WO2012168733A1 - Substituted 8 - amino - imidazo [1, 2-a] pyrazines as antibacterial agents - Google Patents
Substituted 8 - amino - imidazo [1, 2-a] pyrazines as antibacterial agents Download PDFInfo
- Publication number
- WO2012168733A1 WO2012168733A1 PCT/GB2012/051303 GB2012051303W WO2012168733A1 WO 2012168733 A1 WO2012168733 A1 WO 2012168733A1 GB 2012051303 W GB2012051303 W GB 2012051303W WO 2012168733 A1 WO2012168733 A1 WO 2012168733A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- substituted
- mmol
- heteroaryl
- aryl
- Prior art date
Links
- 0 *c1c(*)nc2[n]1ccnc2* Chemical compound *c1c(*)nc2[n]1ccnc2* 0.000 description 9
- SWGILLQQUULMJB-UHFFFAOYSA-N CC(c1nc2ccccc2nc1)=O Chemical compound CC(c1nc2ccccc2nc1)=O SWGILLQQUULMJB-UHFFFAOYSA-N 0.000 description 1
- JATMCAQQSXISOR-UHFFFAOYSA-N Cc(cc1)ccc1S(NCCN)(=O)=O Chemical compound Cc(cc1)ccc1S(NCCN)(=O)=O JATMCAQQSXISOR-UHFFFAOYSA-N 0.000 description 1
- MIHHWBVNJDEABA-UHFFFAOYSA-N Cc(cc1)ccc1S(Nc(cc1)ccc1Nc1ncc[n]2c1ncc2-c1ccc(cccc2)c2c1)(=O)=O Chemical compound Cc(cc1)ccc1S(Nc(cc1)ccc1Nc1ncc[n]2c1ncc2-c1ccc(cccc2)c2c1)(=O)=O MIHHWBVNJDEABA-UHFFFAOYSA-N 0.000 description 1
- ZCOYUCHIJHVYMJ-UHFFFAOYSA-N Cc(cc1)ccc1S(Nc1ccc(CNc2ncc[n]3c2nc(-c2cc4ccccc4cc2)c3)cc1)(=O)=O Chemical compound Cc(cc1)ccc1S(Nc1ccc(CNc2ncc[n]3c2nc(-c2cc4ccccc4cc2)c3)cc1)(=O)=O ZCOYUCHIJHVYMJ-UHFFFAOYSA-N 0.000 description 1
- KTEDMUMDGGOTDY-UHFFFAOYSA-N Cc1ncc[n]2c1nc(-c1ccc(cccc3)c3c1)c2 Chemical compound Cc1ncc[n]2c1nc(-c1ccc(cccc3)c3c1)c2 KTEDMUMDGGOTDY-UHFFFAOYSA-N 0.000 description 1
- UYVIHAGYLHHRKE-UHFFFAOYSA-N Clc1ncc[n]2c1nc(-c1c[s]cc1)c2 Chemical compound Clc1ncc[n]2c1nc(-c1c[s]cc1)c2 UYVIHAGYLHHRKE-UHFFFAOYSA-N 0.000 description 1
- ZWFNCFFGEPMORH-UHFFFAOYSA-N OC(CNc1nccnc1Cl)c(cccc1)c1Oc1ccccc1 Chemical compound OC(CNc1nccnc1Cl)c(cccc1)c1Oc1ccccc1 ZWFNCFFGEPMORH-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
Definitions
- the present invention relates to 2- or 3-substituted 8-amino-imidazo[1 ,2-a]pyrazines and derivatives thereof and methods of making such compounds.
- the present invention also relates to the use of these compounds as antibacterial agents and their use in methods of treating or preventing bacterial infection. Background to the Invention
- Gram-negative bacteria such as Helicobacter pylori, Legionella pneumophilia, Brucella suis, Bartonella henselae and Bordetella pertussis, can cause serious infections in both animals and plants and many resources have been devoted to developing pharmaceutical agents to kill such bacteria.
- these bacterial populations in a host can develop resistance to these pharmaceutical agents and survive, resulting in inefficient treatments which do not kill the bacteria in question.
- microorganisms have evolved a number of macromolecular secretion machineries to translocate proteins and nucleoprotein complexes from the bacterial cytosol to the host cell.
- Five types of secretion systems (l-V) have so far been identified, with a diverse range of functions including: the transfer of plasmid DNA from one cell to another (the major mechanism for the spread of antibiotic resistance genes between pathogenic bacteria); the secretion of proteins toxic to host cells; and the secretion of effector molecules required for the propagation of the microorganism within the host cell.
- Type IV secretion systems are vital for the pathogenicity of these important gram-negative bacteria.
- H. pylori utilizes the Type IV secretion system to translocate the toxic protein CagA into gastric epithelial cells, and in doing so induces a number of changes in the host cell.
- T4SS mediate transfer of plasmid DNAs from one cell to another and, as such, are the primary cause for the spread of antibiotics resistance genes.
- Type IV secretion systems require ATP as an energy source to drive this transport and therefore require a class of ATPases known as VirB1 1 ATPases, which are associated with the inner membrane.
- Z is O or NH
- Ri is selected from substituted alkyl, unsubstituted alkyl, substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
- R 2 and R 3 is H, and the other one of R 2 and R 3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
- L is a direct bond or is selected from: NH
- R 4 and R 5 are independently selected from H, alkyl, halo, alkoxy, alkylthio, hydroxy, cyano, amino and nitro;
- Z is O or NH
- R is selected from substituted alkyl, unsubstituted alkyl, substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
- R 2 and R 3 is H, and the other one of R 2 and R 3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
- L is a direct bond or is selected from:
- n 0 1 2, 3, 4, 5 or 6;
- R 4 and R 5 are independently selected from H, alkyl, halo, alkoxy, alkylthio, hydroxy, cyano, amino and nitro;
- Ri is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
- R 2 and R 3 is H, and the other one of R 2 and R 3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
- L is a direct bond or is selected from:
- R 4 and R 5 are independently selected from H, alkyl, halo, alkoxy, alkylthio, hydroxy, cyano, amino and nitro;
- a third aspect of the invention there is provided a compound as defined according to the second aspect or a pharmaceutically acceptable salt thereof for use in treating or preventing bacterial infection.
- a method of treating or preventing bacterial infection in a subject by administering a therapeutically effective amount of a compound according to the second aspect or a pharmaceutically acceptable salt thereof to a patient in need thereof.
- a fifth aspect of the invention there is provided a use of a compound of the second aspect in the manufacture of a medicament for treating or preventing bacterial infection or inhibiting the spread of antibiotic resistance genes.
- the bacterial infection may be infection with Helicobacter pylori, Legionella pneumophilia, Brucella suis, Bartonella henselae or Bordetella pertussis. Inhibition of conjugation applies to all bacterial Gram-negative pathogens.
- a pharmaceutical composition comprising a therapeutically effective amount of a compound according to the second aspect, optionally one or more other active ingredients pharmaceutically acceptable carrier.
- a seventh aspect of the invention there is provided a method of making a compound of the second aspect which comprises reacting a compound of Formula (II):
- X is a leaving group, for example a halo group (eg chloro), and R 2 and R 3 are as defined above, with a compound of the formula: wherein Ri , L and Z are as above.
- a seventh aspect of the invention there is further provided a method of making a compound of the second aspect which comprises reacting a compound of Formula (II):
- X is a leaving group, for example a halo group (eg chloro), and R 2 and R 3 are as defined above, with a compound of the formula: wherein R-i and L are as above.
- Ri , R 3 , L and Z are as above.
- a method of making the compounds of the second aspect in which R 3 is substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl, comprising:
- Ri , R 3 and L are as above.
- a ninth aspect there is provided a method of making a compound of the second aspect in which R 2 is substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl, comprising:
- Ri , R 2 , L and Z are as above.
- a ninth aspect there is also provided a method of making the compounds of the second aspect in which R 2 is substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl, comprising:
- Ri , R 2 and L are as above.
- a or “an” entity refers to one or more of that entity; for example, a compound refers to one or more compounds or at least one compound.
- a compound refers to one or more compounds or at least one compound.
- the terms “a” (or “an”), “one or more”, and “at least one” can be used interchangeably herein.
- alkyl denotes an unbranched or branched chain, saturated, monovalent hydrocarbon residue containing 1 or more carbon atoms.
- alkyl groups include, but are not limited to, lower alkyl groups include methyl, ethyl, propyl, / ' - propyl, n-butyl, / ' -butyl, i-butyl or pentyl, isopentyl, neopentyl and hexyl.
- alkyl may refer to lower alkyl groups of CrC 6 .
- substituted alkyi denotes an alkyi group containing one or more substituents selected from the group consisting of alkenyl, alkynyl, halogen, hydroxyl, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, arylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl, dialkylaminocarbonyl, alkylthiocarbonyl, alkoxyl, phosphate, phosphonato, phosphinato, cyano, amino, alkyi amino, dialkylamino, arylamino, diarylamino, alkylarylamino, acylamino, alkylcarbonylamino, arylcarbonylamino, carbamoyl, ureido, amidino, imino, sulfhydr
- alkylene as used herein means a divalent unbranched or branched saturated hydrocarbon radical consisting solely of carbon and hydrogen atoms, having 1 or more carbon atoms inclusive, for example lower alkylene groups of Ci-C 6 , unless otherwise indicated.
- alkylene radicals include, but are not limited to, methylene, ethylene, propylene, 2-methylethylene, 3-methylpropylene, 2-ethylethylene, pentylene, hexylene, and the like.
- haloalkyl denotes an unbranched or branched chain alkyi group as defined above wherein 1 , 2, 3 or more hydrogen atoms are substituted by a halogen. Examples are 1 -fluoromethyl, 1-chloromethyl, 1-bromomethyl, 1 -iodomethyl, trifluoromethyl, trichloromethyl, tribromomethyl, triiodomethyl, 1-fluoroethyl, 1- chloroethyl, 1-bromoethyl, 1 -iodoethyl, 2-fluoroethyl, 2-chloroethyl, 2-bromoethyl, 2- iodoethyl, 2,2-dichloroethyl, 3-bromopropyl or 2,2,2-trifluoroethyl.
- fluoroalkyl refers to a "haloalkyl” wherein the halogen is fluorine.
- cydoalkyl denotes a saturated carbocyclic ring containing 3 to 7 carbon atoms, e.g. cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cycloheptyl.
- the cydoalkyl group may be substituted with one or more substituents as defined above with reference to substituted alkyi.
- alkenyl denotes an unsubstituted hydrocarbon chain radical having 2 or more carbon atoms, preferably 2 to 6 carbon atoms and having one or two olefinic double bonds, for example C 2 -C 6 . Examples are vinyl, 1-propenyl, 2-propenyl (allyl) or 2-butenyl (crotyl).
- the alkenyl group may be substituted with one or more substituents as defined above with reference to substituted alkyl.
- alkynyl denotes an unsubstituted hydrocarbon chain radical having 2 or more carbon atoms, preferably 2 to 6 carbon atoms and having one or where possible two triple bonds. Examples are ethynyl, 1 -propynyl, 2-propynyl, 1 - butynyl, 2-butynyl or 3-butynyl.
- the alkynyl group may be substituted with one or more substituents as defined above with reference to substituted alkyl.
- alkoxy denotes an unsubstituted unbranched or branched chain alkyloxy group wherein the "alkyl” portion is as defined above such as methoxy, ethoxy, n-propyloxy, / ' -propyloxy, n-butyloxy, / ' -butyloxy, i-butyloxy, pentyloxy and hexyloxy including their isomers.
- haloalkoxy group as used herein means an O-haloalkyl group, wherein haloalkyl is as defined above.
- haloalkoxy groups include, but are not limited to, 2,2,2-trifluoroethoxy, difluoromethoxy and 1 ,1 , 1 ,3,3, 3-hexafluoro-iso- propoxy.
- thioalkyl or “alkylthio” as used herein refers to a group -SR where R is an alkyl group as defined herein such as methylthio, ethylthio, n-propylthio, i-propylthio and n-butylthio including their isomers.
- alkoxyalkyi refers to the radical R'R"-, wherein R' is an alkoxy radical as defined herein, and R" is an alkylene radical as defined herein with the understanding that the attachment point of the alkoxyalkyi moiety will be on the alkylene radical.
- hydroxyalkyl denotes the radical R'R" where R' is an hydroxy radical and R" is alkylene as defined herein and the attachment point of the hydroxyalkyl radical will be on the alkylene radical.
- acylating agent refers to a reagent which is capable of transferring an acyl moiety as defined previously to another functional group capable of reacting with the acylating agent.
- an alkylcarbonyl is introduced by reaction with an anhydride or an acyl halide.
- anhydride refers to compounds of the general structure RC(0)-0-C(0)R wherein R is as defined in the previous paragraph.
- acyl halide refers to the group RC(0)X wherein X is bromo or chloro.
- an alkoxycarbonyl is introduced by reaction with an alkoxycarbonyl chloride.
- heterocyclylalkyl as used herein means a radical R'R" where R' is an alkylene radical and R" is a heterocyclyl radical as defined herein.
- heterocyclylalkyl radicals include, but are not limited to, tetrahydropyran-2-ylmethyl, 2- piperidinylmethyl, 3-piperidinylmethyl, morpholin-1 -ylpropyl, and the like.
- alkylamino as used herein means a radical-NR'R", wherein R' is hydrogen and R" is an alkyl radical as defined herein.
- dialkylamino as used herein means a radical-NR'R", wherein R' and R" are alkyl radicals as defined herein.
- alkylamino radicals include, but are not limited to, methylamino, ethylamino, cyclopropylmethylamino, dicyclopropylmethylamino, dimethylamino, methylethylamino, diethylamino, di(1 -methylethyl)amino, and the like.
- aryl denotes an optionally substituted monocyclic or polycyclic-aromatic group comprising carbon and hydrogen atoms.
- suitable aryl groups include, but are not limited to, phenyl and naphthyl (e.g. 1 -naphthyl or 2-naphthyl).
- Suitable substituents for aryl are selected from the group consisting of Ci-6 alkyl, Ci -6 alkenyl, Ci -6 alkynyl, Ci -6 haloalkyl, Ci -6 alkoxy, Ci -6 haloalkoxy, Ci -6 alkylthio, arylthio, alkoxycarbonyl, amino, substituted amino for example alkylamino, CONR'R", carbamoyl, carbamate, ureido, amidino, imino, aryl, nitro, halogen and cyano.
- Optionally substituted phenyl in R-i or R 2 or R 3 can be for example 2- phenoxyphenyl, 2-methyl-phenyl, 3-methyl-phenyl, 4-methyl-phenyl, 2,3- dimethylphenyl, 2,4-dimethylphenyl, 2,5-dimethylphenyl, 2,6-dimethylphenyl, 3,4- dimethylphenyl, 3,5-dimethylphenyl, 2,3,4-trimethylphenyl, 3,4,5-trimethylphenyl, 2,3,4,5,6-pentamethylphenyl, 2-cyano-phenyl, 3-cyano-phenyl, 4-cyano-phenyl, 2,3- dicyanophenyl, 2,4-dicyanophenyl, 2,5-dicyanophenyl, 2,6-dicyanophenyl, 3,4- dicyanophenyl, 3,5-dicyanophenyl, 3,6-dicyanophenyl, 2-methoxyphenyl, 3- methoxyphenyl
- heteroaryl or “heteroaromatic” as used herein means a monocyclic or bicyclic radical of 5 to 12 ring atoms having at least one aromatic ring containing four to eight atoms per ring, incorporating one or more N, O, or S heteroatoms, the remaining ring atoms being carbon, with the understanding that the attachment point of said heteroaryl radical will be on said aromatic ring.
- heteroaryl rings have less aromatic character than their all-carbon counter parts. Thus, for the purposes of the invention, a heteroaryl group need only have some degree of aromatic character.
- heteroaryl moieties include monocyclic aromatic heterocycles having 5 to 6 ring atoms and 1 to 3 heteroatoms including but not limited to, pyridinyl, pyrimidinyl, pyrazinyl, pyridazinone, pyrrolyl, pyrazolyl, imidazolyl, triazolinyl, thiophenyl and oxadiaxolinyl which can optionally be substituted with one or more, preferably one or two substituents selected from hydroxy, cyano, alkyl, alkoxy, thio, lower haloalkoxy, alkylthio, halo, haloalkyl, alkylsulfinyl, alkylsulfonyl, halogen, amino, alkylamino, dialkylamino, aminoalkyl, alkylaminoalkyl, dialkylaminoalkyl, nitro, alkoxycarbonyl and carbamoyl, alkylcarb
- heterocyclylalkyl as used herein means a radical -R'R" where R' is an alkylene radical and R" is a heterocyclyl radical as defined herein.
- heterocyclylalkyl radicals include, but are not limited to, 2-piperidinylmethyl, 3- piperidinylmethyl, morpholin-1 -ylpropyl, and the like.
- heterocycle or “heterocyclic” as used herein means a non-aromatic monocyclic or polycyclic ring comprising carbon and hydrogen atoms and one or more N, S, or O heteroatoms.
- a heterocyclic group can have one or more carbon-carbon double bonds or carbon-heteroatom double bonds in the ring as long as the ring is not rendered aromatic by their presence.
- heterocycloalkyl groups include pyrrolidinyl, pyrrolidino, piperidinyl, piperidino, piperazinyl, piperazino, morpholinyl, morpholino, thiomorpholinyl, thiomorpholino.
- a heterocyclic group can be unsubstituted or substituted with one to three suitable substituents selected from hydroxy, cyano, alkyl, alkoxy, thio, lower haloalkoxy, alkylthio, halo, haloalkyl, alkylsulfinyl, alkylsulfonyl, halogen, amino, alkylamino, dialkylamino, aminoalkyl, alkylaminoalkyl, and dialkylaminoalkyl, nitro, alkoxycarbonyl and carbamoyl, alkylcarbamoyl and dialkylcarbamoyl.
- amino refers to-NH 2 ,-NHR and-NR 2 respectively and R is alkyl as defined above.
- the two alkyl groups attached to a nitrogen in a dialkyl moiety can be the same or different.
- aminoalkyl refers to NH2(CH 2 )n-, RHN(CH 2 ) n -, and R2N(CH 2 ) n - respectively wherein n is 1 or more and R is alkyl as defined above.
- halogen means fluorine, chlorine, bromine, or iodine.
- halo encompasses fluoro, chloro, bromo, and iodo.
- alkylthio or "thioalkyl” means an -S-alkyl group, wherein alkyl is as defined above such as meththio, ethylthio, n-propylthio, /-propylthio, n-butylthio, hexylthio, including their isomers.
- alkylsulfinyl as used herein means the radical-S(0)R', wherein R' is alkyl as defined herein. Examples of alkylaminosulfonyl include, but are not limited to methylsulfinyl and /so-propylsulfinyl.
- alkylsulfonyl as used herein means the radical-S(0) 2 R', wherein R' is alkyl as defined herein.
- alkylaminosulfonyl include, but are not limited to methylsulfonyl and /so-propylsulfonyl.
- sulfonylating agent refers to a reagent which is capable of transferring an alkyl sulfonyl moiety as defined previously to another functional group capable of reacting with the sulfonating agent such as a sulfonyl chloride Cl-S0 2 - .
- the present inventors have identified compounds of Formula (I) as being inhibitors of the bacterial ATPase VirB1 1 and its homologs, and thus being useful as antibacterial agents.
- the compounds when used as antibacterial agents reduce the spread of antibiotic resistance.
- R 2 is H; and R 3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl. In another embodiment, R 2 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl; and R 3 is H.
- R 4 and R 5 are independently selected from H, lower alkyl, halo (e.g. chloro, fluoro or bromo), alkoxy, alkylthio, hydroxy, cyano, amino and nitro.
- R 4 and R 5 are each hydrogen.
- Z is NH
- the amino substituent may be in the ortho, meta or para position. In another embodiment, the amino substituent is in the para position. In the embodiment where L is the amino substituent may be in the ortho, meta or para position. In another embodiment, the amino substituent is in the para position.
- R-i is unsubstituted or substituted alkyl.
- Ri is unsubstituted or substituted aryl. In another embodiment, Ri is unsubstituted or substituted heteroaryl.
- Ri is substituted aryl substituted with a poly(ethylene) glycol or PEG moiety.
- the PEG moiety may be coupled to the aryl group via a linker.
- the linker is a carbamate linker.
- the present invention encompasses any linker, for example but not limited to an amide, ester, carbonate, imidate, alkyl, alkenyl or alkynyl linker.
- the PEG moiety is coupled directly to the aryl group.
- the PEG moiety may itself be substituted.
- the PEG moiety may be substituted with an alkenyl, alkynyl or maleido group.
- the PEG moiety may comprise one or more PEG monomers, for example two PEG monomers, three PEG monomers or four PEG monomers.
- the PEG moiety may comprise (PEG)i, (PEG) 2 , (PEG) 3 or (PEG) 4 .
- the compound is not selected from the group consisting of:
- the compound is not selected from the compounds of Table 1 :
- the compound is selected from the group consisting of:
- the compound is selected from the compounds in Table 2. In an embodiment, any one of the compounds of Table 2 may be used in treating or preventing bacterial infection.
- R-i, R 2 and R 3 in the following methods are as defined above. All starting materials in the following general syntheses may be commercially available or obtained by conventional methods known to those skilled in the art. Solvents and reagents were obtained from commercial sources and were used as received unless otherwise stated. Dry solvents were dried over anhydrous columns. Moisture levels were usually ⁇ 15 ppm by Karl Fischer titration. Brine refers to a saturated solution of sodium chloride. Anhydrous magnesium sulfate (MgS0 4 ) or sodium sulfate (Na 2 S0 4 ) were used as drying agents after reaction workup, as indicated. Pet Ether refers to the fraction of light petroleum ether boiling in the range 40-60 °C.
- solvents include: halogenated hydrocarbons, such as dichloromethane, chloroform, carbon tetrachloride and 1 ,2-dichloroethane; ethers, such as diethyl ether, diisopropyl ether, tetrahydrofuran and dioxane; nitriles, such as acetonitrile and benzonitrile; aromatic hydrocarbons, such as benzene, toluene and nitrobenzene; amides, such as formamide, /V,/V-dimethylformamide, and ⁇ /,/V-dimethylacetamide sulfoxides, such as dimethyl sulfoxide and sulfolane; or mixed solvents thereof.
- halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride and 1 ,2-dichloroethane
- ethers such as diethyl ether, diisopropyl
- the synthesis of the amino alcohol was achieved via a-bromination of the aryl ketone with pyridinium tribromide. This was followed by substitution of the a-bromine with an azide, giving the azidoketone which was then reduced to give the corresponding azido-alcohol. Hydrogenation to give the amino alcohol was performed at atmospheric pressure, with the exception of the thiophene analogue which required 3 bar pressure to go to completion. Coupling with 2,3- dichloropyrazine in 1 ,4-dioxane afforded the pyrazinyl-amino alcohol in good yields. Swern oxidation to the ketone was followed by acid-induced cyclisation to form the 3- aryl-8-chloroimidazo[1 ,2-a]pyrazine.
- palladium catalysts which may be suitable include but are not limited to: palladium metal, palladium- carbon, palladium (II) acetate, tris(dibenzylideneacetone)dipalladiumchloroform, [1 ,2- bis(diphenylphosphino)ethane]palladium dichloride, bis(tri-o-toluylphosphine)palladium dichloride, bis(triphenylphosphine)palladium dichloride, tetrakis(triphenylphosphine) palladium or dichloro[1 ,1 '-bis(diphenylphosphino)ferrocene]palladium.
- Pd(dba) 2 is Pd(dba) 2 .
- Examples 1 to 6 were prepared according to this general method.
- Melting points were recorded on a Gallenkamp Melting Point Apparatus and are uncorrected.
- Proton nuclear magnetic resonance ( 1 H NMR) were recorded using Bruker AV400 (400 MHz), AV500 (500 MHz) and AV600 (600MHz) spectrometers as indicated.
- Carbon nuclear magnetic resonance ( 13 C NMR) were recorded using Bruker AV400 (100 MHz), AV500 (125 MHz) and AV600 (150MHz) spectrometers as indicated.
- Spectra were obtained using CDCI 3 , CD 3 OD, CD 2 CI 2 and DMSO-c/ 6 as solvents and chemical shifts are quoted on the ⁇ scale in units of ppm using TMS as an internal standard.
- Infra-Red (IR) spectroscopy was carried out using a PerkinElmer Spectrum 100 FT-IR Spectrometer using thin films. Absorption maxima (v max ) are reported in wavenumbers (cm "1 ).
- HPLC High Performance Liquid Chromatography
- LCMS Liquid Chromatography Mass Spectrometry
- NaHC0 3 sodium hydrogen carbonate
- compositions The present invention provides a pharmaceutical composition comprising a compound of Formula (I) or a pharmaceutically acceptable salt thereof, each as described herein, together with a pharmaceutically acceptable carrier for said compound. Also, the present invention provides a pharmaceutical composition comprising a compound of Formula (I) or a pharmaceutically acceptable salt thereof, each as described herein, further comprising other pharmacologically active agent(s).
- Pharmaceutically acceptable salts of a compound of Formula (I) include the acid addition salts (including disalts) thereof.
- Suitable acid addition salts are formed from acids which form non-toxic salts. Examples include the acetate, adipate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, cyclamate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate, palmitate, palmoate, phosphat
- a pharmaceutically acceptable salt of a compound of Formula (I) may be readily prepared by mixing together solutions of the compound of Formula (I) and the desired acid or base, as appropriate. The salt may precipitate from solution and be collected by filtration or may be recovered by evaporation of the solvent. The degree of ionization in the salt may vary from completely ionized to almost non-ionized.
- Pharmaceutically acceptable salts of the compounds of the invention include both unsolvated and solvated forms.
- solvate is used herein to describe a molecular complex comprising a compound of the invention and one or more pharmaceutically acceptable solvent molecules, for example, ethanol.
- the term 'hydrate' is employed when said solvent is water.
- solvates in accordance with the invention include hydrates and solvates wherein the solvent of crystallization may be isotopically substituted, e.g. D 2 0, c/ 6 -acetone, c/ 6 -DMSO.
- complexes such as clathrates, drug-host inclusion complexes wherein, in contrast to the aforementioned solvates, the drug and host are present in stoichiometric or non-stoichiometric amounts.
- complexes of the drug containing two or more organic and/or inorganic components which may be in stoichiometric or non-stoichiometric amounts.
- the resulting complexes may be ionized, partially ionized, or non-ionized.
- the compounds of Formula (I) may exist in one or more crystalline forms. These polymorphs, including mixtures thereof are also included within the scope of the present invention.
- the compounds of Formula (I) containing one or more asymmetric carbon atoms can exist as two or more stereoisomers.
- Compounds of the invention intended for pharmaceutical use may be administered as crystalline or amorphous products. They may be obtained, for example, as solid plugs, powders, or films by methods such as precipitation, crystallization, freeze-drying, spray drying, or evaporative drying. Microwave or radio frequency drying may be used for this purpose.
- compositions suitable for the delivery of compounds of the present invention and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation may be found, for example, in 'Remington's Pharmaceutical Sciences', 19th Edition (Mack Publishing Company, 1995).
- Oral Administration may involve swallowing, so that the compound enters the gastrointestinal tract, or buccal or sublingual administration may be employed by which the compound enters the blood stream directly from the mouth.
- Formulations suitable for oral administration include solid formulations such as, for example, tablets, capsules containing particulates, liquids, or powders, lozenges (including liquid-filled), chews, multi- and nano-particulates, gels, solid solution, liposome, films (including muco-adhesive), ovules, sprays and liquid formulations.
- Liquid formulations include, for example, suspensions, solutions, syrups and elixirs.
- Such formulations may be employed as fillers in soft or hard capsules and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents.
- a carrier for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents.
- Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet.
- the compounds of the invention may also be used in fast-dissolving, fast-disintegrating dosage forms such as those described in Expert Opinion in Therapeutic Patents. H (6), 981-986 by Liang and Chen (2001 ).
- the drug may make up from about 1 wt% to about 80 wt% of the dosage form, more typically from about 5 wt% to about 60 wt% of the dosage form.
- tablets generally contain a disintegrant.
- disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl-substituted hydroxypropyl cellulose, starch, pregelatinised starch and sodium alginate.
- the disintegrant will comprise from about 1 wt% to about 25 wt%, preferably from about 5 wt% to about 20 wt% of the dosage form.
- Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate.
- lactose monohydrate, spray-dried monohydrate, anhydrous and the like
- mannitol xylitol
- dextrose sucrose
- sorbitol microcrystalline cellulose
- starch dibasic calcium phosphate dihydrate
- Tablets may also optionally comprise surface-active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc.
- surface active agents may comprise from about 0.2 wt% to about 5 wt% of the tablet, and glidants may comprise from about 0.2 wt% to about 1 wt% of the tablet.
- Tablets also generally contain lubricants such as magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate.
- Lubricants generally comprise from about 0.25 wt% to about 10 wt%, preferably from about 0.5 wt% to about 3 wt% of the tablet.
- ingredients include anti-oxidants, colourants, flavouring agents, preservatives and taste-masking agents.
- Exemplary tablets contain up to about 80% drug, from about 10 wt% to about 90 wt% binder, from about 0 wt% to about 85 wt% diluent, from about 2 wt% to about 10 wt% disintegrant, and from about 0.25 wt% to about 10 wt% lubricant.
- Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting.
- the final formulation may comprise one or more layers and may be coated or uncoated; it may even be encapsulated.
- Solid formulations for oral administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- Suitable modified release formulations for the purposes of the invention are described in US Patent No. 6,106,864. Details of other suitable release technologies such as high energy dispersions and osmotic and coated particles are to be found in Verma et al, Pharmaceutical Technology On-line. 25(2), 1-14 (2001 ). The use of chewing gum to achieve controlled release is described in WO00/35298. Parenteral Administration
- the compounds of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ.
- Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular and subcutaneous.
- Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
- Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from about 3 to about 9), but, for some applications, they may be more suitably formulated as a sterile non-aqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
- excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from about 3 to about 9)
- a suitable vehicle such as sterile, pyrogen-free water.
- parenteral formulations under sterile conditions may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
- solubility of compounds of Formula (I) used in the preparation of parenteral solutions may be increased by the use of appropriate formulation techniques, such as the incorporation of solubility- enhancing agents.
- Formulations for parenteral administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- compounds of the invention may be formulated as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound. Examples of such formulations include drug-coated stents and PGLA microspheres.
- the compounds of the invention may also be administered topically to the skin or mucosa, that is, dermally or transdermally.
- Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibres, bandages and microemulsions. Liposomes may also be used.
- Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol.
- Penetration enhancers may be incorporated - see, for example, J Pharm Sci, 88 (10), 955-958 by Finnin and Morgan (October 1999).
- Other means of topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and microneedle or needle-free (e.g. Powderject(TM), Bioject(TM), etc.) injection.
- Formulations for topical administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- the compounds of the invention can also be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an aerosol spray from a pressurized container, pump, spray, atomiser (preferably an atomiser using electrohydrodynamics to produce a fine mist), or nebuliser, with or without the use of a suitable propellant, such as 1 ,1 ,1 ,2-tetrafluoroethane or 1 ,1 ,1 ,2,3,3,3- heptafluoropropane.
- a suitable propellant such as 1 ,1 ,1 ,2-tetrafluoroethane or 1 ,1 ,1 ,2,3,3,3- heptafluoropropane.
- the powder may comprise a bioadhesive agent, for example, chitosan or cyclodextrin.
- the pressurized container, pump, spray, atomizer, or nebuliser contains a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- the drug product Prior to use in a dry powder or suspension formulation, the drug product is micronised to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenization, or spray drying.
- comminuting method such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenization, or spray drying.
- Capsules made, for example, from gelatin or HPMC
- blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the compound of the invention, a suitable powder base such as lactose or starch and a performance modifier such as /-leucine, mannitol, or magnesium stearate.
- the lactose may be anhydrous or in the form of the monohydrate, preferably the latter.
- Other suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
- a suitable solution formulation for use in an atomiser using electrohydrodynamics to produce a fine mist may contain from about 1 ⁇ g to about 20mg of the compound of the invention per actuation and the actuation volume may vary from about 1 ⁇ to about 10 ⁇ .
- a typical formulation may comprise a compound of Formula (I), propylene glycol, sterile water, ethanol and sodium chloride.
- Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
- Suitable flavors such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations of the invention intended for inhaled/intranasal administration.
- Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, poly(DL-lactic-coglycolic acid (PGLA).
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- the dosage unit is determined by means of a valve which delivers a metered amount.
- Units in accordance with the invention are typically arranged to administer a metered dose or "puff' containing from about 1 to about 100 ⁇ g of the compound of Formula (I).
- the overall daily dose will typically be in the range about 50 ⁇ g to about 20 mg which may be administered in a single dose or, more usually, as divided doses throughout the day.
- the compounds of the invention may be combined with soluble macromolecular entities, such as cyclodextrin and suitable derivatives thereof or polyethylene glycol- containing polymers, in order to improve their solubility, dissolution rate, taste-masking, bioavailability and/or stability for use in any of the aforementioned modes of administration.
- soluble macromolecular entities such as cyclodextrin and suitable derivatives thereof or polyethylene glycol- containing polymers
- Drug-cyclodextrin complexes are found to be generally useful for most dosage forms and administration routes. Both inclusion and non-inclusion complexes may be used.
- the cyclodextrin may be used as an auxiliary additive, i.e. as a carrier, diluent, or solubiliser. Most commonly used for these purposes are alpha-, beta- and gamma-cyclodextrins, examples of which may be found in. W091/1 1 172, WO94/02518 and W098/55148.
- kits suitable for coadministration of the compositions may conveniently be combined in the form of a kit suitable for coadministration of the compositions.
- the kit of the invention comprises two or more separate pharmaceutical compositions, at least one of which contains a compound of Formula (I) in accordance with the invention, and means for separately retaining said compositions, such as a container, divided bottle, or divided foil packet.
- An example of such a kit is the familiar blister pack used for the packaging of tablets, capsules and the like.
- the kit of the invention is particularly suitable for administering different dosage forms, for example, oral and parenteral, for administering the separate compositions at different dosage intervals, or for titrating the separate compositions against one another.
- the kit typically comprises directions for administration and may be provided with a so-called memory aid.
- the total daily dose of the compounds of the invention is typically in the range of about 0.05 mg to about 500 mg depending, of course, on the mode of administration, preferred in the range of about 0.1 mg to about 400 mg and more preferred in the range of about 0.5 mg to about 300 mg.
- oral administration may require a total daily dose of from about 1 mg to about 300 mg, while an intravenous dose may only require from about 0.5 mg to about 100 mg.
- the total daily dose may be administered in single or divided doses. These dosages are based on an average human subject having a weight of about 65 kg to about 70 kg. The physician will readily be able to determine doses for subjects whose weight falls outside this range, such as infants and the elderly.
- the reaction was then allowed to slowly warm to RT over a period of 2 1 ⁇ 2 h.
- the reaction was quenched with H 2 0 (50 mL) and organics extracted followed by washing with 2.0 M HCI (2 x 40 mL), sat. NaHC0 3 (1 x 40 mL), H 2 0 (1 x 40 mL) and brine (1 x 40 mL). Drying (MgS0 4 ), filtration and concentration gave a yellow solid (2.31 , 8.37 mmol, 90.0%).
- Steps 1 to 6 were carried out as per Steps 1 to 6 of Example
- Step 1 was carried out as per Step 1 of Example 2 Step 2
- Step 1 was carried out as per Step 1 of Example 3.
- Step 2 is carried out as per Step 1 of Example 3.
- Step 1 was carried out as per Step 1 of Example 4 Step 2
- Steps 1 and 2 were carried out as per Steps 1 and 2 of Example 8 to give compound JS18.
- Tris(dibenzylideneacetone)dipalladium(0) ( ⁇ 1 mg, 1 .1 ⁇ ), 2-Dicyclohexylphosphino- 2'-(/V,/V-dimethylamino)biphenyl ( ⁇ 1 mg, 2.5 ⁇ ) and Sodium-ie f-butoxide ( ⁇ 12.5 mg, 130 ⁇ ) were added to a flask and dissolved in 5 ml anhydrous toluene.
- JS1 1 1 (648 mg, 2.58 mmol), 2-amino-3-chloropyrazine (334.4 mg, 2.58 mmol) and NaHC0 3 (271.0 mg, 3.23 mmol) in ie f-butanol (15 ml) were stirred under reflux for 40 h. The solvent was removed and the resulting residual was taken up in CH 2 CI 2 and washed H 2 0 and brine, dried (MgS0 4 ), filtered and solvent removed. Flash chromatography (Pet Ether; 3:1 to 1 :1 to 1 :3 Pet Ether/EtOAc) afforded the title compound as an orange solid (341 .9 mg, 1 .21 mmol, 47.1 %).
- JS1 14 (100 mg, 0.355 mmol), /V-(4-aminophenyl)-4-methylbenzenesulfonamide (1 1 1 .8 mg, 0.426 mmol), Pd(dba) 2 (2.0 mg, 1 mol%), DavePhos (4.2 mg, 3 mol%) and Cs 2 C0 3 (162.0 mg, 0.497 mmol) were stirred in anhydrous 1 ,4-dioxane under reflux for 40 h. After LCMS confirmed the presence of the product mass, the reaction was cooled to RT and solvent removed. The resulting residue was taken up in CH 2 CI 2 and washed with sat. aq.
- Tris(dibenzylideneacetone)dipalladium(0) ( ⁇ 2 mg, 2.2 ⁇ mol), 2-dicyclohexylphosphino- 2'-(N,N-dimethylamino)biphenyl ( ⁇ 3 mg, 7.6 ⁇ ) and sodium-tertbutoxide ( ⁇ 7 mg, 73 ⁇ ) were added to a flask and dissolved in 5 ml anhydrous toluene.
- 2-(8- chloroimidazo[1 ,2-a]pyrazin-2-yl)quinoline (HK006, 15 mg, 53 ⁇ ) and N-(4- aminophenyl)-4-methylbenzenesulfonamide (15 mg, 57 ⁇ ) were added under argon atmosphere.
- the main fraction was collected in three split subfractions; only the first subfraction was furtherly used; 1 H-NMR, but not LCMS showed that the other subfractions were not appropriate.
- the product was dried under high vacuum after evaporation of the solvent (coevaporation with dichloromethane). 0.8 mg (1 .6 ⁇ , yield: 3%) of the solid product were obtained with minor to moderate impurities and characterized by 1 H-NMR and an IC50 assay. Further characterization of the product was impeded by problems to dissolve the compound, occurrence of new relatively significant impurities and the small amount of the sample.
- Benzene-1 ,4-diamine (3.14 g, 29.0 mmol), was dissolved in anhydrous CH 2 CI 2 (100 ml), triethylamine (1 .62 ml, 1 1.61 mmol) added and the mixtures cooled on ice. MeS0 2 CI (0.449 ml, 5.81 mmol) was added drop wise and the reaction was stirred at RT for 16 h. The mixture was then diluted with CH 2 CI 2 and washed sat. aq. NaHC0 3 . The aqueous layer was then extracted with CH 2 CI 2 (4x) as well as EtOAc (4x).
- JS169 (18 mg, 0.049 mmol) was dissolved in anhydrous pyridine (1 ml) and the mixture was cooled on ice. 4-methylbenzene-1-sulfonyl chloride (1 1 .3 mg, 0.059 mmol) was added and the deep yellow/orange solution was stirred under Ar at RT for 16 h.
- JS1 1 (See Step 1 of Example 7) (1 .12 g, 4.01 mmol) was dissolved in anhydrous DMSO (16 ml). NaSMe (337 mg, 4.81 mmol) was added portion wise and the reaction was stirred at 100 °C for 16 h. The mixture was then cooled to RT, diluted with brine and extracted with CH 2 CI 2 . The organic layer was washed with H 2 0 (5x) and brine (1 x), dried (MgS0 4 ), filtered and solvent removed in vacuo.
- JS130 (1.7662 g, 6.07 mmol) was dissolved in anhydrous CH 2 CI 2 (50 ml) and the mixture was cooled on ice.
- mCPBA (5.231 g, 30.3 mmol) was added in one portion and the reaction continued to stir at RT for 5 h.
- the reaction was partitioned with NaHC0 3 and extracted with CH 2 CI 2 (3x); the combined organic extracts were then washed with brine, dried (MgS0 4 ), filtered and concentrated in vacuo. Flash chromatography (CH 2 CI 2 ; 2% to 5% EtOAc) afforded the title compound as a yellow solid (1 .098 g, 3.398 mmol, 56.0%).
- NaH was pre-activated by stirring NaH (60% in Mineral Oil; 12.4 mg, 0.310 mmol) in anhydrous hexanes (3 ml) for 20 min, removing the solvent using a syringe and drying the contents under high vacuum.
- DMF 0.5 ml
- JS168 81 .4 mg, 0.310 mmol
- JS132 50 mg, 0.155 mmol
- DMF 1 .5 ml
- JS206 (556.5 mg, 5.57 mmol) was dissolved in anhydrous toluene (140 ml).
- JS129 (see Example 23) (1 .509 g, 6.68 mmol) and molecular sieves (4A, 10 sieves) were added and the reaction was stirred under reflux for 16 h. The solvent was removed and the reaction purified via flash chromatography (Toluene; 2:1 Toluene/EtOAc) to afford the title compound as a white solid (663 mg, 2.22 mmol, 39.9%).
- NaH was pre-activated by stirring NaH (60% in Mineral Oil; 88.7 mg, 2.215 mmol) in anhydrous hexanes (25 ml) for 20 min, removing the solvent using a syringe and drying the contents under high vacuum.
- DMF (6 ml) was added followed by JS207 (660 mg, 2.215 mmol) in DMF (8 ml) and the mixture was stirred at RT for 20 min.
- JS132 (357.7 mg, 1 .107 mmol) in DMF (20 ml) was added and the resulting dark brown solution was heated at 100 °C under Argon for 18 h.
- NaH was pre-activated by stirring NaH (60% in Mineral Oil; 70.6 mg, 1 .765 mmol) in anhydrous hexanes (50 ml) for 20 min, removing the solvent using a syringe and drying the contents under high vacuum.
- DMF 5 ml
- JS121 529 mg, 1.765 mmol
- JS132 285 mg, 0.882 mmol
- DMF 13 ml
- JS136 (993 mg, 4.39 mmol) was dissolved in anhydrous toluene (92 ml).
- JS129 (750.6 mg, 3.66 mmol) was added and the reaction was stirred under reflux for 16 h. The reaction was cooled to RT and adsorbed onto silica and purification via flash chromatography (Pet Ether; 2:1 to 1 :1 to 1 :2 Pet Ether/EtOAc) afforded the title compound as a white solid (1 .1306 g, 2.81 mmol, 76.7%).
- NaH was pre-activated by stirring NaH (60% in Mineral Oil; 71 .0 mg, 1 .774 mmol) in anhydrous hexanes (25 ml) for 20 min, removing the solvent using a syringe and drying the contents under high vacuum.
- DMF 5 ml
- JS138 715 mg, 1.774 mmol
- DMF 8 ml
- JS132 286.5 mg, 0.887 mmol
- JS179 (317.9 mg, 0.492 mmol) was dissolved in anhydrous CH 2 CI 2 (15 ml) and cooled on ice. TFA (15 ml) was added and the reaction was stirred at RT for 3 h. Removal of the solvent, with the aid of toluene, followed by flash chromatography (CH 2 CI 2 ; 5% to 10% MeOH/CH 2 CI 2 ).
- the VirB1 1 protein was produced in the E. coli strain BL21 Star(DE3) (Invitrogen) as described previously. The protein concentration was estimated spectroscopically using a NanoDrop (Thermo Scientific) and a calculated extinction coefficient at 280 nm, based on the amino acid composition. The ATPase activity of VirB1 1 was measured, with and without a specific amount of compound present, using an in vitro ATPase colorimetric assay kit (Innova Biosciences). The assay was performed in 96-well ELISA microplates (Greiner Bio-One), using a multipipett robot. All measurements were made in duplicate.
- a volume of 49 ⁇ _ of a substrate/buffer solution (200 mM tris(hydroxymethyl)aminomethane (TRIS), pH 7.5; 5 mM MgCI 2 ; 250 ⁇ ATP; and 10% DMSO) was added to each assigned well, followed by the addition of 1 ⁇ _ of compound (at 0.5; 5 or 50 mM to achieve the final concentrations of 5; 50 and 500 ⁇ in the reaction, respectively) in DMSO (or 1 ⁇ _ DMSO to controls).
- the solutions were mixed carefully by pipetting.
- the reaction was started by the addition of 50 ⁇ _ of 0.106 ⁇ VirB1 1 to each well (except the negative control, see text below), and the reaction plate was directly transferred to 37 °C for 30 min of incubation.
- the reaction was stopped by the addition of the Gold mix according to the standard protocol of the kit.
- the absorbance at 620 nm was measured after 30 min at room temperature.
- the percentage of absorbance relative non-inhibited VirB1 1 was calculated, after subtracting the absorbance value of the negative control.
- the protein was added after the Gold mix, as described in the standard protocol of the kit, which when used as a blank corrects for all free P, not produced by the enzyme during the 30 min incubation at 37 °C.
- VirB1 1 A known inhibitor of VirB1 1 (Microbiology, (2006), 152, 2919-2930, denoted CHIR02) was used as a control inhibitor. A selection of the compounds were assayed as above at additional concentrations ranging between 5 and 200 ⁇ from which IC50 values were calculated, assuming a linear relationship between P, production and absorbance. IC50 was defined as the concentration of compound that produces 50% inhibition. Evaluation of compounds as VirB1 1 ATPase inhibitors
- the VirB1 1 protein was produced recombinantly in Escherichia coli and purified to high purity as described previously.
- the ATPase activity of VirB1 1 was measured by monitoring the release of inorganic phosphate (P,) using an in vitro ATPase assay.
- Examples 1 to 12 were evaluated by performing the ATPase assay with and without compound present at concentrations of 500 ⁇ (or 250 ⁇ ), 50 ⁇ and 5 ⁇ (Fig. 1 ). At the highest concentration, precipitation appeared in the reaction wells of compounds 1 , 2, 5, 8, 9 and 1 1 , which interfered with the measurements. Despite this, a clear inhibitory effect could be seen of the compounds 1 , 3, 4, 5, 6, 1 1 and 12 of which 4, 5, 8, 1 1 and 12 clearly reduced the ATPase activity at 50 ⁇ . IC 50 values, logP and logS values were then measured for a selection of the compounds 1 to 23 (Table 3).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to substituted imidazofi,2-a]pyrazines of Formula (I) and their use as antibacterial agents.
Description
SUBSTITUTED 8 - AMINO - IMIDAZO [1 , 2-A] PYRAZINES
AS ANTIBACTERIAL AGENTS
Field of the Invention The present invention relates to 2- or 3-substituted 8-amino-imidazo[1 ,2-a]pyrazines and derivatives thereof and methods of making such compounds. The present invention also relates to the use of these compounds as antibacterial agents and their use in methods of treating or preventing bacterial infection. Background to the Invention
Gram-negative bacteria, such as Helicobacter pylori, Legionella pneumophilia, Brucella suis, Bartonella henselae and Bordetella pertussis, can cause serious infections in both animals and plants and many resources have been devoted to developing pharmaceutical agents to kill such bacteria. However, through natural selection these bacterial populations in a host can develop resistance to these pharmaceutical agents and survive, resulting in inefficient treatments which do not kill the bacteria in question.
These microorganisms have evolved a number of macromolecular secretion machineries to translocate proteins and nucleoprotein complexes from the bacterial cytosol to the host cell. Five types of secretion systems (l-V) have so far been identified, with a diverse range of functions including: the transfer of plasmid DNA from one cell to another (the major mechanism for the spread of antibiotic resistance genes between pathogenic bacteria); the secretion of proteins toxic to host cells; and the secretion of effector molecules required for the propagation of the microorganism within the host cell.
Type IV secretion systems (T4SS) are vital for the pathogenicity of these important gram-negative bacteria. For example, H. pylori utilizes the Type IV secretion system to translocate the toxic protein CagA into gastric epithelial cells, and in doing so induces a number of changes in the host cell. Also, T4SS mediate transfer of plasmid DNAs from one cell to another and, as such, are the primary cause for the spread of antibiotics resistance genes.
Type IV secretion systems require ATP as an energy source to drive this transport and therefore require a class of ATPases known as VirB1 1 ATPases, which are associated with the inner membrane. Targeting the ATPase activity of VirB1 1 therefore represents an attractive approach to generating novel antibacterial agents. There has been one previous report of inhibition of the cag VirB1 1 -type ATPase Caga (Microbiology, (2006), 152, 2919-2930).
It is therefore an object of the present invention to identify inhibitors of the VirB1 1 ATPase from H. pylori and its homologs in other gram-negative bacteria. These inhibitors should also be well absorbed from the gastrointestinal tract, be metabolically stable and possess favourable pharmacokinetic properties. Furthermore, the ideal drug candidate will exist in a physical form that is stable, non-hygroscopic and easily formulated.
Summary of the Invention
According to a first aspect of the invention there is provided a compound of Formula (I) for use in preventing or treating a bacterial infection:

Z is O or NH;
Ri is selected from substituted alkyl, unsubstituted alkyl, substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
one of R2 and R3 is H, and the other one of R2 and R3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
L is a direct bond or is selected from:
NH
(CH2)n
= 0, 1 , 2, 3, 4, 5 or 6;

(CH2)n
is a 5- or 6-membered nitrogen-containing heteroaryl moiety, optionally containing at least one or more further heteroatom; and
R4 and R5 are independently selected from H, alkyl, halo, alkoxy, alkylthio, hydroxy, cyano, amino and nitro;
or a pharmaceutically acceptable salt, solvate or prodrug thereof. According to a second aspect of the invention there is provided a compound of Formula (I):
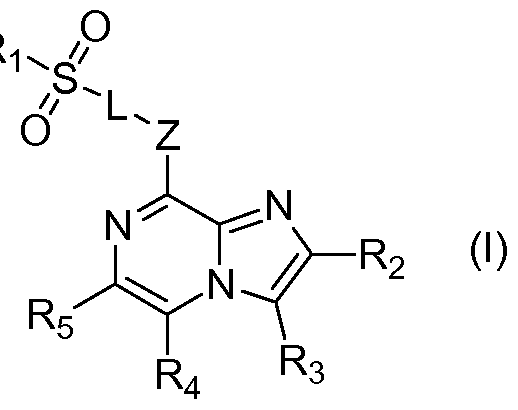
Z is O or NH;
R is selected from substituted alkyl, unsubstituted alkyl, substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
one of R2 and R3 is H, and the other one of R2 and R3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
L is a direct bond or is selected from:
NH
(CH2)n
wherein n = 0 1 2, 3, 4, 5 or 6;

each 0f w ich may optionally be substituted at one or more exocyclic positions; and wherein n = 0, 1 , 2, 3, 4, 5 or 6; wherein
(CH2)n
is a 5- or 6-membered nitrogen-containing heteroaryl moiety, optionally containing at least one or more further heteroatom; and
R4 and R5 are independently selected from H, alkyl, halo, alkoxy, alkylthio, hydroxy, cyano, amino and nitro;
or a pharmaceutically acceptable salt, solvate or prodrug thereof.
According to a second aspect of the present invention there is also provided a compound of Formula (la):

Ri is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
one of R2 and R3 is H, and the other one of R2 and R3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
L is a direct bond or is selected from:

R4 and R5 are independently selected from H, alkyl, halo, alkoxy, alkylthio, hydroxy, cyano, amino and nitro;
or a pharmaceutically acceptable salt, solvate or prodrug thereof. According to a third aspect of the invention there is provided a compound as defined according to the second aspect or a pharmaceutically acceptable salt thereof for use in treating or preventing bacterial infection.
According to a fourth aspect of the invention there is provided a method of treating or preventing bacterial infection in a subject by administering a therapeutically effective amount of a compound according to the second aspect or a pharmaceutically acceptable salt thereof to a patient in need thereof.
According to a fifth aspect of the invention there is provided a use of a compound of the second aspect in the manufacture of a medicament for treating or preventing bacterial infection or inhibiting the spread of antibiotic resistance genes.
The bacterial infection may be infection with Helicobacter pylori, Legionella pneumophilia, Brucella suis, Bartonella henselae or Bordetella pertussis. Inhibition of conjugation applies to all bacterial Gram-negative pathogens.
According to a sixth aspect of the invention there is provided a pharmaceutical composition comprising a therapeutically effective amount of a compound according to
the second aspect, optionally one or more other active ingredients pharmaceutically acceptable carrier.
According to a seventh aspect of the invention, there is provided a method of making a compound of the second aspect which comprises reacting a compound of Formula (II):

wherein X is a leaving group, for example a halo group (eg chloro), and R2 and R3 are as defined above, with a compound of the formula:
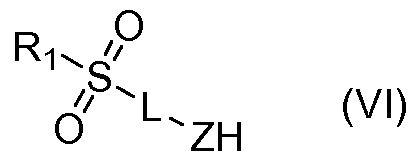 wherein Ri , L and Z are as above.
wherein Ri , L and Z are as above.
According to a seventh aspect of the invention, there is further provided a method of making a compound of the second aspect which comprises reacting a compound of Formula (II):

wherein X is a leaving group, for example a halo group (eg chloro), and R2 and R3 are as defined above, with a compound of the formula:
 wherein R-i and L are as above.
wherein R-i and L are as above.
According to an eighth aspect of the invention there is provided a method of making a compound of the second aspect in which R3 is substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl, comprising:
(i) reacting a compound of Formula (IV)

with 2,3-dichloropyrazine;
(ii) oxidising the product of (i);
(iii) effecting an intramolecular cyclisation of the product of (ii); and
(iv) coupling the product of (iii) with a compound of Formula (VI)

wherein
Ri , R3, L and Z are as above. According to an eighth aspect of the invention there is further provided a method of making the compounds of the second aspect in which R3 is substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl, comprising:
(i) reacting a compound of Formula (IV)

with 2,3-dichloropyrazine;
(ii) oxidising the product of (i);
(iii) effecting an intramolecular cyclisation of the product of (ii); and
(iv) coupling the product of (iii) with a compound of Formula (III)

wherein
Ri , R3 and L are as above.
According to a ninth aspect there is provided a method of making a compound of the second aspect in which R2 is substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl, comprising:
(i) reacting a compound of Formula (V)
 with 2-amino-3-chloropyrazine;
with 2-amino-3-chloropyrazine;
(ii) coupling the product of (i) with a compound of Formula (VI)

wherein
Ri , R2, L and Z are as above.
According to a ninth aspect there is also provided a method of making the compounds of the second aspect in which R2 is substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl, comprising:
(i) reacting a compound of Formula (V)
 with 2-amino-3-chloropyrazine;
with 2-amino-3-chloropyrazine;
(ii) coupling the product of (i) with a compound of Formula (III)

wherein
Ri , R2 and L are as above.
Definitions
The phrase "a" or "an" entity as used herein refers to one or more of that entity; for example, a compound refers to one or more compounds or at least one compound. As such, the terms "a" (or "an"), "one or more", and "at least one" can be used interchangeably herein.
The term "alkyl" as used herein denotes an unbranched or branched chain, saturated, monovalent hydrocarbon residue containing 1 or more carbon atoms. Examples of alkyl groups include, but are not limited to, lower alkyl groups include methyl, ethyl, propyl, /'- propyl, n-butyl, /'-butyl, i-butyl or pentyl, isopentyl, neopentyl and hexyl. For example, alkyl may refer to lower alkyl groups of CrC6.
The term "substituted alkyi" as used herein denotes an alkyi group containing one or more substituents selected from the group consisting of alkenyl, alkynyl, halogen, hydroxyl, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, arylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl, dialkylaminocarbonyl, alkylthiocarbonyl, alkoxyl, phosphate, phosphonato, phosphinato, cyano, amino, alkyi amino, dialkylamino, arylamino, diarylamino, alkylarylamino, acylamino, alkylcarbonylamino, arylcarbonylamino, carbamoyl, ureido, amidino, imino, sulfhydryl, alkylthio, arylthio, thiocarboxylate, sulfate, alkylsulfinyl, sulfonate sulfamoyl, sulfonamido, nitro, trifluoromethyl, azido, heterocyclyl, alkylaryl or an aromatic or heteroaromatic group.
The term "alkylene" as used herein means a divalent unbranched or branched saturated hydrocarbon radical consisting solely of carbon and hydrogen atoms, having 1 or more carbon atoms inclusive, for example lower alkylene groups of Ci-C6, unless otherwise indicated. Examples of alkylene radicals include, but are not limited to, methylene, ethylene, propylene, 2-methylethylene, 3-methylpropylene, 2-ethylethylene, pentylene, hexylene, and the like.
The term "haloalkyl" as used herein denotes an unbranched or branched chain alkyi group as defined above wherein 1 , 2, 3 or more hydrogen atoms are substituted by a halogen. Examples are 1 -fluoromethyl, 1-chloromethyl, 1-bromomethyl, 1 -iodomethyl, trifluoromethyl, trichloromethyl, tribromomethyl, triiodomethyl, 1-fluoroethyl, 1- chloroethyl, 1-bromoethyl, 1 -iodoethyl, 2-fluoroethyl, 2-chloroethyl, 2-bromoethyl, 2- iodoethyl, 2,2-dichloroethyl, 3-bromopropyl or 2,2,2-trifluoroethyl. The term "fluoroalkyl" refers to a "haloalkyl" wherein the halogen is fluorine.
The term "cydoalkyl" as used herein denotes a saturated carbocyclic ring containing 3 to 7 carbon atoms, e.g. cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cycloheptyl. In one embodiment, the cydoalkyl group may be substituted with one or more substituents as defined above with reference to substituted alkyi.
The term "alkenyl" as used herein denotes an unsubstituted hydrocarbon chain radical having 2 or more carbon atoms, preferably 2 to 6 carbon atoms and having one or two olefinic double bonds, for example C2-C6. Examples are vinyl, 1-propenyl, 2-propenyl
(allyl) or 2-butenyl (crotyl). In one embodiment, the alkenyl group may be substituted with one or more substituents as defined above with reference to substituted alkyl.
The term "alkynyl" as used herein denotes an unsubstituted hydrocarbon chain radical having 2 or more carbon atoms, preferably 2 to 6 carbon atoms and having one or where possible two triple bonds. Examples are ethynyl, 1 -propynyl, 2-propynyl, 1 - butynyl, 2-butynyl or 3-butynyl. In one embodiment, the alkynyl group may be substituted with one or more substituents as defined above with reference to substituted alkyl.
The term "alkoxy" as used herein denotes an unsubstituted unbranched or branched chain alkyloxy group wherein the "alkyl" portion is as defined above such as methoxy, ethoxy, n-propyloxy, /'-propyloxy, n-butyloxy, /'-butyloxy, i-butyloxy, pentyloxy and hexyloxy including their isomers.
The term "haloalkoxy group" as used herein means an O-haloalkyl group, wherein haloalkyl is as defined above. Examples of haloalkoxy groups include, but are not limited to, 2,2,2-trifluoroethoxy, difluoromethoxy and 1 ,1 , 1 ,3,3, 3-hexafluoro-iso- propoxy.
The term "thioalkyl" or "alkylthio" as used herein refers to a group -SR where R is an alkyl group as defined herein such as methylthio, ethylthio, n-propylthio, i-propylthio and n-butylthio including their isomers. The term "alkoxyalkyi" as used herein refers to the radical R'R"-, wherein R' is an alkoxy radical as defined herein, and R" is an alkylene radical as defined herein with the understanding that the attachment point of the alkoxyalkyi moiety will be on the alkylene radical. Examples are methoxymethyl, methoxyethyl, methoxypropyl, ethoxymethyl, ethoxyethyl, ethoxypropyl, propyloxypropyl, methoxybutyl, ethoxybutyl, propyloxybutyl, butyloxybutyl, i-butyloxybutyl, methoxypentyl, ethoxypentyl, propyloxypentyl including their isomers.
The terms "hydroxyalkyl" as used herein denotes the radical R'R" where R' is an hydroxy radical and R" is alkylene as defined herein and the attachment point of the hydroxyalkyl radical will be on the alkylene radical. The term "acyl" as used herein denotes a group of formula C(=0)R ("alkylcarbonyl") wherein R is hydrogen, unbranched or branched alkyl containing 1 or more carbon atoms, for example a lower alkyl Ci-C6 group, cycloalkyl containing 3 to 7 carbon atoms, an aryl, an alkoxy, or a NR'R" group. The term acyl includes a group of formula C(=0)OR' ("alkoxycarbonyl") or C(=0)NR'R" ("carbamoyl") where R is an alkyl group as defined hereinabove.
The term "acylating agent" as used herein refers to a reagent which is capable of transferring an acyl moiety as defined previously to another functional group capable of reacting with the acylating agent. Typically an alkylcarbonyl is introduced by reaction with an anhydride or an acyl halide. The term "anhydride" as used herein refers to compounds of the general structure RC(0)-0-C(0)R wherein R is as defined in the previous paragraph. The term "acyl halide" as used herein refers to the group RC(0)X wherein X is bromo or chloro. Typically an alkoxycarbonyl is introduced by reaction with an alkoxycarbonyl chloride. The term "alkoxycarbonyl chloride" as used herein refers to compounds of the general structure ROC(=0)CI. Typically a carbamoyl group is introduced by reaction with an isocyanate. The term "isocyanate" as used herein refers to compounds of the general structure RN=C=0.
The functional group depicted as "-XC(=Y)Z" wherein X and Y are independently O or NR' and Z is C1-6 alkoxy, NR'R", alkyl or alkoxyalkyl preferably refers to "guanidines" (- NR'(=NR") NR'R"), "imidates" (-OC(=NR')alkyl), "amidines" (-NR'C=(NR')alkyl), "carbonates" (-OC(=0)OR), "carbamates" (-OC(=0) NR'R" or -NR'C(=0)OR), "ureas" (-NR'C(=0)NR'R"), "amides" (-NR'C(=0)alkyl) or "esters" (-OC(=0)alkyl) where R' and R" are alkyl groups as defined herein.
The functional group "C(=Y)Z" as used herein refers to esters, amides, imidates and amidines.
The term "heterocyclylalkyl" as used herein means a radical R'R" where R' is an alkylene radical and R" is a heterocyclyl radical as defined herein. Examples of heterocyclylalkyl radicals include, but are not limited to, tetrahydropyran-2-ylmethyl, 2- piperidinylmethyl, 3-piperidinylmethyl, morpholin-1 -ylpropyl, and the like.
The term "alkylamino" as used herein means a radical-NR'R", wherein R' is hydrogen and R" is an alkyl radical as defined herein. The term "dialkylamino" as used herein means a radical-NR'R", wherein R' and R" are alkyl radicals as defined herein. Examples of alkylamino radicals include, but are not limited to, methylamino, ethylamino, cyclopropylmethylamino, dicyclopropylmethylamino, dimethylamino, methylethylamino, diethylamino, di(1 -methylethyl)amino, and the like.
The term "aryl" as used herein denotes an optionally substituted monocyclic or polycyclic-aromatic group comprising carbon and hydrogen atoms. Examples of suitable aryl groups include, but are not limited to, phenyl and naphthyl (e.g. 1 -naphthyl or 2-naphthyl). Suitable substituents for aryl are selected from the group consisting of Ci-6 alkyl, Ci-6 alkenyl, Ci-6 alkynyl, Ci-6 haloalkyl, Ci-6 alkoxy, Ci-6 haloalkoxy, Ci-6 alkylthio, arylthio, alkoxycarbonyl, amino, substituted amino for example alkylamino, CONR'R", carbamoyl, carbamate, ureido, amidino, imino, aryl, nitro, halogen and cyano. Optionally substituted phenyl in R-i or R2 or R3 can be for example 2- phenoxyphenyl, 2-methyl-phenyl, 3-methyl-phenyl, 4-methyl-phenyl, 2,3- dimethylphenyl, 2,4-dimethylphenyl, 2,5-dimethylphenyl, 2,6-dimethylphenyl, 3,4- dimethylphenyl, 3,5-dimethylphenyl, 2,3,4-trimethylphenyl, 3,4,5-trimethylphenyl, 2,3,4,5,6-pentamethylphenyl, 2-cyano-phenyl, 3-cyano-phenyl, 4-cyano-phenyl, 2,3- dicyanophenyl, 2,4-dicyanophenyl, 2,5-dicyanophenyl, 2,6-dicyanophenyl, 3,4- dicyanophenyl, 3,5-dicyanophenyl, 3,6-dicyanophenyl, 2-methoxyphenyl, 3- methoxyphenyl, 4-methoxyphenyl, 2,3-dimethoxyphenyl, 2,4-dimethoxyphenyl, 2,5- dimethoxyphenyl, 2,6-dimethoxyphenyl, 3,4-dimethoxyphenyl, 3,5-dimethoxyphenyl, 3,6-dimethoxyphenyl. In one embodiment, aryl may be substituted with a PEG moiety.
The term "heteroaryl" or "heteroaromatic" as used herein means a monocyclic or bicyclic radical of 5 to 12 ring atoms having at least one aromatic ring containing four to eight atoms per ring, incorporating one or more N, O, or S heteroatoms, the remaining
ring atoms being carbon, with the understanding that the attachment point of said heteroaryl radical will be on said aromatic ring. As well known to those skilled in the art, heteroaryl rings have less aromatic character than their all-carbon counter parts. Thus, for the purposes of the invention, a heteroaryl group need only have some degree of aromatic character.
Examples of heteroaryl moieties include monocyclic aromatic heterocycles having 5 to 6 ring atoms and 1 to 3 heteroatoms including but not limited to, pyridinyl, pyrimidinyl, pyrazinyl, pyridazinone, pyrrolyl, pyrazolyl, imidazolyl, triazolinyl, thiophenyl and oxadiaxolinyl which can optionally be substituted with one or more, preferably one or two substituents selected from hydroxy, cyano, alkyl, alkoxy, thio, lower haloalkoxy, alkylthio, halo, haloalkyl, alkylsulfinyl, alkylsulfonyl, halogen, amino, alkylamino, dialkylamino, aminoalkyl, alkylaminoalkyl, dialkylaminoalkyl, nitro, alkoxycarbonyl and carbamoyl, alkylcarbamoyl and dialkylcarbamoyl and aryloxy such as benzyloxy. Particularly preferred examples of heteroaryl moieties include bicyclic aromatic heterocycles such as quinoxaline and quinoline.
The term "heterocyclylalkyl" as used herein means a radical -R'R" where R' is an alkylene radical and R" is a heterocyclyl radical as defined herein. Examples of heterocyclylalkyl radicals include, but are not limited to, 2-piperidinylmethyl, 3- piperidinylmethyl, morpholin-1 -ylpropyl, and the like.
The term "heterocycle" or "heterocyclic" as used herein means a non-aromatic monocyclic or polycyclic ring comprising carbon and hydrogen atoms and one or more N, S, or O heteroatoms. A heterocyclic group can have one or more carbon-carbon double bonds or carbon-heteroatom double bonds in the ring as long as the ring is not rendered aromatic by their presence. Examples of heterocycloalkyl groups include pyrrolidinyl, pyrrolidino, piperidinyl, piperidino, piperazinyl, piperazino, morpholinyl, morpholino, thiomorpholinyl, thiomorpholino. A heterocyclic group can be unsubstituted or substituted with one to three suitable substituents selected from hydroxy, cyano, alkyl, alkoxy, thio, lower haloalkoxy, alkylthio, halo, haloalkyl, alkylsulfinyl, alkylsulfonyl, halogen, amino, alkylamino, dialkylamino, aminoalkyl, alkylaminoalkyl, and dialkylaminoalkyl, nitro, alkoxycarbonyl and carbamoyl, alkylcarbamoyl and dialkylcarbamoyl.
The terms "amino", "alkylamino" and "dialkylamino" as used herein refer to-NH2,-NHR and-NR2 respectively and R is alkyl as defined above. The two alkyl groups attached to a nitrogen in a dialkyl moiety can be the same or different. The terms "aminoalkyl", "alkylaminoalkyl" and "dialkylaminoalkyl" as used herein refer to NH2(CH2)n-, RHN(CH2)n-, and R2N(CH2)n- respectively wherein n is 1 or more and R is alkyl as defined above.
The term "acyl" or "alkylcarbonyl" as used herein denotes a radical of formula C(=0)R wherein R is hydrogen, unbranched or branched alkyl containing 1 or more carbon atoms, for example a lower alkyl CrC6 group, or a phenyl group.
The term "acylamino" as used herein denotes a radical of formula -NH-C(=0)-R wherein R is hydrogen, unbranched or branched alkyl containing 1 or more carbon atoms, for example a lower alkyl Ci-C6 group, cycloalkyl containing 3 to 7 carbon atoms or an aryl.
The term "halogen" as used herein means fluorine, chlorine, bromine, or iodine. Correspondingly, the meaning of the term "halo" encompasses fluoro, chloro, bromo, and iodo.
The term "alkylthio" or "thioalkyl" means an -S-alkyl group, wherein alkyl is as defined above such as meththio, ethylthio, n-propylthio, /-propylthio, n-butylthio, hexylthio, including their isomers. The term "alkylsulfinyl" as used herein means the radical-S(0)R', wherein R' is alkyl as defined herein. Examples of alkylaminosulfonyl include, but are not limited to methylsulfinyl and /so-propylsulfinyl.
The term "alkylsulfonyl" as used herein means the radical-S(0)2R', wherein R' is alkyl as defined herein. Examples of alkylaminosulfonyl include, but are not limited to methylsulfonyl and /so-propylsulfonyl.
The term "sulfonylating agent" as used herein refers to a reagent which is capable of transferring an alkyl sulfonyl moiety as defined previously to another functional group capable of reacting with the sulfonating agent such as a sulfonyl chloride Cl-S02- . The prefix "carbamoyl" as used herein means the radical -CONH2. The prefix "N- alkylcarbamoyl" and "Ν,Ν-dialkylcarbamoyl" as used herein means the radical CONHR' or CONR'R" respectively wherein the R' and R" groups are independently alkyl as defined herein. Detailed Description of the Invention
As described above, the present inventors have identified compounds of Formula (I) as being inhibitors of the bacterial ATPase VirB1 1 and its homologs, and thus being useful as antibacterial agents. The compounds when used as antibacterial agents reduce the spread of antibiotic resistance.
In an embodiment of the compound of Formula I, R2 is H; and R3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl. In another embodiment, R2 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl; and R3 is H.
In an embodiment, R4 and R5 are independently selected from H, lower alkyl, halo (e.g. chloro, fluoro or bromo), alkoxy, alkylthio, hydroxy, cyano, amino and nitro. For example, R4 and R5 are each hydrogen.
In one embodiment, Z is NH.
In the embodiment where L is
 , the amino substituent may be in the ortho, meta or para position. In another embodiment, the amino substituent is in the para position.
In the embodiment where L is
, the amino substituent may be in the ortho, meta or para position. In another embodiment, the amino substituent is in the para position.
In the embodiment where L is
 the amino substituent may be in the ortho, meta or para position. In another embodiment, the amino substituent is in the para position. In one embodiment, R-i is unsubstituted or substituted alkyl. In another embodiment, Ri is unsubstituted or substituted aryl. In another embodiment, Ri is unsubstituted or substituted heteroaryl.
the amino substituent may be in the ortho, meta or para position. In another embodiment, the amino substituent is in the para position. In one embodiment, R-i is unsubstituted or substituted alkyl. In another embodiment, Ri is unsubstituted or substituted aryl. In another embodiment, Ri is unsubstituted or substituted heteroaryl.
In one embodiment, Ri is substituted aryl substituted with a poly(ethylene) glycol or PEG moiety. The PEG moiety may be coupled to the aryl group via a linker. In one embodiment, the linker is a carbamate linker. However, it would be clear to the skilled person that the present invention encompasses any linker, for example but not limited to an amide, ester, carbonate, imidate, alkyl, alkenyl or alkynyl linker. In one embodiment, the PEG moiety is coupled directly to the aryl group. In a further embodiment, the PEG moiety may itself be substituted. In one embodiment, the PEG moiety may be substituted with an alkenyl, alkynyl or maleido group.
The PEG moiety may comprise one or more PEG monomers, for example two PEG monomers, three PEG monomers or four PEG monomers. Thus, the PEG moiety may comprise (PEG)i, (PEG)2, (PEG)3 or (PEG)4.
In an embodiment, the compound is not selected from the group consisting of:
4-methyl-/V-[3-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide,
4-methyl-/V-[3-(2-phenoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide, /V-[3-(3,4-dimethoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]-4-methyl-benzenesulfonamide,
4-methyl-/V-[3-(3,5-dimethylphenyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide,
4-methyl-/V-[4-[3-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8-
]yl]aminophenyl]benzenesulfonamide,
4-methyl-/V-[4-[3-(3-thienyl)imidazo[1 ,2-a]pyrazin-8- yl]amino]phenyl]benzenesulfonamide.
In an embodiment, the compound is not selected from the compounds of Table 1 :

In an embodiment, the compound is selected from the group consisting of:
4-methyl-/V-[3-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide,
4-methyl-/V-[3-(2-phenoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide, /V-[3-(3,4-dimethoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]-4-methyl-benzenesulfonamide, 4-methyl-/V-[3-(3,5-dimethylphenyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide, 4-methyl-/V-[4-[3-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8- ]yl]aminophenyl]benzenesulfonamide,
4-methyl-/V-[4-[3-(3-thienyl)imidazo[1 ,2-a]pyrazin-8- yl]amino]phenyl]benzenesulfonamide,
4-methyl-/V-[2-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide,
4-methyl-/V-[2-(2-phenoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide, /V-[2-(3,4-dimethoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]-4-methyl-benzenesulfonamide,
/V-[2-(3,5-dimethylphenyl)imidazo[1 ,2-a]pyrazin-8-yl]-4-methyl benzenesulfonamide,
4-methyl-/V-[4-[2-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8- yl]aminophenyl]benzenesulfonamide,
4-methyl-/V-[4-[2-(3-thienyl)imidazo[1 ,2-a]pyrazin-8- yl]aminophenyl]benzenesulfonamide,
4-methyl-/V- [4-[2-(2-phenoxyphenyl)imidazo[1 ,2-a]pyrazin-8- ylaminophenyl]benzenesulfonamide,
4-methyl-/V-(4-(2-(quinoxalin-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)phenyl)benzenesulfonamide,
4-methyl-/V-(4-(2-(quinolin-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)phenyl)benzenesulfonamide,
/V-(4-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8-ylamino)phenyl)methanesulfonamide, 4-methyl-/V-(4-((2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)methyl)phenyl)benzenesulfonamide,
4-methyl-/V-(4-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- yloxy)phenyl)benzenesulfonamide,
4-methyl-/V-(2-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)ethyl)benzenesulfonamide,
4-methyl-/V-(3-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)propyl)benzenesulfonamide,
2-(prop-2-ynyloxy)ethyl-4-(/V-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- yl)sulfamoyl)phenylcarbamate,
2-(allyloxy)ethyl-4-(/V-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- yl)sulfamoyl)phenylcarbamate,
2-(2-(3-(2,5-dioxo-2,5-dihydro-1 H-pyrrol-1-yl)propanamido)ethoxy)ethyl-4-(/V-(2- (naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8-yl)sulfamoyl)phenylcarbamate.
In an embodiment, the compound is selected from the compounds in Table 2.
In an embodiment, any one of the compounds of Table 2 may be used in treating or preventing bacterial infection.



Table 2.
All of the compounds of the Formula (I) can be prepared by the procedures described in the general methods presented below or by the specific methods described in the Examples section. The present invention also encompasses any one or more of these methods for preparing the compounds of Formula (I), in addition to any novel intermediates used therein. It will be clear to the skilled person that many steps in the syntheses of these compounds are analogous and where a particular step is not explicitly described that it is readily apparent from the steps that are described in relation to other compounds.
General Synthesis
The compounds of the present invention wherein Z = NH may be prepared, for example, as shown in the following General Method A for 3-substituted derivatives or General Method B for 2-substituted derivatives.
Unless otherwise indicated, R-i, R2 and R3 in the following methods are as defined above. All starting materials in the following general syntheses may be commercially available or obtained by conventional methods known to those skilled in the art.
Solvents and reagents were obtained from commercial sources and were used as received unless otherwise stated. Dry solvents were dried over anhydrous columns. Moisture levels were usually <15 ppm by Karl Fischer titration. Brine refers to a saturated solution of sodium chloride. Anhydrous magnesium sulfate (MgS04) or sodium sulfate (Na2S04) were used as drying agents after reaction workup, as indicated. Pet Ether refers to the fraction of light petroleum ether boiling in the range 40-60 °C.
Where a solvent is specified, it will be readily understood that there is no restriction on the particular solvent to be used, provided that it has no adverse effect on the reaction or reagents involved and that it can at least partially dissolve the reagents. Examples of suitable solvents include: halogenated hydrocarbons, such as dichloromethane, chloroform, carbon tetrachloride and 1 ,2-dichloroethane; ethers, such as diethyl ether, diisopropyl ether, tetrahydrofuran and dioxane; nitriles, such as acetonitrile and benzonitrile; aromatic hydrocarbons, such as benzene, toluene and nitrobenzene; amides, such as formamide, /V,/V-dimethylformamide, and Λ/,/V-dimethylacetamide sulfoxides, such as dimethyl sulfoxide and sulfolane; or mixed solvents thereof.
Method A

Scheme 1 Reagents: a) NaN3, DMSO, 0 °C to RT, 1.5h; b) NaBH4, MeOH, 0 °C, 1 h.; c) H2, Pd/C, MeOH, RT, 3-16h; d) 3-dichloropyrazine (shown), NEt3, dioxane, reflux, 16- 20 hours; e) (COCI)2, DMSO, NEt3, CH2CI2, -78°C, 3h; f) TFA/TFAA, toluene, room
temp. 72h; g) "sulfonamide" (shown), Pd(dba)2, tBu-XPhos, K2C03, tBuOH, reflux, 16- 48h.
For each 3-substituted derivative, the synthesis of the amino alcohol was achieved via a-bromination of the aryl ketone with pyridinium tribromide. This was followed by substitution of the a-bromine with an azide, giving the azidoketone which was then reduced to give the corresponding azido-alcohol. Hydrogenation to give the amino alcohol was performed at atmospheric pressure, with the exception of the thiophene analogue which required 3 bar pressure to go to completion. Coupling with 2,3- dichloropyrazine in 1 ,4-dioxane afforded the pyrazinyl-amino alcohol in good yields. Swern oxidation to the ketone was followed by acid-induced cyclisation to form the 3- aryl-8-chloroimidazo[1 ,2-a]pyrazine.
In order to install the sulfonamide groups, a Buchwald-Hartwig coupling approach was used. Several different palladium catalysts and ligands were tested.
It will be understood that there are many alternative transition metal catalysts and ligands which could be used to effect this reaction. Examples of palladium catalysts which may be suitable include but are not limited to: palladium metal, palladium- carbon, palladium (II) acetate, tris(dibenzylideneacetone)dipalladiumchloroform, [1 ,2- bis(diphenylphosphino)ethane]palladium dichloride, bis(tri-o-toluylphosphine)palladium dichloride, bis(triphenylphosphine)palladium dichloride, tetrakis(triphenylphosphine) palladium or dichloro[1 ,1 '-bis(diphenylphosphino)ferrocene]palladium. One preferred catalyst is Pd(dba)2.
Examples 1 to 6 were prepared according to this general method.
Method B

Using the a-bromo aryl ketones, the key 8-chloro-2-aryl imidazo[1 ,2-a]pyrazine intermediates were readily prepared by condensation with 2-amino-3-chloropyrazine (Scheme 2). Again, the sulfonamide groups were introduced using Buchwald-Hartwig coupling chemistry, to give Examples 7 - 17 and 29 to 23 in low to moderate yields. However the coupling was very substrate dependent: for the 3,5-dimethylphenyl derivative the previous combination gave no reaction at all, and therefore Pd(dppf)CI2 had to be used as an alternative. It was also found that DavePhos ligand could be used in place of fButyl-XPhos to give the substrates in similar yields.
For compounds wherein Z = O, these can be made by a route exemplified by the synthesis of Example 18 below. Analysis of compounds
Melting points (Mpt) were recorded on a Gallenkamp Melting Point Apparatus and are uncorrected. Proton nuclear magnetic resonance (1H NMR) were recorded using Bruker AV400 (400 MHz), AV500 (500 MHz) and AV600 (600MHz) spectrometers as indicated. Carbon nuclear magnetic resonance (13C NMR) were recorded using Bruker AV400 (100 MHz), AV500 (125 MHz) and AV600 (150MHz) spectrometers as indicated. Spectra were obtained using CDCI3, CD3OD, CD2CI2 and DMSO-c/6 as solvents and chemical shifts are quoted on the δ scale in units of ppm using TMS as an internal standard. Coupling constants (J) are reported in Hz with the following splitting abbreviations: s (singlet), d (doublet), t (triplet), dd (doublet of doublets), bs (broad singlet).
The regiochemistry of the 2- and 3-substituted imidazo[1 ,2-a]pyrazines was confirmed by 2D NMR.
Infra-Red (IR) spectroscopy was carried out using a PerkinElmer Spectrum 100 FT-IR Spectrometer using thin films. Absorption maxima (vmax) are reported in wavenumbers (cm"1).
High Performance Liquid Chromatography (HPLC) was performed using a Varian ProStar instrument; A Chiralpak AD column for normal phase analytical HPLC; DiscoveryBIO wide pore C18-10 (25 cm x 4.6 mm, 10 μηη) for reverse phase analytical HPLC; and a DiscoveryBIO wide pore C18 (25 cm x 21.2 mm, 10 μηη) column for reverse phase preparative HPLC. Each solvent used contained 0.1 % TFA buffer.
Liquid Chromatography Mass Spectrometry (LCMS) was carried out using SQD- Waters Acquity UPLC/SQD with C18 (50 mm x 2.1 mm, 1.7 μπτι) column. A total run time of 5 minutes and flow rate of 0.6 mL/min was used with gradient elution: 95% H20/ 5% MeCN (0 min), 5% H20/ 95% MeCN (3 min), 95% H20/ 5% MeCN (4.5 min). Each solvent contained 0.1 % formic acid buffer. LRMS refers to low resolution mass spectrometry and HRMS refers to high resolution mass spectrometry. Electron Impact/ Chemical lonisation (EI/CI) MS was carried out using MAT900XP (Thermo Finnigan) instrument and electrospray ionization (ESI) accurate mass was determined using Waters LCT Premier XE instrumentation.
Thin layer chromatography (TLC) was carried out using Fluka aluminium backed sheets coated with 60F254 silica gel. Visualisation of the silica plates was achieved using a UV lamp (Amax = 245 nm) and/or potassium permanganate (KMn04 in 1 M NaOH with 5% K2C03). Flash chromatography was carried out using either Geduran (Merck) or ZEOprep (Apollo) Si60 40-63 m silica gel.
Abbreviations In addition to those specified elsewhere, the following abbreviations, designations, and formulae are used throughout:
Cs2C03 = caesium carbonate
DCM = dichloromethane
DMSO = dimethylsulfoxide
Et20 = diethyl ether
Et3N = triethylamine
EtOAc = ethyl acetate
I PA = isopropanol
K2C03 = potassium carbonate
MeCN = acetonitrile
MeOH = methanol
NaHC03 = sodium hydrogen carbonate
Sat. = saturated
RT = room temperature
Pharmaceutical Compositions The present invention provides a pharmaceutical composition comprising a compound of Formula (I) or a pharmaceutically acceptable salt thereof, each as described herein, together with a pharmaceutically acceptable carrier for said compound. Also, the present invention provides a pharmaceutical composition comprising a compound of Formula (I) or a pharmaceutically acceptable salt thereof, each as described herein, further comprising other pharmacologically active agent(s).
Pharmaceutically acceptable salts of a compound of Formula (I) include the acid addition salts (including disalts) thereof. Suitable acid addition salts are formed from acids which form non-toxic salts. Examples include the acetate, adipate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, cyclamate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate, palmitate, palmoate, phosphate/hydrogen phosphate/dihydrogen phosphate, pyroglutamate, saccharate, stearate, succinate, tannate, tartrate, tosylate, trifluoroacetate and xinofoate salts.
For a review on suitable salts, see "Handbook of Pharmaceutical Salts: Properties, Selection, and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002). A pharmaceutically acceptable salt of a compound of Formula (I) may be readily prepared by mixing together solutions of the compound of Formula (I) and the desired acid or base, as appropriate. The salt may precipitate from solution and be collected by filtration or may be recovered by evaporation of the solvent. The degree of ionization in the salt may vary from completely ionized to almost non-ionized. Pharmaceutically acceptable salts of the compounds of the invention include both unsolvated and solvated forms. The term "solvate" is used herein to describe a molecular complex comprising a compound of the invention and one or more pharmaceutically acceptable solvent molecules, for example, ethanol. The term 'hydrate' is employed when said solvent is water.
Pharmaceutically acceptable solvates in accordance with the invention include hydrates and solvates wherein the solvent of crystallization may be isotopically substituted, e.g. D20, c/6-acetone, c/6-DMSO.
Included within the scope of the invention are complexes such as clathrates, drug-host inclusion complexes wherein, in contrast to the aforementioned solvates, the drug and host are present in stoichiometric or non-stoichiometric amounts. Also included are complexes of the drug containing two or more organic and/or inorganic components which may be in stoichiometric or non-stoichiometric amounts.
The resulting complexes may be ionized, partially ionized, or non-ionized. For a review of such complexes, see J Pharm Sci. 64 (8), 1269-1288 by Haleblian (August 1975). The compounds of Formula (I) may exist in one or more crystalline forms. These polymorphs, including mixtures thereof are also included within the scope of the present invention. The compounds of Formula (I) containing one or more asymmetric carbon atoms can exist as two or more stereoisomers.
Included within the scope of the present invention are all stereoisomers of the compounds of Formula (I), including compounds exhibiting more than one type of isomerism, and mixtures of one or more thereof.
Administration
Compounds of the invention intended for pharmaceutical use may be administered as crystalline or amorphous products. They may be obtained, for example, as solid plugs, powders, or films by methods such as precipitation, crystallization, freeze-drying, spray drying, or evaporative drying. Microwave or radio frequency drying may be used for this purpose.
They may be administered alone or in combination with one or more other compounds of the invention or in combination with one or more other drugs (or as any combination thereof). Generally, they will be administered as a pharmaceutical composition or formulation in association with one or more pharmaceutically acceptable carriers or excipients. The term "carrier" or "excipient" is used herein to describe any ingredient other than the compound(s) of the invention. The choice of carrier or excipient will to a large extent depend on factors such as the particular mode of administration, the effect of the excipient on solubility and stability, and the nature of the dosage form. Pharmaceutical compositions suitable for the delivery of compounds of the present invention and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation may be found, for example, in 'Remington's Pharmaceutical Sciences', 19th Edition (Mack Publishing Company, 1995).
Oral Administration The compounds of the invention may be administered orally. Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, or buccal or sublingual administration may be employed by which the compound enters the blood stream directly from the mouth. Formulations suitable for oral administration include solid formulations such as, for example, tablets, capsules containing particulates, liquids, or powders, lozenges (including liquid-filled), chews, multi- and nano-particulates, gels, solid solution, liposome, films (including muco-adhesive), ovules, sprays and liquid formulations.
Liquid formulations include, for example, suspensions, solutions, syrups and elixirs. Such formulations may be employed as fillers in soft or hard capsules and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet.
The compounds of the invention may also be used in fast-dissolving, fast-disintegrating dosage forms such as those described in Expert Opinion in Therapeutic Patents. H (6), 981-986 by Liang and Chen (2001 ).
For tablet dosage forms, depending on dose, the drug may make up from about 1 wt% to about 80 wt% of the dosage form, more typically from about 5 wt% to about 60 wt% of the dosage form. In addition to the drug, tablets generally contain a disintegrant. Examples of disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl-substituted hydroxypropyl cellulose, starch, pregelatinised starch and sodium alginate. Generally, the disintegrant will comprise from about 1 wt% to about 25 wt%, preferably from about 5 wt% to about 20 wt% of the dosage form.
Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate. Tablets may also optionally comprise surface-active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc. When present, surface active agents may comprise from about 0.2 wt% to about 5 wt% of the tablet, and glidants may comprise from about 0.2 wt% to about 1 wt% of the tablet.
Tablets also generally contain lubricants such as magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate. Lubricants generally comprise from about 0.25 wt% to about 10 wt%, preferably from about 0.5 wt% to about 3 wt% of the tablet.
Other possible ingredients include anti-oxidants, colourants, flavouring agents, preservatives and taste-masking agents.
Exemplary tablets contain up to about 80% drug, from about 10 wt% to about 90 wt% binder, from about 0 wt% to about 85 wt% diluent, from about 2 wt% to about 10 wt% disintegrant, and from about 0.25 wt% to about 10 wt% lubricant.
Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting. The final formulation may comprise one or more layers and may be coated or uncoated; it may even be encapsulated.
The formulation of tablets is discussed in "Pharmaceutical Dosage Forms: Tablets, Vol. 1 ", by H. Lieberman and L. Lachman, Marcel Dekker, N.Y., N.Y., 1980 (ISBN 0-8247- 6918-X).
Solid formulations for oral administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
Suitable modified release formulations for the purposes of the invention are described in US Patent No. 6,106,864. Details of other suitable release technologies such as high energy dispersions and osmotic and coated particles are to be found in Verma et al, Pharmaceutical Technology On-line. 25(2), 1-14 (2001 ). The use of chewing gum to achieve controlled release is described in WO00/35298.
Parenteral Administration
The compounds of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ. Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular and subcutaneous. Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques. Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from about 3 to about 9), but, for some applications, they may be more suitably formulated as a sterile non-aqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
The preparation of parenteral formulations under sterile conditions, for example, by lyophilisation, may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art. The solubility of compounds of Formula (I) used in the preparation of parenteral solutions may be increased by the use of appropriate formulation techniques, such as the incorporation of solubility- enhancing agents.
Formulations for parenteral administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release. Thus compounds of the invention may be formulated as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound. Examples of such formulations include drug-coated stents and PGLA microspheres.
Topical Administration
The compounds of the invention may also be administered topically to the skin or mucosa, that is, dermally or transdermally. Typical formulations for this purpose include
gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibres, bandages and microemulsions. Liposomes may also be used. Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol.
Penetration enhancers may be incorporated - see, for example, J Pharm Sci, 88 (10), 955-958 by Finnin and Morgan (October 1999). Other means of topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and microneedle or needle-free (e.g. Powderject(TM), Bioject(TM), etc.) injection.
Formulations for topical administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
Inhaled/lntranasal administration The compounds of the invention can also be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an aerosol spray from a pressurized container, pump, spray, atomiser (preferably an atomiser using electrohydrodynamics to produce a fine mist), or nebuliser, with or without the use of a suitable propellant, such as 1 ,1 ,1 ,2-tetrafluoroethane or 1 ,1 ,1 ,2,3,3,3- heptafluoropropane. For intranasal use, the powder may comprise a bioadhesive agent, for example, chitosan or cyclodextrin. The pressurized container, pump, spray, atomizer, or nebuliser contains a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
Prior to use in a dry powder or suspension formulation, the drug product is micronised to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenization, or spray drying.
Capsules (made, for example, from gelatin or HPMC), blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the compound of the invention, a suitable powder base such as lactose or starch and a performance modifier such as /-leucine, mannitol, or magnesium stearate. The lactose may be anhydrous or in the form of the monohydrate, preferably the latter. Other suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose. A suitable solution formulation for use in an atomiser using electrohydrodynamics to produce a fine mist may contain from about 1 μg to about 20mg of the compound of the invention per actuation and the actuation volume may vary from about 1 μΙ to about 10ΟμΙ. A typical formulation may comprise a compound of Formula (I), propylene glycol, sterile water, ethanol and sodium chloride. Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
Suitable flavors, such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations of the invention intended for inhaled/intranasal administration. Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, poly(DL-lactic-coglycolic acid (PGLA). Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
In the case of dry powder inhalers and aerosols, the dosage unit is determined by means of a valve which delivers a metered amount. Units in accordance with the invention are typically arranged to administer a metered dose or "puff' containing from about 1 to about 100 μg of the compound of Formula (I).
The overall daily dose will typically be in the range about 50 μg to about 20 mg which
may be administered in a single dose or, more usually, as divided doses throughout the day.
Other Technologies
The compounds of the invention may be combined with soluble macromolecular entities, such as cyclodextrin and suitable derivatives thereof or polyethylene glycol- containing polymers, in order to improve their solubility, dissolution rate, taste-masking, bioavailability and/or stability for use in any of the aforementioned modes of administration.
Drug-cyclodextrin complexes, for example, are found to be generally useful for most dosage forms and administration routes. Both inclusion and non-inclusion complexes may be used. As an alternative to direct complexation with the drug, the cyclodextrin may be used as an auxiliary additive, i.e. as a carrier, diluent, or solubiliser. Most commonly used for these purposes are alpha-, beta- and gamma-cyclodextrins, examples of which may be found in. W091/1 1 172, WO94/02518 and W098/55148.
Kit of Parts
Inasmuch as it may be desirable to administer a combination of active compounds, for example, for the purpose of treating a particular disease or condition, it is within the scope of the present invention that two or more pharmaceutical compositions, at least one of which contains a compound in accordance with the invention, may conveniently be combined in the form of a kit suitable for coadministration of the compositions. Thus the kit of the invention comprises two or more separate pharmaceutical compositions, at least one of which contains a compound of Formula (I) in accordance with the invention, and means for separately retaining said compositions, such as a container, divided bottle, or divided foil packet. An example of such a kit is the familiar blister pack used for the packaging of tablets, capsules and the like.
The kit of the invention is particularly suitable for administering different dosage forms, for example, oral and parenteral, for administering the separate compositions at different dosage intervals, or for titrating the separate compositions against one
another. To assist compliance, the kit typically comprises directions for administration and may be provided with a so-called memory aid.
Dosage
For administration to human patients, the total daily dose of the compounds of the invention is typically in the range of about 0.05 mg to about 500 mg depending, of course, on the mode of administration, preferred in the range of about 0.1 mg to about 400 mg and more preferred in the range of about 0.5 mg to about 300 mg. For example, oral administration may require a total daily dose of from about 1 mg to about 300 mg, while an intravenous dose may only require from about 0.5 mg to about 100 mg. The total daily dose may be administered in single or divided doses. These dosages are based on an average human subject having a weight of about 65 kg to about 70 kg. The physician will readily be able to determine doses for subjects whose weight falls outside this range, such as infants and the elderly.
Examples
The following compounds of the invention were synthesized according to the general methods outlined above, with the synthesis of any required precursors using standard transformations provided.
Example 1
4-methyl-A/-[3-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide

Step 1
2-azido-1 -(2-naphthyl)ethanon

2-(bromoacetyl)naphthalene (2.00 g, 8.03 mmol) was dissolved in DMSO (10 mL) and the mixture was cooled on ice such that the temperature was kept below 10°C. Sodium azide (0.630 g, 9.64 mmol) was added in one portion and the reaction was stirred under argon at RT for 90 min. The reaction was quenched with H20 (20 mL), and extracted with EtOAc (3 x 30 mL). The organic layers were combined, washed with H20, dried (Na2S04) and filtered. The solvent was removed in vacuo to give brown/orange oil (1.53 g, 7.25 mmol, 90.3 %) with NMR consistent with literature values. Rf = 0.63 (5:1 Pet Ether/ EtOAc); IR (vmax/cm"1, thin film): 2105 (-N=N+=N" Stretch), 1690 (C=0 Stretch); 1 H NMR (600 MHz, CDCI3): δΗ = 4.73 (s, 2H, 1-H), 7.61 (t, J = 7.6 Hz, 1 H, 7-H), 7.66 (t, J = 7.6 Hz, 1 H, 8-H), 7.91 (d, J = 8.1 Hz, 1 H, 9-H), 7.95(d, J = 8.6 Hz, 1 H, 1 1-H), 7.99(m 2H, 6,12-H), 8.42 (s, 1 H, 4-H); 13C NMR (150 MHz, CDCI3): 5c = 55.0 (C-1 ), 123.3 (C-12), 127.2 (C-7), 127.9 (C-9) 129.0 (C-1 1 ), 129.1 (C-8), 129.6 (C-6), 129.8 (C-4), 131.7, 132.4 (C-5,10), 136.0 (C-3), 193.2 (C-2); LRMS m/z (ΕΓ): 21 1 [M]+, 155 [M-CH2N3]+, 127 [Naphthalene]".
Step 2
2-azido-1 -(2-naphthyl)ethanol
13

2-azido-1-(2-naphthyl)ethanone (2.1 1 g, 10.0 mmol) was dissolved in anhydrous MeOH (100 mL) and cooled on ice. Sodium borohydride (568 mg, 15.0 mmol) was added portion wise and the mixture was stirred on ice under argon for 1 hour until the reaction had gone to completion by TLC. The solvent was removed and the resulting residue was taken up in DCM (100 mL) and carefully washed with H20 (2 x 60 mL) followed by brine (60 mL). The organic extracts were dried over Na2S04, filtered and concentrated in vacuo to give the title compound as a brown oil (2.14 g, 10.0 mmol, 100%). Spectroscopic data was consistent to that previously reported. Rf = 0.65 (3:1 Pet Ether /EtOAc); IR (vmax/cm"1, thin film): 3398 (O-H stretch), 2100 (-N=N+=N" Stretch); 1H NMR (500 MHz, CDCIs): δΗ = 2.70 (bs, 1 H, 13-H), 3.46 - 3.58 (m, 2H, 1 -H), 5.02 (dd, J = 8.1 , 3.9 Hz, 1 H, 2-H), 7.44 (dd J = 8.4, 1 .6 Hz, 1 H, 4-H), 7.49 - 7.52 (m, 2H, 7,8-H), 7.83 - 7.86 (m, 4H, 6,7,11 ,12-H); 13C NMR (125 MHz, CDCI3): 5C = 58.1 (C-1 ), 73.6 (C-2), 123.7 (C-12), 125.1 (C-7), 126.4 (C-9), 126.5 (C-11 ), 127.8 (C-8), 128.1 (C-6), 128.6 (C-4), 133.3 (C-5,10), 138.0 (C-3); LRMS m/z (El+): 221 , 157 [M-CH2N3]+, 147, 129
Step 3
2-amino-1 -(2-naphthyl)ethanol

2-azido-1-(2-naphthyl)ethanol (453 g, 2.13 mmol) was dissolved in anhydrous MeOH (10 mL) and 10% palladium on carbon (45.0 mg, 10% w/w) was added. The reaction mixture was stirred under hydrogen atmosphere until completion as determined by TLC and disappearance of N3 peak by IR. After 3 ½ h, the hydrogen was carefully released,
and the reaction mixture was filtered through celite (pre-washed with MeOH). Solvent removal in vacuo gave the crude compound as a orange oil (383 mg, 2.05 mmol, 96.1 %). Spectroscopic data was consistent with that previously reported. Flash chromatography (100% EtOAc to EtOAc/20% MeOH) afforded the title compound as a white solid (288.6 mg, 73%). Rf = 0.06 (5:1 EtOAc/MeOH); IR (vmax/cm"1, thin film): 3290 (O-H Stretch), 3054 (C-H), 2916 (N-H Stretch), 1599 (N-H Bend); 1H NMR (500 MHz, CDCIs): δΗ = 2.82 (m, 2H, 1 -H), 4.59-4.75 (m, 1 H, 2-H), 7.36-7.40 (m, 3H, 7,8,9- H), 7.74-7.76 (m, 4H, 4,6,11 , 12-H); 13C NMR (125 MHz, CDCI3): 5C = 49.8 (C-1 ), 74.3 (C-2), 123.8 (C-12), 124.5 (C-7), 125.7 (C-9), 126.0 (C-11 ), 127.5 (C-8), 128.1 (C-4), 132.8 (C-5), 133.1 (C-10), 139.7 (C-3); LRMS m/z (ESI+): 229.2 [M+MeCN]+, 21 1 .2 [M+Na]+, 188.1 [M+H]+, 170 [M-OH]+.
Step 4
2-[(3-chloropyrazin-2-yl)amino]-1 -(2-naphthyl)ethanol
2-amino-1-(2-naphthyl)ethanol (289 mg, 1 .55 mmol), 2,3-dichloropyrazine (177 μΙ_, 1.70 mmol) and Et3N (301 μί, 2.16 mmol) were dissolved in 1 ,4-dioxane (3 mL) and the reaction was stirred under reflux, under argon. After 19 h, the reaction was cooled to RT, and the solvent removed in vacuo. The residue was taken up in DCM and washed with H20 (3 x 20 mL) and brine (1 x 20 mL). The organic extracts were dried (Na2S04), filtered and concentrated to give the crude product as an amber oil. Purification was carried out via flash chromatography (100% DCM - 2:1 DCM/EtOAc gradient) to afford the title compound as a yellow oil (295 mg, 0.983 mmol, 63.4%). Rf = 0.64 (2:1 DCM/EtOAc); IR (vmax/crrf1, thin film): 3419 (O-H Stretch), 3054 (C-H Stretch), 2922 (N-H Stretch), 1523 (N-H Bending); 1H NMR (500 MHz, CDCI3): δΗ = 3.65-3.70 (m, 1 H, 1 -H), 3.88 (bs, 1 H, 13-H), 3.92-3.97 (m, 1 H, 1 -H), 5.12 (dd, J = 7.5, 2.8 Hz, 2- H), 5.67 (t, J = 5.3 Hz, 14-H), 7.47-7.50 (m, 3H, 7,8,12-H), 7.59 (d, J = 2.7 Hz, 1 H, 18- H), 7.81-7.84 (m, 3H, 6,9,11 -H), 7.86 (s, 1 H, 4-H), 7.91 (d, J = 2.7 Hz, 1 H, 17-H); 13C NMR (125 MHz, CDCI3): 5C = 49.4 (C-1 ), 73.9 (C-2), 123.9 (C-12), 124.8 (C-7), 126.1 (C-9), 126.4 (C-11 ), 127.8 (C-8), 128.0 (C-6), 128.4 (C-4), 131.3 (C-18), 1332. (C-5),
133.3 (C-10), 135.1 (C-20), 139.4 (C-3), 140.2, (C-17), 151 .5 (C-15); LRMS m/z (ESI+): 300.1 [M(35CI)+H]+, 284.2 [M(37CI)-OH]+, 282.2 [M(35CI)-OH]+; HRMS m/z (ESI"): Found 298.0731 [M(35CI)-H]-; Ci6H13CIN30 requires 298.0747. Step 5
2-[(3-chloropyrazin-2-yl)amino]-1 -(2-naphthyl)ethanone

DMSO (982 μΙ_, 13.9 mmol) was dissolved in anhydrous DCM (60 mL) and the reaction mixture was cooled to and maintained at -78°C. Oxalyl chloride (586 μΙ_, 6.93 mmol) was added drop wise and the mixture was stirred for 20 min. 2-[(3-chloropyrazin-2- yl)amino]-1 -(2-napthyl)ethanol (1 .60 g, 5.33 mmol), dissolved in anhydrous DCM (40 mL) was added drop wise, and stirred for 20 min. Et3N (3.54 mL, 26.6 mmol) was added drop wise and the reaction mixture was allowed to warm to RT over a period of 2 ½ h. The reaction was then quenched with H20 (50 mL) and organics extracted, which were then washed with 2.0 M HCI (2 x 40 mL), sat. NaHC03 (40 mL), H20 (40 mL) and brine (40 mL). The organic were dried (MgS04), filtered and solvent removed to give a yellow/orange solid. Flash chromatography (100:1 to 30:1 DCM/EtOAc) afforded the title compound as a yellow solid (903 mg, 3.03 mmol, 56.9%). Mpt: 160 °C; Rf = 0.30 (30:1 DCM/EtOAc); IR (vmax/cm"1, thin film): 1680 (C=0 stretch); 1 H NMR (500 MHz, CDCI3): δΗ = 5.10 (d, J = 4.3, 2H, 1 -H), 6.54 (s, 1 H, 13-H), 7.58-7.62 (m, 1 H, 7-H), 7.64-7.67 (m, 1 H, 8-H), 7.68 (d, J = 6.1 Hz, 1 H, 17-H), 7.91 (d, J = 8.0 Hz, 1 H, 9- H), 7.96 (d, J = 8.6 Hz, 1 H, 11 -H), 8.00-8.02 (m, 2H, 6,16-H), 8.10 (dd, J = 8.6, 1.8 Hz, 1 H, 12-H), 8.62 (s, 1 H, 4-H); 13C NMR (125 MHz, CDCI3): 5C = 48.4 (C-1 ), 123.4 (C- 12), 127.3 (C-7), 128.0 (C-9), 129.0 (C-8), 129.2 (C-11 ), 129.8 (C-6), 130.1 (C-4), 131.3 (C-17), 131.8 (C-5), 132.6 (C-10), 136.2 (C-3), 139.7 (C-16), 193.8 (C-2); LRMS m/z (ESI+): 300[M(37CI)+H]+, 298 [M(35CI)+H]+, 282[M(37CI)-OH]+, 280[M(35CI)-OH]+; HRMS m/z (ESI"): Found 296.0591 [M(35CI)-H]"; Ci6HnCIN30 requires 296.0591 ; Anal. Calcd. for Ci6H12CIN30: C, 64.54; H, 4.06; N, 14.1 1 . Found C, 64.35; H, 3.94; N, 13.82%
Step 6
8-chloro-3-(2-naphthyl)imidazo[1 ,2-a]pyrazine

2-[(3-chloropyrazin-2-yl)amino]-1-(2-naphthyl)ethanone (903 mg, 3.03 mmol) was dissolved in anhydrous toluene (40 mL) and the mixture was cooled on ice. Trifluoroacetic acid (1 .64 mL, 21.2 mmol) was added and the reaction was allowed to stir on ice for 30 min, followed by the addition of trifluoroacetic anhydride (2.95 mL, 21 .2 mmol). The reaction mixture was then stirred on ice for a further 30 minutes and then at RT for 65 h. The reaction was then diluted with toluene (20 mL) and washed with NaHC03 solution (10% w/v). The organics were dried (MgS04), filtered and concentrated to give crude amber oil. Purification was carried out via flash chromatography (DCM/EtOAc gradient - 80:1 to 10:1 ) to afford the title compound as an off white solid (386 mg, 1 .38 mmol, 45.5%). Mpt: 166 °C; Rf = 0.21 (10:1 DCM/EtOAc); IR (vmax/cm"1, thin film): 3102 (w), 3052 (w), 1333; 1H NMR (500 MHz, CDCIs): δΗ = 7.59 (dd, J = 6.2, 3.2 Hz, 2H, 15,16-H), 7.63 (dd, J = 8.6, 1.7 Hz, 1 H, 12- H), 7.73 (d, J = 4.6 Hz, 1 H, 6-H), 7.91-7.94 (m, 2H, 14,17-H), 8.00 (s, 1 H, 2-H), 8.03 (d, J = 5.8 Hz, 1 H, 11 -H), 8.04 (s, 1 H, 19-H), 8.29 (d, J = 4.6 Hz, 1 H, 5-H); 13C NMR (125 MHz, CDCI3): 5c = 1 16.4 (C-5), 124.7 (C-10), 125.2 (C-12), 127.3 (C-15), 127.4 (C-16), 127.6 (C-19), 128.0 (C-17), 128.2 (C-17), 128.6 (C-6), 129.4 (C-3), 129.7 (C-11 ), 133.4 (C-13), 133.5 (C-18), 134.8 (C-2), 138.4 (C-9), 144.5 (C-8); LRMS m/z (ESI+): 282 [M(37CI)+H]+, 280 [M(35CI)+H]+; HRMS m/z (ESI+): Found 280.0646 [M(35CI)+H]+; Ci6HnCIN3 requires 280.0642; Anal. Calcd for Ci6H10CIN3: C, 68.70; H, 3.60; N, 15.02. Found C, 64.41 ; H, 3.31 ; N, 14.14%
Step 7
4-methyl-A/-[3-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide

4-methyl-W-[3-(2-phenoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide

Step 1
2-bromo-1 -(2-phenoxyphenyl)ethanone

1-(2-phenoxyphenyl)ethanone (2.00 g, 9.42 mmol) was dissolved in chloroform (60 mL) and ethanol (60 mL). Pyridinium tribromide (7.50 g, 23.6 mmol) was added and the reaction was stirred at 50°C for 16 h. The reaction mixture was cooled to RT and the solvents removed in vacuo. The resulting orange slurry was suspended in H20 (30 mL) and extracted with EtOAc (4 x 30 mL). The combined organic extracts were washed with H20 (2 x 20 mL) and brine (1 x 20 mL), dried (Na2S04), filtered and concentrated in vacuo to give a yellow oil. Flash chromatography was carried out (100% Pet ether - 10:1 - 2:1 Pet Ether/DCM) to afford the title compound as a pale yellow oil (2.30 g, 7.90 mmol, 83.9%). Rf = 0.68 (DCM); IR (vmax/cm"1, thin film): 1677 (C=0 stretch), 1598, 1574, 1475, 1446 (Aromatic C=C stretch), 1223 (C-O-C stretch) ; 1H NMR (500 MHz, CDCI3): δΗ = 4.65 (s, 2H, 1 -H), 6.86 (d, J = 8.4 Hz, 1 H, 7-H), 7.09 (d, J = 7.7 Hz, 2H, 10-H), 7.17 (t, J = 7.3 Hz, 1 H, 5-H), 7.22 (t, J = 7.3 Hz, 1 H, 12-H), 7.40-7.47 (m, 3H, 6,11 -H), 7.92 (dd, J = 7.8, 1 .5 Hz, 1 H, 4-H); 13C NMR (125 MHz, CDCI3): 5c = 36.8 (C-1 ), 1 17.6 (C-7), 1 19.5 (C-10), 123.0 (C-5), 124.4 (C-12), 126.2 (C-
3), 129.9 (C-11 ), 131 .3 (C-4), 134.2 (C-6), 155.0 (C-9), 156.5 (C-8), 191 .6 (C-2); LRMS m/z (El+): 292 [M (81 Br)]+, 290 [M (79Br)]+, 212 [M-Br]+, 197 [M-CH2Br]+; HRMS m/z (El+): Found 289.99403 [M(79Br)]+; Ci4Hn Br02 requires 289.99369.
Step 2
2-azido-1 -(2-phenoxyphenyl)ethanone

2-bromo-1-(2-phenoxyphenyl)ethanone (3.07 g, 10.5 mmol) was dissolved in DMSO (15 mL) and the mixture was cooled on ice. Sodium azide (824 mg, 12.68 mmol) was added in one portion and the reaction was stirred under argon at RT for 5 h. An extra portion of sodium azide (200 mg) was added and left to stir overnight. After 15 h, the reaction was quenched with H20 (30 mL), and extracted with EtOAc (4 x 40 mL). The combined organic layers were washed with H20 (5 x 20 mL) and then dried (Na2S04) and filtered. The solvent was removed in vacuo to give brown liquid (2.52 g, 9.96 mmol, 94.9%). Rf = 0.4 (DCM); IR (vmax/cm"1, thin film): 3071 (C-H Stretch), 2100 (- N=N+=N" stretch), 1685 (CO Stretch), 1599, 1475, 1448 (aromatic C=C stretch), 1225 (aromatic C-O-C stretch); 1 H NMR (500 MHz, CDCI3): δΗ = 4.60 (s, 2H, 1 -H), 6.87 (dd, J = 8.3, 0.9 Hz, 1 H, 7-H), 7.04-7.06 (m, 2H, 10,14-H), 7.18-7.23 (m 2H, 5,12-H), 7.40- 7.43 (m, 2H, 11 ,13-H),7.45-7.49 (m, 1 H, 6-H), 7.92 (dd, J = 7.8, 1.5 Hz, 1 H, 4-H); 13C NMR (125 MHz, CDCI3): 5C = 58.9 (C-1 ), 1 18.0 (C-7), 1 19.2 (C-10), 123.2 (C-5), 124.4 (C-12), 126.2 (C-3), 130.0 (C-11 ), 130.8 (C-4), 134.5 (C-6), 155.0 (C-9), 157.0 (C-8), 193.6 (C-2); LRMS m/z (Cl+): 198 [M-CH2N3]+, 85 [COCH2N3]+.
Step 3
2-azido-1 -(2-phenoxyphenyl)ethanol
2-azido-1-(2-phenoxyphenyl)ethanone (2.52 g, 9.96 mmol) was dissolved in anhydrous MeOH (50 mL) and cooled on ice. Sodium borohydride (565 mg, 14.9 mmol) was added portion wise and the mixture was stirred on ice under argon for 1 h until the reaction had gone to completion by TLC. The solvent was removed and the resulting residue was taken up in DCM (50 mL) and carefully washed with H20. An emulsion formed at the interface, and as a result, the mixture was acidified (from approximately pH 12) and the extraction continuted. The combined organic extracts were dried (Na2S04), filtered and concentrated in vacuo to give the title compound as a brown oil (2.47 g, 9.69 mmol, 97.3%). Rf = 0.19 (DCM); IR (vmax/crrf1, thin film): 3413 (O-H Stretch), 3039 (C-H Stretch), 2102 (-N=N+=N" stretch), 1583, 1483, 1452 (aromatic C=C stretch), 1230 (Aromatic C-O-C stretch); 1H NMR (500 MHz, CDCI3): δΗ = 2.54 (bs, 1 H, 15-H), 3.50 (dd, J = 12.5, 8.0 Hz, 1 H, 1 -H), 3.58 (dd, J = 12.5, 3.5 Hz, 1 H, 1 - H), 5.23 (dd, J = 8.0, 3.4 Hz, 1 H, 2-H), 6.84 (dd, J = 8.2, 1 .1 Hz, 1 H, 7-H), 6.97-7.00 (m, 2H, 10-H), 7.13 (tt, J = 7.4, 1 .1 Hz, 1 H, 5-Hj, 7.17 (td, J = 7.5, 1 .0 Hz, 1 H, 12-H), 7.24-7.27 (m, 1 H, 6-H), 7.34-7.37 (m, 2H, 11 -H), 7.58 (dd, J = 7.7, 1.7 Hz, 1 H, 4-H); 13C NMR (125 MHz, CDCI3): 5C = 56.5 (C-1 ), 68.7 (C-2), 1 18.0 (C-7), 1 18.3 (C-10), 123.4 (C-5), 123.5 (C-12), 127.1 (C-4), 129.6 (C-6), 129.6 (C-11 ), 130.8 (C-3), 153.5 (C-9), 156.3 (C-8); LRMS m/z (Cl+): 200 [M-CH2N3]+, 182 [M-OH,CH2N3]+.
Step 4
2-amino-1 -(2-phenoxyphenyl)ethanol
16

2-azido-1-(2-phenoxyphenyl)ethanol (2.46 g, 9.66 mmol) was dissolved in anhydrous MeOH (50 mL) and 10% palladium on carbon (246 mg, 10% w/w) was added. The reaction mixture was stirred under hydrogen atmosphere until completion as determined by TLC and disappearance of N3 peak by IR. After 22 h, the hydrogen was carefully released, and the reaction mixture was filtered through celite (pre-washed with MeOH). Solvent removal in vacuo gave the title compound as a brown oil (2.21 g, 9.66mmol, 99.9%). Rf = 0.0 (DCM); IR (vmax/cm"1, thin film): 3413 (O-H Stretch), 3055 (C-H Stretch), 2983 (N-H Stretch); 1H NMR (600 MHz, CDCI3): δΗ = 2.17 (bs, 3H, 15,16-H), 2.84 (dd, J = 13.2, 7.8 Hz, 1 H, 1 -H), 3.10 (dd, J = 13.2, 4.2 Hz, 1 H, 1 -H), 4.99 (dd, J = 7.8, 3.6 Hz, 1 H, 2-H), 6.82 (dd, J = 8.4, 1.2 Hz, 1 H, 7-H), 6.95 (d, J = 7.8 Hz, 2H, 10-H), 7.08-7.1 1 (m, 1 H, 12-Hj, 7.13-7.15 (m, 1 H, 5-H), 7.21 (td, J = 7.8, 1 .8 Hz, 1 H, 6-H), 7.30-7.33 (m, 2H, 11 -H), 7.57 (dd, J = 7.8, 1 .2 Hz, 1 H, 4-H); 13C NMR (150 MHz, CDCIs): 5c = 47.7 (C-1 ), 69.4 (C-2), 1 18.2 (C-7), 1 18.5 (C-10), 123.4 (C-12), 123.9 (C-5), 127.5 (C-4), 128.6 (C-6), 130.0 (C-11 ), 133.3 (C-3), 153.8 (C-9), 157.2 (C- 8); LRMS m/z (ESI+): 230.1 [M+H]+ 212.1 [M-OH]+, 195.1 [M-OH, NH2]+.
Step 5
2-[(3-chloropyrazin-2-yl)amino]-1 -(2-phenoxyphenyl)ethanol

2-[(3-chloropyrazin-2-yl)amino]-1 -(2-phenoxyphenyl)ethanone

6 5
DMSO (1.13 mL, 15.98 mmol) was dissolved in anhydrous DCM (100 mL) and the mixture was cooled to and maintained at -78°C. Oxalyl chloride (677 μί, 7.99 mmol) was added drop wise and the reaction was stirred for 20 min. 2-[(3-chloropyrazin-2- yl)amino]-1-(2-phenoxyphenyl)ethanol (2.10 g, 6.15 mmol), dissolved in DCM (20 mL), was then added drop wise and after 20 min stirring, Et3N (4.08 mL, 30.7 mmol) was added drop wise. The reaction was then allowed to slowly warm to RT over a period of 2 ½ h. The reaction was quenched with H20 (50 mL) and organics extracted followed by washing with 2.0 M HCI (2 x 40 mL), sat. NaHC03 (1 x 40 mL), H20 (1 x 40 mL) and brine (1 x 40 mL). Drying (MgS04), filtration and concentration gave a brown/orange
oil. Flash chromatography was carried out (DCM/EtOAc - 50:1 to 10:1 gradient) to afford the title compound as a yellow solid (1 .33 g, 3.92 mmol, 63.7%). Mpt: 76-78 °C; Rf = 0.74 (9:1 DCM/EtOAc); IR (vmax/cm"1, thin film): 3423 (O-H Stretch), 3060 (C-H Stretch), 2924 (N-H Stretch); 1H NMR (500 MHz, CDCI3): δΗ = 4.95 (d, J = 4.8 Hz, 1 H, 1 -H), 6.30 (1 H, 15-H), 6.90 (dd, J = 8.4, 0.8 Hz, 1 H, 7-H), 7.1 1-7.13 (m, 2H, 10-H), 7.18-7.23 (m, 2H, 5,12-H), 7.40-7.44 (m, 2H, 11 -H), 7.46-7.49 (m, 1 H, 6-H), 7.59 (d, J = 2.7 Hz, 1 H, 19-H), 7.88 (d, J = 2.7 Hz, 1 H, 18-H), 8.01 (dd, J = 7.9, 1 .8 Hz, 1 H, 4-H); 13C NMR (125 MHz, CDCI3): 5C = 52.0 (C-1 ), 1 18.1 (C-7) 1 19.4 (C-10), 123.0 (C-5), 124.3 (C-12), 126.5 (C-3), 129.9 (C-11 ), 130.7 (C-4), 130.8 (C-19), 134.3 (C-6), 134.8 (C-21 ), 140.0 (C-18), 150.2 (C-16), 155.2 (C-9), 157.2 (C-8), 194.8 (C-2); LRMS m/z (ESI+): 342 [M(37CI)+H]+, 340 [M(35CI)+H]+, 322 [M(35CI)-OH]+; HRMS m/z (ESI+): Found 340.0864 [M(35CI)+H]+; Ci8H15CIN302 requires 340.0853; Anal. Calcd for Ci8H14CIN302: C, 63.63; H, 4.15; N, 12.37. Found C, 63.90; H, 4.26; N, 1 1 .44% Step 7
8-chloro-3-(2-phenoxyphenyl)imidazo[1 ,2-a]pyrazine

2-[(3-chloropyrazin-2-yl)amino]-1-(2-phenoxyphenyl)ethanone (1 .33 g, 3.92 mmol) was dissolved in anhydrous toluene (50 mL) and the mixture was cooled on ice. Trifluoroacetic acid (2.1 1 mL, 24.7 mmol) was added and the reaction was allowed to stir on ice for 30 min, followed by the addition of trifluoroacetic anhydride (3.81 mL, 24.7 mmol). The reaction mixture was then stirred on ice for a further 30 min and then at RT for 68 h. The reaction was then diluted with toluene (50 mL) and washed with NaHC03 solution (10% w/v, 3 x 30 mL) and brine (1 x 40 mL). The organics were dried (MgS04), filtered and concentrated to give crude brown oil. Purification was carried out via flash chromatography (DCM/EtOAc gradient - 50:1 to 5:1 ) to afford the title compound as a yellow sticky solid (1.26 g, 3.92 mmol, 99.9%). Rf = 0.25 (9:1 DCM/EtOAc); IR (vmax/cm"1, thin film): 1460, 1231 (Ar-O-Ar Stretch); 1 H NMR (600 MHz, CDCI3): δΗ = 6.83-6.85 (m, 2H, 18-H), 7.05-7.10 (m, 2H, 14,20-H), 7.23-7.26 (m, 2H, 19-H), 7.29 (td, J = 7.8, 1 .2 Hz, 1 H, 12-H), 7.46-7.49 (m, 1 H, 13-H), 7.53 (dd, J = 7.2,
1.2 Hz, 1 H, 11 -H), 7.70 (d, J = 4.2 Hz, 1 H, 6-H), 7.86 (s, 1 H, 2-H), 8.01 (d, J = 4.2 Hz, 1 H, 5-H); 13C NMR (150 MHz, CDCI3): 5C = 1 18.3 (C-5), 1 18.5 (C-18), 1 18.8 (C-22), 124.2 (C-10,12,20), 126.1 (C-3), 130.0 (C-19), 131.5 (C-13), 132.0 (C-11 ), 135.6 (C-2), 138.4 (C-9), 143.9 (C-8), 155.0 (C-15), 156.0 (C-17); LRMS m/z (El+): 323 [M(37CI)]+, 321 [M(35CI)]+; HRMS m/z (El+): Found: 321 .06586 [M(35CI)]+; Ci8H12CIN30 requires 321.06634.
Step 8
4-methyl-W-[3-(2-phenoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide

All glassware was evacuated and flushed with argon prior to use. 8-chloro-3-(2- phenoxyphenyl)imidazo[1 ,2-a]pyrazine (516 mg, 1.60 mmol), 4-toluenesulfonamide (330 mg, 1.93 mmol), K2C03 (266 mg, 1 .93 mmol), 1 mol% Pd(dba)2 (5.20 mg) and 5 mol% ie f-butyl XPhos (26.0 mg) were weighed into a 50 mL round bottom flask. fBuOH (10 mL) was added and the reaction was stirred under reflux for 40 h. The reaction mixture was cooled to RT, diluted with MeOH and filtered through celite (pre-washed with MeOH). Flash Chromatography was carried out (10:1 DCM/EtOAc to 1 :1 gradient followed by DCM/10% MeOH) to afford the title compound as yellow solid (188 mg, 0.412 mmol 25.8%). Mpt: > 200 °C; Rf = 0.18 (2:1 DCM/EtOAc); IR (vmax/cm"1, thin film): 3243 (N-H), 2917 (C-H Stretch), 1588 (S=0), 1233 (Ar-O-Ar); 1H NMR (600 MHz, CDCIs): δΗ = 2.37 (s, 3H, 27-H), 6.85 (d, J = 7.8 Hz, 2H, 18-H), 7.04 (d, J = 8.4 Hz, 2H, 6, 14H), 7.07 (t, J = 7.2 Hz, 1 H, 20H), 7.23-7.27 (m, 5H, 12, 19, 25-H), 7.43-7.46 (m, 3H, 5, 11 , 13-H), 7.65 (s, 1 H, 2-H), 7.94 (bs, 2H, 24-H), 1 1 .45 (bs, 1 H, 21 -H); 13C NMR (150 MHz, CDCI3): 5c = 21.7 (C-27), 109.5 (C-5), 1 15.1 (C-6), 1 18.6 (C-10), 1 18.8 (C- 18), 1 18.9 (C-14), 124.0 (C-12), 124.2 (C-20), 126.8 (C-24), 128.1 (C-3), 129.4 (C-25), 130.0 (C-19), 131.5 (C-13), 132.1 (C-11 ), 134.6 (C-2), 136.0 (C-9), 139.5 (C-23), 143.2
(C-26), 146.0 (C-8), 155.0 (C-15), 155.9 (C-17); LRMS m/z (ESI+): 457 [M+H]+, 302 [M- C6H5-S02]+. HRMS m/z (ESI"): Found: 455.1 183 [M-H]-; C25H19N403S requires 455.1 178; Anal. Calcd for C25H2oN403S: C, 65.77; H, 4.42; N, 12.27. Found C, 63.37; H, 4.22; N, 1 1 .88%
Example 3
yV-[3-(3,4-dimethoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]-4-methyl- benzenesulfonamide
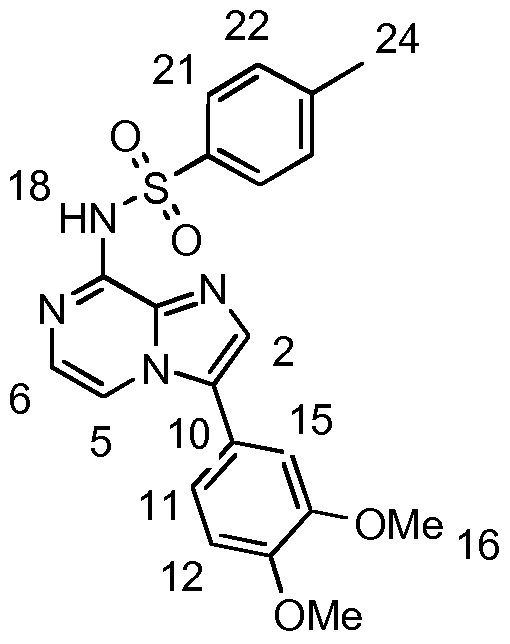
17
Step 1
2-bromo-1 -(3,4-dimethoxyphenyl)eth

3,4-dimethoxyacetophenone (1 .00 g, 5.55 mmol) was dissolved in a 1 :1 mixture of ethanol and chloroform (60 ml_). Pyridinium tribromide (4.46 g, 13.9 mmol) was added and the mixture was stirred at 50°C for 16 h. The reaction was cooled to RT and solvents removed in vacuo to give a sticky orange solid, which was then dissolved in H20 (30 ml_), and organics extracted with EtOAc (3 x 20 mL) and washed with H20 (2 x 20 mL) and brine (20 mL). The organics were dried (MgS04), filtered and concentrated in vacuo to give an orange oil containing a mixture of mono- and di-bromated species (approx 1 :5 to 1 ratio). Flash chromatography (DCM isocratic) afforded the title compound as an off-white solid (752 mg, 2.90 mmol. 52.3%). Spectroscopic data was consistent with that previously reported. Mpt: 62 °C [Lit. 67-70 °C]; Rf = 0.12 (DCM); IR
(vmax/cm-\ thin film): 2940 (C-H stretch), 1679 (C=0 stretch), 1585, 1512, 1465, 1418 (Aromatic C=C stretch), 1241 (C-O-C stretch); 1H NMR (500 MHz, CDCI3): δΗ = 3.93 (s, 3H, 9-H), 3.95 (s, 3H, 10-H), 4.40 (s, 2H, 1 -H), 6.90 (d, J = 8.4 Hz, 1 H, 5-H), 7.53 (d, J = 2.1 Hz, 1 H, 8-H), 7.60 (dd, J = 8.4, 2.1 Hz, 1 H, 4-H); 13C NMR (125 MHz, CDCI3): 5C = 30.5 (C-1 ), 56.1 (C-9), 56.2 (C-10), 1 10.2 (C-5), 1 10.9 (C-8), 123.9 (C-4), 127.1 (C- 3), 149.4 (C-7), 154.1 (C-6), 190.2 (C-2); LRMS m/z (El+): 260 [M(81Br)]+, 258 [M(79Br)]+, 165 [M-CH2Br]+; HRMS m/z (El+): Found 257.98872 [M(79Br)]+; Ci0HnO3Br requires 257.98861 .
Step 2
2-azido-1 -(3,4-dimethoxyphenyl)eth

9
2-bromo-1-(3,4-dimethoxyphenyl)ethanone (2.33 g, 8.61 mmol) was dissolved in anhydrous DMSO (10 mL) and the mixture was cooled on ice. Sodium azide (671 mg, 10.3 mmol) was added in one portion and the reaction was stirred under argon at RT for 2 h. An immediate colour change of yellow to orange was observed on addition of the sodium azide. The reaction was then quenched with H20 (30 mL), and extracted with EtOAc (4 x 40 mL). The combined organic layers were washed with H20 (5 x 20 mL) followed by brine (20 mL) and then dried (MgS04) and filtered. The solvent was removed in vacuo to give an orange solid (1 .87 g, 8.46 mmol, 98.3%) with NMR data comparable with literature values. Mpt: ; Rf = 0.22 (DCM); IR (vmax/cm"1, thin film): 2106 (-N=N+=N" Stretch), 1682 (C=0 Stretch), 1595, 1515 (aromatic C=C stretch), 1264 (Aromatic C-O-C Stretch); 1H NMR (600 MHz, CDCI3): δΗ = 3.94 (s, 3H, 9-H), 3.96 (s, 3H, 10-H), 4.52 (s, 2H, 1 -H), 6.90 (d, J = 8.4 Hz, 1 H, 5-H), 7.47 (dd, J = 8.5, 2.0 Hz, 1 H, 4-H), 7.52 (d, J = 2.1 Hz, 1 H, 8-H); 13C NMR (150 MHz, CDCI3): 5C = 54.5 (C-1 ), 56.1 (C-9), 56.2 (C-10), 1 10.0 (C-8), 1 10.1 (C-5), 122.5 (C-4), 127.5 (C-3), 149.4 (C-7), 154.1 (C-6), 191 .8 (C-2); LRMS m/z (Cl+): 222 [M+H]+, 165 [M-CH2N3]+; HRMS m/z (Cl+): Found 222.08777 [M+H]+; Ci0H12N3O3 requires 222.08787.
Step 3
2-azido-1 -(3,4-dimethoxyphenyl)ethanol
11

9
2-azido-1-(3,4-dimethoxyphenyl)ethanone (1 .86 g, 8.40 mmol) was dissolved in anhydrous Et20 (80 mL). Activated neutral alumina (8 g) and sodium borohydride (635 mg, 16.8 mmol) were added and the suspension was stirred at RT under argon. After 16 h the reaction mixture was filtered and washed with ether. The resulting filtrate was washed with H20 (2 x 30 mL), followed by brine (2 x 30 mL). The combined organic extracts were dried (Na2S04), filtered and concentrated in vacuo to give the title compound as a yellow oil (1.64 g, 7.37 mmol, 87.7 %). Rf = 0.65 (2:1 DCM/EtOAc); IR (vmax/crrf1, thin film): 3497 (O-H Stretch), 2938, 2839 (C-H stretch), 2106 (-N=N+=NT Stretch), 1516 (aromatic C=C stretch), 1263 (Aromatic C-O-C Stretch); 1H NMR (500 MHz, CDCIs): δΗ = 2.67 (bs, 1 H, 11 -H) 3.36 (dd, J = 12.6, 3.8 Hz, 1 H, 1 -H), 3.49 (dd, J = 12.6, 8.3 Hz, 1 H, 1 -H) 3.84 (s, 3H, 9-H), 3.85 (s, 3H, 10-H), 4.78 (dd, J = 8.3, 3.8 Hz, 1 H, 2-H), 6.82 (d, J = 8.2 Hz, 1 H, 5-H), 6.86 (dd, J = 8.2 ,1 .9 Hz, 1 H, 4-H), 6.89 (d, J = 1.9 Hz, 1 H, 8-H); 13C NMR (125 MHz, CDCI3): 5C = 55.6 (C-9,10), 57.7 (C-1 ), 72.9 (C- 2), 108.6 (C-8), 1 10.8 (C-5), 1 17.9 (C-4), 132.0 (C-3), 148.6 (C-7), 148.8 (C-6); LRMS m/z (El+): 223 [M]+,167 [M-CH2N3]+, 139 [M-CH(OH)CH2N3]+.
Step 4
2-amino-1 -(3,4-dimethoxyphenyl)ethanol

9
2-azido-1-(3,4-dimethoxyphenyl)ethanol (1 .58 g, 7.09 mmol) was dissolved in anhydrous MeOH (30 mL) and 10% palladium on carbon (158 mg, 10% w/w) was
added. The reaction mixture was stirred under hydrogen atmosphere until completion as determined by TLC and disappearance of N3 peak by IR. After 2 h, the hydrogen was carefully released, and the reaction mixture was filtered through celite (pre-washed with MeOH). Solvent removal in vacuo gave the crude compound as an orange oil. Flash chromatography (100% EtOAc followed by 100% MeOH), followed by re- dissolving in DCM and filtering to remove silica yielded the title compound as a white solid (880 mg, 4.47 mmol, 63.0 %). Spectroscopic data was consistent that previously reported. Mpt: ;Rf = 0.3 (1 :1 EtOAc/MeOH); IR (vmax/cm"1, thin film): 3362 (O-H Stretch), 2938, 2838 (N-H Stretch); 1 H NMR (500 MHz, CDCI3): δΗ = 2.14 (bs, 3H, 11 ,12-H) 2.80 (dd, J = 12.7, 7.9 Hz, 1 H, 1 -H), 2.96 (dd, J = 12.6, 4.0 Hz, 1 H, 1 -H) 3.86 (s, 3H, 9-H), 3.88 (s, 3H, 10-H), 4.56 (dd, J = 7.9, 4.0 Hz, 1 H, 2-H), 6.82 (d, J = 8.2 Hz, 1 H, 5-H), 6.86 (dd, J = 8.5, 1.8 Hz, 1 H, 4-H), 6.91 (d, J = 1.8 Hz, 1 H, 8-H); 13C NMR (125 MHz, CDCIs): 5c = 48.9 (C-1 ), 55.5 (C-9),55.6 (C-10) 73.8 (C-2), 108.7 (C-8), 1 10.7 (C-5), 1 17.8 (C-4), 134.8 (C-3), 148.1 (C-7), 148.7 (C-6); LRMS m/z (El+): 197 [M]+, 167 [M- CH2NH2]+; HRMS m/z (El+): Found: 197.10492 [M]+; Ci0H15NO3 requires 197.10464
Step 5
2-[(3-chloropyrazin-2-yl)amino]-1 -(3,4-dimethoxyphenyl)ethanol
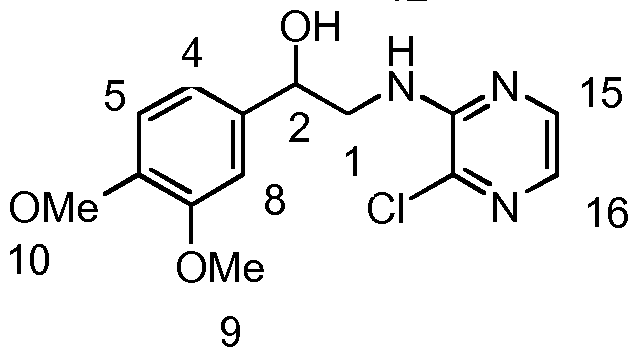
2-amino-1-(3,4-dimethoxyphenyl)ethanol (827 g, 4.20 mmol), 2,3-dichloropyrazine (481 μΙ_, 4.62 mmol) and Et3N (781 μΙ_, 5.88 mmol) were dissolved in 1 ,4-dioxane (8 mL) and the reaction was stirred under reflux, under argon. After 16 h, the reaction was cooled to RT, and the solvent removed in vacuo. The residual brown oil was taken up in DCM and washed with H20 (3 x 30 mL) and brine (1 x 20 mL). The organic extracts were dried (Na2S04), filtered and concentrated to give the crude product as a brown oil. Purification was carried out via flash chromatography (100% DCM - 10:1 - 5:1 DCM/EtOAc gradient) to afford the title compound as a light orange oil (748 mg, 2.42 mmol, 57.6%). Rf = 0.27 (2:1 DCM/EtOAc); IR (vmax/crrf1, thin film): 3377 (O-H Stretch) 2934 (N-H Stretch); 1 H NMR (500 MHz, CDCI3): δΗ = 3.60-3.66 (m, 1 H, 1 -H), 3.83-3.89 (m, 1 H, 1 -H) 3.88 (s, 3H, 9-H), 3.89 (s, 3H, 10-H), 4.93 (dd, J = 7.6, 3.7 Hz, 1 H, 2-H),
5.61 (s, 1 H, 12-H), 6.87 (d, J = 8.2 Hz, 1 H, 5-H), 6.93 (d, J = 8.2 Hz, 1 H, 4-H), 6.97 (s, 1 H, 8-H), 7.62 (d, J = 1.0 Hz, 1 H, 16-H), 7.93 (d, J = 1.0 Hz, 1 H, 15-H); 13C NMR (125 MHz, CDCIs): 5c = 49.0 (C-1 ), 55.6 (C-9,10), 73.2 (C-2), 108.6 (C-8), 1 10.8 (C-5), 1 17.8 (C-4), 130.9 (C-16) 134.1 (C-3),134.7 (C-18),139.6 (C-15) 148.4 (C-7), 148.8 (C- 6), 150.9 (C-13); LRMS m/z (Cl+): 312 [M(37CI)+H]+, 310 [M(35CI)+H]+, 294 [M(37CI)- OH]+ 292 [M(35CI)-OH]+; HRMS m/z (Cl+): Found 310.09562 [M(35CI)+H]+; Ci4H17CIN303 requires 310.09584
Step 6
2-[(3-chloropyrazin-2-yl)amino]-1 -(3,4-dimethoxyphenyl)ethanone

9
DMSO (417 μΙ_, 5.89 mmol) was dissolved in anhydrous DCM (25 mL) and the mixture was cooled to and maintained at -78°C. Oxalyl chloride (249 μί, 2.94 mmol) was added drop wise and the reaction was stirred for 15 min. 2-[(3-chloropyrazin-2- yl)amino]-1 -(3,4-dimethoxyphenyl)ethanol (700 mg, 2.26 mmol), dissolved in DCM (20 mL), was then added drop wise and after 15 min stirring, Et3N (1 .5 mL, 1 1.31 mmol) was added drop wise. The reaction was then allowed to slowly warm to RT over a period of 2 h. The reaction was quenched with H20 (20 mL) and organics extracted followed by washing with 2.0 M HCI (2 x 20 mL), sat.NaHCOs (1 x 20 mL), H20 (1 x 20 mL) and brine (1 x 20 mL). Drying (MgS04), filtration and concentration gave an off white solid. Flash chromatography (DCM - 50:1 to 5:1 DCM/EtOAc gradient) gave the title compound as a white solid (543 mg, 1 .76 mmol, 77.9%). Mpt: 128-130 °C; Rf = 0.31 (5:1 DCM/EtOAc); IR (vmax/cm"1, thin film): 3399 (C-H Stretch), 2936 (N-H Stretch), 1677 (C=0 Stretch); 1H NMR (500 MHz, CDCI3): δΗ = 3.96 (s, 3H, 9-H), 3.97 (s, 3H, 10-H), 4.88 (d, J = 4.2 Hz, 1 H, 1 -H), 6.43 (s, 1 H, 12-H), 6.94 (d, J = 8.2 Hz, 1 H, 5-H), 7.57 (d, J = 1.7 Hz, 1 H, 8-H), 7.64 (d, J = 2.9 Hz, 1 H, 16-H), 7.70 (dd, J = 8.2, 2.0 Hz, 1 H, 4-H), 7.96 (d, J = 2.5 Hz, 1 H, 15-H); 13C NMR (125 MHz, CDCI3): 5C = 47.3 (C-1 ), 55.8 (C-9), 55.9 (C-10) 109.7 (C-8), 1 10.0 (C-5), 122.3 (C-4), 127.3 (C-3), 130.8 (C- 16), 135.1 (C-18), 139.8 (C-15) 149.0 (C-7), 150.1 (C-13), 153.9 (C-6), 192.1 (C-2); LRMS m/z (Cl+): 310 [M(37CI)+H]+, 308 [M(35CI)+H]+; HRMS m/z (Cl+): Found
308.08143 [M(35CI)+H]+; Ci4H15CIN303 requires 308.08109; Anal. Calcd Ci4H14CIN303: C, 54.64; H, 4.59; N, 13.65. Found C, 53.70; H, 4.46; N, 14.01 %
Step 7
8-chloro-3-(3,4-dimethoxyphenyl)imidazo[1 ,2-a]pyrazine

17
2-[(3-chloropyrazin-2-yl)amino]-1-(3,4-dimethoxyphenyl)ethanone (280 mg, 0.912 mmol) was dissolved in anhydrous toluene (20 mL) and the mixture was cooled on ice. Trifluoroacetic acid (490 μΙ_, 6.39 mmol) was added and the reaction was allowed to stir on ice for 30 min, followed by the addition of trifluoroacetic anhydride (887 μΙ_, 6.39 mmol). The reaction mixture was then stirred on ice for a further 30 min and then at RT for 68 h. The reaction was then diluted with toluene (50 mL) and washed with NaHC03 solution (10% w/v, 3 x 30 mL) and brine (1 x 40 mL). The organics were dried (MgS04), filtered and concentrated to give crude yellow solid. Purification was carried out via flash chromatography (9:1 to 2:1 DCM/EtOAc) to afford the title compound as a white solid (46.8 mg, 0.162 mmol, 17.8%). Mpt: > 200 °C; Rf = 0.32 (2:1 DCM/EtOAc); IR (vmax/crrf1 , thin film): 2960 (w), 2924 (w), 1732 (w); 1H NMR (400 MHz, CDCI3): δΗ = 3.93 (s, 3H, 16-H), 3.96 (s, 3H, 17-H), 6.99 (d, J = 2.0 Hz, 1 H, 15-H), 7.03 (d, J = 8.2 Hz, 1 H, 12-H), 7.10 (dd, J = 8.3, 2.0 Hz, 1 H, 11 -H), 7.67 (d, J = 4.6 Hz, 1 H, 6-H), 7.84 (s, 1 H, 2-H), 8.15 (d, J = 4.6 Hz, 1 H, 5-H); 13C NMR (100 MHz, CDCI3): 5C = 56.1 (C- 16), 56.2 (C-17), 1 1 1.4 (C-15), 1 1 1.8 (C-12). 1 16.3 (C-5), 1 19.8 (C-10), 121.0 (C-11 ), 128.2 (C-6), 129.2 (C-3), 134.2 (C-2), 138.0 (C-9), 144.3 (C-8), 149.8 (C-14), 150.1 (C- 13); HRMS m/z (ESI+): Found 290.0683 [M(35CI)+H]+; Ci4H13N302CI requires 290.0696; Anal. Calcd for Ci4H12N302CI: C, 58.04; H, 4.17; N, 14.50.
Step 8
yV-[3-(3,4-dimethoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]-4-methyl- benzenesulfonamide

4-methyl-yV-[3-(3,5-dimethylphenyl)imidazo[1 ,2-a]pyrazin-8- yljbenzenesulfonamide

Step 1
2-bromo-1 -(3,5-dimethylphenyl)ethanone

3,5-dimethyl acetophenone (1 .00 g, 6.75 mmol) was dissolved in chloroform (40 mL) and ethanol (40 mL). Pyridinium tribromide (6.47 g, 20.2 mmol) was added and the reaction was stirred at 50°C for 17 h. The reaction mixture was cooled to RT and the solvents removed in vacuo. The resulting orange slurry was suspended in H20 (30 mL) and extracted with EtOAc (4 x 30 mL). The combined organic extracts were washed with H20 (2 x 20 mL) and brine (1 x 20 mL), dried (Na2S04), filtered and concentrated in vacuo to give a yellow oil. Flash chromatography was carried out (100% Pet Ether - 20:1 - 2:1 Pet Ether/DCM) to afford the title compound as a pale yellow oil (760 g, 3.34 mmol 49.6%). Spectroscopic data was consistent with that previously reported. Rf = 0.31 (1 :1 Pet Ether/DCM); IR (vmax/cm"1, thin film): 2919 (C-H Stretch), 1683 (C=0 Stretch),; 1H NMR (500 MHz, CDCI3): δΗ = 2.38 (s, 6H, 7-H), 4.44 (s, 2H, 1 -H), 7.24 (s, 1 H, 6-H), 7.58 (s, 2H, 4-H); 13C NMR (125 MHz, CDCI3): 5C = 21.3 (C-7), 31.3 (C-1 ), 126.7 (C-4), 134.2 (C-3), 135.7 (C-6), 138.7 (C-5), 191 .7 (C-2); LRMS m/z (ΕΓ): 228[M(81 Br)]+, 226 [M(79Br)]+, 133 [M-CH2Br]+; HRMS m/z (ΕΓ): Found 225.99851 [M(79Br)]+; doHnBrO requires 225.99877.
Step 2
2-azido-1 -(3,5-dimethylphenyl)ethanone

2-bromo-1-(3,5-dimethylphenyl)ethanone (3.44 g, 15.1 mmol) was dissolved in anhydrous DMSO (15 mL) and the mixture was cooled on ice. Sodium azide (1 .18 g, 18.2 mmol) was added in one portion and the reaction was stirred under argon at RT for 16 h. An extra portion of sodium azide (200 mg) was added and the reaction was left to stir for a further 1 h. A colour change from yellow to deep orange was observed. The reaction was then quenched with H20 (30 mL), and extracted with EtOAc (4 x 40 mL). The combined organic layers were washed with H20 (5 x 20 mL) followed by brine (20 mL) and then dried (MgS04) and filtered. The solvent was removed in vacuo to give an orange solid (2.85 g, 15.1 mmol, 99.9%). Rf = 0.59 (DCM); IR (vmax/cm"1, thin film): 2920 (C-H Stretch), 2105 (-N=N+=NT Stretch), 1692 (C=0 Stretch), 1604 (aromatic C=C stretch); 1 H NMR (500 MHz, CDCI3): δΗ = 2.35 (s, 6H, 7-H), 4.52 (s, 2H, 1 -H), 7.20 (s, 1 H, 6-H), 7.40 (s, 2H, 4-H); 13C NMR (125 MHz, CDCI3): 5C = 21.3 (C-7), 55.0 (C-1 ), 125.7 (C-4), 134.5 (C-3), 135.9 (C-6), 138.8 (C-5), 193.6 (C-2); LRMS m/z (Cl+): 132 [M-CH2N3]+
Step 3
2-azido-1 -(3,5-dimethylphenyl)ethanol

2-azido-1-(3,5-dimethylphenyl)ethanone (2.85 g, 15.1 mmol) was dissolved in anhydrous MeOH (60 mL) and cooled on ice. Sodium borohydride (855 mg, 22.6 mmol) was added portion wise and the mixture was stirred on ice under argon for 1 h until the reaction had gone to completion by TLC. The solvent was removed and the resulting orange oil was taken up in DCM (60 mL) and carefully washed with 2.0 M HCI (40 mL), H20 (30 mL) and brine (30 mL). The combined organic extracts were dried (MgS04), filtered and concentrated in vacuo to give the title compound as an orange oil (2.85 g, 14.9 mmol, 98.7%). Rf = 0.27 (DCM); IR (vmax/crrf1, thin film): 3395 (O-H
Stretch), 2918 (C-H Stretch), 2099 (-N=N+=N" Stretch); 1H NMR (500 MHz, CDCI3): δΗ = 2.32 (s, 6H, 7-H), 3.40 (dd, J = 12.6, 3.8 Hz, 1 H, 1 -H), 3.47 (dd, J = 12.6, 8.4 Hz, 1 H, 1 -H), 4.79 (dd, J = 8.4, 3.8 Hz, 1 H, 2-H), 6.96 (s, 1 H, 6-H), 6.97 (s, 2H, 4-H); 13C NMR (125 MHz, CDCIs): 5c = 21.2 (C-7), 58.2 (C-1 ), 73.5 (C-2), 123.8 (C-4), 130.0 (C-6), 138.4 (C-5), 140.7 (C-3); LRMS m/z (Cl+): 192 [M+H]+, 132 [M-OH-N3]+
Step 4
2-amino-1 -(3,5-dimethylphenyl)ethanol

2-azido-1-(3,5-dimethylphenyl)ethanol (2.85 g, 14.9 mmol) was dissolved in anhydrous MeOH (60 mL) and 10% palladium on carbon (285 mg, 10% w/w) was added. The reaction mixture was stirred under hydrogen atmosphere until completion as determined by TLC and disappearance of N3 peak by IR. After 22 h, the hydrogen was carefully released, and the reaction mixture was filtered through celite (pre-washed with MeOH). Solvent removal in vacuo gave the title compound as a sticky green/brown solid (2.37 g, 14.4 mmol, 96.6%). Rf = 0.0 (DCM); IR (vmax/crrf1, thin film): 3289 (O-H Stretch), 3010 (C-H Stretch), 2916, 2861 (N-H Stretch); 1H NMR (600 MHz, CDCI3): δΗ = 2.31 (s, 6H, 7-H), 2.81 (dd, J = 12.6, 7.8 Hz, 1 H, 1 -H), 2.96 (dd, J = 13.2, 4.2 Hz, 1 H, 1 -H), 4.57 (dd, J = 7.8, 4.2 Hz, 1 H, 2-H), 6.91 (s, 1 H, 6-H), 6.96 (s, 2H, 4-H); 13C NMR (150 MHz, CDCI3): 5c = 21.5 (C-7), 49.3 (C-1 ), 74.5 (C-2), 123.8 (C-4), 130.6 (C-6), 138.1 (C-5), 142.5 (C-3); LRMS m/z (ΕΓ): 224, 135 [M-CH2NH2]+, 1 17 [M-CH2OHNH2]+,
Step 5
2-[(3-chloropyrazin-2-yl)ami -1 -(3,5-dimethylphenyl)ethanol

2-amino-1-(3,5-dimethylphenyl)ethanol (2.34 g, 14.2 mmol), 2,3-dichloropyrazine (1 .62 mL, 15.6 mmol) and Et3N (2.76 mL, 19.8 mmol) were dissolved in 1 ,4-dioxane (24 mL) and the reaction was stirred under reflux, under argon. After 16 h, the reaction was
cooled to RT, and the solvent removed in vacuo. The residue was taken up in DCM and washed with H20 (3 x 30 mL) and brine (1 x 20 mL). The organic extracts were dried (MgS04), filtered and concentrated to give the crude product as a brown oil. Purification was carried out via flash chromatography (100% DCM - 30:1 - 10:1 DCM/EtOAc gradient) to afford the title compound as a light orange oil (2.58 g, 9.37 mmol, 65.5%). Rf = 0.37 (9:1 DCM/EtOAc); IR (vmax/cm"1, thin film): 3422 (O-H Stretch), 2921 (C-H Stretch); 1H NMR (600 MHz, CDCI3): δΗ = 2.33 (s, 6H, 7-H), 3.27 (s, 1 H, 8- H) 3.61 (ddd, J = 13.8, 7.8, 4.8 Hz, 1 H, 1 -H), 3.88 (ddd, J = 13.8, 6.6, 3.6 Hz, 1 H, 1 -H), 4.91 (dd, J = 7.9, 2.8 Hz, 1 H, 2-H), 5.62 (bt, 1 H, 9-H), 6.95 (s, 1 H, 6-H), 7.02 (s, 2H, 4- H), 7.62 (d, J = 3.0 Hz. 1 H, 13-H), 7.93 (d, J = 3.0 Hz, 1 H, 12-H); 13C NMR (150 MHz, CDCIs): 5c = 21.5 (C-7), 49.53 (C-1 ), 73.8 (C-2), 123.7 (C-4), 129.7 (C-6), 131.2 (C-13), 135.3 (C-15) 138.4 (C-5), 139.7 (C-12), 142.5 (C-3), 151 .3 (C-10); LRMS m/z (Cl+): 280 [M(37CI)+H]+, 278 [M(35CI)+H]+, 262 [M(37CI)-OH]+, 260 [M(35CI)-OH]+; HRMS m/z (Cl+): Found 278.10589 [M(35CI)+H]+; Ci4H17N3OCI requires 278.10601
Step 6
2-[(3-chloropyrazin-2-yl)amino]-1 -(3,5-dimethylphenyl)ethanone

DMSO (1.71 mL, 24.2 mmol) was dissolved in anhydrous DCM (170 mL) and the mixture was cooled to and maintained at -78°C. Oxalyl chloride (1.02 mL, 12.1 mmol) was added drop wise and the reaction was stirred for 20 min. 2-[(3-chloropyrazin-2- yl)amino]-1 -(3,5-dimethylphenyl)ethanol (2.58 g, 9.30 mmol), dissolved in DCM (30 mL), was then added drop wise and after 20 min stirring, Et3N (6.18 mL, 46.6 mmol) was added drop wise. The reaction was then allowed to slowly warm to RT over a period of 2 ½ h. The reaction was quenched with H20 (50 mL) and organics extracted followed by washing with 2.0 M HCI (2 x 40 mL), sat. NaHC03 (1 x 40 mL), H20 (1 x 40 mL) and brine (1 x 40 mL). Drying (MgS04), filtration and concentration gave a yellow solid (2.31 , 8.37 mmol, 90.0%). Mpt: Decomposed before melting; Rf = 0.76 (9:1 DCM/EtOAc); IR (vmax/crrf1, thin film): 3405 (m), 2916 (w), 1681 (s), 1578 (s), 1497 (s); 1H NMR (500 MHz, CDCI3): δΗ = 2.40 (s, 6H, 7-H), 4.89 (d, J = 4.0 Hz, 1 H, 1 -H), 6.43 (s, 1 H, 8-H), 7.27 (s, 1 H, 6-H), 7.65 (d, J = 3.0 Hz. 1 H, 12-H), 7.67 (s, 2H, 4-H), 7.97
(d, J = 3.0 Hz, 1 H, 11 -H); 13C NMR (125 MHz, CDCI3): 5C = 20.9 (C-7), 47.8 (C-1 ), 125.4 (C-4), 130.9 (C-12), 134.2 (C-3), 135.1 (C-14), 135.5 (C-6), 138.4 (C-5), 139.8 (C-11 ), 150.0 (C-9), 193.6 (C-2); LRMS m/z (ESI"): 276 [M(37CI)-HT/, 274 [M(35CI)-H]"; HRMS m/z (ESI"): Found 274.0762 [M(35CI)-H]"; Ci4H13N3OCI requires 274.0747; Anal. Calcd for d4H14N3OCI: C, 60.98; H, 5.12; N, 15.24. Found C, 60.22; H, 5.02; N, 14.83%
Step 7
8-chloro-3-(3,5-dimethylphenyl)imidazo[1 ,2-a]pyrazine

2-[(3-chloropyrazin-2-yl)amino]-1-(3,5-dimethylphenyl)ethanone (2.29 g, 8.31 mmol) was dissolved in anhydrous toluene (90 mL) and the mixture was cooled on ice. Trifluoroacetic acid (4.48 mL, 58.1 mmol) was added and the reaction was allowed to stir on ice for 30 min, followed by the addition of trifluoroacetic anhydride (8.09 mL, 58.1 mmol). The reaction mixture was then stirred on ice for a further 30 min and then at RT for 68 h. The reaction was then diluted with toluene (50 mL) and washed with NaHC03 solution (10% w/v, 3 x 40 mL) and brine (1 x 40 mL). The organics were dried (MgS04), filtered and concentrated to give crude orange sticky solid. Purification was carried out via flash chromatography (40:1 DCM/EtOAc) to afford the title compound as a yellow solid (531.7 g, 2.06 mmol, 24.8%). Mpt: 178-180 °C; Rf = 0.32 (9:1 DCM/EtOAc); IR (vmax/cm"1, thin film): 2918 (C-H), 1603, 1457, 1336, 922; 1H NMR (600 MHz, CDCI3): δΗ = 2.41 (s, 6H, 14-H), 7.14 (s, 1 H, 13-H), 7.16 (s, 2H, 11 -H), 7.69 (d, J = 4.6 Hz, 1 H, 6-H), 7.87 (s, 1 H, 2-H), 8.21 (d, J = 4.6 Hz, 1 H, 5-H); 13C NMR (150 MHz, CDCI3): 5C = 21 .5 (C-14), 1 16.6 (C-5), 125.9 (C-11 ), 127.4 (C-10), 128.3 (C-6), 129.6 (C-3), 131.3 (C-13), 134.5 (C-2), 138.3 (C-9), 139.5 (C-12), 144.4 (C-8); LRMS m/z (El+): 259 [M(37CI)]+, 257 [M(35CI)]+; HRMS m/z (Cl+): Found: 257.07169 [M(35CI)]+; Ci4H12CIN3 requires 257.07143; Anal. Calcd for Ci4H12CIN3: C, 65.25; H, 4.69; N, 16.30. Found C, 64.67; H, 4.57; N, 16.09%
Step 8
4-methyl-yV-[3-(3,5-dimethylphenyl)imidazo[1 ,2-a]pyrazin-8- yljbenzenesulfonamide

All glassware was evacuated and flushed with argon prior to use. 8-chloro-3-(3,5- dimethylphenyl)imidazo[1 ,2-a]pyrazine (93.8 mg, 0.364 mmol), 4-toluenesulfonamide (74.8 mg, 0.437 mmol), K2C03 (60.4 mg, 0.437 mmol), 1 mol% Pd(dba)2 (2.10 mg) and 5 mol% ie f-butyl XPhos (7.70 mg) were weighed into a 10 mL round bottom flask. fBuOH (2 mL) was added and the reaction was stirred under reflux for 40 h. The reaction mixture was cooled to RT, diluted with MeOH and filtered through celite (pre- washed with MeOH). Flash Chromatography was carried out (9:1 to 1 :1 DCM/ EtOAc gradient followed by DCM/10% MeOH) to afford the title compound as a pale yellow solid (96.2 mg, 0.245 mmol, 67.3%). Mpt: >200 °C; Rf = 0.5 (DCM/10% MeOH); IR (vmax/crrf1, thin film): 3253 (w), 1591 (s); 1H NMR (600 MHz, CD2CI2): δΗ = 2.38 (s, 6H, 14-H), 2.39 (s, 3H, 21 -H), 6.98 (bs, 1 H, 6-H), 7.1 1 (s, 2H, 11 -H), 7.13 (s, 1 H, 13-H), 7.30 (d, J = 8.0 Hz, 2H, 19-H), 7.50 (bs, 1 H, 5-H), 7.64 (s, 1 H, 2-H), 7.90 (bd, J = 6.1 Hz, 2H, 18-H), 1 1.33 (bs, 1 H, 15-H); 13C NMR (150 MHz, CD2CI2): 5C = 21 .4 (C-14), 21 .7 (C-21 ), 108.9 (C-5), 1 15.8 (C-6), 125.9 (C-3), 126.4 (C-11 ), 126.8 (C-18), 129.8 (C-19), 131.4 (C-13), 132.2 (C-10), 133.6 (C-2), 136.1 (C-9), 139.4 (C-12), 139.7 (C- 17), 143.9 (C-20), 146.2 (C-8); LRMS m/z (ESI+): 415 [M+Na]+, 393 [M+H]+; HRMS m/z (ESI+): Found: 393.1366 [M+H]+; C2iH21 N402S requires 393.1385; Anal. Calcd for C2i H20N4O2S: C, 64.27; H, 5.14; N, 14.27. Found C, 63.49; H, 5.06; N, 14.24%
Example 5
4-methyl-yV-[4-[3-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8- ]yl]aminophenyl]benzenesulfonamide
25

Steps 1 to 6 were carried out as per Steps 1 to 6 of Example
Step 7
4-methyl-yV-[4-[3-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8- ]yl]aminophenyl]benzenesulfonamide
25

All glassware was evacuated and flushed with argon prior to use. 8-chloro-3-(2- naphthyl)imidazo[1 ,2-a]pyrazine (283 mg, 1 .01 mmol), /V-(4-aminophenyl)-4- methylbenzenesulfonamide (318 mg, 1 .21 mmol), K2C03 (167 mg, 1.21 mmol), 1 mol%
Pd(dba)2 (5.80 mg) and 5 mol% ie f-butyl XPhos (21.5 mg) were weighed into a 25 mL round bottom flask. fBuOH (6 mL) was added and the reaction was stirred under reflux for 46 h. The reaction mixture was cooled to RT, diluted with MeOH (100 mL) and filtered through celite (pre-washed with MeOH). Flash chromatography (DCM/Et20 100:1 to 8:1 gradient) was carried out to give the title compound as a yellow solid (181 mg, 0.355 mmol, 35.1 %). Mpt: >200 °C; Rf = 0.12 (10:1 DCM/EtOAc); IR (vmax/cm"\ thin film): 3240 (w), 3057 (w), 1623 (m), 1500 (s), 1 154 (s); 1H NMR (600 MHz, CDCI3): δΗ = 2.37 (s, 3H, 31 -H), 7.09 (d, J = 8.9 Hz, 2H, 23-H), 7.17 (s, 1 H, 25-H), 7.21 (d, J = 8.2 Hz, 2H, 29-H), 7.52 (d, J = 4.7 Hz, 1 H, 6-H), 7.58 (dd, J = 5.8, 3.1 Hz, 2H, 15,16-H), 7.64 (dd, J = 8.3, 1 .6 Hz, 1 H, 11 -H), 7.65 (d, J = 8.3 Hz, 2H, 28-H), 7.73 (s, 1 H, 2-H), 7.79-7.80 (m, 3H, 5,22-H), 7.91 (dd, J = 5.9, 3.4 Hz, 2H, 14,17-H), 8.00 (d, J = 8.6 Hz, 1 H, 12-H), 8.03 (s, 1 H, 19-H), 8.39 (bs, 1 H, 20-H); 13C NMR (150 MHz, CDCI3): 5C = 21 .6 (C-31 ), 109.4 (C-5), 120.3 (C-22), 123.7 (C-23), 125.2 (C-10), 125.4 (C-11 ), 127.1 (C-15,16), 127.3 (C-19,28), 127.9 (C-14), 128.1 (C-17), 128.9 (C-3), 129.0 (C-6), 129.3 (C-12), 129.7 (C-29), 130.4 (C-2), 131.2 (C-24), 133.1 (C-9,18), 133.4 (C-13), 136.1 (C-27), 137.2 (C-21 ), 143.7 (C-30), 146.2 (C-8); LRMS m/z (ESI"): 504 [M-H]"; HRMS m/z (ESI"): Found 504.1503 [M-H]"; C29H22N502S requires 504.1494; Anal. Calcd for C29H23N502S: C, 68.89; H, 4.59; N, 13.85. Found C, 66.66; H, 4.51 ; N, 13.1 1 %
Example 6
4-methyl-yV-[4-[3-(3-thienyl)imidazo[1 ,2-a]pyrazin-8- yl]aminophenyl]benzenesulfonamide
20


1-(3-thienyl)ethanone (2.00 g, 15.9 mmol) was dissolved in chloroform (100 mL) and ethanol (100 mL). Pyridinium tribromide (10.1 g, 31 .7 mmol) was added and the reaction was stirred at 50°C for 18 h. The reaction mixture was cooled to RT and the solvents removed in vacuo. The resulting orange slurry was suspended in H20 (40 mL) and extracted with EtOAc (4 x 50 mL). The combined organic extracts were washed with H20 (2 x 30 mL) and brine (1 x 30 mL), dried (MgS04), filtered and concentrated in vacuo to give an amber liquid. Flash chromatography was carried out (3:1 Pet Ether/DCM) to afford the title compound as a white solid (1.61 g, 7.81 mmol, 49.1 %). Spectroscopic data was consistent with that previously reported (Laufer, S, et al Arch Pharm. Pharm. Med Chem., 1997, 330, 307-312). Mpt: 59-60 °C [Lit. 61 .6 °C]; Rf = 0.48 (1 :1 Pet Ether/DCM); IR (vmax/cm"1, thin film): 3094.5 (C-H stretch), 1688.5 (C=0 stretch), 1508, 1415, 1400, 1393, 1380 (Aromatic C=C stretch), 1 181 ; 1H NMR (600 MHz,CDCI3): δΗ = 4.34 (s, 2H, 1 -H), 7.36 (dd, J = 2.9, 5.2 Hz, 1 H, 5-H), 7.58 (dd, J = 5.2, 1.2Hz, 1 H, 4-H), 8.17 (dd, J = 2.9, 1.3 Hz, 1 H, 7-H); 13C NMR (150 MHz, CDCIs): 5c = 31.7 (C-1 ), 127.0 (C-5), 127.4 (C-4), 133.9 (C-7), 138.9 (C-3), 185.7 (C-2); LRMS m/z (Cl+): 207 [M(81 Br)]+, 205 [M(79Br)]+
Step 2
2-azido-1 -(3-thienyl)ethanone

2-bromo-1-(3-thienyl)-1 -ethanone (573 mg, 2.80 mmol) was dissolved in DMSO (3 mL) and the mixture was cooled on ice. Sodium azide (218 mg, 3.35 mmol) was added in one portion and the reaction was stirred under argon at RT for 5 h. The reaction was quenched with H20 (20 mL), and extracted with EtOAc (3 x 30 mL). The organic layers were combined, washed with H20 (5 x 20 mL) followed by brine (20 mL), dried (Na2S04) and filtered. The solvent was removed in vacuo to give brown/orange oil (460 mg, 2.75 mmol, 98.5%). Rf = 0.27 (DCM); IR (vmax/crrf1, thin film): 3105 (C-H
stretch), 2097 (-N=N+=N" Stretch), 1679 (C=0 Stretch), 1508, 1409, 1231 , 1 177; 1 H NMR (500 MHz,CDCI3): δΗ = 4.43 (s, 2H, 1 -H), 7.38 (dd, J = 5.1 , 2.8 Hz, 1 H, 5-H), 7.55 (dd, J = 5.0, 1.2 Hz, 1 H, 4-H), 8.10 (dd, J = 2.9, 1.3 Hz, 1 H, 7-H); 13C NMR (125 MHz, CDCIs): 5c = 55.5 (C-1 ), 126.6 (C-5), 127.2 (C-4), 132.7 (C-7), 139.1 (C-3), 187.6 (C-2); LRMS m/z HRMS m/z (ESI+): Found 168.02354 [M+H]+; C6H6N3OS requires 168.02316.
Step 3
2-azido-1 -(3-thienyl)ethanol

2-azido-1-(3-thienyl)ethanone (278 mg, 1.67 mmol) was dissolved in anhydrous MeOH
(10 mL) and cooled on ice. Sodium borohydride (94.5 mg, 2.45 mmol) was added portion wise and the mixture was stirred on ice under argon for 1 h until the reaction had gone to completion by TLC. The solvent was removed and the resulting residue was taken up in DCM (30 mL) and carefully washed with H20 (2 x 20 mL) followed by brine (20 mL). The organic extracts were (Na2S04), filtered and concentrated in vacuo to give the title compound as a yellow oil (257 mg, 1.52 mmol 91 .2%). Rf = 0.8 (1 :1 Pet Ether/EtOAc); IR (vmax/cm"1, thin film): 3372 (O-H stretch), 3105 (C-H stretch), 2096 (- N=N+=N" Stretch); 1H NMR (500 MHz,CDCI3): δΗ = 3.51-3.54 (m, 2H, 1 -H), 4.98 (dd, J = 4.7,1.9 Hz, 1 H, 2-H), 7.08 (dd, J = 5.0,1.3 Hz, 1 H, 4-H), 7.29-7.30 (m„ 1 H, 7-H), 7.34 (dd, J = 5.0, 3.0 Hz, 1 H, 5-H); 13C NMR (125 MHz, CDCI3): 5C = 57.5 (C-1 ), 69.9 (C-2) 122.0 (C-7), 125.4 (C-4), 126.7 (C-5), 142.0 (C-3); LRMS m/z (Cl+): 152 [M-OH]+, 127 [M-N3]+, 1 13 [M-CH2N3]+; HRMS m/z (Cl+): Found 152.02859 [M-OH]+; C6H6N3S requires 152.02824.
Step 4
2-amino-1 -(3-thienyl)ethanol
8
 2-azido-1-(3-thienyl)ethanol (257 mg, 1 .52 mmol) was dissolved in anhydrous MeOH (10 mL) and 10% palladium on carbon (25.7 mg, 10% w/w) was added. The reaction vessel was then subjected to 3 bar hydrogen for 5 h. After this period the mixture was filtered through celite and concentrated in vacuo to give a pale yellow solid (217 mg, 1.52 mmol, 99.9%). Rf = 0.0 (1 :1 DCM/EtOAc); vmax/cm"1 (thin film): 3194 (O-H stretch), 3091 (N-H), 2921 (C-H); 1H NMR (600 MHz,CDCI3): δΗ = 1 .91 (bs, 3H, 8,9-H), 2.88 (dd, J = 12.6, 7.8 Hz, 1 H, 1 -H), 3.04 (d, J = 10.2 Hz, 1 H, 1 -H), 4.75 (dd, J = 6.6, 3.6 Hz, 1 H, 2-H), 7.08 (d, J = 5.4 Hz, 1 H, 4-H), 7.23 (d, J = 2.4 Hz, 1 H, 7-H), 7.31 (dd, J = 4.8, 3.0 Hz, 1 H, 5-H); 13C NMR (150 MHz, CDCI3): 5C = 48.4 (C-1 ), 70.9 (C-2) 121 .1 (C-7), 125.6 (C-4), 126.3 (C-5), 144.0 (C-3); LRMS m/z (Cl+): 177, 159, 127 [M-NH2]+; (El+): 131 , 127 [M-NH2]+, 1 19, 1 14
2-azido-1-(3-thienyl)ethanol (257 mg, 1 .52 mmol) was dissolved in anhydrous MeOH (10 mL) and 10% palladium on carbon (25.7 mg, 10% w/w) was added. The reaction vessel was then subjected to 3 bar hydrogen for 5 h. After this period the mixture was filtered through celite and concentrated in vacuo to give a pale yellow solid (217 mg, 1.52 mmol, 99.9%). Rf = 0.0 (1 :1 DCM/EtOAc); vmax/cm"1 (thin film): 3194 (O-H stretch), 3091 (N-H), 2921 (C-H); 1H NMR (600 MHz,CDCI3): δΗ = 1 .91 (bs, 3H, 8,9-H), 2.88 (dd, J = 12.6, 7.8 Hz, 1 H, 1 -H), 3.04 (d, J = 10.2 Hz, 1 H, 1 -H), 4.75 (dd, J = 6.6, 3.6 Hz, 1 H, 2-H), 7.08 (d, J = 5.4 Hz, 1 H, 4-H), 7.23 (d, J = 2.4 Hz, 1 H, 7-H), 7.31 (dd, J = 4.8, 3.0 Hz, 1 H, 5-H); 13C NMR (150 MHz, CDCI3): 5C = 48.4 (C-1 ), 70.9 (C-2) 121 .1 (C-7), 125.6 (C-4), 126.3 (C-5), 144.0 (C-3); LRMS m/z (Cl+): 177, 159, 127 [M-NH2]+; (El+): 131 , 127 [M-NH2]+, 1 19, 1 14
Step 5
2-[(3-chloropyrazin-2-yl)ami -1 -(3-thienyl)ethanol

2-amino-1-(3-thienyl)ethanol (232 mg, 1.63 mmol), 2,3-dichloropyrazine (183 μΙ_, 1 .79 mmol), Et3N (316 μΙ_, 2.27 mmol) and 1 ,4-dioxane (2.5 mL) were stirred under reflux, under argon for 18 h. The solvent was removed in vacuo and the residual was taken up in DCM (30 mL), washed with H20 (5 x 10 mL) and brine (10 mL). The organics were dried (MgS04), filtered and solvent removed to give a brown oil. Flash chromatography (DCM followed by 2:1 DCM/EtOAc) afforded the title compound as an orange oil (193 mg, 0.753 mmol, 46.2 %). Rf = 0.46 (2:1 DCM/EtOAc) ; IR (vmax/cnT1, thin film): 3420 (O-H), 3091 (N-H), 2920 (C-H), 1583, 1525; 1H NMR (600 MHz,CDCI3): δΗ = 3.69-3.74 (m, 1 H, 1 -H), 3.96-4.00 (m, 1 H, 1 -H), 5.10 (dd, J = 7.2, 3.0 Hz, 1 H, 2-H), 5.72 (bs, 1 H, 9-H), 7.13 (dd, J = 4.8, 1.2 Hz, 1 H, 4-H), 7.31 -7.32 (m, 1 H, 7-H), 7.35 (dd, J = 5.4, 3.0 Hz, 1 H, 5-H), 7.65 (d, J = 3.0 Hz, 1 H, 13-H), 7.93 (d, J = 2.4 Hz, 12-H); 13C NMR (150 MHz, CDCI3): 5C = 49.0 (C-1 ), 70.4 (C-2), 121.6 (C-7), 125.5 (C-4), 126.7 (C-5), 131.3 (C-13), 135.6 (C-15), 138.8 (C-12), 143.1 (C-3), 150.8 (C-10); LRMS m/z (ESI+): 240 [M(37CI)-OH]+, 238 [M(35CI)-OH]+; HRMS m/z (ESI"): Found 254.0145 [M-H]" ; CioH9N3OSCI requires 254.0155.
Step 6
2-[(3-chloropyrazin-2-yl)ami -1 -(3-thienyl)ethanone

DMSO (128 μί, 1.81 mmol) was dissolved in anhydrous DCM (9 mL) and the mixture was cooled to and maintained at -78°C. Oxalyl chloride (77.0 μΙ_, 0.906 mmol) was added drop wise and the reaction was stirred for 20 min. 2-[(3-chloropyrazin-2- yl)amino]-1-(3-thienyl)ethanol (178 mg, 0.697 mmol), dissolved in DCM (5 mL), was then added drop wise and after 20 min stirring, Et3N (463 μί, 3.48 mmol) was added drop wise. The reaction was then allowed to slowly warm to RT over a period of 2 1/2 h. The reaction was quenched with H20 (20 mL) and organics extracted followed by washing with 2.0 M HCI (2 x 10 mL), sat. NaHC03 (1 x 10 mL), H20 (1 x 10 mL) and brine (1 x 10 mL). Drying (MgS04), filtration and concentration gave an orange sticky solid. Flash chromatography (DCM - 50:1 to 20:1 DCM/EtOAc gradient) gave the title compound as a yellow solid (135 mg, 0.531 mmol, 76.2%). Mpt: 130-134 °C; Rf = 0.85 (2:1 DCM/EtOAc); IR (vmax/crrf1, thin film): 3407 (w), 3107 (w), 2917 (w), 1682 (s), 1582 (s); 1H NMR (600 MHz,CDCI3): δΗ = 4.84 (d, J = 4.4 Hz, 1 H, 1 -H), 6.33(s, 1 H, 9-H), 7.41 (dd, J = 5.0, 2.8 Hz, 1 H, 5-H), 7.65 (dd, J = 5.0, 1.2 Hz, 1 H, 4-H), 7.66 (d, J = 2.8 Hz, 1 H, 13-H), 7.96 (d, J = 2.7 Hz, 12-H), 8.26 (dd, J = 5.4, 3.0 Hz, 1 H, 7-H); 13C NMR (150 MHz, CDCIs): 5c = 49.0 (C-1 ), 70.4 (C-2), 121.6 (C-4), 125.5 (C-7), 126.7 (C-5), 131.3 (C-13), 135.6 (C-15), 138.8 (C-12), 143.1 (C-3), 150.8 (C-10); LRMS m/z (Cl+): 256 [M(37CI)+H]+, 254 [M(35CI)+H]+; HRMS m/z (Cl+): Found 254.01471 [M(35CI)+H]+; CioH9N3OSCI requires 254.01549; Anal. Calcd for Ci0H8N3OSCI: C, 47.34; H, 3.18; N, 16.56. Found C, 48.16; H, 3.41 ; N, 15.78% Step 7
8-chloro-3-(3-thienyl)imidazo[1 ,2-a]pyrazine

4-methyl-yV-[4-[3-(3-thienyl)imidazo[1 ,2-a]pyrazin-8- yl]aminophenyl]benzenesulfonamide
20

All glassware was evacuated and flushed with argon prior to use. 8-chloro-3-(3- thienyl)imidazo[1 ,2-a]pyrazine (35.0 mg, 0.149 mmol), /V-(4-aminophenyl)-4- methylbenzenesulfonamide (47.8 mg, 0.178 mmol), K2C03 (24.6 mg, 0.178 mmol), 1
mol% Pd(dba)2 (0.900 mg) and 5 mol% ie/f-butyl XPhos (3.20 mg) were weighed into a 25 mL round bottom flask. fBuOH (2 mL) was added and the reaction was stirred under reflux for 46 h. The reaction mixture was cooled to RT, diluted with MeOH (100 mL) and filtered through celite (pre-washed with MeOH). Flash chromatography (3:2 Pet Ether/EtOAc) was carried out to give a mixture of the product and starting sulfonamide. Reverse phase preparative HPLC (5% MeCN - 40% 3 min - 70% 25 min - 95% 27 min, hold 2 min - 5% 30 min, hold 2 min) carried out to afford title compound as off white solid (12.4 mg, 0.027 mmol, 18.1 %). Mpt: ; Rf = 0.62 (1 :1 DCM/EtOAc); IR (vmax/crrf1, thin film): 31 12 (w), 1624 (w), 1542 (s), 1506 (s), 1 159 (s); 1 H NMR (600 MHz, CD3OD): δΗ = 2.38 (s, 3H, 26-H), 7.17 (d, J = 5.4 Hz, 1 H, 6-H), 7.28 (d, J = 9.0 Hz, 2H, 17-H), 7.33 (d, J = 8.4 Hz, 2H, 24-H), 7.48 (d, J = 9.0 Hz, 2H, 18-H), 7.49, (dd, J = 5.4 Hz, 1 .2 1 H, 11 -H), 7.72-7.73 (m, 3H, 12-H, 23-H), 7.92 (dd, J = 2.4, 1.2 Hz, 1 H, 14-H), 7.96 (bs, 1 H, 2-H), 8.02 (d, J = 5.4 Hz, 1 H, 5-H); 13C NMR (150 MHz, CD3OD): 5c = 21.4 (C-26), 1 12.2 (C-5), 1 19.8 (C-6), 122.7 (C-17), 126.1 (C-14), 126.6 (C-18), 128.0 (C-11 ), 128.3 (C-10), 128.3 (C-23), 128.9 (C-12), 129.1 (C-3), 130.7 (C-24), 132.1 (C-19), 133.0 (C-9), 134.2 (C-2), 138.2 (C-22), 138.8 (C-16), 145.3 (C-25), 146.2 (C-8); 1H NMR (600 MHz,CDCI3): δΗ = 2.38 (s, 3H, 26-H), 6.69 (s, 1 H, 20-H), 7.07 (d, J = 8.8 Hz, 2H, 18-H), 7.22 (d, J = 8.1 Hz, 2H, 24-H), 7.34 (dd, J = 4.0, 2.0 Hz, 1 H, 11 - H), 7.52 (d, J = 4.7 Hz, 1 H, 6-H), 7.54 - 7.56 (m, 2H, 12,14-H), 7.63 (d, J = 8.3 Hz, 2H, 23-H), 7.64 (s, 1 H, 2-H), 7.71 (d, J = 4.7 Hz, 1 H, 5-H), 7.79 (d, J = 8.8 Hz, 2H, 17-H), 8.34 (bs, 1 H, 15-H); 13C NMR (150 MHz,CDCI3): 21.7 (C-26), 109.7 (C-5), 120.3 (C- 17), 123.6 (C-12), 123.9 (C-18), 124.5 (C-10), 126.9 (C-11 ), 127.4 (C-14), 127.5 (C-2, 23), 128.1 (C-3), 129.1 (C-6), 129.8 (C-24), 131.2 (C-19), 132.9 (C-9), 136.2 (C-22), 137.4 (C-16), 143.9 (C-25), 146.1 (C-8); LRMS m/z (ESI+): 484 [M+Na]+, 462 [M+H]+; HRMS m/z (ESI+): Found 462.1065 [M+H]+; C23H2oN5S202 requires 462.1058; Anal. Calcd for C23H19N5S202: C, 59.9; H, 4.1 ; N, 15.2. Found C, ; H, ; N, %
Example 7
4-methyl-A/-[2-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide

17 16
Step 1
8-chloro-2-(2-naphthyl)imidazo[1 ,2-a]pyrazine
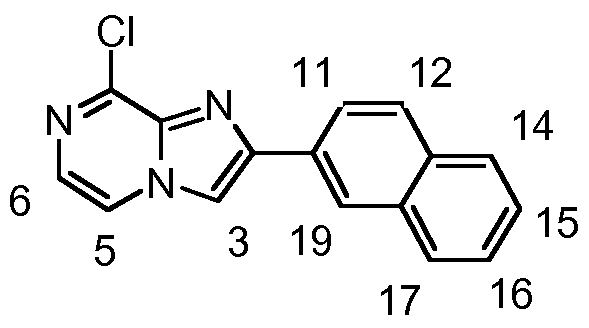
2-(Bromoacetyl)naphthalene (3.14 g, 12.6 mmol), 2-amino-3-chloropyrazine (1 .63 g, 12.6 mmol), NaHC03 (1 .32 g, 16.7 mmol) and fBuOH (60 mL) were stirred under reflux for 40 h. The reaction mixture was cooled to RT and solvent removed in vacuo. The resulting orange solid was taken up in H20 (60 mL) and extracted with DCM (3x 60 mL). The combined organics were washed with H20 (50 mL) and brine (40 mL), dried (MgS04), filtered and concentrated in vacuo to give crude orange solid. On addition of DCM, insoluble material filtered off to give title compound as a cream fluffy solid (1.05 g). Purification of the remaining filtrate via flash chromatography (10:1 to 2:1 Pet Ether/EtOAc gradient) afforded the title compound as a pale orange/brown solid (338 g, Total: 1 .38 g, 4.95 mmol, 39.3%). Mpt: Decomposed before melting; Rf = 0.34 (1 :1 Pet Ether/ethyl acetate); IR (vmax/cm"1, thin film): 3026.3 (w), 2921 .0 (w), 1495.3 (m); 1H NMR (600 MHz, CDCI3): δΗ = 7.51-7.53 (m, 2H, 15,16-H), 7.71 (d, J = 4.5 Hz, 1 H, 6- H), 7.88 (dd, J = 6.7, 2.2 Hz, 1 H, 14-H), 7.93 (d, J = 8.6, 1 H, 12-H) 7.96 (dd, J = 6.7, 2.4 1 H, 17-H), 8.06 (dd, J = 8.6, 1.7 Hz, 1 H, 11 -H) 8.08 (d, J = 4.5 Hz, H, 5-H), 8.14 (s, 1 H, 3-H), 8.57 (s, 1 H, 19-H); 13C NMR (150 MHz, CDCI3): 5C = 1 1 1 .6 (C-3), 1 18.5 (C- 5), 124.1 (C-11 ), 126.0 (C-19), 126.6/126.7 (C-15,16), 127.8 (C-14), 128.2 (C-6), 128.5 (C-17), 128.6 (C-12), 129.4 (C-10), 133.5 (C-18), 133.7 (C-13), 138.3 (C-9), 143.5 (C- 8), 148.2 (C-2); LRMS m/z (ESI+): 282 [M(37CI)+H]+, 280 [M(35CI)+H]+; HRMS m/z (Cl+):
Found 280.06338 [M(35CI)+H]+; Ci6Hn N3CI requires 280.06415; Anal. Calcd Ci6HioN3CI: C, 68.70; H, 3.60; N, 15.02. Found C, 67.53; H, 3.64; N, 13.97%.
Step 2
4-methyl-A/-[2-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide

17 16
All glassware was evacuated and flushed with argon prior to use. 8-chloro-2-(2- naphthyl)imidazo[1 ,2-a]pyrazine (50.0 mg, 0.178 mmol), 4-toluenesulfonamide (36.9 mg, 0.215 mmol), K2C03 (29.7 mg, 0.215 mmol), 1 mol% Pd(dba)2 (1.03 mg) and 5 mol% ie f-butyl XPhos (3.80 mg) were weighed into a 25 mL round bottom flask. fBuOH (3 mL) was added and the reaction was stirred under reflux for 24 h. The reaction mixture was cooled to RT, diluted with MeOH (100 mL) and filtered through celite (pre- washed with MeOH). Flash chromatography (silica pre washed with 1 % Et3N, DCIW EtO Ac-gradient ran) afforded the target compound as a white solid (28.0 mg, 0.068 mmol, 38.2%). Mpt: >200 °C; Rf = 0.32 (1 :1 DCIW EtOAc); IR (vmax/cm"1, thin film): 1 1 16.3, 1 135.4 (S=0 Symmetric Stretch), 1360.9 (S=0 Asymmetric Stretch), 3252.8 (N-H Stretch); 1H NMR (600 MHz, DMSO-d6): δΗ = 2.37 (s, 3H, 26-H), 7.16 (bd, J = 5.2 Hz, 1 H, 6-H), 7.39 (d, J = 8.2 Hz, 2H, 24-H), 7.51 - 7.54 (m, 2H, 15,16-H), 7.86 (bd, J = 5.2 Hz, 1 H, 5-H), 7.89 (d, J = 8.0 Hz, 2H, 23-H), 7.92 (d, J = 7.7 Hz, 1 H, 14-H), 7.98 (d, J = 8.6 Hz, 1 H, 12-H), 8.01 - 8.05 (m, 2H, 11 ,17-H), 8.52 (s, 1 H, 19-H), 8.59 (s, 1 H, 3- H), 1 1 .69 (s, 1 H, NH: 20-H); 13C NMR (150 MHz, DMSO-d6): 5C = 21 .0 (C-26), 1 1 1 .0 (C-5), 1 15.3 (C-3), 1 16.8 (C-6), 123.8 (C-11 ), 124.2 (C-19), 126.2 (C-23), 126.3 (C15/16), 126.6 (C-15/16), 127.7 (C-14), 128.3 (C-17), 128.4 (C-12), 129.5 (C-24), 130.0 (C-10), 132.8 (C-13), 133.2 (C-18), 135.6 (C-9), 140.0 (C-22), 142.7 (C-25), 144.5 (C-8), 145.3 (C-2); LRMS m/z (ESI+): 415 [M+H]+, (ESI"): 413 [M-H]", HRMS m/z
(ESI+): Found 415.1219 [M+H]+; C23H19N402S requires 415.1229; Anal. Calcd. for C23H18N402S: C, 66.65; H, 4.38; N, 13.52. Found C, 65.84; H, 4.38; N, 13.10%.
Example 8
4-methyl-W-[2-(2-phenoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide

19 20
Step 1 was carried out as per Step 1 of Example 2 Step 2
8-chloro-2-(2-phenoxyphenyl)imidazo[1 ,2-a]pyrazine (JS18)
2-bromo-1-(2-phenoxyphenyl)ethanone (438 mg, 1.50 mmol), 2-amino-3- chloropyrazine (195 mg, 1.50 mmol), NaHC03 (158 mg, 1.88 mmol) and fBuOH (9 mL) were stirred under reflux for 48 h. The reaction was cooled to RT and solvent removed in vacuo. The sample was taken up in DCM (25 mL) and washed with H20 (3 x 10 mL). The combined aqueous extracts were further washed with DCM (4 x 10 mL), and the organic extracts were combined, dried (MgS04), filtered and solvent removed. Flash chromatography (1 st column: 25:1 to 5:1 DCM/EtOAc gradient; 2nd column: 3:1 Pet Ether /EtOAc) afforded a pale yellow solid. (40.0 mg, 0.124 mmol, 29.0%). Mpt: > 200 °C; Rf = 0.75 (1 :1 Pet Ether/Ethyl Acetate); IR (vmax/cm"1, thin film): 1071 - 1225
cm-1 (Ar-O-Ar Stretch); 1H NMR (600 MHz, CDCI3): δΗ = 6.97 (dd, J = 1 .5, 8.2 Hz, 1 H, 14-H), 7.07 (d, J = 7.6 Hz, 2H, 18-H), 7.16 (t, J = 7.4 Hz, 1 H, 20-H), 7.29 - 7.34 (m, 2H, 12,13-H), 7.38 (dd, J = 8.6, 7.5 Hz, 2H, 19-H), 7.65 (d, J = 4.5 Hz, 1 H, 6-H), 7.99 (d, J = 4.5 Hz, 1 H, 5-H), 8.31 (s, 1 H, 3-H), 8.59 (dd, J = 7.6, 1 .9 Hz, 1 H, 11 -H); 13C NMR (150 MHz, CDCI3): 5c = 1 15.3 (C-3), 1 18.5 (C-5), 1 18.8 (C-18), 1 19.0 (C-14), 123.7 (C- 20), 123.8 (C-10), 124.1 (C-12), 127.8 (C-6), 129.7 (C-13), 129.9 (C-19), 130.1 (C-11 ), 137.1 (C-9), 143.2 (C-2), 143.3 (C-8), 154.6 (C-15), 156.5 (C-17); LRMS m/z (ESI+): 324 [M(37CI)+H]+, 322 [M(35CI)+H]+; HRMS m/z (Cl+): Found 322.075537 [M(35CI)+H]+; Ci8H13CIN30 requires 322.07471 ; Anal. Calcd. for Ci8H12CIN30: C, 67.19; H, 3.76; N, 13.06. Found C, 65.36; H, 3.66; N, 12.62%.
Step 3
4-methyl-A/-[2-(2-phenoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]benzenesulfonamide

19 20
All glassware was evacuated and flushed with argon prior to use. 8-chloro-2-(2- phenoxyphenyl)imidazo[1 ,2-a]pyrazine (130 mg, 0.405 mmol), 4-toluenesulfonamide (83.1 mg, 0.486 mmol), K2C03 (67.1 mg, 0.127 mmol), Pd(dba)2 (1.30 mg) and 5 mol% ie f-butyl XPhos (6.50 mg) were weighed into a 10 mL round bottom flask. fBuOH (3 mL) was added and the reaction was stirred under reflux for 48 h. The reaction mixture was cooled to RT, diluted with MeOH (100 mL) and filtered through celite (pre-washed with MeOH). Flash chromatography was carried out (100% DCM to 40:1 to 30:1 to 10:1 to 1 :1 DCM/EtOAc gradient) to afford the title compound as a yellow solid (23.9 mg, 0.052 mmol 12.8%). Mpt: >200 °C; Rf = 0.18 (9:1 DCM/EtOAc); IR (vmax/cm"1, thin film): 3245 (N-H Stretch), 1586, 1227 (Ar-O-Ar Stretch), 1 1 15; 1H NMR (600 MHz, CD2CI2): δΗ = 2.43 (s, 3H, 27-H), 6.91 (bs, 1 H, 6-H), 6.96 (d, J = 8.1 Hz, 1 H, 14-H),
7.08 (dd, J = 8.8, 0.7 Hz, 2H, 18-H), 7.17 (t, J = 7.1 Hz, 1 H, 20-H), 7.27 (t, J = 7.3 Hz, 1 H, 12-H), 7.31 - 7.37 (m, 4H, 5,13,25-H), 7.38 - 7.41 (m, 2H, 19-H), 7.91 (bd, J = 5.7 Hz, 2H, 24-H), 8.12 (s, 1 H, 3-H), 8.48 (dd, J = 7.8,1.7 Hz 1 H, 11 -H), 1 1.41 (s, 1 H, 21 - H); 13C NMR (150 MHz, CD2CI2): 5C = 21 .3 (C-27), 1 10.2 (C-5), 1 15.0 (C-6), 1 17.7 (C- 3), 1 18.8 (C-18), 1 18.9 (C-14), 123.7 (C-20), 123.8 (C-10), 123.9 (C-12), 126.2 (C-24), 129,1 (C-11 ), 129.5 (C-15,25), 130.0 (C-19), 134.7 (C-9), 139.4 (C-23), 142.1 (C-2), 143.5 (C-26), 145.5 (C-8), 154.3 (C-15), 156.5 (C-17); LRMS m/z (ESI+): 457 [M+H]+, HRMS m/z (ESI"): Found 455.1 167 [M +H]"; C25H19N403S requires 455.1 178; Anal. Calcd. for C25H2oN403S: C, 65.77; H, 4.42; N, 12.27. Found C, 62.62; H, 4.28; N, 1 1 .40%
Example 9
yV-[2-(3,4-dimethoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]-4-methyl- benzenesulfonamide

Step 1 was carried out as per Step 1 of Example 3. Step 2
8-chloro-2-(3,4-dimethoxyphenyl)imidazo[1 ,2-a]pyrazine
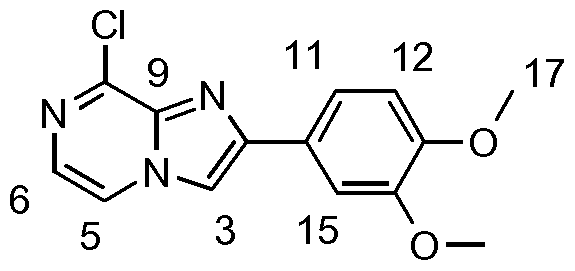
16
A mixture of 2-bromo-1-(3,4-dimethoxyphenyl)ethanone (229 mg, 0.885 mmol), 2- amino-3-chloropyrazine (1 15 mg, 0.885 mmol) and NaHC03 (93.0 mg, 1 .1 1 mmol) in fBuOH (5 mL) were stirred under argon, under reflux for 40 h. After this point, the
reaction mixture was cooled to RT and solvent removed in vacuo. The resulting orange solid was dissolved in DCM (30 mL) and washed with H20 (3 x 30 mL). The aqueous layers were extracted further with DCM (2 x 20 mL) before the combined organics were washed with brine (20 mL), dried (MgS04), filtered and solvent removed to give a light brown solid. Flash chromatography (3:1 Pet Ether /EtOAc) was carried out to afford the title compound (155 mg, 0.536 mmol, 60.6%). Mpt: Decomposed before melting; Rf = 0.1 1 (1 :1 Pet Ether/EtOAc); IR (vmax/cm"1, thin film): 3142 (w), 2933 (w), 2833 (w), 1498 (s); 1 H NMR (600 MHz, CDCI3): δΗ = 3.93 (s, 3H, 16-H), 4.00 (s, 3H, 17-H), 6.93 (d, J = 8.3 Hz, 1 H, 12-H), 7.48 (dd, J = 8.2, 2.1 Hz, 1 H, 11 -H), 7.59 (d, J = 2.1 Hz, 1 H, 15-H), 7.65 (d, J = 4.5, 1 H, 6-H), 7.96 (s, 1 H, 3-H), 8.02 (d, J = 4.5 Hz, 1 H, 5-H); 13C NMR (150 MHz, CDCI3): 5C = 56.0 (C-16), 56.1 (C-17), 109.6 (C-15), 1 10.6 (C-3), 1 1 1.2 (C-12). 1 18.3 (C-5), 1 19.3 (C-11 ), 125.2 (C-10), 128.0 (C-6), 138.0 (C-9), 143.1 (C-8), 148.3 (C-2), 149.3 (C-13). 149.9 (C-14); LRMS m/z (ESI+): 292 [M37CI+H]+, 290 [M(35CI)+H]+; HRMS m/z (ESI+): Found 322.075537 [M(35CI)+H]+; Ci4H13CIN302 requires 290.0696. Anal. Calcd for Ci4H12CIN302: C, 58.04; H, 4.17; N, 14.50. Found C, 56.12; H, 4.105; N, 13.68%
Step 3
yV-[2-(3,4-dimethoxyphenyl)imidazo[1 ,2-a]pyrazin-8-yl]-4-methyl- benzenesulfonamide

All glassware was evacuated and flushed with argon prior to use. 8-chloro-2-(3,4- dimethoxylphenyl)imidazo[1 ,2-a]pyrazine (200 mg, 0.691 mmol), 4-toluenesulfonamide (142 mg, 0.829 mmol), K2C03 (1 15 mg, 0.829 mmol), 1 mol% Pd(dba)2 (2.00 mg) and 5 mol% ie f-butyl XPhos (10.0 mg) were weighed into a 10 mL round bottom flask. fBuOH (4 mL) was added and the reaction was stirred under reflux for 48 h. The reaction mixture was cooled to RT, diluted with MeOH (100 mL) and filtered through
celite (pre-washed with MeOH). Flash chromatography was carried out (100% DCM to 50:1 to 1:9 DCM/EtOAc gradient followed by DCM/10% MeOH) gave the title compound as a yellow sticky sold (5.2 mg, 0.012 mmol, 1.8%). Mpt: >200 °C; Rf = 0.26 (1:1 DCM/EtOAc); IR (vmax/cm"1, thin film): 3274, 3138, 1584; 1H NMR (600 MHz, DMSO-d6): δΗ = 2.36 (s, 3H, 24-H), 3.78 (s, 3H, 16-H), 3.83 (s, 3H, 17-H), 7.02 (d, J = 8.2 Hz, 1H, 12-H), 7.13 (bd, J = 4.9 Hz, 1H, 6-H), 7.37 (d, J = 8.2 Hz, 1H, 22-H), 7.45 (bs, 1H, 11-H), 7.47 (bs, 1H, 15-H), 7.81 (d, J = 5.5 Hz, 1H, 5-H), 7.87 (d, J = 8.0 Hz, 2H, 21-H), 8.41 (s, 1H, 3-H), 11.63 (s, 1H, 18-H); 13C NMR (150 MHz, DMSO-d6): 5C = 21.0 (C-24), 55.5 (C-16), 55.6 (C-17), 108.9 (C-15), 111.0 (C-5), 111.9 (C-12), 114.3 (C-3), 116.6 (C-6) 118.2 (C-11), 125.3 (C-10), 126.1 (C-21), 129.5 (C-22), 135.0 (C-9), 139.8 (C-20), 142.7 (C-23), 144.4 (C-8), 145.6 (C-2), 149.0 (C-14), 149.1 (C-13); LRMS m/z; HRMS m/z (ESI+): Found 423.1115 [M+H]+; C21H21N4O4S requires 423.1127; Anal. Calcd. for C2iH2oN404S: C, 59.42; H, 4.75; N, 13.20. Found C, 58.32; H, 4.73; N, 12.52%
Example 10
yV-[2-(3,5-dimethylphenyl)imidazo[1,2-a]pyrazin-8-yl]-4-methyl benzenesulfonamide

Step 1 was carried out as per Step 1 of Example 4
Step 2
8-chloro-2-(3,5-dimethylphenyl)imidazo[1 ,2-a]pyrazine

A mixture of 2-bromo-1-(3,5-dimethylphenyl)ethanone (271 mg, 1 .19 mmol), 2-amino- 3-chloropyrazine (155 mg, 1 .19 mmol) and NaHC03 (125 mg, 1.49 mmol) in fBuOH (5 mL) were stirred under argon, under reflux for 40 h. After this point, the reaction mixture was cooled to RT and solvent removed in vacuo. The resulting orange solid was dissolved in DCM (30 mL) and washed with H20 (3 x 30 mL). The aqueous layers were extracted further with DCM (2 x 20 mL) before the combined organics were washed with brine (20 mL), dried (MgS04), filtered and solvent removed to give crude orange solid. Flash chromatography (100% DCM to 19:1 DCM/EtOAc) was carried out to afford the title compound (103 mg, 0.400 mmol, 33.6%). Mpt: 158-162 °C; Rf = 0.68 (2:1 DCM/EtOAc); IR (vmax/cm"1, thin film): 2920 (C-H), 1365; 1 H NMR (500 MHz, CDCIs): δΗ = 2.38 (s, 6H, 14-H), 7.05 (s, 1 H, 13-H), 7.62 (s, 2H, 11 -H), 7.66 (d, J = 4.5 Hz, 1 H, 6-H), 7.99 (s, 1 H, 3-H). 8.02 (d, J = 4.5 Hz, 1 H, 5-H); 13C NMR (125 MHz, CDCI3): 6c = 21 .3 (C-14), 1 1 1.3 (C-3), 1 18.4 (C-5), 124.5 (C-11 ), 128.0 (C-6), 131.1 (C- 13), 132.1 (C-10), 138.2 (C-9), 138.6 (C-12), 143.4 (C-8). 148.6 (C-2); LRMS m/z; HRMS m/z (El+): Found 257.07184 [M(35CI)]+; Ci4H12CIN3 requires 257.07142; Anal. Calcd. for d4H12CIN3: C, 65.25; H, 4.69; N, 16.30. Found C, 64.02; H, 4.63; N, 15.76%
Step 3
yV-[2-(3,5-dimethylphenyl)imidazo[1 ,2-a]pyrazin-8-yl]-4-methyl
benzenesulfonamide

A mixture of 8-chloro-2-(3,5-dimethylphenyl)imidazo[1 ,2-a]pyrazine (100 mg, 0.388 mmol), 4-toluenesulfonamide (79.8 mg, 0.466 mmol), 2 mol% Pd(dppf)CI2 (6.30 mg) and Cs2C03 (152 mg, 0.466 mmol) in anhydrous toluene (3 mL) were stirred under reflux, under argon. After 21 h, the reaction was cooled to RT, diluted with toluene and washed with H20 and brine. Further extraction of the aqueous layers was carried out using DCM. The combined organics were dried (MgS04), filtered and solvent removed in vacuo. Flash chromatography (19:1 DCM/EtOAc) afforded the title compound as a yellow solid (43.4 mg, 0.1 1 1 mmol, 28.6%). Mpt: >200 °C; Rf = 0.37 (2:1 DCM/EtOAc); IR (vmax/cm"1, thin film): 2928 (C-H Stretch), 1595; 1 H NMR (600 MHz, DMSO-d6): δΗ = 2.31 (s, 6H, 14-H), 2.36 (s, 3H, 21 -H), 6.97 (s, 1 H, 13-H), 7.13 (t, J = 5.6 Hz, 1 H, 6-H), 7.38 (d, J = 8.1 Hz, 2H, 19-H), 7.54 (s, 2H, 11 -H), 7.81 (d, J = 5.3 Hz, 1 H, 5-H), 7.87 (d, J = 8.0 Hz, 2H, 18-H), 8.42 (s, 1 H, 3-H), 1 1.63 (d, J = 5.2 Hz, 1 H, 15-H); 13C NMR (150 MHz, DMSO-d6): 5C = 21.0 (C-14,21 ), 1 1 1 .0 (C-5), 1 14.8 (C-3), 1 16.7 (C-6), 123.3 (C- 11 ), 126.1 (C-18), 129.5 (C-19), 129.8 (C-13), 132.4 (C-10), 135.2 (C-9), 137.9 (C-12), 139.9 (C-17), 142.6 (C-20), 144.5 (C-8), 145.5 (C-2); LRMS m/z (ESI+): 393 [M+H]+, HRMS m/z (ESI"): Found 391.1236 [M-H]"; C2i H19N402 requires 391 .1229; Anal. Calcd. for C2i H20N4O2: C, 64.27; H, 5.14; N, 14.27. Found: C, 63.08; H, 5.18; N, 13.52% With XPhos:
All glassware was evacuated and flushed with argon prior to use. 8-chloro-2-(3,5- dimethylphenyl)imidazo[1 ,2-a]pyrazine (21.5 mg, 0.0835 mmol), 4-toluenesulfonamide (17.2 mg, 0.100 mmol), K2C03 (13.8 mg, 0.100 mmol), 1 mol% Pd(dba)2 (0.2 mg) and 5 mol% ie f-butyl XPhos (1.1 mg) were weighed into a 10 mL round bottom flask. fBuOH (1 mL) was added and the reaction was stirred under reflux for 48 h. The reaction mixture was cooled to RT, diluted with MeOH (100 mL) and filtered through celite (pre-washed with MeOH). Flash chromatography was carried out (100% DCM to 30:1 DCM/EtOAc gradient), but contamination of the pure product was evident by NMR with not enough sample to re-purify.
Example 11
4-methyl-yV-[4-[2-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8- yl]aminophenyl]benzenesulfonamide
31

17 16 Step 1
8-chloro-2-(2-naphthyl)imidazo[1 ,2-a]pyrazine

2-(Bromoacetyl)naphthalene (3.14 g, 12.6 mmol), 2-amino-3-chloropyrazine (1 .63 g, 12.6 mmol), NaHC03 (1 .32 g, 16.7 mmol) and fBuOH (60 mL) were stirred under reflux for 40 h. The reaction mixture was cooled to RT and solvent removed in vacuo. The resulting orange solid was taken up in H20 (60 mL) and extracted with DCM (3x 60 mL). The combined organics were washed with H20 (50 mL) and brine (40 mL), dried (MgS04), filtered and concentrated in vacuo to give crude orange solid. On addition of DCM, insoluble material filtered off to give title compound as a cream fluffy solid (1.05 g). Purification of the remaining filtrate via flash chromatography (10:1 to 2:1 Pet Ether/EtOAc gradient) afforded the title compound as a pale orange/brown solid (338 g, Total: 1 .38 g, 4.95 mmol, 39.3%). Mpt: Decomposed before melting; Rf = 0.34 (1 :1 Pet Ether/ethyl acetate); IR (vmax/cm"1, thin film): 3026.3 (w), 2921 .0 (w), 1495.3 (m); 1H NMR (600 MHz, CDCI3): δΗ = 7.51-7.53 (m, 2H, 15,16-H), 7.71 (d, J = 4.5 Hz, 1 H, 6- H), 7.88 (dd, J = 6.7, 2.2 Hz, 1 H, 14-H), 7.93 (d, J = 8.6, 1 H, 12-H) 7.96 (dd, J = 6.7, 2.4 1 H, 17-H), 8.06 (dd, J = 8.6, 1.7 Hz, 1 H, 11 -H) 8.08 (d, J = 4.5 Hz, H, 5-H), 8.14 (s,
1 H, 3-H), 8.57 (s, 1 H, 19-H); ldC NMR (150 MHz, CDCI3): 5C = 1 1 1 .6 (C-3), 1 18.5 (C- 5), 124.1 (C-11 ), 126.0 (C-19), 126.6/126.7 (C-15,16), 127.8 (C-14), 128.2 (C-6), 128.5 (C-17), 128.6 (C-12), 129.4 (C-10), 133.5 (C-18), 133.7 (C-13), 138.3 (C-9), 143.5 (C- 8), 148.2 (C-2); LRMS m/z (ESI+): 282 [M(37CI)+H]+, 280 [M(35CI)+H]+; HRMS m/z (Cl+): Found 280.06338 [M(35CI)+H]+; Ci6Hn N3CI requires 280.06415; Anal. Calcd for Ci6H10N3CI: C, 68.70; H, 3.60; N, 15.02. Found C, 67.53; H, 3.64; N, 13.97%.
Step 2
4-methyl-yV-[4-[2-(2-naphthyl)imidazo[1 ,2-a]pyrazin-8- yl]aminophenyl]benzenesulfonamide

17 16
All glassware was evacuated and flushed with argon prior to use. 8-chloro-2-(2- naphthyl)imidazo[1 ,2-a]pyrazine (50.0 mg, 0.1 78 mmol), /V-(4-aminophenyl)-4- methylbenzenesulfonamide (56.3 mg, 0.215 mmol), K2C03 (29.7 mg, 0.215 mmol), 1 mol% Pd(dba)2 (1 .03 mg) and 5 mol% ie f-butyl XPhos (3.80 mg) were weighed into a 25 mL round bottom flask. fBuOH (3 mL) was added and the reaction was stirred under reflux for 24 h. The reaction mixture was cooled to RT, diluted with MeOH (100 mL) and filtered through celite (pre-washed with MeOH). LCMS indicated the correct product mass was present, and flash chromatography (numerous attempts: 2:1 Pet Ether/ EtOAc, 20:1 DCM/ EtOAc, 10:1 DCM/ EtOAc, 5:1 DCM/Et20) followed by reverse phase preparative HPLC (0 min: 10% IPA, 90% H20; 30 min: 90% IPA, 10% H20; 36 min: 90% IPA, 10% H20; 39 min: 10% IPA, 90% H20) to give the title compound (12.4 mg, 0.025 mmol, 14.0%). Mpt: decomposed before melting; Rf = 0.50
(1 :1 Pet Ether/EtOAc); IR (vmax/cm"1, thin film): 1 143 (S=0 symmetric stretch); 1H NMR (600 MHz, CD3OD): δΗ = 2.42 (s, 3H, 31 -H), 7.20 (d, J = 5.2 Hz, 1 H, 6-H), 7.29 (d, J = 6.8 Hz, 2H, 22-H), 7.36 (d, J = 8.0 Hz, 2H, 29-H), 7.52 - 7.56 (m, 2H, 15, 16-H), 7.58 (d, J = 8.8 Hz, 2H, 23-H), 7.75 (d, J = 8.3 Hz, 2H, 28-H), 7.92 (d, J = 7.1 , 1 H, 17-H), 7.96 - 7.99 (m, 3H, 5-H, 12-H, 14-H), 8.13 (dd, J = 1.7, 8.5 Hz, 1 H, 11 -H), 8.53 (s, 1 H, 3-H), 8.56 (s, 1 H, 19-H); 13C NMR (150 MHz, CD3OD): 5C = 21 .4 (C-31 ), 1 13.9 (C-5), 1 15.9 (C-3), 1 15.9 (C-6) 122.9 (C-22), 124.9 (C-11 ), 125.8 (C-23), 126.1 (C-19), 127.6/127.7 (C-15,16), 128.4 (C-28), 128.9 (C-17), 129.3 (C-14), 129.7 (C-12), 130.7 (C-29), 131.0 (C-10), 133.5 (C-9, 24), 135.0 (C-13,18), 137.7 (C-21 ) 138.2 (C-27), 145.2 (C-30), 146.1 (C-8), 148.1 (C-2); LRMS m/z (ESI+): 506 [M+H]+, (ESI"): 504 [M- H]" ; HRMS m/z (ESI+): Found 506.1651 [M+H]+; Czs^NsOzS requires 506.1651 ; Anal. Calcd. For C29H23N502S: C, 68.89; H, 4.59; N, 13.85. Found C, 68.59; H, 4.94; N, 13.25.
Example 12
4-methyl-A/-[4-[2-(3-thienyl)imidazo[1 ,2-a]pyrazin-8- yl]aminophenyl]benzenesulfonamide

5 3 14
2-bromo-1 -(3-thienyl)-1 -ethanone
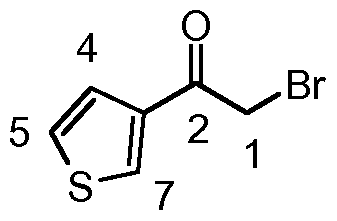
1-(3-thienyl)ethanone (2.00 g, 15.9 mmol) was dissolved in chloroform (100 mL) and ethanol (100 mL). Pyridinium tribromide (10.1 g, 31 .7 mmol) was added and the reaction was stirred at 50°C for 18 h. The reaction mixture was cooled to RT and the solvents removed in vacuo. The resulting orange slurry was suspended in H20 (40 mL) and extracted with EtOAc (4 x 50 mL). The combined organic extracts were washed with H20 (2 x 30 mL) and brine (1 x 30 mL), dried (MgS04), filtered and concentrated in vacuo to give an amber liquid. Flash chromatography was carried out (3:1 Pet Ether/DCM) to afford the title compound as a white solid (1.61 g, 7.81 mmol, 49.1 %). Spectroscopic data was consistent with that previously reported. Mpt: 59-60 °C [Lit. 61.6 °C]; Rf = 0.48 (1 :1 Pet Ether/DCM); IR (vmax/cm"1, thin film): 3094.5 (C-H stretch), 1688.5 (C=0 stretch), 1508, 1415, 1400, 1393, 1380 (Aromatic C=C stretch), 1 181 ; 1H NMR (600 MHz,CDCI3): δΗ = 4.34 (s, 2H, 1 -H), 7.36 (dd, J = 2.9, 5.2 Hz, 1 H, 5-H), 7.58 (dd, J = 5.2, 1 .2Hz, 1 H, 4-H), 8.17 (dd, J = 2.9, 1 .3 Hz, 1 H, 7-H); 13C NMR (150 MHz, CDCIs): 5c = 31.7 (C-1 ), 127.0 (C-5), 127.4 (C-4), 133.9 (C-7), 138.9 (C-3), 185.7 (C-2); LRMS m/z (Cl+): 207 [M(81Br)]+, 205 [M(79Br)]+ Step 2
8-chloro-2-(3-thienyl)imidazo[ -a]pyrazine

2-bromo-1-(3-thienyl)-1 -ethanone (293 mg, 1.43 mmol), 2-amino-3-chloropyrazine (185 mg, 1.43 mmol), NaHC03 (150 mg, 1.79 mmol) and fBuOH (6 mL) were stirred under reflux for 41 h. The reaction was cooled to RT and solvent removed in vacuo. The sample was taken up in water (15 mL) and extracted with DCM (2 x 20 mL). The combined organics were washed with H20 (2 x 15 mL) and brine (15 mL) before they were dried (MgS04), filtered and concentrated to give crude brown/orange solid. Flash chromatography (2:1 Pet Ether/Ethyl acetate) afforded a pale yellow solid. (109 mg, 0.464 mmol, 32.4%). Mpt: decomposed before melting; Rf = 0.33 (1 :1 Pet Ether/Ethyl
Acetate); IR (vmax/cm"1, thin film): 1240 - 1480 (Thiophene Stretch); 1H NMR (600 MHz, CDCIs): δΗ = 7.44 (dd, J = 5.0, 3.0 Hz, 1 H, 12-H), 7.58 (dd, J = 5.0, 1.3 Hz, 1 H, 11 -H), 7.69 (d, J = 4.4 Hz, 1 H, 6-H), 7.93 (s, 1 H, 3-H), 7.96 (dd, J = 3.0, 1.3 Hz, 1 H 14-H), 8.04 (d, J = 4.4 Hz, 1 H, 5-H); 13C NMR (150 MHz, CDCI3): 5C = 1 1 1 .1 (C-3), 1 18.4 (C- 5), 123.6 (C-14), 126.0 (C-12), 126.6 (C-11 ), 128.1 (C-6), 134.1 (C-10), 138.0 (C-9), 143.3 (C-8), 144.3 (C-2); LRMS m/z (ESI+) 238 [M(37CI)+H]+, 236 [M(35CI)+H]+; HRMS m/z (ESI+): Found: 236.0056 [M(35CI)+H]+; Ci0H7CIN3S requires 236.0049; Anal. Calcd. for CioH6CIN3S: C, 50.96; H, 2.57; N, 17.83. Found C, 47.14; H, 2.66; N, 15.26%.
Step 3
4-methyl-W-[4-[2-(3-thienyl)imidazo[1 ,2-a]pyrazin
yl]aminophenyl]benzenesulfonamide

3 14
All glassware was evacuated and flushed with argon prior to use. 8-chloro-2-(3- thienyl)imidazo[1 ,2-a]pyrazine (128 mg, 0.540 mmol), /V-(4-aminophenyl)-4- methylbenzenesulfonamide (171 mg, 0.650 mmol), K2C03 (90.1 mg, 0.652 mmol), 1 mol% Pd(dba)2 (3.12 mg) and 5 mol% ie f-butyl XPhos (1 1.5 mg) were weighed into a 25 mL round bottom flask. fBuOH (5 mL) was added and the reaction was stirred under reflux for 40 h. The reaction mixture was cooled to RT, diluted with MeOH (100 mL) and filtered through celite (pre-washed with MeOH). LCMS indicated the correct product mass was present, and flash chromatography (1 :1 Pet Ether/ EtOAc) was carried to give the title compound as a yellow solid (19.2 mg, 0.042 mmol, 7.8%). Mpt: 158-164 °C; Rf = 0.57 (1 :1 DCM/Et20); IR (vmax/crrf1, thin film): 1 140 cm"1 (S=0 symmetric stretch); 1H NMR (600 MHz, CD3OD): δΗ = 2.41 (s, 3H, 26-H), 7.17 (d, J =
4.5 Hz, 1 H, 6-H), 7.29 (d, J = 8.8 Hz, 2H, 17-H), 7.35 (d, J = 8.0 Hz, 2H, 24-H), 7.53 (d, J = 8.8, 2H, 18-H), 7.57 (dd, J = 4.9, 2.9 Hz, 1 H, 12-H), 7.66 (dd, J = 4.9, 1 .1 Hz, 1 H, 11 -H), 7.75 (d, J = 8.4, 2 H, 23-H), 7.96 (d, J = 5.4, 1 H, 5-H), 7.98 (dd, J = 1.1 , 2.8 Hz, 1 H, 14-H), 8.30 (s, 1 H, 3-H); 13C NMR (150 MHz, CD3OD): 5C = 20.0 (C-26), 1 12.6 (C- 5), 1 14.3 (C-3), 1 18.8 (C-6), 121 .4 (C-17), 122.3 (C-14), 122.6, 124.8 (C-18), 125.5 (C- 11 ), 126.5 (C-12), 127.0 (C-23), 129.3, (C-24), 131.4 (C-16), 134.0 (C-9 +C-10), 136.8 (C-19 + C-22), 143.2 (C-2), 143.8 (C-25), 144.4 (C-8); LRMS m/z (ESI+) 462 [M+H]+; HRMS m/z (ESI+): Found 462.1047 [M+H]+; C23H2oN502S2 requires 462.1058; Anal. Calcd. for CzsHigNsOzSz: C, 59.85; H, 4.15; N, 15.17. Found C, 53.48; H, 3.74; N, 12.89%.
Example 13
4-Methyl-/V- (4-(2-(2-phenoxyphenyl)imidazo[1 ,2-a]pyrazin-8- ylamino)phenyl)benzenesulfonamide (HK007)

Steps 1 and 2 were carried out as per Steps 1 and 2 of Example 8 to give compound JS18.
Step 3
4-Methyl-yV-(4-(2-(2-phenoxyphenyl)imidazo[1 ,2-a]pyrazin-8- ylamino)phenyl)benzenesulfonamide (HK007)

Tris(dibenzylideneacetone)dipalladium(0) (~ 1 mg, 1 .1 μηηοΙ), 2-Dicyclohexylphosphino- 2'-(/V,/V-dimethylamino)biphenyl (~ 1 mg, 2.5 μηηοΙ) and Sodium-ie f-butoxide (~ 12.5 mg, 130 μηηοΙ) were added to a flask and dissolved in 5 ml anhydrous toluene. 8- chloro-2-(2-phenoxyphenyl)imidazo[1 ,2-a]pyrazine (JS18, 30 mg, 93.4 μηιοΙ) and N-(4- aminophenyl)-4-methylbenzenesulfonamide (29 mg, 1 12 μmol) were added under argon atmosphere. The reaction was stirred under reflux (121 °C) for 24 hours. The reaction was cooled to room temperature and solvent removed in vacuo. The product was taken up in dichloromethane (2 ml) and filtered. The filtrate was washed with water (3 x 1 ml), the combined aqueous extracts were further washed with dichloromethane (2 x 1 ml). The solvent was removed in vacuo. Flash chromatography (3:1 Petroleum ether / ethyl acetate) was performed. Solvent was removed from the product fraction in vacuo. The product was taken up in dichloromethane and further purified by a second plastic-free flash chromatography in a Pasteur pipette column (dichloromethane to ethyl acetate gradient). Solvent was removed in vacuo (repeated coevaporation with dichloromethane), and the product was dried under high vacuum. 4.8 mg (8.8 μηηοΙ, yield: 9%) of a yellow, sticky solid were obtained as final product. Mpt: decomposed before melting; Rf = 0.14 (3:1 Petroleum ether / ethyl acetate); IR (vmax/cm"1, thin film): 1229 (C-O-C-stretch), 1489-1542 (Ar-ringvibration); 1H NMR (600 MHz, CDCI3), δΗ (ppm): 2.38 (s, 3H, CH3: 32-H), 6.36 (bs, 1 H, NH: 21 -H), 6.96 (dd, J = 6.7, 2.3 Hz, 1 H, CH: 14-H), 7.06 (d, J = 7.5 Hz, 2H, CH: 18-H), 7.07 (d, J = 8.6 Hz, 1 H, CH: 24-H), 7.14 (t, J = 7.2 Hz, 1 H, CH: 20-H), 7.22 (d, J = 8.3 Hz, 2H, CH: 30-H), 7.21-7.31 (m, 2H, CH: 12-H, CH: 13-H), 7.35-7.38 (m, 3H, CH: 6-H, CH: 19-H), 7.48 (d, J = 4.5 Hz, 1 H, CH: 5- H), 7.62 (d, J = 8.2 Hz, 2H, CH: 29-H), 7.79 (d, J = 8.3 Hz, 2H, CH: 23-H), 8.05 (s, 1 H, CH: 3-H), 8.45 (dd, J = 6.2, 2.2 Hz, 1 H, CH: 1 1-H); 13C-NMR (125 MHz, CDCI3), 5C (ppm): 21 .7 (C-32), 1 1 1.9 (C-5), 1 15.0 (C-3), 1 18.8 (C-18), 1 19.4 (C-14), 120.1 (C-23), 123.7 (C-20), 124.1 (C-24), 124.2 (C-12), 124.8 (C-10), 127.4 (C-29), 127.8 (C-6),
129.1 (C-1 1 ), 129.3 (C-13), 129.7 (C-30), 130.1 (C-19), 130.8 (C-25), 132.6 (C-9), 136.1 (C-28), 137.6 (C-22), 140.0 (C-2), 143.9 (C-8), 145.9 (C-31 ), 154.3 (C-15); LRMS m/z (ES+): 548 [M+H]+; HRMS m/z (ES+): Found 548.1733 [M+H]+; C31 H26N5O2S requires 548.1756; analysis calculated for C31 H25N5O2S: C, 67.99; H, 4.60; N, 12.79; Found C, 65.37; H, 4.57; N, 1 1.38%
Example 14
4-methyl-yV-(4-(2-(quinoxalin-2-yl)imidazo[1 ,2-a]pyrazin
ylamino)phenyl)benzenesulfonamide (JS117)

13 14
Step 1
1 -(quinoxalin-2-yl)ethanone (JS110)

Quinoxaline (1 .1864 g, 9.1 1 mmol), Pyruvic acid (1.90 ml, 27.3 mmol), AgN03 (0.124 g, 0.73 mmol), N2H8S208 (3.12 g, 13.67 mmol) and H2S04 (0.49 ml, 9.1 1 mmol) were stirred in 1 :1 CH2CI2/H20 (150 ml) at 40 °C for 2 ½ h. The solution was then basified via the addition of NaOH and the organics extracted (3x) and washed brine, dried (MgS04), filtered and solvent removed. Flash chromatography (Pet Ether; 3:1 Pet Ether/EtOAc) afforded the title compound as a yellow solid (617.5 mg, 3.91 mmol, 42.9%). Mpt: 70-74 °C [Lit.( J. Org. Chem., 1991 , 56, 2866-2869) 76-97 °C]; Rf = 0.24 (3:1 Pet Ether/EtOAc); IR (vmax/cm"1, thin film): 1689 (CO stretch), 1357; 1H NMR (500 MHz, CDCI3): δΗ = 2.84 (s, 3H, 12-H), 7.82-7.90 (m, 2H, 7,8-H), 8.14-8.21 (m, 2H, 6,9-H), 9.47 (s, 1 H, 3-H); 13C NMR (125 MHz, CDCI3): 5C = 25.6 (C-12), 129.5 (C-6), 130.5 (C-8), 130.8 (C-9), 132.3 (C-7), 141 .1 (C-10), 143.1 (C-3), 143.9 (C-5), 146.6 (C-
2), 199.8 (C-11 ); LRMS m/z (El+): 172 [M]+, 130 [M-Ac]+, 86; HRMS m/z (El+): Found 172.06372; Ci0H8N2O requires 172.0631 1 ; Anal. Calcd. for Ci0H8N2O: C, 69.76; H, 4.68; N, 16.27. Found C, 69.28; H, 4.56; N, 16.00%.
Step 2
2-bromo-1 -(quinoxalin-2-yl)ethanone (JS111 )

Pyridinium tribromide (2.935 g, 9.18 mmol) was added to a stirred solution of JS1 10 (580 mg, 3.67 mmol) in 1 :1 CHCI3/EtOH (60 ml) and the mixture was heated at 50 °C for 16 h. Removal of the solvent in vacuo was followed by addition of H20 and extraction with EtOAc (3x). The combined organic extracts were further washed with H20 and brine, dried (MgS04), filtered and concentrated in vacuo. Flash chromatography (CH2CI2 isocractic) afforded the title compound as a brown solid (656 mg, 2.61 mmol, 71 .2%). Mpt: Decomposed before melting [Lit. (Pharmazie 1983, 38(12), 829-32) 1 12-1 14 °C]; Rf = 0.26 (CH2CI2); IR (vmax/crrf1, thin film): 1708 (CO stretch), 1392, 762 (C-Br Stretch); 1 H NMR (600 MHz, CDCI3): δΗ = 4.96 (s, 2H, 12-H), 7.88-7.90 (m, 1 H, 8-H), 7.93-7.96 (m, 1 H, 7-H), 8.20-8.21 (m, 2H, 6,9-H), 9.53 (s, 1 H, 3-H); 13C NMR (150 MHz, CDCI3): 5C = 31.3 (C-12), 129.7 (C-6), 130.6 (C-9), 131 .3 (C- 8), 133.0 (C-7), 141.0 (C-10), 143.4 (C-3), 144.3 (C-5), 144.7 (C-2), 192.4 (C-11 ); LRMS m/z (El+): 252 [M(81Br)]+, 250 [M(79Br)]+, 142, 1 15 [(81Br)], 1 13 [(79Br)]; HRMS m/z (Ε ): Found 249.97396; Ci0H7BrN2O requires 249.97363; Anal. Calcd. for Ci0H7BrN2O: C, 47.84; H, 2.81 ; N, 1 1 .16. Found C, 47.70; H, 2.68; N, 10.86%.
Step 3
2-(8-chloroimidazo[1 ,2-a]pyrazin-2-yl)quinoxaline (JS1

13 14
JS1 1 1 (648 mg, 2.58 mmol), 2-amino-3-chloropyrazine (334.4 mg, 2.58 mmol) and NaHC03 (271.0 mg, 3.23 mmol) in ie f-butanol (15 ml) were stirred under reflux for 40
h. The solvent was removed and the resulting residual was taken up in CH2CI2 and washed H20 and brine, dried (MgS04), filtered and solvent removed. Flash chromatography (Pet Ether; 3:1 to 1 :1 to 1 :3 Pet Ether/EtOAc) afforded the title compound as an orange solid (341 .9 mg, 1 .21 mmol, 47.1 %). Mpt: >200 °C; Rf = 0.1 1 (1 :1 Pet Ether/EtOAc); IR (vmax/cm"1, thin film): 2924, 1675, 1495, 1354, 1200; 1H NMR (600 MHz, DMSO-d6): δΗ = 7.84 (d, J = 4.5 Hz, 1 H, 6-H), 7.89-7.95 (m, 2H, 14,15-H), 8.15-8.17 (m, 2H, 13,16-H), 8.73 (d, J = 4.4 Hz, 1 H, 5-H), 9.08 (s, 1 H, 3-H), 9.71 (s, 1 H, 19-H); 13C NMR (150 MHz, DMSO-d6): 5C = 1 16.9 (C-3), 121 .0 (C-5), 128.3 (C-6). 129.0 (C-13), 129.1 (C-16), 130.5 (C-15), 131.1 (C-14), 137.7 (C-9), 141.4 (C-12), 141.8 (C-17), 142.2 (C-8), 143.4 (C-19), 143.9 (C-10), 146.4 (C-2); LRMS m/z (CI): 282 [M(35CI)+H]+, 284 [M(37CI)+H]+; HRMS m/z (CI): Found 282.05522; Ci4H9CIN5 requires 282.05465; Anal. Calcd. for Ci4H8CIN5: C, 59.69; H, 2.86; N, 24.86. Found C, 59.88; H, 3.25; N, 20.72%. Step 4
4-methyl-yV-(4-(2-(quinoxalin-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)phenyl)benzenesulfonamide (JS117)

13 14
JS1 14 (100 mg, 0.355 mmol), /V-(4-aminophenyl)-4-methylbenzenesulfonamide (1 1 1 .8 mg, 0.426 mmol), Pd(dba)2 (2.0 mg, 1 mol%), DavePhos (4.2 mg, 3 mol%) and Cs2C03 (162.0 mg, 0.497 mmol) were stirred in anhydrous 1 ,4-dioxane under reflux for 40 h. After LCMS confirmed the presence of the product mass, the reaction was cooled to RT and solvent removed. The resulting residue was taken up in CH2CI2 and washed with sat. aq. NaHC03, H20 and brine, dried (MgS04), filtered and solvent removed. Flash chromatography (Toluene; 0% to 50% EtOAc) afforded the title compound as a light yellow solid (12.3 mg, 0.024 mmol, 6.8%). Mpt: Decomposed before melting; Rf = 0.49 (1 :1 CH2CI2/EtOAc); IR (vmax/cm"1, thin film): 3135, 3061 (Aromatic C-H Stretch), 1495, 1291 , 1201 , 936; 1H NMR (600 MHz, DMSO-d6): δΗ = 2.34 (s, 3H, 31 -H), 7.05
(d, J = 8.9 Hz, 2H, 23-H), 7.35 (d, J = 8.2 Hz, 2H, 29-H), 7.44 (d, J = 4.6 Hz, 1 H, 6-H), 7.64 (d, J = 8.3 Hz, 2H, 28-H), 7.84-7.86 (m, 1 H, 15-H), 7.88-7.90 (m, 3H, 14, 22-H), 8.03 (d, J = 4.6 Hz, 1 H, 5-H), 8.10-8.14 (m, 2H, 13,16-H), 8.80 (s, 1 H, 3-H), 9.61 (s, 1 H, 20-H), 9.78 (s, 1 H, 19-H), 10.06 (bs, 1 H, 25-H); 13C NMR (150 MHz, DMSO-d6): 5C = 21 .0 (C-31 ), 1 12.5 (C-5), 1 15.9 (C-3), 121.1 (C-23), 121 .2 (C-22), 126.8 (C-28), 128.2 (C-6), 128.8 (C-13), 129.1 (C-16), 129.7 (C-29), 129.9 (C-15), 130.9 (C-14), 132.1 (C-24), 133.4 (C-9), 136.6 (C-21 ), 136.7 (C-27), 141.2 (C-10), 141.4 (C-17), 141.5 (C-12), 143.1 (C-30), 143.7 (C-19), 146.1 (C-8), 147.0 (C-2) ; LRMS m/z (ES+): 508 [M+H]+, 530 [M+Na]+; HRMS m/z (ES+): Found 508.1565; C27H22N702S requires 508.1556; Anal. Calcd. for C27H21N702S: C, 63.89; H, 4.17; N, 19.32. Found C, 63.82; H, 4.23; N, 18.30%.
Example 15
4-methyl-W-(4-(2-(quinolin-2-yl)imidazo[1 ,2-a]pyrazin
ylamino)phenyl)benzenesulfonamide (HK010)

Step 1
1 -(quinolin-2-yl)ethanone (HK003)

1.177g Quinoline (9.1 1 mol), 150 ml dichloromethane / water (1 :1 ), pyruvic acid (0.64 ml, 9.1 1 mmol), 0.49 ml H2S04 (9.1 1 mmol), 3.12 g N2H8S208 (13.7 mmol) and 0.13 g AgN03 (0.73 mmol) were added to a flask and stirred for 4 hours at 50 °C; the solution turned from yellow to brown. The solution was neutralized with KOH, and the organic phase separated. The aqueous phase was extracted 4x (2x) with dichloromethane; the united organic phases were washed with basic brine (with KOH) and dried with MgS04. The solvent was removed in vacuo, and flash chromatography (Gradient petroleum
ether to petroleum ether / ethyl acetate 10:1 ) was performed. The product was dried under high vacuum after final solvent removal in vacuo; it was liquid until induction of crystallization by movement of the product containing flask. A yellow solid was obtained as a final product (H K003: 0.1 93g, 1 .1 3 mmol , yield: 12%, 1 H-N M R identical to HK001 ). Mpt: 37.5 °C; Rf = 0.40 (10:1 Petroleum ether / ethyl acetate); IR (vmax/cm"1, thin film): 1688 (C=0-stretch); 1H NMR (600 MHz, CDCI3), δΗ (ppm): 2.86 (s, 3H, CH3: 12-H), 7.63 (ddd, J = 8.0, 7.0, 1.0 Hz, 1 H, CH: 7-H), 7.77 (ddd, J = 8.4, 7.0, 1.3 Hz, 1 H, CH: 8-H), 7.85 (d, J = 8.1 Hz, 1 H, CH: 6-H), 8.1 1 (d, J = 8.5 Hz, 1 H, CH: 3-H), 8.19 (d, J = 8.5 Hz, 1 H, CH: 9-H), 8.24 (d, J = 8.5 Hz, 1 H, CH: 4-H); 13C-NMR (125 MHz, CDCI3), 5c (ppm): 25.7 (C-12), 1 18.1 (C-3), 127.8 (C-6), 128.7 (C-7), 129.7 (C-5), 130.1 (C-8), 130.7 (C-9), 137 (C-4), 147.3 (C-10), 153.3 (C-2), 200.8 (C-11); HRMS m/z (ES+): Found 171 .0675 [M(79Br)]+; CnH9NO requires 171.06786; analysis calculated for Cn H9NO: C, 77.17; H, 5.30; N, 8.18. Found C, 76.85; H, 5.24; N, 8.08%. Step 2
2-bromo-1 -(quinolin-2-yl)ethanone (HK005)

Step 3
2-(8-chloroimidazo[1 ,2-a]pyrazin-2-yl)quinoline (HK006)

0.26 g (1 .04 mmol) 2-bromo-1 -(quinolin-2-yl)ethanone (HK005), 0.1 17 g (0.90 mmol)2- amino-3-chloropyrazine, 0.094 g (1.12 mmol) NaHC03 and 5.4 ml ie f-Butanol were added to a flask and stirred under reflux (105 °C) for 48 hours. The sample was taken up in 15 ml chloromethane and washed with 3 x 10 ml water. The aqueous phases were washed with 3 x 7.5 ml dichloromethane. The combined organic phases were dried with MgS04 and filtered. Flash chromatography (Petroleum ether / ethyl acetate 5:1 to 1 :4), solvent evaporation and drying under high vacuum afforded a white solid with grease as an impurity (50 mg, 0.18 mmol, yield 17%). Mpt: decomp. before melting; Rf = 0.19 (1 :1 Dichloromethane / Ethyl acetate); IR (vmax/cm"1 , thin film): 804 (s), 996 (s), 1287 (s), 1600 (Ar-ring-vib.), 2852 ("grease"- CH2-asymmetric stretch), 2922 ("grease"-CH2-symmetric stretch); 1 H NMR (600 MHz, CD2CI2), δΗ (ppm): 7.57 (t, J = 7.4 Hz, 1 H, CH: 16-H), 7.70 (d, J = 4.3 Hz, 1 H, CH: 12-H), 7.75 (dd, J = 8.3, 6.8 Hz, 1 H, CH: 15- H), 7.89 (d, J = 8.5 Hz, 1 H, CH: 17-H), 8.10 (d, J = 8.2 Hz, 1 H, CH: 14-H), 8.17 (d, J = 4.4 Hz, 1 H, CH: 11 -H), 8.33 (d, J = 8.1 Hz, 1 H, CH: 5-H), 8.44 (d, J = 8.8 Hz, 1 H, CH: 6-H), 8.64 (s, 1 H, CH: 3-H); 13C-NMR (125 MHz, CD2CI2), 5C (ppm): 1 15 (C-11), 1 19.4 (C-3), 1 19.6 (C-16), 127.1 (C-12), 128.2 (C-17), 128.6 (C-13), 128.7 (C- 14), 129.6 (C-5), 130.2 (C-15), 137.3 (C-6), 138.5 (C-9), 144 (C-2), 148.2 (C-10), 148.5 (C-18), 152.2 (C-8); HRMS m/z (ES+): Found 280.051 16 [M(35CI)]+; Ci5H9CIN4 requires 280.05103.
Step 4
4-methyl-yV-(4-(2-(quinolin-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)phenyl)benzenesulfonamide (HK010)
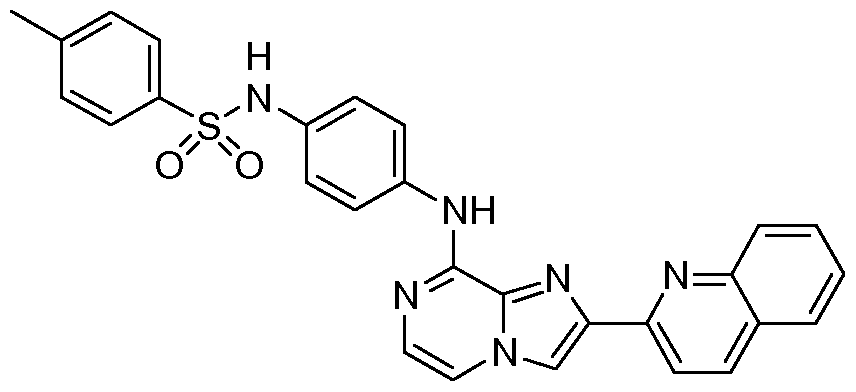
Tris(dibenzylideneacetone)dipalladium(0) (~ 2 mg, 2.2 μmol), 2-dicyclohexylphosphino- 2'-(N,N-dimethylamino)biphenyl (~ 3 mg, 7.6 μηηοΙ) and sodium-tertbutoxide (~ 7 mg, 73 μηηοΙ) were added to a flask and dissolved in 5 ml anhydrous toluene. 2-(8- chloroimidazo[1 ,2-a]pyrazin-2-yl)quinoline (HK006, 15 mg, 53 μηιοΙ) and N-(4- aminophenyl)-4-methylbenzenesulfonamide (15 mg, 57 μηηοΙ) were added under argon atmosphere. The reaction was stirred under reflux (121 °C) for 24 (27) hours. The reaction was cooled to room temperature and solvent removed in vacuo. The product was taken up in 2 ml acetonitrile / water / methanol (2:2:1 ) and < 1 % trifluoroacetic acid. Preparative HPLC was performed: x% H20 + y% MeCN + 0.1 % TFA, x+y « 100; x = 98 (0 min), 65 (4 min), 60 (5 min), 50 (20 min), 2 (21 min), 2 (24 min), 98 (25 min), 98 (29 min). The main fraction was collected in three split subfractions; only the first subfraction was furtherly used; 1 H-NMR, but not LCMS showed that the other subfractions were not appropriate. This HPLC procedure was repeated with this first subfraction: conditions and time points see above, but x = 95, 65, 62, 47, 2, 2, 98, 98. The product was dried under high vacuum after evaporation of the solvent (coevaporation with dichloromethane). 0.8 mg (1 .6 μηηοΙ, yield: 3%) of the solid product were obtained with minor to moderate impurities and characterized by 1 H-NMR and an IC50 assay. Further characterization of the product was impeded by problems to dissolve the compound, occurrence of new relatively significant impurities and the small amount of the sample. Rf = 0.35 (1 :1 Petroleum ether / ethyl acetate); 1 H NMR (600 MHz, d6-DMSO), δΗ (ppm): 0.8 (m, "grease"), 1 .29 (brm, "grease"), 2.34 (s, 3H, CH3: 31 -H), 2.50 (s, solvent peak), 3.32 (s, DHO), 3.35 (s, H20), 7.04 (d, J = 9.42 Hz, «2H), 7.35 (d, J = 8.28 Hz, «2H), 7.42 (d, J = 4.56 Hz, «1 H), 7.61 (d, J = 7.32 Hz, «1 H), 7.64 (t, J = 8.1 Hz, «1 H), 7.68 (m, 5 - 5.5H, impurity?, « toluene?), 7.72 (m, 5 - 5.5H,
impurity?, « toluene?), 7.80 (t, J = 8.0 Hz, «1 H), 7.90 (d, J = 8.82 Hz, «2H), 8.00 (d, J = 7.98 Hz, 1 H), 8.04 (m, 2H), 8.43 (d, J = 8.8 Hz, «1 H), 8.51 (d, J = 8.8 Hz, «1 H), 8.71 (s, «1 H), 9.49 (s, «1 H), 10.04 (s, «1 H); LRMS m/z (both HK008 and HK010, ES+): 530, 529 [M+Na]+, 508, 507 [M+H]+; HRMS m/z (only HK008, ES+): Found 507.1617 [M+H]+ (and an impurity); C28H23N602S requires 507.1603.
Example 16
yV-(4-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)phenyl)methanesulfonamide (JS119)

17 16
Step 1
A/-(4-aminophenyl)methanesulfonamide JS113

Benzene-1 ,4-diamine (3.14 g, 29.0 mmol), was dissolved in anhydrous CH2CI2 (100 ml), triethylamine (1 .62 ml, 1 1.61 mmol) added and the mixtures cooled on ice. MeS02CI (0.449 ml, 5.81 mmol) was added drop wise and the reaction was stirred at RT for 16 h. The mixture was then diluted with CH2CI2 and washed sat. aq. NaHC03. The aqueous layer was then extracted with CH2CI2 (4x) as well as EtOAc (4x). Each of the organic extracts were washed with brine, dried (MgS04), filtered and concentrated in vacuo. Flash chromatography (CH2CI2; 1 % to 2% to 5% MeOH) afforded the title compound as a white solid (843.9 mg, 4.54 mmol, 78.1 %). Mpt: 89 °C [L\t.{Synthetic Commun., 2008, 38: 1909-1916) 1 16-1 17 °C]; Rf = 0.53 (10% MeOH/CH2CI2); IR (vmax/crrf1, thin film): 3459, 3375, 3330 (N-H Stretch), 1625, 1518, 1309, 1 152; 1H NMR
(600 MHz, CD3OD): δΗ = 2.87 (s, 3H, 8-H), 6.71-6.73 (m, 2H, 3-H), 7.02-7.05 (m, 2H, 4-H); 13C NMR (150 MHz, CD3OD): 5C = 36.8 (C-8), 1 15.4 (C-3), 124.4 (C-4), 127.5 (C- 5), 145.8 (C-2); LRMS m/z (CI): 187 [M+H]+, 107; HRMS m/z (CI): Found 187.05387; C7HH N2O2S requires 187.05412; Anal. Calcd. for C7H10N2O2S: C, 45.15; H, 5.41 ; N, 15.04. Found C, 43.53; H, 5.05; N, 13.90%.
Step 2
yV-(4-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)phenyl)methanesulfonamid (JS119)

17 16
All glassware was dried and purged with argon prior to use. Pd2(dba)3 (1 .6 mg, 1 mol%), DavePhos (2.1 mg, 3 mol%) and sodium ie f-butoxide (24.1 mg, 0.250 mmol) were dissolved in anhydrous toluene. JS1 1 (50 mg, 0.179 mmol) and JS1 13 (39.9 mg, 0.215 mmol) were added and the reaction was stirred under reflux, under argon for 20 h. The reaction was cooled to RT and solvent removed, before the residue was taken up in CH2CI2 and washed with NaHC03, H20 and brine, dried (MgS04), filtered and concentrated in vacuo. Flash chromatography (Toluene; 2:1 Toluene/EtOAc) afforded the title compound as an off white solid (5.2 mg, 0.0121 mmol, 6.8%). Rf = 0.32 (1 :1 Toluene/EtOAc); IR (vmax/cm"1, thin film): 3248, 3056, 2926, 2854 (Aromatic C-H and N-H stretch), 1624, 1543, 1508, 1326, 1 152; 1H NMR (600 MHz, CDCI3): δΗ = 3.01 (s, 3H, 27-H), 6.39 (s, 1 H, 25-H), 7.28 (ap.d, J = 8.8 Hz, 2H, 13-H), 7.47 (d, J = 4.6 Hz, 1 H, 6-H), 7.49-7.54 (m, 2H, 15,16-H), 7.60 (d, J = 4.6 Hz, 1 H, 5-H), 7.87 (d J = 7.8 Hz, 1 H, 14-H), 7.92-7.95 (m, 5H, 3,12,17,22-H), 8.02 (dd, J = 8.5, 1.6 Hz, 1 H, 11 -H), 8.18 (s, 1 H, 20-H), 8.48 (s, 1 H, 19-H); 13C NMR (150 MHz, CDCI3): 5C = 39.3 (C-27), 1 1 1 .2 (C-3), 1 1 1.9 (C-5), 120.8 (C-22), 123.3 (C-23), 124.1 (C-11 ), 125.0 (C-19), 126.4 (C- 15), 126.6 (C-16), 127.9 (C-14), 128.2 (C-17), 128.8 (C-12), 130.4 (C-10), 131.1 (C- 24), 133.4 (C-13), 133.7 (C-9,18), 137.7 (C-21 ), 145.1 (C-2), 146.1 (C-8); LRMS m/z
(ES+): 430 [M+H]+; HRMS m/z (ES+): Found 430.1324; C23H2oN502S requires 430.1338;
Example 17
4-methyl-yV-(4-((2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)methyl)p

17 16
Step 1
A/-(4-aminobenzyl)-2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8 -amine (JS169)

17 16
All glassware was dried and purged with argon prior to use. Pd2(dba)3 (1 .6 mg, 1 mol%), DavePhos (2.1 mg, 3 mol%) and sodium ie f-butoxide (24.1 mg, 0.250 mmol) were dissolved in anhydrous toluene. JS1 1 (see Step 1 of Example 7) (50 mg, 0.179 mmol) and 4-(aminomethyl)aniline (24.3 μΙ, 0.215 mmol) were added and the reaction was stirred under reflux, under argon for 20 h. The reaction was cooled to RT and solvent removed, before the residue was taken up in CH2CI2 and washed with NaHC03, H20 and brine, dried (MgS04), filtered and concentrated in vacuo. Flash
chromatography (Pet Ether; 4:1 to 3:1 to 2:1 to 1 :1 to 1 :3 Pet Ether/EtOAc) afforded the title compound as a light yellow oil (20.1 mg, 0.055 mmol, 30.8%). Rf = 0.52 (3:1 EtOAc/Pet Ether); IR (vmax/cm"1, thin film): 3326 (Aromatic C-H stretch), 1619, 1544, 1519; 1 H NMR (600 MHz, CD3OD): δΗ = 4.58 (s, 2H, 21 -H), 6.73 (ap.d, J = 10.9 Hz, 2H, 24-H), 7.19 (ap.d, J = 8.4 Hz, 2H, 23-H), 7.25 (d, J = 4.7 Hz, 1 H, 6-H), 7.44-7.49 (m, 2H, 15,16-H), 7.65 (d, J = 4.7 Hz, 1 H, 5-H), 7.82-7.84 (m, 1 H, 14-H), 7.85-7.88 (m, 2H, 12,17-H), 7.94 (dd, J = 8.5, 1 .6 Hz, 1 H, 11 -H), 8.16 (s, 1 H, 3-H), 8.34 (s, 1 H, 19-H); 13C NMR (150 MHz, CD3OD): 5C = 45.5 (C-21 ), 1 1 1 .5 (C-5), 1 13.1 (C-3), 1 16.7 (C-24), 124.9 (C-11 ), 125.5 (C-19), 127.2 (C-15), 127.5 (C-16), 128.7 (C-14), 129.1 (C-6), 129.2 (C-17), 129.2 (C-22), 129.5 (C-12), 129.9 (C-23), 131 .7 (C-10), 134.6 (C-9,13), 135.0 (C-18), 145.4 (C-2), 148.1 (C-25), 149.8 (C-8); LRMS m/z (ES+): 366 [M+H]+, 273 [M-aniline]+, 261 [M-CH2-aniline]+; HRMS m/z (ES+): Found 366.1716; C23H20N5 requires 366.1719;
Step 2
4-methyl-yV-(4-((2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin
ylamino)methyl)phenyl)benzenesulfonamide (JS173)

JS169 (18 mg, 0.049 mmol) was dissolved in anhydrous pyridine (1 ml) and the mixture was cooled on ice. 4-methylbenzene-1-sulfonyl chloride (1 1 .3 mg, 0.059 mmol) was added and the deep yellow/orange solution was stirred under Ar at RT for 16 h. The solvent was then removed and the crude material was purified via flash chromatography (CH2CI2; 10% to 20% EtOAc) to afford the title compound as an off white solid (8.2 mg, 0.016 mmol, 32.2%).; Rf = 0.44 (20% EtOAc/CH2CI2); IR (vmax/cm"1,
thin film): 3240, 3050 (Aromatic C-H stretches), 2923, 2823 (C-H and N-H stretches), 1544, 1509, 1 136; 1 H NMR (600 MHz, CDCI3): δΗ = 2.47 (s, 3H, 32-H), 4.76 (bs, 2H, 21 -H), 6.49 (bs, 1 H, 20-H), 6.73 (bs, 1 H, 26-H), 7.03 (d, J = 8.5 Hz, 2H, 24-H), 7.22 (d, J = 8.1 Hz, 2H, 30-H), 7.29 (d, J = 8.4 Hz, 2H, 23-H), 7.35 (d, J = 4.5 Hz, 1 H, 6-H), 7.46-7.51 (m, 3H, 5,15,16-H), 7.64 (d, J = 8.3 Hz, 2H, 29-H), 7.83-7.85 (m, 1 H, 14-H), 7.87-7.90 (m, 3H, 3,12,17-H), 7.95 (dd, J = 8.6, 1 .2 Hz, 1 H, 11 -H), 8.40 (s, 1 H, 19-H); 13C NMR (150 MHz, CDCI3): 5C =; LRMS m/z (ES"): 518 [M-H]"; HRMS m/z (ES"): Found 518.1658; C30H24N5O2S requires 518.1651 .
Example 18
4-methyl-yV-(4-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- yloxy)phenyl)benzenesulfonamide (JS175)

17 16
Step 1
8-(methylthio)-2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazine (JS130)
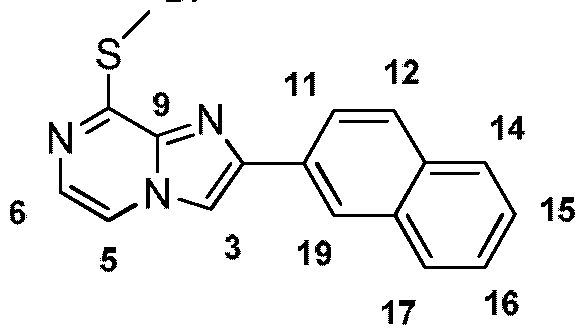
JS1 1 (See Step 1 of Example 7) (1 .12 g, 4.01 mmol) was dissolved in anhydrous DMSO (16 ml). NaSMe (337 mg, 4.81 mmol) was added portion wise and the reaction was stirred at 100 °C for 16 h. The mixture was then cooled to RT, diluted with brine and extracted with CH2CI2. The organic layer was washed with H20 (5x) and brine (1 x), dried (MgS04), filtered and solvent removed in vacuo. Flash chromatography (CH2CI2; 0% to 1 % to 2% EtOAc) afforded the title compound as an off white/yellow solid (989.3 mg, 3.40 mmol, 84.8%). Mpt: 168 °C; Rf = 0.47 (5% EtOAc/CH2CI2); IR (vmax/crrf1, thin
film): 3055 (aromatic C-H stretch), 1470, 1432, 1368; 1H NMR (600 MHz, CDCI3): δΗ = 2.71 (s, 3H, 21 -H), 7.47-7.52 (m, 2H, 15,16-H), 7.72 (d, J = 4.5 Hz, 1 H, 6-H), 7.82 (d, J = 4.5 Hz, 1 H, 5-H), 7.85 (ap.d, J =7.4 Hz, 1 H, 17-H), 7.90 (d, J = 8.5 Hz, 1 H, 12-H), 7.94 (ap.d, J = 7.4 Hz, 1 H, 14-H), 7.98 (s, 1 H, 3-H), 8.04 (dd, J = 8.4, 1 .6 Hz, 1 H, 11 - H), 8.55 (s, 1 H, 19-H); 13C NMR (150 MHz, CDCI3): 5C =12.3 (C-21 ), 1 10.3 (C-3), 1 15.1 (C-5), 124.3 (C-11 ), 125.6 (C-19), 126.4 (C-15,16), 126.5 (C-15,16), 127.9 (C-17), 128.5 (C-12), 128.6 (C-14), 129.0 (C-6), 130.2 (C-10), 133.5 (C-18), 138.9 (C-9), 146.4 (C-2), 154.4 (C-8); LRMS m/z (ES+): 292 [M+H]+; HRMS m/z (ES+): Found 292.0909; Ci7H14N3S requires 292.0908; Anal. Calcd. for Ci7H13N3S: C, 70.08; H, 4.50; N, 14.42. Found C, 69.85; H, 4.28; N, 14.42%.
Step 2
8-(methylsulfonyl)-2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazine (JS132)

17 16
JS130 (1.7662 g, 6.07 mmol) was dissolved in anhydrous CH2CI2 (50 ml) and the mixture was cooled on ice. mCPBA (5.231 g, 30.3 mmol) was added in one portion and the reaction continued to stir at RT for 5 h. The reaction was partitioned with NaHC03 and extracted with CH2CI2 (3x); the combined organic extracts were then washed with brine, dried (MgS04), filtered and concentrated in vacuo. Flash chromatography (CH2CI2; 2% to 5% EtOAc) afforded the title compound as a yellow solid (1 .098 g, 3.398 mmol, 56.0%). Mpt: >200 °C; Rf = 0.22 (10% EtOAc/CH2CI2); IR (vmax/cm-1, thin film): 3121 ,3010 (aromatic C-H stretch), 1312, 1 138; 1H NMR (600 MHz, CDCI3): δΗ = 3.84 (s, 3H, 21 -H), 7.51 -7.54 (m, 2H, 15,16-H), 7.86-7.87 (m,1 H, 14- H), 7.92 (d, J = 8.5 Hz, 1 H, 12-H), 7.95-7.96 (m, 1 H, 17-H), 8.02 (d, J = 4.3 Hz, 1 H, 6- H), 8.08 (dd, J = 8.5, 1 .4 Hz, 1 H, 11 -H), 8.26 (s, 1 H, 3-H), 8.32 (d, J = 4.3 Hz, 1 H, 5-H), 8.59 (s, 1 H, 19-H); 13C NMR (150 MHz, CDCI3): 5C = 41.7 (C-21 ), 1 10.9 (C-3), 122.1 (C-5), 124.3 (C-11 ), 126.7 (C-19), 126.8 (C-16), 127.0 (C-15), 127.9 (C-14), 128.1 (C- 6), 128.7 (C-17), 128.8 (C-12), 129.1 (C-10), 133.5 (C-18), 134.0 (C-13), 136.3 (C-9),
148.6 (C-8), 149.8 (C-2); LRMS m/z (El+): 323 [M]+; HRMS m/z (El+): Found 323.07266; Ci7H13N302S requires 323.07230; Anal. Calcd. for Ci7H13N302S: C, 63.14; H, 4.05; N, 12.99. Found C, 61 .27; H, 3.84; N, 12.48%. Step 3
yV-(4-hydroxyphenyl)-4-methylbenzenesulfonamide (JS168)

Step 4
4-methyl-yV-(4-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- yloxy)phenyl)benzenesulfonamide (JS175)

NaH was pre-activated by stirring NaH (60% in Mineral Oil; 12.4 mg, 0.310 mmol) in anhydrous hexanes (3 ml) for 20 min, removing the solvent using a syringe and drying the contents under high vacuum. DMF (0.5 ml) was added followed by JS168 (81 .4 mg, 0.310 mmol) in DMF (1 ml) and the mixture was stirred at RT for 20 min. JS132 (50 mg, 0.155 mmol) in DMF (1 .5 ml) was added and the resulting deep red solution was heated at 100 °C under Argon for 16 h. The mixture was then diluted with EtOAc and washed with NH4CI (Aq. Sat) and H20 (5x). The combined aqueous layers were then re-extracted with EtOAc (2x), followed by washing the combined organics with brine, drying (MgS04), filtering and concentrating in vacuo. The crude material was purified via flash chromatography (Toluene; 25% EtOAc/Toluene) to give the title compound as an off white-pink solid (36.4 mg, 0.072 mmol, 46.6%). Mpt: 128 °C; Rf = 0.21 (2:1 Toluene/EtOAc); IR (vmax/cm"1, thin film): 3568, 3049 (Aromatic C-H stretch), 1488, 1330, 1 153; 1H NMR (600 MHz, DMSO-d6): δΗ = 2.39 (s, 3H, 31 -H), 7.16 (ap.d, J = 8.9 Hz, 2H, 23-H), 7.22 (ap.d, J = 8.9 Hz, 2H, 22-H), 7.33 (d, J = 4.5 Hz, 1 H, 6-H), 7.39 (d, J = 8.1 Hz, 2H, 29-H), 7.52-7.56 (m, 2H, 15,16-H), 7.70 (d, J = 8.3 Hz, 2H, 28- H), 7.94 (d, J = 7.6 Hz, 1 H, 17-H), 8.01-8.04 (m, 2H, 12,14-H), 8.15 (dd, J = 8.5, 1 .6 Hz, 1 H, 11 -H), 8.32 (d, J = 4.6 Hz, 1 H, 5-H), 8.61 (s, 1 H, 19-H), 8.74 (s, 1 H, 3-H), 10.34 (s, 1 H, 25-H); 13C NMR (150 MHz, DMSO-d6): 5C = 21 .0 (C-31 ), 1 12.9 (C-3), 1 16.9 (C-5), 121.3 (C-23), 122.7 (C-22), 124.0 (C-11 ), 124.5 (C-19), 125.5 (C-6), 126.4 (C-16), 126.6 (C-15), 126.8 (C-28), 127.7 (C-17), 128.3 (C-14), 128.5 (C-12), 129.9 (C- 29), 130.4 (C-10), 132.8 (C-9), 132.9 (C-13), 133.2 (C-18), 135.0 (C-24), 136.7 (C-27), 143.4 (C-30), 145.1 (C-2), 148.7 (C-21 ), 153.1 (C-8); LRMS m/z (ES"): 505 [M-H]-; HRMS m/z (ES"): Found 505.1323; C29H23N403S requires 505.1334; Anal. Calcd. for C29H22N403S: C, 68.76; H, 4.38; N, 4.06. Found C, 65.16; H, 4.38; N, 10.17%.
Example 19
4-methyl-W-(2-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)ethyl)benzenesulfonamide (JS209)

Step 1
A/-(2-aminoethyl)-4-methylbenzenesulfonamide (8TP27)

1 ,2-Diaminoethane (2.0 g, 33 mmol) and triethylamine (6.7 g, 66 mmol) were dissolved in a 2/1 solvent mixture of CH2CI2/THF (50 mL). The resulting solution was stirred at room temperature and 4-methylbenzenesulfonyl chloride (3.2 g, 17 mmol) was added portion-wise over 2 h. The reaction was quenched by addition of 1 M HCI (50 mL). The biphasic mixture was extracted with 1 M HCI (20 mL x 2). The combined aqueous layers were basified with 2 M NaOH (50 mL) and extracted with CH2CI2 (50 mL x 3). The organic layers were combined, dried (MgS04), filtered and concentrated in vacuo to give a colourless solid (2.1 g, 58% yield) suitably pure for subsequent synthetic steps. Rf = 0.22 (15% MeOH in CH2CI2); 1H NMR (600 MHz, CD3OD): δΗ = 2.42 (s, 3H, CH3), 2.65 (t, J = 6.2 Hz, 2H, CH2NH2), 2.89 (t, J = 6.2 Hz, 2H, NHCH2), 7.38 (d, J = 8.2 Hz, 2H, 3-H and 5-H), 7.73 (d, J = 8.2 Hz, 2H, 2-H and 6-H); 13C NMR (150 MHz, CD3OD): 5c = 21.4 (CH3), 42.2 (CH2NH2), 46.4 (NHCH2), 128.0 (C-2 and C-6), 130.8 (C-3 and C-5), 138.8 (C-1 ), 144.7 (C-4);
Step 2
4-methyl-yV-(2-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)ethyl)benzenesulfonamide (JS209)

4-methyl-yV-(3-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)propyl)benzenesulfonamide (JS210)

Step 1
yV-(3-aminopropyl)-4-methylbenzenesulfonamide (8TP28)

1 ,3-Diaminopropane (2.0 g, 27 mmol) and triethylamine (5.5 g, 54 mmol) were dissolved in a 2/1 solvent mixture of CH2CI2/THF (50 mL). The resulting solution was stirred at room temperature and 4-methylbenzenesulfonyl chloride (2.5 g, 13 mmol) was added portion-wise over 2 h. The reaction was quenched by addition of 1 M HCI (50 mL). The biphasic mixture was extracted with 1 M HCI (20 mL x 2). The combined aqueous layers were basified with 2 M NaOH (50 mL) and extracted with CH2CI2 (50 mL x 3). The organic layers were combined, dried (MgS04), filtered and concentrated in vacuo to give a colourless solid (1 .45 g, 49% yield) suitably pure for subsequent synthetic steps. Rf = 0.25 (15% MeOH in CH2CI2); 1H NMR (600 MHz, CD3OD): δΗ = 1.58 (app. qt, J = 6.9 Hz, 2H, NHCH2CH2), 2.43 (s, 3H, CH3), 2.62 (t, J = 6.2 Hz, 2H, CH2NH2), 2.87 (t, J = 6.2 Hz, 2H, NHCH2), 7.38 (d, J = 8.2 Hz, 2H, 3-H and 5-H), 7.72 (d, J = 8.2 Hz, 2H, 2-H and 6-H); 13C NMR (150 MHz, CD3OD): 5C = 21.4 (CH3), 42.2
(CH2NH2 46.4 (NHCH2), 128.0 (C-2 and C-6), 130.8 (C-3 and C-5), 138.8 (C-1 ), 144.7 (C-4);
Step 2
4-methyl-W-(3-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- ylamino)propyl)benzenesulfonamide (JS210)

Example 21
2-(prop-2-ynyloxy)ethyl-4-(yV-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- yl)sulfamoyl)phenylcarbamate (JS208)

2-(prop-2-ynyloxy)ethanol (JS206)
 Ethylene glycol (5 ml, 0.089 mol) was cooled on ice and NaH (60% in mineral oil; 0.894 g, 0.022 mol) was added slowly to form a viscous white paste. Propargyl bromide (80% solution in toluene; 2.49 ml, 0.022 mol) was added drop wise and the reaction mixture was stirred at 45 °C for 3h. The reaction was carefully quenched with H20 and extracted with CHCI3 (3x) and CH2CI2 (5x). The combined organic extracts were dried (MgS04), filtered and concentrated in vacuo. Flash chromatography (Pet Ether; 10% to 50% EtOAc/Pet Ether) afforded the title compound as a light yellow oil (1.0316 g, 0.0103 mmol, 46.9%). Rf = 0.39 (1 :1 Pet Ether/EtOAc); IR (vmax/cm"1, thin film): 3390 (O-H stretch), 3286, 2936, 2668 (C-H stretch), 1354, 1 105, 1065, 1027; 1 H NMR (600 MHz, CDCIs): δΗ = 2.06 (bs, 1 H, 1 -H), 2.46 (J = 2.4 Hz, 1 H, 7-H), 2.65 (ap.t, J = 4.7 Hz, 2H, 3-H), 3.77 (ap.t, J = 4.3 Hz, 2H, 2-H), 4.20 (d, J = 2.3 Hz, 2H, 5-H); 13C NMR (150
MHz, CDCI3): 5c = 58.5 (C-5), 61 .8 (C-2), 71 .3 (C-3), 74.9 (C-7), 79.6 (C-6); LRMS m/z (Cl+): 101 [M+H]+; HRMS m/z (Cl+): Found 101 .06094; C5H902 requires 101 .06025;
Ethylene glycol (5 ml, 0.089 mol) was cooled on ice and NaH (60% in mineral oil; 0.894 g, 0.022 mol) was added slowly to form a viscous white paste. Propargyl bromide (80% solution in toluene; 2.49 ml, 0.022 mol) was added drop wise and the reaction mixture was stirred at 45 °C for 3h. The reaction was carefully quenched with H20 and extracted with CHCI3 (3x) and CH2CI2 (5x). The combined organic extracts were dried (MgS04), filtered and concentrated in vacuo. Flash chromatography (Pet Ether; 10% to 50% EtOAc/Pet Ether) afforded the title compound as a light yellow oil (1.0316 g, 0.0103 mmol, 46.9%). Rf = 0.39 (1 :1 Pet Ether/EtOAc); IR (vmax/cm"1, thin film): 3390 (O-H stretch), 3286, 2936, 2668 (C-H stretch), 1354, 1 105, 1065, 1027; 1 H NMR (600 MHz, CDCIs): δΗ = 2.06 (bs, 1 H, 1 -H), 2.46 (J = 2.4 Hz, 1 H, 7-H), 2.65 (ap.t, J = 4.7 Hz, 2H, 3-H), 3.77 (ap.t, J = 4.3 Hz, 2H, 2-H), 4.20 (d, J = 2.3 Hz, 2H, 5-H); 13C NMR (150
MHz, CDCI3): 5c = 58.5 (C-5), 61 .8 (C-2), 71 .3 (C-3), 74.9 (C-7), 79.6 (C-6); LRMS m/z (Cl+): 101 [M+H]+; HRMS m/z (Cl+): Found 101 .06094; C5H902 requires 101 .06025;
Step 2
2-(prop-2-ynyloxy)ethyl 4-sulfamoylphenylcarbamate (JS207)

JS206 (556.5 mg, 5.57 mmol) was dissolved in anhydrous toluene (140 ml). JS129 (see Example 23) (1 .509 g, 6.68 mmol) and molecular sieves (4A, 10 sieves) were added and the reaction was stirred under reflux for 16 h. The solvent was removed and the reaction purified via flash chromatography (Toluene; 2:1 Toluene/EtOAc) to afford the title compound as a white solid (663 mg, 2.22 mmol, 39.9%). Mpt: 100-102 °C; Rf = 0.17 (2:1 Toluene/EtOAc); IR (vmax/cm"1, thin film): 3350, 3273, 1704 (C=0 Stretch), 1595, 1533, 1314, 1234, 1 152; 1H NMR (600 MHz, DMSO-d6): δΗ = 3.48 (t, J = 2.3 Hz, 1 H, 15-H), 3.69-3.70 (m, 2H, 11 -H), 4.20 (d, J = 2.3 Hz, 2H, 13-H), 4.24-4.26 (m, 2H, 10-H), 7.23 (s, 2H, 1 -H), 7.61 (ap.d, J = 8.8 Hz, 2H, 5-H), 7.72 (ap.d, J = 8.8 Hz, 2H, 4-H), 10.18 (s, 1 H, 7-H); 13C NMR (150 MHz, DMSO-d6): 5C =57.5 (C-13), 63.6 (C-10), 67.3 (C-11 ), 77.5 (C-15), 80.1 (C-14), 1 17.6 (C-5), 126.8 (C-4), 137.6 (C-3), 142.2 (C-6), 1533.3 (C-8); LRMS m/z (ΕΓ): 298 [M]+, 243 [M-OCH2C≡CH]+, 216 [M- CH2CH2OCH2C≡CH]+, 183 [M-C(0)OCH2CH2OCH2C≡CH]+; HRMS m/z (ΕΓ): Found 298.06247; C12H14N205S requires 298.06179; Anal. Calcd. for C12H14N205S: C, 48.31 ; H, 4.73; N, 9.39. Found C, 48.60; H, 4.66; N, 9.12%.
Step 3
2-(prop-2-ynyloxy)ethyl-4-(yV-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- yl)sulfamoyl)phenylcarbamate (JS208)

NaH was pre-activated by stirring NaH (60% in Mineral Oil; 88.7 mg, 2.215 mmol) in anhydrous hexanes (25 ml) for 20 min, removing the solvent using a syringe and drying the contents under high vacuum. DMF (6 ml) was added followed by JS207 (660 mg, 2.215 mmol) in DMF (8 ml) and the mixture was stirred at RT for 20 min. JS132 (357.7 mg, 1 .107 mmol) in DMF (20 ml) was added and the resulting dark brown solution was heated at 100 °C under Argon for 18 h. The solvent was removed and the resulting residue was then taken up in EtOAc and washed with NH4CI (Aq. Sat) and H20 (5x). The combined aqueous layers were then re-extracted with EtOAc (2x), followed by washing the combined organics with brine, drying (MgS04), filtering and concentrating in vacuo. Flash chromatography (Pet Ether; 1 :1 to 1 :2 to 1 :4 to 1 :9 Pet Ether/EtOAc) afforded the title compound as a yellow/orange solid (380.3 mg, 0.703 mmol, 63.5%). Mpt: 184-186 °C; Rf = 0.32 (9:1 EtOAc/Pet Ether); IR (vmax/cm"1, thin film): 3227, 1719, 1582, 1532, 1391 , 1222, 1 141 , 1 1 12; 1H NMR (600 MHz, DMSO-d6): δΗ = 3.46 (t, J = 2.4 Hz, 1 H, 34-H), 3.67-3.69 (m, 2H, 30-H), 4.18 (d, J = 2.4 Hz, 2H, 32-H), 4.23-4.25 (m, 2H, 29-H), 7.16 (t, J = 5.5 Hz, 1 H, 6-H), 7.51-7.54 (m, 2H, 15,16- H), 7.65 (d, J = 8.9 Hz, 2H 24-H), 7.86 (d, J = 5.6 Hz, 1 H, 5-H), 7.92-7.93 (m, 3H, 14,23-H), 7.98 (ap.d, J = 8.6 Hz, 1 H, 12-H), 8.02-8.05 (m, 2H, 11 ,17-H), 8.52 (s, 1 H, 19-H), 8.59 (s, 1 H, 3-H), 10.22 (s, 1 H, 26-H), 1 1.63 (bd, J = 4.7 Hz, 7/20-H); 13C NMR (150 MHz, DMSO-d6): 5C = 57.5 (C-32), 63.7 (C-29), 67.3 (C-30), 77.5 (C-34), 80.1 (C- 33), 1 10.9 (C-3), 1 15.3 (C-3), 1 16.8 (C-6), 1 17.7 (C-24), 123.8 (C-11 ), 124.2 (C-19), 126.3 (C-15/16), 126.6 (C-15/16), 127.4 (C-23), 127.7 (C-14), 128.3 (C-17), 128.4 (C- 12), 130.0 (C-10), 132.8 (C-13), 133.2 (C-18), 135.6 (C-9), 136.9 (C-22), 142.9 (C-25),
144.4 (C-8), 145.2 (C-2), 153.3 (C-27); LRMS m/z (ES+): 542 [M+H]+; HRMS m/z (ES+): Found 542.1512; C28H24N5O5S requires 542.1498.
Example 22
2-(allyloxy)ethyl-4-(yV-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- yl)sulfamoyl)phenylcarbamate (JS152)
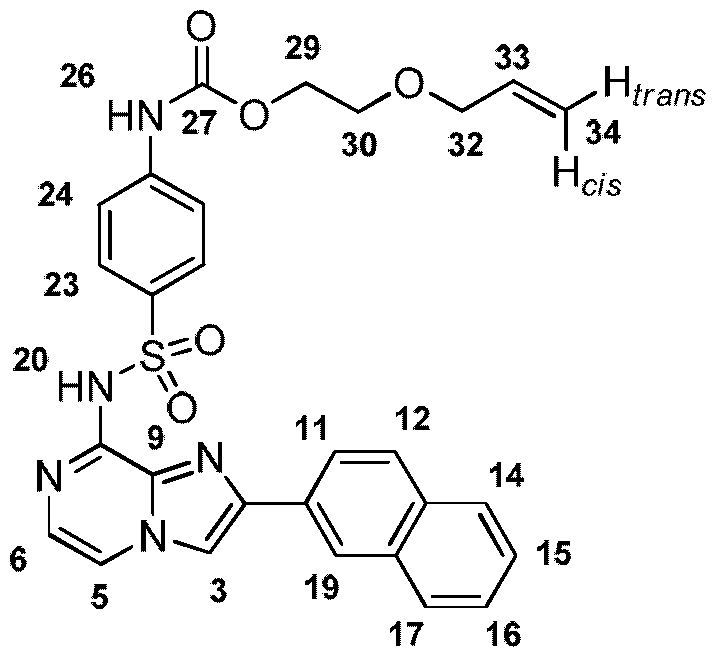
Step 1
2-(allyloxy)ethyl 4-sulfamoylphenylcarbamate (JS121 )
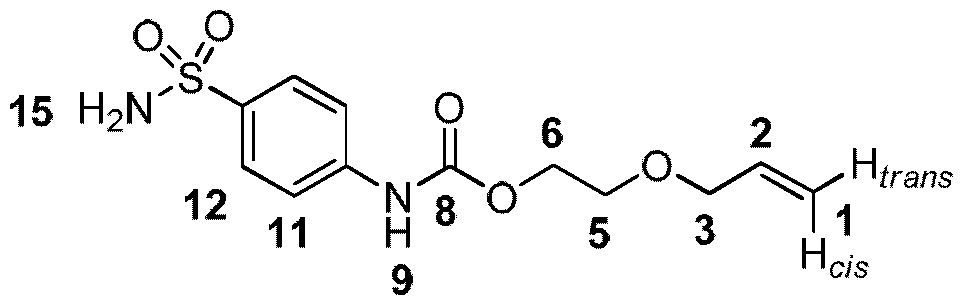
To a suspension of 4-sulfamoylbenzoic acid (2.01 g, 10.0 mmol) in anhydrous toluene (15 ml) was added triethylamine (1.63 ml, 1 1 .7 mmol). Diphenyphosphoryl azide (2.52 ml, 1 1.7 mmol) in anhydrous toluene (10 ml) was added drop wise to the reaction mixture and stirred at RT for 30 min followed by heating at 90 °C for 30 min. 2- (allyloxy)ethanol (1 .28 ml, 12.0 mmol) in anhydrous DMF (6 ml) was then added and the reaction temperature was increased to 90 °C for 16 h. The reaction was cooled to RT and solvent removed in vacuo. The crude material was taken up in EtOAc and washed with bicarb (1 x), H20 (3x) and brine (1 x); dried (MgS04), filtered and concentrated in vacuo to give an orange/brown sticky solid. Flash chromatography (Pet Ether; 2:1 to 1 :1 Pet Ether/EtOAc) afforded the title compound as a white solid
(689.0 mg, 2.30 mmol, 23.0%). Mpt: 128 °C; Rf = 0.41 (2:1 EtOAc/Pet Ether); IR (vmax/cm"1, thin film): 3356, 3298, 3193, 31 10, 2908 (C-H and N-H stretches), 1734 (C=0 stretch), 1596, 1529, 1336, 1314, 1216, 1 151 , 1099, 1055; 1H NMR (600 MHz, DMSO-d6): δΗ = 3.63-3.64 (m, 2H, 5-H), 3.98 - 3.99 (m, 2H, 3-H), 4.24-4.25 (m, 2H, 6- H), 5.16 (ap.dd, J = 10.4, 1 .7 Hz, 1 H, 1 -HCIS), 5.27 (ap.dd, J = 17.3, 1 .8 Hz, 1 H, 1 - Hfrans), 5.85-5.91 (m, 1 H, 2-H), 7.23 (s, 1 H, 15-H), 7.62 (d, J = 8.8 Hz, 2H, 11 -H), 7.73 (ap.d, J = 8.8 Hz, 2H, 12-H), 10.17 (s, 1 H, 9-H); 13C NMR (150 MHz, DMSO-d6): 5C = 63.9 (C-6), 67.7 (C-5), 71.0 (C-3), 1 16.7 (C-1 ), 1 17.7 (C-11 ), 126.8 (C-12), 135.0 (C-2), 137.6 (C-13), 142.3 (C-10), 153.4 (C-8); LRMS m/z (El+): 300 [M]", 243 [M- OCH2CH=CH2]; HRMS m/z (El+): Found 300.07810; Ci2H16N205S requires 300.07744; Anal. Calcd. for Ci2H16N205S: C, 47.99; H, 5.37; N, 9.33. Found C, 48.24; H, 5.27; N, 9.57%.
Step 2
2-(allyloxy)ethyl-4-(yV-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- yl)sulfamoyl)phenylcarbamate (JS152)

Example 23
2-(2-(3-(2,5-dioxo-2, -(yV-(2- (naphthalen-2-yl)im JS199)
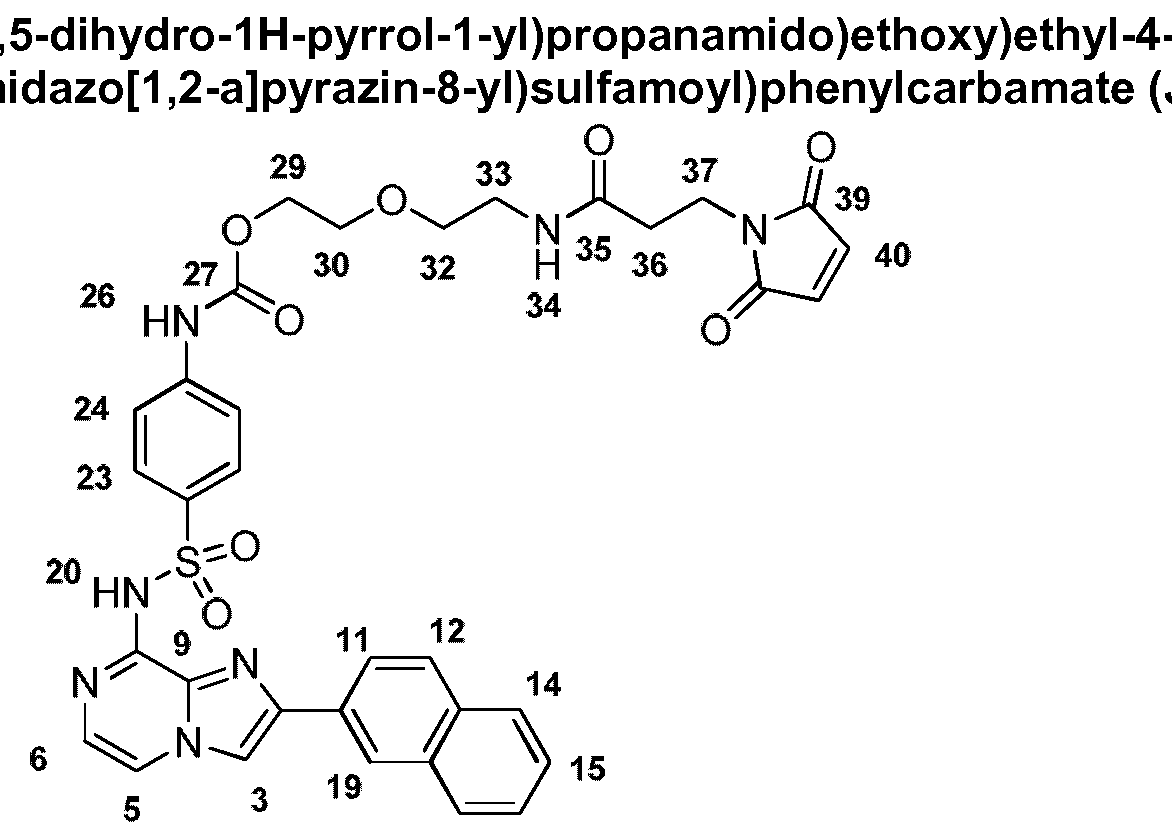
17 16
Step 1
4-sulfamoylbenzoyl azide (JS129)

4-sulfamoylbenzoic acid (1 .00 g, 4.97 mmol), triphenylphosphine (2.61 g, 9.94 mmol) and sodium azide (0.388 g, 5.96 mmol) were suspended in anhydrous acetone (10 ml). To this milky white suspension was added trichloroacetonitrile (0.997 ml, 9.94 mmol) drop wise and the reaction was left to stir at RT for 18 h. The solvent was removed in vacuo and the resulting dark yellow slurry was diluted with CH2CI2 and washed with H20 and brine, dried (MgS04), filtered and concentrated in vacuo. Flash chromatography (Pet Ether; 3:1 to 2:1 to 1 :1 Pet Ether/EtOAc) afforded the title compound as a white solid (906.9 mg, 4.01 mmol, 80.7%). Mpt: 1 18 °C; Rf = 0.23 (1 :1 Pet Ether/EtOAc); IR (vmax/cm"1, thin film): 3362 (aromatic C-H stretch), 3258 (N-H stretch), 2137 (N=N=N stretch), 1687 (CO stretch), 1339 (S=0 stretch, asymmetrical), 1238, 1 155 (S=0 Symmetrical stretch); 1H NMR (600 MHz, CD3OD): δΗ = 8.02 (ap.d, J = 6.9 Hz, 2H, 4-H), 8.18 (ap.d, J = 6.8 Hz, 2H, 3-H); 13C NMR (150 MHz, CD3OD): 5C = 127.6 (C-4), 131.0 (C-3), 134.9 (C-2), 150.2 (C-5), 172.8 (C-1 ); LRMS/HRMS m/z (ES+): no product mass present; Anal. Calcd. for C7H6N403S: C, 37.17; H, 2.67; N, 24.77. Found C, 37.27; H, 2.49; N, 24.40%.
ferf-butyl 2-(2-hydroxyethoxy)ethylcarbamate (JS136)

To a solution of 2-(2-aminoethoxy)ethanol (2 ml, 19.9 mmol) in CHCI3 (20 ml) cooled on ice, was added (Boc)20 (4.35 g, 19.9 mmol) in CHCI3 (20 ml). The reaction was stirred at RT for 90 min, followed by diluting with H20 and then extracting with CHCI3 (3x). The combined organics were dried (MgS04), filtered, concentrated in vacuo, and then
purified via flash chromatography (CH2CI2; 2% to 5% to 10% MeOH) to afford the title compound as a colourless oil (3.93 g, 19.2 mmol, 96.4%). Rf = 0.83 (10% MeOH/CH2CI2); IR (vmax/crrf1, thin film): 3353 (O-H stretch), 2976, 2931 , 2871 (C-H and N-H Stretches), 1687 (CO stretch), 1520, 1366, 1 169, 1 123; 1H NMR (500 MHz, CDCIs): δΗ = 1 .43 (s, 9H, 11 -H), 2.16 (bs, 1 H, 1 -H), 3.32 (t, J = 5.1 Hz, 2H, 6-H), 3.53- 3.57 (m, 4H, 3,5-H), 3.72-3.74 (m, 2H, 2-H), 4.98 (bs, 1 H, 7-H); 13C NMR (125 MHz, CDCI3): 5c = 28.5 (C-11 ), 40.5 (C-6), 61.8 (C-2), 70.4 (C-5), 72.3 (C-3), 79.5 (C-10), 156.2 (C-8); LRMS m/z (Cl+): 206 [M+H]+, 150 [M-fBu]+; HRMS m/z (Cl+): Found 206.1976; C9H2oN04 requires 206.13923.
Step 3
(JS138)

JS136 (993 mg, 4.39 mmol) was dissolved in anhydrous toluene (92 ml). JS129 (750.6 mg, 3.66 mmol) was added and the reaction was stirred under reflux for 16 h. The reaction was cooled to RT and adsorbed onto silica and purification via flash chromatography (Pet Ether; 2:1 to 1 :1 to 1 :2 Pet Ether/EtOAc) afforded the title compound as a white solid (1 .1306 g, 2.81 mmol, 76.7%). Mpt: 94-95 °C; Rf = 0.35 (1 :3 Pet Ether/EtOAc); IR (vmax/cnY1, thin film): 3368, 3332 (Aromatic C-H stretch), 3201 , 3101 , 2976, 2919 (C-H, N-H Stretches), 1703, 1676 (C=0 Stretches), 1522, 1 156; 1H NMR (600 MHz, CD3OD): δΗ = 1.41 (s, 9H, 19-H), 3.22 (t, J = 5.6 Hz, 2H, 14-H), 3.53 (t, J = 5.7 Hz, 2H, 13-H), 3.71 (t, J = 4.7 Hz, 2H, 11 -H), 4.29 (t, J = 4.6 Hz, 2H, 10-H), 7.60 (d, J = 8.8 Hz, 2H, 5-H), 7.81 (ap.d, J = 8.8 Hz, 2H, 4-H); 13C NMR (150 MHz, CD3OD): 5c = 28.8 (C-19), 41 .3 (C-14), 65.4 (C-10), 70.2 (C-11 ), 71 .0 (C-13), 80.2 (C- 18), 1 19.0 (C-5), 128.3 (C-4), 138.7 (C-3), 144.1 (C-6), 155.4 (C-8), 158.2 (C-16); LRMS m/z (ES+): 426 [M+Na]+, 370 [M-fBu+Na]+; HRMS m/z (ES+): Found 426.1306; Ci6H25N307NaS requires 426.131 1 ; Anal. Calcd. for
 C, 47.63; H, 6.25; N, 10.42. Found C, 47.78; H, 6.46; N, 10.20%
Step 4
C, 47.63; H, 6.25; N, 10.42. Found C, 47.78; H, 6.46; N, 10.20%
Step 4
(JS179)

Step 5
2-(2-aminoethoxy)ethyl-4-(yV-(2-(naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8- yl)sulfamoyl)phenylcarbamate (JS178)

17 16
JS179 (317.9 mg, 0.492 mmol) was dissolved in anhydrous CH2CI2 (15 ml) and cooled on ice. TFA (15 ml) was added and the reaction was stirred at RT for 3 h. Removal of the solvent, with the aid of toluene, followed by flash chromatography (CH2CI2; 5% to 10% MeOH/CH2CI2). Mpt: >200 °C; Rf = 0.26 (20% MeOH/CH2CI2); IR (vmax/crTf1 , thin film): 1726, 1675, 1200, 1 120, 1071 ; 1 H NMR (400 MHz, DMSO-d6): δΗ = 2.99 (t, J = 5.3 Hz, 2H, 33-H), 3.62 (t, J = 5.2 Hz, 2H, 32-H), 3.70 (t, J = 4.7 Hz, 2H, 30-H), 4.25 (t, J = 4.4 Hz, 2H, 29-H), 7.1 1 (d, J = 5.0 Hz, 1 H, 6-H), 7.48-7.56 (m, 4H, 15,16,24-H), 7.74 (d, J = 5.0 Hz, 1 H, 5-H), 7.88 (d, J = 8.8 Hz, 2H, 23-H), 7.92 (ap.d, J = 7.4 Hz, 1 H, 14-H), 7.97 (d, J = 8.6 Hz, 1 H, 12-H), 8.00 (ap.d, J = 7.4 Hz, 1 H, 17-H), 8.06 (dd, J = 8.5, 1.6 Hz, 1 H, 11 -H), 8.42 (s, 1 H, 3-H), 8.52 (s, 1 H, 19-H), 9.98 (bs, 1 H, 20-H); 13C NMR (150 MHz, DMSO-d6): 5C = 38.6 (C-33), 63.6 (C-29), 66.7 (C-32), 68.5 (C-30), 1 14.5 (C-3), 1 16.5 (C-6), 1 18.4 (C-24), 123.9 (C-11 ,19), 126.5 (C-15), 126.6 (C-16) 127.7 (C-14,23), 128.2 (C-17), 128.3 (C-12), 132.7 (C-13,18), 133.3 (C-10,22), 142.0 (C-25), 144.1 (C-2) 153.4 (C-27); LRMS m/z (ES+): 547 [M+H]+; HRMS m/z (ES+):
Found 547.1757; C27H27N6O5S requires 547.1764; Anal. Calcd. for C35H30F12N6O13S (4x TFA): C, 41.92; H, 3.02; N, 8.38. Found C, 41 .14; H, 2.98; N, 8.71 %.
Step 6
2-(2-(3-(2,5-dioxo-2,5-dihydro-1 H-pyrrol-1 -yl)propanamido)ethoxy)ethyl-4-(/V-(2- (naphthalen-2-yl)imidazo[1 ,2-a]pyrazin-8-yl)sulfamoyl)phenyl carbamate (JS199)

17 16 3-(2,5-dioxo-2,5-dihydro-1 H-pyrrol-1 -yl)propanoic acid (62.7 mg, 0.371 mmol) and H BTU (210.9 mg, 0.556 mmol) were dissolved in anhydrous DMF (15 ml) and the mixture was purged with Argon. DIPEA (197.4 μΙ, 1.1 1 mmol) was added and a colourless to orange colour change was observed after stirring at RT for 20 min. JS178 (367 mg, 0.556 mmol) in anhydrous DMF (5 ml) was added and the reaction was stirred at RT for 16 h. Removal of the solvent was followed by diluting with EtOAc and washing with H20 (4x) and brine, drying (MgS04), filtering and concentrating in vacuo. Flash chromatography (CH2CI2; 2% to 3% to 4% MeOH/CH2CI2) afforded the title compound as an off white solid (146.2 mg, 0.210 mmol, 56.5%). Mpt: >200 °C; Rf = 0.43 (10% MeOH/CH2CI2); IR (vmax/cm"1, thin film): 3254 (aromatic C-H stretch), 2925 (C-H and N-H Stretches), 1706 (C=0 stretch), 1589, 1527, 1405, 1224, 1 140; 1 H NMR (600 MHz, CD3CN): δΗ = 2.38 (t, J = 7.1 Hz, 2H, 36-H), 3.27 (q, J = 5.5 Hz, 2H, 33-H), 3.48 (t, J = 5.6 Hz, 2H, 32-H), 3.64-3.67 (m, 4H, 20,37-H), 4.24-4.26 (m, 2H, 29-H), 6.58 (bs, 1 H, 34-H), 6.72 (s, 2H, 40-H), 7.06 (bd, J = 5.4 Hz, 1 H, 6-H), 7.51 -7.56 (m, 2H, 15,16-H), 7.62-7.65 (m, 3H, 5,24-H), 7.91-7.93 (m, 3H, 14,23-H), 7.95-7.98 (m, 2H, 12,17-H), 8.03 (dd, J = 8.5, 1 .6 Hz, 1 H, 11 -H), 8.20 (s, 1 H, 3-H), 8.31 (bs, 1 H, 20-H),
8.49 (s, 1 H, 19-H); 13C NMR (150 MHz, CD3CN): 5C = 33.8 (C-37), 33.9 (C-36), 38.4 (C-33), 64.0 (C-29), 68.3 (C-30), 68.9 (C-32), 1 10.3 (C-5) 1 14.5 (C-3), 1 15.8 (C-6) 1 17.6 (C-24), 123.4 (C-11 ), 124.2 (C-19), 126.1 (C-15), 126.2 (C-16), 127.1 (C-23), 127.3 (C-14), 127.9 (C-17), 128.2 (C-12), 129.4 (C-10) 132.9 (C-13), 133.2 (C-18), 133.9 (C-40), 135.4 (C-9), 135.8 (C-22), 145.2 (C-2), 142.5 (C-25), 144.7 (C-8) 153.1 (C-27), 169.8 (C-35), 170.5 (C-39); LRMS m/z (ES+): 698 [M+H]+; HRMS m/z (ES+): Found 698.2010; C34H32N7O8S requires 698.2033.
VirB1 1 protein production and ATPase assay.
The VirB1 1 protein was produced in the E. coli strain BL21 Star(DE3) (Invitrogen) as described previously. The protein concentration was estimated spectroscopically using a NanoDrop (Thermo Scientific) and a calculated extinction coefficient at 280 nm, based on the amino acid composition. The ATPase activity of VirB1 1 was measured, with and without a specific amount of compound present, using an in vitro ATPase colorimetric assay kit (Innova Biosciences). The assay was performed in 96-well ELISA microplates (Greiner Bio-One), using a multipipett robot. All measurements were made in duplicate. A volume of 49 μΙ_ of a substrate/buffer solution (200 mM tris(hydroxymethyl)aminomethane (TRIS), pH 7.5; 5 mM MgCI2; 250 μΜ ATP; and 10% DMSO) was added to each assigned well, followed by the addition of 1 μΙ_ of compound (at 0.5; 5 or 50 mM to achieve the final concentrations of 5; 50 and 500 μΜ in the reaction, respectively) in DMSO (or 1 μΙ_ DMSO to controls). The solutions were mixed carefully by pipetting. The reaction was started by the addition of 50 μΙ_ of 0.106 μΜ VirB1 1 to each well (except the negative control, see text below), and the reaction plate was directly transferred to 37 °C for 30 min of incubation. The reaction was stopped by the addition of the Gold mix according to the standard protocol of the kit. The absorbance at 620 nm was measured after 30 min at room temperature. For each compound, the percentage of absorbance relative non-inhibited VirB1 1 was calculated, after subtracting the absorbance value of the negative control. In the negative control, the protein was added after the Gold mix, as described in the standard protocol of the
kit, which when used as a blank corrects for all free P, not produced by the enzyme during the 30 min incubation at 37 °C.
A known inhibitor of VirB1 1 (Microbiology, (2006), 152, 2919-2930, denoted CHIR02) was used as a control inhibitor. A selection of the compounds were assayed as above at additional concentrations ranging between 5 and 200 μΜ from which IC50 values were calculated, assuming a linear relationship between P, production and absorbance. IC50 was defined as the concentration of compound that produces 50% inhibition. Evaluation of compounds as VirB1 1 ATPase inhibitors
The VirB1 1 protein was produced recombinantly in Escherichia coli and purified to high purity as described previously. The ATPase activity of VirB1 1 was measured by monitoring the release of inorganic phosphate (P,) using an in vitro ATPase assay.
The inhibitory effects of Examples 1 to 12 were evaluated by performing the ATPase assay with and without compound present at concentrations of 500 μΜ (or 250 μΜ), 50 μΜ and 5 μΜ (Fig. 1 ). At the highest concentration, precipitation appeared in the reaction wells of compounds 1 , 2, 5, 8, 9 and 1 1 , which interfered with the measurements. Despite this, a clear inhibitory effect could be seen of the compounds 1 , 3, 4, 5, 6, 1 1 and 12 of which 4, 5, 8, 1 1 and 12 clearly reduced the ATPase activity at 50 μΜ. IC50 values, logP and logS values were then measured for a selection of the compounds 1 to 23 (Table 3).






Table 3. IC5o values
Claims
Claims
1. A compound of Formula (I) for use in preventing or treating a bacterial infection:

wherein:
Z is O or NH;
Ri is selected from substituted alkyl, unsubstituted alkyl, substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
one of R2 and R3 is H, and the other one of R2 and R3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
L is a direct bond or is selected from:
NH
(CH2)n
n = 0, 1 , 2, 3, 4, 5 or 6;

(CH2)n
■"J™ is a 5- or 6-membered nitrogen-containing heteroaryl moiety, optionally containing at least one or more further heteroatom; and
R4 and R5 are independently selected from H, alkyl, halo, alkoxy, alkylthio, hydroxy, cyano, amino and nitro;
or a pharmaceutically acceptable salt, solvate or prodrug thereof.
2. The compound for use as claimed in claim 1 , wherein
Z is O or NH;
R2 is H;
R3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl unsubstituted heteroaryl; and
L is a direct bond.
3. The compound for use as claimed in claim 1 , wherein
Z is O or NH;
R2 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
R3 is H; and
L is a direct bond.
4. The compound for use as claimed in claim 1 , wherein
Z is O or NH;
R2 is H;
R3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl; and
L is selected from:
NH
(CH2)n
, wherein n = 0, 1 , 2, 3, 4, 5 or 6;

each of which may optionally be substituted at one more exocyclic positions and wherein n = 0, 1 , 2, 3, 4, 5 or 6; wherein
(CH2)n
is a 5- or 6-membered nitrogen-containing heteroaryl moiety, optionally containing at least one or more further heteroatom.
5. The compound for use as claimed in claim 1 , wherein:
Z is O or NH;
R2 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
R3 is H; and
L is selected from:
NH
(CH2)n
, wherein n = 0, 1 , 2, 3, 4, 5 or 6;

each of which may optionally be substituted at one or more exocyclic positions, and wherein n = 0, 1 , 2, 3, 4, 5 or 6; wherein
(CH2)n
™" is a 5- or 6-membered nitrogen-containing heteroaryl moiety, optionally containing at least one or more further heteroatom.
6. The compound for use as claimed in any one of claims 1 , 3 or 5, wherein R2 is 2-naphthyl. 7. The compound for use as claimed in any one of claims 1 , 2 or 4, wherein R3 is 2-naphthyl.
8. The compound for use as claimed in any one of the preceding claims, wherein R4 is H.
9. The compound for use as claimed in any one of the preceding claims, wherein R5 is H.
10. The compound for use as claimed in any one of the preceding claims, wherein R is substituted aryl.
1 1 . The compound for use as claimed in any one of the preceding claims, wherein Ri is substituted aryl substituted with a poly(ethylene) glycol moiety.
12. The compound for use as claimed in claim 1 1 , wherein the poly(ethylene) glycol moiety is coupled to the aryl group via a linker.
13. The compound for use as claimed in claim 12, wherein the linker comprises a carbamate moiety.
14. The compound for use as claimed in any one of claims 1 to 1 1 , wherein R-i is substituted aryl directly substituted with a poly(ethylene) glycol moiety.
15. The compound for use as claimed in any one of the preceding claims, wherein the bacterial infection is infection with Helicobacter pylori, Legionella pneumophilia, Brucella suis, Bartonella henselae or Bordetella pertussis.
16. A compound of Formula (I):

wherein:
Z is O or NH;
Ri is selected from substituted alkyl, unsubstituted alkyl, substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
one of R2 and R3 is H, and the other one of R2 and R3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
L is a direct bond or is selected from:
NH
(CH2)n
wherein n = 0, 1 , 2, 3, 4, 5 or 6;

eac o w c may opt ona y e su st tute at one or more exocyclic positions and wherein n = 0, 1 , 2, 3, 4, 5 or 6; wherein
(CH2)n
is a 5- or 6-membered nitrogen-containing heteroaryl moiety, optionally containing at least one or more further heteroatom; and
R4 and R5 are independently selected from H, alkyl, halo, alkoxy, alkylthio, hydroxy, cyano, amino and nitro;
or a pharmaceutically acceptable salt, solvate or prodrug thereof.
17. The compound of claim 16, wherein
Z is O or NH;
R2 is H;
R3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl; and
L is a direct bond.
18. The compound of claim 16, wherein
Z is O or NH;
R2 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
R3 is H; and
L is a direct bond.
19. The compound of claim 16, wherein
Z is O or NH;
R2 is H;
R3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl unsubstituted heteroaryl; and
L is selected from:
NH
(CH2)n
, wherein n = 0, 1 , 2, 3, 4, 5 or 6;
(CH2)n
is a 5- or 6-membered nitrogen-containing heteroaryl moiety, optionally containing at least one or more further heteroatom.
The compound of claim 16, wherein:
Z is O or NH;
R is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl bstituted heteroaryl;
R3 is H; and
L is selected from:
NH
(CH2)n
herein n = 0, 1 , 2, 3, 4, 5 or 6;

™" is a 5- or 6-membered nitrogen-containing heteroaryl moiety, optionally containing at least one or more further heteroatom.
21 . The compound of a as defined by Formula (la):

wherein:
Ri is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
one of R2 and R3 is H, and the other one of R2 and R3 is selected from substituted aryl, unsubstituted aryl, substituted heteroaryl or unsubstituted heteroaryl;
L is selected from:

is a 5- or 6-membered nitrogen-containing heteroaryl moiety, optionally containing at least one or more further heteroatom; and
R4 and R5 are independently selected from H, alkyl, halo, alkoxy, alkylthio, hydroxy, cyano, amino and nitro;
or a pharmaceutically acceptable salt, solvate or prodrug thereof.
22. The compound as claimed in any one of claims 16, 18, 20, or 21 , wherein R2 is 2-naphthyl.
23. The compound as claimed in any one of claims 16, 17, 19, or 21 , wherein R3 is 2-naphthyl. 24. The compound as claimed in any one of claims 16 to 23, wherein R4 is H.
25. The compound as claimed in any one of claims 16 to 24, wherein R5 is H.
26. The compound of any one of claims 16 to 25, wherein R-i is substituted aryl.
27. The compound of claim 26, wherein R-i is substituted aryl substituted with a poly(ethylene) glycol moiety.
28. The compound of claim 27, wherein the poly(ethylene) glycol moiety is coupled to the aryl group via a linker.
29. The compound of claim 28, wherein the linker comprises a carbamate moiety.
30. The compound as claimed in any one of claims 16 to 26, wherein Ri is substituted aryl directly substituted with a poly(ethylene) glycol moiety.
31 . A method of treating or preventing bacterial infection in a subject by administering a therapeutically effective amount of a compound of any one of claims 16 to 30 or a pharmaceutically acceptable salt thereof to a patient in need thereof.
32. The method of claim 31 , wherein the bacterial infection is infection with Helicobacter pylori, Legionella pneumophilia, Brucella suis, Bartonella henselae or Bordetella pertussis. 33. Use of a compound of any one of claims 16 to 30 in the manufacture of a medicament for treating or preventing bacterial infection.
34. The use of the compound of claim 33, wherein the bacterial infection is infection with Helicobacter pylori, Legionella pneumophilia, Brucella suis, Bartonella henselae or Bordetella pertussis. 35. A pharmaceutical composition comprising a therapeutically effective amount of a compound as defined in any one of claims 16 to 30, optionally one or more other active ingredients and a pharmaceutically acceptable carrier.
36. A method of making a compound as claimed in claim 16 which comprises reacting a compound of Formula (II):
X
R4 R3
wherein X is a leaving group, for example a halo group, and R2, R3 , R4 and R5 are as defined in claim 16, with a compound of the formula:
 wherein R-i , L and and Z are as defined in claim 16.
wherein R-i , L and and Z are as defined in claim 16.
37. A method of making the compounds of claims 17, 19 or 21 , comprising:
(i) reacting a compound of Formula (IV)

with 2,3-dichloropyrazine;
(ii) oxidising the product of (i);
(iii) effecting an intramolecular cyclisation of the product of (ii); and
(iv) coupling the product of (iii) with a compound of Formula (VI)

wherein
R-i , R3, L and Z are as defined in claim 16.
38. The method of claim 37, wherein step (iv) is carried out in the presence of a palladium catalyst. 39. A method of making the compounds of claims 18, 20 or 21 , comprising:
(i) reacting a compound of Formula (V)
 with 2-amino-3-chloropyrazine;
with 2-amino-3-chloropyrazine;
coupling the product of (i) with a compound of Formula (VI)

wherein
Ri , R2, L and Z are as defined in claim 16.
40. The method of claim 39, wherein step (iv) is carried out in the presence of a palladium catalyst.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB1109763.1 | 2011-06-10 | ||
| GBGB1109763.1A GB201109763D0 (en) | 2011-06-10 | 2011-06-10 | Compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012168733A1 true WO2012168733A1 (en) | 2012-12-13 |
Family
ID=44357536
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2012/051303 WO2012168733A1 (en) | 2011-06-10 | 2012-06-08 | Substituted 8 - amino - imidazo [1, 2-a] pyrazines as antibacterial agents |
Country Status (2)
| Country | Link |
|---|---|
| GB (1) | GB201109763D0 (en) |
| WO (1) | WO2012168733A1 (en) |
Cited By (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016162318A1 (en) | 2015-04-08 | 2016-10-13 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents and intermediate product |
| WO2017072039A1 (en) | 2015-10-26 | 2017-05-04 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| WO2017093180A1 (en) | 2015-12-01 | 2017-06-08 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| WO2017144341A1 (en) | 2016-02-23 | 2017-08-31 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| WO2017174414A1 (en) | 2016-04-05 | 2017-10-12 | Bayer Cropscience Aktiengesellschaft | Naphthaline-derivatives as pest control agents |
| EP3241830A1 (en) | 2016-05-04 | 2017-11-08 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pesticides |
| WO2018015289A1 (en) | 2016-07-19 | 2018-01-25 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| WO2018033455A1 (en) | 2016-08-15 | 2018-02-22 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| WO2018050825A1 (en) | 2016-09-19 | 2018-03-22 | Bayer Cropscience Aktiengesellschaft | Pyrazolo [1,5-a]pyridine derivatives and their use as pesticides |
| EP3305786A2 (en) | 2018-01-22 | 2018-04-11 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pesticides |
| WO2018065288A1 (en) | 2016-10-07 | 2018-04-12 | Bayer Cropscience Aktiengesellschaft | 2-[2-phenyl-1-(sulfonyl-methyl)-vinyl]-imidazo-[4,5-b] pyridine derivatives and related compounds as pesticides in plant protection |
| WO2018130437A1 (en) | 2017-01-10 | 2018-07-19 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2018130443A1 (en) | 2017-01-10 | 2018-07-19 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| CN108299252A (en) * | 2018-04-04 | 2018-07-20 | 梯尔希(南京)药物研发有限公司 | A kind of synthetic method of Norepinephrine Salonc Acid metabolite |
| WO2018138050A1 (en) | 2017-01-26 | 2018-08-02 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclene derivatives as pest control agents |
| WO2018197257A1 (en) | 2017-04-24 | 2018-11-01 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic-compound derivatives as pest control agents |
| US10308644B2 (en) | 2016-12-22 | 2019-06-04 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2019162174A1 (en) | 2018-02-21 | 2019-08-29 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pest control agents |
| US10618916B2 (en) | 2018-05-11 | 2020-04-14 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10669271B2 (en) | 2018-03-30 | 2020-06-02 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2020127075A1 (en) * | 2018-12-17 | 2020-06-25 | F. Hoffmann-La Roche Ag | Imidazopyrazine derivatives as antibacterials |
| WO2020126954A1 (en) * | 2018-12-17 | 2020-06-25 | F. Hoffmann-La Roche Ag | Novel imidazopyrazine derivatives |
| WO2020126953A1 (en) * | 2018-12-17 | 2020-06-25 | F. Hoffmann-La Roche Ag | Novel imidazopyrazine derivatives as antibacterials |
| WO2020126956A1 (en) * | 2018-12-17 | 2020-06-25 | F. Hoffmann-La Roche Ag | Imidazopyrazine derivatives as antibacterial agents |
| WO2020126952A1 (en) * | 2018-12-17 | 2020-06-25 | F. Hoffmann-La Roche Ag | Imidazopyrazine derivatives as antibacterial agents |
| WO2020173861A1 (en) | 2019-02-26 | 2020-09-03 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pest control agents |
| WO2020173860A1 (en) | 2019-02-26 | 2020-09-03 | Bayer Aktiengesellschaft | Fused bicyclic heterocycle derivatives as pesticides |
| US10793565B2 (en) | 2016-12-22 | 2020-10-06 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10806785B2 (en) | 2016-12-22 | 2020-10-20 | Incyte Corporation | Immunomodulator compounds and methods of use |
| EP3896065A1 (en) | 2015-08-07 | 2021-10-20 | Bayer CropScience Aktiengesellschaft | 2-(het)aryl-substituted condensed heterocycle derivatives as pesticides |
| WO2021219578A1 (en) * | 2020-04-29 | 2021-11-04 | F. Hoffmann-La Roche Ag | Antibacterial 8-phenylamino-3-(pyrazol-4-yl)imidazo[1,2-a]pyrazine derivatives |
| WO2021244997A1 (en) * | 2020-06-01 | 2021-12-09 | F. Hoffmann-La Roche Ag | Novel imidazopyrazne derivatives |
| WO2021249896A1 (en) * | 2020-06-09 | 2021-12-16 | F. Hoffmann-La Roche Ag | Novel imidazopyrazine derivatives |
| WO2021249893A1 (en) * | 2020-06-08 | 2021-12-16 | F. Hoffmann-La Roche Ag | Novel imidazo-pyrazine derivatives |
| US11401279B2 (en) | 2019-09-30 | 2022-08-02 | Incyte Corporation | Pyrido[3,2-d]pyrimidine compounds as immunomodulators |
| US11407749B2 (en) | 2015-10-19 | 2022-08-09 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11465981B2 (en) | 2016-12-22 | 2022-10-11 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11535615B2 (en) | 2015-12-22 | 2022-12-27 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2023285497A1 (en) * | 2021-07-15 | 2023-01-19 | F. Hoffmann-La Roche Ag | Imidazole derivatives and their use as antibiotics |
| US11572366B2 (en) | 2015-11-19 | 2023-02-07 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11608337B2 (en) | 2016-05-06 | 2023-03-21 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11613536B2 (en) | 2016-08-29 | 2023-03-28 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11673883B2 (en) | 2016-05-26 | 2023-06-13 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11718605B2 (en) | 2016-07-14 | 2023-08-08 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11753406B2 (en) | 2019-08-09 | 2023-09-12 | Incyte Corporation | Salts of a PD-1/PD-L1 inhibitor |
| US11760756B2 (en) | 2020-11-06 | 2023-09-19 | Incyte Corporation | Crystalline form of a PD-1/PD-L1 inhibitor |
| US11780836B2 (en) | 2020-11-06 | 2023-10-10 | Incyte Corporation | Process of preparing a PD-1/PD-L1 inhibitor |
| US11866451B2 (en) | 2019-11-11 | 2024-01-09 | Incyte Corporation | Salts and crystalline forms of a PD-1/PD-L1 inhibitor |
| US11866434B2 (en) | 2020-11-06 | 2024-01-09 | Incyte Corporation | Process for making a PD-1/PD-L1 inhibitor and salts and crystalline forms thereof |
| US11873309B2 (en) | 2016-06-20 | 2024-01-16 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008079460A2 (en) * | 2006-09-05 | 2008-07-03 | Emory University | Tyrosine kinase inhibitors for prevention or treatment of infection |
| WO2009024585A2 (en) * | 2007-08-21 | 2009-02-26 | Biofocus Dpi Limited | Imidazo [1,2-a] pyrazine compounds for treatment of viral infections such as hepatitis |
| WO2010069684A1 (en) * | 2008-12-17 | 2010-06-24 | Biomarin Iga, Ltd. | Compounds for treatment of duchenne muscular dystrophy |
-
2011
- 2011-06-10 GB GBGB1109763.1A patent/GB201109763D0/en not_active Ceased
-
2012
- 2012-06-08 WO PCT/GB2012/051303 patent/WO2012168733A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008079460A2 (en) * | 2006-09-05 | 2008-07-03 | Emory University | Tyrosine kinase inhibitors for prevention or treatment of infection |
| WO2009024585A2 (en) * | 2007-08-21 | 2009-02-26 | Biofocus Dpi Limited | Imidazo [1,2-a] pyrazine compounds for treatment of viral infections such as hepatitis |
| WO2010069684A1 (en) * | 2008-12-17 | 2010-06-24 | Biomarin Iga, Ltd. | Compounds for treatment of duchenne muscular dystrophy |
Cited By (65)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016162318A1 (en) | 2015-04-08 | 2016-10-13 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents and intermediate product |
| EP3896066A2 (en) | 2015-08-07 | 2021-10-20 | Bayer CropScience Aktiengesellschaft | 2-(het)aryl-substituted condensed heterocycle derivatives as pesticides |
| EP3896065A1 (en) | 2015-08-07 | 2021-10-20 | Bayer CropScience Aktiengesellschaft | 2-(het)aryl-substituted condensed heterocycle derivatives as pesticides |
| US11407749B2 (en) | 2015-10-19 | 2022-08-09 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2017072039A1 (en) | 2015-10-26 | 2017-05-04 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| US11572366B2 (en) | 2015-11-19 | 2023-02-07 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2017093180A1 (en) | 2015-12-01 | 2017-06-08 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| US11535615B2 (en) | 2015-12-22 | 2022-12-27 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11866435B2 (en) | 2015-12-22 | 2024-01-09 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2017144341A1 (en) | 2016-02-23 | 2017-08-31 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| WO2017174414A1 (en) | 2016-04-05 | 2017-10-12 | Bayer Cropscience Aktiengesellschaft | Naphthaline-derivatives as pest control agents |
| EP3241830A1 (en) | 2016-05-04 | 2017-11-08 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pesticides |
| US11608337B2 (en) | 2016-05-06 | 2023-03-21 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11673883B2 (en) | 2016-05-26 | 2023-06-13 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11873309B2 (en) | 2016-06-20 | 2024-01-16 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11718605B2 (en) | 2016-07-14 | 2023-08-08 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2018015289A1 (en) | 2016-07-19 | 2018-01-25 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| WO2018033455A1 (en) | 2016-08-15 | 2018-02-22 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| US11613536B2 (en) | 2016-08-29 | 2023-03-28 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2018050825A1 (en) | 2016-09-19 | 2018-03-22 | Bayer Cropscience Aktiengesellschaft | Pyrazolo [1,5-a]pyridine derivatives and their use as pesticides |
| WO2018065288A1 (en) | 2016-10-07 | 2018-04-12 | Bayer Cropscience Aktiengesellschaft | 2-[2-phenyl-1-(sulfonyl-methyl)-vinyl]-imidazo-[4,5-b] pyridine derivatives and related compounds as pesticides in plant protection |
| US10806785B2 (en) | 2016-12-22 | 2020-10-20 | Incyte Corporation | Immunomodulator compounds and methods of use |
| US11339149B2 (en) | 2016-12-22 | 2022-05-24 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10308644B2 (en) | 2016-12-22 | 2019-06-04 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11465981B2 (en) | 2016-12-22 | 2022-10-11 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11566026B2 (en) | 2016-12-22 | 2023-01-31 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11787793B2 (en) | 2016-12-22 | 2023-10-17 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10793565B2 (en) | 2016-12-22 | 2020-10-06 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10800768B2 (en) | 2016-12-22 | 2020-10-13 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2018130443A1 (en) | 2017-01-10 | 2018-07-19 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2018130437A1 (en) | 2017-01-10 | 2018-07-19 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2018138050A1 (en) | 2017-01-26 | 2018-08-02 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclene derivatives as pest control agents |
| WO2018197257A1 (en) | 2017-04-24 | 2018-11-01 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic-compound derivatives as pest control agents |
| EP3305786A2 (en) | 2018-01-22 | 2018-04-11 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pesticides |
| WO2019162174A1 (en) | 2018-02-21 | 2019-08-29 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pest control agents |
| US11124511B2 (en) | 2018-03-30 | 2021-09-21 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10669271B2 (en) | 2018-03-30 | 2020-06-02 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| CN108299252A (en) * | 2018-04-04 | 2018-07-20 | 梯尔希(南京)药物研发有限公司 | A kind of synthetic method of Norepinephrine Salonc Acid metabolite |
| US11414433B2 (en) | 2018-05-11 | 2022-08-16 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10618916B2 (en) | 2018-05-11 | 2020-04-14 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10906920B2 (en) | 2018-05-11 | 2021-02-02 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2020126952A1 (en) * | 2018-12-17 | 2020-06-25 | F. Hoffmann-La Roche Ag | Imidazopyrazine derivatives as antibacterial agents |
| WO2020126953A1 (en) * | 2018-12-17 | 2020-06-25 | F. Hoffmann-La Roche Ag | Novel imidazopyrazine derivatives as antibacterials |
| CN113164771A (en) * | 2018-12-17 | 2021-07-23 | 豪夫迈·罗氏有限公司 | Imidazopyrazine derivatives as antibacterial agents |
| JP2022513937A (en) * | 2018-12-17 | 2022-02-09 | エフ.ホフマン-ラ ロシュ アーゲー | Imidazopyrazine derivative as an antibacterial agent |
| JP2022513851A (en) * | 2018-12-17 | 2022-02-09 | エフ.ホフマン-ラ ロシュ アーゲー | Imidazopyrazine derivative as an antibacterial agent |
| WO2020127075A1 (en) * | 2018-12-17 | 2020-06-25 | F. Hoffmann-La Roche Ag | Imidazopyrazine derivatives as antibacterials |
| US20230012368A1 (en) * | 2018-12-17 | 2023-01-12 | Hoffmann-La Roche Inc. | Novel imidazopyrazine derivatives |
| WO2020126954A1 (en) * | 2018-12-17 | 2020-06-25 | F. Hoffmann-La Roche Ag | Novel imidazopyrazine derivatives |
| WO2020126956A1 (en) * | 2018-12-17 | 2020-06-25 | F. Hoffmann-La Roche Ag | Imidazopyrazine derivatives as antibacterial agents |
| WO2020173861A1 (en) | 2019-02-26 | 2020-09-03 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pest control agents |
| WO2020173860A1 (en) | 2019-02-26 | 2020-09-03 | Bayer Aktiengesellschaft | Fused bicyclic heterocycle derivatives as pesticides |
| US11753406B2 (en) | 2019-08-09 | 2023-09-12 | Incyte Corporation | Salts of a PD-1/PD-L1 inhibitor |
| US11401279B2 (en) | 2019-09-30 | 2022-08-02 | Incyte Corporation | Pyrido[3,2-d]pyrimidine compounds as immunomodulators |
| US11866451B2 (en) | 2019-11-11 | 2024-01-09 | Incyte Corporation | Salts and crystalline forms of a PD-1/PD-L1 inhibitor |
| WO2021219578A1 (en) * | 2020-04-29 | 2021-11-04 | F. Hoffmann-La Roche Ag | Antibacterial 8-phenylamino-3-(pyrazol-4-yl)imidazo[1,2-a]pyrazine derivatives |
| WO2021244997A1 (en) * | 2020-06-01 | 2021-12-09 | F. Hoffmann-La Roche Ag | Novel imidazopyrazne derivatives |
| WO2021249893A1 (en) * | 2020-06-08 | 2021-12-16 | F. Hoffmann-La Roche Ag | Novel imidazo-pyrazine derivatives |
| WO2021249896A1 (en) * | 2020-06-09 | 2021-12-16 | F. Hoffmann-La Roche Ag | Novel imidazopyrazine derivatives |
| US11760756B2 (en) | 2020-11-06 | 2023-09-19 | Incyte Corporation | Crystalline form of a PD-1/PD-L1 inhibitor |
| US12084443B2 (en) | 2020-11-06 | 2024-09-10 | Incyte Corporation | Process of preparing a PD-1/PD-L1 inhibitor |
| US11780836B2 (en) | 2020-11-06 | 2023-10-10 | Incyte Corporation | Process of preparing a PD-1/PD-L1 inhibitor |
| US11866434B2 (en) | 2020-11-06 | 2024-01-09 | Incyte Corporation | Process for making a PD-1/PD-L1 inhibitor and salts and crystalline forms thereof |
| WO2023285497A1 (en) * | 2021-07-15 | 2023-01-19 | F. Hoffmann-La Roche Ag | Imidazole derivatives and their use as antibiotics |
| US11858912B2 (en) | 2021-07-15 | 2024-01-02 | Hoffmann-La Roche Inc. | Heterocyclic compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| GB201109763D0 (en) | 2011-07-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2012168733A1 (en) | Substituted 8 - amino - imidazo [1, 2-a] pyrazines as antibacterial agents | |
| US10526291B2 (en) | Potent dual BRD4-kinase inhibitors as cancer therapeutics | |
| US9381192B2 (en) | Indazole inhibitors of the Wnt signal pathway and therapeutic uses thereof | |
| CN104470902B (en) | N-(3-heteroarylaryl)-4-arylarylcarboxamtdes and analogs as hedgehog pathway inhibitors and use thereof | |
| US20200407328A1 (en) | Brd4-kinase inhibitors as cancer therapeutics | |
| JP5150878B2 (en) | Kinase inhibitor | |
| CN108290860B (en) | Iminotetrahydropyrimidinone derivatives as PLASMEPSIN V inhibitors | |
| JP5876503B2 (en) | Imidazopyridine as an antiviral agent for respiratory syncytial virus | |
| CN105189505B (en) | Tricyclic gyrase inhibitor as antibacterial agent | |
| JP2019508462A (en) | 4,6-Dihydropyrrolo [3,4-c] pyrazole-5 (1H) -carbonitrile derivatives for cancer treatment | |
| HUE035612T2 (en) | 1h-pyrazolo[3,4-b]pyridines and therapeutic uses thereof | |
| CA2890981A1 (en) | Amide-substituted heterocyclic compounds useful as modulators of il-12, il-23 and/or ifn.alpha. responses | |
| EP3143021A1 (en) | Pyrazolopyridines and pyrazolopyrimidines | |
| US11858897B2 (en) | Substituted 6,11-dihydro-5H-benzo[b]carbazoles as inhibitors of ALK and SRPK | |
| EP2888242B1 (en) | Substituted 4-pyridones and their use as inhibitors of neutrophil elastase activity | |
| KR20150031318A (en) | Inhibition of enzymes | |
| KR20190104632A (en) | Tricyclic gyrase inhibitors | |
| TW201625620A (en) | Heterocyclic hydroxamic acids as protein deacetylase inhibitors and dual protein deacetylase-protein kinase inhibitors and methods of use thereof | |
| CA3134814A1 (en) | Tyk2 pseudokinase ligands | |
| WO2017083756A1 (en) | Heterocyclic compounds for the treatment of disease | |
| CA2779951A1 (en) | Imidazopyridine derivatives | |
| TW200530245A (en) | Pyrrolo pyrimidine derivatives useful for treating proliferative diseases | |
| CN114907350B (en) | Nitrogen-containing condensed ring compound, preparation method and application | |
| JP7110335B2 (en) | Pyridoquinazoline derivatives useful as protein kinase inhibitors | |
| US20110015395A1 (en) | Pyrazole compounds with inhibitory activity against ros kinase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12726851 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12726851 Country of ref document: EP Kind code of ref document: A1 |