WO2012115159A1 - Polypeptide compound - Google Patents
Polypeptide compound Download PDFInfo
- Publication number
- WO2012115159A1 WO2012115159A1 PCT/JP2012/054301 JP2012054301W WO2012115159A1 WO 2012115159 A1 WO2012115159 A1 WO 2012115159A1 JP 2012054301 W JP2012054301 W JP 2012054301W WO 2012115159 A1 WO2012115159 A1 WO 2012115159A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- ring
- lower alkylene
- formula
- production example
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/50—Cyclic peptides containing at least one abnormal peptide link
- C07K7/54—Cyclic peptides containing at least one abnormal peptide link with at least one abnormal peptide link in the ring
- C07K7/56—Cyclic peptides containing at least one abnormal peptide link with at least one abnormal peptide link in the ring the cyclisation not occurring through 2,4-diamino-butanoic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/10—Anti-acne agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Definitions
- the present inventors have found that the compound of the formula (I) has an excellent antifungal activity and completed the present invention.
- the compound has the following formula at a specific carbon atom on the B ring in the compound of formula (I)
- lower alkyl means linear or branched alkyl having 1 to 6 carbon atoms (hereinafter abbreviated as C 1-6 ), such as methyl, ethyl, n-propyl, isopropyl, n -Butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, n-hexyl and the like. In another embodiment, it is C 1-4 alkyl. Yet another embodiment is C 1-3 alkyl. Yet another embodiment is methyl or ethyl. “Lower alkylene” means a divalent group (C 1-6 alkylene) formed by removing any one hydrogen atom of the above “lower alkyl”, and one embodiment is C 1-4 alkylene. Another embodiment is methylene or ethylene.
- Heterocycle group refers to i) a 3-8 membered, alternatively 5-7 membered monocyclic heterocycle containing 1-4 heteroatoms selected from oxygen, sulfur and nitrogen, And ii) the monocyclic heterocycle is condensed with one or two rings selected from the group consisting of a monocyclic heterocycle, a benzene ring, a C 5-10 cycloalkane and a C 5-10 cycloalkene.
- Certain embodiments of the compound of the present invention represented by the formula (I) or a pharmaceutically acceptable salt thereof are shown below.
- a compound in which the A ring is a monocyclic saturated heterocyclic group.
- a compound in which the A ring is a 6-membered monocyclic heterocyclic group.
- a compound in which the A ring is a monocyclic unsaturated heterocyclic group or a condensed polycyclic unsaturated heterocyclic group.
- a compound in which the A ring is a nitrogen-containing heterocycloalkyl.
- a compound in which the A ring is a 6-membered nitrogen-containing heterocycloalkyl.
- R 2 is —S ( ⁇ O) 2 OH, —CH 2 CH (OH) CH 2 OH, —CH 2 C ( ⁇ O) OH, lower alkyl, or H.
- X is a single bond.
- R 1 is —NH 2
- R 2 is Me
- the A ring is 1,2,3,6-tetrahydropyridyl-4-yl
- X is a single bond
- the B ring is a formula A group represented by (II)
- R B is H
- R c is H
- R D is a group represented by the formula (V)
- the E ring is piperidinyl
- L is a single bond.
- a compound in which the Fg ring is cycloalkyl.
- R 1 is —NH 2
- R 2 is Me
- the A ring is 1,2,3,6-tetrahydropyridyl-4-yl
- R A is H
- X is a single bond and a is a group B ring represented by the formula (II)
- an R B is H
- R c is H
- D ring is a pyrimidine
- a group R D is represented by formula (V)
- L is —CH 2 CH 2 —
- the Fg ring is phenyl.
- Specific examples of the compound included in the present invention include a compound selected from the following groups (85) to (88) or a pharmaceutically acceptable salt thereof.
- geometric isomers may exist depending on the type of substituent.
- the compound of the formula (I) may be described in only one form of an isomer, but the present invention includes other isomers, separated isomers, or those isomers. And mixtures thereof.
- the compound of formula (I) may have an asymmetric carbon atom or axial asymmetry, and optical isomers based on this may exist.
- the present invention also includes separated optical isomers of the compound of formula (I) or a mixture thereof.
- the compound of the formula (I) and salts thereof can be produced by applying various known synthetic methods utilizing characteristics based on the basic structure or the type of substituent. At that time, depending on the type of functional group, it is effective in terms of production technology to replace the functional group with an appropriate protecting group (a group that can be easily converted into the functional group) at the stage from the raw material to the intermediate. There is a case.
- protecting groups include protecting groups described in “Greene's Protective Groups in Organic Synthesis (4th edition, 2006)” by PGM Wuts and TW Greene. These may be appropriately selected according to the reaction conditions. In such a method, after carrying out the reaction by introducing the protective group, the desired compound can be obtained by removing the protective group as necessary.
- Compound (I) can also be obtained by isolating compound (P-1) as a reactive derivative and then reacting with compound (Q).
- reactive derivatives of the carboxylic acid form (P-1) are obtained by reacting with an acid halide obtained by reacting with a halogenating agent (for example, phosphorus oxychloride, thionyl chloride, etc.), isobutyl chloroformate, etc.
- a halogenating agent for example, phosphorus oxychloride, thionyl chloride, etc.
- Examples thereof include mixed acid anhydrides or active esters obtained by condensation with HOBt or the like.
- the reaction between the compound (P-1) and the reactive derivative is performed under cooling to heating, preferably in a solvent inert to the reaction such as DMF, halogenated hydrocarbons, aromatic hydrocarbons, ethers, It can be performed at -20 ° C to 60 ° C. In the reaction, it may be advantageous to cause the reaction to proceed smoothly in the presence of a base (for example, NMM, TEA, DIPEA, DMAP, pyridine, picoline, lutidine, etc.). Pyridine can also serve as a solvent. [Literature] SR Sandler and W. Karo, "Organic Functional Group Preparations", 2nd edition, 1st volume, Academic Press Inc. (2005) (Maruzen) (General production method 2-1)
- the solvent used here include, but are not limited to, alcohols such as MeOH and EtOH, ethers such as diethyl ether, DMF, halogenated hydrocarbons, and mixtures thereof.
- the reducing agent include NaBH (OAc) 3 , NaBH 3 CN, NaBH 4 and the like.
- the compounds of formula (I) are isolated and purified as free compounds, their salts, hydrates, solvates, or crystalline polymorphs.
- the salt of the compound of formula (I) can also be produced by subjecting it to a conventional salt formation reaction. Isolation and purification are performed by applying ordinary chemical operations such as extraction, fractional crystallization, and various fractional chromatography.
- Various isomers can be produced by selecting an appropriate raw material compound, or can be separated by utilizing a difference in physicochemical properties between isomers.
- optical isomers can be obtained by general optical resolution of racemates (for example, fractional crystallization leading to diastereomeric salts with optically active bases or acids, chromatography using chiral columns, etc.). Further, it can also be produced from a suitable optically active raw material compound.
- the above fungi are skin, eyes, hair, nails, oral mucosa, gastrointestinal tract, bronchial, lung, endocardium, brain, meninges, urinary organs, buttocks, oral cavity, eyeball, whole body, kidney, heart, ear canal, It is well known to cause various infectious diseases in bone, nasal cavity, sinus cavity, spleen, liver, subcutaneous tissue, lymphatic vessel, gastrointestinal tract, joint, muscle, tendon, stromal plasma cell in lung, blood vessel and the like.
- Production Example 37 Under ice-cooling, trans-4- [3- (4-bromophenyl) -1,2,4-oxadiazol-5-yl] cyclohexanol (410 mg), benzyl 2,2,2-trichloroethaneimidate ( TfOH (0.0553 mL) was added to a mixture of 545 mg) and CH 2 Cl 2 (6.15 mL). The mixture was warmed to room temperature and stirred for 2 hours. The reaction solution was diluted with EtOAc, and sat. NaHCO 3 aqueous solution was added.
- Example 1 3- ⁇ 1-[(9H-Fluoren-9-ylmethoxy) carbonyl] -1,2,3,6-tetrahydropyridin-4-yl ⁇ -4 '-[5- (trans-4-isobutoxycyclohexyl)-
- a mixture of 1,2,4-oxadiazol-3-yl] biphenyl-4-carboxylic acid (402 mg), HBTU (221.2 mg), DMF (6.00 mL), DIPEA (0.237 mL) was stirred at room temperature for 30 minutes did.
- the compound of Example No. 169 (578 mg) of International Publication WO99 / 40108 was added and stirred at room temperature for 5 hours. In TLC, it was confirmed that the active ester disappeared.
- ODS silica gel chromatography ODS silica gel 15 mL, eluent; hydrochloric acid water of 0% to 65% pH 2.0: MeCN system. Lyophilization was performed to obtain the compound of Example 182 (34.2 mg) as a white powder.
- NMR1 d 6 -DMSO chemical shift values of 1 H-NMR measured in ( ⁇ )
- NMR2 d 6 -DMSO + D 2 O
- ESI and EI represent mass analysis values by electrospray ionization method and mass analysis values by electron ionization method, respectively, and mean positive or positive when ESI and EI data are not described.
- APCI indicates atmospheric pressure chemical ionization mass spectrometry
- APCI / ESI means mass spectrometry values measured simultaneously with APCI and ESI.
- the compound of the formula (I) or a salt thereof has antifungal activity and has various infectious diseases such as dermatophytosis (e.g. ringworm), vermiculitis, candidiasis, geotrichumosis, sandy hair, Aspergillus , Penicillosis, sporotricosis, chromomises, coccidioidomycosis, histoplasmosis, blastosomiasis, paracoccidioidomycosis, pseudoaleseriasis, foot mycosis, fungal keratitis, otomycosis, pneumocystis, fungi It is useful for the prevention and / or treatment of blood pressure.
- infectious diseases such as dermatophytosis (e.g. ringworm), vermiculitis, candidiasis, geotrichumosis, sandy hair, Aspergillus , Penicillosis, sporotricosis, chromomises, cocc
Abstract
[Problem] To provide an antibacterial agent, particularly a novel polypeptide compound having an antifungal activity or a salt thereof. [Solution] The present inventors considered a novel antibacterial agent and found that excellent antifungal activity can be achieved by introducing a benzoyl group having a cyclic substituent at the carbonyl ortho position, as a partial structure for a natural polypeptide compound, thereby completing the present invention. This polypeptide compound has excellent antifungal activity and can be used as a prophylactic and/or a therapeutic agent for various fungal infections.
Description
本発明は、医薬、殊に抗真菌活性を有する新規なポリペプチド化合物又はその製薬学的に許容される塩に関する。
The present invention relates to a pharmaceutical, particularly a novel polypeptide compound having antifungal activity or a pharmaceutically acceptable salt thereof.
真菌による感染症は、皮膚からの感染による表在性皮膚真菌症(白癬菌等)及び呼吸器又は経口からの感染による深在性真菌症(カンジダ菌、アスペルギルス菌等)に大別される。深在性真菌症は免疫不全や抗癌剤投与など様々なリスクファクターを抱える患者に発症し、一度発症すると難治化、予後不良となることがしばしば見られる。特に侵襲性肺アスペルギルス症は発症するとその死亡率は全疾患を通して58%であり、特に骨髄移植患者においては87%にもなる。しかし、有効な治療手段や薬が少なく、標準薬であるボリコナゾールやアムビソームでも有効率は低く60%以下である。そのため、新たに強力な活性を有する抗真菌剤が求められている。
抗真菌化合物として、例えば特許文献1には下記式の化合物が開示されている。 Infectious diseases caused by fungi are roughly classified into superficial dermatomycosis caused by infection from the skin (such as ringworm) and deep mycosis caused by respiratory or oral infection (such as Candida and Aspergillus). Deep mycosis develops in patients with various risk factors such as immunodeficiency and anticancer drug administration, and once it develops, it often becomes intractable and has a poor prognosis. In particular, when invasive pulmonary aspergillosis develops, the mortality rate is 58% throughout the disease, especially 87% in bone marrow transplant patients. However, there are few effective treatment means and drugs, and even the standard drugs voriconazole and ambisome have a low effective rate of 60% or less. Therefore, a new antifungal agent having a strong activity is demanded.
As an antifungal compound, for example, Patent Document 1 discloses a compound represented by the following formula.
抗真菌化合物として、例えば特許文献1には下記式の化合物が開示されている。 Infectious diseases caused by fungi are roughly classified into superficial dermatomycosis caused by infection from the skin (such as ringworm) and deep mycosis caused by respiratory or oral infection (such as Candida and Aspergillus). Deep mycosis develops in patients with various risk factors such as immunodeficiency and anticancer drug administration, and once it develops, it often becomes intractable and has a poor prognosis. In particular, when invasive pulmonary aspergillosis develops, the mortality rate is 58% throughout the disease, especially 87% in bone marrow transplant patients. However, there are few effective treatment means and drugs, and even the standard drugs voriconazole and ambisome have a low effective rate of 60% or less. Therefore, a new antifungal agent having a strong activity is demanded.
As an antifungal compound, for example, Patent Document 1 discloses a compound represented by the following formula.

(式中、R1はH、R2はアシル基、R3はOH等、R4はOH等、R5はH等、R6はH等を示す)。
特許文献2、3、5及び6においても、特許文献1と同様に上記R2に該当する基がアシルと定義されており、R2が種々のベンゾイル基である化合物が開示されている。しかしながら、当該ベンゾイル基の置換基として、カルボニルのオルト位に環基が置換した化合物は開示されていない。
特許文献4には、上記定義のR2に該当する基にベンゾイル基である化合物が開示されており、カルボニルのオルト位にメチル基である化合物が開示されているが、カルボニルのオルト位に環基が置換した化合物は開示されていない。 (Wherein R 1 is H, R 2 is an acyl group, R 3 is OH, R 4 is OH, R 5 is H, R 6 is H, etc.).
In Patent Documents 2, 3, 5 and 6, as in Patent Document 1, a group corresponding to R 2 is defined as acyl, and compounds in which R 2 is various benzoyl groups are disclosed. However, a compound in which a ring group is substituted at the ortho position of the carbonyl is not disclosed as a substituent of the benzoyl group.
Patent Document 4 discloses a compound in which the group corresponding to R 2 defined above is a benzoyl group, and discloses a compound in which a methyl group is present at the ortho position of the carbonyl. Compounds with substituted groups are not disclosed.
特許文献2、3、5及び6においても、特許文献1と同様に上記R2に該当する基がアシルと定義されており、R2が種々のベンゾイル基である化合物が開示されている。しかしながら、当該ベンゾイル基の置換基として、カルボニルのオルト位に環基が置換した化合物は開示されていない。
特許文献4には、上記定義のR2に該当する基にベンゾイル基である化合物が開示されており、カルボニルのオルト位にメチル基である化合物が開示されているが、カルボニルのオルト位に環基が置換した化合物は開示されていない。 (Wherein R 1 is H, R 2 is an acyl group, R 3 is OH, R 4 is OH, R 5 is H, R 6 is H, etc.).
In Patent Documents 2, 3, 5 and 6, as in Patent Document 1, a group corresponding to R 2 is defined as acyl, and compounds in which R 2 is various benzoyl groups are disclosed. However, a compound in which a ring group is substituted at the ortho position of the carbonyl is not disclosed as a substituent of the benzoyl group.
Patent Document 4 discloses a compound in which the group corresponding to R 2 defined above is a benzoyl group, and discloses a compound in which a methyl group is present at the ortho position of the carbonyl. Compounds with substituted groups are not disclosed.
医薬組成物、特に真菌症の予防用及び/又は治療用医薬組成物の有効成分として有用な化合物を提供する。
Provided is a compound useful as an active ingredient of a pharmaceutical composition, in particular, a pharmaceutical composition for preventing and / or treating mycosis.
本発明者らは、優れた真菌症の予防及び/又は治療薬について鋭意検討した結果、式(I)の化合物が優れた抗真菌活性を有することを知見して本発明を完成した。当該化合物は式(I)の化合物中B環上の特定の炭素原子に下式
As a result of intensive studies on an excellent preventive and / or therapeutic agent for mycosis, the present inventors have found that the compound of the formula (I) has an excellent antifungal activity and completed the present invention. The compound has the following formula at a specific carbon atom on the B ring in the compound of formula (I)

で示される基が結合することを特徴とする化合物である。
即ち、本発明は、式(I)の化合物又はその製薬学的に許容される塩、並びに、式(I)の化合物又はその製薬学的に許容される塩、及び賦形剤を含有する医薬組成物に関する。 A compound represented by the formula:
That is, the present invention relates to a compound containing the compound of formula (I) or a pharmaceutically acceptable salt thereof, and a compound containing the compound of formula (I) or a pharmaceutically acceptable salt thereof, and an excipient. Relates to the composition.
即ち、本発明は、式(I)の化合物又はその製薬学的に許容される塩、並びに、式(I)の化合物又はその製薬学的に許容される塩、及び賦形剤を含有する医薬組成物に関する。 A compound represented by the formula:
That is, the present invention relates to a compound containing the compound of formula (I) or a pharmaceutically acceptable salt thereof, and a compound containing the compound of formula (I) or a pharmaceutically acceptable salt thereof, and an excipient. Relates to the composition.

(式中、R1は、-NH2、-OH、-NH-低級アルキレン-NH2、-低級アルキレン-NH-低級アルキル、-低級アルキレン-N(低級アルキル)2、-NH-低級アルキレン-C(=O)OH、又は、-NHCH(OH)C(=O)OH、
R2は、-S(=O)2OH、-CH2CH(OH)CH2OH、-低級アルキレン-C(=O)OH、低級アルキル、又は、H、
A環は、含窒素ヘテロ環、アリール、又はシクロアルキル、
RAは、H、低級アルキル、-低級アルキレン-NH2、-低級アルキレン-NH-低級アルキル、-低級アルキレン-N(低級アルキル)2、-低級アルキレン-OH、-低級アルキレン-O-低級アルキル、-低級アルキレン-C(=O)OH、-低級アルキレン-S(=O) 2OH、-低級アルキレン-S(=O) 2O-低級アルキル、-C(=O)O-低級アルキル、-C(=O)-低級アルキレン-NH2、-C(=O)-低級アルキレン- NH-低級アルキル、-C(=O)-低級アルキレン-N(低級アルキル)2、-C(=O)-NH2で置換されていてもよい低級アルキレン-OH、-C(=NH)-NH2、-CH(低級アルキレン-OH)2、又は、シクロアルキル、
Xは、単結合、低級アルキレン、-NH-、又は、-N(低級アルキル)-
RB及びRCは、それぞれ同一又は異なって、H又はハロゲン、
B環は、下記式(II)、式(III-a)、又は、式(III-b) Wherein R 1 is -NH 2 , -OH, -NH-lower alkylene-NH 2 , -lower alkylene-NH-lower alkyl, -lower alkylene-N (lower alkyl) 2 , -NH-lower alkylene- C (= O) OH or -NHCH (OH) C (= O) OH,
R 2 is -S (= O) 2 OH, -CH 2 CH (OH) CH 2 OH, -lower alkylene-C (= O) OH, lower alkyl, or H,
A ring is a nitrogen-containing heterocycle, aryl, or cycloalkyl,
R A is H, lower alkyl, -lower alkylene-NH 2 , -lower alkylene-NH-lower alkyl, -lower alkylene-N (lower alkyl) 2 , -lower alkylene-OH, -lower alkylene-O-lower alkyl , -Lower alkylene-C (= O) OH, -lower alkylene-S (= O) 2 OH, -lower alkylene-S (= O) 2 O-lower alkyl, -C (= O) O-lower alkyl, -C (= O) - lower alkylene -NH 2, -C (= O) - lower alkylene - NH- lower alkyl, -C (= O) - lower alkylene -N (lower alkyl) 2, -C (= O ) -NH 2 optionally substituted with lower alkylene-OH, —C (═NH) —NH 2 , —CH (lower alkylene-OH) 2 , or cycloalkyl,
X is a single bond, lower alkylene, -NH-, or -N (lower alkyl)-
R B and R C are the same or different, and H or halogen,
The ring B is represented by the following formula (II), formula (III-a), or formula (III-b)
R2は、-S(=O)2OH、-CH2CH(OH)CH2OH、-低級アルキレン-C(=O)OH、低級アルキル、又は、H、
A環は、含窒素ヘテロ環、アリール、又はシクロアルキル、
RAは、H、低級アルキル、-低級アルキレン-NH2、-低級アルキレン-NH-低級アルキル、-低級アルキレン-N(低級アルキル)2、-低級アルキレン-OH、-低級アルキレン-O-低級アルキル、-低級アルキレン-C(=O)OH、-低級アルキレン-S(=O) 2OH、-低級アルキレン-S(=O) 2O-低級アルキル、-C(=O)O-低級アルキル、-C(=O)-低級アルキレン-NH2、-C(=O)-低級アルキレン- NH-低級アルキル、-C(=O)-低級アルキレン-N(低級アルキル)2、-C(=O)-NH2で置換されていてもよい低級アルキレン-OH、-C(=NH)-NH2、-CH(低級アルキレン-OH)2、又は、シクロアルキル、
Xは、単結合、低級アルキレン、-NH-、又は、-N(低級アルキル)-
RB及びRCは、それぞれ同一又は異なって、H又はハロゲン、
B環は、下記式(II)、式(III-a)、又は、式(III-b) Wherein R 1 is -NH 2 , -OH, -NH-lower alkylene-NH 2 , -lower alkylene-NH-lower alkyl, -lower alkylene-N (lower alkyl) 2 , -NH-lower alkylene- C (= O) OH or -NHCH (OH) C (= O) OH,
R 2 is -S (= O) 2 OH, -CH 2 CH (OH) CH 2 OH, -lower alkylene-C (= O) OH, lower alkyl, or H,
A ring is a nitrogen-containing heterocycle, aryl, or cycloalkyl,
R A is H, lower alkyl, -lower alkylene-NH 2 , -lower alkylene-NH-lower alkyl, -lower alkylene-N (lower alkyl) 2 , -lower alkylene-OH, -lower alkylene-O-lower alkyl , -Lower alkylene-C (= O) OH, -lower alkylene-S (= O) 2 OH, -lower alkylene-S (= O) 2 O-lower alkyl, -C (= O) O-lower alkyl, -C (= O) - lower alkylene -NH 2, -C (= O) - lower alkylene - NH- lower alkyl, -C (= O) - lower alkylene -N (lower alkyl) 2, -C (= O ) -NH 2 optionally substituted with lower alkylene-OH, —C (═NH) —NH 2 , —CH (lower alkylene-OH) 2 , or cycloalkyl,
X is a single bond, lower alkylene, -NH-, or -N (lower alkyl)-
R B and R C are the same or different, and H or halogen,
The ring B is represented by the following formula (II), formula (III-a), or formula (III-b)

で示される基、
*は、式(I)におけるXと結合する位置を示し、**は、式(I)におけるRBと結合する位置を示し、
D環は、含窒素ヘテロアリール、又は、アリール、
RDは、下記式(IV)から(VI) A group represented by
* Indicates the position at which the group bonds to X in formula (I), ** indicates the position at which the group bonds to R B in the formula (I),
Ring D is a nitrogen-containing heteroaryl or aryl,
R D represents the following formulas (IV) to (VI)
*は、式(I)におけるXと結合する位置を示し、**は、式(I)におけるRBと結合する位置を示し、
D環は、含窒素ヘテロアリール、又は、アリール、
RDは、下記式(IV)から(VI) A group represented by
* Indicates the position at which the group bonds to X in formula (I), ** indicates the position at which the group bonds to R B in the formula (I),
Ring D is a nitrogen-containing heteroaryl or aryl,
R D represents the following formulas (IV) to (VI)

で示される基、又は、-L-O-R101で示される基、
Lは、低級アルキレン、高級アルキレン、-O-、-O-低級アルキレン、又は、単結合、
E環は、ヘテロ環基、
F環及びF1環は、それぞれ同一又は異なって、シクロアルキル、
Fg環は、シクロアルキル、又は、アリール、
Efg環は、ヘテロ環基、シクロアルキル、又は、アリール、
REは、H、低級アルキル、-O-低級アルキル、又は、ハロゲン、
R101は、低級アルキル、-低級アルキレン-シクロアルキル、又は、-高級アルキレン-シクロアルキル、
RZ1又はRZ2は、それぞれ同一又は異なって、H、ハロゲン、ハロゲン又は低級アルキルで置換されていてもよいアリール、低級アルキル、高級アルキル、-O-低級アルキル、-O-高級アルキル、-O- シクロアルキル、-O-アリール、-O-低級アルキレン-アリール、-高級アルキレン-O-低級アルキル、シクロアルキル、又は、-C(=O)O-低級アルキレン-アリールである。) Or a group represented by -LOR 101 ,
L is lower alkylene, higher alkylene, -O-, -O-lower alkylene, or a single bond,
E ring is a heterocyclic group,
F ring and F1 ring are the same or different and each represents cycloalkyl,
Fg ring is cycloalkyl or aryl,
Efg ring is a heterocyclic group, cycloalkyl or aryl,
R E is H, lower alkyl, -O-lower alkyl, or halogen,
R 101 represents lower alkyl, -lower alkylene-cycloalkyl, or -higher alkylene-cycloalkyl,
R Z1 or R Z2 is the same or different and is optionally substituted with H, halogen, halogen or lower alkyl, aryl, lower alkyl, higher alkyl, —O-lower alkyl, —O-higher alkyl, —O -Cycloalkyl, -O-aryl, -O-lower alkylene-aryl, -higher alkylene-O-lower alkyl, cycloalkyl, or -C (= O) O-lower alkylene-aryl. )
Lは、低級アルキレン、高級アルキレン、-O-、-O-低級アルキレン、又は、単結合、
E環は、ヘテロ環基、
F環及びF1環は、それぞれ同一又は異なって、シクロアルキル、
Fg環は、シクロアルキル、又は、アリール、
Efg環は、ヘテロ環基、シクロアルキル、又は、アリール、
REは、H、低級アルキル、-O-低級アルキル、又は、ハロゲン、
R101は、低級アルキル、-低級アルキレン-シクロアルキル、又は、-高級アルキレン-シクロアルキル、
RZ1又はRZ2は、それぞれ同一又は異なって、H、ハロゲン、ハロゲン又は低級アルキルで置換されていてもよいアリール、低級アルキル、高級アルキル、-O-低級アルキル、-O-高級アルキル、-O- シクロアルキル、-O-アリール、-O-低級アルキレン-アリール、-高級アルキレン-O-低級アルキル、シクロアルキル、又は、-C(=O)O-低級アルキレン-アリールである。) Or a group represented by -LOR 101 ,
L is lower alkylene, higher alkylene, -O-, -O-lower alkylene, or a single bond,
E ring is a heterocyclic group,
F ring and F1 ring are the same or different and each represents cycloalkyl,
Fg ring is cycloalkyl or aryl,
Efg ring is a heterocyclic group, cycloalkyl or aryl,
R E is H, lower alkyl, -O-lower alkyl, or halogen,
R 101 represents lower alkyl, -lower alkylene-cycloalkyl, or -higher alkylene-cycloalkyl,
R Z1 or R Z2 is the same or different and is optionally substituted with H, halogen, halogen or lower alkyl, aryl, lower alkyl, higher alkyl, —O-lower alkyl, —O-higher alkyl, —O -Cycloalkyl, -O-aryl, -O-lower alkylene-aryl, -higher alkylene-O-lower alkyl, cycloalkyl, or -C (= O) O-lower alkylene-aryl. )
また、本発明は、式(I)の化合物又はその塩を含有する真菌症の予防用及び/又は治療用医薬組成物、即ち、式(I)の化合物又はその塩を含有する真菌症の予防剤及び/又は治療剤に関する。
また、本発明は、真菌症の予防用及び/又は治療用医薬組成物の製造のための式(I)の化合物又はその塩の使用、真菌感染症の予防若しくは治療のための式(I)の化合物又はその塩の使用、並びに、式(I)の化合物又はその塩の有効量を患者に投与することからなる真菌症の予防方法及び/又は治療方法に関する。
また、本発明は、真菌症の治療における使用の為の式(I)の化合物に関する。 The present invention also relates to a pharmaceutical composition for preventing and / or treating mycosis comprising a compound of formula (I) or a salt thereof, ie, prevention of mycosis comprising a compound of formula (I) or a salt thereof. The present invention relates to an agent and / or a therapeutic agent.
The present invention also relates to the use of a compound of formula (I) or a salt thereof for the manufacture of a pharmaceutical composition for the prevention and / or treatment of mycosis, formula (I) for the prevention or treatment of fungal infections. And a method for preventing and / or treating mycosis comprising administering to a patient an effective amount of a compound of formula (I) or a salt thereof.
The invention also relates to a compound of formula (I) for use in the treatment of mycosis.
また、本発明は、真菌症の予防用及び/又は治療用医薬組成物の製造のための式(I)の化合物又はその塩の使用、真菌感染症の予防若しくは治療のための式(I)の化合物又はその塩の使用、並びに、式(I)の化合物又はその塩の有効量を患者に投与することからなる真菌症の予防方法及び/又は治療方法に関する。
また、本発明は、真菌症の治療における使用の為の式(I)の化合物に関する。 The present invention also relates to a pharmaceutical composition for preventing and / or treating mycosis comprising a compound of formula (I) or a salt thereof, ie, prevention of mycosis comprising a compound of formula (I) or a salt thereof. The present invention relates to an agent and / or a therapeutic agent.
The present invention also relates to the use of a compound of formula (I) or a salt thereof for the manufacture of a pharmaceutical composition for the prevention and / or treatment of mycosis, formula (I) for the prevention or treatment of fungal infections. And a method for preventing and / or treating mycosis comprising administering to a patient an effective amount of a compound of formula (I) or a salt thereof.
The invention also relates to a compound of formula (I) for use in the treatment of mycosis.
式(I)の化合物又はその塩は、抗真菌活性を有し、真菌症等の予防剤又は治療剤として使用できる。
The compound of formula (I) or a salt thereof has antifungal activity and can be used as a preventive or therapeutic agent for mycosis.
以下、本明細書を詳細に説明する。
本明細書において、「低級アルキル」とは、直鎖又は分枝状の炭素数が1から6(以後、C1-6と略す)のアルキル、例えばメチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、sec-ブチル、tert-ブチル、n-ペンチル、イソペンチル、n-ヘキシル等である。別の態様としては、C1-4アルキルである。さらに別の態様としては、C1-3アルキルである。またさらに別の態様としては、メチル又はエチルである。
「低級アルキレン」は、上記「低級アルキル」の任意の水素原子1個を除去してなる二価基(C1-6アルキレン)を意味し、ある態様としてはC1-4アルキレンである。別の態様としてはメチレン又はエチレンである。 Hereinafter, the present specification will be described in detail.
In the present specification, “lower alkyl” means linear or branched alkyl having 1 to 6 carbon atoms (hereinafter abbreviated as C 1-6 ), such as methyl, ethyl, n-propyl, isopropyl, n -Butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, n-hexyl and the like. In another embodiment, it is C 1-4 alkyl. Yet another embodiment is C 1-3 alkyl. Yet another embodiment is methyl or ethyl.
“Lower alkylene” means a divalent group (C 1-6 alkylene) formed by removing any one hydrogen atom of the above “lower alkyl”, and one embodiment is C 1-4 alkylene. Another embodiment is methylene or ethylene.
本明細書において、「低級アルキル」とは、直鎖又は分枝状の炭素数が1から6(以後、C1-6と略す)のアルキル、例えばメチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、sec-ブチル、tert-ブチル、n-ペンチル、イソペンチル、n-ヘキシル等である。別の態様としては、C1-4アルキルである。さらに別の態様としては、C1-3アルキルである。またさらに別の態様としては、メチル又はエチルである。
「低級アルキレン」は、上記「低級アルキル」の任意の水素原子1個を除去してなる二価基(C1-6アルキレン)を意味し、ある態様としてはC1-4アルキレンである。別の態様としてはメチレン又はエチレンである。 Hereinafter, the present specification will be described in detail.
In the present specification, “lower alkyl” means linear or branched alkyl having 1 to 6 carbon atoms (hereinafter abbreviated as C 1-6 ), such as methyl, ethyl, n-propyl, isopropyl, n -Butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, n-hexyl and the like. In another embodiment, it is C 1-4 alkyl. Yet another embodiment is C 1-3 alkyl. Yet another embodiment is methyl or ethyl.
“Lower alkylene” means a divalent group (C 1-6 alkylene) formed by removing any one hydrogen atom of the above “lower alkyl”, and one embodiment is C 1-4 alkylene. Another embodiment is methylene or ethylene.
「高級アルキル」とは、直鎖又は分枝状の炭素数が7から12(以後、C7-12と略す)のアルキル、例えば、ヘプチル、1-プロピルブチル、オクチル、ノニル、デシル、ドデシルである。別の態様としては、C7-8アルキルである。さらに別の態様としては、ヘプチル、オクチルである。
“Higher alkyl” means linear or branched alkyl having 7 to 12 carbon atoms (hereinafter abbreviated as C 7-12 ), for example, heptyl, 1-propylbutyl, octyl, nonyl, decyl, dodecyl. is there. In another embodiment, it is C 7-8 alkyl. Yet another embodiment is heptyl or octyl.
「高級アルキレン」は、上記「高級アルキル」の任意の水素原子1個を除去してなる二価基(C7-12アルキレン)を意味し、ある態様としてはC7-10アルキレンである。別の態様としてはヘプタンジイル、オクタンジイルである。
“Higher alkylene” means a divalent group (C 7-12 alkylene) formed by removing any one hydrogen atom of the above “higher alkyl”, and as an embodiment, C 7-10 alkylene. Another embodiment is heptanediyl or octanediyl.
「ハロゲン」は、F、Cl、Br、Iを意味する。
「シクロアルキル」とは、C3-10の飽和炭化水素環基であり、架橋を有していてもよい。例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、アダマンチル等である。別の態様としては、C3-8シクロアルキルである。さらに別の態様としては、シクロプロピル、シクロブチル、シクロヘキシル、シクロヘプチル、アダマンチルである。
「アリール」とは、C6-14の単環式~三環式芳香族炭化水素環基であり、例えばフェニル、ナフチルであり、別の態様としてはフェニルである。 “Halogen” means F, Cl, Br, I.
“Cycloalkyl” is a C 3-10 saturated hydrocarbon ring group, which may have a bridge. For example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, adamantyl and the like. In another embodiment, it is C 3-8 cycloalkyl. Yet another embodiment is cyclopropyl, cyclobutyl, cyclohexyl, cycloheptyl, adamantyl.
“Aryl” is a C 6-14 monocyclic to tricyclic aromatic hydrocarbon ring group, for example, phenyl or naphthyl, and in another embodiment, phenyl.
「シクロアルキル」とは、C3-10の飽和炭化水素環基であり、架橋を有していてもよい。例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、アダマンチル等である。別の態様としては、C3-8シクロアルキルである。さらに別の態様としては、シクロプロピル、シクロブチル、シクロヘキシル、シクロヘプチル、アダマンチルである。
「アリール」とは、C6-14の単環式~三環式芳香族炭化水素環基であり、例えばフェニル、ナフチルであり、別の態様としてはフェニルである。 “Halogen” means F, Cl, Br, I.
“Cycloalkyl” is a C 3-10 saturated hydrocarbon ring group, which may have a bridge. For example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, adamantyl and the like. In another embodiment, it is C 3-8 cycloalkyl. Yet another embodiment is cyclopropyl, cyclobutyl, cyclohexyl, cycloheptyl, adamantyl.
“Aryl” is a C 6-14 monocyclic to tricyclic aromatic hydrocarbon ring group, for example, phenyl or naphthyl, and in another embodiment, phenyl.
「ヘテロ環」基とは、i)酸素、硫黄及び窒素から選択されるヘテロ原子を1~4個含有する3~8員の、別の態様としては5~7員の単環式ヘテロ環、並びに、ii)当該単環式ヘテロ環が、単環式へテロ環、ベンゼン環、C5-10シクロアルカン及びC5-10シクロアルケンからなる群より選択される1又は2個の環と縮環し形成される、酸素、硫黄および窒素から選択されるヘテロ原子を1~5個含有する二から三環式ヘテロ環、から選択される環基を意味する。環原子である硫黄又は窒素が酸化されオキシドやジオキシドを形成してもよい。
“Heterocycle” group refers to i) a 3-8 membered, alternatively 5-7 membered monocyclic heterocycle containing 1-4 heteroatoms selected from oxygen, sulfur and nitrogen, And ii) the monocyclic heterocycle is condensed with one or two rings selected from the group consisting of a monocyclic heterocycle, a benzene ring, a C 5-10 cycloalkane and a C 5-10 cycloalkene. A ring group selected from bicyclic to tricyclic heterocycles containing 1 to 5 heteroatoms selected from oxygen, sulfur and nitrogen formed in a ring. Ring atoms such as sulfur or nitrogen may be oxidized to form oxides or dioxides.
「ヘテロ環」基として以下の態様が挙げられる。
(1)単環式飽和へテロ環基
(a)1~4個の窒素原子を含むもの、例えば、アゼパニル、ジアゼパニル、アジリジニル、アゼチジニル、ピロリジニル、イミダゾリジニル、ピペリジル、ピラゾリジニル、ピペラジニル、アゾカニル等;
(b)1~3個の窒素原子、ならびに1~2個の硫黄原子および/または1~2個の酸素原子を含むもの、例えば、チオモルホリニル、チアゾリジニル、イソチアゾリジニル、オキサゾリジニル、モルホリニル等;
(c)1~2個の硫黄原子を含むもの、例えば、テトラヒドロチオピラニル等;
(d)1~2個の硫黄原子および1~2個の酸素原子を含むもの、例えば、オキサチオラニル等;
(e)1~2個の酸素原子を含むもの、例えば、オキシラニル、オキセタニル、ジオキソラニル、テトラヒドロフラニル、テトラヒドロピラニル、1,4-ジオキサニル等; The “heterocyclic” group includes the following embodiments.
(1) Monocyclic saturated heterocyclic group
(a) those containing 1 to 4 nitrogen atoms, such as azepanyl, diazepanyl, aziridinyl, azetidinyl, pyrrolidinyl, imidazolidinyl, piperidyl, pyrazolidinyl, piperazinyl, azocanyl and the like;
(b) those containing 1 to 3 nitrogen atoms and 1 to 2 sulfur atoms and / or 1 to 2 oxygen atoms, such as thiomorpholinyl, thiazolidinyl, isothiazolidinyl, oxazolidinyl, morpholinyl and the like;
(c) those containing 1 to 2 sulfur atoms, such as tetrahydrothiopyranyl;
(d) those containing 1 to 2 sulfur atoms and 1 to 2 oxygen atoms, such as oxathiolanyl;
(e) those containing 1 to 2 oxygen atoms, such as oxiranyl, oxetanyl, dioxolanyl, tetrahydrofuranyl, tetrahydropyranyl, 1,4-dioxanyl and the like;
(1)単環式飽和へテロ環基
(a)1~4個の窒素原子を含むもの、例えば、アゼパニル、ジアゼパニル、アジリジニル、アゼチジニル、ピロリジニル、イミダゾリジニル、ピペリジル、ピラゾリジニル、ピペラジニル、アゾカニル等;
(b)1~3個の窒素原子、ならびに1~2個の硫黄原子および/または1~2個の酸素原子を含むもの、例えば、チオモルホリニル、チアゾリジニル、イソチアゾリジニル、オキサゾリジニル、モルホリニル等;
(c)1~2個の硫黄原子を含むもの、例えば、テトラヒドロチオピラニル等;
(d)1~2個の硫黄原子および1~2個の酸素原子を含むもの、例えば、オキサチオラニル等;
(e)1~2個の酸素原子を含むもの、例えば、オキシラニル、オキセタニル、ジオキソラニル、テトラヒドロフラニル、テトラヒドロピラニル、1,4-ジオキサニル等; The “heterocyclic” group includes the following embodiments.
(1) Monocyclic saturated heterocyclic group
(a) those containing 1 to 4 nitrogen atoms, such as azepanyl, diazepanyl, aziridinyl, azetidinyl, pyrrolidinyl, imidazolidinyl, piperidyl, pyrazolidinyl, piperazinyl, azocanyl and the like;
(b) those containing 1 to 3 nitrogen atoms and 1 to 2 sulfur atoms and / or 1 to 2 oxygen atoms, such as thiomorpholinyl, thiazolidinyl, isothiazolidinyl, oxazolidinyl, morpholinyl and the like;
(c) those containing 1 to 2 sulfur atoms, such as tetrahydrothiopyranyl;
(d) those containing 1 to 2 sulfur atoms and 1 to 2 oxygen atoms, such as oxathiolanyl;
(e) those containing 1 to 2 oxygen atoms, such as oxiranyl, oxetanyl, dioxolanyl, tetrahydrofuranyl, tetrahydropyranyl, 1,4-dioxanyl and the like;
(2)単環式不飽和へテロ環基
(a)1~4個の窒素原子を含むもの、例えば、ピロリル、イミダゾリル、ピラゾリル、ピリジル、ジヒドロピリジル、テトラヒドロピリジニル、ピリミジニル、ピラジニル、ピリダジニル、トリアゾリル、テトラゾリル、トリアジニル、ジヒドロトリアジニル、アゼピニル、1,2,3,6-テトラヒドロピリジル等;
(b)1~3個の窒素原子、ならびに1~2個の硫黄原子および/または1~2個の酸素原子を含むもの、例えば、チアゾリル、イソチアゾリル、チアジアゾリル、ジヒドロチアジニル、オキサゾリル、イソオキサゾリル、オキサジアゾリル、オキサジニル等;
(c)1~2個の硫黄原子を含むもの、例えば、チエニル、チエピニル、ジヒドロジチオピラニル、ジヒドロジチオニル等;
(d)1~2個の硫黄原子および1~2個の酸素原子を含むもの、具体的には、ジヒドロオキサチオピラニル等;
(e)1~2個の酸素原子を含むもの、例えば、フリル、ピラニル、オキセピニル、ジオキソリル等; (2) Monocyclic unsaturated heterocyclic group
(a) those containing 1 to 4 nitrogen atoms, such as pyrrolyl, imidazolyl, pyrazolyl, pyridyl, dihydropyridyl, tetrahydropyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazolyl, tetrazolyl, triazinyl, dihydrotriazinyl, azepinyl 1,2,3,6-tetrahydropyridyl and the like;
(b) those containing 1 to 3 nitrogen atoms and 1 to 2 sulfur atoms and / or 1 to 2 oxygen atoms, such as thiazolyl, isothiazolyl, thiadiazolyl, dihydrothiazinyl, oxazolyl, isoxazolyl, oxadiazolyl, Oxazinyl and the like;
(c) those containing 1 to 2 sulfur atoms, such as thienyl, thiepinyl, dihydrodithiopyranyl, dihydrodithionyl and the like;
(d) those containing 1 to 2 sulfur atoms and 1 to 2 oxygen atoms, specifically, dihydrooxathiopyranyl and the like;
(e) those containing 1 to 2 oxygen atoms, such as furyl, pyranyl, oxepinyl, dioxolyl and the like;
(a)1~4個の窒素原子を含むもの、例えば、ピロリル、イミダゾリル、ピラゾリル、ピリジル、ジヒドロピリジル、テトラヒドロピリジニル、ピリミジニル、ピラジニル、ピリダジニル、トリアゾリル、テトラゾリル、トリアジニル、ジヒドロトリアジニル、アゼピニル、1,2,3,6-テトラヒドロピリジル等;
(b)1~3個の窒素原子、ならびに1~2個の硫黄原子および/または1~2個の酸素原子を含むもの、例えば、チアゾリル、イソチアゾリル、チアジアゾリル、ジヒドロチアジニル、オキサゾリル、イソオキサゾリル、オキサジアゾリル、オキサジニル等;
(c)1~2個の硫黄原子を含むもの、例えば、チエニル、チエピニル、ジヒドロジチオピラニル、ジヒドロジチオニル等;
(d)1~2個の硫黄原子および1~2個の酸素原子を含むもの、具体的には、ジヒドロオキサチオピラニル等;
(e)1~2個の酸素原子を含むもの、例えば、フリル、ピラニル、オキセピニル、ジオキソリル等; (2) Monocyclic unsaturated heterocyclic group
(a) those containing 1 to 4 nitrogen atoms, such as pyrrolyl, imidazolyl, pyrazolyl, pyridyl, dihydropyridyl, tetrahydropyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazolyl, tetrazolyl, triazinyl, dihydrotriazinyl, azepinyl 1,2,3,6-tetrahydropyridyl and the like;
(b) those containing 1 to 3 nitrogen atoms and 1 to 2 sulfur atoms and / or 1 to 2 oxygen atoms, such as thiazolyl, isothiazolyl, thiadiazolyl, dihydrothiazinyl, oxazolyl, isoxazolyl, oxadiazolyl, Oxazinyl and the like;
(c) those containing 1 to 2 sulfur atoms, such as thienyl, thiepinyl, dihydrodithiopyranyl, dihydrodithionyl and the like;
(d) those containing 1 to 2 sulfur atoms and 1 to 2 oxygen atoms, specifically, dihydrooxathiopyranyl and the like;
(e) those containing 1 to 2 oxygen atoms, such as furyl, pyranyl, oxepinyl, dioxolyl and the like;
(3)縮合多環式飽和へテロ環基
(a)1~5個の窒素原子を含むもの、例えば、キヌクリジニル、7-アザビシクロ[2.2.1]ヘプチル、3-アザビシクロ[3.2.2]ノナニル等;
(b)1~4個の窒素原子、ならびに1~3個の硫黄原子および/または1~3個の酸素原子を含むもの、例えば、トリチアジアザインデニル、ジオキソロイミダゾリジニル等;
(c)1~3個の硫黄原子および/または1~3個の酸素原子を含むもの、例えば、2,6-ジオキサビシクロ[3.2.2]オクト-7-イル等; (3) Fused polycyclic saturated hetero ring group
(a) those containing 1 to 5 nitrogen atoms, such as quinuclidinyl, 7-azabicyclo [2.2.1] heptyl, 3-azabicyclo [3.2.2] nonanyl;
(b) those containing 1 to 4 nitrogen atoms, and 1 to 3 sulfur atoms and / or 1 to 3 oxygen atoms, such as trithiadiazaindenyl, dioxoleumidazolidinyl and the like;
(c) those containing 1 to 3 sulfur atoms and / or 1 to 3 oxygen atoms, such as 2,6-dioxabicyclo [3.2.2] oct-7-yl;
(a)1~5個の窒素原子を含むもの、例えば、キヌクリジニル、7-アザビシクロ[2.2.1]ヘプチル、3-アザビシクロ[3.2.2]ノナニル等;
(b)1~4個の窒素原子、ならびに1~3個の硫黄原子および/または1~3個の酸素原子を含むもの、例えば、トリチアジアザインデニル、ジオキソロイミダゾリジニル等;
(c)1~3個の硫黄原子および/または1~3個の酸素原子を含むもの、例えば、2,6-ジオキサビシクロ[3.2.2]オクト-7-イル等; (3) Fused polycyclic saturated hetero ring group
(a) those containing 1 to 5 nitrogen atoms, such as quinuclidinyl, 7-azabicyclo [2.2.1] heptyl, 3-azabicyclo [3.2.2] nonanyl;
(b) those containing 1 to 4 nitrogen atoms, and 1 to 3 sulfur atoms and / or 1 to 3 oxygen atoms, such as trithiadiazaindenyl, dioxoleumidazolidinyl and the like;
(c) those containing 1 to 3 sulfur atoms and / or 1 to 3 oxygen atoms, such as 2,6-dioxabicyclo [3.2.2] oct-7-yl;
(4)縮合多環式不飽和へテロ環基
(a)1~5個の窒素原子を含むもの、例えば、インドリル、イソインドリル、インドリニル、インドリジニル、ベンゾイミダゾリル、ジヒドロベンゾイミダゾリル、テトラヒドロベンゾイミダゾリル、キノリル、テトラヒドロキノリル、イソキノリル、テトラヒドロイソキノリル、インダゾリル、イミダゾピリジル、ベンゾトリアゾリル、テトラゾロピリダジニル、カルバゾリル、アクリジニル、キノキサリニル、ジヒドロキノキサリニル、テトラヒドロキノキサリニル、フタラジニル、ジヒドロインダゾリル、ベンゾピリミジニル、ナフチリジニル、キナゾリニル、シンノリニル、;
(b)1~4個の窒素原子、ならびに1~3個の硫黄原子および/または1~3個の酸素原子を含むもの、例えば、ベンゾチアゾリル、ジヒドロベンゾチアゾリル、ベンゾチアジアゾリル、イミダゾチアゾリル、イミダゾチアジアゾリル、ベンゾオキサゾリル、ジヒドロベンゾオキサゾリル、ジヒドロベンゾオキサジニル、ベンゾオキサジアゾリル、ベンゾイソチアゾリル、ベンゾイソオキサゾリル、イミダゾ[2,1-b][1,3,4]チアジアゾリル等;
(c)1~3個の硫黄原子を含むもの、例えば、ベンゾチエニル、ベンゾジチオピラニル、ジベンゾ[b,d]チエニル等;
(d)1~3個の硫黄原子および1~3個の酸素原子を含むもの、例えば、ベンゾオキサチオピラニル、フェノキサジニル等;
(e)1~3個の酸素原子を含むもの、例えば、ベンゾジオキソリル、ベンゾフラニル、ジヒドロベンゾフラニル、イソベンゾフラニル、クロマニル、クロメニル、ジベンゾ[b,d]フラニル、メチレンジオキシフェニル、エチレンジオキシフェニル等;
など。 (4) Fused polycyclic unsaturated heterocyclic group
(a) those containing 1 to 5 nitrogen atoms, for example, indolyl, isoindolyl, indolinyl, indolizinyl, benzimidazolyl, dihydrobenzimidazolyl, tetrahydrobenzoimidazolyl, quinolyl, tetrahydroquinolyl, isoquinolyl, tetrahydroisoquinolyl, indazolyl, imidazopyridyl, Benzotriazolyl, tetrazolopyridazinyl, carbazolyl, acridinyl, quinoxalinyl, dihydroquinoxalinyl, tetrahydroquinoxalinyl, phthalazinyl, dihydroindazolyl, benzopyrimidinyl, naphthyridinyl, quinazolinyl, cinnolinyl;
(b) those containing 1 to 4 nitrogen atoms and 1 to 3 sulfur atoms and / or 1 to 3 oxygen atoms, for example benzothiazolyl, dihydrobenzothiazolyl, benzothiadiazolyl, imidazothia Zolyl, imidazothiadiazolyl, benzoxazolyl, dihydrobenzoxazolyl, dihydrobenzoxazinyl, benzooxadiazolyl, benzisothiazolyl, benzisoxazolyl, imidazo [2,1-b] [ 1,3,4] thiadiazolyl, etc .;
(c) those containing 1 to 3 sulfur atoms, such as benzothienyl, benzodithiopyranyl, dibenzo [b, d] thienyl, etc .;
(d) those containing 1 to 3 sulfur atoms and 1 to 3 oxygen atoms, such as benzooxathiopyranyl, phenoxazinyl and the like;
(e) those containing 1 to 3 oxygen atoms, such as benzodioxolyl, benzofuranyl, dihydrobenzofuranyl, isobenzofuranyl, chromanyl, chromenyl, dibenzo [b, d] furanyl, methylenedioxyphenyl, Ethylenedioxyphenyl and the like;
Such.
(a)1~5個の窒素原子を含むもの、例えば、インドリル、イソインドリル、インドリニル、インドリジニル、ベンゾイミダゾリル、ジヒドロベンゾイミダゾリル、テトラヒドロベンゾイミダゾリル、キノリル、テトラヒドロキノリル、イソキノリル、テトラヒドロイソキノリル、インダゾリル、イミダゾピリジル、ベンゾトリアゾリル、テトラゾロピリダジニル、カルバゾリル、アクリジニル、キノキサリニル、ジヒドロキノキサリニル、テトラヒドロキノキサリニル、フタラジニル、ジヒドロインダゾリル、ベンゾピリミジニル、ナフチリジニル、キナゾリニル、シンノリニル、;
(b)1~4個の窒素原子、ならびに1~3個の硫黄原子および/または1~3個の酸素原子を含むもの、例えば、ベンゾチアゾリル、ジヒドロベンゾチアゾリル、ベンゾチアジアゾリル、イミダゾチアゾリル、イミダゾチアジアゾリル、ベンゾオキサゾリル、ジヒドロベンゾオキサゾリル、ジヒドロベンゾオキサジニル、ベンゾオキサジアゾリル、ベンゾイソチアゾリル、ベンゾイソオキサゾリル、イミダゾ[2,1-b][1,3,4]チアジアゾリル等;
(c)1~3個の硫黄原子を含むもの、例えば、ベンゾチエニル、ベンゾジチオピラニル、ジベンゾ[b,d]チエニル等;
(d)1~3個の硫黄原子および1~3個の酸素原子を含むもの、例えば、ベンゾオキサチオピラニル、フェノキサジニル等;
(e)1~3個の酸素原子を含むもの、例えば、ベンゾジオキソリル、ベンゾフラニル、ジヒドロベンゾフラニル、イソベンゾフラニル、クロマニル、クロメニル、ジベンゾ[b,d]フラニル、メチレンジオキシフェニル、エチレンジオキシフェニル等;
など。 (4) Fused polycyclic unsaturated heterocyclic group
(a) those containing 1 to 5 nitrogen atoms, for example, indolyl, isoindolyl, indolinyl, indolizinyl, benzimidazolyl, dihydrobenzimidazolyl, tetrahydrobenzoimidazolyl, quinolyl, tetrahydroquinolyl, isoquinolyl, tetrahydroisoquinolyl, indazolyl, imidazopyridyl, Benzotriazolyl, tetrazolopyridazinyl, carbazolyl, acridinyl, quinoxalinyl, dihydroquinoxalinyl, tetrahydroquinoxalinyl, phthalazinyl, dihydroindazolyl, benzopyrimidinyl, naphthyridinyl, quinazolinyl, cinnolinyl;
(b) those containing 1 to 4 nitrogen atoms and 1 to 3 sulfur atoms and / or 1 to 3 oxygen atoms, for example benzothiazolyl, dihydrobenzothiazolyl, benzothiadiazolyl, imidazothia Zolyl, imidazothiadiazolyl, benzoxazolyl, dihydrobenzoxazolyl, dihydrobenzoxazinyl, benzooxadiazolyl, benzisothiazolyl, benzisoxazolyl, imidazo [2,1-b] [ 1,3,4] thiadiazolyl, etc .;
(c) those containing 1 to 3 sulfur atoms, such as benzothienyl, benzodithiopyranyl, dibenzo [b, d] thienyl, etc .;
(d) those containing 1 to 3 sulfur atoms and 1 to 3 oxygen atoms, such as benzooxathiopyranyl, phenoxazinyl and the like;
(e) those containing 1 to 3 oxygen atoms, such as benzodioxolyl, benzofuranyl, dihydrobenzofuranyl, isobenzofuranyl, chromanyl, chromenyl, dibenzo [b, d] furanyl, methylenedioxyphenyl, Ethylenedioxyphenyl and the like;
Such.
「含窒素へテロ環」基とは、上記の「へテロ環」基のうち、(1)(a)、(1)(b)、(2)(a)、(2)(b)、(3)(a)、(3)(b)、(4)(a)及び(4)(b)等のように、少なくとも1個の窒素原子を含んでいるものをいう。
「ヘテロアリール」とは、上記の「へテロ環」基の(2)及び(4)のうち、芳香族性を有する単環式または縮合多環式へテロ環基である。
例えば、ピリジル、ピロリル、ピラジニル、ピリミジニル、ピリダジニル、イミダゾリル、トリアゾリル、トリアジニル、テトラゾリル、チアゾリル、ピラゾリル、イソチアゾリル、オキサゾリル、イソオキサゾリル、チアジアゾリル、オキサジアゾリル、チエニル、フリル等の単環式ヘテロアリール、インドリル、イソインドリル、ベンゾイミダゾリル、インダゾリル、キノリル、イソキノリル、キナゾリニル、キノキサリニル、イミダゾピリジル、ピラゾロピリジル、フタラジニル、ベンゾチアゾリル、ベンゾイソチアゾリル、ベンゾチアジアゾリル、ベンゾオキサゾリル、イミダゾチアゾリル、ベンゾイソオキサゾリル、ベンゾフラニル、イミダゾ[2,1-b][1,3,4]チアジアゾリル、ベンゾチエニル、イミダゾチアジアゾリル、等の二環式ヘテロアリール、カルバゾリル、ジベンゾ[b,d]フラニル、ジベンゾ[b,d]チエニル等の三環式ヘテロアリールであり、別の態様としては、ピリジル、ピリミジニル、チアゾリル、オキサゾリル、イソオキサゾリル、チアジアゾリル、オキサジアゾリル、イミダゾピリジル、ピラゾロピリジル、ベンゾチアジアゾリル、ベンゾオキサゾリル、イミダゾチアゾリル、イミダゾチアジアゾリルであり、さらに別の態様としては、ピリミジニル、オキサゾリル、イミダゾ[2,1-b][1,3,4]チアジアゾリル又は、イミダゾチアジアゾリルである。 The “nitrogen-containing heterocycle” group is one of the above “heterocycle” groups (1) (a), (1) (b), (2) (a), (2) (b), (3) A substance containing at least one nitrogen atom, such as (a), (3) (b), (4) (a) and (4) (b).
“Heteroaryl” is a monocyclic or condensed polycyclic heterocyclic group having aromaticity among (2) and (4) of the above “heterocyclic” group.
For example, pyridyl, pyrrolyl, pyrazinyl, pyrimidinyl, pyridazinyl, imidazolyl, triazolyl, triazinyl, tetrazolyl, thiazolyl, pyrazolyl, isothiazolyl, oxazolyl, isoxazolyl, thiadiazolyl, oxadiazolyl, thienyl, furyl etc. monocyclic heteroaryl, indolyl, isoindolyl, benzoimidyl , Indazolyl, quinolyl, isoquinolyl, quinazolinyl, quinoxalinyl, imidazopyridyl, pyrazolopyridyl, phthalazinyl, benzothiazolyl, benzoisothiazolyl, benzothiadiazolyl, benzoxazolyl, imidazothiazolyl, benzoisoxazolyl, benzofuranyl, Bicyclic [2,1-b] [1,3,4] thiadiazolyl, benzothienyl, imidazothiadiazolyl, etc. Triaryl heteroaryl such as roaryl, carbazolyl, dibenzo [b, d] furanyl, dibenzo [b, d] thienyl, and other embodiments include pyridyl, pyrimidinyl, thiazolyl, oxazolyl, isoxazolyl, thiadiazolyl, oxadiazolyl, imidazo Pyridyl, pyrazolopyridyl, benzothiadiazolyl, benzoxazolyl, imidazothiazolyl, imidazothiadiazolyl, and in yet another embodiment, pyrimidinyl, oxazolyl, imidazo [2,1-b] [1,1-b 3,4] thiadiazolyl or imidazochiadiazolyl.
「ヘテロアリール」とは、上記の「へテロ環」基の(2)及び(4)のうち、芳香族性を有する単環式または縮合多環式へテロ環基である。
例えば、ピリジル、ピロリル、ピラジニル、ピリミジニル、ピリダジニル、イミダゾリル、トリアゾリル、トリアジニル、テトラゾリル、チアゾリル、ピラゾリル、イソチアゾリル、オキサゾリル、イソオキサゾリル、チアジアゾリル、オキサジアゾリル、チエニル、フリル等の単環式ヘテロアリール、インドリル、イソインドリル、ベンゾイミダゾリル、インダゾリル、キノリル、イソキノリル、キナゾリニル、キノキサリニル、イミダゾピリジル、ピラゾロピリジル、フタラジニル、ベンゾチアゾリル、ベンゾイソチアゾリル、ベンゾチアジアゾリル、ベンゾオキサゾリル、イミダゾチアゾリル、ベンゾイソオキサゾリル、ベンゾフラニル、イミダゾ[2,1-b][1,3,4]チアジアゾリル、ベンゾチエニル、イミダゾチアジアゾリル、等の二環式ヘテロアリール、カルバゾリル、ジベンゾ[b,d]フラニル、ジベンゾ[b,d]チエニル等の三環式ヘテロアリールであり、別の態様としては、ピリジル、ピリミジニル、チアゾリル、オキサゾリル、イソオキサゾリル、チアジアゾリル、オキサジアゾリル、イミダゾピリジル、ピラゾロピリジル、ベンゾチアジアゾリル、ベンゾオキサゾリル、イミダゾチアゾリル、イミダゾチアジアゾリルであり、さらに別の態様としては、ピリミジニル、オキサゾリル、イミダゾ[2,1-b][1,3,4]チアジアゾリル又は、イミダゾチアジアゾリルである。 The “nitrogen-containing heterocycle” group is one of the above “heterocycle” groups (1) (a), (1) (b), (2) (a), (2) (b), (3) A substance containing at least one nitrogen atom, such as (a), (3) (b), (4) (a) and (4) (b).
“Heteroaryl” is a monocyclic or condensed polycyclic heterocyclic group having aromaticity among (2) and (4) of the above “heterocyclic” group.
For example, pyridyl, pyrrolyl, pyrazinyl, pyrimidinyl, pyridazinyl, imidazolyl, triazolyl, triazinyl, tetrazolyl, thiazolyl, pyrazolyl, isothiazolyl, oxazolyl, isoxazolyl, thiadiazolyl, oxadiazolyl, thienyl, furyl etc. monocyclic heteroaryl, indolyl, isoindolyl, benzoimidyl , Indazolyl, quinolyl, isoquinolyl, quinazolinyl, quinoxalinyl, imidazopyridyl, pyrazolopyridyl, phthalazinyl, benzothiazolyl, benzoisothiazolyl, benzothiadiazolyl, benzoxazolyl, imidazothiazolyl, benzoisoxazolyl, benzofuranyl, Bicyclic [2,1-b] [1,3,4] thiadiazolyl, benzothienyl, imidazothiadiazolyl, etc. Triaryl heteroaryl such as roaryl, carbazolyl, dibenzo [b, d] furanyl, dibenzo [b, d] thienyl, and other embodiments include pyridyl, pyrimidinyl, thiazolyl, oxazolyl, isoxazolyl, thiadiazolyl, oxadiazolyl, imidazo Pyridyl, pyrazolopyridyl, benzothiadiazolyl, benzoxazolyl, imidazothiazolyl, imidazothiadiazolyl, and in yet another embodiment, pyrimidinyl, oxazolyl, imidazo [2,1-b] [1,1-b 3,4] thiadiazolyl or imidazochiadiazolyl.
「含窒素ヘテロアリール」は、上記「ヘテロアリール」のうち、少なくとも1つの窒素原子を含んでいる単環式または縮合多環式ものをいう。例えば、ある態様として、イミダゾチアジアゾリル、ピリミジニル、又は、オキサジアゾリルがあげられる。また、別の態様として、イミダゾ[2,1-b][1,3,4]チアジアゾリル、ピリミジニル、1,2,4-オキサジアゾリルが挙げられる。
「5乃至6員の単環式含窒素へテロアリール」は、環を構成する原子数が5個又は6個の上記単環式の「含窒素ヘテロアリール」あり、ピリジル、ピリミジニル、チアゾリル、チアジアゾリル、オキサゾール又は、オキサジアゾリル等が挙げられる。
「5員-5員縮合多環式含窒素へテロアリールまたは5員-6員縮合多環式含窒素へテロアリール」は、上記「含窒素ヘテロアリール」のうちベンズイミダゾール、、イミダゾチアジアゾリル、インドール、ベンズオキサゾール、イミダゾ[2,1-b][1,3,4]チアジアゾリル等が挙げられる。 “Nitrogen-containing heteroaryl” refers to a monocyclic or condensed polycyclic ring containing at least one nitrogen atom among the above “heteroaryl”. For example, as an embodiment, imidazothiadiazolyl, pyrimidinyl, or oxadiazolyl can be mentioned. As another embodiment, imidazo [2,1-b] [1,3,4] thiadiazolyl, pyrimidinyl, 1,2,4-oxadiazolyl may be mentioned.
“5- to 6-membered monocyclic nitrogen-containing heteroaryl” is the above monocyclic “nitrogen-containing heteroaryl” having 5 or 6 atoms constituting the ring, and pyridyl, pyrimidinyl, thiazolyl, thiadiazolyl, Examples include oxazole or oxadiazolyl.
"5-membered or 5-membered condensed polycyclic nitrogen-containing heteroaryl or 5-membered or 6-membered condensed polycyclic nitrogen-containing heteroaryl" is a group of benzimidazole, imidazothiadiazolyl, indole among the above "nitrogen-containing heteroaryl". Benzoxazole, imidazo [2,1-b] [1,3,4] thiadiazolyl, and the like.
「5乃至6員の単環式含窒素へテロアリール」は、環を構成する原子数が5個又は6個の上記単環式の「含窒素ヘテロアリール」あり、ピリジル、ピリミジニル、チアゾリル、チアジアゾリル、オキサゾール又は、オキサジアゾリル等が挙げられる。
「5員-5員縮合多環式含窒素へテロアリールまたは5員-6員縮合多環式含窒素へテロアリール」は、上記「含窒素ヘテロアリール」のうちベンズイミダゾール、、イミダゾチアジアゾリル、インドール、ベンズオキサゾール、イミダゾ[2,1-b][1,3,4]チアジアゾリル等が挙げられる。 “Nitrogen-containing heteroaryl” refers to a monocyclic or condensed polycyclic ring containing at least one nitrogen atom among the above “heteroaryl”. For example, as an embodiment, imidazothiadiazolyl, pyrimidinyl, or oxadiazolyl can be mentioned. As another embodiment, imidazo [2,1-b] [1,3,4] thiadiazolyl, pyrimidinyl, 1,2,4-oxadiazolyl may be mentioned.
“5- to 6-membered monocyclic nitrogen-containing heteroaryl” is the above monocyclic “nitrogen-containing heteroaryl” having 5 or 6 atoms constituting the ring, and pyridyl, pyrimidinyl, thiazolyl, thiadiazolyl, Examples include oxazole or oxadiazolyl.
"5-membered or 5-membered condensed polycyclic nitrogen-containing heteroaryl or 5-membered or 6-membered condensed polycyclic nitrogen-containing heteroaryl" is a group of benzimidazole, imidazothiadiazolyl, indole among the above "nitrogen-containing heteroaryl". Benzoxazole, imidazo [2,1-b] [1,3,4] thiadiazolyl, and the like.
「ヘテロシクロアルキル」とは、上記の「へテロ環」基の(1)(a)、(1)(b) 、(3)(a)、(3)(b)又は、上記「ヘテロ環」基の(2)(a)、(2)(b)、(4)(a)、(4)(b)の内、芳香族性を有さないヘテロ環基をいう。例えば、アゼパニル、ジアゼパニル、アジリジニル、アゼチジニル、ピロリジニル、イミダゾリジニル、ピペリジル、1,2,3,6-テトラヒドロピリジル、ピラゾリジニル、ピペラジニル、アゾカニル、チオモルホリニル、チアゾリジニル、イソチアゾリジニル、オキサゾリジニル、モルホリニル、テトラヒドロピリジニル、ジヒドロトリアジニル、テトラヒドロピリジル等の単環ヘテロシクロアルキル、キヌクリジル、7-アザビシクロ[2.2.1]ヘプチル等の二環ヘテロシクロアルキルが挙げられる。
“Heterocycloalkyl” refers to (1) (a), (1) (b), (3) (a), (3) (b) of the above “heterocyclic” group or “heterocyclic” Of the group (2) (a), (2) (b), (4) (a), (4) (b) is a heterocyclic group having no aromaticity. For example, azepanyl, diazepanyl, aziridinyl, azetidinyl, pyrrolidinyl, imidazolidinyl, piperidyl, 1,2,3,6-tetrahydropyridyl, pyrazolidinyl, piperazinyl, azocanyl, thiomorpholinyl, thiazolidinyl, isothiazolidinyl, oxazolidinyl, morpholinyl, morpholinyl, Monocyclic heterocycloalkyl such as dihydrotriazinyl and tetrahydropyridyl, and bicyclic heterocycloalkyl such as quinuclidyl and 7-azabicyclo [2.2.1] heptyl.
「含窒素ヘテロシクロアルキル」とは、上記「ヘテロシクロアルキル」のうち、少なくとも1つの窒素原子を含んでいるものをいう。ある態様としては、ピペリジル、ピペラジニル、テトラヒドロピリジル等である。また別の態様としては、1,2,3,6-テトラヒドロピリジルである。
「6員の含窒素ヘテロシクロアルキル」とは、上記「含窒素ヘテロアルキル」のうち、環を構成する原子数が6個のものをいう。ある態様としては、ピペリジル、ピペラジニル、テトラヒドロピリジル等である。また別の態様としては、1,2,3,6-テトラヒドロピリジルである。
「6員の単環式ヘテロ環」基とは、上記(1)及び(2)のうち環を構成する原子数が6個のものをいう。ある態様としては、ピペリジル、ピペラジニル、1,2,3,6-テトラヒドロピリジル、ピリジル、ピリミジニル、ピラジニル等である。 “Nitrogen-containing heterocycloalkyl” refers to the above “heterocycloalkyl” containing at least one nitrogen atom. Some embodiments include piperidyl, piperazinyl, tetrahydropyridyl and the like. In another embodiment, it is 1,2,3,6-tetrahydropyridyl.
The “6-membered nitrogen-containing heterocycloalkyl” refers to the above-mentioned “nitrogen-containing heteroalkyl” having 6 atoms constituting the ring. Some embodiments include piperidyl, piperazinyl, tetrahydropyridyl and the like. In another embodiment, it is 1,2,3,6-tetrahydropyridyl.
The “6-membered monocyclic heterocycle” group refers to a group having 6 atoms constituting the ring among the above (1) and (2). Some embodiments include piperidyl, piperazinyl, 1,2,3,6-tetrahydropyridyl, pyridyl, pyrimidinyl, pyrazinyl and the like.
「6員の含窒素ヘテロシクロアルキル」とは、上記「含窒素ヘテロアルキル」のうち、環を構成する原子数が6個のものをいう。ある態様としては、ピペリジル、ピペラジニル、テトラヒドロピリジル等である。また別の態様としては、1,2,3,6-テトラヒドロピリジルである。
「6員の単環式ヘテロ環」基とは、上記(1)及び(2)のうち環を構成する原子数が6個のものをいう。ある態様としては、ピペリジル、ピペラジニル、1,2,3,6-テトラヒドロピリジル、ピリジル、ピリミジニル、ピラジニル等である。 “Nitrogen-containing heterocycloalkyl” refers to the above “heterocycloalkyl” containing at least one nitrogen atom. Some embodiments include piperidyl, piperazinyl, tetrahydropyridyl and the like. In another embodiment, it is 1,2,3,6-tetrahydropyridyl.
The “6-membered nitrogen-containing heterocycloalkyl” refers to the above-mentioned “nitrogen-containing heteroalkyl” having 6 atoms constituting the ring. Some embodiments include piperidyl, piperazinyl, tetrahydropyridyl and the like. In another embodiment, it is 1,2,3,6-tetrahydropyridyl.
The “6-membered monocyclic heterocycle” group refers to a group having 6 atoms constituting the ring among the above (1) and (2). Some embodiments include piperidyl, piperazinyl, 1,2,3,6-tetrahydropyridyl, pyridyl, pyrimidinyl, pyrazinyl and the like.
「環基」とは、上記定義のうち、含窒素ヘテロアリール、含窒素ヘテロシクロアルキル、アリール、又は、シクロアルキル等の環状の基を意味し、結合手が二本ある場合には、二価基を示す。
“Ring group” means a cyclic group such as nitrogen-containing heteroaryl, nitrogen-containing heterocycloalkyl, aryl, or cycloalkyl in the above definition, and when there are two bonds, it is divalent. Indicates a group.
本明細書において、「置換されていてもよい」とは、無置換、若しくは置換基を1~5個有していることを意味する。なお、複数個の置換基を有する場合、それらの置換基は同一であっても、互いに異なっていてもよい。
In the present specification, “optionally substituted” means unsubstituted or having 1 to 5 substituents. In addition, when it has a some substituent, those substituents may be the same or may mutually differ.
式(I)で示される本発明化合物又はその製薬学的に許容される塩のある態様を以下に示す。
(1)A環が単環式飽和へテロ環基である化合物。
(2)A環が6員の単環式へテロ環基である化合物。
(3)A環が単環式不飽和へテロ環基または縮合多環式不飽和へテロ環基である化合物。
(4)A環が、含窒素ヘテロシクロアルキルである化合物。
(5)A環が、6員の含窒素ヘテロシクロアルキルである化合物。
(6)A環が、1,2,3,6-テトラヒドロピリジル、又は、ピペリジルで示される基である化合物。
(7)B環が、式(II)で示される基である化合物。
(8)B環が、式(III-a)または式(III-b)で示される基である化合物。 Certain embodiments of the compound of the present invention represented by the formula (I) or a pharmaceutically acceptable salt thereof are shown below.
(1) A compound in which the A ring is a monocyclic saturated heterocyclic group.
(2) A compound in which the A ring is a 6-membered monocyclic heterocyclic group.
(3) A compound in which the A ring is a monocyclic unsaturated heterocyclic group or a condensed polycyclic unsaturated heterocyclic group.
(4) A compound in which the A ring is a nitrogen-containing heterocycloalkyl.
(5) A compound in which the A ring is a 6-membered nitrogen-containing heterocycloalkyl.
(6) A compound in which the A ring is a group represented by 1,2,3,6-tetrahydropyridyl or piperidyl.
(7) A compound in which the B ring is a group represented by the formula (II).
(8) A compound wherein the ring B is a group represented by the formula (III-a) or the formula (III-b).
(1)A環が単環式飽和へテロ環基である化合物。
(2)A環が6員の単環式へテロ環基である化合物。
(3)A環が単環式不飽和へテロ環基または縮合多環式不飽和へテロ環基である化合物。
(4)A環が、含窒素ヘテロシクロアルキルである化合物。
(5)A環が、6員の含窒素ヘテロシクロアルキルである化合物。
(6)A環が、1,2,3,6-テトラヒドロピリジル、又は、ピペリジルで示される基である化合物。
(7)B環が、式(II)で示される基である化合物。
(8)B環が、式(III-a)または式(III-b)で示される基である化合物。 Certain embodiments of the compound of the present invention represented by the formula (I) or a pharmaceutically acceptable salt thereof are shown below.
(1) A compound in which the A ring is a monocyclic saturated heterocyclic group.
(2) A compound in which the A ring is a 6-membered monocyclic heterocyclic group.
(3) A compound in which the A ring is a monocyclic unsaturated heterocyclic group or a condensed polycyclic unsaturated heterocyclic group.
(4) A compound in which the A ring is a nitrogen-containing heterocycloalkyl.
(5) A compound in which the A ring is a 6-membered nitrogen-containing heterocycloalkyl.
(6) A compound in which the A ring is a group represented by 1,2,3,6-tetrahydropyridyl or piperidyl.
(7) A compound in which the B ring is a group represented by the formula (II).
(8) A compound wherein the ring B is a group represented by the formula (III-a) or the formula (III-b).
(9)D環が、含窒素ヘテロアリールである化合物。
(10)D環が、単環式含窒素へテロアリールもしくは縮合多環式含窒素へテロアリールで選択される化合物。
(11)D環が、5乃至6員の単環式含窒素へテロアリールである化合物。
(12)D環が、5員-5員縮合多環式含窒素へテロアリールまたは5員-6員縮合多環式含窒素へテロアリールである化合物。
(13)D環が、イミダゾチアジアゾリル、ピリジル、ピリミジニル、又は、オキサジアゾリルである化合物。
(14)D環がイミダゾ[2,1-b][1,3,4]チアジアゾリルである化合物。
(15)RDが、式(V)または式(VI)で示される基である化合物。
(16)RD中のE環が、単環式飽和ヘテロ環基または単環式不飽和ヘテロ環基である化合物。
(17)RD中のE環が、含窒素ヘテロシクロアルキルである化合物。
(18)RD中のE環が、6員の含窒素ヘテロシクロアルキルである化合物。
(19)RD中のE環が、1,2,3,6-テトラヒドロピリジル、ピペラジニル又はピペリジルである化合物。 (9) The compound in which the D ring is a nitrogen-containing heteroaryl.
(10) A compound wherein the D ring is selected from monocyclic nitrogen-containing heteroaryl or condensed polycyclic nitrogen-containing heteroaryl.
(11) The compound wherein the D ring is a 5- to 6-membered monocyclic nitrogen-containing heteroaryl.
(12) A compound in which the D ring is a 5-membered to 5-membered condensed polycyclic nitrogen-containing heteroaryl or a 5-membered to 6-membered condensed polycyclic nitrogen-containing heteroaryl.
(13) A compound in which the ring D is imidazothiadiazolyl, pyridyl, pyrimidinyl, or oxadiazolyl.
(14) A compound wherein the D ring is imidazo [2,1-b] [1,3,4] thiadiazolyl.
(15) The compound wherein R D is a group represented by formula (V) or formula (VI).
(16) The compound wherein the E ring in R D is a monocyclic saturated heterocyclic group or a monocyclic unsaturated heterocyclic group.
(17) The compound wherein the E ring in R D is nitrogen-containing heterocycloalkyl.
(18) The compound wherein the E ring in R D is a 6-membered nitrogen-containing heterocycloalkyl.
(19) The compound wherein the E ring in R D is 1,2,3,6-tetrahydropyridyl, piperazinyl or piperidyl.
(10)D環が、単環式含窒素へテロアリールもしくは縮合多環式含窒素へテロアリールで選択される化合物。
(11)D環が、5乃至6員の単環式含窒素へテロアリールである化合物。
(12)D環が、5員-5員縮合多環式含窒素へテロアリールまたは5員-6員縮合多環式含窒素へテロアリールである化合物。
(13)D環が、イミダゾチアジアゾリル、ピリジル、ピリミジニル、又は、オキサジアゾリルである化合物。
(14)D環がイミダゾ[2,1-b][1,3,4]チアジアゾリルである化合物。
(15)RDが、式(V)または式(VI)で示される基である化合物。
(16)RD中のE環が、単環式飽和ヘテロ環基または単環式不飽和ヘテロ環基である化合物。
(17)RD中のE環が、含窒素ヘテロシクロアルキルである化合物。
(18)RD中のE環が、6員の含窒素ヘテロシクロアルキルである化合物。
(19)RD中のE環が、1,2,3,6-テトラヒドロピリジル、ピペラジニル又はピペリジルである化合物。 (9) The compound in which the D ring is a nitrogen-containing heteroaryl.
(10) A compound wherein the D ring is selected from monocyclic nitrogen-containing heteroaryl or condensed polycyclic nitrogen-containing heteroaryl.
(11) The compound wherein the D ring is a 5- to 6-membered monocyclic nitrogen-containing heteroaryl.
(12) A compound in which the D ring is a 5-membered to 5-membered condensed polycyclic nitrogen-containing heteroaryl or a 5-membered to 6-membered condensed polycyclic nitrogen-containing heteroaryl.
(13) A compound in which the ring D is imidazothiadiazolyl, pyridyl, pyrimidinyl, or oxadiazolyl.
(14) A compound wherein the D ring is imidazo [2,1-b] [1,3,4] thiadiazolyl.
(15) The compound wherein R D is a group represented by formula (V) or formula (VI).
(16) The compound wherein the E ring in R D is a monocyclic saturated heterocyclic group or a monocyclic unsaturated heterocyclic group.
(17) The compound wherein the E ring in R D is nitrogen-containing heterocycloalkyl.
(18) The compound wherein the E ring in R D is a 6-membered nitrogen-containing heterocycloalkyl.
(19) The compound wherein the E ring in R D is 1,2,3,6-tetrahydropyridyl, piperazinyl or piperidyl.
(20)RD中のEfg環が、含窒素ヘテロシクロアルキルである化合物。
(21)RD中のEfg環が、6員の含窒素ヘテロシクロアルキルである化合物。
(22)RD中のFg環が、シクロアルキルまたはアリールである化合物。
(23)R1が、-NH2、-OH、-NHCH2CH2NH2、-NHCH2C(=O)OH、又は、-NHCH(OH)C(=O)OHである化合物。
(24)R2は、-S(=O)2OH、-CH2CH(OH)CH2OH、-CH2C(=O)OH、低級アルキル、又は、Hである化合物。
(25)RAが、H、低級アルキル、-低級アルキレン-NH2、-低級アルキレン-OH、-低級アルキレン-O-低級アルキル、-低級アルキレン-C(=O)OH、-低級アルキレン-S(=O)OH、-C(=O)OH、-C(=O)O-低級アルキル、-C(=O)-低級アルキレン-NH2、-C(=O)CH(NH2)CH2OH、-CH(CN)-NH2、-CH(-低級アルキレン-OH)2、又は、シクロアルキルである化合物。
(26)Xが、単結合である化合物。
(27)Lが、単結合、低級アルキレン、-O-、又は、-O-低級アルキレン-である化合物。 (20) The compound wherein the Efg ring in R D is nitrogen-containing heterocycloalkyl.
(21) The compound wherein the Efg ring in R D is a 6-membered nitrogen-containing heterocycloalkyl.
(22) The compound wherein the Fg ring in R D is cycloalkyl or aryl.
(23) The compound wherein R 1 is —NH 2 , —OH, —NHCH 2 CH 2 NH 2 , —NHCH 2 C (═O) OH, or —NHCH (OH) C (═O) OH.
(24) The compound wherein R 2 is —S (═O) 2 OH, —CH 2 CH (OH) CH 2 OH, —CH 2 C (═O) OH, lower alkyl, or H.
(25) R A is H, lower alkyl, -lower alkylene-NH 2 , -lower alkylene-OH, -lower alkylene-O-lower alkyl, -lower alkylene-C (= O) OH, -lower alkylene-S (= O) OH, -C ( = O) OH, -C (= O) O- lower alkyl, -C (= O) - lower alkylene -NH 2, -C (= O) CH (NH 2) CH 2 OH, —CH (CN) —NH 2 , —CH (-lower alkylene-OH) 2 , or a compound that is cycloalkyl.
(26) The compound wherein X is a single bond.
(27) The compound wherein L is a single bond, lower alkylene, -O- or -O-lower alkylene-.
(21)RD中のEfg環が、6員の含窒素ヘテロシクロアルキルである化合物。
(22)RD中のFg環が、シクロアルキルまたはアリールである化合物。
(23)R1が、-NH2、-OH、-NHCH2CH2NH2、-NHCH2C(=O)OH、又は、-NHCH(OH)C(=O)OHである化合物。
(24)R2は、-S(=O)2OH、-CH2CH(OH)CH2OH、-CH2C(=O)OH、低級アルキル、又は、Hである化合物。
(25)RAが、H、低級アルキル、-低級アルキレン-NH2、-低級アルキレン-OH、-低級アルキレン-O-低級アルキル、-低級アルキレン-C(=O)OH、-低級アルキレン-S(=O)OH、-C(=O)OH、-C(=O)O-低級アルキル、-C(=O)-低級アルキレン-NH2、-C(=O)CH(NH2)CH2OH、-CH(CN)-NH2、-CH(-低級アルキレン-OH)2、又は、シクロアルキルである化合物。
(26)Xが、単結合である化合物。
(27)Lが、単結合、低級アルキレン、-O-、又は、-O-低級アルキレン-である化合物。 (20) The compound wherein the Efg ring in R D is nitrogen-containing heterocycloalkyl.
(21) The compound wherein the Efg ring in R D is a 6-membered nitrogen-containing heterocycloalkyl.
(22) The compound wherein the Fg ring in R D is cycloalkyl or aryl.
(23) The compound wherein R 1 is —NH 2 , —OH, —NHCH 2 CH 2 NH 2 , —NHCH 2 C (═O) OH, or —NHCH (OH) C (═O) OH.
(24) The compound wherein R 2 is —S (═O) 2 OH, —CH 2 CH (OH) CH 2 OH, —CH 2 C (═O) OH, lower alkyl, or H.
(25) R A is H, lower alkyl, -lower alkylene-NH 2 , -lower alkylene-OH, -lower alkylene-O-lower alkyl, -lower alkylene-C (= O) OH, -lower alkylene-S (= O) OH, -C ( = O) OH, -C (= O) O- lower alkyl, -C (= O) - lower alkylene -NH 2, -C (= O) CH (NH 2) CH 2 OH, —CH (CN) —NH 2 , —CH (-lower alkylene-OH) 2 , or a compound that is cycloalkyl.
(26) The compound wherein X is a single bond.
(27) The compound wherein L is a single bond, lower alkylene, -O- or -O-lower alkylene-.
(28)Lが、単結合、-CH2CH2-、-O-、又は、-O-CH2-である化合物。
(29)RAが、-低級アルキレン-OH、-C(=O)-低級アルキレン-NH2、シクロアルキル、低級アルキル、又は、Hである化合物。
(30)RAが、、-CH2CH2-OHである化合物。
(31)RAが、メチルである化合物。
(32)R1が、-NH2である化合物。
(33)R2が、-S(=O)2OH又はメチルである化合物。
(34)R2が、-S(=O)2OHである化合物。
(35)RBが、Hである化合物。
(36)上記(1)~(35)の化合物に示された基のうち二以上の組み合わせである化合物。
本発明に包含される具体的化合物の例として、以下の化合物(37)から(84)又はそれらの製薬学的に許容される塩が挙げられる。 (28) The compound wherein L is a single bond, —CH 2 CH 2 —, —O—, or —O—CH 2 —.
(29) The compound wherein R A is -lower alkylene-OH, -C (= O) -lower alkylene-NH 2 , cycloalkyl, lower alkyl, or H.
(30) The compound wherein R A is —CH 2 CH 2 —OH.
(31) The compound wherein R A is methyl.
(32) The compound wherein R 1 is —NH 2 .
(33) The compound wherein R 2 is —S (═O) 2 OH or methyl.
(34) The compound wherein R 2 is —S (═O) 2 OH.
(35) The compound wherein R B is H.
(36) A compound which is a combination of two or more of the groups shown in the compounds (1) to (35) above.
Examples of specific compounds encompassed by the present invention include the following compounds (37) to (84) or pharmaceutically acceptable salts thereof.
(29)RAが、-低級アルキレン-OH、-C(=O)-低級アルキレン-NH2、シクロアルキル、低級アルキル、又は、Hである化合物。
(30)RAが、、-CH2CH2-OHである化合物。
(31)RAが、メチルである化合物。
(32)R1が、-NH2である化合物。
(33)R2が、-S(=O)2OH又はメチルである化合物。
(34)R2が、-S(=O)2OHである化合物。
(35)RBが、Hである化合物。
(36)上記(1)~(35)の化合物に示された基のうち二以上の組み合わせである化合物。
本発明に包含される具体的化合物の例として、以下の化合物(37)から(84)又はそれらの製薬学的に許容される塩が挙げられる。 (28) The compound wherein L is a single bond, —CH 2 CH 2 —, —O—, or —O—CH 2 —.
(29) The compound wherein R A is -lower alkylene-OH, -C (= O) -lower alkylene-NH 2 , cycloalkyl, lower alkyl, or H.
(30) The compound wherein R A is —CH 2 CH 2 —OH.
(31) The compound wherein R A is methyl.
(32) The compound wherein R 1 is —NH 2 .
(33) The compound wherein R 2 is —S (═O) 2 OH or methyl.
(34) The compound wherein R 2 is —S (═O) 2 OH.
(35) The compound wherein R B is H.
(36) A compound which is a combination of two or more of the groups shown in the compounds (1) to (35) above.
Examples of specific compounds encompassed by the present invention include the following compounds (37) to (84) or pharmaceutically acceptable salts thereof.
(37)(3)の化合物に示された基、および(10)の化合物に示された基である化合物。
(38)(4)の化合物に示された基、および(10)の化合物に示された基である化合物。
(39)(5)の化合物に示された基、および(10)の化合物に示された基である化合物。
(40)(3)の化合物に示された基、および(11)の化合物に示された基である化合物。
(41)(4)の化合物に示された基、および(11)の化合物に示された基である化合物。
(42)(5)の化合物に示された基、および(11)の化合物に示された基である化合物。
(43)(3)の化合物に示された基、および(12)の化合物に示された基である化合物。
(44)(4)の化合物に示された基、および(12)の化合物に示された基である化合物。
(45)(5)の化合物に示された基、および(12)の化合物に示された基である化合物。
(46)(3)の化合物に示された基、(7)の化合物に示された基、および(10)の化合物に示された基である化合物。
(47)(4)の化合物に示された基、(7)の化合物に示された基、および(10)の化合物に示された基である化合物。
(48)(5)の化合物に示された基、(7)の化合物に示された基、および(10)の化合物に示された基である化合物。
(49)(3)の化合物に示された基、(7)の化合物に示された基、および(11)の化合物に示された基である化合物。
(50)(4)の化合物に示された基、(7)の化合物に示された基、および(11)の化合物に示された基である化合物。 (37) A compound which is a group shown in the compound of (3) and a group shown in the compound of (10).
(38) A compound which is a group shown in the compound of (4) and a group shown in the compound of (10).
(39) A compound which is a group shown in the compound of (5) and a group shown in the compound of (10).
(40) A compound which is a group shown in the compound of (3) and a group shown in the compound of (11).
(41) A compound which is a group shown in the compound of (4) and a group shown in the compound of (11).
(42) A compound which is a group shown in the compound of (5) and a group shown in the compound of (11).
(43) A compound which is a group shown in the compound of (3) and a group shown in the compound of (12).
(44) A compound which is a group shown in the compound of (4) and a group shown in the compound of (12).
(45) A compound which is a group shown in the compound of (5) and a group shown in the compound of (12).
(46) A compound which is the group shown in the compound of (3), the group shown in the compound of (7), and the group shown in the compound of (10).
(47) A compound which is the group shown in the compound of (4), the group shown in the compound of (7), and the group shown in the compound of (10).
(48) A compound which is the group shown in the compound of (5), the group shown in the compound of (7), and the group shown in the compound of (10).
(49) A compound which is the group shown in the compound of (3), the group shown in the compound of (7), and the group shown in the compound of (11).
(50) A compound which is the group shown in the compound of (4), the group shown in the compound of (7), and the group shown in the compound of (11).
(38)(4)の化合物に示された基、および(10)の化合物に示された基である化合物。
(39)(5)の化合物に示された基、および(10)の化合物に示された基である化合物。
(40)(3)の化合物に示された基、および(11)の化合物に示された基である化合物。
(41)(4)の化合物に示された基、および(11)の化合物に示された基である化合物。
(42)(5)の化合物に示された基、および(11)の化合物に示された基である化合物。
(43)(3)の化合物に示された基、および(12)の化合物に示された基である化合物。
(44)(4)の化合物に示された基、および(12)の化合物に示された基である化合物。
(45)(5)の化合物に示された基、および(12)の化合物に示された基である化合物。
(46)(3)の化合物に示された基、(7)の化合物に示された基、および(10)の化合物に示された基である化合物。
(47)(4)の化合物に示された基、(7)の化合物に示された基、および(10)の化合物に示された基である化合物。
(48)(5)の化合物に示された基、(7)の化合物に示された基、および(10)の化合物に示された基である化合物。
(49)(3)の化合物に示された基、(7)の化合物に示された基、および(11)の化合物に示された基である化合物。
(50)(4)の化合物に示された基、(7)の化合物に示された基、および(11)の化合物に示された基である化合物。 (37) A compound which is a group shown in the compound of (3) and a group shown in the compound of (10).
(38) A compound which is a group shown in the compound of (4) and a group shown in the compound of (10).
(39) A compound which is a group shown in the compound of (5) and a group shown in the compound of (10).
(40) A compound which is a group shown in the compound of (3) and a group shown in the compound of (11).
(41) A compound which is a group shown in the compound of (4) and a group shown in the compound of (11).
(42) A compound which is a group shown in the compound of (5) and a group shown in the compound of (11).
(43) A compound which is a group shown in the compound of (3) and a group shown in the compound of (12).
(44) A compound which is a group shown in the compound of (4) and a group shown in the compound of (12).
(45) A compound which is a group shown in the compound of (5) and a group shown in the compound of (12).
(46) A compound which is the group shown in the compound of (3), the group shown in the compound of (7), and the group shown in the compound of (10).
(47) A compound which is the group shown in the compound of (4), the group shown in the compound of (7), and the group shown in the compound of (10).
(48) A compound which is the group shown in the compound of (5), the group shown in the compound of (7), and the group shown in the compound of (10).
(49) A compound which is the group shown in the compound of (3), the group shown in the compound of (7), and the group shown in the compound of (11).
(50) A compound which is the group shown in the compound of (4), the group shown in the compound of (7), and the group shown in the compound of (11).
(51)(5)の化合物に示された基、(7)の化合物に示された基、および(11)の化合物に示された基である化合物。
(52)(3)の化合物に示された基、(7)の化合物に示された基、および(12)の化合物に示された基である化合物。
(53)(4)の化合物に示された基、(7)の化合物に示された基、および(12)の化合物に示された基である化合物。
(54)(5)の化合物に示された基、(7)の化合物に示された基、および(12)の化合物に示された基である化合物。
(55)(6)の化合物に示された基、および(10)の化合物に示された基である化合物。
(56)(6)の化合物に示された基、および(11)の化合物に示された基である化合物。
(57)(6)の化合物に示された基、および(12)の化合物に示された基である化合物。
(58)(6)の化合物に示された基、(7)の化合物に示された基、および(10)の化合物に示された基である化合物。
(59)(6)の化合物に示された基、(7)の化合物に示された基、および(11)の化合物に示された基である化合物。
(60)(6)の化合物に示された基、(7)の化合物に示された基、および(12)の化合物に示された基である化合物。
(61)(7)の化合物に示された基、および(10)の化合物に示された基である化合物。
(62)(7)の化合物に示された基、および(11)の化合物に示された基である化合物。 (51) A compound which is the group shown in the compound of (5), the group shown in the compound of (7), and the group shown in the compound of (11).
(52) A compound which is the group shown in the compound of (3), the group shown in the compound of (7), and the group shown in the compound of (12).
(53) A compound which is the group shown in the compound of (4), the group shown in the compound of (7), and the group shown in the compound of (12).
(54) A compound which is the group shown in the compound of (5), the group shown in the compound of (7), and the group shown in the compound of (12).
(55) A compound which is a group shown in the compound of (6) and a group shown in the compound of (10).
(56) A compound which is a group shown in the compound of (6) and a group shown in the compound of (11).
(57) A compound which is a group shown in the compound of (6) and a group shown in the compound of (12).
(58) A compound which is the group shown in the compound of (6), the group shown in the compound of (7), and the group shown in the compound of (10).
(59) A compound which is the group shown in the compound of (6), the group shown in the compound of (7), and the group shown in the compound of (11).
(60) A compound which is the group shown in the compound of (6), the group shown in the compound of (7), and the group shown in the compound of (12).
(61) A compound which is the group shown in the compound of (7) and the group shown in the compound of (10).
(62) A compound which is a group shown in the compound of (7) and a group shown in the compound of (11).
(52)(3)の化合物に示された基、(7)の化合物に示された基、および(12)の化合物に示された基である化合物。
(53)(4)の化合物に示された基、(7)の化合物に示された基、および(12)の化合物に示された基である化合物。
(54)(5)の化合物に示された基、(7)の化合物に示された基、および(12)の化合物に示された基である化合物。
(55)(6)の化合物に示された基、および(10)の化合物に示された基である化合物。
(56)(6)の化合物に示された基、および(11)の化合物に示された基である化合物。
(57)(6)の化合物に示された基、および(12)の化合物に示された基である化合物。
(58)(6)の化合物に示された基、(7)の化合物に示された基、および(10)の化合物に示された基である化合物。
(59)(6)の化合物に示された基、(7)の化合物に示された基、および(11)の化合物に示された基である化合物。
(60)(6)の化合物に示された基、(7)の化合物に示された基、および(12)の化合物に示された基である化合物。
(61)(7)の化合物に示された基、および(10)の化合物に示された基である化合物。
(62)(7)の化合物に示された基、および(11)の化合物に示された基である化合物。 (51) A compound which is the group shown in the compound of (5), the group shown in the compound of (7), and the group shown in the compound of (11).
(52) A compound which is the group shown in the compound of (3), the group shown in the compound of (7), and the group shown in the compound of (12).
(53) A compound which is the group shown in the compound of (4), the group shown in the compound of (7), and the group shown in the compound of (12).
(54) A compound which is the group shown in the compound of (5), the group shown in the compound of (7), and the group shown in the compound of (12).
(55) A compound which is a group shown in the compound of (6) and a group shown in the compound of (10).
(56) A compound which is a group shown in the compound of (6) and a group shown in the compound of (11).
(57) A compound which is a group shown in the compound of (6) and a group shown in the compound of (12).
(58) A compound which is the group shown in the compound of (6), the group shown in the compound of (7), and the group shown in the compound of (10).
(59) A compound which is the group shown in the compound of (6), the group shown in the compound of (7), and the group shown in the compound of (11).
(60) A compound which is the group shown in the compound of (6), the group shown in the compound of (7), and the group shown in the compound of (12).
(61) A compound which is the group shown in the compound of (7) and the group shown in the compound of (10).
(62) A compound which is a group shown in the compound of (7) and a group shown in the compound of (11).
(63)(7)の化合物に示された基、および(12)の化合物に示された基である化合物。
(64)(3)の化合物に示された基、および(15)の化合物に示された基である化合物。
(65)(4)の化合物に示された基、(7)の化合物に示された基、(11)の化合物に示された基、および(15)の化合物に示された基である化合物。
(66)(4)の化合物に示された基、(7)の化合物に示された基、(11)の化合物に示された基、および(18)の化合物に示された基である化合物。
(67)(4)の化合物に示された基、(7)の化合物に示された基、(11)の化合物に示された基、および(21)の化合物に示された基である化合物。
(68)(4)の化合物に示された基、(7)の化合物に示された基、(12)の化合物に示された基、および(15)の化合物に示された基である化合物。
(69)(4)の化合物に示された基、(7)の化合物に示された基、(12)の化合物に示された基、および(18)の化合物に示された基である化合物。 (63) A compound which is a group shown in the compound of (7) and a group shown in the compound of (12).
(64) A compound which is a group shown in the compound of (3) and a group shown in the compound of (15).
(65) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (11), and a group shown in the compound of (15) .
(66) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (11), and a group shown in the compound of (18) .
(67) A compound shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (11), and a group shown in the compound of (21) .
(68) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (12), and a group shown in the compound of (15) .
(69) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (12), and a group shown in the compound of (18) .
(64)(3)の化合物に示された基、および(15)の化合物に示された基である化合物。
(65)(4)の化合物に示された基、(7)の化合物に示された基、(11)の化合物に示された基、および(15)の化合物に示された基である化合物。
(66)(4)の化合物に示された基、(7)の化合物に示された基、(11)の化合物に示された基、および(18)の化合物に示された基である化合物。
(67)(4)の化合物に示された基、(7)の化合物に示された基、(11)の化合物に示された基、および(21)の化合物に示された基である化合物。
(68)(4)の化合物に示された基、(7)の化合物に示された基、(12)の化合物に示された基、および(15)の化合物に示された基である化合物。
(69)(4)の化合物に示された基、(7)の化合物に示された基、(12)の化合物に示された基、および(18)の化合物に示された基である化合物。 (63) A compound which is a group shown in the compound of (7) and a group shown in the compound of (12).
(64) A compound which is a group shown in the compound of (3) and a group shown in the compound of (15).
(65) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (11), and a group shown in the compound of (15) .
(66) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (11), and a group shown in the compound of (18) .
(67) A compound shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (11), and a group shown in the compound of (21) .
(68) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (12), and a group shown in the compound of (15) .
(69) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (12), and a group shown in the compound of (18) .
(70)(4)の化合物に示された基、(7)の化合物に示された基、(12)の化合物に示された基、および(21)の化合物に示された基である化合物。
(71)(4)の化合物に示された基、(7)の化合物に示された基、(13)の化合物に示された基、および(18)の化合物に示された基である化合物。
(72)(4)の化合物に示された基、(7)の化合物に示された基、(13)の化合物に示された基、および(21)の化合物に示された基である化合物。
(73)(4)の化合物に示された基、(7)の化合物に示された基、(14)の化合物に示された基、および(18)の化合物に示された基である化合物。
(74)(4)の化合物に示された基、(7)の化合物に示された基、(14)の化合物に示された基、および(21)の化合物に示された基である化合物。
(75)(6)の化合物に示された基、および(9)の化合物に示された基である化合物。。 (70) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (12), and a group shown in the compound of (21) .
(71) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (13), and a group shown in the compound of (18) .
(72) A compound shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (13), and a group shown in the compound of (21) .
(73) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (14), and a group shown in the compound of (18) .
(74) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (14), and a group shown in the compound of (21) .
(75) A compound which is a group shown in the compound of (6) and a group shown in the compound of (9). .
(71)(4)の化合物に示された基、(7)の化合物に示された基、(13)の化合物に示された基、および(18)の化合物に示された基である化合物。
(72)(4)の化合物に示された基、(7)の化合物に示された基、(13)の化合物に示された基、および(21)の化合物に示された基である化合物。
(73)(4)の化合物に示された基、(7)の化合物に示された基、(14)の化合物に示された基、および(18)の化合物に示された基である化合物。
(74)(4)の化合物に示された基、(7)の化合物に示された基、(14)の化合物に示された基、および(21)の化合物に示された基である化合物。
(75)(6)の化合物に示された基、および(9)の化合物に示された基である化合物。。 (70) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (12), and a group shown in the compound of (21) .
(71) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (13), and a group shown in the compound of (18) .
(72) A compound shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (13), and a group shown in the compound of (21) .
(73) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (14), and a group shown in the compound of (18) .
(74) A compound which is a group shown in the compound of (4), a group shown in the compound of (7), a group shown in the compound of (14), and a group shown in the compound of (21) .
(75) A compound which is a group shown in the compound of (6) and a group shown in the compound of (9). .
(76)R1が-NH2であり、R2が-S(=O)2OHであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、D環がイミダゾ[2,1-b][1,3,4]チアジアゾリル、ピリジル、ピリミジニル、又は、1,2,4-オキサジアゾリルであり、RDが式(V)で示される基であり、E環が、ピペラジニル又はピペリジルであり、Lが、-CH2CH2-であり、Fg環がフェニルである化合物。
(77)R1が-NH2であり、R2が-S(=O)2OHであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、RAが-CH2CH2-OHであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、D環がイミダゾ[2,1-b][1,3,4]チアジアゾリル、ピリジル、ピリミジニル、又は、1,2,4-オキサジアゾリルであり、RDが式(V)で示される基であり、E環が、ピペラジニル又はピペリジルであり、Lが、-CH2CH2-であり、Fg環がフェニルである化合物。
(78) R1が-NH2であり、R2が-S(=O)2OHであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、RDが式(V)で示される基であり、E環がピペラジルであり、Fg環がフェニルである化合物。 (76) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and X is a single bond , and the a group B ring represented by the formula (II), an R B is H, an R c is H, D ring imidazo [2,1-b] [1,3,4] thiadiazolyl , Pyridyl, pyrimidinyl, or 1,2,4-oxadiazolyl, R D is a group represented by the formula (V), the E ring is piperazinyl or piperidyl, and L is —CH 2 CH 2 — Wherein the Fg ring is phenyl.
(77) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and R A is — CH 2 CH 2 —OH, X is a single bond, B ring is a group represented by formula (II), R B is H, R c is H, and D ring is imidazo [2 , 1-b] [1,3,4] thiadiazolyl, pyridyl, pyrimidinyl, or 1,2,4-oxadiazolyl, R D is a group represented by formula (V), and ring E is piperazinyl or A compound that is piperidyl, L is —CH 2 CH 2 —, and the Fg ring is phenyl.
(78) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and X is a single bond and a is a group B ring represented by the formula (II), an R B is H, an R c is H, a group R D is represented by formula (V), E ring is in a piperazyl A compound in which the Fg ring is phenyl.
(77)R1が-NH2であり、R2が-S(=O)2OHであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、RAが-CH2CH2-OHであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、D環がイミダゾ[2,1-b][1,3,4]チアジアゾリル、ピリジル、ピリミジニル、又は、1,2,4-オキサジアゾリルであり、RDが式(V)で示される基であり、E環が、ピペラジニル又はピペリジルであり、Lが、-CH2CH2-であり、Fg環がフェニルである化合物。
(78) R1が-NH2であり、R2が-S(=O)2OHであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、RDが式(V)で示される基であり、E環がピペラジルであり、Fg環がフェニルである化合物。 (76) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and X is a single bond , and the a group B ring represented by the formula (II), an R B is H, an R c is H, D ring imidazo [2,1-b] [1,3,4] thiadiazolyl , Pyridyl, pyrimidinyl, or 1,2,4-oxadiazolyl, R D is a group represented by the formula (V), the E ring is piperazinyl or piperidyl, and L is —CH 2 CH 2 — Wherein the Fg ring is phenyl.
(77) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and R A is — CH 2 CH 2 —OH, X is a single bond, B ring is a group represented by formula (II), R B is H, R c is H, and D ring is imidazo [2 , 1-b] [1,3,4] thiadiazolyl, pyridyl, pyrimidinyl, or 1,2,4-oxadiazolyl, R D is a group represented by formula (V), and ring E is piperazinyl or A compound that is piperidyl, L is —CH 2 CH 2 —, and the Fg ring is phenyl.
(78) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and X is a single bond and a is a group B ring represented by the formula (II), an R B is H, an R c is H, a group R D is represented by formula (V), E ring is in a piperazyl A compound in which the Fg ring is phenyl.
(79) R1が-NH2であり、R2が-S(=O)2OHであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、RAが-CH2CH2-OHであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、RDが式(V)で示される基であり、E環がピペリジルであり、Lが単結合であり、Fg環がシクロアルキルである化合物。
(80) R1が-NH2であり、R2が-S(=O)2OHであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、RAが-CH2CH2-OHであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、D環がオキサジアゾリルであり、RDが式(V)で示される基であり、E環がピペリジルであり、Lが-CH2CH2-であり、Fg環がシクロアルキルである化合物。
(81) R1が-NH2であり、R2が-S(=O)2OHであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、D環がオキサジアゾリルであり、RDが式(V)で示される基であり、E環がピペリジルであり、Lが単結合であり、Efg環がピペラジンまたはピペリジンである化合物。 (79) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and R A is — CH 2 CH 2 --OH, X is a single bond, the B ring is a group represented by the formula (II), R B is H, R c is H, R D is a formula (V ), E ring is piperidyl, L is a single bond, and Fg ring is cycloalkyl.
(80) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and R A is — CH 2 CH 2 —OH, X is a single bond, B ring is a group represented by formula (II), R B is H, R c is H, and D ring is oxadiazolyl , R D is a group represented by formula (V), E ring is piperidyl, L is —CH 2 CH 2 —, and Fg ring is cycloalkyl.
(81) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and X is a single bond and a is a group B ring represented by the formula (II), R B is H, R c is H, D ring is oxadiazolyl, a group R D is represented by formula (V) A compound in which the E ring is piperidyl, L is a single bond, and the Efg ring is piperazine or piperidine.
(80) R1が-NH2であり、R2が-S(=O)2OHであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、RAが-CH2CH2-OHであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、D環がオキサジアゾリルであり、RDが式(V)で示される基であり、E環がピペリジルであり、Lが-CH2CH2-であり、Fg環がシクロアルキルである化合物。
(81) R1が-NH2であり、R2が-S(=O)2OHであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、D環がオキサジアゾリルであり、RDが式(V)で示される基であり、E環がピペリジルであり、Lが単結合であり、Efg環がピペラジンまたはピペリジンである化合物。 (79) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and R A is — CH 2 CH 2 --OH, X is a single bond, the B ring is a group represented by the formula (II), R B is H, R c is H, R D is a formula (V ), E ring is piperidyl, L is a single bond, and Fg ring is cycloalkyl.
(80) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and R A is — CH 2 CH 2 —OH, X is a single bond, B ring is a group represented by formula (II), R B is H, R c is H, and D ring is oxadiazolyl , R D is a group represented by formula (V), E ring is piperidyl, L is —CH 2 CH 2 —, and Fg ring is cycloalkyl.
(81) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and X is a single bond and a is a group B ring represented by the formula (II), R B is H, R c is H, D ring is oxadiazolyl, a group R D is represented by formula (V) A compound in which the E ring is piperidyl, L is a single bond, and the Efg ring is piperazine or piperidine.
(82) R1が-NH2であり、R2が-S(=O)2OHであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、RAがメチルであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、D環がピリミジニルであり、RDが式(VI)で示される基であり、Lが-O-CH2-であり、Efg環がフェニルである化合物。
(83) R1が-NH2であり、R2がMeであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、RDが式(V)で示される基であり、E環がピペリジニルであり、Lが単結合であり、Fg環がシクロアルキルである化合物。
(84) R1が-NH2であり、R2がMeであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、RAがHであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、D環がピリミジンであり、RDが式(V)で示される基であり、E環がピペラジニルであり、Lが-CH2CH2-であり、Fg環がフェニルである化合物。
本発明に包含される具体的化合物の例として、以下の(85)から(88)に示される群から選択される化合物又はその製薬学的に許容される塩が挙げられる。 (82) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and R A is methyl X is a single bond, B ring is a group represented by formula (II), R B is H, R c is H, D ring is pyrimidinyl, and R D is formula ( A compound represented by VI), wherein L is —O—CH 2 — and the Efg ring is phenyl.
(83) R 1 is —NH 2 , R 2 is Me, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, X is a single bond, and the B ring is a formula A group represented by (II), R B is H, R c is H, R D is a group represented by the formula (V), the E ring is piperidinyl, and L is a single bond. A compound in which the Fg ring is cycloalkyl.
(84) R 1 is —NH 2 , R 2 is Me, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, R A is H, and X is a single bond and a is a group B ring represented by the formula (II), an R B is H, R c is H, D ring is a pyrimidine, a group R D is represented by formula (V) A compound in which the E ring is piperazinyl, L is —CH 2 CH 2 —, and the Fg ring is phenyl.
Specific examples of the compound included in the present invention include a compound selected from the following groups (85) to (88) or a pharmaceutically acceptable salt thereof.
(83) R1が-NH2であり、R2がMeであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、RDが式(V)で示される基であり、E環がピペリジニルであり、Lが単結合であり、Fg環がシクロアルキルである化合物。
(84) R1が-NH2であり、R2がMeであり、A環が1,2,3,6-テトラヒドロピリジル-4-イルであり、RAがHであり、Xが単結合であり、B環が式(II)で示される基であり、RBがHであり、RcがHであり、D環がピリミジンであり、RDが式(V)で示される基であり、E環がピペラジニルであり、Lが-CH2CH2-であり、Fg環がフェニルである化合物。
本発明に包含される具体的化合物の例として、以下の(85)から(88)に示される群から選択される化合物又はその製薬学的に許容される塩が挙げられる。 (82) R 1 is —NH 2 , R 2 is —S (═O) 2 OH, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, and R A is methyl X is a single bond, B ring is a group represented by formula (II), R B is H, R c is H, D ring is pyrimidinyl, and R D is formula ( A compound represented by VI), wherein L is —O—CH 2 — and the Efg ring is phenyl.
(83) R 1 is —NH 2 , R 2 is Me, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, X is a single bond, and the B ring is a formula A group represented by (II), R B is H, R c is H, R D is a group represented by the formula (V), the E ring is piperidinyl, and L is a single bond. A compound in which the Fg ring is cycloalkyl.
(84) R 1 is —NH 2 , R 2 is Me, the A ring is 1,2,3,6-tetrahydropyridyl-4-yl, R A is H, and X is a single bond and a is a group B ring represented by the formula (II), an R B is H, R c is H, D ring is a pyrimidine, a group R D is represented by formula (V) A compound in which the E ring is piperazinyl, L is —CH 2 CH 2 —, and the Fg ring is phenyl.
Specific examples of the compound included in the present invention include a compound selected from the following groups (85) to (88) or a pharmaceutically acceptable salt thereof.
(85)硫酸水素5-{(2R)-2-[(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-アミノ-1-ヒドロキシ-3-オキソプロピル]-2,11,15-トリヒドロキシ-6-[(1R)-1-ヒドロキシエチル]-9-(3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}[1,1'-ビフェニル]-4-カルボキサミド)-16-メチル-5,8,14,19,22,25-ヘキサオキソテトラコサヒドロ-1H-ジピロロ[2,1-c:2',1'-l][1,4,7,10,13,16]ヘキサアザシクロヘンイコシン-23-イル]-2-ヒドロキシエチル}-2-ヒドロキシフェニルエステル。
(86)硫酸水素5-{(2R)-2-[(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-アミノ-1-ヒドロキシ-3-オキソプロピル]-2,11,15-トリヒドロキシ-6-[(1R)-1-ヒドロキシエチル]-9-(3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{5-[4-(2-フェニルエチル)ピペラジン-1-イル]ピリミジン-2-イル}[1,1'-ビフェニル]-4-カルボキサミド)-16-メチル-5,8,14,19,22,25-ヘキサオキソテトラコサヒドロ-1H-ジピロロ[2,1-c:2',1'-l][1,4,7,10,13,16]ヘキサアザシクロヘンイコシン-23-イル]-2-ヒドロキシエチル}-2-ヒドロキシフェニルエステル。
(87)硫酸水素5-[(2R)-2-{(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-アミノ-1-ヒドロキシ-3-オキソプロピル]-9-{4'-(5-{4-[(4,4-ジメチルシクロヘキシル)オキシ]ピペリジン-1-イル}-1,2,4-オキサジアゾール-3-イル)-3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル][1,1'-ビフェニル]-4-カルボキサミド}-2,11,15-トリヒドロキシ-6-[(1R)-1-ヒドロキシエチル]-16-メチル-5,8,14,19,22,25-ヘキサオキソテトラコサヒドロ-1H-ジピロロ[2,1-c:2',1'-l][1,4,7,10,13,16]ヘキサアザシクロヘンイコシン-23-イル}-2-ヒドロキシエチル]-2-ヒドロキシフェニルエステル。
(88)N-{(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-アミノ-1-ヒドロキシ-3-オキソプロピル]-2,11,15-トリヒドロキシ-6-[(1R)-1-ヒドロキシエチル]-23-[(1R)-1-ヒドロキシ-2-(4-ヒドロキシ-3-メトキシフェニル)エチル]-16-メチル-5,8,14,19,22,25-ヘキサオキソテトラコサヒドロ-1H-ジピロロ[2,1-c:2',1'-l][1,4,7,10,13,16]ヘキサアザシクロヘンイコシン-9-イル}-4'-(5-{4-[2-(4-エトキシフェニル)エチル]ピペラジン-1-イル}ピリミジン-2-イル)-3-(1,2,3,6-テトラヒドロピリジン-4-イル)[1,1'-ビフェニル]-4-カルボキサミド。 (85) Hydrogen sulfate 5-{(2R) -2-[(2R, 6S, 9S, 11R, 14aS, 15S, 16S, 20S, 23S, 25aS) -20-[(1R) -3-amino-1- Hydroxy-3-oxopropyl] -2,11,15-trihydroxy-6-[(1R) -1-hydroxyethyl] -9- (3- [1- (2-hydroxyethyl) -1,2,3 , 6-Tetrahydropyridin-4-yl] -4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazole-6 -Yl} [1,1′-biphenyl] -4-carboxamide) -16-methyl-5,8,14,19,22,25-hexaoxotetracosahydro-1H-dipyrolo [2,1-c: 2 ', 1'-l] [1,4,7,10,13,16] hexaazacyclohenicosin-23-yl] -2-hydroxyethyl} -2-hydroxyphenyl ester.
(86) Hydrogen sulfate 5-{(2R) -2-[(2R, 6S, 9S, 11R, 14aS, 15S, 16S, 20S, 23S, 25aS) -20-[(1R) -3-Amino-1- Hydroxy-3-oxopropyl] -2,11,15-trihydroxy-6-[(1R) -1-hydroxyethyl] -9- (3- [1- (2-hydroxyethyl) -1,2,3 , 6-Tetrahydropyridin-4-yl] -4 '-{5- [4- (2-phenylethyl) piperazin-1-yl] pyrimidin-2-yl} [1,1'-biphenyl] -4-carboxamide ) -16-Methyl-5,8,14,19,22,25-hexaoxotetracosahydro-1H-dipyrolo [2,1-c: 2 ', 1'-l] [1,4,7,10 , 13, 16] Hexaazacyclohenicosin-23-yl] -2-hydroxyethyl} -2-hydroxyphenyl ester.
(87) Hydrogen sulfate 5-[(2R) -2-{(2R, 6S, 9S, 11R, 14aS, 15S, 16S, 20S, 23S, 25aS) -20-[(1R) -3-amino-1- Hydroxy-3-oxopropyl] -9- {4 '-(5- {4-[(4,4-dimethylcyclohexyl) oxy] piperidin-1-yl} -1,2,4-oxadiazole-3- Yl) -3- [1- (2-hydroxyethyl) -1,2,3,6-tetrahydropyridin-4-yl] [1,1'-biphenyl] -4-carboxamide} -2,11,15- Trihydroxy-6-[(1R) -1-hydroxyethyl] -16-methyl-5,8,14,19,22,25-hexaoxotetracosahydro-1H-dipyrolo [2,1-c: 2 ' , 1'-l] [1,4,7,10,13,16] hexaazacyclohenicosin-23-yl} -2-hydroxyethyl] -2-hydroxyphenyl ester.
(88) N-{(2R, 6S, 9S, 11R, 14aS, 15S, 16S, 20S, 23S, 25aS) -20-[(1R) -3-Amino-1-hydroxy-3-oxopropyl] -2 , 11,15-Trihydroxy-6-[(1R) -1-hydroxyethyl] -23-[(1R) -1-hydroxy-2- (4-hydroxy-3-methoxyphenyl) ethyl] -16-methyl -5,8,14,19,22,25-hexaoxotetracosahydro-1H-dipirolo [2,1-c: 2 ', 1'-l] [1,4,7,10,13,16] Hexaazacyclohenicosin-9-yl} -4 '-(5- {4- [2- (4-ethoxyphenyl) ethyl] piperazin-1-yl} pyrimidin-2-yl) -3- (1, 2,3,6-Tetrahydropyridin-4-yl) [1,1′-biphenyl] -4-carboxamide.
(86)硫酸水素5-{(2R)-2-[(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-アミノ-1-ヒドロキシ-3-オキソプロピル]-2,11,15-トリヒドロキシ-6-[(1R)-1-ヒドロキシエチル]-9-(3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{5-[4-(2-フェニルエチル)ピペラジン-1-イル]ピリミジン-2-イル}[1,1'-ビフェニル]-4-カルボキサミド)-16-メチル-5,8,14,19,22,25-ヘキサオキソテトラコサヒドロ-1H-ジピロロ[2,1-c:2',1'-l][1,4,7,10,13,16]ヘキサアザシクロヘンイコシン-23-イル]-2-ヒドロキシエチル}-2-ヒドロキシフェニルエステル。
(87)硫酸水素5-[(2R)-2-{(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-アミノ-1-ヒドロキシ-3-オキソプロピル]-9-{4'-(5-{4-[(4,4-ジメチルシクロヘキシル)オキシ]ピペリジン-1-イル}-1,2,4-オキサジアゾール-3-イル)-3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル][1,1'-ビフェニル]-4-カルボキサミド}-2,11,15-トリヒドロキシ-6-[(1R)-1-ヒドロキシエチル]-16-メチル-5,8,14,19,22,25-ヘキサオキソテトラコサヒドロ-1H-ジピロロ[2,1-c:2',1'-l][1,4,7,10,13,16]ヘキサアザシクロヘンイコシン-23-イル}-2-ヒドロキシエチル]-2-ヒドロキシフェニルエステル。
(88)N-{(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-アミノ-1-ヒドロキシ-3-オキソプロピル]-2,11,15-トリヒドロキシ-6-[(1R)-1-ヒドロキシエチル]-23-[(1R)-1-ヒドロキシ-2-(4-ヒドロキシ-3-メトキシフェニル)エチル]-16-メチル-5,8,14,19,22,25-ヘキサオキソテトラコサヒドロ-1H-ジピロロ[2,1-c:2',1'-l][1,4,7,10,13,16]ヘキサアザシクロヘンイコシン-9-イル}-4'-(5-{4-[2-(4-エトキシフェニル)エチル]ピペラジン-1-イル}ピリミジン-2-イル)-3-(1,2,3,6-テトラヒドロピリジン-4-イル)[1,1'-ビフェニル]-4-カルボキサミド。 (85) Hydrogen sulfate 5-{(2R) -2-[(2R, 6S, 9S, 11R, 14aS, 15S, 16S, 20S, 23S, 25aS) -20-[(1R) -3-amino-1- Hydroxy-3-oxopropyl] -2,11,15-trihydroxy-6-[(1R) -1-hydroxyethyl] -9- (3- [1- (2-hydroxyethyl) -1,2,3 , 6-Tetrahydropyridin-4-yl] -4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazole-6 -Yl} [1,1′-biphenyl] -4-carboxamide) -16-methyl-5,8,14,19,22,25-hexaoxotetracosahydro-1H-dipyrolo [2,1-c: 2 ', 1'-l] [1,4,7,10,13,16] hexaazacyclohenicosin-23-yl] -2-hydroxyethyl} -2-hydroxyphenyl ester.
(86) Hydrogen sulfate 5-{(2R) -2-[(2R, 6S, 9S, 11R, 14aS, 15S, 16S, 20S, 23S, 25aS) -20-[(1R) -3-Amino-1- Hydroxy-3-oxopropyl] -2,11,15-trihydroxy-6-[(1R) -1-hydroxyethyl] -9- (3- [1- (2-hydroxyethyl) -1,2,3 , 6-Tetrahydropyridin-4-yl] -4 '-{5- [4- (2-phenylethyl) piperazin-1-yl] pyrimidin-2-yl} [1,1'-biphenyl] -4-carboxamide ) -16-Methyl-5,8,14,19,22,25-hexaoxotetracosahydro-1H-dipyrolo [2,1-c: 2 ', 1'-l] [1,4,7,10 , 13, 16] Hexaazacyclohenicosin-23-yl] -2-hydroxyethyl} -2-hydroxyphenyl ester.
(87) Hydrogen sulfate 5-[(2R) -2-{(2R, 6S, 9S, 11R, 14aS, 15S, 16S, 20S, 23S, 25aS) -20-[(1R) -3-amino-1- Hydroxy-3-oxopropyl] -9- {4 '-(5- {4-[(4,4-dimethylcyclohexyl) oxy] piperidin-1-yl} -1,2,4-oxadiazole-3- Yl) -3- [1- (2-hydroxyethyl) -1,2,3,6-tetrahydropyridin-4-yl] [1,1'-biphenyl] -4-carboxamide} -2,11,15- Trihydroxy-6-[(1R) -1-hydroxyethyl] -16-methyl-5,8,14,19,22,25-hexaoxotetracosahydro-1H-dipyrolo [2,1-c: 2 ' , 1'-l] [1,4,7,10,13,16] hexaazacyclohenicosin-23-yl} -2-hydroxyethyl] -2-hydroxyphenyl ester.
(88) N-{(2R, 6S, 9S, 11R, 14aS, 15S, 16S, 20S, 23S, 25aS) -20-[(1R) -3-Amino-1-hydroxy-3-oxopropyl] -2 , 11,15-Trihydroxy-6-[(1R) -1-hydroxyethyl] -23-[(1R) -1-hydroxy-2- (4-hydroxy-3-methoxyphenyl) ethyl] -16-methyl -5,8,14,19,22,25-hexaoxotetracosahydro-1H-dipirolo [2,1-c: 2 ', 1'-l] [1,4,7,10,13,16] Hexaazacyclohenicosin-9-yl} -4 '-(5- {4- [2- (4-ethoxyphenyl) ethyl] piperazin-1-yl} pyrimidin-2-yl) -3- (1, 2,3,6-Tetrahydropyridin-4-yl) [1,1′-biphenyl] -4-carboxamide.
式(I)の化合物には、置換基の種類によって、幾何異性体が存在しうる。本明細書中、式(I)の化合物が異性体の一形態のみで記載されることがあるが、本発明は、それ以外の異性体も包含し、異性体の分離されたもの、あるいはそれらの混合物も包含する。
また、式(I)の化合物には、不斉炭素原子や軸不斉を有する場合があり、これに基づく光学異性体が存在しうる。本発明は、式(I)の化合物の光学異性体の分離されたもの、あるいはそれらの混合物も包含する。 In the compound of the formula (I), geometric isomers may exist depending on the type of substituent. In the present specification, the compound of the formula (I) may be described in only one form of an isomer, but the present invention includes other isomers, separated isomers, or those isomers. And mixtures thereof.
In addition, the compound of formula (I) may have an asymmetric carbon atom or axial asymmetry, and optical isomers based on this may exist. The present invention also includes separated optical isomers of the compound of formula (I) or a mixture thereof.
また、式(I)の化合物には、不斉炭素原子や軸不斉を有する場合があり、これに基づく光学異性体が存在しうる。本発明は、式(I)の化合物の光学異性体の分離されたもの、あるいはそれらの混合物も包含する。 In the compound of the formula (I), geometric isomers may exist depending on the type of substituent. In the present specification, the compound of the formula (I) may be described in only one form of an isomer, but the present invention includes other isomers, separated isomers, or those isomers. And mixtures thereof.
In addition, the compound of formula (I) may have an asymmetric carbon atom or axial asymmetry, and optical isomers based on this may exist. The present invention also includes separated optical isomers of the compound of formula (I) or a mixture thereof.
さらに、本発明は、式(I)で示される化合物の製薬学的に許容されるプロドラッグも包含する。製薬学的に許容されるプロドラッグとは、加溶媒分解により又は生理学的条件下で、アミノ基、水酸基、カルボキシル基等に変換されうる基を有する化合物である。プロドラッグを形成する基としては、例えば、Prog. Med., 5, 2157-2161(1985)や、「医薬品の開発」(廣川書店、1990年)第7巻 分子設計163-198に記載の基が挙げられる。
Furthermore, the present invention includes a pharmaceutically acceptable prodrug of the compound represented by the formula (I). A pharmaceutically acceptable prodrug is a compound having a group that can be converted to an amino group, a hydroxyl group, a carboxyl group, or the like by solvolysis or under physiological conditions. Examples of groups that form prodrugs include those described in Prog. Med., 5, 2157-2161 (1985) and “Development of pharmaceuticals” (Yodogawa Shoten, 1990), Volume 7, Molecular Design 163-198. Is mentioned.
また、式(I)の化合物の塩とは、式(I)の化合物の製薬学的に許容される塩であり、置換基の種類によって、酸付加塩又は塩基との塩を形成する場合がある。具体的には、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、リン酸等の無機酸や、ギ酸、酢酸、プロピオン酸、シュウ酸、マロン酸、コハク酸、フマル酸、マレイン酸、乳酸、リンゴ酸、マンデル酸、酒石酸、ジベンゾイル酒石酸、ジトルオイル酒石酸、クエン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸、アスパラギン酸、グルタミン酸等の有機酸との酸付加塩、ナトリウム、カリウム、マグネシウム、カルシウム、アルミニウム等の無機塩基、メチルアミン、エチルアミン、エタノールアミン、リシン、オルニチン等の有機塩基との塩、アセチルロイシン等の各種アミノ酸及びアミノ酸誘導体との塩やアンモニウム塩等が挙げられる。
The salt of the compound of the formula (I) is a pharmaceutically acceptable salt of the compound of the formula (I), and may form an acid addition salt or a salt with a base depending on the type of substituent. is there. Specifically, inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, fumaric acid, maleic acid Acid addition with organic acids such as lactic acid, malic acid, mandelic acid, tartaric acid, dibenzoyl tartaric acid, ditoluoyl tartaric acid, citric acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, aspartic acid, glutamic acid Salts, salts with inorganic bases such as sodium, potassium, magnesium, calcium and aluminum, salts with organic bases such as methylamine, ethylamine, ethanolamine, lysine and ornithine, salts with various amino acids and amino acid derivatives such as acetylleucine and ammonium salts Etc.
さらに、本発明は、式(I)の化合物及びその塩の各種の水和物や溶媒和物、及び結晶多形の物質も包含する。また、本発明は、種々の放射性又は非放射性同位体でラベルされた化合物も包含する。
Furthermore, the present invention also includes various hydrates and solvates of the compound of formula (I) and salts thereof, and crystalline polymorphic substances. The present invention also includes compounds labeled with various radioactive or non-radioactive isotopes.
(製造法)
式(I)の化合物及びその塩は、その基本構造あるいは置換基の種類に基づく特徴を利用し、種々の公知の合成法を適用して製造することができる。その際、官能基の種類によっては、当該官能基を原料から中間体へ至る段階で適当な保護基(容易に当該官能基に転化可能な基)に置き換えておくことが製造技術上効果的な場合がある。このような保護基としては、例えば、ウッツ(P. G. M. Wuts)及びグリーン(T. W. Greene)著、「Greene's Protective Groups in Organic Synthesis(第4版、2006年)」に記載の保護基等を挙げることができ、これらの反応条件に応じて適宜選択して用いればよい。このような方法では、当該保護基を導入して反応を行なったあと、必要に応じて保護基を除去することにより、所望の化合物を得ることができる。
また、式(I)の化合物のプロドラッグは、上記保護基と同様、原料から中間体へ至る段階で特定の基を導入、あるいは得られた式(I)の化合物を用いてさらに反応を行なうことで製造できる。反応は通常のエステル化、アミド化、脱水等、当業者に公知の方法を適用することにより行うことができる。
以下、式(I)の化合物の代表的な製造法を説明する。各製法は、当該説明に付した参考文献を参照して行うこともできる。なお、本発明の製造法は以下に示した例には限定されない。 (一般的製法1)

(Production method)
The compound of the formula (I) and salts thereof can be produced by applying various known synthetic methods utilizing characteristics based on the basic structure or the type of substituent. At that time, depending on the type of functional group, it is effective in terms of production technology to replace the functional group with an appropriate protecting group (a group that can be easily converted into the functional group) at the stage from the raw material to the intermediate. There is a case. Examples of such protecting groups include protecting groups described in “Greene's Protective Groups in Organic Synthesis (4th edition, 2006)” by PGM Wuts and TW Greene. These may be appropriately selected according to the reaction conditions. In such a method, after carrying out the reaction by introducing the protective group, the desired compound can be obtained by removing the protective group as necessary.
Further, the prodrug of the compound of the formula (I) introduces a specific group at the stage from the raw material to the intermediate as in the above protecting group, or further reacts with the obtained compound of the formula (I). Can be manufactured. The reaction can be carried out by applying a method known to those skilled in the art, such as ordinary esterification, amidation, dehydration and the like.
Hereinafter, typical production methods of the compound of the formula (I) will be described. Each manufacturing method can also be performed with reference to the reference attached to the said description. In addition, the manufacturing method of this invention is not limited to the example shown below. (General manufacturing method 1)
式(I)の化合物及びその塩は、その基本構造あるいは置換基の種類に基づく特徴を利用し、種々の公知の合成法を適用して製造することができる。その際、官能基の種類によっては、当該官能基を原料から中間体へ至る段階で適当な保護基(容易に当該官能基に転化可能な基)に置き換えておくことが製造技術上効果的な場合がある。このような保護基としては、例えば、ウッツ(P. G. M. Wuts)及びグリーン(T. W. Greene)著、「Greene's Protective Groups in Organic Synthesis(第4版、2006年)」に記載の保護基等を挙げることができ、これらの反応条件に応じて適宜選択して用いればよい。このような方法では、当該保護基を導入して反応を行なったあと、必要に応じて保護基を除去することにより、所望の化合物を得ることができる。
また、式(I)の化合物のプロドラッグは、上記保護基と同様、原料から中間体へ至る段階で特定の基を導入、あるいは得られた式(I)の化合物を用いてさらに反応を行なうことで製造できる。反応は通常のエステル化、アミド化、脱水等、当業者に公知の方法を適用することにより行うことができる。
以下、式(I)の化合物の代表的な製造法を説明する。各製法は、当該説明に付した参考文献を参照して行うこともできる。なお、本発明の製造法は以下に示した例には限定されない。 (一般的製法1)
The compound of the formula (I) and salts thereof can be produced by applying various known synthetic methods utilizing characteristics based on the basic structure or the type of substituent. At that time, depending on the type of functional group, it is effective in terms of production technology to replace the functional group with an appropriate protecting group (a group that can be easily converted into the functional group) at the stage from the raw material to the intermediate. There is a case. Examples of such protecting groups include protecting groups described in “Greene's Protective Groups in Organic Synthesis (4th edition, 2006)” by PGM Wuts and TW Greene. These may be appropriately selected according to the reaction conditions. In such a method, after carrying out the reaction by introducing the protective group, the desired compound can be obtained by removing the protective group as necessary.
Further, the prodrug of the compound of the formula (I) introduces a specific group at the stage from the raw material to the intermediate as in the above protecting group, or further reacts with the obtained compound of the formula (I). Can be manufactured. The reaction can be carried out by applying a method known to those skilled in the art, such as ordinary esterification, amidation, dehydration and the like.
Hereinafter, typical production methods of the compound of the formula (I) will be described. Each manufacturing method can also be performed with reference to the reference attached to the said description. In addition, the manufacturing method of this invention is not limited to the example shown below. (General manufacturing method 1)
本発明化合物(I)は、化合物(Q)と化合物(P-1)との反応により製造できる。本反応は、化合物(Q)のアミノ基と対応する(P-1)のカルボン酸との縮合反応である。本反応では、化合物(Q)と化合物(P-1)とを等量若しくは一方を過剰量用い、これらの混合物を縮合剤の存在下、反応に不活性な溶媒中、冷却下から加熱下、好ましくは-20℃から60℃で、0.1時間から5日間撹拌する。溶媒の例としては、特に限定はされないが、芳香族炭化水素類(トルエンまたはキシレン等)、ハロゲン化炭化水素類(例えばDCM、DCE若しくはクロロホルム等)、エーテル類(例えば、ジエチルエーテル、THFまたはジオキサン等)、非プロトン性溶媒(例えば、DMF、DMSO、EtOAc、MeCN)、又は、水、及びこれらの混合溶媒が挙げられる。縮合剤の例としては、BOP、HBTU、EDCI、DPPA、オキシ塩化リンが挙げられるが、これらに限定されるものではない。添加剤(例えばHOBt等)を用いることが反応に好ましい場合がある。有機塩基(例えばTEA、DIPEAまたはNMM等)、または無機塩基(例えば、K2CO3またはKOH等)の存在下で反応を行うことが、反応を円滑に進行させる上で有利な場合がある。
The compound (I) of the present invention can be produced by reacting the compound (Q) with the compound (P-1). This reaction is a condensation reaction between the amino group of compound (Q) and the corresponding carboxylic acid of (P-1). In this reaction, the compound (Q) and the compound (P-1) are used in an equal amount or in an excess amount, and a mixture of these in the presence of a condensing agent, in a solvent inert to the reaction, under cooling to heating, The mixture is preferably stirred at -20 ° C to 60 ° C for 0.1 hour to 5 days. Examples of the solvent include, but are not limited to, aromatic hydrocarbons (such as toluene or xylene), halogenated hydrocarbons (such as DCM, DCE, or chloroform), ethers (such as diethyl ether, THF, or dioxane). Etc.), an aprotic solvent (for example, DMF, DMSO, EtOAc, MeCN), water, and a mixed solvent thereof. Examples of condensing agents include, but are not limited to, BOP, HBTU, EDCI, DPPA, phosphorus oxychloride. It may be preferable for the reaction to use an additive (eg HOBt). It may be advantageous to carry out the reaction in the presence of an organic base (such as TEA, DIPEA or NMM) or an inorganic base (such as K 2 CO 3 or KOH) in order to facilitate the reaction.
また、化合物(I)は、化合物(P-1)を反応性誘導体として一度単離した後に、化合物(Q)と反応させて得ることもできる。カルボン酸体(P-1)の反応性誘導体の例としては、ハロゲン化剤(例えばオキシ塩化リン、塩化チオニル等)と反応して得られる酸ハロゲン化物、クロロギ酸イソブチル等と反応して得られる混合酸無水物、又は、HOBt等と縮合して得られる活性エステル等が挙げられる。化合物(P-1)と反応性誘導体との反応は、DMF、ハロゲン化炭化水素類、芳香族炭化水素類、エーテル類等の反応に不活性な溶媒中、冷却下から加熱下、好ましくは、-20℃から60℃で行うことができる。なお、反応に際して塩基(例えば、NMM、TEA、DIPEA、DMAP、ピリジン、ピコリン、ルチジン等)の存在下に反応させるのが、反応を円滑に進行させる上で有利な場合がある。また、ピリジンは溶媒を兼ねることもできる。
[文献]S. R. Sandler及びW. Karo著、「Organic Functional Group Preparations」、第2版、第1巻、Academic Press Inc.、1991年
日本化学会編「実験化学講座(第5版)」16巻(2005年)(丸善)
(一般的製法2-1) Compound (I) can also be obtained by isolating compound (P-1) as a reactive derivative and then reacting with compound (Q). Examples of reactive derivatives of the carboxylic acid form (P-1) are obtained by reacting with an acid halide obtained by reacting with a halogenating agent (for example, phosphorus oxychloride, thionyl chloride, etc.), isobutyl chloroformate, etc. Examples thereof include mixed acid anhydrides or active esters obtained by condensation with HOBt or the like. The reaction between the compound (P-1) and the reactive derivative is performed under cooling to heating, preferably in a solvent inert to the reaction such as DMF, halogenated hydrocarbons, aromatic hydrocarbons, ethers, It can be performed at -20 ° C to 60 ° C. In the reaction, it may be advantageous to cause the reaction to proceed smoothly in the presence of a base (for example, NMM, TEA, DIPEA, DMAP, pyridine, picoline, lutidine, etc.). Pyridine can also serve as a solvent.
[Literature] SR Sandler and W. Karo, "Organic Functional Group Preparations", 2nd edition, 1st volume, Academic Press Inc. (2005) (Maruzen)
(General production method 2-1)
[文献]S. R. Sandler及びW. Karo著、「Organic Functional Group Preparations」、第2版、第1巻、Academic Press Inc.、1991年
日本化学会編「実験化学講座(第5版)」16巻(2005年)(丸善)
(一般的製法2-1) Compound (I) can also be obtained by isolating compound (P-1) as a reactive derivative and then reacting with compound (Q). Examples of reactive derivatives of the carboxylic acid form (P-1) are obtained by reacting with an acid halide obtained by reacting with a halogenating agent (for example, phosphorus oxychloride, thionyl chloride, etc.), isobutyl chloroformate, etc. Examples thereof include mixed acid anhydrides or active esters obtained by condensation with HOBt or the like. The reaction between the compound (P-1) and the reactive derivative is performed under cooling to heating, preferably in a solvent inert to the reaction such as DMF, halogenated hydrocarbons, aromatic hydrocarbons, ethers, It can be performed at -20 ° C to 60 ° C. In the reaction, it may be advantageous to cause the reaction to proceed smoothly in the presence of a base (for example, NMM, TEA, DIPEA, DMAP, pyridine, picoline, lutidine, etc.). Pyridine can also serve as a solvent.
[Literature] SR Sandler and W. Karo, "Organic Functional Group Preparations", 2nd edition, 1st volume, Academic Press Inc. (2005) (Maruzen)
(General production method 2-1)

(式中、RA1は-CH(-R')-R''を意味する。R'又はR''は、同一又は異なってH、または低級アルキルを意味する。但しRA1は、-CH(-R')-R''基中の合計炭素数は1から6個である。)
本発明化合物(I-1)は、化合物(P-2)と化合物(P-3-1)とを反応により得ることができる。
この反応では、化合物(P-2)と化合物(P-3-1)とを等量若しくは一方を過剰量用い、これらの混合物を、還元剤の存在下、反応に不活性な溶媒中、-45℃から加熱還流下、好ましくは0℃~室温において、通常0.1時間~5日間撹拌する。ここで用いられる溶媒の例としては、特に限定されないが、MeOH、EtOH等のアルコール類、ジエチルエーテル等のエーテル類、DMF、ハロゲン化炭化水素類及びこれらの混合物が挙げられる。還元剤としては、NaBH(OAc)3、NaBH3CN、NaBH4等が挙げられる。化合物(P-3-1)の例としては、一般式O=C(-R')-R''で表わされるアルデヒド又はケトンの他、1,4-ジオキサン-2,5-ジオール等も用いることができる。モレキュラーシーブス等の脱水剤、又はAcOH、塩酸、チタニウム(IV)イソプロポキシド錯体等の酸存在下で反応を行うことが好ましい場合がある。反応によっては、化合物(P-2)と化合物(P-3-1)との縮合によりイミンが生成し、安定な中間体として単離できる場合がある。そのような場合には、このイミン中間体の還元反応により化合物(I)を得ることができる。また、前記還元剤での処理の代わりに、MeOH、EtOH、EtOAc等の溶媒中、AcOH、塩酸等の酸の存在下又は非存在下で、還元触媒(例えば、パラジウム炭素、ラネーニッケル等)を用いて反応を行うこともできる。この場合、反応を常圧から50気圧の水素雰囲気下で、冷却下から加熱下で行うことが好ましい。
〔文献〕
A. R. Katritzky及びR. J. K. Taylor著、「Comprehensive Organic Functional Group Transformations II」、第2巻、Elsevier Pergamon、2005年
日本化学会編「実験化学講座(第5版)」14巻(2005年)(丸善)
(一般的製法2-2) (Wherein R A1 represents —CH (—R ′) — R ″. R ′ or R ″ are the same or different and represent H or lower alkyl, provided that R A1 represents —CH (The total number of carbon atoms in the (-R ')-R''group is 1 to 6.)
The compound (I-1) of the present invention can be obtained by reacting the compound (P-2) and the compound (P-3-1).
In this reaction, an equivalent amount of compound (P-2) and compound (P-3-1) or an excess amount of one of them is used, and a mixture of these in a solvent inert to the reaction in the presence of a reducing agent, The mixture is stirred at 45 ° C. with heating under reflux, preferably at 0 ° C. to room temperature, usually for 0.1 hour to 5 days. Examples of the solvent used here include, but are not limited to, alcohols such as MeOH and EtOH, ethers such as diethyl ether, DMF, halogenated hydrocarbons, and mixtures thereof. Examples of the reducing agent include NaBH (OAc) 3 , NaBH 3 CN, NaBH 4 and the like. As an example of the compound (P-3-1), 1,4-dioxane-2,5-diol and the like are used in addition to the aldehyde or ketone represented by the general formula O = C (—R ′) — R ″. be able to. It may be preferable to carry out the reaction in the presence of a dehydrating agent such as molecular sieves or an acid such as AcOH, hydrochloric acid, titanium (IV) isopropoxide complex. Depending on the reaction, imine may be generated by condensation between compound (P-2) and compound (P-3-1), and may be isolated as a stable intermediate. In such a case, compound (I) can be obtained by the reduction reaction of this imine intermediate. Further, instead of the treatment with the reducing agent, a reduction catalyst (for example, palladium carbon, Raney nickel, etc.) is used in a solvent such as MeOH, EtOH, EtOAc or the like in the presence or absence of an acid such as AcOH or hydrochloric acid. The reaction can also be carried out. In this case, it is preferable to carry out the reaction under a hydrogen atmosphere at normal pressure to 50 atm and under cooling to heating.
[Reference]
A. R. Katritzky and R. J. K. Taylor, `` Comprehensive Organic Functional Group Transformations II '', Volume 2, Elsevier Pergamon, 2005 The Chemical Society of Japan `` Experimental Chemistry Course (5th Edition) '' Volume 14 (2005) (Maruzen)
(General production method 2-2)
本発明化合物(I-1)は、化合物(P-2)と化合物(P-3-1)とを反応により得ることができる。
この反応では、化合物(P-2)と化合物(P-3-1)とを等量若しくは一方を過剰量用い、これらの混合物を、還元剤の存在下、反応に不活性な溶媒中、-45℃から加熱還流下、好ましくは0℃~室温において、通常0.1時間~5日間撹拌する。ここで用いられる溶媒の例としては、特に限定されないが、MeOH、EtOH等のアルコール類、ジエチルエーテル等のエーテル類、DMF、ハロゲン化炭化水素類及びこれらの混合物が挙げられる。還元剤としては、NaBH(OAc)3、NaBH3CN、NaBH4等が挙げられる。化合物(P-3-1)の例としては、一般式O=C(-R')-R''で表わされるアルデヒド又はケトンの他、1,4-ジオキサン-2,5-ジオール等も用いることができる。モレキュラーシーブス等の脱水剤、又はAcOH、塩酸、チタニウム(IV)イソプロポキシド錯体等の酸存在下で反応を行うことが好ましい場合がある。反応によっては、化合物(P-2)と化合物(P-3-1)との縮合によりイミンが生成し、安定な中間体として単離できる場合がある。そのような場合には、このイミン中間体の還元反応により化合物(I)を得ることができる。また、前記還元剤での処理の代わりに、MeOH、EtOH、EtOAc等の溶媒中、AcOH、塩酸等の酸の存在下又は非存在下で、還元触媒(例えば、パラジウム炭素、ラネーニッケル等)を用いて反応を行うこともできる。この場合、反応を常圧から50気圧の水素雰囲気下で、冷却下から加熱下で行うことが好ましい。
〔文献〕
A. R. Katritzky及びR. J. K. Taylor著、「Comprehensive Organic Functional Group Transformations II」、第2巻、Elsevier Pergamon、2005年
日本化学会編「実験化学講座(第5版)」14巻(2005年)(丸善)
(一般的製法2-2) (Wherein R A1 represents —CH (—R ′) — R ″. R ′ or R ″ are the same or different and represent H or lower alkyl, provided that R A1 represents —CH (The total number of carbon atoms in the (-R ')-R''group is 1 to 6.)
The compound (I-1) of the present invention can be obtained by reacting the compound (P-2) and the compound (P-3-1).
In this reaction, an equivalent amount of compound (P-2) and compound (P-3-1) or an excess amount of one of them is used, and a mixture of these in a solvent inert to the reaction in the presence of a reducing agent, The mixture is stirred at 45 ° C. with heating under reflux, preferably at 0 ° C. to room temperature, usually for 0.1 hour to 5 days. Examples of the solvent used here include, but are not limited to, alcohols such as MeOH and EtOH, ethers such as diethyl ether, DMF, halogenated hydrocarbons, and mixtures thereof. Examples of the reducing agent include NaBH (OAc) 3 , NaBH 3 CN, NaBH 4 and the like. As an example of the compound (P-3-1), 1,4-dioxane-2,5-diol and the like are used in addition to the aldehyde or ketone represented by the general formula O = C (—R ′) — R ″. be able to. It may be preferable to carry out the reaction in the presence of a dehydrating agent such as molecular sieves or an acid such as AcOH, hydrochloric acid, titanium (IV) isopropoxide complex. Depending on the reaction, imine may be generated by condensation between compound (P-2) and compound (P-3-1), and may be isolated as a stable intermediate. In such a case, compound (I) can be obtained by the reduction reaction of this imine intermediate. Further, instead of the treatment with the reducing agent, a reduction catalyst (for example, palladium carbon, Raney nickel, etc.) is used in a solvent such as MeOH, EtOH, EtOAc or the like in the presence or absence of an acid such as AcOH or hydrochloric acid. The reaction can also be carried out. In this case, it is preferable to carry out the reaction under a hydrogen atmosphere at normal pressure to 50 atm and under cooling to heating.
[Reference]
A. R. Katritzky and R. J. K. Taylor, `` Comprehensive Organic Functional Group Transformations II '', Volume 2, Elsevier Pergamon, 2005 The Chemical Society of Japan `` Experimental Chemistry Course (5th Edition) '' Volume 14 (2005) (Maruzen)
(General production method 2-2)

(式中、Lvgは、脱離基を示す。)
なお、本発明化合物(I-2)は(P-2)の化合物を(P-3-2)の化合物でアルキル化することによっても得られる。ここで、脱離基Lvgの例には、ハロゲン、メタンスルホニルオキシ、p-トルエンスルホニルオキシ基等が含まれる。
この反応では、化合物(P-2)と化合物(P-3-2)とを等量若しくは一方を過剰量用い、これらの混合物を、反応に不活性な溶媒中、又は無溶媒下、冷却下から加熱還流下、好ましくは0℃から80℃において、通常0.1時間~5日間撹拌する。ここで用いられる溶媒の例としては、特に限定はされないが、トルエン、キシレン等の芳香族炭化水素類、ジオキサン、THF等のエーテル類、又はDCM、クロロホルム等のハロゲン化炭化水素類、DMF、DMSO、EtOAc、MeCN及びこれらの混合物が挙げられる。Et3N若しくはDIPEA等の有機塩基、又はK2CO3、Na2CO3若しくはKOH等の無機塩基の存在下で反応を行うのが、反応を円滑に進行させる上で有利な場合がある。
〔文献〕
S. R. Sandler及びW. Karo著、「Organic Functional Group Preparations」、第2版、第1巻、Academic Press Inc.、1991年
日本化学会編「実験化学講座(第5版)」14巻(2005年)(丸善)
(一般的製法3) (In the formula, Lvg represents a leaving group.)
The compound (I-2) of the present invention can also be obtained by alkylating the compound (P-2) with the compound (P-3-2). Here, examples of the leaving group Lvg include halogen, methanesulfonyloxy, p-toluenesulfonyloxy group and the like.
In this reaction, an equivalent amount of compound (P-2) and compound (P-3-2) or an excess amount of one of them is used, and a mixture thereof is cooled in a solvent inert to the reaction or without solvent. From 0 to 80 ° C., usually for 0.1 hour to 5 days. Examples of the solvent used here are not particularly limited, but are aromatic hydrocarbons such as toluene and xylene, ethers such as dioxane and THF, or halogenated hydrocarbons such as DCM and chloroform, DMF, DMSO. , EtOAc, MeCN and mixtures thereof. It may be advantageous to carry out the reaction in the presence of an organic base such as Et 3 N or DIPEA, or an inorganic base such as K 2 CO 3 , Na 2 CO 3 or KOH in order to facilitate the reaction.
[Reference]
SR Sandler and W. Karo, "Organic Functional Group Preparations", 2nd edition, 1st volume, Academic Press Inc., 1991, Chemical Society of Japan, "Experimental Chemistry Course (5th edition)" 14th volume (2005) (Maruzen)
(General manufacturing method 3)
なお、本発明化合物(I-2)は(P-2)の化合物を(P-3-2)の化合物でアルキル化することによっても得られる。ここで、脱離基Lvgの例には、ハロゲン、メタンスルホニルオキシ、p-トルエンスルホニルオキシ基等が含まれる。
この反応では、化合物(P-2)と化合物(P-3-2)とを等量若しくは一方を過剰量用い、これらの混合物を、反応に不活性な溶媒中、又は無溶媒下、冷却下から加熱還流下、好ましくは0℃から80℃において、通常0.1時間~5日間撹拌する。ここで用いられる溶媒の例としては、特に限定はされないが、トルエン、キシレン等の芳香族炭化水素類、ジオキサン、THF等のエーテル類、又はDCM、クロロホルム等のハロゲン化炭化水素類、DMF、DMSO、EtOAc、MeCN及びこれらの混合物が挙げられる。Et3N若しくはDIPEA等の有機塩基、又はK2CO3、Na2CO3若しくはKOH等の無機塩基の存在下で反応を行うのが、反応を円滑に進行させる上で有利な場合がある。
〔文献〕
S. R. Sandler及びW. Karo著、「Organic Functional Group Preparations」、第2版、第1巻、Academic Press Inc.、1991年
日本化学会編「実験化学講座(第5版)」14巻(2005年)(丸善)
(一般的製法3) (In the formula, Lvg represents a leaving group.)
The compound (I-2) of the present invention can also be obtained by alkylating the compound (P-2) with the compound (P-3-2). Here, examples of the leaving group Lvg include halogen, methanesulfonyloxy, p-toluenesulfonyloxy group and the like.
In this reaction, an equivalent amount of compound (P-2) and compound (P-3-2) or an excess amount of one of them is used, and a mixture thereof is cooled in a solvent inert to the reaction or without solvent. From 0 to 80 ° C., usually for 0.1 hour to 5 days. Examples of the solvent used here are not particularly limited, but are aromatic hydrocarbons such as toluene and xylene, ethers such as dioxane and THF, or halogenated hydrocarbons such as DCM and chloroform, DMF, DMSO. , EtOAc, MeCN and mixtures thereof. It may be advantageous to carry out the reaction in the presence of an organic base such as Et 3 N or DIPEA, or an inorganic base such as K 2 CO 3 , Na 2 CO 3 or KOH in order to facilitate the reaction.
[Reference]
SR Sandler and W. Karo, "Organic Functional Group Preparations", 2nd edition, 1st volume, Academic Press Inc., 1991, Chemical Society of Japan, "Experimental Chemistry Course (5th edition)" 14th volume (2005) (Maruzen)
(General manufacturing method 3)

(式中、X1は、単結合又は-CH2-を示す。Lvgは前述の定義と同じである。)
化合物(I-3)は、化合物(I)のXがX1の化合物である。化合物(I-1)は、化合物(P-4)と化合物(P-5)とから製造できる。本反応はアリール化合物(P-4)とホウ酸化合物(P-5)とを反応させる、いわゆる鈴木カップリングにより、化合物(I-1)を製造する方法である。反応は無溶媒中、芳香族炭化水素類、エーテル類、ハロゲン化炭化水素類、非プロトン性溶媒等、反応に不活性な溶媒中、室温から加熱還流下に行うことができる。反応は、パラジウム、ホスフィンリガンドおよび金属塩基共存下で行う。パラジウムとしては、Pd(OAc)2等の2価パラジウムや、Pd2(dba)3等の0価パラジウムを使用することができる。ホスフィンリガンドとしては、BINAP、DPPF等の2座配位子、P(But)3等の単座配位子、等を使用することができる。金属塩基としては、K2CO3、Cs2CO3、リン酸カリウム、NaOBut等を使用することができる。ここで脱離基Lvgとしては、ハロゲン、トリフルオロメタンスルホニルオキシ等を含む。
(一般的製法4) (In the formula, X 1 represents a single bond or —CH 2 —. Lvg has the same definition as described above.)
Compound (I-3) is a compound in which X of compound (I) is X 1 . Compound (I-1) can be produced from compound (P-4) and compound (P-5). This reaction is a method for producing compound (I-1) by so-called Suzuki coupling in which an aryl compound (P-4) and a boric acid compound (P-5) are reacted. The reaction can be carried out without heating in a solvent inert to the reaction such as aromatic hydrocarbons, ethers, halogenated hydrocarbons, aprotic solvents, etc., from room temperature under heating and reflux. The reaction is carried out in the presence of palladium, a phosphine ligand and a metal base. As palladium, divalent palladium such as Pd (OAc) 2 and zerovalent palladium such as Pd 2 (dba) 3 can be used. As the phosphine ligand, bidentate ligands such as BINAP and DPPF, monodentate ligands such as P (Bu t ) 3 , and the like can be used. As the metal base, K 2 CO 3, Cs 2 CO 3, potassium phosphate, it can be used NaOBu t like. Here, the leaving group Lvg includes halogen, trifluoromethanesulfonyloxy and the like.
(General manufacturing method 4)
化合物(I-3)は、化合物(I)のXがX1の化合物である。化合物(I-1)は、化合物(P-4)と化合物(P-5)とから製造できる。本反応はアリール化合物(P-4)とホウ酸化合物(P-5)とを反応させる、いわゆる鈴木カップリングにより、化合物(I-1)を製造する方法である。反応は無溶媒中、芳香族炭化水素類、エーテル類、ハロゲン化炭化水素類、非プロトン性溶媒等、反応に不活性な溶媒中、室温から加熱還流下に行うことができる。反応は、パラジウム、ホスフィンリガンドおよび金属塩基共存下で行う。パラジウムとしては、Pd(OAc)2等の2価パラジウムや、Pd2(dba)3等の0価パラジウムを使用することができる。ホスフィンリガンドとしては、BINAP、DPPF等の2座配位子、P(But)3等の単座配位子、等を使用することができる。金属塩基としては、K2CO3、Cs2CO3、リン酸カリウム、NaOBut等を使用することができる。ここで脱離基Lvgとしては、ハロゲン、トリフルオロメタンスルホニルオキシ等を含む。
(一般的製法4) (In the formula, X 1 represents a single bond or —CH 2 —. Lvg has the same definition as described above.)
Compound (I-3) is a compound in which X of compound (I) is X 1 . Compound (I-1) can be produced from compound (P-4) and compound (P-5). This reaction is a method for producing compound (I-1) by so-called Suzuki coupling in which an aryl compound (P-4) and a boric acid compound (P-5) are reacted. The reaction can be carried out without heating in a solvent inert to the reaction such as aromatic hydrocarbons, ethers, halogenated hydrocarbons, aprotic solvents, etc., from room temperature under heating and reflux. The reaction is carried out in the presence of palladium, a phosphine ligand and a metal base. As palladium, divalent palladium such as Pd (OAc) 2 and zerovalent palladium such as Pd 2 (dba) 3 can be used. As the phosphine ligand, bidentate ligands such as BINAP and DPPF, monodentate ligands such as P (Bu t ) 3 , and the like can be used. As the metal base, K 2 CO 3, Cs 2 CO 3, potassium phosphate, it can be used NaOBu t like. Here, the leaving group Lvg includes halogen, trifluoromethanesulfonyloxy and the like.
(General manufacturing method 4)

(式中、X2はNH又はN(Me)を示す。)
化合物(I-4)は、化合物(I)のXがX2の化合物である。化合物(I-2)は、化合物(P-4)と化合物(P-6)との反応により得ることができる。この反応では、化合物(P-4)と化合物(P-6)とを等量若しくは一方を過剰量用い、これらの混合物を、反応に不活性な溶媒中、又は無溶媒下、冷却下から加熱還流下、好ましくは0℃から80℃において、通常0.1時間~5日間撹拌する。ここで用いられる溶媒の例としては、特に限定はされないが、トルエン、キシレン等の芳香族炭化水素類、ジエチルエーテル、THF、ジオキサン、DME等のエーテル類、DCM、1,2-ジクロロエタン、クロロホルム等のハロゲン化炭化水素類、DMF、DMSO、EtOAc、MeCN及びこれらの混合物が挙げられる。Et3N、DIPEA若しくはNMM等の有機塩基、又はK2CO3、Na2CO3若しくはKOH等の無機塩基の存在下で反応を行うのが、反応を円滑に進行させる上で有利な場合がある。
〔文献〕
S. R. Sandler及びW. Karo著、「Organic Functional Group Preparations」、第2版、第1巻、Academic Press Inc.、1991年
日本化学会編「実験化学講座(第5版)」14巻(2005年)(丸善) (In the formula, X 2 represents NH or N (Me).)
Compound (I-4) are compounds X is X 2 of the compound (I). Compound (I-2) can be obtained by reacting compound (P-4) with compound (P-6). In this reaction, compound (P-4) and compound (P-6) are used in an equal amount or in excess of one, and the mixture is heated in a solvent inert to the reaction or in the absence of solvent and from under cooling. The mixture is stirred under reflux, preferably at 0 ° C. to 80 ° C., usually for 0.1 hour to 5 days. Examples of the solvent used here are not particularly limited, but aromatic hydrocarbons such as toluene and xylene, ethers such as diethyl ether, THF, dioxane and DME, DCM, 1,2-dichloroethane, chloroform and the like , Halogenated hydrocarbons, DMF, DMSO, EtOAc, MeCN and mixtures thereof. It may be advantageous to carry out the reaction in the presence of an organic base such as Et 3 N, DIPEA or NMM, or an inorganic base such as K 2 CO 3 , Na 2 CO 3 or KOH, in order to facilitate the reaction. is there.
[Reference]
SR Sandler and W. Karo, "Organic Functional Group Preparations", 2nd edition, 1st volume, Academic Press Inc., 1991, Chemical Society of Japan, "Experimental Chemistry Course (5th edition)" 14th volume (2005) (Maruzen)
化合物(I-4)は、化合物(I)のXがX2の化合物である。化合物(I-2)は、化合物(P-4)と化合物(P-6)との反応により得ることができる。この反応では、化合物(P-4)と化合物(P-6)とを等量若しくは一方を過剰量用い、これらの混合物を、反応に不活性な溶媒中、又は無溶媒下、冷却下から加熱還流下、好ましくは0℃から80℃において、通常0.1時間~5日間撹拌する。ここで用いられる溶媒の例としては、特に限定はされないが、トルエン、キシレン等の芳香族炭化水素類、ジエチルエーテル、THF、ジオキサン、DME等のエーテル類、DCM、1,2-ジクロロエタン、クロロホルム等のハロゲン化炭化水素類、DMF、DMSO、EtOAc、MeCN及びこれらの混合物が挙げられる。Et3N、DIPEA若しくはNMM等の有機塩基、又はK2CO3、Na2CO3若しくはKOH等の無機塩基の存在下で反応を行うのが、反応を円滑に進行させる上で有利な場合がある。
〔文献〕
S. R. Sandler及びW. Karo著、「Organic Functional Group Preparations」、第2版、第1巻、Academic Press Inc.、1991年
日本化学会編「実験化学講座(第5版)」14巻(2005年)(丸善) (In the formula, X 2 represents NH or N (Me).)
Compound (I-4) are compounds X is X 2 of the compound (I). Compound (I-2) can be obtained by reacting compound (P-4) with compound (P-6). In this reaction, compound (P-4) and compound (P-6) are used in an equal amount or in excess of one, and the mixture is heated in a solvent inert to the reaction or in the absence of solvent and from under cooling. The mixture is stirred under reflux, preferably at 0 ° C. to 80 ° C., usually for 0.1 hour to 5 days. Examples of the solvent used here are not particularly limited, but aromatic hydrocarbons such as toluene and xylene, ethers such as diethyl ether, THF, dioxane and DME, DCM, 1,2-dichloroethane, chloroform and the like , Halogenated hydrocarbons, DMF, DMSO, EtOAc, MeCN and mixtures thereof. It may be advantageous to carry out the reaction in the presence of an organic base such as Et 3 N, DIPEA or NMM, or an inorganic base such as K 2 CO 3 , Na 2 CO 3 or KOH, in order to facilitate the reaction. is there.
[Reference]
SR Sandler and W. Karo, "Organic Functional Group Preparations", 2nd edition, 1st volume, Academic Press Inc., 1991, Chemical Society of Japan, "Experimental Chemistry Course (5th edition)" 14th volume (2005) (Maruzen)
式(I)の化合物中、A環の窒素原子が四級アンモニウム塩を形成した化合物は、本発明化合物の含窒素ヘテロシクロアルキル基をN-アルキル化反応させることにより製造できる。
N-アルキル化反応としては、常法のN-アルキル化反応によって行うことができるが、具体的には、本発明化合物を、A環のアミノ基のアルキル化を行うのに十分量のアルキル化剤とDMF、CHCl3、MEK、MeCNまたはTHFなどの反応に不活性な溶媒中、氷冷下から室温下、または場合により加温下攪拌することにより行うことができる。アルキル化剤としては、アルキルハライド、または低級アルキルメタンスルホネート等が挙げられる。
(中間体製法1) Among the compounds of formula (I), a compound in which the nitrogen atom of the A ring forms a quaternary ammonium salt can be produced by N-alkylation reaction of the nitrogen-containing heterocycloalkyl group of the compound of the present invention.
The N-alkylation reaction can be carried out by a conventional N-alkylation reaction. Specifically, the compound of the present invention is alkylated in a sufficient amount to alkylate the amino group of the A ring. The reaction can be carried out by stirring in an inert solvent such as DMF, CHCl 3 , MEK, MeCN, or THF, under ice-cooling to room temperature, or optionally with heating. Examples of the alkylating agent include alkyl halides and lower alkyl methanesulfonates.
(Intermediate production method 1)
N-アルキル化反応としては、常法のN-アルキル化反応によって行うことができるが、具体的には、本発明化合物を、A環のアミノ基のアルキル化を行うのに十分量のアルキル化剤とDMF、CHCl3、MEK、MeCNまたはTHFなどの反応に不活性な溶媒中、氷冷下から室温下、または場合により加温下攪拌することにより行うことができる。アルキル化剤としては、アルキルハライド、または低級アルキルメタンスルホネート等が挙げられる。
(中間体製法1) Among the compounds of formula (I), a compound in which the nitrogen atom of the A ring forms a quaternary ammonium salt can be produced by N-alkylation reaction of the nitrogen-containing heterocycloalkyl group of the compound of the present invention.
The N-alkylation reaction can be carried out by a conventional N-alkylation reaction. Specifically, the compound of the present invention is alkylated in a sufficient amount to alkylate the amino group of the A ring. The reaction can be carried out by stirring in an inert solvent such as DMF, CHCl 3 , MEK, MeCN, or THF, under ice-cooling to room temperature, or optionally with heating. Examples of the alkylating agent include alkyl halides and lower alkyl methanesulfonates.
(Intermediate production method 1)

(式中、Rはカルボキシル基の保護基を意味する。)
化合物(P-1)は、化合物(P-7)からのカルボキシル基の脱保護することにより製造することができる。例えば、加水分解反応により脱保護することができる。
(中間体製法2) (In the formula, R means a protecting group for a carboxyl group.)
Compound (P-1) can be produced by deprotecting the carboxyl group from compound (P-7). For example, it can be deprotected by a hydrolysis reaction.
(Intermediate production method 2)
化合物(P-1)は、化合物(P-7)からのカルボキシル基の脱保護することにより製造することができる。例えば、加水分解反応により脱保護することができる。
(中間体製法2) (In the formula, R means a protecting group for a carboxyl group.)
Compound (P-1) can be produced by deprotecting the carboxyl group from compound (P-7). For example, it can be deprotected by a hydrolysis reaction.
(Intermediate production method 2)

化合物(P-7-1)は化合物(P-7)のXがX1の化合物であり、化合物(P-7-2)は化合物(P-7)のXがX2の化合物である。本反応は化合物(P-8)を出発原料として、化合物(P-5)又は化合物(P-6)とをそれぞれ、反応させることにより、それぞれ、化合物(P-7-1)、化合物(P-7-2)が製造できる。反応条件については、一般的製法3又は一般的製法4と同様の条件で求める化合物を製造することができる。
Compound (P-7-1) is a compound in which X of compound (P-7) is X 1 , and Compound (P-7-2) is a compound in which X of compound (P-7) is X 2 . This reaction is performed by reacting compound (P-5) or compound (P-6) with compound (P-8) as a starting material, respectively, to give compound (P-7-1) and compound (P-6), respectively. -7-2) can be manufactured. With respect to the reaction conditions, the desired compound can be produced under the same conditions as in General Production Method 3 or General Production Method 4.

また化合物(P-7)は化合物(P-9)と(P-10)を反応させることにより、製造できる。YとZはいずれか一方がLvg、他方がボロン酸、またはボロン酸エステルである。反応条件については、一般的製法3と同様の条件で求める化合物を製造することができる。
Compound (P-7) can be produced by reacting compounds (P-9) and (P-10). One of Y and Z is Lvg, and the other is boronic acid or boronic ester. With respect to the reaction conditions, the desired compound can be produced under the same conditions as in General Production Method 3.
なお、上記の全ての反応は、本明細書の製造例若しくは実施例に記載の方法、またはそれに準じた方法を採用することもでき、化合物は、公知の方法、当業者にとって自明である方法、あるいは本明細書の製造例若しくは実施例に記載の方法、またはそれに準じた方法により製造することができる。
In addition, for all the above reactions, the methods described in the production examples or examples of the present specification or methods according thereto can be adopted, and the compounds are known methods, methods obvious to those skilled in the art, Or it can manufacture by the method as described in the manufacture example or Example of this specification, or the method according to it.
(原料の製造法)
本願実施例化合物を製造の際に用いられる一般式(Q)で示される原料化合物は、国際公開WO97/32975号、国際公開WO99/40108号、国際公開WO2000/064927号、国際公開WO2002/072621号、国際公開WO2003/068807号、国際公開WO2009/057568号に示される方法、又は、記載の方法と同様にして製造することができる。 (Raw material production method)
The raw material compound represented by the general formula (Q) used in the production of the compound of the present example is an international publication WO97 / 32975, an international publication WO99 / 40108, an international publication WO2000 / 064927, an international publication WO2002 / 072621. In addition, it can be produced in the same manner as the method shown in International Publication WO2003 / 068807 or International Publication WO2009 / 057568 or the method described.
本願実施例化合物を製造の際に用いられる一般式(Q)で示される原料化合物は、国際公開WO97/32975号、国際公開WO99/40108号、国際公開WO2000/064927号、国際公開WO2002/072621号、国際公開WO2003/068807号、国際公開WO2009/057568号に示される方法、又は、記載の方法と同様にして製造することができる。 (Raw material production method)
The raw material compound represented by the general formula (Q) used in the production of the compound of the present example is an international publication WO97 / 32975, an international publication WO99 / 40108, an international publication WO2000 / 064927, an international publication WO2002 / 072621. In addition, it can be produced in the same manner as the method shown in International Publication WO2003 / 068807 or International Publication WO2009 / 057568 or the method described.
式(I)の化合物は、遊離化合物、その塩、水和物、溶媒和物、あるいは結晶多形の物質として単離され、精製される。式(I)の化合物の塩は、常法の造塩反応に付すことにより製造することもできる。
単離、精製は、抽出、分別結晶化、各種分画クロマトグラフィー等、通常の化学操作を適用して行なわれる。
各種の異性体は、適当な原料化合物を選択することにより製造でき、あるいは異性体間の物理化学的性質の差を利用して分離することができる。例えば、光学異性体は、ラセミ体の一般的な光学分割法(例えば、光学活性な塩基又は酸とのジアステレオマー塩に導く分別結晶化や、キラルカラム等を用いたクロマトグラフィー等)により得られ、また、適当な光学活性な原料化合物から製造することもできる。 The compounds of formula (I) are isolated and purified as free compounds, their salts, hydrates, solvates, or crystalline polymorphs. The salt of the compound of formula (I) can also be produced by subjecting it to a conventional salt formation reaction.
Isolation and purification are performed by applying ordinary chemical operations such as extraction, fractional crystallization, and various fractional chromatography.
Various isomers can be produced by selecting an appropriate raw material compound, or can be separated by utilizing a difference in physicochemical properties between isomers. For example, optical isomers can be obtained by general optical resolution of racemates (for example, fractional crystallization leading to diastereomeric salts with optically active bases or acids, chromatography using chiral columns, etc.). Further, it can also be produced from a suitable optically active raw material compound.
単離、精製は、抽出、分別結晶化、各種分画クロマトグラフィー等、通常の化学操作を適用して行なわれる。
各種の異性体は、適当な原料化合物を選択することにより製造でき、あるいは異性体間の物理化学的性質の差を利用して分離することができる。例えば、光学異性体は、ラセミ体の一般的な光学分割法(例えば、光学活性な塩基又は酸とのジアステレオマー塩に導く分別結晶化や、キラルカラム等を用いたクロマトグラフィー等)により得られ、また、適当な光学活性な原料化合物から製造することもできる。 The compounds of formula (I) are isolated and purified as free compounds, their salts, hydrates, solvates, or crystalline polymorphs. The salt of the compound of formula (I) can also be produced by subjecting it to a conventional salt formation reaction.
Isolation and purification are performed by applying ordinary chemical operations such as extraction, fractional crystallization, and various fractional chromatography.
Various isomers can be produced by selecting an appropriate raw material compound, or can be separated by utilizing a difference in physicochemical properties between isomers. For example, optical isomers can be obtained by general optical resolution of racemates (for example, fractional crystallization leading to diastereomeric salts with optically active bases or acids, chromatography using chiral columns, etc.). Further, it can also be produced from a suitable optically active raw material compound.
式(I)の化合物の薬理活性は、以下の試験により確認した。
試験例1:C. glabrata; Echinocandin 耐性株、カンジダ・グラブラータ(Candida glabrata)FP2135株に対する感受性測定
20mM-HEPES緩衝液(pH 7.3)で緩衝したヒト血清を試験媒体として用いる微量液体希釈法によって、ヒト血清中のMIC(最小阻害濃度)を求めた。細胞数106個/mLの懸濁液を血球計算版で調製し、細胞約5x103~2x104個/mLの接種サイズを得た。マイクロプレートを5%CO2雰囲気下37℃で24時間インキュベートした。本株に対するMICは増殖が完全に阻止されている試験化合物の最低濃度として定義した。上市されているEchinocandin類(Caspofungin、Anidulafungin及びMicafungin)の本株に対するMICは全て>64 μg/mLであった。
試験例2:アスペルギルス・フミガーツス(Aspergillus fumigatus)株に対する感受性測定
アスペルギルス・フミガーツスを用いて、試験例1と同様に測定した。 The pharmacological activity of the compound of formula (I) was confirmed by the following test.
Test Example 1: Sensitivity measurement for C. glabrata; Echinocandin resistant strain, Candida glabrata FP2135 Serum MIC (minimum inhibitory concentration) was determined. A suspension of 10 6 cells / mL was prepared with a hemocytometer and an inoculum size of approximately 5 × 10 3 to 2 × 10 4 cells / mL was obtained. The microplate was incubated for 24 hours at 37 ° C. in a 5% CO 2 atmosphere. The MIC for this strain was defined as the lowest concentration of test compound at which growth was completely inhibited. The MIC for all Echinocandins (Caspofungin, Anidulafungin and Micafungin) on the market was> 64 μg / mL.
Test Example 2: Sensitivity measurement for Aspergillus fumigatus strain Aspergillus fumigatus strain was measured in the same manner as in Test Example 1.
試験例1:C. glabrata; Echinocandin 耐性株、カンジダ・グラブラータ(Candida glabrata)FP2135株に対する感受性測定
20mM-HEPES緩衝液(pH 7.3)で緩衝したヒト血清を試験媒体として用いる微量液体希釈法によって、ヒト血清中のMIC(最小阻害濃度)を求めた。細胞数106個/mLの懸濁液を血球計算版で調製し、細胞約5x103~2x104個/mLの接種サイズを得た。マイクロプレートを5%CO2雰囲気下37℃で24時間インキュベートした。本株に対するMICは増殖が完全に阻止されている試験化合物の最低濃度として定義した。上市されているEchinocandin類(Caspofungin、Anidulafungin及びMicafungin)の本株に対するMICは全て>64 μg/mLであった。
試験例2:アスペルギルス・フミガーツス(Aspergillus fumigatus)株に対する感受性測定
アスペルギルス・フミガーツスを用いて、試験例1と同様に測定した。 The pharmacological activity of the compound of formula (I) was confirmed by the following test.
Test Example 1: Sensitivity measurement for C. glabrata; Echinocandin resistant strain, Candida glabrata FP2135 Serum MIC (minimum inhibitory concentration) was determined. A suspension of 10 6 cells / mL was prepared with a hemocytometer and an inoculum size of approximately 5 × 10 3 to 2 × 10 4 cells / mL was obtained. The microplate was incubated for 24 hours at 37 ° C. in a 5% CO 2 atmosphere. The MIC for this strain was defined as the lowest concentration of test compound at which growth was completely inhibited. The MIC for all Echinocandins (Caspofungin, Anidulafungin and Micafungin) on the market was> 64 μg / mL.
Test Example 2: Sensitivity measurement for Aspergillus fumigatus strain Aspergillus fumigatus strain was measured in the same manner as in Test Example 1.
本発明の幾つかの代表化合物について、上記試験例1の試験結果を下記表1に示した。表中、Exは実施例番号を示した。値は、カンジダ・グラブラータ(C. glabrata; Echinocandin 耐性株)に対するMIC、アスペルギルス・フミガーツス(A. fumigatus)は臨床分離株3株(A.fumigatus 20030, 20024,及び200293株)に対するMECの相乗平均値として示した。
Table 1 below shows the test results of Test Example 1 for some representative compounds of the present invention. In the table, Ex indicates an example number. Values are the MIC against C. glabrata (C. glabrata; Echinocandin resistant strain), and the geometric mean of MEC against Aspergillus fumigatus (A. As shown.

試験例3:ヒト血清中の抗真菌剤感受性測定
20mM-HEPES緩衝液(pH 7.4)で緩衝したヒト血清を試験媒体として用いる微量液体希釈法によって、ヒト血清中のMIC(最小阻害濃度)を求めた。細胞数107個/mLの懸濁液を血球計算版で調製し、細胞約2.5x103~2x105個/mLの接種サイズを得た。マイクロプレートを5%CO2雰囲気下37℃で24時間インキュベートした。Candida属に対するMICは増殖が完全に阻止されている試験化合物の最低濃度、Aspergillus属に対するMECは発育コントロールに比べ充分に増殖が抑制されている試験化合物の最低濃度として定義した。
値は、カンジダ・グラブラータ(C. glabrata; Echinocandin 耐性株)、カンジダ・アルビカンス(C.albicans FP1943)とカンジダ・パラプシローシス(C. parapsilosis 20001)の臨床分離株1菌株に対するMIC、アスペルギルス・フミガーツス(A. fumigatus)は臨床分離株3株(A.fumigatus 20030, 20024,及び200293株)に対するMECの相乗平均値として示した。
Test Example 3: Antifungal susceptibility measurement in human serum MIC (minimum inhibitory concentration) in human serum was determined by a micro liquid dilution method using human serum buffered with 20 mM HEPES buffer (pH 7.4) as a test medium. It was. A suspension with 10 7 cells / mL was prepared with a hemocytometer and an inoculum size of approximately 2.5 × 10 3 to 2 × 10 5 cells / mL was obtained. The microplate was incubated for 24 hours at 37 ° C. in a 5% CO 2 atmosphere. MIC for Candida was defined as the lowest concentration of the test compound at which growth was completely inhibited, and MEC for Aspergillus was defined as the lowest concentration of the test compound at which growth was sufficiently inhibited compared to the growth control.
Values are MIC, Aspergillus fumigatus (A. fumigatus) is shown as the geometric mean value of MEC for 3 clinical isolates (A. fumigatus 20030, 20024, and 200293 strains).
20mM-HEPES緩衝液(pH 7.4)で緩衝したヒト血清を試験媒体として用いる微量液体希釈法によって、ヒト血清中のMIC(最小阻害濃度)を求めた。細胞数107個/mLの懸濁液を血球計算版で調製し、細胞約2.5x103~2x105個/mLの接種サイズを得た。マイクロプレートを5%CO2雰囲気下37℃で24時間インキュベートした。Candida属に対するMICは増殖が完全に阻止されている試験化合物の最低濃度、Aspergillus属に対するMECは発育コントロールに比べ充分に増殖が抑制されている試験化合物の最低濃度として定義した。
値は、カンジダ・グラブラータ(C. glabrata; Echinocandin 耐性株)、カンジダ・アルビカンス(C.albicans FP1943)とカンジダ・パラプシローシス(C. parapsilosis 20001)の臨床分離株1菌株に対するMIC、アスペルギルス・フミガーツス(A. fumigatus)は臨床分離株3株(A.fumigatus 20030, 20024,及び200293株)に対するMECの相乗平均値として示した。
Test Example 3: Antifungal susceptibility measurement in human serum MIC (minimum inhibitory concentration) in human serum was determined by a micro liquid dilution method using human serum buffered with 20 mM HEPES buffer (pH 7.4) as a test medium. It was. A suspension with 10 7 cells / mL was prepared with a hemocytometer and an inoculum size of approximately 2.5 × 10 3 to 2 × 10 5 cells / mL was obtained. The microplate was incubated for 24 hours at 37 ° C. in a 5% CO 2 atmosphere. MIC for Candida was defined as the lowest concentration of the test compound at which growth was completely inhibited, and MEC for Aspergillus was defined as the lowest concentration of the test compound at which growth was sufficiently inhibited compared to the growth control.
Values are MIC, Aspergillus fumigatus (A. fumigatus) is shown as the geometric mean value of MEC for 3 clinical isolates (A. fumigatus 20030, 20024, and 200293 strains).


表中の化合物は以下のとおりである。
化合物Aは米国特許第5569646号の実施例19の化合物。
化合物Bは、WO98/23637号の実施例8の化合物。
化合物Cは、WO96/11210号の実施例100の化合物。
化合物Dは、WO99/40108号の実施例94の化合物。
上記の各試験の結果、表に示された代表的な式(I)の化合物は抗真菌活性を有することが認められた。また当該化合物はC. albicans、A. fumigatusだけでなく C. parapsilosis、更にC. glabrata; Echinocandin 耐性株に対しても抗真菌活性を有していた。
この結果から従来の化合物と比較して抗真菌活性が優れていることが確認された。このことから、式(I)の化合物は種々の真菌感染症、特に下記の真菌による感染症疾患の予防剤又は治療剤として有用しうる。
アクレモニウム;
アスペルギルス(例えばアスペルギルス・クラバタス、アスペルギルス・フラーブス、アスペルギルス・フミガーツス、アスペルギルス・ニデュランス、アスペルギルス・ニガー、アスペルギルス・テレウス、アスペルギルス・ベジルコロル等);
ブラストミセス(例えばブラストミセス・デルマチチジス等);
カンジダ(例えばカンジダ・アルビカンス、カンジダ・グラブラタ、カンジダ・ギリエルモンディ、カンジダ・ケフィール、カンジダ・クルセイ、カンジダ・パラシローシス、カンジダ・ステラトイデア、カンジダ・トロピカリス、カンジダ・ユチリス等);
クラドスポリウム(例えばクラドスポリウム・トリコイデス等);
コクシジオイデス(例えばコクシジオイデス・イミチス等);
クンニングアメラ(例えばクンニングアメラ・エレガンス等);
皮膚糸状菌;
エキソフィアラ(例えばエキソフィアラ・デルマチチジス、エキソフィアラ・スピニフェラ等);
エピデルモフィトン(例えばエピデルモフィトン・フロッコーズム等);
フォンセセア(例えばフォンセセア・ペドロソイ等);
ゲオトリクム(例えばゲオトリクム・カンジドゥム等);
ヒストプラスマ(例えばヒストプラスマ・カプスラーツム変異カプスラーツム等);
マラセジア(例えばマラセジア・フルフール等);
ミクロスポルム(例えばミクロスポルム・カニス、ミクロスポルム・ジプセウム等);
パラコクシジオイデス(例えばパラコクシジオイデス・ブラジリエンシス等);
ペニシリウム(例えばペニシリウム・マーネフェイ等);
フィアロフォラ;
ニューモシスチス(例えばニューモシスチス・ジロヴェチ等);
プソイドアレシェリア(例えばプソイドアレシェリア・ボイジー等);
サッカロミセス(例えばサッカロミセス・セレビジエ等);
スコプラリオプシス;
スポロトリクス(例えばスポロトリクス・シェンキー等);
トリコフィトン(例えばトリコフィトン・メンタグロフィテス、トリコフィトン・ルブルム等)。
別の態様として、式(I)の化合物は下記の真菌感染症、真菌による感染症疾患の予防剤又は治療剤として有用しうる。
アスペルギルス(例えばアスペルギルス・クラバタス、アスペルギルス・フラーブス、アスペルギルス・フミガーツス、アスペルギルス・ニデュランス、アスペルギルス・ニガー、アスペルギルス・テレウス、アスペルギルス・ベジルコロル等);
カンジダ(例えばカンジダ・アルビカンス、カンジダ・グラブラタ、カンジダ・ギリエルモンディ、カンジダ・ケフィール、カンジダ・クルセイ、カンジダ・パラシローシス、カンジダ・ステラトイデア、カンジダ・トロピカリス、カンジダ・ユチリス等);
ニューモシスチス(例えばニューモシスチス・ジロヴェチ等);
プソイドアレシェリア(例えばプソイドアレシェリア・ボイジー等);
サッカロミセス(例えばサッカロミセス・セレビジエ等)。 The compounds in the table are as follows.
Compound A is the compound of Example 19 of US Pat. No. 5,556,646.
Compound B is the compound of Example 8 of WO98 / 23637.
Compound C is the compound of Example 100 of WO96 / 11210.
Compound D is the compound of Example 94 of WO99 / 40108.
As a result of each of the above tests, it was found that the representative compounds of formula (I) shown in the table have antifungal activity. The compound also had antifungal activity against not only C. albicans and A. fumigatus but also C. parapsilosis and C. glabrata; Echinocandin resistant strains.
From these results, it was confirmed that the antifungal activity was superior as compared with conventional compounds. From this, the compound of the formula (I) can be useful as a prophylactic or therapeutic agent for various fungal infections, particularly infection diseases caused by the following fungi.
Acremonium;
Aspergillus (for example, Aspergillus clubatas, Aspergillus flavus, Aspergillus fumigatus, Aspergillus nidulans, Aspergillus niger, Aspergillus tereus, Aspergillus vesilcolor);
Blast Mrs. (eg Blast Mrs. dermatitis);
Candida (e.g. Candida albicans, Candida glabrata, Candida guilliermondi, Candida kefir, Candida crusei, Candida paracylosis, Candida stellatoidea, Candida tropicalis, Candida utilis, etc.);
Cladosporium (eg Cladosporium trichoides);
Coccidioides (eg coccidioides imimitis);
Cunning Amera (eg Cunning Amera Elegance);
Dermatophytes;
Exofiara (eg, Exofiara del machitisis, Exofiara spinifera, etc.);
Epidermophyton (eg Epidermophyton floccum);
Foncea (eg foncea pedrosoi);
Geotricum (eg Geotricum candidum);
Histoplasma (for example, histoplasma capsulatum mutant capsulatsum etc.);
Malassezia (eg, Malassezia furfur, etc.);
Microsporum (eg, microsporum canis, microsporum dipseum, etc.);
Paracoccidioides (eg, paracoccidioides brasiliensis);
Penicillium (eg, Penicillium manefei);
Fialophora;
Pneumocystis (eg, Pneumocystis jiroveci);
Pseudoaleseria (eg pseudoaleseria boise);
Saccharomyces (eg Saccharomyces cerevisiae);
Skoplariopsis;
Sporotrix (eg Sporotrix Schenky, etc.);
Trichophyton (for example, Trichophyton Mentagrophytes, Trichophyton rubrum, etc.).
In another embodiment, the compound of formula (I) may be useful as a prophylactic or therapeutic agent for the following fungal infections and infectious diseases caused by fungi.
Aspergillus (for example, Aspergillus clubatas, Aspergillus flavus, Aspergillus fumigatus, Aspergillus nidulans, Aspergillus niger, Aspergillus tereus, Aspergillus vesilcolor);
Candida (e.g. Candida albicans, Candida glabrata, Candida guilliermondi, Candida kefir, Candida crusei, Candida paracylosis, Candida stellatoidea, Candida tropicalis, Candida utilis, etc.);
Pneumocystis (eg, Pneumocystis jiroveci);
Pseudoaleseria (eg pseudoaleseria boise);
Saccharomyces (eg Saccharomyces cerevisiae).
化合物Aは米国特許第5569646号の実施例19の化合物。
化合物Bは、WO98/23637号の実施例8の化合物。
化合物Cは、WO96/11210号の実施例100の化合物。
化合物Dは、WO99/40108号の実施例94の化合物。
上記の各試験の結果、表に示された代表的な式(I)の化合物は抗真菌活性を有することが認められた。また当該化合物はC. albicans、A. fumigatusだけでなく C. parapsilosis、更にC. glabrata; Echinocandin 耐性株に対しても抗真菌活性を有していた。
この結果から従来の化合物と比較して抗真菌活性が優れていることが確認された。このことから、式(I)の化合物は種々の真菌感染症、特に下記の真菌による感染症疾患の予防剤又は治療剤として有用しうる。
アクレモニウム;
アスペルギルス(例えばアスペルギルス・クラバタス、アスペルギルス・フラーブス、アスペルギルス・フミガーツス、アスペルギルス・ニデュランス、アスペルギルス・ニガー、アスペルギルス・テレウス、アスペルギルス・ベジルコロル等);
ブラストミセス(例えばブラストミセス・デルマチチジス等);
カンジダ(例えばカンジダ・アルビカンス、カンジダ・グラブラタ、カンジダ・ギリエルモンディ、カンジダ・ケフィール、カンジダ・クルセイ、カンジダ・パラシローシス、カンジダ・ステラトイデア、カンジダ・トロピカリス、カンジダ・ユチリス等);
クラドスポリウム(例えばクラドスポリウム・トリコイデス等);
コクシジオイデス(例えばコクシジオイデス・イミチス等);
クンニングアメラ(例えばクンニングアメラ・エレガンス等);
皮膚糸状菌;
エキソフィアラ(例えばエキソフィアラ・デルマチチジス、エキソフィアラ・スピニフェラ等);
エピデルモフィトン(例えばエピデルモフィトン・フロッコーズム等);
フォンセセア(例えばフォンセセア・ペドロソイ等);
ゲオトリクム(例えばゲオトリクム・カンジドゥム等);
ヒストプラスマ(例えばヒストプラスマ・カプスラーツム変異カプスラーツム等);
マラセジア(例えばマラセジア・フルフール等);
ミクロスポルム(例えばミクロスポルム・カニス、ミクロスポルム・ジプセウム等);
パラコクシジオイデス(例えばパラコクシジオイデス・ブラジリエンシス等);
ペニシリウム(例えばペニシリウム・マーネフェイ等);
フィアロフォラ;
ニューモシスチス(例えばニューモシスチス・ジロヴェチ等);
プソイドアレシェリア(例えばプソイドアレシェリア・ボイジー等);
サッカロミセス(例えばサッカロミセス・セレビジエ等);
スコプラリオプシス;
スポロトリクス(例えばスポロトリクス・シェンキー等);
トリコフィトン(例えばトリコフィトン・メンタグロフィテス、トリコフィトン・ルブルム等)。
別の態様として、式(I)の化合物は下記の真菌感染症、真菌による感染症疾患の予防剤又は治療剤として有用しうる。
アスペルギルス(例えばアスペルギルス・クラバタス、アスペルギルス・フラーブス、アスペルギルス・フミガーツス、アスペルギルス・ニデュランス、アスペルギルス・ニガー、アスペルギルス・テレウス、アスペルギルス・ベジルコロル等);
カンジダ(例えばカンジダ・アルビカンス、カンジダ・グラブラタ、カンジダ・ギリエルモンディ、カンジダ・ケフィール、カンジダ・クルセイ、カンジダ・パラシローシス、カンジダ・ステラトイデア、カンジダ・トロピカリス、カンジダ・ユチリス等);
ニューモシスチス(例えばニューモシスチス・ジロヴェチ等);
プソイドアレシェリア(例えばプソイドアレシェリア・ボイジー等);
サッカロミセス(例えばサッカロミセス・セレビジエ等)。 The compounds in the table are as follows.
Compound A is the compound of Example 19 of US Pat. No. 5,556,646.
Compound B is the compound of Example 8 of WO98 / 23637.
Compound C is the compound of Example 100 of WO96 / 11210.
Compound D is the compound of Example 94 of WO99 / 40108.
As a result of each of the above tests, it was found that the representative compounds of formula (I) shown in the table have antifungal activity. The compound also had antifungal activity against not only C. albicans and A. fumigatus but also C. parapsilosis and C. glabrata; Echinocandin resistant strains.
From these results, it was confirmed that the antifungal activity was superior as compared with conventional compounds. From this, the compound of the formula (I) can be useful as a prophylactic or therapeutic agent for various fungal infections, particularly infection diseases caused by the following fungi.
Acremonium;
Aspergillus (for example, Aspergillus clubatas, Aspergillus flavus, Aspergillus fumigatus, Aspergillus nidulans, Aspergillus niger, Aspergillus tereus, Aspergillus vesilcolor);
Blast Mrs. (eg Blast Mrs. dermatitis);
Candida (e.g. Candida albicans, Candida glabrata, Candida guilliermondi, Candida kefir, Candida crusei, Candida paracylosis, Candida stellatoidea, Candida tropicalis, Candida utilis, etc.);
Cladosporium (eg Cladosporium trichoides);
Coccidioides (eg coccidioides imimitis);
Cunning Amera (eg Cunning Amera Elegance);
Dermatophytes;
Exofiara (eg, Exofiara del machitisis, Exofiara spinifera, etc.);
Epidermophyton (eg Epidermophyton floccum);
Foncea (eg foncea pedrosoi);
Geotricum (eg Geotricum candidum);
Histoplasma (for example, histoplasma capsulatum mutant capsulatsum etc.);
Malassezia (eg, Malassezia furfur, etc.);
Microsporum (eg, microsporum canis, microsporum dipseum, etc.);
Paracoccidioides (eg, paracoccidioides brasiliensis);
Penicillium (eg, Penicillium manefei);
Fialophora;
Pneumocystis (eg, Pneumocystis jiroveci);
Pseudoaleseria (eg pseudoaleseria boise);
Saccharomyces (eg Saccharomyces cerevisiae);
Skoplariopsis;
Sporotrix (eg Sporotrix Schenky, etc.);
Trichophyton (for example, Trichophyton Mentagrophytes, Trichophyton rubrum, etc.).
In another embodiment, the compound of formula (I) may be useful as a prophylactic or therapeutic agent for the following fungal infections and infectious diseases caused by fungi.
Aspergillus (for example, Aspergillus clubatas, Aspergillus flavus, Aspergillus fumigatus, Aspergillus nidulans, Aspergillus niger, Aspergillus tereus, Aspergillus vesilcolor);
Candida (e.g. Candida albicans, Candida glabrata, Candida guilliermondi, Candida kefir, Candida crusei, Candida paracylosis, Candida stellatoidea, Candida tropicalis, Candida utilis, etc.);
Pneumocystis (eg, Pneumocystis jiroveci);
Pseudoaleseria (eg pseudoaleseria boise);
Saccharomyces (eg Saccharomyces cerevisiae).
上記の真菌は、皮膚、眼、毛髪、爪、口腔粘膜、胃腸管、気管支、肺、心内膜、脳、髄膜、泌尿器、腟部、口腔、眼球、全身、腎臓、心臓、外耳孔、骨、鼻腔、副鼻腔、脾臓、肝臓、皮下組織、リンパ管、胃腸、関節、筋肉、腱、肺内間質性形質細胞、血管等に種々の感染症を引き起こすことがよく知られている。
The above fungi are skin, eyes, hair, nails, oral mucosa, gastrointestinal tract, bronchial, lung, endocardium, brain, meninges, urinary organs, buttocks, oral cavity, eyeball, whole body, kidney, heart, ear canal, It is well known to cause various infectious diseases in bone, nasal cavity, sinus cavity, spleen, liver, subcutaneous tissue, lymphatic vessel, gastrointestinal tract, joint, muscle, tendon, stromal plasma cell in lung, blood vessel and the like.
従って、式(I)の化合物は、種々の感染症、例えば皮膚糸状菌症(例えば白癬症等)、でん風、カンジダ症、ゲオトリクム症、砂毛症、アスペルギルス症、ペニシリウム症、スポロトリクス症、クロモミセス症、コクシジオイデス症、ヒストプラスマ症、ブラストミセス症、パラコクシジオイデス症、プソイドアレシェリアシス、足菌腫、真菌性角膜炎、耳真菌症、ニューモシスティス症、真菌血症等の予防及び/又は治療に有用である。
Accordingly, the compounds of formula (I) can be used in various infectious diseases such as dermatophytosis (e.g. ringworm, etc.), gallbladder, candidiasis, geotrichumosis, sandy hair, aspergillosis, penicillosis, sporotricosis, Prevention and / or prevention of chromomises, coccidioidomycosis, histoplasmosis, blastosomiasis, paracoccidioidomycosis, pseudoaleseriosis, foot mycosis, fungal keratitis, otomycosis, pneumocystis, fungiemia, etc. Useful for treatment.
式(I)の化合物又はその塩の1種又は2種以上を有効成分として含有する医薬組成物は、当分野において通常用いられている賦形剤、即ち、薬剤用賦形剤や薬剤用担体等を用いて、通常使用されている方法によって調製することができる。
投与は錠剤、丸剤、カプセル剤、顆粒剤、散剤、液剤等による経口投与、又は、関節内、静脈内、筋肉内等の注射剤、坐剤、点眼剤、眼軟膏、経皮用液剤、軟膏剤、経皮用貼付剤、経粘膜液剤、経粘膜貼付剤、吸入剤等による非経口投与のいずれの形態であってもよい。 A pharmaceutical composition containing one or more compounds of the formula (I) or a salt thereof as an active ingredient is an excipient normally used in the art, that is, a pharmaceutical excipient or a pharmaceutical carrier. Can be prepared by a commonly used method.
Administration is orally by tablets, pills, capsules, granules, powders, solutions, etc., or injections such as intra-articular, intravenous, intramuscular, suppositories, eye drops, ophthalmic ointments, transdermal solutions, Any form of parenteral administration such as an ointment, a transdermal patch, a transmucosal liquid, a transmucosal patch, and an inhalant may be used.
投与は錠剤、丸剤、カプセル剤、顆粒剤、散剤、液剤等による経口投与、又は、関節内、静脈内、筋肉内等の注射剤、坐剤、点眼剤、眼軟膏、経皮用液剤、軟膏剤、経皮用貼付剤、経粘膜液剤、経粘膜貼付剤、吸入剤等による非経口投与のいずれの形態であってもよい。 A pharmaceutical composition containing one or more compounds of the formula (I) or a salt thereof as an active ingredient is an excipient normally used in the art, that is, a pharmaceutical excipient or a pharmaceutical carrier. Can be prepared by a commonly used method.
Administration is orally by tablets, pills, capsules, granules, powders, solutions, etc., or injections such as intra-articular, intravenous, intramuscular, suppositories, eye drops, ophthalmic ointments, transdermal solutions, Any form of parenteral administration such as an ointment, a transdermal patch, a transmucosal liquid, a transmucosal patch, and an inhalant may be used.
経口投与のための固体組成物としては、錠剤、散剤、顆粒剤等が用いられる。このような固体組成物においては、1種又は2種以上の有効成分を、少なくとも1種の不活性な賦形剤、例えば乳糖、マンニトール、ブドウ糖、ヒドロキシプロピルセルロース、微結晶セルロース、デンプン、ポリビニルピロリドン、及び/又はメタケイ酸アルミン酸マグネシウム等と混合される。組成物は、常法に従って、不活性な添加剤、例えばステアリン酸マグネシウムのような滑沢剤やカルボキシメチルスターチナトリウム等のような崩壊剤、安定化剤、溶解補助剤を含有していてもよい。錠剤又は丸剤は必要により糖衣又は胃溶性若しくは腸溶性物質のフィルムで被膜してもよい。
経口投与のための液体組成物は、薬剤的に許容される乳濁剤、溶液剤、懸濁剤、シロップ剤又はエリキシル剤等を含み、一般的に用いられる不活性な希釈剤、例えば精製水又はエタノールを含む。当該液体組成物は不活性な希釈剤以外に可溶化剤、湿潤剤、懸濁剤のような補助剤、甘味剤、風味剤、芳香剤、防腐剤を含有していてもよい。 As a solid composition for oral administration, tablets, powders, granules and the like are used. In such solid compositions, one or more active ingredients are combined with at least one inert excipient such as lactose, mannitol, glucose, hydroxypropylcellulose, microcrystalline cellulose, starch, polyvinylpyrrolidone. And / or mixed with magnesium aluminate metasilicate. The composition may contain an inert additive, for example, a lubricant such as magnesium stearate, a disintegrant such as sodium carboxymethyl starch, a stabilizer, and a solubilizing agent according to a conventional method. . If necessary, tablets or pills may be coated with a sugar coating or a film of a gastric or enteric substance.
Liquid compositions for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups or elixirs and the like, and commonly used inert diluents such as purified water. Or it contains ethanol. The liquid composition may contain solubilizers, wetting agents, auxiliaries such as suspending agents, sweeteners, flavors, fragrances and preservatives in addition to the inert diluent.
経口投与のための液体組成物は、薬剤的に許容される乳濁剤、溶液剤、懸濁剤、シロップ剤又はエリキシル剤等を含み、一般的に用いられる不活性な希釈剤、例えば精製水又はエタノールを含む。当該液体組成物は不活性な希釈剤以外に可溶化剤、湿潤剤、懸濁剤のような補助剤、甘味剤、風味剤、芳香剤、防腐剤を含有していてもよい。 As a solid composition for oral administration, tablets, powders, granules and the like are used. In such solid compositions, one or more active ingredients are combined with at least one inert excipient such as lactose, mannitol, glucose, hydroxypropylcellulose, microcrystalline cellulose, starch, polyvinylpyrrolidone. And / or mixed with magnesium aluminate metasilicate. The composition may contain an inert additive, for example, a lubricant such as magnesium stearate, a disintegrant such as sodium carboxymethyl starch, a stabilizer, and a solubilizing agent according to a conventional method. . If necessary, tablets or pills may be coated with a sugar coating or a film of a gastric or enteric substance.
Liquid compositions for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups or elixirs and the like, and commonly used inert diluents such as purified water. Or it contains ethanol. The liquid composition may contain solubilizers, wetting agents, auxiliaries such as suspending agents, sweeteners, flavors, fragrances and preservatives in addition to the inert diluent.
非経口投与のための注射剤は、無菌の水性又は非水性の溶液剤、懸濁剤又は乳濁剤を含有する。水性の溶剤としては、例えば注射用蒸留水又は生理食塩液が含まれる。非水性の溶剤としては、例えばプロピレングリコール、ポリエチレングリコール又はオリーブ油のような植物油、エタノールのようなアルコール類、又はポリソルベート80(局方名)等がある。このような組成物は、さらに等張化剤、防腐剤、湿潤剤、乳化剤、分散剤、安定化剤、又は溶解補助剤を含んでもよい。これらは例えばバクテリア保留フィルターを通す濾過、殺菌剤の配合又は照射によって無菌化される。また、これらは無菌の固体組成物を製造し、使用前に無菌水又は無菌の注射用溶媒に溶解又は懸濁して使用することもできる。
The injection for parenteral administration contains a sterile aqueous or non-aqueous solution, suspension or emulsion. Examples of the aqueous solvent include distilled water for injection or physiological saline. Examples of non-aqueous solvents include propylene glycol, polyethylene glycol or vegetable oils such as olive oil, alcohols such as ethanol, or polysorbate 80 (a pharmacopeia name). Such compositions may further contain isotonic agents, preservatives, wetting agents, emulsifiers, dispersants, stabilizers, or solubilizing agents. These are sterilized by, for example, filtration through a bacteria-retaining filter, blending with a bactericide or irradiation. These can also be used by producing a sterile solid composition and dissolving or suspending it in sterile water or a sterile solvent for injection before use.
外用剤としては、軟膏剤、硬膏剤、クリーム剤、ゼリー剤、パップ剤、噴霧剤、ローション剤、点眼剤、眼軟膏等を包含する。一般に用いられる軟膏基剤、ローション基剤、水性又は非水性の液剤、懸濁剤、乳剤等を含有する。例えば、軟膏又はローション基剤としては、ポリエチレングリコール、プロピレングリコール、白色ワセリン、サラシミツロウ、ポリオキシエチレン硬化ヒマシ油、モノステアリン酸グリセリン、ステアリルアルコール、セチルアルコール、ラウロマクロゴール、セスキオレイン酸ソルビタン等が挙げられる。
External preparations include ointments, plasters, creams, jellies, poultices, sprays, lotions, eye drops, eye ointments and the like. Contains commonly used ointment bases, lotion bases, aqueous or non-aqueous solutions, suspensions, emulsions, and the like. For example, ointments or lotion bases include polyethylene glycol, propylene glycol, white petrolatum, white beeswax, polyoxyethylene hydrogenated castor oil, glyceryl monostearate, stearyl alcohol, cetyl alcohol, lauromacrogol, sorbitan sesquioleate, etc. Can be mentioned.
吸入剤や経鼻剤等の経粘膜剤は固体、液体又は半固体状のものが用いられ、従来公知の方法に従って製造することができる。例えば公知の賦形剤や、更に、pH調整剤、防腐剤、界面活性剤、滑沢剤、安定剤や増粘剤等が適宜添加されていてもよい。投与は、適当な吸入又は吹送のためのデバイスを使用することができる。例えば、計量投与吸入デバイス等の公知のデバイスや噴霧器を使用して、化合物を単独で又は処方された混合物の粉末として、もしくは医薬的に許容し得る担体と組み合わせて溶液又は懸濁液として投与することができる。乾燥粉末吸入器等は、単回又は多数回の投与用のものであってもよく、乾燥粉末又は粉末含有カプセルを利用することができる。あるいは、適当な駆出剤、例えば、クロロフルオロアルカン、ヒドロフルオロアルカン又は二酸化炭素等の好適な気体を使用した加圧エアゾールスプレー等の形態であってもよい。
A transmucosal agent such as an inhalant or a nasal agent is used in a solid, liquid, or semi-solid state, and can be produced according to a conventionally known method. For example, known excipients, and further pH adjusters, preservatives, surfactants, lubricants, stabilizers, thickeners and the like may be appropriately added. For administration, an appropriate device for inhalation or insufflation can be used. For example, using a known device such as a metered dose inhalation device or a nebulizer, the compound is administered alone or as a powder in a formulated mixture or as a solution or suspension in combination with a pharmaceutically acceptable carrier. be able to. The dry powder inhaler or the like may be for single or multiple administration, and a dry powder or a powder-containing capsule can be used. Alternatively, it may be in the form of a pressurized aerosol spray using a suitable propellant, for example, a suitable gas such as chlorofluoroalkane, hydrofluoroalkane or carbon dioxide.
特に真菌感染症の予防及び/又は治療の場合には、前述の非経口投与のための注射剤又は吸入剤や経鼻剤などの経粘膜剤が好ましい。
In particular, in the case of prevention and / or treatment of fungal infections, the aforementioned injections for parenteral administration or transmucosal agents such as inhalants and nasal agents are preferred.
通常経口投与の場合、1日の投与量は、体重当たり約0.001~100 mg/kg、好ましくは10~30 mg/kg、更に好ましくは0.1~10 mg/kgが適当であり、これを1回であるいは2回~4回に分けて投与する。静脈内投与される場合は、1日の投与量は、体重当たり約0.0001~10 mg/kgが適当で、1日1回~複数回に分けて投与する。また、経粘膜剤としては、体重当たり約0.001~100 mg/kgを1日1回~複数回に分けて投与する。投与量は症状、年令、性別等を考慮して個々の場合に応じて適宜決定される。
In general, in the case of oral administration, the appropriate daily dosage is about 0.001 to 100 mg / kg, preferably 10 to 30 mg / kg, more preferably 0.1 to 10 mg / kg per body weight. Or in 2 to 4 divided doses. When administered intravenously, the appropriate daily dose is about 0.0001 to 10 mg / kg per body weight, and is administered once to several times a day. As a transmucosal agent, about 0.001 to 100 mg / kg per body weight is administered once to several times a day. The dose is appropriately determined according to individual cases in consideration of symptoms, age, sex, and the like.
式(I)の化合物は、前述の式(I)の化合物が有効性を示すと考えられる疾患の種々の予防剤又は治療剤と併用することができる。当該併用は、同時投与、或いは別個に連続して、若しくは所望の時間間隔をおいて投与してもよい。同時投与製剤は、配合剤であっても別個に製剤化されていてもよい。
The compound of formula (I) can be used in combination with various prophylactic or therapeutic agents for diseases for which the compound of formula (I) is considered to be effective. The combination may be administered simultaneously, separately separately, or at desired time intervals. The simultaneous administration preparation may be a compounding agent or may be separately formulated.
以下、実施例に基づき、式(I)の化合物の製造法をさらに詳細に説明する。なお、本発明は、下記実施例に記載の化合物に限定されるものではない。また、原料化合物の製法を製造例に、それぞれ示す。また、式(I)の化合物の製造法は、以下に示される具体的実施例の製造法のみに限定されるものではなく、式(I)の化合物はこれらの製造法の組み合わせ、あるいは当業者に自明である方法によっても製造されうる。
Hereinafter, based on an Example, the manufacturing method of the compound of Formula (I) is demonstrated in detail. In addition, this invention is not limited to the compound as described in the following Example. Moreover, the manufacturing method of a raw material compound is shown in a manufacture example, respectively. Further, the production method of the compound of the formula (I) is not limited to the production methods of the specific examples shown below. It can also be produced by methods that are obvious.
また、本明細書、実施例、製造例及び後記表中において、以下の略号を用いることがある。
Ag2O=酸化銀、AgOTf =銀トリフルオロメタンスルホネート、BINAP=1,1'-ビナフタレン-2,2'-ジイルビス(ジフェニルホスフィン)、BOC又はboc=tert-ブチルオキシカルボニル、BOP=ヘキサフルオリン酸ベンゾトリアゾール-1-イルオキシトリス(ジメチルアミノ)ホスホニウム、Boc2O=二炭酸ジtert-ブチル、Bn=ベンジル、DBU=1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン、CH2Cl2又はDCM=ジクロロメタン、CO=一酸化炭素、DCE=ジクロロエタン、DIPEA=N,N-ジイソプロピルエチルアミン、DMAc=N,N-ジメチルアセトアミド、DMF=N,N-ジメチルホルムアミド、DMI=1,3-ジメチル‐2-イミダゾリジノン、DMSO=ジメチルスルホキシド、DPPA=ジフェニルリン酸アジド、DPPP=1,3-ビス(ジフェニルホスフィノ)プロパン、EDCI、WSC=3-エチル-1-(3-ジメチルアミノプロピル)カルボジイミド、Et3N=トリエチルアミン、EtOAc=酢酸エチル、EtOH=エタノール、FMOC =フルオレニルメチルオキシカルボニル、HBTU=o-ベンゾトリアゾール-N,N,N',N'-テトラメチル-ウロニウム-ヘキサフルオロホスフェート、HOBt=1-ヒドロキシベンゾトリアゾール、Hex=ヘキサン、K3PO4=トリポタシウムホスフェート、KOAc=酢酸カリウム、LHMDS=リチウムビストリメチルシリルアミド、MeI=メチルアイオダイド、MS3A=モレキュラーシーブス3オングストローム、MeCN=アセトニトリル、MgSO4=無水硫酸マグネシウム、NIS=N-ヨードスクシンイミド、NMM=N-メチルモルホリン、NMP=N-メチルピロリドン、NaBH3CN=シアン化水素化ホウ素ナトリウム、NaBH(OAc)3=トリアセトキシ水素化ホウ素ナトリウム、NaBH4=水素化ホウ素ナトリウム、Na2S2O3=チオ硫酸ナトリウム、Na2SO4=無水硫酸ナトリウム、NaBH(OAc)3=ナトリウムトリアセトキシボロハイドライド、NaHCO3=炭酸水素ナトリウム、NaOtBu=ナトリウムtertブトキシド、Pd/C=パラジウム炭素、Pd2(dba)3=トリス(ジベンジリデンアセトン)ジパラジウム(0)、PdCl2(dppf)CH2CL2=1,1'-ビス(ジフェニルホスフィノ)フェロセン-パラジウム(II)ジクロリド ジクロロメタンコンプレックス、TBAI=テトラブチルアンモニウムアイオダイド、TEA又はEt3N=トリエチルアミン、TFA=トリフルオロ酢酸、THF=テトラヒドロフラン、TfOH=トリフルオロメチルスルホン酸、AcOH=酢酸、brine=飽和食塩水、n-BuOH=ノルマルブタノール、n-=ノルマル、sat.=飽和、tert-=ターシャリー。 Moreover, the following abbreviations may be used in this specification, an Example, a manufacture example, and a postscript table | surface.
Ag 2 O = silver oxide, AgOTf = silver trifluoromethanesulfonate, BINAP = 1,1′-binaphthalene-2,2′-diylbis (diphenylphosphine), BOC or boc = tert-butyloxycarbonyl, BOP = hexafluoric acid Benzotriazol-1-yloxytris (dimethylamino) phosphonium, Boc 2 O = ditert-butyl dicarbonate, Bn = benzyl, DBU = 1,8-diazabicyclo [5.4.0] undec-7-ene, CH 2 Cl 2 or DCM = dichloromethane, CO = carbon monoxide, DCE = dichloroethane, DIPEA = N, N-diisopropylethylamine, DMAc = N, N-dimethylacetamide, DMF = N, N-dimethylformamide, DMI = 1,3-dimethyl -2-imidazolidinone, DMSO = dimethyl sulfoxide, DPPA = diphenyl phosphate azide, DPPP = 1,3-bis (diphenylphosphino) propane, EDCI, WSC = 3-ethyl-1- (3-dimethylaminopropyl) Carbodiimi Et 3 N = triethylamine, EtOAc = ethyl acetate, EtOH = ethanol, FMOC = fluorenylmethyloxycarbonyl, HBTU = o-benzotriazole-N, N, N ′, N′-tetramethyl-uronium-hexafluoro Phosphate, HOBt = 1-hydroxybenzotriazole, Hex = hexane, K 3 PO 4 = Tripotassium phosphate, KOAc = potassium acetate, LHMDS = lithium bistrimethylsilylamide, MeI = methyl iodide, MS3A = molecular sieve 3 angstrom, MeCN = Acetonitrile, MgSO 4 = anhydrous magnesium sulfate, NIS = N-iodosuccinimide, NMM = N-methylmorpholine, NMP = N-methylpyrrolidone, NaBH 3 CN = sodium cyanoborohydride, NaBH (OAc) 3 = triacetoxyborohydride sodium, NaBH 4 = sodium borohydride, Na 2 S 2 O 3 = thiosulfate isocyanatomethyl Um, Na 2 SO 4 = sodium sulfate anhydride, NaBH (OAc) 3 = sodium triacetoxyborohydride, NaHCO 3 = sodium hydrogen carbonate, NaOtBu = sodium tert-butoxide, Pd / C = palladium on carbon, Pd 2 (dba) 3 = Tris (dibenzylideneacetone) dipalladium (0), PdCl 2 (dppf) CH 2 CL 2 = 1,1′-bis (diphenylphosphino) ferrocene-palladium (II) dichloride dichloromethane complex, TBAI = tetrabutylammonium iodide , TEA or Et 3 N = triethylamine, TFA = trifluoroacetic acid, THF = tetrahydrofuran, TfOH = trifluoromethylsulfonic acid, AcOH = acetic acid, brine = saturated saline, n-BuOH = normal butanol, n- = normal, sat . = Saturated, tert- = tertiary.
Ag2O=酸化銀、AgOTf =銀トリフルオロメタンスルホネート、BINAP=1,1'-ビナフタレン-2,2'-ジイルビス(ジフェニルホスフィン)、BOC又はboc=tert-ブチルオキシカルボニル、BOP=ヘキサフルオリン酸ベンゾトリアゾール-1-イルオキシトリス(ジメチルアミノ)ホスホニウム、Boc2O=二炭酸ジtert-ブチル、Bn=ベンジル、DBU=1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン、CH2Cl2又はDCM=ジクロロメタン、CO=一酸化炭素、DCE=ジクロロエタン、DIPEA=N,N-ジイソプロピルエチルアミン、DMAc=N,N-ジメチルアセトアミド、DMF=N,N-ジメチルホルムアミド、DMI=1,3-ジメチル‐2-イミダゾリジノン、DMSO=ジメチルスルホキシド、DPPA=ジフェニルリン酸アジド、DPPP=1,3-ビス(ジフェニルホスフィノ)プロパン、EDCI、WSC=3-エチル-1-(3-ジメチルアミノプロピル)カルボジイミド、Et3N=トリエチルアミン、EtOAc=酢酸エチル、EtOH=エタノール、FMOC =フルオレニルメチルオキシカルボニル、HBTU=o-ベンゾトリアゾール-N,N,N',N'-テトラメチル-ウロニウム-ヘキサフルオロホスフェート、HOBt=1-ヒドロキシベンゾトリアゾール、Hex=ヘキサン、K3PO4=トリポタシウムホスフェート、KOAc=酢酸カリウム、LHMDS=リチウムビストリメチルシリルアミド、MeI=メチルアイオダイド、MS3A=モレキュラーシーブス3オングストローム、MeCN=アセトニトリル、MgSO4=無水硫酸マグネシウム、NIS=N-ヨードスクシンイミド、NMM=N-メチルモルホリン、NMP=N-メチルピロリドン、NaBH3CN=シアン化水素化ホウ素ナトリウム、NaBH(OAc)3=トリアセトキシ水素化ホウ素ナトリウム、NaBH4=水素化ホウ素ナトリウム、Na2S2O3=チオ硫酸ナトリウム、Na2SO4=無水硫酸ナトリウム、NaBH(OAc)3=ナトリウムトリアセトキシボロハイドライド、NaHCO3=炭酸水素ナトリウム、NaOtBu=ナトリウムtertブトキシド、Pd/C=パラジウム炭素、Pd2(dba)3=トリス(ジベンジリデンアセトン)ジパラジウム(0)、PdCl2(dppf)CH2CL2=1,1'-ビス(ジフェニルホスフィノ)フェロセン-パラジウム(II)ジクロリド ジクロロメタンコンプレックス、TBAI=テトラブチルアンモニウムアイオダイド、TEA又はEt3N=トリエチルアミン、TFA=トリフルオロ酢酸、THF=テトラヒドロフラン、TfOH=トリフルオロメチルスルホン酸、AcOH=酢酸、brine=飽和食塩水、n-BuOH=ノルマルブタノール、n-=ノルマル、sat.=飽和、tert-=ターシャリー。 Moreover, the following abbreviations may be used in this specification, an Example, a manufacture example, and a postscript table | surface.
Ag 2 O = silver oxide, AgOTf = silver trifluoromethanesulfonate, BINAP = 1,1′-binaphthalene-2,2′-diylbis (diphenylphosphine), BOC or boc = tert-butyloxycarbonyl, BOP = hexafluoric acid Benzotriazol-1-yloxytris (dimethylamino) phosphonium, Boc 2 O = ditert-butyl dicarbonate, Bn = benzyl, DBU = 1,8-diazabicyclo [5.4.0] undec-7-ene, CH 2 Cl 2 or DCM = dichloromethane, CO = carbon monoxide, DCE = dichloroethane, DIPEA = N, N-diisopropylethylamine, DMAc = N, N-dimethylacetamide, DMF = N, N-dimethylformamide, DMI = 1,3-dimethyl -2-imidazolidinone, DMSO = dimethyl sulfoxide, DPPA = diphenyl phosphate azide, DPPP = 1,3-bis (diphenylphosphino) propane, EDCI, WSC = 3-ethyl-1- (3-dimethylaminopropyl) Carbodiimi Et 3 N = triethylamine, EtOAc = ethyl acetate, EtOH = ethanol, FMOC = fluorenylmethyloxycarbonyl, HBTU = o-benzotriazole-N, N, N ′, N′-tetramethyl-uronium-hexafluoro Phosphate, HOBt = 1-hydroxybenzotriazole, Hex = hexane, K 3 PO 4 = Tripotassium phosphate, KOAc = potassium acetate, LHMDS = lithium bistrimethylsilylamide, MeI = methyl iodide, MS3A = molecular sieve 3 angstrom, MeCN = Acetonitrile, MgSO 4 = anhydrous magnesium sulfate, NIS = N-iodosuccinimide, NMM = N-methylmorpholine, NMP = N-methylpyrrolidone, NaBH 3 CN = sodium cyanoborohydride, NaBH (OAc) 3 = triacetoxyborohydride sodium, NaBH 4 = sodium borohydride, Na 2 S 2 O 3 = thiosulfate isocyanatomethyl Um, Na 2 SO 4 = sodium sulfate anhydride, NaBH (OAc) 3 = sodium triacetoxyborohydride, NaHCO 3 = sodium hydrogen carbonate, NaOtBu = sodium tert-butoxide, Pd / C = palladium on carbon, Pd 2 (dba) 3 = Tris (dibenzylideneacetone) dipalladium (0), PdCl 2 (dppf) CH 2 CL 2 = 1,1′-bis (diphenylphosphino) ferrocene-palladium (II) dichloride dichloromethane complex, TBAI = tetrabutylammonium iodide , TEA or Et 3 N = triethylamine, TFA = trifluoroacetic acid, THF = tetrahydrofuran, TfOH = trifluoromethylsulfonic acid, AcOH = acetic acid, brine = saturated saline, n-BuOH = normal butanol, n- = normal, sat . = Saturated, tert- = tertiary.
また、特に構造式において、以下の略号を用いることがある。
BOC=ターシャリーブトキシカルボニル、Bn=ベンジル、Et=エチル、FMOC=フルオレニルメチルオキシカルボニル、Me=メチル、OMe=メトキシ、OBn=ベンジルオキシ、OEt=エトキシ、OMe=メトキシ、OiPr=イソプロポキシ、OnBu=ノルマルブチルオキシ、OnPn=ノルマルペンチルオキシ、OnPr=ノルマルプロポキシ、OtBu=ターシャリーブチルオキシ、Tf=トリフルオロメタンスルホナート、Cbz=ベンジルオキシカルボニル、iPr=イソプロピル、nBu=ノルマルブチル、nHep=ノルマルヘプチル、nHex=ノルマルヘキシル、nOct=ノルマルオクチル、nPn=ノルマルペンチル、nPr=ノルマルプロピル、tBu-=ターシャリーブチル。
下表中、(HCl)は塩酸塩として単離したことを示す。 In particular, the following abbreviations may be used in structural formulas.
BOC = tertiary butoxycarbonyl, Bn = benzyl, Et = ethyl, FMOC = fluorenylmethyloxycarbonyl, Me = methyl, OMe = methoxy, OBn = benzyloxy, OEt = ethoxy, OMe = methoxy, OiPr = isopropoxy, OnBu = normal butyloxy, OnPn = normal pentyloxy, OnPr = normal propoxy, OtBu = tertiary butyloxy, Tf = trifluoromethanesulfonate, Cbz = benzyloxycarbonyl, iPr = isopropyl, nBu = normal butyl, nHep = normal heptyl , NHex = normal hexyl, nOct = normal octyl, nPn = normal pentyl, nPr = normal propyl, tBu- = tertiary butyl.
In the table below, (HCl) indicates isolation as a hydrochloride salt.
BOC=ターシャリーブトキシカルボニル、Bn=ベンジル、Et=エチル、FMOC=フルオレニルメチルオキシカルボニル、Me=メチル、OMe=メトキシ、OBn=ベンジルオキシ、OEt=エトキシ、OMe=メトキシ、OiPr=イソプロポキシ、OnBu=ノルマルブチルオキシ、OnPn=ノルマルペンチルオキシ、OnPr=ノルマルプロポキシ、OtBu=ターシャリーブチルオキシ、Tf=トリフルオロメタンスルホナート、Cbz=ベンジルオキシカルボニル、iPr=イソプロピル、nBu=ノルマルブチル、nHep=ノルマルヘプチル、nHex=ノルマルヘキシル、nOct=ノルマルオクチル、nPn=ノルマルペンチル、nPr=ノルマルプロピル、tBu-=ターシャリーブチル。
下表中、(HCl)は塩酸塩として単離したことを示す。 In particular, the following abbreviations may be used in structural formulas.
BOC = tertiary butoxycarbonyl, Bn = benzyl, Et = ethyl, FMOC = fluorenylmethyloxycarbonyl, Me = methyl, OMe = methoxy, OBn = benzyloxy, OEt = ethoxy, OMe = methoxy, OiPr = isopropoxy, OnBu = normal butyloxy, OnPn = normal pentyloxy, OnPr = normal propoxy, OtBu = tertiary butyloxy, Tf = trifluoromethanesulfonate, Cbz = benzyloxycarbonyl, iPr = isopropyl, nBu = normal butyl, nHep = normal heptyl , NHex = normal hexyl, nOct = normal octyl, nPn = normal pentyl, nPr = normal propyl, tBu- = tertiary butyl.
In the table below, (HCl) indicates isolation as a hydrochloride salt.
製造例1
6-クロロ-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]-1,3-ベンゾチアゾール(0.501 g)のトルエン(5 mL)溶液に、4-シクロヘキシル-4-メトキシピペリジン(343 mg)、Pd2(dba)3 (66.3 mg)、2'-(ジクロロヘキシルホスホノ)-N,N-ジメチルビフェニル-2-アミン(42.7 mg)及びNaOtBu (0.195 g)を加え、マイクロウェーブ中、160℃で1時間照射した。反応混合物中に、0.1M HClを加えて数分間攪拌した。有機層は、brineで洗浄し、MgSO4で乾燥した後、減圧下濃縮した。シリカゲルカラムクロマトグラフィー (溶出液; Hexane:CHCl3 = 50:50から30:70まで)で精製し、6-(4-シクロヘキシル-4-メトキシピペリジン-1-イル)-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]-1,3-ベンゾチアゾール(734 mg)を得た。 Production Example 1
To a solution of 6-chloro-2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] -1,3-benzothiazole (0.501 g) in toluene (5 mL) was added 4-cyclohexyl-4-methoxypiperidine. (343 mg), Pd 2 (dba) 3 (66.3 mg), 2 ′-(dichlorohexylphosphono) -N, N-dimethylbiphenyl-2-amine (42.7 mg) and NaOtBu (0.195 g) were added. Irradiated at 160 ° C for 1 hour in the wave. 0.1M HCl was added to the reaction mixture and stirred for several minutes. The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. Purification by silica gel column chromatography (eluent; Hexane: CHCl 3 = 50: 50 to 30:70), 6- (4-cyclohexyl-4-methoxypiperidin-1-yl) -2- [4- (tetrahydro -2H-pyran-2-yloxy) phenyl] -1,3-benzothiazole (734 mg) was obtained.
6-クロロ-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]-1,3-ベンゾチアゾール(0.501 g)のトルエン(5 mL)溶液に、4-シクロヘキシル-4-メトキシピペリジン(343 mg)、Pd2(dba)3 (66.3 mg)、2'-(ジクロロヘキシルホスホノ)-N,N-ジメチルビフェニル-2-アミン(42.7 mg)及びNaOtBu (0.195 g)を加え、マイクロウェーブ中、160℃で1時間照射した。反応混合物中に、0.1M HClを加えて数分間攪拌した。有機層は、brineで洗浄し、MgSO4で乾燥した後、減圧下濃縮した。シリカゲルカラムクロマトグラフィー (溶出液; Hexane:CHCl3 = 50:50から30:70まで)で精製し、6-(4-シクロヘキシル-4-メトキシピペリジン-1-イル)-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]-1,3-ベンゾチアゾール(734 mg)を得た。 Production Example 1
To a solution of 6-chloro-2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] -1,3-benzothiazole (0.501 g) in toluene (5 mL) was added 4-cyclohexyl-4-methoxypiperidine. (343 mg), Pd 2 (dba) 3 (66.3 mg), 2 ′-(dichlorohexylphosphono) -N, N-dimethylbiphenyl-2-amine (42.7 mg) and NaOtBu (0.195 g) were added. Irradiated at 160 ° C for 1 hour in the wave. 0.1M HCl was added to the reaction mixture and stirred for several minutes. The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. Purification by silica gel column chromatography (eluent; Hexane: CHCl 3 = 50: 50 to 30:70), 6- (4-cyclohexyl-4-methoxypiperidin-1-yl) -2- [4- (tetrahydro -2H-pyran-2-yloxy) phenyl] -1,3-benzothiazole (734 mg) was obtained.
製造例1の方法と同様にして、後記表に示す製造例1-1から製造例1-7の化合物を製造した。
In the same manner as in Production Example 1, the compounds of Production Example 1-1 to Production Example 1-7 shown in the table below were produced.
製造例2
メチル 4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}-3-(1,2,3,6-テトラヒドロピリジン-4-イル)ビフェニル-4-カルボキシレート(201 mg)を CH2Cl2(4.26 mL)とMeOH(2.13 mL)の混合溶媒に溶解し、氷冷下で1,4-ジオキサン-2,5-ジオール(79.8 mg)、NaBH(OAc)3(140 mg)を加え、室温で1時間撹拌した。反応液に、0℃下sat.NaHCO3水溶液を加え、CHCl3で抽出した。有機層はbrineで洗浄し、MgSO4で乾燥した後、減圧下濃縮した。得られた残渣をMeCNで洗浄し、メチル 3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-4-カルボキシレート(164 mg)を白色固体として得た。 Production Example 2
Methyl 4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazol-6-yl} -3- (1,2 , 3,6-Tetrahydropyridin-4-yl) biphenyl-4-carboxylate (201 mg) dissolved in a mixed solvent of CH 2 Cl 2 (4.26 mL) and MeOH (2.13 mL), 4-Dioxane-2,5-diol (79.8 mg) and NaBH (OAc) 3 (140 mg) were added, and the mixture was stirred at room temperature for 1 hour. To the reaction solution was added sat.NaHCO 3 aqueous solution at 0 ° C., and the mixture was extracted with CHCl 3 . The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The resulting residue was washed with MeCN and methyl 3- [1- (2-hydroxyethyl) -1,2,3,6-tetrahydropyridin-4-yl] -4 '-{2- [4- (2 -Phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazol-6-yl} biphenyl-4-carboxylate (164 mg) was obtained as a white solid.
メチル 4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}-3-(1,2,3,6-テトラヒドロピリジン-4-イル)ビフェニル-4-カルボキシレート(201 mg)を CH2Cl2(4.26 mL)とMeOH(2.13 mL)の混合溶媒に溶解し、氷冷下で1,4-ジオキサン-2,5-ジオール(79.8 mg)、NaBH(OAc)3(140 mg)を加え、室温で1時間撹拌した。反応液に、0℃下sat.NaHCO3水溶液を加え、CHCl3で抽出した。有機層はbrineで洗浄し、MgSO4で乾燥した後、減圧下濃縮した。得られた残渣をMeCNで洗浄し、メチル 3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-4-カルボキシレート(164 mg)を白色固体として得た。 Production Example 2
Methyl 4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazol-6-yl} -3- (1,2 , 3,6-Tetrahydropyridin-4-yl) biphenyl-4-carboxylate (201 mg) dissolved in a mixed solvent of CH 2 Cl 2 (4.26 mL) and MeOH (2.13 mL), 4-Dioxane-2,5-diol (79.8 mg) and NaBH (OAc) 3 (140 mg) were added, and the mixture was stirred at room temperature for 1 hour. To the reaction solution was added sat.NaHCO 3 aqueous solution at 0 ° C., and the mixture was extracted with CHCl 3 . The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The resulting residue was washed with MeCN and methyl 3- [1- (2-hydroxyethyl) -1,2,3,6-tetrahydropyridin-4-yl] -4 '-{2- [4- (2 -Phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazol-6-yl} biphenyl-4-carboxylate (164 mg) was obtained as a white solid.
製造例2の方法と同様にして、後記表に示す製造例2-1から製造例2-48の化合物を製造した。
In the same manner as in Production Example 2, the compounds of Production Example 2-1 to Production Example 2-48 shown in the table below were produced.
製造例3
2-ブロモ-6-(4-ブロモフェニル)イミダゾ[2,1-b][1,3,4]チアジアゾール(54 g)のDMAc (840 mL)溶液に1-(2-フェニルエチル)ピペラジン(34.3 g)、Et3N(36.5 g)を添加し、バス温90℃で3時間攪拌した。反応混合物を放冷した後、3時間攪拌し、得られた析出物を水に懸濁してろ取し、析出物を水、少量のMeCNで洗浄し、6-(4-ブロモフェニル)-2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール(64.7 g)を得た。 Production Example 3
2-Bromo-6- (4-bromophenyl) imidazo [2,1-b] [1,3,4] thiadiazole (54 g) in DMAc (840 mL) solution with 1- (2-phenylethyl) piperazine ( 34.3 g) and Et 3 N (36.5 g) were added, and the mixture was stirred at a bath temperature of 90 ° C. for 3 hours. The reaction mixture was allowed to cool and then stirred for 3 hours. The resulting precipitate was suspended in water and collected by filtration, and the precipitate was washed with water and a small amount of MeCN to give 6- (4-bromophenyl) -2- [4- (2-Phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazole (64.7 g) was obtained.
2-ブロモ-6-(4-ブロモフェニル)イミダゾ[2,1-b][1,3,4]チアジアゾール(54 g)のDMAc (840 mL)溶液に1-(2-フェニルエチル)ピペラジン(34.3 g)、Et3N(36.5 g)を添加し、バス温90℃で3時間攪拌した。反応混合物を放冷した後、3時間攪拌し、得られた析出物を水に懸濁してろ取し、析出物を水、少量のMeCNで洗浄し、6-(4-ブロモフェニル)-2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール(64.7 g)を得た。 Production Example 3
2-Bromo-6- (4-bromophenyl) imidazo [2,1-b] [1,3,4] thiadiazole (54 g) in DMAc (840 mL) solution with 1- (2-phenylethyl) piperazine ( 34.3 g) and Et 3 N (36.5 g) were added, and the mixture was stirred at a bath temperature of 90 ° C. for 3 hours. The reaction mixture was allowed to cool and then stirred for 3 hours. The resulting precipitate was suspended in water and collected by filtration, and the precipitate was washed with water and a small amount of MeCN to give 6- (4-bromophenyl) -2- [4- (2-Phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazole (64.7 g) was obtained.
製造例3の方法と同様にして、後記表に示す製造例3-1から製造例3-4の化合物を製造した。
In the same manner as in Production Example 3, the compounds of Production Example 3-1 to Production Example 3-4 shown in the table below were produced.
製造例4
3-ブロモ-5-(4-ブロモフェニル)-4,5-ジヒドロ-1,2-オキサゾール(4.70 g)のNMP (70.5 mL) 溶液にピペラジン(2.66 g) を加え130℃にて3時間攪拌した。反応液に水、EtOAcを加えて分配した。有機層をbrineで洗浄し、減圧下濃縮して粗生成物を得た。残渣を CH2Cl2に溶解し、Et3N(2.58 mL)、Boc2O(4.37 g)を加え室温で攪拌した。反応混合物に水、EtOAcを加えて抽出し、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー (CHCl3:EtOAc =98:2)にて精製し、tert-ブチル 4-[5-(4-ブロモフェニル)-4,5-ジヒドロ-1,2-オキサゾール-3-イル]ピペラジン-1-カルボキシレート(3.3 g)を得た。 Production Example 4
Piperazine (2.66 g) was added to a solution of 3-bromo-5- (4-bromophenyl) -4,5-dihydro-1,2-oxazole (4.70 g) in NMP (70.5 mL) and stirred at 130 ° C for 3 hours. did. The reaction solution was partitioned by adding water and EtOAc. The organic layer was washed with brine and concentrated under reduced pressure to obtain a crude product. The residue was dissolved in CH 2 Cl 2 , Et 3 N (2.58 mL) and Boc 2 O (4.37 g) were added, and the mixture was stirred at room temperature. The reaction mixture was extracted with water and EtOAc, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (CHCl 3 : EtOAc = 98: 2) and tert-butyl 4- [5- (4-bromophenyl) -4,5-dihydro-1,2-oxazole -3-yl] piperazine-1-carboxylate (3.3 g) was obtained.
3-ブロモ-5-(4-ブロモフェニル)-4,5-ジヒドロ-1,2-オキサゾール(4.70 g)のNMP (70.5 mL) 溶液にピペラジン(2.66 g) を加え130℃にて3時間攪拌した。反応液に水、EtOAcを加えて分配した。有機層をbrineで洗浄し、減圧下濃縮して粗生成物を得た。残渣を CH2Cl2に溶解し、Et3N(2.58 mL)、Boc2O(4.37 g)を加え室温で攪拌した。反応混合物に水、EtOAcを加えて抽出し、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー (CHCl3:EtOAc =98:2)にて精製し、tert-ブチル 4-[5-(4-ブロモフェニル)-4,5-ジヒドロ-1,2-オキサゾール-3-イル]ピペラジン-1-カルボキシレート(3.3 g)を得た。 Production Example 4
Piperazine (2.66 g) was added to a solution of 3-bromo-5- (4-bromophenyl) -4,5-dihydro-1,2-oxazole (4.70 g) in NMP (70.5 mL) and stirred at 130 ° C for 3 hours. did. The reaction solution was partitioned by adding water and EtOAc. The organic layer was washed with brine and concentrated under reduced pressure to obtain a crude product. The residue was dissolved in CH 2 Cl 2 , Et 3 N (2.58 mL) and Boc 2 O (4.37 g) were added, and the mixture was stirred at room temperature. The reaction mixture was extracted with water and EtOAc, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (CHCl 3 : EtOAc = 98: 2) and tert-butyl 4- [5- (4-bromophenyl) -4,5-dihydro-1,2-oxazole -3-yl] piperazine-1-carboxylate (3.3 g) was obtained.
製造例5A
6-(4-ブロモフェニル)-2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール (20 g)、{3-[1-(tert-ブトキシカルボニル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4-(メトキシカルボニル)フェニル}ボロン酸(23.1 g)のジオキサン(600 mL)及び水 (60 mL)混合溶液にK3PO4(22.6g)、1,2,3,4,5-ペンタフェニル-1'-(ジ-tert-ブチルホスフィノ)フェロセン (1.58 g)の混合物を窒素で置換した。Pd2(dba)3 (488.7 mg) を加え、再度系内を減圧にして窒素で置換した。窒素雰囲気下、混合物を100℃で24時間撹拌した。反応液を放冷し、析出物をろ過した。得られた析出物を水(500 mL)に懸濁させてろ過した。得られた白色粉末をMeCN(300 mL)に懸濁させ加熱還流下30分攪拌し、放冷した。析出物をろ過し、少量のアセトニトリルで洗浄し、tert-ブチル 4-[4-(メトキシカルボニル)-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-3-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(25.3 g)を得た。 Production Example 5A
6- (4-Bromophenyl) -2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazole (20 g), {3- [1- (tert-Butoxycarbonyl) -1,2,3,6-tetrahydropyridin-4-yl] -4- (methoxycarbonyl) phenyl} boronic acid (23.1 g) in dioxane (600 mL) and water (60 mL) Mix the mixed solution of K 3 PO 4 (22.6 g), 1,2,3,4,5-pentaphenyl-1 '-(di-tert-butylphosphino) ferrocene (1.58 g) with nitrogen did. Pd 2 (dba) 3 (488.7 mg) was added, and the system was again depressurized and replaced with nitrogen. The mixture was stirred at 100 ° C. for 24 hours under a nitrogen atmosphere. The reaction solution was allowed to cool and the precipitate was filtered. The resulting precipitate was suspended in water (500 mL) and filtered. The obtained white powder was suspended in MeCN (300 mL), stirred under heating to reflux for 30 minutes, and allowed to cool. The precipitate was filtered, washed with a small amount of acetonitrile, and tert-butyl 4- [4- (methoxycarbonyl) -4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2 , 1-b] [1,3,4] thiadiazol-6-yl} biphenyl-3-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (25.3 g) was obtained.
6-(4-ブロモフェニル)-2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール (20 g)、{3-[1-(tert-ブトキシカルボニル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4-(メトキシカルボニル)フェニル}ボロン酸(23.1 g)のジオキサン(600 mL)及び水 (60 mL)混合溶液にK3PO4(22.6g)、1,2,3,4,5-ペンタフェニル-1'-(ジ-tert-ブチルホスフィノ)フェロセン (1.58 g)の混合物を窒素で置換した。Pd2(dba)3 (488.7 mg) を加え、再度系内を減圧にして窒素で置換した。窒素雰囲気下、混合物を100℃で24時間撹拌した。反応液を放冷し、析出物をろ過した。得られた析出物を水(500 mL)に懸濁させてろ過した。得られた白色粉末をMeCN(300 mL)に懸濁させ加熱還流下30分攪拌し、放冷した。析出物をろ過し、少量のアセトニトリルで洗浄し、tert-ブチル 4-[4-(メトキシカルボニル)-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-3-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(25.3 g)を得た。 Production Example 5A
6- (4-Bromophenyl) -2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazole (20 g), {3- [1- (tert-Butoxycarbonyl) -1,2,3,6-tetrahydropyridin-4-yl] -4- (methoxycarbonyl) phenyl} boronic acid (23.1 g) in dioxane (600 mL) and water (60 mL) Mix the mixed solution of K 3 PO 4 (22.6 g), 1,2,3,4,5-pentaphenyl-1 '-(di-tert-butylphosphino) ferrocene (1.58 g) with nitrogen did. Pd 2 (dba) 3 (488.7 mg) was added, and the system was again depressurized and replaced with nitrogen. The mixture was stirred at 100 ° C. for 24 hours under a nitrogen atmosphere. The reaction solution was allowed to cool and the precipitate was filtered. The resulting precipitate was suspended in water (500 mL) and filtered. The obtained white powder was suspended in MeCN (300 mL), stirred under heating to reflux for 30 minutes, and allowed to cool. The precipitate was filtered, washed with a small amount of acetonitrile, and tert-butyl 4- [4- (methoxycarbonyl) -4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2 , 1-b] [1,3,4] thiadiazol-6-yl} biphenyl-3-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (25.3 g) was obtained.
製造例5Aの方法と同様にして、後記表に示す製造例5A-1から製造例5A-47の化合物を製造した。
In the same manner as in Production Example 5A, the compounds of Production Example 5A-47 were produced from Production Example 5A-1 shown in the table below.
製造例5B
4,4,4',4',5,5,5',5'-オクタメチル-2,2'-ビ-1,3,2-ジオキサボロラン(1.2 g)、KOAc(750 mg)、tert-ブチル 4-{[(トリフルオロメチル)スルホニル]オキシ}-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(1.6 g)に、超音波で20分脱気したジオキサン(20 mL)を加えた。反応系内は、窒素で3回置換した。反応系内に、Pd2(dba)3(68 mg)、ジシクロヘキシル(2',6'-ジメトキシビフェニル-2-イル)ホスフィン(245 mg)を加え、再度、反応系内を窒素で3回置換した。反応混合物は110℃(内温95℃)で3時間攪拌した。室温に戻し、メチル 2-クロロ-4-(テトラヒドロ-2H-ピラン-2-イルオキシ)ベンゾエート(1 g)、K3PO4(750 mg)、H2O(2 mL)を加え、110℃(内温95℃)で2時間攪拌した。反応混合物をEtOAcで希釈した後、水を加え洗浄した。有機層を分離し、brineで洗浄し、MgSO4で乾燥後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー (Hexane/EtOAc系) で精製し、tert-ブチル 4-[2-(メトキシカルボニル)-5-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]-3,6-ジヒドロキシピリジン-1(2H)-カルボキシレート(1.3 g)をオレンジ色油状物として得た。 Production Example 5B
4,4,4 ', 4', 5,5,5 ', 5'-octamethyl-2,2'-bi-1,3,2-dioxaborolane (1.2 g), KOAc (750 mg), tert-butyl To 4-{[(trifluoromethyl) sulfonyl] oxy} -3,6-dihydropyridine-1 (2H) -carboxylate (1.6 g) was added dioxane (20 mL) degassed with ultrasound for 20 minutes. The reaction system was replaced with nitrogen three times. Pd 2 (dba) 3 (68 mg) and dicyclohexyl (2 ', 6'-dimethoxybiphenyl-2-yl) phosphine (245 mg) were added to the reaction system, and the reaction system was again replaced with nitrogen three times. did. The reaction mixture was stirred at 110 ° C. (internal temperature 95 ° C.) for 3 hours. Return to room temperature, add methyl 2-chloro-4- (tetrahydro-2H-pyran-2-yloxy) benzoate (1 g), K 3 PO 4 (750 mg), H 2 O (2 mL), and add 110 ° C. The mixture was stirred at an internal temperature of 95 ° C. for 2 hours. The reaction mixture was diluted with EtOAc and washed with water. The organic layer was separated, washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (Hexane / EtOAc system) and tert-butyl 4- [2- (methoxycarbonyl) -5- (tetrahydro-2H-pyran-2-yloxy) phenyl] -3, 6-Dihydroxypyridine-1 (2H) -carboxylate (1.3 g) was obtained as an orange oil.
4,4,4',4',5,5,5',5'-オクタメチル-2,2'-ビ-1,3,2-ジオキサボロラン(1.2 g)、KOAc(750 mg)、tert-ブチル 4-{[(トリフルオロメチル)スルホニル]オキシ}-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(1.6 g)に、超音波で20分脱気したジオキサン(20 mL)を加えた。反応系内は、窒素で3回置換した。反応系内に、Pd2(dba)3(68 mg)、ジシクロヘキシル(2',6'-ジメトキシビフェニル-2-イル)ホスフィン(245 mg)を加え、再度、反応系内を窒素で3回置換した。反応混合物は110℃(内温95℃)で3時間攪拌した。室温に戻し、メチル 2-クロロ-4-(テトラヒドロ-2H-ピラン-2-イルオキシ)ベンゾエート(1 g)、K3PO4(750 mg)、H2O(2 mL)を加え、110℃(内温95℃)で2時間攪拌した。反応混合物をEtOAcで希釈した後、水を加え洗浄した。有機層を分離し、brineで洗浄し、MgSO4で乾燥後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー (Hexane/EtOAc系) で精製し、tert-ブチル 4-[2-(メトキシカルボニル)-5-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]-3,6-ジヒドロキシピリジン-1(2H)-カルボキシレート(1.3 g)をオレンジ色油状物として得た。 Production Example 5B
4,4,4 ', 4', 5,5,5 ', 5'-octamethyl-2,2'-bi-1,3,2-dioxaborolane (1.2 g), KOAc (750 mg), tert-butyl To 4-{[(trifluoromethyl) sulfonyl] oxy} -3,6-dihydropyridine-1 (2H) -carboxylate (1.6 g) was added dioxane (20 mL) degassed with ultrasound for 20 minutes. The reaction system was replaced with nitrogen three times. Pd 2 (dba) 3 (68 mg) and dicyclohexyl (2 ', 6'-dimethoxybiphenyl-2-yl) phosphine (245 mg) were added to the reaction system, and the reaction system was again replaced with nitrogen three times. did. The reaction mixture was stirred at 110 ° C. (internal temperature 95 ° C.) for 3 hours. Return to room temperature, add methyl 2-chloro-4- (tetrahydro-2H-pyran-2-yloxy) benzoate (1 g), K 3 PO 4 (750 mg), H 2 O (2 mL), and add 110 ° C. The mixture was stirred at an internal temperature of 95 ° C. for 2 hours. The reaction mixture was diluted with EtOAc and washed with water. The organic layer was separated, washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (Hexane / EtOAc system) and tert-butyl 4- [2- (methoxycarbonyl) -5- (tetrahydro-2H-pyran-2-yloxy) phenyl] -3, 6-Dihydroxypyridine-1 (2H) -carboxylate (1.3 g) was obtained as an orange oil.
製造例5Bの方法と同様にして、後記表に示す製造例5B-1から製造例5B-15の化合物を製造した。
In the same manner as in Production Example 5B, the compound of Production Example 5B-15 was produced from Production Example 5B-1 shown in the table below.
製造例5C
1-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]-4-[(4,4-ジメチルシクロヘキシル)オキシ]ピペリジン (243 mg)、{3-[1-(tert-ブトキシカルボニル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4-(メトキシカルボニル)フェニル}ボロン酸 (223 mg)、K3PO4(356 mg)、 パラジウムアセテート(12.6 mg)、トリシクロヘキシルホスフィン(15.7 mg)のジオキサン(4.86 mL)-水(1.22 mL)混合溶液を室温で1時間撹拌した。反応液にEtOAcと水を加えた後、セライト濾過した。ろ液を分配して、有機層をbrineで洗浄した。有機層はMgSO4で乾燥後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(Hexane/EtOAc系)で精製し、tert-ブチル 4-[4'-(5-{4-[(4,4-ジメチルシクロヘキシル)オキシ]ピペリジン-1-イル}-1,2,4-オキサジアゾール-3-イル)-4-(メトキシカルボニル)ビフェニル-3-イル]-3,6-ジヒドロキシピリジン-1(2H)-カルボキシレート (220 mg)を白色固体として得た。 Production Example 5C
1- [3- (4-Bromophenyl) -1,2,4-oxadiazol-5-yl] -4-[(4,4-dimethylcyclohexyl) oxy] piperidine (243 mg), {3- [ 1- (tert-butoxycarbonyl) -1,2,3,6-tetrahydropyridin-4-yl] -4- (methoxycarbonyl) phenyl} boronic acid (223 mg), K 3 PO 4 (356 mg), palladium A mixed solution of acetate (12.6 mg) and tricyclohexylphosphine (15.7 mg) in dioxane (4.86 mL) -water (1.22 mL) was stirred at room temperature for 1 hour. EtOAc and water were added to the reaction solution, followed by filtration through celite. The filtrate was partitioned and the organic layer was washed with brine. The organic layer was dried over MgSO 4 and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (Hexane / EtOAc system) and tert-butyl 4- [4 '-(5- {4-[(4,4-dimethylcyclohexyl) oxy] piperidin-1-yl } -1,2,4-oxadiazol-3-yl) -4- (methoxycarbonyl) biphenyl-3-yl] -3,6-dihydroxypyridine-1 (2H) -carboxylate (220 mg) in white Obtained as a solid.
1-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]-4-[(4,4-ジメチルシクロヘキシル)オキシ]ピペリジン (243 mg)、{3-[1-(tert-ブトキシカルボニル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4-(メトキシカルボニル)フェニル}ボロン酸 (223 mg)、K3PO4(356 mg)、 パラジウムアセテート(12.6 mg)、トリシクロヘキシルホスフィン(15.7 mg)のジオキサン(4.86 mL)-水(1.22 mL)混合溶液を室温で1時間撹拌した。反応液にEtOAcと水を加えた後、セライト濾過した。ろ液を分配して、有機層をbrineで洗浄した。有機層はMgSO4で乾燥後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(Hexane/EtOAc系)で精製し、tert-ブチル 4-[4'-(5-{4-[(4,4-ジメチルシクロヘキシル)オキシ]ピペリジン-1-イル}-1,2,4-オキサジアゾール-3-イル)-4-(メトキシカルボニル)ビフェニル-3-イル]-3,6-ジヒドロキシピリジン-1(2H)-カルボキシレート (220 mg)を白色固体として得た。 Production Example 5C
1- [3- (4-Bromophenyl) -1,2,4-oxadiazol-5-yl] -4-[(4,4-dimethylcyclohexyl) oxy] piperidine (243 mg), {3- [ 1- (tert-butoxycarbonyl) -1,2,3,6-tetrahydropyridin-4-yl] -4- (methoxycarbonyl) phenyl} boronic acid (223 mg), K 3 PO 4 (356 mg), palladium A mixed solution of acetate (12.6 mg) and tricyclohexylphosphine (15.7 mg) in dioxane (4.86 mL) -water (1.22 mL) was stirred at room temperature for 1 hour. EtOAc and water were added to the reaction solution, followed by filtration through celite. The filtrate was partitioned and the organic layer was washed with brine. The organic layer was dried over MgSO 4 and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (Hexane / EtOAc system) and tert-butyl 4- [4 '-(5- {4-[(4,4-dimethylcyclohexyl) oxy] piperidin-1-yl } -1,2,4-oxadiazol-3-yl) -4- (methoxycarbonyl) biphenyl-3-yl] -3,6-dihydroxypyridine-1 (2H) -carboxylate (220 mg) in white Obtained as a solid.
製造例5Cの方法と同様にして、後記表に示す製造例5C-1から製造例5C-27の化合物を製造した。
In the same manner as in Production Example 5C, the compounds of Production Example 5C-27 were produced from Production Example 5C-1 shown in the table below.
製造例5D
tert-ブチル 4-[2-(メトキシカルボニル)-5-{[(トリフルオロメチル)スルホニル]オキシ}フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(441 mg), 4,4,4',4',5,5,5',5'-オクタメチル-2,2'-ビ-1,3,2-ジオキサボロラン(277 mg)、トリフェニルホスフィン(33.3 mg), KOAc(279 mg)、ジオキサン(5 mL)の混合物を入れた反応容器の系内を減圧にして窒素置換した。PdCl2(PPh3)2を加えて再度窒素置換し、バス温100℃で20時間撹拌した。反応混合物を室温に戻し、水を加えて、EtOAcで抽出した。有機層をbrineで洗浄し、MgSO4で乾燥後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーで精製しtert-ブチル 4-[2-(メトキシカルボニル)-5-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート (480 mg)を得た。 Production Example 5D
tert-Butyl 4- [2- (methoxycarbonyl) -5-{[(trifluoromethyl) sulfonyl] oxy} phenyl] -3,6-dihydropyridine-1 (2H) -carboxylate (441 mg), 4,4 , 4 ', 4', 5,5,5 ', 5'-octamethyl-2,2'-bi-1,3,2-dioxaborolane (277 mg), triphenylphosphine (33.3 mg), KOAc (279 mg ) And dioxane (5 mL), and the inside of the reaction vessel containing the mixture was depressurized and purged with nitrogen. PdCl 2 (PPh 3 ) 2 was added and the atmosphere was replaced with nitrogen again, followed by stirring at a bath temperature of 100 ° C. for 20 hours. The reaction mixture was allowed to warm to room temperature, water was added and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography and tert-butyl 4- [2- (methoxycarbonyl) -5- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl ) Phenyl] -3,6-dihydropyridine-1 (2H) -carboxylate (480 mg) was obtained.
tert-ブチル 4-[2-(メトキシカルボニル)-5-{[(トリフルオロメチル)スルホニル]オキシ}フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(441 mg), 4,4,4',4',5,5,5',5'-オクタメチル-2,2'-ビ-1,3,2-ジオキサボロラン(277 mg)、トリフェニルホスフィン(33.3 mg), KOAc(279 mg)、ジオキサン(5 mL)の混合物を入れた反応容器の系内を減圧にして窒素置換した。PdCl2(PPh3)2を加えて再度窒素置換し、バス温100℃で20時間撹拌した。反応混合物を室温に戻し、水を加えて、EtOAcで抽出した。有機層をbrineで洗浄し、MgSO4で乾燥後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーで精製しtert-ブチル 4-[2-(メトキシカルボニル)-5-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート (480 mg)を得た。 Production Example 5D
tert-Butyl 4- [2- (methoxycarbonyl) -5-{[(trifluoromethyl) sulfonyl] oxy} phenyl] -3,6-dihydropyridine-1 (2H) -carboxylate (441 mg), 4,4 , 4 ', 4', 5,5,5 ', 5'-octamethyl-2,2'-bi-1,3,2-dioxaborolane (277 mg), triphenylphosphine (33.3 mg), KOAc (279 mg ) And dioxane (5 mL), and the inside of the reaction vessel containing the mixture was depressurized and purged with nitrogen. PdCl 2 (PPh 3 ) 2 was added and the atmosphere was replaced with nitrogen again, followed by stirring at a bath temperature of 100 ° C. for 20 hours. The reaction mixture was allowed to warm to room temperature, water was added and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography and tert-butyl 4- [2- (methoxycarbonyl) -5- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl ) Phenyl] -3,6-dihydropyridine-1 (2H) -carboxylate (480 mg) was obtained.
製造例5Dの方法と同様にして、後記表に示す製造例5D-1から製造例5D-6の化合物を製造した。
In the same manner as in Production Example 5D, the compounds of Production Example 5D-6 were produced from Production Example 5D-1 shown in the table below.
製造例5E
4-{5-[4-(2-フェニルエチル)ピペラジン-1-イル]ピリミジン-2-イル}フェニル トリフルオロメタンスルホネート (2.1 g)、[3-クロロ-4-(メトキシカルボニル)フェニル]ボロン酸 (1.1 g)、 2.0M Na2CO3水溶液(5.4 mL)、テトラキストリフェニルホスフィンパラジウム(0)、DME (21 mL)を混合し、窒素を吹き込んだ後、加熱還流下で2時間攪拌した。反応液を水にあけた後に、生じた固体をろ取し、水、MeCNで洗浄し、40℃で減圧下乾燥してメチル 3-クロロ-4'-{5-[4-(2-フェニルエチル)ピペラジン-1-イル]ピリミジン-2-イル}ビフェニル-4-カルボキシレート (1.95 g)を得た。 Production Example 5E
4- {5- [4- (2-Phenylethyl) piperazin-1-yl] pyrimidin-2-yl} phenyl trifluoromethanesulfonate (2.1 g), [3-chloro-4- (methoxycarbonyl) phenyl] boronic acid (1.1 g), 2.0 M Na 2 CO 3 aqueous solution (5.4 mL), tetrakistriphenylphosphine palladium (0), and DME (21 mL) were mixed, and nitrogen was blown, followed by stirring for 2 hours with heating under reflux. The reaction mixture was poured into water, and the resulting solid was collected by filtration, washed with water and MeCN, dried under reduced pressure at 40 ° C., and methyl 3-chloro-4 ′-{5- [4- (2-phenyl). Ethyl) piperazin-1-yl] pyrimidin-2-yl} biphenyl-4-carboxylate (1.95 g) was obtained.
4-{5-[4-(2-フェニルエチル)ピペラジン-1-イル]ピリミジン-2-イル}フェニル トリフルオロメタンスルホネート (2.1 g)、[3-クロロ-4-(メトキシカルボニル)フェニル]ボロン酸 (1.1 g)、 2.0M Na2CO3水溶液(5.4 mL)、テトラキストリフェニルホスフィンパラジウム(0)、DME (21 mL)を混合し、窒素を吹き込んだ後、加熱還流下で2時間攪拌した。反応液を水にあけた後に、生じた固体をろ取し、水、MeCNで洗浄し、40℃で減圧下乾燥してメチル 3-クロロ-4'-{5-[4-(2-フェニルエチル)ピペラジン-1-イル]ピリミジン-2-イル}ビフェニル-4-カルボキシレート (1.95 g)を得た。 Production Example 5E
4- {5- [4- (2-Phenylethyl) piperazin-1-yl] pyrimidin-2-yl} phenyl trifluoromethanesulfonate (2.1 g), [3-chloro-4- (methoxycarbonyl) phenyl] boronic acid (1.1 g), 2.0 M Na 2 CO 3 aqueous solution (5.4 mL), tetrakistriphenylphosphine palladium (0), and DME (21 mL) were mixed, and nitrogen was blown, followed by stirring for 2 hours with heating under reflux. The reaction mixture was poured into water, and the resulting solid was collected by filtration, washed with water and MeCN, dried under reduced pressure at 40 ° C., and methyl 3-chloro-4 ′-{5- [4- (2-phenyl). Ethyl) piperazin-1-yl] pyrimidin-2-yl} biphenyl-4-carboxylate (1.95 g) was obtained.
製造例5Eの方法と同様にして、後記表に示す製造例5E-1から製造例5E-29の化合物を製造した。
In the same manner as in Production Example 5E, the compounds of Production Example 5E-29 were produced from Production Example 5E-1 shown in the table below.
製造例5F
4,4,4',4',5,5,5',5'-オクタメチル-2,2'-ビ-1,3,2-ジオキサボロラン(0.125 g)、DIPEA (0.16 mL)および tert-ブチル 4-[3-(メトキシカルボニル)-7-{[(トリフルオロメチル)スルホニルオキシ]オキシ}-2-ナフチル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(0.228 g)に、超音波で20分脱気したジオキサン(2 mL)を加えた。反応系内は、窒素で3回置換した。反応系内に、 ビフェニル-2-イル(ジシクロヘキシル)ホスフィン (0.032 g)、Pd(OAc)2(5 mg)を加え、再度、反応系内を窒素で3回置換した。反応混合物を80℃で1時間攪拌した。室温に戻し、水 (0.8 mL)、K3PO4 (0.28 g)、 トリシクロヘキシルホスフィン(0.025 g)、Pd(OAc)2 (5 mg)および 1-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]-4-[2-(4-フルオロフェニル)エチル]ピペラジン (0.19 g)を加え、80℃で終夜攪拌した。反応混合物をCHCl3(30 mL)で希釈した後、水(20 mL)を加え洗浄した。有機層を分離し、MgSO4で乾燥後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー (CHCl3/MeOH系) で精製し、 tert-ブチル 4-{7-[4-(5-{4-[2-(4-フルオロフェニル)エチル]ピペラジン-1-イル}-1,2,4-オキサジアゾール-3-イル)フェニル]-3-(メトキシカルボニル)-2-ナフチル}-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(0.074 mg)を黄色固体として得た。 Production Example 5F
4,4,4 ', 4', 5,5,5 ', 5'-octamethyl-2,2'-bi-1,3,2-dioxaborolane (0.125 g), DIPEA (0.16 mL) and tert-butyl 4- [3- (methoxycarbonyl) -7-{[(trifluoromethyl) sulfonyloxy] oxy} -2-naphthyl] -3,6-dihydropyridine-1 (2H) -carboxylate (0.228 g) Dioxane (2 mL) degassed by sonication for 20 minutes was added. The reaction system was replaced with nitrogen three times. Biphenyl-2-yl (dicyclohexyl) phosphine (0.032 g) and Pd (OAc) 2 (5 mg) were added to the reaction system, and the reaction system was again substituted with nitrogen three times. The reaction mixture was stirred at 80 ° C. for 1 hour. Return to room temperature, water (0.8 mL), K 3 PO 4 (0.28 g), tricyclohexylphosphine (0.025 g), Pd (OAc) 2 (5 mg) and 1- [3- (4-bromophenyl) -1 , 2,4-oxadiazol-5-yl] -4- [2- (4-fluorophenyl) ethyl] piperazine (0.19 g) was added and stirred at 80 ° C. overnight. The reaction mixture was diluted with CHCl 3 (30 mL), and washed with water (20 mL). The organic layer was separated, dried over MgSO 4 and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (CHCl 3 / MeOH system), and tert-butyl 4- {7- [4- (5- {4- [2- (4-fluorophenyl) ethyl] piperazine- 1-yl} -1,2,4-oxadiazol-3-yl) phenyl] -3- (methoxycarbonyl) -2-naphthyl} -3,6-dihydropyridine-1 (2H) -carboxylate (0.074 mg ) Was obtained as a yellow solid.
4,4,4',4',5,5,5',5'-オクタメチル-2,2'-ビ-1,3,2-ジオキサボロラン(0.125 g)、DIPEA (0.16 mL)および tert-ブチル 4-[3-(メトキシカルボニル)-7-{[(トリフルオロメチル)スルホニルオキシ]オキシ}-2-ナフチル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(0.228 g)に、超音波で20分脱気したジオキサン(2 mL)を加えた。反応系内は、窒素で3回置換した。反応系内に、 ビフェニル-2-イル(ジシクロヘキシル)ホスフィン (0.032 g)、Pd(OAc)2(5 mg)を加え、再度、反応系内を窒素で3回置換した。反応混合物を80℃で1時間攪拌した。室温に戻し、水 (0.8 mL)、K3PO4 (0.28 g)、 トリシクロヘキシルホスフィン(0.025 g)、Pd(OAc)2 (5 mg)および 1-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]-4-[2-(4-フルオロフェニル)エチル]ピペラジン (0.19 g)を加え、80℃で終夜攪拌した。反応混合物をCHCl3(30 mL)で希釈した後、水(20 mL)を加え洗浄した。有機層を分離し、MgSO4で乾燥後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー (CHCl3/MeOH系) で精製し、 tert-ブチル 4-{7-[4-(5-{4-[2-(4-フルオロフェニル)エチル]ピペラジン-1-イル}-1,2,4-オキサジアゾール-3-イル)フェニル]-3-(メトキシカルボニル)-2-ナフチル}-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(0.074 mg)を黄色固体として得た。 Production Example 5F
4,4,4 ', 4', 5,5,5 ', 5'-octamethyl-2,2'-bi-1,3,2-dioxaborolane (0.125 g), DIPEA (0.16 mL) and tert-butyl 4- [3- (methoxycarbonyl) -7-{[(trifluoromethyl) sulfonyloxy] oxy} -2-naphthyl] -3,6-dihydropyridine-1 (2H) -carboxylate (0.228 g) Dioxane (2 mL) degassed by sonication for 20 minutes was added. The reaction system was replaced with nitrogen three times. Biphenyl-2-yl (dicyclohexyl) phosphine (0.032 g) and Pd (OAc) 2 (5 mg) were added to the reaction system, and the reaction system was again substituted with nitrogen three times. The reaction mixture was stirred at 80 ° C. for 1 hour. Return to room temperature, water (0.8 mL), K 3 PO 4 (0.28 g), tricyclohexylphosphine (0.025 g), Pd (OAc) 2 (5 mg) and 1- [3- (4-bromophenyl) -1 , 2,4-oxadiazol-5-yl] -4- [2- (4-fluorophenyl) ethyl] piperazine (0.19 g) was added and stirred at 80 ° C. overnight. The reaction mixture was diluted with CHCl 3 (30 mL), and washed with water (20 mL). The organic layer was separated, dried over MgSO 4 and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (CHCl 3 / MeOH system), and tert-butyl 4- {7- [4- (5- {4- [2- (4-fluorophenyl) ethyl] piperazine- 1-yl} -1,2,4-oxadiazol-3-yl) phenyl] -3- (methoxycarbonyl) -2-naphthyl} -3,6-dihydropyridine-1 (2H) -carboxylate (0.074 mg ) Was obtained as a yellow solid.
製造例5G
窒素雰囲気下、tert-ブチル 4-メチレンピペリジン-1-カルボキシレート(240 mg)のTHF溶液(1.20 mL)に0.5M 9-ボラビシクロ[3.3.1]ノナンTHF溶液(5.00 mL)を加え1時間加熱還流した。反応液にメチル 4-クロロ-2-{[(トリフルオロメチル)スルホニル]オキシ}ベンゾエート(323 mg)、K3PO4(538 mg)、PdCL2(dppf)CH2CL2(62.0 mg)、水(0.845 mL)を加え80℃で12時間撹拌した。反応液に水を加えEtOAcで抽出した。brineで洗浄し、MgSO4で乾燥後、減圧化濃縮し、残渣をシリカゲルカラムクロマトグラフィー(Hex:EtOAc系)で精製し、tert-ブチル 4-[5-クロロ-2-(メトキシカルボニル)ベンジル]ピペリジン-1-カルボキシレート(184 mg)を薄黄色油状物として得た。 Production Example 5G
Under a nitrogen atmosphere, add 0.5M 9-borabicyclo [3.3.1] nonane THF solution (5.00 mL) to a THF solution (1.20 mL) of tert-butyl 4-methylenepiperidine-1-carboxylate (240 mg) and heat for 1 hour. Refluxed. Methyl 4-chloro-2-{[(trifluoromethyl) sulfonyl] oxy} benzoate (323 mg), K 3 PO 4 (538 mg), PdCL 2 (dppf) CH 2 CL 2 (62.0 mg), Water (0.845 mL) was added and stirred at 80 ° C. for 12 hours. Water was added to the reaction solution and extracted with EtOAc. The extract was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (Hex: EtOAc system), and tert-butyl 4- [5-chloro-2- (methoxycarbonyl) benzyl]. Piperidine-1-carboxylate (184 mg) was obtained as a pale yellow oil.
窒素雰囲気下、tert-ブチル 4-メチレンピペリジン-1-カルボキシレート(240 mg)のTHF溶液(1.20 mL)に0.5M 9-ボラビシクロ[3.3.1]ノナンTHF溶液(5.00 mL)を加え1時間加熱還流した。反応液にメチル 4-クロロ-2-{[(トリフルオロメチル)スルホニル]オキシ}ベンゾエート(323 mg)、K3PO4(538 mg)、PdCL2(dppf)CH2CL2(62.0 mg)、水(0.845 mL)を加え80℃で12時間撹拌した。反応液に水を加えEtOAcで抽出した。brineで洗浄し、MgSO4で乾燥後、減圧化濃縮し、残渣をシリカゲルカラムクロマトグラフィー(Hex:EtOAc系)で精製し、tert-ブチル 4-[5-クロロ-2-(メトキシカルボニル)ベンジル]ピペリジン-1-カルボキシレート(184 mg)を薄黄色油状物として得た。 Production Example 5G
Under a nitrogen atmosphere, add 0.5M 9-borabicyclo [3.3.1] nonane THF solution (5.00 mL) to a THF solution (1.20 mL) of tert-butyl 4-methylenepiperidine-1-carboxylate (240 mg) and heat for 1 hour. Refluxed. Methyl 4-chloro-2-{[(trifluoromethyl) sulfonyl] oxy} benzoate (323 mg), K 3 PO 4 (538 mg), PdCL 2 (dppf) CH 2 CL 2 (62.0 mg), Water (0.845 mL) was added and stirred at 80 ° C. for 12 hours. Water was added to the reaction solution and extracted with EtOAc. The extract was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (Hex: EtOAc system), and tert-butyl 4- [5-chloro-2- (methoxycarbonyl) benzyl]. Piperidine-1-carboxylate (184 mg) was obtained as a pale yellow oil.
製造例6
tert-ブチル 4-[5-ヒドロキシ-2-(メトキシカルボニル)フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(5.33 g)、1,1,1-トリフルオロ-N-フェニル-N-[(トリフルオロメチル)スルホニル]メタンスルホンアミド(6.30 g)、及びDIPEA (d=0.742、8 mL)のDMF溶液(50 mL)を室温で5時間撹拌した。反応液にEtOAcを加えて抽出した。有機層は、水、brineで洗浄し、MgSO4で乾燥後、減圧下濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(Hex/EtOAc系)で精製し、tert-ブチル 4-[2-(メトキシカルボニル)-5-{[(トリフルオロメチル)スルホニル]オキシ}フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(6.7 g)を黄色油状物として得た。 Production Example 6
tert-butyl 4- [5-hydroxy-2- (methoxycarbonyl) phenyl] -3,6-dihydropyridine-1 (2H) -carboxylate (5.33 g), 1,1,1-trifluoro-N-phenyl- A DMF solution (50 mL) of N-[(trifluoromethyl) sulfonyl] methanesulfonamide (6.30 g) and DIPEA (d = 0.742, 8 mL) was stirred at room temperature for 5 hours. The reaction mixture was extracted with EtOAc. The organic layer was washed with water and brine, dried over MgSO 4 and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (Hex / EtOAc system), and tert-butyl 4- [2- ( Methoxycarbonyl) -5-{[(trifluoromethyl) sulfonyl] oxy} phenyl] -3,6-dihydropyridine-1 (2H) -carboxylate (6.7 g) was obtained as a yellow oil.
tert-ブチル 4-[5-ヒドロキシ-2-(メトキシカルボニル)フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(5.33 g)、1,1,1-トリフルオロ-N-フェニル-N-[(トリフルオロメチル)スルホニル]メタンスルホンアミド(6.30 g)、及びDIPEA (d=0.742、8 mL)のDMF溶液(50 mL)を室温で5時間撹拌した。反応液にEtOAcを加えて抽出した。有機層は、水、brineで洗浄し、MgSO4で乾燥後、減圧下濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(Hex/EtOAc系)で精製し、tert-ブチル 4-[2-(メトキシカルボニル)-5-{[(トリフルオロメチル)スルホニル]オキシ}フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(6.7 g)を黄色油状物として得た。 Production Example 6
tert-butyl 4- [5-hydroxy-2- (methoxycarbonyl) phenyl] -3,6-dihydropyridine-1 (2H) -carboxylate (5.33 g), 1,1,1-trifluoro-N-phenyl- A DMF solution (50 mL) of N-[(trifluoromethyl) sulfonyl] methanesulfonamide (6.30 g) and DIPEA (d = 0.742, 8 mL) was stirred at room temperature for 5 hours. The reaction mixture was extracted with EtOAc. The organic layer was washed with water and brine, dried over MgSO 4 and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (Hex / EtOAc system), and tert-butyl 4- [2- ( Methoxycarbonyl) -5-{[(trifluoromethyl) sulfonyl] oxy} phenyl] -3,6-dihydropyridine-1 (2H) -carboxylate (6.7 g) was obtained as a yellow oil.
製造例6の方法と同様にして、後記表に示す製造例6-1から製造例6-26の化合物を製造した。
In the same manner as in Production Example 6, the compounds of Production Example 6-1 to Production Example 6-26 shown in the table below were produced.
製造例7
5-[4-(2-フェニルエチル)ピペラジン-1-イル]-1,3-チアゾール-2-アミン (475 mg) 及び2-ブロモ-1-(4-ブロモフェニル)エタノン(460 mg) をn-BuOH(6 mL) 中90℃下撹拌1時間した。反応混合物を室温まで放冷した後、EtOH(5 mL)を加えて析出物をろ取し、得られた固体をEtOHで洗浄し、40℃で減圧下乾燥し、6-(4-ブロモフェニル)-2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3]チアゾール(731mg)を紫色固体として得た。 Production Example 7
5- [4- (2-phenylethyl) piperazin-1-yl] -1,3-thiazol-2-amine (475 mg) and 2-bromo-1- (4-bromophenyl) ethanone (460 mg) The mixture was stirred at 90 ° C. in n-BuOH (6 mL) for 1 hour. The reaction mixture was allowed to cool to room temperature, EtOH (5 mL) was added, the precipitate was collected by filtration, the resulting solid was washed with EtOH, dried at 40 ° C. under reduced pressure, and 6- (4-bromophenyl ) -2- [4- (2-Phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3] thiazole (731 mg) was obtained as a purple solid.
5-[4-(2-フェニルエチル)ピペラジン-1-イル]-1,3-チアゾール-2-アミン (475 mg) 及び2-ブロモ-1-(4-ブロモフェニル)エタノン(460 mg) をn-BuOH(6 mL) 中90℃下撹拌1時間した。反応混合物を室温まで放冷した後、EtOH(5 mL)を加えて析出物をろ取し、得られた固体をEtOHで洗浄し、40℃で減圧下乾燥し、6-(4-ブロモフェニル)-2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3]チアゾール(731mg)を紫色固体として得た。 Production Example 7
5- [4- (2-phenylethyl) piperazin-1-yl] -1,3-thiazol-2-amine (475 mg) and 2-bromo-1- (4-bromophenyl) ethanone (460 mg) The mixture was stirred at 90 ° C. in n-BuOH (6 mL) for 1 hour. The reaction mixture was allowed to cool to room temperature, EtOH (5 mL) was added, the precipitate was collected by filtration, the resulting solid was washed with EtOH, dried at 40 ° C. under reduced pressure, and 6- (4-bromophenyl ) -2- [4- (2-Phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3] thiazole (731 mg) was obtained as a purple solid.
製造例8
5-フルオロ-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピリミジン(7.20 g)のDMSO(72 mL)溶液に 1-(2-フェニルエチル)ピペラジン(9.93 mL)及び K2CO3(7.26 g)を加え140℃で20時間攪拌した。室温まで放冷後、反応混合物に水(100 mL)を加え、生じた沈殿をろ取した。得られた粉末をIPEで洗浄した後に、減圧下40℃で一夜乾燥して、5-[4-(2-フェニルエチル)ピペラジン-1-イル]-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピリミジン(10.06 g)を得た。 Production Example 8
To a solution of 5-fluoro-2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyrimidine (7.20 g) in DMSO (72 mL) was added 1- (2-phenylethyl) piperazine (9.93 mL) and K 2 CO 3 (7.26 g) was added and the mixture was stirred at 140 ° C. for 20 hours. After cooling to room temperature, water (100 mL) was added to the reaction mixture, and the resulting precipitate was collected by filtration. The obtained powder was washed with IPE and dried overnight at 40 ° C. under reduced pressure to give 5- [4- (2-phenylethyl) piperazin-1-yl] -2- [4- (tetrahydro-2H-pyran -2-yloxy) phenyl] pyrimidine (10.06 g) was obtained.
5-フルオロ-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピリミジン(7.20 g)のDMSO(72 mL)溶液に 1-(2-フェニルエチル)ピペラジン(9.93 mL)及び K2CO3(7.26 g)を加え140℃で20時間攪拌した。室温まで放冷後、反応混合物に水(100 mL)を加え、生じた沈殿をろ取した。得られた粉末をIPEで洗浄した後に、減圧下40℃で一夜乾燥して、5-[4-(2-フェニルエチル)ピペラジン-1-イル]-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピリミジン(10.06 g)を得た。 Production Example 8
To a solution of 5-fluoro-2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyrimidine (7.20 g) in DMSO (72 mL) was added 1- (2-phenylethyl) piperazine (9.93 mL) and K 2 CO 3 (7.26 g) was added and the mixture was stirred at 140 ° C. for 20 hours. After cooling to room temperature, water (100 mL) was added to the reaction mixture, and the resulting precipitate was collected by filtration. The obtained powder was washed with IPE and dried overnight at 40 ° C. under reduced pressure to give 5- [4- (2-phenylethyl) piperazin-1-yl] -2- [4- (tetrahydro-2H-pyran -2-yloxy) phenyl] pyrimidine (10.06 g) was obtained.
製造例8の方法と同様にして、後記表に示す製造例8-1から製造例8-26 の化合物を製造した。
In the same manner as in Production Example 8, the compounds of Production Example 8-1 to Production Example 8-26 shown in the table below were produced.
製造例9
5-フルオロ-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピリミジン(500 mg)のNMP(2.5 mL)溶液に(2R)-2-メチルピペラジン(547 mg)及びK2CO3 (1.26 g)を加え140℃で6時間撹拌した。さらに、(2R)-2-メチルピペラジン(547 mg)、K2CO3(1.26 g)を追加し、終夜攪拌した。反応液にbrine(10 mL)、水(10 mL)を加え、EtOAcで3回抽出した。有機層は、brineで4回洗浄し、MgSO4で乾燥後、減圧下濃縮した。得られた残渣の CH2Cl2溶液(3.5 mL)にフェニルアセトアルデヒド(245 μL)、NaBH(OAc)3 (502 mg)を加えて、室温で1時間撹拌した。反応液にsat.NaHCO3水溶液をゆっくりと加え、 CH2Cl2で2回抽出した。有機層はMgSO4で乾燥し、得られた残渣を(Hexane:CHCl3:EtOAc=1:1:1から0:70:30まで)で精製し5-[(3R)-3-メチル-4-(2-フェニルエチル)ピペラジン-1-イル]-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピリミジン(600 mg)を得た。 Production Example 9
To a solution of 5-fluoro-2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyrimidine (500 mg) in NMP (2.5 mL) was added (2R) -2-methylpiperazine (547 mg) and K 2 CO 3 (1.26 g) was added and the mixture was stirred at 140 ° C. for 6 hours. Furthermore, (2R) -2-methylpiperazine (547 mg) and K 2 CO 3 (1.26 g) were added, and the mixture was stirred overnight. To the reaction solution were added brine (10 mL) and water (10 mL), and the mixture was extracted 3 times with EtOAc. The organic layer was washed 4 times with brine, dried over MgSO 4 and concentrated under reduced pressure. Phenylacetaldehyde (245 μL) and NaBH (OAc) 3 (502 mg) were added to a CH 2 Cl 2 solution (3.5 mL) of the obtained residue, and the mixture was stirred at room temperature for 1 hour. A sat. NaHCO 3 aqueous solution was slowly added to the reaction mixture, and the mixture was extracted twice with CH 2 Cl 2 . The organic layer was dried over MgSO 4 and the resulting residue was purified (Hexane: CHCl 3 : EtOAc = 1: 1: 1 to 0:70:30) and purified by 5-[(3R) -3-methyl-4 -(2-Phenylethyl) piperazin-1-yl] -2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyrimidine (600 mg) was obtained.
5-フルオロ-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピリミジン(500 mg)のNMP(2.5 mL)溶液に(2R)-2-メチルピペラジン(547 mg)及びK2CO3 (1.26 g)を加え140℃で6時間撹拌した。さらに、(2R)-2-メチルピペラジン(547 mg)、K2CO3(1.26 g)を追加し、終夜攪拌した。反応液にbrine(10 mL)、水(10 mL)を加え、EtOAcで3回抽出した。有機層は、brineで4回洗浄し、MgSO4で乾燥後、減圧下濃縮した。得られた残渣の CH2Cl2溶液(3.5 mL)にフェニルアセトアルデヒド(245 μL)、NaBH(OAc)3 (502 mg)を加えて、室温で1時間撹拌した。反応液にsat.NaHCO3水溶液をゆっくりと加え、 CH2Cl2で2回抽出した。有機層はMgSO4で乾燥し、得られた残渣を(Hexane:CHCl3:EtOAc=1:1:1から0:70:30まで)で精製し5-[(3R)-3-メチル-4-(2-フェニルエチル)ピペラジン-1-イル]-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピリミジン(600 mg)を得た。 Production Example 9
To a solution of 5-fluoro-2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyrimidine (500 mg) in NMP (2.5 mL) was added (2R) -2-methylpiperazine (547 mg) and K 2 CO 3 (1.26 g) was added and the mixture was stirred at 140 ° C. for 6 hours. Furthermore, (2R) -2-methylpiperazine (547 mg) and K 2 CO 3 (1.26 g) were added, and the mixture was stirred overnight. To the reaction solution were added brine (10 mL) and water (10 mL), and the mixture was extracted 3 times with EtOAc. The organic layer was washed 4 times with brine, dried over MgSO 4 and concentrated under reduced pressure. Phenylacetaldehyde (245 μL) and NaBH (OAc) 3 (502 mg) were added to a CH 2 Cl 2 solution (3.5 mL) of the obtained residue, and the mixture was stirred at room temperature for 1 hour. A sat. NaHCO 3 aqueous solution was slowly added to the reaction mixture, and the mixture was extracted twice with CH 2 Cl 2 . The organic layer was dried over MgSO 4 and the resulting residue was purified (Hexane: CHCl 3 : EtOAc = 1: 1: 1 to 0:70:30) and purified by 5-[(3R) -3-methyl-4 -(2-Phenylethyl) piperazin-1-yl] -2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyrimidine (600 mg) was obtained.
製造例9の方法と同様にして、後記表に示す製造例9-1の化合物を製造した。
In the same manner as in Production Example 9, the compound of Production Example 9-1 shown in the table below was produced.
製造例10
3-[1-(tert-ブトキシカルボニル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-4-カルボン酸(1 g)、BOP(768 mg)、DMF、DIPEA(d=0.742, 662 μL)の混合物を室温で1時間撹拌した。国際公開WO99/40108号の実施例番号169の化合物(1.50 g)を加え、室温で終夜撹拌した。反応液にEtOAc(100 mL)を加え生じた析出物をろ取し,減圧下、3時間乾燥した。この粉末を溶媒(MeCN:H2O=1:1)に溶解した後、1M NaOH水溶液を加えて撹拌しながら水で希釈した。この溶液をODS中圧カラムクロマトグラフィーを用いて精製した後、凍結乾燥し、製造例10の化合物(1.76 g)を得た。 Production Example 10
3- [1- (tert-Butoxycarbonyl) -1,2,3,6-tetrahydropyridin-4-yl] -4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo A mixture of [2,1-b] [1,3,4] thiadiazol-6-yl} biphenyl-4-carboxylic acid (1 g), BOP (768 mg), DMF, DIPEA (d = 0.742, 662 μL) Was stirred at room temperature for 1 hour. The compound of Example No. 169 (1.50 g) of International Publication No. WO99 / 40108 was added and stirred at room temperature overnight. EtOAc (100 mL) was added to the reaction solution, and the resulting precipitate was collected by filtration and dried under reduced pressure for 3 hours. After dissolving this powder in a solvent (MeCN: H 2 O = 1: 1), 1M NaOH aqueous solution was added and diluted with water with stirring. This solution was purified using ODS medium pressure column chromatography and then lyophilized to obtain the compound of Production Example 10 (1.76 g).
3-[1-(tert-ブトキシカルボニル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-4-カルボン酸(1 g)、BOP(768 mg)、DMF、DIPEA(d=0.742, 662 μL)の混合物を室温で1時間撹拌した。国際公開WO99/40108号の実施例番号169の化合物(1.50 g)を加え、室温で終夜撹拌した。反応液にEtOAc(100 mL)を加え生じた析出物をろ取し,減圧下、3時間乾燥した。この粉末を溶媒(MeCN:H2O=1:1)に溶解した後、1M NaOH水溶液を加えて撹拌しながら水で希釈した。この溶液をODS中圧カラムクロマトグラフィーを用いて精製した後、凍結乾燥し、製造例10の化合物(1.76 g)を得た。 Production Example 10
3- [1- (tert-Butoxycarbonyl) -1,2,3,6-tetrahydropyridin-4-yl] -4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo A mixture of [2,1-b] [1,3,4] thiadiazol-6-yl} biphenyl-4-carboxylic acid (1 g), BOP (768 mg), DMF, DIPEA (d = 0.742, 662 μL) Was stirred at room temperature for 1 hour. The compound of Example No. 169 (1.50 g) of International Publication No. WO99 / 40108 was added and stirred at room temperature overnight. EtOAc (100 mL) was added to the reaction solution, and the resulting precipitate was collected by filtration and dried under reduced pressure for 3 hours. After dissolving this powder in a solvent (MeCN: H 2 O = 1: 1), 1M NaOH aqueous solution was added and diluted with water with stirring. This solution was purified using ODS medium pressure column chromatography and then lyophilized to obtain the compound of Production Example 10 (1.76 g).
製造例10の方法と同様にして、後記表に示す製造例10-1から10-24及び製造例10-26から10-33の化合物を製造した。製造例10-25は、製造例10に準じた方法で縮合反応を行ったのち、次いで製造例18の方法に準じて環化させて得た。
単離、精製は、抽出、分別結晶化、各種分画クロマトグラフィー(例えばODSカラムクロマトグラフィーやシリカゲルカラムクロマトグラフィー)等、通常の化学操作及び本明細書の製造例若しくは実施例に記載の方法、またはそれに準じた方法のうち、化合物に適した方法を適用して行なった。 In the same manner as in Production Example 10, the compounds of Production Examples 10-1 to 10-24 and Production Examples 10-26 to 10-33 shown in the table below were produced. Production Example 10-25 was obtained by subjecting the condensation reaction to a method according to Production Example 10 and then cyclizing according to the method of Production Example 18.
Isolation and purification include extraction, fractional crystallization, various fractional chromatography (e.g., ODS column chromatography and silica gel column chromatography), etc., ordinary chemical operations and methods described in the production examples or examples of the present specification, Alternatively, a method suitable for the compound among methods equivalent thereto was applied.
単離、精製は、抽出、分別結晶化、各種分画クロマトグラフィー(例えばODSカラムクロマトグラフィーやシリカゲルカラムクロマトグラフィー)等、通常の化学操作及び本明細書の製造例若しくは実施例に記載の方法、またはそれに準じた方法のうち、化合物に適した方法を適用して行なった。 In the same manner as in Production Example 10, the compounds of Production Examples 10-1 to 10-24 and Production Examples 10-26 to 10-33 shown in the table below were produced. Production Example 10-25 was obtained by subjecting the condensation reaction to a method according to Production Example 10 and then cyclizing according to the method of Production Example 18.
Isolation and purification include extraction, fractional crystallization, various fractional chromatography (e.g., ODS column chromatography and silica gel column chromatography), etc., ordinary chemical operations and methods described in the production examples or examples of the present specification, Alternatively, a method suitable for the compound among methods equivalent thereto was applied.
製造例11
製造例33-24の化合物(451 mg)、BOP(328 mg)、DMF(6.77 mL)、DIPEA(d=0.742, 282.0 μL)の混合物を室温で1時間撹拌した。反応混合物に国際公開WO99/40108号の実施例番号169の化合物(642 mg)を加え室温で終夜撹拌した。反応液にEtOAc(300 mL)を加え生じた析出物をろ取した。ろ取物のMeOH-H2O溶液にPd/C(225.5 mg)のMeOH溶液を加え、H2雰囲気中で9時間撹拌した。さらに反応混合物にPd/C(90.0 mg)を追加して7時間撹拌した。反応液をセライトろ過(H2O:MeCN =1:1)で洗いこんだ。ろ液を軽く減圧下留去した後、得られた溶液にH2O:MeCN=80:20と1M NaOH水溶液を加え、ODSカラムクロマトグラフィー(H2O:MeCN=80:20から20:80まで)で精製し、製造例11の化合物(356 mg)を得た。 Production Example 11
A mixture of the compound of Production Example 33-24 (451 mg), BOP (328 mg), DMF (6.77 mL), and DIPEA (d = 0.742, 282.0 μL) was stirred at room temperature for 1 hour. To the reaction mixture, the compound of Example No. 169 (642 mg) of International Publication WO99 / 40108 was added and stirred at room temperature overnight. EtOAc (300 mL) was added to the reaction solution, and the resulting precipitate was collected by filtration. A MeOH solution of Pd / C (225.5 mg) was added to a MeOH-H 2 O solution of the filtered product, and the mixture was stirred for 9 hours in an H 2 atmosphere. Further, Pd / C (90.0 mg) was added to the reaction mixture, and the mixture was stirred for 7 hours. The reaction solution was washed by Celite filtration (H 2 O: MeCN = 1: 1). After the filtrate was distilled off lightly under reduced pressure, H 2 O: MeCN = 80: 20 and 1M NaOH aqueous solution were added to the resulting solution, and ODS column chromatography (H 2 O: MeCN = 80: 20 to 20:80 The compound of Production Example 11 (356 mg) was obtained.
製造例33-24の化合物(451 mg)、BOP(328 mg)、DMF(6.77 mL)、DIPEA(d=0.742, 282.0 μL)の混合物を室温で1時間撹拌した。反応混合物に国際公開WO99/40108号の実施例番号169の化合物(642 mg)を加え室温で終夜撹拌した。反応液にEtOAc(300 mL)を加え生じた析出物をろ取した。ろ取物のMeOH-H2O溶液にPd/C(225.5 mg)のMeOH溶液を加え、H2雰囲気中で9時間撹拌した。さらに反応混合物にPd/C(90.0 mg)を追加して7時間撹拌した。反応液をセライトろ過(H2O:MeCN =1:1)で洗いこんだ。ろ液を軽く減圧下留去した後、得られた溶液にH2O:MeCN=80:20と1M NaOH水溶液を加え、ODSカラムクロマトグラフィー(H2O:MeCN=80:20から20:80まで)で精製し、製造例11の化合物(356 mg)を得た。 Production Example 11
A mixture of the compound of Production Example 33-24 (451 mg), BOP (328 mg), DMF (6.77 mL), and DIPEA (d = 0.742, 282.0 μL) was stirred at room temperature for 1 hour. To the reaction mixture, the compound of Example No. 169 (642 mg) of International Publication WO99 / 40108 was added and stirred at room temperature overnight. EtOAc (300 mL) was added to the reaction solution, and the resulting precipitate was collected by filtration. A MeOH solution of Pd / C (225.5 mg) was added to a MeOH-H 2 O solution of the filtered product, and the mixture was stirred for 9 hours in an H 2 atmosphere. Further, Pd / C (90.0 mg) was added to the reaction mixture, and the mixture was stirred for 7 hours. The reaction solution was washed by Celite filtration (H 2 O: MeCN = 1: 1). After the filtrate was distilled off lightly under reduced pressure, H 2 O: MeCN = 80: 20 and 1M NaOH aqueous solution were added to the resulting solution, and ODS column chromatography (H 2 O: MeCN = 80: 20 to 20:80 The compound of Production Example 11 (356 mg) was obtained.
製造例12
2-(4-ブロモピリジン-2-イル)-1-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]エタノン オキシム (1.6 g)の CH2Cl2(16.0 mL)懸濁液にDIPEA(d=0.726, 1.71 mL)を加え、氷冷下TFAA(d=1.49, 865 μL)を滴下した。反応混合物を7℃~10 ℃で1時間、水冷下で2時間攪拌した。反応液を50 mLの水で洗浄した後、有機層を、MgSO4で乾燥後、減圧下濃縮した。シリカゲルカラムクロマトグラフィー(溶出液;Hexane:EtOAc=80:20)で精製し、4-ブロモ-2-{3-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]-2H-アジレン-2-イル}ピリジン(1.38 g)を油状物質として得た。 Production Example 12
To a suspension of 2- (4-bromopyridin-2-yl) -1- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] ethanone oxime (1.6 g) in CH 2 Cl 2 (16.0 mL) DIPEA (d = 0.726, 1.71 mL) was added, and TFAA (d = 1.49, 865 μL) was added dropwise under ice cooling. The reaction mixture was stirred at 7 ° C. to 10 ° C. for 1 hour and under water cooling for 2 hours. The reaction mixture was washed with 50 mL of water, and the organic layer was dried over MgSO 4 and concentrated under reduced pressure. Purification by silica gel column chromatography (eluent; Hexane: EtOAc = 80: 20) gave 4-bromo-2- {3- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] -2H-azilene- 2-yl} pyridine (1.38 g) was obtained as an oil.
2-(4-ブロモピリジン-2-イル)-1-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]エタノン オキシム (1.6 g)の CH2Cl2(16.0 mL)懸濁液にDIPEA(d=0.726, 1.71 mL)を加え、氷冷下TFAA(d=1.49, 865 μL)を滴下した。反応混合物を7℃~10 ℃で1時間、水冷下で2時間攪拌した。反応液を50 mLの水で洗浄した後、有機層を、MgSO4で乾燥後、減圧下濃縮した。シリカゲルカラムクロマトグラフィー(溶出液;Hexane:EtOAc=80:20)で精製し、4-ブロモ-2-{3-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]-2H-アジレン-2-イル}ピリジン(1.38 g)を油状物質として得た。 Production Example 12
To a suspension of 2- (4-bromopyridin-2-yl) -1- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] ethanone oxime (1.6 g) in CH 2 Cl 2 (16.0 mL) DIPEA (d = 0.726, 1.71 mL) was added, and TFAA (d = 1.49, 865 μL) was added dropwise under ice cooling. The reaction mixture was stirred at 7 ° C. to 10 ° C. for 1 hour and under water cooling for 2 hours. The reaction mixture was washed with 50 mL of water, and the organic layer was dried over MgSO 4 and concentrated under reduced pressure. Purification by silica gel column chromatography (eluent; Hexane: EtOAc = 80: 20) gave 4-bromo-2- {3- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] -2H-azilene- 2-yl} pyridine (1.38 g) was obtained as an oil.
製造例13
窒素雰囲気下で、5-ブロモ-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フエニル]ピリジン(1.20 g)、4-シクロヘキシル-4-メトキシピペリジン塩酸塩 (1.26 g)、Cs2CO3(5.26 g)のトルエン溶液(12 mL)にPd(dba)2,(413 mg)、BINAP (671 mg)を室温で加えた。反応溶液は、終夜還流した。室温まで冷却し、反応液にCHCl3水を加えて分配した。有機層はbrineで洗浄し、MgSO4で乾燥後、減圧下濃縮し、得られた固体はIPEに懸濁させ、30分間攪拌した。固体はIPEで洗浄及び粉末化し、5-(4-シクロヘキシル-4-メトキシピペリジン-1-イル)-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピリジン(632 mg)を橙色固体として得た。 Production Example 13
Under nitrogen atmosphere, 5-bromo-2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyridine (1.20 g), 4-cyclohexyl-4-methoxypiperidine hydrochloride (1.26 g), Cs 2 Pd (dba) 2 , (413 mg) and BINAP (671 mg) were added to a toluene solution (12 mL) of CO 3 (5.26 g) at room temperature. The reaction solution was refluxed overnight. The mixture was cooled to room temperature, and CHCl 3 water was added to the reaction solution to partition. The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The obtained solid was suspended in IPE and stirred for 30 minutes. The solid was washed and powdered with IPE, and 5- (4-cyclohexyl-4-methoxypiperidin-1-yl) -2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyridine (632 mg) was added. Obtained as an orange solid.
窒素雰囲気下で、5-ブロモ-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フエニル]ピリジン(1.20 g)、4-シクロヘキシル-4-メトキシピペリジン塩酸塩 (1.26 g)、Cs2CO3(5.26 g)のトルエン溶液(12 mL)にPd(dba)2,(413 mg)、BINAP (671 mg)を室温で加えた。反応溶液は、終夜還流した。室温まで冷却し、反応液にCHCl3水を加えて分配した。有機層はbrineで洗浄し、MgSO4で乾燥後、減圧下濃縮し、得られた固体はIPEに懸濁させ、30分間攪拌した。固体はIPEで洗浄及び粉末化し、5-(4-シクロヘキシル-4-メトキシピペリジン-1-イル)-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピリジン(632 mg)を橙色固体として得た。 Production Example 13
Under nitrogen atmosphere, 5-bromo-2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyridine (1.20 g), 4-cyclohexyl-4-methoxypiperidine hydrochloride (1.26 g), Cs 2 Pd (dba) 2 , (413 mg) and BINAP (671 mg) were added to a toluene solution (12 mL) of CO 3 (5.26 g) at room temperature. The reaction solution was refluxed overnight. The mixture was cooled to room temperature, and CHCl 3 water was added to the reaction solution to partition. The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The obtained solid was suspended in IPE and stirred for 30 minutes. The solid was washed and powdered with IPE, and 5- (4-cyclohexyl-4-methoxypiperidin-1-yl) -2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyridine (632 mg) was added. Obtained as an orange solid.
製造例14A
tert-ブチル4-{4-(メトキシカルボニル)-4'-[5-(ピペラジン-1-イル)ピリミジン-2-イル]ビフェニル-3-イル}-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(400 mg)、1-(2-ブロモエチル)-2-メチルベンゼン(215 mg)、K2CO3(199 mg)、NaI(162 mg)、DMI(13.3 mL)の混合物を140℃で20時間攪拌した。冷却後、EtOAcで希釈し、sat.NaHCO3水溶液で洗浄後、MgSO4で乾燥した。溶媒を減圧下濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液;CHCl3:EtOAc=100:0から3:1まで)で精製し、tert-ブチル4-[4-(メトキシカルボニル)-4'-(5-{4-[2-(2-メチルフェニル)エチル]ピペラジン-1-イル}ピリミジン-2-イル)ビフェニル-3-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(160 mg)を得た。 Production Example 14A
tert-butyl 4- {4- (methoxycarbonyl) -4 '-[5- (piperazin-1-yl) pyrimidin-2-yl] biphenyl-3-yl} -3,6-dihydropyridin-1 (2H)- A mixture of carboxylate (400 mg), 1- (2-bromoethyl) -2-methylbenzene (215 mg), K 2 CO 3 (199 mg), NaI (162 mg), DMI (13.3 mL) at 140 ° C Stir for 20 hours. After cooling, it was diluted with EtOAc, washed with sat. NaHCO 3 aqueous solution, and dried over MgSO 4 . After concentrating the solvent under reduced pressure, the residue was purified by silica gel column chromatography (eluent; CHCl 3 : EtOAc = 100: 0 to 3: 1) and tert-butyl 4- [4- (methoxycarbonyl) -4 ′. -(5- {4- [2- (2-Methylphenyl) ethyl] piperazin-1-yl} pyrimidin-2-yl) biphenyl-3-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (160 mg) was obtained.
tert-ブチル4-{4-(メトキシカルボニル)-4'-[5-(ピペラジン-1-イル)ピリミジン-2-イル]ビフェニル-3-イル}-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(400 mg)、1-(2-ブロモエチル)-2-メチルベンゼン(215 mg)、K2CO3(199 mg)、NaI(162 mg)、DMI(13.3 mL)の混合物を140℃で20時間攪拌した。冷却後、EtOAcで希釈し、sat.NaHCO3水溶液で洗浄後、MgSO4で乾燥した。溶媒を減圧下濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液;CHCl3:EtOAc=100:0から3:1まで)で精製し、tert-ブチル4-[4-(メトキシカルボニル)-4'-(5-{4-[2-(2-メチルフェニル)エチル]ピペラジン-1-イル}ピリミジン-2-イル)ビフェニル-3-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(160 mg)を得た。 Production Example 14A
tert-butyl 4- {4- (methoxycarbonyl) -4 '-[5- (piperazin-1-yl) pyrimidin-2-yl] biphenyl-3-yl} -3,6-dihydropyridin-1 (2H)- A mixture of carboxylate (400 mg), 1- (2-bromoethyl) -2-methylbenzene (215 mg), K 2 CO 3 (199 mg), NaI (162 mg), DMI (13.3 mL) at 140 ° C Stir for 20 hours. After cooling, it was diluted with EtOAc, washed with sat. NaHCO 3 aqueous solution, and dried over MgSO 4 . After concentrating the solvent under reduced pressure, the residue was purified by silica gel column chromatography (eluent; CHCl 3 : EtOAc = 100: 0 to 3: 1) and tert-butyl 4- [4- (methoxycarbonyl) -4 ′. -(5- {4- [2- (2-Methylphenyl) ethyl] piperazin-1-yl} pyrimidin-2-yl) biphenyl-3-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (160 mg) was obtained.
製造例14Aの方法と同様にして、後記表に示す製造例14A-1から製造例14A-15の化合物を製造した。単離、精製は、抽出、分別結晶化、各種分画クロマトグラフィー(例えばODSカラムクロマトグラフィーやシリカゲルカラムクロマトグラフィー)等、通常の化学操作及び本明細書の製造例若しくは実施例に記載の方法、またはそれに準じた方法のうち、化合物に適した方法を適用して行なった。
In the same manner as in Production Example 14A, the compounds of Production Example 14A-15 were produced from Production Examples 14A-1 shown in the table below. Isolation and purification include extraction, fractional crystallization, various fractional chromatography (e.g. ODS column chromatography and silica gel column chromatography), etc., ordinary chemical operations and methods described in the production examples or examples of the present specification, Alternatively, a method suitable for the compound was applied among the methods equivalent thereto.
製造例14B
氷冷下、トランス-4-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]シクロヘキサノール(200 mg)、CH2Cl2(2 mL)、AgOTf(310.06 mg)、2,6-ジtert-ブチルピリジン(d=0.870,306.2 μL)の混合物中に1-ヨードプロパン(d=1.743, 109 μL)を加え、10分撹拌した。室温に戻し1時間攪拌した。反応混合物をセライト濾過した後、ろ液を1M 塩酸水、sat.NaHCO3水、水、brineで洗浄し、MgSO4で乾燥後減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;EtOAc/Hexane)で精製し3-(4-ブロモフェニル)-5-(トランス-4-プロポキシシクロヘキシル)-1,2,4-オキサジアゾール (168 mg)を白色固体として得た。 Production Example 14B
Under cooling with ice, trans-4- [3- (4-bromophenyl) -1,2,4-oxadiazol-5-yl] cyclohexanol (200 mg), CH 2 Cl 2 (2 mL), AgOTf ( 1-iodopropane (d = 1.743, 109 μL) was added to a mixture of 310.06 mg) and 2,6-ditert-butylpyridine (d = 0.870, 306.2 μL), and the mixture was stirred for 10 minutes. It returned to room temperature and stirred for 1 hour. The reaction mixture was filtered through celite, and the filtrate was washed with 1M aqueous hydrochloric acid, sat. NaHCO 3 water, water and brine, dried over MgSO 4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent; EtOAc / Hexane), and 3- (4-bromophenyl) -5- (trans-4-propoxycyclohexyl) -1,2,4-oxadiazole (168 mg) Was obtained as a white solid.
氷冷下、トランス-4-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]シクロヘキサノール(200 mg)、CH2Cl2(2 mL)、AgOTf(310.06 mg)、2,6-ジtert-ブチルピリジン(d=0.870,306.2 μL)の混合物中に1-ヨードプロパン(d=1.743, 109 μL)を加え、10分撹拌した。室温に戻し1時間攪拌した。反応混合物をセライト濾過した後、ろ液を1M 塩酸水、sat.NaHCO3水、水、brineで洗浄し、MgSO4で乾燥後減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;EtOAc/Hexane)で精製し3-(4-ブロモフェニル)-5-(トランス-4-プロポキシシクロヘキシル)-1,2,4-オキサジアゾール (168 mg)を白色固体として得た。 Production Example 14B
Under cooling with ice, trans-4- [3- (4-bromophenyl) -1,2,4-oxadiazol-5-yl] cyclohexanol (200 mg), CH 2 Cl 2 (2 mL), AgOTf ( 1-iodopropane (d = 1.743, 109 μL) was added to a mixture of 310.06 mg) and 2,6-ditert-butylpyridine (d = 0.870, 306.2 μL), and the mixture was stirred for 10 minutes. It returned to room temperature and stirred for 1 hour. The reaction mixture was filtered through celite, and the filtrate was washed with 1M aqueous hydrochloric acid, sat. NaHCO 3 water, water and brine, dried over MgSO 4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent; EtOAc / Hexane), and 3- (4-bromophenyl) -5- (trans-4-propoxycyclohexyl) -1,2,4-oxadiazole (168 mg) Was obtained as a white solid.
製造例14Bの方法と同様にして、後記表に示す製造例14B-1から製造例14B-4の化合物を製造した。
In the same manner as in Production Example 14B, the compounds of Production Example 14B-4 were produced from Production Examples 14B-1 shown in the table below.
製造例14C
トランス-4-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]シクロヘキサノール (200 mg)と1.0Mのトリエチルオキソニウム テトラフルオロボレートのジクロロメタン溶液(1 mL)の混合物を室温で終夜撹拌した。反応液に水を加えEtOAcで抽出した。有機層を、水、brineで洗浄しMgSO4で乾燥した後、減圧下濃縮し得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液;Hexane/EtOAc系)で精製し目的物3-(4-ブロモフェニル)-5-(トランス-4-エトキシシクロヘキシル)-1,2,4-オキサジアゾール(173.2 mg)を白色固体として得た。 Production Example 14C
Trans-4- [3- (4-bromophenyl) -1,2,4-oxadiazol-5-yl] cyclohexanol (200 mg) and 1.0 M triethyloxonium tetrafluoroborate in dichloromethane (1 mL ) Was stirred at room temperature overnight. Water was added to the reaction solution and extracted with EtOAc. The organic layer was washed with water and brine, dried over MgSO 4 , and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (eluent: Hexane / EtOAc system) to obtain the desired product 3- (4-bromo Phenyl) -5- (trans-4-ethoxycyclohexyl) -1,2,4-oxadiazole (173.2 mg) was obtained as a white solid.
トランス-4-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]シクロヘキサノール (200 mg)と1.0Mのトリエチルオキソニウム テトラフルオロボレートのジクロロメタン溶液(1 mL)の混合物を室温で終夜撹拌した。反応液に水を加えEtOAcで抽出した。有機層を、水、brineで洗浄しMgSO4で乾燥した後、減圧下濃縮し得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液;Hexane/EtOAc系)で精製し目的物3-(4-ブロモフェニル)-5-(トランス-4-エトキシシクロヘキシル)-1,2,4-オキサジアゾール(173.2 mg)を白色固体として得た。 Production Example 14C
Trans-4- [3- (4-bromophenyl) -1,2,4-oxadiazol-5-yl] cyclohexanol (200 mg) and 1.0 M triethyloxonium tetrafluoroborate in dichloromethane (1 mL ) Was stirred at room temperature overnight. Water was added to the reaction solution and extracted with EtOAc. The organic layer was washed with water and brine, dried over MgSO 4 , and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (eluent: Hexane / EtOAc system) to obtain the desired product 3- (4-bromo Phenyl) -5- (trans-4-ethoxycyclohexyl) -1,2,4-oxadiazole (173.2 mg) was obtained as a white solid.
製造例15
tert-ブチル 4-ヒドロキシピペリジン-1-カルボキシレート(300 mg)のTHF(3.0 mL)溶液に0℃でAg2O(690 mg)を加え、6-ブロモ‐3,3-ジメチルシクロヘキセン(640 mg)のTHF溶液を加えた。室温で終夜攪拌した後ろ過し、ろ液をEtOAcで洗浄した。有機層はbrineで洗浄し、MgSO4で乾燥後、減圧下濃縮した。シリカゲルクロマトグラフィー(溶出液;Hexane/EtOAc系)で精製し、tert-ブチル 4-[(4,4-ジメチルシクロヘキシ-2-エン-1-イル)オキシ]ピペリジン-1-カルボキシレート(121 mg)を無色透明固体として得た。 Production Example 15
To a solution of tert-butyl 4-hydroxypiperidine-1-carboxylate (300 mg) in THF (3.0 mL) was added Ag 2 O (690 mg) at 0 ° C., and 6-bromo-3,3-dimethylcyclohexene (640 mg ) In THF. The mixture was stirred at room temperature overnight and filtered, and the filtrate was washed with EtOAc. The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. Purified by silica gel chromatography (eluent; Hexane / EtOAc system) and tert-butyl 4-[(4,4-dimethylcyclohexyl-2-en-1-yl) oxy] piperidine-1-carboxylate (121 mg ) Was obtained as a colorless transparent solid.
tert-ブチル 4-ヒドロキシピペリジン-1-カルボキシレート(300 mg)のTHF(3.0 mL)溶液に0℃でAg2O(690 mg)を加え、6-ブロモ‐3,3-ジメチルシクロヘキセン(640 mg)のTHF溶液を加えた。室温で終夜攪拌した後ろ過し、ろ液をEtOAcで洗浄した。有機層はbrineで洗浄し、MgSO4で乾燥後、減圧下濃縮した。シリカゲルクロマトグラフィー(溶出液;Hexane/EtOAc系)で精製し、tert-ブチル 4-[(4,4-ジメチルシクロヘキシ-2-エン-1-イル)オキシ]ピペリジン-1-カルボキシレート(121 mg)を無色透明固体として得た。 Production Example 15
To a solution of tert-butyl 4-hydroxypiperidine-1-carboxylate (300 mg) in THF (3.0 mL) was added Ag 2 O (690 mg) at 0 ° C., and 6-bromo-3,3-dimethylcyclohexene (640 mg ) In THF. The mixture was stirred at room temperature overnight and filtered, and the filtrate was washed with EtOAc. The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. Purified by silica gel chromatography (eluent; Hexane / EtOAc system) and tert-butyl 4-[(4,4-dimethylcyclohexyl-2-en-1-yl) oxy] piperidine-1-carboxylate (121 mg ) Was obtained as a colorless transparent solid.
製造例16
tert-ブチル 4-[4-(tert-ブトキシカルボニル)-4'-シアノビフェニル-3-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート (3.2 g)、ヒドロキシルアミン塩酸塩(1038 mg)、NaHCO3 (1751 mg)及びMeOH(48 mL)の混合物を80℃で6時間撹拌した。反応液に水を加え、EtOAcで抽出した。有機層を、水、brineで洗浄しMgSO4で乾燥した後、減圧下濃縮し、tert-ブチル 4-{4'-[アミノ(ヒドロキシイミノ)メチル]-4-(tert-ブトキシカルボニル)ビフェニル-3-イル}-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(3.2 g)を白色固体として得た。 Production Example 16
tert-Butyl 4- [4- (tert-butoxycarbonyl) -4'-cyanobiphenyl-3-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (3.2 g), hydroxylamine hydrochloride (1038 mg), NaHCO 3 (1751 mg) and MeOH (48 mL) were stirred at 80 ° C. for 6 h. Water was added to the reaction mixture, and the mixture was extracted with EtOAc. The organic layer was washed with water and brine, dried over MgSO 4 , concentrated under reduced pressure, and tert-butyl 4- {4 ′-[amino (hydroxyimino) methyl] -4- (tert-butoxycarbonyl) biphenyl- 3-yl} -3,6-dihydropyridine-1 (2H) -carboxylate (3.2 g) was obtained as a white solid.
tert-ブチル 4-[4-(tert-ブトキシカルボニル)-4'-シアノビフェニル-3-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート (3.2 g)、ヒドロキシルアミン塩酸塩(1038 mg)、NaHCO3 (1751 mg)及びMeOH(48 mL)の混合物を80℃で6時間撹拌した。反応液に水を加え、EtOAcで抽出した。有機層を、水、brineで洗浄しMgSO4で乾燥した後、減圧下濃縮し、tert-ブチル 4-{4'-[アミノ(ヒドロキシイミノ)メチル]-4-(tert-ブトキシカルボニル)ビフェニル-3-イル}-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(3.2 g)を白色固体として得た。 Production Example 16
tert-Butyl 4- [4- (tert-butoxycarbonyl) -4'-cyanobiphenyl-3-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (3.2 g), hydroxylamine hydrochloride (1038 mg), NaHCO 3 (1751 mg) and MeOH (48 mL) were stirred at 80 ° C. for 6 h. Water was added to the reaction mixture, and the mixture was extracted with EtOAc. The organic layer was washed with water and brine, dried over MgSO 4 , concentrated under reduced pressure, and tert-butyl 4- {4 ′-[amino (hydroxyimino) methyl] -4- (tert-butoxycarbonyl) biphenyl- 3-yl} -3,6-dihydropyridine-1 (2H) -carboxylate (3.2 g) was obtained as a white solid.
製造16の方法と同様にして、後記表に示す製造例16-1から製造例16-3の化合物を製造した。
In the same manner as in Production 16, the compounds of Production Example 16-1 to Production Example 16-3 shown in the table below were produced.
製造例17
1-[4-(ベンジルオキシ)フェニル]-2-ブロモエタン(2.48 g)、5-ブロモ-2-ピリジンアミン(155 g)、及び95% EtOH(20 mL)の反応混合物を3時間還流し、さらに60℃で12時間攪拌した。放冷した後、析出物をろ取し、2-[4-(ベンジルオキシ)フェニル]-6-ブロモイミダゾ[1,2-a]ピリジン(2.76 g)を固体として得た。 Production Example 17
A reaction mixture of 1- [4- (benzyloxy) phenyl] -2-bromoethane (2.48 g), 5-bromo-2-pyridinamine (155 g), and 95% EtOH (20 mL) was refluxed for 3 hours, The mixture was further stirred at 60 ° C. for 12 hours. After allowing to cool, the precipitate was collected by filtration to give 2- [4- (benzyloxy) phenyl] -6-bromoimidazo [1,2-a] pyridine (2.76 g) as a solid.
1-[4-(ベンジルオキシ)フェニル]-2-ブロモエタン(2.48 g)、5-ブロモ-2-ピリジンアミン(155 g)、及び95% EtOH(20 mL)の反応混合物を3時間還流し、さらに60℃で12時間攪拌した。放冷した後、析出物をろ取し、2-[4-(ベンジルオキシ)フェニル]-6-ブロモイミダゾ[1,2-a]ピリジン(2.76 g)を固体として得た。 Production Example 17
A reaction mixture of 1- [4- (benzyloxy) phenyl] -2-bromoethane (2.48 g), 5-bromo-2-pyridinamine (155 g), and 95% EtOH (20 mL) was refluxed for 3 hours, The mixture was further stirred at 60 ° C. for 12 hours. After allowing to cool, the precipitate was collected by filtration to give 2- [4- (benzyloxy) phenyl] -6-bromoimidazo [1,2-a] pyridine (2.76 g) as a solid.
製造例18
4-ブロモ-N'-({[4-(4-クロロフェニル)シクロヘキシル)カルボニル]オキシ)ベンゼンカルボキシイミダミド(862 mg)及びDMAc(8.62 mL)の混合物を120℃で6時間攪拌した。反応液を室温に戻し水を加え30分撹拌した。析出物をろ取した後、減圧乾燥し、3-(4-ブロモフェニル)-5-[4-(4-クロロフェニル)シクロヘキシル]-1,2,4-オキサジアゾール(732.9 mg)をベージュ色の粉末として得た。 Production Example 18
A mixture of 4-bromo-N ′-({[4- (4-chlorophenyl) cyclohexyl) carbonyl] oxy) benzenecarboxyimidamide (862 mg) and DMAc (8.62 mL) was stirred at 120 ° C. for 6 hours. The reaction solution was returned to room temperature, water was added, and the mixture was stirred for 30 minutes. The precipitate was collected by filtration and dried under reduced pressure to give 3- (4-bromophenyl) -5- [4- (4-chlorophenyl) cyclohexyl] -1,2,4-oxadiazole (732.9 mg) in beige color. Obtained as a powder.
4-ブロモ-N'-({[4-(4-クロロフェニル)シクロヘキシル)カルボニル]オキシ)ベンゼンカルボキシイミダミド(862 mg)及びDMAc(8.62 mL)の混合物を120℃で6時間攪拌した。反応液を室温に戻し水を加え30分撹拌した。析出物をろ取した後、減圧乾燥し、3-(4-ブロモフェニル)-5-[4-(4-クロロフェニル)シクロヘキシル]-1,2,4-オキサジアゾール(732.9 mg)をベージュ色の粉末として得た。 Production Example 18
A mixture of 4-bromo-N ′-({[4- (4-chlorophenyl) cyclohexyl) carbonyl] oxy) benzenecarboxyimidamide (862 mg) and DMAc (8.62 mL) was stirred at 120 ° C. for 6 hours. The reaction solution was returned to room temperature, water was added, and the mixture was stirred for 30 minutes. The precipitate was collected by filtration and dried under reduced pressure to give 3- (4-bromophenyl) -5- [4- (4-chlorophenyl) cyclohexyl] -1,2,4-oxadiazole (732.9 mg) in beige color. Obtained as a powder.
製造例18の方法と同様にして、後記表に示す製造例18-1から製造例18-22の化合物を製造した。
In the same manner as in Production Example 18, the compounds of Production Example 18-1 to Production Example 18-22 shown in the table below were produced.
製造例19
窒素雰囲気下、4-ブロモ-2-{3-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]-2H-アジレン-2-イル}ピリジン(1.36 g)のMeCN(13.6 mL)溶液に FeCl2(46.1 g)を加え油浴(60℃)で2時間加熱攪拌した。室温まで冷却後、MeCNを減圧下濃縮した。得られた残渣をCHCl3(200 mL)に溶解し、水(100 mL)を加えた。セライトでろ過して、不溶物を除いた。ろ液の有機層を分離し、水層をCHCl3(50 mL)で抽出した。抽出液をあわせ、MgSO4で乾燥後、活性炭処理した。ろ過後、ろ液を溶媒を留去した。粗生成物をシリカゲルカラムクロマトグラフィー(溶出液;Hexane:CHCl3= 2:1)で精製し、5-ブロモ-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピラゾロ[1,5-a]ピリジン (860 mg)得た。 Production Example 19
4-Bromo-2- {3- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] -2H-azilen-2-yl} pyridine (1.36 g) in MeCN (13.6 mL) under nitrogen atmosphere FeCl 2 (46.1 g) was added to the mixture, and the mixture was stirred with heating in an oil bath (60 ° C.) for 2 hours. After cooling to room temperature, MeCN was concentrated under reduced pressure. The obtained residue was dissolved in CHCl 3 (200 mL), and water (100 mL) was added. The insoluble matter was removed by filtration through Celite. The organic layer of the filtrate was separated and the aqueous layer was extracted with CHCl 3 (50 mL). The extracts were combined, dried over MgSO 4 and treated with activated carbon. After filtration, the solvent was distilled off from the filtrate. The crude product was purified by silica gel column chromatography (eluent; Hexane: CHCl 3 = 2: 1) to give 5-bromo-2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyrazolo [1 , 5-a] pyridine (860 mg) was obtained.
窒素雰囲気下、4-ブロモ-2-{3-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]-2H-アジレン-2-イル}ピリジン(1.36 g)のMeCN(13.6 mL)溶液に FeCl2(46.1 g)を加え油浴(60℃)で2時間加熱攪拌した。室温まで冷却後、MeCNを減圧下濃縮した。得られた残渣をCHCl3(200 mL)に溶解し、水(100 mL)を加えた。セライトでろ過して、不溶物を除いた。ろ液の有機層を分離し、水層をCHCl3(50 mL)で抽出した。抽出液をあわせ、MgSO4で乾燥後、活性炭処理した。ろ過後、ろ液を溶媒を留去した。粗生成物をシリカゲルカラムクロマトグラフィー(溶出液;Hexane:CHCl3= 2:1)で精製し、5-ブロモ-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピラゾロ[1,5-a]ピリジン (860 mg)得た。 Production Example 19
4-Bromo-2- {3- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] -2H-azilen-2-yl} pyridine (1.36 g) in MeCN (13.6 mL) under nitrogen atmosphere FeCl 2 (46.1 g) was added to the mixture, and the mixture was stirred with heating in an oil bath (60 ° C.) for 2 hours. After cooling to room temperature, MeCN was concentrated under reduced pressure. The obtained residue was dissolved in CHCl 3 (200 mL), and water (100 mL) was added. The insoluble matter was removed by filtration through Celite. The organic layer of the filtrate was separated and the aqueous layer was extracted with CHCl 3 (50 mL). The extracts were combined, dried over MgSO 4 and treated with activated carbon. After filtration, the solvent was distilled off from the filtrate. The crude product was purified by silica gel column chromatography (eluent; Hexane: CHCl 3 = 2: 1) to give 5-bromo-2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyrazolo [1 , 5-a] pyridine (860 mg) was obtained.
製造例20
4-ブロモ-N'-[(トランス-4-メチルシクロヘキシル)カルボニル]ベンゾヒドラジド (300 mg)、P2S5(216 mg)及びTHF(6 mL)の反応混合物を50℃で5時間撹拌した。反応液に水、1M NaOH水溶液(420 μL)を順次加え、室温で15分撹拌した。生じた析出物をろ取し、減圧下乾燥し、シリカゲルカラムクロマトグラフィー(溶出液;CHCl3:EtOAc=20:1)で精製し、2-(4-ブロモフェニル)-5-(トランス-4-メチルシクロヘキシル)-1,3,4-チアジアゾール (223 mg)を白色固体として得た。 Production Example 20
A reaction mixture of 4-bromo-N ′-[(trans-4-methylcyclohexyl) carbonyl] benzohydrazide (300 mg), P 2 S 5 (216 mg) and THF (6 mL) was stirred at 50 ° C. for 5 hours. . Water and 1M NaOH aqueous solution (420 μL) were sequentially added to the reaction solution, and the mixture was stirred at room temperature for 15 minutes. The resulting precipitate was collected by filtration, dried under reduced pressure, and purified by silica gel column chromatography (eluent; CHCl 3 : EtOAc = 20: 1) to give 2- (4-bromophenyl) -5- (trans-4 -Methylcyclohexyl) -1,3,4-thiadiazole (223 mg) was obtained as a white solid.
4-ブロモ-N'-[(トランス-4-メチルシクロヘキシル)カルボニル]ベンゾヒドラジド (300 mg)、P2S5(216 mg)及びTHF(6 mL)の反応混合物を50℃で5時間撹拌した。反応液に水、1M NaOH水溶液(420 μL)を順次加え、室温で15分撹拌した。生じた析出物をろ取し、減圧下乾燥し、シリカゲルカラムクロマトグラフィー(溶出液;CHCl3:EtOAc=20:1)で精製し、2-(4-ブロモフェニル)-5-(トランス-4-メチルシクロヘキシル)-1,3,4-チアジアゾール (223 mg)を白色固体として得た。 Production Example 20
A reaction mixture of 4-bromo-N ′-[(trans-4-methylcyclohexyl) carbonyl] benzohydrazide (300 mg), P 2 S 5 (216 mg) and THF (6 mL) was stirred at 50 ° C. for 5 hours. . Water and 1M NaOH aqueous solution (420 μL) were sequentially added to the reaction solution, and the mixture was stirred at room temperature for 15 minutes. The resulting precipitate was collected by filtration, dried under reduced pressure, and purified by silica gel column chromatography (eluent; CHCl 3 : EtOAc = 20: 1) to give 2- (4-bromophenyl) -5- (trans-4 -Methylcyclohexyl) -1,3,4-thiadiazole (223 mg) was obtained as a white solid.
製造例20の方法と同様にして、後記表に示す製造例20-1の化合物を製造した。
In the same manner as in Production Example 20, the compound of Production Example 20-1 shown in the table below was produced.
製造例21
4-ブロモ-N-[2-(4-シクロヘキシル-4-メトキシピペリジン-1-イル)-2-オキソエチル]ベンズアミド(1.67 g)、ピリジン(16.7 mL)、P2S5(1.70 g)の混合物を100℃で6時間攪拌した。反応液に水を加え、1M NaOH水溶液で塩基性として、15分間攪拌した。反応液を CHCl3で抽出し、1M HCl水で2回、水、brineで順次洗浄し、MgSO4で乾燥後、減圧下濃縮した。得られた残渣にMeOHを加え析出物をろ取した。残渣をMeOHで洗浄後、減圧下乾燥し、1-[2-(4-ブロモフェニル)-1,3-チアゾール-5-イル]-4-シクロヘキシル-4-メトキシピペリジン(1.25 g)をベージュ色固体として得た。 Production Example 21
A mixture of 4-bromo-N- [2- (4-cyclohexyl-4-methoxypiperidin-1-yl) -2-oxoethyl] benzamide (1.67 g), pyridine (16.7 mL), P 2 S 5 (1.70 g) Was stirred at 100 ° C. for 6 hours. Water was added to the reaction solution, and the mixture was made basic with 1M NaOH aqueous solution and stirred for 15 minutes. The reaction solution was extracted with CHCl 3 , washed twice with 1M aqueous HCl, water and brine in that order, dried over MgSO 4 , and concentrated under reduced pressure. MeOH was added to the obtained residue, and the precipitate was collected by filtration. The residue was washed with MeOH and dried under reduced pressure. 1- [2- (4-Bromophenyl) -1,3-thiazol-5-yl] -4-cyclohexyl-4-methoxypiperidine (1.25 g) was beige Obtained as a solid.
4-ブロモ-N-[2-(4-シクロヘキシル-4-メトキシピペリジン-1-イル)-2-オキソエチル]ベンズアミド(1.67 g)、ピリジン(16.7 mL)、P2S5(1.70 g)の混合物を100℃で6時間攪拌した。反応液に水を加え、1M NaOH水溶液で塩基性として、15分間攪拌した。反応液を CHCl3で抽出し、1M HCl水で2回、水、brineで順次洗浄し、MgSO4で乾燥後、減圧下濃縮した。得られた残渣にMeOHを加え析出物をろ取した。残渣をMeOHで洗浄後、減圧下乾燥し、1-[2-(4-ブロモフェニル)-1,3-チアゾール-5-イル]-4-シクロヘキシル-4-メトキシピペリジン(1.25 g)をベージュ色固体として得た。 Production Example 21
A mixture of 4-bromo-N- [2- (4-cyclohexyl-4-methoxypiperidin-1-yl) -2-oxoethyl] benzamide (1.67 g), pyridine (16.7 mL), P 2 S 5 (1.70 g) Was stirred at 100 ° C. for 6 hours. Water was added to the reaction solution, and the mixture was made basic with 1M NaOH aqueous solution and stirred for 15 minutes. The reaction solution was extracted with CHCl 3 , washed twice with 1M aqueous HCl, water and brine in that order, dried over MgSO 4 , and concentrated under reduced pressure. MeOH was added to the obtained residue, and the precipitate was collected by filtration. The residue was washed with MeOH and dried under reduced pressure. 1- [2- (4-Bromophenyl) -1,3-thiazol-5-yl] -4-cyclohexyl-4-methoxypiperidine (1.25 g) was beige Obtained as a solid.
製造例22
グローブボックス中、窒素気流下で充填した三径コルベン中で無水CeCl3 (7.62 g)を細かく粉砕した。三径コルベンをグローブボックスから取り出し、系内をアルゴンで置換した。コルベン中に無水THF を加え、反応混合物を、室温で終夜激しく攪拌した。反応系内に、n-ペンチルマグネシウムブロマイドの2.0Mのジエチルエーテル溶液(15.5 mL)を10分かけて加えて、氷浴を用いて内温を10℃以下に保った。反応混合物は室温で3時間攪拌した。tert-ブチル 4-オキソピペリジン-1-カルボキシレートの THF溶液を反応混合物に15分かけて添加し、氷浴を用いて、内温を10℃以下に保った。反応混合物は、4℃で1.5時間攪拌した。反応混合物中に、10% 酢酸をゆっくり加え、反応混合物はEtOAcで2回抽出した。有機層は、水とbrineで洗浄し、MgSO4で乾燥させた。ろ過後、を液を減圧下濃縮した。残渣は、シリカゲルカラムクロマトグラフィー(シリカゲル 120 g; 溶出液;Hexane:EtOAc = 95:5から75:25まで)で精製し tert-ブチル 4-ヒドロキシ-4-ペンチルピペリジン-1-カルボキシレート(5.16 g)を白色固体として得た。 Production Example 22
In a glove box, anhydrous CeCl 3 (7.62 g) was finely pulverized in a three-diameter Kolben filled under a nitrogen stream. The three-diameter Kolben was taken out from the glove box, and the system was replaced with argon. Anhydrous THF was added in Kolben and the reaction mixture was stirred vigorously at room temperature overnight. To the reaction system, a 2.0M diethyl ether solution (15.5 mL) of n-pentylmagnesium bromide was added over 10 minutes, and the internal temperature was kept at 10 ° C. or lower using an ice bath. The reaction mixture was stirred at room temperature for 3 hours. A THF solution of tert-butyl 4-oxopiperidine-1-carboxylate was added to the reaction mixture over 15 minutes, and the internal temperature was kept at 10 ° C. or lower using an ice bath. The reaction mixture was stirred at 4 ° C. for 1.5 hours. To the reaction mixture 10% acetic acid was added slowly and the reaction mixture was extracted twice with EtOAc. The organic layer was washed with water and brine and dried over MgSO 4 . After filtration, the solution was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (silica gel 120 g; eluent; Hexane: EtOAc = 95: 5 to 75:25) tert-butyl 4-hydroxy-4-pentylpiperidine-1-carboxylate (5.16 g ) Was obtained as a white solid.
グローブボックス中、窒素気流下で充填した三径コルベン中で無水CeCl3 (7.62 g)を細かく粉砕した。三径コルベンをグローブボックスから取り出し、系内をアルゴンで置換した。コルベン中に無水THF を加え、反応混合物を、室温で終夜激しく攪拌した。反応系内に、n-ペンチルマグネシウムブロマイドの2.0Mのジエチルエーテル溶液(15.5 mL)を10分かけて加えて、氷浴を用いて内温を10℃以下に保った。反応混合物は室温で3時間攪拌した。tert-ブチル 4-オキソピペリジン-1-カルボキシレートの THF溶液を反応混合物に15分かけて添加し、氷浴を用いて、内温を10℃以下に保った。反応混合物は、4℃で1.5時間攪拌した。反応混合物中に、10% 酢酸をゆっくり加え、反応混合物はEtOAcで2回抽出した。有機層は、水とbrineで洗浄し、MgSO4で乾燥させた。ろ過後、を液を減圧下濃縮した。残渣は、シリカゲルカラムクロマトグラフィー(シリカゲル 120 g; 溶出液;Hexane:EtOAc = 95:5から75:25まで)で精製し tert-ブチル 4-ヒドロキシ-4-ペンチルピペリジン-1-カルボキシレート(5.16 g)を白色固体として得た。 Production Example 22
In a glove box, anhydrous CeCl 3 (7.62 g) was finely pulverized in a three-diameter Kolben filled under a nitrogen stream. The three-diameter Kolben was taken out from the glove box, and the system was replaced with argon. Anhydrous THF was added in Kolben and the reaction mixture was stirred vigorously at room temperature overnight. To the reaction system, a 2.0M diethyl ether solution (15.5 mL) of n-pentylmagnesium bromide was added over 10 minutes, and the internal temperature was kept at 10 ° C. or lower using an ice bath. The reaction mixture was stirred at room temperature for 3 hours. A THF solution of tert-butyl 4-oxopiperidine-1-carboxylate was added to the reaction mixture over 15 minutes, and the internal temperature was kept at 10 ° C. or lower using an ice bath. The reaction mixture was stirred at 4 ° C. for 1.5 hours. To the reaction mixture 10% acetic acid was added slowly and the reaction mixture was extracted twice with EtOAc. The organic layer was washed with water and brine and dried over MgSO 4 . After filtration, the solution was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (silica gel 120 g; eluent; Hexane: EtOAc = 95: 5 to 75:25) tert-butyl 4-hydroxy-4-pentylpiperidine-1-carboxylate (5.16 g ) Was obtained as a white solid.
製造例22の方法と同様にして、後記表に示す製造例22-1から製造例22-4の化合物を製造した。
In the same manner as in Production Example 22, the compounds of Production Example 22-4 to Production Example 22-4 shown in the table below were produced.
製造例23
2-(4-ブロモフェニル)-1,3,4-オキサジアゾールの酢酸溶液に臭素(251.8 μL)を室温で10分かけて加えた。室温で1.5時間攪拌した。反応液を、50℃に昇温し1.5時間攪拌し、臭素を(114.5 μL)追加し、50℃で5時間攪拌した。反応液に臭素を(114.5 μL)を加え50℃で5時間攪拌した。反応液を室温に戻し、1M NaOH水溶液(200 mL)、EtOAc(200 mL)を加えた。有機層を分離し、1M NaOH水溶液(100 mL)で洗浄した。10%Na2S2O3水溶液、水、brineで洗浄後、MgSO4で乾燥し、減圧下濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(Hexane:EtOAc系)で精製し、 2-ブロモ-5-(4-ブロモフェニル)-1,3,4-オキサジアゾール(404 mg)を白色固体として得た。 Production Example 23
Bromine (251.8 μL) was added to 2- (4-bromophenyl) -1,3,4-oxadiazole in acetic acid over 10 minutes at room temperature. Stir at room temperature for 1.5 hours. The reaction mixture was heated to 50 ° C. and stirred for 1.5 hours, bromine (114.5 μL) was added, and the mixture was stirred at 50 ° C. for 5 hours. Bromine (114.5 μL) was added to the reaction solution, and the mixture was stirred at 50 ° C. for 5 hours. The reaction solution was returned to room temperature, and 1M NaOH aqueous solution (200 mL) and EtOAc (200 mL) were added. The organic layer was separated and washed with 1M aqueous NaOH (100 mL). The residue obtained after washing with 10% Na 2 S 2 O 3 aqueous solution, water and brine, drying over MgSO 4 and concentrating under reduced pressure was purified by silica gel column chromatography (Hexane: EtOAc system). -5- (4-Bromophenyl) -1,3,4-oxadiazole (404 mg) was obtained as a white solid.
2-(4-ブロモフェニル)-1,3,4-オキサジアゾールの酢酸溶液に臭素(251.8 μL)を室温で10分かけて加えた。室温で1.5時間攪拌した。反応液を、50℃に昇温し1.5時間攪拌し、臭素を(114.5 μL)追加し、50℃で5時間攪拌した。反応液に臭素を(114.5 μL)を加え50℃で5時間攪拌した。反応液を室温に戻し、1M NaOH水溶液(200 mL)、EtOAc(200 mL)を加えた。有機層を分離し、1M NaOH水溶液(100 mL)で洗浄した。10%Na2S2O3水溶液、水、brineで洗浄後、MgSO4で乾燥し、減圧下濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(Hexane:EtOAc系)で精製し、 2-ブロモ-5-(4-ブロモフェニル)-1,3,4-オキサジアゾール(404 mg)を白色固体として得た。 Production Example 23
Bromine (251.8 μL) was added to 2- (4-bromophenyl) -1,3,4-oxadiazole in acetic acid over 10 minutes at room temperature. Stir at room temperature for 1.5 hours. The reaction mixture was heated to 50 ° C. and stirred for 1.5 hours, bromine (114.5 μL) was added, and the mixture was stirred at 50 ° C. for 5 hours. Bromine (114.5 μL) was added to the reaction solution, and the mixture was stirred at 50 ° C. for 5 hours. The reaction solution was returned to room temperature, and 1M NaOH aqueous solution (200 mL) and EtOAc (200 mL) were added. The organic layer was separated and washed with 1M aqueous NaOH (100 mL). The residue obtained after washing with 10% Na 2 S 2 O 3 aqueous solution, water and brine, drying over MgSO 4 and concentrating under reduced pressure was purified by silica gel column chromatography (Hexane: EtOAc system). -5- (4-Bromophenyl) -1,3,4-oxadiazole (404 mg) was obtained as a white solid.
製造例23の方法と同様にして、後記表に示す製造例23-1から製造例23-2の化合物を製造した。
In the same manner as in Production Example 23, the compounds of Production Example 23-2 to Production Example 23-2 shown in the table below were produced.
製造例24
アルゴン雰囲気下、4-(4-ブロモフェニル)-1,3-オキサゾール(787 mg)とTHF(17.3 mL)の混合物に内温-65~-60℃で LHMDS (4.7 mL)を8分かけて滴下した。反応混合物をMeCN/ドライアイスバス中、内温-43℃で30分撹拌した。ヘキサクロロエタン(1.66 g)を加えて室温に昇温し終夜撹拌した。反応液にEtOAcと水を加えた。有機層を分離し、0.1M塩酸水、brineで洗浄した。MgSO4で乾燥し濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製し(Hexane/EtOAc系) 4-(4-ブロモフェニル)-2-クロロ-1,3-オキサゾール (630 mg)を白色固体として得た。 Production Example 24
Under an argon atmosphere, LHMDS (4.7 mL) was added to a mixture of 4- (4-bromophenyl) -1,3-oxazole (787 mg) and THF (17.3 mL) at an internal temperature of -65 to -60 ° C over 8 minutes. It was dripped. The reaction mixture was stirred in a MeCN / dry ice bath at an internal temperature of −43 ° C. for 30 minutes. Hexachloroethane (1.66 g) was added, the temperature was raised to room temperature, and the mixture was stirred overnight. EtOAc and water were added to the reaction solution. The organic layer was separated and washed with 0.1M aqueous hydrochloric acid and brine. Dried over MgSO 4 and concentrated. The residue was purified by silica gel column chromatography (Hexane / EtOAc system) to give 4- (4-bromophenyl) -2-chloro-1,3-oxazole (630 mg) as a white solid.
アルゴン雰囲気下、4-(4-ブロモフェニル)-1,3-オキサゾール(787 mg)とTHF(17.3 mL)の混合物に内温-65~-60℃で LHMDS (4.7 mL)を8分かけて滴下した。反応混合物をMeCN/ドライアイスバス中、内温-43℃で30分撹拌した。ヘキサクロロエタン(1.66 g)を加えて室温に昇温し終夜撹拌した。反応液にEtOAcと水を加えた。有機層を分離し、0.1M塩酸水、brineで洗浄した。MgSO4で乾燥し濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製し(Hexane/EtOAc系) 4-(4-ブロモフェニル)-2-クロロ-1,3-オキサゾール (630 mg)を白色固体として得た。 Production Example 24
Under an argon atmosphere, LHMDS (4.7 mL) was added to a mixture of 4- (4-bromophenyl) -1,3-oxazole (787 mg) and THF (17.3 mL) at an internal temperature of -65 to -60 ° C over 8 minutes. It was dripped. The reaction mixture was stirred in a MeCN / dry ice bath at an internal temperature of −43 ° C. for 30 minutes. Hexachloroethane (1.66 g) was added, the temperature was raised to room temperature, and the mixture was stirred overnight. EtOAc and water were added to the reaction solution. The organic layer was separated and washed with 0.1M aqueous hydrochloric acid and brine. Dried over MgSO 4 and concentrated. The residue was purified by silica gel column chromatography (Hexane / EtOAc system) to give 4- (4-bromophenyl) -2-chloro-1,3-oxazole (630 mg) as a white solid.
製造例25
0℃にて1-ベンジル-4-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]ピリジニウムブロミド(1.24 g)のMeOH白色懸濁液にNaBH4(198 mg)を少しずつ加えた。黄色の反応懸濁液を同温で1時間攪拌した。反応混合物中に固体のNH4Clを加え、減圧下溶媒を留去した。残渣に水を加え、EtOAc:THF=4:1の溶液を用いて抽出操作を行った。有機層をbrineで洗浄し、MgSO4で乾燥後、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;Hexane:EtOAc=80:20から60:40まで)で精製し、1-ベンジル-4-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]-1,2,3,6-テトラヒドロピリジン(0.39 g)を黄色油状物として得た。 Production Example 25
To a white suspension of 1-benzyl-4- [3- (4-bromophenyl) -1,2,4-oxadiazol-5-yl] pyridinium bromide (1.24 g) at 0 ° C. in NaBH 4 ( 198 mg) was added in small portions. The yellow reaction suspension was stirred at the same temperature for 1 hour. Solid NH 4 Cl was added to the reaction mixture, and the solvent was distilled off under reduced pressure. Water was added to the residue, and an extraction operation was performed using a solution of EtOAc: THF = 4: 1. The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent; Hexane: EtOAc = 80: 20 to 60:40), and 1-benzyl-4- [3- (4-bromophenyl) -1,2,4-oxa Diazol-5-yl] -1,2,3,6-tetrahydropyridine (0.39 g) was obtained as a yellow oil.
0℃にて1-ベンジル-4-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]ピリジニウムブロミド(1.24 g)のMeOH白色懸濁液にNaBH4(198 mg)を少しずつ加えた。黄色の反応懸濁液を同温で1時間攪拌した。反応混合物中に固体のNH4Clを加え、減圧下溶媒を留去した。残渣に水を加え、EtOAc:THF=4:1の溶液を用いて抽出操作を行った。有機層をbrineで洗浄し、MgSO4で乾燥後、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;Hexane:EtOAc=80:20から60:40まで)で精製し、1-ベンジル-4-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]-1,2,3,6-テトラヒドロピリジン(0.39 g)を黄色油状物として得た。 Production Example 25
To a white suspension of 1-benzyl-4- [3- (4-bromophenyl) -1,2,4-oxadiazol-5-yl] pyridinium bromide (1.24 g) at 0 ° C. in NaBH 4 ( 198 mg) was added in small portions. The yellow reaction suspension was stirred at the same temperature for 1 hour. Solid NH 4 Cl was added to the reaction mixture, and the solvent was distilled off under reduced pressure. Water was added to the residue, and an extraction operation was performed using a solution of EtOAc: THF = 4: 1. The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent; Hexane: EtOAc = 80: 20 to 60:40), and 1-benzyl-4- [3- (4-bromophenyl) -1,2,4-oxa Diazol-5-yl] -1,2,3,6-tetrahydropyridine (0.39 g) was obtained as a yellow oil.
製造例26
5-ブロモ-1,3,4-チアジアゾール-2-アミン(26 g)、2-ブロモ-1-(4-ブロモフェニル)エタノン(40.1 g)のn-BuOHの懸濁液(780 mL)を125℃で20時間攪拌した。反応混合物は、放冷した後EtOH(1600 mL)に注ぎ、2時間攪拌した後、析出した固体をろ取した。固体をEtOHにて洗浄し2-ブロモ-6-(4-ブロモフェニル)イミダゾ[2,1-b][1,3,4]チアジアゾールを白色粉末(38.3 g)を得た。 Production Example 26
A suspension of n-BuOH (780 mL) of 5-bromo-1,3,4-thiadiazol-2-amine (26 g) and 2-bromo-1- (4-bromophenyl) ethanone (40.1 g) was added. The mixture was stirred at 125 ° C. for 20 hours. The reaction mixture was allowed to cool, poured into EtOH (1600 mL), stirred for 2 hours, and the precipitated solid was collected by filtration. The solid was washed with EtOH to give 2-bromo-6- (4-bromophenyl) imidazo [2,1-b] [1,3,4] thiadiazole as a white powder (38.3 g).
5-ブロモ-1,3,4-チアジアゾール-2-アミン(26 g)、2-ブロモ-1-(4-ブロモフェニル)エタノン(40.1 g)のn-BuOHの懸濁液(780 mL)を125℃で20時間攪拌した。反応混合物は、放冷した後EtOH(1600 mL)に注ぎ、2時間攪拌した後、析出した固体をろ取した。固体をEtOHにて洗浄し2-ブロモ-6-(4-ブロモフェニル)イミダゾ[2,1-b][1,3,4]チアジアゾールを白色粉末(38.3 g)を得た。 Production Example 26
A suspension of n-BuOH (780 mL) of 5-bromo-1,3,4-thiadiazol-2-amine (26 g) and 2-bromo-1- (4-bromophenyl) ethanone (40.1 g) was added. The mixture was stirred at 125 ° C. for 20 hours. The reaction mixture was allowed to cool, poured into EtOH (1600 mL), stirred for 2 hours, and the precipitated solid was collected by filtration. The solid was washed with EtOH to give 2-bromo-6- (4-bromophenyl) imidazo [2,1-b] [1,3,4] thiadiazole as a white powder (38.3 g).
製造例27
窒素雰囲気下、2-[(6-ブロモ-1-クロロ-2-ナフチル)オキシ]テトラヒドロ-2H-ピラン (54.5 g、ホウ酸トリイソプロピル(44 mL)、THF(550 mL)を混合させた。2.76M n-BuLiの-ヘキサン溶液(69.4 mL)を内温-65~-60℃で30分かけて滴下し、室温に昇温しながら1時間撹拌した。反応液にMeOH(30 mL)を加え1時間撹拌した。brine(400 mL)を加え、1M塩酸水でpH=4に調整した。EtOAc(1000 mL、500mL)で抽出した。有機層をbrine(400 mL)で洗浄し、MgSO4で乾燥後、減圧下濃縮して得た残渣にヘキサン(600 mL)を加えて析出物をろ取した後、減圧乾燥し、[5-クロロ-6-(テトラヒドロ-2H-ピラン-2-イルオキシ)-2-ナフチル]ボロン酸(46.7 g)を白色固体として得た。 Production Example 27
Under a nitrogen atmosphere, 2-[(6-bromo-1-chloro-2-naphthyl) oxy] tetrahydro-2H-pyran (54.5 g, triisopropyl borate (44 mL), and THF (550 mL) were mixed. 2.76M n-BuLi in hexane (69.4 mL) was added dropwise over 30 minutes at an internal temperature of -65 to -60 ° C., and the mixture was stirred for 1 hour while warming to room temperature. The mixture was added and stirred for 1 hour, and brine (400 mL) was added, and the pH was adjusted with 1 M aqueous hydrochloric acid to pH 4. The mixture was extracted with EtOAc (1000 mL, 500 mL) The organic layer was washed with brine (400 mL), and MgSO 4. After drying with hexane, hexane (600 mL) was added to the residue obtained by concentration under reduced pressure, and the precipitate was collected by filtration and then dried under reduced pressure to give [5-chloro-6- (tetrahydro-2H-pyran-2-yloxy]. ) -2-Naphthyl] boronic acid (46.7 g) was obtained as a white solid.
窒素雰囲気下、2-[(6-ブロモ-1-クロロ-2-ナフチル)オキシ]テトラヒドロ-2H-ピラン (54.5 g、ホウ酸トリイソプロピル(44 mL)、THF(550 mL)を混合させた。2.76M n-BuLiの-ヘキサン溶液(69.4 mL)を内温-65~-60℃で30分かけて滴下し、室温に昇温しながら1時間撹拌した。反応液にMeOH(30 mL)を加え1時間撹拌した。brine(400 mL)を加え、1M塩酸水でpH=4に調整した。EtOAc(1000 mL、500mL)で抽出した。有機層をbrine(400 mL)で洗浄し、MgSO4で乾燥後、減圧下濃縮して得た残渣にヘキサン(600 mL)を加えて析出物をろ取した後、減圧乾燥し、[5-クロロ-6-(テトラヒドロ-2H-ピラン-2-イルオキシ)-2-ナフチル]ボロン酸(46.7 g)を白色固体として得た。 Production Example 27
Under a nitrogen atmosphere, 2-[(6-bromo-1-chloro-2-naphthyl) oxy] tetrahydro-2H-pyran (54.5 g, triisopropyl borate (44 mL), and THF (550 mL) were mixed. 2.76M n-BuLi in hexane (69.4 mL) was added dropwise over 30 minutes at an internal temperature of -65 to -60 ° C., and the mixture was stirred for 1 hour while warming to room temperature. The mixture was added and stirred for 1 hour, and brine (400 mL) was added, and the pH was adjusted with 1 M aqueous hydrochloric acid to pH 4. The mixture was extracted with EtOAc (1000 mL, 500 mL) The organic layer was washed with brine (400 mL), and MgSO 4. After drying with hexane, hexane (600 mL) was added to the residue obtained by concentration under reduced pressure, and the precipitate was collected by filtration and then dried under reduced pressure to give [5-chloro-6- (tetrahydro-2H-pyran-2-yloxy]. ) -2-Naphthyl] boronic acid (46.7 g) was obtained as a white solid.
製造例28
ヒドロキシカーボンイミジック ジブロマイド (5.00 g)のDMF(25 mL)溶液に、5℃で1-ブロモ-4-ビニルベンゼン(5.41 g)を加え、続いてNaHCO3(6.16 g)水溶液を滴下し、終夜攪拌した。反応混合物に水を加え、EtOAcで抽出し、有機層を減圧下濃縮した。得られた残渣はシリカゲルカラムクロマトグラフィー(溶出液;Hexane:EtOAc= 90:10)で精製し、3-ブロモ-5-(4-ブロモフェニル)-4,5-ジヒドロイソキサゾール(7.14 g)を得た。 Production Example 28
Hydroxycarbon imidic dibromide (5.00 g) in DMF (25 mL) was added 1-bromo-4-vinylbenzene (5.41 g) at 5 ° C., followed by dropwise addition of aqueous NaHCO 3 (6.16 g), Stir overnight. Water was added to the reaction mixture, extraction was performed with EtOAc, and the organic layer was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent; Hexane: EtOAc = 90: 10) to give 3-bromo-5- (4-bromophenyl) -4,5-dihydroisoxazole (7.14 g) Got.
ヒドロキシカーボンイミジック ジブロマイド (5.00 g)のDMF(25 mL)溶液に、5℃で1-ブロモ-4-ビニルベンゼン(5.41 g)を加え、続いてNaHCO3(6.16 g)水溶液を滴下し、終夜攪拌した。反応混合物に水を加え、EtOAcで抽出し、有機層を減圧下濃縮した。得られた残渣はシリカゲルカラムクロマトグラフィー(溶出液;Hexane:EtOAc= 90:10)で精製し、3-ブロモ-5-(4-ブロモフェニル)-4,5-ジヒドロイソキサゾール(7.14 g)を得た。 Production Example 28
Hydroxycarbon imidic dibromide (5.00 g) in DMF (25 mL) was added 1-bromo-4-vinylbenzene (5.41 g) at 5 ° C., followed by dropwise addition of aqueous NaHCO 3 (6.16 g), Stir overnight. Water was added to the reaction mixture, extraction was performed with EtOAc, and the organic layer was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent; Hexane: EtOAc = 90: 10) to give 3-bromo-5- (4-bromophenyl) -4,5-dihydroisoxazole (7.14 g) Got.
製造例29
過ヨウ素酸ナトリウム(957 mg)と酢酸アンモニウム(345 mg)の水溶液に tert-ブチル 4-[2-(メトキシカルボニル)-5-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート (1.32 g)のEtOAc溶液を加えて3日間撹拌した。析出物をろ別してEtOAcで洗浄した。ろ液と洗浄液を合わせて分液し、有機層を5%Na2S2O3水溶液、brineで洗浄し、MgSO4で乾燥し、減圧下濃縮した後、粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(溶出液; CHCl3:MeOH=100:0 から90:10まで)にて精製し、{3-[1-(tert-ブトキシカルボニル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4-(メトキシカルボニル)フェニル}ボロン酸(860 mg)を得た。 Production Example 29
To an aqueous solution of sodium periodate (957 mg) and ammonium acetate (345 mg), tert-butyl 4- [2- (methoxycarbonyl) -5- (4,4,5,5-tetramethyl-1,3,2 -Dioxaborolan-2-yl) phenyl] -3,6-dihydropyridine-1 (2H) -carboxylate (1.32 g) in EtOAc was added and stirred for 3 days. The precipitate was filtered off and washed with EtOAc. The filtrate and the washing solution were combined and separated, and the organic layer was washed with 5% Na 2 S 2 O 3 aqueous solution and brine, dried over MgSO 4 and concentrated under reduced pressure to obtain a crude product. The crude product was purified by silica gel column chromatography (eluent; CHCl 3 : MeOH = 100: 0 to 90:10), and {3- [1- (tert-butoxycarbonyl) -1,2,3, 6-Tetrahydropyridin-4-yl] -4- (methoxycarbonyl) phenyl} boronic acid (860 mg) was obtained.
過ヨウ素酸ナトリウム(957 mg)と酢酸アンモニウム(345 mg)の水溶液に tert-ブチル 4-[2-(メトキシカルボニル)-5-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート (1.32 g)のEtOAc溶液を加えて3日間撹拌した。析出物をろ別してEtOAcで洗浄した。ろ液と洗浄液を合わせて分液し、有機層を5%Na2S2O3水溶液、brineで洗浄し、MgSO4で乾燥し、減圧下濃縮した後、粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(溶出液; CHCl3:MeOH=100:0 から90:10まで)にて精製し、{3-[1-(tert-ブトキシカルボニル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4-(メトキシカルボニル)フェニル}ボロン酸(860 mg)を得た。 Production Example 29
To an aqueous solution of sodium periodate (957 mg) and ammonium acetate (345 mg), tert-butyl 4- [2- (methoxycarbonyl) -5- (4,4,5,5-tetramethyl-1,3,2 -Dioxaborolan-2-yl) phenyl] -3,6-dihydropyridine-1 (2H) -carboxylate (1.32 g) in EtOAc was added and stirred for 3 days. The precipitate was filtered off and washed with EtOAc. The filtrate and the washing solution were combined and separated, and the organic layer was washed with 5% Na 2 S 2 O 3 aqueous solution and brine, dried over MgSO 4 and concentrated under reduced pressure to obtain a crude product. The crude product was purified by silica gel column chromatography (eluent; CHCl 3 : MeOH = 100: 0 to 90:10), and {3- [1- (tert-butoxycarbonyl) -1,2,3, 6-Tetrahydropyridin-4-yl] -4- (methoxycarbonyl) phenyl} boronic acid (860 mg) was obtained.
製造例29の方法と同様にして、後記表に示す製造例29-1の化合物を製造した。
In the same manner as in Production Example 29, the compound of Production Example 29-1 shown in the table below was produced.
製造例30
tert-ブチル 4-{3-[4-(5-クロロ-6-{[(トリフルオロメチル)スルホニル]オキシ}-2-ナフチル)フェニル]-1,2,4-オキサジアゾール-5-イル}ピペラジン-1-カルボキシレート(850 mg)、 DMI(56.7 mL)、Pd(OAc)2(34 mg)、Et3N (d=0.726, 935 μL)、プロパン-1,3-ジイルビス(ジフェニルホスフィン(125 mg)及びMeOH(22.6 mL)の反応混合物を、一酸化炭素で充填した。反応混合物は、一酸化炭素雰囲気下で5時間95℃で攪拌した。反応混合物を室温まで冷却し、水(200 mL)を加えた。反応混合物をろ過し、得られた固体を水で洗浄した。固体をCHCl3に溶解させた。有機層はNa2SO4で乾燥後、減圧下濃縮した。残渣をシリカゲルクロマトグラフィー(溶出液;Hexane:CHCl3=50:50からEtOAc:CHCl3=15:75まで)で精製しtert-ブチル 4-(3-{4-[5-クロロ-6-(メトキカルボニル)-2-ナフチル]フェニル}-1,2,4-オキサジアゾール-5-イル)ピペラジン-1-カルボキシレート(440 mg)を白色固体として得た。 Production Example 30
tert-butyl 4- {3- [4- (5-chloro-6-{[(trifluoromethyl) sulfonyl] oxy} -2-naphthyl) phenyl] -1,2,4-oxadiazol-5-yl } Piperazine-1-carboxylate (850 mg), DMI (56.7 mL), Pd (OAc) 2 (34 mg), Et 3 N (d = 0.726, 935 μL), Propane-1,3-diylbis (diphenylphosphine) (125 mg) and MeOH (22.6 mL) were charged with carbon monoxide, which was stirred under a carbon monoxide atmosphere for 5 hours at 95 ° C. The reaction mixture was cooled to room temperature and water ( The reaction mixture was filtered and the resulting solid was washed with water, and the solid was dissolved in CHCl 3. The organic layer was dried over Na 2 SO 4 and concentrated under reduced pressure. Purified by silica gel chromatography (eluent; Hexane: CHCl 3 = 50: 50 to EtOAc: CHCl 3 = 15: 75) and purified by tert-butyl 4- (3- {4- [5-chloro-6- (methoxycarbonyl) ) -2-Naphtyl] phenyl} -1,2,4-oxadiazo 5-yl) piperazine-1-carboxylate (440 mg) as a white solid.
tert-ブチル 4-{3-[4-(5-クロロ-6-{[(トリフルオロメチル)スルホニル]オキシ}-2-ナフチル)フェニル]-1,2,4-オキサジアゾール-5-イル}ピペラジン-1-カルボキシレート(850 mg)、 DMI(56.7 mL)、Pd(OAc)2(34 mg)、Et3N (d=0.726, 935 μL)、プロパン-1,3-ジイルビス(ジフェニルホスフィン(125 mg)及びMeOH(22.6 mL)の反応混合物を、一酸化炭素で充填した。反応混合物は、一酸化炭素雰囲気下で5時間95℃で攪拌した。反応混合物を室温まで冷却し、水(200 mL)を加えた。反応混合物をろ過し、得られた固体を水で洗浄した。固体をCHCl3に溶解させた。有機層はNa2SO4で乾燥後、減圧下濃縮した。残渣をシリカゲルクロマトグラフィー(溶出液;Hexane:CHCl3=50:50からEtOAc:CHCl3=15:75まで)で精製しtert-ブチル 4-(3-{4-[5-クロロ-6-(メトキカルボニル)-2-ナフチル]フェニル}-1,2,4-オキサジアゾール-5-イル)ピペラジン-1-カルボキシレート(440 mg)を白色固体として得た。 Production Example 30
tert-butyl 4- {3- [4- (5-chloro-6-{[(trifluoromethyl) sulfonyl] oxy} -2-naphthyl) phenyl] -1,2,4-oxadiazol-5-yl } Piperazine-1-carboxylate (850 mg), DMI (56.7 mL), Pd (OAc) 2 (34 mg), Et 3 N (d = 0.726, 935 μL), Propane-1,3-diylbis (diphenylphosphine) (125 mg) and MeOH (22.6 mL) were charged with carbon monoxide, which was stirred under a carbon monoxide atmosphere for 5 hours at 95 ° C. The reaction mixture was cooled to room temperature and water ( The reaction mixture was filtered and the resulting solid was washed with water, and the solid was dissolved in CHCl 3. The organic layer was dried over Na 2 SO 4 and concentrated under reduced pressure. Purified by silica gel chromatography (eluent; Hexane: CHCl 3 = 50: 50 to EtOAc: CHCl 3 = 15: 75) and purified by tert-butyl 4- (3- {4- [5-chloro-6- (methoxycarbonyl) ) -2-Naphtyl] phenyl} -1,2,4-oxadiazo 5-yl) piperazine-1-carboxylate (440 mg) as a white solid.
製造例31
Production Example 31
tert-ブチル 4-ヒドロキシ-4-ペンチルピペリジン-1-カルボキシレート(590 mg)をTHFに溶解し、氷冷下NaH(60%油性懸濁、130 mg)を加え、室温で1時間撹拌した。反応液を氷冷し、ヨードメタン(0.925 g)を加えて5分間撹拌し、さらに、室温で5時間撹拌した。反応液に水を加え、EtOAcと水で分配し、水層をEtOAcで2回抽出した。まとめた有機層をbrineで洗浄し、MgSO4で乾燥した後、減圧下濃縮し、tert-ブチル 4-メトキシ-4-ペンチルピペリジン-1-カルボキシレート(590 mg)を無色透明油状物質として得た。
tert-Butyl 4-hydroxy-4-pentylpiperidine-1-carboxylate (590 mg) was dissolved in THF, and NaH (60% oil suspension, 130 mg) was added under ice cooling, followed by stirring at room temperature for 1 hour. The reaction solution was ice-cooled, iodomethane (0.925 g) was added, and the mixture was stirred for 5 minutes, and further stirred at room temperature for 5 hours. Water was added to the reaction mixture, and the mixture was partitioned between EtOAc and water, and the aqueous layer was extracted twice with EtOAc. The combined organic layers were washed with brine, dried over MgSO 4 and concentrated under reduced pressure to obtain tert-butyl 4-methoxy-4-pentylpiperidine-1-carboxylate (590 mg) as a colorless transparent oily substance. .
製造例31の方法と同様にして、後記表に示す製造例31-1から製造例31-5の化合物を製造した。
In the same manner as in Production Example 31, the compounds of Production Example 31-1 to Production Example 31-5 shown in the table below were produced.
製造例32
6-アセトキシ-2-ナフチル酸(5 g)、TBAI(12 g)及びPd(OAc)2(500 mg) のDMF(100 mL)溶液中にNIS(5 g)を加えた。反応溶液を45℃で8時間攪拌した。反応混合物中にNIS(5 g)を加え、45℃で12時間攪拌した。反応混合物中に、NIS(5 g)及びPd(OAc)2(100 mg)を加え、20時間撹拌後、Pd(OAc)2(100mg)及びNIS(5g)を加え、48時間攪拌した。反応混合物を0℃まで冷却し、K2CO3を加えた。反応溶液中に、MeIを加えて、室温で1時間攪拌した。反応混合物をEtOAcで希釈した。有機層をNa2S2O3水溶液、1M NaOH水溶液、H2O、0.05M HCl で2回、H2O、NaHCO3水溶液、brineで洗浄しNa2SO4で乾燥後.減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフイー(溶出液;CHCl3、Hexane/EtOAc)で精製し、メチル 6-アセトキシ-3-ヨード-2-ナフトエート(6.37 g)を不純物の混ざりの黄色固体として得た。 Production Example 32
NIS (5 g) was added to a solution of 6-acetoxy-2-naphthylic acid (5 g), TBAI (12 g) and Pd (OAc) 2 (500 mg) in DMF (100 mL). The reaction solution was stirred at 45 ° C. for 8 hours. NIS (5 g) was added to the reaction mixture, and the mixture was stirred at 45 ° C. for 12 hours. NIS (5 g) and Pd (OAc) 2 (100 mg) were added to the reaction mixture, and after stirring for 20 hours, Pd (OAc) 2 (100 mg) and NIS (5 g) were added and stirred for 48 hours. The reaction mixture was cooled to 0 ° C. and K 2 CO 3 was added. MeI was added to the reaction solution and stirred at room temperature for 1 hour. The reaction mixture was diluted with EtOAc. The organic layer was washed with Na 2 S 2 O 3 aqueous solution, 1M NaOH aqueous solution, H 2 O, 0.05M HCl twice, H 2 O, NaHCO 3 aqueous solution, brine, dried over Na 2 SO 4 and concentrated under reduced pressure. . The obtained residue was purified by silica gel column chromatography (eluent; CHCl 3 , Hexane / EtOAc) to obtain methyl 6-acetoxy-3-iodo-2-naphthoate (6.37 g) as a yellow solid mixed with impurities. It was.
6-アセトキシ-2-ナフチル酸(5 g)、TBAI(12 g)及びPd(OAc)2(500 mg) のDMF(100 mL)溶液中にNIS(5 g)を加えた。反応溶液を45℃で8時間攪拌した。反応混合物中にNIS(5 g)を加え、45℃で12時間攪拌した。反応混合物中に、NIS(5 g)及びPd(OAc)2(100 mg)を加え、20時間撹拌後、Pd(OAc)2(100mg)及びNIS(5g)を加え、48時間攪拌した。反応混合物を0℃まで冷却し、K2CO3を加えた。反応溶液中に、MeIを加えて、室温で1時間攪拌した。反応混合物をEtOAcで希釈した。有機層をNa2S2O3水溶液、1M NaOH水溶液、H2O、0.05M HCl で2回、H2O、NaHCO3水溶液、brineで洗浄しNa2SO4で乾燥後.減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフイー(溶出液;CHCl3、Hexane/EtOAc)で精製し、メチル 6-アセトキシ-3-ヨード-2-ナフトエート(6.37 g)を不純物の混ざりの黄色固体として得た。 Production Example 32
NIS (5 g) was added to a solution of 6-acetoxy-2-naphthylic acid (5 g), TBAI (12 g) and Pd (OAc) 2 (500 mg) in DMF (100 mL). The reaction solution was stirred at 45 ° C. for 8 hours. NIS (5 g) was added to the reaction mixture, and the mixture was stirred at 45 ° C. for 12 hours. NIS (5 g) and Pd (OAc) 2 (100 mg) were added to the reaction mixture, and after stirring for 20 hours, Pd (OAc) 2 (100 mg) and NIS (5 g) were added and stirred for 48 hours. The reaction mixture was cooled to 0 ° C. and K 2 CO 3 was added. MeI was added to the reaction solution and stirred at room temperature for 1 hour. The reaction mixture was diluted with EtOAc. The organic layer was washed with Na 2 S 2 O 3 aqueous solution, 1M NaOH aqueous solution, H 2 O, 0.05M HCl twice, H 2 O, NaHCO 3 aqueous solution, brine, dried over Na 2 SO 4 and concentrated under reduced pressure. . The obtained residue was purified by silica gel column chromatography (eluent; CHCl 3 , Hexane / EtOAc) to obtain methyl 6-acetoxy-3-iodo-2-naphthoate (6.37 g) as a yellow solid mixed with impurities. It was.
製造例33
メチル 3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-4-カルボキシレート(164 mg)のジオキサン溶液に1M NaOH水溶液(2.52 mL)を加え100℃で5時間撹拌した。反応液を室温まで冷却した後、減圧下濃縮し、水、1M 塩酸水を加えpHを3とした。析出物をろ取した。得られた固体はIPEで洗浄し、3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-4-カルボン酸(151 mg)を得た。 Production Example 33
Methyl 3- [1- (2-hydroxyethyl) -1,2,3,6-tetrahydropyridin-4-yl] -4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] 1M NaOH aqueous solution (2.52 mL) was added to a dioxane solution of imidazo [2,1-b] [1,3,4] thiadiazol-6-yl} biphenyl-4-carboxylate (164 mg) and stirred at 100 ° C. for 5 hours. did. The reaction mixture was cooled to room temperature, concentrated under reduced pressure, and adjusted to pH 3 with water and 1M aqueous hydrochloric acid. The precipitate was collected by filtration. The obtained solid was washed with IPE and 3- [1- (2-hydroxyethyl) -1,2,3,6-tetrahydropyridin-4-yl] -4 ′-{2- [4- (2- Phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazol-6-yl} biphenyl-4-carboxylic acid (151 mg) was obtained.
メチル 3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-4-カルボキシレート(164 mg)のジオキサン溶液に1M NaOH水溶液(2.52 mL)を加え100℃で5時間撹拌した。反応液を室温まで冷却した後、減圧下濃縮し、水、1M 塩酸水を加えpHを3とした。析出物をろ取した。得られた固体はIPEで洗浄し、3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-4-カルボン酸(151 mg)を得た。 Production Example 33
Methyl 3- [1- (2-hydroxyethyl) -1,2,3,6-tetrahydropyridin-4-yl] -4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] 1M NaOH aqueous solution (2.52 mL) was added to a dioxane solution of imidazo [2,1-b] [1,3,4] thiadiazol-6-yl} biphenyl-4-carboxylate (164 mg) and stirred at 100 ° C. for 5 hours. did. The reaction mixture was cooled to room temperature, concentrated under reduced pressure, and adjusted to pH 3 with water and 1M aqueous hydrochloric acid. The precipitate was collected by filtration. The obtained solid was washed with IPE and 3- [1- (2-hydroxyethyl) -1,2,3,6-tetrahydropyridin-4-yl] -4 ′-{2- [4- (2- Phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazol-6-yl} biphenyl-4-carboxylic acid (151 mg) was obtained.
製造例33の方法と同様にして、後記表に示す製造例33-1から製造例33-135の化合物を製造した。但し、製造例33-25の化合物は、製造例33の方法に準じて加水分解後、反応液に水を加え生じた析出物を濾取して得た。
単離、精製は、抽出、分別結晶化、各種分画クロマトグラフィー(例えばODSカラムクロマトグラフィーやシリカゲルカラムクロマトグラフィー)等、通常の化学操作及び本明細書の製造例若しくは実施例に記載の方法、またはそれに準じた方法のうち、化合物に適した方法を適用して行なった。 In the same manner as in Production Example 33, the compounds of Production Examples 33-1 to 33-135 shown in the table below were produced. However, the compound of Production Example 33-25 was obtained by hydrolysis according to the method of Production Example 33, and then adding water to the reaction solution to collect the resulting precipitate.
Isolation and purification include extraction, fractional crystallization, various fractional chromatography (e.g., ODS column chromatography and silica gel column chromatography), etc., ordinary chemical operations and methods described in the production examples or examples of the present specification, Alternatively, a method suitable for the compound among methods equivalent thereto was applied.
単離、精製は、抽出、分別結晶化、各種分画クロマトグラフィー(例えばODSカラムクロマトグラフィーやシリカゲルカラムクロマトグラフィー)等、通常の化学操作及び本明細書の製造例若しくは実施例に記載の方法、またはそれに準じた方法のうち、化合物に適した方法を適用して行なった。 In the same manner as in Production Example 33, the compounds of Production Examples 33-1 to 33-135 shown in the table below were produced. However, the compound of Production Example 33-25 was obtained by hydrolysis according to the method of Production Example 33, and then adding water to the reaction solution to collect the resulting precipitate.
Isolation and purification include extraction, fractional crystallization, various fractional chromatography (e.g., ODS column chromatography and silica gel column chromatography), etc., ordinary chemical operations and methods described in the production examples or examples of the present specification, Alternatively, a method suitable for the compound among methods equivalent thereto was applied.
製造例34
0℃で2-(3-イソプロポキシフェニル)エタノール(200 mg)のCH2Cl2溶液(2 mL)中、デス-マーチン ペルヨージナン(565 mg)を加え、室温で2時間攪拌した。反応混合物に、sat.NaHCO3水溶液(5 mL)、sat.Na2S2O3水溶液(5 mL)を加え、CH2Cl2 (5 mL)で2回抽出した。有機層をMgSO4で乾燥後、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;Hexane:EtOAc=90 :10から70:30まで)で精製し、(3-イソプロポキシフェニル)アセトアルデヒド(175 mg)を無色油状物質として得た。 Production Example 34
Dess-Martin periodinane (565 mg) was added to a CH 2 Cl 2 solution (2 mL) of 2- (3-isopropoxyphenyl) ethanol (200 mg) at 0 ° C., and the mixture was stirred at room temperature for 2 hours. To the reaction mixture were added sat. NaHCO 3 aqueous solution (5 mL) and sat. Na 2 S 2 O 3 aqueous solution (5 mL), and the mixture was extracted twice with CH 2 Cl 2 (5 mL). The organic layer was dried over MgSO 4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent; Hexane: EtOAc = 90: 10 to 70:30) to give (3-isopropoxyphenyl) acetaldehyde (175 mg) as a colorless oil.
0℃で2-(3-イソプロポキシフェニル)エタノール(200 mg)のCH2Cl2溶液(2 mL)中、デス-マーチン ペルヨージナン(565 mg)を加え、室温で2時間攪拌した。反応混合物に、sat.NaHCO3水溶液(5 mL)、sat.Na2S2O3水溶液(5 mL)を加え、CH2Cl2 (5 mL)で2回抽出した。有機層をMgSO4で乾燥後、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;Hexane:EtOAc=90 :10から70:30まで)で精製し、(3-イソプロポキシフェニル)アセトアルデヒド(175 mg)を無色油状物質として得た。 Production Example 34
Dess-Martin periodinane (565 mg) was added to a CH 2 Cl 2 solution (2 mL) of 2- (3-isopropoxyphenyl) ethanol (200 mg) at 0 ° C., and the mixture was stirred at room temperature for 2 hours. To the reaction mixture were added sat. NaHCO 3 aqueous solution (5 mL) and sat. Na 2 S 2 O 3 aqueous solution (5 mL), and the mixture was extracted twice with CH 2 Cl 2 (5 mL). The organic layer was dried over MgSO 4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent; Hexane: EtOAc = 90: 10 to 70:30) to give (3-isopropoxyphenyl) acetaldehyde (175 mg) as a colorless oil.
製造例34の方法と同様にして、後記表に示す製造例34-1から製造例34-3の化合物を製造した。
製造例35
DMF(2 mL)中、実施例161の化合物(110 mg)、BOC2O(19.9 mg)及びDIPEA(d=0.742、31.8 μL )の反応混合物を25℃で4時間攪拌した。反応混合物をEtOAc(20 mL)で希釈すると沈殿物が生じ、ろ取した。得られた固体をEtOAcで洗浄して製造例35の化合物(102 mg)を得た。 In the same manner as in Production Example 34, the compounds of Production Example 34-1 to Production Example 34-3 shown in the table below were produced.
Production Example 35
A reaction mixture of the compound of Example 161 (110 mg), BOC 2 O (19.9 mg) and DIPEA (d = 0.742, 31.8 μL) in DMF (2 mL) was stirred at 25 ° C. for 4 hours. The reaction mixture was diluted with EtOAc (20 mL) to form a precipitate that was collected by filtration. The obtained solid was washed with EtOAc to give the compound of Production Example 35 (102 mg).
製造例35
DMF(2 mL)中、実施例161の化合物(110 mg)、BOC2O(19.9 mg)及びDIPEA(d=0.742、31.8 μL )の反応混合物を25℃で4時間攪拌した。反応混合物をEtOAc(20 mL)で希釈すると沈殿物が生じ、ろ取した。得られた固体をEtOAcで洗浄して製造例35の化合物(102 mg)を得た。 In the same manner as in Production Example 34, the compounds of Production Example 34-1 to Production Example 34-3 shown in the table below were produced.
Production Example 35
A reaction mixture of the compound of Example 161 (110 mg), BOC 2 O (19.9 mg) and DIPEA (d = 0.742, 31.8 μL) in DMF (2 mL) was stirred at 25 ° C. for 4 hours. The reaction mixture was diluted with EtOAc (20 mL) to form a precipitate that was collected by filtration. The obtained solid was washed with EtOAc to give the compound of Production Example 35 (102 mg).
製造例36
CH2Cl2 (65 mL)中、メチル 2-クロロ-4-ヒドロキシベンゾエート (8.73 g), 3,4-ジヒドロ-2H-ピラン(11.986 g)及びPPTS(4 g)の混合物を室温で18時間攪拌した。反応混合物をEtOAcで希釈後、sat.NaHCO3水溶液で洗浄した。分離した有機層をbrineで洗浄し、MgSO4で乾燥後、減圧下濃縮し、得られた残渣をシリカゲルカラムクロマト(Hexane:EtOAc=9:1)で精製し、メチル 2-クロロ-4-(テトラヒドロ-2H-ピラン-2-イルオキシ)ベンゾエート(10.6 g)を黄色油状物として得た。 Production Example 36
A mixture of methyl 2-chloro-4-hydroxybenzoate (8.73 g), 3,4-dihydro-2H-pyran (11.986 g) and PPTS (4 g) in CH 2 Cl 2 (65 mL) at room temperature for 18 hours. Stir. The reaction mixture was diluted with EtOAc and washed with sat. NaHCO 3 aqueous solution. The separated organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (Hexane: EtOAc = 9: 1), and methyl 2-chloro-4- ( Tetrahydro-2H-pyran-2-yloxy) benzoate (10.6 g) was obtained as a yellow oil.
CH2Cl2 (65 mL)中、メチル 2-クロロ-4-ヒドロキシベンゾエート (8.73 g), 3,4-ジヒドロ-2H-ピラン(11.986 g)及びPPTS(4 g)の混合物を室温で18時間攪拌した。反応混合物をEtOAcで希釈後、sat.NaHCO3水溶液で洗浄した。分離した有機層をbrineで洗浄し、MgSO4で乾燥後、減圧下濃縮し、得られた残渣をシリカゲルカラムクロマト(Hexane:EtOAc=9:1)で精製し、メチル 2-クロロ-4-(テトラヒドロ-2H-ピラン-2-イルオキシ)ベンゾエート(10.6 g)を黄色油状物として得た。 Production Example 36
A mixture of methyl 2-chloro-4-hydroxybenzoate (8.73 g), 3,4-dihydro-2H-pyran (11.986 g) and PPTS (4 g) in CH 2 Cl 2 (65 mL) at room temperature for 18 hours. Stir. The reaction mixture was diluted with EtOAc and washed with sat. NaHCO 3 aqueous solution. The separated organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (Hexane: EtOAc = 9: 1), and methyl 2-chloro-4- ( Tetrahydro-2H-pyran-2-yloxy) benzoate (10.6 g) was obtained as a yellow oil.
製造例36の方法と同様にして、後記表に示す製造例36-1から製造例36-3の化合物を製造した。
製造例37
氷冷下、トランス-4-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]シクロヘキサノール(410 mg)、ベンジル 2,2,2-トリクロロエタンイミデート(545 mg)、CH2Cl2(6.15 mL)の混合物中にTfOH(0.0553 mL)を加えた。室温に昇温し2時間撹拌した。反応液をEtOAcで希釈し、sat.NaHCO3水溶液を加えた。有機層をbrineで洗浄し、MgSO4で乾燥した後、減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(溶出液;Hexane:EtOAc系)で精製し、5-[トランス-4-(ベンジルオキシ)シクロヘキシル]-3-(4-ブロモフェニル)-1,2,4-オキサジアゾール(293 mg)を白色固体として得た In the same manner as in Production Example 36, the compounds of Production Example 36-1 to Production Example 36-3 shown in the table below were produced.
Production Example 37
Under ice-cooling, trans-4- [3- (4-bromophenyl) -1,2,4-oxadiazol-5-yl] cyclohexanol (410 mg), benzyl 2,2,2-trichloroethaneimidate ( TfOH (0.0553 mL) was added to a mixture of 545 mg) and CH 2 Cl 2 (6.15 mL). The mixture was warmed to room temperature and stirred for 2 hours. The reaction solution was diluted with EtOAc, and sat. NaHCO 3 aqueous solution was added. The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent; Hexane: EtOAc system) to give 5- [trans-4- (benzyloxy) Cyclohexyl] -3- (4-bromophenyl) -1,2,4-oxadiazole (293 mg) was obtained as a white solid
製造例37
氷冷下、トランス-4-[3-(4-ブロモフェニル)-1,2,4-オキサジアゾール-5-イル]シクロヘキサノール(410 mg)、ベンジル 2,2,2-トリクロロエタンイミデート(545 mg)、CH2Cl2(6.15 mL)の混合物中にTfOH(0.0553 mL)を加えた。室温に昇温し2時間撹拌した。反応液をEtOAcで希釈し、sat.NaHCO3水溶液を加えた。有機層をbrineで洗浄し、MgSO4で乾燥した後、減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(溶出液;Hexane:EtOAc系)で精製し、5-[トランス-4-(ベンジルオキシ)シクロヘキシル]-3-(4-ブロモフェニル)-1,2,4-オキサジアゾール(293 mg)を白色固体として得た In the same manner as in Production Example 36, the compounds of Production Example 36-1 to Production Example 36-3 shown in the table below were produced.
Production Example 37
Under ice-cooling, trans-4- [3- (4-bromophenyl) -1,2,4-oxadiazol-5-yl] cyclohexanol (410 mg), benzyl 2,2,2-trichloroethaneimidate ( TfOH (0.0553 mL) was added to a mixture of 545 mg) and CH 2 Cl 2 (6.15 mL). The mixture was warmed to room temperature and stirred for 2 hours. The reaction solution was diluted with EtOAc, and sat. NaHCO 3 aqueous solution was added. The organic layer was washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent; Hexane: EtOAc system) to give 5- [trans-4- (benzyloxy) Cyclohexyl] -3- (4-bromophenyl) -1,2,4-oxadiazole (293 mg) was obtained as a white solid
製造例38
2-[4-(ベンジルオキシ)フェニル]-6-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[1,2-a]ピリジン(270 mg)、10%Pd/C(294 mg)、MeOH(8.1 mL)、THF(5.4 mL)の混合物を3気圧の水素下、室温にて終夜攪拌した。溶媒を減圧下濃縮し、4-{6-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[1,2-a]ピリジン-2-イル}フェノール(194 mg)を灰色固体として得た。 Production Example 38
2- [4- (Benzyloxy) phenyl] -6- [4- (2-phenylethyl) piperazin-1-yl] imidazo [1,2-a] pyridine (270 mg), 10% Pd / C (294 mg), MeOH (8.1 mL), and THF (5.4 mL) were stirred overnight at room temperature under 3 atm of hydrogen. The solvent was concentrated under reduced pressure to give 4- {6- [4- (2-phenylethyl) piperazin-1-yl] imidazo [1,2-a] pyridin-2-yl} phenol (194 mg) as a gray solid Obtained.
2-[4-(ベンジルオキシ)フェニル]-6-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[1,2-a]ピリジン(270 mg)、10%Pd/C(294 mg)、MeOH(8.1 mL)、THF(5.4 mL)の混合物を3気圧の水素下、室温にて終夜攪拌した。溶媒を減圧下濃縮し、4-{6-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[1,2-a]ピリジン-2-イル}フェノール(194 mg)を灰色固体として得た。 Production Example 38
2- [4- (Benzyloxy) phenyl] -6- [4- (2-phenylethyl) piperazin-1-yl] imidazo [1,2-a] pyridine (270 mg), 10% Pd / C (294 mg), MeOH (8.1 mL), and THF (5.4 mL) were stirred overnight at room temperature under 3 atm of hydrogen. The solvent was concentrated under reduced pressure to give 4- {6- [4- (2-phenylethyl) piperazin-1-yl] imidazo [1,2-a] pyridin-2-yl} phenol (194 mg) as a gray solid Obtained.
製造例38の方法と同様にして、後記表に示す製造例38-1の化合物を製造した。
In the same manner as in Production Example 38, the compound of Production Example 38-1 shown in the table below was produced.
製造例39
常圧水素雰囲気下、ベンジル トランス-4-(3-メチルブトキシ)シクロヘキサンカルボキシレート(700 mg)、EtOAc (19.5 mL)、及びPd/C 10%(50% wet 70 mg)の混合物を室温で5時間撹拌した。反応液をセライトろ過してPd/Cを除去し、ろ液を減圧下濃縮し、トランス-4-(3-メトキシブトキシ)シクロヘキサンカルボン酸(470 mg)を白色固体として得た。 Production Example 39
Under normal pressure hydrogen atmosphere, a mixture of benzyl trans-4- (3-methylbutoxy) cyclohexanecarboxylate (700 mg), EtOAc (19.5 mL), and Pd / C 10% (50% wet 70 mg) was stirred at room temperature for 5 hours. Stir for hours. The reaction solution was filtered through Celite to remove Pd / C, and the filtrate was concentrated under reduced pressure to obtain trans-4- (3-methoxybutoxy) cyclohexanecarboxylic acid (470 mg) as a white solid.
常圧水素雰囲気下、ベンジル トランス-4-(3-メチルブトキシ)シクロヘキサンカルボキシレート(700 mg)、EtOAc (19.5 mL)、及びPd/C 10%(50% wet 70 mg)の混合物を室温で5時間撹拌した。反応液をセライトろ過してPd/Cを除去し、ろ液を減圧下濃縮し、トランス-4-(3-メトキシブトキシ)シクロヘキサンカルボン酸(470 mg)を白色固体として得た。 Production Example 39
Under normal pressure hydrogen atmosphere, a mixture of benzyl trans-4- (3-methylbutoxy) cyclohexanecarboxylate (700 mg), EtOAc (19.5 mL), and Pd / C 10% (50% wet 70 mg) was stirred at room temperature for 5 hours. Stir for hours. The reaction solution was filtered through Celite to remove Pd / C, and the filtrate was concentrated under reduced pressure to obtain trans-4- (3-methoxybutoxy) cyclohexanecarboxylic acid (470 mg) as a white solid.
製造例39の方法と同様にして、後記表に示す製造例39-1から製造例39-3の化合物を製造した。
In the same manner as in Production Example 39, the compounds of Production Example 39-1 to Production Example 39-3 shown in the table below were produced.
製造例40
tert-ブチル 4-[4-(メトキシカルボニル)-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-3-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート (287 mg)をジオキサン(3.5 mL)に溶解し、4M HCl/ジオキサン(3.5 mL)を加え、室温で1時間撹拌した。反応混合物をIPEで希釈し、沈殿物をろ取してIPEで洗浄し、減圧下に乾燥した。得られた粉末にCH2Cl2を加えた。有機層はNaHCO3、brineで洗浄し、有機層を減圧下濃縮し、メチル 4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}-3-(1,2,3,6-テトラヒドロピリジン-4-イル)ビフェニル-4-カルボキシレート (201 mg)を得た。 Production Example 40
tert-Butyl 4- [4- (methoxycarbonyl) -4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazole -6-yl} biphenyl-3-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (287 mg) in dioxane (3.5 mL), 4M HCl / dioxane (3.5 mL) was added, Stir at room temperature for 1 hour. The reaction mixture was diluted with IPE, the precipitate was collected by filtration, washed with IPE, and dried under reduced pressure. CH 2 Cl 2 was added to the obtained powder. The organic layer was washed with NaHCO 3 , brine, the organic layer was concentrated under reduced pressure, and methyl 4 ′-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2,1-b] [ 1,3,4] thiadiazol-6-yl} -3- (1,2,3,6-tetrahydropyridin-4-yl) biphenyl-4-carboxylate (201 mg) was obtained.
tert-ブチル 4-[4-(メトキシカルボニル)-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-3-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート (287 mg)をジオキサン(3.5 mL)に溶解し、4M HCl/ジオキサン(3.5 mL)を加え、室温で1時間撹拌した。反応混合物をIPEで希釈し、沈殿物をろ取してIPEで洗浄し、減圧下に乾燥した。得られた粉末にCH2Cl2を加えた。有機層はNaHCO3、brineで洗浄し、有機層を減圧下濃縮し、メチル 4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}-3-(1,2,3,6-テトラヒドロピリジン-4-イル)ビフェニル-4-カルボキシレート (201 mg)を得た。 Production Example 40
tert-Butyl 4- [4- (methoxycarbonyl) -4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazole -6-yl} biphenyl-3-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (287 mg) in dioxane (3.5 mL), 4M HCl / dioxane (3.5 mL) was added, Stir at room temperature for 1 hour. The reaction mixture was diluted with IPE, the precipitate was collected by filtration, washed with IPE, and dried under reduced pressure. CH 2 Cl 2 was added to the obtained powder. The organic layer was washed with NaHCO 3 , brine, the organic layer was concentrated under reduced pressure, and methyl 4 ′-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2,1-b] [ 1,3,4] thiadiazol-6-yl} -3- (1,2,3,6-tetrahydropyridin-4-yl) biphenyl-4-carboxylate (201 mg) was obtained.
製造例40の方法と同様にして、後記表に示す製造例40-1から製造例40-23の化合物を製造した。
製造例40-2は製造例40に準じて反応を行ったのち、反応混合物を減圧下に濃縮し、残渣の粉末として得た。
製造例40-1から40-23の化合物は、製造例40に準じて反応を行ったのち、塩酸塩の化合物は製造例40-2に準じた方法で化合物を単離し、その他の化合物は製造例40に準じた方法で化合物を単離して得た。 In the same manner as in Production Example 40, the compounds of Production Example 40-1 to Production Example 40-23 shown in the table below were produced.
In Production Example 40-2, the reaction was performed according to Production Example 40, and then the reaction mixture was concentrated under reduced pressure to obtain a residual powder.
After the compounds of Production Examples 40-1 to 40-23 were reacted according to Production Example 40, the hydrochloride compound was isolated by the method according to Production Example 40-2, and the other compounds were produced. A compound was isolated and obtained by a method according to Example 40.
製造例40-2は製造例40に準じて反応を行ったのち、反応混合物を減圧下に濃縮し、残渣の粉末として得た。
製造例40-1から40-23の化合物は、製造例40に準じて反応を行ったのち、塩酸塩の化合物は製造例40-2に準じた方法で化合物を単離し、その他の化合物は製造例40に準じた方法で化合物を単離して得た。 In the same manner as in Production Example 40, the compounds of Production Example 40-1 to Production Example 40-23 shown in the table below were produced.
In Production Example 40-2, the reaction was performed according to Production Example 40, and then the reaction mixture was concentrated under reduced pressure to obtain a residual powder.
After the compounds of Production Examples 40-1 to 40-23 were reacted according to Production Example 40, the hydrochloride compound was isolated by the method according to Production Example 40-2, and the other compounds were produced. A compound was isolated and obtained by a method according to Example 40.
製造例41
tert-ブチル 4-{4-(tert-ブトキシカルボニル)-4'-[5-(トランス-4-ブトキシカルボニル)-1,2,4-オキサジアゾール-3-イル]ビフェニル-3-イル}-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(135 mg)及び4M HCl/EtOAc (3 mL)の混合物を室温で2時間撹拌した。さらに5時間撹拌した。濃縮し得た残渣に、ジオキサン、2M Na2CO3水溶液(0.41 mL)、9H-フルオレン-9-イルメチル クロロカーボネート(55.7 mg)を加え室温で4時間撹拌した。反応液に水を加えた。1M 塩酸水でpH=2に調整し、EtOAcで抽出した。反応液は、水、brineで洗浄した。有機層をMgSO4で乾燥後、減圧下濃縮し4'-[5-(トランス-4-ブトキシシクロヘキシル)-1,2,4-オキサジアゾール-3-イル]-3-{1-[(9H-フルオレン-9-イルメトキシ)カルボニル]-1,2,3,6-テトラヒドロピリジン-4-イル}ビフェニル-4-カルボン酸(150 mg)を白色泡状固体として得た。 Production Example 41
tert-butyl 4- {4- (tert-butoxycarbonyl) -4 '-[5- (trans-4-butoxycarbonyl) -1,2,4-oxadiazol-3-yl] biphenyl-3-yl} A mixture of -3,6-dihydropyridine-1 (2H) -carboxylate (135 mg) and 4M HCl / EtOAc (3 mL) was stirred at room temperature for 2 hours. Stir for another 5 hours. Dioxane, 2M Na 2 CO 3 aqueous solution (0.41 mL) and 9H-fluoren-9-ylmethyl chlorocarbonate (55.7 mg) were added to the residue obtained by concentration, and the mixture was stirred at room temperature for 4 hours. Water was added to the reaction solution. The pH was adjusted to 2 with 1M hydrochloric acid and extracted with EtOAc. The reaction solution was washed with water and brine. The organic layer was dried over MgSO 4 , concentrated under reduced pressure, and 4 ′-[5- (trans-4-butoxycyclohexyl) -1,2,4-oxadiazol-3-yl] -3- {1-[( [9H-Fluoren-9-ylmethoxy) carbonyl] -1,2,3,6-tetrahydropyridin-4-yl} biphenyl-4-carboxylic acid (150 mg) was obtained as a white foamy solid.
tert-ブチル 4-{4-(tert-ブトキシカルボニル)-4'-[5-(トランス-4-ブトキシカルボニル)-1,2,4-オキサジアゾール-3-イル]ビフェニル-3-イル}-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(135 mg)及び4M HCl/EtOAc (3 mL)の混合物を室温で2時間撹拌した。さらに5時間撹拌した。濃縮し得た残渣に、ジオキサン、2M Na2CO3水溶液(0.41 mL)、9H-フルオレン-9-イルメチル クロロカーボネート(55.7 mg)を加え室温で4時間撹拌した。反応液に水を加えた。1M 塩酸水でpH=2に調整し、EtOAcで抽出した。反応液は、水、brineで洗浄した。有機層をMgSO4で乾燥後、減圧下濃縮し4'-[5-(トランス-4-ブトキシシクロヘキシル)-1,2,4-オキサジアゾール-3-イル]-3-{1-[(9H-フルオレン-9-イルメトキシ)カルボニル]-1,2,3,6-テトラヒドロピリジン-4-イル}ビフェニル-4-カルボン酸(150 mg)を白色泡状固体として得た。 Production Example 41
tert-butyl 4- {4- (tert-butoxycarbonyl) -4 '-[5- (trans-4-butoxycarbonyl) -1,2,4-oxadiazol-3-yl] biphenyl-3-yl} A mixture of -3,6-dihydropyridine-1 (2H) -carboxylate (135 mg) and 4M HCl / EtOAc (3 mL) was stirred at room temperature for 2 hours. Stir for another 5 hours. Dioxane, 2M Na 2 CO 3 aqueous solution (0.41 mL) and 9H-fluoren-9-ylmethyl chlorocarbonate (55.7 mg) were added to the residue obtained by concentration, and the mixture was stirred at room temperature for 4 hours. Water was added to the reaction solution. The pH was adjusted to 2 with 1M hydrochloric acid and extracted with EtOAc. The reaction solution was washed with water and brine. The organic layer was dried over MgSO 4 , concentrated under reduced pressure, and 4 ′-[5- (trans-4-butoxycyclohexyl) -1,2,4-oxadiazol-3-yl] -3- {1-[( [9H-Fluoren-9-ylmethoxy) carbonyl] -1,2,3,6-tetrahydropyridin-4-yl} biphenyl-4-carboxylic acid (150 mg) was obtained as a white foamy solid.
製造例41の方法と同様にして、後記表に示す製造例41-1から製造例41-2の化合物を製造した。
In the same manner as in Production Example 41, the compounds of Production Example 41-1 to Production Example 41-2 shown in the table below were produced.
製造例42
製造例10-2の化合物(2.38 g)のMeOH/H2O(23.8 mL/23.8 mL)溶液にPd/C 10%, 50% wet (1.19 g)を加え、H2置換して7時間撹拌した。反応液をセライトろ過した。ろ液を軽く減圧留去した後、得られた溶液にH2O:MeCN=80:20溶液と1M NaOH水溶液を加え、ODSカラムクロマトグラフィー(H2O:MeCN=80:20から20:80まで)で精製した。目的物フラクションのMeCNを減圧下留去した後、凍結乾燥し、製造例42の化合物(470 mg)を得た。 Production Example 42
Pd / C 10%, 50% wet (1.19 g) was added to a solution of the compound of Production Example 10-2 (2.38 g) in MeOH / H 2 O (23.8 mL / 23.8 mL), and the resulting mixture was substituted with H 2 and stirred for 7 hours. did. The reaction solution was filtered through celite. After the filtrate was lightly distilled off under reduced pressure, H 2 O: MeCN = 80: 20 solution and 1M NaOH aqueous solution were added to the resulting solution, and ODS column chromatography (H 2 O: MeCN = 80: 20 to 20:80 Purified). The target fraction, MeCN, was distilled off under reduced pressure and then lyophilized to obtain the compound of Production Example 42 (470 mg).
製造例10-2の化合物(2.38 g)のMeOH/H2O(23.8 mL/23.8 mL)溶液にPd/C 10%, 50% wet (1.19 g)を加え、H2置換して7時間撹拌した。反応液をセライトろ過した。ろ液を軽く減圧留去した後、得られた溶液にH2O:MeCN=80:20溶液と1M NaOH水溶液を加え、ODSカラムクロマトグラフィー(H2O:MeCN=80:20から20:80まで)で精製した。目的物フラクションのMeCNを減圧下留去した後、凍結乾燥し、製造例42の化合物(470 mg)を得た。 Production Example 42
Pd / C 10%, 50% wet (1.19 g) was added to a solution of the compound of Production Example 10-2 (2.38 g) in MeOH / H 2 O (23.8 mL / 23.8 mL), and the resulting mixture was substituted with H 2 and stirred for 7 hours. did. The reaction solution was filtered through celite. After the filtrate was lightly distilled off under reduced pressure, H 2 O: MeCN = 80: 20 solution and 1M NaOH aqueous solution were added to the resulting solution, and ODS column chromatography (H 2 O: MeCN = 80: 20 to 20:80 Purified). The target fraction, MeCN, was distilled off under reduced pressure and then lyophilized to obtain the compound of Production Example 42 (470 mg).
製造例42の方法と同様にして、後記表に示す製造例42-1から製造例42-4の化合物を製造した。単離、精製は、抽出、分別結晶化、各種分画クロマトグラフィー(ODSカラムクロマトグラフィーまたはシリカゲルクロマトグラフィー等)等、通常の化学操作及び本明細書の製造例若しくは実施例に記載の方法、またはそれに準じた方法のうち、化合物に適した方法を適用して行なった。
In the same manner as in Production Example 42, the compounds of Production Example 42-1 to Production Example 42-4 shown in the table below were produced. Isolation and purification include extraction, fractional crystallization, various fractional chromatography (ODS column chromatography, silica gel chromatography, etc.) and other normal chemical operations and methods described in the production examples or examples of the present specification, or Of the similar methods, the method suitable for the compound was applied.
製造例43
tert-ブチルピペラジン-1-カルボキシレート(17 g)のCH2Cl2(340 mL)溶液にEt3N(15.3 mL)と2-ニトロベンゼンスルホニルクロリド(22.3 g)を氷冷下に加えて同温で15分、室温で4.5時間撹拌した。反応混合物に0℃で水(150 mL)を加え、EtOAcで3回抽出した。有機層をあわせて、brineで洗浄し、MgSO4で乾燥した。溶媒を減圧下に濃縮し、残渣をシリカゲルクロマトグラフィー(Hexane: EtOAc=70:30 から20:80で溶出)で精製した。フラクションを減圧下に濃縮し、残渣をIPEで粉末化して tert-ブチル4-[(2-ニトロフェニル)スルホニル]ピペラジン-1-カルボキシレート(31.8g)を得た。 Production Example 43
Et 3 N (15.3 mL) and 2-nitrobenzenesulfonyl chloride (22.3 g) were added to a solution of tert-butylpiperazine-1-carboxylate (17 g) in CH 2 Cl 2 (340 mL) under ice-cooling. For 15 minutes and at room temperature for 4.5 hours. Water (150 mL) was added to the reaction mixture at 0 ° C., and the mixture was extracted 3 times with EtOAc. The organic layers were combined, washed with brine and dried over MgSO 4 . The solvent was concentrated under reduced pressure and the residue was purified by silica gel chromatography (eluting with Hexane: EtOAc = 70: 30 to 20:80). The fraction was concentrated under reduced pressure and the residue was triturated with IPE to give tert-butyl 4-[(2-nitrophenyl) sulfonyl] piperazine-1-carboxylate (31.8 g).
tert-ブチルピペラジン-1-カルボキシレート(17 g)のCH2Cl2(340 mL)溶液にEt3N(15.3 mL)と2-ニトロベンゼンスルホニルクロリド(22.3 g)を氷冷下に加えて同温で15分、室温で4.5時間撹拌した。反応混合物に0℃で水(150 mL)を加え、EtOAcで3回抽出した。有機層をあわせて、brineで洗浄し、MgSO4で乾燥した。溶媒を減圧下に濃縮し、残渣をシリカゲルクロマトグラフィー(Hexane: EtOAc=70:30 から20:80で溶出)で精製した。フラクションを減圧下に濃縮し、残渣をIPEで粉末化して tert-ブチル4-[(2-ニトロフェニル)スルホニル]ピペラジン-1-カルボキシレート(31.8g)を得た。 Production Example 43
Et 3 N (15.3 mL) and 2-nitrobenzenesulfonyl chloride (22.3 g) were added to a solution of tert-butylpiperazine-1-carboxylate (17 g) in CH 2 Cl 2 (340 mL) under ice-cooling. For 15 minutes and at room temperature for 4.5 hours. Water (150 mL) was added to the reaction mixture at 0 ° C., and the mixture was extracted 3 times with EtOAc. The organic layers were combined, washed with brine and dried over MgSO 4 . The solvent was concentrated under reduced pressure and the residue was purified by silica gel chromatography (eluting with Hexane: EtOAc = 70: 30 to 20:80). The fraction was concentrated under reduced pressure and the residue was triturated with IPE to give tert-butyl 4-[(2-nitrophenyl) sulfonyl] piperazine-1-carboxylate (31.8 g).
製造例44
tert-ブチル 4-[4-(メトキシカルボニル)-4'-(5-{4-[(2-ニトロフェニル)スルホニル]ピペラジン-1-イル}-1,2,4-オキサジアゾール-3-イル)ビフェニル-3-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(22.5 g)をDMF(225 mL)に溶解し、K2CO3(12.8 g)、4-メチルベンゼンチオール(4.59 g)を加えて室温で一夜撹拌した。反応混合物に水を加え、 CH2Cl2で2回抽出した。有機層を水、brineで洗浄しMgSO4で乾燥後、減圧下濃縮した。残渣をEtOAc約1Lに溶解し、6回水洗後、brineで洗浄しMgSO4で乾燥後、減圧下濃縮した。残渣を中圧シリカゲルカラムクロマトグラフィー(溶出液;Hexane:EtOAc=1:1、次に、CHCl3:MeOH=95:5)で精製し、tert-ブチル 4-[4-(メトキシカルボニル)-4'-(5-ピペラジン-1-イル-1,2,4-オキサジアゾール-3-イル)ビフェニル-3-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート (16.0 g)を淡褐色泡状物質として得た。 Production Example 44
tert-butyl 4- [4- (methoxycarbonyl) -4 '-(5- {4-[(2-nitrophenyl) sulfonyl] piperazin-1-yl} -1,2,4-oxadiazole-3- Yl) biphenyl-3-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (22.5 g) dissolved in DMF (225 mL), K 2 CO 3 (12.8 g), 4-methylbenzenethiol (4.59 g) was added and stirred overnight at room temperature. Water was added to the reaction mixture, and extracted twice with CH 2 Cl 2 . The organic layer was washed with water and brine, dried over MgSO 4 and concentrated under reduced pressure. The residue was dissolved in about 1 L of EtOAc, washed 6 times with water, washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The residue was purified by medium pressure silica gel column chromatography (eluent; Hexane: EtOAc = 1: 1, then CHCl 3 : MeOH = 95: 5) and tert-butyl 4- [4- (methoxycarbonyl) -4 '-(5-Piperazin-1-yl-1,2,4-oxadiazol-3-yl) biphenyl-3-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (16.0 g) Obtained as a light brown foam.
tert-ブチル 4-[4-(メトキシカルボニル)-4'-(5-{4-[(2-ニトロフェニル)スルホニル]ピペラジン-1-イル}-1,2,4-オキサジアゾール-3-イル)ビフェニル-3-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(22.5 g)をDMF(225 mL)に溶解し、K2CO3(12.8 g)、4-メチルベンゼンチオール(4.59 g)を加えて室温で一夜撹拌した。反応混合物に水を加え、 CH2Cl2で2回抽出した。有機層を水、brineで洗浄しMgSO4で乾燥後、減圧下濃縮した。残渣をEtOAc約1Lに溶解し、6回水洗後、brineで洗浄しMgSO4で乾燥後、減圧下濃縮した。残渣を中圧シリカゲルカラムクロマトグラフィー(溶出液;Hexane:EtOAc=1:1、次に、CHCl3:MeOH=95:5)で精製し、tert-ブチル 4-[4-(メトキシカルボニル)-4'-(5-ピペラジン-1-イル-1,2,4-オキサジアゾール-3-イル)ビフェニル-3-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート (16.0 g)を淡褐色泡状物質として得た。 Production Example 44
tert-butyl 4- [4- (methoxycarbonyl) -4 '-(5- {4-[(2-nitrophenyl) sulfonyl] piperazin-1-yl} -1,2,4-oxadiazole-3- Yl) biphenyl-3-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (22.5 g) dissolved in DMF (225 mL), K 2 CO 3 (12.8 g), 4-methylbenzenethiol (4.59 g) was added and stirred overnight at room temperature. Water was added to the reaction mixture, and extracted twice with CH 2 Cl 2 . The organic layer was washed with water and brine, dried over MgSO 4 and concentrated under reduced pressure. The residue was dissolved in about 1 L of EtOAc, washed 6 times with water, washed with brine, dried over MgSO 4 and concentrated under reduced pressure. The residue was purified by medium pressure silica gel column chromatography (eluent; Hexane: EtOAc = 1: 1, then CHCl 3 : MeOH = 95: 5) and tert-butyl 4- [4- (methoxycarbonyl) -4 '-(5-Piperazin-1-yl-1,2,4-oxadiazol-3-yl) biphenyl-3-yl] -3,6-dihydropyridine-1 (2H) -carboxylate (16.0 g) Obtained as a light brown foam.
製造例45
5-[4-(2-フェニルエチル)ピペラジン-1-イル]-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピリミジン(10.05 g)のジオキサン (100 mL)懸濁液に氷冷下、6M 塩酸(18.8 mL)を加え、室温で2時間攪拌した。反応混合物を氷冷下、1N NaOH水溶液でpH=8にした後、生じた固体をろ取、MeCNで洗浄し、4-{5-[4-(2-フェニルエチル)ピペラジン-1-イル]ピリミジン-2-イル}フェノール(8.06 g)をクリーム色粉末として得た。 Production Example 45
Suspension of 5- [4- (2-phenylethyl) piperazin-1-yl] -2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyrimidine (10.05 g) in dioxane (100 mL) Under ice-cooling, 6M hydrochloric acid (18.8 mL) was added, and the mixture was stirred at room temperature for 2 hr. The reaction mixture was adjusted to pH = 8 with 1N NaOH aqueous solution under ice cooling, and the resulting solid was collected by filtration, washed with MeCN, and 4- {5- [4- (2-phenylethyl) piperazin-1-yl] Pyrimidin-2-yl} phenol (8.06 g) was obtained as a cream powder.
5-[4-(2-フェニルエチル)ピペラジン-1-イル]-2-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]ピリミジン(10.05 g)のジオキサン (100 mL)懸濁液に氷冷下、6M 塩酸(18.8 mL)を加え、室温で2時間攪拌した。反応混合物を氷冷下、1N NaOH水溶液でpH=8にした後、生じた固体をろ取、MeCNで洗浄し、4-{5-[4-(2-フェニルエチル)ピペラジン-1-イル]ピリミジン-2-イル}フェノール(8.06 g)をクリーム色粉末として得た。 Production Example 45
Suspension of 5- [4- (2-phenylethyl) piperazin-1-yl] -2- [4- (tetrahydro-2H-pyran-2-yloxy) phenyl] pyrimidine (10.05 g) in dioxane (100 mL) Under ice-cooling, 6M hydrochloric acid (18.8 mL) was added, and the mixture was stirred at room temperature for 2 hr. The reaction mixture was adjusted to pH = 8 with 1N NaOH aqueous solution under ice cooling, and the resulting solid was collected by filtration, washed with MeCN, and 4- {5- [4- (2-phenylethyl) piperazin-1-yl] Pyrimidin-2-yl} phenol (8.06 g) was obtained as a cream powder.
製造例45の方法と同様にして、後記表に示す製造例45-1から製造例45-19の化合物を製造した。
In the same manner as in Production Example 45, the compounds of Production Example 45-1 to Production Example 45-19 shown in the table below were produced.
製造例46
tert-ブチル 4-[2-(メトキシカルボニル)-5-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(2.65 g)、4-メチルベンゼンスルホン酸(328 mg)、MeOH(27 mL)の混合物を室温で3時間撹拌した。反応混合物を水(90 mL)中に注ぎ、EtOAc(45 mL)で抽出した。水層にbrineを加え、EtOAcで再度抽出した。有機層をあわせてbrineで洗浄し、MgSO4で乾燥した後、減圧下濃縮し、tert-ブチル 4-[5-ヒドロキシ-2-(メトキシカルボニル)フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート (2.04 g)を得た。 Production Example 46
tert-Butyl 4- [2- (methoxycarbonyl) -5- (tetrahydro-2H-pyran-2-yloxy) phenyl] -3,6-dihydropyridine-1 (2H) -carboxylate (2.65 g), 4-methyl A mixture of benzenesulfonic acid (328 mg) and MeOH (27 mL) was stirred at room temperature for 3 hours. The reaction mixture was poured into water (90 mL) and extracted with EtOAc (45 mL). Brine was added to the aqueous layer and extracted again with EtOAc. The organic layers were combined, washed with brine, dried over MgSO 4 , concentrated under reduced pressure, and tert-butyl 4- [5-hydroxy-2- (methoxycarbonyl) phenyl] -3,6-dihydropyridine-1 (2H ) -Carboxylate (2.04 g) was obtained.
tert-ブチル 4-[2-(メトキシカルボニル)-5-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート(2.65 g)、4-メチルベンゼンスルホン酸(328 mg)、MeOH(27 mL)の混合物を室温で3時間撹拌した。反応混合物を水(90 mL)中に注ぎ、EtOAc(45 mL)で抽出した。水層にbrineを加え、EtOAcで再度抽出した。有機層をあわせてbrineで洗浄し、MgSO4で乾燥した後、減圧下濃縮し、tert-ブチル 4-[5-ヒドロキシ-2-(メトキシカルボニル)フェニル]-3,6-ジヒドロピリジン-1(2H)-カルボキシレート (2.04 g)を得た。 Production Example 46
tert-Butyl 4- [2- (methoxycarbonyl) -5- (tetrahydro-2H-pyran-2-yloxy) phenyl] -3,6-dihydropyridine-1 (2H) -carboxylate (2.65 g), 4-methyl A mixture of benzenesulfonic acid (328 mg) and MeOH (27 mL) was stirred at room temperature for 3 hours. The reaction mixture was poured into water (90 mL) and extracted with EtOAc (45 mL). Brine was added to the aqueous layer and extracted again with EtOAc. The organic layers were combined, washed with brine, dried over MgSO 4 , concentrated under reduced pressure, and tert-butyl 4- [5-hydroxy-2- (methoxycarbonyl) phenyl] -3,6-dihydropyridine-1 (2H ) -Carboxylate (2.04 g) was obtained.
製造例46の方法と同様にして、後記表に示す製造例46-1から製造例46-5の化合物を製造した。
In the same manner as in Production Example 46, the compounds of Production Example 46-1 to Production Example 46-5 shown in the table below were produced.
製造例47
6-{4-[5-(ベンジルオキシ)ピリミジン-2-イル]フェニル}-1-クロロ-2-ナフチル トリフルオロメタンスルホネート(200 mg), Pd(OAc)2(8.0 mg)及びDPPP(15 mg)のDMSO/MeOH(16 mL/4 mL)溶液にEt3N(d=0.726, 150 μL)を室温で加えた。CO雰囲気下、90℃で4時間攪拌した。反応溶液に水を加えて析出物をろ取して水洗し、減圧乾燥を行い粗粉末を得た。粗粉末をシリカゲルカラムクロマトグラフィー(溶出液;CHCl3:MeOH=99:1)で精製し、メチル 6-{4-[5-(ベンジルオキシ)ピリミジン-2-イル]フェニル}-1-クロロ-2-ナフトエート(143 mg)を得た。 Production Example 47
6- {4- [5- (benzyloxy) pyrimidin-2-yl] phenyl} -1-chloro-2-naphthyl trifluoromethanesulfonate (200 mg), Pd (OAc) 2 (8.0 mg) and DPPP (15 mg ) In DMSO / MeOH (16 mL / 4 mL) was added Et 3 N (d = 0.726, 150 μL) at room temperature. The mixture was stirred at 90 ° C. for 4 hours in a CO atmosphere. Water was added to the reaction solution, and the precipitate was collected by filtration, washed with water, and dried under reduced pressure to obtain a crude powder. The crude powder was purified by silica gel column chromatography (eluent; CHCl 3 : MeOH = 99: 1), and methyl 6- {4- [5- (benzyloxy) pyrimidin-2-yl] phenyl} -1-chloro- 2-Naphthoate (143 mg) was obtained.
6-{4-[5-(ベンジルオキシ)ピリミジン-2-イル]フェニル}-1-クロロ-2-ナフチル トリフルオロメタンスルホネート(200 mg), Pd(OAc)2(8.0 mg)及びDPPP(15 mg)のDMSO/MeOH(16 mL/4 mL)溶液にEt3N(d=0.726, 150 μL)を室温で加えた。CO雰囲気下、90℃で4時間攪拌した。反応溶液に水を加えて析出物をろ取して水洗し、減圧乾燥を行い粗粉末を得た。粗粉末をシリカゲルカラムクロマトグラフィー(溶出液;CHCl3:MeOH=99:1)で精製し、メチル 6-{4-[5-(ベンジルオキシ)ピリミジン-2-イル]フェニル}-1-クロロ-2-ナフトエート(143 mg)を得た。 Production Example 47
6- {4- [5- (benzyloxy) pyrimidin-2-yl] phenyl} -1-chloro-2-naphthyl trifluoromethanesulfonate (200 mg), Pd (OAc) 2 (8.0 mg) and DPPP (15 mg ) In DMSO / MeOH (16 mL / 4 mL) was added Et 3 N (d = 0.726, 150 μL) at room temperature. The mixture was stirred at 90 ° C. for 4 hours in a CO atmosphere. Water was added to the reaction solution, and the precipitate was collected by filtration, washed with water, and dried under reduced pressure to obtain a crude powder. The crude powder was purified by silica gel column chromatography (eluent; CHCl 3 : MeOH = 99: 1), and methyl 6- {4- [5- (benzyloxy) pyrimidin-2-yl] phenyl} -1-chloro- 2-Naphthoate (143 mg) was obtained.
製造例47の方法と同様にして、後記表に示す製造例47-1から製造例47-3の化合物を製造した。
In the same manner as in Production Example 47, the compounds of Production Example 47-1 to Production Example 47-3 shown in the table below were produced.
製造例48
メチル 4-(テトラヒドロ-2H-ピラン-2-イルオキシ)ベンゼン(1.37 g)、4-ブロモ-2-メチルピリジン (1 g)のTHF(20 mL)溶液に氷冷下1M LHMDS(11.6 mL)を加え、室温で終夜攪拌した。反応液に水、EtOAcを加えて抽出した。有機層は、MgSO4で乾燥後、減圧下濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィーで精製し、2-(4-ブロモピリジン-2-イル)-1-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]エタノン(1.91 g)を得た。 Production Example 48
To a solution of methyl 4- (tetrahydro-2H-pyran-2-yloxy) benzene (1.37 g) and 4-bromo-2-methylpyridine (1 g) in THF (20 mL) was added 1M LHMDS (11.6 mL) under ice cooling. In addition, the mixture was stirred overnight at room temperature. Water and EtOAc were added to the reaction solution for extraction. The organic layer was dried over MgSO 4 and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography to obtain 2- (4-bromopyridin-2-yl) -1- [4- (tetrahydro-2H -Pyran-2-yloxy) phenyl] ethanone (1.91 g) was obtained.
メチル 4-(テトラヒドロ-2H-ピラン-2-イルオキシ)ベンゼン(1.37 g)、4-ブロモ-2-メチルピリジン (1 g)のTHF(20 mL)溶液に氷冷下1M LHMDS(11.6 mL)を加え、室温で終夜攪拌した。反応液に水、EtOAcを加えて抽出した。有機層は、MgSO4で乾燥後、減圧下濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィーで精製し、2-(4-ブロモピリジン-2-イル)-1-[4-(テトラヒドロ-2H-ピラン-2-イルオキシ)フェニル]エタノン(1.91 g)を得た。 Production Example 48
To a solution of methyl 4- (tetrahydro-2H-pyran-2-yloxy) benzene (1.37 g) and 4-bromo-2-methylpyridine (1 g) in THF (20 mL) was added 1M LHMDS (11.6 mL) under ice cooling. In addition, the mixture was stirred overnight at room temperature. Water and EtOAc were added to the reaction solution for extraction. The organic layer was dried over MgSO 4 and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography to obtain 2- (4-bromopyridin-2-yl) -1- [4- (tetrahydro-2H -Pyran-2-yloxy) phenyl] ethanone (1.91 g) was obtained.
製造例49
製造例35の化合物(180 mg)の MeOH/DMF(1.8 mL/1.98 mL)の溶液に(ジメトキシメチル)ジメチルアミン(1.53 g)を加えた。反応混合物を25℃で18時間、40℃で8時間攪拌した。反応混合物に1M NaOH水溶液(698.7 μL)を加え、反応混合物は室温で終夜攪拌した。反応液を中和し、水(100 mL)で希釈し、ODSカラムクロマトグラフィー(pH6.8 buffer、 水で洗浄後、50%MeCN)で溶出し、減圧下濃縮した後に凍結乾燥し、製造例49の化合物(157 mg)を得た。 Production Example 49
(Dimethoxymethyl) dimethylamine (1.53 g) was added to a solution of the compound of Production Example 35 (180 mg) in MeOH / DMF (1.8 mL / 1.98 mL). The reaction mixture was stirred at 25 ° C. for 18 hours and at 40 ° C. for 8 hours. To the reaction mixture was added 1M aqueous NaOH (698.7 μL) and the reaction mixture was stirred at room temperature overnight. The reaction solution was neutralized, diluted with water (100 mL), eluted with ODS column chromatography (pH 6.8 buffer, washed with water, 50% MeCN), concentrated under reduced pressure, and freeze-dried. 49 compounds (157 mg) were obtained.
製造例35の化合物(180 mg)の MeOH/DMF(1.8 mL/1.98 mL)の溶液に(ジメトキシメチル)ジメチルアミン(1.53 g)を加えた。反応混合物を25℃で18時間、40℃で8時間攪拌した。反応混合物に1M NaOH水溶液(698.7 μL)を加え、反応混合物は室温で終夜攪拌した。反応液を中和し、水(100 mL)で希釈し、ODSカラムクロマトグラフィー(pH6.8 buffer、 水で洗浄後、50%MeCN)で溶出し、減圧下濃縮した後に凍結乾燥し、製造例49の化合物(157 mg)を得た。 Production Example 49
(Dimethoxymethyl) dimethylamine (1.53 g) was added to a solution of the compound of Production Example 35 (180 mg) in MeOH / DMF (1.8 mL / 1.98 mL). The reaction mixture was stirred at 25 ° C. for 18 hours and at 40 ° C. for 8 hours. To the reaction mixture was added 1M aqueous NaOH (698.7 μL) and the reaction mixture was stirred at room temperature overnight. The reaction solution was neutralized, diluted with water (100 mL), eluted with ODS column chromatography (pH 6.8 buffer, washed with water, 50% MeCN), concentrated under reduced pressure, and freeze-dried. 49 compounds (157 mg) were obtained.
製造例50
tert-ブチル 4-[(4,4-ジメチルシクロヘキス-2-エン-1-イル)オキシ]ピペリジン-1-カルボキシレート (810 mg) のMeOH(8 mL)溶液に10% Pd/C(50% wet 、50 mg) を加えて水素1気圧下、室温で5時間接触還元を行った。反応液をセライトろ過して触媒をろ別し、ろ液を減圧下濃縮して、tert-ブチル 4-[(4,4-ジメチルシクロヘキシル)オキシ]ピペリジン-1-カルボキシレート (500 mg)を無色油状物として得た。 Production Example 50
To a solution of tert-butyl 4-[(4,4-dimethylcyclohex-2-en-1-yl) oxy] piperidine-1-carboxylate (810 mg) in MeOH (8 mL) was added 10% Pd / C (50 % wet, 50 mg) was added, and catalytic reduction was performed at room temperature under 1 atm of hydrogen for 5 hours. The reaction mixture was filtered through Celite to remove the catalyst, and the filtrate was concentrated under reduced pressure to give tert-butyl 4-[(4,4-dimethylcyclohexyl) oxy] piperidine-1-carboxylate (500 mg) colorless. Obtained as an oil.
tert-ブチル 4-[(4,4-ジメチルシクロヘキス-2-エン-1-イル)オキシ]ピペリジン-1-カルボキシレート (810 mg) のMeOH(8 mL)溶液に10% Pd/C(50% wet 、50 mg) を加えて水素1気圧下、室温で5時間接触還元を行った。反応液をセライトろ過して触媒をろ別し、ろ液を減圧下濃縮して、tert-ブチル 4-[(4,4-ジメチルシクロヘキシル)オキシ]ピペリジン-1-カルボキシレート (500 mg)を無色油状物として得た。 Production Example 50
To a solution of tert-butyl 4-[(4,4-dimethylcyclohex-2-en-1-yl) oxy] piperidine-1-carboxylate (810 mg) in MeOH (8 mL) was added 10% Pd / C (50 % wet, 50 mg) was added, and catalytic reduction was performed at room temperature under 1 atm of hydrogen for 5 hours. The reaction mixture was filtered through Celite to remove the catalyst, and the filtrate was concentrated under reduced pressure to give tert-butyl 4-[(4,4-dimethylcyclohexyl) oxy] piperidine-1-carboxylate (500 mg) colorless. Obtained as an oil.
製造例50の方法と同様にして、後記表に示す製造例50-1の化合物を製造した。
In the same manner as in Production Example 50, the compound of Production Example 50-1 shown in the table below was produced.
製造例51
4-ブロモ-2-クロロ安息香酸(15 g)、DIBOC(16.7 g)、DMAP(390 mg)、THF (230 mL)、t-BuOH (75 mL)の混合物を室温で24時間撹拌した。反応液にsat.NaHCO3水を加え、EtOAc(300 mL)で抽出した。有機層を、水、0.1M塩酸水、brineで順次洗浄し、MgSO4で乾燥した後に減圧下濃縮し、tert-ブチル 4-ブロモ-2-クロロベンゾエート(17 g)をオレンジ色の油状物質として得た。 Production Example 51
A mixture of 4-bromo-2-chlorobenzoic acid (15 g), DIBOC (16.7 g), DMAP (390 mg), THF (230 mL), t-BuOH (75 mL) was stirred at room temperature for 24 hours. To the reaction solution was added sat. NaHCO 3 aqueous solution, and the mixture was extracted with EtOAc (300 mL). The organic layer was washed successively with water, 0.1M aqueous hydrochloric acid and brine, dried over MgSO 4 and concentrated under reduced pressure to give tert-butyl 4-bromo-2-chlorobenzoate (17 g) as an orange oil. Obtained.
4-ブロモ-2-クロロ安息香酸(15 g)、DIBOC(16.7 g)、DMAP(390 mg)、THF (230 mL)、t-BuOH (75 mL)の混合物を室温で24時間撹拌した。反応液にsat.NaHCO3水を加え、EtOAc(300 mL)で抽出した。有機層を、水、0.1M塩酸水、brineで順次洗浄し、MgSO4で乾燥した後に減圧下濃縮し、tert-ブチル 4-ブロモ-2-クロロベンゾエート(17 g)をオレンジ色の油状物質として得た。 Production Example 51
A mixture of 4-bromo-2-chlorobenzoic acid (15 g), DIBOC (16.7 g), DMAP (390 mg), THF (230 mL), t-BuOH (75 mL) was stirred at room temperature for 24 hours. To the reaction solution was added sat. NaHCO 3 aqueous solution, and the mixture was extracted with EtOAc (300 mL). The organic layer was washed successively with water, 0.1M aqueous hydrochloric acid and brine, dried over MgSO 4 and concentrated under reduced pressure to give tert-butyl 4-bromo-2-chlorobenzoate (17 g) as an orange oil. Obtained.
実施例1
3-{1-[(9H-フルオレン-9-イルメトキシ)カルボニル]-1,2,3,6-テトラヒドロピリジン-4-イル}-4'-[5-(トランス-4-イソブトキシシクロヘキシル)-1,2,4-オキサジアゾール-3-イル]ビフェニル-4-カルボン酸(402 mg)、HBTU(221.2 mg)、DMF(6.00 mL)、DIPEA(0.237 mL)の混合物を室温で30分撹拌した。国際公開WO99/40108号の実施例番号169の化合物(578 mg)を加え室温で5時間撹拌した。TLCにおいて、活性エステルが消失したことを確認した。ピペリジン(709.3 mg)を加え室温で30分撹拌した。反応液にEtOAcを加え生じた析出物をろ取した。水に懸濁させ1M NaOH水を溶解するまで加えた。その混合物をODSで精製(水で置換したODSシリカゲル80 mLにチャージした後、10、20、30、40%MeCN水で段階的に溶出)した後凍結乾燥し、実施例1の化合物(472 mg)を白色粉末として得た。 Example 1
3- {1-[(9H-Fluoren-9-ylmethoxy) carbonyl] -1,2,3,6-tetrahydropyridin-4-yl} -4 '-[5- (trans-4-isobutoxycyclohexyl)- A mixture of 1,2,4-oxadiazol-3-yl] biphenyl-4-carboxylic acid (402 mg), HBTU (221.2 mg), DMF (6.00 mL), DIPEA (0.237 mL) was stirred at room temperature for 30 minutes did. The compound of Example No. 169 (578 mg) of International Publication WO99 / 40108 was added and stirred at room temperature for 5 hours. In TLC, it was confirmed that the active ester disappeared. Piperidine (709.3 mg) was added and stirred at room temperature for 30 minutes. EtOAc was added to the reaction solution, and the resulting precipitate was collected by filtration. Suspended in water and added until 1M NaOH water was dissolved. The mixture was purified by ODS (charged to 80 mL of ODS silica gel substituted with water, then eluted stepwise with 10, 20, 30, 40% MeCN water) and then lyophilized to give the compound of Example 1 (472 mg ) Was obtained as a white powder.
3-{1-[(9H-フルオレン-9-イルメトキシ)カルボニル]-1,2,3,6-テトラヒドロピリジン-4-イル}-4'-[5-(トランス-4-イソブトキシシクロヘキシル)-1,2,4-オキサジアゾール-3-イル]ビフェニル-4-カルボン酸(402 mg)、HBTU(221.2 mg)、DMF(6.00 mL)、DIPEA(0.237 mL)の混合物を室温で30分撹拌した。国際公開WO99/40108号の実施例番号169の化合物(578 mg)を加え室温で5時間撹拌した。TLCにおいて、活性エステルが消失したことを確認した。ピペリジン(709.3 mg)を加え室温で30分撹拌した。反応液にEtOAcを加え生じた析出物をろ取した。水に懸濁させ1M NaOH水を溶解するまで加えた。その混合物をODSで精製(水で置換したODSシリカゲル80 mLにチャージした後、10、20、30、40%MeCN水で段階的に溶出)した後凍結乾燥し、実施例1の化合物(472 mg)を白色粉末として得た。 Example 1
3- {1-[(9H-Fluoren-9-ylmethoxy) carbonyl] -1,2,3,6-tetrahydropyridin-4-yl} -4 '-[5- (trans-4-isobutoxycyclohexyl)- A mixture of 1,2,4-oxadiazol-3-yl] biphenyl-4-carboxylic acid (402 mg), HBTU (221.2 mg), DMF (6.00 mL), DIPEA (0.237 mL) was stirred at room temperature for 30 minutes did. The compound of Example No. 169 (578 mg) of International Publication WO99 / 40108 was added and stirred at room temperature for 5 hours. In TLC, it was confirmed that the active ester disappeared. Piperidine (709.3 mg) was added and stirred at room temperature for 30 minutes. EtOAc was added to the reaction solution, and the resulting precipitate was collected by filtration. Suspended in water and added until 1M NaOH water was dissolved. The mixture was purified by ODS (charged to 80 mL of ODS silica gel substituted with water, then eluted stepwise with 10, 20, 30, 40% MeCN water) and then lyophilized to give the compound of Example 1 (472 mg ) Was obtained as a white powder.
実施例2から6の化合物は、実施例1の方法と同様にして製造した。
The compounds of Examples 2 to 6 were produced in the same manner as the method of Example 1.
実施例7
製造例42-1の化合物(50 mg)のDMF溶液(0.75 mL)に 3-メトキシベンズアルデヒド(14 mg)、AcOH (5 μL)、NaBH(OAc)3(15 mg)を加え、窒素雰囲気下、室温で15時間撹拌した。反応液にEtOAcを加え生じた析出物をろ取した。得られた混合物に0℃で、TFA(0.5 mL)を加え、同温で0.5時間撹拌した。1M NaOH水溶液 (6.7 mL)-H2O (200 mL)に氷冷下、反応液をゆっくりと加え、そのままODSカラムクロマトグラフィー(H2O:MeCN=80:20から20:80まで)で精製した。目的物のフラクションのMeCNを減圧留去した後、凍結乾燥し、実施例7の化合物を得た。 Example 7
To a DMF solution (0.75 mL) of the compound of Production Example 42-1 (0.75 mL), 3-methoxybenzaldehyde (14 mg), AcOH (5 μL), NaBH (OAc) 3 (15 mg) was added, and under a nitrogen atmosphere, Stir at room temperature for 15 hours. EtOAc was added to the reaction solution, and the resulting precipitate was collected by filtration. TFA (0.5 mL) was added to the obtained mixture at 0 ° C., and the mixture was stirred at the same temperature for 0.5 hour. The reaction mixture is slowly added to 1M NaOH aqueous solution (6.7 mL) -H 2 O (200 mL) under ice-cooling, and purified as it is by ODS column chromatography (H 2 O: MeCN = 80: 20 to 20:80). did. The target fraction of MeCN was distilled off under reduced pressure and then lyophilized to obtain the compound of Example 7.
製造例42-1の化合物(50 mg)のDMF溶液(0.75 mL)に 3-メトキシベンズアルデヒド(14 mg)、AcOH (5 μL)、NaBH(OAc)3(15 mg)を加え、窒素雰囲気下、室温で15時間撹拌した。反応液にEtOAcを加え生じた析出物をろ取した。得られた混合物に0℃で、TFA(0.5 mL)を加え、同温で0.5時間撹拌した。1M NaOH水溶液 (6.7 mL)-H2O (200 mL)に氷冷下、反応液をゆっくりと加え、そのままODSカラムクロマトグラフィー(H2O:MeCN=80:20から20:80まで)で精製した。目的物のフラクションのMeCNを減圧留去した後、凍結乾燥し、実施例7の化合物を得た。 Example 7
To a DMF solution (0.75 mL) of the compound of Production Example 42-1 (0.75 mL), 3-methoxybenzaldehyde (14 mg), AcOH (5 μL), NaBH (OAc) 3 (15 mg) was added, and under a nitrogen atmosphere, Stir at room temperature for 15 hours. EtOAc was added to the reaction solution, and the resulting precipitate was collected by filtration. TFA (0.5 mL) was added to the obtained mixture at 0 ° C., and the mixture was stirred at the same temperature for 0.5 hour. The reaction mixture is slowly added to 1M NaOH aqueous solution (6.7 mL) -H 2 O (200 mL) under ice-cooling, and purified as it is by ODS column chromatography (H 2 O: MeCN = 80: 20 to 20:80). did. The target fraction of MeCN was distilled off under reduced pressure and then lyophilized to obtain the compound of Example 7.
実施例8から32の化合物は、実施例7の方法と同様にして製造した。
The compounds of Examples 8 to 32 were produced in the same manner as in Example 7.
実施例33から36の化合物は、製造例10の方法と同様にして製造した。
The compounds of Examples 33 to 36 were produced in the same manner as in Production Example 10.
実施例37
3-[1-(tert-ブトキシカルボニル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-4-カルボン酸(10.6 g)、BOP(8.14 g)、DMF(212 mL)、DIPEA(d=0.742, 7.01 mL)の混合物を室温で1時間撹拌した。国際公開WO99/40108号の実施例番号169の化合物(15.967 g)を加え室温で終夜撹拌した。EtOAc(3L)中に反応液を加え生じた析出物をろ取し,IPEにて洗浄し、減圧下3時間乾燥した。氷冷下、TFA(d=1.480,130 mL)に得られた粉末加え,2時間撹拌した。氷冷下反応液に CH2Cl2(1 L)を加え、Et3N(d=0.726, 320.8 mL)を、内温 15℃以下になる様に留意しゆっくりと滴下した。反応液を減圧下濃縮し、得られたオイルを氷冷下攪拌しながら、1M NaOH水溶液(30 mL)を含む水(4L)に加えた。さらに、1M NaOH水溶液(20 mL)を 加え、pH=11にした。この溶液をODSカラムクロマトグラフィー(H2Oで洗浄し、 MeCN:pH7 標準緩衝溶液=20%から42%まで)を用いて精製した。目的物を含むフラクションを水に希釈し、1M NaOH水溶液を加えて溶解させた。この溶液をODSカラムクロマトグラフィー(H2Oで洗浄し、MeCN:H2O=4:1)を用いて脱塩し、減圧下濃縮し、凍結乾燥により実施例37の化合物(16.89 g)を得た。 Example 37
3- [1- (tert-Butoxycarbonyl) -1,2,3,6-tetrahydropyridin-4-yl] -4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazol-6-yl} biphenyl-4-carboxylic acid (10.6 g), BOP (8.14 g), DMF (212 mL), DIPEA (d = 0.742, 7.01 mL) was stirred at room temperature for 1 hour. The compound of Example No. 169 (15.967 g) of WO99 / 40108 was added and stirred at room temperature overnight. The reaction mixture was added to EtOAc (3 L), and the resulting precipitate was collected by filtration, washed with IPE, and dried under reduced pressure for 3 hours. Under ice cooling, the powder obtained in TFA (d = 1.480, 130 mL) was added and stirred for 2 hours. CH 2 Cl 2 (1 L) was added to the reaction solution under ice-cooling, and Et 3 N (d = 0.726, 320.8 mL) was slowly added dropwise taking care to keep the internal temperature at 15 ° C. or lower. The reaction solution was concentrated under reduced pressure, and the resulting oil was added to water (4 L) containing 1 M NaOH aqueous solution (30 mL) with stirring under ice cooling. Further, 1M NaOH aqueous solution (20 mL) was added to adjust pH = 11. This solution was purified using ODS column chromatography (washed with H 2 O, MeCN: pH7 standard buffer solution = 20% to 42%). Fractions containing the desired product were diluted in water and dissolved by adding 1M NaOH aqueous solution. This solution was desalted using ODS column chromatography (washed with H 2 O, MeCN: H 2 O = 4: 1), concentrated under reduced pressure, and freeze-dried to give the compound of Example 37 (16.89 g). Obtained.
3-[1-(tert-ブトキシカルボニル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}ビフェニル-4-カルボン酸(10.6 g)、BOP(8.14 g)、DMF(212 mL)、DIPEA(d=0.742, 7.01 mL)の混合物を室温で1時間撹拌した。国際公開WO99/40108号の実施例番号169の化合物(15.967 g)を加え室温で終夜撹拌した。EtOAc(3L)中に反応液を加え生じた析出物をろ取し,IPEにて洗浄し、減圧下3時間乾燥した。氷冷下、TFA(d=1.480,130 mL)に得られた粉末加え,2時間撹拌した。氷冷下反応液に CH2Cl2(1 L)を加え、Et3N(d=0.726, 320.8 mL)を、内温 15℃以下になる様に留意しゆっくりと滴下した。反応液を減圧下濃縮し、得られたオイルを氷冷下攪拌しながら、1M NaOH水溶液(30 mL)を含む水(4L)に加えた。さらに、1M NaOH水溶液(20 mL)を 加え、pH=11にした。この溶液をODSカラムクロマトグラフィー(H2Oで洗浄し、 MeCN:pH7 標準緩衝溶液=20%から42%まで)を用いて精製した。目的物を含むフラクションを水に希釈し、1M NaOH水溶液を加えて溶解させた。この溶液をODSカラムクロマトグラフィー(H2Oで洗浄し、MeCN:H2O=4:1)を用いて脱塩し、減圧下濃縮し、凍結乾燥により実施例37の化合物(16.89 g)を得た。 Example 37
3- [1- (tert-Butoxycarbonyl) -1,2,3,6-tetrahydropyridin-4-yl] -4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazol-6-yl} biphenyl-4-carboxylic acid (10.6 g), BOP (8.14 g), DMF (212 mL), DIPEA (d = 0.742, 7.01 mL) was stirred at room temperature for 1 hour. The compound of Example No. 169 (15.967 g) of WO99 / 40108 was added and stirred at room temperature overnight. The reaction mixture was added to EtOAc (3 L), and the resulting precipitate was collected by filtration, washed with IPE, and dried under reduced pressure for 3 hours. Under ice cooling, the powder obtained in TFA (d = 1.480, 130 mL) was added and stirred for 2 hours. CH 2 Cl 2 (1 L) was added to the reaction solution under ice-cooling, and Et 3 N (d = 0.726, 320.8 mL) was slowly added dropwise taking care to keep the internal temperature at 15 ° C. or lower. The reaction solution was concentrated under reduced pressure, and the resulting oil was added to water (4 L) containing 1 M NaOH aqueous solution (30 mL) with stirring under ice cooling. Further, 1M NaOH aqueous solution (20 mL) was added to adjust pH = 11. This solution was purified using ODS column chromatography (washed with H 2 O, MeCN: pH7 standard buffer solution = 20% to 42%). Fractions containing the desired product were diluted in water and dissolved by adding 1M NaOH aqueous solution. This solution was desalted using ODS column chromatography (washed with H 2 O, MeCN: H 2 O = 4: 1), concentrated under reduced pressure, and freeze-dried to give the compound of Example 37 (16.89 g). Obtained.
実施例38から153および295の化合物は、実施例37の方法と同様にして製造した。
The compounds of Examples 38 to 153 and 295 were prepared in the same manner as in Example 37.
実施例154
製造例35の化合物(100 mg)とグリオキシル酸水和物(30 mg)とMS3A (300 mg)をDMF (1.5 mL)に懸濁させ、終夜室温で攪拌した。MeOHで反応液を希釈し、MS3Aをろ別し、MeOHで洗浄した。ろ液と洗液をあわせ減圧下濃縮した後、残渣にEtOAcを加え、析出した粉末をろ取し、EtOAcで洗浄した。粉末を乾燥後、CH2Cl2に懸濁させ、氷冷下、TFAを加えた。室温に昇温しながら3時間攪拌した。氷冷下、Et3Nを加え、反応を停止させた。減圧下濃縮して得られた残渣に水を加えた。さらに、その懸濁液に1MNaOH水溶液, 10%-MeCN/H2Oを加え溶解させた。その混合物をODS精製(条件:ODSシリカ15 mL、チャージ後、buffer, 10%、20%、30%、40% MeCN/bufferで溶出した(各10R)。フラクションを濃縮後、H2Oで希釈、チャージ後(溶出液;H2O、10% MeCN/H2O、50%MeCN/H2Oの順)溶出した。凍結乾燥し目的物(45 mg)を白色粉末として得た。 Example 154
The compound of Production Example 35 (100 mg), glyoxylic acid hydrate (30 mg) and MS3A (300 mg) were suspended in DMF (1.5 mL) and stirred overnight at room temperature. The reaction was diluted with MeOH, MS3A was filtered off and washed with MeOH. The filtrate and the washing solution were combined and concentrated under reduced pressure, EtOAc was added to the residue, and the precipitated powder was collected by filtration and washed with EtOAc. The powder was dried, suspended in CH 2 Cl 2 , and TFA was added under ice cooling. The mixture was stirred for 3 hours while warming to room temperature. Et 3 N was added under ice cooling to stop the reaction. Water was added to the residue obtained by concentration under reduced pressure. Further, 1 M NaOH aqueous solution, 10% -MeCN / H 2 O was added to the suspension and dissolved. The mixture was purified by ODS (conditions: 15 mL of ODS silica, charged, then eluted with buffer, 10%, 20%, 30%, 40% MeCN / buffer (10R each). The fraction was concentrated and diluted with H 2 O After elution (eluent: H 2 O, 10% MeCN / H 2 O, 50% MeCN / H 2 O in this order), elution was carried out to obtain the desired product (45 mg) as a white powder.
製造例35の化合物(100 mg)とグリオキシル酸水和物(30 mg)とMS3A (300 mg)をDMF (1.5 mL)に懸濁させ、終夜室温で攪拌した。MeOHで反応液を希釈し、MS3Aをろ別し、MeOHで洗浄した。ろ液と洗液をあわせ減圧下濃縮した後、残渣にEtOAcを加え、析出した粉末をろ取し、EtOAcで洗浄した。粉末を乾燥後、CH2Cl2に懸濁させ、氷冷下、TFAを加えた。室温に昇温しながら3時間攪拌した。氷冷下、Et3Nを加え、反応を停止させた。減圧下濃縮して得られた残渣に水を加えた。さらに、その懸濁液に1MNaOH水溶液, 10%-MeCN/H2Oを加え溶解させた。その混合物をODS精製(条件:ODSシリカ15 mL、チャージ後、buffer, 10%、20%、30%、40% MeCN/bufferで溶出した(各10R)。フラクションを濃縮後、H2Oで希釈、チャージ後(溶出液;H2O、10% MeCN/H2O、50%MeCN/H2Oの順)溶出した。凍結乾燥し目的物(45 mg)を白色粉末として得た。 Example 154
The compound of Production Example 35 (100 mg), glyoxylic acid hydrate (30 mg) and MS3A (300 mg) were suspended in DMF (1.5 mL) and stirred overnight at room temperature. The reaction was diluted with MeOH, MS3A was filtered off and washed with MeOH. The filtrate and the washing solution were combined and concentrated under reduced pressure, EtOAc was added to the residue, and the precipitated powder was collected by filtration and washed with EtOAc. The powder was dried, suspended in CH 2 Cl 2 , and TFA was added under ice cooling. The mixture was stirred for 3 hours while warming to room temperature. Et 3 N was added under ice cooling to stop the reaction. Water was added to the residue obtained by concentration under reduced pressure. Further, 1 M NaOH aqueous solution, 10% -MeCN / H 2 O was added to the suspension and dissolved. The mixture was purified by ODS (conditions: 15 mL of ODS silica, charged, then eluted with buffer, 10%, 20%, 30%, 40% MeCN / buffer (10R each). The fraction was concentrated and diluted with H 2 O After elution (eluent: H 2 O, 10% MeCN / H 2 O, 50% MeCN / H 2 O in this order), elution was carried out to obtain the desired product (45 mg) as a white powder.
実施例155
製造例33-55の化合物(50.0 mg)、HBTU(30.2 mg)、DMF(750 μL)、DIPEA(d=0.742, 37.2 μL)の混合物を室温で1時間撹拌した。 国際公開WO03/068807号の製造例20の化合物(63.8 mg) を加え室温で6時間撹拌した。反応液にEtOAcを加え生じた析出物をろ取した。減圧乾燥して得た固体を、氷冷下、TFA中(d=1.480、2.00 mL)に加え3時間撹拌した。反応液を7%MeCN水(100 mL)中に滴下した。その混合物をODSシリカゲルクロマトグラフィーで精製した(水で置換したODSシリカゲル20 mLにチャージ後、0, 5, 10, 15, 20% pH2.0の塩酸水:MeCN系で段階的に溶出した)。凍結乾燥し実施例155の化合物(47.3 mg)を黄色固体として得た。 Example 155
A mixture of the compound of Production Example 33-55 (50.0 mg), HBTU (30.2 mg), DMF (750 μL), and DIPEA (d = 0.742, 37.2 μL) was stirred at room temperature for 1 hour. The compound of Production Example 20 (63.8 mg) of International Publication WO03 / 068807 was added and stirred at room temperature for 6 hours. EtOAc was added to the reaction solution, and the resulting precipitate was collected by filtration. The solid obtained by drying under reduced pressure was added to TFA (d = 1.480, 2.00 mL) under ice cooling and stirred for 3 hours. The reaction solution was added dropwise to 7% MeCN water (100 mL). The mixture was purified by ODS silica gel chromatography (charged to 20 mL of ODS silica gel replaced with water and then eluted stepwise with 0, 5, 10, 15, 20% pH 2.0 hydrochloric acid: MeCN system). Lyophilization was performed to obtain the compound of Example 155 (47.3 mg) as a yellow solid.
製造例33-55の化合物(50.0 mg)、HBTU(30.2 mg)、DMF(750 μL)、DIPEA(d=0.742, 37.2 μL)の混合物を室温で1時間撹拌した。 国際公開WO03/068807号の製造例20の化合物(63.8 mg) を加え室温で6時間撹拌した。反応液にEtOAcを加え生じた析出物をろ取した。減圧乾燥して得た固体を、氷冷下、TFA中(d=1.480、2.00 mL)に加え3時間撹拌した。反応液を7%MeCN水(100 mL)中に滴下した。その混合物をODSシリカゲルクロマトグラフィーで精製した(水で置換したODSシリカゲル20 mLにチャージ後、0, 5, 10, 15, 20% pH2.0の塩酸水:MeCN系で段階的に溶出した)。凍結乾燥し実施例155の化合物(47.3 mg)を黄色固体として得た。 Example 155
A mixture of the compound of Production Example 33-55 (50.0 mg), HBTU (30.2 mg), DMF (750 μL), and DIPEA (d = 0.742, 37.2 μL) was stirred at room temperature for 1 hour. The compound of Production Example 20 (63.8 mg) of International Publication WO03 / 068807 was added and stirred at room temperature for 6 hours. EtOAc was added to the reaction solution, and the resulting precipitate was collected by filtration. The solid obtained by drying under reduced pressure was added to TFA (d = 1.480, 2.00 mL) under ice cooling and stirred for 3 hours. The reaction solution was added dropwise to 7% MeCN water (100 mL). The mixture was purified by ODS silica gel chromatography (charged to 20 mL of ODS silica gel replaced with water and then eluted stepwise with 0, 5, 10, 15, 20% pH 2.0 hydrochloric acid: MeCN system). Lyophilization was performed to obtain the compound of Example 155 (47.3 mg) as a yellow solid.
実施例156から164の化合物は、実施例155の方法と同様にして製造した。
The compounds of Examples 156 to 164 were prepared in the same manner as in Example 155.
実施例165
実施例55の化合物(60mg)のDMF(450 μL)溶液にエチル 3-ブロモプロピオネート(5.45 μL )、粉末K2CO3 (11.6 mg)及びNaI (6.3 mg) を加え、室温で4日間撹拌した。反応液にEtOAcを加え生じた析出物をろ取し、粗エチルエステル中間体を白色粉末(84.3 mg)として得た。粗エチルエステル中間体を水(600 μL)に溶解し、1M NaOH水溶液(420 μL)を加え、室温で2時間撹拌した。1M塩酸で中和後、ODSシリカゲルクロマトグラフィーで精製した(溶出液;MeCN:pH6.86 buffer=27.5:72.5)。 目的物を含むフラクションを集め水で約2倍に希釈した後、ODSカラムクロマトグラフィーにより脱塩し、MeCNを減圧下濃縮した後、凍結乾燥し、実施例165の化合物(15.0 mg) を白色アモルファスとして得た。 Example 165
Ethyl 3-bromopropionate (5.45 μL), powdered K 2 CO 3 (11.6 mg) and NaI (6.3 mg) were added to a solution of the compound of Example 55 (60 mg) in DMF (450 μL), and the mixture was stirred at room temperature for 4 days. Stir. EtOAc was added to the reaction mixture, and the resulting precipitate was collected by filtration to give a crude ethyl ester intermediate as a white powder (84.3 mg). The crude ethyl ester intermediate was dissolved in water (600 μL), 1M aqueous NaOH solution (420 μL) was added, and the mixture was stirred at room temperature for 2 hr. After neutralization with 1M hydrochloric acid, purification was performed by ODS silica gel chromatography (eluent; MeCN: pH6.86 buffer = 27.5: 72.5). Fractions containing the desired product were collected and diluted approximately 2-fold with water, then desalted by ODS column chromatography, and MeCN was concentrated under reduced pressure and lyophilized to obtain the compound of Example 165 (15.0 mg) as a white amorphous product. Got as.
実施例55の化合物(60mg)のDMF(450 μL)溶液にエチル 3-ブロモプロピオネート(5.45 μL )、粉末K2CO3 (11.6 mg)及びNaI (6.3 mg) を加え、室温で4日間撹拌した。反応液にEtOAcを加え生じた析出物をろ取し、粗エチルエステル中間体を白色粉末(84.3 mg)として得た。粗エチルエステル中間体を水(600 μL)に溶解し、1M NaOH水溶液(420 μL)を加え、室温で2時間撹拌した。1M塩酸で中和後、ODSシリカゲルクロマトグラフィーで精製した(溶出液;MeCN:pH6.86 buffer=27.5:72.5)。 目的物を含むフラクションを集め水で約2倍に希釈した後、ODSカラムクロマトグラフィーにより脱塩し、MeCNを減圧下濃縮した後、凍結乾燥し、実施例165の化合物(15.0 mg) を白色アモルファスとして得た。 Example 165
Ethyl 3-bromopropionate (5.45 μL), powdered K 2 CO 3 (11.6 mg) and NaI (6.3 mg) were added to a solution of the compound of Example 55 (60 mg) in DMF (450 μL), and the mixture was stirred at room temperature for 4 days. Stir. EtOAc was added to the reaction mixture, and the resulting precipitate was collected by filtration to give a crude ethyl ester intermediate as a white powder (84.3 mg). The crude ethyl ester intermediate was dissolved in water (600 μL), 1M aqueous NaOH solution (420 μL) was added, and the mixture was stirred at room temperature for 2 hr. After neutralization with 1M hydrochloric acid, purification was performed by ODS silica gel chromatography (eluent; MeCN: pH6.86 buffer = 27.5: 72.5). Fractions containing the desired product were collected and diluted approximately 2-fold with water, then desalted by ODS column chromatography, and MeCN was concentrated under reduced pressure and lyophilized to obtain the compound of Example 165 (15.0 mg) as a white amorphous product. Got as.
実施例166
実施例55の化合物(100 mg) のDMF(750 μL)溶液に1,3-プロパンサルトン(9 mg)を加え室温で20時間撹拌した。1,3-プロパンサルトン(9 mg)を追加し、室温撹拌20時間し、さらに1,3-プロパンサルトン(9 mg)を追加した後、50℃に昇温し、3日間撹拌した。反応液を室温まで放冷後、水(50 mL)を加え、ODSシリカゲルクロマトグラフィー(溶出液;MeCN:pH 6.86 buffer=25:75から27.5:72.5まで)で精製した。 目的物を含むフラクションを集め水で約2倍に薄めた後、ODSカラムクロマトグラフィーで脱塩し、MeCNを減圧下濃縮後、凍結乾燥し、実施例166の化合物(8.46 mg)を白色アモルファスとして得た。 Example 166
1,3-propane sultone (9 mg) was added to a DMF (750 μL) solution of the compound of Example 55 (100 mg), and the mixture was stirred at room temperature for 20 hours. 1,3-propane sultone (9 mg) was added, and the mixture was stirred at room temperature for 20 hours. 1,3-propane sultone (9 mg) was further added, and the mixture was warmed to 50 ° C. and stirred for 3 days. The reaction mixture was allowed to cool to room temperature, water (50 mL) was added, and the mixture was purified by ODS silica gel chromatography (eluent; MeCN: pH 6.86 buffer = 25: 75 to 27.5: 72.5). Fractions containing the desired product were collected, diluted with water approximately twice, and then desalted with ODS column chromatography.MeCN was concentrated under reduced pressure and lyophilized to obtain the compound of Example 166 (8.46 mg) as a white amorphous substance. Obtained.
実施例55の化合物(100 mg) のDMF(750 μL)溶液に1,3-プロパンサルトン(9 mg)を加え室温で20時間撹拌した。1,3-プロパンサルトン(9 mg)を追加し、室温撹拌20時間し、さらに1,3-プロパンサルトン(9 mg)を追加した後、50℃に昇温し、3日間撹拌した。反応液を室温まで放冷後、水(50 mL)を加え、ODSシリカゲルクロマトグラフィー(溶出液;MeCN:pH 6.86 buffer=25:75から27.5:72.5まで)で精製した。 目的物を含むフラクションを集め水で約2倍に薄めた後、ODSカラムクロマトグラフィーで脱塩し、MeCNを減圧下濃縮後、凍結乾燥し、実施例166の化合物(8.46 mg)を白色アモルファスとして得た。 Example 166
1,3-propane sultone (9 mg) was added to a DMF (750 μL) solution of the compound of Example 55 (100 mg), and the mixture was stirred at room temperature for 20 hours. 1,3-propane sultone (9 mg) was added, and the mixture was stirred at room temperature for 20 hours. 1,3-propane sultone (9 mg) was further added, and the mixture was warmed to 50 ° C. and stirred for 3 days. The reaction mixture was allowed to cool to room temperature, water (50 mL) was added, and the mixture was purified by ODS silica gel chromatography (eluent; MeCN: pH 6.86 buffer = 25: 75 to 27.5: 72.5). Fractions containing the desired product were collected, diluted with water approximately twice, and then desalted with ODS column chromatography.MeCN was concentrated under reduced pressure and lyophilized to obtain the compound of Example 166 (8.46 mg) as a white amorphous substance. Obtained.
実施例167
製造例10-19の化合物(115 mg)にTFAを加えて室温下1時間撹拌し、氷冷下でEt3Nを加えた。混合溶液を減圧下濃縮し、20%MeCN水溶液を加えたところ、懸濁液になったため、1M NaOH水溶液を加えてpH 9.5程度にし、完全に溶解させた。この水溶液をODSカラムクロマトグラフィー(溶出液;MeCN:pH 6.8 buffer = 20:80 から80:20まで)で精製した。得られた水溶液を再度ODSカラムクロマトグラフィーを行うことにより、塩を除き、また溶媒をMeCNで置換した。得られた溶液を減圧下濃縮し、残った水溶液を凍結乾燥することで実施例167の化合物(89 mg)を白色粉末として得た。 Example 167
TFA was added to the compound of Production Example 10-19 (115 mg) and stirred at room temperature for 1 hour, and Et 3 N was added under ice cooling. The mixed solution was concentrated under reduced pressure, and a 20% aqueous MeCN solution was added to form a suspension. A 1M NaOH aqueous solution was added to adjust the pH to about 9.5, and the solution was completely dissolved. This aqueous solution was purified by ODS column chromatography (eluent; MeCN: pH 6.8 buffer = 20: 80 to 80:20). The obtained aqueous solution was again subjected to ODS column chromatography to remove salts and replace the solvent with MeCN. The obtained solution was concentrated under reduced pressure, and the remaining aqueous solution was lyophilized to obtain the compound of Example 167 (89 mg) as a white powder.
製造例10-19の化合物(115 mg)にTFAを加えて室温下1時間撹拌し、氷冷下でEt3Nを加えた。混合溶液を減圧下濃縮し、20%MeCN水溶液を加えたところ、懸濁液になったため、1M NaOH水溶液を加えてpH 9.5程度にし、完全に溶解させた。この水溶液をODSカラムクロマトグラフィー(溶出液;MeCN:pH 6.8 buffer = 20:80 から80:20まで)で精製した。得られた水溶液を再度ODSカラムクロマトグラフィーを行うことにより、塩を除き、また溶媒をMeCNで置換した。得られた溶液を減圧下濃縮し、残った水溶液を凍結乾燥することで実施例167の化合物(89 mg)を白色粉末として得た。 Example 167
TFA was added to the compound of Production Example 10-19 (115 mg) and stirred at room temperature for 1 hour, and Et 3 N was added under ice cooling. The mixed solution was concentrated under reduced pressure, and a 20% aqueous MeCN solution was added to form a suspension. A 1M NaOH aqueous solution was added to adjust the pH to about 9.5, and the solution was completely dissolved. This aqueous solution was purified by ODS column chromatography (eluent; MeCN: pH 6.8 buffer = 20: 80 to 80:20). The obtained aqueous solution was again subjected to ODS column chromatography to remove salts and replace the solvent with MeCN. The obtained solution was concentrated under reduced pressure, and the remaining aqueous solution was lyophilized to obtain the compound of Example 167 (89 mg) as a white powder.
実施例168から178の化合物は、実施例167の方法と同様にして製造した。
The compounds of Examples 168 to 178 were prepared in the same manner as in Example 167.
実施例180
実施例37の化合物(50 mg)のMeOH(1.5 mL)溶液に10% HCl/MeOH溶液 (1.48 mL)を加えると懸濁した。反応混合物は室温で攪拌した。反応混合物を減圧下濃縮し、得られた固体をEtOAcを加えて粉末化した。沈殿物をろ取し、EtOAcで洗浄後、乾燥し、実施例180の化合物(29 mg)を得た。 Example 180
A 10% HCl / MeOH solution (1.48 mL) was suspended in a solution of the compound of Example 37 (50 mg) in MeOH (1.5 mL). The reaction mixture was stirred at room temperature. The reaction mixture was concentrated under reduced pressure, and the resulting solid was pulverized by adding EtOAc. The precipitate was collected by filtration, washed with EtOAc, and dried to give the compound of Example 180 (29 mg).
実施例37の化合物(50 mg)のMeOH(1.5 mL)溶液に10% HCl/MeOH溶液 (1.48 mL)を加えると懸濁した。反応混合物は室温で攪拌した。反応混合物を減圧下濃縮し、得られた固体をEtOAcを加えて粉末化した。沈殿物をろ取し、EtOAcで洗浄後、乾燥し、実施例180の化合物(29 mg)を得た。 Example 180
A 10% HCl / MeOH solution (1.48 mL) was suspended in a solution of the compound of Example 37 (50 mg) in MeOH (1.5 mL). The reaction mixture was stirred at room temperature. The reaction mixture was concentrated under reduced pressure, and the resulting solid was pulverized by adding EtOAc. The precipitate was collected by filtration, washed with EtOAc, and dried to give the compound of Example 180 (29 mg).
実施例179及び181の化合物は、実施例180の方法と同様にして製造した。
The compounds of Examples 179 and 181 were prepared in the same manner as in Example 180.
実施例182
実施例156の化合物 (50 mg)、1H-ピラゾール-1-カルボキサミジン塩酸塩(12.9 mg)、DMF(250 μL)、DIPEA(d=0.742, 24.6 μL)の混合物を室温で終夜撹拌した。反応液に水を加え溶解するまでMeCNを加えた。その混合物をODSシリカゲルクロマトグラフィー(ODSシリカゲル15 mL、溶出液;0%から65% pH2.0の塩酸水:MeCN系)精製した。凍結乾燥し、 実施例182の化合物(34.2 mg)を白色粉末として得た。 Example 182
A mixture of the compound of Example 156 (50 mg), 1H-pyrazole-1-carboxamidine hydrochloride (12.9 mg), DMF (250 μL), DIPEA (d = 0.742, 24.6 μL) was stirred at room temperature overnight. MeCN was added to the reaction solution until water was added and dissolved. The mixture was purified by ODS silica gel chromatography (ODS silica gel 15 mL, eluent; hydrochloric acid water of 0% to 65% pH 2.0: MeCN system). Lyophilization was performed to obtain the compound of Example 182 (34.2 mg) as a white powder.
実施例156の化合物 (50 mg)、1H-ピラゾール-1-カルボキサミジン塩酸塩(12.9 mg)、DMF(250 μL)、DIPEA(d=0.742, 24.6 μL)の混合物を室温で終夜撹拌した。反応液に水を加え溶解するまでMeCNを加えた。その混合物をODSシリカゲルクロマトグラフィー(ODSシリカゲル15 mL、溶出液;0%から65% pH2.0の塩酸水:MeCN系)精製した。凍結乾燥し、 実施例182の化合物(34.2 mg)を白色粉末として得た。 Example 182
A mixture of the compound of Example 156 (50 mg), 1H-pyrazole-1-carboxamidine hydrochloride (12.9 mg), DMF (250 μL), DIPEA (d = 0.742, 24.6 μL) was stirred at room temperature overnight. MeCN was added to the reaction solution until water was added and dissolved. The mixture was purified by ODS silica gel chromatography (ODS silica gel 15 mL, eluent; hydrochloric acid water of 0% to 65% pH 2.0: MeCN system). Lyophilization was performed to obtain the compound of Example 182 (34.2 mg) as a white powder.
実施例183
実施例55の化合物(103mg)、1,4-ジオキサン-2,5-ジオール(17.3 mg)、CHCl3 (3 mL)、MeOH (1 mL)、MS3A (154.5 mg)の混合物にNaBH3CN (10 mg)とAcOH (14 μL)を加えた。混合物を室温で3時間撹拌した。MS3Aをろ別して減圧下濃縮した。残渣をMeCN:H2O=約1:10の溶液に懸濁し、1M NaOHを用いてpH8~9(pH試験紙)に調整し溶解させた。この溶液をODS中圧カラムクロマトグラフィー(洗浄:MeCN:H2O=1:9、MeCN:(pH 6.86 標準緩衝溶液)=1:9、溶出:MeCN:(pH 6.86 標準緩衝溶液)= 1:4, 3:7)を用いて精製した。目的物を含むフラクションを集めてH2Oで約1.5倍に希釈し、ODS中圧カラムクロマトグラフィー(洗浄液:MeCN:H2O=1:9、溶出液;MeCN:H2O=4:1)を用いて脱塩・減圧下濃縮した。目的物を含むフラクションを減圧下に濃縮し、水を含む残渣を凍結乾燥して実施例183の化合物(73.8 mg)を粉末として得た。 Example 183
To a mixture of the compound of Example 55 (103 mg), 1,4-dioxane-2,5-diol (17.3 mg), CHCl 3 (3 mL), MeOH (1 mL), MS3A (154.5 mg), NaBH 3 CN ( 10 mg) and AcOH (14 μL) were added. The mixture was stirred at room temperature for 3 hours. MS3A was filtered off and concentrated under reduced pressure. The residue was suspended in a solution of MeCN: H 2 O = about 1:10, adjusted to pH 8-9 (pH test paper) with 1M NaOH, and dissolved. This solution was subjected to ODS medium pressure column chromatography (washing: MeCN: H 2 O = 1: 9, MeCN: (pH 6.86 standard buffer solution) = 1: 9, elution: MeCN: (pH 6.86 standard buffer solution) = 1: 4, 3: 7). Fractions containing the desired product were collected, diluted approximately 1.5 times with H 2 O, and ODS medium pressure column chromatography (washing solution: MeCN: H 2 O = 1: 9, eluent; MeCN: H 2 O = 4: 1) ) And then concentrated under reduced pressure. The fraction containing the desired product was concentrated under reduced pressure, and the residue containing water was lyophilized to obtain the compound of Example 183 (73.8 mg) as a powder.
実施例55の化合物(103mg)、1,4-ジオキサン-2,5-ジオール(17.3 mg)、CHCl3 (3 mL)、MeOH (1 mL)、MS3A (154.5 mg)の混合物にNaBH3CN (10 mg)とAcOH (14 μL)を加えた。混合物を室温で3時間撹拌した。MS3Aをろ別して減圧下濃縮した。残渣をMeCN:H2O=約1:10の溶液に懸濁し、1M NaOHを用いてpH8~9(pH試験紙)に調整し溶解させた。この溶液をODS中圧カラムクロマトグラフィー(洗浄:MeCN:H2O=1:9、MeCN:(pH 6.86 標準緩衝溶液)=1:9、溶出:MeCN:(pH 6.86 標準緩衝溶液)= 1:4, 3:7)を用いて精製した。目的物を含むフラクションを集めてH2Oで約1.5倍に希釈し、ODS中圧カラムクロマトグラフィー(洗浄液:MeCN:H2O=1:9、溶出液;MeCN:H2O=4:1)を用いて脱塩・減圧下濃縮した。目的物を含むフラクションを減圧下に濃縮し、水を含む残渣を凍結乾燥して実施例183の化合物(73.8 mg)を粉末として得た。 Example 183
To a mixture of the compound of Example 55 (103 mg), 1,4-dioxane-2,5-diol (17.3 mg), CHCl 3 (3 mL), MeOH (1 mL), MS3A (154.5 mg), NaBH 3 CN ( 10 mg) and AcOH (14 μL) were added. The mixture was stirred at room temperature for 3 hours. MS3A was filtered off and concentrated under reduced pressure. The residue was suspended in a solution of MeCN: H 2 O = about 1:10, adjusted to pH 8-9 (pH test paper) with 1M NaOH, and dissolved. This solution was subjected to ODS medium pressure column chromatography (washing: MeCN: H 2 O = 1: 9, MeCN: (pH 6.86 standard buffer solution) = 1: 9, elution: MeCN: (pH 6.86 standard buffer solution) = 1: 4, 3: 7). Fractions containing the desired product were collected, diluted approximately 1.5 times with H 2 O, and ODS medium pressure column chromatography (washing solution: MeCN: H 2 O = 1: 9, eluent; MeCN: H 2 O = 4: 1) ) And then concentrated under reduced pressure. The fraction containing the desired product was concentrated under reduced pressure, and the residue containing water was lyophilized to obtain the compound of Example 183 (73.8 mg) as a powder.
実施例184から294の化合物は、実施例183の方法と同様にして製造した。このうち、実施例199は造塩して塩酸塩として得た。
The compounds of Examples 184 to 294 were prepared in the same manner as in Example 183. Of these, Example 199 was salted to obtain a hydrochloride.
The compounds of Examples 184 to 294 were prepared in the same manner as in Example 183. Of these, Example 199 was salted to obtain a hydrochloride.
上述の実施例又は製造例の方法と同様にして、後記表に示す実施例及び製造例の化合物を製造した。製造例化合物および実施例化合物について、それぞれ、構造、物理化学的データ及び製造法を示した。表中、Prは製造例番号、Exは実施例番号を表す。Ref-Prは参考にして製造できる製造例番号を表し、Ref-Exは参考にして製造できる実施例番号を表す。ただし、Ref-Pr中に、参考にして製造できる実施例番号を記載することもあり、その場合には、Ex番号で記載している。Ref-Ex中に、参考にして製造できる製造例番号を記載することもあり、その場合には、Pr番号で記載している。STRの欄には構造式を表している。DATの欄は、1H-NMR及び/又はMSデータを記載した。NMR1 = d6-DMSO中で測定した1H-NMRのケミカルシフト値(δ)、NMR2 = d6-DMSO+D2O中で測定した1H-NMRのケミカルシフト値(δ)、NMR3 = CDCl3中で測定した1H-NMRのケミカルシフト値(δ)を示す。ESI及びEIは、それぞれエレクトロスプレーイオン化法による質量分析値、電子イオン化法による質量分析値を表し、ESIおよびEIデータの+又は未記載の場合にはポジティブを意味する。APCIは、大気圧化学イオン化法質量分析を示し、APCI/ESIはAPCIとESIで同時測定された質量分析値を意味する。
The compounds of Examples and Production Examples shown in the table below were produced in the same manner as in the above Examples or Production Examples. For the production example compounds and the example compounds, structures, physicochemical data and production methods are shown, respectively. In the table, Pr represents the production example number and Ex represents the example number. Ref-Pr represents a production example number that can be produced by reference, and Ref-Ex represents an example number that can be produced by reference. However, in the Ref-Pr, an example number that can be manufactured by reference may be described, and in this case, the Ex number is described. In Ref-Ex, a production example number that can be produced by reference may be described, in which case it is indicated by a Pr number. The column of STR represents the structural formula. In the column of DAT, 1 H-NMR and / or MS data are described. NMR1 = d 6 -DMSO chemical shift values of 1 H-NMR measured in (δ), NMR2 = d 6 -DMSO + D 2 O The chemical shift values of 1 H-NMR measured in ([delta]), in NMR3 = CDCl3 1 shows the 1 H-NMR chemical shift value (δ) measured in ( 1 ). ESI and EI represent mass analysis values by electrospray ionization method and mass analysis values by electron ionization method, respectively, and mean positive or positive when ESI and EI data are not described. APCI indicates atmospheric pressure chemical ionization mass spectrometry, and APCI / ESI means mass spectrometry values measured simultaneously with APCI and ESI.
































































































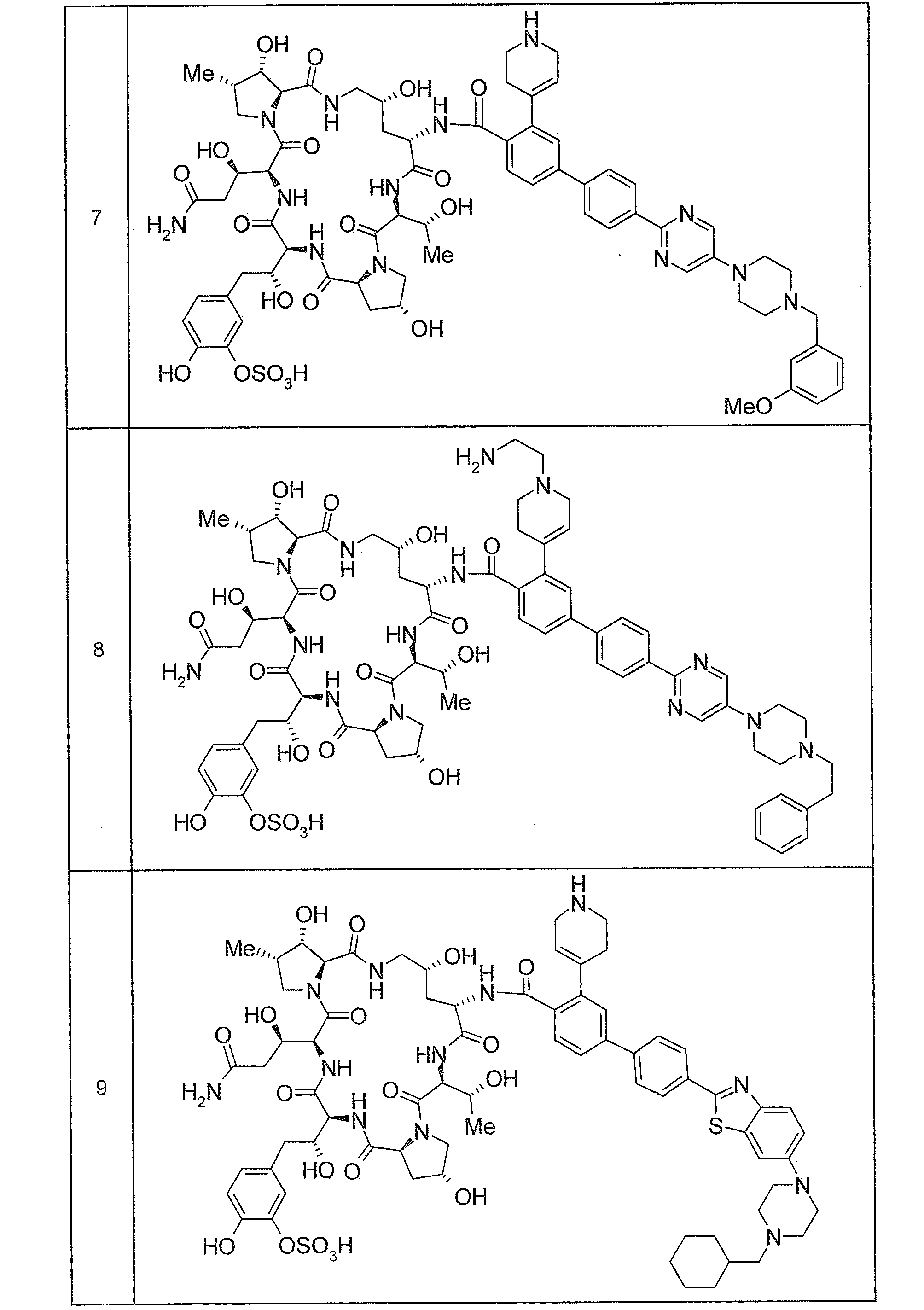




























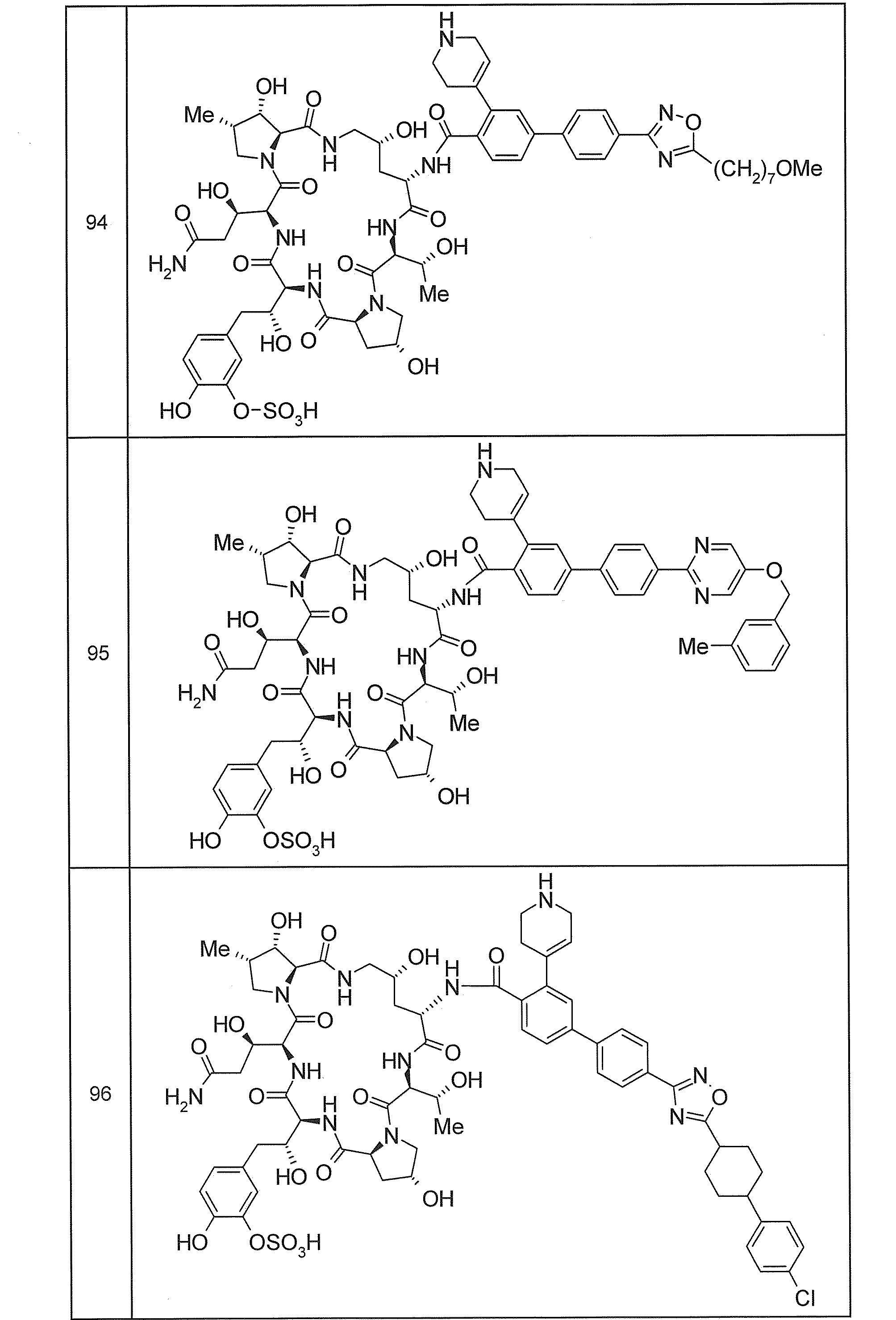











































































式(I)の化合物又はその塩は、抗真菌活性を有し、種々の感染症、例えば皮膚糸状菌症(例えば白癬症等)、でん風、カンジダ症、ゲオトリクム症、砂毛症、アスペルギルス症、ペニシリウム症、スポロトリクス症、クロモミセス症、コクシジオイデス症、ヒストプラスマ症、ブラストミセス症、パラコクシジオイデス症、プソイドアレシェリアシス、足菌腫、真菌性角膜炎、耳真菌症、ニューモシスティス症、真菌血症等の予防及び/又は治療に有用である。
The compound of the formula (I) or a salt thereof has antifungal activity and has various infectious diseases such as dermatophytosis (e.g. ringworm), vermiculitis, candidiasis, geotrichumosis, sandy hair, Aspergillus , Penicillosis, sporotricosis, chromomises, coccidioidomycosis, histoplasmosis, blastosomiasis, paracoccidioidomycosis, pseudoaleseriasis, foot mycosis, fungal keratitis, otomycosis, pneumocystis, fungi It is useful for the prevention and / or treatment of blood pressure.
Claims (21)
- 式(I)の化合物又はその製薬学的に許容される塩。

(式中、R1は、-NH2、-OH、-NH-低級アルキレン-NH2、-低級アルキレン-NH-低級アルキル、-低級アルキレン-N(低級アルキル)2、-NH-低級アルキレン-C(=O)OH、又は、-NHCH(OH)C(=O)OH、
R2は、-S(=O)2OH、-CH2CH(OH)CH2OH、-低級アルキレン-C(=O)OH、低級アルキル、又は、H、
A環は、含窒素ヘテロ環基、アリール、又はシクロアルキル、
RAは、H、-低級アルキル、-低級アルキレン-NH2、-低級アルキレン-NH-低級アルキル、-低級アルキレン-N(低級アルキル)2、-低級アルキレン-OH、-低級アルキレン-O-低級アルキル、-低級アルキレン-C(=O)OH、-低級アルキレン-S(=O)2OH、-低級アルキレン-S(=O)2O-低級アルキル、-C(=O)O-低級アルキル、-C(=O)-低級アルキレン-NH2、-C(=O)-低級アルキレン- NH-低級アルキル、-C(=O)-低級アルキレン-N(低級アルキル)2、-C(=O)-NH2で置換されていてもよい低級アルキレン-OH、-C(=NH)-NH2、-CH(低級アルキレン-OH)2、又は、シクロアルキル、
Xは、単結合、低級アルキレン、-NH-、又は、-N(低級アルキル)-
RB及びRCは、それぞれ同一又は異なって、H又はハロゲン、
B環は、下記式(II)、式(III-a)、又は、式(III-b)、

で示される基であり、
*は、式(I)におけるXと結合する位置を示し、**は、式(I)におけるRBと結合する位置を示し、
D環は、含窒素ヘテロアリールであり、
RDは、下記式(IV)から(VI)
で示される基、又は、-L-O-R101で示される基、
Lは、低級アルキレン、高級アルキレン、-O-、-O-低級アルキレン、又は、単結合、
E環は、ヘテロ環基、
F環及びF1環は、それぞれ同一又は異なって、シクロアルキル、
Fg環は、シクロアルキル、又は、アリール、
Efg環は、ヘテロ環基、シクロアルキル、又は、アリール、
REは、H、低級アルキル、-O-低級アルキル、又は、ハロゲン、
R101は、低級アルキル、-低級アルキレン-シクロアルキル、又は、-高級アルキレン-シクロアルキル、
RZ1又はRZ2は、それぞれ同一又は異なって、H、ハロゲン、ハロゲン又は低級アルキルで置換されていてもよいアリール、低級アルキル、高級アルキル、-O-低級アルキル、-O-高級アルキル、-O- シクロアルキル、-O-アリール、-O-低級アルキレン-アリール、-高級アルキレン-O-低級アルキル、シクロアルキル、又は、-C(=O)O-低級アルキレン-アリールである。) A compound of formula (I) or a pharmaceutically acceptable salt thereof.
Wherein R 1 is -NH 2 , -OH, -NH-lower alkylene-NH 2 , -lower alkylene-NH-lower alkyl, -lower alkylene-N (lower alkyl) 2 , -NH-lower alkylene- C (= O) OH or -NHCH (OH) C (= O) OH,
R 2 is -S (= O) 2 OH, -CH 2 CH (OH) CH 2 OH, -lower alkylene-C (= O) OH, lower alkyl, or H,
A ring is a nitrogen-containing heterocyclic group, aryl, or cycloalkyl,
R A is H, -lower alkyl, -lower alkylene-NH 2 , -lower alkylene-NH-lower alkyl, -lower alkylene-N (lower alkyl) 2 , -lower alkylene-OH, -lower alkylene-O-lower Alkyl, -lower alkylene-C (= O) OH, -lower alkylene-S (= O) 2 OH, -lower alkylene-S (= O) 2 O-lower alkyl, -C (= O) O-lower alkyl , -C (= O) -lower alkylene-NH 2 , -C (= O) -lower alkylene-NH-lower alkyl, -C (= O) -lower alkylene-N (lower alkyl) 2 , -C (= O) -lower alkylene optionally substituted with —NH 2 , —C (═NH) —NH 2 , —CH (lower alkylene-OH) 2 , or cycloalkyl,
X is a single bond, lower alkylene, -NH-, or -N (lower alkyl)-
R B and R C are the same or different, and H or halogen,
Ring B is represented by the following formula (II), formula (III-a), or formula (III-b),
A group represented by
* Indicates the position at which the group bonds to X in formula (I), ** indicates the position at which the group bonds to R B in the formula (I),
Ring D is a nitrogen-containing heteroaryl,
R D represents the following formulas (IV) to (VI)
Or a group represented by -LOR 101 ,
L is lower alkylene, higher alkylene, -O-, -O-lower alkylene, or a single bond,
E ring is a heterocyclic group,
F ring and F1 ring are the same or different and each represents cycloalkyl,
Fg ring is cycloalkyl or aryl,
Efg ring is a heterocyclic group, cycloalkyl or aryl,
R E is H, lower alkyl, -O-lower alkyl, or halogen,
R 101 represents lower alkyl, -lower alkylene-cycloalkyl, or -higher alkylene-cycloalkyl,
R Z1 or R Z2 is the same or different and is optionally substituted with H, halogen, halogen or lower alkyl, aryl, lower alkyl, higher alkyl, —O-lower alkyl, —O-higher alkyl, —O -Cycloalkyl, -O-aryl, -O-lower alkylene-aryl, -higher alkylene-O-lower alkyl, cycloalkyl, or -C (= O) O-lower alkylene-aryl. ) - A環が単環式飽和へテロ環基又は単環式不飽和ヘテロ環である、請求項1に記載の化合物又はその製薬学的に許容される塩。 The compound according to claim 1 or a pharmaceutically acceptable salt thereof, wherein the A ring is a monocyclic saturated heterocyclic group or a monocyclic unsaturated heterocyclic ring.
- A環が6員の含窒素へテロシクロアルキルである、請求項2に記載の化合物又はその製薬学的に許容される塩。 The compound according to claim 2 or a pharmaceutically acceptable salt thereof, wherein the A ring is a 6-membered nitrogen-containing heterocycloalkyl.
- D環が、単環式含窒素へテロアリールまたは縮合多環式含窒素へテロアリールである、請求項2又は3に記載の化合物又はその製薬学的に許容される塩。 The compound or a pharmaceutically acceptable salt thereof according to claim 2 or 3, wherein the ring D is a monocyclic nitrogen-containing heteroaryl or a condensed polycyclic nitrogen-containing heteroaryl.
- D環が、5乃至6員の単環式含窒素へテロアリールである、請求項2または3に記載の化合物又はその製薬学的に許容される塩。 The compound according to claim 2 or 3, or a pharmaceutically acceptable salt thereof, wherein the D ring is a 5- to 6-membered monocyclic nitrogen-containing heteroaryl.
- D環が、5員-5員縮合多環式含窒素へテロアリールまたは5員-6員縮合多環式含窒素へテロアリールである、請求項2または3に記載の化合物又はその製薬学的に許容される塩。 The compound according to claim 2 or 3, or a pharmaceutically acceptable salt thereof, wherein the ring D is a 5-membered to 5-membered condensed polycyclic nitrogen-containing heteroaryl or a 5-membered to 6-membered condensed polycyclic nitrogen-containing heteroaryl. Salt.
- B環が、式(II)で示される基である、請求項1から6の何れかに記載の化合物又はその製薬学的に許容される塩。 The compound or a pharmaceutically acceptable salt thereof according to any one of claims 1 to 6, wherein the B ring is a group represented by the formula (II).
- B環が式(II)で示される基であり、D環が単環式含窒素へテロアリールまたは縮合多環式含窒素へテロアリールである、請求項1から3の何れかに記載の化合物又はその製薬学的に許容される塩。 The compound according to any one of claims 1 to 3, wherein the ring B is a group represented by the formula (II), and the ring D is a monocyclic nitrogen-containing heteroaryl or a condensed polycyclic nitrogen-containing heteroaryl, or a compound thereof A pharmaceutically acceptable salt.
- B環が、式(II)で示される基であり、D環が、5員-5員縮合多環式含窒素へテロアリールである、請求項1から3の何れかに記載の化合物又はその製薬学的に許容される塩。 The compound according to any one of claims 1 to 3, wherein the B ring is a group represented by the formula (II), and the D ring is a 5-membered to 5-membered condensed polycyclic nitrogen-containing heteroaryl, or a pharmaceutical thereof A chemically acceptable salt.
- B環が、式(II)で示される基であり、D環が、5乃至6員の単環式含窒素へテロアリールである、請求項1から3に記載の化合物又はその製薬学的に許容される塩。 The compound according to claim 1 or 3, or a pharmaceutically acceptable salt thereof, wherein B ring is a group represented by formula (II) and D ring is a 5- to 6-membered monocyclic nitrogen-containing heteroaryl. Salt.
- RDが、式(V)で示される基である、請求項2に記載の化合物又はその製薬学的に許容される塩。 The compound according to claim 2 or a pharmaceutically acceptable salt thereof, wherein R D is a group represented by the formula (V).
- RDが、式(VI)で示される基である、請求項2に記載の化合物又はその製薬学的に許容される塩。 The compound according to claim 2 or a pharmaceutically acceptable salt thereof, wherein R D is a group represented by formula (VI).
- RD中のE環が含窒素ヘテロシクロアルキルである、請求項11に記載の化合物又はその製薬学的に許容される塩。 The compound according to claim 11 or a pharmaceutically acceptable salt thereof, wherein the E ring in R D is nitrogen-containing heterocycloalkyl.
- RD中のE環が6員の含窒素飽和ヘテロシクロアルキルである、請求項13に記載の化合物又はその製薬学的に許容される塩。 The compound according to claim 13 or a pharmaceutically acceptable salt thereof, wherein the E ring in R D is a 6-membered nitrogen-containing saturated heterocycloalkyl.
- 硫酸水素5-{(2R)-2-[(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-アミノ-1-ヒドロキシ-3-オキソプロピル]-2,11,15-トリヒドロキシ-6-[(1R)-1-ヒドロキシエチル]-9-(3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{2-[4-(2-フェニルエチル)ピペラジン-1-イル]イミダゾ[2,1-b][1,3,4]チアジアゾール-6-イル}[1,1'-ビフェニル]-4-カルボキサミド)-16-メチル-5,8,14,19,22,25-ヘキサオキソテトラコサヒドロ-1H-ジピロロ[2,1-c:2',1'-l][1,4,7,10,13,16]ヘキサアザシクロヘンイコシン-23-イル]-2-ヒドロキシエチル}-2-ヒドロキシフェニルエステル、
硫酸水素5-{(2R)-2-[(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-アミノ-1-ヒドロキシ-3-オキソプロピル]-2,11,15-トリヒドロキシ-6-[(1R)-1-ヒドロキシエチル]-9-(3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル]-4'-{5-[4-(2-フェニルエチル)ピペラジン-1-イル]ピリミジン-2-イル}[1,1'-ビフェニル]-4-カルボキサミド)-16-メチル-5,8,14,19,22,25-ヘキサオキソテトラコサヒドロ-1H-ジピロロ[2,1-c:2',1'-l][1,4,7,10,13,16]ヘキサアザシクロヘンイコシン-23-イル]-2-ヒドロキシエチル}-2-ヒドロキシフェニルエステル、
硫酸水素5-[(2R)-2-{(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-アミノ-1-ヒドロキシ-3-オキソプロピル]-9-{4'-(5-{4-[(4,4-ジメチルシクロヘキシル)オキシ]ピペリジン-1-イル}-1,2,4-オキサジアゾール-3-イル)-3-[1-(2-ヒドロキシエチル)-1,2,3,6-テトラヒドロピリジン-4-イル][1,1'-ビフェニル]-4-カルボキサミド}-2,11,15-トリヒドロキシ-6-[(1R)-1-ヒドロキシエチル]-16-メチル-5,8,14,19,22,25-ヘキサオキソテトラコサヒドロ-1H-ジピロロ[2,1-c:2',1'-l][1,4,7,10,13,16]ヘキサアザシクロヘンイコシン-23-イル}-2-ヒドロキシエチル]-2-ヒドロキシフェニルエステル及びN-{(2R,6S,9S,11R,14aS,15S,16S,20S,23S,25aS)-20-[(1R)-3-アミノ-1-ヒドロキシ-3-オキソプロピル]-2,11,15-トリヒドロキシ-6-[(1R)-1-ヒドロキシエチル]-23-[(1R)-1-ヒドロキシ-2-(4-ヒドロキシ-3-メトキシフェニル)エチル]-16-メチル-5,8,14,19,22,25-ヘキサオキソテトラコサヒドロ-1H-ジピロロ[2,1-c:2',1'-l][1,4,7,10,13,16]ヘキサアザシクロヘンイコシン-9-イル}-4'-(5-{4-[2-(4-エトキシフェニル)エチル]ピペラジン-1-イル}ピリミジン-2-イル)-3-(1,2,3,6-テトラヒドロピリジン-4-イル)[1,1'-ビフェニル]-4-カルボキサミドからなる群より選択された化合物又はその製薬学的に許容される塩。
Hydrogen sulfate 5-{(2R) -2-[(2R, 6S, 9S, 11R, 14aS, 15S, 16S, 20S, 23S, 25aS) -20-[(1R) -3-Amino-1-hydroxy-3 -Oxopropyl] -2,11,15-trihydroxy-6-[(1R) -1-hydroxyethyl] -9- (3- [1- (2-hydroxyethyl) -1,2,3,6- Tetrahydropyridin-4-yl] -4 '-{2- [4- (2-phenylethyl) piperazin-1-yl] imidazo [2,1-b] [1,3,4] thiadiazol-6-yl} [1,1'-biphenyl] -4-carboxamide) -16-methyl-5,8,14,19,22,25-hexaoxotetracosahydro-1H-dipirolo [2,1-c: 2 ', 1 '-l] [1,4,7,10,13,16] hexaazacyclohenicosin-23-yl] -2-hydroxyethyl} -2-hydroxyphenyl ester,
Hydrogen sulfate 5-{(2R) -2-[(2R, 6S, 9S, 11R, 14aS, 15S, 16S, 20S, 23S, 25aS) -20-[(1R) -3-Amino-1-hydroxy-3 -Oxopropyl] -2,11,15-trihydroxy-6-[(1R) -1-hydroxyethyl] -9- (3- [1- (2-hydroxyethyl) -1,2,3,6- Tetrahydropyridin-4-yl] -4 '-{5- [4- (2-phenylethyl) piperazin-1-yl] pyrimidin-2-yl} [1,1'-biphenyl] -4-carboxamide) -16 -Methyl-5,8,14,19,22,25-hexaoxotetracosahydro-1H-dipyrolo [2,1-c: 2 ', 1'-l] [1,4,7,10,13, 16] hexaazacyclohenicosin-23-yl] -2-hydroxyethyl} -2-hydroxyphenyl ester,
Hydrogen sulfate 5-[(2R) -2-{(2R, 6S, 9S, 11R, 14aS, 15S, 16S, 20S, 23S, 25aS) -20-[(1R) -3-Amino-1-hydroxy-3 -Oxopropyl] -9- {4 '-(5- {4-[(4,4-dimethylcyclohexyl) oxy] piperidin-1-yl} -1,2,4-oxadiazol-3-yl)- 3- [1- (2-hydroxyethyl) -1,2,3,6-tetrahydropyridin-4-yl] [1,1'-biphenyl] -4-carboxamide} -2,11,15-trihydroxy- 6-[(1R) -1-hydroxyethyl] -16-methyl-5,8,14,19,22,25-hexaoxotetracosahydro-1H-dipyrolo [2,1-c: 2 ', 1' -l] [1,4,7,10,13,16] hexaazacyclohenicosin-23-yl} -2-hydroxyethyl] -2-hydroxyphenyl ester and N-{(2R, 6S, 9S, 11R, 14aS, 15S, 16S, 20S, 23S, 25aS) -20-[(1R) -3-Amino-1-hydroxy-3-oxopropyl] -2,11,15-trihydroxy-6-[(1R ) -1-hydroxyethyl] -23-[(1R) -1-hydroxy-2- (4-hydroxy-3-methoxyphenyl) ethyl] -16-methyl-5,8,14,19,22,25- Hexa Xotetracosahydro-1H-dipyrolo [2,1-c: 2 ', 1'-l] [1,4,7,10,13,16] hexaazacyclohenicosin-9-yl} -4' -(5- {4- [2- (4-Ethoxyphenyl) ethyl] piperazin-1-yl} pyrimidin-2-yl) -3- (1,2,3,6-tetrahydropyridin-4-yl) [ A compound selected from the group consisting of 1,1′-biphenyl] -4-carboxamide or a pharmaceutically acceptable salt thereof.
- 請求項1に記載の化合物又はその塩、及び製薬学的に許容される賦形剤を含有する医薬組成物。 A pharmaceutical composition comprising the compound according to claim 1 or a salt thereof, and a pharmaceutically acceptable excipient.
- 真菌感染症の予防用若しくは治療用医薬組成物である請求項16に記載の医薬組成物。 The pharmaceutical composition according to claim 16, which is a pharmaceutical composition for preventing or treating fungal infection.
- 真菌感染症の予防若しくは治療用医薬組成物の製造のための請求項1に記載の化合物又はその塩の使用。 Use of the compound according to claim 1 or a salt thereof for the manufacture of a pharmaceutical composition for preventing or treating fungal infections.
- 真菌感染症の予防若しくは治療のための請求項1に記載の化合物又はその塩の使用。 Use of the compound according to claim 1 or a salt thereof for prevention or treatment of fungal infection.
- 真菌感染症の予防若しくは治療のための請求項1に記載の化合物又はその塩。 The compound according to claim 1 or a salt thereof for the prevention or treatment of fungal infection.
- 請求項1に記載の化合物又はその塩の有効量を対象に投与することからなる真菌感染症の予防若しくは治療方法。 A method for preventing or treating a fungal infection comprising administering an effective amount of the compound or salt thereof according to claim 1 to a subject.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011035450A JP2014088326A (en) | 2011-02-22 | 2011-02-22 | Polypeptide compound |
| JP2011-035450 | 2011-02-22 | ||
| PCT/JP2012/054172 WO2012115118A1 (en) | 2011-02-22 | 2012-02-21 | Polypeptide compound |
| JPPCT/JP2012/054172 | 2012-02-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012115159A1 true WO2012115159A1 (en) | 2012-08-30 |
Family
ID=46720894
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/054172 WO2012115118A1 (en) | 2011-02-22 | 2012-02-21 | Polypeptide compound |
| PCT/JP2012/054301 WO2012115159A1 (en) | 2011-02-22 | 2012-02-22 | Polypeptide compound |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/054172 WO2012115118A1 (en) | 2011-02-22 | 2012-02-21 | Polypeptide compound |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JP2014088326A (en) |
| TW (1) | TW201247215A (en) |
| WO (2) | WO2012115118A1 (en) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9353072B2 (en) * | 2012-10-31 | 2016-05-31 | Purdue Research Foundation | Antimicrobial substituted thiazoles and methods of use |
| EP2860177A3 (en) * | 2013-09-20 | 2015-06-10 | Bayer Intellectual Property GmbH | Synthesis of functionalized arenes |
| GB201318886D0 (en) * | 2013-10-25 | 2013-12-11 | Givaudan Sa | Improvements i or relating to organic compounds |
| US10562869B2 (en) | 2014-03-17 | 2020-02-18 | Remynd Nv | Oxadiazole compounds |
| CA3043066A1 (en) | 2018-05-14 | 2019-11-14 | Apotex Inc. | Processes for the preparation of ribociclib and intermediates thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06340693A (en) * | 1993-05-17 | 1994-12-13 | Fujisawa Pharmaceut Co Ltd | New polypeptide compound and its salt |
| WO2002068456A1 (en) * | 2001-02-26 | 2002-09-06 | Fujisawa Pharmaceutical Co., Ltd. | Echinocandin derivatives, pharmaceutical compositions containing same and use thereof as drugs |
| WO2003068807A2 (en) * | 2002-02-11 | 2003-08-21 | Fujisawa Pharmaceutical Co., Ltd. | Echinocandin cyclic peptide derivatives |
-
2011
- 2011-02-22 JP JP2011035450A patent/JP2014088326A/en not_active Withdrawn
-
2012
- 2012-02-21 WO PCT/JP2012/054172 patent/WO2012115118A1/en active Application Filing
- 2012-02-22 TW TW101105873A patent/TW201247215A/en unknown
- 2012-02-22 WO PCT/JP2012/054301 patent/WO2012115159A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06340693A (en) * | 1993-05-17 | 1994-12-13 | Fujisawa Pharmaceut Co Ltd | New polypeptide compound and its salt |
| WO2002068456A1 (en) * | 2001-02-26 | 2002-09-06 | Fujisawa Pharmaceutical Co., Ltd. | Echinocandin derivatives, pharmaceutical compositions containing same and use thereof as drugs |
| WO2003068807A2 (en) * | 2002-02-11 | 2003-08-21 | Fujisawa Pharmaceutical Co., Ltd. | Echinocandin cyclic peptide derivatives |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2014088326A (en) | 2014-05-15 |
| WO2012115118A1 (en) | 2012-08-30 |
| TW201247215A (en) | 2012-12-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2009273105B2 (en) | Azole compound | |
| TW201026681A (en) | New(3-oxo)pyridazin-4-ylurea derivatives | |
| KR101682638B1 (en) | Tetrahydroisoquinolin-1-one derivative or salt thereof | |
| WO2012115159A1 (en) | Polypeptide compound | |
| EP2141147A1 (en) | Ornithine derivative | |
| WO2012036278A1 (en) | Glycine transporter inhibitor | |
| JP2007523868A (en) | Substituted arylthiourea derivatives useful as inhibitors of viral replication | |
| KR20200062243A (en) | Griseofulvin compounds and pharmaceutical uses | |
| WO2023087611A1 (en) | Salt of ebastine, preparation method therefor and application thereof | |
| US9266840B2 (en) | Bicyclic heterocyclic compound | |
| TW470645B (en) | Compound for inhibiting or preventing HIV infection, its preparation and pharmaceutical composition comprising same | |
| MXPA03011441A (en) | Pyrrolidine oxadiazole- and thiadiazole derivatives. | |
| JP2022507117A (en) | A novel compound for the treatment of respiratory diseases | |
| RU2731618C2 (en) | Nonsteroidal modulators of glucocorticoid receptors for local drug delivery | |
| JPWO2009057568A1 (en) | Polypeptide compounds | |
| EP3150598B1 (en) | Substituted tropane derivatives | |
| JP2004083511A (en) | Acrylamide derivative | |
| AU2017293946A1 (en) | Indoline derivatives and method for using and producing the same | |
| JP2006511575A (en) | Cyclopentyl substituted glutaramide compounds as endopeptidase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12749705 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12749705 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |