WO2011094580A2 - Chelated copper for use in the preparation of conjugated oligonucleotides - Google Patents
Chelated copper for use in the preparation of conjugated oligonucleotides Download PDFInfo
- Publication number
- WO2011094580A2 WO2011094580A2 PCT/US2011/022977 US2011022977W WO2011094580A2 WO 2011094580 A2 WO2011094580 A2 WO 2011094580A2 US 2011022977 W US2011022977 W US 2011022977W WO 2011094580 A2 WO2011094580 A2 WO 2011094580A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- linker
- independently
- occurrence
- solid support
- Prior art date
Links
- 0 O=C(CCCCCOc1cc(-c2ccccn2)nc(-c2ccccn2)c1)NC*1(C2)C=C2CC1 Chemical compound O=C(CCCCCOc1cc(-c2ccccn2)nc(-c2ccccn2)c1)NC*1(C2)C=C2CC1 0.000 description 4
- HVRBCXZGTURXBT-UHFFFAOYSA-N Cc1n[n](C)nn1 Chemical compound Cc1n[n](C)nn1 HVRBCXZGTURXBT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/165—Polymer immobilised coordination complexes, e.g. organometallic complexes
- B01J31/1658—Polymer immobilised coordination complexes, e.g. organometallic complexes immobilised by covalent linkages, i.e. pendant complexes with optional linking groups, e.g. on Wang or Merrifield resins
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/165—Polymer immobilised coordination complexes, e.g. organometallic complexes
- B01J31/1658—Polymer immobilised coordination complexes, e.g. organometallic complexes immobilised by covalent linkages, i.e. pendant complexes with optional linking groups, e.g. on Wang or Merrifield resins
- B01J31/1683—Polymer immobilised coordination complexes, e.g. organometallic complexes immobilised by covalent linkages, i.e. pendant complexes with optional linking groups, e.g. on Wang or Merrifield resins the linkage being to a soluble polymer, e.g. PEG or dendrimer, i.e. molecular weight enlarged complexes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/18—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms
- B01J31/1805—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms the ligands containing nitrogen
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/18—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms
- B01J31/1805—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms the ligands containing nitrogen
- B01J31/181—Cyclic ligands, including e.g. non-condensed polycyclic ligands, comprising at least one complexing nitrogen atom as ring member, e.g. pyridine
- B01J31/1815—Cyclic ligands, including e.g. non-condensed polycyclic ligands, comprising at least one complexing nitrogen atom as ring member, e.g. pyridine with more than one complexing nitrogen atom, e.g. bipyridyl, 2-aminopyridine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/04—1,2,3-Triazoles; Hydrogenated 1,2,3-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2231/00—Catalytic reactions performed with catalysts classified in B01J31/00
- B01J2231/30—Addition reactions at carbon centres, i.e. to either C-C or C-X multiple bonds
- B01J2231/32—Addition reactions to C=C or C-C triple bonds
- B01J2231/324—Cyclisations via conversion of C-C multiple to single or less multiple bonds, e.g. cycloadditions
- B01J2231/327—Dipolar cycloadditions
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/02—Compositional aspects of complexes used, e.g. polynuclearity
- B01J2531/0238—Complexes comprising multidentate ligands, i.e. more than 2 ionic or coordinative bonds from the central metal to the ligand, the latter having at least two donor atoms, e.g. N, O, S, P
- B01J2531/0241—Rigid ligands, e.g. extended sp2-carbon frameworks or geminal di- or trisubstitution
- B01J2531/0244—Pincer-type complexes, i.e. consisting of a tridentate skeleton bound to a metal, e.g. by one to three metal-carbon sigma-bonds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/02—Compositional aspects of complexes used, e.g. polynuclearity
- B01J2531/0238—Complexes comprising multidentate ligands, i.e. more than 2 ionic or coordinative bonds from the central metal to the ligand, the latter having at least two donor atoms, e.g. N, O, S, P
- B01J2531/0258—Flexible ligands, e.g. mainly sp3-carbon framework as exemplified by the "tedicyp" ligand, i.e. cis-cis-cis-1,2,3,4-tetrakis(diphenylphosphinomethyl)cyclopentane
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/10—Complexes comprising metals of Group I (IA or IB) as the central metal
- B01J2531/16—Copper
Definitions
- Oligonucleotide compounds have important therapeutic applications in medicine. Oligonucleotides can be used to silence genes that are responsible for a particular disease. Gene- silencing prevents formation of a protein by inhibiting translation. Importantly, gene-silencing agents are a promising alternative to traditional small, organic compounds that inhibit the function of the protein linked to the disease. siR A, antisense RNA, and micro-RNA are oligonucleotides that prevent the formation of proteins by gene-silencing.
- nucleic acid based therapies holds significant promise, providing solutions to medical problems that could not be addressed with current, traditional medicines.
- the location and sequences of an increasing number of disease-related genes are being identified, and clinical testing of nucleic acid-based therapeutics for a variety of diseases is now underway.
- the invention relates to chelated copper on solid support enables copper free click conjugation of ligands and drug carriers (nano particles, polymers and other drug carrier system) to nucleic acids (siR A, ssRNA, microRNAs, antisense, aptamer, decoy etc.).
- the chelated immobilized copper on solid support catalyzes click reaction of completely deprotected oligonucleotides with ligands of choice; and it makes the medium free of floating copper.
- oligonucleotides with functionalized ligands are challenging due to free floating Cu ion which ion-pair/coordinate with the oligonucleotide.
- the separation of copper completely from the product during purification is extremely difficult after conjugation by using conventional click reagent due to chelation or ion-pairing of free copper with the nucleic acid.
- the immobilized copper catalysts described herein are very efficient and recyclable.
- the chelated copper on solid support of the present invention features a compound of formula A:
- Y1-Y4 are each independently CR , CR 2, N, NR , O, and S; r, s and t are each indenpendently 1-6; Rioo is independently for each occurrence OH, OR P , or solid support; where R P is independently for each occurrence occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl or optionally substituted heteroaryl; R N is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group; each linker can be the same or different; and provided that at least one Rioo is bound to a solid support.
- Representative chelated copper of formula (A) is selected from the group consisting of:
- the chelated copper on solid support of the present invention features a compound of formula B:
- Xi-X 6 are each independently CR P , CR P 2 , N, NR N , O, and S; r, s and t are each indenpendently 1-6; Rioo is independently for each occurrence OH, OR P , or solid support; where R P is independently for each occurrence occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl or optionally substituted heteroaryl; R N is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group; each linker can be the same or different; and provided that at least one Rioo is bound to a solid support.
- the chelated copper on solid support of the present invention features a compound of formula C:
- X 1 -X4 are each independently CR P , CR P 2 , N, NR N , O, and S; where R P is independently for each occurrence occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl or optionally substituted heteroaryl; R N is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group; and ⁇ is a solid support.
- a representative chelated copper of formula (C) is in one embodiment, the chelated copper on solid support of the present invention features a compound of formula D:
- Y 1 -Y4 are each independently N, NR N , O, and S; where R 1 00 is independently for each occurrence OH, OR P , or solid support; R N is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group; each linker can be the same or different; each linker can be the same or different; and provided that at least one R 1 00 is bound to a solid support.
- solid support refers to a flexible or non-flexible support that is suitable for carrying said immobilized chelated copper.
- Said solid support may be homogenous or inhomogeneous.
- said solid support may consist of different materials having the same or different properties with respect to flexibility and immobilization, for instance, or said solid support may consist of one material exhibiting a plurality of properties also comprising flexibility and immobilization properties.
- Such supports are well known in the art and comprise, inter alia, commercially available column materials, polystyrene beads, latex beads, magnetic beads, colloid metal particles, glass and/or silicon chips and surfaces, nitrocellulose strips, membranes, sheets, duracytes, wells and walls of reaction trays, plastic tubes etc.
- Examples of well-known carriers include glass, polystyrene, polyvinyl chloride, polypropylene, polyethylene, polycarbonate, dextran, nylon, amyloses, natural and modified celluloses,
- Said solid support may comprise glass-, polypropylene- or silicon-chips, membranes oligonucleotide-conjugated beads or bead arrays.
- Supports used in solid phase synthesis are typically substantially inert and nonreactive with the solid phase synthesis reagents. Methods of using solid supports in solid phase synthesis are well known in the art and may include, but are not limited to, those described in U.S. Pat. Nos. 4,415,732, 4,458,066; 4,500,707, 4,668,777; 4,973,679, and 5,132,418 issued to Caruthers, and U.S. Pat. No. 4,725,677 and Re. 34,069 issued to Koster, each of which are herein incorporated by reference in their entirety for all purposes.
- the solid support is selected from polystyrenes with different degree of cross linking, glass support (controlled pore glass) or silica support, PEGylated polystyrene, other swellable or non-swellable polymer supports for e.g, NittoPhase from NittoDenko and the like, including liquid polymers such as PEGs and Ionic liquids.
- the chelated copper catalyst is immobilized on soluble or insoluble solid support so that the product is free of any copper catalyst.
- the invention features a process for preparing a ligand conjugated oligonucleotide comprising a step of contacting a nucleoside or
- the invention features a process for preparing a ligand conjugated oligonucleotide comprising a step of contacting a nucleoside or
- oligonucleotide containing an alkyne moiety with ligand containing an azide moiety in the presence of a solid supported copper catalyst of formula (A), (B), (C), or (D).
- the invention features a process for preparing a ligand conjugated oligonucleotide comprising a step of contacting a nucleoside or oligonucleotide containing an azide moiety with ligand containing an alkyne moiety in the presence of a solid supported copper catalyst of formula (A), (B), (C), or (D).
- the invention features a process for preparing a ligand conjugated oligonucleotide comprising a step of contacting a nucleoside or
- oligonucleotide containing an azide moiety with ligand containing an alkyne moiety in the presence of a solid supported copper catalyst is oligonucleotide containing an azide moiety with ligand containing an alkyne moiety in the presence of a solid supported copper catalyst.
- the invention features a method for preparing compound having the structure shown in formula (I) by using a chelated copper catalyst of formula (A), (B), (C), or (D);
- X is O, S, NR N or CR P 2 ;
- B is independently for each occurrence hydrogen, optionally substituted natural or non-natural nucleobase, optionally substituted triazole or optionally substituted tetrazole; NH-C(0)-0-C(CH 2 Bi) 3 , NH-C(0)-NH-C(CH 2 Bi) 3 ; where Bi is halogen, mesylate, N 3 , CN, optionally substituted triazole or optionally substituted tetrazole;
- R 1 , R 2 , R 3 , R 4 and R 5 are each independently for each occurrence H, OR 6 , F, N(R N ) 2 , N 3 , CN, -J-linker-N 3 , -J-linker-CN, -J-linker-C ⁇ R 8 , -J-linker-cycloalkyne, -J- linker-RL, or -J-linker-Q-linker-R L ;
- J is absent, O, S, NR N , OC(0)NH, NHC(0)0, C(0)NH, NHC(O), NHSO, NHS0 2 , NHS0 2 NH, OC(O), C(0)0, OC(0)0, NHC(0)NH, NHC(S)NH, OC(S)NH, OP(N(R P ) 2 )0, or OP(N(R P ) 2 );
- R 6 is independently for each occurrence hydrogen, hydroxyl protecting group, optionally substituted alkyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted alkenyl, optionally substituted heteroaryl, polyethyleneglycol (PEG), a phosphate, a diphosphate, a triphosphate, a phosphonate, a phosphonothioate, a phosphonodithioate, a phosphorothioate, a phosphorothiolate, a phosphorodithioate, a phosphorothiolothionate, a phosphodiester, a phosphotriester, an activated phosphate group, an activated phosphite group, a
- phosphoramidite a solid support, -P(Z 1 )(Z 2 )-0-nucleoside, -P(Z 1 )(Z 2 )-0-oligonucleotide, -P ⁇ XZ ⁇ -formula (I), -PCZ ⁇ CO-linker-Q-linker-R ⁇ -O-nucleoside, P ⁇ XO- linker-R L )- O-nucleoside, -P(Z 1 )(0-linker-N 3 )-0-nucleoside, P ⁇ XO-linker-CNXO-nucleoside, P(Z 1 )(0-linker-C ⁇ R 8 )-0-nucleoside, P(Z 1 )(0-linker-cycloalkyne)-0-nucleoside, - PiZ ⁇ CO-linker-Q-linker-R ⁇ -O-oligonucleotide, P ⁇ XO-linker-R ⁇ -O-oligonucle
- R N is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group;
- R P is independently for each occurrence occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl or optionally substituted heteroaryl; absent or independently for each occurrence
- R L is hydrogen or a ligand
- R 8 is N or CR 9 ;
- R 9 is H, optionally substituted alkyl or silyl
- Z 1 and Z 2 are each independently for each occurrence O, S or optionally substituted alkyl;
- R 1 , R 2 , R 3 , R 4 and R 5 is -J-Linker-Q-Linker-R L or - Linker-Q-R L when B is an unsubstitued natural base.
- the invention features a method for preparing a compound having the structure shown in formula (II) by using a chelated copper catalyst of formula
- a and B are independently for each occurrence hydrogen, protecting group, optionally substituted aliphatic, optionally substituted aryl, optionally substituted heteroaryl, polyethyleneglycol (PEG), a phosphate, a diphosphate, a triphosphate, a phosphonate, a phosphonothioate, a phosphonodithioate, a phosphorothioate, a phosphorothiolate, a phosphorodithioate, a phosphorothiolothionate, a phosphodiester, a phosphotriester, an activated phosphate group, an activated phosphite group, a phosphoramidite, a solid support, -P(Z 1 )(Z 2 )-0-nucleoside, or -P(Z 1 )(Z 2 )-0- oligonucleotide; wherein Z 1 and Z 2 are each independently for each occurrence O, S or optionally substituted alkyl;
- Ji and J 2 are independently O, S, NR N , optionally substituted alkyl, OC(0)NH, NHC(0)0, C(0)NH, NHC(O), OC(O), C(0)0, OC(0)0, NHC(0)NH, NHC(S)NH, OC(S)NH, OP(N(R P ) 2 )0, or OP(N(R P ) 2 );
- the cyclic group is cyclic group or acyclic group; preferably, the cyclic group is selected from pyrrolidinyl, pyrazolinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, piperidinyl, piperazinyl, [l,3]dioxolane, oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl, isothiazolidinyl, quinoxalinyl, pyridazinonyl, tetrahydrofuryl and and decalin; preferably, the acyclic group is selected from serinol backbone or diethanolamine backbone;
- Lio and Ln are independently absent or a linker.
- the invention features a method for preparing a compound having the structure shown in formula (III) by using a chelated copper catalyst of formula (A), (B), (C), or (D);
- R and R of the same compound are connected together a "locked" compound similar to a locked nucleic acid (LNA).
- LNA locked nucleic acid
- R 1 and R 4 are H.
- R 1 is -0-Linker-Q-Linker-R L , -OC(0)N(R N )-Linker-Q- Linker-R L or -Linker-Q-Linker-R L , B is H.
- the R 2 and R 4 of the same compound are connected together to form a "locked” compound similar to a locked nucleic acid (LNA).
- LNA locked nucleic acid
- R 1 when B is hydrogen, R 1 is -0-Linker-Q-Linker-R L , - OC(0)N(R 7 )-Linker-Q-Linker-R L or -Linker-Q-Linker-R L .
- B is H.
- B is pyrimidine substituted at C5 position.
- R 2 is OR 6 and R 3 is -0-Linker-Q-Linker-R L , -OC(0)N(R N )- Linker-Q-Linker-R L or -Linker-Q-Linker-R L and R L is present.
- R 3 is OR 6 and R 2 is -0-Linker-Q-Linker-R L , -OC(0)N(R N )- Linker-Q-Linker-R L or -Linker-Q-Linker-R L and R L is present.
- R 2 is OH
- R 9 is H.
- R 5 is -0-Linker-Q-Linker-R L , -OC(0)N(R N )-Linker-Q- Linker-R L or -Linker-Q-Linker-R L and R L is present.
- R 5 is -OC(0)NH(CH 2 )fC ⁇ CR 9 , and f is 1-20.
- R 4 is -OC(0)NH(CH 2 )fC ⁇ CR 9 , and f is 1-20.
- R 3 is -OC(0)NH(CH 2 )iC ⁇ CR 9 , and f is 1-20.
- R 2 is -OC(0)NH(CH 2 )iC ⁇ CR 9 , and f is 1-20.
- R 1 is -OC(0)NH(CH 2 )iC ⁇ CR 9 , and f is 1-20.
- B is a nucleobase substituted with -OC(0)NH(CH 2 ) f C ⁇ CH, and f is 1-20.
- B is a nucleobase substituted with
- B is a nucleobase substituted with In one embodiment, B is a nucleobase substituted with
- R 5 is -0-(CH 2 ) f C ⁇ CR 9 , and f is 1-20.
- R 4 is -0-(CH 2 ) t C ⁇ CR 9 , and f is 1-20.
- R 3 is -0-(CH 2 ) f C ⁇ CR 9 , and f is 1-20.
- R 2 is -0-(CH 2 ) t C ⁇ CR 9 , and f is 1-20.
- R 1 is -0-(CH 2 )iC ⁇ CR 9 , and f is 1-20.
- B is a nucleobase substituted with -0-(CH 2 ) t C ⁇ CH, and f
- B is nucleobase substituted with
- f is 1, 2, 3, 4 or 5. In a preferred embodiment, f is 1.
- Q is
- click-carrier compound of formula (I) when click-carrier compound of formula (I) is at the 5'- terminal end of an oligonucleotide, the oligonucleotides is linked at the R 5 position of the click-carrier compound.
- click-carrier compound of formula (I) when click-carrier compound of formula (I) is at the 3'- terminal end of an oligonucleotide, the oligonucleotides is linked at the R 3 or R 2 position of the click-carrier compound.
- the R 5 position of the compound is linked to the 3'- or 2 '-position of an oligonucleotide on one side and the R 2 or R 3 position of the compound is linked to the 5 '-position of an oligonucleotide on the other side.
- the two different click-carrier compounds comprise complementary functional groups and are clicked together to each other.
- complementarty functional groups are at R 5 position of one click-compound and R 2 or R 3 position of the second compound. In one embodiment, the complementarty functional groups are at R 5 position of one click-compound and R 5 position of the second compound. In one embodiment, complementarty functional groups are at R 2 or R 3 position of one click-compound and R 2 or R 3 position of the second compound.
- B can form part of the click-carrier that connects the linker to the carrier.
- the -linker-Q-linker-R L can be present at the C2, C6, C7 or C8 position of a purine nucleobase or at the C2, C5 or C6 position of a pyrimidine nucleobase.
- the linker can be directly attached to the nucleobase or indirectly through one or more intervening groups such as O, N, S, C(O), C(0)0 C(0)NH.
- B in the click-carrier described above, is uracilyl or a universal base, e.g., an aryl moiety, e.g., phenyl, optionally having additional substituents, e.g., one or more fluoro groups.
- the invention features a method for preparing a compound having the structure shown in formula (IV) by using a chelated copper catalyst of formula (A), (B), (C), or (D);
- R 10 and R 20 are independently for each occurrence hydrogen, optionally substituted aliphatic, optionally substituted aryl, or optionally substituted heteroaryl; R 1 , R 2 , R 3 , R 4 , R 5 , and X are as defined in the first embodiment.
- the invention features a method for preparing a compound having the structure shown in formula (V) by using a chelated copper catalyst of formula (A), (B), (C), or (D);
- the invention features a method for preparing a compound having the structure shown in formula (VI) by using a chelated copper catalyst of formula (A), (B), (C), or (D);
- E is absent or C(O), C(0)0, C(0)NH, C(S), C(S)NH, SO, S0 2 , or
- R 11 , R 12 , R 13 , R 14 , R 15 , R 16 , R 17 , and R 18 are each independently for each occurrence H, -CH 2 OR a , or OR R a and R b are each independently for each occurrence hydrogen, hydroxyl protecting group, optionally substituted alkyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted alkenyl, optionally substituted heteroaryl, polyethyleneglycol (PEG), a phosphate, a diphosphate, a triphosphate, a phosphonate, a phosphonothioate, a phosphonodithioate, a
- PEG polyethyleneglycol
- phosphorothioate a phosphorothiolate, a phosphorodithioate, a phosphorothiolothionate, a phosphodiester, a phosphotriester, an activated phosphate group, an activated phosphite group, a phosphoramidite, a solid support, -P(Z 1 )(Z 2 )-0-nucleoside, -P(Z 1 )(Z 2 )-0- oligonucleotide, -P ⁇ XZ ⁇ -formula (I), -P Z ⁇ O-linker-Q-linker-R ⁇ -O-nucleoside, - P(Z 1 )(0-linker-N 3 )-0-nucleoside, P ⁇ XO-linker-CNXO-nucleoside, P ⁇ XO-linker- C ⁇ R 8 )-0-nucleoside, P(Z 1 )(0-linker-cycloalkyne)-0-
- R 30 is independently for each occurrence -linker-Q-linker-R L , -linker-R L or R 31 ;
- R L is hydrogen or a ligand
- R 5 is N or CR 9 ;
- R 9 is H, optionally substituted alkyl or silyl;
- R 31 is -C(0)CH(N(R 32 ) 2 )(CH 2 ) h N(R 32 ) 2 ;
- R 32 is independently for each occurrence H, -linker-Q-linker-R L , -linker- R L or f and h are independently for each occurrence 1 -20;
- Z 1 and Z 2 are each independently for each occurrence O, S or optionally substituted alkyl.
- R 11 is -CH 2 OR a and R 3 is OR b ; or R 11 is - CH 2 OR a and R 9 is OR b ; or R 11 is -CH 2 OR a and R 17 is OR b ; or R 13 is -CH 2 OR a and R 11 is OR b ; or R 13 is -CH 2 OR a and R 15 is OR b ; or R 13 is -CH 2 OR a and R 17 is OR b .
- CH 2 OR a and OR b may be geminally substituted.
- R 11 is -CH 2 OR a and R 17 is OR b .
- the pyrroline- and 4- hydroxyproline-based compounds may therefore contain linkages (e.g., carbon-carbon bonds) wherein bond rotation is restricted about that particular linkage, e.g. restriction resulting from the presence of a ring.
- linkages e.g., carbon-carbon bonds
- CH 2 OR a and OR b may be cis or trans with respect to one another in any of the pairings delineated above Accordingly, all cis/trans isomers are expressly included.
- the compounds may also contain one or more asymmetric centers and thus occur as racemates and racemic mixtures, single
- R 11 is CH 2 OR a and R 9 is OR b .
- R b is a solid support.
- R 30 is -C(0)(CH 2 ) f NHC(0)(CH 2 ) g C ⁇ CR 9 ; wherein f and g are independently 1 - 20. wherein f is 1-20. L; wherein f is 1-20.
- erein f is 1-20.
- R is
- R 30 is R 1 .
- R 31 is -C(0)CH(N(R 32 )2)(CH 2 ) 4 N(R 32 ) 2 and at least one R 32 is -C(0)(CH 2 )fC ⁇ R 8 or -linker-Q-linker-R L and R L is present.
- R 1 is -C(0)CH(N(R 2 ) 2 )(CH 2 ) 4 NH 2 and at least one R 32 is -C(0)(CH 2 )fC ⁇ R 8 or -linker-Q-linker-R L and R L L i . ⁇ s . present.
- R 32 is -C(0)(CH 2 )iC ⁇ R 8 .
- R is -C(0)(CH 2 ) 3 C ⁇ H.
- R 31 is -C(0)CH(NH 2 )(CH 2 ) 4 NH 2 .
- acyclic sugar replacement -based compounds e.g., replacement based click-carrier compounds
- ribose replacement compound subunit (RRMS) compound compounds are also referred to herein as ribose replacement compound subunit (RRMS) compound compounds.
- Preferred acyclic carriers can have the structure shown in formula (III) or formula (IV) below.
- the invention features a method for preparing a compound having the structure shown in formula (VII) by using a chelated copper catalyst of formula (A), (B), (C), or (D);
- W is absent, O, S and N(R N ) , where R N is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group;
- E is absent or C(O), C(0)0, C(0)NH, C(S), C(S)NH, SO, S0 2 , or S0 2 NH;
- R a and R b are each independently for each occurrence hydrogen, hydroxyl protecting group, optionally substituted alkyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted alkenyl, optionally substituted heteroaryl, polyethyleneglycol (PEG), a phosphate, a diphosphate, a triphosphate, a phosphonate, a phosphonothioate, a phosphonodithioate, a
- PEG polyethyleneglycol
- phosphorothioate a phosphorothiolate, a phosphorodithioate, a phosphorothiolothionate, a phosphodiester, a phosphotriester, an activated phosphate group, an activated phosphite group, a phosphoramidite, a solid support, -P(Z 1 )(Z 2 )-0-nucleoside, -P(Z 1 )(Z 2 )-0- oligonucleotide, -P ⁇ XZ ⁇ -formula (I), ⁇ (Z ⁇ -linker-Q-linker-R ⁇ -O-nucleoside, - P(Z 1 )(0-linker-N 3 )-0-nucleoside, P(Z 1 )(0-linker-CN)-0-nucleoside, P(Z 1 )(0-linker- C ⁇ R 8 )-0-nucleoside, P(Z 1 )(0-linker-cycloalky
- R 30 is independently for each occurrence -linker-Q-linker-R L , -linker-R L or R 31 ;
- R L is hydrogen or a ligand
- R 8 is N or CR 9
- R 9 is H, optionally substituted alkyl or silyl
- R 31 is -C(0)CH(N(R 32 ) 2 )(CH 2 ) h N(R 32 ) 2 ;
- R 32 is independently for each occurrence H, -linker-Q-linker-R L or R 31 ;
- f and h are independently for each occurrence 1 -20;
- Z 1 and Z 2 are each independently for each occurrence O, S or optionally substituted alkyl
- r, s and t are each independently for each occurrence 0, 1, 2 or 3.
- the tertiary carbon can be either the R or S configuration.
- x and y are one and z is zero (e.g. carrier is based on serinol).
- the acyclic carriers can optionally be substituted, e.g. with hydroxy, alkoxy, perhaloalky.
- linker means an organic moiety that connects two parts of a compound.
- Linkers typically comprise a direct bond or an atom such as oxygen or sulfur, a unit such as NR 1 , C(O), C(0)NH, SO, S0 2 , S0 2 NH or a chain of atoms, such as substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, arylalkyl, arylalkenyl, arylalkynyl, heteroarylalkyl,
- heteroarylalkenyl heteroarylalkynyl, heterocyclylalkyl, heterocyclylalkenyl,
- heterocyclylalkynyl aryl, heteroaryl, heterocyclyl, cycloalkyl, cycloalkenyl
- alkylarylalkyl alkylarylalkenyl, alkylarylalkynyl, alkenylarylalkyl, alkenylarylalkenyl, alkenylarylalkynyl, alkynylarylalkyl, alkynylarylalkenyl, alkynylarylalkynyl,
- alkylheteroarylalkyl alkylheteroarylalkenyl, alkylheteroarylalkynyl,
- alkenylheteroarylalkyl alkenylheteroarylalkenyl, alkenylheteroarylalkynyl,
- alkynylheteroarylalkyl alkynylheteroarylalkenyl, alkynylheteroarylalkynyl,
- alkylheterocyclylalkyl alkylheterocyclylalkenyl, alkylhererocyclylalkynyl,
- alkenylheterocyclylalkyl alkenylheterocyclylalkenyl, alkenylheterocyclylalkynyl, alkynylheterocyclylalkyl, alkynylheterocyclylalkenyl, alkynylheterocyclylalkynyl, alkylaryl, alkenylaryl, alkynylaryl, alkylheteroaryl, alkenylheteroaryl, alkynylhereroaryl, which one or more methylenes can be interrupted or terminated by O, S, S(O), S0 2 , N(R 1 ) 2 , C(O), substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocyclic; where R 1 is hydrogen, acyl, aliphatic or substituted aliphatic. It is understood that a linker can clea
- P, R and T are each independently for each occurrence absent, CO, NH, O, S, OC(O), NHC(O), CH 2 , CH 2 NH, CH 2 0; NHC a )C(0), -C(0)-CH(R a )-NH-, -C(O)-
- Qi is independently for each occurrence absent, -(CH 2 ) n -, -C(R 100 )(R 200 )(CH 2 ) n -, - (CH 2 ) n C(R 100 )(R 200 )-, -(CH 2 CH 2 0) m CH 2 CH 2 -, or -(CH 2 CH 2 0) m CH 2 CH 2 NH-;
- R a is H or an amino acid side chain
- R 100 and R 200 are each independently for each occurrence H, C3 ⁇ 4, OH, SH or N(R X ) 2 ;
- R x is independently for each occurrence H, methyl, ethyl, propyl, isopropyl, butyl or benzyl;
- q is independently for each occurrence 0-20;
- n is independently for each occurrence 1-20;
- n is independently for each occurrence 0-50.
- the linker has the structure -[(P-Qi-R) q -X-(P'-Qi '-R') q '] q "-T, wherein:
- Qi and Qi ' are each independently for each occurrence absent, -(CH 2 ) n -, - C(R 100 )(R 200 )(CH 2 ) n -, -(CH 2 ) n C(R 100 )(R 200 )-, -(CH 2 CH 2 0) m CH 2 CH 2 -, or -
- X is a cleavable linker
- R a is H or an amino acid side chain
- R 100 and R 200 are each independently for each occurrence H, C3 ⁇ 4, OH, SH or
- R is independently for each occurrence H, methyl, ethyl, propyl, isopropyl, butyl or benzyl;
- q, q' and q' are each independently for each occurrence 0-20;
- n is independently for each occurrence 1-20;
- n is independently for each occurrence 0-50.
- the linker comprises at least one cleavable linker.
- a cleavable linker is one which is sufficiently stable outside the cell, but which upon entry into a target cell is cleaved to release the two parts the linker is holding together.
- the cleavable linker is cleaved at least 10 times or more, preferably at least 100 times faster in the target cell or under a first reference condition (which can, e.g., be selected to mimic or represent intracellular conditions) than in the blood of a subject, or under a second reference condition (which can, e.g., be selected to mimic or represent conditions found in the blood or serum).
- Cleavable linkers are susceptible to cleavage agents, e.g., pH, redox potential or the presence of degradative molecules. Generally, cleavage agents are more prevalent or found at higher levels or activities inside cells than in serum or blood. Examples of such degradative agents include: redox agents which are selected for particular substrates or which have no substrate specificity, including, e.g., oxidative or reductive enzymes or reductive agents such as mercaptans, present in cells, that can degrade a redox cleavable linker by reduction; esterases; endosomes or agents that can create an acidic environment, e.g., those that result in a pH of five or lower; enzymes that can hydrolyze or degrade an acid cleavable linker by acting as a general acid, peptidases (which can be substrate specific), and phosphatases.
- redox agents which are selected for particular substrates or which have no substrate specificity, including, e.g.,
- a cleavable linker such as a disulfide bond can be susceptible to pH.
- the pH of human serum is 7.4, while the average intracellular pH is slightly lower, ranging from about 7.1-7.3.
- Endosomes have a more acidic pH, in the range of 5.5-6.0, and lysosomes have an even more acidic pH at around 5.0.
- Some sapcers will have a linker that is cleaved at a preferred pH, thereby releasing the iRNA agent from the carrier oligomer inside the cell, or into the desired compartment of the cell.
- a spacer can include a linker that is cleavable by a particular enzyme.
- the type of linker incorporated into a spacer can depend on the cell to be targeted by the iRNA agent.
- an iRNA agent that targets an mRNA in liver cells can be linked to the carrier oligomer through a spacer that includes an ester group.
- Liver cells are rich in esterases, and therefore the tether will be cleaved more efficiently in liver cells than in cell types that are not esterase-rich. Cleavage of the sapcer releases the iRNA agent from the carrier oligomer, thereby potentially enhancing silencing activity of the iRNA agent.
- Other cell-types rich in esterases include cells of the lung, renal cortex, and testis.
- Spacers that contain peptide bonds can be used when the iRNA agents are targeting cell types rich in peptidases, such as liver cells and synoviocytes.
- an iRNA agent targeted to synoviocytes such as for the treatment of an inflammatory disease (e.g., rheumatoid arthritis) can be linked to a carrier oligomer through spacer that comprises a peptide bond.
- the suitability of a candidate cleavable linker can be evaluated by testing the ability of a degradative agent (or condition) to cleave the candidate linker. It will also be desirable to also test the candidate cleavable linker for the ability to resist cleavage in the blood or when in contact with other non-target tissue, e.g., tissue the iR A agent would be exposed to when administered to a subject.
- tissue e.g., tissue the iR A agent would be exposed to when administered to a subject.
- the evaluations can be carried out in cell free systems, in cells, in cell culture, in organ or tissue culture, or in whole animals. It may be useful to make initial evaluations in cell- free or culture conditions and to confirm by further evaluations in whole animals.
- useful candidate compounds are cleaved at least 2, 4, 10 or 100 times faster in the cell (or under in vitro conditions selected to mimic intracellular conditions) as compared to blood or serum (or under in vitro conditions selected to mimic extracellular conditions).
- cleavable linkers are redox cleavable linkers that are cleaved upon reduction or oxidation.
- An example of reductively cleavable linker is a disulphide linker (-S-S-).
- a candidate cleavable linker is a suitable "reductively cleavable linker," or for example is suitable for use with a particular iRNA moiety and particular targeting agent one can look to methods described herein.
- a candidate can be evaluated by incubation with dithiothreitol (DTT), or other reducing agent using reagents know in the art, which mimic the rate of cleavage which would be observed in a cell, e.g., a target cell.
- DTT dithiothreitol
- the candidates can also be evaluated under conditions which are selected to mimic
- candidate compounds are cleaved by at most 10% in the blood.
- useful candidate compounds are degraded at least 2, 4, 10 or 100 times faster in the cell (or under in vitro conditions selected to mimic intracellular conditions) as compared to blood (or under in vitro conditions selected to mimic extracellular conditions).
- the rate of cleavage of candidate compounds can be determined using standard enzyme kinetics assays under conditions chosen to mimic intracellular media and compared to conditions chosen to mimic extracellular media. Phosphate-based cleavable linkers
- Phosphate-based linkers are cleaved by agents that degrade or hydro lyze the phosphate group.
- An example of an agent that cleaves phosphate groups in cells are enzymes such as phosphatases in cells.
- Examples of phosphate-based linkers are -O- P(0)(ORk)-0-, -0-P(S)(ORk)-0-, -0-P(S)(SRk)-0-, -S-P(0)(ORk)-0-, -0-P(0)(ORk)- S-, -S-P(0)(ORk)-S-, -0-P(S)(ORk)-S-, -S-P(S)(ORk)-0-, -0-P(0)(Rk)-0-, -O- P(S)(Rk)-0-, -S-P(0)(Rk)-0-, -S-P(0)(Rk)-0-, -S-P(S)(Rk)-0-, -S
- Preferred embodiments are -0-P(0)(OH)-0-, -0-P(S)(OH)-0-, -0-P(S)(SH)-0-, -S- P(0)(OH)-0-, -0-P(0)(OH)-S-, -S-P(0)(OH)-S-, -0-P(S)(OH)-S-, -S-P(S)(OH)-0-, -O- ⁇ (0)( ⁇ )-0-, -0-P(S)(H)-0-, -S-P(0)(H)-0-, -S-P(S)(H)-0-, -S-P(0)(H)-S-, -0-P(S)(H)- S-.
- a preferred embodiment is -0-P(0)(OH)-0-.
- Acid cleavable linkers are linkers that are cleaved under acidic conditions.
- acid cleavable linkers are cleaved in an acidic environment with a pH of about 6.5 or lower (e.g., about 6.0, 5.5, 5.0, or lower), or by agents such as enzymes that can act as a general acid.
- specific low pH organelles such as endosomes and lysosomes can provide a cleaving environment for acid cleavable linkers.
- acid cleavable linkers include but are not limited to hydrazones, esters, and esters of amino acids.
- a preferred embodiment is when the carbon attached to the oxygen of the ester (the alkoxy group) is an aryl group, substituted alkyl group, or tertiary alkyl group such as dimethyl pentyl or t-butyl.
- Ester-based linkers are cleaved by enzymes such as esterases and amidases in cells.
- ester-based cleavable linkers include but are not limited to esters of alkylene, alkenylene and alkynylene groups.
- Ester cleavable linkers have the general formula -C(0)0-, or -OC(O)-. These candidates can be evaluated using methods analogous to those described above.
- Peptide-based linkers are cleaved by enzymes such as peptidases and proteases in cells.
- Peptide-based cleavable linkers are peptide bonds formed between amino acids to yield oligopeptides (e.g., dipeptides, tripeptides etc.) and polypeptides.
- Peptide-based cleavable groups do not include the amide group (-C(O)NH-).
- the amide group can be formed between any alkylene, alkenylene or alkynelene.
- a peptide bond is a special type of amide bond formed between amino acids to yield peptides and proteins.
- the peptide based cleavage group is generally limited to the peptide bond (i.e., the amide bond) formed between amino acids yielding peptides and proteins and does not include the entire amide functional group.
- Peptide cleavable linkers have the general formula - NHCHR 1 C(0)NHCHR 2 C(0)-, where R 1 and R 2 are the R groups of the two adjacent amino acids. These candidates can be evaluated using methods analogous to those described above.
- the synthesis methods of the present invention utilize click chemistry to conjugate the ligand to the click-carrier compound.
- Click chemistry techniques are described, for example, in the following references, which are incorporated herein by reference in their entirety:
- a 1,5- disubstituted 1,2,3-triazole can be formed using azide and alkynyl reagents (Kraniski, A.; Fokin, V.V. and Sharpless, K.B. Org. Lett. (2004) 6: 1237-1240. Hetero-Diels-Alder reactions or 1,3-dipolar cycloaddition reaction could also be used (see for example Padwa, A. ⁇ ,3-Dipolar Cycloaddition Chemistry: Volume I, John Wiley, New York, (1984) 1-176; Jorgensen, K. A. Angew. Chem., Int. Ed. (2000) 39: 3558-3588 and Tietze, L. F. and Kettschau, G. Top. Curr. Chem. (1997) 189: 1-120)
- the required copper(I) species are added directly as cuprous salts, for example Cul, CuOTf.C 6 H 6 or [Cu(CH 3 CN)4][PF 6 ], usually with stabilizing ligands (see for example Tornoe, C. W.; Christensen, C. and Meldal, M. J. Org. Chem. (2002) 67: 3057- 3064; Chan, T. R. et al, Org. Lett. (2004) 6: 2853-2855; Lewis, W.G. et al, J. Am.
- the reaction is extremely straightforward.
- the azide and alkyne are usually mixed together in water and a co-solvent such as tert-butanol, THF, DMSO, toluene or DMF.
- the water/co-solvent are usually in a 1 : 1 to 1 :9 ratio.
- the reactions are usually run overnight although mild heating shortens reaction times (Sharpless, W. D.; Wu, P.; Hansen, T. V.; and Li, J.G. J. Chem. Ed. (2005) 82: 1833).
- Aqueous systems can also use copper(I) species directly such that a reducing agent is not needed.
- the reactions conditions then usually require acetonitrile as a co-solvent (although not essential (Chan, T.
- the click reaction may be performed thermally. In one aspect, the click reaction is performed at slightly elevated temperatures between 25°C and 100°C. In one aspect, the reaction may be performed between 25°C and 75°C, or between 25°C and 65°C, or between 25°C and 50°C. In one embodiment, the reaction is performed at room temperature. In another aspect, the click reaction may also be performed using a microwave oven. The microwave assisted click reaction may be carried out in the presence or absence of copper.
- the invention provides a method for coupling a click-carrier compound to a ligand through a click reaction.
- the click reaction is a cycloaddition reaction of azide with alkynyl group and catalyzed by copper.
- the equal molar amount of alkyne and azide are mixed together in DCM/MeOH (10: 1 to 1 : 1 ratio v/v) and 0.05-0.5 mol% each of [Cu(CH 3 CN) 4 ][PF 6 ] and copper are added the reaction.
- DCM/MeOH ratio is 5 : 1 to 1 : 1.
- DCM/MeOH ratio is 4: 1.
- equal molar amounts of [Cu(CH 3 CN) 4 ][PF 6 ] and copper are added.
- 0.05- 0.25mol% each of [Cu(CH 3 CN) 4 ][PF 6 ] and copper are added to the reaction.
- 0.05 mol%, 0.1 mol%, 0.15 mol%, 0.2 mol% or 0.25 mol% each of [Cu(CH 3 CN) 4 ][PF 6 ] and copper are added to the reaction.
- prodrug indicates a therapeutic agent that is prepared in an inactive form that is converted to an active form (i.e., drug) within the body or cells thereof by the action of endogenous enzymes or other chemicals and/or conditions.
- prodrug versions of the oligonucleotides of the invention are prepared as SATE [(S- acetyl-2-thioethyl)phosphate] derivatives according to the methods disclosed in WO 93/24510 to Gosselin et al, published Dec. 9, 1993 or in WO 94/26764 and U.S. Pat. No. 5,770,713 to Imbach et al.
- pharmaceutically acceptable salts refers to physiologically and pharmaceutically acceptable salts of the oligomeric compounds of the invention: i.e., salts that retain the desired biological activity of the parent compound and do not impart undesired toxico logical effects thereto.
- pharmaceutically acceptable salts for oligonucleotides, preferred examples of pharmaceutically acceptable salts and their uses are further described in U.S. Pat. No. 6,287,860, which is incorporated herein in its entirety.
- a wide variety of entities can be coupled to the oligonucleotide, e.g. the iRNA agent, using the "click" reaction.
- Preferred entities can be coupled to the oligonucleotide at various places, for example, 3 '-end, 5 '-end, and/or at internal positions.
- the ligand is attached to the iRNA agent via an intervening linker.
- the ligand may be present on a compound when said compound is incorporated into the growing strand.
- the ligand may be incorporated via coupling to a "precursor" compound after said "precursor" compound has been incorporated into the growing strand.
- a compound having, e.g., an azide terminated linker (i.e., having no associated ligand), e.g., -linker-N3 may be incorporated into a growing sense or antisense strand.
- a ligand having an alkyne e.g. terminal acetylene, e.g. ligand-C ⁇ CH
- the compound linker comprises an alkyne, e.g. terminal acetylene
- the ligand comprises an azide functionality for the "click" reaction to take place.
- the azide or alkyne functionalities can be incorporated into the ligand by methods known in the art.
- moieties carrying azide or alkyne functionalities can be linked to the ligand or a functional group on the ligand can be transformed into an azide or alkyne.
- the conjugation of the ligand to the precursor compound takes place while the oligonucleotide is still attached to the solid support.
- the precursor carrying oligonucleotide is first deprotected but not purified before the ligand conjugation takes place.
- the precursor compound carrying oligonucleotide is first deprotected and purified before the ligand conjugation takes place.
- the "click" reaction is carried out under microwave.
- a ligand alters the distribution, targeting or lifetime of an iR A agent into which it is incorporated.
- a ligand provides an enhanced affinity for a selected target, e.g., molecule, cell or cell type, compartment, e.g., a cellular or organ compartment, tissue, organ or region of the body, as, e.g., compared to a species absent such a ligand.
- Preferred ligands will not take part in duplex pairing in a duplexed nucleic acid.
- Preferred ligands can have endosomolytic properties.
- the endosomolytic ligands promote the lysis of the endosome and/or transport of the composition of the invention, or its components, from the endosome to the cytoplasm of the cell.
- the endosomolytic ligand may be a polyanionic peptide or peptidomimetic which shows pH-dependent membrane activity and fusogenicity.
- the endosomolytic ligand assumes its active conformation at endosomal pH.
- the "active" conformation is that conformation in which the endosomolytic ligand promotes lysis of the endosome and/or transport of the composition of the invention, or its components, from the endosome to the cytoplasm of the cell.
- Exemplary endosomolytic ligands include the GALA peptide (Subbarao et al, Biochemistry, 1987, 26: 2964-2972), the EALA peptide (Vogel et al, J. Am. Chem. Soc, 1996, 118: 1581-1586), and their derivatives (Turk et al, Biochem. Biophys. Acta, 2002, 1559: 56-68).
- the endosomolytic component may contain a chemical group (e.g., an amino acid) which will undergo a change in charge or protonation in response to a change in pH.
- the endosomolytic component may be linear or branched. Exemplary primary sequences of peptide based endosomo lytic ligands are shown in table 1.
- Table 1 List of peptides with endosomo lytic activity.
- Preferred ligands can improve transport, hybridization, and specificity properties and may also improve nuclease resistance of the resultant natural or modified
- oligoribonucleotide or a polymeric molecule comprising any combination of compounds described herein and/or natural or modified ribonucleotides.
- Ligands in general can include therapeutic modifiers, e.g., for enhancing uptake; diagnostic compounds or reporter groups e.g., for monitoring distribution; cross-linking agents; and nuclease-resistance conferring moieties.
- therapeutic modifiers e.g., for enhancing uptake
- diagnostic compounds or reporter groups e.g., for monitoring distribution
- cross-linking agents e.g., for monitoring distribution
- nuclease-resistance conferring moieties lipids, steroids, vitamins, sugars, proteins, peptides, polyamines, and peptide mimics.
- Ligands can include a naturally occurring substance, such as a protein (e.g., human serum albumin (HSA), low-density lipoprotein (LDL), high-density lipoprotein (HDL), or globulin); an carbohydrate (e.g., a dextran, pullulan, chitin, chitosan, inulin, cyclodextrin or hyaluronic acid); or a lipid.
- the ligand may also be a recombinant or synthetic molecule, such as a synthetic polymer, e.g., a synthetic polyamino acid, an oligonucleotide (e.g. an aptamer).
- polyamino acids examples include polyamino acid is a polylysine (PLL), poly L-aspartic acid, poly L-glutamic acid, styrene-maleic acid anhydride copolymer, poly(L-lactide-co-glycolied) copolymer, divinyl ether-maleic anhydride copolymer, N-(2-hydroxypropyl)methacrylamide copolymer (HMPA), polyethylene glycol (PEG), polyvinyl alcohol (PVA), polyurethane, poly(2-ethylacryllic acid), N-isopropylacrylamide polymers, or polyphosphazine.
- PLL polylysine
- poly L-aspartic acid poly L-glutamic acid
- styrene-maleic acid anhydride copolymer poly(L-lactide-co-glycolied) copolymer
- divinyl ether-maleic anhydride copolymer divinyl ether-
- polyamines include: polyethylenimine, polylysine (PLL), spermine, spermidine, polyamine, pseudopeptide-polyamine, peptidomimetic polyamine, dendrimer polyamine, arginine, amidine, protamine, cationic lipid, cationic porphyrin, quaternary salt of a polyamine, or an alpha helical peptide.
- Ligands can also include targeting groups, e.g., a cell or tissue targeting agent, e.g., a lectin, glycoprotein, lipid or protein, e.g., an antibody, that binds to a specified cell type such as a kidney cell.
- a cell or tissue targeting agent e.g., a lectin, glycoprotein, lipid or protein, e.g., an antibody, that binds to a specified cell type such as a kidney cell.
- a targeting group can be a thyrotropin, melanotropin, lectin, glycoprotein, surfactant protein A, Mucin carbohydrate, multivalent lactose, multivalent galactose, N-acetyl-galactosamine, N-acetyl-gulucosamine multivalent mannose, multivalent fucose, glycosylated polyaminoacids, multivalent galactose, transferrin, bisphosphonate, polyglutamate, polyaspartate, a lipid, cholesterol, a steroid, bile acid, folate, vitamin B12, biotin, an RGD peptide, an RGD peptide mimetic or an aptamer.
- Table 2 shows some examples of targeting ligands and their associated receptors.
- ligands include dyes, intercalating agents (e.g. acridines), cross-linkers (e.g. psoralene, mitomycin C), porphyrins (TPPC4, texaphyrin, Sapphyrin), polycyclic aromatic hydrocarbons (e.g., phenazine, dihydrophenazine), artificial endonucleases (e.g.
- intercalating agents e.g. acridines
- cross-linkers e.g. psoralene, mitomycin C
- porphyrins TPPC4, texaphyrin, Sapphyrin
- polycyclic aromatic hydrocarbons e.g., phenazine, dihydrophenazine
- artificial endonucleases e.g.
- EDTA lipophilic molecules, e.g, cholesterol, cholic acid, adamantane acetic acid, 1-pyrene butyric acid, dihydrotestosterone, 1 ,3-Bis- 0(hexadecyl)glycerol, geranyloxyhexyl group, hexadecylglycerol, borneol, menthol, 1 ,3- propanediol, heptadecyl group, palmitic acid, myristic acid,03-(oleoyl)lithocholic acid, 03-(oleoyl)cholenic acid, dimethoxytrityl, or phenoxazine)and peptide conjugates (e.g., antennapedia peptide, Tat peptide), alkylating agents, phosphate, amino, mercapto, PEG (e.g., PEG-40K), MPEG, [MPEG] 2 , polyamino, alkyl,
- biotin e.g., aspirin, vitamin E, folic acid
- transport/absorption facilitators e.g., aspirin, vitamin E, folic acid
- synthetic ribonucleases e.g., imidazole, bisimidazole, histamine, imidazole clusters, acridine-imidazole conjugates, Eu3+ complexes of
- Mannose-6-phosphate Mannose-6-phosphate receptor
- Ligands can be proteins, e.g., glycoproteins, or peptides, e.g., molecules having a specific affinity for a co-ligand, or antibodies e.g., an antibody, that binds to a specified cell type such as a cancer cell, endothelial cell, or bone cell.
- Ligands may also include hormones and hormone receptors. They can also include non-peptidic species, such as lipids, lectins, carbohydrates, vitamins, cofactors, multivalent lactose, multivalent galactose, N-acetyl-galactosamine, N-acetyl-gulucosamine multivalent mannose, multivalent fucose, or aptamers.
- the ligand can be, for example, a lipopolysaccharide, an activator of p38 MAP kinase, or an activator of NF-KB.
- the ligand can be a substance, e.g, a drug, which can increase the uptake of the iR A agent into the cell, for example, by disrupting the cell's cytoskeleton, e.g., by disrupting the cell's microtubules, microfilaments, and/or intermediate filaments.
- the drug can be, for example, taxon, vincristine, vinblastine, cytochalasin, nocodazole, japlakinolide, latrunculin A, phalloidin, swinholide A, indanocine, or myoservin.
- the ligand can increase the uptake of the iRNA agent into the cell by activating an inflammatory response, for example.
- exemplary ligands that would have such an effect include tumor necrosis factor alpha (TNF alpha), interleukin-1 beta, or gamma interferon.
- the ligand is a lipid or lipid-based molecule.
- a lipid or lipid-based molecule preferably binds a serum protein, e.g., human serum albumin (HSA).
- HSA binding ligand allows for distribution of the conjugate to a target tissue, e.g., a non- kidney target tissue of the body.
- the target tissue can be the liver, including parenchymal cells of the liver.
- Other molecules that can bind HSA can also be used as ligands. For example, neproxin or aspirin can be used.
- a lipid or lipid-based ligand can (a) increase resistance to degradation of the conjugate, (b) increase targeting or transport into a target cell or cell membrane, and/or (c) can be used to adjust binding to a serum protein, e.g., HSA.
- a serum protein e.g., HSA.
- a lipid based ligand can be used to modulate, e.g., control the binding of the conjugate to a target tissue.
- a lipid or lipid-based ligand that binds to HSA more strongly will be less likely to be targeted to the kidney and therefore less likely to be cleared from the body.
- a lipid or lipid-based ligand that binds to HSA less strongly can be used to target the conjugate to the kidney.
- the lipid based ligand binds HSA.
- it binds HSA with a sufficient affinity such that the conjugate will be preferably distributed to a non-kidney tissue.
- the affinity not be so strong that the HSA-ligand binding cannot be reversed.
- the lipid based ligand binds HSA weakly or not at all, such that the conjugate will be preferably distributed to the kidney.
- Other moieties that target to kidney cells can also be used in place of or in addition to the lipid based ligand.
- the ligand is a moiety, e.g., a vitamin, which is taken up by a target cell, e.g., a proliferating cell.
- a target cell e.g., a proliferating cell.
- vitamins include vitamin A, E, and K.
- B vitamin e.g., folic acid, B12, riboflavin, biotin, pyridoxal or other vitamins or nutrients taken up by cancer cells.
- HAS low density lipoprotein
- HDL high-density lipoprotein
- the ligand is a cell-permeation agent, preferably a helical cell- permeation agent.
- the agent is amphipathic.
- An exemplary agent is a peptide such as tat or antennopedia. If the agent is a peptide, it can be modified, including a peptidylmimetic, invertomers, non-peptide or pseudo-peptide linkages, and use of D- amino acids.
- the helical agent is preferably an alpha- helical agent, which preferably has a lipophilic and a lipophobic phase.
- the ligand can be a peptide or peptidomimetic.
- a peptidomimetic also referred to herein as an oligopeptido mimetic is a molecule capable of folding into a defined three-dimensional structure similar to a natural peptide.
- the attachment of peptide and peptidomimetics to iRNA agents can affect pharmacokinetic distribution of the iR A, such as by enhancing cellular recognition and absorption.
- the peptide or peptidomimetic moiety can be about 5-50 amino acids long, e.g., about 5, 10, 15, 20, 25, 30, 35, 40, 45, or 50 amino acids long (see Table 3, for example).
- a peptide or peptidomimetic can be, for example, a cell permeation peptide, cationic peptide, amphipathic peptide, or hydrophobic peptide (e.g., consisting primarily of Tyr, Trp or Phe).
- the peptide moiety can be a dendrimer peptide, constrained peptide or crosslinked peptide.
- the peptide moiety can include a hydrophobic membrane translocation sequence (MTS).
- An exemplary hydrophobic MTS-containing peptide is RFGF having the amino acid sequence
- RFGF analogue e.g., amino acid sequence
- AALLPVLLAAP containing a hydrophobic MTS can also be a targeting moiety.
- the peptide moiety can be a "delivery" peptide, which can carry large polar molecules including peptides, oligonucleotides, and protein across cell membranes.
- Antennapedia protein (RQIKIWFQNRRMKWK ) have been found to be capable of functioning as delivery peptides.
- a peptide or peptidomimetic can be encoded by a random sequence of DNA, such as a peptide identified from a phage-display library, or one-bead-one-compound (OBOC) combinatorial library (Lam et al., Nature, 354:82-84, 1991).
- the peptide or peptidomimetic tethered to an iRNA agent via an incorporated compound unit is a cell targeting peptide such as an arginine-glycine- aspartic acid (RGD)-peptide, or RGD mimic.
- a peptide moiety can range in length from about 5 amino acids to about 40 amino acids.
- the peptide moieties can have a structural modification, such as to increase stability or direct conformational properties. Any of the structural modifications described below can be utilized.
- An RGD peptide moiety can be used to target a tumor cell, such as an endothelial tumor cell or a breast cancer tumor cell (Zitzmann et al., Cancer Res., 62:5139-43, 2002).
- An RGD peptide can facilitate targeting of an iRNA agent to tumors of a variety of other tissues, including the lung, kidney, spleen, or liver (Aoki et al., Cancer Gene Therapy 8:783-787, 2001).
- the RGD peptide will facilitate targeting of an iRNA agent to the kidney.
- the RGD peptide can be linear or cyclic, and can be modified, e.g., glycosylated or methylated to facilitate targeting to specific tissues.
- a glycosylated RGD peptide can deliver an iRNA agent to a tumor cell expressing yB3 (Haubner et al., Jour. Nucl. Med., 42:326-336, 2001).
- RGD containing peptides and peptidomimetics can target cancer cells, in particular cells that exhibit an I v 3 ⁇ 4 integrin.
- RGD one can use other moieties that target the I v -3 ⁇ 4 integrin ligand. Generally, such ligands can be used to control proliferating cells and
- conjugates of this type lignads that targets PECAM-1, VEGF, or other cancer gene, e.g., a cancer gene described herein.
- a "cell permeation peptide” is capable of permeating a cell, e.g., a microbial cell, such as a bacterial or fungal cell, or a mammalian cell, such as a human cell.
- a microbial cell-permeating peptide can be, for example, an a-helical linear peptide (e.g., LL-37 or Ceropin PI), a disulfide bond-containing peptide (e.g., a -defensin, ⁇ -defensin or bactenecin), or a peptide containing only one or two dominating amino acids (e.g., PR-39 or indolicidin).
- a cell permeation peptide can also include a nuclear localization signal (NLS).
- NLS nuclear localization signal
- a cell permeation peptide can be a bipartite amphipathic peptide, such as MPG, which is derived from the fusion peptide domain of HIV- 1 gp41 and the NLS of SV40 large T antigen (Simeoni et al, Nucl. Acids Res. 31 :2717-2724, 2003).
- a targeting peptide tethered to an iRNA agent and/or the carrier oligomer can be an amphipathic a-helical peptide.
- exemplary amphipathic a- helical peptides include, but are not limited to, cecropins, lycotoxins, paradaxins, buforin, CPF, bombinin-like peptide (BLP), cathelicidins, ceratotoxins, S.
- clava peptides hagfish intestinal antimicrobial peptides (HFIAPs), magainines, brevinins-2, dermaseptins, melittins, pleurocidin, H 2 A peptides, Xenopus peptides, esculentinis-1, and caerins.
- HFIAPs hagfish intestinal antimicrobial peptides
- magainines brevinins-2, dermaseptins, melittins, pleurocidin
- H 2 A peptides Xenopus peptides, esculentinis-1, and caerins.
- H 2 A peptides Xenopus peptides
- esculentinis-1 esculentinis-1
- caerins a number of factors will preferably be considered to maintain the integrity of helix stability.
- a maximum number of helix stabilization residues will be utilized (e.g., leu, ala, or lys)
- the capping residue will be considered (for example Gly is an exemplary N-capping residue and/or C-terminal amidation can be used to provide an extra H-bond to stabilize the helix.
- Formation of salt bridges between residues with opposite charges, separated by i ⁇ 3, or i ⁇ 4 positions can provide stability.
- cationic residues such as lysine, arginine, homo-arginine, ornithine or histidine can form salt bridges with the anionic residues glutamate or aspartate.
- Peptide and peptidomimetic ligands include those having naturally occurring or modified peptides, e.g., D or L peptides; ⁇ , ⁇ , or ⁇ peptides; N-methyl peptides;
- azapeptides peptides having one or more amide, i.e., peptide, linkages replaced with one or more urea, thiourea, carbamate, or sulfonyl urea linkages; or cyclic peptides.
- the targeting ligand can be any ligand that is capable of targeting a specific receptor. Examples are: folate, GalNAc, GalNAc 3 , galactose, mannose, mannose-6P, clusters of sugars such as GalNAc cluster, mannose cluster, galactose cluster, or an apatamer. A cluster is a combination of two or more sugar units.
- the targeting ligands also include integrin receptor ligands, Chemokine receptor ligands, transferrin, biotin, serotonin receptor ligands, PSMA, endothelin, GCPII, somatostatin, LDL and HDL ligands.
- the ligands can also be based on nucleic acid, e.g., an aptamer.
- the aptamer can be unmodified or have any combination of modifications disclosed herein.
- Endosomal release agents include imidazoles, poly or oligoimidazoles, PEIs, peptides, fusogenic peptides, polycaboxylates, polyacations, masked oligo or poly cations or anions, acetals, polyacetals, ketals/polyketyals, orthoesters, polymers with masked or unmasked cationic or anionic charges, dendrimers with masked or unmasked cationic or anionic charges.
- PK modulator stands for pharmacokinetic modulator.
- PK modulator include lipophiles, bile acids, steroids, phospholipid analogues, peptides, protein binding agents, PEG, vitamins etc.
- Examplary PK modulator include, but are not limited to, cholesterol, fatty acids, cholic acid, lithocholic acid, dialkylglycerides, diacylglyceride,
- Oligonucleotides that comprise a number of phosphorothioate linkages are also known to bind to serum protein, thus short oligonucleotides, e.g. oligonucleotides of about 5 bases, 10 bases, 15 bases or 20 bases, comprising multiple of phosphorothioate linkages in the backbaone are also amenable to the present invention as ligands (e.g. as PK modulating ligands).
- aptamers that bind serum components are also amenable to the present invention as PK modulating ligands.
- the ligands can all have same properties, all have different properties or some ligands have the same properties while others have different properties.
- a ligand can have targeting properties, have endosomolytic activity or have PK modulating properties.
- all the ligands have different properties.
- the compound comprising the ligand can be present in any position of an oligonucleotide, e.g. an iRNA agent.
- click-carrier compound can be present at the terminus such as a 5' or 3' terminal of the iRNA agent.
- Click-carrier compounds can also present at an internal postion of the iRNA agent.
- click-carrier compounds can be incorporated into one or both strands.
- the sense strand of the double-stranded iRNA agent comprises the click-carrier compound.
- the antisense strand of the double-stranded iRNA agent comprises the click-carrier compound.
- lignads can be conjugated to nucleobases, sugar moieties, or internucleosidic linkages of nucleic acid molecules. Conjugation to purine nucleobases or derivatives thereof can occur at any position including, endocyclic and exocyclic atoms. In some embodiments, the 2-, 6-, 7-, or 8-positions of a purine nucleobase are attached to a conjugate moiety. Conjugation to pyrimidine nucleobases or derivatives thereof can also occur at any position. In some embodiments, the 2-, 5-, and 6-positions of a pyrimidine nucleobase can be substituted with a conjugate moiety.
- Conjugation to sugar moieties of nucleosides can occur at any carbon atom.
- Example carbon atoms of a sugar moiety that can be attached to a conjugate moiety include the 2', 3', and 5' carbon atoms.
- the ⁇ position can also be attached to a conjugate moiety, such as in an abasic residue.
- Internucleosidic linkages can also bear conjugate moieties.
- the conjugate moiety can be attached directly to the phosphorus atom or to an O, N, or S atom bound to the phosphorus atom.
- amine- or amide-containing internucleosidic linkages e.g., PNA
- the conjugate moiety can be attached to the nitrogen atom of the amine or amide or to an adjacent carbon atom.
- an oligomeric compound is attached to a conjugate moiety by contacting a reactive group (e.g., OH, SH, amine, carboxyl, aldehyde, and the like) on the oligomeric compound with a reactive group on the conjugate moiety.
- a reactive group e.g., OH, SH, amine, carboxyl, aldehyde, and the like
- one reactive group is electrophilic and the other is nucleophilic.
- an electrophilic group can be a carbonyl-containing functionality and a nucleophilic group can be an amine or thiol.
- Methods for conjugation of nucleic acids and related oligomeric compounds with and without linkers are well described in the literature such as, for example, in Manoharan in Antisense Research and
- oligonucleotide refers to a polymer or oligomer of nucleotide or nucleoside monomers consisting of naturally occurring bases, sugars and intersugar (backbone) linkages.
- oligonucleotide also includes polymers or oligomers comprising non-naturally occurring monomers, or portions thereof, which function similarly. Such modified or substituted oligonucleotides are often preferred over native forms because of properties such as, for example, enhanced cellular uptake and increased stability in the presence of nucleases.
- the nucleic acids used herein can be single-stranded or double-stranded.
- a single stranded oligonucleotide may have double stranded regions and a double stranded oligonucleotide may have regions of single-stranded regions.
- double- stranded DNA include structural genes, genes including control and termination regions, and self-replicating systems such as viral or plasmid DNA.
- double-stranded RNA include siRNA and other RNA interference reagents.
- Single-stranded nucleic acids include, e.g., antisense oligonucleotides, ribozymes, microRNAs, aptamers, antagomirs, triplex-forming oligonucleotides and single-stranded RNAi agents.
- Oligonucleotides of the present invention may be of various lengths. In particular embodiments, oligonucleotides may range from about 10 to 100 nucleotides in length. In various related embodiments, oligonucleotides, single-stranded, double-stranded, and triple-stranded, may range in length from about 10 to about 50 nucleotides, from about 20 to about 50 nucleotides, from about 15 to about 30 nucleotides, from about 20 to about 30 nucleotides in length.
- the oligonucleotides of the invention may comprise any oligonucleotide modification described herein and below. In certain instances, it may be desirable to modify one or both strands of a dsRNA.
- the two strands will include different modifications. Multiple different modifications can be included on each of the strands.
- the modifications on a given strand may differ from each other, and may also differ from the various modifications on other strands.
- one strand may have a modification, e.g., a modification described herein, and a different strand may have a different modification, e.g., a different modification described herein.
- one strand may have two or more different modifications, and the another strand may include a modification that differs from the at least two modifications on the other strand.
- oligonucleotides of the invention comprises 5 '
- nucleotides 1 and 2 having cationic modifications via C-5 position of pyrimidines, 2-Position of Purines, N2-G, G-clamp, 8- position of purines, 6-position of purines, internal nucleotides having a 2'-F sugar with base modifications (Pseudouridine, G-clamp, phenoxazine, pyridopyrimidines, gem2'- Me-up/2'-F-down), 3 '-end with two purines with novel 2'-substituted MOE analogs, 5'- end nucleotides with novel 2'-substituted MOE analogs, 5 '-end having a 3'-F and a 2'-5'- linkage, 4 '-substituted nucleoside at the nucleotide 1 at 5 '-end and the substituent is cationic, alkyl, alkoxyalkyl,
- the invention provides double-stranded ribonucleic acid (dsRNA) molecules for inhibiting the expression of the target gene (alone or in combination with a second dsRNA for inhibiting the expression of a second target gene) in a cell or mammal, wherein the dsRNA comprises an antisense strand comprising a region of complementarity which is complementary to at least a part of an mR A formed in the expression of the target gene, and wherein the region of complementarity is less than 30 nucleotides in length, generally 19-24 nucleotides in length, and wherein said dsRNA, upon contact with a cell expressing said target gene, inhibits the expression of said target gene.
- dsRNA double-stranded ribonucleic acid
- the dsR A comprises two RNA strands that are sufficiently complementary to hybridize to form a duplex structure.
- the duplex structure is between 15 and 30, more generally between 18 and 25, yet more generally between 19 and 24, and most generally between 19 and 21 base pairs in length.
- longer dsR As of between 25 and 30 base pairs in length are preferred.
- shorter dsRNAs of between 10 and 15 base pairs in length are preferred.
- the dsRNA is at least 21 nucleotides long and includes a sense RNA strand and an antisense RNA strand, wherein the antisense RNA strand is 25 or fewer nucleotides in length, and the duplex region of the dsRNA is 18-25 nucleotides in length, e.g., 19-24 nucleotides in length.
- the region of complementarity to the target sequence is between 15 and 30, more generally between 18 and 25, yet more generally between 19 and 24, and most generally between 19 and 21 nucleotides in length.
- the dsRNA of the invention may further comprise one or more single-stranded nucleotide overhang(s).
- the target gene is a human target gene.
- the target gene is selected from the group consisting of Factor VII, Eg5, PCSK9, TPX2, apoB, SAA, TTR, RSV, PDGF beta gene, Erb-B gene, Src gene, CR gene, GRB2 gene, RAS gene, MEK gene, JNK gene, RAF gene, Erkl/2 gene,
- PCNA(p21) gene MYB gene, JUN gene, FOS gene, BCL-2 gene, Cyclin D gene, VEGF gene, EGFR gene, Cyclin A gene, Cyclin E gene, WNT-1 gene, beta-catenin gene, c- MET gene, PKC gene, NFKB gene, STAT3 gene, survivin gene, Her2/Neu gene, topoisomerase I gene, topoisomerase II alpha gene, p73 gene, p21(WAFl/CIPl) gene, p27(KIPl) gene, PPM ID gene, RAS gene, caveolin I gene, MIB I gene, MTAI gene, M68 gene, mutations in tumor suppressor genes, p53 tumor suppressor gene, and combinations thereof.
- dsRNAs comprising a duplex structure of between 20 and 23, but specifically 21, base pairs have been hailed as particularly effective in inducing RNA interference (Elbashir et al, EMBO 2001, 20:6877-6888). However, others have found that shorter or longer dsRNAs can be effective as well.
- the dsRNAs of the invention can comprise at least one strand of a length of minimally 21 nt. It can be reasonably expected that shorter dsRNAs comprising a known sequence minus only a few nucleotides on one or both ends may be similarly effective as compared to the dsRNAs of the lengths described above.
- dsRNAs comprising a partial sequence of at least 15, 16, 17, 18, 19, 20, or more contiguous nucleotides, and differing in their ability to inhibit the expression of the target gene by not more than 5, 10, 15, 20, 25, or 30 % inhibition from a dsRNA comprising the full sequence, are contemplated by the invention. Further dsRNAs that cleave within the target sequence can readily be made using the target gene sequence and the target sequence provided.
- Double-stranded and single-stranded oligonucleotides that are effective in inducing RNA interference are also referred to as siRNA, RNAi agent and/or iRNA agent. These RNA interference inducing oligonucleotides associate with a cytoplasmic multi-protein complex known as RNAi-induced silencing complex (RISC).
- RISC RNAi-induced silencing complex
- single-stranded and double stranded RNAi agents are sufficiently long that they can be cleaved by an endogenous molecule, e.g. by Dicer, to produce smaller oligonucleotides that can enter the RISC machinery and participate in RISC mediated cleavage of a target sequence, e.g. a target mRNA.
- the present invention further includes RNAi agents that target within the sequence targeted by one of the agents of the present invention.
- a second RNAi agent is said to target within the sequence of a first RNAi agent if the second RNAi agent cleaves the message anywhere within the mRNA that is complementary to the antisense strand of the first RNAi agent.
- Such a second agent will generally consist of at least 15 contiguous nucleotides coupled to additional nucleotide sequences taken from the region contiguous to the selected sequence in the target gene.
- the dsRNA of the invention can contain one or more mismatches to the target sequence. In a preferred embodiment, the dsRNA of the invention contains no more than 3 mismatches. If the antisense strand of the dsRNA contains mismatches to a target sequence, it is preferable that the area of mismatch not be located in the center of the region of complementarity. If the antisense strand of the dsRNA contains mismatches to the target sequence, it is preferable that the mismatch be restricted to 5 nucleotides from either end, for example 5, 4, 3, 2, or 1 nucleotide from either the 5' or 3' end of the region of complementarity.
- the dsRNA generally does not contain any mismatch within the central 13 nucleotides.
- the methods described within the invention can be used to determine whether a dsRNA containing a mismatch to a target sequence is effective in inhibiting the expression of the target gene. Consideration of the efficacy of dsRNAs with mismatches in inhibiting expression of the target gene is important, especially if the particular region of complementarity in the target gene is known to have polymorphic sequence variation within the population.
- the sense-strand comprises a mismatch to the antisense strand.
- the mismatch is at the 5 nucleotides from the 3 '-end, for example 5, 4, 3, 2, or 1 nucleotide from the end of the region of complementarity.
- the mismatch is located in the target cleavage site region.
- the sense strand comprises no more than 1, 2, 3, 4 or 5 mismatches to the antisense strand.
- the sense strand comprises no more than 3 mismatches to the antisense strand.
- the sense strand comprises a nucleobase modification, e.g. an optionally substituted natural or non-natural nucleobase, a universal nucleobase, in the target cleavage site region.
- a nucleobase modification e.g. an optionally substituted natural or non-natural nucleobase, a universal nucleobase, in the target cleavage site region.
- target cleavage site herein means the backbone linkage in the target gene, e.g. target mRNA, or the sense strand that is cleaved by the RISC mechanism by utilizing the iRNA agent.
- target cleavage site region comprises at least one or at least two nucleotides on both side of the cleavage site.
- the target cleavage site is the backbone linkage in the sense strand that would get cleaved if the sense strand itself was the target to be cleaved by the RNAi mechanism.
- the target cleavage site can be determined using methods known in the art, for example the 5'- RACE assay as detailed in Soutschek et ah, Nature (2004) 432, 173-178.
- the cleavage site region for a conical double stranded RNAi agent comprising two 21 -nucleotides long strands (wherin the strands form a double stranded region of 19 consective basepairs having 2-nucleotide single stranded overhangs at the 3'-ends)
- the cleavage site region corresponds to postions 9-12 from the 5'-end of the sense strand.
- At least one end of the dsRNA has a single-stranded nucleotide overhang of 1 to 4, generally 1 or 2 nucleotides.
- the single- stranded overhang has the sequence 5'-GCNN-3', wherein N is independently for each occuurence, A, G, C, U, dT, dU or absent.
- dsR As having at least one nucleotide overhang have unexpectedly superior inhibitory properties than their blunt-ended counterparts. Moreover, the present inventors have discovered that the presence of only one nucleotide overhang strengthens the interference activity of the dsRNA, without affecting its overall stability.
- the dsRNA having only one overhang has proven particularly stable and effective in vivo, as well as in a variety of cells, cell culture mediums, blood, and serum.
- the single- stranded overhang is located at the 3'-terminal end of the antisense strand or, alternatively, at the 3 '-terminal end of the sense strand.
- the dsRNA may also have a blunt end, generally located at the 5 '-end of the antisense strand.
- the antisense strand of the dsRNA has a nucleotide overhang at the 3 '-end, and the 5 '-end is blunt.
- the antisense strand of the dsRNA has 1-10 nucleotides overhangs each at the 3 ' end and the 5 ' end over the sense strand. In one embodiment, the sense strand of the dsRNA has 1-10 nucleotides overhangs each at the 3' end and the 5 ' end over the antisense strand.
- the dsRNAs of the invention may comprise any oligonucleotide modification described herein and below.
- the two strands will include different modifications. Multiple different modifications can be included on each of the strands.
- the modifications on a given strand may differ from each other, and may also differ from the various modifications on other strands.
- one strand may have a modification, e.g., a modification described herein
- a different strand may have a different modification, e.g., a different modification described herein.
- one strand may have two or more different modifications
- the another strand may include a modification that differs from the at least two modifications on the other strand.
- the dsRNA is chemically modified to enhance stability.
- one or more of the nucleotides in the overhang is replaced with a nucleoside thiophosphate.
- the present invention also includes dsRNA compounds which are chimeric compounds.
- "Chimeric" dsRNA compounds or “chimeras,” in the context of this invention are dsRNA compounds, particularly dsR As, which contain two or more chemically distinct regions, each made up of at least one monomer unit, i.e., a nucleotide in the case of an dsRNA compound.
- These dsRNAs typically contain at least one region wherein the dsRNA is modified so as to confer upon the dsRNA increased resistance to nuclease degradation, increased cellular uptake, and/or increased binding affinity for the target nucleic acid.
- An additional region of the dsRNA may serve as a substrate for enzymes capable of cleaving RNA:DNA or RNA:RNA hybrids.
- RNase H is a cellular endonuclease which cleaves the RNA strand of an
- RNA:DNAduplex Activation of RNase H, therefore, results in cleavage of the RNA target, thereby greatly enhancing the efficiency of dsRNA inhibition of gene expression.
- the present invention also includes dsRNAs wherein the two strands are linked together.
- the two strands can be linked together by a polynucleotide linker such as (dT) n ; wherein n is 4-10, and thus forming a hairpin.
- the two strands can also be linked together by a non-nucleosidic linker, e.g. a linker described herein. It will be appreciated by one of skill in the art that any oligonucleotide chemical modifications or variations describe herein can be used in the polynucleotide linker.
- Hairpin RNAi agents will have a duplex region equal to or at least 17, 18, 19, 29, 21, 22, 23, 24, or 25 nucleotide pairs.
- the duplex region will may be equal to or less than 200, 100, or 50, in length. In one embodiment, ranges for the duplex region are 10-35, 12-30, 12-25, 15-30, 17 to 23, 19 to 23, and 19 to 21 nucleotides pairs in length.
- the hairpin may have a single strand overhang or terminal unpaired region, in some embodiments at the 3', and in one embodiment on the antisense side of the hairpin. In one embodiment, the overhangs are 2-3 nucleotides in length.
- the RNAi agents of the invention can target more than one RNA region.
- an RNAi agent can include a first and second sequence that are sufficiently complementary to each other to hybridize.
- the first sequence can be complementary to a first target RNA region and the second sequence can be complementary to a second target RNA region.
- the first and second sequences of the RNAi agent can be on different RNA strands, and the mismatch between the first and second sequences can be less than 50%, 40%, 30%, 20%, 10%, 5%, or 1%.
- the first and second sequences of the RNAi agent can be on the same RNA strand, and in a related embodiment more than 50%, 60%, 70%, 80%, 90%, 95%, or 1% of the RNAi agent can be in bimolecular form.
- the first and second sequences of the RNAi agent can be fully complementary to each other.
- the first target RNA region can be encoded by a first gene and the second target RNA region can encoded by a second gene, or the first and second target RNA regions can be different regions of an RNA from a single gene.
- the first and second sequences can differ by at least 1 nucleotide.
- the first and second target RNA regions can be on transcripts encoded by first and second sequence variants, e.g., first and second alleles, of a gene.
- the sequence variants can be mutations, or polymorphisms, for example.
- the first target RNA region can include a nucleotide substitution, insertion, or deletion relative to the second target RNA region, or the second target RNA region can a mutant or variant of the first target region.
- the first and second target RNA regions can comprise viral or human RNA regions.
- the first and second target RNA regions can also be on variant transcripts of an oncogene or include different mutations of a tumor suppressor gene transcript.
- the first and second target RNA regions can correspond to hot-spots for genetic variation.
- the double stranded oligonucleotides can be optimized for RNA interference by increasing the propensity of the duplex to disassociate or melt (decreasing the free energy of duplex association), in the region of the 5' end of the antisense strand.
- This can be accomplished, e.g., by the inclusion of modifications or modified nucleosides which increase the propensity of the duplex to disassociate or melt in the region of the 5' end of the antisense strand. It can also be accomplished by inclusion of modifications or modified nucleosides or attachment of a ligand that increases the propensity of the duplex to disassociate of melt in the region of the 5 'end of the antisense strand. While not wishing to be bound by theory, the effect may be due to promoting the effect of an enzyme such as helicase, for example, promoting the effect of the enzyme in the proximity of the 5 ' end of the antisense strand.
- Modifications which increase the tendency of the 5' end of the antisense strand in the duplex to dissociate can be used alone or in combination with other modifications described herein, e.g., with modifications which decrease the tendency of the 3' end of the antisense in the duplex to dissociate.
- modifications which decrease the tendency of the 3' end of the antisense in the duplex to dissociate can be used alone or in combination with other modifications described herein, e.g., with modifications which increase the tendency of the 5' end of the antisense in the duplex to dissociate.
- Nucleic acid base pairs can be ranked on the basis of their propensity to promote dissociation or melting (e.g., on the free energy of association or dissociation of a particular pairing, the simplest approach is to examine the pairs on an individual pair basis, though next neighbor or similar analysis can also be used).
- A:U is preferred over G:C
- G:U is preferred over G:C
- mismatches e.g., non-canonical or other than canonical pairings are preferred over canonical (A:T, A:U, G:C) pairings
- pairings which include a universal base are preferred over canonical pairings.
- pairings which decrease the propensity to form a duplex are used at 1 or more of the positions in the duplex at the 5 ' end of the antisense strand.
- the terminal pair (the most 5' pair in terms of the antisense strand), and the subsequent 4 base pairing positions (going in the 3 ' direction in terms of the antisense strand) in the duplex are preferred for placement of modifications to decrease the propensity to form a duplex. More preferred are placements in the terminal most pair and the subsequent 3, 2, or 1 base pairings.
- At least 1, and more preferably 2, 3, 4, or 5 of the base pairs from the 5 '-end of antisense strand in the duplex be chosen independently from the group of: A:U, G:U, I:C, mismatched pairs, e.g., non-canonical or other than canonical pairings or pairings which include a universal base.
- at least one, at least 2, or at least 3 base-pairs include a universal base.
- Modifications or changes which promote dissociation are preferably made in the sense strand, though in some embodiments, such modifications/changes will be made in the antisense strand.
- Nucleic acid base pairs can also be ranked on the basis of their propensity to promote stability and inhibit dissociation or melting (e.g., on the free energy of association or dissociation of a particular pairing, the simplest approach is to examine the pairs on an individual pair basis, though next neighbor or similar analysis can also be used).
- G:C is preferred over A:U
- Watson-Crick matches A:T, A:U, G:C
- A:T, A:U, G:C are preferred over non-canonical or other than canonical pairings
- analogs that increase stability are preferred over Watson-Crick matches (A:T, A:U, G:C), e.g.
- 2-amino-A:U is preferred over A:U
- 2-thio U or 5 Me-thio-U:A are preferred over U:A
- G-clamp an analog of C having 4 hydrogen bonds
- :G is preferred over C:G
- guanadinium-G-clamp:G is preferred over C:G
- psuedo uridine: A is preferred over U:A
- sugar modifications e.g., 2' modifications, e.g., 2'F, ENA, or LNA, which enhance binding are preferred over non-modified moieties and can be present on one or both strands to enhance stability of the duplex.
- pairings which increase the propensity to form a duplex are used at 1 or more of the positions in the duplex at the 3 ' end of the antisense strand.
- the terminal pair (the most 3 ' pair in terms of the antisense strand), and the subsequent 4 base pairing positions (going in the 5 ' direction in terms of the antisense strand) in the duplex are preferred for placement of modifications to decrease the propensity to form a duplex. More preferred are placements in the terminal most pair and the subsequent 3, 2, or 1 base pairings.
- At least 1, and more preferably 2, 3, 4, or 5 of the pairs of the recited regions be chosen independently from the group of: G:C, a pair having an analog that increases stability over Watson-Crick matches (A:T, A:U, G:C), 2-amino- A:U, 2-thio U or 5 Me-thio-U:A, G-clamp (an analog of C having 4 hydrogen bonds):G, guanadinium-G-clamp:G, psuedo uridine:A, a pair in which one or both subunits has a sugar modification, e.g., a 2' modification, e.g., 2'F, ENA, or LNA, which enhance binding.
- at least one, at least, at least 2, or at least 3, of the base pairs promote duplex stability.
- the at least one, at least 2, or at least 3, of the base pairs are a pair in which one or both subunits has a sugar modification, e.g., a 2' modification, e.g., 2'- ⁇ 9-Me (2'- ⁇ 9-methyl), 2'- ⁇ 9-MOE (2'- ⁇ 9-methoxyethyl), 2'-F, 2'-0- [2-(methylamino)-2-oxoethyl] (2'-0-NMA), 2'-S-methyl, 2'-0-CH 2 -(4'-C) (LNA) and 2'-0-CH 2 CH 2 -(4'-C) (ENA), which enhances binding.
- a sugar modification e.g., a 2' modification, e.g., 2'- ⁇ 9-Me (2'- ⁇ 9-methyl), 2'- ⁇ 9-MOE (2'- ⁇ 9-methoxyethyl), 2'-F, 2'-0- [2-(methylamino)
- G-clamps and guanidinium G-clamps are discussed in the following references: Holmes and Gait, "The Synthesis of 2'-0-Methyl G-Clamp Containing Oligonucleotides and Their Inhibition of the HIV-1 Tat-TAR Interaction," Nucleosides, Nucleotides & Nucleic Acids, 22: 1259-1262, 2003; Holmes et al., "Steric inhibition of human immunodeficiency virus type-1 Tat-dependent trans-activation in vitro and in cells by oligonucleotides containing 2'-0-methyl G-clamp ribonucleoside analogues," Nucleic Acids Research, 31 :2759-2768, 2003; Wilds, et al., "Stural basis for recognition of guanosine by a synthetic tricyclic cytosine analogue: Guanidinium G-clamp," Helvetica Chimica Acta, 86:966-978, 2003; Rajeev
- oligonucleotides containing the tricyclic cytosine analogues phenoxazine and 9-(2- aminoethoxy)-phenoxazine ("G-clamp") and origins of their nuclease resistance properties," Biochemistry, 41 :1323-7, 2002; Flanagan, et al., "A cytosine analog that confers enhanced potency to antisense oligonucleotides," Proceedings Of The National Academy Of Sciences Of The United States Of America, 96:3513-8, 1999.
- an oligonucleotide can be modified to both decrease the stability of the antisense 5 'end of the duplex and increase the stability of the antisense 3' end of the duplex. This can be effected by combining one or more of the stability decreasing modifications in the antisense 5 ' end of the duplex with one or more of the stability increasing modifications in the antisense 3' end of the duplex.
- the single-stranded oligonucleotides of the present invention also comprise nucleotide sequence that is substantially complementary to a "sense" nucleic acid encoding a gene expression product, e.g., complementary to the coding strand of a double-stranded cDNA molecule or complementary to an RNA sequence, e.g., a pre- mR A, mRNA, miRNA, or pre-miRNA.
- the region of complementarity is less than 30 nucleotides in length, and at least 15 nucleotides in length.
- the single stranded oligonucleotides are 10 to 25 nucleotides in length ⁇ e.g., 11, 12, 13, 14, 15, 16, 18, 19, 20, 21, 22, 23, or 24 nucleotides in length). In one embodiment the strand is 25-30 nucleotides.
- Single strands having less than 100% complementarity to the target mRNA, RNA or DNA are also embraced by the present invention.
- These single-stranded oligonucleotides are also referred to as antisense, antagomir and antimir oligonucleotides.
- the single-stranded oligonucleotide can hybridize to a complementary RNA, and prevent access of the translation machinery to the target RNA transcript, thereby preventing protein synthesis.
- the single-stranded oligonucleotide can also hybridize to a complementary RNA and the RNA target can be subsequently cleaved by an enzyme such as RNase H. Degradation of the target RNA prevents translation.
- Single-stranded oligonucleotides including those described and/or identified as single stranded siRNAs, microRNAs or mirs which may be used as targets or may serve as a template for the design of oligonucleotides of the invention are taught in, for example, Esau, et al. US Publication #20050261218 (USSN: 10/909125) entitled
- MicroRNAs are a highly conserved class of small RNA molecules that are transcribed from DNA in the genomes of plants and animals, but are not translated into protein. Pre-microRNAs are processed into miRNAs. Processed microRNAs are single stranded -17-25 nucleotide (nt) RNA molecules that become incorporated into the RNA-induced silencing complex (RISC) and have been identified as key regulators of development, cell proliferation, apoptosis and differentiation. They are believed to play a role in regulation of gene expression by binding to the 3'- untranslated region of specific mRNAs. RISC mediates down-regulation of gene expression through translational inhibition, transcript cleavage, or both. RISC is also implicated in transcriptional silencing in the nucleus of a wide range of eukaryotes.
- RISC RNA-induced silencing complex
- miRNA sequences identified to date is large and growing, illustrative examples of which can be found, for example, in: "miRBase: microRNA sequences, targets and gene nomenclature” Griffiths- Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. NAR, 2006, 34, Database Issue, D140-D144; "The microRNA Registry” Griffiths- Jones S. NAR, 2004, 32, Database Issue, D109-D111; and also on the worldwide web at http://microma.dot.sanger.dot.ac.dot.uk/sequences/ .
- Antagomirs are RNA-like oligonucleotides that harbor various modifications for RNAse protection and pharmacologic properties, such as enhanced tissue and cellular uptake. They differ from normal RNA by, for example, complete 2'-O-methylation of sugar, phosphorothioate backbone and, for example, a cholesterol-moiety at 3'-end. Antagomirs may be used to efficiently silence endogenous miRNAs by forming duplexes comprising the antagomir and endogenous miRNA, thereby preventing miRNA-induced gene silencing.
- antagomir-mediated miRNA silencing is the silencing of miR-122, described in Krutzfeldt et al, Nature, 2005, 438: 685-689, which is expressly incorporated by reference herein in its entirety.
- Antagomir RNAs may be synthesized using standard solid phase oligonucleotide synthesis protocols. See US Patent
- An antagomir can include ligand-conjugated monomer subunits and monomers for oligonucleotide synthesis. Exemplary monomers are described in U.S. Application No. 10/916,185, filed on August 10, 2004.
- An antagomir can have a ZXY structure, such as is described in PCT Application No. PCT/US2004/07070 filed on March 8, 2004.
- An antagomir can be complexed with an amphipathic moiety. Exemplary amphipathic moieties for use with oligonucleotide agents are described in PCT Application
- Single stranded siRNAs are known and are described in US publication US 2006/0166910 and hereby incorporated by herein by its entirety.
- the single-stranded RNA molecule has a length from 15-29 nucleotides.
- the RNA-strand may have a 3'hydroxyl group. In some cases, however, it may be preferable to modify the 3' end to make it resistant against 3' to 5' exonucleases.
- Tolerated 3'- modifications are for example terminal 2'-deoxy nucleotides, 3' phosphate, 2',3'-cyclic phosphate, C3 (or C6, C7, C12) amino linker, thiol linkers, carboxyl linkers, non- nucleotidic spacers (C3, C6, C9, C12, abasic, triethylene glycol, hexaethylene glycol), biotin, fluoresceine, etc.
- Single stranded siRNAs of the invention include at least one of the following motifs: 5' phosphorothioate or 5'-phosphorodithioate, nucleotides 1 and 2 having cationic modifications via C-5 position of pyrimidines, 2-Position of Purines, N2- G, G-clamp, 8-position of purines, 6-position of purines, internal nucleotides having a 2'- F sugar with base modifications (Pseudouridine, G-clamp, phenoxazine,
- pyridopyrimidines gem2'-Me-up/2'-F-down
- 3 '-end with two purines with novel 2'- substituted MOE analogs 5 '-end nucleotides with novel 2'-substituted MOE analogs, 5'- end having a 3'-F and a 2'-5 '-linkage
- Ribozymes are oligonucleotides having specific catalytic domains that possess endonuclease activity (Kim and Cech, Proc Natl Acad Sci U S A. 1987 Dec;84(24):8788- 92; Forster and Symons, Cell. 1987 Apr 24;49(2):211-20). At least six basic varieties of naturally-occurring enzymatic RNAs are known presently.
- enzymatic nucleic acids act by first binding to a target RNA. Such binding occurs through the target binding portion of an enzymatic nucleic acid which is held in close proximity to an enzymatic portion of the molecule that acts to cleave the target RNA. Thus, the enzymatic nucleic acid first recognizes and then binds a target RNA through
- Ribozymes may be designed as described in Int. Pat. Appl. Publ. No. WO 93/23569 and Int. Pat. Appl. Publ. No. WO 94/02595, each specifically incorporated herein by reference, and synthesized to be tested in vitro and in vivo, as described therein.
- Aptamers are nucleic acid or peptide molecules that bind to a particular molecule of interest with high affinity and specificity (Tuerk and Gold, Science 249:505 (1990); Ellington and Szostak, Nature 346:818 (1990)).
- DNA or RNA aptamers have been successfully produced which bind many different entities from large proteins to small organic molecules. See Eaton, Curr. Opin. Chem. Biol. 1: 10-16 (1997), Famulok, Curr. Opin. Struct. Biol. 9:324-9(1999), and Hermann and Patel, Science 287:820-5 (2000).
- Aptamers may be RNA or DNA based.
- aptamers are engineered through repeated rounds of in vitro selection or equivalent ly, SELEX (systematic evolution of ligands by exponential enrichment) to bind to various molecular targets such as small molecules, proteins, nucleic acids, and even cells, tissues and organisms.
- the aptamer may be prepared by any known method, including synthetic, recombinant, and purification methods, and may be used alone or in combination with other aptamers specific for the same target.
- the term "aptamer” specifically includes "secondary aptamers” containing a consensus sequence derived from comparing two or more known aptamers to a given target.
- Nucleic acids of the present invention may be immunostimulatory, including immunostimulatory oligonucleotides (single-or double-stranded) capable of inducing an immune response when administered to a subject, which may be a mammal or other patient.
- the immune response may be an innate or an adaptive immune response.
- the immune system is divided into a more innate immune system, and acquired adaptive immune system of vertebrates, the latter of which is further divided into humoral cellular components.
- the immune response may be mucosal.
- Immunostimulatory nucleic acids are considered to be non-sequence specific when it is not required that they specifically bind to and reduce the expression of a target polynucleotide in order to provoke an immune response.
- immunostimulatory nucleic acids may comprise a seuqence correspondign to a region of a naturally occurring gene or mR A, but they may still be considered non-sequence specific immunostimulatory nucleic acids.
- the immunostimulatory nucleic acid or oligonucleotide comprises at least one CpG dinucleotide.
- the oligonucleotide or CpG dinucleotide may be unmethylated or methylated.
- the immunostimulatory nucleic acid comprises at least one CpG dinucleotide having a methylated cytosine.
- the nucleic acid comprises a single CpG dinucleotide, wherein the cytosine in said CpG dinucleotide is methylated.
- the immunostimulatory nucleic acid or oligonucleotide comprises capable of inducing an anti- viral or an antibacterial response, in particular, the induction of type I IFN, IL-18 and/or IL- ⁇ by modulating RIG-I.
- oligonucleotides bearing the consensus binding sequence of a specific transcription factor can be used as tools for manipulating gene expression in living cells.
- This strategy involves the intracellular delivery of such "decoy oligonucleotides", which are then recognized and bound by the target factor. Occupation of the transcription factor's DNA-binding site by the decoy renders the transcription factor incapable of subsequently binding to the promoter regions of target genes. Decoys can be used as therapeutic agents, either to inhibit the expression of genes that are activated by a transcription factor, or to upregulate genes that are suppressed by the binding of a transcription factor. Examples of the utilization of decoy oligonucleotides may be found in Mann et al, J. Clin. Invest., 2000, 106: 1071-1075, which is expressly incorporated by reference herein, in its entirety.
- Ul adaptor inhibit polyA sites and are bifunctional oligonucleotides with a target domain complementary to a site in the target gene's terminal exon and a 'Ul domain' that binds to the Ul smaller nuclear RNA component of the Ul snRNP (Goraczniak, et al, 2008, Nature Biotechnology, 27(3), 257-263, which is expressly incorporated by reference herein, in its entirety).
- Ul snRNP is a ribonucleoprotein complex that functions primarily to direct early steps in spliceosome formation by binding to the pre- mRNA exon- intron boundary (Brown and Simpson, 1998, Annu Rev Plant Physiol Plant Mol Biol 49:77-95).
- oligonucleotides of the invention are Ul adaptors.
- the Ul adaptor can be administered in combination with at least one other iRNA agent.
- Unmodified oligonucleotides may be less than optimal in some applications, e.g., unmodified oligonucleotides can be prone to degradation by e.g., cellular nucleases. Nucleases can hydro lyze nucleic acid phosphodiester bonds. However, chemical modifications to one or more of the above oligonucleotide components can confer improved properties, and, e.g., can render oligonucleotides more stable to nucleases.
- Modified nucleic acids and nucleotide surrogates can include one or more of:
- modification of the 3' end or 5' end of the oligonucelotide e.g., removal, modification or replacement of a terminal phosphate group or conjugation of a moiety, e.g., a fluorescently labeled moiety, to either the 3" or 5' end of oligonucleotide;
- oligonucleotides are polymers of subunits or monomers
- many of the modifications described herein can occur at a position which is repeated within an oligonucleotide, e.g., a modification of a nucleobase, a sugar, a phosphate moiety, or the non-bridging oxygen of a phosphate moiety. It is not necessary for all positions in a given oligonucleotide to be uniformly modified, and in fact more than one of the aforementioned modifications may be incorporated in a single oligonucleotide or even at a single nucleoside within an oligonucleotide.
- the modification will occur at all of the subject positions in the oligonucleotide but in many, and in fact in most cases it will not.
- a modification may only occur at a 3' or 5' terminal position, may only occur in the internal region, may only occur in a terminal regions, e.g. at a position on a terminal nucleotide or in the last 2, 3, 4, 5, or 10 nucleotides of an oligonucleotide.
- modification may occur in a double strand region, a single strand region, or in both.
- a modification may occur only in the double strand region of an oligonucleotide or may only occur in a single strand region of an oligonucleotide.
- a phosphorothioate modification at a non-bridging oxygen position may only occur at one or both termini, may only occur in a terminal regions, e.g., at a position on a terminal nucleotide or in the last 2, 3, 4, 5, or 10 nucleotides of a strand, or may occur in double strand and single strand regions, particularly at termini.
- the 5' end or ends can be phosphorylated.
- a modification described herein may be the sole modification, or the sole type of modification included on multiple nucleotides, or a modification can be combined with one or more other modifications described herein.
- the modifications described herein can also be combined onto an oligonucleotide, e.g. different nucleotides of an oligonucleotide have different modifications described herein.
- Modifications can include, e.g., the use of modifications at the 2' OH group of the ribose sugar, e.g., the use of deoxyribonucleotides, e.g., deoxythymidine, instead of ribonucleotides, and modifications in the phosphate group, e.g., phosphothioate modifications.
- Overhangs need not be homologous with the target sequence.
- the phosphate group is a negatively charged species. The charge is distributed equally over the two non-bridging oxygen atoms. However, the phosphate group can be modified by replacing one of the oxygens with a different substituent. One result of this modification to R A phosphate backbones can be increased resistance of the
- oligoribonucleotide to nucleo lytic breakdown.
- modified phosphate groups include phosphorothioate,
- one of the non-bridging phosphate oxygen atoms in the phosphate backbone moiety can be replaced by any of the following: S, Se, BR 3 (R is hydrogen, alkyl, aryl), C (i.e. an alkyl group, an aryl group, etc...), H, NR 2 (R is hydrogen, alkyl, aryl), or OR (R is alkyl or aryl).
- the phosphorous atom in an unmodified phosphate group is achiral.
- the stereogenic phosphorous atom can possess either the "R" configuration (herein Rp) or the "S” configuration (herein Sp).
- Phosphorodithioates have both non-bridging oxygens replaced by sulfur.
- the phosphorus center in the phosphorodithioates is achiral which precludes the formation of oligoribonucleotides diastereomers.
- modifications to both non-bridging oxygens, which eliminate the chiral center, e.g. phosphorodithioate formation may be desirable in that they cannot produce diastereomer mixtures.
- the non-bridging oxygens can be independently any one of S, Se, B, C, H, N, or OR (R is alkyl or aryl).
- the phosphate linker can also be modified by replacement of bridging oxygen, (i.e. oxgen that links the phosphate to the nucleoside), with nitrogen (bridged
- the replacement can occur at the either linking oxygen or at both the linking oxygens.
- the bridging oxygen is the 3 '-oxygen of a nucleoside, replcament with carbobn is preferred.
- replcament with nitrogen is preferred.
- the phosphate group can be replaced by non-phosphorus containing connectors. While not wishing to be bound by theory, it is believed that since the charged
- phosphodiester group is the reaction center in nucleolytic degradation, its replacement with neutral structural mimics should impart enhanced nuclease stability.
- moieties which can replace the phosphate group include methyl phosphonate, hydroxy lamino, siloxane, carbonate, carboxymethyl, carbamate, amide, thioether, ethylene oxide linker, sulfonate, sulfonamide, thioformacetal, formacetal, oxime, methyleneimino, methylenemethylimino, methylenehydrazo,
- Preferred replacements include the methylenecarbonylamino and methylenemethylimino groups.
- Modified phosphate linkages where at least one of the oxygens linked to the phosphate has been replaced or the phosphate group has been replaced by a non- phosphorous group are also referred to as "non-phosphodiester backbone linkage.”
- Oligonucleotide- mimicking scaffolds can also be constructed wherein the phosphate linker and ribose sugar are replaced by nuclease resistant nucleoside or nucleotide surrogates. While not wishing to be bound by theory, it is believed that the absence of a repetitively charged backbone diminishes binding to proteins that recognize polyanions (e.g. nucleases). Again, while not wishing to be bound by theory, it can be desirable in some embodiment, to introduce alterations in which the bases are tethered by a neutral surrogate backbone. Examples include the mophilino, cyclobutyl, pyrrolidine and peptide nucleic acid (PNA) nucleoside surrogates. A preferred surrogate is a PNA surrogate.
- An oligonucleotide can include modification of all or some of the sugar groups of the nucleic acid.
- the 2' hydroxyl group (OH) can be modified or replaced with a number of different "oxy" or "deoxy” substituents. While not being bound by theory, enhanced stability is expected since the hydroxyl can no longer be deprotonated to form a 2'-alkoxide ion.
- the 2'-alkoxide can catalyze degradation by intramolecular nucleophilic attack on the linker phosphorus atom.
- polyethyleneglycols PEG
- LNA locked nucleic acids
- O-AMINE NH 2 ; alkylamino, dialkylamino, heterocyclyl, arylamino, diaryl amino, heteroaryl amino, or diheteroaryl amino, ethylene diamine, polyamino
- AMINE NH 2 ; alkylamino, dialkylamino, heterocyclyl, arylamino, diaryl amino, heteroaryl amino, or diheteroaryl amino, ethylene diamine, polyamino
- AMINE NH 2 ; alkylamino, dialkylamino, heterocyclyl, arylamino, diaryl amino, heteroaryl amino, or diheteroaryl amino, ethylene diamine, polyamino).
- MOE methoxyethyl group
- an oligonucleotide can include nucleotides containing e.g., arabinose, as the sugar.
- the monomer can have an alpha linkage at the 1 ' position on the sugar, e.g., alpha- nucleosides.
- Oligonucleotides can also include "abasic" sugars, which lack a nucleobase at C- . These abasic sugars can also be further containing modifications at one or more of the constituent sugar atoms.
- Oligonucleotides can also contain one or more sugars that are in the L form, e.g.
- Preferred substitutents are 2'- ⁇ 9-Me (2'- ⁇ 9-methyl), 2'- ⁇ 9-MOE ( -0- methoxyethyl), 2'-F, 2'-0-[2-(methylamino)-2-oxoethyl] (2'- ⁇ 9-NMA), 2'-5-methyl, 2'- 0-CH 2 -(4'-C) (LNA), 2'-0-CH 2 CH 2 -(4'-C) (ENA), 2'-0-aminopropyl (2'-0-AP), 2'-0- dimethylaminoethyl (2'-0-DMAOE), 2'-0-dimethylaminopropyl (2'-0-DMAP) and 2'-0- dimethylaminoethyloxyethyl (2'-0-DMAEOE).
- the 3' and 5' ends of an oligonucleotide can be modified. Such modifications can be at the 3' end, 5' end or both ends of the molecule. They can include modification or replacement of an entire terminal phosphate or of one or more of the atoms of the phosphate group.
- the 3' and 5' ends of an oligonucleotide can be conjugated to other functional molecular entities such as labeling moieties, e.g., fluorophores ⁇ e.g., pyrene, TAMRA, fluorescein, Cy3 or Cy5 dyes) or protecting groups (based e.g., on sulfur, silicon, boron or ester).
- the functional molecular entities can be attached to the sugar through a phosphate group and/or a linker.
- the terminal atom of the linker can connect to or replace the linking atom of the phosphate group or the C-3' or C-5' O, N, S or C group of the sugar.
- the linker can connect to or replace the terminal atom of a nucleotide surrogate ⁇ e.g., PNAs).
- this array can substitute for a hairpin R A loop in a hairpin-type RNA agent.
- Terminal modifications useful for modulating activity include modification of the 5' end with phosphate or phosphate analogs.
- antisense strands of dsRNAs are 5 ' phosphorylated or include a phosphoryl analog at the 5 ' prime terminus.
- 5'-phosphate modifications include those which are compatible with RISC mediated gene silencing. Modifications at the 5 '-terminal end can also be useful in stimulating or inhibiting the immune system of a subject.
- Suitable modifications include: 5 * -monophosphate ((HO)2(0)P-0-5 * ); 5 * -diphosphate ((HO)2(0)P-0-P(HO)(0)-0-5 * ); 5'- triphosphate ((HO)2(0)P-0-(HO)(0)P-0-P(HO)(0)-0-5 * ); 5 * -guanosine cap (7- methylated or non-methylated) (7m-G-0-5 * -(HO)(0)P-0-(HO)(0)P-0-P(HO)(0)-0-5 * ); 5'-adenosine cap (Appp), and any modified or unmodified nucleotide cap structure (N-O- 5 * -(HO)(0)P-0-(HO)(0)P-0-P(HO)(0)-0-5 * ); 5 * -monothiophosphate (phosphorothioate; (HO)2(S)P-0-5 * ); 5 * -monodithiophosphate (phosphorodithioate
- Terminal modifications can also be useful for monitoring distribution, and in such cases the preferred groups to be added include fluorophores, e.g., fluorscein or an Alexa dye, e.g., Alexa 488. Terminal modifications can also be useful for enhancing uptake, useful modifications for this include cholesterol. Terminal modifications can also be useful for cross-linking an RNA agent to another moiety; modifications useful for this include mitomycin C.
- Adenine, guanine, cytosine and uracil are the most common bases found in RNA. These bases can be modified or replaced to provide RNA's having improved properties.
- nuclease resistant oligoribonucleotides can be prepared with these bases or with synthetic and natural nucleobases (e.g., inosine, thymine, xanthine, hypoxanthine, nubularine, isoguanisine, or tubercidine) and any one of the above modifications.
- substituted or modified analogs of any of the above bases and “universal bases” can be employed.
- Examples include 2-(halo)adenine, 2-(alkyl)adenine, 2- (propyl)adenine, 2-(amino)adenine, 2-(aminoalkyll)adenine, 2-(aminopropyl)adenine, 2- (methylthio)-N 6 -(isopentenyl)adenine, 6-(alkyl)adenine, 6-(methyl)adenine,
- 3- (deaza)-5-(aza)cytosine 3-(alkyl)cytosine, 3-(methyl)cytosine, 5-(alkyl)cytosine, 5- (alkynyl)cytosine, 5-(halo)cytosine, 5-(methyl)cytosine, 5-(propynyl)cytosine,
- phenanthracenyl pyrenyl, stilbenyl, tetracenyl, pentacenyl, difluorotolyl, 4-(fluoro)-6- (methyl)benzimidazole, 4-(methyl)benzimidazole, 6-(azo)thymine, 2-pyridinone,
- Modifications to oligonucleotides can also include attachment of one or more cationic groups to the sugar, base, and/or the phosphorus atom of a phosphate or modified phosphate backbone moiety.
- a cationic group can be attached to any atom capable of substitution on a natural, unusual or universal base.
- a preferred position is one that does not interfere with hybridization, i.e., does not interfere with the hydrogen bonding interactions needed for base pairing.
- a cationic group can be attached e.g., through the C2' position of a sugar or analogous position in a cyclic or acyclic sugar surrogate.
- AMINE NH 2 ; alkylamino, dialkylamino, heterocyclyl, arylamino, diaryl amino, heteroaryl amino,or diheteroaryl amino).
- Some modifications may preferably be included on an oligonucleotide at a particular location, e.g., at an internal position of a strand, or on the 5' or 3' end of an oligonucleotide.
- a preferred location of a modification on an oligonucleotide may confer preferred properties on the agent.
- preferred locations of particular modifications may confer optimum gene silencing properties, or increased resistance to endonuclease or exonuclease activity.
- One or more nucleotides of an oligonucleotide may have a 2'-5' linkage.
- One or more nucleotides of an oligonucleotide may have inverted linkages, e.g. 3'-3', 5'-5', 2'- 2' or 2'-3' linkages.
- a double-stranded oligonucleotide may include at least one 5'-uridine-adenine-3' (5'-UA-3') dinucleotide wherein the uridine is a 2'-modified nucleotide, or a terminal 5'- uridine-guanine-3 ' (5'-UG-3') dinucleotide, wherein the 5 '-uridine is a 2'-modified nucleotide, or a terminal 5'-cytidine-adenine-3' (5'-CA-3') dinucleotide, wherein the 5'- cytidine is a 2'-modified nucleotide, or a terminal 5'-uridine-uridine-3' (5'-UU-3') dinucleotide, wherein the 5 '-uridine is a 2'-modified nucleotide, or a terminal 5'- cytidine-cytidine-3 ' (5'-CC-3') dinu
- oligoribonucleotides and oligoribonucleosides used in accordance with this invention may be synthesized with solid phase synthesis, see for example
- Methyloligoribonucleotide- s synthesis and applications, Chapter 4, Phosphorothioate oligonucleotides, Chapter 5, Synthesis of oligonucleotide phosphorodithioates, Chapter 6, Synthesis of oligo-2'-deoxyribonucleoside methylphosphonates, and. Chapter 7,
- Oligodeoxynucleotides containing modified bases are described in Martin, P., Helv. Chim. Acta, 1995, 78, 486-504; Beaucage, S. L. and Iyer, R. P., Tetrahedron, 1992, 48, 2223-2311 and Beaucage, S. L. and Iyer, R. P., Tetrahedron, 1993, 49, 6123-6194, or references referred to therein. Modification described in WO 00/44895, WOOl/75164, or WO02/44321 can be used herein. The disclosure of all publications, patents, and published patent applications listed herein are hereby incorporated by reference.
- phosphinate oligoribonucleotides The preparation of phosphinate oligoribonucleotides is described in U.S. Pat. No. 5,508,270. The preparation of alkyl phosphonate oligoribonucleotides is described in U.S. Pat. No. 4,469,863. The preparation of phosphoramidite oligoribonucleotides is described in U.S. Pat. No. 5,256,775 or U.S. Pat. No. 5,366,878. The preparation of phosphotriester oligoribonucleotides is described in U.S. Pat. No. 5,023,243. The preparation of borano phosphate oligoribonucleotide is described in U.S. Pat. Nos.
- MMI linked oligoribonucleosides also identified herein as MMI linked oligoribonucleosides, methylenedimethylhydrazo linked
- oligoribonucleosides also identified herein as MDH linked oligoribonucleosides
- methylenecarbonylamino linked oligonucleosides also identified herein as amide-3 linked oligoribonucleosides
- methyleneaminocarbonyl linked oligonucleosides also identified herein as amide-4 linked oligoribonucleosides as well as mixed backbone compounds having, as for instance, alternating MMI and PO or PS linkages can be prepared as is described in U.S. Pat. Nos.
- Formacetal and thioformacetal linked oligoribonucleosides can be prepared as is described in U.S. Pat. Nos. 5,264,562 and 5,264,564.
- Ethylene oxide linked oligoribonucleosides can be prepared as is described in U.S. Pat. No. 5,223,618.
- Siloxane replacements are described in Cormier .F. et al. Nucleic Acids Res. 1988, 16, 4583. Carbonate replacements are described in Tittensor, J.R.
- Cyclobutyl sugar surrogate compounds can be prepared as is described in U.S. Pat. No. 5,359,044. Pyrrolidine sugar surrogate can be prepared as is described in U.S. Pat. No. 5,519,134. Morpholino sugar surrogates can be prepared as is described in U.S. Pat. Nos. 5,142,047 and 5,235,033, and other related patent disclosures.
- Peptide Nucleic Acids (PNAs) are known per se and can be prepared in accordance with any of the various procedures referred to in Peptide Nucleic Acids (PNA): Synthesis, Properties and Potential Applications, Bioorganic & Medicinal Chemistry, 1996, 4, 5-23. They may also be prepared in accordance with U.S. Pat. No. 5,539,083.
- N-2 substitued purine nucleoside amidites can be prepared as is described in U.S. Pat. No. 5,459,255.
- 3-Deaza purine nucleoside amidites can be prepared as is described in U.S. Pat. No. 5,457,191.
- 5,6-Substituted pyrimidine nucleoside amidites can be prepared as is described in U.S. Pat. No. 5,614,617.
- 5-Propynyl pyrimidine nucleoside amidites can be prepared as is described in U.S. Pat. No. 5,484,908. Additional references are disclosed in the above section on base modifications
- the oligonucleotide compounds of the invention can be prepared using solution- phase or solid-phase organic synthesis.
- Organic synthesis offers the advantage that the oligonucleotide strands comprising non-natural or modified nucleotides can be easily prepared. Any other means for such synthesis known in the art may additionally or alternatively be employed. It is also known to use similar techniques to prepare other oligonucleotides, such as the phosphorothioates, phosphorodithioates and alkylated derivatives.
- the double-stranded oligonucleotide compounds of the invention may be prepared using a two-step procedure. First, the individual strands of the double-stranded molecule are prepared separately. Then, the component strands are annealed.
- the oligonucleotide can be prepared in a solution (e.g., an aqueous and/or organic solution) that is appropriate for formulation.
- a solution e.g., an aqueous and/or organic solution
- the iRNA preparation can be precipitated and redissolved in pure double- distilled water, and lyophilized. The dried iRNA can then be resuspended in a solution appropriate for the intended formulation process.
- Pat. No. 5,457, 191 drawn to modified nucleobases based on the 3-deazapurine ring system and methods of synthesis thereof; U.S. Pat. No. 5,459,255, drawn to modified nucleobases based on N-2 substituted purines; U.S. Pat. No. 5,521,302, drawn to processes for preparing oligonucleotides having chiral phosphorus linkages; U.S. Pat. No. 5,539,082, drawn to peptide nucleic acids; U.S. Pat. No. 5,554,746, drawn to oligonucleotides having ⁇ -lactam backbones; U.S. Pat. No.
- 5,587,469 drawn to oligonucleotides having N-2 substituted purines
- U.S. Pat. No. 5,587,470 drawn to oligonucleotides having 3-deazapurines
- U.S. Pat. Nos. 5,602,240, and 5,610,289 drawn to backbone-modified oligonucleotide analogs
- U.S. Pat. Nos. 6,262,241, and 5,459,255 drawn to, inter alia, methods of synthesizing 2'- fluoro -oligonucleotides .
- the invention relates to a method for inhibiting the expression of a target gene in a cell or organism.
- the method includes administering the inventive oligonucleotide, e.g. antisense, aptamer, antagomir, or an iRNA agent; or a pharmaceutical composition containing the said oligonucleotide to a cell or an organism, such as a mammal, such that expression of the target gene is silenced.
- inventive oligonucleotide e.g. antisense, aptamer, antagomir, or an iRNA agent
- a pharmaceutical composition containing the said oligonucleotide to a cell or an organism, such as a mammal, such that expression of the target gene is silenced.
- Compositions and methods for inhibiting the expression of a target gene using the inventive oligonucleotide, e.g. an iRNA agent can be performed as described in the preceding sections.
- a pharmaceutical composition containing the inventive oligonucleotide may be administered by any means known in the art including, but not limited to oral or parenteral routes, including intravenous, intramuscular, intraperitoneal, subcutaneous, transdermal, airway (aerosol), ocular, rectal, vaginal, and topical
- the pharmaceutical compositions are administered by intravenous or intraparenteral infusion or injection.
- the pharmaceutical compositions can also be administered
- the methods for inhibiting the expression of a target gene can be applied to any gene one wishes to silence, thereby specifically inhibiting its expression, provided the cell or organism in which the target gene is expressed includes the cellular machinery which effects R A interference.
- genes which can be targeted for silencing include, without limitation, developmental genes including but not limited to adhesion molecules, cyclin kinase inhibitors, Wnt family members, Pax family members, Winged helix family members, Hox family members, cytokines/lymphokines and their receptors, growth/differentiation factors and their receptors, and neurotransmitters and their receptors; (2) oncogenes including but not limited to ABLI, BCL1, BCL2, BCL6, CBFA2, CBL, CSFIR, ERBA, ERBB, EBRB2, ETS1, ETS1, ETV6, FGR, FOS, FYN, HCR, HRAS, JUN, KRAS, LCK, LYN, MDM2, MLL, MYB, MYC, MYCL
- inventive oligonucleotides e.g. iR A agent
- inventive oligonucleotides are useful in a wide variety of in vitro applications.
- Such in vitro applications include, for example, scientific and commercial research (e.g., elucidation of physiological pathways, drug discovery and development), and medical and veterinary diagnostics.
- the method involves the introduction of the oligonucleotide, e.g. an iRNA agent, into a cell using known techniques (e.g., absorption through cellular processes, or by auxiliary agents or devices, such as electroporation and lipofection), then maintaining the cell for a time sufficient to obtain degradation of an mRNA transcript of the target gene.
- nucleoside includes nucleotides as well as nucleoside and nucleotide analogs, and modified nucleosides such as amino modified nucleosides.
- nucleoside includes non-naturally occurring analog structures. Thus for example the individual units of a peptide nucleic acid, each containing a base, are referred to herein as a nucleoside.
- aliphatic refers to a straight or branched hydrocarbon radical containing up to twenty four carbon atoms wherein the saturation between any two carbon atoms is a single, double or triple bond.
- An aliphatic group preferably contains from 1 to about 24 carbon atoms, more typically from 1 to about 12 carbon atoms.
- Suitable aliphatic groups include, but are not limited to, linear or branched, substituted or unsubstituted alkyl, alkenyl, alkynyl groups and hybrids thereof such as (cycloalkyl)alkyl, (cycloalkenyl)alkyl or (cycloalkyl)alkenyl.
- the straight or branched chain of an aliphatic group may be interrupted with one or more heteroatoms that include nitrogen, oxygen, sulfur and phosphorus.
- Such aliphatic groups interrupted by heteroatoms include without limitation polyalkoxys, such as polyalkylene glycols, polyamines, and polyimines, for example.
- Aliphatic groups as used herein may optionally include further substitutent groups.
- alkyl refers to saturated and unsaturated non-aromatic hydrocarbon chains that may be a straight chain or branched chain, containing the indicated number of carbon atoms (these include without limitation propyl, allyl, or propargyl), which may be optionally inserted with N, O, or S.
- C1-C20 indicates that the group may have from 1 to 20 (inclusive) carbon atoms in it.
- alkoxy refers to an -O-alkyl radical.
- alkylene refers to a divalent alkyl (i.e., -R-). The term
- alkylenedioxo refers to a divalent species of the structure -O-R-O-, in which R represents an alkylene.
- aminoalkyl refers to an alkyl substituted with an amino.
- mercapto refers to an -SH radical.
- thioalkoxy refers to an -S-alkyl radical.
- cyclic as used herein includes a cycloalkyl group and a heterocyclic group. Any suitable ring position of the cyclic group may be covalently linked to the defined chemical structure.
- acyclic may describe any carrier that is branched or unbranched, and does not form a closed ring.
- aryl refers to a 6-carbon monocyclic or 10-carbon bicyclic aromatic ring system wherein 0, 1, 2, 3, or 4 atoms of each ring may be substituted by a substituent. Examples of aryl groups include phenyl, naphthyl and the like.
- arylalkyl or the term “aralkyl” refers to alkyl substituted with an aryl.
- arylalkoxy refers to an alkoxy substituted with aryl.
- cycloalkyl as employed herein includes saturated and partially unsaturated cyclic hydrocarbon groups having 3 to 12 carbons, for example, 3 to 8 carbons, and, for example, 3 to 6 carbons, wherein the cycloalkyl group additionally may be optionally substituted.
- Cycloalkyl groups include, without limitation, decalin, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cycloheptyl, and cyclooctyl.
- heteroaryl refers to an aromatic 5-8 membered monocyclic, 8-12 membered bicyclic, or 11-14 membered tricyclic ring system having 1-3 heteroatoms if monocyclic, 1-6 heteroatoms if bicyclic, or 1-9 heteroatoms if tricyclic, said heteroatoms selected from O, N, or S (e.g., carbon atoms and 1-3, 1-6, or 1-9 heteroatoms of N, O, or S if monocyclic, bicyclic, or tricyclic, respectively), wherein 0, 1, 2, 3, or 4 atoms of each ring may be substituted by a substituent.
- heteroaryl groups include pyridyl, furyl or furanyl, imidazolyl, benzimidazolyl, pyrimidinyl, thiophenyl or thienyl, quinolinyl, indolyl, thiazolyl, and the like.
- heteroarylalkyl or the term “heteroaralkyl” refers to an alkyl substituted with a heteroaryl.
- heteroarylalkoxy refers to an alkoxy substituted with heteroaryl.
- heterocycloalkyl and “heterocyclic” can be used interchangeably and refer to a non-aromatic 3-, 4-, 5-, 6- or 7-membered ring or a bi- or tri-cyclic group fused system, where (i) each ring contains between one and three heteroatoms independently selected from oxygen, sulfur and nitrogen, (ii) each 5-membered ring has 0 to 1 double bonds and each 6-membered ring has 0 to 2 double bonds, (iii) the nitrogen and sulfur heteroatoms may optionally be oxidized, (iv) the nitrogen heteroatom may optionally be quaternized, (iv) any of the above rings may be fused to a benzene ring, and (v) the remaining ring atoms are carbon atoms which may be optionally oxo-substituted.
- heterocycloalkyl groups include, but are not limited to, [l,3]dioxolane, pyrrolidinyl, pyrazolinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, piperidinyl, piperazinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl, isothiazolidinyl, quinoxalinyl, pyridazinonyl, and tetrahydrofuryl.
- Such heterocyclic groups may be further substituted to give substituted heterocyclic.
- oxo refers to an oxygen atom, which forms a carbonyl when attached to carbon, an N-oxide when attached to nitrogen, and a sulfoxide or sulfone when attached to sulfur.
- acyl refers to an alkylcarbonyl, cycloalkylcarbonyl, arylcarbonyl, heterocyclylcarbonyl, or heteroarylcarbonyl substituent, any of which may be further substituted by substituents.
- sil as used herein is represented by the formula -SiA 1 A 2 A 3 , where A 1 , A 2 , and A 3 can be, independently, hydrogen or a substituted or unsubstituted alkyl, cycloalkyl, alkoxy, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, or heteroaryl group as described herein.
- substituted refers to the replacement of one or more hydrogen radicals in a given structure with the radical of a specified substituent including, but not limited to: halo, alkyl, alkenyl, alkynyl, aryl, heterocyclyl, thiol, alkylthio, arylthio, alky It hio alky 1, arylthioalkyl, alkylsulfonyl, alkylsulfonylalkyl, arylsulfonylalkyl, alkoxy, aryloxy, aralkoxy, aminocarbonyl, alkylaminocarbonyl, arylaminocarbonyl,
- aralkoxycarbonyl carboxylic acid, sulfonic acid, sulfonyl, phosphonic acid, aryl, heteroaryl, heterocyclic, and aliphatic. It is understood that the substituent may be further substituted.
- Adenine, guanine, cytosine and uracil are the most common bases found in R A. These bases can be modified or replaced to provide R A's having improved properties.
- nuclease resistant oligoribonucleotides can be prepared with these bases or with synthetic and natural nucleobases ⁇ e.g., inosine, thymine, xanthine, hypoxanthine, nubularine, isoguanisine, or tubercidine) and any one of the above modifications.
- substituted or modified analogs of any of the above bases and "universal bases” can be employed.
- Examples include 2-aminoadenine, 2-fluoroadenine, 6-methyl and other alkyl derivatives of adenine and guanine, 2-propyl and other alkyl derivatives of adenine and guanine, 5-halouracil and cytosine, 5-propynyl uracil and cytosine, 6-azo uracil, cytosine and thymine, 5-uracil (pseudouracil), 4-thiouracil, 5-halouracil, 5-(2- aminopropyl)uracil, 5-amino allyl uracil, 8-halo, amino, thiol, thioalkyl, hydroxyl and other 8-substituted adenines and guanines, 5 -trifluoromethyl and other 5-substituted uracils and cytosines, 7-methylguanine, 5-substi
- non-natural nucleobase refers any one of the following: 2- methyladeninyl, N6-methyladeninyl, 2-methylthio-N6-methyladeninyl, N6- isopentenyladeninyl, 2-methylthio-N6-isopentenyladeninyl, N6-(cis- hydroxyisopentenyl)adeninyl, 2-methylthio-N6-(cis-hydroxyisopentenyl) adeninyl, N6- glycinylcarbamoyladeninyl, N6-threonylcarbamoyladeninyl, 2-methylthio-N6-threonyl carbamoyladeninyl, N6-methyl-N6-threonylcarbamoyladeninyl, N6- hydroxynorvalylcarbamoyladeninyl, 2-methylthio-N6-hydroxynorvalylcarbamoyladeninyl, 2-methyl
- Method B To a solution of PEG-azide (2, 0.28 g, 0.75 mmol, 3.0 eq) and tris- propargylamine (0.032 g, 0.25 mmol, 1.0 eq) in t-butanol: water (1 : 1) were added sodium ascorbate (0.015 g, 0.075 mmol, 0.3 eq) followed by CuS0 4 .5H 2 0 (0.0018g, 0.0075 mmol, 0.03 eq) and the reaction was continued under microwave conditions at 80 °C, TLC and LC Mass shows complete consumption of the starting materials after lh. Reaction mixture was diluted with the dichloromethane, separated the organic layer and washed with water. Combined organics were dried on MgSC ⁇ , concentrated, purified on column chromatography using dichloromethane and methanol (10%) as gradients to get pure benzyl protected triazolyl ligand (6) as an oil.
- Recyclable ligand 9 Using ligand 9 in the repeated click reaction cycles gave quantitative yield of the product.
- Example 5 Chelated copper mediated click reaction of nucleoside with an azide 13
- Compound 14 has been prepared by using similar experimental conditions used for 12, using alkyne 10 (0.05 g, 0.08 mmol, 1.0 eq), azide 13 (0.04 g, 0.08 mmol, 1.0 eq) and Cu-complex 9 (0.05 g, 0.01 mmol, 0.12 eq) in DMF: DCM (2: 1) get pure trizole compound (14). Calc. mass for 1152.6; found 1152.5.
- Compound 17 has been prepared by using similar experimental conditions used for 12, using oligo alkyne 15 (35.6 ⁇ in stock solution, 1.0 eq), azide 16 (71.0 ⁇ , 2.0 eq) and Cu-complex 9 (9.8 ⁇ , 0.2 eq) in DMF gave complete conversion of the product 17 in overnight at room temperature. Calc. mass for Compound 17 is 7402, observed 7402.
- Example 7 Chelated copper mediated click reaction of Oligonucleotide 15 with azide 18
- Compound 19 has been prepared by using similar experimental conditions used for 12, using oligo alkyne 15 (35.6 ⁇ in stock solution, 1.0 eq), azide 18 (71.0 ⁇ , 2.0 eq) and Cu-complex 9 (9.8 ⁇ , 0.2 eq) in DMF gave conversion of the product 19 in overnight at room temperature. Calc. mass for Compound 19 is 7341, observed 7342.
- Compound 21 has been prepared by using similar experimental conditions used for 12, using oligo alkyne 15 (35.6 ⁇ in stock solution, 1.0 eq), azide 20 (71.0 ⁇ , 2.0 eq) and Cu-complex 9 (17.8 ⁇ , 0.5 eq) in DMF gave complete conversion of the product 21 in overnight at room temperature. Calc. mass for Compound 21 is 7235, observed 7235.
- Ligand 25 was loaded on amino methyl polystyrene resin as similar conditions mentioned above using compound 25 (0.65 g, 0.55 mmol, 1.2 eq), HBTU (0.26 g, 0.69 mmol, 1.5 eq), DIPEA (0.2 mL, 1.3 mmol, 3.0 eq) and aminomethyl polystyrene resin (230 2.0 g, 0.46 mmol, 1.0), after complete dry, gave solid supported ligand 26 in 2.1 g yield.
- Example 12 Click reaction of nucleoside 10 with azide 11 using Catalysts II
Abstract
In one embodiment, the invention features a process for preparing ligand conjugated oligonucleotides comprising a step of contacting a nucleoside or oligonucleotide containing an alkyne moiety with ligand containing an azide moiety in the presence of a solid supported copper catalyst of formula A, B, C or D.
Description
Chelated Copper for use in the Preparation of
Conjugated Oligonucleotides
PRIORITY CLAIM
This application claims priority to U.S. Provisional Application No. 61/299,302, filed January 28, 2010.
FIELD OF INVENTION
The present invention relates to the field of conjugation of ligands to
oligonucleotides with "click" chemistry in the presence of solid supported Cu(I) catalyst.
BACKGROUND
Oligonucleotide compounds have important therapeutic applications in medicine. Oligonucleotides can be used to silence genes that are responsible for a particular disease. Gene- silencing prevents formation of a protein by inhibiting translation. Importantly, gene-silencing agents are a promising alternative to traditional small, organic compounds that inhibit the function of the protein linked to the disease. siR A, antisense RNA, and micro-RNA are oligonucleotides that prevent the formation of proteins by gene-silencing.
The opportunity to use of nucleic acid based therapies holds significant promise, providing solutions to medical problems that could not be addressed with current, traditional medicines. The location and sequences of an increasing number of disease- related genes are being identified, and clinical testing of nucleic acid-based therapeutics for a variety of diseases is now underway.
Despite the different synthetic strategies developed for conjugation of various ligands to the oligonucleotides, the synthesis of ligand-oligonucleotide conjugates is anything but trivial and requires extensive expertise in organic chemistry and solid-phase synthesis. A real advance would be to use a coupling reaction that can be utilized for a large variety of ligands and oligonucleotides. The Huisgen 1,3-dipolar cycloaddition of alkynes and azides, the "click" reaction, is especially attractive for irreversible coupling of two molecules under mild conditions. The "click" chemistry has recently emerged as
an efficient strategy to conjugate carbohydrates, peptides and proteins, fluorescent labels and lipids to oligonucleotides. Therefore, there is a clear need for new reagents that can be utilize for "click" chemistry for conjugation of ligands to oligonucleotides. The present invention is directed to this very important end.
SUMMARY
The invention relates to chelated copper on solid support enables copper free click conjugation of ligands and drug carriers (nano particles, polymers and other drug carrier system) to nucleic acids (siR A, ssRNA, microRNAs, antisense, aptamer, decoy etc.). The chelated immobilized copper on solid support catalyzes click reaction of completely deprotected oligonucleotides with ligands of choice; and it makes the medium free of floating copper. Conventional click conjugation on completely deprotected
oligonucleotides (RNA or DNA) with functionalized ligands are challenging due to free floating Cu ion which ion-pair/coordinate with the oligonucleotide. The separation of copper completely from the product during purification is extremely difficult after conjugation by using conventional click reagent due to chelation or ion-pairing of free copper with the nucleic acid. In addition, the immobilized copper catalysts described herein are very efficient and recyclable.
DETAILED DESCRIPTION
In one embodiment, the chelated copper on solid support of the present invention features a compound of formula A:

(A)
wherein Y1-Y4 are each independently CR , CR 2, N, NR , O, and S; r, s and t are each indenpendently 1-6; Rioo is independently for each occurrence OH, ORP, or solid support; where RP is independently for each occurrence occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl or optionally substituted heteroaryl; RN is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group; each linker can be the same or different; and provided that at least one Rioo is bound to a solid support.
Representative chelated copper of formula (A) is selected from the group consisting of:

and
In one embodiment, the chelated copper on solid support of the present invention features a compound of formula B:

(B)
wherein Xi-X6 are each independently CRP, CRP 2, N, NRN, O, and S; r, s and t are each indenpendently 1-6; Rioo is independently for each occurrence OH, ORP, or solid support; where RP is independently for each occurrence occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl or optionally substituted heteroaryl; RN is independently
for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group; each linker can be the same or different; and provided that at least one Rioo is bound to a solid support.
In one embodiment, the chelated copper on solid support of the present invention features a compound of formula C:

(C)
wherein X1-X4 are each independently CRP, CRP 2, N, NRN, O, and S; where RP is independently for each occurrence occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl or optionally substituted heteroaryl; RN is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group; and ^ is a solid support.
A representative chelated copper of formula (C) is
 In one embodiment, the chelated copper on solid support of the present invention features a compound of formula D:
In one embodiment, the chelated copper on solid support of the present invention features a compound of formula D:

(D)
wherein is a bond or absent; Y1-Y4 are each independently N, NRN, O, and S; where R100 is independently for each occurrence OH, ORP, or solid support; RN is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group; each linker can be the same or different; each linker can be the same or different; and provided that at least one R100 is bound to a solid support.
The term "solid support" as used herein refers to a flexible or non-flexible support that is suitable for carrying said immobilized chelated copper. Said solid support may be homogenous or inhomogeneous. For example, said solid support may consist of different materials having the same or different properties with respect to flexibility and immobilization, for instance, or said solid support may consist of one material exhibiting a plurality of properties also comprising flexibility and immobilization properties. Such supports are well known in the art and comprise, inter alia, commercially available column materials, polystyrene beads, latex beads, magnetic beads, colloid metal particles, glass and/or silicon chips and surfaces, nitrocellulose strips, membranes, sheets,
duracytes, wells and walls of reaction trays, plastic tubes etc. Examples of well-known carriers include glass, polystyrene, polyvinyl chloride, polypropylene, polyethylene, polycarbonate, dextran, nylon, amyloses, natural and modified celluloses,
polyacrylamides, agaroses, and magnetite. Said solid support may comprise glass-, polypropylene- or silicon-chips, membranes oligonucleotide-conjugated beads or bead arrays. Supports used in solid phase synthesis are typically substantially inert and nonreactive with the solid phase synthesis reagents. Methods of using solid supports in solid phase synthesis are well known in the art and may include, but are not limited to, those described in U.S. Pat. Nos. 4,415,732, 4,458,066; 4,500,707, 4,668,777; 4,973,679, and 5,132,418 issued to Caruthers, and U.S. Pat. No. 4,725,677 and Re. 34,069 issued to Koster, each of which are herein incorporated by reference in their entirety for all purposes.
In one embodiment, the solid support is selected from polystyrenes with different degree of cross linking, glass support (controlled pore glass) or silica support, PEGylated polystyrene, other swellable or non-swellable polymer supports for e.g, NittoPhase from NittoDenko and the like, including liquid polymers such as PEGs and Ionic liquids.
In one embodiment, the chelated copper catalyst is immobilized on soluble or insoluble solid support so that the product is free of any copper catalyst.
In one embodiment, the invention features a process for preparing a ligand conjugated oligonucleotide comprising a step of contacting a nucleoside or
oligonucleotide containing an alkyne moiety with ligand containing an azide moiety in the presence of a solid supported copper catalyst.
In one embodiment, the invention features a process for preparing a ligand conjugated oligonucleotide comprising a step of contacting a nucleoside or
oligonucleotide containing an alkyne moiety with ligand containing an azide moiety in the presence of a solid supported copper catalyst of formula (A), (B), (C), or (D).
In one embodiment, the invention features a process for preparing a ligand conjugated oligonucleotide comprising a step of contacting a nucleoside or
oligonucleotide containing an azide moiety with ligand containing an alkyne moiety in the presence of a solid supported copper catalyst of formula (A), (B), (C), or (D).
In one embodiment, the invention features a process for preparing a ligand conjugated oligonucleotide comprising a step of contacting a nucleoside or
oligonucleotide containing an azide moiety with ligand containing an alkyne moiety in the presence of a solid supported copper catalyst.
In one aspect, the invention features a method for preparing compound having the structure shown in formula (I) by using a chelated copper catalyst of formula (A), (B), (C), or (D);

Formula (I)
wherein:
X is O, S, NRN or CRP 2;
B is independently for each occurrence hydrogen, optionally substituted natural or non-natural nucleobase, optionally substituted triazole or optionally substituted tetrazole; NH-C(0)-0-C(CH2Bi)3, NH-C(0)-NH-C(CH2Bi)3; where Bi is halogen, mesylate, N3, CN, optionally substituted triazole or optionally substituted tetrazole;
R1, R2, R3, R4 and R5 are each independently for each occurrence H, OR6, F, N(RN)2, N3, CN, -J-linker-N3, -J-linker-CN, -J-linker-C≡R8, -J-linker-cycloalkyne, -J- linker-RL, or -J-linker-Q-linker-RL;
J is absent, O, S, NRN, OC(0)NH, NHC(0)0, C(0)NH, NHC(O), NHSO, NHS02, NHS02NH, OC(O), C(0)0, OC(0)0, NHC(0)NH, NHC(S)NH, OC(S)NH, OP(N(RP)2)0, or OP(N(RP)2);
R6 is independently for each occurrence hydrogen, hydroxyl protecting group, optionally substituted alkyl, optionally substituted aryl, optionally substituted cycloalkyl,
optionally substituted aralkyl, optionally substituted alkenyl, optionally substituted heteroaryl, polyethyleneglycol (PEG), a phosphate, a diphosphate, a triphosphate, a phosphonate, a phosphonothioate, a phosphonodithioate, a phosphorothioate, a phosphorothiolate, a phosphorodithioate, a phosphorothiolothionate, a phosphodiester, a phosphotriester, an activated phosphate group, an activated phosphite group, a
phosphoramidite, a solid support, -P(Z1)(Z2)-0-nucleoside, -P(Z1)(Z2)-0-oligonucleotide, -P^XZ^-formula (I), -PCZ^CO-linker-Q-linker-R^-O-nucleoside, P^XO- linker-RL)- O-nucleoside, -P(Z1)(0-linker-N3)-0-nucleoside, P^XO-linker-CNXO-nucleoside, P(Z1)(0-linker-C≡R8)-0-nucleoside, P(Z1)(0-linker-cycloalkyne)-0-nucleoside, - PiZ^CO-linker-Q-linker-R^-O-oligonucleotide, P^XO-linker-R^-O-oligonucleotide, P(Z1)(0-linker-N3)-0-oligonucleotide, -P^XO-linker-CNXO-oligonucleotide, P(Zl)(0- linker-C≡R8)-0-oligonucleotide, P(Z1)(0-linker-cycloalkyne)-0-oligonucleotide, -P(Z1)(- linker-Q-linker-RL)-0-nucleoside, -P^X^inker-Q-R^-O-nucleoside, -P^X-linker-^)- O-nucleoside, P^X^inker-CNXO-nucleoside, P(Z1)(-linker-C≡R8)-0-nucleoside, P(Z1)(-linker-cycloalkyne)-0-nucleoside, -P(Z1)(-linker-Q-linker-RL)-0-oligonucleotide, -P^X-linker-R^-O-oligonucleotide, P(Z1)(-linker-N3)-0-oligonucleotide, -P(Zl)(- linker-CN)-0-oligonucleotide, P(Z1)(-linker-C≡R8)-0-oligonucleotide or P^X-linker- cycloalkyne)-0-oligonucleotide;
RN is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group;
RP is independently for each occurrence occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl or optionally substituted heteroaryl;
absent or independently for each occurrence


RL is hydrogen or a ligand;
R8 is N or CR9;
R9 is H, optionally substituted alkyl or silyl;
Z1 and Z2 are each independently for each occurrence O, S or optionally substituted alkyl;
provided that at least one of R1, R2, R3, R4 and R5 is -J-Linker-Q-Linker-RL or - Linker-Q-RL when B is an unsubstitued natural base.
In one embodiment, the invention features a method for preparing a compound having the structure shown in formula (II) by using a chelated copper catalyst of formula
(A), (B), (C), or (D);

A and B are independently for each occurrence hydrogen, protecting group, optionally substituted aliphatic, optionally substituted aryl, optionally substituted heteroaryl, polyethyleneglycol (PEG), a phosphate, a diphosphate, a triphosphate, a phosphonate, a phosphonothioate, a phosphonodithioate, a phosphorothioate, a phosphorothiolate, a phosphorodithioate, a phosphorothiolothionate, a phosphodiester, a phosphotriester, an activated phosphate group, an activated phosphite group, a
phosphoramidite, a solid support, -P(Z1)(Z2)-0-nucleoside, or -P(Z1)(Z2)-0- oligonucleotide; wherein Z1 and Z2 are each independently for each occurrence O, S or optionally substituted alkyl;
Ji and J2 are independently O, S, NRN, optionally substituted alkyl, OC(0)NH, NHC(0)0, C(0)NH, NHC(O), OC(O), C(0)0, OC(0)0, NHC(0)NH, NHC(S)NH, OC(S)NH, OP(N(RP)2)0, or OP(N(RP)2);
^ carrier J .
is cyclic group or acyclic group; preferably, the cyclic group is selected from pyrrolidinyl, pyrazolinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, piperidinyl, piperazinyl, [l,3]dioxolane, oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl, isothiazolidinyl, quinoxalinyl, pyridazinonyl, tetrahydrofuryl and and decalin; preferably, the acyclic group is selected from serinol backbone or diethanolamine backbone;
Q is independently for each occurrence


Lio and Ln are independently absent or a linker.
In one embodiment, the invention features a method for preparing a compound having the structure shown in formula (III) by using a chelated copper catalyst of formula (A), (B), (C), or (D);
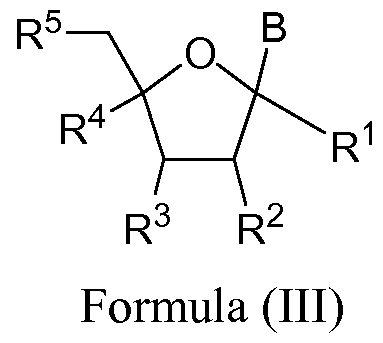
In one embodiment, the R and R of the same compound are connected together a "locked" compound similar to a locked nucleic acid (LNA).
In one embodiment, R1 and R4 are H.
In one embodiment, R1 is -0-Linker-Q-Linker-RL, -OC(0)N(RN)-Linker-Q- Linker-RL or -Linker-Q-Linker-RL, B is H.
In one embodiment, the R2 and R4 of the same compound are connected together to form a "locked" compound similar to a locked nucleic acid (LNA).
In one embodiment, when B is hydrogen, R1 is -0-Linker-Q-Linker-RL, - OC(0)N(R7)-Linker-Q-Linker-RL or -Linker-Q-Linker-RL.
In one embodiment, B is H.
In one embodiment, B is pyrimidine substituted at C5 position.
In one embodiment, R2 is OR6 and R3 is -0-Linker-Q-Linker-RL, -OC(0)N(RN)- Linker-Q-Linker-RL or -Linker-Q-Linker-RL and RL is present.
In one embodiment, R3 is OR6 and R2 is -0-Linker-Q-Linker-RL, -OC(0)N(RN)- Linker-Q-Linker-RL or -Linker-Q-Linker-RL and RL is present.
In one embodiment, R2 is OH.
In one embodiment, R9 is H.
In one embodiment, R5 is -0-Linker-Q-Linker-RL, -OC(0)N(RN)-Linker-Q- Linker-RL or -Linker-Q-Linker-RL and RL is present.
In one embodiment, R5 is -OC(0)NH(CH2)fC≡CR9, and f is 1-20.
In one embodiment, R4 is -OC(0)NH(CH2)fC≡CR9, and f is 1-20.
In one embodiment, R3 is -OC(0)NH(CH2)iC≡CR9, and f is 1-20.
In one embodiment, R2 is -OC(0)NH(CH2)iC≡CR9, and f is 1-20.
In one embodiment, R1 is -OC(0)NH(CH2)iC≡CR9, and f is 1-20.
In one embodiment, B is a nucleobase substituted with -OC(0)NH(CH2)fC≡CH, and f is 1-20.

In one embodiment, B is a nucleobase substituted with

In one embodiment, B is a nucleobase substituted with
 In one embodiment, B is a nucleobase substituted with
In one embodiment, B is a nucleobase substituted with
Linker
Linker-RL.
In one embodiment, R5 is -0-(CH2)fC≡CR9, and f is 1-20.
In one embodiment, R4 is -0-(CH2)tC≡CR9, and f is 1-20.
In one embodiment, R3 is -0-(CH2)fC≡CR9, and f is 1-20.
In one embodiment, R2 is -0-(CH2)tC≡CR9, and f is 1-20.
In one embodiment, R1 is -0-(CH2)iC≡CR9, and f is 1-20.
In one embodiment, B is a nucleobase substituted with -0-(CH2)tC≡CH, and f
In one embodiment, and f is 1-20.
In one embodiment, and f is 1-20.
In one embodiment, and f is 1-20.
In one embodiment, and f is 1-20.
In one embodiment,
 and f is 1-20.
In one embodiment, B is nucleobase substituted with
and f is 1-20.
In one embodiment, B is nucleobase substituted with

In one embodiment, f is 1, 2, 3, 4 or 5. In a preferred embodiment, f is 1.
In one embodiment Q
In one embodiment, Q
In one embodiment, Q
In one embodiment, Q
 is
is
In one embodiment, when click-carrier compound of formula (I) is at the 5'- terminal end of an oligonucleotide, the oligonucleotides is linked at the R5 position of the click-carrier compound.
In one embodiment, when click-carrier compound of formula (I) is at the 3'- terminal end of an oligonucleotide, the oligonucleotides is linked at the R3 or R2 position of the click-carrier compound.
In one embodiment, when the click-carrier compound of formula (I) is not at a terminal position in an oligonucleotide, the R5 position of the compound is linked to the
3'- or 2 '-position of an oligonucleotide on one side and the R2 or R3 position of the compound is linked to the 5 '-position of an oligonucleotide on the other side.
In one embodiment, the two different click-carrier compounds comprise complementary functional groups and are clicked together to each other. In one embodiment, complementarty functional groups are at R5 position of one click-compound and R2 or R3 position of the second compound. In one embodiment, the complementarty functional groups are at R5 position of one click-compound and R5 position of the second compound. In one embodiment, complementarty functional groups are at R2 or R3 position of one click-compound and R2 or R3 position of the second compound.
In some embodiments, B can form part of the click-carrier that connects the linker to the carrier. For example, the -linker-Q-linker-RL can be present at the C2, C6, C7 or C8 position of a purine nucleobase or at the C2, C5 or C6 position of a pyrimidine nucleobase. The linker can be directly attached to the nucleobase or indirectly through one or more intervening groups such as O, N, S, C(O), C(0)0 C(0)NH. In certain embodiments, B, in the click-carrier described above, is uracilyl or a universal base, e.g., an aryl moiety, e.g., phenyl, optionally having additional substituents, e.g., one or more fluoro groups.
In one embodiment, the invention features a method for preparing a compound having the structure shown in formula (IV) by using a chelated copper catalyst of formula (A), (B), (C), or (D);

Formula (IV)
wherein R10 and R20 are independently for each occurrence hydrogen, optionally substituted aliphatic, optionally substituted aryl, or optionally substituted heteroaryl; R1, R2, R3, R4, R5, and X are as defined in the first embodiment.
In one embodiment, the invention features a method for preparing a compound having the structure shown in formula (V) by using a chelated copper catalyst of formula (A), (B), (C), or (D);

Formula (V) wherein Bi is halogen, N3, CN, optionally substituted triazole or optionally substituted tetrazole; R1, R2, R3, R4, and R5 are as defined in the first embodiment.
In one embodiment, the invention features a method for preparing a compound having the structure shown in formula (VI) by using a chelated copper catalyst of formula (A), (B), (C), or (D);

Formula (VI)
wherein E is absent or C(O), C(0)0, C(0)NH, C(S), C(S)NH, SO, S02, or
SO2NH:
R11, R12, R13, R14, R15, R16, R17, and R18 are each independently for each occurrence H, -CH2ORa, or OR
Ra and Rb are each independently for each occurrence hydrogen, hydroxyl protecting group, optionally substituted alkyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted alkenyl, optionally substituted heteroaryl, polyethyleneglycol (PEG), a phosphate, a diphosphate, a triphosphate, a phosphonate, a phosphonothioate, a phosphonodithioate, a
phosphorothioate, a phosphorothiolate, a phosphorodithioate, a phosphorothiolothionate, a phosphodiester, a phosphotriester, an activated phosphate group, an activated phosphite group, a phosphoramidite, a solid support, -P(Z1)(Z2)-0-nucleoside, -P(Z1)(Z2)-0- oligonucleotide, -P^XZ^-formula (I), -P Z^ O-linker-Q-linker-R^-O-nucleoside, - P(Z1)(0-linker-N3)-0-nucleoside, P^XO-linker-CNXO-nucleoside, P^XO-linker- C≡R8)-0-nucleoside, P(Z1)(0-linker-cycloalkyne)-0-nucleoside, -P^XO- linker-RL)-0- oligonucleotide, -PCZ^CO-linker-Q-linker-R^-O-oligonucleotide, -P^XO-linker-R^-O- oligonucleotide, P(Z1)(0-linker-N3)-0-oligonucleotide, -P^XO-linker-CNXO- oligonucleotide, P(Z1)(0-linker-C≡R8)-0-oligonucleotide, P(Z1)(0-linker-cycloalkyne)- O-oligonucleotide, -P^X^inker-Q^nker-R^-O-nucleoside, P^X-linker- RL)-0- nucleoside, -P(Z1)(-linker-N3)-0-nucleoside, P^X^inker-CNXO-nucleoside, Ρ(Ζ!)(- linker-C≡R8)-0-nucleoside, P(Z1)(-linker-cycloalkyne)-0-nucleoside, -P(Z1)(-linker-Q- linker-RL)-0-oligonucleotide, (Z^-linker- RL)-0-oligonucleotide, P(Z1)(-linker-N3)-0- oligonucleotide, -P^X^inker-CNXO-oligonucleotide, P(Z1)(-linker-C≡R8)-0- oligonucleotide or P(Z1)(-linker-cycloalkyne)-0-oligonucleotide;
R30 is independently for each occurrence -linker-Q-linker-RL, -linker-RL or R31;
absent or independently for each occurrence


RL is hydrogen or a ligand;
R5 is N or CR9;
R9 is H, optionally substituted alkyl or silyl;
R31 is -C(0)CH(N(R32)2)(CH2)hN(R32)2;
R32 is independently for each occurrence H, -linker-Q-linker-RL, -linker- RL or f and h are independently for each occurrence 1 -20; and
Z1 and Z2 are each independently for each occurrence O, S or optionally substituted alkyl.
For the pyrroline-based click-carriers, R11 is -CH2ORa and R3 is ORb; or R11 is - CH2ORa and R9 is ORb; or R11 is -CH2ORa and R17 is ORb; or R13 is -CH2ORa and R11 is ORb; or R13 is -CH2ORa and R15 is ORb; or R13 is -CH2ORa and R17 is ORb. In certain embodiments, CH2ORa and ORb may be geminally substituted. For the 4- hydroxyproline-based carriers, R11 is -CH2ORa and R17 is ORb. The pyrroline- and 4- hydroxyproline-based compounds may therefore contain linkages (e.g., carbon-carbon bonds) wherein bond rotation is restricted about that particular linkage, e.g. restriction resulting from the presence of a ring. Thus, CH2ORa and ORb may be cis or trans with respect to one another in any of the pairings delineated above Accordingly, all cis/trans isomers are expressly included. The compounds may also contain one or more asymmetric centers and thus occur as racemates and racemic mixtures, single
enantiomers, individual diastereomers and diastereomeric mixtures. All such isomeric forms of the compounds are expressly included (e.g., the centers bearing CH2ORa and ORb can both have the R configuration; or both have the S configuration; or one center can have the R configuration and the other center can have the S configuration and vice versa).
In one embodiment, R11 is CH2ORa and R9 is ORb.
In one embodiment, Rb is a solid support.
In one embodiment, R30 is -C(0)(CH2)fNHC(0)(CH2)gC≡CR9; wherein f and g are independently 1 - 20.
wherein f is 1-20. L; wherein f is 1-20.
erein f is 1-20.

In one em o ment, R is

In one em o iment, is
In one preferred embodiment, R30 is R 1.
In one preferred embodiment, R31 is -C(0)CH(N(R32)2)(CH2)4N(R32)2 and at least one R32 is -C(0)(CH2)fC≡R8 or -linker-Q-linker-RL and RL is present.
In one preferred embodiment, R 1 is -C(0)CH(N(R 2)2)(CH2)4NH2 and at least one R32 is -C(0)(CH2)fC≡R8 or -linker-Q-linker-RL and RL L i .·s . present.
In one embodiment, R32 is -C(0)(CH2)iC≡R8.
In one embodiment, R is -C(0)(CH2)3C≡H.
In one embodiment, R31 is -C(0)CH(NH2)(CH2)4NH2.
In one embodiment features acyclic sugar replacement -based compounds, e.g., replacement based click-carrier compounds, are also referred to herein as ribose
replacement compound subunit (RRMS) compound compounds. Preferred acyclic carriers can have the structure shown in formula (III) or formula (IV) below.
In one aspect, the invention features a method for preparing a compound having the structure shown in formula (VII) by using a chelated copper catalyst of formula (A), (B), (C), or (D);

Formula (VII) wherein:
W is absent, O, S and N(RN) , where RN is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group;
E is absent or C(O), C(0)0, C(0)NH, C(S), C(S)NH, SO, S02, or S02NH;
Ra and Rb are each independently for each occurrence hydrogen, hydroxyl protecting group, optionally substituted alkyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted alkenyl, optionally substituted heteroaryl, polyethyleneglycol (PEG), a phosphate, a diphosphate, a triphosphate, a phosphonate, a phosphonothioate, a phosphonodithioate, a
phosphorothioate, a phosphorothiolate, a phosphorodithioate, a phosphorothiolothionate, a phosphodiester, a phosphotriester, an activated phosphate group, an activated phosphite group, a phosphoramidite, a solid support, -P(Z1)(Z2)-0-nucleoside, -P(Z1)(Z2)-0- oligonucleotide, -P^XZ^-formula (I), ^(Z^^-linker-Q-linker-R^-O-nucleoside, - P(Z1)(0-linker-N3)-0-nucleoside, P(Z1)(0-linker-CN)-0-nucleoside, P(Z1)(0-linker- C≡R8)-0-nucleoside, P(Z1)(0-linker-cycloalkyne)-0-nucleoside, -P(Z!)(0- linker-RL)-0- oligonucleotide, -P(Z1)(0-linker-Q-linker-RL)-0-oligonucleotide, -P(Z1)(0-linker-RL)-0-
oligonucleotide, P(Z1)(0-linker-N3)-0-oligonucleotide, -P^XO-linker-CNVO- oligonucleotide, P(Z1)(0-linker-C≡R8)-0-oligonucleotide, P(Z1)(0-linker-cycloalkyne)- O-oligonucleotide, -P Z^ -linker-Q-linker-R^-O-nucleoside, P^X-linker- RL)-0- nucleoside, -P(Z1)(-linker-N3)-0-nucleoside, P^X^inker-CNVO-nucleoside, Ρ(Ζ!)(- linker-C≡R8)-0-nucleoside, P(Z1)(-linker-cycloalkyne)-0-nucleoside, -P(Z1)(-linker-Q- linker-RL)-0-oligonucleotide, (Z^-linker- RL)-0-oligonucleotide, P(Z1)(-linker-N3)-0- oligonucleotide, -P^X^inker-CNXO-oligonucleotide, P(Z1)(-linker-C≡R8)-0- oligonucleotide or P(Z1)(-linker-cycloalkyne)-0-oligonucleotide;
R30 is independently for each occurrence -linker-Q-linker-RL, -linker-RL or R31;
absent or independently for each occurrence


RL is hydrogen or a ligand;
R8 is N or CR9
R9 is H, optionally substituted alkyl or silyl;
R31 is -C(0)CH(N(R32)2)(CH2)hN(R32)2;
R32 is independently for each occurrence H, -linker-Q-linker-RL or R31;
f and h are independently for each occurrence 1 -20;
Z1 and Z2 are each independently for each occurrence O, S or optionally substituted alkyl; and
r, s and t are each independently for each occurrence 0, 1, 2 or 3.
When r and s are different, then the tertiary carbon can be either the R or S configuration. In preferred embodiments, x and y are one and z is zero (e.g. carrier is
based on serinol). The acyclic carriers can optionally be substituted, e.g. with hydroxy, alkoxy, perhaloalky.
Other carrier compounds amenable to the invention are described in copending applications USSN: 10/916,185, filed August 10, 2004; USSN: 10/946,873, filed
September 21, 2004; USSN: 10/985,426, filed November 9, 2004; USSN: 10/833,934, filed August 3, 2007; USSN: 11/115,989 filed April 27, 2005 and USSN: 11/119,533, filed April 29, 2005, which are incorporated by reference in their entireties for all purposes.
LINKERS
The term "linker" means an organic moiety that connects two parts of a compound. Linkers typically comprise a direct bond or an atom such as oxygen or sulfur, a unit such as NR1, C(O), C(0)NH, SO, S02, S02NH or a chain of atoms, such as substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, arylalkyl, arylalkenyl, arylalkynyl, heteroarylalkyl,
heteroarylalkenyl, heteroarylalkynyl, heterocyclylalkyl, heterocyclylalkenyl,
heterocyclylalkynyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, cycloalkenyl,
alkylarylalkyl, alkylarylalkenyl, alkylarylalkynyl, alkenylarylalkyl, alkenylarylalkenyl, alkenylarylalkynyl, alkynylarylalkyl, alkynylarylalkenyl, alkynylarylalkynyl,
alkylheteroarylalkyl, alkylheteroarylalkenyl, alkylheteroarylalkynyl,
alkenylheteroarylalkyl, alkenylheteroarylalkenyl, alkenylheteroarylalkynyl,
alkynylheteroarylalkyl, alkynylheteroarylalkenyl, alkynylheteroarylalkynyl,
alkylheterocyclylalkyl, alkylheterocyclylalkenyl, alkylhererocyclylalkynyl,
alkenylheterocyclylalkyl, alkenylheterocyclylalkenyl, alkenylheterocyclylalkynyl, alkynylheterocyclylalkyl, alkynylheterocyclylalkenyl, alkynylheterocyclylalkynyl, alkylaryl, alkenylaryl, alkynylaryl, alkylheteroaryl, alkenylheteroaryl, alkynylhereroaryl, which one or more methylenes can be interrupted or terminated by O, S, S(O), S02, N(R1)2, C(O), substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocyclic; where R1 is hydrogen, acyl, aliphatic or substituted aliphatic. It is understood that a linker can cleavable or non-cleavable.
In one embodiment, the linker is represented by structure

wherein:
P, R and T are each independently for each occurrence absent, CO, NH, O, S, OC(O), NHC(O), CH2, CH2NH, CH20; NHC a)C(0), -C(0)-CH(Ra)-NH-, -C(O)-

Qi is independently for each occurrence absent, -(CH2)n-, -C(R100)(R200)(CH2)n-, - (CH2)nC(R100)(R200)-, -(CH2CH20)mCH2CH2-, or -(CH2CH20)mCH2CH2NH-;
Ra is H or an amino acid side chain;
R100 and R200 are each independently for each occurrence H, C¾, OH, SH or N(RX)2;
Rx is independently for each occurrence H, methyl, ethyl, propyl, isopropyl, butyl or benzyl;
q is independently for each occurrence 0-20;
n is independently for each occurrence 1-20; and
m is independently for each occurrence 0-50.
In one embodiment, the linker has the structure -[(P-Qi-R)q-X-(P'-Qi '-R')q']q"-T, wherein:
P, R, T, P', R' and are each independently for each occurrence absent, CO, NH, O, S, OC(O), NHC(O), CH2, CH2NH, CH20; NHCH(Ra)C(0), -C(0)-CH(Ra)-NH-,
-C(0)-(optionally substituted alkyl)-NH-, CH=N-0 ,

S-S, acetal, ketal,

Qi and Qi ' are each independently for each occurrence absent, -(CH2)n-, - C(R100)(R200)(CH2)n-, -(CH2)nC(R100)(R200)-, -(CH2CH20)mCH2CH2-, or -
(CH2CH20)mCH2CH2NH-;
X is a cleavable linker;
Ra is H or an amino acid side chain;
R100 and R200 are each independently for each occurrence H, C¾, OH, SH or
N(RA)2;
R is independently for each occurrence H, methyl, ethyl, propyl, isopropyl, butyl or benzyl;
q, q' and q' are each independently for each occurrence 0-20;
n is independently for each occurrence 1-20; and
m is independently for each occurrence 0-50.
In one embodiment, the linker comprises at least one cleavable linker. Cleavable Linkers
A cleavable linker is one which is sufficiently stable outside the cell, but which upon entry into a target cell is cleaved to release the two parts the linker is holding together. In a preferred embodiment, the cleavable linker is cleaved at least 10 times or more, preferably at least 100 times faster in the target cell or under a first reference condition (which can, e.g., be selected to mimic or represent intracellular conditions) than in the blood of a subject, or under a second reference condition (which can, e.g., be selected to mimic or represent conditions found in the blood or serum).
Cleavable linkers are susceptible to cleavage agents, e.g., pH, redox potential or the presence of degradative molecules. Generally, cleavage agents are more prevalent or found at higher levels or activities inside cells than in serum or blood. Examples of such
degradative agents include: redox agents which are selected for particular substrates or which have no substrate specificity, including, e.g., oxidative or reductive enzymes or reductive agents such as mercaptans, present in cells, that can degrade a redox cleavable linker by reduction; esterases; endosomes or agents that can create an acidic environment, e.g., those that result in a pH of five or lower; enzymes that can hydrolyze or degrade an acid cleavable linker by acting as a general acid, peptidases (which can be substrate specific), and phosphatases.
A cleavable linker, such as a disulfide bond can be susceptible to pH. The pH of human serum is 7.4, while the average intracellular pH is slightly lower, ranging from about 7.1-7.3. Endosomes have a more acidic pH, in the range of 5.5-6.0, and lysosomes have an even more acidic pH at around 5.0. Some sapcers will have a linker that is cleaved at a preferred pH, thereby releasing the iRNA agent from the carrier oligomer inside the cell, or into the desired compartment of the cell.
A spacer can include a linker that is cleavable by a particular enzyme. The type of linker incorporated into a spacer can depend on the cell to be targeted by the iRNA agent. For example, an iRNA agent that targets an mRNA in liver cells can be linked to the carrier oligomer through a spacer that includes an ester group. Liver cells are rich in esterases, and therefore the tether will be cleaved more efficiently in liver cells than in cell types that are not esterase-rich. Cleavage of the sapcer releases the iRNA agent from the carrier oligomer, thereby potentially enhancing silencing activity of the iRNA agent. Other cell-types rich in esterases include cells of the lung, renal cortex, and testis.
Spacers that contain peptide bonds can be used when the iRNA agents are targeting cell types rich in peptidases, such as liver cells and synoviocytes. For example, an iRNA agent targeted to synoviocytes, such as for the treatment of an inflammatory disease (e.g., rheumatoid arthritis), can be linked to a carrier oligomer through spacer that comprises a peptide bond.
In general, the suitability of a candidate cleavable linker can be evaluated by testing the ability of a degradative agent (or condition) to cleave the candidate linker. It will also be desirable to also test the candidate cleavable linker for the ability to resist cleavage in the blood or when in contact with other non-target tissue, e.g., tissue the
iR A agent would be exposed to when administered to a subject. Thus one can determine the relative susceptibility to cleavage between a first and a second condition, where the first is selected to be indicative of cleavage in a target cell and the second is selected to be indicative of cleavage in other tissues or biological fluids, e.g., blood or serum. The evaluations can be carried out in cell free systems, in cells, in cell culture, in organ or tissue culture, or in whole animals. It may be useful to make initial evaluations in cell- free or culture conditions and to confirm by further evaluations in whole animals. In preferred embodiments, useful candidate compounds are cleaved at least 2, 4, 10 or 100 times faster in the cell (or under in vitro conditions selected to mimic intracellular conditions) as compared to blood or serum (or under in vitro conditions selected to mimic extracellular conditions).
Redox cleavable linkers
One class of cleavable linkers are redox cleavable linkers that are cleaved upon reduction or oxidation. An example of reductively cleavable linker is a disulphide linker (-S-S-). To determine if a candidate cleavable linker is a suitable "reductively cleavable linker," or for example is suitable for use with a particular iRNA moiety and particular targeting agent one can look to methods described herein. For example, a candidate can be evaluated by incubation with dithiothreitol (DTT), or other reducing agent using reagents know in the art, which mimic the rate of cleavage which would be observed in a cell, e.g., a target cell. The candidates can also be evaluated under conditions which are selected to mimic
blood or serum conditions. In a preferred embodiment, candidate compounds are cleaved by at most 10% in the blood. In preferred embodiments, useful candidate compounds are degraded at least 2, 4, 10 or 100 times faster in the cell (or under in vitro conditions selected to mimic intracellular conditions) as compared to blood (or under in vitro conditions selected to mimic extracellular conditions). The rate of cleavage of candidate compounds can be determined using standard enzyme kinetics assays under conditions chosen to mimic intracellular media and compared to conditions chosen to mimic extracellular media.
Phosphate-based cleavable linkers
Phosphate-based linkers are cleaved by agents that degrade or hydro lyze the phosphate group. An example of an agent that cleaves phosphate groups in cells are enzymes such as phosphatases in cells. Examples of phosphate-based linkers are -O- P(0)(ORk)-0-, -0-P(S)(ORk)-0-, -0-P(S)(SRk)-0-, -S-P(0)(ORk)-0-, -0-P(0)(ORk)- S-, -S-P(0)(ORk)-S-, -0-P(S)(ORk)-S-, -S-P(S)(ORk)-0-, -0-P(0)(Rk)-0-, -O- P(S)(Rk)-0-, -S-P(0)(Rk)-0-, -S-P(S)(Rk)-0-, -S-P(0)(Rk)-S-, -0-P(S)( Rk)-S-.
Preferred embodiments are -0-P(0)(OH)-0-, -0-P(S)(OH)-0-, -0-P(S)(SH)-0-, -S- P(0)(OH)-0-, -0-P(0)(OH)-S-, -S-P(0)(OH)-S-, -0-P(S)(OH)-S-, -S-P(S)(OH)-0-, -O- Ρ(0)(Η)-0-, -0-P(S)(H)-0-, -S-P(0)(H)-0-, -S-P(S)(H)-0-, -S-P(0)(H)-S-, -0-P(S)(H)- S-. A preferred embodiment is -0-P(0)(OH)-0-. These candidates can be evaluated using methods analogous to those described above.
Acid cleavable linkers
Acid cleavable linkers are linkers that are cleaved under acidic conditions. In preferred embodiments acid cleavable linkers are cleaved in an acidic environment with a pH of about 6.5 or lower (e.g., about 6.0, 5.5, 5.0, or lower), or by agents such as enzymes that can act as a general acid. In a cell, specific low pH organelles, such as endosomes and lysosomes can provide a cleaving environment for acid cleavable linkers. Examples of acid cleavable linkers include but are not limited to hydrazones, esters, and esters of amino acids. Acid cleavable groups can have the general formula -C=NN- C(0)0, or -OC(O). A preferred embodiment is when the carbon attached to the oxygen of the ester (the alkoxy group) is an aryl group, substituted alkyl group, or tertiary alkyl group such as dimethyl pentyl or t-butyl. These candidates can be evaluated using methods analogous to those described above.
Ester-based linkers
Ester-based linkers are cleaved by enzymes such as esterases and amidases in cells. Examples of ester-based cleavable linkers include but are not limited to esters of
alkylene, alkenylene and alkynylene groups. Ester cleavable linkers have the general formula -C(0)0-, or -OC(O)-. These candidates can be evaluated using methods analogous to those described above.
Peptide-based cleaving groups
Peptide-based linkers are cleaved by enzymes such as peptidases and proteases in cells. Peptide-based cleavable linkers are peptide bonds formed between amino acids to yield oligopeptides (e.g., dipeptides, tripeptides etc.) and polypeptides. Peptide-based cleavable groups do not include the amide group (-C(O)NH-). The amide group can be formed between any alkylene, alkenylene or alkynelene. A peptide bond is a special type of amide bond formed between amino acids to yield peptides and proteins. The peptide based cleavage group is generally limited to the peptide bond (i.e., the amide bond) formed between amino acids yielding peptides and proteins and does not include the entire amide functional group. Peptide cleavable linkers have the general formula - NHCHR1C(0)NHCHR2C(0)-, where R1 and R2 are the R groups of the two adjacent amino acids. These candidates can be evaluated using methods analogous to those described above.
"Click" reaction
The synthesis methods of the present invention utilize click chemistry to conjugate the ligand to the click-carrier compound. Click chemistry techniques are described, for example, in the following references, which are incorporated herein by reference in their entirety:
Kolb, H. C; Finn, M. G. and Sharpless, K. B. Angew. Chem., Int. Ed. (2001) 40: 2004-2021.
Kolb, H. C. and Shrapless, K. B. Drug Disc. Today (2003) 8: 112-1137. Rostovtsev, V. V.; Green L. G.; Fokin, V. V. and Shrapless, K.B. Angew. Chem., Int. Ed. (2002) 41 : 2596-2599.
Tornoe, C. W.; Christensen, C. and Meldal, M. J. Org. Chem. (2002) 67: 3057-3064.
Wang, Q. et al, J. Am. Chem. Soc. (2003) 125: 3192-3193..
Lee, .V.et al, J. Am. Chem. Soc. (2003) 125: 9588-9589.
Lewis, W.G. et al, Angew. Chem., Int. Ed. (2002) 41 : 1053-1057.
Manetsch, R. et al, J. Am. Chem. Soc. (2004) 126: 12809-12818.
Mocharla, V.P. et al, Angew. Chem., Int. Ed. (2005) 44: 116-120.
Although other click chemistry functional groups can be utilized, such as those described in the above references, the use of cyclo addition reactions is preferred, particularly the reaction of azides with alkynyl groups. In the presence of Cu(I) salts, terminal alkynes and azides undergo 1,3 -dipolar cycloaddition forming 1 ,4-disubsstituted 1,2,3-triazoles. In the presence of Ru(II) salts (e.g. Cu*RuCl(PPh3)2), terminal alkynes and azides under go 1,3-dipolar cycloaddition forming 1,5-disubstituted 1,2,3-triazoles (Folkin, V.V. et al, Org. Lett. (2005) 127: 15998-15999). Alternatively, a 1,5- disubstituted 1,2,3-triazole can be formed using azide and alkynyl reagents (Kraniski, A.; Fokin, V.V. and Sharpless, K.B. Org. Lett. (2004) 6: 1237-1240. Hetero-Diels-Alder reactions or 1,3-dipolar cycloaddition reaction could also be used (see for example Padwa, A. \,3-Dipolar Cycloaddition Chemistry: Volume I, John Wiley, New York, (1984) 1-176; Jorgensen, K. A. Angew. Chem., Int. Ed. (2000) 39: 3558-3588 and Tietze, L. F. and Kettschau, G. Top. Curr. Chem. (1997) 189: 1-120)
The choice of azides and alkynes as coupling partners is particularly
advantageous as they are essentially non-reactive towards each other (in the absence of copper) and are extremely tolerant of other functional groups and reaction conditions. This chemical compatibility helps ensure that many different types of azides and alkynes may be coupled with each other with a minimal amount of side reactions.
The required copper(I) species are added directly as cuprous salts, for example Cul, CuOTf.C6H6 or [Cu(CH3CN)4][PF6], usually with stabilizing ligands (see for example Tornoe, C. W.; Christensen, C. and Meldal, M. J. Org. Chem. (2002) 67: 3057- 3064; Chan, T. R. et al, Org. Lett. (2004) 6: 2853-2855; Lewis, W.G. et al, J. Am.
Chem. Soc. (2004) 126: 9152-9153; Mantovani, G. et al, Chem. Comm. (2005) 2089- 2091; Diez-Gonzalez, S. et al, Chem. Eur. J. (2006) 12: 7558-7564 and Candelon, N. et
al, Chem. Comm. (2008) 741-743), or more often generated from copper (II) salts with reducing agents (Rostovtsev, V. V. et al, Angew. Chem. (2002) 114: 2708— 2711 and Angew. Chem., Int. Ed. (2002) 41 : 2596-2599). Metallic copper (for example see Himo, F. et al, J. Am. Chem. Soc. (2005) 127: 210-216) or clusters (for example see Pachon, L. D. et al, Adv. Synth. Catal (2005) 347: 811-815 and Molteni, G. et al, New J. Chem (2006) 30: 1137- 1139) can also be employed. Chassaing et al, recently reported copper(I) zeolites as catalysts for the azide-alkyne cycloaddition (Chem. Eur. J. (2008) 14: 6713-6721). As copper(I) salts are prone to redox process, nitrogen- or phosphorous- based ligands must be added to protect and stabilize the active copper catalyst during the cycloaddition reaction.
The reaction is extremely straightforward. The azide and alkyne are usually mixed together in water and a co-solvent such as tert-butanol, THF, DMSO, toluene or DMF. The water/co-solvent are usually in a 1 : 1 to 1 :9 ratio. The reactions are usually run overnight although mild heating shortens reaction times (Sharpless, W. D.; Wu, P.; Hansen, T. V.; and Li, J.G. J. Chem. Ed. (2005) 82: 1833). Aqueous systems can also use copper(I) species directly such that a reducing agent is not needed. The reactions conditions then usually require acetonitrile as a co-solvent (although not essential (Chan, T. Pv.; Hilgraf, R.; Shrapless, K. B. and Fokin, V.V. Org. Lett. (2004) 6: 2853)) and a nitrogen base, such as triethylamine, 2,6-lutidine, pyridine and diisopropylamine. In this case copper(I) species is supplied as Cul, CuOTf.C6H6 or [Cu(CH3CN)4][PF6]
(Rostoctsev, V. V.; Green L. G.; Fokin, V. V. and Shrapless, K.B. Angew. Chem., Int. Ed. (2002) 41 : 2596-2599).
Although the water-based methods are attractive for many applications, solvent based azide-alkyne cycloaddition methods have found utility in situations when solubility and/or other problems arise, for example see:
Malkoch, M. et al, Macromolecules (2005) 38: 3663.
Gujadhur, R.; Venkataraman, D. and J. T. Kintigh. J.T. Tet. Lett. (2001)
42: 4791.
Laurent, B.A. and Grayson, S.M. J. Am. Chem. Soc. (2006) 128: 4238. Opsteen, J. A.; van Hest, J. C. M. Chem. Commun. (2005) 57.
Tsarevsky, N. V.; Sumerlin, B. S. and Matyjaszewski, K. Macromolecules (2005) 38: 3558.
Johnson, J. A. et al, J. Am. Chem. Soc. (2006) 128: 6564.
Sumerlin, B. S. et al, Macromolecules (2005) 38: 7540.
Gao, H. F. and Matyjaszewski, K. Macromolecules (2006) 39: 4960.
Gao, H. et al, Macromolecules (2005) 38: 8979.
Vogt, A. P. and Sumerlin, B.S. Macromolecules (2006) 39: 5286.
Lutz, J. F.; Borner, H. G. and Weichenhan, K. Macromol. Rapid Comm. (2005) 26: 514.
Mantovani, G.; Ladmiral, V.; Tao, L. and Haddleton, D. M. Chem. Comm. (2005) 2089.
The click reaction may be performed thermally. In one aspect, the click reaction is performed at slightly elevated temperatures between 25°C and 100°C. In one aspect, the reaction may be performed between 25°C and 75°C, or between 25°C and 65°C, or between 25°C and 50°C. In one embodiment, the reaction is performed at room temperature. In another aspect, the click reaction may also be performed using a microwave oven. The microwave assisted click reaction may be carried out in the presence or absence of copper.
In one aspect, the invention provides a method for coupling a click-carrier compound to a ligand through a click reaction. In a preferred embodiment, the click reaction is a cycloaddition reaction of azide with alkynyl group and catalyzed by copper. In one embodiment the equal molar amount of alkyne and azide are mixed together in DCM/MeOH (10: 1 to 1 : 1 ratio v/v) and 0.05-0.5 mol% each of [Cu(CH3CN)4][PF6] and copper are added the reaction. In one embodiment DCM/MeOH ratio is 5 : 1 to 1 : 1. In a preferred embodiment, DCM/MeOH ratio is 4: 1. In one embodiment, equal molar amounts of [Cu(CH3CN)4][PF6] and copper are added. In a preferred embodiment, 0.05- 0.25mol% each of [Cu(CH3CN)4][PF6] and copper are added to the reaction. In a more preferred embodiment, 0.05 mol%, 0.1 mol%, 0.15 mol%, 0.2 mol% or 0.25 mol% each of [Cu(CH3CN)4][PF6] and copper are added to the reaction.
The term "prodrug" indicates a therapeutic agent that is prepared in an inactive form that is converted to an active form (i.e., drug) within the body or cells thereof by the action of endogenous enzymes or other chemicals and/or conditions. In particular, prodrug versions of the oligonucleotides of the invention are prepared as SATE [(S- acetyl-2-thioethyl)phosphate] derivatives according to the methods disclosed in WO 93/24510 to Gosselin et al, published Dec. 9, 1993 or in WO 94/26764 and U.S. Pat. No. 5,770,713 to Imbach et al.
The term "pharmaceutically acceptable salts" refers to physiologically and pharmaceutically acceptable salts of the oligomeric compounds of the invention: i.e., salts that retain the desired biological activity of the parent compound and do not impart undesired toxico logical effects thereto. For oligonucleotides, preferred examples of pharmaceutically acceptable salts and their uses are further described in U.S. Pat. No. 6,287,860, which is incorporated herein in its entirety.
Ligands
A wide variety of entities can be coupled to the oligonucleotide, e.g. the iRNA agent, using the "click" reaction. Preferred entities can be coupled to the oligonucleotide at various places, for example, 3 '-end, 5 '-end, and/or at internal positions.
In preferred embodiments, the ligand is attached to the iRNA agent via an intervening linker. The ligand may be present on a compound when said compound is incorporated into the growing strand. In some embodiments, the ligand may be incorporated via coupling to a "precursor" compound after said "precursor" compound has been incorporated into the growing strand. For example, a compound having, e.g., an azide terminated linker (i.e., having no associated ligand), e.g., -linker-N3 may be incorporated into a growing sense or antisense strand. In a subsequent operation, i.e., after incorporation of the precursor compound into the strand, a ligand having an alkyne, e.g. terminal acetylene, e.g. ligand-C≡CH, can subsequently be attached to the precursor compound by the "click" reaction. Alternatively, the compound linker comprises an alkyne, e.g. terminal acetylene; and the ligand comprises an azide functionality for the "click" reaction to take place. The azide or alkyne functionalities can be incorporated
into the ligand by methods known in the art. For example, moieties carrying azide or alkyne functionalities can be linked to the ligand or a functional group on the ligand can be transformed into an azide or alkyne. In one embodiment, the conjugation of the ligand to the precursor compound takes place while the oligonucleotide is still attached to the solid support. In one embodiment, the precursor carrying oligonucleotide is first deprotected but not purified before the ligand conjugation takes place. In one
embodiment, the precursor compound carrying oligonucleotide is first deprotected and purified before the ligand conjugation takes place. In certain embodiments, the "click" reaction is carried out under microwave.
In preferred embodiments, a ligand alters the distribution, targeting or lifetime of an iR A agent into which it is incorporated. In preferred embodiments a ligand provides an enhanced affinity for a selected target, e.g., molecule, cell or cell type, compartment, e.g., a cellular or organ compartment, tissue, organ or region of the body, as, e.g., compared to a species absent such a ligand. Preferred ligands will not take part in duplex pairing in a duplexed nucleic acid.
Preferred ligands can have endosomolytic properties. The endosomolytic ligands promote the lysis of the endosome and/or transport of the composition of the invention, or its components, from the endosome to the cytoplasm of the cell. The endosomolytic ligand may be a polyanionic peptide or peptidomimetic which shows pH-dependent membrane activity and fusogenicity. In certain embodiments, the endosomolytic ligand assumes its active conformation at endosomal pH. The "active" conformation is that conformation in which the endosomolytic ligand promotes lysis of the endosome and/or transport of the composition of the invention, or its components, from the endosome to the cytoplasm of the cell. Exemplary endosomolytic ligands include the GALA peptide (Subbarao et al, Biochemistry, 1987, 26: 2964-2972), the EALA peptide (Vogel et al, J. Am. Chem. Soc, 1996, 118: 1581-1586), and their derivatives (Turk et al, Biochem. Biophys. Acta, 2002, 1559: 56-68). In certain embodiments, the endosomolytic component may contain a chemical group (e.g., an amino acid) which will undergo a change in charge or protonation in response to a change in pH. The endosomolytic
component may be linear or branched. Exemplary primary sequences of peptide based endosomo lytic ligands are shown in table 1.
Table 1: List of peptides with endosomo lytic activity.

n, norleucine
References
1. Subbarao et al. (1987) Biochemistry 26: 2964-2972.
2. Vogel, et al (1996) J. Am. Chem. Soc. 118: 1581-1586
3. Turk, et al. (2002) Biochim. Biophys. Acta 1559: 56-68.
4. Plank, et al. (1994) J. Biol. Chem. 269: 12918-12924.
5. Mastrobattista, et al. (2002) J. Biol. Chem. 277:27135-43.
6. Oberhauser, et al. (1995) Deliv. Strategies Antisense Oligonucleotide Ther. 247-66.
Preferred ligands can improve transport, hybridization, and specificity properties and may also improve nuclease resistance of the resultant natural or modified
oligoribonucleotide, or a polymeric molecule comprising any combination of compounds described herein and/or natural or modified ribonucleotides.
Ligands in general can include therapeutic modifiers, e.g., for enhancing uptake; diagnostic compounds or reporter groups e.g., for monitoring distribution; cross-linking
agents; and nuclease-resistance conferring moieties. General examples include lipids, steroids, vitamins, sugars, proteins, peptides, polyamines, and peptide mimics.
Ligands can include a naturally occurring substance, such as a protein (e.g., human serum albumin (HSA), low-density lipoprotein (LDL), high-density lipoprotein (HDL), or globulin); an carbohydrate (e.g., a dextran, pullulan, chitin, chitosan, inulin, cyclodextrin or hyaluronic acid); or a lipid. The ligand may also be a recombinant or synthetic molecule, such as a synthetic polymer, e.g., a synthetic polyamino acid, an oligonucleotide (e.g. an aptamer). Examples of polyamino acids include polyamino acid is a polylysine (PLL), poly L-aspartic acid, poly L-glutamic acid, styrene-maleic acid anhydride copolymer, poly(L-lactide-co-glycolied) copolymer, divinyl ether-maleic anhydride copolymer, N-(2-hydroxypropyl)methacrylamide copolymer (HMPA), polyethylene glycol (PEG), polyvinyl alcohol (PVA), polyurethane, poly(2-ethylacryllic acid), N-isopropylacrylamide polymers, or polyphosphazine. Example of polyamines include: polyethylenimine, polylysine (PLL), spermine, spermidine, polyamine, pseudopeptide-polyamine, peptidomimetic polyamine, dendrimer polyamine, arginine, amidine, protamine, cationic lipid, cationic porphyrin, quaternary salt of a polyamine, or an alpha helical peptide.
Ligands can also include targeting groups, e.g., a cell or tissue targeting agent, e.g., a lectin, glycoprotein, lipid or protein, e.g., an antibody, that binds to a specified cell type such as a kidney cell. A targeting group can be a thyrotropin, melanotropin, lectin, glycoprotein, surfactant protein A, Mucin carbohydrate, multivalent lactose, multivalent galactose, N-acetyl-galactosamine, N-acetyl-gulucosamine multivalent mannose, multivalent fucose, glycosylated polyaminoacids, multivalent galactose, transferrin, bisphosphonate, polyglutamate, polyaspartate, a lipid, cholesterol, a steroid, bile acid, folate, vitamin B12, biotin, an RGD peptide, an RGD peptide mimetic or an aptamer. Table 2 shows some examples of targeting ligands and their associated receptors.
Other examples of ligands include dyes, intercalating agents (e.g. acridines), cross-linkers (e.g. psoralene, mitomycin C), porphyrins (TPPC4, texaphyrin, Sapphyrin), polycyclic aromatic hydrocarbons (e.g., phenazine, dihydrophenazine), artificial endonucleases (e.g. EDTA), lipophilic molecules, e.g, cholesterol, cholic acid,
adamantane acetic acid, 1-pyrene butyric acid, dihydrotestosterone, 1 ,3-Bis- 0(hexadecyl)glycerol, geranyloxyhexyl group, hexadecylglycerol, borneol, menthol, 1 ,3- propanediol, heptadecyl group, palmitic acid, myristic acid,03-(oleoyl)lithocholic acid, 03-(oleoyl)cholenic acid, dimethoxytrityl, or phenoxazine)and peptide conjugates (e.g., antennapedia peptide, Tat peptide), alkylating agents, phosphate, amino, mercapto, PEG (e.g., PEG-40K), MPEG, [MPEG]2, polyamino, alkyl, substituted alkyl, radiolabeled markers, enzymes, haptens (e.g. biotin), transport/absorption facilitators (e.g., aspirin, vitamin E, folic acid), synthetic ribonucleases (e.g., imidazole, bisimidazole, histamine, imidazole clusters, acridine-imidazole conjugates, Eu3+ complexes of
tetraazamacrocycles), dinitrophenyl, HRP, or AP.
Table 2: Liver targeting Ligands and their associated receptors
Liver Cells Ligand Receptor
1) Parenchymal Cell Galactose ASGP-R
(PC) (Hepatocytes) (Asiologlycoprotein receptor)
Gal NAc (n-acetyl-galactosamine) ASPG-R (Gal Ac
Receptor)
Lactose
Asialofetuin ASPG-r
2) Sinusoidal Hyaluronan Hyaluronan receptor Endothelial Cell (SEC)
Procollagen Procollagen receptor
Negatively charged molecules Scavenger receptors Mannose Mannose receptors
N-acetyl Glucosamine Scavenger receptors Immuno globulins Fc Receptor
LPS CD 14 Receptor
Insulin Receptor mediated
transcytosis
Transferrin Receptor mediated
transcytosis
Albumins Non-specific
Sugar- Albumin conjugates
Mannose-6-phosphate Mannose-6-phosphate receptor
3) Kupffer Cell (KC) Mannose Mannose receptors
Fucose Fucose receptors
Albumins Non-specific
Mannose-albumin conjugates
Ligands can be proteins, e.g., glycoproteins, or peptides, e.g., molecules having a specific affinity for a co-ligand, or antibodies e.g., an antibody, that binds to a specified cell type such as a cancer cell, endothelial cell, or bone cell. Ligands may also include hormones and hormone receptors. They can also include non-peptidic species, such as lipids, lectins, carbohydrates, vitamins, cofactors, multivalent lactose, multivalent galactose, N-acetyl-galactosamine, N-acetyl-gulucosamine multivalent mannose,
multivalent fucose, or aptamers. The ligand can be, for example, a lipopolysaccharide, an activator of p38 MAP kinase, or an activator of NF-KB.
The ligand can be a substance, e.g, a drug, which can increase the uptake of the iR A agent into the cell, for example, by disrupting the cell's cytoskeleton, e.g., by disrupting the cell's microtubules, microfilaments, and/or intermediate filaments. The drug can be, for example, taxon, vincristine, vinblastine, cytochalasin, nocodazole, japlakinolide, latrunculin A, phalloidin, swinholide A, indanocine, or myoservin.
The ligand can increase the uptake of the iRNA agent into the cell by activating an inflammatory response, for example. Exemplary ligands that would have such an effect include tumor necrosis factor alpha (TNF alpha), interleukin-1 beta, or gamma interferon.
In one aspect, the ligand is a lipid or lipid-based molecule. Such a lipid or lipid- based molecule preferably binds a serum protein, e.g., human serum albumin (HSA). An HSA binding ligand allows for distribution of the conjugate to a target tissue, e.g., a non- kidney target tissue of the body. For example, the target tissue can be the liver, including parenchymal cells of the liver. Other molecules that can bind HSA can also be used as ligands. For example, neproxin or aspirin can be used. A lipid or lipid-based ligand can (a) increase resistance to degradation of the conjugate, (b) increase targeting or transport into a target cell or cell membrane, and/or (c) can be used to adjust binding to a serum protein, e.g., HSA.
A lipid based ligand can be used to modulate, e.g., control the binding of the conjugate to a target tissue. For example, a lipid or lipid-based ligand that binds to HSA more strongly will be less likely to be targeted to the kidney and therefore less likely to be cleared from the body. A lipid or lipid-based ligand that binds to HSA less strongly can be used to target the conjugate to the kidney.
In a preferred embodiment, the lipid based ligand binds HSA. Preferably, it binds HSA with a sufficient affinity such that the conjugate will be preferably distributed to a non-kidney tissue. However, it is preferred that the affinity not be so strong that the HSA-ligand binding cannot be reversed.
In another preferred embodiment, the lipid based ligand binds HSA weakly or not at all, such that the conjugate will be preferably distributed to the kidney. Other moieties that target to kidney cells can also be used in place of or in addition to the lipid based ligand.
In another aspect, the ligand is a moiety, e.g., a vitamin, which is taken up by a target cell, e.g., a proliferating cell. These are particularly useful for treating disorders characterized by unwanted cell proliferation, e.g., of the malignant or non-malignant type, e.g., cancer cells. Exemplary vitamins include vitamin A, E, and K. Other exemplary vitamins include are B vitamin, e.g., folic acid, B12, riboflavin, biotin, pyridoxal or other vitamins or nutrients taken up by cancer cells. Also included are HAS, low density lipoprotein (LDL) and high-density lipoprotein (HDL).
In another aspect, the ligand is a cell-permeation agent, preferably a helical cell- permeation agent. Preferably, the agent is amphipathic. An exemplary agent is a peptide such as tat or antennopedia. If the agent is a peptide, it can be modified, including a peptidylmimetic, invertomers, non-peptide or pseudo-peptide linkages, and use of D- amino acids. The helical agent is preferably an alpha- helical agent, which preferably has a lipophilic and a lipophobic phase.
The ligand can be a peptide or peptidomimetic. A peptidomimetic (also referred to herein as an oligopeptido mimetic) is a molecule capable of folding into a defined three-dimensional structure similar to a natural peptide. The attachment of peptide and peptidomimetics to iRNA agents can affect pharmacokinetic distribution of the iR A, such as by enhancing cellular recognition and absorption. The peptide or peptidomimetic moiety can be about 5-50 amino acids long, e.g., about 5, 10, 15, 20, 25, 30, 35, 40, 45, or 50 amino acids long (see Table 3, for example).
Table 3. Exemplary Cell Permeation Peptides

272: 16010, 1997
Signal GALFLGWLGAAGSTMGAWSQPKKKRKV Chaloin et al., Sequence- Biochem.
based peptide Biophys. Res.
Commun., 243:601, 1998
PVEC LLIILRRRIR QAHAHSK Elmquist et al.,
Exp. Cell Res., 269:237, 2001
Transportan GWTLNSAGYLLKINLKALAALAKKIL Pooga et al.,
FASEB J., 12:67, 1998
Amphiphilic KLALKLALKALKAALKLA Oehlke et al., model peptide Mol. Ther., 2:339,
2000
Arg9 RRRRRRRRR Mitchell et al., J.
Pept. Res., 56:318, 2000
Bacterial cell KFFKFFKFFK
wall
permeating
LL-37 LLGDFFR SKEKIGKEFKRIVQRIKDFLRNL
VPRTES
Cecropin PI SWLSKTAKKLENSAKKRISEGIAIAIQGGPR a-defensin ACYCRIPACIAGERRYGTCIYQGRLWAFCC b-defensin DHYNCVSSGGQCLYSACPIFTKIQGTCYRG
KAKCCK )
Bactenecin RKCRIVVIRVCR
PPv-39 RRRPRPPYLPRPRPPPFFPPRLPPRIPPGFPPR
FPPRFPGKR-NH2
Indolicidin ILPWKWPWWPWRR-NH2
A peptide or peptidomimetic can be, for example, a cell permeation peptide, cationic peptide, amphipathic peptide, or hydrophobic peptide (e.g., consisting primarily of Tyr, Trp or Phe). The peptide moiety can be a dendrimer peptide, constrained peptide or crosslinked peptide. In another alternative, the peptide moiety can include a
hydrophobic membrane translocation sequence (MTS). An exemplary hydrophobic MTS-containing peptide is RFGF having the amino acid sequence
AAVALLPAVLLALLAP. An RFGF analogue (e.g., amino acid sequence
AALLPVLLAAP) containing a hydrophobic MTS can also be a targeting moiety. The peptide moiety can be a "delivery" peptide, which can carry large polar molecules including peptides, oligonucleotides, and protein across cell membranes. For example, sequences from the HIV Tat protein (GRKKRRQRRRPPQ) and the Drosophila
Antennapedia protein (RQIKIWFQNRRMKWK ) have been found to be capable of functioning as delivery peptides. A peptide or peptidomimetic can be encoded by a random sequence of DNA, such as a peptide identified from a phage-display library, or one-bead-one-compound (OBOC) combinatorial library (Lam et al., Nature, 354:82-84, 1991). Preferably the peptide or peptidomimetic tethered to an iRNA agent via an incorporated compound unit is a cell targeting peptide such as an arginine-glycine- aspartic acid (RGD)-peptide, or RGD mimic. A peptide moiety can range in length from about 5 amino acids to about 40 amino acids. The peptide moieties can have a structural modification, such as to increase stability or direct conformational properties. Any of the structural modifications described below can be utilized.
An RGD peptide moiety can be used to target a tumor cell, such as an endothelial tumor cell or a breast cancer tumor cell (Zitzmann et al., Cancer Res., 62:5139-43, 2002). An RGD peptide can facilitate targeting of an iRNA agent to tumors of a variety of other tissues, including the lung, kidney, spleen, or liver (Aoki et al., Cancer Gene Therapy 8:783-787, 2001). Preferably, the RGD peptide will facilitate targeting of an iRNA agent to the kidney. The RGD peptide can be linear or cyclic, and can be modified, e.g., glycosylated or methylated to facilitate targeting to specific tissues. For example, a glycosylated RGD peptide can deliver an iRNA agent to a tumor cell expressing yB3 (Haubner et al., Jour. Nucl. Med., 42:326-336, 2001).
Peptides that target markers enriched in proliferating cells can be used. E.g., RGD containing peptides and peptidomimetics can target cancer cells, in particular cells that exhibit an Iv¾ integrin. Thus, one could use RGD peptides, cyclic peptides containing RGD, RGD peptides that include D-amino acids, as well as synthetic RGD
mimics. In addition to RGD, one can use other moieties that target the Iv-¾ integrin ligand. Generally, such ligands can be used to control proliferating cells and
angiogeneis. Preferred conjugates of this type lignads that targets PECAM-1, VEGF, or other cancer gene, e.g., a cancer gene described herein.
Table 4. Azide modified peptides.

A "cell permeation peptide" is capable of permeating a cell, e.g., a microbial cell, such as a bacterial or fungal cell, or a mammalian cell, such as a human cell. A microbial cell-permeating peptide can be, for example, an a-helical linear peptide (e.g., LL-37 or Ceropin PI), a disulfide bond-containing peptide (e.g., a -defensin, β-defensin or bactenecin), or a peptide containing only one or two dominating amino acids (e.g., PR-39 or indolicidin). A cell permeation peptide can also include a nuclear localization signal (NLS). For example, a cell permeation peptide can be a bipartite amphipathic peptide, such as MPG, which is derived from the fusion peptide domain of HIV- 1 gp41 and the NLS of SV40 large T antigen (Simeoni et al, Nucl. Acids Res. 31 :2717-2724, 2003).
In one embodiment, a targeting peptide tethered to an iRNA agent and/or the carrier oligomer can be an amphipathic a-helical peptide. Exemplary amphipathic a- helical peptides include, but are not limited to, cecropins, lycotoxins, paradaxins, buforin, CPF, bombinin-like peptide (BLP), cathelicidins, ceratotoxins, S. clava peptides, hagfish intestinal antimicrobial peptides (HFIAPs), magainines, brevinins-2, dermaseptins, melittins, pleurocidin, H2A peptides, Xenopus peptides, esculentinis-1, and caerins. A number of factors will preferably be considered to maintain the integrity of helix stability.
For example, a maximum number of helix stabilization residues will be utilized (e.g., leu, ala, or lys), and a minimum number helix destabilization residues will be utilized (e.g., proline, or cyclic compoundic units. The capping residue will be considered (for example Gly is an exemplary N-capping residue and/or C-terminal amidation can be used to provide an extra H-bond to stabilize the helix. Formation of salt bridges between residues with opposite charges, separated by i ± 3, or i ± 4 positions can provide stability. For example, cationic residues such as lysine, arginine, homo-arginine, ornithine or histidine can form salt bridges with the anionic residues glutamate or aspartate.
Peptide and peptidomimetic ligands include those having naturally occurring or modified peptides, e.g., D or L peptides; α, β, or γ peptides; N-methyl peptides;
azapeptides; peptides having one or more amide, i.e., peptide, linkages replaced with one or more urea, thiourea, carbamate, or sulfonyl urea linkages; or cyclic peptides.
The targeting ligand can be any ligand that is capable of targeting a specific receptor. Examples are: folate, GalNAc, GalNAc3, galactose, mannose, mannose-6P, clusters of sugars such as GalNAc cluster, mannose cluster, galactose cluster, or an apatamer. A cluster is a combination of two or more sugar units. The targeting ligands also include integrin receptor ligands, Chemokine receptor ligands, transferrin, biotin, serotonin receptor ligands, PSMA, endothelin, GCPII, somatostatin, LDL and HDL ligands. The ligands can also be based on nucleic acid, e.g., an aptamer. The aptamer can be unmodified or have any combination of modifications disclosed herein.
Endosomal release agents include imidazoles, poly or oligoimidazoles, PEIs, peptides, fusogenic peptides, polycaboxylates, polyacations, masked oligo or poly cations or anions, acetals, polyacetals, ketals/polyketyals, orthoesters, polymers with masked or unmasked cationic or anionic charges, dendrimers with masked or unmasked cationic or anionic charges.
PK modulator stands for pharmacokinetic modulator. PK modulator include lipophiles, bile acids, steroids, phospholipid analogues, peptides, protein binding agents, PEG, vitamins etc. Examplary PK modulator include, but are not limited to, cholesterol, fatty acids, cholic acid, lithocholic acid, dialkylglycerides, diacylglyceride,
phospholipids, sphingo lipids, naproxen, ibuprofen, vitamin E, biotin etc.
Oligonucleotides that comprise a number of phosphorothioate linkages are also known to bind to serum protein, thus short oligonucleotides, e.g. oligonucleotides of about 5 bases, 10 bases, 15 bases or 20 bases, comprising multiple of phosphorothioate linkages in the backbaone are also amenable to the present invention as ligands (e.g. as PK modulating ligands).
In addition, aptamers that bind serum components (e.g. serum proteins) are also amenable to the present invention as PK modulating ligands.
Other ligands amenable to the invention are described in copending applications USSN: 10/916,185, filed August 10, 2004; USSN: 10/946,873, filed September 21, 2004; USSN: 10/833,934, filed August 3, 2007; USSN: 11/115,989 filed April 27, 2005 and USSN: 11/944,227 filed November 21, 2007, which are incorporated by reference in their entireties for all purposes.
When two or more ligands are present, the ligands can all have same properties, all have different properties or some ligands have the same properties while others have different properties. For example, a ligand can have targeting properties, have endosomolytic activity or have PK modulating properties. In a preferred embodiment, all the ligands have different properties.
The compound comprising the ligand, e.g. the click-carrier compound, can be present in any position of an oligonucleotide, e.g. an iRNA agent. In some embodiments, click-carrier compound can be present at the terminus such as a 5' or 3' terminal of the iRNA agent. Click-carrier compounds can also present at an internal postion of the iRNA agent. For double- stranded iRNA agents, click-carrier compounds can be incorporated into one or both strands. In some embodiments, the sense strand of the double-stranded iRNA agent comprises the click-carrier compound. In other embodiments, the antisense strand of the double-stranded iRNA agent comprises the click-carrier compound.
In some embodiments, lignads can be conjugated to nucleobases, sugar moieties, or internucleosidic linkages of nucleic acid molecules. Conjugation to purine nucleobases or derivatives thereof can occur at any position including, endocyclic and exocyclic atoms. In some embodiments, the 2-, 6-, 7-, or 8-positions of a purine nucleobase are attached to a conjugate moiety. Conjugation to pyrimidine nucleobases or derivatives
thereof can also occur at any position. In some embodiments, the 2-, 5-, and 6-positions of a pyrimidine nucleobase can be substituted with a conjugate moiety. Conjugation to sugar moieties of nucleosides can occur at any carbon atom. Example carbon atoms of a sugar moiety that can be attached to a conjugate moiety include the 2', 3', and 5' carbon atoms. The Γ position can also be attached to a conjugate moiety, such as in an abasic residue. Internucleosidic linkages can also bear conjugate moieties. For phosphorus- containing linkages (e.g., phosphodiester, phosphorothioate, phosphorodithiotate, phosphoroamidate, and the like), the conjugate moiety can be attached directly to the phosphorus atom or to an O, N, or S atom bound to the phosphorus atom. For amine- or amide-containing internucleosidic linkages (e.g., PNA), the conjugate moiety can be attached to the nitrogen atom of the amine or amide or to an adjacent carbon atom.
There are numerous methods for preparing conjugates of oligomeric compounds. Generally, an oligomeric compound is attached to a conjugate moiety by contacting a reactive group (e.g., OH, SH, amine, carboxyl, aldehyde, and the like) on the oligomeric compound with a reactive group on the conjugate moiety. In some embodiments, one reactive group is electrophilic and the other is nucleophilic.
For example, an electrophilic group can be a carbonyl-containing functionality and a nucleophilic group can be an amine or thiol. Methods for conjugation of nucleic acids and related oligomeric compounds with and without linkers are well described in the literature such as, for example, in Manoharan in Antisense Research and
Applications, Crooke and LeBleu, eds., CRC Press, Boca Raton, Fla., 1993, Chapter 17, which is incorporated herein by reference in its entirety.
Representative United States patents that teach the preparation of oligonucleotide conjugates include, but are not limited to, U.S. Pat. Nos. 4,828,979; 4,948,882; 5,218, 105; 5,525,465; 5,541,313; 5,545,730; 5,552,538; 5,578, 717, 5,580,731; 5,580,731; 5,591,584; 5,109,124; 5,118, 802; 5,138,045; 5,414,077; 5,486,603; 5,512,439; 5,578, 718; 5,608,046; 4,587,044; 4,605,735; 4,667,025; 4,762, 779; 4,789,737; 4,824,941; 4,835,263; 4,876,335; 4,904, 582; 4,958,013; 5,082,830; 5,112,963; 5,214,136; 5,082, 830; 5,112,963; 5,149,782; 5,214,136; 5,245,022; 5,254, 469; 5,258,506; 5,262,536; 5,272,250; 5,292,873; 5,317, 098; 5,371,241, 5,391,723; 5,416,203, 5,451,463; 5,510,
475; 5,512,667; 5,514,785; 5,565,552; 5,567,810; 5,574, 142; 5,585,481; 5,587,371; 5,595,726; 5,597,696; 5,599, 923; 5,599,928; 5,672,662; 5,688,941; 5,714,166; 6,153, 737; 6,172,208; 6,300,319; 6,335,434; 6,335,437; 6,395, 437; 6,444,806; 6,486,308; 6,525,031; 6,528,631; 6,559, 279; each of which is herein incorporated by reference.
Olison ucleotides
In the context of this invention, the term "oligonucleotide" refers to a polymer or oligomer of nucleotide or nucleoside monomers consisting of naturally occurring bases, sugars and intersugar (backbone) linkages. The term "oligonucleotide" also includes polymers or oligomers comprising non-naturally occurring monomers, or portions thereof, which function similarly. Such modified or substituted oligonucleotides are often preferred over native forms because of properties such as, for example, enhanced cellular uptake and increased stability in the presence of nucleases.
The nucleic acids used herein can be single-stranded or double-stranded. A single stranded oligonucleotide may have double stranded regions and a double stranded oligonucleotide may have regions of single-stranded regions. Examples of double- stranded DNA include structural genes, genes including control and termination regions, and self-replicating systems such as viral or plasmid DNA. Examples of double-stranded RNA include siRNA and other RNA interference reagents. Single-stranded nucleic acids include, e.g., antisense oligonucleotides, ribozymes, microRNAs, aptamers, antagomirs, triplex-forming oligonucleotides and single-stranded RNAi agents.
Oligonucleotides of the present invention may be of various lengths. In particular embodiments, oligonucleotides may range from about 10 to 100 nucleotides in length. In various related embodiments, oligonucleotides, single-stranded, double-stranded, and triple-stranded, may range in length from about 10 to about 50 nucleotides, from about 20 to about 50 nucleotides, from about 15 to about 30 nucleotides, from about 20 to about 30 nucleotides in length.
The oligonucleotides of the invention may comprise any oligonucleotide modification described herein and below. In certain instances, it may be desirable to modify one or both strands of a dsRNA. In some cases, the two strands will include different modifications. Multiple different modifications can be included on each of the strands. The modifications on a given strand may differ from each other, and may also differ from the various modifications on other strands. For example, one strand may have a modification, e.g., a modification described herein, and a different strand may have a different modification, e.g., a different modification described herein. In other cases, one strand may have two or more different modifications, and the another strand may include a modification that differs from the at least two modifications on the other strand.
In one embodiment, oligonucleotides of the invention comprises 5 '
phosphorothioate or 5'-phosphorodithioate, nucleotides 1 and 2 having cationic modifications via C-5 position of pyrimidines, 2-Position of Purines, N2-G, G-clamp, 8- position of purines, 6-position of purines, internal nucleotides having a 2'-F sugar with base modifications (Pseudouridine, G-clamp, phenoxazine, pyridopyrimidines, gem2'- Me-up/2'-F-down), 3 '-end with two purines with novel 2'-substituted MOE analogs, 5'- end nucleotides with novel 2'-substituted MOE analogs, 5 '-end having a 3'-F and a 2'-5'- linkage, 4 '-substituted nucleoside at the nucleotide 1 at 5 '-end and the substituent is cationic, alkyl, alkoxyalkyl, thioether and the like , 4 '-substitution at the 3 '-end of the strand, and combinations thereof.
Double-stranded oligonucleotides
In one embodiment, the invention provides double-stranded ribonucleic acid (dsRNA) molecules for inhibiting the expression of the target gene (alone or in combination with a second dsRNA for inhibiting the expression of a second target gene) in a cell or mammal, wherein the dsRNA comprises an antisense strand comprising a region of complementarity which is complementary to at least a part of an mR A formed in the expression of the target gene, and wherein the region of complementarity is less than 30 nucleotides in length, generally 19-24 nucleotides in length, and wherein said dsRNA, upon contact with a cell expressing said target gene, inhibits the expression of
said target gene. The dsR A comprises two RNA strands that are sufficiently complementary to hybridize to form a duplex structure. Generally, the duplex structure is between 15 and 30, more generally between 18 and 25, yet more generally between 19 and 24, and most generally between 19 and 21 base pairs in length. In one embodiment, longer dsR As of between 25 and 30 base pairs in length are preferred. In one embodiment, shorter dsRNAs of between 10 and 15 base pairs in length are preferred. In another embodiment, the dsRNA is at least 21 nucleotides long and includes a sense RNA strand and an antisense RNA strand, wherein the antisense RNA strand is 25 or fewer nucleotides in length, and the duplex region of the dsRNA is 18-25 nucleotides in length, e.g., 19-24 nucleotides in length.
Similarly, the region of complementarity to the target sequence is between 15 and 30, more generally between 18 and 25, yet more generally between 19 and 24, and most generally between 19 and 21 nucleotides in length. The dsRNA of the invention may further comprise one or more single-stranded nucleotide overhang(s).
In a preferred embodiment, the target gene is a human target gene. In one embodiment, the target gene is selected from the group consisting of Factor VII, Eg5, PCSK9, TPX2, apoB, SAA, TTR, RSV, PDGF beta gene, Erb-B gene, Src gene, CR gene, GRB2 gene, RAS gene, MEK gene, JNK gene, RAF gene, Erkl/2 gene,
PCNA(p21) gene, MYB gene, JUN gene, FOS gene, BCL-2 gene, Cyclin D gene, VEGF gene, EGFR gene, Cyclin A gene, Cyclin E gene, WNT-1 gene, beta-catenin gene, c- MET gene, PKC gene, NFKB gene, STAT3 gene, survivin gene, Her2/Neu gene, topoisomerase I gene, topoisomerase II alpha gene, p73 gene, p21(WAFl/CIPl) gene, p27(KIPl) gene, PPM ID gene, RAS gene, caveolin I gene, MIB I gene, MTAI gene, M68 gene, mutations in tumor suppressor genes, p53 tumor suppressor gene, and combinations thereof.
The skilled person is well aware that dsRNAs comprising a duplex structure of between 20 and 23, but specifically 21, base pairs have been hailed as particularly effective in inducing RNA interference (Elbashir et al, EMBO 2001, 20:6877-6888). However, others have found that shorter or longer dsRNAs can be effective as well. In
the embodiments described above the dsRNAs of the invention can comprise at least one strand of a length of minimally 21 nt. It can be reasonably expected that shorter dsRNAs comprising a known sequence minus only a few nucleotides on one or both ends may be similarly effective as compared to the dsRNAs of the lengths described above. Hence, dsRNAs comprising a partial sequence of at least 15, 16, 17, 18, 19, 20, or more contiguous nucleotides, and differing in their ability to inhibit the expression of the target gene by not more than 5, 10, 15, 20, 25, or 30 % inhibition from a dsRNA comprising the full sequence, are contemplated by the invention. Further dsRNAs that cleave within the target sequence can readily be made using the target gene sequence and the target sequence provided.
Double-stranded and single-stranded oligonucleotides that are effective in inducing RNA interference are also referred to as siRNA, RNAi agent and/or iRNA agent. These RNA interference inducing oligonucleotides associate with a cytoplasmic multi-protein complex known as RNAi-induced silencing complex (RISC). In many embodiments, single-stranded and double stranded RNAi agents are sufficiently long that they can be cleaved by an endogenous molecule, e.g. by Dicer, to produce smaller oligonucleotides that can enter the RISC machinery and participate in RISC mediated cleavage of a target sequence, e.g. a target mRNA.
The present invention further includes RNAi agents that target within the sequence targeted by one of the agents of the present invention. As used herein a second RNAi agent is said to target within the sequence of a first RNAi agent if the second RNAi agent cleaves the message anywhere within the mRNA that is complementary to the antisense strand of the first RNAi agent. Such a second agent will generally consist of at least 15 contiguous nucleotides coupled to additional nucleotide sequences taken from the region contiguous to the selected sequence in the target gene.
The dsRNA of the invention can contain one or more mismatches to the target sequence. In a preferred embodiment, the dsRNA of the invention contains no more than 3 mismatches. If the antisense strand of the dsRNA contains mismatches to a target sequence, it is preferable that the area of mismatch not be located in the center of the region of complementarity. If the antisense strand of the dsRNA contains mismatches to
the target sequence, it is preferable that the mismatch be restricted to 5 nucleotides from either end, for example 5, 4, 3, 2, or 1 nucleotide from either the 5' or 3' end of the region of complementarity. For example, for a 23 nucleotide dsRNA strand which is complementary to a region of the target gene, the dsRNA generally does not contain any mismatch within the central 13 nucleotides. The methods described within the invention can be used to determine whether a dsRNA containing a mismatch to a target sequence is effective in inhibiting the expression of the target gene. Consideration of the efficacy of dsRNAs with mismatches in inhibiting expression of the target gene is important, especially if the particular region of complementarity in the target gene is known to have polymorphic sequence variation within the population.
In certain embodiment, the sense-strand comprises a mismatch to the antisense strand. In some embodiments, the mismatch is at the 5 nucleotides from the 3 '-end, for example 5, 4, 3, 2, or 1 nucleotide from the end of the region of complementarity. In some embodiments, the mismatch is located in the target cleavage site region. In one embodiment, the sense strand comprises no more than 1, 2, 3, 4 or 5 mismatches to the antisense strand. In preferred embodiments, the sense strand comprises no more than 3 mismatches to the antisense strand.
In one embodiment, the sense strand comprises a nucleobase modification, e.g. an optionally substituted natural or non-natural nucleobase, a universal nucleobase, in the target cleavage site region.
The "target cleavage site" herein means the backbone linkage in the target gene, e.g. target mRNA, or the sense strand that is cleaved by the RISC mechanism by utilizing the iRNA agent. And the "target cleavage site region" comprises at least one or at least two nucleotides on both side of the cleavage site. For the sense strand, the target cleavage site is the backbone linkage in the sense strand that would get cleaved if the sense strand itself was the target to be cleaved by the RNAi mechanism. The target cleavage site can be determined using methods known in the art, for example the 5'- RACE assay as detailed in Soutschek et ah, Nature (2004) 432, 173-178. As is well understood in the art, the cleavage site region for a conical double stranded RNAi agent comprising two 21 -nucleotides long strands (wherin the strands form a double stranded
region of 19 consective basepairs having 2-nucleotide single stranded overhangs at the 3'-ends), the cleavage site region corresponds to postions 9-12 from the 5'-end of the sense strand.
In one embodiment, at least one end of the dsRNA has a single-stranded nucleotide overhang of 1 to 4, generally 1 or 2 nucleotides. In one embodiment, the single- stranded overhang has the sequence 5'-GCNN-3', wherein N is independently for each occuurence, A, G, C, U, dT, dU or absent. dsR As having at least one nucleotide overhang have unexpectedly superior inhibitory properties than their blunt-ended counterparts. Moreover, the present inventors have discovered that the presence of only one nucleotide overhang strengthens the interference activity of the dsRNA, without affecting its overall stability. dsRNA having only one overhang has proven particularly stable and effective in vivo, as well as in a variety of cells, cell culture mediums, blood, and serum. Generally, the single- stranded overhang is located at the 3'-terminal end of the antisense strand or, alternatively, at the 3 '-terminal end of the sense strand. The dsRNA may also have a blunt end, generally located at the 5 '-end of the antisense strand. Generally, the antisense strand of the dsRNA has a nucleotide overhang at the 3 '-end, and the 5 '-end is blunt.
In one embodiment, the antisense strand of the dsRNA has 1-10 nucleotides overhangs each at the 3 ' end and the 5 ' end over the sense strand. In one embodiment, the sense strand of the dsRNA has 1-10 nucleotides overhangs each at the 3' end and the 5 ' end over the antisense strand.
The dsRNAs of the invention may comprise any oligonucleotide modification described herein and below. In certain instances, it may be desirable to modify one or both strands of a dsRNA. In some cases, the two strands will include different modifications. Multiple different modifications can be included on each of the strands. The modifications on a given strand may differ from each other, and may also differ from the various modifications on other strands. For example, one strand may have a modification, e.g., a modification described herein, and a different strand may have a different modification, e.g., a different modification described herein. In other cases, one
strand may have two or more different modifications, and the another strand may include a modification that differs from the at least two modifications on the other strand.
In one embodiment, the dsRNA is chemically modified to enhance stability. In one preferred embodiment, one or more of the nucleotides in the overhang is replaced with a nucleoside thiophosphate.
The present invention also includes dsRNA compounds which are chimeric compounds. "Chimeric" dsRNA compounds or "chimeras," in the context of this invention, are dsRNA compounds, particularly dsR As, which contain two or more chemically distinct regions, each made up of at least one monomer unit, i.e., a nucleotide in the case of an dsRNA compound. These dsRNAs typically contain at least one region wherein the dsRNA is modified so as to confer upon the dsRNA increased resistance to nuclease degradation, increased cellular uptake, and/or increased binding affinity for the target nucleic acid. An additional region of the dsRNA may serve as a substrate for enzymes capable of cleaving RNA:DNA or RNA:RNA hybrids. By way of example, RNase H is a cellular endonuclease which cleaves the RNA strand of an
RNA:DNAduplex. Activation of RNase H, therefore, results in cleavage of the RNA target, thereby greatly enhancing the efficiency of dsRNA inhibition of gene expression.
The present invention also includes dsRNAs wherein the two strands are linked together. The two strands can be linked together by a polynucleotide linker such as (dT)n; wherein n is 4-10, and thus forming a hairpin. The two strands can also be linked together by a non-nucleosidic linker, e.g. a linker described herein. It will be appreciated by one of skill in the art that any oligonucleotide chemical modifications or variations describe herein can be used in the polynucleotide linker.
Hairpin RNAi agents will have a duplex region equal to or at least 17, 18, 19, 29, 21, 22, 23, 24, or 25 nucleotide pairs. The duplex region will may be equal to or less than 200, 100, or 50, in length. In one embodiment, ranges for the duplex region are 10-35, 12-30, 12-25, 15-30, 17 to 23, 19 to 23, and 19 to 21 nucleotides pairs in length. The hairpin may have a single strand overhang or terminal unpaired region, in some embodiments at the 3', and in one embodiment on the antisense side of the hairpin. In one embodiment, the overhangs are 2-3 nucleotides in length.
The RNAi agents of the invention can target more than one RNA region. For example, an RNAi agent can include a first and second sequence that are sufficiently complementary to each other to hybridize. The first sequence can be complementary to a first target RNA region and the second sequence can be complementary to a second target RNA region. The first and second sequences of the RNAi agent can be on different RNA strands, and the mismatch between the first and second sequences can be less than 50%, 40%, 30%, 20%, 10%, 5%, or 1%. The first and second sequences of the RNAi agent can be on the same RNA strand, and in a related embodiment more than 50%, 60%, 70%, 80%, 90%, 95%, or 1% of the RNAi agent can be in bimolecular form. The first and second sequences of the RNAi agent can be fully complementary to each other.
The first target RNA region can be encoded by a first gene and the second target RNA region can encoded by a second gene, or the first and second target RNA regions can be different regions of an RNA from a single gene. The first and second sequences can differ by at least 1 nucleotide.
The first and second target RNA regions can be on transcripts encoded by first and second sequence variants, e.g., first and second alleles, of a gene. The sequence variants can be mutations, or polymorphisms, for example. The first target RNA region can include a nucleotide substitution, insertion, or deletion relative to the second target RNA region, or the second target RNA region can a mutant or variant of the first target region.
The first and second target RNA regions can comprise viral or human RNA regions. The first and second target RNA regions can also be on variant transcripts of an oncogene or include different mutations of a tumor suppressor gene transcript. In addition, the first and second target RNA regions can correspond to hot-spots for genetic variation.
The double stranded oligonucleotides can be optimized for RNA interference by increasing the propensity of the duplex to disassociate or melt (decreasing the free energy of duplex association), in the region of the 5' end of the antisense strand This can be accomplished, e.g., by the inclusion of modifications or modified nucleosides which increase the propensity of the duplex to disassociate or melt in the region of the 5' end of
the antisense strand. It can also be accomplished by inclusion of modifications or modified nucleosides or attachment of a ligand that increases the propensity of the duplex to disassociate of melt in the region of the 5 'end of the antisense strand. While not wishing to be bound by theory, the effect may be due to promoting the effect of an enzyme such as helicase, for example, promoting the effect of the enzyme in the proximity of the 5 ' end of the antisense strand.
Modifications which increase the tendency of the 5' end of the antisense strand in the duplex to dissociate can be used alone or in combination with other modifications described herein, e.g., with modifications which decrease the tendency of the 3' end of the antisense in the duplex to dissociate. Likewise, modifications which decrease the tendency of the 3' end of the antisense in the duplex to dissociate can be used alone or in combination with other modifications described herein, e.g., with modifications which increase the tendency of the 5' end of the antisense in the duplex to dissociate.
Nucleic acid base pairs can be ranked on the basis of their propensity to promote dissociation or melting (e.g., on the free energy of association or dissociation of a particular pairing, the simplest approach is to examine the pairs on an individual pair basis, though next neighbor or similar analysis can also be used). In terms of promoting dissociation: A:U is preferred over G:C; G:U is preferred over G:C; I:C is preferred over G:C (I=inosine); mismatches, e.g., non-canonical or other than canonical pairings are preferred over canonical (A:T, A:U, G:C) pairings; pairings which include a universal base are preferred over canonical pairings.
It is preferred that pairings which decrease the propensity to form a duplex are used at 1 or more of the positions in the duplex at the 5 ' end of the antisense strand. The terminal pair (the most 5' pair in terms of the antisense strand), and the subsequent 4 base pairing positions (going in the 3 ' direction in terms of the antisense strand) in the duplex are preferred for placement of modifications to decrease the propensity to form a duplex. More preferred are placements in the terminal most pair and the subsequent 3, 2, or 1 base pairings. It is preferred that at least 1, and more preferably 2, 3, 4, or 5 of the base pairs from the 5 '-end of antisense strand in the duplex be chosen independently from the group of: A:U, G:U, I:C, mismatched pairs, e.g., non-canonical or other than canonical
pairings or pairings which include a universal base. In a preferred embodiment at least one, at least 2, or at least 3 base-pairs include a universal base.
Modifications or changes which promote dissociation are preferably made in the sense strand, though in some embodiments, such modifications/changes will be made in the antisense strand.
Nucleic acid base pairs can also be ranked on the basis of their propensity to promote stability and inhibit dissociation or melting (e.g., on the free energy of association or dissociation of a particular pairing, the simplest approach is to examine the pairs on an individual pair basis, though next neighbor or similar analysis can also be used). In terms of promoting duplex stability: G:C is preferred over A:U, Watson-Crick matches (A:T, A:U, G:C) are preferred over non-canonical or other than canonical pairings, analogs that increase stability are preferred over Watson-Crick matches (A:T, A:U, G:C), e.g. 2-amino-A:U is preferred over A:U, 2-thio U or 5 Me-thio-U:A, are preferred over U:A, G-clamp (an analog of C having 4 hydrogen bonds) :G is preferred over C:G, guanadinium-G-clamp:G is preferred over C:G, psuedo uridine: A, is preferred over U:A, sugar modifications, e.g., 2' modifications, e.g., 2'F, ENA, or LNA, which enhance binding are preferred over non-modified moieties and can be present on one or both strands to enhance stability of the duplex.
It is preferred that pairings which increase the propensity to form a duplex are used at 1 or more of the positions in the duplex at the 3 ' end of the antisense strand. The terminal pair (the most 3 ' pair in terms of the antisense strand), and the subsequent 4 base pairing positions (going in the 5 ' direction in terms of the antisense strand) in the duplex are preferred for placement of modifications to decrease the propensity to form a duplex. More preferred are placements in the terminal most pair and the subsequent 3, 2, or 1 base pairings. It is preferred that at least 1, and more preferably 2, 3, 4, or 5 of the pairs of the recited regions be chosen independently from the group of: G:C, a pair having an analog that increases stability over Watson-Crick matches (A:T, A:U, G:C), 2-amino- A:U, 2-thio U or 5 Me-thio-U:A, G-clamp (an analog of C having 4 hydrogen bonds):G, guanadinium-G-clamp:G, psuedo uridine:A, a pair in which one or both subunits has a sugar modification, e.g., a 2' modification, e.g., 2'F, ENA, or LNA, which enhance
binding. In some embodiments, at least one, at least, at least 2, or at least 3, of the base pairs promote duplex stability.
In a preferred embodiment the at least one, at least 2, or at least 3, of the base pairs are a pair in which one or both subunits has a sugar modification, e.g., a 2' modification, e.g., 2'-<9-Me (2'-<9-methyl), 2'-<9-MOE (2'-<9-methoxyethyl), 2'-F, 2'-0- [2-(methylamino)-2-oxoethyl] (2'-0-NMA), 2'-S-methyl, 2'-0-CH2-(4'-C) (LNA) and 2'-0-CH2CH2-(4'-C) (ENA), which enhances binding.
G-clamps and guanidinium G-clamps are discussed in the following references: Holmes and Gait, "The Synthesis of 2'-0-Methyl G-Clamp Containing Oligonucleotides and Their Inhibition of the HIV-1 Tat-TAR Interaction," Nucleosides, Nucleotides & Nucleic Acids, 22: 1259-1262, 2003; Holmes et al., "Steric inhibition of human immunodeficiency virus type-1 Tat-dependent trans-activation in vitro and in cells by oligonucleotides containing 2'-0-methyl G-clamp ribonucleoside analogues," Nucleic Acids Research, 31 :2759-2768, 2003; Wilds, et al., "Structural basis for recognition of guanosine by a synthetic tricyclic cytosine analogue: Guanidinium G-clamp," Helvetica Chimica Acta, 86:966-978, 2003; Rajeev, et al, "High- Affinity Peptide Nucleic Acid Oligomers Containing Tricyclic Cytosine Analogues," Organic Letters, 4:4395-4398, 2002; Ausin, et al., "Synthesis of Amino- and Guanidino-G-Clamp PNA Monomers," Organic Letters, 4:4073-4075, 2002; Maier et al., "Nuclease resistance of
oligonucleotides containing the tricyclic cytosine analogues phenoxazine and 9-(2- aminoethoxy)-phenoxazine ("G-clamp") and origins of their nuclease resistance properties," Biochemistry, 41 :1323-7, 2002; Flanagan, et al., "A cytosine analog that confers enhanced potency to antisense oligonucleotides," Proceedings Of The National Academy Of Sciences Of The United States Of America, 96:3513-8, 1999.
As is discussed above, an oligonucleotide can be modified to both decrease the stability of the antisense 5 'end of the duplex and increase the stability of the antisense 3' end of the duplex. This can be effected by combining one or more of the stability decreasing modifications in the antisense 5 ' end of the duplex with one or more of the stability increasing modifications in the antisense 3' end of the duplex.
Single-stranded oligonucleotides
The single-stranded oligonucleotides of the present invention also comprise nucleotide sequence that is substantially complementary to a "sense" nucleic acid encoding a gene expression product, e.g., complementary to the coding strand of a double-stranded cDNA molecule or complementary to an RNA sequence, e.g., a pre- mR A, mRNA, miRNA, or pre-miRNA. The region of complementarity is less than 30 nucleotides in length, and at least 15 nucleotides in length. Generally, the single stranded oligonucleotides are 10 to 25 nucleotides in length {e.g., 11, 12, 13, 14, 15, 16, 18, 19, 20, 21, 22, 23, or 24 nucleotides in length). In one embodiment the strand is 25-30 nucleotides. Single strands having less than 100% complementarity to the target mRNA, RNA or DNA are also embraced by the present invention. These single-stranded oligonucleotides are also referred to as antisense, antagomir and antimir oligonucleotides.
The single-stranded oligonucleotide can hybridize to a complementary RNA, and prevent access of the translation machinery to the target RNA transcript, thereby preventing protein synthesis. The single-stranded oligonucleotide can also hybridize to a complementary RNA and the RNA target can be subsequently cleaved by an enzyme such as RNase H. Degradation of the target RNA prevents translation.
Single-stranded oligonucleotides, including those described and/or identified as single stranded siRNAs, microRNAs or mirs which may be used as targets or may serve as a template for the design of oligonucleotides of the invention are taught in, for example, Esau, et al. US Publication #20050261218 (USSN: 10/909125) entitled
"Oligonucleotides and compositions for use in modulation small non-coding RNAs" the entire contents of which is incorporated herein by reference. It will be appreciated by one of skill in the art that any oligonucleotide chemical modifications or variations describe herein also apply to single stranded oligonucleotides.
MicroRNAs (miRNAs or mirs) are a highly conserved class of small RNA molecules that are transcribed from DNA in the genomes of plants and animals, but are not translated into protein. Pre-microRNAs are processed into miRNAs. Processed microRNAs are single stranded -17-25 nucleotide (nt) RNA molecules that become incorporated into the RNA-induced silencing complex (RISC) and have been identified
as key regulators of development, cell proliferation, apoptosis and differentiation. They are believed to play a role in regulation of gene expression by binding to the 3'- untranslated region of specific mRNAs. RISC mediates down-regulation of gene expression through translational inhibition, transcript cleavage, or both. RISC is also implicated in transcriptional silencing in the nucleus of a wide range of eukaryotes.
The number of miRNA sequences identified to date is large and growing, illustrative examples of which can be found, for example, in: "miRBase: microRNA sequences, targets and gene nomenclature" Griffiths- Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. NAR, 2006, 34, Database Issue, D140-D144; "The microRNA Registry" Griffiths- Jones S. NAR, 2004, 32, Database Issue, D109-D111; and also on the worldwide web at http://microma.dot.sanger.dot.ac.dot.uk/sequences/ .
Antagomirs are RNA-like oligonucleotides that harbor various modifications for RNAse protection and pharmacologic properties, such as enhanced tissue and cellular uptake. They differ from normal RNA by, for example, complete 2'-O-methylation of sugar, phosphorothioate backbone and, for example, a cholesterol-moiety at 3'-end. Antagomirs may be used to efficiently silence endogenous miRNAs by forming duplexes comprising the antagomir and endogenous miRNA, thereby preventing miRNA-induced gene silencing. An example of antagomir-mediated miRNA silencing is the silencing of miR-122, described in Krutzfeldt et al, Nature, 2005, 438: 685-689, which is expressly incorporated by reference herein in its entirety. Antagomir RNAs may be synthesized using standard solid phase oligonucleotide synthesis protocols. See US Patent
Application Ser. Nos. 11/502,158 and 11/657,341 (the disclosure of each of which are incorporated herein by reference).
An antagomir can include ligand-conjugated monomer subunits and monomers for oligonucleotide synthesis. Exemplary monomers are described in U.S. Application No. 10/916,185, filed on August 10, 2004. An antagomir can have a ZXY structure, such as is described in PCT Application No. PCT/US2004/07070 filed on March 8, 2004. An antagomir can be complexed with an amphipathic moiety. Exemplary amphipathic moieties for use with oligonucleotide agents are described in PCT Application
No. PCT/US2004/07070, filed on March 8, 2004.
Single stranded siRNAs (ss siRNAs) are known and are described in US publication US 2006/0166910 and hereby incorporated by herein by its entirety.
Preferably, the single-stranded RNA molecule has a length from 15-29 nucleotides. The RNA-strand may have a 3'hydroxyl group. In some cases, however, it may be preferable to modify the 3' end to make it resistant against 3' to 5' exonucleases. Tolerated 3'- modifications are for example terminal 2'-deoxy nucleotides, 3' phosphate, 2',3'-cyclic phosphate, C3 (or C6, C7, C12) amino linker, thiol linkers, carboxyl linkers, non- nucleotidic spacers (C3, C6, C9, C12, abasic, triethylene glycol, hexaethylene glycol), biotin, fluoresceine, etc. Single stranded siRNAs of the invention include at least one of the following motifs: 5' phosphorothioate or 5'-phosphorodithioate, nucleotides 1 and 2 having cationic modifications via C-5 position of pyrimidines, 2-Position of Purines, N2- G, G-clamp, 8-position of purines, 6-position of purines, internal nucleotides having a 2'- F sugar with base modifications (Pseudouridine, G-clamp, phenoxazine,
pyridopyrimidines, gem2'-Me-up/2'-F-down), 3 '-end with two purines with novel 2'- substituted MOE analogs, 5 '-end nucleotides with novel 2'-substituted MOE analogs, 5'- end having a 3'-F and a 2'-5 '-linkage, 4 '-substituted nucleoside at the nucleotide 1 at 5'- end and the substituent is cationic, alkyl, alkoxyalkyl, thioether and the like , 4'- substitution at the 3 '-end of the strand, and combinations thereof.
Ribozymes are oligonucleotides having specific catalytic domains that possess endonuclease activity (Kim and Cech, Proc Natl Acad Sci U S A. 1987 Dec;84(24):8788- 92; Forster and Symons, Cell. 1987 Apr 24;49(2):211-20). At least six basic varieties of naturally-occurring enzymatic RNAs are known presently. In general, enzymatic nucleic acids act by first binding to a target RNA. Such binding occurs through the target binding portion of an enzymatic nucleic acid which is held in close proximity to an enzymatic portion of the molecule that acts to cleave the target RNA. Thus, the enzymatic nucleic acid first recognizes and then binds a target RNA through
complementary base-pairing, and once bound to the correct site, acts enzymatically to cut the target RNA. Strategic cleavage of such a target RNA will destroy its ability to direct synthesis of an encoded protein. After an enzymatic nucleic acid has bound and cleaved
its RNA target, it is released from that R A to search for another target and can repeatedly bind and cleave new targets.
Methods of producing a ribozyme targeted to any target sequence are known in the art. Ribozymes may be designed as described in Int. Pat. Appl. Publ. No. WO 93/23569 and Int. Pat. Appl. Publ. No. WO 94/02595, each specifically incorporated herein by reference, and synthesized to be tested in vitro and in vivo, as described therein.
Aptamers are nucleic acid or peptide molecules that bind to a particular molecule of interest with high affinity and specificity (Tuerk and Gold, Science 249:505 (1990); Ellington and Szostak, Nature 346:818 (1990)). DNA or RNA aptamers have been successfully produced which bind many different entities from large proteins to small organic molecules. See Eaton, Curr. Opin. Chem. Biol. 1: 10-16 (1997), Famulok, Curr. Opin. Struct. Biol. 9:324-9(1999), and Hermann and Patel, Science 287:820-5 (2000). Aptamers may be RNA or DNA based. Generally, aptamers are engineered through repeated rounds of in vitro selection or equivalent ly, SELEX (systematic evolution of ligands by exponential enrichment) to bind to various molecular targets such as small molecules, proteins, nucleic acids, and even cells, tissues and organisms. The aptamer may be prepared by any known method, including synthetic, recombinant, and purification methods, and may be used alone or in combination with other aptamers specific for the same target. Further, as described more fully herein, the term "aptamer" specifically includes "secondary aptamers" containing a consensus sequence derived from comparing two or more known aptamers to a given target.
Immuno stimulatory Oligonucleotides
Nucleic acids of the present invention may be immunostimulatory, including immunostimulatory oligonucleotides (single-or double-stranded) capable of inducing an immune response when administered to a subject, which may be a mammal or other patient. The immune response may be an innate or an adaptive immune response. The immune system is divided into a more innate immune system, and acquired adaptive immune system of vertebrates, the latter of which is further divided into humoral cellular components. In particular embodiments, the immune response may be mucosal.
Immunostimulatory nucleic acids are considered to be non-sequence specific when it is not required that they specifically bind to and reduce the expression of a target polynucleotide in order to provoke an immune response. Thus, certain
immunostimulatory nucleic acids may comprise a seuqence correspondign to a region of a naturally occurring gene or mR A, but they may still be considered non-sequence specific immunostimulatory nucleic acids.
In one embodiment, the immunostimulatory nucleic acid or oligonucleotide comprises at least one CpG dinucleotide. The oligonucleotide or CpG dinucleotide may be unmethylated or methylated. In another embodiment, the immunostimulatory nucleic acid comprises at least one CpG dinucleotide having a methylated cytosine. In one embodiment, the nucleic acid comprises a single CpG dinucleotide, wherein the cytosine in said CpG dinucleotide is methylated. Methods of immune stimulation using single stranded oligonucleotides and immune stimulatory oligonucleotides.
The immunostimulatory nucleic acid or oligonucleotide comprises capable of inducing an anti- viral or an antibacterial response, in particular, the induction of type I IFN, IL-18 and/or IL-Ιβ by modulating RIG-I.
Other oligonucleotides
Because transcription factors recognize their relatively short binding sequences, even in the absence of surrounding genomic DNA, short oligonucleotides bearing the consensus binding sequence of a specific transcription factor can be used as tools for manipulating gene expression in living cells. This strategy involves the intracellular delivery of such "decoy oligonucleotides", which are then recognized and bound by the target factor. Occupation of the transcription factor's DNA-binding site by the decoy renders the transcription factor incapable of subsequently binding to the promoter regions of target genes. Decoys can be used as therapeutic agents, either to inhibit the expression of genes that are activated by a transcription factor, or to upregulate genes that are suppressed by the binding of a transcription factor. Examples of the utilization of decoy
oligonucleotides may be found in Mann et al, J. Clin. Invest., 2000, 106: 1071-1075, which is expressly incorporated by reference herein, in its entirety.
Ul adaptor inhibit polyA sites and are bifunctional oligonucleotides with a target domain complementary to a site in the target gene's terminal exon and a 'Ul domain' that binds to the Ul smaller nuclear RNA component of the Ul snRNP (Goraczniak, et al, 2008, Nature Biotechnology, 27(3), 257-263, which is expressly incorporated by reference herein, in its entirety). Ul snRNP is a ribonucleoprotein complex that functions primarily to direct early steps in spliceosome formation by binding to the pre- mRNA exon- intron boundary (Brown and Simpson, 1998, Annu Rev Plant Physiol Plant Mol Biol 49:77-95). Nucleotides 2-11 of the 5'end of Ul snRNA base pair bind with the 5'ss of the pre mRNA. In one embodiment, oligonucleotides of the invention are Ul adaptors. In one embodiment, the Ul adaptor can be administered in combination with at least one other iRNA agent.
Oligonucleotide modifications
Unmodified oligonucleotides may be less than optimal in some applications, e.g., unmodified oligonucleotides can be prone to degradation by e.g., cellular nucleases. Nucleases can hydro lyze nucleic acid phosphodiester bonds. However, chemical modifications to one or more of the above oligonucleotide components can confer improved properties, and, e.g., can render oligonucleotides more stable to nucleases.
Modified nucleic acids and nucleotide surrogates can include one or more of:
(i) alteration, e.g., replacement, of one or both of the non-linking phosphate oxygens and/or of one or more of the linking phosphate oxygens in the phosphodiester backbone linakge.
(ii) alteration, e.g., replacement, of a constituent of the ribose sugar, e.g., of the 2' hydroxyl on the ribose sugar;
(iii) wholesale replacement of the phosphate moiety with "dephospho" linkers;
(iv) modification or replacement of a naturally occurring base with a non-natural base;
(v) replacement or modification of the ribose-phosphate backbone;
(vi) modification of the 3' end or 5' end of the oligonucelotide, e.g., removal, modification or replacement of a terminal phosphate group or conjugation of a moiety, e.g., a fluorescently labeled moiety, to either the 3" or 5' end of oligonucleotide; and
(vii) modification of the sugar (e.g., six membered rings).
The terms replacement, modification, alteration, and the like, as used in this context, do not imply any process limitation, e.g., modification does not mean that one must start with a reference or naturally occurring ribonucleic acid and modify it to produce a modified ribonucleic acid bur rather modified simply indicates a difference from a naturally occurring molecule.
As oligonucleotides are polymers of subunits or monomers, many of the modifications described herein can occur at a position which is repeated within an oligonucleotide, e.g., a modification of a nucleobase, a sugar, a phosphate moiety, or the non-bridging oxygen of a phosphate moiety. It is not necessary for all positions in a given oligonucleotide to be uniformly modified, and in fact more than one of the aforementioned modifications may be incorporated in a single oligonucleotide or even at a single nucleoside within an oligonucleotide.
In some cases the modification will occur at all of the subject positions in the oligonucleotide but in many, and in fact in most cases it will not. By way of example, a modification may only occur at a 3' or 5' terminal position, may only occur in the internal region, may only occur in a terminal regions, e.g. at a position on a terminal nucleotide or in the last 2, 3, 4, 5, or 10 nucleotides of an oligonucleotide. A
modification may occur in a double strand region, a single strand region, or in both. A modification may occur only in the double strand region of an oligonucleotide or may only occur in a single strand region of an oligonucleotide. E.g., a phosphorothioate modification at a non-bridging oxygen position may only occur at one or both termini, may only occur in a terminal regions, e.g., at a position on a terminal nucleotide or in the last 2, 3, 4, 5, or 10 nucleotides of a strand, or may occur in double strand and single strand regions, particularly at termini. The 5' end or ends can be phosphorylated.
A modification described herein may be the sole modification, or the sole type of modification included on multiple nucleotides, or a modification can be combined with one or more other modifications described herein. The modifications described herein can also be combined onto an oligonucleotide, e.g. different nucleotides of an oligonucleotide have different modifications described herein.
In some embodiments it is particularly preferred, e.g., to enhance stability, to include particular nucleobases in overhangs, or to include modified nucleotides or nucleotide surrogates, in single strand overhangs, e.g., in a 5' or 3' overhang, or in both. E.g., it can be desirable to include purine nucleotides in overhangs. In some
embodiments all or some of the bases in a 3' or 5' overhang will be modified, e.g., with a modification described herein. Modifications can include, e.g., the use of modifications at the 2' OH group of the ribose sugar, e.g., the use of deoxyribonucleotides, e.g., deoxythymidine, instead of ribonucleotides, and modifications in the phosphate group, e.g., phosphothioate modifications. Overhangs need not be homologous with the target sequence.
Specific modifications are discussed in more detail below.
The Phosphate Group
The phosphate group is a negatively charged species. The charge is distributed equally over the two non-bridging oxygen atoms. However, the phosphate group can be modified by replacing one of the oxygens with a different substituent. One result of this modification to R A phosphate backbones can be increased resistance of the
oligoribonucleotide to nucleo lytic breakdown. Thus while not wishing to be bound by theory, it can be desirable in some embodiments to introduce alterations which result in either an uncharged linker or a charged linker with unsymmetrical charge distribution.
Examples of modified phosphate groups include phosphorothioate,
phosphoroselenates, borano phosphates, borano phosphate esters, hydrogen
phosphonates, phosphoroamidates, alkyl or aryl phosphonates and phosphotriesters. In one embodiment, one of the non-bridging phosphate oxygen atoms in the phosphate backbone moiety can be replaced by any of the following: S, Se, BR3 (R is hydrogen,
alkyl, aryl), C (i.e. an alkyl group, an aryl group, etc...), H, NR2 (R is hydrogen, alkyl, aryl), or OR (R is alkyl or aryl). The phosphorous atom in an unmodified phosphate group is achiral. However, replacement of one of the non-bridging oxygens with one of the above atoms or groups of atoms renders the phosphorous atom chiral; in other words a phosphorous atom in a phosphate group modified in this way is a stereogenic center. The stereogenic phosphorous atom can possess either the "R" configuration (herein Rp) or the "S" configuration (herein Sp).
Phosphorodithioates have both non-bridging oxygens replaced by sulfur. The phosphorus center in the phosphorodithioates is achiral which precludes the formation of oligoribonucleotides diastereomers. Thus, while not wishing to be bound by theory, modifications to both non-bridging oxygens, which eliminate the chiral center, e.g. phosphorodithioate formation, may be desirable in that they cannot produce diastereomer mixtures. Thus, the non-bridging oxygens can be independently any one of S, Se, B, C, H, N, or OR (R is alkyl or aryl).
The phosphate linker can also be modified by replacement of bridging oxygen, (i.e. oxgen that links the phosphate to the nucleoside), with nitrogen (bridged
phosphoroamidates), sulfur (bridged phosphorothioates) and carbon (bridged
methylenephosphonates). The replacement can occur at the either linking oxygen or at both the linking oxygens. When the bridging oxygen is the 3 '-oxygen of a nucleoside, replcament with carbobn is preferred. When the bridging oxygen is the 5 '-oxygen of a nucleoside, replcament with nitrogen is preferred.
Replacement of the Phosphate Group
The phosphate group can be replaced by non-phosphorus containing connectors. While not wishing to be bound by theory, it is believed that since the charged
phosphodiester group is the reaction center in nucleolytic degradation, its replacement with neutral structural mimics should impart enhanced nuclease stability. Again, while not wishing to be bound by theory, it can be desirable, in some embodiment, to introduce alterations in which the charged phosphate group is replaced by a neutral moiety.
Examples of moieties which can replace the phosphate group include methyl phosphonate, hydroxy lamino, siloxane, carbonate, carboxymethyl, carbamate, amide, thioether, ethylene oxide linker, sulfonate, sulfonamide, thioformacetal, formacetal, oxime, methyleneimino, methylenemethylimino, methylenehydrazo,
methylenedimethylhydrazo and methyleneoxymethylimino. Preferred replacements include the methylenecarbonylamino and methylenemethylimino groups.
Modified phosphate linkages where at least one of the oxygens linked to the phosphate has been replaced or the phosphate group has been replaced by a non- phosphorous group, are also referred to as "non-phosphodiester backbone linkage."
Replacement of Ribophosphate Backbone
Oligonucleotide- mimicking scaffolds can also be constructed wherein the phosphate linker and ribose sugar are replaced by nuclease resistant nucleoside or nucleotide surrogates. While not wishing to be bound by theory, it is believed that the absence of a repetitively charged backbone diminishes binding to proteins that recognize polyanions (e.g. nucleases). Again, while not wishing to be bound by theory, it can be desirable in some embodiment, to introduce alterations in which the bases are tethered by a neutral surrogate backbone. Examples include the mophilino, cyclobutyl, pyrrolidine and peptide nucleic acid (PNA) nucleoside surrogates. A preferred surrogate is a PNA surrogate.
Sugar modifications
An oligonucleotide can include modification of all or some of the sugar groups of the nucleic acid. E.g., the 2' hydroxyl group (OH) can be modified or replaced with a number of different "oxy" or "deoxy" substituents. While not being bound by theory, enhanced stability is expected since the hydroxyl can no longer be deprotonated to form a 2'-alkoxide ion. The 2'-alkoxide can catalyze degradation by intramolecular nucleophilic attack on the linker phosphorus atom. Again, while not wishing to be bound by theory, it can be desirable to some embodiments to introduce alterations in which alkoxide formation at the 2' position is not possible.
Examples of "oxy"-2' hydroxyl group modifications include alkoxy or aryloxy (OR, e.g., R = H, alkyl, cycloalkyl, aryl, aralkyl, heteroaryl or sugar);
polyethyleneglycols (PEG), 0(CH2CH20)nCH2CH2OR; "locked" nucleic acids (LNA) in which the 2' hydroxyl is connected, e.g., by a methylene bridge, to the 4' carbon of the same ribose sugar; O-AMINE (AMINE = NH2; alkylamino, dialkylamino, heterocyclyl, arylamino, diaryl amino, heteroaryl amino, or diheteroaryl amino, ethylene diamine, polyamino) and aminoalkoxy, 0(CH2)nAMINE, {e.g., AMINE = NH2; alkylamino, dialkylamino, heterocyclyl, arylamino, diaryl amino, heteroaryl amino, or diheteroaryl amino, ethylene diamine, polyamino). It is noteworthy that oligonucleotides containing only the methoxyethyl group (MOE), (OCH2CH2OCH3, a PEG derivative), exhibit nuclease stabilities comparable to those modified with the robust phosphorothioate modification.
"Deoxy" modifications include hydrogen {i.e. deoxyribose sugars, which are of particular relevance to the overhang portions of partially ds RNA); halo {e.g., fluoro); amino {e.g. NH2; alkylamino, dialkylamino, heterocyclyl, arylamino, diaryl amino, heteroaryl amino, diheteroaryl amino, or amino acid); NH(CH2CH2NH)nCH2CH2- AMINE (AMINE = NH2; alkylamino, dialkylamino, heterocyclyl, arylamino, diaryl amino, heteroaryl amino,or diheteroaryl amino), -NHC(0)R (R = alkyl, cycloalkyl, aryl, aralkyl, heteroaryl or sugar), cyano; mercapto; alkyl-thio-alkyl; thioalkoxy; and alkyl, cycloalkyl, aryl, alkenyl and alkynyl, which may be optionally substituted with e.g., an amino functionality.
The sugar group can also contain one or more carbons that possess the opposite stereochemical configuration than that of the corresponding carbon in ribose. Thus, an oligonucleotide can include nucleotides containing e.g., arabinose, as the sugar. The monomer can have an alpha linkage at the 1 ' position on the sugar, e.g., alpha- nucleosides. Oligonucleotides can also include "abasic" sugars, which lack a nucleobase at C- . These abasic sugars can also be further containing modifications at one or more of the constituent sugar atoms. Oligonucleotides can also contain one or more sugars that are in the L form, e.g. L-nucleosides.
Preferred substitutents are 2'-<9-Me (2'-<9-methyl), 2'-<9-MOE ( -0- methoxyethyl), 2'-F, 2'-0-[2-(methylamino)-2-oxoethyl] (2'-<9-NMA), 2'-5-methyl, 2'- 0-CH2-(4'-C) (LNA), 2'-0-CH2CH2-(4'-C) (ENA), 2'-0-aminopropyl (2'-0-AP), 2'-0- dimethylaminoethyl (2'-0-DMAOE), 2'-0-dimethylaminopropyl (2'-0-DMAP) and 2'-0- dimethylaminoethyloxyethyl (2'-0-DMAEOE).
Terminal Modifications
The 3' and 5' ends of an oligonucleotide can be modified. Such modifications can be at the 3' end, 5' end or both ends of the molecule. They can include modification or replacement of an entire terminal phosphate or of one or more of the atoms of the phosphate group. E.g., the 3' and 5' ends of an oligonucleotide can be conjugated to other functional molecular entities such as labeling moieties, e.g., fluorophores {e.g., pyrene, TAMRA, fluorescein, Cy3 or Cy5 dyes) or protecting groups (based e.g., on sulfur, silicon, boron or ester). The functional molecular entities can be attached to the sugar through a phosphate group and/or a linker. The terminal atom of the linker can connect to or replace the linking atom of the phosphate group or the C-3' or C-5' O, N, S or C group of the sugar. Alternatively, the linker can connect to or replace the terminal atom of a nucleotide surrogate {e.g., PNAs).
When a linker/phosphate-functional molecular entity-linker/phosphate array is interposed between two strands of a dsR A, this array can substitute for a hairpin R A loop in a hairpin-type RNA agent.
Terminal modifications useful for modulating activity include modification of the 5' end with phosphate or phosphate analogs. E.g., in preferred embodiments antisense strands of dsRNAs, are 5 ' phosphorylated or include a phosphoryl analog at the 5 ' prime terminus. 5'-phosphate modifications include those which are compatible with RISC mediated gene silencing. Modifications at the 5 '-terminal end can also be useful in stimulating or inhibiting the immune system of a subject. Suitable modifications include: 5*-monophosphate ((HO)2(0)P-0-5*); 5*-diphosphate ((HO)2(0)P-0-P(HO)(0)-0-5*); 5'- triphosphate ((HO)2(0)P-0-(HO)(0)P-0-P(HO)(0)-0-5*); 5*-guanosine cap (7-
methylated or non-methylated) (7m-G-0-5*-(HO)(0)P-0-(HO)(0)P-0-P(HO)(0)-0-5*); 5'-adenosine cap (Appp), and any modified or unmodified nucleotide cap structure (N-O- 5*-(HO)(0)P-0-(HO)(0)P-0-P(HO)(0)-0-5*); 5*-monothiophosphate (phosphorothioate; (HO)2(S)P-0-5*); 5*-monodithiophosphate (phosphorodithioate; (HO)(HS)(S)P-0-5*), 5'- phosphorothiolate ((HO)2(0)P-S-5'); any additional combination of oxgen/sulfur replaced monophosphate, diphosphate and triphosphates (e.g. 5'-alpha-thiotriphosphate, 5 '-beta-thio triphosphate, 5'-gamma-thiotriphosphate, etc.), 5'-phosphoramidates
((HO)2(0)P-NH-5*, (HO)(NH2)(0)P-0-5*), 5*-alkylphosphonates (R=alkyl=methyl, ethyl, isopropyl, propyl, etc., e.g. RP(OH)(0)-0-5*-, (OH)2(0)P-5*-CH2-), 5*- alkyletherphosphonates (R=alkylether=methoxymethyl (MeOCH2-), ethoxymethyl, etc., e.g. RP(OH)(0)-0-5'-). Other embodiments include replacement of oxygen/sulfur with BH3, BH3 " and/or Se.
Terminal modifications can also be useful for monitoring distribution, and in such cases the preferred groups to be added include fluorophores, e.g., fluorscein or an Alexa dye, e.g., Alexa 488. Terminal modifications can also be useful for enhancing uptake, useful modifications for this include cholesterol. Terminal modifications can also be useful for cross-linking an RNA agent to another moiety; modifications useful for this include mitomycin C.
End-caps for exonuclease protection
Nucleobases
Adenine, guanine, cytosine and uracil are the most common bases found in RNA. These bases can be modified or replaced to provide RNA's having improved properties. For example, nuclease resistant oligoribonucleotides can be prepared with these bases or with synthetic and natural nucleobases (e.g., inosine, thymine, xanthine, hypoxanthine, nubularine, isoguanisine, or tubercidine) and any one of the above modifications.
Alternatively, substituted or modified analogs of any of the above bases and "universal bases" can be employed. Examples include 2-(halo)adenine, 2-(alkyl)adenine, 2- (propyl)adenine, 2-(amino)adenine, 2-(aminoalkyll)adenine, 2-(aminopropyl)adenine,
2- (methylthio)-N6-(isopentenyl)adenine, 6-(alkyl)adenine, 6-(methyl)adenine,
7- (deaza)adenine, 8-(alkenyl)adenine, 8-(alkyl)adenine, 8-(alkynyl)adenine,
8- (amino)adenine, 8-(halo)adenine, 8-(hydroxyl)adenine, 8-(thioalkyl)adenine, 8- (thiol)adenine, N6-(isopentyl)adenine, N6-(methyl)adenine, N6, N6-(dimethyl)adenine, 2- (alkyl)guanine,2-(propyl)guanine, 6-(alkyl)guanine, 6-(methyl)guanine, 7-(alkyl)guanine,
7- (methyl)guanine, 7-(deaza)guanine, 8-(alkyl)guanine, 8-(alkenyl)guanine,
8- (alkynyl)guanine, 8-(amino)guanine, 8-(halo)guanine, 8-(hydroxyl)guanine,
8-(thioalkyl)guanine, 8-(thiol)guanine, N-(methyl)guanine, 2-(thio)cytosine,
3- (deaza)-5-(aza)cytosine, 3-(alkyl)cytosine, 3-(methyl)cytosine, 5-(alkyl)cytosine, 5- (alkynyl)cytosine, 5-(halo)cytosine, 5-(methyl)cytosine, 5-(propynyl)cytosine,
5-(propynyl)cytosine, 5-(trifluoromethyl)cytosine, 6-(azo)cytosine, N4-(acetyl)cytosine,
3 - (3 -amino-3 -carboxypropyl)uracil, 2-(thio)uracil, 5 -(methyl)-2-(thio)uracil,
5 -(methylaminomethyl)-2-(thio)uracil, 4-(thio)uracil, 5 -(methyl)-4-(thio)uracil,
5-(methylaminomethyl)-4-(thio)uracil, 5-(methyl)-2,4-(dithio)uracil,
5-(methylaminomethyl)-2,4-(dithio)uracil, 5-(2-aminopropyl)uracil, 5-(alkyl)uracil, 5- (alkynyl)uracil, 5-(allylamino)uracil, 5-(aminoallyl)uracil, 5-(aminoalkyl)uracil,
5-(guanidiniumalkyl)uracil, 5-(l,3-diazole-l-alkyl)uracil, 5-(cyanoalkyl)uracil, 5- (dialkylaminoalkyl)uracil, 5-(dimethylaminoalkyl)uracil, 5-(halo)uracil, 5- (methoxy)uracil, uracil-5-oxyacetic acid, 5-(methoxycarbonylmethyl)-2-(thio)uracil, 5 -(methoxycarbonyl-methyl)uracil, 5 -(propynyl)uracil, 5 -(propynyl)uracil,
5-(trifluoromethyl)uracil, 6-(azo)uracil, dihydrouracil, N3-(methyl)uracil, 5-uracil (i.e., pseudouracil), 2-(thio)pseudouracil,4-(thio)pseudouracil,2,4-(dithio)psuedouracil,5- (alkyl)pseudouracil, 5-(methyl)pseudouracil, 5-(alkyl)-2-(thio)pseudouracil, 5-(methyl)- 2-(thio)pseudouracil, 5-(alkyl)-4-(thio)pseudouracil, 5-(methyl)-4-(thio)pseudouracil, 5- (alkyl)-2,4-(dithio)pseudouracil, 5-(methyl)-2,4-(dithio)pseudouracil, 1 -substituted pseudouracil, 1 -substituted 2(thio)-pseudouracil, 1 -substituted 4-(thio)pseudouracil, 1 -substituted 2,4-(dithio)pseudouracil, 1 -(aminocarbonylethylenyl)-pseudouracil, 1 -(aminocarbonylethylenyl)-2(thio)-pseudouracil, 1 -(aminocarbonylethylenyl)-
4- (thio)pseudouracil, l-(aminocarbonylethylenyl)-2,4-(dithio)pseudouracil,
1 -(aminoalkylaminocarbonylethylenyl)-pseudouracil, 1 -(aminoalkylamino-
carbonylethylenyl)-2(thio)-pseudouracil, 1 -(amino alkylaminocarbonylethylenyl)-
4- (thio)pseudouracil, l-(aminoalkylaminocarbonylethylenyl)-2,4-(dithio)pseudouracil, 1 ,3-(diaza)-2-(oxo)-phenoxazin- 1 -yl, 1 -(aza)-2-(thio)-3-(aza)-phenoxazin- 1 -yl, 1 ,3- (diaza)-2-(oxo)-phenthiazin- 1 -yl, 1 -(aza)-2-(thio)-3-(aza)-phenthiazin- 1 -yl, 7-substituted 1 ,3-(diaza)-2-(oxo)-phenoxazin- 1 -yl, 7-substituted 1 -(aza)-2-(thio)-3-(aza)-phenoxazin- 1 - yl, 7-substituted l,3-(diaza)-2-(oxo)-phenthiazin-l-yl, 7-substituted l-(aza)-2-(thio)-3- (aza)-phenthiazin- 1 -yl, 7-(aminoalkylhydroxy)- 1 ,3 -(diaza)-2-(oxo)-phenoxazin- 1 -yl, 7- (aminoalkylhydroxy)- 1 -(aza)-2-(thio)-3 -(aza)-phenoxazin- 1 -yl, 7-(aminoalkylhydroxy)-
1 ,3-(diaza)-2-(oxo)-phenthiazin- 1 -yl, 7-(aminoalkylhydroxy)- 1 -(aza)-2-(thio)-3-(aza)- phenthiazin- 1 -yl, 7-(guanidiniumalkylhydroxy)- 1 ,3-(diaza)-2-(oxo)-phenoxazin- 1 -yl, 7- (guanidiniumalkylhydroxy)- 1 -(aza)-2-(thio)-3 -(aza)-phenoxazin- 1 -yl, 7- (guanidiniumalkyl-hydroxy)- 1 ,3-(diaza)-2-(oxo)-phenthiazin- 1 -yl, 7- (guanidiniumalkylhydroxy)- 1 -(aza)-2-(thio)-3-(aza)-phenthiazin- 1 -yl, 1 ,3,5-(triaza)-2,6- (dioxa)-naphthalene, inosine, xanthine, hypoxanthine, nubularine, tubercidine, isoguanisine, inosinyl, 2-aza-inosinyl, 7-deaza-inosinyl, nitroimidazolyl, nitropyrazolyl, nitrobenzimidazolyl, nitroindazolyl, aminoindolyl, pyrrolopyrimidinyl, 3- (methyl)isocarbostyrilyl, 5 -(methyl)isocarbostyrilyl, 3 -(methyl)-7-
(propynyl)isocarbostyrilyl, 7-(aza)indolyl, 6-(methyl)-7-(aza)indolyl, imidizopyridinyl, 9- (methyl)-imidizopyridinyl, pyrrolopyrizinyl, isocarbostyrilyl, 7- (propynyl)isocarbostyrilyl, propynyl-7-(aza)indolyl, 2,4,5-(trimethyl)phenyl, 4- (methyl)indolyl, 4,6-(dimethyl)indolyl, phenyl, napthalenyl, anthracenyl,
phenanthracenyl, pyrenyl, stilbenyl, tetracenyl, pentacenyl, difluorotolyl, 4-(fluoro)-6- (methyl)benzimidazole, 4-(methyl)benzimidazole, 6-(azo)thymine, 2-pyridinone,
5- nitroindole, 3-nitropyrrole, 6-(aza)pyrimidine, 2-(amino)purine, 2,6-(diamino)purine, 5-substituted pyrimidines, N2-substituted purines, N6-substituted purines, 06-substituted purines, substituted 1,2,4-triazoles, or any O-alkylated or N-alkylated derivatives thereof;
Further purines and pyrimidines include those disclosed in U.S. Pat. No.
3,687,808, hereby incorporated by refernence, those disclosed in the Concise
Encyclopedia Of Polymer Science And Engineering, pages 858-859, Kroschwitz, J. I., ed.
John Wiley & Sons, 1990, and those disclosed by Englisch et al., Angewandte Chemie, International Edition, 1991, 30, 613.
Cationic Groups
Modifications to oligonucleotides can also include attachment of one or more cationic groups to the sugar, base, and/or the phosphorus atom of a phosphate or modified phosphate backbone moiety. A cationic group can be attached to any atom capable of substitution on a natural, unusual or universal base. A preferred position is one that does not interfere with hybridization, i.e., does not interfere with the hydrogen bonding interactions needed for base pairing. A cationic group can be attached e.g., through the C2' position of a sugar or analogous position in a cyclic or acyclic sugar surrogate.
Cationic groups can include e.g., protonated amino groups, derived from e.g., O-AMINE (AMINE = NH2; alkylamino, dialkylamino, heterocyclyl, arylamino, diaryl amino, heteroaryl amino, or diheteroaryl amino, ethylene diamine, polyamino); aminoalkoxy, e.g., 0(CH2)nAMINE, (e.g., AMINE = NH2; alkylamino, dialkylamino, heterocyclyl, arylamino, diaryl amino, heteroaryl amino, or diheteroaryl amino, ethylene diamine, polyamino); amino (e.g. NH2; alkylamino, dialkylamino, heterocyclyl, arylamino, diaryl amino, heteroaryl amino, diheteroaryl amino, or amino acid); or
NH(CH2CH2NH)nCH2CH2-AMINE (AMINE = NH2; alkylamino, dialkylamino, heterocyclyl, arylamino, diaryl amino, heteroaryl amino,or diheteroaryl amino).
End-caps for exonuclease protection
Placement within an oligonucleotide
Some modifications may preferably be included on an oligonucleotide at a particular location, e.g., at an internal position of a strand, or on the 5' or 3' end of an oligonucleotide. A preferred location of a modification on an oligonucleotide, may confer preferred properties on the agent. For example, preferred locations of particular modifications may confer optimum gene silencing properties, or increased resistance to endonuclease or exonuclease activity.
One or more nucleotides of an oligonucleotide may have a 2'-5' linkage. One or more nucleotides of an oligonucleotide may have inverted linkages, e.g. 3'-3', 5'-5', 2'- 2' or 2'-3' linkages.
A double-stranded oligonucleotide may include at least one 5'-uridine-adenine-3' (5'-UA-3') dinucleotide wherein the uridine is a 2'-modified nucleotide, or a terminal 5'- uridine-guanine-3 ' (5'-UG-3') dinucleotide, wherein the 5 '-uridine is a 2'-modified nucleotide, or a terminal 5'-cytidine-adenine-3' (5'-CA-3') dinucleotide, wherein the 5'- cytidine is a 2'-modified nucleotide, or a terminal 5'-uridine-uridine-3' (5'-UU-3') dinucleotide, wherein the 5 '-uridine is a 2'-modified nucleotide, or a terminal 5'- cytidine-cytidine-3 ' (5'-CC-3') dinucleotide, wherein the 5'-cytidine is a 2'-modified nucleotide, or a terminal 5'-cytidine-uridine-3' (5'-CU-3') dinucleotide, wherein the 5'- cytidine is a 2'-modified nucleotide, or a terminal 5'-uridine-cytidine-3' (5'-UC-3') dinucleotide, wherein the 5 '-uridine is a 2'-modified nucleotide. Double-stranded oligonucleotides including these modifications are particularly stabilized against endonuclease activity.
General References
The oligoribonucleotides and oligoribonucleosides used in accordance with this invention may be synthesized with solid phase synthesis, see for example
"Oligonucleotide synthesis, a practical approach", Ed. M. J. Gait, IRL Press, 1984;
"Oligonucleotides and Analogues, A Practical Approach", Ed. F. Eckstein, IRL Press, 1991 (especially Chapter 1, Modern machine-aided methods of oligodeoxyribonucleotide synthesis, Chapter 2, Oligoribonucleotide synthesis, Chapter 3, 2'-0—
Methyloligoribonucleotide- s: synthesis and applications, Chapter 4, Phosphorothioate oligonucleotides, Chapter 5, Synthesis of oligonucleotide phosphorodithioates, Chapter 6, Synthesis of oligo-2'-deoxyribonucleoside methylphosphonates, and. Chapter 7,
Oligodeoxynucleotides containing modified bases. Other particularly useful synthetic procedures, reagents, blocking groups and reaction conditions are described in Martin, P., Helv. Chim. Acta, 1995, 78, 486-504; Beaucage, S. L. and Iyer, R. P., Tetrahedron, 1992, 48, 2223-2311 and Beaucage, S. L. and Iyer, R. P., Tetrahedron, 1993, 49, 6123-6194, or
references referred to therein. Modification described in WO 00/44895, WOOl/75164, or WO02/44321 can be used herein. The disclosure of all publications, patents, and published patent applications listed herein are hereby incorporated by reference.
Phosphate Group References
The preparation of phosphinate oligoribonucleotides is described in U.S. Pat. No. 5,508,270. The preparation of alkyl phosphonate oligoribonucleotides is described in U.S. Pat. No. 4,469,863. The preparation of phosphoramidite oligoribonucleotides is described in U.S. Pat. No. 5,256,775 or U.S. Pat. No. 5,366,878. The preparation of phosphotriester oligoribonucleotides is described in U.S. Pat. No. 5,023,243. The preparation of borano phosphate oligoribonucleotide is described in U.S. Pat. Nos.
5,130,302 and 5,177,198. The preparation of 3'-Deoxy-3'-amino phosphoramidate oligoribonucleotides is described in U.S. Pat. No. 5,476,925. 3'-Deoxy-3'- methylenephosphonate oligoribonucleotides is described in An, H, et al. J. Org. Chem. 2001, 66, 2789-2801. Preparation of sulfur bridged nucleotides is described in Sproat et al. Nucleosides Nucleotides 1988, 7,651 and Crosstick et al. Tetrahedron Lett. 1989, 30, 4693.
Sugar Group References
Modifications to the 2' modifications can be found in Verma, S. et al. Annu. Rev. Biochem. 1998, 67, 99-134 and all references therein. Specific modifications to the ribose can be found in the following references: 2'-fluoro (Kawasaki et. al., J. Med. Chem., 1993, 36, 831-841), 2'-MOE (Martin, P. Helv. Chim. Acta 1996, 79, 1930-1938), "UNA" (Wengel, J. Acc. Chem. Res. 1999, 32, 301-310).
Replacement of the Phosphate Group References
Methylenemethylimino linked oligoribonucleosides, also identified herein as MMI linked oligoribonucleosides, methylenedimethylhydrazo linked
oligoribonucleosides, also identified herein as MDH linked oligoribonucleosides, and methylenecarbonylamino linked oligonucleosides, also identified herein as amide-3
linked oligoribonucleosides, and methyleneaminocarbonyl linked oligonucleosides, also identified herein as amide-4 linked oligoribonucleosides as well as mixed backbone compounds having, as for instance, alternating MMI and PO or PS linkages can be prepared as is described in U.S. Pat. Nos. 5,378,825, 5,386,023, 5,489,677 and in published PCT applications PCT/US92/04294 and PCT/US92/04305 (published as WO 92/20822 WO and 92/20823, respectively). Formacetal and thioformacetal linked oligoribonucleosides can be prepared as is described in U.S. Pat. Nos. 5,264,562 and 5,264,564. Ethylene oxide linked oligoribonucleosides can be prepared as is described in U.S. Pat. No. 5,223,618. Siloxane replacements are described in Cormier .F. et al. Nucleic Acids Res. 1988, 16, 4583. Carbonate replacements are described in Tittensor, J.R. J. Chem. Soc. C 1971, 1933. Carboxymethyl replacements are described in Edge, M.D. et al. J. Chem. Soc. Perkin Trans. 1 1972, 1991. Carbamate replacements are described in Stirchak, E.P. Nucleic Acids Res. 1989, 17, 6129.
Replacement of the Phosphate-Ribose Backbone References
Cyclobutyl sugar surrogate compounds can be prepared as is described in U.S. Pat. No. 5,359,044. Pyrrolidine sugar surrogate can be prepared as is described in U.S. Pat. No. 5,519,134. Morpholino sugar surrogates can be prepared as is described in U.S. Pat. Nos. 5,142,047 and 5,235,033, and other related patent disclosures. Peptide Nucleic Acids (PNAs) are known per se and can be prepared in accordance with any of the various procedures referred to in Peptide Nucleic Acids (PNA): Synthesis, Properties and Potential Applications, Bioorganic & Medicinal Chemistry, 1996, 4, 5-23. They may also be prepared in accordance with U.S. Pat. No. 5,539,083.
Terminal Modification References
Terminal modifications are described in Manoharan, M. et al. Antisense and Nucleic Acid Drug Development 12, 103-128 (2002) and references therein.
Bases References
N-2 substitued purine nucleoside amidites can be prepared as is described in U.S. Pat. No. 5,459,255. 3-Deaza purine nucleoside amidites can be prepared as is described in U.S. Pat. No. 5,457,191. 5,6-Substituted pyrimidine nucleoside amidites can be prepared as is described in U.S. Pat. No. 5,614,617. 5-Propynyl pyrimidine nucleoside amidites can be prepared as is described in U.S. Pat. No. 5,484,908. Additional references are disclosed in the above section on base modifications
Oligonucleotide production
The oligonucleotide compounds of the invention can be prepared using solution- phase or solid-phase organic synthesis. Organic synthesis offers the advantage that the oligonucleotide strands comprising non-natural or modified nucleotides can be easily prepared. Any other means for such synthesis known in the art may additionally or alternatively be employed. It is also known to use similar techniques to prepare other oligonucleotides, such as the phosphorothioates, phosphorodithioates and alkylated derivatives. The double-stranded oligonucleotide compounds of the invention may be prepared using a two-step procedure. First, the individual strands of the double-stranded molecule are prepared separately. Then, the component strands are annealed.
Regardless of the method of synthesis, the oligonucleotide can be prepared in a solution (e.g., an aqueous and/or organic solution) that is appropriate for formulation. For example, the iRNA preparation can be precipitated and redissolved in pure double- distilled water, and lyophilized. The dried iRNA can then be resuspended in a solution appropriate for the intended formulation process.
Teachings regarding the synthesis of particular modified oligonucleotides may be found in the following U.S. patents or pending patent applications: U.S. Pat. Nos. 5,138,045 and 5,218, 105, drawn to polyamine conjugated oligonucleotides; U.S. Pat. No. 5,212,295, drawn to compounds for the preparation of oligonucleotides having chiral phosphorus linkages; U.S. Pat. Nos. 5,378,825 and 5,541 ,307, drawn to oligonucleotides having modified backbones; U.S. Pat. No. 5,386,023, drawn to backbone-modified oligonucleotides and the preparation thereof through reductive coupling; U.S. Pat. No. 5,457, 191 , drawn to modified nucleobases based on the 3-deazapurine ring system and
methods of synthesis thereof; U.S. Pat. No. 5,459,255, drawn to modified nucleobases based on N-2 substituted purines; U.S. Pat. No. 5,521,302, drawn to processes for preparing oligonucleotides having chiral phosphorus linkages; U.S. Pat. No. 5,539,082, drawn to peptide nucleic acids; U.S. Pat. No. 5,554,746, drawn to oligonucleotides having β-lactam backbones; U.S. Pat. No. 5,571,902, drawn to methods and materials for the synthesis of oligonucleotides; U.S. Pat. No. 5,578,718, drawn to nucleosides having alkylthio groups, wherein such groups may be used as linkers to other moieties attached at any of a variety of positions of the nucleoside; U.S. Pat. Nos. 5,587,361 and 5,599,797, drawn to oligonucleotides having phosphorothioate linkages of high chiral purity; U.S. Pat. No. 5,506,351, drawn to processes for the preparation of 2'-0-alkyl guanosine and related compounds, including 2,6-diaminopurine compounds; U.S. Pat. No. 5,587,469, drawn to oligonucleotides having N-2 substituted purines; U.S. Pat. No. 5,587,470, drawn to oligonucleotides having 3-deazapurines; U.S. Pat. No. 5,223,168, and U.S. Pat. No. 5,608,046, both drawn to conjugated 4'-desmethyl nucleoside analogs; U.S. Pat. Nos. 5,602,240, and 5,610,289, drawn to backbone-modified oligonucleotide analogs; and U.S. Pat. Nos. 6,262,241, and 5,459,255, drawn to, inter alia, methods of synthesizing 2'- fluoro -oligonucleotides .
It is not necessary for all positions in a given compound to be uniformly modified, and in fact more than one type of modification may be incorporated in a single oligonucleotide compound or even in a single nucleotide thereof.
Methods for inhibiting expression of a target gene
In yet another aspect, the invention relates to a method for inhibiting the expression of a target gene in a cell or organism. In one embodiment, the method includes administering the inventive oligonucleotide, e.g. antisense, aptamer, antagomir, or an iRNA agent; or a pharmaceutical composition containing the said oligonucleotide to a cell or an organism, such as a mammal, such that expression of the target gene is silenced. Compositions and methods for inhibiting the expression of a target gene using the inventive oligonucleotide, e.g. an iRNA agent, can be performed as described in the preceding sections.
In this embodiment, a pharmaceutical composition containing the inventive oligonucleotide may be administered by any means known in the art including, but not limited to oral or parenteral routes, including intravenous, intramuscular, intraperitoneal, subcutaneous, transdermal, airway (aerosol), ocular, rectal, vaginal, and topical
(including buccal and sublingual) administration. In preferred embodiments, the pharmaceutical compositions are administered by intravenous or intraparenteral infusion or injection. The pharmaceutical compositions can also be administered
intraparenchymally, intrathecally, and/or by stereotactic injection.
The methods for inhibiting the expression of a target gene can be applied to any gene one wishes to silence, thereby specifically inhibiting its expression, provided the cell or organism in which the target gene is expressed includes the cellular machinery which effects R A interference. Examples of genes which can be targeted for silencing include, without limitation, developmental genes including but not limited to adhesion molecules, cyclin kinase inhibitors, Wnt family members, Pax family members, Winged helix family members, Hox family members, cytokines/lymphokines and their receptors, growth/differentiation factors and their receptors, and neurotransmitters and their receptors; (2) oncogenes including but not limited to ABLI, BCL1, BCL2, BCL6, CBFA2, CBL, CSFIR, ERBA, ERBB, EBRB2, ETS1, ETS1, ETV6, FGR, FOS, FYN, HCR, HRAS, JUN, KRAS, LCK, LYN, MDM2, MLL, MYB, MYC, MYCL1, MYCN, NRAS, PIM1, PML, RET, SRC, TALI, TCL3 and YES; (3) tumor suppresser genes including but not limited to APC, BRCA1, BRCA2, MADH4, MCC, NF1, NF2, RBI, TP53 and WT1; and (4) enzymes including but not limited to ACP desaturases and hydroxylases, ADP-glucose pyrophorylases, ATPases, alcohol dehydrogenases, amylases, amyloglucosidases, catalases, cellulases, cyclooxygenases, decarboxylases, dextrinases, DNA and RNA polymerases, galactosidases, glucanases, glucose oxidases, GTPases, helicases, hemicellulases, integrases, invertases, isomerases, kinases, lactases, lipases, lipoxygenases, lysozymes, pectinesterases, peroxidases, phosphatases, phospholipases, phosphorylases, polygalacturonases, proteinases and peptideases, pullanases, recombinases, reverse transcriptases, topoisomerases, and xylanases.
In addition to in vivo gene inhibition, the skilled artisan will appreciate that the inventive oligonucleotides, e.g. iR A agent, of the present invention are useful in a wide variety of in vitro applications. Such in vitro applications, include, for example, scientific and commercial research (e.g., elucidation of physiological pathways, drug discovery and development), and medical and veterinary diagnostics. In general, the method involves the introduction of the oligonucleotide, e.g. an iRNA agent, into a cell using known techniques (e.g., absorption through cellular processes, or by auxiliary agents or devices, such as electroporation and lipofection), then maintaining the cell for a time sufficient to obtain degradation of an mRNA transcript of the target gene.
Definitions
The term "nucleoside" includes nucleotides as well as nucleoside and nucleotide analogs, and modified nucleosides such as amino modified nucleosides. In addition, "nucleoside" includes non-naturally occurring analog structures. Thus for example the individual units of a peptide nucleic acid, each containing a base, are referred to herein as a nucleoside.
The term "aliphatic," as used herein, refers to a straight or branched hydrocarbon radical containing up to twenty four carbon atoms wherein the saturation between any two carbon atoms is a single, double or triple bond. An aliphatic group preferably contains from 1 to about 24 carbon atoms, more typically from 1 to about 12 carbon atoms. Suitable aliphatic groups include, but are not limited to, linear or branched, substituted or unsubstituted alkyl, alkenyl, alkynyl groups and hybrids thereof such as (cycloalkyl)alkyl, (cycloalkenyl)alkyl or (cycloalkyl)alkenyl. The straight or branched chain of an aliphatic group may be interrupted with one or more heteroatoms that include nitrogen, oxygen, sulfur and phosphorus. Such aliphatic groups interrupted by heteroatoms include without limitation polyalkoxys, such as polyalkylene glycols, polyamines, and polyimines, for example. Aliphatic groups as used herein may optionally include further substitutent groups.
The term "alkyl" refers to saturated and unsaturated non-aromatic hydrocarbon chains that may be a straight chain or branched chain, containing the indicated number of
carbon atoms (these include without limitation propyl, allyl, or propargyl), which may be optionally inserted with N, O, or S. For example, C1-C20 indicates that the group may have from 1 to 20 (inclusive) carbon atoms in it. The term "alkoxy" refers to an -O-alkyl radical. The term "alkylene" refers to a divalent alkyl (i.e., -R-). The term
"alkylenedioxo" refers to a divalent species of the structure -O-R-O-, in which R represents an alkylene. The term "aminoalkyl" refers to an alkyl substituted with an amino. The term "mercapto" refers to an -SH radical. The term "thioalkoxy" refers to an -S-alkyl radical.
The term "cyclic " as used herein includes a cycloalkyl group and a heterocyclic group. Any suitable ring position of the cyclic group may be covalently linked to the defined chemical structure.
The term "acyclic" may describe any carrier that is branched or unbranched, and does not form a closed ring.
The term "aryl" refers to a 6-carbon monocyclic or 10-carbon bicyclic aromatic ring system wherein 0, 1, 2, 3, or 4 atoms of each ring may be substituted by a substituent. Examples of aryl groups include phenyl, naphthyl and the like. The term "arylalkyl" or the term "aralkyl" refers to alkyl substituted with an aryl. The term "arylalkoxy" refers to an alkoxy substituted with aryl.
The term "cycloalkyl" as employed herein includes saturated and partially unsaturated cyclic hydrocarbon groups having 3 to 12 carbons, for example, 3 to 8 carbons, and, for example, 3 to 6 carbons, wherein the cycloalkyl group additionally may be optionally substituted. Cycloalkyl groups include, without limitation, decalin, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cycloheptyl, and cyclooctyl.
The term "heteroaryl" refers to an aromatic 5-8 membered monocyclic, 8-12 membered bicyclic, or 11-14 membered tricyclic ring system having 1-3 heteroatoms if monocyclic, 1-6 heteroatoms if bicyclic, or 1-9 heteroatoms if tricyclic, said heteroatoms selected from O, N, or S (e.g., carbon atoms and 1-3, 1-6, or 1-9 heteroatoms of N, O, or S if monocyclic, bicyclic, or tricyclic, respectively), wherein 0, 1, 2, 3, or 4 atoms of each ring may be substituted by a substituent. Examples of heteroaryl groups include pyridyl,
furyl or furanyl, imidazolyl, benzimidazolyl, pyrimidinyl, thiophenyl or thienyl, quinolinyl, indolyl, thiazolyl, and the like. The term "heteroarylalkyl" or the term "heteroaralkyl" refers to an alkyl substituted with a heteroaryl. The term
"heteroarylalkoxy" refers to an alkoxy substituted with heteroaryl.
The term "heterocycloalkyl" and "heterocyclic" can be used interchangeably and refer to a non-aromatic 3-, 4-, 5-, 6- or 7-membered ring or a bi- or tri-cyclic group fused system, where (i) each ring contains between one and three heteroatoms independently selected from oxygen, sulfur and nitrogen, (ii) each 5-membered ring has 0 to 1 double bonds and each 6-membered ring has 0 to 2 double bonds, (iii) the nitrogen and sulfur heteroatoms may optionally be oxidized, (iv) the nitrogen heteroatom may optionally be quaternized, (iv) any of the above rings may be fused to a benzene ring, and (v) the remaining ring atoms are carbon atoms which may be optionally oxo-substituted.
Representative heterocycloalkyl groups include, but are not limited to, [l,3]dioxolane, pyrrolidinyl, pyrazolinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, piperidinyl, piperazinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl, isothiazolidinyl, quinoxalinyl, pyridazinonyl, and tetrahydrofuryl. Such heterocyclic groups may be further substituted to give substituted heterocyclic.
The term "oxo" refers to an oxygen atom, which forms a carbonyl when attached to carbon, an N-oxide when attached to nitrogen, and a sulfoxide or sulfone when attached to sulfur.
The term "acyl" refers to an alkylcarbonyl, cycloalkylcarbonyl, arylcarbonyl, heterocyclylcarbonyl, or heteroarylcarbonyl substituent, any of which may be further substituted by substituents.
The term "silyl" as used herein is represented by the formula -SiA1 A2 A3, where A1, A2, and A3 can be, independently, hydrogen or a substituted or unsubstituted alkyl, cycloalkyl, alkoxy, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, or heteroaryl group as described herein.
The term "substituted" refers to the replacement of one or more hydrogen radicals in a given structure with the radical of a specified substituent including, but not limited to: halo, alkyl, alkenyl, alkynyl, aryl, heterocyclyl, thiol, alkylthio, arylthio,
alky It hio alky 1, arylthioalkyl, alkylsulfonyl, alkylsulfonylalkyl, arylsulfonylalkyl, alkoxy, aryloxy, aralkoxy, aminocarbonyl, alkylaminocarbonyl, arylaminocarbonyl,
alkoxycarbonyl, aryloxycarbonyl, haloalkyl, amino, trifluoromethyl, cyano, nitro, alkylamino, arylamino, alkylaminoalkyl, arylaminoalkyl, aminoalkylamino, hydroxy, alkoxyalkyl, carboxyalkyl, alkoxycarbonylalkyl, aminocarbonylalkyl, acyl,
aralkoxycarbonyl, carboxylic acid, sulfonic acid, sulfonyl, phosphonic acid, aryl, heteroaryl, heterocyclic, and aliphatic. It is understood that the substituent may be further substituted.
The Bases
Adenine, guanine, cytosine and uracil are the most common bases found in R A. These bases can be modified or replaced to provide R A's having improved properties. E.g., nuclease resistant oligoribonucleotides can be prepared with these bases or with synthetic and natural nucleobases {e.g., inosine, thymine, xanthine, hypoxanthine, nubularine, isoguanisine, or tubercidine) and any one of the above modifications.
Alternatively, substituted or modified analogs of any of the above bases and "universal bases" can be employed. Examples include 2-aminoadenine, 2-fluoroadenine, 6-methyl and other alkyl derivatives of adenine and guanine, 2-propyl and other alkyl derivatives of adenine and guanine, 5-halouracil and cytosine, 5-propynyl uracil and cytosine, 6-azo uracil, cytosine and thymine, 5-uracil (pseudouracil), 4-thiouracil, 5-halouracil, 5-(2- aminopropyl)uracil, 5-amino allyl uracil, 8-halo, amino, thiol, thioalkyl, hydroxyl and other 8-substituted adenines and guanines, 5 -trifluoromethyl and other 5-substituted uracils and cytosines, 7-methylguanine, 5-substituted pyrimidines, 6-azapyrimidines and N-2, N-6 and 0-6 substituted purines, including 2-aminopropyladenine, 5-propynyluracil and 5-propynylcytosine, dihydrouracil, 3-deaza-5-azacytosine, 2-aminopurine, 5- alkyluracil, 7-alkylguanine, 5-alkyl cytosine,7-deazaadenine, N6, N6-dimethyladenine, 2,6-diaminopurine, 5-amino-allyl-uracil, N3-methyluracil, substituted 1,2,4-triazoles, 2- pyridinone, 5-nitroindole, 3-nitropyrrole, 5-methoxyuracil, uracil-5-oxyacetic acid, 5- methoxycarbonylmethyluracil, 5 -methyl-2-thiouracil, 5 -methoxycarbonylmethyl-2- thiouracil, 5-methylaminomethyl-2-thiouracil, 3-(3-amino-3carboxypropyl)uracil, 3- methylcytosine, 5-methylcytosine, N4-acetyl cytosine, 2-thiocytosine, N6-methyladenine,
N6-isopentyladenine, 2-methylthio-N6-isopentenyladenine, N-methylguanines, or O- alkylated bases. Further purines and pyrimidines include those disclosed in U.S. Pat. No. 3,687,808, those disclosed in the Concise Encyclopedia Of Polymer Science And Engineering, pages 858-859, Kroschwitz, J. I., ed. John Wiley & Sons, 1990, and those disclosed by Englisch et al., Angewandte Chemie, International Edition, 1991, 30, 613.
The term "non-natural" nucleobase refers any one of the following: 2- methyladeninyl, N6-methyladeninyl, 2-methylthio-N6-methyladeninyl, N6- isopentenyladeninyl, 2-methylthio-N6-isopentenyladeninyl, N6-(cis- hydroxyisopentenyl)adeninyl, 2-methylthio-N6-(cis-hydroxyisopentenyl) adeninyl, N6- glycinylcarbamoyladeninyl, N6-threonylcarbamoyladeninyl, 2-methylthio-N6-threonyl carbamoyladeninyl, N6-methyl-N6-threonylcarbamoyladeninyl, N6- hydroxynorvalylcarbamoyladeninyl, 2-methylthio-N6-hydroxynorvalyl
carbamoyladeninyl, N6,N6-dimethyladeninyl, 3-methylcytosinyl, 5-methylcytosinyl, 2- thiocytosinyl, 5-formylcytosinyl, N4-methylcytosinyl, 5-hydroxymethylcytosinyl, 1- methylguaninyl, N2-methylguaninyl, 7-methylguaninyl, N2,N2-dimethylguaninyl, N2,7- dimethylguaninyl, N2,N2,7-trimethylguaninyl, 1-methylguaninyl, 7-cyano-7- deazaguaninyl, 7-aminomethyl-7-deazaguaninyl, pseudouracilyl, dihydrouracilyl, 5- methyluracilyl, 1-methylpseudouracilyl, 2-thiouracilyl, 4-thiouracilyl, 2-thiothyminyl 5- methyl-2-thiouracilyl, 3-(3-amino-3-carboxypropyl)uracilyl, 5-hydroxyuracilyl, 5- methoxyuracilyl, uracilyl 5-oxyacetic acid, uracilyl 5-oxyacetic acid methyl ester, 5- (carboxyhydroxymethyl)uracilyl, 5-(carboxyhydroxymethyl)uracilyl methyl ester, 5- methoxycarbonylmethyluracilyl, 5 -methoxycarbonylmethyl-2-thiouracilyl, 5 - amino methyl-2-thiouracilyl, 5 -methylamino methyluracilyl, 5 -methylamino methyl-2- thiouracilyl, 5 -methylamino methyl-2-selenouracilyl, 5-carbamoylmethyluracilyl, 5- carboxymethylamino methyluracilyl, 5 -carboxymethylaminomethyl-2-thiouracilyl, 3 - methyluracilyl, l-methyl-3-(3-amino-3-carboxypropyl) pseudouracilyl, 5- carboxymethyluracilyl, 5 -methyldihydrouracilyl, 3 -methylpseudouracilyl,
EQUIVALENTS
Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents to the specific embodiments of the invention described herein. It is, therefore, to be understood that the foregoing embodiments are presented by way of example only and that, within the scope of the appended claims and equivalents thereto, the invention may be practiced otherwise than as specifically described and claimed.
EXAMPLES
The invention now being generally described, it will be more readily understood by reference to the following examples, which are included merely for purposes of illustration of certain aspects and embodiments of the invention, and are not intended to limit the invention. Thus, the invention should in way be construed as being limited to the following examples, but rather, should be construed to encompass any and all variations which become evident as a result of the teaching provided herein.
Example 1. Design and Synthesis of Novel Solid Supported Cu-Ligand for Click-Chemistry
Scheme I:

3
Preparation of Compound 2: To a solution of azido-PEG acid (1, 2.0 g, 6.88 mmol, 1.0 eq) and benzyl bromide (1.76 g, 10.32 mmol, 1.5 eq) in acetone was added K2C03 (2.84 g, 20.64 mmol, 3.0 eq) as solid and the reaction was refluxed for overnight.
After completion of the reaction by TLC, was cooled to room temperature, filtered K2CO3, evaporated the solvent, re-dissolved in the ethylacetate, washed with water. Organic layer was separated, dried on Na2S04, concentrated and purified on column chromatography using hexane and ethylacetate (50%) as gradients to get pure benzyl protected PEG azide (3) as an oil in 70% yield.
1H NMR (400 MHz, CDC13) δ 7.44 - 7.22 (m, 5H), 5.13 (s, 2H), 3.77 (t, J = 6.5, 2H), 3.71 - 3.53 (m, 14H), 3.37 (t, J = 5.1, 2H), 2.65 (t, J = 6.5, 2H); calc. mass for C18H27N3O6 is 381.4; found 404.2 (+Na).
Preparation of compound 3: To a solution of compound (2, 0.85 g, 2.25 mmol, 3.0 eq) and tris propargylamine (0.09 g, 0.75 mmol, 1.0 eq) in 4: 1 mixture of dichloromethane :methanol at room temperature were added tetrakis methylcyano phosphorous hexaflouro copper (I)complex (0.16 g, 0.45 mmol, 0.6 eq) and elemental Cu(0) (0.02 g, 0.45 mmol, 0.6 eq). Reaction was continued stirring at room temperature for overnight, after completion of the reaction, solvent was evaporated and re-dissolved in the dichloromethane, washed with water. Combined organic layers were dried over MgS04, filtered, concentrated and purified on the column chromatography using dichloromethane and methanol (10%) as gradients to get pure triazolyl ligand (3) as copper complex.
Calc. mass for C63H90CuNioOi8 is 1338.9; found 1339.5.
Example 2. Click Reaction using Cu-Complex 3:

Preparation of compound 5: To a mixture of azide (4, 0.07 g, 0.2 mmol, 1.0 eq) and N,N-dimethyl propargylamine (0.016 g, 0.2 mmol, 1.0 eq) in t-butanol: water (1 : 1) was added tris-(PEG triazolyl) Cu-complex (3, 0.07 g, 0.07 mmol, 0.3 eq) and the
reaction was continued under microwave conditions at 80 °C, and complete consumption of the starting materials were observed after 30 minutes.
Calc. mass for C27H54N4 is 434.74; found 435.4.
Example 3.
Scheme II: Preparation of immobilized Cu Catalyst I (9)

Preparation of Compound 6:
Method A: To a mixture of PEG-azide (2, 0.7 g, 1.83 mmol, 3.0 eq) and tris- propargylamine (0.08 g, 0.61 mmol, 1.0 eq) in t-butanol: water (1 : 1) were added sodium ascorbate (0.036 g, 0.18 mmol, 0.3 eq) followed by CuS04.5H20 (0.004g, 0.018 mmol, 0.03 eq) at room temperature and the reaction was continued for overnight. TLC shows complete consumption of the starting materials, reaction mixture was diluted with excess amount of dichloromethane, separated the organic layer and washed with water. Combined organics were dried on MgS04, concentrated, purified on column chromatography using dichloromethane and methanol (10%) as gradients to get pure benzyl protected triazolyl ligand (6) as an oil in 88% yield.
Method B: To a solution of PEG-azide (2, 0.28 g, 0.75 mmol, 3.0 eq) and tris- propargylamine (0.032 g, 0.25 mmol, 1.0 eq) in t-butanol: water (1 : 1) were added sodium ascorbate (0.015 g, 0.075 mmol, 0.3 eq) followed by CuS04.5H20 (0.0018g, 0.0075
mmol, 0.03 eq) and the reaction was continued under microwave conditions at 80 °C, TLC and LC Mass shows complete consumption of the starting materials after lh. Reaction mixture was diluted with the dichloromethane, separated the organic layer and washed with water. Combined organics were dried on MgSC^, concentrated, purified on column chromatography using dichloromethane and methanol (10%) as gradients to get pure benzyl protected triazolyl ligand (6) as an oil.
1H NMR (400 MHz, CDC13) δ 7.86 (s, 3H), 7.41 - 7.19 (m, 15H), 5.12 (s, 6H), 4.52 (t, J = 5.3, 6H), 3.87 (t, J = 5.3, 6H), 3.76 (dd, J = 7.7, 5.2, 12H), 3.60 (d, J = 3.4, 36H), 2.64 (t, J= 6.5, 6H). Calc. mass for C63H9oNioOi8 is 1274.6; found 1275.5.
Preparation of Compound 7: To a solution of benzyl protected ligand (6, 0.6g, 0.47 mmol) in methanol was added catalytic amount of 10% Pd-C, removed air and purged with the argon, repeated the cycles two times and continued the reaction at room temperature under ¾ atmosphere for overnight. After completion of the reaction was filtered through small bed of celite, washed with methanol, combined washings were concentrated and further dried on vacuum to get pure ligand (7) as oil in 97%> yields.
1H NMR (400 MHz, CDC13) δ 8.08 (s, 3H), 4.57 (t, J = 5.1, 6H), 3.99 (s, 5H), 3.90 (t, J = 5.1, 6H), 3.75 (t, J = 6.1, 6H), 3.65 - 3.54 (m, 35H), 3.47 (s, 5H), 2.56 (t, J = 6.1, 6H). Calc. mass for C42H72N10O18 is 1004.5; found 1005.3
Preparation of solid supported ligand (8): To a solution of compound 7 (0.88 g, 0.87 mmol), HBTU (0.16 g, 0.43 mmol), DIPEA (0.24 mL, 1.13 mmol) in DMF was added aminomethyl polystyrene resin (230 μητηιοΐ^, 1.52 g, 0.35 mmol) and the mixture was continued on shaker at room temperature for overnight. Filtered the solvent, washed the resin with DCM: methanol (9: 1, 2x), DCM (4x), ether (2x) and dried on vacuum gave solid supported triazolyl ligand (8).
Preparation of solid supported trizolyl ligand-Cu complex (9): To a solution of tetrakis methylcyano phosphorous hexaflouro copper (I) complex (0.08 g, 0.23 mmol) in DMF was added ligand 8 (0.5 g, 0.11 mmol) and kept on shaker at room temperature for overnight. Reaction was filtered and the resin was washed with DCM: methanol (9: 1, 2x), DCM (4x), ether (2x) and further dried on vacuum to get Cu-complex (9). Loading of the Cu on resin was analyzed by ICP/OES, which gave 222 μιηοΐ/g, (97%>).
Example 4
Chelated copper mediated click reaction of nucleoside with an azide 11

12
Preparation of Compound 12: To a mixture of alkyne 10 (0.05 g, 0.08 mmol, 1.0 eq) and azide 11 (0.03 g, 0.08 mmol, 1.0 eq) in DMF: DCM (2: 1) was added Cu- complex 9 (0.05 g, 0.01 mmol, 0.12 eq) and the mixture was continued on shaker for overnight. After completion of the reaction was filtered, washed with DCM (2x), the solvent mixture was poured onto ice, extracted with DCM, separated the organic and washed with excess amount of water. Combined organics were dried on MgS04, evaporated and the mixture is purified by column chromatography using dichloromethane: methanol (5%) as gradients to get pure trizole compound (12).
Calc. mass for CsgHygNjOs: 949.5; found 948.5 (M"1).
Recyclable ligand 9: Using ligand 9 in the repeated click reaction cycles gave quantitative yield of the product.
Example 5. Chelated copper mediated click reaction of nucleoside with an azide 13

Preparation of Compound 14: Compound 14 has been prepared by using similar experimental conditions used for 12, using alkyne 10 (0.05 g, 0.08 mmol, 1.0 eq), azide 13 (0.04 g, 0.08 mmol, 1.0 eq) and Cu-complex 9 (0.05 g, 0.01 mmol, 0.12 eq) in DMF: DCM (2: 1) get pure trizole compound (14). Calc. mass for
 1152.6; found 1152.5.
1152.6; found 1152.5.
Example 6. Chelated copper mediated click reaction of Oligonucleotide 15 with azide 16

Preparation of Compound 17: Compound 17 has been prepared by using similar experimental conditions used for 12, using oligo alkyne 15 (35.6 μιηοΐ in stock solution, 1.0 eq), azide 16 (71.0 μιηοΐ, 2.0 eq) and Cu-complex 9 (9.8 μιηοΐ, 0.2 eq) in DMF gave complete conversion of the product 17 in overnight at room temperature. Calc. mass for Compound 17 is 7402, observed 7402.
Example 7: Chelated copper mediated click reaction of Oligonucleotide 15 with azide 18

Cu cat. on solid support (9)
DMF, rt, overnight

19
Preparation of Compound 19: Compound 19 has been prepared by using similar experimental conditions used for 12, using oligo alkyne 15 (35.6 μιηοΐ in stock solution, 1.0 eq), azide 18 (71.0 μιηοΐ, 2.0 eq) and Cu-complex 9 (9.8 μιηοΐ, 0.2 eq) in DMF gave conversion of the product 19 in overnight at room temperature. Calc. mass for Compound 19 is 7341, observed 7342.
Example 8: Chelated copper mediated click reaction of Oligonucleotide 15 with azide 20

Cu cat. on solid support (9)
DMF, rt, overnight

Preparation of Compound 21: Compound 21 has been prepared by using similar experimental conditions used for 12, using oligo alkyne 15 (35.6 μιηοΐ in stock solution, 1.0 eq), azide 20 (71.0 μιηοΐ, 2.0 eq) and Cu-complex 9 (17.8 μιηοΐ, 0.5 eq) in DMF gave complete conversion of the product 21 in overnight at room temperature. Calc. mass for Compound 21 is 7235, observed 7235.
Example 9. Preparation of immobilized Cu Catalyst II:

27, catalyst II
Preparation of Compound 23: To the mixture of tris-tripropargylamine (1.3 g, 10.1 mmol, 3.0 eq) and azide 22 (1.0 g, 3.36 mmol, 1.0 eq) in tert-butanol : H20 (1 : 1) at room temperature was added sodium ascorbate (0.2 g, 1.0 mmol, 0.3 eq) followed by CuS04.5H20 (0.02 g, 0.1 mmol, 0.03 eq) and continued the reaction for overnight. After Completion of the reaction, evaporated the organic solvent, dissolved in the dichloromethane and washed with water. Combined organics were dried on MgSC , concentrated and purified by column chromatography using DCM : MeOH (5%) as gradients to get pure mono triazolyl acid 23 (0. 7 g, 48%) and bis triazolyl compound 24 (0.2 g, 8%).
Preparation of Compound 25: To a solution of PEG-azide 2 (0.66 g, 1.75 mmol, 2.5 eq) and mono triazolyl acid 23 (0.3 g, 0.7 mmol, 1.0 eq) in t-butanol: water (1 : 1) were added sodium ascorbate (0.041 g, 0.21 mmol, 0.3 eq) followed by CuS04.5H20 (0.005g, 0.021 mmol, 0.03 eq) and the reaction was continued under microwave conditions at 100
°C, TLC and LC Mass shows complete consumption of the starting materials after lh. Reaction mixture was diluted with the dichloromethane, separated the organic layer and washed with water. Combined organics were dried on MgS04, concentrated, purified on column chromatography using dichloromethane and methanol (5%) as gradients to get pure tris-triazolyl ligand 25 as a white solid in 82% (0.7 g) yields.
Preparation of Compound 26: Ligand 25 was loaded on amino methyl polystyrene resin as similar conditions mentioned above using compound 25 (0.65 g, 0.55 mmol, 1.2 eq), HBTU (0.26 g, 0.69 mmol, 1.5 eq), DIPEA (0.2 mL, 1.3 mmol, 3.0 eq) and aminomethyl polystyrene resin (230
 2.0 g, 0.46 mmol, 1.0), after complete dry, gave solid supported ligand 26 in 2.1 g yield.
2.0 g, 0.46 mmol, 1.0), after complete dry, gave solid supported ligand 26 in 2.1 g yield.
Preparation of Cu Complex 27: Repeated by exact procedure mentioned above using tetrakis methylcyano phosphorous hexaflouro copper (I) complex (0.08 g, 0.23 mmol, 2.0 eq), ligand 26 (0.5 g, 0.11 mmol, 1.0 eq) in DMF, after completion of the reaction was filtered and the resin was washed with DCM: methanol (9: 1, 2x), DCM (4x), ether (2x) and further dried on vacuum to get Cu-complex (27). Loading of the Cu on resin is analyzed by elemental analysis.
Example 10. Preparation of immobilized Cu Catalyst III:
Synthesis of Terpyridine ligand:

Preparation of Compound 29: ε-caprolactone was added dropwise to a stirred suspension of powdered KOH (1.0 g, 18.65 mmol, 5.0 eq) in DMSO at 60 oC. After 30
minutes, 4'-chloro-2,2',6',2"-terpyridine (28, 1.0 g, 3.73 mmol, 1.0 eq) was added as solid. The reaction mixture was stirred at 60 oC for 48h and then poured into water. Concentrated HCl wa added dropwise to the solution until precipitation of white solid formed (pH -6.0). Precipitation was filtered and the product was dried on vacuum, which gave pure white solid 29 in 93% (1.26 g) yields.
Preparation of Compound 30: Repeated exactly as earlier using compound 29 (0.12 g, 0.34 mmol, 1.5 eq), HBTU (0.13 g, 0.34 mmol, 1.5 eq), DIPEA (0.12 mL, 0.7 mmol, 3.0 eq) and aminomethyl polystyrene resin (230 μητηιοΐ^, 1.0 g, 0.23 mmol, 1.0), after complete dry, gave solid supported ligand 30 in 0.9 g yield.
Preparation of Cu Complex 31: Repeated by exact procedure mentioned above using tetrakis methylcyano phosphorous hexaflouro copper (I) complex (0.08 g, 0.23 mmol, 2.0 eq), ligand 30 (0.5 g, 0.11 mmol, 1.0 eq) in DMF, after completion of the reaction was filtered and the resin was washed with DCM: methanol (9: 1, 2x), DCM (4x), ether (2x) and further dried on vacuum to get Cu-complex (31). Loading of the Cu on resin is analyzed by elemental analysis.
Example 11. Preparation of immobilized Cu Catalyst IV:


32, catalyst IV
Preparation of Cu Complex 32: Repeated by exact procedure mentioned above using Cu(I)Br (0.016 g, 0.11 mmol, 2.0 eq), ligand 26 (0.25 g, 0.057 mmol, 1.0 eq) in
DMF, after completion of the reaction was filtered and the resin was washed with DCM: methanol (9: 1 , 2x), DCM (4x), ether (2x) and further dried on vacuum to get Cu-complex (32). Loading of the Cu on resin is analyzed by elemental analysis.
Example 12: Click reaction of nucleoside 10 with azide 11 using Catalysts II,
III:

Preparation of Compound 12: Repeated exact procedure as earlier for the click reactions using catalyst II and III by using a mixture of alkyne 10 (0.03 g, 0.05 mmol, 1.0 eq) and azide 11 (0.017 g, 0.05 mmol, 1.0 eq) in DMF: DCM (2: 1) and Cu-complex II and III (0.025 g, 0.005 mmol, 0.1 eq) to get the product 12 in complete conversion.
Calc. mass for CsgHygNjOs: 949.5; found 948.5 (M"1).
Claims
1. A chelated copper on solid support of formula A:

(A)
wherein Y1-Y4 are each independently CRP, CRP 2, N, NRN, O, and S; r, s and t are each indenpendently 1-6; R100 is independently for each occurrence OH, ORP, or solid support; RP is independently for each occurrence occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl or optionally substituted heteroaryl; RN is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group; and each linker can be the same or different; provided that at least one R100 is bound to a solid support.
2. The chelated copper solid support of claim 1, selected from group consisting of:

3. A chelated copper on solid support of formula B:
O^R-ioo

(B)
wherein X1-X5 are each independently CRP, CRP 2, N, NRN, O, and S; r, s and t are each indenpendently 1-6; Rioo is independently for each occurrence OH, ORP, or solid support; RP is independently for each occurrence occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl or optionally substituted heteroaryl; RN is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group; and each linker can be the same or different; provided that at least one Rioo is bound to a solid support.
4. A chelated copper on solid support of formula C:

(C)
wherein X1-X4 are each independently CRP, CRP 2, N, NRN, O, and S; J is absent, O, S, NRN, OC(0)NH, NHC(0)0, C(0)NH, NHC(O), NHSO, NHS02, NHS02NH, OC(O), C(0)0, OC(0)0, NHC(0)NH, NHC(S)NH, OC(S)NH, OP(N(RP)2)0, or OP(N(RP)2); RP is independently for each occurrence occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl or optionally substituted heteroaryl; RN is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group; and ® is a solid support.
5. A chelated copper on solid support of formula D:

(D)
wherein is a bond or absent; Y1-Y4 are each independently N, NRN, O, and S;
Rioo is independently for each occurrence OH, ORP, or solid support; Rp is independently for each occurrence occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl or optionally substituted heteroaryl; RN is independently for each occurrence H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted aralkyl, optionally substituted heteroaryl or an amino protecting group; and each linker can be the same or different; provided that at least one Rioo is bound to a solid support.
6. A process for preparing a ligand conjugated oligonucleotide comprising the step of contacting a nucleoside or oligonucleotide containing an alkyne moiety with ligand containing an azide moiety in the presence of a solid supported copper catalyst.
7. The process of claim 6, wherein the ligand is selected from the group consisting of lipids, steroids, vitamins, sugars, proteins, peptides, polyamines, peptide mimics, multivalent lactose, multivalent galactose, N-acetyl-galactosamine, N-acetyl- gulucosamine multivalent mannose, multivalent fucose, glycosylated polyaminoacids, multivalent galactose, transferrin, bisphosphonate, polyglutamate, polyaspartate, a lipid, cholesterol, a steroid, bile acid, folate, vitamin B12, biotin, an RGD peptide, an RGD peptide mimetic and an aptamer.
8. The process of claim 6, wherein the solid support copper catalyst is the chelated copper on solid support of claims 1, 3, or 4.
9. A process for preparing a ligand-conjugated oligonucleotide comprising the step of contacting a nucleoside or oligonucleotide containing an azide moiety with ligand containing an alkyne moiety in the presence of a solid supported copper catalyst.
10. The process of claim 9, wherein the ligand is selected from the group consisting of lipids, steroids, vitamins, sugars, proteins, peptides, polyamines, peptide mimics, multivalent lactose, multivalent galactose, N-acetyl-galactosamine, N-acetyl- gulucosamine multivalent mannose, multivalent fucose, glycosylated polyaminoacids, multivalent galactose, transferrin, bisphosphonate, polyglutamate, polyaspartate, a lipid, cholesterol, a steroid, bile acid, folate, vitamin B12, biotin, an RGD peptide, an RGD peptide mimetic and an aptamer.
11. The process of claim 9, wherein the solid support copper catalyst is the chelated copper on solid support of claims 1, 3, or 4.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US29930210P | 2010-01-28 | 2010-01-28 | |
| US61/299,302 | 2010-01-28 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2011094580A2 true WO2011094580A2 (en) | 2011-08-04 |
| WO2011094580A3 WO2011094580A3 (en) | 2012-06-28 |
Family
ID=44201128
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/022977 WO2011094580A2 (en) | 2010-01-28 | 2011-01-28 | Chelated copper for use in the preparation of conjugated oligonucleotides |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011094580A2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014172885A1 (en) * | 2013-04-26 | 2014-10-30 | Guang Ming Innovation Company (Wuhan) | Group 9 transition metal catalysts and process for use of same |
| US10010562B2 (en) * | 2013-11-06 | 2018-07-03 | Merck Sharp & Dohme Corp. | Dual molecular delivery of oligonucleotides and peptide containing conjugates |
Citations (117)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4415732A (en) | 1981-03-27 | 1983-11-15 | University Patents, Inc. | Phosphoramidite compounds and processes |
| US4458066A (en) | 1980-02-29 | 1984-07-03 | University Patents, Inc. | Process for preparing polynucleotides |
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US4500707A (en) | 1980-02-29 | 1985-02-19 | University Patents, Inc. | Nucleosides useful in the preparation of polynucleotides |
| US4587044A (en) | 1983-09-01 | 1986-05-06 | The Johns Hopkins University | Linkage of proteins to nucleic acids |
| US4605735A (en) | 1983-02-14 | 1986-08-12 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| US4667025A (en) | 1982-08-09 | 1987-05-19 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| US4668777A (en) | 1981-03-27 | 1987-05-26 | University Patents, Inc. | Phosphoramidite nucleoside compounds |
| US4725677A (en) | 1983-08-18 | 1988-02-16 | Biosyntech Gmbh | Process for the preparation of oligonucleotides |
| US4762779A (en) | 1985-06-13 | 1988-08-09 | Amgen Inc. | Compositions and methods for functionalizing nucleic acids |
| US4824941A (en) | 1983-03-10 | 1989-04-25 | Julian Gordon | Specific antibody to the native form of 2'5'-oligonucleotides, the method of preparation and the use as reagents in immunoassays or for binding 2'5'-oligonucleotides in biological systems |
| US4828979A (en) | 1984-11-08 | 1989-05-09 | Life Technologies, Inc. | Nucleotide analogs for nucleic acid labeling and detection |
| US4835263A (en) | 1983-01-27 | 1989-05-30 | Centre National De La Recherche Scientifique | Novel compounds containing an oligonucleotide sequence bonded to an intercalating agent, a process for their synthesis and their use |
| US4876335A (en) | 1986-06-30 | 1989-10-24 | Wakunaga Seiyaku Kabushiki Kaisha | Poly-labelled oligonucleotide derivative |
| US4904582A (en) | 1987-06-11 | 1990-02-27 | Synthetic Genetics | Novel amphiphilic nucleic acid conjugates |
| US4948882A (en) | 1983-02-22 | 1990-08-14 | Syngene, Inc. | Single-stranded labelled oligonucleotides, reactive monomers and methods of synthesis |
| US4958013A (en) | 1989-06-06 | 1990-09-18 | Northwestern University | Cholesteryl modified oligonucleotides |
| US4973679A (en) | 1981-03-27 | 1990-11-27 | University Patents, Inc. | Process for oligonucleo tide synthesis using phosphormidite intermediates |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US5082830A (en) | 1988-02-26 | 1992-01-21 | Enzo Biochem, Inc. | End labeled nucleotide probe |
| US5109124A (en) | 1988-06-01 | 1992-04-28 | Biogen, Inc. | Nucleic acid probe linked to a label having a terminal cysteine |
| US5112963A (en) | 1987-11-12 | 1992-05-12 | Max-Planck-Gesellschaft Zur Foerderung Der Wissenschaften E.V. | Modified oligonucleotides |
| US5118802A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | DNA-reporter conjugates linked via the 2' or 5'-primary amino group of the 5'-terminal nucleoside |
| US5130302A (en) | 1989-12-20 | 1992-07-14 | Boron Bilogicals, Inc. | Boronated nucleoside, nucleotide and oligonucleotide compounds, compositions and methods for using same |
| US5132418A (en) | 1980-02-29 | 1992-07-21 | University Patents, Inc. | Process for preparing polynucleotides |
| US5138045A (en) | 1990-07-27 | 1992-08-11 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5142047A (en) | 1985-03-15 | 1992-08-25 | Anti-Gene Development Group | Uncharged polynucleotide-binding polymers |
| USRE34069E (en) | 1983-08-18 | 1992-09-15 | Biosyntech Gmbh | Process for the preparation of oligonucleotides |
| US5149782A (en) | 1988-08-19 | 1992-09-22 | Tanox Biosystems, Inc. | Molecular conjugates containing cell membrane-blending agents |
| WO1992020822A1 (en) | 1991-05-21 | 1992-11-26 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| US5177198A (en) | 1989-11-30 | 1993-01-05 | University Of N.C. At Chapel Hill | Process for preparing oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5212295A (en) | 1990-01-11 | 1993-05-18 | Isis Pharmaceuticals | Monomers for preparation of oligonucleotides having chiral phosphorus linkages |
| US5214136A (en) | 1990-02-20 | 1993-05-25 | Gilead Sciences, Inc. | Anthraquinone-derivatives oligonucleotides |
| US5218105A (en) | 1990-07-27 | 1993-06-08 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5223168A (en) | 1989-12-12 | 1993-06-29 | Gary Holt | Surface cleaner and treatment |
| US5223618A (en) | 1990-08-13 | 1993-06-29 | Isis Pharmaceuticals, Inc. | 4'-desmethyl nucleoside analog compounds |
| US5235033A (en) | 1985-03-15 | 1993-08-10 | Anti-Gene Development Group | Alpha-morpholino ribonucleoside derivatives and polymers thereof |
| US5245022A (en) | 1990-08-03 | 1993-09-14 | Sterling Drug, Inc. | Exonuclease resistant terminally substituted oligonucleotides |
| US5254469A (en) | 1989-09-12 | 1993-10-19 | Eastman Kodak Company | Oligonucleotide-enzyme conjugate that can be used as a probe in hybridization assays and polymerase chain reaction procedures |
| US5256775A (en) | 1989-06-05 | 1993-10-26 | Gilead Sciences, Inc. | Exonuclease-resistant oligonucleotides |
| US5258506A (en) | 1984-10-16 | 1993-11-02 | Chiron Corporation | Photolabile reagents for incorporation into oligonucleotide chains |
| US5262536A (en) | 1988-09-15 | 1993-11-16 | E. I. Du Pont De Nemours And Company | Reagents for the preparation of 5'-tagged oligonucleotides |
| US5264564A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences | Oligonucleotide analogs with novel linkages |
| US5264562A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| WO1993023569A1 (en) | 1992-05-11 | 1993-11-25 | Ribozyme Pharmaceuticals, Inc. | Method and reagent for inhibiting viral replication |
| WO1993024510A1 (en) | 1992-05-25 | 1993-12-09 | Centre National De La Recherche Scientifique (Cnrs) | Phosphotriester-type biologically active compounds |
| US5272250A (en) | 1992-07-10 | 1993-12-21 | Spielvogel Bernard F | Boronated phosphoramidate compounds |
| WO1994002595A1 (en) | 1992-07-17 | 1994-02-03 | Ribozyme Pharmaceuticals, Inc. | Method and reagent for treatment of animal diseases |
| US5292873A (en) | 1989-11-29 | 1994-03-08 | The Research Foundation Of State University Of New York | Nucleic acids labeled with naphthoquinone probe |
| US5317098A (en) | 1986-03-17 | 1994-05-31 | Hiroaki Shizuya | Non-radioisotope tagging of fragments |
| US5359044A (en) | 1991-12-13 | 1994-10-25 | Isis Pharmaceuticals | Cyclobutyl oligonucleotide surrogates |
| US5366878A (en) | 1990-02-15 | 1994-11-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of RNA and production of encoded polypeptides |
| WO1994026764A1 (en) | 1993-05-12 | 1994-11-24 | Centre National De La Recherche Scientifique (Cnrs) | Triester phosphorothioate oligonucleotides and method of preparation |
| US5371241A (en) | 1991-07-19 | 1994-12-06 | Pharmacia P-L Biochemicals Inc. | Fluorescein labelled phosphoramidites |
| US5386023A (en) | 1990-07-27 | 1995-01-31 | Isis Pharmaceuticals | Backbone modified oligonucleotide analogs and preparation thereof through reductive coupling |
| US5391723A (en) | 1989-05-31 | 1995-02-21 | Neorx Corporation | Oligonucleotide conjugates |
| US5414077A (en) | 1990-02-20 | 1995-05-09 | Gilead Sciences | Non-nucleoside linkers for convenient attachment of labels to oligonucleotides using standard synthetic methods |
| US5451463A (en) | 1989-08-28 | 1995-09-19 | Clontech Laboratories, Inc. | Non-nucleoside 1,3-diol reagents for labeling synthetic oligonucleotides |
| US5457191A (en) | 1990-01-11 | 1995-10-10 | Isis Pharmaceuticals, Inc. | 3-deazapurines |
| US5459255A (en) | 1990-01-11 | 1995-10-17 | Isis Pharmaceuticals, Inc. | N-2 substituted purines |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| US5484908A (en) | 1991-11-26 | 1996-01-16 | Gilead Sciences, Inc. | Oligonucleotides containing 5-propynyl pyrimidines |
| US5486603A (en) | 1990-01-08 | 1996-01-23 | Gilead Sciences, Inc. | Oligonucleotide having enhanced binding affinity |
| US5489677A (en) | 1990-07-27 | 1996-02-06 | Isis Pharmaceuticals, Inc. | Oligonucleoside linkages containing adjacent oxygen and nitrogen atoms |
| US5506351A (en) | 1992-07-23 | 1996-04-09 | Isis Pharmaceuticals | Process for the preparation of 2'-O-alkyl guanosine and related compounds |
| US5508270A (en) | 1993-03-06 | 1996-04-16 | Ciba-Geigy Corporation | Nucleoside phosphinate compounds and compositions |
| US5510475A (en) | 1990-11-08 | 1996-04-23 | Hybridon, Inc. | Oligonucleotide multiple reporter precursors |
| US5512667A (en) | 1990-08-28 | 1996-04-30 | Reed; Michael W. | Trifunctional intermediates for preparing 3'-tailed oligonucleotides |
| US5512439A (en) | 1988-11-21 | 1996-04-30 | Dynal As | Oligonucleotide-linked magnetic particles and uses thereof |
| US5514785A (en) | 1990-05-11 | 1996-05-07 | Becton Dickinson And Company | Solid supports for nucleic acid hybridization assays |
| US5519134A (en) | 1994-01-11 | 1996-05-21 | Isis Pharmaceuticals, Inc. | Pyrrolidine-containing monomers and oligomers |
| US5525465A (en) | 1987-10-28 | 1996-06-11 | Howard Florey Institute Of Experimental Physiology And Medicine | Oligonucleotide-polyamide conjugates and methods of production and applications of the same |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| US5539083A (en) | 1994-02-23 | 1996-07-23 | Isis Pharmaceuticals, Inc. | Peptide nucleic acid combinatorial libraries and improved methods of synthesis |
| US5541307A (en) | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| US5545730A (en) | 1984-10-16 | 1996-08-13 | Chiron Corporation | Multifunctional nucleic acid monomer |
| US5554746A (en) | 1994-05-16 | 1996-09-10 | Isis Pharmaceuticals, Inc. | Lactam nucleic acids |
| US5565552A (en) | 1992-01-21 | 1996-10-15 | Pharmacyclics, Inc. | Method of expanded porphyrin-oligonucleotide conjugate synthesis |
| US5571902A (en) | 1993-07-29 | 1996-11-05 | Isis Pharmaceuticals, Inc. | Synthesis of oligonucleotides |
| US5574142A (en) | 1992-12-15 | 1996-11-12 | Microprobe Corporation | Peptide linkers for improved oligonucleotide delivery |
| US5578718A (en) | 1990-01-11 | 1996-11-26 | Isis Pharmaceuticals, Inc. | Thiol-derivatized nucleosides |
| US5580731A (en) | 1994-08-25 | 1996-12-03 | Chiron Corporation | N-4 modified pyrimidine deoxynucleotides and oligonucleotide probes synthesized therewith |
| US5585481A (en) | 1987-09-21 | 1996-12-17 | Gen-Probe Incorporated | Linking reagents for nucleotide probes |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5587470A (en) | 1990-01-11 | 1996-12-24 | Isis Pharmaceuticals, Inc. | 3-deazapurines |
| US5587371A (en) | 1992-01-21 | 1996-12-24 | Pharmacyclics, Inc. | Texaphyrin-oligonucleotide conjugates |
| US5595726A (en) | 1992-01-21 | 1997-01-21 | Pharmacyclics, Inc. | Chromophore probe for detection of nucleic acid |
| US5597696A (en) | 1994-07-18 | 1997-01-28 | Becton Dickinson And Company | Covalent cyanine dye oligonucleotide conjugates |
| US5599797A (en) | 1991-10-15 | 1997-02-04 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5599923A (en) | 1989-03-06 | 1997-02-04 | Board Of Regents, University Of Tx | Texaphyrin metal complexes having improved functionalization |
| US5602240A (en) | 1990-07-27 | 1997-02-11 | Ciba Geigy Ag. | Backbone modified oligonucleotide analogs |
| US5608046A (en) | 1990-07-27 | 1997-03-04 | Isis Pharmaceuticals, Inc. | Conjugated 4'-desmethyl nucleoside analog compounds |
| US5610289A (en) | 1990-07-27 | 1997-03-11 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| US5614617A (en) | 1990-07-27 | 1997-03-25 | Isis Pharmaceuticals, Inc. | Nuclease resistant, pyrimidine modified oligonucleotides that detect and modulate gene expression |
| US5672662A (en) | 1995-07-07 | 1997-09-30 | Shearwater Polymers, Inc. | Poly(ethylene glycol) and related polymers monosubstituted with propionic or butanoic acids and functional derivatives thereof for biotechnical applications |
| US5688941A (en) | 1990-07-27 | 1997-11-18 | Isis Pharmaceuticals, Inc. | Methods of making conjugated 4' desmethyl nucleoside analog compounds |
| US5714166A (en) | 1986-08-18 | 1998-02-03 | The Dow Chemical Company | Bioactive and/or targeted dendrimer conjugates |
| WO2000044895A1 (en) | 1999-01-30 | 2000-08-03 | Roland Kreutzer | Method and medicament for inhibiting the expression of a defined gene |
| US6153737A (en) | 1990-01-11 | 2000-11-28 | Isis Pharmaceuticals, Inc. | Derivatized oligonucleotides having improved uptake and other properties |
| US6172208B1 (en) | 1992-07-06 | 2001-01-09 | Genzyme Corporation | Oligonucleotides modified with conjugate groups |
| US6262241B1 (en) | 1990-08-13 | 2001-07-17 | Isis Pharmaceuticals, Inc. | Compound for detecting and modulating RNA activity and gene expression |
| US6287860B1 (en) | 2000-01-20 | 2001-09-11 | Isis Pharmaceuticals, Inc. | Antisense inhibition of MEKK2 expression |
| US6300319B1 (en) | 1998-06-16 | 2001-10-09 | Isis Pharmaceuticals, Inc. | Targeted oligonucleotide conjugates |
| WO2001075164A2 (en) | 2000-03-30 | 2001-10-11 | Whitehead Institute For Biomedical Research | Rna sequence-specific mediators of rna interference |
| US6335437B1 (en) | 1998-09-07 | 2002-01-01 | Isis Pharmaceuticals, Inc. | Methods for the preparation of conjugated oligomers |
| US6335434B1 (en) | 1998-06-16 | 2002-01-01 | Isis Pharmaceuticals, Inc., | Nucleosidic and non-nucleosidic folate conjugates |
| US6395437B1 (en) | 1999-10-29 | 2002-05-28 | Advanced Micro Devices, Inc. | Junction profiling using a scanning voltage micrograph |
| WO2002044321A2 (en) | 2000-12-01 | 2002-06-06 | MAX-PLANCK-Gesellschaft zur Förderung der Wissenschaften e.V. | Rna interference mediating small rna molecules |
| US6444806B1 (en) | 1996-04-30 | 2002-09-03 | Hisamitsu Pharmaceutical Co., Inc. | Conjugates and methods of forming conjugates of oligonucleotides and carbohydrates |
| US6486308B2 (en) | 1995-04-03 | 2002-11-26 | Epoch Biosciences, Inc. | Covalently linked oligonucleotide minor groove binder conjugates |
| US6528631B1 (en) | 1993-09-03 | 2003-03-04 | Isis Pharmaceuticals, Inc. | Oligonucleotide-folate conjugates |
| US6559279B1 (en) | 2000-09-08 | 2003-05-06 | Isis Pharmaceuticals, Inc. | Process for preparing peptide derivatized oligomeric compounds |
| US20050261218A1 (en) | 2003-07-31 | 2005-11-24 | Christine Esau | Oligomeric compounds and compositions for use in modulation small non-coding RNAs |
| US20060166910A1 (en) | 2002-07-10 | 2006-07-27 | Max-Planck-Gesellschaft Zur Foerderung Der Wissenschaften E.V. | Rna-interference by single-stranded rna molecules |
| US11598905B2 (en) | 2020-03-19 | 2023-03-07 | Korea Institute Of Science And Technology | Inverted nanocone structure for optical device and method of producing the same |
| US11953305B2 (en) | 2019-02-04 | 2024-04-09 | Detnet South Africa (Pty) Ltd | Detonator installation including a controller |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0507123D0 (en) * | 2005-04-08 | 2005-05-11 | Isis Innovation | Method |
-
2011
- 2011-01-28 WO PCT/US2011/022977 patent/WO2011094580A2/en active Application Filing
Patent Citations (131)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4458066A (en) | 1980-02-29 | 1984-07-03 | University Patents, Inc. | Process for preparing polynucleotides |
| US4500707A (en) | 1980-02-29 | 1985-02-19 | University Patents, Inc. | Nucleosides useful in the preparation of polynucleotides |
| US5132418A (en) | 1980-02-29 | 1992-07-21 | University Patents, Inc. | Process for preparing polynucleotides |
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US4415732A (en) | 1981-03-27 | 1983-11-15 | University Patents, Inc. | Phosphoramidite compounds and processes |
| US4668777A (en) | 1981-03-27 | 1987-05-26 | University Patents, Inc. | Phosphoramidite nucleoside compounds |
| US4973679A (en) | 1981-03-27 | 1990-11-27 | University Patents, Inc. | Process for oligonucleo tide synthesis using phosphormidite intermediates |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US4667025A (en) | 1982-08-09 | 1987-05-19 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| US4789737A (en) | 1982-08-09 | 1988-12-06 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives and production thereof |
| US4835263A (en) | 1983-01-27 | 1989-05-30 | Centre National De La Recherche Scientifique | Novel compounds containing an oligonucleotide sequence bonded to an intercalating agent, a process for their synthesis and their use |
| US4605735A (en) | 1983-02-14 | 1986-08-12 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| US5541313A (en) | 1983-02-22 | 1996-07-30 | Molecular Biosystems, Inc. | Single-stranded labelled oligonucleotides of preselected sequence |
| US4948882A (en) | 1983-02-22 | 1990-08-14 | Syngene, Inc. | Single-stranded labelled oligonucleotides, reactive monomers and methods of synthesis |
| US4824941A (en) | 1983-03-10 | 1989-04-25 | Julian Gordon | Specific antibody to the native form of 2'5'-oligonucleotides, the method of preparation and the use as reagents in immunoassays or for binding 2'5'-oligonucleotides in biological systems |
| USRE34069E (en) | 1983-08-18 | 1992-09-15 | Biosyntech Gmbh | Process for the preparation of oligonucleotides |
| US4725677A (en) | 1983-08-18 | 1988-02-16 | Biosyntech Gmbh | Process for the preparation of oligonucleotides |
| US4587044A (en) | 1983-09-01 | 1986-05-06 | The Johns Hopkins University | Linkage of proteins to nucleic acids |
| US5118802A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | DNA-reporter conjugates linked via the 2' or 5'-primary amino group of the 5'-terminal nucleoside |
| US5552538A (en) | 1984-10-16 | 1996-09-03 | Chiron Corporation | Oligonucleotides with cleavable sites |
| US5545730A (en) | 1984-10-16 | 1996-08-13 | Chiron Corporation | Multifunctional nucleic acid monomer |
| US5578717A (en) | 1984-10-16 | 1996-11-26 | Chiron Corporation | Nucleotides for introducing selectably cleavable and/or abasic sites into oligonucleotides |
| US5258506A (en) | 1984-10-16 | 1993-11-02 | Chiron Corporation | Photolabile reagents for incorporation into oligonucleotide chains |
| US4828979A (en) | 1984-11-08 | 1989-05-09 | Life Technologies, Inc. | Nucleotide analogs for nucleic acid labeling and detection |
| US5235033A (en) | 1985-03-15 | 1993-08-10 | Anti-Gene Development Group | Alpha-morpholino ribonucleoside derivatives and polymers thereof |
| US5142047A (en) | 1985-03-15 | 1992-08-25 | Anti-Gene Development Group | Uncharged polynucleotide-binding polymers |
| US4762779A (en) | 1985-06-13 | 1988-08-09 | Amgen Inc. | Compositions and methods for functionalizing nucleic acids |
| US5317098A (en) | 1986-03-17 | 1994-05-31 | Hiroaki Shizuya | Non-radioisotope tagging of fragments |
| US4876335A (en) | 1986-06-30 | 1989-10-24 | Wakunaga Seiyaku Kabushiki Kaisha | Poly-labelled oligonucleotide derivative |
| US5714166A (en) | 1986-08-18 | 1998-02-03 | The Dow Chemical Company | Bioactive and/or targeted dendrimer conjugates |
| US4904582A (en) | 1987-06-11 | 1990-02-27 | Synthetic Genetics | Novel amphiphilic nucleic acid conjugates |
| US5585481A (en) | 1987-09-21 | 1996-12-17 | Gen-Probe Incorporated | Linking reagents for nucleotide probes |
| US5525465A (en) | 1987-10-28 | 1996-06-11 | Howard Florey Institute Of Experimental Physiology And Medicine | Oligonucleotide-polyamide conjugates and methods of production and applications of the same |
| US5112963A (en) | 1987-11-12 | 1992-05-12 | Max-Planck-Gesellschaft Zur Foerderung Der Wissenschaften E.V. | Modified oligonucleotides |
| US5082830A (en) | 1988-02-26 | 1992-01-21 | Enzo Biochem, Inc. | End labeled nucleotide probe |
| US5109124A (en) | 1988-06-01 | 1992-04-28 | Biogen, Inc. | Nucleic acid probe linked to a label having a terminal cysteine |
| US5149782A (en) | 1988-08-19 | 1992-09-22 | Tanox Biosystems, Inc. | Molecular conjugates containing cell membrane-blending agents |
| US5262536A (en) | 1988-09-15 | 1993-11-16 | E. I. Du Pont De Nemours And Company | Reagents for the preparation of 5'-tagged oligonucleotides |
| US5512439A (en) | 1988-11-21 | 1996-04-30 | Dynal As | Oligonucleotide-linked magnetic particles and uses thereof |
| US5599923A (en) | 1989-03-06 | 1997-02-04 | Board Of Regents, University Of Tx | Texaphyrin metal complexes having improved functionalization |
| US5391723A (en) | 1989-05-31 | 1995-02-21 | Neorx Corporation | Oligonucleotide conjugates |
| US5256775A (en) | 1989-06-05 | 1993-10-26 | Gilead Sciences, Inc. | Exonuclease-resistant oligonucleotides |
| US5416203A (en) | 1989-06-06 | 1995-05-16 | Northwestern University | Steroid modified oligonucleotides |
| US4958013A (en) | 1989-06-06 | 1990-09-18 | Northwestern University | Cholesteryl modified oligonucleotides |
| US5451463A (en) | 1989-08-28 | 1995-09-19 | Clontech Laboratories, Inc. | Non-nucleoside 1,3-diol reagents for labeling synthetic oligonucleotides |
| US5254469A (en) | 1989-09-12 | 1993-10-19 | Eastman Kodak Company | Oligonucleotide-enzyme conjugate that can be used as a probe in hybridization assays and polymerase chain reaction procedures |
| US5264564A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences | Oligonucleotide analogs with novel linkages |
| US5264562A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| US5292873A (en) | 1989-11-29 | 1994-03-08 | The Research Foundation Of State University Of New York | Nucleic acids labeled with naphthoquinone probe |
| US5177198A (en) | 1989-11-30 | 1993-01-05 | University Of N.C. At Chapel Hill | Process for preparing oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5223168A (en) | 1989-12-12 | 1993-06-29 | Gary Holt | Surface cleaner and treatment |
| US5130302A (en) | 1989-12-20 | 1992-07-14 | Boron Bilogicals, Inc. | Boronated nucleoside, nucleotide and oligonucleotide compounds, compositions and methods for using same |
| US5486603A (en) | 1990-01-08 | 1996-01-23 | Gilead Sciences, Inc. | Oligonucleotide having enhanced binding affinity |
| US5587469A (en) | 1990-01-11 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides containing N-2 substituted purines |
| US5578718A (en) | 1990-01-11 | 1996-11-26 | Isis Pharmaceuticals, Inc. | Thiol-derivatized nucleosides |
| US5212295A (en) | 1990-01-11 | 1993-05-18 | Isis Pharmaceuticals | Monomers for preparation of oligonucleotides having chiral phosphorus linkages |
| US5587470A (en) | 1990-01-11 | 1996-12-24 | Isis Pharmaceuticals, Inc. | 3-deazapurines |
| US5521302A (en) | 1990-01-11 | 1996-05-28 | Isis Pharmaceuticals, Inc. | Process for preparing oligonucleotides having chiral phosphorus linkages |
| US6153737A (en) | 1990-01-11 | 2000-11-28 | Isis Pharmaceuticals, Inc. | Derivatized oligonucleotides having improved uptake and other properties |
| US5457191A (en) | 1990-01-11 | 1995-10-10 | Isis Pharmaceuticals, Inc. | 3-deazapurines |
| US5459255A (en) | 1990-01-11 | 1995-10-17 | Isis Pharmaceuticals, Inc. | N-2 substituted purines |
| US5366878A (en) | 1990-02-15 | 1994-11-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of RNA and production of encoded polypeptides |
| US5414077A (en) | 1990-02-20 | 1995-05-09 | Gilead Sciences | Non-nucleoside linkers for convenient attachment of labels to oligonucleotides using standard synthetic methods |
| US5214136A (en) | 1990-02-20 | 1993-05-25 | Gilead Sciences, Inc. | Anthraquinone-derivatives oligonucleotides |
| US5514785A (en) | 1990-05-11 | 1996-05-07 | Becton Dickinson And Company | Solid supports for nucleic acid hybridization assays |
| US5610289A (en) | 1990-07-27 | 1997-03-11 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| US5489677A (en) | 1990-07-27 | 1996-02-06 | Isis Pharmaceuticals, Inc. | Oligonucleoside linkages containing adjacent oxygen and nitrogen atoms |
| US5386023A (en) | 1990-07-27 | 1995-01-31 | Isis Pharmaceuticals | Backbone modified oligonucleotide analogs and preparation thereof through reductive coupling |
| US5378825A (en) | 1990-07-27 | 1995-01-03 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs |
| US5541307A (en) | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| US5602240A (en) | 1990-07-27 | 1997-02-11 | Ciba Geigy Ag. | Backbone modified oligonucleotide analogs |
| US5608046A (en) | 1990-07-27 | 1997-03-04 | Isis Pharmaceuticals, Inc. | Conjugated 4'-desmethyl nucleoside analog compounds |
| US5218105A (en) | 1990-07-27 | 1993-06-08 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5688941A (en) | 1990-07-27 | 1997-11-18 | Isis Pharmaceuticals, Inc. | Methods of making conjugated 4' desmethyl nucleoside analog compounds |
| US5138045A (en) | 1990-07-27 | 1992-08-11 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5614617A (en) | 1990-07-27 | 1997-03-25 | Isis Pharmaceuticals, Inc. | Nuclease resistant, pyrimidine modified oligonucleotides that detect and modulate gene expression |
| US5567810A (en) | 1990-08-03 | 1996-10-22 | Sterling Drug, Inc. | Nuclease resistant compounds |
| US5245022A (en) | 1990-08-03 | 1993-09-14 | Sterling Drug, Inc. | Exonuclease resistant terminally substituted oligonucleotides |
| US6262241B1 (en) | 1990-08-13 | 2001-07-17 | Isis Pharmaceuticals, Inc. | Compound for detecting and modulating RNA activity and gene expression |
| US5223618A (en) | 1990-08-13 | 1993-06-29 | Isis Pharmaceuticals, Inc. | 4'-desmethyl nucleoside analog compounds |
| US5512667A (en) | 1990-08-28 | 1996-04-30 | Reed; Michael W. | Trifunctional intermediates for preparing 3'-tailed oligonucleotides |
| US5510475A (en) | 1990-11-08 | 1996-04-23 | Hybridon, Inc. | Oligonucleotide multiple reporter precursors |
| WO1992020822A1 (en) | 1991-05-21 | 1992-11-26 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| WO1992020823A1 (en) | 1991-05-21 | 1992-11-26 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs |
| US5371241A (en) | 1991-07-19 | 1994-12-06 | Pharmacia P-L Biochemicals Inc. | Fluorescein labelled phosphoramidites |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5599797A (en) | 1991-10-15 | 1997-02-04 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5484908A (en) | 1991-11-26 | 1996-01-16 | Gilead Sciences, Inc. | Oligonucleotides containing 5-propynyl pyrimidines |
| US5359044A (en) | 1991-12-13 | 1994-10-25 | Isis Pharmaceuticals | Cyclobutyl oligonucleotide surrogates |
| US5587371A (en) | 1992-01-21 | 1996-12-24 | Pharmacyclics, Inc. | Texaphyrin-oligonucleotide conjugates |
| US5565552A (en) | 1992-01-21 | 1996-10-15 | Pharmacyclics, Inc. | Method of expanded porphyrin-oligonucleotide conjugate synthesis |
| US5595726A (en) | 1992-01-21 | 1997-01-21 | Pharmacyclics, Inc. | Chromophore probe for detection of nucleic acid |
| WO1993023569A1 (en) | 1992-05-11 | 1993-11-25 | Ribozyme Pharmaceuticals, Inc. | Method and reagent for inhibiting viral replication |
| WO1993024510A1 (en) | 1992-05-25 | 1993-12-09 | Centre National De La Recherche Scientifique (Cnrs) | Phosphotriester-type biologically active compounds |
| US6172208B1 (en) | 1992-07-06 | 2001-01-09 | Genzyme Corporation | Oligonucleotides modified with conjugate groups |
| US5272250A (en) | 1992-07-10 | 1993-12-21 | Spielvogel Bernard F | Boronated phosphoramidate compounds |
| WO1994002595A1 (en) | 1992-07-17 | 1994-02-03 | Ribozyme Pharmaceuticals, Inc. | Method and reagent for treatment of animal diseases |
| US5506351A (en) | 1992-07-23 | 1996-04-09 | Isis Pharmaceuticals | Process for the preparation of 2'-O-alkyl guanosine and related compounds |
| US5574142A (en) | 1992-12-15 | 1996-11-12 | Microprobe Corporation | Peptide linkers for improved oligonucleotide delivery |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| US5508270A (en) | 1993-03-06 | 1996-04-16 | Ciba-Geigy Corporation | Nucleoside phosphinate compounds and compositions |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| US5770713A (en) | 1993-05-12 | 1998-06-23 | Centre National De La Recherche Scientifique | Phosphorothioate triester oligonucleotides and method of preparation |
| WO1994026764A1 (en) | 1993-05-12 | 1994-11-24 | Centre National De La Recherche Scientifique (Cnrs) | Triester phosphorothioate oligonucleotides and method of preparation |
| US5571902A (en) | 1993-07-29 | 1996-11-05 | Isis Pharmaceuticals, Inc. | Synthesis of oligonucleotides |
| US6528631B1 (en) | 1993-09-03 | 2003-03-04 | Isis Pharmaceuticals, Inc. | Oligonucleotide-folate conjugates |
| US5519134A (en) | 1994-01-11 | 1996-05-21 | Isis Pharmaceuticals, Inc. | Pyrrolidine-containing monomers and oligomers |
| US5599928A (en) | 1994-02-15 | 1997-02-04 | Pharmacyclics, Inc. | Texaphyrin compounds having improved functionalization |
| US5539083A (en) | 1994-02-23 | 1996-07-23 | Isis Pharmaceuticals, Inc. | Peptide nucleic acid combinatorial libraries and improved methods of synthesis |
| US5554746A (en) | 1994-05-16 | 1996-09-10 | Isis Pharmaceuticals, Inc. | Lactam nucleic acids |
| US5597696A (en) | 1994-07-18 | 1997-01-28 | Becton Dickinson And Company | Covalent cyanine dye oligonucleotide conjugates |
| US5591584A (en) | 1994-08-25 | 1997-01-07 | Chiron Corporation | N-4 modified pyrimidine deoxynucleotides and oligonucleotide probes synthesized therewith |
| US5580731A (en) | 1994-08-25 | 1996-12-03 | Chiron Corporation | N-4 modified pyrimidine deoxynucleotides and oligonucleotide probes synthesized therewith |
| US6486308B2 (en) | 1995-04-03 | 2002-11-26 | Epoch Biosciences, Inc. | Covalently linked oligonucleotide minor groove binder conjugates |
| US5672662A (en) | 1995-07-07 | 1997-09-30 | Shearwater Polymers, Inc. | Poly(ethylene glycol) and related polymers monosubstituted with propionic or butanoic acids and functional derivatives thereof for biotechnical applications |
| US6444806B1 (en) | 1996-04-30 | 2002-09-03 | Hisamitsu Pharmaceutical Co., Inc. | Conjugates and methods of forming conjugates of oligonucleotides and carbohydrates |
| US6525031B2 (en) | 1998-06-16 | 2003-02-25 | Isis Pharmaceuticals, Inc. | Targeted Oligonucleotide conjugates |
| US6300319B1 (en) | 1998-06-16 | 2001-10-09 | Isis Pharmaceuticals, Inc. | Targeted oligonucleotide conjugates |
| US6335434B1 (en) | 1998-06-16 | 2002-01-01 | Isis Pharmaceuticals, Inc., | Nucleosidic and non-nucleosidic folate conjugates |
| US6335437B1 (en) | 1998-09-07 | 2002-01-01 | Isis Pharmaceuticals, Inc. | Methods for the preparation of conjugated oligomers |
| WO2000044895A1 (en) | 1999-01-30 | 2000-08-03 | Roland Kreutzer | Method and medicament for inhibiting the expression of a defined gene |
| US6395437B1 (en) | 1999-10-29 | 2002-05-28 | Advanced Micro Devices, Inc. | Junction profiling using a scanning voltage micrograph |
| US6287860B1 (en) | 2000-01-20 | 2001-09-11 | Isis Pharmaceuticals, Inc. | Antisense inhibition of MEKK2 expression |
| WO2001075164A2 (en) | 2000-03-30 | 2001-10-11 | Whitehead Institute For Biomedical Research | Rna sequence-specific mediators of rna interference |
| US6559279B1 (en) | 2000-09-08 | 2003-05-06 | Isis Pharmaceuticals, Inc. | Process for preparing peptide derivatized oligomeric compounds |
| WO2002044321A2 (en) | 2000-12-01 | 2002-06-06 | MAX-PLANCK-Gesellschaft zur Förderung der Wissenschaften e.V. | Rna interference mediating small rna molecules |
| US20060166910A1 (en) | 2002-07-10 | 2006-07-27 | Max-Planck-Gesellschaft Zur Foerderung Der Wissenschaften E.V. | Rna-interference by single-stranded rna molecules |
| US20050261218A1 (en) | 2003-07-31 | 2005-11-24 | Christine Esau | Oligomeric compounds and compositions for use in modulation small non-coding RNAs |
| US11953305B2 (en) | 2019-02-04 | 2024-04-09 | Detnet South Africa (Pty) Ltd | Detonator installation including a controller |
| US11598905B2 (en) | 2020-03-19 | 2023-03-07 | Korea Institute Of Science And Technology | Inverted nanocone structure for optical device and method of producing the same |
Non-Patent Citations (90)
| Title |
|---|
| "Angew. Chem.", vol. 41, 2002, pages: 2596 - 2599 |
| "Concise Encyclopedia Of Polymer Science And Engineering", 1990, JOHN WILEY & SONS, pages: 858 - 859 |
| "Manoharan in Antisense Research and Applications", 1993, CRC PRESS |
| "Oligonucleotide synthesis, a practical approach", 1984, IRL PRESS |
| "Oligonucleotides and Analogues, A Practical Approach", 1991, IRL PRESS |
| "Peptide Nucleic Acids (PNA): Synthesis, Properties and Potential Applications", BIOORGANIC & MEDICINAL CHEMISTRY, vol. 4, 1996, pages 5 - 23 |
| AN, H ET AL., J. ORG. CHEM., vol. 66, 2001, pages 2789 - 2801 |
| AOKI ET AL., CANCER GENE THERAPY, vol. 8, 2001, pages 783 - 787 |
| AUSIN ET AL.: "Synthesis of Amino- and Guanidino-G-Clamp PNA Monomers", ORGANIC LETTERS, vol. 4, 2002, pages 4073 - 4075, XP002515121, DOI: doi:10.1021/OL026815P |
| BEAUCAGE, S. L.; IYER, R. P., TETRAHEDRON, vol. 48, 1992, pages 2223 - 2311 |
| BEAUCAGE, S. L.; IYER, R. P., TETRAHEDRON, vol. 49, 1993, pages 6123 - 6194 |
| BROWN; SIMPSON, ANNU REV PLANT PHYSIOL PLANT MOL BIOL, vol. 49, 1998, pages 77 - 95 |
| CANDELON, N. ET AL., CHEM. COMM., 2008, pages 741 - 743 |
| CHAN, T. R. ET AL., ORG. LETT., vol. 6, 2004, pages 2853 - 2855 |
| CHAN, T. R.; HILGRAF, R.; SHRAPLESS, K. B.; FOKIN, V.V., ORG. LETT., vol. 6, 2004, pages 2853 |
| CHASSAING ET AL.: "recently reported copper(I) zeolites as catalysts for the azide-alkyne cycloaddition", CHEM. EUR. J., vol. 14, 2008, pages 6713 - 6721 |
| CORMIER,J.F. ET AL., NUCLEIC ACIDS RES., vol. 16, 1988, pages 4583 |
| CROSSTICK ET AL., TETRAHEDRON LETT., vol. 30, 1989, pages 4693 |
| DIEZ-GONZALEZ, S. ET AL., CHEM. EUR. J., vol. 12, 2006, pages 7558 - 7564 |
| EATON, CURR. OPIN. CHEM. BIOL., vol. 1, 1997, pages 10 - 16 |
| EDGE, M.D. ET AL., J. CHEM. SOC. PERKIN TRANS., vol. 1, 1972, pages 1991 |
| ELBASHIR ET AL., EMBO, vol. 20, 2001, pages 6877 - 6888 |
| ELLINGTON; SZOSTAK, NATURE, vol. 346, no. 818, 1990 |
| ENGLISCH ET AL.: "Angewandte Chemie", vol. 30, 1991, pages: 613 |
| FAMULOK, CURR. OPIN. STRUCT. BIOL., vol. 9, 1999, pages 324 - 329 |
| FLANAGAN ET AL.: "A cytosine analog that confers enhanced potency to antisense oligonucleotides", PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA, vol. 96, 1999, pages 3513 - 3518 |
| FOLKIN, V.V. ET AL., ORG. LETT., vol. 127, 2005, pages 15998 - 15999 |
| FORSTER; SYMONS, CELL, vol. 49, no. 2, 24 April 1987 (1987-04-24), pages 211 - 220 |
| GAO, H. ET AL., MACROMOLECULES, vol. 38, 2005, pages 8979 |
| GAO, H. F.; MATYJASZEWSKI, K., MACROMOLECULES, vol. 39, 2006, pages 4960 |
| GORACZNIAK ET AL., NATURE BIOTECHNOLOGY, vol. 27, no. 3, 2008, pages 257 - 263 |
| GUJADHUR, R.; VENKATARAMAN, D.; J. T. KINTIGH. J.T., TET. LETT., vol. 42, 2001, pages 4791 |
| HAUBNER ET AL., JOUR. NUCL. MED., vol. 42, 2001, pages 326 - 336 |
| HERMANN; PATEL, SCIENCE, vol. 287, 2000, pages 820 - 825 |
| HIMO, F. ET AL., J. AM. CHEM. SOC., vol. 127, 2005, pages 210 - 216 |
| HOLMES ET AL.: "Steric inhibition of human immunodeficiency virus type- Tat-dependent trans-activation in vitro and in cells by oligonucleotides containing 2'-O-methyl G-clamp ribonucleoside analogues", NUCLEIC ACIDS RESEARCH, vol. 31, 2003, pages 2759 - 2768, XP002354372, DOI: doi:10.1093/nar/gkg384 |
| JOHNSON, J. A. ET AL., J. AM. CHEM. SOC., vol. 128, 2006, pages 6564 |
| JORGENSEN, K. A.: "Angew. Chem.", vol. 39, 2000, pages: 3558 - 3588 |
| KAWASAKI, J MED. CHEM., vol. 36, 1993, pages 831 - 841 |
| KIM; CECH, PROC NATL ACAD SCI USA., vol. 84, no. 24, December 1987 (1987-12-01), pages 8788 - 8792 |
| KOLB, H. C.; FINN, M. G.; SHARPLESS, K. B.: "Angew. Chem.", vol. 40, 2001, pages: 2004 - 2021 |
| KOLB, H. C.; SHRAPLESS, K. B.: "The growing impact of click chemistry on drug discovery", DRUG DISC. TODAY, vol. 8, no. 24, 15 December 2003 (2003-12-15), pages 1128 - 1137, XP002377521, DOI: doi:10.1016/S1359-6446(03)02933-7 |
| KRANISKI, A.; FOKIN, V.V.; SHARPLESS, K.B., ORG. LETT., vol. 6, 2004, pages 1237 - 1240 |
| KRUTZFELDT ET AL., NATURE, vol. 438, 2005, pages 685 - 689 |
| LAM ET AL., NATURE, vol. 354, 1991, pages 82 - 84 |
| LAURENT, B.A.; GRAYSON, S.M., J. AM. CHEM. SOC., vol. 128, 2006, pages 4238 |
| LEE, L.V. ET AL., J. AM. CHEM. SOC., vol. 125, 2003, pages 9588 - 9589 |
| LEWIS, W.G. ET AL., J. AM. CHEM. SOC., vol. 126, 2004, pages 9152 - 9153 |
| LEWIS, W.G. ET AL.: "Angew. Chem.", vol. 41, 2002, pages: 1053 - 1057 |
| LUTZ, J. F.; BORNER, H. G.; WEICHENHAN, K., MACROMOL. RAPID COMM., vol. 26, 2005, pages 514 |
| MAIER ET AL.: "Nuclease resistance of oligonucleotides containing the tricyclic cytosine analogues phenoxazine and 9-(2-aminoethoxy)-phenoxazine ("G-clamp") and origins of their nuclease resistance properties", BIOCHEMISTRY, vol. 41, 2002, pages 1323 - 1327 |
| MALKOCH, M. ET AL., MACROMOLECULES, vol. 38, 2005, pages 3663 |
| MANETSCH, R. ET AL., J. AM. CHEM. SOC., vol. 126, 2004, pages 12809 - 12818 |
| MANN ET AL., J. CLIN. INVEST., vol. 106, 2000, pages 1071 - 1075 |
| MANOHARAN, M. ET AL., ANTISENSE AND NUCLEIC ACID DRUG DEVELOPMENT, vol. 12, 2002, pages 103 - 128 |
| MANTOVANI, G. ET AL., CHEM. COMM., 2005, pages 2089 - 2091 |
| MANTOVANI, G.; LADMIRAL, V.; TAO, L.; HADDLETON, D. M., CHEM. COMM., 2005, pages 2089 |
| MARTIN, P., HELV. CHIM. ACTA, vol. 78, 1995, pages 486 - 504 |
| MARTIN, P., HELV. CHIM. ACTA, vol. 79, 1996, pages 1930 - 1938 |
| MOCHARLA, V.P. ET AL.: "Angew. Chem.", vol. 44, 2005, pages: 116 - 120 |
| MOLTENI, G. ET AL., NEWJ. CHEM, vol. 30, 2006, pages 1137 - 1139 |
| NUCLEOSIDES, NUCLEOTIDES & NUCLEIC ACIDS, vol. 22, 2003, pages 1259 - 1262 |
| OPSTEEN, J. A.; VAN HEST, J. C. M., CHEM. COMMUN., 2005, pages 57 |
| PACHON, L. D. ET AL., ADV. SYNTH. CATAL., vol. 347, 2005, pages 811 - 815 |
| PADWA, A.: "1,3-Dipolar Cycloaddition Chemistry", vol. 1, 1984, JOHN WILEY, pages: 1 - 176 |
| RAJEEV ET AL.: "High-Affinity Peptide Nucleic Acid Oligomers Containing Tricyclic Cytosine Analogues", ORGANIC LETTERS, vol. 4, 2002, pages 4395 - 4398, XP008156256, DOI: doi:10.1021/ol027026a |
| ROSTOCTSEV, V. V.; GREEN L. G.; FOKIN, V. V.; SHRAPLESS, K.B.: "Angew. Chem.", vol. 41, 2002, pages: 2596 - 2599 |
| ROSTOVTSEV, V. V. ET AL., ANGEW. CHEM., vol. 114, 2002, pages 2708 - 2711 |
| ROSTOVTSEV, V. V.; GREEN L. G.; FOKIN, V. V.; SHRAPLESS, K.B.: "Angew. Chem.", vol. 41, 2002, pages: 2596 - 2599 |
| SHARPLESS, W. D.; WU, P.; HANSEN, T. V.; LI, J.G., J. CHEM. ED., vol. 82, 2005, pages 1833 |
| SIMEONI ET AL., NUCL. ACIDS RES., vol. 31, 2003, pages 2717 - 2724 |
| SOUTSCHEK ET AL., NATURE, vol. 432, 2004, pages 173 - 178 |
| SPROAT ET AL., NUCLEOSIDES NUCLEOTIDES, vol. 7, 1988, pages 651 |
| STIRCHAK, E.P., NUCLEIC ACIDS RES., vol. 17, 1989, pages 6129 |
| SUBBARAO ET AL., BIOCHEMISTRY, vol. 26, 1987, pages 2964 - 2972 |
| SUMERLIN, B. S. ET AL., MACROMOLECULES, vol. 38, 2005, pages 7540 |
| TIETZE, L. F.; KETTSCHAU, G., TOP. CURR. CHEM., vol. 189, 1997, pages 1 - 120 |
| TITTENSOR, J.R., J. CHEM. SOC. C, 1971, pages 1933 |
| TOMOE, C. W.; CHRISTENSEN, C.; MELDAL, M., J. ORG. CHEM., vol. 67, 2002, pages 3057 - 3064 |
| TORNOE, C. W.; CHRISTENSEN, C.; MELDAL, M., J. ORG. CHEM., vol. 67, 2002, pages 3057 - 3064 |
| TSAREVSKY, N. V.; SUMERLIN, B. S.; MATYJASZEWSKI, K., MACROMOLECULES, vol. 38, 2005, pages 3558 |
| TUERK; GOLD, SCIENCE, vol. 249, 1990, pages 505 |
| TURK ET AL., BIOCHEM. BIOPHYS. ACTA, vol. 1559, 2002, pages 56 - 68 |
| VERMA, S. ET AL., ANNU. REV. BIOCHEM., vol. 67, 1998, pages 99 - 134 |
| VOGEL ET AL., J. AM. CHEM. SOC., vol. 118, 1996, pages 1581 - 1586 |
| VOGT, A. P.; SUMERLIN, B.S., MACROMOLECULES, vol. 39, 2006, pages 5286 |
| WANG, Q. ET AL., J. AM. CHEM. SOC., vol. 125, 2003, pages 3192 - 3193 |
| WENGEL, J., ACC. CHEM. RES., vol. 32, 1999, pages 301 - 310 |
| WILDS ET AL.: "Structural basis for recognition of guanosine by a synthetic tricyclic cytosine analogue: Guanidinium G-clamp", HELVETICA CHIMICA ACTA, vol. 86, 2003, pages 966 - 978 |
| ZITZMANN ET AL., CANCER RES., vol. 62, 2002, pages 5139 - 5143 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014172885A1 (en) * | 2013-04-26 | 2014-10-30 | Guang Ming Innovation Company (Wuhan) | Group 9 transition metal catalysts and process for use of same |
| CN105531028A (en) * | 2013-04-26 | 2016-04-27 | 光明创新(武汉)有限公司 | Group 9 transition metal catalysts and process for use of same |
| CN105531028B (en) * | 2013-04-26 | 2019-06-25 | 光明创新(武汉)有限公司 | 9th group 4 transition metal catalyst and its application method |
| US10010562B2 (en) * | 2013-11-06 | 2018-07-03 | Merck Sharp & Dohme Corp. | Dual molecular delivery of oligonucleotides and peptide containing conjugates |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2011094580A3 (en) | 2012-06-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2708171C (en) | Folate conjugates | |
| AU2016202354B2 (en) | Chemical modifications of monomers and oligonucleotides with cycloaddition | |
| US10550386B2 (en) | Monomers and oligonucleotides comprising cycloaddition adduct(s) | |
| US9725479B2 (en) | 5′-end derivatives | |
| AU2009234266B2 (en) | Site-specific delivery of nucleic acids by combining targeting ligands with endosomolytic components | |
| WO2010006237A2 (en) | Phosphorothioate oligonucleotides non-nucleosidic phosphorothiotes as delivery agents for irna agents | |
| WO2011163121A1 (en) | Multifunctional copolymers for nucleic acid delivery | |
| KR20240010762A (en) | Modified double-stranded rna agents | |
| US20130203836A1 (en) | 2' and 5' modified monomers and oligonucleotides | |
| WO2009120878A2 (en) | Non-natural ribonucleotides, and methods of use thereof | |
| WO2011094580A2 (en) | Chelated copper for use in the preparation of conjugated oligonucleotides | |
| AU2017206154A1 (en) | Site-specific delivery of nucleic acids by combining targeting ligands with endosomolytic components |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11706662 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11706662 Country of ref document: EP Kind code of ref document: A2 |