WO2012142237A1 - Fused imidazole derivatives useful as ido inhibitors - Google Patents
Fused imidazole derivatives useful as ido inhibitors Download PDFInfo
- Publication number
- WO2012142237A1 WO2012142237A1 PCT/US2012/033245 US2012033245W WO2012142237A1 WO 2012142237 A1 WO2012142237 A1 WO 2012142237A1 US 2012033245 W US2012033245 W US 2012033245W WO 2012142237 A1 WO2012142237 A1 WO 2012142237A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- imidazo
- isoindol
- ethyl
- cyclohexyl
- ethanol
- Prior art date
Links
- 0 CC1(*)C=CC(c(nc2)c[n]2[Tl])=C(C=O)C=C1 Chemical compound CC1(*)C=CC(c(nc2)c[n]2[Tl])=C(C=O)C=C1 0.000 description 5
- STOKPMKFLFMGTB-UHFFFAOYSA-N CC12C(F)=CC=CC1c1cnc[n]1C2CC(C(CC1)CCC1O)O Chemical compound CC12C(F)=CC=CC1c1cnc[n]1C2CC(C(CC1)CCC1O)O STOKPMKFLFMGTB-UHFFFAOYSA-N 0.000 description 1
- BZKQJSLASWRDNE-UHFFFAOYSA-N CCOC(C(CC1)CCC1O)=O Chemical compound CCOC(C(CC1)CCC1O)=O BZKQJSLASWRDNE-UHFFFAOYSA-N 0.000 description 1
- PFPLBJQGBUCUPK-UHFFFAOYSA-N CCOC(C(CC1)CCC1OC)=O Chemical compound CCOC(C(CC1)CCC1OC)=O PFPLBJQGBUCUPK-UHFFFAOYSA-N 0.000 description 1
- WRFXUJBZOJLWBB-UHFFFAOYSA-N CCOC(CC(c1ccccc1-1)[n]2c-1cnc2)=O Chemical compound CCOC(CC(c1ccccc1-1)[n]2c-1cnc2)=O WRFXUJBZOJLWBB-UHFFFAOYSA-N 0.000 description 1
- VJRHMPFUEKSHGV-ORLJMPNPSA-N CS(NCCNC([C@H](CC1)CC[C@@H]1C(CC(c1ccccc1-1)[n]2c-1cnc2)O)=O)(=O)=O Chemical compound CS(NCCNC([C@H](CC1)CC[C@@H]1C(CC(c1ccccc1-1)[n]2c-1cnc2)O)=O)(=O)=O VJRHMPFUEKSHGV-ORLJMPNPSA-N 0.000 description 1
- AVUZOXPRZPEVIP-SSCNNSSESA-N OC(CC(c1c-2cccc1)[n]1c-2cnc1)[C@H](CC1)CC[C@@H]1C(O)=O Chemical compound OC(CC(c1c-2cccc1)[n]1c-2cnc1)[C@H](CC1)CC[C@@H]1C(O)=O AVUZOXPRZPEVIP-SSCNNSSESA-N 0.000 description 1
- YTRRAUACYORZLX-UHFFFAOYSA-N OC(CC(c1ccccc1-1)[n]2c-1cnc2)C1CCCCC1 Chemical compound OC(CC(c1ccccc1-1)[n]2c-1cnc2)C1CCCCC1 YTRRAUACYORZLX-UHFFFAOYSA-N 0.000 description 1
- VUDZQMQFFPJPQH-UHFFFAOYSA-N OC(CC1)CCC1C(CC(c1c-2cccc1F)[n]1c-2cnc1)=O Chemical compound OC(CC1)CCC1C(CC(c1c-2cccc1F)[n]1c-2cnc1)=O VUDZQMQFFPJPQH-UHFFFAOYSA-N 0.000 description 1
- KECJSORXVGLSAH-UHFFFAOYSA-N OC(CCC(OC(CC(c1ccccc1-1)[n]2c-1cnc2)C1CCCCC1)=O)=O Chemical compound OC(CCC(OC(CC(c1ccccc1-1)[n]2c-1cnc2)C1CCCCC1)=O)=O KECJSORXVGLSAH-UHFFFAOYSA-N 0.000 description 1
- VQGLVXRSMONFQT-UHFFFAOYSA-N OCCC(c1ccccc1-1)[n]2c-1cnc2 Chemical compound OCCC(c1ccccc1-1)[n]2c-1cnc2 VQGLVXRSMONFQT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4188—1,3-Diazoles condensed with other heterocyclic ring systems, e.g. biotin, sorbinil
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
Definitions
- the present disclosure relates to compounds and methods for inhibition of indoleamine 2,3-dioxygenase; further the disclosure relates to method of treatment of diseases and disorders mediated by indoleamine 2,3-dioxygenase.
- Tryptophan is an essential amino acid required for the biosynthesis of proteins, niacin and the neurotransmitter 5-hydroxytryptamine (serotonin).
- the enzyme indoleamine 2,3-dioxygenase also known as INDO or IDO
- IDO indoleamine 2,3-dioxygenase
- IFN-y stimulation induces activation of IDO, which leads to a depletion of Trp, thereby arresting the growth of Trp-dependent intracellular pathogens such as Toxoplasma gondii and Chlamydia trachomatis.
- IDO activity also has an antiproliferative effect on many tumor cells, and IDO induction has been observed in vivo during rejection of allogeneic tumors, indicating a possible role for this enzyme in the tumor rejection process.
- IDO is involved in induction of immune tolerance.
- Studies of mammalian pregnancy, tumor resistance, chronic infections and autoimmune diseases have shown that cells expressing IDO can suppress T-cell responses and promote tolerance. Accelerated Trp catabolism has been observed in diseases and disorders associated with cellular immune activation, such as infection, malignancy, autoimmune diseases and AIDS, as well as during pregnancy. It was proposed that IDO is induced chronically by HIV infection, and is further increased by opportunistic infections, and that the chronic loss of Trp initiates mechanisms responsible for cachexia, dementia and diarrhea and possibly immunosuppression of AIDS patients (Brown, et ah, 1991, Adv. Exp. Med. Biol, 294: 425-35).
- IDO inhibition can enhance the levels of virus-specific T cells and, concomitantly, reduce the number of virally infected macrophages in a mouse model of HIV (Portula et ah, 2005, Blood, 106:2382-90).
- IDO is believed to play a role in the immunosuppressive processes that prevent fetal rejection in utero. More than 40 years ago, it was observed that, during pregnancy, the genetically disparate mammalian conceptus survives in spite of what would be predicted by tissue transplantation immunology (Medawar, 1953, Symp. Soc. Exp. Biol. 7: 320-38). Anatomic separation of mother and fetus and antigenic immaturity of the fetus cannot fully explain fetal allograft survival. Recent attention has focused on immunologic tolerance of the mother.
- the mammalian conceptus appears to suppress T-cell activity and defends itself against rejection, and blocking tryptophan catabolism during murine pregnancy allows maternal T cells to provoke fetal allograft rejection (Munn, et al, 1998, Science 281 : 1 191-3).
- IDO inhibitor 1-MT
- chemotherapeutic agents can synergize with chemotherapeutic agents to reduce tumor growth in mice, suggesting that IDO inhibition may also enhance the anti-tumor activity of conventional cytotoxic therapies (Muller et ah, 2005, Nature Med., 1 1 :312-9).
- One mechanism contributing to immunologic unresponsiveness toward tumors may be presentation of tumor antigens by tolerogenic host APCs.
- APCs human IDO-expressing antigen-presenting cells (APCs) that coexpressed CD 123 (IL3RA) and CCR6 and inhibited T-cell proliferation have also been described.
- TDLNs mouse tumor-draining lymph nodes
- pDCs plasmacytoid dendritic cells
- IDO degrades the indole moiety of tryptophan, serotonin and melatonin, and initiates the production of neuroactive and immunoregulatory metabolites, collectively known as kynurenines.
- kynurenines neuroactive and immunoregulatory metabolites
- IDO expressed by dendritic cells (DCs) can greatly affect T-cell proliferation and survival. IDO induction in DCs could be a common mechanism of deletional tolerance driven by regulatory T cells.
- tryptophan metabolism and kynurenine production might represent a crucial interface between the immune and nervous systems (Grohmann, et al, 2003, Trends Immunol, 24: 242-8).
- Small molecule inhibitors of IDO are being developed to treat or prevent IDO-related diseases such as those described above.
- PCT Publication WO 99/29310 reports methods for altering T cell-mediated immunity comprising altering local extracellular concentrations of tryptophan and tryptophan metabolites, using an inhibitor of IDO such as 1-methyl-DL-tryptophan, p-(3-benzofuranyl)-DL-alanine, p-[3-benzo(b)thienyl]-DL-alanine, and 6-nitro-L-tryptophan) (Munn, 1999).
- IDO Inhibitors of IDO can be used to activate T cells and therefore enhance T cell activation when the T cells are suppressed by pregnancy, malignancy or a virus such as HIV. Inhibition of IDO may also be an important treatment strategy for patients with neurological or neuropsychiatric diseases or disorders such as depression.
- the compounds, compositions and methods herein help meet the current need for IDO modulators.
- the invention com rises compounds according to the formula (I),
- R 1 , R 2 , n and a are each defined herein.
- the invention comprises compounds according to the formula (II),
- R 1 , R 3 , R c , and n are each defined herein.
- compositions comprising a pharmaceutically acceptable excipient, diluent, or carrier, and a compound according to formula (I) or (II).
- methods are provided for (a) modulating an activity of indoleamine 2,3-dioxygenase comprising contacting an indoleamine 2,3-dioxygenase with a modulation effective amount of a compound according to formula (I) or (II), or a pharmaceutical composition comprising a compound according to formula (I) or (II); (b) treating indoleamine 2,3-dioxygenase (IDO) mediated immunosuppression in a subject in need thereof, comprising administering an effective indoleamine 2,3-dioxygenase inhibiting amount of a compound according to formula (I) or (II), or a pharmaceutical composition comprising a compound according to formula (I) or (II); (c) treating a medical condition that benefits from the inhibition of
- Figure 1 shows the absolute configuration of a diasteromer of the HBr salt of compound 1417 as confirmed by X-ray crystallography.
- the invention provides com ounds of formula (I),
- bond a is a single or double bond
- n 0, 1 , 2, 3, or 4;
- each R 1 is independently halogen, cyano, nitro, Ci- 6 alkyl, -OR, -N(R) 2 , -SR, -C(0)OR, -C(0)N(R) 2 , -C(0)R, -S(0)R, -S(0)OR, -S(0)N(R) 2 , -S(0) 2 R, -S(0) 2 OR, -S(0) 2 N(R) 2 , -OC(0)R, -OC(0)OR, -OC(0)N(R) 2 , -N(R)C(0)R, -N(R)C(0)OR, or -N(R)C(0)N(R) 2 ;
- R 2 is -Ci_4alkyl-R A or -C 2 _4alkenyl-R 3 when bond a is a single bond;
- R B is hydrogen, Ci_ 6 alkyl, Ci_ 6 haloalkyl, -Ci_ 6 alkyl-R B1 , -C(0)R 3 , or -S(0) 2 R 3 , -C(0)(CH 2 ) ! . 4 COOR, -C(0)CH(NH 2 )(R D ), -S(0) 2 OR 3 , -S(0) 2 N(R 3 ) 2 , -CH 2 - OP(0) 2 (OR) 2 , or -P(0)(OR 3 ) 2 , wherein
- R B1 is cyano, nitro, d_ 6 alkyl, Ci_ 6 haloalkyl, -OR, -N(R) 2 , -SR, -C(0)OR, -C(0)N(R) 2 , -C(0)R, -S(0)R, -S(0)OR, -S(0)N(R) 2 , -S(0) 2 R, -S(0) 2 OR, -S(0) 2 N(R) 2 , -OC(0)R, -OC(0)OR, -OC(0)N(R) 2 , -N(R)C(0)R, -N(R)C(0)OR, or -N(R)C(0)N(R) 2 ;
- R D is hydrogen, methyl, -CH(CH 3 ) 2 , -CH 2 CH(CH 3 ) 2 , -CH(CH 3 )(CH 2 CH 3 ), benzyl, 4-hydroxybenzyl, -CH 2 (3-indolyl), -CH 2 SH
- each R 3 is independently hydrogen, Ci- 6 alkyl, aryl, heteroaryl, C 3 _ 8 cycloalkyl, C 3 _ 8 cycloalkenyl, 3- 10 membered heterocyclyl, arylCi- 6 alkyl-, heteroarylCi-6 alkyl-, C 3 _ 8 cycloalkylCi_ 6 alkyl-, C 3 _ 8 cycloalkenylCi_ 6 alkyl-, or (3- 10 membered heterocyclyl)Ci_ 6 alkyl-,
- aryl, heteroaryl, arylCi_ 6 alkyl-, and heteroarylCi_ 6 alkyl- groups are each optionally substituted by one, two, three, or four R 31 groups;
- each R 31 is independently halogen, cyano, nitro, Ci- 6 alkyl, -Ci_ 6 alkyl-R 33 , Ci_ 6 haloalkyl, -OR, -N(R) 2 , -SR, -C(0)OR, -C(0)N(R) 2 , -C(0)R,- -S(0)R, -S(0)OR, -S(0)N(R) 2 , -S(0) 2 R, -S(0) 2 OR, -S(0) 2 N(R) 2 , -OC(0)R, -OC(0)OR, -OC(0)N(R) 2 , -N(R)C(0)R, -N(R)C(0)OR, -N(R)C(0)N(R) 2 , wherein
- R 33 is cyano, -OR, -N(R) 2 , -SR, -C(0)OR, -C(0)N(R) 2 , -C(0)R, -S(0)R, -S(0)OR, -S(0)N(R) 2 , -S(0) 2 R, -S(0) 2 OR, -S(0) 2 N(R) 2 , -OC(0)R, -OC(0)OR, -OC(0)N(R) 2 , -N(R)C(0)R, -N(R)C(0)OR, or -N(R)C(0)N(R) 2 ;
- each R 34 is independently hydrogen, halogen, Ci_ 6 alkyl,
- R c is hydrogen or Ci- 6 alkyl
- each R is independently hydrogen or R , wherein
- R 10 is Ci- 6 alkyl, aryl, heteroaryl, C3- 8 cycloalkyl, C3_ 8 cycloalkenyl, 3-10 membered heterocyclyl, arylCi- 6 alkyl, heteroarylCi_ 6 aikyl-, C3-8 cycloalkylCi- 6 alkyl-, C3- 8 cycloalkenylCi_ 6 alkyl-, or (3-10 membered heterocyclyl)Ci_ 6 alkyl-, each R 10 optionally substituted by one, two, three, or four groups that are each independently halogen, cyano, nitro, Ci_ 6 alkyl, Ci_ 6 haloalkyl, -OR 11 , -N(R U ) 2 , -SR 11 , -C(0)OR n , -C(0)N(R n ) 2 , -C(0)R u , -S(0)R n , -S(0)OR n
- R B is additionally -C(0)N(H)R 3 or -C(0)(CH 2 )!_ 4 (NR)COOR;
- R 3 is additionally (heteroaryl)-(3-10 membered heterocyclyl)-,
- R 34 is additionally cyano or - Ci- 6 alkyl-OR; and/or
- R 10 is additionally optionally substituted by -N(R u )S(0)2R n or -C(O)-(3-10 membered heterocyclyl);
- the invention further comprises subgenera of formula (I) and formula ( ⁇ ) in which the substituents are selected as any and all combinations of one or more of structural formula
- Structural Formula I is one of formulae (la) - (Ih);
- n and R 1 are selected from one of the following groups (la) - (lu);
- n is 1, 2, 3, or 4, and each R 1 is as defined for formula (I).
- (lb) n is 0, 1, 2, or 3, and each R 1 is as defined for formula (I).
- n 0, 1, or 2 and each R 1 is independently halogen, -OR, -N(R) 2 , or -SR.
- n 0, 1, or 2 and each R 1 is independently halogen, -OR 0 , -N(R°) 2 , or -SR°, wherein each R° is independently hydrogen or Ci- 6 alkyl.
- n is 0, 1, or 2 and each R 1 is independently fluoro, chloro, hydroxy, or methoxy.
- n is 0, 1, or 2 and each R 1 is independently halogen,
- (lh) n is 0, 1, or 2 and each R 1 is independently fluoro or chloro.
- n 0 or 1 and R 1 is as defined for formula (I),
- (lj) n is 0 or 1 and R 1 is halogen, -OR, -N(R) 2 , or -SR.
- (Ik) n is 0 or 1 and R 1 is halogen, -OR 0 , -N(R°) 2 , or -SR°;wherein each R° is independently hydrogen or Ci_ 6 alkyl.
- n is 0 or 1 and R 1 is fluoro, chloro, hydroxy, or methoxy.
- n 0 or 1 and R 1 is fluoro or chloro.
- (lp) n is 1 and R 1 is halogen, -OR, -N(R) 2 , or -SR;
- n 1 and R 1 is halogen, -OR 0 , -N(R°) 2 , or -SR°;wherein each R° is independently hydrogen or Ci_ 6 alkyl.
- (lr) n is 1 and R 1 is fluoro, chloro, hydroxy, or methoxy.
- R 2 is selected from one of the following groups (2a) - (21);
- R 2 is -Ci_ 4 alkyl-R A .
- R 2 is -Ci_ 2 alkyl-R A .
- R 2 is -CH 2 -R A , -CH 2 CH 2 -R A , or -C(H)(CH 3 )CH 2 -R A .
- R 2 is -CH 2 -R A .
- R 2 is -CH 2 CH 2 -R A .
- R 2 is -C(H)(CH 3 )CH 2 -R A
- R A is selected from one of the following groups (3a) - (3n);
- R A is -CN, -C(0)OR 3 , or -C(0)N(R 3 )(R c ).
- R A is -C(0)R 3 or -C(OR B )(R 3 )(R c ).
- R A is -C( HR B )(R 3 )(R C ), wherein R B is hydrogen, C 1-6 alkyl, or -C(0)Ci_ 6 alkyl.
- R A is -C( H 2 )(R 3 )(R C ).
- R A is -C(0)OR 3 .
- R A is -C(0)N(R 3 )(R c ).
- R A is -C(0)R 3 .
- R A is -C(OR B )(R 3 )(R c ).
- R A is -C(OH)(R 3 )(R c ).
- R A is -CH(OH)(R 3 ).
- R A is -CN, -C(0)R 3 , -C(0)OR 3 , -C(0)N(R 3 )(R c ), -C(OR B )(R 3 )(R c ),
- R A is -C(0)R 3 or -C(OR B )(R 3 )(R c ), wherein R B is hydrogen and R c is hydrogen or
- R A is -C(OR B )(R 3 )(R c ), wherein R B is hydrogen and R c is hydrogen or Ci- 6 alkyl.
- R B is selected from one of the following groups (4a) - (4k);
- R B is hydrogen, Ci_ 6 alkyl, Ci_ 6 haloalkyl, -Ci_ 6 alkyl-R B1 , -C(0)(CH 2 ) ! _ 4 COOR B2 , -C(0)C(NH 2 )R D , -P(0 3 )(R B2 ) 2 , -CH 2 -OP(0) 2 (OR) 2 , wherein R D is the side chain of natural alpha amino acids , -C(0)R 3 , or -S(0) 2 R 3 , wherein R B1 is cyano, nitro, Ci_ 6 alkyl, Ci_ 6 haloalkyl, -OR B2 , -N(R B2 ) 2 , -SR B2 , -C(0)OR B2 , -C(0)N(R B2 ) 2 , -C(0)R B2 , -S(0)R B2 , -S(0)OR B2 , -S(0)N(R B2 ,
- R B is hydrogen, Ci_ 6 alkyl, Ci_ 6 haloalkyl, -Ci_ 6 alkyl-R B1 , -C(0)R 3 , or -S(0) 2 R 3 , wherein R B1 is cyano, nitro, Ci_ 6 alkyl, Ci_ 6 haloalkyl, -OR B2 , -N(R B2 ) 2 , -SR B2 , -C(0)OR B2 , -C(0)N(R B2 ) 2 , -C(0)R B2 , -S(0)R B2 , -S(0)OR B2 , -S(0)N(R B2 ) 2 , -S(0) 2 R B2 , -S(0) 2 OR B2 , -S(0) 2 N(R B2 ) 2 , -OC(0)R B2 , -OC(0)OR B2 , -OC(0)N(R B2 ) 2 , -N(R B2 )C ,
- R B is hydrogen, Ci_ 6 alkyl, or -Ci- 6 alkyl-R B1 , wherein R B1 is cyano, nitro, Ci_ 6 alkyl, Ci_ 6 haloalkyl, -OR B2 , -N(R B2 ) 2 , -SR B2 , -C(0)OR B2 , -C(0)N(R B2 ) 2 , -C(0)R B2 , -S(0)R B2 , -S(0)OR B2 , -S(0)N(R B2 ) 2 , -S(0) 2 R B2 , -S(0) 2 OR B2 , -S(0) 2 N(R B2 ) 2 , -OC(0)R B2 , -OC(0)OR B2 , -OC(0)N(R B2 ) 2 , -N(R B2 )C(0)R B2 , -N(R B2 )C(0)OR B2 , or -N(
- R B is hydrogen, Ci- 6 alkyl, or -Ci- 6 alkyl-R B1 , wherein R B1 is cyano,
- each R B2 is independently hydrogen or Ci- 6 alkyl.
- R B is -Ci_ 6 alkyl-R B1 , wherein R B1 is cyano, -C(0)OR B2 , -C(0)N(R B2 ) 2 , -C(0)R B2 ,
- each R B2 is independently hydrogen or Ci- 6 alkyl.
- R B is hydrogen, Ci- 6 alkyl, Ci- 6 haloalkyl, -Ci- 6 alkyl-R B1 , -C(0)R B2 , or -S(0) 2 R B2 , wherein R B1 is -C(0)OR B3 , -C(0)N(R B3 ) 2 , -S(0) 2 OR B3 , or -S(0) 2 N(R 3 ) 2 , R B2 is Ci_ 6 alkyl; and R B3 is hydrogen or Ci_6 alkyl.
- R B is hydrogen, Ci- 6 alkyl, or Ci_ 6 haloalkyl.
- R B is hydrogen or Ci_ 6 alkyl
- R B is hydrogen
- R B is d-ealkyl
- R B is hydrogen, -C(0)R B2 , -C(0)(CH 2 ) ! _ 4 COOR B2 , -C(0)C( H 2 )R D , -P(0)(OR B2 ) 2 , -CH 2 -OP(0) 2 (OR) 2 , -S(0) 2 R B2 , -C(0)N(R B2 ) 2 , -S(0) 2 OR B2 , -S(0) 2 N(R 3 ) 2 , wherein and R B2 is hydrogen or Ci_6 alkyl.
- R c is selected from one of the following groups (5a) - (5g);
- R c is hydrogen or Ci_ 4 alkyl.
- R c is hydrogen or Ci- 2 alkyl.
- R is hydrogen or methyl.
- R c is hydrogen
- R c is Ci_ 6 alkyl.
- R c is Ci_ 4 alkyl.
- R 3 is selected from one of the following groups (6a) - (6z);
- R 3 is phenyl or a five or six membered heteroaryl, each optionally substituted by one or two R 31 groups.
- R 3 is , wherein bond a is a single bond or a double bond; m is 0, 1, or 2; p is 0 or 1 ; and wherein
- R 35 is hydrogen, Ci_ 6 alkyl, -C(0)R, -S(0) 2 R, -C(0)OR, -C(0)N(R) 2 , -S(0) 2 OR, or -S(0) 2 N(R) 2 ;
- R 35 is hydrogen, Ci_ 6 alkyl, -C(0)R, -S(0) 2 R, -C(0)OR, -C(0)N(R) 2 , -S(0) 2 OR, or -S(0) 2 N(R) 2 ;
- R 3 is hydrogen, Ci- 6 alkyl, aryl, heteroaryl, C3_ 8 cycloalkyl, C3_ 8 cycloalkenyl, 3-10 membered heterocyclyl, or C3_ 8 cycloalkylCi- 6 alkyl, wherein
- the aryl and heteroaryl groups are each optionally substituted by one or two R 31 groups;
- each R 31 is independently halogen, cyano, nitro, Ci- 6 alkyl, -Ci_ 6 alkyl-R 33 , Ci_ 6 haloalkyl, -OR, -N(R) 2 , -SR, -C(0)OR, -C(0)N(R) 2 , -C(0)R, -S(0)R, -S(0)OR, -S(0)N(R) 2 , -S(0) 2 R, -S(0) 2 OR, -S(0) 2 N(R) 2 , -OC(0)R, -OC(0)OR, -OC(0)N(R) 2 , -N(R)C(0)R, -N(R)C(0)OR, -N(R)C(0)N(R) 2 , wherein R 33 is -OR, -N(R) 2 , or -SR; and
- R 3 is hydrogen, Ci_ 6 alkyl, aryl, heteroaryl, C3_ 8 cycloalkyl, C3_ 8 cycloalkenyl, 3-10 membered heterocyclyl, or C3_ 8 cycloalkylCi_ 6 alkyl-, wherein
- the aryl and heteroaryl groups are each optionally substituted by one or two R 31 groups;
- each R is independently halogen, cyano, nitro, Ci- 6 alkyl, -Ci- 6 alkyl-R , Ci_ 6 haloalkyl, -OR, -N(R) 2 , -SR, -C(0)OR, -C(0)N(R) 2 , -C(0)R, -S(0)R, -S(0)OR, -S(0)N(R) 2 , -S(0) 2 R, -S(0) 2 OR, -S(0) 2 N(R) 2 , -OC(0)R, -OC(0)OR, -OC(0)N(R) 2 , -N(R)C(0)R, -N(R)C(0)OR, -N(R)C(0)N(R) 2 , wherein R 33 is -OR, -N(R) 2 , or -SR; and
- the aryl and heteroaryl are each optionally substituted by one, two, three, or four R 31 groups.
- R 3 is phenyl, cyclopentyl, cyclohexyl, cyclohex-l-en-l-yl, cyclohex-3-en-l-yl, furan-
- each R is independently hydrogen, Ci_ 6 alkyl, Ci- 6 haloalkyl, aryl, heteroaryl, C3_ 8 cycloalkyl, C3_ 8 cycloalkenyl, 3-10 membered heterocyclyl, arylCi- 6 alkyl, heteroarylCi_ 6 alkyl-, C3-8 cycloalkylCi- 6 alkyl-, C 3 _ 8 cycloalkenylCi_ 6 alkyl-, or (3-10 membered heterocyclyl)Ci_ 6 alkyl-.
- each R is independently hydrogen, Ci_ 6 alkyl, Ci- 6 haloalkyl, phenyl, 5- or 6-membered heteroaryl, C3_ 8 cycloalkyl, C3_ 8 cycloalkenyl,
- Particular embodiments of this aspect of the invention include compounds of any one of the formulae (I), ( ⁇ ), and (la) - (Id), each as defined in each of the following rows, wherein each entry is a group number as defined above (e.g., (Is) refers to n is 1 and each R 1 is halogen), and a dash "-" indicates that the variable is as defined for formula (I) or ( ⁇ ) or defined according to any one of the applicable variable definitions (la)-(6z) [e.g., when R c is a dash, it can be either as defined for Formula (I) or ( ⁇ ) or any one of definitions (5a)-(5g)] :
- the invention rovides the compound according to formula (II),
- n 0 or 1 ;
- each R 1 is independently halogen, -OR, -N(R) 2 , or -SR;
- the aryl and heteroaryl groups are each optionally substituted by one or two R 31 groups;
- each R 31 is independently halogen, cyano, nitro, Ci- 6 alkyl,
- -Ci_ 6 alkyl-R C 1-6 haloalkyl, -OR, -N(R) 2 , -SR, -C(0)OR, -C(0)N(R) 2 , -C(0)R, -S(0)R, -S(0)OR, -S(0)N(R) 2 , -S(0) 2 R, -S(0) 2 OR, -S(0) 2 N(R) 2 , -OC(0)R, -OC(0)OR, -OC(0)N(R) 2 , -N(R)C(0)R, -N(R)C(0)OR, -N(R)C(0)N(R) 2 , wherein R 33 is -OR, -N(R) 2 , or -SR;
- R is hydrogen or Ci_ 6 alkyl
- each R is independently hydrogen or R 10 , wherein
- R 10 is Ci- 6 alkyl, aryl, heteroaryl, C3_ 8 cycloalkyl, C3_ 8 cycloalkenyl, 3-10 membered heterocyclyl, arylCi- 6 alkyl, heteroarylCi- 6 alkyl-, C3-8 cycloalkylCi- 6 alkyl-, C3- 8 cycloalkenylCi_ 6 alkyl-, or (3-10 membered heterocyclyl)Ci_ 6 alkyl-, each R 10 optionally substituted by one, two, three, or four groups that are each independently halogen, cyano, nitro, C 1-6 alkyl, Ci_ 6 haloalkyl, -OR 11 , -N(R U ) 2 , -SR 11 , -C(0)OR n , -C(0)N(R n ) 2 , -C(0)R u , -S(0)R n , -S(0)OR n ,
- the compounds of formula (II) further include those compounds where,
- R 3 is additionally (heteroaryl)-(3-10 membered heterocyclyl)-;
- R 34 is additionally cyano or - Ci- 6 alkyl-OR; and/or
- R 10 is additionally optionally substituted by -N(R u )S(0) 2 R n or -C(O)-(3-10 membered heterocyclyl);
- the invention further comprises subgenera of formula (II) or ( ⁇ ) in which the substituents are selected as any and all combinations of one or more of structural formula (II), n, R 1 , R 3 , and R c as defined herein, including without limitation, the following:
- Structural Formula II is on f formulae (Ila) - (lid):
- n and R 1 are selected from one of the following groups (7a) - (7i):
- n is 0 or 1 and R 1 is halogen, -OR 0 , -N(R°) 2 , or -SR°; wherein each R° is independently hydrogen or C h alky 1.
- n is 0 or 1 and R 1 is fluoro, chloro, hydroxy, or methoxy.
- n is 0 or 1 and R 1 is halogen.
- n is 0 or 1 and R 1 is fluoro or chloro.
- n is 1 and R 1 is halogen, -OR 0 , -N(R°) 2 , or -SR°; wherein each R° is independently hydrogen or
- n is 1 and R 1 is fluoro, chloro, hydroxy, or methoxy.
- n 1 and R 1 is fluoro or chloro.
- R c is selected from one of the following groups (8a) - (8g):
- R c is hydrogen or Ci- 4 alkyl.
- R c is hydrogen or
- R c is hydrogen or methyl.
- R c is hydrogen
- R c is Ci-ealkyl.
- R is Ci-4 lkyl.
- R 3 is selected from one of the following groups (9a) - (9x);
- R 3 is phenyl or a five or six membered heteroaryl, each optionally substituted by one or two R 31 groups.
- R 3 is , wherein bond a is a single bond or a double bond; m is 0, 1, or 2; p is 0 or 1 ; and wherein
- R 35 is hydrogen, Ci_ 6 alkyl, -C(0)R, -S(0) 2 R, -C(0)OR, -C(0)N(R) 2 , -S(0) 2 OR, or -S(0) 2 N(R) 2 ;
- R 35 is hydrogen, Ci_ 6 alkyl, -C(0)R, -S(0) 2 R, -C(0)OR, -C(0)N(R) 2 , -S(0) 2 OR, or -S(0) 2 N(R) 2 ;
- each R 36 is independently hydrogen, halogen, Ci_ 6 alkyl,
- the aryl and heteroaryl groups are each optionally substituted by one or two R 31 groups;
- each R 31 is independently halogen, cyano, nitro, Ci_ 6 alkyl, -Ci_ 6 alkyl-R 33 , Ci_ 6 haloalkyl, -OR, -N(R) 2 , -SR, -C(0)OR, -C(0)N(R) 2 , -C(0)R, -S(0)R, -S(0)OR, -S(0)N(R) 2 , -S(0) 2 R, -S(0) 2 OR, -S(0) 2 N(R) 2 , -OC(0)R, -OC(0)OR, -OC(0)N(R) 2 , -N(R)C(0)R, -N(R)C(0)OR, -N(R)C(0)N(R) 2 , wherein R 33 is -OR, -N(R) 2 , or -SR; and
- the aryl and heteroaryl are each optionally substituted by one, two, three, or four R 31 groups.
- each R is independently hydrogen, Ci_ 6 alkyl, Ci- 6 haloalkyl, aryl, heteroaryl, C3_ 8 cycloalkyl, C3_ 8 cycloalkenyl, 3-10 membered heterocyclyl, arylCi- 6 alkyl, heteroarylCi_ 6 alkyl-, C3-8 cycloalkylCi- 6 alkyl-, C3- 8 cycloalkenylCi_ 6 alkyl-, or (3-10 membered heterocyclyl)Ci_ 6 alkyl-.
- each R is independently hydrogen, Ci- 6 alkyl, Ci_ 6 haloalkyl, phenyl, 5- or 6-membered heteroaryl, C3_ 8 cycloalkyl, C3_ 8 cycloalkenyl, 3-8 membered heterocyclyl, benzyl, (5- or 6-membered heteroaryl)Ci_ 6 alkyl-, C3-8 cycloalkylCi_ 6 alkyl-, C3- 8 cycloalkenylCi- 6 alkyl-, or (3-8 membered heterocyclyl)Ci- 6 alkyl-.
- Particular embodiments of this aspect of the invention include compounds of any one of the formulae (II), (IF), and (Ha) - (Ild), each as defined in each of the following rows, wherein each entry is a group number as defined above and a dash "-" indicates that the variable is as defined for formula (II), or ( ⁇ ), or defined according to any one of the applicable variable definitions (7a)-(9t) [e.g., when R c is a dash, it can be either as defined for Formula (II), or (IF), or any one of definitions (8a)-(8g)]:
- the present disclosure provides compounds that are 1 - -methyl-
- the present disclosure provides compounds and pharmaceutical compositions comprising the compounds according to any one of the preceding aspects of the invention or any embodiment thereof, together with a pharmaceutically acceptable excipient, diluent, or carrier.
- the invention provides methods for treating indoleamine 2,3-dioxygenase (IDO) mediated immunosuppression in a subject in need thereof, comprising administering an effective indoleamine 2,3-dioxygenase inhibiting amount of a compound or a pharmaceutical composition according to any of the preceding aspects of the invention or any embodiment thereof.
- IDO indoleamine 2,3-dioxygenase
- the immunosuppression is associated with an infectious disease, or cancer.
- the immunosuppression is associated with an infectious disease and the infectious disease is a viral infection selected from the group consisting of: hepatitis C virus (HCV), human papilloma virus (HPV), cytomegalovirus (CMV), Epstein-Barr virus (EBV), poliovirus, varicella zoster virus, coxsackie virus, human immunodeficiency virus (HIV).
- HCV hepatitis C virus
- HPV human papilloma virus
- CMV cytomegalovirus
- EBV Epstein-Barr virus
- poliovirus varicella zoster virus
- coxsackie virus coxsackie virus
- human immunodeficiency virus HCV
- the immunosuppression is immunosupression associated with HIV-1 infection.
- the immunosuppression is associated with a cancer.
- the immunosuppression is tumor-specific immunosuppression associated with cancer.
- the immunosuppression is associated with a cancer, wherein the cancer is colon, pancreas, breast, prostate, lung, brain, ovary, cervix, testes, renal, head, or neck cancer, or lymphoma, leukemia, or melanoma.
- the invention provides the use of compounds described by any one of the preceding aspects (and any embodiment thereof), as defined above, for the preparation of a medicament for the treatment of medical conditions that benefit from the inhibition of enzymatic activity of indoleamine-2,3-dioxygenase.
- Medical conditions contemplated in this aspect include all the conditions described herein.
- the invention provides a use of compounds described by any one of the preceding aspects (and any embodiment thereof), as defined above, for the preparation of a medicament to stimulate T cell proliferation or to reverse an immunologic state of anergy or immunosuppression.
- the anergy or immunosuppression is caused by expression of the enzyme indoleamine-2,3 -dioxygenase.
- the invention provides the use of compounds described by any one of the preceding aspects (and any embodiment thereof), as defined above, for the preparation of a medicament for the treatment of immunosuppression associated with cancer, infectious diseases, or viral infections.
- the invention provides the use of compounds described in to any one of the preceding aspects (and any embodiment thereof), as defined above, for the preparation of a medicament for the treatment of tumor-specific immunosuppression associated with cancer.
- the cancer is cancer of the colon, pancreas, breast, prostate, lung, brain, ovary, cervix, testes, renal, or head and neck, lymphoma, leukemia, melanoma, and the like.
- the invention provides the use of compounds described in any of the preceding aspects (and any embodiment thereof), as defined above, and embodiments thereof as defined above, for the preparation of a medicament for the treatment of infectious diseases where the infectious disease is a viral infection.
- the viral infection is selected from the group consisting of: influenza, hepatitis C virus (HCV), human papilloma virus (HPV), cytomegalovirus (CMV), Epstein-Barr virus (EBV), varicella zoster virus, poliovirus, coxsackie virus, and human immunodeficiency virus (HIV). More preferably, the viral infection is human immunodeficiency virus (HIV).
- alkyl group can be both a monovalent radical or divalent radical; in the latter case, it would be apparent to one skilled in the art that an additional hydrogen atom is removed from a monovalent alkyl radical to provide a suitable divalent moiety.
- alkenyl as used herein, means a straight or branched chain hydrocarbon containing from 2 to 10 carbons, unless otherwise specified, and containing at least one carbon-carbon double bond.
- alkenyl include, but are not limited to, ethenyl, 2-propenyl, 2-methyl-2-propenyl, 3-butenyl, 4-pentenyl, 5-hexenyl, 2-heptenyl, 2-methyl-l-heptenyl, 3-decenyl, and 3,7-dimethylocta-2,6-dienyl.
- alkoxy means an alkyl group, as defined herein, appended to the parent molecular moiety through an oxygen atom.
- Representative examples of alkoxy include, but are not limited to, methoxy, ethoxy, propoxy, 2-propoxy, butoxy, tert-butoxy, pentyloxy, and hexyloxy.
- alkyl as used herein, means a straight or branched chain hydrocarbon containing from 1 to 10 carbon atoms, unless otherwise specified.
- Representative examples of alkyl include, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec -butyl, iso-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, 3-methylhexyl,
- alkyl group is a linking group between two other moieties, then it may also be a straight or branched chain; examples include, but are not limited to -CH 2 -, -CH 2 CH 2 -, -CH 2 CH 2 CHC(CH 3 )-, -CH 2 CH(CH 2 CH 3 )CH 2 -.
- aryl means a phenyl (i.e., monocyclic aryl), or a bicyclic ring system containing at least one phenyl ring or an aromatic bicyclic ring containing only carbon atoms in the aromatic bicyclic ring system.
- the bicyclic aryl can be azulenyl, naphthyl, or a phenyl fused to a monocyclic cycloalkyl, a monocyclic cycloalkenyl, or a monocyclic heterocyclyl.
- the bicyclic aryl is attached to the parent molecular moiety through any carbon atom contained within the phenyl portion of the bicyclic system, or any carbon atom with the napthyl or azulenyl ring.
- the fused monocyclic cycloalkyl or monocyclic heterocyclyl portions of the bicyclic aryl are optionally substituted with one or two oxo and/or thia groups.
- bicyclic aryls include, but are not limited to, azulenyl, naphthyl, dihydroinden-l-yl, dihydroinden-2-yl, dihydroinden-3-yl, dihydroinden-4-yl, 2,3-dihydroindol-4-yl, 2,3-dihydroindol-5-yl, 2,3-dihydroindol-6-yl,
- the bicyclic aryl is (i) naphthyl or (ii) a phenyl ring fused to either a 5 or 6 membered monocyclic cycloalkyl, a 5 or 6 membered monocyclic cycloalkenyl, or a 5 or 6 membered monocyclic heterocyclyl, wherein the fused cycloalkyl, cycloalkenyl, and heterocyclyl groups are optionally substituted with one or two groups which are independently oxo or thia.
- arylalkyl means an aryl group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- Representative examples of arylalkyl include, but are not limited to, benzyl, 2-phenylethyl, 3-phenylpropyl, and 2-naphth-2-ylethyl.
- cycloalkyl as used herein, means a monocyclic or a bicyclic cycloalkyl ring system.
- Monocyclic ring systems are cyclic hydrocarbon groups containing from 3 to 8 carbon atoms, where such groups can be saturated or unsaturated, but not aromatic. In certain embodiments, cycloalkyl groups are fully saturated. Examples of monocyclic cycloalkyls include cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cycloheptyl, and cyclooctyl.
- Bicyclic cycloalkyl ring systems are bridged monocyclic rings or fused bicyclic rings.
- Bridged monocyclic rings contain a monocyclic cycloalkyl ring where two non-adjacent carbon atoms of the monocyclic ring are linked by an alkylene bridge of between one and three additional carbon atoms (i.e., a bridging group of the form -(CH 2 )w-, where w is 1, 2, or 3).
- bicyclic ring systems include, but are not limited to, bicyclo[3.1.1]heptane, bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane, bicyclo[3.2.2]nonane, bicyclo[3.3.1]nonane, and bicyclo[4.2.1]nonane.
- Fused bicyclic cycloalkyl ring systems contain a monocyclic cycloalkyl ring fused to either a phenyl, a monocyclic cycloalkyl, a monocyclic cycloalkenyl, a monocyclic heterocyclyl, or a monocyclic heteroaryl.
- the bridged or fused bicyclic cycloalkyl is attached to the parent molecular moiety through any carbon atom contained within the monocyclic cycloalkyl ring.
- Cycloalkyl groups are optionally substituted with one or two groups which are independently oxo or thia.
- the fused bicyclic cycloalkyl is a 5 or 6 membered monocyclic cycloalkyl ring fused to either a phenyl ring, a 5 or 6 membered monocyclic cycloalkyl, a 5 or 6 membered monocyclic cycloalkenyl, a 5 or 6 membered monocyclic heterocyclyl, or a 5 or 6 membered monocyclic heteroaryl, wherein the fused bicyclic cycloalkyl is optionally substituted by one or two groups which are independently oxo or thia.
- Cycloalkenyl refers to a monocyclic or a bicyclic cycloalkenyl ring system.
- Monocyclic ring systems are cyclic hydrocarbon groups containing from 3 to 8 carbon atoms, where such groups are unsaturated (i.e., containing at least one annular carbon-carbon double bond), but not aromatic. Examples of monocyclic ring systems include cyclopentenyl and cyclohexenyl.
- Bicyclic cycloalkenyl rings are bridged monocyclic rings or a fused bicyclic rings.
- Bridged monocyclic rings contain a monocyclic cycloalkenyl ring where two non-adjacent carbon atoms of the monocyclic ring are linked by an alkylene bridge of between one and three additional carbon atoms (i.e., a bridging group of the form -(CH 2 ) W -, where w is 1, 2, or 3).
- alkylene bridge of between one and three additional carbon atoms
- bicyclic cycloalkenyls include, but are not limited to, norbornenyl and bicyclo[2.2.2]oct-2-enyl.
- Fused bicyclic cycloalkenyl ring systems contain a monocyclic cycloalkenyl ring fused to either a phenyl, a monocyclic cycloalkyl, a monocyclic cycloalkenyl, a monocyclic heterocyclyl, or a monocyclic heteroaryl.
- the bridged or fused bicyclic cycloalkenyl is attached to the parent molecular moiety through any carbon atom contained within the monocyclic cycloalkenyl ring.
- Cycloalkenyl groups are optionally substituted with one or two groups which are independently oxo or thia.
- halo or halogen as used herein, means -CI, -Br, -I or -F.
- haloalkyl means at least one halogen, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- Representative examples of haloalkyl include, but are not limited to, chloromethyl, 2-fluoroethyl, trifluoromethyl, pentafluoroethyl, and 2-chloro-3-fluoropentyl.
- heteroaryl means a monocyclic heteroaryl or a bicyclic ring system containing at least one heteroaromatic ring.
- the monocyclic heteroaryl can be a 5 or 6 membered ring.
- the 5 membered ring consists of two double bonds and one, two, three or four nitrogen atoms and optionally one oxygen or sulfur atom.
- the 6 membered ring consists of three double bonds and one, two, three or four nitrogen atoms.
- the 5 or 6 membered heteroaryl is connected to the parent molecular moiety through any carbon atom or any nitrogen atom contained within the heteroaryl.
- monocyclic heteroaryl include, but are not limited to, furyl, imidazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, oxazolyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, pyrazolyl, pyrrolyl, tetrazolyl, thiadiazolyl, thiazolyl, thienyl, triazolyl, and triazinyl.
- the bicyclic heteroaryl consists of a monocyclic heteroaryl fused to a phenyl, a monocyclic cycloalkyl, a monocyclic cycloalkenyl, a monocyclic heterocyclyl, or a monocyclic heteroaryl.
- the fused cycloalkyl or heterocyclyl portion of the bicyclic heteroaryl group is optionally substituted with one or two groups which are independently oxo or thia.
- the bicyclic heteroaryl contains a fused cycloalkyl, cycloalkenyl, or heterocyclyl ring
- the bicyclic heteroaryl group is connected to the parent molecular moiety through any carbon or nitrogen atom contained within the monocyclic heteroaryl portion of the bicyclic ring system.
- the bicyclic heteroaryl is a monocyclic heteroaryl fused to a phenyl ring or a monocyclic heteroaryl, then the bicyclic heteroaryl group is connected to the parent molecular moiety through any carbon atom or nitrogen atom within the bicyclic ring system.
- bicyclic heteroaryl include, but are not limited to, benzimidazolyl, benzofuranyl, benzothienyl, benzoxadiazolyl, benzoxathiadiazolyl, benzothiazolyl, cinnolinyl, 5,6-dihydroquinolin-2-yl,
- the fused bicyclic heteroaryl is a 5 or 6 membered monocyclic heteroaryl ring fused to either a phenyl ring, a 5 or 6 membered monocyclic cycloalkyl, a 5 or 6 membered monocyclic cycloalkenyl, a 5 or 6 membered monocyclic heterocyclyl, or a 5 or 6 membered monocyclic heteroaryl, wherein the fused cycloalkyl, cycloalkenyl, and heterocyclyl groups are optionally substituted with one or two groups which are independently oxo or thia.
- heteroarylalkyl and "-alkylheteroaryl” as used herein, means a heteroaryl, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- heteroarylalkyl include, but are not limited to, fur-3-ylmethyl, lH-imidazol-2-ylmethyl, lH-imidazol-4-ylmethyl, l-(pyridin-4-yl)ethyl, pyridin-3-ylmethyl, pyridin-4-ylmethyl, pyrimidin-5-ylmethyl, 2-(pyrimidin-2-yl)propyl, thien-2-ylmethyl, and thien-3-ylmethyl.
- heterocyclyl as used herein, means a monocyclic heterocycle or a bicyclic heterocycle.
- the monocyclic heterocycle is a 3, 4, 5, 6 or 7 membered ring containing at least one heteroatom independently selected from the group consisting of O, N, and S where the ring is saturated or unsaturated, but not aromatic.
- the 3 or 4 membered ring contains 1 heteroatom selected from the group consisting of O, N and S.
- the 5 membered ring can contain zero or one double bond and one, two or three heteroatoms selected from the group consisting of O, N and S.
- the 6 or 7 membered ring contains zero, one or two double bonds and one, two or three heteroatoms selected from the group consisting of O, N and S.
- the monocyclic heterocycle is connected to the parent molecular moiety through any carbon atom or any nitrogen atom contained within the monocyclic heterocycle.
- monocyclic heterocycle include, but are not limited to, azetidinyl, azepanyl, aziridinyl, diazepanyl, 1,3-dioxanyl, 1,3-dioxolanyl, 1,3-dithiolanyl, 1,3-dithianyl, imidazolinyl, imidazolidinyl, isothiazolinyl, isothiazolidinyl, isoxazolinyl, isoxazolidinyl, morpholinyl, oxadiazolinyl, oxadiazolidinyl, oxazolinyl, oxazolidinyl, piperazinyl, piperidinyl, pyranyl, pyrazolinyl, pyrazolidinyl, pyrrolinyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydrothienyl, thiadiazol
- the bicyclic heterocycle is a monocyclic heterocycle fused to either a phenyl, a monocyclic cycloalkyl, a monocyclic cycloalkenyl, a monocyclic heterocycle, or a monocyclic heteroaryl.
- the bicyclic heterocycle is connected to the parent molecular moiety through any carbon atom or any nitrogen atom contained within the monocyclic heterocycle portion of the bicyclic ring system.
- bicyclic heterocyclyls include, but are not limited to, 2,3-dihydrobenzofuran-2-yl, 2,3-dihydrobenzofuran-3-yl, indolin-l-yl, indolin-2-yl, indolin-3-yl, 2,3-dihydrobenzothien-2-yl, decahydroquinolinyl, decahydroisoquinolinyl, octahydro-lH-indolyl, and octahydrobenzofuranyl.
- Heterocyclyl groups are optionally substituted with one or two groups which are independently oxo or thia.
- the bicyclic heterocyclyl is a 5 or 6 membered monocyclic heterocyclyl ring fused to phenyl ring, a 5 or 6 membered monocyclic cycloalkyl, a 5 or 6 membered monocyclic cycloalkenyl, a 5 or 6 membered monocyclic heterocyclyl, or a 5 or 6 membered monocyclic heteroaryl, wherein the bicyclic heterocyclyl is optionally substituted by one or two groups which are independently oxo or thia.
- hydroxy as used herein, means an -OH group.
- nitro as used herein, means a -N0 2 group.
- saturated means the referenced chemical structure does not contain any multiple carbon-carbon bonds.
- a saturated cycloalkyl group as defined herein includes cyclohexyl, cyclopropyl, and the like.
- spiro refers to a cyclic moiety formed by the subsituted atom and two available substitutable postions on that same atom.
- spiro-cyclopentyl group is the R group attached to the parent cyclohexyl ring by two single bonds.
- R is a spiro-heterocyclyl group
- such compounds include where the spiro-l,3-dioxolanyl ring is the R group attached to the parent cyclohexyl ring by two single bonds.
- unsaturated means the referenced chemical structure contains at least one multiple carbon-carbon bond, but is not aromatic.
- a unsaturated cycloalkyl group as defined herein includes cyclohexenyl, cyclopentenyl, cyclohexadienyl, and the like.
- an ex vivo cell can be part of a tissue sample excised from an organism such as a mammal.
- an in vitro cell can be a cell in a cell culture.
- an in vivo cell is a cell living in an organism such as a mammal.
- contacting refers to the bringing together of indicated moieties in an in vitro system or an in vivo system.
- "contacting" the IDO enzyme with a compound includes the administration of a compound described herein to an individual or patient, such as a human, having IDO, as well as, for example, introducing a compound into a sample containing a cellular or purified preparation containing the IDO enzyme.
- the term "individual” or “patient,” used interchangeably, refers to any animal, including mammals, preferably mice, rats, other rodents, rabbits, dogs, cats, swine, cattle, sheep, horses, or primates, and most preferably humans.
- the phrase "therapeutically effective amount” refers to the amount of active compound or pharmaceutical agent that elicits the biological or medicinal response that is being sought in a tissue, system, animal, individual or human by a researcher, veterinarian, medical doctor or other clinician.
- a therapeutically effective amount can be an amount suitable for (1) preventing the disease; for example, preventing a disease, condition or disorder in an individual who may be predisposed to the disease, condition or disorder but does not yet experience or display the pathology or symptomatology of the disease;
- inhibiting the disease for example, inhibiting a disease, condition or disorder in an individual who is experiencing or displaying the pathology or symptomatology of the disease, condition or disorder; or
- ameliorating the disease for example, ameliorating a disease, condition or disorder in an individual who is experiencing or displaying the pathology or symptomatology of the disease, condition or disorder (i.e., reversing the pathology and/or symptomatology) such as decreasing the severity of disease.
- treatment means (i) ameliorating the referenced disease state, for example, ameliorating a disease, condition or disorder in an individual who is experiencing or displaying the pathology or symptomatology of the disease, condition or disorder (i.e., reversing or improving the pathology and/or symptomatology) such as decreasing the severity of disease; or (ii) eliciting the referenced biological effect (e.g., IDO modulation or tryptophan degradation inhibition).
- ameliorating the referenced disease state for example, ameliorating a disease, condition or disorder in an individual who is experiencing or displaying the pathology or symptomatology of the disease, condition or disorder (i.e., reversing or improving the pathology and/or symptomatology) such as decreasing the severity of disease; or (ii) eliciting the referenced biological effect (e.g., IDO modulation or tryptophan degradation inhibition).
- Manifestation of amelioration of a disease condition with underlying IDO-mediated immunosuppression may require the concomitant or sequential administration of additional therapeutic agents, such as antineoplastic agents in the case of cancer, or antiretroviral agents in the case of viral diseases.
- additional therapeutic agents such as antineoplastic agents in the case of cancer, or antiretroviral agents in the case of viral diseases.
- administration of IDO inhibitors for the treatment of cancer does not always produce a direct antitumor effect when used as a single agent.
- chemotherapeutic drugs antagonistineoplastic
- the antitumor effect observed is higher than the sum of effects of each agent alone.
- catalytic pocket As used herein, the terms “catalytic pocket”, “catalytic site”, “active site” collectively and indistinctly refer to a region of the enzyme that contains amino acid residues responsible for the substrate binding (charge, hydrophobicity, steric hindrance) and catalytic amino acid residues which act as proton donors or acceptors or are responsible for binding a cofactor and participate in the catalysis of a chemical reaction.
- pharmaceutically acceptable salt refers to both pharmaceutically acceptable acid and base addition salts and solvates.
- Such pharmaceutically acceptable salts include salts of acids such as hydrochloric, phosphoric, hydrobromic, sulfuric, sulfinic, formic, toluenesulfonic, methanesulfonic, nitric, benzoic, citric, tartaric, maleic, hydroiodic, alkanoic such as acetic, HOOC-(CH 2 ) n -COOH where n is 0-4, and the like.
- Non-toxic pharmaceutical base addition salts include salts of bases such as sodium, potassium, calcium, ammonium, and the like. Those skilled in the art will recognize a wide variety of non-toxic pharmaceutically acceptable addition salts.
- the compounds and pharmaceutical compositions described herein can modulate activity of the enzyme indoleamine-2,3-dioxygenase (IDO).
- IDO indoleamine-2,3-dioxygenase
- modulate is meant to refer to an ability to decrease activity of an enzyme or receptor.
- compounds described herein can be used in methods of modulating IDO by contacting the enzyme with any one or more of the compounds or compositions described herein.
- the compounds described herein can act as inhibitors of IDO.
- the compounds described herein can be used to modulate activity of IDO in cell or in an individual in need of modulation of the enzyme by administering a modulating (e.g., inhibiting) amount of a compound described herein.
- methods of inhibiting the degradation of tryptophan and preventing the production of N-formylkynurenine in a system containing cells expressing IDO such as a tissue, living organism, or cell culture comprise administering an effective amount of a compound or pharmaceutical composition provided herein. Methods of measuring tryptophan levels and tryptophan degradation are routine in the art.
- IDO-mediated immunosuppression has been associated with, for example, cancers, tumor growth, metastasis, infectious diseases (e.g., viral infection), viral replication, etc.
- Example tumor-specific immunosuppression associated with cancers treatable by the methods herein include immunosuppression associated with cancer of the colon, pancreas, breast, prostate, lung, brain, ovary, cervix, testes, renal, head and neck, lymphoma, leukemia, melanoma, and the like.
- a patient undergoing or having completed a course of chemotherapy and/or radiation therapy for the treatment of a disease state can benefit from administering to the patient a therapeutically effective amount of a compound or composition recited herein for inhibiting immunosuppression resulting from the disease state and/or treatment thereof.
- IDO-mediated immunosuppression associated with viral infection is associated with a viral infection selected from the group consisting of: influenza, hepatitis C virus (HCV), human papilloma virus (HPV), cytomegalovirus (CMV), Epstein-Barr virus (EBV), poliovirus, varicella zoster virus, coxsackie virus, human immunodeficiency virus (HIV).
- a viral infection selected from the group consisting of: influenza, hepatitis C virus (HCV), human papilloma virus (HPV), cytomegalovirus (CMV), Epstein-Barr virus (EBV), poliovirus, varicella zoster virus, coxsackie virus, human immunodeficiency virus (HIV).
- Example diseases can include any disease, disorder or condition that is directly or indirectly linked to expression or activity of the IDO enzyme, such as over expression or abnormal activity.
- An IDO-associated disease can also include any disease, disorder or condition that can be prevented, ameliorated, or cured by modulating enzyme activity.
- IDO-associated diseases include cancer, viral infection such as HIV infection, depression, neurodegenerative disorders such as Alzheimer's disease and Huntington's disease, trauma, age-related cataracts, organ transplantation (e.g., organ transplant rejection), and autoimmune diseases including asthma, rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease, psoriasis and systemic lupus erythematosus or.
- Example cancers treatable by the methods herein include cancer of the colon, pancreas, breast, prostate, lung, brain, ovary, cervix, testes, renal, head and neck, lymphoma, leukemia, melanoma, and the like.
- One or more additional pharmaceutical agents for treatment methods such as, for example, anti-viral agents, chemotherapeutics or other anti-cancer agents, immune enhancers, immunosuppressants, radiation, anti-tumor and anti-viral vaccines, cytokine therapy (e.g., IL2, GM-CSF, etc.), and/or tyrosine kinase inhibitors can be used in combination with the compounds and pharmaceutical compositions described herein for treatment of IDO-associated diseases, disorders or conditions (as noted above) or for enhancing the effectiveness of the treatment of a disease state or condition, such as cancer.
- the agents can be combined with the present compounds in a single dosage form, or the agents can be administered simultaneously or sequentially as separate dosage forms.
- Therapeutic agents that constitute the standard of care for a particular cancer type or infectious disease are expected to benefit when combined with IDO inhibitors of the present invention.
- the tumor is sensitive to the cytotoxic effects of the chemotherapeutic agent in order to stimulate the release of antigens that will eventually mediate an immune response that will be enhanced by addition of IDO inhibitors to the combination treatment.
- a person of skill in the art will know how to select such chemotherapeutic agent based on the clinical characteristics and known sensititivity of each tumor to different antineoplastic agents.
- Suitable antiviral agents contemplated for use in combination with the compounds described herein can comprise nucleoside and nucleotide reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors and other antiviral drugs.
- NRTIs nucleoside and nucleotide reverse transcriptase inhibitors
- NRTIs non-nucleoside reverse transcriptase inhibitors
- protease inhibitors and other antiviral drugs.
- Example suitable NRTIs include zidovudine (AZT); didanosine (ddl); zalcitabine (ddC); stavudine (d4T); lamivudine (3TC); abacavir (1592U89); adefovir dipivoxil [bis(POM)-PMEA]; lobucavir (BMS-180194); BCH- 10652; emitricitabine [(-)-FTC]; beta-L-FD4 (also called beta-L-D4C and named beta-L-2',3'-dicleoxy-5-fluoro-cytidene); DAPD, ((-)-beta-D-2,6,-diamino-purine dioxolane); and lodenosine (FddA).
- Typical suitable NNRTIs include nevirapine (BI-RG-587); delaviradine (BHAP, U-90152); efavirenz (DMP-266
- protease inhibitors include saquinavir (Ro 31-8959); ritonavir (ABT-538); indinavir (MK-639); nelfnavir (AG-1343); amprenavir (141W94); lasinavir (BMS-234475); DMP-450; BMS-2322623; ABT-378; and AG-1549.
- Other antiviral agents include hydroxyurea, ribavirin, IL-2, IL-12, pentafuside and Yissum Project No. 11607.
- Suitable chemotherapeutic or other anti-cancer agents include, for example, alkylating agents (including, without limitation, nitrogen mustards, ethylenimine derivatives, alkyl sulfonates, nitrosoureas and triazenes) such as uracil mustard, chlormethine, cyclophosphamide (CytoxanTM), ifosfamide, melphalan, chlorambucil, pipobroman, triethylene-melamine, triethylenethiophosphoramine, busulfan, carmustine, lomustine, streptozocin, dacarbazine, and temozolomide.
- alkylating agents including, without limitation, nitrogen mustards, ethylenimine derivatives, alkyl sulfonates, nitrosoureas and triazenes
- alkylating agents including, without limitation, nitrogen mustards, ethylenimine derivatives, alkyl sulfonates, nitrosoure
- Suitable chemotherapeutic or other anti-cancer agents include, for example, antimetabolites (including, without limitation, folic acid antagonists, pyrimidine analogs, purine analogs and adenosine deaminase inhibitors) such as methotrexate, 5-fluorouracil, floxuridine, cytarabine, 6-mercaptopurine, 6-thioguanine, fludarabine phosphate, pentostatine, and gemcitabine.
- antimetabolites including, without limitation, folic acid antagonists, pyrimidine analogs, purine analogs and adenosine deaminase inhibitors
- methotrexate including, without limitation, folic acid antagonists, pyrimidine analogs, purine analogs and adenosine deaminase inhibitors
- methotrexate including, without limitation, folic acid antagonists, pyrimidine analogs, purine analogs and adenosine deaminase inhibitors
- Suitable chemotherapeutic or other anti-cancer agents further include, for example, certain natural products and their derivatives (for example, vinca alkaloids, antitumor antibiotics, enzymes, lymphokines and epipodophyllotoxins) such as vinblastine, vincristine, vindesine, bleomycin, dactinomycin, daunorubicin, doxorubicin, epirubicin, idarubicin, ara-C, paclitaxel (TaxolTM), docetaxel, mithramycin, deoxyco-formycin, mitomycin-C, L-asparaginase, interferons (especially IFN-a), etoposide, and teniposide.
- certain natural products and their derivatives for example, vinca alkaloids, antitumor antibiotics, enzymes, lymphokines and epipodophyllotoxins
- vinblastine vincristine, vindesine
- bleomycin dactinomycin
- cytotoxic agents include navelbene, CPT-1 1, anastrazole, letrazole, capecitabine, reloxafine, cyclophosphamide, ifosamide, and droloxafine.
- cytotoxic agents such as epidophyllotoxin; an antineoplastic enzyme; a topoisomerase inhibitor; procarbazine; mitoxantrone; platinum coordination complexes such as cis-platin and carboplatin; biological response modifiers; growth inhibitors; antihormonal therapeutic agents; leucovorin; tegafur; and haematopoietic growth factors.
- anti-cancer agent(s) include antibody therapeutics such as trastuzumab (Herceptin), antibodies to costimulatory molecules such as CTLA-4,4-lBB and PD-1, or antibodies to cytokines (IL-10, TGF- ⁇ , etc.).
- trastuzumab Herceptin
- costimulatory molecules such as CTLA-4,4-lBB and PD-1
- cytokines IL-10, TGF- ⁇ , etc.
- anti-cancer agents also include those that block immune cell migration such as antagonists to chemokine receptors, including CCR2, CCR4 and CCR6.
- anti-cancer agents also include those that augment the immune system such as adjuvants or adoptive T cell transfer.
- Anti-cancer vaccines include dendritic cells, synthetic peptides, DNA vaccines and recombinant viruses.
- compositions described herein generally comprise a combination of a compound described herein and a pharmaceutically acceptable carrier, diluent, or excipient. Such compositions are substantially free of non-pharmaceutically acceptable components, i.e., contain amounts of non-pharmaceutically acceptable components lower than permitted by US regulatory requirements at the time of filing this application.
- the composition if the compound is dissolved or suspended in water, the composition further optionally comprises an additional pharmaceutically acceptable carrier, diluent, or excipient.
- the pharmaceutical compositions described herein are solid pharmaceutical compositions (e.g., tablet, capsules, etc.).
- compositions can be prepared in a manner well known in the pharmaceutical art, and can be administered by a variety of routes, depending upon whether local or systemic treatment is desired and upon the area to be treated. Administration may be topical (including ophthalmic and to mucous membranes including intranasal, vaginal and rectal delivery), pulmonary (e.g., by inhalation or insufflation of powders or aerosols, including by nebulizer; intratracheal, intranasal, epidermal and transdermal), ocular, oral or parenteral.
- topical including ophthalmic and to mucous membranes including intranasal, vaginal and rectal delivery
- pulmonary e.g., by inhalation or insufflation of powders or aerosols, including by nebulizer; intratracheal, intranasal, epidermal and transdermal
- ocular oral or parenteral.
- Methods for ocular delivery can include topical administration (eye drops), subconjunctival, periocular or intravitreal injection or introduction by balloon catheter or ophthalmic inserts surgically placed in the conjunctival sac.
- Parenteral administration includes intravenous, intraarterial, subcutaneous, intraperitoneal or intramuscular injection or infusion; or intracranial, e.g., intrathecal or intraventricular, administration.
- Parenteral administration can be in the form of a single bolus dose, or may be, for example, by a continuous perfusion pump.
- Pharmaceutical compositions and formulations for topical administration may include transdermal patches, ointments, lotions, creams, gels, drops, suppositories, sprays, liquids and powders. Conventional pharmaceutical carriers, aqueous, powder or oily bases, thickeners and the like may be necessary or desirable.
- compositions can contain, as the active ingredient, one or more of the compounds described herein above in combination with one or more pharmaceutically acceptable carriers.
- the active ingredient is typically mixed with an excipient, diluted by an excipient or enclosed within such a carrier in the form of, for example, a capsule, sachet, paper, or other container.
- the excipient serves as a diluent, it can be a solid, semi-solid, or liquid material, which acts as a vehicle, carrier or medium for the active ingredient.
- compositions can be in the form of tablets, pills, powders, lozenges, sachets, cachets, elixirs, suspensions, emulsions, solutions, syrups, aerosols (as a solid or in a liquid medium), ointments containing, for example, up to 10% by weight of the active compound, soft and hard gelatin capsules, suppositories, sterile injectable solutions, and sterile packaged powders.
- the active compound in preparing a formulation, can be milled to provide the appropriate particle size prior to combining with the other ingredients. If the active compound is substantially insoluble, it can be milled to a particle size of less than 200 mesh. If the active compound is substantially water soluble, the particle size can be adjusted by milling to provide a substantially uniform distribution in the formulation, e.g. about 40 mesh.
- excipients include lactose, dextrose, sucrose, sorbitol, mannitol, starches, gum acacia, calcium phosphate, alginates, tragacanth, gelatin, calcium silicate, microcrystalline cellulose, polyvinylpyrrolidone, cellulose, water, syrup, and methyl cellulose.
- the formulations can additionally include: lubricating agents such as talc, magnesium stearate, and mineral oil; wetting agents; emulsifying and suspending agents; preserving agents such as methyl- and propylhydroxy-benzoates; sweetening agents; and flavoring agents.
- the compositions described herein can be formulated so as to provide quick, sustained or delayed release of the active ingredient after administration to the patient by employing procedures known in the art.
- compositions can be formulated in a unit dosage form, each dosage containing from about 5 to about 100 mg, more usually about 10 to about 30 mg, of the active ingredient.
- unit dosage forms refers to physically discrete units suitable as unitary dosages for human subjects and other mammals, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect, in association with a suitable pharmaceutical excipient.
- the active compound can be effective over a wide dosage range and is generally administered in a pharmaceutically effective amount. It will be understood, however, that the amount of the compound actually administered will usually be determined by a physician, according to the relevant circumstances, including the condition to be treated, the chosen route of administration, the actual compound administered, the age, weight, and response of the individual patient, the severity of the patient's symptoms, and the like.
- the principal active ingredient is mixed with a pharmaceutical excipient to form a solid preformulation composition containing a homogeneous mixture of a compound described herein.
- a solid preformulation composition containing a homogeneous mixture of a compound described herein.
- the active ingredient is typically dispersed evenly throughout the composition so that the composition can be readily subdivided into equally effective unit dosage forms such as tablets, pills and capsules.
- This solid preformulation is then subdivided into unit dosage forms of the type described above containing from, for example, 0.1 to about 500 mg of the active ingredient of a compound described herein.
- the tablets or pills can be coated or otherwise compounded to provide a dosage form affording the advantage of prolonged action.
- the tablet or pill can comprise an inner dosage and an outer dosage component, the latter being in the form of an envelope over the former.
- the two components can be separated by an enteric layer which serves to resist disintegration in the stomach and permit the inner component to pass intact into the duodenum or to be delayed in release.
- enteric layers or coatings such materials including a number of polymeric acids and mixtures of polymeric acids with such materials as shellac, cetyl alcohol, and cellulose acetate.
- liquid forms in which the compounds and compositions can be incorporated for administration orally or by injection include aqueous solutions, suitably flavored syrups, aqueous or oil suspensions, and flavored emulsions with edible oils such as cottonseed oil, sesame oil, coconut oil, or peanut oil, as well as elixirs and similar pharmaceutical vehicles.
- compositions for inhalation or insufflation include solutions and suspensions in pharmaceutically acceptable, aqueous or organic solvents, or mixtures thereof, and powders.
- the liquid or solid compositions may contain suitable pharmaceutically acceptable excipients as described supra.
- the compositions are administered by the oral or nasal respiratory route for local or systemic effect.
- Compositions in can be nebulized by use of inert gases. Nebulized solutions may be breathed directly from the nebulizing device or the nebulizing device can be attached to a face masks tent, or intermittent positive pressure breathing machine. Solution, suspension, or powder compositions can be administered orally or nasally from devices which deliver the formulation in an appropriate manner.
- compositions can be administered to a patient already suffering from a disease in an amount sufficient to cure or at least partially arrest the symptoms of the disease and its complications. Effective doses will depend on the disease condition being treated as well as by the judgment of the attending clinician depending upon factors such as the severity of the disease, the age, weight and general condition of the patient, and the like.
- compositions administered to a patient can be in the form of pharmaceutical compositions described above. These compositions can be sterilized by conventional sterilization techniques, or may be sterile filtered. Aqueous solutions can be packaged for use as is, or lyophilized, the lyophilized preparation being combined with a sterile aqueous carrier prior to administration.
- the pH of the compound preparations typically will be between 3 and 11, more preferably from 5 to 9 and most preferably from 7 to 8. It will be understood that use of certain of the foregoing excipients, carriers, or stabilizers will result in the formation of pharmaceutical salts.
- the therapeutic dosage of the compounds can vary according to, for example, the particular use for which the treatment is made, the manner of administration of the compound, the health and condition of the patient, and the judgment of the prescribing physician.
- the proportion or concentration of a compound described herein in a pharmaceutical composition can vary depending upon a number of factors including dosage, chemical characteristics (e.g., hydrophobicity), and the route of administration.
- the compounds described herein can be provided in an aqueous physiological buffer solution containing about 0.1 to about 10% w/v of the compound for parenteral administration. Some typical dose ranges are from about 1 ⁇ g/kg to about 1 g/kg of body weight per day.
- the dose range is from about 0.01 mg kg to about 100 mg/kg of body weight per day.
- the dosage is likely to depend on such variables as the type and extent of progression of the disease or disorder, the overall health status of the particular patient, the relative biological efficacy of the compound selected, formulation of the excipient, and its route of administration. Effective doses can be extrapolated from dose-response curves derived from in vitro or animal model test systems.
- the compounds described herein can also be formulated in combination with one or more additional active ingredients which can include any pharmaceutical agent such as anti-viral agents, vaccines, antibodies, immune enhancers, immune suppressants, anti-inflammatory agents and the like.
- Another aspect relates to fluorescent dye, spin label, heavy metal or radio-labeled derivatives of the compounds described herein that would be useful not only in imaging but also in assays, both in vitro and in vivo, for localizing and quantitating the IDO enzyme in tissue samples, including human, and for identifying IDO enzyme ligands by inhibition binding of a labeled compound. Accordingly, further provided are IDO enzyme assays that contain such labeled compounds.
- isotopically-labeled compounds of the compounds described herein are isotopically-labeled compounds of the compounds described herein.
- An “isotopically” or “radio-labeled” compound is a compound described herein where one or more atoms are replaced or substituted by an atom having an atomic mass or mass number different from the atomic mass or mass number typically found in nature (i.e., naturally occurring).
- Suitable radionuclides that may be include but are not limited to 2H (also written as D for deuterium), 3H (also written as T for tritium), n C, 13 C, 14 C, 13 N, 15 N, 15 0, 17 0, 18 0, 18 F, 35 S, 36 C1, 82 Br, 75 Br, 76 Br, 77 Br, 123 I, 124 I, 125 I and 131 I.
- the radionuclide that is incorporated in the instant radio-labeled compounds will depend on the specific application of that radio-labeled compound. For example, for in vitro IDO enzyme labeling and competition assays, compounds that incorporate H, C, Br, I, I, S or will generally be most useful. For radio-imaging applications n C, 18 F, 125 I, 123 I, 124 I, 131 I, 75 Br, 76 Br or 77 Br will generally be most useful.
- a "radio-labeled” or “labeled compound” is a compound that has incorporated at least one radionuclide.
- the radionuclide is selected from the group consisting of 3 H, 14 C, 125 1, 35 S and 82 Br.
- a radio-labeled compound described herein can be used in a screening assay to identify/evaluate compounds.
- a newly synthesized or identified compound i.e., test compound
- the ability of a test compound to compete with the radio-labeled compound for binding to the IDO enzyme directly correlates to its binding affinity.
- kits useful for example, in the treatment or prevention of IDO-associated diseases or disorders, obesity, diabetes and other diseases referred to herein which include one or more containers containing a pharmaceutical composition comprising a therapeutically effective amount of a compound described herein.
- kits can further include, if desired, one or more of various conventional pharmaceutical kit components, such as, for example, containers with one or more pharmaceutically acceptable carriers, additional containers, etc., as will be readily apparent to those skilled in the art.
- Instructions, either as inserts or as labels, indicating quantities of the components to be administered, guidelines for administration, and/or guidelines for mixing the components, can also be included in the kit.
- l H NMR spectra were obtained with a Bruker DRX400, Varian VXR400 or VXR300. l H NMR spectra were reported in parts per million ( ⁇ ) relative to TMS (0.0), DMSO-d 6 (2.50) or CD 3 OD (4.80) as an internal reference. All *H NMR spectra were taken in CDCI 3 unless otherwise indicated.
- the following starting materials were prepared according to their literature procedures: E)-ethyl 3-(2-iodophenyl)acrylate (Synth. Comm. 2007, 37, 2989-2994), 2-chloro-6-iodobenzaldehyde (J. Agric. Food Chem.
- intermediate A can be achieved by allowing 2-iodobenzaldehydes to react with substituted methyl ketones in the presence of a base to afford 3-(2-iodophenyl)prop-2-en-l- ones. Negishi cross-coupling of the resulting 3-(2-iodophenyl)prop-2-en-l-ones with 4- iodo-l-trityl-lH- imidazole, also leads to intermediate A.
- the solvent was distilled off under reduced pressure and the crude was partitioned between CH2CI2 (50 mL) and satd. NH4CI (30 mL). The organic layer was collected and the aqueous layer was extracted with CH2CI2 (2 x 30 mL). The organic layer was washed with brine, dried (Na 2 S0 4 ) and concentrated under reduced pressured to obtain the crude product. The crude was purified by column chromatography on silica gel to afford 21 as a clear oil (280 mg, 24%).
- the solution was stirred at 50 °C overnight.
- the solvent was distilled off under reduced pressure and the crude was partitioned between CH2CI2 (50 mL) and satd. NH4CI (30 mL).
- the organic layer was collected and the aqueous layer was extracted with CH2CI2 (2 x 30 mL).
- the organic layer was washed with brine, dried (Na 2 S0 4 ) and concentrated under reduced pressured to obtain the crude product.
- the crude was purified by column chromatography on silica gel to afford 22 as colorless oil (800 mg, 65%).
- the pure diastereomers were obtained from the racemic mixture of 1363 using preparative chiral supercritical fluid chromatography (SFC) technique, using a AD-H column (Regis Technologies, Inc.) in methanohCC (24:76).
- SFC preparative chiral supercritical fluid chromatography
- Example 52 1 -(( 15,35)-3 -(tert-Butyldimethylsilyloxy)-3 -cyclohexyl- 1 -(2- iodophenyl)propyl)-lH- imidazole
- the solvent was distilled off under reduced pressure and the crude was diluted with sat'd NH 4 C1 (80 mL), water (100 mL) and EtOAc (100 mL). The solution was partitioned in a separatory funnel and the organic layer was collected. The aqueous layer was extracted with EtOAc (3 x 150 mL) and the combined organic fractions were washed with brine and dried over Na 2 S0 4 . The solution was filtered and concentrated under reduced pressure to afford the crude product. The crude was stirred in a mixture of acetic acid (20 mL) and MeOH (170 mL) at 90 °C for 1.5 h.
- Expression vectors for human indoleamine-2,3-dioxygenase (IDO) protein were prepared by amplification of a 1219 bp fragment of the sequence present in vector phID06His cDNA with primers 5 '-ggagcatgctaATGGCACACGCTATGGAAAAC-3 ' and 5 ' -gagagatctACCTTCCTTCAAAAGGGATTTC-3 ' and cloning the Sphl-Bglll 1213 bp fragment into pQE70 (Qiagen), to yield vector pQE70-hIDO.
- This construct adds 2 extra amino acids and a 6-Histidine tag to the C-terminus of the natural human IDO protein while preserving intact the natural start codon and N-terminus amino acid sequence.
- the amplified allele of human IDO shows two polymorphisms with respect to the sequence deposited in accession file P14902 of SwissProt database. These polymorphisms result in a P1 10S and El 19G amino acid changes.
- Plasmid pQE70-hIDO was transformed into M15(pREP4) cells (Qiagen) and clones were selected in LB-agar plates supplemented with carbenicillin 50 ⁇ g/mL and kanamycin 30 ⁇ g/mL. Protein expression was carried out by growing an overnight culture of the M15pREP4/pQE70-hIDO clone in 100 mL LB supplemented with 100 ⁇ g/mL carbenicillin, 50 ⁇ g/mL kanamycin and 50 ⁇ g/mL of L-tryptophan (LBCKT medium). 40 mL of this culture were inoculated into 750 mL of LBCKT for 4 hours at 37 °C.
- This culture was diluted 1 : 10 into LBCKT medium and cultured for another 2 hours at 37 °C until OD600 was higher than 0.8. At this point the cultures were inoculated with Hemin to 7 ⁇ and L-Tryptophan to 75 ⁇ g/mL and incubated at 37 °C for 2 h. Induction of protein expression was carried out by supplementing the cultures with IPTG to 1 mM, PMSF to 200 ⁇ , EDTA to 1 mM and L-tryptophan to 50 ⁇ g/mL. Incubation was continued for additional 16 h at 25 °C. Cells were collected by centrifugation, and the cell pellets were washed with PBS buffer supplemented with 200 ⁇ PMSF and 1 mM EDTA and stored at -80 °C until protein purification.
- the filtered supernatant was loaded onto a 60 mL phosphocellulose column equilibrated with 50 mM potassium phosphate buffer pH 6.5 (KPB) at 1-3 mL/min.
- KPB potassium phosphate buffer pH 6.5
- the column was washed with 3 volumes of 50 mM KPB, 3 volumes of 100 mM KPB and the protein was eluted with 15 volumes of a linear gradient of 100-500 mM KPB.
- Fractions were collected and IDO activity assay was performed by measuring kynurenine production.
- the IC5 0 values for each compound were determined by testing the activity of IDO in a mixture containing 50 mM potassium phosphate buffer at pH 6.5; 70 nM purified human IDO protein, 200 ⁇ L-tryptophan, 20 mM ascorbate, 20 ⁇ methylene blue, 0.1% DMSO.
- the inhibitors were initially diluted in DMSO at 100 mM and were diluted in potassium phosphate 50 mM, added to the reaction mixture at final concentrations raging from 1 mM to 5 nM and preincubated with the enzyme for 5 min at 25 °C.
- the reaction was started by addition of L-tryptophan to 200 ⁇ and incubated 15 min at 37 °C.
- the reaction was stopped by addition of 0.5 vol of 30% trichloroacetic acid and incubated 30 min at 60 °C to hydrolyze N-formylkynurenine to kynurenine.
- the reaction was centrifuged at 3400 g for 5 min to remove precipitated protein and the supernatant was reacted with 2% (w/v) of p-dimethylaminobenzaldehyde in acetic acid.
- the reaction was incubated 10 min at 25 °C and read at 480 nm in a spectrophotometer.
- Control samples with no IDO inhibitor, or with no IDO enzyme or with the reference inhibitors 1-methyl-tryptophan (200 ⁇ ) and menadione (1.2 ⁇ ) were used as controls to set the parameters for the non-linear regressions necessary for determination of the IC5 0 for each compound.
- Nonlinear regressions and determination of the IC5 0 values were performed using the GraphPad Prism 4 software. Compounds with an IC5 0 of less than 500 ⁇ were considered as active inhibitors in this assay.
- 293-T-RExTM cells constitutively express a tet operator binding repressor protein and are maintained in DMEM, 10 % FBS, IX Penicillin+Streptomycin, 2 mM L-glutamine, 5 ⁇ g/mL blasticidin at 37 °C with a 5% CO 2 in air atmosphere and typically split prior to confluency.
- Cells were passed by splitting the culture 1/10- by removing media by aspiration, washing IX with PBS, incubating with 0.25% trypsin/EDTA until the cells detach, disbursing the cells in fresh growth media, and plating at 1/10 dilutions in fresh growth media.
- cells are detached from the plate as described above, collected by centrifugation, resuspended in freeze medium (growth medium, 10%DMSO), stored in 1.8 mL cyropreservation vials ( ⁇ 2-5 X 106 cells per vial), in liquid nitrogen vapor storage tanks.
- freeze medium growth medium, 10%DMSO
- IDOl- expressing 293-T-RexTM cell lines were generated by stable transfection of plasmid pcDNA-tetO-IDO expressing human IDO or murine IDO under the control of the doxycycline-inducible CMV-tet promoter.
- Transfected cells were selected in DBZ medium (DMEM, 10 % FBS, IX Penicillin + Streptomycin, 2 mM L-glutamine, 5 ⁇ g/mL blasticidin and 25 ⁇ g/mL Zeocin) at 37 °C with a 5% CO 2 in air atmosphere. Individual clones were isolated by limiting dilution cloning from these populations.
- IDO-293-T-Rex cells were harvested and resuspended in DBZ media at 10 6 cells/mL, and split into poly-D-lysine coated 96-well plates at 100,000 cells per well. 100 ⁇ , of Neutral medium (DBZ medium, 200 ⁇ L-tryptophan) or Induction media (Neutral medium supplemented with 5 ⁇ doxycycline) are added to the cells and incubated 28 h at 37 °C.
- medium is removed and replaced with Induction or Neutral medium containing different concentrations of each inhibitor (1 mM to 0.5 nM).
- the cells incubated in Neutral medium serve as negative control of the assay.
- the cells incubated in Induction medium and without inhibitor serve as the positive control of the assay.
- the incubation is carried out for 16 h at 37 °C in a cell culture incubator. 200 ⁇ ⁇ of medium are transferred to U-bottom polypropylene 96-well plates containing 25 ⁇ , of 30% TCA, incubated 30 minutes at 60 °C and centrifuged at 3400 g for 5 minutes.
- cell viability is measured via a WST-1 assay (Roche) according to instructions from the manufacturer. Briefly, after the incubation with each compound, medium is aspirated and replaced with 100 mL of WST-1 reagent, and incubated 30 min at 37 °C. Absorbance at 540 nm is correlated with the number of viable cells. Determination of IC5 0 (Kynurenine assay) or LD5 0 (WST-1 assay) is performed via non-linear regression analysis using GraphPad Prism software.
- Human monocytes were collected from peripheral mononuclear cells by leukoapheresis and cultured overnight at 10 6 cells/well in a 96-well plate in RPMI 1640 medium supplemented with 10% fetal calf serum and 2 mM L-glutamine. Adherent cells were retained and cultured for 7 days with 200 ng/ml IL-4, 100 ng/ml GM-CSF. Cells were matured for 2 days with a cytokine cocktail containing TNF-a, IL- ⁇ , IL-6 and PGE2 for additional 2 days to induce dendritic cell maturation.
- T cell proliferation was measured by BrdU incorporation assay after an overnight pulse with BrdU labeling mix (Roche Molecular Biochemicals). At the en of the pulse, the cells were fixed and incubated with 100 ⁇ L/well anti-BrdU-POD antibody following the instructions from the manufacturer. Plates were read in a microplate reader.
- testing of IDO inhibitors in an in vitro mouse model of IDO-mediated suppression of T cell proliferation is performed by the following procedure.
- C57bl6 mice are inoculated with lxlO 6 B78H1-GMCSF tumor cells in the right flank. After 10-12 days, tumor draining lymph nodes are collected and cells are stained with anti-CD 1 1c and anti-B220 monoclonal antibodies. Cells are sorted by high-speed fluorescence activated cell sorting and the CDl lc+/B220+ plasmacytoid dendritic cells are collected and seeded at 2000 cells/well in 96 well V-bottom plates.
- Splenocytes are collected from BM3 transgenic mice (in CBA background) and collected by nylon wool enrichment.
- BM3 T cells (10 5 cells/well) are added to each well in 200 ⁇ L of RPMI, 10% FCS, 50 ⁇ M ⁇ -mercaptoetanol.
- T cells are obtained from spleens of OT-I transgenic mice and added to the culture in combination with OVA peptide.
- IDO inhibitors are added dissolved in RPMI at final concentrations ranging from 1 mM to 10 nM. After 3 days of stimulation, cells are pulsed by 16 h with BrdU or 3 H-thymidine.
- Human IDO IC5 0 this is the concentration of the compound at which we observe 50% of enzymatic activity using recombinant human IDO under the assay conditions described in one of the examples; [0210] IC 50 values are reported in ranges: A: ⁇ 1 ⁇ , B: 1 - 10 ⁇ , C: 10 - 100 ⁇ ; D: > 100 ⁇ .
- In vivo anti-tumor efficacy can be tested using modified tumor allograft protocols. For instance, it has been described in the literature that IDO inhibition can syngerize with cytotoxic chemotherapy in immune-competent mice. Due to different susceptibilities of different tumor cell lines to chemotherapeutic drugs and to immune mediated rejection, each IDO inhibitor is tested alone and in combination with 2 different chemotherapeutic drugs in 4 different animal tumor models, represented by 4 different mouse tumor cell lines, of different tissue origin (colorectal, bladder, mammary and lung carcinoma), implanted subcutaneously in syngeneic strains of mice. These cell lines have been selected based on their known susceptibility to chemotherapeutic drugs, their partial response to IDO inhibitors as single agents, their presumed pattern of IDO expression according to their tissue of origin, and their ability to elicit an immune reaction.
- the following chemotherapeutic drugs are used, at the indicated doses.
- the maximum tolerated dose for the following chemotherapeutic agents in mice depends on the formulation, concentration, frequency of administration, route of administration and number of doses.
- the chemotherapeutic drugs administered in conjunction with each IDO inhibitor drug are: 1] Paclitaxel: 20 mg/kg/day i.p, every 4 days, 4 times (q4dx4) (in Cremophor); 2] Doxorubicin: 5 mg/kg, once a week for 3 weeks (q7dx3); 3] Cyclophosphamide (CTX): 100 mg/kg, LP., every 4 days, 4 times (q4dx4); 4] Gemcitabine: 80 mg/kg every 4 days, 4 times, i.p. (q4dx4).
- All animals receive a subcutaneous injection of a tumor forming dose of live tumor cells ( ⁇ 50000 - 1000000 cells) suspended in 0.1 mL of PBS or saline on day 1. Subcutaneous injection forms a localized tumor that allows monitoring tumor growth over time.
- IDO inhibitor drugs begins at day 5-8 after tumor inoculation. Dosing, route of administration, dosing frequency varies depending on the toxicity and pharmacokinetics profile of each drug. Duration of the treatment is 2 weeks. Most preferably, drug is administered continuously via oral gavage or dissolution in the drinking water. Alternatively, subcutaneous slow release pellets or osmotic pumps containing 100 mg of each drug are implanted under the skin by surgical procedure. IDO inhibitor drug are administered at the maximum tolerated dose or at a concentration corresponding to the LD5 0 .
- FIG. 1-2 An example of antitumor activity is shown in Figures 1-2 (for Cpd# 1357) and Figures 3-4 (for Cpd# 1304).
- 200000 LLC murine tumor cells were injected subcutaneously into syngeneic C57B16 mice on day 0.
- Each treatement group consists of 10 mice.
- mice were optionally treated with cyclophosphamide 100 mg/kg by intraperitoneal injection on days 9, 13 and 15 post-tumor innoculation, either as a single agent or in combination with compound 1304.
- the results of these tests indicate that compounds 1357 and 1304 have a significant antitumor effect either as a single agent or when administered in combination with chemotherapy.
- the therapeutic effect is observed as a reduced rate of tumor growth, which has an impact on median survival time and in overall survival fraction.
- Figure 1 shows the average tumor volume over time of two groups of 10 mice each.
- the control group was treated with vehicle, while the treatment groups received osmotic pumps with compound 1357 as described above.
- the tumor volumes were fitted to an exponential growth equation and the fitted parameters were compared using GraphPad software. The data indicate a statistically significant differences between the two curves (pO.0001).
- Figure 2 shows the survival plot of the same groups of mice described in Figure 1. The logrank test indicates a statistically significant difference in median survival time when animals were treated with compound 1357 as a single agent.
- Figure 3 shows the average tumor volume over time of four groups of 10 mice each.
- the control group was treated with vehicle, while the treatment groups received either cyclophosphamide chemotherapy, osmotic pumps with compound 1304, or a combination therapy of cyclophosphamide with compound 1304.
- the data shows that this tumor is very sensitive to the effects of treatment with compound 1304 either as a single agent or in combination with chemotherapy.
- Figure 4 shows the survival plot of the same groups of mice described in Figure 3.
- the logrank test indicated a statistically significant difference in median survival time when animals were treated with compound 1304, either as a single agent or in combination with cyclophosphamide.
- the long term survival fraction observed for treatment with 1304 is exceptionally high, with 70-80% of the mice being tumor free after 60 days.
Abstract
Description































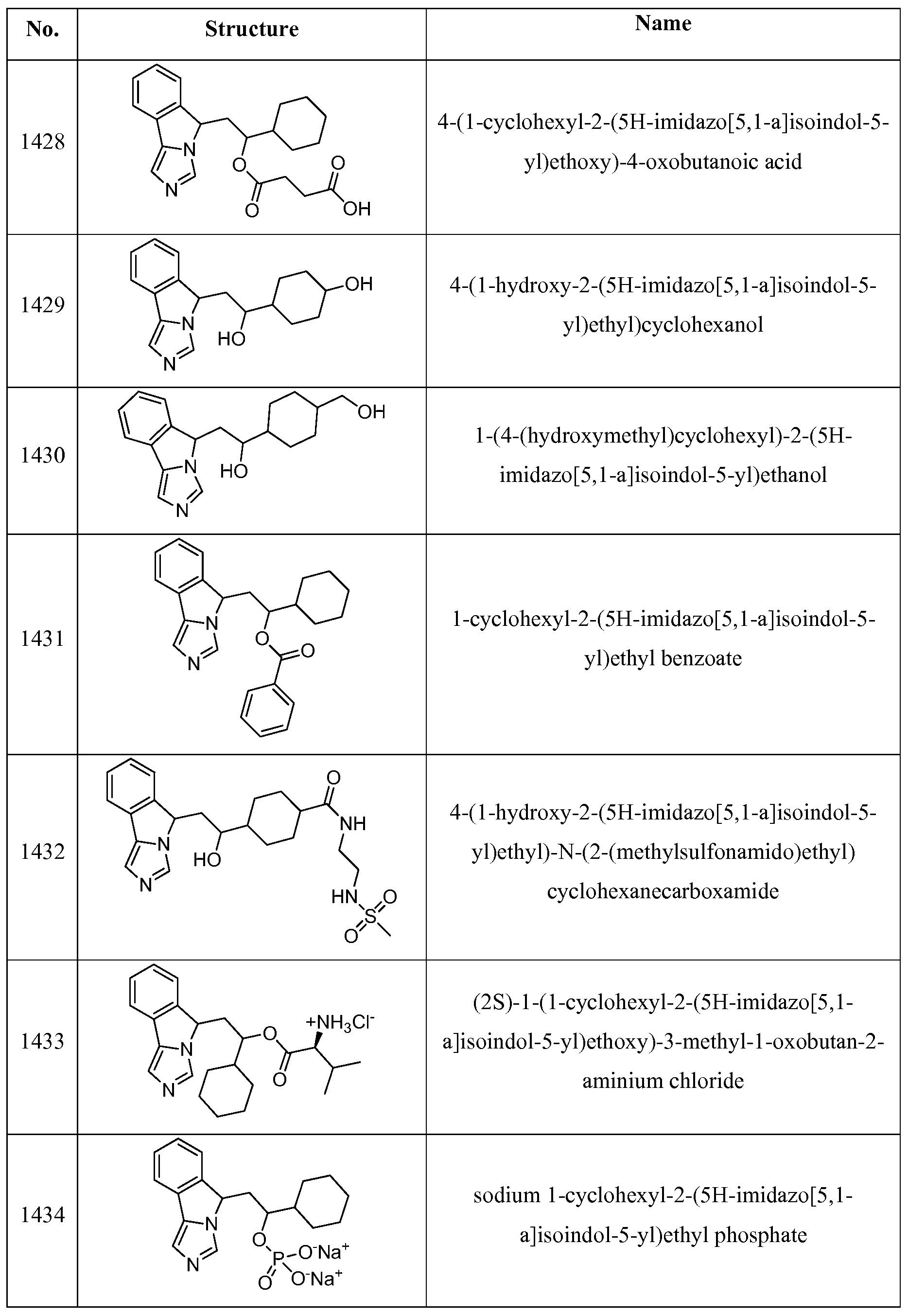







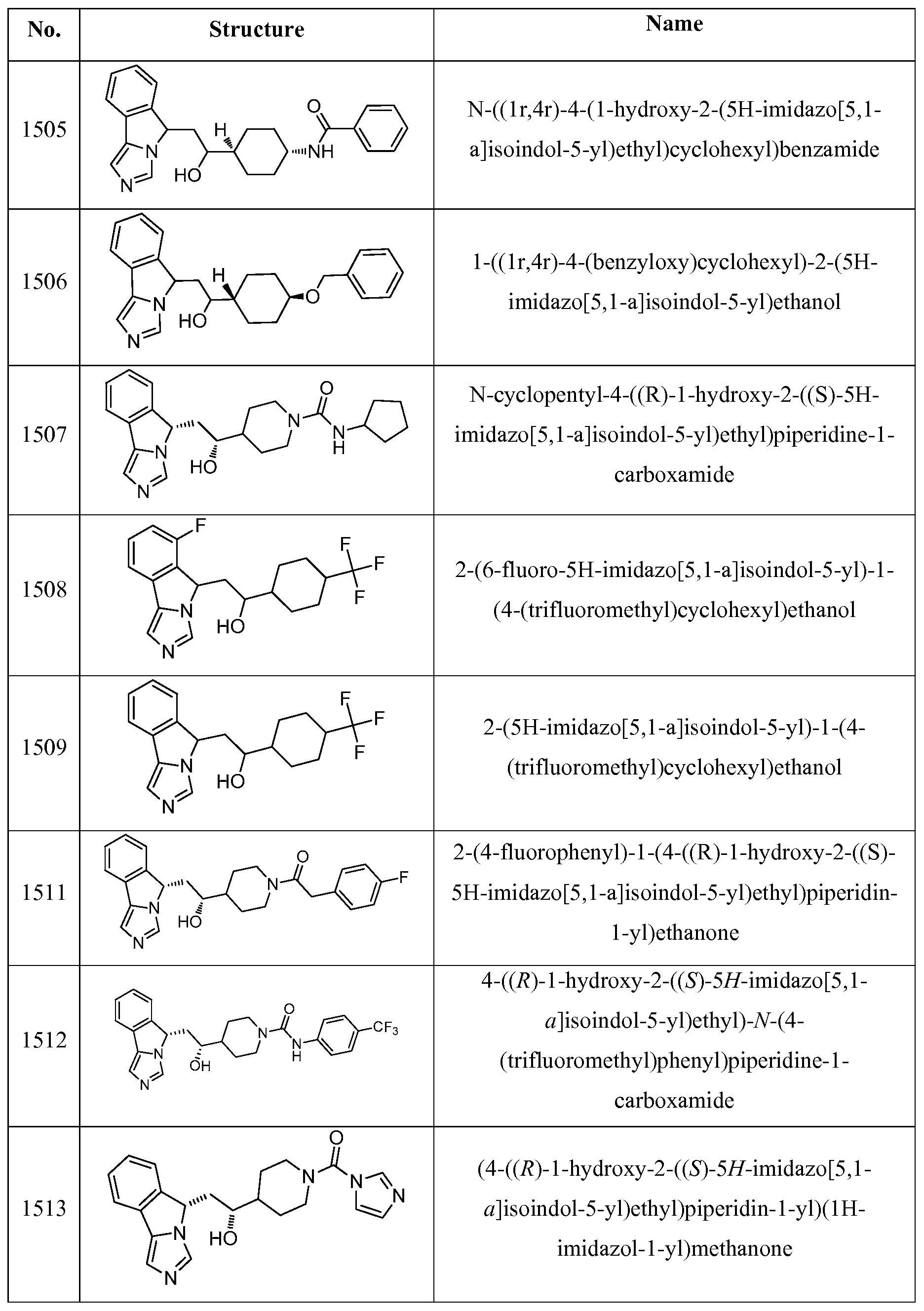









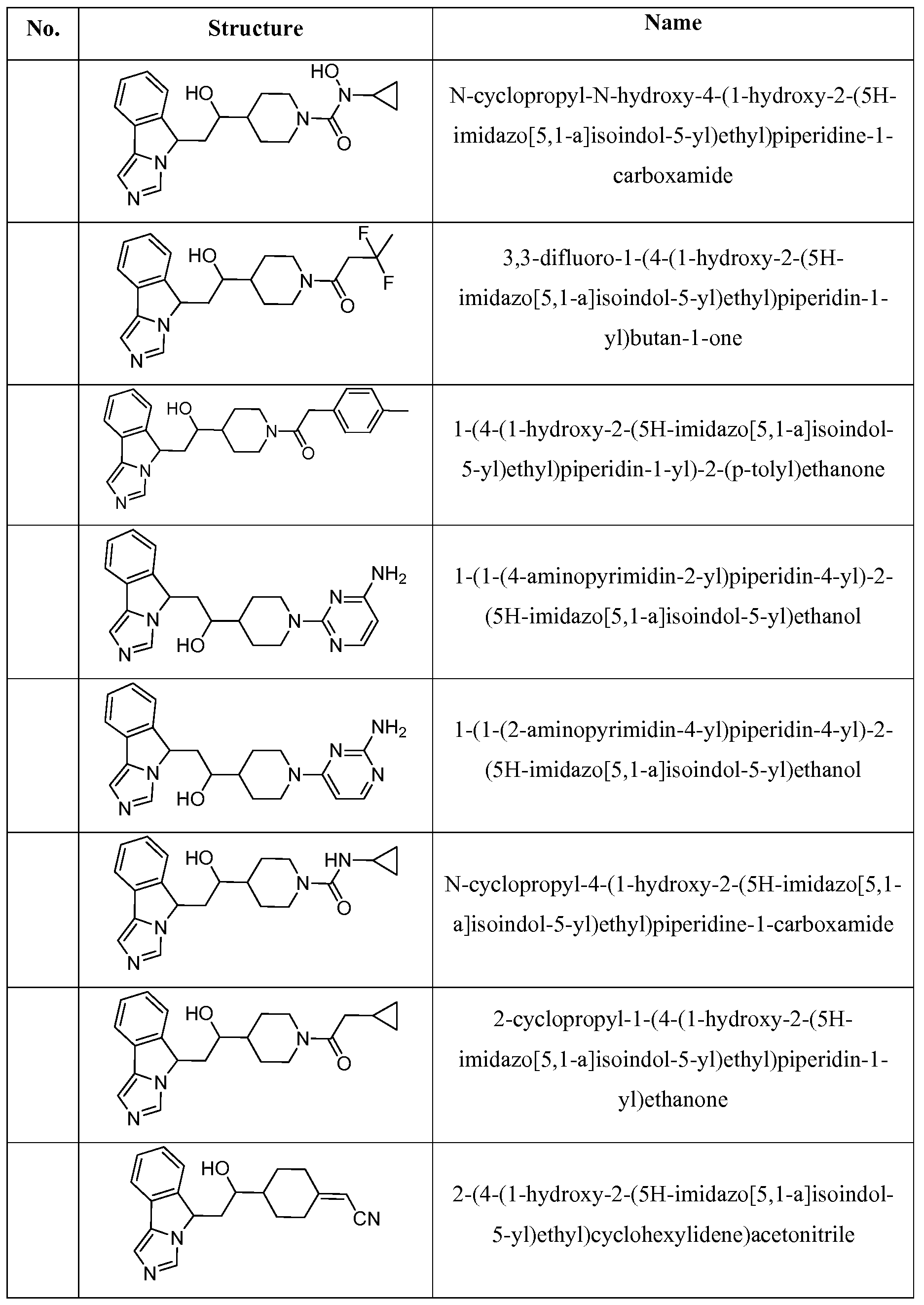
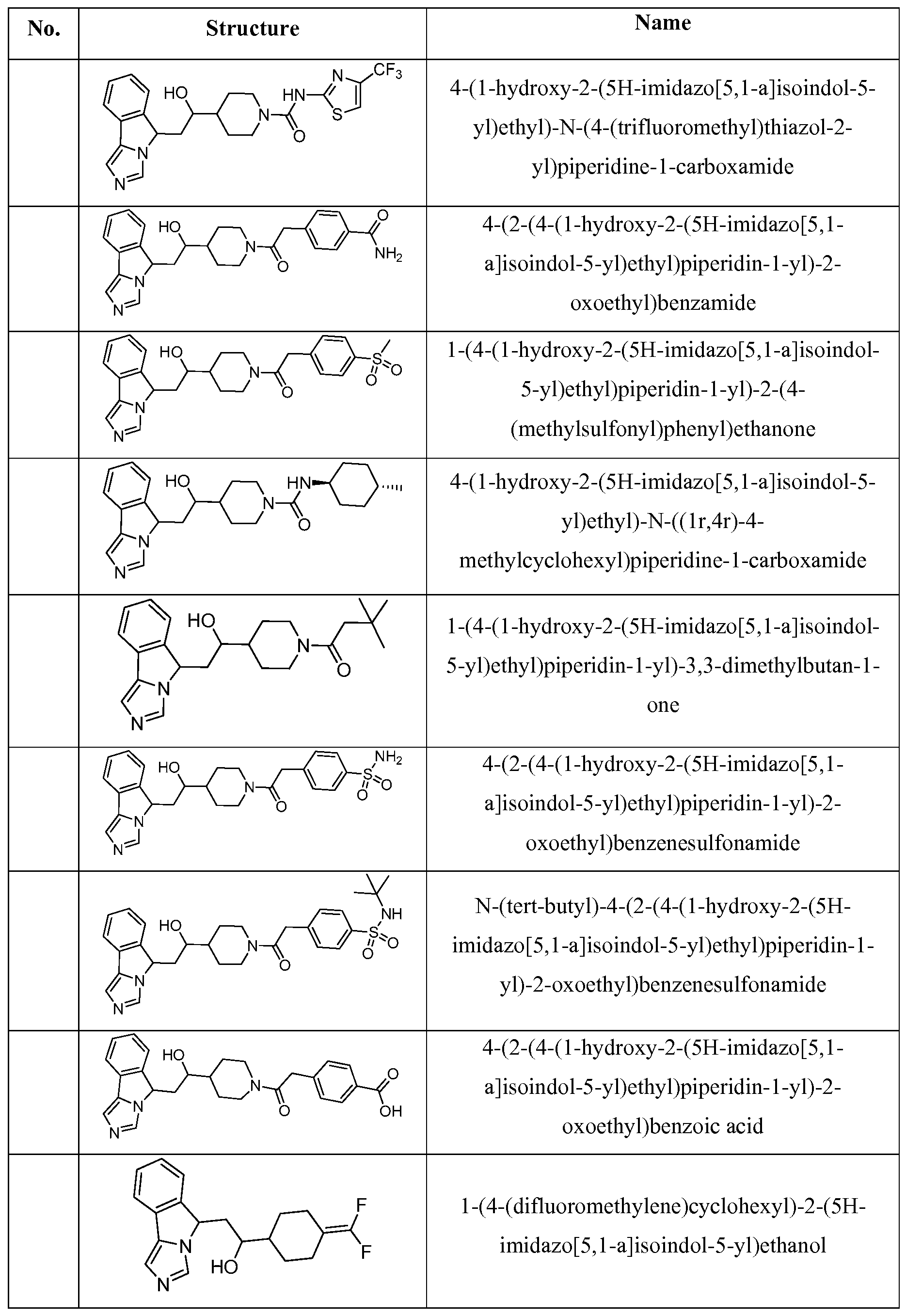




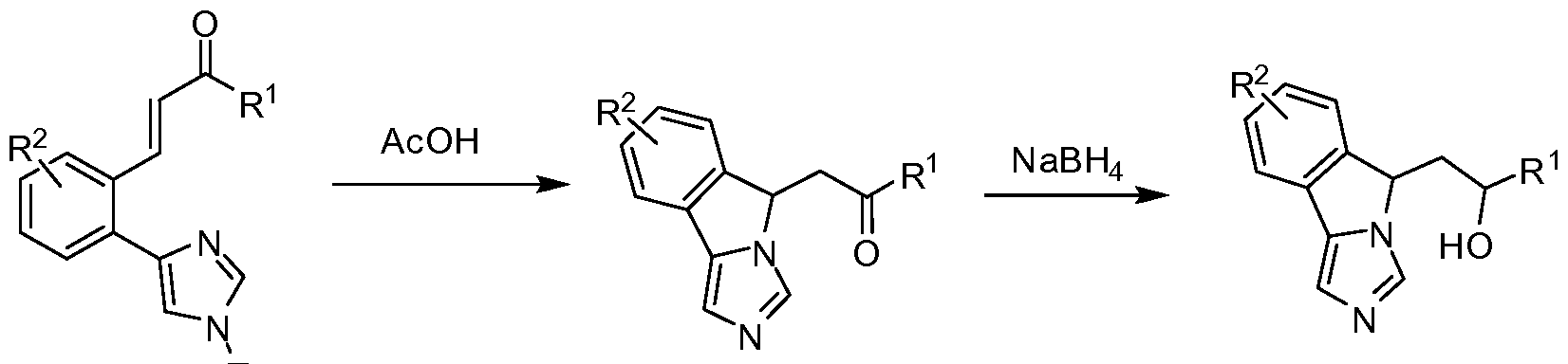















































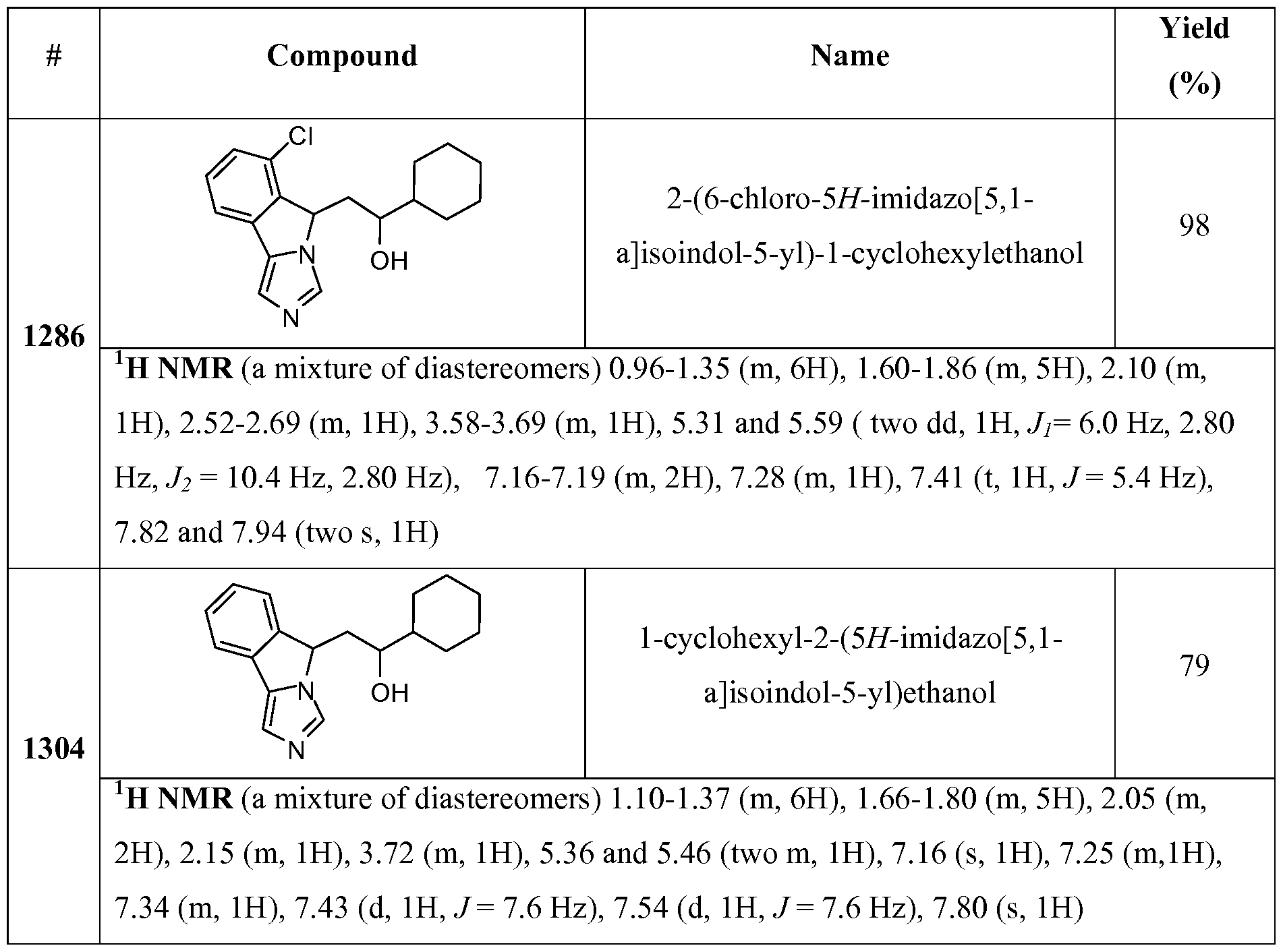



































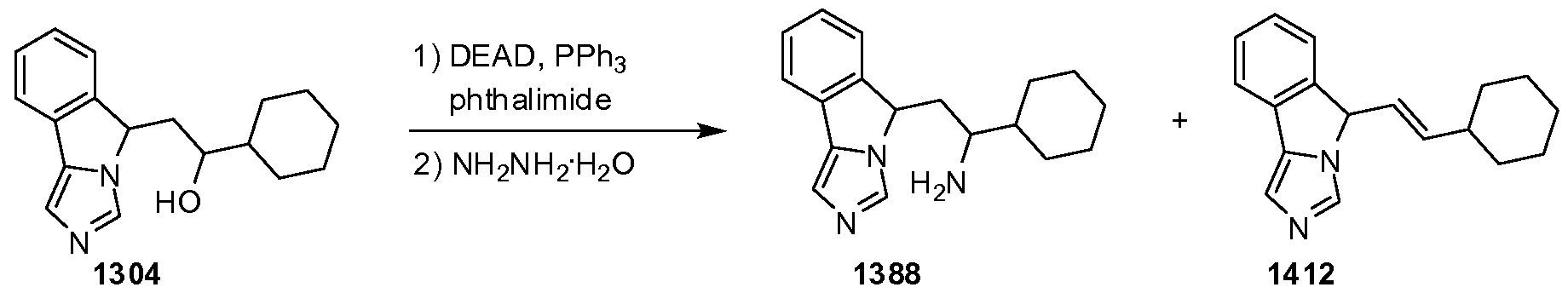
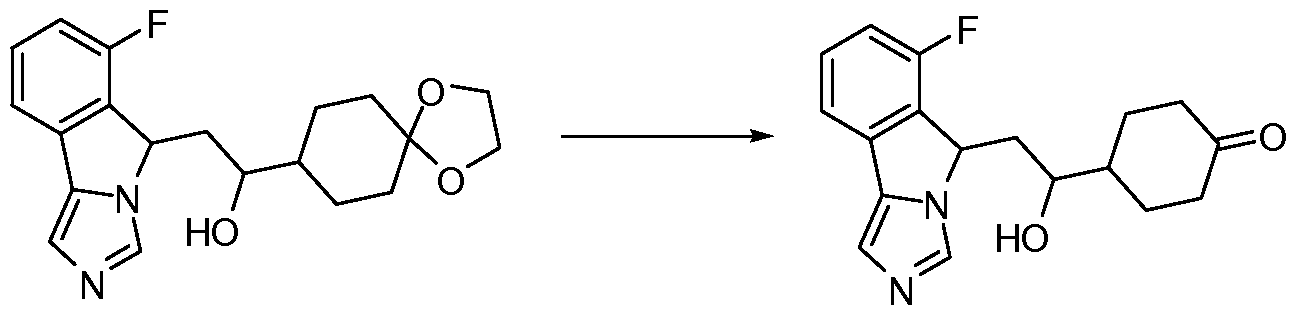



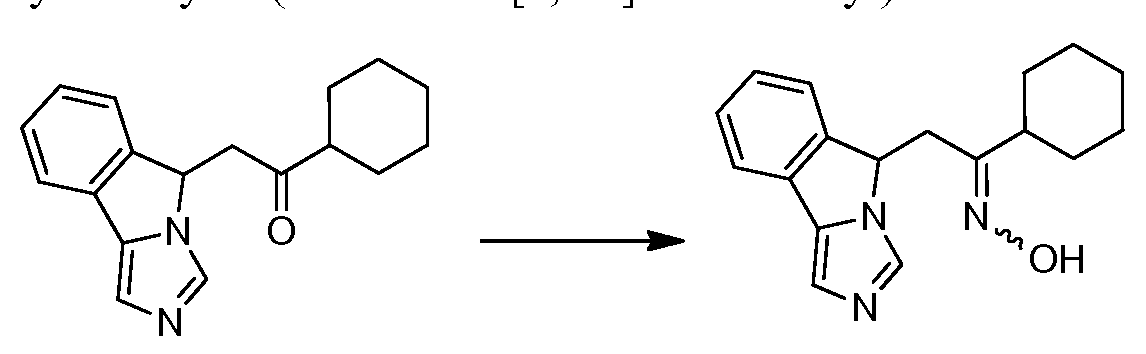






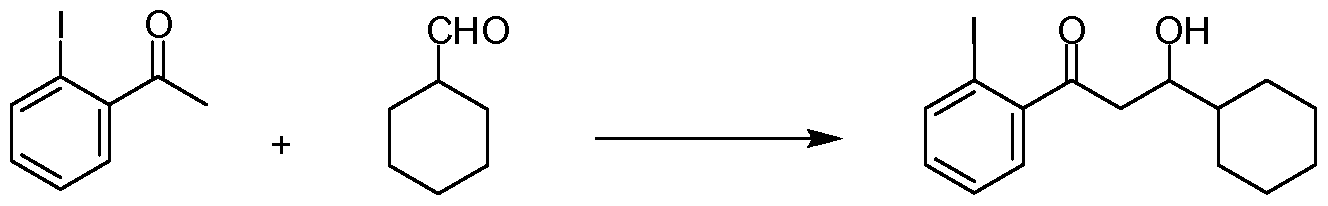






































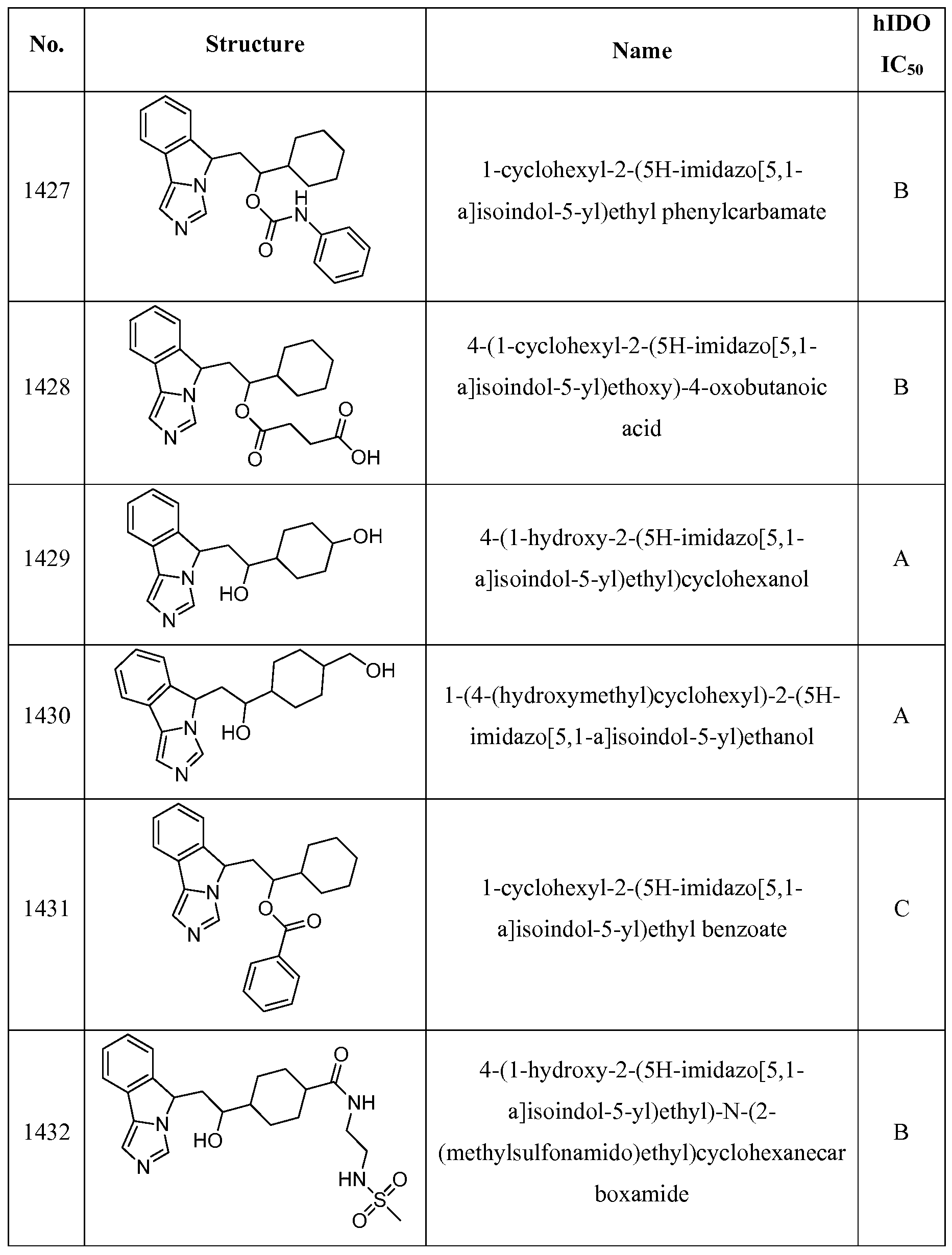








Claims








Priority Applications (27)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020137030438A KR102164443B1 (en) | 2011-04-15 | 2012-04-12 | Fused imidazole derivatives useful as ido inhibitors |
| CA2833296A CA2833296C (en) | 2011-04-15 | 2012-04-12 | Ido inhibitors |
| NZ616457A NZ616457B2 (en) | 2011-04-15 | 2012-04-12 | Fused imidazole derivatives useful as ido inhibitors |
| MEP-2016-82A ME02417B (en) | 2011-04-15 | 2012-04-12 | Fused imidazole derivatives useful as ido inhibitors |
| ES12715295.7T ES2569665T3 (en) | 2011-04-15 | 2012-04-12 | Condensed imidazole derivatives useful as IDO inhibitors |
| EP12715295.7A EP2697227B1 (en) | 2011-04-15 | 2012-04-12 | Fused imidazole derivatives useful as ido inhibitors |
| MX2013012021A MX340442B (en) | 2011-04-15 | 2012-04-12 | Fused imidazole derivatives useful as ido inhibitors. |
| SI201230515A SI2697227T1 (en) | 2011-04-15 | 2012-04-12 | Fused imidazole derivatives useful as ido inhibitors |
| JP2014505274A JP2014511876A (en) | 2011-04-15 | 2012-04-12 | IDO inhibitor |
| AU2012242871A AU2012242871C1 (en) | 2011-04-15 | 2012-04-12 | Fused imidazole derivatives useful as IDO inhibitors |
| DK12715295.7T DK2697227T3 (en) | 2011-04-15 | 2012-04-12 | DEHYDRATED imidazole USED AS IDO INHIBITORS |
| RU2013150811A RU2613579C2 (en) | 2011-04-15 | 2012-04-12 | Ido inhibitors |
| BR112013026494A BR112013026494A2 (en) | 2011-04-15 | 2012-04-12 | compound, pharmaceutical composition, method for treating condition |
| RS20160285A RS54723B1 (en) | 2011-04-15 | 2012-04-12 | Fused imidazole derivatives useful as ido inhibitors |
| EP17205515.4A EP3348558A1 (en) | 2011-04-15 | 2012-04-12 | Compositions comprising fused imidazole derivatives useful as ido inhibitors |
| CN201280018684.7A CN103547579B (en) | 2011-04-15 | 2012-04-12 | Fused imidazole derivatives useful as ido inhibitors |
| IL228862A IL228862A (en) | 2011-04-15 | 2013-10-14 | נגזרות אימידאזול ממוזגות לשימוש כמונעי ido |
| US14/053,440 US9260434B2 (en) | 2011-04-15 | 2013-10-14 | Fused imidazole derivatives useful as IDO inhibitors |
| HK14107271.9A HK1193822A1 (en) | 2011-04-15 | 2014-07-16 | Fused imidazole derivatives useful as ido inhibitors ido |
| US14/794,193 US9388191B2 (en) | 2011-04-15 | 2015-07-08 | Fused imidazole derivatives useful as IDO inhibitors |
| IL241846A IL241846A (en) | 2011-04-15 | 2015-09-24 | Fused imidazole derivatives useful as ido inhibitors |
| HRP20160369TT HRP20160369T1 (en) | 2011-04-15 | 2016-04-12 | Fused imidazole derivatives useful as ido inhibitors |
| SM201600130T SMT201600130B (en) | 2011-04-15 | 2016-05-04 | USEFUL IMIDAZOLIC DERIVATIVES USEFUL AS IDO INHIBITORS |
| US15/181,062 US9850248B2 (en) | 2011-04-15 | 2016-06-13 | IDO inhibitors |
| IL246515A IL246515B (en) | 2011-04-15 | 2016-06-28 | Fused imidazole derivatives useful as ido inhibitors |
| US15/800,190 US10233190B2 (en) | 2011-04-15 | 2017-11-01 | IDO inhibitors |
| US16/269,681 US20190225618A1 (en) | 2011-04-15 | 2019-02-07 | IDO Inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161475788P | 2011-04-15 | 2011-04-15 | |
| US61/475,788 | 2011-04-15 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/053,440 Continuation US9260434B2 (en) | 2011-04-15 | 2013-10-14 | Fused imidazole derivatives useful as IDO inhibitors |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2012142237A1 true WO2012142237A1 (en) | 2012-10-18 |
| WO2012142237A8 WO2012142237A8 (en) | 2012-11-22 |
Family
ID=45976556
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2012/033245 WO2012142237A1 (en) | 2011-04-15 | 2012-04-12 | Fused imidazole derivatives useful as ido inhibitors |
Country Status (30)
| Country | Link |
|---|---|
| US (5) | US9260434B2 (en) |
| EP (3) | EP2697227B1 (en) |
| JP (4) | JP2014511876A (en) |
| KR (1) | KR102164443B1 (en) |
| CN (2) | CN105111210B (en) |
| AU (4) | AU2012242871C1 (en) |
| BR (1) | BR112013026494A2 (en) |
| CA (1) | CA2833296C (en) |
| CL (1) | CL2013002990A1 (en) |
| CO (1) | CO6862146A2 (en) |
| CY (1) | CY1117440T1 (en) |
| DK (2) | DK2697227T3 (en) |
| ES (2) | ES2569665T3 (en) |
| HK (3) | HK1193822A1 (en) |
| HR (2) | HRP20160369T1 (en) |
| HU (2) | HUE027316T2 (en) |
| IL (3) | IL228862A (en) |
| LT (1) | LT3018132T (en) |
| ME (1) | ME02417B (en) |
| MX (1) | MX340442B (en) |
| NO (1) | NO2694640T3 (en) |
| NZ (2) | NZ708090A (en) |
| PE (2) | PE20181023A1 (en) |
| PL (2) | PL2697227T3 (en) |
| PT (1) | PT3018132T (en) |
| RS (2) | RS56992B1 (en) |
| RU (2) | RU2017107026A (en) |
| SI (2) | SI2697227T1 (en) |
| SM (1) | SMT201600130B (en) |
| WO (1) | WO2012142237A1 (en) |
Cited By (200)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103382187A (en) * | 2013-08-06 | 2013-11-06 | 信实生物医药(上海)有限公司 | 3-chloro-7(5)-bromo-benzo-isoxazole compounding method |
| WO2014159248A1 (en) | 2013-03-14 | 2014-10-02 | Newlink Genetics Corporation | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization |
| WO2015100282A1 (en) | 2013-12-24 | 2015-07-02 | Bristol-Myers Squibb Company | Tricyclic compounds as anticancer agents |
| WO2015187835A2 (en) | 2014-06-06 | 2015-12-10 | Bristol-Myers Squibb Company | Antibodies against glucocorticoid-induced tumor necrosis factor receptor (gitr) and uses thereof |
| US9260434B2 (en) | 2011-04-15 | 2016-02-16 | Newlink Genetics Corporation | Fused imidazole derivatives useful as IDO inhibitors |
| WO2016037026A1 (en) * | 2014-09-05 | 2016-03-10 | Merck Patent Gmbh | Cyclohexyl-ethyl substituted diaza- and triaza-tricyclic compounds as indole-amine-2,3-dioxygenase (ido) antagonists for the treatment of cancer |
| WO2016051181A1 (en) * | 2014-10-01 | 2016-04-07 | Redx Pharma Plc | 4h-imidazo[1,5-a]indole derivatives and their use as indoleamine 2,3-dioxygenase (ido) and/or tryptophan 2,3-dioxygenase (td02) modulators |
| WO2016059412A1 (en) * | 2014-10-15 | 2016-04-21 | Redx Pharma Plc | 6,7-heterocyclic fused 5h-pyrrolo[1,2-c]imidazole derivatives and their use as indoleamine 2,3-dioxygenase (ido) and/or tryptophan 2,3-dioxygenase (td02) modulators |
| WO2016081748A2 (en) | 2014-11-21 | 2016-05-26 | Bristol-Myers Squibb Company | Antibodies against cd73 and uses thereof |
| JP2016518329A (en) * | 2013-03-14 | 2016-06-23 | キュラデブ ファーマ プライベート リミテッド | Inhibitors of the kynurenine pathway |
| WO2016106266A1 (en) | 2014-12-22 | 2016-06-30 | Bristol-Myers Squibb Company | TGFβ RECEPTOR ANTAGONISTS |
| WO2016127052A1 (en) | 2015-02-05 | 2016-08-11 | Bristol-Myers Squibb Company | Cxcl11 and smica as predictive biomarkers for efficacy of anti-ctla4 immunotherapy |
| WO2016131381A1 (en) * | 2015-02-16 | 2016-08-25 | Shanghai De Novo Pharmatech Co. Ltd. | Fused-ring compounds, pharmaceutical composition and uses thereof |
| WO2016140884A1 (en) | 2015-03-02 | 2016-09-09 | Rigel Pharmaceuticals, Inc. | TGF-β INHIBITORS |
| WO2016161269A1 (en) | 2015-04-03 | 2016-10-06 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase for the treatment of cancer |
| WO2016162505A1 (en) | 2015-04-08 | 2016-10-13 | F-Star Biotechnology Limited | Her2 binding agent therapies |
| WO2016165613A1 (en) | 2015-04-12 | 2016-10-20 | Hangzhou Innogate Pharma Co., Ltd. | Heterocycles useful as ido and tdo inhibitors |
| WO2016168149A1 (en) | 2015-04-13 | 2016-10-20 | Five Prime Therapeutics, Inc. | Combination therapy for cancer |
| WO2016169421A1 (en) * | 2015-04-21 | 2016-10-27 | 江苏恒瑞医药股份有限公司 | Imidazo isoindole derivative, preparation method therefor and medical use thereof |
| WO2016183114A1 (en) | 2015-05-11 | 2016-11-17 | Bristol-Myers Squibb Company | Tricyclic compounds as anticancer agents |
| WO2016183118A1 (en) | 2015-05-12 | 2016-11-17 | Bristol-Myers Squibb Company | Tricyclic compounds as anticancer agents |
| WO2016183115A1 (en) | 2015-05-12 | 2016-11-17 | Bristol-Myers Squibb Company | 5h-pyrido[3,2-b]indole compounds as anticancer agents |
| WO2016196228A1 (en) | 2015-05-29 | 2016-12-08 | Bristol-Myers Squibb Company | Antibodies against ox40 and uses thereof |
| WO2017004016A1 (en) | 2015-06-29 | 2017-01-05 | The Rockefeller University | Antibodies to cd40 with enhanced agonist activity |
| WO2017009842A2 (en) | 2015-07-16 | 2017-01-19 | Biokine Therapeutics Ltd. | Compositions and methods for treating cancer |
| WO2017019757A1 (en) | 2015-07-28 | 2017-02-02 | Bristol-Myers Squibb Company | Tgf beta receptor antagonists |
| WO2017019175A1 (en) | 2015-07-24 | 2017-02-02 | Newlink Genetics Corporation | Salts and prodrugs of 1-methyl-d-tryptophan |
| WO2017035118A1 (en) | 2015-08-25 | 2017-03-02 | Bristol-Myers Squibb Company | Tgf beta receptor antagonists |
| WO2017079117A1 (en) | 2015-11-02 | 2017-05-11 | Five Prime Therapeutics, Inc. | Cd80 extracellular domain polypeptides and their use in cancer treatment |
| WO2017087678A2 (en) | 2015-11-19 | 2017-05-26 | Bristol-Myers Squibb Company | Antibodies against glucocorticoid-induced tumor necrosis factor receptor (gitr) and uses thereof |
| WO2017091580A1 (en) | 2015-11-23 | 2017-06-01 | Five Prime Therapeutics, Inc. | Predicting response to cancer treatment with fgfr2 inhibitors |
| WO2017106291A1 (en) | 2015-12-15 | 2017-06-22 | Bristol-Myers Squibb Company | Cxcr4 receptor antagonists |
| WO2017107979A1 (en) | 2015-12-24 | 2017-06-29 | Genentech, Inc. | Tdo2 inhibitors |
| WO2017134555A1 (en) | 2016-02-02 | 2017-08-10 | Emcure Pharmaceuticals Limited | Derivatives of pyrroloimidazole or analogues thereof which are useful for the treatment of inter alia cancer |
| WO2017140835A1 (en) | 2016-02-19 | 2017-08-24 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for the treatment of obesity |
| WO2017140272A1 (en) * | 2016-02-19 | 2017-08-24 | 正大天晴药业集团股份有限公司 | Tricyclic compound acting as immunomodulator |
| WO2017152085A1 (en) | 2016-03-04 | 2017-09-08 | Bristol-Myers Squibb Company | Combination therapy with anti-cd73 antibodies |
| WO2017149469A1 (en) | 2016-03-03 | 2017-09-08 | Emcure Pharmaceuticals Limited | Heterocyclic compounds useful as ido and/or tdo modulators |
| CN107176956A (en) * | 2017-05-31 | 2017-09-19 | 成都海博锐药业有限公司 | A kind of IDO inhibitor compound, Pharmaceutical composition, purposes |
| GB2548542A (en) * | 2015-06-16 | 2017-09-27 | Redx Pharma Plc | Compounds |
| WO2017184619A2 (en) | 2016-04-18 | 2017-10-26 | Celldex Therapeutics, Inc. | Agonistic antibodies that bind human cd40 and uses thereof |
| WO2017197055A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| WO2017197036A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Spirocyclic degronimers for target protein degradation |
| WO2017197046A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | C3-carbon linked glutarimide degronimers for target protein degradation |
| WO2017198159A1 (en) * | 2016-05-16 | 2017-11-23 | 鲁南制药集团股份有限公司 | Imidazole derivative containing bridge ring |
| WO2017213919A1 (en) | 2016-06-10 | 2017-12-14 | Eli Lilly And Company | 1-tetrahydropyranylcarbonyl-2,3-dihydro-1h-indole compounds for treating cancer |
| WO2018013818A2 (en) | 2016-07-14 | 2018-01-18 | Bristol-Myers Squibb Company | Antibodies against tim3 and uses thereof |
| WO2018017633A1 (en) | 2016-07-21 | 2018-01-25 | Bristol-Myers Squibb Company | TGF Beta RECEPTOR ANTAGONISTS |
| WO2018054365A1 (en) | 2016-09-24 | 2018-03-29 | Beigene, Ltd. | NOVEL 5 or 8-SUBSTITUTED IMIDAZO [1, 5-a] PYRIDINES AS SELECTIVE INHIBITORS OF INDOLEAMINE AND/OR TRYPTOPHANE 2, 3-DIOXYGENASES |
| WO2018071873A2 (en) | 2016-10-13 | 2018-04-19 | Juno Therapeutics, Inc. | Immunotherapy methods and compositions involving tryptophan metabolic pathway modulators |
| WO2018083635A2 (en) | 2016-11-04 | 2018-05-11 | Auckland Uniservices Limited | Tricyclic heterocyclic derivatives and uses thereof |
| WO2018132279A1 (en) | 2017-01-05 | 2018-07-19 | Bristol-Myers Squibb Company | Tgf beta receptor antagonists |
| WO2018175954A1 (en) | 2017-03-23 | 2018-09-27 | F. Hoffmann-La Roche Ag | Synthesis of imidazo[5,1-a]isoindole derivative useful as ido inhibitors |
| WO2018183608A1 (en) | 2017-03-31 | 2018-10-04 | Five Prime Therapeutics, Inc. | Combination therapy for cancer using anti-gitr antibodies |
| WO2018187613A2 (en) | 2017-04-07 | 2018-10-11 | Bristol-Myers Squibb Company | Anti-icos agonist antibodies and uses thereof |
| WO2018195397A2 (en) | 2017-04-21 | 2018-10-25 | Kyn Therapeutics | Indole ahr inhibitors and uses thereof |
| WO2018201014A1 (en) | 2017-04-28 | 2018-11-01 | Five Prime Therapeutics, Inc. | Methods of treatment with cd80 extracellular domain polypeptides |
| WO2018209049A1 (en) | 2017-05-12 | 2018-11-15 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| CN108884103A (en) * | 2016-02-19 | 2018-11-23 | 正大天晴药业集团股份有限公司 | Three and cycle compound as immunomodulator |
| WO2019006047A1 (en) | 2017-06-28 | 2019-01-03 | Genentech, Inc. | Tdo2 and ido1 inhibitors |
| WO2019006283A1 (en) | 2017-06-30 | 2019-01-03 | Bristol-Myers Squibb Company | Amorphous and crystalline forms of ido inhibitors |
| WO2019005559A1 (en) | 2017-06-28 | 2019-01-03 | Genentech, Inc. | Tdo2 and ido1 inhibitors |
| WO2019023459A1 (en) | 2017-07-28 | 2019-01-31 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| WO2019029507A1 (en) * | 2017-08-08 | 2019-02-14 | 江苏恒瑞医药股份有限公司 | Preparation method for imidazoisoindole derivatives |
| WO2019034725A1 (en) | 2017-08-17 | 2019-02-21 | Idorsia Pharmaceuticals Ltd | Inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan 2,3-dioxygenase |
| WO2019046498A1 (en) | 2017-08-31 | 2019-03-07 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| WO2019046496A1 (en) | 2017-08-31 | 2019-03-07 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| WO2019046500A1 (en) | 2017-08-31 | 2019-03-07 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| WO2019074822A1 (en) | 2017-10-09 | 2019-04-18 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2019074824A1 (en) | 2017-10-09 | 2019-04-18 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2019074887A1 (en) | 2017-10-10 | 2019-04-18 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| WO2019075090A1 (en) | 2017-10-10 | 2019-04-18 | Tilos Therapeutics, Inc. | Anti-lap antibodies and uses thereof |
| WO2019079261A1 (en) | 2017-10-16 | 2019-04-25 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| WO2019089921A1 (en) | 2017-11-01 | 2019-05-09 | Bristol-Myers Squibb Company | Immunostimulatory agonistic antibodies for use in treating cancer |
| WO2019090198A1 (en) | 2017-11-06 | 2019-05-09 | Bristol-Myers Squibb Company | Isofuranone compounds useful as hpk1 inhibitors |
| US10323004B2 (en) | 2016-05-04 | 2019-06-18 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2019133747A1 (en) | 2017-12-27 | 2019-07-04 | Bristol-Myers Squibb Company | Anti-cd40 antibodies and uses thereof |
| WO2019136112A1 (en) | 2018-01-05 | 2019-07-11 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2019138107A1 (en) | 2018-01-15 | 2019-07-18 | Idorsia Pharmaceuticals Ltd | Inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan 2,3-dioxygenase |
| WO2019140229A1 (en) | 2018-01-12 | 2019-07-18 | Bristol-Myers Squibb Company | Antibodies against tim3 and uses thereof |
| WO2019160884A1 (en) | 2018-02-13 | 2019-08-22 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| TWI671302B (en) * | 2016-12-20 | 2019-09-11 | 大陸商深圳微芯生物科技有限責任公司 | Fused imidazole compounds that may inhibit indoleamine 2,3-dioxygenase |
| WO2019173587A1 (en) | 2018-03-08 | 2019-09-12 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| WO2019183040A1 (en) | 2018-03-21 | 2019-09-26 | Five Prime Therapeutics, Inc. | ANTIBODIES BINDING TO VISTA AT ACIDIC pH |
| WO2019200256A1 (en) | 2018-04-12 | 2019-10-17 | Bristol-Myers Squibb Company | Anticancer combination therapy with cd73 antagonist antibody and pd-1/pd-l1 axis antagonist antibody |
| WO2019204257A1 (en) | 2018-04-16 | 2019-10-24 | Arrys Therapeutics, Inc. | Ep4 inhibitors and use thereof |
| WO2019213340A1 (en) | 2018-05-03 | 2019-11-07 | Bristol-Myers Squibb Company | Uracil derivatives as mer-axl inhibitors |
| US10508085B2 (en) | 2016-09-22 | 2019-12-17 | Plexxikon Inc. | Compounds and methods for IDO and TDO modulation, and indications therefor |
| WO2019243832A1 (en) | 2018-06-22 | 2019-12-26 | Bicycletx Limited | Bicyclic peptide ligands specific for nectin-4 |
| WO2020006016A1 (en) | 2018-06-27 | 2020-01-02 | Bristol-Myers Squibb Company | Naphthyridinone compounds useful as t cell activators |
| WO2020006018A1 (en) | 2018-06-27 | 2020-01-02 | Bristol-Myers Squibb Company | Substituted naphthyridinone compounds useful as t cell activators |
| US10525035B2 (en) | 2014-12-18 | 2020-01-07 | Lankenau Institute For Medical Research | Methods and compositions for the treatment of retinopathy and other ocular diseases |
| WO2020010177A1 (en) | 2018-07-06 | 2020-01-09 | Kymera Therapeutics, Inc. | Tricyclic crbn ligands and uses thereof |
| WO2020014327A2 (en) | 2018-07-11 | 2020-01-16 | Five Prime Therapeutics, Inc. | Antibodies binding to vista at acidic ph |
| WO2020014132A2 (en) | 2018-07-09 | 2020-01-16 | Five Prime Therapeutics, Inc. | Antibodies binding to ilt4 |
| US10544099B2 (en) | 2016-05-04 | 2020-01-28 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2020023355A1 (en) | 2018-07-23 | 2020-01-30 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2020023356A1 (en) | 2018-07-23 | 2020-01-30 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| CN110872289A (en) * | 2015-04-10 | 2020-03-10 | 百济神州(北京)生物科技有限公司 | Novel 8-substituted imidazo [1,5-a ] pyridines as IDO1 and/or TDO inhibitors |
| WO2020051424A1 (en) | 2018-09-07 | 2020-03-12 | Pic Therapeutics | Eif4e inhibitors and uses thereof |
| WO2020076969A2 (en) | 2018-10-10 | 2020-04-16 | Tilos Therapeutics, Inc. | Anti-lap antibody variants and uses thereof |
| US10633342B2 (en) | 2016-05-04 | 2020-04-28 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2020102501A1 (en) | 2018-11-16 | 2020-05-22 | Bristol-Myers Squibb Company | Anti-nkg2a antibodies and uses thereof |
| EP3670659A1 (en) | 2018-12-20 | 2020-06-24 | Abivax | Biomarkers, and uses in treatment of viral infections, inflammations, or cancer |
| WO2020132561A1 (en) | 2018-12-20 | 2020-06-25 | C4 Therapeutics, Inc. | Targeted protein degradation |
| US10696650B2 (en) | 2017-08-17 | 2020-06-30 | Ikena Oncology, Inc. | AHR inhibitors and uses thereof |
| US10696648B2 (en) | 2016-05-04 | 2020-06-30 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2020187998A1 (en) | 2019-03-19 | 2020-09-24 | Fundació Privada Institut D'investigació Oncològica De Vall Hebron | Combination therapy with omomyc and an antibody binding pd-1 or ctla-4 for the treatment of cancer |
| US10793563B2 (en) | 2018-01-29 | 2020-10-06 | Merck Patent Gmbh | GCN2 inhibitors and uses thereof |
| WO2020201753A1 (en) | 2019-04-02 | 2020-10-08 | Bicycletx Limited | Bicycle toxin conjugates and uses thereof |
| WO2020231766A1 (en) | 2019-05-13 | 2020-11-19 | Bristol-Myers Squibb Company | AGONISTS OF ROR GAMMAt |
| WO2020231713A1 (en) | 2019-05-13 | 2020-11-19 | Bristol-Myers Squibb Company | AGONISTS OF ROR GAMMAt |
| WO2020243423A1 (en) | 2019-05-31 | 2020-12-03 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| US10874743B2 (en) | 2017-12-26 | 2020-12-29 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| WO2021005222A1 (en) | 2019-07-11 | 2021-01-14 | Idorsia Pharmaceuticals Ltd | Inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan 2,3-dioxygenase |
| WO2021026179A1 (en) | 2019-08-06 | 2021-02-11 | Bristol-Myers Squibb Company | AGONISTS OF ROR GAMMAt |
| WO2021041588A1 (en) | 2019-08-28 | 2021-03-04 | Bristol-Myers Squibb Company | Substituted pyridopyrimidinonyl compounds useful as t cell activators |
| WO2021055698A1 (en) | 2019-09-19 | 2021-03-25 | Bristol-Myers Squibb Company | Antibodies binding to vista at acidic ph |
| US10959986B2 (en) | 2018-08-29 | 2021-03-30 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| US10988477B2 (en) | 2018-01-29 | 2021-04-27 | Merck Patent Gmbh | GCN2 inhibitors and uses thereof |
| WO2021101919A1 (en) | 2019-11-19 | 2021-05-27 | Bristol-Myers Squibb Company | Compounds useful as inhibitors of helios protein |
| US11021481B2 (en) | 2019-09-13 | 2021-06-01 | Nimbus Saturn, Inc. | Substituted isoindolin-1-ones and 2,3-dihydro-1h-pyrrolo[3,4-c]pyridin-1-ones as HPK1 antagonists |
| WO2021108528A1 (en) | 2019-11-26 | 2021-06-03 | Ikena Oncology, Inc. | Polymorphic carbazole derivatives and uses thereof |
| WO2021108288A1 (en) | 2019-11-26 | 2021-06-03 | Bristol-Myers Squibb Company | Salts/cocrystals of (r)-n-(4-chlorophenyl)-2-((1s,4s)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide |
| US11046649B2 (en) | 2018-07-17 | 2021-06-29 | Board Of Regents, The University Of Texas System | Compounds useful as inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan dioxygenase |
| WO2021133750A1 (en) | 2019-12-23 | 2021-07-01 | Bristol-Myers Squibb Company | Substituted bicyclic piperidine derivatives useful as t cell activators |
| WO2021133749A1 (en) | 2019-12-23 | 2021-07-01 | Bristol-Myers Squibb Company | Substituted piperazine derivatives useful as t cell activators |
| WO2021133751A1 (en) | 2019-12-23 | 2021-07-01 | Bristol-Myers Squibb Company | Substituted quinazolinyl compounds useful as t cell activators |
| WO2021133752A1 (en) | 2019-12-23 | 2021-07-01 | Bristol-Myers Squibb Company | Substituted heteroaryl compounds useful as t cell activators |
| WO2021133748A1 (en) | 2019-12-23 | 2021-07-01 | Bristol-Myers Squibb Company | Substituted quinolinonyl piperazine compounds useful as t cell activators |
| WO2021141907A1 (en) | 2020-01-06 | 2021-07-15 | Hifibio (Hong Kong) Limited | Anti-tnfr2 antibody and uses thereof |
| WO2021139682A1 (en) | 2020-01-07 | 2021-07-15 | Hifibio (Hk) Limited | Anti-galectin-9 antibody and uses thereof |
| US11066383B2 (en) | 2016-05-04 | 2021-07-20 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2021146370A1 (en) | 2020-01-15 | 2021-07-22 | Blueprint Medicines Corporation | Map4k1 inhibitors |
| WO2021178488A1 (en) | 2020-03-03 | 2021-09-10 | PIC Therapeutics, Inc. | Eif4e inhibitors and uses thereof |
| US11117889B1 (en) | 2018-11-30 | 2021-09-14 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| WO2021183428A1 (en) | 2020-03-09 | 2021-09-16 | Bristol-Myers Squibb Company | Antibodies to cd40 with enhanced agonist activity |
| WO2021194914A1 (en) | 2020-03-23 | 2021-09-30 | Bristol-Myers Squibb Company | Substituted oxoisoindoline compounds for the treatment of cancer |
| WO2021207449A1 (en) | 2020-04-09 | 2021-10-14 | Merck Sharp & Dohme Corp. | Affinity matured anti-lap antibodies and uses thereof |
| US11149011B2 (en) | 2018-03-20 | 2021-10-19 | Plexxikon Inc. | Compounds and methods for IDO and TDO modulation, and indications therefor |
| US11173145B2 (en) | 2017-01-17 | 2021-11-16 | Board Of Regents, The University Of Texas System | Compounds useful as inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan dioxygenase |
| WO2021231732A1 (en) | 2020-05-15 | 2021-11-18 | Bristol-Myers Squibb Company | Antibodies to garp |
| WO2021258010A1 (en) | 2020-06-19 | 2021-12-23 | Gossamer Bio Services, Inc. | Oxime compounds useful as t cell activators |
| US11242393B2 (en) | 2018-03-23 | 2022-02-08 | Bristol-Myers Squibb Company | Antibodies against MICA and/or MICB and uses thereof |
| WO2022033419A2 (en) | 2020-08-10 | 2022-02-17 | Shanghai Xbh Biotechnology Co., Ltd. | Compositions and methods for treating autoimmune diseases and cancers by targeting igsf8 |
| US11253525B2 (en) | 2018-08-29 | 2022-02-22 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2022038158A1 (en) | 2020-08-17 | 2022-02-24 | Bicycletx Limited | Bicycle conjugates specific for nectin-4 and uses thereof |
| WO2022081718A1 (en) | 2020-10-14 | 2022-04-21 | Five Prime Therapeutics, Inc. | Anti-c-c chemokine receptor 8 (ccr8) antibodies and methods of use thereof |
| US11351164B2 (en) | 2016-08-26 | 2022-06-07 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2022120353A1 (en) | 2020-12-02 | 2022-06-09 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| WO2022120354A1 (en) | 2020-12-02 | 2022-06-09 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| US11358948B2 (en) | 2017-09-22 | 2022-06-14 | Kymera Therapeutics, Inc. | CRBN ligands and uses thereof |
| WO2022133083A1 (en) | 2020-12-16 | 2022-06-23 | Gossamer Bio Services, Inc. | Compounds useful as t cell activators |
| WO2022148979A1 (en) | 2021-01-11 | 2022-07-14 | Bicycletx Limited | Methods for treating cancer |
| WO2022169921A1 (en) | 2021-02-04 | 2022-08-11 | Bristol-Myers Squibb Company | Benzofuran compounds as sting agonists |
| WO2022167457A1 (en) | 2021-02-02 | 2022-08-11 | Liminal Biosciences Limited | Gpr84 antagonists and uses thereof |
| WO2022167445A1 (en) | 2021-02-02 | 2022-08-11 | Liminal Biosciences Limited | Gpr84 antagonists and uses thereof |
| WO2022171745A1 (en) | 2021-02-12 | 2022-08-18 | F. Hoffmann-La Roche Ag | Bicyclic tetrahydroazepine derivatives for the treatment of cancer |
| EP4052705A1 (en) | 2021-03-05 | 2022-09-07 | Universität Basel Vizerektorat Forschung | Compositions for the treatment of ebv associated diseases or conditions |
| WO2022184930A2 (en) | 2021-03-05 | 2022-09-09 | Universität Basel | Compositions for the treatment of ebv associated diseases or conditions |
| WO2022192145A1 (en) | 2021-03-08 | 2022-09-15 | Blueprint Medicines Corporation | Map4k1 inhibitors |
| WO2022197641A1 (en) | 2021-03-15 | 2022-09-22 | Rapt Therapeutics, Inc. | 1h-pyrazolo[3,4-d]pyrimidin-6-yl-amine derivatives as hematopoietic progenitor kinase 1 (hpk1) modulators and/or inhibitors for the treatment of cancer and other diseases |
| WO2022212876A1 (en) | 2021-04-02 | 2022-10-06 | The Regents Of The University Of California | Antibodies against cleaved cdcp1 and uses thereof |
| WO2022216644A1 (en) | 2021-04-06 | 2022-10-13 | Bristol-Myers Squibb Company | Pyridinyl substituted oxoisoindoline compounds |
| WO2022216573A1 (en) | 2021-04-05 | 2022-10-13 | Bristol-Myers Squibb Company | Pyridinyl substituted oxoisoindoline compounds for the treatment of cancer |
| US11472788B2 (en) | 2017-11-25 | 2022-10-18 | Beigene, Ltd. | Benzoimidazoles as selective inhibitors of indoleamine 2,3-dioxygenases |
| WO2022221866A1 (en) | 2021-04-16 | 2022-10-20 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| US11485743B2 (en) | 2018-01-12 | 2022-11-01 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| US11485750B1 (en) | 2019-04-05 | 2022-11-01 | Kymera Therapeutics, Inc. | STAT degraders and uses thereof |
| US11512080B2 (en) | 2018-01-12 | 2022-11-29 | Kymera Therapeutics, Inc. | CRBN ligands and uses thereof |
| WO2023288254A1 (en) | 2021-07-14 | 2023-01-19 | Blueprint Medicines Corporation | Heterocyclic compounds as map4k1 inhibitors |
| WO2023288264A1 (en) | 2021-07-15 | 2023-01-19 | Blueprint Medicines Corporation | Map4k1 inhibitors |
| US11591332B2 (en) | 2019-12-17 | 2023-02-28 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| WO2023028235A1 (en) | 2021-08-25 | 2023-03-02 | PIC Therapeutics, Inc. | Eif4e inhibitors and uses thereof |
| WO2023028238A1 (en) | 2021-08-25 | 2023-03-02 | PIC Therapeutics, Inc. | Eif4e inhibitors and uses thereof |
| US11623932B2 (en) | 2017-09-22 | 2023-04-11 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| WO2023114984A1 (en) | 2021-12-17 | 2023-06-22 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| US11685750B2 (en) | 2020-06-03 | 2023-06-27 | Kymera Therapeutics, Inc. | Crystalline forms of IRAK degraders |
| WO2023122772A1 (en) | 2021-12-22 | 2023-06-29 | Gossamer Bio Services, Inc. | Oxime derivatives useful as t cell activators |
| WO2023122778A1 (en) | 2021-12-22 | 2023-06-29 | Gossamer Bio Services, Inc. | Pyridazinone derivatives useful as t cell activators |
| WO2023122777A1 (en) | 2021-12-22 | 2023-06-29 | Gossamer Bio Services, Inc. | Oxime derivatives useful as t cell activators |
| US11707457B2 (en) | 2019-12-17 | 2023-07-25 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| WO2023150186A1 (en) | 2022-02-01 | 2023-08-10 | Arvinas Operations, Inc. | Dgk targeting compounds and uses thereof |
| WO2023173053A1 (en) | 2022-03-10 | 2023-09-14 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| WO2023173057A1 (en) | 2022-03-10 | 2023-09-14 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| EP4249066A2 (en) | 2014-12-23 | 2023-09-27 | Bristol-Myers Squibb Company | Antibodies to tigit |
| WO2023211889A1 (en) | 2022-04-25 | 2023-11-02 | Ikena Oncology, Inc. | Polymorphic compounds and uses thereof |
| WO2023230205A1 (en) | 2022-05-25 | 2023-11-30 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| US11857535B2 (en) | 2020-07-30 | 2024-01-02 | Kymera Therapeutics, Inc. | Methods of treating mutant lymphomas |
| WO2024028364A1 (en) | 2022-08-02 | 2024-02-08 | Liminal Biosciences Limited | Aryl-triazolyl and related gpr84 antagonists and uses thereof |
| WO2024028363A1 (en) | 2022-08-02 | 2024-02-08 | Liminal Biosciences Limited | Heteroaryl carboxamide and related gpr84 antagonists and uses thereof |
| WO2024028365A1 (en) | 2022-08-02 | 2024-02-08 | Liminal Biosciences Limited | Substituted pyridone gpr84 antagonists and uses thereof |
| WO2024033458A1 (en) | 2022-08-11 | 2024-02-15 | F. Hoffmann-La Roche Ag | Bicyclic tetrahydroazepine derivatives |
| WO2024033388A1 (en) | 2022-08-11 | 2024-02-15 | F. Hoffmann-La Roche Ag | Bicyclic tetrahydrothiazepine derivatives |
| WO2024033457A1 (en) | 2022-08-11 | 2024-02-15 | F. Hoffmann-La Roche Ag | Bicyclic tetrahydrothiazepine derivatives |
| WO2024036101A1 (en) | 2022-08-09 | 2024-02-15 | Bristol-Myers Squibb Company | Tertiary amine substituted bicyclic compounds useful as t cell activators |
| WO2024036100A1 (en) | 2022-08-08 | 2024-02-15 | Bristol-Myers Squibb Company | Substituted tetrazolyl compounds useful as t cell activators |
| WO2024033389A1 (en) | 2022-08-11 | 2024-02-15 | F. Hoffmann-La Roche Ag | Bicyclic tetrahydrothiazepine derivatives |
| US11926625B2 (en) | 2021-03-05 | 2024-03-12 | Nimbus Saturn, Inc. | HPK1 antagonists and uses thereof |
| US11932624B2 (en) | 2020-03-19 | 2024-03-19 | Kymera Therapeutics, Inc. | MDM2 degraders and uses thereof |
Families Citing this family (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9693957B2 (en) | 2011-07-08 | 2017-07-04 | The University Of North Carolina At Chapel Hill | Metal bisphosphonate nanoparticles for anti-cancer therapy and imaging and for treating bone disorders |
| GB201311888D0 (en) | 2013-07-03 | 2013-08-14 | Glaxosmithkline Ip Dev Ltd | Novel compounds |
| GB201311891D0 (en) | 2013-07-03 | 2013-08-14 | Glaxosmithkline Ip Dev Ltd | Novel compound |
| CN106256830B (en) * | 2015-06-18 | 2019-03-08 | 成都海创药业有限公司 | A kind of deuterated IDO inhibitor and its preparation method and application |
| CN105037371A (en) * | 2015-06-30 | 2015-11-11 | 西华大学 | Deuterated indoleamine-2,3-dioxygenase inhibitor |
| GB201511790D0 (en) | 2015-07-06 | 2015-08-19 | Iomet Pharma Ltd | Pharmaceutical compound |
| EP3337476A4 (en) | 2015-08-19 | 2019-09-04 | Arvinas, Inc. | Compounds and methods for the targeted degradation of bromodomain-containing proteins |
| CN106478634B (en) * | 2015-09-01 | 2020-05-22 | 尚华医药科技(江西)有限公司 | Fused imidazole compounds, preparation method, pharmaceutical composition and use thereof |
| US10308647B2 (en) | 2015-10-29 | 2019-06-04 | Scifluor Life Sciences, Inc. | Fused imidazole derivatives as IDO/TDO inhibitors |
| WO2017080934A1 (en) * | 2015-11-09 | 2017-05-18 | F. Hoffmann-La Roche Ag | Screening assay to identify id01 and/or tdo modulators |
| CN107056785B (en) * | 2016-01-02 | 2021-06-22 | 杭州英创医药科技有限公司 | Heterocyclic compounds as IDO and TDO inhibitors |
| CN105732643A (en) * | 2016-04-18 | 2016-07-06 | 苏州大学 | Conjugate and preparation method thereof and application to preparation of IDO enzyme inhibitor and non-steroidal anti-inflammatory drugs |
| CN107312005B (en) * | 2016-04-27 | 2021-12-17 | 上海翰森生物医药科技有限公司 | Fused imidazole derivative with IDO/TDO inhibitory activity and preparation method and application thereof |
| CN107556316B (en) * | 2016-06-30 | 2021-11-12 | 鲁南制药集团股份有限公司 | Bridged ring-containing imidazole derivatives |
| CN107383012B (en) * | 2016-05-16 | 2021-09-28 | 鲁南制药集团股份有限公司 | Bicyclic imidazole alcohol derivatives |
| CN105902542A (en) * | 2016-05-16 | 2016-08-31 | 张阳 | Application of conjugate in preparation of medicine for treating cardiovascular diseases |
| CN106957318B (en) * | 2016-05-19 | 2019-12-10 | 中国科学院上海有机化学研究所 | Condensed polycyclic indoline compound, preparation method, pharmaceutical composition and application thereof |
| US11246877B2 (en) | 2016-05-20 | 2022-02-15 | The University Of Chicago | Nanoparticles for chemotherapy, targeted therapy, photodynamic therapy, immunotherapy, and any combination thereof |
| CN107488179A (en) * | 2016-06-11 | 2017-12-19 | 鲁南制药集团股份有限公司 | Imidazoles 01 derivatives containing bridged ring |
| CN107556315B (en) * | 2016-06-30 | 2021-08-31 | 鲁南制药集团股份有限公司 | Imidazole derivatives containing four-membered rings |
| CN107663159A (en) * | 2016-07-29 | 2018-02-06 | 上海迪诺医药科技有限公司 | Polycyclic compound, its pharmaceutical composition and application |
| WO2018028491A1 (en) * | 2016-08-09 | 2018-02-15 | 苏州国匡医药科技有限公司 | Indoleamine2,3-dioxygenase inhibitors and uses thereof in pharmacy |
| WO2018045966A1 (en) * | 2016-09-12 | 2018-03-15 | 广州必贝特医药技术有限公司 | Imidazole-containing fused tricyclic compounds and applications thereof |
| CN108778332B (en) * | 2016-10-21 | 2019-10-18 | 苏州盛迪亚生物医药有限公司 | PD-1 antibody is combined with IDO inhibitor is preparing the purposes in anti-tumor drug |
| TW201815793A (en) * | 2016-10-21 | 2018-05-01 | 江蘇恆瑞醫藥股份有限公司 | Crystalline form of free alkali of imidazo isoindole derivative and a preparation method thereof |
| CN106474468B (en) * | 2016-11-23 | 2020-03-27 | 中国医学科学院医学生物学研究所 | Compound adjuvant, vaccine containing compound adjuvant and preparation method of vaccine |
| WO2018106579A1 (en) * | 2016-12-06 | 2018-06-14 | Albert Einstein College Of Medicine, Inc. | Drug targeting of human indoleamine 2,3-dioxygenase |
| DK3559009T3 (en) | 2016-12-22 | 2021-05-03 | Calithera Biosciences Inc | COMPOSITIONS AND METHODS FOR INHIBITATION OF ARGINASE ACTIVITY |
| CN108239091B (en) * | 2016-12-26 | 2021-08-13 | 中国医学科学院药物研究所 | Resolution of 1-cyclohexyl-2- (5H-imidazo [5,1-a ] isoindol) ethyl-1-one |
| JP7364552B2 (en) | 2017-08-02 | 2023-10-18 | ザ ユニバーシティ オブ シカゴ | Nanoscale metal-organic layers and metal-organic nanoplates for X-ray-induced photodynamic therapy, radiotherapy, radiodynamic therapy, chemotherapy, immunotherapy, and any combination thereof |
| CN109384791B (en) * | 2017-08-09 | 2020-09-11 | 江苏恒瑞医药股份有限公司 | Crystal form of imidazo isoindole derivative free alkali and preparation method thereof |
| US11236107B2 (en) | 2017-08-18 | 2022-02-01 | Chia Tai Tianqing Pharmaceutical Group Co., Ltd. | Crystal of tricyclic compound |
| CN107501272B (en) * | 2017-09-05 | 2020-03-31 | 中国药科大学 | Imidazoisoindole IDO1 inhibitor, and preparation method and application thereof |
| JP2020534289A (en) | 2017-09-14 | 2020-11-26 | ランケナー インスティテュート フォー メディカル リサーチ | Methods and compositions for the treatment of cancer |
| WO2019141095A1 (en) * | 2018-01-19 | 2019-07-25 | 四川科伦博泰生物医药股份有限公司 | Amidine and guanidine derivative, preparation method therefor and medical use thereof |
| KR20210066857A (en) * | 2018-09-27 | 2021-06-07 | 쉔젠 칩스크린 바이오사이언스 씨오., 엘티디. | Quinoline derivatives having indoleamine-2,3-dioxygenase inhibitory activity |
| CN111333653A (en) * | 2019-12-16 | 2020-06-26 | 山东大学 | ICD inducer-IDO inhibitor conjugate, preparation method and application |
| WO2021133920A1 (en) | 2019-12-23 | 2021-07-01 | Kymera Therapeutics, Inc. | Smarca degraders and uses thereof |
| CN111803635B (en) * | 2020-06-17 | 2023-03-14 | 中国医学科学院基础医学研究所 | Application of small molecule inhibitor in treating respiratory viral pneumonia |
| US11839659B2 (en) | 2020-07-02 | 2023-12-12 | Northwestern University | Proteolysis-targeting chimeric molecules (PROTACs) that induce degradation of indoleamine 2,3-dioxygenase (IDO) protein |
| WO2022174269A1 (en) | 2021-02-15 | 2022-08-18 | Kymera Therapeutics, Inc. | Irak4 degraders and uses thereof |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4584013A (en) | 1983-05-18 | 1986-04-22 | Ciba-Geigy Corporation | Cyclohexanedionecarboxylic acid derivatives with herbicidal and plant growth regulating properties |
| US5807892A (en) | 1994-09-30 | 1998-09-15 | Alcon Laboratories, Inc. | Use of certain prostaglandin analogues to treat glaucoma and ocular hypertension |
| WO1999029310A2 (en) | 1997-12-05 | 1999-06-17 | Medical College Of Georgia Research Institute, Inc. | Regulation of t cell-mediated immunity by tryptophan and its analogs |
| WO2003087347A1 (en) | 2002-04-12 | 2003-10-23 | Medical College Of Georgia Research Institute, Inc. | Antigen-presenting cell populations and their use as reagents for enhancing or reducing immune tolerance |
| WO2004094409A1 (en) | 2003-03-27 | 2004-11-04 | Lankenau Institute For Medical Research | Novel ido inhibitors and methods of use |
| US20040234623A1 (en) | 2003-04-01 | 2004-11-25 | Medical College Of Georgia Research Institute, Inc. | Use of inhibitors of indoleamine-2,3-dioxygenase in combination with other therapeutic modalities |
| US20060025383A1 (en) | 2004-02-03 | 2006-02-02 | Neil Wishart | Aminobenzoxazoles as therapeutic agents |
| US20080306084A1 (en) | 2007-04-30 | 2008-12-11 | Gruenenthal Gmbh | Substituted Amide Compounds |
| WO2009132238A2 (en) * | 2008-04-24 | 2009-10-29 | Newlink Genetics | Ido inhibitors |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2606783C (en) * | 2005-05-10 | 2014-03-25 | Incyte Corporation | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| US7705022B2 (en) | 2005-10-27 | 2010-04-27 | Lankenau Institute For Medical Research | IDO inhibitors and methods of use thereof |
| JP5294874B2 (en) | 2005-12-20 | 2013-09-18 | インサイト・コーポレイション | N-hydroxyamidino heterocycle as modulator of indoleamine 2,3-dioxygenase |
| US8389568B2 (en) | 2007-03-16 | 2013-03-05 | Lankenau Institute For Medical Research | IDO inhibitors and methods of use thereof |
| WO2009073620A2 (en) | 2007-11-30 | 2009-06-11 | Newlink Genetics | Ido inhibitors |
| WO2009085185A1 (en) | 2007-12-19 | 2009-07-09 | Amgen Inc. | Fused pyridine, pyrimidine and triazine compounds as cell cycle inhibitors |
| PT2824100T (en) | 2008-07-08 | 2018-05-10 | Incyte Holdings Corp | 1,2,5-oxadiazoles as inhibitors of indoleamine 2,3-dioxygenase |
| ES2601226T3 (en) | 2009-10-28 | 2017-02-14 | Newlink Genetics Corporation | Imidazole derivatives as IDO inhibitors |
| NO2694640T3 (en) * | 2011-04-15 | 2018-03-17 | ||
| AU2013348167A1 (en) | 2012-11-20 | 2015-05-28 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of indoleamine 2,3-dioxygenase |
| WO2014141110A2 (en) | 2013-03-14 | 2014-09-18 | Curadev Pharma Pvt. Ltd. | Aminonitriles as kynurenine pathway inhibitors |
| HUE039473T2 (en) | 2013-03-14 | 2019-01-28 | Curadev Pharma Private Ltd | Inhibitors of the kynurenine pathway |
| EP2976332B1 (en) | 2013-03-14 | 2018-01-31 | Newlink Genetics Corporation | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization. |
| JP6313416B2 (en) | 2013-03-15 | 2018-04-18 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | IDO inhibitor |
| WO2014150646A1 (en) | 2013-03-15 | 2014-09-25 | Bristol-Myers Squibb Company | Ido inhibitors |
| ES2719327T3 (en) | 2013-07-01 | 2019-07-09 | Bristol Myers Squibb Co | IDO inhibitors |
| CN105517999B (en) | 2013-07-11 | 2019-06-28 | 百时美施贵宝公司 | IDO inhibitor |
-
2012
- 2012-04-05 NO NO12767723A patent/NO2694640T3/no unknown
- 2012-04-12 RS RS20180223A patent/RS56992B1/en unknown
- 2012-04-12 CN CN201510262906.5A patent/CN105111210B/en active Active
- 2012-04-12 DK DK12715295.7T patent/DK2697227T3/en active
- 2012-04-12 PL PL12715295.7T patent/PL2697227T3/en unknown
- 2012-04-12 LT LTEP15197593.5T patent/LT3018132T/en unknown
- 2012-04-12 ME MEP-2016-82A patent/ME02417B/en unknown
- 2012-04-12 PL PL15197593T patent/PL3018132T3/en unknown
- 2012-04-12 PT PT151975935T patent/PT3018132T/en unknown
- 2012-04-12 RU RU2017107026A patent/RU2017107026A/en unknown
- 2012-04-12 MX MX2013012021A patent/MX340442B/en active IP Right Grant
- 2012-04-12 CN CN201280018684.7A patent/CN103547579B/en active Active
- 2012-04-12 NZ NZ708090A patent/NZ708090A/en unknown
- 2012-04-12 HU HUE12715295A patent/HUE027316T2/en unknown
- 2012-04-12 SI SI201230515A patent/SI2697227T1/en unknown
- 2012-04-12 RS RS20160285A patent/RS54723B1/en unknown
- 2012-04-12 EP EP12715295.7A patent/EP2697227B1/en active Active
- 2012-04-12 AU AU2012242871A patent/AU2012242871C1/en active Active
- 2012-04-12 ES ES12715295.7T patent/ES2569665T3/en active Active
- 2012-04-12 WO PCT/US2012/033245 patent/WO2012142237A1/en active Application Filing
- 2012-04-12 KR KR1020137030438A patent/KR102164443B1/en active IP Right Grant
- 2012-04-12 PE PE2017002751A patent/PE20181023A1/en not_active Application Discontinuation
- 2012-04-12 EP EP17205515.4A patent/EP3348558A1/en not_active Withdrawn
- 2012-04-12 RU RU2013150811A patent/RU2613579C2/en active
- 2012-04-12 JP JP2014505274A patent/JP2014511876A/en not_active Withdrawn
- 2012-04-12 CA CA2833296A patent/CA2833296C/en active Active
- 2012-04-12 PE PE2013002332A patent/PE20141124A1/en active IP Right Grant
- 2012-04-12 ES ES15197593.5T patent/ES2660831T3/en active Active
- 2012-04-12 BR BR112013026494A patent/BR112013026494A2/en not_active Application Discontinuation
- 2012-04-12 HU HUE15197593A patent/HUE038586T2/en unknown
- 2012-04-12 EP EP15197593.5A patent/EP3018132B1/en active Active
- 2012-04-12 DK DK15197593.5T patent/DK3018132T3/en active
- 2012-04-12 NZ NZ723271A patent/NZ723271A/en unknown
- 2012-04-12 SI SI201231193T patent/SI3018132T1/en unknown
-
2013
- 2013-10-14 IL IL228862A patent/IL228862A/en active IP Right Grant
- 2013-10-14 US US14/053,440 patent/US9260434B2/en active Active
- 2013-10-15 CL CL2013002990A patent/CL2013002990A1/en unknown
- 2013-11-14 CO CO13268287A patent/CO6862146A2/en active IP Right Grant
-
2014
- 2014-07-16 HK HK14107271.9A patent/HK1193822A1/en unknown
- 2014-12-10 AU AU2014274564A patent/AU2014274564B2/en active Active
- 2014-12-22 JP JP2014258630A patent/JP5837673B2/en active Active
-
2015
- 2015-07-08 US US14/794,193 patent/US9388191B2/en active Active
- 2015-09-24 IL IL241846A patent/IL241846A/en active IP Right Grant
- 2015-11-05 JP JP2015217361A patent/JP6145491B2/en active Active
-
2016
- 2016-04-12 HR HRP20160369TT patent/HRP20160369T1/en unknown
- 2016-04-27 CY CY20161100355T patent/CY1117440T1/en unknown
- 2016-05-04 SM SM201600130T patent/SMT201600130B/en unknown
- 2016-06-13 US US15/181,062 patent/US9850248B2/en active Active
- 2016-06-28 IL IL246515A patent/IL246515B/en active IP Right Grant
- 2016-10-11 HK HK16111731.3A patent/HK1223371A1/en unknown
- 2016-12-20 AU AU2016277574A patent/AU2016277574B2/en active Active
-
2017
- 2017-05-15 JP JP2017096307A patent/JP2017149769A/en not_active Withdrawn
- 2017-11-01 US US15/800,190 patent/US10233190B2/en active Active
-
2018
- 2018-02-26 HR HRP20180335TT patent/HRP20180335T1/en unknown
- 2018-04-18 AU AU2018202706A patent/AU2018202706A1/en not_active Abandoned
- 2018-12-03 HK HK18115442.2A patent/HK1256355A1/en unknown
-
2019
- 2019-02-07 US US16/269,681 patent/US20190225618A1/en not_active Abandoned
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4584013A (en) | 1983-05-18 | 1986-04-22 | Ciba-Geigy Corporation | Cyclohexanedionecarboxylic acid derivatives with herbicidal and plant growth regulating properties |
| US5807892A (en) | 1994-09-30 | 1998-09-15 | Alcon Laboratories, Inc. | Use of certain prostaglandin analogues to treat glaucoma and ocular hypertension |
| WO1999029310A2 (en) | 1997-12-05 | 1999-06-17 | Medical College Of Georgia Research Institute, Inc. | Regulation of t cell-mediated immunity by tryptophan and its analogs |
| WO2003087347A1 (en) | 2002-04-12 | 2003-10-23 | Medical College Of Georgia Research Institute, Inc. | Antigen-presenting cell populations and their use as reagents for enhancing or reducing immune tolerance |
| EP1501918A1 (en) | 2002-04-12 | 2005-02-02 | Medical College Of Georgia Research Institute, Inc. | Antigen-presenting cell populations and their use as reagents for enhancing or reducing immune tolerance |
| WO2004094409A1 (en) | 2003-03-27 | 2004-11-04 | Lankenau Institute For Medical Research | Novel ido inhibitors and methods of use |
| US20040234623A1 (en) | 2003-04-01 | 2004-11-25 | Medical College Of Georgia Research Institute, Inc. | Use of inhibitors of indoleamine-2,3-dioxygenase in combination with other therapeutic modalities |
| US20060025383A1 (en) | 2004-02-03 | 2006-02-02 | Neil Wishart | Aminobenzoxazoles as therapeutic agents |
| US20080306084A1 (en) | 2007-04-30 | 2008-12-11 | Gruenenthal Gmbh | Substituted Amide Compounds |
| WO2009132238A2 (en) * | 2008-04-24 | 2009-10-29 | Newlink Genetics | Ido inhibitors |
Non-Patent Citations (17)
| Title |
|---|
| "Physicians' Desk Reference", 1996, MEDICAL ECONOMICS COMPANY |
| BIOORG. MED. CHEM. LETT., vol. 18, 2008, pages 5280 - 5284 |
| BROWN ET AL., ADV. EXP. MED. BIOL., vol. 294, 1991, pages 425 - 35 |
| CHEM. - EUR. J., vol. 10, 2004, pages 5233 - 5242 |
| GROHMANN ET AL., TRENDS IMMUNOL., vol. 24, 2003, pages 242 - 8 |
| J. AGRIC. FOOD CHEM., vol. 56, 2008, pages 5247 - 5253 |
| KUMAR S ET AL: "Structure based development of phenylimidazole-derived inhibitors of indoleamine 2,3-dioxygenase", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 51, no. 16, 28 August 2008 (2008-08-28), pages 4968 - 4977, XP002558480, ISSN: 0022-2623, [retrieved on 20080730], DOI: 10.1021/JM800512Z * |
| LOGAN ET AL., IMMUNOLOGY, vol. 105, 2002, pages 478 - 87 |
| MEDAWAR, SYMP. SOC. EXP. BIOL., vol. 7, 1953, pages 320 - 38 |
| MULLER, NATURE MED., vol. 11, 2005, pages 312 - 9 |
| MUNN ET AL., J. CLIN. INVEST., vol. 114, no. 2, 2004, pages 280 - 90 |
| MUNN ET AL., SCIENCE, vol. 281, 1998, pages 1191 - 3 |
| MUNN ET AL., SCIENCE, vol. 297, 2002, pages 1867 - 70 |
| PHOSPHORUS, SULFUR SILICON RELAT. ELEM., vol. I55, 1999, pages 67 - 80 |
| PORTULA ET AL., BLOOD, vol. 106, 2005, pages 2382 - 90 |
| SYNTH. COMM., vol. 37, 2007, pages 2989 - 2994 |
| UYTTENHOVE ET AL., NATURE MED., vol. 9, 2003, pages 1269 - 74 |
Cited By (305)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9260434B2 (en) | 2011-04-15 | 2016-02-16 | Newlink Genetics Corporation | Fused imidazole derivatives useful as IDO inhibitors |
| US10233190B2 (en) | 2011-04-15 | 2019-03-19 | Newlink Genetics Corporation | IDO inhibitors |
| US9850248B2 (en) | 2011-04-15 | 2017-12-26 | Newlink Genetics Corporation | IDO inhibitors |
| CN105189466A (en) * | 2013-03-14 | 2015-12-23 | 新联基因公司 | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization |
| JP2016519056A (en) * | 2013-03-14 | 2016-06-30 | ニューリンク ジェネティクス コーポレイション | Inhibitors of immunosuppression mediated by tryptophan metabolism |
| EP3366678A1 (en) | 2013-03-14 | 2018-08-29 | Newlink Genetics Corporation | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization |
| US9981973B2 (en) | 2013-03-14 | 2018-05-29 | Newlink Genetics Corporation | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization |
| RU2667509C2 (en) * | 2013-03-14 | 2018-09-21 | Ньюлинк Джинетикс Корпорейшин | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization |
| AU2014241079C1 (en) * | 2013-03-14 | 2017-07-06 | Newlink Genetics Corporation | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization |
| US9815811B2 (en) | 2013-03-14 | 2017-11-14 | Curadev Pharma, Pvt. Ltd. | Inhibitors of the kynurenine pathway |
| US10294212B2 (en) | 2013-03-14 | 2019-05-21 | Curadev Pharma, Pvt. Ltd. | Inhibitors of the kynurenine pathway |
| US9617272B2 (en) | 2013-03-14 | 2017-04-11 | Newlink Genetics Corporation | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization |
| JP2016518329A (en) * | 2013-03-14 | 2016-06-23 | キュラデブ ファーマ プライベート リミテッド | Inhibitors of the kynurenine pathway |
| JP2017052786A (en) * | 2013-03-14 | 2017-03-16 | ニューリンク ジェネティクス コーポレイション | Inhibitor of immunosuppression mediated by tryptophan metabolism |
| AU2014241079A1 (en) * | 2013-03-14 | 2015-09-03 | Newlink Genetics Corporation | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization |
| EP2970173A4 (en) * | 2013-03-14 | 2016-08-03 | Curadev Pharma Private Ltd | Inhibitors of the kynurenine pathway |
| WO2014159248A1 (en) | 2013-03-14 | 2014-10-02 | Newlink Genetics Corporation | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization |
| AU2014241079B2 (en) * | 2013-03-14 | 2016-12-15 | Newlink Genetics Corporation | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization |
| CN103382187B (en) * | 2013-08-06 | 2015-06-03 | 信实生物医药(上海)有限公司 | 3-chloro-7(5)-bromo-benzo-isoxazole compounding method |
| CN103382187A (en) * | 2013-08-06 | 2013-11-06 | 信实生物医药(上海)有限公司 | 3-chloro-7(5)-bromo-benzo-isoxazole compounding method |
| EP3466949A1 (en) | 2013-12-24 | 2019-04-10 | Bristol-Myers Squibb Company | Tricyclic compound as anticancer agents |
| WO2015100282A1 (en) | 2013-12-24 | 2015-07-02 | Bristol-Myers Squibb Company | Tricyclic compounds as anticancer agents |
| EP3610924A1 (en) | 2014-06-06 | 2020-02-19 | Bristol-Myers Squibb Company | Antibodies against glucocorticoid-induced tumor necrosis factor receptor (gitr) and uses thereof |
| WO2015187835A2 (en) | 2014-06-06 | 2015-12-10 | Bristol-Myers Squibb Company | Antibodies against glucocorticoid-induced tumor necrosis factor receptor (gitr) and uses thereof |
| EP3998079A1 (en) | 2014-06-06 | 2022-05-18 | Bristol-Myers Squibb Company | Antibodies against glucocorticoid-induced tumor necrosis factor receptor (gitr) and uses thereof |
| CN107074859B (en) * | 2014-09-05 | 2021-08-06 | 默克专利有限公司 | Cyclohexylethyl substituted diaza-and triaza-tricyclics as indoleamine-2, 3-dioxygenase (IDO) antagonists for the treatment of cancer |
| US10329297B2 (en) | 2014-09-05 | 2019-06-25 | Merck Patent Gmbh | Compounds for the inhibition of indoleamine-2,3-dioxygenase |
| AU2015311826B2 (en) * | 2014-09-05 | 2019-05-23 | Merck Patent Gmbh | Cyclohexyl-ethyl substituted diaza- and triaza-tricyclic compounds as indole-amine-2,3-dioxygenase (IDO) antagonists for the treatment of cancer |
| CN107074859A (en) * | 2014-09-05 | 2017-08-18 | 默克专利有限公司 | Indoleamine 2, the diaza and three aza-tricycle compounds of the cyclohexyl-ethyl substitution of 3 dioxygenases (IDO) antagonist are used as treating cancer |
| US9771370B2 (en) | 2014-09-05 | 2017-09-26 | Merck Patent Gmbh | Compounds for the inhibition of indoleamine-2,3-dioxygenase |
| WO2016037026A1 (en) * | 2014-09-05 | 2016-03-10 | Merck Patent Gmbh | Cyclohexyl-ethyl substituted diaza- and triaza-tricyclic compounds as indole-amine-2,3-dioxygenase (ido) antagonists for the treatment of cancer |
| WO2016051181A1 (en) * | 2014-10-01 | 2016-04-07 | Redx Pharma Plc | 4h-imidazo[1,5-a]indole derivatives and their use as indoleamine 2,3-dioxygenase (ido) and/or tryptophan 2,3-dioxygenase (td02) modulators |
| WO2016059412A1 (en) * | 2014-10-15 | 2016-04-21 | Redx Pharma Plc | 6,7-heterocyclic fused 5h-pyrrolo[1,2-c]imidazole derivatives and their use as indoleamine 2,3-dioxygenase (ido) and/or tryptophan 2,3-dioxygenase (td02) modulators |
| EP3725808A1 (en) | 2014-11-21 | 2020-10-21 | Bristol-Myers Squibb Company | Antibodies against cd73 and uses thereof |
| WO2016081748A2 (en) | 2014-11-21 | 2016-05-26 | Bristol-Myers Squibb Company | Antibodies against cd73 and uses thereof |
| US10525035B2 (en) | 2014-12-18 | 2020-01-07 | Lankenau Institute For Medical Research | Methods and compositions for the treatment of retinopathy and other ocular diseases |
| US11564907B2 (en) | 2014-12-18 | 2023-01-31 | Lankenau Institute For Medical Research | Methods and compositions for the treatment of retinopathy and other ocular diseases |
| US11376236B2 (en) | 2014-12-18 | 2022-07-05 | Lankenau Institute For Medical Research | Methods and compositions for the treatment of retinopathy and other ocular diseases |
| US11564906B2 (en) | 2014-12-18 | 2023-01-31 | Lankenau Institute For Medical Research | Methods and compositions for the treatment of retinopathy and other ocular diseases |
| WO2016106266A1 (en) | 2014-12-22 | 2016-06-30 | Bristol-Myers Squibb Company | TGFβ RECEPTOR ANTAGONISTS |
| EP4249066A2 (en) | 2014-12-23 | 2023-09-27 | Bristol-Myers Squibb Company | Antibodies to tigit |
| WO2016127052A1 (en) | 2015-02-05 | 2016-08-11 | Bristol-Myers Squibb Company | Cxcl11 and smica as predictive biomarkers for efficacy of anti-ctla4 immunotherapy |
| AU2016222140B2 (en) * | 2015-02-16 | 2019-10-24 | Shanghai De Novo Pharmatech Co., Ltd. | Fused-ring compounds, pharmaceutical composition and uses thereof |
| WO2016131381A1 (en) * | 2015-02-16 | 2016-08-25 | Shanghai De Novo Pharmatech Co. Ltd. | Fused-ring compounds, pharmaceutical composition and uses thereof |
| WO2016140884A1 (en) | 2015-03-02 | 2016-09-09 | Rigel Pharmaceuticals, Inc. | TGF-β INHIBITORS |
| WO2016161269A1 (en) | 2015-04-03 | 2016-10-06 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase for the treatment of cancer |
| US9790169B2 (en) | 2015-04-03 | 2017-10-17 | Bristol-Myers Squibb Company | IDO inhibitors |
| US10399933B2 (en) | 2015-04-03 | 2019-09-03 | Bristol-Myers Squibb Company | Inhibitors of indoleamine-2,3-dioxygenase for the treatment of cancer |
| US10167254B2 (en) | 2015-04-03 | 2019-01-01 | Bristol-Myers Squibb Company | IDO inhibitors |
| WO2016161279A1 (en) | 2015-04-03 | 2016-10-06 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase for the treatment of cancer |
| US10399932B2 (en) | 2015-04-03 | 2019-09-03 | Bristol-Myers Squibb Company | Inhibitors of indoleamine-2,3-dioxygenase for the treatment of cancer |
| WO2016162505A1 (en) | 2015-04-08 | 2016-10-13 | F-Star Biotechnology Limited | Her2 binding agent therapies |
| TWI698435B (en) * | 2015-04-10 | 2020-07-11 | 英屬開曼群島商百濟神州有限公司 | NOVEL 5 or 8-SUBSTITUTED IMIDAZO[1,5-a]PYRIDINES AS INDOLEAMINE AND/OR TRYPTOPHANE 2,3-DIOXYGENASES |
| CN110872289A (en) * | 2015-04-10 | 2020-03-10 | 百济神州(北京)生物科技有限公司 | Novel 8-substituted imidazo [1,5-a ] pyridines as IDO1 and/or TDO inhibitors |
| US10358451B2 (en) | 2015-04-12 | 2019-07-23 | Hangzhou Innogate Pharma Co., Ltd. | Heterocycles useful as IDO and TDO inhibitors |
| WO2016165613A1 (en) | 2015-04-12 | 2016-10-20 | Hangzhou Innogate Pharma Co., Ltd. | Heterocycles useful as ido and tdo inhibitors |
| EP3964527A2 (en) | 2015-04-13 | 2022-03-09 | Five Prime Therapeutics, Inc. | Combination therapy for cancer |
| WO2016168149A1 (en) | 2015-04-13 | 2016-10-20 | Five Prime Therapeutics, Inc. | Combination therapy for cancer |
| WO2016169421A1 (en) * | 2015-04-21 | 2016-10-27 | 江苏恒瑞医药股份有限公司 | Imidazo isoindole derivative, preparation method therefor and medical use thereof |
| RU2717577C2 (en) * | 2015-04-21 | 2020-03-24 | Цзянсу Хэнжуй Медицин Ко., Лтд. | Imidazoisoindole derivative, method for production thereof and medical use |
| US10899764B2 (en) | 2015-04-21 | 2021-01-26 | Jiangsu Hengrui Medicine Co., Ltd. | Imidazo isoindole derivative, preparation method therefor and medical use thereof |
| WO2016183114A1 (en) | 2015-05-11 | 2016-11-17 | Bristol-Myers Squibb Company | Tricyclic compounds as anticancer agents |
| WO2016183115A1 (en) | 2015-05-12 | 2016-11-17 | Bristol-Myers Squibb Company | 5h-pyrido[3,2-b]indole compounds as anticancer agents |
| WO2016183118A1 (en) | 2015-05-12 | 2016-11-17 | Bristol-Myers Squibb Company | Tricyclic compounds as anticancer agents |
| WO2016196228A1 (en) | 2015-05-29 | 2016-12-08 | Bristol-Myers Squibb Company | Antibodies against ox40 and uses thereof |
| GB2548542A (en) * | 2015-06-16 | 2017-09-27 | Redx Pharma Plc | Compounds |
| WO2017004016A1 (en) | 2015-06-29 | 2017-01-05 | The Rockefeller University | Antibodies to cd40 with enhanced agonist activity |
| WO2017009842A2 (en) | 2015-07-16 | 2017-01-19 | Biokine Therapeutics Ltd. | Compositions and methods for treating cancer |
| EP3943098A2 (en) | 2015-07-16 | 2022-01-26 | Biokine Therapeutics Ltd. | Compositions and methods for treating cancer |
| EP3744340A2 (en) | 2015-07-16 | 2020-12-02 | Biokine Therapeutics Ltd. | Compositions and methods for treating cancer |
| EP3954369A1 (en) | 2015-07-24 | 2022-02-16 | Lumos Pharma, Inc. | Salts and prodrugs of 1-methyl-d-tryptophan |
| US11485705B2 (en) | 2015-07-24 | 2022-11-01 | Lumos Pharma, Inc. | Salts and prodrugs of 1-methyl-d-tryptophan |
| US10207990B2 (en) | 2015-07-24 | 2019-02-19 | Newlink Genetics Corporation | Salts and prodrugs of 1-methyl-D-tryptophan |
| US9732035B2 (en) | 2015-07-24 | 2017-08-15 | Newlink Genetics Corporation | Salts and prodrugs of 1-methyl-D-tryptophan |
| WO2017019175A1 (en) | 2015-07-24 | 2017-02-02 | Newlink Genetics Corporation | Salts and prodrugs of 1-methyl-d-tryptophan |
| EP3613420A1 (en) | 2015-07-24 | 2020-02-26 | Newlink Genetics Corporation | Salts and prodrugs of 1-methyl-d-tryptophan |
| WO2017019757A1 (en) | 2015-07-28 | 2017-02-02 | Bristol-Myers Squibb Company | Tgf beta receptor antagonists |
| WO2017035118A1 (en) | 2015-08-25 | 2017-03-02 | Bristol-Myers Squibb Company | Tgf beta receptor antagonists |
| WO2017079117A1 (en) | 2015-11-02 | 2017-05-11 | Five Prime Therapeutics, Inc. | Cd80 extracellular domain polypeptides and their use in cancer treatment |
| WO2017087678A2 (en) | 2015-11-19 | 2017-05-26 | Bristol-Myers Squibb Company | Antibodies against glucocorticoid-induced tumor necrosis factor receptor (gitr) and uses thereof |
| WO2017091577A1 (en) | 2015-11-23 | 2017-06-01 | Five Prime Therapeutics, Inc. | Fgfr2 inhibitors alone or in combination with immune stimulating agents in cancer treatment |
| WO2017091580A1 (en) | 2015-11-23 | 2017-06-01 | Five Prime Therapeutics, Inc. | Predicting response to cancer treatment with fgfr2 inhibitors |
| WO2017106291A1 (en) | 2015-12-15 | 2017-06-22 | Bristol-Myers Squibb Company | Cxcr4 receptor antagonists |
| JP7227005B2 (en) | 2015-12-24 | 2023-02-21 | ジェネンテック, インコーポレイテッド | TDO2 inhibitor |
| WO2017107979A1 (en) | 2015-12-24 | 2017-06-29 | Genentech, Inc. | Tdo2 inhibitors |
| US10800780B2 (en) | 2015-12-24 | 2020-10-13 | Genentech, Inc. | TDO2 Inhibitors |
| US20190016726A1 (en) * | 2015-12-24 | 2019-01-17 | Genentech, Inc. | TDO2 Inhibitors |
| JP2019504039A (en) * | 2015-12-24 | 2019-02-14 | ジェネンテック, インコーポレイテッド | TDO2 inhibitor |
| US20220306635A1 (en) * | 2015-12-24 | 2022-09-29 | Genentech, Inc. | TDO2 Inhibitors |
| CN110072864A (en) * | 2015-12-24 | 2019-07-30 | 基因泰克公司 | TDO2 inhibitor |
| CN109071548A (en) * | 2016-02-02 | 2018-12-21 | 埃姆库瑞医药品有限公司 | It can be used for treating the pyrroles's benzimidazole derivative or its analog of especially cancer |
| WO2017134555A1 (en) | 2016-02-02 | 2017-08-10 | Emcure Pharmaceuticals Limited | Derivatives of pyrroloimidazole or analogues thereof which are useful for the treatment of inter alia cancer |
| EP3418282A4 (en) * | 2016-02-19 | 2019-01-09 | Chai Tai Tianqing Pharmaceutical Group Co., Ltd. | Tricyclic compound serving as immunomodulator |
| WO2017140835A1 (en) | 2016-02-19 | 2017-08-24 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for the treatment of obesity |
| WO2017140272A1 (en) * | 2016-02-19 | 2017-08-24 | 正大天晴药业集团股份有限公司 | Tricyclic compound acting as immunomodulator |
| US10487088B2 (en) | 2016-02-19 | 2019-11-26 | Chia Tai Tianqing Pharmaceutical Group Co., Ltd. | Tricyclic compound serving as immunomodulator |
| CN108884103A (en) * | 2016-02-19 | 2018-11-23 | 正大天晴药业集团股份有限公司 | Three and cycle compound as immunomodulator |
| CN108884104B (en) * | 2016-02-19 | 2021-01-15 | 正大天晴药业集团股份有限公司 | Tricyclic compounds as immunomodulators |
| CN108884103B (en) * | 2016-02-19 | 2021-01-15 | 正大天晴药业集团股份有限公司 | Tricyclic compounds as immunomodulators |
| CN108884104A (en) * | 2016-02-19 | 2018-11-23 | 正大天晴药业集团股份有限公司 | Three and cycle compound as immunomodulator |
| TWI743088B (en) * | 2016-02-19 | 2021-10-21 | 大陸商正大天晴藥業集團股份有限公司 | Triadic compounds as immunomodulators |
| WO2017149469A1 (en) | 2016-03-03 | 2017-09-08 | Emcure Pharmaceuticals Limited | Heterocyclic compounds useful as ido and/or tdo modulators |
| WO2017152085A1 (en) | 2016-03-04 | 2017-09-08 | Bristol-Myers Squibb Company | Combination therapy with anti-cd73 antibodies |
| WO2017184619A2 (en) | 2016-04-18 | 2017-10-26 | Celldex Therapeutics, Inc. | Agonistic antibodies that bind human cd40 and uses thereof |
| US10544099B2 (en) | 2016-05-04 | 2020-01-28 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| US11066383B2 (en) | 2016-05-04 | 2021-07-20 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| US10323004B2 (en) | 2016-05-04 | 2019-06-18 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| US10696648B2 (en) | 2016-05-04 | 2020-06-30 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| US10633342B2 (en) | 2016-05-04 | 2020-04-28 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2017197055A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| WO2017197036A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Spirocyclic degronimers for target protein degradation |
| WO2017197046A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | C3-carbon linked glutarimide degronimers for target protein degradation |
| WO2017198159A1 (en) * | 2016-05-16 | 2017-11-23 | 鲁南制药集团股份有限公司 | Imidazole derivative containing bridge ring |
| TWI671294B (en) * | 2016-06-10 | 2019-09-11 | 美商美國禮來大藥廠 | 2,3-dihydro-1h-indole compounds |
| US9872853B2 (en) | 2016-06-10 | 2018-01-23 | Eli Lilly And Company | 2,3-dihydro-1H-indole compounds |
| WO2017213919A1 (en) | 2016-06-10 | 2017-12-14 | Eli Lilly And Company | 1-tetrahydropyranylcarbonyl-2,3-dihydro-1h-indole compounds for treating cancer |
| US10759786B2 (en) | 2016-06-10 | 2020-09-01 | Eli Lilly And Company | 1-tetrahydropyranylcarbonyl-2,3-dihydro-1H-indole compounds for treating cancer |
| WO2018013818A2 (en) | 2016-07-14 | 2018-01-18 | Bristol-Myers Squibb Company | Antibodies against tim3 and uses thereof |
| WO2018017633A1 (en) | 2016-07-21 | 2018-01-25 | Bristol-Myers Squibb Company | TGF Beta RECEPTOR ANTAGONISTS |
| US11351164B2 (en) | 2016-08-26 | 2022-06-07 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| US10508085B2 (en) | 2016-09-22 | 2019-12-17 | Plexxikon Inc. | Compounds and methods for IDO and TDO modulation, and indications therefor |
| US10882856B2 (en) | 2016-09-24 | 2021-01-05 | Beigene, Ltd. | 5 or 8-substituted imidazo [1,5-a] pyridines as selective inhibitors of indoleamine and/or tryptophane 2,3-dioxygenases |
| WO2018054365A1 (en) | 2016-09-24 | 2018-03-29 | Beigene, Ltd. | NOVEL 5 or 8-SUBSTITUTED IMIDAZO [1, 5-a] PYRIDINES AS SELECTIVE INHIBITORS OF INDOLEAMINE AND/OR TRYPTOPHANE 2, 3-DIOXYGENASES |
| US11896615B2 (en) | 2016-10-13 | 2024-02-13 | Juno Therapeutics, Inc. | Immunotherapy methods and compositions involving tryptophan metabolic pathway modulators |
| EP4190335A1 (en) | 2016-10-13 | 2023-06-07 | Juno Therapeutics, Inc. | Immunotherapy methods and compositions involving tryptophan metabolic pathway modulators |
| WO2018071873A2 (en) | 2016-10-13 | 2018-04-19 | Juno Therapeutics, Inc. | Immunotherapy methods and compositions involving tryptophan metabolic pathway modulators |
| WO2018083635A2 (en) | 2016-11-04 | 2018-05-11 | Auckland Uniservices Limited | Tricyclic heterocyclic derivatives and uses thereof |
| US11028064B2 (en) | 2016-11-04 | 2021-06-08 | Auckland Uniservices Limited | Tricyclic heterocyclic derivatives and uses thereof |
| TWI671302B (en) * | 2016-12-20 | 2019-09-11 | 大陸商深圳微芯生物科技有限責任公司 | Fused imidazole compounds that may inhibit indoleamine 2,3-dioxygenase |
| EP3560928A4 (en) * | 2016-12-20 | 2020-09-16 | Shenzhen Chipscreen Biosciences Co., Ltd. | Fused imidazole compound having indoleamine 2,3-dioxygenase inhibitory activity |
| WO2018132279A1 (en) | 2017-01-05 | 2018-07-19 | Bristol-Myers Squibb Company | Tgf beta receptor antagonists |
| US11173145B2 (en) | 2017-01-17 | 2021-11-16 | Board Of Regents, The University Of Texas System | Compounds useful as inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan dioxygenase |
| WO2018175954A1 (en) | 2017-03-23 | 2018-09-27 | F. Hoffmann-La Roche Ag | Synthesis of imidazo[5,1-a]isoindole derivative useful as ido inhibitors |
| WO2018183608A1 (en) | 2017-03-31 | 2018-10-04 | Five Prime Therapeutics, Inc. | Combination therapy for cancer using anti-gitr antibodies |
| WO2018187613A2 (en) | 2017-04-07 | 2018-10-11 | Bristol-Myers Squibb Company | Anti-icos agonist antibodies and uses thereof |
| WO2018195397A2 (en) | 2017-04-21 | 2018-10-25 | Kyn Therapeutics | Indole ahr inhibitors and uses thereof |
| US11358969B2 (en) | 2017-04-21 | 2022-06-14 | Ikena Oncology, Inc. | Indole AHR inhibitors and uses thereof |
| US10570138B2 (en) | 2017-04-21 | 2020-02-25 | Kyn Therapeutics | Indole AHR inhibitors and uses thereof |
| US10689388B1 (en) | 2017-04-21 | 2020-06-23 | Ikena Oncology, Inc. | Indole AHR inhibitors and uses thereof |
| WO2018201014A1 (en) | 2017-04-28 | 2018-11-01 | Five Prime Therapeutics, Inc. | Methods of treatment with cd80 extracellular domain polypeptides |
| US11066392B2 (en) | 2017-05-12 | 2021-07-20 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2018209049A1 (en) | 2017-05-12 | 2018-11-15 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| CN107176956B (en) * | 2017-05-31 | 2019-11-12 | 成都海博锐药业有限公司 | A kind of IDO inhibitor compound, Pharmaceutical composition, purposes |
| CN107176956A (en) * | 2017-05-31 | 2017-09-19 | 成都海博锐药业有限公司 | A kind of IDO inhibitor compound, Pharmaceutical composition, purposes |
| WO2019006047A1 (en) | 2017-06-28 | 2019-01-03 | Genentech, Inc. | Tdo2 and ido1 inhibitors |
| US11827639B2 (en) | 2017-06-28 | 2023-11-28 | Genentech, Inc. | TDO2 and IDO1 inhibitors |
| WO2019005559A1 (en) | 2017-06-28 | 2019-01-03 | Genentech, Inc. | Tdo2 and ido1 inhibitors |
| WO2019006283A1 (en) | 2017-06-30 | 2019-01-03 | Bristol-Myers Squibb Company | Amorphous and crystalline forms of ido inhibitors |
| US11236049B2 (en) | 2017-06-30 | 2022-02-01 | Bristol-Myers Squibb Company | Amorphous and crystalline forms of IDO inhibitors |
| WO2019023459A1 (en) | 2017-07-28 | 2019-01-31 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| CN109983019A (en) * | 2017-08-08 | 2019-07-05 | 江苏恒瑞医药股份有限公司 | A kind of preparation method of imidazo isoindoles derivative |
| WO2019029507A1 (en) * | 2017-08-08 | 2019-02-14 | 江苏恒瑞医药股份有限公司 | Preparation method for imidazoisoindole derivatives |
| CN109983019B (en) * | 2017-08-08 | 2021-12-21 | 江苏恒瑞医药股份有限公司 | Preparation method of imidazo isoindole derivative |
| US11555026B2 (en) | 2017-08-17 | 2023-01-17 | Ikena Oncology, Inc. | AHR inhibitors and uses thereof |
| WO2019034725A1 (en) | 2017-08-17 | 2019-02-21 | Idorsia Pharmaceuticals Ltd | Inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan 2,3-dioxygenase |
| US10696650B2 (en) | 2017-08-17 | 2020-06-30 | Ikena Oncology, Inc. | AHR inhibitors and uses thereof |
| US11267824B2 (en) | 2017-08-17 | 2022-03-08 | Idorsia Pharmaceuticals Ltd | Inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan 2,3-dioxygenase |
| WO2019046498A1 (en) | 2017-08-31 | 2019-03-07 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| WO2019046500A1 (en) | 2017-08-31 | 2019-03-07 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| WO2019046496A1 (en) | 2017-08-31 | 2019-03-07 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| US11623932B2 (en) | 2017-09-22 | 2023-04-11 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| US11358948B2 (en) | 2017-09-22 | 2022-06-14 | Kymera Therapeutics, Inc. | CRBN ligands and uses thereof |
| WO2019074822A1 (en) | 2017-10-09 | 2019-04-18 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| US11649212B2 (en) | 2017-10-09 | 2023-05-16 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2019074824A1 (en) | 2017-10-09 | 2019-04-18 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| US11203592B2 (en) | 2017-10-09 | 2021-12-21 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2019075090A1 (en) | 2017-10-10 | 2019-04-18 | Tilos Therapeutics, Inc. | Anti-lap antibodies and uses thereof |
| WO2019074887A1 (en) | 2017-10-10 | 2019-04-18 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| US11230601B2 (en) | 2017-10-10 | 2022-01-25 | Tilos Therapeutics, Inc. | Methods of using anti-lap antibodies |
| WO2019079261A1 (en) | 2017-10-16 | 2019-04-25 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| WO2019089921A1 (en) | 2017-11-01 | 2019-05-09 | Bristol-Myers Squibb Company | Immunostimulatory agonistic antibodies for use in treating cancer |
| WO2019090198A1 (en) | 2017-11-06 | 2019-05-09 | Bristol-Myers Squibb Company | Isofuranone compounds useful as hpk1 inhibitors |
| US11472788B2 (en) | 2017-11-25 | 2022-10-18 | Beigene, Ltd. | Benzoimidazoles as selective inhibitors of indoleamine 2,3-dioxygenases |
| US10874743B2 (en) | 2017-12-26 | 2020-12-29 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US11318205B1 (en) | 2017-12-26 | 2022-05-03 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US11723980B2 (en) | 2017-12-26 | 2023-08-15 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| WO2019133747A1 (en) | 2017-12-27 | 2019-07-04 | Bristol-Myers Squibb Company | Anti-cd40 antibodies and uses thereof |
| US11306149B2 (en) | 2017-12-27 | 2022-04-19 | Bristol-Myers Squibb Company | Anti-CD40 antibodies and uses thereof |
| US11952427B2 (en) | 2017-12-27 | 2024-04-09 | Bristol-Myers Squibb Company | Anti-CD40 antibodies and uses thereof |
| US11447449B2 (en) | 2018-01-05 | 2022-09-20 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2019136112A1 (en) | 2018-01-05 | 2019-07-11 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2019140229A1 (en) | 2018-01-12 | 2019-07-18 | Bristol-Myers Squibb Company | Antibodies against tim3 and uses thereof |
| US11485743B2 (en) | 2018-01-12 | 2022-11-01 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| US11512080B2 (en) | 2018-01-12 | 2022-11-29 | Kymera Therapeutics, Inc. | CRBN ligands and uses thereof |
| WO2019138107A1 (en) | 2018-01-15 | 2019-07-18 | Idorsia Pharmaceuticals Ltd | Inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan 2,3-dioxygenase |
| US10988477B2 (en) | 2018-01-29 | 2021-04-27 | Merck Patent Gmbh | GCN2 inhibitors and uses thereof |
| US10793563B2 (en) | 2018-01-29 | 2020-10-06 | Merck Patent Gmbh | GCN2 inhibitors and uses thereof |
| WO2019160884A1 (en) | 2018-02-13 | 2019-08-22 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| WO2019173587A1 (en) | 2018-03-08 | 2019-09-12 | Bristol-Myers Squibb Company | Cyclic dinucleotides as anticancer agents |
| US11149011B2 (en) | 2018-03-20 | 2021-10-19 | Plexxikon Inc. | Compounds and methods for IDO and TDO modulation, and indications therefor |
| WO2019183040A1 (en) | 2018-03-21 | 2019-09-26 | Five Prime Therapeutics, Inc. | ANTIBODIES BINDING TO VISTA AT ACIDIC pH |
| US11242393B2 (en) | 2018-03-23 | 2022-02-08 | Bristol-Myers Squibb Company | Antibodies against MICA and/or MICB and uses thereof |
| WO2019200256A1 (en) | 2018-04-12 | 2019-10-17 | Bristol-Myers Squibb Company | Anticancer combination therapy with cd73 antagonist antibody and pd-1/pd-l1 axis antagonist antibody |
| WO2019204257A1 (en) | 2018-04-16 | 2019-10-24 | Arrys Therapeutics, Inc. | Ep4 inhibitors and use thereof |
| WO2019213340A1 (en) | 2018-05-03 | 2019-11-07 | Bristol-Myers Squibb Company | Uracil derivatives as mer-axl inhibitors |
| WO2019243832A1 (en) | 2018-06-22 | 2019-12-26 | Bicycletx Limited | Bicyclic peptide ligands specific for nectin-4 |
| US11912792B2 (en) | 2018-06-22 | 2024-02-27 | Bicycletx Limited | Bicyclic peptide ligands specific for nectin-4 |
| US11180531B2 (en) | 2018-06-22 | 2021-11-23 | Bicycletx Limited | Bicyclic peptide ligands specific for Nectin-4 |
| US11453702B2 (en) | 2018-06-22 | 2022-09-27 | Bicycletx Limited | Bicyclic peptide ligands specific for Nectin-4 |
| WO2019243833A1 (en) | 2018-06-22 | 2019-12-26 | Bicycletx Limited | Bicyclic peptide ligands specific for nectin-4 |
| WO2020006018A1 (en) | 2018-06-27 | 2020-01-02 | Bristol-Myers Squibb Company | Substituted naphthyridinone compounds useful as t cell activators |
| WO2020006016A1 (en) | 2018-06-27 | 2020-01-02 | Bristol-Myers Squibb Company | Naphthyridinone compounds useful as t cell activators |
| US11897882B2 (en) | 2018-07-06 | 2024-02-13 | Kymera Therapeutics, Inc. | Tricyclic crbn ligands and uses thereof |
| WO2020010177A1 (en) | 2018-07-06 | 2020-01-09 | Kymera Therapeutics, Inc. | Tricyclic crbn ligands and uses thereof |
| US11292792B2 (en) | 2018-07-06 | 2022-04-05 | Kymera Therapeutics, Inc. | Tricyclic CRBN ligands and uses thereof |
| WO2020014132A2 (en) | 2018-07-09 | 2020-01-16 | Five Prime Therapeutics, Inc. | Antibodies binding to ilt4 |
| US11401328B2 (en) | 2018-07-09 | 2022-08-02 | Five Prime Therapeutics, Inc. | Antibodies binding to ILT4 |
| WO2020014327A2 (en) | 2018-07-11 | 2020-01-16 | Five Prime Therapeutics, Inc. | Antibodies binding to vista at acidic ph |
| US11046649B2 (en) | 2018-07-17 | 2021-06-29 | Board Of Regents, The University Of Texas System | Compounds useful as inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan dioxygenase |
| WO2020023355A1 (en) | 2018-07-23 | 2020-01-30 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2020023356A1 (en) | 2018-07-23 | 2020-01-30 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| US10959986B2 (en) | 2018-08-29 | 2021-03-30 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| US11253525B2 (en) | 2018-08-29 | 2022-02-22 | Bristol-Myers Squibb Company | Inhibitors of indoleamine 2,3-dioxygenase and methods of their use |
| WO2020051424A1 (en) | 2018-09-07 | 2020-03-12 | Pic Therapeutics | Eif4e inhibitors and uses thereof |
| WO2020076969A2 (en) | 2018-10-10 | 2020-04-16 | Tilos Therapeutics, Inc. | Anti-lap antibody variants and uses thereof |
| US11130802B2 (en) | 2018-10-10 | 2021-09-28 | Tilos Therapeutics, Inc. | Anti-lap antibody variants |
| WO2020102501A1 (en) | 2018-11-16 | 2020-05-22 | Bristol-Myers Squibb Company | Anti-nkg2a antibodies and uses thereof |
| US11352350B2 (en) | 2018-11-30 | 2022-06-07 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US11807636B2 (en) | 2018-11-30 | 2023-11-07 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US11117889B1 (en) | 2018-11-30 | 2021-09-14 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| EP3670659A1 (en) | 2018-12-20 | 2020-06-24 | Abivax | Biomarkers, and uses in treatment of viral infections, inflammations, or cancer |
| WO2020127853A1 (en) | 2018-12-20 | 2020-06-25 | Abivax | Biomarkers, and uses in treatment of viral infections, inflammations, or cancer |
| WO2020132561A1 (en) | 2018-12-20 | 2020-06-25 | C4 Therapeutics, Inc. | Targeted protein degradation |
| WO2020187998A1 (en) | 2019-03-19 | 2020-09-24 | Fundació Privada Institut D'investigació Oncològica De Vall Hebron | Combination therapy with omomyc and an antibody binding pd-1 or ctla-4 for the treatment of cancer |
| WO2020201753A1 (en) | 2019-04-02 | 2020-10-08 | Bicycletx Limited | Bicycle toxin conjugates and uses thereof |
| US11485750B1 (en) | 2019-04-05 | 2022-11-01 | Kymera Therapeutics, Inc. | STAT degraders and uses thereof |
| US11746120B2 (en) | 2019-04-05 | 2023-09-05 | Kymera Therapeutics, Inc. | Stat degraders and uses thereof |
| WO2020231713A1 (en) | 2019-05-13 | 2020-11-19 | Bristol-Myers Squibb Company | AGONISTS OF ROR GAMMAt |
| WO2020231766A1 (en) | 2019-05-13 | 2020-11-19 | Bristol-Myers Squibb Company | AGONISTS OF ROR GAMMAt |
| WO2020243423A1 (en) | 2019-05-31 | 2020-12-03 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| WO2021005222A1 (en) | 2019-07-11 | 2021-01-14 | Idorsia Pharmaceuticals Ltd | Inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan 2,3-dioxygenase |
| WO2021026179A1 (en) | 2019-08-06 | 2021-02-11 | Bristol-Myers Squibb Company | AGONISTS OF ROR GAMMAt |
| WO2021041588A1 (en) | 2019-08-28 | 2021-03-04 | Bristol-Myers Squibb Company | Substituted pyridopyrimidinonyl compounds useful as t cell activators |
| US11021481B2 (en) | 2019-09-13 | 2021-06-01 | Nimbus Saturn, Inc. | Substituted isoindolin-1-ones and 2,3-dihydro-1h-pyrrolo[3,4-c]pyridin-1-ones as HPK1 antagonists |
| US11034694B2 (en) | 2019-09-13 | 2021-06-15 | Nimbus Saturn, Inc. | Substituted isoindolin-1-ones and 2,3-dihydro-1H-pyrrolo[3,4-c]pyridin-1-ones as HPK1 antagonists |
| US11548890B1 (en) | 2019-09-13 | 2023-01-10 | Nimbus Saturn, Inc. | HPK1 antagonists and uses thereof |
| US11078201B2 (en) | 2019-09-13 | 2021-08-03 | Nimbus Saturn, Inc. | Substituted isoindolin-1-ones and 2,3-dihydro-1H-pyrrol[3,4-c]pyridin-1-ones as HPK1 antagonists |
| US11028085B2 (en) | 2019-09-13 | 2021-06-08 | Nimbus Saturn, Inc. | Substituted isoindolin-1-ones and 2,3-dihydro-1h-pyrrolo[3,4-c]pyridin-1-ones as hpk1 antagonists |
| WO2021055698A1 (en) | 2019-09-19 | 2021-03-25 | Bristol-Myers Squibb Company | Antibodies binding to vista at acidic ph |
| WO2021101919A1 (en) | 2019-11-19 | 2021-05-27 | Bristol-Myers Squibb Company | Compounds useful as inhibitors of helios protein |
| WO2021108288A1 (en) | 2019-11-26 | 2021-06-03 | Bristol-Myers Squibb Company | Salts/cocrystals of (r)-n-(4-chlorophenyl)-2-((1s,4s)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide |
| US11591339B2 (en) | 2019-11-26 | 2023-02-28 | Ikena Oncology, Inc. | Solid forms of (R)-N-(2-(5-fluoropyridin-3-yl)-8-isopropylpyrazolo[ 1,5-a][1,3,5]triazin-4-yl)-2,3,4,9-tetrahydro-1H-carbazol-3-amine maleate as aryl hydrocarbon receptor (AHR) inhibitors |
| WO2021108528A1 (en) | 2019-11-26 | 2021-06-03 | Ikena Oncology, Inc. | Polymorphic carbazole derivatives and uses thereof |
| US11779578B2 (en) | 2019-12-17 | 2023-10-10 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US11707457B2 (en) | 2019-12-17 | 2023-07-25 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| US11591332B2 (en) | 2019-12-17 | 2023-02-28 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| WO2021133750A1 (en) | 2019-12-23 | 2021-07-01 | Bristol-Myers Squibb Company | Substituted bicyclic piperidine derivatives useful as t cell activators |
| WO2021133752A1 (en) | 2019-12-23 | 2021-07-01 | Bristol-Myers Squibb Company | Substituted heteroaryl compounds useful as t cell activators |
| WO2021133748A1 (en) | 2019-12-23 | 2021-07-01 | Bristol-Myers Squibb Company | Substituted quinolinonyl piperazine compounds useful as t cell activators |
| WO2021133751A1 (en) | 2019-12-23 | 2021-07-01 | Bristol-Myers Squibb Company | Substituted quinazolinyl compounds useful as t cell activators |
| WO2021133749A1 (en) | 2019-12-23 | 2021-07-01 | Bristol-Myers Squibb Company | Substituted piperazine derivatives useful as t cell activators |
| WO2021141907A1 (en) | 2020-01-06 | 2021-07-15 | Hifibio (Hong Kong) Limited | Anti-tnfr2 antibody and uses thereof |
| WO2021139682A1 (en) | 2020-01-07 | 2021-07-15 | Hifibio (Hk) Limited | Anti-galectin-9 antibody and uses thereof |
| WO2021146370A1 (en) | 2020-01-15 | 2021-07-22 | Blueprint Medicines Corporation | Map4k1 inhibitors |
| WO2021178488A1 (en) | 2020-03-03 | 2021-09-10 | PIC Therapeutics, Inc. | Eif4e inhibitors and uses thereof |
| WO2021183428A1 (en) | 2020-03-09 | 2021-09-16 | Bristol-Myers Squibb Company | Antibodies to cd40 with enhanced agonist activity |
| US11932624B2 (en) | 2020-03-19 | 2024-03-19 | Kymera Therapeutics, Inc. | MDM2 degraders and uses thereof |
| WO2021194914A1 (en) | 2020-03-23 | 2021-09-30 | Bristol-Myers Squibb Company | Substituted oxoisoindoline compounds for the treatment of cancer |
| WO2021207449A1 (en) | 2020-04-09 | 2021-10-14 | Merck Sharp & Dohme Corp. | Affinity matured anti-lap antibodies and uses thereof |
| WO2021231732A1 (en) | 2020-05-15 | 2021-11-18 | Bristol-Myers Squibb Company | Antibodies to garp |
| US11685750B2 (en) | 2020-06-03 | 2023-06-27 | Kymera Therapeutics, Inc. | Crystalline forms of IRAK degraders |
| WO2021258010A1 (en) | 2020-06-19 | 2021-12-23 | Gossamer Bio Services, Inc. | Oxime compounds useful as t cell activators |
| US11857535B2 (en) | 2020-07-30 | 2024-01-02 | Kymera Therapeutics, Inc. | Methods of treating mutant lymphomas |
| WO2022033419A2 (en) | 2020-08-10 | 2022-02-17 | Shanghai Xbh Biotechnology Co., Ltd. | Compositions and methods for treating autoimmune diseases and cancers by targeting igsf8 |
| WO2022038158A1 (en) | 2020-08-17 | 2022-02-24 | Bicycletx Limited | Bicycle conjugates specific for nectin-4 and uses thereof |
| WO2022081718A1 (en) | 2020-10-14 | 2022-04-21 | Five Prime Therapeutics, Inc. | Anti-c-c chemokine receptor 8 (ccr8) antibodies and methods of use thereof |
| WO2022120353A1 (en) | 2020-12-02 | 2022-06-09 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| WO2022120354A1 (en) | 2020-12-02 | 2022-06-09 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| WO2022133083A1 (en) | 2020-12-16 | 2022-06-23 | Gossamer Bio Services, Inc. | Compounds useful as t cell activators |
| WO2022148979A1 (en) | 2021-01-11 | 2022-07-14 | Bicycletx Limited | Methods for treating cancer |
| WO2022167445A1 (en) | 2021-02-02 | 2022-08-11 | Liminal Biosciences Limited | Gpr84 antagonists and uses thereof |
| WO2022167457A1 (en) | 2021-02-02 | 2022-08-11 | Liminal Biosciences Limited | Gpr84 antagonists and uses thereof |
| WO2022169921A1 (en) | 2021-02-04 | 2022-08-11 | Bristol-Myers Squibb Company | Benzofuran compounds as sting agonists |
| WO2022171745A1 (en) | 2021-02-12 | 2022-08-18 | F. Hoffmann-La Roche Ag | Bicyclic tetrahydroazepine derivatives for the treatment of cancer |
| EP4052705A1 (en) | 2021-03-05 | 2022-09-07 | Universität Basel Vizerektorat Forschung | Compositions for the treatment of ebv associated diseases or conditions |
| WO2022184930A2 (en) | 2021-03-05 | 2022-09-09 | Universität Basel | Compositions for the treatment of ebv associated diseases or conditions |
| US11926625B2 (en) | 2021-03-05 | 2024-03-12 | Nimbus Saturn, Inc. | HPK1 antagonists and uses thereof |
| WO2022192145A1 (en) | 2021-03-08 | 2022-09-15 | Blueprint Medicines Corporation | Map4k1 inhibitors |
| WO2022197641A1 (en) | 2021-03-15 | 2022-09-22 | Rapt Therapeutics, Inc. | 1h-pyrazolo[3,4-d]pyrimidin-6-yl-amine derivatives as hematopoietic progenitor kinase 1 (hpk1) modulators and/or inhibitors for the treatment of cancer and other diseases |
| WO2022212876A1 (en) | 2021-04-02 | 2022-10-06 | The Regents Of The University Of California | Antibodies against cleaved cdcp1 and uses thereof |
| WO2022216573A1 (en) | 2021-04-05 | 2022-10-13 | Bristol-Myers Squibb Company | Pyridinyl substituted oxoisoindoline compounds for the treatment of cancer |
| WO2022216644A1 (en) | 2021-04-06 | 2022-10-13 | Bristol-Myers Squibb Company | Pyridinyl substituted oxoisoindoline compounds |
| WO2022221866A1 (en) | 2021-04-16 | 2022-10-20 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| WO2023288254A1 (en) | 2021-07-14 | 2023-01-19 | Blueprint Medicines Corporation | Heterocyclic compounds as map4k1 inhibitors |
| WO2023288264A1 (en) | 2021-07-15 | 2023-01-19 | Blueprint Medicines Corporation | Map4k1 inhibitors |
| WO2023028238A1 (en) | 2021-08-25 | 2023-03-02 | PIC Therapeutics, Inc. | Eif4e inhibitors and uses thereof |
| WO2023028235A1 (en) | 2021-08-25 | 2023-03-02 | PIC Therapeutics, Inc. | Eif4e inhibitors and uses thereof |
| WO2023114984A1 (en) | 2021-12-17 | 2023-06-22 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| WO2023122777A1 (en) | 2021-12-22 | 2023-06-29 | Gossamer Bio Services, Inc. | Oxime derivatives useful as t cell activators |
| WO2023122778A1 (en) | 2021-12-22 | 2023-06-29 | Gossamer Bio Services, Inc. | Pyridazinone derivatives useful as t cell activators |
| WO2023122772A1 (en) | 2021-12-22 | 2023-06-29 | Gossamer Bio Services, Inc. | Oxime derivatives useful as t cell activators |
| WO2023150186A1 (en) | 2022-02-01 | 2023-08-10 | Arvinas Operations, Inc. | Dgk targeting compounds and uses thereof |
| WO2023173053A1 (en) | 2022-03-10 | 2023-09-14 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| WO2023173057A1 (en) | 2022-03-10 | 2023-09-14 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| WO2023211889A1 (en) | 2022-04-25 | 2023-11-02 | Ikena Oncology, Inc. | Polymorphic compounds and uses thereof |
| WO2023230205A1 (en) | 2022-05-25 | 2023-11-30 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| WO2024028363A1 (en) | 2022-08-02 | 2024-02-08 | Liminal Biosciences Limited | Heteroaryl carboxamide and related gpr84 antagonists and uses thereof |
| WO2024028364A1 (en) | 2022-08-02 | 2024-02-08 | Liminal Biosciences Limited | Aryl-triazolyl and related gpr84 antagonists and uses thereof |
| WO2024028365A1 (en) | 2022-08-02 | 2024-02-08 | Liminal Biosciences Limited | Substituted pyridone gpr84 antagonists and uses thereof |
| WO2024036100A1 (en) | 2022-08-08 | 2024-02-15 | Bristol-Myers Squibb Company | Substituted tetrazolyl compounds useful as t cell activators |
| WO2024036101A1 (en) | 2022-08-09 | 2024-02-15 | Bristol-Myers Squibb Company | Tertiary amine substituted bicyclic compounds useful as t cell activators |
| WO2024033389A1 (en) | 2022-08-11 | 2024-02-15 | F. Hoffmann-La Roche Ag | Bicyclic tetrahydrothiazepine derivatives |
| WO2024033457A1 (en) | 2022-08-11 | 2024-02-15 | F. Hoffmann-La Roche Ag | Bicyclic tetrahydrothiazepine derivatives |
| WO2024033388A1 (en) | 2022-08-11 | 2024-02-15 | F. Hoffmann-La Roche Ag | Bicyclic tetrahydrothiazepine derivatives |
| WO2024033458A1 (en) | 2022-08-11 | 2024-02-15 | F. Hoffmann-La Roche Ag | Bicyclic tetrahydroazepine derivatives |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2016277574B2 (en) | Fused imidazole derivatives useful as IDO inhibitors | |
| US9981973B2 (en) | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization | |
| NZ616457B2 (en) | Fused imidazole derivatives useful as ido inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12715295 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014505274 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2833296 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 002332-2013 Country of ref document: PE Ref document number: MX/A/2013/012021 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2012242871 Country of ref document: AU Date of ref document: 20120412 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2013-000565 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15056299 Country of ref document: CO Ref document number: 13268287 Country of ref document: CO Ref document number: 2012715295 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2013150811 Country of ref document: RU Kind code of ref document: A Ref document number: 20137030438 Country of ref document: KR Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112013026494 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 241846 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2016/0285 Country of ref document: RS |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 246515 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 112013026494 Country of ref document: BR Kind code of ref document: A2 Effective date: 20131014 |