WO2011153588A1 - Viral polymerase inhibitors - Google Patents
Viral polymerase inhibitors Download PDFInfo
- Publication number
- WO2011153588A1 WO2011153588A1 PCT/AU2011/000713 AU2011000713W WO2011153588A1 WO 2011153588 A1 WO2011153588 A1 WO 2011153588A1 AU 2011000713 W AU2011000713 W AU 2011000713W WO 2011153588 A1 WO2011153588 A1 WO 2011153588A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- cyclohexyl
- amino
- carbonyl
- indazole
- Prior art date
Links
- 0 CC(C)(C)OC(N*C1OC1N**)=O Chemical compound CC(C)(C)OC(N*C1OC1N**)=O 0.000 description 7
- SVZMBEPDSIVQNL-UHFFFAOYSA-N CC(C)(C)OC(NC1(CCC1)C(Nc(cc1)ccc1C#N)=O)=O Chemical compound CC(C)(C)OC(NC1(CCC1)C(Nc(cc1)ccc1C#N)=O)=O SVZMBEPDSIVQNL-UHFFFAOYSA-N 0.000 description 1
- ROVVUKFHORPDSM-UHFFFAOYSA-N CC(C)(C)OC(NC1(CCC1)C(O)=O)=O Chemical compound CC(C)(C)OC(NC1(CCC1)C(O)=O)=O ROVVUKFHORPDSM-UHFFFAOYSA-N 0.000 description 1
- YBAZINRZQSAIAY-UHFFFAOYSA-N Nc(cc1)ccc1C#N Chemical compound Nc(cc1)ccc1C#N YBAZINRZQSAIAY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
- C07D231/56—Benzopyrazoles; Hydrogenated benzopyrazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the present invention relates to viral polymerase inhibitors, in particular inhibitors of viral polymerases within the Flaviviridae family such as hepatitis C virus (HCV), processes for their preparation and their use in the treatment of Flaviviridae viral infections such as Hepatitis C virus (HCV) infections.
- HCV hepatitis C virus
- the Flaviviridae are a group of positive single-stranded RNA viruses with a genome size from 9-15 kb.
- the Flaviviridae consist of various genera including: Hepaciviruses (this genus contains only one species, the Hepatitis C virus (HCV), which is composed of many genotypes and subtypes); Flaviviruses (this genus includes the Dengue virus, Japanese Tick-Borne and the Yellow Fever virus and there are some additional Flaviviruses that are unclassified) and Pestiviruses (this genus includes three serotypes of bovine viral diarrhoea virus, but no known human pathogens).
- HCV Hepatitis C virus
- Hepatitis C virus is a major cause of viral hepatitis and has infected more than 200 million people worldwide.
- Hepatitis C virus has a positive-strand RNA genome enclosed in a nucleocapsid and lipid envelope.
- the HCV genome is approximately 9.6 kb in length and encodes a polyprotein of about 3,000 amino acids.
- genotypes 1 a and 1 b account for about 75 % of cases, and genotypes 2 and 3 for 10-20 % of cases.
- HCV is the most common chronic bloodborne infection, affecting approximately 3.2 million persons.
- SOC Current standard of care
- peg-IFN pegylated interferon
- SOC therapy the recommended duration of treatment depends on the genotype. For patients with genotype 1 , a 48-week course is recommended, and the success rate for achieving a sustained viral response (SVR) using SOC therapy is approximately 50%.
- SVR sustained viral response
- SOC therapy In addition to being prolonged and having limited efficacy, this SOC therapy is associated with serious side effects, such as fatigue, influenza-like symptoms, depression and suicide with peg-I FN, and haemolytic anaemia with ribavirin. There are also several contraindications to SOC therapy including pregnancy, depression, anaemia, HCV related decompensated cirrhosis, alcohol/substance abuse and autoimmune disorders.
- ADAC Antiviral Drugs Advisory Committee
- telaprevir and boceprevir have demonstrated significant effectiveness in improving cure rates over current SOC therapy and are likely to improve the current SOC.
- telaprevir and boceprevir have issues with adverse effects.
- Telaprevir is associated with rash and anaemia, and boceprevir with anaemia and dysgeusia (dysfunction of the sense of taste).
- both of these new agents are limited to use in genotype 1 .
- the anticipated new SOC (boceprevir/telaprevir plus peg-IFN and ribavirin) remains unsuitable for those intolerant to or with contraindications to peg- I FN/ribavirin.
- the HCV genome possesses structural (core) and non-structural (NS2, NS3, NS4A, NS4B, NS5A and NS5B) proteins.
- the non-structural proteins are involved in viral genomic replication, with the initial synthesis of RNA carried out by NS5B RNA dependent RNA polymerase.
- the NS5B protein is a key target for anti-HCV therapy, as it is essential for HCV replication and has no human host equivalent. This protein has been well characterised and is a validated target for drug discovery.
- the inventors have found a new class of NS5B polymerase inhibitors for the treatment of HCV infections.
- Zi, Z 2 ,Z 3 and Z 4 are each independently selected from C-R 1 , C-R a , N and N + -0 " wherein one of Zi, Z 2 ,Z 3 and Z 4 is C-R 1 , preferably Z 2 is C-R 1 and Zi, Z 3 and Z 4 are each independently selected from C-R a , N and N + -0 ⁇ , preferably C-R a and N;
- R a is independently selected from H, optionally substituted d. 6 alkyl, optionally substituted C 2 - 6 alkenyl, halo, haloC -6 alkyl, CHF 2 , OCHF 2 , CF 3 , OCF 3 , CN, OH, optionally substituted d_ 6 alkoxy, N0 2 , NH 2 , NH(d_ 6 alkyl), N(d_ 6 alkyl) 2 , NHC(0)d_ 6 alkyl, NHS0 2 C(0)NH 2 ,
- R 1 is (CH 2 ) m R 4 , (CH 2 ) m -C(0)R 4 , (CH 2 ) m -OC(0)R 4 (CH 2 ) m -NR 5 C(0)R 4 , (CH 2 ) m -NR 5 S(0) 2 R 4 or (CH 2 ) m -S(0) 2 R 4 , preferably (CH 2 ) m -C(0)R 4 ;
- R 2 is selected from optionally substituted (CH 2 ) m C 3 . 8 cycloalkyl, optionally substituted (CH 2 ) m heterocyclyl, optionally substituted (CH 2 ) m aryl and optionally substituted
- R 3 is selected from optionally substituted C 1 . 6 alkyl, optionally substituted C 2 . 6 alkenyl, optionally substituted (CH 2 ) m C 3 . 8 cycloalkyl, optionally substituted (CH 2 ) m C 3 . 8 cycloalkenyl, optionally substituted (CH 2 ) m heterocyclyl, optionally substituted (CH 2 ) m aryl and optionally substituted (CH 2 ) m heteroaryl;
- R 4 is (CH 2 ) m R 6 , (CH 2 ) m OR 6 or (CH 2 ) m NR 5 R 6 preferably (CH 2 ) m OR 6 or (CH 2 ) m NR 5 R 6 , most preferably (CH 2 ) m NR 5 R 6 ;
- R 5 is independently selected from H, optionally substituted d. 6 alkyl and optionally substituted C 2 . 6 alkenyl, preferably H;
- R 6 is selected from H, optionally substituted d. 6 alkyl, optionally substituted C 2 . 6 alkenyl, optionally substituted (CH 2 ) m C 3 . 8 cycloalkyl, optionally substituted (CH 2 ) m C 3 .
- R 7 and R 8 are independently selected from H, optionally substituted C 1 . 6 alkyl, optionally substituted C 2 . 6 alkenyl, optionally substituted (CH 2 ) m C 3 . 8 cycloalkyl, optionally substituted (CH 2 ) m C 3 . 8 cycloalkenyl, optionally substituted (CH 2 ) m heterocyclyl, optionally substituted (CH 2 ) m aryl, optionally substituted (CH 2 ) m heteroaryl, the side chain of an amino acid or an alkyl ester thereof or R 7 and R 8 together with the atom or adjacent atoms to which they are attached form an optionally substituted C 3 .
- R 9 is selected from R 10 , (CH 2 ) m C(0)-R 1 °, (CH 2 ) m C(0)NR 5 R 10 , (CH 2 ) m C(0)NR 5 C(0)-R 10 , (CH 2 ) m C(0)NR 5 S0 2 -R 1 ° (CH 2 ) m C(0)NR 5 R 10 , (CH 2 ) m NR 5 R 10 , (CH 2 ) m NR 5 C(0)-R 10 ,
- R 10 is H, OH, -A-(Q) protest or 0-A-(Q) n ;
- A is selected from optionally substituted C ⁇ alkyl, optionally substituted C 2 . 6 alkenyl, optionally substituted (CH 2 ) m C 3 - 8 cycloalkyl, optionally substituted (CH 2 ) m aryl, optionally substituted (CH 2 ) m heterocyclyl and optionally substituted (CH 2 ) m heteroaryl; and
- Q is selected from optionally substituted C ⁇ alkyl, optionally substituted C 2 . 6 alkenyl, halo, haloCi -6 alkyl, CHF 2 , OCHF 2 , CF 3 , OCF 3 , CN, OH, optionally substituted Ci. 6 alkoxy, N0 2 , NH 2 , NH(Ci_ 6 alkyl), NiCi-salkylk, NHC(0)Ci-6alk l, NHS0 2 C(0)NH 2 , C(0)NH(Ci-6alkyl), C(0)N(Ci.
- n in each instance is independently 0, 1 , 2 or 3, preferably 0 or 1 , more preferably 0;
- n is independently 0, 1 , 2, 3, 4 or 5, preferably 0, 1 , 2 or 3, more preferably 0, 1 or 2, most preferably 0 or 1 ; and v is independently 1 , 2 or 3, preferably 1 .
- Z'i , Z' 2 , Z' 3 and Z' 4 are each independently selected from C-X, C-R a , N and N + -0 " wherein one of ⁇ , Z' 2 ,Z' 3 and Z' 4 is C-X, preferably Z' 2 is C-X and Z , Z' 3 and Z' 4 are each independently selected from C-R a , N and N + -0 " ;
- X is (R 1 ) t C0 2 H or (R 1 ) t C0 2 d_ 3 alkyl, preferably (R 1 ) t C0 2 H;
- R 1 , R 2 , R 3 and R a are as defined in formula (I).
- the compounds of formula (I) are inhibitors of HCV.
- the compounds of formula (I) inhibit RNA synthesis by the RNA dependent RNA polymerase of HCV (the NS5B protein encoded by HCV).
- NS5B polymerase inhibitors have been clinically validated as potential antiviral agents for the treatment of HCV infection.
- a pharmaceutical agent comprising the compound of formula (I) defined above.
- the pharmaceutical agent may be an antiviral agent.
- a viral polymerase inhibitor in particular a HCV polymerase inhibitor such as a NS5B polymerase inhibitor comprising the compound of formula (I) defined above.
- the compound of formula (I) as a viral polymerase inhibitor in particular a HCV polymerase inhibitor such as a NS5B polymerase inhibitor.
- the compound of formula (I) defined above for use as a viral polymerase inhibitor in particular a HCV polymerase inhibitor such as a NS5B polymerase inhibitor.
- the compound of formula (I) may be administered in the form of a pharmaceutical composition together with a pharmaceutically acceptable carrier.
- a pharmaceutical composition comprising the compound of formula (I) and a pharmaceutically acceptable carrier.
- the pharmaceutical composition additionally comprises a therapeutically effective amount of one or more antiviral agents such as at least one other anti-HCV agent.
- a method for the treatment of a Flaviviridae viral infection such as a HCV infection which comprises administering an effective amount of the compound of formula (I) or the pharmaceutical composition defined above to a subject in need thereof.
- a seventh aspect there is provided a method of inhibiting the RNA-dependent RNA polymerase activity of the enzyme NS5B, encoded by HCV, comprising exposing the enzyme NS5B to an effective amount of the compound of formula (I) defined above.
- a method of inhibiting HCV replication comprising exposing a cell infected with HCV to an effective amount of the compound of formula (I) defined above.
- the present invention relates to compounds of formula (I) which inhibit viral polymerases and are useful in the treatment of Flaviviridae viral infections, particularly, hepatitis C (HCV).
- HCV hepatitis C
- the present invention relates to compounds of formula (I), salts, N-oxides, racemates, enantiomers and isomers thereof as defined above.
- Z 2 is C-R 1 and any one of Zi , Z 3 and Z 4 is N and the remaining two are C-R a , preferably CH thereby forming a fused pyridinyl ring.
- Z 2 is C-R 1 and both Z 1 and Z 4 are N and Z 3 is C-R a , preferably CH, thereby forming a fused pyrazinyl ring.
- Z 2 is C-R 1 and both Z 1 and Z 3 are N and Z 4 is C-R a , preferably CH, thereby forming a fused pyrimidinyl ring.
- Z 2 is C-R 1 and Z 1 is C-R a , preferably CH, and Z 3 and Z 4 are N thereby forming a fused pyridazinyl ring.
- Z 2 is C-R 1 and Zy , Z 3 and Z 4 are each C-R a , preferably CH, thereby forming a fused phenyl ring.
- R 2 is an optionally substituted (CH 2 ) m aryl or optionally substituted (CH 2 ) m heteroaryl, preferably m is 0, aryl is optionally substituted phenyl more preferably unsubstituted or para-substituted phenyl and heteroaryl is 6- membered heteroaryl containing nitrogen, preferably selected from optionally substituted pyridinyl, pyrazinyl, pyrimidinyl and pyridazinyl more preferably optionally substituted pyridinyl, even more preferably unsubstituted pyridinyl, most preferably unsubstituted 2- pyridinyl.
- R 3 is an optionally substituted (CH 2 ) m C3- 8 cycloalkyl, an optionally substituted (CH 2 )mC 3 - 8 cycloalkenyl or an optionally substituted (CH 2 ) m aryl.
- m is 0 and R 3 is phenyl, cyclopentyl, cyclohexyl or cyclohexenyl, preferably cyclohexyl.
- R 2 is an optionally substituted aryl, preferably phenyl or an optionally substituted heteroaryl, preferably a 6-membered heteroaryl containing nitrogen, preferably selected from optionally substituted pyridinyl, pyrazinyl, pyrimidinyl and pyridazinyl more preferably pyridinyl even more preferably unsubstituted pyridinyl and R 3 is an optionally substituted C 3 - 8 cycloalkyl or optionally substituted C 3 .
- scycloalkenyl preferably optionally substituted C 3 . 8 cycloalkyl, more preferably cyclohexyl.
- R 1 is (CH 2 ) m C(0)R 4
- R 4 is (CH 2 ) m NR 5 R 6
- R 5 is H
- R 6 is selected from H, optionally substituted C 1-3 alkyl, R 9 and [C(R 7 )(R 8 )] V (R 9 ) wherein v is 1 .
- m in each instance is 0 and R 6 is R 9 or [C(R 7 )(R 8 )] V (R 9 ), most preferably
- R 7 and R 8 are independently selected from H, optionally substituted C ⁇ alkyl, preferably C 1 . 3 alkyl such as methyl, ethyl, propyl and / ' so-propyl, most preferably methyl; optionally substituted (CH 2 ) m C 3 .
- R 7 and R 8 are independently selected from H, optionally substituted methyl, an optionally substituted side chain of an amino acid, or R 7 and R 8 together with the carbon atom to which they are attached form an optionally substituted C 3 . 6 cycloalkyl, or R 7 and R 8 together with the carbon atom to which they are attached form an optionally substituted 4-6-membered heterocyclyl containing oxygen and/or nitrogen.
- R 9 is selected from R 10 , (CH 2 ) m C(0)-R 10 , (CH 2 ) m C(0)NR 5 R 1 °, (CH 2 ) m S0 2 -R 10 and (CH 2 ) m S0 2 NR 5 R 10 where m is 0 or 1 , preferably 0.
- R 6 is [C(R 7 )(R 8 )] V (R 9 )
- R 9 is (CH 2 ) m C(0)-R 10 or
- R is H, OH, -A-(Q) n or 0-A-(Q) n where A is optionally substituted d_ 3 alkyl and n is 0.
- R 10 is -A-(Q) n where n is 0, 1 or 2, preferably 0 or 1 and A is selected from optionally substituted (CH 2 )mC 3 - 8 cycloalkyl, optionally substituted (CH 2 ) m aryl, optionally substituted (CH 2 ) m heterocyclyl and optionally substituted (CH 2 ) m heteroaryl, preferably m is 0 or 1 , most preferably 0.
- R 10 is optionally substituted aryl preferably phenyl, optionally substituted CH 2 aryl preferably benzyl, optionally substituted heterocyclyl preferably 5- or 6- membered heterocyclyl or optionally substituted heteroaryl, preferably 5-, 6- or 9-membered heteroaryl.
- Optionally substituted phenyl and optionally substituted 5-, 6- or 9-membered heteroaryls are particularly preferred.
- Particularly preferred 5-membered heteroaryls contain nitrogen and/or sulphur and include optionally substituted thiazolyl.
- Particularly preferred 6-membered heteroaryls contain nitrogen and include optionally substituted pyridinyl, optionally substituted pyridazinyl, optionally substituted pyrimidinyl and optionally substituted pyrazinyl.
- Particularly preferred 9- membered heteroaryls contain nitrogen and/or oxygen and/or sulphur and include optionally substituted benzofuranyl, optionally substituted benzoxazolyl, optionally substituted benzothiazolyl, optionally substituted benzimidazolyl including N-methyl derivatives thereof, optionally substituted indolyl including N-methyl derivatives thereof and optionally substituted pyrazolepyrimidinyl including N-methyl derivatives thereof.
- Q is selected from optionally substituted d_
- alkyl optionally substituted C 2 . 3 alkenyl, halo, halod. 3 alkyl, CHF 2 , CF 3 , CN, OH, NH 2 , NH(d_ 3 alkyl), N(d_ 3 alkyl) 2 , NHC(0)d_ 3 alkyl, NHS0 2 C(0)NH 2 , C(0)NH(Ci_ 3 alkyl),
- Suitable optional substituents in the case of optionally substituted d_ 3 alkyl, optionally substituted C 2 . 3 alkenyl, optionally substituted phenyl, optionally substituted heterocyclyl and optionally substituted heteroaryl include d_ 3 alkyl (such as methyl, ethyl, propyl), halo (particularly CI), CN, amino (NH 2 ), alkylamino (i.e. NHCi. 3 alkyl such as NHCH 3 ), dialkylamino (i.e.
- N(Ci_ 3 alkyl) 2 such as N(CH 3 ) 2
- C0 2 H esters
- esters such as C0 2 CH 3 , C0 2 CH 2 CH 3
- amides such as CONH 2 , CONHCi_ 3 alkyl e.g.
- W is CH or N
- Yi is independently selected from optionally substituted d. 6 alkyl, optionally substituted C 2 . 6 alkenyl, halo, haloCi_ 6 alkyl, CHF 2 , OCHF 2 , CF 3 , OCF 3 , CN, OH, optionally substituted d. 6 alkoxy including alkoxyaryl such as benzyloxy, N0 2 , NH 2 , NH(d- 6 alkyl), N(Ci-6alkyl) 2 , NHC(0)d_ 6 alkyl, NHS0 2 C(0)NH 2 , C(0)NH(Ci-6alkyl), C(0)N(d.
- n is independently 0, 1 , 2, 3, 4 or 5, preferably 0, 1 , 2 or 3, more preferably 0, 1 or 2, most preferably 0 or 1.
- Y 1 is selected from optionally substituted d_ 6 alkyl, halo, OH, optionally substituted d. 6 alkoxy and alkoxyaryl.
- Y 1 is selected from F, CI, Br, I, CH 3 , CHF 2 , CF 3 , CH 2 CH 3 , CH 2 CH 2 CH 3 , OH, OCH 3 , OCH 2 -phenyl (benzyloxy), OCHF 2 and OCF 3 , even more preferably F, OH, OCH 3 , OCHF 2 and OCH 2 phenyl.
- Y 2 is independently selected from optionally substituted d. 6 alkyl, optionally substituted C 2 . 6 alkenyl, halo, haloCi. 6 alkyl, CHF 2 , OCHF 2 , CF 3 , OCF 3 , CN, OH, optionally substituted d_ 6 alkoxy, N0 2 , NH 2 , NH(d_ 6 alkyl), N(d_ 6 alkyl) 2 , NHC(0)d_ 6 alkyl, NHS0 2 C(0)NH 2 , C(0)NH(d. 6 alkyl), C(0)N(d_ 6 alkyl) 2 , C(0)NHS0 2 (d_ 6 alkyl), C(0)N(d.
- R 2 is an unsubstituted 6-membered heteroaryl containing nitrogen, preferably pyridinyl, more preferably 2-pyridinyl or phenyl optionally substituted with (Yi) n wherein and n are as defined for formula (la).
- R 3 is cyclohexyl optionally substituted with (Y 2 ) n wherein Y 2 and n are as defined for formula (la-i), preferably R 3 is unsubstituted cyclohexyl.
- R 2 is an unsubstituted 6-membered heteroaryl containing nitrogen, preferably pyridinyl, more preferably 2-pyridinyl or phenyl optionally substituted with (Yi) n wherein and n are as defined for formula (la).
- R 3 is cyclohexyl optionally substituted with (Y 2 ) n wherein Y 2 and n are as defined for formula (la-i), preferably R 3 is unsubstituted cyclohexyl.
- R 2 is an unsubstituted 6-membered heteroaryl containing nitrogen, preferably pyridinyl, more preferably 2-pyridinyl or phenyl optionally substituted with (Yi) n wherein Y 1 and n are as defined for formula (la).
- R 3 is cyclohexyl optionally substituted with (Y 2 ) n wherein Y 2 and n are as defined for formula (la-i), preferably R 3 is unsubstituted cyclohexyl.
- Wi, W 2 , W 3 , W 4 and W 5 are each independently selected from CH and N or together with an adjacent ring member join to form a fused 5-membered heterocyclic moiety.
- W 2 , W 3 , W 4 and W 5 are each CH thereby forming an optionally substituted phenyl.
- any one of W 2 , W 3 , W 4 and W 5 is N and the remaining are CH thereby forming an optionally substituted pyridinyl.
- any two of W, , W 2 , W 3 , W 4 and W 5 are N and the remaining are CH thereby forming an optionally substituted pyrazinyl, pyrimidinyl or pyridazinyl.
- W 2 , W 3 , W 4 and W 5 are each CH wherein any two adjacent ring members join to form a fused 5-membered heterocyclic moiety.
- the fused 5-membered heterocyclic moiety is selected from furan to form an optionally substituted benzofuranyl, oxazole to form an optionally substituted benzoxazolyl, pyrrole or N-methyl pyrrole to form an optionally substituted indolyl or N-methyl indolyl and imidazole or N-methyl imidazole to form an optionally substituted benzimidazolyl or N-methyl benzimidazolyl.
- any two of W 2 , W 3 , W 4 and W 5 are N and the remaining are CH wherein any two adjacent ring members join to form a fused 5-membered heterocyclic moiety.
- the fused 5-membered heterocyclic moiety is pyrazole thereby forming an optionally substituted pyrazolopyrimidinyl.
- R 2 is an unsubstituted 6-membered heteroaryl containing nitrogen, preferably pyridinyl, more preferably 2-pyridinyl or phenyl optionally substituted with (Yi) n wherein and n are as defined for formula (la).
- R 3 is cyclohexyl optionally substituted with (Y 2 ) n where Y 2 and n are as defined for formula (la-i).
- R 7 and R 8 are each independently H, optionally substituted C ⁇ alkyl (particularly optionally substituted methyl such as CH 3 , CH 2 OH, CH 2 amino including CH 2 NHCH 3 and CH 2 N(CH 3 ) 2 and optionally substituted CH 2 heteroaryl (particularly 5-membered heteroaryls such as optionally substituted CH 2 thiazolyl and CH 2 imidazolyl) or the optionally substituted side chain of an L- or D- amino acid (such as Glycine (-H), Alanine (-CH 3 ), Valine (-CH(CH 3 ) 2 ), Serine (-CH 2 OH), Tryptophan (-CH 2 -(3-1 H-indolyl)), Histidine (-CH 2 -(5-1 H-imidazolyl)), particularly Alanine (methyl) wherein optional substituents include but are not limited to OH (such as Tryptophan-OH), benzyl (such as N- benzyl Histidine) and benzy
- R 7 and R 8 together with the carbon atom to which they are attached form an optionally substituted C 3 . 8 cycloalkyl, preferably C 3 . 6 cycloalkyl such as cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl, most preferably cyclobutyl.
- R 7 and R 8 together with the carbon atom to which they are attached form an optionally substituted C 3 . 8 heterocyclyl, preferably a 4-6-membered heterocyclyl such as azetidinyl, pyrrolidinyl, piperidinyl, tetrahydrofuranyl and tetrahydropyranyl.
- Suitable optional substituents for the heterocycles containing nitrogen include methyl, such as N-methyl.
- R 7 or R 8 and R 5 together with the adjacent atoms to which they are attached form an optionally substituted 5-6-membered-heterocyclyl containing nitrogen such as pyrrolidine and piperidine.
- Q is selected from halo (CI, Br, F, I preferably F and I , most preferably F), CF 3 , CN, C0 2 R 5 , CON(R 5 ) 2 , CON(R 5 )S0 2 R 5 , N(R 5 ) 2 , optionally substituted aryl such as phenyl, optionally substituted 5-6-membered heteroaryl such as pyridyl, pyridinone, pyrimidinyl, pyrazolyl, thiazolyl and tetrazolyl, optionally substituted 9- membered heteroaryl such as benzodioxalane, optionally substituted C 6 alkyl (preferably Ci.
- R 5 in each instance is independently H or d. 3 alkyl (preferably methyl or ethyl, more preferably methyl).
- Suitable optional substituents include but are not limited to halo, N(R 5 ) 2 , alkylhalo such as CF 3 , CN, C0 2 R 5 , CON(R 5 ) 2 , CON(R 5 )S0 2 R 5 and Ci_ 3 alkyl preferably methyl. More particularly, suitable optional substituents for aryl and heteroaryl include halo, N(R 5 ) 2 ,
- C0 2 R 5 and C ⁇ alkyl preferably methyl such as N-methyl and suitable optional substituents for Ci_ 6 alkyl and C 2 .
- 6 alkenyl include halo, alkyl halo such as CF 3 , CN, C0 2 R 5 , CON(R 5 ) 2 , and CON(R 5 )S0 2 R 5 .
- C h alky refers to optionally substituted straight chain or branched chain hydrocarbon groups having from 1 to 6 carbon atoms. Examples include methyl, ethyl, propyl, /sopropyl, butyl, / ' sobutyl, sec-butyl, ferf-butyl, pentyl, neopentyl and hexyl. "C ⁇ alkyl” including methyl, ethyl, propyl and /sopropyl is preferred with methyl being particularly preferred.
- C 2 . 6 alkenyl refers to optionally substituted straight chain or branched chain hydrocarbon groups having at least one double bond of either E or Z stereochemistry where applicable and 2 to 6 carbon atoms. Examples include vinyl, 1 -propenyl, 1- and 2- butenyl and 2-methyl-2-propenyl. "C 2 . 3 alkenyl” including ethenyl and propenyl is preferred with ethenyl being particularly preferred.
- C 3 . 8 cycloalkyl refers to non-aromatic cyclic groups having from 3 to 8 carbon atoms, including cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
- C ⁇ cycloalkyl such as cyclobutyl, cyclopentyl and cyclohexyl are preferred.
- C 3 . 8 cycloalkenyl refers to an unsaturated C 3 . 8 cycloalkyl having at least one double bond such as cyclohexenyl.
- C ⁇ alkoxy refers to an alkyl group as defined above covalently bound via an O linkage containing 1 to 6 carbon atoms, such as methoxy, ethoxy, propoxy, isoproxy, butoxy, tert-butoxy and pentoxy. "C ⁇ alkoxy” including methoxy, ethoxy and propoxy is preferred with methoxy being particularly preferred.
- aryl refers to a carbocyclic (non-heterocyclic) aromatic ring or mono-, bi- or tri-cyclic ring system.
- the aromatic ring or ring system is generally composed of 6 to 10 carbon atoms.
- aryl groups include but are not limited to phenyl, biphenyl, naphthyl and tetrahydronaphthyl. 6-membered aryls such as phenyl are preferred.
- alkylaryl refers to C ⁇ alkylaryl such as benzyl.
- alkoxyaryl refers to
- 6alkyloxyaryl such as benzyloxy.
- heterocyclyl refers to a monovalent moiety obtained by removing a hydrogen atom from a ring atom of a heterocyclic compound, which moiety has from 3 to 7 ring atoms (unless otherwise specified), of which from 1 to 4 are ring heteroatoms.
- each ring has from 3 to 7 ring atoms, of which from 1 to 4 are ring heteroatoms.
- the prefixes denote the number of ring atoms, or range of number of ring atoms, whether carbon atoms or heteroatoms.
- C 5 _ 6 heterocyclyl refers to a heterocyclyl group having 5 or 6 ring atoms, that is, a 5- or 6- membered heterocyclyl group.
- groups of heterocyclyl groups include C 3 . 7 heterocyclyl, C 5 . 7 heterocyclyl, and C 5 -6 heterocyclyl.
- Examples of monocyclic heterocyclyl groups include, but are not limited to, those containing one nitrogen atom such as aziridine (3-membered ring), azetidine (4-membered ring), pyrrolidine (tetrahydropyrrole), pyrroline (e.g., 3-pyrroline, 2,5-dihydropyrrole), 2H- pyrrole or 3H-pyrrole (isopyrrole, isoazole) (5-membered rings) , piperidine, dihydropyridine, tetrahydropyridine (6-membered rings), azepine (7-membered ring); those containing two nitrogen atoms such as imidazoline, pyrazolidine (diazolidine), imidazoline, pyrazoline (dihydropyrazole) (5-membered rings), piperazine (6-membered ring); those containing one oxygen atom such as oxirane (3-membered ring), oxetan
- tetrahydroisoxazole dihydroisoxazole (5-membered rings), morpholine, tetrahydrooxazine, dihydrooxazine, oxazine (6-membered rings); those containing one nitrogen and one sulfur atom such as thiazoline, thiazolidine (5-membered rings), thiomorpholine (6-membered ring); those containing two nitrogen and one oxygen atom such as oxadiazine (6-membered ring); those containing one oxygen and one sulfur such as: oxathiole (5-membered ring) and oxathiane (thioxane) (6-membered ring); and those containing one nitrogen, one oxygen and one sulfur atom such as oxathiazine (6-membered ring).
- heteroaryl is used herein to denote a heterocyclic group having aromatic character and embraces aromatic monocyclic ring systems and polycyclic (e.g. bicyclic) ring systems containing one or more aromatic rings.
- the term covers polycyclic ring systems in which all of the fused rings are aromatic as well as ring systems where one or more rings are non-aromatic, provided that at least one ring is aromatic.
- the group may be attached to another moiety by the aromatic ring, or by a non-aromatic ring.
- heteroaryl groups are monocyclic and bicyclic groups containing from five to ten ring members.
- the heteroaryl group can be, for example, a five membered or six membered monocyclic ring or a bicyclic structure formed from fused five and six membered rings or two fused six membered rings or two fused five membered rings.
- Each ring may contain up to about four heteroatoms typically selected from nitrogen, sulphur and oxygen.
- the heteroaryl ring will contain up to 4 heteroatoms, more typically up to 3 heteroatoms, more usually up to 2, for example a single heteroatom.
- the heteroaryl ring contains at least one ring nitrogen atom.
- the nitrogen atoms in the heteroaryl rings can be basic, as in the case of an imidazole or pyridine, or essentially non-basic as in the case of an indole or pyrrole nitrogen.
- the number of basic nitrogen atoms present in the heteroaryl group, including any amino group substituents of the ring, will be less than five.
- Examples of 5 membered heteroaryl groups include but are not limited to pyrrole, furan, imidazole, furazan, oxazole, oxadiazole, oxatriazole, isoxazole, thiazole, isothiazole, pyrazole, triazole and tetrazole groups.
- 6 membered heteroaryl groups include but are not limited to pyridine, pyrazine, pyridazine, pyrimidine, triazine, pyran, oxazine, dioxine, thiazine and thiadiazine.
- a bicyclic heteroaryl group may be, for example, a group selected from: a) a benzene ring fused to a 5- or 6-membered ring containing 1 , 2 or 3 ring heteroatoms; b) a pyridine ring fused to a 5- or 6-membered ring containing 1 , 2 or 3 ring heteroatoms; c) a pyrimidine ring fused to a 5- or 6-membered ring containing 1 or 2 ring heteroatoms; d) a pyrrole ring fused to a 5- or 6-membered ring containing 1 , 2 or 3 ring heteroatoms; e) a pyrazole ring fused to a 5- or 6-membered ring containing 1 or 2 ring heteroatoms; f) an imidazole ring fused to a 5- or 6-membered ring containing 1 or 2 ring heteroatoms; g) an oxazole ring fused to a 5-
- bicyclic heteroaryl groups containing a five membered ring fused to another five membered ring include but are not limited to imidazothiazole (e.g. imidazo[2, 1-b]thiazole) and imidazoimidazole (e.g. imidazo[1 ,2-a]imidazole).
- imidazothiazole e.g. imidazo[2, 1-b]thiazole
- imidazoimidazole e.g. imidazo[1 ,2-a]imidazole
- bicyclic heteroaryl groups containing a six membered ring fused to a five membered ring include but are not limited to benzofuran, benzothiophene, benzimidazole, benzoxazole, isobenzoxazole, benzisoxazole, benzothiazole,
- benzisothiazole isobenzofuran, indole, isoindole, indolizine, indoiine, isoindoline, purine (e.g., adenine, guanine), indazole, pyrazolopyrimidine (e.g. pyrazolo[1 ,5-a]pyrimidine), benzodioxole and pyrazolopyridine (e.g. pyrazolo[1 ,5-a]pyridine) groups.
- a further example of a six membered ring fused to a five membered ring is a pyrrolopyridine group such as a pyrrolo[2,3-b]pyridine group.
- bicyclic heteroaryl groups containing two fused six membered rings include but are not limited to quinoline, isoquinoline, chroman, thiochroman, chromene, isochromene, isochroman, benzodioxan, quinolizine, benzoxazine, benzodiazine, pyridopyridine, quinoxaline, quinazoline, cinnoline, phthalazine, naphthyridine and pteridine groups.
- heteroaryl groups containing an aromatic ring and a non-aromatic ring include tetrahydronaphthalene, tetrahydroisoquinoline, tetrahydroquinoline,
- side chain of an amino acid refers to any side chain that may be present in natural (L-) or unnatural (D-) amino acids.
- amino acid side chain moieties derived from unnatural amino acids are -(CH 2 )2-C(0)-0-C(CH 3 )3 (glutamic acid t- butyl ester), -(CH 2 ) 4 -NH-C(0)-0- C(CH 3 ) 3 (Ne-(tert-butoxycarbonyl)-lysine), -(CH 2 ) 3 -NH-C(0)NH 2 (citrulline), -CH 2 -CH 2 OH (homoserine) and -(CH 2 ) 2 -CH 2 NH 2 (ornithine).
- Examples can also include alkyl, alkenyl, alkynyl, aryl, saturated and unsaturated heterocycles (functionalized and unfunctionalized).
- the term 'amino-acid side chain moiety' can also include a number of unnatural amide and sulfonamide, aryl and heteroaryl side chains.
- halo refers to fluoro, chloro, bromo or iodo.
- R 7 and R 8 together with the atom or adjacent atoms to which they are attached form an optionally substituted C 3 . 8 cycloalkyl or an optionally substituted C 3 . 8 heterocyclyl refers to either R 7 and R 8 being on the same atom and joining together to form optionally substituted C 3 . 8 cycloalkyl or an optionally substituted C 3 . 8 heterocyclyl or R 7 and R 8 being on separate adjacent atoms and joining together to form optionally substituted C 3 . 8 cycloalkyl or an optionally substituted C 3 . 8 heterocyclyl.
- R 7 and R 8 are on separate adjacent atoms, it will be appreciated that these atoms may be the same (e.g. two carbon atoms) or different (e.g. a carbon atom and a nitrogen atom).
- optionally substituted refers to a group that may or may not be further substituted with one or more groups selected from Ci_ 6 alkyl, C 3 . 8 cycloalkyl, C 2 . 6 alkenyl, C 2 . 6 alkynyl, aryl, CF 3 , OCF 3 , CHF 2 , OCHF 2 , heterocyclyl, heteroaryl, halo, haloCi_ 6 alkyl, haloC 3 . 6 cycloalkyl, haloC 2 . 6 alkenyl, haloC 2 .
- aryl such as phenyl
- haloalkyl such as CHF 2 and CF 3
- heterocyclyl such as 5- and 6-membered heterocyclyl
- heteroaryl such
- alkylamino such as NHCi_ 3 alkyl
- d-sdialkylamino such as N(Ci- 3 alkyl) 2 , NH(OH)
- acylamino such as C(0)NH 2
- acylC ⁇ aalkylamino such as C(0)NHCi_ 3 alkyl and C(0)N(Ci- 3 alkyl) 2 wherein each optional substituent may be further optionally substituted.
- the compounds of the invention may also be prepared as salts which are pharmaceutically acceptable, but it will be appreciated that non-pharmaceutically acceptable salts also fall within the scope of the present invention, since these are useful as
- pharmaceutically acceptable salts include salts of pharmaceutically acceptable cations such as sodium, potassium, lithium, calcium, magnesium, ammonium and alkylammonium; acid addition salts of pharmaceutically acceptable inorganic acids such as hydrochloric, orthophosphoric, sulfuric, phosphoric, nitric, carbonic, boric, sulfamic and hydrobromic acids; or salts of pharmaceutically acceptable organic acids such as acetic, propionic, butyric, tartaric, maleic, hydroxymaleic, fumaric, citric, lactic, mucic, gluconic, benzoic, succinic, oxalic, phenylacetic, methanesulfonic, trihalomethanesulfonic, toluenesulfonic,
- Salts of amine groups may also comprise quaternary ammonium salts in which the amino nitrogen atom carries a suitable organic group such as an alkyl, alkenyl, alkynyl or aralkyl moiety.
- the salts may be formed by conventional means, such as by reacting the free base form of the compound with one or more equivalents of the appropriate acid.
- a reference to a pharmaceutically acceptable salt includes the solvent addition forms or crystal forms thereof, particularly solvates or polymorphs.
- Solvates contain either stoichiometric or non-stoichiometric amounts of a solvent, and may be formed during the process of crystallization with pharmaceutically acceptable solvents such as water, alcohols such as methanol, ethanol or isopropyl alcohol, DMSO, acetonitrile, dimethyl formamide (DMF) and the like with the solvate forming part of the crystal lattice by either non-covalent binding or by occupying a hole in the crystal lattice. Hydrates are formed when the solvent is water, alcoholates are formed when the solvent is alcohol.
- Solvates of the compounds of the present invention can be conveniently prepared or formed during the processes described herein.
- the compounds of the present invention can exist in unsolvated as well as solvated forms.
- the solvated forms are considered equivalent to the unsolvated forms for the purposes of the compounds and methods provided herein.
- the compounds of the present invention can exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like.
- the solvated forms of the compounds of the present invention are also considered to be disclosed herein.
- compounds of formula (I) may possess a chiral centre and may therefore exist as an isomer such as a racemate or an R- or S- enantiomer.
- the compounds may therefore be used as a purified enantiomer or diastereomer, or as a mixture of any ratio thereof.
- This invention also encompasses prodrugs of the compounds of formula (I).
- pro-drug is used herein in its broadest sense to include those compounds which are converted in vivo to the compound of formula (I). Use of the prodrug strategy optimises the delivery of the drug to its site of action.
- compounds of formula (I) having free amino, amido, hydroxyl, or carboxylic acid groups can be converted into prodrugs.
- Prodrugs include compounds wherein carbonates, carbamates, amide and alkyl esters which are covalently bonded to the above substituents of compounds of the present invention through a carbonyl carbon prodrug sidechain.
- Prodrugs may also include N-oxides of ring nitrogen atoms in formula (I).
- RNA dependent RNA polymerase of HCV RNA dependent RNA polymerase of HCV
- HCV NS5B polymerase which is the viral RNA-dependent RNA polymerase (RdRp) that is responsible for viral replications.
- RdRp viral RNA-dependent RNA polymerase
- HCV NS5B protein is released from a polyprotein and is involved in the synthesis of double-stranded RNA from a single-stranded viral RNA genome. It is believed that the replication and/or reproduction of HCV virus may be inhibited or prevented through the inhibition of NS5B polymerase and suppress or prevent the formation of the double- stranded HCV RNA.
- the compounds of formula (I) act by specific inhibition of NS5B polymerase, the compounds may be tested for the lack of inhibitory activity in an assay measuring the activity of an RNA-dependent RNA polymerase other than HCV polymerase or in a DNA dependent RNA polymerase assay.
- the invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I) and a pharmaceutically acceptable carrier.
- the pharmaceutical composition may further comprise or be administered in combination with one or more other antiviral agents such as ribavirin, an antiviral nucleoside, polymerase inhibitor, protease inhibitor and/or inhibitor of viral entry, assembly or egress.
- the composition may also additionally comprise at least one immunomodulatory agent for example an interferon or interferon derivative and/or an inhibitor of inosine-5'- monophosphate dehydrogenase (IMPDH).
- IMPDH inosine-5'- monophosphate dehydrogenase
- composition is intended to include the formulation of an active ingredient with conventional carriers and excipients, and also with encapsulating materials as the carrier, to give a capsule in which the active ingredient (with or without other carriers) is surrounded by the encapsulation carrier.
- Any carrier must be “pharmaceutically acceptable” meaning that it is compatible with the other ingredients of the composition and is not deleterious to a subject.
- compositions of the present invention may contain other therapeutic agents as described above, and may be formulated, for example, by employing conventional solid or liquid vehicles or diluents, as well as pharmaceutical additives of a type appropriate to the mode of desired administration (for example, excipients, binders, preservatives, stabilizers, flavours, etc.) according to techniques such as those well known in the art of pharmaceutical formulation (See, for example, Remington: The Science and Practice of Pharmacy, 21st Ed. , 2005, Lippincott Williams & Wilkins).
- the pharmaceutical composition includes those suitable for oral, rectal, nasal, topical (including buccal and sub-lingual), vaginal or parenteral (including intramuscular, sub-cutaneous and intravenous) administration or in a form suitable for administration by inhalation or insufflation.
- the compounds of the invention may thus be placed into the form of pharmaceutical compositions and unit dosages thereof, and in such form may be employed as solids, such as tablets or filled capsules, or liquids such as solutions, suspensions, emulsions, elixirs, or capsules filled with the same, all for oral use, in the form of suppositories for rectal administration ; or in the form of sterile injectable solutions for parenteral (including subcutaneous) use.
- compositions and unit dosage forms thereof may comprise conventional ingredients in conventional proportions, with or without additional active compounds or principles, and such unit dosage forms may contain any suitable effective amount of the active ingredient commensurate with the intended daily dosage range to be employed.
- pharmaceutically acceptable carriers can be either solid or liquid. Solid form preparations include powders, tablets, pills, capsules, cachets, suppositories, and dispensable granules.
- a solid carrier can be one or more substances which may also act as diluents, flavouring agents, solubilisers, lubricants, suspending agents, binders, preservatives, tablet disintegrating agents, or an encapsulating material.
- Suitable carriers are magnesium carbonate, magnesium stearate, talc, sugar, lactose, pectin, dextrin, starch, gelatin, tragacanth, methylcellulose, sodium
- carboxymethylcellulose a low melting wax, cocoa butter, and the like.
- preparation is intended to include the formulation of the active compound with
- encapsulating material as carrier providing a capsule in which the active component, with or without carriers, is surrounded by a carrier, which is thus in association with it.
- a carrier which is thus in association with it.
- cachets and lozenges are included. Tablets, powders, capsules, pills, cachets, and lozenges can be used as solid forms suitable for oral administration.
- Liquid form preparations include solutions, suspensions, and emulsions, for example, water or water-propylene glycol solutions.
- parenteral injection liquid preparations can be formulated as solutions in aqueous polyethylene glycol solution.
- Sterile liquid form compositions include sterile solutions, suspensions, emulsions, syrups and elixirs.
- the active ingredient can be dissolved or suspended in a
- pharmaceutically acceptable carrier such as sterile water, sterile organic solvent or a mixture of both.
- compositions according to the present invention may thus be formulated for parenteral administration (e. g. by injection, for example bolus injection or continuous infusion) and may be presented in unit dose form in ampoules, pre-filled syringes, small volume infusion or in multi-dose containers with an added preservative.
- the compositions may take such forms as suspensions, solutions, or emulsions in oily or aqueous vehicles, and may contain formulation agents such as suspending, stabilising and/or dispersing agents.
- the active ingredient may be in powder form, obtained by aseptic isolation of sterile solid or by lyophilisation from solution, for constitution with a suitable vehicle, e.g. sterile, pyrogen-free water, before use.
- compositions suitable for injectable use include sterile injectable solutions or dispersions, and sterile powders for the extemporaneous preparation of sterile injectable solutions. They should be stable under the conditions of manufacture and storage and may be preserved against oxidation and the contaminating action of microorganisms such as bacteria or fungi.
- the solvent or dispersion medium for the injectable solution or dispersion may contain any of the conventional solvent or carrier systems for the compounds, and may contain, for example, water, ethanol, polyol (for example, glycerol, propylene glycol and liquid polyethylene glycol, and the like), suitable mixtures thereof, and vegetable oils.
- polyol for example, glycerol, propylene glycol and liquid polyethylene glycol, and the like
- compositions suitable for injectable use may be delivered by any appropriate route including intravenous, intramuscular, intracerebral, intrathecal, epidural injection or infusion.
- Sterile injectable solutions are prepared by incorporating the active compounds in the required amount in the appropriate solvent with various other ingredients such as these enumerated above, as required, followed by filtered sterilization.
- dispersions are prepared by incorporating the various sterilised active ingredient into a sterile vehicle which contains the basic dispersion medium and the required other ingredients from those enumerated above.
- preferred methods of preparation are vacuum drying or freeze-drying of a previously sterile-filtered solution of the active ingredient plus any additional desired ingredients.
- the active ingredients When the active ingredients are suitably protected they may be orally administered, for example, with an inert diluent or with an assimilable edible carrier, or it may be enclosed in hard or soft shell gelatin capsule, or it may be compressed into tablets, or it may be incorporated directly with the food of the diet.
- the active compound may be incorporated with excipients and used in the form of ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups, wafers, and the like.
- the amount of active compound in therapeutically useful compositions should be sufficient that a suitable dosage will be obtained.
- the tablets, troches, pills, capsules and the like may also contain the components as listed hereafter: a binder such as gum, acacia, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid and the like; a lubricant such as magnesium stearate; and a sweetening agent such a sucrose, lactose or saccharin may be added or a flavouring agent such as peppermint, oil of wintergreen, or cherry flavouring.
- a binder such as gum, acacia, corn starch or gelatin
- excipients such as dicalcium phosphate
- a disintegrating agent such as corn starch, potato starch, alginic acid and the like
- a lubricant such as magnesium stearate
- a sweetening agent such as sucrose, lactose or saccharin may be added or a flavouring agent such as peppermint, oil of winter
- any material used in preparing any dosage unit form should be pharmaceutically pure and substantially non-toxic in the amounts employed.
- the active compound (s) may be incorporated into sustained-release preparations and formulations, including those that allow specific delivery of the active peptide to specific regions of the gut.
- Aqueous solutions suitable for oral use can be prepared by dissolving the active component in water and adding suitable colorants, flavours, stabilising and thickening agents, as desired.
- Aqueous suspensions suitable for oral use can be made by dispersing the finely divided active component in water with viscous material, such as natural or synthetic gums, resins, methylcellulose, sodium carboxymethylcellulose, or other well known suspending agents.
- Pharmaceutically acceptable carriers and/or diluents include any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents and the like.
- solid form preparations that are intended to be converted, shortly before use, to liquid form preparations for oral administration.
- liquid forms include solutions, suspensions, and emulsions.
- These preparations may contain, in addition to the active component, colorants, flavours, stabilisers, buffers, artificial and natural sweeteners, dispersants, thickeners, solubilising agents, and the like.
- the compounds according to the invention may be formulated as ointments, creams or lotions, or as a transdermal patch.
- Ointments and creams may, for example, be formulated with an aqueous or oily base with the addition of suitable thickening and/or gelling agents.
- Lotions may be formulated with an aqueous or oily base and will in general also contain one or more emulsifying agents, stabilising agents, dispersing agents, suspending agents, thickening agents, or colouring agents.
- Formulations suitable for topical administration in the mouth include lozenges comprising active agent in a flavoured base, usually sucrose and acacia or tragacanth; pastilles comprising the active ingredient in an inert base such as gelatin and glycerin or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier.
- Solutions or suspensions are applied directly to the nasal cavity by conventional means, for example with a dropper, pipette or spray.
- the formulations may be provided in single or multidose form. In the latter case of a dropper or pipette, this may be achieved by the patient administering an appropriate, predetermined volume of the solution or suspension.
- the compounds according to the invention may be encapsulated with cyclodextrins, or formulated with other agents expected to enhance delivery and retention in the nasal mucosa.
- Administration to the respiratory tract may also be achieved by means of an aerosol formulation in which the active ingredient is provided in a pressurised pack with a suitable propellant such as a chlorofluorocarbon (CFC) for example dichlorodifluoromethane, trichlorofluoromethane, or dichlorotetrafluoroethane, carbon dioxide, or other suitable gas.
- a suitable propellant such as a chlorofluorocarbon (CFC) for example dichlorodifluoromethane, trichlorofluoromethane, or dichlorotetrafluoroethane, carbon dioxide, or other suitable gas.
- CFC chlorofluorocarbon
- the aerosol may conveniently also contain a surfactant such as lecithin.
- a surfactant such as lecithin.
- the dose of drug may be controlled by provision of a metered valve.
- the active ingredients may be provided in the form of a dry powder, for example a powder mix of the compound in a suitable powder base such as lactose, starch, starch derivatives such as hydroxypropylmethyl cellulose and polyvinylpyrrolidone (PVP).
- a powder base such as lactose, starch, starch derivatives such as hydroxypropylmethyl cellulose and polyvinylpyrrolidone (PVP).
- PVP polyvinylpyrrolidone
- the powder carrier will form a gel in the nasal cavity.
- the powder composition may be presented in unit dose form for example in capsules or cartridges of, e. g. gelatin, or blister packs from which the powder may be administered by means of an inhaler.
- the compound In formulations intended for administration to the respiratory tract, including intranasal formulations, the compound will generally have a small particle size for example of the order of 5 to 10 microns or less. Such a particle size may be obtained by means known in the art, for example by micronisation.
- formulations adapted to give sustained release of the active ingredient may be employed.
- the pharmaceutical preparations are preferably in unit dosage forms.
- the preparation is subdivided into unit doses containing appropriate quantities of the active component.
- the unit dosage form can be a packaged preparation, the package containing discrete quantities of preparation, such as packeted tablets, capsules, and powders in vials or ampoules.
- the unit dosage form can be a capsule, tablet, cachet, or lozenge itself, or it can be the appropriate number of any of these in packaged form.
- Dosage unit form refers to physically discrete units suited as unitary dosages for the subjects to be treated; each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier.
- the specification for the dosage unit forms are dictated by and directly dependent on (a) the unique characteristics of the active material and the particular therapeutic effect to be achieved, and (b) the limitations inherent in the art of compounding such an active material for the treatment of a HCV viral infection in living subjects having a diseased condition in which bodily health is impaired.
- the invention also includes the compounds in the absence of carrier where the compounds are in unit dosage form.
- the compounds of formula (I) may be used in the treatment of a Flaviviridae viral infection such as a HCV infection.
- the term "treatment" means affecting a subject, tissue or cell to obtain a desired pharmacological and/or physiological effect and includes: (a) inhibiting the viral infection, i.e. arresting its development or further development; (b) relieving or ameliorating the effects of the viral infection, i.e. cause regression of the effects of the viral infection; (c) reducing the incidence or the viral infection or (d) preventing the infection from occurring in a subject, tissue or cell predisposed to the viral infection disease or at risk thereof, but has not yet been diagnosed with a protective pharmacological and/or physiological effect so that the viral infection does not develop or occur in the subject, tissue or cell.
- the prevention of hepatitis C means, for example, administration of a
- subject refers to any animal, in particular mammals such as humans having a disease or condition which requires treatment with the compound of formula (I).
- administering refers to providing the compound or pharmaceutical composition of the invention to a subject suffering from or at risk of the diseases or conditions to be treated or prevented.
- viral infection refers to the introduction of a virus into cells or tissues, e.g., hepatitis C virus (HCV). In general, the introduction of a virus is also associated with replication. Viral infection may be determined by measuring virus antibody titer in samples of a biological fluid, such as blood, using, e.g., enzyme immunoassay. Other suitable diagnostic methods include molecular based techniques, such as RT-PC , direct hybrid capture assay, nucleic acid sequence based amplification, and the like. A virus may infect an organ, e.g., liver, and cause disease, e.g. , hepatitis, cirrhosis, chronic liver disease and hepatocellular carcinoma.
- HCV hepatitis C virus
- Flaviviridae virus refers to a virus of the family Flaviviridae, which family includes the Hepacivirus Flavivirus and Pestivirus or hepatitis C-like virus genera. A representative species of the genus of hepatitis C-like viruses is hepatitis C virus. Dosages
- terapéuticaally effective amount refers to the amount of the compound of formula (I) that will elicit the biological or medical response of a subject, tissue or cell that is being sought by the researcher, veterinarian, medical doctor or other clinician.
- an appropriate dosage level will generally be about 0.01 to 500 mg per kg subject body weight per day which can be administered in single or multiple doses.
- the dosage level will be about 0.1 to about 250 mg/kg per day; more preferably about 0.5 to about 100 mg/kg per day.
- a suitable dosage level may be about 0.01 to 250 mg/kg per day, about 0.05 to 100 mg/kg per day, or about 0.1 to 50 mg/kg per day. Within this range the dosage may be 0.05 to 0.5, 0.5 to 5 or 5 to 50 mg/kg per day.
- the dosage may be selected, for example to any dose within any of these ranges, for therapeutic efficacy and/or symptomatic adjustment of the dosage to the subject to be treated
- X is (R 1 ) t C0 2 H or (R 1 ) t C0 2 Ci. 3 alkyl, t is 0 or 1 ; ⁇ , Z' 3 and Z' 4 are each
- R 1 , R 2 and R 3 are as defined in formula (I).
- a process for producing a compound of formula (Ib-ii) comprising the step of reacting a compound of formula (II) with an amino precursor of general formula NHR 5 R 6 under amide coupling conditions.
- Suitable amide coupling conditions will be understood by those skilled in the art and have been described in many references such as Advanced Organic Chemistry - 4 th Edition, March J., John Wiley & Sons Inc, New York,1992.
- Preparative HPLC was carried out using either a Gilson 322 pump with a Gilson 215 liquid handler and a HP1 100 PDA detector or an Agilent 1200 Series mass detected preparative LCMS using a Varian XRs C-18 100 x 21 .2 mm column. Unless otherwise specified, the HPLC systems employed Phenomenex C8(2) columns using either acetonitrile or acetonitrile containing 0.06% TFA in water or water containing 0.1 % TFA.
- DABCO 33-LV 33% solution by weight of 1 ,4-Diazabicyclo[2.2.2]octane in propylene glycol DIPEA: N,N-Diisopropylethylamine
- HATU 2-(7-Aza-1 H-benzotriazole-1 -yl)-1 , 1 ,3,3-tetramethyluronium hexafluorophosphate
- NaHMDS sodium hexamethyldisilazane (sodium bis(trimethylsilyl)amide)
- TLC thin-layer chromatography
- TMSCN trimethylsilyl cyanide
- Intermediate v was reacted with an appropriate aromatic or cycloalkenyl boronic acid or ester under standard palladium-catalysed cross-coupling conditions to give intermediate (vi).
- Basic hydrolysis of the nitrile of vi afforded the carboxylic acid (vii), and the alkene was reduced to the saturated roduct (viii) with Pd/C and ammonium formate.
- Step a To an ice-cooled, stirred suspension of 2-amino-4-bromobenzoic acid (i) (10 g, 46.3 mmol) in water was added concentrated HCI (aq) (88 mL) at such a rate so as to maintain the internal temperature at 5 °C. The reaction mixture was then heated at 80 °C for 30 min and cooled to 0 °C, whereupon a solution of sodium nitrite (3.83 g, 55.6 mmol) in water (10 mL) was added dropwise.
- concentrated HCI aq
- reaction mixture was allowed to stir for 1 h and then added dropwise to a stirred solution of sodium azide (2.95 g, 45.4 mmol) and sodium ethoxide (55.9 g, 681 .6 mmol) in water (88 mL) at RT.
- the resultant reaction mixture was stirred at room temperature for 3 h.
- Step b To the stirred solution of 2-azido-4-bromobenzoic acid (ii) (9.8 g, 40.5 mmol) in DMF (50 mL) at 0 °C was added HATU (18.5 g, 48.6 mmol). The reaction mixture was stirred for 15 min whereupon DIPEA (14.3 mL, 60.7 mmol) was added followed by aniline (4.52 g, 48.6 mmol). The reaction mixture was then allowed to warm to RT and the progress was monitored by TLC and LCMS analysis. After 4 h the reaction mixture was poured onto cold water (500 mL) and the resultant suspension then stirred for 15 min.
- Step c A mixture of 2-azido-4-bromo-/V-phenylbenzamide (iii) (4.7 g, 14.8 mmol) and phosphorus oxychloride (25 mL) was heated at 95 °C for 2 h and then cooled to RT. The reaction mixture was then concentrated in vacuo and the residue was then added dropwise to an ice-cold solution of saturated sodium carbonate (30 mL) such that the temperature of the resultant mixture did not rise above 5 °C.
- Step d To a stirred solution of 6-bromo-3-chloro-2-phenyl-2/-/-indazole (iv) in NMP (10 x 10 mL batches) at T was added copper (I) cyanide (10 x 1.45 g, 162.9 mmol per batch) and the reaction mixtures were then heated at 175 °C. After 1.5 h the combined reaction mixtures were cooled to RT and poured into aqueous saturated FeCI 3 (100 mL). The organics were extracted into ethyl acetate (3 x 75 mL) and the combined organic layers were then washed with water (3 x 50 mL), dried (Na 2 S0 4 ) and concentrated in vacuo.
- Step e Argon was bubbled through a solution of dioxane-water (50 mL) for 0.5 h whereupon 3-chloro-2-phenyl-2A7-indazole-6-carbonitrile (v) was added to the de-gassed medium. To this suspension was added cyclohexyl boronic acid pinacol ester (3.21 g, 15.4 mmol) and the reaction mixture was then stirred for 10 min. Triphenyl phosphine (1 .07 g, 4.1 mmol) and palladium (II) acetate (0.46 g, 2.1 mmol) were then added followed by addition of sodium carbonate (2.54 g, 20.5 mmol).
- reaction mixture was heated at reflux and monitored by LCMS analysis. After 1 h, the reaction mixture was concentrated in vacuo and the aqueous layer was extracted into ethyl acetate (4 x 100 mL). The combined organic layers were dried (Na 2 S0 4 ) and concentrated in vacuo.
- Step f To a stirred suspension of 3-(cyclohex-1-en-1 -yl)-2-phenyl-2/-/-indazole-6-carbonitrile (vi) (910 mg, 3.0 mmol) in ethanol:water (1 : 1 v/v, 40 mL) was added sodium hydroxide (1.8 g, 45.0 mmol) and the reaction mixture was then heated at reflux and monitored by TLC and LCMS analysis. After 4 h, the reaction mixture was cooled to RT and the volatiles were removed in vacuo. The residue was acidified with 2N aqueous HCI to pH 2 and the resulting precipitate was then separated by filtration.
- Step g To a stirred suspension of 3-(cyclohex-1 -en-1 -yl)-2-phenyl-2/-/-indazole-6-carboxylic acid (1 ) (750 mg, 2.4 mmol) in methanol (20 mL) was added 10% Pd/C under an atmosphere of nitrogen. Ammonium formate (1.48 g, 23.6 mmol) was then added and the reaction mixture was heated to reflux and monitored by LCMS analysis. Upon complete consumption of the starting materials, the reaction mixture was filtered through a pad of Celite® and the residue was washed with methanol (2 x 20 mL).
- the pyrazolopyrimidine (ii) was prepared by reaction of ethyl 2,4- dichloropyrimidine-5-carboxylate with an aromatic hydrazine.
- the chloride of ii was converted to the nitrile via reaction with a metal cyanide.
- the nitrile of iii was converted to a carboxylic ester via a Pinner reaction (reaction with an alcohol under acid catalysis).
- the alcohol of iv was converted to a triflate which was reacted with an appropriate cycloalkenyl boronic acid or ester under standard palladium-catalysed cross-coupling conditions to give intermediate vi.
- the carboxylic ester of vi was hydrolysed under basic conditions to give intermediate vii, and the alkene was reduced to the saturated product viii via palladium catal sed hydrogenation.
- Step a Ethyl 2,4-dichloropyrimidine-5-carboxylate (1.0 g, 4.5 mmol) in EtOH (8 mL) had phenylhydrazine (0.45 mL, 4.5 mmol) added and was stirred at 60 °C. After 1.5 h, the reaction was cooled to room temperature and 1 M HCI (8 mL) was added. The precipitate was filtered off, washed with a little EtOH/H 2 0 (1 : 1 ) and dried to give 6
- Step b A solution of 6-chloro-2-phenyl-2H-pyrazolo[3,4-d]pyrimidin-3-ol (0.37 g, 1 .5 mmol) in DMSO (2 mL) was added to a mixture of NaCN (0.15 g, 3.0 mmol) and DABCO 33-LV (0.59 mL, 1.8 mmol) in DMSO (1 mL) and H 2 0 (2 mL).
- Step c (3-hydroxy-2-phenyl-2A7-pyrazolo[3,4-c ]pyrimidin-6-yl)acetonitrile (78 mg, 0.33 mmol) in a microwave vial (10-20 mL) under Ar was suspended in saturated HCI in EtOH (4 mL). The vial was sealed and then heated at 80 °C behind a blast screen. After 16 h the reaction was cooled to room temperature and carefully vented with a needle before removing the cap. The reaction mixture was pipetted into saturated NaHC0 3 (8 mL). The EtOH was removed on the rotary evaporator and saturated NaHC0 3 was added to the mixture until a solution formed ( ⁇ 4 mL).
- Step d To ethyl 3-hydroxy-2-phenyl-2H-pyrazolo[3,4- ]pyrimidine-6-carboxylate (1 1 mg, 37 pmol) in dry THF (0.5 mL) under Ar was added triethylamine (10 ⁇ , 45 ⁇ ), followed by N-phenyltrifluoromethanesulphonimide (18 mg, 45 ⁇ ) and the resulting solution was stirred at room temperature for 16 h. The reaction was quenched with saturated NH 4 CI (1 mL) and extracted with EtOAc (2 x 2 mL). The combined organics were dried over MgS0 4 , filtered and concentrated.
- Step e A mixture of ethyl 2-phenyl-3- ⁇ [(trifluoromethyl)sulfonyl]oxy ⁇ -2/-/-pyrazolo[3,4- c ]pyrimidine-6-carboxylate (53 mg, 0.13 mmol) in a microwave vial in toluene (0.8 mL) and H 2 0 (50 ⁇ L) had Ar bubbled through for 5 min. Cyclohexeneboronic acid, pinacol ester (0.10 mL, 0.51 mmol) was added, followed by K 2 C0 3 (74 mg, 0.51 mmol) and Pd(PPh 3 ) 4 (18 mg, 13 ⁇ ).
- Step f A mixture of ethyl 3-(cyclohex-1 -en-1-yl)-2-phenyl-2H-pyrazolo[3,4-c ]pyrimidine-6- carboxylate (18 mg, 51 pmol) and LiOH hydrate (14 mg, 0.31 mmol) in dioxane (0.80 mL), iPrOH (0.80 mL), and H2O (0.64 mL) was stirred at room temperature for 30 min. The rection mixture was acidified with 1 M HCI and extracted with EtOAc (3 x 4 mL).
- Step g 3-(cyclohex-1-en-1 -yl)-2-phenyl-2A7-pyrazolo[3,4-c ]pyrimidine-6-carboxylic acid (16 mg, 49 pmol) in MeOH (2 mL) and EtOAc (0.50 mL) had 10% Pd/C (3.3 mg, 20% by mass) added.
- the reaction flask was evacuated and filled with H 2 (x3) then stirred at room temperature under H 2 for 1 h.
- the reaction mixture was filtered through a syringe filter (0.45 pm), washed through with MeOH and the filtrate was concentrated.
- the nitrile of iv was converted to a carboxylic ester via a Pinner reaction (reaction with an alcohol under acid catalysis).
- the alkene of vii was reduced to the saturated product viii with Pd/C and ammonium formate and ester hydrolysis under basic conditions afforded the carboxylic acid ix.
- Step a To a solution of methyl 4,6-dichloropyridine-3-carboxylate (1 .0 g, 4.9 mmol) in anhydrous ethanol (10 mL) was added phenylhydrazine (0.50 mL, 5.1 mmol), followed by triethylamine (1.7 mL, 12 mmol). The reaction mixture was heated under microwave irradiation at 120 oC for 40 min.
- Step b To a solution of methyl 6-chloro-4-(2-phenylhydrazinyl)pyridine-3-carboxylate (0.55 g, 2.0 mmol) in anhydrous DMA (11 ml_) was added zinc cyanide (0.15 g, 1 .3 mmol). The reaction mixture was evacuated and flushed with argon (x 2), then zinc (16 mg, 0.24 mmol), Pd 2 (dba) 3 (30 mg, 0.033 mmol) and dppf (33 mg, 0.060 mmol) were added and the mixture was again flushed with argon (x2). The mixture was heated at 120 °C for 1 .5 h.
- Step c To a solution of methyl 6-cyano-4-(2-phenylhydrazinyl)pyridine-3-carboxylate (0.32 g, 1.1 mmol) in anhydrous DMF (10 ml_) that was degassed with argon was added 1 M NaHMDS in THF (1 .6 mL, 1.6 mmol). The reaction was heated at 95 °C for 15 min then cooled in ice/water. After 30 min at 0 °C 1 M aqueous acetic acid (5 mL) was added and reaction mixture was evaporated to dryness.
- Step d A solution of 3-hydroxy-2-phenyl-2/-/-pyrazolo[4,3-c]pyridine-6-carbonitrile (0.1 1 g, 0.44 mmol) in phosphorus oxychloride (7 mL) was heated at 120 °C for 45 min, then left at room temperature overnight. The reaction was then poured into ice water/ethyl acetate (1 :1 ) and the aqueous layer adjusted to pH 5-7 with 7 M NaOH.
- Step e A solution of 3-chloro-2-phenyl-2H-pyrazolo[4,3-c]pyridine-6-carbonitrile (0.21 g, 0.82 mmol) in dioxane/water (2: 1 , 30 mL total) was de-gassed with argon and split into two batches. To each batch was added sodium carbonate (65 mg, 0.78 mmol) and the mixture was stirred at room temperature for 5 min with sonication.
- Step f A solution of 3-(cyclohex-1 -en-1 -yl)-2-phenyl-2/-/-pyrazolo[4,3-c]pyridine-6-carbonitrile (65 mg, 0.22 mmol) in freshly prepared saturated HCI in ethanol (12 mL) was heated in a sealed tube at 80 °C for 6 h then left overnight at room temperature. The reaction mixture was poured into ice/water and extracted with ethyl acetate (x 3). The combined organics were dried over MgS0 4 , filtered and evaporated to dryness.
- Step g To a solution of ethyl 3-(cyclohex-1 -en-1-yl)-2-phenyl-2H-pyrazolo[4,3-c]pyridine-6- carboxylate (31 mg, 80 pmol) in methanol (2 mL) was added ammonium formate (26 mg, 0.41 mmol) and the reaction mixture was de-gassed with argon. 10% Pd/C (13 mg, -40% by mass) was added and the mixture was heated at reflux for 3 h. The reaction mixture was decanted from the Pd catalyst and the catalyst was washed with methanol (x 2). The combined organics were evaporated to dryness.
- Step h A solution of ethyl 3-cyclohexyl-2-phenyl-2H-pyrazolo[4,3-c]pyridine-6-carboxylate (10 mg, 0.029 mmol) and KOH (39 mg, 0.70 mmol) in 1 ,4-dioxane/water (1 : 1 , 1 .5 mL) was stirred at room temperature for 1.75 h. The reaction was quenched with water (3.5 mL) and glacial acetic acid (0.08 mL) and co-evaporated to dryness with acetonitrile.
- Cyclisation to v was achieved by heating iv in phosphorus oxychloride.
- the W-oxide of v was prepared via oxidation with m-chloroperbenzoic acid. Reaction of vi with TMSCN under basic conditions afforded vii.
- the nitrile was hydrolysed to a carboxylic acid under acidic conditions, and esterification was achieved with thionyl chloride and an alcohol to give ix.
- the chloride of x was reacted with a cycloalkenyl boronic acid or ester under standard palladium-catalysed cross-coupling conditions and the carboxylic ester was hydrolysed under basic conditions to give x.
- the alkene of x was reduced to the saturated product xi with Pd/C and ammonium formate.
- Step a To stirred solution of 2-chloronicotinic acid (50g, 0.32 mol) in DCM was added DMAP (78 g, 0.63 mol), aniline (44 g, 0.48 mol) and EDCI.HCI (92 g, 0.48 mol). The reaction was stirred overnight at room temperature, then diluted with water and the product extracted with DCM. The organic layer was washed with 1 M aqueous acetic acid then saturated sodium bicarbonate and concentrated in vacuo to give 2-chloro-/V-phenylnicotinamide (48 g, 65%).
- Step b To a stirred solution of 2-chloro-/V-phenylnicotinamide (48 g, 0.21 mol) in ethanol (120 ml) was added hydrazine hydrate (155 g, 3.1 mol) and the reaction mixture was stirred for 48 h at room temperature. The reaction mixture was diluted with water and the precipitated product was collected by filtration and dried to give 2-hydrazinyl-/V- phenylnicotinamide (35 g, 74%). ESI-MS m/z calculated for [M+H] + : 229.2; found: 229.0.
- Step c To a ice cold solution of 2-hydrazinyl-W-phenylnicotinamide (35 g, 0.15 mol) in 4 M HCI (105 ml_) was added a solution of sodium nitrite (21 g, 0.31 mol) in water (175 ml_) at 0- 5 °C and the reaction was stirred for 30 min at 0 °C when precipitation occurred. The reaction mixture was filtered to give 2-azido-/V-phenylnicotinamide (32 g, 87%). ESI-MS m/z calculated for [M+H] + : 240.1 ; found: 240.0.
- Step d A solution of 2-azido-/V-phenylnicotinamide (32 g, 0.15 mol) in phosphorus oxychloride (320 ml) was heated at reflux for 24 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. The residue was diluted with ice water and adjusted to pH ⁇ 8 with aqueous sodium bicarbonate. The product was extracted with ethyl acetate and the organic layer concentrated in vacuo. The crude product was purified by column chromatography to give 3-chloro-2-phenyl-2/-/-pyrazolo[3,4-6]pyridine (22 g, 72%). ESI-MS m/z calculated for [M+H] + : 230.0; found: 230.3.
- Step e To a suspension of 3-chloro-2-phenyl-2/-/-pyrazolo[3,4-£>]pyridine (22 g, 96 mmol) in dichloromethane (440 mL) was added m-chloroperbenzoic acid (50 g, 0.29 mol) and the reaction mixture was stirred at room temperature for 2 h. The reaction mixture was quenched with saturated sodium bicarbonate, the product was extracted with
- Step f To a solution of 3-chloro-2-phenyl-2H-pyrazolo[3,4-6]pyridine-7-oxide (17g, 0.69 mol) in DMF (255 mL) was added DIPEA (24 mL, 0.14 mol) and TMSCN (14 g, 0.14 mol) and the reaction was heated at 90 °C for 2 h. The reaction mixture was poured into ice water and the precipitate was collected by filtration. The crude product was purified by column
- Step g A suspension of 3-chloro-2-phenyl-2/-/-pyrazolo[3,4-0]pyridine-6-carbonitrile (5.0 g, 20 mmol) in 70% sulphuric acid (50 mL) was heated to 81 °C for 4 h. The reaction was poured into ice water and the precipitate was collected by filtration. The crude product was purified by acid base treatment to give 3-chloro-2-phenyl-2H-pyrazolo[3,4-b]pyridine-6- carboxylic acid (3.8 g, 71 %) as off white solid.
- ESI-MS m/z calculated for [M+H] + : 274.0; found: 274.0.
- Step h To a solution of 3-chloro-2-phenyl-2H-pyrazolo[3,4-b]pyridine-6-carboxylic acid (3.8 g, 0.14 mol) in ethanol (76 mL) was added thionyl chloride (5.8 g, 0.49 mol) at 0 °C and the reaction was heated at reflux for 4 h. The reaction mixture was cooled and concentration of the solvent in vacuo gave crude product which was purified by column chromatography to give ethyl 3-chloro-2-phenyl-2H-pyrazolo[3,4-0]pyridine-6-carboxylate (2.0 g, 48%). ESI-MS m/z calculated for [M+H] + : 302.1 ; found: 302.1.
- Step i To a solution of ethyl 3-chloro-2-phenyl-2H-pyrazolo[3,4-b]pyridine-6-carboxylate (2.0 g, 6.6 mmol) in 1 ,4-dioxane (20 mL) was added 2-(cyclohex-1-en-1-yl)-4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolane (2.1 g, 9.9 mmol) and a solution of sodium carbonate (1 .4 g, 13 mmol) in water (6 mL).
- the mixture was purged with nitrogen and stirred for 30 min at room temperature, then palladium acetate (0.30 g, 1 .3 mmol) and triphenylphosphine (0.70 g, 26 mmol) were added and the reaction was heated at 80 °C for 2 h. Then potassium hydroxide (1.1 g, 20 mmol) was added and the reaction mixture was stirred for 1 hr at 80 °C. The reaction mixture was cooled to room temperature and concentrated. The residue was dissolved in water and the aqueous layer was washed with ether. The aqueous layer was adjusted to pH ⁇ 4 using 1 M HCI and the product was extracted with ethyl acetate.
- Step j To a solution of 3-(cyclohex-1-en-1 -yl)-2-phenyl-2H-pyrazolo[3,4-ib]pyridine-6- carboxylic acid (1.0 g, 3.1 mmol) in methanol (20 mL) was added 10% Pd/C (0.50 g, 50% by mass) and ammonium formate (2.0 g, 31 mmol) and the reaction was heated at 50 °C for 30 min. The reaction mixture was cooled to room temperature and filtered through Celite®. The filtrate was concentrated and the residue was dissolved in water and the aqueous adjusted to pH ⁇ 4 with 1 M HCI.
- Step a To a solution of 2-[4-(benzyloxy)phenyl]-3-(cyclohex-1 -en-1 -yl)-2/-/-indazole-6- carboxylic acid (xxiii) (prepared according to Method A using 4-(benzyloxy)aniline hydrochloride in place of aniline at Step b) (1.0 g, 2.35 mmol) in methanol (15 mL) was added 10% Pd/C (0.45 g, 0.4 mmol, 45% w/w) and ammonium formate (1 .33 g, 21.1 mmol). The reaction mixture was then heated at reflux for 1 h, cooled to RT and filtered through a pad of Celite®.
- Step b To a stirred suspension of 3-cyclohexyl-2-(4-hydroxyphenyl)-2/-/-indazole-6- carboxylic acid (11 ) (200 mg, 0.59 mmol) in methanol (8 mL) was added concentrated sulphuric acid (1 .6 mL) at RT. The reaction mixture was then heated at reflux for 1 h and the volatiles were then removed in vacuo. The residue was diluted with water (5 mL) and the organics were extracted into ethyl acetate (3 x 15 mL) and dried (Na 2 S0 4 ).
- Step c To a stirred solution of methyl 3-cyclohexyl-2-(4-hydroxyphenyl)-2A7-indazole-6- carboxylate (12) (80 mg, 0.22 mmol) in DMF ( 2 ml_) was added potassium carbonate (94 mg, 0.68 mmol) and methyl iodide (64 mg, 0.45 mmol). The reaction mixture was then stirred at RT for 2 h. After completion of the reaction, the mixture was diluted with water (8 ml_) and extracted with ethyl acetate (3 x 15 ml_). The combined organic layer was dried (Na 2 S0 4 ) and concentrated in vacuo to give the crude residue.
- Step d To a stirred solution of methyl 3-cyclohexyl-2-(4-methoxyphenyl)-2/-/-indazole-6- carboxylate (13) (55 mg, 0.15 mmol) in ethanohwater (1 : 1 v/v, 2 mL) was added sodium hydroxide (1 10 mg, 2.75 mmol) and the reaction mixture was heated at reflux for 1 h. Upon cooling to RT, the reaction mixture was acidified with 1 N aqueous HCI and the organics were extracted into ethyl acetate (3 x 10 mL).
- Rea ent (vii) was prepared according to the followin procedure:
- Step a To a stirred solution of 1 -[(ferf-butoxycarbonyl)amino]cyclobutanecarboxylic acid
- Step b To a stirred solution of ferf-butyl ⁇ 1-[(4-iodophenyl)carbamoyl]cyclobutyl ⁇ carbamate
- Step c 4M HCI in dioxane (2 mL) was added to ethyl (2E)-3- ⁇ 4-[( ⁇ 1-[(ferf- butoxycarbonyl)amino]cyclobutyl ⁇ carbonyl)amino]phenyl ⁇ prop-2-enoate (xi) (350 mg, 0.90 mmol) and the resultant suspension stirred for 5 h at RT. The volatiles were then removed in vacuo and the residue was then was dissolved in water (7 mL) and washed with ethyl acetate (2 x 10 mL).
- aqueous layer was basified by addition of saturated solution of sodium bicarbonate and the organics were then extracted into ethyl acetate (3 x 20 mL). The combined organics were washed with brine (50 mL), dried (Na 2 S0 4 ) and concentrated in vacuo.
- Step b ferf-Butyl ⁇ (2R)-1-[(4-iodophenyl)amino]-1-oxopropan-2-yl ⁇ carbamate (xv) (201 mg, 0.52 mmol), pyridine-4-boronic acid (99 mg, 0.81 mmol) and sodium carbonate (1 1 1 mg, 1 .05 mmol) in a mixture of dioxane (2 mL) and water (2 mL) was degassed with a stream of argon. Palladium acetate (25 mg, 0.1 1 mmol) and triphenylphosphine (58 mg, 0.22 mmol) were added and the reaction was heated at 100°C for 2 h.
- the crude reaction mixture was purified by automated flash chromatography (Biotage SP4, 12 g Grace RevelerisTM C-18 silica gel cartridge) eluting with acetonitrile:water (0% to 100%) to yield ierf-butyl [(2R)-1 - oxo-1 - ⁇ [4-(pyridin-4-yl)phenyl]amino ⁇ propan-2-yl]carbamate (xvi) (92 mg, 52 %); ESI-MS m/z calculated for [M+H] + : 342.2; found: 342.0.
- Step c A suspension of ferf-butyl [(2R)-1 -oxo-1- ⁇ [4-(pyridin-4-yl)phenyl]amino ⁇ propan-2- yl]carbamate (xvi) (92 mg, 0.27 mmol) in 4M HCI in dioxane (4 mL) was stirred at RT for 1.5 h. Concentration of the reaction mixture gave W-[4-(pyridin-4-yl)phenyl]-D-alaninamide hydrochloride (xii), which was used unpurified, ESI-MS m/z calculated for [M+H] + : 242.1 ; found: 242.0.
- Rea ent (xvii) was prepared as follows:
- Step a To a solution of W-Boc-1 -aminocyclobutanecarboxylic acid (xviii) (1.00 g, 4.65 mmol) and methyl-4-aminobenzoate (xix) (0.85 g, 5.62 mmol) in dry DMF (10 mL) under Ar was added DIPEA (1.21 mL, 6.80 mmol) added, followed by HATU (2.13 g, 5.60 mmol). The reaction mixture was stirred at RT under argon and monitored by LCMS.
- reaction mixture was concentrated to dryness in vacuo, dissolved in DMSO (10 mL) and purified by automated column chromatography (Biotage SP4, 60 g Biotage SNAPTM C-18 silica gel cartridge) eluting with acetonitrile:water (0% to 100%). The relevant fractions were concentrated in vacuo and the residue was dissolved in ethyl acetate (50 mL) and washed with 1 M aqueous HCI (30 mL), saturated sodium bicarbonate solution (30 mL) and brine (30 mL).
- Step b Methyl 4-[( ⁇ 1-[(ferf-butoxycarbonyl)amino]cyclobutyl ⁇ carbonyl)amino] benzoate (xx) (325 mg, 0.93 mmol) and lithium hydroxide hydrate (238 mg, 5.67 mmol) in dioxane (3 mL), /so-propanol (3 mL), and water (3.4 mL) were stirred at 60 °C for 45 min. The reaction mixture was then cooled to RT, acidified with 1 M aqueous HCI and then concentrated in vacuo.
- Step c To a solution of 4-[( ⁇ 1 -[(ierf-butoxycarbonyl)amino]cyclobutyl ⁇ carbonyl)
- Step d [1 -( ⁇ 4-[(Methylsulfonyl)carbamoyl]phenyl ⁇ carbamoyl)cyclobutyl]carbamate (xxii) (245 mg, 0.60 mmol) was suspended in 4M HCI in dioxane (4 mL) and the reaction mixture was stirred at RT for 1 .5 h.
- Step a A solution of 1-(ferf-butoxycarbonyl)-4- ⁇ [(9H-fluoren-9-ylmethoxy)- carbonyl]amino ⁇ piperidine-4-carboxylic acid (0.330 g, 0.64 mmol) in 4 M HCI-dioxane (6 mL) was stirred at room temperature. After 1.25 h the residue was co-evaporated with diethyl ether (x2) and dried under high vacuum to afford 4- ⁇ [(9A7-fluoren-9- ylmethoxy)carbonyl]amino ⁇ piperidine-4-carboxylic acid hydrochloride (ii) (0.35 g) as a yellow foam.
- ESI-MS m/z calculated for [M+H] + : 367.2; found: 367.0.
- Step b To a suspension of 4- ⁇ [(9H-fluoren-9-ylmethoxy)carbonyl]amino ⁇ piperidine-4- carboxylic acid hydrochloride (0.35 g, 0.86 mmol) in MeOH (5 mL) at room temperature in a medium pressure vessel was added an aqueous solution of formaldehyde (37%, 70 pL, 0.94 mmol), followed by catalytic amounts of 10% Pd/C (90 mg, 0.10 mmol). The reaction was stirred at room temperature at 6-7 Bar under an atmosphere of hydrogen for a total of 27.5 h, during which time there were two further additions of formaldehyde and one addition of 10% Pd/C.
- Q is a carboxylic ester
- the ester can be hydrolysed to a carboxylic acid under basic conditions.
- some standard transformations such as cleavage of common protecting groups may be employed. The final
- Step a To a stirred solution of 3-cyclohexyl-2-phenyl-2/-/-indazole-6-carboxylic acid (2) (200 mg, 0.62 mmol) and ethyl (2E)-3-(4- ⁇ [(1 -aminocyclobutyl)carbonyl]amino ⁇ phenyl)prop-2- enoate hydrochloride (vii) (180 mg, 0.62 mmol) in DMF (15 mL) was added HATU (356 mg, 0.94 mmol) followed by DIPEA (326 mL, 1.87 mmol). The reaction mixture was then stirred at RT for 16 h and then poured onto water (30 mL). The organics were extracted into ethyl acetate (3 x 30 mL) and the combined organics dried (MgS0 4 ) and concentrated in vacuo. The residue was purified by automated column chromatography (Biotage SP4,
- Step a To a solution of /V-[4-(pyridin-4-yl)phenyl]-D-alaninamide hydrochloride (xii) (9 mg, 0.037 mmol) in dry DMF (500 L) under argon was added 3-cyclohexyl-2-phenyl-2A7- indazole-6-carboxylic acid (2) (1 1 mg, 0.034 mmol) and DIPEA (17 ⁇ _, 0.098 mmol), followed by HATU (15 mg, 0.039 mmol). The solution was stirred under argon at RT for 3.5 h.
- Step a To a stirred solution of 3-cyclohexyl-2-phenyl-2/-/-indazole-6-carboxylic acid (2) (102.9 mg, 0.32 mmol) and 4- ⁇ [(1 -aminocyclobutyl)carbonyl]amino ⁇ -A- (methylsulfonyl)benzamide hydrochloride (xvii) (120.9 mg, 0.39 mmol) in dry DMF (2 mL) under argon was added DIPEA (170 pL, 0.95 mmol) followed by HATU (148.9 mg, 0.39 mmol). The mixture was stirred at RT for 3 h.
- reaction mixture was then diluted with ethyl acetate (5 mL) and washed with 1 M HCI (2 x 2 mL), saturated sodium bicarbonate solution (2 mL), brine (2 mL) and then concentrated in vacuo.
- Step a To a solution of 1-amino-W-(4-cyanophenyl)cyclobutanecarboxamide hydrochloride (xxiv) (70 mg, 0.325 mmol) in dry DMF (1 imL) under argon was added 3-cyclohexyl-2- phenyl-2A7-indazole-6-carboxylic acid (2) prepared according to Method A (86 mg, 0.268 mmol) and DIPEA (150 ⁇ _, 0.861 mmol), followed by HATU (125 mg, 0.327 mmol). The solution was stirred under argon at T for 16 h.
- Step b To a stirred solution of W- ⁇ 1 -[(4-cyanophenyl)carbamoyl]cyclobutyl ⁇ -3-cyclohexyl-2- phenyl-2A7-indazole-6-carboxamide (16) (21 mg, 0.41 mmol) in methanol (400 pL) and THF (1 mL) was added 28% aqueous ammonia (200 pL) and 30% aqueous hydrogen peroxide (200 )L). The reaction was heated at 60°C for 4h.
- Step a To a solution of /V-Boc-1 -aminocyclobutanecarboxylic acid (xxvi) (501 mg, 2.33 mmol) and 4-aminobenzonitrile (xxv) (330 mg, 2.79 mmol) in dry DMF (5 mL) under argon was added DIPEA (610 3.50 mmol) added, followed by HATU (1 .07 g, 2.81 mmol). The reaction mixture was stirred at RT under argon for 16 h.
- DIPEA 610 3.50 mmol
- reaction mixture was then concentrated to dryness in vacuo, dissolved in DMSO (5 mL) and purified by automated column chromatography (Biotage SP4, 60 g Biotage SNAPTM C-18 silica gel cartridge) eluting with acetonitrile:water (0% to 100%).
- the relevant fractions were concentrated in vacuo and the residue was dissolved in ethyl acetate (50 mL) and washed with 1 M aqueous HCI (30 mL), saturated sodium bicarbonate solution (30 mL) and brine (30 mL).
- Step b ferf-butyl ⁇ 1 -[(4-cyanophenyl)carbamoyl]cyclobutyl ⁇ carbamate (xxvii) (76 mg, 0.24 mmol) was suspended in 4M HCI in dioxane (2 mL) and the reaction mixture was stirred at RT for 1 .5 h. The volatiles were removed in vacuo and the residue 1 -amino-W-(4- cyanophenyl)cyclobutanecarboxamide hydrochloride (xxiv) was used without purification; ESI-MS m/z calculated for [M+H] + : 216.1 ; found: 215.9.
- Step a To a stirred solution of 3-cyclohexyl-2-phenyl-2/-/-indazole-6-carboxylic acid (2) (42 mg, 0.13 mmol) and ethyl (2E)-3- ⁇ 4-[(0-benzyl-L-seryl)amino]phenyl ⁇ prop-2-enoate hydrochloride (1:1) (i) (64 mg, 0.16 mmol) in dry DMF (0.8 mL) was added DIPEA (91 ⁇ _,
- Step b To a solution of ethyl (2E)-3-[4-( ⁇ 0-benzyl-/V-[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]-L-seryl ⁇ amino)phenyl]prop-2-enoate (40 mg, 60 ⁇ ) in dichloromethane (0.5 mL) cooled to -78 °C was added dropwise 1 M boron tribromide in dichloromethane (0.18 mL, 0.18 mmol). The resulting mixture was removed from the cold bath and after 30 min the reaction was quenched with water and extracted with ethyl acetate.
- Step c To a solution of ethyl (2E)-3-[4-( ⁇ /V-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]- L-seryl ⁇ amino)phenyl]prop-2-enoate (6.0 mg, 10 pmol) in ethanol (0.5 mL) was added a solution of potassium carbonate (14 mg) in water (0.5 mL). The resulting mixture was heated at 80 °C for 2 h. The reaction was then cooled to room temperature and quenched with 0.5 M HCI. The precipitate was collected by filtration, washed with water and dried in a desiccator.
- Step a To a solution of 1-aminocyclobutanecarboxylic acid ethyl ester (xxviii) (69 mg, 0.38 mmol) in dry DMF (1 mL) under argon was added 3-cyclohexyl-2-phenyl-2H-indazole-6- carboxylic acid (2) (101 mg, 0.32 mmol) and DIPEA (200 pL, 1 .15 mmol), followed by HATU (155 mg, 0.41 mmol). The solution was stirred under argon at RT for 1 .5 h.
- Step b To a stirred solution of ethyl 1 - ⁇ [(3-cyclohexyl-2-phenyl-2/-/-indazol-6- yl)carbonyl]amino ⁇ cyclobutanecarboxylate (xxix) (122 mg, 0.27 mmol) in 1 ,4-dioxane (1 mL) and /so-propanol (1 mL) was added a solution of lithium hydroxide (74 mg, 1 .76 mmol) in water (0.8 mL). The mixture was then stirred at 60°C for 1 h whereupon the reaction mixture was acidified to pH 1 with 1 M aqueous HCI and then concentrated in vacuo.
- Step c To a stirred solution of 1- ⁇ [(3-cyclohexyl-2-phenyl-2/-/-indazol-6- yl)carbonyl]amino ⁇ cyclobutanecarboxylic acid (18) (5 mg, 0.012 mmol) in dry DMF (250 ⁇ ) under argon was added 4-fluoroaniline (2 ⁇ , 0.021 mmol), followed by DIPEA (7 ⁇ , 0.040 mmol) and then HATU (6 mg, 0.016 mmol). The reaction was stirred at RT for 1 h and then concentrated in vacuo.
- Step a To a solution of 3-cyclohexyl-2-(pyridin-2-yl)-2/-/-indazole-6-carboxylic acid (0.40 g, 1 .2 mmol) and D-alanine methyl ester hydrochloride (0.19 g, 1.4 mmol) in dry DMF (3 ml.) was added DIPEA (0.87 ml_, 5.0 mmol), followed by HATU (0.62 g, 1.6 mmol). After 17 h, the reaction was quenched with 1 M HCI and diluted with water. The product was initially a gum, which became a solid upon standing.
- Step b To a mixture of methyl W- ⁇ [3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6-yl]carbonyl ⁇ -D- alaninate (0.21 g, 0.52 mmol) in ethanol (3 mL) was added a solution of potassium carbonate (0.71 g, 5.2 mmol) in water (3 mL). The resulting mixture was heated at 70 °C for 1 h. The reaction was cooled to room temperature and quenched with 1 M HCI and ice.
- Step c To a solution of N- ⁇ [3-cyclohexyl-2-(pyridin-2-yl)-2/-/-indazol-6-yl]carbonyl ⁇ -D-alanine (0.12 g, 0.31 mmol) and 4-aminophenylboronic acid hydrochloride (64 mg, 0.37 mmol) in dry DMF (2 mL) was added DIPEA (0.21 mL, 1 .2 mmol), followed by HATU (0.15 g, 0.40 mmol). After 18 h the reaction was quenched with 1 M HCI and diluted with water.
- Step d To a mixture of ⁇ 4-[(W- ⁇ [3-cyclohexyl-2-(pyridin-2-yl)-2/-/-indazol-6-yl]carbonyl ⁇ -D- alanyl)amino]phenyl ⁇ boronic acid (60 mg, 0.12 mmol), 5-bromo-2-chloropyrimidine (23 mg, 0.12 mmol) and potassium carbonate (32 mg, 0.23 mmol) in 1 ,4-dioxane (1 mL) was added water (0.6 mL) followed by PdCI 2 dppf.DCM (10 mg, 12 ⁇ ) and the mixture was heated at 90 °C for 2 h under argon.
- Step e A mixture of W-[(2R)-1 - ⁇ [4-(2-chloropyrimidin-5-yl)phenyl]amino ⁇ -1 -oxopropan-2-yl]-3- cyclohexyl-2-(pyridin-2-yl)-2/-/-indazole-6-carboxamide (12 mg, 21 mol), methylamine hydrochloride (9.0 mg, 0.13 mmol) and DIPEA (36 ⁇ , 0.21 mmol) in 2-propanol (0.5 mL) were heated under microwave irradiation for 2 h at 150 °C. The reaction mixture was quenched with ice water, and the precipitation collected by filtration.
- 6-carboxamide 2.39 (m, 2H), 2.26 - 1.69 (m, 9H), 1.55 - 1.15 (m, 3H).
- Step a To a stirred solution of ethyl (2E)-3-(4- ⁇ [(1- ⁇ [(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino ⁇ cyclobutyl)carbonyl]amino ⁇ phenyl)prop-2-enoate (3) (100 mg, 0.169 mmol) in 1 ,4-dioxane (1 .5 mL) and /so-propanol (1 .5 mL) was added a solution of lithium hydroxide (43 mg, 1 .016 mmol) in water (1 mL).
- Step b To a stirred solution of (2E)-3-(4- ⁇ [(1 - ⁇ [(3-Cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino ⁇ cyclobutyl)carbonyl]amino ⁇ phenyl)prop-2-enoic acid (4) (8 mg, 0.014 mmol) in ethyl acetate (0.5 mL) was added 10% Pd/C ( ⁇ 2 mg) and the mixture was stirred under a hydrogen atmosphere at RT for 24 h. The mixture was diluted with ethyl acetate (2mL) and filtered.
- Step a To a solution of 3-cyclohexyl-2-phenyl-2H-indazole-6-carboxylic acid (2) (200 mg, 0.62 mmol) in dichloromethane was added oxalyl chloride (158 ⁇ , 1 .87 mmol) and a catalytic amount of DMF. The mixture was then purged with ammonia gas for 30 min.
- Step a To a stirred solution of (2E)-3-(4- ⁇ [(1 - ⁇ [(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino ⁇ cyclobutyl)carbonyl]amino ⁇ phenyl)prop-2-enoic acid (4) (49 mg, 0.09 mmol) in DMF (3 mL) was added methylamine hydrochloride (16 mg, 0.237 mmol) followed by HATU (42 mg, 0.1 1 mmol) and DIPEA (40 pL, 0.22 mmol). The reaction mixture was then stirred at RT for 5 days and then concentrated to a volume of ⁇ 1 mL in vacuo.
- Step a To a stirred solution of 3-(cyclohex-1 -en-1 -yl)-2-phenyl-2/-/-indazole-6-carboxylic acid (1 ) (300 mg, 0.94 mmol) in DMF (10 mL) at 0 °C was added EDCI (360 mg, 1.88 mmol) and DMAP (230 mg, 1.88 mmol) followed by W,W-dimethylsulfamamide (175 mg, 1.41 mmol).
- Step b To a stirred suspension of 3-(cyclohex-1 -en-1 -yl)-/V-(dimethylsulfamoyl)-2-phenyl- 2A7-indazole-6-carboxamide (8) (245 mg, 0.58 mmol) in methanol (20 mL) was added 10% Pd/C (245 mg, 2.31 mmol, 100% wt. by wt.) under an atmosphere of nitrogen. Ammonium formate (2.156 g, 34.12 mmol) was then added and the reaction mixture was heated to reflux and monitored by LCMS analysis.
- reaction mixture Upon complete consumption of the starting material (4 h), the reaction mixture was filtered through a pad of Celite® and the residue was washed with methanol (2 x 10 mL). The combined filtrates were concentrated in vacuo and the residue was purified by automated column chromatography (Combi-FlashTM, silica gel) eluting with ethyl acetate: hexane (2:1 ). Further purification by crystallization from
- HCV polymerase reactions were carried out using a modified method of Howe et al. , Antimicrobial Agents and Chemotherapy 2004 48(12): 4813-4821. Reactions contained 50 nM NS5bA21 , 20 mM Tris-HCI pH 7.5, 5 rtiM MgCI 2 , 5 mM MnCI 2 , 3 mM DTT, 0.05% BSA, 0.2 U/pL RNasin, 10 pg/mL Poly(rC) template, GTP (at Km) and 0.05 pCi/pL 33 P-GTP in a total reaction volume of 50 pL.
- IC 50 ( ⁇ ) values determined in accordance with the general ranges: A: ⁇ 0.99 ⁇ ; ⁇ : 1.0-9.99 ⁇ ; C: 10.0-49.99pM; D: > 50 ⁇ .
- the IC 50 values for the majority of compounds in the A range were determined to ⁇ 500nM.
- the IC 50 values for compounds 5, 7, 25, 30, 32, 34, 36, 37, 40, 41 , 45, 46, 51 , 56, 59, 68, 70-74, 76, 78, 80, 83, 85, 88, 92, 94-96, 99, 106, 1 12, 1 15, 124, 125, 135, 138-141 , 148, 151 , 155, 158, 174, 175, 182, 183, 198, 199, 201-204 were determined to be ⁇ 500nM.
- Table 1 HCV polymerase values for selected compounds with IC 50 values ⁇ 100nM.
- a genotype 1 b subgenomic replicon cell line based on Blight et ai, Science 2000 290: 1972-1974, modified to express a Renilla luciferase reporter gene was used to assess antiviral activity of test compounds.
- Cell cultures were maintained in a sub-confluent state in DMEM with glutamine, 10% heat-inactivated foetal bovine serum and Geneticin.
- DMEM dimethyl methyl bovine serum
- Geneticin fetal bovine serum
- cells were seeded at a concentration of 7000 cells/well into 96 well tissue culture trays in culture media lacking Geneticin.
- Compounds were tested in a three fold dilutions series for example starting from 50 ⁇ . After 72 hours incubation at 37°C and 5% C0 2 , enilla luciferase activity was quantified. The compound concentration that reduced luciferase activity by 50% (EC 50 ) was calculated using non-linear regression.
- EC 50 values for selected compounds were determined in accordance with the general ranges: A: ⁇ 0.99 ⁇ ; ⁇ : 1.0-9.99 ⁇ ; C: 10.0-49.99 ⁇ .
- A Compounds 4, 49, 62, 96, 101 , 1 12, 120, 124, 133, 137, 138, 140, 141 , 146, 148, 151 , 160, 164, 165, 172, 174, 178, 182, 191 , 195, 200.
- Cytotoxicity of compounds against genotype 1 b subgenomic replicon cells were assessed using the MTT assay (Watanabe et al. , Journal of Virological Methods 1994 48:257-265). Plates were prepared as described for the HCV Replicon assay and cytotoxicity of the test article was evaluated after three days. MTT was added to assay plates followed by three hour incubation at 37 ° C. Wells were aspirated to dryness and the formazan dye dissolved by the addition of 100% isopropanol. Absorbencies were read in a spectrophotometer at two wavelengths (540 nm and 690 nm). The compound concentration that reduced cell viability by 50% (CC 50 ) is calculated using non-linear regression.
Abstract
The present invention relates to viral polymerase inhibitors of formula (I), salts, N-oxides, racemates, enantiomers and isomers thereof, processes for their preparation and their use in the treatment of Flaviviridae viral infections such as Hepatitis C virus (HCV) infections.
Description
VIRAL POLYMERASE INHIBITORS
FIELD
The present invention relates to viral polymerase inhibitors, in particular inhibitors of viral polymerases within the Flaviviridae family such as hepatitis C virus (HCV), processes for their preparation and their use in the treatment of Flaviviridae viral infections such as Hepatitis C virus (HCV) infections.
BACKGROUND
The Flaviviridae are a group of positive single-stranded RNA viruses with a genome size from 9-15 kb. The Flaviviridae consist of various genera including: Hepaciviruses (this genus contains only one species, the Hepatitis C virus (HCV), which is composed of many genotypes and subtypes); Flaviviruses (this genus includes the Dengue virus, Japanese Tick-Borne and the Yellow Fever virus and there are some additional Flaviviruses that are unclassified) and Pestiviruses (this genus includes three serotypes of bovine viral diarrhoea virus, but no known human pathogens).
Hepatitis C virus (HCV) is a major cause of viral hepatitis and has infected more than 200 million people worldwide. Hepatitis C virus has a positive-strand RNA genome enclosed in a nucleocapsid and lipid envelope. The HCV genome is approximately 9.6 kb in length and encodes a polyprotein of about 3,000 amino acids. There are at least six major genotypes, which have different geographic distributions. In the United States (US), for example, genotypes 1 a and 1 b account for about 75 % of cases, and genotypes 2 and 3 for 10-20 % of cases. In the US, HCV is the most common chronic bloodborne infection, affecting approximately 3.2 million persons. After infection with HCV, approximately 75-85% of people develop chronic infection, whilst 60-70% develop chronic liver disease. Of these, 5-20% go on to develop cirrhosis over a period of 20-30 years, and, finally, 1-5% succumb to the consequences of chronic infection (liver cancer/cirrhosis).
Current standard of care (SOC) consists of weekly injections of pegylated interferon (peg-IFN) in combination with daily oral ribavirin. There is little difference in the severity of disease or outcome in patients infected with the different genotypes; however, those with genotypes 2 and 3 are almost three times more likely than those with genotype 1 to respond to therapy. In addition, when using SOC therapy, the recommended duration of treatment depends on the genotype. For patients with genotype 1 , a 48-week course is recommended, and the success rate for achieving a sustained viral response (SVR) using SOC therapy is approximately 50%. SVR is usually defined as an undetectable viral load 6 months following the end of treatment. In addition to being prolonged and having limited efficacy, this SOC therapy is associated with serious side effects, such as fatigue, influenza-like symptoms,
depression and suicide with peg-I FN, and haemolytic anaemia with ribavirin. There are also several contraindications to SOC therapy including pregnancy, depression, anaemia, HCV related decompensated cirrhosis, alcohol/substance abuse and autoimmune disorders.
In April 201 1 , the FDA Antiviral Drugs Advisory Committee (ADAC) recommended the approval of the first direct acting antiviral (DAA) HCV drugs, telaprevir (Vertex
Pharmaceuticals) and boceprevir (Merck), for use in combination with peg-IFN and ribavirin in genotype 1 chronic HCV infection. Both drugs are NS3/4A protease inhibitors which block a viral enzyme involved in HCV replication. Telaprevir and boceprevir have demonstrated significant effectiveness in improving cure rates over current SOC therapy and are likely to improve the current SOC. However, both telaprevir and boceprevir have issues with adverse effects. Telaprevir is associated with rash and anaemia, and boceprevir with anaemia and dysgeusia (dysfunction of the sense of taste). In addition, both of these new agents are limited to use in genotype 1 . The anticipated new SOC (boceprevir/telaprevir plus peg-IFN and ribavirin) remains unsuitable for those intolerant to or with contraindications to peg- I FN/ribavirin.
The HCV genome possesses structural (core) and non-structural (NS2, NS3, NS4A, NS4B, NS5A and NS5B) proteins. The non-structural proteins are involved in viral genomic replication, with the initial synthesis of RNA carried out by NS5B RNA dependent RNA polymerase. The NS5B protein is a key target for anti-HCV therapy, as it is essential for HCV replication and has no human host equivalent. This protein has been well characterised and is a validated target for drug discovery.
Due to the limited tolerability, efficacy, side effects and concern over the emergence of resistance there is an ongoing need to find alternative agents for the treatment of HCV. SUMMARY
The inventors have found a new class of NS5B polymerase inhibitors for the treatment of HCV infections.
According to a first aspect there is provided a compound of formula (I), salts, N- oxides, racemates, enantiomers and isomers thereof:

(I)
wherein
Zi, Z2 ,Z3 and Z4 are each independently selected from C-R1 , C-Ra, N and N+-0" wherein one of Zi, Z2 ,Z3 and Z4 is C-R1 , preferably Z2 is C-R1 and Zi, Z3 and Z4 are each independently selected from C-Ra, N and N+-0~, preferably C-Ra and N;
Ra is independently selected from H, optionally substituted d.6alkyl, optionally substituted C2-6alkenyl, halo, haloC -6alkyl, CHF2, OCHF2, CF3, OCF3, CN, OH, optionally substituted d_ 6alkoxy, N02, NH2, NH(d_6alkyl), N(d_6alkyl)2, NHC(0)d_6alkyl, NHS02 C(0)NH2,
C(0)NH(d.6alkyl), C(0)N(d_6alkyl)2, C(0)NHS02(d_6alkyl), C(0)N(d.6alkyl)S02(d_6alkyl), SO3H, OS02(d_6alkyl), NS02(d_6alkyl), S02(d_6alkyl), S02NH2, S02NH(d_6alkyl), S02N(Ci_ 6alkyl)2, C02H, C(0)d.6alkyl, C02d.6alkyl, optionally substituted (CH2)mC3.8cycloalkyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheterocyclyl and optionally substituted (CH2)mheteroaryl, preferably H;
R1 is (CH2)mR4, (CH2)m-C(0)R4, (CH2)m-OC(0)R4 (CH2)m-NR5C(0)R4, (CH2)m-NR5S(0)2R4 or (CH2)m-S(0)2R4, preferably (CH2)m-C(0)R4;
R2 is selected from optionally substituted (CH2)mC3.8cycloalkyl, optionally substituted (CH2)mheterocyclyl, optionally substituted (CH2)maryl and optionally substituted
(CH2)mheteroaryl;
R3 is selected from optionally substituted C1.6alkyl, optionally substituted C2.6alkenyl, optionally substituted (CH2)mC3.8cycloalkyl, optionally substituted (CH2)mC3.8cycloalkenyl, optionally substituted (CH2)mheterocyclyl, optionally substituted (CH2)maryl and optionally substituted (CH2)mheteroaryl;
R4 is (CH2)mR6, (CH2)mOR6or (CH2)mNR5R6 preferably (CH2)mOR6or (CH2)mNR5R6, most preferably (CH2)mNR5R6;
R5 is independently selected from H, optionally substituted d.6alkyl and optionally substituted C2.6alkenyl, preferably H;
R6 is selected from H, optionally substituted d.6alkyl, optionally substituted C2.6alkenyl, optionally substituted (CH2)mC3.8cycloalkyl, optionally substituted (CH2)mC3.8cycloalkenyl, optionally substituted (CH2)mheterocyclyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheteroaryl, R9, [C(R7)(R8)]V(R9), C(0)-[C(R7)(R8)]v(R9), C02- [C(R7)(R8)]V(R9), C(0)NR5-[C(R7)(R8)]v(R9), NR5-[C(R7)(R8)]V(R9), NR5C(0)-[C(R7)(R8)]v(R9), NR5S02-[C(R7)(R8)]v(R9), S02-[C(R7)(R8)]v(R9) and S02NR5-[C(R7)(R8)]v(R9);
R7 and R8 are independently selected from H, optionally substituted C1.6alkyl, optionally substituted C2.6alkenyl, optionally substituted (CH2)mC3.8cycloalkyl, optionally substituted (CH2)mC3.8cycloalkenyl, optionally substituted (CH2)mheterocyclyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheteroaryl, the side chain of an amino acid or an alkyl ester thereof or R7 and R8 together with the atom or adjacent atoms to which they are attached form an optionally substituted C3.8cycloalkyl or an optionally substituted C3_
8heterocyclyl or R7 or R8 and R5 together with the adjacent atoms to which they are attached form an optionally substituted C3-8cycloalkyl or an optionally substituted C3-8heterocyclyl; R9 is selected from R10, (CH2)mC(0)-R1°, (CH2)mC(0)NR5R10, (CH2)mC(0)NR5C(0)-R10, (CH2)mC(0)NR5S02-R1° (CH2)mC(0)NR5R10, (CH2)mNR5R10, (CH2)mNR5C(0)-R10,
(CH2)mNR5C(0)NR5R1°, (CH2)mNR5S02-R10, (CH2)mS02-R10, (CH2)mS02NR5R10 and (CH2)mS02NR5C(0)-R10;
R10 is H, OH, -A-(Q)„ or 0-A-(Q)n;
A is selected from optionally substituted C^alkyl, optionally substituted C2.6alkenyl, optionally substituted (CH2)mC3-8cycloalkyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheterocyclyl and optionally substituted (CH2)mheteroaryl; and
Q is selected from optionally substituted C^alkyl, optionally substituted C2.6alkenyl, halo, haloCi-6alkyl, CHF2, OCHF2, CF3, OCF3, CN, OH, optionally substituted Ci.6alkoxy, N02, NH2, NH(Ci_6alkyl), NiCi-salkylk, NHC(0)Ci-6alk l, NHS02 C(0)NH2, C(0)NH(Ci-6alkyl), C(0)N(Ci.6alkyl)2, C(0)NHS02(C1_6alkyl), qO d-ealky SO^d-ealkyl), S03H, OS02(d_ ealkyl), NS02(d_6alkyl), S02(d_6alkyl), S02NH2, S02NH(C1.6alkyl), S02N(d_6alkyl)2, C02H, C(0)Ci.6alkyl, C02Ci-6alkyl, optionally substituted (CH2)mC3.8cycloalkyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheterocyclyl and optionally substituted
(CH2)mheteroaryl; and
in each instance (CH2)m may be optionally substituted;
m in each instance is independently 0, 1 , 2 or 3, preferably 0 or 1 , more preferably 0;
n is independently 0, 1 , 2, 3, 4 or 5, preferably 0, 1 , 2 or 3, more preferably 0, 1 or 2, most preferably 0 or 1 ; and v is independently 1 , 2 or 3, preferably 1 .
In a second aspect there is provided a process for producing a compound of general formula (I) when R1 is C(0)R4 and R4 is NR5R6 comprising reacting a compound of formula (I I) with an amino precursor of general formula NHR5R6 under amide coupling conditions

(Π)
wherein
Z'i , Z'2, Z'3 and Z'4 are each independently selected from C-X, C-Ra, N and N+-0" wherein one of ΖΊ , Z'2 ,Z'3 and Z'4 is C-X, preferably Z'2 is C-X and Z , Z'3 and Z'4 are each independently selected from C-Ra, N and N+-0";
X is (R1)tC02H or (R1)tC02d_3alkyl, preferably (R1)tC02H;
t is 0 or 1 ; and
R1 , R2, R3 and Ra are as defined in formula (I).
The compounds of formula (I) are inhibitors of HCV. In particular, the compounds of formula (I) inhibit RNA synthesis by the RNA dependent RNA polymerase of HCV (the NS5B protein encoded by HCV). NS5B polymerase inhibitors have been clinically validated as potential antiviral agents for the treatment of HCV infection.
In a third aspect, there is provided a pharmaceutical agent comprising the compound of formula (I) defined above.
There is also provided use of the compound of formula (I) defined above as a pharmaceutical agent.
There is further provided the compound of formula (I) defined above for use as a pharmaceutical agent.
The pharmaceutical agent may be an antiviral agent.
In a fourth aspect, there is provided a viral polymerase inhibitor in particular a HCV polymerase inhibitor such as a NS5B polymerase inhibitor comprising the compound of formula (I) defined above.
There is also provided use of the compound of formula (I) as a viral polymerase inhibitor in particular a HCV polymerase inhibitor such as a NS5B polymerase inhibitor.
There is further provided the compound of formula (I) defined above for use as a viral polymerase inhibitor in particular a HCV polymerase inhibitor such as a NS5B polymerase inhibitor.
The compound of formula (I) may be administered in the form of a pharmaceutical composition together with a pharmaceutically acceptable carrier.
In a fifth aspect, there is provided a pharmaceutical composition comprising the compound of formula (I) and a pharmaceutically acceptable carrier.
According to one embodiment, the pharmaceutical composition additionally comprises a therapeutically effective amount of one or more antiviral agents such as at least one other anti-HCV agent.
In a sixth aspect, there is provided a method for the treatment of a Flaviviridae viral infection such as a HCV infection which comprises administering an effective amount of the compound of formula (I) or the pharmaceutical composition defined above to a subject in need thereof.
There is also provided use of the compound of formula (I) or the pharmaceutical composition as defined above in the manufacture of a medicament for use in the treatment of a Flaviviridae viral infection such as a HCV infection.
There is further provided use of the compound of formula (I) or the pharmaceutical composition as defined above in the treatment of a Flaviviridae viral infection such as a HCV infection.
There is still further provided the compound of the formula (I) or the pharmaceutical composition defined above for use in the treatment of a Flaviviridae viral infection such as a HCV infection.
I n a seventh aspect, there is provided a method of inhibiting the RNA-dependent RNA polymerase activity of the enzyme NS5B, encoded by HCV, comprising exposing the enzyme NS5B to an effective amount of the compound of formula (I) defined above.
I n an eighth aspect, there is provided a method of inhibiting HCV replication comprising exposing a cell infected with HCV to an effective amount of the compound of formula (I) defined above.
DETAILED DESCRIPTION
The present invention relates to compounds of formula (I) which inhibit viral polymerases and are useful in the treatment of Flaviviridae viral infections, particularly, hepatitis C (HCV).
Compounds
The present invention relates to compounds of formula (I), salts, N-oxides, racemates, enantiomers and isomers thereof as defined above.
I n one embodiment of formula (I) Z2 is C-R1 and any one of Zi , Z3 and Z4 is N and the remaining two are C-Ra, preferably CH thereby forming a fused pyridinyl ring.
I n another embodiment of formula (I) Z2 is C-R1 and both Z1 and Z4 are N and Z3 is C-Ra, preferably CH, thereby forming a fused pyrazinyl ring.
I n yet another embodiment of formula (I ) Z2 is C-R1 and both Z1 and Z3 are N and Z4 is C-Ra, preferably CH, thereby forming a fused pyrimidinyl ring.
I n still another embodiment of formula (I) Z2 is C-R1 and Z1 is C-Ra, preferably CH, and Z3 and Z4 are N thereby forming a fused pyridazinyl ring.
I n yet another embodiment of formula (I ) Z2 is C-R1 and Zy , Z3 and Z4 are each C-Ra, preferably CH, thereby forming a fused phenyl ring.
I n one embodiment of formula (I) R2 is an optionally substituted (CH2)maryl or optionally substituted (CH2)mheteroaryl, preferably m is 0, aryl is optionally substituted phenyl more preferably unsubstituted or para-substituted phenyl and heteroaryl is 6- membered heteroaryl containing nitrogen, preferably selected from optionally substituted pyridinyl, pyrazinyl, pyrimidinyl and pyridazinyl more preferably optionally substituted
pyridinyl, even more preferably unsubstituted pyridinyl, most preferably unsubstituted 2- pyridinyl.
In another embodiment, R3 is an optionally substituted (CH2)mC3-8cycloalkyl, an optionally substituted (CH2)mC3-8cycloalkenyl or an optionally substituted (CH2)maryl.
Preferably m is 0 and R3 is phenyl, cyclopentyl, cyclohexyl or cyclohexenyl, preferably cyclohexyl.
In a particularly preferred embodiment R2 is an optionally substituted aryl, preferably phenyl or an optionally substituted heteroaryl, preferably a 6-membered heteroaryl containing nitrogen, preferably selected from optionally substituted pyridinyl, pyrazinyl, pyrimidinyl and pyridazinyl more preferably pyridinyl even more preferably unsubstituted pyridinyl and R3 is an optionally substituted C3-8cycloalkyl or optionally substituted C3.
scycloalkenyl, preferably optionally substituted C3.8cycloalkyl, more preferably cyclohexyl.
In yet another embodiment R1 is (CH2)mC(0)R4, R4 is (CH2)mNR5R6, R5 is H and R6 is selected from H, optionally substituted C1-3alkyl, R9 and [C(R7)(R8)]V(R9) wherein v is 1 . Preferably m in each instance is 0 and R6 is R9 or [C(R7)(R8)]V(R9), most preferably
[C(R7)(R8)]V(R9).
In one embodiment R7 and R8 are independently selected from H, optionally substituted C^alkyl, preferably C1.3alkyl such as methyl, ethyl, propyl and /'so-propyl, most preferably methyl; optionally substituted (CH2)mC3.6cycloalkyl where m is 0 or 1 ; optionally substituted (CH2)m-4-6-membered heterocyclyl where m is 0 or 1 ; optionally substituted (CH2)mphenyl where m is 0 or 1 ; optionally substituted (CH2)m-5-6-membered-heteroaryl where m is 0 or 1 ; the side chain of an amino acid or an alkyl ester thereof; or R7 and R8 together with the atom to which they are attached form an optionally substituted C3.
6cycloalkyl or an optionally substituted 4-6-membered heterocyclyl; or R7 or R8 and R5 together with the adjacent atoms to which they are attached form an optionally substituted 5- 6-membered-heterocyclyl.
In a preferred embodiment R7 and R8 are independently selected from H, optionally substituted methyl, an optionally substituted side chain of an amino acid, or R7 and R8 together with the carbon atom to which they are attached form an optionally substituted C3. 6cycloalkyl, or R7 and R8 together with the carbon atom to which they are attached form an optionally substituted 4-6-membered heterocyclyl containing oxygen and/or nitrogen.
In one embodiment R9 is selected from R10, (CH2)mC(0)-R10, (CH2)mC(0)NR5R1°, (CH2)mS02-R10 and (CH2)mS02NR5R10 where m is 0 or 1 , preferably 0. In a particularly preferred embodiment when R6 is [C(R7)(R8)]V(R9), R9 is (CH2)mC(0)-R10 or
(CH2)mC(0)NR5R10, preferably (CH2)mC(0)NR5R10, most preferably C(0)NHR10.
In one embodiment R is H, OH, -A-(Q)n or 0-A-(Q)n where A is optionally substituted d_3alkyl and n is 0.
In another embodiment R10 is -A-(Q)n where n is 0, 1 or 2, preferably 0 or 1 and A is selected from optionally substituted (CH2)mC3-8cycloalkyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheterocyclyl and optionally substituted (CH2)mheteroaryl, preferably m is 0 or 1 , most preferably 0. Preferably R10 is optionally substituted aryl preferably phenyl, optionally substituted CH2aryl preferably benzyl, optionally substituted heterocyclyl preferably 5- or 6- membered heterocyclyl or optionally substituted heteroaryl, preferably 5-, 6- or 9-membered heteroaryl. Optionally substituted phenyl and optionally substituted 5-, 6- or 9-membered heteroaryls are particularly preferred. Particularly preferred 5-membered heteroaryls contain nitrogen and/or sulphur and include optionally substituted thiazolyl. Particularly preferred 6-membered heteroaryls contain nitrogen and include optionally substituted pyridinyl, optionally substituted pyridazinyl, optionally substituted pyrimidinyl and optionally substituted pyrazinyl. Particularly preferred 9- membered heteroaryls contain nitrogen and/or oxygen and/or sulphur and include optionally substituted benzofuranyl, optionally substituted benzoxazolyl, optionally substituted benzothiazolyl, optionally substituted benzimidazolyl including N-methyl derivatives thereof, optionally substituted indolyl including N-methyl derivatives thereof and optionally substituted pyrazolepyrimidinyl including N-methyl derivatives thereof.
In a particular embodiment when n is 1 , Q is selected from optionally substituted d_
3alkyl, optionally substituted C2.3alkenyl, halo, halod.3alkyl, CHF2, CF3, CN, OH, NH2, NH(d_3alkyl), N(d_3alkyl)2, NHC(0)d_3alkyl, NHS02 C(0)NH2, C(0)NH(Ci_3alkyl),
C(0)N(Ci_3alkyl)2, C(0)NHS02(Ci_3alkyl), C02H, C02Ci.3alkyl, optionally substituted phenyl, optionally substituted heterocyclyl preferably 5- and 6- membered heterocycyl including morpholinyl and pyrrolidinyl and optionally substituted heteroaryl preferably 5-, 6- and 9- membered heteroaryl including tetrazolyl, thiazolyl, pyrazolyl, imidazolyl, pyridinyl, pyrimidinyl, pyridinone and benzodioxalane. Suitable optional substituents in the case of optionally substituted d_3alkyl, optionally substituted C2.3alkenyl, optionally substituted phenyl, optionally substituted heterocyclyl and optionally substituted heteroaryl include d_ 3alkyl (such as methyl, ethyl, propyl), halo (particularly CI), CN, amino (NH2), alkylamino (i.e. NHCi.3alkyl such as NHCH3), dialkylamino (i.e. N(Ci_3alkyl)2 such as N(CH3)2), C02H, esters (such as C02CH3, C02CH2CH3) and amides (such as CONH2, CONHCi_3alkyl e.g.
CONHCH3 and CON(d_3alkyl)2 e.g. CON(CH3)2).
In a further embodiment of formula (I) there is provided a compound of formula (la):
 wherein Zi , Z3, Z4, R1 and R3 are as previously defined;
wherein Zi , Z3, Z4, R1 and R3 are as previously defined;
W is CH or N;
Yi is independently selected from optionally substituted d.6alkyl, optionally substituted C2. 6alkenyl, halo, haloCi_6alkyl, CHF2, OCHF2, CF3, OCF3, CN, OH, optionally substituted d. 6alkoxy including alkoxyaryl such as benzyloxy, N02, NH2, NH(d-6alkyl), N(Ci-6alkyl)2, NHC(0)d_6alkyl, NHS02 C(0)NH2, C(0)NH(Ci-6alkyl), C(0)N(d.6alkyl)2, C(0)NHS02(d_ ealkyl), C(0)N(C1.6alkyl)S02(Ci_6alkyl), S03H, OS02(d_6alkyl), NS02(d_6alkyl), S02(Ci_ ealkyl), S02NH2, S02NH(d_6alkyl), S02N(d_6alkyl)2, C02H, C(0)d_6alkyl, C02d_6alkyl, optionally substituted (CH2)mC3-8cycloalkyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheterocyclyl and optionally substituted (CH2)mheteroaryl; and
n is independently 0, 1 , 2, 3, 4 or 5, preferably 0, 1 , 2 or 3, more preferably 0, 1 or 2, most preferably 0 or 1.
In one embodiment Y1 is selected from optionally substituted d_6alkyl, halo, OH, optionally substituted d.6alkoxy and alkoxyaryl. Preferably Y1 is selected from F, CI, Br, I, CH3, CHF2, CF3, CH2CH3, CH2CH2CH3, OH, OCH3, OCH2-phenyl (benzyloxy), OCHF2 and OCF3, even more preferably F, OH, OCH3, OCHF2 and OCH2phenyl.
In a further preferred embodiment of formula (la) there is provided a compound of formula (la-i):

(la-i)
wherein Zi , Z3, Z4, R1, W and n are as previously defined; and
Y2 is independently selected from optionally substituted d.6alkyl, optionally substituted C2. 6alkenyl, halo, haloCi.6alkyl, CHF2, OCHF2, CF3, OCF3, CN, OH, optionally substituted d_ 6alkoxy, N02, NH2, NH(d_6alkyl), N(d_6alkyl)2, NHC(0)d_6alkyl, NHS02 C(0)NH2, C(0)NH(d.6alkyl), C(0)N(d_6alkyl)2, C(0)NHS02(d_6alkyl), C(0)N(d.6alkyl)S02(d_6alkyl),
SO3H, OSOziCi-ealkyl), NSO^C^alkyl), S02(d_6alkyl), S02NH2, SOzNHid^alkyl), S02N(d_ 6alkyl)2, C02H, C(0)Ci.6alkyl, C02Ci.6alkyl, optionally substituted (CH2)mC3-8cycloalkyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheterocyclyl and optionally substituted (CH2)mheteroaryl. Preferably Y2 is absent.
In another embodiment of formula (I) there is provided a compound of formula (lb):

wherein Zi , Z3, Z4, R2, R3 and R4 are as previously defined.
In a further embodiment of formula (lb) there is provided a compound of formula (Ib-i):

(Ib-i)
wherein Zi , Z3, Z4, R2, R3 and R5 are as previously defined.
In a preferred embodiment of formula (Ib-i), R2 is an unsubstituted 6-membered heteroaryl containing nitrogen, preferably pyridinyl, more preferably 2-pyridinyl or phenyl optionally substituted with (Yi)n wherein and n are as defined for formula (la). In still a further preferred embodiment, R3 is cyclohexyl optionally substituted with (Y2)n wherein Y2 and n are as defined for formula (la-i), preferably R3 is unsubstituted cyclohexyl.
In another embodiment of formula (lb) there is provided a compound of formula (Ib-ii):
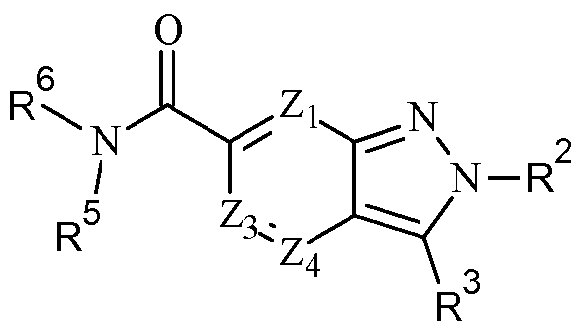
(Ib-ii)
wherein Z^ Z3, Z4, R2, R3, R5 and R6 are as previously defined.
In a preferred embodiment of formula (Ib-ii), R2 is an unsubstituted 6-membered heteroaryl containing nitrogen, preferably pyridinyl, more preferably 2-pyridinyl or phenyl optionally substituted with (Yi)n wherein and n are as defined for formula (la). In still a further preferred embodiment, R3 is cyclohexyl optionally substituted with (Y2)n wherein Y2 and n are as defined for formula (la-i), preferably R3 is unsubstituted cyclohexyl.
In another embodiment of formula (lb) there is provided a compound of formula
(Ib-iii):

(Ib-iii)
wherein Zi , Z3, Z4, R2, R3, R5, R7, R8 and R9 are as previously defined and each m is independently 0, 1 , 2 or 3, preferably 0, 1 or 2, more preferably 0 or 1 , most preferably 0. In a preferred embodiment of formula (Ib-iii), R2 is an unsubstituted 6-membered heteroaryl containing nitrogen, preferably pyridinyl, more preferably 2-pyridinyl or phenyl optionally substituted with (Yi)n wherein Y1 and n are as defined for formula (la). In still a further preferred embodiment, R3 is cyclohexyl optionally substituted with (Y2)n wherein Y2 and n are as defined for formula (la-i), preferably R3 is unsubstituted cyclohexyl.
In a particularly preferred embodiment of formula (Ib-iii) there is provided a compound of formula (Ib-iv):

(Ib-iv)
wherein Zi , Z3, Z4, R2, R3, R7, R8, m, n and Q are as previously defined; and
Wi, W2, W3, W4 and W5 are each independently selected from CH and N or together with an adjacent ring member join to form a fused 5-membered heterocyclic moiety.
In one embodiment W2, W3, W4 and W5 are each CH thereby forming an optionally substituted phenyl.
In another embodiment any one of W2, W3, W4 and W5 is N and the remaining are CH thereby forming an optionally substituted pyridinyl.
In yet another embodiment any two of W, , W2, W3, W4 and W5 are N and the remaining are CH thereby forming an optionally substituted pyrazinyl, pyrimidinyl or pyridazinyl.
In another embodiment W2, W3, W4 and W5 are each CH wherein any two adjacent ring members join to form a fused 5-membered heterocyclic moiety. In a particular embodiment the fused 5-membered heterocyclic moiety is selected from furan to form an optionally substituted benzofuranyl, oxazole to form an optionally substituted benzoxazolyl, pyrrole or N-methyl pyrrole to form an optionally substituted indolyl or N-methyl indolyl and imidazole or N-methyl imidazole to form an optionally substituted benzimidazolyl or N-methyl benzimidazolyl.
In still another embodiment any two of W2, W3, W4 and W5 are N and the remaining are CH wherein any two adjacent ring members join to form a fused 5-membered heterocyclic moiety. In a particular embodiment the fused 5-membered heterocyclic moiety is pyrazole thereby forming an optionally substituted pyrazolopyrimidinyl.
In a further embodiment of formula (Ib-iv), R2 is an unsubstituted 6-membered heteroaryl containing nitrogen, preferably pyridinyl, more preferably 2-pyridinyl or phenyl optionally substituted with (Yi)n wherein and n are as defined for formula (la). In still a further preferred embodiment, R3 is cyclohexyl optionally substituted with (Y2)n where Y2 and n are as defined for formula (la-i).
In yet another embodiment R7 and R8 are each independently H, optionally substituted C^alkyl (particularly optionally substituted methyl such as CH3, CH2OH, CH2amino including CH2NHCH3 and CH2N(CH3)2 and optionally substituted CH2heteroaryl (particularly 5-membered heteroaryls such as optionally substituted CH2thiazolyl and CH2imidazolyl) or the optionally substituted side chain of an L- or D- amino acid (such as Glycine (-H), Alanine (-CH3), Valine (-CH(CH3)2), Serine (-CH2OH), Tryptophan (-CH2-(3-1 H-indolyl)), Histidine (-CH2-(5-1 H-imidazolyl)), particularly Alanine (methyl) wherein optional substituents include but are not limited to OH (such as Tryptophan-OH), benzyl (such as N- benzyl Histidine) and benzyloxy (such as N-benzyloxy Histidine)).
In still another embodiment R7 and R8 together with the carbon atom to which they are attached form an optionally substituted C3.8cycloalkyl, preferably C3.6cycloalkyl such as cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl, most preferably cyclobutyl. Suitable optional substituents include but are not limited to halo such as F, OH, =0, benzyloxy and NHCH2CH20H.
In yet another embodiment R7 and R8 together with the carbon atom to which they are attached form an optionally substituted C3.8heterocyclyl, preferably a 4-6-membered heterocyclyl such as azetidinyl, pyrrolidinyl, piperidinyl, tetrahydrofuranyl and
tetrahydropyranyl. Suitable optional substituents for the heterocycles containing nitrogen include methyl, such as N-methyl.
In still another embodiment R7 or R8 and R5 together with the adjacent atoms to which they are attached form an optionally substituted 5-6-membered-heterocyclyl containing nitrogen such as pyrrolidine and piperidine.
In still another embodiment Q is selected from halo (CI, Br, F, I preferably F and I , most preferably F), CF3, CN, C02R5, CON(R5)2, CON(R5)S02R5, N(R5)2, optionally substituted aryl such as phenyl, optionally substituted 5-6-membered heteroaryl such as pyridyl, pyridinone, pyrimidinyl, pyrazolyl, thiazolyl and tetrazolyl, optionally substituted 9- membered heteroaryl such as benzodioxalane, optionally substituted C 6alkyl (preferably Ci.3alkyl), and optionally substituted C2-6alkenyl (preferably C2.3alkenyl). Preferably R5 in each instance is independently H or d.3alkyl (preferably methyl or ethyl, more preferably methyl). Suitable optional substituents include but are not limited to halo, N(R5)2, alkylhalo such as CF3, CN, C02R5, CON(R5)2, CON(R5)S02R5 and Ci_3alkyl preferably methyl. More particularly, suitable optional substituents for aryl and heteroaryl include halo, N(R5)2,
C02R5, and C^alkyl preferably methyl such as N-methyl and suitable optional substituents for Ci_6alkyl and C2.6alkenyl include halo, alkyl halo such as CF3, CN, C02R5, CON(R5)2, and CON(R5)S02R5.
The term "Chalky!" refers to optionally substituted straight chain or branched chain hydrocarbon groups having from 1 to 6 carbon atoms. Examples include methyl, ethyl, propyl, /sopropyl, butyl, /'sobutyl, sec-butyl, ferf-butyl, pentyl, neopentyl and hexyl. "C^alkyl" including methyl, ethyl, propyl and /sopropyl is preferred with methyl being particularly preferred.
The term "C2.6alkenyl" refers to optionally substituted straight chain or branched chain hydrocarbon groups having at least one double bond of either E or Z stereochemistry where applicable and 2 to 6 carbon atoms. Examples include vinyl, 1 -propenyl, 1- and 2- butenyl and 2-methyl-2-propenyl. "C2.3alkenyl" including ethenyl and propenyl is preferred with ethenyl being particularly preferred.
The term "C3.8cycloalkyl" refers to non-aromatic cyclic groups having from 3 to 8 carbon atoms, including cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl. C^cycloalkyl such as cyclobutyl, cyclopentyl and cyclohexyl are preferred.
The term "C3.8cycloalkenyl" refers to an unsaturated C3.8cycloalkyl having at least one double bond such as cyclohexenyl.
The term "C^alkoxy" refers to an alkyl group as defined above covalently bound via an O linkage containing 1 to 6 carbon atoms, such as methoxy, ethoxy, propoxy, isoproxy,
butoxy, tert-butoxy and pentoxy. "C^alkoxy" including methoxy, ethoxy and propoxy is preferred with methoxy being particularly preferred.
The term "aryl" refers to a carbocyclic (non-heterocyclic) aromatic ring or mono-, bi- or tri-cyclic ring system. The aromatic ring or ring system is generally composed of 6 to 10 carbon atoms. Examples of aryl groups include but are not limited to phenyl, biphenyl, naphthyl and tetrahydronaphthyl. 6-membered aryls such as phenyl are preferred. The term "alkylaryl" refers to C^alkylaryl such as benzyl. The term "alkoxyaryl" refers to
6alkyloxyaryl such as benzyloxy.
The term "heterocyclyl", refers to a monovalent moiety obtained by removing a hydrogen atom from a ring atom of a heterocyclic compound, which moiety has from 3 to 7 ring atoms (unless otherwise specified), of which from 1 to 4 are ring heteroatoms.
Preferably, each ring has from 3 to 7 ring atoms, of which from 1 to 4 are ring heteroatoms.
In this context, the prefixes (e.g. C3.7, C5.6, etc.) denote the number of ring atoms, or range of number of ring atoms, whether carbon atoms or heteroatoms. For example, the term "C5_6heterocyclyl", as used herein, pertains to a heterocyclyl group having 5 or 6 ring atoms, that is, a 5- or 6- membered heterocyclyl group. Examples of groups of heterocyclyl groups include C3.7 heterocyclyl, C5.7 heterocyclyl, and C5-6 heterocyclyl.
Examples of monocyclic heterocyclyl groups include, but are not limited to, those containing one nitrogen atom such as aziridine (3-membered ring), azetidine (4-membered ring), pyrrolidine (tetrahydropyrrole), pyrroline (e.g., 3-pyrroline, 2,5-dihydropyrrole), 2H- pyrrole or 3H-pyrrole (isopyrrole, isoazole) (5-membered rings) , piperidine, dihydropyridine, tetrahydropyridine (6-membered rings), azepine (7-membered ring); those containing two nitrogen atoms such as imidazoline, pyrazolidine (diazolidine), imidazoline, pyrazoline (dihydropyrazole) (5-membered rings), piperazine (6-membered ring); those containing one oxygen atom such as oxirane (3-membered ring), oxetane (4-membered ring), oxolane (tetrahydrofuran), oxole (dihydrofuran) (5-membered rings), oxane (tetrahydropyran), dihydropyran, pyran (6-membered rings), oxepin (7-membered ring); those containing two oxygen atoms such as dioxolane (5-membered ring), dioxane (6-membered ring), and dioxepane (7-membered ring); those containing three oxygen atoms such as trioxane (6- membered ring); those containing one sulfur atom such as thiirane (3-membered ring), thietane (4-membered ring), thiolane (tetrahydrothiophene) (5-membered ring), thiane (tetrahydrothiopyran) (6-membered ring), thiepane (7-membered ring); those containing one nitrogen and one oxygen atom such as tetrahydrooxazole, dihydrooxazole,
tetrahydroisoxazole, dihydroisoxazole (5-membered rings), morpholine, tetrahydrooxazine, dihydrooxazine, oxazine (6-membered rings); those containing one nitrogen and one sulfur atom such as thiazoline, thiazolidine (5-membered rings), thiomorpholine (6-membered
ring); those containing two nitrogen and one oxygen atom such as oxadiazine (6-membered ring); those containing one oxygen and one sulfur such as: oxathiole (5-membered ring) and oxathiane (thioxane) (6-membered ring); and those containing one nitrogen, one oxygen and one sulfur atom such as oxathiazine (6-membered ring).
The term "heteroaryl" is used herein to denote a heterocyclic group having aromatic character and embraces aromatic monocyclic ring systems and polycyclic (e.g. bicyclic) ring systems containing one or more aromatic rings. The term covers polycyclic ring systems in which all of the fused rings are aromatic as well as ring systems where one or more rings are non-aromatic, provided that at least one ring is aromatic. In polycyclic systems containing both aromatic and non-aromatic rings fused together, the group may be attached to another moiety by the aromatic ring, or by a non-aromatic ring.
Examples of heteroaryl groups are monocyclic and bicyclic groups containing from five to ten ring members. The heteroaryl group can be, for example, a five membered or six membered monocyclic ring or a bicyclic structure formed from fused five and six membered rings or two fused six membered rings or two fused five membered rings. Each ring may contain up to about four heteroatoms typically selected from nitrogen, sulphur and oxygen. The heteroaryl ring will contain up to 4 heteroatoms, more typically up to 3 heteroatoms, more usually up to 2, for example a single heteroatom. In one embodiment, the heteroaryl ring contains at least one ring nitrogen atom. The nitrogen atoms in the heteroaryl rings can be basic, as in the case of an imidazole or pyridine, or essentially non-basic as in the case of an indole or pyrrole nitrogen. In general the number of basic nitrogen atoms present in the heteroaryl group, including any amino group substituents of the ring, will be less than five.
Examples of 5 membered heteroaryl groups include but are not limited to pyrrole, furan, imidazole, furazan, oxazole, oxadiazole, oxatriazole, isoxazole, thiazole, isothiazole, pyrazole, triazole and tetrazole groups.
Examples of 6 membered heteroaryl groups include but are not limited to pyridine, pyrazine, pyridazine, pyrimidine, triazine, pyran, oxazine, dioxine, thiazine and thiadiazine.
A bicyclic heteroaryl group may be, for example, a group selected from: a) a benzene ring fused to a 5- or 6-membered ring containing 1 , 2 or 3 ring heteroatoms; b) a pyridine ring fused to a 5- or 6-membered ring containing 1 , 2 or 3 ring heteroatoms; c) a pyrimidine ring fused to a 5- or 6-membered ring containing 1 or 2 ring heteroatoms; d) a pyrrole ring fused to a 5- or 6-membered ring containing 1 , 2 or 3 ring heteroatoms; e) a pyrazole ring fused to a 5- or 6-membered ring containing 1 or 2 ring heteroatoms; f) an imidazole ring fused to a 5- or 6-membered ring containing 1 or 2 ring heteroatoms; g) an oxazole ring fused to a 5- or 6-membered ring containing 1 or 2 ring heteroatoms; h) an isoxazole ring fused to a 5- or 6-membered ring containing 1 or 2 ring heteroatoms; i) a thiazole ring fused
to a 5- or 6-membered ring containing 1 or 2 ring heteroatoms; j) an isothiazole ring fused to a 5- or 6-membered ring containing 1 or 2 ring heteroatoms; k) a thiophene ring fused to a 5- or 6-membered ring containing 1 , 2 or 3 ring heteroatoms; I) a furan ring fused to a 5- or 6- membered ring containing 1 , 2 or 3 ring heteroatoms; m) a cyclohexyl ring fused to a 5- or 6- membered ring containing 1 , 2 or 3 ring heteroatoms; and n) a cyclopentyl ring fused to a 5- or 6-membered ring containing 1 , 2 or 3 ring heteroatoms.
Particular examples of bicyclic heteroaryl groups containing a five membered ring fused to another five membered ring include but are not limited to imidazothiazole (e.g. imidazo[2, 1-b]thiazole) and imidazoimidazole (e.g. imidazo[1 ,2-a]imidazole).
Particular examples of bicyclic heteroaryl groups containing a six membered ring fused to a five membered ring include but are not limited to benzofuran, benzothiophene, benzimidazole, benzoxazole, isobenzoxazole, benzisoxazole, benzothiazole,
benzisothiazole, isobenzofuran, indole, isoindole, indolizine, indoiine, isoindoline, purine (e.g., adenine, guanine), indazole, pyrazolopyrimidine (e.g. pyrazolo[1 ,5-a]pyrimidine), benzodioxole and pyrazolopyridine (e.g. pyrazolo[1 ,5-a]pyridine) groups. A further example of a six membered ring fused to a five membered ring is a pyrrolopyridine group such as a pyrrolo[2,3-b]pyridine group.
Particular examples of bicyclic heteroaryl groups containing two fused six membered rings include but are not limited to quinoline, isoquinoline, chroman, thiochroman, chromene, isochromene, isochroman, benzodioxan, quinolizine, benzoxazine, benzodiazine, pyridopyridine, quinoxaline, quinazoline, cinnoline, phthalazine, naphthyridine and pteridine groups.
Examples of heteroaryl groups containing an aromatic ring and a non-aromatic ring include tetrahydronaphthalene, tetrahydroisoquinoline, tetrahydroquinoline,
dihydrobenzothiophene, dihydrobenzofuran, 2,3-dihydro- benzo[1 ,4]dioxine,
benzo[1 ,3]dioxole, 4,5,6,7-tetrahydrobenzofuran, indoiine, isoindoline and indane groups.
The term "side chain of an amino acid" refers to any side chain that may be present in natural (L-) or unnatural (D-) amino acids. Examples of amino acid side chain moieties derived from natural amino acids, with the amino acids from which they are derived shown in brackets, are -H (Glycine), -CH3 (Alanine), -CH(CH3)2 (Valine), -CH2CH(CH3)2 (Leucine), - CH(CH3)CH2CH3 (Isoleucine), -(CH2)4NH2 (Lysine), -(CH2)3NHC(=NH)NH2 (Arginine), -CH2- (5-1 H-imidazolyl) (Histidine), -CH2CONH2 (Asparagine), -CH2CH2CONH2 (Glutamine), - CH2COOH (Aspartic acid), -CH2CH2COOH (Glutamic acid), -CH2-phenyl (Phenylalanine), - CH2-(4-OH-phenyl) (Tyrosine), -CH2-(3-1 H-indolyl) (Tryptophan), -CH2SH (Cysteine), - CH2CH2SCH3 (Methioine), -CH2OH (Serine), -CH(OH)CH3 (Threonine) and the cyclic side chain pyrrolidinyl (Proline) whereby the covalent bond between the nitrogen and carbon in
the pyrrolidinyl ring forms the backbone. Examples of amino acid side chain moieties derived from unnatural amino acids, with the amino acids from which they are derived shown in brackets, are -(CH2)2-C(0)-0-C(CH3)3 (glutamic acid t- butyl ester), -(CH2)4-NH-C(0)-0- C(CH3)3 (Ne-(tert-butoxycarbonyl)-lysine), -(CH2)3-NH-C(0)NH2 (citrulline), -CH2-CH2OH (homoserine) and -(CH2)2-CH2NH2 (ornithine). Examples can also include alkyl, alkenyl, alkynyl, aryl, saturated and unsaturated heterocycles (functionalized and unfunctionalized). The term 'amino-acid side chain moiety' can also include a number of unnatural amide and sulfonamide, aryl and heteroaryl side chains.
The term "halo" refers to fluoro, chloro, bromo or iodo.
The term "R7 and R8 together with the atom or adjacent atoms to which they are attached form an optionally substituted C3.8 cycloalkyl or an optionally substituted C3.8 heterocyclyl" refers to either R7 and R8 being on the same atom and joining together to form optionally substituted C3.8 cycloalkyl or an optionally substituted C3.8 heterocyclyl or R7 and R8 being on separate adjacent atoms and joining together to form optionally substituted C3.8 cycloalkyl or an optionally substituted C3.8 heterocyclyl. When R7 and R8 are on separate adjacent atoms, it will be appreciated that these atoms may be the same (e.g. two carbon atoms) or different (e.g. a carbon atom and a nitrogen atom).
The term "optionally substituted" refers to a group that may or may not be further substituted with one or more groups selected from Ci_6 alkyl, C3.8cycloalkyl, C2.6alkenyl, C2. 6alkynyl, aryl, CF3, OCF3, CHF2, OCHF2, heterocyclyl, heteroaryl, halo, haloCi_6alkyl, haloC3. 6cycloalkyl, haloC2.6alkenyl, haloC2.6alkynyl, haloaryl, haloheterocyclyl, haloheteroaryl, hydroxy, C^alkylOH, C2.6alkenylOH, C2.6alkynylOH, Ci_6 alkoxy, C2.6alkenyloxy, C2.
6alkynyloxy, aryloxy, heterocyclyloxy, heteroaryloxy, carboxy, C^alkylcarboxy, haloCi. 6alkoxy, haloC2.6alkenyloxy, haloC2.6alkynyloxy, haloaryloxy, haloheterocyclyloxy, haloheteroaryloxy, nitro, nitroC^, alkyl, nitroC2.6alkenyl, nitroaryl, nitroheterocyclyl, nitroheteroaryl, azido, amino, C1.6alkylamino, C^dialkylamino, C2.6alkenylamino, C2.
6alkynylamino, arylamino, heterocyclamino, heteroarylamino, NH(OH) acyl, C^alkylacyl, C2. 6alkenylacyl, C2.6alkynylacyl, arylacyl, heterocyclylacyl, heteroarylacyl, acylamino, acylCi. 6alkylamino, acylC^dialkylamino, acyloxy, acyloxyC^alkyl, acylC^alkoxy, aldehydo, Ci_ 6alkylsulphonyl, arylsulphonyl, C^alkylsulphonylamino, arylsulphonylamino, Ci.
6alkylsulphonyloxy, arylsulphonyloxy, C1.6alkylsulphenyl, C2.6alklysulphenyl, arylsulphenyl, carboalkoxy, carboaryloxy, mercapto, Ci_6alkylthio, arylthio, acylthio, cyano and the like. Preferred optional substituents include but are not limited to C^alkyl, C2.3alkenyl, aryl (such as phenyl), haloalkyl (such as CHF2 and CF3), heterocyclyl (such as 5- and 6-membered heterocyclyl), heteroaryl (such as 5-, 6- and 9-membered heteroaryl), cyano, halo, hydroxy, C^alkoxy, C^alkoxyOH, C^alkoxyhalo (such as OCHF2 and OCF3), =0, carboxy (such as
C02H), C^alkylcarboxy, acyloxyC^alkyl (such as CiO C^alkyl), S02Ci_3alkyl, S02aryl, alkylcarbamates (such as t-butylcarbamate), amino (NH2), C1.3alkylamino (such as NHCi_ 3alkyl), d-sdialkylamino (such as N(Ci-3alkyl)2, NH(OH), acylamino (such as C(0)NH2) and acylC^aalkylamino (such as C(0)NHCi_3alkyl and C(0)N(Ci-3alkyl)2) wherein each optional substituent may be further optionally substituted.
The compounds of the invention may also be prepared as salts which are pharmaceutically acceptable, but it will be appreciated that non-pharmaceutically acceptable salts also fall within the scope of the present invention, since these are useful as
intermediates in the preparation of pharmaceutically acceptable salts. Examples of pharmaceutically acceptable salts include salts of pharmaceutically acceptable cations such as sodium, potassium, lithium, calcium, magnesium, ammonium and alkylammonium; acid addition salts of pharmaceutically acceptable inorganic acids such as hydrochloric, orthophosphoric, sulfuric, phosphoric, nitric, carbonic, boric, sulfamic and hydrobromic acids; or salts of pharmaceutically acceptable organic acids such as acetic, propionic, butyric, tartaric, maleic, hydroxymaleic, fumaric, citric, lactic, mucic, gluconic, benzoic, succinic, oxalic, phenylacetic, methanesulfonic, trihalomethanesulfonic, toluenesulfonic,
benzenesulfonic, isethionic, salicylic, sulphanilic, aspartic, glutamic, edetic, stearic, palmitic, oleic, lauric, pantothenic, tannic, ascorbic, valeric and orotic acids. Salts of amine groups may also comprise quaternary ammonium salts in which the amino nitrogen atom carries a suitable organic group such as an alkyl, alkenyl, alkynyl or aralkyl moiety.
The salts may be formed by conventional means, such as by reacting the free base form of the compound with one or more equivalents of the appropriate acid.
It should be understood that a reference to a pharmaceutically acceptable salt includes the solvent addition forms or crystal forms thereof, particularly solvates or polymorphs. Solvates contain either stoichiometric or non-stoichiometric amounts of a solvent, and may be formed during the process of crystallization with pharmaceutically acceptable solvents such as water, alcohols such as methanol, ethanol or isopropyl alcohol, DMSO, acetonitrile, dimethyl formamide (DMF) and the like with the solvate forming part of the crystal lattice by either non-covalent binding or by occupying a hole in the crystal lattice. Hydrates are formed when the solvent is water, alcoholates are formed when the solvent is alcohol. Solvates of the compounds of the present invention can be conveniently prepared or formed during the processes described herein. In addition, the compounds of the present invention can exist in unsolvated as well as solvated forms. In general, the solvated forms are considered equivalent to the unsolvated forms for the purposes of the compounds and methods provided herein.
Additionally, the compounds of the present invention can exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like. The solvated forms of the compounds of the present invention are also considered to be disclosed herein.
It will be understood that compounds of formula (I) may possess a chiral centre and may therefore exist as an isomer such as a racemate or an R- or S- enantiomer. The compounds may therefore be used as a purified enantiomer or diastereomer, or as a mixture of any ratio thereof.
This invention also encompasses prodrugs of the compounds of formula (I).
The term "pro-drug" is used herein in its broadest sense to include those compounds which are converted in vivo to the compound of formula (I). Use of the prodrug strategy optimises the delivery of the drug to its site of action. In one embodiment, compounds of formula (I) having free amino, amido, hydroxyl, or carboxylic acid groups can be converted into prodrugs. Prodrugs include compounds wherein carbonates, carbamates, amide and alkyl esters which are covalently bonded to the above substituents of compounds of the present invention through a carbonyl carbon prodrug sidechain. Prodrugs may also include N-oxides of ring nitrogen atoms in formula (I).
Viral Polymerase Inhibition
The ability of the compounds of formula I to inhibit RNA synthesis by the RNA dependent RNA polymerase of HCV (NS5B) can be demonstrated by any assay capable of measuring RNA dependent RNA polymerase activity. A suitable assay is described in the examples.
While the invention is described with particular reference to compounds having inhibitory activity against a HCV NS5B polymerase, it will be understood that other polymerases can, if desired, be substituted in whole or in part for the HCV polymerase herein described. For example, one microbial polymerase target is HCV NS5B polymerase which is the viral RNA-dependent RNA polymerase (RdRp) that is responsible for viral replications. HCV NS5B protein, is released from a polyprotein and is involved in the synthesis of double-stranded RNA from a single-stranded viral RNA genome. It is believed that the replication and/or reproduction of HCV virus may be inhibited or prevented through the inhibition of NS5B polymerase and suppress or prevent the formation of the double- stranded HCV RNA.
To demonstrate that the compounds of formula (I) act by specific inhibition of NS5B polymerase, the compounds may be tested for the lack of inhibitory activity in an assay measuring the activity of an RNA-dependent RNA polymerase other than HCV polymerase or in a DNA dependent RNA polymerase assay.
Pharmaceutical Compositions
The invention also provides a pharmaceutical composition comprising a compound of formula (I) and a pharmaceutically acceptable carrier.
The pharmaceutical composition may further comprise or be administered in combination with one or more other antiviral agents such as ribavirin, an antiviral nucleoside, polymerase inhibitor, protease inhibitor and/or inhibitor of viral entry, assembly or egress. The composition may also additionally comprise at least one immunomodulatory agent for example an interferon or interferon derivative and/or an inhibitor of inosine-5'- monophosphate dehydrogenase (IMPDH).
The term "composition" is intended to include the formulation of an active ingredient with conventional carriers and excipients, and also with encapsulating materials as the carrier, to give a capsule in which the active ingredient (with or without other carriers) is surrounded by the encapsulation carrier. Any carrier must be "pharmaceutically acceptable" meaning that it is compatible with the other ingredients of the composition and is not deleterious to a subject. The compositions of the present invention may contain other therapeutic agents as described above, and may be formulated, for example, by employing conventional solid or liquid vehicles or diluents, as well as pharmaceutical additives of a type appropriate to the mode of desired administration (for example, excipients, binders, preservatives, stabilizers, flavours, etc.) according to techniques such as those well known in the art of pharmaceutical formulation (See, for example, Remington: The Science and Practice of Pharmacy, 21st Ed. , 2005, Lippincott Williams & Wilkins).
The pharmaceutical composition includes those suitable for oral, rectal, nasal, topical (including buccal and sub-lingual), vaginal or parenteral (including intramuscular, sub-cutaneous and intravenous) administration or in a form suitable for administration by inhalation or insufflation.
The compounds of the invention, together with a conventional adjuvant, carrier, or diluent, may thus be placed into the form of pharmaceutical compositions and unit dosages thereof, and in such form may be employed as solids, such as tablets or filled capsules, or liquids such as solutions, suspensions, emulsions, elixirs, or capsules filled with the same, all for oral use, in the form of suppositories for rectal administration ; or in the form of sterile injectable solutions for parenteral (including subcutaneous) use.
Such pharmaceutical compositions and unit dosage forms thereof may comprise conventional ingredients in conventional proportions, with or without additional active compounds or principles, and such unit dosage forms may contain any suitable effective amount of the active ingredient commensurate with the intended daily dosage range to be employed.
For preparing pharmaceutical compositions from the compounds of the present invention, pharmaceutically acceptable carriers can be either solid or liquid. Solid form preparations include powders, tablets, pills, capsules, cachets, suppositories, and dispensable granules. A solid carrier can be one or more substances which may also act as diluents, flavouring agents, solubilisers, lubricants, suspending agents, binders, preservatives, tablet disintegrating agents, or an encapsulating material.
Suitable carriers are magnesium carbonate, magnesium stearate, talc, sugar, lactose, pectin, dextrin, starch, gelatin, tragacanth, methylcellulose, sodium
carboxymethylcellulose, a low melting wax, cocoa butter, and the like. The term
"preparation" is intended to include the formulation of the active compound with
encapsulating material as carrier providing a capsule in which the active component, with or without carriers, is surrounded by a carrier, which is thus in association with it. Similarly, cachets and lozenges are included. Tablets, powders, capsules, pills, cachets, and lozenges can be used as solid forms suitable for oral administration.
Liquid form preparations include solutions, suspensions, and emulsions, for example, water or water-propylene glycol solutions. For example, parenteral injection liquid preparations can be formulated as solutions in aqueous polyethylene glycol solution.
Sterile liquid form compositions include sterile solutions, suspensions, emulsions, syrups and elixirs. The active ingredient can be dissolved or suspended in a
pharmaceutically acceptable carrier, such as sterile water, sterile organic solvent or a mixture of both.
The compositions according to the present invention may thus be formulated for parenteral administration (e. g. by injection, for example bolus injection or continuous infusion) and may be presented in unit dose form in ampoules, pre-filled syringes, small volume infusion or in multi-dose containers with an added preservative. The compositions may take such forms as suspensions, solutions, or emulsions in oily or aqueous vehicles, and may contain formulation agents such as suspending, stabilising and/or dispersing agents. Alternatively, the active ingredient may be in powder form, obtained by aseptic isolation of sterile solid or by lyophilisation from solution, for constitution with a suitable vehicle, e.g. sterile, pyrogen-free water, before use.
Pharmaceutical forms suitable for injectable use include sterile injectable solutions or dispersions, and sterile powders for the extemporaneous preparation of sterile injectable solutions. They should be stable under the conditions of manufacture and storage and may be preserved against oxidation and the contaminating action of microorganisms such as bacteria or fungi.
The solvent or dispersion medium for the injectable solution or dispersion may
contain any of the conventional solvent or carrier systems for the compounds, and may contain, for example, water, ethanol, polyol (for example, glycerol, propylene glycol and liquid polyethylene glycol, and the like), suitable mixtures thereof, and vegetable oils.
Pharmaceutical forms suitable for injectable use may be delivered by any appropriate route including intravenous, intramuscular, intracerebral, intrathecal, epidural injection or infusion.
Sterile injectable solutions are prepared by incorporating the active compounds in the required amount in the appropriate solvent with various other ingredients such as these enumerated above, as required, followed by filtered sterilization. Generally, dispersions are prepared by incorporating the various sterilised active ingredient into a sterile vehicle which contains the basic dispersion medium and the required other ingredients from those enumerated above. In the case of sterile powders for the preparation of sterile injectable solutions, preferred methods of preparation are vacuum drying or freeze-drying of a previously sterile-filtered solution of the active ingredient plus any additional desired ingredients.
When the active ingredients are suitably protected they may be orally administered, for example, with an inert diluent or with an assimilable edible carrier, or it may be enclosed in hard or soft shell gelatin capsule, or it may be compressed into tablets, or it may be incorporated directly with the food of the diet. For oral therapeutic administration, the active compound may be incorporated with excipients and used in the form of ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups, wafers, and the like.
The amount of active compound in therapeutically useful compositions should be sufficient that a suitable dosage will be obtained.
The tablets, troches, pills, capsules and the like may also contain the components as listed hereafter: a binder such as gum, acacia, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid and the like; a lubricant such as magnesium stearate; and a sweetening agent such a sucrose, lactose or saccharin may be added or a flavouring agent such as peppermint, oil of wintergreen, or cherry flavouring. When the dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier.
Various other materials may be present as coatings or to otherwise modify the physical form of the dosage unit. For instance, tablets, pills, or capsules may be coated with shellac, sugar or both. A syrup or elixir may contain the active compound, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and flavouring such as cherry or orange flavour. Of course, any material used in preparing any dosage unit form should be pharmaceutically pure and substantially non-toxic in the amounts employed. In
addition, the active compound (s) may be incorporated into sustained-release preparations and formulations, including those that allow specific delivery of the active peptide to specific regions of the gut.
Aqueous solutions suitable for oral use can be prepared by dissolving the active component in water and adding suitable colorants, flavours, stabilising and thickening agents, as desired. Aqueous suspensions suitable for oral use can be made by dispersing the finely divided active component in water with viscous material, such as natural or synthetic gums, resins, methylcellulose, sodium carboxymethylcellulose, or other well known suspending agents.
Pharmaceutically acceptable carriers and/or diluents include any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents and the like.
Also included are solid form preparations that are intended to be converted, shortly before use, to liquid form preparations for oral administration. Such liquid forms include solutions, suspensions, and emulsions. These preparations may contain, in addition to the active component, colorants, flavours, stabilisers, buffers, artificial and natural sweeteners, dispersants, thickeners, solubilising agents, and the like.
For topical administration to the epidermis the compounds according to the invention may be formulated as ointments, creams or lotions, or as a transdermal patch. Ointments and creams may, for example, be formulated with an aqueous or oily base with the addition of suitable thickening and/or gelling agents. Lotions may be formulated with an aqueous or oily base and will in general also contain one or more emulsifying agents, stabilising agents, dispersing agents, suspending agents, thickening agents, or colouring agents.
Formulations suitable for topical administration in the mouth include lozenges comprising active agent in a flavoured base, usually sucrose and acacia or tragacanth; pastilles comprising the active ingredient in an inert base such as gelatin and glycerin or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier.
Solutions or suspensions are applied directly to the nasal cavity by conventional means, for example with a dropper, pipette or spray. The formulations may be provided in single or multidose form. In the latter case of a dropper or pipette, this may be achieved by the patient administering an appropriate, predetermined volume of the solution or suspension.
In the case of a spray, this may be achieved for example by means of a metering atomising spray pump. To improve nasal delivery and retention the compounds according to
the invention may be encapsulated with cyclodextrins, or formulated with other agents expected to enhance delivery and retention in the nasal mucosa.
Administration to the respiratory tract may also be achieved by means of an aerosol formulation in which the active ingredient is provided in a pressurised pack with a suitable propellant such as a chlorofluorocarbon (CFC) for example dichlorodifluoromethane, trichlorofluoromethane, or dichlorotetrafluoroethane, carbon dioxide, or other suitable gas.
The aerosol may conveniently also contain a surfactant such as lecithin. The dose of drug may be controlled by provision of a metered valve.
Alternatively the active ingredients may be provided in the form of a dry powder, for example a powder mix of the compound in a suitable powder base such as lactose, starch, starch derivatives such as hydroxypropylmethyl cellulose and polyvinylpyrrolidone (PVP). Conveniently the powder carrier will form a gel in the nasal cavity. The powder composition may be presented in unit dose form for example in capsules or cartridges of, e. g. gelatin, or blister packs from which the powder may be administered by means of an inhaler.
In formulations intended for administration to the respiratory tract, including intranasal formulations, the compound will generally have a small particle size for example of the order of 5 to 10 microns or less. Such a particle size may be obtained by means known in the art, for example by micronisation.
When desired, formulations adapted to give sustained release of the active ingredient may be employed.
The pharmaceutical preparations are preferably in unit dosage forms. In such form, the preparation is subdivided into unit doses containing appropriate quantities of the active component. The unit dosage form can be a packaged preparation, the package containing discrete quantities of preparation, such as packeted tablets, capsules, and powders in vials or ampoules. Also, the unit dosage form can be a capsule, tablet, cachet, or lozenge itself, or it can be the appropriate number of any of these in packaged form.
It is especially advantageous to formulate parenteral compositions in dosage unit form for ease of administration and uniformity of dosage. Dosage unit form as used herein refers to physically discrete units suited as unitary dosages for the subjects to be treated; each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier. The specification for the dosage unit forms are dictated by and directly dependent on (a) the unique characteristics of the active material and the particular therapeutic effect to be achieved, and (b) the limitations inherent in the art of compounding such an active material for the treatment of a HCV viral infection in living subjects having a diseased condition in which bodily health is impaired.
The invention also includes the compounds in the absence of carrier where the compounds are in unit dosage form.
Methods of treatment
The compounds of formula (I) may be used in the treatment of a Flaviviridae viral infection such as a HCV infection.
Generally, the term "treatment" means affecting a subject, tissue or cell to obtain a desired pharmacological and/or physiological effect and includes: (a) inhibiting the viral infection, i.e. arresting its development or further development; (b) relieving or ameliorating the effects of the viral infection, i.e. cause regression of the effects of the viral infection; (c) reducing the incidence or the viral infection or (d) preventing the infection from occurring in a subject, tissue or cell predisposed to the viral infection disease or at risk thereof, but has not yet been diagnosed with a protective pharmacological and/or physiological effect so that the viral infection does not develop or occur in the subject, tissue or cell.
The prevention of hepatitis C means, for example, administration of a
pharmaceutical agent to a subject found to carry a HCV by a test and the like but without a symptom of infection, or to a subject who shows an improved disease state of hepatitis after a treatment of hepatitis C, but who still carries a HCV and is associated with a risk of recurrence of hepatitis.
The term "subject" as used herein refers to any animal, in particular mammals such as humans having a disease or condition which requires treatment with the compound of formula (I).
The term "administering" refers to providing the compound or pharmaceutical composition of the invention to a subject suffering from or at risk of the diseases or conditions to be treated or prevented.
The term "viral infection" refers to the introduction of a virus into cells or tissues, e.g., hepatitis C virus (HCV). In general, the introduction of a virus is also associated with replication. Viral infection may be determined by measuring virus antibody titer in samples of a biological fluid, such as blood, using, e.g., enzyme immunoassay. Other suitable diagnostic methods include molecular based techniques, such as RT-PC , direct hybrid capture assay, nucleic acid sequence based amplification, and the like. A virus may infect an organ, e.g., liver, and cause disease, e.g. , hepatitis, cirrhosis, chronic liver disease and hepatocellular carcinoma.
The term "Flaviviridae virus" refers to a virus of the family Flaviviridae, which family includes the Hepacivirus Flavivirus and Pestivirus or hepatitis C-like virus genera. A representative species of the genus of hepatitis C-like viruses is hepatitis C virus.
Dosages
The term "therapeutically effective amount" refers to the amount of the compound of formula (I) that will elicit the biological or medical response of a subject, tissue or cell that is being sought by the researcher, veterinarian, medical doctor or other clinician.
In the prevention or treatment of HCV infections or diseases an appropriate dosage level will generally be about 0.01 to 500 mg per kg subject body weight per day which can be administered in single or multiple doses. Preferably, the dosage level will be about 0.1 to about 250 mg/kg per day; more preferably about 0.5 to about 100 mg/kg per day. A suitable dosage level may be about 0.01 to 250 mg/kg per day, about 0.05 to 100 mg/kg per day, or about 0.1 to 50 mg/kg per day. Within this range the dosage may be 0.05 to 0.5, 0.5 to 5 or 5 to 50 mg/kg per day. The dosage may be selected, for example to any dose within any of these ranges, for therapeutic efficacy and/or symptomatic adjustment of the dosage to the subject to be treated
It will be understood that the specific dose level and frequency of dosage for any particular subject may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the subject undergoing therapy.
Method of Preparation
Compounds of formula (I) may be generally synthesised via a common synthetic intermediate of general formula (II):

(II)
wherein X is (R1)tC02H or (R1 )tC02Ci.3alkyl, t is 0 or 1 ; ΖΊ , Z'3 and Z'4 are each
independently selected from C-Ra, N and N+-0"; and R1 , R2 and R3 are as defined in formula (I).
Accordingly, in one aspect there is provided a process for preparing a compound of formula (I) via an intermediate of general formula (II).
Intermediates of formula (II) having a carboxylic acid moiety, C02H may be used to access a diversity of compounds of formula (I) with side chains R1 , such as those shown in Scheme 1 , via amide coupling. Side chains comprising a carboxylic acid moiety may also
undergo further amide coupling thereby providing access to a diversity of extended side chains.

Scheme 1
Accordingly, in one embodiment there is provided a process for producing a compound of formula (Ib-ii) comprising the step of reacting a compound of formula (II) with an amino precursor of general formula NHR5R6 under amide coupling conditions. Suitable amide coupling conditions will be understood by those skilled in the art and have been described in many references such as Advanced Organic Chemistry - 4th Edition, March J., John Wiley & Sons Inc, New York,1992.
In some instances, compounds of formula (II) have also demonstrated antiviral activity. Accordingly, compounds of formula (II) will be understood to be intermediates and/or final compounds as the case may be. EXAMPLES
The invention will now be described with reference to the following non-limiting examples.
Synthetic Methods
1 H NMR spectra were recorded on either a Bruker Avance DRX 400, AC 200 or AM 300 spectrometer. Spectra were recorded in CDCI3, acetone-c/6, CD3OD (MeOD) or DMSO-c/6 using the residual solvent peak as a reference. Chemical shifts are reported on the δ scale in parts per million (ppm) using the following conventions to assign the multiplicity: s (singlet), d (doublet), t (triplet), q (quartet), qt (quintet), m (multiplet) and prefixed br (broad).
Mass spectra (ESI) were recorded on either a Micromass Platform QMS or Thermo Finnigan LCQ Advantage spectrometer. Flash chromatography was performed on 40-63pm silica gel 60 (Merck No. 9385). Automated flash chromatography was performed either on a Combi- Flash™ purification system using Combi-Flash™ silica gel columns or on a Biotage SP4 purification system using either GraceResolv™ silica gel cartridges, Grace Reveleris™ C-18 reverse phase silica gel cartridges or Biotage SNAP™ C-18 reverse phase silica gel cartridges. Preparative HPLC was carried out using either a Gilson 322 pump with a Gilson 215 liquid handler and a HP1 100 PDA detector or an Agilent 1200 Series mass detected preparative LCMS using a Varian XRs C-18 100 x 21 .2 mm column. Unless otherwise specified, the HPLC systems employed Phenomenex C8(2) columns using either acetonitrile or acetonitrile containing 0.06% TFA in water or water containing 0.1 % TFA.
During the reactions a number of the moieties may need to be protected. Suitable protecting groups are well known in industry and have been described in many references such as "Greene's Protective Groups in Organic Synthesis" Fourth Edition, Peter G.M. Wuts and Theodora W. Greene, Wiley-lnterscience, 2007 .
The abbreviations used in the Examples are as follows unless indicated otherwise:
Boc: i-butoxycarbonyl
Cbz: benzyloxycarbonyl
DABCO 33-LV: 33% solution by weight of 1 ,4-Diazabicyclo[2.2.2]octane in propylene glycol DIPEA: N,N-Diisopropylethylamine
DMAP: Λ/,/V-dimethylaminopyridine
DMF: dimethylformamide
DMSO: dimethylsulfoxide
EDCI: W-(3-dimethylaminopropyl)-W-ethylcarbodiimide
ESI: electrospray ionisation
HATU: 2-(7-Aza-1 H-benzotriazole-1 -yl)-1 , 1 ,3,3-tetramethyluronium hexafluorophosphate
HPLC: high performance liquid chromatography
LCMS: liquid chromatography coupled mass spectrometry
MS: mass spectrometry
NaHMDS: sodium hexamethyldisilazane (sodium bis(trimethylsilyl)amide)
NMP: W-Methyl-2-pyrrolidone
NMR: nuclear magnetic resonance
RT: room temperature
TFA: trifluoroacetic acid
THF: tetrahydrofuran
TLC: thin-layer chromatography
TMSCN: trimethylsilyl cyanide
Method A: Synthesis of Core Intermediates (of General Formula II)
Route (a): 2-azido-4-bromobenzoic acid (ii) was prepared by reaction of 2-amino-4- bromobenzoic acid (i) with a metal nitrite under acidic conditions, followed by reaction with a basic solution of sodium azide. The carboxylic acid of ii was coupled with an aromatic amine using standard peptide coupling conditions (eg. HATU, DIPEA in DMF) to give intermediate (iii). Cyclisation to (iv) was achieved by heating iii in phosphorus oxychloride, and the bromide of iv was converted to the nitrile via reaction with a metal cyanide. Intermediate v was reacted with an appropriate aromatic or cycloalkenyl boronic acid or ester under standard palladium-catalysed cross-coupling conditions to give intermediate (vi). Basic hydrolysis of the nitrile of vi afforded the carboxylic acid (vii), and the alkene was reduced to the saturated roduct (viii) with Pd/C and ammonium formate.

= Ph, 2-pyridyl, 3-pyridyl, 4-fluorophenyl, 4-difluoromethoxyphenyl, 4-benzyloxyphenyl. Example: Synthesis of intermediate compounds 3-(Cyclohex-1-en-1-yl)-2-phenyl-2H- indazole-6-carboxylic acid (1) and 3-Cyclohexyl-2-phenyl-2H-indazole-6-carboxylic acid (2)


2
Step a: To an ice-cooled, stirred suspension of 2-amino-4-bromobenzoic acid (i) (10 g, 46.3 mmol) in water was added concentrated HCI (aq) (88 mL) at such a rate so as to maintain the internal temperature at 5 °C. The reaction mixture was then heated at 80 °C for 30 min and cooled to 0 °C, whereupon a solution of sodium nitrite (3.83 g, 55.6 mmol) in water (10 mL) was added dropwise. The reaction mixture was allowed to stir for 1 h and then added dropwise to a stirred solution of sodium azide (2.95 g, 45.4 mmol) and sodium ethoxide (55.9 g, 681 .6 mmol) in water (88 mL) at RT. The resultant reaction mixture was stirred at room temperature for 3 h. After the reaction was complete (by TLC) the resultant precipitate was separated by filtration, washed with water (3 x 50 mL) and dried in vacuo to give 2- azido-4-bromobenzoic acid (ii) as an off-white powder (10.12 g, 90 %); 1 H-NMR (400 MHz, CDCI3) δ 7.35-7.43 (m, 2H), 7.92-7.99 (m, 1 H).
Step b: To the stirred solution of 2-azido-4-bromobenzoic acid (ii) (9.8 g, 40.5 mmol) in DMF (50 mL) at 0 °C was added HATU (18.5 g, 48.6 mmol). The reaction mixture was stirred for 15 min whereupon DIPEA (14.3 mL, 60.7 mmol) was added followed by aniline (4.52 g, 48.6 mmol). The reaction mixture was then allowed to warm to RT and the progress was monitored by TLC and LCMS analysis. After 4 h the reaction mixture was poured onto cold water (500 mL) and the resultant suspension then stirred for 15 min. The precipitate was separated by filtration and washed with water (3 x 100 mL) and dried in vacuo overnight to give 2-azido-4-bromo-/V-phenylbenzamide (iii) as an off-white powder (12.1 g, 94%); 1H- NMR (400 MHz, CDCI3) δ 7.1 1-7.20 (m, 1 H), 7.33-7.45 (m, 4H), 7.60-7.70 (2H, m), 8.09- 8.17 (1 H, m), 9.24 (1 H, brs).
Step c: A mixture of 2-azido-4-bromo-/V-phenylbenzamide (iii) (4.7 g, 14.8 mmol) and phosphorus oxychloride (25 mL) was heated at 95 °C for 2 h and then cooled to RT. The reaction mixture was then concentrated in vacuo and the residue was then added dropwise
to an ice-cold solution of saturated sodium carbonate (30 mL) such that the temperature of the resultant mixture did not rise above 5 °C. The solid so obtained was filtered, dried in vacuo and was then purified by column chromatography on silica gel eluting with ethyl acetate: hexane (gradient elution 1 :49 to 3:97 v/v) to give 6-bromo-3-chloro-2-phenyl-2/-/- indazole (iv) as an off-white powder (3.35 g, 73 %); ESI-MS m/z calculated for [M+H]+: 308.96; found: 308.95.
Step d: To a stirred solution of 6-bromo-3-chloro-2-phenyl-2/-/-indazole (iv) in NMP (10 x 10 mL batches) at T was added copper (I) cyanide (10 x 1.45 g, 162.9 mmol per batch) and the reaction mixtures were then heated at 175 °C. After 1.5 h the combined reaction mixtures were cooled to RT and poured into aqueous saturated FeCI3 (100 mL). The organics were extracted into ethyl acetate (3 x 75 mL) and the combined organic layers were then washed with water (3 x 50 mL), dried (Na2S04) and concentrated in vacuo. The crude product was purified by column chromatography on silica gel eluting with dichloromethane to give 3-chloro-2-phenyl-2H-indazole-6-carbonitrile (v) (9.4 g, 54 % overall); ESI-MS m/z calculated for [M+H]+: 254.05; found: 254.05.
Step e: Argon was bubbled through a solution of dioxane-water (50 mL) for 0.5 h whereupon 3-chloro-2-phenyl-2A7-indazole-6-carbonitrile (v) was added to the de-gassed medium. To this suspension was added cyclohexyl boronic acid pinacol ester (3.21 g, 15.4 mmol) and the reaction mixture was then stirred for 10 min. Triphenyl phosphine (1 .07 g, 4.1 mmol) and palladium (II) acetate (0.46 g, 2.1 mmol) were then added followed by addition of sodium carbonate (2.54 g, 20.5 mmol). The reaction mixture was heated at reflux and monitored by LCMS analysis. After 1 h, the reaction mixture was concentrated in vacuo and the aqueous layer was extracted into ethyl acetate (4 x 100 mL). The combined organic layers were dried (Na2S04) and concentrated in vacuo. The residue was purified by column chromatography on silica gel by gradient elution (10-20% ethyl acetate in hexane) to give 3- (cyclohex-1-en-1 -yl)-2-phenyl-2/-/-indazole-6-carbonitrile (vi) as an off-white powder (2.21 g, 71 %); ESI-MS m/z calculated for [M+H]+: 300.15; found: 300.05.
Step f: To a stirred suspension of 3-(cyclohex-1-en-1 -yl)-2-phenyl-2/-/-indazole-6-carbonitrile (vi) (910 mg, 3.0 mmol) in ethanol:water (1 : 1 v/v, 40 mL) was added sodium hydroxide (1.8 g, 45.0 mmol) and the reaction mixture was then heated at reflux and monitored by TLC and LCMS analysis. After 4 h, the reaction mixture was cooled to RT and the volatiles were removed in vacuo. The residue was acidified with 2N aqueous HCI to pH 2 and the resulting precipitate was then separated by filtration. The residue was washed with water (2 x 20 mL) and dried in vacuo to give 3-(cyclohex-1 -en-1 -yl)-2-phenyl-2/-/-indazole-6-carboxylic acid (1 ) as a white solid (900 mg, 93 %); ESI-MS m/z calculated for [M+H]+: 319.14; found: 319.10;
1 H-NMR (400 MHz, CDCI3) δ 8.64 (s, 1 H), 7.79-7.65 (m, 4H), 7.57-7.44 (m, 3H), 6.20 (brs, 1 H), 2.38-2.25 (m, 2H), 2.03-1.92 (m, 2H), 1.77-1 .60 (m, 4H).
Step g: To a stirred suspension of 3-(cyclohex-1 -en-1 -yl)-2-phenyl-2/-/-indazole-6-carboxylic acid (1 ) (750 mg, 2.4 mmol) in methanol (20 mL) was added 10% Pd/C under an atmosphere of nitrogen. Ammonium formate (1.48 g, 23.6 mmol) was then added and the reaction mixture was heated to reflux and monitored by LCMS analysis. Upon complete consumption of the starting materials, the reaction mixture was filtered through a pad of Celite® and the residue was washed with methanol (2 x 20 mL). The combined filtrates were concentrated in vacuo and the residue was purified by automated column chromatography (Combi-Flash™, silica gel) eluting with ethyl acetate:hexane (3:7 to 7: 15 v/v) to give 3- cyclohexyl-2-phenyl-2/-/-indazole-6-carboxylic acid (2) as a white solid (519 mg, 68 %); ESI- MS m/z calculated for [M+H]+: 321.16; found: 321 .20; 1H-NMR (400 MHz, CDCI3) δ 8.59 (s, 1 H), 7.92 (d, J 8.9 Hz, 1 H), 7.70 (dd, J 8.9 Hz, J 1.4 Hz, 1 H), 7.53-7.57 (m, 3H), 7.45-7.49 (m, 2H), 2.90-2.99 (m, 1 H), 1.71 -2.00 (m, 7H), 1.17-1 .41 (m, 3H).
Similarly prepared were the following compounds.

Route (b): The pyrazolopyrimidine (ii) was prepared by reaction of ethyl 2,4- dichloropyrimidine-5-carboxylate with an aromatic hydrazine. The chloride of ii was converted to the nitrile via reaction with a metal cyanide. The nitrile of iii was converted to a carboxylic ester via a Pinner reaction (reaction with an alcohol under acid catalysis). The alcohol of iv was converted to a triflate which was reacted with an appropriate cycloalkenyl boronic acid or ester under standard palladium-catalysed cross-coupling conditions to give intermediate vi. The carboxylic ester of vi was hydrolysed under basic conditions to give
intermediate vii, and the alkene was reduced to the saturated product viii via palladium catal sed hydrogenation.

Example: Synthesis of intermediate compound 3-cyclohexyl-2-phenyl-2H-pyrazolo[3,4- d rimidine-6-carboxylic acid

Step a: Ethyl 2,4-dichloropyrimidine-5-carboxylate (1.0 g, 4.5 mmol) in EtOH (8 mL) had phenylhydrazine (0.45 mL, 4.5 mmol) added and was stirred at 60 °C. After 1.5 h, the reaction was cooled to room temperature and 1 M HCI (8 mL) was added. The precipitate was filtered off, washed with a little EtOH/H20 (1 : 1 ) and dried to give 6
chloro-2-phenyl-2H-pyrazolo[3,4-d]pyrimidin-3-ol (ii) (0.37 g, 33%) as an off-white sold. ESI- MS m/z calculated for [M+H]+: 247.0; found: 247.1.
Step b: A solution of 6-chloro-2-phenyl-2H-pyrazolo[3,4-d]pyrimidin-3-ol (0.37 g, 1 .5 mmol) in DMSO (2 mL) was added to a mixture of NaCN (0.15 g, 3.0 mmol) and DABCO 33-LV (0.59 mL, 1.8 mmol) in DMSO (1 mL) and H20 (2 mL). This was stirred at 60 °C for 4 h. The reaction was cooled to room temperature and 1 M HCI (8 mL) was added. The precipitate was collected by filtration and washed with H20. The crude product was dissolved in DMSO (15 mL) and purified by reverse-phase column chromatography by gradient elution (0-100% MeCN in water) to give (3-hydroxy-2-phenyl-2/-/-pyrazolo[3,4-c ]pyrimidin-6-yl)acetonitrile (iii) (0.14 g, 41 %) as an off-white solid. ESI-MS m/z calculated for [M+H]+: 227.1 ; found: 236.1 . Step c: (3-hydroxy-2-phenyl-2A7-pyrazolo[3,4-c ]pyrimidin-6-yl)acetonitrile (78 mg, 0.33 mmol) in a microwave vial (10-20 mL) under Ar was suspended in saturated HCI in EtOH (4 mL). The vial was sealed and then heated at 80 °C behind a blast screen. After 16 h the reaction was cooled to room temperature and carefully vented with a needle before removing the cap. The reaction mixture was pipetted into saturated NaHC03 (8 mL). The EtOH was removed on the rotary evaporator and saturated NaHC03 was added to the mixture until a solution formed (~ 4 mL). The product in the solution was purified by reverse-phase column chromatography by gradient elution (0-100% MeCN in water) to give ethyl 3-hydroxy-2- phenyl-2/-/-pyrazolo[3,4-c/]pyrimidine-6-carboxylate (iv) (78 mg, 84%) as a white solid. ESI- MS m/z calculated for [M+H]+: 285.1 ; found: 285.0.
Step d: To ethyl 3-hydroxy-2-phenyl-2H-pyrazolo[3,4- ]pyrimidine-6-carboxylate (1 1 mg, 37 pmol) in dry THF (0.5 mL) under Ar was added triethylamine (10 μί, 45 μηηοΙ), followed by N-phenyltrifluoromethanesulphonimide (18 mg, 45 μηηοΙ) and the resulting solution was stirred at room temperature for 16 h. The reaction was quenched with saturated NH4CI (1 mL) and extracted with EtOAc (2 x 2 mL). The combined organics were dried over MgS04, filtered and concentrated. The residue was dissolved in DCM (1 mL) and purified by column chromatography on silica gel by gradient elution (0-40% ethyl acetate in hexane) to give ethyl 2-phenyl-3-{[(trifluoromethyl)sulfonyl]oxy}-2/-/-pyrazolo[3,4-c/]pyrimidine-6-carboxylate (v) (11 mg, 69%) as a white solid. ESI-MS m/z calculated for [M+H]+: 417.3; found: 417.0. Step e: A mixture of ethyl 2-phenyl-3-{[(trifluoromethyl)sulfonyl]oxy}-2/-/-pyrazolo[3,4- c ]pyrimidine-6-carboxylate (53 mg, 0.13 mmol) in a microwave vial in toluene (0.8 mL) and H20 (50 \ L) had Ar bubbled through for 5 min. Cyclohexeneboronic acid, pinacol ester (0.10 mL, 0.51 mmol) was added, followed by K2C03 (74 mg, 0.51 mmol) and Pd(PPh3)4 (18 mg, 13 μηηοΙ). The reaction was heated under microwave irradiation for 40 min at 105 °C. EtOH (3 mL) was added and the reaction filtered through a syringe filter (0.45 μΜ), washed through with EtOH and concentrated. The crude product was dissolved in DCM (2 mL) and purified by column chromatography on silica gel by gradient elution (0-30% ethyl acetate in hexane) to give ethyl 3-(cyclohex-1 -en-1-yl)-2-phenyl-2H-pyrazolo[3,4-c ]pyrimidine-6-
carboxylate (vi) (23 mg, 52 %) as a white solid. ESI-MS m/z calculated for [M+H]+: 349.4; found: 349.1 .
Step f: A mixture of ethyl 3-(cyclohex-1 -en-1-yl)-2-phenyl-2H-pyrazolo[3,4-c ]pyrimidine-6- carboxylate (18 mg, 51 pmol) and LiOH hydrate (14 mg, 0.31 mmol) in dioxane (0.80 mL), iPrOH (0.80 mL), and H2O (0.64 mL) was stirred at room temperature for 30 min. The rection mixture was acidified with 1 M HCI and extracted with EtOAc (3 x 4 mL). The combined organics were dried over MgS04, filtered and concentrated to give 3-(cyclohex-1 - en-1 -yl)-2-phenyl-2/-/-pyrazolo[3,4-c ]pyrimidine-6-carboxylic acid (vii) (16 mg, 95 %) as a white solid. ESI-MS m/z calculated for [M+H]+: 321 .3; found: 321 .0.
Step g: 3-(cyclohex-1-en-1 -yl)-2-phenyl-2A7-pyrazolo[3,4-c ]pyrimidine-6-carboxylic acid (16 mg, 49 pmol) in MeOH (2 mL) and EtOAc (0.50 mL) had 10% Pd/C (3.3 mg, 20% by mass) added. The reaction flask was evacuated and filled with H2 (x3) then stirred at room temperature under H2 for 1 h. The reaction mixture was filtered through a syringe filter (0.45 pm), washed through with MeOH and the filtrate was concentrated. The crude product was dissolved in DMSO (1 mL) and purified by reverse-phase column chromatography by gradient elution (0-100% MeCN in water) to give 3-cyclohexyl-2-phenyl-2/-/-pyrazolo[3,4- c ]pyrimidine-6-carboxylic acid
(viii) (8.0 mg, 51 %) as a white solid. ESI-MS m/z calculated for [M+H]+: 323.4; found: 323.1. 1 H NMR (400 MHz, MeOD) δ 9.50 (s, 1 H), 8.34 (dd, J = 8.7, 1.1 Hz, 2H), 7.64 - 7.52 (m, 2H), 7.44 - 7.34 (m, 1 H), 3.31 - 3.18 (m, 1 H), 2.27 - 2.15 (m, 2H), 2.03 - 1.93 (m, 2H), 1 .92 - 1 .80 (m, 3H), 1.67 - 1.53 (m, 2H), 1 .53 - 1.40 (m, 1 H).
Route (c): The hydrazynylpyrimidine ii was prepared by reaction of methyl 4,6- dichloropyridine-3-carboxylate with an aromatic hydrazine. The chloride of ii was converted to the nitrile via reaction with a metal cyanide. Cyclisation of iv was achieved by heating iii under basic conditions, and the alcohol was converted to a chloride with phosphorus oxychloride. Intermediate v was reacted with an appropriate cycloalkenyl boronic acid or ester under standard palladium-catalysed cross-coupling conditions to give intermediate vi. The nitrile of iv was converted to a carboxylic ester via a Pinner reaction (reaction with an alcohol under acid catalysis). The alkene of vii was reduced to the saturated product viii with Pd/C and ammonium formate and ester hydrolysis under basic conditions afforded the carboxylic acid ix.

viii ix

Step a: To a solution of methyl 4,6-dichloropyridine-3-carboxylate (1 .0 g, 4.9 mmol) in anhydrous ethanol (10 mL) was added phenylhydrazine (0.50 mL, 5.1 mmol), followed by triethylamine (1.7 mL, 12 mmol). The reaction mixture was heated under microwave irradiation at 120 oC for 40 min. The reaction mixture was evaporated to dryness and the crude product was purified by column chromatography on silica gel by gradient elution (20- 100% ethyl acetate in hexane) to give methyl 6-chloro-4-(2-phenylhydrazinyl)pyridine-3- carboxylate (ii) (0.55 g, 41 %) as an oil. ESI-MS m/z calculated for [M+H]+: 278.1 ; found: 278.0.
Step b: To a solution of methyl 6-chloro-4-(2-phenylhydrazinyl)pyridine-3-carboxylate (0.55 g, 2.0 mmol) in anhydrous DMA (11 ml_) was added zinc cyanide (0.15 g, 1 .3 mmol). The reaction mixture was evacuated and flushed with argon (x 2), then zinc (16 mg, 0.24 mmol), Pd2(dba)3 (30 mg, 0.033 mmol) and dppf (33 mg, 0.060 mmol) were added and the mixture was again flushed with argon (x2). The mixture was heated at 120 °C for 1 .5 h. The reaction mixture was cooled to room temperature and partitioned between ethyl acetate and water and the aqueous was extracted with ethyl acetate (x 2). The combined organics were washed with brine, dried over MgS04, filtered and concentrated. The crude product was purified by column chromatography on silica gel by gradient elution (20-33% ethyl acetate in hexane) to give methyl 6-cyano-4-(2-phenylhydrazinyl)pyridine-3-carboxylate (iii) (0.32 g, 59%) as a pale yellow solid. ESI-MS m/z calculated for [M+H]+: 269.1 ; found: 269.0.
Step c: To a solution of methyl 6-cyano-4-(2-phenylhydrazinyl)pyridine-3-carboxylate (0.32 g, 1.1 mmol) in anhydrous DMF (10 ml_) that was degassed with argon was added 1 M NaHMDS in THF (1 .6 mL, 1.6 mmol). The reaction was heated at 95 °C for 15 min then cooled in ice/water. After 30 min at 0 °C 1 M aqueous acetic acid (5 mL) was added and reaction mixture was evaporated to dryness. The crude product was purified by reverse- phase column chromatography by gradient elution (0-100% MeCN in water) to give 3- hydroxy-2-phenyl-2H-pyrazolo[4,3-c]pyridine-6-carbonitrile (iv) (0.22 g, 82%) as a white solid. ESI-MS m/z calculated for [M+H]+: 237.1 ; found: 237.2.
Step d: A solution of 3-hydroxy-2-phenyl-2/-/-pyrazolo[4,3-c]pyridine-6-carbonitrile (0.1 1 g, 0.44 mmol) in phosphorus oxychloride (7 mL) was heated at 120 °C for 45 min, then left at room temperature overnight. The reaction was then poured into ice water/ethyl acetate (1 :1 ) and the aqueous layer adjusted to pH 5-7 with 7 M NaOH. The organic layer was washed with a small amount of water and brine, dried over MgS04, filtered and evaporated to dryness to give 3-chloro-2-phenyl-2/-/-pyrazolo[4,3-c]pyridine-6-carbonitrile (v) (0.14 g, quant) as a beige solid. ESI-MS m/z calculated for [M+H]+: 255.0; found: 255.2.
Step e: A solution of 3-chloro-2-phenyl-2H-pyrazolo[4,3-c]pyridine-6-carbonitrile (0.21 g, 0.82 mmol) in dioxane/water (2: 1 , 30 mL total) was de-gassed with argon and split into two batches. To each batch was added sodium carbonate (65 mg, 0.78 mmol) and the mixture was stirred at room temperature for 5 min with sonication. 2-(cyclohexen-1 -yl)-4,4,5,5- tetramethyl-1 ,3,2-dioxaborolane (0.10 g, 0.50 mmol) was added to each batch, followed by Pd(PPh3)4 (80 mg, 0.069 mmol) and the mixtures were heated at 120 °C for 1 .5 h. The two batches were combined, filtered and the precipitate washed with water. The filtrate was diluted with water and extracted with ethyl acetate (x 3). The combined organics were washed with brine, dried over MgS04, filtered and evaporated to dryness. The crude product was purified by column chromatography on silica gel by gradient elution (20-50% ethyl
acetate in hexane) to give 3-(cyclohex-1-en-1 -yl)-2-phenyl-2/-/-pyrazolo[4,3-c]pyridine-6- carbonitrile (vi) (0.19 g, 75%) as a waxy solid. ESI-MS m/z calculated for [M+H]+: 301 .1 ; found: 301 .3.
Step f: A solution of 3-(cyclohex-1 -en-1 -yl)-2-phenyl-2/-/-pyrazolo[4,3-c]pyridine-6-carbonitrile (65 mg, 0.22 mmol) in freshly prepared saturated HCI in ethanol (12 mL) was heated in a sealed tube at 80 °C for 6 h then left overnight at room temperature. The reaction mixture was poured into ice/water and extracted with ethyl acetate (x 3). The combined organics were dried over MgS04, filtered and evaporated to dryness. The crude product was purified by column chromatography on silica gel by gradient elution (20-50% ethyl acetate in hexane) to give ethyl 3-(cyclohex-1 -en-1 -yl)-2-phenyl-2H-pyrazolo[4,3-c]pyridine-6-carboxylate (vii) (31 mg, 38%) as a white solid. ESI-MS m/z calculated for [M+H]+: 348.2; found: 348.1 . Step g: To a solution of ethyl 3-(cyclohex-1 -en-1-yl)-2-phenyl-2H-pyrazolo[4,3-c]pyridine-6- carboxylate (31 mg, 80 pmol) in methanol (2 mL) was added ammonium formate (26 mg, 0.41 mmol) and the reaction mixture was de-gassed with argon. 10% Pd/C (13 mg, -40% by mass) was added and the mixture was heated at reflux for 3 h. The reaction mixture was decanted from the Pd catalyst and the catalyst was washed with methanol (x 2). The combined organics were evaporated to dryness. The crude product was purified by column chromatography on silica gel by gradient elution (20-50% ethyl acetate in hexane) to give ethyl 3-cyclohexyl-2-phenyl-2/-/-pyrazolo[4,3-c]pyridine-6-carboxylate (viii) (10 mg, 36%) as a white solid. ESI-MS m/z calculated for [M+H]+: 350.2; found: 350.1 .
Step h: A solution of ethyl 3-cyclohexyl-2-phenyl-2H-pyrazolo[4,3-c]pyridine-6-carboxylate (10 mg, 0.029 mmol) and KOH (39 mg, 0.70 mmol) in 1 ,4-dioxane/water (1 : 1 , 1 .5 mL) was stirred at room temperature for 1.75 h. The reaction was quenched with water (3.5 mL) and glacial acetic acid (0.08 mL) and co-evaporated to dryness with acetonitrile. The crude product was purified by reverse-phase column chromatography by gradient elution (0-100% MeCN in water) to give 3-cyclohexyl-2-phenyl-2H-pyrazolo[4,3-c]pyridine-6-carboxylic acid (ix) (3.4 mg, 37%) as a white solid. ESI-MS m/z calculated for [M+H]+: 322.2; found: 322.1. 1 H-NMR (400 MHz, CD3OD) δ 9.36 (s, 1 H), 8.20 (s, 1 H), 7.66-7.63 (m, 3H), 7.56-7.53 (m, 2H), 3.03-2.95 (m, 1 H), 2.02-1.96 (m, 4H), 1.90-1 .86 (m, 2H), 1.78-1.75 (m, 1 H), 1.50-1.39 (m, 1 H), 1 .21-1.33 (m, 2H).
Route (d): 2-chloronicotinic acid was converted to the amide ii via an amide coupling with an appropriate aromatic amine. The chloride of ii was converted to the hydrazine iii with hydrazine hydrate and the azide iv was formed by reaction of iii with sodium nitrite.
Cyclisation to v was achieved by heating iv in phosphorus oxychloride. The W-oxide of v was prepared via oxidation with m-chloroperbenzoic acid. Reaction of vi with TMSCN under basic conditions afforded vii. The nitrile was hydrolysed to a carboxylic acid under acidic
conditions, and esterification was achieved with thionyl chloride and an alcohol to give ix. The chloride of x was reacted with a cycloalkenyl boronic acid or ester under standard palladium-catalysed cross-coupling conditions and the carboxylic ester was hydrolysed under basic conditions to give x. The alkene of x was reduced to the saturated product xi with Pd/C and ammonium formate.

Example: Synthesis of 3-cyclohexyl-2-phenyl-2H-pyrazolo[3,4-b]pyridine-6-carboxylic acid

Step a: To stirred solution of 2-chloronicotinic acid (50g, 0.32 mol) in DCM was added DMAP (78 g, 0.63 mol), aniline (44 g, 0.48 mol) and EDCI.HCI (92 g, 0.48 mol). The reaction was stirred overnight at room temperature, then diluted with water and the product extracted with DCM. The organic layer was washed with 1 M aqueous acetic acid then saturated sodium bicarbonate and concentrated in vacuo to give 2-chloro-/V-phenylnicotinamide (48 g, 65%).
Step b: To a stirred solution of 2-chloro-/V-phenylnicotinamide (48 g, 0.21 mol) in ethanol (120 ml) was added hydrazine hydrate (155 g, 3.1 mol) and the reaction mixture was stirred for 48 h at room temperature. The reaction mixture was diluted with water and the precipitated product was collected by filtration and dried to give 2-hydrazinyl-/V- phenylnicotinamide (35 g, 74%). ESI-MS m/z calculated for [M+H]+: 229.2; found: 229.0. Step c: To a ice cold solution of 2-hydrazinyl-W-phenylnicotinamide (35 g, 0.15 mol) in 4 M HCI (105 ml_) was added a solution of sodium nitrite (21 g, 0.31 mol) in water (175 ml_) at 0- 5 °C and the reaction was stirred for 30 min at 0 °C when precipitation occurred. The reaction mixture was filtered to give 2-azido-/V-phenylnicotinamide (32 g, 87%). ESI-MS m/z calculated for [M+H]+: 240.1 ; found: 240.0.
Step d: A solution of 2-azido-/V-phenylnicotinamide (32 g, 0.15 mol) in phosphorus oxychloride (320 ml) was heated at reflux for 24 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. The residue was diluted with ice water and adjusted to pH ~8 with aqueous sodium bicarbonate. The product was extracted with ethyl acetate
and the organic layer concentrated in vacuo. The crude product was purified by column chromatography to give 3-chloro-2-phenyl-2/-/-pyrazolo[3,4-6]pyridine (22 g, 72%). ESI-MS m/z calculated for [M+H]+: 230.0; found: 230.3.
Step e: To a suspension of 3-chloro-2-phenyl-2/-/-pyrazolo[3,4-£>]pyridine (22 g, 96 mmol) in dichloromethane (440 mL) was added m-chloroperbenzoic acid (50 g, 0.29 mol) and the reaction mixture was stirred at room temperature for 2 h. The reaction mixture was quenched with saturated sodium bicarbonate, the product was extracted with
dichloromethane and the organic layer concentration in vacuo. The crude product was crystallized from ethanol to give 3-chloro-2-phenyl-2H-pyrazolo[3,4-0]pyridine-7-oxide (17 g, 72%). ESI-MS m/z calculated for [M+H]+: 246.0; found: 246.1.
Step f: To a solution of 3-chloro-2-phenyl-2H-pyrazolo[3,4-6]pyridine-7-oxide (17g, 0.69 mol) in DMF (255 mL) was added DIPEA (24 mL, 0.14 mol) and TMSCN (14 g, 0.14 mol) and the reaction was heated at 90 °C for 2 h. The reaction mixture was poured into ice water and the precipitate was collected by filtration. The crude product was purified by column
chromatography to give 3-chloro-2-phenyl-2/-/-pyrazolo[3,4-0]pyridine-6-carbonitrile (5.0 g, 28%). ESI-MS m/z calculated for [M+H]+: 255.0; found: 255.2.
Step g: A suspension of 3-chloro-2-phenyl-2/-/-pyrazolo[3,4-0]pyridine-6-carbonitrile (5.0 g, 20 mmol) in 70% sulphuric acid (50 mL) was heated to 81 °C for 4 h. The reaction was poured into ice water and the precipitate was collected by filtration. The crude product was purified by acid base treatment to give 3-chloro-2-phenyl-2H-pyrazolo[3,4-b]pyridine-6- carboxylic acid (3.8 g, 71 %) as off white solid. ESI-MS m/z calculated for [M+H]+: 274.0; found: 274.0.
Step h: To a solution of 3-chloro-2-phenyl-2H-pyrazolo[3,4-b]pyridine-6-carboxylic acid (3.8 g, 0.14 mol) in ethanol (76 mL) was added thionyl chloride (5.8 g, 0.49 mol) at 0 °C and the reaction was heated at reflux for 4 h. The reaction mixture was cooled and concentration of the solvent in vacuo gave crude product which was purified by column chromatography to give ethyl 3-chloro-2-phenyl-2H-pyrazolo[3,4-0]pyridine-6-carboxylate (2.0 g, 48%). ESI-MS m/z calculated for [M+H]+: 302.1 ; found: 302.1.
Step i: To a solution of ethyl 3-chloro-2-phenyl-2H-pyrazolo[3,4-b]pyridine-6-carboxylate (2.0 g, 6.6 mmol) in 1 ,4-dioxane (20 mL) was added 2-(cyclohex-1-en-1-yl)-4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolane (2.1 g, 9.9 mmol) and a solution of sodium carbonate (1 .4 g, 13 mmol) in water (6 mL). The mixture was purged with nitrogen and stirred for 30 min at room temperature, then palladium acetate (0.30 g, 1 .3 mmol) and triphenylphosphine (0.70 g, 26 mmol) were added and the reaction was heated at 80 °C for 2 h. Then potassium hydroxide (1.1 g, 20 mmol) was added and the reaction mixture was stirred for 1 hr at 80 °C. The reaction mixture was cooled to room temperature and concentrated. The residue was
dissolved in water and the aqueous layer was washed with ether. The aqueous layer was adjusted to pH ~4 using 1 M HCI and the product was extracted with ethyl acetate. The solvent was concentrated in vacuo and the crude product was stirred in ethanol and filtered to give 3-(cyclohex-1 -en-1 -yl)-2-phenyl-2/-/-pyrazolo[3,4-0]pyridine-6-carboxylic acid (1.1 g, 50%). ESI-MS m/z calculated for [M+H]+: 320.1 ; found: 320.0.
Step j: To a solution of 3-(cyclohex-1-en-1 -yl)-2-phenyl-2H-pyrazolo[3,4-ib]pyridine-6- carboxylic acid (1.0 g, 3.1 mmol) in methanol (20 mL) was added 10% Pd/C (0.50 g, 50% by mass) and ammonium formate (2.0 g, 31 mmol) and the reaction was heated at 50 °C for 30 min. The reaction mixture was cooled to room temperature and filtered through Celite®. The filtrate was concentrated and the residue was dissolved in water and the aqueous adjusted to pH ~4 with 1 M HCI. The product was extracted with ethyl acetate and the organic concentrated in vacuo to give a crude product that was stirred in ethanol and filtered. The precipitate was dissolved in dichloromethane and concentrated to give 3-cyclohexyl-2- phenyl-2H-pyrazolo[3,4-0]pyridine-6-carboxylic acid (0.40 g, 40%) as a white solid. ESI-MS m/z calculated for [M+H]+: 322.1 ; found: 322.2. 1H NM (400 MHz, c^-DMSO) δ 8.74 (d, J = 10.6 Hz, 1 H), 8.69 (d, J = 10.6 Hz, 1 H), 7.70-7.57 (m, 5H), 2.99-2.88 (m, 1 H), 1 .98-1.63 (m, 7H), 1.46-1.30 (m, 1 H), 1.27-1.12 (m, 2H).
Route (e): Synthesis of 3-Cyclohexyl-2-(4-hydroxyphenyl)-2H-indazole-6-carboxylic acid (11), Methyl 3-cyclohexyl-2-(4-hydroxyphenyl)-2H-indazole-6-carboxylate (12), Methyl 3- cyclohexyl-2-(4-methoxyphenyl)-2H-indazole-6-carboxylate (13), and 3-(Cyclohex-1-en-1-yl)- 2-(4-methoxyphenyl)-2H-indazole-6-carboxylic acid (14)



14
Step a: To a solution of 2-[4-(benzyloxy)phenyl]-3-(cyclohex-1 -en-1 -yl)-2/-/-indazole-6- carboxylic acid (xxiii) (prepared according to Method A using 4-(benzyloxy)aniline hydrochloride in place of aniline at Step b) (1.0 g, 2.35 mmol) in methanol (15 mL) was added 10% Pd/C (0.45 g, 0.4 mmol, 45% w/w) and ammonium formate (1 .33 g, 21.1 mmol). The reaction mixture was then heated at reflux for 1 h, cooled to RT and filtered through a pad of Celite®. The residue was washed with methanol (30 mL) and the combined filtrate washings were then concentrated in vacuo. The residue was purified by preparative HPLC to give 3-cyclohexyl-2-(4-hydroxyphenyl)-2/-/-indazole-6-carboxylic acid (11 ) as a white solid (450 mg, 57 %); ESI-MS m/z calculated for [M+H]+: 337.15; found: 337.25. 1H NMR (400
MHz, ο^-ΟΜεθ) δ 8.13 (5, 1 H), 8.91 (d, J = 8.2 Hz, 1 H), 8.54 (d, J = 9.8 Hz, 1 H), 7.30 (d, J = 8.2 Hz, 2H), 6.94 (d, J = 9.8 Hz, 2H), 2.94-2.78 (m, 1 H), 1.98-1.60 (m, 7H), 1.45-1 .09 (m , 3H).
Step b: To a stirred suspension of 3-cyclohexyl-2-(4-hydroxyphenyl)-2/-/-indazole-6- carboxylic acid (11 ) (200 mg, 0.59 mmol) in methanol (8 mL) was added concentrated sulphuric acid (1 .6 mL) at RT. The reaction mixture was then heated at reflux for 1 h and the volatiles were then removed in vacuo. The residue was diluted with water (5 mL) and the organics were extracted into ethyl acetate (3 x 15 mL) and dried (Na2S04). Upon removal of the volatiles in vacuo, the residue was triturated with ethyl acetate and dried in vacuo to give
methyl 3-cyclohexyl-2-(4-hydroxyphenyl)-2/-/-indazole-6-carboxylate (12) as an off-white solid (125 mg, 62 %); ESI-MS m/z calculated for [M+H]+: 351 .17; found: 351 .25. 1H NMR (400 MHz, CDCI3) δ 8.50 (s, 1 H), 7.91 (d, J = 1 1.1 Hz, 1 H), 7.70 (d, J = 1 1.1 Hz, 1 H), 7.27 (d, J = 9.8 Hz, 2H), 6.90 (d, J = 9.8 Hz, 2H), 3.97 (s, 3H), 3.00-2.86 (m, 1 H), 2.00-1 .74 (m, 7H), 1.40-1.22 (m, 3H).
Step c: To a stirred solution of methyl 3-cyclohexyl-2-(4-hydroxyphenyl)-2A7-indazole-6- carboxylate (12) (80 mg, 0.22 mmol) in DMF ( 2 ml_) was added potassium carbonate (94 mg, 0.68 mmol) and methyl iodide (64 mg, 0.45 mmol). The reaction mixture was then stirred at RT for 2 h. After completion of the reaction, the mixture was diluted with water (8 ml_) and extracted with ethyl acetate (3 x 15 ml_). The combined organic layer was dried (Na2S04) and concentrated in vacuo to give the crude residue. The crude product was combined with another batch of crude product derived from 20 mg (0.06 mmol) of methyl 3- cyclohexyl-2-(4-hydroxyphenyl)-2/-/-indazole-6-carboxylate (12) according to the procedure described above and purified by column chromatography on silica gel by eluting with ethyl acetate: hexane (gradient elution from 1 : 19 to 1 :9 v/v) to give methyl 3-cyclohexyl-2-(4- methoxyphenyl)-2A7-indazole-6-carboxylate (13) as a white solid (80 mg, 80 % overall yield), ESI-MS m/z calculated for [M+H]+: 365.19; found: 365.20. 1H NMR (400 MHz, CDCI3) δ 8.88 (d, J = 12.0 Hz, 1 H), 8.47 (s, 1 H), 7.66 (d, J = 10.2 Hz, 1 H), 7.40 (d, J = 10.2 Hz, 2H), 7.06 (d, J = 10.2 Hz, 2H), 3.95 (s, 3H), 3.90 (s, 3H), 3.00-2.89 (m, 1 H), 1 .99-1.74 (m, 7H), 1.43- 1 .20 (m, 3H).
Step d: To a stirred solution of methyl 3-cyclohexyl-2-(4-methoxyphenyl)-2/-/-indazole-6- carboxylate (13) (55 mg, 0.15 mmol) in ethanohwater (1 : 1 v/v, 2 mL) was added sodium hydroxide (1 10 mg, 2.75 mmol) and the reaction mixture was heated at reflux for 1 h. Upon cooling to RT, the reaction mixture was acidified with 1 N aqueous HCI and the organics were extracted into ethyl acetate (3 x 10 mL). The combined organics were dried (Na2S04), concentrated in vacuo and the residue then purified by trituration with ethyl acetate in hexane (15%, 2 x 4 mL) to give 3-(cyclohex-1 -en-1 -yl)-2-(4-methoxyphenyl)-2/-/-indazole-6- carboxylic acid (14) as a white solid (20 mg, 38 %); ESI-MS m/z calculated for [M+H]+: 351.17; found: 351.20; 1H-NMR (400 MHz, CDCI3) δ 8.60 (s, 1 H), 7.91 (d, J = 9.9 Hz, 1 H), 7.70 (d, J = 9.9 Hz, 1 H), 7.41 (d, J = 8.7 Hz, 2H), 7.07 (d, J = 8.7 Hz, 2H), 3.92 (s, 3H), 3.03- 2.90 (m, 1 H), 2.00-1 .72 (m, 7H), 1 .44-1.22 (m, 3H).
Method B: Synthesis of Precursor Coupling Intermediates
Route (a): An amine (i) was coupled with a N-boc protected amino acid (which may have common side-chain protecting groups), using standard peptide coupling conditions (eg. HATU, DIPEA in DMF). If intermediate iii has X as a halide or boronic acid or ester, standard cross-coupling conditions with an appropriate aryl or vinyl compound were
employed to give extended an intermediate (iv). The W-boc group of iii or iv is deprotected usin HCI in dioxane in most cases.

v
Example: Synthesis of the reagent ethyl (2E)-3-(4-{[(1-aminocyclobutyl)carbonyl] amino}phenyl)prop-2-enoate hydrochloride (vii)
Rea ent (vii) was prepared according to the followin procedure:

VIII IX

vii
XI
Step a: To a stirred solution of 1 -[(ferf-butoxycarbonyl)amino]cyclobutanecarboxylic acid
(ix) (100 mg, 0.46 mmol) in DMF (15 mL) at 0 °C was added HATU (265 mg, 0.70 mmol), 4- iodoaniline (viii) (1 12 mg, 0.51 mmol) and DIPEA (0.124 mL, 0.70 mmol) . The reaction mixture was allowed to warm to RT and stirred for 15 h and then quenched with the addition of ice. The precipitate was separated by filtration, washed with hexane (3 x 10 mL) and dried in a stream of air to give ferf-butyl {1 -[(4-iodophenyl)carbamoyl]cyclobutyl}carbamate (x) as an off-white powder (1 15mg, 60 %); ESI-MS m/z calculated for [M+H-C(CH3)3]+: 361.00; found: 361 .05.
Step b: To a stirred solution of ferf-butyl {1-[(4-iodophenyl)carbamoyl]cyclobutyl} carbamate
(x) (100 mg, 0.24 mmol) and ethyl acrylate (52 pL, 0.48 mmol) in degassed DMF (5 was added palladium (II) acetate (8 mg, 0.04 mmol), triphenylphosphine (19 mg, 0.07 mmol) and triethylamine (67 pL, 0.48 mmol) at RT. The reaction mixture was then heated at 85 °C for
15h, cooled to RT and filtered through a plug of celite ®. The filtrate was diluted with ethyl acetate (20 mL) and the combined organics washed with water (15 mL), brine and dried (Na2S04). The volatiles were removed in vacuo and the residue was purified by automated column chromatography (Combi-Flash™) on silica gel eluting with ethyl acetate:hexane (3:7 v/v) to give ethyl (2E)-3-{4-[({1 -[(ferf-butoxycarbonyl)amino]cyclobutyl
carbonyl)amino]phenyl}prop-2-enoate (xi) as an off-white solid (58 mg, 62%); ESI-MS m/z calculated for [M+Na]+: 41 1 .19; found: 41 1.35 [M+Na]+
Step c: 4M HCI in dioxane (2 mL) was added to ethyl (2E)-3-{4-[({1-[(ferf- butoxycarbonyl)amino]cyclobutyl}carbonyl)amino]phenyl}prop-2-enoate (xi) (350 mg, 0.90 mmol) and the resultant suspension stirred for 5 h at RT. The volatiles were then removed in vacuo and the residue was then was dissolved in water (7 mL) and washed with ethyl acetate (2 x 10 mL). The aqueous layer was basified by addition of saturated solution of sodium bicarbonate and the organics were then extracted into ethyl acetate (3 x 20 mL). The combined organics were washed with brine (50 mL), dried (Na2S04) and concentrated in vacuo. The residue was purified by trituration with ethyl actete (1 mL) to give ethyl (2E)-3-(4- {[(l-aminocyclobutyl)carbonyl] amino}phenyl)prop-2-enoate hydrochloride (vii) (730 mg, 75 %) as a white solid; ESI-MS m/z calculated for [M+H]+: 289.15; found: 289.15; 1 H-NMR (400 MHz, CDCI3) δ 9.46 (s, 1 H), 7.60-7.74 (m, 3H), 7.51 (d, J 10.0 Hz, 2H), 6.35 (d, J 14.3 Hz, 1 H), 4.26 (q, J 7.1 Hz, 2H), 2.74-2.91 (m, 2H), 2.00-2.16 (m, 2H), 1.84-1 .97 (m, 2H), 1 .33 (t, J 7.1 Hz, 3H).
Example: Synthesis of the reagent N-[4-(Pyridin-4-yl)phenyl]-D-alaninamide hydrochloride (xii)
Reagent (xii) was synthesised as follows:

AND Enantiomer AND Enantiomer
Step a: To a stirred solution of N-(ferf-butoxycarbonyl)-D-alanine (xiii) (1.0 g, 5.3 mmol) in dry DMF (10 mL) under argon was added 4-iodoaniline (xiv) (1.4 g, 6.39 mmol) and DIPEA (1.4 mL, 8.0 mmol), followed by HATU (2.41 g, 6.34 mmol). The solution was stirred under argon at RT for 1 h. Ethyl acetate (100 mL) was added and this was washed with 1 M aqueous HCI (2 x 40 mL), aqueous sodium bicarbonate (40 mL) and brine (40 mL) then dried (MgS04) and concentrated in vacuo. The residue was purified by automated flash chromatography (Biotage SP4, 60 g Biotage SNAP™ C-18 silica gel cartridge) eluting with acetonitrile:water (0% to 100%) to yield ferf-butyl {(2R)-1-[(4-iodophenyl)amino]-1 - oxopropan-2-yl}carbamate (xv) (1.75 g, 84 %); ESI-MS m/z calculated for [M+H]+: 391.1 ; found: 390.6.
Step b: ferf-Butyl {(2R)-1-[(4-iodophenyl)amino]-1-oxopropan-2-yl}carbamate (xv) (201 mg, 0.52 mmol), pyridine-4-boronic acid (99 mg, 0.81 mmol) and sodium carbonate (1 1 1 mg, 1 .05 mmol) in a mixture of dioxane (2 mL) and water (2 mL) was degassed with a stream of argon. Palladium acetate (25 mg, 0.1 1 mmol) and triphenylphosphine (58 mg, 0.22 mmol) were added and the reaction was heated at 100°C for 2 h. The crude reaction mixture was purified by automated flash chromatography (Biotage SP4, 12 g Grace Reveleris™ C-18 silica gel cartridge) eluting with acetonitrile:water (0% to 100%) to yield ierf-butyl [(2R)-1 - oxo-1 -{[4-(pyridin-4-yl)phenyl]amino}propan-2-yl]carbamate (xvi) (92 mg, 52 %); ESI-MS m/z calculated for [M+H]+: 342.2; found: 342.0.
Step c: A suspension of ferf-butyl [(2R)-1 -oxo-1-{[4-(pyridin-4-yl)phenyl]amino}propan-2- yl]carbamate (xvi) (92 mg, 0.27 mmol) in 4M HCI in dioxane (4 mL) was stirred at RT for 1.5 h. Concentration of the reaction mixture gave W-[4-(pyridin-4-yl)phenyl]-D-alaninamide hydrochloride (xii), which was used unpurified, ESI-MS m/z calculated for [M+H]+: 242.1 ; found: 242.0.
Example: Synthesis of the reagent 4-{[(1-aminocyclobutyl)carbonyl]amino}-N- (methylsulfonyl)benzamide hydrochloride (xvii)
Rea ent (xvii) was prepared as follows:

Step a: To a solution of W-Boc-1 -aminocyclobutanecarboxylic acid (xviii) (1.00 g, 4.65 mmol) and methyl-4-aminobenzoate (xix) (0.85 g, 5.62 mmol) in dry DMF (10 mL) under Ar
was added DIPEA (1.21 mL, 6.80 mmol) added, followed by HATU (2.13 g, 5.60 mmol). The reaction mixture was stirred at RT under argon and monitored by LCMS. After 16 h, the reaction mixture was concentrated to dryness in vacuo, dissolved in DMSO (10 mL) and purified by automated column chromatography (Biotage SP4, 60 g Biotage SNAP™ C-18 silica gel cartridge) eluting with acetonitrile:water (0% to 100%). The relevant fractions were concentrated in vacuo and the residue was dissolved in ethyl acetate (50 mL) and washed with 1 M aqueous HCI (30 mL), saturated sodium bicarbonate solution (30 mL) and brine (30 mL). The organics were dried (MgS04) and concentrated in vacuo to give methyl 4-[({1 - [(iert-butoxycarbonyl)amino]cyclobutyl}carbonyl) amino]benzoate (xx) as an off-white solid (691 mg, 43 %); ESI-MS m/z calculated for [M+H]+: 349.18; found: 348.76.
Step b: Methyl 4-[({1-[(ferf-butoxycarbonyl)amino]cyclobutyl}carbonyl)amino] benzoate (xx) (325 mg, 0.93 mmol) and lithium hydroxide hydrate (238 mg, 5.67 mmol) in dioxane (3 mL), /so-propanol (3 mL), and water (3.4 mL) were stirred at 60 °C for 45 min. The reaction mixture was then cooled to RT, acidified with 1 M aqueous HCI and then concentrated in vacuo. The residue was dissolved in DMSO (5 mL) and water (5 mL) and purified by automated column chromatography (Biotage SP4, 60 g Biotage SNAP™ C-18 silica gel cartridge) eluting with acetonitrile:water (0% to 100%) to give 4-[({1-[(ferf- butoxycarbonyl)amino]cyclobutyl}carbonyl) amino]benzoic acid (xxi) as a white solid (21 1 mg, 68 %); ESI-MS m/z calculated for {[M+H]+-[C(CH3)]}+: 279.10; found: 278.85.
Step c: To a solution of 4-[({1 -[(ierf-butoxycarbonyl)amino]cyclobutyl}carbonyl)
amino]benzoic acid (xxi) (21 1 mg, 0.63 mmol) in dry DMF (5 mL) under argon was added DIPEA (0.33 mL, 1 .89 mmol) followed by HATU (295 mg, 0.76 mmol) and the resultant solution was stirred at RT for 30 min. In the meantime, to a solution of
methanesulphonamide (31 1 mg, 3.27 mmol) in dry THF (4 mL) under argon was added sodium hydride (60 % dispersion in mineral oil, 102 mg, 2.55 mmol) the resultant reaction mixture was stirred for 30 min. To this suspension was added slowly the solution containing HATU and 4-[({1-[(ferf-butoxycarbonyl)amino] cyclobutyl}carbonyl)amino]benzoic acid (xxi) in DMF and DIPEA and the combined reaction mixture was stirred for 2 h. Water (2 mL) was added to the reaction mixture and the volatiles were removed in vacuo. The residue was dissolved in DMSO (4 mL) and water (4 mL) then purified by automated column chromatography (Biotage SP4, 60 g Biotage SNAP™ C-18 silica gel cartridge) eluting with acetonitrile:water (0% to 100%) to give ierf-butyl [1 -({4-
[(methylsulfonyl)carbamoyl]phenyl}carbamoyl) cyclobutyl]carbamate (xxii) as a white solid (245 mg, 95 %); ESI-MS m/z calculated for {[M+H]+-[C(CH3)]}+: 356.09; found: 355.88. Step d: [1 -({4-[(Methylsulfonyl)carbamoyl]phenyl}carbamoyl)cyclobutyl]carbamate (xxii) (245 mg, 0.60 mmol) was suspended in 4M HCI in dioxane (4 mL) and the reaction mixture
was stirred at RT for 1 .5 h. The volatiles were removed in vacuo and the residue 4-{[(1- aminocyclobutyl)carbonyl]amino}-A/-(methylsulfonyl)benzamide hydrochloride (xvii) was used without purification; ESI-MS m/z calculated for [M+H]+: 312.10; found: 31 1 .89.
Route (b): An W-protected aziridine, pyrrolidine or piperidine (i) (where the W-protecting group is Boc or Cbz) is deprotected then reductive amination with formaldehyde gives the N- meth l derivative iii.

Step b: To a suspension of 4-{[(9H-fluoren-9-ylmethoxy)carbonyl]amino}piperidine-4- carboxylic acid hydrochloride (0.35 g, 0.86 mmol) in MeOH (5 mL) at room temperature in a medium pressure vessel was added an aqueous solution of formaldehyde (37%, 70 pL, 0.94 mmol), followed by catalytic amounts of 10% Pd/C (90 mg, 0.10 mmol). The reaction was stirred at room temperature at 6-7 Bar under an atmosphere of hydrogen for a total of 27.5 h, during which time there were two further additions of formaldehyde and one addition of 10% Pd/C. The catalyst was removed by filtration through a 0.45 μΜ filter and washed with MeOH. The filtrate was concentrated in vacuo and azeotroped with MeOH/toluene. The residue was dried under high vacuum to give 4-{[(9H-fluoren-9-ylmethoxy)carbonyl]amino}- 1 -methylpiperidine-4-carboxylic acid (iii) (0.31 g, 94% over two steps) as a yellow solid. ESI- MS m/z calculated for [M+H]+: 381.2; found: 381.1.
Method C: Coupling
Route (a): A functionalised amino acid (i) prepared according to Method B Route (a) is coupled to a compound of general formula II (ii) (prepared according to Method A), under standard peptide coupling conditions. When Q is a carboxylic ester, the ester can be hydrolysed to a carboxylic acid under basic conditions. After coupling, some standard transformations such as cleavage of common protecting groups may be employed. The final


2
VII

Step a: To a stirred solution of 3-cyclohexyl-2-phenyl-2/-/-indazole-6-carboxylic acid (2) (200 mg, 0.62 mmol) and ethyl (2E)-3-(4-{[(1 -aminocyclobutyl)carbonyl]amino} phenyl)prop-2- enoate hydrochloride (vii) (180 mg, 0.62 mmol) in DMF (15 mL) was added HATU (356 mg, 0.94 mmol) followed by DIPEA (326 mL, 1.87 mmol). The reaction mixture was then stirred at RT for 16 h and then poured onto water (30 mL). The organics were extracted into ethyl acetate (3 x 30 mL) and the combined organics dried (MgS04) and concentrated in vacuo. The residue was purified by automated column chromatography (Biotage SP4,
GraceResolv™ silica gel 12 g cartridge) eluting with ethyl acetate:hexane (20% to 80 %) to give ethyl (2E)-3-(4-{[(1-{[(3-cyclohexyl-2-phenyl-2/-/-indazol-6-
yl)carbonyl]amino}cyclobutyl)carbonyl]amino}phenyl)prop-2-enoate (3) as a white powder (226 mg, 61 %); ESI-MS m/z calculated for [M+H]+: 591.30; found: 591.12.
1H NMR (400 MHz, c -DMSO) δ 9.70 (s, 1Η), 8.97 (s, 1Η), 8.41 (s, 1H), 8.01 (d, J = 9.8Hz, 1H), 7.73-7.49 (m, 11H), 6.50 (d, J= 16.4 Hz, 1H), 4.15 (q, J = 9.23 Hz, 2H), 2.96-2.84 (m, 1H), 2.77-2.65 (m, 2H), 2.44-2.31 (m, 2H), 2.02-1.64 (m, 9H), 1.47-1.31 (m, 1H), 1.30-1.11 (m, 5H).
Similarly prepared were the following compounds.

using N-Boc-D-alanine instead of N-Boc-1-aminocyclobutanecarboxylic acid and methyl 4- aminocinnamate instead of ethyl 4-aminocinnamate; ' using methyl or ethyl 4-aminobenzoate instead of 4-aminobenzonitrile.
Example: Synthesis of 3-Cyclohexyl-N-[(2R)-1-oxo-1-{[4-(pyridin-4-yl)phenyl]amino}propan- 2-yl]-2-phenyl-2H-indazole-6-carboxamide (7)

AND Enantiomer
Step a: To a solution of /V-[4-(pyridin-4-yl)phenyl]-D-alaninamide hydrochloride (xii) (9 mg, 0.037 mmol) in dry DMF (500 L) under argon was added 3-cyclohexyl-2-phenyl-2A7- indazole-6-carboxylic acid (2) (1 1 mg, 0.034 mmol) and DIPEA (17 μΙ_, 0.098 mmol), followed by HATU (15 mg, 0.039 mmol). The solution was stirred under argon at RT for 3.5 h. Ethyl acetate (2 mL) was added and this was washed with 1 M aqueous HCI (1 mL), aqueous sodium bicarbonate (1 mL) and brine (1 mL) then dried (MgS04) and concentrated in vacuo. The residue was purified by preparative HPLC to yield 3-cyclohexyl-W-[(2R)-1-oxo- 1 -{[4-(pyridin-4-yl)phenyl]amino}propan-2-yl]-2-phenyl-2H-indazole-6-carboxamide (7) (2 mg, 1 1 %); ESI-MS m/z calculated for [M+H]+: 544.3; found: 544.3; 1H-NMR (400 MHz, acetone- cfe) δ 9.69 (s, 1 H), 8.60-8.63 (m, 2H), 8.33 (s, 1 H), 8.10 (brd, J 7.1 Hz, 1 H), 8.05 (d, J 9.1 Hz, 1 H), 7.85-7.90 (m, 2H), 7.75-7.80 (m, 2H), 7.58-7.70 (m, 8H), 4.82-4.91 (m, 1 H), 3.02-3.1 1 (m, 1 H), 1 .70-2.04 (m, 7H), 1.60 (d, J 7.1 Hz, 3H),
1 .23-1.51 (m, 3H).
Similarly prepared were the following compounds.
No Name [ +H]+ NMR
ESI-MS
m/z
24,a) N-[(2R)-1-{[4-(1 ,3-benzodioxol- 587.2 Ή NMR (400 MHz, Acetone) δ 9.53
5-yl)phenyl]amino}-1- (s, 1 H), 8.32 (s, 1 H), 8.04 (d, J = oxopropan-2-yl]-3-cyclohexyl- 8.9 Hz, 2H), 7.77 (d, J = 8.1 Hz,
2-phenyl-2H-indazole-6- 2H), 7.70 - 7.51 (m, 8H), 7.18 - carboxamide 7.10 (m, 2H), 6.92 (d, J = 7.9 Hz,
1 H), 6.04 (s, 2H), 4.91 - 4.80 (m, 1 H), 3.13 - 3.00 (m, 1 H), 2.13 - 1.70 (m, 7H), 1 .59 (d, J = 7.1 Hz, 3H), 1 .51 - 1.21 (m, 3H).

using 3,4-methylenedioxyphenyl boronic acid instead of pyridine-4-boronic acid;
3-boronic acid instead of pyridine-4-boronic acid.
Example: Synthesis of 3-cyclohexyl-N-[1 -({4-[(methylsulfonyl)carbamoyl]phenyl} carbamoyl) cyclobutyl]-2-phenyl-2H-indazole-6-carboxamide (10)

Step a: To a stirred solution of 3-cyclohexyl-2-phenyl-2/-/-indazole-6-carboxylic acid (2) (102.9 mg, 0.32 mmol) and 4-{[(1 -aminocyclobutyl)carbonyl]amino}-A- (methylsulfonyl)benzamide hydrochloride (xvii) (120.9 mg, 0.39 mmol) in dry DMF (2 mL) under argon was added DIPEA (170 pL, 0.95 mmol) followed by HATU (148.9 mg, 0.39 mmol). The mixture was stirred at RT for 3 h. The reaction mixture was then diluted with ethyl acetate (5 mL) and washed with 1 M HCI (2 x 2 mL), saturated sodium bicarbonate solution (2 mL), brine (2 mL) and then concentrated in vacuo. This crude material was dissolved in DMSO (2 mL) and purified by automated column chromatography (Biotage SP4, 12 g Biotage SNAP™ C-18 silica gel cartridge) eluting with acetonitrile:water (0% to 100%) to give 3-cyclohexyl-W-[1 -({4-[(methylsulfonyl)carbamoyl]phenyl}carbamoyl)cyclobutyl]-2- phenyl-2H-indazole-6-carboxamide (10) as a white solid (80 mg, 42%); ESI-MS m/z calculated for [M+H]+: 614.24; found: 614.14; 1 H-NMR (400 MHz, acetone- 4) δ 9.92 (s, 1 H),
8.50 (s, 1 H), 8.36 (s, 1 H), 8.04 (dd, J 9 Hz, 0.68Hz, 1 H), 7.99 (d, J 8.8 Hz, 2H), 7.81 (d, J 8.8 Hz, 2H), 7.59-7.69 (m, 6H), 3.36 (s, 3H), 3.02-3.10 (m, 1 H), 2.88-2.95 (m, 2H), 2.45-2.52 (m, 2H), 1.72-2.04 (m, 9H), 1.23-1.50 (m, 3H).
Example: Synthesis of N-{1-[(4-cyanophenyl)carbamoyl]cyclobutyl}-3-cyclohexyl-2-phenyl- 2H-indazole-6-carboxamide acid (16) and N-{1-[(4-carbamoylphenyl)carbamoyl]cyclobutyl}- 3-c clohexyl-2-phenyl-2H-indazole-6-carboxamide (17)

Step a: To a solution of 1-amino-W-(4-cyanophenyl)cyclobutanecarboxamide hydrochloride (xxiv) (70 mg, 0.325 mmol) in dry DMF (1 imL) under argon was added 3-cyclohexyl-2- phenyl-2A7-indazole-6-carboxylic acid (2) prepared according to Method A (86 mg, 0.268 mmol) and DIPEA (150 μΙ_, 0.861 mmol), followed by HATU (125 mg, 0.327 mmol). The solution was stirred under argon at T for 16 h. Ethyl acetate (3 mL) was added and this was washed with 1 M aqueous HCI (2 x 1 mL), aqueous sodium bicarbonate (1 mL) and brine (1 mL) then dried (MgS04) and concentrated in vacuo. The residue was purified by automated flash chromatography (Biotage SP4, Grace esolv™ silica gel 12 g cartridge) eluting with methanohdichloromethane (0% to 20 %) to yield A/-{1-[(4- cyanophenyl)carbamoyl]cyclobutyl}-3-cyclohexyl-2-phenyl-2/-/-indazole-6-carboxamide acid (16) as a white solid (35 mg, 25 %); ESI-MS m/z calculated for [M+H]+: 518.3; found: 518.2; 1 H-NMR (400 MHz, CDCI3) δ 10.39 (bs, 1 H), 8.20 (s, 1 H), 7.94 (d, J 8.8 Hz, 1 H), 7.71 -7.75 (m, 2H), 7.53-7.62 (m, 5H), 7.45-7.49 (m, 3H), 7.20 (bs, 1 H), 2.93-3.02 (m, 3H), 2.38-2.48 (m, 2H), 1 .99-2.15 (m, 2H), 1.69-1.98 (m, 7H), 1.21 -1 .44 (m, 3H).
Step b: To a stirred solution of W-{1 -[(4-cyanophenyl)carbamoyl]cyclobutyl}-3-cyclohexyl-2- phenyl-2A7-indazole-6-carboxamide (16) (21 mg, 0.41 mmol) in methanol (400 pL) and THF (1 mL) was added 28% aqueous ammonia (200 pL) and 30% aqueous hydrogen peroxide (200 )L). The reaction was heated at 60°C for 4h. More 28% aqueous ammonia (100 pL) and 30% aqueous hydrogen peroxide (100 pL) were added at this time and heating was continued at 60 °C for 2 h. The solution was cooled to RT, quenched with aqueous
ammonium chloride (1 mL) and then extracted with ethyl acetate (3 x 2 mL). The combined organic portions were dried (MgS04) and concentrated in vacuo. The residue was purified by automated flash chromatography (Biotage SP4, 12 g Grace eveleris™ C-18 silica gel cartridge) eluting with acetonitrile:water (0% to 100%) to yield Λ/-{1 -[(4- carbamoylphenyl)carbamoyl]cyclobutyl}-3-cyclohexyl-2-phenyl-2/-/-indazole-6-carboxamide (17) as a white solid (17 mg, 78 %); ESI-MS m/z calculated for [M+H]+: 536.27; found: 536.13; 1 H-NMR (400 MHz, acetone-cfe) δ 9.82 (s, 1 H), 8.53 (s, 1 H), 8.37 (s, 1 H), 8.03 (d, J 9 Hz, 1 H), 7.90 (d, J 8.7 Hz, 2H), 7.74 (d, J 8.7 Hz, 2H), 7.58-7.69 (m, 6H), 7.37 (br s, 1 H), 6.50 (br s, 1 H), 3.02-3.10 (m, 1 H), 2.85-2.94 (m, 2H), 2.45-2.55 (m, 2H), 1.72-2.10 (m, 9H), 1 .23-1.49 (m, 3H).
N.B. The reagent 1 -amino-/V-(4-cyanophenyl)cyclobutanecarboxamide hydrochloride (xxiv) was prepared as follows:

Step a: To a solution of /V-Boc-1 -aminocyclobutanecarboxylic acid (xxvi) (501 mg, 2.33 mmol) and 4-aminobenzonitrile (xxv) (330 mg, 2.79 mmol) in dry DMF (5 mL) under argon was added DIPEA (610 3.50 mmol) added, followed by HATU (1 .07 g, 2.81 mmol). The reaction mixture was stirred at RT under argon for 16 h. The reaction mixture was then concentrated to dryness in vacuo, dissolved in DMSO (5 mL) and purified by automated column chromatography (Biotage SP4, 60 g Biotage SNAP™ C-18 silica gel cartridge) eluting with acetonitrile:water (0% to 100%). The relevant fractions were concentrated in vacuo and the residue was dissolved in ethyl acetate (50 mL) and washed with 1 M aqueous HCI (30 mL), saturated sodium bicarbonate solution (30 mL) and brine (30 mL). The organics were dried (MgS04) and concentrated in vacuo to give ferf-butyl {1 -[(4- cyanophenyl)carbamoyl]cyclobutyl}carbamate (xxvii) as an off-white solid (127 mg, 17 %); ESI-MS m/z calculated for [M+H]+: 316.2; found: 315.9.
Step b: ferf-butyl {1 -[(4-cyanophenyl)carbamoyl]cyclobutyl}carbamate (xxvii) (76 mg, 0.24 mmol) was suspended in 4M HCI in dioxane (2 mL) and the reaction mixture was stirred at
RT for 1 .5 h. The volatiles were removed in vacuo and the residue 1 -amino-W-(4- cyanophenyl)cyclobutanecarboxamide hydrochloride (xxiv) was used without purification; ESI-MS m/z calculated for [M+H]+: 216.1 ; found: 215.9.
Similarly prepared were the following compounds.

(cyanomethyl)phenyl]carbamoy (s, 1H), 8.06 (d, J = 8.3Hz, 1H), l}cyclobutyl)-3-cyclohexyl-2- 7.75-7.63 (m, 3H), 7.63-7.51 phenyl-2H-indazole-6- (m, 5H), 7.33 (d, J= 8.6 Hz, 2H), carboxamide 3.87 (s, 2H), 3.06-2.78 (m, 3H),
2.57-2.34 (m, 2H), 2.24-1.67 (m, 9H), 1.56-1.16 (m, 3H).
41(÷) N-(1-{[4-(2-amino-2- 550.1 Ή NMR (400 MHz, MeOD) δ 8.28 oxoethyl)phenyl]carbamoyl}cycl (s, 1H), 8.06 (d, J = 8.9Hz, 1H), obutyl)-3-cyclohexyl-2-phenyl- 7.72-7.62 (m, 3H), 7.62-7.46 2H-indazole-6-carboxamide (m, 5H), 7.27 (d, J= 8.7 Hz, 2H),
3.49 (s, 2H), 3.09-2.79 (m, 3H), 2.55-2.37 (m, 2H), 2.22-1.69 (m, 9H), 1.53-1.12 (m, 3H). using 4-iodoaniline instead of 4-aminobenzonitrile; using 4-iodoaniline instead of 4- aminobenzonitrile and N-Boc-D-alanine instead of N-Boc-1-aminocyclobutanecarboxylic acid; ^ using ethyl 4-aminophenylacetate instead of 4-aminobenzonitrile; ^ using 4-(1 H-tetrazol-5- yl)aniline instead of 4-aminobenzonitrile; (e) using methyl 4-aminobenzoate instead of 4- aminobenzonitrile; () using 4-aminobenzyl cyanide instead of 4-aminobenzonitrile.
Example: Synthesis of (2E)-3-[4-({N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-L- ser l}amino)phenyl]prop-2-enoic acid (121)
Step a: To a stirred solution of 3-cyclohexyl-2-phenyl-2/-/-indazole-6-carboxylic acid (2) (42 mg, 0.13 mmol) and ethyl (2E)-3-{4-[(0-benzyl-L-seryl)amino]phenyl}prop-2-enoate hydrochloride (1:1) (i) (64 mg, 0.16 mmol) in dry DMF (0.8 mL) was added DIPEA (91 μΙ_,
0.52 mmol), followed by HATU (60 mg, 0.16 mmol). After 17 h at room temperature the
reaction was quenched with 1 M HCI and diluted with water. The precipitate was collected by filtration and dried in a desiccator, affording ethyl (2E)-3-[4-({0-benzyl-W-[(3-cyclohexyl-2- phenyl-2/-/-indazol-6-yl)carbonyl]-L-seryl}amino)phenyl]prop-2-enoate (ii) (80 mg, 91 %) as a pale brown solid which became a gum upon standing.
Step b: To a solution of ethyl (2E)-3-[4-({0-benzyl-/V-[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]-L-seryl}amino)phenyl]prop-2-enoate (40 mg, 60 μηηοΙ) in dichloromethane (0.5 mL) cooled to -78 °C was added dropwise 1 M boron tribromide in dichloromethane (0.18 mL, 0.18 mmol). The resulting mixture was removed from the cold bath and after 30 min the reaction was quenched with water and extracted with ethyl acetate. The organic was washed with saturated NaHC03 then brine, dried (MgS04), filtered and concentrated in vacuo. The crude product was purified by reverse-phase chromatography (55-100% MeCN in water+0.1 % formic acid) to give ethyl (2E)-3-[4-({/V-[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]-L-seryl}amino)phenyl]prop-2-enoate (iii) (13 mg, 38%) as a white solid. ESI-MS m/z calculated for [M+H]+: 581 .3; found: 581.3.
Step c: To a solution of ethyl (2E)-3-[4-({/V-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]- L-seryl}amino)phenyl]prop-2-enoate (6.0 mg, 10 pmol) in ethanol (0.5 mL) was added a solution of potassium carbonate (14 mg) in water (0.5 mL). The resulting mixture was heated at 80 °C for 2 h. The reaction was then cooled to room temperature and quenched with 0.5 M HCI. The precipitate was collected by filtration, washed with water and dried in a desiccator. The crude product was purified by reverse phase chromatography to give (2E)-3- [4-({/V-[(3-cyclohexyl-2-phenyl-2/-/-indazol-6-yl)carbonyl]-L-seryl}amino)phenyl]prop-2-enoic acid (121 ) (3.0 mg, 53%) as a white solid. ESI-MS m/z calculated for [M+H]+: 553.2; found: 553.3. 1H NMR (400 MHz, MeOD) δ 8.27 (d, J = 0.8 Hz, 1 H), 8.15 - 7.96 (m, 1 H), 7.84 - 7.44 (m, 1 1 H), 6.45 (d, J = 15.9 Hz, 1 H), 4.81 - 4.42 (m, 1 H), 4.05 (d, J = 5.5 Hz, 2H), 3.06 - 2.86 (m, 1 H), 2.15 - 1 .68 (m, 7H), 1 .61 - 1 .07 (m, 3H).
The following compounds were similarly prepared in accordance with general Method C Route (a) and with reference to the Examples previously described.


[(2R)-4-({4-[(1E)-3- 1 H), 8.04 (d, J = 8.9 Hz, 1 H), 7.72 - 7.62
(methylamino)-3- (m, 5H), 7.62 - 7.43 (m, 6H), 6.53 (d, J = oxoprop-1-en-1- 15.7 Hz, 1H), 4.71 - 4.57 (m, 1H), 3.06 - yl]phenyl}amino)-4- 2.92 (m, 1H), 2.92 - 2.85 (m, 3H), 2.85 - oxobutan-2-yl]-2- 2.59 (m, 2H), 2.12-1.69 (m, 7H), 1.53 - phenyl-2H-indazole- 1.19 (m, 6H).
6-carboxamide
(2E)-3-[4-({[1-({[3- 564.1 1H NMR (400 MHz, c -DMSO) δ 12.14 cyclohexyl-2- (brs, 1H), 9.72 (s, 1H), 9.07 (s, 1H), 8.73- (pyridin-2-yl)-2H- 8.67 (m, 1H), 8.44 (s, 1H), 8.17 (td, J = indazol-6- 15.6 and 1.9 Hz, 1H), 8.04 (d, J= 9.0 Hz, yl]carbonyl}amino)cy 1 H), 7.91 (d, J = 8.0 Hz, 1 H), 7.74-7.58 clobutyl]carbonyl}am (m, 5H), 7.56-7.46 (m, 2H), 6.41 (d, J = ino)phenyl]prop-2- 15.9 Hz, 1H), 3.63-3.50 (m, 1H), 2.81- enoic acid 2.66 (m, 2H), 2.45-2.31 (m, 2H), 2.07- 1.65 (m,9H), 1.50-1.20 (m, 3H).
(2E)-3-(4-{[(1-{[(3- 577.2 1H NMR (400 M Hz, MeOD) δ 8.24 (s, cyclohexyl-2-phenyl- 1H), 8.03 (d, J = 8.9 Hz, 1H), 7.69 - 7.49
2H-indazol-6- (m, 11H), 6.43 (d, J = 16.0 Hz, 1H), 3.03 yl)carbonyl]amino}cy - 2.92 (m, 1 H), 2.54 - 2.43 (m, 2H), 2.25 - clopentyl)carbonyl]a 2.15 (m,2H), 2.08- 1.72 (m, 11H), 1.50- mino}phenyl)prop-2- 1.20 (m, 6H).
enoic acid
3-cyclohexyl-N-(1- 538.2 1H NMR (400 MHz, c -DMSO) δ 9.56 (s, «2- 1H), 9.29 (s, 1H), 8.48 (s, 2H), 8.40 (s,
(dimethylamino)pyri 1H), 8.00 (d, J= 9.0 Hz, 1H), 7.70-7.52 midin-5- (m, 6H), 3.08 (s, 6H), 2.98-2.84 (m, 1H), yl]carbamoyl}cyclob 2.76-2.63 (m, 2H), 2.44-2.30 (m, 2H), utyl)-2-phenyl-2H- 2.05-1.62 (m, 9H), 1.49-1.31 (m, 1H), indazole-6- 1.30-1.21 (m, 2H).
carboxamide
(2E)-3-(4-{[(1-{[(3- 591.2 1H NMR (400 MHz, Acetone) δ 9.81 (br cyclohexyl-2-phenyl- s, 1 H), 8.32 - 8.23 (m, 1 H), 8.02 (dd, J =
2H-indazol-6- 8.9, 0.8 Hz, 1H), 7.80-7.49 (m, 12H), yl)carbonyl]amino}cy 6.43 (brd, J= 16.0 Hz, 1H), 3.11 -2.99 clohexyl)carbonyl]a (m, 1H), 2.47 (d, J= 13.5 Hz, 2H), 2.10- mino}phenyl)prop-2- 1.79 (m, 8H), 1.78 - 1.64 (m, 6H), 1.49 - enoic acid 1.36 (m,2H), 1.35-1.20 (m, 2H).
4-{[(1-{[(3- 538.1 1H NMR (400 MHz, c -DMSO) δ 9.82 (s, cyclohexyl-2-phenyl- 1H), 9.27 (s, 1H), 8.69 (d, J= 8.2 Hz,
2H-pyrazolo[3,4- 1H), 7.85 (brd, J= 7.8 Hz, 2H), 7.77-7.58 b] pyridin-6- (m, 8H), 3.00-2.90 (m, 1H), 2.83-2.72 (m, yl)carbonyl]amino}cy 2H), 2.44-2.33 (m, 2H), 2.05-1.62 (m, clobutyl)carbonyl]am 9H), 1.47-1.32 (m, 1H), 1.28-1.12 (m, ino}benzoic acid 2H).
(2E)-3-[4-({N-[(3- 551.2 1H NMR (400 MHz, c -DMSO) δ 9.99 (s, cyclohexyl-2-phenyl- 1H), 9.14 (s, 1H), 8.42 (s, 1H), 8.01 (d, J
2H-indazol-6- = 9.2 Hz, 1H), 7.83-7.50 (m, 9H), 7.44 yl)carbonyl]-2- (dd, J = 8.7 and 1.5 Hz, 1 H), 2.96-2.85 methylalanyl}amino) (m, 1H), 2.78-2.66 (m, 2H), 2.45-2.31 (m, phenyl]prop-2-enoic 2H), 2.06-1.74 (m, 8H), 1.68 (brd, J = acid 12.3 Hz, 1H), 1.47-1.32 (m, 1H), 1.31-











carboxamide
162(c)'(9) N-(1-{[4-(2- 616.3 1 H NMR (400 MHz, MeOD) δ 8.55 (s, aminopyrimidin-5- 2H), 8.29 (s, 1 H), 8.06 (d, J = 8.9 Hz, yl)phenyl] carbamoyl 1 H), 7.68 (d, J = 8.7 Hz, 2H), 7.59 (dd, J
}cyclobutyl)-3- = 8.9, 1 .5 Hz, 1 H), 7.54 (d, J = 8.8 Hz, cyclohexyl-2-(4- 2H), 7.47 (d, J = 8.9 Hz, 2H), 7.19 (d, J = methoxyphenyl)-2H- 8.9 Hz, 2H), 3.95 (s, 3H), 3.09 - 2.82 (m, indazole-6- 3H), 2.62 - 2.39 (m, 2H), 2.25 - 1 .70 (m, carboxamide 9H), 1.56 - 1 .16 (m, 3H).
6g(c),(g) N-(1-{[4-(6- 615.3 1 H NMR (400 MHz, MeOD) δ 8.29 (s, aminopyridin-3- 1 H), 8.17 (s, 1 H), 8.06 (d, J = 8.9 Hz, yl)phenyl] carbamoyl 1 H), 7.77 (dd, J = 8.7, 2.4 Hz, 1 H), 7.67
}cyclobutyl)-3- - 7.56 (m, 3H), 7.49 (dd, J = 16.8, 8.8 cyclohexyl-2-(4- Hz, 4H), 7.19 (d, J = 8.9 Hz, 2H), 6.69 (d, methoxyphenyl)-2H- J = 8.7 Hz, 1 H), 3.95 (s, 3H), 2.94 (dt, J = indazole-6- 14.3, 10.3 Hz, 3H), 2.59 - 2.41 (m, 2H), carboxamide 2.23 - 1.71 (m, 9H), 1 .55 - 1.19 (m, 3H).
170(c) 3-cyclohexyl-2-(4- 630.3 1 H NMR (400 MHz, MeOD) δ 8.29 (s, methoxyphenyl)-N- 1 H), 8.06 (d, J = 9.0 Hz, 1 H), 7.71 (dt, J (1 -{[4-(1-methyl-2- = 1 1.0, 8.9 Hz, 5H), 7.59 (dd, J = 8.9, 1.5 oxo-1 ,2- Hz, 1 H), 7.47 (d, J = 8.9 Hz, 2H), 7.19 (d, dihydropyridin-4- J = 8.9 Hz, 2H), 6.76 (ddd, J = 15.0, 9.3, yl)phenyl] carbamoyl 3.9 Hz, 2H), 3.95 (s, 3H), 3.62 (s, 3H), }cyclobutyl)-2H- 3.05 - 2.82 (m, 3H), 2.56 - 2.42 (m, 2H), indazole-6- 2.24 - 1.71 (m, 9H), 1 .56 - 1.17 (m, 3H). carboxamide
171 (c) 3-cyclohexyl-N-[1- 644.2 1 H NMR (400 MHz, MeOD) δ 8.59 (s,
({4-[2- 2H), 8.29 (s, 1 H), 8.10 - 8.02 (m, 1 H),
(dimethylamino)pyri 7.72 - 7.64 (m, 2H), 7.60 (dd, J = 8.9, midin-5- 1 .5 Hz, 1 H), 7.57 - 7.51 (m, 2H), 7.51 - yl]phenyl}carbamoyl 7.43 (m, 2H), 7.24 - 7.15 (m, 2H), 3.95
)cyclobutyl]-2-(4- (s, 3H), 3.24 (s, 6H), 3.07 - 2.84 (m, 3H), methoxyphenyl)-2H- 2.57 - 2.41 (m, 2H), 2.23 - 1.72 (m, 9H), indazole-6- 1 .54 - 1.20 (m, 3H).
carboxamide
-|72(c .(g) N-(4-{[4-(2- 630.2 1 H NMR (400 MHz, MeOD) δ 8.71 (d, J = aminopyrimidin-5- 3.8 Hz, 1 H), 8.55 (s, 2H), 8.32 (s, 1 H), yl)phenyl] carbamoyl 8.17 (td, J = 7.9, 1 .8 Hz, 1 H), 8.10 (d, J = }-1 -methylpiperidin- 9.2 Hz, 1 H), 7.86 (d, J = 8.0 Hz, 1 H), 4-yl)-3-cyclohexyl-2- 7.72 - 7.61 (m, 3H), 7.61 - 7.51 (m, 3H), (pyridin-2-yl)-2H- 3.63 - 3.44 (m, 1 H), 3.31 - 3.02 (m, 4H), indazole-6- 2.74 (s, 3H), 2.53 (brs, 4H), 2.17 - 1 .74 carboxamide (m, 7H), 1.54 - 1 .27 (m, 3H).
1 73(m) 3-cyclohexyl-2-(4- 606.2 1 H NMR (400 MHz, MeOD) δ 8.28 (d, J = methoxyphenyl)-N- 1 .3 Hz, 1 H), 8.04 (d, J = 8.9 Hz, 1 H),
[1 -({4-[(1 E)-3- 7.70 - 7.42 (m, 8H), 7.19 (d, J = 8.9 Hz,
(methylamino)-3- 2H), 6.53 (d, J = 15.7 Hz, 1 H), 3.95 (s, oxoprop-1-en-1- 3H), 3.03 - 2.82 (m, 6H), 2.55 - 2.41 (m, yl]phenyl}carbamoyl 2H), 2.22 - 1 .70 (m, 9H), 1 .37 (m, J =
)cyclobutyl]-2H- 38.1 , 12.9 Hz, 3H).
indazole-6- carboxamide





^ then ester or nitrile hydrolysis; ' ' then chloro substitution with DMF (reaction-by-product); then Suzuki coupling; 'd' then W-alkyl or W-methylamine substitution from chloride; ^ then amino substitution from chloride; ^ then aminolysis under high temperature and pressure; ^ then deprotection e.g. benzyl deprotection (e.g. (BBr3)) or boc deprotection; ^ then chloro reduction; ^ diastereoisomeric separation; Method B Route (b) step b then C Route (a); Method B Route (a) then Cbz deprotection then reduction amination then C Route (a); ^ B Route (a) and/or (b); then Method E; (n) then Method C Route (a); (0) then oxidation (Dess Martin); (pl then flurorination; (q) then reductive amination.
Route (b): An amino acid ester (i) was coupled with a compound of formula II (ii) using standard peptide coupling conditions. The ester is hydrolysed under basic conditions and coupled with an aromatic or aliphatic amine using standard peptide coupling conditions to give v. After the coupling step c, the final compound have common protecting groups removed under standard conditions. In some cases when step c was incomplete under the reaction conditions described below, the reaction can be heated or sodium hydride added to increase conversion to product. The final compounds were purified by column
chromatography if required.

Example: Synthesis of 1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutanecarboxylic acid (18) and 3-Cyclohexyl-N-{1 -[(4- fluorophenyl)carbamoyl]cyclobutyl}-2-phenyl-2H-indazole-6-carboxamide (19)


Step a: To a solution of 1-aminocyclobutanecarboxylic acid ethyl ester (xxviii) (69 mg, 0.38 mmol) in dry DMF (1 mL) under argon was added 3-cyclohexyl-2-phenyl-2H-indazole-6- carboxylic acid (2) (101 mg, 0.32 mmol) and DIPEA (200 pL, 1 .15 mmol), followed by HATU (155 mg, 0.41 mmol). The solution was stirred under argon at RT for 1 .5 h. Ethyl acetate (5 mL) was added and this was washed with 1 M aqueous HCI (2 x 2 mL), aqueous sodium bicarbonate (2 mL) and brine (2 mL) then dried (MgS04) and concentrated in vacuo and the residue ethyl 1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutanecarboxylate (xxix) was used without purification; ESI-MS m/z calculated for [M+H]+: 446.2; found: 446.3.
Step b: To a stirred solution of ethyl 1 -{[(3-cyclohexyl-2-phenyl-2/-/-indazol-6- yl)carbonyl]amino}cyclobutanecarboxylate (xxix) (122 mg, 0.27 mmol) in 1 ,4-dioxane (1 mL) and /so-propanol (1 mL) was added a solution of lithium hydroxide (74 mg, 1 .76 mmol) in water (0.8 mL). The mixture was then stirred at 60°C for 1 h whereupon the reaction mixture was acidified to pH 1 with 1 M aqueous HCI and then concentrated in vacuo. The residue was purified by automated column chromatography (Biotage SP4, 12 g Grace Reveleris™ C-18 silica gel cartridge) eluting with acetonitrile:water (0% to 100%) to give 1 -{[(3- cyclohexyl-2-phenyl-2/-/-indazol-6-yl)carbonyl]amino}cyclobutanecarboxylic acid (18) as a white solid (67 mg, 59 %); ESI-MS m/z calculated for [M+H]+: 418.2; found: 418.2.
Step c: To a stirred solution of 1-{[(3-cyclohexyl-2-phenyl-2/-/-indazol-6- yl)carbonyl]amino}cyclobutanecarboxylic acid (18) (5 mg, 0.012 mmol) in dry DMF (250 μί) under argon was added 4-fluoroaniline (2 μί, 0.021 mmol), followed by DIPEA (7 μί, 0.040 mmol) and then HATU (6 mg, 0.016 mmol). The reaction was stirred at RT for 1 h and then concentrated in vacuo. Ethyl acetate (5 mL) was added and this was washed with 1 M aqueous HCI (2 x 2 mL), aqueous sodium bicarbonate (2 mL) and brine (2 mL) then dried (MgS04) and concentrated in vacuo. The residue was purified by automated flash chromatography (Biotage SP4, GraceResolv™ silica gel 4 g cartridge) eluting with ethyl
acetate:dichloromethane (0% to 50 %) to yield 3-cyclohexyl-/V-{1-[(4- fluorophenyl)carbamoyl]cyclobutyl}-2-phenyl-2/-/-indazole-6-carboxamide (19) (4 mg, 65 %) as a white solid; ESI-MS m/z calculated for [M+H]+: 51 1.10; found: 51 1.25; 1H-NMR (400 MHz, acetone-c 6) δ 9.62 (s, 1 H), 8.43 (s, 1 H), 8.35 (dd, J 1 .40 Hz, 1 Hz, 1 H), 8.04 (dd, J 8.9 Hz, 0.8 Hz, 1 H), 7.59-7.69 (m, 8H), 7.03-7.09 (m, 2H), 3.02-3.10 (m, 1 H), 2.86-2.93 (m, 2H), 2.43-2.51 (m, 2H), 1 .72-2.05 (m, 9H), 1 .24-1.50 (m, 3H).
Similarly prepared were the following compounds.

Example: Synthesis of 3-cyclohexyl^-[(2R)-1-({4-[2-(methylamino)pyrimidiri-5- yl henyl}amino)-1-oxopropan-2-yl]-2-(pyridin-2-yl)-2H-m^ (202)

202
Step a: To a solution of 3-cyclohexyl-2-(pyridin-2-yl)-2/-/-indazole-6-carboxylic acid (0.40 g, 1 .2 mmol) and D-alanine methyl ester hydrochloride (0.19 g, 1.4 mmol) in dry DMF (3 ml.) was added DIPEA (0.87 ml_, 5.0 mmol), followed by HATU (0.62 g, 1.6 mmol). After 17 h, the reaction was quenched with 1 M HCI and diluted with water. The product was initially a gum, which became a solid upon standing. The precipitate was collected by filtration, washed with water and dried under high vacuum to give methyl /V-{[3-cyclohexyl-2-(pyridin- 2-yl)-2H-indazol-6-yl]carbonyl}-D-alaninate (iii) (0.50 g, quant) as a white solid. ESI-MS m/z calculated for [M+H]+: 407.2; found: 407.3.
Step b: To a mixture of methyl W-{[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6-yl]carbonyl}-D- alaninate (0.21 g, 0.52 mmol) in ethanol (3 mL) was added a solution of potassium carbonate (0.71 g, 5.2 mmol) in water (3 mL). The resulting mixture was heated at 70 °C for 1 h. The reaction was cooled to room temperature and quenched with 1 M HCI and ice. The precipitate was collected by filtration, washed with water and dried under high vacuum to give W-{[3-cyclohexyl-2-(pyridin-2-yl)-2/-/-indazol-6-yl]carbonyl}-D-alanine (iv) (0.18 g, 90%) as a white solid. ESI-MS m/z calculated for [M+H] +: 393.2; found: 393.2.
Step c: To a solution of N-{[3-cyclohexyl-2-(pyridin-2-yl)-2/-/-indazol-6-yl]carbonyl}-D-alanine (0.12 g, 0.31 mmol) and 4-aminophenylboronic acid hydrochloride (64 mg, 0.37 mmol) in dry DMF (2 mL) was added DIPEA (0.21 mL, 1 .2 mmol), followed by HATU (0.15 g, 0.40 mmol). After 18 h the reaction was quenched with 1 M HCI and diluted with water. The precipitate was collected by filtration, washed with water and dried under high vacuum to give {4-[(W- {[3-cyclohexyl-2-(pyridin-2-yl)-2/-/-indazol-6-yl]carbonyl}-D-alanyl)amino]phenyl} boronic acid (v) (0.12 g, 77%) as a beige solid. ESI-MS m/z calculated for [M+H] +: 512.2; found: 512.2. Step d: To a mixture of {4-[(W-{[3-cyclohexyl-2-(pyridin-2-yl)-2/-/-indazol-6-yl]carbonyl}-D- alanyl)amino]phenyl}boronic acid (60 mg, 0.12 mmol), 5-bromo-2-chloropyrimidine (23 mg, 0.12 mmol) and potassium carbonate (32 mg, 0.23 mmol) in 1 ,4-dioxane (1 mL) was added water (0.6 mL) followed by PdCI2dppf.DCM (10 mg, 12 μιτιοΙ) and the mixture was heated at 90 °C for 2 h under argon. The reaction was cooled to room temperature, DMSO (1 mL) was added and the mixture was filtered through a 0.45 pm syringe filter. The solution was purified by reverse-phase chromatography (gradient elution, 10-100% MeCN in water) to give W-[(2R)-1 -{[4-(2-chloropyrimidin-5-yl)phenyl]amino}-1-oxopropan-2-yl]-3-cyclohexyl-2- (pyridin-2-yl)-2A7-indazole-6-carboxamide (vi) (36 mg, 52%) as an off white solid. ESI-MS m/z calculated for [M+H] +: 580.2; found: 580.2.
Step e: A mixture of W-[(2R)-1 -{[4-(2-chloropyrimidin-5-yl)phenyl]amino}-1 -oxopropan-2-yl]-3- cyclohexyl-2-(pyridin-2-yl)-2/-/-indazole-6-carboxamide (12 mg, 21 mol), methylamine hydrochloride (9.0 mg, 0.13 mmol) and DIPEA (36 μί, 0.21 mmol) in 2-propanol (0.5 mL) were heated under microwave irradiation for 2 h at 150 °C. The reaction mixture was quenched with ice water, and the precipitation collected by filtration. The solid was suspended in a 1 : 1 mixture of cold water and 1 M HCI and sonicated. The precipitate was collected by filtration and washed with water. The solid was dissolved in a minimum volume of methanol and lyophilised with water (x 2) to give 3-cyclohexyl-W-[(2R)-1-({4-[2- (methylamino)pyrimidin-5-yl]phenyl}amino)-1-oxopropan-2-yl]-2-(pyridin-2-yl)-2/-/-indazole-6- carboxamide (202) (3.0 mg, 25%) as a white solid. ESI-MS m/z calculated for [M+H] +: 575.3; found: 575.2; 1H NMR (400 MHz, MeOD) δ 8.70 (d, J = 3.8 Hz, 1 H), 8.62 (s, 2H), 8.28 (s, 1 H), 8.21 - 8.12 (m, 1 H), 8.09 (d, J = 9.0 Hz, 1 H), 7.86 (d, J = 8.0 Hz, 1 H), 7.80 - 7.71
(m, 2H), 7.67 - 7.61 (m, 1 H), 7.61 - 7.53 (m, 3H), 4.82 - 4.72 (m, 1 H), 3.56 - 3.43 (m, 1 H),
3.02 (s, 3H), 2.15 - 1.75 (m, 7H), 1 .63 (d, J = 7.2 Hz, 3H), 1 .53 - 1.29 (m, 3H).
The following compounds were similarly prepared in accordance with general Method C
Route (b) and with reference to the Examples previously described.


61.8, 12.9 Hz, 3H).
)1ic) 3-cyclohexyl-N-(1-{[2- 590.2 1H NMR (400 MHz, MeOD) δ 9.67 (br s,
(methylcarbamoyl)-l - 1H), 8.31 (s, 1H), 8.07 (d, J =8.9 Hz, benzofuran-5- 1 H), 8.00 - 7.90 (m, 1 H), 7.72 - 7.53 (m, yl]carbamoyl}cyclobutyl) 8H), 7.45 (s, 1H), 3.05 (s, 3H), 3.08 -
-2-phenyl-2H-indazole- 2.84 (m, 3H), 2.60 - 2.40 (m, 2H), 2.26 -
6-carboxamide 1.69 (m, 9H), 1.55-1.17 (m, 3H).
)5 6-{[(1-{[(3-cyclohexyl-2- 578.1 1H NMR (400 MHz, MeOD) δ 9.65 (s, phenyl-2H-indazol-6- 1H), 8.79 (s, 1H), 8.52 (s, 1H), 8.34 (s, yl)carbonyl]amino}cyclo 1H), 8.12-7.92 (m, 1H), 7.78-7.46 (m, butyl )carbonyl]amino}py 7H), 3.08 - 2.84 (m, 3H), 2.61 - 2.37 (m, razolo[1 ,5-a]pyrimidine- 2H), 2.23-1.68 (m, 9H), 1.53-1.10 (m, 3-carboxylic acid 3H).
100 6-{[(1-{[(3-cyclohexyl-2- 577.1 1H NMR (400 MHz, MeOD) δ 8.31 (s, phenyl-2H-indazol-6- 1H), 8.16-7.98 (m, 2H), 7.78-7.30 (m, yl)carbonyl]amino}cyclo 9H), 3.07 - 2.86 (m, 3H), 2.50 (dd, J = butyl )carbonyl]amino}-1- 20.5, 9.1 Hz, 2H), 2.22 -1.71 (m, 9H), benzofuran-2-carboxylic 1.52-1.21 (m, 3H).
acid
I01(c) 3-cyclohexyl-N-(1-{[2- 590.1 1H NMR (400 MHz, MeOD) δ 8.31 (s,
(methylcarbamoyl)-l - 1H), 8.19 (s, 1H), 8.07 (d, J =8.9 Hz, benzofuran-6- 1 H), 7.73 - 7.51 (m, 7H), 7.42 (d, J = 1.0 yl]carbamoyl}cyclobutyl) Hz, 1 H), 7.33 (dd, J = 8.5, 1.8 Hz, 1 H),
-2-phenyl-2H-indazole- 3.07 - 2.81 (m, 3H), 2.97 (s, 3H), 2.58 -
6-carboxamide 2.39 (m, 2H), 2.26 - 1.69 (m, 9H), 1.55 - 1.15 (m, 3H).
104 ethyl 6-{[(1-{[(3- 605.2 1H NMR (400 MHz, MeOD) δ 8.31 (s, cyclohexyl-2-phenyl-2H- 1H), 8.19 (brs, 1H), 8.04 (d, J = 8.9 Hz, indazol-6- 1H), 7.79-7.48 (m, 8H), 4.50 (q, J = 7.1 yl)carbonyl]amino}cyclo Hz, 2H), 3.09 - 2.88 (m, 3H), 2.62 - 2.42 butyl )carbonyl]amino}- (m, 2H), 2.24-1.69 (m, 9H), 1.55-1.17 1 H-benzimidazole-2- (m, 6H).
carboxylate
109 6-{[(1-{[(3-cyclohexyl-2- 577.0 1H NMR (400 MHz, MeOD) δ 8.32 (s, phenyl-2H-indazol-6- 1 H), 8.23 (br s, 1 H), 8.07 (d, J = 8.9 Hz, yl)carbonyl]amino}cyclo 1H), 7.80-7.40 (m, 8H), 3.11 -2.72 (m, butyl )carbonyl]amino}- 3H), 2.60 - 2.34 (m, 2H), 2.25 - 1.64 (m, 1 H-benzimidazole-2- 9H), 1.52-1.08 (m, 3H).
carboxylic acid



 2.04 - 1.88 (m, 4H), 1.88 - 1.76 (m, 2H),
2.04 - 1.88 (m, 4H), 1.88 - 1.76 (m, 2H),
1.76 - 1.66 (m, 1 H), 1.48 - 1.20 (m, 3H).
159 ethyl (2E)-3-[4-({[1-({[3- 621.3 1H NMR (400 MHz, MeOD) δ 8.31 - 8.23 cyclohexyl-2-(4- (m, 1 H), 8.03 (dd, J = 8.9, 0.7 Hz, 1 H), methoxyphenyl)-2H- 7.66 (dd, J = 9.5, 7.5 Hz, 3H), 7.61 - indazol-6- 7.52 (m, 3H), 7.50 - 7.40 (m, 2H), 7.24 - yl]carbonyl}amino)cyclo 7.11 (m, 2H), 6.46 (d, J= 16.0 Hz, 1H), butyl]carbonyl}amino)ph 4.25 (q, J = 7.1 Hz, 2H), 3.94 (s, 3H), enyl]prop-2-enoate 3.06 - 2.81 (m, 3H), 2.55 - 2.38 (m, 2H),
2.20- 1.71 (m, 9H), 1.53- 1.17 (m, 6H).
160 (2E)-3-[4-({[1-({[3- 593.3 1H NMR (400 MHz, MeOD) δ 8.28 (s, cyclohexyl-2-(4- 1 H), 8.05 (d, J = 8.8 Hz, 1 H), 7.72 - 7.53 methoxyphenyl)-2H- (m, 6H), 7.52 - 7.41 (m, 2H), 7.25 - 7.12 indazol-6- (m, 2H), 6.43 (d, J = 15.9 Hz, 1H), 3.95 yl]carbonyl}amino)cyclo (s, 3H), 2.94 (ddd, J= 18.4, 14.0, 10.4 butyl]carbonyl}amino)ph Hz, 3H), 2.60 - 2.38 (m, 2H), 2.25 - 1.71 enyl]prop-2-enoic acid (m, 9H), 1.56- 1.18 (m, 3H).
161 5-({[1-({[3-cyclohexyl-2- 607.2 1H NMR (400 MHz, MeOD) δ 8.30 (s,
(4-methoxyphenyl)-2H- 1H), 8.06 (d, J= 8.3 Hz, 1H), 7.94 (s, indazol-6- 1H), 7.67-7.33 (m, 6H), 7.19 (d, J = 9.0 yl]carbonyl}amino)cyclo Hz, 2H), 3.95 (s, 3H), 3.04 - 2.85 (m, butyl]carbonyl}amino)-1- 3H), 2.50 (dd, J = 20.4, 8.9 Hz, 2H), 2.23 benzofuran-2-carboxylic - 1.70 (m, 9H), 1.35 (t, J = 30.3 Hz, 3H). acid
163(h) N-(1-{[4-(2- 587.2 1H NMR (400 MHz, MeOD) δ 8.74 - 8.66 aminopyrimidin-5- (m, 1 H), 8.55 (s, 2H), 8.33 (s, 1 H), 8.23 - yl)phenyl]carbamoyl}cyc 8.13 (m, 1H), 8.09 (d, J= 9.0 Hz, 1H), lobutyl)-3-cyclohexyl-2- 7.86 (d, J = 8.0 Hz, 1H), 7.74 - 7.49 (m,
(pyridin-2-yl)-2H- 6H), 3.60 - 3.42 (m, 1 H), 3.05 - 2.81 (m, indazole-6-carboxamide 2H), 2.62 - 2.41 (m, 2H), 2.31 - 1.73 (m,
9H), 1.56- 1.24 (m, 3H).
164 ethyl 5-({[1-({[3- 635.2 1H NMR (400 MHz, MeOD) δ 8.22 (s, cyclohexyl-2-(4- 1 H), 8.01 - 7.91 (m, 2H), 7.52 (dd, J = methoxyphenyl)-2H- 14.8, 8.9 Hz, 4H), 7.40 (d, J= 8.9 Hz, indazol-6- 2H), 7.12 (d, J = 8.9 Hz, 2H), 4.36 (q, J = yl]carbonyl}amino)cyclo 7.1 Hz, 2H), 3.88 (s, 3H), 2.98 - 2.77 (m, butyl]carbonyl}amino)-1- 3H), 2.53-2.33 (m, 2H), 2.17- 1.61 (m, benzofuran-2- 9H), 1.35 (t, J = 7.1 Hz, 3H), 1.30- 1.13 carboxylate (m, 3H).
165(c) 3-cyclohexyl-2-(4- 620.2 1H NMR (400 MHz, MeOD) δ 8.30 (s, methoxyphenyl)-N-( 1 - 1 H), 8.04 (d, J = 8.9 Hz, 1 H), 7.97 (d, J =
{[2-(methylcarbamoyl)- 1.2 Hz, 1H), 7.62-7.41 (m, 6H), 7.19 (d,
1-benzofuran-5- J = 8.9 Hz, 2H), 3.95 (s, 3H), 3.04 - 2.87 yl]carbamoyl}cyclobutyl) (m, 3H), 2.97 (s, 3H), 2.57 - 2.42 (m,
-2H-indazole-6- 2H), 2.21 - 1.73 (m, 9H), 1.54 - 1.22 (m, carboxamide 3H).

 nitro reduction.
Method D: Hydrolysis and/or Alkene Reduction
nitro reduction.
Method D: Hydrolysis and/or Alkene Reduction
Example: Synthesis of (2E)-3-(4-{[(1-{[(3-Cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl] amino}phenyl)prop-2-enoic acid (4) and ethyl 3-(4- {[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}phenyl)propanoic acid (5)

Step a: To a stirred solution of ethyl (2E)-3-(4-{[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}phenyl)prop-2-enoate (3) (100 mg, 0.169 mmol) in 1 ,4-dioxane (1 .5 mL) and /so-propanol (1 .5 mL) was added a solution of lithium hydroxide (43 mg, 1 .016 mmol) in water (1 mL). The resultant solution was then stirred at RT for 18 h whereupon the reaction mixture was concentrated in vacuo. The residue was dissolved in water (5 mL) and the resultant solution was acidified to pH 1 with 1 M HCI (aq). The organics were then extracted into ethyl acetate (2 x 10 mL), dried (MgS04) and the volatiles removed in vacuo. The residue was purified by automated column chromatography (Biotage SP4, 12 g Grace Reveleris™ C-18 silica gel cartridge) eluting with
acetonitrile:water (0% to 100%) to give (2E)-3-(4-{[(1 -{[(3-Cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}phenyl)prop-2-enoic acid (4) as a white solid (64 mg, 67 %); ESI-MS m/z calculated for [M+H]+: 563.27; found: 563.1 1 . 1H NMR (400 MHz, c -DMSO) δ 12.26 (s, 1 H), 9.69 (s, 1 H), 8.98 (s, 1 H), 8.41 (s, 1 H), 8.02 (d, J = 8.6 Hz, 1 H), 7.72-7.44 (m, 10H), 6.41 (d, J = 17.1 Hz, 1 H), 2.99-2.86 (m, 1 H), 2.80-2.66 (m, 2H), 2.46-2.30 (m, 2H), 2.08-1 .81 (m, 9H), 1 .49-1.32 (m, 1 H), 1 .31-1.1 1 (m, 2H).
Step b: To a stirred solution of (2E)-3-(4-{[(1 -{[(3-Cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}phenyl)prop-2-enoic acid (4) (8 mg, 0.014 mmol) in ethyl acetate (0.5 mL) was added 10% Pd/C (~2 mg) and the mixture was stirred under a hydrogen atmosphere at RT for 24 h. The mixture was diluted with ethyl acetate (2mL) and filtered. The filtrate was concentrated in vacuo and the sample was lyophilised to give ethyl 3-(4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6-
yl)carbonyl]amino}cyclobutyl)carbonyl]amino}phenyl)propanoic acid (5) as a white solid (6.1 mg, 76%); ESI-MS m/z calculated for [M+H]+: 565.28; found: 565.05; 1 H-NMR (400 MHz, CDCI3) δ 9.74 (s, 1 H), 8.15 (s, 1 H), 7.92 (d, J 8.8 Hz, 1 H), 7.43-7.57 (m, 8H), 7.15 (d, J 8.4 Hz, 2H), 6.86 (s, 1 H), 2.89-2.97 (m, 5H), 2.64 (t, J 7.7 Hz, 2H), 2.36-2.43 (m, 2H), 1 .26-2.1 1 (m, 12H).
Method E: Amide Formation
Route (a) Primary Amide Formation: Synthesis of 3-Cyclohexyl-2-phenyl-2H-indazole-6- carboxamide (15)

Route (b) Amide Formation: The carboxylic acid i is converted to the W-methyl amide ii under

Example: Synthesis of 3-Cyclohexyl-N-[1-({4-[(1E)-3-(methylamino)-3-oxoprop-1-en-1- yl] henyl}carbamoyl)cyclobutyl]-2-phenyl-2H-indazole-6-carboxamide (6)

Similarly prepared were the following compounds.

using 2M methylamine in THF instead of methylamine hydrochloride.
Method F: Acyl Sulfonamoyl Formation
Example: Synthesis of 3-(cyclohex-1 -en-1 -yl)-N-(dimethylsulfamoyl)-2-phenyl-2H-indazole- 6-carboxamide (8) and 3-Cyclohexyl-N-(dimethylsulfamoyl)-2-phenyl-2H-indazole-6- carboxamide (9)

Step a: To a stirred solution of 3-(cyclohex-1 -en-1 -yl)-2-phenyl-2/-/-indazole-6-carboxylic acid (1 ) (300 mg, 0.94 mmol) in DMF (10 mL) at 0 °C was added EDCI (360 mg, 1.88 mmol) and DMAP (230 mg, 1.88 mmol) followed by W,W-dimethylsulfamamide (175 mg, 1.41 mmol). The reaction mixture was then allowed to warm to RT and stirred for 16 h whereupon the reaction mixture was concentrated in vacuo and residue purified by automated column chromatography (Combi-Flash™, silica gel) eluting with ethyl acetate: hexane (2:1 v/v) to give 3-(cyclohex-1 -en-1-yl)-/V-(dimethylsulfamoyl)-2-phenyl-2/-/-indazole-6-carboxamide (8) as a white solid (251 mg, 62 %); ESI-MS m/z calculated for [M+H]+: 425.16; found: 425.30. 1 H NMR (400 MHz, cf-DMSO) δ 1 1.91 (brs, 1 H), 8.39 (s, 1 H), 7.78 (d, J = 8.9 Hz, 1 H), 7.70- 7.51 (m, 6H), 6.14 (brs, 1 H), 2.94 (s, 6H), 2.30-2.16 (m, 2H), 2.00-1.90 (m, 2H), 1.69-1.52 (m, 4H).
Step b: To a stirred suspension of 3-(cyclohex-1 -en-1 -yl)-/V-(dimethylsulfamoyl)-2-phenyl- 2A7-indazole-6-carboxamide (8) (245 mg, 0.58 mmol) in methanol (20 mL) was added 10% Pd/C (245 mg, 2.31 mmol, 100% wt. by wt.) under an atmosphere of nitrogen. Ammonium formate (2.156 g, 34.12 mmol) was then added and the reaction mixture was heated to reflux and monitored by LCMS analysis. Upon complete consumption of the starting material (4 h), the reaction mixture was filtered through a pad of Celite® and the residue was washed with methanol (2 x 10 mL). The combined filtrates were concentrated in vacuo and the residue was purified by automated column chromatography (Combi-Flash™, silica gel) eluting with ethyl acetate: hexane (2:1 ). Further purification by crystallization from
hexane:ethyl acetate gave 3-cyclohexyl-/V-(dimethylsulfamoyl)-2-phenyl-2/-/-indazole-6- carboxamide (9) as a white solid (78 mg, 31 %); ESI-MS m/z calculated for [M+H]+: 427.18; found: 427.25; 1 H-NMR (400 MHz, DMSO-d6) δ 1 1.89 (brs, 1 H), 8.35 (s, 1 H), 8.08 (d, J = 7.7 Hz, 1 H), 7.73-7.41 (m, 6H), 3.00-2.81 (m, 7H), 1.98-1 .62 (m, 7H), 1.48-1 .30 (m, 1 H), 1.29- 1 .1 1 (m, 2H).
Similarly prepared were the following compounds.

using ethanesulfonamide instead of N, N-dimethylsulfamamide.
Biological Data
HCV Polymerase Inhibition Assay
HCV polymerase reactions were carried out using a modified method of Howe et al. , Antimicrobial Agents and Chemotherapy 2004 48(12): 4813-4821. Reactions contained 50 nM NS5bA21 , 20 mM Tris-HCI pH 7.5, 5 rtiM MgCI2, 5 mM MnCI2, 3 mM DTT, 0.05% BSA, 0.2 U/pL RNasin, 10 pg/mL Poly(rC) template, GTP (at Km) and 0.05 pCi/pL 33P-GTP in a total reaction volume of 50 pL. Compounds were tested in a three fold dilutions series for example starting from 50 pM. Reactions were initiated with the addition of GTP and terminated after 1 hour with 50 pL ice cold 0.2 M EDTA. Terminated reactions were transferred to DEAE 96-well filter plates, unincorporated nucleotides washed from the filters and 50 pL scintillation fluid added prior to reading on a scintillation counter. The compound concentration that reduced 33P-GTP incorporation 50% (IC50) was calculated using nonlinear regression.
Representative data for compounds of the invention in the HCV polymerase inhibition assay was obtained and IC50 (μΜ) values determined in accordance with the general ranges: A: < 0.99μΜ; Β: 1.0-9.99μΜ; C: 10.0-49.99pM; D: > 50μΜ.
A: Compounds 4, 5, 6, 7, 10, 14, 16, 17, 25, 30, 31 , 32, 34, 35, 36, 37, 39, 40, 41 , 44-48,
50-60, 62-65, 67-78, 80-83, 85-102, 104, 106-204.
B: Compounds 2, 9, 18, 19, 23, 38, 49, 61 , 66, 84, 103, 105.
C: Compounds 8, 1 1 , 15, 21 , 22, 26, 27, 28, 29, 43, 79.
D: Compounds 1 , 3, 12, 13, 20, 24, 28, 33, 42.
Further, the IC50 values for the majority of compounds in the A range were determined to <500nM. For example, the IC50 values for compounds 5, 7, 25, 30, 32, 34, 36, 37, 40, 41 , 45, 46, 51 , 56, 59, 68, 70-74, 76, 78, 80, 83, 85, 88, 92, 94-96, 99, 106, 1 12,
1 15, 124, 125, 135, 138-141 , 148, 151 , 155, 158, 174, 175, 182, 183, 198, 199, 201-204 were determined to be <500nM.
Representative values for compounds with IC50values <100nM are provided in Table
1 .
Table 1 : HCV polymerase values for selected compounds with IC50values <100nM.

HCV replicon assays
A genotype 1 b subgenomic replicon cell line based on Blight et ai, Science 2000 290: 1972-1974, modified to express a Renilla luciferase reporter gene was used to assess antiviral activity of test compounds. Cell cultures were maintained in a sub-confluent state in DMEM with glutamine, 10% heat-inactivated foetal bovine serum and Geneticin.
For assay, cells were seeded at a concentration of 7000 cells/well into 96 well tissue culture trays in culture media lacking Geneticin. Compounds were tested in a three fold dilutions series for example starting from 50 μΜ. After 72 hours incubation at 37°C and 5% C02, enilla luciferase activity was quantified. The compound concentration that reduced luciferase activity by 50% (EC50) was calculated using non-linear regression.
EC50 values for selected compounds were determined in accordance with the general ranges: A: < 0.99μΜ; Β: 1.0-9.99μΜ; C: 10.0-49.99μΜ.
A: Compounds 4, 49, 62, 96, 101 , 1 12, 120, 124, 133, 137, 138, 140, 141 , 146, 148, 151 , 160, 164, 165, 172, 174, 178, 182, 191 , 195, 200.
B: Compounds 6, 7, 17, 25, 29, 30, 31 , 37, 46, 55, 61 , 63, 65, 67, 70-73, 80, 85-88, 90-91 , 97, 102, 104, 107-1 1 1 , 115-1 17, 122, 123, 125, 126, 129-131 , 134, 143, 149, 150, 153, 154, 156-159, 161-163, 166-170, 173, 175- 177, 179-181 , 183, 185, 189, 190, 194, 197, 198, 201 , 202, 204.
C: Compounds 5, 9, 14, 15, 19, 28, 32, 34, 35, 36, 39, 41 , 42, 44, 45, 47, 48, 53, 54, 59, 64, 68, 69, 74, 75, 77, 78, 82, 94, 100, 106, 1 13, 1 19, 121 , 127, 128, 145, 155, 188, 192, 193.
Cytotoxicity analysis
Cytotoxicity of compounds against genotype 1 b subgenomic replicon cells were assessed using the MTT assay (Watanabe et al. , Journal of Virological Methods 1994 48:257-265). Plates were prepared as described for the HCV Replicon assay and cytotoxicity of the test article was evaluated after three days. MTT was added to assay plates followed by three hour incubation at 37°C. Wells were aspirated to dryness and the formazan dye dissolved by the addition of 100% isopropanol. Absorbencies were read in a spectrophotometer at two wavelengths (540 nm and 690 nm). The compound concentration that reduced cell viability by 50% (CC50) is calculated using non-linear regression.
The reference in this specification to any prior publication, or information derived from it, or to any matter which is know, is not, and should not be taken as an
acknowledgement or admission or any form of suggestion that that prior publication, or information derived from it, or known matter forms part of the common general knowledge in the field of endeavour to which this specification relates.
Throughout this specification and the claims which follow, unless the context requires otherwise, the word "comprise", and variations such as "comprises" and "comprising", will be understood to imply the inclusion of a stated integer or step or group of integers or steps but not the exclusion of any other integer or step or group of integers or steps.
It will be understood to persons skilled in the art of the invention that many modifications may be made without departing from the spirit and scope of the invention.
Claims
1. A compound of formula (I):

Zi, Z2 ,Z3 and Z4 are each independently selected from C-R1 , C-Ra , N and N+-0" wherein one of Zi, Z2 ,Z3 and Z4 is C-R1 and Ra is independently selected from H, optionally substituted C^alkyl, optionally substituted C2-6alkenyl, halo, haloC^alkyl, CHF2, OCHF2, CF3, OCF3, CN, OH, optionally substituted d_6alkoxy, N02, NH2, NH(Ci-6alkyl), N(C1-6alkyl)2, NHC(0)d_6alkyl, NHS02 C(0)NH2, C(0)NH(Ci-6alkyl), C(0)N(Ci.6alkyl)2, C(0)NHS02(d_ ealkyl), C(0)N(C1.6alkyl)S02(Ci_6alkyl), S03H, OSO^d-ealkyl), NS02(d_6alkyl), S02(Ci_ ealkyl), S02NH2, S02NH(C1_6alkyl), S02N(C1_6alkyl)2, C02H, C(0)Ci-6alkyl, C02Ci_6alkyl, optionally substituted (CH2)mC3-8cycloalkyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheterocyclyl and optionally substituted (CH2)mheteroaryl;
R1 is (CH2)mR4, (CH2)m-C(0)R4, (CH2)m-OC(0)R4 (CH2)m-NR5C(0)R4, (CH2)m-NR5S(0)2R4 or (CH2)m-S(0)2R4;
R2 is selected from optionally substituted (CH2)mC3.8cycloalkyl, optionally substituted (CH2)mheterocyclyl, optionally substituted (CH2)maryl and optionally substituted
(CH2)mheteroaryl;
R3 is selected from optionally substituted C1.6alkyl, optionally substituted C2.6alkenyl, optionally substituted (CH2)mC3.8cycloalkyl, optionally substituted (CH2)mC3.8cycloalkenyl, optionally substituted (CH2)mheterocyclyl, optionally substituted (CH2)maryl and optionally substituted (CH2)mheteroaryl;
R4 is (CH2)mR6, (CH2)mOR6or (CH2)mNR5R6;
R5 is independently selected from H, optionally substituted C^alkyl and optionally substituted C2.6alkenyl;
R6 is selected from H, optionally substituted Ci_6alkyl, optionally substituted C2.6alkenyl, optionally substituted (CH2)mC3.8cycloalkyl, optionally substituted (CH2)mC3.8cycloalkenyl, optionally substituted (CH2)mheterocyclyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheteroaryl, R9, [C(R7)(R8)]V(R9), C(0)-[C(R7)(R8)]v(R9), C02- [C(R7)(R8)]V(R9), C(0)NR5-[C(R7)(R8)]v(R9), NR5-[C(R7)(R8)]V(R9), NR5C(0)-[C(R7)(R8)]v(R9), NR5S02-[C(R7)(R8)]v(R9), S02-[C(R7)(R8)]v(R9) and S02NR5-[C(R7)(R8)]v(R9);
R7 and R8 are independently selected from H, optionally substituted C1.6alkyl, optionally substituted C2.6alkenyl, optionally substituted (CH2)mC3-8cycloalkyl, optionally substituted (CH2)mC3.8cycloalkenyl, optionally substituted (CH2)mheterocyclyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheteroaryl, the side chain of an amino acid or an a Iky I ester thereof
or R7 and R8 together with the atom or adjacent atoms to which they are attached form an optionally substituted C3-8cycloalkyl or an optionally substituted C3-8heterocyclyl or R7 and R8 and R5 together with the adjacent atoms to which they are attached form an optionally substituted C3.8cycloalkyl or an optionally substituted C3.8heterocyclyl;
R9 is selected from R10, (CH2)mC(0)-R10, (CH2)mC(0)NR5R10, (CH2)mC(0)NR5C(0)-R10, (CH2)mC(0)NR5S02-R10 (CH2)mC(0)NR5R10, (CH2)mNR5R10, (CH2)mNR5C(0)-R10,
(CH2)mNR5C(0)NR5R1°, (CH2)mNR5S02-R10, (CH2)mS02-R10, (CH2)mS02NR5R10 and (CH2)mS02NR5C(0)-R10;
R10 is H, OH, -A-(Q)n or 0-A-(Q)n;
A is selected from optionally substituted C^alkyl, optionally substituted C2.6alkenyl, optionally substituted (CH2)mC3.8cycloalkyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheterocyclyl and optionally substituted (CH2)mheteroaryl; and
Q is selected from optionally substituted C^alkyl, optionally substituted C2.6alkenyl, halo, haloCi_6alkyl, CHF2, OCHF2, CF3, OCF3, CN, OH, optionally substituted Ci.6alkoxy, N02, NH2, NH(Ci_6alkyl), Nid-ealkyl^, NHCiOX^alkyl, NHS02 C(0)NH2, C^NHtd-ealkyl), CiOJNid-ealkyl^, C(0)NHS02(C1_6alkyl), qO d-ealky SO^d-ealkyl), S03H, OS02(d_ 6alkyl), NS02(d_6alkyl), SO^C^alkyl), S02NH2, S02NH(C1.6alkyl), SOzNid-ealkyl^, C02H, C(0)C1.6alkyl, CO^-ealkyl, optionally substituted (CH2)mC3.8cycloalkyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheterocyclyl and optionally substituted
(CH2)mheteroaryl; and
in each instance (CH2)m may be optionally substituted;
m in each instance is independently 0, 1 , 2 or 3;
n is independently 0, 1 , 2, 3, 4 or 5; and
v is independently 1 , 2 or 3;
or its salts, N-oxides, racemates, enantiomers and isomers thereof
2. A compound according to claim 1 wherein Z2 is C-R1 and Zi, Z3 and Z4 are each independently selected from C-Ra and N
3. A compound according to claim 1 or claim 2 wherein R1 is (CH2)m-C(0)R4.
4. A compound according to claim 3 wherein R4 is (CH2)mNR5R6 or (CH2)m0R6.
5. A compound according to claim 3 wherein R4 is (CH2)mNR5R6, wherein R5 is H and R6 is selected from H, optionally substituted C1-3alkyl, R9 and [C(R7)(R8)]V(R9) wherein v is 1.
6. A compound according to any one of claims 1 to 5 wherein R2 is an optionally substituted (CH2)maryl or optionally substituted (CH2)mheteroaryl.
7. A compound according to any one of claims 1 to 6 wherein R3 is an optionally substituted (CH2)mC3-8cycloalkyl, optionally substituted (CH2)mC3-8cycloalkenyl or optionally substituted (CH2)maryl.
8. A compound according to any one of claims 1 to 7 wherein R7 and R8 are independently selected from H, optionally substituted Ci_6alkyl, optionally substituted
(CH2)mC3-6cycloalkyl wherein m is 0 or 1 , optionally substituted (CH2)m-4-6-membered heterocyclyl wherein m is 0 or 1 , optionally substituted (CH2)mphenyl where m is 0 or 1 , optionally substituted (CH2)m-5-6-membered-heteroaryl wherein m is 0 or 1 , and the side chain of an amino acid or an alkyl ester thereof; or R7 and R8 together with the atom to which they are attached form an optionally substituted C3.6cycloalkyl or an optionally substituted 4- 6-membered heterocyclyl.
9. A compound according to any one of claims 1 to 8 wherein R9 is selected from R10, (CH2)mC(0)-R10, (CH2)mC(0)NR5R10, (CH2)mS02-R10 and (CH2)mS02NR5R10 wherein m is 0 or 1.
10. A compound according to any one of claims 1 to 9 wherein R6 is
[C(R7)(R8)]V(R9) and R9 is (CH2)mC(0)R10 or (CH2)mC(0)NR5R10.
1 1. A compound according to any one of claims 1 to 10 wherein R10 is H, OH, -A- (Q)n or 0-A-(Q)n wherein A is optionally substituted Ci_3alkyl and n is 0.
12. A compound according to any one of claims 1 to 10 wherein R10 is -A-(Q)n wherein n is 0, 1 or 2 and A is selected from optionally substituted (CH2)mC3.8cycloalkyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheterocyclyl and optionally substituted (CH2)mheteroaryl.
13. A compound according to claim 12 wherein n is 1 and Q is selected from optionally substituted C^alkyl, optionally substituted C2.3alkenyl, halo, haloC^alkyl, CHF2, CF3, CN, OH, NH2, NH(d_3alkyl), NHS02 C(0)NH2,
C(0)NH(d.3alkyl), C(0)N(Ci.3alkyl)2, C(0)NHS02(d_3alkyl), C02H, C02d.3alkyl, optionally substituted phenyl, optionally substituted heterocyclyl and optionally substituted heteroaryl.
14. A compound according to claim 1 which is a compound of formula (la): 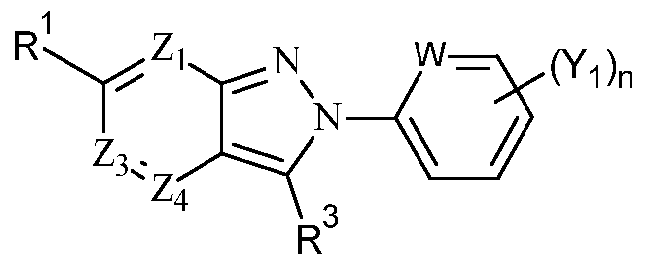
(la)
wherein Zi , Z3, Z4, 1 and R3 are as defined in claim 1 ;
W is CH or N;
Yi is independently selected from optionally substituted C^alkyl, optionally substituted C2. 6alkenyl, halo, haloCi_6alkyl, CHF2, OCHF2, CF3, OCF3, CN, OH, optionally substituted d. 6alkoxy, N02, NH2, NH(d_6alkyl), N(d-6alkyl)2, NHC(0)d_6alkyl, NHS02 C(0)NH2,
C(0)NH(d.6alkyl), C(0)N(d_6alkyl)2, C(0)NHS02(d_6alkyl), C(0)N(d.6alkyl)S02(d_6alkyl), S03H, OS02(d_6alkyl), NS02(d_6alkyl), S02(d_6alkyl), S02NH2, S02NH(d_6alkyl), S02N(Ci_ 6alkyl)2, C02H, C(0)d.6alkyl, C02d.6alkyl, optionally substituted (CH2)mC3-8cycloalkyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheterocyclyl and optionally substituted (CH2)mheteroaryl; and
n is independently 0, 1 , 2, 3, 4 or 5;
or its salts, N-oxides, racemates, enantiomers and isomers thereof.
15. A compound according to claim 14 which is a compound of formula (la-i):

(la-i)
wherein Zi , Z3, Z4, R1, Y1, W and n are as defined in claim 14; and
Y2 is independently selected from optionally substituted d.6alkyl, optionally substituted C2- 6alkenyl, halo, halod_6alkyl, CHF2, OCHF2, CF3, OCF3, CN, OH, optionally substituted d- 6alkoxy, N02, NH2, NH(d_6alkyl), N(d_6alkyl)2, NHC(0)d_6alkyl, NHS02 C(0)NH2,
C(0)NH(d.6alkyl), C(0)N(d_6alkyl)2, C(0)NHS02(d_6alkyl), C(0)N(d.6alkyl)S02(d_6alkyl), SO3H, OS02(d_6alkyl), NS02(d_6alkyl), S02(d_6alkyl), S02NH2, S02NH(d_6alkyl), S02N(Ci_ 6alkyl)2, C02H, C(0)d.6alkyl, C02d.6alkyl, optionally substituted (CH2)md.8cycloalkyl, optionally substituted (CH2)maryl, optionally substituted (CH2)mheterocyclyl and optionally substituted (CH2)mheteroaryl,
or its salts, N-oxides, racemates, enantiomers and isomers thereof.
16. A compound according to claim 1 which is a compound of formula (lb): 
(lb)
wherein Zi , Z3, Z4, R2, R3 and R4 are as defined in claim 1 ;
or its salts, N-oxides, racemates, enantiomers and isomers thereof.
17. A compound according to claim 16 which is a compound of formula (Ib-i)

(I b-i)
wherein Zi , Z3, Z4, R2, R3 and R5 are as defined in claim 1 ;
or its salts, N-oxides, racemates, enantiomers and isomers thereof.
18. A compound according to claim 16 which is a compound of formula (Ib-ii):
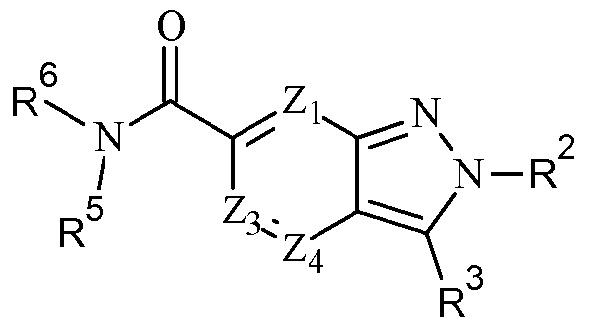
(Ib-ii)
wherein Z^ Z3, Z4, R2, R3, R5 and R6 are as defined in claim 1 ;
or its salts, N-oxides, racemates, enantiomers and isomers thereof.
19. A compound according to claim 16 which is a compound of formula (Ib-iii)

(Ib-iii) wherein Ζ , Z3, Z4, R2, R3, R5, R7, R8 and R9 are as defined in claim 1 and each m is independently 0, 1 , 2 or 3;
or its salts, N-oxides, racemates, enantiomers and isomers thereof.
20. A compound according to claim 19 which is a compound of formula (Ib-iv):

(Ib-iv)
wherein Zi , Z3, Z4, R2, R3, R7, R8, m, n and Q are as defined in claim 1 ; and
Wi, W2, W3, W4 and W5 are each independently selected from CH and N or together with an adjacent ring member join to form a fused 5-membered heterocyclic moiety;
or its salts, N-oxides, racemates, enantiomers and isomers thereof.
21. A compound according to claim 1 selected from the group consisting of
1 ) 3-(cyclohex-1 -en-1 -yl)-2-phenyl-2H-indazole-6-carboxylic acid;
2) 3-cyclohexyl-2-phenyl-2H-indazole-6-carboxylic acid;
3) ethyl (2E)-3-(4-{[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}phenyl)prop-2-enoate;
4) (2E)-3-(4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}phenyl)prop-2-enoic acid;
5) 3-(4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}phenyl)propanoic acid;
6) 3-cyclohexyl-N-[1 -({4-[(1 E)-3-(methylamino)-3-oxoprop-1-en-1 - yl]phenyl}carbamoyl)cyclobutyl]-2-phenyl-2H-indazole-6-carboxamide;
7) 3-cyclohexyl-N-[(2R)-1-oxo-1 -{[4-(pyridin-4-yl)phenyl]amino}propan-2-yl]-2- phenyl-2H-indazole-6-carboxamide;
8) 3-(cyclohex-1 -en-1 -yl)-N-(dimethylsulfamoyl)-2-phenyl-2H-indazole-6- carboxamide;
9) 3-cyclohexyl-N-(dimethylsulfamoyl)-2-phenyl-2H-indazole-6-carboxamide;
10) 3-cyclohexyl-N-[1 -({4- [(methylsulfonyl)carbamoyl]phenyl}carbamoyl)cyclobutyl]-2-phenyl-2H- indazole-6-carboxamide;
1 1 ) 3-cyclohexyl-2-(4-hydroxyphenyl)-2H-indazole-6-carboxylic acid;
12) methyl 3-cyclohexyl-2-(4-hydroxyphenyl)-2H-indazole-6-carboxylate; 13) methyl 3-cyclohexyl-2-(4-methoxyphenyl)-2H-indazole-6-carboxylate;
14) 3-cyclohexyl-2-(4-methoxyphenyl)-2H-indazole-6-carboxylic acid;
15) 3-cyclohexyl-2-phenyl-2H-indazole-6-carboxamide;
16) N-{1 -[(4-cyanophenyl)carbamoyl]cyclobutyl}-3-cyclohexyl-2-phenyl-2H- indazole-6-carboxamide;
17) N-{1 -[(4-carbamoylphenyl)carbamoyl]cyclobutyl}-3-cyclohexyl-2-phenyl-2H- indazole-6-carboxamide;
18) 1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutanecarboxylic acid;
19) 3-cyclohexyl-N-{1 -[(4-fluorophenyl)carbamoyl]cyclobutyl}-2-phenyl-2H- indazole-6-carboxamide;
20) 3-cyclohexyl-N-{1 -[(4-iodophenyl)carbamoyl]cyclobutyl}-2-phenyl-2H- indazole-6-carboxamide;
21 ) N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]glycine;
22) 3-(cyclohex-1 -en-1 -yl)-N-(ethylsulfonyl)-2-phenyl-2H-indazole-6- carboxamide;
23) 3-cyclohexyl-N-(ethylsulfonyl)-2-phenyl-2H-indazole-6-carboxamide;
24) N-[(2R)-1 -{[4-(1 ,3-benzodioxol-5-yl)phenyl]amino}-1 -oxopropan-2-yl]-3- cyclohexyl-2-phenyl-2H-indazole-6-carboxamide;
25) 3-cyclohexyl-N-[(2R)-1-oxo-1 -{[4-(pyridin-3-yl)phenyl]amino}propan-2-yl]-2- phenyl-2H-indazole-6-carboxamide;
26) 3-cyclohexyl-N-{(2R)-1 -[(4-iodophenyl)amino]-1 -oxopropan-2-yl}-2-phenyl- 2H-indazole-6-carboxamide;
27) N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-D-alanine;
28) 3-cyclohexyl-N-methyl-2-phenyl-2H-indazole-6-carboxamide;
29) ethyl N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]glycinate;
30) methyl (2E)-3-[4-({N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-D- alanyl}amino)phenyl]prop-2-enoate;
31 ) (2E)-3-[4-({N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-D- alanyl}amino)phenyl]prop-2-enoic acid;
32) 3-cyclohexyl-N-[1 -({4-[3-(methylamino)-3- oxopropyl]phenyl}carbamoyl)cyclobutyl]-2-phenyl-2H-indazole-6- carboxamide;
33) 2,3-diphenyl-2H-indazole-6-carboxylic acid;
34) ethyl (4-{[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}phenyl)acetate; 35) 3-cyclohexyl-2-phenyl-N-(1 -{[4-(1 H-tetrazol-5- yl)phenyl]carbamoyl}cyclobutyl)-2H-indazole-6-carboxamide;
36) methyl 4-{[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]annino}benzoate;
37) 4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]annino}benzoic acid;
38) 3-cyclopentyl-2-phenyl-2H-indazole-6-carboxylic acid;
39) (4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]annino}phenyl)acetic acid;
40) N-(1 -{[4-(cyanomethyl)phenyl]carbamoyl}cyclobutyl)-3-cyclohexyl-2-phenyl- 2H-indazole-6-carboxamide;
41 ) N-(1 -{[4-(2-amino-2-oxoethyl)phenyl]carbamoyl}cyclobutyl)-3-cyclohexyl-2- phenyl-2H-indazole-6-carboxamide;
42) 3-cyclohexyl-N-{1 -[(4-fluorobenzyl)carbamoyl]cyclobutyl}-2-phenyl-2H- indazole-6-carboxamide;
43) 3-cyclohexyl-N-(1 -{[4-methyl-3-(trifluoromethyl)phenyl]carbamoyl}cyclobutyl)- 2-phenyl-2H-indazole-6-carboxamide;
44) 2-[4-(benzyloxy)phenyl]-3-cyclohexyl-2H-indazole-6-carboxylic acid;
45) N-{1 -[(6-cyanopyridin-3-yl)carbamoyl]cyclobutyl}-3-cyclohexyl-2-phenyl-2H- indazole-6-carboxamide;
46) N-{1 -[(6-carbamoylpyridin-3-yl)carbamoyl]cyclobutyl}-3-cyclohexyl-2-phenyl- 2H-indazole-6-carboxamide;
47) 3-cyclohexyl-2-phenyl-N-[1 -(pyrazin-2-ylcarbamoyl)cyclobutyl]-2H-indazole- 6-carboxamide;
48) ethyl 1 -[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]piperidine-4-carboxylate;
49) ethyl 5-{[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]annino}-1-benzofurari-2-carboxylate;
50) 4-({[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]annino}nnethyl)benzoic acid;
51 ) 3-cyclohexyl-2-phenyl-N-(1 -{[6-(1 H-tetrazol-5-yl)pyridin-3- yl]carbamoyl}cyclobutyl)-2H-indazole-6-carboxamide;
52) 3-cyclohexyl-2-phenyl-N-(1 -{[5-(1 H-tetrazol-5-yl)pyridin-2- yl]carbamoyl}cyclobutyl)-2H-indazole-6-carboxamide;
53) N-{1 -[(5-carbamoylpyridin-2-yl)carbamoyl]cyclobutyl}-3-cyclohexyl-2-phenyl- 2H-indazole-6-carboxamide; 54) 5-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]annino}-1-benzofurari-2-carboxylic acid;
55) ethyl N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-5-hydroxy-L- tryptophanate;
56) N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-5-hydroxy-L-tryptophan;
57) (2E)-3-[4-({N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-L- alanyl}amino)phenyl]prop-2-enoic acid;
58) (2E)-3-[4-({N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-L- valyl}amino)phenyl]prop-2-enoic acid;
59) ethyl (2E)-3-[4-({[1-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}annino)phenyl]prop-2-enoate;
60) ethyl (2E)-3-(4-{[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclopentyl)carbonyl]amino}phenyl)prop-2-enoate;
61 ) 3-cyclohexyl-N-[(2R)-4-({4-[(1 E)-3-(methylamino)-3-oxoprop-1 -en-1 - yl]phenyl}amino)-4-oxobutan-2-yl]-2-phenyl-2H-indazole-6-carboxamide;
62) (2E)-3-[4-({[1-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}annino)phenyl]prop-2-enoic acid;
63) (2E)-3-(4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclopentyl)carbonyl]amino}phenyl)prop-2-enoic acid;
64) 3-cyclohexyl-N-(1 -{[2-(dimethylamino)pyrimidin-5-yl]carbamoyl}cyclobutyl)-2- phenyl-2H-indazole-6-carboxamide;
65) (2E)-3-(4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclohexyl)carbonyl]amino}phenyl)prop-2-enoic acid;
66) 4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-pyrazolo[3,4-b]pyridin-6- yl)carbonyl]amino}cyclobutyl)carbonyl]annino}benzoic acid;
67) (2E)-3-[4-({N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-2- methylalanyl}amino)phenyl]prop-2-enoic acid;
68) (2E)-3-[4-({N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-3-(1 ,3- thiazol-4-yl)-L-alanyl}amino)phenyl]prop-2-enoic acid;
69) (2E)-3-(4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclopropyl)carbonyl]amino}phenyl)prop-2-enoic acid;
70) 3-cyclohexyl-N-(1 -{[2-(1 -methyl-1 H-pyrazol-4-yl)pyrimidin-5- yl]carbamoyl}cyclobutyl)-2-phenyl-2H-indazole-6-carboxamide;
71 ) 3-cyclohexyl-N-(1 -{[2-(1 ,5-dimethyl-1 H-pyrazol-4-yl)pyrimidin-5- yl]carbamoyl}cyclobutyl)-2-phenyl-2H-indazole-6-carboxamide; 72) 3-cyclohexyl-2-phenyl-N-(1 -{[2-(1 -propyl-1 H-pyrazol-4-yl)pyrinnidin-5- yl]carbamoyl}cyclobutyl)-2H-indazole-6-carboxamide;
73) 3-cyclohexyl-2-phenyl-N-(1 -{[2-(1 H-pyrazol-4-yl)pyrimidin-5- yl]carbamoyl}cyclobutyl)-2H-indazole-6-carboxamide;
74) 3-cyclohexyl-N-(1 -{[2-(methylamino)pyrimidin-5-yl]carbamoyl}cyclobutyl)-2- phenyl-2H-indazole-6-carboxamide;
75) 3-cyclohexyl-N-{1 -[(2-{[2-(dimethylamino)ethyl]amino}pyrimidin-5- yl)carbamoyl]cyclobutyl}-2-phenyl-2H-indazole-6-carboxamide;
76) (2E)-3-[4-({3-[(benzyloxy)methyl]-N-[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]histidyl}amino)phenyl]prop-2-enoic acid;
77) N-(5-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}pyrimidin-2-yl)glycine;
78) N-{1 -[(2-aminopyrimidin-5-yl)carbamoyl]cyclobutyl}-3-cyclohexyl-2-phenyl- 2H-indazole-6-carboxamide;
79) (2E)-3-{4-[({1 -[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]piperidin-3- yl}carbonyl)amino]phenyl}prop-2-enoic acid;
80) ethyl (2E)-3-(4-{[(3R)-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)butanoyl]amino}phenyl)prop-2-enoate;
81 ) (2E)-3-(4-{[(3R)-3-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}butanoyl]amino}phenyl)prop-2-enoic acid;
82) 4-({[3-(benzyloxy)-1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl]carbonyl}annino)benzoic acid;
83) (2E)-3-(4-{[(3R)-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)butanoyl]amino}phenyl)prop-2-enoic acid;
84) (2E)-3-(4-{[(4-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}tetrahydro-2H-pyran-4-yl)acetyl]amino}phenyl)prop-2- enoic acid;
85) ethyl 4-{[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]amino}-3- hydroxycyclobutyl)carbonyl]amino}benzoate;
86) (2E)-3-[4-({[1-({[3-cyclohexyl-2-(4-fluorophenyl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}annino)phenyl]prop-2-enoic acid;
87) (2E)-3-{4-[({1 -[({3-cyclohexyl-2-[4-(difluoromethoxy)phenyl]-2H-indazol-6- yl}carbonyl)amino]cyclobutyl}carbonyl) amino]phenyl}prop-2-enoic acid;
88) ethyl 5-{[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}-1-methyl-1 H-indole-2- carboxylate; 89) 3-cyclohexyl-2-phenyl-N-[1 -(pyrimidin-5-ylcarbamoyl)cyclobutyl]-2H-indazole- 6-carboxamide;
90) 5-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}-1-methyl-1 H-indole-2- carboxylic acid;
91 ) 3-cyclohexyl-N-(1 -{[2-(methylcarbamoyl)-1 -benzofuran-5- yl]carbamoyl}cyclobutyl)-2-phenyl-2H-indazole-6-carboxamide;
92) 4-{[(3R)-3-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}butanoyl]amino}benzoic acid;
93) 1-(5-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cydobutyl)carbonyl]amino}pyrimidin-2-yl)-L-proline;
94) (2E)-3-[4-({[1-({[3-cyclohexyl-2-(pyridin-3-yl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}annino)phenyl]prop-2-enoic acid;
95) 6-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}pyrazolo[1 ,5-a]pyrimidine-3- carboxylic acid;
96) ethyl (2E)-3-[4-({[(3R)-3-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}-1-methylpyrrolidin-3-yl]carbonyl}amino)phenyl]prop-2- enoate;
97) (2E)-3-[4-({[(3R)-3-{[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]amino}- 1-methylpyrrolidin-3-yl]carbonyl}amino)phenyl]prop-2-enoic acid;
98) 4-{[(trans-1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]amino}-3- hydroxycyclobutyl)carbonyl]amino}benzoic acid;
99) 4-{[(cis-1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]amino}-3- hydroxycyclobutyl)carbonyl]amino}benzoic acid;
100) 6-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]annino}-1-benzofurari-2-carboxylic acid;
101 ) 3-cyclohexyl-N-(1 -{[2-(methylcarbamoyl)-1 -benzofuran-6- yl]carbamoyl}cyclobutyl)-2-phenyl-2H-indazole-6-carboxamide;
102) (2E)-3-(4-{[(4-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}tetrahydro-2H-pyran-4-yl)carbonyl]amino}phenyl)prop-2- enoic acid;
103) (2E)-3-(4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-pyrazolo[4,3-c]pyridin-6- yl)carbonyl]amino}cyclobutyl)carbonyl]annino}phenyl)prop-2-enoic acid;
104) ethyl 6-{[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}-1 l-l-benzimidazole-2- carboxylate;
105) (2E)-3-(4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-pyrazolo[3,4-d]pyrimidin-6- yl)carbonyl]amino}cyclobutyl)carbonyl]annino}phenyl)prop-2-enoic acid;
106) 4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]annino}-2-fluorobenzoic acid;
107) 4-({[1 -({[3-cyclohexyl-2-(4-f luorophenyl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}annino)benzoic acid;
108) 4-[({1 -[({3-cyclohexyl-2-[4-(difluoromethoxy)phenyl]-2H-indazol-6- yl}carbonyl)amino]cyclobutyl}carbonyl)annino]benzoic acid;
109) 6-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}-1 H-benzimidazole-2-carboxylic acid;
1 10) ethyl 5-{[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}-1-methyl-1 H-benzimidazole-2- carboxylate;
1 1 1 ) ethyl 6-{[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}cyclobutyl)carbonyl]amino}-1-methyl-1 H-benzimidazole-2- carboxylate;
1 12) ethyl (2E)-3-[4-({[(3 )-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)-1-methylpyrrolidin-3-yl]carbonyl}amino)phenyl]prop-2- enoate;
1 13) 4-({[3-(benzyloxy)-1 -({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}annino)benzoic acid;
1 14) 4-({N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-L- alanyl}amino)benzoic acid;
1 15) ethyl (2E)-3-[4-({[(3R)-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)tetrahydrofuran-3-yl]carbonyl}amino)phenyl]prop-2-enoate;
1 16) (2E)-3-[4-({[(3R)-3-{[(3-cyclohexyl-2-phenyl-2H-indazol-6- yl)carbonyl]amino}tetrahydrofuran-3-yl]carbonyl}amino)phenyl]prop-2-enoic acid;
1 17) (2E)-3-[4-({[(3R)-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)tetrahydrofuran-3-yl]carbonyl}amino)phenyl]prop-2-enoic acid;
1 18) 4-({N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-3-(dimethylamino)-L- alanyl}amino)benzoic acid; 1 19) 4-{[(4-{[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]amino}-1 - methylpiperidin-4-yl)carbonyl]amino}benzoic acid;
120) N-{1 -[(4-carbamoylphenyl)carbamoyl]cyclobutyl}-3-cyclohexyl-2-(pyridin-2- yl)-2H-indazole-6-carboxamide;
121 ) (2E)-3-[4-({N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-L- seryl}amino)phenyl]prop-2-enoic acid;
122) N-{1 -[(2-carbamoyl-1 -benzofuran-6-yl)carbamoyl]cyclobutyl}-3-cyclohexyl-2- phenyl-2H-indazole-6-carboxamide;
123) ethyl 5-({[1-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}amino)-1-methyl-1 H-indole-2- carboxylate;
124) ethyl 5-({[1-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}annino)-1-benzofurari-2-carboxylate;
125) ethyl 6-({[1-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}annino)-1-benzofurari-2-carboxylate;
126) 5-({[1 -({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}armino)-1-inethyl-1 l-l-indole-2- carboxylic acid;
127) 5-({[1 -({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}annino)-1-benzofurari-2-carboxylic acid;
128) 6-({[1 -({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}annino)-1-benzofurari-2-carboxylic acid;
129) N-{1 -[(6-cyanopyridin-3-yl)carbamoyl]cyclobutyl}-3-cyclohexyl-2-(pyridin-2- yl)-2H-indazole-6-carboxamide;
130) 3-cyclohexyl-N-(1 -{[1-methyl-2-(methylcarbamoyl)-1 H-indol-5- yl]carbamoyl}cyclobutyl)-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
131 ) 3-cyclohexyl-N-[(2S)-1 -{[2-(methylcarbamoyl)-1-benzofuran-5-yl]amino}-1 - oxopropan-2-yl]-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
132) 3-cyclohexyl-N-(1 -{[1-methyl-2-(methylcarbamoyl)-1 H-indol-5- yl]carbamoyl}cyclobutyl)-2-phenyl-2H-indazole-6-carboxamide;
133) 3-cyclohexyl-N-(1 -{[2-(methylcarbamoyl)-1 -benzofuran-6- yl]carbamoyl}cyclobutyl)-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
134) N-{1 -[(6-carbamoylpyridin-3-yl)carbamoyl]cyclobutyl}-3-cyclohexyl-2-(pyridin- 2-yl)-2H-indazole-6-carboxamide; 135) 5-[(N-{[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6-yl]carbonyl}-L- alanyl)amino]-1 -benzofuran-2-carboxylic acid;
136) 4-({1-benzyl-N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-L- histidyl}amino)benzoic acid;
137) ethyl 5-({[4-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6-yl]carbonyl}amino)-1 - methylpiperidin-4-yl]carbonyl}amino)-1-benzofuran-2-carboxylate;
138) ethyl (2E)-3-[4-({[4-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)-1-methylpiperidin-4-yl]carbonyl}amino)phenyl]prop-2- enoate;
139) (2E)-3-[4-({1 -benzyl-N-[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]-L- histidyl}amino)phenyl]prop-2-enoic acid;
140) ethyl 5-({[(3S)-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)tetrahydrofuran-3-yl]carbonyl}amino)-1-benzofuran-2- carboxylate;
141 ) ethyl 5-({[(3S)-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)-1-methylpyrrolidin-3-yl]carbonyl}amino)-1-benzofuran-2- carboxylate;
142) 5-({[4-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6-yl]carbonyl}amino)-1 - methylpiperidin-4-yl]carbonyl}amino)-1-benzofuran-2-carboxylic acid;
143) (2E)-3-[4-({[4-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6-yl]carbonyl}amino)- 1-methylpiperidin-4-yl]carbonyl}amino)phenyl]prop-2-enoic acid;
144) 5-({[(3S)-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)tetrahydrofuran-3-yl]carbonyl}amino)-1-benzofuran-2- carboxylic acid;
145) 5-({[(3S)-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6-yl]carbonyl}amino)-1- methylpyrrolidin-3-yl]carbonyl}amino)-1-benzofuran-2-carboxylic acid;
146) N-[(2 )-1 -{[4-(2-aminopyrimidin-5-yl)phenyl]aiTiino}-1-oxopropan-2-yl]-3- cyclohexyl-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
147) methyl 5-({[(3R)-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)tetrahydrofuran-3-yl]carbonyl}amino)-1 ,3-benzoxazole-2- carboxylate;
148) 3-cyclohexyl-N-(1 -methyl-4-{[2-(methylcarbamoyl)-1 -benzof uran-5- yl]carbamoyl}piperidin-4-yl)-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
149) 3-cyclohexyl-N-[1 -methyl-4-({4-[(1 E)-3-(methylamino)-3-oxoprop-1-en-1 - yl]phenyl}carbamoyl)piperidin-4-yl]-2-(pyridin-2-yl)-2H-indazole-6- carboxamide; 150) 3-cyclohexyl-N-[(3S)-3-{[2-(methylcarbamoyl)-1-benzofuran-5- yl]carbamoyl}tetrahydrofuran-3-yl]-2-(pyridin-2-yl)-2H-indazole-6- carboxamide;
151 ) 3-cyclohexyl-N-[(3S)-1 -methyl-3-{[2-(methylcarbamoyl)-1-benzofuran-5- yl]carbamoyl}pyrrolidin-3-yl]-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
152) 5-({[4-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6-yl]carbonyl}amino)-1 - methylpiperidin-4-yl]carbonyl}amino)-1-methyl-1 H-indole-2-carboxylic acid;
153) (2E)-3-[4-({[(3S)-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)-1-methylpyrrolidin-3-yl]carbonyl}amino)phenyl]prop-2- enoic acid;
154) ethyl 5-({[(3 )-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)tetrahydrofuran-3-yl]carbonyl}amino)-1-methyl-1 H-indole- 2-carboxylate;
155) N-[(3R)-3-(1 ,3-benzoxazol-5-ylcarbamoyl)tetrahydrofuran-3-yl]-3-cyclohexyl- 2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
156) 5-({[(3R)-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)tetrahydrofuran-3-yl]carbonyl}amino)-1-methyl-1 H-indole- 2-carboxylic acid;
157) 3-cyclohexyl-N-[(3R)-3-{[1-methyl-2-(methylcarbamoyl)-1 H-indol-5- yl]carbamoyl}tetrahydrofuran-3-yl]-2-(pyridin-2-yl)-2H-indazole-6- carboxamide;
158) 5-({[(3R)-3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6- yl]carbonyl}amino)tetrahydrofuran-3-yl]carbonyl}amino)-N-methyl-1 ,3- benzoxazole-2-carboxamide;
159) ethyl (2E)-3-[4-({[1-({[3-cyclohexyl-2-(4-methoxyphenyl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl} amino)phenyl]prop-2-enoate;
160) (2E)-3-[4-({[1-({[3-cyclohexyl-2-(4-methoxyphenyl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}annino)phenyl]prop-2-enoic acid;
161 ) 5-({[1 -({[3-cyclohexyl-2-(4-methoxyphenyl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}annino)-1-benzofurari-2-carboxylic acid;
162) N-(1 -{[4-(2-aminopyrimidin-5-yl)phenyl]carbamoyl}cyclobutyl)-3-cyclohexyl-2- (4-methoxyphenyl)-2H-indazole-6-carboxamide;
163) N-(1 -{[4-(2-aminopyrimidin-5-yl)phenyl]carbamoyl}cyclobutyl)-3-cyclohexyl-2- (pyridin-2-yl)-2H-indazole-6-carboxamide;
164) ethyl 5-({[1-({[3-cyclohexyl-2-(4-methoxyphenyl)-2H-indazol-6- yl]carbonyl}amino)cyclobutyl]carbonyl}annino)-1-benzofurari-2-carboxylate;
165) 3-cyclohexyl-2-(4-methoxyphenyl)-N-(1 -{[2-(methylcarbamoyl)-1 -benzofuran- 5-yl]carbamoyl}cyclobutyl)-2H-indazole-6-carboxamide;
166) (2E)-3-[4-({[3-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6-yl]carbonyl}amino)- 1-methylazetidin-3-yl]carbonyl}amino)phenyl]prop-2-enoic acid;
167) 3-cyclohexyl-N-[1 -methyl-3-({4-[(1 E)-3-(methylamino)-3-oxoprop-1-en-1 - yl]phenyl}carbamoyl)azetidin-3-yl]-2-(pyridin-2-yl)-2H-indazole-6- carboxamide;
168) 3-cyclohexyl-N-(1 -methyl-3-{[2-(methylcarbamoyl)-1 -benzof uran-5- yl]carbamoyl}azetidin-3-yl)-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
169) N-(1 -{[4-(6-aminopyridin-3-yl)phenyl]carbamoyl}cyclobutyl)-3-cyclohexyl-2-(4- methoxyphenyl)-2H-indazole-6-carboxamide;
170) 3-cyclohexyl-2-(4-methoxyphenyl)-N-(1 -{[4-(1-methyl-2-oxo-1 ,2- dihydropyridin-4-yl)phenyl]carbamoyl}cyclobutyl)-2H-indazole-6- carboxamide;
171 ) 3-cyclohexyl-N-[1 -({4-[2-(dimethylamino)pyrimidin-5- yl]phenyl}carbamoyl)cyclobutyl]-2-(4-methoxyphenyl)-2H-indazole-6- carboxamide;
172) N-(4-{[4-(2-aminopyrimidin-5-yl)phenyl]carbamoyl}-1 -methylpiperidin-4-yl)-3- cyclohexyl-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
173) 3-cyclohexyl-2-(4-methoxyphenyl)-N-[1 -({4-[(1 E)-3-(methylamino)-3-oxoprop- 1-en-1 -yl]phenyl}carbamoyl)cyclobutyl]-2H-indazole-6-carboxamide;
174) N-[(2S)-1-{[4-(2-aminopyrimidin-5-yl)phenyl]amino}-1-oxopropan-2-yl]-3- cyclohexyl-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
175) ethyl 4-{[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]amino}-3- oxocyclobutyl)carbonyl]amino}benzoate;
176) 3-cyclohexyl-N-[(2 )-1-({4-[2-(dimethylamino)pyrimidin-5-yl]phenyl}amino)-1 - oxopropan-2-yl]-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
177) 3-cyclohexyl-2-(4-methoxyphenyl)-N-(1 -{[4-(pyrimidin-5- yl)phenyl]carbamoyl}cyclobutyl)-2H-indazole-6-carboxamide;
178) N-[(2R)-1 -{[4-(2-aminopyrimidin-5-yl)phenyl]aiTiino}-1-oxopropan-2-yl]-3- cyclohexyl-2-(4-methoxyphenyl)-2H-indazole-6-carboxamide;
179) tert-butyl (5-{4-[(N-{[3-cyclohexyl-2-(4-methoxyphenyl)-2H-indazol-6- yl]carbonyl}-D-alanyl)amino]phenyl}pyrimidin-2-yl)carbamate;
180) 3-cyclohexyl-N-[(2R)-1-{[4-(1 -methyl-2-oxo-1 ,2-dihydropyridin-4- yl)phenyl]amino}-1-oxopropan-2-yl]-2-(pyridin-2-yl)-2H-indazole-6- carboxamide;
181 ) 3-cyclohexyl-N-[(2R)-1-({4-[2-(dimethylamino)-1 ,3-thiazol-4-yl]phenyl}amino)- 1-oxopropan-2-yl]-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
182) methyl 5-{4-[(N-{[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6-yl]carbonyl}-D- alanyl)amino]phenyl}pyridine-3-carboxylate;
183) 3-cyclohexyl-N-[(2R)-1-oxo-1 -{[4-(pyrimidin-5-yl)phenyl]amino}propan-2-yl]-2- (pyridin-2-yl)-2H-indazole-6-carboxamide;
184) N-{(2R)-1 -[(4'-aminobiphenyl-4-yl)amino]-1-oxopropan-2-yl}-3-cyclohexyl-2- (pyridin-2-yl)-2H-indazole-6-carboxamide;
185) 3-cyclohexyl-N-[(2R)-1-({4-[2-(morpholin-4-yl)-1 ,3-thiazol-4-yl]phenyl}amino)-
1- oxopropan-2-yl]-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
186) 4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]amino}-3- fluorocyclobutyl)carbonyl]amino}benzoic acid;
187) 4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]amino}-3- oxocyclobutyl)carbonyl]amino}benzoic acid;
188) 3-cyclohexyl-N-[(2R)-1-oxo-1 -{[4-(6-oxo-1 ,6-dihydropyridin-3- yl)phenyl]amino}propan-2-yl]-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
189) N-[(2R)-1 -{[4-(5-aminopyridin-3-yl)phenyl]amino}-1 -oxopropan-2-yl]-3- cyclohexyl-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
190) ethyl 4-({[1-({[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6-yl]carbonyl}amino)-3- hydroxycyclobutyl]carbonyl}amino)benzoate;
191 ) N-[(2R)-1 -{[4-(6-aminopyridin-3-yl)phenyl]amino}-1 -oxopropan-2-yl]-3- cyclohexyl-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
192) 4-{[(1 -{[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]amino}-3,3- difluorocyclobutyl)carbonyl]amino}benzoic acid;
193) 3-cyclohexyl-N-{(2S)-3-hydroxy-1 -[(4-iodophenyl)amino]-1-oxopropan-2-yl}-2- (pyridin-2-yl)-2H-indazole-6-carboxamide;
194) 3-cyclohexyl-N-[(2R)-1-{[4-(6-methylpyridin-3-yl)phenyl]amino}-1-oxopropan-
2- yl]-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
195) N-[(2S)-1-{[4-(2-aminopyrimidin-5-yl)phenyl]amino}-3-hydroxy-1 -oxopropan- 2-yl]-3-cyclohexyl-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
196) 5-{4-[(N-{[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6-yl]carbonyl}-D- alanyl)amino]phenyl}pyridine-3-carboxylic acid;
197) N-[(2R)-1 -{[4-(2-chloro-1 -methyl-1 H-imidazol-5-yl)phenyl]amino}-1 - oxopropan-2-yl]-3-cyclohexyl-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
198) 3-cyclohexyl-N-[(2R)-1-{[4-(2-methylpyrimidin-5-yl)phenyl]amino}-1- oxopropan-2-yl]-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
199) 4-{[(1-{[(3-cyclohexyl-2-phenyl-2H-indazol-6-yl)carbonyl]amino}-3-[(2- hydroxyethyl)amino]cyclobutyl)carbonyl]amino}benzoic acid;
200) N-[(2R)-1-({4-[6-(acetylamino)pyridin-3-yl]phenyl}amino)-1-oxopropan-2-yl]-3- cyclohexyl-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
201 ) N-[(2R)-1-{[4-(2-aminopyridin-4-yl)phenyl]amino}-1-oxopropan-2-yl]-3- cyclohexyl-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
202) 3-cyclohexyl-N-[(2R)-1-({4-[2-(methylamino)pyrimidin-5-yl]phenyl}amino)-1 - oxopropan-2-yl]-2-(pyridin-2-yl)-2H-indazole-6-carboxamide;
203) 3-cyclohexyl-N-{(2R)-1-[(5-{[4-(hydroxyamino)phenyl]sulfonyl}-1 ,3-thiazol-2- yl)amino]-1 -oxopropan-2-yl}-2-(pyridin-2-yl)-2H-indazole-6-carboxamide; and
204) (5-{4-[(N-{[3-cyclohexyl-2-(pyridin-2-yl)-2H-indazol-6-yl]carbonyl}-D- alanyl)amino]phenyl}pyrimidin-2-yl)acetic acid;
or salts, W-oxides, racemates, enantiomers and isomers thereof.
22. A process for producing a compound of general formula (I) according to claim 1 wherein R1 is C(0)R4 and R4 is NR5R6 comprising reacting a compound of formula (II) with an amino precursor of general formula NHR5R6 under amide coupling conditions

wherein
Z'i , Z'2, Z'3 and Z'4 are each independently selected from C-X, C-Ra , N and N+-0" wherein one of Z'i, Z'2 ,Z'3 and Z'4 is C-X;
X is (R1)tC02H or (R^COaC^alkyl;
t is 0 or 1 ; and
R1 , R2, R3 and Ra are as defined in claim 1 ;
or its salts, N-oxides, racemates, enantiomers and isomers thereof.
23. A compound of formula (II) as defined in claim 22 or its salts, N-oxides, racemates, enantiomers and isomers thereof.
24. A compound of formula (II) according to claim 23 selected from the group consisting of:
3-(cyclohex-1-en-1-yl)-2-phenyl-2H-indazole-6-carboxylic acid;
3-cyclohexyl-2-phenyl-2/-/-indazole-6-carboxylic acid;
2,3-diphenyl-2H-indazole-6-carboxylic acid; 3-cyclopentyl-2-phenyl-2H-indazole-6-carboxylic acid;
2- [4-(benzyloxy)phenyl]-3-cyclohexyl-2H-indazole-6-carboxylic acid;
ethyl 3-(cyclohex-1 -en-1 -yl)-2-phenyl-2/-/-pyrazolo[3,4-d]pyrimidine-6-carboxylate;
3- (cyclohex-1-en-1 -yl)-2-phenyl-2/-/-pyrazolo[3,4-c/]pyrimidine-6-carboxylic acid;
3-cyclohexyl-2-phenyl-2H-pyrazolo[3,4-a^pyrimidine-6-carboxylic acid;
ethyl 3-(cyclohex-1 -en-1 -yl)-2-phenyl-2/-/-pyrazolo[4,3-c]pyridine-6-carboxylate;
ethyl 3-cyclohexyl-2-phenyl-2/-/-pyrazolo[4,3-c]pyridine-6-carboxylate;
3-cyclohexyl-2-phenyl-2/-/-pyrazolo[4,3-c]pyridine-6-carboxylic acid;
3-(cyclohex-1-en-1 -yl)-2-phenyl-2/-/-pyrazolo[3,4-0]pyridine-6-carboxylic acid;
3-cyclohexyl-2-phenyl-2/-/-pyrazolo[3,4-£>]pyridine-6-carboxylic acid;
2- [4-(benzyloxy)phenyl]-3-(cyclohex-1 -en-1 -yl)-2/-/-indazole-6-carboxylic acid;
3- cyclohexyl-2-(4-hydroxyphenyl)-2/-/-indazole-6-carboxylic acid;
methyl 3-cyclohexyl-2-(4-hydroxyphenyl)-2H-indazole-6-carboxylate;
methyl 3-cyclohexyl-2-(4-methoxyphenyl)-2/-/-indazole-6-carboxylate; and
3-(cyclohex-1-en-1 -yl)-2-(4-methoxyphenyl)-2/-/-indazole-6-carboxylic acid;
or salts, W-oxides, racemates, enantiomers and isomers thereof.
25. A pharmaceutical agent comprising the compound of formula (I) as defined in any one of claims 1 to 21 or its salts, W-oxides, racemates, enantiomers and isomers thereof.
26. The pharmaceutical agent according to claim 25 which is an antiviral agent.
27. A viral polymerase inhibitor comprising the compound of formula (I) as defined in any one of claims 1 to 21 or its salts, W-oxides, racemates, enantiomers and isomers thereof.
28. The viral polymerase inhibitor according to claim 27 which is a HCV polymerase inhibitor.
29. A pharmaceutical composition comprising the compound of formula (I) as defined in any one of claims 1 to 21 or its salts, W-oxides, racemates, enantiomers and isomers thereof and a pharmaceutically acceptable carrier.
30. The pharmaceutical composition according to claim 29 which additionally comprises a therapeutically effective amount of one or more antiviral agents.
31. A method for the treatment of a Flaviviridae viral infection which comprises administering an effective amount of the compound of formula (I) as defined in any one of claims 1 to 21 or its salts, W-oxides, racemates, enantiomers and isomers thereof or the pharmaceutical composition as defined in claim 29 or claim 30 to a subject in need thereof.
32. The method of treatment according to claim 31 wherein the Flaviviridae viral infection is a Hepatitis C virus (HCV) infection.
33. A method of inhibiting the RNA-dependent RNA polymerase activity of the enzyme NS5B, encoded by HCV, comprising exposing the enzyme NS5B to an effective amount of the compound of formula (I) as defined in any one of claims 1 to 21 or its salts, N- oxides, racemates, enantiomers and isomers thereof.
34. A method of inhibiting HCV replication comprising exposing a cell infected with HCV to an effective amount of the compound of formula (I) as defined in any one of claims 1 to 21 or its salts, W-oxides, racemates, enantiomers and isomers thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US35338210P | 2010-06-10 | 2010-06-10 | |
| US61/353,382 | 2010-06-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011153588A1 true WO2011153588A1 (en) | 2011-12-15 |
Family
ID=45097403
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/AU2011/000713 WO2011153588A1 (en) | 2010-06-10 | 2011-06-08 | Viral polymerase inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011153588A1 (en) |
Cited By (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103214377A (en) * | 2013-03-22 | 2013-07-24 | 苏州康润医药有限公司 | Synthesis process of 1, 3, 5-triaminobenzene |
| WO2013152391A1 (en) * | 2012-04-11 | 2013-10-17 | Biota Scientific Management Pty Ltd | Viral polymerase inhibitors |
| US20130317000A1 (en) * | 2012-05-22 | 2013-11-28 | Xenon Pharmaceuticals Inc. | N-substituted benzamides and methods of use thereof |
| WO2014068988A1 (en) * | 2012-10-31 | 2014-05-08 | Raqualia Pharma Inc. | Pyrazolopyridine derivatives as ttx-s blockers |
| CN105503865A (en) * | 2015-12-24 | 2016-04-20 | 南京华威医药科技开发有限公司 | Novel pyrazolopyridine antineoplastic compound |
| US9481677B2 (en) | 2011-10-31 | 2016-11-01 | Xenon Pharmaceuticals Inc. | Biaryl ether sulfonamides and their use as therapeutic agents |
| WO2016174183A1 (en) | 2015-04-30 | 2016-11-03 | Bayer Pharma Aktiengesellschaft | Combinations of inhibitors of irak4 with inhibitors of btk |
| US9493429B2 (en) | 2013-03-15 | 2016-11-15 | Genentech, Inc. | Substituted benzoxazoles and methods of use thereof |
| US9546164B2 (en) | 2013-11-27 | 2017-01-17 | Genentech, Inc. | Substituted benzamides and methods of use thereof |
| US9550775B2 (en) | 2013-03-14 | 2017-01-24 | Genentech, Inc. | Substituted triazolopyridines and methods of use thereof |
| US9630929B2 (en) | 2011-10-31 | 2017-04-25 | Xenon Pharmaceuticals Inc. | Benzenesulfonamide compounds and their use as therapeutic agents |
| US9695166B2 (en) | 2014-05-05 | 2017-07-04 | Global Blood Therapeutics, Inc. | Pyrazolopyridine pyrazolopyrimidine and related compounds |
| EP3195865A1 (en) | 2016-01-25 | 2017-07-26 | Bayer Pharma Aktiengesellschaft | Combinations of inhibitors of irak4 with inhibitors of btk |
| WO2017148902A1 (en) | 2016-03-03 | 2017-09-08 | Bayer Pharma Aktiengesellschaft | New 2-substituted indazoles, methods for producing same, pharmaceutical preparations that contain same, and use of same to produce drugs |
| EP3219329A1 (en) | 2016-03-17 | 2017-09-20 | Bayer Pharma Aktiengesellschaft | Combinations of copanlisib |
| WO2017186693A1 (en) | 2016-04-29 | 2017-11-02 | Bayer Pharma Aktiengesellschaft | Synthesis of indazoles |
| WO2017186703A1 (en) | 2016-04-29 | 2017-11-02 | Bayer Pharma Aktiengesellschaft | Polymorphic form of n-{6-(2-hydroxypropan-2-yl)-2-[2-(methylsulphonyl)ethyl]-2h-indazol-5-yl}-6-(trifluoromethyl)pyridine-2-carboxamide |
| WO2017207481A1 (en) | 2016-06-01 | 2017-12-07 | Bayer Animal Health Gmbh | Substituted indazoles ueful for treatment and prevention of allergic and/or inflammatory diseases in animals |
| WO2017207385A1 (en) | 2016-05-31 | 2017-12-07 | Bayer Pharma Aktiengesellschaft | Substituted 3-methylindazoles, methods for the production thereof, pharmaceutical preparations containing same, and use thereof for the production of medicaments |
| US10005724B2 (en) | 2014-07-07 | 2018-06-26 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| CN108276406A (en) * | 2018-04-04 | 2018-07-13 | 苏州大学 | The synthetic method of polycyclic 2- hydrogen pyrazole compound |
| US10071957B2 (en) | 2012-07-06 | 2018-09-11 | Genentech, Inc. | N-substituted benzamides and methods of use thereof |
| US10179767B2 (en) | 2015-05-22 | 2019-01-15 | Genentech, Inc. | Substituted benzamides and methods of use thereof |
| US10308634B2 (en) | 2014-11-26 | 2019-06-04 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, methods for the production thereof, pharmaceutical preparations that contain said new substituted indazoles, and use of said new substituted indazoles to produce drugs |
| US10457654B2 (en) | 2016-10-17 | 2019-10-29 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| WO2020127685A1 (en) * | 2018-12-19 | 2020-06-25 | Leo Pharma A/S | Amino-acid anilides as small molecule modulators of il-17 |
| US10766858B2 (en) | 2016-03-30 | 2020-09-08 | Genentech, Inc. | Substituted benzamides and methods of use thereof |
| US10787446B2 (en) | 2015-09-28 | 2020-09-29 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| US10793550B2 (en) | 2017-03-24 | 2020-10-06 | Genentech, Inc. | 4-piperidin-n-(pyrimidin-4-yl)chroman-7-sulfonamide derivatives as sodium channel inhibitors |
| US10899732B2 (en) | 2015-11-25 | 2021-01-26 | Genentech, Inc. | Substituted benzamides useful as sodium channel blockers |
| US10947251B2 (en) | 2018-03-30 | 2021-03-16 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| US11028075B2 (en) | 2018-02-26 | 2021-06-08 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| US11130726B2 (en) | 2015-08-27 | 2021-09-28 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| US11161850B2 (en) | 2018-07-05 | 2021-11-02 | Incyte Corporation | Fused pyrazine derivatives as A2A / A2B inhibitors |
| US11168089B2 (en) | 2018-05-18 | 2021-11-09 | Incyte Corporation | Fused pyrimidine derivatives as A2A / A2B inhibitors |
| US11390624B2 (en) | 2019-01-29 | 2022-07-19 | Incyte Corporation | Pyrazolopyridines and triazolopyridines as A2A / A2B inhibitors |
| WO2022267673A1 (en) | 2021-06-21 | 2022-12-29 | 上海勋和医药科技有限公司 | Sulfoximide substituted indazole irak4 kinase inhibitor, preparation method therefor, and use thereof |
| US11673894B2 (en) | 2018-02-27 | 2023-06-13 | Incyte Corporation | Imidazopyrimidines and triazolopyrimidines as A2A / A2B inhibitors |
| US11786528B2 (en) | 2018-06-04 | 2023-10-17 | Exscientia Ltd. | Pyrazolopyrimidine compounds as adenosine receptor antagonists |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003000682A1 (en) * | 2001-06-25 | 2003-01-03 | Merck & Co., Inc. | (pyrimidyl)(phenyl)substituted fused heteroaryl p38 inhibiting and pkg kinase inhibiting compounds |
| WO2005014543A1 (en) * | 2003-08-06 | 2005-02-17 | Japan Tobacco Inc. | Condensed ring compound and use thereof as hcv polymerase inhibitor |
| WO2009095752A1 (en) * | 2008-01-29 | 2009-08-06 | Glenmark Pharmaceuticals, S.A. | Fused pyrazole derivatives as cannabinoid receptor modulators |
-
2011
- 2011-06-08 WO PCT/AU2011/000713 patent/WO2011153588A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003000682A1 (en) * | 2001-06-25 | 2003-01-03 | Merck & Co., Inc. | (pyrimidyl)(phenyl)substituted fused heteroaryl p38 inhibiting and pkg kinase inhibiting compounds |
| WO2005014543A1 (en) * | 2003-08-06 | 2005-02-17 | Japan Tobacco Inc. | Condensed ring compound and use thereof as hcv polymerase inhibitor |
| WO2009095752A1 (en) * | 2008-01-29 | 2009-08-06 | Glenmark Pharmaceuticals, S.A. | Fused pyrazole derivatives as cannabinoid receptor modulators |
Non-Patent Citations (2)
| Title |
|---|
| BEAULIEU, P. L. ET AL.: "Improved replicon cellular activity of non-nucleoside allosteric inhibitors of HCV NS5B polymerase: From benzimidazole to indole scaffolds", BIOORG. MED. CHEM. LETT., vol. 16, no. 19, 2006, pages 4987 - 4993 * |
| GOULET, S. ET AL.: "Discovery of benzimidazole-diamide finger loop (Thumb Pocket I) allosteric inhibitors of HCV NS5B polymerase: Implementing parallel synthesis for rapid linker optimization", BIOORG. MED. CHEM. LETT., vol. 20, no. 1, 2010, pages 196 - 200 * |
Cited By (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9771376B2 (en) | 2000-05-22 | 2017-09-26 | Genentech, Inc. | N-substituted benzamides and methods of use thereof |
| US9630929B2 (en) | 2011-10-31 | 2017-04-25 | Xenon Pharmaceuticals Inc. | Benzenesulfonamide compounds and their use as therapeutic agents |
| US9481677B2 (en) | 2011-10-31 | 2016-11-01 | Xenon Pharmaceuticals Inc. | Biaryl ether sulfonamides and their use as therapeutic agents |
| WO2013152391A1 (en) * | 2012-04-11 | 2013-10-17 | Biota Scientific Management Pty Ltd | Viral polymerase inhibitors |
| US9447080B2 (en) | 2012-04-11 | 2016-09-20 | Biota Scientific Management Pty Ltd. | Viral polymerase inhibitors |
| US20130317000A1 (en) * | 2012-05-22 | 2013-11-28 | Xenon Pharmaceuticals Inc. | N-substituted benzamides and methods of use thereof |
| US8933236B2 (en) * | 2012-05-22 | 2015-01-13 | Xenon Pharmaceuticals Inc. | N-substituted benzamides and methods of use thereof |
| US8952169B2 (en) | 2012-05-22 | 2015-02-10 | Xenon Pharmaceuticals Inc. | N-substituted benzamides and methods of use thereof |
| JP2015518863A (en) * | 2012-05-22 | 2015-07-06 | ジェネンテック, インコーポレイテッド | N-substituted benzamides and their use in the treatment of pain |
| US10071957B2 (en) | 2012-07-06 | 2018-09-11 | Genentech, Inc. | N-substituted benzamides and methods of use thereof |
| JP2015534941A (en) * | 2012-10-31 | 2015-12-07 | ラクオリア創薬株式会社 | Pyrazolopyridine derivatives as TTX-S blockers |
| US20150291582A1 (en) * | 2012-10-31 | 2015-10-15 | Raqualia Pharma Inc. | Pyrazolopyridine derivatives as ttx-s blockers |
| CN104812755A (en) * | 2012-10-31 | 2015-07-29 | 拉夸里亚创药株式会社 | Pyrazolopyridine derivatives as TTX-S blockers |
| WO2014068988A1 (en) * | 2012-10-31 | 2014-05-08 | Raqualia Pharma Inc. | Pyrazolopyridine derivatives as ttx-s blockers |
| US9944634B2 (en) | 2012-10-31 | 2018-04-17 | Raqualia Pharma Inc. | Pyrazolopyridine derivatives as TTX-S blockers |
| US9550775B2 (en) | 2013-03-14 | 2017-01-24 | Genentech, Inc. | Substituted triazolopyridines and methods of use thereof |
| US9493429B2 (en) | 2013-03-15 | 2016-11-15 | Genentech, Inc. | Substituted benzoxazoles and methods of use thereof |
| CN103214377B (en) * | 2013-03-22 | 2014-12-31 | 苏州康润医药有限公司 | Synthesis process of 1, 3, 5-triaminobenzene |
| CN103214377A (en) * | 2013-03-22 | 2013-07-24 | 苏州康润医药有限公司 | Synthesis process of 1, 3, 5-triaminobenzene |
| US9546164B2 (en) | 2013-11-27 | 2017-01-17 | Genentech, Inc. | Substituted benzamides and methods of use thereof |
| US9694002B2 (en) | 2013-11-27 | 2017-07-04 | Genentech, Inc. | Substituted benzamides and methods of use thereof |
| US9695166B2 (en) | 2014-05-05 | 2017-07-04 | Global Blood Therapeutics, Inc. | Pyrazolopyridine pyrazolopyrimidine and related compounds |
| US10005724B2 (en) | 2014-07-07 | 2018-06-26 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| US11149002B2 (en) | 2014-07-07 | 2021-10-19 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| US10125098B2 (en) | 2014-07-07 | 2018-11-13 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| US10526285B2 (en) | 2014-07-07 | 2020-01-07 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| US10308634B2 (en) | 2014-11-26 | 2019-06-04 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, methods for the production thereof, pharmaceutical preparations that contain said new substituted indazoles, and use of said new substituted indazoles to produce drugs |
| EP4260909A2 (en) | 2014-11-26 | 2023-10-18 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, process for their preparation, pharmaceutical preparations containing them and their use in the preparation of drugs |
| US10793545B2 (en) | 2014-11-26 | 2020-10-06 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, methods for the production thereof, pharmaceutical preparations that contain said new substituted indazoles, and use of said new substituted indazoles to produce drugs |
| EP3674298A1 (en) | 2014-11-26 | 2020-07-01 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, process for their preparation, pharmaceutical formulations containing them, and their use for the preparation of medicaments |
| WO2016174183A1 (en) | 2015-04-30 | 2016-11-03 | Bayer Pharma Aktiengesellschaft | Combinations of inhibitors of irak4 with inhibitors of btk |
| US10179767B2 (en) | 2015-05-22 | 2019-01-15 | Genentech, Inc. | Substituted benzamides and methods of use thereof |
| US11130726B2 (en) | 2015-08-27 | 2021-09-28 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| US10787446B2 (en) | 2015-09-28 | 2020-09-29 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| US10899732B2 (en) | 2015-11-25 | 2021-01-26 | Genentech, Inc. | Substituted benzamides useful as sodium channel blockers |
| CN105503865A (en) * | 2015-12-24 | 2016-04-20 | 南京华威医药科技开发有限公司 | Novel pyrazolopyridine antineoplastic compound |
| EP3195865A1 (en) | 2016-01-25 | 2017-07-26 | Bayer Pharma Aktiengesellschaft | Combinations of inhibitors of irak4 with inhibitors of btk |
| US10435396B2 (en) | 2016-03-03 | 2019-10-08 | Bayer Pharma Aktiegesellschaft | 2-substituted indazoles, methods for producing same, pharmaceutical preparations that contain same, and use of same to produce drugs |
| WO2017148902A1 (en) | 2016-03-03 | 2017-09-08 | Bayer Pharma Aktiengesellschaft | New 2-substituted indazoles, methods for producing same, pharmaceutical preparations that contain same, and use of same to produce drugs |
| EP3219329A1 (en) | 2016-03-17 | 2017-09-20 | Bayer Pharma Aktiengesellschaft | Combinations of copanlisib |
| WO2017157792A1 (en) | 2016-03-17 | 2017-09-21 | Bayer Pharma Aktiengesellschaft | Combinations of copanlisib |
| US11203572B2 (en) | 2016-03-30 | 2021-12-21 | Genentech, Inc. | Substituted benzamides and methods of use thereof |
| US10766858B2 (en) | 2016-03-30 | 2020-09-08 | Genentech, Inc. | Substituted benzamides and methods of use thereof |
| US10759758B2 (en) | 2016-04-29 | 2020-09-01 | Bayer Pharma Aktiengesellschaft | Polymorphic form of N-{6-(hydroxypropan-2-yl)-2-[2-(methylsulphonyl)ethyl]-2H-indazol-5-yl}-6-(trifluoromethyl)pyridine-2-carboxamide |
| WO2017186703A1 (en) | 2016-04-29 | 2017-11-02 | Bayer Pharma Aktiengesellschaft | Polymorphic form of n-{6-(2-hydroxypropan-2-yl)-2-[2-(methylsulphonyl)ethyl]-2h-indazol-5-yl}-6-(trifluoromethyl)pyridine-2-carboxamide |
| EP4275754A2 (en) | 2016-04-29 | 2023-11-15 | Bayer Pharma Aktiengesellschaft | Polymorphic form of n-{6-(2-hydroxypropan-2-yl)-2-[2-(methylsulphonyl)ethyl]-2h-indazol-5-yl}-6-(trifluoromethyl)pyridine-2-carboxamide |
| EP4275755A2 (en) | 2016-04-29 | 2023-11-15 | Bayer Pharma Aktiengesellschaft | Polymorphic form of n-{6-(2-hydroxypropan-2-yl)-2-[2-(methylsulphonyl)ethyl]-2h-indazol-5-yl}-6-(trifluoromethyl)pyridine-2-carboxamide |
| US10501437B2 (en) | 2016-04-29 | 2019-12-10 | Bayer Pharma Aktiengesellschaft | Crystalline forms of N-[2-(3-Hydroxy-3-methylbutyl)-6-(2-hydroxypropan-2-yl)-2H-indazol-5-yl]-6-(trifluoromethyl)pyridine-2-carboxamide |
| US10501417B2 (en) | 2016-04-29 | 2019-12-10 | Bayer Pharma Aktiengesellschaft | Synthesis of indazoles |
| WO2017186689A1 (en) | 2016-04-29 | 2017-11-02 | Bayer Pharma Aktiengesellschaft | Synthesis of indazoles |
| WO2017186700A1 (en) | 2016-04-29 | 2017-11-02 | Bayer Pharma Aktiengesellschaft | Crystalline forms of n-[2-(3-hydroxy-3-methylbutyl)-6-(2-hydroxypropan-2-yl)-2h-indazol-5-yl]-6-(trifluoromethyl)pyridine-2-carboxamide |
| WO2017186693A1 (en) | 2016-04-29 | 2017-11-02 | Bayer Pharma Aktiengesellschaft | Synthesis of indazoles |
| US10633365B2 (en) | 2016-04-29 | 2020-04-28 | Bayer Pharma Aktiengesellschaft | Synthesis of indazoles |
| WO2017207385A1 (en) | 2016-05-31 | 2017-12-07 | Bayer Pharma Aktiengesellschaft | Substituted 3-methylindazoles, methods for the production thereof, pharmaceutical preparations containing same, and use thereof for the production of medicaments |
| WO2017207481A1 (en) | 2016-06-01 | 2017-12-07 | Bayer Animal Health Gmbh | Substituted indazoles ueful for treatment and prevention of allergic and/or inflammatory diseases in animals |
| US10457654B2 (en) | 2016-10-17 | 2019-10-29 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| US10793550B2 (en) | 2017-03-24 | 2020-10-06 | Genentech, Inc. | 4-piperidin-n-(pyrimidin-4-yl)chroman-7-sulfonamide derivatives as sodium channel inhibitors |
| US11028075B2 (en) | 2018-02-26 | 2021-06-08 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| US11673894B2 (en) | 2018-02-27 | 2023-06-13 | Incyte Corporation | Imidazopyrimidines and triazolopyrimidines as A2A / A2B inhibitors |
| US10947251B2 (en) | 2018-03-30 | 2021-03-16 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| CN108276406A (en) * | 2018-04-04 | 2018-07-13 | 苏州大学 | The synthetic method of polycyclic 2- hydrogen pyrazole compound |
| US11168089B2 (en) | 2018-05-18 | 2021-11-09 | Incyte Corporation | Fused pyrimidine derivatives as A2A / A2B inhibitors |
| US11873304B2 (en) | 2018-05-18 | 2024-01-16 | Incyte Corporation | Fused pyrimidine derivatives as A2A/A2B inhibitors |
| US11786528B2 (en) | 2018-06-04 | 2023-10-17 | Exscientia Ltd. | Pyrazolopyrimidine compounds as adenosine receptor antagonists |
| US11161850B2 (en) | 2018-07-05 | 2021-11-02 | Incyte Corporation | Fused pyrazine derivatives as A2A / A2B inhibitors |
| US11377425B1 (en) | 2018-12-19 | 2022-07-05 | Leo Pharma A/S | Small molecule modulators of IL-17 |
| WO2020127685A1 (en) * | 2018-12-19 | 2020-06-25 | Leo Pharma A/S | Amino-acid anilides as small molecule modulators of il-17 |
| US11390624B2 (en) | 2019-01-29 | 2022-07-19 | Incyte Corporation | Pyrazolopyridines and triazolopyridines as A2A / A2B inhibitors |
| US11884665B2 (en) | 2019-01-29 | 2024-01-30 | Incyte Corporation | Pyrazolopyridines and triazolopyridines as A2A / A2B inhibitors |
| WO2022267673A1 (en) | 2021-06-21 | 2022-12-29 | 上海勋和医药科技有限公司 | Sulfoximide substituted indazole irak4 kinase inhibitor, preparation method therefor, and use thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2011153588A1 (en) | Viral polymerase inhibitors | |
| JP6349360B2 (en) | Pyridine-2-amide useful as a CB2 agonist | |
| KR101387274B1 (en) | Inhibitors of hepatitis c virus replication | |
| CA2718528C (en) | Substituted 4-hydroxypyrimidine-5-carboxamides | |
| US20030232860A1 (en) | Medicine comprising dicyanopyridine derivative | |
| AU2013302725A1 (en) | 4-heteroaryl substituted benzoic acid compounds as RORgammaT inhibitors and uses thereof | |
| WO2008141385A1 (en) | Viral polymerase inhibitors | |
| WO2006000085A1 (en) | Hepatitis c inhibitor peptide analogs | |
| EP3414234A1 (en) | Bruton's tyrosine kinase inhibitors | |
| JP2009502845A (en) | Macrocyclic inhibitor of hepatitis C virus | |
| AU2014234906B2 (en) | Cycloalkyl nitrile pyrazolo pyridones as Janus kinase inhibitors | |
| ES2292126T3 (en) | USED 4-CARBOXIPIRAZOL DERIVATIVES AS ANTIVIRAL AGENTS. | |
| WO2012174312A2 (en) | Benzimidazole derivatives as antiviral agents | |
| AU2014234907B2 (en) | Geminally substituted cyanoethylpyrazolo pyridones as Janus kinase inhibitors | |
| JP2008545756A (en) | 1-methyl-1H-pyrazole-4-carboxamides useful as cancer chemotherapeutic agents | |
| CN114258391A (en) | Pyrrole compounds | |
| WO2013036994A1 (en) | Compounds for the treatment of hcv | |
| JP5826856B2 (en) | Viral polymerase inhibitor | |
| JPWO2004101514A1 (en) | Cyanofluoropyrrolidine derivatives | |
| JP2022529466A (en) | Bicyclic and tricyclic compounds | |
| US9447080B2 (en) | Viral polymerase inhibitors | |
| JP2005530802A (en) | Acyl bicyclic derivatives of pyrrole | |
| AU2021290652A1 (en) | Amido compounds | |
| EP4282862A1 (en) | Flavivirus inhibitors | |
| WO2020219808A1 (en) | Dicarbamate inhibitors of ns5a for treatment of hepatitis c virus infections and related diseases |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11791751 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11791751 Country of ref document: EP Kind code of ref document: A1 |