WO2012027240A1 - Fused tricyclic inhibitors of mammalian target of rapamycin - Google Patents
Fused tricyclic inhibitors of mammalian target of rapamycin Download PDFInfo
- Publication number
- WO2012027240A1 WO2012027240A1 PCT/US2011/048549 US2011048549W WO2012027240A1 WO 2012027240 A1 WO2012027240 A1 WO 2012027240A1 US 2011048549 W US2011048549 W US 2011048549W WO 2012027240 A1 WO2012027240 A1 WO 2012027240A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- mmol
- pyrimidin
- inhibitors
- compounds
- Prior art date
Links
- 0 CC(C)(c(cc1)ncc1-c1c2nc(C(CC3)C*(CC4)C4N3C(c3nnc[n]3)=O)c(COCN3)c3[n]2nc1)O Chemical compound CC(C)(c(cc1)ncc1-c1c2nc(C(CC3)C*(CC4)C4N3C(c3nnc[n]3)=O)c(COCN3)c3[n]2nc1)O 0.000 description 9
- QENRNDGNMJFSHD-UHFFFAOYSA-N C(C1)C2NC(C3)C12CC3c(c(CCCN1)c1[n]1nc2)nc1c2-c(cc1)cnc1-c1ccccc1 Chemical compound C(C1)C2NC(C3)C12CC3c(c(CCCN1)c1[n]1nc2)nc1c2-c(cc1)cnc1-c1ccccc1 QENRNDGNMJFSHD-UHFFFAOYSA-N 0.000 description 1
- BTDGSORKBGMZNT-HDICACEKSA-N C[C@H](C1)O[C@@H](C)CN1c(c(CCCN1)c1[n]1nc2)nc1c2-c(cc1)cnc1-c1ccccc1 Chemical compound C[C@H](C1)O[C@@H](C)CN1c(c(CCCN1)c1[n]1nc2)nc1c2-c(cc1)cnc1-c1ccccc1 BTDGSORKBGMZNT-HDICACEKSA-N 0.000 description 1
- FGYYMUJSIBREQI-UHFFFAOYSA-N N#CCC(C1CCSCC1)=O Chemical compound N#CCC(C1CCSCC1)=O FGYYMUJSIBREQI-UHFFFAOYSA-N 0.000 description 1
- BYXITCVDAIFWLI-UHFFFAOYSA-N NCc1cc(C2CCSCC2)nc2ccn[n]12 Chemical compound NCc1cc(C2CCSCC2)nc2ccn[n]12 BYXITCVDAIFWLI-UHFFFAOYSA-N 0.000 description 1
- JVVRJMXHNUAPHW-UHFFFAOYSA-N Nc1ccn[nH]1 Chemical compound Nc1ccn[nH]1 JVVRJMXHNUAPHW-UHFFFAOYSA-N 0.000 description 1
- SWMDEFGWDTWDNE-UHFFFAOYSA-N OCC(N(C(CC1)C2)C1CC2c(c(CCCN1)c1[n]1nc2)nc1c2-c(cc1)cnc1-c1ccccc1)=O Chemical compound OCC(N(C(CC1)C2)C1CC2c(c(CCCN1)c1[n]1nc2)nc1c2-c(cc1)cnc1-c1ccccc1)=O SWMDEFGWDTWDNE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/14—Ortho-condensed systems
Definitions
- Rictor-mTOR complex is a rapamycin-insensitive complex that phosphorlates AKT. Although the precise mechanism by which rapamycin inhibitis mTOR function is not well understood, rapamycin partially inhibits mTOR function through mTORCl . Since mTORC2 is involved in the regulation of cell survival and actin cytoskeletal organization in a rapamycin-independent manner, complete inhibition of mTOR function through inhibition of both mTORCl and mTORC2 may lead to a broader spectrum antitumor activity and/or better efficacy than through inhibition of mTORCl alone.
- the compounds of this invention are useful in the inhibition of mTOR kinase, and are illustrated by a compound of the formula:
- R 2 is hydrogen, halo, cyano, NR y R z , OR y , C 1-6 alkyl or Ci -6 haloalkyl;
- R 3 is C 3 personally8 cycloalkyi, C 3- 8 cycloalkenyl, aryl, heteroaryl or heterocyclyl, O(heterocyclyl), SO m (heterocyclyl), C 3-6 alkyl, (Ci -6 alkyl)NHR 5 , (C w alkyl)NHCOR x , wherein said cycloalkyi, cycloalkenyl, aryl, heteroaryl and heterocyclyl groups are optionally substituted with one to three substituents independently selected from the group consisting of R 5 , halo, cyano, C 1-( s alkyl, C 1-6 haloalkyl, OR y , 0(Ci -6 haloalkyl), 0(Ci -6 alkyl)OR y , C(0)R y , C(0)OR y ,
- R s is C3.8 cycloalkyl, C 3 ,g cycloalkenyl, aryl, heteroaryl or heterocyclyl, wherein said cycloalkyl, cycloalkenyl, aryl, heteroaryl and heterocyclyl groups are optionally substituted with one to three substituents independently selected from the group consisting of halo, cyano, Ci_6 alkyl, C I-6 haloalkyl, OR y , 0(C, -6 haloalkyl), C(0)R y , C(0)OR y , SO m R y and NR y R z ;
- R 6 is selected from the group consisting of hydrogen, halo, cyano, Ci -6 alkyl, Ci -6 haloalkyl, (C 1-6 alkyl)OR y , OR y , 0(Ci portrait 6 haloalkyl), C(0)R C(0)OR , SO m R y and NR y R z ;
- D is CR y R z or C(O).
- R is C 3- 8 cycloalkyl or heterocyclyl, wherein said cycloalkyl and heterocyclyl groups are optionally substituted with one to three substituents independently selected from the group consisting of halo, cyano, Ci -6 alkyl, OR y , C(0)R y , and
- Y is absent or CR y R z ;
- the compounds of the present invention may have asymmetric centers, chiral axes, and chiral planes (as described in: E.L. Eliel and S.H. Wilen, Stereochemistry of Carbon Compounds, John Wiley & Sons, New York, 1994, pages 1119-1190), and occur as racemates, racemic mixtures, and as individual diastereomers, with all possible isomers and mixtures thereof, including optical isomers, all such stereoisomers being included in the present invention.
- the compounds disclosed herein may exist as tautomers and both tautomeric forms are intended to be encompassed by the scope of the invention, even though only one tautomeric structure is depicted.
- Isotopically-enriched compounds within generic Formula I can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
- any variable e.g. Ry
- any variable e.g. Ry
- its definition on each occurrence is independent at every other occurrence.
- combinations of substituents and variables are permissible only if such combinations result in stable
- substituents and substitution patterns on the compounds of the instant invention can be selected by one of ordinary skill in the art to provide compounds that are chemically stable and that can be readily synthesized by techniques known in the art, as well as those methods set forth below, from readily available starting materials. If a substituent is itself substituted with more than one group, it is understood that these multiple groups may be on the same carbon or on different carbons, so long as a stable structure results.
- the phrase "optionally substituted with one or more substituents” should be taken to be equivalent to the phrase “optionally substituted with at least one substituent” and in such cases another embodiment will have from zero to three substituents.
- heterocycle or “heterocyclyl” as used herein is intended to mean a 3- to 10-membered aromatic or nonaromatic heterocycle containing from 1 to 4 heteroatoms selected from the group consisting of O, N and S, and includes bicyclic groups.
- heterocyclic is also considered to be synonymous with the terms “heterocycle” and “heterocyclyl” and is understood as also having the definitions set forth herein.
- Heterocyclyl therefore includes the above mentioned heteroaryls, as well as dihydro and tetrathydro analogs thereof.
- tetrahydrothiopyranyl tetrahydroisoquinolinyl, tetrazolyl, tetrazolopyridyl, thiadiazolyl, thiazolyl, thienyl, triazolyl, 1 ,4-dioxanyl, hexahydroazepinyl, piperazinyl, piperidinyl, pyridin- 2-onyl, pyrrolidinyl, morpholinyl, thiomorpholinyl, dihydrobenzoimidazolyl,
- dihydrooxadiazolyl dihydrooxazolyl, dihydropyrazinyl, dihydropyrazolyl, dihydropyridinyl, dihydropyrimidinyl, dihydropyrrolyl, dihydroquinolinyl, dihydrotetrazolyl, dihydrothiadiazolyl, dihydrothiazolyl, dihydrothienyl, dihydrotriazolyl, dihydroazetidinyl, dioxidothiomorpholinyl, methylenedioxybenzoyl, tetrahydrofuranyl, and tetrahydrothienyl, and N-oxides thereof.
- a compound of the instant invention may also be useful for treating cancer in combination with the following therapeutic agents: abarelix (Plenaxis depot®); aldesleukin (Prokine®); Aldesleukin (Proleukin®); Alemtuzumabb (Campath®); alitretinoin (Panretin®); allopurinol (Zyloprim®); altretamine (Hexalen®); amifostine (Ethyol®); anastrozole
- Arimidex® arsenic trioxide (Trisenox®); asparaginase (Elspar®); azacitidine (Vidaza®); bendamustine hydrochloride (Treanda®); bevacuzimab (Avastin®); bexarotene capsules
- Meethosarb® capecitabine (Xeloda®); carboplatin (Paraplatin®); carmustine (BCNU®, BiCNU®); carmustine (Gliadel®); carmustine with Polifeprosan 20 Implant (Gliadel Wafer®); celecoxib (Celebrex®); cetuximab (Erbitux®); chlorambucil (Leukeran®); cisplatin
- Platinum® cladribine (Leustatin®, 2-CdA®); clofarabine (Clolar®); cyclophosphamide (Cytoxan®, Neosar®); cyclophosphamide (Cytoxan Injection®); cyclophosphamide (Cytoxan Tablet®); cytarabine (Cytosar-U®); cytarabine liposomal (DepoCyt®); dacarbazine (DTIC- Dome®); dactinomycin, actinomycin D (Cosmegen®); dalteparin sodium injection
- fludarabine Fludarabine
- fiuorouracil 5-FU
- fulvestrant Fludarabine
- gefitinib Iressa®
- geldanamycin gemcitabine
- gemtuzumab ozogamicin Mylotarg®
- goserelin acetate Zoladex Implant®
- goserelin acetate Zoladex®
- histrelin acetate Histrelin implant®
- hydroxyurea Hydrea®
- Ibritumomab Tiuxetan Zevalin®
- cytotoxic/cytostatic agent an antiproliferative agent, a prenyl-protein transferase inhibitor, an HMG-CoA reductase inhibitor, an HIV protease inhibitor, a reverse transcriptase inhibitor, an angiogenesis inhibitor, a PPAR- ⁇ agonist, a PPAR- ⁇ agonist, an inhibitor of inherent multidrug resistance, an anti-emetic agent, an agent useful in the treatment of anemia, an agent useful in the treatment of neutropenia, an immunologic-enhancing drug, an inhibitor of cell proliferation and survival signaling, an apoptosis inducing agent, a bisphosphonate, an aromatase inhibitor, an siR A therapeutic ⁇ -secretase inhibitors, agents that interfere with receptor tyrosine kinases (RTKs), an agent that interferes with a cell cycle checkpoint and any of the therapeutic agents listed above.
- RTKs receptor tyrosine kinases
- compounds of the instant invention may also be applicable to any one or more of the therapeutic agents to be used in the combination treatment (hereinafter refered to as the "second therapeutic agent").
- the specific dosage and dosage schedule of this second therapeutic agent can further vary, and the optimal dose, dosing schedule and route of administration will be determined based upon the specific second therapeutic agent that is being used.
- a total treatment period can be decided for a compound of the instant invention.
- the second therapeutic agent can be administered prior to onset of treatment with a compound of the instant invention or following treatment with a compound of the mstant invention.
- anti-cancer treatment can be administered during the period of administration of a compound of the instant invention but does not need to occur over the entire treatment period of a compound of the instant invention.
- terapéuticaally effective amount means that amount of active compound or pharmaceutical agent that elicits the biological or medicinal response in a tissue, system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician.
- treating cancer refers to administration to a mammal afflicted with a cancerous condition and refers to an effect that alleviates the cancerous condition by killing the cancerous cells, but also to an effect that results in the inhibition of growth and/or metastasis of the cancer.
- the angiogenesis inhibitor to be used as the second compound is selected from a tyrosine kinase inhibitor, an inhibitor of epidermal-derived growth factor, an inhibitor of fibroblast-derived growth factor, an inhibitor of platelet derived growth factor, an MMP (matrix metalloprotease) inhibitor, an integrin blocker, interferon-ct, interleukin-12, pentosan poly sulfate, a cyclooxygenase inhibitor, carboxyamidotriazole, combretastatin A-4, squalamine, 6-0-chloroacetyl-carbonyl)-fumagillol, thalidomide, angiostatin, troponin- 1, or an antibody to VEGF.
- a tyrosine kinase inhibitor an inhibitor of epidermal-derived growth factor, an inhibitor of fibroblast-derived growth factor, an inhibitor of platelet derived growth factor, an MMP (matrix metalloprotease) inhibitor, an integrin
- antiproliferative agent a prenyl-protein transferase inhibitor, an HMG-CoA reductase inhibitor, an HIV protease inhibitor, a reverse transcriptase inhibitor, an angiogenesis inhibitor, a PPAR- ⁇ agonist, a PPAR- ⁇ agonist; an inhibitor of cell proliferation and survival signaling, a bisphosphonate, an aromatase inhibitor, an siRNA therapeutic and an agent that interferes with a cell cycle checkpoint.
- a method of treating or preventing a disease in which angiogenesis is implicated which is comprised of administering to a mammal in need of such treatment a therapeutically effective amount of a compound of the present invention.
- Other inhibitors of MET may also be administered for this method of treatment.
- Ocular neovascular diseases which may result in certain forms of blindness, are examples of conditions where much of the resulting tissue damage can be attributed to aberrant infiltration of blood vessels in the eye.
- Ophthalmic pharmaceutical compositions that are adapted for topical administration to the eye may be in the form of solutions, suspensions, ointments, creams or as a solid insert.
- Ophthalmic formulations of this compound may contain from 0.01 ppm to 1% and especially 0.1 ppm to 1% of medicament.
- For a single dose from between 0.01 to 5000 ng, preferably 0.1 to 500 ng, and especially 1 to 100 ng of the compound can be applied to the human eye.
- Formulations useful for intravitreal administration are similar to saline solutions described previously for intravenous administration.
- the compounds of this invention may be prepared by employing reactions as shown in the following schemes, in addition to other standard manipulations that are known in the literature or exemplified in the experimental procedures.
- the illustrative schemes below are not limited by the compounds listed or by any particular substituents employed for illustrative purposes. Substituent numbering as shown in the schemes does not necessarily correlate to that used in the claims and often, for clarity, a single substituent is shown attached to the compound where multiple substituents are allowed under the definitions of the instant invention hereinabove.
- DIBAL diisobutylaluminum hydride
- NSAID non-steroidal anti-inflammatory drug
- PdCl 2 (dppf) 2 1 , 1 '-Bis(diphenylphosphino)ferrocene- palladium(II)dichloride
- TBTU ⁇ -(benzotriazol- 1 -yl)-N, ⁇ , ⁇ ', N'-tetramethyluronium tetrafluoroborate
- Enol ether intermediate III is treated with an appropriate acid, such as TFA, to afford the corresponding dihydropyridone IV.
- Dihydropyridone IV is treated with an appropriate reducing reagent such as first by NaB3 ⁇ 4 then TFA Et 3 SiH to afford the
- Diamino compounds IX is treated with CDI in appropriate solvent such as pyridine to afford the substituted urea X (General Scheme 4).
- Step 1 Preparation of 3-oxo-3-(tetrahydro-2H-thiopyran-4-yl)propanenitrile
- Step 2 Preparation of 5-(tetrahydro-2H-thiopyran-4-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo [1,5 -a]pyrimidin-7-amine
- Step 7 Preparation of 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenyIpyridin-3-yl)- 5-(tetrahydro-l , 1 -dioxido-2H-thiopyran-4-yI)pyrazolo[ 1 ,5-a]pyrimidine-6-carbaldehyde
- Step 9 Preparation of 6-((methylamino)methyl)-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-l,l- dioxido-2H-thiopyran-4-yl)pyrazolo [ 1 , 5 -a]pyrimidin-7-amine
- Step 10 Preparation of 3-methyl-7-(6-phenylpyridin-3-yl)-5-(tetrahydro-l,l-dioxido-2H- thiopyran-4-yl)-3 ,4-dihydro-pyrazolo[ 1 ,5-a]pyrimido [5 ,4-e] pyrimidin-2( 1 H)-one
- CDI (10 mg) was added to a mixture of 6-((methylamino)methyl)-3-(6- phenylpyridin-3-yl)-5-(tetrahydro- 1 ,1 -dioxido-2H-thiopyran-4-yl)pyrazolo[ 1 ,5-a]pyrimidu>7- amine (3.2 mg) in pyridine (0.2 mL).
- Step 2 Preparation of 7-(qumolin-3-yl)-5-(tetrahydro-l,l-dioxido-2H-thiopyran-4-yl)-2,4- dihydro-lH-[ 1 ,3]oxazino[5,4-e]pyrazolo[ 1 ,5-a]pyrimidine
- Step 4 Preparation of trans- ⁇ -tert-Butyl 4-(7-aminopyrazolo[l,5-a]pyrimidin-5- yl)cyclohexanecarboxylate
- Step 6 Preparation of trans-l-tert-Butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3- iodopyrazolo[ 1 ,5-a]pyrimidin-5-yl)cyclohexanecarboxylate
- Step 7 Preparation of traw-tert-Butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)pyrazolo[l,5-a]pyrimidin-5-yl)cyclohexanecarboxylate
- Step 8 Preparation of traiw-tert-Butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6- bromo-3-(6-phenylpyridin-3-yl)pyrazolo[l,5-a]pyrimidin-5-yl)cyclohexanecarboxylate
- N-bromosuccinimide (71 mg, 0.394 mmol) was added to a solution of trans-teri- butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3-yl)pyrazolo[l,5- a]pyrimidin-5-yl)cyclohexanecarboxylate (240 mg, 0.329 mmol) in acetonitrile (3 mL) and dichloromethane (2 ml). The resulting solution is stirred at room temperature for 1 hour.
- Step 9 tert-Butyl 4-(7-(bis((2-(trimethylsilyl)emoxy)methyl)amino)-3-(6-phenylpyridin-3-yl)- 6-vinylpyrazolo [ 1 ,5-a]pyrimidin ⁇ 5 -yl)cyclohexanecarboxylate
- Tributyl(vinyl)tin (271 mg, 0.855 mmol) and Pd(PPh 3 ) 4 (33 mg, 0.029 mmol) was added to a solution of traTO-tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6- bromo-3 -(6-phenylpyridin-3 -yl)pyrazolo [ 1 , 5 -a] pyrimidin-5 -yl)cyclohexanecarboxy late (230 mg, 0.285 mmol) in 1,4-dioxane (2 mL).
- the reaction is heated to 90°C under argon for 16 hours. After 16 hours, the solvent is removed in vacuo and the residue is purified via silica gel chromatography (0% to 30% ethyl acetate in hexanes gradient) to yield the title compound as yellow oil.
- Step 10 Preparation of terf-Butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-(l 5 2- dihydroxyethyl)-3-(6-phenylpyridin-3-yl)pyrazolo[l,5-a]pyrimidin-5- yl)cyclohexanecarboxylate
- Step 11 Preparation of tra w-4-(4-(Hydroxymethyl)-7-(6-phenylpyridin-3-yl)-2 ? 4 ⁇ dihydro-l H- [1 ,3]oxazino[5,4-e]pyrazolo[l ,5-a]pyrimidin-5-yl)cyclohexanecarboxylic acid
- Step 1 Preparation of pyrazolo[l,5-a]pyrimidine ⁇ 5,7-diol
- Step 2 Preparation of 5,7-dichloropyrazolo[l,5-a]pyrimidine To pyrazolo[l,5-a]pyrimidine-5,7-dioI (9.6 g, 63.5 mmol) in a 500 mL flask was added P0C1 3 (125 mL, 1341.1 mmol). The flask was then cooled to 0°C and N 5 N- dimethylaniline (22 mL, 173.6 mmol) was carefully added. On warming to room temperature, the reaction was then heated at 60°C under an atmosphere of argon for 16 hours. On cooling, the reaction mixture was concentrated in vacuo to give a brown viscous liquid.
- Step 4 Preparation of 5-chloro-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5- aj pyrimidin-7-amine
- Step 5 Preparation of tert-butyl 3 ⁇ (trifluoromethylsulfonyloxy)-8-azabicyclo[3.2.1]oct-3-ene ⁇ 8-carboxylate
- Step 6 Preparation of tert-butyl 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-8- azabicyclo[3.2. l]oct-2-ene-8-carboxylate
- Step 8 Preparation of anti & syn - tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl) amino)pyrazolo[l ,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1 ]oct-2-ene-8-carboxylate
- Step 9 Preparation of tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3- iodopyrazolo[ 1 ,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1 ]octane-8-carboxylate
- Step 10 Preparation of fert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)pyrazolo[l,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1]octane-8-carboxylate
- Step 2 Preparation of tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-(2- hy droxypropan-2-yl)pyridin-3 -yl)pyrazolo [1,5-a] pyrimidin- 5 -yI)-8 -azabicyclo [3.2.1 ]octane- 8 - carboxylate
- Step 1 Preparation of 8-azabicyclo[3.2.1]octan-3-yl)-3-(6-phenylpyridin-3-yl)-8,9- dihydropyrazolo[l,5-a]pyrido[3 ! 2-e]pyrimidin-6(7H)-one
- a]pyrimidin ⁇ 5-yl)-8-azabicyclo[3.2A]octane ⁇ 8-carboxylate (834 mg, 0.49 mmol), tributyl(l- ethoxyvinyl)tin (356 mg, 0.98 mmol), tetrakis(triphenylphosphine)palladium (56.9 mg, 0.049 mmol) in dioxane (6 mL) was degassed with argon for five minutes.
- Step 2 Preparation of 8-azabicyclo[3.2.1]octan-3-yl)-3-(6-phenylpyridin-3-yl)-6 J 7 J 8,9- tetrahydropyrazolo[l,5-a]pyrido[3,2 ⁇ e]pyrimidine
- Step 2 Preparation of 5-((2R,6S)-2,6-dimet ylmorpholino)-3-(6-phenylpyridin-3- yl)pyrazolo [ 1 , 5-a]pyrimidin ⁇ 7-amine
- Step 3 Preparation of 6-bromo-5-((2R,6S)-2,6-dimethylmorpholino)-3-(6-phenylpyridin-3- yl)pyrazolo[l ,5-a]pyrimidin-7-amine
- the mTOR assay buffer contains 10 mM hepes (pH 7.4), 50 raM NaCl, 100 Mg/ml BSA, 50 mM B-glycerophosphate, 10 mM MnC12 and 0.5 mM DTT. 20 ng of mTOR enzyme is preincubated with the compound for 10 minutes. 5 ⁇ ATP and 0.1 ⁇ GSTS6K is added. The reaction is incubated for one hour at 30°C. Anti phospho p70S6K (about 1.7 ng/well) and anti GSTXL665 (1 :1 Ratio with the substrate GSTS6K) are added after incubating. The plates are read at least 2 hours after adding the anti phospho p70S6K and the anti GSTXL665.
- This in vitro assay utilizes recombinant His-CHKl expressed in the baculovirus expression system as an enzyme source and a biotinylated peptide based on CDC25C as substrate (biotin-RSGLYRSPSMPENLNRPR).
- Staurosporine 100 g: CALBIOCHEM, Cat. # 569397
- ICgti DETERMINATIONS Dose-response curves were plotted from inhibition data generated, each in duplicate, from 8 point serial dilutions of inhibitory compounds.
- Concentration of compound was plotted against % kinase activity, calculated by CPM of treated samples divided by CPM of untreated samples. To generate IC50 values, the dose- response curves were then fitted to a standard sigmoidal curve and IC50 values were derived by nonlinear regression analysis.
- Selected Thiazole Derivatives of the present invention were tested using this assay and provided IC50 values ranging from about 1 nM to about 5500 nM.
- BACULOVIRUS CONSTRUCTIONS Cyclin E was cloned into pVLl 393 (Pharmingen, La Jolla, California) by PCR, with the addition of 5 histidine residues at the amino-terminal end to allow purification on nickel resin. The expressed protein was approximately 45kDa.
- CD 2 was cloned into pVL1393 by PCR, with the addition of a haemaglutinin epitope tag at the carboxy-terminal end (YDVPDYAS). The expressed protein was approximately 34kDa in size.
- ENZYME PRODUCTION Recombinant baculoviruses expressing cyclin E and CDK2 were co-infected into SF9 cells at an equal multiplicity of infection (MOI ⁇ S), for 48 hrs. Cells were harvested by centrifugation at 1000 RPM for 10 minutes, then pellets lysed on ice for 30 minutes in five times the pellet volume of lysis buffer containing 50mM Tris pH 8.0, 150mM NaCl, 1% NP40, ImM DTT and protease inhibitors (Roche Diagnostics GmbH, Mannheim,
- Cyclin E/CDK2 kinase assays can be performed as described below in low protein binding 96-well plates (Corning Inc, Corning, New York).
- Enzyme is diluted to a final concentration of 50 ] ⁇ Jmh in kinase buffer containing 50mM Tris pH 8.0, 10 mM MgCl 2; l mM DTT, and 0.1 raM sodium orthovanadate.
- the substrate used in these reactions is a biotinylated peptide derived from Histone HI (from Amersham, UK). The substrate is thawed on ice and diluted to 2 ⁇ in kinase buffer. Test compounds are diluted in 10% DMSO to desirable concentrations. For each kinase reaction, 20 ⁇ xL of the 50 ⁇ g/mL enzyme solution (1 ⁇ g of enzyme) and 20 ⁇ of the 2 ⁇ substrate solution are mixed, then combined with 10 ⁇ of diluted compound in each well for testing. The kinase reaction is initiated by addition of 50 ⁇ of 2 ⁇ ATP and 0.1 ⁇ of 33P-ATP (from
- ICgn DETERMINATIONS Dose-response curves are plotted from inhibition data generated, each in duplicate, from 8 point serial dilutions of inhibitory compounds. Concentration of compound is plotted against % kinase activity, calculated by CPM of treated samples divided by CPM of untreated samples. To generate IC50 values, the dose-response curves are then fitted to a standard sigmoidal curve and IC 5 o values can be derived using nonlinear regression analysis.
Abstract
This invention relates to novel fused tricyclic compounds that are inhibitors of mammalian Target of Rapamycin (mTOR) kinase, which is also known as FRAP, RAFT, RAPT or SEP, and are useful in the treatment of cellular proliferative diseases, for example cancer and other proliferative disorders.
Description
FUSED TRICYCLIC INHIBITORS OF MAMMALIAN TARGET OF RAPAMYCIN
BACKGROUND OF THE INVENTION
This invention relates to novel fused tricyclic compounds that are inhibitors of mammalian Target of Rapamycin (mTOR) kinase, which is also known as FRAP, RAFT, RAPT or SEP, and are useful in the treatment of cellular proliferative diseases, for example cancer and other proliferative disorders.
The mammalian target of rapamycin (mTOR) is a central regulator of cell growth and proliferation and plays a gatekeeper role in the control of cell cycle progression. mTOR mediates mitgenic signals from P13K/AKT through to the downstream targets S6K1 and 4E-BP1 and to Ser 473 on AKT. Recently, it has been shown that mTOR exists in two complexes. Rator-mTOR complex (mTORCl) is a rapamycin- sensitive complex that phosphorylates S6K1 (ribosomal S6 kinase 1) and 4E-BP1 (eukaryotic translation initiation factor 4E-binding protein). Rictor-mTOR complex (mTORC2) is a rapamycin-insensitive complex that phosphorlates AKT. Although the precise mechanism by which rapamycin inhibitis mTOR function is not well understood, rapamycin partially inhibits mTOR function through mTORCl . Since mTORC2 is involved in the regulation of cell survival and actin cytoskeletal organization in a rapamycin-independent manner, complete inhibition of mTOR function through inhibition of both mTORCl and mTORC2 may lead to a broader spectrum antitumor activity and/or better efficacy than through inhibition of mTORCl alone.
There exists a need in the art for small-molecule compounds having desirable physiochemical properties that are useful for treating cancer and other proliferative disorders. Specifically, there exists a need for small molecule inhibitors of mTOR kinase that block signaling through mTORCl and mTORC2 for treating cancer and other cell proliferative diseases.
SUMMARY OF THE INVENTION
The present invention relates to novel fused tricyclic derivatives, that are useful for treating cancer and other cellular proliferative diseases, for treating disorders associated with mTOR activity, and for inhibiting the mTOR kinase. The compounds of the invention may be illustrated by the Formula I:

The compounds of this invention are useful in the inhibition of mTOR kinase, and are illustrated by a compound of the formula:

E is CRyRz, O, C(0), NRy or SOm;
R1 is hydrogen, halo, -C(0)R5, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3.g cycloalkyi, C3.8 cycloalkenyl, aryl, heteroaryl or heterocyclyl, wherein said alkyl, alkenyl, alkynyl, cycloalkyi, cycloalkenyl, aryl, heteroaryl and heterocyclyl groups are optionally substituted with one to three substituents independently selected from the group consisting of Rs, halo, cyano, C1-6 alkyl, Cw haloalkyl, (CRyRz)mORy, (CRyRz)mNRyRz, C(0)Ry, C(0)ORy, SOmRy and
C(0)NRyRz;
R2 is hydrogen, halo, cyano, NRyRz, ORy, C1-6 alkyl or Ci-6 haloalkyl;
R3 is C3„8 cycloalkyi, C3-8 cycloalkenyl, aryl, heteroaryl or heterocyclyl, O(heterocyclyl), SOm(heterocyclyl), C3-6 alkyl, (Ci-6 alkyl)NHR5, (Cw alkyl)NHCORx, wherein said cycloalkyi, cycloalkenyl, aryl, heteroaryl and heterocyclyl groups are optionally substituted with one to three substituents independently selected from the group consisting of R5, halo, cyano, C1-(s alkyl, C1-6 haloalkyl, ORy, 0(Ci-6 haloalkyl), 0(Ci-6 alkyl)ORy, C(0)Ry, C(0)ORy,
C(0)CRxRyRz, SOmRy, NRyRz, C(0)NRyRz, C(0)heteroaryl and NHCORy;

Wherein U is N or CH;
X is absent, CRyRz, O or C(O);
Y is absent, CRyRz, O, or C(O);
L is absent or (CRyRz)n;
T is absent or (CRyRz)n;
Z is absent or (CRyRz)n;
R4 is hydrogen, halo, C\.$ alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyi, C3-8 cycloalkenyl, aryl, heteroaryl or heterocyclyl, wherein said alkyl, alkenyl, alkynyl, cycloalkyi, cycloalkenyl, aryl, heteroaryl, and heterocyclyl groups are optionally substituted with one to three
substituents independently selected from the group consisting of R , halo, cyano, Ci-6 alkyl, Cj. 6 haloalkyl, ORy, 0(C1-6 haloalkyl), C(0)Ry, C(0)ORy, SOmRy, C(0)NRyRz and NRyRz;
Rs is C3.8 cycloalkyl, C3,g cycloalkenyl, aryl, heteroaryl or heterocyclyl, wherein said cycloalkyl, cycloalkenyl, aryl, heteroaryl and heterocyclyl groups are optionally substituted with one to three substituents independently selected from the group consisting of halo, cyano, Ci_6 alkyl, CI-6 haloalkyl, ORy, 0(C,-6 haloalkyl), C(0)Ry, C(0)ORy, SOmRy and NRyRz;
R6 is selected from the group consisting of hydrogen, halo, cyano, Ci-6 alkyl, Ci-6 haloalkyl, (C1-6 alkyl)ORy, ORy, 0(Ci„6 haloalkyl), C(0)R C(0)OR , SOmRy and NRyRz;
R7 is selected from the group consisting of hydrogen, halo, cyano, C1-($ alkyl, Ci-6 haloalkyl, ORy, 0(Ci-6 haloalkyl), C(0)Ry, C(0)ORy, SOmRy and NR Rz;
Rx is hydrogen or C1-6 alkyl;
Ry is hydrogen, Ci-6 alkyl or C1-6 haloalkyl, wherein said alkyl group is optionally substituted with one to three hydroxyl;
Rz is hydrogen, C1-6 alkyl or Cj.6 haloalkyl, wherein said alkyl group is optionally substituted with one to three hydroxyl;
m is an integer from zero to two;
n is an integer from zero to two;
or a pharmaceutically acceptable salt thereof.
In a class of the invention, D is CRyRz or C(O).
In a class of the invention, E is CRyRz, O or NRy.
In a class of the invention R1 is heteroaryl, wherein said heteroaryl group is optionally substituted with one to three substituents independently selected from the group consisting of R5, halo, cyano, Cj.6 alkyl, Ci.6 haloalkyl, (CRyRz)mORy, (CRyRz)mNRyRz,
C(0)Ry, C(0)ORy, SOmRy and C(0)NRyRz.
In a class of the invention, R is hydrogen.
In a class of the invention,R is C3-8 cycloalkyl or heterocyclyl, wherein said cycloalkyl and heterocyclyl groups are optionally substituted with one to three substituents independently selected from the group consisting of halo, cyano, Ci-6 alkyl, ORy, C(0)Ry, and
C(0)ORy;

Y is absent or CRyRz;
L is absent or (CRyRz)„;
T is absent or (CRyRz)n;
Z is absent or (CRyRz)n.
In a class of the invention, R6 is selected from the group consisting of hydrogen, halo, Ci.6 alkyl and (Ci-6 alkyl)ORy.
In a class of the invention, R7 is hydrogen.
Specific examples of the compounds of the instant invention include, but are not limited to:


or a pharmaceutically acceptable salt or stereoisomer thereof.
The compounds of the present invention may have asymmetric centers, chiral axes, and chiral planes (as described in: E.L. Eliel and S.H. Wilen, Stereochemistry of Carbon Compounds, John Wiley & Sons, New York, 1994, pages 1119-1190), and occur as racemates, racemic mixtures, and as individual diastereomers, with all possible isomers and mixtures thereof, including optical isomers, all such stereoisomers being included in the present invention. In addition, the compounds disclosed herein may exist as tautomers and both tautomeric forms are intended to be encompassed by the scope of the invention, even though only one tautomeric structure is depicted.
In the compounds of generic Formula I, the atoms may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature. The present invention is meant to include all suitable isotopic variations of the compounds of generic Formula I. For example, different isotopic forms of hydrogen (H) include protium (1H) and deuterium (2H). Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples.
Isotopically-enriched compounds within generic Formula I can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
When any variable (e.g. Ry) occurs more than one time in any constituent, its definition on each occurrence is independent at every other occurrence. Also, combinations of substituents and variables are permissible only if such combinations result in stable
compounds. Lines drawn into the ring systems from substituents represent that the indicated bond may be attached to any of the substitutable ring atoms. If the ring system is polycyclic, it is intended that the bond be attached to any of the suitable carbon atoms on the proximal ring only.
It is understood that substituents and substitution patterns on the compounds of the instant invention can be selected by one of ordinary skill in the art to provide compounds that are chemically stable and that can be readily synthesized by techniques known in the art, as well as those methods set forth below, from readily available starting materials. If a substituent is itself substituted with more than one group, it is understood that these multiple groups may be on the same carbon or on different carbons, so long as a stable structure results. The phrase "optionally substituted with one or more substituents" should be taken to be equivalent to the phrase "optionally substituted with at least one substituent" and in such cases another embodiment will have from zero to three substituents.
As used herein, "alkyl" is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms. For example, Cl-ClO, as in "C1-C10 alkyl" is defined to include groups having 1, 2, 3, 4, 5, 6, 7, 8, 9 or 10 carbons in a linear or branched arrangement. For example, "Cj-Cio alkyl" specifically includes methyl, ethyl, ^-propyl, /-propyl, iz-butyl, t-butyl, /-butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, and so on. The term "cycloalkyl" means a monocyclic saturated aliphatic hydrocarbon group having the specified number of carbon atoms. For example, "cycloalkyl" includes cyclopropyl, methyl-cyclopropyl, 2,2-dimethyl-cyclobutyl, 2-ethyl-cycIopentyl, cyclohexyl, and so on. In an embodiment of the invention the term "cycloalkyl" includes the groups described immediately above and further includes monocyclic unsaturated aliphatic hydrocarbon groups. For example, "cycloalkyl" as defined in this embodiment includes cyclopropyl, methyl-cyclopropyl, 2,2-dimethyl-cyclobutyl, 2-ethyl-cyclopentyl, cyclohexyl, cyclopentenyl, cyclobutenyl and so on.
The term "haloalkyl" means an alkyl radical as defined above, unless otherwise specified, that is substituted with one to five, preferably one to three halogen. Representative examples include, but are not limited to trifluoromethyl, dichloroethyl, and the like.
"Alkoxy" represents either a cyclic or non-cyclic alkyl group of indicated number of carbon atoms attached through an oxygen bridge. "Alkoxy" therefore encompasses the definitions of alkyl and cycloalkyl above.
If no number of carbon atoms is specified, the term "alkenyl" refers to a non- aromatic hydrocarbon radical, straight or branched, containing from 2 to 10 carbon atoms and at least 1 carbon to carbon double bond. Preferably 1 carbon to carbon double bond is present,
and up to 4 non-aromatic carbon-carbon double bonds may be present. Thus, "C2-Cg alkenyl" means an alkenyl radical having from 2 to 6 carbon atoms. Alkenyl groups include ethenyl, propenyl, butenyl and cyclohexenyl. As described above with respect to alkyl, the straight, branched or cyclic portion of the alkenyl group may contain double bonds and may be substituted if a substituted alkenyl group is indicated.
The term "alkynyl" refers to a hydrocarbon radical straight or branched, containing from 2 to 10 carbon atoms, unless otherwise specified, containing at least 1 carbon to carbon triple bond. Up to 3 carbon-carbon triple bonds may be present. Thus, "C2-C6 alkynyl" means an alkynyl radical having from 2 to 6 carbon atoms. Alkynyl groups include ethynyl, propynyl and butynyl. As described above with respect to alkyl, the straight, branched or cyclic portion of the alkynyl group may contain triple bonds and may be substituted if a substituted alkynyl group is indicated.
In certain instances, substituents may be defined with a range of carbons that includes zero, such as (Co-C6)alkylene-aryL If aryl is taken to be phenyl, this definition would include phenyl itself as well as -CtfePh, -CH2CH2Ph, CH(CH3)CH2CH(CH3)Ph, and so on.
As used herein, "aryl" is intended to mean any stable monocyclic or bicyclic carbon ring of up to 7 atoms in each ring, wherein at least one ring is aromatic. Examples of such aryl elements include phenyl, naphthyl, tetrahydronaphthyl, indanyl and biphenyl. In cases where the aryl substituent is bicyclic and one ring is non-aromatic, it is understood that attachment is via the aromatic ring.
The term "heteroaryl," as used herein, represents a stable monocyclic or bicyclic ring of up to 7 atoms in each ring, wherein at least one ring is aromatic and contains from 1 to 4 heteroatoms selected from the group consisting of O, N and S. Heteroaryl groups within the scope of this definition include but are not limited to: acridinyl, carbazolyl, cinnolinyl, quinoxalinyl, pyrrazolyl, indolyi, benzotriazolyl, furanyl, thienyl, benzothienyl, benzofuranyl, benzimidazolonyl, benzoxazolonyl, quinolinyl, isoquinolinyl, dihydroisoindolonyl, imidazopyridinyl, isoindolonyl, mdazolyl, oxazolyl, oxadiazolyl, isoxazolyl, indolyi, pyrazinyl, pyridazinyl, pyridinyl, pyrimidinyl, pyrrolyl, tetrahydroquinoline. As with the definition of heterocycle below, "heteroaryl" is also understood to include the N-oxide derivative of any nitrogen-containing heteroaryl. In cases where the heteroaryl substituent is bicyclic and one ring is non-aromatic or contains no heteroatoms, it is understood that attachment is via the aromatic ring or via the heteroatom containing ring, respectively.
The term "heterocycle" or "heterocyclyl" as used herein is intended to mean a 3- to 10-membered aromatic or nonaromatic heterocycle containing from 1 to 4 heteroatoms selected from the group consisting of O, N and S, and includes bicyclic groups. For the purposes of this invention, the term "heterocyclic" is also considered to be synonymous with the terms "heterocycle" and "heterocyclyl" and is understood as also having the definitions set forth herein. "Heterocyclyl" therefore includes the above mentioned heteroaryls, as well as
dihydro and tetrathydro analogs thereof. Further examples of "heterocyclyl" include, but are not limited to the following: azetidinyl, benzoimidazolyl, benzofuranyl, benzofurazanyl, benzopyrazolyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, carbazolyl, carbolinyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazolyl, oxooxazolidinyl, oxazolyl, oxazoline, oxopiperazinyl, oxopyrrolidinyl, oxomorpholinyl, isoxazolme, oxetanyl, pyranyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridopyridinyl, pyridazinyl, pyridyl, pyrimidyl, pyrrolyl, quinazolinyl, quinolyl, quinoxalinyl, tetrahydropyranyl, tetrahydrofuranyl,
tetrahydrothiopyranyl, tetrahydroisoquinolinyl, tetrazolyl, tetrazolopyridyl, thiadiazolyl, thiazolyl, thienyl, triazolyl, 1 ,4-dioxanyl, hexahydroazepinyl, piperazinyl, piperidinyl, pyridin- 2-onyl, pyrrolidinyl, morpholinyl, thiomorpholinyl, dihydrobenzoimidazolyl,
dihydrobenzofuranyl, dihydrobenzothiophenyl, dihydrobenzoxazolyl, dihydrofuranyl, dihydroimidazolyl, dihydroindolyl, dihydroisooxazolyl, dihydroisothiazolyl,
dihydrooxadiazolyl, dihydrooxazolyl, dihydropyrazinyl, dihydropyrazolyl, dihydropyridinyl, dihydropyrimidinyl, dihydropyrrolyl, dihydroquinolinyl, dihydrotetrazolyl, dihydrothiadiazolyl, dihydrothiazolyl, dihydrothienyl, dihydrotriazolyl, dihydroazetidinyl, dioxidothiomorpholinyl, methylenedioxybenzoyl, tetrahydrofuranyl, and tetrahydrothienyl, and N-oxides thereof.
Attachment of a heterocyclyl substituent can occur via a carbon atom or via a heteroatom.
As appreciated by those of skill in the art, "halo" or "halogen" as used herein is intended to include chloro, fluoro, bromo and iodo.
The alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl and heterocyclyl substituents may be substituted or unsubstituted, unless specifically defined otherwise. For example, a (Ci-C(>)alkyl may be substituted with one, two Or three substituents selected from
OH, oxo, halogen, alkoxy, dialkylamino, or heterocyclyl, such as morpholinyl, piperidinyl, and so on. In this case, if one substituent is oxo and the other is OH, the following are included in the definition: -CO)CH2CH(OH)CH3, -(C=0)OH, -CH2(OH)CH2CH(0), and so on.
Included in the instant invention is the free form of compounds of the instant invention, as well as the pharmaceutically acceptable salts and stereoisomers thereof. The term "free form" refers to the amine compounds in non-salt form. The encompassed
pharmaceutically acceptable salts not only include the salts exemplified for the specific compounds described herein, but also all the typical pharmaceutically acceptable salts of the free form of compounds of the instant invention. The free form of the specific salt compounds described may be isolated using techniques known in the art. For example, the free form may be regenerated by treating the salt with a suitable dilute aqueous base solution such as dilute aqueous NaOH, potassium carbonate, ammonia and sodium bicarbonate. The free forms may differ from their respective salt forms somewhat in certain physical properties, such as solubility in polar solvents, but the acid and base salts are otherwise pharmaceutically equivalent to their respective free forms for purposes of the invention.
The pharmaceutically acceptable salts of the instant compounds can be synthesized from the compounds of this invention which contain a basic or acidic moiety by conventional chemical methods. Generally, the salts of the basic compounds are prepared either by ion exchange chromatography or by reacting the free base with stoichiometric amounts or with an excess of the desired salt-forming inorganic or organic acid in a suitable solvent or various combinations of solvents. Similarly, the salts of the acidic compounds are formed by reactions with the appropriate inorganic or organic base.
Thus, pharmaceutically acceptable salts of the compounds of this invention include the conventional non-toxic salts of the compounds of this invention as formed by reacting a basic instant compound with an inorganic or organic acid. For example,
conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like, as well as salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxy-benzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, trifiuoroacetic and the like.
When the compound of the present invention is acidic, suitable "pharmaceutically acceptable salts" refers to salts prepared form pharmaceutically acceptable non-toxic bases including inorganic bases and organic bases. Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium and sodium salts. Salts derived from
pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion exchange resins, such as arginine, betaine caffeine, choline, Ν,Ν1- dibenzylethylenediamine, diethylamin, 2-diethylaminoethanol, 2-dimethylaminoethanoI, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine,
glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamme tripropylamine, tromethamme and the like. When the compound of the present invention is acidic, the term "free form" refers to the compound in its non-salt form, such that the acidic functionality is still protonated.
The preparation of the pharmaceutically acceptable salts described above and other typical pharmaceutically acceptable salts is more fully described by Berg et at,
"Pharmaceutical Salts," J. Pharm. Set, 1977:66: 1-19.
It will also be noted that the compounds of the present invention may potentially be internal salts or zwitterions, since under physiological conditions a deprotonated acidic moiety in the compound, such as a carboxyl group, may be anionic, and this electronic charge
might then be balanced off internally against the cationic charge of a protonated or alkylated basic moiety, such as a quaternary nitrogen atom. An isolated compound having internally balance charges, and thus not associated with a intermolecular counterion, may also be considered the "free form" of a compound.
Utilities
The compounds of the invention are useful to bind to and/or modulate the activity of mTOR kinase. In an embodiment, the compounds of the instant invention inhibit the activity of mTORCl . In another embodiment, the compounds of the instant invention inhibit the activity of mTORC2. In another embodiment, the compounds of the instant invention inhibit the activity of both mTORC 1 and mTORC2. In this context, modulate means either increasing or decreasing kinase activity of mTOR. In an embodiment, the compounds of the instant invention inhibit the kinase activity of mTOR.
The compounds of the invention find use in a variety of applications. As will be appreciated by those skilled in the art, the kinase activity of mTOR may be modulated in a variety of ways; that is, one can affect the phosphorylation/activation of mTOR either by modulating the initial phosphorylation of the protein or by modulating the autophosphorylation of the other active sites of the protein. Alternatively, the kinase activity of mTOR may be modulated by affecting the binding of a substrate of mTOR phosphorylation.
The compounds of the invention are used to treat or prevent cellular
proliferation diseases. Disease states which can be treated by the methods and compositions provided herein include, but are not limited to, cancer (further discussed below), autoimmune disease, viral disease, fungal disease, neurological/neurodegenerative disorders, arthritis, inflammation, anti-proliferative (e.g. ocular retinopathy), neuronal, alopecia, cardiovascular disease, graft rejection, inflammatory bowel disease, proliferation induced after medical procedures, including, but not limited to, surgery, angioplasty, and the like. It is appreciated that in some cases the cells may not be in a hyper- or hypoproliferation state (abnormal state) and still require treatment. Thus, in one embodiment, the invention herein includes application to cells or individuals which are afflicted or may eventually become afflicted with any one of these disorders or states.
The compounds, compositions and methods provided herein are particularly deemed useful for the treatment and prevention of cancer including solid tumors such as skin, breast, brain, cervical carcinomas, testicular carcinomas, etc. In an embodiment, the instant compounds are useful for treating cancer. In particular, cancers that may be treated by the compounds, compositions and methods of the invention include, but are not limited to:
Cardiac: sarcoma (angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma), myxoma, rhabdomyoma, fibroma, lipoma and teratoma; Lung: bronchogenic carcinoma (squamous cell, undifferentiated small cell, undifferentiated large cell, adenocarcinoma), alveolar (bronchiolar) carcinoma, bronchial adenoma, sarcoma, lymphoma, chondromatous hamartoma,
mesothelioma; Gastrointestinal: esophagus (squamous cell carcinoma, adenocarcinoma, leiomyosarcoma, lymphoma), stomach (carcinoma, lymphoma, leiomyosarcoma), pancreas (ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma, carcinoid tumors, vipoma), small bowel (adenocarcinoma, lymphoma, carcinoid tumors, Karposi's sarcoma, leiomyoma, hemangioma, lipoma, neurofibroma, fibroma), large bowel (adenocarcinoma, tubular adenoma, villous adenoma, hamartoma, leiomyoma); Genitourinary tract: kidney (adenocarcinoma, Wilm's tumor [nephroblastoma], lymphoma, leukemia,), bladder and urethra (squamous cell carcinoma, transitional cell carcinoma, adenocarcinoma), prostate (adenocarcinoma, sarcoma), testis (seminoma, teratoma, embryonal carcinoma, teratocarcinoma, choriocarcinoma, sarcoma, interstitial cell carcinoma, fibroma, fibroadenoma, adenomatoid tumors, lipoma); Liver:
hepatoma (hepatocellular carcinoma), cholangiocarcinoma, hepatoblastoma, angiosarcoma, hepatocellular adenoma, hemangioma; Bone: osteogenic sarcoma (osteosarcoma),
fibrosarcoma, malignant fibrous histiocytoma, chondrosarcoma, Ewing's sarcoma, malignant lymphoma (reticulum cell sarcoma), multiple myeloma, malignant giant cell tumor chordoma, osteoclironfroma (osteocartilaginous exostoses), benign chondroma, chondroblastoma, chondromyxofibroma, osteoid osteoma and giant cell tumors; Nervous system: skull (osteoma, hemangioma, granuloma, xanthoma, osteitis deformans), meninges (meningioma,
meningiosarcoma, gliomatosis), brain (astrocytoma, medulloblastoma, glioma, ependymoma, germinoma [pinealoma], glioblastoma multiform, oligodendroglioma, schwannoma, retinoblastoma, congenital tumors), spinal cord (neurofibroma, meningioma, glioma, sarcoma); Gynecological: uterus (endometrial carcinoma), cervix (cervical carcinoma, pre-tumor cervical dysplasia), ovaries (ovarian carcinoma [serous cystadenocarcinoma, mucinous
cystadenocarcinoma, unclassified carcinoma], granulosa-thecal cell tumors, Sertoli-Leydig cell tumors, dysgerminoma, malignant teratoma), vulva (squamous cell carcinoma, intraepithelial carcinoma, adenocarcinoma, fibrosarcoma, melanoma), vagina (clear cell carcinoma, squamous cell carcinoma, botryoid sarcoma (embryonal rhabdomyosarcoma), fallopian tubes (carcinoma); Hematologic: blood (myeloid leukemia [acute and chronic], acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myeloma, myelodysplastic syndrome), Hodgkin's disease, non-Hodgkin's lymphoma [malignant lymphoma]; Skin:
malignant melanoma, basal cell carcinoma, squamous cell carcinoma, Karposi's sarcoma, moles dysplastic nevi, lipoma, angioma, dermatofibroma, keloids, psoriasis; and Adrenal glands: neuroblastoma. Thus, the term "cancerous cell" as provided herein, includes a cell afflicted by any one of the above-identified conditions. In an embodiment of the invention, cancers that may be treated by the compounds, compositions and methods of the invention include, in addition to the cancers listed above: Lung: bronchogenic carcinoma (non-small cell lung); Gastrointestinal: rectal, colorectal and colon; Genitourinary tract: kidney (papillary renal cell carcinoma); and Skin: head and neck squamous cell carcinoma.
In another embodiment, the compounds of the instant invention are useful for treating or preventing cancer selected from: head and neck squamous cell carcinomas, histiocytic lymphoma, lung adenocarcinoma, small cell lung cancer, non-small cell lung cancer, pancreatic cancer, papillary renal cell carcinoma, liver cancer, gastric cancer, colon cancer, multiple myeloma, glioblastomas and breast carcinoma. In yet another embodiment, the compounds of the instant invention are useful for treating or preventing cancer selected from: histiocytic lymphoma, lung adenocarcinoma, small cell lung cancer, pancreatic cancer, liver cancer, gastric cancer, colon cancer, multiple myeloma, glioblastomas and breast carcinoma. In still another embodiment, the compounds of the instant invention are useful for treating cancer selected from: histiocytic lymphoma, lung adenocarcinoma, small cell lung cancers, pancreatic cancer, liver cancer, gastric cancer, colon cancer, multiple myeloma, glioblastomas and breast carcinoma.
In another embodiment, the compounds of the instant invention are useful for the prevention or modulation of the metastases of cancer cells and cancer. In particular, the compounds of the instant invention are useful to prevent or modulate the metastases of ovarian cancer, childhood hepatocellular carcinoma, metastatic head and neck squamous cell carcinomas, gastric cancers, breast cancer, colorectal cancer, cervical cancer, lung cancer, nasopharyngeal cancer, pancreatic cancer, glioblastoma and sarcomas.
The compounds of this invention may be administered to mammals, preferably humans, either alone or in combination with pharmaceutically acceptable carriers, excipients or diluents, in a pharmaceutical composition, according to standard pharmaceutical practice. The compounds can be administered orally or parenterally, including the intravenous,
intramuscular, intraperitoneal, subcutaneous, rectal and topical routes of administration.
The pharmaceutical compositions containing the active ingredient may be in a form suitable for oral use, for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, or syrups or elixirs. Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations. Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
These excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, microcrystalline cellulose, sodium crosscarmellose, corn starch, or alginic acid; binding agents, for example starch, gelatin, polyvinyl-pyrrolidone or acacia, and lubricating agents, for example, magnesium stearate, stearic acid or talc. The tablets may be uncoated or they may be coated by known techniques to mask the unpleasant taste of the drug
or delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period. For example, a water soluble taste masking material such as hydroxypropyl-methylcellulose or hydroxypropylcellulose, or a time delay material such as ethyl cellulose, cellulose acetate butyrate may be employed.
Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water soluble carrier such as polyethyleneglycol or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
Aqueous suspensions contain the active material in admixture with excipients suitable for the manufacture of aqueous suspensions. Such excipients are suspending agents, for example sodium carboxymethylcellulose, methylcellulose, hydroxypropylmethyl-cellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia; dispersing or wetting agents may be a naturally-occurring phosphatide, for example lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbitan monooleate. The aqueous suspensions may also contain one or more preservatives, for example ethyl, or n-propyl p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose, saccharin or aspartame.
Oily suspensions may be formulated by suspending the active ingredient in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in mineral oil such as liquid paraffin. The oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as those set forth above, and flavoring agents may be added to provide a palatable oral preparation. These compositions may be preserved by the addition of an anti-oxidant such as butylated hydroxyanisol or alpha- tocopherol.
Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those already mentioned above.
Additional excipients, for example sweetening, flavoring and coloring agents, may also be present. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
The pharmaceutical compositions of the invention may also be in the form of an oil-in- water emulsion. The oily phase may be a vegetable oil, for example olive oil or arachis oil, or a mineral oil, for example liquid paraffin or mixtures of these. Suitable emulsifying agents may be naturally occurring phosphatides, for example soy bean lecithin, and esters or partial esters derived from fatty acids and hexitoi anhydrides, for example sorbitan monooleate, and condensation products of the said partial esters with ethylene oxide, for example polyoxyethylene sorbitan monooleate. The emulsions may also contain sweetening, flavoring agents, preservatives and antioxidants.
Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative, flavoring and coloring agents and antioxidant.
The pharmaceutical compositions may be in the form of a sterile injectable aqueous solution. Among the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
The sterile injectable preparation may also be a sterile injectable oil-in-water microemulsion where the active ingredient is dissolved in the oily phase. For example, the active ingredient may be first dissolved in a mixture of soybean oil and lecithin. The oil solution then introduced into a water and glycerol mixture and processed to form a
microemulation.
The injectable solutions or microemulsions may be introduced into a patient's blood stream by local bolus injection. Alternatively, it may be advantageous to administer the solution or microemulsion in such a way as to maintain a constant circulating concentration of the instant compound. In order to maintain such a constant concentration, a continuous intravenous delivery device may be utilized. An example of such a device is the Deltec CADD-PLUS™ model 5400 intravenous pump.
The pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleagenous suspension for intramuscular and subcutaneous administration. This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above. The sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, for example as a solution in 1 ,3-butane diol. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose any bland fixed oil may be employed including synthetic mono- or diglycerides. In addition, fatty acids such as oleic acid find use in the preparation of injectables.
Compounds of Formula I may also be administered in the form of suppositories for rectal administration of the drug. These compositions can be prepared by mixing the drug with a suitable non-irritating excipient which is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug. Such materials
include cocoa butter, glycerinated gelatin, hydrogenated vegetable oils, mixtures of
polyethylene glycols of various molecular weights and fatty acid esters of polyethylene glycol
For topical use, creams, ointments, jellies, solutions or suspensions, etc, containing the compound of Formula I are employed. (For purposes of this application, topical application shall include mouth washes and gargles.)
The compounds for the present invention can be administered in intranasal form via topical use of suitable intranasal vehicles and delivery devices, or via transdermal routes, using those forms of transdermal skin patches well known to those of ordinary skill in the art. To be administered in the form of a transdermal delivery system, the dosage administration will, of course, be continuous rather than intermittent throughout the dosage regimen.
Compounds of the present invention may also be delivered as a suppository employing bases such as cocoa butter, glycerinated gelatin, hydrogenated vegetable oils, mixtures of
polyethylene glycols of various molecular weights and fatty acid esters of polyethylene glycol.
The dosage regimen utilizing the compounds of the instant invention can be selected in accordance with a variety of factors including type, species, age, weight, sex and the type of cancer being treated; the severity (i.e., stage) of the cancer to be treated; the route of administration; the renal and hepatic function of the patient; and the particular compound or salt thereof employed. An ordinarily skilled physician or veterinarian can readily determine and prescribe the effective amount of the drug required to treat, for example, to prevent, inhibit (fully or partially) or arrest the progress of the disease.
In one exemplary application, a suitable amount of compound is administered to a mammal undergoing treatment for cancer. Administration occurs in an amount between about 0.1 mg kg of body weight to about 60 mg kg of body weight per day, preferably of between 0.5 mg/kg of body weight to about 40 mg kg of body weight per day.
In a further example, compounds of the instant invention can be administered in a total daily dose of up to 1000 mg. Compounds of the instant invention can be administered once daily (QD), or divided into multiple daily doses such as twice daily (BID), and three times daily (TID). Compounds of the instant invention can be administered at a total daily dosage of up to 1000 mg, e.g., 200 mg, 300 mg, 400 mg, 600 mg, 800 mg or 1000 mg, which can be administered in one daily dose or can be divided into multiple daily doses as described above.
In addition, the administration can be continuous, i.e., every day, or
intermittently. The terms "intermittent" or "intermittently" as used herein means stopping and starting at either regular or irregular intervals. For example, intermittent administration of a compound of the instant invention may be administration one to six days per week or it may mean administration in cycles (e.g. daily administration for two to eight consecutive weeks, then a rest period with no administration for up to one week) or it may mean administration on alternate days.
In addition, the compounds of the instant invention may be administered according to any of the schedules described above, consecutively for a few weeks, followed by a rest period. For example, the compounds of the instant invention may be administered according to any one of the schedules described above from two to eight weeks, followed by a rest period of one week, or twice daily at a dose of 100 - 500 mg for three to five days a week. In another particular embodiment, the compounds of the instant invention may be administered three times daily for two consecutive weeks, followed by one week of rest.
The instant compounds are also useful in combination with known therapeutic agents and anti-cancer agents. For example, instant compounds are useful in combination with known anti-cancer agents. Combinations of the presently disclosed compounds with other anticancer or chemotherapeutic agents are within the scope of the invention. Examples of such agents can be found in Cancer Principles and Practice of Oncology by V.T. Devita and S. Hellman (editors), 6th edition (February 15, 2001), Lippincott Williams & Wilkins Publishers. A person of ordinary skill in the art would be able to discern which combinations of agents would be useful based on the particular characteristics of the drugs and the cancer involved. Such anti-cancer agents include, but are not limited to, the following: estrogen receptor modulators, androgen receptor modulators, retinoid receptor modulators, cytotoxic/cytostatic agents, antiproliferative agents, prenyl-protein transferase inhibitors, HMG-CoA reductase inhibitors and other angiogenesis inhibitors, inhibitors of cell proliferation and survival signaling, apoptosis inducing agents and agents that interfere with cell cycle checkpoints. The instant compounds are particularly useful when co-administered with radiation therapy.
In an embodiment, the instant compounds are also useful in combination with known anti-cancer agents including the following: estrogen receptor modulators, androgen receptor modulators, retinoid receptor modulators, cytotoxic agents, antiproliferative agents, prenyl-protein transferase inhibitors, HMG-CoA reductase inhibitors, HIV protease inhibitors, reverse transcriptase inhibitors, and other angiogenesis inhibitors.
"Estrogen receptor modulators" refers to compounds that interfere with or inhibit the binding of estrogen to the receptor, regardless of mechanism. Examples of estrogen receptor modulators include, but are not limited to, tamoxifen, raloxifene, idoxifene,
LY353381, LY117081, toremifene, fulvestrant, 4-[7-(2,2-dimethyl-l-oxopropoxy-4-methyl-2- [4-[2-(l -piperidinyl)ethoxy]phenyl]-2H- 1 -benzopyran-3-yl]-phenyl-2,2-dimethylpropanoate, 4,4'-dihydroxybenzophenone-2,4-dinitrophenyl-hydrazone, and SH646.
"Androgen receptor modulators" refers to compounds which interfere or inhibit the binding of androgens to the receptor, regardless of mechanism. Examples of androgen receptor modulators include finasteride and other 5ot-reductase inhibitors, nilutamide, flutamide, bicalutamide, liarozole, and abiraterone acetate.
"Retinoid receptor modulators" refers to compounds which interfere or inhibit the binding of retinoids to the receptor, regardless of mechanism. Examples of such retinoid
receptor modulators include bexarotene, tretinoin, 13-cis-retinoic acid, 9-cis-retinoic acid, a- difluoromethylornithine, ILX23-7553, trans-N-(4'-hydroxyphenyl) retinamide, and N-4- carboxyphenyl retinamide.
"Cytotoxic/cytostatic agents" refer to compounds which cause cell death or inhibit cell proliferation primarily by interfering directly with the cell's functioning or inhibit or interfere with cell mytosis, including alkylating agents, tumor necrosis factors, intercalators, hypoxia activatable compounds, microtubule inliibitors/microtubule-stabilizing agents, inhibitors of mitotic kinesins, inhibitors of histone deacetylase, inhibitors of kinases involved in mitotic progression, antimetabolites; biological response modifiers; hormonal/anti-hormonal therapeutic agents, haematopoietic growth factors, monoclonal antibody targeted therapeutic agents, topoisomerase inhibitors, proteasome inhibitors and ubiquitin ligase inhibitors.
Examples of cytotoxic agents include, but are not limited to, sertenef, cachectin, ifosfamide, tasonermin, lonidamine, carboplatin, altretamine, prednimustine, dibromodulcitol, ranimustine, fotemustine, nedaplatin, oxaliplatin, temozolomide, heptaplatin, estramustine, improsulfan tosilate, trofosfamide, nimustine, dibrospidium chloride, pumitepa, lobaplatin, satraplatin, proftromycin, cisplatin, irofulven, dexifosfamide, cis-aminedichloro(2-methyl- pyridine)platinum, benzylguanine, glufosfamide, GPX100, (trans, trans, trans)-bis-mu-(hexane- 1 ,6-diamine)-mu-[diamine-platinum(II)]bis [diamine(chloro)platinum (II)]tetrachloride, diarizidinylspermine, arsenic trioxide, l-(l l-dodecylamino-10-hydroxyundecyl)-3,7- dimethylxanthine, zorubicin, idarabicin, daunorubicin, bisantrene, mitoxantrone, pirarubicin, pinafide, valrubicin, amrubicin, antineoplaston, 3'-deamino-3'-morpholino-13-deoxo-10- hydroxycarminomycin, annamycin, galarubicin, elinafide, MEN 10755, and 4-demethoxy-3- deamino-3-aziridinyl-4-methylsulphonyl-daunorubicin (see WO 00/50032).
An example of a hypoxia activatable compound is tirapazamine.
Examples of proteasome inhibitors include but are not limited to lactacystin and bortezomib.
Examples of microtubule inhibitors/microtubule-stabilising agents include paclitaxel, vindesine sulfate, S'^'-didehydro^'-deoxy-S'-norvincaleukoblastine, docetaxol, rhizoxin, dolastatin, mivobulin isethionate, auristatin, cemadotin, RPRl 09881, BMS 184476, vinflunine, cryptophycin, 2,3,4,5,6-pentafluoro-N-(3-fluoro-4-methoxyphenyl) benzene sulfonamide, anhydro vinblastine, N,N-dimethyl-L-valyl-L-valyl-N-methyl-L-valyl-L-prolyl-L- proline-t-butylamide, TDX258, the epothilones (see for example U.S. Pat. Nos. 6,284,781 and 6,288,237) and BMS188797.
Some examples of topoisomerase inhibitors are topotecan, hycaptamine, irinotecan, rabitecan, 6-ethoxypropionyl-3 ' ,4 ' -0-exo-benzylidene-chartreusin, 9-methoxy-N,N- dimethyl-5-nitropyrazolo[3,4,5-kl]acridine-2-(6H) propanamine, 1 -amino-9-ethyl-5-fluoro-2,3- dihydro-9-hydroxy-4-methyl-lH,12H-benzo[de]pyrano[3',4':b,7]-indolizino[l,2b]quinoline- 10,13(9H,15H)dione, lurtotecan, 7-[2-(N-isopropylamino)ethyl]-(20S)camptothecin, BNP1350,
BNPI1100, BN80915, BN80942, etoposide phosphate, tenyposide, sobuzoxane, 2'~
dimethylamino-2'-deoxy-etoposide, GL331 , N-[2-(dimethylamino)ethyl]-9-hydroxy-5,6- dimethyl-6H-pyrido[4,3-b]carbazole-l-carboxamide, asulacrine, (5a, 5aB, 8aa,9b)-9-[2-[N-[2- (dimethylamino)ethyl] -N-n ethylamino] ethyl] - 5 - [4-hydro0xy-3 , 5-dimethoxyphenyl] - 5,5a,6,8,8as9-hexohydrofuro(3',4':6J7)naphtho(2,3~d)-l,3-dioxol-6-one, 2,3-(methylenedioxy)- 5-raethyl-7-hydroxy-8-rnethoxybenzo[c]-phenanthridinium, 6,9-bis[(2- aminoethy^ammojbenzoIglisoguinoline-S^O-dione, 5-(3-aminopropylamino)-7,10-dihydroxy- 2-(2-hydroxyethylaminomethyi)-6H-pyrazolo[4J5,l-de]acridin-6-one, N-[l- [2(diethylamino)ethylamino]~7-memoxy N-(2- (dimethyIamino)ethyl)acridine-4-carboxamide, 6-[[2-(dimethylamino)ethyl]amino]-3-hydroxy- 7H-indeno[2,l-c] quinolin-7-one, and dimesna.
Examples of inhibitors of mitotic kinesins, and in particular the human mitotic kinesin KSP, are described in PCT Publications WO 01/30768, WO 01/98278, WO
03/050,064, WO 03/050,122, WO 03/049,527, WO 03/049,679, WO 03/049,678,
WO04/039774, WO03/079973, WO03/099211, WO03/105855, WO03/106417,
WO04/037171, WO04/058148, WO04/058700, WO04/126699, WO05/018638,
WO05/019206, WO05/019205, WO05/018547, WO05/017190, US2005/0176776.. In an embodiment inhibitors of mitotic kinesins include, but are not limited to inhibitors of KSP, inhibitors of M XP1, inhibitors of CENP-E, inhibitors of MCAK, inhibitors of Kifl4, inhibitors of Mphosp and inhibitors of Rab6-KIFL.
Examples of "histone deacetylase inhibitors" include, but are not limited to, SAHA, TSA, oxamflatin, PXD101, MG98, valproic acid and scriptaid. Further reference to other histone deacetylase inhibitors may be found in the following manuscript; Miller, T.A. et al. J. Med. Chem. 46(24): 5097-5116 (2003).
"Inhibitors of kinases involved in mitotic progression" include, but are not limited to, inhibitors of aurora kinase, inhibitors of Polo-like kinases (PLK) (in particular inhibitors of PLK-1), inhibitors of bub-1 and inhibitors of bub-RL
"Antiproliferative agents" includes antisense RNA and DNA oligonucleotides such as G3139, ODN698, RVASKRAS, GEM231, and INX3001, and antimetabolites such as enocitabine, carmofur, tegafur, pentostatin, doxif ridine, trimetrexate, fludarabine, capecitabine, galocitabine, cytarabine ocfosfate, fosteabine sodium hydrate, raltitrexed, paltitrexid, emitefur, tiazofurin, decitabine, nolatrexed, pemetrexed, nelzarabine, 2'-deoxy-2'- methylidenecytidine, 2 ' -fluoromethylene-2' -deoxycytidine, N- [5-(2,3 -dihydro- benzofuryl)sulfonyl3-N'-(3,4-dichlorophenyl)urea, N6-[4-deoxy-4-[N2-t2(E)54(E)- tetradecadienoyl]glycylamino]-L-glycero-B-L-manno-heptopyranosyl]adenine, aplidine, ecteinascidin, troxacitabine, 4-[2-amino-4-oxo-4J6,758-tetrahydro-3H-pyrimidino[5,4- b][l,4]thiazin-6-yl-(S)-ethyl]-2,5-thienoyl-L-glutamic acid, aminopterin, 5-flurouracil, alanosine, 11 -acetyl- 8 -(carbamoyloxymethyl)-4-formyl-6-methoxy- 14-oxa- 1,11-
diazatetracyclo(7.4.1.0.0)-tetradeca-2,4,6-trien-9-yl acetic acid ester, swainsonine, lometrexol, dexrazoxane, methioninase, 2'-cyano-2'-deoxy-N4-palmitoyl-l-B-D-arabino furanosyl cytosine and 3-aminopyridine-2-carboxaldehyde thiosemicarbazone.
Examples of monoclonal antibody targeted therapeutic agents include those therapeutic agents which have cytotoxic agents or radioisotopes attached to a cancer cell specific or target cell specific monoclonal antibody. Examples include Bexxar.
"HMG-CoA reductase inhibitors" refers to inhibitors of 3-hydroxy-3- methylglutaryl-CoA reductase. Examples of HMG-CoA reductase inhibitors that may be used include but are not limited to lovastatin (MEVACOR®; see U.S. Pat. Nos. 4,231,938,
4,294,926 and 4,319,039), simvastatin (ZOCOR®; see U.S. Pat. Nos. 4,444,784, 4,820,850 and 4,916,239), pravastatin (PRAVACBOL®; see U.S. Pat. Nos. 4,346,227, 4,537,859, 4,410,629, 5,030,447 and 5,180,589), fluvastatin (LESCOL®; see U.S. Pat. Nos. 5,354,772, 4,911,165, 4,929,437, 5,189,164, 5,118,853, 5,290,946 and 5,356,896) and atorvastatin (LIPITOR®; see U.S. Pat. Nos. 5,273,995, 4,681,893, 5,489,691 and 5,342,952). The structural formulas of these and additional HMG-CoA reductase inhibitors that may be used in the instant methods are described at page 87 of M. Yalpani, "Cholesterol Lowering Drugs", Chemistry & Industry, pp. 85-89 (5 February 1996) and US Patent Nos. 4,782,084 and 4,885,314. The term HMG- CoA reductase inhibitor as used herein includes all pharmaceutically acceptable lactone and open-acid forms (i.e., where the lactone ring is opened to form the free acid) as well as salt and ester forms of compounds which have HMG-CoA reductase inhibitory activity, and therefor the use of such salts, esters, open-acid and lactone forms is included within the scope of this invention.
"Prenyl-protein transferase inhibitor" refers to a compound which inhibits any one or any combination of the prenyl-protein transferase enzymes, including farnesyl-protein transferase (FPTase), geranylgeranyl-protein transferase type I (GGPTase-I), and
geranylgeranyl-protein transferase type-II (GGPTase-II, also called Rab GGPTase).
Examples of prenyl-protein transferase inhibitors can be found in the following publications and patents: WO 96/30343, WO 97/18813, WO 97/21701, WO 97/23478, WO 97/38665, WO 98/28980, WO 98/29119, WO 95/32987, U.S. Pat. No. 5,420,245, U.S. Pat. No. 5,523,430, U.S. Pat. No. 5,532,359, U.S. Pat. No. 5,510,510, U.S. Pat. No. 5,589,485, U.S. Pat. No. 5,602,098, European Patent Publ. 0 618 221, European Patent Publ. 0 675 112, European Patent Publ. 0 604 181, European Patent Publ. 0 696 593, WO 94/19357, WO 95/08542, WO 95/11917, WO 95/12612, WO 95/12572, WO 95/10514, U.S. Pat. No. 5,661,152, WO
95/10515, WO 95/10516, WO 95/24612, WO 95/34535, WO 95/25086, WO 96/05529, WO 96/06138, WO 96/06193, WO 96/16443, WO 96/21701, WO 96/21456, WO 96/22278, WO 96/2461 1, WO 96/24612, WO 96/05168, WO 96/05169, WO 96/00736, U.S. Pat. No.
5,571,792, WO 96/17861, WO 96/33159, WO 96/34850, WO 96/34851, WO 96/30017, WO 96/30018, WO 96/30362, WO 96/30363, WO 96/3111 1, WO 96/31477, WO 96/31478,
WO 96/31501, WO 97/00252, WO 97/03047, WO 97/03050, WO 97/04785, WO 97/02920, WO 97/17070, WO 97/23478, WO 97/26246, WO 97/30053, WO 97/44350, WO 98/02436, and U.S. Pat. No. 5,532,359. For an example of the role of a prenyl-protein transferase inhibitor on angiogenesis see European J. of Cancer, Vol. 35, No. 9, pp.1394-1401 (1999).
"Angiogenesis inhibitors" refers to compounds that inhibit the formation of new blood vessels, regardless of mechanism. Examples of angiogenesis inhibitors include, but are not limited to, tyrosine kinase inhibitors, such as inhibitors of the tyrosine kinase receptors Flt- 1 (VEGFR1) and Flk-l/KDR (VEGFR2), inhibitors of epidermal-derived, fibroblast-derived, or platelet derived growth factors, MMP (matrix metalloprotease) inhibitors, integrin blockers, interferon-ot, interleukin- 12, pentosan polysulfate, cyclooxygenase inhibitors, including nonsteroidal anti-inflammatories (NSAIDs) like aspirin and ibuprofen as well as selective cyclooxy-genase-2 inhibitors like celecoxib and rofecoxib (PNAS, Vol. 89, p. 7384 (1992); JNCI, Vol. 69, p. 475 (1982); Arch. OpthaimoL, Vol. 108, p.573 (1990); Anat. Rec, Vol. 238, p. 68 (1994); FEBS Letters, Vol. 372, p. 83 (1995); Clin, Orthop. Vol. 313, p. 76 (1995); J. Mol. Endocrinol, Vol. 16, p.107 (1996); Jpn. J. Pharmacol, Vol. 75, p. 105 (1997); Cancer Res., Vol. 57, p. 1625 (1997); Cell, Vol. 93, p. 705 (1998); Intl. J. Mol. Med., Vol. 2, p. 715 (1998); J. Biol Chem., Vol. 274, p. 9116 (1999)), steroidal anti-inflammatories (such as corticosteroids, mineralocorticoids, dexamethasone, prednisone, prednisolone, methylpred, betamethasone), carboxyamidotriazole, combretastatin A-4, squalamine, 6-O-chloroacetyl- carbonyl)-fumagillol, thalidomide, angiostatin, troponin- 1 , angiotensin II antagonists (see
Fernandez et al., J. Lab. Clin. Med. 105:141-145 (1985)), and antibodies to VEGF (see, Nature Biotechnology, Vol. 17, pp.963-968 (October 1999); Kim et al., Nature, 362, 841-844 (1993); WO 00/44777; and WO 00/ 1186).
Other therapeutic agents that modulate or inhibit angiogenesis and may also be used in combination with the compounds of the instant invention include agents that modulate or inhibit the coagulation and fibrinolysis systems (see review in Clin. Chem. La. Med. 38:679- 692 (2000)). Examples of such agents that modulate or inhibit the coagulation and fibrinolysis pathways include, but are not limited to, heparin (see Thromb. Haemost. 80:10-23 (1998)), low molecular weight heparins and carboxypeptidase U inhibitors (also known as inhibitors of active thrombin activatable fibrinolysis inhibitor [TAFIa]) (see Thrombosis Res. 101 :329-354 (2001)). TAFIa inhibitors have been described in PCT Publication WO 03/013,526 and U,S, Ser. No. 60/349,925 (filed January 18, 2002).
"Agents that interfere with cell cycle checkpoints" refer to compounds that inhibit protein kinases that transduce cell cycle checkpoint signals, thereby sensitizing the cancer cell to DNA damaging agents. Such agents include inhibitors of ATR, ATM, the Chkl and Chk2 kinases and cdk and cdc kinase inhibitors and are specifically exemplified by 7- hydroxystaurosporin, flavopiridol, CYC202 (Cyclacel) and BMS-387032.
"Agents that interfere with receptor tyrosine kinases (RTKs)" refer to
compounds that inhibit RTKs and therefore mechanisms involved in oncogenesis and tumor progression. Such agents include inhibitors of c-Kit, Eph, PDGF, Flt3 and c-Met. Further agents include inhibitors of RTKs as described by Bume- Jensen and Hunter, Nature, 411 :355- 365, 2001.
"Inhibitors of cell proliferation and survival signaling pathway" refer to pharmaceutical agents that inhibit cell surface receptors and signal transduction cascades downstream of those surface receptors. Such agents include inhibitors of inhibitors of EGFR (for example gefitinib and erlotinib), inhibitors of ERB-2 (for example trastuzumab), inhibitors of IGFR, inhibitors of cytokine receptors, inhibitors of MET, PI3K kinase family inhibitors (for example LY294002) including inhibitos of PI3K-a, PI3K-b, PI3K-g and PI3K-ds
serine/threonine kinases (including but not limited to inhibitors of Akt such as described in WO 02/083064, WO 02/083139, WO 02/083140, US 2004-0116432, WO 02/083138, US 2004- 0102360, WO 03/086404, WO 03/086279, WO 03/086394, WO 03/084473, WO 03/086403, WO 2004/041162, WO 2004/096131, WO 2004/096129, WO 2004/096135, WO 2004/096130, WO 2005/100356, WO 2005/100344), inhibitors of Raf kinase (for example BAY-43-9006 ), MAP kinase pathway inhibitors, bRAF inhibitors, inhibitors of MEK (for example CI- 1040 and PD-098059), ERK inhibitors and inhibitors of mTOR (for example Wyeth CCI-779). Such agents include small molecule inhibitor compounds and antibody antagonists.
Specific anti-IGF-l antibodies include, but are not limited to, dalotuzumab, figitumumab, cixutumumab, SHC 717454, Roche R1507, EM164 or Amgen AMG479.
The mTOR inhibitors in current clinical development are structural analogs of rapamycin. The mTOR inhibitors of the instant invention include ridaforolimus, temsirolimus, everolimus, a rapamycin-analog and combinations thereof.
Ridaforolimus, also known as AP 23573, MK-8669 and deforolimus, is a unique, non-prodrug analog of rapmycin that has antiproliferative activity in a broad range of human tumor cell lines in vitro and in murine tumor xenograft models utilizing human tumor cell lines. Ridaforolimus has been administered to patients with advanced cancer and is currently in clinical development for various advanced malignancies, including studies in patients with advanced soft tissue or bone sarcomas. Thus far, these trials have demonstrated that ridaforolimus is generally well-tolerated with a predictable and manageable adverse even profile, and possess anti-tumor activity in a broad range of cancers. A description and preparation of ridaforolimus is described in U.S. Patent No. 7,091,213 to Ariad Gene
Therapeutics, Inc., which is hereby incorporated by reference in its entirety.
Temsirolimus, also known as Torisel®, is currently marketed for the treatment of renal cell carcinoma. A description and preparation of temsirolimus is described in U.S. Patent No. 5,362,718 to American Home Products Corporation, which is hereby incorporated by reference in its entirety.
Everolimus, also known as Certican® or RAD001, marketed by Novartis, has greater stability and enhanced solubility in organic solvents, as well as more favorable pharmokinetics with fewer side effects than rapamycin (sirolimus). Everolimus has been used in conjunction with microemulsion cyclosporin (Neoral®, Novartis) to increase the efficacy of the immunosuppressive regime.
"Apoptosis inducing agents" include activators of TNF receptor family members (including the TRAIL receptors).
The invention also encompasses combinations with NSAID's which are selective COX-2 inhibitors. For purposes of this specification NSAID's which are selective inhibitors of COX-2 are defined as those which possess a specificity for inhibiting COX-2 over COX-1 of at least 100 fold as measured by the ratio of IC50 for COX-2 over IC50 for COX-1 evaluated by cell or microsomal assays. Such compounds include, but are not limited to those disclosed in U.S. Pat. 5,474,995, U.S. Pat. 5,861,419, U.S. Pat 6,001,843, U.S. Pat. 6,020,343, U.S. Pat. 5,409,944, U.S. Pat. 5,436,265, U.S. Pat. 5,536,752, U.S. Pat. 5,550,142, U.S. Pat. 5,604,260, U.S. 5,698,584, U.S. Pat. 5,710,140, WO 94/15932, U.S. Pat. 5,344,991, U.S. Pat. 5,134,142, U.S. Pat. 5,380,738, U.S. Pat. 5,393,790, U.S. Pat 5,466,823, U.S. Pat 5,633,272, and U.S. Pat. 5,932,598, all of which are hereby incorporated by reference.
Inhibitors of COX-2 that are particularly useful in the instant method of treatment are: 3-phenyl-4-(4-(methylsulfonyl)phenyl)-2-(5H)-furanone; and 5-chloro-3-(4- methylsulfonyl)-phenyl-2-(2-methyl-5-pyridinyl)pyridine; or a pharmaceutically acceptable salt thereof.
Compounds that have been described as specific inhibitors of COX-2 and are therefore useful in the present invention include, but are not limited to: parecoxib,
CELEBREX® and BEXTRA® or a pharmaceutically acceptable salt thereof.
Other examples of angiogenesis inhibitors include, but are not limited to, endostatin, ukrain, ranpirnase, IM862, 5-methoxy-4-[2-methyl-3-(3-methyl-2- butenyl)oxiranyl] - 1 -oxaspiro [2,5] oct-6-yl(chloroacetyl)carbamate , acetyldinanalme, 5 -amino- 1 - [[3 ,5-dichloro-4-(4-chlorobenzoyl)-phenyl]methyl]- 1 H- 1 ,2,3-triazole-4-carboxamide,CM 101, squalamine, combretastatin, RPI4610, NX31838, sulfated mannopentaose phosphate, 7,7- (carbonyl-bis[imino-N-methyl-4,2-pyi olocarbonylimmo| -methyl-4,2-pyrrole]- carbonylimino]-bis-(l,3-naphthalene disulfonate), and 3-[(2,4-dimethylpyrrol-5~yl)methylene]~ 2-indolinone (SU5416).
As used above, "integrin blockers" refers to compounds which selectively antagonize, inhibit or counteract binding of a physiological ligand to the νβ3 integrin, to compounds which selectively antagonize, inhibit or counteract binding of a physiological ligand to the νβ5 integrin, to compounds which antagonize, inhibit or counteract binding of a physiological ligand to both the ανβ3 integrin and the νβ5 integrin, and to compounds which antagonize, inhibit or counteract the activity of the particular integrin(s) expressed on capillary
endothelial cells. The term also refers to antagonists of the ανβ6> <*νβ8> «ΐβΐ> α2βΐ, α5βΐ, 6βΐ and « β4 integrins. The term also refers to antagonists of any combination of ανβ3, νβ5, ανβ6> «νβ8> «·ΐ βΐ, α2βΐ, «5 ΐ, «6 ΐ and 4 integrins.
Some specific examples of tyrosine kinase inhibitors include N- (trifliioromethylpheny^-S-methylisoxazol^-carboxamide, 3-[(2,4-dimethylpyrrol-5~ yl)methylidenyl)indolin-2-one, 17-(allylamino)-l 7-demethoxygeldanamycin, 4-(3-chloro-4- fluorophenylamino)-7-methoxy-6-[3-(4-morpholinyl)propoxyl]quinazoline, N-(3- ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine; BIBX1382, 2,3,9,10,11,12- hexahydro- 10-(hydroxymethyl)- 10-hydroxy-9-methyl-9, 12-epoxy- 1 H-diindolo[ 1 ,2,3 - fg^'^'J '-klJ yrrolotS^-iJtljejbenzodiazoci -l-one, SH268, genistein, imatinib (STI571), CEP2563, 4-(3-chlorophenylamino)-5,6-dimethyl-7H-pyrrolo[2,3-d]pyrimidinemethane sulfonate, 4-(3-bromo-4-hydroxyphenyl)amino-6,7-dimethoxyquinazoline, 4-(4'- hydroxyphenyl)amino-6,7-dimethoxyquinazoline, SU6668, STI571 A, N-4-chlorophenyl-4-(4- pyridylmethyl)-l-phthalazinamine, and EMD121974.
Combinations with compounds other than anti-cancer compounds are also encompassed in the instant methods. For example, combinations of the instantly claimed compounds with PPAR-γ (i.e., PPAR-gamma) agonists and PPAR-δ (i.e., PPAR-delta) agonists are useful in the treatment of certain malingnancies. PPAR-γ and PPAR-6 are the nuclear peroxisome proliferator-activated receptors γ and δ. The expression of PPAR-γ on endothelial cells and its involvement in angiogenesis has been reported in the literature (see J Cardiovasc. Pharmacol. 1998; 31.909-913; J Biol. Chem. 1999;274:9116-9121; Invest. Ophthalmol Vis. Sci. 2000; 41 :2309-2317). More recently, PPAR-γ agonists have been shown to inhibit the angiogenic response to VEGF in vitro; both troglitazone and rosiglitazone maleate inhibit the development of retinal neovascularization in mice. (Arch. Ophthamol. 2001 ; 119:709-717). Examples of PPAR-γ agonists and PPAR- γ/α agonists include, but are not limited to, thiazolidinediones (such as DRF2725, CS-011, troglitazone, rosiglitazone, and pioglitazone), fenofibrate, gemfibrozil, clofibrate, GW2570, SB219994, AR-H039242, JTT-501, MCC-555, GW2331, GW409544, NN2344, KRP297, NP0110, DRF41S8, NN622, GI262570,
PNU182716, DRF552926, 2-[(5,7-dipropyl-3-trifluoromethyl-l ,2-benzisoxazol-6-yl)oxy]-2- methylpropionic acid (disclosed in USSN 09/782,856), and 2(R)-7-(3-(2-chloro-4-(4- fluorophenoxy) phenoxy)propoxy)-2-ethylchromane-2-carboxylic acid (disclosed in USSN 60/235,708 and 60/244,697).
Another embodiment of the instant invention is the use of the presently disclosed compounds in combination with gene therapy for the treatment of cancer. For an overview of genetic strategies to treating cancer see Hall et al (Am J Hum Genet 61 :785-789, 1997) and Kufe et al (Cancer Medicine, 5th Ed, pp 876-889, BC Decker, Hamilton 2000). Gene therapy can be used to deliver any tumor suppressing gene. Examples of such genes include, but are not limited to, p53, which can be delivered via recombinant virus-mediated
gene transfer (see U.S. Pat. No. 6,069,134, for example), a uPA/uPAR antagonist ("Adenovirus-Mediated Delivery of a uPA/uPAR Antagonist Suppresses Angiogenesis- Dependent Tumor Growth and Dissemination in Mice," Gene Therapy, August
1998;5(8):1105-13), and interferon gamma (J Immunol 2000;164:217-222).
The compounds of the instant invention may also be administered in combination with an inhibitor of inherent multidrug resistance (MDR), in particular MDR associated with high levels of expression of transporter proteins. Such MDR inhibitors include inhibitors of p-glycoprotein (P-gp), such as LY335979, XR9576, OC144-093, R101922, VX853 and PSC833 (valspodar).
A compound of the present invention may be employed in conjunction with anti-emetic agents to treat nausea or emesis, including acute, delayed, late-phase, and anticipatory emesis, which may result from the use of a compound of the present invention, alone or with radiation therapy. For the prevention or treatment of emesis, a compound of the present invention may be used in conjunction with other anti-emetic agents, especially neurokinin- 1 receptor antagonists, 5HT3 receptor antagonists, such as ondansetron, granisetron, tropisetron, and zatisetron, GABAB receptor agonists, such as baclofen, a corticosteroid such as Decadron (dexamethasone), Kenalog, Aristocort, Nasalide, Preferid, Benecorten or others such as disclosed in U.S.Patent Nos. 2,789,118, 2,990,401, 3,048,581, 3,126,375, 3,929,768, 3,996,359, 3,928,326 and 3,749,712, an antidopaminergic, such as the phenothiazines (for example prochlorperazine, fluphenazine, thioridazine and mesoridazine), metoclopramide or dronabinol. In an embodiment, an anti-emesis agent selected from a neurokinin- 1 receptor antagonist, a 5HT3 receptor antagonist and a corticosteroid is administered as an adjuvant for the treatment or prevention of emesis that may result upon administration of the instant compounds.
Neurokinin- 1 receptor antagonists of use in conjunction with the compounds of the present invention are fully described, for example, in U.S. Pat. Nos. 5,162,339, 5,232,929, 5,242,930, 5,373,003, 5,387,595, 5,459,270, 5,494,926, 5,496,833, 5,637,699, 5,719,147;
European Patent Publication Nos. EP 0 360 390, 0 394 989, 0 428 434, 0 429 366, 0 430 771, 0 436 334, 0 443 132, 0 482 539, 0 498 069, 0 499 313, 0 512 901, 0 512 902, 0 514 273, 0 514 274, 0 514 275, 0 514 276, 0 515 681, 0 517 589, 0 520 555, 0 522 808, 0 528 495, 0 532 456, 0 533 280, 0 536 817, 0 545 478, 0 558 156, 0 577 394, 0 585 913,0 590 152, 0 599 538, 0 610 793, 0 634 402, 0 686 629, 0 693 489, 0 694 535, 0 699 655, 0 699 674, 0 707 006, 0 708 101, 0 709 375, 0 709 376, 0 714 891, 0 723 959, 0 733 632 and 0 776 893; PCT International Patent Publication Nos. WO 90/05525, 90/05729, 91/09844, 91/18899, 92/01688, 92/06079, 92/12151, 92/15585, 92/17449, 92/20661, 92/20676, 92/21677, 92/22569, 93/00330, 93/00331, 93/01159, 93/01165, 93/01169, 93/01170, 93/06099, 93/09116, 93/10073, 93/14084, 93/14113, 93/18023, 93/19064, 93/21 155, 93/21181 , 93/23380, 93/24465, 94/00440, 94/01402, 94/02461, 94/02595, 94/03429, 94/03445, 94/04494, 94/04496, 94/05625, 94/07843, 94/08997, 94/10165,
94/10167, 94/10168, 94/10170, 94/11368, 94/13639, 94/13663, 94/14767, 94/15903, 94/19320, 94/19323, 94/20500, 94/26735, 94/26740, 94/29309, 95/02595, 95/04040, 95/04042, 95/06645, 95/07886, 95/07908, 95/08549, 95/11880, 95/14017, 95/15311, 95/16679, 95/17382, 95/18124, 95/18129, 95/19344, 95/20575, 95/21819, 95/22525, 95/23798, 95/26338, 95/28418, 95/30674, 95/30687, 95/33744, 96/05181, 96/05193, 96/05203, 96/06094, 96/07649, 96/10562, 96/16939, 96/18643, 96/20197, 96/21661, 96/29304, 96/29317, 96/29326, 96/29328, 96/31214, 96/32385, 96/37489, 97/01553, 97/01554, 97/03066, 97/08144, 97/14671, 97/17362, 97/18206, 97/19084, 97/19942 and 97/21702; and in British Patent Publication Nos. 2 266 529, 2 268 931, 2 269 170, 2 269 590, 2 271 774, 2 292 144, 2 293 168, 2 293 169, and 2 302 689. The preparation of such compounds is fully described in the aforementioned patents and publications, which are incorporated herein by reference.
In an embodiment, the neurokinin- 1 receptor antagonist for use in conjunction with the compounds of the present invention is selected from: 2-( )-(l-(R)-(3,5- bis(trifiuoromethyl)-phenyl)ethoxy)-3-(S)-(4-fluorophenyl)-4-(3-(5-oxo-lH,4H-l,2,4- triazolo)methyl)morpholine, or a pharmaceutically acceptable salt thereof, which is described in U.S. Pat. No. 5,719,147.
A compound of the instant invention may also be useful for treating or preventing cancer, including bone cancer, in combination with bisphosphonates (understood to include bisphosphonates, diphosphonates, bisphosphonic acids and diphosphonic acids).
Examples of bisphosphonates include but are not limited to: etidronate (Didronel), pamidronate (Aredia), alendronate (Fosamax), risedronate (Actonel), zoledronate (Zometa), ibandronate (Boniva), incadronate or cimadronate, clodronate, EB-1053, minodronate, neridronate, piridronate and tiludronate including any and all pharmaceutically acceptable salts, derivatives, hydrates and mixtures thereof.
A compound of the instant invention may also be administered with an agent useful in the treatment of anemia. Such an anemia treatment agent is, for example, a continuous eythropoiesis receptor activator (such as epoetin alfa).
A compound of the instant invention may also be administered with an agent useful in the treatment of neutropenia. Such a neutropenia treatment agent is, for example, a hematopoietic growth factor which regulates the production and function of neutrophils such as a human granulocyte colony stimulating factor, (G-CSF). Examples of a G-CSF include filgrastim.
A compound of the instant invention may also be administered with an immunologic-enhancing drug, such as levamisole, isoprinosine and Zadaxin.
A compound of the instant invention may also be useful for treating or preventing cancer, including bone cancer, in combination with bisphosphonates (understood to include bisphosphonates, diphosphonates, bisphosphonic acids and diphosphonic acids).
Examples of bisphosphonates include but are not limited to: etidronate (Didronel), pamidronate
(Aredia), alendronate (Fosamax), risedronate (Actonel), zoledronate (Zometa), ibandronate (Boniva), incadronate or cimadronate, clodronate, EB-1053, minodronate, neridronate, piridronate and tiludronate including any and all pharmaceutically acceptable salts, derivatives, hydrates and mixtures thereof,
A compound of the instant invention may also be useful for treating or preventing breast cancer in combination with aromatase inhibitors. Examples of aromatase inhibitors include but are not limited to: anastrozole, letrozole and exemestane.
A compound of the instant invention may also be useful for treating or preventing cancer in combination with siRNA therapeutics.
The compounds of the instant invention may also be administered in combination with γ-secretase inhibitors and/or inhibitors of NOTCH signaling. Such inhibitors include compounds described in WO 01/90084, WO 02/30912, WO 01/70677, WO
03/013506, WO 02/36555, WO 03/093252, WO 03/093264, WO 03/093251, WO 03/093253, WO 2004/039800, WO 2004/039370, WO 2005/030731, WO 2005/014553, USSN
10/957,251, WO 2004/089911, WO 02/081435, WO 02/081433, WO 03/018543, WO
2004/031137, WO 2004/031139, WO 2004/031138, WO 2004/101538, WO 2004/101539 and WO 02/47671 (including LY-450139).
A compound of the instant invention may also be useful for treating or preventing cancer in combination with PARP inhibitors.
A compound of the instant invention may also be useful for treating cancer in combination with the following therapeutic agents: abarelix (Plenaxis depot®); aldesleukin (Prokine®); Aldesleukin (Proleukin®); Alemtuzumabb (Campath®); alitretinoin (Panretin®); allopurinol (Zyloprim®); altretamine (Hexalen®); amifostine (Ethyol®); anastrozole
(Arimidex®); arsenic trioxide (Trisenox®); asparaginase (Elspar®); azacitidine (Vidaza®); bendamustine hydrochloride (Treanda®); bevacuzimab (Avastin®); bexarotene capsules
(Targretin®); bexarotene gel (Targretin®); bleomycin (Blenoxane®); bortezomib (Velcade®); brefeldin A; busulfan intravenous (Busulfex®); busulfan oral (Myleran®); calusterone
(Methosarb®); capecitabine (Xeloda®); carboplatin (Paraplatin®); carmustine (BCNU®, BiCNU®); carmustine (Gliadel®); carmustine with Polifeprosan 20 Implant (Gliadel Wafer®); celecoxib (Celebrex®); cetuximab (Erbitux®); chlorambucil (Leukeran®); cisplatin
(Platinol®); cladribine (Leustatin®, 2-CdA®); clofarabine (Clolar®); cyclophosphamide (Cytoxan®, Neosar®); cyclophosphamide (Cytoxan Injection®); cyclophosphamide (Cytoxan Tablet®); cytarabine (Cytosar-U®); cytarabine liposomal (DepoCyt®); dacarbazine (DTIC- Dome®); dactinomycin, actinomycin D (Cosmegen®); dalteparin sodium injection
(Fragmin®); Darbepoetin alfa (Aranesp®); dasatinib (Sprycel®); daunorubicin liposomal (DanuoXome®); daunorubicin, daunomycin (Daunorubicin®); daunorubicin, daunomycin (Cerubidine®); degarelix (Firmagon®); Denileukin diftitox (Ontak®); dexrazoxane
(Zinecard®); dexrazoxane hydrochloride (Totect®); didemnin B; 17-DMAG; docetaxel
(Taxotere®); doxorubicin (Adriamycin PFS®); doxorubicin (Adriamycin®, ubex®);
doxorubicin (Adriamycin PFS Injection®); doxorubicin liposomal (Doxil®); dromostanolone propionate (Dromostanolone ®); dromostanolone propionate (Masterone Injection®);
eculizumab injection (Soliris®); Elliott's B Solution (Elliott's B Solution®); eltrombopag (Promacta®); epirubicin (Ellence®); Epoetin alfa (epogen®); erlotinib (Tarceva®);
estramustine (Emcyt®); ethinyl estradiol; etoposide phosphate (Etopophos®); etoposide, VP- 16 (Vepesid®); everolimus tablets (Afmitor®); exemestane (Aromasin®); ferumoxytol (Feraheme Injection®); Filgrastim (Neupogen®); floxuridine (intraarterial) (FUDR®);
fludarabine (Fludara®); fiuorouracil, 5-FU (Adrucil®); fulvestrant (Faslodex®); gefitinib (Iressa®); geldanamycin; gemcitabine (Gemzar®); gemtuzumab ozogamicin (Mylotarg®); goserelin acetate (Zoladex Implant®); goserelin acetate (Zoladex®); histrelin acetate (Histrelin implant®); hydroxyurea (Hydrea®); Ibritumomab Tiuxetan (Zevalin®); idarubicin
(Idamycin®); ifosfamide (IFEX®); imatinib mesylate (Gleevec®); interferon alfa 2a (Roferon A®); Interferon alfa-2b (Intron A®); iobenguane 1 123 injection (AdreView®); irinotecan (Camptosar®); ixabepilone (Ixempra®); lapatinib tablets (Tykerb®); lenalidomide
(Revlimid®); letrozole (Fernara®); leucovorin (Wellcovorin®, Leucovorin®); Leuprolide Acetate (Eligard®); levamisole (Ergamisol®); lomustine, CCNU (CeeBU®); meclorethamine, nitrogen mustard (Mustargen®); megestrol acetate (Megace®); melphalan, L-PAM
(Alkeran®); mercaptopurine, 6-MP (Purinethol®); mesna (Mesnex®); mesna (Mesnex tabs®); methotrexate (Methotrexate®); methoxsalen (Uvadex®); 8-methoxypsoralen; mitomycin C
(Mutamycin®); mitotane (Lysodren®); mitoxantrone (Novantrone®); mitramycin; nandrolone phenpropionate (DuraboHn-50®); nelarabine (Arranon®); nilotinib (Tasigna®); Nofetumomab (Verluma®); ofatumumab (Arzerra®); Oprelvekin (Neumega®); oxaliplatin (Eloxatin®); paclitaxel (Paxene®); paclitaxel (Taxol®); paclitaxel protein-bound particles (Abraxane®); palifermin (Kepivance®); pamidronate (Aredia®); panitumumab (Vectibix®); pazopanib tablets (Votrienttm®); pegademase (Adagen (Pegademase Bovine)®); pegaspargase
(Oncaspar®); Pegfilgrastim (Neulasta®); pemetrexed disodium (Alimta®); pentostatin (Nipent®); pipobroman (Vercyte®); plerixafor (Mozobil®); plicamycin, mithramycin
(Mithracin®); porfimer sodium (Photofrin®); pralatrexate injection (Folotyn®); procarbazine (Matulane®); quinacrine (Atabrine®); rapamycin; Rasburicase (Elitek®); raloxifene hydrochloride (Evista®); Rituximab (Rituxan®); romidepsin (Istodax®); romiplostim
(Nplate®); sargramostim (Leukine®); Sargramostim (Prokine®); sorafenib (Nexavar®);
streptozocin (Zanosar®); sunitinib maleate (Sutent®); talc (Sclerosol®); tamoxifen
(Nolvadex®); temozolomide (Temodar®); temsirolimus (Torisel®); teniposide, VM-26 (Vumon®); testolactone (Teslac®); thioguanine, 6-TG (Thioguanine®); thiopurine; thiotepa (Thioplex®); topotecan (Hycamtin®); toremifene (Fareston®); Tositumomab (Bexxar®); Tositumomab/I-131 tositumomab (Bexxar®); trans-retinoic acid; Trastuzumab (Herceptin®); tretinoin, ATRA (Vesanoid®); triethylenemelamine; Uracil Mustard (Uracil Mustard
Capsules®); valrubicin (Valstar®); vinblastine (Velban®); vincristine (Oncovin®); vinorelbine (Navelbine®); vorinostat (Zolinza®); wortmannin; and zoledronate (Zometa®).
Thus, the scope of the instant invention encompasses the use of the instantly claimed compounds in combination with a second compound selected from: an estrogen receptor modulator, an androgen receptor modulator, retinoid receptor modulator, a
cytotoxic/cytostatic agent, an antiproliferative agent, a prenyl-protein transferase inhibitor, an HMG-CoA reductase inhibitor, an HIV protease inhibitor, a reverse transcriptase inhibitor, an angiogenesis inhibitor, a PPAR-γ agonist, a PPAR-δ agonist, an inhibitor of inherent multidrug resistance, an anti-emetic agent, an agent useful in the treatment of anemia, an agent useful in the treatment of neutropenia, an immunologic-enhancing drug, an inhibitor of cell proliferation and survival signaling, an apoptosis inducing agent, a bisphosphonate, an aromatase inhibitor, an siR A therapeutic γ-secretase inhibitors, agents that interfere with receptor tyrosine kinases (RTKs), an agent that interferes with a cell cycle checkpoint and any of the therapeutic agents listed above.
Any one or more of the specific dosages and dosage schedules of the
compounds of the instant invention, may also be applicable to any one or more of the therapeutic agents to be used in the combination treatment (hereinafter refered to as the "second therapeutic agent").
Moreover, the specific dosage and dosage schedule of this second therapeutic agent can further vary, and the optimal dose, dosing schedule and route of administration will be determined based upon the specific second therapeutic agent that is being used.
Of course, the route of administration of the compounds of the instant invention is independent of the route of administration of the second therapeutic agent. In an
embodiment, the administration for a compound of the instant invention is oral administration. In another embodiment, the administration for a compound of the instant invention is intravenous administration. Thus, in accordance with these embodiments, a compound of the instant invention is administered orally or intravenously, and the second therapeutic agent can be administered orally, parenterally, intraperitoneally, intravenously, intraarterially, transdermally, sublingually, intramuscularly, rectally, transbuccally, intranasally, liposomally, via inhalation, vaginally, intraoccularly, via local delivery by catheter or stent, subcutaneously, intraadiposally, intraarticularly, intrathecally, or in a slow release dosage form.
In addition, a compound of the instant invention and second therapeutic agent may be administered by the same mode of administration, i.e. both agents administered e.g. orally, by IV. However, it is also within the scope of the present invention to administer a compound of the instant invention by one mode of administration, e.g. oral, and to administer the second therapeutic agent by another mode of administration, e.g. IV or any other ones of the administration modes described hereinabove.
The first treatment procedure, administration of a compound of the instant invention, can take place prior to the second treatment procedure, i.e., the second therapeutic agent, after the treatment with the second therapeutic agent, at the same time as the treatment with the second therapeutic agent, or a combination thereof. For example, a total treatment period can be decided for a compound of the instant invention. The second therapeutic agent can be administered prior to onset of treatment with a compound of the instant invention or following treatment with a compound of the mstant invention. In addition, anti-cancer treatment can be administered during the period of administration of a compound of the instant invention but does not need to occur over the entire treatment period of a compound of the instant invention.
The term "administration" and variants thereof (e.g., "administering" a compound) in reference to a compound of the invention means introducing the compound or a prodrug of the compound into the system of the animal in need of treatment. When a compound of the invention or prodrug thereof is provided in combination with one or more other active agents (e.g., a cytotoxic agent, etc.), "administration" and its variants are each understood to include concurrent and sequential introduction of the compound or prodrug thereof and other agents.
As used herein, the term "composition" is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
The term "therapeutically effective amount" as used herein means that amount of active compound or pharmaceutical agent that elicits the biological or medicinal response in a tissue, system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician.
The term "treating cancer" or "treatment of cancer" refers to administration to a mammal afflicted with a cancerous condition and refers to an effect that alleviates the cancerous condition by killing the cancerous cells, but also to an effect that results in the inhibition of growth and/or metastasis of the cancer.
In an embodiment, the angiogenesis inhibitor to be used as the second compound is selected from a tyrosine kinase inhibitor, an inhibitor of epidermal-derived growth factor, an inhibitor of fibroblast-derived growth factor, an inhibitor of platelet derived growth factor, an MMP (matrix metalloprotease) inhibitor, an integrin blocker, interferon-ct, interleukin-12, pentosan poly sulfate, a cyclooxygenase inhibitor, carboxyamidotriazole, combretastatin A-4, squalamine, 6-0-chloroacetyl-carbonyl)-fumagillol, thalidomide, angiostatin, troponin- 1, or an antibody to VEGF. In an embodiment, the estrogen receptor modulator is tamoxifen or raloxifene.
Also included in the scope of the claims is a method of treating cancer that comprises administering a therapeutically effective amount of a compound of Formula I in combination with radiation therapy and/or in combination with a compound selected from: an estrogen receptor modulator, an androgen receptor modulator, retinoid receptor modulator, a cytotoxic/cytostatic agent, an antiproliferative agent, a prenyl-protein transferase inhibitor, an HMG-CoA reductase inhibitor, an HIV protease inhibitor, a reverse transcriptase inhibitor, an angiogenesis inhibitor, a PPAR-γ agonist, a PPAR-δ agonist, an inhibitor of inherent multidrug resistance, an anti-emetic agent, an agent useful in the treatment of anemia, an agent useful in the treatment of neutropenia, an immunologic-enhancing drug, an inhibitor of cell proliferation and survival signaling, an apoptosis inducing agent, a bisphosphonate, an aromatase inhibitor, an siRNA therapeutic and an agent that interferes with a cell cycle checkpoint.
And yet another embodiment of the invention is a method of treating cancer that comprises administering a therapeutically effective amount of a compound of Formula I in combination with paclitaxel or trastuzumab.
The invention further encompasses a method of treating or preventing cancer that comprises administering a therapeutically effective amount of a compound of Formula I in combination with a COX-2 inhibitor.
The instant invention also includes a pharmaceutical composition useful for treating or preventing cancer that comprises a therapeutically effective amount of a compound of Formula I and a compound selected from: an estrogen receptor modulator, an androgen receptor modulator, a retinoid receptor modulator, a cytotoxic/cytostatic agent, an
antiproliferative agent, a prenyl-protein transferase inhibitor, an HMG-CoA reductase inhibitor, an HIV protease inhibitor, a reverse transcriptase inhibitor, an angiogenesis inhibitor, a PPAR-γ agonist, a PPAR-δ agonist; an inhibitor of cell proliferation and survival signaling, a bisphosphonate, an aromatase inhibitor, an siRNA therapeutic and an agent that interferes with a cell cycle checkpoint.
Further included within the scope of the invention is a method of treating or preventing a disease in which angiogenesis is implicated, which is comprised of administering to a mammal in need of such treatment a therapeutically effective amount of a compound of the present invention. Other inhibitors of MET may also be administered for this method of treatment. Ocular neovascular diseases, which may result in certain forms of blindness, are examples of conditions where much of the resulting tissue damage can be attributed to aberrant infiltration of blood vessels in the eye. The undesirable infiltration can be triggered by ischemic retinopathy, such as that resulting from diabetic retinopathy, retinopathy of prematurity, retinal vein occlusions, etc., or by degenerative diseases, such as the choroidal neovascularization observed in age-related macular degeneration. Inhibiting the growth of blood vessels by administration of the present compounds would therefore prevent the infiltration of blood vessels and prevent or treat diseases where angiogenesis is implicated,
such as ocular diseases like retinal vascularization, diabetic retinopathy, age-related macular degeneration, and the like.
Routes of systemic administration of the compounds of the present invention described above may be utilized in the treatment of such ocular neo ascular diseases. Other routes of ocular administration may also be employed, such as topical, periocular, intravitreal and the like. Intravitreal implants coated with a drug:polymer matrix may also be employed.
Ophthalmic pharmaceutical compositions that are adapted for topical administration to the eye may be in the form of solutions, suspensions, ointments, creams or as a solid insert. Ophthalmic formulations of this compound may contain from 0.01 ppm to 1% and especially 0.1 ppm to 1% of medicament. For a single dose, from between 0.01 to 5000 ng, preferably 0.1 to 500 ng, and especially 1 to 100 ng of the compound can be applied to the human eye. Formulations useful for intravitreal administration are similar to saline solutions described previously for intravenous administration.
These and other aspects of the invention will be apparent from the teachings contained herein.
SCHEMES AND EXAMPLES
The compounds of this invention may be prepared by employing reactions as shown in the following schemes, in addition to other standard manipulations that are known in the literature or exemplified in the experimental procedures. The illustrative schemes below, therefore, are not limited by the compounds listed or by any particular substituents employed for illustrative purposes. Substituent numbering as shown in the schemes does not necessarily correlate to that used in the claims and often, for clarity, a single substituent is shown attached to the compound where multiple substituents are allowed under the definitions of the instant invention hereinabove.
Examples provided are intended to assist in a further understanding of the invention. Particular materials employed, species and conditions are intended to be illustrative of the invention and not limiting of the reasonable scope thereof.
The abbreviations used herein have the following tabulated meanings.
Abbreviations not tabulated below have their meanings as commonly used unless specifically stated otherwise.

DMA N,N-dimethylacetamide
DIEA N,N-Diisopropylethylamine
DBU 1 ,8-diazabicyclo [5.4.0]undec-7-ene
DCM dichloromethane
DIAD diisopropyl azodicarboxylate
DIBAL = diisobutylaluminum hydride
DIPEA - N,N-Diisopropylethylamine
DMAP 4-(dimethylamino)pyridine
DME dimethoxyethane
DMF N,N-dimethylformamide
DMFDMA = dimethylformamide dimethyl acetal
DPPA diphenylphosphoryl azide
EDC 3-(ethyliminomethyleneamino)-N,N-dimethyl-propan-l- amine
EDC1 dichloroethane
Et3N triethylamine
EtOAc ethyl acetate
GST glutathione transferase
HC1 hydrochloric acid
HMDS hexamethyldisilazide
HOBt 1 -hydroxybenzotriazole
hr hour
KOAc = Potassium acetate
K3P04 = Potassium phosphate
LDA lithium diisopropyla ide
metachloroperbenzoic acid
MMPP monoperoxyphthalic acid
Mn02 manganese dioxide
MPLC = medium pressure liquid chromatography
MPPM monoperoxyphthalic acid, magnesium salt 6]¾0
Ms methanesulfonyl = mesyl = S02Me
MsO methane sulfonate = mesylate
μψ microwave
NaH sodium hydride
Na2C<¾ - Sodium bicarbonate
NaOEt - sodium ethoxide
NBS N-bromo succinimide
NIS N-Iodosuccinimide
NMP = N-Methyl-2-pyrrolidone
NSAID = non-steroidal anti-inflammatory drug
NH4OH = Ammonium hydroxide
o-Tol ortho-tolyl
OXONE® - 2 HS05*KHS04* 2S04
PCC pyridinium chlorochromate
PDC pyridinium dichromate
prjg = phosphodiesterase
Pd(PPh3)4 = tetrakis(triphenylphosphine)palladium
PdCl2(dppf)2 = 1 , 1 '-Bis(diphenylphosphino)ferrocene- palladium(II)dichloride
Ph phenyl
Phe benzenediyl
PnNTfa - N-phenyl bis-trifluoromethanesulfommide
PMB para-methoxybenzyl
POCI3 = phosphoryl chloride
PPA polyphosphoric acid
Pye pyridinediyl
r.t. room temperature
Rac. = racemic
SAM aminosulfonyl or sulfonamide or SO2 H2
SEM 2-(trimethy 1 silyl)ethoxymethoxy
SPA scintillation proximity assay
TBAF = tetra-n-butylammonium fluoride
TBTU = < -(benzotriazol- 1 -yl)-N, Ν,Ν', N'-tetramethyluronium tetrafluoroborate
TEA triethylamine
Th 2- or 3-thienyl
THP tetrahydropyran
TFA trifluoroacetic acid
TFAA = trifluoroacetic acid anhydride
THF = tetrahydrofuran
Thi thiophenediyl
TLC thin layer chromatography
TMS-CN = trimethylsilyl cyanide
TMSI trimethylsilyl iodide
Tz 1H (or 2H)-tetrazol-5-yl
XPhos - 2-dicyclohexylphosphino-2'>4'}6,-triisopropylbiphenyl
C3H5 = allyl
Alkyl Group Abbreviations

General Scheme 1

tv
Substituted pyrazolo[l,5-a]pyrimidine I is brominated by reacting with N- bromosuccinimide (NBS) at room temperature in an appropriate solvent or solvent mixture such as DCM, DCM-CH3CN and DMF to provide the corresponding bromide intermediate II (General Scheme 1). Bromide intermediate II is heated with tributyl(l-ethoxyvinyl)stannane in
an appropriate solvent or solvent mixture such asl54-dioxane at or around 100°C, in the presence of an appropriate catalyst such as Pd(PPh3)4, to afford the corresponding enol ether interemediate III. Enol ether intermediate III is treated with an appropriate acid, such as TFA, to afford the corresponding dihydropyridone IV. Dihydropyridone IV is treated with an appropriate reducing reagent such as first by NaB¾ then TFA Et3SiH to afford the
corresponding tetrahydropyridine V.
General Scheme 2

IV VI
Dihydropyridone IV is treated with appropriate reagents to afford the corresponding substituted tetrahydropyridine VI (General Scheme 2).
General Scheme 3

SEM-protected hydroxyl compound VII is treated with appropriate acid such as TFA or HCl in H20 to afford the corresponding substituted l ,3-oxazinane VII (General Scheme 3).
General Scheme 4

Example 1
Preparation of 3 -methyl-7-(6-phenylpyridin-3-yl)-5-(tetrahydro- 1 , 1 -dioxido-2H-thiopyran-4- yl)-3,4-dihydro-pyrazolo[l,5-a]pyrimido[5,4-e] pyrimidin-2(lH)-one
Step 1 : Preparation of 3-oxo-3-(tetrahydro-2H-thiopyran-4-yl)propanenitrile

A solution of CH3CN (2.54 mL, 48.7 mmol) in 10 mL of THF was added dropwise to a solution of "BuLi (48.7 mmol) in 15 mL of THF at -78°C. After stirring for 1 h at -78°C, a solution of methyl tetrahydro-2H-thiopyran-4-carboxylate (3.90 g, 24.3 mmol) in 10 mL of THF was added dropwise and the resulting reaction mixture was stirred at -78°C for 1 h, and then slowly warmed to -40°C in 2 h. The reaction was quench with 1 N HCl (pH<l). THF was removed and the residue was extracted with EtO Ac, washed with brine, dried over Na2S04, and concentrated. The crude product was purified by a Si02 column (0-50%
EtOAc Hexanes) to afford 3-oxo-3-(tetrahydro-2H-thiopyran-4-yl)propanenitrile as a light brownish oil. LCMS tR = 1.29 Min (5 min run, UV254m«)- Mass calculated for, M+ 1694.1 , observed LC/MS m/z 170.1 (M+H)
Step 2: Preparation of 5-(tetrahydro-2H-thiopyran-4-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo [1,5 -a]pyrimidin-7-amine

A mixture of 3-aminopyrazole (1.61 g, 19.3 mmol) and 3-oxo-3-(tetrahydro-2H- thiopyran-4-yl)propanenitrile (3.60 g, 21.3 mmol) in acetic acid (40 mL) was heated at 100°C in a sealed tube overnight. After cooling to room temperature, all the volatiles were removed under reduced pressure to afford 5-(tetrahydro-2H-thiopyran-4-yl)pyrazolo[l,5-a]pyrimidin-7- amine as a light brownish oil. LCMS tR = 0.59 Min (5 min run, UV254nm). Mass calculated for, M+ 234.1, observed LC/MS m/z 235.1 (M+H).
5-(tetrahydro-2H-thiopyran-4-yl)pyrazolo[l ,5-a]pyrimidin-7-amine was dissolve in DCM (60 mL), and then SEMC1 (11.9 mL, 67.6 mmol) and DIPEA (23.5 mL, 135 mmol) were added. The resulting reaction mixture was stirred at 45°C for 1 h. After cooling to room temperature, all the volatiles were removed under reduced pressure. The residue was diluted with EtOAc (300 mL), filtered and washed with EtOAc. The filtrate was concentrated and the crude product was purified by a Si02 column (0-30%) to afford 5-(tetrahydro-2H-thiopyran-4- yl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l ,5-a]pyrimidin-7-amine as light brownish oil. LCMS tR ^3. 7 Min (5 min run, UV2S4nm)- Mass calculated for, M+ 494.2, observed LC/MS m/z 495.2 (M+H).
Step 3: Preparation of 3-iodo-5-(tetrahydro-lJl-dioxido-2H-thiopyran-4-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[ 1 ,5-a]pyrimidin-7-amine

Na2S04. Evaporation afforded crude 5-(tetrahydro~l,l-dioxido-2H-thiopyran-4-yl)-N,N-bis((2- (trimethylsiIyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-amine which was used for next step without further purification: LCMS tR = 2.69 Min (5 min run, UV254nni)- Mass calculated for, M+ 526.2, observed m/z 527.2 (M+H).
NIS (852 mg, 3.79 mmoL) was added to a solution of crude 5-(tetrahydro- 1,1- dioxido-2H-thiopyran-4-yl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5- a]pyrimidin-7-amine in C3¾CN (18 mL). The mixture was stirred at room temperature for lh. Concentration and purification by column chromatography afforded 3-iodo-5-(tetrahydro-l,l- dioxido-2H-thiopyran-4-yl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5- a]pyrimidin-7-amine: LCMS tR = 2.87 Min (5 min run, UV254nm). Mass calculated for, M+ 652.1, observed m/z 653.0 (M+H).
Step 4: Preparation of 3~(6-phenylpyridin-3~yl)-5-(tetrahydro- 1,1 -dioxido-2H-thiopyran-4-yl)- N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-amine

2-phenyl-5-(4;4,5>5 etramethyl-l,352-dioxaborolan-2-yl)pyridine (616 mg, 2.19 mmoL), K3PO4 (1073 mg, 5.06 mmol), and PdCI2(dppf) · CH2C12 (138 mg, 0.17 mmol) was added to a solution of 3-iodo-5-(tetrahydro-l,l-dioxido-2H-thiopyran-4-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[lf5~a]pyrimidin-7-amine (1 100 mg, 1.69 mmol) in dioxane (15 mL) and H20 (3 mL). The resulting solution was stirred at 90° C under argon overnight. The mixture was diluted with ¾0 and then extracted with ethyl acetate (x2). The combined organic layers were washed with brine and dried with Na2S04. Evaporation and purification by column chromatography afforded 3-(6-phenylpyridin-3-yl)-5-(tetrahydro-l,l- dioxido-2H-thiopyran-4-yl)-N?N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo [1,5- a]pyrimidin-7-amine: LCMS t = 2.69 Min (5 min run, UV254nm)- Mass calculated for, M+ 679.3, observed rn/z 680.3 (M+H).
Step 5: Preparation of 6-bromo-3-(6-phenylpyridin-3-yl)-5-(tetrahydro
thiopyran-4-yl)-N,N-bis((2-(trim

NBS (176 mg, 0.98 mmoL) was added to a solution of 3-(6-phenylpyridin-3-yl)- 5-(tetrahydro- 1 , 1 -dioxido-2H-thiopyran-4-yl)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7 -amine (640 mg, 0.94 mmoL) in CH3CN (10 mL). After stirring at room temperature for 30 min, the mixture was concentrated in vacuo. Purification by column cliromatography afforded 6-bromo-3-(6-phenylpyridin-3-yl)- 5-(tetrahydro- 1 , 1 -dioxido-2H-thiopyran-4-yl)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-amine: LCMS IR = 3.03 Min (5 min run, UV254nm). Mass calculated for, M+ 757.2, observed m/z 758.0 (M+H).
Step 6: Preparation of 3-(6-phenylpyridin-3-yl)-5-(tetrahydro-l,l-dioxido-2H-thiopyran-4-yl)~ N,N-bis((2-(trimethylsilyl)ethoxy)methyl)-6-vinylpyrazolo [ 1 J5-a]pyrimidin~7-amine

A degassed mixture of 6-bromo-3~(6-phenylpyridin-3-yl)-5-(tetrahydro-l,l- dioxido-2H-thiopyran-4-yl)-NsN-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5- a]pyrimidin-7-amine (203 mg , 0.27mmol), Pd(PPh3)4 (31 mg, 0.027 mmoL),
tributyl(vinyl)stannane (255 mg, 0.80 mmoL) in CH3CN (6 mL) was heated at 150 C under microwave condition for 60 min. The reaction mixture was cooled to room temperature, filtered through 9:1 Si02:KF plug and concentrated in vacuo. Purification by column chromatography afforded 3-(6-phenylpyridin-3~yl)-5-(tetrahydro-l,l-dioxido-2H-thiopyran~4- yl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)~6~vinylpyrazolo[l,5-a]pyrimidin-7-amine:
LCMS tR = 2.82 Min (5 min run, UV254nm)- Mass calculated for, M+ 705.3, observed m/z 706.3 (M+H).
Step 7: Preparation of 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenyIpyridin-3-yl)- 5-(tetrahydro-l , 1 -dioxido-2H-thiopyran-4-yI)pyrazolo[ 1 ,5-a]pyrimidine-6-carbaldehyde

To 3-(6-phenylpyridin-3-yl)-5-(tetrahydro-l,l-dioxido-2H-thiopyran-4-yl)-N,N- bis((2-(trimethylsilyl)ethoxy)methyl)-6-vinylpyrazolo[l,5-a]pyrimidin-7-amine (189 mg, 0.26 mmol) in 1,4-doixane (3 mL) was added 2.5 wt% Os04 in 1,4-dioxane (168 L, 0.013 mmol), 2,6-lutidine (265 L, 2.68 mmol) and H20 (1 mL) and the resulting mixture was stirred at room temperature for 20 minutes. NaI04 (287 mg, 1.34 mmol) was then added and stirring at room temperature continued for 4 days. Saturated Na2S2C>3 solution (5 mL) was added and the mixture stirred for 10 minutes. Orgamcs were then extracted with CH2C12 (2 x), dried (Na2S(>4) and concentrated in vacuo to 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)-5-(tetrahydro-l J
carbaldehyde, which was used without further purification: LCMS % = 2.87 Min (5 min run, UV254nm). Mass calculated for, M+ 707.2, observed m/z 708.3 (M+H).
Step 8: Preparation of (7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3- yl)-5-(tetrahydro-l,l~dioxido-2H-thiopyran^

NaBH4 (30 mg, 0.79 mmoL) was added to a solution of 7-(bis((2- (trimethyIsilyl)ethoxy)methyl)amino)-3 -(6-phenylpyridin-3-yl)-5-(tetrahydro- 1 , 1 -dioxido-2H- thiopyran-4-yl)pyrazolo[l,5-a]pyrimidine-6-carbaldehyde (53 mg, 0.075 mmoL) i MeOH (2 mL). After stirring at room temperature for lh, the mixture was concentrated to dry.
Purification by column chromatography afforded (7-(bis((2-
(trimethylsilyl)ethoxy)methyl)amino)-3 -(6-phenylpyridin-3 -yl)-5 -(tetrahydro- 1 , 1 -dioxido-2H- thiopyran-4-yl)pyrazolo[l55-a]pyrimidin-6-yl)methanol: LCMS t& = 2.82 Min (5 min run, UV254nm). Mass calculated for, M+ 709.3, observed m/z 710.2 (M+H)
Step 9: Preparation of 6-((methylamino)methyl)-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-l,l- dioxido-2H-thiopyran-4-yl)pyrazolo [ 1 , 5 -a]pyrimidin-7-amine

Et3N (41.7 μΙ_, 0.29 mmoL), then MsCl (17.4 μΙ_, 0.22 mmoL) was added to a solution of (7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3-yl)-5- (tetrahydro- 1 , 1 -dioxido-2H-thiopyran-4-yl)pyrazolo[ 1 ,5-a]pyrimidin-6-yl)methanol (52 mg,
0.073 mmoL) in THF (2 mL) at 0°C. After stirring at room temperature for lh, N¾ in MeOH (2 mL) was added dropwise and the mixture was stirred at room temperature overnight. The mixture was concentrated to afford crude methyl amine, which was then treated with 50% TFA in ¾0 (2 mL) for lh. Concentration and purification afforded 6-((methylamino)methyl)-3-(6- phenylpyridin-3 -yl)-5~(tetrahydro- 1 , 1 -dioxido-2H-thiopyran-4-yl)pyrazolo [ 1 , 5 -a] pyrimidin-7- amine: LCMS IR = 2.1 Min (10 min run, UV254NM). Mass calculated for, M+ 462.1, observed m/z 463.1 (M+H).
Step 10: Preparation of 3-methyl-7-(6-phenylpyridin-3-yl)-5-(tetrahydro-l,l-dioxido-2H- thiopyran-4-yl)-3 ,4-dihydro-pyrazolo[ 1 ,5-a]pyrimido [5 ,4-e] pyrimidin-2( 1 H)-one

CDI (10 mg) was added to a mixture of 6-((methylamino)methyl)-3-(6- phenylpyridin-3-yl)-5-(tetrahydro- 1 ,1 -dioxido-2H-thiopyran-4-yl)pyrazolo[ 1 ,5-a]pyrimidu>7- amine (3.2 mg) in pyridine (0.2 mL). After stirring at room temperature overnight, the mixture was concentrated and purified by prep-LC to afford 3-methyl-7-(6-phenylpyridin-3-yl)-5- (tetrahydro- 1 , 1 -dioxido-2H-thiopyran-4-yl)- 3 ,4-dihydro-pyrazolo[ 1 , 5 -a]pyrimido [5 ,4-e] pyrimidin-2(lH)-one, LCMS tR = 3.08 Min (10 min run, U ^nm)- Mass calculated for, M+ 488.1, observed m/z 489.1 (M+H).
Example 2
Preparation of 7-(quinolin-3 -yl)-5 -(tetrahydro- 1 , 1 -dioxido-2H-thiopyran-4-y l)-2,4-dihydro- 1 H-
[l,3]oxazino[554-e]pyrazolo[l;5-a]pyrimidine
Step 1 : Preparation of (7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quinolin-3-yl)-5- (tetrahydro-1 , 1 -dioxido-2H-thiopyran-4-yl)pyrazolo[l ,5-a]pyrimidin-6-yl)methanol

Step 2: Preparation of 7-(qumolin-3-yl)-5-(tetrahydro-l,l-dioxido-2H-thiopyran-4-yl)-2,4- dihydro-lH-[ 1 ,3]oxazino[5,4-e]pyrazolo[ 1 ,5-a]pyrimidine

(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quinolin-3-yl)-5-(tetrahydro- l,l-dioxido-2H-thiopyran-4-yl)pyrazolo[l,5-a]pyrimidin-6-yl)methanol (7.3 mg, 0.011 mmoL) was treated with 50% TFA in ¾0 (1 mL) at room temperature until the disappearance of starting material in LCMS. Concentration and purification by prep-LC afforded 7-(quinolin-3- yl)-5-(tetrahydro-l5l-dioxido-2H-thiopyran-4-yl)-2,4-dihydro-lH-[l,3]oxazino[5,4- e]pyrazolo[l,5-a]pyrimidine: LCMS t& = 2.80 Min (10 min run, UV254nm). Mass calculated for, M+ 435.1, observed m/z 436.0 (M+H).
Example 3
Preparation of tm«i'-4-(4-(Hydroxymethyl)-7-(6-phenylpyridin-3 -yl)-2,4-dihydro- 1 H- [ 1 ,3]oxazino[5,4-e]pyrazolo[ 1 ,5-a]pyrimidin-5-yl)cycIohexanecarboxylic acid
Stepl : Preparation of iram'-Cyclohexane- -dicarboxylic acid monomethyl ester

To a solution of dimethyl cyclohexane-l,4-dicarboxylate (80 g, 400 mmol) in MeOH (200 mL) at reflux was added dropwise a solution of KOH (22.4 g, 400 mmol)in MeOH (200 mL) and the resulting reaction mixture was heated under reflux for additional 8 h. After cooling to room temperature, MeOH was evaporated. The resulting solid was taken up in water (600 mL), and then washed with EtOAc (100 mLX3). The aqueous layer was acidified to pH 6 with 6 N HC1. The precipitated white solid was collected and washed with water and dried to afford the desired product as a white solid.
Step 2: Preparation of trans-l-tert-Butyl 4-methyl cyclohexane-l,4-dicarboxylate

To a suspension of trara-cyclohexane-1 ,4-dicarboxylic acid monomethyl ester (16.5 g, 88.6 mmol) in DCM (90 niL) at room temperature was added 10 drops of DMF, followed by oxalyl chloride (38.7 rnL, 5 eq). Then the reaction mixture was warmed to room temperature and stirred for 2 h (turned clear gradually and stopped bubbling after about 1 h). All the volatiles were removed under reduced pressure. To the oily residue was added ¾uOH (90 mL) and pyridine (30 mL). The resulting mixture was stirred at room temperature overnight- All the volatiles were removed under reduced pressure and the residue was diluted with EtOAc, washed with water and brine, dried over Na2S04, and concentrated. The crude mixture was purified by a Si02 column (0-20% EtOAc/Hexanes, Rf = 0.5 in 20% EtOAc, PMA staining) to afford the desired product as a white solid.
Step 3: Preparation of trans-tert-Bx yl 4-(2-cyanoacetyl)cyclohexanecarboxylate

C3¾CN (16.49 mL, 316 mmol) was added dropwise to a solution of "BuLi (2.2 eq) in 300 mL of THF at -78°C. After stirring for 1 h at -78°C, a solution of

4-methyl cyclohexane-l? 4-dicarboxylate (35.2 g, 145 mmol) in 150 mL of THF was added dropwise during a period of 1 h and the resulting reaction mixture was stirred at -78°C for 1 h, and then slowly warmed to room temperature overnight. The reaction was quenched with IN HC1 solution to pH=7. After filtration, THF was removed and the residue was diluted with EtOAc. The organic layer was separated and washed with brine, dried over Na2S04? and concentrated. The crude product was purified by a Si02 column (0-30% EtOAc/Hexanes, Rf - 0.5 in 30% EtOAc, PMA staining) to afford the desired compound as a white solid.
Step 4: Preparation of trans-\-tert-Butyl 4-(7-aminopyrazolo[l,5-a]pyrimidin-5- yl)cyclohexanecarboxylate

A mixture of 3-aminopyrazole (5.16 g, 62.1 mmol) and trans-tert-butyl 4-(2- cyanoacetyl)cycIohexanecarboxylate (13.0 g, 51.8 mmol) in toluene (70 mL) was heated at 1 15°C in a sealed tube overnight. After cooling to room temperature, all the volatiles were removed under reduced pressure to afford the crude product as light brown oil, which was used without further purification.
Step 5: Preparation of transA-tert- ntyl 4-(7-(bis((2-
(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[l,5-a]pyrimidin-5-yl)cyclohexanecarboxylate

To a solution of trans-l~tert~butyl 4-(7-aminopyrazolo[l,5-a]pyrimidin-5- yl)cyclohexanecarboxylate (51.8 mmol) in CH2CI2 (50 mL) was added SEMCI (32 mL, 181.3 mmol), followed by DIPEA (63.2 mL, 362.6 mmol). The resulting reaction mixture was stirred at 45°C for 2 h. After cooling to room temperature, all the volatiles were removed under reduced pressure. The residue was diluted with EtOAc, washed with H20 and brine, and concentrated. The crude product was purified by a Si02 column (0-20% EtOAc/Hexane, Rf = 0.25 in 20% EtOAc) to afford the desired product as a brownish oil.
Step 6: Preparation of trans-l-tert-Butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3- iodopyrazolo[ 1 ,5-a]pyrimidin-5-yl)cyclohexanecarboxylate

To a solution of trans-l-tert-Qutyl 4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)pyrazolo[l,5-a]pyrimidin-5-yl)cyclohexanecarboxylate (21.9 g, 38 mmol) in CH3CN was added NIS (8.56 g, 38 mmol). The resulting solution was stirred at room temperature for 1 h. The reaction was quenched with Na2S203 (sat.). CH3CN was removed under reduced pressure. The residue was dissolved in EtOAc, washed with H20
and brine, dried over Na2S04, and concentrated. The crude product was purified by a Si02 column (0-20% EtOAc/Hexanes, Rf = 0.6 in 20% EtOAc) to afford the desired product as a light brown oil.
Step 7: Preparation of traw-tert-Butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)pyrazolo[l,5-a]pyrimidin-5-yl)cyclohexanecarboxylate

2-phenyl-5-(4,4,5,5 etramethyl-lJ3,2-dioxaborolan-2-yl)pyridine (313 mg, 1.11 mmol), K3P04 (544 mg, 2.56 mmol), and PdCl2(dppf) · CH2C12 (70 mg, 0.085 mmol) was added to a solution of trans-l-tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3- iodopyrazoIo[l,5-a]pyrimidin-5-yl)cyclohexanecarboxylate (600 mg, 0.85 mmol) in dioxane (4 mL). To this suspension was added distilled H20 (0.4 mL). The resulting solution is stirred at 100°C under argon for 18 hours. The reaction mixture was concentrated in vacuo and then purified via silica gel chromatography (0% to 60% ethyl acetate in hexanes gradient) to yield the title compound as yellow oil.
Step 8: Preparation of traiw-tert-Butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6- bromo-3-(6-phenylpyridin-3-yl)pyrazolo[l,5-a]pyrimidin-5-yl)cyclohexanecarboxylate

N-bromosuccinimide (71 mg, 0.394 mmol) was added to a solution of trans-teri- butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3-yl)pyrazolo[l,5- a]pyrimidin-5-yl)cyclohexanecarboxylate (240 mg, 0.329 mmol) in acetonitrile (3 mL) and dichloromethane (2 ml). The resulting solution is stirred at room temperature for 1 hour. The reaction is reduced in vacuo and the resulting oil is then purified via silica gel chromatography (0% to 30% ethyl acetate in hexanes gradient) to yield the title compound as yellow oil.
Step 9: tert-Butyl 4-(7-(bis((2-(trimethylsilyl)emoxy)methyl)amino)-3-(6-phenylpyridin-3-yl)- 6-vinylpyrazolo [ 1 ,5-a]pyrimidin~5 -yl)cyclohexanecarboxylate

Tributyl(vinyl)tin (271 mg, 0.855 mmol) and Pd(PPh3)4 (33 mg, 0.029 mmol) was added to a solution of traTO-tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6- bromo-3 -(6-phenylpyridin-3 -yl)pyrazolo [ 1 , 5 -a] pyrimidin-5 -yl)cyclohexanecarboxy late (230 mg, 0.285 mmol) in 1,4-dioxane (2 mL). The reaction is heated to 90°C under argon for 16 hours. After 16 hours, the solvent is removed in vacuo and the residue is purified via silica gel chromatography (0% to 30% ethyl acetate in hexanes gradient) to yield the title compound as yellow oil.
Step 10: Preparation of terf-Butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-(l52- dihydroxyethyl)-3-(6-phenylpyridin-3-yl)pyrazolo[l,5-a]pyrimidin-5- yl)cyclohexanecarboxylate

To a slurry of tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)-6-vinylpyrazolo[l ,5-a]pyrimidin-5-yl)cyclohexanecarboxylate (176 mg, 0.233 mmol) in a mixture of acetone (3 ml), water (1 ml) and acetonitrile (1 ml) was added N- methylmorpholine-N~oxide (0.096 ml, 50 wt% in water, 0.466 mmol), followed by osmium tetroxide (0.145 ml, 2.5 wt% in tert-butanol, 0.012 mmol) at room temperature. The resulting mixture was stirred for 16 hrs at room temperature. Organic solvents were evaporated and the aqueous residue was extracted with ethyl acetate (50 ml, three times), dried over Na2S045 and concentrated. The crude residue was purified via silica gel chromatography (0% to 60% ethyl acetate in hexanes gradient) to yield the title compound as yellow oil.
Step 11 : Preparation of tra w-4-(4-(Hydroxymethyl)-7-(6-phenylpyridin-3-yl)-2?4~dihydro-l H- [1 ,3]oxazino[5,4-e]pyrazolo[l ,5-a]pyrimidin-5-yl)cyclohexanecarboxylic acid
tert-Butyl 4-(7-(bis((2-(trimethylsilyI)et oxy)methyl)amino)-6-( 1 ,2- dihydroxyethyl)-3 -(6-phenylpyridin-3 -yl)pyrazolo [1,5 -a]pyr imidin- 5 - yl)cyclohexanecarboxylate (140 mg, 0.177 mmol) in 1,4-dioxane (1 mL) was treated with 4N HCl(aq) (1 mL). The resulting solution was stirred at 65°C for 3 hours. The reaction solution was concentrated and purified using HPLC to give the titled compound. LC-MS: 486.21
[M+H]. LC/MS RT = 3.16 min.
Example 4
Preparation of (3 -(7-(6-phenylpyridin-3 -yl)-2,4-dihydro- 1 H- [ 1 ,3] oxazino [5 ,4-e]pyrazolo [1,5- a]pyrimidin-5-yl)-8-azabicycIo[3.2.1]octan-8-yl)(4H-l,2,4-triazoI-3-yl)methanone
Step 1 : Preparation of pyrazolo[l,5-a]pyrimidine~5,7-diol

To lH-pyrazol-3 -amine (123 g, 148.0 mmol) in EtOH (50 mL) was added diethyl malonate (25.0 mL, 164.7 mmol), 21 wt% NaOEt in EtOH (110 mL, 294.6 mmol) and additional EtOH (50 mL). The resulting mixture was then heated at 80°C under an atmosphere of argon for 16 hours, at which time the reaction was allowed to cool to room temperature. The reaction mixture was then concentrated in vacuo until almost dry, before H20 (500 mL) was added. Vigorous stirring aided the dissolving of solids, at which time cone. HCI was added until pH~2 was attained (solid precipitate formed). The precipitate was collected and dried by vacuum filtration giving pyrazolo[l,5-a]pyrimidine~5,7-diol as a tan solid.
Step 2: Preparation of 5,7-dichloropyrazolo[l,5-a]pyrimidine
 To pyrazolo[l,5-a]pyrimidine-5,7-dioI (9.6 g, 63.5 mmol) in a 500 mL flask was added P0C13 (125 mL, 1341.1 mmol). The flask was then cooled to 0°C and N5 N- dimethylaniline (22 mL, 173.6 mmol) was carefully added. On warming to room temperature, the reaction was then heated at 60°C under an atmosphere of argon for 16 hours. On cooling, the reaction mixture was concentrated in vacuo to give a brown viscous liquid. This brown viscous liquid was carefully poured onto ice and allowed to warm to room temperature overnight. To the brown solution was carefully added saturated NaHC03 solution until no further effervescence was observed and pH ~ 8 was attained. Organics were then extracted with CH2C12 (4 x 50 mL), dried (NajSO,,) and concentrated in vacuo to give a brown liquid (29.8 g). Gradient column chromatography on silica eluting with 50% CH2Cl2/hexanes (to elute aniline) followed by 75% CH2Cl2/hexanes (to elute product) gave 5,7- dichloropyrazolo[l,5-a]pyrimidine as a white solid.
To pyrazolo[l,5-a]pyrimidine-5,7-dioI (9.6 g, 63.5 mmol) in a 500 mL flask was added P0C13 (125 mL, 1341.1 mmol). The flask was then cooled to 0°C and N5 N- dimethylaniline (22 mL, 173.6 mmol) was carefully added. On warming to room temperature, the reaction was then heated at 60°C under an atmosphere of argon for 16 hours. On cooling, the reaction mixture was concentrated in vacuo to give a brown viscous liquid. This brown viscous liquid was carefully poured onto ice and allowed to warm to room temperature overnight. To the brown solution was carefully added saturated NaHC03 solution until no further effervescence was observed and pH ~ 8 was attained. Organics were then extracted with CH2C12 (4 x 50 mL), dried (NajSO,,) and concentrated in vacuo to give a brown liquid (29.8 g). Gradient column chromatography on silica eluting with 50% CH2Cl2/hexanes (to elute aniline) followed by 75% CH2Cl2/hexanes (to elute product) gave 5,7- dichloropyrazolo[l,5-a]pyrimidine as a white solid.
Step 3: Preparation of 5-chloropyrazolo[l,5-a3pyrimidin-7~amine

To 5,7-dichloropyrazolo[l,5-a]pyrimidine (7.6 g, 40.4 mmol) in a sealed vessel was added NH4OH (100 mL). The vessel was then sealed and heated at 85°C for 2.5 hours, at which time the consistency of the white solid had changed (from foamy white solid to free- flowing white solid). The vessel was removed from the heat source and allowed to cool to room temperature overnight. On cooling, the contents of the vessel were collected and dried by vacuum filtration giving 5-chloropyrazolo[l,5~a]pyrimidin-7-amine as a yellow-tinged white solid.
Step 4: Preparation of 5-chloro-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5- aj pyrimidin-7-amine

To 5-chloropyrazolo[l,5-a]pyrimidin-7-amine (6.7 g, 39.7 mmol) in CH2C12 (30 mL) was added N,N-diisopropylethylamine (48.0 mL, 275.6 mmol) followed by 2- (trimethylsilyl)ethoxymethyl chloride (25.0 mL, 141.7 mmol). The reaction was heated at 45°C for 3 hours before being allowed to cool to room temperature. The reaction mixture was then poured into a separatory runnel containing -100 mL saturated NaHC03 solution and CH2C12
(50 mL). Organics were then extracted with CH2C12 (4 x 50 mL), dried (N-½S04) and concentrated in vacuo to give a thick orange liquid (33.8 g). Gradient column chromatography on silica eluting with 5% to 15% EtOAc/hexanes gave crude 5-chloro-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-amine as a colorless liquid.
Step 5: Preparation of tert-butyl 3~(trifluoromethylsulfonyloxy)-8-azabicyclo[3.2.1]oct-3-ene~ 8-carboxylate

To a solution of N-Boc-nortropinone (6 g, 26.6 mmol) in THF (70 ml) at -78°C was added LDA (2 M in haptane/THF/ethyl benzene, 20ml, 40 mmol) slowly and the reaction mixture was stirred for 10 min. A solution of N~phenylbis(trifluoromethanesulfonimide) (10.5 g, 29.3 mmol) in THF (48 ml) was added. The reaction mixture was stirred at -78°C for 30 min and the cooling bath was removed to warm it up to room temperature for 1.5 h until all N-Boc- nortropinone was utilized. Saturated NH4C1 solution (-10 mL) was added and stirring was continued for 5 minutes before the reaction mixture was transferred to a separatory funnel using EtOAc (150 mL). The organics were then extracted with EtOAc (2 x 125 ml), and washed with water (2 x 30 ml), brine (1 x 30 ml), and dried over MgS04 The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with
EtOAc/Hexanes (0-35%) gave the desired product, tert-butyl 3-(trifluoromethylsulfonyloxy)-8- azabicyclo[3.2. l]oct-3-ene-8-carboxylate.
Step 6: Preparation of tert-butyl 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-8- azabicyclo[3.2. l]oct-2-ene-8-carboxylate

A mixture of tert-butyl 3-(trifluoromethylsulfonyloxy)-8-azabicyclo[3.2.1 ]oct-3- ene-8-carboxylate (10.1 g, 28.4 mmol), bis(pinacolato)diboron (8.7 g, 34.1 mmol), KOAc (8.4 g, 85.3 mmol), PdCl2(dppf)2.CH2Cl2 (1.4g, Ummol), and dppf (1 g, 1.8 mmol) in dioxane (170 ml) was flushed with argon and stirred at 80°C for 16 h. On cooling, the solvent was evaporated, and the crude residue was redissolved in EtOAc (500 ml), washed with water (1 x 125 ml), brine (1 x 125 ml), and dried over MgS04. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with EtOAc/Hexanes (0- 40%) gave the desired product, tert-butyl 3-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-8- azabicyc3o[3.2.1]oct-2-ene-8-carboxylate.
Step 7; Preparation of tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)
amino)pyrazolo[l ,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1 ]oct-2-ene-8-carboxylate

To 5 -chloro-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo [ 1 , 5 -a] pyrimidin- 7-amine (11.1 g, 25.8 mmol) in DME (200 mL) was added tert-butyl 3-(4,45555-tetramethyI- l,3,2-dioxaborolan-2-yI)-8-azabicycio[3.2.1]oct-2-ene-8-carboxylate (9.5 g, 28.4 mmol), PdCl2(dppf)2 (2.1 g, 2.6 mmol) and 2M N¾C03 (100 ml). The reaction was heated at 100°C for 16 hours, at which time LC MS analysis confirmed full consumption of starting material. On cooling, H?0 (80 ml) and EtOAc (200 ml) were added and the organics were extracted with EtOAc (2 x 250 ml), dried (N¾S04) and concentrated in vacuo to give a crude product.
Gradient column chromatography on silica eluting with 10% to 60% EtOAc/hexanes(0-50%) gave tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl) amino)pyrazolo[l ,5-ajpyrimidin-5- yl)-8-azabicyclo[3.2.1 ]oct-2-ene-8-carboxylate.
Step 8: Preparation of anti & syn - tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl) amino)pyrazolo[l ,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1 ]oct-2-ene-8-carboxylate

A mixture of tert-butyl 3-(7-(bis((2- (trimethy lsilyl)ethoxy)methyl)amino)pyrazolo[ 1 , 5 -a] pyrimidin- 5 -yl)- 8 -azabicyclo [3.2.1 ] oct-3 - ene-8-carboxylate (12.2 g, 20.3 mmol), 10% Pd C (2.1 g) in EtOAc (175 ml) was stirred at 45°C under hydrogen (balloon pressure) for 16 hours. After filtration and concentration, the crude mixture of two isomers was purified by gradient column chromatography on silica eluting with EtOAc/Hexanes (0-35%) to give the slightly impure "syn" product (6.24 g, Rf = 0.6 in 25% EtOAc/Hexanes) and the "anti" product (5.44 g, Rf = 0.5 in 25% EtOAc/Hexanes) which was used in the following reaction sequences.
Step 9: Preparation of tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3- iodopyrazolo[ 1 ,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1 ]octane-8-carboxylate

To the "anti" teri-butyl 3-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)pyrazolo[ 1 ,5-a]pyrimidm-5-yl)-8- azabicyclo[3.2.1]octane-8-carboxylate (6.04 g, 10 mmol) in CH3CN (40 mL) and DCM (40 mL) was added N-iodosuccinimide (2.5 g, 1 1 mmol) portionwise and the resulting mixture was stirred at room temperature for 1.5 h, at which time LC/MS confirmed full conversion of starting material to product. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with EtOAc/Hexanes (0-50%) gave tert-butyl 3- (7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[l,5-a]pyrimidin-5-yl)-8- azabicyclo [3.2.1] octane- 8-carboxylate.
Step 10: Preparation of fert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)pyrazolo[l,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1]octane-8-carboxylate

To tert-butyl 3-(7-(bis((2-(trimethyIsilyl)ethoxy)methyl)amino)-3- iodopyrazolo[l ,5-a3pyrimidin-5-yl)-8-azabicyclo[3.2. l]octane-8-carboxylate (2 g, 2.7 mmol) in dioxane (22 mL) and H20 (5.5 mL) was added 2-phenyl-5~(4,4,5,5-tetramethyl-l ,3,2- dioxaborolan-2-yl)pyridine (1.2 g, 4.1 mmol), PdCl2(dppf)-CH2Cl2 (0.3 g, 0.3 mmol) and K2C03 (1.2 g, 8.2 mmol). The reaction was heated at 100°C for 15 hours, at which time LC/MS analysis confirmed full consumption of starting material. On cooling, H20 (40 ml) and EtOAc (100 mL) were added and organics were extracted with EtOAc (2 x 75 ml), dried (Na2S04) and concentrated in vacuo to crude. Gradient column chromatography on silica eluting with 0 to 50% EtOAc/hexanes gave tert-butyl 3-(7-(bis((2-
(trimefhylsilyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3--yl)pyrazolo[l;5-a]pyrimidin-5-yl)- 8-azabicyclo[3.2.1]octane-8-carboxylate.
Step 11 : Preparation of feri-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-
3-(6-phenylpyridin-3-yl)pyrazolo[l,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1]octane-8- carboxylate
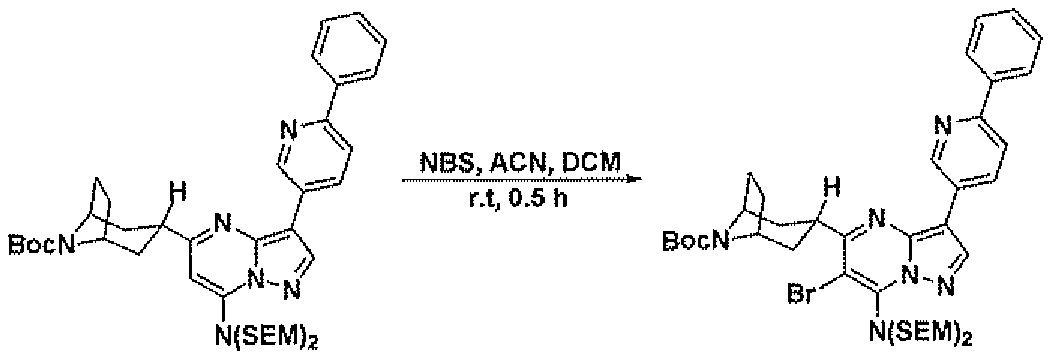
To a mixture of terf-butyl 3-(7-(bis((2-(trimethylsiIyl)ethoxy)met yl)amino)-3- (6-phenylpyridin-3-yl)pyrazolo[ 1 J5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1 ]octane-8-carboxylate (1.7 g, 2.3 mmol) in CH3CN (10 mL) and dichloromethane (10 mL) was added N~
bromosuccinimide (0.45 g, 2.5 mmol) portionwise and the resulting mixture was stirred at room temperature for 0.5 h, at which time LC/MS confirmed full conversion of starting material to product. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with EtOAc/Hexanes (0-50%) gave tert-butyl 3-(7-
(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3 -(6-phenylpyridin~3-yi)pyrazolo [1,5- a]pyrimidin-5-yl)-8-azabicyclo[3.2.1 ]octane-8-carboxylate.
Step 12: Preparation of tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)-6-vinylpyrazolo[ 1 ,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1 ]octane-8- carboxylate

A mixture of ter -butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6- bromo-3-(6-phenylpyridin-3-yl)pyrazolo[l ?5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1 ]octane-8- carboxylate (836 mg, 1.0 mmol), vinylboronic acid pinacol ester (0.17 mL, 1 mmol), tetrakis(triphenylphosphine)palladium (0.23 g, 0.2 mmol), bis(tri-i-butylphophine)palladium (0) (100 mg, 0.2 mmol), 2 M Na2C03 (5 mL) and dioxane (15 mL) was degassed with Ar briefly and heated with stirring at 100 C in a capped vessel for 16 hours. After cooling, the mixture was diluted with EtOAc (20 mL) and water (5 mL). Organic layer was isolated and dried over MgS04. Solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with EtOAc/Hexanes (0-30%) gave the title product.
Step 13: Preparation of tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-formyl-
3-(6-phenylpyridin-3-yl)pyrazolo[l,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1]octane-8- carboxylate

To a solution of terf-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3- (6-phenylpyridin-3-yl)-6-vinylpyrazolo[l ,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2. l]octane-8- carboxylate (128 rag, 0.16 mmol) in dioxane (3 mL) was added 2,6-Lutidine (74 L), water (0.5 mL) and Os04 (2.5% in t-BuOH, 154 μΐ, 0.012 mmol). After stirring for 20 minutes,
NaI04 (103 mg,) was added and the resulting mixture was stirred overnight. The reaction was quenched with saturated Na2S203 (5 mL) and stirred for additional 20 minutes. The aqueous layer was isolated, extracted with dichloromethane three times. Combined organic layers were dried over MgSC Solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with EtOAc/Hexanes (0-30%) gave the title product.
Step 14: Preparation of tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6- (hydroxymethyl)-3-(6-phenylpyridin-3-yl)pyrazolo[l,5-a)pyrimidin-5-yl)-8-

To a solution of tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6- formyl-3-(6-phenylpyridin-3-yl)pyrazolo[l,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1]octane-8 carboxylate (477 rag, 0.61 mmol) in MeOH (4 mL) was added NaBH4 (23 mg, 0.61 mmol). After stirring for half hour, 10% NaOH (2 mL) was added and concentrated. The residue was extracted with dichloromethane three times and combined organic layers were dried over MgS04. Solvent was removed in vacuo and the residue was purified by column
chromatography on silica gel. Elution with EtOAc/Hexanes (0-40%) gave the title product.
Step 15; Preparation of 5-(8-azabicyclo[3.2.1]octan-3-yl)-7-(6-phenylpyridin-3-yl)-2,4-dihydro- lH-fl^JoxazinotS^-ejpyrazolotl^-aJpyriniidine

Step 16: Preparation of (3-(7-(6-phenylpyridin-3-yl)-2,4-dihydro-lH-[l,3]oxazino[5,4- e]pyrazolo[l ,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1 ]octan-8-yl)(4H-l ,2s4-triazol-3- yi)methanone

A mixture of lH-1 ,2,4-triazole-3-carboxylic acid (8 mg, 0.07 mmol), EDCI (20 mg, 0.1 mmol), and 1-hydroxybenzotriazole (9 mg, 0.07 mmol) in DMF (1 mL) was slightly warmed up to a homogeneous solution which was added into a stirred solution of 5~(8- azabicyclo [3.2.1 ]octan-3 -yl)-7-(6-phenylpyridin-3 -yl)-2,4-dihydro- 1 H-[ 1 ,3]oxazino[5 ,4- e]pyrazolo[l,5-a]pyrimidine TFA salt (0.07 mmol) and N,N-diisopropylethylamine (37 μΐ,, 0.2 mmol) in DMF (2mL). It was stirred further for 10 min at room temperature. This crude compound was purified by HPLC to afford (3-(7-(6-phenylpyridin-3-yl)-2,4-dihydro-l FIJI, 3]oxazino[5,4-e]pyrazolo[l,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2. l]octan-8-yl)(4H-l?2,4- triazol-3-yl)methanone. LCMS t = 3.20 Min (10 min run, UV254nm). Mass calculated for M+ H 534.0, observed LC/MS m/z 534.0 (M+H).
Example 5
Preparation of (3-(7-(6-(4-methoxyphenyl)pyridin-3-yl)-2,4-dihydro-l H- [l,3]oxazino[554-e]pyrazolo[l,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2.1]octan-8-yl)(4H-l,2,4- triazol-3 -yl)methanone

By applying the chemistry described in Example 5, steps 10- 16, and using suitable starting materials, (3-(7-(6-(4-methoxyphenyl)pyridin-3-yl)-2,4-dihydro-lH- [1 ,3]oxazino[5,4-e]pyrazolo[l ,5-a]pyrimidin-5-yl)~8-azabicyclo[3.2. l]octan-8-yl)(4H-l ,2,4- triazol-3-yl)methanone was synthesized. LCMS tR = 3.20 Min (10 min run, UV254nm). Mass calculated for M+ H 564.0, observed LC/MS m/z 564.0 (M+H).
Example 6
Preparation of 3-(7-(6-(2-hydroxypropan-2-yl)pyridin-3-yl)-2,4-dihydro-lH- [l,3]oxazino[5,4-e]pyrazolo[l,5-a pyrimidin-5-yl)-8-azabicyclo[3.2J]octan-8-yl)(lH-l,2,4 triazol-3 -yl)methanone
Stepl: Preparation of tert-butyl 3-(3-(6-acetylpyridin-3-yl)-7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)pyrazolo[l,5»a]pyrimidin-5-yl)-8- azabicyclo[3.2.1 ]octane-8-carboxylate

To l-(5-bromopyridin-2-yl)ethanone (2g, lO.lmmol) in Dioxane (80 ml) was added bis(pinacolato)diboron (3.3g, 13.1mmol), PdCl2(d pf)2.CH2Cl2 (0.8 g, lmmol) and KOAc (3 g, 30.2 mmol). It was then degassed with Argon for five minute before heating at 100 °C for 16 hours, at which time LC MS analysis confirmed full consumption of starting material. On cooling, the solvent was rotoevaporated, and the crude was redissolved in DCM
(500ml), washed with water (1 x 125ml), brine (1 x 125ml), and dried over MgS04. Solvent was removed in vacuo and the crude compound l-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan- 2-yl)pyridin-2-yl)ethanone was used for the next step without any further purification.
To l-(5-(4,4,5,5-tetramethyl-l5352-dioxaborolan-2-yl)pyridin-2-yl)ethanone (Crude) in Dioxane (52 ml) and H20 (13 ml) was added tert-butyl 3-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3 -iodopyrazolo [1,5 -a]pyrimidin-5-yl)- 8 - azabicyclo[3.2.1]octane-8-carboxylate 3 (4.9g, 6.7mmol), PdCl2(dppf)2.CH2Cl2 (0.5 g,
OJrnmol) and 2CO3 (2.8 g, 20.1 mmol). The reaction was heated at 100°C for 16 hours, at which time LC/MS analysis confirmed full consumption of starting material. On cooling, the solvent was evaporated, and the crude was redissolved in DCM (500ml), washed with water (1 x 125ml), brine (1 x 125ml), dried (MgSO*) and concentrated in vacuo to crude. Gradient column chromatography on silica eluting with 0 to 50% EtOAc/hexanes gave the desired tert- butyl 3-(3-(6-acerylpyridin-3-yl)-7-(bis((2-(trimethylsilyl)emoxy)methyl)amino)pyrazolo[ 1 ,5- a]pyrimidin-5-yl)-8-azabicyclo[3.2.1]octane-8-carboxylate.
Step 2; Preparation of tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-(2- hy droxypropan-2-yl)pyridin-3 -yl)pyrazolo [1,5-a] pyrimidin- 5 -yI)-8 -azabicyclo [3.2.1 ]octane- 8 - carboxylate

To a solution of tert-butyl 3-(3-(6-acetylpyridin-3-yl)-7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)pyrazolo [1,5-a] pyrimidin- 5 -yl)-8- azabicyclo[3.2.1]octane-8-carboxylate (2.9g, 4.1mmol) in THF (75ml) at -78°C was added MeMgBr (1 ,4M in toluene/THF, 5.8ml, 8.1mmol) slowly. The reaction mixture was stirred at - 78°C for 10 min and the cooling bath was removed to warm it up to room temperature for 2.5 h. Saturated NH4CI solution (~10mL) was added and stirring continued for 5 minutes before the reaction mixture was transferred to a separatory funnel using EtOAc (150mL). Organics were then extracted with EtOAc (2 x 125ml), and washed with water (2 x 30ml), brine (1 x 30ml), and dried over MgSO-t. Solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with EtOAc/Hexanes (0-50%) gave desired product, tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-(2-hydroxypropan-2- yl)pyridin-3-yl)pyrazolo[l ,5-a]pyrimidin-5-yl)-8-azabicyclo[3.2. l]octane-8-carboxylate.

By applying the chemistry described in Example 5, steps 11-16, and using suitable starting materials, 3-(7-(6-(2-hydroxypropan-2-yl)pyridin-3-yl)-2,4-dihydro-lH- [ 1 ,3]oxazino [5 ?4-e]pyrazolo [ 1 ,5-a]pyrimidin-5 -yl)-8-azabicyclo [3.2.1 ] octan-8-yl)( 1 H- 1 ,2,4- triazol-3-yl)methanone was synthesized. LCMS: 1.93 mins, m/z = 516.0 (MH+).
Example 7
Preparation of (3-(3-(6-phenyIpyridin-3-yl)-6,7,8,9-tetrahydropyrazolo[l ,5-a]pyrido[3,2- e]pyrimidin- 5 -yl)-8-azabicyclo[3.2.13 octan- 8 -y!)(4H- 1 ,2,4-triazol-3 -yl)methanone
Step 1 : Preparation of 8-azabicyclo[3.2.1]octan-3-yl)-3-(6-phenylpyridin-3-yl)-8,9- dihydropyrazolo[l,5-a]pyrido[3!2-e]pyrimidin-6(7H)-one

A mixture of compound tert-butyl 3-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-6-bromo
a]pyrimidin~5-yl)-8-azabicyclo[3.2A]octane~8-carboxylate (834 mg, 0.49 mmol), tributyl(l- ethoxyvinyl)tin (356 mg, 0.98 mmol), tetrakis(triphenylphosphine)palladium (56.9 mg, 0.049 mmol) in dioxane (6 mL) was degassed with argon for five minutes. It was then heated at 100°C in a sealed tube for 16 h, at which time LC/MS analysis confirmed full consumption of starting material On cooling, the solvent was evaporated, and the crude residue was redissolved in EtOAc, washed with 0.5 M F solution, brine (1 x 25 mL), and dried over MgS04. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with EtOAc/Hexanes gave tert-butyl 3-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)~6- ( 1 -ethoxyvinyl) -3-( 6-phenylpyridin-3~yl)pyrazolo[l , 5- a]pyrimidin-5~yl)-8-azabicyclo[3.2.1]octane-8-carboxylate.
The enol ether was then treated with 20% TFA CH2C12 (10 mL) for 2h at room temperature. Concentration afforded 8-azabicyclo[3.2.1]octan-3-yl)-3-(6-phenylpyridin-3-yl)- 8,9-dihydropyrazolo[l,5-a]pyrido[3,2-e]pyrimidin-6(7H)-one as an TFA salt. LCMS tR = 0.67
Min (5 min run, UV 254mn). Mass calculated for, M+ 450.2, observed LC/MS m/z 451.0 (M+H).
Step 2: Preparation of 8-azabicyclo[3.2.1]octan-3-yl)-3-(6-phenylpyridin-3-yl)-6J7J8,9- tetrahydropyrazolo[l,5-a]pyrido[3,2~e]pyrimidine

At 0°C, NaBH4 (50 mg, 0.79 mmol) was added to a solution of 5-((lR,3s,5S)-8- azabicyclo [3.2.1 ]octan-3 -yl)-3-(6-pheny!pyridin- 3 -yl)-8,9-dihydropyrazolo [1,5 -ajpyrido [3,2- e]pyrimidin-6(7H)-one (78.9 mg, 0.18 mmoL) in MeOH (5 mL). The mixture was stirred at room temperature for 2h, the solvent was removed in vacuo and at 0°C, TFA (lmL) and then Et3SiH (0.5 mL) was added to the residue. After stirring at room temperature for 2h and concentration to dryness, the crude 8-azabicyclo[3.2.13octan-3-yl)-3-(6-phenylpyridin-3-yl)- 6,7,8,9-tetrahydropyrazolo[l,5-a]pyrido[3,2-e]pyrimidine was used for next step without further purification. LCMS t = 0.93 Min (5 min run, UV254nm)- Mass calculated for, M+ 436.2, observed LC/MS m/z 437.3 (M+H).
Step 3: Preparation of (3-(3-(6-phenylpyridin-3-yl)-6,7,8,9-tetrahydropyrazolo[l!5- a]pyrido[3,2-e l-3-yl)methanone

A mixture of 8-azabicyclo[3.2. l]octan-3-yl)-3-(6-phenylpyridin-3-yl)-6,7,8,9- tetrahydropyrazolo[l,5-a]pyrido[3,2-e]pyrimidine (73 mg, 0.17 mmoL), lH-l,2,4-triazole-3- carboxylic acid (22.7 mg, 0.20 mmoL), EDC (64.0 mg, 0.33mmoL), HOBt (45.2 mg, 0.33 mmoL) and DIEA (173.9 μί, 1.00 mmoL) in DMF (2 mL) was stirred at room temperature for lh. Purification with prep-LC provided (3-(3-(6-phenylpyridin-3-yl)-6,7>8,9- tetrahydropyrazolo[l ,5-a]pyrido[3,2-e]pyrimidin-5-yl)-8-azabicyclo[3.2.1 ]octan-8-yl)(4H- lf2,4-triazol-3-yl)methanone: LCMS tR = 2.48 Min (10 min run, UV254nm). Mass calculated for, M+ 531.2, observed LC/MS m/z 532.0 (M+H).
Example 8
Preparation of 2-hydroxy-l-(3-(3-(6-phenylpyridin-3-yl)-6,7,8,9- tetrahydropyrazolo[l,5-a]pyrido[3,2-e]pyrimidin-5-yl)-8-azabicyclo[3.2.1]octa

By applying the chemistry described in Example 7, steps 3, and using suitable starting materials, 2-hydroxy-l-(3-(3-(6-phenylpyridin-3-yl)-6,7,8,9-tetrahydropyrazolo[l ,5- a3pyrido[3J2-e]pyrimidin-5-yl)-8-azabicyclo[3.2.1]octan-8-yl)ethanone was synthesized: LCMS tR = 2.49 Min (10 min run, UV254nm). Mass calculated for, M+ 494.2, observed LC/MS m/z 495.0 (M+H).
Example 9
Preparation of (2R,6S)-2,6-dimethyl-4-(3-(6-phenylpyridin-3-yl)-6J7,8,9- tetrahydropyrazolo[l,5-a]pyrido[3,2-e]pyrimidin-5-yl)morpholine
Stepl : Preparation of 5-chloro-3-(6-phenylpyridin-3-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo [ 1 ,5-a]pyrimidin-7-am ine

2-phenyl-5-(4J4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridine (675 mg, 2.38 mmol), K3P04 (1264 mg, 5.96 mmol), and PdCl2(dppf)-CH2Cl2 (162 mg, 0.20 mmol) was added to a solution of 5-chloro-3-iodo-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5- a]pyrimidin-7-amine (1101 mg, 1.98 mmol) in dioxane (18 mL) and ¾0 (3 mL). The resulting solution was stirred at 70° C under argon overnight. The mixture was diluted with H20 and then extracted with ethyl acetate (x2). The combined organic layers were washed with brine and dried with Na2SC>4. Evaporation and purification by column chromatography afforded 5-chloro-3-(6-phenylpyridin-3-yl)-NJN-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5-
a]pyrimidin-7-amine: LCMS tR = 3.36 Min (5 min run, UV254nm). Mass calculated for, M+ 581.2, observed LC/MS m z 582.2 (M+H).
Step 2: Preparation of 5-((2R,6S)-2,6-dimet ylmorpholino)-3-(6-phenylpyridin-3- yl)pyrazolo [ 1 , 5-a]pyrimidin~7-amine

A mixture of 5-chloro-3-(6-phenylpyridin-3-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[l ,5-a]pyrimidin-7-amine (582 mg, 1.0 mmol), NaHC03 (336 mg, 4.0 mmol), and (2S,6R)-2,6-dimethylmorpholine (230mg, 2.0 mmol) in NMP (6 mL was heated at 130°C for 4h. On cooling, the mixture was diluted with H20 and then extracted with ethyl acetate (x2). The combined organic layers were washed with brine and dried with Na2S(¾. Evaporation and purification by column chromatography afforded 5-((2R,6S)-2,6- dimethylmorpholino)-3-(6-phenylpyridin-3-yl)pyrazolo[ 1 ,5-a]pyrimidin-7-amine: LCMS tR = 1.61 Min (5 min run, UV254lBn). Mass calculated for, M+ 660.3, observed LC/MS m/z 661.3 (M+H).
Step 3: Preparation of 6-bromo-5-((2R,6S)-2,6-dimethylmorpholino)-3-(6-phenylpyridin-3- yl)pyrazolo[l ,5-a]pyrimidin-7-amine

To a solution of 5-((2R,6S)-2,6-dimethylmorpholino)~3-(6~phenylpyridin-3- yl)pyrazolo[l,5-a]pyrimidin-7-amine (655 mg, 0.99 mmol) in dichloromethane (10 mL) was added N-bromosuccinimide (21 1.9 mg, 1.19 mmol) portionwise and the resulting mixture was stirred at room temperature for 1 h, at which time LC/MS confirmed full conversion of starting material to product. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel to afford 6-bromo-5-((2R,6S)-2,6-dimethylmorpholino)-3-(6-
phenylpyridin-3-yl)pyrazolo[l,5-a]pyrimidin-7-amine. LCMS tR = 1.84 Min (5 min run,
UV254nm). Mass calculated for, M+ 738.2, observed LC/MS m/z 739.2 (M+H).

By applying the chemistry described in Example 7, steps 1-2, and using suitable starting materials, (2R,6S)-2,6-dimethyl-4-(3-(6-phenylpyridin-3-yl)-6,7,8,9~
tetrahydropyrazolo[l J5-a]pyrido[3,2-e]pyrimidin-5-yl)morpholine was synthesized. LCMS tR = 3.04 Min (10 min run, UV25 nm). Mass calculated for, M+ 440.2, observed LC/MS m/z 440.98 (M+H).
Pharmaceutical Composition
As a specific embodiment of this invention, 100 mg of 3-methyl-7-(6- phenylpyridin-3-yl)-5-(tetrahydro-l,l-dioxido-2H-thiopyran-4-yl)-3f4-dihydro-pyrazolo[l,5- a]pyrimido[5s4-e] pyrimidin-2(l H)-one is formulated with sufficient finely divided lactose to provide a total amount of 580 to 590 mg to fill a size 0, hard-gelatin capsule.
The compounds of the instant invention described in the Examples were tested by the assays described below and were found to have mTOR inhibitory activity. Other assays are known in the literature and could be readily performed by those of skill in the art. mTOR Kinase Assay
The mTOR assay buffer contains 10 mM hepes (pH 7.4), 50 raM NaCl, 100 Mg/ml BSA, 50 mM B-glycerophosphate, 10 mM MnC12 and 0.5 mM DTT. 20 ng of mTOR enzyme is preincubated with the compound for 10 minutes. 5 μΜ ATP and 0.1 μΜ GSTS6K is added. The reaction is incubated for one hour at 30°C. Anti phospho p70S6K (about 1.7 ng/well) and anti GSTXL665 (1 :1 Ratio with the substrate GSTS6K) are added after incubating. The plates are read at least 2 hours after adding the anti phospho p70S6K and the anti GSTXL665.
ICso DETERMINATIONS: Dose-response curves were plotted from inhibition data generated, each in duplicate, from 8 point serial dilutions of inhibitory compounds.
Concentration of compound was plotted against % kinase activity, calculated by CPM of
treated samples divided by CPM of untreated samples. To generate IC50 values, the dose- response curves were then fitted to a standard sigmoidal curve and IC50 values were derived by nonlinear regression analysis. CH 1 in vitro Kinase Assay
This in vitro assay utilizes recombinant His-CHKl expressed in the baculovirus expression system as an enzyme source and a biotinylated peptide based on CDC25C as substrate (biotin-RSGLYRSPSMPENLNRPR).
Materials and Reagents:
1) CDC25C Ser 216 C-term Biotinylated peptide substrate (25 mg), stored at -20° C, Custom Synthesis by Research Genetics: biotin-RSGLYRSPSMPENLNRPR 2595.4 MW
2) His-CHKl In House lot P976, 235 g/n L, stored at -80° C.
3) D-PBS (without CaCl and MgCl): GIBCO, Cat.# 14190-144
4) SPA beads: Amersham, Cat SPQ0032: 500 mg/vial
Add 10 mL of D-PBS to 500 mg of SPA beads to make a working concentration of 50 mg mL. Store at 4 ^C. Use within 2 week after hydration.
5) 96- Well White Microplate with Bonded GF/B filter: Packard, Cat.# 6005177
6) Top seal-A 96 well Adhesive Film: Perkin Elmer, Cat J 6005185
7) 96-well Non-Binding White Polystyrene Plate: Corning, Cat. # 6005177
8) MgCl2: Sigma, Cat J M-8266
9) DTT: Promega, CatJ V3155
10) ATP, stored at 4 °C: Sigma, CatJ A-5394
11) γ33ρ-ΑΤΡ, 1000-3000 Ci/mMol: Amersham, CatJ AH9968
12) NaCl: Fisher Scientific, CatJ BP358-212
13) H3PO4 85% Fisher, CatJ A242-500
14) Tris-HCL pH 8.0: Bio-Whittaker, Cat. # 16-015 V
15) Staurosporine, 100 g: CALBIOCHEM, Cat. # 569397
16) Hypure Cell Culture Grade Water, 500 mL: HyClone, CatJ SH30529.02
Reaction Mixtures:
1) Kinase Buffer: 50 mM Tris pH 8.0; 10 mM MgCl2; 1 mM DTT
2) His-CHKl, In House Lot P976, MW ~30KDa, stored at -80°C.
6 nM is required to yield positive controls of -5,000 CPM. For 1 plate (100 reaction): dilute 8 \L of 235 μg mL (7.83 μΜ) stock in 2 mL Kinase Buffer. This makes a 31 nM mixture. Add 20 uL/well. This makes a final reaction concentration of 6 nM.
3) CDC25C Biotinylated peptide.
Dilute CDC25C to 1 mg/mL (385 μΜ) stock and store at -20°C. For 1 plate (100 reactions): dilute 10 μϋ of 1 mg/mL peptide stock in 2 mL Kinase Buffer. This gives a 1.925 μΜ mix. Add 20 μΕ/κ ΰΐίοη. This makes a final reaction concentration of 385 nM.
4) ATP Mix.
For 1 plate (100 reactions): dilute 10 μΐ, of 1 mM ATP (cold) stock and 2 pL fresh P33-ATP (20 μθ) in 5 mL Kinase Buffer. This gives a 2 μΜ ATP (cold) solution; add 50 μΐνννεΐΐ to start the reaction. Final volume is 100 μΐ,/reaction so the final reaction concentrations will be 1 μΜ ATP (cold) and O^Ci/reaction.
5) Stop Solution:
For 1 plate add: To 10 mL Wash Buffer 2 (2M NaCl 1% H3PO4): lmL SPA bead slurry (50 mg); Add 100 μΙΤννεΙΙ
6) Wash buffer 1: 2 M NaCl
7) Wash buffer 2: 2 M NaCl, 1% H3P04
Assay Procedure:

* Total reaction volume for assay.** Final reaction volume at termination of reaction (after addition of stop solution).
1) Dilute test compounds to desired concentrations in water/ 10% DMSO - this will give a final DMSO concentration of 1% in the reaction. Dispense 10 μΐΐ,/reaction to appropriate wells. Add 10 μΤ 10% DMSO to positive (CHK1+CDC25C+ATP) and negative (CHKl+ATP only) control wells.
2) Thaw enzyme on ice ~ dilute enzyme to proper concentration in kinase buffer (see
Reaction Mixtures) and dispense 20 μΐ, to each well.
3) Thaw the Biotinylated substrate on ice and dilute in kinase buffer (see Reaction Mixtures). Add 20 pL/well except to negative control wells. Instead, add 20 pL Kinase Buffer to these wells.
4) Dilute ATP (cold) and P33-ATP in kinase buffer (see Reaction Mixtures). Add 50 μΙΛνεΙΙ to start the reaction.
5) Allow the reaction to run for 2 hours at room temperature.
6) Stop reaction by adding 100 μΐ, of the SPA beads/stop solution (see Reaction Mixtures) and leave to incubate for 15 minutes before harvest
7) Place a blank Packard GF/B filter plate into the vacuum filter device (Packard plate harvester) and aspirate 200 mL water through to wet the system.
8) Take out the blank and put in the Packard GF/B filter plate.
9) Aspirate the reaction through the filter plate.
10) Wash: 200 mL each wash; IX with 2M NaCl; IX with 2M NaCI/ 1% H3PO4
11 ) Allow filter plate to dry 15 minutes.
12) Put TopSeal-A adhesive on top of filter plate.
13) Run filter plate in Top Count
Settings: Data mode: CPM
Radio nuclide: Manual SPA:P33
Scintillator: Liq/plast
Energy Range: Low
ICgti DETERMINATIONS: Dose-response curves were plotted from inhibition data generated, each in duplicate, from 8 point serial dilutions of inhibitory compounds.
Concentration of compound was plotted against % kinase activity, calculated by CPM of treated samples divided by CPM of untreated samples. To generate IC50 values, the dose- response curves were then fitted to a standard sigmoidal curve and IC50 values were derived by nonlinear regression analysis.
Selected Thiazole Derivatives of the present invention were tested using this assay and provided IC50 values ranging from about 1 nM to about 5500 nM.
CDK2 Kinase Assay
BACULOVIRUS CONSTRUCTIONS: Cyclin E was cloned into pVLl 393 (Pharmingen, La Jolla, California) by PCR, with the addition of 5 histidine residues at the amino-terminal end to allow purification on nickel resin. The expressed protein was approximately 45kDa. CD 2 was cloned into pVL1393 by PCR, with the addition of a haemaglutinin epitope tag at the carboxy-terminal end (YDVPDYAS). The expressed protein was approximately 34kDa in size.
ENZYME PRODUCTION: Recombinant baculoviruses expressing cyclin E and CDK2 were co-infected into SF9 cells at an equal multiplicity of infection (MOI^S), for 48 hrs. Cells were harvested by centrifugation at 1000 RPM for 10 minutes, then pellets lysed on ice for 30 minutes in five times the pellet volume of lysis buffer containing 50mM Tris pH 8.0, 150mM NaCl, 1% NP40, ImM DTT and protease inhibitors (Roche Diagnostics GmbH, Mannheim,
Germany). Lysates were spun down at 15000 RPM for 10 minutes and the supernatant retained. 5mL of nickel beads (for one liter of SF9 cells) were washed three times in lysis buffer (Qiagen GmbH, Germany). Imidazole was added to the baculovirus supernatant to a final concentration
of 20mM5 then incubated with the nickel beads for 45 minutes at 4°C. Proteins were eluted with lysis buffer containing 250mM imidazole. Eluate was dialyzed about 15 hours in 2 liters of kinase buffer containing 50mM Tris pH 8.0, ImM DTT, lOmM MgCl2, ΙΟΟμΜ sodium orthovanadate and 20% glycerol. Enzyme was stored in aliquots at -70°C,
In Vitro Cyclin E/CDK2 Kinase Assays
Cyclin E/CDK2 kinase assays can be performed as described below in low protein binding 96-well plates (Corning Inc, Corning, New York).
Enzyme is diluted to a final concentration of 50 ] ^Jmh in kinase buffer containing 50mM Tris pH 8.0, 10 mM MgCl2;l mM DTT, and 0.1 raM sodium orthovanadate.
The substrate used in these reactions is a biotinylated peptide derived from Histone HI (from Amersham, UK). The substrate is thawed on ice and diluted to 2 μΜ in kinase buffer. Test compounds are diluted in 10% DMSO to desirable concentrations. For each kinase reaction, 20 \xL of the 50 μg/mL enzyme solution (1 μg of enzyme) and 20 μΐ of the 2 μΜ substrate solution are mixed, then combined with 10 Ε of diluted compound in each well for testing. The kinase reaction is initiated by addition of 50 μΕ of 2 μΜ ATP and 0.1 μθ of 33P-ATP (from
Amersham, UK). The reaction iss allowed to run for 1 hour at room temperature, then is stopped by adding 200 \xL of stop buffer containing 0.1% Triton X-100, 1 mM ATP, 5mM EDTA, and 5 mg/mL streptavidine coated SPA beads (from Amersham, UK) for 15 minutes. The SPA beads are then captured onto a 96-well GF/B filter plate (Packard/Perkin Elmer Life Sciences) using a Filtermate universal harvester (Packard Perkin Elmer Life Sciences.). Nonspecific signals are eliminated by washing the beads twice with 2M NaCl then twice with 2 M NaCl with 1% phosphoric acid. The radioactive signal can then be measured using, for example, a TopCount 96 well liquid scintillation counter (from Packard/Perkin Elmer Life Sciences).
ICgn DETERMINATIONS: Dose-response curves are plotted from inhibition data generated, each in duplicate, from 8 point serial dilutions of inhibitory compounds. Concentration of compound is plotted against % kinase activity, calculated by CPM of treated samples divided by CPM of untreated samples. To generate IC50 values, the dose-response curves are then fitted to a standard sigmoidal curve and IC5o values can be derived using nonlinear regression analysis.
Compounds of the present invention exhibit mTOR IC50 values of about 1 nM to about 5500 nM, CHK1 IC50 values of about 100 nM to about 55000 nM, and CDK2 IC50 values of about 800 nM to about 30000 nM. In all cases, the compounds are much more selective for mTOR over CHK1 and CDK2. The following table shows the activity data for an illustrative list of compounds of the invention.



+++IC50>100nM
++ IC50= 10-lOOnM
+ IC50<10nM
Claims
1. A compound of the formula:

E is CRyRz, O, C(0), NRy or SOm;
R! is hydrogen, halo, -C(0)R5, Ci-6 alkyl, C2-6 alkenyl, C -6 alkynyi, C3-8 cycloalkyl, C3 -8 cycloalkenyl, aryl, heteroaryl or heterocyclyl, wherein said alkyl, alkenyl, alkynyi, cycloalkyl, cycloalkenyl, aryl, heteroaryl and heterocyclyl groups are optionally substituted with one to three substituents independently selected from the group consisting of R5, halo, cyano, C)-6 alkyl, C1-6 haloalkyl, (CRyRz)mORy, (CRyRz)mNRyRz, C(0)Ry, C(0)ORy, SOmRy and
C(0)NRyR2;
R2 is hydrogen, halo, cyano, NRyRz, ORy, Ci- alkyl or C1-6 haloalkyl;
R3 is C3-8 cycloalkyl, C3-8 cycloalkenyl, aryl, heteroaryl or heterocyclyl, 0(heterocyclyl), SOm(heterocyclyl), Cj.6 alkyl, (C1-6 alkyl)NHR5, (CI-6 alkyl)NHCORx, wherein said cycloalkyl, cycloalkenyl, aryl, heteroaryl and heterocyclyl groups are optionally substituted with one to three substituents independently selected from the group consisting of R5, halo, cyano, Ci-6 alkyl, C w haloalkyl, ORy, 0(C1-6 haloalkyl), 0(Ci-6 alkyl)ORy, C(0)R C(0)ORy,
C(0)CRxRyRz, SOmRy, NRyRz, C(0)NRyRz, C(0)heteroaryl and NHCORy;

Wherein U is N or CH;
X is absent, CRyRz, O or C(O);
Y is absent, CRyRz, O, or C(O);
L is absent or (CRyRz)n;
T is absent or (CRyRz)n; Z is absent or (CRyRz)n;
R4 is hydrogen, halo, C1-6 alkyl, C2.6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, aryl, heteroaryl or heterocyclyl, wherein said alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, aryl, heteroaryl, and heterocyclyl groups are optionally substituted with one to three
substituents independently selected from the group consisting of R5, halo, cyano, C1-6 alkyl, Cf. 6 haloalkyl, ORy, 0(Ci-6 haloalkyl), C(0)Ry, C(0)ORy, SOmRy, C(0)NRyRz and NRyRz;
R5 is C3-g cycloalkyl, C3-g cycloalkenyl, aryl, heteroaryl or heterocyclyl, wherein said cycloalkyl, cycloalkenyl, aryl, heteroaryl and heterocyclyl groups are optionally substituted with one to three substituents independently selected from the group consisting of halo, cyano, Ci-6 alkyl, Ci-6 haloalkyl, ORy, 0(d-6 haloalkyl), C(0)R C(0)ORy, SOmRy and NRyRz;
R6 is selected from the group consisting of hydrogen, halo, cyano, Ci-6 alkyl, Cj-6 haloalkyl, (Ci-6 alkyl)ORy, ORy, 0(d-6 haloalkyl), C(0)R C(0)ORy, SOmRy and NRyRz;
R7 is selected from the group consisting of hydrogen, halo, cyano, C1-6 alkyl, C1-6 haloalkyl, ORy, 0(Ci-6 haloalkyl), C(0)Ry, C(0)ORy, SOmRy and NRyRz; Rx is hydrogen or C1-6 alkyl;
Ry is hydrogen, C1-6 alkyl or Ci-6 haloalkyl, wherein said alkyl group is optionally substituted with one to three hydroxyl;
Rz is hydrogen, C]-6 alkyl or Ci-6 haloalkyl, wherein said alkyl group is optionally substituted with one to three hydroxyl; m is an integer from zero to two;
n is an integer from zero to two;
or a pharmaceutically acceptable salt thereof.
2. The compound of Claim 1 wherein D is CRyRz or C(O); E is CRyRz, O or NRy; or a pharmaceutically acceptable salt thereof.
3. The compound of Claim 2 wherein R1 is heteroaryl, wherein said heteroaryl group is optionally substituted with one to three substituents independently selected from the group consisting of R5, halo, cyano, C^ alkyl, Ci.6 haloalkyl, (CRyRz)mORy,
(CRyRz)mNRyRz, C(0)Ry, C(0)ORy > SOmRy and C(0)NRyRz; or a pharmaceutically acceptable salt thereof.
4. The compound of Claim 3 wherein R2 is hydrogen, or a pharmaceutically acceptable salt thereof.
5. The compound of Claim 4 wherein
R is C3.8 cycloalkyl or heterocyclyl, wherein said cycloalkyl and heterocyclyl groups are optionally substituted with one to three substituents independently selected from the group consisting of halo, cyano, Ci-6 alkyl, OR , C(0)Ry, and C(0)ORy;

Y is absent or CRyRz;
L is absent or (CRyRz)n;
T is absent or (CRyRz)n;
Z is absent or (CRyRz)n;
or a pharmaceutically acceptable salt thereof.
6. The compound of Claim 5 wherein R6 is selected from the group consisting of hydrogen, halo, Ci-6 alkyl and (C1-6 alkyl)ORy; R7 is hydrogen; or a
pharmaceutically acceptable salt thereof.
7. The compound of Claim 1 selected from:

Or a stereoisomer thereof;
or a pharmaceutically acceptable salt thereof;
or a pharmaceutically acceptable salt of the stereoisomer thereof.
8. A pharmaceutical composition comprising a pharmaceutically effective amount of the compound according to any one of Claims 1 to 7, and a pharmaceutically acceptable carrier.
9. A compound according to any one of Claims 1 to 8 for the treatment prevention of proliferative disorders or cancer in a mammal.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US37596710P | 2010-08-23 | 2010-08-23 | |
| US61/375,967 | 2010-08-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012027240A1 true WO2012027240A1 (en) | 2012-03-01 |
Family
ID=45723757
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/048549 WO2012027240A1 (en) | 2010-08-23 | 2011-08-22 | Fused tricyclic inhibitors of mammalian target of rapamycin |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2012027240A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102633803A (en) * | 2012-05-03 | 2012-08-15 | 盛世泰科生物医药技术(苏州)有限公司 | Synthetic method of 5,7-dichloro pyrazole [1,5-a] pyrimidine |
| CN114423756A (en) * | 2019-06-28 | 2022-04-29 | 上海瑛派药业有限公司 | Substituted fused heteroaromatic bicyclic compounds as kinase inhibitors and uses thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020160984A1 (en) * | 1998-05-15 | 2002-10-31 | Guilford Pharmaceuticals Inc. | Fused tricyclic compounds, methods and compositions for inhibiting parp activity |
| US20040106642A1 (en) * | 2000-10-25 | 2004-06-03 | Jorg Senn-Bilfinger | Polysubstituted imidazopyridines as gastric secretion inhibitors |
| US20050004159A1 (en) * | 1999-12-13 | 2005-01-06 | Eisai Co., Ltd. | Tricyclic fused heterocyclic compound, process for preparing it and medicament comprising it |
| US7393860B1 (en) * | 1998-09-23 | 2008-07-01 | Altana Pharma Ag | Tetrahydropyridoethers |
-
2011
- 2011-08-22 WO PCT/US2011/048549 patent/WO2012027240A1/en active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020160984A1 (en) * | 1998-05-15 | 2002-10-31 | Guilford Pharmaceuticals Inc. | Fused tricyclic compounds, methods and compositions for inhibiting parp activity |
| US7393860B1 (en) * | 1998-09-23 | 2008-07-01 | Altana Pharma Ag | Tetrahydropyridoethers |
| US20050004159A1 (en) * | 1999-12-13 | 2005-01-06 | Eisai Co., Ltd. | Tricyclic fused heterocyclic compound, process for preparing it and medicament comprising it |
| US20040106642A1 (en) * | 2000-10-25 | 2004-06-03 | Jorg Senn-Bilfinger | Polysubstituted imidazopyridines as gastric secretion inhibitors |
| US20040214852A1 (en) * | 2000-10-25 | 2004-10-28 | Altana Pharma Ag | Polysubstituted imidazopyridines |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102633803A (en) * | 2012-05-03 | 2012-08-15 | 盛世泰科生物医药技术(苏州)有限公司 | Synthetic method of 5,7-dichloro pyrazole [1,5-a] pyrimidine |
| CN114423756A (en) * | 2019-06-28 | 2022-04-29 | 上海瑛派药业有限公司 | Substituted fused heteroaromatic bicyclic compounds as kinase inhibitors and uses thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2584903B1 (en) | Novel heterocyclic compounds as erk inhibitors | |
| EP2770987B1 (en) | Novel compounds that are erk inhibitors | |
| EP2613782B1 (en) | Indazole derivatives useful as erk inhibitors | |
| US9169260B2 (en) | Amidopyrazole inhibitors of interleukin receptor-associated kinases | |
| WO2012036997A1 (en) | Fused pyrazole derivatives as novel erk inhibitors | |
| CA2726317A1 (en) | Inhibitors of akt activity | |
| US9351965B2 (en) | Indazole derivatives useful as ERK inhibitors | |
| EP2032141B1 (en) | Inhibitors of janus kinases | |
| EP2621925B1 (en) | Fused tricyclic inhibitors of mammalian target of rapamycin | |
| EP2953470A1 (en) | 2,6,7 substituted purines as hdm2 inhibitors | |
| WO2021126728A1 (en) | Prmt5 inhibitors | |
| EP2608668B1 (en) | Fused tricyclic inhibitors of mammalian target of rapamycin | |
| WO2012027240A1 (en) | Fused tricyclic inhibitors of mammalian target of rapamycin | |
| WO2012047569A1 (en) | Fused tricyclic inhibitors of mammalian target of rapamycin | |
| WO2021247809A1 (en) | PYRAZOLO[4,3-d]PYRIMIDINE DERIVATIVES AND METHODS OF USE THEREOF FOR THE TREATMENT OF CELLULAR PROLIFERATIVE DISORDERS | |
| US20230114091A1 (en) | Prmt5 inhibitors | |
| WO2012030633A1 (en) | Tyrosine kinase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11820441 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11820441 Country of ref document: EP Kind code of ref document: A1 |