WO2012012803A2 - Methods for detection of rare subpopulations of cells and highly purified compositions of cells - Google Patents
Methods for detection of rare subpopulations of cells and highly purified compositions of cells Download PDFInfo
- Publication number
- WO2012012803A2 WO2012012803A2 PCT/US2011/045232 US2011045232W WO2012012803A2 WO 2012012803 A2 WO2012012803 A2 WO 2012012803A2 US 2011045232 W US2011045232 W US 2011045232W WO 2012012803 A2 WO2012012803 A2 WO 2012012803A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- cell
- marker
- rpe
- stain
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
- G01N33/56966—Animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/34—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving hydrolase
- C12Q1/42—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving hydrolase involving phosphatase
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
Definitions
- embryonic stem cells may be differentiated into retinal pigment epithelium cells and transplanted into patients for the prevention or treatment of retinai disease, as disclosed in U.S. Patents 7,795,025, 7,794,704, and 7,736,896, U.S. Scr. No. 12/682.71 2, US Provisional Applications 60/998,668, 60/998,766. 61 /009,9 1 1 , 61/009,908, and 61/262,002. WO/2009/05 1671 , and WO 201 1/063005, each of which is incorporated by reference herein in its entirety.
- Another convention method of detecting stem cells can have somewhat greater sensitivity than RT-PCR for detection of stem cells because individual cells are considered rather than bulk extracts.
- preparation of cells for flow cytometry involves steps that result in loss of cells, such as cell permeabilization, washing, and sieving cells to ensure a single cell suspension. Due to this cell loss, there is concern whether the
- subpopulation of cells that reach the flow cytometer are truly representative of the initial population of cells. For example, because stem cells may have a tendency be relatively "sticky” compared to other cell types, they may be preferentially lost at the cell sieving stage. Moreover, due to their small size, small clumps of stem cells may pass through a sieve together and be miscounted as a single event in the flow cytometer, leading to undercounting of stem ceils in the preparation. Furthermore, with flow cytometry assays, gating of the cel l population is typically required to exclude cell debris, cell aggregates, and non-specific background "noise".
- the target cells will comprise rare cells, i.e., cells which are present in very minute amounts in a cell population, preferably which cell population is to be used for cell therapy.
- these rare cells will comprise cells which if administered to a subject could cause an adverse reaction r disease.
- viraily infected e.g., HIV, hepatitis, et al.
- other diseased or aberrant cells such as cancerous, precancerous and cancer stem cells
- certain immune cells such as T lymphocytes, and the like which if administered to a recipient, could result in infection, disease, or other adverse reaction such as an adverse immune reaction ⁇ e.g., GVHD), or result in the proliferation of undesired cells.
- the subject methods may be used to confirm that a donor cell containing population, such as bone marrow or pancreatic cells derived from an autologous or heterologous donor, which is to be used in cell therapy or transplant is devoid of cells that may cause cancer, viral infection, or an adverse immune reaction.
- examples are provided using these methods to test for the presence of residual pluripotent cells in a population of cells to be used for transplantation therapy.
- Experiments in which defined numbers of ES cells were added to the cell population demonstrate sufficient sensitivity to detect as few as 5 ES cells out of a population of 600,000 cells, thus showing the capability to detect target ceils as rare as 0.0008% of the total cell population.
- a trained operator can examine on the order of between one and ten million cells per hour. Because arbitrarily large numbers of ceils can be examined, the method has the potential to have unlimited sensitivity,
- the present disclosure provides a method of detecting the presence of target cells in a cell population, comprising:
- the first stain may be observable under visible light and under ultraviolet light.
- the first marker may be alkaline phosphatase.
- Th first stain may comprise an antibody directly or indirectly coupled to a colored reagent or an enzyme capable of producing a colored reagent.
- the colored reagent may comprise gold particles, silver particles, or latex particles.
- the first stain may comprise an antibody directly or indirectly coupled to gold particles and said method may further comprise forming a silver precipitate on said gold particles.
- the first marker may be selected from the group consisting of: alkaline phosphatase, Oct -4, Nanog, Stage-specific embryonic antigen-3 (SSEA-3 ), Stage-specific embryonic antigen-4 (SSEA-4), TRA- 1 -60, TRA- 3 -8 1 , TRA-2-49/6E, Sox 2. growth and differentiation factor 3 (GDF3), reduced expression 1 (REX 1 ), fibroblast growth factor 4 (FGF4), embryonic cell-specific gene 1 (ESG1 ), developmental pluri potency-associated 2 (DPPA2), DPPA4.
- SSEA-3 Stage-specific embryonic antigen-3
- SSEA-4 Stage-specific embryonic antigen-4
- TRA- 1 -60 Stage-specific embryonic antigen-4
- TRA- 3 -8 1 TRA-2-49/6E
- Sox 2 Sox 2. growth and differentiation factor 3 (GDF3), reduced expression 1 (REX 1 ), fibroblast growth factor 4 (FGF4), embryonic cell-specific gene 1 (ESG1 ), developmental pl
- telomerase reverse transcriptase hTERT
- SALL4 E-CADHERIN
- Cluster designation 30 CD3Q
- Cripto TDGF-1
- GCTM-2 Genesis
- Germ cell nuclear factor a malignant fibroblasts
- SCF Stem cell factor
- the first stain may comprise a first enzyme selected from the group consisting of: alkaline phosphatase, beta galactosidase, and peroxidase.
- the peroxidase may be horseradish peroxidase.
- the first enzyme may be expressed by target cells.
- the first enzyme may be directly or indirectly coupled to a primary or secondary antibody.
- the first stain may comprise alkaline phosphatase.
- the first stain may further comprise an alkaline phosphatase substrate selected from the group consisting of: napthol AS-BI phosphate; 5-bromo-4-chloro-3-indolyl phosphate (BCiP) and Nitro Blue Tetrazolium (NBT); BCIP reagent and INTX reagent; naphtho! AS-BI and fast red violet LB; teirazolium salts; diazo compounds; VECTOR® Red; VECTOR® Blue; VECTOR® Black; and p-Nitropheny!phosphate (pNPP).
- an alkaline phosphatase substrate selected from the group consisting of: napthol AS-BI phosphate; 5-bromo-4-chloro-3-indolyl phosphate (BCiP) and Nitro Blue Tetrazolium (NBT); BCIP reagent and INTX reagent; naphtho! AS-BI and fast red violet LB; teirazolium salts; diazo compounds; VECTOR®
- the first stain may comprise peroxidase.
- the first stain may further comprise a peroxidase substrate selected from the group consisting of: 3 ,3 ' ,5 ,5 ' -Tctrameth y 1 benzi dine ( TMB); 3,3'-Diaminobenzidine (DAB); 3- Amino-9-EthylCarbazole (AEC); 4-Chloro- l -naphthol: 2,2'-azino-bis(3-ethylbenzthiazoline-6- sulphonic acid) (ABTS); 2,3,5-Triphenyltetrazolium chloride; 2-Chloro-5.5-dimet.hyl- 1 ,3- cyclohexanedione; 3.3' 5.5'-Tetramethylbenzidine: 3.3'-Diaminobenzidine tetrahydroch!oride; 3- Nitroteirazolium blue chloride; 4- A mi nophth a) h yd razi de ; 4-Chloro-
- the first stain may further comprise a beta galactosidase substrate selected from the group consisting of: 1 -Metby]-3-indoly]-p-D-galactopyranoside; 2- itrophenyl ⁇ -D- ga!aciopyranosid; 4- Met b y !
- a beta galactosidase substrate selected from the group consisting of: 1 -Metby]-3-indoly]-p-D-galactopyranoside; 2- itrophenyl ⁇ -D- ga!aciopyranosid; 4- Met b y !
- the target cell may be an embryonic stem cell and said second marker may be selected from the group consisting of; alkaline phosphatase, Oct-4, Nanog, Stage-specific embryonic antigen-3 (SSEA-3), Stage-specific embryonic antigen-4 (SSEA-4), TRA - 1 -60.
- TRA 1 -8 1.
- DPPA2 developmental pluripotency-associated 2
- DPPA4 telomerase reverse transcriptase
- hTERT telomerase reverse transcriptase
- SALL4 E-CADHERIN
- CD30 Cluster designation 30
- Cripto TDGF- 1
- GCTM-2 GCTM-2. Genesis, Germ cell nuclear factor, and Stem cell factor (SCF or c-Kil ligand).
- the second stain may comprise a primary antibody, which may comprise a fluorescent label.
- the second stain may further comprise a secondary antibody, which may comprise a fluorescent label.
- the fluorescent label may be selected from the group consisting of: Alexa Fluor
- fluorescein isothiocyanate FITC
- Texas Red Texas Red
- SYBR Green Green fluorescent protein
- TRIT tetramethyl rhodamine isothiol
- NBD 7-nitroben/-2-oxa- 1 .3-diazole
- Texas Red dye phthalic acid, terephthaiic acid, isophthalic acid, cresyl fast violet, cresyl blue violet, brilliant cresyl blue, para-aminobenzoic acid, erythrosine, biotin, digoxigenin, 5-carboxy-4',5'-dichloro-2 ' ,7'-dimethoxy fluorescein, TET (6-carboxy-2',4,7,7'-tetrachlorofluorescein), HEX 6-carboxy-2',4,4',5 ' ,7,7 - hexachlorofluorescein), Joe ⁇ 6-carboxy
- the cell population may comprise cells of a species selected from the group consisting of: antelopes, bovines, camels, cats, chevrotains (mouse deer), chimpanzee, cow, deer, dog, giraffes, goat, gui nea pig. hamster, hippopotamuses, horse, hum n, mouse, non-human primate, ovine, peccaries, pig, pronghorn, rabbit, rat, rhesus macaque, rhinoceroses, sheep, tapirs, and ungulates.
- a species selected from the group consisting of: antelopes, bovines, camels, cats, chevrotains (mouse deer), chimpanzee, cow, deer, dog, giraffes, goat, gui nea pig. hamster, hippopotamuses, horse, hum n, mouse, non-human primate, ovine, peccaries,
- the method may further compri e determining the approximate number of cells in the eel! population.
- At least 90% of the cells in said cell population may be examined under visible light to detect cells that are positive for said first marker, and each cell that is positive for said first marker may be examined under ultraviolet light to determine whether that cel l is positive for said second marker.
- the cell population may contain any number of cells, depending on the intended use.
- a cell population may contain at least 10 s cells, at least 10 6 cells, at least 10 7 cells, at least 10 8 cells, at least I 0 9 cells, at least 10 10 cells, or between 10 5 and 10 10 cells.
- the first marker and second marker may be embryonic stem ceil markers.
- the population of cells may be produced by in vivo or in vitro differentiation of embryonic stem cells.
- the target cell may be an embryonic stem cell or induced pluripotent (iPS) cell.
- iPS induced pluripotent
- the cell population may further comprise cells differentiated from an embryonic stem cell or i PS ceil.
- the cells differentiated from an embryonic stem cell or iPS cell may be RPE cells.
- the RPE DCis may be produced by any of the methods disclosed in U.S. Patents 7,795,025, 7,794.704, and 7,736,896, U.S. Ser. No. 1 2/682,712, WO/2009/051671 , and WO 201 1/063005, each of which is incorporated by reference herein in its entirety,
- the target cell may be selected from specific types of cells including by way of example: adipocyte; bone marrow fibroblast; cardiomyocyte; chondrocyte; differentiated RBC and WBC lineages; andothelial bone marrow fibroblasts; ectoderm; ectoderm progenitor;
- ernbryoid body embryonal carcinoma (EC); embryonic stem (ES); endoderm; endothelial; hematopoietic cells; hematopoietic stem cell (HSC), satellite, endothelial progenitor; hepatocyte; keratinocyte; mesenchymal; mesencyhmai stem cell (MSG); mesoderm: MSG progenitor;
- the first marker and the second marker may be markers associated with said target cell as listed in Table 1.
- the target cell may comprise a ceil involved in a specific disease, e.g., a cancerous or
- precancerous cell or a cancer stem cell which is desirably eliminated from a cell population potentially for transplant or cell therapy such as bone marrow.
- the target cell may comprise an adult stem cell that gives rise to ceils of a specific lineage.
- the method may further comprise prior to step (a) culturing the cell population under conditions that favor maintenance cells of the target cell type, and the target celt may be an embryonic stem cell or induced pluripotent (iPS) cell, and the culture conditions may comprise embryonic stem cell media and/or the presence of mouse embryonic fibroblast feeder cells.
- iPS induced pluripotent
- the method may further comprise plating a second cell population comprising the target cell type and analyzing said second cell population by the same method as said cell population of step (a), thereby establishing the limit of detection of said method. Only cells of said target cell type are added to said second population, or the second cell population may comprise a mixture of a first group of cells and a second group of cells, wherein the first group of cells is of the same type as the target cell type.
- the second group of cells may have essentially the same constituenc as said cell population of step (a).
- the ratio of the number of cells in said first group of cells and said second group of cells may be selected from the group consisting of: 1. TO; 1:100; 1:1.000; 1:10,000; 1:100,000; 1:1,000,000; 1:10,000,000; 1:100,000,000;
- the target cell may be selected from a virally infected cell, cancerous or precancerous cell, cancer stem cell, and an immune cell.
- the cel l population putanve!y containing the target cell may comprise bone marrow, blood cells, or pancreatic cells.
- the cell population putatively containing the target cell may have previously been treated by cell sorting, irradiation, chemotherapy or another means to remove the target cell.
- Another exemplary embodiment provides a composition comprising somatic cells derived from stem cells that may be essentially free of said stem cells.
- compositions comprising cells and an indicator that indicates the number or fraction of target cells (such as stem cells) present, wherein the value of said indicator may be determined by applying the methods described herein to a cell population representative of the cells in said composition.
- the composition may comprise RPE cells, which may be differentiated from piuripotent stem cells, such as embryonic stem cells, iPS cells, blastomeres. inner mass cells, or oocytes which may be parthenogenet i cal I y activated.
- piuripotent stem cells may be recombinant or genetically engineered (e.g., engineered to express a desired therapeutic protein or to elimi ate the expression of a gene involved in a genetic deficiency such as macul r degeneration.)
- the RPE cells may be formulated and used to treat retinal degenerative diseases.
- piuripotent stem cell-deri ved RPE cells can be used in screening assays to identify agents that modulate RPE cell survival (in vitro and/or in vivo), to study RPE cell maturation, or to identify agents that modulate RPE cell maturation.
- composition may comprise a substantially purified preparation of human RPE cells differentiated from human piuripotent stem cells, wherein the RPE cells express, at the mRNA and protein level, RPE-65, Bestrophin, PEDF, CRALBP, Otx2, and ⁇ ⁇ F, and wherein the cells substantially lack expression of Oct-4, NANOG, and Rex- 1 .
- the RPE cells comprise differentiated RPE cells and mature differentiated RPE cells, and wherein at least the mature differentiated RPE cells further express, at the mRNA and protein level, PAX2, pa -6, and tyrosinase.
- the RPE cells are differentiated from human ES cells or human i PS cells.
- the composition may comprise at least 10 5 cells, at least 10 6 cells, at least 10 7 cells, at least 10 s ceils, at least 10 9 cells, at least 10 !0 cells, or between 10 5 and 10 10 ceils.
- the composition may comprise cryopreserved cells, in one embodiment, the composition may comprise a cryopreserved preparation comprising at least about 10 4 human RPE cells, wherein the preparation is a substantially purified preparation of human RPE cells derived from human pluripotent stem cells, and wherein the RPE cells express RPE-65,
- the RPE cells may be
- cryopreserved by a method comprising: (a) culturing RPE cells, (b) harvesting said RPE cells, (c) centrifuging said RPE cells, (d) resuspending said RPE cells in 10% DMSO/90% FBS solution; and (e) freezing said RPE cells.
- Another exemplary embodiment provides a method of detecting the presence of human embryonic stem cel ls in a cell population, comprising:
- the cell population may comprise RPE cells differentiated from pluripotent cells by the methods described in U.S. Patents 7,795,025, 7,794,704, and 7,736,896, U.S. Ser. No. 12/682,712, US Provisional Applications 60/998,668, 60/998,766, 61/009,91 1 , 61/009,908, and 1 /262,002, WO/2009/05 1671 , and WO 201 1/063005, each of which is incorporated by- reference herein in its entirety.
- the target cells are embryonic stem cells, induced pluripotent stem (iPS) cells, adult stem cells, hematopoietic cells, fetal stem cells, mesenchymal stem cells, postpartum stem cells, multipotent stem cells, or embryonic germ cells.
- the pluripotent stem ceils may be mammalian pluripotent stem cells.
- the pluripotent stem cel ls may be human pluripotent stem cells including but not limited to human embryonic stem (hES) cells, human induced pluripotent stem (iPS) cells, human adult stem cells, human hematopoietic stem cells, human fetal stem cells, human mesenchymal stem cells, human postpartum stem cells, human multipotent stem cells, or human embryonic germ cells.
- the pluripotent stem ceils may be a hES cell line listed in the European Human Embryonic Stem Cell Registry - hESCreg.
- the hES cell line may be a blastomere-derived hES cell line such as MA09 or another cell li e derived by the methods described in U.S. Patents 7,893,3 15 or 7,838.727. each of which is incorporated by reference herein in its entirety.
- the invention provides for the use of a pharmaceutical preparation of RPE cells in the manufacture of a medicament for the treatment of retina! degeneration.
- the invention provides a method of treating retinal degeneration comprising administering an effective amount of RPE cells described herein.
- the retinal degeneration is due to choroideremia, diabetic retinopathy, age- related macular degeneration, retinal detachment, retinitis pigmentosa, or Stargardt's Disease.
- the preparation is transplanted in a suspension, matrix, gel, colloid, scaffold, or substrate.
- the preparation is administered by injection into the subrctinal space of the eye,
- the effective amount is at least about 20,000-200,000
- the effective amount is at least about 20,000, 50,000, 75,000, 100,000, 125,000, 150,000, 175,000, 180,000, 1 85,000, 1 0,000, or 200,000 RPE cells, [0067]
- the method further comprising monitoring the efficacy of the method by measuring electroretinograni responses, optomotor acuity threshold, or luminance threshold in the subject.
- the preparation is substantially free of ES cell, viral, bacterial, or fungal contamination
- the RPE cells are functional RPE cells capable of integrating into the retina upon transplantation.
- the RPE cells are functional RPE cells capable of integrating into the retina upon transplantation.
- RPE cells improve visual acuity following transplantation.
- the present invention provides methods for the treatment of eye disorders.
- these methods involve the use of RPE cells to treat or ameliorate the symptoms of eye disorders, particularly eye disorders caused or exacerbated, in whole or in part, by damage to or breakdown of the endogenous RPE layer (e.g. , retinal degeneration).
- the RPE cells described herein are substantially free of genetic mutations that may lead to retinal degeneration.
- the RPE cells may be transplanted with a biocompatible polymer such as poly!actic acid, po 1 y (1 acti c -c'oglyc I i c acid), 50:50 PDLGA, 85: 15 PDLGA, and IN ION GTR® biodegradable membrane (mixture of biocompatible polymers).
- a biocompatible polymer such as poly!actic acid, po 1 y (1 acti c -c'oglyc I i c acid), 50:50 PDLGA, 85: 15 PDLGA, and IN ION GTR® biodegradable membrane (mixture of biocompatible polymers).
- the RPE cells adhere to Bruch's membrane after transplantation, establish polarity, and integrate into the receipt's tissue.
- the RPE cells may improve visual acuity after
- the RPE cells may substantially improve visual acuity after transplantation.
- the RPE cells lack substantial expression of embryonic stem cell markers including but not limited to Oet-4. NANQG. Rex- 1 , alkaline phosphatase, Sox 2. TDGF- 1 , DPPA-2. and DPPA-4.
- the RPE cells express RPE cell markers including but not limited to RPE65, CRALBP. PEDF, Besirophin, MITF, Otx2, AX 2, Pax-6, and tyrosinase.
- the RPE cells express at least one gene, wherein expression of the at least one gene is increased in the RPE cells relative to expression in human ES cells.
- the RPE cells show increased alpha i rue grin subun.it expression.
- the alpha integrin subunit is alpha 1 , 2, 3, 4, 5, 6, or 9.
- the expression is mRNA expression, protein expression, or both mRNA and protein expression.
- the resuspension and centrifugatton steps may be repeated at least 1. 2, 3, 4, or 5 times.
- the RPE product is transported to the clinical site within at least about 1 , 2, 3, 4, 5, 6, 7. 8, 9, or 10 hours of completion of step (e).
- the vials may be labeled.
- the present invention also provides a method for a providing RPE ceil preparation for sale comprising (a) producing RPE cells and (b) preparing said RPE cell preparations for transfer to a customer.
- the method may comprise cryopreserving the RPE cells.
- the method comprises offering said R E cell preparations for sale.
- the method comprises advertising the RPE cell preparations.
- the invention contemplates any combination of the aspects and embodiments described above or below.
- preparations of RPE cells comprising any combination of differentiated RPE cells and mature RPE cells can be used i the treatment of any of the conditions described herein.
- methods described herein for producing RPE cells using human embryonic stem cells as a starting material may be similarly performed using any human pluri potent stem as a starting material.
- FIG. 1 is a representative micrograph showing detection of liES ceils (Oct-4 positive and Alkaline Phosphatase positive) among differentiated cells. GFP expression confirms hES cel l identity.
- FIG. 2A-C RNA was extracted from mixed populations of RPE cells and hES cells (containing 0%, 0.01 %, 0.1 %, 1 %, 10%, or 100% hES cells as indicated). cDNA was then synthesized and relative gene expression was assayed in triplicate replicates normalized to the beta actin signal present in each sample. Gene expression profiling was performed by RT-qPCR using Applied Biosysiems StepOne Plus with software version 2.1 and TaqMan gene expression assays from Li e Technologies following the manufacturer ' s recommended cycle conditions for comparative relative quantification . Data are expressed as the mean +/- standard deviation for three replicated with P-values determined by T-Tcsiing. Results are presented for (A) OCT4, (B) anog, and (C) SOX 2 expression.
- cells are plated at high density but most preferably in no greater density than a monolayer (to simplify microscopic observation of the cells), which may be on top of a monolayer of feeder cells, and then stained for the presence of two or more characteristic markers of the target ceil type, including a first marker which is detectable under visible light and a second marker which is detectable under ultraviolet light.
- the plated cells are then microscopically observed under visible light to detect cells expressing the first marker. Starting from one corner of the plated cells, the operator (or an automated system) scans across the entire plate and back in overlapping tracks, thus ensuring that each ceil is examined.
- the unstained cells are visible and the focal plane can be adjusted as needed to ensure that the cells remain in focus as the plated cells are moved through the field of view.
- Use of visible light also prevents photobleachiiig that can occur under ultraviolet light. Scanning under visible light can be performed with relatively low magnification, which increases the number of cells that are observed in each microscopic field and increases throughput, Cells that are positive for the first marker are then examined under ultraviolet light to determine whether those cells also express the second marker.
- alkaline phosphatase which may be used with ES cells as the marker used to "scan" the whole plate in visual light
- a cell is positive for the alkaline phosphatase, it can be either positive or negative for the second marker, and therefore, ceils positive for the first and second marker are considered an hES cell.
- the first marker stains the target cells robustly (even though it may stain other cells as well) to avoid missing the chance to observe cells that may be positive for the second marker but have low or undetectable expression of the first marker.
- positive cells are photographed so that comparison of photographs can avoid double-counting of cells thai are observed in overlapping fields.
- the total number of plated cells is determined by counting all cells in each of one or more representative microscopic fields and dividing the number of cells counted by the fraction of the total plated area that was counted. Finally, the number of target cells detected is expressed as a proportion of the total number of cells examined.
- markers are chosen to distinguish target cells from the other cell types in the population.
- the target cells are hES cells and the other cell type in the population is human RPE cells then the first marker may be alkaline phosphatase and the second marker may be Oct-4.
- Other exemplary hES cell markers that may be used with these methods include: Nanog, Stage-specific embryonic antigen-3 (SSEA-3), Stage-specific embryonic antigen-4 (SSEA-4), TRA- 1 -60, TRA- 1 -8 1 . TRA-2-49/6E.
- E CADHE I Cluster designation 30 (CD30), Cripto (TDGF- 1 ), GCTM-2. Genesis, Germ cell nuclear factor, and Stem cell factor (SCF or c-Kit ligand).
- the present methods utilize at least two markers and may utilize, three, four, five, six, etc. markers which may provide further confirmation of whether a cell is of a target type.
- the target cell is a cancer stem cell and is detected by staining for one or more of the markers disclosed in one of the following U.S.
- the cells are plated under conditions thai favor the maintenance of the target cell type.
- the target cell type is more likely to be retained in the culture and the sensitivity of the method should be improved.
- the cells may be plated under conditions known to maintain hES cell in the pluripotent state.
- Exemplary culture conditions include plating on feeder cells, which may be mitotieally inactivated by pre-treatment with irradiation or mitomycin C or other methods known in the art, or in media conditioned by feeder cells.
- feeder cell types include pri mary fibroblast cultures such as murine embryonic fibroblast (MEF), human adult skin fibroblasts, STO cells, and others.
- MEF murine embryonic fibroblast
- STO cells human adult skin fibroblasts
- Culture conditions for feeder-free maintenance of ES cells are also known in the art and may include matri el, laminin.
- ES cell culture media including feeder-free culture media, are described in Carpenter et al., Dev Dyn. 2004 Feb;229(2):243-58; Xu et ah, Nat Biotechnol. 2001 Oct; 19( 10):97 1 -4; Rosier et ah, Dev Dyn. 2004 Feb;229f2):259-74; and Amit et ah, Biol Reprod. 2004 Mar;70 ⁇ 3):837-45;
- the method is validated to determine its sensitivity, for example by
- spiking experiments in which defined numbers of cells of a target cell type are added to a population, which is then assayed using the subject methods to detect ceils of the target cell type.
- Use of populations containing defined numbers of target cells permits determination of the minimum number of target cells that must be present or the limit of detection of the assay, e.g., the minimum percentage of the population that must, on average, be of the target cell type before target cells are detected by the assay.
- the limit of detection may be expressed as a ratio or fraction that indicates the minimum proportion f cells of the target cell type that can be detected in the assay (e.g., 5 in 600,000 cells or 0.0008%).
- the method may include a step of determining a correction factor that can be used to correct the sensitivity or limit of detection of the assay.
- the target cells may be lost during culturirig prior to performing the assay (e.g., target cells may differentiate into other cell types).
- defined numbers of cells of the target type may be plated under the same conditions as in the spiking experiments, but in the absence of the other type of ceils in the population (though if feeder cells are part of the culture conditions then they may be present).
- the cells may be stained with a species-specific antibody that specifically identi ies cells of the target cells' species, which would detect cells that have lost the specific markers of target cells.
- the culture can be examined to determine the fraction of target cells that retained the expression of the target cell markers used in the assay, thereby determining a "correction factor" that establishes the relationship between the number of cells plated and the number that retain the phenotypes used for detection. For example, if it is determined that 50% of the target cells lose expression of the assayed target cell markers under these conditions, then the effective number of target cells is (e.g., used in calculating the limit of detection in spiking experiments) is reduced accordingly.
- the corrected limit of detection of the assay may be computed to be five cells in ten million to reflect the number of target cells that actually remain present in the population.
- the method preferably employs sample preparations that maximize the number of target cells that remain present in the population and retain expression f the target cell markers used for their detection.
- An additional exemplary embodiment provides a method of identifying a culture condition that preserves expression of a selected marker by target cells, comprising plating target cells under varying culture conditions, identifying the fraction of target cells that retain expression of one or more markers characteristic of target cells under each culture condition, and identifying the culture conditions that retains a greater fraction as a culture condition that preserves expression of a selected marker by target cells.
- the present disclosure demonstrates that for detecting hES cells, the retention of hES cell markers (and therefore the sensitivity of the assay) is greatly improved by using hES cell culture conditions (culture on MEFs in hES cell culture media) rather than RPE cell culture conditions (absence of MEFs and a medium in which hES cells differentiate).
- the markers may be detected using at least one stain that is detectable under visible light and another marker that is detectable under ultraviolet light. Markers detectable under visible light may be detectable based on an enzymatic activity. Exemplary enzymes that can produce colored products detectable under visible light include alkaline phosphatase, peroxidases (including horseradish peroxidase), and ⁇ -galactosidases. These enzymes may be expressed by target cells, or may be coupled to a molecule that binds to a target cells (such as a primary or secondary antibody).
- alkaline phosphatase which may be stained based on its enzymatic activity.
- the alkaline phosphatase may be endogenously expressed, coupled to an antibody, or both.
- substrates can be used for alkaline phosphatase staining to produce a product detectable under visible light, including: napthol AS-BI phosphate as substrate and fast red violet dye as the colorimetric read-out (e.g., using the Stemgent® Alkaline Phosphatase (AP) Staining Kit II); Bromo-Chloro-Indolylphosphate (BCIP) and Nitro Blue Tetrazoiium (NBT/Thiazoiyl Bluc/Nitro BT) (e.g., Alkaline Phosphatase Blue Microwell Substrate (SIGMA-ALDRICH®)); BCIP reagent and INTX reagent (SIGMA-ALDRICH®); naphthol AS-BI and fast red violet LB (e.g...
- napthol AS-BI phosphate as substrate and fast red violet dye as the colorimetric read-out
- BCIP Bromo-Chloro-Indolylphosphat
- Leukocyte Alkaline Phosphatase kit SIGMA-ALDRICH®
- substrates which form precipitates are based on either reduction of tetrazoiium salts or the production of colored diazo compounds (e.g.. VECTOR® Red, VECTOR® Blue. Vector® Black, p- itrophenylphosphate. BCIP/NBT AP Substrate Kit, and 5-bromo-4-chloro-3-indolyi phosphate/nitroblue tetrazoiium).
- an alkaline phosphatase inhibitor such as Levaniisole ⁇ ( S ) - 6 - p h e n y 1 - 2 , 3 , 5 , 6 - tetrahydroimidazo[2, l -b][ i ,3]thiazole) or a heat treatment, may be used to inhibit undesircd background alkaline phosphatase activity.
- peroxidase such as horseradish peroxidase (HRP), which may be stained based on its enzymatic activity.
- HRP horseradish peroxidase
- Peroxidase may be endogenously expressed, coupled to an antibody, or both. Peroxidase can catalyze the conversion of ehromogenic substrates into colored molecules.
- Exemplary Peroxidase substrates include 3.3 * ,5.5 ' -Tetranicthylben/_idinc ⁇ TMB); 3.3 ' -Diami noben/.idine (DAB); 3-Amino-9- EthytCarbazole (AEC); 4-Chioro- l -naphthol; 2,2'-azino ⁇ bis(3-ethy!benzthiazoiine-6-sulphonic acid) (ABTS); 2,3,5 -Triphenykeirazoliu chloride; 2-Chlom-5,5-dimethyl- 1 .3- cyciohexancdione; 3,3',5,5'-Tetramethylbenzidine; 3,3 '-Diuminoben/idinc tetrahydroch lori de ; 3- Nitrotetrazoliuni blue chloride; 4-Arniriophthalhydrazide; 4-Chloro - 1 -napht
- ⁇ -galactosidase Another marker that is detectable under visible light that may be used in embodiments of the present disclosure is ⁇ -galactosidase, which may be stained based on its enzymatic activity, ⁇ -galactosidase may be endogenously expressed, coupled to an antibody, or both, ⁇ -galactosidase can catalyze the conversion of chromogenic substrates into colored molecules.
- Exemplary ⁇ -gafactosidase substrates include: l -Methyl-3-indolyI ⁇ -D- galactopyranoside; 2-Nitropnenyl ⁇ -D-galactopyranosid; 4-Methy tu mbel I ifery 1 ⁇ -D- galactopyranoside; 4-Nitrophenyl ⁇ -D-galactopyranoside; 5-Bromo-3-indolyl ⁇ -D- galactopyranoside; 5-Bromo-4-chioro-3-indolyl ⁇ -D-gaiactopyranoside; 5-Bromo-6-chloro-3- indolyl-p-D-galactopyranoside; 6-Bromo-2-naphthyi ⁇ -D-galactopyranoside; 6-Chloro-3- indolyl-P-D-galactopyranoside; Fluorescein di(
- a marker can be directly or indirectly coupled to the antibody.
- indirect coupling include avidin/biotin coupling, couplin via a secondary antibody, and combinations thereof.
- cells may be stained with a primary antibody that binds a target-specific antigen, and a secondary antibody that binds the primary antibody or a molecule coupled to the primary antibody can be coupled to a detectable marker.
- Use of indirect coupling can improve signal to noise ratio, for example by reduci ng background binding and/or providing signal ampli ication.
- stains detectable under visible light include particles including gold, silver, or latex.
- particles may be directly or indirectly coupled to primary or secondary antibodies or otherwise bound to target cells.
- staining may be further enhanced using immunogold-silver staining, in which cells are stained with an antibody coupled to colloidal gold, and the gold particles are revealed using a silver precipitation reaction method (Holgate et al., J Histochem Cytochem. 1983 Jui;3 1 (7):938-44).
- Exemplary embodiments o the present method utilize antibodies coupled to a fluorescent molecule, such as ethidium bromide, SYBR Green, fluorescein isothiocyanate (FiTC), DyLight Floors, green fluorescent protein (GFP), TRiT (tetramethyl rhodamine isothiol).
- a fluorescent molecule such as ethidium bromide, SYBR Green, fluorescein isothiocyanate (FiTC), DyLight Floors, green fluorescent protein (GFP), TRiT (tetramethyl rhodamine isothiol).
- NBD (7-ni£robenz-2-oxa- 1 ,3-diazole), Texas Red dye, phthalic acid, terephthalic acid, isophthalic acid, cresyl fast violet, cresyl blue violet, brilliant cresy] blue, para-aminobenzoic acid, erythrosine, biotin, digoxigenin, 5-carboxy-4',5'-dichloiO-2',7'-dimethoxy fluorescein, TET (6-carboxy-2',4,7,7'-tetrachlorofluorescein), HEX (6-carboxy-2',4,4',5',7,7'- hexachlorofluorescein), Joe (6-carboxy-4'.5'-dich loro-2'.7'-dimethox fluorescein) 5-carboxy- 2'.4',5',7'-tetrachlorofluorescein, 5-earboxyfluoresc
- Quantum dot also includes alloyed quantum dots, such as ZnSSe, ZnSeTe, ZnSTe, CdSSe, CdSeTe, ScSTe, HgSSe, HgSeTe, HgSTe, ZnCdS, ZnCdSe, ZnCdTe, Znl lgS, ZnHgSe, ZnHgTe, CdHgS. CdHgSe, CdHgTe, ZnCdSSe, ZoHgSSe, ZnCdSeTe. ZnHgSeTe. CdHgSSe, CdHgSeTe, InGaAs, GaAlAs, and InGaN.
- alloyed quantum dots such as ZnSSe, ZnSeTe, ZnSTe, CdSSe, CdSeTe, ScSTe, HgSSe, HgSeTe, HgSTe, ZnCd
- Alloyed quantum dots and methods for making the same are disclosed, for example, in US Application Publication No. 2005/00121 82 and PCT Publication WO 2005/001889.
- the present disclosure provides exemplary embodiments illustrating highly sensiti ve detection human embryonic stem cells in a differentiated RPE cell population, but can readily be adapted to detect other cell types or with other species than human. For example, these methods may be used with other cell types including other stem cell types, cancer cells, feeder cells, xenogeneic cells, or other any other cell type that expresses characteristic markers.
- these methods may be used with human, non-human primates, antelopes, bovines, camels, cats, chevrotains (mouse deer), chimpanzee, cow, deer, dog, giraffes, goat, guinea pig, hamster, hippopotamuses, horse, human, mouse, non-human primate, ovine, peccaries, pig, pronghorn, rabbit, rat, rhesus macaque, rhinoceroses, sheep, tapirs, and ungulates, or any other mammalian or non-mammalian eel! type.
- Target cel ls may be identified by expression of one or more stem cell markers. It may be desirable to test for expression of one or more target cell markers (e.g., two markers, three markers, four markers, etc.) to provide further assurance that a cell expressing a target cell marker is in fact a target cell.
- a "cocktail" of antibodies to different markers may be each coupled (whether directly or indirectly) to the same label or to different labels.
- a cocktail of antibodies to different markers may each contain a binding motif that binds the same label (e.g., each may contain an Fc of the same species that is recognized by the same secondary antibody, or each may be biotinylated and specifically bound by the same avidin-coupled label ).
- two or more different antibodies or cocktails of antibodies may be utilized.
- the cells are stained using at least two labels that can be distinguished from one another, thereby permitting identification of cells that express at ieast two different markers of the target cell types.
- Cells may also be stained using at least three, four, five, or more different labels that can be distinguished from one another, thereby permitting detection of cells that express greater numbers of markers of the target cell type.
- a ceil may be identified as a eel! of the target type if it expresses a preselected number of markers or certain preselected combinations of markers.
- alkaline phosphatase may be used as a suitable marker for detecting hES cells within a population of RPE, even though other cell types also express alkaline phosphatase, because alkaline phosphatase is not normally expressed by RPE.
- Exemplary embryonic stem cell markers i n clude alkaline phosphatase, Oct-4,
- Stage-specific embryonic antigen-3 SSEA-3
- Stage-specific embryonic antigen-4 SSEA-4
- TRA- 1 -60 TRA- 1 -81
- TRA-2-49/6E Sox2.
- growth and di ferentiation factor 3 GDF3
- reduced expression I REX 1
- FGF4 embryonic cell-specific gene 1
- ESG1 embryonic cell-specific gene 1
- DPPA2 developmental pluripotency-associated 2
- DPPA4 telomerase reverse transcriptase
- SALL4 E-CADHERIN
- Cluster designation 30 CD30
- Cripto TDGF- 1
- GCTM-2 Genesis
- Germ ceil nuclear factor Germ ceil nuclear factor
- SCF or c-Kit ligand SCF or c-Kit ligand
- cells may only be considered positive for a given marker if that marker exhibits a characteristic localization or pattern within the ceil. For instance, a cell may be considered "positive” if a cytoskeietal marker is present in the
- cells may be cultured under suitable conditions (e.g., as adherent cultures) to establish the
- Suitable culture conditions and time for cytoskeleton assembly (or other processes to establish subcellular organization) that may be necessary for robust detection of a given marker are readily determined by those of ordinary skill in the art. Additionally, markers may readily be chosen which decrease or eliminate the need for adherent culture as a precondition to robust staining. For example, non-adherent cell populations may be stained after Cytospin-like procedure (in which cells may lose their normal morphology while being "squashed" onto slides by centrifugal force); suitable markers for use under such circumstances are well known or readily identified.
- osteoblast marker of bone formation
- BMPR Bone important for the differentiation of committed morphogenetie Mesenchymal stem mesenchymal cell types from mesenchymal stem protein receptor and progenitor cells and progenitor cells; BMPR identifies early (BMPR) mesenchymal lineages (stem and progenitor cells)
- WBC lymphocyte
- CD34 cell (HSC) ; satellite,
- CD34 also identifies muscle satellite, a muscle endothelial progenitor
- CD34 Scal + Lin " Mesencyhma! stem Identifies MSCs, which can differentiate into prof i !e cell (MSG) adipocyte, osteocyte, chondrocyte, and myocyte
- CD38 Present on WBC lineages. Selection of CD347CD38 " cells allows lineages for purification of HSC populations.
- hematopoietic progenitor cells identifies HSC and MSC; binding by fetal calf serum (PCS) enhances proliferation of ES cells, HSCs, MSCs, and hematopoietic progenitor cells
- CFU assay detects the ability of a single stem cell
- CFU red blood cell lineages
- RBC red blood cell
- WBC white blood cell
- Bone marrow to a colony of mu!tipotent fibroblastic cells such forming unit (CFU- fibroblast identi ied cells are precursors of differentiated F)
- Lin-negative cells Lineage surface Differentiated RBC detection of Lin-negative cells assists in the antigen (Lin) and WBC lineages purification of HSC and hematopoietic progenitor populations
- WBC subtypes granulocyte and macrophage
- Cell-surface protein immunoglobulin superfami ly
- Muc- 18+ cells are mesenchymal precursors
- BM bone marrow
- Stro- 1 + cells assists in isolating mesenchymal ( mesenchymal)
- Stro- 1 antigen precursor cells which are multipotent cells that precursor cells
- myocytes myocytes, fibroblasts, chondrocytes, and blood ceils
- GFAP acidic protein Astrocyte Protein specifically produced by astrocyte
- Dendrite-specific MAP protein found specifically associated protein- Neuron
- Oligodendrocyte located in the myelin sheath surrounding neuronal protein (MPB)
- Neurofilament Important structural protein for neuron identifies
- EB Embryoid body
- Neuronal protein located in synapses indicates that
- Tau Neuron Type of MAP helps maintain structure of the axon
- Cytokeratin 19 CK 1 identifies specific pancreatic epithelial cells
- the RPE cells may express RPE cell markers.
- the expression level of the RPE cell genes RPE65, PAX2, PAX6. and tyrosinase, bestrophin, PEDF, CRALBP, Oix2, and MITF may be equivalent to that in naturally occurring RPE cells.
- the level of maturity of the RPE cells may assessed by expression of at least one of PAX2, PAX6, and tyrosinase, or their respective expression levels.
- the RPE cells may not express ES cell markers.
- the expression levels of the ES cell genes Oct-4, NANOG, and/or Rex- 1 may be about 100-1000 fold lower in RPE cells than in ES cel ls.
- the RPE cells may substantially lack expression of ES cell markers including but not limited to octamer-binding transcription factor 4 (Oct-4, a.k.a., POU5F1 ), stage specific embryonic antigens ( SSEA)-3 and SSEA-4, tumor rejection antigen (TRA 1- 1 -60. TRA- 1 -80, alkaline phosphatase, NANOG, and Rex- 1 .
- TRA 1- 1 -60. TRA- 1 -80 alkaline phosphatase
- NANOG alkaline phosphatase
- Rex- 1 alkaline phosphatase
- the present invention provides preparations of RPE cells and an indicator that indicates the number or fraction of pluri potent cells present, wherein the value of said indicator may be determined by applying the methods described herein to a cell population representative of the cells in said preparation.
- the invention described herein provides RPE cells, substantially purified populations of RPE cells, pharmaceutical preparations comprising RPE cells, and cryopreserved preparations of the RPE cells.
- the RPE cells described herein may be
- the RPE cells may be mammalian, including, human RPE cells.
- the invention also provides human RPE cells, a substantially purified population of human RPE cells, pharmaceutical preparations comprising human RPE cells, and cryopreserved preparations of the human RPE cells.
- the preparation may be a preparation comprising human embryonic stem cell-derived RPE cells, human iPS cell-derived RPE ceils, and substantially purified (with respect to non-RPE cells) preparations comprising differentiated ES- derived RPE cells,
- the RPE cel l populations may include differentiated RPE cells of varying levels of maturity, or may be substantially pure with respect to differentiated RPE cells of a particular level of maturity.
- the RPE cells may be a substantially puri fied preparation comprising RPE cells of varying levels of maturity/pigmentation.
- the substantially purified culture of RPE ceils may contain both differentiated RPE cells and mature differentiated RPE cells.
- the level of pigment may vary.
- the mature RPE cells may be distinguished visually from the RPE cells based on the increased level of pigmentation and the more columnar shape.
- the substantially purified preparation f RPE cells compri ses RPE cells of differing levels of maturity (e.g.
- differentiated RPE cells and mature differentiated RPE cells may be variabi lity across the preparation with respect to expression of markers indicative of pigmentation.
- the pigmentation of the RPE cells in the cell culture may be homogeneous. Further, the pigmentation of the RPE cells in the cell culture may be heterogeneous, and the culture of RPE cells may comprise both differentiated RPE cells and mature RPE cells.
- Preparations comprising RPE cells include preparations that are substantially pure, with respect to non-RPE cell types, but which contain a mi ture of differentiated RPE cells and mature differentiated RPE cells. Preparations comprising RPE cells also include
- the percentage of mature differentiated RPE cells in the culture may be reduced by decreasing the density of the culture.
- the methods described herein may further comprise subculturing a population of mature RPE cells to produce a culture containing a smaller percentage of mature RPE cells.
- the number of RPE cells in the preparation includes differentiated RPE cells, regardless of level of maturity and regardless of the relative percentages of differentiated R E cells and mature differentiated RP cells.
- the number of RPE cells in the preparation refers to the number of either differentiated E cells or mature RPE cells.
- the preparation may comprise at least about 75%, 80%, 85%, 90%, 91 %, 92%, 93%, 94%, 95% ⁇ , 96%, 97%, 98%, 99%, or 100% differentiated RPE cells.
- the preparation may comprise at least about 75%, 80%, 85%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% mature RPE cells.
- the RPE ceil preparation may comprise a mixed population of differentiated RPE cells and mature RPE cells.
- the invention provides a cell culture comprising human RPE cells which are pigmented and express at ieast one gene that is not expressed in a ceil that is not a human R E.
- RPE cells may have substantially the same expression of RPE65, PEDF, CRALBP. and bestrophin as a natural human RPE cell.
- the RPE cells may vary, depending on level of maturity, with respect to expression of one or more of PAX 2, Pax-6, MITF , and/or tyrosinase. Note that changes in pigmentation post-differentiation also correlate with changes in PAX2 expression.
- Mature RPE cells may be distinguished from R E cells by the level of pigmentation, level of expression of PAX2, Pax-6, and/or tyrosinase.
- mature RPE cells may have a higher level of pigmentation or a higher level of expression of PAX2, Pax-6, and/or tyrosinase compared to RPE cells.
- the preparations may be substantially purified, with respect to non-RPE cells, comprising at least about 75%, 80%, 85%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% RPE cells.
- the RPE cell preparation may be essentially free of non-RPE cells or consist of RPE cells.
- the substantially purified preparation of RPE cells may comprise less than about 25%, 20%, 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, or 1 % non- RPE cell type.
- the RPE cell preparation may comprise less than about 25%, 20%, 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1 %, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1 %, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, 0.01 %, 0.009%,
- the RPE ceil preparations may be substantially pure, both with respect to non- RPE cells and with respect to RPE cells of other levels of maturity.
- the preparations may be substantially purified, with respect to non-RPE cells, and enriched for mature PE cells.
- RPE cell preparations enriched for mature RPE cells at least about 30%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, 99%, 99%, or 100% of the RPE cells are mature RPE cells.
- the preparations may be substantially purified, with respect to non-RPE cells, and enriched for differentiated RPE cells rather than mature RPE cells. For example, at least about 30%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% of the RPE cells may be differentiated RPE cells rather than mature RPE cells.
- the RPE cell preparations may comprise at least about 1 x 10 3 , 2x 10 ' ⁇ 3 ] 0 ⁇ 4xl0 3 , 5x10 3 , 6xI0 3 , 7 l0 3 , 8xl0 3 , 9xl0 3 , I lO 4 , 2xlG 4 , 3xl0 4 , 4x10 4 , 5xl0 4 , 6x l0 4 , 7x!0 4 , 8xl0 4 , 9 l0 4 , IxlO 5 , 2xl0 5 , 3xl0 5 , 4 l0 5 , 5xl0 5 , 6 10 5 , 7xl0 5 , 8xl0 5 , 9 l0 5 , IxlO 6 , 2xl0 6 , 3xl0 6 , 4xl0 6 , SxlO 6 , 6xl0 6 , 7xl0 6 , 8xl0 6 , Sx
- the RPE cell preparations may comprise at least about 5.000-10,000, 50,000-100,000, 100,000-200,000, 200,000-500.000, 300,000-500,000, or 400,000-500,000 RPE cells.
- the RPE cell preparation may comprise at least, about 20,000- 50,000 RPE cells.
- the RPE cell preparation may comprise at least about 5,000, 10,000, 20,000, 30,000, 40,000, 50,000, 60,000, 70,000, 75,000, 80,000, 100,000, or 500,000 RPE cells.
- the RPE cell preparations may comprise at least about 1 x 10 J , 2x 10 3 , 3x 10 ' ⁇ IxlO 4 , 2 l0 4 .3xl0 4 , 4xl0 4 , 5xl0 4 , 6 l0 4 , 7 l0 4 , 8xl0 4 , 9xl0 4 , IxlO 5 , 2xl0 5 .3x10 s , 4x10 s , 5x10 s .6xl0 5 , 7 10 s .8xl0 5 , 9x10 ⁇ IxlO 6 , 2xl0 6 , 3xl0 6 , 4x 10 6 ,5xl 0 6 , 6xl0 6 , 7x 10 6 , 8x 10 6 , 9x 10 6 , I lO 7 , 2x 10 7 , 3x10 7 , 4x 10 7 , 5x 10 7 , 6x !
- the RPE cell preparations may comprise at least about 5,000-10,000, 50,000-100,000, 100,000-200,000, 200,000-500,000, 300,000-500,000, or 400,000-500,000 RPE cel!s/rnL.
- the RPE cell preparation may comprise at least about 20.000- 50,000 RPE cells/mL.
- the RPE cell preparatioo may comprise at least about 5,000, 10,000, 20,000, 30,000, 40,000, 50,000, 60,000, 75,000, 80,000, 100,000, or 500,000 RPE cells/mL.
- the preparations described herein may be substantially free of bacteria), viral, or fungal contamination or infection, including but not limited to the presence of HIV-1, HiV-2, HBV, HCV, CMV, llTLV-l, HTLV-2, parvovirus B 19, Epstein-Barr virus, or herpesvirus 6.
- the preparations described herein may be substantially free of mycoplasma contamination or infection.
- the RPE cells described herein may also act as functional RPE cells after transplantation where the RPE cells form a monolayer between the neurosensory retina and the 2
- the RPE cells may also supply nutrients to adjacent photoreceptors and dispose of shed photoreceptor outer segments by phagocytosis. Additionally, the RPE cells described herein may have undergone less senescence than cells derived from eye donors (e.g., the RPE cells are "younger" than those of eye donors). This allows the RPE cell described herein to have a longer useful lifespan than cells derived from eye donors.
- the preparations comprising RPE cells may be prepared in accordance with Good Manufacturing Practices (GMP) (e.g., the preparations are GMP-compliant) and/or current Good Tissue Practices (GTP) (e.g., the preparations may be GTP-co repliant.)
- GMP Good Manufacturing Practices
- GTP current Good Tissue Practices
- the present invention also provides substantially purified cultures of RPE cells, including human RPE cells and an indicator that indicates the number or fraction of pluripotcnt cells present, wherein the value of said indicator may be determined by applying the methods described herein to a cell population representative of the cells in said cultures.
- the RPE cultures described herein may comprise at least about 1,000; 2,000; 3,000; 4,000; 5,000; 6,000; 7,000; 8,000; or 9,000 RPE cells.
- the culture may comprise at least about IxlO 4 , 2x.l0 4 , 3xl0 4 , 4x 10 4 , 5x10 4 , 6 l0 4 , 7xl0 4 , 8xl0 4 , 9 l0 4 , IxlO 5 , 2 l0 5 , 3x10 s , 4x 10 5 , 5x 10 s , 6x 10 5 , 7x 10 5 , 8x 10 5 , 9x 10 5 , 1x10 6 , 2x 10 6 , 3x 10 6 , 4x 10 6 , 5x 10 6 , 6x 10 6 , 7x 10 6 , 8x 10 6 , 9 l0 6 , lx 10 7 , 2xl0 7 , 3xl0 7 , 4x10 ⁇ 5xl0 7 .6xl0 7 .7xl0 7 , 8xl0 7 , 9xl0 7 , 1x10 s , 2 10 s
- the RPE cells may be further cultured to produce a culture of mature RPE cells.
- the RPE cells may be matured, and the R E cells may be further cultured in, for example MDBK-MM medium until the desired level of maturation is obtained. This may be determined by monitoring the increase in pigmentation level during maturation.
- MDBK-MM medium a functionally equivalent or similar medium, may be used. Regardless of the particular medium used to mature the RPE cells, the medium may optionally be
- R E cells and mature RPE cells are differentiated RPE cells.
- mature RPE cells are characterized by increased level of pigment in comparison to differentiated RPE cells. The level of maturit and pigmentation may U 2011/045232
- a culture of RPE cells may be further cultured to produce mature RPE cells.
- the density of a culture containing maiore RPE cells may be decreased to decrease the percentage of mature differentiated RPE ceils and increase the percentage of differentiated RPE cells.
- the RPE ceils may be identified by comparing the messenger RNA transcripts of such cells with cells derived in vivo. An aliquot of ceils is taken at various intervals during the differentiation of embryonic stem cells to RPE cells and assayed for the expression of any of the markers described above. These characteristic distinguish differentiated RPE ceils.
- the RPE cell culture may be a substantially purified culture comprising at least about 30%, 35%, 40%, or 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91 , 92%, 93 %, 94%, 95% ⁇ , 96%, 97%, 98% ⁇ , 99%, or 100% differentiated RPE cells.
- the substantially purified culture may comprise at least about 30%, 35%, 40%, or 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% mature differentiated RPE cells.
- the RPE cell cultures may be prepared in accordance with Good Manufacturing Practices (GMP) (e.g., the cultures are GMP-compliant) and/or current Good Tissue Practices (GTP) (e.g., the cultures may be GTP-eomp!iant. )
- GMP Good Manufacturing Practices
- GTP Good Tissue Practices
- RPE cells may be frozen for storage. For example, portion of an RPE cell population may be analyzed using the methods described herein to detect the presence of any residual hES cells in the population, and the remainder of the population frozen for storage, thereby permitting determination of the approximate concentration of hES cells in the cell population.
- the frozen RPE cells may be associated with an indicator that indicates the number or concentration of hES cells detected in the population or otherwise indicates the result of the assay, e.g., an indicator that indicates that the cells have "passed" the test for hES cel ts contamination because the detected number or concentration of hES cells below an established release threshold, indicating that the ceils are considered suitable for use.
- the RPE cells may be stored by any appropriate method known in the art (e.g. , cryogenicalJy frozen ) and may be frozen at any temperature appropriate for storage of the cells.
- the cells may be frozen at about -20°C, -80°C. ⁇ 120°C, - 130 J C, -1 35°C, -140°C, -150°C, -16() J C. -170°C, - 180"C ⁇ -i9G°C, -196X, at any other temperature appropriate for storage of cells.
- Cryogeiiically frozen cells may be stored in appropriate containers and prepared for storage to reduce risk of cell damage and maximize the likelihood that the cells will survive thawing.
- RPE cells may be cryopreserved immediately following differentiation, following in vitro maturation, or after some period of time in culture. The RPE cells may also be maintained at room temperature, or refrigerated at, for example, about 4 ' C.
- the RPE cells may be harvested, washed in buffer or media, counted, concentrated (via centrifugation), formulated in freezing media (e.g., 90% FBS/10% DMSO). or any combination of these steps.
- the RPE cells may be seeded in several culture vessels and serially expanded. As the RPE cells are harvested and maintained in FBS at about 4°C while several flasks of RPE cells are combined into a single lot.
- the RPE cells may be also washed with saline solution (e.g., DPBS) at least 1, 2, 3, 4, or 5 times.
- saline solution e.g., DPBS
- the RPE cells may be cryopreserved after dystrophin is organized at the cell membrane and PAX6 expression is low.
- the vials may be labeled, with a primary and/or secondary label.
- the information on the label may include the type of cell (e.g., hRPE cells), the lot number and date, the number of cells (e.g., IxlO 6 ce!l /mL), the expiration date (e.g., recommended date by which the vial should be used), manufacture information (e.g., name and address), warnings, and the storage means (e.g., storage in liquid nitrogen).
- Cryopreserved RPE cell preparations described herein may comprise at least about 50,000-100.000 RPE cells.
- the cryopreserved RPE cell preparations may also comprise at least about 20.000-500,000 RPE cells, Also, the cryopreserved RPE cell preparations may comprise at least about 5,000, 10,000, 20,000, 30,000, 40,000, 50,000, 60,000, 75,000, 80,000, or 100,000 RPE cells.
- the cryopreserved RPE cell preparations ma comprise at least about 1,000, 2,000, 3,000, 4,000, 5,000, 10,000, 20,000, 30,000, 40,000, 50,000, 60,000, 75,000, 80,000, 100,000, or 500,000 RPE cells.
- the cryopreserved RPE cell preparations may comprise at least about 1,000, 2,000, 3,000, 4,000, 5,000, 6,000, 7,000, 8,000, 9,000, IxlO 4 , 2xl0 4 .3 l0 4 , 4x 10 4 , 5x 10 4 , 6x 10 4 , 7x 10 4 , 8x10 4 , 9x 10 4 , 1 x 10 5 , 2x 10 ' ⁇ 3x 10 5 , 4x 10 5 , 5x 10 5 , 6x 10 5 , 7x 10 " ⁇ 8xl0 5 , 9xl0 5 , IxlO 6 , 2xl0 6 , 3xl0 6 , 4xl0 6 , 5xl0 6 , 6xt0 6 , 7xl0 6 , 8xl0 6 , 9xl0 6 , IxlO 7 , 2xl0 7 , 3xl0 7 , 4x 10 7 , 5x 10 7 ,
- the RPE cells of the cryopreserved RPE cell preparations may be mammalian RPE cells, including human RPE cells.
- cryopreserved RPE ce!I preparations described herein may comprise at least about 50,000-100,000 RPE cells/mL.
- the cryopreserved RPE cell preparations may also comprise at least about 20,000-500,000 RPE cells/mL.
- the cryopreserved RPE cell preparations may comprise at least about 5,000, 10,000, 20,000, 30,000, 40,000, 50,000, 60,000, 75,000, 80,000, and 100,000 RPE cells/mL.
- the cryopreserved RPE cell preparations may comprise at least about 1,000, 2,000, 3,000, 4,000, 5,000, 10,000, 20,000, 30,000, 40,000, 50,000, 60,000, 75,000, 80,000, 100,000, or 500,000 RPE ee!ls/mL,
- the cryopreserved RPE cell preparations may comprise at least about 1,000, 2,000, 3,000, 4,000, 5,000, 6,000, 7,000, 8,000, 9,000, lxlO 4 , 2 l0 , 4 l0 4 , 5xl0 4 .6xl0 4 , 7xl0 4 , 8xl0 4 , 9xl0 4 , 1x10 ' , 2x10 s .3x10 s , 4xl0 5 , 6 l0 5 , 7 l0 5 , 8x10 s , 9x10 s , lxlO 6 , 2xl0 6 , 3xl0 6 ,4 l0
- the RPE cells of the invention may be recovered from storage following cryopreservation.
- the RPE cells recovered from cryopreservation also maintain their viability and differentiation status. For example, at least about 65%, 70%, 75%, 80%, 81%, 82%, 837c, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% of the RPE cells may retain viability and differentiation following cryopreservation.
- the RPE cells of the invention may be cryopreserved and maintain their viability after being stored for at least about 1 , 2, 3, 4, 5, 6, or 7 days.
- the RPE cells of the invention may also be cryopreserved and maintain their viability after being stored for at least about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, or 12 months.
- the RPE cells of the invention may be cryopreserved and maintain their viability after being stored for at least about 1 , 2, 3, 4, 5, 6, or 7 years.
- the RPE cells of the invention may be cryopreserved for at least about 4 years and show at least about 80% viability.
- the cryopreservation preparation comprising RPE cells may be
- Cel l populations analyzed by the subject methods may be produced from piuripotent stem cells.
- Cell types that may be produced include, but are not limited to, RPE cells, RPE progenitor cells, iris pigmented epithelial (IPE) cells, and other vi sion associated neural cells, such as internuncial neurons (e.g. , "relay" neurons of the inner nuclear layer (INL)) and amacrine cells.
- IPE iris pigmented epithelial
- INL inner nuclear layer
- retinal cells, rods, cones, and corneal cells may be produced. Cells providing the vasculature of the eye may also be produced by the methods described herein.
- PAX6 acts synergistically with PAX ' 2 to terminally differentiate mature RPE via the coordination of M1TF and Otx2 to transcribe RPE-specifie genes such as Tyrosinase (Tyr), and downstream targets such as RPE-65, Bestrophin, CRALBP, and PEDF. See WO 2009/05 1671 , Figure 1.
- the RPE cells described herein may be di fferentiated from piuripotent stem cells, such as human embryonic stem cells, and may be molecularly distinct from embryonic stem cells, adult-derived RPE cells, and fetal-derived RPE cells.
- the manufacturing process steps described herein may impart distinctive structural and functional characteristics to the final RPE cell product such that these cells closely resemble native RPE cells and are distinct from fetal derived RPE cells or RPE cell lines (e.g. , APRE 19).
- An exemplary method for producing a RPE cell comprises: (a) providing piuripotent stem cells; (b) culturing the piuripotent stem cells as embryoid bodies in nutrient rich. low protein medium, wherein the medium optionally comprises serum free B-27 supplement; (c) culturing the embryoid bodies as an adherent culture in nutrient rich, low protein medium, wherein the medium optionally comprises serum free B-27 supplement; (d) culturing the adherent culture of cells of (c) in nutrient rich, low protein medium, wherein the medium does not comprise serum free B-27 supplement; (e) culturing the cells of (d) in medium capable of supporting growth of high-density somatic cell culture, whereby RPE cells appear in the culture of cells; (f) dissociating cells or clumps of cells from the culture of (e), preferably mechanically or chemically (e.g., using a protease or other enzyme, or another dissociation medium); (g) selecting the RPE cells appear
- the invention also provides a method for producing a mature retinal pigment epithelial (RPE) cell comprising: (a) providing pluripotent stem cel ls; (b) culturing the pluripotent stem cells as embryoid bodies in nutrient rich, low protein medium, wherein the medium optionally comprises serum free B-27 supplement; (c) culturing the embryoid bodies as an adherent culture in nutrient rich, low protein medium, wherein the medium optionally comprises serum free B-27 supplement; (d) culturing the adherent culture of cells of step (c) in nutrient rich, low protein medium, wherein the medium does not comprise serum free B-27 supplement; (e) culturing the cells of (d) in medium capable of supporting growth of high- density somatic cell culture, whereby RPE cells appear in the culture of cells; (f) dissociating cells or clumps of cells from the culture of (e), preferably mechanically or chemically (e.g., using a proteas
- the cells may be cultured for at least about 1 -10 weeks.
- the cells may be cultured for at least about 3-6 weeks.
- the cells may be cultured for between about 1 days and 50 days, for example, for at least about 1 -3, 3-1, 7, 4-9, 7-10, 7-12, 8-1 1 , 9-12, 7-14, 14-21 , and 3 ⁇ 45 days.
- the cells may be cultured for about 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10. 1 1.
- the cells may be cultured for about 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 1 1 , 12, 13, 14, 15, 16, 17, 18, 19, 20, 2 1 , 22. 23, or 24 hours.
- the cells may be cultured for 2-4 and 3-6 hours.
- the cells may be cultured for the same period of time at each step or for differing periods of time at one or more of the steps. Additionally, any of the above articulated method steps may be repeated to produce more RPE cells (e.g. , scaled up to produce large numbers of RPE cells).
- the RPE cells may begin to differentiate from amongst cells in the adherent culture of EBs.
- RPE ceils may be visually recognized based on their cobblestone morphology and the initial appearance of pigmentation. As RPE cells continue to differentiate, clusters of RPE ceils may be observed.
- Mechanical or enzymatic methods may be used to select R E cells from amongst clusters of non-RPE cells in a culture of embryoid body, or to facilitate sub-culture of adherent cells.
- Exemplary mechanical methods include, but are not limited to, titration with a pipette or cutting with a pulled needle.
- Exemplary enzymatic methods include, but are not limited to, any enzymes appropriate for disassociating cells (e.g. , trypsin (e.g., Trypsin/EDTA), collagenase (e.g. , collagenase B, collagenase IV), dispase, papain, mixture of collagenase and dispase, a mixture of collagenase and trypsin).
- a non-enzymatic solution may be used to disassociate the cells, such as a high EDTA-containing solution e.g., Hanks-based cell disassociation buffer.
- the RPE cells may be di fferentiated from the embryoid bodies. Isolating RPE cells from the EBs allows for the expansion of the RPE cells in an enriched culture in vitro.
- RPE cells may be obtained form EBs grown for less than 90 days. Further, RPE cells may arise in human EBs grown for at least about 7-14 days, 14—28 days, 28-45 days, or 45-90 days.
- the medium used to culture piuripotent stem cells, embryoid bodies, and RPE cells may be removed and/or replaced with the same or different media at any interval. For example, the medium may be removed and/or replaced after at least about 0-7 days, 7—10 days, 10-14 days, 14-28 days, or 28-90 days. Further, the medium may be replaced at least daily, every other day, or at least every 3 days.
- RPE cells may be dissociated from each other and from non-RPE cells using mechanical and/or chemical (including enzymatic) methods. A suspension of RPE cells may then be transferred to fresh medium and a fresh culture vessel to provide an enriched population of RPE cells.
- RPE cells may be seJected from the dissociated cells and cultured separately to produce a substantially purified culture of RPE cells. RPE cells are selected based on characteristics associated with RPE cells.
- RPE cells can be recognized by cobblestone cellular morphology and pigmentation, in addition, there are several known markers of the RPE, including cellular red naldehyde-bi ruling protein (CRALBP), a cytoplasmic protein thai i also found in apical microvilli; RPE65, a cytoplasmic protein involved in retinoid metabolism; bestrophin, the product of the Best vitel!i orm macular dystrophy gene (VMD2), and pigment epithelium derived factor ( PEDF), a 48kD secreted protein with angiostatic properties.
- CRALBP cellular red naldehyde-bi ruling protein
- RPE65 a cytoplasmic protein involved in retinoid metabolism
- bestrophin the product of the Best vitel!i orm macular dystrophy gene (VMD2)
- PEDF pigment epithelium derived factor
- the messenger RNA transcripts of these markers may be assayed using PCR (e.g
- the RPE cells may also be selected based on cell function, such as by
- phagocytosis of shed rod and cone outer segments or phagocytosis of another substrate, such as polystyrene beads
- absorption of stray light vitamin A metabolism
- regeneration of retinoids and tissue repair.
- Evaluation may also be performed by testing in vivo function after RPE cell implantation i to a suitable host animal (such as a human or non-human animal suffering from a naturally occurring or induced condition of retinal degeneration), e.g., using behavioral tests, fluorescent angiography, histology, tight junctions conductivity, or evaluation using electron microscopy.
- the enriched cultures of RPE cells may be cultured in appropriate medium, for example, EGM-2 medium.
- EGM-2 medium This, or a functionally equivalent or similar medium, may be supplemented with a growth factor or agent (e.g., bFGF, heparin, hydrocortisone, vascular endothelial growth factor, recombinant insulin-like growth factor, ascorbic acid, or human epidermal growth factor).
- a growth factor or agent e.g., bFGF, heparin, hydrocortisone, vascular endothelial growth factor, recombinant insulin-like growth factor, ascorbic acid, or human epidermal growth factor.
- the RPE cells may be phenotypically stable over a long period of time in culture (e.g. , >6 weeks).
- pluripotent stem cells include but are not limited to embryonic stem, ceils, embryo-derived stem cells, and induced pluripotent stem cells, regardless of the method by which the pluripotent stem cells are derived.
- Pluripotent stem cells may be generated using, for example, methods known in the art.
- Exemplary pluripotent stem cells include embryonic stem cells derived from the inner cell mass (ICM) of blastocyst stage embryos, as well as embryonic stem cells derived from one or more blastomeres of a cleavage stage or morula stage embryo (optionally without destroying the remainder of the embryo).
- Such embryonic stem cells may be generated from embryonic material produced by fertilization or by- asexual means, including somatic cell nuclear transfer (SCNT), parthenogenesis, cellular reprogramming. and androgenesis.
- SCNT somatic cell nuclear transfer
- suitable pluripotent stem cells include but are not limited to human embryonic stem cells, human embryo-derived stem ceils, and human induced pluripotent stem cells, regardless of the method by which the pluripotent stem cells are derived.
- the pluripotent stem cells may be cultured as a suspension culture to produce embryoid bodies (EBs).
- the embryoid bodies may be cultured in suspension for about 7-14 days. However, in certain embodiments, the EBs may be cultured in suspension for fewer than 7 days (less than 7, 6, 5, 4, 3, 2, or less than 1 day) or greater than 14 days.
- the EBs may be cultured in medium supplemented with B-27 supplement.
- the EBs may be transferred to produce an adherent culture.
- the EBs may be plated onto gelatin coated plates in medium.
- the EBs may be cultured in the same type of media as when grown in suspension.
- the media may not supplemented with B-27 supplement when the cells are cultured as an adherent culture.
- the medium is supplemented with B-27 initially (e.g. , for less than or equal to about 7 days), but then subsequently cultured in the absence of B-27 for the remainder of the period as an adherent culture.
- the EBs may be cultured as an adherent culture for at least about 14-28. However, in certain embodiments, the EBs may be cultured as an adherent culture for fewer than about 14 days (less than 14, 13. 12, 1 1 , 10, 9, 8, 7, 6, 5, 4, 3, 2, or less than 1 day) or greater than about 28 days.
- Human embryonic stem (hES) cells may be used as a pluri otent stem cell in the methods described herein.
- Human embryonic stem cells (hES) include progeny of the inner cell mass (ICM) of a blastocyst or cells derived from another source, and may remain pluripotent virtually indefinitely.
- the hES cells may be derived from one or more blastomeres of an early cleavage stage embryo, optionally without destroying or without harming the embryo.
- the hES cells may be produced using nuclear transfer.
- the hES cells may also be induced piuripotent cells (iPS cells) which are described in further detail below.
- cr oprc served hES cells may be used.
- the hES cells may be cultured in any way known in the art. such as in the presence or absence of feeder cells.
- the hES cells may be cultured in MDBK-GM, hESC Medium, INVITROGEN® Stern Cell Media, OptiPro SFM, VP SFM , EGM-2, or MDBK MM.
- MDBK-GM hESC Medium
- INVITROGEN® Stern Cell Media OptiPro SFM
- VP SFM VP SFM
- EGM-2 EGM-2
- MDBK MM MDBK MM.
- Stem Cell Information Culture of Human Embryonic Stem Cells (hESC) [NIH website, 2OI O3.
- the hES cells may be used and maintained in accordance with GMP standards.
- hES cells When grown in culture on a feeder layer in defined conditions hES cells maintain a specific morphology, forming flat colonies comprised of small, tightly packed cells with a high ratio of nucleus to cytoplasm, clear boundaries between the cells, and sharp, refractile colony borders.
- hES cells express a set of molecular markers, such as Octamer binding protein 4 (Oct -4. a.k.a., Pou5fl ), stage specific embryonic antigens (SSEA)-3 and SSEA-4, tumor rejection antigen (TRA)- l -60, TRA- 1 -80. alkaline phosphatase, NANQG. and Rex- 1 .
- hES cells in culture may be induced to differentiate.
- hES cells may be differentiated into human RPE under the defined conditions described herein.
- Human embryonic stem cells that may be used include, but are not limited to,
- Additional exemplary ceil lines include NED 1 , NED 2, NEDS, NED4, and NEDS. See also NIH Human Embryonic Stem Cell Registry.
- An exemplary human embryonic stem cel l line that may be used is MA09 cells. The isolation and preparation of MA09 cells was previously described in Klimanskaya. et al. (2006) "Human Embryonic Stem Cell lines Derived from Single Blastomeres.” Nature 444; 481 -485.
- the hES cells may be initially co-cultivated with murine embryonic feeder cells (MEF) cells.
- the MEF cells may be mitotically inactivated by exposure to mitomycin C prior to seeding hES cells in co-culture, and thus the MEFs do not propagate in culture.
- hES cell cultures may be examined microscopically and colonies containing non-hES cell morphology may be picked and discarded, e.g., using a stem cell cutting tool, by laser ablation, or other means.
- no additional MEF cells are used in the process.
- the time between MEF removal and RPE cells described herein harvest may be a minimum of at least one, two, three, four, or five passages and at least about 5. 10, 20, 30, 40, 50, 60, 70, 80, 90, or 100 days in MEF- free cell culture.
- the time between MEF removal and harvesting the RPE ceils may also be a minimum of at least about 3 passages and at least about 80-90 days in MEF-free cell culture. Due to the methods of production described herein, the RPE cell cultures and preparations described herein may be substantially free of mouse embryo fibroblasts (MEF) and human embryonic stem cells (hES).
- MEF mouse embryo fibroblasts
- hES human embryonic stem cells
- pluripotent stem cells include induced pluripotent stem ceils (iPS cells) generated by reprogramming a somatic cell by expressing or inducing expression of a combination of factors ("reprogramming factors").
- iPS cells may be generated using fetal, postnatal, newborn, juvenile, or adult somatic cells.
- iPS cells may be obtained from a cell bank.
- i S cells may be newly generated by methods known in the art prior to
- iPS cells may be an initial step in the production of differentiated cells.
- iPS cells may be specifically generated using material from a particular patient or matched donor with the goal of generating tissue- matched RPE cells.
- iPS cells can be produced from cells that are not substantially immunogenic in an intended recipient, e.g., produced from autologous cells or from cells histocompatible to an intended recipient.
- the induced pluripotent stem cell may be produced by expressing or inducing the expression of one or more reprogramming factors in a somatic cell.
- the somatic cell is a fibroblast, such as a dermal fibroblast, synovial fibroblast, or lung fibroblast, or a non- fibroblastic somatic cell.
- the somatic cell is reprogram tried by expressing at least 1 , 2, 3, 4, 5.
- the reprogramming factors may be selected from Oct 3/4, Sox2, NANOG, Lin28, c-M c, and Klf4. Expression of the reprogramming factors may be induced by contacting the somatic cells with at least one agent, such as a small organic molecule agents, that induce expression of reprogramming factors.
- the somatic cell may also be reprogrammed using a combinatorial approach wherein the reprogramming factor is expressed (e.g., using a virai vector, plasmid, and the like) and the expression of the reprogramming factor is induced (e.g., using a small organic molecule. )
- reprogramming factors may be expressed in the somatic cell by infection using a viral vector, such as a retroviral vector or a lentivirai vector.
- reprogramming factors may be expressed in the somatic cell using a non -integrative vector, such as an episomal plasmi l.
- the factors may be expressed in the cells using electroporation, transfection, or transformation of the somatic ceils with the vectors.
- electroporation, transfection, or transformation of the somatic ceils with the vectors For example, in mouse cells, expression of four factors (Oct3/4, Sox2, c-myc, and KIf4) using integrative viral vectors is sufficient to reprogram a somatic cell.
- expression of four factors (Oct3/4, Sox 2, NANOG, and Lin ' 28) usin integrative viral vectors is sufficient to reprogram a somatic cell.
- the cells may be cultured. Over time, cells with ES characteristics appear in the culture dish. The cells may be chosen and subcultured based on, for example, ES morphology, or based on expression of a selectable or detectable marker. The cells may be cultured to produce a culture of cells that resemble ES cells— these are putative iPS cells.
- the cells may be tested in one or more assays of pluri potency.
- the DC ls may be tested for expression of ES cell markers; the cells may be evaluated for ability to produce teratomas when transplanted into SCID mice; the cells may be evaluated for ability to differentiate to produce cell types of all three germ layers.
- Human embryonic stem (hES) cells may be derived from a library of human embryonic stem cells.
- the library of human embryonic stem cells may comprise stem cells, each of which is hemizygous, homozygous, or nullizygous for at least one MHC allele present in a human population, wherein each member of said library of stem cells is hemizygous, homozygous, or nullizygous for a different set of MHC alleles relative to the remaining members of the library.
- the library of human embryonic stem cells may comprise stem cells that are hemizygous, homozygous, or nullizygous for all MHC alleles present in a human population.
- stem cells that are homozygous for one or more histocompatibility antigen genes include cells that are nullizygous for one or more (and in some embodiments, all) such genes.
- Nullizygous for a genetic locus means that the gene is null at that locus (i.e., both alleles of that gene are deleted or inactive.)
- a hES cell may comprise modifications to one of the alleles of sister
- modifications such as gene targeting, may be used to modify the genes in the MHC complex.
- the modified alleles of the MHC complex in the cells may be subsequently engineered to be homozygous so that identical alleles are present on sister chromosomes.
- Methods such as loss of heterozygosity (LOH) may be utilized to engineer cells to have homozygous alleles in the MHC complex.
- LOH loss of heterozygosity
- one or more genes in a set of MHC genes from a parental allele can be targeted to generate hemizygous cells.
- the other set of MHC genes can be removed by gene targeting or LOH to make a null line.
- This null line can be used further as the embryonic cell line in which to drop arrays of the HI , A genes, or individual genes, to make a hemizygous or homozygous bank with an otherwise uniform genetic background.
- Stem cells that are nuilizygous for all MHC genes may be produced by standard methods known in the art, such as, for example, gene targeting and/or loss of heterozygosity (LOH). See, for example, United States Patent Application Publications 2004/0091936, 2003/0217374 and 2003/0232430, and U.S. Provisional Patent Application Number 60/729, 173.
- the present invention relates to methods of obtaining differentiated cells (such as RPE cells), including a library of differentiated cells having reduced MHC complexity.
- differentiated cells such as RPE cells
- Differentiated cells with reduced MHC complexity may be used to increase the supply of available cells for therapeutic applications as it may eliminate the difficulties associated with patient matching.
- Such cells may be derived from stem cells that are engineered to be hemizygous or homozygous for genes of the MHC complex.
- the invention also provides a library of differentiated cells (such as RPE cells and/or RPE lineage cells), wherein several lines of ES cells are selected and differentiated into the differentiated cells. These differentiated cells may be used for a patient i need of a cell- based therapy.
- the invention also provides a library of differentiated cells, each of which is hemizygous, homozygous, or nuilizygous for at least one MHC allele present in a human population, wherein each member of said library of differentiated cells is hemizygous, homozygous, or nuilizygous for a di fferent set of HC alleles relative to the remaining members of the library.
- the invention provides a library of human differentiated cells that are
- any medium that is capable of supporting high-density cultures may be used in the methods described herein, such as medium for viral, bacteria!, or eukaryotic cell culture.
- the medium may be high nutrient, protein-free medium or high nutrient, low protein medium.
- the medium also may include nutrient components such as albumin, B-27 supplement, ethano!amine, fetuin, glutamine, insulin, peptone, purified lipoprotein material, sodium selenite, transferrin, vitamin A, vitamin C, or vitamin E.
- nutrient rich, low protein medium may be any medium which supports the growth of cells in culture and has a low protein content.
- nutrient rich, low protein media includes but is not limited to MDBK-GM, OptiPro SFM, VP-SFM.
- DM EM , RPM1 Media 1640, IDMEM.
- MEM F- 12 nutrient mixture
- F- 10 nutrient mixture EGM-2 F- 10 nutrient mixture
- DMEM/F- 1 2 media media 1999, or
- the nutrient rich, low protein medium may be a medium that does not support the growth or maintenance of embryonic stem cells
- the medium may contain at least about 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 1 1 %, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2.5%, 2%, 1 .5%, 1 %, 0.75%, 0.5%, 0.25%, 0.20%, 0.10%, 0.05%, 0.02%, 0.016%, 0.015%, or 0.010% animal-derived protein ⁇ e.g. , 10% FBS).
- reference to the percentage of protein present in low protein medium refers to the medium alone and does not account for protein present in, for example, B-27 supplement.
- the low protein or protein free medium are supplemented with serum free B-27 supplement.
- Nutrient components of B27 supplement may comprise biotin, L-carnitine, corticosterone, ethanolamine, D+-galactose, reduced glutathione, linoleic acid, linolenic acid, progesterone, putrescine, rednyl acetate, selenium, triodo- 1 -thyronine (T3), DL-alpha-iocopherol (vitamin E), DL-alpha-tocopherol acetate, bovine serum albumin, catalase, insulin, superoxide dismutase, and transferrin.
- protein free refers to the medium prior to addition of B-27.
- Growth factors, agents, and other supplements described herein may be used alone or in combination with other factors, agents, or supplements for inclusion in media.
- the medium may also contain supplements such as heparin, hydrocortisone, ascorbic acid, serum (e.g., fetal bovine serum), or a growth matrix (e.g., extracellular matrix from bovine corneal epithelium, MATRIGEL® (basement membrane matrix), or gciatin).
- supplements such as heparin, hydrocortisone, ascorbic acid, serum (e.g., fetal bovine serum), or a growth matrix (e.g., extracellular matrix from bovine corneal epithelium, MATRIGEL® (basement membrane matrix), or gciatin).
- fibronectin fibronectin, proteolytic fragments of fibronectin, laminin, thrombospondin, aggrecan, and syndezan.
- the culture media may be supplemented with one or more factors or agents.
- Growth factors that may be used include, for example, EGF, FGF, VEGF. and recombinant insulin-like growth factor. Growth factors that may be used in the present invention also include 6Ckine (recombinant), activin A, a-interferon. alpha-interfcron, amphiregulin. angiogenin, ⁇ -endothelial cell growth factor, beta cc!iulin, ⁇ - interferon, brain derived neurotrophic factor, cardiotrophin- 1.
- 6Ckine recombinant
- activin A a-interferon. alpha-interfcron, amphiregulin. angiogenin, ⁇ -endothelial cell growth factor, beta cc!iulin, ⁇ - interferon, brain derived neurotrophic factor, cardiotrophin- 1.
- ciliary neurotrophic factor cytoki ne-induced neutrophil chcmoattraetant- 1 , endothelial cell growth supplement, eotaxin, epidermal growth factor, epithelial neutrophil activating peptide-78, erythropoiten, estrogen receptor-a, estrogen receptor- ⁇ , fibroblast growth factor (acidic/basic, heparin stabilized, recombinant), FLT-3/FLK-2 ligand (FLT-3 ligand), gamma-interferon, glial cell line-derived neurotrophic factor,
- Gly-His-Lys granulocyte colony-stimulating factor, granulocyte macrophage colony-stimulating factor, GRO-alpha/MGSA, GRO-B, GRO-gamma, HCC- 1 , heparin-binding epidermal growth factor like growth factor, hepatocytc growth factor, heregulin-alpha (EGF domain), insulin growth factor binding protein- 1 , insulin-like growth factor binding protein- 1 /IGF- 1 complex, insulin-like growth factor, insulin-like growth factor II.
- NEF 2.5S nerve growth factor
- 7S-NGF macrophage inflammatory protein- 1 ⁇
- macrophage inflammatory protein-2 macrophage inflammatory protein-3 a
- macrophage inflammatory protein-3p ⁇ monocyte chemotactic protein- 1 .
- monocyte chemotactic protein-2 monocyte chemotactic protein-3, neurotrophin-3.
- neurotrophiri-4 NGF- ⁇ (human or rat recombinant), oncostatin M (human or mouse recombinant), pituitary extract, placenta growth factor, platelet-derived endothelial cell growth factor, platelet-derived growth factor, pleiotrophin, rantes, stem cell factor, stromal cell- derived factor J B/pre-B cell growth stimulating factor, thrombopoetin, transforming growth factor alpha, transforming growth factor- ⁇ , transforming growth f actor- ⁇ 2, transformin growth factor-p3, transforming growtb-factor-pS, tumor necrosis factor (a and ⁇ ), and vascular endothelial growth factor.
- Agents that may be used according to the present invention include cytokines such as itiierferon-a, interferon ⁇ a A/D, interferon- ⁇ , i nterferon- ⁇ . i n t e rf e ro n - ⁇ - inducible protein- ! O. interleukin- S , interleukin-2. interleukin-3. interleuki n-4, i nterleukin-5, interleukin-6, interleukin-7, interieukin-8, interleuk.in-9, interleukin- i ( ) . interleukin- ! 1 , interleukin- 12.
- cytokines such as itiierferon-a, interferon ⁇ a A/D, interferon- ⁇ , i nterferon- ⁇ . i n t e rf e ro n - ⁇ - inducible protein- ! O. interleukin- S
- interleukin- 13 interleukin- 15, interleukin- 17, kerati oocyte growth factor, leptin, leukemia inhibitory factor, macrophage colony-stimulating factor, and macrophage inflammatory protein- 1.
- the culture media may be supplemented with hormones and hormone antagonists, including but not limited to 17B-estradioL adrenocorticotropic hormone, adrenomedullin, alpha- melanocyte stimulatin hormone, chorionic gonadotropin, corticosteroid-binding globulin, corticosterone, dexamethasone, estriol, follicle stimulating hormone, gastrin 1 , glucagon, gonadotropin, hydrocortisone, insulin, insulin-like growth factor binding protein, L-3,3',5'- triiodothyronine, L-3, 3'.5' -triiodothyronine, leptin, leutinizing hormone, L-thyroxine, melatonin, MZ-4.
- hormones and hormone antagonists including but not limited to 17B-estradioL adrenocorticotropic hormone, adrenomedullin, alpha- melanocyte stimulat
- the culture media may be supplemented with antibodies to various factors including but not limited to anti-low density lipoprotein receptor antibody, anti-progesterone receptor, internal antibody, anti-alpha interferon receptor chain 2 antibody, anti-c-c cheniokine receptor 1 antibody, anti -CD 1 18 antibody.
- anti- interleukin-6 human recombinant antibody anti-inter!eukin- 1 human recombinant antibody, anti- inter leu kin -2 human recombinant antibody, anti-!eptin mouse recombinant antibody, anti-nerve growth factor receptor ami body, anti- ⁇ , chicken antibody, anti-parathyroid hormone-like protein antibody, anti-plate!et-derived growth factor receptor antibody, anti-p!atelet-derivcd growth factor receptor-B antibody, anti-platelet-deri ved growth factor-alpha antibody, anti- progesterone receptor antibody, anti-retinoic acid receptor-alpha antibody, anti-thyroid hormone nuclear receptor antibody, anti-thyroid hormone nuciear receptor-alpha 1/Bi antibody, anti- transfesferin receptor/CD71 antibody, anti-transforming growth factor-alpha antibody, anti- transforming growth factor-B3 antibody, anti -rumor necrosis factor-alpha antibody, and anti- vascular endothelial growth factor antibody.
- Exemplary growth media suitable for use in the methods described herein are- listed in Table 2.
- the retinal pigment epithelium is the pigmented cell layer outside the neurosensory retina between the underlying choroid (the layer of blood vessels behind the retina) and overlying retinal visual cells (e.g., photoreceptors— rods and cones).
- the RPE is critical to the function and health of photoreceptors and the retina.
- the RPE maintains photoreceptor function by recycling photopigments, delivering, metabolizing, and storing vitamin A, phagocvtosing rod photoreceptor outer segments, transporting iron and small molecules between the retina and choroid, maintaining Bruch's membrane and absorbing stray light to allow better image resolution.
- Mature RPE is characterized by its cobblestone cellular morphology of black pigmented cells and RPE cell markers including cellular retinaldehyde-binding protein
- CRALBP retinaldehyde-binding protein
- RPE65 a 65 kD cytoplasmic protein involved in retinoid metabolism
- bcstrophin a membrane localized 68 kD product of the Best vitelliform macular dystrophy gene (VMD2) (Marmorstein, et al.
- PNAS 97(23): 12758-12763 pigment epi thelium derived factor
- PEDF pigment epi thelium derived factor
- Degeneration of the RPE can cause retinal detachment, retinal dysplasia, or retinal atrophy that is associated with a number of vision-altering ailments that result in photoreceptor damage and blindness, such as choroideremia.
- diabetic retinopathy diabetic retinopathy, macular degeneration (including age-related macular degeneration), retinitis pigmentosa, and Stargardt' s Disease (fundus flavimaculatus).
- Choroideremia Choroideremia.
- Choroiderernia is an X-linked recessive retinal degenerati v disease that leads to the degeneration of the choriocapillaris, the retinal pigment epithelium, and the photoreceptor of the eye caused by mutations in the CHM gene, which encodes the Rab escort protein- 1 ( REP- 1 ). Genetics Home Reference (U.S. National Library of Medicine)
- Diabetic Retinopathy is the most common diabetic eye disease nd a leading cause of blindness in the United States. Diabetic retinopathy is caused by changes in the blood vessels of the retina. Generally, diabetic retinopathy may only be controiied or slowed with surgery but not treated, and the patient usually continues to suffer from vision problems. Therefore, there exists a need for improved diabetic retinopathy therapies. "Diabetic Retinopathy” (MayoClinic.org) [February 1 1 , 2010],
- AMD Age-related macular degeneration
- CNV choroidal neovascularizations
- Neovascular or exudative AMD causes vision loss due to abnormal blood vessel growth (choroidal neovascularization) in the choriocapillaris, through Bruch's membrane, ultimately leading to blood and protein leakage below the macula. Bleeding. leaking, and scarring from these blood vessels eventually cause irreversible damage to the photoreceptors and rapid vision loss if left untreated.
- Current treatments for macular degeneration include anti-angiogenic therapy with ranibizumab (LUCE TIS®) or bevacizumab (AVASTIN®), photocoagulation (laser surgery), photodynamic therapy with verteporfin ⁇ ViSUDYNE® ), and submacular hemorrhage displacement biny. "Macular Degeneration.” (MayoClinic.org) [October 2010]. However, the goal of these therapies is to stem further vision loss and, unfortunately, existing damage cannot be reversed. Therefore, a great need exists for the treatment of macular degeneration.
- Retinitis Pigmentosa RP
- Retinitis pigmentosa RP is a group of inherited diseases that damage the photoreceptors (e.g., rods and cones) in the retina affecting
- RP autosomal recessive RP
- RPE65 retinaJdeh de binding protein
- the progression of RP is slow and varies from patient to patient. Patients with RP ail suffer some vision loss, with night blindness as a typical early symptom followed by tunnel vision, and some may lose all sight.
- Retinal Detachment i ncludin rhegmatogenous retinal detachment, exudative, serous, or secondary retinal detachment, and fractional retinal detachment, is a disorder of the eye in which the retina peels away from its underlying layer of support tissue, which can lead to vision loss and blindness. See Ghazi and Green (2002) Eye 16: 41 1-42 1 ; Facts About Retinal Detachment [NEI Health Information] (October 2010). A need exists for a treatment for retinal detachment.
- Stargardt's Disease (fundus flavimaculatus ).
- Stargardt's Disease ( fundus flavimaculatus) is a type of macular degeneration, including both an autosomal recessive and a dominant form, that causes a progressive loss of central vision of both eyes. See Gass and Hummer ( 1999) Retina 19(4): 297-301 and Aaberg ( 1986) Tr. Am. Ophth. Soc. LXXIV: 453- 487.
- Gouras, et al. (2002) Investigative Ophthalmology & Visual Science 43( 10): 3307-31 1 describes the transplantation of RPE cells from normal mice info transgenic RPE65 mice (a mouse model of retinal degeneration). Gouras discloses that the transplantation of healthy RPE cells slowed the retinal degeneration in the RPE6 " " mice but after 3.7 weeks, its salubrious effect began to diminish. Treumer, et al. (2007) Br J Opthalmol 91 : 349-353 describes the
- RPE cells have been suggested as a possible therapy for treating Parkinson's disease, a chronic degenerative disease of the brain. The disease is caused by degeneration of specialized neuronal cells in the region of the basal ganglia. The death of dopaminergic neurons results in reduced synthesis of dopamine, an important neurotransmitter, in patients with Parkinson's disease.
- RPE cells sourced from human donors has several intractable problems.
- First is the shortage of eye donors, and the current need is beyond what could be met by donated eye tissue.
- Third, donated RPE cells are derived from cadavers, which may not be of sufficient quality as to be useful in transplantation.
- cadaver-sourced RPE may have age-associated defects including being close to senesce.
- RPE cells derived from fetal tissue have shown a very low proliferative potential.
- cadaver-sourced RPE cells vary widely from batch to batch and must be characterized for safety before transplantation. See, e.g., Irina Klimanskaya. Retinal Pigment Epithelium Derived From
- Autologous RPE cells have also been attempted in limited trials but has proven unsatisfactory.
- Autologous cells are fundamentally limited because the autologous cells carry the same genetic propensities that may have lead to the development of AMD. See, e.g., Binder, el al. (2007) Progress in Retinal and Eve Research 26(5): 5 16-554.
- autologous RPE cells have limited proliferative capacity (particularly because AMD most frequently develops in older patients) which limits their utility in therapeutic applications (e.g.. the RPE cells may not transplant well and are less l ikely to last long enough for more complete recovery of vision).
- Embryonic Stem Cells derived RPE Cells hESC-RPE cells
- hES Human embryonic stem cells
- numerous problems continue to plague their use as therapeutics, including the risk of teratoma formation and the need for powerful immunosuppressive drugs to overcome the problems with immune rejection, For example, Wang, et al. (2010) Transplantation describes a study where mouse embryonic stem cells were differentiated into RPE cells and then
- GTP Current Good Manufacturing Practices
- GTP Current Good Tissue Practices
- Patents and applications owned by the assignee of the present applications have disclosed methods of producing RPE cells through differentiation of embryonic stem cells in U.S. Patents 7,795,025, 7,794,704, and 7,736,896, U.S. Ser. No. 12/682,712, as well as PCT Application no. PCT US 10/57056, filed Nov. 17, 2010 (now published as WO 201 1 /063005) and entitled "Methods of Producing Human RPE Cells and Pharmaceutical Preparations of Human RPE Cells" (Attorney Docket No. 75820.001020), and U.S. Provisional Patent Application No. 61/262,002, filed November 17, 2009 (Attorney Docket No, 75820.001000), each of which is hereby incorporated by reference in its entirety.
- the RPE cells and pharmaceutically preparations comprising RPE cells produced by the methods described herein may be used for cell-based treatments.
- the invention provides methods for treating a condition involving retinal degeneration comprising administering an effecti ve amount of a pharmaceutical preparation comprising RPE cells, wherein the RPE cells are derived from pluripotent stem cells in vitro.
- Conditions involving retinal degeneration include, for example, choroideremia, diabetic retinopathy, retinal atrophy, retinal detachment, retinal dysplasia, and retinitis pigmentosa.
- the RPE cells described herein may also be used in methods for treating macular degeneration including but are not limited to age related macular degeneration (dry or wet).
- RPE cells described herein may also be used in methods of treating Parkinson's disease (PD).
- PD Parkinson's disease
- a common feature of cell transplantation is low graft survival, for example, in many ceil transplantation studies there tends to be a loss of cells immediately following transplantation (e.g. , within the first week). This loss of cells does not appear to be due to rejection of the transplanted cells but rather an inability of a certain percentage of the cells to be retained at the transplant site. This lack of cell retention is most likely due to a number of factors such as the failure of the cells to attach to an underlying structure, a lack of sufficient nutrients, or physical stresses at the transplant site. Following this initial drop-off of cell number, the cell survival at various time after transplantation can vary considerably from study to study. Thus, although some studies show a steady decline in numbers, other show results where the grafted cells can reach a stable number. However, an important factor in considering the success of a transplantation is the percentage of recipients with surviving grafts following cell transplant.
- the RPE cells in the pharmaceutical preparations described herein may survive long term following transplantation.
- the RPE cells may survive at least about 1 , 2, 3, 4. 5, 6, 7, 8, 9, or 10 days.
- the RPE cells may survive at least about 1 , 2, 3. 4, 5, 6, 7, 8, 9, or 10 weeks; at least about 1 , 2, 3, 4, 5, 6, 7, 8, 9, or 10 months; or at least about 1 , 2, 3, 4, 5, 6, 7, 8, 9, or 10 years.
- the RPE cells may survive throughout the lifespan of the receipt of the transplant.
- At least 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, 96, 97, 98, 99, or 100% of the receipts of RPE cells described herein may show survival of the transplanted RPE cells. Further, the RPE cells described herein may successfully incorporate into the RPE layer in the transplantation receipt, forming a semi- continuous line of cells and retain expression of key RPE molecular markers (e.g. , RPE65 and 2
- the RPE cells described herein may also attach to the Bruch's membrane, forming a stable RPE layer in the transplantation receipt, Also, the RPE ce!Ls described herein are substantially free of ES cells and the transplantation receipts doe not show abnormal growth or tumor formation at the transplantation site.
- the methods of treating a patient suffering from a condition associated with retinal degeneration may comprise administering a composition of the invention locally (e.g. , by intraocular injection or insertion of a matrix comprising the pharmaceutical preparation of the invention), intraocular administration of pharmaceutical preparation of the invention include, for example, delivery into the vitreous body, transcorneally, sub-conjunctival, jiixiascieral, posterior scleral, and sub-tenon portions of the eye. See, for example, U.S. Patent Nos, 7,794.704;
- the invention also provides a method of administering human RPE cells that have been derived from reduced-complexity embryonic stem cells to a patient.
- This method may comprise: (a) identifying a patient that needs treatment involving administering human RPE cells to him or her; (b) identifying MHC proteins expressed on the surface of the patient's cells;
- step (c) providing a library of human RPE cells of reduced MHC complexity made by the method for producing RPE cells of the present invention; (d) selecting the RPE cells from the library that match this patient's MHC proteins on his or her cells; (e) administering any of the cells from step (d) to said patient.
- This method may be performed in a regional center, such as, for example, a hospital, a clinic, a physician's office, and other health care facilities. Further, the RPE cells selected as a match for the patient, if stored in small cell numbers, may be expanded prior to patient treatment.
- the RPE cells may be cultured under conditions to increase the expression of alpha integrin subunits 1-6 or 9 as compared to uncultured RPE cells or other RPE cell preparations prior to transplantation.
- the RPE cells described herein may be cultured to elevate the expression level of alpha integrin subunits 1 , 2, 3, 4, 5, 6, or 9.
- the RPE cells described herein may be cultured under conditions that promote the expression of alpha integrin subunits 1 -6.
- the RPE cells may be cultured with imegrin-activating agents including but not limited to manganese and the activating monoclonal antibody (mAb) TS2/ 16. See Afshari, et al. Brain (2010) 133(2): 448-464.
- the particular treatment regimen, route of administration, and adjuvant therapy may be tailored based on the particular condition, the severity of the condition, and the patient's overall health.
- Administration of the pharmaceutical preparations comprising RPE cells may be effective to reduce the severity of the symptoms and/or to prevent further degeneration in the patient's condition.
- administration of a pharmaceutical preparation comprising RPE cells may improve the patient's visual acuity.
- administration of the RPE cells may be effective to fully restore any vision loss or other symptoms.
- the RPE cell administration may treat the symptoms of injuries to the endogenous RPE layer.
- the RPE cells may be formulated with a pharmaceutically acceptable carrier.
- RPE cells may be administered alone or as a component of a pharmaceutical formulation.
- the subject compounds may be formulated for administration in any convenient way for use in medicine.
- Pharmaceutical preparations suitable for administration may comprise the RPE cells, in combination with one or more pharmaceutically acceptable sterile isotonic aqueous or nonaqueous solutions (e.g. , balanced salt solution (BSS)), dispersions, suspensions or emulsions, or sterile powders which may be reconstituted into sterile injectable solutions or dispersions just prior to use, which may contain antioxidants, buffers, bacteriostats, solutes or suspending or thickening agents.
- sterile isotonic aqueous or nonaqueous solutions e.g. , balanced salt solution (BSS)
- dispersions suspensions or emulsions
- sterile powders which may be reconstituted into sterile injectable solutions or dispersions just prior to use
- the pharmaceutical preparations for use in this invention may be in a pyrogen-free, physiologically acceptable form.
- the preparation comprising RPE cells used in the methods described herein may be transplanted in a suspension, gel, colloid, slurry, or mixture. Further, the preparation may desirably be encapsulated or injected in a viscous form into the vitreous humor for delivery to the site of retinal or choroidal damage. Also, at the time of injection, cryoprescrved RPE cells may be may be resuspended with commercially available balanced salt solution to achieve the desired osmolality and concentration for administration by subretinal injection.
- the RPE cells of the invention may be delivered in a pharmaceutically acceptable ophthalmic formulation by intraocular injection.
- the solution When administering the formulation by jntravitreal injection, for example, the solution may be concentrated so that minimized volumes may be delivered. Concentrations for injections may be at any amount that is effective and nontoxic, depending upon the factors described herein.
- the pharmaceutical preparations of RPE cells for treatment of a patient may be formulated at doses of at least about 10 4 cells/mL.
- the RPE cell preparations for treatment of a patient are formulated at doses of at least about 1G ⁇ lO 4 , 10 5 , 10 6 , ID 7 , 10 s , 10% or 10 10 RPE cells/mL.
- the RPE ceils may be formulated in a pharmaceutically acceptable carrier or excipient.
- the pharmaceutical preparations of RPE ceils described herein may comprise at least about 1 ,000; 2,000; 3,000; 4,000; 5,000; 6,000; 7,000; 8,000; or 9,000 RPE cells.
- the pharmaceutical preparations of RPE cells may comprise at least about IxlO 4 , 2x 1(T, 3x10 "* , 4x 10 4 , 5x 10 4 , 6x 10 4 , 7x 10 4 .8x10 4 , 9x 10 4 , 1 x 10 5 , 2x 10 5 , 3x 10 5 , 4x 10 5 , 5x 10 5 , 6x 10 5 , 7x 10 5 , 8xl0 5 , 9x1 ⁇ IxlO 6 , 2x10°, 3xl0 6 , 4xl0 6 , 5 l0 6 , 6xl0 6 , 7xl0 6 , 8xl0 6 , 9xl0 6 , IxlO 7 , 2 l0 7 , 3x
- the pharmaceutical preparations of RPE cells may comprise at least about Ixl0 2 - ⁇ l l0 ⁇ Ixl0 2 -lxl0 4 , lxl0 4 -lx 10 ⁇ or l l0 3 -lxl.0 6 RPE cells.
- the pharmaceutical preparations of RPE cells may comprise at least about 10,000, 20,000, 25,000, 50,000, 75,000, 100,000, 125,000, 150,000, 175,000, 180,000, 185,000, 190,000, or 200,000 RPE cells.
- the pharmaceutical preparation of RPE cells may comprise at least about 20.000-200,000 RPE cells in a volume at least about 50-200 ⁇ L ⁇ .
- the pharmaceutical preparation of RPE cells may comprise at least, about 180,000 RPE cells in a volume at least about 150 ⁇ .
- RPE cells may be formulated for delivery in a pharmaceutically acceptable ophthalmic vehicle, such that the preparation is maintained in contact with the ocular surface for a sufficient time period to allow the cells to penetrate the affected regions of the eye, as for example, the anterior chamber, posterior chamber, vitreous body, aqueous humor, vitreous humor, cornea, iris/ciliary, lens, choroid, retina, sclera, suprachoridal space, conjunctiva, subconjunctival space, episcleral space, intracorneal space, epicorneal space, pars plana, surgically-induced avascular regions, or the macula.
- a pharmaceutically acceptable ophthalmic vehicle such that the preparation is maintained in contact with the ocular surface for a sufficient time period to allow the cells to penetrate the affected regions of the eye, as for example, the anterior chamber, posterior chamber, vitreous body, aqueous humor, vitreous humor, cornea, iris/ciliary, lens, choroid, retina, sclera
- the volume of preparation administered according to the methods described herein may dependent on factors such as the mode of administration, number of RPE cells, age and weight of the patient, and type and severity of the disease being treated. If administered by injection, the volume of a pharmaceutical preparations of RPE cells of the invention may be from at least about 1, .1.5, 2, 2.5, 3, 4, or 5 mL. The volume may be at least about 1-2 rriL.
- the volume of a pharmaceutical preparations of RPE ceils of the invention may be at least about 1,2,3,4,5,6,7, 8, 9.10, II, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31.32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67 ,68, 69, 70, 71,72, 73, 74, 75, 76, 77, 78, 79, 80.81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 1, 92.93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108,
- the volume of a preparation of the invention may be from at least about 10-50, 20-50, 25-50, or 1-200 ⁇ _.
- the volume of a preparation of the invention may be at least about 10, 20, 30, 40, 50, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, or 200 ⁇ .
- the preparation may comprise at least about 1x10 ' , 2x10 " , 3x10 " , 4x10 " , 8x10', 9x10 ; , 3xl0 4 , 2xl0 4 , 3xl0 4 , 4x10 4 , 5xl0 4 , 6xl0 4 , 7xl0 4 , 8xl0 4 , or 9 l0 4 RPE cells per ⁇ _.
- the preparation may comprise 2000 RPE cells per ⁇ . for example, 100,000 RPE cells per 50 iiL or 180,000 RPE cells per 90 ⁇ _.
- the method of treating retinal degeneration may further comprise administration of an immunosuppressant, immunosuppressants that may be used include but are not limited to anti-lymphocyte globulin (ALG) polyclonal antibody, anti- thymocyte globulin (ATG) polyclonal antibody, azathioprine, BASILIX1MAB® (anti-IL-2Rcc receptor antibody), cyclosporin
- cyclosporin A cyclosporin A
- DACLIZUMAB® anti-IL-2Ru receptor antibody
- everolimus mycophenolic acid
- RITUXIMAB® anti-CD20 antibody
- sirolimus sirolimus
- tacrolimus tacrolimus.
- immunosuppressants may be dosed at least about. I, 2, 4, 5, 6, 7, 8, 9, or 10 mg/kg. When immunosuppressants are used, they may be administered systemically or locally, and they may be administered prior to, concomitantly with, or following administration of the RPE cells.
- Immunosuppressive therapy continues for weeks, months, years, or indefinitely following administration of RPE cells.
- the patient may be administered 5 mg/kg cyclosporin for 6 weeks following administration of the RPE cells.
- the method of treatment of retinal degeneration may comprise the administration of a single dose of RPE ceils. Also, the methods of treatment described herein may comprise a course of therapy where RPE cells are administered multiple times over some period,
- Exemplary courses of treatment may comprise weekly, biweekly, monthly, quarterly, biannually, or yearly treatments.
- treatment may proceed in phases whereby multiple doses are required initially (e.g. , daily doses for the first week), and subsequently fewer and less frequent doses are needed.
- the RPE cells may be delivered one or more times periodically throughout the li e of a patient.
- the RPE cells may be delivered once per year, once every 6-12 months, once every 3-6 months, once every 1-3 months, or once every 1-4 weeks.
- more frequent administration may be desirable for certain conditions or disorders.
- the RPE cells may be administered one time, or one or more times periodically throughout the lifetime of the patient, as necessary for the particular patient and disorder or condition being treated. Simi larly contemplated is a therapeutic regimen that changes over time. For example, more frequent treatment may be needed at the outset (e.g. , daily or weekly treatment). Over time, as the patient's condition improves, less frequent treatment or even no further treatment may be needed,
- the methods described herein may further comprises the step of monitoring the efficacy of treatment or prevention by measuring electroretinograrn responses, optomotor acuity threshold, or luminance threshold in the subject.
- the method may also comprise monitoring the efficacy of treatment or prevention by monitoring immunogenicity of the cells or migration of the cells in the eye.
- the RPE cells may be used in the manufacture of a medicament to treat retinal degeneration.
- the invention also encompasses the use of the preparation comprising RPE cells in the treatment of blindness.
- the preparations comprising human RPE cells may used to treat retinal degeneration associated with a number of vision-altering ailments that result in photoreceptor damage and blindness, such as, diabetic retinopathy, macular degeneration (including age-related macular degeneration, e.g. , wet age-related macular degeneration and dry age-related macular degeneration), retinitis pigmentosa, and Siargardt ' s Disease (fundus flavimaculatus).
- the preparation may comprise at least about 5,000-500,000 RPE celis ⁇ e.g., 100,00 RPE cells) which may be administered to the retina to treat retinal degeneration associated with a number of vision-altering ailments that result in photoreceptor damage and blindness, such as, diabetic retinopathy, macular degeneration (including age-related macular degeneration), retinitis pigmentosa, and Stargardt's Disease (fundus flavimaculatus).
- the RPE cells provided herein may be human RPE cells.
- the human cells may be used in human patients, as well as in animal models or animal patients.
- the human celis may be tested in mouse, rat, cat, dog, or non-human primate models of retinal degeneration.
- the human celis may be used therapeutically to treat animals in need thereof, such as in veterinary medicine.
- the pharmaceutical preparation may be formulated in a pharmaceutically acceptable carrier according to the route of administration.
- the preparation may be formulated to be administered to the subretinal space of the eye.
- the preparation comprising RPE cells may be administered to one eye or both eyes in the same patient. The administration to both eyes may be sequential or simultaneous.
- the preparation comprising RPE cells may be formulated as a suspension, solution, slurry, gel, or colloid.
- RPE cells of the invention may be administered locally by injection ⁇ e.g. , i niravitreal injection), or as part of a device or implant (e.g. , an implant).
- the preparation may be administered by injection into the subretinal space of the eye.
- the preparation may be administered transcorneally.
- the cells of the present invention may be transplanted into the subretinal space by using vitrectomy surgery.
- RPE cells may be may be resuspended with commercially available balanced salt solution to achieve the desired osmolality and concentration for administration by subretinal i njection.
- the RPE cells may be added to buffered and electrolyte balanced aqueous solutions, buffered and eiectrolyte balanced aqueous solutions with a lubricating polymer, mineral oil or petrolatum-based ointment, other oils, liposomes, cylcodextrins. sustained release polymers or gels.
- a lubricating polymer mineral oil or petrolatum-based ointment, other oils, liposomes, cylcodextrins. sustained release polymers or gels.
- the methods described herein may comprise a step of administering RPE cells of the invention as an implant or device.
- the device is bioerodibie implant for treating a medical condition of the eye comprising an active agent dispersed within a biodegradable polymer matrix, wherein at least about 75% of the particles of the active agent have a diameter of less than about 10 ⁇ .
- the bioerodibie implant is sized for implantation in an ocular region.
- the ocular region may be any one or more of the anterior chamber, the posterior chamber, the vitreous cavity, the choroid, the suprachoroidal space, the conjunctiva, the subconjunctival space, the episcleral space, the intracorneal space, the epicorneal space, the sclera, the pars plana, surgically-induced avascular regions, the macula, and the retina.
- the biodegradable polymer may be, for example, a poly(lactic-co-glycolic)acid (PLGA) copolymer, biodegradable poly(DL-lactic-co-glycolic acid) films, or PLLA/PLGA polymer substrates.
- the ratio of lactic to glycolic acid monomers in the polymer is about 25/75, 40/60, 50/50, 60/40, 75/25 weight percentage, more preferably about 50/50.
- the PLGA copolymer may be about 20, 30, 40, 50, 60, 70, 80 to about 90 percent by weight of the bioerodibie implant.
- the PLGA copolymer may be from about 30 to about 50 percent by weight, preferably about 40 percent by weight of the bioerodibie implant.
- the RPE cells may be transplanted in conjunction with a biocompatible polymer such as poly lactic acid, po i y ( I ae t ie - o-gl ⁇ col i c acid), 50:50 PDLGA, 85: 1 PDLGA, and INION GTR® biodegradable membrane (mixture of biocompatible polymers).
- a biocompatible polymer such as poly lactic acid, po i y ( I ae t ie - o-gl ⁇ col i c acid), 50:50 PDLGA, 85: 1 PDLGA, and INION GTR® biodegradable membrane (mixture of biocompatible polymers). See U.S. Patent No. 6,331 ,313; 7,462,471 ; and 7,625,582.
- the invention provides a method for screening to identify agents that modulate RPE cell maturity.
- RPE ceils differentiated from human ES cells may be used to screen for agents that promote RPE maturation, identified agents may be used, alone or in combination with RPE cells, as part of a treatment regimen. Alternatively, identified agents may be used as part of a culture method to improve the survival of RPE cells differentiated in vitro.
- the RPE cells may be used a research too! in settings such as a pharmaceutical, chemical, or biotechnology company, a hospital, or an academic or research institution.
- Such uses include the use of RPE cells differentiated from embryonic stem cells in screening assays to identify, for example, agents that may be u ed to promote RPE survival in vitro or in vivo, or that may be used to promote RPE maturation, identified agents may be studied in vitro or in animal models to evaluate, for example, their potential use alone or in combination with RPE cells.
- the invention provides a method for identifying agents that promote RPE maturation comprising providing a RPE cell, contacting said RPE cell with an agent, assessing said RPE cell for signs of maturity, and then identifying an agent that promotes RPE maturation when said agent causes RPE cell to show signs of maturity.
- the signs of maturity may be pigmentation level, gene expression levels, and morphology as discussed herein.
- Certain aspects of the present invention pertain to the production of RPE cells to reach commercial quantities.
- the RPE cells may be produced on a large scale, stored if necessary, and supplied to hospitals, clinicians or other healthcare facilities.
- RPE cells may be harvested, purified, and optionally stored prior to a patient's treatment.
- RPE cells may optionally be patient specific or specifically selected based on HLA or other immunologic profile. For example, once a patient presents with an indication such as. for example, diabetic retinopathy, macular degeneration (including age-related macular degeneration), retinitis pigmentosa, retinal atrophy, retinal detachment, retinal dysplasia, and Stargardt's Disease (fundus flavimaculatus).
- RPE ceils may be ordered and provided in a timely- manner.
- the present invention relates to methods of producing RPE cells to attain cells on a commercial scale, cell preparations comprising RPE cells derived from said methods, as well as methods of providing (i.e. , producing, optionally storing, and selling) RPE cells to hospitals and clinicians.
- the production of differentiated RPE cells or mature differentiated RPE cells may be scaled up for commercial use.
- the present invention aiso provides for methods of conducting a pharmaceutical business comprising establishi ng a distribution system for distributing the preparation for sale or may include establishing a sales group for marketing the pharmaceutical preparation.
- the present invention provides methods of supplying RPE cells to hospitals, healthcare centers, and clinicians, whereby RPE cells produced by the methods disclosed herein are stored, ordered on demand by a hospital, healthcare center, or clinician, and administered to a patient in need of RPE cell therapy.
- a hospital, healthcare center, or clinician orders RPE cells based on patient specific data, RPE cells are produced according to the patient's specifications and subsequently supplied to the hospital or clinician placing the order. For example, after a particular RPE cell preparation is chosen to be suitable for a patient, it is thereafter expanded to reach appropriate quantities for patient treatment.
- the invention provides a method of conducting a pharmaceutical business, comprising the step of providing RPE cell preparations that are homozygous for at least one histocompatibility antigen, wherein cells are chosen from a bank of such cells comprising a library of RPE cells that may be expanded by the methods disclosed herein, wherein each RPE cell preparation is hemizygous or homozygous for at least one MHC allele present in the human population, and wherein said bank of RPE cells comprises cells that are each hemizygous or homozygous for a different set of MHC alleles relative to the other members in the bank of cells.
- heterozygosity may be used to generate the hemizygous or homozygous MHC allele stem cells used to derive the RPE cells.
- the present invention a!so includes methods of obtaining human ES cells from a patient and then generating and expanding RPE cells derived from the ES cel ls. These RPE cells may be stored. In addition, these RPE cells may be used to treat the patient from which the ES were obtained or a relative of that patient.
- human RPE cells may be reliably differentiated and expanded from human ES cells under well-defined and reproducible conditions— representing an inexhaustible source of cells for patients with retinal degenerative disorders.
- concentration of these cells would not be limited by availability, but rather could be titrated to the precise clinical requirements of the individual. Repeated infusion or transplantation of the same cell population over the lifetime of the patient would also be possible if deemed necessary by the physician.
- the abi ity to create banks of matching or reduced-complexity H I .
- a hES lines from which RPE cells could be produced could potentially reduce or eliminate the need for immunosuppressive drugs and/or immunomodulatory protocols altogether.
- Effective amount refers broadly to the amouni of a compound or cells that, when administered to a patient for treating a disease, is sufficient to effect such treatment for the disease.
- the effective amount may be an amount effective for prophylaxis, and/or an amount effective for prevention.
- the effective amount may be an amount effective to reduce, an amount effective to prevent the incidence of signs/symptoms, to reduce the severity of the incidence of signs/symptoms, to eliminate the incidence of signs/symptoms, to slow the development of the incidence of signs/symptoms, to prevent the de velopment of the incidence of signs/symptoms, and/or effect prophylaxis of the incidence of signs/symptoms.
- the "effective amount” may vary depending on the disease and its severity and the age, weight, medical history. susceptibility, and preexisting conditions, of the patient to be treated.
- the term "effective amount '* is synonymous with "therapeutically effective amount” for purposes of this invention.
- Embryo or “embryonic,” as used herein refers broadly to a developi ng cell mass that has not implanted into the uterine membrane of a maternal host.
- An “embryonic cell” is a cell isolated from or contained in an embryo. This also includes blastomeres, obtained as early as the two-cell stage, and aggregated blastomeres.
- Embryonic stem cells refers broadly to cells derived from the inner cell mass of blastocysts or morulae that have been serially passaged as cell lines.
- the ES cells may be derived from fertilization of an egg cell with sperm or DNA, nuclear transfer, parthenogenesis, or by means to generate ES cells with homozygosity in the HLA region.
- ES cells may also refer to cells derived from a zygote, blastomeres, or blastocyst-staged mammalian embryo produced by the fusion of a sperm and egg cell, nuclear transfer, parthenogenesis, or the reprogramming of chromatin and subsequent incorporation of the reprograrnrned chromatin into a plasma membrane to produce a cell.
- Embryonic stem cells regardless of their source or the particular method used to produce them, can be identified based on the: (i) ability to differentiate into cells of all three germ layers, (ii) expression of at least Oct- 4 and alkaline phosphatase, and (iii) ability to produce teratomas when transplanted into immunocompromised animals.
- the term also includes cells isolated from one or more blastomeres of an embryo, preferably without destroying the remainder of the embryo.
- the term also includes cells produced by somatic cell nuclear transfer, even when non-embryonic cells are used in the process.
- ES cells may be derived from fertilization of an egg cell with sperm or DNA, nuclear transfer, parthenogenesis, or by means to generate ES cells with homozygosity in the HLA region.
- ES cells are also cells derived from a zygote, blastomeres, or blastocyst-staged mammalian embryo produced by the fusion of a sperm and egg cell, nuclear transfer, parthenogenesis, or the reprogramming of chromatin and subsequent incorporation of the reprograrnrned chromatin into a plasma membrane to produce a cell.
- Human embryonic stem cells of the present invention may include, but are not limited to, MA01 , MA09, ACT-4, No. 3, H i , H7, H9, H14 and ACT30 embryonic stem cells, in certain embodiments, human ES cells used to produce RPE cells are derived and maintained in accordance with GMP standards.
- EDC Embryo-derived ceils
- blastocyst-dcrived cells include those of the inner cell mass, embryonic shield, or epib!ast, or other pluri potent stem cells of the early embryo, including primitive endoderm. ectoderm, and mesoderm and their derivatives.
- EDC also including blastomeres and cell masses from aggregated single blastomeres or embryos from varying stages of development, but excludes human embryonic stem cells that have been passaged as cell lines,
- Macular degeneration refers broadly to diseases characterized by a progressive loss of central vision associated with abnormalities of Bruch's membrane, the neural retina, and the retinal pi ment epithelium. Macu lar degeneration diseases include but are not limited to age- related macular degeneration, North Carolina macular dystrophy, Sorsby's fundus dystrophy, Stargardi's disease, pattern dystrophy, Best disease, malattia leventinese, Doyne's honeycomb choroiditis, dominant dm sen. and radial drusen.
- Pluri potent stem cell refers broadly to a cell capable of prolonged or virtually indefinite proliferation in vitro while retaining their undifferentiated state, exhibiting a stable (preferably normal ) karyotype, and having the capacity to differentiate into all three germ layers (i.e., ectoderm, mesoderm and endoderm) under the appropriate conditions.
- Pluripotent embryonic stem cells refers broadly cells that: (a) are capable of inducing teratomas when transplanted in immunodeficient (SOD ) mice; (b) are capable of differentiating to cell types of all three germ layers (e.g., ectodermal, mesodermal, and endodermal cell types); and (c) express at least one molecular embryonic stem eel) markers (e.g., express Oct-4, alkaline phosphatase, SSEA 3 surface antigen, SSEA 4 surface antigen, NANOG, TRA 1 60, TRA 1 81 , SOX2, REX 1 ).
- SOD immunodeficient
- Exemplary pluripotent stem cells can be generated using, for example, methods known in the art.
- Exemplary pluripotent stern cells include embryonic stem cells derived from the I CM of blastocyst stage embryos, as well as embryonic stem cells derived from one or more blastomeres of a cleavage stage or morula stage embryo (optionally without destroying the remainder of the embryo).
- Such embryonic stem cells can be generated from embryonic material produced by fertilization or by asexual means, including somatic cel l nuclear transfer (SCNT). parthenogenesis, and androgenesis.
- SCNT somatic cel l nuclear transfer
- pluripotent stem cells include induced pluripotent stem cells (iPS cells) generated by reprogramming a somatic cell by expressing or inducing expression of a combination of factors (herein referred to as reprogramming factors), iPS cells can be generated using fetal, postnatal, newborn, juvenile, or adult somatic cells.
- factors that can be used to reprogram somatic cells to pluripotent stem cells include, for example, a combination of Oct4 (sometimes referred to as Oct 3/4), Sox2, c-Myc, and K!f4.
- factors thai can be used to reprogram somatic cells to pluripotent stem cells include, for example, a combination of Oct-4, Sox2, Nanog, and Lin28.
- somatic cells are reprogram med by expressing at least 2 reprogramming factors, at least three reprogrammin factors, or four reprogramming factors.
- additional reprogramming factors are identified and used alone or in combination with one or more known reprogramming factors to reprogram a somatic cell to a pluripotent stem cell.
- IPS ceils typically can be identified by expression of the same markers as embryonic stem cells, though a particular iPS cell line may vary in its expression profile.
- RPE cell differentiated from a pluripotent stem cell using a method of the invention.
- the term is used generically to refer to differentiated RPE cells, regardless of the level of maturity of the cells, and thus may encompass RPE cells of various levels of maturity.
- RPE cells can be visually recognized by their cobblestone morphology and the initial appearance of pigment.
- RPE cells can also be identified molecularly based on substantial lack of expression of embryonic stem cell markers such as Oct- 4 and NANOG, as well as based on the expression of RPE markers such as RPE-65. PEDF, CRALBP, and bestrophin.
- RPE cells refers to RPE cel ls differentiated in vitro from pluripotent stem cells.
- Mature RPE cell and “mature differentiated RPE cell,” as used herein, may be used interchangeably throughout to refer broadly to changes that occur following initial differentiating of RPE cells. Specifically, although RPE cells can be recognized, in part, based on initial appearance of pigment, after differentiation mature RPE ceils can be recognized based on enhanced pigmentation.
- Pigmented refers broadly to any level of pigmentation, for example, the pigmentation that initial occurs when RPE cells differentiate from ES cells.
- Pigmentation may vary with eel! density and the maturity of the differentiated RPE cells.
- the pigmentation of a RPE cel l may be the same as an average RPE cell after terminal di fferentiation of the RPE cell.
- the pigmentation of a RPE cell may be more pigmented than the average RPE cell after terminal differentiation of the RPE cel l.
- the pigmentation of a RPE cel l may be less pigmented than the average RPE cell after terminal differentiation.
- "Signs" of disease refers broadly to any abnormality indicative of disease, discoverable on examination of the patient; an objective indication of disease, in contrast to a symptom, which is a subjective indication of disease.
- Symptoms of disease refers broadly to any morbid phenomenon or departure from the normal in structure, function, or sensation, experienced by the patient and indicative of disease.
- “Therapy,” “therapeutic,” “treating,” or “treatment”, as used herein, refers broadly to treating a disease, arresting or reducing the development of the disease or its clinical symptoms, and/or relieving the disease, causing regression of the disease or its clinical symptoms.
- Therapy encompasses prophylaxis, prevention, treatment, cure, remedy, reduction, alleviation, and/or providing relief from a disease, signs, and/or symptoms of a disease. Therapy encompasses an alleviation of signs and/or symptoms in patients with ongoing disease signs and/or symptoms (e.g., blindness, retinal deterioration.) Therapy also encompasses
- Prophylaxis includes preventing disease occurring subsequent to treatment of a disease in a patient or reducing the incidence or severity of the disease in a patient.
- the term “reduced”, for purpose of therapy, refers broadly to the clinical significant reduction in signs and/or symptoms.
- Therapy includes treating relapses or recurrent signs and/or symptoms (e.g., retinal degeneration, loss of vision.) Therapy encompasses but is not limited to precluding the appearance of signs and/or symptoms anytime as well as reducing existing signs and/or symptoms and eliminating existing signs and/or symptoms.
- Therapy includes treating chronic disease (“maintenance") and acute disease.
- treatment includes treating or preventing relapses or the recurrence of signs and/or symptoms (e.g., blindness, retinal degeneration).
- hRPE retinal pigment epithelium
- This example describes an assay having extraordinar sensitivity for detection of pluripotent hES cells in a cell population. Using this assay, cells differentiated from hES cells were tested to determine whether any residual hES cells remained in the population. This assay can provide valuable safety information because any residual hES cells have the potential to form tumors upon implantation into a recipient.
- li is currently accepted in scientific literature that pluripotent hES cells (which can form teratomas in vivo) express a known set of molecular markers, and Oct-4 expression is critical for retention of pluripotency while ioss of this marker is associated with cellular differentiation. Any cell expressing this marker at a detectable level may be seen as pluripotent and thus undesired in the final product. A cell that has lost Oct-4 expression can be seen as no longer pluripotent. In this assay, cells were first examined for positive alkaline phosphatase staining under visible light, then examined for Oct-4 expression by immunofluorescence.
- hRPE Human retinal pigment epithelium
- the hRPE cells differentiated from the MA09 hES cell line which was originated from a single human blastomere as described in US Pat. No.
- hES cells were seeded into two different types of culture conditions: either RPE culture conditions (specifically, plating on gelatin coated 4- well plates in EGM@-2 (Lonza Biologies, Basel, Switzerland) medium according to the normal procedure for culturing hRPE cells), or hES cell culture conditions (conditions that support the growth and maintenance of hES cel ls which in this experiment were plating on MEF and using conditioned hES-GM ; see Table 2).
- the hES cells used were a GFP-positive hES cell line (HI or WA01 ). By using this GFP-positive hES cell line, it was possible to detect the presence of cells which have derived from the hES cells whether the cells remained pluripotent or differentiated.
- the cells were cultured to permit attachment to the substrate, then the cells were fixed with 2% paraformaldehyde, pcrmeabilized with 0.1 % NP-40 substitute (which may be done immediately or up to several weeks after fixation, e.g., after about 24 hours), and stained for alkaline phosphatase (AP) and Oct-4.
- AP was detected using VECTOR® Blue (Vector Laboratories) according to the manufacturer's instructions, which produces a stain that is detectable under visible light.
- Oct-4 was detected using immunofluorescence (using essentially the same methods as described in Example 7, below).
- FIG. 1 Representative photomicrographs are presented in FIG. 1 .
- the top panels show GFP-positive (left) and Oct-4 positive (right) cells mixed with RPE cells at a 1 : 10 ratio ( 10%).
- DAPI staining bottom left panel shows all cells present in the field (both RPE and hES) and bottom right panel shows alkaline phosphatase staining. Magnification x200.
- AP staining is detectable under visible light, as primary screening because it allowed us to rapidly examine every cell in the population (200,000 cells in this example).
- Cells that were AP-positive were examined under ultraviolet light for Oct-4 staining and GFP expression.
- Cells positive for AP, Oct-4, and GFP are pluripotent, while cells that are GFP positive and Oct-4 negative are differentiated progeny of pluripotent cells.
- Some AP positive cells were GFP negative and Oct-4 negative, indicating that AP can be used to rapidly identify potential hES cells, and those cells can then be examined for a second hES cell marker (Oct-4 in this example) to identify hES cells.
- alkaline phosphatase was more sensitive that Oct-4 allowing the detection as few 5 cells in approximately 178.000.
- alkaline phosphatase assay was chosen as the primary screening method.
- AP is not a unique marker of hES cells, its expression identifies potential hES cells that can be confirmed with a more definitive marker, like Oct-4. False AP positives were seen (GFP or Oct-4 negative), and Oct-4 is used as a confirmatory assay.
- GFP or Oct-4 negative
- Oct-4 is used as a confirmatory assay.
- With 0.001 % hES cells a total of 5 ceils in the three wells stained positive for alkaline phosphatase and were GFP positive.
- the limit of detection for the immunofluorcscent detection of hES cells in RPE cell cultures was concluded to be 5/600,000 cell (0.0008%).
- This example describes the results of an in vitro spiking study, its purpose was to show that all added hESC, even at high percentage, are differentiating under RPE culture conditions over a course of 6 weeks or 2 passages - which is less than the time separating RPE at harvest for cryoprcservation from hESC or differentiated EB .
- Cultures contained mixtures of hESC and RPE, with 1 %, 10%, or 20% hES cells.
- the ceil populations were evaluated by staining and Q-P R at 3 eeks (p i ) and 6 weeks (p2).
- the EGM-2 medium is conducive for the proliferation of hRPE cells.
- the 4-well control plates were fixed with 2% paraformaldehyde or harvested (for qPCR analysis) on Day 3, to assess the initial appearance of these cultures.
- the remaining 6- well plates were cultured until con fluency at which time the medium was changed to MDBK-MM that is utilized for the maintenance of hRPE cells. After approximately 3 weeks, the cultures were either fixed or harvested (for qPCR) or passaged into 6-well plates or 4-well plates.
- the plates were cultured for an additional 3 weeks before they were either fixed or harvested for qPCR. This culturing simulated the manufacturing process from the isolation of the differentiated RPE colonies to the final product.
- hES ceils added to the cultures were MA09 cells.
- the fixed plates were stained for hES cell marker, Oet-4 using indirect immunofluorescence followed with Alexa-conj ugated secondary antibodies (identification as a red emitted light) and DAP1 for visualization of the nuclei and examined under an inverted fluorescent microscope.
- Alkaline phosphatase activity was determined on the same plates using VECTOR® Blue kit (Vector Laboratories, Burlingame. CA). Additionally, live cultures were examined under the phase contrast microscope or usin Hoffman Modulation Optics (HMC) for irregularities in the monolayer.
- HMC Hoffman Modulation Optics
- the hES cells added to the cultures were H l -GFPp cells which constitutively express GFP. These hES cells and their progeny are readily detectable due to their GFP expression, permitting detection of the ES cells or their during the entire culturing period regardless of whether they remained pluripotent or differentiated.
- q C was performed using the TaqM Gene Expression Assay (Applied Biosystems) assaying for expression of Oct-4, Nanog, Rex- 1 and Sox2. Beta-actin gene expression was used for normalization.
- Study 3 was identical to study 2, except that the MA09- PE cells underwent an additional passage in culture before being mixed with the hES H l -GFPp cells.
- hES cells behaved in a typical embryonic stem cell ma ner, i.e. they differentiated, forming three-dimensional structures with some free-floating embryoid body-like aggregates.
- pure RPE cultures demonstrated typical for RPE growth behavior, i.e. they became elongated and less pigmented during the proliferative phase and regained epithelial polygonal pigmented appearance after the monolayer was established and the medium was changed to MDBK-MM.
- Tables 4 through 8 summarize the morphological observations and staining results for these cultures.
- - means undetectable
- ? means signal detectable but not convincingly specific
- + means signal detectable
- ++ means signal very strong
- +++ means signal strong and specific.
- hESC and their derivatives are detectable by microscopic observation in both early and late cultures of RPE cells even when added at 1 % concentration. Colonies of hES cells (more or less differentiated) are visible at day 3 when RPE cells are still flat and under-confluent. After three weeks in culture, microscopic observation reveals large cell masses derived from hESC that are very di ferent from RPE morphology and easily noticeable.
- Oct-4 staining is detectable at day 3. in 1 % hESC cultures, Oct-4 positive DC!s were more difficult to detect but i n 10% and 20% these cells were readily detected.
- GFP-positive cells were present in all examined samples and their morphology was indicative of various ES cell derivatives but did not resemble ES cells.
- phosphatase at this stage the cells are under-confluent and grow as a monolayer and have not deposited too much of extracellular matrix which creates background, so presence of any single ceil or small cluster should be easy to detect.
- a sample size of at least 200,000 cells, preferably in triplicates would give greater sensitivity.
- hES cells when grown under the conditions that do not support their pluripotent state (such as on feeder cells, defined matrix, or defined media) differentiate very quickly. This differentiation is associated with the loss of molecular markers of pluripotency, such as Oct-4 and alkaline phosphatase.
- RPE culture conditions do not support undifferentiated growth of hES cells, therefore it was postulated (and confirmed by these experiments) that at the time when the mi ture of initially pluripotent hES cells with RPE cells is fixed, many of hES cells would already have lost or down regulated Oct-4 expression and thus become undetectable.
- the cells were either cultured in hES cell culture media on mouse embryonic fibroblast feeder cells (MEFs), or in endothelial growth media supplemented with 2% FES (EGM-2) on gelatin.
- EGM-2 is a medium that is conducive to RPE proliferation but does not maintain the pluripotent state of hES cells.
- the cells were cultured for only 16 hours before being fixed. The cells were then stained for alkaline phosphatase and Oct-4 and microscopically examined as described in Example 2. For the cel l populations plated in ES cell media on feeder cells, about 80% of the GFP-positive ceils were positive for AP and Oct-4.
- This example shows the relative insensitivity of real-time PGR for detecting rare cell sub-populations.
- the qRT-PCR LOD was assessed using mixtures of MA09-RPE cells and MA09 hES cells. Cryopreserved MA09-RPE cells were thawed and counted, and hES MA09 were also thawed and counted (using alkaline phosphatase staining to determine the number o pluripotent hES cells present). Mixtures were then formed of 400,000 cells with 100%, 10%, 1 %, 0.1 %, 0.01 %, or 0% of the cells being hES cells and the remainder being MA09-RPE.
- the RNeasy RNA isolation kit from Qiagen was used to extract R.N A from the cell mixtures resulting in a final volume of 30 ⁇ RNA per sample.
- cDNA was then synthesized from 10 ⁇ . of R A with the Quantitect cDNA synthesis kit from Qiagen resulting in a final volume of 20 ⁇ cDNA.
- One ⁇ of cDNA was then tested for relative gene expression in triplicate replicates normalized to the beta actin signal present in each sample.
- Gene expression analysis was performed using the Applied Biosystems StepOne Plus with software version 2.1 and TaqMan gene expression assays from Li fe Technologies following the manufacturer's recommended cycle conditions for comparative Ct relative quantification .
- FIG. 2A-C graphically illustrates the relative gene expression (detected by qRT- PCR ) for (A) Oct-4; (B) Nanog; and (C) SOX2. Statistically significant differences between the 2011/045232
- This example further demonstrates the extraordinarily sensitivity of detection of rare hES cells within a population of RPE cells using staining methods.
- These experiments measure the sensitivity of an assay for detecting undifferentiated (piuripotent ) hESC in a population of RPE.
- RPE were spiked with defined, relatively low numbers of hESC (both H 1 and MA09 in different experiments) onto MEF in hESC medium for minimal time th t allows the cells to attach and spread, so they remain adherent during immunostaining procedure.
- These conditions (MEF, conditioned hESC-GM medium, minimal exposure to RPE) preserved pluripotency of hESC. After 14- 36 h the plates were fixed and stained, then examined.
- Nuclei per field were counted in several random fields for total cell number per well examined. Because AP staining is expressed in other cell types in addition to piuripotent cells. AP ⁇ /Oct4- cells were likely originated from MEF; thus, each AP-positive cell was examined for Oct-4, and double- positive cells were counted as hES cells. As determined by comparison of Oct-4 and GFP, there was a small loss of piuripotent cells to differentiation, but under these optimized for hESC conditions this loss is minima) (the loss was much higher without MEF and in RPE medium).
- the cultures were then stained with an anti-human nuclei (anti-HuNu) antibody to permit detection of the human cells on top of the MEFs and remaining undifferentiated hES cells were counted.
- anti-HuNu anti-human nuclei
- These measurements determined how many undifferentiated hES cells would remain when plated in low density but under the best culture conditions (plating in hES medium on MEFs).
- the number of undifferentiated hES cells detected in these control plates were used as the denominator when calculating the percentage of undifferentiated cells that were detected. When 5 hES cells per 600,000 were introduced as calculated, at least two double-positive were detected. When such low numbers of contaminating cells are introduced, "noise" effects can increase in prominence, as cell loss during manipulations such as pipetting is constant and is not correlated with decreasing percentage of contaminating cells.
- the assay conditions were modified to incorporate the use of MEF feeder cells and hES conditioned growth medium to promote the persistence of hES pluripoteni markers during the course of the assay.
- AP was retained as a primary screening tool because the positive signal (blue cell staining) can be seen in a bright field, allowing the operator to remain in the plane of focus.
- scanning cells for fluorescence requires viewing cel ls under fluorescent light, which may result in the cells deviating from the plane of focus and may lead to missing a positive signal
- AP staining allows the operator to examine every cell among millions to pinpoint AP positive ceils for subsequent examination to confirm Oct-4 expression.
- a positive response was defined as a minimum of two double stained ceils in RPE preparations spiked at a calculated ratio of 1.6 MA09 hES cells for every 2 million RPE cell.
- the LOD for detecting contaminating hES is therefore at a level of 0.00008%. Note that this is an order of magnitude more sensitive than thai previously reported for the GFP- hES studies. At least two factors account for the enhanced sensitivity 1 ) the use of MEF and hES conditioned media to retain pi uri potency of the hES and 2 ⁇ increasing the number of cel l s inspected increases the sensitivity of the assay.
- Target cells are detected using a first antibody coupled directly or indirectly to Alkaline Phosphatase (AP) and a second antibody coupled to a fluorescent label.
- AP Alkaline Phosphatase
- This method may be used to detect rare cells of a type that do not express AP (or insufficiently express AP that are insufficient for robust detection). Additionally, this method may be used to detect rare cells that endogenously express AP (e.g., ES ceils), in which instance the antibody coupled to AP would be expected to increase AP staining intensity.
- the first and second antibodies specifically bind to different markers of the rare cell type.
- the antibodies specifically bind two different ES cell markers such as alkaline phosphatase, Oct-4, Nanog, Stage-specific embryonic antigen-3 (SSEA-3), Stage-specific embryonic antigen- 4 (SSEA-4 ), TRA-1 - ⁇ , TRA- 1 -81 .
- TRA-2-49/6E Stage-specific embryonic antigen-3
- SSEA-4 Stage-specific embryonic antigen- 4
- TRA-1 - ⁇ TRA- 1 -81
- TRA-2-49/6E Sox2, growth and differentiation factor 3 (GDF3), reduced expression 1 (REX I ), fibroblast growth factor 4 (FGF4), embryonic cell- specific gene 1 (ESG I ), developmental pluri potency-associated 2 ( DPPA2 ).
- GDF3 growth and differentiation factor 3
- REX I reduced expression 1
- FGF4 fibroblast growth factor 4
- ESG I embryonic cell- specific gene 1
- DPPA2 developmental pluri potency-associated 2
- DPPA4 telomerase reverse transcriptase
- SALL4 E-CADHERIN
- CD30 Cripto
- GCTM-2 GCTM-2
- Genesis Germ cell nuclear factor
- SCF Stem cell factor
- Respective coupling of the antibodies to AP and the fluorescent label may be direct or indirect, e.g., through a secondary antibody, actin/biotin affinity, and other methods of indirect coupling.
- the ceils are then stained for alkaline phosphatase activity to permit visualization of ceils that bound to the first antibody under visible light.
- the cell population is then examined under visible light as described in the preceding examples to identify AP-positive cells; AP-positive cells are then examined under ultraviolet light to detect cells that are also labeled by the second antibody. Cells that bound both the first and second antibody are identified as cells of the target type.
- the cell population is plated under conditions that favor growth and/or survival of the target cell type.
- the total number of cells examined is estimated, for example by counting the numbers of cells in random fields and scaling up to the total area counted, and a predetermi ed minimum number of cells may be examined to ensure a desired level of sensitivity.
- counting of the cell population may be performed in the presence of a label that distinguishes the cell population from the feeder cells (e.g., a species-specific antibody or lineage-specific antibody, where the feeder cells are of a different species or type than the cell population), which may be performed in a duplicate well.
- This example describes a further exemplary methods of detecting target cells within a population.
- the cell population is stained to detect two different target ceil markers using a first and second stain that are visually distinguishable from one another.
- the cells may be labeled with a first and second antibody, each coupled directly or indirectly to two different enzymes that catalyze reactions that give visible products (e.g. , selected from among alkaline phosphatase, beta galactosidase, and a peroxidase such as horse radish peroxidase ).
- the cells are then contacted with substrates of the two different enzymes to catalyze reactions that gives a visible product, wherein the substrates are chosen such that their products are different colors from one another.
- the products may be the same color as one another but chosen to have distinguishable staining patterns (e.g., one coupled to an antibody that stains nuclei and the other coupled to an antibody that stains the cytoplasmic membrane).
- one of the enzymes may be alkaline phosphatase and its substrate may be Vector Red or Vector Blue (producing red and blue products, respectively ), and the other enzyme may be a peroxidase and its substrate may be 3,3'-Diaminobenzidine (DAB) (producing a dark brown product).
- DAB 3,3'-Diaminobenzidine
- beta galactosidase with the substrate X-Gal (5-Bromo-4-chloro-3-indolyl ⁇ -D-ga!actopyranoside) (producing a blue product) which may readi ly be used together with peroxidasc/DAB (brown product) or
- AP/Vector Red red product
- Other suitable combinations of enzyme/substrate combinations that produce visually distinguishable products are readily identifiable through routine experimentation.
- An advantage of this method is that the rare cells may be detected using visible light only without the need for examination under fluorescent light. Additionally, if desired, a further fluorescent label may be utilized (e.g., coupled to an antibody to a third marker ) thereby permitting further verification of whether detected cells (i.e., labeled by the first and/or second label) by examination under ultraviolet light.
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Biomedical Technology (AREA)
- Cell Biology (AREA)
- Analytical Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Zoology (AREA)
- Physics & Mathematics (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Food Science & Technology (AREA)
- Tropical Medicine & Parasitology (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Wood Science & Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Virology (AREA)
- Biophysics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Investigating, Analyzing Materials By Fluorescence Or Luminescence (AREA)
- Sampling And Sample Adjustment (AREA)
- Investigating Or Analysing Materials By Optical Means (AREA)
- Materials For Medical Uses (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Abstract
Description


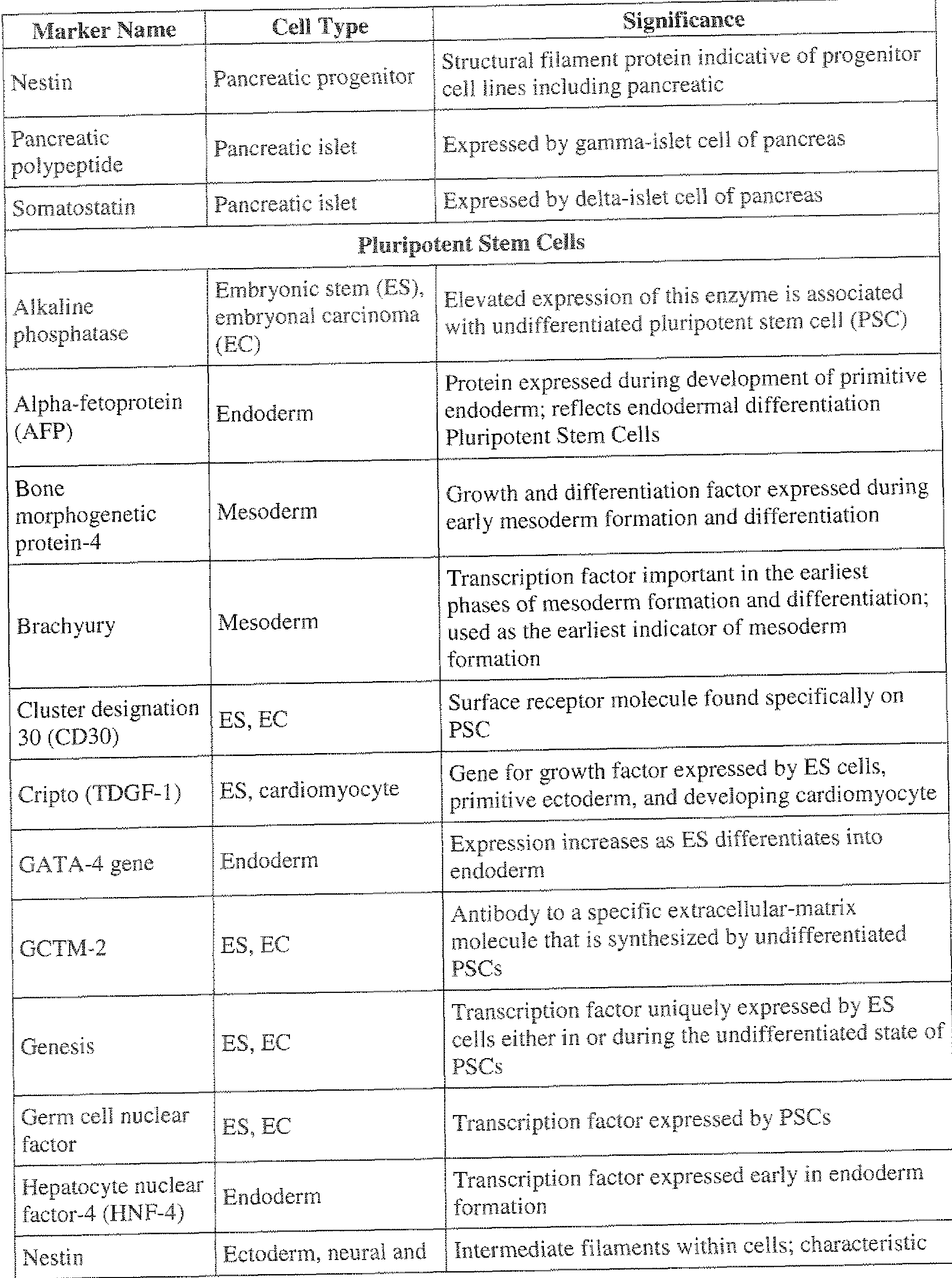
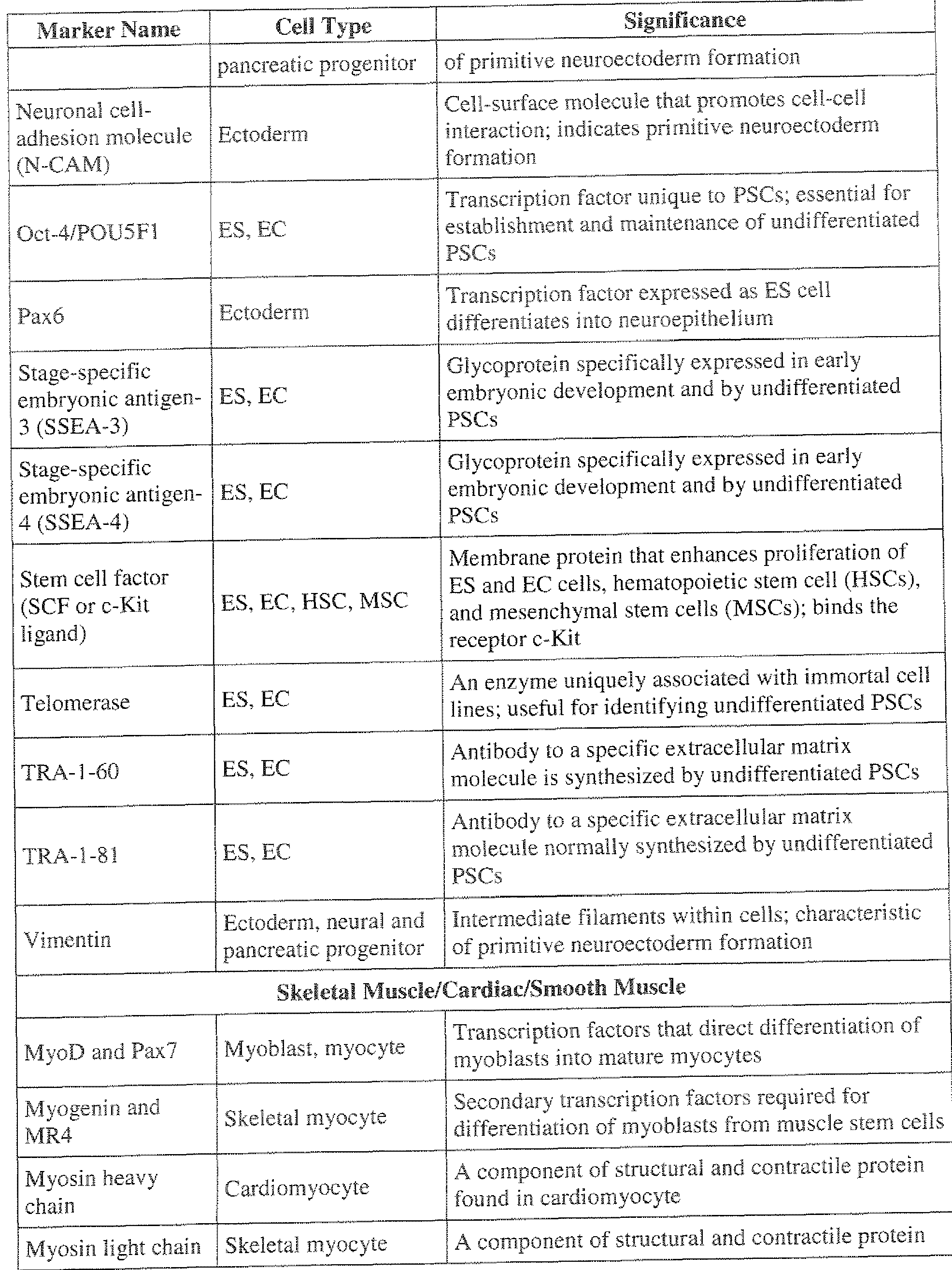







Claims
Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES11810518T ES2874953T3 (en) | 2010-07-23 | 2011-07-25 | Methods for the detection of rare subpopulations of cells and highly purified compositions of cells |
| JP2013520901A JP6184865B2 (en) | 2010-07-23 | 2011-07-25 | Methods and highly purified compositions for detecting rare subpopulations of cells |
| EP21163768.1A EP3875599B1 (en) | 2010-07-23 | 2011-07-25 | Methods for detection of rare subpopulations of cells and highly purified compositions of cells |
| DK11810518.8T DK2596119T3 (en) | 2010-07-23 | 2011-07-25 | METHODS FOR DETECTING RARE SUBPOPULATIONS OF CELLS AND HIGHLY PURIFIED COMPOSITIONS OF CELLS |
| AU2011280878A AU2011280878B2 (en) | 2010-07-23 | 2011-07-25 | Methods for detection of rare subpopulations of cells and highly purified compositions of cells |
| PL11810518T PL2596119T3 (en) | 2010-07-23 | 2011-07-25 | Methods for detection of rare subpopulations of cells and highly purified compositions of cells |
| US13/811,708 US11739366B2 (en) | 2010-07-23 | 2011-07-25 | Methods for detection of rare subpopulations of cells and highly purified compositions of cells |
| CA2806127A CA2806127C (en) | 2010-07-23 | 2011-07-25 | Methods for detection of rare subpopulations of cells and highly purified compositions of cells |
| EP11810518.8A EP2596119B8 (en) | 2010-07-23 | 2011-07-25 | Methods for detection of rare subpopulations of cells and highly purified compositions of cells |
| EP24212880.9A EP4524568A3 (en) | 2010-07-23 | 2011-07-25 | Methods for detection of rare subpopulations of cells and highly purified compositions of cells |
| US18/347,930 US12545945B2 (en) | 2010-07-23 | 2023-07-06 | Methods for detection of rare subpopulations of cells and highly purified composition of cells |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US36703810P | 2010-07-23 | 2010-07-23 | |
| US61/367,038 | 2010-07-23 | ||
| US41477010P | 2010-11-17 | 2010-11-17 | |
| US61/414,770 | 2010-11-17 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/811,708 A-371-Of-International US11739366B2 (en) | 2010-07-23 | 2011-07-25 | Methods for detection of rare subpopulations of cells and highly purified compositions of cells |
| US18/347,930 Continuation US12545945B2 (en) | 2010-07-23 | 2023-07-06 | Methods for detection of rare subpopulations of cells and highly purified composition of cells |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2012012803A2 true WO2012012803A2 (en) | 2012-01-26 |
| WO2012012803A3 WO2012012803A3 (en) | 2012-11-01 |
Family
ID=45497502
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/045232 Ceased WO2012012803A2 (en) | 2010-07-23 | 2011-07-25 | Methods for detection of rare subpopulations of cells and highly purified compositions of cells |
Country Status (11)
| Country | Link |
|---|---|
| US (2) | US11739366B2 (en) |
| EP (3) | EP2596119B8 (en) |
| JP (5) | JP6184865B2 (en) |
| AU (1) | AU2011280878B2 (en) |
| CA (1) | CA2806127C (en) |
| DK (2) | DK3875599T3 (en) |
| ES (2) | ES3013241T3 (en) |
| HU (1) | HUE054517T2 (en) |
| PL (2) | PL2596119T3 (en) |
| PT (2) | PT3875599T (en) |
| WO (1) | WO2012012803A2 (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB2496969A (en) * | 2011-11-14 | 2013-05-29 | Advanced Cell Tech Inc | Retinal pigment epithelial cells |
| JP2013208104A (en) * | 2012-03-02 | 2013-10-10 | Yamaguchi Univ | Serum-free medium for culturing digestive system cancer stem cell and method for proliferating digestive system cancer stem cell using the same |
| US9040038B2 (en) | 2004-01-23 | 2015-05-26 | Ocata Therapeutics, Inc. | Modalities for the treatment of degenerative diseases of the retina |
| US9040770B2 (en) | 2004-01-23 | 2015-05-26 | Ocata Therapeutics, Inc. | Modalities for the treatment of degenerative diseases of the retina |
| CN104833805A (en) * | 2015-03-09 | 2015-08-12 | 武汉格蓝丽富科技有限公司 | Circulating tumor cell detection and identification kit and application thereof |
| JP2015533122A (en) * | 2012-10-09 | 2015-11-19 | サンバイオ,インコーポレイティド | Methods and compositions for retinal degeneration treatment |
| CN107076724A (en) * | 2013-03-29 | 2017-08-18 | 国立大学法人三重大学 | Organism coloring agent |
| US9752118B2 (en) | 2011-12-06 | 2017-09-05 | Astellas Institute For Regenerative Medicine | Method of directed differentiation producing corneal endothelial cells from neural crest stem cells by PDGFB and DKK2, compositions thereof, and uses thereof |
| US10077424B2 (en) | 2007-10-12 | 2018-09-18 | Astellas Institute For Regenerative Medicine | Methods of producing RPE cells and compositions of RPE cells |
| WO2018225705A1 (en) | 2017-06-05 | 2018-12-13 | テルモ株式会社 | Method for producing cell culture |
| US10485829B2 (en) | 2009-11-17 | 2019-11-26 | Astellas Institute For Regenerative Medicine | Methods of producing human RPE cells and pharmaceutical preparations of human RPE cells |
| WO2020067438A1 (en) | 2018-09-27 | 2020-04-02 | 国立大学法人大阪大学 | Sheeting method for pluripotent stem cell-derived cells |
| CN112461807A (en) * | 2020-11-26 | 2021-03-09 | 山西大学 | Application of carbon quantum dots in targeted nucleolus wash-free imaging |
| WO2021071011A1 (en) * | 2019-10-10 | 2021-04-15 | 고려대학교 산학협력단 | Stem cell-derived mature cardiomyocytes and cardiovascular disease model using same |
| WO2021086911A1 (en) | 2019-10-30 | 2021-05-06 | Astellas Institute For Regenerative Medicine | Methods for producing retinal pigment epithelium cells |
| US11422125B2 (en) | 2015-03-23 | 2022-08-23 | Astellas Institute For Regenerative Medicine | Assays for potency of human retinal pigment epithelium (RPE) cells and photoreceptor progenitors |
| US11739366B2 (en) | 2010-07-23 | 2023-08-29 | Astellas Institute For Regenerative Medicine | Methods for detection of rare subpopulations of cells and highly purified compositions of cells |
| US12247071B2 (en) | 2016-12-21 | 2025-03-11 | Amgen Inc. | Anti-TNF alpha antibody formulations |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2932664T3 (en) | 2009-11-12 | 2023-01-23 | Technion Res & Dev Foundation | Culture media, cell cultures and methods of culturing pluripotent stem cells in the undifferentiated state |
| WO2015045962A1 (en) * | 2013-09-26 | 2015-04-02 | コニカミノルタ株式会社 | Method for determining quantity of biological material in tissue section |
| US10975353B2 (en) * | 2014-06-10 | 2021-04-13 | Polybiocept Gmbh | Culture medium for cellular immunotherapy |
| JP6271407B2 (en) * | 2014-12-17 | 2018-01-31 | 富士フイルム株式会社 | Cell evaluation integration method |
| JP6459568B2 (en) * | 2015-01-30 | 2019-01-30 | コニカミノルタ株式会社 | Target cell detection method and target cell detection system |
| KR102651291B1 (en) | 2015-08-18 | 2024-03-26 | 아스텔라스 인스티튜트 포 리제너러티브 메디슨 | Clinical formulations |
| WO2017100498A1 (en) | 2015-12-11 | 2017-06-15 | The Johns Hopkins University | Isolation of fusion-competent myoblasts and therapeutic applications thereof related to muscular dystrophy |
| CN106053196B (en) * | 2016-05-23 | 2019-02-15 | 无锡帝科电子材料股份有限公司 | A kind of dispersing method of photovoltaic front side silver paste silver powder testing graininess |
| CN106525541B (en) * | 2016-09-20 | 2019-01-18 | 广东东阳光药业有限公司 | A kind of detection method of fresh cordyceps sinensis cell viability |
Citations (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7A (en) | 1836-08-10 | Thomas Blanchard | Machine for boring holes and cutting lanyard-scores in deadeyes | |
| US6207299B1 (en) | 1996-10-10 | 2001-03-27 | Sollac | Sheet metal with an aluminum-containing coating having low emissivity |
| WO2001051616A2 (en) | 2000-01-11 | 2001-07-19 | Geron Corporation | Techniques for growth and differentiation of human pluripotent stem cells |
| US6322901B1 (en) | 1997-11-13 | 2001-11-27 | Massachusetts Institute Of Technology | Highly luminescent color-selective nano-crystalline materials |
| US6331313B1 (en) | 1999-10-22 | 2001-12-18 | Oculex Pharmaceticals, Inc. | Controlled-release biocompatible ocular drug delivery implant devices and methods |
| US20020119565A1 (en) | 2000-08-03 | 2002-08-29 | Regents Of The University Of Michigan | Isolation and use of solid tumor stem cells |
| US6576291B2 (en) | 2000-12-08 | 2003-06-10 | Massachusetts Institute Of Technology | Preparation of nanocrystallites |
| US6649138B2 (en) | 2000-10-13 | 2003-11-18 | Quantum Dot Corporation | Surface-modified semiconductive and metallic nanoparticles having enhanced dispersibility in aqueous media |
| US20030217374A1 (en) | 2002-01-15 | 2003-11-20 | Advanced Cell Technology | Cloning B and T lymphocytes |
| US20030232430A1 (en) | 2001-11-26 | 2003-12-18 | Advanced Cell Technology | Methods for making and using reprogrammed human somatic cell nuclei and autologous and isogenic human stem cells |
| US6682596B2 (en) | 2000-12-28 | 2004-01-27 | Quantum Dot Corporation | Flow synthesis of quantum dot nanocrystals |
| US20040091936A1 (en) | 2002-05-24 | 2004-05-13 | Michael West | Bank of stem cells for producing cells for transplantation having HLA antigens matching those of transplant recipients, and methods for making and using such a stem cell bank |
| US6800480B1 (en) | 1997-10-23 | 2004-10-05 | Geron Corporation | Methods and materials for the growth of primate-derived primordial stem cells in feeder-free culture |
| US6815064B2 (en) | 2001-07-20 | 2004-11-09 | Quantum Dot Corporation | Luminescent nanoparticles and methods for their preparation |
| WO2005001889A2 (en) | 2003-05-07 | 2005-01-06 | Indiana University Research & Technology Corporation | Alloyed semiconductor quantum dots and concentration-gradient alloyed quantum dots, series comprising the same and methods related thereto |
| US20050012182A1 (en) | 2003-07-19 | 2005-01-20 | Samsung Electronics Co., Ltd. | Alloy type semiconductor nanocrystals and method for preparing the same |
| US6943153B1 (en) | 1999-03-15 | 2005-09-13 | The Regents Of The University Of California | Use of recombinant gene delivery vectors for treating or preventing diseases of the eye |
| US6943145B2 (en) | 1997-09-10 | 2005-09-13 | University Of Florida | Compounds and method for the prevention and treatment of diabetic retinopathy |
| US20070160974A1 (en) | 2006-01-10 | 2007-07-12 | South Eastern Sydney And Illawarra Area Health Service | Human embryonic stem cell clones |
| US20070297983A1 (en) | 2006-03-16 | 2007-12-27 | Soldano Ferrone | Inhibition of breast carcinoma stem cell growth and metastasis |
| US20080064099A1 (en) | 2001-08-23 | 2008-03-13 | Reliance Life Sciences Pvt Ltd. | Isolation of Inner Cell Mass for the Establishment of Human Embryonic Stem Cell (hESC) Lines |
| US20080064049A1 (en) | 2005-10-31 | 2008-03-13 | The Regents Of The University Of Michigan | Compositions and methods for treating and diagnosing cancer |
| US20080178305A1 (en) | 2000-08-03 | 2008-07-24 | The Regents Of The University Of Michigan | Isolation And Use Of Solid Tumor Stem Cells |
| US20080175870A1 (en) | 2007-01-22 | 2008-07-24 | Mather Jennie P | Human Cancer Stem Cells |
| US20080194022A1 (en) | 2000-08-03 | 2008-08-14 | Clarke Michael F | Isolation and use of solid tumor stem cells |
| US7462471B2 (en) | 1993-02-01 | 2008-12-09 | Massachusetts Institute Of Technology | Porous biodegradable polymeric materials for cell transplantation |
| WO2009051671A1 (en) | 2007-10-12 | 2009-04-23 | Advanced Cell Technology, Inc. | Improved methods of producing rpe cells and compositions of rpe cells |
| US20090275128A1 (en) | 2004-09-08 | 2009-11-05 | Thomson James A | Medium and culture of embryonic stem cells |
| US7625582B2 (en) | 2000-11-29 | 2009-12-01 | Allergan, Inc. | Methods for reducing or preventing transplant rejection in the eye and intraocular implants for use therefor |
| US7736896B2 (en) | 2004-01-23 | 2010-06-15 | Advanced Cell Technology, Inc. | Methods for producing enriched populations of human retinal pigment epithelium cells |
| US7794704B2 (en) | 2004-01-23 | 2010-09-14 | Advanced Cell Technology, Inc. | Methods for producing enriched populations of human retinal pigment epithelium cells for treatment of retinal degeneration |
| US7893315B2 (en) | 2004-11-04 | 2011-02-22 | Advanced Cell Technology, Inc. | Derivation of embryonic stem cells and embryo-derived cells |
| WO2011063005A2 (en) | 2009-11-17 | 2011-05-26 | Advanced Cell Technology, Inc. | Methods of producing human rpe cells and pharmaceutical preparations of human rpe cells |
Family Cites Families (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6172197B1 (en) | 1991-07-10 | 2001-01-09 | Medical Research Council | Methods for producing members of specific binding pairs |
| US5780300A (en) | 1995-09-29 | 1998-07-14 | Yale University | Manipulation of non-terminally differentiated cells using the notch pathway |
| EP0880584A1 (en) | 1996-02-16 | 1998-12-02 | University Of Edinburgh | Cytokine expressed by dia/lif-deficient embryonic stem cells for the inhibition of differentiation |
| DE19609838A1 (en) * | 1996-03-13 | 1997-09-18 | Boehringer Mannheim Gmbh | Methods and test strips for the determination of an analyte |
| AU2884499A (en) * | 1998-03-02 | 1999-09-20 | Compucyte Corp. | Selective cell analysis |
| JP2005505746A (en) | 2001-02-02 | 2005-02-24 | ダナ−ファーバー キャンサー インスチチュート インコーポレーテッド | Rare event detection system |
| EP1322957B1 (en) * | 2001-05-04 | 2009-08-12 | Biosite Incorporated | Diagnostic markers of acute coronary syndromes and methods of use thereof |
| US7425448B2 (en) * | 2001-07-12 | 2008-09-16 | Geron Corporation | Cardiomyocyte precursors from human embryonic stem cells |
| US7662625B2 (en) | 2002-07-02 | 2010-02-16 | Cancer Research Technology Limited | Methods for detecting the differentiation status of cells using 5T4 antigen expression |
| WO2004045374A2 (en) * | 2002-11-14 | 2004-06-03 | Ethicon Endo-Surgery, Inc. | Methods and devices for detecting tissue cells |
| US20050032126A1 (en) | 2003-03-03 | 2005-02-10 | Coombs James H. | Methods and apparatus for use in detection and quantitation of various cell types and use of optical bio-disc for performing same |
| US20050118713A1 (en) * | 2003-05-12 | 2005-06-02 | Nikolai Strelchenko | Morula derived embryonic stem cells |
| KR100569168B1 (en) * | 2003-08-08 | 2006-04-07 | (주)아비코아생명공학연구소 | Cultivation method of algae garden stem cells and algae garden stem cells obtained by |
| US20240174978A1 (en) | 2004-01-23 | 2024-05-30 | Astellas Institute For Regenerative Medicine | Modalities for the treatment of degenerative diseases of the retina |
| US20190282622A1 (en) | 2004-01-23 | 2019-09-19 | Astellas Institute For Regenerative Medicine | Modalities for the treatment of degenerative diseases of the retina |
| EP2479256A1 (en) | 2004-11-04 | 2012-07-25 | Advanced Cell Technology, Inc. | Derivation of embryonic stem cells |
| CZ301148B6 (en) * | 2006-01-25 | 2009-11-18 | Univerzita Karlova V Praze | Method of culturing human mezenchymal stem cells, particularly for facilitating fracture healing process, and bioreactor for carrying out the method |
| KR100832592B1 (en) * | 2006-08-17 | 2008-05-27 | 박현숙 | Coculture of Stem Cells and Feeder Cells Using Polymer Membranes |
| US8097588B2 (en) * | 2007-02-08 | 2012-01-17 | Sanford-Burnham Medical Research Institute | Trophinin-binding peptides and uses thereof |
| CN103555654B (en) | 2007-04-18 | 2016-04-20 | 哈达锡特医学研究服务及发展有限公司 | Stem cell-derived retinal pigment epithelium |
| JP2008307007A (en) * | 2007-06-15 | 2008-12-25 | Bayer Schering Pharma Ag | Human pluripotent stem cell induced from human tissue-originated undifferentiated stem cell after birth |
| HU0700675D0 (en) | 2007-10-15 | 2007-12-28 | Mta Tamogatott Kutatohelyek Ir | Method for monitoring stem cell differentiation |
| US20090123439A1 (en) * | 2007-11-09 | 2009-05-14 | The Jackson Laboratory | Diagnostic and prognosis methods for cancer stem cells |
| US20090233324A1 (en) * | 2008-03-11 | 2009-09-17 | Kopf-Sill Anne R | Methods for Diagnosing Cancer Using Samples Collected From A Central Vein Location or an Arterial Location |
| PL2596119T3 (en) | 2010-07-23 | 2021-12-06 | Astellas Institute For Regenerative Medicine | Methods for detection of rare subpopulations of cells and highly purified compositions of cells |
| US20120258451A1 (en) | 2011-04-08 | 2012-10-11 | Advanced Cell Technology, Inc. | Laser isolation of viable cells |
| TW202434266A (en) | 2011-11-14 | 2024-09-01 | 安斯泰來再生醫藥協會 | Pharmaceutical preparations of human rpe cells and uses thereof |
| CA3188112A1 (en) | 2015-03-23 | 2016-09-29 | Astellas Institute For Regenerative Medicine | Improved assays for potency of human retinal pigment epithelium (rpe) cells and photoreceptor progenitors |
-
2011
- 2011-07-25 PL PL11810518T patent/PL2596119T3/en unknown
- 2011-07-25 JP JP2013520901A patent/JP6184865B2/en active Active
- 2011-07-25 EP EP11810518.8A patent/EP2596119B8/en active Active
- 2011-07-25 AU AU2011280878A patent/AU2011280878B2/en active Active
- 2011-07-25 WO PCT/US2011/045232 patent/WO2012012803A2/en not_active Ceased
- 2011-07-25 US US13/811,708 patent/US11739366B2/en active Active
- 2011-07-25 ES ES21163768T patent/ES3013241T3/en active Active
- 2011-07-25 EP EP24212880.9A patent/EP4524568A3/en active Pending
- 2011-07-25 PT PT211637681T patent/PT3875599T/en unknown
- 2011-07-25 DK DK21163768.1T patent/DK3875599T3/en active
- 2011-07-25 PT PT118105188T patent/PT2596119T/en unknown
- 2011-07-25 PL PL21163768.1T patent/PL3875599T3/en unknown
- 2011-07-25 HU HUE11810518A patent/HUE054517T2/en unknown
- 2011-07-25 DK DK11810518.8T patent/DK2596119T3/en active
- 2011-07-25 EP EP21163768.1A patent/EP3875599B1/en active Active
- 2011-07-25 ES ES11810518T patent/ES2874953T3/en active Active
- 2011-07-25 CA CA2806127A patent/CA2806127C/en active Active
-
2016
- 2016-12-05 JP JP2016235929A patent/JP6630657B2/en active Active
-
2019
- 2019-12-09 JP JP2019221824A patent/JP7125382B2/en active Active
-
2022
- 2022-03-22 JP JP2022045743A patent/JP7412470B2/en active Active
-
2023
- 2023-07-06 US US18/347,930 patent/US12545945B2/en active Active
- 2023-12-26 JP JP2023219452A patent/JP7788436B2/en active Active
Patent Citations (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7A (en) | 1836-08-10 | Thomas Blanchard | Machine for boring holes and cutting lanyard-scores in deadeyes | |
| US7462471B2 (en) | 1993-02-01 | 2008-12-09 | Massachusetts Institute Of Technology | Porous biodegradable polymeric materials for cell transplantation |
| US6207299B1 (en) | 1996-10-10 | 2001-03-27 | Sollac | Sheet metal with an aluminum-containing coating having low emissivity |
| US6943145B2 (en) | 1997-09-10 | 2005-09-13 | University Of Florida | Compounds and method for the prevention and treatment of diabetic retinopathy |
| US6800480B1 (en) | 1997-10-23 | 2004-10-05 | Geron Corporation | Methods and materials for the growth of primate-derived primordial stem cells in feeder-free culture |
| US6322901B1 (en) | 1997-11-13 | 2001-11-27 | Massachusetts Institute Of Technology | Highly luminescent color-selective nano-crystalline materials |
| US6943153B1 (en) | 1999-03-15 | 2005-09-13 | The Regents Of The University Of California | Use of recombinant gene delivery vectors for treating or preventing diseases of the eye |
| US6331313B1 (en) | 1999-10-22 | 2001-12-18 | Oculex Pharmaceticals, Inc. | Controlled-release biocompatible ocular drug delivery implant devices and methods |
| WO2001051616A2 (en) | 2000-01-11 | 2001-07-19 | Geron Corporation | Techniques for growth and differentiation of human pluripotent stem cells |
| US20020119565A1 (en) | 2000-08-03 | 2002-08-29 | Regents Of The University Of Michigan | Isolation and use of solid tumor stem cells |
| US20060051325A1 (en) | 2000-08-03 | 2006-03-09 | Regents Of The University Of Michigan | Isolation and use of solid tumor stem cells |
| US20040037815A1 (en) | 2000-08-03 | 2004-02-26 | Clarke Michael F. | Isolation and use of solid tumor stem cells |
| US20070190647A1 (en) | 2000-08-03 | 2007-08-16 | Regents Of The University Of Michigan | Isolation and use of solid tumor stem cells |
| US20070231325A1 (en) | 2000-08-03 | 2007-10-04 | The Regents Of The University Of Michigan | Prospective identification and characterization of breast cancer stem cells |
| US20080194022A1 (en) | 2000-08-03 | 2008-08-14 | Clarke Michael F | Isolation and use of solid tumor stem cells |
| US20080178305A1 (en) | 2000-08-03 | 2008-07-24 | The Regents Of The University Of Michigan | Isolation And Use Of Solid Tumor Stem Cells |
| US20070259969A1 (en) | 2000-08-03 | 2007-11-08 | The Regents Of The University Of Michigan | Isolation And Use Of Solid Tumor Stem Cells |
| US20060073125A1 (en) | 2000-08-03 | 2006-04-06 | Regents Of The University Of Michigan | Isolation and use of solid tumor stem cells |
| US20070212737A1 (en) | 2000-08-03 | 2007-09-13 | Regents Of The University Of Michigan | Isolation and use of solid tumor stem cells |
| US20090004205A1 (en) | 2000-08-03 | 2009-01-01 | The Regents Of The University Of Michigan | Prospective identification and characterization of breast cancer stem cells |
| US6649138B2 (en) | 2000-10-13 | 2003-11-18 | Quantum Dot Corporation | Surface-modified semiconductive and metallic nanoparticles having enhanced dispersibility in aqueous media |
| US7625582B2 (en) | 2000-11-29 | 2009-12-01 | Allergan, Inc. | Methods for reducing or preventing transplant rejection in the eye and intraocular implants for use therefor |
| US6576291B2 (en) | 2000-12-08 | 2003-06-10 | Massachusetts Institute Of Technology | Preparation of nanocrystallites |
| US6682596B2 (en) | 2000-12-28 | 2004-01-27 | Quantum Dot Corporation | Flow synthesis of quantum dot nanocrystals |
| US6815064B2 (en) | 2001-07-20 | 2004-11-09 | Quantum Dot Corporation | Luminescent nanoparticles and methods for their preparation |
| US20080064099A1 (en) | 2001-08-23 | 2008-03-13 | Reliance Life Sciences Pvt Ltd. | Isolation of Inner Cell Mass for the Establishment of Human Embryonic Stem Cell (hESC) Lines |
| US20030232430A1 (en) | 2001-11-26 | 2003-12-18 | Advanced Cell Technology | Methods for making and using reprogrammed human somatic cell nuclei and autologous and isogenic human stem cells |
| US20030217374A1 (en) | 2002-01-15 | 2003-11-20 | Advanced Cell Technology | Cloning B and T lymphocytes |
| US20040091936A1 (en) | 2002-05-24 | 2004-05-13 | Michael West | Bank of stem cells for producing cells for transplantation having HLA antigens matching those of transplant recipients, and methods for making and using such a stem cell bank |
| WO2005001889A2 (en) | 2003-05-07 | 2005-01-06 | Indiana University Research & Technology Corporation | Alloyed semiconductor quantum dots and concentration-gradient alloyed quantum dots, series comprising the same and methods related thereto |
| US20050012182A1 (en) | 2003-07-19 | 2005-01-20 | Samsung Electronics Co., Ltd. | Alloy type semiconductor nanocrystals and method for preparing the same |
| US7794704B2 (en) | 2004-01-23 | 2010-09-14 | Advanced Cell Technology, Inc. | Methods for producing enriched populations of human retinal pigment epithelium cells for treatment of retinal degeneration |
| US7795025B2 (en) | 2004-01-23 | 2010-09-14 | Advanced Cell Technology, Inc. | Methods for producing enriched populations of human retinal pigment epithelium cells |
| US7736896B2 (en) | 2004-01-23 | 2010-06-15 | Advanced Cell Technology, Inc. | Methods for producing enriched populations of human retinal pigment epithelium cells |
| US20090275128A1 (en) | 2004-09-08 | 2009-11-05 | Thomson James A | Medium and culture of embryonic stem cells |
| US7893315B2 (en) | 2004-11-04 | 2011-02-22 | Advanced Cell Technology, Inc. | Derivation of embryonic stem cells and embryo-derived cells |
| US20100169990A1 (en) | 2005-10-31 | 2010-07-01 | The Regents Of The University Of Michigan | Compositions and methods for treating and diagnosing cancer |
| US20080064049A1 (en) | 2005-10-31 | 2008-03-13 | The Regents Of The University Of Michigan | Compositions and methods for treating and diagnosing cancer |
| US20070160974A1 (en) | 2006-01-10 | 2007-07-12 | South Eastern Sydney And Illawarra Area Health Service | Human embryonic stem cell clones |
| US20070297983A1 (en) | 2006-03-16 | 2007-12-27 | Soldano Ferrone | Inhibition of breast carcinoma stem cell growth and metastasis |
| US20080175870A1 (en) | 2007-01-22 | 2008-07-24 | Mather Jennie P | Human Cancer Stem Cells |
| WO2009051671A1 (en) | 2007-10-12 | 2009-04-23 | Advanced Cell Technology, Inc. | Improved methods of producing rpe cells and compositions of rpe cells |
| WO2011063005A2 (en) | 2009-11-17 | 2011-05-26 | Advanced Cell Technology, Inc. | Methods of producing human rpe cells and pharmaceutical preparations of human rpe cells |
Non-Patent Citations (13)
| Title |
|---|
| AFSHARI ET AL., BRAIN, vol. 133, no. 2, 2010, pages 448 - 464 |
| AMIT ET AL., BIOL REPROD., vol. 70, no. 3, March 2004 (2004-03-01), pages 837 - 45 |
| CARPENTER ET AL., DEV DYN., vol. 229, no. 2, February 2004 (2004-02-01), pages 259 - 74 |
| HOLGATE ET AL., J HISTOCHEM CYTOCHEM., vol. 31, no. 7, July 1983 (1983-07-01), pages 938 - 44 |
| HUTALA ET AL.: "In vitro biocompatibility of degradable biopolymers in cell line cultures from various ocular tissues: Direct contact studies", JOURNAL OF BIOMEDICAL MATERIALS RESEARCH, vol. 83A, no. 2, 2007, pages 407 - 413 |
| IDELSON, CELL STEM CELL, vol. 5, 2009, pages 396 - 408 |
| KLIMANSKAYA ET AL.: "Human Embryonic Stem Cell lines Derived from Single Bastomeres", NATURE, vol. 444, 2006, pages 481 - 485, XP009076989, DOI: 10.1038/nature05142 |
| LU ET AL., J BIOMATER SCI POLYM ED, vol. 9, 1998, pages 1187 - 205 |
| LU ET AL., STEM CELLS, vol. 27, 2009, pages 2126 - 2135 |
| QU ET AL.: "Alternative Routes toward High Quality CdSe Nanocrystals", NANO LETT., vol. 1, no. 6, 2001, pages 333 - 337, XP002343572, DOI: 10.1021/nl0155532 |
| TOMITA ET AL., STEM CELLS, vol. 23, 2005, pages 1579 - 88 |
| WANG ET AL., TRANSPLANTATION, 2010 |
| XU ET AL., NAT BIOTECHNOL., vol. 19, no. 10, October 2001 (2001-10-01), pages 971 - 4 |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9193950B2 (en) | 2004-01-23 | 2015-11-24 | Ocata Therapeutics, Inc. | Modalities for the treatment of degenerative diseases of the retina |
| US9181524B2 (en) | 2004-01-23 | 2015-11-10 | Ocata Therapeutics, Inc. | Modalities for the treatment of degenerative diseases of the retina |
| US9040038B2 (en) | 2004-01-23 | 2015-05-26 | Ocata Therapeutics, Inc. | Modalities for the treatment of degenerative diseases of the retina |
| US9040770B2 (en) | 2004-01-23 | 2015-05-26 | Ocata Therapeutics, Inc. | Modalities for the treatment of degenerative diseases of the retina |
| US9040039B2 (en) | 2004-01-23 | 2015-05-26 | Ocata Therapeutics, Inc. | Modalities for the treatment of degenerative diseases of the retina |
| US9045732B2 (en) | 2004-01-23 | 2015-06-02 | Ocata Therapeutics, Inc. | Modalities for the treatment of degenerative diseases of the retina |
| US9080150B2 (en) | 2004-01-23 | 2015-07-14 | Ocata Therapeutics, Inc. | Modalities for the treatment of degenerative diseases of the retina |
| US9562217B2 (en) | 2004-01-23 | 2017-02-07 | Astellas Institute For Regenerative Medicine | Modalities for the treatment of degenerative diseases of the retina |
| US9730962B2 (en) | 2004-01-23 | 2017-08-15 | Astellas Institute For Regenerative Medicine | Modalities for the treatment of degenerative diseases of the retina |
| US9650607B2 (en) | 2004-01-23 | 2017-05-16 | Astellas Institute For Regenerative Medicine | Modalities for the treatment of degenerative diseases of the retina |
| US9649340B2 (en) | 2004-01-23 | 2017-05-16 | Astellas Institute For Regenerative Medicine | Methods for producing enriched populations of human retinal pigment epithelium cells |
| US10077424B2 (en) | 2007-10-12 | 2018-09-18 | Astellas Institute For Regenerative Medicine | Methods of producing RPE cells and compositions of RPE cells |
| US11850261B2 (en) | 2009-11-17 | 2023-12-26 | Astellas Institute For Regenerative Medicine | Methods of producing human RPE cells and pharmaceutical preparations of human RPE cells |
| US10485829B2 (en) | 2009-11-17 | 2019-11-26 | Astellas Institute For Regenerative Medicine | Methods of producing human RPE cells and pharmaceutical preparations of human RPE cells |
| US12545945B2 (en) | 2010-07-23 | 2026-02-10 | Astellas Institute For Regenerative Medicine | Methods for detection of rare subpopulations of cells and highly purified composition of cells |
| US11739366B2 (en) | 2010-07-23 | 2023-08-29 | Astellas Institute For Regenerative Medicine | Methods for detection of rare subpopulations of cells and highly purified compositions of cells |
| EP3563860A1 (en) | 2011-11-14 | 2019-11-06 | Astellas Institute for Regenerative Medicine | Pharmaceutical preparations of human rpe cells and uses thereof |
| GB2496969A (en) * | 2011-11-14 | 2013-05-29 | Advanced Cell Tech Inc | Retinal pigment epithelial cells |
| AU2017225146B2 (en) * | 2011-11-14 | 2019-01-17 | Astellas Institute For Regenerative Medicine | Pharmaceutical preparations of human RPE cells and uses thereof |
| AU2017225146C1 (en) * | 2011-11-14 | 2019-08-22 | Astellas Institute For Regenerative Medicine | Pharmaceutical preparations of human RPE cells and uses thereof |
| US12049642B2 (en) | 2011-12-06 | 2024-07-30 | Astellas Institute For Regenerative Medicine | Corneal endothelial cells with increased cell density and compositions thereof |
| US9752118B2 (en) | 2011-12-06 | 2017-09-05 | Astellas Institute For Regenerative Medicine | Method of directed differentiation producing corneal endothelial cells from neural crest stem cells by PDGFB and DKK2, compositions thereof, and uses thereof |
| JP2013208104A (en) * | 2012-03-02 | 2013-10-10 | Yamaguchi Univ | Serum-free medium for culturing digestive system cancer stem cell and method for proliferating digestive system cancer stem cell using the same |
| US9855299B2 (en) | 2012-10-09 | 2018-01-02 | Sanbio, Inc. | Methods and compositions for treatment of retinal degeneration |
| JP2016210813A (en) * | 2012-10-09 | 2016-12-15 | サンバイオ,インコーポレイティド | Methods and compositions for treatment of retinal degeneration |
| JP2015533122A (en) * | 2012-10-09 | 2015-11-19 | サンバイオ,インコーポレイティド | Methods and compositions for retinal degeneration treatment |
| CN107076724A (en) * | 2013-03-29 | 2017-08-18 | 国立大学法人三重大学 | Organism coloring agent |
| CN104833805A (en) * | 2015-03-09 | 2015-08-12 | 武汉格蓝丽富科技有限公司 | Circulating tumor cell detection and identification kit and application thereof |
| US11422125B2 (en) | 2015-03-23 | 2022-08-23 | Astellas Institute For Regenerative Medicine | Assays for potency of human retinal pigment epithelium (RPE) cells and photoreceptor progenitors |
| US11680941B2 (en) | 2015-03-23 | 2023-06-20 | Astellas Institute For Regenerative Medicine | Assays for potency of human retinal pigment epithelium (RPE) cells and photoreceptor progenitors |
| US12247071B2 (en) | 2016-12-21 | 2025-03-11 | Amgen Inc. | Anti-TNF alpha antibody formulations |
| WO2018225705A1 (en) | 2017-06-05 | 2018-12-13 | テルモ株式会社 | Method for producing cell culture |
| WO2020067438A1 (en) | 2018-09-27 | 2020-04-02 | 国立大学法人大阪大学 | Sheeting method for pluripotent stem cell-derived cells |
| WO2021071011A1 (en) * | 2019-10-10 | 2021-04-15 | 고려대학교 산학협력단 | Stem cell-derived mature cardiomyocytes and cardiovascular disease model using same |
| WO2021086911A1 (en) | 2019-10-30 | 2021-05-06 | Astellas Institute For Regenerative Medicine | Methods for producing retinal pigment epithelium cells |
| CN112461807A (en) * | 2020-11-26 | 2021-03-09 | 山西大学 | Application of carbon quantum dots in targeted nucleolus wash-free imaging |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12545945B2 (en) | Methods for detection of rare subpopulations of cells and highly purified composition of cells | |
| AU2011280878A1 (en) | Methods for detection of rare subpopulations of cells and highly purified compositions of cells | |
| US20250134932A1 (en) | Pharmaceutical preparations of human rpe cells and uses thereof | |
| KR20180091125A (en) | Methods of producing human rpe cells and pharmaceutical preparations of human rpe cells | |
| KR20220020422A (en) | Methods of treating retinal diseases | |
| HK40122464A (en) | Methods for detection of rare subpopulations of cells and highly purified compositions of cells | |
| HK40056946A (en) | Methods for detection of rare subpopulations of cells and highly purified compositions of cells | |
| HK40056946B (en) | Methods for detection of rare subpopulations of cells and highly purified compositions of cells | |
| HK40015631A (en) | Pharmaceutical preparations of human rpe cells and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11810518 Country of ref document: EP Kind code of ref document: A2 |
|
| ENP | Entry into the national phase |
Ref document number: 2806127 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2013520901 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2011280878 Country of ref document: AU Date of ref document: 20110725 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011810518 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13811708 Country of ref document: US |