WO2011069197A1 - Inhalable formulations - Google Patents
Inhalable formulations Download PDFInfo
- Publication number
- WO2011069197A1 WO2011069197A1 PCT/AU2010/001656 AU2010001656W WO2011069197A1 WO 2011069197 A1 WO2011069197 A1 WO 2011069197A1 AU 2010001656 W AU2010001656 W AU 2010001656W WO 2011069197 A1 WO2011069197 A1 WO 2011069197A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- particles
- particle
- excipient
- microns
- respiratory
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
- A61K9/0073—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy
- A61K9/0075—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy for inhalation via a dry powder inhaler [DPI], e.g. comprising micronized drug mixed with lactose carrier particles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/137—Arylalkylamines, e.g. amphetamine, epinephrine, salbutamol, ephedrine or methadone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/167—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the nitrogen of a carboxamide group directly attached to the aromatic ring, e.g. lidocaine, paracetamol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/565—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids not substituted in position 17 beta by a carbon atom, e.g. estrane, estradiol
- A61K31/568—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids not substituted in position 17 beta by a carbon atom, e.g. estrane, estradiol substituted in positions 10 and 13 by a chain having at least one carbon atom, e.g. androstanes, e.g. testosterone
- A61K31/569—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids not substituted in position 17 beta by a carbon atom, e.g. estrane, estradiol substituted in positions 10 and 13 by a chain having at least one carbon atom, e.g. androstanes, e.g. testosterone substituted in position 17 alpha, e.g. ethisterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/57—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/57—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone
- A61K31/573—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone substituted in position 21, e.g. cortisone, dexamethasone, prednisone or aldosterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/58—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids containing heterocyclic rings, e.g. danazol, stanozolol, pancuronium or digitogenin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
Definitions
- the present invention relates to inhalable formulations, to methods of manufacturing inhalable formulations and to pharmaceutical compositions containing inhalable formulations.
- DPI Dry powder inhalation
- DPI therapy depends on a number of factors including the biological aspect of the active ingredient, the physicochemical properties of the formulation, and the performance of the inhaler.
- the efficiency of dose delivery of dry powders also depends on the particle size, size distribution, shape and surface morphology of the powder.
- drug particles having a size between 1 and 5 microns have a high surface area to mass ratio and therefore tend to be highly cohesive resulting in poor aerosolisation efficiency and thus respiratory deposition.
- their delivery into the lung is traditionally enhanced when they are blended with larger and coarser inert carrier materials.
- the aim is for the drug particles to be freed from the carriers and to enter and penetrate the lung while the carriers themselves impact in the upper airways and are ingested.
- DPI therapy may deliver a single active or may deliver a combination of actives.
- Combination inhalation therapy have been used to treat respiratory diseases and combination therapies include an inhaled corticosteroid (ICS) and a long-acting ⁇ 2- agonist (LABA) which offers the advantages of convenience to the patients along with synergistic pharmacological actions, leading to better patient compliance and therapeutic outcomes.
- ICS inhaled corticosteroid
- LAA long-acting ⁇ 2- agonist
- APIs active pharmaceutical ingredients
- MDI suspension metered dose inhaler
- differences in the particle properties may cause differential suspension characteristics of the APIs and wall loss to the canister, resulting in variable product performance. Wall loss can potentially be minimized by non-sticky coating of the canister wall.
- disparities in the formulations have been dealt with by separating them in two canisters, it would make the MDI more bulky and potentially require more effort to actuate.
- DPI formulations rely on the use of blends in which the API particles adhere on the carrier lactose surface. Lactose is the major carrier used in DPIs and its performance is highly variable, depending on its amount of fines, surface roughness, polymorphic form, production batch, and grade.
- the present inventors have found that it is possible to form inhalable particles from two or more active agents together with an excipient which is at least partially in a crystalline form, and that these particles may have advantageous properties, especially when used in a dry particle inhaler (DPI).
- DPI dry particle inhaler
- a particle which is of respirable size and which contains two or more active agents and an excipient at least partially in a crystalline form.
- inhalable formulations comprised particles of active agent(s) of inhalable size together with a larger non-respirable carrier particle.
- the present invention has found that it is possible to form respirable sized particles containing two or more active agents and an excipient at least partially in a crystalline form.
- the ability to form uniform particles of a defined size containing both the actives and excipient is of great use because it allows for more uniform particle formation providing targeted delivery of active agents to patients using more manageable particles.
- an inhalable particle comprising two or more active agents and an excipient which is at least partially in crystalline form, wherein the particle is of respirable size.
- the present invention is useful in inhalable formulations which will generally be made up of many particles. Therefore, there will generally be a large number of particles in a given composition. This collection of particles will comprise at least one but generally a large number of the particles disclosed herein. Accordingly, in a further embodiment of the present invention there is provided inhalable particles comprising two or more active agents and an excipient which is at least partially in a crystalline form, wherein the particles are of respirable size.
- At least 1%, at least 2%, at least 5%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, at least 98%, at least 99%, or 100% of the particles in the total collection of particles are particles according to the invention as disclosed herein.
- the present invention has identified a novel particle of respirable size comprising active agent(s) and excipients as disclosed herein.
- the particles contain an excipient. This may be thought of as a bulking agent.
- the excipient is present to assist with formulation and/or performance of the inhalable particles rather than to have a physiological effect itself on the patient.
- the excipient may itself have a physiological effect.
- excipients like mannitol are encompassed by the present invention. Therefore, mannitol may be thought of as an excipient or bulking agent for the purposes of the present invention regardless of the fact that mannitol has been shown to have a physiological effect on the body.
- the traditional carrier particle system required the active agents to be milled to respirable size and then mixed with a larger carrier particle. Typical active doses were relatively small (on the milligram scale) and it was not possible to deliver such a small dose to a patient. It was therefore necessary to use additional excipients to 'bulk out' the total composition so as to facilitate manufacture and delivery.
- the total formulation that was prepared could be of a manageable size but the patient still received the required dose of active agents.
- using larger particles assisted in formulation because it avoided some of the difficulties associated with small particles on the 1-3 micron scale, like aggregation and poor flow properties. However, there were difficulties with this approach because the active agents did not always separate from the larger carriers during delivery in a predictable manner.
- the present invention aims, at least in its preferred forms to overcome some or all of these difficulties by combining all of the actives and the excipient together in a single particle. This has a number of advantages. Putting multiple actives together into a single particle means that both actives will be delivered to the same target site at the same time. It avoids the problems associated with different actives having different aerodynamic characteristics.
- the actives and excipient together means that all of the particles of the composition contain the active agents rather than a small amount of active particles and a large amount of non- functioning carrier or bulking agents. Furthermore, the particles are at least partially ' crystalline. It is possible for the first time to prepare particles of defined uniform shapes which may improve and standardise their aerodynamic performance. The crystallinity may also help with stability and long term storage for a number of reasons including: avoiding or minimising the transformation of amorphous particles into more crystalline forms with the associated degradation in the particle morphology, and improved surface properties which may reduce or minimise aggregation or other unwanted surface phenomenon like undesirable interactions with the containers.
- particles of the present invention can be formed from a combination of at least one active agent and an excipient, where the particles are at least partially, or substantially hollow. This has advantages because a hollow particle, when compared against an equivalent sized solid particle, will have a lower density which can lead to improvements in aerodynamic and aerosolisation properties. This can be seen when considering the aerodynamic diameter of a particle which depends on both the physical diameter and density of the particle.
- the dynamic shape factor accounts for the effect of shape on particle motion. It is the ratio of the resistance force experienced by the non-spherical particle moving in air to that of a sphere with the same volume and velocity.
- hollow inhalable particles comprising one or more active agents and an excipient which is at least partially in a crystalline form, wherein the particles are of respirable size.
- Particular embodiments include two or more active agents.
- an inhalable particle comprising one or more active agents and an excipient which is at least partially in crystalline form, wherein the particle is of respirable size and is hollow.
- the particles of the present invention consist essentially of a single or two or more active agents and an excipient.
- respirable it is meant a particle which has an aerodynamic diameter of less than 20 microns. This allows for targeted delivery of particles to comprising two or more active agents and an excipient which is at least partially in a crystalline form to areas of the respiratory tract. Particles are considered respirable if they can be inhaled and deposited onto one or more of the oropharynx and upper airways (including the trachea), lower airways (including the bronchus and bronchioles) or deposited in the alveoli. In particular embodiments the respirable particles can be deposited onto the lower airways. This provides particles which can be used in inhalation therapy. The present invention allows for better design of particles with advantageous size distributions. Thus, it is possible to prepare particles which are easy to handle and which can deliver active agents to areas with better specificity and success.
- the excipient may be selected from any excipient which may at least partially be in crystalline form in the particle and which may be suitable for pulmonary administration.
- exemplary excipients include sugars, sugar alcohols, amino acids and other excipients.
- the excipient may be selected from sugars and sugar alcohols (including mannitol, sucrose, glucose, trehalose, lactose, dextrose, sorbitol, maltilol, maltodextrin); amino acids (including glycine, leucine, trileucine, arginine, threonine, phenylalanine, aspartic acid); and other excipients such as sodium chloride, poly-lactic glycolic acid or poly ethylene glycol.
- a particular excipient is mannitol.
- a further particular excipient is glucose. Both excipients have a relatively low Tg. This means that the excipients will be more likely to be in crystalline form in the particle.
- the excipient is chosen on the basis that it has a Tg ⁇ 150°C. In a further embodiment the excipient is chosen on the basis that it has a Tg ⁇ 100°C, Tg ⁇ 60°C or Tg ⁇ 50°C.
- Tg values are ⁇ 40°C, ⁇ 35°C, ⁇ 30°C, ⁇ 25°C, ⁇ 20°C, ⁇ 15°C, ⁇ 10°C, ⁇ 5°C, ⁇ 0°C, ⁇ - 5°C, ⁇ -01°C, ⁇ -15°C, ⁇ -20°C, ⁇ -25°C.
- the particles may contain a single excipient. In further embodiments, the particles may contain two or more excipients. The particles may contain 1 , 2, 3, 4, 5, 6, 7, 8 or more excipients.
- the excipient is present at least partially in a crystalline form.
- the excipient is present in a majority crystalline form.
- the excipient is present in about 1%, about 2%, about 3%, about 4%, about 5%, about 6%, about 7%, about 8%, about 9%, about 10% , about 20%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, about 95%, or about 100% crystalline form.
- a specific embodiment of the invention has an excipient comprising mannitol which is present in a majority crystalline form, which may be about 50%, about 60%, about 70%, about 80%, about 90%, about 95%, or about 100%-crystalline form.
- At least one active agent is in crystalline form in the particle.
- an active is present in about 10%, about 20%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, or about 100% crystalline form.
- two or more actives are present in about 10%, about 20%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, or about 100% crystalline form.
- the particles comprise about at least 10% excipient.
- the particles comprise about at least 20% excipient.
- the particles comprise about at least 30% excipient.
- the particles comprise about at least 40% excipient.
- the particles comprise about at least 50% excipient. In a yet further embodiment, the particles comprise about at least 60% excipient. In a yet further embodiment, the particles comprise about at least 70% excipient. In a yet further embodiment, the particles comprise about at least 80% excipient. In a yet further embodiment, the particles comprise about at least 90% excipient. In a yet further embodiment, the particles comprise about at least 95% excipient. In a yet further embodiment, the particles comprise about at least 99% excipient.
- Particular embodiments contain at least about 40% excipient. Further particular embodiments contain at least 50% excipient. Yet further particular embodiments contain about at least 80% excipient.
- Increasing the amount of excipient may increase the degree of crystallinity of the particles.
- the amount of excipient may be chosen to achieve the desired degree of crystallinity. There may be a linear relationship between the amount of excipient and the degree of crystallinity. However, for some particles there may be a non-linear relationship between the degree of crystallinity and the percentage amount of excipient.
- the percentage degree of crystallinity is greater than or equal to the percentage amount of excipient present in the particle.
- a particle containing 80% excipient would be at least 80% or more crystalline.
- the percentage degree of crystallinity is greater than the percentage amount of excipient present in the particle.
- the percentage degree of crystallinity exceeds the percentage amount of excipient present in the particle.
- the percentage degree of crystallinity is at least 1 % greater than the percentage amount of excipient.
- a particle with 80% excipient would have at least 81% crystallinity.
- the percentage degree of crystallinity is at least 2%, at least 3%, at least 4%, at least 5%, at least 6%, at least 7%, at least 8%, at least 9%, at least 10%, at least 15%, at least 20%, at least 25%, or at least 30%, greater than the percentage amount of excipient.
- higher percentage crystallinity values vs percentage excipient have not been seen.
- the present invention provides for the first time a system to produce highly crystalline particles combining an active and an excipient. By forming the components together it is possible to achieve improved particle performance due, at least in part, to the crystalline nature of the particles.
- inhalable particles of respirable size comprising one or more active agents and an excipient wherein the percentage crystallinity of the particle is greater than the percentage amount of excipient present in the particle.
- Particular embodiments include two or more active agents.
- an inhalable particle of respirable size comprising one or more active agents and an excipient wherein the percentage crystallinity of the particle is greater than the percentage amount of excipient present in the particle.
- the amount of active agent present in the particle can influence the physical properties of the particle, for example the morphology or aerodynamic performance of the particles. Having a small amount of actives present in the overall composition will mean that the particle is primarily made up of the excipient and the physical characteristics of the excipient may dominate the characteristics of the particle as a whole. For example, if the excipient adopts a predominantly crystalline structure and is present in sufficient amounts in the particle as a whole then it may force an otherwise amorphous active agent into a crystalline composition. However, if the active agents are predominantly present in the composition then they may dominate the overall properties of the particle. The amount of each active will also have an effect on the physiological properties of the particle.
- the total amount of active agents present in the particle is about 1%. In further embodiments, the total amount of active agents present is about 2%, about 3%, about 4%, about 5%, about 6%, about 7%, about 8%, about 9%, about 10%, about 11%, about 12%, about 13%, about 14%, about 15%, about 16%, about 17%, about 18%, about 19%, about 20%, about 25%, about 30%, about 35%, about 40%, about 45%, about 50%, about 55%, about 60%, about 65%, about 70%, about 75%, about 80%, about 85%, or about 90%. In a further embodiment, the amount of actives present in the particle is up to about 50%. In yet further embodiments, the amount of actives present in the particle is up to about 40%, up to about 30%, up to about 20%, up to about 15%, or up to about 10%.
- the present invention provides particles which can be used in inhalation therapy. It is therefore important that a sufficient quantity of the particles are of respirable size. This allows sufficient quantity of the active agents to reach the target areas of the patient. Such target areas include one or more of the alveoli, the lower airways including the bronchus and bronchioles, the upper airways, for example the trachea and/or the oropharynx. Appropriate selection of the particle characteristics allows for targeting delivery to particular areas of the respiratory tract. When designing respirable particles it is desirable to be able to carefully control the particle characteristics to ensure that sufficient particles are of the required respirable size so that enough of the active agent is delivered to the required area.
- particles of the present invention When forming particles of the present invention it is appreciated that a large number of physical particles are formed and that a large number of particles are used in an inhalation treatment. When considering large numbers of particles it is expected that there will be a distribution of individual particles sizes, ranging from very small through to large. There may be a normal distribution of particle sizes with a percentage of the particles within a given size range. Thus, when considering the particles of the present invention the particle size may be thought of as the average particle size. Thus, for example, if the particles are said to be about 1-3 microns in size, this represents an average particle size, with some particles being smaller or larger than the average particle size.
- the present invention is intended to cover particles which, on average, have a size of about 7 microns.
- this average size may be the mean particle size.
- the particle size may be the median particle size.
- the present invention provides particles of respirable size.
- the actual diameters of the particles in a sample will range depending on factors like particle composition and method of synthesis.
- the distribution of particle sizes can be chosen to achieve the desired delivery of the active agents to the target areas in the respiratory tract.
- a percentage of the particles may be of respirable size.
- a powder formed using the particles of the present invention may contain 10% particles of respirable size with the remaining particles being non-respirable. It is generally preferable that the amount of respirable particles is maximised. The more particles that can reach the target delivery area, a lower overall dose of active agents may be needed. It is generally preferred to minimise the dose provided to a patient.
- about 1% of the particles (do.oi) are of respirable size.
- the particles may be measured in terms of their aerodynamic diameter.
- aerodynamic diameter (d ae ) P° '5 d p , where p is the density of the particle and d p the physical diameter of the particle.
- the advantage of considering an aerodynamic diameter is that it differentiates between otherwise similar physically sized particles that have different aerodynamic properties. As an example, a hollow sphere will have a lower density than an equally sized solid sphere.
- the aerodynamic diameter of the two spheres will be different with the hollow sphere being lower, even though the physical diameter is the same. Equally, seemingly differently shaped or sized particles may have the same aerodynamic diameter.
- An advantage of considering the aerodynamic diameter of a collection of particles is that it can be measured empirically in an impactor and it is not necessary to look at each particle individually to measure its physical size. The inhalation properties of the particles are assessed and the average aerodynamic properties of the particles determined to arrive at the average aerodynamic diameter of the particles are obtained. A typical measurement will identify what percentage of the total particles are within various aerodynamic diameter size ranges.
- the average aerodynamic diameter of the particles is between from about 0.01 to about 20 microns. In a further embodiment, the average particle size is between from about 0.1 to about 20 microns. In a yet further embodiment, the average particle size is between from about 0.1 to about 15 microns, about 0.2 to about 15 microns, about 0.2 to about 10 microns, about 0.5 to about 10 microns, about 0.8 to about 10 microns, about 1 to about 10 microns, about 1 to about 9 microns, about 1 to about 8 microns, about 1 to about 7 microns, about 1 to about 6 microns, about 1 to about 5 microns, about 1 to about 4 microns, about 1 to about 3 microns, from about 1 to about 2 microns.
- Particular average aerodynamic diameter ranges are between from about 1 micron to about 5 microns, between from about 1 micron to about 4 microns, and between from about 1 micron to about 3 microns. In a yet further embodiment, the average aerodynamic diameter is ⁇ about 10 microns,
- the average aerodynamic diameter is about 1 micron, about 2 microns, about 3 microns, about 4 microns, about 5 microns, about 6 microns, about 7 microns, or about 8 microns.
- the average aerodynamic diameter is from about 5-20 microns (large particles) which may deposit in the oropharynx and upper airways, for example the trachea. In a yet further embodiment, the average aerodynamic diameter is from about 1-5 microns (small particles) which may deposit in the lower airways, for example the bronchus and bronchioles. In a yet further embodiment, the average aerodynamic diameter is from about 0.01 to about 1 micron (submicron particles) which may deposit in the alveoli.
- the average aerodynamic diameter is from about 1 to about 3 microns.
- about 90% of the particles (do.9) have an average size of ⁇ about 10 microns, ⁇ about 9 microns, ⁇ about 8 microns, ⁇ about 7 microns, ⁇ about 6 microns, ⁇ about 5 microns, ⁇ about 4 microns, ⁇ about 3 microns, ⁇ about 2 microns,
- 80% of the particles (do.g), 70% of the particles (do.7), 60% of the particles (do.6), 50% of the particles (do.5), 40% of the particles (do. 4 ), 30% of the particles (do. 3 ), 20% of the particles (do 2), or 10% of the particles (do.i), are less than at least one of the abovementioned sizes.
- the particles can have an average size of 2 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; 3 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; 4 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; 5 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; 6 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; 7 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; 8 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron;
- the present invention specifically encompasses the situation where a percentage of the particles are of an average size as outlined above.
- Specific percentages include about 1% of the particles (do.oi), about 5% of the particles (do.os). about 10% (do.i), about 20% (do. 2 ), about 30% (do. 3 ), about 40% (do. ), about 50% (do. 5 ), about 60% (doe), about 70% (do.?), about 80% (dog), about 90% (do. 9 ), about 95% (do.95), about 98% (do.98), about 99% (do.w). or about 100% (di) of the particles are of an aerodynamic diameter outlined above.
- the fine particle dose which is the total mass of particles with aerodynamic diameters ⁇ 5 micron.
- the fine particle fraction is > about 10%.
- the FPF is > about 20%, > about 30%, > about 40%, . > about 50%, > about 60%, > about 70%, > about 80%, > about 90%, > about 95%, or about 100%. The greater the FPF, the more active agent reaches the target area. Particular FPF are > 10 % of the loaded dose, > 30 % of the loaded dose, or > 50 % of the loaded dose.
- the aerodynamic diameter instead of the aerodynamic diameter, it is possible to measure the specific dimensions of particles and to determine their average size. This may be the average particle diameter, including the mean or median diameter. These embodiments are not limited to spherical particles and can be used to measure irregular particles. It may be considered the average length, width and or height of the particles.
- the present invention can allow for defined doses to reach the same target areas, for example the same cells. This may have advantages in terms of therapy because targeted therapy with defined doses is possible. This may also allow for synergistic effects between the combination products because they may, for example, be simultaneously deposited the same target cell hi the lung epithelium. This has distinct advantages over physical mixtures of individual active agents.
- the particles, compositions methods and uses of the present invention allow for delivery of more than one active to the same target site, for example the same target cell in the lung epithelium.
- components may be added to the particles to allow for controlled release. This may be achieved by further coating the particles with a polymer.
- the particles may further contain surfactants or polymers. These may be used to control crystal growth during co-precipitation in some techniques, such as by anti-solvent.
- Other excipients may be pH modifiers, antioxidants, and flavouring agents.
- the active agents and the excipient are evenly mixed throughout the particle.
- one active agent is predominantly present on the surface of the particle.
- two or more actives are predominantly present on the surface of the particle.
- one active agent is predominantly present in the interior of the particle.
- two or more actives are predominantly present in the interior of the particle.
- the present invention encompasses the situation where the active agents and excipient are homogeneously mixed throughout the particle and also the situation where the components are not homogeneously mixed.
- the shape of the particles may affect the properties and or performance of combination products made using them.
- the shape of the particles can have an effect on their aerosolisation properties.
- the shape of the particles can have an effect on their handling properties.
- substantially uniform and/or substantially spherical particles may have improved aerosolisation properties and may be less likely to stick together. These particles may have improved flow properties which may make capsule and/or device filling easier.
- the particles are predominantly spherical.
- the particles are ovoid.
- the particles are predominantly ellipsoidal.
- the particles are predominantly needle or fibre shaped.
- the particles are predominantly plate like or flaked.
- the particles are predominantly pyramidal.
- the particles are spiky.
- the particles are irregularly shaped.
- the particles have a substantially uniform shape.
- the surface conditions of the particles may be affected by the formation conditions and also by the selection of active agents.
- the surface of the particles may be substantially uniform.
- the surface may be dimpled.
- the surface may contain crystals shaped particles adhered to the surface.
- the surface may contain a clay like material.
- the surface may be smooth.
- the surface may be roughened.
- the surface may be corrugated.
- the surface may have plate like materials on the surface.
- the surface may have spikes on the particle surface.
- the particles are at least partially, or are substantially hollow. This has advantages in terms of the aerodynamic diameter of the particles and allows for larger particles which can still deliver active agents to the lower airways (or elsewhere). The ability to form larger particles with the same aerodynamic performance has advantages because larger particles are easier to handle.
- the individual components do not homogeneously mix. Without wishing to be bound by theory, one reason for this could be that the individual components have different solubilities, and/or different rates of crystallisation! This could lead to a non-uniform distribution of component in the final particle. For example, if the particle contains two active agents, both of which are more soluble than the excipient in the initial solution, then as the particle is formed the excipient comes out of solution first and adopts a predominantly crystalline form while the active agents remain in solution.
- the excipient crystallises at the liquid-air interface you may end up with a predominance of excipient on or near the surface of the particle with the active agents forming predominantly at or near the centre of the particle since the solution will dry from the outside-in for a given droplet as it dries into a particle.
- the active agents are less soluble than the excipient then they may come out of solution before the excipient.
- the active agents may be on or near the surface of the particle with the inside of the particle being predominantly excipient.
- the active agents may be fully formed on the surface of the particle or may be embedded to some degree or fully in the outer surface of the particle.
- one of the active agents is less soluble than the excipient where as one active is more soluble. Under these circumstances it may be that one active is predominantly found on or near the surface of the particle and the other particle is predominantly found at or near the centre of the particle.
- the particle may contain one or more actives predominantly on or near the surface of the particle.
- two actives are found on or near the surface of the particle.
- the particle may contain one or more actives predominantly at or near the centre of the particle. According to a further embodiment, two actives are found predominantly at or near the centre of the particle. According to yet further embodiments, the particle may contain one or more actives predominantly on or near the surface of the particle and one or more actives predominantly at or near the centre of the particle.
- the particle may contain excipient predominantly on or near the surface of the particle.
- the particle may contain excipient predominantly at or near the centre of the particle.
- the particle may contain a substantially homogeneous mixture of active agents and excipient.
- the present invention includes particles containing two or more active agents.
- the particles contain two active agents.
- the particles contain 3 active agents.
- the particles contain 4 active agents.
- the particles contain a single active agent.
- the particles contain an inhaled corticosteroid (ICS) and a long-acting P2-agonist (LABA).
- drugs which can be used include acetonide, albuterol, albuterol sulfate, beclomethasone, budesonide, cortisone, cromolyn, cromolyn sodium, dexamethasone, flunisolide, fluticasone, formoterol, formoterol fumarate, hydrocortisone, pratropium, ipratropium / albuterol, levalbuterol HC1, metaproterenol, methylprednisolone, mometasone, montelukast, nedocromil, nedocromil sodium, omalizumab, pirbuterol, prednisolone, propionate, salbutamol, salmeterol, salmeterol xinafoate, terbutaline, theophylline, tiotropium, triamcinolone, zafirlukast or zileuton.
- “Drugs”, for the purposes of the invention, include a variety of pharmaceutically active ingredients, such as, for example, those which are useful in inhalation therapy.
- the term “drug” is to be broadly construed and include, without limitation, actives, drugs and bioactive agents, as well as biopharmaceuticals.
- drug is interchangeable with the term medicament or active agent.
- Appropriate drugs may thus be selected from, for example, analgesics, (e.g., codeine, dihydromorphine, ergotamine, fentanyl or morphine); anginal preparations, (e.g., diltiazem); anti-allergies, (e.g., cromoglicate, ketotifen or nedocromil); antiinfectives (e.g., cephalosporins, penicillins, streptomycin, sulphonamides, tetracyclines and pentamidine); antihistamines, (e.g., methapyrilene); antiinflammatories, (e.g., antiinflammatory steroids, beclomethasone (e.g.
- beclomethasone dipropionate fluticasone (e.g. fluticasone propionate), flunisolide, budesonide, rofleponide, mometasone (e.g. mometasone furoate), ciclesonide, triamcinolone (e.g.
- the medicaments may be used in the form of salts, (e.g., as alkali metal or amine salts or as acid addition salts) or as esters (e.g., lower alkyl esters) or as solvates (e.g., hydrates) to optimize the activity and/or stability of the medicament.
- the medicaments may be used in the form of a pure isomer, for example, R-salbutamol or R-formoterol.
- Particular medicaments for administration using pharmaceutical formulations in accordance with the invention include anti-allergies, bronchodilators, beta agonists (e.g., long-acting beta agonists), and anti-inflammatory steroids of use in the treatment of respiratory conditions, as defined herein, by inhalation therapy, for example, cromoglicate (e.g. as the sodium salt), salbutamol (e.g. as the free base or the sulphate salt), salmeterol (e.g. as the xinafoate salt), bitolterol, formoterol (e.g. as the fumarate salt), terbutaline (e.g.
- cromoglicate e.g. as the sodium salt
- salbutamol e.g. as the free base or the sulphate salt
- salmeterol e.g. as the xinafoate salt
- bitolterol e.g. as the fumarate salt
- terbutaline e.g.
- a beclomethasone ester e.g. the dipropionate
- a fluticasone ester e.g. the propionate
- a mometasone ester e.g., the furoate
- budesonide e.g. the hydrochloride salt
- Medicaments useful in erectile dysfunction treatment e.g., PDE-V inhibitors such as vardenafil
- hydrochloride along with alprostadil and sildenafil citrate
- alprostadil and sildenafil citrate may also be employed. It should be understood that the drugs that may be used in conjunction with the inhaler are not limited to those described herein.
- antibiotics erythromycin oleandomycin kitasamycin spiramycin josamycin midecamycin crarythromycin tetracycline antibiotics chlortetracycline oxytetracycline tetracycline doxycycline minocycline chloramphenicol thiamphnicol lincomycin clidamycin fosfomycin pyridonecarboxylic acids nalidixic acid pipemidic acid norfloxacin ofloxacin
- agent/analgesic agent acetoaminophen aminopyrine etc. hypoglycemic agent tolbutamide gliclazide etc. anticancer agent methotrexate etc.
- Combinations of two or more agents selected from the group consisting of salmeterol, especially salmeterol xinafoate, salbutamol, fluticasone propionate, formoterol, budesonide, beclomethasone dipropionate and physiologically acceptable salts and solvates thereof are specifically exemplified.
- the particle contains a combination of two active ingredients known for the treatment and/or prophylaxis of respiratory disorders.
- the particle may comprise formoterol (e.g. as the fumarate) and budesonide.
- the particle may comprise salmeterol (e.g. as the xinafoate salt) and fluticasone (e.g. as the propionate ester).
- the particle may comprise salbutamol (e.g. as free base or sulphate salt) and beclomethasone (as the dipropionate ester).
- the particles of the present invention may be used directly in inhalable compositions. Alternatively, they may be combined with additional components, carriers and/or components which are therapeutically acceptable.
- an inhalable composition comprising particles as disclosed herein, together with a therapeutically acceptable carrier.
- a dry powder inhaler containing particles as defined herein.
- a dry powder inhaler containing a composition as defined herein.
- the particles may be suitable for use in a metered dose inhaler (MDI).
- MDI metered dose inhaler
- an MDI containing particles as defined herein there is provided an MDI containing a composition as defined herein.
- the particles may be suitable for use in a nebuliser.
- a nebuliser containing particles as defined herein there is provided a nebuliser containing a composition as defined herein.
- the particles may be used directly without further modifications or further components. In particular, in a DPI there is no need for a propellant.
- the particles may be part of a composition containing additional components, for example a therapeutically acceptable carrier.
- the composition may further comprise a propellant, for example a hydrofluorocarbon propellant.
- a propellant for example a hydrofluorocarbon propellant.
- Example propellants include HFA 134a and 227ea.
- the composition may further include water, salts (for example for tonicity adjustment), pH modifiers (for example weak acids), and/or polymers for viscosity adjustment.
- the present invention encompasses any way to make the particles.
- the present invention encompasses any method to dry a solution to form particles.
- Example methods of manufacture include: spray drying an aqueous solution of the components; co-precipitation by anti-solvents, co-precipitation by other means (for example by chemical reaction including pH changes) followed by drying including spray drying; co-precipitation by super-critical fluid precipitation; mechanofusion (coating crystalline particles with additional drugs onto the surface), optionally with annealing afterwards to crystallise an amorphous surface; or freeze drying, spray drying an organic solution; spray drying a solution containing a co-solvent mixture of water and organic solvent(s); spray freeze drying (this is different to simple spray drying or freeze drying a solution, involving spraying a solution into liquid nitrogen, the frozen droplets are then dried in a freeze dryer).
- the solvent does not need to be aqueous, it can be totally organic or a mixture of water and organic solvent(s).
- a co-solvent system could comprise water and ethanol.
- an organic solvent/water mixture could be used and this may be used to increase the crystallinity of the particles compared to an aqueous solution alone.
- the particles are formed by spray drying.
- the active agents together with the excipient may be dissolved in an aqueous solution.
- This solution may be, for example, water or a mixture of water and an alcohol such as ethanol.
- the feed solution may then be spray dried.
- Crystallization depends on Tg of the components and also other factors including the material and systems.
- One additional factor is the inlet temperature of the spray drying equipment.
- the inlet temperature is higher than Tg of the excipient that is expected to be crystalline.
- the outlet temperature will be lower than the inlet temperature.
- a method of delivering a combination of two or more active agents to a patient in need thereof comprising administering particles as defined herein.
- a method of delivering a combination of two or more active agents to a patient in need thereof comprising inhaling particles as defined herein.
- the present invention comprises a method of delivering a combination of two or more active agents to a patient, the method comprising forming particles as defined herein and aerosolising the particles to be suitable for inhalation by the patient.
- the present invention comprises particles as defined herein for use as a medicament.
- the present invention may be used to treat a wide array of diseases and/or conditions. Particular conditions include a respiratory or non-respiratory condition.
- a method of treating a patient having a respiratory or non-respiratory condition comprising administering particles as defined herein.
- the present invention comprises use of the particles as defined herein in the manufacture of a medicament for treatment of a respiratory or non-respiratory condition of a patient.
- the present invention comprises particles as defined herein for use in the treatment of a respiratory or non-respiratory condition of a patient. According to a further aspect, the present invention provides the use of particles as defined herein for the manufacture of a medicament for the treatment of a respiratory or non-respiratory condition.
- the present invention provides a method of delivering a medicament to a patient in need thereof comprising administering particles as defined herein.
- the present invention provides a pharmaceutical composition comprising particles as defined herein.
- the present invention provides particles as defined herein for use as a medicament.
- the present invention provides the use of particles as defined herein as a medicament.
- the present invention provides particles as defined herein for use in the treatment of respiratory or non-respiratory conditions.
- the present invention provides the use of particles as defined herein in the treatment of respiratory or non-respiratory conditions.
- Diseases or conditions for which the present invention may apply include respiratory or non-respiratory conditions.
- Respiratory conditions include, COPD, bronchitis, allergy, rhinitis, cystic fibrosis, pulmonary infection, tuberculosis, influenza, other lung infections, lung cancer and asthma.
- Non-respiratory conditions include diabetes, hypertension, hypercholesterolaemia, gout, infections (bacterial or viral), fever, pairi (neurological or muscular).
- the respiratory condition is COPD. In a further embodiment, the respiratory condition is bronchitis. In a yet further embodiment, the respiratory condition is allergy. In a yet further embodiment, the respiratory condition is rhinitis. In a yet further embodiment, the respiratory condition is cystic fibrosis. In a yet further embodiment, the respiratory condition is pulmonary infection. In a yet further embodiment, the respiratory condition is asthma.
- COPD COPD
- asthma COPD
- the medicament can be manufactured using the methods as defined herein.
- the word “comprise”, or variations such as “comprises” or “comprising”, will be understood to imply the inclusion of a stated element, integer or step, or group of elements, integers or steps, but not the exclusion of any other element, integer or step, or group of elements, integers or steps.
- budesonide (a) budesonide, (b) formoterol fumarate dihydrate, (c) fluticasone propionate, (d) salmeterol xinafoate, (e) mannitol
- B/F/M-SD and its raw materials, (a) B/F/M-SD, (b) mannitol, (c) budesonide, (d) formoterol fumarate dihydrate
- F/S/M-SD and its raw materials (e) F/S/M-SD, (f) mannitol, (g) fluticasone propionate, (h) salmeterol xinafoate
- B/F/M-SD spray dried budesonide/formoterol fumarate dehydrate/mannitol
- F/S/M-SD spray dried fluticasone propionate/salmeterol xinafoate/mannitol
- M-SD spray dried mannitol
- B/F/M-SD spray dried budesonide/formoterol/mannitol fumarate dehydrate
- F/S/M-SD spray dried fluticasone propionate/salmeterol xinafoate/mannitol
- M-SD spray dried mannitol
- Fig. 8 shows cross section images of B/F/M-SD (budesonide / formoterol fumarate dihydrate / mannitol spray dried) particles.
- FIG. 9 Cross section images of F/S/M-SD (fluticasone propionate / salmterol xinafoate / mannitol spray dried) particles.
- the following description describes methods of forming particles of the present invention from mannitol and active agents selected budesonide / fluticasone propionate, and formoterol fumarate dihydrate / salmeterol xinafoate. It will be appreciated that other components, including other active agents and/or other excipients, such as other sugars, can be utilised to form suitable carrier particles.
- the particles as formed herein can be used to form compositions suitable for inhalation by a patient requiring the drug.
- the drug can be suitable for treating respiratory conditions such as cystic fibrosis, COPD, bronchitis, allergy, rhinitis and asthma. It will also be appreciated that the drug can be suitable for delivery via the lungs and be used to treat non-pulmonary conditions.
- the following information is intended to demonstrate how the aerodynamic diameter can be measured. This provides a method to determine the aerodynamic diameter of a given particle sample.
- the aerodynamic particle size is measured by dispersing 5 mg of the powder from an Aerolizer ® through a Next Generation Impactor (British Pharmacopoeia Apparatus E) without the pre-separator at 60 and 100 L min air flow for 4 and 2.4 s, respectively.
- the experimental procedure largely follows that detailed in the British Pharmacopoeia.
- the flow rate and solenoid valve timer are adjusted appropriately prior to sampling.
- a known amount of powder may be weighed into a Size 3 hydroxypropyl methylcellulose capsule and loaded into the capsule compartment in the Aerolizer ® .
- the capsule is pieced and the inhaler is inserted into the induction port via a rubber adaptor flush with the inhaler mouthpiece to discharge the dose. Repeat the sampling procedure with more loaded capsules if necessary.
- the number of discharges should be minimised (typically ⁇ 10) to obtain sufficient powder for an accurate and precise quantification.
- the powder deposits of actives and excipient in the capsule, Aerolizer ® , rubber adaptor, induction port, and all impactor stages of the NGI are assayed chemically.
- Suitable assay methods include, but are not limited to, the following: ultraviolet spectrophotometry, high performance liquid chromatography (HPLC), gas chromatography (GC), or liquid chromatography-mass spectrometry (LC-MS). Both actives should deposit concurrently on all assayed parts.
- the loaded dose is defined as the total amount of powder weighed into the capsules for aerosol sampling.
- the fine particle dose (FPD) is the total mass of particles with aerodynamic diameters ⁇ 5 ⁇ .
- the fine particle fraction (FPF) is calculated by dividing the FPD by the loaded dose.
- the examples show particles made from a combination of an inhaled corticosteroid (ICS) and a long-acting p2-agonist (LABA). Preparation of spray dried particles
- ICS, LABA and mannitol were dissolved in ethanol/water to prepare the feed solutions for spray drying.
- Spray drying was performed using a B-290 mini spray dryer (Buchi Labortechnik AG, Falwil, Switzerland) with operating conditions detailed in Table 2.
- the spray dried powder samples were kept in a glass desiccator containing silica gel at 22 °C until used. (mg/mL)
- Particle size distributions of the powders were determined by laser diffraction using Mastersizer 2000 (Malvern Instruments, Worcs, UK). The powders were dispersed through the measurement zone with compressed air at 4 bars of pressure by a Scirocco 2000 dry powder feeder (Malvern Instruments, Worcs, UK). The particle refractive index and absorption were 1.52 and 0.1, respectively. The dispersant refractive index was 1.000 for air. All measurements were conducted in triplicate. Drug quantification
- Drug content in the spray dried powders after preparation and in the aerosol samples obtained from the dispersion study were determined using high performance liquid chromatography (HPLC) (Model LC-20; Shimadzu, Kyoto, Japan).
- HPLC high performance liquid chromatography
- a LiChrosphere 60 ® RP-select B column (4 x 125 mm, 5 ⁇ ) (Merck, Darmstadt, Germany) was used as the stationary phase with the mobile phase comprising methanol, water and acetic acid (550:450:1).
- a flow rate of 1 mL/min and UV absorption wavelength at 230 nm was employed for parallel detection of both drugs (retention times were 4.9 min and 15.8 min for formoterol fumarate dehydrate and budesonide, respectively).
- an Intersil ODS2 column (4.6 x 200 mm, 5 ⁇ ) (Capital HPLC, Scotland, UK) was used as stationary phase with methanol and 0.6% ammonium acetate buffer (75:25) being the mobile phase.
- a flow rate of 1 mL min and wavelength 228 nm was used for parallel detection of salmeterol xinafoate and fluticasone propionate, at retention times of 2.7 min and 7.1 min, respectively.
- Mannitol content in the powders was determined by HPLC using refractive index detection as described previously (20). Briefly, a CI 8 Radial-Pack column (Waters, USA) was used as stationary phase with deionized water as mobile phase running at 1 mL min.
- DSC Differential scanning calorimetry
- TGA Thermal gravimetric analysis
- X-ray powder diffraction (XRD) measurement was carried out at room temperature using an X-ray diffractometer (Model D5000; Siemens, Kunststoff, Germany). Cu a radiation at 30mA and 40kV was used with an angular increment of 0.04° at 3 sec per step covering a 2 ⁇ range of 5-50°. Morphological observation
- the morphology of the spray-dried particles was investigated using scanning electron microscopy (SEM). Samples were deposited on carbon sticky tape and mounted on a SEM stubs, followed by sputter coating with gold (15 nm thick) on a 550X sputter coater (Quorum Emitech, Kent, UK). The specimens were then imaged using a field emission SEM (Zeiss Ultra Plus; Carl Zeiss SMT AG, Oberkochen, Germany) at 1.90- 1.99 kV using an in-lens detector. Internal structure observation
- Focused-ion-beam (FIB) milling of the particles was performed in an FIB/scanning electron microscope dual beam system (Nova 200, FEI, USA) using a similar procedure described previously, but without the cleaning step (21).
- the powder samples were coated with a 50 nm layer of platinum.
- Vertical cutting of the sample was performed at a stage tilt of 52 degrees with an accelerating voltage of 10 kV and a beam current of 19 pA. Milling times were kept under 5 min. The milled samples were later examined at 2 kV to examine the cut surface.
- Atomic force microscopy Atomic force microscopy
- Particle-particle cohesion force was evaluated using the colloid probe microscopy technique. Individual particle was mounted onto the apex of V-shaped tipless atomic force microscopy (AFM) cantilever (NP-0 silicon nitride cantilevers with gold reflective coating, nominal spring constant 0.58 nN; Veeco Inc., New York, USA) using a micromanipulation technique described elsewhere. The force of adhesion between each probe and particles mounted on a thermoplastic adhesive (Tempfix ® ; Piano, Wetzlar, Germany) was investigated using force-volume imaging.
- AFM tipless atomic force microscopy
- XPS X-ray photoelectron spectroscopy
- the instrument comprises a Thermo VG ESCALAB250 spectrometer with a non-monochromatic Al alpha (1486.6 eV) X-ray source. Analysis was carried out under a pass energy of 20 eV. The powders were pressed onto double sided conductive adhesive tapes. Experimental molar percentages of all elements except hydrogen were derived from the XPS peak areas as described elsewhere (30). The molar percentages (At%) were multiplied by the appropriate atomic mass to obtain weight percentages (Wt%) (Table 5 and Table 7).
- the surface elemental composition of the raw materials (mannitol, budesonide, formoterol fumarate dihydrate, fluticasone propionate and salmeterol xinafoate) and the three spray dried powders (M-SD, B/F M-SD and F/S/ - SD) were measured.
- the type of molecules present on the particle surface can be deduced from the detected elements and elemental percentages.
- the expected elemental percentages of the spray dried powders were calculated from the percentages measured on the raw materials. All components of the ternary powders were assumed to be distributed evenly on the surface for the calculation. The expected elemental percentages are shown in Table 6 and Table 7.
- the expected Wt% of an element in the powder was the sum of the products obtained by multiplying the Wt of that element in the raw materials by the Wt% of the corresponding materials in the powder (Table 6). Differences between the experimental and expected percentages suggest an over- or under-abundance of certain types of molecules on the surface.
- the dispersion test was performed for 2.4 sec at 100 IJmin and 4 sec at 60 L/min, with the flow rate measured by a flow meter (Model 3063; TSI Incorporated, Minnesota, USA). Dispersions were performed in triplicate for each powder. FPF was defined as the mass fraction of particles ⁇ 5.0 urn with respect to the loaded dose in the capsules. The cut-off diameters of the NGI stages at 100 L min were calculated with the cut-off adjustment equations given in Appendix XII C of the British Pharmacopoeia.
- the drug content of each component in the powder was determined by HPLC to be close to 100 % (Table 4). In addition, the drug content ratios of the two combination powders were confirmed to be the same as those of the feed solutions.
- M-SD spray dried mannitol
- B/F/M-SD spray dried budesonide / formoterol fumarate dihydrate / mannitol
- F/S/M-SD spray dried fluticasone propionate /salmeterol xinafoate / mannitol
- B/F/M-SD also had a smooth surface but the packing of mannitol crystals was not observed. Small crystal-shaped particles adhered onto the surface. The surface of F/S/M-SD particles seemed roughly covered with mixture of clay-like material and small particles. It also had many dimples on the surface.
- this inhomogeneity could be caused by the difference of solubility of mannitol and drugs. Solidification of drugs and mannitol would have occurred at the different time points during the spray drying process as the solvent evaporated. It could be that the active agents solidified first on the surface of the droplets and then mannitol came out inside of the outer shell of drugs. As the particles are mainly composed of mannitol, the crystallinity of drugs could not be examined using XRD and DSC.
- Focus ion beam-scanning electron, microscopy is similar to scanning electron microscopy (SEM) but it uses a liquid metal source as the filament to produce a finely focused beam of energetic metal ions that can be applied for both imaging and milling.
- the particle is cut with ion beam and its cross section is viewed under SEM.
- condition adjustment can be difficult when the specimen is sensitive because it can easily deteriorate under the ion or electron beam. Since the ion beam removes material from the specimen surface, mild conditions preferable to reduce the deteriorating effect on the specimen. However, it becomes more difficult to obtain clear images at lower accelerating voltages and lower beam currents. Therefore, the mildest condition that can give acceptably clear image should be applied.
- FIB-SEM revealed partially hollow interior structures (Figure 2).
- the partially hollow interior structures may lead to a lower particle density and smaller aerodynamic diameter resulting in better dispersibility. Additional FIB-SEM images are shown in figures 8 and 9.
- XRD patterns showed that both B/F/M-SD and F/S/M-SD contained a-mannitol, while M-SD contained ⁇ -mannitol ( Figure 5). Although ICS contents were approaching the detection limit of XRD, the patterns were able to reveal the major diffraction peaks of budesonide at 2 ⁇ angles of 6.1, 12.0, 15.5 & 16.0 degrees and fluticasone propionate at 10.0, 13.0, 14.8 & 16.2 degrees.
- the inhalable particle size range and consistent drug contents show the potential applicability of ICS/LABA/mannitol powder for DPI formulations.
- the consistent drug contents indicate no selective adsorption or loss of the drugs in the spray drying line, nor segregation of the drugs in the powders.
- the drug content ratios are considered to be constant throughout the whole particle size range based on the concomitant deposition of ICS/LABA shown in the aerosol data ( Figure 6 and Figure 7).
- the high and similar FPF values of B F M-SD and F/S/M-SD are in accordance with the inhalable particle size range (Table 3) as well as similar AFM force values for the powders (Table 8).
- the relatively high FPF in the aerosol also showed that the powders were not particularly cohesive, hence a good dispersion could be achieved even at a low air flow rate with minimal inhalation effort.
- Using a comfortable inspiratory effort would generate 105 L min through the Aerolizer.
- Increasing the flow from 60 to 100 L/min simply reduces the capsule and device retention by providing more energy to empty the powder from the inhaler, while augmenting the deposition on the throat and Stages 1 and 2 of the impactor by increasing the air velocity for impaction at those sites.
- Co-precipitates of fluticasone propionate and salmeterol xinafoate showed a FPF of 22% when formulated with lubricant and 36% with lactose carrier.
- the ICS/LABA/mannitol co-spray dried powders in the present study are similar to mannitol in their cohesive force (Table 8) and FPF values. Surface roughness of these particles is likely to have contributed to the low cohesiveness and high dispersibiliry.
- Another contributing factor may be the crystallinity of the mannitol and possibly of the ICSs (as confirmed by DSC & XRD). Compared with previous studies, the inlet temperature of the spray drying process was higher, hence allowing glass transition and subsequent crystallization of the drugs.
- the ICS/LABA mannitol system provides an innovative approach for combination formulations at appropriate doses without the need of physical blending.
- the powders showed high aerosol performance and uniform deposition of the two drugs. Storage stability is an important consideration for product development. Preliminary results showed that compared to the initial values, there was no significant difference (P ⁇ 0.05) in the dispersion of the B/F/M-SD powder after storage over silica gel at 22 °C for 11 weeks (FPF at 60 L/min: 53.7 ⁇ 1.5% for formoterol fumarate dihydrate and 53.4 ⁇ 1.7% for budesonide). Additional Spray Dried Powders
- the drug solutions are shown in Table 9 below and were spray dried using the conditions as those for B/F/M-SD.
- Respirable-sized (D50 of 2 ⁇ ) crystalline mannitol particles containing two drugs with at least one confirmed to be also crystalline were successfully obtained from co-spray drying two different ternary systems containing budesonide/formoterol furnarate dihydrate/mannitol and fluticasone propionate/salmeterol xinafoate/mannitol.
- the powders When dispersed using an Aerolizer at 60 and 100 IJmin, the powders showed a concomitant in vitro deposition patterns of ICS LABA with a FPF of 54 - 62 %.
- the aerosol performance can be due to the low interparticulate force, resulting from a combination of the rough surface and crystalline nature of the particles.
- the particles of the present invention provide an alternative simple method for effective one-step processing of combination formulations for inhalation.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pulmonology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Otolaryngology (AREA)
- Emergency Medicine (AREA)
- Pain & Pain Management (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Respirable inhalable particles comprising active agent(s) and excipients in at least a partially crystalline form and their uses thereof.
Description
INHALABLE FORMULATIONS
The present invention relates to inhalable formulations, to methods of manufacturing inhalable formulations and to pharmaceutical compositions containing inhalable formulations.
Dry powder inhalation (DPI) drug therapy has been used for many years to treat respiratory conditions such as chronic obstructive pulmonary disease (COPD), bronchitis, allergy, rhinitis and asthma. Compared to oral drug intake, only relatively small doses are needed for effective therapy as first pass metabolism is significantly reduced. Such small doses reduce the body's exposure to the drug and minimise side effects. Systemic adverse effects are also reduced as topical lung delivery takes the drug directly to the site of action. Lower dosage regimens may also provide considerable cost savings, particularly where expensive therapeutic agents are concerned. There is also growing interest in the use of lung drug delivery to treat non- pulmonary conditions.
The success of DPI therapy depends on a number of factors including the biological aspect of the active ingredient, the physicochemical properties of the formulation, and the performance of the inhaler. The efficiency of dose delivery of dry powders also depends on the particle size, size distribution, shape and surface morphology of the powder.
While considered optimal, drug particles having a size between 1 and 5 microns have a high surface area to mass ratio and therefore tend to be highly cohesive resulting in poor aerosolisation efficiency and thus respiratory deposition. As such, their delivery into the lung is traditionally enhanced when they are blended with larger and coarser inert carrier materials. Upon inhalation, the aim is for the drug particles to be freed from the carriers and to enter and penetrate the lung while the carriers themselves impact in the upper airways and are ingested.
DPI therapy may deliver a single active or may deliver a combination of actives. Combination inhalation therapy have been used to treat respiratory diseases and combination therapies include an inhaled corticosteroid (ICS) and a long-acting β2- agonist (LABA) which offers the advantages of convenience to the patients along with
synergistic pharmacological actions, leading to better patient compliance and therapeutic outcomes.
However, there are a number of technical challenges for combination products which contain two or more active pharmaceutical ingredients (APIs) in the same powder or propellant formulation. These problems can be amplified for APIs present at highly different doses or stored in an unwanted environment. Currently marketed combination products contain the APIs in a single formulation and dose container. The technical issues stem from the need of dealing with two APIs simultaneously.
In suspension metered dose inhaler (MDI) formulations, differences in the particle properties may cause differential suspension characteristics of the APIs and wall loss to the canister, resulting in variable product performance. Wall loss can potentially be minimized by non-sticky coating of the canister wall. Although disparities in the formulations have been dealt with by separating them in two canisters, it would make the MDI more bulky and potentially require more effort to actuate. It is possible in an MDI to have a solution formulation if all the APIs are soluble and chemically stable in the propellant, or to have one drug dissolved while another drug suspended in the propellant. In contrast, DPI formulations rely on the use of blends in which the API particles adhere on the carrier lactose surface. Lactose is the major carrier used in DPIs and its performance is highly variable, depending on its amount of fines, surface roughness, polymorphic form, production batch, and grade.
Unpredictable or preferential adhesion of one API over the other on the lactose, along with self-agglomeration of one or both drugs, will cause content segregation and dose non-uniformity. Any difference in the particle properties between the APIs as manifested in different interactions between the APIs, carrier and container surface, may lead to changes in the fine particle fraction, device retention and dose variability. Thus there is a need for better DPI formulations and vehicles to allow for effective dosing of multiple APIs.
Any discussion of documents, acts, materials, devices, articles or the like which has been included in the present specification is solely for the purpose of providing a context for the present invention. It is not to be taken as an admission that any or all of these matters form part of the prior art base or were common general knowledge in the
field relevant to the present invention as it existed before the priority date of each claim of this application.
The present inventors have found that it is possible to form inhalable particles from two or more active agents together with an excipient which is at least partially in a crystalline form, and that these particles may have advantageous properties, especially when used in a dry particle inhaler (DPI).
For the first time, a particle has been provided which is of respirable size and which contains two or more active agents and an excipient at least partially in a crystalline form. Traditionally, inhalable formulations comprised particles of active agent(s) of inhalable size together with a larger non-respirable carrier particle. The present invention has found that it is possible to form respirable sized particles containing two or more active agents and an excipient at least partially in a crystalline form. The ability to form uniform particles of a defined size containing both the actives and excipient is of great use because it allows for more uniform particle formation providing targeted delivery of active agents to patients using more manageable particles. Accordingly, in a first embodiment of the present invention there is provided an inhalable particle comprising two or more active agents and an excipient which is at least partially in crystalline form, wherein the particle is of respirable size.
The present invention is useful in inhalable formulations which will generally be made up of many particles. Therefore, there will generally be a large number of particles in a given composition. This collection of particles will comprise at least one but generally a large number of the particles disclosed herein. Accordingly, in a further embodiment of the present invention there is provided inhalable particles comprising two or more active agents and an excipient which is at least partially in a crystalline form, wherein the particles are of respirable size.
In embodiments of the present invention, at least 1%, at least 2%, at least 5%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, at least 98%, at least 99%, or 100% of the particles in the total collection of particles are particles according to the invention as disclosed herein.
Thus, the present invention has identified a novel particle of respirable size comprising active agent(s) and excipients as disclosed herein. The particles contain an excipient. This may be thought of as a bulking agent. The excipient is present to assist with formulation and/or performance of the inhalable particles rather than to have a physiological effect itself on the patient. However, in some examples, the excipient may itself have a physiological effect. Nevertheless, because it is being used to achieve the desirable formulation and/or performance characteristics for the particles, rather than for the physiological effects it causes on the patient, it is considered an excipient. For the avoidance of doubt, excipients like mannitol are encompassed by the present invention. Therefore, mannitol may be thought of as an excipient or bulking agent for the purposes of the present invention regardless of the fact that mannitol has been shown to have a physiological effect on the body.
The traditional carrier particle system required the active agents to be milled to respirable size and then mixed with a larger carrier particle. Typical active doses were relatively small (on the milligram scale) and it was not possible to deliver such a small dose to a patient. It was therefore necessary to use additional excipients to 'bulk out' the total composition so as to facilitate manufacture and delivery. By mixing the active agents with the larger carrier particles the total formulation that was prepared could be of a manageable size but the patient still received the required dose of active agents. Furthermore, using larger particles assisted in formulation because it avoided some of the difficulties associated with small particles on the 1-3 micron scale, like aggregation and poor flow properties. However, there were difficulties with this approach because the active agents did not always separate from the larger carriers during delivery in a predictable manner. There were particular problems associated with combination products because different actives may adhere to the carrier particles with different affinities and it was difficult to ensure that the patient received the required doses of each agent. Equally, the different active particles may be differently shaped thereby giving rise to very different flight properties during delivery. This may cause differences in delivery dose and/or location. Thus, to date it has been difficult to deliver accurate doses to specific target sites in the respiratory tract.
The present invention aims, at least in its preferred forms to overcome some or all of these difficulties by combining all of the actives and the excipient together in a single particle. This has a number of advantages. Putting multiple actives together into a single particle means that both actives will be delivered to the same target site at the same time. It avoids the problems associated with different actives having different aerodynamic characteristics. This allows for highly targeted delivery of multiple actives, for example multiple actives targeting the same cell. By combining the actives and excipient together means that all of the particles of the composition contain the active agents rather than a small amount of active particles and a large amount of non- functioning carrier or bulking agents. Furthermore, the particles are at least partially ' crystalline. It is possible for the first time to prepare particles of defined uniform shapes which may improve and standardise their aerodynamic performance. The crystallinity may also help with stability and long term storage for a number of reasons including: avoiding or minimising the transformation of amorphous particles into more crystalline forms with the associated degradation in the particle morphology, and improved surface properties which may reduce or minimise aggregation or other unwanted surface phenomenon like undesirable interactions with the containers.
It has also been found that particles of the present invention can be formed from a combination of at least one active agent and an excipient, where the particles are at least partially, or substantially hollow. This has advantages because a hollow particle, when compared against an equivalent sized solid particle, will have a lower density which can lead to improvements in aerodynamic and aerosolisation properties. This can be seen when considering the aerodynamic diameter of a particle which depends on both the physical diameter and density of the particle. The aerodynamic diameter can be approximated by considering the following formulae:
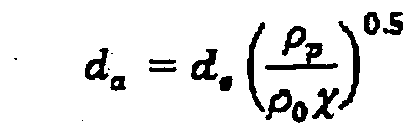 where d„ = aerodynamic diameter, de = equivalent volume diameter of a sphere (diameter of the sphere with the same volume as the non-spherical particle), pp = density of the particle, pp - unit density (1 g/cm3), and χ = dynamic shape factor. The dynamic shape factor accounts for the effect of shape on particle motion. It is the ratio of the resistance force experienced by the non-spherical particle moving in air to that of a sphere with the same volume and velocity. Thus, by forming hollow particles it is
possible to achieve a lower overall aerodynamic diameter for a given particle size or to achieve the same aerodynamic diameter with a larger particle. Because larger particles are easier to formulate and control there are distinct advantages in having hollow particles which allow for increases in the actual particle size without a corresponding increase in the aerodynamic diameter.
where d„ = aerodynamic diameter, de = equivalent volume diameter of a sphere (diameter of the sphere with the same volume as the non-spherical particle), pp = density of the particle, pp - unit density (1 g/cm3), and χ = dynamic shape factor. The dynamic shape factor accounts for the effect of shape on particle motion. It is the ratio of the resistance force experienced by the non-spherical particle moving in air to that of a sphere with the same volume and velocity. Thus, by forming hollow particles it is
possible to achieve a lower overall aerodynamic diameter for a given particle size or to achieve the same aerodynamic diameter with a larger particle. Because larger particles are easier to formulate and control there are distinct advantages in having hollow particles which allow for increases in the actual particle size without a corresponding increase in the aerodynamic diameter.
Thus, according to a further embodiment of the present invention there is provided hollow inhalable particles comprising one or more active agents and an excipient which is at least partially in a crystalline form, wherein the particles are of respirable size. Particular embodiments include two or more active agents. Thus, in a further embodiment, there is provided an inhalable particle comprising one or more active agents and an excipient which is at least partially in crystalline form, wherein the particle is of respirable size and is hollow. In certain embodiments, the particles of the present invention consist essentially of a single or two or more active agents and an excipient. Throughout this specification, the term "consist essentially of or "consisting essentially of is intended to exclude elements which would materially affect the properties of the claimed composition. By respirable, it is meant a particle which has an aerodynamic diameter of less than 20 microns. This allows for targeted delivery of particles to comprising two or more active agents and an excipient which is at least partially in a crystalline form to areas of the respiratory tract. Particles are considered respirable if they can be inhaled and deposited onto one or more of the oropharynx and upper airways (including the trachea), lower airways (including the bronchus and bronchioles) or deposited in the alveoli. In particular embodiments the respirable particles can be deposited onto the lower airways. This provides particles which can be used in inhalation therapy. The present invention allows for better design of particles with advantageous size distributions. Thus, it is possible to prepare particles which are easy to handle and which can deliver active agents to areas with better specificity and success.
In one embodiment the excipient may be selected from any excipient which may at least partially be in crystalline form in the particle and which may be suitable for pulmonary administration. Exemplary excipients include sugars, sugar alcohols, amino acids and other excipients. The excipient may be selected from sugars and sugar alcohols (including mannitol, sucrose, glucose, trehalose, lactose, dextrose, sorbitol,
maltilol, maltodextrin); amino acids (including glycine, leucine, trileucine, arginine, threonine, phenylalanine, aspartic acid); and other excipients such as sodium chloride, poly-lactic glycolic acid or poly ethylene glycol. A particular excipient is mannitol. A further particular excipient is glucose. Both excipients have a relatively low Tg. This means that the excipients will be more likely to be in crystalline form in the particle. In a particular embodiment, the excipient is chosen on the basis that it has a Tg < 150°C. In a further embodiment the excipient is chosen on the basis that it has a Tg < 100°C, Tg < 60°C or Tg < 50°C. Particular Tg values are < 40°C, < 35°C, < 30°C, < 25°C, < 20°C, < 15°C, < 10°C, < 5°C, < 0°C, < - 5°C, < -01°C, < -15°C, < -20°C, < -25°C.
The particles may contain a single excipient. In further embodiments, the particles may contain two or more excipients. The particles may contain 1 , 2, 3, 4, 5, 6, 7, 8 or more excipients.
The excipient is present at least partially in a crystalline form. In particular embodiments the excipient is present in a majority crystalline form. In some embodiments, the excipient is present in about 1%, about 2%, about 3%, about 4%, about 5%, about 6%, about 7%, about 8%, about 9%, about 10% , about 20%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, about 95%, or about 100% crystalline form. A specific embodiment of the invention has an excipient comprising mannitol which is present in a majority crystalline form, which may be about 50%, about 60%, about 70%, about 80%, about 90%, about 95%, or about 100%-crystalline form.
In some embodiments, at least one active agent is in crystalline form in the particle. In an embodiment, an active is present in about 10%, about 20%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, or about 100% crystalline form. In a further embodiment, two or more actives are present in about 10%, about 20%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, or about 100% crystalline form.
In some embodiments, the particles comprise about at least 10% excipient. In a further embodiment, the particles comprise about at least 20% excipient. In a yet further embodiment, the particles comprise about at least 30% excipient. In a yet further embodiment, the particles comprise about at least 40% excipient. In a yet further embodiment, the particles comprise about at least 50% excipient. In a yet further embodiment, the particles comprise about at least 60% excipient. In a yet further embodiment, the particles comprise about at least 70% excipient. In a yet further embodiment, the particles comprise about at least 80% excipient. In a yet further embodiment, the particles comprise about at least 90% excipient. In a yet further embodiment, the particles comprise about at least 95% excipient. In a yet further embodiment, the particles comprise about at least 99% excipient.
Particular embodiments contain at least about 40% excipient. Further particular embodiments contain at least 50% excipient. Yet further particular embodiments contain about at least 80% excipient.
Increasing the amount of excipient may increase the degree of crystallinity of the particles. Thus, the amount of excipient may be chosen to achieve the desired degree of crystallinity. There may be a linear relationship between the amount of excipient and the degree of crystallinity. However, for some particles there may be a non-linear relationship between the degree of crystallinity and the percentage amount of excipient.
In one embodiment, the percentage degree of crystallinity is greater than or equal to the percentage amount of excipient present in the particle. For example, a particle containing 80% excipient would be at least 80% or more crystalline. In a further embodiment, the percentage degree of crystallinity is greater than the percentage amount of excipient present in the particle. In this embodiment, the percentage degree of crystallinity exceeds the percentage amount of excipient present in the particle. In a particular embodiment, the percentage degree of crystallinity is at least 1 % greater than the percentage amount of excipient. For example, a particle with 80% excipient would have at least 81% crystallinity. In further embodiments, the percentage degree of crystallinity is at least 2%, at least 3%, at least 4%, at least 5%, at least 6%, at least 7%, at least 8%, at least 9%, at least 10%, at least 15%, at least 20%, at least 25%, or at least 30%, greater than the percentage amount of excipient.
To date, higher percentage crystallinity values vs percentage excipient have not been seen. The present invention provides for the first time a system to produce highly crystalline particles combining an active and an excipient. By forming the components together it is possible to achieve improved particle performance due, at least in part, to the crystalline nature of the particles.
Thus, according to a further embodiment of the present invention there is provided inhalable particles of respirable size comprising one or more active agents and an excipient wherein the percentage crystallinity of the particle is greater than the percentage amount of excipient present in the particle. Particular embodiments include two or more active agents. Thus, in a further embodiment, there is provided an inhalable particle of respirable size comprising one or more active agents and an excipient wherein the percentage crystallinity of the particle is greater than the percentage amount of excipient present in the particle.
The amount of active agent present in the particle can influence the physical properties of the particle, for example the morphology or aerodynamic performance of the particles. Having a small amount of actives present in the overall composition will mean that the particle is primarily made up of the excipient and the physical characteristics of the excipient may dominate the characteristics of the particle as a whole. For example, if the excipient adopts a predominantly crystalline structure and is present in sufficient amounts in the particle as a whole then it may force an otherwise amorphous active agent into a crystalline composition. However, if the active agents are predominantly present in the composition then they may dominate the overall properties of the particle. The amount of each active will also have an effect on the physiological properties of the particle.
In one embodiment, the total amount of active agents present in the particle is about 1%. In further embodiments, the total amount of active agents present is about 2%, about 3%, about 4%, about 5%, about 6%, about 7%, about 8%, about 9%, about 10%, about 11%, about 12%, about 13%, about 14%, about 15%, about 16%, about 17%, about 18%, about 19%, about 20%, about 25%, about 30%, about 35%, about 40%, about 45%, about 50%, about 55%, about 60%, about 65%, about 70%, about 75%, about 80%, about 85%, or about 90%. In a further embodiment, the amount of actives present in the particle is up to about 50%. In yet further embodiments, the amount of
actives present in the particle is up to about 40%, up to about 30%, up to about 20%, up to about 15%, or up to about 10%.
The present invention provides particles which can be used in inhalation therapy. It is therefore important that a sufficient quantity of the particles are of respirable size. This allows sufficient quantity of the active agents to reach the target areas of the patient. Such target areas include one or more of the alveoli, the lower airways including the bronchus and bronchioles, the upper airways, for example the trachea and/or the oropharynx. Appropriate selection of the particle characteristics allows for targeting delivery to particular areas of the respiratory tract. When designing respirable particles it is desirable to be able to carefully control the particle characteristics to ensure that sufficient particles are of the required respirable size so that enough of the active agent is delivered to the required area. If it is desired to be able to deliver agents to bronchus and bronchioles, then appropriate particle design is needed to allow the particles to be carried to the target area and not deposit in the upper airways. Equally, in certain circumstances, it may be desirable to specifically target the upper airways and again, appropriate particle design is needed. The present invention provides a reliable and reproducible way to design particles of respirable size.
When forming particles of the present invention it is appreciated that a large number of physical particles are formed and that a large number of particles are used in an inhalation treatment. When considering large numbers of particles it is expected that there will be a distribution of individual particles sizes, ranging from very small through to large. There may be a normal distribution of particle sizes with a percentage of the particles within a given size range. Thus, when considering the particles of the present invention the particle size may be thought of as the average particle size. Thus, for example, if the particles are said to be about 1-3 microns in size, this represents an average particle size, with some particles being smaller or larger than the average particle size. Thus, when mention is made of 7 micron particles, the present invention is intended to cover particles which, on average, have a size of about 7 microns. Obviously, some variation in particle size is to be expected when forming particles of this size and particles with an average size similar to that of the present invention are intended to fall within the scope of the invention.
In some embodiments, this average size may be the mean particle size. In further embodiments, the particle size may be the median particle size.
The present invention provides particles of respirable size. The actual diameters of the particles in a sample will range depending on factors like particle composition and method of synthesis. The distribution of particle sizes can be chosen to achieve the desired delivery of the active agents to the target areas in the respiratory tract.
It is possible to separate out the different size fractions of a given sample or it may be possible to select synthesis conditions to favour particular size ranges. This may allow for pre-selected size distributions. As an example, 20%, 40% or 50% of the particles in a given sample may be within the range of 1-5 micron. Therefore, in embodiments of the present invention a percentage of the particles may be of respirable size. For example, a powder formed using the particles of the present invention may contain 10% particles of respirable size with the remaining particles being non-respirable. It is generally preferable that the amount of respirable particles is maximised. The more particles that can reach the target delivery area, a lower overall dose of active agents may be needed. It is generally preferred to minimise the dose provided to a patient. This reduces the likelihood of adverse patient events including unwanted side effects and also reduces the cost of the medicine because a smaller amount of active is needed. For example, if an inhalable medicine needs to deliver lmg of an active agent to the lower airways and 1% of the particles are of the required respirable size then it may be necessary to have a significantly higher total amount of the active agent in the dose to ensure that lmg reaches the lower airways. In contrast, if 50% of the particles are of the required respirable size then a much lower total dose is needed because half of the particles should reach the target area. It is therefore advantageous if the amount of respirable particles in the composition is maximised.
Thus, according to an aspect of the present invention about 1% of the particles (do.oi) are of respirable size. In a further embodiment, about 5% of the particles (do.os), about 10% (do. , about 20% (do ), about 30% (do.3), about 40% (do.4), about 50% (do.5), about 60% (do e), about 70% (do.7), about 80% (do.8), about 90% (do.9), about 95% (do.95), about 98% (do.98), about 99% (do.99), or about 100% (di) of the particles are of respirable size.
The particles may be measured in terms of their aerodynamic diameter. Aerodynamic diameter provides a useful measurement of inhalable particles and takes into account factors that affect their aerodynamic properties. Aerodynamic diameter can be used to compare particles of differing physical size and takes into account the density of the particle as well as its size. The aerodynamic diameter can be approximated by considering the following formulae: aerodynamic diameter (dae) = P°'5dp, where p is the density of the particle and dp the physical diameter of the particle. The advantage of considering an aerodynamic diameter is that it differentiates between otherwise similar physically sized particles that have different aerodynamic properties. As an example, a hollow sphere will have a lower density than an equally sized solid sphere. The aerodynamic diameter of the two spheres will be different with the hollow sphere being lower, even though the physical diameter is the same. Equally, seemingly differently shaped or sized particles may have the same aerodynamic diameter. An advantage of considering the aerodynamic diameter of a collection of particles is that it can be measured empirically in an impactor and it is not necessary to look at each particle individually to measure its physical size. The inhalation properties of the particles are assessed and the average aerodynamic properties of the particles determined to arrive at the average aerodynamic diameter of the particles are obtained. A typical measurement will identify what percentage of the total particles are within various aerodynamic diameter size ranges.
In one embodiment the average aerodynamic diameter of the particles is between from about 0.01 to about 20 microns. In a further embodiment, the average particle size is between from about 0.1 to about 20 microns. In a yet further embodiment, the average particle size is between from about 0.1 to about 15 microns, about 0.2 to about 15 microns, about 0.2 to about 10 microns, about 0.5 to about 10 microns, about 0.8 to about 10 microns, about 1 to about 10 microns, about 1 to about 9 microns, about 1 to about 8 microns, about 1 to about 7 microns, about 1 to about 6 microns, about 1 to about 5 microns, about 1 to about 4 microns, about 1 to about 3 microns, from about 1 to about 2 microns.
Particular average aerodynamic diameter ranges are between from about 1 micron to about 5 microns, between from about 1 micron to about 4 microns, and between from about 1 micron to about 3 microns.
In a yet further embodiment, the average aerodynamic diameter is < about 10 microns,
< about 9 microns, < about 8 microns, < about 7 microns, < about 6 microns, < about 5 microns, < about 4 microns, < about 3 microns, < about 2 microns, < about 1 micron. In a yet further embodiment, the average aerodynamic diameter is about 1 micron, about 2 microns, about 3 microns, about 4 microns, about 5 microns, about 6 microns, about 7 microns, or about 8 microns.
In a yet further embodiment, the average aerodynamic diameter is from about 5-20 microns (large particles) which may deposit in the oropharynx and upper airways, for example the trachea. In a yet further embodiment, the average aerodynamic diameter is from about 1-5 microns (small particles) which may deposit in the lower airways, for example the bronchus and bronchioles. In a yet further embodiment, the average aerodynamic diameter is from about 0.01 to about 1 micron (submicron particles) which may deposit in the alveoli.
In a particular embodiment, the average aerodynamic diameter is from about 1 to about 3 microns. In a yet further embodiment, about 90% of the particles (do.9) have an average size of < about 10 microns, < about 9 microns, < about 8 microns, < about 7 microns, < about 6 microns, < about 5 microns, < about 4 microns, < about 3 microns, < about 2 microns,
< about 1 micron. In a further embodiment 80% of the particles (do.g), 70% of the particles (do.7), 60% of the particles (do.6), 50% of the particles (do.5), 40% of the particles (do.4), 30% of the particles (do.3), 20% of the particles (do 2), or 10% of the particles (do.i), are less than at least one of the abovementioned sizes.
In a further embodiment, the particles can have an average size of 2 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; 3 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; 4 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; 5 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; 6 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; 7 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; 8 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; 9 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron; or 10 microns +/- 1 micron, +/- 0.5 micron, +/- 0.2 micron, or +/- 0.1 micron.
As has been discussed above, it may be that only a percentage of the total particles are of respirable size. Thus, the present invention specifically encompasses the situation where a percentage of the particles are of an average size as outlined above. Specific percentages include about 1% of the particles (do.oi), about 5% of the particles (do.os). about 10% (do.i), about 20% (do.2), about 30% (do.3), about 40% (do. ), about 50% (do.5), about 60% (doe), about 70% (do.?), about 80% (dog), about 90% (do.9), about 95% (do.95), about 98% (do.98), about 99% (do.w). or about 100% (di) of the particles are of an aerodynamic diameter outlined above.
When considering inhalable particles, one measurement is to look at the fine particle dose (FPD) which is the total mass of particles with aerodynamic diameters < 5 micron. It is also possible to consider the fine particle fraction (FPF), which is calculated by dividing the FPD by the loaded dose. In an embodiment the fine particle fraction (FPF) is > about 10%. In a further embodiment, the FPF is > about 20%, > about 30%, > about 40%, . > about 50%, > about 60%, > about 70%, > about 80%, > about 90%, > about 95%, or about 100%. The greater the FPF, the more active agent reaches the target area. Particular FPF are > 10 % of the loaded dose, > 30 % of the loaded dose, or > 50 % of the loaded dose.
In an alternative embodiment, instead of the aerodynamic diameter, it is possible to measure the specific dimensions of particles and to determine their average size. This may be the average particle diameter, including the mean or median diameter. These embodiments are not limited to spherical particles and can be used to measure irregular particles. It may be considered the average length, width and or height of the particles.
Because the actives are all present in a single respirable particle, the present invention can allow for defined doses to reach the same target areas, for example the same cells. This may have advantages in terms of therapy because targeted therapy with defined doses is possible. This may also allow for synergistic effects between the combination products because they may, for example, be simultaneously deposited the same target cell hi the lung epithelium. This has distinct advantages over physical mixtures of individual active agents.
Thus, in one embodiment, the particles, compositions methods and uses of the present invention allow for delivery of more than one active to the same target site, for example the same target cell in the lung epithelium. In a further embodiment, components may be added to the particles to allow for controlled release. This may be achieved by further coating the particles with a polymer. This could be achieved, for example, by mechanofusion or forming the particles with polymer included in the composition. In a yet further embodiment, the particles may further contain surfactants or polymers. These may be used to control crystal growth during co-precipitation in some techniques, such as by anti-solvent. Other excipients may be pH modifiers, antioxidants, and flavouring agents. In one aspect of the present invention the active agents and the excipient are evenly mixed throughout the particle. In a further embodiment, one active agent is predominantly present on the surface of the particle. . In a yet further embodiment, two or more actives are predominantly present on the surface of the particle. In a yet further embodiment, one active agent is predominantly present in the interior of the particle. In a yet further embodiment, two or more actives are predominantly present in the interior of the particle.
Thus, the present invention encompasses the situation where the active agents and excipient are homogeneously mixed throughout the particle and also the situation where the components are not homogeneously mixed.
The shape of the particles may affect the properties and or performance of combination products made using them. In particular, the shape of the particles can have an effect on their aerosolisation properties. Furthermore, the shape of the particles can have an effect on their handling properties. For example, substantially uniform and/or substantially spherical particles may have improved aerosolisation properties and may be less likely to stick together. These particles may have improved flow properties which may make capsule and/or device filling easier. In one aspect of the present invention the particles are predominantly spherical. In a further aspect the particles are ovoid. In a further aspect the particles are
predominantly ellipsoidal. In a yet further aspect the particles are predominantly needle or fibre shaped. In a yet further aspect the particles are predominantly plate like or flaked. In a yet further aspect the particles are predominantly pyramidal. In a yet further aspect the particles are spiky. In a yet further aspect the particles are irregularly shaped. In a further aspect the particles have a substantially uniform shape.
The surface conditions of the particles may be affected by the formation conditions and also by the selection of active agents. In some embodiments, the surface of the particles may be substantially uniform. In some embodiments, the surface may be dimpled. In some embodiments, the surface may contain crystals shaped particles adhered to the surface. In some embodiments, the surface may contain a clay like material. In further embodiments, the surface may be smooth. In yet further embodiments, the surface may be roughened. In yet further embodiments, the surface may be corrugated. In yet further embodiments, the surface may have plate like materials on the surface. In yet further embodiments, the surface may have spikes on the particle surface.
In a particular embodiment the particles are at least partially, or are substantially hollow. This has advantages in terms of the aerodynamic diameter of the particles and allows for larger particles which can still deliver active agents to the lower airways (or elsewhere). The ability to form larger particles with the same aerodynamic performance has advantages because larger particles are easier to handle.
It may be that as the dry particles are formed from solution, the individual components do not homogeneously mix. Without wishing to be bound by theory, one reason for this could be that the individual components have different solubilities, and/or different rates of crystallisation! This could lead to a non-uniform distribution of component in the final particle. For example, if the particle contains two active agents, both of which are more soluble than the excipient in the initial solution, then as the particle is formed the excipient comes out of solution first and adopts a predominantly crystalline form while the active agents remain in solution. If the excipient crystallises at the liquid-air interface you may end up with a predominance of excipient on or near the surface of the particle with the active agents forming predominantly at or near the centre of the particle since the solution will dry from the outside-in for a given droplet as it dries into a particle.
Alternatively, if the active agents are less soluble than the excipient then they may come out of solution before the excipient. Thus, the active agents may be on or near the surface of the particle with the inside of the particle being predominantly excipient. The active agents may be fully formed on the surface of the particle or may be embedded to some degree or fully in the outer surface of the particle.
Equally, it may be that one of the active agents is less soluble than the excipient where as one active is more soluble. Under these circumstances it may be that one active is predominantly found on or near the surface of the particle and the other particle is predominantly found at or near the centre of the particle.
Other factors may have the same effect, for example the rates of crystallisation or any other factor which will dictate at what stage a given component comes out of the liquid phase.
Thus, according to embodiments of the present invention the particle may contain one or more actives predominantly on or near the surface of the particle. According to a further embodiment, two actives are found on or near the surface of the particle.
According to yet further embodiments, the particle may contain one or more actives predominantly at or near the centre of the particle. According to a further embodiment, two actives are found predominantly at or near the centre of the particle. According to yet further embodiments, the particle may contain one or more actives predominantly on or near the surface of the particle and one or more actives predominantly at or near the centre of the particle.
According to yet further embodiments of the present invention the particle may contain excipient predominantly on or near the surface of the particle.
According to yet further embodiments, the particle may contain excipient predominantly at or near the centre of the particle. According to yet further embodiments, the particle may contain a substantially homogeneous mixture of active agents and excipient.
By selecting appropriate solvent conditions it may be possible to design a specific particle composition taking into account the solubility and other factors for the components of the particle.
The present invention includes particles containing two or more active agents. In one embodiment, the particles contain two active agents. In a further embodiment, the particles contain 3 active agents. In a yet further embodiment the particles contain 4 active agents.
Equally, in certain embodiments, the particles contain a single active agent.
Examples of active agents or drugs which can be used with the present invention include beta-2 agonists, anticholinergics, mast cell stabilisers, steroids, methylxanthines, inhaled corticosteroids, theophylline, leukotriene modifiers long- acting beta-2 agonists, short-acting beta-2 agonists and/or systemic corticosteroids. In a particular embodiment, the particles contain an inhaled corticosteroid (ICS) and a long-acting P2-agonist (LABA). Specific examples of drugs which can be used include acetonide, albuterol, albuterol sulfate, beclomethasone, budesonide, cortisone, cromolyn, cromolyn sodium, dexamethasone, flunisolide, fluticasone, formoterol, formoterol fumarate, hydrocortisone, pratropium, ipratropium / albuterol, levalbuterol HC1, metaproterenol, methylprednisolone, mometasone, montelukast, nedocromil, nedocromil sodium, omalizumab, pirbuterol, prednisolone, propionate, salbutamol, salmeterol, salmeterol xinafoate, terbutaline, theophylline, tiotropium, triamcinolone, zafirlukast or zileuton.
"Drugs", for the purposes of the invention, include a variety of pharmaceutically active ingredients, such as, for example, those which are useful in inhalation therapy. In general, the term "drug" is to be broadly construed and include, without limitation, actives, drugs and bioactive agents, as well as biopharmaceuticals.
The term "drug" is interchangeable with the term medicament or active agent. Appropriate drugs may thus be selected from, for example, analgesics, (e.g., codeine, dihydromorphine, ergotamine, fentanyl or morphine); anginal preparations, (e.g.,
diltiazem); anti-allergies, (e.g., cromoglicate, ketotifen or nedocromil); antiinfectives (e.g., cephalosporins, penicillins, streptomycin, sulphonamides, tetracyclines and pentamidine); antihistamines, (e.g., methapyrilene); antiinflammatories, (e.g., antiinflammatory steroids, beclomethasone (e.g. beclomethasone dipropionate), fluticasone (e.g. fluticasone propionate), flunisolide, budesonide, rofleponide, mometasone (e.g. mometasone furoate), ciclesonide, triamcinolone (e.g. triamcinolon acetonide), 6a, 9a- difluoro- 1 1 β-hydroxy- 16a-methyl-3-oxo-l 7a-propionyloxy-androsta- 1 ,4-diene- 17β- carbothioic acid, S-(2-oxo-tetrahydro-furan-3-yl)ester), (6a, 1 lb,16a,17a)-6,9-difluoro- 17- { [(fluoromethyl)thio]carbonyl } - 11 -hydroxy- 16-methyl-3 -oxoandrosta- 1 ,4-dien- 17- yl 2-furoate, and (6a,l lb,16a,17a)-6,9-difluoro-17-{[(fluoromethyl)thio]carbonyl}-l 1 - hydroxy- 16-methyl-3-oxoandrosta-l,4-dien-17-yl 4-methyl-l ,3-thiazole-5- carboxylate); antitussives, (e.g., noscapine); bronchodilators, (e.g., albuterol (e.g., as sulphate), salbutamol (e.g., as the free base or the sulphate salt), salmeterol (e.g., as xinafoate), ephedrine, adrenaline, fenoterol (e.g., as hydrobromide), bitolterol, formoterol (e.g., as fumarate), isoprenaline, metaproterenol, phenylephrine, phenylpropanolamine, pirbuterol (e.g., as acetate), reproterol (e.g., as hydrochloride), rimiterol, terbutaline (e.g., as sulphate), isoetharine, tulobuterol, 4-hydroxy-7-[2-[[2- [[3-(2-(henylemoxy)propyl]sulfonylurea]emyl]-amino]emyl-2(3H)-benzothia2olone), 3-(4-{[6-({(2R)-2-hydroxy-2-[4-hydroxy-3-(hydroxymethyl)
phenyl]ethyl}amino)hexyl]oxy}butyl)benzenesulfonamide, 3-(3-{[7-({(2R)-2-hydroxy- 2-[4-hydroxy-3-(hydroxymethyl)phenyl]ethyl}amino)heptyl]oxyjpropyl)
benzenesulfonamide, 4- {( 1 R)-2-[(6- {2-[(2,6-dichlorobenzyl)oxy]
ethoxy}hexyl)amino]-l -hydroxyethyl} -2-(hydroxymethyl)phenol, 2-hydroxy-5-(( 1 R)- 1 -hydroxy-2-{[2-(4- {[(2R)-2-hydroxy-2- phenylethyl]amino}phenyl)ethyl]amino}ethyl)phenylformamide, and 8-hydroxy-5- {( 1 R)- 1 -hydroxy-2-[(2- {4-[(6-methoxy- 1 , 1 '-biphenyl-3- yl)amino]phenyl}ethyl)amino]ethyl}quinolin-2(lH)-one); diuretics, (e.g., amiloride) ; anticholinergics, (e.g., ipatropium (e.g., as bromide), tiotropium, atropine or oxitropium); hormones, (e.g., cortisone, hydrocortisone or prednisolone); xanthines, (e.g., aminophylline, choline theophyllinate, lysine theophyllinate or theophylline); therapeutic proteins and peptides, (e.g., insulin).
In addition to those stated above, it will be clear to a person skilled in the art that, where appropriate, the medicaments may be used in the form of salts, (e.g., as alkali metal or amine salts or as acid addition salts) or as esters (e.g., lower alkyl esters) or as solvates (e.g., hydrates) to optimize the activity and/or stability of the medicament. It
will be further clear to a person skilled in the art that where appropriate, the medicaments may be used in the form of a pure isomer, for example, R-salbutamol or R-formoterol. Particular medicaments for administration using pharmaceutical formulations in accordance with the invention include anti-allergies, bronchodilators, beta agonists (e.g., long-acting beta agonists), and anti-inflammatory steroids of use in the treatment of respiratory conditions, as defined herein, by inhalation therapy, for example, cromoglicate (e.g. as the sodium salt), salbutamol (e.g. as the free base or the sulphate salt), salmeterol (e.g. as the xinafoate salt), bitolterol, formoterol (e.g. as the fumarate salt), terbutaline (e.g. as the sulphate salt), 3-(4-{[6-({(2R)-2-hydroxy-2-[4- hydroxy-3-(hydroxymethyl)phenyl]ethyl}amino)hexyl]oxy}butyl)benzenesulfonamide, 3-(3-{[7-({(2R)-2-hydroxy-2-[4-hydroxy-3-
(hydroxymethyl)phenyl]ethyl}amino)heptyl]oxy}propyl)benzenesulfonamide, 4-{(l f?)-2-[(6-{2-[(2,6-dichlorobenzyl)oxy] ethoxy}hexyl)amino]-l -hydroxyethyl}-2- (hydroxymethyl)phenol, 2-hydroxy-5-((l R)- l-hydroxy-2-{[2-(4-{[(2R)-2-hydroxy-2- phenylethyl]amino}phenyl)ethyl]amino'} ethyl)phenylformamide, 8-hydroxy-5-{(l R)- 1 -hydroxy-2-[(2-{4-[(6-methoxy-l ,V- biphenyl-3- yl)amino]phenyl}ethyl)amino]ethyI}quinolin-2(l H)-one, reproterol (e.g. as the hydrochloride salt), a beclomethasone ester (e.g. the dipropionate), a fluticasone ester (e.g. the propionate), a mometasone ester (e.g., the furoate), budesonide,
dexamethasone, flunisplide, triamcinolone, tripredane, (22R)-6a,9a- difluoro-11 β, 21 - dihydroxy- 16a, 17a-propylmethylenedioxy-4-pregnen-3 ,20-dione. Medicaments useful in erectile dysfunction treatment (e.g., PDE-V inhibitors such as vardenafil
hydrochloride, along with alprostadil and sildenafil citrate) may also be employed. It should be understood that the drugs that may be used in conjunction with the inhaler are not limited to those described herein.
Further specific actives include:
Anti asthmatic / COPD B-agonist orciprenaline
metaproterenol clorprenaline salbutamol terbutaline (sulphate) fenoterol soterenol trimetoquinol salmeterol
formoterol fumarate montelukast procaterol hydrochloride isoproterenol xanthine derivative caffeine
teophylline theobromine aminophylline anti-allergic agent cromoglicate
ketotifen
repirinast oxatomide tranilast
ibudirast
antihistamine agent tefenadine
mequitazine anticholinergic
agent ipratropium (bromide) atropine
methylbromide oxitropium bromide beclometasone steroid (dipropionate)
fluticasone
(propionate) budesonide mometasone furoate ciclesonide dexamethasone
Expectorants bromhexine
acetylcysteine methylcysteine ethylcysteine
Respiratory stimulants doxapram
dimo holamine
Antitussives codeine
dihydrocodeine dextromethorphan dimemorphan
cloperastine
pentoxyverine oxeladine benzonatate
Antibiotics
Sulfa drugs ciprofloxacine
sulfisoxazole sulfisomidine sulfamethoxazole sulfamine sulfamonomethoxine antitubercular
agent isoniazid
isonicotinic acid hydrazide p-aminosalicylic acid rifampicin ethambutol cycloserine ethionamide beta-lactam
antibiotic Penicillins methicillin
cloxacillin ampicillin amoxicillin carbenicillin piperacillin benzylpenicillin sulbenicillin
Cephems cephalosporin C cephalothin cephaloridine cefazolin cephalexin cefradine cefaclor cefotiam
cefroxime cefotaxime ceftizoxime cefmenoxime
cefoperazone ceftazidime
Carbapenems imipenem Monobactams azthreonam
carumonam β-Lactamase
inhibitor clavulanic acid sulbactam sultamicillin amino
glycosides tobramycin gentamicin kanamycin streptomycin amikacin dibekacin colomycin neomycin paromomycin bekanamycin arbekacin macrolide
antibiotics erythromycin oleandomycin kitasamycin spiramycin josamycin midecamycin crarythromycin tetracycline antibiotics chlortetracycline oxytetracycline tetracycline doxycycline minocycline chloramphenicol thiamphnicol lincomycin clidamycin fosfomycin pyridonecarboxylic acids nalidixic acid pipemidic acid
 norfloxacin
ofloxacin
norfloxacin
ofloxacin
lomefioxacin enoxacin ciprofloxacin tosufloxacin
Anti virus zanamivir hydrate amantadine idoxuridine cytarabine vidarabine acyclovir ribavirin zidobudine antifungal agents griseofulvin flucytosine trichomycin amphotericin B nystatin econazole miconazole clotrimazole fluconazole terconazole tolnaftate pyrrolnitrin butenafine antipyretic
agent/analgesic agent acetoaminophen aminopyrine etc. hypoglycemic agent tolbutamide gliclazide etc. anticancer agent methotrexate etc.
Combinations of two or more agents selected from the group consisting of salmeterol, especially salmeterol xinafoate, salbutamol, fluticasone propionate, formoterol, budesonide, beclomethasone dipropionate and physiologically acceptable salts and solvates thereof are specifically exemplified.
In one embodiment, the particle contains a combination of two active ingredients known for the treatment and/or prophylaxis of respiratory disorders. In a particular
embodiment the particle may comprise formoterol (e.g. as the fumarate) and budesonide. In a further embodiment the particle may comprise salmeterol (e.g. as the xinafoate salt) and fluticasone (e.g. as the propionate ester). In a yet further embodiment the particle may comprise salbutamol (e.g. as free base or sulphate salt) and beclomethasone (as the dipropionate ester).
Further particular combinations include:
mometasone furoate/formoterol fumarate;
mometasone furoate and salmeterol xinafoate;
beclomethasone dipropionate and formoterol fumarate;
beclomethasone dipropionate and salmeterol xinafoate;
fluticasone propionate and formoterol fumarate;
budesonide and salmeterol xinafoate;
salbutamol sulfate and fluticasone propionate;
salbutamol sulfate and budesonide;
terbutaline sulfate and beclomethasone dipropionate;
terbutaline sulfate and fluticasone propionate; or
terbutaline sulfate and budesonide. The particles of the present invention may be used directly in inhalable compositions. Alternatively, they may be combined with additional components, carriers and/or components which are therapeutically acceptable.
Thus, in a further aspect of the present invention there is provided an inhalable composition comprising particles as disclosed herein, together with a therapeutically acceptable carrier.
In a yet further embodiment, there is provided a dry powder inhaler (DPI) containing particles as defined herein. In a yet further embodiment, there is provided a dry powder inhaler (DPI) containing a composition as defined herein.
The particles may be suitable for use in a metered dose inhaler (MDI). Thus, in a yet further embodiment, there is provided an MDI containing particles as defined herein. In a yet further embodiment, there is provided an MDI containing a composition as defined herein.
The particles may be suitable for use in a nebuliser. Thus, in a yet further embodiment, there is provided a nebuliser containing particles as defined herein. In a yet further embodiment, there is provided a nebuliser containing a composition as defined herein. In some embodiments, (for example with DPIs) the particles may be used directly without further modifications or further components. In particular, in a DPI there is no need for a propellant.
Alternatively, the particles may be part of a composition containing additional components, for example a therapeutically acceptable carrier.
The composition may further comprise a propellant, for example a hydrofluorocarbon propellant. Example propellants include HFA 134a and 227ea. The composition may further include water, salts (for example for tonicity adjustment), pH modifiers (for example weak acids), and/or polymers for viscosity adjustment.
The present invention encompasses any way to make the particles. Particularly, the present invention encompasses any method to dry a solution to form particles. Example methods of manufacture include: spray drying an aqueous solution of the components; co-precipitation by anti-solvents, co-precipitation by other means (for example by chemical reaction including pH changes) followed by drying including spray drying; co-precipitation by super-critical fluid precipitation; mechanofusion (coating crystalline particles with additional drugs onto the surface), optionally with annealing afterwards to crystallise an amorphous surface; or freeze drying, spray drying an organic solution; spray drying a solution containing a co-solvent mixture of water and organic solvent(s); spray freeze drying (this is different to simple spray drying or freeze drying a solution, involving spraying a solution into liquid nitrogen, the frozen droplets are then dried in a freeze dryer).
In a yet further embodiment there is provided a method of manufacturing particles comprising:
- forming a solution containing two or more active agents together with an excipient in a solvent
- drying the solvent to form particles that have an at least partially crystalline form.
The solvent does not need to be aqueous, it can be totally organic or a mixture of water and organic solvent(s). For example, a co-solvent system could comprise water and ethanol. Equally, an organic solvent/water mixture could be used and this may be used to increase the crystallinity of the particles compared to an aqueous solution alone.
In one embodiment, the particles are formed by spray drying.
In one embodiment, the active agents together with the excipient may be dissolved in an aqueous solution. This solution may be, for example, water or a mixture of water and an alcohol such as ethanol. The feed solution may then be spray dried.
Crystallization depends on Tg of the components and also other factors including the material and systems. One additional factor is the inlet temperature of the spray drying equipment. In a particular embodiment, the inlet temperature is higher than Tg of the excipient that is expected to be crystalline. In general, the outlet temperature will be lower than the inlet temperature.
In a further embodiment there is provided a method of delivering a combination of two or more active agents to a patient in need thereof comprising administering particles as defined herein.
In a yet further embodiment there is provided a method of delivering a combination of two or more active agents to a patient in need thereof comprising inhaling particles as defined herein.
According to yet another aspect, the present invention comprises a method of delivering a combination of two or more active agents to a patient, the method comprising forming particles as defined herein and aerosolising the particles to be suitable for inhalation by the patient.
Such aerosolisation techniques are well known in the art and all suitable methods are encompasses herein.
According to yet another aspect, the present invention comprises particles as defined herein for use as a medicament.
The present invention may be used to treat a wide array of diseases and/or conditions. Particular conditions include a respiratory or non-respiratory condition.
Thus, according to one aspect of the present invention there is provided a method of treating a patient having a respiratory or non-respiratory condition, said method comprising administering particles as defined herein.
According to yet another aspect, the present invention comprises use of the particles as defined herein in the manufacture of a medicament for treatment of a respiratory or non-respiratory condition of a patient.
According to yet another aspect, the present invention comprises particles as defined herein for use in the treatment of a respiratory or non-respiratory condition of a patient. According to a further aspect, the present invention provides the use of particles as defined herein for the manufacture of a medicament for the treatment of a respiratory or non-respiratory condition.
According to a further aspect, the present invention provides a method of delivering a medicament to a patient in need thereof comprising administering particles as defined herein.
According to a further aspect, the present invention provides a pharmaceutical composition comprising particles as defined herein.
According to a further aspect, the present invention provides particles as defined herein for use as a medicament.
According to a further aspect, the present invention provides the use of particles as defined herein as a medicament.
According to a further aspect, the present invention provides particles as defined herein for use in the treatment of respiratory or non-respiratory conditions. According to a further aspect, the present invention provides the use of particles as defined herein in the treatment of respiratory or non-respiratory conditions.
Diseases or conditions for which the present invention may apply include respiratory or non-respiratory conditions. Respiratory conditions include, COPD, bronchitis, allergy, rhinitis, cystic fibrosis, pulmonary infection, tuberculosis, influenza, other lung infections, lung cancer and asthma. Non-respiratory conditions include diabetes, hypertension, hypercholesterolaemia, gout, infections (bacterial or viral), fever, pairi (neurological or muscular).
In one embodiment, the respiratory condition is COPD. In a further embodiment, the respiratory condition is bronchitis. In a yet further embodiment, the respiratory condition is allergy. In a yet further embodiment, the respiratory condition is rhinitis. In a yet further embodiment, the respiratory condition is cystic fibrosis. In a yet further embodiment, the respiratory condition is pulmonary infection. In a yet further embodiment, the respiratory condition is asthma.
Particular conditions are COPD and asthma.
The medicament can be manufactured using the methods as defined herein. Throughout this specification the word "comprise", or variations such as "comprises" or "comprising", will be understood to imply the inclusion of a stated element, integer or step, or group of elements, integers or steps, but not the exclusion of any other element, integer or step, or group of elements, integers or steps. Brief Description of Drawings
By way of example only, preferred embodiments of the invention are now described with reference to the following drawings, in which: Figure 1. SEM of spray dried powder
(a), (b): spray dried mannitol (M-SD), (c), (d): spray dried budesonide/formoterol fumarate dihydrate/mannitol (B/F/M-SD), (e), (f): spray dried fluticasone propionate/salmeterol xinafoate/mannitol (F/S/M-SD) Figure 2. FIB-SEM of spray dried powder
(a): spray dried budesonide/formoterol fumarate dihydrate/mannitol (B/F/M-SD), (b): spray dried fluticasone propionate/salmeterol xinafoate/mannitol (F/S/M-SD)
Figure 3. Chemical structure of drugs and mannitol
(a) budesonide, (b) formoterol fumarate dihydrate, (c) fluticasone propionate, (d) salmeterol xinafoate, (e) mannitol
Figure 4. DSC curves of co-spray dried powders and raw materials
Left: B/F/M-SD and its raw materials, (a) B/F/M-SD, (b) mannitol, (c) budesonide, (d) formoterol fumarate dihydrate
Right: F/S/M-SD and its raw materials, (e) F/S/M-SD, (f) mannitol, (g) fluticasone propionate, (h) salmeterol xinafoate
Figure 5. XRD patterns of spray dried powders
(a) fluticasone propionate/salmeterol xinafoate/mannitol (F/S/M-SD), (b) budesonide/formoterol fumarate dihydrate/mannitol (B/F/M-SD), (c) mannitol (M-SD)
Figure 6. In vitro deposition of spray dried powder at 100 L/min
FPF shows average ± SD (n=3). (a) spray dried budesonide/formoterol fumarate dehydrate/mannitol (B/F/M-SD), (b): spray dried fluticasone propionate/salmeterol xinafoate/mannitol (F/S/M-SD), (c) spray dried mannitol (M-SD). Note: The LABA values are not available as their peaks overlapped with those of the HPMC capsule in the HPLC analysis. Deposition in the micro-orifice collector (MOC) was too low to be detected.
Figure 7. In vitro deposition of spray dried powder, at 60 L/min
FPF shows average ± SD (n=3). (a) spray dried budesonide/formoterol/mannitol fumarate dehydrate (B/F/M-SD), (b): spray dried fluticasone propionate/salmeterol xinafoate/mannitol (F/S/M-SD), (c) spray dried mannitol (M-SD)
Fig. 8 shows cross section images of B/F/M-SD (budesonide / formoterol fumarate dihydrate / mannitol spray dried) particles.
(a), (b): SEM photos from FIB-SEM
(c)-(e): SEM photos from high resolution FESEM
Fig. 9 Cross section images of F/S/M-SD (fluticasone propionate / salmterol xinafoate / mannitol spray dried) particles.
(a)-(c): SEM photos from FIB-SEM
(d), (e): SEM photos from high resolution FESEM
Figure 10. FESEM of spray dried powder S/B/M-SD (salbutamol sulfate / beclomethasone dipropionate / mannitol)
Figure 11. FESEM of spray dried powder M/F/M-SD (mometasone furoate / formoterol fumarate / mannitol)
The following description describes methods of forming particles of the present invention from mannitol and active agents selected budesonide / fluticasone propionate, and formoterol fumarate dihydrate / salmeterol xinafoate. It will be appreciated that other components, including other active agents and/or other excipients, such as other sugars, can be utilised to form suitable carrier particles. The particles as formed herein can be used to form compositions suitable for inhalation by a patient requiring the drug. The drug can be suitable for treating respiratory conditions such as cystic fibrosis, COPD, bronchitis, allergy, rhinitis and asthma. It will also be appreciated that the drug can be suitable for delivery via the lungs and be used to treat non-pulmonary conditions.
Respirable particles - aerodynamic diameter
The following information is intended to demonstrate how the aerodynamic diameter can be measured. This provides a method to determine the aerodynamic diameter of a given particle sample.
The aerodynamic particle size is measured by dispersing 5 mg of the powder from an Aerolizer® through a Next Generation Impactor (British Pharmacopoeia Apparatus E) without the pre-separator at 60 and 100 L min air flow for 4 and 2.4 s, respectively. The experimental procedure largely follows that detailed in the British Pharmacopoeia.
Essentially, the flow rate and solenoid valve timer are adjusted appropriately prior to sampling. A known amount of powder may be weighed into a Size 3 hydroxypropyl methylcellulose capsule and loaded into the capsule compartment in the Aerolizer®. The capsule is pieced and the inhaler is inserted into the induction port via a rubber adaptor flush with the inhaler mouthpiece to discharge the dose. Repeat the sampling
procedure with more loaded capsules if necessary. The number of discharges should be minimised (typically < 10) to obtain sufficient powder for an accurate and precise quantification. The powder deposits of actives and excipient in the capsule, Aerolizer®, rubber adaptor, induction port, and all impactor stages of the NGI are assayed chemically. Suitable assay methods include, but are not limited to, the following: ultraviolet spectrophotometry, high performance liquid chromatography (HPLC), gas chromatography (GC), or liquid chromatography-mass spectrometry (LC-MS). Both actives should deposit concurrently on all assayed parts. The loaded dose is defined as the total amount of powder weighed into the capsules for aerosol sampling. The fine particle dose (FPD) is the total mass of particles with aerodynamic diameters < 5 μπι. The fine particle fraction (FPF) is calculated by dividing the FPD by the loaded dose.
Materials Budesonide (EP grade) was purchased from Yicheng Chemical Corp. (Jiangsu, China), fluticasone propionate (BP grade), formoterol fumarate dihydrate (EP grade) and salmeterol xinafoate (EP grade) from Jai Radhe Sales (Gujarat, India), and mannitol (EP grade) from Roquette (Lestrem, France). Water was purified by a Modulab II DI Unit (Siemens Water Technologies Corp., Pennsylvania, USA). All organic solvents were supplied by Biolab Ltd. (Victoria, Australia) and were of analytical grade.
The examples show particles made from a combination of an inhaled corticosteroid (ICS) and a long-acting p2-agonist (LABA). Preparation of spray dried particles
According to the compositions in Table 1, ICS, LABA and mannitol were dissolved in ethanol/water to prepare the feed solutions for spray drying. Spray drying was performed using a B-290 mini spray dryer (Buchi Labortechnik AG, Falwil, Switzerland) with operating conditions detailed in Table 2. The spray dried powder samples were kept in a glass desiccator containing silica gel at 22 °C until used.
(mg/mL)
B/F/M-SD F/S/M-SD M-SD
Budesonide
Fluticasone propionate 0.8
Formoterol fumarate dihydrate
Salmeterol xinafoate 0.08
Mannitol 7.12 20
Solvent 50%EtOH (v/v) 75%EtOH (v/v) 50%EtOH (v/v)
Table 1 Composition of feed solution for spray drying
B F/M-SD F/S/M-SD M-SD
Inlet temperature 130 °C 130 °C 130 °C
Feed rate (mL/min) 2.5 5 2.5
Aspiration (%) 100 100 100
Atomization (Liter/hr) 742 473 473
Gas Nitrogen Nitrogen Nitrogen
Yield (%) 58 69 86
Table 2 Spray drying condition
Particle size determination
Particle size distributions of the powders were determined by laser diffraction using Mastersizer 2000 (Malvern Instruments, Worcs, UK). The powders were dispersed through the measurement zone with compressed air at 4 bars of pressure by a Scirocco 2000 dry powder feeder (Malvern Instruments, Worcs, UK). The particle refractive index and absorption were 1.52 and 0.1, respectively. The dispersant refractive index was 1.000 for air. All measurements were conducted in triplicate. Drug quantification
Drug content in the spray dried powders after preparation and in the aerosol samples obtained from the dispersion study were determined using high performance liquid chromatography (HPLC) (Model LC-20; Shimadzu, Kyoto, Japan). For assays of budesonide and formoterol fumarate dihydrate, a LiChrosphere 60® RP-select B column (4 x 125 mm, 5 μιη) (Merck, Darmstadt, Germany) was used as the stationary phase with the mobile phase comprising methanol, water and acetic acid (550:450:1). A flow rate of 1 mL/min and UV absorption wavelength at 230 nm was employed for
parallel detection of both drugs (retention times were 4.9 min and 15.8 min for formoterol fumarate dehydrate and budesonide, respectively). For fluticasone propionate and salmeterol xinafoate, an Intersil ODS2 column (4.6 x 200 mm, 5 μπι) (Capital HPLC, Scotland, UK) was used as stationary phase with methanol and 0.6% ammonium acetate buffer (75:25) being the mobile phase. A flow rate of 1 mL min and wavelength 228 nm was used for parallel detection of salmeterol xinafoate and fluticasone propionate, at retention times of 2.7 min and 7.1 min, respectively.
Mannitol quantification
Mannitol content in the powders was determined by HPLC using refractive index detection as described previously (20). Briefly, a CI 8 Radial-Pack column (Waters, USA) was used as stationary phase with deionized water as mobile phase running at 1 mL min.
Thermal analysis
Differential scanning calorimetry (DSC) was performed on a DSC instrument (Model 82 le; Mettler Toledo, Greifensee, Switzerland). A known amount (8 - 13 mg) of each sample was weighed into a 40 aluminium crucible with a vent hole. The samples were heated from 20 to 350 °C Under 250 cmVmin nitrogen purge at a scan rate of 10 °C/min.
Thermal gravimetric analysis (TGA) was carried out on a thermogravimetric analyser (Model 2050; TA Instruments, Delaware, USA). Each sample (10 - 15 mg) was placed on an open platinum pan and was measured under 90 cmVmin nitrogen purge at a scan rate of 10 °C/min from 20 to 120 °C.
X-ray powder diffraction
For analysis of crystallinity and polymorphism, X-ray powder diffraction (XRD) measurement was carried out at room temperature using an X-ray diffractometer (Model D5000; Siemens, Munich, Germany). Cu a radiation at 30mA and 40kV was used with an angular increment of 0.04° at 3 sec per step covering a 2Θ range of 5-50°.
Morphological observation
The morphology of the spray-dried particles was investigated using scanning electron microscopy (SEM). Samples were deposited on carbon sticky tape and mounted on a SEM stubs, followed by sputter coating with gold (15 nm thick) on a 550X sputter coater (Quorum Emitech, Kent, UK). The specimens were then imaged using a field emission SEM (Zeiss Ultra Plus; Carl Zeiss SMT AG, Oberkochen, Germany) at 1.90- 1.99 kV using an in-lens detector. Internal structure observation
Focused-ion-beam (FIB) milling of the particles was performed in an FIB/scanning electron microscope dual beam system (Nova 200, FEI, USA) using a similar procedure described previously, but without the cleaning step (21). The powder samples were coated with a 50 nm layer of platinum. Vertical cutting of the sample was performed at a stage tilt of 52 degrees with an accelerating voltage of 10 kV and a beam current of 19 pA. Milling times were kept under 5 min. The milled samples were later examined at 2 kV to examine the cut surface. Atomic force microscopy
Particle-particle cohesion force was evaluated using the colloid probe microscopy technique. Individual particle was mounted onto the apex of V-shaped tipless atomic force microscopy (AFM) cantilever (NP-0 silicon nitride cantilevers with gold reflective coating, nominal spring constant 0.58 nN; Veeco Inc., New York, USA) using a micromanipulation technique described elsewhere. The force of adhesion between each probe and particles mounted on a thermoplastic adhesive (Tempfix®; Piano, Wetzlar, Germany) was investigated using force-volume imaging. Scanning of force-volume was conducted over a 10 μιη x 10 μιη area using the following settings: approach retraction cycle 3 μπι, cycle rate 8 Hz, and constant compliance distance 60 nm. The matrix of force of adhesion and topographical data were obtained using custom-written software. Topographical data was used to identify each particle and individual force curves taken diagonally across each particle. Five particles were analysed for each probe to collect the force curves (n > 43 per probe) and three probes were used for each sample.
Surface composition evaluation
Surface elemental composition was measured using X-ray photoelectron spectroscopy (XPS). The instrument comprises a Thermo VG ESCALAB250 spectrometer with a non-monochromatic Al alpha (1486.6 eV) X-ray source. Analysis was carried out under a pass energy of 20 eV. The powders were pressed onto double sided conductive adhesive tapes. Experimental molar percentages of all elements except hydrogen were derived from the XPS peak areas as described elsewhere (30). The molar percentages (At%) were multiplied by the appropriate atomic mass to obtain weight percentages (Wt%) (Table 5 and Table 7). The surface elemental composition of the raw materials (mannitol, budesonide, formoterol fumarate dihydrate, fluticasone propionate and salmeterol xinafoate) and the three spray dried powders (M-SD, B/F M-SD and F/S/ - SD) were measured. The type of molecules present on the particle surface can be deduced from the detected elements and elemental percentages. The expected elemental percentages of the spray dried powders were calculated from the percentages measured on the raw materials. All components of the ternary powders were assumed to be distributed evenly on the surface for the calculation. The expected elemental percentages are shown in Table 6 and Table 7. Essentially, the expected Wt% of an element in the powder was the sum of the products obtained by multiplying the Wt of that element in the raw materials by the Wt% of the corresponding materials in the powder (Table 6). Differences between the experimental and expected percentages suggest an over- or under-abundance of certain types of molecules on the surface.
In vitro dispersion study
Dispersibility of the spray-dried powders was assessed using a Next Generation Impactor (NGI) with a, United State Pharmacopoeia (USP) throat. The method followed the procedure specified for DPIs in the British Pharmacopoeia. Prior to testing, all eight stages were coated with silicon grease (Slipicone; DC Products Pty Ltd., Victoria, Australia) to minimize particle bounce. A sample (5.00 ± 0.10 mg) of each powder was weighed into a size 3 hydroxypropyl methylcellulose capsule (Capsugel, West Ryde, Australia) which was then loaded into an Aerolizer® (Novartis Pharmaceuticals, North Ryde, Australia) and pierced. After connecting the Aerolizer® to the USP throat via a mouthpiece adapter, the dispersion test was performed for 2.4 sec at 100 IJmin and 4
sec at 60 L/min, with the flow rate measured by a flow meter (Model 3063; TSI Incorporated, Minnesota, USA). Dispersions were performed in triplicate for each powder. FPF was defined as the mass fraction of particles < 5.0 urn with respect to the loaded dose in the capsules. The cut-off diameters of the NGI stages at 100 L min were calculated with the cut-off adjustment equations given in Appendix XII C of the British Pharmacopoeia.
Results Preparation of spray dried powders
Spray dried powders of mannitol alone (M-SD) or ternary systems of ICS, LABA and mannitol (B/F/M-SD and F/S/M-SD) were obtained with > 50% yield (Table 2). All three powders had a similar particle size distribution in the inhalable range with volumetric median diameters (D50) of 2 μπι (Table 3).
(μπι)
B/F/M-SD F/S/M-SD M-SD
D10 0.63±0.04 0.79±0.02 0.75±0.00
D50 2.01±0.02 2.02±0.02 2.01±0.01
D90 5.00±0.64 4.28±0.04 4.22±0.02
Span 2.17±0.29 1.73±0.01 1.73±0.00
Average ± standard deviation (n=3)
Table 3 Particle size of spray dried powder
The drug content of each component in the powder was determined by HPLC to be close to 100 % (Table 4). In addition, the drug content ratios of the two combination powders were confirmed to be the same as those of the feed solutions.
(% of Ideal)
B/F/M-SD F/S/M-SD
Budesonide 100.6±0.8 -
Fluticasone propionate - 99.9±0.4
Formoterol fumarate dihydrate 99.9±1.3 -
Salmeterol xinafoate - 99.8±0.3
Average ± standard deviation (n=3)
Table 4 Drug content measured by HPLC
Evaluation of physico-chemical properties of the powders
Particles of all three powders were spherical but with different surface characteristics, as shown in the SEM images (Figure 1). Most distinctively, B F/M-SD particles possessed smaller prismatic projections on the surface. The surface of the F/S M-SD particles showed an irregular mosaic structure comprising small particles and pores, whereas the M-SD particle surface was smoother and more uniform. In both B/F M-SD and F/S/M-SD, FIB-SEM revealed partially hollow interior structures (Figure 2). Scanning electron microscopy (SEM) was carried out on particles made according to the present invention and the results are included in Figure 1. Particles of spray dried mannitol (M-SD), spray dried budesonide / formoterol fumarate dihydrate / mannitol (B/F/M-SD), and spray dried fluticasone propionate /salmeterol xinafoate / mannitol (F/S/M-SD) were compared. The images show that the particles of all the three samples are spherical but the surface conditions of them were different. M-SD showed the spherical particles with a smooth surface and packed crystals of mannitol were observed on the surface. M-SD spray dried from both 50 and 75% ethanol did not show small particles adhered onto the surface as the other two combination powders described later. B/F/M-SD also had a smooth surface but the packing of mannitol crystals was not observed. Small crystal-shaped particles adhered onto the surface. The surface of F/S/M-SD particles seemed roughly covered with mixture of clay-like material and small particles. It also had many dimples on the surface. These observations might suggest that the components of the B/F/M-SD and F/S/M-SD particles are not homogeneously mixed.
While not wishing to be bound by theory, this inhomogeneity could be caused by the difference of solubility of mannitol and drugs. Solidification of drugs and mannitol would have occurred at the different time points during the spray drying process as the solvent evaporated. It could be that the active agents solidified first on the surface of the droplets and then mannitol came out inside of the outer shell of drugs. As the particles are mainly composed of mannitol, the crystallinity of drugs could not be examined using XRD and DSC.
Focus ion beam-scanning electron, microscopy (FIB-SEM) is similar to scanning electron microscopy (SEM) but it uses a liquid metal source as the filament to produce
a finely focused beam of energetic metal ions that can be applied for both imaging and milling.
Under FIB-SEM, the particle is cut with ion beam and its cross section is viewed under SEM. One difficulty that arises out of this procedure is that condition adjustment can be difficult when the specimen is sensitive because it can easily deteriorate under the ion or electron beam. Since the ion beam removes material from the specimen surface, mild conditions preferable to reduce the deteriorating effect on the specimen. However, it becomes more difficult to obtain clear images at lower accelerating voltages and lower beam currents. Therefore, the mildest condition that can give acceptably clear image should be applied.
In this experiment, a separate SEM (field emission SEM, ZEISS Ultra) was used for observation. Thus, a transmission electron microscopy grid for defining the position was placed on the stage with a thin layer of double-sided adhesive carbon tape. The sample powder was sprinkled on the grid and coated with platinum/gold (30 nm thick). The coated sample was set in the FIB-SEM (Quanta 200) chamber and milling (vertical cutting) was performed at the eucentric height with stage tilt of 52° as the ion column and electron column faces each other at the angle of 52°. Cleaning of the cross section followed the milling to reduce the water falling effect. The cut particles were observed under SEM.
In both B/F M-SD and F/S/M-SD, FIB-SEM revealed partially hollow interior structures (Figure 2). The partially hollow interior structures may lead to a lower particle density and smaller aerodynamic diameter resulting in better dispersibility. Additional FIB-SEM images are shown in figures 8 and 9.
Further examination of the particles by XPS reflected the chemical composition of the surface (Table 5 and Table 7). The carbon oxygen ratio of the spray dried mannitol is consistent with the theoretical value which assumes even distribution of each component in the particles. However, in co-spray dried ICS LABA/mannitoI the carbon/oxygen ratio is higher than the theoretical value (Table 7). The ratios of nitrogen, sulphur and fluorine, which are specific to the drugs (Figure 3), are also higher than theoretical ratios (Table 7).
Compound Element At% Wt%
Mannitol Carbon 54% 47%
C6H1406 Oxygen 46% 53%
Carbon/Oxygen ratio - 0.87
Budesonide Carbon 78% 73%
C25H34O6 Oxygen 22% 27%
Carbon/Oxygen ratio - 2.7
Formoterol fumarate Carbon 73% 68% dihydrate
2[Cl9H2 N204]-C4H404-2H20 Oxygen 21% 26%
Nitrogen 5.6% 6.0%
Carbon/Oxygen ratio - 2.6
Fluticasone propionate Carbon 74% 65%
C25H31F3O5S Oxygen 15% 18%
Sulfur 2.3% 5.4%
Fluorine 8.6% 12%
Carbon/Oxygen ratio - 3.6
Salmeterol xinafoate Carbon 80% 75%
C25H37N04-C, iH803 Oxygen 18% 23%
Nitrogen 2.0% 2.2%
Carbon/Oxygen ratio - 3.3
Table 5 Surface composition of the raw materials
Raw material Element
Wt% in Wt% in Wt% in Expected Wt% in powder raw powder powder material
M-SD Mannitol 100% Carbon 47% 47% Carbon 47%
C6Hi406 . Oxygen 53% 53% Oxygen 53%
Total 100%
BFM- Mannitol 91.76% Carbon 47% 43% Carbon 49%
SD
C6Hi406 Oxygen 53% 49% Oxygen 51%
Budesonide 8% Carbon 73% 5.8% Nitrogen 0.014%
C25H34O6 Oxygen 27% 2.2%
Formoterol 0.24 Carbon 68% 0.16%
fumarate
dihydrate
- Oxygen 26% 0.062%

Nitrogen 6.0% 0.014%
Total 100%
FSM- Mannitol 89% Carbon 47% 42% Carbon 49%
SD
C6H,406 Oxygen 53% 47% Oxygen 49%
Fluticasone 10% Carbon 65% 6.5% Nitrogen 0.22% propionate
C25H31F3O5S Oxygen 18% .1.8% Sulfur 0.54%
Sulfur 5.4% 0.54% Fluorine 1.2%
Fluorine 12% 1.2%
Salmeterol 1% Carbon 75% 0.75%
xinafoate
C25H37N04- Oxygen 23% 0.23%
CHgOs
Nitrogen 2.2% 0.22%
Total 100%
Table 6 Calculation of expected elemental percentages in the spray dried powders
Experimental Expected
Sample Element
At% Wt% Wt%
M-SD Carbon 56% 49% 47%
Oxygen 44% 51% 53%
Carbon/Oxygen ratio 0.94 0.87
B/F/M-SD Carbon 64% 57% 49%
Oxygen 36% 42% 51%
Nitrogen 0.32% 0.33% 0.014%
Carbon/Oxygen ratio 1.4 0.95
F/S/M-SD Carbon 67% 59% 49%
Oxygen 28% 33% 49%
Nitrogen 0.53% 0.55% 0.022%
Sulfur 0.93% 2.2% 0.54%
Fluorine 3.4% 4.7% 1.2%
Carbon/Oxygen ratio 1.8 0.98
Nitrogen/Sulfur ratio 0.25 0.041
Nitrogen/Fluorine ratio 0.12 0.019
Sulfur/Fluorine ratio 0.47 0.45
Table 7 Surface composition of the spray dried powders
Regardless of the surface differences, there is no statistically significant difference in the cohesion force between the three samples measured using AFM with the larger variation of B/F/M-SD compared to the other two (Table 8). The cohesion force of these samples ranged between 15.2 - 20.7 nN.
Spray dried mannitol (M-SD) 20.7±0.9
Co-spray dried budesonide/formoterol fumarate dihydrate/mannitol (B/F/M-SD) 15.2±4.5
Co-spray, dried fluticasone propionate/salmeterol xinafoate/mannitol (F/S/M-SD) 18.7± 1.0 Average ± standard deviation (n=3)
Table 8 Cohesion force DSC curves of B/F/M-SD and F/S/M-SD showed a large melting peak of mannitol at 167 and 169 °C (Figure 4), which is expected as mannitol is the major molar component (> 90 %). There were also smaller endothermic peaks at 243 °C and 249 "C, most likely attributed to the melting of budesonide and fluticasone propionate,
respectively. No thermal events relating to LABA were observed. All three powders had a very low residual solvent content, as indicated by the weight loss of 0.062%, 0.063%, and 0.045% for M-SD, B/F M-SD, and F/S/M-SD, respectively, in the TGA. XRD patterns showed that both B/F/M-SD and F/S/M-SD contained a-mannitol, while M-SD contained β-mannitol (Figure 5). Although ICS contents were approaching the detection limit of XRD, the patterns were able to reveal the major diffraction peaks of budesonide at 2Θ angles of 6.1, 12.0, 15.5 & 16.0 degrees and fluticasone propionate at 10.0, 13.0, 14.8 & 16.2 degrees.
In vitro dispersion study
Both B/F/M-SD and F/S/M-SD. showed concomitant depositions of ICS and LABA within the device, adaptor, throat and the impactor stages, indicating that the drugs followed each other closely in the powder and or aerosol (Figure 6 and Figure 7). LABA remained in the capsule was not quantified because of the overlapping of HPLC peaks with HPMC capsule. All of the three formulations had relatively high FPF values (> 50%) at 60 L min, which was not significantly different to those at 100 L/min. Discussion
The inhalable particle size range and consistent drug contents show the potential applicability of ICS/LABA/mannitol powder for DPI formulations. The consistent drug contents indicate no selective adsorption or loss of the drugs in the spray drying line, nor segregation of the drugs in the powders. The drug content ratios are considered to be constant throughout the whole particle size range based on the concomitant deposition of ICS/LABA shown in the aerosol data (Figure 6 and Figure 7).
The surface projections of ICS/LABA/mannitol particles have well-defined shapes, suggesting that they are crystalline (Figure 1). The different surface features of the B/F/M-SD and F/S/M-SD particles are likely due to separation of the drug components and mannitol by differential solidification in the droplets during spray drying. The larger variation of surface cohesion force in the B/F/M-SD powder might reflect the rougher surface morphology of the particles compared to the other two (Table 8). The results are consistent with previous findings which showed lower force variation with
smoother surfaces. The partially hollow interior structures (Figure 2) would lead to a lower particle density and smaller aerodynamic diameter.
The enrichment of drugs on the particle surface is suggested by the discrepancy between the experimental and theoretical ratios of each element in the XPS results (Table 7). Since the drugs have higher carbon/oxygen ratios than mannitol (Table 5), the higher carbon/oxygen experimental ratio implies at least one of the drugs preferably resides on the surface. This is corroborated by the higher than theoretical ratios of the elements (nitrogen, sulphur and fluorine) specific to the drugs (Figure 3, Table 7). Considering sulphur and fluorine exist only in fluticasone propionate and nitrogen only in salmeterol xinafoate (Figure 3), the six-times higher experimental ratios of nitrogen/sulphur and nitrogen/fluorine (Table 7) than the theoretical values indicate that salmeterol xinafoate resides on the surface more preferably than fluticasone propionate, possibly because of the lower solubility of salmeterol in the ethanol/water system. On the other hand, the consistency between the experimental and theoretical values of the sulphur/fluorine ratio is expected as these two elements are present in the same drug (i.e., fluticasone).
As ICS and LABA are poorly water soluble, while mannitol is water soluble, a separation of components could have occurred as the solvent was evaporating with the ethanol-water ratio changing in the process. Initially all the components were dissolved in the feed solution before spray drying. As drying began, the most volatile component (i.e. alcohol) would evaporate faster from the droplet (until it reached the azeotropic point), forcing the hydrophobic drugs to precipitate first (at least partly) on the surface of the alcohol-deficient droplets and subsequently on the dried particles.
The melting temperatures of budesonide and fluticasone propionate shown by DSC (243 °C and 249 °C, respectively) were reduced in the presence of the large amount of mannitol. On the other hand, thermal events of LABA were not detectable by the DSC due to their low contents (formoterol fumarate dihydrate 0.24 wt % and salmeterol xinafoate 1.0 wt %). These drugs may exist as crystalline or amorphous solids in the particles.
Since the spray drying parameters were similar for all the powders, the different polymorph (a form shown by XRD) of mannitol in the combination formulations compared with the pure mannitol (a form) must be due to the presence of the drugs,
even though their contents were only 8.24% (B/F M-SD) and 11% (F/S/M-SD). Under normal storage conditions, both the a and β forms are considered stable. Furthermore, in corroboration with the SEM and DSC data, the XRD findings confirmed the crystalline nature of ICS in the particles.
The high and similar FPF values of B F M-SD and F/S/M-SD are in accordance with the inhalable particle size range (Table 3) as well as similar AFM force values for the powders (Table 8). The relatively high FPF in the aerosol also showed that the powders were not particularly cohesive, hence a good dispersion could be achieved even at a low air flow rate with minimal inhalation effort. Using a comfortable inspiratory effort would generate 105 L min through the Aerolizer. Increasing the flow from 60 to 100 L/min simply reduces the capsule and device retention by providing more energy to empty the powder from the inhaler, while augmenting the deposition on the throat and Stages 1 and 2 of the impactor by increasing the air velocity for impaction at those sites.
Co-precipitates of fluticasone propionate and salmeterol xinafoate showed a FPF of 22% when formulated with lubricant and 36% with lactose carrier. Previous studies also reported that co-spray dried powder of budesonide and formoterol fumarate dihydrate showed a low emitted dose (19.7-41.7%) and FPF (12.1%) due to low flowability and high adhesiveness of the powder. The ICS/LABA/mannitol co-spray dried powders in the present study are similar to mannitol in their cohesive force (Table 8) and FPF values. Surface roughness of these particles is likely to have contributed to the low cohesiveness and high dispersibiliry. Another contributing factor may be the crystallinity of the mannitol and possibly of the ICSs (as confirmed by DSC & XRD). Compared with previous studies, the inlet temperature of the spray drying process was higher, hence allowing glass transition and subsequent crystallization of the drugs.
The ICS/LABA mannitol system provides an innovative approach for combination formulations at appropriate doses without the need of physical blending. The powders showed high aerosol performance and uniform deposition of the two drugs. Storage stability is an important consideration for product development. Preliminary results showed that compared to the initial values, there was no significant difference (P < 0.05) in the dispersion of the B/F/M-SD powder after storage over silica gel at 22 °C for 11 weeks (FPF at 60 L/min: 53.7 ± 1.5% for formoterol fumarate dihydrate and 53.4 ± 1.7% for budesonide).
Additional Spray Dried Powders
Two further drug solutions were prepared:
1) Salbutamol sulfate / beclomethasone dipropionate / mannitol (S B/M-SD)
2) Mometasone furoate / formoterol furnarate / mannitol (M/F/M-SD)
The drug solutions are shown in Table 9 below and were spray dried using the conditions as those for B/F/M-SD.
Concentration (mg/mL) S/B/M-SD M/F/M-SD
Salbutamol sulfate 0.8
Beclomethasone dipropionate 0.4
Mometasone furoate 0.8
Formoterol furnarate 0.02
Terbutaline sulfate
Budesonide
Mannitol 18.8 19.18
Solvent 50% v/v 50% v/v
Ethanol Ethanol
Table 9 - Drug Solutions
The SEM images of the particles are shown in Figures 10 and 11. The surfaces of S/B/M and M/F/M particles were corrugated. Conclusions
Respirable-sized (D50 of 2 μηι) crystalline mannitol particles containing two drugs with at least one confirmed to be also crystalline were successfully obtained from co-spray drying two different ternary systems containing budesonide/formoterol furnarate dihydrate/mannitol and fluticasone propionate/salmeterol xinafoate/mannitol. When dispersed using an Aerolizer at 60 and 100 IJmin, the powders showed a concomitant
in vitro deposition patterns of ICS LABA with a FPF of 54 - 62 %. The aerosol performance can be due to the low interparticulate force, resulting from a combination of the rough surface and crystalline nature of the particles. The particles of the present invention provide an alternative simple method for effective one-step processing of combination formulations for inhalation.
It will be appreciated by persons skilled in the art that numerous variations and/or modifications may be made to the invention as shown in the specific embodiments without departing from the scope of the invention as broadly described. The present embodiments are, therefore, to be considered in all respects as illustrative and not restrictive.
Claims
1. An inhalable particle comprising two or more active agents and an excipient which is at least partially in crystalline form, wherein the particle is of respirable size.
2. An inhalable particle comprising one or more active agent(s) and an excipient which is at least partially in crystalline form, wherein the particle is of respirable size and is hollow.
3. An inhalable particle of respirable size comprising one or more active agent(s) and an excipient wherein the percentage crystallinity of the particle is greater than the percentage amount of excipient present in the particle.
4. An inhalable particle according to claim 2 or claim 3 comprising two or more active agents.
5. Inhalable particles comprising one or more particles according to any one of claims 1 to 4.
6. Particles according to claim 5 comprising at least 1%, at least 2%, at least 5%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, at least 98%, at least 99%, or 100% of the particles according to any one of claims 1 to 4.
7. Particle(s) according to any preceding claim wherein the excipient is present in a majority crystalline form.
8. Particle(s) according to any preceding claim wherein the excipient is selected from sugars and sugar alcohols (including mannitol, sucrose, glucose, lactose, dextrose, sorbitol, maltilol, maltodextrin); amino acids (including glycine, leucine, trileucine, arginine, threonine, phenylalanine, aspartic acid); and other excipients such as sodium chloride, poly-lactic glycolic acid or poly ethylene glycol.
9. Particle(s) according to any preceding claim wherein the excipient is mannitol.
10. Particle(s) according to any preceding claim wherein the excipient is present in about 10%, about 20%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, about 95%, or about 100% crystalline form.
5 11. Particle(s) according to any preceding claim wherein the at least one active agent is present in about 1%, about 2%, about 3%, about 4%, about 5%, about 6%, about 7%, about 8%, about 9%, about 10%, about 20%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, or about 100% crystalline form.
10 12. Particle(s) according to any preceding claim wherein the particles comprise about 50% excipient, about 60% excipient, about 70% excipient, about 80% excipient, about 90% excipient, about 95% excipient, or about 99% excipient.
13. Particles according to any one of claims 5 to 10 wherein the average 15 aerodynamic diameter of the particles is between from about 0.01 to about 20 microns, or between between from about 0.1 to about 20 microns, about 0.1 to about 15 microns, about 0.2 to about 15 microns, about 0.2 to about 10 microns, about 0.5 to about 10 microns, about 0.8 to about 10 microns, about 1 to about 10 microns, about 1 to about 9 microns, about 1 to about 8 microns, about ί to about 7 microns, about 1 to about 6 20 microns, about 1 to about 5 microns, about 1 to about 4 microns, about 1 to about 3 microns, or from about 1 to about 2 microns.
14. Particles according to any one of claims 5 to 1 1 wherein about 1% of the particles (do.oi), about 5% of the particles (do.os), about 10% (do.i), about 20% (do.2),
25 about 30% (doj), about 40% (do.4), about 50% (do.5), about 60% (doe), about 70% (do.7), about 80% (do g), about 90% (do.9), about 95% (do.95), about 98% (do.98), about 99% (do.99), or about 100% (di) of the particles are of respirable size.
15. Particles according to any one of claims 5 to 12 wherein the fine particle 30 fraction (FPF) is > about 10%, > about 20%, > about 30%, > about 40%, > about 50%,
> about 60%, > about 70%, > about 80%, > about 90%, > about 95%, or about 100%.
16. Particle(s) according to any preceding claim wherein:
- the active agents and the excipient are evenly mixed throughout the 35 particle; - one active agent is predominantly present on and/or near the surface of the particle;
- two or more actives are predominantly present on and/or near the surface of the particle;
5 - one active agent is predominantly present in and/or near the interior of the particle; or
two or more actives are predominantly present in and/or near the interior of the particle.
10 17. Particle(s) according to any preceding claim wherein the particle(s) are:
predominantly spherical;
substantially ovoid;
substantially ellipsoidal;
predominantly plate like or flaked;
15 predominantly pyramidal;
predominantly needle or fibre shaped;
spiky;
irregularly shaped; or
have a substantially uniform shape.
20
18. Particle(s) according to any preceding claim wherein the surface of the particle(s) are:
substantially uniform;
dimpled;
25 contain crystals shaped particles adhered to the surface;
contain a clay like material;
contain plate like or flaked particles adhered to the surface; smooth;
roughened;
30 spiky and/or
corrugated.
19. Particle(s) according to any preceding claim wherein the particle(s) are hollow.
35 20. Particle(s) according to any preceding claim wherein the active agents are selected from beta-2 agonists, anticholinergics, mast cell stabilisers, steroids, 1305617 1.doc methylxanthines, inhaled corticosteroids, theophylline, leukotriene modifiers long- acting beta-2 agonists, short-acting beta-2 agonists and or systemic corticosteroids.
21. Particles according to any preceding claim wherein the active agents are selected 5 from an inhaled corticosteroid (ICS) and a long-acting 2-agonist (LABA), including:
formoterol and budesonide;
salmeterol and fluticasone;
salbutamol and beclomethasone;
mometasone furoate/formoterol fumarate;
10 mometasone furoate and salmeterol xinafoate;
beclomethasone dipropionate and formoterol fumarate;
beclomethasone dipropionate and salmeterol xinafoate;
fluticasone propionate and formoterol fumarate;
budesonide and salmeterol xinafoate;
15 salbutamol sulfate and fluticasone propionate;
salbutamol sulfate and budesonide;
terbutaline sulfate and beclomethasone dipropionate;
terbutaline sulfate and fluticasone propionate; or
terbutaline sulfate and budesonide.
20
22. Particles according to any preceding claim wherein the particles have been formed by spray drying.
23. A dry powder inhaler (DPI) containing particles as claimed in any of claims 5 to 25 22.
24. A method of delivering a combination of two or more active agents to a patient in need thereof comprising administering particles as defined in any one of claims 5 to 22.
30
25. A method of treating a patient having a respiratory or non-respiratory condition, said method comprising administering particles as defined in any one of claims 5 to 22.
26. A method according to claim 25 wherein the respiratory or non-respiratory 35 conditions are selected from: Respiratory conditions including: COPD, bronchitis, allergy, rhinitis, cystic fibrosis, pulmonary infection, tuberculosis, influenza, other lung infections, lung cancer and asthma; or
Non-respiratory conditions including: diabetes, hypertension, 5 hypercholesterolaemia, gout, infections (bacterial or viral), fever, pain
(neurological or muscular).
27. Particles according to any one of claims 5 to 22 for use in the treatment of a respiratory or non-respiratory condition.
10
28. Use of particles as defined in any one of claims 5 to 22 for the manufacture of a medicament for the treatment of a respiratory or non-respiratory condition.
29. A method of delivering a medicament to a patient in need thereof comprising 15 administering particles as defined in any one of claims 5 to 22.
30. A pharmaceutical composition comprising particles according to any one of claims 5 to 22.
20 31. Particles according to any one of claims 5 to 22 for use as a medicament.
32. Use of particles according to any one of claims 5 to 22 as a medicament.
33. Particles according to any one of claims 5 to 22 for use in the treatment of 25 respiratory or non-respiratory conditions.
34. Use of particles according to any one of claims 5 to 22 in the treatment of respiratory or non-respiratory conditions.
30 35. Particles according to claim 33 or use according to claim 34 wherein the respiratory or non-respiratory conditions are selected from:
Respiratory conditions including: COPD, bronchitis, allergy, rhinitis, cystic fibrosis, pulmonary infection, tuberculosis, influenza, other lung infections, lung cancer and asthma; or Non-respiratory conditions including: diabetes, hypertension, hypercholesterolaemia, gout, infections (bacterial or viral), fever, pain (neurological or muscular).
36. Particle, particles, compositions, uses or methods substantially as hereinbefore described with reference to the accompanying examples and/or figures, excluding comparative examples if present.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2010800633023A CN102811715A (en) | 2009-12-08 | 2010-12-08 | Inhalable Formulations |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2009905978 | 2009-12-08 | ||
| AU2009905978A AU2009905978A0 (en) | 2009-12-08 | Inhalable formulations |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011069197A1 true WO2011069197A1 (en) | 2011-06-16 |
Family
ID=44145030
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/AU2010/001656 WO2011069197A1 (en) | 2009-12-08 | 2010-12-08 | Inhalable formulations |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN102811715A (en) |
| WO (1) | WO2011069197A1 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8765725B2 (en) | 2012-05-08 | 2014-07-01 | Aciex Therapeutics, Inc. | Preparations of hydrophobic therapeutic agents, methods of manufacture and use thereof |
| WO2016090260A1 (en) | 2014-12-04 | 2016-06-09 | Microdose Therapeutx, Inc. | Inhalation monitoring system and method |
| US9815865B2 (en) | 2013-01-07 | 2017-11-14 | Nicox Ophthalmics, Inc. | Preparations of hydrophobic therapeutic agents, methods of manufacture and use thereof |
| US10174071B2 (en) | 2012-05-08 | 2019-01-08 | Nicox Ophthalmics, Inc. | Preparations of hydrophobic therapeutic agents, methods of manufacture and use thereof |
| WO2019060595A1 (en) | 2017-09-20 | 2019-03-28 | Teva Branded Pharmaceutical Products R&D, Inc. | Dry powder inhalable medicament comprising glycopyrronium |
| WO2020020957A1 (en) * | 2018-07-27 | 2020-01-30 | Chiesi Farmaceutici S.P.A. | Novel carrier particles for dry powder formulations for inhalation |
| WO2020022975A3 (en) * | 2017-12-29 | 2020-02-27 | Neutec Ar-Ge Sanayi Ve Ticaret Anonim Sirketi | New pharmaceutical compositions in the treatment of copd |
| EP3288541B1 (en) * | 2015-05-01 | 2020-09-02 | Board of Regents, The University of Texas System | Multidrug brittle matrix compositions |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ITMI20130571A1 (en) * | 2013-04-10 | 2014-10-11 | Zambon Spa | PHARMACEUTICAL COMPOSITION CONTAINING BUDESONIDE AND FORMOTEROL |
| MY191712A (en) * | 2014-10-08 | 2022-07-09 | Zambon Spa | Composition comprising at least one dry powder obtained by spray drying to increase the stability of the formulation |
| CN111658661B (en) * | 2020-06-12 | 2021-06-01 | 深圳大佛药业股份有限公司 | Salbutamol sulfate inhalation preparation and preparation method thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008134817A1 (en) * | 2007-05-03 | 2008-11-13 | The University Of Sydney | Composite carriers for dry powder inhalation therapy |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001517692A (en) * | 1997-09-29 | 2001-10-09 | インヘール セラピューティック システムズ, インコーポレイテッド | Stabilized preparation for use in a nebulizer |
| WO2002015880A2 (en) * | 2000-08-25 | 2002-02-28 | Merck Patent Gmbh | Powdered mannitol and mannitol-containing compositions |
-
2010
- 2010-12-08 WO PCT/AU2010/001656 patent/WO2011069197A1/en active Application Filing
- 2010-12-08 CN CN2010800633023A patent/CN102811715A/en active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008134817A1 (en) * | 2007-05-03 | 2008-11-13 | The University Of Sydney | Composite carriers for dry powder inhalation therapy |
Non-Patent Citations (2)
| Title |
|---|
| ELVERSSON, J. ET AL.: "Particle Size and Density in Spray Drying - Effects of Carbohydrate Properties", JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 94, no. 9, September 2005 (2005-09-01), pages 2049 - 2060, XP002674366, DOI: doi:10.1002/JPS.20418 * |
| STECKEL, H. ET AL.: "A novel spray-drying technique to produce low density particles for pulmonary delivery", INTERNATIONAL JOURNAL OF PHARMACEUTICS, vol. 278, 2004, pages 187 - 195, XP055121369, DOI: doi:10.1016/j.ijpharm.2004.03.010 * |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10954263B2 (en) | 2012-05-08 | 2021-03-23 | Nicox Ophthalmics, Inc | Preparations of hydrophobic therapeutic agents, methods of manufacture and use thereof |
| US9822142B2 (en) | 2012-05-08 | 2017-11-21 | Nicox Ophthalmics, Inc. | Preparations of hydrophobic therapeutic agents, methods of manufacture and use thereof |
| US10174071B2 (en) | 2012-05-08 | 2019-01-08 | Nicox Ophthalmics, Inc. | Preparations of hydrophobic therapeutic agents, methods of manufacture and use thereof |
| US8765725B2 (en) | 2012-05-08 | 2014-07-01 | Aciex Therapeutics, Inc. | Preparations of hydrophobic therapeutic agents, methods of manufacture and use thereof |
| US9815865B2 (en) | 2013-01-07 | 2017-11-14 | Nicox Ophthalmics, Inc. | Preparations of hydrophobic therapeutic agents, methods of manufacture and use thereof |
| WO2016090260A1 (en) | 2014-12-04 | 2016-06-09 | Microdose Therapeutx, Inc. | Inhalation monitoring system and method |
| EP3583899A1 (en) | 2014-12-04 | 2019-12-25 | Norton (Waterford) Limited | Inhalation monitoring system and method |
| EP3838319A1 (en) | 2014-12-04 | 2021-06-23 | Norton (Waterford) Limited | Inhalation monitoring system and method |
| AU2015393953B2 (en) * | 2015-05-01 | 2021-09-02 | Board Of Regents, The University Of Texas System | Multidrug brittle matrix compositions |
| EP3288541B1 (en) * | 2015-05-01 | 2020-09-02 | Board of Regents, The University of Texas System | Multidrug brittle matrix compositions |
| WO2019060595A1 (en) | 2017-09-20 | 2019-03-28 | Teva Branded Pharmaceutical Products R&D, Inc. | Dry powder inhalable medicament comprising glycopyrronium |
| WO2020022975A3 (en) * | 2017-12-29 | 2020-02-27 | Neutec Ar-Ge Sanayi Ve Ticaret Anonim Sirketi | New pharmaceutical compositions in the treatment of copd |
| WO2020020957A1 (en) * | 2018-07-27 | 2020-01-30 | Chiesi Farmaceutici S.P.A. | Novel carrier particles for dry powder formulations for inhalation |
| JP2021531948A (en) * | 2018-07-27 | 2021-11-25 | シエシー ファルマセウティチィ ソシエタ ペル アチオニ | New carrier particles for dry powder formulations for inhalation |
| US11813360B2 (en) | 2018-07-27 | 2023-11-14 | Chiesi Farmaceutici S.P.A. | Carrier particles for dry powder formulations for inhalation |
| JP7439084B2 (en) | 2018-07-27 | 2024-02-27 | シエシー ファルマセウティチィ ソシエタ ペル アチオニ | Novel carrier particles for dry powder formulations for inhalation |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102811715A (en) | 2012-12-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2011069197A1 (en) | Inhalable formulations | |
| JP6082049B2 (en) | Dry powder formulation of particles containing two or more active ingredients for the treatment of obstructive or inflammatory airway diseases | |
| JP5936353B2 (en) | Method for improving the crystallinity of fluticasone particles | |
| AU2017203258B2 (en) | Respirable agglomerates of porous carrier particles and micronized drug | |
| ES2368482T3 (en) | PHARMACEUTICAL FORMULATIONS FOR DRY POWDER INHALERS. | |
| ES2275669T3 (en) | FORMULATIONS TO USE IN INHALING DEVICES. | |
| KR102285066B1 (en) | Inhalation particles comprising a combination of an anticholinergic, a corticosteroid and a beta-adrenergic | |
| JP2010500356A (en) | Method for producing lactose | |
| KR20180050320A (en) | Targeted delivery of the spray-dried formulation to the lungs | |
| Westmeier et al. | Combination particles containing salmeterol xinafoate and fluticasone propionate: Formulation and aerodynamic assessment | |
| WO2016176552A1 (en) | Dry power formulations for inhalation | |
| JPWO2003077891A1 (en) | Powder pharmaceutical composition for inhalation and method for producing the same | |
| WO2008134817A1 (en) | Composite carriers for dry powder inhalation therapy | |
| JP2012524817A (en) | Aggregate formulation comprising an active pharmaceutical agent having a target particle size |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080063302.3 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10835297 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10835297 Country of ref document: EP Kind code of ref document: A1 |