WO2012006369A2 - Immunisation of large mammals with low doses of rna - Google Patents
Immunisation of large mammals with low doses of rna Download PDFInfo
- Publication number
- WO2012006369A2 WO2012006369A2 PCT/US2011/043096 US2011043096W WO2012006369A2 WO 2012006369 A2 WO2012006369 A2 WO 2012006369A2 US 2011043096 W US2011043096 W US 2011043096W WO 2012006369 A2 WO2012006369 A2 WO 2012006369A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- rna
- liposomes
- immunogen
- virus
- dose
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/117—Nucleic acids having immunomodulatory properties, e.g. containing CpG-motifs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
- A61K39/155—Paramyxoviridae, e.g. parainfluenza virus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/39—Medicinal preparations containing antigens or antibodies characterised by the immunostimulating additives, e.g. chemical adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0008—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition
- A61K48/0025—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition wherein the non-active part clearly interacts with the delivered nucleic acid
- A61K48/0041—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition wherein the non-active part clearly interacts with the delivered nucleic acid the non-active part being polymeric
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0083—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the administration regime
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/53—DNA (RNA) vaccination
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/55—Medicinal preparations containing antigens or antibodies characterised by the host/recipient, e.g. newborn with maternal antibodies
- A61K2039/552—Veterinary vaccine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55555—Liposomes; Vesicles, e.g. nanoparticles; Spheres, e.g. nanospheres; Polymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55566—Emulsions, e.g. Freund's adjuvant, MF59
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/18011—Paramyxoviridae
- C12N2760/18511—Pneumovirus, e.g. human respiratory syncytial virus
- C12N2760/18534—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2770/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses positive-sense
- C12N2770/00011—Details
- C12N2770/36011—Togaviridae
- C12N2770/36111—Alphavirus, e.g. Sindbis virus, VEE, EEE, WEE, Semliki
- C12N2770/36141—Use of virus, viral particle or viral elements as a vector
- C12N2770/36143—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2820/00—Vectors comprising a special origin of replication system
- C12N2820/60—Vectors comprising a special origin of replication system from viruses
Definitions
- This invention is in the field of non- viral delivery of RNA for immunisation.
- Reference 1 delivered 50 ⁇ g of lipoplexed mRNA or DNA to mice, but also used intraglossal ⁇ g and 10 ⁇ g doses to analyse luciferase expression in tongue tissue.
- Reference 2 delivered 12 ⁇ g of mRNA encoding influenza virus nucleoprotein to mice.
- Reference 3 delivered O. ⁇ g, ⁇ g or 10 ⁇ g of self-replicating RNA encoding ⁇ -galactosidase to mice.
- Reference 4 delivered 10 ⁇ g of self-replicating RNA encoding rabies virus glycoprotein to mice.
- Reference 5 delivered a total of 2 ⁇ g or 4 ⁇ g of DNA encoding influenza haemagglutinin to humans, but did not deliver RNA.
- RNA encoding an immunogen is delivered to a large mammal at a dose of 0. ⁇ g/kg to l ⁇ g/kg.
- a dose of ⁇ 0.94 ⁇ g/kg is immunogenic in cattle.
- Prior art studies have used lOOng to 10 ⁇ g RNA in mice which, with a ⁇ 20g body weight, is 5 ⁇ g/kg to 500 ⁇ g/kg.
- the invention provides a method of raising an immune response in a large mammal, comprising administering to the mammal a dose of between 2 ⁇ g and 10( ⁇ g of immunogen-encoding RNA.
- the invention also provides an immunogen-encoding RNA for use in an in vivo method of raising an immune response in a large mammal, wherein the method comprises administering between 2 ⁇ g and 10( ⁇ g of the RNA to the mammal.
- the invention also provides the use of an immunogen-encoding RNA in the manufacture medicament for raising an in vivo immune response in a large mammal, wherein the medicament has between 2 ⁇ g and 10( ⁇ g of immunogen-encoding RNA per unit dose.
- the invention also provides a pharmaceutical composition for a large mammal, comprising between 2 ⁇ g and 10( ⁇ g of immunogen-encoding RNA per unit dose.
- a pharmaceutical composition for a large mammal comprising between 2 ⁇ g and 10( ⁇ g of immunogen-encoding RNA per unit dose.
- concentration of the immunogen-encoding RNA will thus be between 4 ⁇ g/ml and 200 ⁇ g/ml.
- the invention also provides a unit dose of a pharmaceutical composition for administration to a large mammal, wherein the unit dose comprises between 2 ⁇ g and 100 ⁇ g of immunogen-encoding RNA.
- the invention also provides a delivery device ⁇ e.g. syringe, nebuliser, sprayer, inhaler, dermal patch, etc.) containing a pharmaceutical composition for administration to a large mammal, wherein the composition in the device contains between 2 ⁇ g and 100 ⁇ g of immunogen-encoding RNA.
- a delivery device e.g. syringe, nebuliser, sprayer, inhaler, dermal patch, etc.
- the invention also provides a hermetically sealed container containing a pharmaceutical composition for administration to a large mammal, wherein the composition in the container contains between 2 ⁇ g and 100 ⁇ g of immunogen-encoding RNA.
- the invention also provides a method of raising an immune response in a large mammal, comprising administering to the mammal between O. ⁇ g and 1.5 ⁇ RNA per kg of the mammal's body weight.
- the invention also provides an immunogen-encoding RNA for use in an in vivo method of raising an immune response in a large mammal, wherein the method comprises administering between O. ⁇ g and 1 ⁇ g RNA per kg of the mammal's body weight.
- the invention also provides the use of an immunogen-encoding RNA in the manufacture medicament for raising an in vivo immune response in a large mammal, wherein the medicament has between O. ⁇ g and l ⁇ g of immunogen-encoding RNA per kg of the mammal's body weight.
- RNA messenger RNA
- the site of administration will usually be muscle tissue, such as skeletal muscle.
- Alternatives to intramuscular administration include, but are not limited to: intradermal, intranasal, intraocular, subcutaneous, intraperitoneal, intravenous, interstitial, buccal, transdermal, or sublingual administration. Intradermal and intramuscular administration are two preferred routes.
- Administration can be achieved in various ways. For instance, injection via a needle (e.g. a hypodermic needle) can be used, particularly for intramuscular, subcutaneous, intraocular, intraperitoneal or intravenous administration. Needle-free injection can be used as an alternative.
- a needle e.g. a hypodermic needle
- Needle-free injection can be used as an alternative.
- Intramuscular injection is the preferred way of administering RNA according to the invention. Injection into the upper arm, deltoid or thigh muscle (e.g. anterolateral thigh) is typical.
- deltoid or thigh muscle e.g. anterolateral thigh
- the administration site can include both immune cells (such as macrophages e.g. bone marrow derived macrophages), dendritic cells (e.g. bone marrow derived plasmacytoid dendritic cells and/or bone marrow derived myeloid dendritic cells), monocytes (e.g. human peripheral blood monocytes), etc.) and non-immune cells (such as muscle cells, which may be multinucleated and may be arranged into fascicles, and/or fibroblasts).
- the immune cells can be present at the time of administration, but will usually infiltrate the site after administration.
- the tissue damage caused by invasive administration e.g. caused by a needle at the administration site
- immune cells can be present at the time of administration, but will usually infiltrate the site after administration.
- the tissue damage caused by invasive administration e.g. caused by a needle at the administration site
- immune cells can be present at the time of administration, but will usually infiltrate the site after administration
- RIG-I-like receptor family i.e. RLRs
- RLR-3 LGP2
- the RNA can also be translated in the immune and/or non-immune cells, leading to expression of the immunogen, and ultimately to presentation of the expressed immunogen via the MHC system.
- the cells can also secrete type I interferons and/or pro-inflammatory cytokines to provide a local adjuvant effect.
- the RNA can be delivered as naked RNA (e.g. merely as an aqueous solution of RNA) but, to enhance both entry to immune and non-immune cells and also subsequent intercellular effects, and also to reduce the amount of RNA required for a good immunogenic effect, the RNA is preferably administered in combination with a delivery system, such as a particulate or emulsion delivery system.
- liposomes are a preferred delivery system.
- RNA encoding an immunogen is delivered to a large mammal at a dose of between 2 ⁇ g and 100 ⁇ g.
- the dose can be between 5 ⁇ g and 75 ⁇ g, between 6 ⁇ g and 50g, between 7 ⁇ g and 25 ⁇ g, between 8 ⁇ g and 20 ⁇ g, or between 9 ⁇ g and 15 ⁇ g.
- RNA encoding an immunogen is delivered to a large mammal at a dose of between O. ⁇ g RNA per kg of body weight to l ⁇ g RNA per kg of body weight.
- the dose can be between 0 ⁇ g/kg to l ⁇ g/kg, between 0.3 ⁇ g/kg to l . ⁇ g/kg, between 0 ⁇ g/kg to l .( ⁇ g/kg, between 0 ⁇ g/kg to l .( ⁇ g/kg, or between 0 ⁇ g/kg to l ⁇ g/kg.
- Specific doses can be O. ⁇ g/kg, 0.15 ⁇ g/kg, 0.2 ⁇ g/kg, 0.25 ⁇ g/kg, 0 ⁇ g/kg, 0.4 ⁇ g/kg, 0.5 ⁇ g/kg, ⁇ g/kg, or l ⁇ g/kg.
- Suitable classes of phospholipid include, but are not limited to, phosphatidyl ethanolamines, phosphatidylcholines, phosphatidylserines, and phosphatidyl-glycerols, and some useful phospholipids are listed in Table 1.
- Useful cationic lipids include, but are not limited to, dioleoyl trimethylammonium propane (DOTAP), l ,2-distearyloxy-N,N-dimethyl-3-aminopropane (DSDMA), 1 ,2-dioleyloxy-N,Ndimethyl- 3-aminopropane (DODMA), l ,2-dilinoleyloxy-N,N-dimethyl-3-aminopropane (DLinDMA), 1 ,2- dilinolenyloxy-N,N-dimethyl-3-aminopropane (DLenDMA).
- DOTAP dioleoyl trimethylammonium propane
- DSDMA distearyloxy-N,N-dimethyl-3-aminopropane
- DODMA 1 ,2-dioleyloxy-N,Ndimethyl- 3-aminopropane
- DLinDMA 1-dilinolenyl
- Zwitterionic lipids include, but are not limited to, acyl zwitterionic lipids and ether zwitterionic lipids.
- useful zwitterionic lipids are DPPC, DOPC and dodecylphosphocholine.
- the lipids can be saturated or unsaturated. The use of at least one unsaturated lipid for preparing liposomes is preferred. If an unsaturated lipid has two tails, both tails can be unsaturated, or it can have one saturated tail and one unsaturated tail.
- Liposomes can be formed from a single lipid or from a mixture of lipids.
- a mixture may comprise (i) a mixture of anionic lipids (ii) a mixture of cationic lipids (iii) a mixture of zwitterionic lipids (iv) a mixture of anionic lipids and cationic lipids (v) a mixture of anionic lipids and zwitterionic lipids (vi) a mixture of zwitterionic lipids and cationic lipids or (vii) a mixture of anionic lipids, cationic lipids and zwitterionic lipids.
- a mixture may comprise both saturated and unsaturated lipids.
- a mixture may comprise DSPC (zwitterionic, saturated), DlinDMA (cationic, unsaturated), and/or DMG (anionic, saturated).
- DSPC zwitterionic, saturated
- DlinDMA cationic, unsaturated
- DMG anionic, saturated
- the hydrophilic portion of a lipid can be PEGylated (i.e. modified by covalent attachment of a polyethylene glycol). This modification can increase stability and prevent non-specific adsorption of the liposomes.
- lipids can be conjugated to PEG using techniques such as those disclosed in reference 6 and 7.
- Various lengths of PEG can be used e.g. between 0.5-8kDa.
- a mixture of DSPC, DlinDMA, PEG-DMG and cholesterol is used in the examples.
- Liposomes are usually divided into three groups: multilamellar vesicles (MLV); small unilamellar vesicles (SUV); and large unilamellar vesicles (LUV).
- MLVs have multiple bilayers in each vesicle, forming several separate aqueous compartments.
- SUVs and LUVs have a single bilayer encapsulating an aqueous core; SUVs typically have a diameter ⁇ 50nm, and LUVs have a diameter >50nm.
- Liposomes useful with of the invention are ideally LUVs with a diameter in the range of 50-220nm.
- compositions comprising a population of LUVs with different diameters: (i) at least 80% by number should have diameters in the range of 20-220nm, (ii) the average diameter (Zav, by intensity) of the population is ideally in the range of 40-200nm, and/or (iii) the diameters should have a polydispersity index ⁇ 0.2.
- the liposome/RNA complexes of reference 1 are expected to have a diameter in the range of 600-800nm and to have a high polydispersity.
- RNA is preferably encapsulated within the liposomes, and so the liposome forms a outer layer around an aqueous RNA-containing core. This encapsulation has been found to protect RNA from RNase digestion.
- the liposomes can include some external RNA (e.g. on the surface of the liposomes), but at least half of the RNA (and ideally all of it) is encapsulated.
- RNA molecules can form microparticles to encapsulate or adsorb RNA.
- the use of a substantially non-toxic polymer means that a recipient can safely receive the particles, and the use of a biodegradable polymer means that the particles can be metabolised after delivery to avoid long-term persistence.
- Useful polymers are also sterilisable, to assist in preparing pharmaceutical grade formulations.
- Suitable non-toxic and biodegradable polymers include, but are not limited to, poly(a-hydroxy acids), polyhydroxy butyric acids, polylactones (including polycaprolactones), polydioxanones, polyvalerolactone, polyorthoesters, polyanhydrides, polycyanoacrylates, tyrosine-derived polycarbonates, polyvinyl-pyrrolidinones or polyester-amides, and combinations thereof.
- the microparticles are formed from poly(a-hydroxy acids), such as a poly(lactides) (“PLA”), copolymers of lactide and glycolide such as a poly(D,L-lactide-co-glycolide) (“PLG”), and copolymers of D,L-lactide and caprolactone.
- PLG polymers include those having a lactide/glycolide molar ratio ranging, for example, from 20:80 to 80:20 e.g. 25:75, 40:60, 45:55, 50:50, 55:45, 60:40, 75:25.
- Useful PLG polymers include those having a molecular weight between, for example, 5,000-200,000 Da e.g. between 10,000-100,000, 20,000-70,000, 30,000- 40,000, 40,000-50,000 Da.
- microparticles ideally have a diameter in the range of 0.02 ⁇ to 8 ⁇ .
- a composition comprising a population of microparticles with different diameters at least 80% by number should have diameters in the range of 0.03-7 ⁇ .
- a microparticle may include a cationic surfactant and/or lipid e.g. as disclosed in references 14 & 15.
- An alternative way of making polymeric microparticles is by molding and curing e.g. as disclosed in reference 16.
- Microparticles of the invention can have a zeta potential of between 40-100 mV.
- RNA can be adsorbed to the microparticles, and adsorption is facilitated by including cationic materials (e.g. cationic lipids) in the microparticle.
- cationic materials e.g. cationic lipids
- Oil-in-water emulsions are known for adjuvanting influenza vaccines e.g. the MF59TM adjuvant in the FLUADTM product, and the AS03 adjuvant in the PREPANDRIXTM product.
- RNA delivery according to the present invention can utilise an oil-in-water emulsion, provided that the emulsion includes one or more cationic molecules.
- a cationic lipid can be included in the emulsion to provide a positive droplet surface to which negatively-charged RNA can attach.
- the emulsion comprises one or more oils.
- Suitable oil(s) include those from, for example, an animal (such as fish) or a vegetable source.
- the oil is ideally biodegradable (metabolisable) and biocompatible.
- Sources for vegetable oils include nuts, seeds and grains. Peanut oil, soybean oil, coconut oil, and olive oil, the most commonly available, exemplify the nut oils.
- Jojoba oil can be used e.g. obtained from the jojoba bean.
- Seed oils include safflower oil, cottonseed oil, sunflower seed oil, sesame seed oil and the like.
- corn oil is the most readily available, but the oil of other cereal grains such as wheat, oats, rye, rice, teff, triticale and the like may also be used.
- 6-10 carbon fatty acid esters of glycerol and 1 ,2-propanediol, while not occurring naturally in seed oils, may be prepared by hydrolysis, separation and esterification of the appropriate materials starting from the nut and seed oils. Fats and oils from mammalian milk are metabolisable and so may be used. The procedures for separation, purification, saponification and other means necessary for obtaining pure oils from animal sources are well known in the art.
- cod liver oil cod liver oil
- shark liver oils and whale oil such as spermaceti exemplify several of the fish oils which may be used herein.
- a number of branched chain oils are synthesized biochemically in 5-carbon isoprene units and are generally referred to as terpenoids.
- Squalane the saturated analog to squalene
- Fish oils, including squalene and squalane are readily available from commercial sources or may be obtained by methods known in the art.
- oils are the tocopherols, particularly in combination with squalene.
- the oil phase of an emulsion includes a tocopherol
- any of the ⁇ , ⁇ , ⁇ , ⁇ , ⁇ or ⁇ tocopherols can be used, but a-tocopherols are preferred.
- D-a-tocopherol and DL-a-tocopherol can both be used.
- a preferred a-tocopherol is DL-a-tocopherol.
- An oil combination comprising squalene and a tocopherol e.g. DL-a-tocopherol
- the oil in the emulsion may comprise a combination of oils e.g. squalene and at least one further oil.
- the aqueous component of the emulsion can be plain water (e.g. w.f.i.) or can include further components e.g. solutes. For instance, it may include salts to form a buffer e.g. citrate or phosphate salts, such as sodium salts.
- Typical buffers include: a phosphate buffer; a Tris buffer; a borate buffer; a succinate buffer; a histidine buffer; or a citrate buffer.
- a buffered aqueous phase is preferred, and buffers will typically be included in the 5-20mM range.
- the emulsion also includes a cationic lipid.
- this lipid is a surfactant so that it can facilitate formation and stabilisation of the emulsion.
- Useful cationic lipids generally contains a nitrogen atom that is positively charged under physiological conditions e.g. as a tertiary or quaternary amine. This nitrogen can be in the hydrophilic head group of an amphiphilic surfactant.
- Useful cationic lipids include, but are not limited to: l,2-dioleoyloxy-3-(trimethylammonio)propane (DOTAP), 3'-[N- (N',N'-Dimethylaminoethane)-carbamoyl]Cholesterol (DC Cholesterol), dimethyldioctadecyl- ammonium (DDA e.g. the bromide), l,2-Dimyristoyl-3-Trimethyl-AmmoniumPropane (DMTAP), dipalmitoyl(C16:0)trimethyl ammonium propane (DPTAP), distearoyltrimethylammonium propane (DSTAP).
- DOTAP l,2-dioleoyloxy-3-(trimethylammonio)propane
- DC Cholesterol dimethyldioctadecyl- ammonium
- DMTAP dipalmitoyl(C16:0)trimethyl ammonium
- benzalkonium chloride BAK
- benzethonium chloride cetramide (which contains tetradecyltrimethylammonium bromide and possibly small amounts of dedecyltrimethylammonium bromide and hexadecyltrimethyl ammonium bromide)
- cetylpyridinium chloride CPC
- cetyl trimethylammonium chloride CAC
- ⁇ , ⁇ ', ⁇ '-polyoxyethylene (lO)-N- tallow-1,3 -diaminopropane dodecyltrimethylammonium bromide, hexadecyltrimethyl-ammonium bromide, mixed alkyl-trimethyl-ammonium bromide, benzyldimethyldodecylammonium chloride, benzyldimethylhexadecyl-ammonium chloride, benzyltrimethylammonium methoxide, cetyl
- cetylpyridinium bromide and cetylpyridinium chloride N-alkylpiperidinium salts, dicationic bolaform electrolytes (Ci 2 Me 6 ; C 12 BU 6 ), dialkylglycetylphosphorylcholine, lysolecithin, L-a dioleoyl-phosphatidylethanolamine, cholesterol hemisuccinate choline ester, lipopolyamines, including but not limited to dioctadecylamidoglycylspermine (DOGS), dipalmitoyl phosphatidyl ethanol-amidospermine (DPPES), lipopoly-L (or D)- lysine (LPLL, LPDL), poly (L (or D)-lysine conjugated to N- glutarylphosphatidylethanolamine, didodecyl glutamate ester with pendant amino group (Ci 2 GluPhC n N + ), ditetradecyl glutamate este
- the cationic lipid is preferably biodegradable (metabolisable) and biocompatible.
- an emulsion can include a non-ionic surfactant and/or a zwitterionic surfactant.
- surfactants include, but are not limited to: the polyoxyethylene sorbitan esters surfactants (commonly referred to as the Tweens), especially polysorbate 20 and polysorbate 80; copolymers of ethylene oxide (EO), propylene oxide (PO), and/or butylene oxide (BO), sold under the DOWFAXTM tradename, such as linear EO/PO block copolymers; octoxynols, which can vary in the number of repeating ethoxy (oxy- 1 ,2-ethanediyl) groups, with octoxynol-9 (Triton X-100, or t-octylphenoxypolyethoxyethanol) being of particular interest; (octylphenoxy)polyethoxyethanol (IGEPAL CA-630/NP-40); phospholipid
- Preferred surfactants for including in the emulsion are polysorbate 80 (Tween 80; polyoxyethylene sorbitan monooleate), Span 85 (sorbitan trioleate), lecithin and Triton X-100. Mixtures of these surfactants can be included in the emulsion e.g. Tween 80/Span 85 mixtures, or Tween 80/Triton-X100 mixtures.
- a combination of a polyoxyethylene sorbitan ester such as polyoxyethylene sorbitan monooleate (Tween 80) and an octoxynol such as t-octylphenoxy- polyethoxyethanol (Triton X-100) is also suitable.
- Another useful combination comprises laureth 9 plus a polyoxyethylene sorbitan ester and/or an octoxynol.
- Useful mixtures can comprise a surfactant with a HLB value in the range of 10-20 (e.g. polysorbate 80, with a HLB of 15.0) and a surfactant with a HLB value in the range of 1-10 (e.g. sorbitan trioleate, with a HLB of 1.8).
- Preferred amounts of oil (% by volume) in the final emulsion are between 2-20% e.g. 5-15%, 6-14%, 7-13%), 8-12%).
- a squalene content of about 4-6% or about 9-11% is particularly useful.
- Preferred amounts of surfactants (% by weight) in the final emulsion are between 0.001%) and 8%o.
- polyoxyethylene sorbitan esters such as polysorbate 80 0.2 to 4%, in particular between 0.4-0.6%, between 0.45-0.55%, about 0.5% or between 1.5-2%, between 1.8-2.2%, between 1.9-2.1%), about 2%, or 0.85-0.95%), or about 1%; sorbitan esters (such as sorbitan trioleate) 0.02 to 2%, in particular about 0.5% or about ⁇ %; octyl- or nonylphenoxy polyoxyethanols (such as Triton X-100) 0.001 to 0.1%, in particular 0.005 to 0.02%; polyoxyethylene ethers (such as laureth 9) 0.1 to 8%, preferably 0.1 to 10% and in particular 0.1 to 1% or about 0.5%.
- polyoxyethylene sorbitan esters such as polysorbate 80
- sorbate 80 0.2 to 4%, in particular between 0.4-0.6%, between 0.45-0.55%, about 0.5% or between 1.5-2%, between 1.8-2.2%,
- the absolute amounts of oil and surfactant, and their ratio, can be varied within wide limits while still forming an emulsion.
- a skilled person can easily vary the relative proportions of the components to obtain a desired emulsion, but a weight ratio of between 4: 1 and 5: 1 for oil and surfactant is typical (excess oil).
- the oil droplet size (diameter).
- the most effective emulsions have a droplet size in the submicron range.
- the droplet sizes will be in the range 50-750nm.
- the average droplet size is less than 250nm e.g. less than 200nm, less than 150nm.
- the average droplet size is usefully in the range of 80-180nm.
- at least 80%o (by number) of the emulsion's oil droplets are less than 250 nm in diameter, and preferably at least 90%o.
- Apparatuses for determining the average droplet size in an emulsion, and the size distribution are commercially available. These these typically use the techniques of dynamic light scattering and/or single-particle optical sensing e.g. the AccusizerTM and NicompTM series of instruments available from Particle Sizing Systems (Santa Barbara, USA), or the ZetasizerTM instruments from Malvern Instruments (UK), or the Particle Size Distribution Analyzer instruments from Horiba (Kyoto, Japan).
- the distribution of droplet sizes has only one maximum i.e. there is a single population of droplets distributed around an average (mode), rather than having two maxima.
- Preferred emulsions have a polydispersity of ⁇ 0.4 e.g. 0.3, 0.2, or less.
- Suitable emulsions with submicron droplets and a narrow size distribution can be obtained by the use of microfluidisation. This technique reduces average oil droplet size by propelling streams of input components through geometrically fixed channels at high pressure and high velocity. These streams contact channel walls, chamber walls and each other. The results shear, impact and cavitation forces cause a reduction in droplet size. Repeated steps of microfluidisation can be performed until an emulsion with a desired droplet size average and distribution are achieved.
- thermal methods can be used to cause phase inversion, as disclosed in reference 19. These methods can also provide a submicron emulsion with a tight particle size distribution.
- Preferred emulsions can be filter sterilised i. e. their droplets can pass through a 220nm filter. As well as providing a sterilisation, this procedure also removes any large droplets in the emulsion.
- the cationic lipid in the emulsion is DOTAP.
- the cationic oil-in-water emulsion may comprise from about 0.5 mg/ml to about 25 mg/ml DOTAP.
- the cationic oil-in-water emulsion may comprise DOTAP at from about 0.5 mg/ml to about 25 mg/ml, from about 0.6 mg/ml to about 25 mg/ml, from about 0.7 mg/ml to about 25 mg/ml, from about 0.8 mg/ml to about 25 mg/ml, from about 0.9 mg/ml to about 25 mg/ml, from about 1.0 mg/ml to about 25 mg/ml, from about 1.1 mg/ml to about 25 mg/ml, from about 1.2 mg/ml to about 25 mg/ml, from about 1.3 mg/ml to about 25 mg/ml, from about 1.4 mg/ml to about 25 mg/ml, from about 1.5 mg/ml to about 25 mg/ml, from about
- the cationic oil-in-water emulsion comprises from about 0.8 mg/ml to about 1.6 mg/ml DOTAP, such as 0.8 mg/ml, 1.2 mg/ml, 1.4 mg/ml or 1.6 mg/ml.
- the cationic lipid is DC Cholesterol.
- the cationic oil-in-water emulsion may comprise DC Cholesterol at from about 0.1 mg/ml to about 5 mg/ml DC Cholesterol.
- the cationic oil-in-water emulsion may comprise DC Cholesterol from about 0.1 mg/ml to about 5 mg/ml, from about 0.2 mg/ml to about 5 mg/ml, from about 0.3 mg/ml to about 5 mg/ml, from about 0.4 mg/ml to about 5 mg/ml, from about 0.5 mg/ml to about 5 mg/ml, from about 0.62 mg/ml to about 5 mg/ml, from about 1 mg/ml to about 5 mg/ml, from about 1.5 mg/ml to about 5 mg/ml, from about 2 mg/ml to about 5 mg/ml, from about 2.46 mg/ml to about 5 mg/ml, from about 3 mg/ml to about 5 mg/ml,
- the cationic lipid is DDA.
- the cationic oil-in-water emulsion may comprise from about 0.1 mg/ml to about 5 mg/ml DDA.
- the cationic oil-in-water emulsion may comprise DDA at from about 0.1 mg/ml to about 5 mg/ml, from about 0.1 mg/ml to about 4.5 mg/ml, from about 0.1 mg/ml to about 4 mg/ml, from about 0.1 mg/ml to about 3.5 mg/ml, from about 0.1 mg/ml to about 3 mg/ml, from about 0.1 mg/ml to about 2.5 mg/ml, from about 0.1 mg/ml to about 2 mg/ml, from about 0.1 mg/ml to about 1.5 mg/ml, from about 0.1 mg/ml to about 1.45 mg/ml, from about 0.2 mg/ml to about 5 mg/ml, from about 0.3 mg/ml to about 5 mg/ml, from about 0.4 mg/m
- the cationic oil-in-water emulsion may comprise DDA at about 20 mg/ml, about 21 mg/ml, about 21.5 mg/ml, about 21.6 mg/ml, about 25 mg/ml.
- the cationic oil-in-water emulsion comprises from about 0.73 mg/ml to about 1.45 mg/ml DDA, such as 1.45 mg/ml.
- Certain preferred compositions of the invention for administration to a patient comprise squalene, span 85, polysorbate 80, and DOTAP.
- squalene may be present at 5-15mg/ml; span 85 may be present at 0.5-2mg/ml; polysorbate 80 may be present at 0.5-2mg/ml; and DOTAP may be present at 0.1-lOmg/ml.
- the emulsion can include the same amount (by volume) of span 85 and polysorbate 80.
- the emulsion can include more squalene than surfactant.
- the emulsion can include more squalene than DOTAP.
- the invention involves in vivo delivery of RNA which encodes an immunogen.
- the RNA can trigger innate immunity pathways and is also translated, leading to expression of the immunogen.
- RNA is +-stranded, and so it can be translated without needing any intervening replication steps such as reverse transcription.
- Preferred +-stranded RNAs are self-replicating.
- a self-replicating RNA molecule (replicon) can, when delivered to a mammalian cell even without any proteins, lead to the production of multiple daughter RNAs by transcription from itself (via an antisense copy which it generates from itself).
- a self-replicating RNA molecule is thus typically a +-strand molecule which can be directly translated after delivery to a cell, and this translation provides a RNA-dependent RNA polymerase which then produces both antisense and sense transcripts from the delivered RNA.
- the delivered RNA leads to the production of multiple daughter RNAs.
- RNAs may be translated themselves to provide in situ expression of an encoded immunogen, or may be transcribed to provide further transcripts with the same sense as the delivered RNA which are translated to provide in situ expression of the immunogen.
- the overall results of this sequence of transcriptions is a huge amplification in the number of the introduced replicon RNAs and so the encoded immunogen becomes a major polypeptide product of the cells.
- RNA replicon One suitable system for achieving self-replication is to use an alphavirus-based RNA replicon. These +-stranded replicons are translated after delivery to a cell to give of a replicase (or replicase- transcriptase). The replicase is translated as a polyprotein which auto-cleaves to provide a replication complex which creates genomic— strand copies of the +-strand delivered RNA. These— strand transcripts can themselves be transcribed to give further copies of the +-stranded parent RNA and also to give a subgenomic transcript which encodes the immunogen. Translation of the subgenomic transcript thus leads to in situ expression of the immunogen by the infected cell.
- a replicase or replicase- transcriptase
- the replicase is translated as a polyprotein which auto-cleaves to provide a replication complex which creates genomic— strand copies of the +-strand delivered RNA.
- These— strand transcripts can themselves be transcribed to give further copies of the
- a preferred self-replicating RNA molecule thus encodes (i) a RNA-dependent RNA polymerase which can transcribe RNA from the self-replicating RNA molecule and (ii) an immunogen.
- the polymerase can be an alphavirus replicase e.g. comprising one or more of alphavirus proteins nsPl, nsP2, nsP3 and nsP4.
- RNA molecule of the invention does not encode alphavirus structural proteins.
- a preferred self-replicating RNA can lead to the production of genomic RNA copies of itself in a cell, but not to the production of RNA- containing virions.
- the inability to produce these virions means that, unlike a wild-type alphavirus, the self-replicating RNA molecule cannot perpetuate itself in infectious form.
- RNA molecule useful with the invention may have two open reading frames.
- the first (5') open reading frame encodes a replicase; the second (3') open reading frame encodes an immunogen.
- the RNA may have additional (e.g. downstream) open reading frames e.g. to encode further immunogens (see below) or to encode accessory polypeptides.
- a self-replicating RNA molecule can have a 5' sequence which is compatible with the encoded replicase.
- Self-replicating RNA molecules can have various lengths but they are typically 5000-25000 nucleotides long e.g. 8000-15000 nucleotides, or 9000-12000 nucleotides. Thus the RNA is longer than seen in siRNA delivery.
- a RNA molecule useful with the invention may have a 5' cap (e.g. a 7-methylguanosine). This cap can enhance in vivo translation of the RNA.
- the 5' nucleotide of a RNA molecule useful with the invention may have a 5' triphosphate group. In a capped RNA this may be linked to a 7-methylguanosine via a 5'-to-5' bridge. A 5' triphosphate can enhance RIG-I binding.
- a RNA molecule may have a 3' poly-A tail. It may also include a poly-A polymerase recognition sequence (e.g. AAUAAA) near its 3' end.
- AAUAAA poly-A polymerase recognition sequence
- RNA molecule useful with the invention will typically be single-stranded.
- Single-stranded RNAs can generally initiate an adjuvant effect by binding to TLR7, TLR8, RNA helicases and/or PKR.
- RNA delivered in double-stranded form can bind to TLR3, and this receptor can also be triggered by dsRNA which is formed either during replication of a single-stranded RNA or within the secondary structure of a single-stranded RNA.
- RNA molecule useful with the invention can conveniently be prepared by in vitro transcription (IVT).
- IVT can use a (cDNA) template created and propagated in plasmid form in bacteria, or created synthetically (for example by gene synthesis and/or polymerase chain-reaction (PCR) engineering methods).
- a DNA-dependent RNA polymerase such as the bacteriophage T7, T3 or SP6 RNA polymerases
- Appropriate capping and poly-A addition reactions can be used as required (although the replicon's poly-A is usually encoded within the DNA template).
- RNA polymerases can have stringent requirements for the transcribed 5' nucleotide(s) and in some embodiments these requirements must be matched with the requirements of the encoded replicase, to ensure that the IVT -transcribed RNA can function efficiently as a substrate for its self-encoded replicase.
- the self-replicating RNA can include (in addition to any 5' cap structure) one or more nucleotides having a modified nucleobase.
- the RNA can comprise m5C (5-methylcytidine), m5U (5-methyluridine), m6A (N6-methyladenosine), s2U (2-thiouridine), Um (2'-0-methyluridine), mlA (1-methyladenosine); m2A (2-methyladenosine); Am (2'-0- methyladenosine); ms2m6A (2-methylthio-N6-methyladenosine); i6A (N6-isopentenyladenosine); ms2i6A (2-methylthio-N6isopentenyladenosine); io6A (N6-(cis-hydroxyisopentenyl)adenosine); ms2io6A (2-methylthio-N6-(cis-hydroxyisopenten
- a self-replicating RNA can include one or more modified pyrimidine nucleobases, such as pseudouridine and/or 5-methylcytosine residues.
- the RNA includes no modified nucleobases, and may include no modified nucleotides i.e. all of the nucleotides in the RNA are standard A, C, G and U ribonucleotides (except for any 5' cap structure, which may include a 7'-methylguanosine).
- the RNA may include a 5' cap comprising a 7'-methylguanosine, and the first 1, 2 or 3 5' ribonucleotides may be methylated at the 2' position of the ribose.
- a RNA used with the invention ideally includes only phosphodiester linkages between nucleosides, but in some embodiments it can contain phosphoramidate, phosphorothioate, and/or methylphosphonate linkages.
- administered RNA includes fewer than 10 different species of RNA e.g. 5, 4, 3, or 2 different species; most preferably, a composition includes a single RNA species i.e. all RNA molecules in the composition (e.g. within a liposome) have the same sequence and same length.
- RNA molecules used with the invention encode a polypeptide immunogen. After administration of the RNA the immunogen is translated in vivo and can elicit an immune response in the recipient.
- the immunogen may elicit an immune response against a bacterium, a virus, a fungus or a parasite (or, in some embodiments, against an allergen; and in other embodiments, against a tumor antigen).
- the immune response may comprise an antibody response (usually including IgG) and/or a cell-mediated immune response.
- the polypeptide immunogen will typically elicit an immune response which recognises the corresponding bacterial, viral, fungal or parasite (or allergen or tumour) polypeptide, but in some embodiments the polypeptide may act as a mimotope to elicit an immune response which recognises a bacterial, viral, fungal or parasite saccharide.
- the immunogen will typically be a surface polypeptide e.g. an adhesin, a hemagglutinin, an envelope glycoprotein, a spike glycoprotein, etc.
- RNA molecules can encode a single polypeptide immunogen or multiple polypeptides. Multiple immunogens can be presented as a single polypeptide immunogen (fusion polypeptide) or as separate polypeptides. If immunogens are expressed as separate polypeptides then one or more of these may be provided with an upstream IRES or an additional viral promoter element. Alternatively, multiple immunogens may be expressed from a polyprotein that encodes individual immunogens fused to a short autocatalytic protease ⁇ e.g. foot-and-mouth disease virus 2A protein), or as inteins.
- the RNA encodes an immunogen.
- the invention does not encompass RNA which encodes a firefly luciferase or which encodes a fusion protein of E.coli ⁇ -galactosidase or which encodes a green fluorescent protein (GFP).
- GFP green fluorescent protein
- the RNA is not total mouse thymus RNA.
- useful immunogens include, but are not limited to, membrane proteins such as adhesins, autotransporters, toxins, iron acquisition proteins, and factor H binding protein. A combination of three useful polypeptides is disclosed in reference 23.
- Streptococcus pneumoniae useful polypeptide immunogens are disclosed in reference 24. These include, but are not limited to, the RrgB pilus subunit, the beta-N-acetyl-hexosaminidase precursor (spr0057), spr0096, General stress protein GSP-781 (spr2021, SP2216), serine/threonine kinase StkP (SP1732), and pneumococcal surface adhesin PsaA.
- Streptococcus pyogenes include, but are not limited to, the polypeptides disclosed in references 25 and 26.
- Bordetella pertussis Useful pertussis immunogens include, but are not limited to, pertussis toxin or toxoid (PT), filamentous haemagglutinin (FHA), pertactin, and agglutinogens 2 and 3.
- Staphylococcus aureus Useful immunogens include, but are not limited to, the polypeptides disclosed in reference 27, such as a hemolysin, esxA, esxB, ferrichrome-binding protein (sta006) and/or the staOl l lipoprotein.
- Clostridium tetani the typical immunogen is tetanus toxoid.
- Cornynebacterium diphtheriae the typical immunogen is diphtheria toxoid.
- Haemophilus influenzae Useful immunogens include, but are not limited to, the polypeptides disclosed in references 28 and 29.
- Streptococcus agalactiae useful immunogens include, but are not limited to, the polypeptides disclosed in reference 25.
- Chlamydia trachomatis Useful immunogens include, but are not limited to, PepA, LcrE, ArtJ, DnaK, CT398, OmpH-like, L7/L12, OmcA, AtoS, CT547, Eno, HtrA and MurG ⁇ e.g. as disclosed in reference 30.
- LcrE [31] and HtrA [32] are two preferred immunogens.
- Chlamydia pneumoniae Useful immunogens include, but are not limited to, the polypeptides disclosed in reference 33.
- Helicobacter pylori Useful immunogens include, but are not limited to, CagA, VacA, NAP, and/or urease [34].
- Escherichia coli Useful immunogens include, but are not limited to, immunogens derived from enterotoxigenic E. coli (ETEC), enteroaggregative E. coli (EAggEC), diffusely adhering E. coli (DAEC), enteropathogenic E. coli (EPEC), extraintestinal pathogenic E. coli (ExPEC) and/or enterohemorrhagic E. coli (EHEC).
- ExPEC strains include uropathogenic E.coli (UPEC) and meningitis/sepsis-associated E.coli (MNEC).
- UPEC uropathogenic E.coli
- MNEC meningitis/sepsis-associated E.coli
- Useful UPEC polypeptide immunogens are disclosed in references 35 and 36.
- Useful MNEC immunogens are disclosed in reference 37.
- a useful immunogen for several E.coli types is AcfD [38].
- Yersinia pestis Useful immunogens include, but are not limited to, those disclosed in references 39 and 40.
- Brucella such as B. abortus, B.canis, B.melitensis, B.neotomae, B.ovis, B.suis, B.pinnipediae.
- Francisella such as F.novicida, F.philomiragia, F.tularensis .
- Salmonella typhi Salmonella typhi
- Useful immunogens can be from an influenza A, B or C virus, such as the hemagglutinin, neuraminidase or matrix M2 proteins. Where the immunogen is an influenza A virus hemagglutinin it may be from any subtype e.g. HI, H2, H3, H4, H5, H6, H7, H8, H9, H10, Hl l, H12, H13, H14, H15 or H16.
- Viral immunogens include, but are not limited to, those derived from Pneumoviruses (e.g. respiratory syncytial virus, RSV), Rubulaviruses (e.g. mumps virus), Paramyxoviruses (e.g. parainfluenza virus), Metapneumoviruses and Morbilliviruses (e.g. measles virus).
- Pneumoviruses e.g. respiratory syncytial virus, RSV
- Rubulaviruses e.g. mumps virus
- Paramyxoviruses e.g. parainfluenza virus
- Metapneumoviruses e.g. measles virus
- Poxviridae Viral immunogens include, but are not limited to, those derived from Orthopoxvirus such as Variola vera, including but not limited to, Variola major and Variola minor.
- Viral immunogens include, but are not limited to, those derived from Picornaviruses, such as Enteroviruses, Rhinoviruses, Heparnavirus, Cardioviruses and Aphthoviruses.
- the enterovirus is a poliovirus e.g. a type 1, type 2 and/or type 3 poliovirus.
- the enterovirus is an EV71 enterovirus.
- the enterovirus is a coxsackie A or B virus.
- Bunyavirus Viral immunogens include, but are not limited to, those derived from an Orthobunyavirus, such as California encephalitis virus, a Phlebovirus, such as Rift Valley Fever virus, or a Nairovirus, such as Crimean-Congo hemorrhagic fever virus.
- Heparnavirus Viral immunogens include, but are not limited to, those derived from a Heparnavirus, such as hepatitis A virus (HAV).
- HAV hepatitis A virus
- Viral immunogens include, but are not limited to, those derived from a filovirus, such as an Ebola virus (including a Zaire, Ivory Coast, Reston or Sudan ebolavirus) or a Marburg virus.
- a filovirus such as an Ebola virus (including a Zaire, Ivory Coast, Reston or Sudan ebolavirus) or a Marburg virus.
- Viral immunogens include, but are not limited to, those derived from a Togavirus, such as a Rubivirus, an Alphavirus, or an Arterivirus. This includes rubella virus.
- Flavivirus Viral immunogens include, but are not limited to, those derived from a Flavivirus, such as Tick-borne encephalitis (TBE) virus, Dengue (types 1, 2, 3 or 4) virus, Yellow Fever virus, Japanese encephalitis virus, Kyasanur Forest Virus, West Nile encephalitis virus, St.
- TBE Tick-borne encephalitis
- Dengue types 1, 2, 3 or 4
- Yellow Fever virus Japanese encephalitis virus
- Kyasanur Forest Virus Kyasanur Forest Virus
- West Nile encephalitis virus St.
- Viral immunogens include, but are not limited to, those derived from a Pestivirus, such as Bovine viral diarrhea (BVDV), Classical swine fever (CSFV) or Border disease (BDV).
- BVDV Bovine viral diarrhea
- CSFV Classical swine fever
- BDV Border disease
- Viral immunogens include, but are not limited to, those derived from a Hepadnavirus, such as Hepatitis B virus.
- a composition can include hepatitis B virus surface antigen (HBsAg).
- a composition can include an immunogen from a hepatitis C virus, delta hepatitis virus, hepatitis E virus, or hepatitis G virus.
- Viral immunogens include, but are not limited to, those derived from a Rhabdovirus, such as a Lyssavirus ⁇ e.g. a Rabies virus) and Vesiculovirus (VSV).
- a Rhabdovirus such as a Lyssavirus ⁇ e.g. a Rabies virus
- VSV Vesiculovirus
- Viral immunogens include, but are not limited to, those derived from Calciviridae, such as Norwalk virus (Norovirus), and Norwalk-like Viruses, such as Hawaii Virus and Snow Mountain Virus.
- Coronavirus include, but are not limited to, those derived from a SARS coronavirus, avian infectious bronchitis (IBV), Mouse hepatitis virus (MHV), and Porcine transmissible gastroenteritis virus (TGEV).
- the coronavirus immunogen may be a spike polypeptide.
- Retrovirus Viral immunogens include, but are not limited to, those derived from an Oncovirus, a Lentivirus ⁇ e.g. HIV-1 or HIV-2) or a Spumavirus.
- Viral immunogens include, but are not limited to, those derived from an Orthoreovirus, a Rotavirus, an Orbivirus, or a Coltivirus.
- Parvovirus Viral immunogens include, but are not limited to, those derived from Parvovirus B19.
- Viral immunogens include, but are not limited to, those derived from a human herpesvirus, such as, by way of example only, Herpes Simplex Viruses (HSV) ⁇ e.g. HSV types 1 and 2), Varicella-zoster virus (VZV), Epstein-Barr virus (EBV), Cytomegalovirus (CMV), Human Herpesvirus 6 (HHV6), Human Herpesvirus 7 (HHV7), and Human
- HSV Herpes Simplex Viruses
- VZV Varicella-zoster virus
- EBV Epstein-Barr virus
- CMV Cytomegalovirus
- HHV6 Human Herpesvirus 6
- HHV7 Human Herpesvirus 7
- Herpesvirus 8 (HHV8).
- Viral immunogens include, but are not limited to, those derived from Papillomaviruses and Polyomaviruses.
- the (human) papillomavirus may be of serotype 1, 2, 4, 5, 6, 8, 11, 13, 16, 18, 31, 33, 35, 39, 41, 42, 47, 51, 57, 58, 63 or 65 e.g. from one or more of serotypes 6, 11, 16 and/or 18.
- Viral immunogens include those derived from adenovirus serotype 36 (Ad-36).
- the immunogen elicits an immune response against a virus which infects fish, such as: infectious salmon anemia virus (ISAV), salmon pancreatic disease virus (SPDV), infectious pancreatic necrosis virus (IPNV), channel catfish virus (CCV), fish lymphocystis disease virus (FLDV), infectious hematopoietic necrosis virus (IHNV), koi herpesvirus, salmon picorna-like virus (also known as picorna-like virus of atlantic salmon), landlocked salmon virus (LSV), atlantic salmon rotavirus (ASR), trout strawberry disease virus (TSD), coho salmon tumor virus (CSTV), or viral hemorrhagic septicemia virus (VHSV).
- infectious salmon anemia virus ISAV
- SPDV salmon pancreatic disease virus
- IPNV infectious pancreatic necrosis virus
- CCV channel catfish virus
- FLDV fish lymphocystis disease virus
- IHNV infectious hematopoietic necrosis virus
- Fungal immunogens may be derived from Dermatophytres, including: Epidermophyton floccusum, Microsporum audouini, Microsporum canis, Microsporum distortum, Microsporum equinum, Microsporum gypsum, Microsporum nanum, Trichophyton concentricum, Trichophyton equinum, Trichophyton gallinae, Trichophyton gypseum, Trichophyton megnini, Trichophyton mentagrophytes, Trichophyton quinckeanum, Trichophyton rubrum, Trichophyton schoenleini, Trichophyton tonsurans, Trichophyton verrucosum, T. verrucosum var. album, var.
- the immunogen elicits an immune response against a parasite from the Plasmodium genus, such as P. falciparum, P.vivax, P.malariae or P. ovale.
- the invention may be used for immunising against malaria.
- the immunogen elicits an immune response against a parasite from the Caligidae family, particularly those from the Lepeophtheirus and Caligus genera e.g. sea lice such as Lepeophtheirus salmonis or Caligus rogercresseyi.
- the immunogen elicits an immune response against: pollen allergens (tree-, herb, weed-, and grass pollen allergens); insect or arachnid allergens (inhalant, saliva and venom allergens, e.g. mite allergens, cockroach and midges allergens, hymenopthera venom allergens); animal hair and dandruff allergens (from e.g. dog, cat, horse, rat, mouse, etc.); and food allergens (e.g. a gliadin).
- pollen allergens tree-, herb, weed-, and grass pollen allergens
- insect or arachnid allergens inhalant, saliva and venom allergens, e.g. mite allergens, cockroach and midges allergens, hymenopthera venom allergens
- animal hair and dandruff allergens from e.g. dog, cat, horse
- venom allergens including such originating from stinging or biting insects such as those from the taxonomic order of Hymenoptera including bees (Apidae), wasps (Vespidea), and ants (Formicoidae).
- the immunogen is a tumor antigen selected from: (a) cancer-testis antigens such as NY-ESO-1, SSX2, SCP1 as well as RAGE, BAGE, GAGE and MAGE family polypeptides, for example, GAGE-1, GAGE-2, MAGE-1, MAGE-2, MAGE-3, MAGE-4, MAGE-5, MAGE-6, and MAGE- 12 (which can be used, for example, to address melanoma, lung, head and neck, NSCLC, breast, gastrointestinal, and bladder tumors; (b) mutated antigens, for example, p53 (associated with various solid tumors, e.g., colorectal, lung, head and neck cancer), p21/Ras (associated with, e.g., melanoma, pancreatic cancer and colorectal cancer), CDK4 (associated with, e.g., melanoma), MUMl (associated with, e.g., melanoma), caspase
- melanoma-melanocyte differentiation antigens such as MART-l/Melan A, gplOO, MC1R, melanocyte-stimulating hormone receptor, tyrosinase, tyrosinase related protein- 1/TRPl and tyrosinase related protein-2/TRP2 (associated with, e.g., melanoma);
- prostate associated antigens such as PAP, PSA, PSMA, PSH-P1, PSM-P1, PSM-P2, associated with e.g., prostate cancer;
- immunoglobulin idiotypes associated with myeloma and B cell lymphomas, for example).
- tumor immunogens include, but are not limited to, pi 5, Hom/Mel-40, H-Ras, E2A-PRL, H4-RET, IGH-IGK, MYL-RAR, Epstein Barr virus antigens, EBNA, human papillomavirus (HPV) antigens, including E6 and E7, hepatitis B and C virus antigens, human T-cell lymphotropic virus antigens, TSP-180, pl85erbB2, pl80erbB-3, c-met, mn-23Hl, TAG-72-4, CA 19-9, CA 72-4, CAM 17.1, NuMa, K-ras, pl6, TAGE, PSCA, CT7, 43-9F, 5T4, 791 Tgp72, beta-HCG, BCA225, BTAA, CA 125, CA 15-3 (CA 27.29YBCAA), CA 195, CA 242, CA-50, CAM43, CD68 ⁇ KP1, CO-029,
- RNA will be administered as a component in a pharmaceutical composition for immunising subjects against various diseases.
- compositions will typically include a pharmaceutically acceptable carrier in addition to the RNA, often as part of a delivery system as described above. A thorough discussion of pharmaceutically acceptable carriers is available in reference 41.
- a pharmaceutical composition of the invention may include one or more small molecule immunopotentiators.
- the composition may include a TLR2 agonist (e.g. Pam3CSK4), a TLR4 agonist (e.g. an aminoalkyl glucosaminide phosphate, such as E6020), a TLR7 agonist (e.g. imiquimod), a TLR8 agonist (e.g. resiquimod) and/or a TLR9 agonist (e.g. IC31).
- a TLR2 agonist e.g. Pam3CSK4
- a TLR4 agonist e.g. an aminoalkyl glucosaminide phosphate, such as E6020
- TLR7 agonist e.g. imiquimod
- a TLR8 agonist e.g. resiquimod
- TLR9 agonist e.g. IC31
- RNA is encapsulated
- such agonist(s) are also encapsulated with the RNA, but in other embodiments they are unencapsulated.
- a RNA is adsorbed to a particle
- such agonist(s) are also adsorbed with the RNA, but in other embodiments they are unadsorbed.
- compositions of the invention may include the particles in plain water (e.g. w.f.i.) or in a buffer e.g. a phosphate buffer, a Tris buffer, a borate buffer, a succinate buffer, a histidine buffer, or a citrate buffer.
- Buffer salts will typically be included in the 5-20mM range.
- compositions of the invention may have a pH between 5.0 and 9.5 e.g. between 6.0 and 8.0.
- compositions of the invention may include sodium salts (e.g. sodium chloride) to give tonicity.
- sodium salts e.g. sodium chloride
- a concentration of 10+2 mg/ml NaCl is typical e.g. about 9 mg/ml.
- compositions of the invention may include metal ion chelators. These can prolong RNA stability by removing ions which can accelerate phosphodiester hydrolysis.
- a composition may include one or more of EDTA, EGTA, BAPTA, pentetic acid, etc..
- chelators are typically present at between 10-500 ⁇ e.g. O. lmM.
- a citrate salt, such as sodium citrate, can also act as a chelator, while advantageously also providing buffering activity.
- compositions of the invention may have an osmolality of between 200 mOsm/kg and 400 mOsm/kg, e.g. between 240-360 mOsm/kg, or between 290-310 mOsm/kg.
- compositions of the invention may include one or more preservatives, such as thiomersal or 2-phenoxyethanol.
- preservatives such as thiomersal or 2-phenoxyethanol.
- Mercury-free compositions are preferred, and preservative-free vaccines can be prepared.
- compositions of the invention are preferably sterile.
- Pharmaceutical compositions of the invention are preferably non-pyrogenic e.g. containing ⁇ 1 EU (endotoxin unit, a standard measure) per dose, and preferably ⁇ 0.1 EU per dose.
- compositions of the invention are preferably gluten free.
- compositions of the invention may be prepared in unit dose form.
- a unit dose may have a volume of between 0.1-1.0ml e.g. about 0.5ml.
- compositions may be prepared as injectables, either as solutions or suspensions.
- the composition may be prepared for pulmonary administration e.g. by an inhaler, using a fine spray.
- the composition may be prepared for nasal, aural or ocular administration e.g. as spray or drops. Injectables for intramuscular administration are typical.
- the RNA content of compositions of the invention is expressed in terms of the amount of RNA per unit dose. RNA is readily quantified using available techniques.
- RNAs are not delivered in combination with ribosomes and so pharmaceutical compositions of the invention are ribosome-free.
- RNA delivery according to the invention is for eliciting an immune response in vivo against an immunogen of interest.
- the immune response is preferably protective and preferably involves antibodies and/or cell-mediated immunity.
- the method may raise a booster response.
- RNA-containing compositions are immunogenic, and are more preferably vaccine compositions.
- Vaccines according to the invention may either be prophylactic ⁇ i.e. to prevent infection) or therapeutic ⁇ i.e. to treat infection), but will typically be prophylactic.
- the mammal immunised according to the present invention is a large mammal, such as a human or a large veterinary mammal ⁇ e.g. horses, cattle, deer, goats, pigs, camels, antelope, elephants).
- the human is preferably a child ⁇ e.g. a toddler or infant) or a teenager; where the vaccine is for therapeutic use, the human is preferably a teenager or an adult.
- a vaccine intended for children may also be administered to adults e.g. to assess safety, dosage, immunogenicity, etc.
- Vaccines prepared according to the invention may be used to treat both children and adults.
- a human patient may be less than 1 year old, less than 5 years old, 1-5 years old, 5-15 years old, 15-55 years old, or at least 55 years old.
- Preferred patients for receiving the vaccines are the elderly ⁇ e.g. >50 years old, >60 years old, and preferably >65 years), the young ⁇ e.g. ⁇ 5 years old), hospitalised patients, healthcare workers, armed service and military personnel, pregnant women, the chronically ill, or immunodeficient patients.
- the vaccines are not suitable solely for these groups, however, and may be used more generally in a population.
- compositions of the invention will generally be administered directly to a patient.
- Direct delivery may be accomplished by parenteral injection (e.g. subcutaneously, intraperitoneally, intravenously, intramuscularly, or to the interstitial space of a tissue; unlike reference 1 , intraglossal injection is not typically used with the present invention), or mucosally, such as by rectal, oral (e.g. tablet, spray), vaginal, topical, transdermal or transcutaneous, intranasal, ocular, aural, pulmonary or other mucosal administration.
- Injection may be via a needle (e.g. a hypodermic needle), but needle-free injection may alternatively be used.
- a typical intramuscular dose is 0.5 ml.
- the invention may be used to elicit systemic and/or mucosal immunity, preferably to elicit an enhanced systemic and/or mucosal immunity.
- Dosage can be by a single unit dose schedule or a multiple unit dose schedule. Multiple doses may be used in a primary immunisation schedule and/or in a booster immunisation schedule. In a multiple dose schedule the various doses may be given by the same or different routes e.g. a parenteral prime and mucosal boost, a mucosal prime and parenteral boost, etc. Multiple doses will typically be administered at least 1 week apart (e.g. about 2 weeks, about 3 weeks, about 4 weeks, about 6 weeks, about 8 weeks, about 10 weeks, about 12 weeks, about 16 weeks, etc.). In one embodiment, unit doses may be administered approximately 6 weeks, 10 weeks and 14 weeks after birth, e.g.
- two primary unit doses are administered about two months apart, e.g. about 7, 8 or 9 weeks apart, followed by one or more booster unit doses about 6 months to 1 year after the second primary dose, e.g. about 6, 8, 10 or 12 months after the second primary dose.
- three primary doses are administered about two months apart, e.g. about 7, 8 or 9 weeks apart, followed by one or more booster doses about 6 months to 1 year after the third primary dose, e.g. about 6, 8, 10, or 12 months after the third primary dose.
- the RNA includes no modified nucleotides (see above). In other embodiments the RNA can optionally include at least one modified nucleotide, provided that one or more of the following features (already disclosed above) is also required:
- the liposome comprises DSDMA, DODMA, DLinDMA and/or DLenDMA.
- the hydrophilic portion of a lipid in the liposome is PEGylated.
- RNA is encapsulated in a liposome
- at least 80% by number of the liposomes have diameters in the range of 20-220nm.
- the microparticle is a non-toxic and biodegradable polymer microparticle.
- the microparticles have a diameter in the range of 0.02 ⁇ to 8 ⁇ .
- RNA is delivered with a microparticle
- at least 80% by number of the microparticles have a diameter in the range of 0.03-7 ⁇ .
- the composition is lyophilised.
- the emulsion comprises a biodegradable oil (e.g. squalene).
- a biodegradable oil e.g. squalene
- the emulsion includes one or more cationic molecules e.g. one or more cationic lipids.
- the RNA has a 3' poly-A tail, and the immunogen can elicits an immune response in vivo against a bacterium, a virus, a fungus or a parasite.
- RNA is delivered in combination with a metal ion chelator with a delivery system selected from (i) liposomes (ii) non-toxic and biodegradable polymer microparticles (iii) cationic submicron oil-in-water emulsions.
- composition comprising X may consist exclusively of X or may include something additional e.g. X + Y.
- TLR7 is the Toll-like receptor 7. It is a single membrane-spanning receptor which plays a key role in the innate immune system.
- Known TLR7 agonists include e.g. imiquimod. "TLR7" is the approved HGNC name for the gene encoding this receptor, and its unique HGNC ID is HGNC: 15631.
- the RefSeq sequence for the human TLR7 gene is GI: 67944638.
- TLR8 is the Toll-like receptor 8. It is a single membrane-spanning receptor which plays a key role in the innate immune system.
- Known TLR8 agonists include e.g. resiquimod. "TLR8" is the approved HGNC name for the gene encoding this receptor, and its unique HGNC ID is HGNC: 15632.
- the RefSeq sequence for the human TLR8 gene is GL20302165.
- RLR-1 The RIG-I-like receptor family includes various RNA helicases which play key roles in the innate immune system[49].
- RLR-1 also known as RIG-I or retinoic acid inducible gene I
- RLR-1 helicase has two caspase recruitment domains near its N-terminus.
- the approved HGNC name for the gene encoding the RLR-1 helicase is "DDX58" (for DEAD (Asp-Glu- Ala-Asp) box polypeptide 58) and the unique HGNC ID is HGNC: 19102.
- the RefSeq sequence for the human RLR-1 gene is GL77732514.
- RLR-2 (also known as MDA5 or melanoma differentiation-associated gene 5) also has two caspase recruitment domains near its N-terminus.
- the approved HGNC name for the gene encoding the RLR-2 helicase is "IFIH1" (for interferon induced with helicase C domain 1) and the unique HGNC ID is HGNC: 18873.
- the RefSeq sequence for the human RLR-2 gene is GI: 27886567.
- RLR- 3 (also known as LGP2 or laboratory of genetics and physiology 2) has no caspase recruitment domains.
- the approved HGNC name for the gene encoding the RLR-3 helicase is "DHX58" (for DEXH (Asp-Glu- X-His) box polypeptide 58) and the unique HGNC ID is HGNC:29517.
- the RefSeq sequence for the human RLR-3 gene is GI: 149408121.
- FIG. 1 shows a gel with stained RNA. Lanes show (1) markers (2) naked replicon (3) replicon after
- FIG. 2 is an electron micrograph of liposomes.
- FIG. 3 shows protein expression (as relative light units, RLU) at days 1, 3 and 6 after delivery of RNA as a virion-packaged replicon (squares), naked RNA (triangles), or as microparticles (circles).
- FIG. 4 shows a gel with stained RNA. Lanes show (1) markers (2) naked replicon (3) replicon encapsulated in liposome (4) liposome treated with RNase then subjected to phenol/chloroform extraction.
- FIG. 6 shows protein expression at days 1, 3 and 6 after delivery of four different doses of liposome- encapsulated RNA.
- FIG. 7 shows anti-F IgG titers in animals receiving virion-packaged replicon (VRP or VSRP), ⁇ g naked RNA, and ⁇ g liposome-encapsulated RNA.
- FIG. 8 shows anti-F IgG titers in animals receiving VRP, ⁇ g naked RNA, and O.lg or ⁇ g liposome-encapsulated RNA.
- FIG. 9 shows neutralising antibody titers in animals receiving VRP or either O.lg or ⁇ g liposome- encapsulated RNA.
- FIG. 10 shows expression levels after delivery of a replicon as naked RNA (circles), liposome- encapsulated RNA (triangle & square), or as a lipoplex (inverted triangle).
- FIG. 11 shows F-specific IgG titers (2 weeks after second dose) after delivery of a replicon as naked RNA (0.01- ⁇ g), liposome-encapsulated RNA (0.01- ⁇ g), or packaged as a virion (VRP, 10 6 infectious units or IU).
- FIG. 12 shows F-specific IgG titers (circles) and PRNT titers (squares) after delivery of a replicon as naked RNA ( ⁇ g), liposome-encapsulated RNA (0.1 or ⁇ g), or packaged as a virion (VRP, 10 6 IU). Titers in naive mice are also shown. Solid lines show geometric means.
- FIG. 13 shows intracellular cytokine production after restimulation with synthetic peptides representing the major epitopes in the F protein, 4 weeks after a second dose. The y-axis shows the % cytokine+ of CD8+CD4-.
- replicons are used below. In general these are based on a hybrid alphavirus genome with non-structural proteins from Venezuelan equine encephalitis virus (VEEV), a packaging signal from Sindbis virus, and a 3' UTR from Sindbis virus or a VEEV mutant.
- VEEV Venezuelan equine encephalitis virus
- the replicon is about lOkb long and has a poly-A tail.
- Plasmid DNA encoding alphavirus replicons (named: pT7-mVEEV-FL.RSVF or A317; pT7- mVEEV-SEAP or A306; pSP6-VCR-GFP or A50) served as a template for synthesis of RNA in vitro.
- the replicons contain the alphavirus genetic elements required for RNA replication but lack those encoding gene products necessary for particle assembly; the structural proteins are instead replaced by a protein of interest (either a reporter, such as SEAP or GFP, or an immunogen, such as full-length RSV F protein) and so the replicons are incapable of inducing the generation of infectious particles.
- a bacteriophage (T7 or SP6) promoter upstream of the alphavirus cDNA facilitates the synthesis of the replicon RNA in vitro and a hepatitis delta virus (HDV) ribozyme immediately downstream of the poly(A)-tail generates the correct 3 '-end through its self-cleaving activity.
- HDV hepatitis delta virus
- run-off transcripts were synthesized in vitro using T7 or SP6 bacteriophage derived DNA-dependent RNA polymerase. Transcriptions were performed for 2 hours at 37°C in the presence of 7.5 mM (T7 RNA polymerase) or 5 mM (SP6 RNA polymerase) of each of the nucleoside triphosphates (ATP, CTP, GTP and UTP) following the instructions provided by the manufacturer (Ambion). Following transcription the template DNA was digested with TURBO DNase (Ambion).
- RNA was precipitated with LiCl and reconstituted in nuclease-free water.
- Uncapped RNA was capped post-transcriptionally with Vaccinia Capping Enzyme (VCE) using the ScriptCap m7G Capping System (Epicentre Biotechnologies) as outlined in the user manual; replicons capped in this way are given the "v" prefix e.g. vA317 is the A317 replicon capped by VCE.
- Post-transcriptionally capped RNA was precipitated with LiCl and reconstituted in nuclease-free water. The concentration of the RNA samples was determined by measuring OD 2 60nm- Integrity of the in vitro transcripts was confirmed by denaturing agarose gel electrophoresis.
- Microparticles were made using 500mg of PLG RG503 (50:50 lactide/glycolide molar ratio, MW ⁇ 30kDa) and 20mg DOTAP using an Omni Macro Homogenizer. The particle suspension was shaken at 150rpm overnight and then filtered through a 40 ⁇ sterile filter for storage at 2-8 °C. Self- replicating RNA was adsorbed to the particles. To prepare 1 mL of PLG/RNA suspension the required volume of PLG particle suspension was added to a vial and nuclease-free water was added to bring the volume to 900 ⁇ . ⁇ RNA (10 ⁇ g/mL) was added dropwise to the PLG suspension, with constant shaking.
- PLG/RNA was incubated at room temperature for 30 min. For 1 mL of reconstituted suspension, 45mg mannitol, 15mg sucrose and 250-500 ⁇ g of PVA were added. The vials were frozen at -80°C and lyophilized.
- RNA adsorption To evaluate RNA adsorption, ⁇ particle suspension was centrifuged at 10,000 rpm for 5 min and supernatant was collected. PLG/RNA was reconstituted using lmL nuclease-free water. To ⁇ particle suspension (1 ⁇ g RNA), lmg heparin sulfate was added. The mixture was vortexed and allowed to sit at room temperature for 30 min for RNA desorption. Particle suspension was centrifuged and supernatant was collected.
- RNAse stability ⁇ ⁇ particle suspension was incubated with 6.4mAU of RNase A at room temperature for 30 min. RNAse was inactivated with 0.126mAU of Proteinase K at 55°C for 10 min. lmg of heparin sulfate was added to desorb the RNA followed by centrifugation. The supernatant samples containing RNA were mixed with formaldehyde load dye, heated at 65°C for 10 min and analyzed using a 1% denaturing gel (460ng RNA loaded per lane).
- An oil-in-water emulsion was prepared by microfluidising squalene, span 85, polysorbate 80, and varying amounts of DOTAP. Briefly, oil soluble components (squalene, span 85, cationic lipids, lipid surfactants) were combined in a beaker, lipid components were dissolved in organic solvent. The resulting lipid solution was added directly to the oil phase. The solvent was allowed to evaporate at room temperature for 2 hours in a fume hood prior to combining the aqueous phase and homogenizing the sample to provide a homogeneous feedstock. The primary emulsions were passed three to five times through a Microfluidizer with an ice bath cooling coil. The batch samples were removed from the unit and stored at 4°C.
- This emulsion is thus similar to the commercial MF59 adjuvant, but supplemented by a cationic DOTAP to provide a cationic nanoemulsion ("CNE").
- CNE17 The final composition of emulsion "CNE17” was squalene (4.3% by weight), span 85 (0.5% by weight), polysorbate 80 (0.5%) by weight), DOTAP (1.4mg/ml), in lOmM citrate buffer, pH 6.5.
- a RNA solution is diluted to the appropriate concentration in RNase free water and then added directly into an equal volume of emulsion while vortexing lightly. The solution is allowed to sit at room temperature for approximately 2 hours to allow adsorption. The resulting solution is diluted to the required RNA concentration prior to administration.
- RNA was encapsulated in liposomes made by the method of references 1 1 and 50.
- the liposomes were made of 10% DSPC (zwitterionic), 40% DlinDMA (cationic), 48% cholesterol and 2% PEG- conjugated DMG (2kDa PEG). These proportions refer to the %> moles in the total liposome.
- DlinDMA (l ,2-dilinoleyloxy-N,N-dimethyl-3-aminopropane) was synthesized using the procedure of reference 6.
- DSPC (l ,2-Diastearoyl-sn-glycero-3-phosphocholine) was purchased from Genzyme. Cholesterol was obtained from Sigma- Aldrich.
- PEG-conjugated DMG (1 ,2-dimyristoyl-sn-glycero- 3-phosphoethanolamine-N-[methoxy(polyethylene glycol), ammonium salt), DOTAP (1 ,2-dioleoyl- 3-trimethylammonium-propane, chloride salt) and DC-chol (3 -[N-(N',N'-dimethylaminoethane)- carbamoyl] cholesterol hydrochloride) were from Avanti Polar Lipids.
- lipids were dissolved in ethanol (2ml), a RNA replicon was dissolved in buffer (2ml, lOOmM sodium citrate, pH 6) and these were mixed with 2ml of buffer followed by 1 hour of equilibration. The mixture was diluted with 6ml buffer then filtered. The resulting product contained liposomes, with -95%) encapsulation efficiency.
- fresh lipid stock solutions were prepared in ethanol.
- 37 mg of DlinDMA, 1 1.8 mg of DSPC, 27.8 mg of cholesterol and 8.07 mg of PEG-DMG were weighed and dissolved in 7.55 mL of ethanol.
- the freshly prepared lipid stock solution was gently rocked at 37°C for about 15 min to form a homogenous mixture.
- 755 iL of the stock was added to 1.245 mL ethanol to make a working lipid stock solution of 2 mL. This amount of lipids was used to form liposomes with 250 ⁇ g RNA.
- RNA working solution was also prepared from a stock solution of ⁇ g ⁇ L in 100 mM citrate buffer (pH 6). Three 20 mL glass vials (with stir bars) were rinsed with RNase Away solution (Molecular BioProducts) and washed with plenty of MilliQ water before use to decontaminate the vials of RNases. One of the vials was used for the RNA working solution and the others for collecting the lipid and RNA mixes (as described later). The working lipid and RNA solutions were heated at 37°C for 10 min before being loaded into 3cc luer-lok syringes. 2 mL citrate buffer (pH 6) was loaded in another 3 cc syringe.
- RNA and the lipids were connected to a T mixer (PEEKTM 500 ⁇ ID junction, Idex Health Science) using FEP tubing (fluorinated ethylene-propylene; all FEP tubing used had a 2mm internal diameter and a 3mm outer diameter; obtained from Idex Health Science).
- the outlet from the T mixer was also FEP tubing.
- the third syringe containing the citrate buffer was connected to a separate piece of tubing. All syringes were then driven at a flow rate of 7 mL/min using a syringe pump. The tube outlets were positioned to collect the mixtures in a 20 mL glass vial (while stirring).
- the stir bar was taken out and the ethanol/aqueous solution was allowed to equilibrate to room temperature for 1 h. 4 ml of the mixture was loaded into a 5 cc syringe, which was connected to a piece of FEP tubing and in another 5 cc syringe connected to an equal length of FEP tubing, an equal amount of 100 mM citrate buffer (pH 6) was loaded. The two syringes were driven at 7mL/min flow rate using the syringe pump and the final mixture collected in a 20 mL glass vial (while stirring).
- the mixture collected from the second mixing step were passed through a Mustang Q membrane (an anion-exchange support that binds and removes anionic molecules, obtained from Pall Corporation).
- a Mustang Q membrane an anion-exchange support that binds and removes anionic molecules, obtained from Pall Corporation.
- 4 mL of 1 M NaOH, 4 mL of 1 M NaCl and 10 mL of 100 mM citrate buffer (pH 6) were successively passed through it. Liposomes were warmed for 10 min at 37°C before passing through the membrane.
- liposomes were concentrated to 2 mL and dialyzed against 10-15 volumes of IX PBS using by tangential flow filtration before recovering the final product.
- TFF system and hollow fiber filtration membranes were purchased from Spectrum Labs (Rancho Dominguez) and were used according to the manufacturer's guidelines. Polysulfone hollow fiber filtration membranes with a 100 kD pore size cutoff and 8 cm 2 surface area were used. For in vitro and in vivo experiments formulations were diluted to the required RNA concentration with IX PBS.
- FIG. 2 shows an example electron micrograph of liposomes prepared by these methods.
- These liposomes contain encapsulated RNA encoding full-length RSV F antigen. Dynamic light scattering of one batch showed an average diameter of 141nm (by intensity) or 78nm (by number). The percentage of encapsulated RNA and RNA concentration were determined by Quant-iT RiboGreen RNA reagent kit (Invitrogen), following manufacturer's instructions. The ribosomal RNA standard provided in the kit was used to generate a standard curve. Liposomes were diluted lOx or lOOx in IX TE buffer (from kit) before addition of the dye.
- liposomes were diluted lOx or lOOx in IX TE buffer containing 0.5% Triton X before addition of the dye (to disrupt the liposomes and thus to assay total RNA). Thereafter an equal amount of dye was added to each solution and then -180 iL of each solution after dye addition was loaded in duplicate into a 96 well tissue culture plate. The fluorescence (Ex 485 nm, Em 528 nm) was read on a microplate reader. All liposome formulations were dosed in vivo based on the encapsulated amount of RNA.
- RNA from liposomes was shown to protect RNA from RNase digestion. Experiments used 3.8mAU of RNase A per microgram of RNA, incubated for 30 minutes at room temperature. RNase was inactivated with Proteinase K at 55°C for 10 minutes. A 1 : 1 v/v mixture of sample to 25:24: 1 v/v/v, phenol: chloroform: isoamyl alcohol was then added to extract the RNA from the lipids into the aqueous phase. Samples were mixed by vortexing for a few seconds and then placed on a centrifuge for 15 minutes at 12k RPM. The aqueous phase (containing the RNA) was removed and used to analyze the RNA.
- FIG. 1 shows that RNase completely digests RNA in the absence of encapsulation (lane 3). RNA is undetectable after encapsulation (lane 4), and no change is seen if these liposomes are treated with RNase (lane 4). After RNase-treated liposomes are subjected to phenol extraction, undigested RNA is seen (lane 6).
- RNA Even after 1 week at 4°C the RNA could be seen without any fragmentation (FIG. 4, arrow). Protein expression in vivo was unchanged after 6 weeks at 4 °C and one freeze-thaw cycle. Thus liposome-encapsulated RNA is stable.
- RNA a reporter enzyme SEAP; secreted alkaline phosphatase
- SEAP secreted alkaline phosphatase
- Expression levels were measured in sera diluted 1 :4 in IX Phospha-Light dilution buffer using a chemiluminescent alkaline phosphate substrate. 8-10 week old BALB/c mice (5/group) were injected intramuscularly on day 0, 50 ⁇ 1 per leg with O. ⁇ g or ⁇ g RNA dose. The same vector was also administered without the liposomes (in RNase free IX PBS) at ⁇ g. Virion-packaged replicons were also tested.
- Virion-packaged replicons used herein were obtained by the methods of reference 51, where the alphavirus replicon is derived from the mutant VEEV or a chimera derived from the genome of VEEV engineered to contain the 3' UTR of Sindbis virus and a Sindbis virus packaging signal (PS), packaged by co-electroporating them into BHK cells with defective helper RNAs encoding the Sindbis virus capsid and glycoprotein genes.
- PS Sindbis virus packaging signal
- encapsulation increased SEAP levels by about 1 ⁇ 2 log at the dose, and at day 6 expression from a O. ⁇ g encapsulated dose matched levels seen with ⁇ g unencapsulated dose.
- day 3 expression levels exceeded those achieved with VRPs (squares).
- VRPs squares
- RNA was formulated in the liposomes relative to the naked RNA control, even at a lOx lower dose. Expression was also higher relative to the VRP control, but the kinetics of expression were very different (see FIG. 5). Delivery of the RNA with electroporation resulted in increased expression relative to the naked RNA control, but these levels were lower than with liposomes.
- the replicon was administered in encapsulated form (with two different purification protocols, O. ⁇ g RNA), or mixed with the liposomes after their formation (a non-encapsulated "lipoplex", O. ⁇ g RNA), or as naked RNA ( ⁇ g).
- FIG. 10 shows that the lipoplex gave the lowest levels of expression, showing that shows encapsulation is essential for potent expression.
- SEAP experiments showed a clear dose response in vivo, with expression seen after delivery of as little as lng RNA (FIG. 6).
- FIG. 7 shows anti-F IgG titers 2 weeks after the second dose, and the liposomes clearly enhance immunogenicity.
- FIG. 8 shows titers 2 weeks later, by which point there was no statistical difference between the encapsulated RNA at O. ⁇ g, the encapsulated RNA at or the VRP group.
- Neutralisation titers (measured as 60% plaque reduction, "PRNT60") were not significantly different in these three groups 2 weeks after the second dose (FIG. 9).
- FIG. 12 shows both IgG and PRNT titers 4 weeks after the second dose.
- FIG. 13 confirms that the RNA elicits a robust CD8 T cell response.
- Further experiments compared F-specific IgG titers in mice receiving VRP, O. ⁇ g liposome- encapsulated RNA, or ⁇ ig ⁇ liposome-encapsulated RNA. Titer ratios (VRP liposome) at various times after the second dose were as follows:
- mice were used to see if host defence responses (innate or adaptive immunity) might limit the immune response to encoded antigens at higher RNA doses.
- RNA was also prepared from a stock solution of - in 100 mM citrate buffer (pH 6). Three 20 mL glass vials (with stir bars) were rinsed with RNase Away solution (Molecular BioProducts) and washed with plenty of MilliQ water before use to decontaminate the vials of RNases. One of the vials was used for the RNA working solution and the others for collecting the lipid and RNA mixes (as described later).
- TFF system and hollow fiber filtration membranes were purchased from Spectrum Labs and were used according to the manufacturer's guidelines.
- Polyethersulfone (PES) hollow fiber filtration membranes (part number P-Cl-lOOE-100-OlN) with a 100 kD pore size cutoff and 20 cm 2 surface area were used.
- PES Polyethersulfone
- mice 5 animals per group, were given bilateral intramuscular vaccinations (50 per leg) on days 0 and 21 with:
- Group 4 a mixture of self-replicating RSV-F RNA (vA317, O. ⁇ g) and self-replicating GFP RNA (vA17, 10 ⁇ ⁇ )
- Group 5 a mixture of self-replicating RSV-F RNA (vA317, O. ⁇ g) and replication-defective GFP RNA (vA336, l( ⁇ g)
- Group 6 a mixture of self-replicating RSV-F RNA formulated in liposomes (vA317, 0.1 ⁇ g) and self-replicating GFP RNA (vA17, l( ⁇ g)
- Group 7 a mixture of self-replicating RSV-F RNA formulated in liposomes (vA317, 0.1 ⁇ g) and replication-defective GFP RNA (vA336, l0 ⁇ g)
- Group 8 a mixture of self-replicating RSV-F RNA formulated in liposomes (vA317, 0.1 ⁇ g) and self-replicating GFP RNA formulated in liposomes (vA17, 1 ⁇ g)
- Group 9 a mixture of self-replicating RSV-F RNA formulated in liposomes (vA317, 0.1 ⁇ g) and replication-defective GFP RNA formulated in liposomes (vA336, 1 ⁇ g)
- Serum was collected for antibody analysis on days 14, 35 and 51.
- F-specific specific serum IgG titers were measured; if an individual animal had a titer of ⁇ 25 (limit of detection), it was assigned a titer of 5.
- spleens were harvested from mice at day 51 for T cell analysis, to determine cells which were cytokine-positive and specific for RSV F51-66 peptide (CD4+) or for RSV F peptides F85-93 and F249-258 (CD8+).
- IgG titers were as follows in the 10 groups and in non-immunised control mice:
- RSV serum neutralization titers at day 51 were as follows:
- Animals showing RSV F-specific CD4+ splenic T cells on day 51 were as follows, where a number (% positive cells) is given only if the stimulated response was statistically significantly above zero:
- mice were given bilateral intramuscular vaccinations (50 per leg) on day 0. Animals, 35 total, were divided into 7 groups (5 animals per group) and were immunised as follows:
- Group 3 were given bilateral intramuscular vaccinations (50 ⁇ ⁇ per leg) on day 0 with RNA (vA306, 0.1 ⁇ g, SEAP) formulated in liposomes mixed with non-replicating RNA (vA336, 1.0 ⁇ g, GFP) formulated in liposomes.
- RNA vA306, 0.1 ⁇ g, SEAP
- vA336, 1.0 ⁇ g, GFP non-replicating RNA
- Group 4 were given bilateral intramuscular vaccinations (50 ⁇ ⁇ per leg) on day 0 with RNA (vA306, 0.1 ⁇ g, SEAP) formulated in liposomes mixed with non-replicating RNA (vA336*, 1.0 ⁇ g, GFP) formulated in liposomes.
- RNA vA306, 0.1 ⁇ g, SEAP
- vA336* non-replicating RNA
- Serum SEAP activity (relative light units) at days 0, 3 and 6 were as follows (GMT):
- Replication-competent RNA encoding GFP suppressed the expression of SEAP more than replication-defective GFP RNA, suggesting a strong host defence response against replicating RNA which leads to suppression of SEAP expression. It is possible that interferons induced in response to the GFP RNA suppressed the expression of SEAP. Under the host response/suppression model, blocking host recognition of RNA would be expected to lead to increased SEAP expression, but 5' methylation of U residues in the GFP RNA was not associated with increased SEAP, suggesting that host recognition of RNA was insensitive to 5' methylation.
- a typical mouse delivery volume for intramuscular injection is 50 ⁇ into the hind leg, which is a relatively high volume for a mouse leg muscle.
- a human intramuscular dose of ⁇ 0.5ml is relatively small. If immunogenicity in mice would be volume-dependent then the replicon vaccines' efficacy might be due, at least in part, on hydrodynamic forces, which would not be encouraging for use of the same vaccines in humans and larger animals.
- the vA317 replicon was delivered to BALB/c mice, 10 per group, by bilateral intramuscular vaccinations (5 or 50 per leg) on day 0 and 21 :
- Group 1 received naked replicon, 0 ⁇ g in 50 ⁇ ⁇ per leg
- Group 2 received naked replicon, 0.2 ⁇ g in 5 ⁇ ⁇ per leg
- Group 3 received emulsion-formulated replicon (0.2 ⁇ g, 50 ⁇ ⁇ per leg)
- Group 5 received liposome-formulated replicon (0.2 ⁇ g, 50 ⁇ ⁇ per leg)
- Group 6 received liposome-formulated replicon (0.2 ⁇ g, 5 ⁇ ⁇ per leg)
- F-specific serum IgG GMTs were:
- RNA vaccines encoded human RSV F whereas the "Triangle 4" vaccine contains bovine RSV F, but the RSV F protein is highly conserved between BRSV and HRSV.
- the liposomes had the same proportion of DlinDMA, DSPC, cholesterol and PEG-DMG as mentioned above.
- Fresh lipid stock solutions in ethanol were prepared. 37 mg of DlinDMA, 1 1.8 mg of DSPC, 27.8 mg of Cholesterol and 8.07 mg of PEG-DMG were weighed and dissolved in 7.55 mL of ethanol. The freshly prepared lipid stock solution was gently rocked at 37°C for about 15 min to form a homogenous mixture. Then, 226.7 ⁇ ⁇ of the stock was added to 1.773 mL ethanol to make a working lipid stock solution of 2 mL.
- RNA was also prepared from a stock solution of - ⁇ g ⁇ L in 100 mM citrate buffer (pH 6). Three 20 mL glass vials (with stir bars) were rinsed with RNase Away solution (Molecular BioProducts) and washed with plenty of MilliQ water before use to decontaminate the vials of RNAses. One of the vials was used for the RNA working solution and the others for collecting the lipid and RNA mixes (as described later). The working lipid and RNA solutions were heated at 37°C for 10 min before being loaded into 3cc syringes. 2 mL of citrate buffer (pH 6) was loaded in another 3 cc syringe.
- RNA and the lipids were connected to a T mixer (PEEKTM 500 ⁇ ID junction) using FEP tubing.
- the outlet from the T mixer was also FEP tubing.
- the third syringe containing the citrate buffer was connected to a separate piece of FEP tubing. All syringes were then driven at a flow rate of 7 mL/min using a syringe pump.
- the tube outlets were positioned to collect the mixtures in a 20 mL glass vial (while stirring). The stir bar was taken out and the ethanol/aqueous solution was allowed to equilibrate to room temperature for 1 h.
- FIG. 14A shows F-specific IgG titers over the first 63 days.
- the RNA replicon was immunogenic in the cows using both delivery systems, although it gave lower titers than the licensed vaccine. All vaccinated cows showed F-specific antibodies after the second dose, and titers were very stable from the period of 2 to 6 weeks after the second dose (and were particularly stable for the RNA vaccines). The titers with the liposome delivery system were more tightly clustered than with the emulsion.
- FIG. 14B shows F-specific serum IgG titers (GMT) over 210 days, and measured values up to day 202 were as follows:
- RNA replicon RSV vaccines in large animals, with two of the five calves in the emulsion-adjuvanted group demonstrating good neutralizing antibody titers after the third vaccination, as measured by the complement-independent HRSV neutralization assay.
- the emulsion-adjuvanted vaccines appear to be more immunogenic than the liposome-adjuvanted vaccines, one complicating factor is that the material used for the second liposome dose was not freshly prepared, and the same lot of RNA showed a decrease in potency in a mouse immunogenicity study. Therefore it is possible that the liposome-adjuvanted vaccine would have been more immunogenic if fresh material had been used for all vaccinations.
- RNA vaccines in large animals is particularly important in light of the loss in potency observed previously with DNA-based vaccines when moving from small animal models to larger animals and humans.
- a typical dose for a cow DNA vaccine would be 0.5-1 mg [52,53] and so it is very encouraging that immune responses were induced with only 66 ⁇ g of RNA.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Virology (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Epidemiology (AREA)
- Biomedical Technology (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Mycology (AREA)
- Biochemistry (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Physics & Mathematics (AREA)
- Pulmonology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Abstract
Description




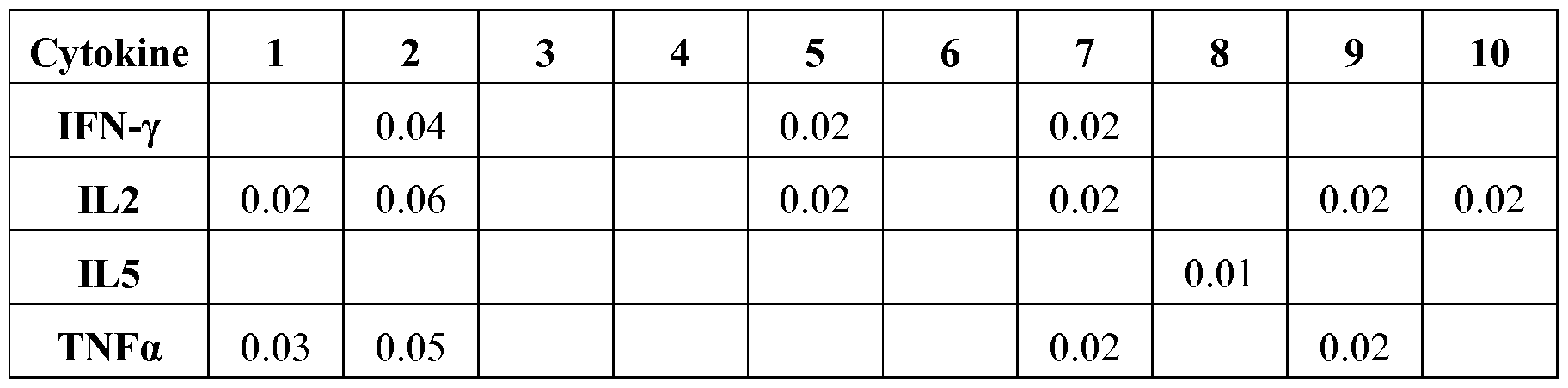




Claims
Priority Applications (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11736497.6A EP2591114B1 (en) | 2010-07-06 | 2011-07-06 | Immunisation of large mammals with low doses of rna |
| CA2804492A CA2804492A1 (en) | 2010-07-06 | 2011-07-06 | Immunisation of large mammals with low doses of rna |
| JP2013518811A JP5940064B2 (en) | 2010-07-06 | 2011-07-06 | Immunization of large mammals with low doses of RNA |
| DK11736497.6T DK2591114T3 (en) | 2010-07-06 | 2011-07-06 | Immunization of large mammals with low doses of RNA |
| SI201130919A SI2591114T1 (en) | 2010-07-06 | 2011-07-06 | Immunisation of large mammals with low doses of rna |
| US13/808,153 US10487332B2 (en) | 2010-07-06 | 2011-07-06 | Immunisation of large mammals with low doses of RNA |
| ES11736497.6T ES2586580T3 (en) | 2010-07-06 | 2011-07-06 | Immunization of large mammals with low doses of RNA |
| HRP20160805TT HRP20160805T1 (en) | 2010-07-06 | 2011-07-06 | Immunisation of large mammals with low doses of rna |
| CY20161100707T CY1117819T1 (en) | 2010-07-06 | 2016-07-19 | Immunization of large mammals with low doses of RNA |
| US16/656,929 US11655475B2 (en) | 2010-07-06 | 2019-10-18 | Immunisation of large mammals with low doses of RNA |
| US17/512,258 US11913001B2 (en) | 2010-07-06 | 2021-10-27 | Immunisation of large mammals with low doses of RNA |
| US18/080,164 US11905514B2 (en) | 2010-07-06 | 2022-12-13 | Immunisation of large mammals with low doses of RNA |
| US18/065,267 US11851660B2 (en) | 2010-07-06 | 2022-12-13 | Immunisation of large mammals with low doses of RNA |
| US18/065,230 US11739334B2 (en) | 2010-07-06 | 2022-12-13 | Immunisation of large mammals with low doses of RNA |
| US18/065,243 US11773395B1 (en) | 2010-07-06 | 2022-12-13 | Immunization of large mammals with low doses of RNA |
| US18/065,256 US11845925B2 (en) | 2010-07-06 | 2022-12-13 | Immunisation of large mammals with low doses of RNA |
| US18/080,150 US11891608B2 (en) | 2010-07-06 | 2022-12-13 | Immunization of large mammals with low doses of RNA |
| US18/411,482 US20240417740A1 (en) | 2010-07-06 | 2024-01-12 | Immunisation of large mammals with low doses of rna |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US36179410P | 2010-07-06 | 2010-07-06 | |
| US61/361,794 | 2010-07-06 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/808,153 A-371-Of-International US10487332B2 (en) | 2010-07-06 | 2011-07-06 | Immunisation of large mammals with low doses of RNA |
| US16/656,929 Continuation US11655475B2 (en) | 2010-07-06 | 2019-10-18 | Immunisation of large mammals with low doses of RNA |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2012006369A2 true WO2012006369A2 (en) | 2012-01-12 |
| WO2012006369A3 WO2012006369A3 (en) | 2012-08-23 |
Family
ID=44629097
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/043096 Ceased WO2012006369A2 (en) | 2010-07-06 | 2011-07-06 | Immunisation of large mammals with low doses of rna |
Country Status (13)
| Country | Link |
|---|---|
| US (10) | US10487332B2 (en) |
| EP (1) | EP2591114B1 (en) |
| JP (1) | JP5940064B2 (en) |
| CA (2) | CA2804492A1 (en) |
| CY (1) | CY1117819T1 (en) |
| DK (1) | DK2591114T3 (en) |
| ES (1) | ES2586580T3 (en) |
| HR (1) | HRP20160805T1 (en) |
| HU (1) | HUE029284T2 (en) |
| PL (1) | PL2591114T3 (en) |
| PT (1) | PT2591114T (en) |
| SI (1) | SI2591114T1 (en) |
| WO (1) | WO2012006369A2 (en) |
Cited By (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012031043A1 (en) | 2010-08-31 | 2012-03-08 | Novartis Ag | Pegylated liposomes for delivery of immunogen-encoding rna |
| WO2013006825A1 (en) | 2011-07-06 | 2013-01-10 | Novartis Ag | Liposomes having useful n:p ratio for delivery of rna molecules |
| US8664194B2 (en) | 2011-12-16 | 2014-03-04 | Moderna Therapeutics, Inc. | Method for producing a protein of interest in a primate |
| US8710200B2 (en) | 2011-03-31 | 2014-04-29 | Moderna Therapeutics, Inc. | Engineered nucleic acids encoding a modified erythropoietin and their expression |
| WO2014108515A1 (en) | 2013-01-10 | 2014-07-17 | Novartis Ag | Influenza virus immunogenic compositions and uses thereof |
| US8822663B2 (en) | 2010-08-06 | 2014-09-02 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| WO2014140211A1 (en) | 2013-03-15 | 2014-09-18 | Novartis Ag | Rna purification methods |
| WO2014152211A1 (en) | 2013-03-14 | 2014-09-25 | Moderna Therapeutics, Inc. | Formulation and delivery of modified nucleoside, nucleotide, and nucleic acid compositions |
| US8980864B2 (en) | 2013-03-15 | 2015-03-17 | Moderna Therapeutics, Inc. | Compositions and methods of altering cholesterol levels |
| US9149506B2 (en) | 2012-04-02 | 2015-10-06 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding septin-4 |
| US9254311B2 (en) | 2012-04-02 | 2016-02-09 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of proteins |
| US9334328B2 (en) | 2010-10-01 | 2016-05-10 | Moderna Therapeutics, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US9464124B2 (en) | 2011-09-12 | 2016-10-11 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| US9572897B2 (en) | 2012-04-02 | 2017-02-21 | Modernatx, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| US9587003B2 (en) | 2012-04-02 | 2017-03-07 | Modernatx, Inc. | Modified polynucleotides for the production of oncology-related proteins and peptides |
| WO2017070622A1 (en) * | 2015-10-22 | 2017-04-27 | Modernatx, Inc. | Respiratory syncytial virus vaccine |
| US9950065B2 (en) | 2013-09-26 | 2018-04-24 | Biontech Rna Pharmaceuticals Gmbh | Particles comprising a shell with RNA |
| US10022436B2 (en) | 2016-01-11 | 2018-07-17 | Verndari, Inc. | Microneedle compositions and methods of using same |
| WO2018170260A1 (en) * | 2017-03-15 | 2018-09-20 | Modernatx, Inc. | Respiratory syncytial virus vaccine |
| EP3292873B1 (en) | 2013-02-22 | 2019-05-01 | CureVac AG | Combination of vaccination and inhibition of the pd-1 pathway |
| US10323076B2 (en) | 2013-10-03 | 2019-06-18 | Modernatx, Inc. | Polynucleotides encoding low density lipoprotein receptor |
| EP3365007A4 (en) * | 2015-10-22 | 2019-07-03 | ModernaTX, Inc. | VACCINE AGAINST BROAD SPECTRUM INFLUENZA VIRUS |
| EP3364980A4 (en) * | 2015-10-22 | 2019-07-10 | ModernaTX, Inc. | Nucleic acid vaccines for varicella zoster virus (vzv) |
| EP3364981A4 (en) * | 2015-10-22 | 2019-08-07 | ModernaTX, Inc. | VACCINE AGAINST THE CYTOMEGALOVIRUS HUMAN |
| EP2591103B1 (en) | 2010-07-06 | 2019-08-28 | GlaxoSmithKline Biologicals SA | Delivery of rna to different cell types |
| US10449244B2 (en) | 2015-07-21 | 2019-10-22 | Modernatx, Inc. | Zika RNA vaccines |
| US10493143B2 (en) | 2015-10-22 | 2019-12-03 | Modernatx, Inc. | Sexually transmitted disease vaccines |
| US10653767B2 (en) | 2017-09-14 | 2020-05-19 | Modernatx, Inc. | Zika virus MRNA vaccines |
| EP3718565A1 (en) * | 2015-10-22 | 2020-10-07 | ModernaTX, Inc. | Respiratory virus vaccines |
| US10815291B2 (en) | 2013-09-30 | 2020-10-27 | Modernatx, Inc. | Polynucleotides encoding immune modulating polypeptides |
| US10925958B2 (en) | 2016-11-11 | 2021-02-23 | Modernatx, Inc. | Influenza vaccine |
| US11045540B2 (en) | 2017-03-15 | 2021-06-29 | Modernatx, Inc. | Varicella zoster virus (VZV) vaccine |
| US11058762B2 (en) | 2011-07-06 | 2021-07-13 | Glaxosmithkline Biologicals Sa | Immunogenic compositions and uses thereof |
| US11103578B2 (en) | 2016-12-08 | 2021-08-31 | Modernatx, Inc. | Respiratory virus nucleic acid vaccines |
| EP3134131B1 (en) | 2014-04-23 | 2021-12-22 | ModernaTX, Inc. | Nucleic acid vaccines |
| US11235052B2 (en) | 2015-10-22 | 2022-02-01 | Modernatx, Inc. | Chikungunya virus RNA vaccines |
| US11291635B2 (en) | 2010-07-06 | 2022-04-05 | Glaxosmithkline Biological Sa | Virion-like delivery particles for self-replicating RNA molecules |
| US11291682B2 (en) | 2010-07-06 | 2022-04-05 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US20220125723A1 (en) | 2010-07-06 | 2022-04-28 | Glaxosmithkline Biologicals Sa | Lipid formulations with viral immunogens |
| US11351242B1 (en) | 2019-02-12 | 2022-06-07 | Modernatx, Inc. | HMPV/hPIV3 mRNA vaccine composition |
| US11364292B2 (en) | 2015-07-21 | 2022-06-21 | Modernatx, Inc. | CHIKV RNA vaccines |
| EP2611467B1 (en) | 2010-08-31 | 2022-07-20 | GlaxoSmithKline Biologicals SA | Small liposomes for delivery of immunogen-encoding rna |
| US11458195B2 (en) | 2013-02-22 | 2022-10-04 | Curevac Ag | Combination of vaccination and inhibition of the PD-1 pathway |
| US11471525B2 (en) | 2020-02-04 | 2022-10-18 | Curevac Ag | Coronavirus vaccine |
| WO2022259191A1 (en) | 2021-06-09 | 2022-12-15 | Glaxosmithkline Biologicals Sa | Release assay for determining potency of self-amplifying rna drug product and methods for using |
| US11547764B2 (en) | 2011-06-08 | 2023-01-10 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US11576961B2 (en) | 2017-03-15 | 2023-02-14 | Modernatx, Inc. | Broad spectrum influenza virus vaccine |
| EP4159741A1 (en) | 2014-07-16 | 2023-04-05 | ModernaTX, Inc. | Method for producing a chimeric polynucleotide encoding a polypeptide having a triazole-containing internucleotide linkage |
| US11639370B2 (en) | 2010-10-11 | 2023-05-02 | Glaxosmithkline Biologicals Sa | Antigen delivery platforms |
| US11655475B2 (en) | 2010-07-06 | 2023-05-23 | Glaxosmithkline Biologicals Sa | Immunisation of large mammals with low doses of RNA |
| US11752206B2 (en) | 2017-03-15 | 2023-09-12 | Modernatx, Inc. | Herpes simplex virus vaccine |
| US11793843B2 (en) | 2019-01-10 | 2023-10-24 | Janssen Biotech, Inc. | Prostate neoantigens and their uses |
| US11872280B2 (en) | 2020-12-22 | 2024-01-16 | CureVac SE | RNA vaccine against SARS-CoV-2 variants |
| US11878060B2 (en) | 2016-08-07 | 2024-01-23 | Novartis Ag | mRNA-mediated immunization methods |
| US11896636B2 (en) | 2011-07-06 | 2024-02-13 | Glaxosmithkline Biologicals Sa | Immunogenic combination compositions and uses thereof |
| US11911453B2 (en) | 2018-01-29 | 2024-02-27 | Modernatx, Inc. | RSV RNA vaccines |
| EP4117725A4 (en) * | 2020-03-09 | 2024-05-29 | Arcturus Therapeutics, Inc. | CORONAVIRUS VACCINE COMPOSITIONS AND METHODS |
| WO2024125597A1 (en) | 2022-12-14 | 2024-06-20 | Providence Therapeutics Holdings Inc. | Compositions and methods for infectious diseases |
| US12018289B2 (en) | 2019-11-18 | 2024-06-25 | Janssen Biotech, Inc. | Vaccines based on mutant CALR and JAK2 and their uses |
| US12070495B2 (en) | 2019-03-15 | 2024-08-27 | Modernatx, Inc. | HIV RNA vaccines |
| US12102720B2 (en) | 2011-06-08 | 2024-10-01 | Translate Bio, Inc. | Cleavable lipids |
| US12128113B2 (en) | 2016-05-18 | 2024-10-29 | Modernatx, Inc. | Polynucleotides encoding JAGGED1 for the treatment of Alagille syndrome |
| US12133899B2 (en) | 2020-04-22 | 2024-11-05 | BioNTech SE | Coronavirus vaccine |
| US12186389B2 (en) | 2022-10-28 | 2025-01-07 | Glaxosmithkline Biologicals Sa | Nucleic acid base vaccine against emerging SARS-CoV-2 variants |
| US12186387B2 (en) | 2021-11-29 | 2025-01-07 | BioNTech SE | Coronavirus vaccine |
| US12295997B2 (en) | 2020-07-06 | 2025-05-13 | Janssen Biotech, Inc. | Prostate neoantigens and their uses |
Families Citing this family (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2648950C2 (en) | 2011-10-03 | 2018-04-02 | Модерна Терапьютикс, Инк. | Modified nucleosides, nucleotides and nucleic acids and their application |
| US9512456B2 (en) | 2012-08-14 | 2016-12-06 | Modernatx, Inc. | Enzymes and polymerases for the synthesis of RNA |
| US8906668B2 (en) | 2012-11-23 | 2014-12-09 | Seres Health, Inc. | Synergistic bacterial compositions and methods of production and use thereof |
| CA3212215A1 (en) | 2012-11-23 | 2014-05-30 | Seres Therapeutics, Inc. | Synergistic bacterial compositions and methods of production and use thereof |
| WO2014081507A1 (en) | 2012-11-26 | 2014-05-30 | Moderna Therapeutics, Inc. | Terminally modified rna |
| HK1218560A1 (en) | 2013-02-04 | 2017-02-24 | Seres Therapeutics, Inc. | Compositions and methods |
| CA2899925A1 (en) | 2013-02-04 | 2014-08-07 | Seres Therapeutics, Inc. | Compositions and methods for inhibition of pathogenic bacterial growth |
| EP2968391A1 (en) | 2013-03-13 | 2016-01-20 | Moderna Therapeutics, Inc. | Long-lived polynucleotide molecules |
| EP2967077A4 (en) | 2013-03-15 | 2016-09-14 | Seres Therapeutics Inc | MICROBIAL COMPOSITIONS AND ASSOCIATED METHODS BASED ON A NETWORK |
| EP3074027B1 (en) | 2013-11-25 | 2024-12-18 | Seres Therapeutics, Inc. | Synergistic bacterial compositions and methods of production and use thereof |
| US9956282B2 (en) | 2013-12-16 | 2018-05-01 | Seres Therapeutics, Inc. | Bacterial compositions and methods of use thereof for treatment of immune system disorders |
| AU2016336344A1 (en) | 2015-10-05 | 2018-04-19 | Modernatx, Inc. | Methods for therapeutic administration of messenger ribonucleic acid drugs |
| US12491261B2 (en) | 2016-10-26 | 2025-12-09 | Acuitas Therapeutics, Inc. | Lipid nanoparticle formulations |
| MX2019013259A (en) | 2017-05-08 | 2020-01-13 | Gritstone Oncology Inc | Alphavirus neoantigen vectors. |
| JP2020524143A (en) | 2017-06-15 | 2020-08-13 | インフェクシャス ディズィーズ リサーチ インスティチュート | Nanostructured lipid carriers, stable emulsions, and uses thereof |
| EP3668527A1 (en) | 2017-08-14 | 2020-06-24 | Seres Therapeutics, Inc. | Compositions and methods for treating cholestatic disease |
| CA3073020A1 (en) | 2017-08-16 | 2019-02-21 | Acuitas Therapeutics, Inc. | Lipids for use in lipid nanoparticle formulations |
| JP2021501185A (en) | 2017-10-30 | 2021-01-14 | セレス セラピューティクス インコーポレイテッド | Compositions and Methods for Treating Antibiotic Resistance |
| EP3735271A4 (en) | 2018-01-04 | 2022-06-15 | Iconic Therapeutics, Inc. | ANTI-TISSUE FACTOR ANTIBODIES, ANTIBODY-DRUG CONJUGATE AND METHODS THEREOF |
| US11814464B2 (en) | 2019-04-29 | 2023-11-14 | Yale University | Poly(amine-co-ester) polymers and polyplexes with modified end groups and methods of use thereof |
| MX2021014525A (en) | 2019-05-30 | 2022-03-17 | Gritstone Bio Inc | MODIFIED ADENOVIRUSES. |
| US20220313815A1 (en) | 2019-06-20 | 2022-10-06 | Janssen Sciences Ireland Unlimited Company | Self-replicating rna molecules for hepatitis b virus (hbv) vaccines and uses thereof |
| KR20230013237A (en) | 2020-03-09 | 2023-01-26 | 다이나박스 테크놀로지 코퍼레이션 | Herpes zoster vaccine containing a TLR9 agonist |
| AU2021271300A1 (en) | 2020-05-11 | 2023-02-02 | Janssen Pharmaceuticals, Inc. | RNA replicon encoding a stabilized corona virus spike protein |
| EP4168471A1 (en) | 2020-06-19 | 2023-04-26 | Yale University | Poly(amine-co-ester) polymers with modified end groups and enhanced pulmonary delivery |
| WO2022016070A1 (en) | 2020-07-16 | 2022-01-20 | Acuitas Therapeutics, Inc. | Cationic lipids for use in lipid nanoparticles |
| KR20230046313A (en) | 2020-08-06 | 2023-04-05 | 그릿스톤 바이오, 인코포레이티드 | Multi-epitope vaccine cassette |
| US12178921B2 (en) | 2020-08-14 | 2024-12-31 | Arcturus Therapeutics, Inc. | Method of lyophilizing lipid nanoparticles |
| CZ310613B6 (en) | 2020-09-23 | 2026-01-28 | Ústav organické chemie a biochemie AV ČR, v. v. i. | Lipidoids for the transfection of nucleic acids and their use |
| CZ310443B6 (en) | 2021-07-19 | 2025-06-25 | Ústav organické chemie a biochemie AV ČR, v. v. i. | Cyclohexane lipidoids for nucleic acid transfection and their use |
| JP2025502630A (en) * | 2021-12-08 | 2025-01-28 | イエール・ユニバーシテイ | Nanoparticle immunogenic compositions and vaccination methods |
| CA3242402A1 (en) | 2021-12-16 | 2023-06-22 | Acuitas Therapeutics, Inc. | Lipids for use in lipid nanoparticle formulations |
| US12115217B2 (en) | 2022-11-18 | 2024-10-15 | Trustees Of Boston University | Self-replicating RNA and uses thereof |
| JP2025099281A (en) * | 2023-12-21 | 2025-07-03 | 株式会社リュージュサイエンス | Cationic polysaccharide copolymer adjuvant, and vaccine |
Citations (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002002606A2 (en) | 2000-07-03 | 2002-01-10 | Chiron S.P.A. | Immunisation against chlamydia pneumoniae |
| WO2002034771A2 (en) | 2000-10-27 | 2002-05-02 | Chiron Srl | Nucleic acids and proteins from streptococcus groups a & b |
| WO2003018054A1 (en) | 2001-08-31 | 2003-03-06 | Chiron Srl. | Helicobacter pylori vaccination |
| WO2005002619A2 (en) | 2003-06-26 | 2005-01-13 | Chiron Corporation | Immunogenic compositions for chlamydia trachomatis |
| WO2005032582A2 (en) | 2003-07-31 | 2005-04-14 | Chiron Corporation | Immunogenic compositions for streptococcus pyogenes |
| WO2005111066A2 (en) | 2004-05-14 | 2005-11-24 | Chiron Srl | Polypeptides from non-typeable haemophilus influenzae |
| WO2005113782A1 (en) | 2004-05-18 | 2005-12-01 | Alphavax, Inc. | Tc-83-derived alphavirus vectors, particles and methods |
| WO2005121348A1 (en) | 2004-06-07 | 2005-12-22 | Protiva Biotherapeutics, Inc. | Lipid encapsulated interfering rna |
| WO2006089264A2 (en) | 2005-02-18 | 2006-08-24 | Novartis Vaccines And Diagnostics Inc. | Proteins and nucleic acids from meningitis/sepsis-associated escherichia coli |
| WO2006091517A2 (en) | 2005-02-18 | 2006-08-31 | Novartis Vaccines And Diagnostics Inc. | Immunogens from uropathogenic escherichia coli |
| WO2006110413A2 (en) | 2005-03-30 | 2006-10-19 | Novartis Vaccines And Diagnostics Inc. | Haemophilus influenzae type b |
| WO2006138004A2 (en) | 2005-05-12 | 2006-12-28 | Novartis Vaccines And Diagnostics, Inc. | Immunogenic compositions for chlamydia trachomatis |
| US20070014805A1 (en) | 2005-07-07 | 2007-01-18 | Sanofi Pasteur | Immuno-adjuvant emulsion |
| WO2007049155A2 (en) | 2005-10-25 | 2007-05-03 | Novartis Vaccines And Diagnostics Srl | Compositions comprising yersinia pestis antigens |
| WO2008020330A2 (en) | 2006-08-16 | 2008-02-21 | Novartis Ag | Immunogens from uropathogenic escherichia coli |
| US20080057080A1 (en) | 2004-05-18 | 2008-03-06 | Vical Incorporated | Influenza virus vaccine composition and methods of use |
| US20080085870A1 (en) | 2002-12-23 | 2008-04-10 | Vical Incorporated | Codon-optimized polynucleotide-based vaccines against human cytomegalovirus infection |
| WO2009016515A2 (en) | 2007-08-01 | 2009-02-05 | Novartis Ag | Compositions comprising pneumococcal antigens |
| WO2009031043A2 (en) | 2007-09-04 | 2009-03-12 | Novartis Ag | Compositions comprising yersinia pestis antigens |
| WO2009104092A2 (en) | 2008-02-22 | 2009-08-27 | Novartis Ag | Escherichia coli immunogens with improved solubility |
| WO2009109860A2 (en) | 2008-03-06 | 2009-09-11 | Novartis Ag | Mutant forms of chlamydia htra |
| WO2009132206A1 (en) | 2008-04-25 | 2009-10-29 | Liquidia Technologies, Inc. | Compositions and methods for intracellular delivery and release of cargo |
| WO2010119343A2 (en) | 2009-04-14 | 2010-10-21 | Novartis Ag | Compositions for immunising against staphylococcus aureus |
| WO2011005799A2 (en) | 2009-07-06 | 2011-01-13 | Novartis Ag | Self replicating rna molecules and uses thereof |
Family Cites Families (218)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6090406A (en) | 1984-04-12 | 2000-07-18 | The Liposome Company, Inc. | Potentiation of immune responses with liposomal adjuvants |
| US4853228A (en) | 1987-07-28 | 1989-08-01 | Micro-Pak, Inc. | Method of manufacturing unilamellar lipid vesicles |
| US6867195B1 (en) | 1989-03-21 | 2005-03-15 | Vical Incorporated | Lipid-mediated polynucleotide administration to reduce likelihood of subject's becoming infected |
| ES2116269T3 (en) | 1989-03-21 | 1998-07-16 | Vical Inc | EXPRESSION OF EXOGENOUS SEQUENCES OF POLYNUCLEOTIDES IN A VERTEBRATE. |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5279833A (en) | 1990-04-04 | 1994-01-18 | Yale University | Liposomal transfection of nucleic acids into animal cells |
| US5264618A (en) | 1990-04-19 | 1993-11-23 | Vical, Inc. | Cationic lipids for intracellular delivery of biologically active molecules |
| FR2676072B1 (en) | 1991-05-03 | 1994-11-18 | Transgene Sa | RNA DELIVERY VECTOR. |
| US5693535A (en) | 1992-05-14 | 1997-12-02 | Ribozyme Pharmaceuticals, Inc. | HIV targeted ribozymes |
| US5750390A (en) | 1992-08-26 | 1998-05-12 | Ribozyme Pharmaceuticals, Inc. | Method and reagent for treatment of diseases caused by expression of the bcl-2 gene |
| EP0646178A1 (en) | 1992-06-04 | 1995-04-05 | The Regents Of The University Of California | expression cassette with regularoty regions functional in the mammmlian host |
| WO1994002595A1 (en) | 1992-07-17 | 1994-02-03 | Ribozyme Pharmaceuticals, Inc. | Method and reagent for treatment of animal diseases |
| US5474914A (en) | 1992-07-29 | 1995-12-12 | Chiron Corporation | Method of producing secreted CMV glycoprotein H |
| US20020102273A1 (en) | 1995-08-08 | 2002-08-01 | Robert B. Grieve | Use of alphavirus expression vectors to produce parasite anitgens |
| EP0702516A4 (en) | 1993-06-01 | 1998-04-22 | Life Technologies Inc | GENETIC IMMUNIZATION USING CATIONIC LIPIDS |
| US6015686A (en) | 1993-09-15 | 2000-01-18 | Chiron Viagene, Inc. | Eukaryotic layered vector initiation systems |
| JPH08511956A (en) | 1994-04-07 | 1996-12-17 | アクゾ・ノベル・エヌ・ベー | Freeze-dried composition containing RNA |
| US5993850A (en) | 1994-09-13 | 1999-11-30 | Skyepharma Inc. | Preparation of multivesicular liposomes for controlled release of encapsulated biologically active substances |
| US5885613A (en) | 1994-09-30 | 1999-03-23 | The University Of British Columbia | Bilayer stabilizing components and their use in forming programmable fusogenic liposomes |
| AU4594996A (en) | 1994-11-30 | 1996-06-19 | Chiron Viagene, Inc. | Recombinant alphavirus vectors |
| US5965434A (en) | 1994-12-29 | 1999-10-12 | Wolff; Jon A. | Amphipathic PH sensitive compounds and delivery systems for delivering biologically active compounds |
| US5792462A (en) | 1995-05-23 | 1998-08-11 | University Of North Carolina At Chapel Hill | Alphavirus RNA replicon systems |
| US5981501A (en) | 1995-06-07 | 1999-11-09 | Inex Pharmaceuticals Corp. | Methods for encapsulating plasmids in lipid bilayers |
| US7422902B1 (en) | 1995-06-07 | 2008-09-09 | The University Of British Columbia | Lipid-nucleic acid particles prepared via a hydrophobic lipid-nucleic acid complex intermediate and use for gene transfer |
| AU727923B2 (en) | 1995-09-27 | 2001-01-04 | Government Of The United States Of America, As Represented By The Secretary Of The Department Of Health And Human Services, The | Production of infectious respiratory syncytial virus from cloned nucleotide sequences |
| EP0880360B1 (en) | 1996-02-12 | 2002-10-09 | Cobra Therapeutics Limited | Novel methods of vaccination and vaccines therefore comprising a nucleic acid encoding a first epitope and a peptide containing a second epitope |
| DE19605548A1 (en) | 1996-02-15 | 1997-09-04 | Boehringer Ingelheim Int | Composition for transfection of higher eukaryotic cells |
| US6451592B1 (en) | 1996-04-05 | 2002-09-17 | Chiron Corporation | Recombinant alphavirus-based vectors with reduced inhibition of cellular macromolecular synthesis |
| WO1998000110A1 (en) | 1996-07-03 | 1998-01-08 | University Of Pittsburgh | Emulsion formulations for hydrophilic active agents |
| AU728581B2 (en) | 1996-09-13 | 2001-01-11 | Lipoxen Limited | Liposomes |
| US7384923B2 (en) | 1999-05-14 | 2008-06-10 | Lipoxen Technologies Limited | Liposomes |
| US6395302B1 (en) | 1996-11-19 | 2002-05-28 | Octoplus B.V. | Method for the preparation of microspheres which contain colloidal systems |
| CA2289702C (en) | 1997-05-14 | 2008-02-19 | Inex Pharmaceuticals Corp. | High efficiency encapsulation of charged therapeutic agents in lipid vesicles |
| US6048546A (en) | 1997-07-31 | 2000-04-11 | Sandia Corporation | Immobilized lipid-bilayer materials |
| US6060308A (en) | 1997-09-04 | 2000-05-09 | Connaught Laboratories Limited | RNA respiratory syncytial virus vaccines |
| EP1034290A4 (en) * | 1997-11-28 | 2004-09-15 | Queensland Inst Med Res | Flavivirus expression and delivery system |
| US6009406A (en) | 1997-12-05 | 1999-12-28 | Square D Company | Methodology and computer-based tools for re-engineering a custom-engineered product line |
| GB9726555D0 (en) | 1997-12-16 | 1998-02-11 | Smithkline Beecham Plc | Vaccine |
| US6432925B1 (en) | 1998-04-16 | 2002-08-13 | John Wayne Cancer Institute | RNA cancer vaccine and methods for its use |
| EP2311436A1 (en) * | 1998-04-27 | 2011-04-20 | Altus Pharmaceuticals Inc. | Stabilized protein crystals, formulations containing them and methods of making them |
| WO2000000616A2 (en) | 1998-06-29 | 2000-01-06 | U.S. Medical Research Institute Of Infectious Diseases | Marburg virus vaccines |
| WO2000003683A2 (en) | 1998-07-20 | 2000-01-27 | Inex Pharmaceuticals Corporation | Liposomal encapsulated nucleic acid-complexes |
| WO2000039302A2 (en) | 1998-12-31 | 2000-07-06 | Chiron Corporation | Improved expression of hiv polypeptides and production of virus-like particles |
| GB9908309D0 (en) | 1999-04-12 | 1999-06-02 | Phares Pharm Res Nv | Lipid aggregate forming compositions and their use |
| ATE289630T1 (en) | 1999-09-09 | 2005-03-15 | Curevac Gmbh | TRANSFER OF MRNAS USING POLYCATIONIC COMPOUNDS |
| DE60038622D1 (en) | 1999-10-20 | 2008-05-29 | Univ Johns Hopkins Med | CHIMERIC IMMUNOGENIC COMPOSITIONS AND NUCLEIC ACID CODING THEREFOR |
| US8541008B2 (en) | 1999-11-19 | 2013-09-24 | Los Angeles Biomedical Research Institute At Harbor-Ucla Medical Center | Pharmaceutical compositions and methods to vaccinate against candidiasis |
| US20030212022A1 (en) | 2001-03-23 | 2003-11-13 | Jean-Marie Vogel | Compositions and methods for gene therapy |
| US7149665B2 (en) | 2000-04-03 | 2006-12-12 | Browzwear International Ltd | System and method for simulation of virtual wear articles on virtual models |
| EP1287015A1 (en) | 2000-04-18 | 2003-03-05 | Human Genome Sciences, Inc. | Extracellular matrix polynucleotides, polypeptides, and antibodies |
| CN1254234C (en) | 2000-06-09 | 2006-05-03 | 莱古伦公司 | Encapsulation of plasmid DNA (LIPOGENEST™) and therapeutic agent containing nuclear localization signal/fusogenic peptide conjugate into directed liposome complexes |
| AU2001290520A1 (en) | 2000-08-01 | 2002-02-13 | The Johns Hokpins University | Intercellular transport protein linked to an antigen as a molecular vaccine |
| EP1363938B1 (en) * | 2000-08-03 | 2013-12-11 | Johns Hopkins University | Molecular vaccine linking an endoplasmic reticulum chaperone polypeptide to an antigen |
| US20040142474A1 (en) | 2000-09-14 | 2004-07-22 | Expression Genetics, Inc. | Novel cationic lipopolymer as a biocompatible gene delivery agent |
| MXPA03002640A (en) | 2000-09-28 | 2003-06-19 | Chiron Corp | Microparticles for delivery of the heterologous nucleic acids. |
| US7731975B2 (en) | 2001-01-31 | 2010-06-08 | The United States Of America As Represented By The Secretary Of The Army | Chimeric filovirus glycoprotein |
| AU2002242016A1 (en) | 2001-02-01 | 2002-08-12 | The Johns Hopkins University | Nucleic acid derived vaccine that encodes an antigen linked to a polypeptide that promotes antigen presentation |
| US20030040498A1 (en) | 2001-03-14 | 2003-02-27 | Ansardi David Calvert | Oncolytic RNA replicons |
| AU2002306736A1 (en) * | 2001-03-16 | 2002-10-03 | Johns Hopkins University | A replication-defective alphavirus vaccine linking antigen with an immunogenicity-potentiating polypeptide and a method of delivery the same |
| JP2004535388A (en) | 2001-04-30 | 2004-11-25 | ターゲティッド ジェネティクス コーポレイション | Lipid-containing drug delivery conjugates and methods for their production |
| US20030077251A1 (en) | 2001-05-23 | 2003-04-24 | Nicolas Escriou | Replicons derived from positive strand RNA virus genomes useful for the production of heterologous proteins |
| ATE490267T1 (en) | 2001-06-05 | 2010-12-15 | Curevac Gmbh | STABILIZED MRNA WITH INCREASED G/C CONTENT CODING A VIRAL ANTIGEN |
| WO2003023026A1 (en) | 2001-09-06 | 2003-03-20 | Alphavax, Inc. | Alphavirus replicon vector systems |
| US20050163832A1 (en) | 2002-02-13 | 2005-07-28 | Vladimir Torchilin | Intracellular delivery of therapeutic agents |
| DE10207177A1 (en) | 2002-02-19 | 2003-09-04 | Novosom Ag | Optionally cationic lipids |
| US7901708B2 (en) | 2002-06-28 | 2011-03-08 | Protiva Biotherapeutics, Inc. | Liposomal apparatus and manufacturing methods |
| ES2279127T3 (en) | 2002-07-05 | 2007-08-16 | Lipoxen Technologies Limited | PROCEDURE TO INCREASE AN IMMUNE RESPONSE OF NUCLEIC ACID VACCINATION. |
| EP1530585A2 (en) | 2002-08-22 | 2005-05-18 | Cytos Biotechnology AG | Inducible alphaviral/orip based gene expression system |
| EP2330194A3 (en) | 2002-09-13 | 2011-10-12 | Replicor, Inc. | Non-sequence complementary antiviral oligonucleotides |
| US20040208848A1 (en) | 2002-12-13 | 2004-10-21 | Smith Jonathan F. | Multi-antigenic alphavirus replicon particles and methods |
| US8338583B2 (en) | 2003-02-04 | 2012-12-25 | Bar-Ilan University | Snornai-small nucleolar RNA degradation by RNA interference in trypanosomatids |
| US20040228842A1 (en) | 2003-02-27 | 2004-11-18 | Shan Lu | Compositions and methods for cytomegalovirus treatment |
| CN1791678A (en) | 2003-03-20 | 2006-06-21 | 阿尔法瓦克斯公司 | Improved alphavirus replicons and helper constructs |
| US7731967B2 (en) | 2003-04-30 | 2010-06-08 | Novartis Vaccines And Diagnostics, Inc. | Compositions for inducing immune responses |
| CA2527301A1 (en) | 2003-05-30 | 2004-12-09 | Nippon Shinyaku Co., Ltd. | Oligonucleic acid-bearing composite and pharmaceutical composition containing the composite |
| DE602004021260D1 (en) | 2003-07-11 | 2009-07-09 | Alphavax Inc | CYTOMEGALOVIRUS VACCINES BASED ON ALPHAVIRUS |
| US7368537B2 (en) * | 2003-07-15 | 2008-05-06 | Id Biomedical Corporation Of Quebec | Subunit vaccine against respiratory syncytial virus infection |
| ES2559828T3 (en) | 2003-07-16 | 2016-02-16 | Protiva Biotherapeutics Inc. | RNA interference encapsulated in lipids |
| EP1512393A1 (en) | 2003-09-08 | 2005-03-09 | BOEHRINGER INGELHEIM PHARMA GMBH & CO. KG | Process for the production of homogeneous liposomes and lipoplexes |
| WO2005046621A2 (en) | 2003-11-12 | 2005-05-26 | The United States Of America As Represented By The Secretary Of The Navy | Enhancement of vaccine-induced immune responses and protection by heterologous boosting with alphavirus replicon vaccines |
| US7303881B2 (en) | 2004-04-30 | 2007-12-04 | Pds Biotechnology Corporation | Antigen delivery compositions and methods of use |
| GB0411428D0 (en) | 2004-05-21 | 2004-06-23 | Got A Gene Ab | Vectors |
| US20090104226A1 (en) | 2004-05-21 | 2009-04-23 | Novartis Vaccines And Diagnostics Inc. | Alphavirus Vectors for Respiratory Pathogen Vaccines |
| US7745651B2 (en) | 2004-06-07 | 2010-06-29 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods of use |
| CA2572921C (en) | 2004-07-09 | 2017-01-03 | The University Of North Carolina At Chapel Hill | Replication-competent and propagation-defective venezuelan equine encephalitis (vee) viral adjuvants |
| WO2006007712A1 (en) | 2004-07-19 | 2006-01-26 | Protiva Biotherapeutics, Inc. | Methods comprising polyethylene glycol-lipid conjugates for delivery of therapeutic agents |
| EP1794183A2 (en) | 2004-10-01 | 2007-06-13 | Novartis Vaccines and Diagnostics S.r.l. | Hepatitis c virus replication system |
| EP1811960A2 (en) | 2004-11-19 | 2007-08-01 | Novosom AG | Improvements in or relating to pharmaceutical compositions for local administration |
| GB2421025A (en) | 2004-12-09 | 2006-06-14 | Oxxon Therapeutics Ltd | HSV vaccination vectors |
| US7404969B2 (en) | 2005-02-14 | 2008-07-29 | Sirna Therapeutics, Inc. | Lipid nanoparticle based compositions and methods for the delivery of biologically active molecules |
| JP5042863B2 (en) | 2005-02-14 | 2012-10-03 | サーナ・セラピューティクス・インコーポレイテッド | Lipid nanoparticle-based compositions and methods for delivering biologically active molecules |
| AU2006219717B2 (en) | 2005-03-02 | 2009-05-21 | The Secretary Of State For Defence | Pharmaceutical composition |
| GB0504436D0 (en) | 2005-03-03 | 2005-04-06 | Glaxosmithkline Biolog Sa | Vaccine |
| US7618393B2 (en) | 2005-05-03 | 2009-11-17 | Pharmajet, Inc. | Needle-less injector and method of fluid delivery |
| WO2007014754A1 (en) | 2005-08-02 | 2007-02-08 | I.D.M. Immuno-Designed Molecules | Process for the preparation of liposomal formulations |
| US7951384B2 (en) | 2005-08-05 | 2011-05-31 | University Of Massachusetts | Virus-like particles as vaccines for paramyxovirus |
| EP3611266B1 (en) | 2005-08-23 | 2022-11-09 | The Trustees of the University of Pennsylvania | Rna containing modified nucleosides and methods of use thereof |
| EP1764089A1 (en) | 2005-09-15 | 2007-03-21 | Novosom AG | Serum stable liposomes comprising amphoter II lipid mixtures |
| DE102005046490A1 (en) | 2005-09-28 | 2007-03-29 | Johannes-Gutenberg-Universität Mainz | New nucleic acid molecule comprising promoter, a transcriptable nucleic acid sequence, a first and second nucleic acid sequence for producing modified RNA with transcriptional stability and translational efficiency |
| ATE493405T1 (en) | 2005-09-29 | 2011-01-15 | Elan Pharm Inc | PYRIMIDINYLAMIDE COMPOUNDS THAT INHIBIT LEUKOCYTE ADHESION MEDIATED BY VLA-4 |
| CA2626253A1 (en) | 2005-10-18 | 2007-04-26 | Novartis Vaccines And Diagnostics, Inc. | Mucosal and systemic immunizations with alphavirus replicon particles |
| JP2007112768A (en) | 2005-10-24 | 2007-05-10 | Kyoto Univ | Liver-directed liposome composition |
| CA2630220C (en) | 2005-11-22 | 2020-10-13 | Doris Coit | Norovirus and sapovirus antigens |
| WO2008051245A2 (en) | 2005-12-02 | 2008-05-02 | Novartis Ag | Nanoparticles for use in immunogenic compositions |
| EP2004141A2 (en) | 2006-03-17 | 2008-12-24 | Novosom AG | An efficient method for loading amphoteric liposomes with nucleic acid active substances |
| EP3031469B1 (en) | 2006-06-07 | 2023-08-23 | The Trustees Of Princeton University | Cytomegalovirus surface protein complex for use in vaccines and as a drug target |
| US7915399B2 (en) | 2006-06-09 | 2011-03-29 | Protiva Biotherapeutics, Inc. | Modified siRNA molecules and uses thereof |
| PL2035581T3 (en) | 2006-06-21 | 2013-01-31 | Scripps Research Inst | Dna composition against tumor stromal antigen fap and methods of use thereof |
| NZ575901A (en) | 2006-09-12 | 2012-04-27 | Alphavax Inc | Alphavirus replicon particles matched to protein antigens as immunological adjuvants |
| DE102007001370A1 (en) | 2007-01-09 | 2008-07-10 | Curevac Gmbh | RNA-encoded antibodies |
| US20100015218A1 (en) | 2007-02-16 | 2010-01-21 | Vasant Jadhav | Compositions and methods for potentiated activity of biologically active molecules |
| WO2008109806A2 (en) | 2007-03-08 | 2008-09-12 | Massachusetts Institute Of Technology | Electrostatic coating of particles for drug delivery |
| US8877206B2 (en) | 2007-03-22 | 2014-11-04 | Pds Biotechnology Corporation | Stimulation of an immune response by cationic lipids |
| AU2008232891B2 (en) | 2007-03-29 | 2012-01-12 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for inhibiting expression of a gene from the Ebola |
| US8748591B2 (en) | 2007-04-17 | 2014-06-10 | The Board Of Regents Of The University Of Texas System | Chimeric sindbis-western equine encephalitis virus and uses thereof |
| CA2686735A1 (en) | 2007-05-04 | 2008-11-13 | Mdrna, Inc. | Amino acid lipids and uses thereof |
| MX2009012635A (en) | 2007-05-23 | 2012-09-13 | Mannkind Corp | Multicistronic vectors and methods for their design. |
| DE102007029471A1 (en) | 2007-06-20 | 2008-12-24 | Novosom Ag | New optional cationic sterols |
| DK2947149T3 (en) | 2007-06-21 | 2018-06-06 | Alphavax Inc | ALPHAVIRUS REPLICATION PARTICLES FOR USE IN VACCINATION |
| US8202518B2 (en) | 2007-07-04 | 2012-06-19 | Ribovax Biotechnologies S.A. | Antibodies against human cytomegalovirus (HCMV) |
| WO2009026328A2 (en) | 2007-08-21 | 2009-02-26 | Immune Disease Institute, Inc. | Methods of delivery of agents to leukocytes and endothelial cells |
| US20090162395A1 (en) | 2007-09-26 | 2009-06-25 | Crowe Jr James E | Vaccine for rsv and mpv |
| EP2042193A1 (en) | 2007-09-28 | 2009-04-01 | Biomay AG | RNA Vaccines |
| JP5627464B2 (en) | 2007-11-26 | 2014-11-19 | ノバルティス アーゲー | How to generate alphavirus particles |
| EP2067749A1 (en) | 2007-11-29 | 2009-06-10 | Total Petrochemicals France | Process for purification of an aqueous phase containing polyaromatics |
| WO2009074861A2 (en) | 2007-12-10 | 2009-06-18 | Powderject Research Limited | Improved vaccine |
| WO2009086558A1 (en) | 2008-01-02 | 2009-07-09 | Tekmira Pharmaceuticals Corporation | Improved compositions and methods for the delivery of nucleic acids |
| US20110165223A1 (en) | 2008-01-02 | 2011-07-07 | The Johns Hopkins University | Antitumor Immunization by Liposomal Delivery of Vaccine to the Spleen |
| AU2009238175C1 (en) | 2008-04-15 | 2023-11-30 | Arbutus Biopharma Corporation | Novel lipid formulations for nucleic acid delivery |
| WO2009127230A1 (en) | 2008-04-16 | 2009-10-22 | Curevac Gmbh | MODIFIED (m)RNA FOR SUPPRESSING OR AVOIDING AN IMMUNOSTIMULATORY RESPONSE AND IMMUNOSUPPRESSIVE COMPOSITION |
| WO2009132131A1 (en) | 2008-04-22 | 2009-10-29 | Alnylam Pharmaceuticals, Inc. | Amino lipid based improved lipid formulation |
| US20100040650A1 (en) | 2008-05-30 | 2010-02-18 | Crowe Jr James E | Virus-Like paramyxovirus particles and vaccines |
| EP2130912A1 (en) | 2008-06-04 | 2009-12-09 | Institut für Viruskrankeiten und Immunprophylaxe | Pestivirus replicons providing an RNA-based viral vector system |
| EP2310494A1 (en) | 2008-06-25 | 2011-04-20 | ProBioGen AG | Cell line for propagation of highly attenuated alphaviruses |
| JP5712126B2 (en) | 2008-06-25 | 2015-05-07 | ノバルティス アーゲー | Rapid response to delayed boost immunization |
| JP2010025644A (en) | 2008-07-16 | 2010-02-04 | Kochi Univ Of Technology | Coloration reagent of nitrate ions and method for detecting and quantifying nitrate ions using it |
| EP2303923B1 (en) | 2008-07-16 | 2015-06-24 | Institute for Research in Biomedicine | Human cytomegalovirus neutralising antibodies and use thereof |
| PE20141435A1 (en) | 2008-07-16 | 2014-10-22 | Inst Research In Biomedicine | NEUTRALIZING ANTIBODIES OF HUMAN CYTOMEGALOVIRUS |
| CA2733147A1 (en) | 2008-08-06 | 2010-02-11 | Novartis Ag | Microparticles for use in immunogenic compositions |
| CL2008002322A1 (en) | 2008-08-07 | 2009-06-05 | Univ Concepcion | Veterinary pharmaceutical formulation comprising a viral vector system consisting of a recombinant RNA particle encoding a cu / zn superoxide dismutase from the pathogenic bacterium of bovine brucella abortus, and at least one arn alphavirus belonging to the family of the semliki forest virus (sfv) , useful as a vaccine. |
| WO2010019718A2 (en) | 2008-08-13 | 2010-02-18 | California Institute Of Technology | Carrier nanoparticles and related compositions, methods and systems |
| US20110177122A1 (en) | 2008-09-26 | 2011-07-21 | The United States Of America, As Represented By The Secretary, Dept. Of Health & Human Services | Dna prime/activated vaccine boost immunization to influenza virus |
| CN104119242B (en) | 2008-10-09 | 2017-07-07 | 泰米拉制药公司 | The amino lipids of improvement and the method for delivering nucleic acid |
| ES2646630T3 (en) | 2008-11-07 | 2017-12-14 | Massachusetts Institute Of Technology | Amino alcoholic lipidoids and uses thereof |
| HUE037082T2 (en) | 2008-11-10 | 2018-08-28 | Arbutus Biopharma Corp | New lipids and preparations for the delivery of therapeutic agents |
| US20120093855A1 (en) | 2008-11-18 | 2012-04-19 | Ligocyte Pharmaceuticals, Inc. | RSV F VLPs AND METHODS OF MANUFACTURE AND USE THEREOF |
| EP3243504A1 (en) | 2009-01-29 | 2017-11-15 | Arbutus Biopharma Corporation | Improved lipid formulation |
| JP5889783B2 (en) | 2009-05-05 | 2016-03-22 | テクミラ ファーマシューティカルズ コーポレイションTekmira Pharmaceuticals Corporation | Methods for delivering oligonucleotides to immune cells |
| MX342785B (en) | 2009-06-10 | 2016-10-12 | Alnylam Pharmaceuticals Inc | IMPROVED LIPID FORMULATION. |
| ES2613498T3 (en) | 2009-07-01 | 2017-05-24 | Protiva Biotherapeutics Inc. | New lipid formulations for the delivery of therapeutic agents to solid tumors |
| WO2011001780A1 (en) | 2009-07-02 | 2011-01-06 | コニカミノルタホールディングス株式会社 | Method for producing liposomes by two-stage emulsification method using outer aqueous phase containing specific dispersing agent, method for producing liposome dispersion or dry powder thereof using the method for producing liposomes, and liposome dispersion or dry powder thereof produced thereby |
| EP4218799A1 (en) | 2009-07-15 | 2023-08-02 | GlaxoSmithKline Biologicals S.A. | Rsv f protein compositions and methods for making same |
| WO2011007359A2 (en) * | 2009-07-16 | 2011-01-20 | Vaxil Biotherapeutics Ltd. | Antigen specific multi epitope -based anti-infective vaccines |
| BR112012002291A2 (en) | 2009-07-31 | 2016-11-29 | Ethris Gmbh | "polyribonucleotide with a sequence encoding a protein or protein fragment, implant, and process for selecting nucleotide sequences" |
| JO3257B1 (en) | 2009-09-02 | 2018-09-16 | Novartis Ag | Vehicles and installations as TLR |
| ES2443952T3 (en) | 2009-09-02 | 2014-02-21 | Novartis Ag | Immunogenic compositions that include modulators of TLR activity |
| US20110070260A1 (en) | 2009-09-09 | 2011-03-24 | Baric Ralph S | Multivalent Immunogenic Compositions Against Noroviruses and Methods of Use |
| CA2816925C (en) | 2009-11-04 | 2023-01-10 | The University Of British Columbia | Nucleic acid-containing lipid particles and related methods |
| US20110112353A1 (en) | 2009-11-09 | 2011-05-12 | Circulite, Inc. | Bifurcated outflow cannulae |
| HUE042177T2 (en) | 2009-12-01 | 2019-06-28 | Translate Bio Inc | Steroid derivative for the delivery of mrna in human genetic diseases |
| WO2011071860A2 (en) | 2009-12-07 | 2011-06-16 | Alnylam Pharmaceuticals, Inc. | Compositions for nucleic acid delivery |
| KR20240136456A (en) | 2009-12-07 | 2024-09-13 | 더 트러스티스 오브 더 유니버시티 오브 펜실베니아 | Rna preparations comprising purified modified rna for reprogramming cells |
| CA2784568A1 (en) | 2009-12-18 | 2011-06-23 | The University Of British Columbia | Lipid particles for delivery of nucleic acids |
| WO2011076807A2 (en) | 2009-12-23 | 2011-06-30 | Novartis Ag | Lipids, lipid compositions, and methods of using them |
| ES2536429T3 (en) | 2010-01-24 | 2015-05-25 | Novartis Ag | Irradiated biodegradable polymer microparticles |
| US20130195961A1 (en) | 2010-03-09 | 2013-08-01 | Kejian Yang | Novel mucosal vaccination approach for herpes simplex virus type-2 |
| BR112012025364A2 (en) | 2010-04-07 | 2015-09-22 | Novartis Ag | parvovirus b19 virus-like particle generation method |
| US9192661B2 (en) | 2010-07-06 | 2015-11-24 | Novartis Ag | Delivery of self-replicating RNA using biodegradable polymer particles |
| US9770463B2 (en) | 2010-07-06 | 2017-09-26 | Glaxosmithkline Biologicals Sa | Delivery of RNA to different cell types |
| EP2590625B1 (en) | 2010-07-06 | 2017-09-20 | GlaxoSmithKline Biologicals SA | Cationic oil-in-water emulsions |
| AU2011276328C1 (en) | 2010-07-06 | 2016-01-21 | Novartis Ag | Norovirus derived immunogenic compositions and methods |
| RS63817B1 (en) | 2010-07-06 | 2023-01-31 | Glaxosmithkline Biologicals Sa | Virion-like delivery particles for self-replicating rna molecules |
| PL2591114T3 (en) | 2010-07-06 | 2017-08-31 | Glaxosmithkline Biologicals Sa | Immunisation of large mammals with low doses of rna |
| HUE047796T2 (en) | 2010-07-06 | 2020-05-28 | Glaxosmithkline Biologicals Sa | Introduction of RNA to activate multiple immune pathways |
| HUE026646T2 (en) | 2010-07-06 | 2016-07-28 | Glaxosmithkline Biologicals Sa | Liposomes with lipids having an advantageous pka-value for rna delivery |
| US8898852B2 (en) | 2010-08-04 | 2014-12-02 | Honeywell International Inc. | Air burst to clear detection window |
| WO2012019168A2 (en) | 2010-08-06 | 2012-02-09 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| RS63890B1 (en) | 2010-08-31 | 2023-02-28 | Glaxosmithkline Biologicals Sa | Small liposomes for delivery of immunogen-encoding rna |
| ES2727583T3 (en) | 2010-08-31 | 2019-10-17 | Glaxosmithkline Biologicals Sa | Lipids suitable for liposomal administration of RNA encoding proteins |
| PT3970742T (en) | 2010-08-31 | 2022-06-27 | Glaxosmithkline Biologicals Sa | Pegylated liposomes for delivery of immunogen-encoding rna |
| US20130164289A1 (en) | 2010-09-09 | 2013-06-27 | Virginia Commonwealth University | Human cytomegalovirus vaccine |
| RU2013120302A (en) | 2010-10-01 | 2014-11-20 | Модерна Терапьютикс, Инк. | DESIGNED NUCLEIC ACIDS AND WAYS OF THEIR APPLICATION |
| JP2013544504A (en) | 2010-10-11 | 2013-12-19 | ノバルティス アーゲー | Antigen delivery platform |
| MX341185B (en) | 2010-10-25 | 2016-08-09 | Stepan Co | Quaternized fatty amines, amidoamines, and their derivatives from natural oil metathesis. |
| AU2012211278B2 (en) | 2011-01-26 | 2016-11-10 | Glaxosmithkline Biologicals Sa | RSV immunization regimen |
| EP2691101A2 (en) | 2011-03-31 | 2014-02-05 | Moderna Therapeutics, Inc. | Delivery and formulation of engineered nucleic acids |
| DK2707385T3 (en) | 2011-05-13 | 2017-11-20 | Glaxosmithkline Biologicals Sa | RSV F præfusionsantigener |
| WO2012158736A1 (en) | 2011-05-17 | 2012-11-22 | modeRNA Therapeutics | Engineered nucleic acids and methods of use thereof for non-human vertebrates |
| EP3354644A1 (en) | 2011-06-08 | 2018-08-01 | Translate Bio, Inc. | Cleavable lipids |
| US11058762B2 (en) | 2011-07-06 | 2021-07-13 | Glaxosmithkline Biologicals Sa | Immunogenic compositions and uses thereof |
| US20140141070A1 (en) | 2011-07-06 | 2014-05-22 | Andrew Geall | Liposomes having useful n:p ratio for delivery of rna molecules |
| SG10201605512WA (en) | 2011-07-06 | 2016-09-29 | Novartis Ag | Oil-in-water emulsions that contain nucleic acids |
| WO2013006838A1 (en) | 2011-07-06 | 2013-01-10 | Novartis Ag | Immunogenic combination compositions and uses thereof |
| CA2840965C (en) | 2011-07-06 | 2021-03-02 | Novartis Ag | Cationic oil-in-water emulsions |
| EP3508220A1 (en) | 2011-08-31 | 2019-07-10 | GlaxoSmithKline Biologicals S.A. | Pegylated liposomes for delivery of immunogen-encoding rna |
| EP3384938A1 (en) | 2011-09-12 | 2018-10-10 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| RU2648950C2 (en) | 2011-10-03 | 2018-04-02 | Модерна Терапьютикс, Инк. | Modified nucleosides, nucleotides and nucleic acids and their application |
| BR112014008694A2 (en) | 2011-10-11 | 2017-06-20 | Novartis Ag | recombinant polycistronic nucleic acid molecules |
| WO2013054199A2 (en) | 2011-10-12 | 2013-04-18 | Novartis Ag | Cmv antigens and uses thereof |
| US20140378538A1 (en) | 2011-12-14 | 2014-12-25 | Moderma Therapeutics, Inc. | Methods of responding to a biothreat |
| CN104114572A (en) | 2011-12-16 | 2014-10-22 | 现代治疗公司 | Modified nucleosides, nucleotides and nucleic acid compositions |
| JP2015510495A (en) | 2011-12-21 | 2015-04-09 | モデルナ セラピューティクス インコーポレイテッドModerna Therapeutics,Inc. | Methods for extending the viability or longevity of an organ or organ graft |
| CA2868391A1 (en) | 2012-04-02 | 2013-10-10 | Stephane Bancel | Polynucleotides comprising n1-methyl-pseudouridine and methods for preparing the same |
| AU2013243948A1 (en) | 2012-04-02 | 2014-10-30 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of proteins associated with human disease |
| WO2014081507A1 (en) | 2012-11-26 | 2014-05-30 | Moderna Therapeutics, Inc. | Terminally modified rna |
| EP2943221A1 (en) | 2013-01-10 | 2015-11-18 | Novartis AG | Influenza virus immunogenic compositions and uses thereof |
| US9504747B2 (en) | 2013-03-08 | 2016-11-29 | Novartis Ag | Lipids and lipid compositions for the delivery of active agents |
| WO2014160243A1 (en) | 2013-03-14 | 2014-10-02 | The Trustees Of The University Of Pennsylvania | Purification and purity assessment of rna molecules synthesized with modified nucleosides |
| EP2971010B1 (en) | 2013-03-14 | 2020-06-10 | ModernaTX, Inc. | Formulation and delivery of modified nucleoside, nucleotide, and nucleic acid compositions |
| US8980864B2 (en) | 2013-03-15 | 2015-03-17 | Moderna Therapeutics, Inc. | Compositions and methods of altering cholesterol levels |
| ES2895651T3 (en) | 2013-12-19 | 2022-02-22 | Novartis Ag | Lipids and lipid compositions for the administration of active agents |
| EP4286012A3 (en) | 2015-09-17 | 2024-05-29 | ModernaTX, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| IL307179A (en) | 2015-10-28 | 2023-11-01 | Acuitas Therapeutics Inc | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| MX2019005470A (en) | 2016-11-10 | 2019-11-21 | Translate Bio Inc | Improved ice-based lipid nanoparticle formulation for delivery of mrna. |
| US11045540B2 (en) | 2017-03-15 | 2021-06-29 | Modernatx, Inc. | Varicella zoster virus (VZV) vaccine |
| MX2021005969A (en) | 2018-11-21 | 2021-09-14 | Translate Bio Inc | TREATMENT OF CYSTIC FIBROSIS BY DELIVERY OF NEBULIZED mRNA ENCODING CFTR. |
| US12533318B2 (en) | 2019-08-30 | 2026-01-27 | Glaxosmithkline Biologicals Sa | Jet mixing lipid nanoparticle manufacturing process |
| WO2022137133A1 (en) | 2020-12-22 | 2022-06-30 | Curevac Ag | Rna vaccine against sars-cov-2 variants |
-
2011
- 2011-07-06 PL PL11736497T patent/PL2591114T3/en unknown
- 2011-07-06 HU HUE11736497A patent/HUE029284T2/en unknown
- 2011-07-06 WO PCT/US2011/043096 patent/WO2012006369A2/en not_active Ceased
- 2011-07-06 PT PT117364976T patent/PT2591114T/en unknown
- 2011-07-06 CA CA2804492A patent/CA2804492A1/en active Pending
- 2011-07-06 EP EP11736497.6A patent/EP2591114B1/en not_active Revoked
- 2011-07-06 HR HRP20160805TT patent/HRP20160805T1/en unknown
- 2011-07-06 JP JP2013518811A patent/JP5940064B2/en active Active
- 2011-07-06 DK DK11736497.6T patent/DK2591114T3/en active
- 2011-07-06 US US13/808,153 patent/US10487332B2/en active Active
- 2011-07-06 SI SI201130919A patent/SI2591114T1/en unknown
- 2011-07-06 ES ES11736497.6T patent/ES2586580T3/en active Active
- 2011-07-06 CA CA3169291A patent/CA3169291A1/en active Pending
-
2016
- 2016-07-19 CY CY20161100707T patent/CY1117819T1/en unknown
-
2019
- 2019-10-18 US US16/656,929 patent/US11655475B2/en active Active
-
2021
- 2021-10-27 US US17/512,258 patent/US11913001B2/en active Active
-
2022
- 2022-12-13 US US18/080,150 patent/US11891608B2/en active Active
- 2022-12-13 US US18/065,230 patent/US11739334B2/en active Active
- 2022-12-13 US US18/065,243 patent/US11773395B1/en active Active
- 2022-12-13 US US18/080,164 patent/US11905514B2/en active Active
- 2022-12-13 US US18/065,256 patent/US11845925B2/en active Active
- 2022-12-13 US US18/065,267 patent/US11851660B2/en active Active
-
2024
- 2024-01-12 US US18/411,482 patent/US20240417740A1/en active Pending
Patent Citations (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002002606A2 (en) | 2000-07-03 | 2002-01-10 | Chiron S.P.A. | Immunisation against chlamydia pneumoniae |
| WO2002034771A2 (en) | 2000-10-27 | 2002-05-02 | Chiron Srl | Nucleic acids and proteins from streptococcus groups a & b |
| WO2003018054A1 (en) | 2001-08-31 | 2003-03-06 | Chiron Srl. | Helicobacter pylori vaccination |
| US20080085870A1 (en) | 2002-12-23 | 2008-04-10 | Vical Incorporated | Codon-optimized polynucleotide-based vaccines against human cytomegalovirus infection |
| WO2005002619A2 (en) | 2003-06-26 | 2005-01-13 | Chiron Corporation | Immunogenic compositions for chlamydia trachomatis |
| WO2005032582A2 (en) | 2003-07-31 | 2005-04-14 | Chiron Corporation | Immunogenic compositions for streptococcus pyogenes |
| WO2005111066A2 (en) | 2004-05-14 | 2005-11-24 | Chiron Srl | Polypeptides from non-typeable haemophilus influenzae |
| WO2005113782A1 (en) | 2004-05-18 | 2005-12-01 | Alphavax, Inc. | Tc-83-derived alphavirus vectors, particles and methods |
| US20080057080A1 (en) | 2004-05-18 | 2008-03-06 | Vical Incorporated | Influenza virus vaccine composition and methods of use |
| WO2005121348A1 (en) | 2004-06-07 | 2005-12-22 | Protiva Biotherapeutics, Inc. | Lipid encapsulated interfering rna |
| WO2006089264A2 (en) | 2005-02-18 | 2006-08-24 | Novartis Vaccines And Diagnostics Inc. | Proteins and nucleic acids from meningitis/sepsis-associated escherichia coli |
| WO2006091517A2 (en) | 2005-02-18 | 2006-08-31 | Novartis Vaccines And Diagnostics Inc. | Immunogens from uropathogenic escherichia coli |
| WO2006110413A2 (en) | 2005-03-30 | 2006-10-19 | Novartis Vaccines And Diagnostics Inc. | Haemophilus influenzae type b |
| WO2006138004A2 (en) | 2005-05-12 | 2006-12-28 | Novartis Vaccines And Diagnostics, Inc. | Immunogenic compositions for chlamydia trachomatis |
| US20070014805A1 (en) | 2005-07-07 | 2007-01-18 | Sanofi Pasteur | Immuno-adjuvant emulsion |
| WO2007049155A2 (en) | 2005-10-25 | 2007-05-03 | Novartis Vaccines And Diagnostics Srl | Compositions comprising yersinia pestis antigens |
| WO2008020330A2 (en) | 2006-08-16 | 2008-02-21 | Novartis Ag | Immunogens from uropathogenic escherichia coli |
| WO2009016515A2 (en) | 2007-08-01 | 2009-02-05 | Novartis Ag | Compositions comprising pneumococcal antigens |
| WO2009031043A2 (en) | 2007-09-04 | 2009-03-12 | Novartis Ag | Compositions comprising yersinia pestis antigens |
| WO2009104092A2 (en) | 2008-02-22 | 2009-08-27 | Novartis Ag | Escherichia coli immunogens with improved solubility |
| WO2009109860A2 (en) | 2008-03-06 | 2009-09-11 | Novartis Ag | Mutant forms of chlamydia htra |
| WO2009132206A1 (en) | 2008-04-25 | 2009-10-29 | Liquidia Technologies, Inc. | Compositions and methods for intracellular delivery and release of cargo |
| WO2010119343A2 (en) | 2009-04-14 | 2010-10-21 | Novartis Ag | Compositions for immunising against staphylococcus aureus |
| WO2011005799A2 (en) | 2009-07-06 | 2011-01-13 | Novartis Ag | Self replicating rna molecules and uses thereof |
Non-Patent Citations (32)
| Title |
|---|
| ARSHADY & GUYOT: "Functional Polymer Colloids and Microparticles", vol. 4, 2002, CITUS BOOKS |
| AUSUBEL ET AL.: "Short protocols in molecular biology, 5th edition", 2002, CURRENT PROTOCOLS |
| BIRDI, K.S.: "Handbook of Surface and Colloidal Chemistry", 1997, CRC PRESS |
| BOXUS ET AL., J VIROL, vol. 81, 2007, pages 6879 - 89 |
| COHEN & BERNSTEIN: "Microparticulate Systems for the Delivery of Proteins and Vaccines", 1996, CRC PRESS |
| D.M. WEIR AND C.C. BLACKWELL,: "Handbook of Experimental Immunology", vol. I-IV, 1986, BLACKWELL SCIENTIFIC PUBLICATIONS |
| DEERING ET AL.: "Nucleic acid vaccines: prospects for non-viral delivery ofmRNA vaccines", EXPERT OPIN. DRUG DELIV., vol. 11, no. 6, 2014, pages 1 - 15, XP009183535 |
| EL OUAHABI ET AL., FEBS LETTS, vol. 380, 1996, pages 108 - 12 |
| GEALL ET AL.: "Nonviral delivery of self-amplifying RNA vaccines", PNAS, vol. 109, 2012, pages 14604 - 14609, XP002683929 |
| GENNARO: "Remington: The Science and Practice of Pharmacy. 20th edition", 2000 |
| GIULIANI ET AL., PROC NATL ACAD SCI USA, vol. 103, no. 29, 2006, pages 10834 - 9 |
| GREGORIADIS: "Liposome Technology", vol. I, II, I, 2006, ED. GREGORIADIS). INFORMA HEALTHCARE |
| HEYES ET AL., J CONTROLLED RELEASE, vol. 107, 2005, pages 276 - 87 |
| JEFFS ET AL., PHARMACEUTICAL RESEARCH, vol. 22, no. 3, 2005, pages 362 - 372 |
| JOHANNING ET AL., NUCLEIC ACIDS RES, vol. 23, 1995, pages 1495 - 1501 |
| JONES ET AL., VACCINE, vol. 27, 2009, pages 2506 - 12 |
| MARTINON ET AL., EUR J IMMUNOL, vol. 22, 1993, pages 1719 - 22 |
| MAURER ET AL., BIOPHYSICAL JOURNAL, vol. 80, 2001, pages 2310 - 2326 |
| NEWTON & GRAHAM: "PCR (Introduction to Biotechniques Series, 2nd ed.", 1997, SPRINGER VERLAG |
| O'HAGAN ET AL., J VIROLOGY, vol. 75, 2001, pages 9037 - 9043 |
| PERRI ET AL., J VIROL, vol. 77, 2003, pages 10394 - 10403 |
| REAM ET AL.,: "Molecular Biology Techniques: An Intensive Laboratory Course", 1998, ACADEMIC PRESS |
| REN ET AL.: "Immunogene therapy of recurrent glioblastoma multiforme with a liposomally encapsulated replication-incompetent Semliki forest virus vector carrying the human interleukin-12 gene - a phase I/II clinical protocol", J. NEUROONCOL, vol. 64, 2003, pages 147 - 154, XP055249047 |
| S. COLOWICK AND N. KAPLAN,: "Methods In Enzymology", ACADEMIC PRESS, INC. |
| SAMBROOK ET AL.: "Molecular Cloning: A Laboratory Manual, 3rd edition", 2001, COLD SPRING HARBOR LABORATORY PRESS |
| SAXCNA ET AL., VET MICROBIOL, vol. 136, 2009, pages 36 - 44 |
| SINGH ET AL., PHARMACEUTICAL RESEARCH, vol. 20, 2003, pages 247 - 251 |
| TAYLOR ET AL., VACCINE, vol. 23, 2005, pages 1242 - 50 |
| UCHEGBU & SCHATZLEIN: "Polymers in Drug Delivery", 2006, CRC PRESS |
| WEISSIG: "Liposomes: Methods and Protocols, Volume 1: Pharmaceutical Nanocarriers: Methods and Protocols", vol. 1, 2009, HUMANA PRESS |
| YING ET AL., NATURE MEDICINE, vol. 5, 1999, pages 823 - 27 |
| YONEYAMA, FUJITA, CYTOKINE & GROWTH FACTOR REVIEWS, vol. 18, 2007, pages 545 - 51 |
Cited By (192)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11739334B2 (en) | 2010-07-06 | 2023-08-29 | Glaxosmithkline Biologicals Sa | Immunisation of large mammals with low doses of RNA |
| US11766401B2 (en) | 2010-07-06 | 2023-09-26 | Glaxosmithkline Biologicals Sa | Methods of administering lipid formulations with immunogens |
| US11730754B2 (en) | 2010-07-06 | 2023-08-22 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11717529B2 (en) | 2010-07-06 | 2023-08-08 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11905514B2 (en) | 2010-07-06 | 2024-02-20 | Glaxosmithkline Biological Sa | Immunisation of large mammals with low doses of RNA |
| US11707482B2 (en) | 2010-07-06 | 2023-07-25 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11026964B2 (en) | 2010-07-06 | 2021-06-08 | Glaxosmithkline Biologicals Sa | Delivery of RNA to different cell types |
| US12186333B2 (en) | 2010-07-06 | 2025-01-07 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11696923B2 (en) | 2010-07-06 | 2023-07-11 | Glaxosmithkline Biologicals, Sa | Delivery of RNA to trigger multiple immune pathways |
| US11690863B2 (en) | 2010-07-06 | 2023-07-04 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11690865B2 (en) | 2010-07-06 | 2023-07-04 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11690861B2 (en) | 2010-07-06 | 2023-07-04 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11690864B2 (en) | 2010-07-06 | 2023-07-04 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11850305B2 (en) | 2010-07-06 | 2023-12-26 | Glaxosmithkline Biologicals Sa | Method of making lipid formulations with RNA encoding immunogens |
| US11690862B1 (en) | 2010-07-06 | 2023-07-04 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11666534B2 (en) | 2010-07-06 | 2023-06-06 | Glaxosmithkline Biologicals Sa | Methods of administering lipid formulations with viral immunogens |
| US11655475B2 (en) | 2010-07-06 | 2023-05-23 | Glaxosmithkline Biologicals Sa | Immunisation of large mammals with low doses of RNA |
| US11759475B2 (en) | 2010-07-06 | 2023-09-19 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11638693B2 (en) | 2010-07-06 | 2023-05-02 | Glaxosmithkline Biologicals Sa | Vaccine for eliciting immune response comprising RNA encoding an immunogen and lipid formulations comprising mole percentage of lipids |
| US11638694B2 (en) | 2010-07-06 | 2023-05-02 | Glaxosmithkline Biologicals Sa | Vaccine for eliciting immune response comprising lipid formulations and RNA encoding multiple immunogens |
| US11883534B2 (en) | 2010-07-06 | 2024-01-30 | Glaxosmithkline Biologicals Sa | Immunisation with lipid formulations with RNA encoding immunogens |
| US11596645B2 (en) | 2010-07-06 | 2023-03-07 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11851660B2 (en) | 2010-07-06 | 2023-12-26 | Glaxosmithkline Biologicals Sa | Immunisation of large mammals with low doses of RNA |
| US11913001B2 (en) | 2010-07-06 | 2024-02-27 | Glaxosmithkline Biologicals Sa | Immunisation of large mammals with low doses of RNA |
| US11865080B2 (en) | 2010-07-06 | 2024-01-09 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11773395B1 (en) | 2010-07-06 | 2023-10-03 | Glaxosmithkline Biologicals Sa | Immunization of large mammals with low doses of RNA |
| US11786467B2 (en) | 2010-07-06 | 2023-10-17 | Glaxosmithkline Biologicals Sa | Lipid formulations with immunogens |
| US11291635B2 (en) | 2010-07-06 | 2022-04-05 | Glaxosmithkline Biological Sa | Virion-like delivery particles for self-replicating RNA molecules |
| US11291682B2 (en) | 2010-07-06 | 2022-04-05 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11857681B2 (en) | 2010-07-06 | 2024-01-02 | Glaxosmithkline Biologicals Sa | Lipid formulations with RNA encoding immunogens |
| US20220125723A1 (en) | 2010-07-06 | 2022-04-28 | Glaxosmithkline Biologicals Sa | Lipid formulations with viral immunogens |
| US11891608B2 (en) | 2010-07-06 | 2024-02-06 | Glaxosmithkline Biologicals Sa | Immunization of large mammals with low doses of RNA |
| US11324770B2 (en) | 2010-07-06 | 2022-05-10 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11839686B2 (en) | 2010-07-06 | 2023-12-12 | Glaxosmithkline Biologicals Sa | Lipid formulations with viral immunogens |
| EP2591103B1 (en) | 2010-07-06 | 2019-08-28 | GlaxoSmithKline Biologicals SA | Delivery of rna to different cell types |
| EP4005592B1 (en) | 2010-07-06 | 2022-10-12 | GlaxoSmithKline Biologicals S.A. | Virion-like delivery particles for self-replicating rna molecules |
| US11845925B2 (en) | 2010-07-06 | 2023-12-19 | Glaxosmithkline Biologicals Sa | Immunisation of large mammals with low doses of RNA |
| US11857562B2 (en) | 2010-07-06 | 2024-01-02 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US9937233B2 (en) | 2010-08-06 | 2018-04-10 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US9181319B2 (en) | 2010-08-06 | 2015-11-10 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| US8822663B2 (en) | 2010-08-06 | 2014-09-02 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| EP4008357B1 (en) | 2010-08-31 | 2022-12-28 | GlaxoSmithKline Biologicals SA | Small liposomes for delivery of immunogen-encoding rna |
| EP4226941B1 (en) | 2010-08-31 | 2024-10-23 | GlaxoSmithKline Biologicals SA | Pegylated liposomes for delivery of immunogen-encoding rna |
| EP2611467B1 (en) | 2010-08-31 | 2022-07-20 | GlaxoSmithKline Biologicals SA | Small liposomes for delivery of immunogen-encoding rna |
| EP4066819B1 (en) | 2010-08-31 | 2023-03-01 | GlaxoSmithKline Biologicals SA | Small liposomes for delivery of immunogen-encoding rna |
| EP4043040B1 (en) | 2010-08-31 | 2023-01-11 | GlaxoSmithKline Biologicals SA | Small liposomes for delivery of immunogen-encoding rna |
| EP2611461B1 (en) | 2010-08-31 | 2022-03-09 | GlaxoSmithKline Biologicals SA | Pegylated liposomes for delivery of immunogen-encoding rna |
| EP3981427B1 (en) | 2010-08-31 | 2022-05-25 | GlaxoSmithKline Biologicals S.A. | Pegylated liposomes for delivery of immunogen-encoding rna |
| EP3970742B1 (en) | 2010-08-31 | 2022-05-25 | GlaxoSmithKline Biologicals S.A. | Pegylated liposomes for delivery of immunogen-encoding rna |
| WO2012031043A1 (en) | 2010-08-31 | 2012-03-08 | Novartis Ag | Pegylated liposomes for delivery of immunogen-encoding rna |
| EP4066856B1 (en) | 2010-08-31 | 2022-12-07 | GlaxoSmithKline Biologicals SA | Pegylated liposomes for delivery of immunogen-encoding rna |
| EP4066857B1 (en) | 2010-08-31 | 2022-12-21 | GlaxoSmithKline Biologicals SA | Pegylated liposomes for delivery of immunogen-encoding rna |
| US11759422B2 (en) | 2010-08-31 | 2023-09-19 | Glaxosmithkline Biologicals Sa | Pegylated liposomes for delivery of immunogen-encoding RNA |
| EP4066855B1 (en) | 2010-08-31 | 2022-12-28 | GlaxoSmithKline Biologicals SA | Pegylated liposomes for delivery of immunogen-encoding rna |
| US9334328B2 (en) | 2010-10-01 | 2016-05-10 | Moderna Therapeutics, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US9657295B2 (en) | 2010-10-01 | 2017-05-23 | Modernatx, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US10064959B2 (en) | 2010-10-01 | 2018-09-04 | Modernatx, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US11639370B2 (en) | 2010-10-11 | 2023-05-02 | Glaxosmithkline Biologicals Sa | Antigen delivery platforms |
| US9950068B2 (en) | 2011-03-31 | 2018-04-24 | Modernatx, Inc. | Delivery and formulation of engineered nucleic acids |
| US8710200B2 (en) | 2011-03-31 | 2014-04-29 | Moderna Therapeutics, Inc. | Engineered nucleic acids encoding a modified erythropoietin and their expression |
| US11951180B2 (en) | 2011-06-08 | 2024-04-09 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US11951181B2 (en) | 2011-06-08 | 2024-04-09 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11951179B2 (en) | 2011-06-08 | 2024-04-09 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US11547764B2 (en) | 2011-06-08 | 2023-01-10 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US12102720B2 (en) | 2011-06-08 | 2024-10-01 | Translate Bio, Inc. | Cleavable lipids |
| US12121592B2 (en) | 2011-06-08 | 2024-10-22 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11730825B2 (en) | 2011-06-08 | 2023-08-22 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| EP3821879A1 (en) * | 2011-07-06 | 2021-05-19 | GlaxoSmithKline Biologicals S.A. | Liposomes having useful n:p ratio for delivery of rna molecules |
| US11896636B2 (en) | 2011-07-06 | 2024-02-13 | Glaxosmithkline Biologicals Sa | Immunogenic combination compositions and uses thereof |
| US11058762B2 (en) | 2011-07-06 | 2021-07-13 | Glaxosmithkline Biologicals Sa | Immunogenic compositions and uses thereof |
| WO2013006825A1 (en) | 2011-07-06 | 2013-01-10 | Novartis Ag | Liposomes having useful n:p ratio for delivery of rna molecules |
| EP2729126B1 (en) * | 2011-07-06 | 2020-12-23 | GlaxoSmithKline Biologicals SA | Liposomes having useful n:p ratio for delivery of rna molecules |
| EP4115875A1 (en) * | 2011-07-06 | 2023-01-11 | GlaxoSmithKline Biologicals S.A. | Liposomes having useful n:p ratio for delivery of rna molecules |
| EP4014966A1 (en) * | 2011-07-06 | 2022-06-22 | GlaxoSmithKline Biologicals S.A. | Liposomes having useful n:p ratio for delivery of rna molecules |
| EP4115876A1 (en) * | 2011-07-06 | 2023-01-11 | GlaxoSmithKline Biologicals S.A. | Liposomes having useful n:p ratio for delivery of rna molecules |
| US9464124B2 (en) | 2011-09-12 | 2016-10-11 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| US10022425B2 (en) | 2011-09-12 | 2018-07-17 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US10751386B2 (en) | 2011-09-12 | 2020-08-25 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US8680069B2 (en) | 2011-12-16 | 2014-03-25 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of G-CSF |
| US9271996B2 (en) | 2011-12-16 | 2016-03-01 | Moderna Therapeutics, Inc. | Formulation and delivery of PLGA microspheres |
| EP4144378A1 (en) | 2011-12-16 | 2023-03-08 | ModernaTX, Inc. | Modified nucleoside, nucleotide, and nucleic acid compositions |
| US9186372B2 (en) | 2011-12-16 | 2015-11-17 | Moderna Therapeutics, Inc. | Split dose administration |
| US8754062B2 (en) | 2011-12-16 | 2014-06-17 | Moderna Therapeutics, Inc. | DLIN-KC2-DMA lipid nanoparticle delivery of modified polynucleotides |
| US8664194B2 (en) | 2011-12-16 | 2014-03-04 | Moderna Therapeutics, Inc. | Method for producing a protein of interest in a primate |
| EP2791160B1 (en) | 2011-12-16 | 2022-03-02 | ModernaTX, Inc. | Modified mrna compositions |
| US9255129B2 (en) | 2012-04-02 | 2016-02-09 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding SIAH E3 ubiquitin protein ligase 1 |
| US9149506B2 (en) | 2012-04-02 | 2015-10-06 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding septin-4 |
| US9192651B2 (en) | 2012-04-02 | 2015-11-24 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of secreted proteins |
| US9233141B2 (en) | 2012-04-02 | 2016-01-12 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of proteins associated with blood and lymphatic disorders |
| US9254311B2 (en) | 2012-04-02 | 2016-02-09 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of proteins |
| US9301993B2 (en) | 2012-04-02 | 2016-04-05 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding apoptosis inducing factor 1 |
| US9303079B2 (en) | 2012-04-02 | 2016-04-05 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| US9878056B2 (en) | 2012-04-02 | 2018-01-30 | Modernatx, Inc. | Modified polynucleotides for the production of cosmetic proteins and peptides |
| US9828416B2 (en) | 2012-04-02 | 2017-11-28 | Modernatx, Inc. | Modified polynucleotides for the production of secreted proteins |
| US9827332B2 (en) | 2012-04-02 | 2017-11-28 | Modernatx, Inc. | Modified polynucleotides for the production of proteins |
| US9814760B2 (en) | 2012-04-02 | 2017-11-14 | Modernatx, Inc. | Modified polynucleotides for the production of biologics and proteins associated with human disease |
| US9782462B2 (en) | 2012-04-02 | 2017-10-10 | Modernatx, Inc. | Modified polynucleotides for the production of proteins associated with human disease |
| US9675668B2 (en) | 2012-04-02 | 2017-06-13 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding hepatitis A virus cellular receptor 2 |
| US9572897B2 (en) | 2012-04-02 | 2017-02-21 | Modernatx, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| US9587003B2 (en) | 2012-04-02 | 2017-03-07 | Modernatx, Inc. | Modified polynucleotides for the production of oncology-related proteins and peptides |
| JP2016506416A (en) * | 2013-01-10 | 2016-03-03 | ノバルティス アーゲー | Influenza virus immunogenic compositions and uses thereof |
| WO2014108515A1 (en) | 2013-01-10 | 2014-07-17 | Novartis Ag | Influenza virus immunogenic compositions and uses thereof |
| CN104902925A (en) * | 2013-01-10 | 2015-09-09 | 诺华股份有限公司 | Influenza virus immunogenic compositions and uses thereof |
| US12447209B2 (en) | 2013-02-22 | 2025-10-21 | Cure Vac SE | Combination of vaccination and inhibition of the PD-1 pathway |
| EP3292873B1 (en) | 2013-02-22 | 2019-05-01 | CureVac AG | Combination of vaccination and inhibition of the pd-1 pathway |
| US11458195B2 (en) | 2013-02-22 | 2022-10-04 | Curevac Ag | Combination of vaccination and inhibition of the PD-1 pathway |
| WO2014152211A1 (en) | 2013-03-14 | 2014-09-25 | Moderna Therapeutics, Inc. | Formulation and delivery of modified nucleoside, nucleotide, and nucleic acid compositions |
| EP3521429A1 (en) | 2013-03-15 | 2019-08-07 | GlaxoSmithKline Biologicals SA | Rna purification methods |
| WO2014140211A1 (en) | 2013-03-15 | 2014-09-18 | Novartis Ag | Rna purification methods |
| US8980864B2 (en) | 2013-03-15 | 2015-03-17 | Moderna Therapeutics, Inc. | Compositions and methods of altering cholesterol levels |
| US10576146B2 (en) | 2013-09-26 | 2020-03-03 | Biontech Rna Pharmaceuticals Gmbh | Particles comprising a shell with RNA |
| US11660338B2 (en) | 2013-09-26 | 2023-05-30 | BioNTech SE | Particles comprising a shell with RNA |
| US9950065B2 (en) | 2013-09-26 | 2018-04-24 | Biontech Rna Pharmaceuticals Gmbh | Particles comprising a shell with RNA |
| US10815291B2 (en) | 2013-09-30 | 2020-10-27 | Modernatx, Inc. | Polynucleotides encoding immune modulating polypeptides |
| US10323076B2 (en) | 2013-10-03 | 2019-06-18 | Modernatx, Inc. | Polynucleotides encoding low density lipoprotein receptor |
| EP3981437B1 (en) | 2014-04-23 | 2024-10-09 | ModernaTX, Inc. | Nucleic acid vaccines |
| EP4023249A1 (en) | 2014-04-23 | 2022-07-06 | ModernaTX, Inc. | Nucleic acid vaccines |
| EP4501318A2 (en) | 2014-04-23 | 2025-02-05 | ModernaTX, Inc. | Nucleic acid vaccines |
| EP4023249B1 (en) | 2014-04-23 | 2024-10-09 | ModernaTX, Inc. | Nucleic acid vaccines |
| EP3134131B1 (en) | 2014-04-23 | 2021-12-22 | ModernaTX, Inc. | Nucleic acid vaccines |
| EP3981437A1 (en) | 2014-04-23 | 2022-04-13 | ModernaTX, Inc. | Nucleic acid vaccines |
| EP4159741A1 (en) | 2014-07-16 | 2023-04-05 | ModernaTX, Inc. | Method for producing a chimeric polynucleotide encoding a polypeptide having a triazole-containing internucleotide linkage |
| US11364292B2 (en) | 2015-07-21 | 2022-06-21 | Modernatx, Inc. | CHIKV RNA vaccines |
| US10702597B2 (en) | 2015-07-21 | 2020-07-07 | Modernatx, Inc. | CHIKV RNA vaccines |
| US10449244B2 (en) | 2015-07-21 | 2019-10-22 | Modernatx, Inc. | Zika RNA vaccines |
| US11007260B2 (en) | 2015-07-21 | 2021-05-18 | Modernatx, Inc. | Infectious disease vaccines |
| EP3364981A4 (en) * | 2015-10-22 | 2019-08-07 | ModernaTX, Inc. | VACCINE AGAINST THE CYTOMEGALOVIRUS HUMAN |
| CN108472354A (en) * | 2015-10-22 | 2018-08-31 | 摩登纳特斯有限公司 | Respiratory syncytial virus vaccines |
| US12551734B2 (en) | 2015-10-22 | 2026-02-17 | Modernatx, Inc. | Betacoronavirus mRNA vaccines |
| US11643441B1 (en) | 2015-10-22 | 2023-05-09 | Modernatx, Inc. | Nucleic acid vaccines for varicella zoster virus (VZV) |
| US12521577B2 (en) | 2015-10-22 | 2026-01-13 | Modernatx, Inc. | Betacoronavirus RNA vaccines |
| WO2017070622A1 (en) * | 2015-10-22 | 2017-04-27 | Modernatx, Inc. | Respiratory syncytial virus vaccine |
| US12409347B2 (en) | 2015-10-22 | 2025-09-09 | Modernatx, Inc. | Betacoronavirus mRNA vaccines |
| US12403335B2 (en) | 2015-10-22 | 2025-09-02 | Modernatx, Inc. | Betacoronavirus MRNA vaccines |
| AU2016341311B2 (en) * | 2015-10-22 | 2023-11-16 | Modernatx, Inc. | Respiratory syncytial virus vaccine |
| US12403336B2 (en) | 2015-10-22 | 2025-09-02 | Modernatx, Inc. | Betacorona virus mRNA vaccines |
| EP4349404A3 (en) * | 2015-10-22 | 2024-06-19 | ModernaTX, Inc. | Respiratory virus vaccines |
| US12208288B2 (en) | 2015-10-22 | 2025-01-28 | Modernatx, Inc. | Betacoronavirus RNA vaccines |
| EP4011451A1 (en) * | 2015-10-22 | 2022-06-15 | ModernaTX, Inc. | Metapneumovirus mrna vaccines |
| EP3718565A1 (en) * | 2015-10-22 | 2020-10-07 | ModernaTX, Inc. | Respiratory virus vaccines |
| EP3718565B1 (en) | 2015-10-22 | 2022-04-27 | ModernaTX, Inc. | Respiratory virus vaccines |
| US11278611B2 (en) | 2015-10-22 | 2022-03-22 | Modernatx, Inc. | Zika virus RNA vaccines |
| US11872278B2 (en) | 2015-10-22 | 2024-01-16 | Modernatx, Inc. | Combination HMPV/RSV RNA vaccines |
| EP4349405A3 (en) * | 2015-10-22 | 2024-06-19 | ModernaTX, Inc. | Respiratory virus vaccines |
| EP3365007A4 (en) * | 2015-10-22 | 2019-07-03 | ModernaTX, Inc. | VACCINE AGAINST BROAD SPECTRUM INFLUENZA VIRUS |
| US11235052B2 (en) | 2015-10-22 | 2022-02-01 | Modernatx, Inc. | Chikungunya virus RNA vaccines |
| EP3364980A4 (en) * | 2015-10-22 | 2019-07-10 | ModernaTX, Inc. | Nucleic acid vaccines for varicella zoster virus (vzv) |
| US10933127B2 (en) | 2015-10-22 | 2021-03-02 | Modernatx, Inc. | Betacoronavirus mRNA vaccine |
| US10493143B2 (en) | 2015-10-22 | 2019-12-03 | Modernatx, Inc. | Sexually transmitted disease vaccines |
| US10363303B2 (en) | 2016-01-11 | 2019-07-30 | Verndari, Inc. | Microneedle compositions and methods of using same |
| US10022436B2 (en) | 2016-01-11 | 2018-07-17 | Verndari, Inc. | Microneedle compositions and methods of using same |
| US12128113B2 (en) | 2016-05-18 | 2024-10-29 | Modernatx, Inc. | Polynucleotides encoding JAGGED1 for the treatment of Alagille syndrome |
| US11878060B2 (en) | 2016-08-07 | 2024-01-23 | Novartis Ag | mRNA-mediated immunization methods |
| US12409218B2 (en) | 2016-11-11 | 2025-09-09 | Modernatx, Inc. | Influenza vaccine |
| US11696946B2 (en) | 2016-11-11 | 2023-07-11 | Modernatx, Inc. | Influenza vaccine |
| US12318443B2 (en) | 2016-11-11 | 2025-06-03 | Modernatx, Inc. | Influenza vaccine |
| US10925958B2 (en) | 2016-11-11 | 2021-02-23 | Modernatx, Inc. | Influenza vaccine |
| US11103578B2 (en) | 2016-12-08 | 2021-08-31 | Modernatx, Inc. | Respiratory virus nucleic acid vaccines |
| US11464848B2 (en) | 2017-03-15 | 2022-10-11 | Modernatx, Inc. | Respiratory syncytial virus vaccine |
| US11576961B2 (en) | 2017-03-15 | 2023-02-14 | Modernatx, Inc. | Broad spectrum influenza virus vaccine |
| WO2018170260A1 (en) * | 2017-03-15 | 2018-09-20 | Modernatx, Inc. | Respiratory syncytial virus vaccine |
| US11918644B2 (en) | 2017-03-15 | 2024-03-05 | Modernatx, Inc. | Varicella zoster virus (VZV) vaccine |
| US11752206B2 (en) | 2017-03-15 | 2023-09-12 | Modernatx, Inc. | Herpes simplex virus vaccine |
| US11045540B2 (en) | 2017-03-15 | 2021-06-29 | Modernatx, Inc. | Varicella zoster virus (VZV) vaccine |
| US11207398B2 (en) | 2017-09-14 | 2021-12-28 | Modernatx, Inc. | Zika virus mRNA vaccines |
| US10653767B2 (en) | 2017-09-14 | 2020-05-19 | Modernatx, Inc. | Zika virus MRNA vaccines |
| US12453766B2 (en) | 2018-01-29 | 2025-10-28 | Modernatx, Inc. | RSV RNA vaccines |
| US11911453B2 (en) | 2018-01-29 | 2024-02-27 | Modernatx, Inc. | RSV RNA vaccines |
| US11793843B2 (en) | 2019-01-10 | 2023-10-24 | Janssen Biotech, Inc. | Prostate neoantigens and their uses |
| US12582683B2 (en) | 2019-01-10 | 2026-03-24 | Janssen Biotech, Inc. | Prostate neoantigens and their uses |
| US11351242B1 (en) | 2019-02-12 | 2022-06-07 | Modernatx, Inc. | HMPV/hPIV3 mRNA vaccine composition |
| US12070495B2 (en) | 2019-03-15 | 2024-08-27 | Modernatx, Inc. | HIV RNA vaccines |
| US12018289B2 (en) | 2019-11-18 | 2024-06-25 | Janssen Biotech, Inc. | Vaccines based on mutant CALR and JAK2 and their uses |
| US12194089B2 (en) | 2020-02-04 | 2025-01-14 | CureVac SE | Coronavirus vaccine |
| US11596686B2 (en) | 2020-02-04 | 2023-03-07 | CureVac SE | Coronavirus vaccine |
| US11471525B2 (en) | 2020-02-04 | 2022-10-18 | Curevac Ag | Coronavirus vaccine |
| US11964011B2 (en) | 2020-02-04 | 2024-04-23 | CureVac SE | Coronavirus vaccine |
| US12390523B2 (en) | 2020-02-04 | 2025-08-19 | CureVac SE | Coronavirus vaccine |
| US11576966B2 (en) | 2020-02-04 | 2023-02-14 | CureVac SE | Coronavirus vaccine |
| US11964012B2 (en) | 2020-02-04 | 2024-04-23 | CureVac SE | Coronavirus vaccine |
| EP4117725A4 (en) * | 2020-03-09 | 2024-05-29 | Arcturus Therapeutics, Inc. | CORONAVIRUS VACCINE COMPOSITIONS AND METHODS |
| US12220455B2 (en) | 2020-03-09 | 2025-02-11 | Arcturus Therapeutics, Inc. | Coronavirus vaccine compositions and methods |
| US12390524B2 (en) | 2020-03-09 | 2025-08-19 | Arcturus Therapeutics, Inc. | Compositions and methods for inducing immune responses |
| US12133899B2 (en) | 2020-04-22 | 2024-11-05 | BioNTech SE | Coronavirus vaccine |
| US12295997B2 (en) | 2020-07-06 | 2025-05-13 | Janssen Biotech, Inc. | Prostate neoantigens and their uses |
| US11918643B2 (en) | 2020-12-22 | 2024-03-05 | CureVac SE | RNA vaccine against SARS-CoV-2 variants |
| US11872280B2 (en) | 2020-12-22 | 2024-01-16 | CureVac SE | RNA vaccine against SARS-CoV-2 variants |
| WO2022259191A1 (en) | 2021-06-09 | 2022-12-15 | Glaxosmithkline Biologicals Sa | Release assay for determining potency of self-amplifying rna drug product and methods for using |
| US12208136B2 (en) | 2021-11-29 | 2025-01-28 | BioNTech SE | Coronavirus vaccine |
| US12186387B2 (en) | 2021-11-29 | 2025-01-07 | BioNTech SE | Coronavirus vaccine |
| US12186389B2 (en) | 2022-10-28 | 2025-01-07 | Glaxosmithkline Biologicals Sa | Nucleic acid base vaccine against emerging SARS-CoV-2 variants |
| WO2024125597A1 (en) | 2022-12-14 | 2024-06-20 | Providence Therapeutics Holdings Inc. | Compositions and methods for infectious diseases |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11739334B2 (en) | Immunisation of large mammals with low doses of RNA | |
| US11759475B2 (en) | Delivery of RNA to trigger multiple immune pathways | |
| EP3670658A2 (en) | Delivery of rna to different cell types |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11736497 Country of ref document: EP Kind code of ref document: A2 |
|
| ENP | Entry into the national phase |
Ref document number: 2013518811 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2804492 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011736497 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13808153 Country of ref document: US |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11736497 Country of ref document: EP Kind code of ref document: A2 |