WO2012044993A1 - Compounds, pharmaceutical compositions, and methods of treating or preventing neurodegenerative diseases or disorders - Google Patents
Compounds, pharmaceutical compositions, and methods of treating or preventing neurodegenerative diseases or disorders Download PDFInfo
- Publication number
- WO2012044993A1 WO2012044993A1 PCT/US2011/054325 US2011054325W WO2012044993A1 WO 2012044993 A1 WO2012044993 A1 WO 2012044993A1 US 2011054325 W US2011054325 W US 2011054325W WO 2012044993 A1 WO2012044993 A1 WO 2012044993A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- aryl
- alkyl
- heterocyclyl
- group
- halo
- Prior art date
Links
- IOVAEPYUPBLCKS-UHFFFAOYSA-N CN(C=NN1C)C1=S Chemical compound CN(C=NN1C)C1=S IOVAEPYUPBLCKS-UHFFFAOYSA-N 0.000 description 1
- AXVZFRBSCNEKPQ-UHFFFAOYSA-N CNCCc(cc1)ccc1O Chemical compound CNCCc(cc1)ccc1O AXVZFRBSCNEKPQ-UHFFFAOYSA-N 0.000 description 1
- DNBWGFKLIBQQSL-UHFFFAOYSA-N CNCc1ccncc1 Chemical compound CNCc1ccncc1 DNBWGFKLIBQQSL-UHFFFAOYSA-N 0.000 description 1
- OOTKJPZEEVPWCR-UHFFFAOYSA-N CNCc1ncccc1 Chemical compound CNCc1ncccc1 OOTKJPZEEVPWCR-UHFFFAOYSA-N 0.000 description 1
- VERCQLOBLOLFMW-UHFFFAOYSA-N CNc1ncncc1 Chemical compound CNc1ncncc1 VERCQLOBLOLFMW-UHFFFAOYSA-N 0.000 description 1
- OBGGYGVYXBIDBL-UHFFFAOYSA-N CS(c1cccc(C(c(c(Cl)ncn2)c2Cl)=O)c1)(=O)=O Chemical compound CS(c1cccc(C(c(c(Cl)ncn2)c2Cl)=O)c1)(=O)=O OBGGYGVYXBIDBL-UHFFFAOYSA-N 0.000 description 1
- XOHZHMUQBFJTNH-UHFFFAOYSA-N C[n]1nnnc1S Chemical compound C[n]1nnnc1S XOHZHMUQBFJTNH-UHFFFAOYSA-N 0.000 description 1
- QQQUBGHUBGEPBN-UHFFFAOYSA-N C[n]1nnnc1Sc1c(c(-c(cc2OC=C3)ccc2C3=O)c[s]2)c2ncn1 Chemical compound C[n]1nnnc1Sc1c(c(-c(cc2OC=C3)ccc2C3=O)c[s]2)c2ncn1 QQQUBGHUBGEPBN-UHFFFAOYSA-N 0.000 description 1
- FTLDAKDJOCMDTQ-UHFFFAOYSA-O C[n]1nnnc1Sc1c(c(-c(cccc2)c2[SH+](C)=O)c[s]2)c2ncn1 Chemical compound C[n]1nnnc1Sc1c(c(-c(cccc2)c2[SH+](C)=O)c[s]2)c2ncn1 FTLDAKDJOCMDTQ-UHFFFAOYSA-O 0.000 description 1
- WXSJGULLUWHLNF-UHFFFAOYSA-N C[n]1nnnc1Sc1c(c(-c2cc(cccc3)c3cc2)c[s]2)c2ncn1 Chemical compound C[n]1nnnc1Sc1c(c(-c2cc(cccc3)c3cc2)c[s]2)c2ncn1 WXSJGULLUWHLNF-UHFFFAOYSA-N 0.000 description 1
- PWOMEIYJUFGQQR-UHFFFAOYSA-N C[n]1nnnc1Sc1c(c(-c2cnc(N)nc2)c[s]2)c2ncn1 Chemical compound C[n]1nnnc1Sc1c(c(-c2cnc(N)nc2)c[s]2)c2ncn1 PWOMEIYJUFGQQR-UHFFFAOYSA-N 0.000 description 1
- YTHCPCCDAVEGMX-UHFFFAOYSA-N C[n]1nnnc1Sc1ncnc(Cl)c1C(c1cccc(S(C)(=O)=O)c1)=O Chemical compound C[n]1nnnc1Sc1ncnc(Cl)c1C(c1cccc(S(C)(=O)=O)c1)=O YTHCPCCDAVEGMX-UHFFFAOYSA-N 0.000 description 1
- OQVAOEIMSKZGAL-UHFFFAOYSA-N Cc([o]cc1)c1SC Chemical compound Cc([o]cc1)c1SC OQVAOEIMSKZGAL-UHFFFAOYSA-N 0.000 description 1
- XJZLQJXMQCOCFK-UHFFFAOYSA-N Cc1n[n](Cc2c(C)nc(NC)[s]2)c(NC)c1 Chemical compound Cc1n[n](Cc2c(C)nc(NC)[s]2)c(NC)c1 XJZLQJXMQCOCFK-UHFFFAOYSA-N 0.000 description 1
- VJRHHVGPZXCQSS-UHFFFAOYSA-N Cc1nnc(NC)[s]1 Chemical compound Cc1nnc(NC)[s]1 VJRHHVGPZXCQSS-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- Neurodegenerative diseases impose a heavy burden on the patient as well as on the patient's family.
- Huntington's disease affects 3-10 subjects in 100,000 individuals in Western Europe and North America. In most cases, the onset of the disease occurs in midlife, between the ages of 35 and 50 years. The disease progresses over time and is invariably fatal 15-20 years after the onset of the first symptoms. Initial symptoms of this disease include clumsiness, difficulties with smooth eye pursuit, and slight uncontrolled and awkward movements. These motor disturbances associated with the loss of voluntary movement coordination progress slowly and become more severe with time and the patients gradually lose their capacity to move and eventually communicate. Progression of the disease is accompanied by neuronal death in the striatum and, to a lesser extent, in the cerebral cortex. Presently, there is little treatment to prevent or palliate the progress of Huntington disease.
- Huntington's disease is neurodegenerative disorder due to a dominantly acting expansion of a CAG trinucleotide repeat in exon 1 of the Huntington ⁇ HTT) gene resulting in the production of an altered (mutant) protein Huntingtin having a long chain of
- polyglutamine tract (polyQ). Clinical and statistical analyses have shown that an increased number of polyQ repetition correlates with the probability of developing the disease, with 36 to 40 being the accepted cut off number for developing the disorder with high probability. It is known that polyQ repetitions impact the physical properties of the mutants producing protein aggregates that precipitate forming inclusion bodies, which are toxic to the neuronal cells.
- the invention provides a compound of formula (I):
- R 1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, heterocyclyl aryl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of heterocyclyl aryl, aryl heterocyclyl, and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R 1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono Q-Q alkyl, carboxy Ci-C 6 alkyl, dicarboxy Q- alkyl, dicarboxy halo Ci-C
- CR 3 N(OR 4 ), N 3 , NR 3 R 4 , N(OH)R 3 , C(0)R 3 , C(S)R 3 , C0 2 R 3 , C(0)SR 3 , C(0)NR 3 R 4 , C(S)NR 3 R 4 , C(0)N(OH)R 3 , C(S)N(OH)R 3 , NR 3 C(0)R 4 , NR 3 C(S)R 4 , N(OH)C(0)R 3 , N(OH)C(S)R 3 , NR 3 C0 2 R 4 , N(OH)C0 2 R 3 , NR 3 C(0)SR 4 , NR 3 C(0)NR 4 R 5 , NR 3 C(S)NR 4 R 5 , N(OH)C(0)NR 3 R 4 , N(OH)C(S)NR 3 R 4 , NR 3 R 4 , N + R 3 R 4 R 5 , NR 3 C(0)N(OH)R 4 ,
- R 2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C 6 alkyl, carboxy CpC alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo Ci-C 6 alkyl, sulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C 6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
- R 3 -R 5 are independently hydrogen, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 3 -C 4 cycloalkyl, C 3 -C 4 cycloalkenyl, C6-C 2 o aryl C -C alkyl, C 6 -C 2 o aryl, heterocyclyl, or heterocyclyl alkyl;
- X is a bond, S, O, or NH
- Y is S or NH
- Z is CH or N
- the invention also provides pharmaceutical compositions comprising such compounds of formula (I) or pharmaceutically acceptable salts thereof, and methods of treating or preventing neurodegenerative disorders or diseases, for example, polyglutamine diseases such as Huntington's disease.
- the invention also provides methods for inhibiting the PIP5K2C kinase, for modulating the activity of a mutant Huntingtin protein, for treating or preventing a neurodegenerative disease, and/or for inhibiting the activation of caspase 9 in a cell, for example, in a patient.
- Figure 1 depicts a reaction scheme to prepare certain compounds in accordance with an embodiment of the invention.
- Figure 2 depicts another reaction scheme to prepare compounds in accordance with an embodiment of the invention.
- Figure 3 depicts the number of viable cells in FBS and serum free medium (SFM) for wild type and mutant striatal cells, showing that the mutant cells do not survive the stress of the serum free medium. Shown is cell growth of wild type and Huntington mutant STHdh Q1 1 1/n i striatal cells upon serum deprivation. ATP levels were quantitated using ATPlite, a luciferase-based ATP quantitation assay, after 24 h incubation. Data are represented in RLU counts. Serum deprivation has a greater effect on STHdh ⁇ 111711 1 striatal cells (24% of FBS activity after 24 h) than STHdh Q7/7 cells (68% of FBS activity after 24 h).
- Figure 4 shows that compound 49 in accordance with an embodiment of the invention protects against hydrogen peroxide induced cell death of striatal cells.
- Figure 5 shows that compound 49 in accordance with an embodiment of the invention protects against beta-amyloid induced cell death of striatal cells.
- Figure 6A depicts Western blots of the control showing caspase 9 activation and the downstream production of caspase 3 during mutant cell apoptosis.
- Caspase 9 ans caspase 3 activation after 16 h treatment under normal growth conditions (DMEM + 10% FBS).
- Figure 6B depicts Western blots showing that caspase 9 activation and production of caspase 3 are inhibited by compound 14 (F7) in serum free medium and that the compound inhibits cell apoptosis in accordance with an embodiment of the invention.
- Caspase 9 ans caspase 3 activation after 16 h treatment under serum deprived conditions.
- CT DMSO (vehicle) treatment.
- a and M are two other compounds that affect the Huntington cellular phenotype whose identies are not disclosed by the assay provider.
- the arrows indicate the specific activity of compound 14 under serum deprived conditions.
- Figure 7 shows that PCI 2 cells express PIK5K2C kinase.
- the present invention provides compounds, compositions, and methods for treating or preventing neurodegenerative disorders.
- Embodiments of the invention are predicated on the concept of inhibiting posttranscriptional modifications of the mutant genes responsible for such disorders.
- the invention provides a compound of formula (I):
- R 1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, heterocyclyl aryl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of heterocyclyl aryl, aryl heterocyclyl, and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R 1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono Q-Q alkyl, carboxy Ci-C 6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo Ci
- CR 3 N(OR 4 ), N 3 , NR 3 R 4 , N(OH)R 3 , C(0)R 3 , C(S)R 3 , C0 2 R 3 , C(0)SR 3 , C(0)NR 3 R 4 , C(S)NR 3 R 4 , C(0)N(OH)R 3 , C(S)N(OH)R 3 , NR 3 C(0)R 4 , NR 3 C(S)R 4 , N(OH)C(0)R 3 , N(OH)C(S)R 3 , NR 3 C0 2 R 4 , N(OH)C0 2 R 3 , NR 3 C(0)SR 4 , NR 3 C(0)NR 4 R 5 , NR 3 C(S)NR 4 R 5 , N(OH)C(0)NR 3 R 4 , N(OH)C(S)NR 3 R 4 , NR 3 R 4 , N + R 3 R R 5 , NR 3 C(0)N(OH)R 4 ,
- R is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C 6 alkyl, carboxy Ci-C 6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo C]-C 6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureid
- R 3 -R 5 are independently hydrogen, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C3-C4 cycloalkyl, C3-C4 cycloalkenyl, C 6 -C 2 o aryl Ci-C 6 alkyl, C 6 -C 20 aryl, heterocyclyl, or heterocyclyl alkyl;
- X is a bond, S, O, or NH
- Y is S or NH
- Z is CH or N
- R 1 is tetrazolyl or a substituted tetrazolyl, then R is not halo, heterocyclyl, heterocyclyl aryl, aryl heterocyclyl, aryl, substituted aryl, substituted heterocyclyl, substituted heterocyclyl aryl, or substituted aryl heterocyclyl.
- X is a bond, S or NH.
- R 1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R 1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloalkyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono Cj- C 6 alkyl, carboxy Ci-C 6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo Ci-C 6 alkyl,
- R 1 is alkyl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R 1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloalkyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino,
- R 1 is alkyl, aryl, arylalkyl, heterocyclyl, or heterocyclylalkyl, optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloalkyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR 3 , S(0)R 3 , and S0 2 R 3 .
- R 1 is selected from the group consisting of alkyl, aryl, arylalkyl, diazolyl, thiazolyl, triazolyl, thiadiazolyl, oxadiazolyl, pyridyl, pyridylmethyl, pyridazinyl, furanyl, benzodiazolyl, pyridinotriazolyl, and benzothiazolyl, each of which is optionally substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloalkyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamin
- R 1 is selected from the group consisting of alkyl, aryl, arylalkyl, diazolyl, thiazolyl, triazolyl, thiadiazolyl, oxadiazolyl, pyridyl, pyridylmethyl, pyridazinyl, furanyl, benzodiazolyl, pyridinotriazolyl, benzothiazolyl, each of which is optionally substituted with a substituent selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloalkyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dial
- R 1 is selected from the group consisting of alkyl, phenyl, phenylalkyl, diazolyl, thiazolyl, triazolyl, thiadiazolyl, oxadiazolyl, pyridyl, pyridylmethyl, pyridazinyl, furanyl, benzodiazolyl, pyridinotriazolyl, and benzothiazolyl, each of which is optionally substituted with an alkyl, hydroxy, or amino substituent.
- R 1 is selected from the group consisting of alkyl, phenyl, phenylalkyl, diazolyl, thiazolyl, triazolyl, thiadiazolyl, oxadiazolyl, pyridyl, pyridylmethyl, pyridazinyl, furanyl, benzodiazolyl, pyridinotriazolyl, and benzothiazolyl, each of which is optionally substituted with an alkyl, or amino substituent.
- X-R 1 of the compound or salt of the invention is selected from the group consisting of:
- Y is S.
- Z is CH.
- X is S.
- R is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono CpC 6 alkyl, carboxy Ci-C 6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo Ci-C 6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino
- each of said R 2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Cj-C 6 alkyl, carboxy Ci-C 6 alkyl, dicarboxy Q-Qs alkyl, dicarboxy halo Ci-C 6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Cj-C 6 alkyl, alkylthio, acyl, acyloxy,
- R 2 is selected from the group consisting of halo, aryl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, and S0 2 R 3 ; wherein each of said R 2 groups aryl, aryl heterocyclyl, and aryl heterocyclyl aryl are unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono d-C 6 alkyl, carboxy Ci-C 6 alkyl, dicarboxy C]-C 6 alkyl, dicarboxy halo Cj-C 6 alkyl, sulfonyl, alkylsulfon
- R 2 is selected from the group consisting of phenyl, pyrimidinyl, thiophenyl, pyrrolyl, benzofuranyl, naphthyl, methylenedioxyphenyl, 4-oxo-4H- chromenyl, dibenzothiophenyl, bromo, and iodo, optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C 6 alkyl, carboxy Ci-C 6 alkyl, dicarboxy Cj-C 6 alkyl, dicarboxy halo Ci-C 6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro d-C 6 alkyl, alkylthio, acyl, acyloxy, thioacyl,
- the invention also provides the following
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound or salt as described above and a pharmaceutically acceptable carrier.
- Phosphoinositide is a collective term for phosphatidylinositol (Ptdlns) and its phosphorylated derivatives. Ptdlns contains a 1 o-myo-inositol phosphate group linked to diacylglycerol. Pis regulate many biological processes, including cell proliferation, cell survival, differentiation, signal transduction, cytoskeleton organization and membrane trafficking. The activities of specific Pis are regulated by controlling their levels in the cell, which is achieved by intricate networks of proteins that control their synthesis, transport and degradation.
- PI synthesis is regulated by PI kinases and phosphatidylinositol phosphate (PIP) kinases (PIPKs), and PI degradation is governed by lipid phosphatases, while their transport is mediated by Ptdlns transfer proteins (PITPs).
- PIP phosphatidylinositol phosphate
- PITPs Ptdlns transfer proteins
- the hydroxy groups at positions 3, 4 and 5 on the inositol ring can be phophorylated by the lipid kinases: Ptdlns 3-kinases (PI3Ks), Ptdlns 4-kinases (PI4Ks), Ptdlns 5-kinases (PI5Ks) and PIPKs.
- PIP phosphatidylinositol phosphate
- PIPKs phosphatidylinositol phosphate
- category there are known two classes: the 68 kDa ⁇ 5 ⁇ ( ⁇ , ⁇ and ⁇ ) that uses PtdIns4P to generate PtdIns(4,5)P 2 and the 53 kDa ⁇ 4 ⁇ ( , ⁇ and ⁇ ) that phosphorylates PtdIns3P and PtdIns5P to produce PtdIns(3,4)P 2 and PtdIns(4,5)P 2 .
- the PIPK isoform ⁇ 4 ⁇ (also known as PIP5K2C) has a restricted CNS expression profile, which is limited to neurons, particularly the cerebellar Purkinje cells, pyramidal cells of the hippocampus, large neuronal cell types in the cerebral cortex including pyramidal cells, and mitral cells in the olfactory bulb and is not expressed in cerebellar, hippocampal formation, or olfactory bulb granule cells.
- ⁇ 4 ⁇ has a vesicular distribution and shows partial colocalization with markers of cellular compartments of the endomembrane trafficking pathway.
- the ⁇ 4 ⁇ isoform expression is established after day 7 of postnatal development. It is believed that ⁇ 4 ⁇ plays a role in neuron function, specifically in the regulation of vesicular transport, in specific regions of the developed brain.
- D-m o-Inositol 1 ,4,5-trisphosphate is a cellular second messenger.
- Phospholipase C catalyses hydrolysis of phosphatidylinositol 4,5-bisphosphate (PtdIns-4,5- P2) to Ins(l,4,5)P3 and diacyl glycerol.
- Ins(l,4,5)P3 interacts specifically with a tetrameric receptor-operated Ca 2+ channel on the endoplasmic reticulum to mobilize Ca 2+ stores in stimulated cells.
- Ins(l ,4,5)P3 mediates the agonist-induced response via this rise in intracellular Ca 2+ concentration.
- Phosphatidylinositol 4,5-bisphosphate (PtdIns-4,5-P2) is not only a precursor of Ins(l ,4,5)P3 but it has been shown that the inositol 1,4,5-trisphosphate receptor (InsP3R) forms a stable inhibitory complex with endogenous PtdIns-4,5-P2.
- InsP3R inositol 1,4,5-trisphosphate receptor
- the levels of calcium in mitochondria are low under normal physiologic conditions; however, elevations in mitochondria calcium occur when intracellular concentrations rise during and after prolonged activation of calcium conductance. Prolonged increases in intracellular calcium result in a plethora of harmful effects to the cell, such as excitotoxic events, activation of calcium-dependent enzymes, apoptosis and mitochondrial failure. Imbalances in calcium homeostasis and regulation have been implicated in the pathogenesis of many neurodegenerative diseases, including
- InsP3R InsP3R
- PtdIns-4,5-P2 in unstimulated cells, some fraction of InsP3R is constitutively inhibited due to interaction with PtdIns-4,5-P2 in the juxtaposed membrane (resting state).
- These InsP3R are unable to open because of the spatial constraint imposed by the topology of InsP3R-PIP2 interaction.
- Agonist stimulation leads to activation of PLC, cleavage of InsP3R-tethered PtdIns-4,5-P2, and release of the spatial clamp on the InsP3R (signal transduction step).
- PLC simultaneously removes the inhibitor (PtdIns-4,5-P2) and generates the activator (InsP3) of the InsP3R, leading to Ca 2+ wave initiation.
- This compartmentalized signaling mechanism is responsible for Ca 2+ wave initiation in specialized trigger zones; whereas Ca 2+ wave propagation through the cell is sustained by direct Ca 2+ feedback on the InsP3R.
- Preferential coupling between PLC linked hormonal receptors and InsP3R have been demonstrated previously in some intact cell preparations. It appears that integrity of this
- poly-Q HTT association with InsP3Rl causes sensitization of InsP3Rl to activation by InsP3 in planar lipid bilayers and in primary striatal neurons.
- Poly- Q HTT activates Ca 2+ -permeable NR2B-containing NMDA receptors.
- a cytosolic C-terminal tail of InsP(3)Rl has neuroprotective effects in a Huntington's disease model.
- ⁇ 4 ⁇ has a specific neuronal and vesicular distribution and that the PIP5K2C gene has high levels of expression in the hippocampal formation, primarily in the stratum pyramidale and extending into the stratum radiatum of CA1-CA3, and were excluded from the dentate gyrus.
- Huntington's patients show a neuronal reduction in the CA1 area of the Hippocampus and Huntington's mice show a reduced hippocampal neurogenesis.
- the invention provides a method of inhibiting the PIP5K2C kinase in a cell in need thereof comprising administering to the cell an effective amount of a compound of formula (I):
- R 1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, heterocyclyl aryl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl, heterocyclyl aryl, and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R 1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono Cj-C 6 alkyl, carboxy Q-C6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy Ci-C
- CR 3 N(OR 4 ), N 3 , NR 3 R 4 , N(OH)R 3 , C(0)R 3 , C(S)R 3 , C0 2 R 3 , C(0)SR 3 , C(0)NR 3 R 4 , C(S)NR 3 R 4 , C(0)N(OH)R 3 , C(S)N(OH)R 3 , NR 3 C(0)R 4 , NR 3 C(S)R 4 , N(OH)C(0)R 3 , N(OH)C(S)R 3 , NR 3 C0 2 R 4 , N(OH)C0 2 R 3 , NR 3 C(0)SR 4 , NR 3 C(0)NR 4 R 5 , NR 3 C(S)NR 4 R 5 , N(OH)C(0)NR 3 R 4 , N(OH)C(S)NR 3 R 4 , NR 3 R 4 , N + R 3 R 4 R 5 , NR 3 C(0)N(OH)R 4 ,
- R 2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C 6 alkyl, carboxy Ci-C 6 alkyl, dicarboxy Cj-C 6 alkyl, dicarboxy halo Ci-C 6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, urei
- R 3 -R 5 are independently hydrogen, Q-C6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 3 -C 4 cycloalkyl, C 3 -C 4 cycloalkenyl, C6-C 2 o aryl C)-C 6 alkyl, C6-C 20 aryl, heterocyclyl, or heterocyclyl alkyl;
- X is a bond, S, O, or NH
- Y is S or NH
- Z is CH or N
- X is S
- Y is NH
- Z is N
- R 1 is methyl tetrazolyl
- R 2 is phenyl, optionally substituted with methylsulfonyl.
- the invention further provides a method of treating and/or preventing a neurodegenerative disease or disorder in an animal comprising administering to the animal an effective amount of a compound of formula (I):
- R 1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, heterocyclyl aryl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl, heterocyclyl aryl, and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R 1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono Ci-C 6 alkyl, carboxy Ci-C 6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo
- CR 3 N(OR 4 ), N 3 , NR 3 R 4 , N(OH)R 3 , C(0)R 3 , C(S)R 3 , C0 2 R 3 , C(0)SR 3 , C(0)NR 3 R 4 , C(S)NR 3 R 4 , C(0)N(OH)R 3 , C(S)N(OH)R 3 , NR 3 C(0)R 4 , NR 3 C(S)R 4 , N(OH)C(0)R 3 , N(OH)C(S)R 3 , NR 3 C0 2 R 4 , N(OH)C0 2 R 3 , NR 3 C(0)SR 4 , NR 3 C(0)NR 4 R 5 , NR 3 C(S)NR 4 R 5 , N(OH)C(0)NR 3 R 4 , N(OH)C(S)NR 3 R 4 , NR 3 R 4 , N + R 3 R 4 R 5 , NR 3 C(0)N(OH)R 4 ,
- R is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C 6 alkyl, carboxy Ci-C 6 alkyl, dicarboxy C]-C 6 alkyl, dicarboxy halo Ci-C 6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
- each of said R groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C 6 alkyl, carboxy C]-C 6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo Ci-C 6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C 6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
- R -R are independently hydrogen, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 3 -C cycloalkyl, C 3 -C 4 cycloalkenyl, C 6 -C 2 o aryl Ci-C 6 alkyl, C 6 -C 2 o aryl, heterocyclyl, or heterocyclyl alkyl;
- X is a bond, S, O, or NH
- Y is S or NH
- Z is CH or N
- neurodegenerative diseases or disorders include brain trauma, spinal cord trauma, trauma to the peripheral nervous system, Alzheimer's disease, Pick's disease, diffuse Lewy body disease, progressive supranuclear palsy (Steel-Richardson syndrome), multisystem degeneration (Shy-Drager syndrome), motor neuron diseases including amyotrophic lateral sclerosis, degenerative ataxias, cortical basal degeneration, ALS- Parkinson's-Dementia complex of Guam, subacute sclerosing panencephalitis, Huntington's disease, Parkinson's disease, synucleinopathies, primary progressive aphasia, striatonigral degeneration, Machado-Joseph disease/spinocerebellar ataxia type 3 and olivopontocerebellar degenerations, Gilles De La Tourette's disease, bulbar and pseudobulbar palsy, spinal and spinobulbar muscular atrophy (Kennedy's disease), primary lateral sclerosis, familial-Richard
- the neurodegenerative disease or disorder is selected from the group consisting of Dentatorubropallidoluysian atrophy, Huntington's disease, Spinobulbar muscular atrophy, Spinocerebellar ataxia Type 1 , Spinocerebellar ataxia Type 2,
- Spinocerebellar ataxia Type 3 Spinocerebellar ataxia Type 6, Spinocerebellar ataxia Type 7, Spinocerebellar ataxia Type 17, Cockayne Syndrome, hepatolenticular degeneration, Lafora Disease, Menkes Kinky Hair Syndrome, neurofibromatosis, Tourette Syndrome, Tuberous Sclerosis Amyotrophic Lateral Sclerosis, muscular atrophy, poliomyelitis, Parkinson's Disease, Prion diseases, Creutzfeldt-Jacob Syndrome, Kuru, Scrapie, and Alzheimer's Disease.
- the neurodegenerative disease or disorder is a polyglutamine disease, for example, spinal and bulbar muscular atrophy, Huntington's disease,
- Dentatorubropallidoluysian atrophy Spinocerebellar ataxia 1, 2, 6, 7, and 17, SCA 3/MJD.
- the invention further provides a method of modulating the activity of a mutant Huntingtin protein in a cell comprising administering to the cell an effective amount of a compound of formula (I):
- R 1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, heterocyclyl aryl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl, heterocyclyl aryl, and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R 1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono Ci-C 6 alkyl, carboxy C]-C 6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy Ci-C
- CR 3 N(OR 4 ), N 3 , NR 3 R 4 , N(OH)R 3 , C(0)R 3 , C(S)R 3 , C0 2 R 3 , C(0)SR 3 , C(0)NR 3 R 4 , C(S)NR 3 R 4 , C(0)N(OH)R 3 , C(S)N(OH)R 3 , NR 3 C(0)R 4 , NR 3 C(S)R 4 , N(OH)C(0)R 3 , N(OH)C(S)R 3 , NR 3 C0 2 R 4 , N(OH)C0 2 R 3 , NR C(0)SR 4 , NR 3 C(0)NR 4 R 5 , NR 3 C(S)NR 4 R 5 , N(OH)C(0)NR 3 R 4 , N(OH)C(S)NR 3 R 4 , NR 3 R 4 , N + R 3 R 4 R 5 , NR 3 C(0)N(OH)R 4 ,
- R 2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C 6 alkyl, carboxy Cj-C 6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo Ci-C 6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, urei
- each of said R 2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono C
- R 3 -R 5 are independently hydrogen, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C alkynyl, C 3 -C 4 cycloalkyl, C 3 -C 4 cycloalkenyl, C 6 -C 20 aryl Ci-C 6 alkyl, C 6 -C 2 o aryl, heterocyclyl, or heterocyclyl alkyl;
- X is a bond, S, O, or NH
- Y is S or NH
- Z is CH or N
- Compounds in accordance with an embodiment inhibit the activation of caspase 9. Accordingly, the compounds of the invention inhibit the activation of apoptosis of neuronal cells. Again, without wishing to be bound by theory or mechanism, it is believed there are three main mechanisms for apoptotic activation. The first one involves the mitochondria released of the Apoptosis-inducing factor (AIF) and its translocation to the nucleus to trigger DNA destruction. This pathway is caspase-independent. The second mechanism is triggered by external activators such us TNF-a, Lymphotoxin or the Fas Ligand and involves the activation of caspase 8.
- AIF Apoptosis-inducing factor
- the third mechanism depends on intrinsic signals and is commonly activated in case of cellular damage (from radical species, toxic chemicals, ionization, etc.).
- This pathway involves Bax translocation to the mitochondrion, Bcl-2 inhibition causing cytochrome C leak out, binding of cytochrome C with Apaf-1 (apoptotic protease activating factor- 1) and aggregation to foma apoptosomes, cleavage of pro-caspase 9 by the apoptosome to form active caspase 9, activation of caspase 3 and caspase 7 by caspase 9, and caspase 3-7 digestion of cytoplasmic structural proteins, degradation of nuclear DNA and cell
- Compounds in accordance with an embodiment of the invention block the activation of the intrinsic apoptotic pathway, or prevent the production of caspase 9 and its downstream target caspase 3.
- Activation of the apoptotic intrinsic pathway is found in numerous diseases Huntington disease, ALS, Niemann-Pick disease type C, Lysosomal Cell Death, Ischemia, viral and bacteria infection, and ceramide induced neuronal death.
- the present invention provides a method of inhibiting the activation of caspase 9 in a cell in need thereof comprising administering an effective amount of a compound of formula (I):
- R 1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, heterocyclyl aryl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl, heterocyclyl aryl, and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R 1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono -Q alkyl, carboxy Ci-C 6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo Ci
- CR 3 N(OR 4 ), N 3 , NR 3 R 4 , N(OH)R 3 , C(0)R 3 , C(S)R 3 , C0 2 R 3 , C(0)SR 3 , C(0)NR 3 R 4 , C(S)NR 3 R 4 , C(0)N(OH)R 3 , C(S)N(OH)R 3 , NR 3 C(0)R 4 , NR 3 C(S)R 4 , N(OH)C(0)R 3 , N(OH)C(S)R 3 , NR 3 C0 2 R 4 , N(OH)C0 2 R 3 , NR 3 C(0)SR 4 , NR 3 C(0)NR 4 R 5 , NR 3 C(S)NR 4 R 5 , N(OH)C(0)NR 3 R 4 , N(OH)C(S)NR 3 R 4 , NR 3 R 4 , N + R 3 R 4 R 5 , NR 3 C(0)N(OH)R 4 ,
- R 2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C 6 alkyl, carboxy Ci-C 6 alkyl, dicarboxy Cj-C 6 alkyl, dicarboxy halo C]-C 6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, 3 3 3 aldehydo
- R 3 -R 5 are independently hydrogen, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 3 -C 4 cycloalkyl, C 3 -C 4 cycloalkenyl, C 6 -C 20 aryl Q- alkyl, C 6 -C 2 o aryl, heterocyclyl, or heterocyclyl alkyl;
- X is a bond, S, O, or NH
- Y is S or NH
- Z is CH or N
- X is a bond, S or NH.
- R 1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R 1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono CpC 6 alkyl, carboxy C]-C 6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo
- R 1 is alkyl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R 1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino,
- R 1 is alkyl, aryl, arylalkyl, heterocyclyl, or heterocyclylalkyl, optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR 3 , S(0)R 3 , and S0 2 R 3 .
- R 1 is selected from the group consisting of alkyl, aryl, arylalkyl, pyrazolyl, thiazolyl, triazolyl, tetrazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyridinylmethyl, pyrimidinyl, furanyl, triazolopyridinyl, benzimidazolyl, and benzothiazolyl, each of which is optionally substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryl
- R 1 is selected from the group consisting of alkyl, aryl, arylalkyl, pyrazolyl, thiazolyl, triazolyl, tetrazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyridinylmethyl, pyrimidinyl, furanyl, triazolopyridinyl, benzimidazolyl, and benzothiazolyl, each of which is optionally substituted with a substituent selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy,
- R 1 is selected from the group consisting of alkyl, aryl, arylalkyl, pyrazolyl, thiazolyl, triazolyl, tetrazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyridinylmethyl, pyrimidinyl, furanyl, triazolopyridinyl, benzimidazolyl, and benzothiazolyl, each of which is optionally substituted with an alkyl, hydroxy, or amino substituent.
- -X-R 1 is selected from the group consisting of:
- Y is S.
- Z is CH.
- X is S.
- R is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Q-Q alkyl, carboxy Ci-C 6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo Q-C 6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino
- each of said R 2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Cj-C alkyl, carboxy Cj-C 6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo Ci-C 6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C 6 alkyl, alkylthio, acyl, acyloxy,
- R is selected from the group consisting of aryl, heterocyclyl, aryl heterocyclyl, or aryl
- heterocyclyl aryl wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl
- aryl are fused or unfused to each other, halo, and S0 2 R ; wherein each of said R groups aryl, aryl heterocyclyl, and aryl heterocyclyl aryl are unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C 6 alkyl, carboxy Ci-C 6 alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo Ci-C 6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureid
- R 2 is selected from the group consisting of phenyl, pyrimidinyl, thiophenyl, pyrrolyl, benzofuranyl, naphthyl, 4-oxo-4H-chromenyl, dibenzothiophenyl, bromo, and iodo, each of which other than bromo and iodo, is optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C 6 alkyl, carboxy Q-Q alkyl, dicarboxy Ci-C 6 alkyl, dicarboxy halo Ci-C 6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl,
- R is selected from the group consisting of:
- R 2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl
- heterocyclyl or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Q-Q alkyl, carboxy Ci-C 6 alkyl, dicarboxy C]-C 6 alkyl, dicarboxy halo Ci-C 6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR 3 , S(0)R 3 , S0 2 R
- the cell is in an animal.
- the animal is a human.
- the cell is in an animal.
- the term "animal” refers to any member of the animal kingdom. In embodiments, “animal” refers to a human at any stage of development. In embodiments, “animal” includes mammals, birds, reptiles, amphibians, fish, and worms.
- the non-human animal is a mammal, e.g., a rodent, a mouse, a rat, a rabbit, a monkey, a dog, a cat, a sheep, cattle, a primate, or a pig.
- the animal may also be a transgenic animal, genetically engineered animal, or a clone.
- the subject is a mammal.
- mammals include, but are not limited to, the order Rodentia, such as mice, and the order Logomorpha, such as rabbits. It is preferred that the mammals are from the order Carnivora, including Felines (cats) and Canines (dogs). It is more preferred that the mammals are from the order Artiodactyla, including Bovines (cows) and Swines (pigs) or of the order
- Perssodactyla including Equines (horses). It is most preferred that the mammals are of the order Primates, Ceboids, or Simioids (monkeys) or of the order Anthropoids (humans and apes). An especially preferred mammal is the human. Furthermore, the subject can be the unborn offspring of any of the forgoing hosts, especially mammals (e.g., humans), in which case any screening of the subject or cells of the subject, or administration of compounds to the subject or cells of the subject, can be performed in vitro. In accordance with an embodiment, the animal is a human.
- alkyl means a straight-chain or branched alkyl substituent containing from, for example, 1 to about 12 carbon atoms, from 1 to about 6 carbon atoms, from 1 to 4 carbon atoms, or from 1 to 3 carbon atoms.
- substituents include methyl, ethyl, propyl, isopropyl, n- butyl, sec-butyl, isobutyl, fcri-butyl, pentyl, isoamyl, hexyl, and the like.
- alkylene means a straight-chain or branched alkyl substituent containing from, for example, 1 to about 12 carbon atoms, from 1 to about 6 carbon atoms, from 1 to 3 carbon atoms, and is optionally connected to one, two or more substituents at two or more different positions on the alkylene group.
- alkenyl means a linear alkenyl substituent containing at least one carbon-carbon double bond and from, for example, about 2 to about 12 carbon atoms (branched alkenyls are about 3 to about 12 carbons atoms), from about 2 to about 6 carbon atoms (branched alkenyls are preferably from about 3 to about 7 carbon atoms), or from about 3 to about 4 carbon atoms.
- substituents include vinyl, propenyl, isopropenyl, n-butenyl, sec-butenyl, isobutenyl, tert-butenyl, pentenyl, isopentenyl, hexenyl, and the like.
- alkenylene means a straight-chain or branched alkenyl substituent containing from, for example, 2 to about 12 carbon atoms, 2 to 6 carbon atoms, or from 2 to about 4 carbon atoms, and is connected to one, two or more substituents at two or more different positions on the alkenylene group.
- alkynyl means a linear alkynyl substituent containing at least one carbon-carbon triple bond and from, for example, 2 to about 12 carbon atoms (branched alkynyls are about 3 to about 13 carbons atoms), from 2 to about 6 carbon atoms (branched alkynyls are preferably from about 3 to about 7 carbon atoms), or from about 2 to about 4 carbon atoms.
- substituents include ethynyl, propynyl, isopropynyl, rc-butynyl, sec-butynyl, isobutynyl, ierZ-butynyl, pentynyl, isopentynyl, hexynyl, and the like.
- alkynylene means a straight-chain or branched alkynyl substituent containing from, for example, 2 to about 12 carbon atoms, from 2 to about 6 carbon atoms, or from 2 to about 4 carbon atoms, and is connected to two or more substituents at two or more different positions on the alkynylene group.
- cycloalkyl means a cyclic alkyl substituent containing from, for example, about 3 to about 8 carbon atoms, from about 4 to about 7 carbon atoms, or from about 4 to about 6 carbon atoms.
- substituents include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, and the like.
- cycloalkenyl as used herein, means the same as the term “cycloalkyl,” however one or more double bonds are present. Examples of such substituents include cyclopentenyl and cyclohexenyl.
- the cyclic alkyl groups may be unsubstituted or further substituted with alky] groups such as methyl groups, ethyl groups, and the like.
- aryl refers to an unsubstituted or substituted aromatic carbocyclic moiety, as commonly understood in the art, and includes monocyclic and polycyclic aromatics such as, for example, phenyl, biphenyl, naphthyl, anthracenyl, pyrenyl, and the like.
- heteroaryl refers to an aromatic group containing one or more heteroatoms selected from the group consisting of O, N, P, and S and combinations thereof.
- the heteroaryl group can be 4 to 7 membered ring.
- Non-limiting examples of heteroaryl groups include furanyl, thiopheneyl, pyrrolyl, pyrazolyl, imidazolyl, 1 ,2,3-triazolyl, 1 ,2,4- triazolyl, isoxazolyl, oxazolyl, isothiazolyl, thiazolyl, pyridinyl, pyrimidinyl, pyrazinyl, triazinyl, benzofuranyl, benzothiopheneyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl, benzoxazolinyl, benzothiazolinyl, and quinazolinyl.
- heterocyclyl refers to a monocyclic or bicyclic 5-, 6-, or 7-membered ring system containing one or more heteroatoms selected from the group consisting of O, N, P, S, and combinations thereof.
- the heterocyclyl group can be any suitable heterocyclyl group and can be an aliphatic heterocyclyl group, an aromatic heterocyclyl group, or a combination thereof.
- the heterocyclyl group can be a monocyclic heterocyclyl group or a bicyclic heterocyclyl group.
- Bicyclic heterocyclyl groups include monocylic heterocyclyl rings fused to a C 6 -Cio aryl ring.
- both ring systems can be aliphatic or aromatic, or one ring system can be aromatic and the other ring system can be aliphatic as in, for example, dihydrobenzofuran.
- the heterocyclyl group is an aromatic heterocyclyl or heteroaryl group.
- the heterocyclyl or heteroaryl group is optionally substituted with 1 , 2, 3, 4, or 5 substituents as recited herein, wherein the optional substituent can be present at any open position on the heterocyclyl or heteroaryl group.
- any chemical group e.g., alkyl, alkylamino, etc.
- any chemical group e.g., alkyl, alkylamino, etc.
- any sub-range thereof e.g., 1 -2 carbon atoms, 1-3 carbon atoms, 1-4 carbon atoms, 1-5 carbon atoms, 1-6 carbon atoms, 1-7 carbon atoms, 1-8 carbon atoms, 1-9 carbon atoms, 1-10 carbon atoms, 1-1 1 carbon atoms,
- 6-10 carbon atoms e.g., C 6 -Cio
- any chemical group e.g., aryl
- 6-10 carbon atoms 6-9 carbon atoms, 6-8 carbon atoms, 6-7 carbon atoms, 7-10 carbon atoms, 7-9 carbon atoms, 7-8 carbon atoms, 8-10 carbon atoms, and/or 8-9 carbon atoms, etc., as appropriate).
- Any of the amino acid fragments can be in the natural form or synthetic form, i.e., D, L, or D/L form.
- the compounds of the invention in any of the embodiments can be prepared by the reaction schemes illustrated in Schemes 1 -3.
- the phrase "pharmaceutically acceptable salt” is intended to include nontoxic salts synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two. Generally, nonaqueous media such as ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred. Lists of suitable salts are found in Remington 's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA, 1990, p. 1445, and Journal of Pharmaceutical Science, 66, 2-19 (1977).
- Suitable bases include inorganic bases such as alkali and alkaline earth metal bases, e.g., those containing metallic cations such as sodium, potassium, magnesium, calcium and the like.
- suitable bases include sodium hydroxide, potassium hydroxide, sodium carbonate, and potassium carbonate.
- Suitable acids include inorganic acids such as hydrochloric acid, hydrobromic acid, hydriodic acid, sulfuric acid, phosphoric acid, and the like, and organic acids such as p-toluenesulfonic, methanesulfonic acid, ethane sulfonic acid, benzenesulfonic acid, oxalic acid, p-bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic acid, acetic acid, propionic acid, glycolic acid, glucaric acid, glucuronic acid, citric acid, gluconic acid, hydroxymaleic acid, fumaric acid, maleic acid, malic acid, 4-aminosalicylic acid, cinnamic acid, acetoxybenzoic acid, succinic acid, tartaric acid, ascorbic acid, fatty acids, long chain fatty acids, salicylic acid, alpha amino acids, 2-hydroxymethane sulfonic
- Preferred pharmaceutically acceptable salts of inventive compounds having an acidic moiety include sodium and potassium salts.
- Preferred pharmaceutically acceptable salts of inventive compounds having a basic moiety include hydrochloride and hydrobromide salts.
- the compounds of the present invention containing an acidic or basic moiety are useful in the form of the free base or acid or in the form of a pharmaceutically acceptable salt thereof.
- solvates refers to a molecular complex wherein the solvent molecule, such as the crystallizing solvent, is incorporated into the crystal lattice.
- the solvent incorporated in the solvate is water, the molecular complex is called a hydrate.
- Pharmaceutically acceptable solvates include hydrates, alcoholates such as methanolates and ethanolates, acetonitrilates and the like. These compounds can also exist in polymorphic forms.
- the compounds of the invention may also be administered as prodrugs.
- prodrug denotes a derivative of a compound, which derivative, when administered to warm-blooded animals, e.g. humans, is converted into the compound (drug).
- the enzymatic and/or chemical hydrolytic cleavage of the compounds of the present invention occurs in such a manner that the proven drug form (parent carboxylic acid drug) is released, and the moiety or moieties split off remain nontoxic or are metabolized so that nontoxic metabolic products are produced.
- a carboxylic acid group can be esterified, e.g., with a methyl group or ethyl group to yield an ester.
- an ester is administered to a subject, the ester is cleaved, enzymatically or non-enzymatically, reductively, oxidatively, or
- An anionic group can be esterified with moieties (e.g., acyloxymethyl esters) which are cleaved to reveal an intermediate compound which subsequently decomposes to yield the active compound.
- moieties e.g., acyloxymethyl esters
- the prodrugs can be prepared in situ during the isolation and purification of the compounds, or by separately reacting the purified compound with a suitable derivatizing agent.
- hydroxy groups can be converted into esters via treatment with a carboxylic acid in the presence of a catalyst.
- cleavable alcohol prodrug moieties include substituted or unsubstituted, branched or unbranched lower alkyl ester moieties, e.g., ethyl esters, lower alkenyl esters, di-lower alkylamino lower-alkyl esters, e.g.,
- acylamino lower alkyl esters acyloxy lower alkyl esters (e.g., pivaloyloxymethyl ester)
- aryl esters e.g., phenyl ester, aryl-lower alkyl esters, e.g., benzyl ester, optionally substituted, e.g., with methyl, halo, or methoxy substituents aryl and aryl- lower alkyl esters, amides, lower-alkyl amides, di-lower alkyl amides, and hydroxy amides.
- the present invention is further directed to a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable carrier and at least one compound or salt described herein.
- the pharmaceutically acceptable carrier be one that is chemically inert to the active compounds and one that has no detrimental side effects or toxicity under the conditions of use.
- the compound or pharmaceutical composition can be administered by any suitable route, oral or parenteral, including intravenous, subcutaneous, intraarterial, intraperitoneal, ophthalmic, intramuscular, buccal, rectal, vaginal, intraorbital, intracerebral, intracranial, intraspinal, intraventricuclar, intrathecal, intracisternal, intracapsular, intrapulmonary, intranasal, transmucosal, transdermal, or via inhalation. Accordingly, there is a wide variety of suitable formulations of the pharmaceutical composition of the present invention. The formulations may also be applied topically.
- compositions for parenteral administration that comprise a solution or suspension of the inventive compound or salt dissolved or suspended in an acceptable carrier suitable for parenteral administration, including aqueous and non-aqueous isotonic sterile injection solutions.
- Such solutions can contain anti-oxidants, buffers, bacteriostats, and solutes that render the formulation isotonic with the blood of the intended recipient, and aqueous and non-aqueous sterile suspensions that can include suspending agents, solubilizers, thickening agents, stabilizers, and preservatives.
- the compound or salt of the present invention may be administered in a physiologically acceptable diluent in a pharmaceutical carrier, such as a sterile liquid or mixture of liquids, including water, saline, aqueous dextrose and related sugar solutions, an alcohol, such as ethanol, isopropanol, or hexadecyl alcohol, glycols, such as propylene glycol or polyethylene glycol, dimethylsulfoxide, glycerol ketals, such as 2,2-dimethyl-l ,3-dioxolane-4-methanol, ethers, such as poly(ethyleneglycol) 400, an oil, a fatty acid, a fatty acid ester or glyceride, or an acetylated fatty acid glyceride with or without the addition of a pharmaceutically acceptable surfactant, such as a soap or a detergent, suspending agent, such as pectin, carbomers, methylcellulose, hydroxypropylmethylcellulose, or carb
- Oils useful in parenteral formulations include petroleum, animal, vegetable, or synthetic oils. Specific examples of oils useful in such formulations include peanut, soybean, sesame, cottonseed, corn, olive, petrolatum, and mineral. Suitable fatty acids for use in parenteral formulations include oleic acid, stearic acid, and isostearic acid. Ethyl oleate and isopropyl myristate are examples of suitable fatty acid esters.
- Suitable soaps for use in parenteral formulations include fatty alkali metal, ammonium, and triethanolamine salts
- suitable detergents include (a) cationic detergents such as, for example, dimethyl dialkyl ammonium halides, and alkyl pyridinium halides, (b) anionic detergents such as, for example, alkyl, aryl, and olefin sulfonates, alkyl, olefin, ether, and monoglyceride sulfates, and sulfosuccinates, (c) nonionic detergents such as, for example, fatty amine oxides, fatty acid alkanolamides, and polyoxyethylenepolypropylene copolymers, (d) amphoteric detergents such as, for example, alkyl-beta-aminopropionates, and 2-alkyl-imidazoline quaternary ammonium salts, and (e) mixtures thereof.
- the parenteral formulations can contain preservatives and buffers.
- such compositions may contain one or more nonionic surfactants having a hydrophile-lipophile balance (HLB) of from about 12 to about 17.
- HLB hydrophile-lipophile balance
- the quantity of surfactant in such formulations will typically range from about 5 to about 15% by weight.
- Suitable surfactants include polyethylene sorbitan fatty acid esters, such as sorbitan monooleate and the high molecular weight adducts of ethylene oxide with a hydrophobic base, formed by the condensation of propylene oxide with propylene glycol.
- parenteral formulations can be presented in unit-dose or multi-dose sealed containers, such as ampoules and vials, and can be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid excipient, for example, water, for injections, immediately prior to use.
- sterile liquid excipient for example, water
- Extemporaneous injection solutions and suspensions can be prepared from sterile powders, granules, and tablets of the kind previously described.
- Topical formulations including those that are useful for transdermal drug release, are well-known to those of skill in the art and are suitable in the context of the invention for application to skin.
- Topically applied compositions are generally in the form of liquids, creams, pastes, lotions and gels. Topical administration includes application to the oral mucosa, which includes the oral cavity, oral epithelium, palate, gingival, and the nasal mucosa.
- the composition contains at least one active component and a suitable vehicle or carrier. It may also contain other components, such as an anti-irritant.
- the carrier can be a liquid, solid or semi-solid.
- the composition is an aqueous solution.
- the composition can be a dispersion, emulsion, gel, lotion or cream vehicle for the various components.
- the primary vehicle is water or a biocompatible solvent that is substantially neutral or that has been rendered substantially neutral.
- the liquid vehicle can include other materials, such as buffers, alcohols, glycerin, and mineral oils with various emulsifiers or dispersing agents as known in the art to obtain the desired pH, consistency and viscosity.
- the compositions can be produced as solids, such as powders or granules. The solids can be applied directly or dissolved in water or a biocompatible solvent prior to use to form a solution that is substantially neutral or that has been rendered substantially neutral and that can then be applied to the target site.
- the vehicle for topical application to the skin can include water, buffered solutions, various alcohols, glycols such as glycerin, lipid materials such as fatty acids, mineral oils, phosphoglycerides, collagen, gelatin and silicone based materials.
- Formulations suitable for oral administration can consist of (a) liquid solutions, such as a therapeutically effective amount of the inventive compound dissolved in diluents, such as water, saline, or orange juice, (b) capsules, sachets, tablets, lozenges, and troches, each containing a predetermined amount of the active ingredient, as solids or granules, (c) powders, (d) suspensions in an appropriate liquid, and (e) suitable emulsions.
- Liquid formulations may include diluents, such as water and alcohols, for example, ethanol, benzyl alcohol, and the polyethylene alcohols, either with or without the addition of a
- Capsule forms can be of the ordinary hard- or soft-shelled gelatin type containing, for example, surfactants, lubricants, and inert fillers, such as lactose, sucrose, calcium phosphate, and corn starch.
- Tablet forms can include one or more of lactose, sucrose, mannitol, corn starch, potato starch, alginic acid, microcrystalline cellulose, acacia, gelatin, guar gum, colloidal silicon dioxide, croscarmellose sodium, talc, magnesium stearate, calcium stearate, zinc stearate, stearic acid, and other excipients, colorants, diluents, buffering agents, disintegrating agents, moistening agents, preservatives, flavoring agents, and pharmacologically compatible excipients.
- Lozenge forms can comprise the active ingredient in a flavor, usually sucrose and acacia or tragacanth, as well as pastilles comprising the active ingredient in an inert base, such as gelatin and glycerin, or sucrose and acacia, emulsions, gels, and the like containing, in addition to the active ingredient, such excipients as are known in the art.
- a flavor usually sucrose and acacia or tragacanth
- pastilles comprising the active ingredient in an inert base, such as gelatin and glycerin, or sucrose and acacia, emulsions, gels, and the like containing, in addition to the active ingredient, such excipients as are known in the art.
- the compound or salt of the present invention can be made into aerosol formulations to be administered via inhalation.
- the compounds are preferably supplied in finely divided form along with a surfactant and propellant. Typical percentages of active compound are 0.01%-20% by weight, preferably 1%-10%.
- the surfactant must, of course, be nontoxic, and preferably soluble in the propellant.
- Such surfactants are the esters or partial esters of fatty acids containing from 6 to 22 carbon atoms, such as caproic, octanoic, lauric, palmitic, stearic, linoleic, linolenic, olesteric and oleic acids with an aliphatic polyhydric alcohol or its cyclic anhydride.
- Mixed esters such as mixed or natural glycerides may be employed.
- the surfactant may constitute 0.1%-20% by weight of the composition, preferably 0.25%-5%. The balance of the composition is ordinarily propellant.
- a carrier can also be included as desired, e.g., lecithin for intranasal delivery.
- aerosol formulations can be placed into acceptable pressurized propellants, such as dichlorodifluoromethane, propane, nitrogen, and the like. They also may be formulated as pharmaceuticals for non-pressured preparations, such as in a nebulizer or an atomizer. Such spray formulations may be used to spray mucosa.
- pressurized propellants such as dichlorodifluoromethane, propane, nitrogen, and the like.
- non-pressured preparations such as in a nebulizer or an atomizer.
- Such spray formulations may be used to spray mucosa.
- the compound or salt of the present invention may be made into suppositories by mixing with a variety of bases, such as emulsifying bases or water-soluble bases.
- bases such as emulsifying bases or water-soluble bases.
- Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams, or spray formulas containing, in addition to the active ingredient, such carriers as are known in the art to be appropriate.
- the compound or salt of the present invention may be formulated as inclusion complexes, such as cyclodextrin inclusion complexes, or liposomes.
- inclusion complexes such as cyclodextrin inclusion complexes, or liposomes.
- Liposomes serve to target the compounds to a particular tissue, such as lymphoid tissue or cancerous hepatic cells. Liposomes can also be used to increase the half-life of the inventive compound.
- Liposomes useful in the present invention include emulsions, foams, micelles, insoluble monolayers, liquid crystals, phospholipid dispersions, lamellar layers and the like.
- the active agent to be delivered is incorporated as part of a liposome, alone or in conjunction with a suitable chemotherapeutic agent.
- liposomes filled with a desired inventive compound or salt thereof can be directed to the site of a specific tissue type, hepatic cells, for example, where the liposomes then deliver the selected compositions.
- Liposomes for use in the invention are formed from standard vesicle-forming lipids, which generally include neutral and negatively charged phospholipids and a sterol, such as cholesterol. The selection of lipids is generally guided by consideration of, for example, liposome size and stability of the liposomes in the blood stream.
- a liposome suspension containing a compound or salt of the present invention may be administered intravenously, locally, topically, etc. in a dose that varies according to the mode of administration, the agent being delivered, and the stage of disease being treated.
- Treatment refers to a therapeutic intervention that ameliorates a sign or symptom of a disease or pathological condition after it has begun to develop.
- the term “ameliorating,” with reference to a disease or pathological condition refers to any observable beneficial effect of the treatment.
- the beneficial effect can be evidenced, for example, by a delayed onset of clinical symptoms of the disease in a susceptible subject, a reduction in severity of some or all clinical symptoms of the disease, a slower progression of the disease, an improvement in the overall health or well-being of the subject, or by other parameters well known in the art that are specific to the particular disease.
- the observable benefit of a treatment can be a low level of benefit of about 10%, about 20%, or about 30%, or about 40%> of the full benefit, or it can be a medium level benefit of about 50%, 60%, 70%, or about 80%, or the benefit can also be a high level decrease of about 90%>, about 95%>, about 99%, or about 99.9%, or complete cure.
- “Inhibiting” means decreasing the effect or activity of a given entity, which can be a low level of decrease of about 10%, about 20%, or about 30%, or about 40% of the full effect or activity, or the decrease can be a medium level decrease of about 50%, 60%, 70%, or about 80%, or the decrease can also be a high level decrease of about 90%, about 95%, about 99%, or about 99.9%, or complete elimination of the effect or activity.
- Preventing refers to reducing the probability of developing a disease or disorder in a healthy animal or in an animal at risk for developing such disease or disorder or delaying the onset of the the disease or disorder.
- Moduleating can refer to decreasing or increasing, and in an embodiment, refers to decreasing.
- dosages will be between 0.01 mg/kg and 250 mg/kg of the subject's body weight, and more typically between about 0.05 mg/kg and 100 mg/kg, such as from about 0.2 to about 80 mg/kg, from about 5 to about 40 mg/kg or from about 10 to about 30 mg/kg of the subject's body weight.
- unit dosage forms can be formulated based upon the suitable ranges recited above and the subject's body weight.
- the term "unit dosage form” as used herein refers to a physically discrete unit of therapeutic agent appropriate for the subject to be treated.
- dosages are calculated based on body surface area and from about 1 mg/m 2 to about 200 mg/m 2 , such as from about 5 mg/m 2 to about 100 mg/m 2 will be administered to the subject per day.
- administration of the therapeutically effective amount of the compound or compounds involves administering to the subject from about 5 mg/m 2 to about 50 mg/m 2 , such as from about 10 mg/m 2 to about 40 mg/m 2 per day. It is currently believed that a single dosage of the compound or compounds is suitable, however a therapeutically effective dosage can be supplied over an extended period of time or in multiple doses per day.
- unit dosage forms also can be calculated using a subject's body surface area based on the suitable ranges recited above and the desired dosing schedule.
- the compounds or salts thereof described herein for methods of preventing a neurodegenerative disease or disorder can be about 0.001 to about 1 mg/kg body weight of the subject , for example, about 0.001 mg, 0.002 mg, 0.005 mg, 0.010 mg, 0.015 mg, 0.020 mg, 0.025 mg, 0.050 mg, 0.075 mg, 0.1 mg, 0.15 mg, 0.2 mg, 0.25 mg, 0.5 mg, 0.75 mg, or 1 mg/kg body weight per day.
- the dose of the compounds or salts described herein for methods of treating the disorder can be about 1 to about 1000 mg/kg body weight of the subject being treated per day, for example, about 1 mg, 2 mg, 5 mg, 10 mg, 15 mg, 0.020 mg, 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 250 mg, 500 mg, 750 mg, or 1000 mg/kg body weight per day.
- 6-Bromothieno[2,3-d]pyrimidin-4(3H)-one (7.02 g, 30.4 mmol, 1.0 equiv) was taken up in 100 mL POCI3 and refluxed for 6 h. Upon completion, the reaction was concentrated in vacuo and the residue was taken up in 10 mL acetonitrile and heated to reflux for 10 min. The acetonitrile solution was then poured in a flask and cooled. The ppt was then filtered and washed with hexane to give 6-bromo-4-chlorothieno[2,3-d]pyrimidine (5.51 g, 72.7 % yield) as a tan solid. ⁇ NMR (400 MHz, CHLOROFORM-d) ⁇ ppm 8.80 (s, 1 H), 7.47 (s, 1 H).
- EtOAc layer was separated, dried (MgS0 4 ), filtered, and purified by flash Si0 2 (5 to 100% EtOAc/hexanes) column chromatography to provide (4,6- dichloropyrimidin-5-yl)(3-(methylthio)phenyl)methanol (3.8 g, 13 mmol, 67 % yield).
- This example demonstrates that a correlation was established between a multiplexed high-throughput and high content screening assay that sequentially measured cytotoxicity and protein aggregation for screening of compound libraries.
- This assay can be used to identify compounds that either modulate aggregation and/or cytotoxicity in cellular models of neurodegenerative diseases.
- a stable PC12 cell line containing a gene fusion of Exon 1 of the Huntingtin gene linked to GFP under the control of the inducible ecdysone promoter were used as the cell- based model of Huntington Disease for high throughput screening.
- Exon 1 of the Huntingtin gene econtained an expansion of 103 polyglutamines (Q103HTT) which, when expressed, induced cell death and formation of distinct, bright GFP aggregates. The amount of cell death and the size and intensity of GFP aggregates increased with time and induction level. Cell death was quantified by measurement of ATP content in the cells. A maximal 40-50% of cell death was observed when the Huntington gene was induced in this cell line.
- the cell health or membrane integrity was measured by the protease release method.
- Cells with perturbed membrane integrity release proteins and cofactors from the cytosol into the media, a dead cell protease being one of them.
- Cytox Glo kit contains a cell impermeable pro-luciferin substrate that becomes activated upon cleavage by such protease.
- cells with alered membrane permeability release this protease and generate an increase in the luminescent signal with reliability (averabe S/B of 9.6 and Z' of 0.80).
- Rat pheochromocytoma PC 12 cells harboring HTT Q 103 or Q25 fujsed to GFP under tebufenozide (Sigma Aldrich) indudction were supplied by the Eric Schweizer lab (UCLA).
- Cells were maintained in Phenol red free DMEM (Invitrogen) at 37°C under a humidified atmosphere containing 5% C0 2 and 95% air.
- the medium contained + 5% supplemented calf serum, + 5% horse serum (sera from HyClone), 230 ug/ml Geneticin and lx pen/strep 2 mM L-glutamine. Cells were passaged when they reached 85 to 90 % co fluency.
- Cells were pelated at the density of 1000 to 1500 cells/well in black, clear bottom, tissue culture treated, microclear 1536 well plates (Aurora Biotechnologies) in 5 ul/well in phenol red free DMEM containing 2% serum (1 % each) without geneticin using a Multidrop Combi dispenser (Thermo Scientific).
- Tebufenozide (Sigma Aldrich) inducer at a 200 nM final concentration and test compounds were sequentially added to the cells using a Kalypsys pintool transfer station at a volume of 23 nl/well after the cells were incubated for 24 hrs.
- Compound libraries and Tebufenozide were dissolved in DMSO and the final concentration of DMSO in the cell plates was 0.46% v/v. Plates were incubated at 37°C in 5 % C02 for 48 hours before being assayed.
- Table 1 Protection of striatal cells against cytotoxicity and aggregation by certain compounds.
- Table 2 shows that treatment with these two compounds substantially reduced cell death caused by serum deprivation selectivity for the mutant STHdh ⁇ 1 1 1/1 1 1 Huntington striatal cells.
- FIG. 1 depicts a reaction scheme used for the synthesis of embodiments of the compounds of the invention.
- commercially available thieno[2,3- d]pyrimidin-4(3H)-one 1 was regioselectively halogenated in the 6-position with bromine in acetic acid to obtain compound 2.
- Transformation of the pyrimidinone into 6-bromo-4- chlorothieno[2,3-d]pyrimidine 3 was carried out by the use of phosphorus oxychloride. LDA induced bromo rearrangement and bromo-iodo replacement promoted by ethyl Grignard yielded intermediate 5.
- Fig. 2 depicts a reaction scheme to synthesize additional embodiments, e.g., pyrrazolopyrimidines, of the compounds of the invention.
- Tables 3-5 set forth biological activity data of compounds including their ability (Kd) to inhibit PIP5 2C, in accordance with an embodiment of the invention.
- This Example illustrates that compound 49 in accordance with an embodiment of the invention is able to protect primary neurons against H 2 0 2 and beta-amyloid induced cell death.
- Fig. 4 and 5 show that compound 49 is able to prevent cell death induced by H 2 0 2 and beta-amyloid.
- This Example illustrates that a compound in accordance with an embodiment of the invention is able to block the activation of the intrinsic apoptotic pathway, preventing the production of caspase 9 and its downstream target caspase 3.
- Figs. 6 A and 6B show that compound 14 (F7) protects the cells from serum free medium induced apoptotic cell death by inhibiting the activation of caspase 9 and of its downstream target caspase 3.
Abstract
Disclosed are compounds of the formula (I): wherein R1, R2, X, Y, and Z as described herein, or pharmaceutically acceptable salts thereof, for use in treating or preventing neurodegenerative diseases and disorders such as Huntington's disease. Also disclosed are pharmaceutical compositions comprising such compounds or pharmaceutically acceptable salts thereof and a pharmaceutically acceptable carrier and methods of treating or preventing neurodegenerative diseases and disorders.
Description
COMPOUNDS, PHARMACEUTICAL COMPOSITIONS, AND METHODS OF TREATING OR PREVENTING NEURODEGENERATIVE DISEASES OR DISORDERS
CROSS-REFERENCE TO A RELATED APPLICATION
[0001] The present application claims the benefit of United States Provisional Patent
Application No. 61/388,482, filed September 30, 2010, the disclosure of which is incorporated by reference in its entirety.
BACKGROUND OF THE INVENTION
[0002] Neurodegenerative diseases impose a heavy burden on the patient as well as on the patient's family. For example, Huntington's disease affects 3-10 subjects in 100,000 individuals in Western Europe and North America. In most cases, the onset of the disease occurs in midlife, between the ages of 35 and 50 years. The disease progresses over time and is invariably fatal 15-20 years after the onset of the first symptoms. Initial symptoms of this disease include clumsiness, difficulties with smooth eye pursuit, and slight uncontrolled and awkward movements. These motor disturbances associated with the loss of voluntary movement coordination progress slowly and become more severe with time and the patients gradually lose their capacity to move and eventually communicate. Progression of the disease is accompanied by neuronal death in the striatum and, to a lesser extent, in the cerebral cortex. Presently, there is little treatment to prevent or palliate the progress of Huntington disease.
[0003] Huntington's disease is neurodegenerative disorder due to a dominantly acting expansion of a CAG trinucleotide repeat in exon 1 of the Huntington {HTT) gene resulting in the production of an altered (mutant) protein Huntingtin having a long chain of
polyglutamine tract (polyQ). Clinical and statistical analyses have shown that an increased number of polyQ repetition correlates with the probability of developing the disease, with 36 to 40 being the accepted cut off number for developing the disorder with high probability. It is known that polyQ repetitions impact the physical properties of the mutants producing protein aggregates that precipitate forming inclusion bodies, which are toxic to the neuronal cells.
[0004] While there are some proposals to treat neurodegenerative disorders, e.g., US 2010/0092456 Al disclosing the use of RNAi or antisense mechanisms, there is an unmet
need for finding small molecules suitable for treating and/or preventing neurodegenerative disorders such as Huntington's disease.
BRIEF SUMMARY OF THE INVENTION
The invention provides a compound of formula (I):

CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4,
NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
R2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy CpC alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino,
guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R3, S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4); wherein each of said R2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Cj-C6 alkyl, dicarboxy halo Ci-C alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Q-Q alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
wherein R3-R5 are independently hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C4 cycloalkyl, C3-C4 cycloalkenyl, C6-C2o aryl C -C alkyl, C6-C2o aryl, heterocyclyl, or heterocyclyl alkyl;
X is a bond, S, O, or NH;
Y is S or NH;
Z is CH or N;
or a pharmaceutically acceptable salt thereof.
[0006] The invention also provides pharmaceutical compositions comprising such compounds of formula (I) or pharmaceutically acceptable salts thereof, and methods of treating or preventing neurodegenerative disorders or diseases, for example, polyglutamine diseases such as Huntington's disease. The invention also provides methods for inhibiting the PIP5K2C kinase, for modulating the activity of a mutant Huntingtin protein, for treating or preventing a neurodegenerative disease, and/or for inhibiting the activation of caspase 9 in a cell, for example, in a patient.
BRIEF DESCRIPTION OF THE SEVERAL VIEWS OF THE DRAWING(S)
[0007] Figure 1 depicts a reaction scheme to prepare certain compounds in accordance with an embodiment of the invention.
[0008] Figure 2 depicts another reaction scheme to prepare compounds in accordance with an embodiment of the invention.
[0009] Figure 3 depicts the number of viable cells in FBS and serum free medium (SFM) for wild type and mutant striatal cells, showing that the mutant cells do not survive the stress of the serum free medium. Shown is cell growth of wild type and Huntington mutant STHdhQ1 1 1/n i striatal cells upon serum deprivation. ATP levels were quantitated using ATPlite, a luciferase-based ATP quantitation assay, after 24 h incubation. Data are represented in RLU counts. Serum deprivation has a greater effect on STHdh^111711 1 striatal cells (24% of FBS activity after 24 h) than STHdhQ7/7 cells (68% of FBS activity after 24 h).
[0010] Figure 4 shows that compound 49 in accordance with an embodiment of the invention protects against hydrogen peroxide induced cell death of striatal cells.
[0011] Figure 5 shows that compound 49 in accordance with an embodiment of the invention protects against beta-amyloid induced cell death of striatal cells.
[0012] Figure 6A depicts Western blots of the control showing caspase 9 activation and the downstream production of caspase 3 during mutant cell apoptosis. Caspase 9 ans caspase 3 activation after 16 h treatment under normal growth conditions (DMEM + 10% FBS). Figure 6B depicts Western blots showing that caspase 9 activation and production of caspase 3 are inhibited by compound 14 (F7) in serum free medium and that the compound inhibits cell apoptosis in accordance with an embodiment of the invention. Caspase 9 ans caspase 3 activation after 16 h treatment under serum deprived conditions. CT = DMSO (vehicle) treatment. A and M are two other compounds that affect the Huntington cellular phenotype whose identies are not disclosed by the assay provider. The arrows indicate the specific activity of compound 14 under serum deprived conditions.
[0013] Figure 7 shows that PCI 2 cells express PIK5K2C kinase.
DETAILED DESCRIPTION OF THE INVENTION
[0014] The present invention provides compounds, compositions, and methods for treating or preventing neurodegenerative disorders. Embodiments of the invention are
predicated on the concept of inhibiting posttranscriptional modifications of the mutant genes responsible for such disorders.
[0015] In an embodiment, the invention provides a compound of formula (I):

CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R R5, NR3C(0)N(OH)R4,
NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
R is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo C]-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3,
C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4); wherein each of said R groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Cj-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy CpQ alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Cj-C6 alkyl, alkyl thio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R , S02NR3R4, S02N(OH)R3, CR =NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
wherein R3-R5 are independently hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C4 cycloalkyl, C3-C4 cycloalkenyl, C6-C2o aryl Ci-C6 alkyl, C6-C20 aryl, heterocyclyl, or heterocyclyl alkyl;
X is a bond, S, O, or NH;
Y is S or NH;
Z is CH or N;
or a pharmaceutically acceptable salt thereof;
with the proviso that when X and Y are S, Z is CH, and R1 is tetrazolyl or a substituted tetrazolyl, then R is not halo, heterocyclyl, heterocyclyl aryl, aryl heterocyclyl, aryl, substituted aryl, substituted heterocyclyl, substituted heterocyclyl aryl, or substituted aryl heterocyclyl.
[0016] In a particular embodiment of the above compound or salt, X is a bond, S or NH.
[0017] In any of the embodiments, R1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R1 is unsubstituted or optionally independently substituted with one or more substituents
selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloalkyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono Cj- C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, and S02R3.
[0018] In a particular embodiment of the compound or salt of the invention, R1 is alkyl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloalkyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, and S02R3.
[0019] In an embodiment of the compounds or salts of the invention, R1 is alkyl, aryl, arylalkyl, heterocyclyl, or heterocyclylalkyl, optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloalkyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, and S02R3.
[0020] In another embodiment of the compounds or salts of the invention, R1 is selected from the group consisting of alkyl, aryl, arylalkyl, diazolyl, thiazolyl, triazolyl, thiadiazolyl, oxadiazolyl, pyridyl, pyridylmethyl, pyridazinyl, furanyl, benzodiazolyl, pyridinotriazolyl, and benzothiazolyl, each of which is optionally substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloalkyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , and S02R .
[0021] In another embodiment of the compounds or salts of the invention, R1 is selected from the group consisting of alkyl, aryl, arylalkyl, diazolyl, thiazolyl, triazolyl, thiadiazolyl, oxadiazolyl, pyridyl, pyridylmethyl, pyridazinyl, furanyl, benzodiazolyl, pyridinotriazolyl,
benzothiazolyl, each of which is optionally substituted with a substituent selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloalkyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, and trialkylamino.
[0022] In another embodiment of the compounds or salts of the invention, R1 is selected from the group consisting of alkyl, phenyl, phenylalkyl, diazolyl, thiazolyl, triazolyl, thiadiazolyl, oxadiazolyl, pyridyl, pyridylmethyl, pyridazinyl, furanyl, benzodiazolyl, pyridinotriazolyl, and benzothiazolyl, each of which is optionally substituted with an alkyl, hydroxy, or amino substituent.
[0023] In yet another embodiment of the compounds or salts of the invention, R1 is selected from the group consisting of alkyl, phenyl, phenylalkyl, diazolyl, thiazolyl, triazolyl, thiadiazolyl, oxadiazolyl, pyridyl, pyridylmethyl, pyridazinyl, furanyl, benzodiazolyl, pyridinotriazolyl, and benzothiazolyl, each of which is optionally substituted with an alkyl, or amino substituent.
[0024] In accordance with any of the embodiments above, X-R1 of the compound or salt of the invention is selected from the group consisting of:


[0025] In accordance with an embodiment of the compounds or salts of the invention, Y is S.
[0026] In any of the embodiments of the compounds or salts of the invention, Z is CH.
[0027] In any of the embodiments of the compounds or salts of the invention, X is S.
[0028] In any of the embodiments of the compounds or salts of the invention, R is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono CpC6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4,
S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5,
NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and
P(0)(OR3)(OR4); wherein each of said R2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Cj-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Q-Qs alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Cj-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4).
[0029] In an embodiment, R2 is selected from the group consisting of halo, aryl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, and S02R3; wherein each of said R2 groups aryl, aryl heterocyclyl, and aryl heterocyclyl aryl are unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono d-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy C]-C6 alkyl, dicarboxy halo Cj-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Q-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, and an anionic group.
[0030] In a further embodiment, R2 is selected from the group consisting of phenyl, pyrimidinyl, thiophenyl, pyrrolyl, benzofuranyl, naphthyl, methylenedioxyphenyl, 4-oxo-4H- chromenyl, dibenzothiophenyl, bromo, and iodo, optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Cj-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro d-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, and an anionic group.


In accordance with an embodiment, the invention also provides the following


or pharmaceutically acceptable salts thereof.
[0033] In accordance with an embodiment, the invention provides a pharmaceutical composition comprising a compound or salt as described above and a pharmaceutically acceptable carrier.
[0034] Without wishing to be bound to any particular theory or mechanism, the following is provided. Phosphoinositide (PI) is a collective term for phosphatidylinositol (Ptdlns) and its phosphorylated derivatives. Ptdlns contains a 1 o-myo-inositol phosphate group linked to diacylglycerol. Pis regulate many biological processes, including cell proliferation, cell survival, differentiation, signal transduction, cytoskeleton organization and membrane trafficking. The activities of specific Pis are regulated by controlling their levels in the cell, which is achieved by intricate networks of proteins that control their synthesis, transport and degradation.
[0035] PI synthesis is regulated by PI kinases and phosphatidylinositol phosphate (PIP) kinases (PIPKs), and PI degradation is governed by lipid phosphatases, while their transport is mediated by Ptdlns transfer proteins (PITPs). The hydroxy groups at positions 3, 4 and 5 on the inositol ring can be phophorylated by the lipid kinases: Ptdlns 3-kinases (PI3Ks), Ptdlns 4-kinases (PI4Ks), Ptdlns 5-kinases (PI5Ks) and PIPKs. Within the
phosphatidylinositol phosphate (PIP) kinases (PIPKs) category, there are known two classes: the 68 kDa ΡΙΡ5Κ(α, β and γ) that uses PtdIns4P to generate PtdIns(4,5)P2 and the 53 kDa ΡΙΡ4Κ( , β and γ) that phosphorylates PtdIns3P and PtdIns5P to produce PtdIns(3,4)P2 and PtdIns(4,5)P2. The PIPK isoform ΡΙΡ4Κγ (also known as PIP5K2C) has a restricted CNS
expression profile, which is limited to neurons, particularly the cerebellar Purkinje cells, pyramidal cells of the hippocampus, large neuronal cell types in the cerebral cortex including pyramidal cells, and mitral cells in the olfactory bulb and is not expressed in cerebellar, hippocampal formation, or olfactory bulb granule cells. In neurons expressing this enzyme, ΡΙΡ4Κγ has a vesicular distribution and shows partial colocalization with markers of cellular compartments of the endomembrane trafficking pathway. The ΡΙΡ4Κγ isoform expression is established after day 7 of postnatal development. It is believed that ΡΙΡ4Κγ plays a role in neuron function, specifically in the regulation of vesicular transport, in specific regions of the developed brain.
[0036] D-m o-Inositol 1 ,4,5-trisphosphate (Ins(l ,4,5)P3, is a cellular second messenger. Phospholipase C catalyses hydrolysis of phosphatidylinositol 4,5-bisphosphate (PtdIns-4,5- P2) to Ins(l,4,5)P3 and diacyl glycerol. Ins(l,4,5)P3 interacts specifically with a tetrameric receptor-operated Ca2+ channel on the endoplasmic reticulum to mobilize Ca2+ stores in stimulated cells. Ins(l ,4,5)P3 mediates the agonist-induced response via this rise in intracellular Ca2+ concentration.
[0037] Phosphatidylinositol 4,5-bisphosphate (PtdIns-4,5-P2) is not only a precursor of Ins(l ,4,5)P3 but it has been shown that the inositol 1,4,5-trisphosphate receptor (InsP3R) forms a stable inhibitory complex with endogenous PtdIns-4,5-P2. The interaction between PtdIns-4,5-P2 and InsP3R does not occur in the membrane and a novel model of
compartmentalized Ca2+ signaling has been proposed. The functional link between InsP3R and PtdIns-4,5-P2 provides a basis for a local, rapid, and efficient coupling between phospholipase C activation, PtdIns-4,5-P2 hydrolysis, and intracellular Ca2+ wave initiation in neuronal cells.
[0038] Most aspects of neuronal activity are regulated either directly or indirectly by calcium signals, which vary in their temporal and spatial characteristics. Therefore, free intracellular levels of calcium are precisely controlled in the cell. Cellular events ranging in time scale from the sub-millisecond triggering of neurotransmitter release at presynaptic terminals to changes in gene expression in the nucleus all require the second messenger actions of calcium. Calcium enters the cells via voltage-gated and agonist-induced ion channels and cytosolic calcium levels are further increased by calcium-induced and inositol- (1 ,4,5) triphosphate-induced release from intracellular stores. Diffusion of calcium within the cytoplasm relies on the binding to calcium-binding proteins, which ultimately are responsible for the activation of physiological processes. The levels of calcium in mitochondria are low
under normal physiologic conditions; however, elevations in mitochondria calcium occur when intracellular concentrations rise during and after prolonged activation of calcium conductance. Prolonged increases in intracellular calcium result in a plethora of harmful effects to the cell, such as excitotoxic events, activation of calcium-dependent enzymes, apoptosis and mitochondrial failure. Imbalances in calcium homeostasis and regulation have been implicated in the pathogenesis of many neurodegenerative diseases, including
Huntington's disease. Experimental evidence from HD mouse models and lymphoblasts of HD patients indicates that perturbations in calcium signaling can lead to excitotoxic damage and apoptosis. It is believed that mutant htt can affect calcium signaling by enhancing inositol-( 1 ,4,5) triphosphate-induced intracellular calcium release, stimulating glutamate receptor activity and destabilizing mitochondrial calcium balance. In addition, an improper distribution of calcium among intracellular pools can induce neuronal damage in HD.
[0039] It is believed that in unstimulated cells, some fraction of InsP3R is constitutively inhibited due to interaction with PtdIns-4,5-P2 in the juxtaposed membrane (resting state). These InsP3R are unable to open because of the spatial constraint imposed by the topology of InsP3R-PIP2 interaction. Agonist stimulation leads to activation of PLC, cleavage of InsP3R-tethered PtdIns-4,5-P2, and release of the spatial clamp on the InsP3R (signal transduction step). PLC simultaneously removes the inhibitor (PtdIns-4,5-P2) and generates the activator (InsP3) of the InsP3R, leading to Ca2+ wave initiation. This compartmentalized signaling mechanism is responsible for Ca2+ wave initiation in specialized trigger zones; whereas Ca2+ wave propagation through the cell is sustained by direct Ca2+ feedback on the InsP3R. Preferential coupling between PLC linked hormonal receptors and InsP3R have been demonstrated previously in some intact cell preparations. It appears that integrity of this
2+ preferential coupling depends on correct spatial arrangement between intracellular Ca release stores and PLC-linked receptors, which is maintained by intact actin cytoskeleton network. This direct coupling model is in agreement with the phenomenon of
compartmentalized Ca2+ signaling reported in these papers.
[0040] Furthermore, poly-Q HTT association with InsP3Rl causes sensitization of InsP3Rl to activation by InsP3 in planar lipid bilayers and in primary striatal neurons. Poly- Q HTT activates Ca2+-permeable NR2B-containing NMDA receptors. Moreover, a cytosolic C-terminal tail of InsP(3)Rl has neuroprotective effects in a Huntington's disease model.
[0041] It is further believed that ΡΙΡ4Κγ has a specific neuronal and vesicular distribution and that the PIP5K2C gene has high levels of expression in the hippocampal formation,
primarily in the stratum pyramidale and extending into the stratum radiatum of CA1-CA3, and were excluded from the dentate gyrus. Huntington's patients show a neuronal reduction in the CA1 area of the Hippocampus and Huntington's mice show a reduced hippocampal neurogenesis.
[0042] Accordingly, in accordance with an embodiment, the invention provides a method of inhibiting the PIP5K2C kinase in a cell in need thereof comprising administering to the cell an effective amount of a compound of formula (I):

wherein R1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, heterocyclyl aryl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl, heterocyclyl aryl, and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono Cj-C6 alkyl, carboxy Q-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Cj-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4, S02N(OH)R3, CR3=NR4,
CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4,
NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
R2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Cj-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy,
thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR 3 , S(0)R 3 , S02R 3 , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4); wherein each of said R2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Q-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
wherein R3-R5 are independently hydrogen, Q-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C4 cycloalkyl, C3-C4 cycloalkenyl, C6-C2o aryl C)-C6 alkyl, C6-C20 aryl, heterocyclyl, or heterocyclyl alkyl;
X is a bond, S, O, or NH;
Y is S or NH;
Z is CH or N;
or a pharmaceutically acceptable salt thereof.
[0043] In embodiments of the compounds or salts, X is S, Y is NH, and Z is N, R1 is methyl tetrazolyl, and R2 is phenyl, optionally substituted with methylsulfonyl.
[0044] It is also contemplated that inhibition of the PIP5K2C kinase would lead to a decrease in Ptdlns derivative Ptdlns (4,5)P2 which has been implicated in blocking neurite growth. Accordingly, in accordance with the invention, inhibiting the PIP5K2C kinase is expected to stimulate neural outgrowth in neurodegenerative disorders.
[0045] The invention further provides a method of treating and/or preventing a neurodegenerative disease or disorder in an animal comprising administering to the animal an effective amount of a compound of formula (I):

CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4,
NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
R is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy C]-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R ,
S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4,
NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and
3 4 2
P(0)(OR )(OR ); wherein each of said R groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy C]-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
wherein R -R are independently hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C cycloalkyl, C3-C4 cycloalkenyl, C6-C2o aryl Ci-C6 alkyl, C6-C2o aryl, heterocyclyl, or heterocyclyl alkyl;
X is a bond, S, O, or NH;
Y is S or NH;
Z is CH or N;
or a pharmaceutically acceptable salt thereof.
[0046] Examples of neurodegenerative diseases or disorders include brain trauma, spinal cord trauma, trauma to the peripheral nervous system, Alzheimer's disease, Pick's disease, diffuse Lewy body disease, progressive supranuclear palsy (Steel-Richardson syndrome), multisystem degeneration (Shy-Drager syndrome), motor neuron diseases including amyotrophic lateral sclerosis, degenerative ataxias, cortical basal degeneration, ALS- Parkinson's-Dementia complex of Guam, subacute sclerosing panencephalitis, Huntington's disease, Parkinson's disease, synucleinopathies, primary progressive aphasia, striatonigral degeneration, Machado-Joseph disease/spinocerebellar ataxia type 3 and olivopontocerebellar degenerations, Gilles De La Tourette's disease, bulbar and pseudobulbar palsy, spinal and spinobulbar muscular atrophy (Kennedy's disease), primary lateral sclerosis, familial spastic
paraplegia, Werdnig-Hoffman disease, Kugelberg-Welander disease, Tay-Sach's disease, Sandhoff disease, familial spastic disease, Wohlfart- ugelberg-Welander disease, spastic paraparesis, progressive multifocal leukoencephalopathy, and prion diseases (including Creutzfeldt-Jakob, Gerstmann-Straussler-Scheinker disease, Kura and fatal familial insomnia, age-related dementia, vascular dementia, diffuse white matter disease (Binswanger's disease), dementia of endocrine or metabolic origin, dementia of head trauma and diffuse brain damage, dementia pugilistica or frontal lobe dementia, neurodegenerative disorders resulting from cerebral ischemia or infarction including embolic occlusion and thrombotic occlusion as well as intracranial hemorrhage of any type, intracranial and intravertebral lesions, hereditary cerebral angiopathy, normeuropathic hereditary amyloid, Down's syndrome,
macroglobulinemia, secondary familial Mediterranean fever, Muckle-Wells syndrome, multiple myeloma, pancreatic- and cardiac-related amyloidosis, chronic hemodialysis arthropathy, or Finnish and Iowa amyloidosis.
[0047] In an embodiment, the neurodegenerative disease or disorder is selected from the group consisting of Dentatorubropallidoluysian atrophy, Huntington's disease, Spinobulbar muscular atrophy, Spinocerebellar ataxia Type 1 , Spinocerebellar ataxia Type 2,
Spinocerebellar ataxia Type 3, Spinocerebellar ataxia Type 6, Spinocerebellar ataxia Type 7, Spinocerebellar ataxia Type 17, Cockayne Syndrome, hepatolenticular degeneration, Lafora Disease, Menkes Kinky Hair Syndrome, neurofibromatosis, Tourette Syndrome, Tuberous Sclerosis Amyotrophic Lateral Sclerosis, muscular atrophy, poliomyelitis, Parkinson's Disease, Prion diseases, Creutzfeldt-Jacob Syndrome, Kuru, Scrapie, and Alzheimer's Disease.
[0048] In an embodiment, the neurodegenerative disease or disorder is a polyglutamine disease, for example, spinal and bulbar muscular atrophy, Huntington's disease,
Dentatorubropallidoluysian atrophy, Spinocerebellar ataxia 1, 2, 6, 7, and 17, SCA 3/MJD.
[0049] The invention further provides a method of modulating the activity of a mutant Huntingtin protein in a cell comprising administering to the cell an effective amount of a compound of formula (I):

CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4,
NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
R2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Cj-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR 3 , S(0)R 3 , S02R 3 , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and
P(0)(OR3)(OR4); wherein each of said R2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono C|-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
wherein R3-R5 are independently hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C alkynyl, C3-C4 cycloalkyl, C3-C4 cycloalkenyl, C6-C20 aryl Ci-C6 alkyl, C6-C2o aryl, heterocyclyl, or heterocyclyl alkyl;
X is a bond, S, O, or NH;
Y is S or NH;
Z is CH or N;
or a pharmaceutically acceptable salt thereof.
[0050] Compounds in accordance with an embodiment inhibit the activation of caspase 9. Accordingly, the compounds of the invention inhibit the activation of apoptosis of neuronal cells. Again, without wishing to be bound by theory or mechanism, it is believed there are three main mechanisms for apoptotic activation. The first one involves the mitochondria released of the Apoptosis-inducing factor (AIF) and its translocation to the nucleus to trigger DNA destruction. This pathway is caspase-independent. The second mechanism is triggered by external activators such us TNF-a, Lymphotoxin or the Fas Ligand and involves the activation of caspase 8. The third mechanism depends on intrinsic signals and is commonly activated in case of cellular damage (from radical species, toxic chemicals, ionization, etc.). This pathway involves Bax translocation to the mitochondrion, Bcl-2 inhibition causing cytochrome C leak out, binding of cytochrome C with Apaf-1 (apoptotic protease activating factor- 1) and aggregation to foma apoptosomes, cleavage of pro-caspase 9 by the apoptosome to form active caspase 9, activation of caspase 3 and caspase 7 by caspase 9, and caspase 3-7 digestion of cytoplasmic structural proteins, degradation of nuclear DNA and cell
phagocytosis.
[0051] Compounds in accordance with an embodiment of the invention block the activation of the intrinsic apoptotic pathway, or prevent the production of caspase 9 and its downstream target caspase 3. Activation of the apoptotic intrinsic pathway is found in
numerous diseases Huntington disease, ALS, Niemann-Pick disease type C, Lysosomal Cell Death, Ischemia, viral and bacteria infection, and ceramide induced neuronal death.
[0052] Accordingly, the present invention provides a method of inhibiting the activation of caspase 9 in a cell in need thereof comprising administering an effective amount of a compound of formula (I):
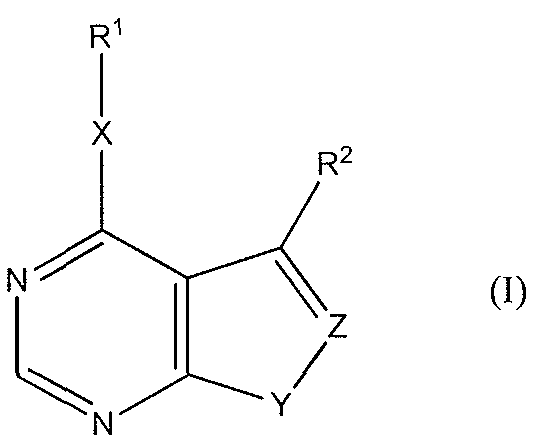
CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4,
NR3C(S)N(OH)R4, NR3S02R4, NHS02NR R4, NR3S02NHR4, and P(0)(OR3)(OR4);
R 2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Cj-C6 alkyl, dicarboxy halo C]-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR R4, NR3S02NHR4, and P(0)(OR3)(OR4); wherein each of said R2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono C]-C6 alkyl, carboxy Cj-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
wherein R3-R5 are independently hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C4 cycloalkyl, C3-C4 cycloalkenyl, C6-C20 aryl Q- alkyl, C6-C2o aryl, heterocyclyl, or heterocyclyl alkyl;
X is a bond, S, O, or NH;
Y is S or NH;
Z is CH or N;
or a pharmaceutically acceptable salt thereof.
[0053] In embodiments of the compounds or salts of the above methods, X is a bond, S or NH.
[0054] In embodiments of the compounds or salts of the above methods, R1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl,
arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono CpC6 alkyl, carboxy C]-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo C]-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , and S02R .
[0055] In embodiments of the compounds or salts of the above methods, R1 is alkyl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, and S02R3.
[0056] In embodiments of the compounds or salts of the above methods, R1 is alkyl, aryl, arylalkyl, heterocyclyl, or heterocyclylalkyl, optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, and S02R3.
[0057] In embodiments of the compounds or salts of the above methods, R1 is selected from the group consisting of alkyl, aryl, arylalkyl, pyrazolyl, thiazolyl, triazolyl, tetrazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyridinylmethyl, pyrimidinyl, furanyl, triazolopyridinyl, benzimidazolyl, and benzothiazolyl, each of which is optionally substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, and S02R3.
[0058] In embodiments of the compounds or salts of the above methods, R1 is selected from the group consisting of alkyl, aryl, arylalkyl, pyrazolyl, thiazolyl, triazolyl, tetrazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyridinylmethyl, pyrimidinyl, furanyl, triazolopyridinyl,
benzimidazolyl, and benzothiazolyl, each of which is optionally substituted with a substituent selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, and trialkylamino.
[0059] In embodiments of the compounds or salts of the above methods, R1 is selected from the group consisting of alkyl, aryl, arylalkyl, pyrazolyl, thiazolyl, triazolyl, tetrazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyridinylmethyl, pyrimidinyl, furanyl, triazolopyridinyl, benzimidazolyl, and benzothiazolyl, each of which is optionally substituted with an alkyl, hydroxy, or amino substituent.
[0060] In embodiments of the compounds or salts of the above methods, -X-R1 is selected from the group consisting of:


[0061] In embodiments of the compounds or salts of the above methods, Y is S.
[0062] In any of the embodiments of the compounds or salts of the above methods, Z is CH.
[0063] In any of the embodiments of the compounds or salts of the above methods, X is S.
[0064] In any of the embodiments of the compounds or salts of the above methods, R is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Q-Q alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Q-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4,
S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5,
NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4); wherein each of said R2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl,
phosphono Cj-C alkyl, carboxy Cj-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4).
[0065] In any of the embodiments of the compounds or salts of the above methods, R is selected from the group consisting of aryl, heterocyclyl, aryl heterocyclyl, or aryl
heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl
3 2
aryl, are fused or unfused to each other, halo, and S02R ; wherein each of said R groups aryl, aryl heterocyclyl, and aryl heterocyclyl aryl are unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, and an anionic group.
[0066] In embodiments of the compounds or salts of the above methods, R2 is selected from the group consisting of phenyl, pyrimidinyl, thiophenyl, pyrrolyl, benzofuranyl, naphthyl, 4-oxo-4H-chromenyl, dibenzothiophenyl, bromo, and iodo, each of which other than bromo and iodo, is optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Q-Q alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, and an anionic group.
[0067] In embodiments of the compounds or salts of the above methods, R is selected from the group consisting of:


[0068] In an embodiment of the compound of formula (I) or in any of the methods above, R2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl
heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Q-Q alkyl, carboxy Ci-C6 alkyl, dicarboxy C]-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5,
NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4); wherein each of said R2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4,
NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4).
[0069] In embodiments of the above methods, the cell is in an animal. In embodiments of the above methods, the animal is a human.
[0070] In accordance with an embodiment, the cell is in an animal. The term "animal" refers to any member of the animal kingdom. In embodiments, "animal" refers to a human at any stage of development. In embodiments, "animal" includes mammals, birds, reptiles, amphibians, fish, and worms. In certain embodiments, the non-human animal is a mammal, e.g., a rodent, a mouse, a rat, a rabbit, a monkey, a dog, a cat, a sheep, cattle, a primate, or a pig. The animal may also be a transgenic animal, genetically engineered animal, or a clone.
[0071] Preferably, the subject is a mammal. For purposes of the present invention, mammals include, but are not limited to, the order Rodentia, such as mice, and the order Logomorpha, such as rabbits. It is preferred that the mammals are from the order Carnivora, including Felines (cats) and Canines (dogs). It is more preferred that the mammals are from the order Artiodactyla, including Bovines (cows) and Swines (pigs) or of the order
Perssodactyla, including Equines (horses). It is most preferred that the mammals are of the order Primates, Ceboids, or Simioids (monkeys) or of the order Anthropoids (humans and apes). An especially preferred mammal is the human. Furthermore, the subject can be the unborn offspring of any of the forgoing hosts, especially mammals (e.g., humans), in which case any screening of the subject or cells of the subject, or administration of compounds to the subject or cells of the subject, can be performed in vitro. In accordance with an embodiment, the animal is a human.
[0072] Referring now to terminology used generically herein, the term "alkyl" means a straight-chain or branched alkyl substituent containing from, for example, 1 to about 12 carbon atoms, from 1 to about 6 carbon atoms, from 1 to 4 carbon atoms, or from 1 to 3 carbon atoms. Examples of such substituents include methyl, ethyl, propyl, isopropyl, n- butyl, sec-butyl, isobutyl, fcri-butyl, pentyl, isoamyl, hexyl, and the like.
[0073] The term "alkylene," as used herein, means a straight-chain or branched alkyl substituent containing from, for example, 1 to about 12 carbon atoms, from 1 to about 6 carbon atoms, from 1 to 3 carbon atoms, and is optionally connected to one, two or more substituents at two or more different positions on the alkylene group.
[0074] The term "alkenyl," as used herein, means a linear alkenyl substituent containing at least one carbon-carbon double bond and from, for example, about 2 to about 12 carbon atoms (branched alkenyls are about 3 to about 12 carbons atoms), from about 2 to about 6 carbon atoms (branched alkenyls are preferably from about 3 to about 7 carbon atoms), or from about 3 to about 4 carbon atoms. Examples of such substituents include vinyl, propenyl, isopropenyl, n-butenyl, sec-butenyl, isobutenyl, tert-butenyl, pentenyl, isopentenyl, hexenyl, and the like.
[0075] The term "alkenylene," as used herein, means a straight-chain or branched alkenyl substituent containing from, for example, 2 to about 12 carbon atoms, 2 to 6 carbon atoms, or from 2 to about 4 carbon atoms, and is connected to one, two or more substituents at two or more different positions on the alkenylene group.
[0076] The term "alkynyl," as used herein, means a linear alkynyl substituent containing at least one carbon-carbon triple bond and from, for example, 2 to about 12 carbon atoms (branched alkynyls are about 3 to about 13 carbons atoms), from 2 to about 6 carbon atoms (branched alkynyls are preferably from about 3 to about 7 carbon atoms), or from about 2 to about 4 carbon atoms. Examples of such substituents include ethynyl, propynyl, isopropynyl, rc-butynyl, sec-butynyl, isobutynyl, ierZ-butynyl, pentynyl, isopentynyl, hexynyl, and the like.
[0077] The term "alkynylene," as used herein, means a straight-chain or branched alkynyl substituent containing from, for example, 2 to about 12 carbon atoms, from 2 to about 6 carbon atoms, or from 2 to about 4 carbon atoms, and is connected to two or more substituents at two or more different positions on the alkynylene group.
[0078] The term "cycloalkyl," as used herein, means a cyclic alkyl substituent containing from, for example, about 3 to about 8 carbon atoms, from about 4 to about 7 carbon atoms, or from about 4 to about 6 carbon atoms. Examples of such substituents include cyclopropyl,
cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, and the like. The term
"cycloalkenyl," as used herein, means the same as the term "cycloalkyl," however one or more double bonds are present. Examples of such substituents include cyclopentenyl and cyclohexenyl. The cyclic alkyl groups may be unsubstituted or further substituted with alky] groups such as methyl groups, ethyl groups, and the like.
[0079] The term "aryl" refers to an unsubstituted or substituted aromatic carbocyclic moiety, as commonly understood in the art, and includes monocyclic and polycyclic aromatics such as, for example, phenyl, biphenyl, naphthyl, anthracenyl, pyrenyl, and the like. An aryl moiety generally contains from, for example, 6 to 30 carbon atoms, preferably from 6 to 18 carbon atoms, more preferably from 6 to 14 carbon atoms and most preferably from 6 to 10 carbon atoms. It is understood that the term aryl includes carbocyclic moieties that are planar and comprise 4n+2 π electrons, according to Huckel's Rule, wherein n = 1, 2, or 3.
[0080] The term "heteroaryl" refers to an aromatic group containing one or more heteroatoms selected from the group consisting of O, N, P, and S and combinations thereof. The heteroaryl group can be 4 to 7 membered ring. Non-limiting examples of heteroaryl groups include furanyl, thiopheneyl, pyrrolyl, pyrazolyl, imidazolyl, 1 ,2,3-triazolyl, 1 ,2,4- triazolyl, isoxazolyl, oxazolyl, isothiazolyl, thiazolyl, pyridinyl, pyrimidinyl, pyrazinyl, triazinyl, benzofuranyl, benzothiopheneyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl, benzoxazolinyl, benzothiazolinyl, and quinazolinyl.
[0081] The term "heterocyclyl," as used herein, refers to a monocyclic or bicyclic 5-, 6-, or 7-membered ring system containing one or more heteroatoms selected from the group consisting of O, N, P, S, and combinations thereof. The heterocyclyl group can be any suitable heterocyclyl group and can be an aliphatic heterocyclyl group, an aromatic heterocyclyl group, or a combination thereof. The heterocyclyl group can be a monocyclic heterocyclyl group or a bicyclic heterocyclyl group. Bicyclic heterocyclyl groups include monocylic heterocyclyl rings fused to a C6-Cio aryl ring. When the heterocyclyl group is a bicyclic heterocyclyl group, both ring systems can be aliphatic or aromatic, or one ring system can be aromatic and the other ring system can be aliphatic as in, for example, dihydrobenzofuran. Preferably, the heterocyclyl group is an aromatic heterocyclyl or heteroaryl group.
[0082] The heterocyclyl or heteroaryl group is optionally substituted with 1 , 2, 3, 4, or 5 substituents as recited herein, wherein the optional substituent can be present at any open position on the heterocyclyl or heteroaryl group.
[0083] Whenever a range of the number of atoms in a structure is indicated (e.g., a
C1-C12, Ci-Cg, Ci-C6, C1 -C4, or C2-C12, C2-C8, C2-C6, C2-C4 alkyl, alkenyl, alkynyl, etc.), it is specifically contemplated that any sub-range or individual number of carbon atoms falling within the indicated range also can be used. Thus, for instance, the recitation of a range of 1- 8 carbon atoms (e.g., Ci-C8), 1-6 carbon atoms (e.g., Ci -C6), 1-4 carbon atoms (e.g., Ci-C4), 1-3 carbon atoms (e.g., Ci-C3), or 2-8 carbon atoms (e.g., C2-C8) as used with respect to any chemical group (e.g., alkyl, alkylamino, etc.) referenced herein encompasses and specifically describes 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 1 1, and/or 12 carbon atoms, as appropriate, as well as any sub-range thereof (e.g., 1 -2 carbon atoms, 1-3 carbon atoms, 1-4 carbon atoms, 1-5 carbon atoms, 1-6 carbon atoms, 1-7 carbon atoms, 1-8 carbon atoms, 1-9 carbon atoms, 1-10 carbon atoms, 1-1 1 carbon atoms, 1-12 carbon atoms, 2-3 carbon atoms, 2-4 carbon atoms, 2-5 carbon atoms, 2-6 carbon atoms, 2-7 carbon atoms, 2-8 carbon atoms, 2-9 carbon atoms, 2-10 carbon atoms, 2-1 1 carbon atoms, 2-12 carbon atoms, 3-4 carbon atoms, 3-5 carbon atoms, 3- 6 carbon atoms, 3-7 carbon atoms, 3-8 carbon atoms, 3-9 carbon atoms, 3-10 carbon atoms, 3-11 carbon atoms, 3-12 carbon atoms, 4-5 carbon atoms, 4-6 carbon atoms, 4-7 carbon atoms, 4-8 carbon atoms, 4-9 carbon atoms, 4-10 carbon atoms, 4-11 carbon atoms, and/or 4- 12 carbon atoms, etc., as appropriate). Similarly, the recitation of a range of 6-10 carbon atoms (e.g., C6-Cio) as used with respect to any chemical group (e.g., aryl) referenced herein encompasses and specifically describes 6, 7, 8, 9, and/or 10 carbon atoms, as appropriate, as well as any sub-range thereof (e.g., 6-10 carbon atoms, 6-9 carbon atoms, 6-8 carbon atoms, 6-7 carbon atoms, 7-10 carbon atoms, 7-9 carbon atoms, 7-8 carbon atoms, 8-10 carbon atoms, and/or 8-9 carbon atoms, etc., as appropriate).
[0084] The term "halo" or "halogen," as used herein, means a substituent selected from Group VIIA, such as, for example, fluorine, bromine, chlorine, and iodine.
[0085] Any of the compounds of the invention can be in any stereochemical
conformation, i.e., R, S, or R/S form. Thus, all possible enantiomers and diastereoisomers are encompassed by the present invention. Any of the amino acid fragments can be in the natural form or synthetic form, i.e., D, L, or D/L form.
[0086] The compounds of the invention in any of the embodiments can be prepared by the reaction schemes illustrated in Schemes 1 -3.
[0087] The phrase "pharmaceutically acceptable salt" is intended to include nontoxic salts synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two. Generally, nonaqueous media such as ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred. Lists of suitable salts are found in Remington 's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA, 1990, p. 1445, and Journal of Pharmaceutical Science, 66, 2-19 (1977).
[0088] Suitable bases include inorganic bases such as alkali and alkaline earth metal bases, e.g., those containing metallic cations such as sodium, potassium, magnesium, calcium and the like. Non-limiting examples of suitable bases include sodium hydroxide, potassium hydroxide, sodium carbonate, and potassium carbonate. Suitable acids include inorganic acids such as hydrochloric acid, hydrobromic acid, hydriodic acid, sulfuric acid, phosphoric acid, and the like, and organic acids such as p-toluenesulfonic, methanesulfonic acid, ethane sulfonic acid, benzenesulfonic acid, oxalic acid, p-bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic acid, acetic acid, propionic acid, glycolic acid, glucaric acid, glucuronic acid, citric acid, gluconic acid, hydroxymaleic acid, fumaric acid, maleic acid, malic acid, 4-aminosalicylic acid, cinnamic acid, acetoxybenzoic acid, succinic acid, tartaric acid, ascorbic acid, fatty acids, long chain fatty acids, salicylic acid, alpha amino acids, 2-hydroxymethane sulfonic acid, ethane 1 ,2-disulfonic acid, naphthalene-2-sulfonic acid, 4-methylbenzene sulfonic acid, sulfo acids, phospho acids, embonic acid, nicotinic acid, N-substituted sulfamic acids. Preferred pharmaceutically acceptable salts of inventive compounds having an acidic moiety include sodium and potassium salts. Preferred pharmaceutically acceptable salts of inventive compounds having a basic moiety (e.g., a dimethylaminoalkyl group) include hydrochloride and hydrobromide salts. The compounds of the present invention containing an acidic or basic moiety are useful in the form of the free base or acid or in the form of a pharmaceutically acceptable salt thereof.
[0089] It should be recognized that the particular counterion forming a part of any salt of this invention is usually not of a critical nature, so long as the salt as a whole is
pharmacologically acceptable and as long as the counterion does not contribute undesired qualities to the salt as a whole.
[0090] It is further understood that the above compounds and salts may form solvates, or exist in a substantially uncomplexed form, such as the anhydrous form. As used herein, the term "solvate" refers to a molecular complex wherein the solvent molecule, such as the crystallizing solvent, is incorporated into the crystal lattice. When the solvent incorporated in the solvate is water, the molecular complex is called a hydrate. Pharmaceutically acceptable solvates include hydrates, alcoholates such as methanolates and ethanolates, acetonitrilates and the like. These compounds can also exist in polymorphic forms.
[0091] The compounds of the invention may also be administered as prodrugs. The term "prodrug" denotes a derivative of a compound, which derivative, when administered to warm-blooded animals, e.g. humans, is converted into the compound (drug). The enzymatic and/or chemical hydrolytic cleavage of the compounds of the present invention occurs in such a manner that the proven drug form (parent carboxylic acid drug) is released, and the moiety or moieties split off remain nontoxic or are metabolized so that nontoxic metabolic products are produced. For example, a carboxylic acid group can be esterified, e.g., with a methyl group or ethyl group to yield an ester. When an ester is administered to a subject, the ester is cleaved, enzymatically or non-enzymatically, reductively, oxidatively, or
hydrolytically, to reveal the anionic group. An anionic group can be esterified with moieties (e.g., acyloxymethyl esters) which are cleaved to reveal an intermediate compound which subsequently decomposes to yield the active compound.
[0092] The prodrugs can be prepared in situ during the isolation and purification of the compounds, or by separately reacting the purified compound with a suitable derivatizing agent. For example, hydroxy groups can be converted into esters via treatment with a carboxylic acid in the presence of a catalyst. Examples of cleavable alcohol prodrug moieties include substituted or unsubstituted, branched or unbranched lower alkyl ester moieties, e.g., ethyl esters, lower alkenyl esters, di-lower alkylamino lower-alkyl esters, e.g.,
dimethylaminoethyl ester, acylamino lower alkyl esters, acyloxy lower alkyl esters (e.g., pivaloyloxymethyl ester), aryl esters, e.g., phenyl ester, aryl-lower alkyl esters, e.g., benzyl ester, optionally substituted, e.g., with methyl, halo, or methoxy substituents aryl and aryl- lower alkyl esters, amides, lower-alkyl amides, di-lower alkyl amides, and hydroxy amides.
[0093] The present invention is further directed to a pharmaceutical composition comprising a pharmaceutically acceptable carrier and at least one compound or salt described herein.
[0094] It is preferred that the pharmaceutically acceptable carrier be one that is chemically inert to the active compounds and one that has no detrimental side effects or toxicity under the conditions of use.
[0095] The choice of carrier will be determined in part by the particular compound of the present invention chosen, as well as by the particular method used to administer the composition.
[0096] The compound or pharmaceutical composition can be administered by any suitable route, oral or parenteral, including intravenous, subcutaneous, intraarterial, intraperitoneal, ophthalmic, intramuscular, buccal, rectal, vaginal, intraorbital, intracerebral, intracranial, intraspinal, intraventricuclar, intrathecal, intracisternal, intracapsular, intrapulmonary, intranasal, transmucosal, transdermal, or via inhalation. Accordingly, there is a wide variety of suitable formulations of the pharmaceutical composition of the present invention. The formulations may also be applied topically.
[0097] For example, in an embodiment, the invention provides compositions for parenteral administration that comprise a solution or suspension of the inventive compound or salt dissolved or suspended in an acceptable carrier suitable for parenteral administration, including aqueous and non-aqueous isotonic sterile injection solutions.
[0098] Overall, the requirements for effective pharmaceutical carriers for parenteral compositions are well known to those of ordinary skill in the art. See, e.g., Banker and Chalmers, eds., Pharmaceutics and Pharmacy Practice, J. B. Lippincott Company,
Philadelphia, pp. 238-250 (1982), and Toissel, ASHP Handbook on Injectable Drugs, 4th ed., pp. 622-630 (1986). Such solutions can contain anti-oxidants, buffers, bacteriostats, and solutes that render the formulation isotonic with the blood of the intended recipient, and aqueous and non-aqueous sterile suspensions that can include suspending agents, solubilizers, thickening agents, stabilizers, and preservatives. The compound or salt of the present invention may be administered in a physiologically acceptable diluent in a pharmaceutical carrier, such as a sterile liquid or mixture of liquids, including water, saline, aqueous dextrose and related sugar solutions, an alcohol, such as ethanol, isopropanol, or hexadecyl alcohol, glycols, such as propylene glycol or polyethylene glycol, dimethylsulfoxide, glycerol ketals, such as 2,2-dimethyl-l ,3-dioxolane-4-methanol, ethers, such as poly(ethyleneglycol) 400, an oil, a fatty acid, a fatty acid ester or glyceride, or an acetylated fatty acid glyceride with or without the addition of a pharmaceutically acceptable surfactant, such as a soap or a detergent, suspending agent, such as pectin, carbomers, methylcellulose,
hydroxypropylmethylcellulose, or carboxymethylcellulose, or emulsifying agents and other pharmaceutical adjuvants.
[0099] Oils useful in parenteral formulations include petroleum, animal, vegetable, or synthetic oils. Specific examples of oils useful in such formulations include peanut, soybean, sesame, cottonseed, corn, olive, petrolatum, and mineral. Suitable fatty acids for use in parenteral formulations include oleic acid, stearic acid, and isostearic acid. Ethyl oleate and isopropyl myristate are examples of suitable fatty acid esters.
[0100] Suitable soaps for use in parenteral formulations include fatty alkali metal, ammonium, and triethanolamine salts, and suitable detergents include (a) cationic detergents such as, for example, dimethyl dialkyl ammonium halides, and alkyl pyridinium halides, (b) anionic detergents such as, for example, alkyl, aryl, and olefin sulfonates, alkyl, olefin, ether, and monoglyceride sulfates, and sulfosuccinates, (c) nonionic detergents such as, for example, fatty amine oxides, fatty acid alkanolamides, and polyoxyethylenepolypropylene copolymers, (d) amphoteric detergents such as, for example, alkyl-beta-aminopropionates, and 2-alkyl-imidazoline quaternary ammonium salts, and (e) mixtures thereof.
[0101] The parenteral formulations can contain preservatives and buffers. In order to minimize or eliminate irritation at the site of injection, such compositions may contain one or more nonionic surfactants having a hydrophile-lipophile balance (HLB) of from about 12 to about 17. The quantity of surfactant in such formulations will typically range from about 5 to about 15% by weight. Suitable surfactants include polyethylene sorbitan fatty acid esters, such as sorbitan monooleate and the high molecular weight adducts of ethylene oxide with a hydrophobic base, formed by the condensation of propylene oxide with propylene glycol. The parenteral formulations can be presented in unit-dose or multi-dose sealed containers, such as ampoules and vials, and can be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid excipient, for example, water, for injections, immediately prior to use. Extemporaneous injection solutions and suspensions can be prepared from sterile powders, granules, and tablets of the kind previously described.
[0102] Topical formulations, including those that are useful for transdermal drug release, are well-known to those of skill in the art and are suitable in the context of the invention for application to skin. Topically applied compositions are generally in the form of liquids, creams, pastes, lotions and gels. Topical administration includes application to the oral mucosa, which includes the oral cavity, oral epithelium, palate, gingival, and the nasal mucosa. In some embodiments, the composition contains at least one active component and a
suitable vehicle or carrier. It may also contain other components, such as an anti-irritant. The carrier can be a liquid, solid or semi-solid. In embodiments, the composition is an aqueous solution. Alternatively, the composition can be a dispersion, emulsion, gel, lotion or cream vehicle for the various components. In one embodiment, the primary vehicle is water or a biocompatible solvent that is substantially neutral or that has been rendered substantially neutral. The liquid vehicle can include other materials, such as buffers, alcohols, glycerin, and mineral oils with various emulsifiers or dispersing agents as known in the art to obtain the desired pH, consistency and viscosity. It is possible that the compositions can be produced as solids, such as powders or granules. The solids can be applied directly or dissolved in water or a biocompatible solvent prior to use to form a solution that is substantially neutral or that has been rendered substantially neutral and that can then be applied to the target site. In embodiments of the invention, the vehicle for topical application to the skin can include water, buffered solutions, various alcohols, glycols such as glycerin, lipid materials such as fatty acids, mineral oils, phosphoglycerides, collagen, gelatin and silicone based materials.
[0103] Formulations suitable for oral administration can consist of (a) liquid solutions, such as a therapeutically effective amount of the inventive compound dissolved in diluents, such as water, saline, or orange juice, (b) capsules, sachets, tablets, lozenges, and troches, each containing a predetermined amount of the active ingredient, as solids or granules, (c) powders, (d) suspensions in an appropriate liquid, and (e) suitable emulsions. Liquid formulations may include diluents, such as water and alcohols, for example, ethanol, benzyl alcohol, and the polyethylene alcohols, either with or without the addition of a
pharmaceutically acceptable surfactant, suspending agent, or emulsifying agent. Capsule forms can be of the ordinary hard- or soft-shelled gelatin type containing, for example, surfactants, lubricants, and inert fillers, such as lactose, sucrose, calcium phosphate, and corn starch. Tablet forms can include one or more of lactose, sucrose, mannitol, corn starch, potato starch, alginic acid, microcrystalline cellulose, acacia, gelatin, guar gum, colloidal silicon dioxide, croscarmellose sodium, talc, magnesium stearate, calcium stearate, zinc stearate, stearic acid, and other excipients, colorants, diluents, buffering agents, disintegrating agents, moistening agents, preservatives, flavoring agents, and pharmacologically compatible excipients. Lozenge forms can comprise the active ingredient in a flavor, usually sucrose and acacia or tragacanth, as well as pastilles comprising the active ingredient in an inert base,
such as gelatin and glycerin, or sucrose and acacia, emulsions, gels, and the like containing, in addition to the active ingredient, such excipients as are known in the art.
[0104] The compound or salt of the present invention, alone or in combination with other suitable components, can be made into aerosol formulations to be administered via inhalation. The compounds are preferably supplied in finely divided form along with a surfactant and propellant. Typical percentages of active compound are 0.01%-20% by weight, preferably 1%-10%. The surfactant must, of course, be nontoxic, and preferably soluble in the propellant. Representative of such surfactants are the esters or partial esters of fatty acids containing from 6 to 22 carbon atoms, such as caproic, octanoic, lauric, palmitic, stearic, linoleic, linolenic, olesteric and oleic acids with an aliphatic polyhydric alcohol or its cyclic anhydride. Mixed esters, such as mixed or natural glycerides may be employed. The surfactant may constitute 0.1%-20% by weight of the composition, preferably 0.25%-5%. The balance of the composition is ordinarily propellant. A carrier can also be included as desired, e.g., lecithin for intranasal delivery. These aerosol formulations can be placed into acceptable pressurized propellants, such as dichlorodifluoromethane, propane, nitrogen, and the like. They also may be formulated as pharmaceuticals for non-pressured preparations, such as in a nebulizer or an atomizer. Such spray formulations may be used to spray mucosa.
[0105] Additionally, the compound or salt of the present invention may be made into suppositories by mixing with a variety of bases, such as emulsifying bases or water-soluble bases. Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams, or spray formulas containing, in addition to the active ingredient, such carriers as are known in the art to be appropriate.
[0106] It will be appreciated by one of ordinary skill in the art that, in addition to the aforedescribed pharmaceutical compositions, the compound or salt of the present invention may be formulated as inclusion complexes, such as cyclodextrin inclusion complexes, or liposomes. Liposomes serve to target the compounds to a particular tissue, such as lymphoid tissue or cancerous hepatic cells. Liposomes can also be used to increase the half-life of the inventive compound. Liposomes useful in the present invention include emulsions, foams, micelles, insoluble monolayers, liquid crystals, phospholipid dispersions, lamellar layers and the like. In these preparations, the active agent to be delivered is incorporated as part of a liposome, alone or in conjunction with a suitable chemotherapeutic agent. Thus, liposomes filled with a desired inventive compound or salt thereof, can be directed to the site of a specific tissue type, hepatic cells, for example, where the liposomes then deliver the selected
compositions. Liposomes for use in the invention are formed from standard vesicle-forming lipids, which generally include neutral and negatively charged phospholipids and a sterol, such as cholesterol. The selection of lipids is generally guided by consideration of, for example, liposome size and stability of the liposomes in the blood stream. A variety of methods are available for preparing liposomes, as described in, for example, Szoka et al., Ann. Rev. Biophys. Bioeng., 9, 467 (1980), and U.S. Patents 4,235,871, 4,501 ,728, 4,837,028, and 5,019,369. For targeting to the cells of a particular tissue type, a ligand to be
incorporated into the liposome can include, for example, antibodies or fragments thereof specific for cell surface determinants of the targeted tissue type. A liposome suspension containing a compound or salt of the present invention may be administered intravenously, locally, topically, etc. in a dose that varies according to the mode of administration, the agent being delivered, and the stage of disease being treated.
[0107] "Treatment" or "treating" refers to a therapeutic intervention that ameliorates a sign or symptom of a disease or pathological condition after it has begun to develop. As used herein, the term "ameliorating," with reference to a disease or pathological condition, refers to any observable beneficial effect of the treatment. The beneficial effect can be evidenced, for example, by a delayed onset of clinical symptoms of the disease in a susceptible subject, a reduction in severity of some or all clinical symptoms of the disease, a slower progression of the disease, an improvement in the overall health or well-being of the subject, or by other parameters well known in the art that are specific to the particular disease. The observable benefit of a treatment can be a low level of benefit of about 10%, about 20%, or about 30%, or about 40%> of the full benefit, or it can be a medium level benefit of about 50%, 60%, 70%, or about 80%, or the benefit can also be a high level decrease of about 90%>, about 95%>, about 99%, or about 99.9%, or complete cure.
[0108] "Inhibiting" means decreasing the effect or activity of a given entity, which can be a low level of decrease of about 10%, about 20%, or about 30%, or about 40% of the full effect or activity, or the decrease can be a medium level decrease of about 50%, 60%, 70%, or about 80%, or the decrease can also be a high level decrease of about 90%, about 95%, about 99%, or about 99.9%, or complete elimination of the effect or activity.
[0109] "Preventing" refers to reducing the probability of developing a disease or disorder in a healthy animal or in an animal at risk for developing such disease or disorder or delaying the onset of the the disease or disorder.
[0110] "Modulating" can refer to decreasing or increasing, and in an embodiment, refers to decreasing.
[0111] The therapeutically effective amount of the compound or compounds
administered can vary depending upon the desired effects and the factors noted above.
Typically, dosages will be between 0.01 mg/kg and 250 mg/kg of the subject's body weight, and more typically between about 0.05 mg/kg and 100 mg/kg, such as from about 0.2 to about 80 mg/kg, from about 5 to about 40 mg/kg or from about 10 to about 30 mg/kg of the subject's body weight. Thus, unit dosage forms can be formulated based upon the suitable ranges recited above and the subject's body weight. The term "unit dosage form" as used herein refers to a physically discrete unit of therapeutic agent appropriate for the subject to be treated.
[0112] Alternatively, dosages are calculated based on body surface area and from about 1 mg/m2 to about 200 mg/m2, such as from about 5 mg/m2 to about 100 mg/m2 will be administered to the subject per day. In particular embodiments, administration of the therapeutically effective amount of the compound or compounds involves administering to the subject from about 5 mg/m2 to about 50 mg/m2, such as from about 10 mg/m2 to about 40 mg/m2 per day. It is currently believed that a single dosage of the compound or compounds is suitable, however a therapeutically effective dosage can be supplied over an extended period of time or in multiple doses per day. Thus, unit dosage forms also can be calculated using a subject's body surface area based on the suitable ranges recited above and the desired dosing schedule.
[0113] In an embodiment, the compounds or salts thereof described herein for methods of preventing a neurodegenerative disease or disorder can be about 0.001 to about 1 mg/kg body weight of the subject , for example, about 0.001 mg, 0.002 mg, 0.005 mg, 0.010 mg, 0.015 mg, 0.020 mg, 0.025 mg, 0.050 mg, 0.075 mg, 0.1 mg, 0.15 mg, 0.2 mg, 0.25 mg, 0.5 mg, 0.75 mg, or 1 mg/kg body weight per day. In accordance with an embodiment, the dose of the compounds or salts described herein for methods of treating the disorder can be about 1 to about 1000 mg/kg body weight of the subject being treated per day, for example, about 1 mg, 2 mg, 5 mg, 10 mg, 15 mg, 0.020 mg, 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 250 mg, 500 mg, 750 mg, or 1000 mg/kg body weight per day.
[0114] The following examples further illustrate the invention but, of course, should not be construed as in any way limiting its scope.
EXAMPLE 1
[0115] General Materials and Methods: All commercially available reagents and solvents were purchased and used without further purification. All microwave reactions were carried out in a sealed microwave vial equipped with a magnetic stir bar and heated in a Biotage Initiator Microwave Synthesizer. All compounds for biological testing were purified using a Waters semi-preparative HPLC equipped with a Phenomenex Luna® CI 8 reverse phase (5 micron, 30 x 75 mm) column having a flow rate of 45 mL/min. The mobile phase was a mixture of acetonitrile and H20 each containing 0.1% trifluoroacetic acid. During purification, a gradient of 30% to 80% acetonitrile over 8 minutes was used with fraction collection triggered by UV detection (220 nM). Pure fractions passed through PL-HC03 MP SPE (Varian) to remove trifluoroacetic acid and concentrated under vacuum on a lyophilizer. Ή spectra were recorded using an Inova 400 (100) MHz spectrometer (Varian).
[0116] General Synthetic Procedures
[0117] The following general procedures were used to synthesize compounds, or intermediates thereof, according to embodiments of the invention:
[0118] 6-bromothieno [2,3-d] pyrimidin-4(3H)-one (4)

[0119] To thieno[2,3-d]pyrimidin-4(3H)-one (5 g, 32.9 mmol, 1.0 equiv) in 50 mL acetic acid was added bromine (5 mL, 2.95 equiv, 97 mmol). The mixture stirred at r.t. for 4 h. The resulting solid was collected by filtration, washed with water, and dried in vacuo to give 6- bromothieno[2,3-d]pyrimidin-4(3H)-one (7.02 g, 92 % yield) as a tan solid. ]H NMR (400 MHz, DMSO- 6) δ ppm 12.61 (br. s., 1 H), 8.1 1 (d, J=3.52 Hz, 1 H), 7.47 - 7.56 (m, 1 H).
[0120] 6-bromo-4-chlorothieno[2,3-d]pyrimidine (5)

[0121] 6-Bromothieno[2,3-d]pyrimidin-4(3H)-one (7.02 g, 30.4 mmol, 1.0 equiv) was taken up in 100 mL POCI3 and refluxed for 6 h. Upon completion, the reaction was concentrated in vacuo and the residue was taken up in 10 mL acetonitrile and heated to reflux for 10 min. The acetonitrile solution was then poured in a flask and cooled. The ppt was then filtered and washed with hexane to give 6-bromo-4-chlorothieno[2,3-d]pyrimidine (5.51
g, 72.7 % yield) as a tan solid. Ή NMR (400 MHz, CHLOROFORM-d) δ ppm 8.80 (s, 1 H), 7.47 (s, 1 H).
[0122] 5-bromo-4-chlorothieno[2,3-d rimidine (6)

[0123] To diisopropylamine (1.028 ml, 7.21 mmol, 1.8 equiv) in 10 mL THF at 0 °C was added n-butyl lithium (3.76 ml, 6.01 mmol, 1.5 equiv). After 1 h, the LDA solution was transferred to a solution of 6-bromo-4-chlorothieno[2,3-d]pyrimidine (1.0 g, 4.01 mmol, 1.0 equiv) in 35 mL THF at -78 °C under nitrogen. The solution stirred for 1 h at -78 °C after which a mixture of 1.25 mL water and 5 mL THF was added slowly. The mixture was then warmed to 0 °C, poured into 60 mL water, and extracted with dichloromethane. The combined organic extracts were then dried over Na2SC> , filtered, and concentrated in vacuo to give a yellow solid which was chromatographed with 20% EtOAc/Hexanes gradient elution to give 5-bromo-4-chlorothieno[2,3-d]pyrimidine (671 mg, 67.1 % yield) as a tan solid. Ή NMR (400 MHz, chloroform- ) δ ppm 8.85 (s, 1 H), 7.64 (s, 1 H).
[0124] 4-chloro-5-iodothieno[2,3-d] rimidine (7)

[0125] Isopropylmagnesium chloride (1.614 ml, 3.23 mmol, 1.2 equiv) was added slowly to 5-bromo-4-chlorothieno[2,3-d]pyrimidine (671 mg, 2.69 mmol, 1.0 equiv) in 10 mL THF at 0 °C under nitrogen. The mixture was allowed to stir for 15 min after which a solution of iodine (819 mg, 3.23 mmol, 1.2 equiv) in 1.5 mL THF was added. The mixture stirred for 20 min. Upon completion, the reaction was quenched with sat. NH4CI. It was then extracted with EtOAc. The combined organic extracts were then washed with Na2S2C>3, water, and brine, dried over MgSC^and concentrated to give 4-chloro-5-iodothieno[2,3-d]pyrimidine
(496 mg, 62.2 % yield) as a tan solid. The crude product was taken on without further purification.
[0126] General procedure for the synthesis of 4-chloro-5-aryl-thieno[2,3- d]pyrimidine (8)

[0127] A microwave vial was charged with 4-chloro-5-iodothieno[2,3-d]pyrimidine (1.0 mmol, 1.0 equiv), arylboronic acid (1.3 mmol, 1.3 equiv), sodium carbonate (1.3 mmol, 1.3 equiv), and Dichlorobis(triphenylphosphine)-palladium(II) (0.05 mmol, 0.05 equiv). To it was added 6 mL DME followed by 0.4 mL water. The mixture was heated to 120 °C for 20 min in the microwave. Upon completion the mixture was taken up in water and EtOAc. The layers were separated and the aqueous layer was extracted with EtOAc. The combined organic extracts were washed with water and brine, dried over MgS04, and concentrated in vacuo to give crude 4-chloro-5-arylthieno[2,3-d]pyrimidine. The crude product was taken on without further purification.
[0128] 4-chloro-5-(3-(methyIsulfon I)phenyI)thieno[2,3-d]pyrimidine (8a)

[0129] A microwave vial was charged with 4-chloro-5-iodothieno[2,3-d]pyrimidine (265 mg, 0.894 mmol, 1.0 equiv), (3-Methylsulfonylphenyl)boronic acid (232 mg, 1.162 mmol, 1.3 equiv), sodium carbonate (123 mg, 1.162 mmol, 1.3 equiv), and
Dichlorobis(triphenylphosphine)-palladium(II) (31.4 mg, 0.045 mmol, 0.05 equiv). To it was added 6 mL DME followed by 0.4 mL water. The mixture was heated to 120 °C for 20 min in the microwave. Upon completion the mixture was taken up in water and EtOAc. The layers were separated and the aqueous layer was extracted with EtOAc. The combined organic extracts were washed with water and brine, dried over MgS04, and concentrated in vacuo to give crude 4-chloro-5-(3-(methylsulfonyl)phenyl)thieno[2,3-d]pyrimidine as a tan solid. The crude product was taken on without further purification.
[0130] General procedure for the synthesis of 5-aryl-4-(arylthio)thieno[2,3- d]pyrimidines (9)

[0132] 2-methyl-5-(5-(3-(methylsulfonyl)phenyl)thieno[2,3-d]pyrimidin-4-ylthio)- 1,3,4-oxadiazole

[0133] Ή NMR (400 MHz, DMSO-J6) δ ppm 8.81 (s, 1 H), 8.10 - 8.13 (m, 2 H), 8.06 (dt, J=7.83, 1.56 Hz, 1 H), 7.93 (dt, J=7.63, 1.37 Hz, 1 H), 7.81 (t, J=7.83 Hz, 1 H), 3.28 (s., 3 H), 2.53 (s, 3 H); LCMS: (electrospray +ve), mlz 405.0 (MH)+; HPLC: tR = 4.80 min, UV254 = 100%. HRMS (ESI): m/z calcd for Ci6H13N403S3 [M+H]+ 405.0145, found
405.0154.
[0134] 4-(l,3,4-thiadiazol-2-ylthio)-5-(3-(methylsulfonyl)phenyl)thieno[2,3- d]pyrimidine

[0135] H NMR (400 MHz, DMSO-J6) δ ppm 10.1 1 - 10.14 (m, 1 H), 9.29 - 9.35 (m, 1
H), 8.40 - 8.50 (m, 3 H), 8.26 - 8.31 (m, 1 H), 8.12 - 8.20 (m, 1 H), 3.55 - 3.62 (m, 3 H);
LCMS: (electrospray +ve), mlz 407.0 (MH)+; HPLC: tR = 4.92 min, UV254 = 100%. HRMS
(ESI): m/z calcd for C15Hi 1N402S4 [M+H]+ 406.9761 , found 406.9772.
[0136] 4-(2-methylfuran-3-ylthio)-5-(3-(methylsulfonyl)phenyl)thieno[2,3- d]pyrimidine

[0137] Ή NMR (400 MHz, DMSO-t¾ δ ppm 8.76 (s, 1 H), 8.10 (t, J=1.66 Hz, 1 H), 7.96 - 8.05 (m, 2 H), 7.91 (d, J=7.63 Hz, 1 H), 7.77 (t, J=7.83 Hz, 1 H), 7.62 (d, J=1.96 Hz, 1 H), 6.39 (d, J=1.96 Hz, 1 H), 3.26 (s, 3 H), 2.15 (s, 3 H); LCMS: (electrospray +ve), mlz 403.0 (MH)+; HPLC: tR = 5.74 min, UV254 = 100%. HRMS (ESI): m/z calcd for
Ci8Hi5N203S3 [M+H]+ 403.0240, found 403.0258.
[0138] 5-(3-(methylsulfonyl)phenyl)-4-(thiazol-2-ylthio)thieno[2,3-d]pyrimidine

[0139] Ή NMR (400 MHz, DMSO- ) δ ppm 8.90 (s, 1 H), 8.03 - 8.12 (m, 3 H), 7.98 (d, J=3.52 Hz, 1 H), 7.88 - 7.95 (m, 2 H), 7.80 (t, J=7.73 Hz, 1 H), 3.27 (s, 3 H); LCMS:
(electrospray +ve), mlz 406.0 (MH)+; HPLC: tR = 5.33 min, UV254 = 100%. HRMS (ESI): m/z calcd for Ci6Hi2N302S4 [M+H]+ 405.9807, found 405.9821.
[0140] 5-(3-(methylsulfonyl)phenyl)-4-(triazolylthio)thieno[2,3-d]pyrimidine

[0141] 1H NMR (400 MHz, DMSO- 6) δ ppm 8.70 (br. s., 1 H), 7.99 - 8.09 (m, 4 H), 7.90 (dt, J=7.73, 1.42 Hz, 1 H), 7.78 (t, J=7.73 Hz, 2 H), 3.26 (s, 3 H); LCMS: (electrospray +ve), mlz 390.0 (MH)+; HPLC: tR = 3.98 min, UV254 = 100%. HRMS (ESI): m/z calcd for Ci5Hi2N502S3 [M+H]+ 390.0148, found 390.0158.
[0142] 5-(3-(methylsulfonyl)phenyl)-4-(4-methylthiazol-2-ylthio)thieno[2,3- djpyrimidine

[0143] Ή NMR (400 MHz, DMSO-i 6) 5 ppm 8.91 (s, 1 H), 8.02 - 8.10 (m, 3 H), 7.90 (d, J=7.43 Hz, 1 H), 7.79 (t, J=7.63 Hz, 1 H), 7.51 (t, J=0.98 Hz, 1 H), 3.27 (s, 3 H), 2.31 (s, 3 H); LCMS: (electrospray +ve), mlz 420.0 (MH)+; HPLC: tR = 5.60 min, UV254 - 100%. HRMS (ESI): m/z calcd for C,7Hi4N302S4 [M+H]+ 419.9964, found 419.9978.
[0144] 5-(3-(methylsulfonyl)phen l)-4-(pyridin-4-ylthio)thieno[2,3-d]pyrimidine

[0145] Ή NMR (400 MHz, DMSO- 6) δ ppm 8.81 (s, 1 H), 8.52 - 8.59 (m, 2 H), 8.04 - 8.09 (m, 2 H), 8.01 (ddd, J=8.17, 1.52, 1.27 Hz, 1 H), 7.88 (dt, J=7.63, 1.37 Hz, 1 H), 7.74 (t, J=7.73 Hz, 1 H), 7.43 - 7.50 (m, 2 H), 3.26 (s, 3 H); LCMS: (electrospray +ve), mlz 400.0 (MH)+; HPLC: tR = 3.62 min, UV254 = 100%. HRMS (ESI): m/z calcd for d8Hi4N302S3 [M+H]+ 400.0243, found 400.0254.
[0146] 2-(5-(3-(methylsuIfonyl)phenyl)thieno[2,3-d]pyrimidin-4-yIthio)oxazoIo[4,5- b] pyridine

[0147] H NMR (400 MHz, DMSO-J6) δ ppm 8.83 (s, 1 H), 8.55 (dd, J=4.79, 1.47 Hz, 1
H), 8.18 (dd, J=8.22, 1.56 Hz, 1 H), 8.09 - 8.15 (m, 2 H), 8.00 - 8.06 (m, 1 H), 7.87 (ddd,
J=7.97, 1.42, 1.17 Hz, 1 H), 7.72 - 7.77 (m, 1 H), 7.49 (dd, J=8.22, 4.89 Hz, 1 H), 3.27 (s, 3
H); LCMS: (electrospray +ve), mlz 441.0 (MH)+; HPLC: tR = 4.87 min, UV254 = 100%.
HRMS (ESI): m/z calcd for C]9Hi3N403S3 [M+H]+ 441.0145, found 441.0156.
[0148] 4-(lH-benzo[d]imidazol-2-ylthio)-5-(3-(methylsulfonyl)phenyl)thieno[2,3- djpyrimidine

[0149] Ή NMR (400 MHz, DMSO-</6) δ ppm 8.76 (s, 1 H), 8.02 - 8.12 (m, 3 H), 7.93 (d, J=7.63 Hz, 1 H), 7.81 (t, J=7.73 Hz, 1 H), 7.54 (dd, J=5.97, 3.23 Hz, 2 H), 7.22 (dd, J=6.06, 3.13 Hz, 2 H), 3.28 (s, 3 H); LCMS: (electrospray +ve), mlz 439.0 (MH)+; HPLC: tR = 4.60 min, UV254 = 100%. HRMS (ESI): m/z calcd for C2oH15N402S3 [M+H]+ 439.0352, found 439.0366.
[0150] 4-(l-methyl-lH-tetrazol-5-ylthio)-5-(3-(methylsulfonyl)phenyl)thieno[2,3- djpyrimidine

[0151] H NMR (400 MHz, DMSO-<¾) δ ppm 8.73 (d, J=1.17 Hz, 1 H), 8.20 (d, J=1.37 Hz, 1 H), 8.1 1 (d, J=1.17 Hz, 1 H), 8.06 (d, J=7.83 Hz, 1 H), 7.95 - 8.02 (m, 1 H), 7.76 - 7.88 (m, 1 H), 3.93 (d, J=1.17 Hz, 3 H), 3.29 (s, 3 H); LCMS: (electrospray +ve), mlz 405.0 (MH)+; HPLC: tR = 4.83 min, UV254 = 100%. HRMS (ESI): m/z calcd for Ci5Hi3N602S3 [M+H]+ 405.0257, found 405.0265.
[0152] 2-(5-(3-(methylsulfonyl)phenyl)thieno[2,3-d]pyrimidin-4-yl)- [l,2,4]triazolo[4,3-a]pyridine-3(2 -thione

[0153] H NMR (400 MHz, DMSO-d6) δ ppm 8.56 (s, 1 H), 8.36 (dd, J=6.95, 0.88 Hz, 1 H), 8.26 (s, 1 H), 8.1 1 (s, 1 H), 8.02 - 8.09 (m, 2 H), 7.91 (dd, J=9.19, 0.98 Hz, 1 H), 7.84 (t, J=7.83 Hz, 1 H), 7.47 - 7.57 (m, 1 H), 7.05 (t, J=6.75 Hz, 1 H), 3.30 (s, 3 H); LCMS:
(electrospray +ve), mlz 440.0 (MH)+; HPLC: tR = 4.47 min, UV254
m/z calcd for C,9H14N502S3 [M+H]+ 440.0304, found 440.0314.
[0154] 4-([l,2,4]triazolo[4,3-a]pyridin-3-ylthio)-5-(3- (methylsulfonyl)phenyl)thieno[2, -d]pyrimidine

[0155] Ή NMR (400 MHz, DMSO-i¾ δ ppm 9.36 (s, 1 H), 8.26 (s, 1 H), 7.91 (dd, J=7.14, 0.88 Hz, 1 H), 7.71 (s, 1 H), 7.37 - 7.52 (m, 3 H), 7.22 (d, J=7.63 Hz, 1 H), 7.06 (t, J=7.73 Hz, 1 H), 6.94 (dd, J=7.24, 6.26 Hz, 1 H), 3.12 (s, 3 H); LCMS: (electrospray +ve), mlz 440.0 (MH)+; HPLC: tR = 4.90 min, UV254 = 100%. HRMS (ESI): m/z calcd for
C19Hi4N502S3 [M+H]+ 440.0304, found 440.0310.
[0156] 4-methyl-l-(5-(3-(methylsulfonyl)phenyl)thieno[2,3-d]pyrimidin-4-yl)-lH- l,2,4-triazole-5(4H)-thione

[0157] Ή NMR (400 MHz, DMSO-< 6) δ ppm 8.79 (s, 1 H), 8.74 (s, 1 H), 8.16 (s, 1 H),
8.01 - 8.1 1 (m, 2 H), 7.94 - 7.99 (m, 1 H), 7.81 (t, J=7.73 Hz, 1 H), 3.50 (s, 3 H), 3.29 (s, 3
H); LCMS: (electrospray +ve), mlz 404.0 (MH)+; HPLC: tR = 4.06 min, UV254 = 100%.
HRMS (ESI): m/z calcd for C16Hi4N502S3 [M+H]+ 404.0304, found 404.0316.
[0158] 4-(4-methyl-4H-l,2,4-triazol-3-ylthio)-5-(3-(methylsulfonyl)phenyI)thieno[2,3- d]pyrimidine

[0159] Ή NMR (400 MHz, DMSO-</6) δ ppm 9.28 (s, 1 H), 8.40 (s, 1 H), 8.23 (s, 1 H), 7.84 (d, J=7.83 Hz, 1 H), 7.69 (s, 1 H), 7.49 (t, J=7.83 Hz, 1 H), 7.36 (d, J=7.83 Hz, 1 H),
3.20 (s, 3 H), 3.10 (s, 3 H); LCMS: (electrospray +ve), mlz 404.0 (MH) ; HPLC: tR = 4.41 min, UV254 = 100%. HRMS (ESI): m/z calcd for C16H14N5O2S3 [M+H]+ 404.0303, found 404.0310.
[0160] 4-(benzo[d]thiazoI-2-yIthio)-5-(3-(methylsulfonyl)phenyl)thieno[2,3- d]pyrimidine
[0161] Ή NMR (400 MHz, DMSO-i/6) δ ppm 9.02 (s, 1 H), 8.04 - 8.15 (m, 3 H), 7.89 -
7.96 (m, 2 H), 7.81 (t, J=7.73 Hz, 1 H), 7.41 - 7.55 (m, 3 H), 3.24 - 3.32 (m, 3 H); LCMS:
(electrospray +ve), m/z 456.0 (MH)+; HPLC: tR = 6.38 min, UV254 = 100%. HRMS (ESI): m/z calcd for C20Hi4N3O2S4 [M+H]+ 455.9965, found 455.9978.
[0162] 4-(lH-l,2,4-triazol-3-ylthio)-5-(3-(methylsulfonyl)phenyl)thieno[2,3- djpyrimidine
[0163] 1H NMR (400 MHz, DMSO-c 6) δ ppm 8.70 (br. s., 1 H), 7.99 - 8.11 (m, 3 H), 7.90 (dt, J=7.63, 1.37 Hz, 2 H), 7.79 (t, J=7.73 Hz, 2 H), 3.23 - 3.29 (m, 3 H); LCMS:
(electrospray +ve), m/z 390.0 (MH)+; HPLC: tR = 4.01 min, UV2S4 = 100%. HRMS (ESI): m/z calcd for C15Hi2N502S3 [M+H]+ 390.0148, found 390.0159.
[0164] 4-(l-methyl-l H-tetr azol-5- lthio)-5-phenylthieno [2,3-d] pyrimidine
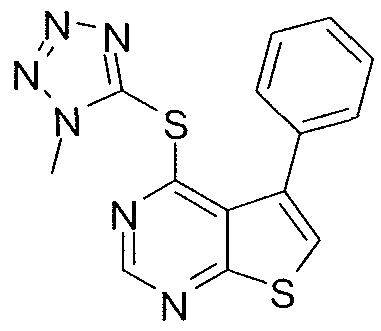
[0165] Ή NMR (400 MHz, DMSO- 6) δ ppm 8.74 (s, 1 H), 7.99 (s, 1 H), 7.61 (m, 2 H), 7.57 (m, 3 H), 3.94 (s., 3 H); LCMS: (electrospray +ve), mlz 327.1 (MH)+; HPLC: tR = 3.47 min, UV254 = 100%.
[0166] General procedure for the synthesis of 5-aryl-4-amino-thieno[2,3- djpyrimidines:
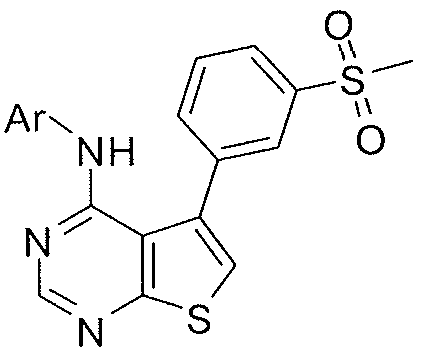
[0167] The same procedure was followed as with the above compounds substituting arylamines for arylthiols to get 5-aryl-4-aminothieno[2,3-d]pyrimidines as a tan solids after HPLC purification (20-50% yields).
[0168] 5-(3-(methylsulfonyl) henyl)-N-(pyridin-4-yl)thieno [2,3-d] pyrimidin-4-amine

[0169] 1H NM (400 MHz, DMSO-d6) δ ppm 9.28 (s, 1 H), 8.61 (s, 1 H), 8.35 (s, 1 H), 8.13 (d, J=7.63 Hz, 2 H), 7.85 (td, J=3.03, 1.37 Hz, 2 H), 7.58 (ddd, J=7.87, 1.42, 1.27 Hz, 1 H), 7.45 - 7.53 (m, 1 H), 6.46 (d, J=7.63 Hz, 2 H), 3.17 (s, 3 H); LCMS: (electrospray +ve), m/z 383.1 (MH)+; HPLC: tR = 3.1 1 min, UV254 = 100%. HRMS (ESI): m/z calcd for Ci8H15N402S2 [M+H]+ 383.0631 , found 383.0645.
[0170] 4-(2-(5-(3-(methylsulfonyl)phenyl)thieno [2,3-d] pyrimidin-4- ylamino)ethyl)phenol

[0171] Ή NMR (400 MHz, DMSO-<¾ δ ppm 9.14 (br. s., 1 H), 8.45 (s, 1 H), 7.92 - 8.00 (m, J=3.67, 1.92, 1.92, 1.92, 1.92 Hz, 2 H), 7.59 - 7.67 (m, 3 H), 6.77 - 6.87 (m, 2 H), 6.55 - 6.65 (m, 2 H), 5.59 (t, J=5.38 Hz, 1 H), 3.49 - 3.60 (m, 2 H), 3.25 (s, 3 H), 2.63 (t, 2 H); LCMS: (electrospray +ve), m/z 426.1 (MH)+; HPLC: tR = 4.44 min, UV254 = 100%. HRMS (ESI): m/z calcd for C21H20N3O3S2 [M+H]+ 426.0941 , found 426.0956.
[0172] 5-ethyl-N-(5-(3-(methylsulfonyl)phenyl)thieno[2,3-d]pyrimidin-4-yl)-l,3,4- oxadiazol-2-amine

[0173] Ή NMR (400 MHz, DMSO- 6) δ ppm 8.54 (s, 1 H), 8.09 (s, 1 H), 7.74 - 7.94 (m,
3 H), 7.60 - 7.71 (m, 1 H), 3.20 - 3.24 (m, 3 H), 2.67 (q, J=7.56 Hz, 2 H), 1.09 - 1.18 (m, 3
H); LCMS: (electrospray +ve), mlz 402.1 (MH)+; HPLC: tR = 5.02 min, UV254 = 100%.
HRMS (ESI): m/z calcd for C]7Hi6N503S2 [M+H]+ 402.0689, found 402.0703.
[0174] 5-(3-(methylsulfonyl)phenyl)-N-(pyridin-4-ylmethyl)thieno[2,3-d]pyrimidin-
4-amine

[0175] Ή NMR (400 MHz, DMSO- 6) δ ppm 8.62 (s, 1 H), 8.32 - 8.38 (m, 1 H), 8.05 (t, J=1.66 Hz, 1 H), 7.89 - 8.01 (m, 2 H), 7.67 - 7.81 (m, 4 H), 6.61 (s, 1 H), 4.74 (s, 2 H), 3.20 (s, 3 H); LCMS: (electrospray +ve), mlz 397.1 (MH)+; HPLC: tR = 3.46 min, UV254 = 100%. HRMS (ESI): m/z calcd for C 19H17N4O2S2 [M+H]+ 397.0788, found 397.0798.
[0176] 5-(3-(methylsulfonyl)phenyl)-N-(pyridin-2-ylmethyl)thieno[2,3-d]pyrimidin-
4-amine

[0177] H NMR (400 MHz, DMSO-<¾) δ ppm 8.42 (s, 1 H), 8.30 - 8.35 (m, 1 H), 8.00 - 8.08 (m, 2 H), 7.74 - 7.96 (m, 3 H), 7.69 (s, 1 H), 7.47 (s, 1 H), 7.31 - 7.39 (m, 1 H), 6.67 (s, 1 H), 4.69 (s, 2 H), 3.10 - 3.19 (m, 3 H); LCMS: (electrospray +ve), mlz 397.1 (MH)+; HPLC:
tR = 3.65 min, UV254 = 100%. HRMS (ESI): m/z calcd for C19H17N402S2 [M+H]+ 397.0788, found 397.0801.
[0178] Nl-(5-(3-(methylsulfonyl)phenyI)thieno[2,3-d]pyrimidin-4-yl)ethane-l,2- diamine

[0179] Ή NMR (400 MHz, DMSO-</6) 6 ppm 8.46 (s, 1 H), 8.06 (s, 1 H), 7.94 - 7.99 (m, 1 H), 7.89 (s, 1 H), 7.77 (d, J=7.83 Hz, 1 H), 7.70 (s, 1 H), 7.62 - 7.68 (m, 2 H), 3.55 - 3.66 (m, 2 H), 3.24 (s, 3 H), 2.92 - 3.02 (m, 2 H); LCMS: (electrospray +ve), mlz 349.1 (MH)+; HPLC: tR = 3.21 min, UV254 = 100%. HRMS (ESI): m/z calcd for C15H17N402S2 [M+H]+ 349.0787, found 349.0797.
[0180] 2-(5-(3-(methylsuIfonyl henyl)thieno[2,3-d]pyrimidin-4-ylamino)ethanol

[0181] Ή NMR (400 MHz, DMSO-<¾) δ ppm 8.41 (s, 1 H), 7.94 - 8.03 (m, 2 H), 7.81 - 7.86 (m, 1 H), 7.71 - 7.78 (m, 1 H), 7.64 (s, 1 H), 3.39 - 3.45 (m, 3 H), 2.93 (br. s., 4 H); LCMS: (electrospray +ve), mlz 350.1 (MH)+; HPLC: tR = 3.52 min, UV254 = 100%. HRMS (ESI): m/z calcd for C15H16N303S2 [M+H]+ 350.0628, found 350.0637.
[0182] 5-(3-(methyIsulfonyl)phenyl)-N-(thiazol-2-yl)thieno[2,3-d]pyrimidin-4-amine

[0183] Ή NMR (400 MHz, DMSO-c¾) δ ppm 8.72 (br. s., 1 H), 8.07 (t, J=l .57 Hz, 1 H), 7.90 - 7.99 (m, 2 H), 7.81 (d, J=3.33 Hz, 1 H), 7.72 (d, J=3.52 Hz, 1 H), 7.29 (br. s., 1 H), 7.03 (br. s., 1 H), 3.23 - 3.27 (m, 3 H); LCMS: (electrospray +ve), mlz 389.0 (MH)+; HPLC:
tR = 5.00 min, UV254 = 100%. HRMS (ESI): m/z calcd for Ci6Hi3N402S3 [M+H]+ 389.0194, found 389.0207.
[0184] 5-(3-(methylsulfonyl)phenyl)-N-(4-methylthiazol-2-yl)thieno[2,3-d]pyrimidin- 4-amine

[0185] Ή NMR (400 MHz, DMSO-i 6) δ ppm 8.70 (s, 1 H), 8.07 (t, J=l .66 Hz, 1 H), 7.88 - 8.00 (m, 2 H), 7.67 - 7.83 (m, 2 H), 7.47 (d, J=9.00 Hz, 1 H), 6.49 - 6.63 (m, 1 H), 3.28 (s, 3 H), 2.12 (s, 3 H); LCMS: (electrospray +ve), mlz 403.0 (MH)+; HPLC: tR = 5.15 min, UV254 = 100%. HRMS (ESI): m/z calcd for Ci7H15N402S3 [M+H]+ 403.0352, found
403.0361.
[0186] Nl-(5-(3-(methylsulfonyl)phenyl)thieno[2,3-d]pyrimidin-4-yl)propane-l,3- diamine
[0187] Ή NMR (400 MHz, DMSO-</6) δ ppm 8.43 (s, 1 H), 7.94 - 8.01 (m, 2 H), 7.83 (d, J=7.83 Hz, 1 H), 7.75 (t, J=8.02 Hz, 1 H), 7.67 (s, 3 H), 5.91 (t, J=5.58 Hz, 1 H), 3.42 (q, J=6.26 Hz, 2 H), 3.25 (s, 3 H), 2.70 - 2.82 (m, 2 H), 1.71 - 1.82 (m, 2 H); LCMS:
(electrospray +ve), mlz 363.1 (MH)+; HPLC: tR = 3.22 min, UV254 = 100%. HRMS (ESI): m/z calcd for Ci6H19N402S2 [M+H]+ 363.0944, found 363.0954.
[0188] 5-(3-(methylsulfonyl)phenyl)-N-(pyrimidin-4-yl)thieno [2,3-d] pyrimidin-4- amine

[0190] 5-(3-(methylsulfonyl)phenyl)-N-(5-methylthiazol-2-yl)thieno[2,3-d]pyrimidin-
4-amine

[0191] 1H NMR (400 MHz, DMSO- ) δ ppm 8.67 (br. s., 1 H), 8.05 (d, J=l .76 Hz, 1 H), 7.92 (dd, J=8.02, 1.37 Hz, 2 H), 7.69 (br. s., 2 H), 6.97 (br. s., 1 H), 3.21 - 3.27 (m, 6 H); LCMS: (electrospray +ve), mlz 403.0 (MH)+; HPLC: tR = 5.13 min, UV254 = 100%. HRMS (ESI): m/z calcd for C)7Hi5N402S3 [M+H]+ 403.0352, found 403.0365.
[0192] N-(l,3-dimethyl-lH-pyrazol-5-yl)-5-(3-(methylsulfonyl)phenyl)thieno[2,3- d] pyrimidin-4-amine

[0193] Ή NMR (400 MHz, DMSO- ) δ ppm 8.51 (s, 1 H), 8.03 (s, 1 H), 7.91 (dd, J=l 5.06, 7.83 Hz, 2 H), 7.79 - 7.85 (m, 2 H), 7.74 (t, J=7.83 Hz, 1 H), 5.87 (s, 1 H), 3.37 (s, 3 H), 3.17 - 3.24 (m, 3 H), 2.02 - 2.09 (m, 3 H); LCMS: (electrospray +ve), mlz 400.0 (MH)+; HPLC: tR = 4.34 min, UV254 = 100%. HRMS (ESI): m/z calcd for Ci8H18N502S2 [M+H]+ 400.0896, found 400.0909.
[0194] N-(5-ethyl-l,3,4-thiadiazol-2-yl)-5-(3-(methylsuIfonyI)phenyl)thieno[2,3- d]pyrimidin-4-amine

[0195] Ή NMR (400 MHz, DMSO- 6) δ ppm 8.74 (s, 1 H), 8.02 (s, 1 H), 7.85 - 7.99 (m, 2 H), 7.76 - 7.85 (m, 1 H), 7.69 (br. s., 1 H), 3.25 (s, 3 H), 2.83 (d, J=7.43 Hz, 2 H), 1.20 - 1.28 (m, 3 H); LCMS: (electrospray +ve), mlz 418.0 (MH)+; HPLC: tR = 5.36 min, UV254 = 100%. HRMS (ESI): m/z calcd for Ci7H16N502S3 [M+H]+ 418.0461 , found 418.0479.
[0196] N-(5-methyl-l,3,4-thiadiazol-2-yl)-5-(3-(methylsulfonyl)phenyl)thieno[2,3- d] pyrimidin-4-amine

[0197] !H NMR (400 MHz, DMSO-i/6) δ ppm 8.72 (br. s., 1 H), 8.03 (br. s., 1 H), 7.90 (br. s., 2 H), 7.74 - 7.83 (m, 1 H), 7.68 (br. s., 1 H), 3.34 (s, 3 H), 2.48 (br. s., 3 H); LCMS: (electrospray +ve), mlz 404.0 (MH)+; HPLC: tR = 4.95 min, UV254 = 100%. HRMS (ESI): m/z calcd for Ci6Hi4N502S3 [M+H]+ 404.0304, found 404.0312.
[0198] Synthesis of 4-(l-Methyl-lH-tetrazol-5-ylthio)-3-(3-(methylsulfonyl)phenyl)- 1 H-pyrazolo [3,4-d] pyrimidine :
[0199] (4,6-Dichloropyrimidin-5-yl)(3-(methyIthio)phenyl)methanol

[0200] A two-necked flask with a stirrer bar was fitted with a vigreux. Magnesium turnings (1.86 g, 76 mmol) were added and the flask was flame dried with a stream of nitrogen with vigorous stirring. The flask was cooled and THF was added, followed by 0.1 ml of 1 ,2-dibromoethane (0.1 ml). (3-Bromophenyl)(methyl)sulfane (3.88 g, 19.10 mmol) was taken in a syringe and one third of the entire amount was added. The initiation of the
Grignard reaction was observed (warming of flask and reflux). The rest of the amount was added slowly to maintain a steady reflux. After the temperature appeared to subside the
solution of the Grignard reagent was transferred via syringe to a premixed solution of 4,6- dichloropyrimidine-5-carbaldehyde (3.38 g, 19.1 mmol) in THF at 0 °C under nitrogen. The reaction was allowed to warm to rt. A small amount of sat aq NH4C1 was added (white precipitation was observed), followed by water, the mixture was extracted with EtOAc, and brine was added. The EtOAc layer was separated, dried (MgS04), filtered, and purified by flash Si02 (5 to 100% EtOAc/hexanes) column chromatography to provide (4,6- dichloropyrimidin-5-yl)(3-(methylthio)phenyl)methanol (3.8 g, 13 mmol, 67 % yield).
0201] (4,6-Dichloropyrimidin-5-yl)(3-(methylsulfonyl)phenyl)methanone

[0202] (4,6-Dichloropyrimidin-5-yl)(3-(methylthio)phenyl)methanol (3.38 g, 11.2 mmol) was dissolved in DCM (100 ml) and treated with sodium bicarbonate (0.943 g, 1 1.2 mmol), and then Dess-Martin Periodinane (5.0 g, 12 mmol). The mixture was stirred for 45 min and then filtered through a pad of silica gel. The filtrate was concentrated, diluted with DCM (100 ml) again and treated with mCPBA (5.03 g, 22.4 mmol). Slight effervescence was observed. A white precipitate (ppt) was formed which was filtered off. The filtrate also had some white ppt. This was diluted with water and more DCM was added till most of it had dissolved. The organic layer was separated, dried (MgS04), filtered, and adsorbed onto silica gel. Flash Si02 column chromatography (5 to 100% EtOAc and then EtOAc) provided (4,6- dichloropyrimidin-5-yl)(3-(methylsulfonyl)phenyl)methanone (3.22 g, 9.72 mmol, 87 % yield).
[0203] (4-chloro-6-(l-methyl-lH-tetrazol-5-yIthio)pyrimidin-5-yl)(3-(methylsulfonyl) phenyl)methanone

[0204] A mixture of (4,6-dichloropyrimidin-5-yl)(3-(methylsulfonyl)phenyl)methanone (358 mg, 1.08 mmol) and potassium carbonate(149 mg, 1.08 mmol) in DMF (5 ml) was treated with a solution of 1 -methyl- lH-tetrazole-5-thiol (94 mg, 0.81 mmol) in DMF (1 mL). The reaction was stirred overnight and concentrated. LCMS indicated approximately equal
amounts of mono and di addition product. Purification by silica gel flash column
chromatography (1 : 1 hexanes ethyl acetate) provided pure (4-chloro-6-(l -methyl- 1H- tetrazol-5-ylthio)pyrimidin-5-yl)(3-(methylsulfonyl)phenyl)methanone (1 19 mg, 0.290 mmol, 26.8 % yield).
[0205] 4-(l-Methyl-lH-tetrazol-5-ylthio)-3-(3-(methylsulfonyl)phenyl)-lH- pyrazolo[3,4-d]pyrimidine

[0206] A solution of (4-Chloro-6-(l-methyl-lH-tetrazol-5-ylthio)pyrimidin-5-yl)(3- (methylsulfonyl)phenyl)methanone (105 mg, 0.256 mmol) in THF (3 ml) was treated at 0 °C with hydrazine monohydrate (0.014 ml, 0.281 mmol) and warmed gradually to rt. The reaction was not complete, so added another 3 equivalents at 0 °C and then warmed to rt. The reaction was concentrated and purified by flash silica gel chromatography to obtain 4-(l- methyl-lH-tetrazol-5-ylthio)-3-(3-(methylsulfonyl)phenyl)-lH-pyrazolo[3,4-d]pyrimidine (93 mg, 0.24 mmol, 94 % yield).
[0207] Ή NMR (400 MHz, CHLOROFORM-*/) δ ppm 3.15 (s, 3 H), 4.09 (s, 3 H), 7.77 (t, J=7.8 Hz, 1 H), 8.09 (m, 2 H), 8.36 (t, J=1.9 Hz, 1 H), 8.58 (s, 1 H), 13.02 (br s, 1 H). 0208] 4-(l-Methyl-lH-tetrazol-5-ylthio)-3-phenyl-lH-pyrazolo[3,4-d]pyrimidine

[0209] This was generated in a similar manner to 4-(l-methyl-lH-tetrazol-5-ylthio)-3-(3- (methylsulfonyl)phenyl)-lH-pyrazolo[3,4-d]pyrimidine.
[0210] LC-MS: rt (min) = 3.23. 1H NMR (400 MHz, DMSO-i/6) δ ppm 4.01 (s, 3 H), 7.62 (m, 3 H), 7.84 (m, 2 H), 8.63 (s, 1 H).
EXAMPLE 2
[0211] This example demonstrates that a correlation was established between a multiplexed high-throughput and high content screening assay that sequentially measured cytotoxicity and protein aggregation for screening of compound libraries. This assay can be used to identify compounds that either modulate aggregation and/or cytotoxicity in cellular models of neurodegenerative diseases. Some of the assays employed herein are described below.
[0212] A stable PC12 cell line containing a gene fusion of Exon 1 of the Huntingtin gene linked to GFP under the control of the inducible ecdysone promoter were used as the cell- based model of Huntington Disease for high throughput screening. Exon 1 of the Huntingtin gene econtained an expansion of 103 polyglutamines (Q103HTT) which, when expressed, induced cell death and formation of distinct, bright GFP aggregates. The amount of cell death and the size and intensity of GFP aggregates increased with time and induction level. Cell death was quantified by measurement of ATP content in the cells. A maximal 40-50% of cell death was observed when the Huntington gene was induced in this cell line.
[0213] The cell health or membrane integrity was measured by the protease release method. Cells with perturbed membrane integrity release proteins and cofactors from the cytosol into the media, a dead cell protease being one of them. Cytox Glo kit contains a cell impermeable pro-luciferin substrate that becomes activated upon cleavage by such protease. Thus, cells with alered membrane permeability release this protease and generate an increase in the luminescent signal with reliability (averabe S/B of 9.6 and Z' of 0.80).
[0214] Rat pheochromocytoma PC 12 cells harboring HTT Q 103 or Q25 fujsed to GFP under tebufenozide (Sigma Aldrich) indudction were supplied by the Eric Schweizer lab (UCLA). Cells were maintained in Phenol red free DMEM (Invitrogen) at 37°C under a humidified atmosphere containing 5% C02 and 95% air. The medium contained + 5% supplemented calf serum, + 5% horse serum (sera from HyClone), 230 ug/ml Geneticin and lx pen/strep 2 mM L-glutamine. Cells were passaged when they reached 85 to 90 % co fluency. Cells were pelated at the density of 1000 to 1500 cells/well in black, clear bottom, tissue culture treated, microclear 1536 well plates (Aurora Biotechnologies) in 5 ul/well in phenol red free DMEM containing 2% serum (1 % each) without geneticin using a Multidrop Combi dispenser (Thermo Scientific). Tebufenozide (Sigma Aldrich) inducer at a 200 nM final concentration and test compounds were sequentially added to the cells using a Kalypsys pintool transfer station at a volume of 23 nl/well after the cells were incubated for 24 hrs.
Compound libraries and Tebufenozide were dissolved in DMSO and the final concentration of DMSO in the cell plates was 0.46% v/v. Plates were incubated at 37°C in 5 % C02 for 48 hours before being assayed.
[0215] For ATP quantitation, 3 ul of ATPLite (PerkinElmer) was added to each well. The plates were spun for 1 minute at 1500 PRM to remove bubbles and luminescent signal was recorded on a Viewlux CCD Plate reader (30 second integration time). Compounds were normalized against induced (200 nM tebufenozide considered maximum cell death) and uninduced (considered min. cell death). Controls were; Columns 1-3 DSMO only, Column 2 Tebufenozide 16 point dose response from 1 uM to 0 uM, Column 4 (and the rest of the plate) received 200 nM tebufenozide. The multiplexed assay also measured aggregate status.
[0216] The results obtained on certain compounds are set forth in Table 1. AC50 values were measured from protease release assay.
Table 1 : Protection of striatal cells against cytotoxicity and aggregation by certain compounds.


[0217] To confirm the activity of lead compounds in an orthogonal assay, the capacity of the compounds to prevent cell death induced by serum deprivation in mouse wild type (wt)
STHdhQ7 7 and mutant STHdhQ1 1 1 /1 " Huntington striatal cells was tested. The cells were plated in normal serum conditions or in serum deprivation conditions and treated at the same time with 30 μΜ solutions of our two initial hits. Cells health was quantitated by measuring ATP levels in cells. Thus, the activity of two of the above compounds D and F was tested. It is well known, that in contrast to wild type, transfected polyQ transfected striatal cells are less resistant to stress conditions such us serum deprivation. Upon treatment with a concentration of 30 μΜ of compound F or compound D, it was observed, as set forth in Table 2, that compounds of this series not only prevented the cell death of mutant neuronal but also promoted their growth. See also Figure 3 which shows the STHdhQ1 I 1/1 1 1 Huntington striatal cells were more susceptible to the serum deprivation challenge than the wild type cells.
Table 2 shows that treatment with these two compounds substantially reduced cell death caused by serum deprivation selectivity for the mutant STHdh^1 1 1/1 1 1 Huntington striatal cells.
Table 2. Rate of growth of transfected wt and mutant Huntington striatal cells in the presence active compounds. Data are expressed as the AVG ± STDV of the ratio of the rlu of treatment versus relative control.

EXAMPLE 3
[0218] This Example illustrates a method of preparing compounds in accordance with an embodiment of the invention. Fig. 1 depicts a reaction scheme used for the synthesis of embodiments of the compounds of the invention. Thus, commercially available thieno[2,3- d]pyrimidin-4(3H)-one 1 was regioselectively halogenated in the 6-position with bromine in
acetic acid to obtain compound 2. Transformation of the pyrimidinone into 6-bromo-4- chlorothieno[2,3-d]pyrimidine 3 was carried out by the use of phosphorus oxychloride. LDA induced bromo rearrangement and bromo-iodo replacement promoted by ethyl Grignard yielded intermediate 5. Regioselective Suzuki coupling and halogen displacement with several nucleophiles yielded final compound 7. Fig. 2 depicts a reaction scheme to synthesize additional embodiments, e.g., pyrrazolopyrimidines, of the compounds of the invention.
EXAMPLE 4
[0219] Tables 3-5 set forth biological activity data of compounds including their ability (Kd) to inhibit PIP5 2C, in accordance with an embodiment of the invention.
Table 3.

39.81 31.62 8.69 39.81 3.55
F
24 22.39 19.95 12.59 35.48 6.31 25 10.00 25.12 7.49 14.13 6.31
16 14.13 25.12 7.94 19.95 12.59
26 25.12 31.62 19.95 null 14.12
27 12.59 12.59 12.59 22.39 11.22
 15.85 19.95 15.85 39.81 28.18
15.85 19.95 15.85 39.81 28.18
28 25.12 35.48 15.85 null
7.94 22.39 19.95 35.48
12.59 15.85 10.00 35.48 39.81
Br
>100 44.67

35 31.62 31.62 15.85 35.48 11.22 40 ^ _ 31.62 19.95 15.85 39.81 14.12
79.43 31.62 ~ j " 12.59 25.12 7,49
39 12.59 39.81
25.12 25.12 15.85 null 6.30
12.59 12.59
42 I 10.00 10.00 10.00
NO,
43 Ό 39.81 25.12
X' 14.13 25.12 25.12
Table. 3 (cont'd) n
ACi0 AC AC AC
8.91 15.85 15.85 6.31
N02
NH2
1.26 null 3.98 null 0.25 null nu,i 5.62
NH2
31.62 39.81 null nuu 44.67 5.01 15.85 7.94 ND

25.12 10.00 12.59
COOtBu
N 39.81 35.48 25.12

5.61 10.00 15.85 2.82

1 15 10.00 19.95 7.94 12.59
;r°i 7.94 7.94 10.00
o
s- 14.13 19.95 39.81
7.94 6.31 7.94 1.12
12.59 15.85 15.85
 Table 4. (cont'd)
Table 4. (cont'd)
Compound ^ P1P5K2C No. d
(μΜ)

85 0~\ 6.31

91 NH, 39.81

-N
93 56.23
-NH,
94 > 100
Table 5

PIP5K2C
Kd (u )

EXAMPLE 5
[0220] This Example illustrates that compound 49 in accordance with an embodiment of the invention is able to protect primary neurons against H202 and beta-amyloid induced cell death. Fig. 4 and 5 show that compound 49 is able to prevent cell death induced by H202 and beta-amyloid.
EXAMPLE 6
[0221] This Example illustrates that a compound in accordance with an embodiment of the invention is able to block the activation of the intrinsic apoptotic pathway, preventing the production of caspase 9 and its downstream target caspase 3. Figs. 6 A and 6B show that compound 14 (F7) protects the cells from serum free medium induced apoptotic cell death by inhibiting the activation of caspase 9 and of its downstream target caspase 3.
EXAMPLE 7
[0222] This Example shows that PC 12 cells tested in Example 2 express PIP5K2C gene. qPCR was employed in this measurement. See Fig. 7.
EXAMPLE 8
[0223] This Example illustrates that a sulfonyl group at the meta position on the phenyl ring protects compound 52 from cytochrome P450 oxidation. Table 6 shows the microsome
stability of the compound after 60 minutes incubation in the presence and absence of
NADPH.
Table 6.
Plus Minus
NADPH NADPH
Parent Parent
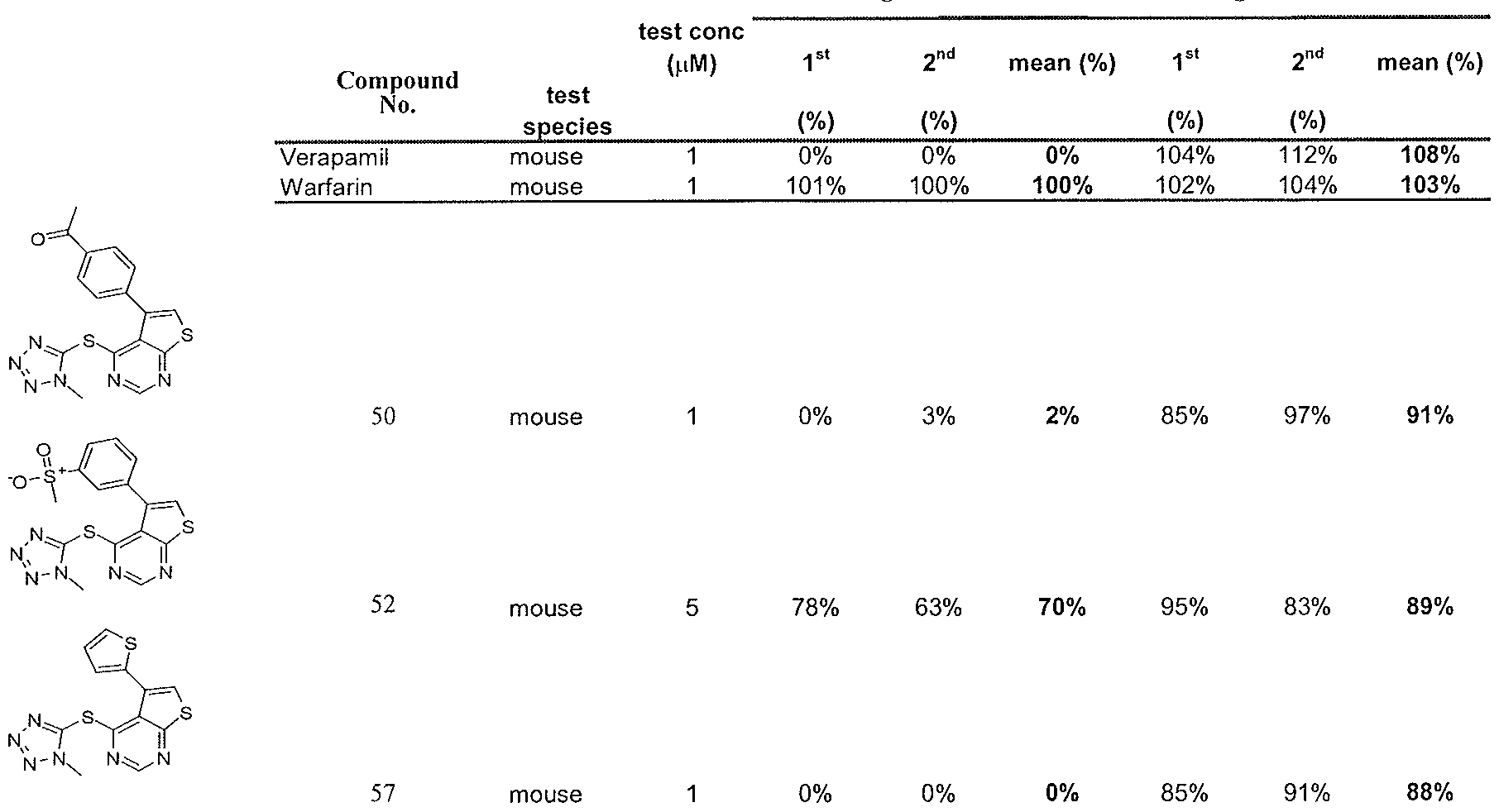
[0224] All references, including publications, patent applications, and patents, cited herein are hereby incorporated by reference to the same extent as if each reference were individually and specifically indicated to be incorporated by reference and were set forth in its entirety herein.
[0225] The use of the terms "a" and "an" and "the" and similar referents in the context of describing the invention (especially in the context of the following claims) are to be construed to cover both the singular and the plural, unless otherwise indicated herein or clearly contradicted by context. The terms "comprising," "having," "including," and "containing" are to be construed as open-ended terms (i.e., meaning "including, but not limited to,") unless otherwise noted. Recitation of ranges of values herein are merely intended to serve as a shorthand method of referring individually to each separate value falling within the range, unless otherwise indicated herein, and each separate value is incorporated into the specification as if it were individually recited herein. All methods described herein can be performed in any suitable order unless otherwise indicated herein or otherwise clearly contradicted by context. The use of any and all examples, or exemplary
language (e.g., "such as") provided herein, is intended merely to better illuminate the invention and does not pose a limitation on the scope of the invention unless otherwise claimed. No language in the specification should be construed as indicating any non-claimed element as essential to the practice of the invention.
[0226] Embodiments of this invention are described herein, including the best mode known to the inventors for carrying out the invention. Variations of those embodiments may become apparent to those of ordinary skill in the art upon reading the foregoing description. The inventors expect skilled artisans to employ such variations as appropriate, and the inventors intend for the invention to be practiced otherwise than as specifically described herein. Accordingly, this invention includes all modifications and equivalents of the subject matter recited in the claims appended hereto as permitted by applicable law. Moreover, any combination of the above-described elements in all possible variations thereof is
encompassed by the invention unless otherwise indicated herein or otherwise clearly contradicted by context.
Claims
CLAIM(S):
1. A compound of formula (I):

CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4,
NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
R2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R ,
S02NR3R4, S02N(OH)R3, CR3=NR4, CR3-N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4,
NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4); wherein each of said R2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono C]-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
wherein R3-R5 are independently hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C4 cycloalkyl, C3-C4 cycloalkenyl, C6-C20 aryl C]-C6 alkyl, C6-C2o aryl, heterocyclyl, or heterocyclyl alkyl;
X is a bond, S, O, or NH;
Y is S or NH;
Z is CH or N;
or a pharmaceutically acceptable salt thereof;
with the proviso that when X and Y are S, Z is CH, and R1 is tetrazolyl or a substituted tetrazolyl, then R2 is not halo, heterocyclyl, heterocyclyl aryl, aryl heterocyclyl, aryl, substituted aryl, substituted heterocyclyl, substituted heterocyclyl aryl, or substituted aryl heterocyclyl.
2. The compound or salt of claim 1 , wherein X is a bond, S or NH.
3. The compound or salt of claim 1 or 2, wherein R1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R1 is unsubstituted or optionally independently substituted with
one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy C]-C alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, and S02R3.
4. The compound or salt of any one of claims 1 to 3, wherein R1 is alkyl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, and S02R3.
5. The compound or salt of any one of claims 1 to 4, wherein R1 is alkyl, aryl, arylalkyl, heterocyclyl, or heterocyclylalkyl, optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, and S02R3.
6. The compound or salt of any one of claims 1 to 5, wherein R1 is selected from the group consisting of alkyl, aryl, arylalkyl, diazolyl, thiazolyl, triazolyl, thiadiazolyl, oxadiazolyl, pyridyl, pyridylmethyl, pyridazinyl, furanyl, benzodiazolyl, pyridinotriazolyl, benzothiazolyl, each of which is optionally substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , and S02R .
7. The compound or salt of any one of claims 1 to 6, wherein R1 is selected from the group consisting of alkyl, aryl, arylalkyl, diazolyl, thiazolyl, triazolyl, thiadiazolyl, oxadiazolyl, pyridyl, pyridylmethyl, pyridazinyl, furanyl, benzodiazolyl, pyridinotriazolyl, benzothiazolyl, each of which is optionally substituted with a substituent selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido,
3 3 3
aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , and S02R .
8. The compound or salt of any one of claims 1 to 7, wherein R1 is selected from the group consisting of alkyl, phenyl, phenylalkyl, diazolyl, thiazolyl, triazolyl, thiadiazolyl, oxadiazolyl, pyridyl, pyridylmethyl, pyridazinyl, furanyl, benzodiazolyl, pyridinotriazolyl, benzothiazolyl, each of which is optionally substituted with an alkyl, hydroxy, or amino substituent.
9. The compound or salt of any one of claims 1 to 8, wherein X-R is selected from the group consisting of:


10. The compound or salt of any one of claims 1 to 8, wherein Y is S.
1 1. The compound or salt of claim 10, wherein Z is CH.
12. The compound or salt of claim 10 or 1 1 , wherein X is S.
13. The compound or salt of any one of claims 1 to 12, wherein R2 is selected from the group consisting of halo, aryl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, and S02R ; wherein each of said R groups aryl, aryl heterocyclyl, and aryl heterocyclyl aryl are unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono CrC6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, and an anionic group.
14. The compound or salt of any one of claims 1 to 13, wherein R2 is selected from the group consisting of phenyl, pyrimidinyl, thiophenyl, pyrrolyl, benzofuranyl, naphthyl, methylenedioxyphenyl, 4-oxo-4H-chromenyl, dibenzothiophenyl, bromo, and iodo, each of which, other than bromo and iodo, is optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Q-Q alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Q-Q alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, and an anionic group.
15. The compound of any one of claims 1 to 14, wherein R2 is selected from the group consisting of:


16. A pharmaceutical composition comprising a compound or salt of any one of claims 1-15 or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier.
17. A method of inhibiting the PIP5K2C kinase in a cell in need thereof comprising administering to the cell an effective amount of a compound of formula (I):

wherein R1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, heterocyclyl aryl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl, heterocyclyl aryl, and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono Ci-C alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo C]-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4, S02N(OH)R3, CR3=NR4,
CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4,
NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
R2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4); wherein each of said R2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR 3 , S(0)R 3 , S02R 3 , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
wherein R3-R5 are independently hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C4 cycloalkyl, C3-C4 cycloalkenyl, C6-C2o aryl Ci-C6 alkyl, C6-C2o aryl, heterocyclyl, or heterocyclyl alkyl;
X is a bond, S, O, or NH;
Y is S or NH;
Z is CH or N;
or a pharmaceutically acceptable salt thereof.
18. A method of modulating the activity of a mutant Huntingtin protein in a cell comprising administering to the cell an effective amount of a compound of formula (I):

wherein R1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, heterocyclyl aryl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl, heterocyclyl aryl, and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo CrC6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4, S02N(OH)R3, CR3=NR4,
CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4,
NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
R2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy CrC6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(GH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and
P(0)(OR3)(OR4); wherein each of said R2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Cj-C6 alkyl, dicarboxy halo C]-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Cj-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
wherein R3-R5 are independently hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C4 cycloalkyl, C3-C4 cycloalkenyl, C6-C2o aryl Ci-C6 alkyl, C6-C2o aryl, heterocyclyl, or heterocyclyl alkyl;
X is a bond, S, O, or NH;
Y is S or NH;
Z is CH or N;
or a pharmaceutically acceptable salt thereof.
19. A method of treating or preventing a neurodegenerative disease in an animal comprising administering to the animal an effective amount of a compound of formula (I):

. wherein R1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, heterocyclyl aryl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl, heterocyclyl aryl, and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl,
dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4, S02N(OH)R3, CR3=NR4,
CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4,
NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
R2 is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo C]-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4); wherein each of said R2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy C]-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino,
3 3 3 aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR , S(0)R , S02R , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5,
NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
wherein R3-R5 are independently hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C4 cycloalkyl, C3-C4 cycloalkenyl, C6-C2o aryl Ci-C6 alkyl, C6-C20 aryl, heterocyclyl, or heterocyclyl alkyl;
X is a bond, S, O, or NH;
Y is S or NH;
Z is CH or N;
or a pharmaceutically acceptable salt thereof.
20. The method of claim 19, wherein the neurodegenerative disease is selected from the group consisting of Dentatorubropallidoluysian atrophy, Huntington's disease, Spinobulbar muscular atrophy, Spinocerebellar ataxia Type 1 , Spinocerebellar ataxia Type 2, Spinocerebellar ataxia Type 3, Spinocerebellar ataxia Type 6, Spinocerebellar ataxia Type 7, Spinocerebellar ataxia Type 17, Cockayne Syndrome, hepatolenticular degeneration, Lafora Disease, Menkes Kinky Hair Syndrome, neurofibromatosis, Tourette Syndrome, Tuberous Sclerosis Amyotrophic Lateral Sclerosis, muscular atrophy, poliomyelitis, Parkinson's Disease, Prion diseases, Creutzfeldt-Jacob Syndrome, Kuru, Scrapie, and Alzheimer's Disease.
21. The method of claim 19, wherein the neurodegenerative disease is a polyglutamine disease.
22. A method of inhibiting the activation of caspase 9 in a cell in need thereof comprising administering an effective amount of a compound of formula (I):

CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4,
NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
R is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy CpQ alkyl, dicarboxy halo C]-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR 3 , S(0)R 3 , S02R 3 , S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and
P(0)(OR 3 )(OR 4 ); wherein each of said R 2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Q-C alkyl, carboxy Cj-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Cj-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4,
NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4);
wherein R3-R5 are independently hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C4 cycloalkyl, C3-C4 cycloalkenyl, C6-C2o aryl Ci-C6 alkyl, C6-C2o aryl, heterocyclyl, or heterocyclyl alkyl;
X is a bond, S, O, or NH;
Y is S or NH;
Z is CH or N;
or a pharmaceutically acceptable salt thereof.
23. The method of any one of claims 17 to 22, wherein X is a bond, S or NH.
24. The method of any one of claims 17 to 23, wherein R1 is alkyl, aryl, arylalkyl, heterocyclyl, heterocyclyl alkyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Q-Q alkyl, dicarboxy Q-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR 3 , S(0)R 3 , and S02R 3.
25. The method of any one claims 17 to 24, wherein R1 is alkyl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, wherein each of said R1 is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, and S02R3.
26. The method of any one of claims 17 to 25, wherein R1 is alkyl, aryl, arylalkyl, heterocyclyl, or heterocyclylalkyl, optionally independently substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, and S02R3.
27. The method of any one of claims 17 to 26, wherein R1 is selected from the group consisting of alkyl, aryl, arylalkyl, pyrazolyl, fhiazolyl, triazolyl, tetrazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyridinylmethyl, pyrimidinyl, furanyl, triazolopyridinyl, benzoimidazolyl, and benzothiazolyl, each of which is optionally substituted with one or more substituents selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, and S02R3.
28. The method of any one of claims 17 to 27, wherein R! is selected from the group consisting of alkyl, aryl, arylalkyl, pyrazolyl, fhiazolyl, triazolyl, tetrazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyridinylmethyl, pyrimidinyl, furanyl, triazolopyridinyl, benzoimidazolyl, and benzothiazolyl, each of which is optionally substituted with a substituent selected from the group consisting of alkyl, aryl, arylalkyl, halo, haloalkyl, dihaloalkyl, trihaloakyl, hydroxy, mercapto, alkoxy, nitro, carboxy, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, and S02R3.
29. The method of any one of claims 18 to 29, wherein R! is selected from the group consisting of alkyl, aryl, arylalkyl, pyrazolyl, fhiazolyl, triazolyl, tetrazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyridinylmethyl, pyrimidinyl, furanyl, triazolopyridinyl, benzoimidazolyl, and benzothiazolyl, each of which is optionally substituted with an alkyl, hydroxy, or amino substituent.
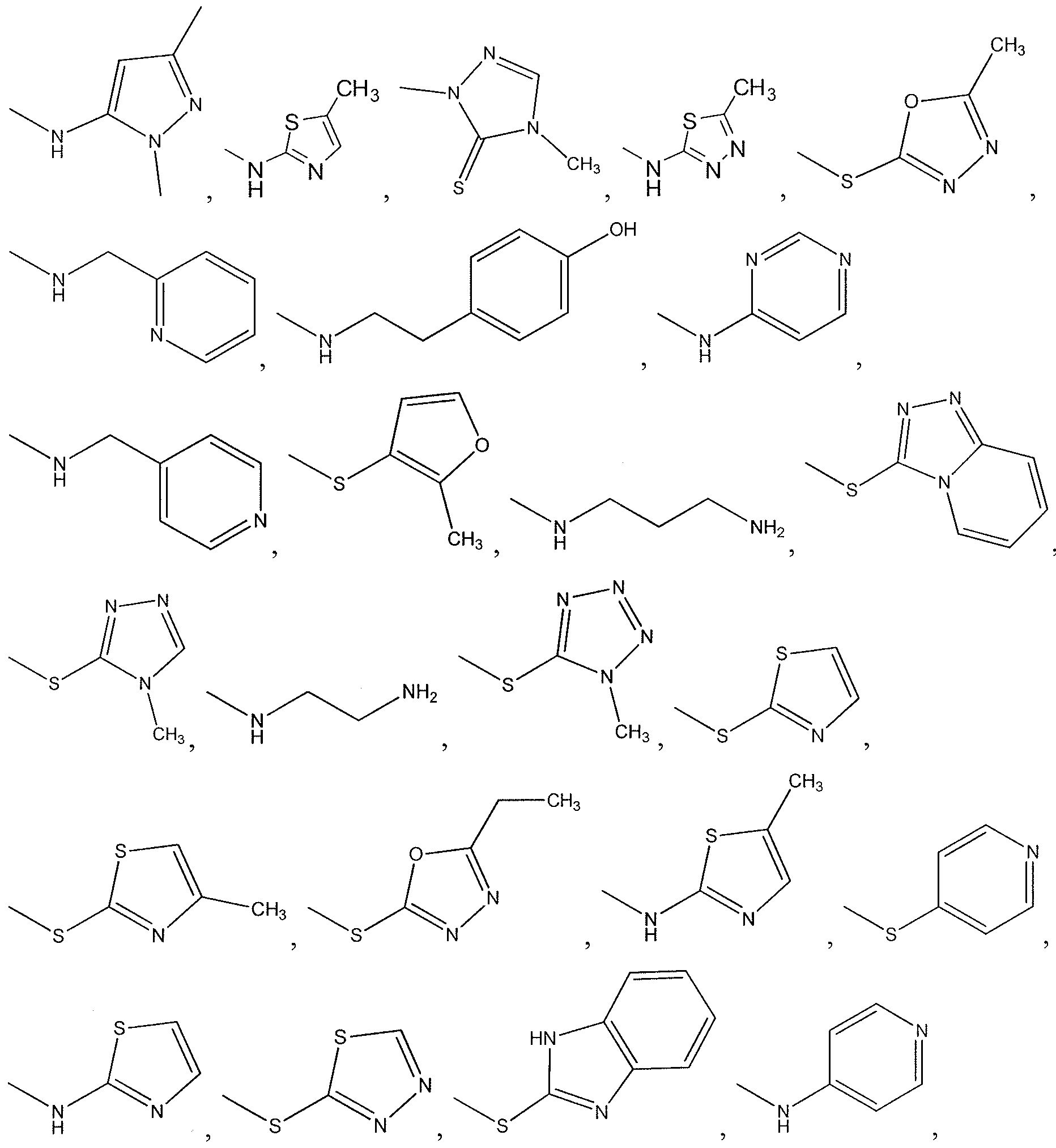

31. The method of any one of claims claim 17 to 26, wherein Y is S.
32. The method of claim 31 , wherein Z is CH.
33. The method of claim 31 or 32, where X is S.
34. The method of any one of claims 22 to 33, wherein R2 is selected from the group consisting of aryl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, and S02R3; wherein each of said R2 groups aryl, aryl heterocyclyl, and aryl heterocyclyl aryl are unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Ci-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo CpC6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Q-Q alkyl, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, and an anionic group.
35. The method of any one of claims 22 to 34, wherein R is selected from the group consisting of phenyl, pyrimidinyl, thiophenyl, pyrrolyl, benzofuranyl, naphthyl, 4-oxo- 4H-chromenyl, dibenzothiophenyl, bromo, and iodo, each of which, other than bromo and iodo, is optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Q- C6 alkyl, carboxy Cj-C alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, trifluoro Ci-C6 alkyl, alkylthio, acyl, acyloxy, thioacyl,
acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, and an anionic group.
36. The method of any one of claims 22 to 35, wherein R2 is selected from the group consisting of:


37. The method of any one of claims 17 to 36, wherein the cell is in an animal.
38. The method of claim 37, wherein the animal is a human.
39. The compound or salt of claim 1 , wherein X is S, Y is NH, and Z is N, R1 is methyl tetrazolyl, and R is phenyl, optionally substituted with methylsulfonyl.
40. A compound selected from the group consisting of the following compounds:


or pharmaceutically acceptable salts thereof.
41. A pharmaceutical composition comprising a compound or salt of claim 40 or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier.
42. The compound or salt of any one of claims 1 to 16 or 40 for use in inhibiting the PIP5K2C kinase, in modulating the activity of a mutant Huntingtin protein, in treating or preventing a neurodegenerative disease, or in inhibiting the activation of caspase 9 in a patient or cell in need thereof.
43. The compound of claim 1 or method of any one of claims 17-33, wherein R is selected from the group consisting of halo, aryl, arylalkyl, heterocyclyl, aryl heterocyclyl, or aryl heterocyclyl aryl, wherein the aryl and heterocyclyl of aryl heterocyclyl and aryl heterocyclyl aryl, are fused or unfused to each other, halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Cj-C6 alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, tnalkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4,
S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5,
NR3C(0)N(OH)R4, NR3C(S)N(OH)R4, NR3S02R4, NHS02NR R4, NR S02NHR4, and
P(0)(OR3)(OR4); wherein each of said R2 groups aryl, arylalkyl, and heterocyclyl is unsubstituted or optionally independently substituted with one or more substituents selected from the group consisting of halo, hydroxy, mercapto, carboxy, phosphoryl, phosphonyl, phosphono Ci-C6 alkyl, carboxy Cj-Q alkyl, dicarboxy Ci-C6 alkyl, dicarboxy halo Ci-C6 alkyl, sulfonyl, alkylsulfonyl, cyano, nitro, alkoxy, alkylthio, acyl, acyloxy, thioacyl, acylthio, aryloxy, amino, alkylamino, dialkylamino, trialkylamino, guanidino, aldehydo, ureido, aminocarbonyl, a cationic group, an anionic group, SR3, S(0)R3, S02R3, S02NR3R4, S02N(OH)R3, CR3=NR4, CR3=N(OR4), N3, NR3R4, N(OH)R3, C(0)R3, C(S)R3, C02R3, C(0)SR3, C(0)NR3R4, C(S)NR3R4, C(0)N(OH)R3, C(S)N(OH)R3, NR3C(0)R4, NR3C(S)R4, N(OH)C(0)R3, N(OH)C(S)R3, NR3C02R4, N(OH)C02R3, NR3C(0)SR4, NR3C(0)NR4R5, NR3C(S)NR4R5, N(OH)C(0)NR3R4, N(OH)C(S)NR3R4, NR3R4, N+R3R4R5, NR3C(0)N(OH)R4,
NR3C(S)N(OH)R4, NR3S02R4, NHS02NR3R4, NR3S02NHR4, and P(0)(OR3)(OR4).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US38848210P | 2010-09-30 | 2010-09-30 | |
| US61/388,482 | 2010-09-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012044993A1 true WO2012044993A1 (en) | 2012-04-05 |
Family
ID=44802398
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/054325 WO2012044993A1 (en) | 2010-09-30 | 2011-09-30 | Compounds, pharmaceutical compositions, and methods of treating or preventing neurodegenerative diseases or disorders |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2012044993A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104725397A (en) * | 2013-12-23 | 2015-06-24 | 法国施维雅药厂 | Thienopyrimidine derivatives, a process for their preparation and pharmaceutical compositions containing them |
| JP2017519826A (en) * | 2014-07-03 | 2017-07-20 | チェーチャン ジュウツォウ ファーマサイエンス アンド テクノロジー カンパニー リミテッド | Ibutinib intermediate compound, production method and use thereof |
| CN107709334A (en) * | 2015-06-23 | 2018-02-16 | 法国施维雅药厂 | New amino acid derivativges, its preparation method and include their Pharmaceutical composition |
| JP2019529514A (en) * | 2015-10-05 | 2019-10-17 | ザ トラスティーズ オブ コロンビア ユニバーシティー イン ザ シティー オブ ニューヨーク | Activator of clearance of protein aggregates containing autophagy flux and phospholipase D and tau and method of treating proteinosis |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4235871A (en) | 1978-02-24 | 1980-11-25 | Papahadjopoulos Demetrios P | Method of encapsulating biologically active materials in lipid vesicles |
| US4501728A (en) | 1983-01-06 | 1985-02-26 | Technology Unlimited, Inc. | Masking of liposomes from RES recognition |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5019369A (en) | 1984-10-22 | 1991-05-28 | Vestar, Inc. | Method of targeting tumors in humans |
| US6600028B1 (en) * | 1997-04-02 | 2003-07-29 | Amersham Pharmacia Biotech Uk Limited | Tricyclic base analogues |
| WO2004092123A2 (en) * | 2003-04-10 | 2004-10-28 | Microbia, Inc. | Inhibitors of fungal invasion |
| WO2004111057A1 (en) * | 2003-06-11 | 2004-12-23 | Xention Discovery Limited | Thienopyrimidine derivatives as potassium channel inhibitors |
| WO2006071988A1 (en) * | 2004-12-23 | 2006-07-06 | Memory Pharmaceuticals Corp. | Thienopyrimidine derivatives as phosphodiesterase 10 inhibitors |
| WO2007046809A1 (en) * | 2005-10-21 | 2007-04-26 | Dow Agrosciences Llc | Thieno-pyrimidine compounds having fungicidal activity |
| WO2007102679A1 (en) * | 2006-03-06 | 2007-09-13 | Je Il Pharmaceutical Co., Ltd. | Novel thienopyrimidine derivatives or pharmaceutically acceptable salts thereof, process for the preparation thereof and pharmaceutical composition comprising the same |
| WO2008014263A2 (en) * | 2006-07-24 | 2008-01-31 | Tetralogic Pharmaceuticals Corporation | Dimeric iap antagonists |
| US20100063047A1 (en) * | 2008-09-10 | 2010-03-11 | Kalypsys, Inc. | Aminopyrimidine inhibitors of histamine receptors for the treatment of disease |
| US20100092456A1 (en) | 2006-05-31 | 2010-04-15 | Louise Bisset | Methods of treatment |
-
2011
- 2011-09-30 WO PCT/US2011/054325 patent/WO2012044993A1/en active Application Filing
Patent Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4235871A (en) | 1978-02-24 | 1980-11-25 | Papahadjopoulos Demetrios P | Method of encapsulating biologically active materials in lipid vesicles |
| US4501728A (en) | 1983-01-06 | 1985-02-26 | Technology Unlimited, Inc. | Masking of liposomes from RES recognition |
| US5019369A (en) | 1984-10-22 | 1991-05-28 | Vestar, Inc. | Method of targeting tumors in humans |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US6600028B1 (en) * | 1997-04-02 | 2003-07-29 | Amersham Pharmacia Biotech Uk Limited | Tricyclic base analogues |
| WO2004092123A2 (en) * | 2003-04-10 | 2004-10-28 | Microbia, Inc. | Inhibitors of fungal invasion |
| WO2004111057A1 (en) * | 2003-06-11 | 2004-12-23 | Xention Discovery Limited | Thienopyrimidine derivatives as potassium channel inhibitors |
| WO2006071988A1 (en) * | 2004-12-23 | 2006-07-06 | Memory Pharmaceuticals Corp. | Thienopyrimidine derivatives as phosphodiesterase 10 inhibitors |
| WO2007046809A1 (en) * | 2005-10-21 | 2007-04-26 | Dow Agrosciences Llc | Thieno-pyrimidine compounds having fungicidal activity |
| WO2007102679A1 (en) * | 2006-03-06 | 2007-09-13 | Je Il Pharmaceutical Co., Ltd. | Novel thienopyrimidine derivatives or pharmaceutically acceptable salts thereof, process for the preparation thereof and pharmaceutical composition comprising the same |
| US20100092456A1 (en) | 2006-05-31 | 2010-04-15 | Louise Bisset | Methods of treatment |
| WO2008014263A2 (en) * | 2006-07-24 | 2008-01-31 | Tetralogic Pharmaceuticals Corporation | Dimeric iap antagonists |
| US20100063047A1 (en) * | 2008-09-10 | 2010-03-11 | Kalypsys, Inc. | Aminopyrimidine inhibitors of histamine receptors for the treatment of disease |
Non-Patent Citations (6)
| Title |
|---|
| "Pharmaceutics and Pharmacy Practice", 1982, J. B. LIPPINCOTT COMPANY, pages: 238 - 250 |
| "Remington's Pharmaceutical Sciences", 1990, MACK PUBLISHING COMPANY, pages: 1445 |
| HOZIEN Z A ET AL: "SYNTHESIS AND APPLICATION OF SOME NEW THIENOPYRIMIDINE DERIVATIVES AS ANTIMICROBIAL AGENTS", SYNTHETIC COMMUNICATIONS, TAYLOR & FRANCIS GROUP, PHILADELPHIA, PA, vol. 26, no. 20, 1 January 1996 (1996-01-01), pages 3733 - 3755, XP002908881, ISSN: 0039-7911, DOI: 10.1080/00397919608003791 * |
| JOURNAL OF PHARMACEUTICAL SCIENCE, vol. 66, 1977, pages 2 - 19 |
| SZOKA ET AL., ANN. REV. BIOPHYS. BIOENG., vol. 9, 1980, pages 467 |
| TOISSEL: "ASHP Handbook on Injectable Drugs", 1986, pages: 622 - 630 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104725397A (en) * | 2013-12-23 | 2015-06-24 | 法国施维雅药厂 | Thienopyrimidine derivatives, a process for their preparation and pharmaceutical compositions containing them |
| JP2017519826A (en) * | 2014-07-03 | 2017-07-20 | チェーチャン ジュウツォウ ファーマサイエンス アンド テクノロジー カンパニー リミテッド | Ibutinib intermediate compound, production method and use thereof |
| CN107709334A (en) * | 2015-06-23 | 2018-02-16 | 法国施维雅药厂 | New amino acid derivativges, its preparation method and include their Pharmaceutical composition |
| CN107709334B (en) * | 2015-06-23 | 2021-04-13 | 法国施维雅药厂 | Novel amino acid derivatives, process for their preparation and pharmaceutical compositions containing them |
| JP2019529514A (en) * | 2015-10-05 | 2019-10-17 | ザ トラスティーズ オブ コロンビア ユニバーシティー イン ザ シティー オブ ニューヨーク | Activator of clearance of protein aggregates containing autophagy flux and phospholipase D and tau and method of treating proteinosis |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11884680B2 (en) | Bromodomain inhibitors | |
| EP3240785B1 (en) | Small molecule inhibitors of lactate dehydrogenase and methods of use thereof | |
| KR101650981B1 (en) | Heterocyclic compounds for the inhibiting of pask | |
| US10676468B2 (en) | N-(3-heteroarylaryl)-4-arylarylcarboxamides and analogs as hedgehog pathway inhibitors and use thereof | |
| TWI423978B (en) | Purinone derivatives as hm74a agonists | |
| TWI675026B (en) | Fused ring derivative, preparation method thereof, intermediate, pharmaceutical composition and application thereof | |
| TW201341386A (en) | Pyrazolo[1,5-a]pyrimidine-based compounds, compositions comprising them, and methods of their use | |
| AU2013265326A1 (en) | Substituted pyrazoloquinazolinones and pyrroloquinazolinones as allosteric modulators of group II metabotropic glutamate receptors | |
| JP2014500331A (en) | 2-Arylimidazo [1,2-b] pyridazine, 2-phenylimidazo [1,2-a] pyridine and 2-phenylimidazo [1,2-a] pyrazine derivatives | |
| EA028815B1 (en) | Substituted xanthines and methods of use thereof | |
| EP3556761B1 (en) | Pyrrolo-aromatic heterocyclic compound, preparation method therefor, and medical use thereof | |
| CN102753551A (en) | 2-arylimidazole derivatives as pde10a enzyme inhibitors | |
| US9738656B2 (en) | Pyranodipyridine compound | |
| US20160031863A1 (en) | Histone deacetylase inhibitors and compositions and methods of use thereof | |
| WO2012044993A1 (en) | Compounds, pharmaceutical compositions, and methods of treating or preventing neurodegenerative diseases or disorders | |
| EP2970316A2 (en) | Substituted xanthines and methods of use thereof | |
| JP2022527025A (en) | Compositions for the treatment of neurodegenerative diseases and mitochondrial diseases and their usage | |
| WO2015066697A1 (en) | Fused morpholinopyrimidines and methods of use thereof | |
| EP2825537A1 (en) | Dihydropyridopyrimidine and dihydronaphthyridine derivatives as tyrosine kinase inhibitors of especially vegf and pdgf | |
| KR20240046530A (en) | Sulfonamide derivatives, methods for their preparation and medical uses thereof | |
| EP2825166A1 (en) | Method of treating ophthalmic conditions with kinase inhibitors | |
| CN109206435B (en) | Thieno [3,2-d ] pyrimidine compound and preparation method and medical application thereof | |
| CN111825694B (en) | Condensed-cyclic compound, preparation method and application thereof | |
| US20230131192A1 (en) | 1H-PYRAZOLO[4,3-d]PYRIMIDINE COMPOUNDS AS TOLL-LIKE RECEPTOR 7 (TLR7) AGONISTS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11770610 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11770610 Country of ref document: EP Kind code of ref document: A1 |