC(=O)C1=CC=CC(N)=N1 OHIHEJTUXNQOPM-UHFFFAOYSA-N 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 229960005127 montelukast Drugs 0.000 description 1
- 230000000877 morphologic effect Effects 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 201000002674 obstructive nephropathy Diseases 0.000 description 1
- VTRAEEWXHOVJFV-UHFFFAOYSA-N olmesartan Chemical compound CCCC1=NC(C(C)(C)O)=C(C(O)=O)N1CC1=CC=C(C=2C(=CC=CC=2)C=2NN=NN=2)C=C1 VTRAEEWXHOVJFV-UHFFFAOYSA-N 0.000 description 1
- 229960005117 olmesartan Drugs 0.000 description 1
- 230000008816 organ damage Effects 0.000 description 1
- 230000004783 oxidative metabolism Effects 0.000 description 1
- UHBGYFCCKRAEHA-UHFFFAOYSA-N p-methylbenzamide Natural products CC1=CC=C(C(N)=O)C=C1 UHBGYFCCKRAEHA-UHFFFAOYSA-N 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 229940056360 penicillin g Drugs 0.000 description 1
- 239000008191 permeabilizing agent Substances 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 210000002381 plasma Anatomy 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 231100000857 poor renal function Toxicity 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- ZNNZYHKDIALBAK-UHFFFAOYSA-M potassium thiocyanate Chemical compound [K+].[S-]C#N ZNNZYHKDIALBAK-UHFFFAOYSA-M 0.000 description 1
- 229940116357 potassium thiocyanate Drugs 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 229960001455 quinapril Drugs 0.000 description 1
- JSDRRTOADPPCHY-HSQYWUDLSA-N quinapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CC2=CC=CC=C2C1)C(O)=O)CC1=CC=CC=C1 JSDRRTOADPPCHY-HSQYWUDLSA-N 0.000 description 1
- 229960001404 quinidine Drugs 0.000 description 1
- 229960003401 ramipril Drugs 0.000 description 1
- HDACQVRGBOVJII-JBDAPHQKSA-N ramipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@@H]2CCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 HDACQVRGBOVJII-JBDAPHQKSA-N 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 239000012925 reference material Substances 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 210000005084 renal tissue Anatomy 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- YIDDLAAKOYYGJG-UHFFFAOYSA-N selonsertib Chemical compound CC(C)N1C=NN=C1C1=CC=CC(NC(=O)C=2C(=CC(C)=C(C=2)N2C=C(N=C2)C2CC2)F)=N1 YIDDLAAKOYYGJG-UHFFFAOYSA-N 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 230000035882 stress Effects 0.000 description 1
- QWCJHSGMANYXCW-UHFFFAOYSA-N sulfaphenazole Chemical compound C1=CC(N)=CC=C1S(=O)(=O)NC1=CC=NN1C1=CC=CC=C1 QWCJHSGMANYXCW-UHFFFAOYSA-N 0.000 description 1
- 229960004818 sulfaphenazole Drugs 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 238000004885 tandem mass spectrometry Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- PHWBOXQYWZNQIN-UHFFFAOYSA-N ticlopidine Chemical compound ClC1=CC=CC=C1CN1CC(C=CS2)=C2CC1 PHWBOXQYWZNQIN-UHFFFAOYSA-N 0.000 description 1
- 229960005001 ticlopidine Drugs 0.000 description 1
- 229960005371 tolbutamide Drugs 0.000 description 1
- 229960003741 tranylcypromine Drugs 0.000 description 1
- 150000008523 triazolopyridines Chemical class 0.000 description 1
- 210000000626 ureter Anatomy 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4436—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a heterocyclic ring having sulfur as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/56—Amides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to a novel compound for use in the treatment of ASK1- mediated diseases.
- the invention also relates to intermediates for its preparation and to pharmaceutical compositions containing said novel compound.
- Apoptosis signal -regulating kinase 1 is a member of the mitogen-activated protein kinase kinase kinase ("MAP3K") family that activates the c-Jun N-terminal protein kinase (“JNK”) and p38 MAP kinase (Ichijo, H., Nishida, E., e, K., Dijke, P. T., Saitoh, M., Moriguchi, T., Matsumoto, K., Miyazono, K., and Gotoh, Y. (1997) Science, 275, 90-94).
- MAP3K mitogen-activated protein kinase kinase kinase kinase
- JNK c-Jun N-terminal protein kinase
- JNK c-Jun N-terminal protein kinase
- JNK c-Jun N-terminal protein kinase
- ASK1 is activated by a variety of stimuli including oxidative stress, reactive oxygen species (ROS), LPS, TNF-a, FasL, ER stress, and increased intracellular calcium concentrations (Hattori, K., Naguro, I., Runchel, C, and Ichijo, H. (2009) Cell Comm. Signal. 7: 1-10; Takeda, K., Noguchi, T., Naguro, I., and Ichijo, H. (2007) Annu. Rev. Pharmacol. Toxicol. 48: 1-8.27; Nagai, H., Noguchi, T., Takeda, K., and Ichijo, I. (2007) J. Biochem. Mol. Biol. 40: 1-6).
- ROS reactive oxygen species
- ASK1 activation and signaling have been reported to play an important role in a broad range of diseases including neurodegenerative, cardiovascular, inflammatory,
- ASK1 has been implicated in mediating organ damage following ischemia and reperfasion of the heart, brain, and kidney (Watanabe et al. (2005) BBRC 333, 562-567; Zhang et al, (2003) Life Sci 74-37-43; Terada et al. (2007) BBRC 364: 1043-49).
- ROS are reported be associated with increases of inflammatory cytokine production, fibrosis, apoptosis, and necrosis in the kidney.
- Sah DK Winocour P, Farrington K. Oxidative stress in early diabetic nephropathy: fueling the fire. Nat Rev Endocrinol 201 1 Mar;7(3): 176- 184; Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001 Dec 13; 414(6865):813-820; Mimura I, Nangaku M.
- oxidative stress facilitates the formation of advanced glycation end-products (AGEs) that cause further renal injury and production of ROS.
- AGEs advanced glycation end-products
- ROS ROS
- Tubulointerstitial fibrosis in the kidney is a strong predictor of progression to renal failure in patients with chronic kidney diseases (Schainuck LI, et al. Structural-functional correlations in renal disease. Part II: The correlations. Hum Pathol 1970; 1 : 631-641.).
- UUO Unilateral ureteral obstruction
- TGF- ⁇ transforming growth factor beta
- myofibroblasts which secrete matrix proteins such as collagen and fibronectin.
- the UUO model can be used to test for a drug's potential to treat chronic kidney disease by inhibiting renal fibrosis (Chevalier et al., Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy, Kidney International (2009) 75, 1 145-1152.
- ASK1 inhibitors have the potential to remedy or improve the lives of patients in need of treatment for diseases or conditions such as neurodegenerative, cardiovascular, inflammatory, autoimmune, and metabolic disorders.
- ASK1 inhibitors have the potential to treat cardio-renal diseases, including kidney disease, diabetic kidney disease, chronic kidney disease, fibrotic diseases (including lung and kidney fibrosis), respiratory diseases (including chronic obstructive pulmonary disease (COPD) and acute lung injury), acute and chronic liver diseases.
- COPD chronic obstructive pulmonary disease
- U.S. Publication No. 2007/0276050 describes methods for identifying AS 1 inhibitors useful for preventing and/or treating cardiovascular disease and methods for preventing and/or treating cardiovascular disease in an animal.
- WO2009027283 discloses triazolopyridine compounds, methods for preparation thereof and methods for treating autoimmune disorders, inflammatory diseases, cardiovascular diseases and neurodegenerative diseases.
- U.S. Patent Publication No. 2001/00095410A1 published January 13, 201 1, discloses compounds useful as ASK-1 inhibitors.
- U.S. Patent Publication No. 2001/00095410A1 relates to compounds of Formula (I):
- alkyl, cycloalkyl, heterocyclyl, phenyl, and phenoxy are optionally substituted by 1, 2, or 3 substituents selected from alkyl, cycloalkyl, alkoxy, hydroxyl, and halo;
- R 6 and R 7 are independently selected from the group consisting of hydrogen, Q- Ci5 alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl, all of which are optionally substituted with 1-3 substituents selected from halo, alkyl, mono- or dialkylamino, alkyl or aryl or heteroaryl amide, -CN, lower alkoxy, -CF 3 , aryl, and heteroaryl; or
- R is hydrogen, halo, cyano, alkoxy, or alkyl optionally substituted by halo;
- heteroaryl or heterocyclyl moiety includes at least one ring nitrogen atom
- X 5 and X 6 or X 6 and X 7 are joined to provide optionally substituted fused aryl or optionally substituted fused heteroaryl;
- X 5 , X 6 , X 7 , and X 8 are C(R 4 );
- At least one of X 2 , X 3 , X 4 , X 5 , X 6 , X 7 and X 8 is N.
- the present invention relates to a compound of the formula:
- the invention relates to the use of a compound of formula (I) in the treatment of a disease in a patient in need of treatment with an ASK1 inhibitor.
- the invention in another embodiment, relates to a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof, and one or more pharmaceutically acceptable carriers.
- the invention is a method of treating diabetic nephropathy, or complications of diabetes, comprising administering a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof, to a patient in need thereof.
- the invention relates to a method of treating kidney disease, or diabetic kidney disease comprising administering a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof, to a patient in need thereof.
- the invention in another embodiment, relates to a method of treating kidney fibrosis, lung fibrosis, or idiopathic pulmonary fibrosis (IPF) comprising administering a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof, to a patient in need thereof.
- the invention relates to a method of treating diabetic kidney disease, diabetic nephropathy, kidney fibrosis, liver fibrosis, or lung fibrosis comprising administering a therapeutically effective amount of a compound or salt of forumia (I), to a patient in need thereof.
- a method of treating diabetic kidney disease, diabetic nephropathy, kidney fibrosis, liver fibrosis, or lung fibrosis comprising administering a therapeutically effective amount of a compound or salt of forumia (I), to a patient in need thereof.
- the invention relates to intermediates useful for the synthesis of the compound of formula (I).
- the invention relates to the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof for the treatment of chronic kidney disease.
- the invention relates to the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof for the treatment of diabetic kidney disease.
- the invention relates to the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of chronic kidney disease. In yet another embodiment, the invention relates to the compound of formula (I) for use in therapy.
- Figure 1 is a bar graph showing the levels of Collagen IV in the kidney cortex of rats subjected to seven days of unilateral ureteral obstruction and treated with either vehicle, or compound of formula (I) at 1, 3, 10, or 30 mg/kg b.i.d. per day.
- Figure 2 shows representative images of kidney cortex sections stained with alpha- smooth muscle actin (a marker of activated myofibroblasts) from rats subjected to seven days of unilateral ureteral obstruction and treated with either vehicle, or compound of formula (I) at 1, 3, 10, or 30 mg/kg b.i.d. per day.
- alpha- smooth muscle actin a marker of activated myofibroblasts
- chronic kidney disease refers to progressive loss of kidney function over time typically months or even years.
- Chronic kidney disease is diagnosed by a competent care giver using appropriate information, tests or markers known to one of skill in the art.
- Chronic kidney disease includes by implication kidney disease.
- kidney disease refers to kidney disease caused by diabetes, exacerbated by diabetes, or co-presenting with diabetes. It is a form of chronic kidney disease occurring in approximately 30% of patients with diabetes. It is defined as diabetes with the presence of albuminuria and/or impaired renal function (i.e. decreased glomerular filtration rate ( See. de B, I, et al. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA 201 1 Jun 22; 305(24):2532-2539).
- pharmaceutically acceptable salt refers to salts of pharmaceutical compounds e.g. compound of formula (I) that retain the biological effectiveness and properties of the underlying compound, and which are not biologically or otherwise undesirable.
- acid addition salts and base addition salts are acid addition salts and base addition salts.
- Pharmaceutically acceptable acid addition salts may be prepared from inorganic and organic acids.
- salts derived from inorganic acids include but are not limited to hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
- Salts derived from organic acids include but are not limited to maleic acid, fumaric acid, tartaric acid, p- toluene-sulfonic acid, and the like.
- Bases useful for forming base addition salts are known to one of skill in the art.
- An example of a pharmaceutically acceptable salt of the compound of formula (I) is the hydrochloride salt of the compound of formula (I).
- pharmaceutically acceptable carrier includes excipients or agents such as solvents, diluents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents and the like that are not deleterious to the compound of the invention or use thereof.
- excipients or agents such as solvents, diluents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents and the like that are not deleterious to the compound of the invention or use thereof.
- the use of such carriers and agents to prepare compositions of pharmaceutically active substances is well known in the art (see, e.g., Remington's Pharmaceutical Sciences, Mace Publishing Co., Philadelphia, PA 17th Ed. (1985); and Modern Pharmaceutics, Marcel Dekker, Inc. 3rd Ed. (G.S. Banker & C.T. Rhodes, Eds.)
- cardiovascular diseases refers to diseases, related to the function of the kidney, that are caused or exacerbated by cardiovascular problems such as, for example, high blood pressure or hypertension. It is believed that hypertension is a major contributor to kidney disease.
- respiratory diseases refers to diseases including chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF).
- COPD chronic obstructive pulmonary disease
- IPF idiopathic pulmonary fibrosis
- therapeutically effective amount refers to an amount of the compound of formula (I) that is sufficient to effect treatment as defined below, when administered to a patient (particularly a human) in need of such treatment in one or more doses.
- the therapeutically effective amount will vary, depending upon the patient, the disease being treated, the weight and/or age of the patient, the severity of the disease, or the manner of administration, as determined by a qualified prescriber or care giver.
- treatment or “treating” means administering a compound or pharmaceutically acceptable salt of formula (I) for the purpose of:
- the invention relates to the use of the compound of formula (I) in treating chronic kidney disease comprising administering a therapeutically effective amount to a patient in need thereof.
- the invention relates to the use of the compound of formula (I) in treating diabetic kidney disease comprising administering a therapeutically effective amount to a patient in need thereof.
- the invention relates to the use of the compound of formula (I) in treating lung or kidney fibrosis comprising administering a therapeutically effective amount to a patient in need thereof.
- the half maximal inhibitory concentration (IC 5 o) of a therapeutic agent is the concentration of a therapeutic agent necessary to produce 50% of the maximum inhibition against a target enzyme. It is a desirable goal to discover a therapeutic agent, for example a compound that inhibits apoptosis signal-regulating kinase (ASK1) with a low IC 50 . In this manner, undesirable side effects are minimized by the ability to use a lower dose of the therapeutic agent to inhibit the ASK1 enzyme.
- a therapeutic agent for example a compound that inhibits apoptosis signal-regulating kinase (ASK1) with a low IC 50 . In this manner, undesirable side effects are minimized by the ability to use a lower dose of the therapeutic agent to inhibit the ASK1 enzyme.
- 3 ⁇ 4 is used to describe the affinity between a ligand (such as a therapeutic agent) and the corresponding kinase or receptor; i.e. a measure of how tightly a therapeutic agent binds to a particular kinase, for example the apoptosis signal-regulating kinase ASK1.
- a lower 3 ⁇ 4 is generally preferred in drug development.
- EC 5 0 is the concentration of a drug that achieves 50% maximal efficacy in the cell.
- the EC50 value translates to the concentration of a compound in the assay medium necessary to achieve 50% of the maximum efficacy.
- a lower EC 5 0 is generally preferred for drug development.
- a useful unit of measure associated with EC 5 0 is the protein binding adjusted EC 50 (PB adj -ECso as used herein). This value measures the amount of a drag e.g. compound of formula (I) correlated to the fraction of the drag that is unbound to protein which provides 50% maximal efficacy. This value measures the efficacy of the drug corrected for or correlated to the amount of drug that is available at the target site of action.
- Another desirable property is having a compound with a low cell membrane efflux ratio as determined by CACO cell permeability studies.
- An efflux ratio ((B/A) / (A/B)) less than 3.0 is preferred.
- a compound with a ratio greater than 3 is expected to undergo active rapid efflux from the cell and may not have sufficient duration in the cell to achieve maximal efficacy.
- Another desirable goal is to discover a drag that exhibits minimal off-target inhibition.
- a drug that minimally inhibits the Cyp450 (cytochrome p450) enzymes More particularly, a drug that is a weak inhibitor of cyp3A4, the most important of the P450 enzymes, is desired.
- a weak inhibitor is a compound that causes at least 1.25-fold but less than 2-fold increase in the plasma AUC values, or 20-50% decrease in clearance
- a measure useful for comparing cyp3 A4 inhibition among drug candidates is the ratio of Cyp3A4 inhibition and the protein binding adjusted EC 50 . This value gives an indication of the relative potential for cyp inhibition corrected for the protein binding adjusted EC 50 which is specific to each drag. A higher ratio in this measure is preferred as indicative of lower potential for cyp3A4 inhibition.
- objects of the present invention include but are not limited to the provision of a compound of formula (I) or pharmaceutically acceptable salt thereof, and methods of using the compound of formula (I) for the treatment of kidney disease, chronic kidney disease, diabetic kidney disease, diabetic nephropathy, kidney fibrosis or lung fibrosis.
- ACE angiotensin converting enzyme
- ARB angiontesin II receptor blockers
- the benefit of combination may be increased efficacy and/or reduced side effects for a component as the dose of that component may be adjusted down to reduce its side effects while benefiting from its efficacy augmented by the efficacy of the compound of formula (I) and/or other active component(s).
- Patients presenting with chronic kidney disease treatable with ASKI inhibitors such as compound of formula (I), may also exhibit conditions that benefit from co- administration (as directed by a qualified caregiver) of a therapeutic agent or agents that are antibiotic, analgesic, antidepressant and/or anti-anxiety agents in combination with compound of formula (I).
- Combination treatments may be administered simultaneously or one after the other within intervals as directed by a qualified caregiver or via a fixed dose (all active ingreditents are combined into the a single dosage form e.g. tablet) presentation of two or more active agents.
- the compound of the present invention may be administered in the form of a
- compositions that contain, as the active ingredient, the compound of formula (I), or a
- compositions may be administered alone or in combination with other therapeutic agents.
- Compositions may be prepared for delivery as solid tablets, capsules, caplets, ointments, skin patches, sustained release, fast disintegrating tablets, inhalation preparations, etc.
- Typical pharmaceutical compositions are prepared and/or administered using methods and/or processes well known in the pharmaceutical art (see, e.g., Remington's
- Formulations for combination treatments comprising the compound of formula (I) may be presented as fixed dose formulations e.g. tablets, elixirs, liquids, ointments, inhalants, gels, etc., using procedures known to one of skill in the art.
- compositions of the compound of formula (I) may be administered in either single or multiple doses by routes including, for example, rectal, buccal, intranasal and transdermal routes; by intra-arterial injection, intravenously, intraperitoneally, parenterally, intramuscularly, subcutaneously, orally, topically, as an inhalant, or via an impregnated or coated device such as a stent, for example, or an artery-inserted cylindrical polymer.
- routes of administration include oral, parental and intravenous administration.
- each dosage unit preferably contains from 1 mg to 500 mg of the compound of formula (I).
- a more preferred dose is from 1 mg to 250 mg of the compound of formula (I).
- Particularly preferred is a dose of the compound of formula (I) ranging from about 20 mg twice a day to about 50 mg twice a day. It will be understood, however, that the amount of the compound actually administered usually will be determined by a physician in light of the relevant circumstances including the condition to be treated, the chosen route of administration, co-administration compound if applicable, the age, weight, response of the individual patient, the severity of the patient's symptoms, and the like.
- 5-(4-cyclopropyl- 1 H-imidazol- 1 -yl)-N-(6-(4-isopropyl-4H- 1 ,2,4-triazol-3 -yl)pyridin-2-yl)-2- fluoro-4-methylbenzamide also known as 5-((4-cyclopropyl-lH-imdazol-l-yl)-2-fluoro-N-(6-(4- isopropyl-4H- 1 ,2,4-triazole-3 -yl)pyridine-2-yl)-4-methylbenzamide.
- the compound of the invention may be prepared using methods disclosed herein or modifications thereof which will be apparent given the disclosure herein.
- the synthesis of the compound of the invention may be accomplished as described in the following example. If available, reagents may be purchased commercially, e.g. from Sigma Aldrich or other chemical suppliers. Alternatively, reagents may be prepared using reaction schemes and methods known to one of skill in the art.
- solvent refers to a solvent inert under the conditions of the reaction being described in conjunction therewith (including, for example, benzene, toluene, acetonitrile, tetrahydrofuran (THF), dimethylformamide (DMF), chloroform, methylene chloride (or dichloromethane), diethyl ether, petroleum ether (PE), methanol, pyridine, ethyl acetate (EA) and the like.
- solvents used in the reactions of the present invention are inert organic solvents, and the reactions are carried out under an inert gas, preferably nitrogen.
- Compound C is a key intermediate for the synthesis of the compound of formula (I).
- an object of the present invention is also the provision of the intermediate compound C,
- the starting 5-bromo-4-fluoro-2-methylaniline (1) (20g, 98 mmol) was dissolved in anhydrous 1-methylpyrrolidinone (100 mL), and copper (I) cyanide (17.6g, 196 mmol) was added. The reaction was heated to 180°C for 3 hours, cooled to room temperature, and water (300 mL) and concentrated ammonium hydroxide (300 mL) added. The mixture was stirred for 30 minutes and extracted with EA (3 x 200 mL). The combined extracts were dried over magnesium sulfate, and the solvent was removed under reduced pressure.
- Step 6 Preparation of 5-(4-cvclopropyl- 1 H-imidazol- 1 -yl)-2-fluoro-N-(6-(4-isopropyl-4H- l,2,4-triazol-3-yl)pyridin-2-yl)-4-methylbenzamide - formula (I)
- ASKl Apoptosis Signal-Regulating Kinase 1 TR-FRET Kinase Assay (Biochemical IC 50 )
- the ability of compounds to inhibit ASKl kinase activity was determined using a time resolved fluorescence resonance energy transfer [TR-FRET] assay utilizing biotinylated myelin basic protein [biotin-MBP] as the protein substrate.
- TR-FRET time resolved fluorescence resonance energy transfer
- biotin-MBP biotinylated myelin basic protein
- a Beckman Biomek FX liquid handling robot was utilized to spot 2 ⁇ 11 of compounds in 2.44% aqueous DMSO into low volume 384-well polypropylene plates [Nunc, #267460] to give a final concentration of between ⁇ and 0.5nM compound in the kinase assay.
- a Deerac Fluidics Equator was used to dispense 3 ⁇ 11 of [Upstate Biotechnologies, #14-606, or the equivalent protein prepared in-house] and 0.1665ng/mL biotin-MBP [Upstate Biotechnologies, #13-111] in buffer (85mM MOPS, pH 7.0, 8.5mM Mg-acetate, 5% glycerol, 0.085% NP-40, 1.7mM DTT and 1.7mg/mL BSA) into the plates containing the spotted compounds.
- buffer 85mM MOPS, pH 7.0, 8.5mM Mg-acetate, 5% glycerol, 0.085% NP-40, 1.7mM DTT and 1.7mg/mL BSA
- the enzyme was allowed to pre-incubate with compound for 20 minutes prior to initiating the kinase reaction with the addition of 5 ⁇ 11 300 ⁇ ATP in buffer (50mM
- the Biomek FX was then used to transfer 1 ⁇ /well of each completed kinase reaction to the wells of an OptiPlate-1536 white polystyrene plate [PerkinElmer, #6004299] that contained 5 ⁇ , ⁇ 11 detection reagents (1.1 InM Eu-W1024 labeled anti-phosphothreonine antibody [PerkinElmer, #AD0094] and 55.56nM streptavidin allophycocyanin [PerkinElmer, #CR130-100] in lx LANCE detection buffer [PerkinElmer, #CR97-100]).
- the TR-FRET signal was then read on a Perkin Elmer Envision plate reader after incubating the plates at ambient temperature for 2 hours.
- the 100% inhibition positive control wells were generated by switching the order of addition of the EDTA and ATP solutions described above. These wells and 0% inhibition wells containing spots of 2.44% DMSO at the beginning of the assay were used in calculating the % inhibition for the test compounds.
- the assay measures the phosphorylation level of a biotinylated peptide substrate by the ASKl kinase using HTRF detection (6.1).
- This is a competitive, time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay, based on HTRF® KinEASETM-STK manual from Cisbio (6.1).
- Test compound 1 ⁇ STK3 peptide substrate, 4 nM of ASKl kinase are incubated with 10 mM MOP buffer, pH.
- the fluorescence is measured at 615 nm (Cryptate) and 665 nm (XL665) and a ratio of 665 nm/615 nm is calculated for each well.
- the resulting TR-FRET level (a ratio of 665 nm/615 nm) is proportional to the phosphorylation level.
- the degree of phosphorylation of peptide substrate was linear with time and concentration for the enzyme.
- the assay system yielded consistent results with regard to K m and specific activities for the enzyme. For inhibition experiments (IC 5 0 values), activities were performed with constant concentrations of ATP, peptide and several fixed concentrations of inhibitors. Staurosporine, the nonselective kinase inhibitor, was used as the positive control. All enzyme activity data are reported as an average of quadruplicate determination.
- x and y represent the concentration of inhibitors and enzyme activity, respectively.
- Enzyme activity is expressed as the amount of Phosphate incorporated into substrate peptide from ATP.
- Range is the maximum y range (no inhibitor, DMSO control) and s is a slope factor (6.2).
- the compound of formula (I) exhibited an IC50 of 3.2nM under this test condition.
- ASK1 Apoptosis Signal-Regulating Kinase 1
- Cell-based assay Cellular EC 5 0
- the cellular potency of compounds was assayed in cells stably expressing an AP- l :luciferase reporter construct (293/APl -Luc cells - Panomics Inc., 6519 Dumbarton Circle, Fremont, CA).
- Cells were infected with an adenovirus expressing kinase active AS 1 (631- 1381 of rat ASK1 cDNA), which will activate the AP-1 transcription factor and increase the expression of luciferase.
- Inhibitors of AS 1 will decrease the enzyme activity of AS 1 and therefore decrease the activity of AP-1 transcription factor and the expression of luciferase. 1.
- Aspirate media wash gently with ⁇ 12 mL sterile D-PBS, aspirate.
- Aspirate media from cell pellet add 20 to 30 mL CGM, resuspend pellet by pipeting 6X, pass through cell strainer to disperse clumps (if necessary), and count cells with hemocytometer.
- Process plates as follows: Set plates in laminar flow hood & uncover for 30 minutes at room temperature to cool.
- the 100% inhibition positive control wells were generated by infecting cells with an adenovirus expressing catalytically inactive ASK1 mutant with lysine to argine mutation at residue 709. Result
- the compound of formula (I) exhibits an EC 5 0 of 2.0 nM.
- kinase-tagged T7 phage strains were prepared in an E. coli host derived from the BL21 strain. E. coli were grown to log-phase and infected with T7 phage and incubated with shaking at 32°C until lysis. The lysates were centrifuged and filtered to remove cell debris. The remaining kinases were produced in HEK-293 cells and subsequently tagged with DNA for qPCR detection. Streptavidin-coated magnetic beads were treated with biotinylated small molecule ligands for 30 minutes at room temperature to generate affinity resins for kinase assays.
- the liganded beads were blocked with excess biotin and washed with blocking buffer
- Binding reactions were assembled by combining kinases, liganded affinity beads, and test compounds in lx binding buffer (20% SeaBlock, 0.17x PBS, 0.05% Tween 20, 6 mM DTT). All reactions were performed in polystyrene 96-well plates in a final volume of 0.135 mL. The assay plates were incubated at room temperature with shaking for 1 hour and the affinity beads were washed with wash buffer (lx PBS, 0.05% Tween 20).
- elution buffer lx PBS, 0.05% Tween 20, 0.5 ⁇ non-biotinylated affinity ligand
- Binding constants were calculated with a standard dose-response curve using the Hill equation.
- the compound of formula (I) exhibited a 3 ⁇ 4 of 0.24 nM. This data suggests that the compound of formula (I) binds potently to ASK1 receptor in the absence of ATP. Determination of Percent of Compound bound to Plasma
- ImL Teflon dialysis cells from Harvard Apparatus (Holliston, Mass, USA) were used in these experiments. Prior to the study, dialysis membrane was soaked for approximately one hour in 0.133 M phosphate buffer, pH 7.4. A nominal concentration of 2 ⁇ of compound was spiked into ImL of plasma or ImL of cell culture media. The total volume of liquid on each side of the cell was ImL. After 3 hours equilibration in a 37°C water bath, samples from each side of the cell were aliquoted into the appropriate vials containing either ImL of human plasma (cell culture media), or buffer. Sample vials were weighed and recorded.
- % Unbound 100(C f /C t ) where C f and C t are the post-dialysis buffer and plasma concentrations, respectively.
- the percent unbound measured in human plasma for the compound of formula (I) is 11.94 %
- Caco-2 cells were maintained in Dulbecco's Modification of Eagle's Medium (DMEM) with sodium pyruvate, Glutmax supplemented with 1% Pen/Strep, 1% NEAA and 10% fetal bovine serum in an incubator set at 37°C, 90% humidity and 5% C(3 ⁇ 4.
- Caco-2 cells between passage 62 and 72 were seeded at 2100 cells/well and were grown to confluence over at least 21- days on 24 well PET (polyethylene-terephthalate) plates (BD Biosciences).
- the receiver well contained HBSS buffer (lOmM HEPES, 15mM Glucose with pH adjusted to pH 6.5) supplemented with 1 % BSA pH adjusted to pH 7.4.
- TEER values were read to test membrane integrity. Buffers containing test compounds were added and 100 ⁇ of solution was taken at 1 and 2 hrs from the receiver compartment. Removed buffer was replaced with fresh buffer and a correction is applied to all calculations for the removed material. The experiment was carried out in replicate. All samples were
- % Recovery 100 x ((V r x R 12 o) + (V d x D I20 ))/ (V d x D 0 )
- dRIdt is the slope of the cumulative concentration in the receiver compartment versus time in ⁇ /s based on receiver concentrations measured at 60 and 120 minutes.
- V r and Vj is the volume in the receiver and donor compartment in cm 3 , respectively.
- A is the area of the cell monolayer (0.33 cm ).
- Do and D 12 o is the measured donor concentration at the beginning and end of the experiment, respectively.
- Rj 2 o is the receiver concentration at the end of the experiment (120 minutes).
- the compound of formula (I) was observed to have a CACO A ⁇ B value of 27; and a CACO B ⁇ A value of 35 resulting in a efflux ratio (B ⁇ A)/(A ⁇ B) of 1.3. Determination of Metabolic Stability in Hepatic Microsomal Fraction:
- NADPH NADPH
- UDP glucuronic acid UDPGA
- each reaction mixture was: 3 ⁇ test compound, 1 mg microsomal protein/mL, 5 mM UDPGA, 23.4 ⁇ g/mL alamethicin, 1.25 mM NADP, 3.3 mM glucose-6-phosphate, 0.4 U/mL glucose-6-phosphate dehydrogenase and 3.3 mM MgCl 2 in 50 mM potassium phosphate buffer, pH 7.4.
- 25 ⁇ aliquots of the reaction mixture were transferred to plates containing 250 ⁇ of IS/Q (quenching solution containing internal standard). After quenching, the plates were centrifuged at 3000 x g for 30 minutes, and 10 ⁇ . aliquots of the supernatant were analyzed using LC/MS to obtain analyte/internal standard peak area ratios.
- the predicted hepatic clearance was calculated as follows ⁇ reference 1 :
- Predicted hepatic extraction was then calculated by comparison of predicted hepatic clearance to hepatic blood flow.
- a compound was considered stable if the reduction of substrate concentration was ⁇ 10% over the course of the incubation (corresponding to an extrapolated half-life of > 395 min in microsomal fractions and > 39.5 hr in hepatocytes).
- the predicted hepatic clearance in human as determined from in vitro experiments in microsomal fractions is 0.1 L/h/kg.
- test compound for IV administration the test compound was formulated in 60:40 PEG 400:water with 1 equivalent HC1 at 0.5 mg/mL.
- the formulation was a solution.
- test compound for PO (oral) administration, the test compound was formulated in 5/75/10/10 ethanol/PG/solutol/water at 2.5 mg/mL.
- the formulation was a solution.
- IV and PO dosing groups each consisted of 3 male SD rats. At dosing, the animals generally weighed between 0.317 and 0.355 kg. The animals were fasted overnight prior to dose administration and up to 4 hr after dosing. Dosing
- test compound was administered by intravenous infusion over 30 minutes.
- the rate of infusion was adjusted according to the body weight of each animal to deliver a dose of 1 mg/kg at 2 mL/kg.
- test article was administered by oral gavage at 2 mL/kg for a dose of 5.0 mg/kg.
- Serial venous blood samples (approximately 0.4 mL each) were taken at specified time points after dosing from each animal.
- the blood samples were collected into VacutainerTM tubes (Becton-Disekinson Corp, New Jersey, USA) containing EDTA as the anti-coagulant and were immediately placed on wet ice pending centrifugation for plasma.
- the compound of formula (I) exhibited a CL of 0.09 L/hr/kg; an oral bioavailability of 75 %; ti/2 of 5.07 hr and a Vss of 0.55 L/kg in rats.
- CYPIA CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 (2 substrates).
- Test compound (0.1 ⁇ - 25 ⁇ ) is incubated with human liver microsomes and NADPH in the presence of a cytochrome P450 isoform-specific probe substrate.
- a cytochrome P450 isoform-specific probe substrate For the CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 specific reactions, the metabolites are monitored by mass spectrometry. CYPIA activity is monitored by measuring the formation of a fluorescent metabolite. A decrease in the formation of the metabolite compared to the vehicle control is used to calculate an IC50 value (test compound concentration which produces 50% inhibition).
- the selective CYP1A inhibitor, alpha-naphthoflavone, is screened alongside the test compounds as a positive control.
- the selective CYP2B6 inhibitor, ticlopidine is screened alongside the test compounds as a positive control.
- the selective CYP2C8 inhibitor, montelukast is screened alongside the test compounds as a positive control.
- the selective CYP2C19 inhibitor, tranylcypromine, is screened alongside the test compounds as a positive control.
- the selective CYP2D6 inhibitor, quinidine is screened alongside the test compounds as a positive control.
- the selective CYP3A4 inhibitor, ketoconazole is screened alongside the test compounds as a positive control.
- CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 incubations the reactions are terminated by methanol.
- the samples are then centrifuged, and the supernatants are combined, for the simultaneous analysis of 4- hydroxytolbutamide, 4-hydroxymephenytoin, dextrorphan, and 1-hydroxymidazolam by LC- MS/MS.
- Hydroxybupropion, 6a-hydroxypaclitaxel and 6B-hydroxytestosterone are analysed separately by LC-MS/MS.
- a decrease in the formation of the metabolites compared to vehicle control is used to calculate an IC50 value (test compound concentration which produces 50% inhibition).
- UUO right ureter
- Rats were allowed to recover in a clean, heated cage before being returned to normal vivarium conditions. Rats were administered compounds at the dose described above twice daily (at 12 hour intervals) for the subsequent 7 days. On day 7 following surgery, rats were anesthetized with isoflurane and serum, plasma, and urine collected. Animals were then euthanized, the kidneys harvested, and renal cortical biopsies collected for morphological, histological, and biochemical analysis. All tissues for biochemical analysis are flash- frozen in liquid nitrogen and stored at -80°C, tissues for histological analysis were fixed in 10% neutral buffered formalin
- Renal fibrosis was evaluated by measuring the amount of collagen IV in the kidney by an
- the compound of formula (I) was found to significantly reduce kidney Collagen IV induction (figure 1) and accumulation of alpha-smooth muscle positive myofibroblasts (figure 2) at doses of 3 to 30 mg/kg.
- the compound of formula (I) has an EC 50 that is comparable to that of Compound A.
- the compound of formula (I) has a functional IC 5 0 that is comparable to ICso s for compounds A and B.
- the compound of formula (I) has a protein binding adjusted EC 50 that is 4 times lower than that of compound A and 33 times lower than that of compound B.
- the compound of formula (I) is a weaker Cyp3 A4 inhibitor compared to compounds A and B.
- the compound of Formula (I) has a CYP3A4 IC50/ PBAdj.ECso value that is 43 times higher than that for compound of formula A, and 92 times higher than for the compound of formula B.
- the compound of formula (I) has a Rat CL value that is 2.7 times lower than that for compound of formula A, and 3.6 times lower than that for the compound of formula B.
- the compound of formula (I) has a percent bioavailability in rats that is 6.8 times higher than compound A and 1.5 times higher than compound B.
- the compound of formula (I) has a half life in rats that is 8.6 times longer than that of compound A and 3.9 times longer than that of compound B.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Physiology (AREA)
- Nutrition Science (AREA)
- Obesity (AREA)
- Virology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pulmonology (AREA)
- Gastroenterology & Hepatology (AREA)
- Biomedical Technology (AREA)
- Physical Education & Sports Medicine (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines Containing Plant Substances (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pyridine Compounds (AREA)
Abstract
Description









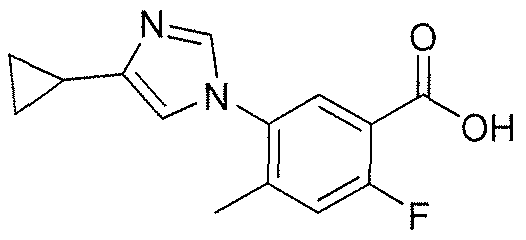






Claims




Priority Applications (35)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MX2014009075A MX2014009075A (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor. |
| CN201380006957.0A CN104080771B (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal regulating kinase inhibitor |
| JP2014554828A JP5770950B2 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| KR1020187037026A KR20190000898A (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| KR1020147023533A KR101582065B1 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| KR1020157036387A KR101707764B1 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| PL13703675T PL2807151T3 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| BR112014018355-4A BR112014018355B1 (en) | 2012-01-27 | 2013-01-24 | COMPOUND INHIBITING KINASE REGULATING SIGNAL OF APOPTOSIS, PHARMACEUTICAL COMPOSITION INCLUDING SUCH COMPOUND, USE AND INTERMEDIATE COMPOUNDS OF THE SAME |
| SI201330109A SI2807151T1 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| MEP-2016-5A ME02322B (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| AP2014007836A AP2014007836A0 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| AU2013201586A AU2013201586C1 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| KR1020177003620A KR101799739B1 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| RS20160019A RS54492B1 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| SG11201404348UA SG11201404348UA (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| MDA20140082A MD4601B1 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| KR1020177032774A KR20170128622A (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| EA201491283A EA024746B1 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| DK13703675.2T DK2807151T3 (en) | 2012-01-27 | 2013-01-24 | APOPTOSESIGNALREGULERENDE kinase inhibitor |
| EP13703675.2A EP2807151B1 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| MX2015009422A MX365954B (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor. |
| ES13703675.2T ES2558203T3 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal regulatory kinase inhibitor |
| CA2862030A CA2862030C (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
| UAA201407926A UA112098C2 (en) | 2012-01-27 | 2013-01-24 | REGULATORY APPOTOTIC KINAS SIGNAL INHIBITOR |
| IL233598A IL233598A (en) | 2012-01-27 | 2014-07-10 | Apoptosis signal-regulating kinase inhibitor |
| MA37202A MA35861B1 (en) | 2012-01-27 | 2014-07-11 | Kinase inhibitor regulating the signals of apoptosis |
| ZA2014/05260A ZA201405260B (en) | 2012-01-27 | 2014-07-17 | Apoptosis signal-regulating kinase inhibitor |
| IN6174DEN2014 IN2014DN06174A (en) | 2012-01-27 | 2014-07-22 | |
| PH12014501697A PH12014501697A1 (en) | 2012-01-27 | 2014-07-25 | Apoptosis signal-regulating kinase inhibitor |
| CR20140405A CR20140405A (en) | 2012-01-27 | 2014-08-29 | QUINASA INHIBITOR REGULATOR SIGNAL APOPTOSIS |
| HK15104864.8A HK1204324A1 (en) | 2012-01-27 | 2015-05-21 | Apoptosis signal-regulating kinase inhibitor |
| IL239311A IL239311B (en) | 2012-01-27 | 2015-06-10 | Apoptosis signal-regulating kinase inhibitor |
| HRP20151399TT HRP20151399T1 (en) | 2012-01-27 | 2015-12-22 | Apoptosis signal-regulating kinase inhibitor |
| SM201600047T SMT201600047B (en) | 2012-01-27 | 2016-02-15 | INHIBITOR OF CHINASES REGULATING THE SIGNALS OF APOPTOSIS |
| IL257965A IL257965A (en) | 2012-01-27 | 2018-03-07 | Apoptosis signal-regulating kinase inhibitor |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261591710P | 2012-01-27 | 2012-01-27 | |
| US61/591,710 | 2012-01-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013112741A1 true WO2013112741A1 (en) | 2013-08-01 |
Family
ID=47684038
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2013/022997 WO2013112741A1 (en) | 2012-01-27 | 2013-01-24 | Apoptosis signal-regulating kinase inhibitor |
Country Status (40)
| Country | Link |
|---|---|
| US (7) | US8742126B2 (en) |
| EP (1) | EP2807151B1 (en) |
| JP (1) | JP5770950B2 (en) |
| KR (5) | KR20190000898A (en) |
| CN (2) | CN109438418B (en) |
| AP (1) | AP2014007836A0 (en) |
| AR (2) | AR089818A1 (en) |
| AU (1) | AU2013201586C1 (en) |
| BR (1) | BR112014018355B1 (en) |
| CA (3) | CA2862030C (en) |
| CL (1) | CL2014001988A1 (en) |
| CO (1) | CO7061073A2 (en) |
| CR (1) | CR20140405A (en) |
| CY (1) | CY1117201T1 (en) |
| DK (1) | DK2807151T3 (en) |
| EA (1) | EA024746B1 (en) |
| ES (1) | ES2558203T3 (en) |
| HK (1) | HK1204324A1 (en) |
| HR (1) | HRP20151399T1 (en) |
| HU (1) | HUE028641T2 (en) |
| IL (3) | IL233598A (en) |
| IN (1) | IN2014DN06174A (en) |
| MA (1) | MA35861B1 (en) |
| MD (1) | MD4601B1 (en) |
| ME (1) | ME02322B (en) |
| MX (3) | MX365954B (en) |
| NZ (1) | NZ739339A (en) |
| PE (1) | PE20141822A1 (en) |
| PH (1) | PH12014501697A1 (en) |
| PL (1) | PL2807151T3 (en) |
| PT (1) | PT2807151E (en) |
| RS (1) | RS54492B1 (en) |
| SG (1) | SG11201404348UA (en) |
| SI (1) | SI2807151T1 (en) |
| SM (1) | SMT201600047B (en) |
| TW (4) | TWI561514B (en) |
| UA (1) | UA112098C2 (en) |
| UY (1) | UY34573A (en) |
| WO (1) | WO2013112741A1 (en) |
| ZA (1) | ZA201405260B (en) |
Cited By (96)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015081127A2 (en) | 2013-11-26 | 2015-06-04 | Gilead Sciences, Inc. | Therapies for treating myeloproliferative disorders |
| WO2015109286A1 (en) | 2014-01-20 | 2015-07-23 | Gilead Sciences, Inc. | Therapies for treating cancers |
| US20160166556A1 (en) * | 2014-08-13 | 2016-06-16 | Gilead Sciences, Inc. | Methods of treating pulmonary hypertension |
| WO2016105453A1 (en) * | 2014-12-23 | 2016-06-30 | Gilead Sciences, Inc. | Solid forms of an ask1 inhibitor |
| WO2016126552A1 (en) | 2015-02-03 | 2016-08-11 | Gilead Sciences, Inc. | Combination therapies for treating cancers |
| WO2016141092A1 (en) | 2015-03-04 | 2016-09-09 | Gilead Sciences, Inc. | Toll-like receptor modulating 4,6-diamino-pyrido[3,2-d]pyrimidine compounds |
| US9585886B2 (en) | 2010-12-16 | 2017-03-07 | Calchan Limited | ASK1 inhibiting pyrrolopyrimidine derivatives |
| WO2017059224A2 (en) | 2015-10-01 | 2017-04-06 | Gilead Sciences, Inc. | Combination of a btk inhibitor and a checkpoint inhibitor for treating cancers |
| CN106714841A (en) * | 2014-09-24 | 2017-05-24 | 吉利德科学公司 | Methods of treating liver disease |
| WO2017106556A1 (en) | 2015-12-17 | 2017-06-22 | Gilead Sciences, Inc. | Tank-binding kinase inhibitor compounds |
| WO2017152062A1 (en) | 2016-03-04 | 2017-09-08 | Gilead Sciences, Inc. | Compositions and combinations of autotaxin inhibitors |
| WO2017177179A1 (en) | 2016-04-08 | 2017-10-12 | Gilead Sciences, Inc. | Compositions and methods for treating cancer, inflammatory diseases and autoimmune diseases |
| WO2017215590A1 (en) | 2016-06-13 | 2017-12-21 | I-Mab | Anti-pd-l1 antibodies and uses thereof |
| JP2018501265A (en) * | 2014-12-23 | 2018-01-18 | ギリアード サイエンシーズ, インコーポレイテッド | Process for preparing an ASK1 inhibitor |
| WO2018026835A1 (en) | 2016-08-04 | 2018-02-08 | Gilead Sciences, Inc. | Cobicistat for use in cancer treatments |
| WO2018045144A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | Toll like receptor modulator compounds |
| WO2018045150A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | 4,6-diamino-pyrido[3,2-d]pyrimidine derivaties as toll like receptor modulators |
| WO2018085069A1 (en) | 2016-11-03 | 2018-05-11 | Gilead Sciences, Inc. | Combination of a bcl-2 inhibitor and a bromodomain inhibitor for treating cancer |
| WO2018097977A1 (en) | 2016-11-22 | 2018-05-31 | Gilead Sciences, Inc. | Crystalline forms of a phosphate complex of a bet inhibitor |
| WO2018133866A1 (en) * | 2017-01-22 | 2018-07-26 | 福建广生堂药业股份有限公司 | Pyridine derivative as ask1 inhibitor and preparation method and use thereof |
| WO2018137598A1 (en) | 2017-01-24 | 2018-08-02 | I-Mab | Anti-cd73 antibodies and uses thereof |
| WO2018156895A1 (en) | 2017-02-24 | 2018-08-30 | Gilead Sciences, Inc. | Inhibitors of bruton's tyrosine kinase |
| WO2018156901A1 (en) | 2017-02-24 | 2018-08-30 | Gilead Sciences, Inc. | Inhibitors of bruton's tyrosine kinase |
| WO2018195321A1 (en) | 2017-04-20 | 2018-10-25 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2018209354A1 (en) | 2017-05-12 | 2018-11-15 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| WO2018231851A1 (en) | 2017-06-13 | 2018-12-20 | Gilead Sciences, Inc. | Methods of treating liver fibrosis |
| WO2019050794A1 (en) * | 2017-09-08 | 2019-03-14 | Eli Lilly And Company | Imidazolidine compounds |
| WO2019047686A1 (en) * | 2017-09-07 | 2019-03-14 | Eli Lilly And Company | Cyclobutyl-imidazolidinone compounds |
| US10246439B2 (en) | 2017-05-25 | 2019-04-02 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| US10253018B2 (en) | 2017-05-25 | 2019-04-09 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| WO2019094777A1 (en) | 2017-11-13 | 2019-05-16 | Gilead Sciences, Inc. | Compositions and methods for identifying and treating liver diseases and monitoring treatment outcomes |
| US10370352B2 (en) | 2017-09-07 | 2019-08-06 | Eli Lilly And Company | Cyclobutyl-imidazolidinone compounds |
| WO2019160882A1 (en) | 2018-02-13 | 2019-08-22 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2019193543A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides |
| WO2019193533A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'2'-cyclic dinucleotides |
| WO2019193542A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides |
| US10450301B2 (en) | 2017-05-25 | 2019-10-22 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| WO2019204609A1 (en) | 2018-04-19 | 2019-10-24 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2019211799A1 (en) | 2018-05-03 | 2019-11-07 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide |
| WO2019213244A1 (en) | 2018-05-02 | 2019-11-07 | Enanta Pharmaceuticals, Inc. | Tetrazole containing apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| WO2019217780A1 (en) | 2018-05-11 | 2019-11-14 | Phosphorex, Inc. | Microparticles and nanoparticles having negative surface charges |
| WO2019222112A1 (en) | 2018-05-14 | 2019-11-21 | Gilead Sciences, Inc. | Mcl-1 inhibitors |
| WO2020006429A1 (en) * | 2018-06-28 | 2020-01-02 | Hepatikos Therapeutics, Llc | Ask1 isoindolin-1-one inhibitors and methods of use thereof |
| WO2020014643A1 (en) | 2018-07-13 | 2020-01-16 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2020015721A1 (en) * | 2018-07-20 | 2020-01-23 | 福建广生堂药业股份有限公司 | Crystal form as ask1 inhibitor, preparation method therefor, and application thereof |
| US10597382B2 (en) | 2017-08-28 | 2020-03-24 | Enanta Pharmaceuticals, Inc. | Tetrazole containing apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| WO2020072656A1 (en) | 2018-10-03 | 2020-04-09 | Gilead Sciences, Inc. | Imidozopyrimidine derivatives |
| WO2020080741A1 (en) | 2018-10-18 | 2020-04-23 | Cj Healthcare Corporation | Novel n-(isopropyl-triazolyl)pyridinyl)-heteroaryl-carboxamide derivatives and use thereof |
| WO2020086556A1 (en) | 2018-10-24 | 2020-04-30 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2020092621A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| WO2020092528A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds having hpk1 inhibitory activity |
| US10683289B2 (en) | 2018-05-02 | 2020-06-16 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| EP3572401A4 (en) * | 2017-01-22 | 2020-08-26 | Fujian Cosunter Pharmaceutical Co., Ltd. | Ask1 inhibitor and preparation method and use thereof |
| WO2020178770A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020178769A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020178768A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| WO2020214663A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020214652A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020237025A1 (en) | 2019-05-23 | 2020-11-26 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| WO2020255038A1 (en) | 2019-06-18 | 2020-12-24 | Janssen Sciences Ireland Unlimited Company | Combination of hepatitis b virus (hbv) vaccines and pyridopyrimidine derivatives |
| WO2020261294A1 (en) * | 2019-06-26 | 2020-12-30 | Mankind Pharma Ltd. | Novel apoptosis signal-regulating kinase 1 inhibitors |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| US10968199B2 (en) | 2018-08-22 | 2021-04-06 | Enanta Pharmaceuticals, Inc. | Cycloalkyl-containing apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| US10966999B2 (en) | 2017-12-20 | 2021-04-06 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| WO2021076908A1 (en) | 2019-10-18 | 2021-04-22 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| WO2021087064A1 (en) | 2019-10-31 | 2021-05-06 | Forty Seven, Inc. | Anti-cd47 and anti-cd20 based treatment of blood cancer |
| WO2021096860A1 (en) | 2019-11-12 | 2021-05-20 | Gilead Sciences, Inc. | Mcl1 inhibitors |
| WO2021130638A1 (en) | 2019-12-24 | 2021-07-01 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| WO2021163064A2 (en) | 2020-02-14 | 2021-08-19 | Jounce Therapeutics, Inc. | Antibodies and fusion proteins that bind to ccr8 and uses thereof |
| USRE48711E1 (en) | 2009-07-13 | 2021-08-31 | Gilead Sciences, Inc. | Apoptosis signal-regulating kinase inhibitors |
| WO2021222522A1 (en) | 2020-05-01 | 2021-11-04 | Gilead Sciences, Inc. | Cd73 inhibiting 2,4-dioxopyrimidine compounds |
| EP3789384A4 (en) * | 2018-04-28 | 2021-12-01 | Shenzhen Chipscreen Biosciences Co., Ltd. | Formamide compound, preparation method therefor and application thereof |
| US11203610B2 (en) | 2017-12-20 | 2021-12-21 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| US11345699B2 (en) | 2018-11-19 | 2022-05-31 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| US11466033B2 (en) | 2019-03-25 | 2022-10-11 | Enanta Pharmaceuticals, Inc. | Substituted pyridines as apoptosis signal-regulating kinase 1 inhibitors |
| WO2022221304A1 (en) | 2021-04-14 | 2022-10-20 | Gilead Sciences, Inc. | CO-INHIBITION OF CD47/SIRPα BINDING AND NEDD8-ACTIVATING ENZYME E1 REGULATORY SUBUNIT FOR THE TREATMENT OF CANCER |
| WO2022245671A1 (en) | 2021-05-18 | 2022-11-24 | Gilead Sciences, Inc. | Methods of using flt3l-fc fusion proteins |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| CN115844887A (en) * | 2022-12-28 | 2023-03-28 | 中国人民解放军空军军医大学 | Application of Selonsertib in preparation of medicine for treating cancer |
| WO2023077030A1 (en) | 2021-10-29 | 2023-05-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2023076983A1 (en) | 2021-10-28 | 2023-05-04 | Gilead Sciences, Inc. | Pyridizin-3(2h)-one derivatives |
| WO2023107954A1 (en) | 2021-12-08 | 2023-06-15 | Dragonfly Therapeutics, Inc. | Antibodies targeting 5t4 and uses thereof |
| WO2023107956A1 (en) | 2021-12-08 | 2023-06-15 | Dragonfly Therapeutics, Inc. | Proteins binding nkg2d, cd16 and 5t4 |
| WO2023122615A1 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023122581A2 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023147418A1 (en) | 2022-01-28 | 2023-08-03 | Gilead Sciences, Inc. | Parp7 inhibitors |
| EP4245756A1 (en) | 2022-03-17 | 2023-09-20 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023183817A1 (en) | 2022-03-24 | 2023-09-28 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| WO2023196784A1 (en) | 2022-04-05 | 2023-10-12 | Gilead Sciences, Inc. | Combinations of antibody therapies for treating colorectal cancer |
| WO2023205719A1 (en) | 2022-04-21 | 2023-10-26 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2024006929A1 (en) | 2022-07-01 | 2024-01-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2024064668A1 (en) | 2022-09-21 | 2024-03-28 | Gilead Sciences, Inc. | FOCAL IONIZING RADIATION AND CD47/SIRPα DISRUPTION ANTICANCER COMBINATION THERAPY |
| WO2024137852A1 (en) | 2022-12-22 | 2024-06-27 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
Families Citing this family (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UY34573A (en) | 2012-01-27 | 2013-06-28 | Gilead Sciences Inc | QUINASE INHIBITOR REGULATING THE APOPTOSIS SIGNAL |
| WO2015187499A1 (en) | 2014-06-03 | 2015-12-10 | Gilead Sciences, Inc. | Use of an ask1 inhibitor for the treatment of liver disease, optionally in combination with a loxl2 inhibitor |
| KR20240008398A (en) | 2015-07-06 | 2024-01-18 | 길리애드 사이언시즈, 인코포레이티드 | Cot modulators and methods of use thereof |
| EP3355871A1 (en) * | 2015-10-02 | 2018-08-08 | Gilead Sciences, Inc. | Combinations of the btk inhibitor gs-4059 with inhibitors selected from a jak, ask1, brd and/or mmp9 inhibitor to treat cancer, allergic disorders, autoimmune diseases or inflammatory diseases |
| JP2019533706A (en) | 2016-11-11 | 2019-11-21 | ギリアード サイエンシーズ, インコーポレイテッド | How to treat liver disease |
| AR110252A1 (en) | 2016-11-30 | 2019-03-13 | Gilead Sciences Inc | FUSIONED HETEROCYCLIC COMPOUNDS AS INHIBITORS OF THE QUINASA CAM |
| WO2018157277A1 (en) | 2017-02-28 | 2018-09-07 | Eli Lilly And Company | Isoquinolin and naphthydrin compounds |
| TW201833108A (en) * | 2017-03-03 | 2018-09-16 | 大陸商江蘇豪森藥業集團有限公司 | Amide derivatives inhibitors, preparation methods and uses thereof |
| JOP20180017A1 (en) | 2017-03-14 | 2019-01-30 | Gilead Sciences Inc | Apoptosis signal-regulating kinase inhibitor |
| US20180311244A1 (en) | 2017-03-28 | 2018-11-01 | Gilead Sciences, Inc. | Methods of treating liver disease |
| KR20190132515A (en) | 2017-04-12 | 2019-11-27 | 길리애드 사이언시즈, 인코포레이티드 | How to treat liver disease |
| TW201908306A (en) * | 2017-07-18 | 2019-03-01 | 大陸商南京聖和藥業股份有限公司 | Heterocyclic compound acting as ask inhibitor and use thereof |
| CN109456308B (en) * | 2017-09-06 | 2022-11-29 | 南京圣和药业股份有限公司 | Heterocyclic compounds as ASK inhibitors and their use |
| WO2019051265A1 (en) | 2017-09-08 | 2019-03-14 | Fronthera U.S. Pharmaceuticals Llc | Apoptosis signal-regulating kinase inhibitors and uses thereof |
| WO2019072130A1 (en) * | 2017-10-12 | 2019-04-18 | 深圳市塔吉瑞生物医药有限公司 | 1, 2, 4-triazole compound |
| CN107793400B (en) * | 2017-10-31 | 2020-01-31 | 武汉九州钰民医药科技有限公司 | Pyridine compound and application thereof in preparation of drugs for treating liver diseases |
| US11434249B1 (en) | 2018-01-02 | 2022-09-06 | Seal Rock Therapeutics, Inc. | ASK1 inhibitor compounds and uses thereof |
| CN110294742B (en) * | 2018-03-21 | 2023-01-31 | 山东轩竹医药科技有限公司 | Fused ring ASK1 inhibitor and application thereof |
| US10919911B2 (en) | 2018-04-12 | 2021-02-16 | Terns, Inc. | Tricyclic ASK1 inhibitors |
| CN111170995A (en) * | 2018-11-09 | 2020-05-19 | 山东轩竹医药科技有限公司 | ASK1 inhibitor and application thereof |
| CN109608441A (en) * | 2018-12-26 | 2019-04-12 | 都创(上海)医药科技有限公司 | A kind of synthesis technology of ASK1 inhibitor |
| SI3911647T1 (en) | 2019-01-15 | 2024-04-30 | Gilead Sciences, Inc. | Isoxazole compound as fxr agonist and pharmaceutical compositions comprising same |
| CN109813915A (en) * | 2019-02-15 | 2019-05-28 | 浠思(上海)生物技术有限公司 | Utilize the method for HTRF one-step method screening kinase inhibitor |
| JP2022519906A (en) | 2019-02-19 | 2022-03-25 | ギリアード サイエンシーズ, インコーポレイテッド | Solid form of FXR agonist |
| KR20240135055A (en) | 2019-03-11 | 2024-09-10 | 길리애드 사이언시즈, 인코포레이티드 | Formulations of a compound and uses thereof |
| TWI770527B (en) | 2019-06-14 | 2022-07-11 | 美商基利科學股份有限公司 | Cot modulators and methods of use thereof |
| AR119594A1 (en) | 2019-08-09 | 2021-12-29 | Gilead Sciences Inc | THIENOPYRIMIDINE DERIVATIVES AS ACC INHIBITORS AND USES THEREOF |
| CN114728168B (en) | 2019-11-15 | 2024-04-09 | 吉利德科学公司 | Triazole carbamate pyridylsulfonamides as LPA receptor antagonists and their use |
| CN111018831B (en) * | 2019-11-21 | 2022-05-31 | 中国药科大学 | Apoptosis signal-regulating kinase inhibitor and application thereof |
| US20230105984A1 (en) | 2019-12-23 | 2023-04-06 | Svetlana Marukian | Compositions and methods for the treatment of liver diseases and disorders |
| WO2021202224A1 (en) | 2020-03-30 | 2021-10-07 | Gilead Sciences, Inc. | Solid forms of (s)-6-(((1-(bicyclo[1.1.1]pentan-1-yl)-1h-1,2,3-triazol-4-yl)2-methyl-1-oxo-1,2- dihydroisoquinolin-5-yl)methyl)))amino)8-chloro-(neopentylamino)quinoline-3-carb onitrile a cot inhibitor compound |
| US11845737B2 (en) | 2020-04-02 | 2023-12-19 | Gilead Sciences, Inc. | Process for preparing a Cot inhibitor compound |
| EP4161936A1 (en) | 2020-06-03 | 2023-04-12 | Gilead Sciences, Inc. | Lpa receptor antagonists and uses thereof |
| CN113831323A (en) * | 2020-06-24 | 2021-12-24 | 武汉人福创新药物研发中心有限公司 | Aromatic heterocyclic amide compound and application thereof |
| CN111808079A (en) * | 2020-08-05 | 2020-10-23 | 中国药科大学 | Indole ASK1 small molecule inhibitor and preparation method and application thereof |
| CN114470217B (en) * | 2020-11-24 | 2023-06-20 | 深圳微芯生物科技股份有限公司 | Pharmaceutical composition for preventing and treating tissue injury caused by metabolic abnormality or inflammation |
| WO2022192428A1 (en) | 2021-03-11 | 2022-09-15 | Gilead Sciences, Inc. | Glp-1r modulating compounds |
| US20230079863A1 (en) | 2021-03-29 | 2023-03-16 | Gilead Sciences, Inc. | Khk inhibitors |
| CN114246866B (en) * | 2021-04-28 | 2023-09-12 | 深圳微芯生物科技股份有限公司 | Medicine for treating chronic kidney disease and application thereof |
| EP4337641A1 (en) | 2021-05-11 | 2024-03-20 | Gilead Sciences, Inc. | Lpa receptor antagonists and uses thereof |
| EP4337654A1 (en) | 2021-05-13 | 2024-03-20 | Gilead Sciences, Inc. | Lpa receptor antagonists and uses thereof |
| TW202304435A (en) | 2021-06-04 | 2023-02-01 | 美商基利科學股份有限公司 | Methods of treating nash |
| TW202311256A (en) | 2021-06-18 | 2023-03-16 | 美商基利科學股份有限公司 | Il-31 modulators for treating fxr-induced pruritis |
| AU2022405082A1 (en) | 2021-12-08 | 2024-07-11 | Gilead Sciences, Inc. | Lpa receptor antagonists and uses thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070276050A1 (en) | 2006-02-27 | 2007-11-29 | Gilead Sciences, Inc. | Methods for identifying ASK1 inhibitors useful for preventing and/or treating cardiovascular diseases |
| WO2009027283A1 (en) | 2007-08-31 | 2009-03-05 | Merck Serono S.A. | Triazolopyridine compounds and their use as ask inhibitors |
| US20110009410A1 (en) | 2009-07-13 | 2011-01-13 | Gilead Sciences, Inc. | Apoptosis signal-regulating kinase inhibitors |
| WO2012003387A1 (en) * | 2010-07-02 | 2012-01-05 | Gilead Sciences, Inc. | Apoptosis signal-regulating kinase inhibitors |
Family Cites Families (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998034946A1 (en) | 1997-02-12 | 1998-08-13 | Massachusetts Institute Of Technology | Daxx, a novel fas-binding protein that activates jnk and apoptosis |
| US6881555B2 (en) | 1999-03-19 | 2005-04-19 | Aventis Pharmaceuticals Inc. | AKT nucleic acids, polypeptides, and uses thereof |
| WO2000056866A2 (en) | 1999-03-19 | 2000-09-28 | Aventis Pharmaceuticals Products Inc. | Akt-3 nucleic acids, polypeptides, and uses thereof |
| WO2001095410A1 (en) * | 2000-06-07 | 2001-12-13 | Andelman Marc D | Fluid and electrical connected flow-through electrochemical cells, system and method |
| US20070237770A1 (en) | 2001-11-30 | 2007-10-11 | Albert Lai | Novel compositions and methods in cancer |
| WO2005009470A1 (en) | 2003-07-28 | 2005-02-03 | Osaka Industrial Promotion Organization | Remedy for cardiac failure comprising ask1 inhibitor as the active ingredient and method of screening the same |
| WO2006036941A2 (en) | 2004-09-27 | 2006-04-06 | Kosan Biosciences Incorporated | Specific kinase inhibitors |
| JP2008519850A (en) | 2004-11-10 | 2008-06-12 | シンタ ファーマシューティカルズ コーポレーション | IL-12 modulating compound |
| EP1677113A1 (en) | 2004-12-29 | 2006-07-05 | Max-Delbrück-Centrum für Molekulare Medizin (MDC) | Method for the identification of protein-protein interactions in disease related protein networks |
| US20100029619A1 (en) | 2006-08-04 | 2010-02-04 | Takeda Pharmaceutical Company Limted | Fused heterocyclic compound |
| WO2008042867A2 (en) | 2006-09-29 | 2008-04-10 | Emiliem Inc. | Modulators of multiple kinases |
| EP2104734A2 (en) | 2006-12-08 | 2009-09-30 | Asuragen, INC. | Mir-20 regulated genes and pathways as targets for therapeutic intervention |
| EP1942104A1 (en) * | 2006-12-20 | 2008-07-09 | sanofi-aventis | Heteroarylcyclopropanecarboxamides and their use as pharmaceuticals |
| US8104727B2 (en) | 2007-03-12 | 2012-01-31 | Denis Joanisse | Adjustable article support |
| US20090069288A1 (en) | 2007-07-16 | 2009-03-12 | Breinlinger Eric C | Novel therapeutic compounds |
| US8461163B2 (en) | 2008-03-31 | 2013-06-11 | Takeda Pharmaceutical Company Limited | Substituted N-(pyrazolo[1,5-a]pyrimidin-5-yl)amides as inhibitors of apoptosis signal-regulating kinase 1 |
| US8178555B2 (en) | 2008-06-24 | 2012-05-15 | Takeda Pharmaceutical Company Limited | Apoptosis signal-regulating kinase 1 inhibitors |
| WO2010045470A2 (en) | 2008-10-15 | 2010-04-22 | Dana-Farber Cancer Institute, Inc. | Compositions, kits, and methods for identification, assessment, prevention, and therapy of hepatic disorders |
| WO2010056982A2 (en) | 2008-11-17 | 2010-05-20 | The George Washington University | Compositions and methods for identifying autism spectrum disorders |
| EP2411516A1 (en) | 2009-03-27 | 2012-02-01 | Merck Sharp&Dohme Corp. | RNA INTERFERENCE MEDIATED INHIBITION OF APOPTOSIS SIGNAL-REGULATING KINASE 1 (ASK1) GENE EXPRESSION USING SHORT INTERFERING NUCLEIC ACID (siNA) |
| WO2011041293A1 (en) | 2009-09-30 | 2011-04-07 | Takeda Pharmaceutical Company Limited | Pyrazolo [1, 5-a] pyrimidine derivatives as apoptosis signal-regulating kinase 1 inhibitors |
| RS53176B (en) | 2010-02-03 | 2014-06-30 | Takeda Pharmaceutical Company Limited | Apoptosis signal-regulating kinase 1 inhibitors |
| JP2013518909A (en) | 2010-02-08 | 2013-05-23 | キナゲン,インク. | Therapeutic methods and compositions involving allosteric kinase inhibition |
| EP2550266B1 (en) | 2010-03-24 | 2018-05-09 | Amitech Therapeutics Solutions, Inc. | Heterocyclic compounds useful for kinase inhibition |
| US20130236441A1 (en) | 2010-11-18 | 2013-09-12 | University Of Delaware | Methods of treating and preventing thrombotic diseases using ask1 inhibitors |
| JP5927201B2 (en) | 2010-12-16 | 2016-06-01 | カルチャン リミテッド | ASK1-inhibited pyrrolopyrimidine derivatives |
| US20140141986A1 (en) | 2011-02-22 | 2014-05-22 | David Spetzler | Circulating biomarkers |
| WO2012170711A1 (en) | 2011-06-07 | 2012-12-13 | Caris Life Sciences Luxembourg Holdings, S.A.R.L | Circulating biomarkers for cancer |
| TWI622583B (en) | 2011-07-01 | 2018-05-01 | 基利科學股份有限公司 | Fused heterocyclic compounds as ion channel modulators |
| US8924765B2 (en) | 2011-07-03 | 2014-12-30 | Ambiq Micro, Inc. | Method and apparatus for low jitter distributed clock calibration |
| UY34573A (en) | 2012-01-27 | 2013-06-28 | Gilead Sciences Inc | QUINASE INHIBITOR REGULATING THE APOPTOSIS SIGNAL |
| PT3237404T (en) | 2014-12-23 | 2021-01-19 | Gilead Sciences Inc | Processes for preparing ask1 inhibitors |
| MA41252A (en) | 2014-12-23 | 2017-10-31 | Gilead Sciences Inc | SOLID FORMS OF AN ASK 1 INHIBITOR |
-
2013
- 2013-01-11 UY UY0001034573A patent/UY34573A/en not_active Application Discontinuation
- 2013-01-24 PT PT137036752T patent/PT2807151E/en unknown
- 2013-01-24 KR KR1020187037026A patent/KR20190000898A/en not_active Application Discontinuation
- 2013-01-24 UA UAA201407926A patent/UA112098C2/en unknown
- 2013-01-24 EP EP13703675.2A patent/EP2807151B1/en active Active
- 2013-01-24 ES ES13703675.2T patent/ES2558203T3/en active Active
- 2013-01-24 RS RS20160019A patent/RS54492B1/en unknown
- 2013-01-24 SG SG11201404348UA patent/SG11201404348UA/en unknown
- 2013-01-24 EA EA201491283A patent/EA024746B1/en unknown
- 2013-01-24 CA CA2862030A patent/CA2862030C/en active Active
- 2013-01-24 PE PE2014001153A patent/PE20141822A1/en active IP Right Grant
- 2013-01-24 AP AP2014007836A patent/AP2014007836A0/en unknown
- 2013-01-24 JP JP2014554828A patent/JP5770950B2/en active Active
- 2013-01-24 KR KR1020157036387A patent/KR101707764B1/en active IP Right Grant
- 2013-01-24 MD MDA20140082A patent/MD4601B1/en not_active IP Right Cessation
- 2013-01-24 HU HUE13703675A patent/HUE028641T2/en unknown
- 2013-01-24 MX MX2015009422A patent/MX365954B/en unknown
- 2013-01-24 CA CA3026700A patent/CA3026700C/en active Active
- 2013-01-24 DK DK13703675.2T patent/DK2807151T3/en active
- 2013-01-24 NZ NZ739339A patent/NZ739339A/en unknown
- 2013-01-24 WO PCT/US2013/022997 patent/WO2013112741A1/en active Application Filing
- 2013-01-24 PL PL13703675T patent/PL2807151T3/en unknown
- 2013-01-24 KR KR1020177032774A patent/KR20170128622A/en not_active Application Discontinuation
- 2013-01-24 CA CA3026738A patent/CA3026738C/en active Active
- 2013-01-24 BR BR112014018355-4A patent/BR112014018355B1/en active IP Right Grant
- 2013-01-24 KR KR1020177003620A patent/KR101799739B1/en active IP Right Grant
- 2013-01-24 AU AU2013201586A patent/AU2013201586C1/en active Active
- 2013-01-24 MX MX2014009075A patent/MX2014009075A/en active IP Right Grant
- 2013-01-24 CN CN201811215092.XA patent/CN109438418B/en active Active
- 2013-01-24 US US13/748,901 patent/US8742126B2/en active Active
- 2013-01-24 KR KR1020147023533A patent/KR101582065B1/en active IP Right Grant
- 2013-01-24 CN CN201380006957.0A patent/CN104080771B/en active Active
- 2013-01-24 ME MEP-2016-5A patent/ME02322B/en unknown
- 2013-01-24 SI SI201330109A patent/SI2807151T1/en unknown
- 2013-01-25 TW TW103125752A patent/TWI561514B/en active
- 2013-01-25 TW TW107127139A patent/TW201916877A/en unknown
- 2013-01-25 TW TW105128669A patent/TWI634890B/en active
- 2013-01-25 TW TW102102991A patent/TWI487523B/en active
- 2013-01-25 AR ARP130100241A patent/AR089818A1/en active IP Right Grant
-
2014
- 2014-04-16 US US14/254,342 patent/US9254284B2/en active Active
- 2014-04-16 US US14/254,359 patent/US9333197B2/en active Active
- 2014-07-10 IL IL233598A patent/IL233598A/en active IP Right Grant
- 2014-07-11 MA MA37202A patent/MA35861B1/en unknown
- 2014-07-17 ZA ZA2014/05260A patent/ZA201405260B/en unknown
- 2014-07-22 IN IN6174DEN2014 patent/IN2014DN06174A/en unknown
- 2014-07-25 MX MX2019007348A patent/MX2019007348A/en unknown
- 2014-07-25 CL CL2014001988A patent/CL2014001988A1/en unknown
- 2014-07-25 PH PH12014501697A patent/PH12014501697A1/en unknown
- 2014-08-26 CO CO14186994A patent/CO7061073A2/en unknown
- 2014-08-29 CR CR20140405A patent/CR20140405A/en unknown
-
2015
- 2015-05-21 HK HK15104864.8A patent/HK1204324A1/en unknown
- 2015-06-10 IL IL239311A patent/IL239311B/en active IP Right Grant
- 2015-12-22 HR HRP20151399TT patent/HRP20151399T1/en unknown
-
2016
- 2016-02-15 SM SM201600047T patent/SMT201600047B/en unknown
- 2016-02-16 CY CY20161100129T patent/CY1117201T1/en unknown
- 2016-04-27 US US15/140,204 patent/US9750730B2/en active Active
-
2017
- 2017-04-19 US US15/491,689 patent/US9873682B2/en active Active
- 2017-12-13 US US15/840,429 patent/US20180099950A1/en not_active Abandoned
-
2018
- 2018-03-07 IL IL257965A patent/IL257965A/en unknown
-
2019
- 2019-01-11 US US16/246,199 patent/US10508100B2/en active Active
- 2019-11-19 AR ARP190103385A patent/AR117113A2/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070276050A1 (en) | 2006-02-27 | 2007-11-29 | Gilead Sciences, Inc. | Methods for identifying ASK1 inhibitors useful for preventing and/or treating cardiovascular diseases |
| WO2009027283A1 (en) | 2007-08-31 | 2009-03-05 | Merck Serono S.A. | Triazolopyridine compounds and their use as ask inhibitors |
| US20110009410A1 (en) | 2009-07-13 | 2011-01-13 | Gilead Sciences, Inc. | Apoptosis signal-regulating kinase inhibitors |
| WO2011008709A1 (en) * | 2009-07-13 | 2011-01-20 | Gilead Sciences, Inc. | Apoptosis signal-regulating kinase inhibitors |
| WO2012003387A1 (en) * | 2010-07-02 | 2012-01-05 | Gilead Sciences, Inc. | Apoptosis signal-regulating kinase inhibitors |
Non-Patent Citations (19)
| Title |
|---|
| "Remington's Pharmaceutical Sciences 17th Ed.", 1985, MACE PUBLISHING CO. |
| BERGE, JOURNAL OFPHARMACEUTICAL SCIENCE, vol. 66, no. 1, January 1977 (1977-01-01) |
| BROWNLEE M.: "Biochemistry and molecular cell biology of diabetic complications", NATURE, vol. 414, no. 6865, 13 December 2001 (2001-12-13), pages 813 - 820 |
| CHEVALIER ET AL.: "Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy", KIDNEY INTERNATIONAL, vol. 75, 2009, pages 1145 - 1152 |
| DE B, I ET AL.: "Temporal trends in the prevalence of diabetic kidney disease in the United States", JAMA, vol. 305, no. 24, 22 June 2011 (2011-06-22), pages 2532 - 2539 |
| G.S. BANKER & C.T. RHODES,: "Modern Pharmaceutics 3rd Ed.", MARCEL DEKKER, INC. |
| HATTORI, K.; NAGURO, I.; RUNCHEL, C.; ICHIJO, H., CELL COMM. SIGNAL., vol. 7, 2009, pages 1 - 10 |
| HUNG KY ET AL.: "N- acetylcysteine-mediated antioxidation prevents hyperglycemia-induced apoptosis and collagen synthesis in rat mesangial cells", AM JNEPHROL, vol. 29, no. 3, 2009, pages 192 - 202 |
| ICHIJO, H.; NISHIDA, E.; IRIE, K.; DIJKE, P. T.; SAITOH, M.; MORIGUCHI, T.; MATSUMOTO, K.; MIYAZONO, K.; GOTOH, Y., SCIENCE, vol. 275, 1997, pages 90 - 94 |
| MIMURA I; NANGAKU M.; 2010 NOV: "The suffocating kidney: tubulointerstitial hypoxia in end-stage renal disease", NAT REV NEPHROL, vol. 6, no. 11, pages 667 - 678 |
| NAGAI, H.; NOGUCHI, T.; TAKEDA, K.; ICHIJO, I., J. BIOCHEM. MOL. BIOL., vol. 40, 2007, pages 1 - 6 |
| PETER G.M. WUTS; THEODORA W. GREENE: "Protective Groups in Organic Chemistry, 2nd edition,", 1991, WILEY AND SONS |
| SCHAINUCK LI ET AL.: "Structural-functional correlations in renal disease. Part II: The correlations", HUM PATHOL, vol. 1, 1970, pages 631 - 641 |
| SINGH DK; WINOCOUR P; FARRINGTON K.: "Oxidative stress in early diabetic nephropathy: fueling the fire", NAT REV ENDOCRINOL, vol. 7, no. 3, March 2011 (2011-03-01), pages 176 - 184 |
| STAMBE ET AL.: "The Role ofp38 Mitogen-Activated Protein Kinase Activation in Renal Fibrosis", JAM SOC NEPHROL, vol. 15, 2004, pages 370 - 379 |
| TAKEDA, K.; NOGUCHI, T.; NAGURO, I.; ICHIJO, H., ANNU. REV. PHARMACOL. TOXICOL., vol. 48, 2007, pages 1 - 8,27 |
| TERADA ET AL., BBRC, vol. 364, 2007, pages 1043 - 49 |
| WATANABE ET AL., BBRC, vol. 333, 2005, pages 562 - 567 |
| ZHANG ET AL., LIFE SCI, vol. 74, 2003, pages 37 - 43 |
Cited By (151)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| USRE48711E1 (en) | 2009-07-13 | 2021-08-31 | Gilead Sciences, Inc. | Apoptosis signal-regulating kinase inhibitors |
| US9585886B2 (en) | 2010-12-16 | 2017-03-07 | Calchan Limited | ASK1 inhibiting pyrrolopyrimidine derivatives |
| WO2015081127A2 (en) | 2013-11-26 | 2015-06-04 | Gilead Sciences, Inc. | Therapies for treating myeloproliferative disorders |
| WO2015109286A1 (en) | 2014-01-20 | 2015-07-23 | Gilead Sciences, Inc. | Therapies for treating cancers |
| US20160166556A1 (en) * | 2014-08-13 | 2016-06-16 | Gilead Sciences, Inc. | Methods of treating pulmonary hypertension |
| CN106714841A (en) * | 2014-09-24 | 2017-05-24 | 吉利德科学公司 | Methods of treating liver disease |
| US10238636B2 (en) | 2014-09-24 | 2019-03-26 | Gilead Sciences, Inc. | Methods of treating liver disease |
| JP2017528471A (en) * | 2014-09-24 | 2017-09-28 | ギリアード サイエンシーズ, インコーポレイテッド | How to treat liver disease |
| KR101971387B1 (en) * | 2014-12-23 | 2019-04-22 | 길리애드 사이언시즈, 인코포레이티드 | Solid forms of an ask1 inhibitor |
| JP2017538774A (en) * | 2014-12-23 | 2017-12-28 | ギリアード サイエンシーズ, インコーポレイテッド | Solid form of ASK1 inhibitor |
| JP2019011371A (en) * | 2014-12-23 | 2019-01-24 | ギリアード サイエンシーズ, インコーポレイテッド | Solid form of ASK1 inhibitor |
| KR20170096036A (en) * | 2014-12-23 | 2017-08-23 | 길리애드 사이언시즈, 인코포레이티드 | Solid forms of an ask1 inhibitor |
| CN107108574A (en) * | 2014-12-23 | 2017-08-29 | 吉利德科学公司 | The solid form of ASK1 inhibitor |
| TWI731591B (en) * | 2014-12-23 | 2021-06-21 | 美商基利科學股份有限公司 | Solid forms of an ask1 inhibitor |
| AU2019200333B2 (en) * | 2014-12-23 | 2020-10-29 | Gilead Sciences, Inc. | Solid forms of an ASK1 inhibitor |
| WO2016105453A1 (en) * | 2014-12-23 | 2016-06-30 | Gilead Sciences, Inc. | Solid forms of an ask1 inhibitor |
| EP4265250A3 (en) * | 2014-12-23 | 2024-01-10 | Gilead Sciences, Inc. | Solid forms of an ask1 inhibitor |
| US11261161B2 (en) | 2014-12-23 | 2022-03-01 | Gilead Sciences, Inc. | Processes for preparing ASK1 inhibitors |
| JP2018501265A (en) * | 2014-12-23 | 2018-01-18 | ギリアード サイエンシーズ, インコーポレイテッド | Process for preparing an ASK1 inhibitor |
| US9643956B2 (en) | 2014-12-23 | 2017-05-09 | Gilead Sciences, Inc. | Solid forms of an ASK1 inhibitor |
| US9907790B2 (en) | 2014-12-23 | 2018-03-06 | Gilead Sciences, Inc. | Solid forms of an ASK1 inhibitor |
| US10604490B2 (en) | 2014-12-23 | 2020-03-31 | Gilead Sciences, Inc. | Processes for preparing ASK1 inhibitors |
| TWI689504B (en) * | 2014-12-23 | 2020-04-01 | 美商基利科學股份有限公司 | Solid forms of an ask1 inhibitor |
| US10946004B2 (en) | 2014-12-23 | 2021-03-16 | Gilead Sciences, Inc. | Solid forms of an ASK1 inhibitor |
| AU2015371629B2 (en) * | 2014-12-23 | 2019-01-31 | Gilead Sciences, Inc. | Solid forms of an ASK1 inhibitor |
| WO2016126552A1 (en) | 2015-02-03 | 2016-08-11 | Gilead Sciences, Inc. | Combination therapies for treating cancers |
| EP3722297A1 (en) | 2015-03-04 | 2020-10-14 | Gilead Sciences, Inc. | Toll-like receptor modulating 4,6-diamino-pyrido[3,2-d]pyrimidine compounds |
| EP3321265A1 (en) | 2015-03-04 | 2018-05-16 | Gilead Sciences, Inc. | 4,6-diamino-pyrido[3,2-d]pyrimidine compounds and their utilisation as modulators of toll-like receptors |
| WO2016141092A1 (en) | 2015-03-04 | 2016-09-09 | Gilead Sciences, Inc. | Toll-like receptor modulating 4,6-diamino-pyrido[3,2-d]pyrimidine compounds |
| WO2017059224A2 (en) | 2015-10-01 | 2017-04-06 | Gilead Sciences, Inc. | Combination of a btk inhibitor and a checkpoint inhibitor for treating cancers |
| WO2017106556A1 (en) | 2015-12-17 | 2017-06-22 | Gilead Sciences, Inc. | Tank-binding kinase inhibitor compounds |
| WO2017152062A1 (en) | 2016-03-04 | 2017-09-08 | Gilead Sciences, Inc. | Compositions and combinations of autotaxin inhibitors |
| WO2017177179A1 (en) | 2016-04-08 | 2017-10-12 | Gilead Sciences, Inc. | Compositions and methods for treating cancer, inflammatory diseases and autoimmune diseases |
| WO2017215590A1 (en) | 2016-06-13 | 2017-12-21 | I-Mab | Anti-pd-l1 antibodies and uses thereof |
| WO2018026835A1 (en) | 2016-08-04 | 2018-02-08 | Gilead Sciences, Inc. | Cobicistat for use in cancer treatments |
| WO2018045144A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | Toll like receptor modulator compounds |
| WO2018045150A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | 4,6-diamino-pyrido[3,2-d]pyrimidine derivaties as toll like receptor modulators |
| WO2018085069A1 (en) | 2016-11-03 | 2018-05-11 | Gilead Sciences, Inc. | Combination of a bcl-2 inhibitor and a bromodomain inhibitor for treating cancer |
| WO2018097977A1 (en) | 2016-11-22 | 2018-05-31 | Gilead Sciences, Inc. | Crystalline forms of a phosphate complex of a bet inhibitor |
| EP3572401A4 (en) * | 2017-01-22 | 2020-08-26 | Fujian Cosunter Pharmaceutical Co., Ltd. | Ask1 inhibitor and preparation method and use thereof |
| AU2018209574B2 (en) * | 2017-01-22 | 2021-04-08 | Fujian Akeylink Biotechnology Co., Ltd. | Pyridine derivative as ASK1 inhibitor and preparation method and use thereof |
| WO2018133866A1 (en) * | 2017-01-22 | 2018-07-26 | 福建广生堂药业股份有限公司 | Pyridine derivative as ask1 inhibitor and preparation method and use thereof |
| US11040968B2 (en) | 2017-01-22 | 2021-06-22 | Fujian Cosunter Pharmaceutical Co., Ltd. | Pyridine derivative as ASK1 inhibitor and preparation method and use thereof |
| EA037005B1 (en) * | 2017-01-22 | 2021-01-26 | Фуцзянь Косантер Фармасьютикал Ко., Лтд. | Pyridine derivative as ask1 inhibitor and preparation method and use thereof |
| WO2018137598A1 (en) | 2017-01-24 | 2018-08-02 | I-Mab | Anti-cd73 antibodies and uses thereof |
| EP4050030A1 (en) | 2017-01-24 | 2022-08-31 | I-Mab Biopharma US Limited | Anti-cd73 antibodies and uses thereof |
| WO2018156895A1 (en) | 2017-02-24 | 2018-08-30 | Gilead Sciences, Inc. | Inhibitors of bruton's tyrosine kinase |
| US10314844B2 (en) | 2017-02-24 | 2019-06-11 | Gilead Sciences, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US10370381B2 (en) | 2017-02-24 | 2019-08-06 | Gilead Sciences, Inc. | Inhibitors of bruton'S tyrosine kinase |
| WO2018156901A1 (en) | 2017-02-24 | 2018-08-30 | Gilead Sciences, Inc. | Inhibitors of bruton's tyrosine kinase |
| EP4026835A2 (en) | 2017-04-20 | 2022-07-13 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2018195321A1 (en) | 2017-04-20 | 2018-10-25 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| US11560368B2 (en) | 2017-05-12 | 2023-01-24 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| US12018017B2 (en) | 2017-05-12 | 2024-06-25 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| WO2018209354A1 (en) | 2017-05-12 | 2018-11-15 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| US10683279B2 (en) | 2017-05-12 | 2020-06-16 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| US10988458B2 (en) | 2017-05-12 | 2021-04-27 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| US10450301B2 (en) | 2017-05-25 | 2019-10-22 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| US10253018B2 (en) | 2017-05-25 | 2019-04-09 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| US10246439B2 (en) | 2017-05-25 | 2019-04-02 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| WO2018231851A1 (en) | 2017-06-13 | 2018-12-20 | Gilead Sciences, Inc. | Methods of treating liver fibrosis |
| US10495648B2 (en) | 2017-06-13 | 2019-12-03 | Gilead Sciences, Inc. | Methods of treating liver fibrosis |
| US10597382B2 (en) | 2017-08-28 | 2020-03-24 | Enanta Pharmaceuticals, Inc. | Tetrazole containing apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| WO2019047686A1 (en) * | 2017-09-07 | 2019-03-14 | Eli Lilly And Company | Cyclobutyl-imidazolidinone compounds |
| US10370352B2 (en) | 2017-09-07 | 2019-08-06 | Eli Lilly And Company | Cyclobutyl-imidazolidinone compounds |
| WO2019050794A1 (en) * | 2017-09-08 | 2019-03-14 | Eli Lilly And Company | Imidazolidine compounds |
| CN110997657A (en) * | 2017-09-08 | 2020-04-10 | 伊莱利利公司 | Imidazolidine compounds |
| US11124496B2 (en) | 2017-09-08 | 2021-09-21 | Eli Lilly And Company | Imidazolidine compounds |
| WO2019094777A1 (en) | 2017-11-13 | 2019-05-16 | Gilead Sciences, Inc. | Compositions and methods for identifying and treating liver diseases and monitoring treatment outcomes |
| US11203610B2 (en) | 2017-12-20 | 2021-12-21 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| US10966999B2 (en) | 2017-12-20 | 2021-04-06 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| EP4227302A1 (en) | 2018-02-13 | 2023-08-16 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2019160882A1 (en) | 2018-02-13 | 2019-08-22 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| US11149052B2 (en) | 2018-04-06 | 2021-10-19 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′-cyclic dinucleotides |
| US11292812B2 (en) | 2018-04-06 | 2022-04-05 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′-cyclic dinucleotides |
| WO2019193543A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides |
| WO2019193533A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'2'-cyclic dinucleotides |
| WO2019193542A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides |
| WO2019204609A1 (en) | 2018-04-19 | 2019-10-24 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| EP3789384A4 (en) * | 2018-04-28 | 2021-12-01 | Shenzhen Chipscreen Biosciences Co., Ltd. | Formamide compound, preparation method therefor and application thereof |
| US10683289B2 (en) | 2018-05-02 | 2020-06-16 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| US11008304B2 (en) | 2018-05-02 | 2021-05-18 | Enanta Pharmaceuticals, Inc. | Tetrazole containing apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| US11834436B2 (en) | 2018-05-02 | 2023-12-05 | Enanta Pharmaceuticals, Inc. | Tetrazole containing apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| WO2019213244A1 (en) | 2018-05-02 | 2019-11-07 | Enanta Pharmaceuticals, Inc. | Tetrazole containing apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| WO2019211799A1 (en) | 2018-05-03 | 2019-11-07 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide |
| WO2019217780A1 (en) | 2018-05-11 | 2019-11-14 | Phosphorex, Inc. | Microparticles and nanoparticles having negative surface charges |
| EP4029868A1 (en) | 2018-05-14 | 2022-07-20 | Gilead Sciences, Inc. | Mcl-1 inhibitors |
| WO2019222112A1 (en) | 2018-05-14 | 2019-11-21 | Gilead Sciences, Inc. | Mcl-1 inhibitors |
| US10654833B2 (en) | 2018-06-28 | 2020-05-19 | Hepatikos Therapeutics, Llc | ASK1 isoindolin-1-one inhibitors and methods of use thereof |
| WO2020006429A1 (en) * | 2018-06-28 | 2020-01-02 | Hepatikos Therapeutics, Llc | Ask1 isoindolin-1-one inhibitors and methods of use thereof |
| WO2020014643A1 (en) | 2018-07-13 | 2020-01-16 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| EP4234030A2 (en) | 2018-07-13 | 2023-08-30 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| JP2021531343A (en) * | 2018-07-20 | 2021-11-18 | 福建広生堂薬業股▲ふん▼有限公司Fujian Cosunter Pharmaceutical Co., Ltd. | Crystal form as an ASK1 inhibitor, its production method and utilization |
| JP7096460B2 (en) | 2018-07-20 | 2022-07-06 | 福建▲広▼生中霖生物科技有限公司 | Crystal form as an ASK1 inhibitor, its production method and utilization |
| TWI710561B (en) * | 2018-07-20 | 2020-11-21 | 大陸商福建廣生堂藥業股份有限公司 | Crystal form as ask1 inhibitor and preparation method and application thereof |
| US11814382B2 (en) | 2018-07-20 | 2023-11-14 | Fujian Akeylink Biotechnology Co., Ltd. | Crystal form as ASK1 inhibitor and preparation method and application thereof |
| WO2020015721A1 (en) * | 2018-07-20 | 2020-01-23 | 福建广生堂药业股份有限公司 | Crystal form as ask1 inhibitor, preparation method therefor, and application thereof |
| US10968199B2 (en) | 2018-08-22 | 2021-04-06 | Enanta Pharmaceuticals, Inc. | Cycloalkyl-containing apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| WO2020072656A1 (en) | 2018-10-03 | 2020-04-09 | Gilead Sciences, Inc. | Imidozopyrimidine derivatives |
| WO2020080741A1 (en) | 2018-10-18 | 2020-04-23 | Cj Healthcare Corporation | Novel n-(isopropyl-triazolyl)pyridinyl)-heteroaryl-carboxamide derivatives and use thereof |
| KR102298546B1 (en) | 2018-10-18 | 2021-09-08 | 에이치케이이노엔 주식회사 | Novel N-(isopropyl-triazolyl)pyridinyl)-heteroaryl-carboxamide derivatives and use thereof |
| KR20200044269A (en) * | 2018-10-18 | 2020-04-29 | 에이치케이이노엔 주식회사 | Novel N-(isopropyl-triazolyl)pyridinyl)-heteroaryl-carboxamide derivatives and use thereof |
| US12077523B2 (en) | 2018-10-18 | 2024-09-03 | Hk Inno.N Corporation | N-(isopropyl-triazolyl)pyridinyl)-heteroaryl-carboxamide derivatives and use thereof |
| WO2020086556A1 (en) | 2018-10-24 | 2020-04-30 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2020092528A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds having hpk1 inhibitory activity |
| WO2020092621A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| EP4371987A1 (en) | 2018-10-31 | 2024-05-22 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| US11345699B2 (en) | 2018-11-19 | 2022-05-31 | Enanta Pharmaceuticals, Inc. | Apoptosis signal-regulating kinase 1 inhibitors and methods of use thereof |
| WO2020178768A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| WO2020178769A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides and prodrugs thereof |
| US11766447B2 (en) | 2019-03-07 | 2023-09-26 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| WO2020178770A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| US11466033B2 (en) | 2019-03-25 | 2022-10-11 | Enanta Pharmaceuticals, Inc. | Substituted pyridines as apoptosis signal-regulating kinase 1 inhibitors |
| WO2020214663A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020214652A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020237025A1 (en) | 2019-05-23 | 2020-11-26 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| WO2020255038A1 (en) | 2019-06-18 | 2020-12-24 | Janssen Sciences Ireland Unlimited Company | Combination of hepatitis b virus (hbv) vaccines and pyridopyrimidine derivatives |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| WO2020261294A1 (en) * | 2019-06-26 | 2020-12-30 | Mankind Pharma Ltd. | Novel apoptosis signal-regulating kinase 1 inhibitors |
| EP4349413A2 (en) | 2019-10-18 | 2024-04-10 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| WO2021076908A1 (en) | 2019-10-18 | 2021-04-22 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| WO2021087064A1 (en) | 2019-10-31 | 2021-05-06 | Forty Seven, Inc. | Anti-cd47 and anti-cd20 based treatment of blood cancer |
| WO2021096860A1 (en) | 2019-11-12 | 2021-05-20 | Gilead Sciences, Inc. | Mcl1 inhibitors |
| WO2021130638A1 (en) | 2019-12-24 | 2021-07-01 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| EP4445902A2 (en) | 2019-12-24 | 2024-10-16 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| WO2021163064A2 (en) | 2020-02-14 | 2021-08-19 | Jounce Therapeutics, Inc. | Antibodies and fusion proteins that bind to ccr8 and uses thereof |
| US11692038B2 (en) | 2020-02-14 | 2023-07-04 | Gilead Sciences, Inc. | Antibodies that bind chemokine (C-C motif) receptor 8 (CCR8) |
| WO2021222522A1 (en) | 2020-05-01 | 2021-11-04 | Gilead Sciences, Inc. | Cd73 inhibiting 2,4-dioxopyrimidine compounds |
| WO2022221304A1 (en) | 2021-04-14 | 2022-10-20 | Gilead Sciences, Inc. | CO-INHIBITION OF CD47/SIRPα BINDING AND NEDD8-ACTIVATING ENZYME E1 REGULATORY SUBUNIT FOR THE TREATMENT OF CANCER |
| WO2022245671A1 (en) | 2021-05-18 | 2022-11-24 | Gilead Sciences, Inc. | Methods of using flt3l-fc fusion proteins |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2023076983A1 (en) | 2021-10-28 | 2023-05-04 | Gilead Sciences, Inc. | Pyridizin-3(2h)-one derivatives |
| WO2023077030A1 (en) | 2021-10-29 | 2023-05-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2023107956A1 (en) | 2021-12-08 | 2023-06-15 | Dragonfly Therapeutics, Inc. | Proteins binding nkg2d, cd16 and 5t4 |
| WO2023107954A1 (en) | 2021-12-08 | 2023-06-15 | Dragonfly Therapeutics, Inc. | Antibodies targeting 5t4 and uses thereof |
| WO2023122615A1 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023122581A2 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023147418A1 (en) | 2022-01-28 | 2023-08-03 | Gilead Sciences, Inc. | Parp7 inhibitors |
| WO2023178181A1 (en) | 2022-03-17 | 2023-09-21 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| EP4245756A1 (en) | 2022-03-17 | 2023-09-20 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023183817A1 (en) | 2022-03-24 | 2023-09-28 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| WO2023196784A1 (en) | 2022-04-05 | 2023-10-12 | Gilead Sciences, Inc. | Combinations of antibody therapies for treating colorectal cancer |
| WO2023205719A1 (en) | 2022-04-21 | 2023-10-26 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2024006929A1 (en) | 2022-07-01 | 2024-01-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2024064668A1 (en) | 2022-09-21 | 2024-03-28 | Gilead Sciences, Inc. | FOCAL IONIZING RADIATION AND CD47/SIRPα DISRUPTION ANTICANCER COMBINATION THERAPY |
| WO2024137852A1 (en) | 2022-12-22 | 2024-06-27 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| CN115844887A (en) * | 2022-12-28 | 2023-03-28 | 中国人民解放军空军军医大学 | Application of Selonsertib in preparation of medicine for treating cancer |
| CN115844887B (en) * | 2022-12-28 | 2023-09-08 | 中国人民解放军空军军医大学 | Use of Selonsertib in preparation of medicines for treating cancers |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10508100B2 (en) | Apoptosis signal-regulating kinase inhibitor | |
| AU2017201650B2 (en) | Apoptosis signal-regulating kinase inhibitor | |
| OA19542A (en) | Apoptosis Signal-Regulating Kinase Inhibitor. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013201586 Country of ref document: AU |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13703675 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 233598 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2862030 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 001153-2014 Country of ref document: PE |
|
| ENP | Entry into the national phase |
Ref document number: 2014554828 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014001988 Country of ref document: CL Ref document number: 201491283 Country of ref document: EA Ref document number: 12014501697 Country of ref document: PH Ref document number: MX/A/2014/009075 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013703675 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20140082 Country of ref document: MD Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A 2014 0082 Country of ref document: MD |
|
| ENP | Entry into the national phase |
Ref document number: 20147023533 Country of ref document: KR Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014018355 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14186994 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201407926 Country of ref document: UA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2014-000405 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 239311 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2016/0019 Country of ref document: RS |
|
| ENP | Entry into the national phase |
Ref document number: 112014018355 Country of ref document: BR Kind code of ref document: A2 Effective date: 20140725 |